




IGF-1 Has Plaque-Stabilizing Effects in Atherosclerosis by Altering Vascular Smooth Muscle Cell Phenotype
Jan H von der Thüsen
Keren S Borensztajn
Silvia Moimas
Sandra van Heiningen
Peter Teeling
Theo JC van Berkel
Erik AL Biessen
Address reprint requests to: Jan von der Thüsen, Department of Histopathology, Royal Brompton Hospital, Sydney Street, London SW3 6NP, United Kingdomj.vonderthuesen@rbht.nhs.uk
Accepted 2010 Oct 19.
Abstract
Insulin-like growth factor-1 (IGF-1) signaling is important for the maintenance of plaque stability in atherosclerosis due to its effects on vascular smooth muscle cell (vSMC) phenotype. To investigate this hypothesis, we studied the effects of the highly inflammatory milieu of the atherosclerotic plaque on IGF-1 signaling and stability-related phenotypic parameters of murine vSMCsin vitro, and the effects of IGF-1 supplementation on plaque phenotype in an atherosclerotic mouse model. M1-polarized, macrophage-conditioned medium inhibited IGF-1 signaling by ablating IGF-1 and increasing IGF-binding protein 3, increased vSMC apoptosis, and decreased proliferation. Expression ofα-actin andcol3a1 genes was strongly attenuated by macrophage-conditioned medium, whereas expression of matrix-degrading enzymes was increased. Importantly, all of these effects could be corrected by supplementation with IGF-1.In vivo, treatment with the stable IGF-1 analog Long R3 IGF-1 in apolipoprotein E knockout mice reduced stenosis and core size, and doubled cap/core ratio in early atherosclerosis. In advanced plaques, Long R3 IGF-1 increased the vSMC content of the plaque by more than twofold and significantly reduced the rate of intraplaque hemorrhage. We believe that IGF-1 in atherosclerotic plaques may have a role in preventing plaque instability, not only by modulating smooth muscle cell turnover, but also by altering smooth muscle cell phenotype.
Atherosclerosis is an inflammatory process,1 of which the cellular components can affect plaque stability. Thus, macrophages are generally considered to have detrimental effects on plaque stability through the elaboration of various proinflammatory factors and matrix-degrading enzymes.2,3 Conversely, an at least partially protective role has been attributed to vascular smooth muscle cells (vSMCs) due to their contribution to the size and structural integrity of the fibrous cap. The plaque content and phenotypic characteristics of plaque vSMCs are therefore considered to be important determinants of plaque stability. Hence, vSMC apoptosis has been identified as a potential contributory factor in plaque rupture.4–6 This has been corroborated by experiments in our group, in which adenovirus-mediated transfer of the proapoptotic genesp537 andFasL8 to cap smooth muscle cells induced vulnerable plaque morphology and features of plaque rupture in a murine model of atherosclerosis.
In the atherosclerotic plaque, both turnover and phenotype of smooth muscle cells are regulated by a host of local stimulatory and inhibitory signals. These include components of the extracellular matrix and locally produced paracrine and autocrine factors, but also systemically active circulating hormones such as insulin-like growth factor (IGF)-1 and IGF-2, 2 members of the insulin-like growth factor axis.9 IGF-1 in particular has received considerable attention as a potentially important factor in atherogenesis, although its overall role remains contested. On a tissue level, IGF-1, its receptor (IGF-1R), and various IGF-1 binding proteins are known to be expressed by cells in human atherosclerotic plaques, including vSMCs.10,11 Clinically, serum levels of IGF-112,13 and its most important systemic binding protein, IGFBP-3,12 have been found to be correlated with carotid atherosclerosis, whereas Juul et al14 have demonstrated a significantly increased risk of ischemic heart disease in patients with low serum IGF-1 and high IGFBP-3 levels. Recently, Sukhanov et al15 showed that IGF-1 may have antiatherogenic properties in apolipoprotein E–deficient (ApoE−/−) mice by reducing macrophage infiltration. With respect to IGF-1 signaling in smooth muscle cells, the atherosclerotic plaque appears to harbor largely inhibitory conditions, possibly due to the immersion of these cells in the highly inflammatory milieu created by resident macrophages.16,17 Most research to date has been focused on the effects of the modulation of IGF-1 signaling on apoptosis of vSMCs, and little is known about its effect on other phenotypic parameters. Thus, in addition to influencing the level of apoptosis in plaque vSMCs, a reduction of IGF-1 signaling might also lead to impaired maintenance of the extracellular matrix of the lesional cap by these cells, thereby diminishing plaque stability.
To investigate the potential of phenotypic modulation of vSMCs by an alteration in IGF-1 signaling to achieve stabilization of the atherosclerotic plaque, we incubated murine arterial vSMCs with medium conditioned by activated macrophages, and measured the effect on various parameters of the IGF-1 system, as well as their productive and degradative capacity for collagenous matrix. The effect of IGF-1 on atherosclerosisin vivo was assessed by continuous subcutaneous administration of the IGF-1 analog Long R3 IGF-1 in a model of accelerated atherogenesis in apoE−/− mice.
Materials and Methods
Animals
All animal work was approved by the regulatory authority of Leiden University and performed in compliance with Dutch government guidelines. Twelve- to 14-week-old female apoE-deficient mice (9× back-crossed onto C57bl/6 background, Jackson Labs, Bar Harbor, ME) and C57bl/6 mice (Broekman, Someren, the Netherlands) were kept on a regular dark/light cycle, and fed regular chow or a semisynthetic Western-type diet containing 15% (w/w) cacao butter and 0.25% (w/w) cholesterol (Diet W; Special Diet Services, Witham, UK).
Cell Culture
The cell culturing reagents were from Cambrex Corporation (Verviers, Belgium), the culturing equipment from Costar (Schiphol Rijk, the Netherlands), human IGF-1 and TNF-α were from Sigma-Aldrich (Saint Louis, MO), and Long R3 IGF-1 from Gropep (Adelaide, Australia). Anti-PDGF-BB neutralizing antibody was from Millipore (Billerica, MA) and used in a concentration of 20 μg/ml.
Smooth muscle cells were isolated from the aortas of C57Bl/6 mice by collagenase digestion as described previously by Michon et al.18 The cells were cultured in wells coated with gelatin [0.1% w/v in phosphate-buffered saline (PBS)], and maintained in culture medium consisting of Dulbecco's Modified Eagle's Medium (DMEM) with 10% normal calf serum (NCS), penicillin (100 U/ml), streptomycin (100 μg/ml), and L-Glu (2 mmol/L) (used at passage numbers 19–30). Cells were washed 3 times with PBS, and serum starved for 4 hours before stimulation.
RAW 264.7 cells, a murine macrophage cell line, were cultured in DMEM containing 10% fetal calf serum (FCS), penicillin (100 U/ml), streptomycin (100 μg/ml), and L-Glu (2 mmol/L). Conditioned medium (CM) was produced as follows: RAW cells were grown to 70% confluency and then incubated with lipopolysaccharide (LPS, 15 ng/ml; Sigma) for 6 hours in medium as described above. The cells were then washed twice with PBS and subsequently allowed to proliferate for 24 hours in standard medium (containing no LPS), following which the supernatant was isolated and stored at −80°C until further use. Nonconditioned medium was used as a control. Smooth muscle cells were serum starved for 4 hours and subsequently incubated with conditioned medium or control medium for 24 hours before harvesting for gene expression and protein synthesis (quantitative PCR and Western blot) analysis.
The rates of apoptosis were inferred from a propidium iodide (PI) or PI/Annexin V assay (Santa Cruz, Heidelberg, Germany) as follows. After trypsinization, cells were centrifuged (1600 ×g) for 5 minutes at 4°C, the supernatant discarded, and the pellet resuspended in PBS-EDTA (1 mmol/L). A total of 800 μl of 100% ethanol was added, and cells were fixed at −20°C. The suspension was again centrifuged (2400 ×g) for 5 minutes at 4°C, and the cells were washed with PBS-EDTA 1 mmol/L. The sediment was resuspended in prodidium iodide/RNase (7.5 μmol/L and 25 μg/L, respectively) in PBS-EDTA (1 mmol/L), followed by FACS (fluorescence-activated cell sorter) analysis (FACSCalibur, Becton Dickinson, Franklin Lakes, NJ). Alternatively, resuspended cells were incubated with propidium iodide and annexin V before FACS analysis.
