




Controllable activation of nanoscale dynamics in a disordered protein alters binding kinetics
David JE Callaway
Tsutomu Matsui
Thomas Weiss
Laura R Stingaciu
Christopher B Stanley
William T Heller
Zimei Bu
Corresponding Authors: David J E Callawaydcallaway@ccny.cuny.edu. Zimei Buzbu@ccny.cuny.edu
Issue date 2017 Apr 7.
Abstract
Phosphorylation of S339/S340 in the disordered tail of the multi-domain scaffolding protein NHERF1 affects the intracellular localization and trafficking of NHERF1 assembled signaling complexes. Using neutron spin echo spectroscopy (NSE), we show salt concentration dependent excitation of nanoscale motion at the tip of the C-terminal tail in the phosphomimic S339D/S340D mutant. The “tip of the whip” that is unleashed is near the S339/S340 phosphorylation site and flanks the hydrophobic Ezrin-binding motif. Further, we show that the kinetic association rate constant of the binding of the S339D/S340D mutant to the FERM domain of Ezrin is sensitive to buffer salt concentration, correlating with the excited nanoscale dynamics. The results demonstrate that nanoscale dynamics of an intrinsically disordered protein influences the binding kinetics of signaling partners. NSE can pinpoint the nanoscale dynamics changes in a highly specific manner.
Keywords: nanoscale protein motion, disordered protein, protein binding kinetics, neutron spin echo spectroscopy, protein dynamics
Graphical abstract
INTRODUCTION
A large number of cellular signaling proteins are disordered or contain long stretches of disordered regions under native conditions [1–4]. Phosphorylation in such disordered regions plays vital roles in protein-protein interactions and in cell signal transduction [5–7]. Phosphorylation modulates the interactions of signaling partners by direct recognition or by acting as a conformational switch to affect the active site [8,9]. However, the effects of phosphorylation on protein dynamics is largely unknown. Here we report that phosphomimetic mutation in the disordered C-terminal tail of the multidomain scaffolding protein NHERF1 affects nanoscale protein dynamics, which correlates with the change in binding kinetics.
The scaffolding NHERF1 participates in regulating the cell surface localization, signaling complex assembly, and endocytosis of a number of physiologically important transmembrane proteins in epithelial cells [10]. The transmembrane proteins that interacts with NHERF1 include ion transporters, G-protein coupled receptors, cell adhesion molecules, and tyrosine kinase receptors [11–15]. NHERF1 is also necessary for assembling microvillus structure in epithelial cells [16–18]. NHERF1 knockout mice have defective auditory hair cell structure and exhibit impaired hearing [19].
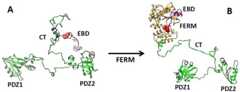
NHERF1 contains two modular PDZ domains and a C-terminal tail, seeFig. 1 [20–22]. The two PDZ domains of NHERF1, which are connected by a flexible linker, bind to a PDZ-binding motif residing in the cytoplasmic tails of transmembrane receptors or channels. The C-terminal end of NHERF1 possesses a 14-residue Ezrin-binding domain (EBD) that binds to the FERM domain of Ezrin [23–25], hence NHERF1 is also called Ezrin binding protein 50, EBP50. Ezrin is a member of the Ezrin-Radixin-Moesin family of membrane-cytoskeleton adapter proteins that link the cell membrane components to the actin cytoskeleton network [10]. Upon activation, the N-terminal FERM domain of Ezrin binds to NHERF1 while the C-terminal tail binds to cytoskeletal actin filaments [26,27]. Previous studies show that Ezrin allosterically modulates NHERF1 to assemble membrane signaling complexes [20,28,29]. Together, NHERF1 and Ezrin dynamically link membrane proteins to the cytoskeletal actin network, which regulates the intracellular trafficking, assembly, and signaling of membrane protein complexes [15–17,30–33].
Figure 1.

Domain organization of NHERF1. The full-length protein consists of PDZ1, PDZ2, disordered C-terminal tail (CT), EBD (magenta), and S339/S340 (red).Shown is the activation of “tip of whip” motion in the phosphomimic mutant revealed by NSE. The graphics were generated using the program UCSF Chimera [71].
Previous NMR study shows that the long C-terminal tail of NHERF1, including the EBD, is largely disordered, and that EBD fluctuates between disordered and transient helical conformations, seeFig. 1 [22]. Crystallography studies show that upon binding to FERM, EBD adopts a helical conformation and docks to the FERM domain with high affinity [24,25]. EBD thus undergoes a disorder to order transition upon binding to FERM, likely through both a conformation selection and induced fit mechanisms [4].
The disordered C-terminal tail of NHERF1 contains several serine residues that can be phosphorylated in cells [34–36]. In particular, PKC phosphorylates S339 and S340 [21,37]. Phosphorylation at S339/S340 modulates the ability of NHERF1 to assemble several signaling complexes, and affects the localization, mobility and intracellular trafficking of NHERF1 and NHERF1 assembled transmembrane proteins [16,30,31,38]. S339 and S340 are located N-terminal to the EBD flanking the hydrophobic motif that docks to the FERM domain of ERM proteins [25]. However, it remains a puzzle as how phosphorylation affects the molecular function of NHERF1, and specifically its dynamics.
Here using neutron spin echo spectroscopy (NSE), small angle X-ray and neutron scattering, and surface plasmon resonance, we show that the phosphomimic mutation S339D/S340D changes the nanoscale dynamics in NHERF1, and alters the kinetics of NHERF1 in assembling complexes. NSE measures macromolecular dynamics on nanosecond-to-microsecond timescales and on nanometer length scales, thus the name nanoscale dynamics. Nanoscale protein motion is a dynamic regime that is difficult to access by experimental methods. NSE fills an important spatial-temporal dynamic niche to determine the dynamics of proteins and protein complexes [39]. With the aid of theoretical analysis, we demonstrate that NSE is highly sensitive to the nanoscale dynamics change caused by the phosphomimetic mutation and buffer salt concentration. We explicitly show, by NSE, the specific section of the protein that is activated by phosphorylation and controlled by salt concentration. This is the first experimental demonstration of a nanoscale motion that is directly controllable by phosphomimic mutation and salt concentration.
RESULTS
Effects of phosphomimic mutation on NHERF1 conformation
In order to clarify if NHERF1 has an oligomer fraction in solution, we have performed online size-exclusion chromatography small angle X-ray scattering (SEC-SAXS) to analyze the conformation of wild-type NHERF1(wt) and the phosphomimic NHERF1(S339D/S340D). In SEC-SAXS, scattering is performed immediately after the protein is eluted out of a fast protein liquid chromatography size exclusion column. SEC-SAXS is thus more able to detect the presence of oligomers or aggregates in a protein solution than a classical SAXS experiment. As shown inFig. 2A, there is only one peak eluted from the SEC column, and the radius of gyration Rg of NHERF1(wt) remains unchanged over the SEC peak, suggesting the absence of oligomerization or intermolecular interference effects. This result confirms our previous findings that NHERF1(wt) is monomeric [20,28]. Rg of NHERF1(S339D/S340D) decreases slightly at the peak tail position as compared to the peak front, seeFig. 2B, suggesting the presence of marginally attractive intermolecular interference effects in NHERF1(S339D/S340D) at high concentrations. Comparing Rg, the length distribution function P(r), and the maximum dimension Dmax obtained from the SEC-SAXS data at the post-peak position indicates that NHERF1(S339D/S340D) is slightly more expanded than NHERF1(wt), seeTable 1 andFig. 2C&2D.
