WO2025096489A1 - Ubiquitin specific processing protease 1 (usp1) compounds - Google Patents
Ubiquitin specific processing protease 1 (usp1) compoundsDownload PDFInfo
- Publication number
- WO2025096489A1 WO2025096489A1PCT/US2024/053492US2024053492WWO2025096489A1WO 2025096489 A1WO2025096489 A1WO 2025096489A1US 2024053492 WUS2024053492 WUS 2024053492WWO 2025096489 A1WO2025096489 A1WO 2025096489A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- cancer
- alkyl
- alkenyl
- alkynyl
- cycloalkyl
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Classifications
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
Definitions
- UBIQUITIN SPECIFIC PROCESSING PROTEASE 1(USP1) COMPOUNDS CROSS REFERENCE TO RELATED APPLICATIONS
- USP1UBIQUITIN SPECIFIC PROCESSING PROTEASE 1
- USP1COMPOUNDS CROSS REFERENCE TO RELATED APPLICATIONS
- This applicationclaims the benefit of U.S. Provisional Application No. 63/594,480, filed, October 31, 2023, the entire content of which is hereby incorporated herein by reference.
- FIELDThis invention relates to compounds which are inhibitors of ubiquitin-specific- processing protease 1 (USP1) useful for treating diseases including, among others, cancer, autoimmune and inflammatory disorders.
- the inventionfurther pertains to pharmaceutical compositions containing at least one compound according to the invention that are useful for the treatment of conditions related to the inhibition of USP1 in a mammal.
- Ubiquitinationis important in the regulation of many cellular functions and cellular homeostasis.
- the conjugation of ubiquitin to a target proteinis a multistep process involving the sequential action of a ubiquitin activating enzyme (E1), a ubiquitin- conjugating enzyme (E2), and a ubiquitin protein-ligase (E3).
- E1ubiquitin activating enzyme
- E2ubiquitin- conjugating enzyme
- E3ubiquitin protein-ligase
- the ubiquitin tagscan mediate non-covalent interactions of the ubiquitinated substrate with other proteins bearing different types of ubiquitin-binding motifs.
- a family of enzymes, termed deubiquitinasesact on ubiquitinated substrates to catalyze the removal of ubiquitin moieties.
- USP1ubiquitin-specific protease 1
- USP1is a regulator of several important steps in the DNA damage response, particularly in the Fanconi anemia pathway, and in the process of translesion synthesis. USP1 has also been reported to contribute to the repair of double-strand DNA breaks through homologous recombination. In addition, USP1 has been reported to deubiquitinate and stabilize members of the family of inhibitors of DNA binding (ID) proteins, ID1, ID2 and ID3.
- IDDNA binding
- the present disclosureprovides compounds that modulate the expression or activity of USP1.
- the disclosurealso provides compositions, including pharmaceutical compositions, kits that include the compounds, and methods of using (or administering) and making the compounds.
- the compounds provided hereinare useful in treating diseases, disorders, or conditions that are mediated by USP1.
- the disclosurealso provides compounds for use in therapy.
- the disclosurefurther provides compounds for use in a method of treating a disease, disorder, or condition that is mediated by USP1.
- the disclosureprovides uses of the compounds in the manufacture of a medicament for the treatment of a disease, disorder or condition that is mediated by (or mediated, at least in part, by) USP1.
- R 1is selected from 5-6 membered heteroaryl, optionally substituted with one to four halo, hydroxy, amino, -C(O)R a , -C(O)OR b , -C(O)NR a R b , -N(R a )C(O)R b , -S(O)NR a R b , - S(O) 2 NR a R b , -S(O)R g , -S(O) 2 R g , -NR a R b , -OR a , -SR b , C 1-6 alkyl, C 2-6 alkenyl, C 2-6 alkynyl and C3-8 cycloalkyl; wherein each C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl and C3-8 cycloalkyl; wherein each C1-6 alkyl, C2-6 alkenyl, C2-6 alky
- pharmaceutical compositionscomprising a pharmaceutically acceptable carrier and at least one of the compounds disclosed herein.
- the present applicationalso provides methods for the inhibition of USP1 comprising administering a therapeutically effective amount of at least one of Formula I.
- the present applicationalso provides a method for treating proliferative, metabolic, allergic, autoimmune and inflammatory diseases, comprising administering to a host in need of such treatment a therapeutically effective amount of at least one of the compounds disclosed herein.
- the compounds of Formula I or a pharmaceutically acceptable salt thereofmay be used to treat cancers that are mediated by, dependent on or associated with USP1 activity.
- the diseaseis a solid tumor.