Cellular viability was determined by MTT assay (Sigma). In the control condition, 1 × 104 cells per well were seeded onto 96-well plates in DMEM supplemented with 10% FCS. To determine the effect of CM on cell viability, 104 cells per well were seeded directly in CM, or CM supplemented with the indicated concentrations of IGF-1. Cell viability was determined by incubating 0.5 mg/ml MTT directly added to the media for a minimum of 2 hours at 37°C. Subsequently, the media were aspirated, and the cells were lysed in isopropanol/0.04 N HCl. The OD550 of this solution was determined using an enzyme-linked immunosorbent assay (ELISA) reader (BioTek, Winooski, VT). Cellular viability was expressed as percentage of viability in the control condition.
Proliferation was measured by incubation with 50 μl/ml of3H-thymidine stock (20–25 Ci/mmol) in culture medium for 24 hours, following which the cells were washed 3 times with PBS, 0.1 M NaOH was added, and the lysate was analyzed in a beta counter (Perkin-Elmer, Boston, MA).
RNA Extraction and Real-Time Quantitative PCR
Forin vitro RNA extraction, TRIZOL reagent (GibcoBRL, Gaithersburg, MD) was used according to the manufacturer's instructions, and tissue RNA isolation was carried out as previously described.19 RNA levels were determined by use of the Ribogreen low-range assay (Promega, Madison, WI), with a calibration curve of 0 to 50 ng/ml tRNA in TE DEPC buffer. The read-out was performed in a 96-well plate reader and an excitation wave length of 492 nm and an emission wave length of 535 nm. RT-PCR was carried out as previously described,19 using reagents supplied by Eurogentec (Seraing, Belgium) except for oligodT and primers, which were from Proligo (Hamburg, Germany). The products were diluted in TE DEPC buffer to a concentration of 5 ng/μL. Real-time PCR was carried out on an ABI Prism 7700 SDS apparatus using Sybr Green dye (Eurogentec, Seraing, Belgium) for quantification. Primer pairs were constructed using Primer Express 1.5 software (seeTable 1). In all experiments, the amount of cDNA used was between 12.5 and 25 ng, in a volume of 25 μL, including 10 μL of SYBR mix (consisting of MgCl2, dNTP, Hot Gold Star enzyme, MilliQ water, and SYBR Green I in working concentrations in DMSO) and 0.5 μl of the relevant primer solution (15 pmol/μl) and MilliQ water to render the final volume.
Table 1.
Primer Pairs Used for Quantitative RT-PCR
| Gene | Sequence | Template nucleotide positions | Product length (bp) | GenBank |
|---|---|---|---|---|
| IGF-1 | Fwd 5′-TTGCTTCCGGAGCTGTGATC-3′ | 497–516 | 79 | AF440694 |
| Rev 5′-GATAGAGCGGGCTGCTTTTG-3′ | 575–556 | |||
| IGF-1-R type I | Fwd 5′-AGACGGCTTCTCTGCAGTAAACA-3′ | 4865–4887 | 90 | AF056187 |
| Rev 5′-CAGTTAAGGGTTCGGGTAAAGGA-3′ | 4954–4932 | |||
| IGFBP-3 | Fwd 5′-GCTGGTGTGTGGACAAGTATGG-3′ | 738–759 | 73 | NM_008343 |
| Rev 5′-AGGCAATGTACGTCGTCTTTCC-3′ | 810–789 | |||
| IGFBP-4 | Fwd 5′-GCAACTTCCACCCCAAACAGT-3′ | 848–868 | 79 | NM_010517 |
| Rev 5′-CCTGTCTTCCGATCCACACA-3′ | 926–907 | |||
| SM α-actin | Fwd 5′-TCCCTGGAGAAGAGCTACGAACT-3′ | 779–801 | 120 | NM_007392 |
| Rev 5′-GATGCCCGCTGACTCCAT-3′ | 898–881 | |||
| Col3a1 | Fwd 5′-TGGACCAGCAGGAACTAATGG-3′ | 1455–1475 | 75 | M18933 |
| Rev 5′-CCCTTCGCACCGTTCTTG-3′ | 1529–1512 | |||
| MMP-13 | Fwd 5′-CAGAGCACTACTTGAAATCATACTACCAT-3′ | 129–157 | 82 | NM_008607 |
| Rev 5′-TCAACTGTGGAGGTCACTGTAGACTT-3′ | 210–185 | |||
| MMP-3 | Fwd 5′-TCCTGATGTTGGTGGCTTCA-3′ | 323–342 | 71 | NM_010809 |
| Rev 5′-TCCTGTAGGTGATGTGGGATTTC-3′ | 393–371 | |||
| β-actin | Fwd 5′-AACCGTGAAAAGATGACCCAGAT-3′ | 422–444 | 75 | X03672 |
| Rev 5′-CACAGCCTGGATGGCTACGTA-3′ | 496–476 | |||
| 36B4 | Fwd 5′-GGACCCGAGAAGACCTCCTT-3′ | 512–531 | 85 | X15267 |
| Rev 5′-GCACATCACTCAGAATTTCAATGG-3′ | 596–573 | |||
| HPRT | Fwd 5′-TTGCTCGAGATGTCATGAAGGA-3′ | 292–313 | 91 | J00423 |
| Rev 5′-AGCAGGTCAGCAAAGAACTTATAG-3′ | 382–359 | |||
| GAPDH | Fwd 5′-TCCATGACAACTTTGGCATTG-3′ | 530–550 | 103 | NM_008084 |
| Rev 5′-TCACGCCACAGCTTTCCA-3′ | 632–615 |
cDNA amplification was achieved with successive incubation for 2 minutes at 50°C, 10 minutes at 95°C, 40 cycles of 15 seconds at 95°C and 1 minute at 60°C, and 2 minutes at 95°C. After PCR amplification, dissociation curves were constructed to confirm formation of the intended PCR products. Expression levels of target genes were related to the averaged expression levels of 3 housekeeping genes:HPRT,GAPDH, and36B4. Measurements were carried out in triplicate.
Western Blot
Cells were lysed in Laemmli lysis buffer, incubated for 5 minutes at 95°C, and whole-cell lysates were separated by 10% sodium dodecyl-sulfate-polyacrylamide gel electrophoresis. After electrophoresis, proteins were transferred onto immobilon-P PVDF membranes (Millipore). Membranes were incubated overnight at 4°C with primary antibodies to α-actin, β-actin, tubulin, collagen 1A1, procollagen 1A1 and 3A1, and TIMP-1 (all Santa Cruz Biotechnology, Santa Cruz, CA), or to phospho(P-)p42/44 MAPK, P-Akt, P-mTor, P-p70S6K, total p42/44 MAPKinase, total mTor, total Akt, and total p70S6K (all from Cell Signaling Technology, Beverly, MA) in TBS with 0.1% Tween (TBS-T)/1% BSA. All secondary HRP-conjugated antibodies were from DakoCytomation (Glostrup, Denmark). Blots were imaged using Lumilight Plus ECL substrate (Roche, Basel, Switzerland) on a GeneGnome imager (Syngene, Cambridge, UK). Signal quantification was done using the GeneTools program from Syngene, using raw volumes as measure.
Zymography
A 7.5% SDS-PAGE denaturing gel was prepared containing 0.1 mg/ml of collagen. Specimens (25 μl) were applied in sample buffer, and after 2 hours of electrophoresis at 125 V, the gel was incubated in renaturing buffer for 1 hour. After 16 hours of agitation in developing buffer, the gel was developed with Coomassie blue R-250 (0.5%) for 30 minutes.