Figure 2.

SEC-SAXS results of NHERF1(wt) and NHERF1(DM) in 150 mM NaCl, 20 mM Tris-HCl, pH=7.5, 5 mM DTT, 0.5 mM EDTA. I(0) and Rg vs. image number (proportional to elution time) of (A) NHERF1(wt) and (B) NHERF1(S339D/S340D). (C) SAXS profile of NHERF1(wt) (filled black square) and NHERF1(S339D/S340D) (open circle) collected after the SEC peak. Lines are fit when computing P(r). (D) P(r) functions of NHERF1(wt) (black) and NHERF1(S339D/S340D) (red).
Table 1.
Rg and Dmax of NHERF1(wt) and NHERF1(S339D/S340D) from SAXS and SANS.
| Rg (Å) | Dmax (Å) | |||
|---|---|---|---|---|
| 300 mM NaCl | 150 mM NaCl | 300 mM NaCl | 150 mM NaCl | |
| NHERF1(wt) (SEC-SAXS) | 40.4±0.4 | 135±5 | ||
| NHERF1(DM) (SEC-SAXS) | 42.0±0.3 Å | 145±5 | ||
| dNHERF1(wt) (SANS) | 42.2±0.7 Å | 145±5 | ||
| dNHERF1(DM) (SANS) | 43.2±0.5 Å | 44.6±0.9 Å | 150±5 | 155±5 |
The small angle neutron scattering (SANS) experiments performed on deuterateddNHERF1(wt) anddNHERF1(S339D/S340D) in H2O buffer show similar results as SEC-SAXS, seeFig. 3 andTable 1. Rg and Dmax of thedNHERF1(S339D/S340D) are slightly larger than those of the deuterateddNHERF1(wt), seeFig. 3B. Together, SEC-SAXS and SANS show that, although the conformational changes are detectable within experimental error, the effects of phosphomimic mutation on the ensemble averaged Rg, Dmax, and P(r) function are not dramatic.
Figure 3.

SANS results of deuterateddNHERF1(wt) anddNHERF1(S339D/S340D) in H2O buffer. (A) I(Q) vs. Q profile. Lines are fit when computing P(r). (B) P(r) function. The SANS data were collected in 20 mM Tris-HCl, 1 mM DTT, 0.5 mM EDTA at the 150 and 300 mM NaCl concentrations specified in the graph.
Phosphomimic mutation excites nanoscale dynamics at the tip of the disordered tail of NHERF1
We have performed neutron spin echo spectroscopy (NSE) experiments to determine the nanoscale dynamics of NHERF1(S339D/S340D) on 1–70 nanosecond time scales and in Q range 0.05–0.175 Å−1. The NSE experiments were performed at the physiological salt concentration 150 mM NaCl, and 300 mM NaCl, respectively, seeFig. 4A&4B for the NSE spectra. The Deff(Q) values, shown inFig. 4C, are obtained from the NSE spectra andEq. 1.
Figure 4.

NSE spectra at different Q values for NHERF1(S339D/S340D) in (A) 150 mM NaCl, and (B) 150 mM NaCl, 20 mM dTris D2O (pD=7.5) buffer solution. The lines are a single exponential fit to the spectra. (C) Deff(Q) as a function of Q for NHERF1(S339D/S340D) in 300 mM NaCl (Black) and 150 mM NaCl (red) solution. Open black circles are the center-of-mass diffusion constant Do. The black lines are Deff(Q) calculated assuming rigid-body diffusion. Red lines are calculated Deff(Q) assuming motion at the “tip of whip” shown inFig. 1.
Notably, Deff(Q) of NHERF1(S339D/S340D) in 150 mM NaCl is higher than that in 300 mM NaCl solution, suggesting the activation of internal motion in the phosphomimic mutant at lower buffer salt concentration. Deff(Q) of NHERF1(S339D/S340D) in 300 mM NaCl is comparable to that of NHERF1(wt). We previously have shown that Deff(Q) of NHERF1(wt) is of that of a rigid-body motion with no internal motion [29]. The Deff(Q) data reveals activated nanoscale motion in the phosphomimic mutant as compared to the rigid wild-type protein, and shows that nanoscale motion in the phosphomimic mutant is highly sensitive to salt concentration.
We have developed a theoretical framework to analyze the NSE data (Eq.2), and have shown how to extract protein nanoscale dynamics in a highly sensitive way from NSE data by measuring the effective diffusion constant Deff(Q) and comparing it with theoretical calculations [29,40]. The analysis requires structural coordinates of the full-length protein. NHERF1 is a multidomain protein with a significantly disordered C-terminal tail and two PDZ domains connected by a flexible linker. There is no high-resolution structure of the full-length NHERF1. We have determined the atomic structures of the individual PDZ1, PDZ2, as well as the disordered C-tail using high-resolution NMR [41]. With the available SANS data and atomic coordinates of the PDZ domains, structural models of the full-length NHERF1(wt) or NHERF1(S339D/S340D) are first generated using the program EOM [42]. Atoms were then added to the Cα-carbon in the disordered linker and C-terminal tail using the program SABBAC [43]. The generated structural models are then refined against the SANS data by back calculating the scattering curves, seeFig. S1. Those calculated scattering profiles that have a good fit to the experimental SANS data (χ2 ≤1) were selected to compute Deff(Q). Such a generated all-atomic structural model represents an ensemble-averaged conformation of the full-length NHERF1(wt) or NHERF1(S339D/S340D).
The center-of-mass diffusion constant Do of NHERF1(wt) measured by pulsed field NMR in 150 mM NaCl D2O buffer is Do=2.4 Å2/ns [29]. Dynamic light scattering shows that the center-of-mass diffusion constants of NHERF1(wt) and NHERF1(S339D/S340D) in H2O buffer are the same within experimental error [21]. The Do=2.4 Å2/ns value was thus used in our calculation of Deff(Q) of both NHERF1(wt) and NHERF1(S339D/S340D). With the available all-atomic structural models of NHERF1(wt) and NHERF1(S339D/S340D) and Do, we were able to compute Deff(Q).
