






Oxygen as Acceptor
Vitaliy B Borisov
Michael I Verkhovsky
Address correspondence to Vitaliy B. Borisov,bor@genebee.msu.su
Received 2015 Aug 19; Revision requested 2015 Sep 14; Collection date 2015 Dec.
Abstract
Like most bacteria,Escherichia coli has a flexible and branched respiratory chain that enables the prokaryote to live under a variety of environmental conditions, from highly aerobic to completely anaerobic. In general, the bacterial respiratory chain is composed of dehydrogenases, a quinone pool, and reductases. Substrate-specific dehydrogenases transfer reducing equivalents from various donor substrates (NADH, succinate, glycerophosphate, formate, hydrogen, pyruvate, and lactate) to a quinone pool (menaquinone, ubiquinone, and dimethylmenoquinone). Then electrons from reduced quinones (quinols) are transferred by terminal reductases to different electron acceptors. Under aerobic growth conditions, the terminal electron acceptor is molecular oxygen. A transfer of electrons from quinol to O2 is served by two major oxidoreductases (oxidases), cytochromebo3 encoded bycyoABCDE and cytochromebd encoded bycydABX. Terminal oxidases of aerobic respiratory chains of bacteria, which use O2 as the final electron acceptor, can oxidize one of two alternative electron donors, either cytochromec or quinol. This review compares the effects of different inhibitors on the respiratory activities of cytochromebo3 and cytochromebd inE. coli. It also presents a discussion on the genetics and the prosthetic groups of cytochromebo3 and cytochromebd. TheE. coli membrane contains three types of quinones that all have an octaprenyl side chain (C40). It has been proposed that thebo3 oxidase can have two ubiquinone-binding sites with different affinities.
“What’s new” in the revised article: The revised article comprises additional information about subunit composition of cytochromebd and its role in bacterial resistance to nitrosative and oxidative stresses. Also, we present the novel data on the electrogenic function ofappBCX-encoded cytochromebd-II, a secondbd-type oxidase that had been thought not to contribute to generation of a proton motive force inE. coli, although its spectral properties closely resemble those ofcydABX-encoded cytochromebd.
TWO TYPES OF METABOLISM, TWO TYPES OF OXIDASES
Anaerobiosis versus Aerobiosis inEscherichia coli
Like most bacteria,Escherichia coli has a flexible and branched respiratory chain that enables the prokaryote to live under a variety of environmental conditions, from highly aerobic to completely anaerobic.E. coli induces the expression of those respiratory components that are best suited to a particular environment. In general, the bacterial respiratory chain is composed of dehydrogenases, a quinone pool, and reductases. Substrate-specific dehydrogenases transfer reducing equivalents from various donor substrates (NADH, succinate,α-glycerophosphate, formate, hydrogen, pyruvate, and lactate) to a quinone pool (menaquinone, ubiquinone, and dimethylmenoquinone). Then electrons from reduced quinones (quinols) are transferred by terminal reductases to different electron acceptors. Under aerobic growth conditions, the terminal electron acceptor is molecular oxygen. A transfer of electrons from quinol to O2 is served by two major oxidoreductases (oxidases), cytochromebo3 and cytochromebd (it is worth noting that accumulated evidence over the past few years also suggests the contribution of a secondbd-type oxidase, cytochromebd-II, to the electron transfer and membrane potential generation). When oxygen is not available (under anaerobic conditions), alternative terminal electron acceptors, including nitrate, nitrite, dimethyl sulfoxide, trimethylamineN-oxide, and fumarate, can be used, and the reaction is catalyzed by nitrate reductases, nitrite reductase, dimethyl sulfoxide reductases, trimethylamineN-oxide reductase, and fumarate reductase, respectively (reviewed in references1 and2).
Two Types of Quinol Oxidases Only
Terminal oxidases of aerobic respiratory chains of bacteria, which use O2 as the final electron acceptor, can oxidize one of two alternative electron donors, either cytochromec or quinol. Oxidases utilizing cytochromec are called cytochromec oxidases, whereas oxidases oxidizing quinol are referred to as quinol oxidases (3,4,5,6,7,8,9,10,11,12,13). Cytochromec oxidases cannot directly accept reducing equivalents from quinol. For this purpose, there is an additional, middle component of the respiratory chain between dehydrogenase and oxidase, the cytochromebc1 complex, which enables the transfer of electrons from quinol to cytochromec. Respiratory chains of many aerobic bacteria, such asParacoccus denitrificans andAzotobacter vinelandii, contain thebc1 complex and both types of terminal oxidase, cytochromec and quinol oxidases. For instance,P. denitrificans has one quinol oxidase (ba3) and two cytochromec oxidases (aa3 andcbb3) (14,15).A. vinelandii has two quinol oxidases (bo3 andbd) and one cytochromec oxidase (cbb3) (reviewed in references16 and17). As shown inFig. 1, the aerobic respiratory chain ofE. coli is much simpler than those ofP. denitrificans andA. vinelandii. It lacks the cytochromebc1 complex and any cytochromec oxidase but contains instead three quinol oxidases,bo3,bd, andbd-II (reviewed in references17,18,19,20, and21).
Figure 1.

Simplified view of theE. coli respiratory chain under aerobic and microaerobic conditions. The two NADH-quinone oxidoreductases called NDH-I and NDH-II and succinate-quinone oxidoreductase (SQR) transfer reducing equivalents to ubiquinone-8 (UQ-8) to yield reduced UQ-8, ubiquinol-8. Three quinol-oxygen oxidoreductases, cytochromebo3 (CyoABCD), cytochromebd (CydABX), and cytochromebd-II (AppBCX), oxidize ubiquinol-8 and reduce O2 to 2H2O. CydABX and possibly AppBCX oxidize menaquinol-8. NDH-I, CyoABCD, CydABX, and AppBCX are coupled (ΔμH+ generators); NDH-II and SQR are uncoupled (no ΔμH+ generation). The energetic efficiency of each enzyme is indicated as the number of protons delivered to the periplasmic side of the membrane per electron (H+/e− ratio).doi:10.1128/ecosalplus.ESP-0012-2015.f1
Physiological Functions
Cytochromebo3 predominates under high aeration, whereas cytochromebd is expressed under low oxygen tension (22,23,24) (Table 1). It is of interest that the cytochromebo3 level increases about 150-fold during aerobic growth, but the cytochromebd level falls only 3-fold, i.e., the change in the cytochromebd level in response to oxygen is much smaller than that of the cytochromebo3 level (23). Both oxidases catalyze the oxidation of ubiquinol-8 to allow cellular respiration with oxygen as the terminal electron acceptor (25). Cytochromebd can also oxidize menaquinol-8 (26,27), which replaces ubiquinol-8 upon a change of growth conditions from aerobic to anaerobic (2).
Table 1.
Properties of cytochromebo3 and cytochromebd inE. coli
| Property | Cytochromebo3 | Cytochromebd |
|---|---|---|
| Level of O2 tension for expressiona | High | Low |
| Catalyzed reaction of oxidationb | Ubiquinol-8 → ubiquinone-8 | Ubi(mena)quinol-8 → ubi(mena)quinone-8 |
| Catalyzed reaction of reductionb | O2 → 2H2O | O2 → 2H2O |
| Energetic efficiency (H+/e− ratio)c | 2 (true proton pump) | 1 |
| KD (O2) (μM)d | >300 | 0.28 |
| ApparentKm for O2 (μM)e | 0.016–2.9 | 0.003–2 |
| ApparentKm for nonphysiological reductants (mM):f | ||
| Ubiquinol-1 | 0.05 | 0.23 |
| Menadiol | 38 | 1.67 |
| TMPD (N,N,N′,N′-tetramethyl-p-phenylenediamine) | 9.5 | 18.2 |
| Operon encoding oxidaseg | cyoABCDE | cydABX |
| Subunits (mass [kDa])h | CyoA (33.5) | CydA (57) |
| CyoB (75) | CydB (43) | |
| CyoC (20.5) | CydX (4) | |
| CyoD (12) | ||
| Redox cofactors (Em value[s] [mV])i | Hemeb (+180, +280) | Hemeb558 (+176) |
| Hemeo3 (+180, +280) | Hemeb595 (+168) | |
| CuB (+370) | Hemed (+258) |
Both cytochromebo3 and cytochromebd are primary generators of a transmembrane gradient of electrochemical H+ potentials (ΔμH+), because the reaction arising from the transfer of reducing equivalents from quinol to O2 is coupled directly to transmembrane charge separation (28,29,32,50,51,52,53,54). The energy conserved in the form of ΔμH+ can be used by theE. coli cell for ATP synthesis, the transport of nutrients, and other useful work. Thus, the main function of both oxidases is energy conservation. The two enzymes, however, are different in their bioenergetic efficiencies (transmembrane proton translocation ratios, or the number of protons delivered to the periplasmic side of the membrane per electron [H+/e− ratios]), with an H+/e− ratio of 2 for cytochromebo3 and an H+/e− ratio of 1 for cytochromebd (28,29) (Table 1). This difference is because cytochromebo3 is a true proton pump, whereas cytochromebd is not capable of proton pumping (28,29). As sources of oxidizing power, cytochromebo3 and cytochromebd can support disulfide bond formation upon protein folding catalyzed by the DsbA-DsbB system (55).
Apart from energy conservation, cytochromebd endowsE. coli with a number of specific physiological functions. Cytochromebd can serve as an oxygen scavenger and inhibit the degradation of O2-sensitive enzymes present under anaerobic and microaerophilic conditions (56). In a recent systematic mutational analysis to elucidate the contribution of the respiratory pathways to the abilities of commensal and pathogenic (enterohemorrhagic)E. coli strains to colonize a streptomycin-treated mouse intestine, mutants lacking cytochromebd failed to colonize whereas cytochromebo3 was found not to be necessary for colonization (57).
The cytochromebd contents increase not only at low oxygen concentrations, but also under some unfavorable conditions, such as alkaline pH (58), high temperature (59,60), the presence of uncouplers-protonophores (58,61,62), and low concentrations of cyanide (63) in growth media. Mutants defective in cytochromebd are sensitive to H2O2 (60) and a self-produced extracellular factor that inhibits their growth (64,65). Mutants that cannot synthesize cytochromebd are also unable to exit from the stationary phase and resume aerobic growth at 37°C (66,67). The expression of cytochromebd, instead of cytochromebo3, may enhance bacterial tolerance to oxidative and nitrosative stresses (68,69,70,71). In particular, cytochromebd contributes to bacterial resistance to peroxynitrite (71,72), nitric oxide (68,69,70,71,73,74,75,76,77), carbon monoxide (78,79), and hydrogen peroxide (59,70,71,80,81,82,83,84,85,86).
Inhibitors
Table 2 compares the effects of different inhibitors on the respiratory activities of cytochromebo3 and cytochromebd inE. coli. Inhibitors of the quinol oxidases can be divided into two groups: quinol-like compounds acting at a quinol-binding site(s) and heme ligands (e.g., cyanide, azide, and NO) acting at the oxygen-binding/reducing site. A specific feature of cytochromebd is that it is much less sensitive to cyanide and azide than cytochromebo3 (32) (Table 2). The lower sensitivity of cytochromebd to anionic heme ligands may be a result of an elevated electron density on the central ion of iron due to the breaking of the circle conjugate π-electron structure in thed-type porphyrin ring and/or may point to a more hydrophobic environment for the O2-reducing site of cytochromebd than for that of cytochromebo3. It is of interest that the quinolone-type compound aurachin D and its derivatives in submicromolar concentrations specifically inhibit cytochromebd but virtually do not affect cytochromebo3 (87). These outcomes may indicate some differences in the specific structures of quinol-binding sites in cytochromebo3 and cytochromebd. It has been shown recently that cytochromebd inE. coli is a bacterial membrane target for a cationic cyclic decapeptide, gramicidin S (50% inhibitory concentration, ∼5.3μM) (Table 2), although it has been generally accepted that the main target of gramicidin S is the membrane lipid bilayer rather than the protein components (88). This finding can provide new insight for the molecular design and development of novel gramicidin S-based antibiotics. The effect of gramicidin S on cytochromebd and some other membrane-bound proteins may be the alteration of the protein structure through binding to the hydrophobic protein surface (88).
Table 2.
Effects of inhibitors on quinol oxidase activities of cytochromebo3 and cytochromebd inE. coli
| Inhibitora | Concentration, inhibition for: | |
|---|---|---|
| Cytochromebo3 | Cytochromebd | |
| KCN | 10μM | 2 mM |
| NaN3 | 15 mM | 400 mM |
| H2O2 | 300 mM | 120 mM |
| HOQNO (2-n-heptyl-4-hydroxyquinolineN-oxide) | 2μM | 7μM |
| ZnSO4 | 1μM | 60μM |
| Piericidin A | 2μM | 15μM |
| Antimicin A | 50μM, 18% | 50μM, 80% |
| UHDBT (undecylhydroxydioxobenzothiazole) | 20μM, 97% | 20μM, 18% |
| (1,5-Dimethylhexyl)quinazolinamide | 100μM, 23% | 100μM, 88% |
| (1-Methyldecyl)quinazolinamide | 100μM, 24% | 100μM, 85% |
| Stigmatellin | 200μM, 94% | 200μM, 14% |
| Nigericin | 100μM, 35% | 100μM, 44% |
| Dibromothymoquinone | 100μM, 82% | 100μM, 38% |
| Aurachin A | 700μM, 56% | 700μM, 27% |
| Aurachin C | 214 nM, 90% | 214 nM, 90% |
| Aurachin D | 400 nM, 5% | 400 nM, 93% |
| NO | << 10−8 M | 100 nM |
| PCP | 200μM | |
| TTFA | 1 mM, 35% | |
| Gramicidin S | 189μM | 5.3μM |
The concentrations shown for KCN, NaN3, H2O2, HOQNO, ZnSO4, and piericidin A (32) and gramicidin S (88) are the concentrations required for 50% inhibition of the ubiquinol-1 oxidase activities of the purified cytochromesbo3 andbd. For PCP (pentachlorophenol) and NO (nitric oxide), the inhibition constants (Ki values) are shown (73,89). For TTFA (2-thenoyl trifluoroacetone), the data shown are the concentrations yielding the indicated percent inhibition of the ubiquinol-1 oxidase activity of purified cytochromebd (47). For other inhibitors, the data shown are the concentrations yielding the indicated percent inhibition of the duroquinol oxidase activities of the membranes containing either cytochromebo3 or cytochromebd (87).
GENETICS
Oxidase Encoding
Cytochromebo3
Cytochromebo3 is composed of four different subunits (90,91,92) encoded by thecyoABCDE operon (39,45) (Table 1). ThecyoABCDE operon, located at 10.2 min on theE. coli genetic map (39,93), has been cloned and sequenced (44). Subunits I (75 kDa), II (33.5 kDa), and III (20.5 kDa) of cytochromebo3 appeared to be homologous to the counterparts of the eukaryotic and prokaryoticaa3-type cytochromec oxidases (45) and were referred to ascyoB,cyoA, andcyoC gene products, respectively, as determined by protein sequencing (46) and other approaches (44,94,95). Thus, cytochromebo3 is a member of the heme-copper terminal oxidase superfamily (45,96). ThecyoD gene encodes subunit IV (12 kDa) (95,97,98), which is homologous to the counterpart in cytochromec oxidases from Gram-positive bacteria and terminal quinol oxidases but unrelated to eukaryotic cytochromec oxidases (96,99). ThecyoE gene, located at the 3′ end of thecyo operon, encodes no subunit of cytochromebo3 but does encode the enzyme that catalyzes the transformation of hemeB (protoporphyrin IX, or protoheme) to hemeO (uppercase letters in heme designations highlight the chemical nature of hemes, as opposed to protein-bound hemes) by attaching a long farnesyl side chain to the former (100,101,102). HemeO is specifically required for the binuclear oxygen-reducing site of cytochromebo3. Subunit I (CyoB) carries all three metal redox cofactors: low-spin hemeb, high-spin hemeo3, and a copper ion (CuB) (3,94,103,104). Hemeo3 and CuB form the heme-copper binuclear center for dioxygen reduction. Unlikeaa3-type cytochrome oxidase, subunit II (CyoA) does not have any metal redox cofactors. Subunits III (CyoC) and IV (CyoD) can be removed from cytochromebo3 without any loss of catalytic activity (38) but seem to be required for the assembly of the metal redox cofactors in subunit I (19,105).
Cytochromebd
Until recently, cytochromebd was thought to be a two-subunit oxidase (106) encoded by thecydAB operon (39,40,41), located at 16.6 min on theE. coli genetic map (39,93), and it was cloned (107) and sequenced (41). The molecular masses of subunit I (CydA) and subunit II (CydB) determined by sodium dodecyl sulfate-polyacrylamide gel electrophoresis, 57 and 43 kDa (47), respectively, are consistent with those determined based on the corresponding DNA sequences, 58 and 42.5 kDa (41). Neither of these two major subunits of cytochromebd has homology to any subunit of other known respiratory chain oxidases, such as cytochromebo3 and cytochromec oxidases (41,108). However, very recently it has been reported that cytochromebd has an additional polypeptide named CydX (42,43). This small (4 kDa) protein seems to be the third subunit of cytochromebd. CydX is required for maintenance of the cytochromebd activity and contributes to stabilization of the hemes (42,43,80,109,110). Thus it is now clear that cytochromebd is a three-subunit oxidase encoded by thecydABX operon (21,42,43). Cytochromebd contains no copper atoms and does not pump protons (28,29,32,47,50). Therefore, cytochromebd is not a member of the heme-copper terminal oxidase superfamily. Cytochromebd subunits carry three hemes:b558,b595, andd (106,111). Hemeb558 is located on subunit I (CydA), whereas hemesb595 andd are likely to be in the area of the subunit contact (112). CydA can be expressed and purified without CydB by using mutant strains defective incydB (113). The purified CydA retains hemeb558 but lacks hemesb595 andd (113). In addition to thecydABX operon, two other genes,cydC andcydD of thecydCD operon, located at 19 min on theE. coli genetic map (114,115,116), are essential for the assembly of cytochromebd (114,116,117,118). CydC and CydD, however, are not subunits of cytochromebd. It was shown previously thatcydCD encodes a heterodimeric ATP-binding cassette-type transporter (ABC transporter) that is a transport system for the thiol-containing redox-active molecules cysteine and glutathione (21,119).
Regulation of Gene Expression
Under high oxygen tension,E. coli expresses cytochromebo3 (encoded bycyoABCDE), whereas cytochromebd (encoded bycydABX) is moderately repressed (22,23,24). The expression of thecyoABCDE andcydABX operons is controlled by the two global transcriptional regulators Arc and Fnr (2,23,120,121,122,123,124,125,126). Arc is a two-component regulatory system that includes ArcA, a cytosolic response regulator, and ArcB, a transmembrane histidine kinase sensor. ArcA controls several hundred genes (127) and responds to the oxidation state of the quinone pool, which is sensed by ArcB (128). ArcB is activated in the course of the transition from aerobic to microaerobic growth and remains active during anaerobic growth. Upon stimulation, ArcB autophosphorylates and then transphosphorylates ArcA (128,129). Under microaerobic conditions (i.e., oxygen tension of 2 to 15% of air saturation), the increased level of phosphorylated ArcA represses thecyoABCDE operon and activates thecydABX operon (130).
Another global regulator, Fnr (an oxygen-labile transcription factor regulating hundreds of genes), controls the induction of anaerobic processes inE. coli (131,132). The Fnr protein has an Fe-S cluster which serves as a redox sensor. The levels of the Fnr protein are similar under both aerobic and anaerobic conditions (122,133), but the protein is active only during anaerobic growth. The active Fnr protein represses bothcyoABCDE andcydABX operons during a transition to anaerobic conditions (i.e., oxygen tension of less than 2% of air saturation) (124,125,133).
PROSTHETIC GROUPS
Quinones
Quinones are lipophilic molecules dissolved within the lipid bilayer of the cytoplasmic membrane. TheE. coli membrane contains three types of quinones that all have an octaprenyl side chain (C40). These are a benzoquinone, ubiquinone, and two naphthoquinones, menaquinone and dimethylmenaquinone. Both cytochromebo3 and cytochromebd catalyze the two-electron oxidation of ubiquinol-8 to ubiquinone-8 (Fig. 2) (apparent midpoint redox potential [Em] = +110 mV [2]), coupled to the four-electron reduction of O2 to 2H2O (Em = +820 mV). Cytochromebd can also oxidize menaquinol-8 to menaquinone-8 (Fig. 2) (Em = −80 mV [2]) (26,27). Ubiquinone-8 is predominant during aerobic growth but is replaced by menaquinone-8 upon the transition from aerobiosis to anaerobiosis (2,134,135). It was shown previously that cytochromebo3 contains a tightly bound ubiquinone (136). The presence or absence of bound quinone in solubilized cytochromebd depends on the purification protocol (20). In some preparations of purified cytochromebd, there is no quinone (32,47,51,137), whereas others clearly contain bound quinone (52,54,138).
Figure 2.

Structures of ubiquinone-8, the reduced ubiquinone-8 (ubiquinol-8), menaquinone-8, and the reduced menaquinone-8 (menaquinol-8).doi:10.1128/ecosalplus.ESP-0012-2015.f2
Cytochromebo3 Prosthetic Groups
Cytochromebo3 contains three redox-active metal groups: a low-spin hemeb, which is involved in quinol oxidation, hemeo3, and CuB; the latter two groups compose a binuclear center, which is the site of the binding of O2 and its reduction to water. The chemical cofactors are hemeb, corresponding to protoporphyrin IX (protoheme, or hemeB); hemeo3, corresponding to the protoheme with a long farnesyl side chain attached (hemeO); and the CuB center, represented by the Cu atom ligated by three histidine residues. In the three-dimensional (3D) structure of cytochromebo3, all prosthetic groups were found to be within subunit I (139). In addition to the metal centers, there is also one tightly bound ubiquinone cofactor. Although the X-ray structure did not show any bound quinone in the crystallized enzyme, the site-directed mutagenesis studies identified residues that modulate the properties of the bound quinone (140,141,142).