Cell Migration Assay
Cells were grown to 70% confluence and, before experimentation, labeled for 1 hour with 10 μmol/L CellTracker Green in serum-free medium. The dye was fixed by a 1-hour incubation in medium with 10% FCS. Subsequently, cells were washed and detached with 2 mmol/L EDTA in PBS. Next, cells were resuspended in serum-free medium and transferred to 8 μmol/L pore size HTS FluoroBlok Cell Culture Inserts (BD Falcon, Franklin Lakes, NJ). CM supplemented or not with the indicated concentrations of IGF-1 or Long R3 IGF-1 was added to the bottom well. Fluorescence values representing migrating cells on the bottom side of the insert were read during 76 cycles (each cycle comprising 4 readings spanning 2 minutes) at 37°C on a BioTek Reader. The raw fluorescence data were expressed as percentage of migration toward FCS; the data were then plotted with GraphPad Prism-4 (GraphPad Software, La Jolla, CA) using nonlinear regression fit.
In Vivo Experiments
For quantitative PCR of IGF-1 signaling-related genes in aortic tissue, 5 female apoE-deficient mice were fed regular chowad libitum for 12 weeks. Mice were then anesthetized by subcutaneous injection of ketamine (75 mg/kg; Eurovet, Bladel, the Netherlands), droperidol (1 mg/kg), fluanisone (0.75 mg/kg), and fentanyl (0.04 mg/kg; all from Janssen-Cilag, Tilburg, the Netherlands). Blood was drawn from the inferior caval vein for analysis of serum lipids. Subsequently, a 10-minute whole-body perfusion was performed using ice-cold phosphate-buffered saline, containing 1 mmol/L EDTA, administered by heart puncture (left ventricle). After perfusion, organs were excised, frozen in liquid N2, and kept at −80°C until RNA isolation. The aorta was prepared free of periadventitial fatin situ. The aortic arch was isolated from the base (most proximal to the heart) and severed from the descending aorta just behind the left subclavian artery. The descending aorta was harvested from the arch down to the bifurcation.
To determine the effect of continuous administration of Long R3 IGF-1 [an IGF-1 analog with an extended N-terminal tail and Arg for Glu substation at position 3, leading to a prolonged half-life (20 to 30 hours) due to decreased binding to IGF binding proteins] on early atherogenesis or advanced atherosclerotic lesions, mice were placed on a Western-type diet 2 weeks before lesion induction by insertion of perivascular carotid collars as previously described.20 Sterile osmotic minipumps (Alzet, type 2004, Durect Corporation, Cupertino, CA) were placed subcutaneously under isoflurane anesthesia 1 (“early” atherosclerosis) or 5 (“advanced” atherosclerosis) weeks after collar placement, respectively. Administration of Long R3 IGF-1 (n = 11 orn = 14, respectively) or control (10 mmol/L HCl) buffer (n = 15 orn = 12, respectively) by minipumps (0.25 μg/hour) was continued for 4 weeks before harvesting. During treatment, plasma Long R3 IGF-1 levels were measured by a human ELISA kit (Gropep). Plasma total cholesterol levels were measured spectrophotometrically using enzymatic procedures, and triglyceride levels were quantified using a commercially available kit (Roche Diagnostics). In both assays, Precipath standardized serum (Roche, Basel, Switzerland) was used as an internal standard. Artery harvesting for histological analysis was preceded byin situ perfusion fixation with formalin as previously described20 and tissues snap-frozen in liquid nitrogen. The specimens were stored at −20°C until further use.
Histology
The carotid arteries were sectioned in a proximal direction from the carotid bifurcation in transverse 5-μm cryosections, and mounted in order on a parallel series of slides. Cryosections were routinely stained with hematoxylin (Sigma Diagnostics) and eosin (Merck Diagnostica, Darmstadt, Germany), Masson's trichrome (Accustain kit, Sigma), and picrosirius red (Direct red 80, Sigma) according to the manufacturers' instructions. Corresponding sections on separate slides were stained immunohistochemically with antibodies against a macrophage-specific antigen (MOMA-2, polyclonal rat IgG2b; Research Diagnostics Inc, Flanders, NJ), α-smooth muscle cell actin (clone 1A4; Sigma), or an isotype-specific control antibody. Goat anti-mouse IgG peroxidase conjugate (Nordic, Tilburg, the Netherlands) and goat anti-rat IgG alkaline phosphatase conjugate (Sigma) were used as secondary antibodies, with 3,3′-diamino-benzidine and nitro blue tetrazolium as enzyme substrates (all Sigma). The images were digitized and analyzed as previously described.6,20 The point of maximal stenosis of each vessel was determined by analysis of sections at 50-μm intervals. At this point (on average, ≈0.5 mm proximal to the collar), morphometry was performed using LeicaQwin software (Leica Microsystems, Wetzlar, Germany). The intimal surface area was calculated by subtracting the free lumen area from the area circumscribed by the internal elastic lamina. The intima was subdivided into a fibrocellular cap and a necrotic core on the basis of extracellular matrix staining by hematoxylin and eosin, which was confirmed by staining for collagen by picrosirius red and Masson's trichrome. Actin-positive and MOMA-2–positive areas were determined by computer-assisted color-gated measurement and related to the total intimal surface area (Leica Qwin).
Enzyme Linked Immunoabsorbant Assay (ELISA)
TNFα and PDGF-BB concentrations in CM were determined by OptEIA (Pharmingen, San Diego, CA) and Quantikine (R&D Systems, Minneapolis, MN) immunoassays, respectively. IGF-1 and IGFBP-3 concentrations in plasma of the animals, conditioned medium, and VSMCs supernatant were measured using specific ELISAs (IGF-1 and IGFBP-3 Quantikine, R&D systems) according to the manufacturer's instructions.
Statistics
Values are expressed as mean ± SEM. A 2-tailed Student'st-test or 1-way analysis of variance Newman-Keuls tests were used in the comparison of continuous data. A level ofP < 0.05 was considered significant. Frequency data analysis was carried out by means of the Fisher's exact test.
Results
Organ Expression of IGF-1 Signaling Molecules
As expected, the liver was found to have the highest level of gene expression for IGF-1 and the primary IGF-1 binding protein in the circulation, IGFBP-3 (Figure 1). Nonetheless, detectable levels of expression of genes encoding these proteins were also found in the atherosclerotic aorta, and to a lesser extent also in isolated vSMCs. The reverse was seen for IGF-R and IGFBP-4, the genes of which were found to be more highly expressed in the aorta than in liver.
Figure 1.

Quantitative RT-PCR for IGF-1 signaling–related proteins on tissue isolates of ApoE−/− mice aged 12 weeks. IGF-1 and the primary IGF-1 binding protein in the circulation, IGFBP-3, were found to be highly expressed by the liver, and less so by the aorta and isolated vascular smooth muscle cells (vSMCs) (material from 5 mice pooled per group). The reverse was seen for IGF-R and IGFBP-4. Asc, ascending; Desc, descending.
In Vitro vSMC Expression of IGF-1 Signaling Molecules
A highly significant reduction in IGF-1 (−99%) and to a lesser extent IGF-R (−62%) expression in vSMCs in culture was seen following addition of CM (Figure 2, A and B;P < 0.001 andP < 0.01, respectively). IGFBP-3 expression was markedly enhanced following incubation with CM (Figure 2C;P < 0.001), and the inverse pattern was seen with respect to IGFBP-4 (−92%;Figure 2D). To correct for potential effects of TNFα in CM, we compared the effect of CM with control medium supplemented with the equivalent concentration (7.2 ng/ml) of TNFα. Whereas TNFα reduced IGF-1 and IGFBP-4 expression significantly, it did so to a lesser extent than CM (Figure 2, A and D;P < 0.001 andP < 0.01, resp.). By contrast, TNFα had no detectable effect on IGF-R or IGFBP-3, in contrast to CM (Figure 2, B and C). In an additional experiment, vSMCs were incubated with LPS in nonconditioned medium (15 ng/ml), which had no appreciable effects on the expression of IGF-1, IGFBP-3 or IGFBP-4 compared with controls (data not shown). Supernatant of SMC contained 50.6 ng/ml of IGF-1, and less than the detection level of the ELISA used for IGFBP-3. The conditioned medium of macrophages contained 119.4 ng/ml of IGF-1 and, again, no significant IGFBP-3 (Figure 2E).
Figure 2.