Comparing our calculations with the experimental NSE data shows that Deff(Q) of NHERF1(S339D/S340D) in 300 mM NaCl is that of rigid-body motion, similar to that of NHERF1(wt) in 150 mM NaCl, seeFig. 4C. However, in 150 mM NaCl, Deff(Q) of NHERF1(S339D/S340D) cannot be described as rigid-body motion, seeFig. 4C. Our calculations indicate that unleashing the “tip of the whip” near the position of S339/S340 and allowing EBD to be a separate body can best describe the experimental Deff(Q) of NHERF1(S339D/S340D) at 150 mM NaCl. Incorporating motion in other parts of the protein, such as in the linker between PDZ1 and PDZ2 or in the N-terminal portion of the C-tail, does not produce theoretical Deff(Q) values that are in agreement with experimental value, seeFig. S2. Thus, NSE analysis via Deff(Q) can specifically pinpoint the location of the internal motion in NHERF1 that is activated by phosphomimic mutation.
Phosphomimic mutation and salt concentration affect the kinetics of NHERF1 binding to FERM, which cannot be explained by electrostatic forces alone
Using surface plasmon resonance (SPR), we have determined the affinity and the kinetics of NHERF1 binding to FERM at various NaCl concentrations while keeping other buffer compositions unchanged.Fig. 5 shows that the equilibrium dissociation constant Kd of NHERF1(wt) is in general smaller than that of NHERF1(S339D/S340D) at different salt concentrations. For example, for NHERF1(wt) at I=0.16 (in 150 mM NaCl buffer), Kd=51±5 nM [20]. At I=0.31 (in 300 mM NaCl buffer), Kd of NHERF1(wt) increases modestly by 2.3 fold as compared to that at I=0.16. Kd of NHERF1(S339D/S340D) increases 6.3 fold at I=0.31 as compared to that at I=0.16. The result show that Kd of NHERF1(wt) is less dependent on the buffer ionic strength I than that of the NHERF1(S339D/S340D) mutant.
Figure 5.

Equilibrium dissociation constant Kd of NHERF1(wt) (black symbols) and NHERF1(S339D/S340D) (red symbols) binding to FERM at different ionic strength I from SPR. The different symbols are from different sets of SPR measurements of different ligand immobilization levels. Representative sensorgrams used to determine Kd are shown inFig. S3.
The kinetics of a protein-protein interaction can be obtained by analyzing the shape of the SPR sensorgrams [44–46]. The response curves of the binding of NHERF1(wt) or NHERF1(S339D/S340D) to FERM can better be fit by a two-state kinetic model (Eq.3) than a single state 1:1 binding model or any other binding models, seeFig. S4. At 150 mM NaCl (i.e. at ionic strength I=0.16M), ka1 is in the order of 106 M−1 s−1 for both NHERF1(wt) and NHERF1(S339D/S340D), seeFig. 6A. The ka1 values of NHERF1 to FERM are comparable to those of other intrinsically disordered proteins measured by solution-based kinetic experimental methods [47–49]. For both NHERF1(wt) and NHERF1(S339D/S340D), the dissociation rate constant kd1 is essentially independent of salt concentration, seeFig. S6A.
Figure 6.

(A) Kinetic association rate constant ka1 of NHERF1(wt) (black) and NHERF1(S339D/S340D) (red) binding to FERM from SPR. The different symbols are from different sets of SPR measurements of different ligand immobilization levels. Typical sensorgrams are shown inFig. S4. (B) logka1 vs. logγ±, where logγ± is calculated from the Debye–Hückel equation (Eq. 4).
Fig. 6A shows the ionic strength dependence of ka1 for NHERF1(wt) and NHERF1(S339D/S340D) to FERM binding, respectively. Notably, ka1 of the NHERF1(S339D/S340D) mutant is more subject to buffer ionic strength changes than the wild-type protein. However, ka1 of the wild-type NHERF1 is not as sensitive to the buffer ionic strengths as the S339D/S340D mutant. For instance, at I=0.31M, ka1 of the mutant reduces 13 fold as compared to that at I=0.16M. At I=0.16M, ka1 of NHERF1(S339D/S340D) is similar to that of NHERF1(wt). In the entire range of buffer ionic strengths measured, ka1 of NHERF1(S339D/S340D) decreases about two order of magnitude from low to high ionic strength, while that of NHERF1(wt) decrease about 5.6 fold.
Following the analysis of Schreiber and Fersht in determining the electrostatic force dependent association rate constant of barnase with barstar [50],Fig. 6B plots logka1 as a function of logγ± of NHERF1(wt) and NHERF1(S339D/S340D) to FERM binding, where γ± is the mean activity coefficient of the buffer, and logγ± is given by the Debye–Hückel equation, seeEq. 4. A linear dependence of logka1 on logγ± suggests that the association rate constants decrease with increasing ionic strength. NHERF1(wt) and NHERF1(S339D/S340D) have different slopes and intercepts. The larger slope suggests that the ka1 of NHERF1(S339D/S340D) is more susceptible to the buffer ionic strength changes than the wild-type protein.
If electrostatic interactions are the only factor that affects ka1, logka1 vs logγ± inFig. 6B will have different slopes but converge to the same value when extrapolating to logγ±=−0.277 [50,51]. According to the Debye–Hückel relationship (Eq.4), logγ± approaches the asymptotic value −0.277 when the ionic strength I is infinitely large. At infinitely large ionic strength I, all the electrostatic charges are screened. Thus, at logγ±=−0.277, the association rate constant approaches the basal value ka0 in the absence of electrostatic force. For the interaction of barnase and barstar in which electrostatic force is the predominant factor that influences the binding kinetics, the basal association rate constant ka0 converge at logγ±=−0.277 [50,51].
However, for NHERF1(wt) and NHERF1(S339D/S340D), logka1 does not converge at the asymptote logγ±=−0.277. A linear fit inFig. 6B gives logka10=4.70 and a basal rate constant ka10=5.06×104 1/Ms for NHERF1(wt) at logγ± = −0.277, whereas for NHERF1(S339D/S340D) logka10=1.68 and ka10=47.9 1/MS. Thus, the Debye–Hückel analysis implies that the change in association rate constant at different ionic strengths cannot be explained by electrostatic effects alone. This results correlates with our NSE results that indicate significant changes in nanoscale dynamics of NHERF1(S339D/S340D) as we lowered the buffer salt concentration.
Analyzing the crystal structures of FERM in complex to the last 14 EBD residues of the NHERF1 C-terminal domain suggests that the complex predominantly utilizes hydrophobic interactions for high affinity binding [24,25], seeFig. S5. The structures of the complex thus explain why the binding kinetics of NHERF1(wt) to FERM is not sensitive to salt concentration. Further, in the structure of the complex, S339 and S340 are located in the loop flanking the hydrophobic binding motif. S339/S340 are also in proximity to the negatively charged surface of the FERM domain, seeFig S5. The phosphomimic mutation introduces negative electrostatic charges, which should have generated repulsive interactions with the negatively charged surface of FERM, and thus reduce ka1 at low salt concentrations. However, such repulsive effects are likely countered by the increased nanoscale motions in the phosphomimic mutant, which allow NHERF1(S339D/S340D) to increase ka1 at low salt concentration. At higher salt concentration of 300 mM NaCl, the electrostatic interactions are screened, and the reduced nanoscale motion in NHERF1(S339D/S340D) is largely responsible for the reduced ka1 to bind FERM. Thus, for NHERF1(S339D/S340D), the sensitivity of ka1 to the salt concentration correlates with the change in nanoscale motion in the mutant.