Hemeb
Subunit I of thebo3 oxidase contains 15 transmembrane helices. Hemeb is located between helices II and X at a depth of about one-third of the membrane thickness from the P (positive) side and oriented perpendicular to the membrane plane, such that its propionates are exposed to the P side of the membrane. In both reduced (S [spin quantum number] = 0) and oxidized (S =1/2) states (143,144), the low-spin heme iron is bound to the four nitrogen atoms of the porphyrin ring and to the two conserved histidines of subunit I (H106I and H421I; subscript “I” identifies the subunit where the residue is). Hemeb is a direct electron donor for the catalytic binuclear site. It transfers electrons obtained from the bound ubiquinone. The Em of this heme is about +280 mV (without redox interaction) (48). The optical spectrum of this heme is rather characteristic for low-spin six-coordinate hemeb, with theα-band of the reduced heme at 562 nm in the absolute and the difference (reduced-minus-oxidized) absorption spectra. The maxima of theβ- and Soret bands of the reduced-minus-oxidized hemeb spectrum are at 530 and 430 nm, respectively. At a high level of cytochromebo3 expression, hemeB in the low-spin heme site can be replaced by hemeO (145). Such heme alteration does not decrease the enzyme activity but lowers the functionalKm of the enzyme for oxygen (36).
Hemeo3
Hemeo3 is the oxygen-binding heme, and the subscript 3 has been used historically to indicate this feature by analogy to the other heme-copper oxidases. Hemeo3 is a high-spin heme in both the fully reduced ferrous state (S = 2) (146) and the oxidized ferric state (S =5/2) (147). Depending on the conditions, hemeo3 may be penta- or hexacoordinate (147): the permanent bonds of the heme iron include four bonds with nitrogen atoms of the porphyrin ring and one extra bond with the conserved H419I from helix X at the same depth in the membrane as hemeb. Fivefold coordination of the heme iron leaves one side of the heme empty and available for the binding of ligands such as O2. This free coordination points toward the CuB, together with which hemeo3 forms the bimetallic catalytic site where the reduction of oxygen to water takes place. The spectrum of hemeo3 is characteristic of high-spin hemes. It has a broadα-band centered at 560 nm in the reduced-minus-oxidized spectrum with small extinction and an intense Soret band with the maximum at 430 nm (145). The redox properties of the high-spin hemeo3 are very similar to those of hemeb (48). Both of these hemes have a redox potential of about 280 mV when the neighboring heme is oxidized, but the reduction of the neighbor results in a 100-mV lowering of the heme potential (redox interaction).
CuB
The second partner in the binuclear catalytic site is CuB. The oxidized CuB is a tetragonal center (148,149); it has three permanent axial histidine imidazole ligands and one mobile oxygen ligand with an exchangeable proton(s) (148,149). The imidazole ligands originate from H284I in helix VI and from H333I and H334I, both located in a loop fragment between helices VII and VIII. This redox center is often called the invisible Cu site because it does not show any changes in optical spectra upon enzyme reduction and oxidation. Usually, the electron paramagnetic resonance (EPR) signal from the oxidized Cu can be easily detected, but the close proximity of the iron atom of hemeo3 results in strong magnetic interaction and the absence of any detectable spectrum of CuB. Such strong magnetic interaction, however, can help to define the Em of the tetragonal center. The reduction of the Cu ion brakes magnetic interaction with the high-spin heme, and the appearance of a high-spin EPR signal can give information on the reduction level of CuB. With such an approach, an Em of +370 mV for CuB was obtained (48).
Bound ubiquinone
It has been proposed that thebo3 oxidase can have two ubiquinone-binding sites with different affinities. The bound ubiquinone in the site with high affinity for ubiquinone (the QH site) can be considered to be the enzyme cofactor (136,150,151,152). At the same time, in the low-affinity (QL) site, fast exchange of the ubiquinone molecules occurs, and it was proposed that electrons are transferred from the QL site to the next electron acceptor (hemeb) via the QH site (136,153). A functional study of mutants obtained by site-directed mutagenesis was used to create a model for the possible QH binding site, which is located in subunit I, close to hemeb (139). According to this model, the QH site is predicted to form up to four hydrogen bonds with D75 and R71 to the 1-carbonyl oxygen and with H98 and Q101 to the 4-carbonyl oxygen. EPR spectroscopy demonstrated that the QH site stabilizes a semiquinone anion radical of bound ubiquinone (154,155,156).
Cytochromebd Prosthetic Groups
Cytochromebd is composed of three subunits, I (CydA), II (CydB), and III (CydX), which are typical integral membrane proteins. The subunits carry three metal-containing redox centers, such as two protoheme IX groups (hemesb558 andb595) and a chlorin molecule (hemed) (Fig. 3), which are proposed to be in 1:1:1 stoichiometry per the enzyme complex, but no copper ion (157,158,159,160,161). Hemeb558 appears to be located within subunit I. Subunits I and II are required for the assembly of hemeb595 and hemed, suggesting that these two hemes may reside at the subunit interface (112). Both hemeb558 and hemed are presumed to be oriented with their heme planes perpendicular to the membrane plane. Hemeb595 is possibly oriented with its heme plane at ∼55° with respect to the plane of the membrane (162).
Figure 3.

Structures of hemeB (protoheme IX), hemeO, and hemeD (chlorin), which are redox cofactors of cytochromebo3 and/or cytochromebd fromE. coli.doi:10.1128/ecosalplus.ESP-0012-2015.f3
Hemeb558
Hemeb558 was shown to be located on subunit I. Although subunits I and II of the isolated cytochromebd complex cannot be split apart without denaturing the enzyme, some genetic approaches have allowed subunit I to be synthesized in the absence of subunit II (113). Antibodies directed against subunit I (163,164), as well as selective proteolysis of this subunit (165,166), inhibit the ubiquinol oxidase activity of cytochromebd. These findings suggest that hemeb558 is associated with subunit I and involved in quinol oxidation. Theα-band of the reduced hemeb558 reveals a peak at 560 to 562 nm in the absolute and the difference (reduced-minus-oxidized) absorption spectra at room temperature. The maximum of theβ-band of the reduced heme is at 531 to 532 nm (167,168). The difference absorption spectrum of hemeb558 in the Soret region reveals a maximum at 429 nm and a minimum at 413 nm (168). The positions of these bands were confirmed upon redox titration by separating the composite difference absorption spectra of the enzyme into the contributions of the individual heme components (111,169) as well as by the detailed spectroelectrochemical redox titration and numerical modeling of the data (168). Low temperature (77 K) shifts all bands by 1 to 4 nm to the blue (167). Hemeb558 is a low-spin hexacoordinate (170,171,172), and amino acid residues H186 and M393 of subunit I were identified as its axial ligands (173,174,175). The location of hemeb558 is predicted to be near the periplasmic surface (176,177).
Hemeb595
The band with a maximum at ∼595 nm in the difference (reduced-minus-“air-oxidized”) absorption spectrum of cytochromebd was long ascribed to hemea1 because of the relatively bathochromic position of the extremum (178). Subsequently, it was established that the prosthetic group of this component is not hemea but protoheme IX (47,111), whereupon the component has been called hemeb595. Magnetic circular dichroism (MCD) studies confirmed such a conclusion (170,172). The difference absorption spectrum of hemeb595 in the visible region was resolved from a set of composite spectra of the isolated cytochromebd recorded at different redox potentials (111,169). It turned out that this spectrum is similar to that of catalases and peroxidases, containing pentacoordinate (high-spin) protoheme IX (111,168). Hemeb595 shows anα-band at 594 nm and aβ-band at 561 nm in the difference absorption spectrum (168). A trough at 643 nm in the difference spectrum of hemeb595 (168) is indicative of the disappearance of a charge transfer to the ligand band, characteristic of the oxidized high-spin hemeb, as in the case of peroxidases. Theγ-band of ferrous hemeb595 in the absolute absorption spectrum is characterized by a maximum at ∼440 nm, as clearly revealed by femtosecond spectroscopy (179). The difference absorption spectrum of hemeb595 in the Soret region shows a maximum at 439 nm and a minimum at 400 nm (168). Hemeb595 is a high-spin pentacoordinate (170,172) ligated by the histidine (H19) of subunit I (180) and located on the periplasmic surface (176,177). The role of hemeb595 remains obscure. It is proposed that hemeb595 participates in the reduction of oxygen, forming, together with hemed, a diheme oxygen-reducing site somewhat similar to the heme-Cu oxygen-reducing site in theaa3- andbo3-type oxidases (52,170,171,179,181,182,183,184,185,186,187,188,189). In favor of this hypothesis is the finding that the circular dichroism (CD) spectrum of the reduced wild-type cytochromebd in the Soret band shows strong excitonic interaction between ferrous hemesd andb595 (171). Modeling the excitonic interactions in absorption and CD spectra yields an estimate of the Fe-to-Fe distance between hemed and hemeb595 to be about 10 Å (171). In the opinion of other researchers, the function of hemeb595 is limited to the transfer of an electron from hemeb558 to hemed (190,191). Some authors believe that hemeb595 can form a second site capable of reacting with oxygen (35,157).
Hemed
Hemed is a chlorin-type molecule (192). Theα-band of the reduced hemed in the absolute absorption spectrum shows a peak at 628 to 630 nm (89). However, under usual conditions, hemed is in the stable oxygenated (oxygen-ligated ferrous) form, which is characterized by a band with a maximum at 647 to 650 nm in the absolute absorption spectrum (193). The reduced-minus-oxidized difference spectrum of hemed in the Soret region shows a maximum at 430 nm and a minimum at 405 nm (168). The spectral contribution of hemed to the complexγ-band is much smaller than those of either hemesb (168). Hemed is predicted to be located near the periplasmic surface (176,177). This heme is the place for capturing and reducing O2 to 2H2O. Being free of external ligands, hemed seems to be in the high-spin state. The protein ligand of hemed is not known. The data on the nature of the hemed axial ligand are controversial. Authors of resonance Raman and electron nuclear double resonance studies have claimed that it cannot be an ordinary histidine, cysteine, or tyrosinate but is either a weakly coordinating protein donor or a water molecule (180,194,195). In contrast, EPR studies have indicated that the hemed axial ligand is histidine in an anomalous condition or another nitrogenous amino acid residue (196). Finally, it has been reported recently that the highly conserved glutamate 99 of subunit I may be a candidate for such a role (137).
The millimolar extinction coefficients used commonly for the determination of theE. coli cytochromebd concentration are listed inTable 3.
Table 3.
Extinction coefficients used for determination of theE. coli cytochromebd concentration
| Absorption spectrum | Heme(s) | Wavelength paira (nm) | Δεb (mM−1·cm−1) | Reference |
|---|---|---|---|---|
| Difference spectra | ||||
| Fully reduced minus as prepared | d | 628–607 | 10.8 | 170 |
| d | 628–651* | 27.9 | 182 | |
| d | 628–649* | 18.8 | 32 | |
| b558 | 561–580 | 21 | 182 | |
| b595 | 595–606.5 | 1.9 | 182 | |
| All | 429–700† | 303 | 182 | |
| Fully reduced CO bound minus fully reduced | d | 642–622 | 12.6 | 32 |
| d | 643–623 | 13.2 | 54 | |
| Absolute spectra | ||||
| Fully reduced | d | 628–670 | 25 | 52 |
| As prepared | All | 414–700† | 223 | 182 |
Values marked with an asterisk or dagger cannot be recommended for the determination of the cytochromebd concentration since, for those marked with an asterisk (*), the as-prepared enzyme contains various amounts of the ferrous hemed-oxy complex that absorbs at 649 to 651 nm and, for those marked with a dagger (†), the intensity of the Soret band is variable depending on the purity of the preparation.
Δε, extinction coefficient.
Redox Potentials of Hemes in Cytochromebd
The Em values of hemesb558,b595, andd in theE. coli cytochromebd solubilized inn-dodecyl-β-d-maltoside at pH 7.0 are +176, +168, and +258 mV, respectively (49) (Table 1). Similar values were reported for cytochromebd contained in bacterial membranes (158,159,197), reconstituted in liposomes (198), or solubilized in Tween 20 or Triton X-100, in which the enzyme is active (198). Notably, the Em value of hemeb558 can depend on the detergent used for solubilization (198). In particular, octylglucoside and cholate cause a large decrease in the Em value of hemeb558 that also correlates with the reversible inactivation of the enzyme (198). The Em values of all three heme components of cytochromebd are sensitive to pH values between 5.8 and 8.3, with dependence of −61 mV per pH unit for hemed and −40 mV per pH unit for hemesb558 andb595, indicating that the reduction of thebd complex is accompanied by enzyme protonation (198). The spectroelectrochemical redox titration and spectral modeling show the strong redox interaction between hemeb558 and hemeb595, while the interaction between hemed and either hemesb is insignificant (168). Of interest, in the absence of hemed the interaction potential between hemeb558 and hemeb595 is much larger compared with the situation when hemed is present (168).
STRUCTURE OFbo3 ANDbd OXIDASES
3D Structure ofbo3 Oxidase
The 3D structure of thebo3 oxidase fromE. coli at 3.5-Å resolution was reported in 2000 (139). The structure confirms that the overall architecture of this complex is very similar to those of all other members of the heme-copper oxidase superfamily. The whole integral protein contains 25 transmembrane helices, of which subunit I has 15, subunit II has only 2, subunit III has 5, and subunit IV has 3 helices. All known enzyme cofactors were found in subunit I. The transmembrane helices of this subunit are not perpendicular to the membrane plane but are tilted about 20 to 35° against it. When viewed from the top (P side), they are arranged in an anticlockwise direction and form three semicircular arcs, organized in a quasithreefold axis of symmetry. Three pores are formed in the center of the arcs. One pore houses the binuclear center (hemeo3 and CuB) of the oxidase and includes the proton-conductive K channel directed from the binuclear center toward the N (negative) side of the membrane. Another pore retains hemeb; the last pore is devoid of cofactors but is used for the proton-conductive D channel.
Hemeb is located at a depth of about one-third of the membrane thickness from the P side and oriented perpendicular to the membrane plane, such that its propionates are pointing toward the P side of the membrane (Fig. 4). The hemeo3 is located at the same depth as hemeb, the plane of hemeo3 is also perpendicular to the membrane, and the propionates point toward the P side in a manner similar to hemea. Hemeb and hemeo3 are facing each other at an angle of 104 to 108o. At an ∼5-Å distance from the iron of hemeo3, a copper atom designated CuB is located. In addition to this Cu atom that is coordinated by the three histidines, there is another important structure identified in all heme-copper oxidases and represented by the covalently bound H284I and Y288. The three histidines and the tyrosine form a conjugated π-electron system around the CuB center. Cytochromebo3 has been proposed to have two ubiquinone-binding sites, one with high (QH) and one with low (QL) affinity for ubiquinone (136). It has also been postulated that electrons are transferred from the QL site to hemeb via the ubiquinone bound at the QH site. Site-directed mutagenesis studies (140,142) have identified residues that modulate the properties of the QH site. The model of a QH quinone-binding site including R71, D75, H98, and Q101 residues is also supported by the results of X-ray crystallography (139).
Figure 4.

Structure of cytochromebo3 fromE. coli. Only two main subunits are shown: subunit I in gray and subunit II in yellow. Hemes are shown in red (hemeo3 on the right and hemeb on the left). The cyan sphere near hemeo3 represents the CuB center. The amino acid residues of possible proton-conducting pathways, the D (red-tag) and K (blue-tag) channels, in subunit I are shown. The most likely position of the membrane is depicted by the gray background.doi:10.1128/ecosalplus.ESP-0012-2015.f4
The protein medium itself cannot facilitate proton delivery toward the binuclear center or across the membrane, and in order to overcome this limitation, the oxidase has special proton-conductive structures. It is proposed that these structures are based on chains of hydrogen bonds between hydrogen-bonding protein side groups (polar and/or protonatable) and water molecules, whereby the proton is transferred via a Grotthuss type mechanism. At least two proton-conductive channels have been identified primarily by site-directed mutagenesis (199,200,201), and these findings were later confirmed by X-ray crystallography (139). One of them is the so-called K pathway, named after the highly conserved lysine 362 (199,200), which is situated approximately halfway through the channel (Fig. 4). This pathway starts with S315 and S299 and continues through conserved residues K362I and T359I toward the hydroxyethyl farnesyl side chain of hemeo3 and Y288I in the proximity of the binuclear center. The other channel is named D after the highly conserved D135I (201,202), which is situated near the surface of the enzyme on the N side. D135I, together with T211I, forms a mouth that leads via polar residues N124I, N142I, S145I, T149I, T201I, and T204I to E286I (Fig. 4), which is an important residue for the proton-pumping mechanism.
Proposed Structure of Cytochromebd
To date, the X-ray structure of cytochromebd is not available; however, the findings from conventional studies of the protein topology in the membrane suggest that all three hemes are located near the periplasmic side of the membrane (176,177).Figure 5 shows topological models of subunits I (CydA) and II (CydB) of cytochromebd fromE. coli (27). Both subunits are integral membrane proteins showing no sequence homology to the subunits of the heme-copper oxidase superfamily (e.g., cytochromebo3). Subunit I consists of nine transmembrane helices, with the N terminus in the periplasm and the C terminus in the cytoplasm (176). Subunit II is composed of eight transmembrane helices, with both N and C termini in the cytoplasm (176). Newly discovered subunit III, the 37 amino acid CydX protein (42,43,109), is proposed to consist of the only membrane-spanning helix, with the N terminus in the cytoplasm and the C terminus in the periplasm (21,80,109). Subunit I contains a large hydrophilic domain, which is called the Q-loop, connecting transmembrane helices VI and VII. As shown by many experimental approaches, the Q-loop is involved in quinol binding and oxidation (163,164,165,166,203,204). Thus, the quinol-oxidizing site in cytochromebd is located on the periplasmic side of the membrane. It is worth mentioning that the size of the Q-loop is taken into account to categorize the bacterial cytochromesbd (20,172).
Figure 5.
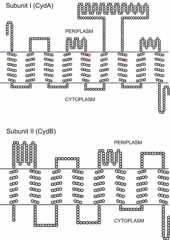
Proposed topology of the CydA and CydB subunits of cytochromebd fromE. coli. The axial ligands of hemeb595 (H19) and hemeb558 (H186 and M393) in the CydA subunit are shown in purple and red, respectively. The protein sequence data have been taken from information available athttp://genolist.pasteur.fr/Colibri/. The alignment has been made by using the TOPO2 program available athttp://www.sacs.ucsf.edu/TOPO2. The model is very similar to that reported in reference27.doi:10.1128/ecosalplus.ESP-0012-2015.f5
Based on 815 sequences of the cytochromebd gene available, it was possible to search for highly conserved residues in the corresponding protein (27). It was shown previously that subunit II evolved significantly faster than subunit I, with the result that subunit II exhibits greater sequence diversity (205). A number of residues in subunit I are totally (>99%) conserved in the 815 sequences (27). These residues include H19 (the hemeb595 axial ligand [180]), H186 and M393 (the hemeb558 axial ligands [173,174,175]), K252 and E257 (involved in quinol binding [204]), R448 (having an unknown function), and E99, E107, and S140 (proposed to be components of a proton channel [54,176] and important for binding in the hemed-hemeb595 diheme site [27,137]). Slightly less conserved (95 to 99%) are E445 (required for charge compensation at the hemeb595-hemed oxygen-reducing site upon the full reduction of oxygen by two electrons [52]), N148 (a plausible component of a proton channel), and R9 (having an unknown function) (27). Somewhat less conserved (∼85%) are R391 (which stabilizes the reduced form of hemeb558 [206]) and D239 (having an unknown function); however, these residues are totally conserved within the group of cytochromesbd with a long Q-loop, to which theE. coli enzyme belongs (27). Other conserved residues are glycines, prolines, phenylalanines, and tryptophans, which may play structural roles. There is only one totally (>99%) conserved residue (W57) in subunit II (27). Residues R100, D29, and D120 of subunit II are totally conserved within the family of long-Q-loop cytochromebd, and the residue at position 58 in subunit II (according to the numbering for theE. coli enzyme) is either an aspartate or a glutamate. The N-terminal portion of subunit II is thought to be involved in the binding of hemed and hemeb595 (27,207).
A multiple sequence alignment of 299 homologues of the newly found CydX subunit reveals the conserved amino acid motif that includes the residues Y3, W6, G9, and E/D25 (109). Of these residues, the first three are part of the predicted transmembrane α-helix being localized to the same side of the helix (109). The latter suggests that this is possibly the side of the α-helix of CydX that interacts with the other subunits in cytochromebd (109).
Since the active site of O2 reduction is located near the periplasmic surface and protons for H2O production are taken from the bacterial cytoplasm, there must be at least one transmembrane proton-conducting pathway to convey protons from the cytoplasm to the hemeb595-hemed site (51,52,54,176) (Fig. 6).
Figure 6.
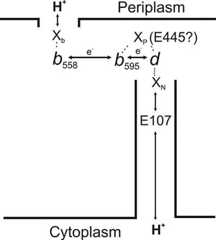
Schematic model of electron and proton transfer pathways in cytochromebd fromE. coli. There are two protonatable groups, XP and XN, redox coupled to the hemeb595-hemed active site. A highly conserved residue, E445, was proposed to be either the XP group or the gateway in a channel that connects XP with the cytoplasm or the periplasm (52). A strictly conserved E107 residue is a part of the channel mediating proton transfer to XN from the cytoplasm (54). Xb, a group at the periplasmic side of the membrane that picks up and releases a proton as hemeb558, is reduced and oxidized.doi:10.1128/ecosalplus.ESP-0012-2015.f6
COMPARISON OF AFFINITIES OF THE TWO OXIDASES FOR OXYGEN
InE. coli, the affinity of cytochromebd for oxygen, i.e., dissociation constant for O2,KD (O2) of 0.28 μM (31), is about 1,000-fold higher than that of cytochromebo3,KD (O2) >0.3 mM (30), which allows us to consider thebd andbo3 enzymes as the high- and low-affinity oxidases, respectively. Such a striking difference in theKD (O2) values correlates with the following facts.
Cytochromebo3 predominates under high aeration, whereas cytochromebd is expressed maximally under low aeration (22,23,24).