Effect of CM on expression of IGF-1, IGF-R, IGFBP-3, and IGFBP-4 in vSMCs as measured by quantitative RT-PCR. Conditioned medium (CM) reduced the expression of IGF-1, IGF-R, and IGFBP-4 while increasing IGFBP-3 levels (A–D). Whereas TNFα (at an equivalent concentration of 7.2 ng/ml) reduced IGF-1 and IGFBP-4 expression significantly, it did so to a lesser extent than CM; TNFα had no effect on IGF-R or IGFBP-3. Data are means ± SEM,n = 3–4, and expressed as relative ratio (normalized against the control).E: Concentration of IGF-1 and IGFBP3 in macrophage conditioned medium and vascular smooth muscle cell (VSMC) supernatant. Data are mean of each sample measured in duplicate for each condition. <detection indicates below detection limit of the ELISA used. **P < 0.01. ***P < 0.001.
Cell Turnover Studies
CM was found to increase the fraction of vSMCs in apoptosis by more than fivefold, from 1.4% to 7.7% (P < 0.001,Figure 3A). Addition of IGF-1 was able to almost completely rescue cells from CM-induced apoptosis at a concentration of 90 ng/ml or 120 ng/ml (Figure 3B), reflected in a reciprocal increase in viability (Figure 3C). The rate of proliferation was decreased by 89% (P < 0.001,Figure 3D) following incubation with CM, but addition of IGF-1 was not found to augment proliferation (Figure 3E). Equivalent levels of TNFα did not influence apoptosis or proliferation. There was an increased potency of Long R3 IGF-1 as compared with IGF-1 on proliferation (data not shown).
Figure 3.

Effect of CM on vSMC turnover. Conditioned mediumj (CM) caused a marked increase in apoptosis, which could be reverted by addition of IGF-1 at 90 and 120 ng/ml (A andB; propidium iodide assay,n = 3–4 andn = 1, respectively). There was a mirrored increase in cellular viability, as determined by MTT assay (C,n = 3). Proliferation determined by3H-thymidine uptake was virtually abolished by CM (D,n = 3–4); however, this effect could not be corrected by IGF-1 (E,n = 4). TNFα did not influence apoptosis or proliferation. Data are means ± SEM, and absolute values or expressed as relative ratio (normalized against the control). *P < 0.05, ***P < 0.001, ****P < 0.0001.
Expression of Extracellular Matrix Regulating Factors
mRNA levels of α-actin and col3a1 were strongly attenuated by CM (P < 0.001 andP < 0.005, resp.), whereas this effect was also notable but less dramatic with TNFα (Figure 4, A and B). The matrix-degrading enzymes matrix metalloproteinase (MMP)-13 and MMP-3 were more highly expressed after exposure to CM, again an effect that could only be partially reproduced by TNFα (Figure 4, C and D). Expression of MMP-9 was unaltered (data not shown), whereas expression of the endogenous MMP inhibitor TIMP-1 was mildly induced by CM (P < 0.001), but not by TNFα (Figure 4E). All of these effects, except TIMP-1, could be at least partially abrogated by addition of IGF-1, in a dose range from 60 ng/ml to 120 ng/ml (Figure 5, A–E). Moreover, Western blot analysis showed that supplementation of the conditioned medium with the same concentration range of IGF-1 induced a dose-dependant production of collagen 1A1, α-actin, and TIMP-1 (Figures 6, A–D), as well as procollagen 1A1 and procollagen 3A1 (Figure 6E). These effects were only partially PDGF-BB dependent, as addition of a PDGF-BB–neutralizing antibody to block the effect of PDGF-BB present in CM (659.7 pg/ml) was ineffective in reverting the effect of IGF-1 on α-actin and TIMP-1 protein expression (data not shown). These results prompted us to examine the phosphorylation of Akt/PI3K pathways, which are known to be involved in cell survival and protein synthesis. As shown inFigure 7, comparison of the phosphospecific signal with the corresponding loading control shows that addition of 120 ng/ml IGF-1 in the CM induces a rapid (already after 30 minutes) and sustained (up to 5 hours) phosphorylation of mTor, as compared with the conditioned medium supplemented with PBS. Accordingly, the phosphorylation induced by addition of IGF-1 to the CM of Akt and p70S6Kinase, which are 2 downstream mTor targets (the latter of which is also involved in protein synthesis), were also sustained for the same time frame, which suggests a robust activation induced by IGF-1 of the Akt pathway. In addition, the phosphorylation of p42/44 MAPKinase, which is a downstream target of PI3K and is known to be involved both in protein synthesis and cell survival, was also enhanced and sustained. Taken together, these results strongly suggest prolonged activation of the Akt/PI3K pathways by addition of IGF-1 to the CM, and are consistent with our other findings of increased protein synthesis and cell survival induced by this growth factor.
Figure 4.

Effect of CM on mRNA levels for α-actin, col3a1, MMP-13, MMP-3, and TIMP-1 in vSMCs as measured by quantitative RT-PCR. Conditioned medium (CM) was seen to markedly reduce the expression of α-actin and col3a1 (A andB) and to increase that of matrix-degrading enzymes MMP-13 and MMP-3 (C andD), while increasing, although only modestly, the expression of the MMP inhibitory geneTIMP-1 (E). Data are means ± SEM,n = 3–4, and expressed as relative ratio (normalized against the control). *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.005.
Figure 5.

Effect of IGF-1 on CM-induced modulation of mRNA levels for α-actin, col3a1, MMP-13, MMP-3, and TIMP-1 in vascular smooth muscle cells (vSMCs) as measured by quantitative RT-PCR. IGF-1 attenuated the effects of conditioned medium (CM) on expression of α-actin, col3a1, MMP-13, and MMP-3 in a dose range from 60 ng/ml to 120 ng/ml (A–D). There was a nonsignificant increase of TIMP-1 (E). Data are means ± SEM,n = 3–4, and expressed as relative ratio (normalized against the control). *P < 0.05, **P < 0.01, ****P < 0.005.
Figure 6.

Western blot analyses of vSMC phenotype. The effects of IGF-1 on mRNA expression were closely mirrored by stimulatory effects on protein levels of α-actin, collagen 1A1, and TIMP-1 following conditioned medium (CM) incubation (densitometric data inA–C, *P < 0.05), expressed as normalized ratio against CM, relative to household protein level (β actin); original Western blot in (D) (on cell lysates of VSMC incubated for 24 hours in macrophage CM, or CM supplemented with 60, 90, or 120 ng/ml of IGF-1. Pictures are representative of an experiment that was repeated twice). In a separate experiment (E), procollagen 1A1 and procollagen 3A1 protein levels were also found to be up-regulated relative to household protein level (tubulin). AU, arbitrary units; vSMC, vascular smooth muscle cell.
Figure 7.

Western blot analyses of supplementation of CM with IGF-1 on vSMC intracellular signaling. Effect of conditioned medium (CM) supplemented with 120 ng/ml IGF-1 (+) or PBS (−) on the phosphorylation of Akt, mTor, p70 S6 kinase, and p42/44 MAP kinase. Vascular smooth muscle cells (vSMCs) were incubated in the indicated conditions for 0 minutes, 30 minutes, 60 minutes, 120 minutes, 300 minutes, or 1440 minutes. Total Akt, mTor, p70S6kinase, and p42/44 MAP kinase serve as loading control.
Functional Consequences of IGF-1 Supplementation to CM on VSMC migration
We observed a reduction in collagenolytic capacity of the vSMC supernatant in a zymography assay following addition of IGF-1 at concentrations of 60 ng/ml and 120 ng/ml (Figure 8A). Functionally, using a modified Boyden chamber assay, we observed that, even at the highest concentration (120 ng/ml), a gradient of IGF-1 in CM did not enhance cell movement, whereas Long R3 IGF-1 increased cell migration to an extent comparable to the 10% FCS condition (Figure 8B).
Figure 8.

Functional assays of vSMCs. There was an overall reduction of collagenolytic activity of IGF-1–treated vSMCs as measured by zymography (A,arrow, lighter shade indicates more collagen break-down), compared with CM containing no IGF-1.B: Cell migration assay of vascular smooth muscle cells (vSMCs) toward medium containing 10% FCS (condition FCS), conditioned medium (CM), or conditioned medium supplemented with the indicated concentrations of IGF-1 or Long R3 IGF-1. Results represent the mean ± SEM (n = 3) and are representative of 2 independent experiments performed in duplicate.