- R 1is selected from 5-6 membered heteroaryl, optionally substituted with one to four halo, hydroxy, amino, -C(O)R a , -C(O)OR b , -C(O)NR a R b , -N(R a )C(O)R b , -S(O)NR a R b , - S(O) 2 NR a R b , -S(O)R g , -S(O) 2 R g , -NR a R b , -OR a , -SR b , C 1-6 alkyl, C 2-6 alkenyl, C 2-6 alkynyl and C3-8 cycloalkyl; wherein each C1-6 al
- R 1is selected from: ; salt, stereoisomer, mixture of stereoisomers thereof.
- R 1is selected from: ; salt, stereoisomer, mixture of stereoisomers thereof.
- G 2is selected from: or deuterated analog thereof.
- a compound of Table A or a pharmaceutically acceptable salt thereofmay be used to treat cancers that are mediated by, dependent on or associated with USP1 activity.
- the diseaseis a solid tumor.
- the solid tumoris selected from prostate cancer, pancreatic cancer, bladder cancer, colorectal cancer, breast cancer, renal cancer, hepatocellular cancer, lung cancer, ovarian cancer, cervical cancer, gastric cancer, esophageal cancer, head and neck cancer, melanoma, neuroendocrine cancers, CNS cancers, brain tumors (e.g., glioma, anaplastic oligodendroglioma, adult glioblastoma multiforme, and adult anaplastic astrocytoma), bone cancer, or soft tissue sarcoma.
- the solid tumoris from non- small cell lung cancer or small-cell lung cancer.
- Cis- and trans-geometric isomers of the compounds of the present inventionare described and may be isolated as a mixture of isomers or as separated isomeric forms.
- the present compoundscan be isolated in optically active or racemic forms. It is well known in the art how to prepare optically active forms, such as by resolution of racemic forms or by synthesis from optically active starting materials. All chiral, (enantiomeric and diastereomeric) and racemic forms and all geometric isomeric forms of a structure are intended, unless the specific stereochemistry or isomer form is specifically indicated.
- any variablee.g., R 3
- its definition at each occurrenceis independent of its definition at every other occurrence.
- a groupmay optionally be substituted with up to two R 3 groups and R 3 at each occurrence is selected independently from the definition of R 3 .
- R 3at each occurrence is selected independently from the definition of R 3 .
- combinations of substituents and/or variablesare permissible only if such combinations result in stable compounds.
- a bond to a substituentis shown to cross a bond connecting two atoms in a ring, then such substituent may be bonded to any atom on the ring.
- substituentWhen a substituent is listed without indicating the atom via which such substituent is bonded to the rest of the compound of a given formula, then such substituent may be bonded via any atom in such substituent.
- a dash "-" that is not between two letters or symbolsis used to indicate a point of attachment for a substituent. For example, -CONH 2 is attached through the carbon atom.
- a dash at the front or end of a chemical groupis a matter of convenience; chemical groups may be depicted with or without one or more dashes without losing their ordinary meaning.
- a wavy line drawn through a line in a structureindicates a point of attachment of a group. Unless chemically or structurally required, no directionality is indicated or implied by the order in which a chemical group is written or named.
- optionally substitutedin reference to a particular moiety of the compound of Formula I (e.g., an optionally substituted heteroaryl group) refers to a moiety having 0, 1, 2, or more substituents.
- optionally substituted alkylencompasses both “alkyl” and “substituted alkyl” as defined below. It will be understood by those skilled in the art, with respect to any group containing one or more substituents, that such groups are not intended to introduce any substitution or substitution patterns that are sterically impractical, synthetically non-feasible and/or inherently unstable.
- the term “at least one chemical entity”is interchangeable with the term "a compound”.
- alkylindicates that the alkyl group has from 1 to 6 carbon atoms.
- alkyl or alkyleneis intended to include both branched and straight-chain saturated aliphatic hydrocarbon groups having the specified number of carbon atoms.
- C1-10 alkyl(or alkylene), is intended to include C1, C2, C3, C4, C 5 , C 6 , C 7 , C 8 , C 9 , and C 10 alkyl groups.
- C 1 -C 6 alkyldenotes alkyl having 1 to 6 carbon atoms.
- Alkyl groupscan be unsubstituted or substituted so that one or more of its hydrogens are replaced by another chemical group.
- Example alkyl groupsinclude, but are not limited to, methyl (Me), ethyl (Et), propyl (e.g., n-propyl and isopropyl), butyl (e.g., n-butyl, isobutyl, t-butyl), pentyl (e.g., n-pentyl, isopentyl, neopentyl), and the like.
- alkenylor “alkenylene” is intended to include hydrocarbon chains of either straight or branched configuration and having one or more double carbon-carbon bonds that may occur in any stable point along the chain.