A peculiar feature of cytochromebd is that it is purified mainly as a stable oxygenated (oxy)complex (hemeb5583+-hemeb5953+-hemed2+-O2) characterized by an absorption peak at 645 to 650 nm (193,208,209,210). The fact that a stable oxycomplex can be generated by the reversible binding of oxygen to the one-electron-reduced cytochromebd can be used for direct measuring of theKD (O2) of cytochromebd (31,49). This is not the case for cytochromebo3 or any other heme-copper oxidase, which in any redox state under normal conditions does not form a stable oxycomplex; therefore, theKD (O2) for cytochromebo3 can be determined only indirectly (30). For cytochromebo3, theKD (O2) is more than 100-fold higher than the apparentKm for O2, which allows us to conclude that thebo3 oxidase is designed to trap O2 kinetically by reducing it to an oxoferryl species (36). Because of its ability to function efficiently under microaerobic conditions, cytochromebd is required for commensal and pathogenicE. coli strains to colonize mouse intestine (57). It turns out thatE. coli mutants lacking cytochromebd, with high affinity for O2, are eliminated by competition with wild-type strains competent in respiration and that cytochromebo3, with low affinity for O2, is not necessary for colonization (57). The colonization defects of the cytochromebd mutants challenge the traditional view that the intestine is anaerobic (211) but support the hypothesis that a microaerobic niche is critical for both establishing and maintainingE. coli in the intestine (57).
BINDING OF LIGANDS (OTHER THAN OXYGEN)
Ligand Binding to the Cytochromebo3 Binuclear Center
Ligands that bind to the binuclear center of cytochromebo3 can be divided into two classes: uncharged molecules like CO and NO, which preferably bind to the binuclear center in the reduced form and induce the transition of the high-spin heme to the low-spin state, making it six coordinate, and ionizable molecules, like HCN and NaN3, or formate, which preferably bind to the oxidized heme. The binding of the second group of ligands can result in a different spin state for hemeo3. Some of the ligands can bind either to hemeo3 only or to a site between the two metals forming the binuclear center. The dynamics of ligand exchange can be used to characterize the possible dynamics of the binding of the substrate and the partial products of the reaction generated during the catalytic cycle.
CO and NO binding
The molecules of CO and NO mimic the oxygen molecule and can be bound to the catalytic site of cytochromebo3. This binding occurs with the high-spin heme of the reduced binuclear center. Carbon monoxide reacts with the reduced enzyme with a stoichiometry of 1:1, and theKD for this reaction was determined to be 1.7 × 10−6 M (147). The value of a second-order rate constant for association (kon) is 6.1 × 104 M−1·s−1.
The CO ligation of hemeo3 results in a blue shift of the heme absorption bands. The characteristic CO-binding spectrum has two small peaks in the visible part of the spectrum at 530 and 570 nm and a pronounced spectral shift in the Soret region, with the maximumλ of ∼415 nm and the minimumλ of ∼430 nm.
The photolysis of the reduced CO-bound enzyme at low temperatures results in the dislocation of the iron-bound CO to CuB, where it can be recognized by the specific C≡O stretch at ∼2,065 cm−1 due to CO bound to copper (212). At room temperature, the photodissociation of CO from the heme iron and its subsequent binding to CuB is an extremely fast reaction, and then CO remains bound to CuB for only a short time (two-component dissociation with the time constants of ∼14 and 140μs [213]). After carbon monoxide dissociation from the binuclear site, the rebinding to the heme iron via CuB occurs at a much lower rate (214). Yoshikawa et al. (215) have reported the crystallographic structures of the reduced bovine enzyme in the presence and absence of CO. While this X-ray study did not find any significant changes in the structure of CuB relative to the fully oxidized bovine enzyme, the extended X-ray absorption fine-structure (148) investigation of the first shell of CuB showed a dramatic change upon the addition of CO, which involves the dissociation of one of the CuB histidine ligands and its possible replacement by a chloride ion.
Fully reduced cytochromebo3 also binds NO with a stoichiometry of 1:1 and very high affinity (KD < 10−8 M) (216), forming the ferrous nitrosyl (Fe2+-NO) species, as determined by EPR spectroscopy (147). The hemeo3-NO complex yields well-resolved EPR signals from the14N atoms of both NO itself and the proximal histidine ligand of hemeo3, showing nuclear hyperfine coupling (147).
Reaction of cytochromebo3 with cyanide
Cyanide reacts almost exclusively with oxidized cytochromebo3. The reaction is manifested by a red shift of the Soret band from approximately 411 to 415 nm (217). A characteristic feature of the ligand-binding reaction in theα-band region is the loss of the broad charge transfer band at 630 nm. This band is attributable to the fully oxidized binuclear center in which hemeo3 is in a high-spin state. The results of EPR studies (217) seem to indicate that these absorption changes occur because the ferric high-spin hemeo3 becomes a ferric low-spin ligand complex. Because of the magnetic coupling with CuB2+, the EPR spectrum (gz [constant of magnetization] = 3.3) is observed only when the copper of the binuclear center is reduced. This signal is characteristic of the conversion of the ferric high-spin heme into the low-spin state by the binding of strong-field ligands such as cyanide (218). Cytochromebo3 binds a single equivalent of cyanide (KD, 1 × 10−6 to 2 × 10−6 M) with monophasic kinetics (219) andkon of 37 M−1·s−1 (at pH 6.0) (220). This rate constant is slightly pH dependent and increases about 1.8-fold over the pH range between 6.0 and 8.5 (220). Room-temperature MCD spectra in the near-infrared region (221), results from infrared and EPR studies (222), and also resonance Raman detection (223) of the product of the reaction of the oxidized cytochromebo3 with cyanide have led us to propose the bridging structure of the cyanide complex to be as follows: Feo33+—C=N—CuB2+, where Feo3 is the iron of hemeo3. It was also shown that the reduction of the enzyme results in the release of the CuB ligation and the formation of an Feo32+—C=N moiety.
Reaction of cytochromebo3 with azide
Azide binds to cytochromebo3 with a stoichiometry of 1:1; theKD for this reaction is about 2 × 10−5 M (224). Contrary to the addition of cyanide, the addition of azide to the oxidized isolated enzyme causes a relatively rapid but small blue shift in the Soret band from approximately 411 to 409 nm. Simultaneously, the broad charge transfer band at 630 nm becomes more pronounced and shifts its maximum to 640 nm. These small changes induced in the electronic absorption spectrum are consistent with hemeo becoming hexacoordinate but remaining high spin, which can also be seen by MCD spectroscopy in the range of 350 to 1,100 nm (221,224). The kinetics of azide binding is an order of magnitude faster than that observed for the binding of cyanide. The calculatedkon for the binding of azide to cytochromebo3 is about 800 M−1·s−1 at pH 7.5. Thekon shows a marked increase upon acidification, indicating that the active species is electroneutral hydrazoic acid. Analyses of EPR, electronic, and MCD spectra (219) were used to prove that, unlike cyanide, azide does not bind to hemeo3 but rather to the CuB site, whereas the resolved 3D structure of the bovine cytochromec oxidase in the presence of azide revealed the bridging structure of the complex (Fea33+—N=N=N—CuB2+) (215). The Fourier transform infrared (FTIR) study of the bovine enzyme complex (225) showed a major infrared band at 2,051 cm−1. Subsequently, an FTIR spectroscopic study of theE. coli bo3 oxidase (226) showed an infrared band at 2,041 cm−1, which was assigned to the bridging structure. Discrepancy in the ligation geometry found by the different methods was resolved by detailed analysis of the FTIR spectroscopy of the complex of cytochromebo3 with asymmetrically15N-labeled azide (227). The experiments showed time-dependent evolution of the geometry of azide binding. In the air-oxidized form, a major infrared azide antisymmetric stretching band corresponds to the bridging geometry. An additional band developing at 2,062.5 cm−1 during longer incubation reflects the appearance of the CuB2+—N=N=N structure. In addition, the partial reduction of the oxidase withβ-NADH caused the appearance of new infrared bands indicating the emergence of the Feo3+—N=N=N configuration (227).
Ligand Binding to Cytochromebd
Since hemesd andb595 in cytochromebd are in the high-spin pentacoordinate state, these redox centers can potentially bind ligands. It has to be expected that the reduced form of thebd enzyme can bind mainly electroneutral molecules like O2, CO, and NO and that the oxidized cytochromebd binds ligands in the anionic form, such as cyanide and azide. It appears that hemed binds ligands readily but that the ligand reactivity of hemeb595 is minor (170,184,189). It was reported previously that hemeb558, although a low-spin hexacoordinate, may also bind ligands (e.g., CO and cyanide) to some extent (170,189). Such marginal reactivity is due possibly to the weakening of the bond of the methionine axial ligand (M393) with the hemeb558 iron caused by the isolation procedure and/or protein denaturation (189).
CO binding
CO brings about a red shift of the hemed band, with a maximum at 643 to 644 nm, a minimum near 624 nm, and a peak at 540 nm. In the Soret band, CO binding to fully reduced cytochromebd results in an absorption decrease and minima at 430 and 442 to 445 nm. Absorption perturbations in the Soret band and at 540 nm occur in parallel with the changes at 630 nm and reach saturation at 3 to 5μM CO. The peak at 540 nm is probably either theβ-band of the hemed-CO complex or part of its splitα-band (189). A peculiar W-shaped curve in the Soret region of the difference spectrum can be caused by a small band shift for unligated hemeb595 induced by CO interaction with the nearby hemed (179,185). Only a small fraction of hemeb595 in cytochromebd binds CO at room temperature or low temperatures (from −70 to −100°C) (170,184). CO binding with about 15% of hemeb595 in the membrane-bound cytochromebd at a cryogenic temperature (4 K) was observed with the aid of FTIR spectroscopy (181). The apparentKD for the CO complex with fully reduced cytochromebd appeared to be ∼80 nM (189). This value is markedly higher than that for cytochromebo3 (1.7μM) (147). The fully reduced cytochromebd can form a photosensitive hemed-CO complex, and following flash photolysis, CO recombines with ferrous hemed proportionally to the CO concentration, withkon of 8 × 107 M−1·s−1 (161) (Table 4). This value is much higher than that for cytochromebo3 (6.1 × 104 M−1·s−1) (147).
Table 4.
Kinetic and thermodynamic parameters for the reaction ofE. coli cytochromebd with O2, CO, and NO at room temperature
| Value for ligandg: | ||||||
|---|---|---|---|---|---|---|
| Parameter | O2 | CO | NO | |||
| MV O2 | R-O2 | MV CO | R-CO | MV NO | R-NO | |
| kon (M−1 s−1) | 2 × 109b,e | 8 × 107b | ||||
| koff (s−1)f | 78 ± 0.5a | 4.2 ± 0.34a | 6.0 ± 0.2a | 0.036 ± 0.003a | 0.133 ± 0.005a | |
| KD (nM) | 280c | 80d | ||||
Data from reference75.
Data from reference161.
Data from reference31.
Data from reference189.
Data from reference53.
koff values are means ± standard deviations.
R-O2, R-CO, and R-NO are complexes of fully reduced cytochromebd with O2, CO, and NO, respectively. MV O2, MV CO, and MV NO are complexes of one-electron-reduced cytochromebd with O2, CO, and NO, respectively.
CO can also react with one-electron-reduced oxygen-free cytochromebd ofE. coli, forming a mixed-valence (MV) CO compound in which bothb-type hemes remain oxidized (hemeb5583+-hemeb5953+-hemed2+-CO) (75,179,185,188). The hemed α-band is also positioned at 635 to 636 nm in the absolute absorption spectrum.
It was proposed that the redox state of theb-type hemes in cytochromebd, presumably that of hemeb595, controls the status of the pathway for ligand transfer between hemed and the bulk phase (75,179). Two observations allowed us to draw such a conclusion.
Flash photolysis of the CO complex of the fully reduced cytochromebd results in the complete photodissociation of the CO molecule into the bulk. If the experiment is repeated with the MV CO complex, a significant part of the CO flashed off hemed2+ (up to 70%) gets trapped inside the protein and undergoes geminate recombination with hemed2+ on a subnanosecond time scale (179).
The apparent off rate constant for the spontaneous dissociation (koff) of CO from hemed2+ is markedly lower for the MV state of cytochromebd than for the fully reduced state of thebd oxidase (75) (Table 4).
Interaction with some nitrogen-containing ligands
A number of small nitrogen-containing molecules can react with fully reduced cytochromebd fromE. coli. NO3−, NO2−, N2O32− (trioxodinitrate), NH2OH, and NO, when added to membranes containing cytochromebd or the purified enzyme, give the decrease in amplitude and shift the 630-nm band of ferrous hemed to 641 to 645 nm (73,75,157,170,196,228,229,230,231). The common peak position was interpreted as all ligands yielding the same or very similar heme-nitrosyl compounds (231). A red shift of theα-band of hemed2+, observed upon the interaction of nitrite with the fully reduced membrane-bound cytochromebd, was accompanied by the slower formation of a trough at 438 nm in the difference (nitrite-treated-minus-reduced) absorption spectrum. These changes in the Soret band were ascribed to the formation of the product of the interaction of hemeb595 with nitrite (hemeb5952+-NO) (157).
Cytochromebd can also form a stable complex with NO in an MV state, in which ligand-bound hemed is reduced (to hemed2+) while the other two hemes (b558 andb595) are oxidized (75,196). The rates of NO dissociation from hemed2+ of cytochromebd in both the fully reduced and MV states were determined previously (75). In the fully reduced state, NO dissociates from hemed2+ at an unusually high rate, 0.133 s−1 (75), which is ∼30-fold higher thankoff measured for the ferrous hemea3 of the mitochondrial cytochromec oxidase, 0.004 s−1 (232). These data are consistent with the proposal that in heme-copper oxidases, CuB acts as a gate, controlling ligand binding to the heme in the active site (233). Another remarkable feature of NO dissociation from cytochromebd is that thekoff value for the MV state (0.036 s−1), although still quite high, is significantly lower than that measured for the fully reduced enzyme (75) (Table 4). The same effect was observed with CO (see above) (75). These data show that the redox state of hemeb595 controls the kinetic barrier for ligand dissociation from the active site of cytochromebd. The unusually high rate of NO dissociation from cytochromebd may explain the observation (73) that the NO-poisoned cytochromebd recovers respiratory function much more rapidly than a heme-copper oxidase.
Of interest, cytochromebd is not inactivated by up to 100 μM peroxynitrite, another nitrogen-containing harmful reactive species (72). Furthermore, cytochromebd in turnover with O2 is able to rapidly metabolize peroxynitrite with an apparent turnover rate increasing at higher concentrations of the reducing substrates (72). Thus, cytochromebd appears to be highly resistant to peroxynitrite damage (71,72). It is suggested that the expression ofbd-type instead of heme-copper-type oxidases enhances bacterial tolerance to nitrosative and oxidative stresses, thus promoting the colonization of host intestine or other microaerobic environments (68,69,70,71,72,75,76).
Reaction with cyanide
An earlier spectrophotometric study of the reaction of cytochromebd fromE. coli with KCN was performed with the membrane vesicles (234) and was confined to measurements in theα-band. That work revealed a decay of the absorbance at 650 nm induced by cyanide, and this finding was interpreted at that time to represent the disappearance of the free form of ferric hemed (234). This result was reinterpreted later as the decay of the ferrous hemed oxycomplex (167). Cyanide, interacting with the oxygenated form of the isolated cytochromebd, brings about significant absorption changes in theγ-region: a maximum at 434 to 437 nm and a minimum near 405 to 410 nm in the difference (KCN-treated-minus-oxygenated) spectrum. There is also considerable increase in the MCD signal in the Soret region. These data were interpreted to indicate the ligand-induced transition of high-spin hemeb595 to the low-spin state (235). Subsequently, a simple and fast method for the conversion of the oxygenated enzyme into the fully oxidized form with the use of lipophilic electron acceptors was developed (193). This approach enabled researchers to study the interaction of cyanide and other ligands with the homogenous oxidized preparation of cytochromebd. It was found that the addition of KCN to the fully oxidized cytochromebd brings about some absorption changes in the visible range of the difference absorption spectrum (the 624-nm peak is most pronounced) (170). These changes suggest the reaction of the ligand with hemed. A typical bathochromic shift of theγ-band is also observed. While the changes in the visible region are virtually saturated at 0.5 mM KCN, the Soret band effect continues to grow, indicating a second low-affinity ligand-binding site (170). The MCD spectrum of the fully oxidized cytochromebd is dominated by an asymmetric signal in the Soret region. Submillimolar cyanide has no effect on the initial MCD spectrum. KCN at 50 mM induces minor changes of the MCD signal in the Soret band, which can be modeled as the transition of a part of the low-spin hemeb558 (15 to 20%) to its low-spin cyanocomplex (170). There is no evidence of the interaction of high-spin ferric hemeb595 with the ligand. The apparent discrepancy between data on the interactions of cyanide with oxygenated (235) and fully oxidized (170) forms of thebd enzyme may derive from the fact that, in the former case, there seemed to be partial reduction of hemeb595 associated with ligand-induced electron transfer from hemed rather than a change of the hemeb595 spin state. On the basis of EPR spectra, Tsubaki et al. (182) proposed that the treatment of air-oxidized cytochromebd with cyanide results in a cyanide-bridging species with a hemed3+—C=N—hemeb5953+ structure. However, Tsubaki et al. (182) did not account for the electron released from hemed upon cyanide binding to as-prepared cytochromebd.
Interaction with H2O2
The addition of excess H2O2 toE. coli membranes containing cytochromebd (236) and the purified enzyme in the as-prepared (183,209) or the fully oxidized (51,183,237) state gives rise to an absorption band at ∼680 nm. The reaction of H2O2 with fully oxidized cytochromebd also induces a bathochromic shift of theγ-band (183,237). H2O2 binds to ferric hemed with an apparentKD of 30μM, but it seems not to interact with hemeb595 (183,237). The fully ferric cytochromebd reacts with peroxide withkon of 600 M−1·s−1. The decay of the H2O2-induced spectral changes upon the addition of catalase (at a rate of ∼10−3 s−1) is about 20-fold slower than expected for the dissociation of H2O2 from the complex, with hemed assuming a simple reversible binding of peroxide (koff =KD ×kon ∼ 2 × 10−2 s−1) (237). This finding suggests that the interaction of H2O2 with cytochromebd is essentially irreversible, giving rise to the oxoferryl state of hemed (237). The assignment of compound 680 to the oxoferryl state of hemed is confirmed by resonance Raman spectroscopy data (160). The results of resonance Raman spectroscopy studies suggest that hemed is in the high-spin pentacoordinate state when it is oxygenated (Fe2+-O2) or exists as an oxoferryl species (Fe4+-O2−) or in a compound with cyanide (238).
Remarkably, the isolated cytochromebd displays some peroxidase enzymatic activity (84). Moreover, cytochromebd, either purified or overexpressed in a catalase-deficientE. coli strain, shows a significant catalase activity that is insensitive to NO (71,85,86). Therefore, one may conclude that thisbd-type terminal oxidase actually contributes to protectE. coli from H2O2 stress.
PROPOSED MECHANISMS OF FUNCTIONING
Mechanism of Cytochromebo3 Functioning
Cytochromebo3 catalyzes the final step ofE. coli respiration—the oxidation of ubiquinol-8 and the reduction of molecular oxygen. The reduction of one dioxygen molecule to water requires four electrons, which are supplied by two molecules of ubiquinol on the P side, and four protons, taken up from the N side of the membrane. The reduction of oxygen to water is an exergonic process, coupled with the release of large amounts of energy. This energy is conserved in the form of ΔμH+. The formation of ΔμH+ by cytochromebo3 is based on two principles: vectorial chemistry and proton pumping. Since the protons and electrons for oxygen reduction to water are taken from different sides of the membrane, the reduction results in the net transfer of four charges across the membrane. At the same time, the enzyme is able not only to catalyze the oxygen reduction, but also to utilize the released energy for proton pumping. This process was discovered in 1977 (239) by the demonstration that the reduction of molecular oxygen to water by mitochondrial cytochromec oxidase is linked to the pumping of additional four protons across the membrane dielectric (239). Later, such functional ability was shown for cytochromebo3 (240). Hence, the overall reaction catalyzed by cytochromebo3 can be described by the following equation:
where Q is ubiquinone, QH2 is ubiquinol, H+N is proton taken from the N side of the membrane (the cytosol), and H+P is proton released to the P side of the membrane (the periplasmic space). The mechanism coupling electron transfer reactions with the transmembrane proton translocation is still under debate. Let’s look through the main elements of this mechanism.
Electron transfer reactions in cytochromebo3
The general sequence of electron transfer in thebo3 oxidase is well established. The ubiquinone molecule occupying the QH-binding site has a stable semiquinone form (241), which ensures the coupling of a one-electron redox reaction to a two-electron donor. A pulse radiolysis study showed that the quinone bound at the QH site is important for the rapid reduction of hemeb but not for rapid electron transfer from hemeb to the hemeo3-CuB binuclear center (242). The rate constant of electron transfer between semiquinone and hemeb was found to be 1.5 × 103 s−1 (242).
In the next step, the low-spin heme delivers electrons to the binuclear site in a controlled fashion that is coupled to proton translocation across the membrane (243). The rate of this electron transfer without coupling to the reaction with protons has been studied extensively, in particular, by the photodissociation of CO from the oxygen-binding heme in the so-called mixed valence form of the enzyme, where only the binuclear-site metals are initially reduced (244,245). (Note that the term “mixed valence” means two-electron-reduced form for cytochromebo3 but one-electron-reduced form for cytochromebd.) Primarily, it was found that this rate is about 3μs (3 × 105 s−1), which is too low to correspond to the pure electron tunneling, especially since this rate is independent of pH and substitution with heavy water (246,247) and therefore may not be presumed to be linked to proton transfer. The obtained value is, however, about three orders of magnitude lower than the value predicted by the empirical electron transfer theory (248,249). The results of recent femtosecond experiments show that the electron transfer from CO-dissociated ferrous hemeo3 to the low-spin ferric hemeb takes place at a rate of 8.3 × 108 s−1 (1.2 ns).
The final step of electron transfer reactions between hemeo3 and CuB has not been a subject for experimental determination, but, from our modern understanding of electron transfer reactions, the rate of transfer between the two metal atoms in the binuclear catalytic site can be predicted to be in the order of picoseconds, taking into account the very small distance (∼5 Å) between these atoms.