In Vivo Effects of Long R3 IGF-1 on Early and Advanced Atherosclerosis
Plasma Long R3 IGF-1 levels were in the effective range throughout the duration of the experiments (reaching 13.1 ng/ml at the time of sacrifice), and these levels did not alter animal weight or serum glucose and lipid levels (data not shown). Long R3 IGF-1 did not significantly alter absolute plaque or media surface area in both early and advanced atherosclerosis, but was found to decrease lumen stenosis by 31% and core size by 31% (Figure 9, A–I andFigure 10A). Cap/core ratio was increased by 106.5% in early atherosclerosis, and unaltered in advanced plaques. Long R3 IGF-1 caused a trend toward an increase in ASMA (α-smooth muscle actin)-positive smooth muscle cell content in an early setting, and more than doubled ASMA content in advanced plaques. Levels of endogenous IGF-1 and IGFBP-3 were unaffected by the treatment (Figure 10, B and C).
Figure 9.
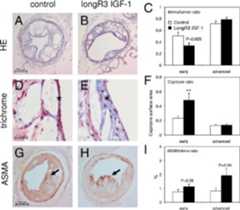
Morphology of collar-induced plaques. Long R3 IGF-1 led to a 31.3% reduction in lumen stenosis, expressed as intima/lumen ratio [C; control,n = 15, Long R3 IGF-1,n = 11; representative hematoxylin and eosin (HE) stains inA andB (magnification ×100)] and a 31.1% reduction in core size (F, **P < 0.01).D andE: Representative trichrome stains of lesional caps (marked with anasterisk, magnification ×400) in early atherogenesis. Representative ASMA-stained advanced plaques are depicted in (G andH,arrows indicate positive staining in the lesional cap). Quantitative assessment showed a more than twofold increase in ASMA-positive smooth muscle cell content in advanced plaques (I; control,n = 12, Long R3 IGF-1,n = 14). Data are means ± SEM.
Figure 10.

Morphometric data of collar-induced plaques. Morphometric data are summarized in tabular format inA. Concentration of IGF-1 and IGFBP3 in plasma of control of Long R3 IGF-1 treated mice as measured by ELISA in the early atherosclerosis experiment are represented inB andC. Long R3 IGF-1 does not change the plasma levels (ng/ml) of IGF-1 and IGFBP-3. Mice received Long R3 IGF-1 (filled bars) or control buffer (open bars) via a subcutaneous osmotic minipump and circulating levels of IGF-1 and IGFBP3 in the plasma were measured. Data are mean ± SE ofn = 15 mice in the group Long R3 IGF-1 andn = 9 mice in the control group.
In addition to these morphometric parameters, we also found pathophysiological evidence for increased plaque stability in the vessels of Long R3 IGF-1–treated animals in a reduced rate of intraplaque hemorrhage (Figure 11).
Figure 11.
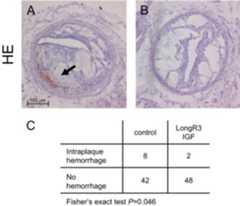
Effect of Long R3 IGF-1 on plaque stability. Long R3 IGF-1 treatment significantly reduced the occurrence of intraplaque hemorrhage in all animals analyzed [A andB; representative hematoxylin and eosin (HE) stains (magnification ×100), hemorrhage marked with anarrow]. When analyzed in a 2 × 2 contingency table with the aid of Fisher's exact test, this effect was indeed significant (C; 4% vs. 16%, respectively).
Discussion
Although the overall role of IGF-1 in atherosclerosis remains unclear, it has recently emerged that in the context of plaque stability, IGF-1 may have a potentially stabilizing role through its antiapoptotic effects on smooth muscle cells. IGF-1 is an important mitogenic, antiapoptotic, and promigratory factor for vSMCs.21–24 Induction of smooth muscle cell apoptosis by oxLDL, on the other hand, is mediated by down-regulation of IGF-1R,25,26 whereas overexpression of IGF-1R by adenoviral transduction can counteract oxLDL-induced apoptosis.27 IGF-1 is seen as an archetypal fibrogenic factor for fibroblasts in various types of wound healing and inflammatory fibrotic conditions, but its role in the modulation of smooth muscle cell phenotype remains largely unknown. Rat intestinal smooth muscle cells have been shown to up-regulate expression of IGFBP-3, -4, and -5 and of type I collagen following exposure to IGF-1.28–30 Also, IGF-1 induces lower levels of collagenase activity in colonic smooth muscle cells, which could affect the turnover of extracellular matrix by inhibiting the degradation side of the equilibrium.29 Similar effects could conceivably occur in vSMCs, and IGF-1 has indeed been found to augment the expression of elastin by bovine, and of collagen by porcine vSMCs, respectively.30,31
We speculated that the pro-oxidant and proinflammatory extracellular milieu prevailing in the atherosclerotic plaque could play a role in the down-regulation of IGF-1 signaling that has been described in previous studies,16,17 and that interaction with another main cellular plaque constituent, the macrophage, could be of special interest. In this study, we initially confirmed the expression of various components of the IGF-1 signaling pathway in murine atherosclerotic aortas. Thus, although IGF-1 and its most important binding protein in the circulation (IGFBP-3) were, as expected, primarily produced by the liver,IGF-R andIGFBP-4 were also found to be significantly expressed in both the ascending and the descending aorta. The expression of both of these genes was found to decrease with the age of the lesions (from 6 to 21 weeks, data not shown). In the case ofIGF-R, this conforms to earlier human data, in which expression ofIGF-R was found to be reduced in atherosclerotic vessels compared with nondiseased arteries.16,17 The decrease in IGFBP-4 with maturation of lesions is unexpected, however, and may be partly explained by the fact that IGFBP-4 is primarily produced by vSMCs in the plaque. It is especially the vSMC population that is reduced in advanced plaques, concomitantly conferring an unstable plaque phenotype. Alternatively, the macrophage-rich milieu of the plaque may selectively attenuate IGFBP-4, as confirmed in ourin vitro studies.
In vitro, medium conditioned by LPS-stimulated, and thus M1-polarized, murine macrophages was found to influence several aspects of IGF-1 signaling in vSMCs. The overall balance of these effects could be seen as inhibitory with a decreased expression of IGF-1 and IGF-R and an increase in IGFBP-3, albeit partly offset by a decrease in IGFBP-4. It remains to be clarified as to what relative extent these effects can be attributed to specific cytokines or other, possibly yet undefined, macrophage-derived intercellular mediators. Thus, although TNFα is both a known major proatherogenic molecule and an effective modulator of IGF-1 and IFGBP-3 expression in vascular smooth muscle,32 the observed effects can only be partially attributed to TNFα elaborated by LPS-stimulated macrophages in the case of IGF-1 and IGFBP-4. Furthermore, a TNFα-mediated effect is unlikely concerning IGF-R and IGFBP-3. Other macrophage-derived cytokines and non-cytokine mediators have also been found to play an important role in the regulation of the expression of IGF-1, IGF-R1, and the IGFBPs.11,33 Whatever the role of these individual factors in the regulation of IGF-1 signaling may be, the overall effect appears to be almost completely inhibitory toward IGF-1 signaling. The latter was confirmed in cell turnover experiments, in which CM increased apoptosis by more than fivefold. This effect was not attributable to TNFα and may be at least partially mediated by attenuated IGF-1 signaling, as addition of IGF-1 substantially counteracted the effect of CM on apoptosis. Furthermore, CM virtually abolished proliferative activity in vSMCs, whereas TNFα had no effect.