For both NHERF1(wt) and NHERF1(S339D/S340D), the general trend of ka2 is to decrease with increasing ionic strength (Fig. S6B), whereas the effects on kd2 of are modest (Fig. S6C). The origin of the slower kinetic binding mode ka2 or kd2 is likely due to the large conformational changes in NHERF1 after the engagement of NHERF1(wt) or NHERF1(S339D/S340D) with FERM. Our previous contrast variation SANS studies indicate that NHERF1(wt) undergoes significant conformational changes upon binding to FERM, with significant rearrangement of the two PDZ domains and the opening of the disordered C-terminal tail [28,29]. Alternatively, the slow binding mode may be due to a subsequent binding interaction, as implicated in the crystal structure of a 31-residue tail (a.a. 321–358) of NHERF1 bound to the Moesin FERM domain [24]. This crystal structure shows that residue 321–330 are in contact with the F2 subdomain of FERM besides the last 14 C-terminal residues that bind tightly to the F3 subdomain. The S339 and S340 phosphorylation sites are located in the loop between the two binding patches.
DISCUSSION
The NSE experiments and our theoretical calculations reveal the excitation of a “tip of the whip” motion in the disordered tail of NHERF1, which depends on phosphomimic mutation and salt concentration. The internal dynamics changes can be precisely located by comparing the NSE experimental data with our theoretical calculations for various lengths of the “tip of the whip”. Combining NSE experiments and theoretical calculations thus allows us to specifically identify, in a very sensitive fashion, which region becomes activated by the phosphomimic mutation.
The phosphomimic mutation becomes a rigid body once again when we increase the salt concentration to 300 mM NaCl, which reduces the electrostatic effects by increasing the dielectric constant of the buffer. The results indicate that the electrostatic charges introduced by phosphomimic mutation cause a shift in protein internal dynamics, which can be reversed by increasing salt concentration. Thus, nanoscale motion in the disordered C-tail can be manipulated by phosphomimicry and by altering buffer salt conditions.
Correlation between binding kinetics and nanoscale motion
The kinetics binding results show that the association rate constant of NHERF1(S339D/S340D) binding to FERM is susceptible to the influence of buffer salt concentration, with a particularly enhanced association rate constant ka1 at low salt concentration. At low buffer ionic strength, because of the activation of the nanoscale motion in the C-tip of NHERF1(S339D/S340D) as shown by NSE, the protein can overcome the negatively charged repulsive interaction with the surface of FERM, and have similar ka1 as the wild-type protein. A high buffer ionic strength, the rigid NHERF1(S339D/S340D) has a significantly reduced ka1 as compared to that in low salt buffer solution. Comparing the kinetics binding and the NSE results at low and high salt concentration thus suggests that the altered binding kinetics correlates with the changes in the nanoscale dynamics.
Intrinsically disordered proteins can be relatively rigid
The full-length NHERF1 and, in particular, the C-terminal tail have all the traits of an intrinsically disordered protein [1]. The largely disordered C-terminal tail can sample a variety of conformations as shown in our previous NMR study and the SAXS and SANS data [22,28]. However, the motion of the full-length wild-type protein as seen by NSE is that of a rigid-body [29,52]. The disordered C-tail of the wild-type NHERF1 as viewed by NSE, on nanometer length scale and nanosecond time scale, is a structural disorder of an ensemble of rigid and heterogeneous conformations. Only in the phosphomimic mutant of NHERF1 and at relatively low salt solution, the nanoscale protein motion at the C-terminal tip is excited. Depending on the spatial and temporal observation, a distinction between static disorder and dynamic disorder of intrinsically disordered proteins has been proposed [53]. Our results show that NSE can distinguish such static disorder from dynamic order on nanoscales.
Phosphorylation has diverse effects on the structure and function of proteins. It has been shown that phosphorylation modulates protein-protein interactions by altering the electrostatics and chemistry of the recognition site directly, or by inducing small or large structural changes that affect the recognition site allosterically [8,9,54,55]. However, a large-scale statistical study shows that the majority of phosphorylated proteins only undergo subtle conformational changes with the global root-mean-square deviation of less than 2Å [56]. Our SAXS and SANS results indicate that the effect of phosphomimic S339/S340D mutation on the overall conformation of NHERF1 is subtle and small, which corroborate the statistical study [56]. Future study should examine the effect of protein kinase phosphorylation on the dynamics and binding kinetics of full-length NHERF1.
In summary, this study reveals that phosphomimic mutation and salt concentration alter the nanoscale dynamics and target-binding kinetics in the intrinsically disordered tail of NHERF1. The altered kinetics of a phosphorylated NHERF1 binding to Ezrin could modulate the dynamic attachment of the NHERF1-assembled membrane signaling complexes to the cytoskeletal actin network, thus influencing the intracellular mobility and localization of membrane signaling complexes. Being able to reveal the nanoscale protein dynamics in the disordered tail provides a mechanistic explanation for the effects of phosphorylation on the cellular functions of NHERF1, demonstrating that controllable nanoscale protein motions inspire mesoscale function and assembly.
MATERIALS AND METHODS
Protein expression and purification
The bacterial expression and purification of the full-length wild-type of human NHERF1 and the FERM domain of Ezrin (residue 1-298) are described previously [20,28]. Site-directed mutagenesis of NHERF1 was performed as described [21]. The deuterated proteins used in the SANS experiments were expressed in M9 minimum medium containing 85% D2O and purified as described previously [28,29].
Online size-exclusion chromatography small angle X-ray scattering (SEC-SAXS) experiments
The SEC-SAXS experiments were performed at SSRL (Stanford Synchrotron Radiation Light Source) Bio-SAXS beamline 4-2 in a similar manner as recently reported [57–59], seeTable S1 for SEC-SAXS setup parameters. Briefly, a 100 μl of the protein sample at 6.9 mg/ml NHERF1(wt) or 11.0 mg/ml NHERF1(S339D/S340D) was applied to a Superdex 200 PC3.2/300 column (GE Healthcare, Wisconsin, USA). The SEC running buffer and the protein buffer was 150 mM NaCl, 20 mM Tris-HCl (pH=7.5), 0.1 mM EDTA. 5mM Dithiothreitol was added to both sample and running buffer solution in order to alleviate radiation damages. Totally 600 images were recorded with 1 second exposure every 5 seconds at 0.05 ml/min flow rate. The program SasTool (http://ssrl.slac.stanford.edu/~saxs/analysis/sastool.htm) was employed for data reduction including scaling, azimuthal integration, averaging and background subtraction. The first 100 images at the early part of the void volume were averaged and used as a buffer-scattering profile for the background subtraction. After taking the first 100 images or so, the X-ray shutter was closed until main elution peak so as to keep the sample cell clean from radiation-damaged sample. The data were then presented as I(Q) vs. Q, where Q=4πsin(θ)/λ, 2θ the scattering angle, and λ the wavelength of the X-ray. The scriptfplcplots, available at SSRL beamline 4-2, was used for consecutive Guinier analysis, implemented in the programAUTORG [60], and assessing data quality (e.g. radiation damage and cleanness of sample cell) by providing Rg, I(0) and an experimental intensity at a low Q angle. Since marginal inter-particle interactions (concentration dependence) were observed over the peak, the average profiles, image number: 500th–504th for NHERF1(wt) and 520th–524th for NHERF1(S339D/S340D) were generated, scaled and merged for further analyses. The length distribution function was generated using the program GNOM [61].