The suggested electron transfer sequence in cytochromebo3 can be described by the following equation:
Proton transfer reactions in cytochromebo3
The proton transfer pathways in cytochromebo3 have been investigated much less than the electron transfer pathways. There are no well-identified places for proton localization during the transfer of protons across the membrane, except the glutamate residue at position 286 (250,251,252) in the middle of the membrane. The quality of the X-ray structure of thebo3 oxidase was not sufficient to resolve individual water molecules in the proton-conducting channels. However, a very high level of structural homology to the other members of the heme-copper superfamily allows us to draw some conclusions about proton movements in the protein milieu including structural water arrays (253,254) in two well-defined proton channels. Both of these channels serve as proton delivery pathways and cross about two-thirds of the membrane dielectric from the cytoplasmic side to the binuclear catalytic site. The functional separation of the channel is not yet completely clear. Originally, models of the proposed molecular mechanism of proton pumping predicted the existence of two proton-conductive structures with different functions. One channel was proposed to be responsible for the translocation of “pumped” protons and the other for the uptake of “chemical” protons for water formation (255,256). The resolved structure revealed the existence of two such channels (named the D and K channels), and, originally (257), it was proposed that the D channel was responsible for the translocation of pumped protons and that the K channel was used for the uptake of chemical protons for water formation. However, more recent results indicate that the D channel is involved in the uptake not only of all four pumped protons, but also of two chemical protons used in the oxidative part of the catalytic cycle (258) and that the K channel is responsible for the uptake of another two chemical protons during the reductive part of the cycle (259,260). The proton translocation mechanism also requires two proton exit channels—one for proton release upon the oxidation of bound ubiquinol and the other for the release of the pumped protons. The 3D structure did not show clear exit pathways. However, the exit pathways should be much shorter than the D and K channels, only one-third of the membrane dielectric. In addition, in the well-resolved structures of bovine (261) andRhodobacter sphaeroides (254)aa3 oxidases, areas rich in structural water molecules, which can serve as exit channels, were found above the heme propionates. The results of site-directed mutagenesis studies suggested that the exit for pumped protons may start at conserved residues R481 and R482 (262), which are hydrogen bonded to the Δ-propionates of the hemes, and then continue further through the chains of mobile water molecules.
Cytochromebo3 catalytic cycle
The catalytic cycle for the reduction of oxygen to water is a rather complex process. One enzyme turnover includes the delivery of four electrons and four protons to the catalytic site, the binding of oxygen in this site, the translocation of four protons across the membrane, and the release of the product (two water molecules). This complexity is the reason why the real-time measurement of a single catalytic cycle shows a large number of intermediate states of the enzyme.
The reaction starts from the very fast (kon = 1.6 × 108 M−1·s−1 [30,263]) formation of an oxygen adduct, so-called compound A, which is characterized by an oxygen molecule bound to a high-spin hemeo3, as in hemoglobin.
Unlike that of hemoglobin, the lifetime of this intermediate is very short. In 24μs (263), the bound oxygen accepts electrons from CuB and hemeb and a proton, which results in O—O bond splitting and the formation of a peroxy intermediate. This compound was named “peroxy” because it can be produced by an oxidase containing only two electrons per enzyme molecule, which formally corresponds to the reduction of O2 to peroxide. However, more recent examination by kinetic resonance Raman spectroscopy (264) and mass spectrometry (265) clearly demonstrated that the oxygen-oxygen bond is already broken and that hemeo3 is in the oxoferryl state (Feo34+=O) with another oxygen atom being bound to CuB as a hydroxide ion. The “peroxy” state can also be generated directly in the reaction of the oxidizedbo3 with H2O2 (266), and the resulting spectrum has a characteristic peak at 582 nm and a shoulder at 550 nm.
Upon the arrival of the next electron and the proton, the “peroxy” form (30) is converted into a ferryl intermediate. The results of time-resolved resonance Raman studies showed the formation of a ferryl intermediate with a rate constant of about 2 × 104 s−1 (267,268). A very similar rate was also detected by visible spectroscopy (30,269,270). The stable ferryl intermediate can be obtained as the end product in the reaction of the fully reduced enzyme with oxygen when no bound ubiquinone is in the QH site (269). Such an enzyme contains only three of the four electrons required for complete O2 reduction, and so the catalytic cycle stalls at the ferryl state. In the visible spectroscopy, this state is characterized by a spectrum with peaks at 557 and about 420 nm.
The presence of bound ubiquinone at the QH site of cytochromebo3 (150) increases the electron capacitance of the enzyme and allows the reaction to proceed further to yield the oxidized form. The rate of formation of the oxidized state in the single-turnover experiments was estimated to be 0.3 × 103 s−1 by recording the electron and proton transfer reactions (271).
Mechanism of ΔμH+ formation by cytochromebo3
The oxidation of ubiquinol (Em of redox couple Q/QH2 ∼ +0.1 V) by oxygen (Em of redox couple O2/H2O ∼ +0.8 V) is linked with free energy release in the order of ∼0.7 V. The proton motive force created by the respiratory chain on theE. coli cellular membrane is about −0.2 V (272,273). It is clear that excess free energy (∼0.5 V) can be used for the translocation of more than one charge across the membrane. Indeed, measurement of the stoichiometry (28) of the proton transfer by thebo3 oxidase on theE. coli cellular membrane showed that two protons are transferred per each electron used for the O2 reduction. Of these two charges, the first is driven by the vectorial chemistry and the second is driven by the proton pump (240). The vectorial chemistry includes the oxidation of the ubiquinol molecule and the reduction of dioxygen, with the release and uptake, respectively, of one proton per electron. The charge separation in this case is achieved by the separation in the space of these two events.
The protons extracted from QH2 owing to its oxidation by hemeb are released to the P side, and the protons for the O2 reduction in the binuclear site are taken up from the N side of the membrane. Because hemeb and the binuclear site are located at the same depth in the membrane, the release and uptake of the protons correspond to the translocation of one complete charge across the membrane. A second charge is transferred by the proton pump, the mechanism of which is still unclear. Several proposals for the proton translocation machinery of the heme-copper oxidases have been presented during the past 30 years since the discovery of this machinery in mitochondria (239) and bacteria (240,274), but a definitive explanation of how it functions has not yet been presented. A high level of structural homology between different members of the heme-copper oxidase superfamily argues for similarity in the molecular mechanism of the transmembrane proton translocation. For cytochromec oxidase, it was shown that, in the continuous-turnover regimen, the catalytic cycle consists of four sequential proton translocation steps (275,276). During each step, the delivery of an electron to the binuclear site initiates a proton pump cycle, which is likely to occur by essentially the same mechanism every time when an electron arrives. There are a number of models explaining how the coupling between electron transfer and proton translocation occurs (277,278,279,280,281). Despite the different postulations regarding the drivers of these processes, the models are quite similar and employ electrostatic coupling between the movements of electrons and protons in the low-dielectric medium of the membrane protein. The delivery of an electron to the binuclear center drives proton pumping across the hydrophobic barrier. This translocation is followed by the uptake of the chemical proton to the active site, which leads to the release of the pumped proton out of the protein at the P side.
Mechanism of Cytochromebd Functioning
Under physiological conditions, cytochromebd can oxidize ubiquinol-8 and menaquinol-8.In vitro, thebd enzyme can also utilize shorter-chained ubiquinols, menadiol, duroquinol, and artificial electron donors such asN,N,N′,N′-tetramethyl-p-phenylendiamine (TMPD) (in the presence of excess ascorbate). Of thein vitro substrates, ubiquinol-1 (plus excess dithiothreitol) shows the highest turnover numbers (34,198). The apparentKm values for some reductants are shown inTable 1. As noted above, the activity of the purifiedbd enzyme strongly depends on the nature of the detergent in which the enzyme is solubilized. Cytochromebd is inactive in octylglucoside or cholate but shows high activity in Tween 20 or Triton X-100 (198) orN-lauroyl-sarcosine (73). The ubiquinol-1 oxidase activity of cytochromebd has a broad optimum above pH 7.5 but decreases at more acidic pH values (198). Cytochromebd possesses three distinct active sites—for quinol oxidation, TMPD oxidation, and oxygen reduction. All three sites seem to be located at or close to the periplasmic surface of the membrane. Electrons donated from quinol transfer to hemeb558 and then to theb595-d diheme site, whereas electrons donated from TMPD transfer directly to theb595-d site, bypassing the quinol-binding site and hemeb558 (165).
Mechanism of ΔμH+ formation by cytochromebd
Cytochromebd was shown to generate a transmembrane electric potential both in single turnover (51,52,53,54) and under multiple-turnover conditions (32,50,282) but without invoking a proton pump (H+/e− ratio, ∼1 [28,29,106];q/e− ratio [the number of charges translocated across the membrane per electron], ∼1 [256]). When reconstituted into liposomes, cytochromebd generates an uncoupler-sensitive transmembrane voltage difference with a value of 160 to 180 mV (negative inside) (32,50). The ubi(mena)quinol molecule generated by the dehydrogenases of the respiratory chain can diffuse laterally within the bilayer, finding its way into the quinol oxidase site located near the outer side of the membrane. Upon the oxidation of a quinol, two protons are released into the periplasmic space and two electrons are transferred through hemeb558 to the hemeb595-hemed oxygen-reducing site also located near the periplasmic surface of the membrane. The four protons used for O2 reduction are taken up from the cytoplasm. Single-turnover electrometric experiments showed that membrane potential generation is associated with electron transfer from hemeb558 to theb595-d active site (51,52,53,54). However, since all three hemes are likely to be located at the same depth of the membrane, close to the periplasmic side (176,177), the electron transfer itself cannot be electrogenic (51). Rather, vectorial proton movement from the cytoplasm toward the active site on the opposite (periplasmic) side of the membrane, coupled with a redox reaction between hemesb andd, must occur (51,52,53,54). The latter means that there must be a proton-conducting channel connecting the cytoplasm to theb595-d active site (51,52,54) (Fig. 6). Thus, the generated potential must result primarily from protons moving from the cytoplasm to the O2-reducing site on the opposite side of the membrane.
Reaction of cytochromebd with oxygen: sequence of catalytic intermediates
For cytochromebd, the following species were detected: fully ferrous (32,47), fully ferric (193,209), MV O2 bound (193,208,209,210,283), fully reduced O2 bound (AR) (51), oxoferryl (160,283), and peroxy (53). AR and the peroxy intermediate are transient, whereas the others can be generated in relatively stable forms. In addition, a recent stopped-flow multiwavelength absorption spectroscopy study of the isolated cytochromebd at steady state revealed one electron-reduced species with the electron being on hemeb558 (283).
The reaction of the fully ferrous cytochromebd with oxygen was studied with the use of the flow-flash method (284) by means of spectroscopic and electrometric techniques (51,52,53,54). Recording absorption spectra and membrane potential development with 1-μs time resolution allowed us (i) to observe the sequence of the catalytic intermediates and (ii) to establish which catalytic steps are linked to electric potential generation (53).
The scheme describing the reaction is shown inFig. 7. The initial complex of the fully ferrous cytochromebd with CO is photolyzed in the presence of oxygen. The unligated ferrous enzyme generated by the CO photolysis binds O2 very rapidly, forming the ferrous hemed oxyspecies (AR). The transition of the unligated fully ferrous enzyme to AR is not electrogenic, and its rate is proportional to [O2] (kon = 1.9 × 109 M−1·s−1 [53,161]). The AR formation is followed by electron transfer from hemeb595 to form the peroxy intermediate. The AR→peroxy transition occurs at a rate of 2.2 × 105 s−1 (4.5μs) and is also nonelectrogenic (53). Thus, electron transfer from hemeb595 to hemed is not coupled with membrane potential generation (52,53). The peroxy intermediate might be a true peroxy complex of ferric hemed (53). If this is the case, the bound peroxide is likely not to be in the anionic form but at least singly protonated. The proton may come from one of the two protonatable groups linked to theb595-d diheme site upon its oxidation (52). Alternatively, the peroxy intermediate could be an oxoferryl species with a π-cation radical on the porphyrin ring of hemed and one electron on hemeb558 (285,286). The peroxy intermediate is further converted into the ferryl intermediate at a rate of 2.1 × 104 s−1 (48μs). This conversion is accompanied by the oxidation of hemeb558. The formation of the ferryl intermediate is coupled to the generation of a membrane potential (51,52,53,54). At the ferryl intermediate stage, theb-type hemes are in a ferric state and hemed is in an oxoferryl state. When cytochromebd contains bound quinol, the reaction proceeds further to form the oxidized enzyme. The transition from the ferryl intermediate to the oxidized enzyme occurs at a rate of 0.9 × 103 s−1 (1.1 ms) and is electrogenic (52,53,54).
Figure 7.

Cytochromebd reaction scheme. The three rhombuses represent hemesb558,b595, andd, respectively. The minus signs and red backgrounds in the rhombuses denote that the heme is in the ferrous state. R-CO, R, AR, P, F, O are fully ferrous CO bound, fully ferrous unligated, fully ferrous O2 bound, peroxy, oxoferryl, and fully ferric species, respectively. Transient peroxy species (P) discovered by Belevich et al. (53) is shown as a true peroxy complex of ferric hemed. It is possible, however, that P is an oxoferryl form with a π-cation radical on the porphyrin ring of hemed (285,286).doi:10.1128/ecosalplus.ESP-0012-2015.f7
Under turnover conditions, the oxoferryl and MV O2-bound species dominate, with a small fraction of cytochromebd containing ferric hemed and one electron on hemeb558 (283). The fully oxidized species is possibly not part of the normal catalytic cycle of cytochromebd (287).
Role of hemeb595
Upon the addition of a ligand (e.g., CO or cyanide) to cytochromebd, most of hemeb595 does not bind the ligand (170,172,184,187,188,189). It is likely that hemeb595, although in the high-spin pentacoordinate state, is resistant to interaction with the classical ligands of the high-spin iron-porphyrin complexes. It cannot be excluded that despite the high-spin pentacoordinate state of the iron-porphyrin group, the specific features of the protein environment are such that this redox cofactor is protected from interaction with ligands. In such a case, the participation of hemeb595 in O2 reduction in cooperation with hemed is unlikely and the role of hemeb595 is limited to the transfer of an electron to hemed. Another possible explanation that we favor is the following.
Both hemeb595 and hemed potentially can bind ligands.
The hemes are located very close to each other, forming a diheme active site.
The spatial proximity of hemesb595 andd results in steric restrictions, allowing such a diheme site to bind only one ligand molecule.
Hemed has a higher affinity for ligands than hemeb595, in which case the final result observed upon the addition of a ligand will always be the binding of the ligand to hemed, whereas hemeb595 will remain mainly in the free state (170,183,184,189).
The data on the redox coupling of the two hemes to the same ionizable groups (52) and the migration of CO within the protein from hemed to hemeb595 at cryogenic temperatures (181) are in agreement with this proposal. Modeling the excitonic interactions in absorption and CD spectra of cytochromebd yields an estimate of the Fed-to-Feb595 distance of about 10 Å (171). This distance is markedly larger than that between the Fe-CuB pair in heme-copper oxidases (4 to 5 Å). If this is the case, hemeb595 cannot be functionally identical to CuB. A possible role of hemeb595, apart from electron delivery to hemed and/or to hemed-bound oxygen intermediates, would be as a binding site for hydroxide produced from hemed-bound oxygen upon the reductive cleavage of the O—O bond (171).
CYTOCHROMEbd-II (appBCX)
Originally, it had been reported that a second cytochromebd inE. coli (cytochromebd-II) is encoded byappBC genes (also have been calledcyxAB orcbdAB) (288). TheappBC genes, located at 22 min on theE. coli genetic map, are upstream from a pH 2.5 acid phosphatase (appA) gene (288). TheappBC andappA genes constitute the complex operon. TheappB andappC genes encode 58.1- and 42.4-kDa integral membrane proteins, respectively. The deduced amino acid sequences ofappB andappC gene products show homologies of 60 and 57%, respectively, to the sequences of subunit I (CydA) and subunit II (CydB) of the major cytochromebd (encoded bycydABX) (288). A mutant lacking thecyo andcyd operons and a functionalappC gene is unable to grow aerobically in rich medium, in contrast to a mutant lacking only thecyo andcyd operons, suggesting thatappBC genes encode a third terminal oxidase inE. coli (288). The expression of theappBC-appA operon is induced by entry into the stationary phase and phosphate starvation (289). TheappBC genes are also induced by anaerobic growth, and this induction is controlled by transcriptional regulators AppY and ArcA but is independent of Fnr, in contrast to the induction of thecyd operon (289,290). Only very recently it has become clear that cytochromebd-II, like the major cytochromebd, constitutes the third subunit, appX, encoded by theappX gene (21,43,109). Thus, inE. coli cytochromebd-II is a trisubunit protein encoded by theappCBX locus (21,43,109). Cytochromebd-II is likely to function under even more oxygen-limiting conditions thancydABX-encoded cytochromebd (290). Cytochromebd-II has been extracted and purified (29,291). With the use of sodium dodecyl sulfate-polyacrylamide gel electrophoresis, two polypeptides, 43 kDa (subunit I) and 27 kDa (subunit II), were resolved from the preparation (291). These subunits show no cross-reactivity to subunit-specific polyclonal antibodies directed against the two major subunits ofcydABX-encoded cytochromebd (291). The spectral properties of cytochromebd-II closely resemble those ofcydABX-encoded cytochromebd. Of the quinols tested in the work of Sturr et al. (291), cytochromebd-II utilizes menadiol as the preferred substrate (although ubiquinol-1, the most efficient in vitro substrate forcydABX-encoded cytochromebd, was not examined in that study). The TMPD oxidase activity of cytochromebd-II is much more sensitive to cyanide than that ofcydABX-encoded cytochromebd (291). The electron flux through cytochromebd-II appeared to be significant (292,293). Bekker et al. reported that cytochromebd-II does not contribute to the production of the proton motive force (H+/e− = 0) (292). That conclusion was made based on the growth properties of the strain that lacks NDH-I, cytochromebo3 and cytochromebd under glucose-limited conditions in continuous culture (292). Later, however, Borisov et al., using cytochromebd from the same strain, directly measured generation of a proton motive force (29). Intact cells, spheroplasts, membrane vesicles, and the purified enzyme were tested (29). The authors showed clearly that the catalytic turnover of cytochromebd-II does produce a proton motive force with the same energetic efficiency ascydABX-encoded cytochromebd (H+/e− = 1) (29). Reexamining the data reported in reference292, Sharma et al. (294) confirmed the H+/e− stoichiometry of 1, determined by Borisov et al. (29). Nonetheless, cytochromebd-II still remains poorly studied.
ACKNOWLEDGMENTS
Studies in our laboratories were supported by the Biocentrum Helsinki, the Sigrid Jusélius Foundation, a grant from the Academy of Finland (to M.I.V.), and the Russian Foundation for Basic Research grants 08-04-00093, 11-04-00031, 14-04-00153, and 15-04-06266 (to V.B.B.).