Collagen deposition in the later stages of atherosclerosis is required for structural support in the plaque cap to prevent the development of unstable lesions.34 The effect of MMPs on plaque progression may be bimodal depending on their relative activity in permitting migration of destabilizing (macrophage) or stabilizing (vSMC) cells into the plaque, and destabilization of plaques due to the degradation of lesional matrix.35–37 Various MMPs have been implicated in plaque destabilization, including MMP-1,38 MMP-339 and MMP-9.37,40 From the oncology field, we have in recent years learned that IGF-1 can influence the expression of various matrix-degrading enzymes by tumor cells.41–43 The influence of IGF-1 on the expression of MMPs in the atherosclerotic plaque, however, remains largely uncharted. Risinger et al have found IGF-1 to have a stimulatory effect on MMP-2 production by vSMCs.44 By contrast, growth hormone replacement has been shown to decrease plasma levels of MMP-2 and MMP-9 in growth hormone–deficient individuals, an effect that is thought to be mediated by IGF-1 and to contribute to the reduction in vascular mortality in these patients.45 In our study, the overall effect of CM was found to be inhibitory toward collagen production and procollagenolytic, with CM effecting a decrease in the expression of α-actin and Col3a1 and an increase in the collagenolytic enzymes MMP-13 (the main interstitial murine collagenase, functionally equivalent to human MMP-1) and MMP-3. These effects were to some extent attributable to TNFα, but counteracted by IGF-1. CM caused a minor increase in expression of TIMP-1, but IGF-1 administration further enhanced the expression of this endogenous inhibitor of metalloproteinase activity. On balance, the effects of IGF-1 were anticollagenolytic, as evidenced by zymography data. A direct stimulation of migratory capacity of vSMCs by IGF-1 has previously been described,46 and to further examine the functional consequences on addition of IGF-1, or Long R3 IGF-1, we used a modified Boyden chamber assay. Interestingly, we observed that, although Long R3 IGF-1 restored cell migration to a level similar to 10% FCS in this assay, IGF-1 added to CM did not induce a significant difference as compared to the CM condition. As we have also shown, by means of ELISA, that IGFBP-3 levels in conditioned medium and vSMC supernatant are negligible, the increased effect on migration by Long R3 IGF-1 is likely to be independent of its increased bioavailabilityin vivo (due to the much reduced affinity of Long R3 IGF-1 for inhibitory IGFBP-3).
There is an apparent lack of consistency in the individual findings of differential expression with respect to phenotype modulation by IGF-1, as on the one hand, these may be seen as typical of a differentiated phenotype (increased α-smooth muscle actin expression) and on the other of a synthetically active, migratory phenotype, as described above. This could be due to the concomitant up-regulation of contractile cytoskeletal proteins through activation of the PI3 kinase pathway and induction of a synthetic phenotype by activation of the MAPK pathway, as we found both of these pathways to be activated by addition of IGF-1.
In vivo, we found divergent, largely stabilizing IGF-1–mediated effects in different stages of atherosclerosis. In early atherosclerosis, Long R3 IGF-1 was seen, not only to decrease plaque burden by significantly reducing lumen stenosis, but also to alter plaque morphology, by decreasing necrotic core size and increasing the cap/core ratio, which is generally considered to be a favorable indicator of plaque stability.34 In advanced plaques, as is perhaps to be expected due to the already large size and mature, complex morphology of these lesions, Long R3 IGF-1 did not have an effect on overall plaque burden or cap size. It did, however, increase the content of ASMA-expressing smooth muscle cells in the plaque, which again could be viewed as a sign of improved plaque stability. This is corroborated by the fact that the rate of intraplaque hemorrhage was significantly reduced by treatment with Long R3 IGF-1. These findings are in line with the skewing toward a stabilizing vSMC phenotype suggested by thein vitro data.
Several issues should be kept in mind when interpreting the data. First, one might argue that macrophage CM is a complex mixture of components, which might render it difficult from which to gain significant mechanistic insights into the observed effects. This model is, however, commonly accepted to simulate the microenvironment of the atherosclerotic milieu forin vitro experiments and has been validated by different groups in a variety of studies. Several factors produced by macrophages in the atherosclerotic plaque have been found to modulate IGF-1 signaling. This, for instance, identified oxLDL16,17 and TNFα32 as potential culprits in the modulation of IGF-1 in atherosclerotic lesions.
Second, for quantitative assessment of IGF-1 signaling-related proteins in tissue homogenates, we used the RT-PCR approach rather than protein measurement. This was motivated by the fact that the targets of interest are likely to be present at relatively low levels in the various murine tissues, which due to their sheer size are likely to contain at best only modest amounts of protein, in practicality often precluding their quantitative detection by Western blot/ELISA. Also, rather than determining absolute levels of different proteins, we were primarily interested in delineating relative variation in organ-specific expression patterns for defined targets. We thus specifically opted for this approach as we have previously found RT-PCR to be a valuable tool in correlating, though admittedly not equating, to relative expression levels in such experiments.19,47,48
Finally, one might consider whether a meaningful morphometric analysis of the plaque can be performed on parameters like cap/core ratio. Relative thinning of the cap and an increase in the lesional necrotic core are (in addition to inflammation) currently seen as the best histological indicators of plaque vulnerability to rupture in humans.49 In previous studies of murine carotid plaques, we have found that the determination of the cap/core ratio is highly reproducible, primarily due to the use of ancillary stains such as Masson's trichrome and picrosirius red, which highlight the collagenous areas of the lesion.7,8,37,48 The thus derived cap/core ratio was found to show close correlation with signs of plaque rupture in a model of p53-induced plaque destabilization.7 We therefore believe that this is a technique that, in our hands, yields reproducible and biologically relevant results.
In conclusion, we believe that the above results indicate that IGF-1 in atherosclerosis may have a role in preventing plaque instability, not only by modulating smooth muscle cell turnover toward a proapoptotic and antiproliferative state, but also by skewing the balance of collagen-rich matrix production and degradation toward a more stable extracellular compartment. Although potentially detrimental in long-term use due to its alleged procarcinogenic effects,50 IGF-1 treatment may thus hold promise in short-term plaque phenotype modulation, especially when combined with targeting strategies aimed at specifically directing treatment to the vasculature.
Footnotes
Supported by a NWO-VENI grant (J.vd.T.); by the Dutch Heart Foundation (Molecular Cardiology Program grant M93.001 (J.v.d.T. and E.B.); by the Established Investigatorship grant 2003T201 (E.B.); and by Dr. E. Dekker Programme Junior Staff Member Fellowship grant 2008T050 (J.v.d.T.).