Small angle neutron scattering (SANS) experiments
SANS data were collected using the EQ-SANS instrument [62], which is a time-of-flight SANS instrument located at the Spallation Neutron Source (SNS) of Oak Ridge National Laboratory. A single instrument configuration with a 4 m sample-to-detector distance was employed. The instrument choppers ran at 60 Hz and were set to provide a minimum wavelength of 2.5 Å. The beam was defined with a 25 mm diameter source aperture and a 10 mm diameter sample aperture. The configuration spans a Q-range from ~0.01 Å−1 to ~0.40 Å−1, which probes the length scales required to see the complex and facilitates subtraction of the solvent background from the scattering signal from the samples. Mantid [63] was used to reduce the data from the samples and from the backgrounds using standard procedures that correct for incident flux spectrum, sample transmission and detector sensitivity, as well as the detector dark current, which represents electronic noise and natural sources of radiation. Then, the data were azimuthally averaged the data to provide I(Q) vs. Q. The sample scattering was then corrected for the solvent scattering by subtracting the 1D profiles to produce the final, reduced data. Details of SANS data analysis to extract protein conformational changes are described in previous studies [27,64–66].
Neutron spin echo experiments
The NSE experiment was performed at SNS-NSE instrument, JCNS1 outstation to Spallation Neutron Source, Oak Ridge National Laboratory [67]. NSE spectra were measured at a maximum incident wavelength of 9Å, accessing a dynamical range of ≤0.1 tmax ≤70 ns (Fouriertimes) for several momentum transfers, Q, between 0.05Å−1 and 0.12Å−1. As an elastic reference measurement we used a Graphite powder sample loaded in an identical quartz container and measured with the same instrument setup. The samples were loaded in top loader quartz containers (Hellma cells) of 40×30×4 mm and NSE spectra were taken at 283.15K. Solvent/background measurement and transmission measurements were also necessary for proper data reduction. The data reduction was performed with the standard ECHODET software package of the NSE-SNS instrument [67]. The momentum transfer (Q) ranges between 0.052 and 0.169 Å−1.
Theoretical analysis of NSE data
We have developed a theoretical framework to analyze and interpret the NSE data and to extract internal nanoscale motion. Details of the theoretical framework can be found in our previous publications [29,40,52]. The effective diffusion constant Deff(Q) as a function of Q is:
| Eq. 1 |
Thus Γ(Q) is the initial decay rate of the NSE spectra I(Q,t)/I(Q,0) and can be extracted by a single-exponential fit of our data (seeFig. 4). The first cumulant of the effective diffusion constant Deff(Q) is [40]:
| Eq. 2 |
Here, bj is the scattering length of a subunit j, HT is the translational mobility tensor, and HR is the rotational mobility tensor. The coordinates of the various subunits are taken relative to the center offriction of the protein, kBT is the usual temperature factor; andLj=rj ×Q is the torque vector for each coordinate. The effective diffusion constant depends strongly upon the mobility tensor, particularly manifest in the way a polysyndetonic protein is parsed into domains. The Q dependence of the effective diffusion constant Deff(Q) is thus a highly sensitive probe of active internal dynamic modes and correlations [29,68–70].
Surface plasmon resonance experiments
Surface plasmon resonance (SPR) experiments were performed on a X100 instrument (GE Healthcare Life Sciences). Before the binding experiments, the Biacore CM5 Biosensor chips was activated by N-hydroxysuccinimide and N-ethyl-N′-[3-(diethylamino) propyl] carbodimide (GE Healthcare Life Sciences). The ligand, which is the FERM domain of Ezrin dissolved at 5 μg/ml in 10mM sodium acetate pH=4.9, was injected to coat the activated sensor chip surface in one of the two flow cells. Uncrosslinked ligand was washed away, and uncoated sites were blocked by 1 M ethanolamine, pH=8.5. The control flow cell was activated and blocked without ligand injection. The analytes (NHERF1 wild-type protein or mutant) were dissolved in HBS-EP buffer containing 10 mM HEPES buffer, pH=7.4, 3 mM EDTA and 0.005% surfactant polysorbate 20 and different NaCl concentrations, respectively. The analytes, of increasing concentrations, were injected over the FERM coated surfaces at 30 μl/min for 180 seconds. The dissociation time is 800 seconds. At the end of injection-dissociation cycle, the sensor chip was regenerated with 4.0 M MgCl2, 50 mM triethylamine (pH=9.15), and HBS-EP buffer. SPR experiments were performed at three different ligand immobilization levels and in HBS-EP buffers containing 10, 25, 50, 100, 150, 200, 250, 300, 400 mM NaCl. All SPR experiments were performed at 15 °C.
The SPR response curves were obtained using the BIA Evaluation Software. The response curves of NHERF1(wt) or NHERF1(S339D/S340D) were fit by a two-state kinetic binding model. In the two-state model, the analyte A binds to the ligand L to form an initial complex (AL), and then undergoes subsequent conformational change or binding to form the final complex (A*L):
| Eq. 3 |
where A is the analyte NHERF1(wt) or NHERF1(S339D/S340D) and L is the ligand FERM immobilized on the CM5 sensor chip.
The association rate constant ka1 was plotted against logγ± that is calculate by the Debye–Hückel equation:
| Eq.4 |
where γ± is mean activity coefficient, I the ionic strength of NaCl in 10 mM Hepes buffer (pH=7.4), 3 mM EDTA, 0.5 mM DTT, a≈5.6Å the effective diameter of the ion, and z+ and z− are the number of positive and negative charges of the ion, respectively. The value of constants A and B inEq. 4 are 0.5085 and 0.3281, respectively.
The binding responses in the steady-state region of the sensorgrams (Req) were also plotted against analyte concentration (C) to determine the overall equilibrium binding affinity. The data were fit to the non-linear equation:
| Eq. 5 |
Where Rmax is the maximum binding response, andKd is the dissociation constant.
Supplementary Material
Research highlights.
Nanoscale protein dynamics in an intrinsically disordered protein affects protein-protein interactions in a largely unknown fashion because it is experimentally difficult to probe protein dynamics on nanoscales.
Employing the novel technique of neutron spin echo spectroscopy (NSE), we are able to reveal nanoscale dynamics changes in a disordered protein.
Our theoretical analysis shows that NSE pinpoints the nanoscale dynamics changes in a highly specific manner.
Debye–Hückel analysis indicates that changes in kinetic association rate constant at different ionic strengths cannot be explained by electrostatic effects alone.
This study shows that nanoscale dynamics in a disordered protein significantly influences the binding kinetics of signaling partners.