REFERENCES
- 1.Gennis RB, Stewart V. 1996. Respiration, p 217–261.In Neidhardt FC, Curtiss R III, Ingraham JL, Lin ECC, Low KB, Magasanik B, Reznikoff WS, Riley M, Schaechter M, Umbarger HE (ed),Escherichia coli and Salmonella: Cellular and Molecular Biology, 2nd ed. ASM Press, Washington, DC. [Google Scholar]
- 2.Unden G, Bongaerts J. 1997. Alternative respiratory pathways ofEscherichia coli: energetics and transcriptional regulation in response to electron acceptors.Biochim Biophys Acta1320:217–234.[PubMed] 10.1016/S0005-2728(97)00034-0 [DOI] [PubMed] [Google Scholar]
- 3.Garcia-Horsman JA, Barquera B, Rumbley J, Ma J, Gennis RB. 1994. The superfamily of heme-copper respiratory oxidases.J Bacteriol176:5587–5600.[PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pereira MM, Santana M, Teixeira M. 2001. A novel scenario for the evolution of haem-copper oxygen reductases.Biochim Biophys Acta1505:185–208.[PubMed] 10.1016/S0005-2728(01)00169-4 [DOI] [PubMed] [Google Scholar]
- 5.Brunori M, Giuffrè A, Sarti P. 2005. Cytochromec oxidase, ligands and electrons.J Inorg Biochem99:324–336.[PubMed] 10.1016/j.jinorgbio.2004.10.011 [DOI] [PubMed] [Google Scholar]
- 6.Azarkina N, Borisov V, Konstantinov AA. 1997. Spontaneous spectral changes of the reduced cytochromebd.FEBS Lett416:171–174.[PubMed] 10.1016/S0014-5793(97)01196-4 [DOI] [PubMed] [Google Scholar]
- 7.Azarkina N, Siletsky S, Borisov V, von Wachenfeldt C, Hederstedt L, Konstantinov AA. 1999. A cytochromebb’-type quinol oxidase inBacillus subtilis strain 168.J Biol Chem274:32810–32817.[PubMed] 10.1074/jbc.274.46.32810 [DOI] [PubMed] [Google Scholar]
- 8.Gavrikova EV, Grivennikova VG, Borisov VB, Cecchini G, Vinogradov AD. 2009. Assembly of a chimeric respiratory chain from bovine heart submitochondrial particles and cytochromebd terminal oxidase ofEscherichia coli.FEBS Lett583:1287–1291.[PubMed] 10.1016/j.febslet.2009.03.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Siletsky SA, Konstantinov AA. 2012. Cytochromec oxidase: charge translocation coupled to single-electron partial steps of the catalytic cycle.Biochim Biophys Acta1817:476–488.[PubMed] 10.1016/j.bbabio.2011.08.003 [DOI] [PubMed] [Google Scholar]
- 10.Siletsky SA. 2013. Steps of the coupled charge translocation in the catalytic cycle of cytochromec oxidase.Front Biosci18:36–57.[PubMed] 10.2741/4086 [DOI] [PubMed] [Google Scholar]
- 11.Borisov VB. 2002. Defects in mitochondrial respiratory complexes III and IV, and human pathologies.Mol Aspects Med23:385–412.[PubMed] 10.1016/S0098-2997(02)00013-4 [DOI] [PubMed] [Google Scholar]
- 12.Borisov VB. 2004. Mutations in respiratory chain complexes and human diseases.Ital J Biochem53:34–40.[PubMed] [PubMed] [Google Scholar]
- 13.Szundi I, Kittredge C, Choi SK, McDonald W, Ray J, Gennis RB, Einarsdottir O. 2014. Kinetics and intermediates of the reaction of fully reducedEscherichia coli bo3 ubiquinol oxidase with O2.Biochemistry53:5393–5404.[PubMed] 10.1021/bi500567m [DOI] [PubMed] [Google Scholar]
- 14.de Gier J-WL, Lübben M, Reijnders WNM, Tipker CA, van Spanning RJM, Stouthamer AH, van der Oost J. 1994. The terminal oxidase ofParacoccus denitrificans.Mol Microbiol13:183–196.[PubMed] 10.1111/j.1365-2958.1994.tb00414.x [DOI] [PubMed] [Google Scholar]
- 15.Richardson DJ. 2000. Bacterial respiration: a flexible process for a changing environment.Microbiology146:551–571.[PubMed] 10.1099/00221287-146-3-551 [DOI] [PubMed] [Google Scholar]
- 16.Bertsova YV, Demin OV, Bogachev AV. 2005. Respiratory protection of nitrogenase complex inAzotobacter vinelandii.Uspekhi Biolog Khim45:205–234. (in Russian) [Google Scholar]
- 17.Poole RK, Cook GM. 2000. Redundancy of aerobic respiratory chains in bacteria? Routes, reasons and regulation.Adv Microb Physiol43:165–224.[PubMed] 10.1016/S0065-2911(00)43005-5 [DOI] [PubMed] [Google Scholar]
- 18.Anraku Y, Gennis RB. 1987. The aerobic respiratory chain ofEscherichia coli.Trends Biochem Sci12:262–266. 10.1016/0968-0004(87)90131-9 [DOI] [Google Scholar]
- 19.Mogi T, Tsubaki M, Hori H, Miyoshi H, Nakamura H, Anraku Y. 1998. Two terminal quinol oxidase families inEscherichia coli: variations on molecular machinery for dioxygen reduction.J Biochem Mol Biol Biophys2:79–110. [Google Scholar]
- 20.Borisov VB, Gennis RB, Hemp J, Verkhovsky MI. 2011. The cytochromebd respiratory oxygen reductases.Biochim Biophys Acta1807:1398–1413.[PubMed] 10.1016/j.bbabio.2011.06.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Holyoake LV, Poole RK, Shepherd M. 2015. The CydDC family of transporters and their roles in oxidase assembly and homeostasis.Adv Microb Physiol66:1–53.[PubMed] 10.1016/bs.ampbs.2015.04.002 [DOI] [PubMed] [Google Scholar]
- 22.Rice CW, Hempfling WP. 1978. Oxygen-limited continuous culture and respiratory energy conservation inEscherichia coli.J Bacteriol134:115–124.[PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cotter PA, Chepuri V, Gennis RB, Gunsalus RP. 1990. Cytochromeo (cyoABCDE) andd (cydAB) oxidase gene expression inEscherichia coli is regulated by oxygen, pH, and thefnr gene product.J Bacteriol172:6333–6338.[PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fu H-A, Iuchi S, Lin ECC. 1991. The requirement of ArcA and Fnr for peak expression of thecyd operon inEscherichia coli under microaerobic conditions.Mol Gen Genet226:209–213.[PubMed] 10.1007/BF00273605 [DOI] [PubMed] [Google Scholar]
- 25.Sharma P, Teixeira de Mattos MJ, Hellingwerf KJ, Bekker M. 2012. On the function of the various quinone species inEscherichia coli.FEBS J279:3364–3373.[PubMed] 10.1111/j.1742-4658.2012.08608.x [DOI] [PubMed] [Google Scholar]
- 26.Zhang J, Oettmeier W, Gennis RB, Hellwig P. 2002. FTIR spectroscopic evidence for the involvement of an acidic residue in quinone binding in cytochromebd fromEscherichia coli.Biochemistry41:4612–4617.[PubMed] 10.1021/bi011784b [DOI] [PubMed] [Google Scholar]
- 27.Yang K, Zhang J, Vakkasoglu AS, Hielscher R, Osborne JP, Hemp J, Miyoshi H, Hellwig P, Gennis RB. 2007. Glutamate 107 in subunit I of the cytochromebd quinol oxidase fromEscherichia coli is protonated and near the hemed/hemeb595 binuclear center.Biochemistry46:3270–3278.[PubMed] 10.1021/bi061946+ [DOI] [PubMed] [Google Scholar]
- 28.Puustinen A, Finel M, Haltia T, Gennis RB, Wikström M. 1991. Properties of the two terminal oxidases ofEscherichia coli.Biochemistry30:3936–3942.[PubMed] 10.1021/bi00230a019 [DOI] [PubMed] [Google Scholar]
- 29.Borisov VB, Murali R, Verkhovskaya ML, Bloch DA, Han H, Gennis RB, Verkhovsky MI. 2011. Aerobic respiratory chain ofEscherichia coli is not allowed to work in fully uncoupled mode.Proc Natl Acad Sci USA108:17320–17324.[PubMed] 10.1073/pnas.1108217108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Svensson M, Nilsson T. 1993. Flow-flash study of the reaction between cytochromebo and oxygen.Biochemistry32:5442–5447.[PubMed] 10.1021/bi00071a021 [DOI] [PubMed] [Google Scholar]
- 31.Belevich I, Borisov VB, Konstantinov AA, Verkhovsky MI. 2005. Oxygenated complex of cytochromebd fromEscherichia coli: stability and photolability.FEBS Lett579:4567–4570.[PubMed] 10.1016/j.febslet.2005.07.011 [DOI] [PubMed] [Google Scholar]
- 32.Kita K, Konishi K, Anraku Y. 1984. Terminal oxidases ofEscherichia coli aerobic respiratory chain. II. Purification and properties of cytochromeb558-d complex from cells grown with limited oxygen and evidence of branched electron-carrying systems.J Biol Chem259:3375–3381.[PubMed] [PubMed] [Google Scholar]
- 33.Kolonay JF, Jr., Moshiri F, Gennis RB, Kaysser TM, Maier RJ. 1994. Purification and characterization of the cytochromebd complex fromAzotobacter vinelandii: comparison to the complex fromEscherichia coli.J Bacteriol176:4177–4181.[PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jünemann S, Butterworth PJ, Wrigglesworth JM. 1995. A suggested mechanism for the catalytic cycle of cytochromebd terminal oxidase based on kinetic analysis.Biochemistry34:14861–14867.[PubMed] 10.1021/bi00045a029 [DOI] [PubMed] [Google Scholar]
- 35.D’mello R, Hill S, Poole RK. 1996. The cytochromebd quinol oxidase inEscherichia coli has an extremely high oxygen affinity and two-oxygen-binding haems: implications for regulation of activityin vivo by oxygen inhibition.Microbiology142:755–763.[PubMed] 10.1099/00221287-142-4-755 [DOI] [PubMed] [Google Scholar]
- 36.Verkhovsky MI, Morgan JE, Puustinen A, Wikström M. 1996. Kinetic trapping of oxygen in cell respiration.Nature380:268–270.[PubMed] 10.1038/380268a0 [DOI] [PubMed] [Google Scholar]
- 37.D’Mello R, Hill S, Poole RK. 1995. The oxygen affinity of cytochromebo’ inEscherichia coli determined by the deoxygenation of oxyleghemoglobin and oxymyoglobin: Km values for oxygen are in the submicromolar range.J Bacteriol177:867–870.[PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kita K, Konishi K, Anraku Y. 1984. Terminal oxidases ofEscherichia coli aerobic respiratory chain. I. Purification and properties of cytochromeb562-o complex from cells in the early exponential phase of aerobic growth.J Biol Chem259:3368–3374.[PubMed] [PubMed] [Google Scholar]
- 39.Calhoun MW, Newton G, Gennis RB. 1991.E. coli map. Physical map locations of genes encoding components of the aerobic respiratory chain ofEscherichia coli.J Bacteriol173:1569–1570.[PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kranz RG, Barassi CA, Miller MJ, Green GN, Gennis RB. 1983. Immunological characterization of anE. coli strain which is lacking cytochromed.J Bacteriol156:115–121.[PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Green GN, Fang H, Lin R-J, Newton G, Mather M, Georgiou CD, Gennis RB. 1988. The nucleotide sequence of thecyd locus encoding the two subunits of the cytochromed terminal oxidase complex ofEscherichia coli.J Biol Chem263:13138–13143.[PubMed] [PubMed] [Google Scholar]
- 42.VanOrsdel CE, Bhatt S, Allen RJ, Brenner EP, Hobson JJ, Jamil A, Haynes BM, Genson AM, Hemm MR. 2013. TheEscherichia coli CydX protein is a member of the CydAB cytochromebd oxidase complex and is required for cytochromebd oxidase activity.J Bacteriol195:3640–3650.[PubMed] 10.1128/JB.00324-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hoeser J, Hong S, Gehmann G, Gennis RB, Friedrich T. 2014. Subunit CydX ofEscherichia coli cytochromebd ubiquinol oxidase is essential for assembly and stability of the di-heme active site.FEBS Lett588:1537–1541.[PubMed] 10.1016/j.febslet.2014.03.036 [DOI] [PubMed] [Google Scholar]
- 44.Au DC-T, Gennis RB. 1987. Cloning of thecyo locus encoding the cytochromeo terminal oxidase complex ofEscherichia coli.J Bacteriol169:3237–3242.[PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chepuri V, Lemieux LJ, Au DC-T, Gennis RB. 1990. The sequence of thecyo operon indicates substantial structural similarities between the cytochromeo ubiquinol oxidase ofEscherichia coli and theaa3-type family of the cytochromec oxidases.J Biol Chem265:11185–11192.[PubMed] [PubMed] [Google Scholar]
- 46.Minghetti KC, Goswitz VC, Gabriel NE, Hill JJ, Barassi C, Georgiou CD, Chan SI, Gennis RB. 1992. Modified, large-scale purification of the cytochromeo complex ofEscherichia coli yields a two heme/one copper terminal oxidase with high specific activity.Biochemistry31:6917–6924.[PubMed] 10.1021/bi00145a008 [DOI] [PubMed] [Google Scholar]
- 47.Miller MJ, Gennis RB. 1983. The purification and characterization of the cytochromed terminal oxidase complex of theEscherichia coli aerobic respiratory chain.J Biol Chem258:9159–9165.[PubMed] [PubMed] [Google Scholar]
- 48.Salerno JC, Bolgiano B, Poole RK, Gennis RB, Ingledew WJ. 1990. Heme-copper and heme-heme interactions in the cytochromebo-containing quinol oxidase ofEscherichia coli.J Biol Chem265:4364–4368.[PubMed] [PubMed] [Google Scholar]
- 49.Belevich I, Borisov VB, Bloch DA, Konstantinov AA, Verkhovsky MI. 2007. Cytochromebd fromAzotobacter vinelandii: evidence for high-affinity oxygen binding.Biochemistry46:11177–11184.[PubMed] 10.1021/bi700862u [DOI] [PubMed] [Google Scholar]
- 50.Miller MJ, Gennis RB. 1985. The cytochromed complex is a coupling site in the aerobic respiratory chain ofEscherichia coli.J Biol Chem260:14003–14008.[PubMed] [PubMed] [Google Scholar]
- 51.Jasaitis A, Borisov VB, Belevich NP, Morgan JE, Konstantinov AA, Verkhovsky MI. 2000. Electrogenic reactions of cytochromebd.Biochemistry39:13800–13809.[PubMed] 10.1021/bi001165n [DOI] [PubMed] [Google Scholar]
- 52.Belevich I, Borisov VB, Zhang J, Yang K, Konstantinov AA, Gennis RB, Verkhovsky MI. 2005. Time-resolved electrometric and optical studies on cytochromebd suggest a mechanism of electron-proton coupling in the di-heme active site.Proc Natl Acad Sci USA102:3657–3662.[PubMed] 10.1073/pnas.0405683102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Belevich I, Borisov VB, Verkhovsky MI. 2007. Discovery of the true peroxy intermediate in the catalytic cycle of terminal oxidases by real-time measurement.J Biol Chem282:28514–28519.[PubMed] 10.1074/jbc.M705562200 [DOI] [PubMed] [Google Scholar]
- 54.Borisov VB, Belevich I, Bloch DA, Mogi T, Verkhovsky MI. 2008. Glutamate 107 in subunit I of cytochromebd fromEscherichia coli is part of a transmembrane intraprotein pathway conducting protons from the cytoplasm to the hemeb595/hemed active site.Biochemistry47:7907–7914.[PubMed] 10.1021/bi800435a [DOI] [PubMed] [Google Scholar]
- 55.Bader M, Muse W, Ballou DP, Gassner C, Bardwell JCA. 1999. Oxidative protein folding is driven by the electron transport system.Cell98:217–227.[PubMed] 10.1016/S0092-8674(00)81016-8 [DOI] [PubMed] [Google Scholar]
- 56.Hill S, Viollet S, Smith AT, Anthony C. 1990. Roles for entericd-type cytochrome oxidase in N2 fixation and microaerobiosis.J Bacteriol172:2071–2078.[PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jones SA, Chowdhury FZ, Fabich AJ, Anderson A, Schreiner DM, House AL, Autieri SM, Leatham MP, Lins JJ, Jorgensen M, Cohen PS, Conway T. 2007. Respiration ofEscherichia coli in the mouse intestine.Infect Immun75:4891–4899.[PubMed] 10.1128/IAI.00484-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Avetisyan AV, Bogachev AV, Murtasina RA, Skulachev VP. 1992. Involvement of ad-type oxidase in the Na+-motive respiratory chain ofEscherichia coli growing under low ΔμH+ conditions.FEBS Lett306:199–202.[PubMed] 10.1016/0014-5793(92)80999-W [DOI] [PubMed] [Google Scholar]
- 59.Wall D, Delaney JM, Fayet O, Lipinska B, Yamamoto T, Georgopoulos C. 1992.arc-Dependent thermal regulation and extragenic suppression of theEscherichia coli cytochromed operon.J Bacteriol174:6554–6562.[PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Delaney JM, Wall D, Georgopoulos C. 1993. Molecular characterization of theEscherichia coli htrD gene: Cloning, sequence, regulation, and involvement with cytochromed oxidase.J Bacteriol175:166–175.[PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bogachev AV, Murtazina RA, Skulachev VP. 1993. Cytochromed induction inEscherichia coli growing under unfavorable conditions.FEBS Lett336:75–78.[PubMed] 10.1016/0014-5793(93)81612-4 [DOI] [PubMed] [Google Scholar]
- 62.Bogachev AV, Murtazina RA, Shestopalov AI, Skulachev VP. 1995. Induction of theEscherichia coli cytochromed by low ΔμH+ and by sodium ions.Eur J Biochem232:304–308.[PubMed] 10.1111/j.1432-1033.1995.tb20812.x [DOI] [PubMed] [Google Scholar]
- 63.Ashcroft JR, Haddock BA. 1975. Synthesis of alternative membrane-bound redox carriers during aerobic growth ofEscherichia coli in the presence of potassium cyanide.Biochem J148:349–352.[PubMed] 10.1042/bj1480349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Macinga DR, Rather PN. 1996.aarD, aProvidencia stuartii homologue ofcydD: role in 2′-N-acetyltransferase expression, cell morphology and growth in the presence of an extracellular factor.Mol Microbiol19:511–520. 10.1046/j.1365-2958.1996.385912.x [DOI] [PubMed] [Google Scholar]
- 65.Cook GM, Loder C, Soballe B, Stafford GP, Membrillo-Hernandez J, Poole RK. 1998. A factor produced byEscherichia coli K-12 inhibits the growth ofE. coli mutants defective in the cytochromebd quinol oxidase complex: enterochelin rediscovered.Microbiology144:3297–3308.[PubMed] 10.1099/00221287-144-12-3297 [DOI] [PubMed] [Google Scholar]
- 66.Siegele DA, Kolter R. 1993. Isolation and characterization of anEscherichia coli mutant defective in resuming growth after starvation.Genes Dev7:2629–2640.[PubMed] 10.1101/gad.7.12b.2629 [DOI] [PubMed] [Google Scholar]
- 67.Siegele DA, Imlay KR, Imlay JA. 1996. The stationary-phase-exit defect ofcydC (surB) mutants is due to the lack of a functional terminal cytochrome oxidase.J Bacteriol178:6091–6096.[PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Forte E, Borisov VB, Konstantinov AA, Brunori M, Giuffrè A, Sarti P. 2007. Cytochromebd, a key oxidase in bacterial survival and tolerance to nitrosative stress.Ital J Biochem56:265–269.[PubMed] [PubMed] [Google Scholar]
- 69.Giuffrè A, Borisov VB, Mastronicola D, Sarti P, Forte E. 2012. Cytochromebd oxidase and nitric oxide: From reaction mechanisms to bacterial physiology.FEBS Lett586:622–629.[PubMed] 10.1016/j.febslet.2011.07.035 [DOI] [PubMed] [Google Scholar]
- 70.Giuffrè A, Borisov VB, Arese M, Sarti P, Forte E. 2014. Cytochromebd oxidase and bacterial tolerance to oxidative and nitrosative stress.Biochim Biophys Acta1837:1178–1187.[PubMed] 10.1016/j.bbabio.2014.01.016 [DOI] [PubMed] [Google Scholar]
- 71.Borisov VB, Forte E, Siletsky SA, Arese M, Davletshin AI, Sarti P, Giuffrè A. 2015. Cytochromebd protects bacteria against oxidative and nitrosative stress: a potential target for next-generation antimicrobial agents.Biochemistry (Mosc)80:565–575.[PubMed] 10.1134/S0006297915050077 [DOI] [PubMed] [Google Scholar]
- 72.Borisov VB, Forte E, Siletsky SA, Sarti P, Giuffrè A. 2015. Cytochromebd fromEscherichia coli catalyzes peroxynitrite decomposition.Biochim Biophys Acta1847:182–188.[PubMed] 10.1016/j.bbabio.2014.10.006 [DOI] [PubMed] [Google Scholar]
- 73.Borisov VB, Forte E, Konstantinov AA, Poole RK, Sarti P, Giuffrè A. 2004. Interaction of the bacterial terminal oxidase cytochromebd with nitric oxide.FEBS Lett576:201–204.[PubMed] 10.1016/j.febslet.2004.09.013 [DOI] [PubMed] [Google Scholar]
- 74.Borisov VB, Forte E, Sarti P, Brunori M, Konstantinov AA, Giuffrè A. 2006. Nitric oxide reacts with the ferryl-oxo catalytic intermediate of the CuB-lacking cytochromebd terminal oxidase.FEBS Lett580:4823–4826.[PubMed] 10.1016/j.febslet.2006.07.072 [DOI] [PubMed] [Google Scholar]
- 75.Borisov VB, Forte E, Sarti P, Brunori M, Konstantinov AA, Giuffrè A. 2007. Redox control of fast ligand dissociation fromEscherichia coli cytochromebd.Biochem Biophys Res Commun355:97–102.[PubMed] 10.1016/j.bbrc.2007.01.118 [DOI] [PubMed] [Google Scholar]
- 76.Mason MG, Shepherd M, Nicholls P, Dobbin PS, Dodsworth KS, Poole RK, Cooper CE. 2009. Cytochromebd confers nitric oxide resistance toEscherichia coli.Nat Chem Biol5:94–96.[PubMed] 10.1038/nchembio.135 [DOI] [PubMed] [Google Scholar]
- 77.Borisov VB, Forte E, Giuffrè A, Konstantinov A, Sarti P. 2009. Reaction of nitric oxide with the oxidized di-heme and heme-copper oxygen-reducing centers of terminal oxidases: Different reaction pathways and end-products.J Inorg Biochem103:1185–1187.[PubMed] 10.1016/j.jinorgbio.2009.06.002 [DOI] [PubMed] [Google Scholar]