References
- 1.Libby P. Inflammation in atherosclerosis. Nature. 2002;420:868–874. doi: 10.1038/nature01323. [DOI] [PubMed] [Google Scholar]
- 2.Stoll G., Bendszus M. Inflammation and atherosclerosis: novel insights into plaque formation and destabilization. Stroke. 2006;37:1923–1932. doi: 10.1161/01.STR.0000226901.34927.10. [DOI] [PubMed] [Google Scholar]
- 3.Dollery C.M., Libby P. Atherosclerosis and proteinase activation. Cardiovasc Res. 2006;69:625–635. doi: 10.1016/j.cardiores.2005.11.003. [DOI] [PubMed] [Google Scholar]
- 4.Littlewood T.D., Bennett M.R. Apoptotic cell death in atherosclerosis. Curr Opin Lipidol. 2003;14:469–475. doi: 10.1097/00041433-200310000-00007. [DOI] [PubMed] [Google Scholar]
- 5.Clarke M., Bennett M. The emerging role of vascular smooth muscle cell apoptosis in atherosclerosis and plaque stability. Am J Nephrol. 2006;26:531–535. doi: 10.1159/000097815. [DOI] [PubMed] [Google Scholar]
- 6.Clarke M.C., Figg N., Maguire J.J., Davenport A.P., Goddard M., Littlewood T.D., Bennett M.R. Apoptosis of vascular smooth muscle cells induces features of plaque vulnerability in atherosclerosis. Nat Med. 2006;12:1075–1080. doi: 10.1038/nm1459. [DOI] [PubMed] [Google Scholar]
- 7.von der Thusen J.H., van Vlijmen B.J., Hoeben R.C., Kockx M.M., Havekes L.M., van Berkel T.J., Biessen E.A. Induction of atherosclerotic plaque rupture in apolipoprotein E−/− mice after adenovirus-mediated transfer of p53. Circulation. 2002;105:2064–2070. doi: 10.1161/01.cir.0000015502.97828.93. [DOI] [PubMed] [Google Scholar]
- 8.Zadelaar A.S., von der Thusen J.H., Boesten L.S., Hoeben R.C., Kockx M.M., Versnel M.A., van Berkel T.J., Havekes L.M., Biessen E.A., van Vlijmen B.J. Increased vulnerability of advanced atherosclerosis in ApoE-deficient mice following adenovirus-mediated Fas ligand gene transfer. Atherosclerosis. 2005;183:244–250. doi: 10.1016/j.atherosclerosis.2005.03.044. [DOI] [PubMed] [Google Scholar]
- 9.Bayes-Genis A., Conover C.A., Schwartz R.S. The insulin-like growth factor axis: a review of atherosclerosis and restenosis. Circ Res. 2000;86:125–130. doi: 10.1161/01.res.86.2.125. [DOI] [PubMed] [Google Scholar]
- 10.Grant M.B., Wargovich T.J., Ellis E.A., Tarnuzzer R., Caballero S., Estes K., Rossing M., Spoerri P.E., Pepine C. Expression of IGF-I, IGF-I receptor and IGF binding proteins-1, -2, -3, -4 and -5 in human atherectomy specimens. Regul Pept. 1996;67:137–144. doi: 10.1016/s0167-0115(96)00124-3. [DOI] [PubMed] [Google Scholar]
- 11.Delafontaine P., Song Y.H., Li Y. Expression, regulation, and function of IGF-1, IGF-1R, and IGF-1 binding proteins in blood vessels. Arterioscler Thromb Vasc Biol. 2004;24:435–444. doi: 10.1161/01.ATV.0000105902.89459.09. [DOI] [PubMed] [Google Scholar]
- 12.Kawachi S., Takeda N., Sasaki A., Kokubo Y., Takami K., Sarui H., Hayashi M., Yamakita N., Yasuda K. Circulating insulin-like growth factor-1 and insulin-like growth factor binding protein-3 are associated with early carotid atherosclerosis. Arterioscler Thromb Vasc Biol. 2005;25:617–621. doi: 10.1161/01.ATV.0000154486.03017.35. [DOI] [PubMed] [Google Scholar]
- 13.Hietaniemi M., Poykko S.M., Ukkola O., Paivansalo M., Antero Kesaniemi Y. IGF-I concentrations are positively associated with carotid artery atherosclerosis in women. Ann Med. 2005;37:373–382. doi: 10.1080/07853890510011967. [DOI] [PubMed] [Google Scholar]
- 14.Juul A., Scheike T., Davidsen M., Gyllenborg J., Jorgensen T. Low serum insulin-like growth factor I is associated with increased risk of ischemic heart disease: a population-based case-control study. Circulation. 2002;106:939–944. doi: 10.1161/01.cir.0000027563.44593.cc. [DOI] [PubMed] [Google Scholar]
- 15.Sukhanov S., Higashi Y., Shai S.Y., Vaughn C., Mohler J., Li Y., Song Y.H., Titterington J., Delafontaine P. IGF-1 reduces inflammatory responses, suppresses oxidative stress and decreases atherosclerosis progression in Apoe-deficient mice. Arterioscler Thromb Vasc Biol. 2007;27:2684–2690. doi: 10.1161/ATVBAHA.107.156257. [DOI] [PubMed] [Google Scholar]
- 16.Okura Y., Brink M., Zahid A.A., Anwar A., Delafontaine P. Decreased expression of insulin-like growth factor-1 and apoptosis of vascular smooth muscle cells in human atherosclerotic plaque. J Mol Cell Cardiol. 2001;33:1777–1789. doi: 10.1006/jmcc.2001.1441. [DOI] [PubMed] [Google Scholar]
- 17.Patel V.A., Zhang Q.J., Siddle K., Soos M.A., Goddard M., Weissberg P.L., Bennett M.R. Defect in insulin-like growth factor-1 survival mechanism in atherosclerotic plaque-derived vascular smooth muscle cells is mediated by reduced surface binding and signaling. Circ Res. 2001;88:895–902. doi: 10.1161/hh0901.090305. [DOI] [PubMed] [Google Scholar]
- 18.Michon I.N., Penning L.C., Molenaar T.J., van Berkel T.J., Biessen E.A., Kuiper J. The effect of TGF-beta receptor binding peptides on smooth muscle cells. Biochem Biophys Res Commun. 2002;293:1279–1286. doi: 10.1016/S0006-291X(02)00378-9. [DOI] [PubMed] [Google Scholar]
- 19.'t Hoen P.A., Van der Lans C.A., Van Eck M., Bijsterbosch M.K., Van Berkel T.J., Twisk J. Aorta of ApoE-deficient mice responds to atherogenic stimuli by a prelesional increase and subsequent decrease in the expression of antioxidant enzymes. Circ Res. 2003;93:262–269. doi: 10.1161/01.RES.0000082978.92494.B1. [DOI] [PubMed] [Google Scholar]
- 20.von der Thusen J.H., van Berkel T.J., Biessen E.A. Induction of rapid atherogenesis by perivascular carotid collar placement in apolipoprotein E-deficient and low-density lipoprotein receptor-deficient mice. Circulation. 2001;103:1164–1170. doi: 10.1161/01.cir.103.8.1164. [DOI] [PubMed] [Google Scholar]
- 21.Arnqvist H.J., Bornfeldt K.E., Chen Y., Lindstrom T. The insulin-like growth factor system in vascular smooth muscle: interaction with insulin and growth factors. Metabolism. 1995;44:58–66. doi: 10.1016/0026-0495(95)90222-8. [DOI] [PubMed] [Google Scholar]
- 22.Scheidegger K.J., Du J., Delafontaine P. Distinct and common pathways in the regulation of insulin-like growth factor-1 receptor gene expression by angiotensin II and basic fibroblast growth factor. J Biol Chem. 1999;274:3522–3530. doi: 10.1074/jbc.274.6.3522. [DOI] [PubMed] [Google Scholar]
- 23.Delafontaine P., Anwar A., Lou H., Ku L. G-protein coupled and tyrosine kinase receptors: evidence that activation of the insulin-like growth factor I receptor is required for thrombin-induced mitogenesis of rat aortic smooth muscle cells. J Clin Invest. 1996;97:139–145. doi: 10.1172/JCI118381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Du J., Brink M., Peng T., Mottironi B., Delafontaine P. Thrombin regulates insulin-like growth factor-1 receptor transcription in vascular smooth muscle: characterization of the signaling pathway. Circ Res. 2001;88:1044–1052. doi: 10.1161/hh1001.090840. [DOI] [PubMed] [Google Scholar]
- 25.Scheidegger K.J., James R.W., Delafontaine P. Differential effects of low density lipoproteins on insulin-like growth factor-1 (IGF-1) and IGF-1 receptor expression in vascular smooth muscle cells. J Biol Chem. 2000;275:26864–26869. doi: 10.1074/jbc.M002887200. [DOI] [PubMed] [Google Scholar]
- 26.Higashi Y., Peng T., Du J., Sukhanov S., Li Y., Itabe H., Parthasarathy S., Delafontaine P. A redox-sensitive pathway mediates oxidized LDL-induced downregulation of insulin-like growth factor-1 receptor. J Lipid Res. 2005;46:1266–1277. doi: 10.1194/jlr.M400478-JLR200. [DOI] [PubMed] [Google Scholar]