Acknowledgments
This research was funded in part by NIH R01HL086496, R01DK105811, and 2G12 RR003060 from the National Center for Research Resources to CCNY.
Use of the Stanford Synchrotron Radiation Light Source, SLAC National Accelerator Laboratory, is supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Contract No. DE-AC02-76SF00515. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research, and by the National Institutes of Health, National Institute of General Medical Sciences (including P41GM103393). The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of NIGMS or NIH.
A portion of the research conducted at ORNL’s Spallation Neutron Source was sponsored by the Scientific User Facilities Division, Office of Basic Energy Sciences, U.S. Department of Energy.
The authors acknowledge Carrie Gao and Malcolm Cochran for technical support during SANS and NSE measurements.
ABBREVIATIONS
- Intrinsically disordered protein
- EBD
Ezrin-binding domain
- NSE
neutron spin echo spectroscopy
- SANS
small angle neutron scattering
- SEC-SAXS
size-exclusion small angle X-ray scattering
- SPR
surface plasmon resonance
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Van Der Lee R, Buljan M, Lang B, Weatheritt RJ, Daughdrill GW, Dunker AK, et al. Classification of intrinsically disordered regions and proteins. Chem Rev. 2014;114:6589–631. doi: 10.1021/cr400525m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tompa P, Fersht A. Structure and function of intrinsically disordered proteins. CRC Press; 2010. [Google Scholar]
- 3.Uversky VN, Oldfield CJ, Dunker AK. Intrinsically disordered proteins in human diseases: introducing the D2 concept. Annu Rev Biophys. 2008;37:215–46. doi: 10.1146/annurev.biophys.37.032807.125924. [DOI] [PubMed] [Google Scholar]
- 4.Sugase K, Dyson HJ, Wright PE. Mechanism of coupled folding and binding of an intrinsically disordered protein. Nature. 2007;447:1021–5. doi: 10.1038/nature05858. [DOI] [PubMed] [Google Scholar]
- 5.Borg M, Mittag T, Pawson T, Tyers M, Forman-Kay JD, Chan HS. Polyelectrostatic interactions of disordered ligands suggest a physical basis for ultrasensitivity. Proc Natl Acad Sci U S A. 2007;104:9650–5. doi: 10.1073/pnas.0702580104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nishi H, Fong JH, Chang C, Teichmann SA, Panchenko AR. Regulation of protein-protein binding by coupling between phosphorylation and intrinsic disorder: analysis of human protein complexes. Mol Biosyst. 2013;9:1620–6. doi: 10.1039/c3mb25514j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Iakoucheva LM, Radivojac P, Brown CJ, O’Connor TR, Sikes JG, Obradovic Z, et al. The importance of intrinsic disorder for protein phosphorylation. Nucleic acids research. 2004;32:1037–49. doi: 10.1093/nar/gkh253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nussinov R, Tsai C-J, Xin F, Radivojac P. Allosteric post-translational modification codes. Trends in biochemical sciences. 2012;37:447–55. doi: 10.1016/j.tibs.2012.07.001. [DOI] [PubMed] [Google Scholar]
- 9.Johnson LN. The regulation of protein phosphorylation. Biochemical Society Transactions. 2009;37:627–41. doi: 10.1042/BST0370627. [DOI] [PubMed] [Google Scholar]
- 10.Fehon RG, McClatchey AI, Bretscher A. Organizing the cell cortex: the role of ERM proteins. Nat Rev Mol Cell Biol. 2010;11:276–87. doi: 10.1038/nrm2866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ardura JA, Friedman PA. Regulation of G protein-coupled receptor function by Na+/H+ exchange regulatory factors. Pharmacol Rev. 2011;63:882–900. doi: 10.1124/pr.110.004176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Loureiro CA, Matos AM, Dias-Alves Â, Pereira JF, Uliyakina I, Barros P, et al. A molecular switch in the scaffold NHERF1 enables misfolded CFTR to evade the peripheral quality control checkpoint. Science signaling. 2015;8:ra48-ra. doi: 10.1126/scisignal.aaa1580. [DOI] [PubMed] [Google Scholar]
- 13.Seidler U, Singh AK, Cinar A, Chen M, Hillesheim J, Hogema B, et al. The role of the NHERF family of PDZ scaffolding proteins in the regulation of salt and water transport. Ann N Y Acad Sci. 2009;1165:249–60. doi: 10.1111/j.1749-6632.2009.04046.x. [DOI] [PubMed] [Google Scholar]
- 14.Fanelli T, Cardone RA, Favia M, LG, Zaccolo M, Monterisi S, et al. Beta-oestradiol rescues DeltaF508CFTR functional expression in human cystic fibrosis airway CFBE41o- cells through the up-regulation of NHERF1. Biol Cell. 2008;100:399–412. doi: 10.1042/BC20070095. [DOI] [PubMed] [Google Scholar]
- 15.Favia M, Guerra L, Fanelli T, Cardone RA, Monterisi S, Di Sole F, et al. Na+/H+ exchanger regulatory factor 1 overexpression-dependent increase of cytoskeleton organization is fundamental in the rescue of F508del cystic fibrosis transmembrane conductance regulator in human airway CFBE41o- cells. Mol Biol Cell. 2010;21:73–86. doi: 10.1091/mbc.E09-03-0185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Garbett D, Bretscher A. PDZ interactions regulate rapid turnover of the scaffolding protein EBP50 in microvilli. The Journal of Cell Biology. 2012;198:195–203. doi: 10.1083/jcb.201204008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Garbett D, Sauvanet C, Viswanatha R, Bretscher A. The tails of apical scaffolding proteins EBP50 and E3KARP regulate their localization and dynamics. Mol Biol Cell. 2013 doi: 10.1091/mbc.E13-06-0330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Viswanatha R, Bretscher A, Garbett D. Dynamics of ezrin and EBP50 in regulating microvilli on the apical aspect of epithelial cells. Biochemical Society transactions. 2014;42:189. doi: 10.1042/BST20130263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kamiya K, Michel V, Giraudet F, Riederer B, Foucher I, Papal S, et al. An unusually powerful mode of low-frequency sound interference due to defective hair bundles of the auditory outer hair cells. Proc Natl Acad Sci U S A. 2014;111:9307–12. doi: 10.1073/pnas.1405322111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li J, Dai Z, Jana D, Callaway DJ, Bu Z. Ezrin controls the macromolecular complexes formed between an adapter protein Na+/H+ exchanger regulatory factor and the cystic fibrosis transmembrane conductance regulator. J Biol Chem. 2005;280:37634–43. doi: 10.1074/jbc.M502305200. [DOI] [PubMed] [Google Scholar]