- 78.Jesse HE, Nye TL, McLean S, Green J, Mann BE, Poole RK. 2013. Cytochromebd-I inEscherichia coli is less sensitive than cytochromesbd-II orbo” to inhibition by the carbon monoxide-releasing molecule, CORM-3:N-acetylcysteine reduces CO-RM uptake and inhibition of respiration.Biochim Biophys Acta1834:1693–1703.[PubMed] 10.1016/j.bbapap.2013.04.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Tinajero-Trejo M, Jesse HE, Poole RK. 2013. Gasotransmitters, poisons, and antimicrobials: it’s a gas, gas, gas!F1000Prime Rep5:28.[PubMed] 10.12703/P5-28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sun YH, de Jong MF, den Hartigh AB, Roux CM, Rolan HG, Tsolis RM. 2012. The small protein CydX is required for function of cytochromebd oxidase inBrucella abortus.Front Cell Infect Microbiol2:47.[PubMed] 10.3389/fcimb.2012.00047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lindqvist A, Membrillo-Hernandez J, Poole RK, Cook GM. 2000. Roles of respiratory oxidases in protectingEscherichia coli K12 from oxidative stress.Antonie Van Leeuwenhoek78:23–31.[PubMed] 10.1023/A:1002779201379 [DOI] [PubMed] [Google Scholar]
- 82.Edwards SE, Loder CS, Wu G, Corker H, Bainbridge BW, Hill S, Poole RK. 2000. Mutation of cytochromebd quinol oxidase results in reduced stationary phase survival, iron deprivation, metal toxicity and oxidative stress inAzotobacter vinelandii.FEMS Microbiol Lett185:71–77.[PubMed] 10.1111/j.1574-6968.2000.tb09042.x [DOI] [PubMed] [Google Scholar]
- 83.Korshunov S, Imlay JA. 2010. Two sources of endogenous hydrogen peroxide inEscherichia coli.Mol Microbiol75:1389–1401.[PubMed] 10.1111/j.1365-2958.2010.07059.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Borisov VB, Davletshin AI, Konstantinov AA. 2010. Peroxidase activity of cytochromebd fromEscherichia coli.Biochemistry (Mosc)75:428–436.[PubMed] 10.1134/S000629791004005X [DOI] [PubMed] [Google Scholar]
- 85.Borisov VB, Forte E, Davletshin A, Mastronicola D, Sarti P, Giuffrè A. 2013. Cytochromebd oxidase fromEscherichia coli displays high catalase activity: an additional defense against oxidative stress.FEBS Lett587:2214–2218.[PubMed] 10.1016/j.febslet.2013.05.047 [DOI] [PubMed] [Google Scholar]
- 86.Forte E, Borisov VB, Davletshin A, Mastronicola D, Sarti P, Giuffrè A. 2013. Cytochromebd oxidase and hydrogen peroxide resistance inMycobacterium tuberculosis.MBio4:e01006–01013.[PubMed] 10.1128/mBio.01006-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Meunier B, Madgwick SA, Reil E, Oettmeier W, Rich PR. 1995. New inhibitors of the quinol oxidation sites of bacterial cytochromesbo andbd.Biochemistry34:1076–1083.[PubMed] 10.1021/bi00003a044 [DOI] [PubMed] [Google Scholar]
- 88.Mogi T, Ui H, Shiomi K, Omura S, Kita K. 2008. Gramicidin S identified as a potent inhibitor for cytochromebd-type quinol oxidase.FEBS Lett582:2299–2302.[PubMed] 10.1016/j.febslet.2008.05.031 [DOI] [PubMed] [Google Scholar]
- 89.Borisov VB. 1996. Cytochromebd: structure and properties.Biochemistry (Mosc)61:565–574. [Google Scholar]
- 90.Matsushita K, Patel L, Gennis RB, Kaback HR. 1983. Reconstitution of active transport in proteoliposomes containing cytochromeso oxidase andlac carrier protein purified fromE. coli.Proc Natl Acad Sci USA80:4889–4893.[PubMed] 10.1073/pnas.80.16.4889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Matsushita K, Patel L, Kaback HR. 1984. Cytochromeo type oxidase fromEscherichia coli. Characterization of the enzyme and mechanism of electrochemical proton gradient generation.Biochemistry23:4703–4714.[PubMed] 10.1021/bi00315a028 [DOI] [PubMed] [Google Scholar]
- 92.Georgiou C, Cokic P, Carter K, Webster DA, Gennis RB. 1988. Relationships between membrane-bound cytochromeo fromVitreoscilla and that ofEscherichia coli.Biochim Biophys Acta933:179–183.[PubMed] 10.1016/0005-2728(88)90068-0 [DOI] [PubMed] [Google Scholar]
- 93.Bachmann BJ. 1990. Linkage map ofEscherichia coli K-12, edition 8.Microbiol Rev54:130–197.[PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Nakamura H, Yamoto I, Anraku Y, Lemieux L, Gennis RB. 1990. Expression ofcyoA andcyoB demonstrates that the CO-binding heme component of theE. coli cytochromeo complex is in subunit I.J Biol Chem265:11193–11197.[PubMed] [PubMed] [Google Scholar]
- 95.Nakamura H, Saiki K, Mogi T, Anraku Y. 1997. Assignment and functional roles of thecyoABCDE gene products required for theEscherichia coli bo-type quinol oxidase.J Biochem (Tokyo)122:415–421.[PubMed] 10.1093/oxfordjournals.jbchem.a021769 [DOI] [PubMed] [Google Scholar]
- 96.Saraste M. 1990. Structural features of cytochrome oxidase.Q Rev Biophys23:331–366.[PubMed] 10.1017/S0033583500005588 [DOI] [PubMed] [Google Scholar]
- 97.Chepuri V, Gennis RB. 1990. The use of gene fusions to determine the topology of all of the subunits of the cytochromeo terminal oxidase complex ofEscherichia coli.J Biol Chem265:12978–12986.[PubMed] [PubMed] [Google Scholar]
- 98.Saiki K, Mogi T, Tsubaki M, Hori H, Anraku Y. 1997. Exploring subunit-subunit interactions in theEscherichia coli bo-type ubiquinol oxidase by extragenic suppressor mutation analysis.J Biol Chem272:14721–14726.[PubMed] 10.1074/jbc.272.23.14721 [DOI] [PubMed] [Google Scholar]
- 99.Castresana J, Lübben M, Saraste M, Higgins DG. 1994. Evolution of cytochrome oxidase, an enzyme older than atmospheric oxygen.EMBO J13:2516–2525.[PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Saiki K, Mogi T, Anraku Y. 1992. Heme O biosynthesis inEscherichia coli: theCYOE gene in the cytochromebo operon encodes a protoheme IX farnesyltransferase.Biochem Biophys Res Commun189:1491–1497. 10.1016/0006-291X(92)90243-E [DOI] [PubMed] [Google Scholar]
- 101.Saiki K, Mogi T, Ogura K, Anraku Y. 1993.In vitro heme O synthesis by thecyoE gene product fromEscherichia coli.J Biol Chem268:26041–26045.[PubMed] [PubMed] [Google Scholar]
- 102.Mogi T, Saiki K, Anraku Y. 1994. Biosynthesis and functional role of haem O and haem A.Mol Microbiol14:391–398.[PubMed] 10.1111/j.1365-2958.1994.tb02174.x [DOI] [PubMed] [Google Scholar]
- 103.Minagawa J, Mogi T, Gennis RB, Anraku Y. 1992. Identification of heme ligands in subunit I of the cytochromebo complex inEscherichia coli.J Biol Chem267:2096–2104.[PubMed] [PubMed] [Google Scholar]
- 104.Lemieux LJ, Calhoun MW, Thomas JW, Ingledew WJ, Gennis RB. 1992. Determination of the ligands of the low-spin heme of the cytochromeO ubiquinol oxidase complex using site-directed mutagenesis.J Biol Chem267:2105–2113.[PubMed] [PubMed] [Google Scholar]
- 105.Saiki K, Nakamura H, Mogi T, Anraku Y. 1996. Probing a role of subunit IV of theEscherichia coli bo-type ubiquinol oxidase by deletion and cross-linking analyses.J Biol Chem271:15336–15340.[PubMed] 10.1074/jbc.271.26.15336 [DOI] [PubMed] [Google Scholar]
- 106.Miller MJ, Hermodson M, Gennis RB. 1988. The active form of the cytochromed terminal oxidase complex ofEscherichia coli is a heterodimer containing one copy of each of the two subunits.J Biol Chem263:5235–5240.[PubMed] [PubMed] [Google Scholar]
- 107.Green GN, Kranz JE, Gennis RB. 1984. Cloning thecyd gene locus coding for the cytochromed complex ofEscherichia coli.Gene32:99–106.[PubMed] 10.1016/0378-1119(84)90037-4 [DOI] [PubMed] [Google Scholar]
- 108.Poole RK. 1994. Oxygen reactions with bacterial oxidases and globins: binding, reduction and regulation.Anthonie van Leeuwenhoek65:289–310.[PubMed] 10.1007/BF00872215 [DOI] [PubMed] [Google Scholar]
- 109.Allen RJ, Brenner EP, VanOrsdel CE, Hobson JJ, Hearn DJ, Hemm MR. 2014. Conservation analysis of the CydX protein yields insights into small protein identification and evolution.BMC Genomics15:946.[PubMed] 10.1186/1471-2164-15-946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Chen H, Luo Q, Yin J, Gao T, Gao H. 2015. Evidence for requirement of CydX in function but not assembly of the cytochromebd oxidase inShewanella oneidensis.Biochim Biophys Acta1850:318–328.[PubMed] 10.1016/j.bbagen.2014.10.005 [DOI] [PubMed] [Google Scholar]
- 111.Lorence RM, Koland JG, Gennis RB. 1986. Coulometric and spectroscopic analysis of the purified cytochromed complex ofEscherichia coli: evidence for the identification of “cytochromea1” as cytochromeb595.Biochemistry25:2314–2321.[PubMed] 10.1021/bi00357a003 [DOI] [PubMed] [Google Scholar]
- 112.Newton G, Gennis RB. 1991. In vivo assembly of the cytochromed terminal oxidase complex ofEscherichia coli from genes encoding the two subunits expressed on separate plasmids.Biochim Biophys Acta1089:8–12.[PubMed] 10.1016/0167-4781(91)90077-Y [DOI] [PubMed] [Google Scholar]
- 113.Green GN, Lorence RM, Gennis RB. 1986. Specific overproduction and purification of the cytochromeb558 component of the cytochromed complex fromEscherichia coli.Biochemistry25:2309–2314.[PubMed] 10.1021/bi00357a002 [DOI] [PubMed] [Google Scholar]
- 114.Georgiou CD, Fang H, Gennis RB. 1987. Identification of thecydC locus required for the expression of the functional form of the cytochromed terminal oxidase complex inEscherichia coli.J Bacteriol169:2107–2112.[PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Poole RK, Williams HD, Downie JA, Gibson F. 1989. Mutations affecting the cytochromed-containing oxidase complex ofEscherichia coli K12: Identification and mapping of a fourth locus,cydD.J Gen Microbiol135:1865–1874.[PubMed] 10.1099/00221287-135-7-1865 [DOI] [PubMed] [Google Scholar]
- 116.Poole RK, Hatch L, Cleeter MWJ, Gibson F, Cox GB, Wu G. 1993. Cytochromebd biosynthesis inEscherichia coli: the sequences of thecydC andcydD genes suggest that they encode the components of an ABC membrane transporter.Mol Microbiol10:421–430.[PubMed] 10.1111/j.1365-2958.1993.tb02673.x [DOI] [PubMed] [Google Scholar]
- 117.Bebbington KJ, Williams HD. 1993. Investigation of the role of thecydD gene product in production of a functional cytochromed oxidase inEscherichia coli.FEMS Microbiol Lett112:19–24.[PubMed] 10.1111/j.1574-6968.1993.tb06417.x [DOI] [PubMed] [Google Scholar]
- 118.Poole RK, Gibson F, Wu G. 1994. ThecydD gene product, component of a heterodimeric ABC transporter, is required for assembly of periplasmic cytochromec and of cytochromebd inEscherichia coli.FEMS Microbiol Lett117:217–224.[PubMed] 10.1111/j.1574-6968.1994.tb06768.x [DOI] [PubMed] [Google Scholar]
- 119.Pittman MS, Robinson HC, Poole RK. 2005. A bacterial glutathione transporter (Escherichia coli CydDC) exports reductant to the periplasm.J Biol Chem280:32254–32261.[PubMed] 10.1074/jbc.M503075200 [DOI] [PubMed] [Google Scholar]
- 120.Iuchi S, Chepuri V, Fu HA, Gennis RB, Lin EC. 1990. Requirement for terminal cytochromes in generation of the aerobic signal for thearc regulatory system inEscherichia coli: study utilizing deletions andlac fusions ofcyo andcyd.J Bacteriol172:6020–6025.[PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Cotter PA, Gunsalus RP. 1992. Contribution of thefnr andarcA gene products in coordinate regulation of cytochromeo andd oxidase (cyoABCDE andcydAB) genes inEscherichia coli.FEMS Microbiol Lett91:31–36. 10.1111/j.1574-6968.1992.tb05179.x [DOI] [PubMed] [Google Scholar]
- 122.Gunsalus RP. 1992. Control of electron flow inEscherichia coli: coordinated transcription of respiratory pathway genes.J Bacteriol174:7069–7074.[PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Tseng C-P, Albrecht J, Gunsalus RP. 1996. Effect of microaerophilic cell growth conditions on expression of the aerobic (cyoABCDE andcydAB) and anaerobic (narGHJI,frdABCD, anddmsABC) respiratory pathway genes inEscherichia coli.J Bacteriol178:1094–1098.[PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Cotter PA, Melville SB, Albrecht JA, Gunsalus RP. 1997. Aerobic regulation of cytochromed oxidase (cydAB) operon expression inEscherichia coli: roles of Fnr and ArcA in repression and activation.Mol Microbiol25:605–615.[PubMed] 10.1046/j.1365-2958.1997.5031860.x [DOI] [PubMed] [Google Scholar]
- 125.Govantes F, Albrecht JA, Gunsalus RP. 2000. Oxygen regulation of theEscherichia coli cytochromed oxidase (cydAB) operon: roles of multiple promoters and the Fnr-1 and Fnr-2 binding sites.Mol Microbiol37:1456–1469.[PubMed] 10.1046/j.1365-2958.2000.02100.x [DOI] [PubMed] [Google Scholar]
- 126.Shalel-Levanon S, San KY, Bennett GN. 2005. Effect of oxygen, and ArcA and FNR regulators on the expression of genes related to the electron transfer chain and the TCA cycle inEscherichia coli.Metab Eng7:364–374.[PubMed] 10.1016/j.ymben.2005.07.001 [DOI] [PubMed] [Google Scholar]
- 127.Lynch AS, Lin ECC. 1996. Responses to molecular oxygen, p 1526–1538.In Neidhardt FCea (ed),Escherichia coli and Salmonella typhimurium: Cellular and Molecular Biology, 2nd ed. ASM Press, Washington, DC. [Google Scholar]
- 128.Georgellis D, Kwon O, Lin EC. 2001. Quinones as the redox signal for the arc two-component system of bacteria.Science292:2314–2316.[PubMed] 10.1126/science.1059361 [DOI] [PubMed] [Google Scholar]
- 129.Georgellis D, Kwon O, Lin EC. 1999. Amplification of signaling activity of the arc two-component system ofEscherichia coli by anaerobic metabolites. An in vitro study with different protein modules.J Biol Chem274:35950–35954.[PubMed] 10.1074/jbc.274.50.35950 [DOI] [PubMed] [Google Scholar]
- 130.Alexeeva S, Hellingwerf KJ, Teixeira de Mattos MJ. 2003. Requirement of ArcA for redox regulation inEscherichia coli under microaerobic but not anaerobic or aerobic conditions.J Bacteriol185:204–209.[PubMed] 10.1128/JB.185.1.204-209.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Kiley PJ, Beinert H. 1998. Oxygen sensing by the global regulator, FNR: the role of the iron-sulfur cluster.FEMS Microbiol Rev22:341–352.[PubMed] 10.1111/j.1574-6976.1998.tb00375.x [DOI] [PubMed] [Google Scholar]
- 132.Overton TW, Griffiths L, Patel MD, Hobman JL, Penn CW, Cole JA, Constantinidou C. 2006. Microarray analysis of gene regulation by oxygen, nitrate, nitrite, FNR, NarL and NarP during anaerobic growth ofEscherichia coli: new insights into microbial physiology.Biochem Soc Trans34:104–107.[PubMed] 10.1042/BST0340104 [DOI] [PubMed] [Google Scholar]
- 133.Becker S, Holighaus G, Gabrielczyk T, Unden G. 1996. O2 as the regulatory signal for FNR-dependent gene regulation inEscherichia coli.J Bacteriol178:4515–4521.[PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Collins MD, Jones D. 1981. Distribution of isoprenoid quinone structural types in bacteria and their taxonomic implication.Microbiol Rev45:316–354.[PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Shestopalov AI, Bogachev AV, Murtazina RA, Viryasov MB, Skulachev VP. 1997. Aeration-dependent changes in composition of the quinone pool inEscherichia coli. Evidence of post-transcriptional regulation of the quinone biosynthesis.FEBS Lett404:272–274.[PubMed] 10.1016/s0014-5793(97)00143-9 [DOI] [PubMed] [Google Scholar]
- 136.Sato-Watanabe M, Mogi T, Ogura T, Kitagawa T, Miyoshi H, Iwamura H, Anraku Y. 1994. Identification of a Novel Quinone Binding Site in the Cytochromebo Complex fromEscherichia coli.J Biol Chem269:28908–28912.[PubMed] [PubMed] [Google Scholar]
- 137.Mogi T, Endou S, Akimoto S, Morimoto-Tadokoro M, Miyoshi H. 2006. Glutamates 99 and 107 in transmembrane helix III of subunit I of cytochromebd are critical for binding of the hemeb595-d binuclear center and enzyme activity.Biochemistry45:15785–15792.[PubMed] 10.1021/bi0615792 [DOI] [PubMed] [Google Scholar]
- 138.Belevich I, Bloch DA, Belevich N, Wikström M, Verkhovsky MI. 2007. Exploring the proton pump mechanism of cytochromec oxidase in real time.Proc Natl Acad Sci USA104:2685–2690.[PubMed] 10.1073/pnas.0608794104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Abramson J, Riistama S, Larsson G, Jasaitis A, Svensson-Ek M, Laakkonen L, Puustinen A, Iwata S, Wikström M. 2000. The structure of the ubiquinol oxidase fromEscherichia coli and its ubiquinone binding site.Nat Struct Biol7:910–917.[PubMed] 10.1038/82824 [DOI] [PubMed] [Google Scholar]
- 140.Hellwig P, Mogi T, Tomson FL, Gennis RB, Iwata J, Miyoshi H, Mantele W. 1999. Vibrational modes of ubiquinone in cytochromebo3 fromEscherichia coli identified by Fourier transform infrared difference spectroscopy and specific13C labeling.Biochemistry38:14683–14689.[PubMed] 10.1021/bi991267h [DOI] [PubMed] [Google Scholar]
- 141.Ma J, Tsatsos PH, Zaslavsky D, Barquera B, Thomas JW, Katsonouri A, Puustinen A, Wikström M, Brzezinski P, Alben JO, Gennis RB. 1999. Glutamate-89 in subunit II of cytochromebo3 fromEscherichia coli is required for the function of the heme-copper oxidase.Biochemistry38:15150–15156.[PubMed] 10.1021/bi991764y [DOI] [PubMed] [Google Scholar]
- 142.Hellwig P, Yano T, Ohnishi T, Gennis RB. 2002. Identification of the residues involved in stabilization of the semiquinone radical in the high-affinity ubiquinone binding site in cytochromebo3 fromEscherichia coli by site-directed mutagenesis and EPR spectroscopy.Biochemistry41:10675–10679.[PubMed] 10.1021/bi012146w [DOI] [PubMed] [Google Scholar]
- 143.Babcock GT, Callahan PM, Ondrias MR, Salmeen I. 1981. Coordination geometries and vibrational properties of cytochromesa anda3 in cytochrome oxidase from soret excitation Raman spectroscopy.Biochemistry20:959–966.[PubMed] 10.1021/bi00507a049 [DOI] [PubMed] [Google Scholar]
- 144.Salerno JC, Bolgiano B, Ingledew WJ. 1989. Potentiometric titration of cytochrome-bo type quinol oxidase ofEscherichia coli: evidence for heme-heme and copper-heme interaction.FEBS Lett247:101–105.[PubMed] 10.1016/0014-5793(89)81249-9 [DOI] [PubMed] [Google Scholar]
- 145.Puustinen A, Morgan JE, Verkhovsky M, Thomas JW, Gennis RB, Wikström M. 1992. The low spin heme site of cytochromeo fromE. coli is promiscuous with respect to heme type.Biochemistry31:10363–10369.[PubMed] 10.1021/bi00157a026 [DOI] [PubMed] [Google Scholar]
- 146.Babcock GT, Vickery LE, Palmer G. 1976. Electronic state of heme in cytochrome oxidase. I. Magnetic circular dichroism of the isolated enzyme and its derivatives.J Biol Chem251:7907–7919.[PubMed] [PubMed] [Google Scholar]
- 147.Cheesman MR, Watmough NJ, Pires CA, Turner R, Brittain T, Gennis RB, Greenwood C, Thomson AJ. 1993. Cytochromebo fromEscherichia coli: identification of haem ligands and reaction of the reduced enzyme with carbon monoxide.Biochem J289:709–718.[PubMed] 10.1042/bj2890709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Ralle M, Verkhovskaya ML, Morgan JE, Verkhovsky MI, Wikström M, Blackburn NJ. 1999. Coordination of CuB in reduced and CO-liganded states of cytochromebo3 fromEscherichia coli. Is chloride ion a cofactor?Biochemistry38:7185–7194.[PubMed] 10.1021/bi982885l [DOI] [PubMed] [Google Scholar]
- 149.Osborne JP, Cosper NJ, Stalhandske CM, Scott RA, Alben JO, Gennis RB. 1999. Cu XAS shows a change in the ligation of CuB upon reduction of cytochromebo3 fromEscherichia coli.Biochemistry38:4526–4532.[PubMed] 10.1021/bi982278y [DOI] [PubMed] [Google Scholar]
- 150.Sato-Watanabe M, Mogi T, Miyoshi H, Iwamura H, Matsushita K, Adachi O, Anraku Y. 1994. Structure-function studies on the ubiquinol oxidation site of the cytochromebo complex fromEscherichia coli usingp-benzoquinones and substituted phenols.J Biol Chem269:28899–28907.[PubMed] [PubMed] [Google Scholar]