- 27.Li Y., Higashi Y., Itabe H., Song Y.H., Du J., Delafontaine P. Insulin-like growth factor-1 receptor activation inhibits oxidized LDL-induced cytochrome C release and apoptosis via the phosphatidylinositol 3 kinase/Akt signaling pathway. Arterioscler Thromb Vasc Biol. 2003;23:2178–2184. doi: 10.1161/01.ATV.0000099788.31333.DB. [DOI] [PubMed] [Google Scholar]
- 28.Zimmermann E.M., Li L., Hou Y.T., Cannon M., Christman G.M., Bitar K.N. IGF-I induces collagen and IGFBP-5 mRNA in rat intestinal smooth muscle. Am J Physiol. 1997;273:G875–G882. doi: 10.1152/ajpgi.1997.273.4.G875. [DOI] [PubMed] [Google Scholar]
- 29.Zeeh J.M., Riley N.E., Hoffmann P., Reinshagen M., Goebell H., Gerken G. Expression of insulin-like growth factor binding proteins and collagen in experimental colitis in rats. Eur J Gastroenterol Hepatol. 2001;13:851–858. doi: 10.1097/00042737-200107000-00014. [DOI] [PubMed] [Google Scholar]
- 30.Xin X., Hou Y.T., Li L., Schmiedlin-Ren P., Christman G.M., Cheng H.L., Bitar K.N., Zimmermann E.M. IGF-I increases IGFBP-5 and collagen alpha1(I) mRNAs by the MAPK pathway in rat intestinal smooth muscle cells. Am J Physiol Gastrointest Liver Physiol. 2004;286:G777–G783. doi: 10.1152/ajpgi.00293.2003. [DOI] [PubMed] [Google Scholar]
- 31.Badesch D.B., Lee P.D., Parks W.C., Stenmark K.R. Insulin-like growth factor I stimulates elastin synthesis by bovine pulmonary arterial smooth muscle cells. Biochem Biophys Res Commun. 1989;160:382–387. doi: 10.1016/0006-291x(89)91667-7. [DOI] [PubMed] [Google Scholar]
- 32.Anwar A., Zahid A.A., Scheidegger K.J., Brink M., Delafontaine P. Tumor necrosis factor-alpha regulates insulin-like growth factor-1 and insulin-like growth factor binding protein-3 expression in vascular smooth muscle. Circulation. 2002;105:1220–1225. doi: 10.1161/hc1002.105187. [DOI] [PubMed] [Google Scholar]
- 33.Jia G., Cheng G., Soundararajan K., Agrawal D.K. Insulin-like growth factor-I receptors in atherosclerotic plaques of symptomatic and asymptomatic patients with carotid stenosis: effect of IL-12 and IFN-gamma. Am J Physiol Heart Circ Physiol. 2007;292:H1051–H1057. doi: 10.1152/ajpheart.00801.2006. [DOI] [PubMed] [Google Scholar]
- 34.Virmani R., Burke A.P., Farb A., Kolodgie F.D. Pathology of the vulnerable plaque. J Am Coll Cardiol. 2006;47:C13–C18. doi: 10.1016/j.jacc.2005.10.065. [DOI] [PubMed] [Google Scholar]
- 35.Newby A.C. Dual role of matrix metalloproteinases (matrixins) in intimal thickening and atherosclerotic plaque rupture. Physiol Rev. 2005;85:1–31. doi: 10.1152/physrev.00048.2003. [DOI] [PubMed] [Google Scholar]
- 36.Johnson J.L., George S.J., Newby A.C., Jackson C.L. Divergent effects of matrix metalloproteinases 3, 7, 9, and 12 on atherosclerotic plaque stability in mouse brachiocephalic arteries. Proc Natl Acad Sci U S A. 2005;102:15575–15580. doi: 10.1073/pnas.0506201102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.de Nooijer R., Verkleij C.J., von der Thusen J.H., Jukema J.W., van der Wall E.E., van Berkel T.J., Baker A.H., Biessen E.A. Lesional overexpression of matrix metalloproteinase-9 promotes intraplaque hemorrhage in advanced lesions but not at earlier stages of atherogenesis. Arterioscler Thromb Vasc Biol. 2006;26:340–346. doi: 10.1161/01.ATV.0000197795.56960.64. [DOI] [PubMed] [Google Scholar]
- 38.Nikkari S.T., O'Brien K.D., Ferguson M., Hatsukami T., Welgus H.G., Alpers C.E., Clowes A.W. Interstitial collagenase (MMP-1) expression in human carotid atherosclerosis. Circulation. 1995;92:1393–1398. doi: 10.1161/01.cir.92.6.1393. [DOI] [PubMed] [Google Scholar]
- 39.Wu T.C., Leu H.B., Lin W.T., Lin C.P., Lin S.J., Chen J.W. Plasma matrix metalloproteinase-3 level is an independent prognostic factor in stable coronary artery disease. Eur J Clin Invest. 2005;35:537–545. doi: 10.1111/j.1365-2362.2005.01548.x. [DOI] [PubMed] [Google Scholar]
- 40.Fukuda D., Shimada K., Tanaka A., Kusuyama T., Yamashita H., Ehara S., Nakamura Y., Kawarabayashi T., Iida H., Yoshiyama M., Yoshikawa J. Comparison of levels of serum matrix metalloproteinase-9 in patients with acute myocardial infarction versus unstable angina pectoris versus stable angina pectoris. Am J Cardiol. 2006;97:175–180. doi: 10.1016/j.amjcard.2005.08.020. [DOI] [PubMed] [Google Scholar]
- 41.Zhang D., Samani A.A., Brodt P. The role of the IGF-I receptor in the regulation of matrix metalloproteinases, tumor invasion and metastasis. Horm Metab Res. 2003;35:802–808. doi: 10.1055/s-2004-814143. [DOI] [PubMed] [Google Scholar]
- 42.Long L., Navab R., Brodt P. Regulation of the Mr 72,000 type IV collagenase by the type I insulin-like growth factor receptor. Cancer Res. 1998;58:3243–3247. [PubMed] [Google Scholar]
- 43.Grzmil M., Hemmerlein B., Thelen P., Schweyer S., Burfeind P. Blockade of the type I IGF receptor expression in human prostate cancer cells inhibits proliferation and invasion, up-regulates IGF binding protein-3, and suppresses MMP-2 expression. J Pathol. 2004;202:50–59. doi: 10.1002/path.1492. [DOI] [PubMed] [Google Scholar]
- 44.Risinger G.M., Jr., Hunt T.S., Updike D.L., Bullen E.C., Howard E.W. Matrix metalloproteinase-2 expression by vascular smooth muscle cells is mediated by both stimulatory and inhibitory signals in response to growth factors. J Biol Chem. 2006;281:25915–25925. doi: 10.1074/jbc.M513513200. [DOI] [PubMed] [Google Scholar]
- 45.Randeva H.S., Lewandowski K.C., Komorowski J., Murray R.D., O'Callaghan C.J., Hillhouse E.W., Stepien H., Shalet S.M. Growth hormone replacement decreases plasma levels of matrix metalloproteinases (2 and 9) and vascular endothelial growth factor in growth hormone-deficient individuals. Circulation. 2004;109:2405–2410. doi: 10.1161/01.CIR.0000129763.51060.77. [DOI] [PubMed] [Google Scholar]
- 46.Pukac L., Huangpu J., Karnovsky M.J. Platelet-derived growth factor-BB, insulin-like growth factor-I, and phorbol ester activate different signaling pathways for stimulation of vascular smooth muscle cell migration. Exp Cell Res. 1998;242:548–560. doi: 10.1006/excr.1998.4138. [DOI] [PubMed] [Google Scholar]
- 47.von der Thüsen J.H., Kuiper J., Fekkes M.L., De Vos P., Van Berkel T.J., Biessen E.A. Attenuation of atherogenesis by systemic and local adenovirus-mediated gene transfer of interleukin-10 in LDLr−/− mice. FASEB J. 2001;15:2730–2732. doi: 10.1096/fj.01-0483fje. [DOI] [PubMed] [Google Scholar]
- 48.de Nooijer R., Verkleij C.J., von der Thüsen J.H., Jukema J.W., van der Wall E.E., van Berkel T.J., Baker A.H., Biessen E.A. Lesional overexpression of matrix metalloproteinase-9 promotes intraplaque hemorrhage in advanced lesions but not at earlier stages of atherogenesis. Arterioscler Thromb Vasc Biol. 2006;26:340–346. doi: 10.1161/01.ATV.0000197795.56960.64. [DOI] [PubMed] [Google Scholar]
- 49.Finn A.V., Nakano M., Narula J., Kolodgie F.D., Virmani R. Concept of vulnerable/unstable plaque. Arterioscler Thromb Vasc Biol. 2010;30:1282–1292. doi: 10.1161/ATVBAHA.108.179739. [DOI] [PubMed] [Google Scholar]
- 50.Moschos S.J., Mantzoros C.S. The role of the IGF system in cancer: from basic to clinical studies and clinical applications. Oncology. 2002;63:317–332. doi: 10.1159/000066230. [DOI] [PubMed] [Google Scholar]