- 21.Li J, Poulikakos PI, Dai Z, Testa JR, Callaway DJ, Bu Z. Protein kinase C phosphorylation disrupts Na+/H+ exchanger regulatory factor 1 autoinhibition and promotes cystic fibrosis transmembrane conductance regulator macromolecular assembly. J Biol Chem. 2007;282:27086–99. doi: 10.1074/jbc.M702019200. [DOI] [PubMed] [Google Scholar]
- 22.Bhattacharya S, Dai Z, Li J, Baxter S, Callaway DJ, Cowburn D, et al. A conformational switch in the scaffolding protein NHERF1 controls autoinhibition and complex formation. J Biol Chem. 2010;285:9981–94. doi: 10.1074/jbc.M109.074005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Reczek D, Berryman M, Bretscher A. Identification of EBP50: A PDZ-containing phosphoprotein that associates with members of the ezrin-radixin-moesin family. J Cell Biol. 1997;139:169–79. doi: 10.1083/jcb.139.1.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Finnerty CM, Chambers D, Ingraffea J, Faber HR, Karplus PA, Bretscher A. The EBP50-moesin interaction involves a binding site regulated by direct masking on the FERM domain. J Cell Sci. 2004;117:1547–52. doi: 10.1242/jcs.01038. [DOI] [PubMed] [Google Scholar]
- 25.Terawaki S, Maesaki R, Hakoshima T. Structural basis for NHERF recognition by ERM proteins. Structure. 2006;14:777–89. doi: 10.1016/j.str.2006.01.015. [DOI] [PubMed] [Google Scholar]
- 26.Fievet BT, Gautreau A, Roy C, Del Maestro L, Mangeat P, Louvard D, et al. Phosphoinositide binding and phosphorylation act sequentially in the activation mechanism of ezrin. J Cell Biol. 2004;164:653–9. doi: 10.1083/jcb.200307032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jayasundar JJ, Ju JH, He L, Liu D, Meilleur F, Zhao J, et al. Open Conformation of Ezrin Bound to Phosphatidylinositol 4,5-Bisphosphate and to F-actin Revealed by Neutron Scattering. J Biol Chem. 2012;287:37119–33. doi: 10.1074/jbc.M112.380972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li J, Callaway DJ, Bu Z. Ezrin induces long-range interdomain allostery in the scaffolding protein NHERF1. J Mol Biol. 2009;392:166–80. doi: 10.1016/j.jmb.2009.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Farago B, Li J, Cornilescu G, Callaway DJ, Bu Z. Activation of nanoscale allosteric protein domain motion revealed by neutron spin echo spectroscopy. Biophys J. 2010;99:3473–82. doi: 10.1016/j.bpj.2010.09.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Garbett D, LaLonde DP, Bretscher A. The scaffolding protein EBP50 regulates microvillar assembly in a phosphorylation-dependent manner. J Cell Biol. 2010;191:397–413. doi: 10.1083/jcb.201004115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang B, Means CK, Yang Y, Mamonova T, Bisello A, Altschuler DL, et al. Ezrin-anchored PKA Coordinates Phosphorylation-dependent Disassembly of a NHERF1 Ternary Complex to Regulate Hormone-sensitive Phosphate Transport. J Biol Chem. 2012 doi: 10.1074/jbc.M112.369405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ardura JA, Wang B, Watkins SC, Vilardaga J-P, Friedman PA. Dynamic Na+-H+ exchanger regulatory factor-1 association and dissociation regulate parathyroid hormone receptor trafficking at membrane microdomains. Journal of Biological Chemistry. 2011 doi: 10.1074/jbc.M111.264978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sneddon WB, Syme CA, Bisello A, Magyar CE, Rochdi MD, Parent J-L, et al. Activation-independent parathyroid hormone receptor internalization is regulated by NHERF1 (EBP50) Journal of Biological Chemistry. 2003;278:43787–96. doi: 10.1074/jbc.M306019200. [DOI] [PubMed] [Google Scholar]
- 34.Hall RA, Spurney RF, Premont RT, Rahman N, Blitzer JT, Pitcher JA, et al. G protein-coupled receptor kinase 6A phosphorylates the Na(+)/H(+) exchanger regulatory factor via a PDZ domain-mediated interaction. J Biol Chem. 1999;274:24328–34. doi: 10.1074/jbc.274.34.24328. [DOI] [PubMed] [Google Scholar]
- 35.He J, Lau AG, Yaffe MB, Hall RA. Phosphorylation and cell cycle-dependent regulation of Na+/H+ exchanger regulatory factor-1 by Cdc2 kinase. J Biol Chem. 2001;276:41559–65. doi: 10.1074/jbc.M106859200. [DOI] [PubMed] [Google Scholar]
- 36.Lau AG, Hall RA. Oligomerization of NHERF-1 and NHERF-2 PDZ domains: differential regulation by association with receptor carboxyl-termini and by phosphorylation. Biochemistry. 2001;40:8572–80. doi: 10.1021/bi0103516. [DOI] [PubMed] [Google Scholar]
- 37.Fouassier L, Nichols MT, Gidey E, McWilliams RR, Robin H, Finnigan C, et al. Protein kinase C regulates the phosphorylation and oligomerization of ERM binding phosphoprotein 50. Exp Cell Res. 2005;306:264–73. doi: 10.1016/j.yexcr.2005.02.011. [DOI] [PubMed] [Google Scholar]
- 38.Chen JY, Lin YY, Jou TS. Phosphorylation of EBP50 negatively regulates [beta]-PIX-dependent Rac1 activity in anoikis. Cell Death Differ. 2012;19:1027–37. doi: 10.1038/cdd.2012.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Callaway DJ, Bu Z. Visualizing the nanoscale: protein internal dynamics and neutron spin echo spectroscopy. Curr Opin Struct Biol. 2016;42:1–5. doi: 10.1016/j.sbi.2016.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bu Z, Biehl R, Monkenbusch M, Richter D, Callaway DJ. Coupled protein domain motion in Taq polymerase revealed by neutron spin-echo spectroscopy. Proc Natl Acad Sci U S A. 2005;102:17646–51. doi: 10.1073/pnas.0503388102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bhattacharya S, Ju JH, Orlova N, Khajeh JA, Cowburn D, Bu Z. Ligand-Induced Dynamic Changes in Extended PDZ Domains from NHERF1. J Mol Biol. 2013 doi: 10.1016/j.jmb.2013.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bernado P, Mylonas E, Petoukhov MV, Blackledge M, Svergun DI. Structural characterization of flexible proteins using small-angle X-ray scattering. J Am Chem Soc. 2007;129:5656–64. doi: 10.1021/ja069124n. [DOI] [PubMed] [Google Scholar]
- 43.Maupetit J, Gautier R, Tuffery P. SABBAC: online Structural Alphabet-based protein BackBone reconstruction from Alpha-Carbon trace. Nucleic Acids Res. 2006;34:W147–51. doi: 10.1093/nar/gkl289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McDonnell JM. Surface plasmon resonance: towards an understanding of the mechanisms of biological molecular recognition. Current opinion in chemical biology. 2001;5:572–7. doi: 10.1016/s1367-5931(00)00251-9. [DOI] [PubMed] [Google Scholar]
- 45.Schuck P. Reliable determination of binding affinity and kinetics using surface plasmon resonance biosensors. Current opinion in biotechnology. 1997;8:498–502. doi: 10.1016/s0958-1669(97)80074-2. [DOI] [PubMed] [Google Scholar]