- 151.Lin MT, Baldansuren A, Hart R, Samoilova RI, Narasimhulu KV, Yap LL, Choi SK, O’Malley PJ, Gennis RB, Dikanov SA. 2012. Interactions of intermediate semiquinone with surrounding protein residues at the QH site of wild-type and D75H mutant cytochromebo3 fromEscherichia coli.Biochemistry51:3827–3838.[PubMed] 10.1021/bi300151q [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Bossis F, De Grassi A, Palese LL, Pierri CL. 2014. Prediction of high- and low-affinity quinol-analogue-binding sites in theaa3 andbo3 terminal oxidases fromBacillus subtilis andEscherichia coli.Biochem J461:305–314.[PubMed] 10.1042/BJ20140082 [DOI] [PubMed] [Google Scholar]
- 153.Yap LL, Lin MT, Ouyang H, Samoilova RI, Dikanov SA, Gennis RB. 2010. The quinone-binding sites of the cytochromebo3 ubiquinol oxidase fromEscherichia coli.Biochim Biophys Acta1797:1924–1932.[PubMed] 10.1016/j.bbabio.2010.04.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Ingledew WJ, Ohnishi T, Salerno JC. 1995. Studies on a stabilisation of ubisemiquinone byEscherichia coli quinol oxidase, cytochromebo.Eur J Biochem227:903–908.[PubMed] 10.1111/j.1432-1033.1995.tb20217.x [DOI] [PubMed] [Google Scholar]
- 155.Sato-Watanabe M, Itoh S, Mogi T, Matsuura K, Miyoshi H, Anraku Y. 1995. Stabilization of a semiquinone radical at the high-affinity quinone-binding site (QH) of theEscherichia coli bo-type ubiquinol oxidase.FEBS Lett374:265–269.[PubMed] 10.1016/0014-5793(95)01125-X [DOI] [PubMed] [Google Scholar]
- 156.Lin MT, Shubin AA, Samoilova RI, Narasimhulu KV, Baldansuren A, Gennis RB, Dikanov SA. 2011. Exploring by pulsed EPR the electronic structure of ubisemiquinone bound at the QH site of cytochromebo3 fromEscherichia coli with in vivo13C-labeled methyl and methoxy substituents.J Biol Chem286:10105–10114.[PubMed] 10.1074/jbc.M110.206821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Rothery RA, Houston AM, Ingledew WJ. 1987. The respiratory chain of anaerobically grownEscherichia coli: reactions with nitrite and oxygen.J Gen Microbiol133:3247–3255.[PubMed] 10.1099/00221287-133-11-3247 [DOI] [PubMed] [Google Scholar]
- 158.Meinhardt SW, Gennis RB, Ohnishi T. 1989. EPR studies of the cytochrome-d complex ofEscherichia coli.Biochim Biophys Acta975:175–184.[PubMed] 10.1016/S0005-2728(89)80216-6 [DOI] [PubMed] [Google Scholar]
- 159.Rothery R, Ingledew WJ. 1989. The cytochromes of anaerobically grownEscherichia coli.Biochem J262:437–443. 10.1042/bj2610437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Kahlow MA, Zuberi TM, Gennis RB, Loehr TM. 1991. Identification of a ferryl intermediate ofEscherichia coli cytochromed terminal oxidase by Resonance Raman spectroscopy.Biochemistry30:11485–11489.[PubMed] 10.1021/bi00113a001 [DOI] [PubMed] [Google Scholar]
- 161.Hill BC, Hill JJ, Gennis RB. 1994. The room temperature reaction of carbon monoxide and oxygen with the cytochromebd quinol oxidase fromEscherichia coli.Biochemistry33:15110–15115.[PubMed] 10.1021/bi00254a021 [DOI] [PubMed] [Google Scholar]
- 162.Ingledew WJ, Rothery RA, Gennis RB, Salerno JC. 1992. The orientation of the three haems of thein situ ubiquinol oxidase, cytochromebd, ofEscherichia coli.Biochem J282:255–259.[PubMed] 10.1042/bj2820255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Dueweke TJ, Gennis RB. 1990. Epitopes of monoclonal antibodies which inhibit ubiquinol oxidase activity ofEscherichia coli cytochromed complex localize a functional domain.J Biol Chem265:4273–4277.[PubMed] [PubMed] [Google Scholar]
- 164.Kranz RG, Gennis RB. 1984. Characterization of the cytochromed terminal oxidase complex ofEscherichia coli using polyclonal and monoclonal antibodies.J Biol Chem259:7998–8003.[PubMed] [PubMed] [Google Scholar]
- 165.Lorence RM, Carter K, Gennis RB, Matsushita K, Kaback HR. 1988. Trypsin proteolysis of the cytochromed complex ofEscherichia coli selectively inhibits ubiquinol oxidase activity while not affectingN,N,N′,N′-tetramethyl-p-phenylenediamine oxidase activity.J Biol Chem11:5271–5276. [PubMed] [Google Scholar]
- 166.Dueweke TJ, Gennis RB. 1991. Proteolysis of the cytochromed complex with trypsin and chymotrypsin localizes a quinol oxidase domain.Biochemistry30:3401–3406.[PubMed] 10.1021/bi00228a007 [DOI] [PubMed] [Google Scholar]
- 167.Poole RK. 1988. Bacterial cytochrome oxidases, p 231–291.In Anthony C (ed),Bacterial Energy Transduction. Academic Press, London. [Google Scholar]
- 168.Bloch DA, Borisov VB, Mogi T, Verkhovsky MI. 2009. Heme/heme redox interaction and resolution of individual optical absorption spectra of the hemes in cytochromebd fromEscherichia coli.Biochim Biophys Acta1787:1246–1253.[PubMed] 10.1016/j.bbabio.2009.05.003 [DOI] [PubMed] [Google Scholar]
- 169.Koland JG, Miller MJ, Gennis RB. 1984. Potentiometric analysis of the purified cytochromed terminal oxidase complex fromEscherichia coli.Biochemistry23:1051–1056. 10.1021/bi00301a003 [DOI] [Google Scholar]
- 170.Borisov V, Arutyunyan AM, Osborne JP, Gennis RB, Konstantinov AA. 1999. Magnetic circular dichroism used to examine the interaction ofEscherichia coli cytochromebd with ligands.Biochemistry38:740–750.[PubMed] 10.1021/bi981908t [DOI] [PubMed] [Google Scholar]
- 171.Arutyunyan AM, Borisov VB, Novoderezhkin VI, Ghaim J, Zhang J, Gennis RB, Konstantinov AA. 2008. Strong excitonic interactions in the oxygen-reducing site ofbd-type oxidase: the Fe-to-Fe distance between hemesd andb595 is 10 Å.Biochemistry47:1752–1759.[PubMed] 10.1021/bi701884g [DOI] [PubMed] [Google Scholar]
- 172.Arutyunyan AM, Sakamoto J, Inadome M, Kabashima Y, Borisov VB. 2012. Optical and magneto-optical activity of cytochromebd fromGeobacillus thermodenitrificans.Biochim Biophys Acta1817:2087–2094.[PubMed] 10.1016/j.bbabio.2012.06.009 [DOI] [PubMed] [Google Scholar]
- 173.Fang H, Lin R-J, Gennis RB. 1989. Location of heme axial ligands in the cytochromed terminal oxidase complex ofEscherichia coli determined by site-directed mutagenesis.J Biol Chem264:8026–8032.[PubMed] [PubMed] [Google Scholar]
- 174.Spinner F, Cheesman MR, Thomson AJ, Kaysser T, Gennis RB, Peng Q, Peterson J. 1995. The haemb558 component of the cytochromebd quinol oxidase complex fromEscherichia coli has histidine-methionine axial ligation.Biochem J308:641–644.[PubMed] 10.1042/bj3080641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Kaysser TM, Ghaim JB, Georgiou C, Gennis RB. 1995. Methionine-393 is an axial ligand of the hemeb558 component of the cytochromebd ubiquinol oxidase fromEscherichia coli.Biochemistry34:13491–13501.[PubMed] 10.1021/bi00041a029 [DOI] [PubMed] [Google Scholar]
- 176.Osborne JP, Gennis RB. 1999. Sequence analysis of cytochromebd oxidase suggests a revised topology for subunits I.Biochim Biophys Acta1410:32–50.[PubMed] 10.1016/S0005-2728(98)00171-6 [DOI] [PubMed] [Google Scholar]
- 177.Zhang J, Barquera B, Gennis RB. 2004. Gene fusions with β-lactamase show that subunit I of the cytochromebd quinol oxidase fromE. coli has nine transmembrane helices with the O2 reactive site near the periplasmic surface.FEBS Lett561:58–62.[PubMed] 10.1016/S0014-5793(04)00125-5 [DOI] [PubMed] [Google Scholar]
- 178.Poole RK. 1983. Bacterial cytochrome oxidases: a structurally and functionally diverse group of electron transfer proteins.Biochim Biophys Acta726:205–243.[PubMed] 10.1016/0304-4173(83)90006-X [DOI] [PubMed] [Google Scholar]
- 179.Vos MH, Borisov VB, Liebl U, Martin J-L, Konstantinov AA. 2000. Femtosecond resolution of ligand-heme interactions in the high-affinity quinol oxidasebd: A di-heme active site?Proc Natl Acad Sci USA97:1554–1559.[PubMed] 10.1073/pnas.030528197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Sun J, Kahlow MA, Kaysser TM, Osborne JP, Hill JJ, Rohlfs RJ, Hille R, Gennis RB, Loehr TM. 1996. Resonance Raman spectroscopic identification of a histidine ligand ofb595 and the nature of the ligation of chlorind in the fully reducedEscherichia coli cytochromebd oxidase.Biochemistry35:2403–2412.[PubMed] 10.1021/bi9518252 [DOI] [PubMed] [Google Scholar]
- 181.Hill JJ, Alben JO, Gennis RB. 1993. Spectroscopic evidence for a heme-heme binuclear center in the cytochromebd ubiquinol oxidase fromEscherichia coli.Proc Natl Acad Sci USA90:5863–5867.[PubMed] 10.1073/pnas.90.12.5863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Tsubaki M, Hori H, Mogi T, Anraku Y. 1995. Cyanide-binding site ofbd-type ubiquinol oxidase fromEscherichia coli.J Biol Chem270:28565–28569.[PubMed] 10.1074/jbc.270.48.28565 [DOI] [PubMed] [Google Scholar]
- 183.Borisov VB, Gennis RB, Konstantinov AA. 1995. Interaction of cytochromebd fromEscherichia coli with hydrogen peroxide.Biochemistry (Mosc)60:231–239. [Google Scholar]
- 184.Borisov VB, Sedelnikova SE, Poole RK, Konstantinov AA. 2001. Interaction of cytochromebd with carbon monoxide at low and room temperatures: evidence that only a small fraction of hemeb595 reacts with CO.J Biol Chem276:22095–22099.[PubMed] 10.1074/jbc.M011542200 [DOI] [PubMed] [Google Scholar]
- 185.Borisov VB, Liebl U, Rappaport F, Martin J-L, Zhang J, Gennis RB, Konstantinov AA, Vos MH. 2002. Interactions between hemed and hemeb595 in quinol oxidasebd fromEscherichia coli: a photoselection study using femtosecond spectroscopy.Biochemistry41:1654–1662.[PubMed] 10.1021/bi0158019 [DOI] [PubMed] [Google Scholar]
- 186.Rappaport F, Zhang J, Vos MH, Gennis RB, Borisov VB. 2010. Heme-heme and heme-ligand interactions in the di-heme oxygen-reducing site of cytochromebd fromEscherichia coli revealed by nanosecond absorption spectroscopy.Biochim Biophys Acta1797:1657–1664.[PubMed] 10.1016/j.bbabio.2010.05.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Borisov VB, Verkhovsky MI. 2013. Accommodation of CO in the di-heme active site of cytochromebd terminal oxidase fromEscherichia coli.J Inorg Biochem118:65–67.[PubMed] 10.1016/j.jinorgbio.2012.09.016 [DOI] [PubMed] [Google Scholar]
- 188.Siletsky SA, Zaspa AA, Poole RK, Borisov VB. 2014. Microsecond time-resolved absorption spectroscopy used to study CO compounds of cytochromebd fromEscherichia coli.PLoS One9:e95617. 10.1371/journal.pone.0095617.[PubMed] 10.1371/journal.pone.0095617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Borisov VB. 2008. Interaction ofbd-type quinol oxidase fromEscherichia coli and carbon monoxide: hemed binds CO with high affinity.Biochemistry (Mosc)73:14–22.[PubMed] 10.1134/S0006297908010021 [DOI] [PubMed] [Google Scholar]
- 190.Poole RK, Williams HD. 1987. Proposal that the function of the membrane-bound cytochromea1-like haemoprotein (cytochromeb-595) inEscherichia coli is a direct electron donation to cytochromed.FEBS Lett217:49–52.[PubMed] 10.1016/0014-5793(87)81240-1 [DOI] [PubMed] [Google Scholar]
- 191.Hata-Tanaka A, Matsuura K, Itoh S, Anraku Y. 1987. Electron flow and heme-heme interaction between cytochromesb-558,b-595 andd in a terminal oxidase ofEscherichia coli.Biochim Biophys Acta893:289–295.[PubMed] 10.1016/0005-2728(87)90050-8 [DOI] [PubMed] [Google Scholar]
- 192.Timkovich R, Cork MS, Gennis RB, Johnson PY. 1985. Proposed structure of hemed, a prosthetic group of bacterial terminal oxidases.J Am Chem Soc107:6069–6075. 10.1021/ja00307a041 [DOI] [Google Scholar]
- 193.Borisov VB, Smirnova IA, Krasnosel’skaya IA, Konstantinov AA. 1994. Oxygenated cytochromebd fromEscherichia coli can be converted into the oxidized form by lipophilic electron acceptors.Biochemistry (Mosc)59:437–443. [PubMed] [Google Scholar]
- 194.Jiang FS, Zuberi TM, Cornelius JB, Clarkson RB, Gennis RB, Belford RL. 1993. Nitrogen and proton ENDOR of cytochromed, hemin, and metmyoglobin in frozen solutions.J Am Chem Soc115:10293–10299. 10.1021/ja00075a052 [DOI] [Google Scholar]
- 195.Hirota S, Mogi T, Anraku Y, Gennis RB, Kitagawa T. 1995. Resonance Raman study on axial ligands of heme irons in cytochromebd-type ubiquinol oxidase fromEscherichia coli.Biospectroscopy1:305–311. 10.1002/bspy.350010502 [DOI] [Google Scholar]
- 196.Hori H, Tsubaki M, Mogi T, Anraku Y. 1996. EPR study of NO complex ofbd-type ubiquinol oxidase fromEscherichia coli.J Biol Chem271:9254–9258.[PubMed] 10.1074/jbc.271.16.9254 [DOI] [PubMed] [Google Scholar]
- 197.Pudek MR, Bragg PD. 1976. Redox potentials of the cytochromes in the respiratory chain of aerobically grownEscherichia coli.Arch Biochem Biophys174:546–552.[PubMed] 10.1016/0003-9861(76)90382-9 [DOI] [PubMed] [Google Scholar]
- 198.Lorence RM, Miller MJ, Borochov A, Faiman-Weinberg R, Gennis RB. 1984. Effects of pH and detergent on the kinetic and electrochemical properties of the purified cytochromed terminal oxidase complex ofEscherichia coli.Biochim Biophys Acta790:148–153.[PubMed] 10.1016/0167-4838(84)90218-8 [DOI] [PubMed] [Google Scholar]
- 199.Hosler JP, Ferguson-Miller S, Calhoun MW, Thomas JW, Hill J, Lemieux L, Ma J, Georgiou C, Fetter J, Shapleigh J, Tecklenburg MMJ, Babcock GT, Gennis RB. 1993. Insight into the active-site structure and function of cytochrome oxidase by analysis of site-directed mutants of bacterial cytochromeaa3 and cytochromebo.J Bioenerg Biomembr25:121–136.[PubMed] 10.1007/BF00762854 [DOI] [PubMed] [Google Scholar]
- 200.Thomas JW, Lemieux LJ, Alben JO, Gennis RB. 1993. Site-directed mutagenesis of highly conserved residues in helix VIII of subunit I of the cytochromebo ubiquinol oxidase fromEscherichia coli: an amphipathic transmembrane helix that may be important in conveying protons to the binuclear center.Biochemistry32:11173–11180.[PubMed] 10.1021/bi00092a029 [DOI] [PubMed] [Google Scholar]
- 201.Thomas JW, Puustinen A, Alben JO, Gennis RB, Wikström M. 1993. Substitution of asparagine for aspartate-135 in subunit I of the cytochromebo ubiquinol oxidase ofEscherichia coli eliminates proton-pumping activity.Biochemistry32:10923–10928.[PubMed] 10.1021/bi00091a048 [DOI] [PubMed] [Google Scholar]
- 202.Fetter JR, Qian J, Shapleigh J, Thomas JW, Garcia-Horsman A, Schmidt E, Hosler J, Babcock GT, Gennis RB, Ferguson-Miller S. 1995. Possible proton relay pathways in cytochromec oxidase.Proc Natl Acad Sci USA92:1604–1608.[PubMed] 10.1073/pnas.92.5.1604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Matsumoto Y, Murai M, Fujita D, Sakamoto K, Miyoshi H, Yoshida M, Mogi T. 2006. Mass spectrometric analysis of the ubiquinol-binding site in cytochromebd fromEscherichia coli.J Biol Chem281:1905–1912.[PubMed] 10.1074/jbc.M508206200 [DOI] [PubMed] [Google Scholar]
- 204.Mogi T, Akimoto S, Endou S, Watanabe-Nakayama T, Mizuochi-Asai E, Miyoshi H. 2006. Probing the ubiquinol-binding site in cytochromebd by site-directed mutagenesis.Biochemistry45:7924–7930.[PubMed] 10.1021/bi060192w [DOI] [PubMed] [Google Scholar]
- 205.Hao W, Golding GB. 2006. Asymmetrical evolution of cytochromebd subunits.J Mol Evol62:132–142.[PubMed] 10.1007/s00239-005-0005-7 [DOI] [PubMed] [Google Scholar]
- 206.Zhang J, Hellwig P, Osborne JP, Gennis RB. 2004. Arginine 391 in subunit I of the cytochromebd quinol oxidase fromEscherichia coli stabilizes the reduced form of the hemes and is essential for quinol oxidase activity.J Biol Chem279:53980–53987.[PubMed] 10.1074/jbc.M408626200 [DOI] [PubMed] [Google Scholar]
- 207.Oden KL, Gennis RB. 1991. Isolation and characterization of a new class of cytochromed terminal oxidase mutants ofEscherichia coli.J Bacteriol173:6174–6183.[PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Poole RK, Kumar C, Salmon I, Chance B. 1983. The 650 nm chromophore inEscherichia coli is an ‘Oxy-’ or oxygenated compound, not the oxidized form of cytochrome oxidased: a hypothesis.J Gen Microbiol129:1335–1344. 10.1099/00221287-129-5-1335 [DOI] [PubMed] [Google Scholar]
- 209.Lorence RM, Gennis RB. 1989. Spectroscopic and quantitative analysis of the oxygenated and peroxy states of the purified cytochromed complex ofEscherichia coli.J Biol Chem264:7135–7140.[PubMed] [PubMed] [Google Scholar]
- 210.Kahlow MA, Loehr TM, Zuberi TM, Gennis RB. 1993. The oxygenated complex of cytochromed terminal oxidase: direct evidence for Fe-O2 coordination in a chlorin-containing enzyme by resonance Raman spectroscopy.J Am Chem Soc115:5845–5846. 10.1021/ja00066a071 [DOI] [Google Scholar]
- 211.Backhed F, Ley RE, Sonnenburg JL, Peterson DA, Gordon JI. 2005. Host-bacterial mutualism in the human intestine.Science307:1915–1920.[PubMed] 10.1126/science.1104816 [DOI] [PubMed] [Google Scholar]
- 212.Alben JO, Moh PP, Fiamingo FG, Altschuld RA. 1981. Cytochrome Oxidase (a3) Heme and Copper observed by low-temperature Fourier transform infrared spectroscopy of the CO complex.Proc Natl Acad Sci USA78:234–237.[PubMed] 10.1073/pnas.78.1.234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Bailey JA, Tomson FL, Mecklenburg SL, MacDonald GM, Katsonouri A, Puustinen A, Gennis RB, Woodruff WH, Dyer RB. 2002. Time-resolved step-scan Fourier transform infrared spectroscopy of the CO adducts of bovine cytochromec oxidase and of cytochromebo3 fromEscherichia coli.Biochemistry41:2675–2683.[PubMed] 10.1021/bi010823g [DOI] [PubMed] [Google Scholar]
- 214.Woodruff WH. 1993. Coordination dynamics of heme-copper oxidases. The ligand shuttle and the control and coupling of electron transfer and proton translocation.J Bioenerg Biomemb25:177–188.[PubMed] 10.1007/BF00762859 [DOI] [PubMed] [Google Scholar]
- 215.Yoshikawa S, Shinzawa-Itoh K, Nakashima R, Yaono R, Yamashita E, Inoue N, Yao M, Fei MJ, Libeu CP, Mizushima T, Yamaguchi H, Tomizaki T, Tsukihara T. 1998. Redox-coupled crystal structural changes in bovine heart cytochromec oxidase.Science280:1723–1729.[PubMed] 10.1126/science.280.5370.1723 [DOI] [PubMed] [Google Scholar]
- 216.Butler C, Forte E, Maria Scandurra F, Arese M, Giuffrè A, Greenwood C, Sarti P. 2002. Cytochromebo3 fromEscherichia coli: the binding and turnover of nitric oxide.Biochem Biophys Res Commun296:1272–1278.[PubMed] 10.1016/S0006-291X(02)02074-0 [DOI] [PubMed] [Google Scholar]
- 217.Ingledew WJ, Horrocks J, Salerno JC. 1993. Ligand binding to the haem-copper binuclear catalytic site of cytochromebo, a respiratory quinol oxidase fromEscherichia coli.Eur J Biochem212:657–664.[PubMed] 10.1111/j.1432-1033.1993.tb17703.x [DOI] [PubMed] [Google Scholar]
- 218.Van Gelder BF, Beinert H. 1969. Studies of the heme components of cytochromec oxidase by EPR spectroscopy.Biochim Biophys Acta189:1–24.[PubMed] 10.1016/0005-2728(69)90219-9 [DOI] [PubMed] [Google Scholar]
- 219.Watmough NJ, Cheesman MR, Butler CS, Little RH, Greenwood C, Thomson AJ. 1998. The dinuclear center of cytochromebo3 fromEscherichia coli.J Bioenerg Biomembr30:55–62.[PubMed] 10.1023/A:1020507511285 [DOI] [PubMed] [Google Scholar]
- 220.Moody AJ, Mitchell R, Jeal AE, Rich PR. 1997. Comparison of the ligand-binding properties of native and copper-less cytochromesbo fromEscherichia coli.Biochem J324:743–752.[PubMed] 10.1042/bj3240743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Cheesman MR, Watmough NJ, Gennis RB, Greenwood C, Thomson AJ. 1994. Magnetic-circular-dichroism studies ofEscherichia coli cytochromebo identification of high-spin ferric, low-spin ferric and ferryl [Fe(IV)] forms of hemeo.Eur J Biochem219:595–602.[PubMed] 10.1111/j.1432-1033.1994.tb19975.x [DOI] [PubMed] [Google Scholar]
- 222.Tsubaki M, Mogi T, Hori H, Sato-Watanabe M, Anraku Y. 1996. Infrared and EPR studies on cyanide binding to the heme-copper binuclear center of cytochromebo-type ubiquinol oxidase fromEscherichia coli.J Biol Chem271:4017–4022.[PubMed] 10.1074/jbc.271.16.9254 [DOI] [PubMed] [Google Scholar]
- 223.Pinakoulaki E, Vamvouka M, Varotsis C. 2004. Resonance Raman detection of the Fe2+-C-N modes in heme-copper oxidases: a probe of the active site.Inorg Chem43:4907–4910.[PubMed] 10.1021/ic035216r [DOI] [PubMed] [Google Scholar]
- 224.Little RH, Cheesman MR, Thomson AJ, Greenwood C, Watmough NJ. 1996. Cytochromebo fromEscherichia coli: binding of azide to CuB.Biochemistry35:13780–13787.[PubMed] 10.1021/bi961221d [DOI] [PubMed] [Google Scholar]
- 225.Tsubaki M. 1993. Fourier-transform infrared study of azide binding to the Fea3-CuB binuclear site of bovine heart cytochromec oxidase: new evidence for a redox-linked conformational change at the binuclear site.Biochemistry32:174–182.[PubMed] 10.1021/bi00052a023 [DOI] [PubMed] [Google Scholar]
- 226.Tsubaki M, Mogi T, Anraku Y, Hori H. 1993. Structure of the heme-copper binuclear center of the cytochromebo complex ofEscherichia coli: WPR and Fourier transform infrared spectroscopic studies.Biochemistry32:6065–6072.[PubMed] 10.1021/bi00074a018 [DOI] [PubMed] [Google Scholar]
- 227.Tsubaki M, Mogi T, Hori H. 1999. Fourier-transform infrared studies on azide-binding to the binuclear center of theEscherichia coli bo-type ubiquinol oxidase.FEBS Lett449:191–195.[PubMed] 10.1016/S0014-5793(99)00423-8 [DOI] [PubMed] [Google Scholar]
- 228.Hubbard JAM, Hughes MN, Poole RK. 1983. Nitrite, but not silver, ions induce spectral changes inEscherichia coli cytochromed.FEBS Lett164:241–243.[PubMed] 10.1016/0014-5793(83)80293-2 [DOI] [PubMed] [Google Scholar]
- 229.Hubbard JAM, Hughes MN, Poole RK. 1985. Reactions of some nitrogen oxyanions and nitric oxide with cytochrome oxidased from oxygen-limitedEscherichia coli K12, p 231–236.In Poole RK, Dow CS (ed),Microbial Gas Metabolism: Mechanistic, Metabolic and Biotechnological Aspects. Academic Press, London.[PubMed] [Google Scholar]
- 230.Bonner FT, Hughes MN, Poole RK, Scott RI. 1991. Kinetics of the reactions of trioxodinitrate and nitrite ions with cytochromed inEscherichia coli.Biochim Biophys Acta1056:133–138.[PubMed] 10.1016/S0005-2728(05)80279-8 [DOI] [PubMed] [Google Scholar]
- 231.Jünemann S. 1997. Cytochromebd terminal oxidase.Biochim Biophys Acta1321:107–127.[PubMed] 10.1016/S0005-2728(97)00046-7 [DOI] [PubMed] [Google Scholar]
- 232.Sarti P, Giuffrè A, Forte E, Mastronicola D, Barone MC, Brunori M. 2000. Nitric oxide and cytochromec oxidase: mechanisms of inhibition and NO degradation.Biochem Biophys Res Commun274:183–187.[PubMed] 10.1006/bbrc.2000.3117 [DOI] [PubMed] [Google Scholar]
- 233.Lemon DD, Calhoun MW, Gennis RB, Woodruff WH. 1993. The gateway to the active site of heme-copper oxidases.Biochemistry32:11953–11956.[PubMed] 10.1021/bi00096a002 [DOI] [PubMed] [Google Scholar]
- 234.Pudek MR, Bragg PD. 1974. Inhibition by cyanide of the respiratory chain oxidases ofEscherichia coli.Arch Biochem Biophys164:682–693.[PubMed] 10.1016/0003-9861(74)90081-2 [DOI] [PubMed] [Google Scholar]
- 235.Krasnoselskaya I, Arutjunjan AM, Smirnova I, Gennis R, Konstantinov AA. 1993. Cyanide-reactive sites in cytochromebd complex fromE. coli.FEBS Lett327:279–283.[PubMed] 10.1016/0014-5793(93)81004-J [DOI] [PubMed] [Google Scholar]
- 236.Poole RK, Williams HD. 1988. Formation of the 680-nm-absorbing form of the cytochromebd oxidase complex ofEscherichia coli by reaction of hydrogen peroxide with the ferric form.FEBS Lett231:243–246.[PubMed] 10.1016/0014-5793(88)80740-3 [DOI] [PubMed] [Google Scholar]
- 237.Borisov V, Gennis R, Konstantinov AA. 1995. Peroxide complex of cytochromebd: kinetics of generation and stability.Biochem Mol Biol Int37:975–982.[PubMed] [PubMed] [Google Scholar]
- 238.Sun J, Osborne JP, Kahlow MA, Kaysser TM, Gennis RB, Loehr TM. 1995. Resonance Raman studies ofEscherichia coli cytochromebd oxidase. Selective enhancement of the three heme chromophores of the “as-isolated” enzyme and characterization of the cyanide adduct.Biochemistry34:12144–12151.[PubMed] 10.1021/bi00038a007 [DOI] [PubMed] [Google Scholar]
- 239.Wikström M. 1977. Proton pump coupled to cytochromec oxidase in mitochondria.Nature266:271–273.[PubMed] 10.1038/266271a0 [DOI] [PubMed] [Google Scholar]
- 240.Puustinen A, Finel M, Virkki M, Wikström M. 1989. Cytochromeo (bo) is a proton pump inParacoccus denitrificans andEscherichia coli.FEBS Lett249:163–167.[PubMed] 10.1016/0014-5793(89)80616-7 [DOI] [PubMed] [Google Scholar]
- 241.Yap LL, Samoilova RI, Gennis RB, Dikanov SA. 2006. Characterization of the exchangeable protons in the immediate vicinity of the semiquinone radical at the QH site of the cytochromebo3 fromEscherichia coli.J Biol Chem281:16879–16887.[PubMed] 10.1074/jbc.M602544200 [DOI] [PubMed] [Google Scholar]
- 242.Kobayashi K, Tagawa S, Mogi T. 2000. Transient formation of ubisemiquinone radical and subsequent electron transfer process in theEscherichia coli cytochromebo.Biochemistry39:15620–15625.[PubMed] 10.1021/bi0014094 [DOI] [PubMed] [Google Scholar]
- 243.Belevich I, Verkhovsky MI, Wikström M. 2006. Proton-coupled electron transfer drives the proton pump of cytochromec oxidase.Nature440:829–832.[PubMed] 10.1038/nature04619 [DOI] [PubMed] [Google Scholar]
- 244.Morgan JE, Verkhovsky MI, Puustinen A, Wikström M. 1993. Intramolecular electron transfer in cytochromeo ofEscherichia coli: events following the photolysis of fully and partially reduced CO-bound forms of thebo3 andoo3 enzymes.Biochemistry32:11413–11418.[PubMed] 10.1021/bi00093a019 [DOI] [PubMed] [Google Scholar]
- 245.Jasaitis A, Johansson MP, Wikström M, Vos MH, Verkhovsky MI. 2007. Nanosecond electron tunneling between the hemes in cytochromebo3.Proc Natl Acad Sci USA104:20811–20814.[PubMed] 10.1073/pnas.0709876105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Oliveberg M, Malmstrцm BG. 1991. Internal electron transfer in cytochromec oxidase: evidence for a rapid equilibrium between cytochromea and the bimetallic site.Biochemistry30:7053–7057.[PubMed] 10.1021/bi00243a003 [DOI] [PubMed] [Google Scholar]
- 247.Hallen S, Brzezinski P, Malmstrom BG. 1994. Internal electron transfer in cytochromec oxidase is coupled to the protonation of a group close to the bimetallic site.Biochemistry33:1467–1472.[PubMed] 10.1021/bi00172a024 [DOI] [PubMed] [Google Scholar]
- 248.Moser CC, Keske JM, Warncke K, Farid RS, Dutton PL. 1992. Nature of biological electron transfer.Nature355:796–802.[PubMed] 10.1038/355796a0 [DOI] [PubMed] [Google Scholar]
- 249.Page CC, Moser CC, Dutton PL. 2003. Mechanism for electron transfer within and between proteins.Curr Opin Chem Biol7:551–556.[PubMed] 10.1016/j.cbpa.2003.08.005 [DOI] [PubMed] [Google Scholar]
- 250.Svensson-Ek M, Thomas JW, Gennis RB, Nilsson T, Brzezinski P. 1996. Kinetics of electron and proton transfer during the reaction of wild type and helix VI mutants of cytochromebo3 with oxygen.Biochemistry35:13673–13680.[PubMed] 10.1021/bi961466q [DOI] [PubMed] [Google Scholar]
- 251.Verkhovskaya ML, Garcia-Horsman A, Puustinen A, Rigaud J-L, Morgan JE, Verkhovsky MI, Wikström M. 1997. Glutamic acid 286 in subunit I of cytochromebo3 is involved in proton translocation.Proc Natl Acad Sci USA94:10128–10131.[PubMed] 10.1073/pnas.94.19.10128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Watmough NJ, Katsonouri A, Little RH, Osborne JP, Furlong-Nickels E, Gennis RB, Brittain T, Greenwood C. 1997. A conserved glutamic acid in helix VI of cytochromebo3 influences a key step in oxygen reduction.Biochemistry36:13736–13742.[PubMed] 10.1021/bi971434i [DOI] [PubMed] [Google Scholar]
- 253.Shinzawa-Itoh K, Aoyama H, Muramoto K, Terada H, Kurauchi T, Tadehara Y, Yamasaki A, Sugimura T, Kurono S, Tsujimoto K, Mizushima T, Yamashita E, Tsukihara T, Yoshikawa S. 2007. Structures and physiological roles of 13 integral lipids of bovine heart cytochromec oxidase.EMBO J26:1713–1725.[PubMed] 10.1038/sj.emboj.7601618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Qin L, Hiser C, Mulichak A, Garavito RM, Ferguson-Miller S. 2006. Identification of conserved lipid/detergent-binding sites in a high-resolution structure of the membrane protein cytochromec oxidase.Proc Natl Acad Sci USA103:16117–16122.[PubMed] 10.1073/pnas.0606149103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Morgan JE, Verkhovsky MI, Wikström M. 1994. The histidine cycle: a new model for proton translocation in the respiratory heme-copper oxidases.J Bioenerg Biomembr26:599–608.[PubMed] 10.1007/BF00831534 [DOI] [PubMed] [Google Scholar]
- 256.Wikström M, Bogachev A, Finel M, Morgan JE, Puustinen A, Raitio M, Verkhovskaya M, Verkhovsky MI. 1994. Mechanism of proton translocation by the respiratory oxidases. The histidine cycle.Biochim Biophys Acta1187:106–111.[PubMed] 10.1016/0005-2728(94)90093-0 [DOI] [PubMed] [Google Scholar]
- 257.Iwata S, Ostermeier C, Ludwig B, Michel H. 1995. Structure at 2.8 Å resolution of cytochromec oxidase fromParacoccus denitrificans.Nature376:660–669.[PubMed] 10.1038/376660a0 [DOI] [PubMed] [Google Scholar]
- 258.Konstantinov AA, Siletsky S, Mitchell D, Kaulen A, Gennis RB. 1997. The roles of the two proton input channels in cytochromec oxidase fromRhodobacter sphaeroides probed by the effects of site-directed mutations on time-resolved electrogenic intraprotein proton transfer.Proc Natl Acad Sci USA94:9085–9090.[PubMed] 10.1073/pnas.94.17.9085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Hosler JP, Shapleigh JP, Kim Y, Pressler M, Georgiou C, Babcock GT, Alben JO, Ferguson-Miller S, Gennis RB. 1996. Polar residues in helix VIII of subunit I of cytochromec oxidase influence the activity and the structure of the active site.Biochemistry35:10776–10783.[PubMed] 10.1021/bi9606511 [DOI] [PubMed] [Google Scholar]
- 260.Wikström M, Jasaitis A, Backgren C, Puustinen A, Verkhovsky MI. 2000. The role of the D- and K-pathways of proton transfer in the function of the haem-copper oxidases.Biochim Biophys Acta1459:514–520.[PubMed] 10.1016/S0005-2728(00)00191-2 [DOI] [PubMed] [Google Scholar]
- 261.Tsukihara T, Shimokata K, Katayama Y, Shimada H, Muramoto K, Aoyama H, Mochizuki M, Shinzawa-Itoh K, Yamashita E, Yao M, Ishimura Y, Yoshikawa S. 2003. The low-spin heme of cytochromec oxidase as the driving element of the proton-pumping process.Proc Natl Acad Sci USA100:15304–15309.[PubMed] 10.1073/pnas.2635097100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Puustinen A, Wikström M. 1999. Proton exit from the heme-copper oxidase ofEscherichia coli.Proc Natl Acad Sci USA96:35–37.[PubMed] 10.1073/pnas.96.1.35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Verkhovsky MI, Morgan JE, Puustinen A, Wikström M. 1996. The “ferrous-oxy” intermediate in the reaction of dioxygen with fully reduced cytochromesaa3 andbo3.Biochemistry35:16241–16246.[PubMed] 10.1021/bi961433a [DOI] [PubMed] [Google Scholar]
- 264.Proshlyakov DA, Pressler MA, Babcock GT. 1998. Dioxygen activation and bond cleavage by mixed-valence cytochromec oxidase.Proc Natl Acad Sci USA95:8020–8025.[PubMed] 10.1073/pnas.95.14.8020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Fabian M, Wong WW, Gennis RB, Palmer G. 1999. Mass spectrometric determination of dioxygen bond splitting in the “peroxy” intermediate of cytochromec oxidase.Proc Natl Acad Sci USA96:13114–13117.[PubMed] 10.1073/pnas.96.23.13114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Morgan JE, Verkhovsky MI, Puustinen A, Wikström M. 1995. Identification of a “peroxy” intermediate in cytochromebo3 ofEscherichia coli.Biochemistry34:15633–15637.[PubMed] 10.1021/bi00048a005 [DOI] [PubMed] [Google Scholar]
- 267.Hirota S, Mogi T, Ogura T, Hirano T, Anraku Y, Kitagawa T. 1994. Observation of the Fe-O2 and FeIV=0 stretching Raman bands for dioxygen reduction intermediates of cytochromebo isolated fromEscherichia coli.FEBS Lett352:67–70.[PubMed] 10.1016/0014-5793(94)00919-8 [DOI] [PubMed] [Google Scholar]
- 268.Wang J, Rumbley J, Ching YC, Takahashi S, Gennis RB, Rousseau DL. 1995. Reaction of cytochromebo3 with oxygen: extra redox center(s) are present in the protein.Biochemistry34:15504–15511.[PubMed] 10.1021/bi00047a016 [DOI] [PubMed] [Google Scholar]
- 269.Puustinen A, Verkhovsky MI, Morgan JE, Belevich NP, Wikström M. 1996. Reaction of theEscherichia coli quinol oxidase cytochromebo3 with dioxygen: the role of a bound ubiquinone molecule.Proc Natl Acad Sci USA93:1545–1548.[PubMed] 10.1073/pnas.93.4.1545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Orii Y, Mogi T, Kawasaki M, Anraku Y. 1994. Facilitated intramolecular electron transfer in cytochromebo-type ubiquinol oxidase initiated upon reaction of the fully reduced enzyme with dioxygen.FEBS Lett352:151–154.[PubMed] 10.1016/0014-5793(94)00939-2 [DOI] [PubMed] [Google Scholar]
- 271.Svensson Ek M, Brzezinski P. 1997. Oxidation of ubiquinol by cytochromebo3 fromEscherichia coli: kinetics of electron and proton transfer.Biochemistry36:5425–5431.[PubMed] 10.1021/bi962478e [DOI] [PubMed] [Google Scholar]
- 272.Kinoshita N, Unemoto T, Kobayashi H. 1984. Proton motive force is not obligatory for growth ofEscherichia coli.J Bacteriol160:1074–1077.[PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Verkhovskaya ML, Verkhovsky MI, Wikström M. 1996. K+-dependent Na+ transport driven by respiration inEscherichia coli cells and membrane vesicles.Biochim Biophys Acta1273:207–216.[PubMed] 10.1016/0005-2728(95)00142-5 [DOI] [PubMed] [Google Scholar]
- 274.van Verseveld HW, Krab K, Stouthamer AH. 1981. Proton pump coupled to cytochromec oxidase inParacoccus denitrificans.Biochim Biophys Acta635:525–534.[PubMed] 10.1016/0005-2728(81)90111-0 [DOI] [PubMed] [Google Scholar]
- 275.Bloch D, Belevich I, Jasaitis A, Ribacka C, Puustinen A, Verkhovsky MI, Wikström M. 2004. The catalytic cycle of cytochromec oxidase is not the sum of its two halves.Proc Natl Acad Sci USA101:529–533.[PubMed] 10.1073/pnas.0306036101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Verkhovsky MI, Belevich I, Bloch DA, Wikström M. 2006. Elementary steps of proton translocation in the catalytic cycle of cytochrome oxidase.Biochim Biophys Acta1757:401–407.[PubMed] 10.1016/j.bbabio.2006.05.026 [DOI] [PubMed] [Google Scholar]
- 277.Popovic DM, Stuchebrukhov AA. 2004. Proton pumping mechanism and catalytic cycle of cytochromec oxidase: coulomb pump model with kinetic gating.FEBS Lett566:126–130.[PubMed] 10.1016/j.febslet.2004.04.016 [DOI] [PubMed] [Google Scholar]
- 278.Siegbahn PEM, Blomberg MRA, Blomberg ML. 2003. Theoretical study of the energetics of proton pumping and oxygen reduction in cytochrome oxidase.J Phys Chem B107:10946–10955. 10.1021/jp035486v [DOI] [Google Scholar]
- 279.Siletsky SA, Pawate AS, Weiss K, Gennis RB, Konstantinov AA. 2004. Transmembrane charge separation during the ferryl-oxo -> oxidized transition in a nonpumping mutant of cytochromec oxidase.J Biol Chem279:52558–52565.[PubMed] 10.1074/jbc.M407549200 [DOI] [PubMed] [Google Scholar]
- 280.Wikström M, Verkhovsky MI. 2007. Mechanism and energetics of proton translocation by the respiratory heme-copper oxidases.Biochim Biophys Acta1767:1200–1214.[PubMed] 10.1016/j.bbabio.2007.06.008 [DOI] [PubMed] [Google Scholar]
- 281.Faxen K, Gilderson G, Adelroth P, Brzezinski P. 2005. A mechanistic principle for proton pumping by cytochromec oxidase.Nature437:286–289.[PubMed] 10.1038/nature03921 [DOI] [PubMed] [Google Scholar]
- 282.Koland JG, Miller MJ, Gennis RB. 1984. Reconstitution of the membrane-bound, ubiquinone-dependent pyruvate oxidase respiratory chain ofEscherichia coli with the cytochromed terminal oxidase.Biochemistry23:445–453.[PubMed] 10.1021/bi00298a008 [DOI] [PubMed] [Google Scholar]
- 283.Borisov VB, Forte E, Sarti P, Giuffrè A. 2011. Catalytic intermediates of cytochromebd terminal oxidase at steady-state: Ferryl and oxy-ferrous species dominate.Biochim Biophys Acta1807:503–509.[PubMed] 10.1016/j.bbabio.2011.02.007 [DOI] [PubMed] [Google Scholar]
- 284.Gibson Q, Greenwood C. 1963. Reactions of cytochrome oxidase with oxygen and carbon monoxide.Biochem J86:541–555.[PubMed] 10.1042/bj0860541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Paulus A, Rossius SG, Dijk M, de Vries S. 2012. Oxoferryl-porphyrin radical catalytic intermediate in cytochromebd oxidases protects cells from formation of reactive oxygen species.J Biol Chem287:8830–8838.[PubMed] 10.1074/jbc.M111.333542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Al-Attar S, de Vries S. 2013. Energy transduction by respiratory metallo-enzymes: from molecular mechanism to cell physiology.Coord Chem Rev257:64–80. 10.1016/j.ccr.2012.05.022 [DOI] [Google Scholar]
- 287.Yang K, Borisov VB, Konstantinov AA, Gennis RB. 2008. The fully oxidized form of the cytochromebd quinol oxidase fromE. coli does not participate in the catalytic cycle: direct evidence from rapid kinetics studies.FEBS Lett582:3705–3709.[PubMed] 10.1016/j.febslet.2008.09.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Dassa J, Fsihi H, Marck C, Dion M, Kieffer-Bontemps M, Boquet PL. 1991. A new oxygen-regulated operon inEscherichia coli comprises the genes for a putative third cytochrome oxidase and for pH 2.5 acid phosphatase (appA).Mol Gen Genet229:341–352.[PubMed] 10.1007/BF00267454 [DOI] [PubMed] [Google Scholar]
- 289.Atlung T, Brondsted L. 1994. Role of the transcriptional activator AppY in regulation of thecyxappA operon ofEscherichia coli by anaerobiosis, phosphate starvation, and growth phase.J Bacteriol176:5414–5422.[PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Brondsted L, Atlung T. 1996. Effect of growth conditions on expression of the acid phosphatase (cyx-appA) operon and theappY gene, which encodes a transcriptional activator ofEscherichia coli.J Bacteriol178:1556–1564.[PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Sturr MG, Krulwich TA, Hicks DB. 1996. Purification of a cytochromebd terminal oxidase encoded by theEscherichia coli app locus from a Δcyo Δcyd strain complemented by genes fromBacillus firmus OF4.J Bacteriol176:1742–1749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Bekker M, de Vries S, Ter Beek A, Hellingwerf KJ, de Mattos MJ. 2009. Respiration ofEscherichia coli can be fully uncoupled via the nonelectrogenic terminal cytochromebd-II oxidase.J Bacteriol191:5510–5517.[PubMed] 10.1128/JB.00562-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Shepherd M, Sanguinetti G, Cook GM, Poole RK. 2010. Compensations for diminished terminal oxidase activity inEscherichia coli: cytochromebd-II-mediated respiration and glutamate metabolism.J Biol Chem285:18464–18472.[PubMed] 10.1074/jbc.M110.118448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Sharma P, Hellingwerf KJ, Teixeira de Mattos MJ, Bekker M. 2012. Uncoupling of substrate-level phosphorylation inEscherichia coli during glucose-limited growth.Appl Environ Microbiol78:6908–6913.[PubMed] 10.1128/AEM.01507-12 [DOI] [PMC free article] [PubMed] [Google Scholar]