- 46.Myszka DG. Kinetic analysis of macromolecular interactions using surface plasmon resonance biosensors. Current opinion in biotechnology. 1997;8:50–7. doi: 10.1016/s0958-1669(97)80157-7. [DOI] [PubMed] [Google Scholar]
- 47.Dogan J, Gianni S, Jemth P. The binding mechanisms of intrinsically disordered proteins. Physical Chemistry Chemical Physics. 2014;16:6323–31. doi: 10.1039/c3cp54226b. [DOI] [PubMed] [Google Scholar]
- 48.Rogers JM, Steward A, Clarke J. Folding and binding of an intrinsically disordered protein: fast, but not ‘diffusion-limited’. Journal of the American Chemical Society. 2013;135:1415–22. doi: 10.1021/ja309527h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Huang Y, Liu Z. Kinetic advantage of intrinsically disordered proteins in coupled folding–binding process: a critical assessment of the “fly-casting” mechanism. Journal of molecular biology. 2009;393:1143–59. doi: 10.1016/j.jmb.2009.09.010. [DOI] [PubMed] [Google Scholar]
- 50.Schreiber G, Fersht AR. Rapid, electrostatically assisted association of proteins. Nature structural & molecular biology. 1996;3:427–31. doi: 10.1038/nsb0596-427. [DOI] [PubMed] [Google Scholar]
- 51.Schreiber G, Haran G, Zhou H-X. Fundamental aspects of protein– protein association kinetics. Chem Rev. 2009;109:839–60. doi: 10.1021/cr800373w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bu Z, Callaway DJ. Proteins MOVE! Protein dynamics and long-range allostery in cell signaling. Adv Protein Chem Struct Biol. 2011;83:163–221. doi: 10.1016/B978-0-12-381262-9.00005-7. [DOI] [PubMed] [Google Scholar]
- 53.Tompa P, Fuxreiter M. Fuzzy complexes: polymorphism and structural disorder in protein-protein interactions. Trends in Biochemical Sciences. 33:2–8. doi: 10.1016/j.tibs.2007.10.003. [DOI] [PubMed] [Google Scholar]
- 54.Nishi H, Shaytan A, Panchenko AR. Physicochemical mechanisms of protein regulation by phosphorylation. Front Genet. 2014;5 doi: 10.3389/fgene.2014.00270. doi:10.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lenz P, Swain PS. An entropic mechanism to generate highly cooperative and specific binding from protein phosphorylations. Current biology. 2006;16:2150–5. doi: 10.1016/j.cub.2006.09.013. [DOI] [PubMed] [Google Scholar]
- 56.Xin F, Radivojac P. Post-translational modifications induce significant yet not extreme changes to protein structure. Bioinformatics. 2012;28:2905–13. doi: 10.1093/bioinformatics/bts541. [DOI] [PubMed] [Google Scholar]
- 57.Matsui T, Gu S, Lam K-h, Carter LG, Rummel A, Mathews II, et al. Structural basis of the pH-dependent assembly of a botulinum neurotoxin complex. Journal of molecular biology. 2014;426:3773–82. doi: 10.1016/j.jmb.2014.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Edwards AL, Matsui T, Weiss TM, Khosla C. Architectures of whole-module and bimodular proteins from the 6-deoxyerythronolide B synthase. Journal of molecular biology. 2014;426:2229–45. doi: 10.1016/j.jmb.2014.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Martel A, Liu P, Weiss TM, Niebuhr M, Tsuruta H. An integrated high-throughput data acquisition system for biological solution X-ray scattering studies. Journal of synchrotron radiation. 2012;19:431–4. doi: 10.1107/S0909049512008072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Petoukhov MV, Konarev PV, Kikhney AG, Svergun DI. ATSAS 2.1–towards automated and web-supported small-angle scattering data analysis. Applied Crystallography. 2007;40:s223–s8. [Google Scholar]
- 61.Semenyuk AV, Svergun DI. GNOM - A program Package for small-angle scattering data-processing. J Appl Cryst. 1991;24:537–40. [Google Scholar]
- 62.Zhao JK, Gao CY, Liu D. The extended Q-range small-angle neutron scattering diffractometer at the SNS. Journal of Applied Crystallography. 2010;43:1068–77. [Google Scholar]
- 63.Arnold O, Bilheux J-C, Borreguero J, Buts A, Campbell SI, Chapon L, et al. Mantid—data analysis and visualization package for neutron scattering and μ SR experiments. Nuclear Instruments and Methods in Physics Research Section A: Accelerators, Spectrometers, Detectors and Associated Equipment. 2014;764:156–66. [Google Scholar]
- 64.Ali Khajeh J, Ju JH, Atchiba M, Allaire M, Stanley C, Heller WT, et al. Molecular conformation of the full-length tumor suppressor NF2/Merlin--a small-angle neutron scattering study. J Mol Biol. 2014;426:2755–68. doi: 10.1016/j.jmb.2014.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chen X, Khajeh JA, Ju JH, Gupta YK, Stanley CB, Do C, et al. Phosphatidylinositol 4,5-bisphosphate clusters the cell adhesion molecule CD44 and assembles a specific CD44-Ezrin heterocomplex, as revealed by small angle neutron scattering. J Biol Chem. 2015;290:6639–52. doi: 10.1074/jbc.M114.589523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ho DL, Byrnes WM, Ma WP, Shi Y, Callaway DJ, Bu Z. Structure-specific DNA-induced conformational changes in Taq polymerase revealed by small angle neutron scattering. J Biol Chem. 2004;279:39146–54. doi: 10.1074/jbc.M404565200. [DOI] [PubMed] [Google Scholar]
- 67.Ohl M, Monkenbusch M, Arend N, Kozielewski T, Vehres G, Tiemann C, et al. The spin-echo spectrometer at the Spallation Neutron Source (SNS) Nuclear Instruments and Methods in Physics Research Section A: Accelerators, Spectrometers, Detectors and Associated Equipment. 2012;696:85–99. [Google Scholar]
- 68.Callaway DJ, Bu Z. Nanoscale protein domain motion and long-range allostery in signaling proteins- a view from neutron spin echo sprectroscopy. Biophys Rev. 2015;7:165–74. doi: 10.1007/s12551-015-0162-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Callaway DJ, Farago B, Bu Z. Nanoscale protein dynamics: A new frontier for neutron spin echo spectroscopy. The European physical journal E, Soft matter. 2013;36:9891. doi: 10.1140/epje/i2013-13076-1. [DOI] [PubMed] [Google Scholar]
- 70.Bu Z, Cook J, Callaway DJ. Dynamic regimes and correlated structural dynamics in native and denatured alpha-lactalbumin. J Mol Biol. 2001;312:865–73. doi: 10.1006/jmbi.2001.5006. [DOI] [PubMed] [Google Scholar]
- 71.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, et al. UCSF Chimera - A Visualization System for Exploratory Research and Analysis. J Comput Chem. 2004;25:1605–12. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.