- C2-6 alkenyl(or alkenylene), is intended to include C 2 , C 3 , C 4 , C 5 , and C 6 alkenyl groups.
- alkenylexamples include, but are not limited to, ethenyl, 1-propenyl, 2-propenyl, 2-butenyl, 3-butenyl, 2-pentenyl, 3- pentenyl, 4-pentenyl, 2-hexenyl, 3-hexenyl, 4-hexenyl, 5-hexenyl, 2-methyl-2-propenyl, 4- methyl-3-pentenyl, and the like.
- alkynylor “alkynylene” is intended to include hydrocarbon chains of either straight or branched configuration and having one or more triple carbon-carbon bonds that may occur in any stable point along the chain.
- C 2-6 alkynyl(or alkynylene), is intended to include C2, C3, C4, C5, and C6 alkynyl groups; such as ethynyl, propynyl, butynyl, pentynyl, hexynyl and the like.
- CO2the designation
- alkylis used together with another group, such as in “arylalkyl", this conjunction defines with more specificity at least one of the substituents that the substituted alkyl will contain.
- arylalkylrefers to a substituted alkyl group as defined above where at least one of the substituents is an aryl, such as benzyl.
- aryl(C 0-4 )alkylincludes a substituted lower alkyl having at least one aryl substituent and also includes an aryl directly bonded to another group, i.e., aryl(C0)alkyl.
- heteroarylalkylrefers to a substituted alkyl group as defined above where at least one of the substituents is a heteroaryl.
- alkenyl, alkynyl, alkylene, alkenylene, or alkynylene groupthese groups are substituted with one to three substituents as defined above for substituted alkyl groups.
- alkoxyrefers to an oxygen atom substituted by alkyl or substituted alkyl, as defined herein.
- alkoxyincludes the group -O-C1-6alkyl such as methoxy, ethoxy, propoxy, isopropoxy, n-butoxy, sec-butoxy, tert-butoxy, pentoxy, 2-pentyloxy, isopentoxy, neopentoxy, hexoxy, 2-hexoxy, 3-hexoxy, 3- methylpentoxy, and the like.
- Lower alkoxyrefers to alkoxy groups having one to four carbons.
- cycloalkylrefers to cyclized alkyl groups, including mono-, bi- or poly- cyclic ring systems.
- C3-7 cycloalkylis intended to include C3, C4, C5, C6, and C7 cycloalkyl groups.
- Example cycloalkyl groupsinclude, but are not limited to, cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, norbornyl, and the like.
- “carbocycle” or “carbocyclic residue”is intended to mean any stable 3-, 4-, 5-, 6-, or 7-membered monocyclic or bicyclic or 7-, 8-, 9-, 10-, 11-, 12-, or 13-membered bicyclic or tricyclic ring, any of which may be saturated, partially unsaturated, unsaturated or aromatic.
- carbocyclesinclude, but are not limited to, cyclopropyl, cyclobutyl, cyclobutenyl, cyclopentyl, cyclopentenyl, cyclohexyl, cycloheptenyl, cycloheptyl, cycloheptenyl, adamantyl, cyclooctyl, cyclooctenyl, cyclooctadienyl, [3.3.0]bicyclooctane, [4.3.0]bicyclononane, [4.4.0]bicyclodecane, [2.2.2]bicyclooctane, fluorenyl, phenyl, naphthyl, indanyl, adamantyl, anthracenyl, and tetrahydronaphthyl (tetralin).
- bridged ringsare also included in the definition of carbocycle (e.g., [2.2.2]bicyclooctane).
- carbocyclese.g., [2.2.2]bicyclooctane
- Preferred carbocyclesare cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, and phenyl.
- carbocycleWhen the term “carbocycle” is used, it is intended to include “aryl”.
- a bridged ringoccurs when one or more carbon atoms link two non-adjacent carbon atoms.
- Preferred bridgesare one or two carbon atoms. It is noted that a bridge always converts a monocyclic ring into a bicyclic ring.
- arylrefers to monocyclic or bicyclic aromatic hydrocarbon groups having 6 to 12 carbon atoms in the ring portion, such as phenyl, and naphthyl groups, each of which may be substituted.
- cycloalkylincludes cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl, bicyclooctyl, etc., as well as the following ring systems: , .
- haloalkylmeans a substituted alkyl having one or more halo substituents.
- haloalkylincludes mono, bi, and trifluoromethyl.
- haloalkoxymeans an alkoxy group having one or more halo substituents.
- haloalkoxyincludes OCF 3 .
- heterocyclerefers to substituted and unsubstituted 3- to 7-membered monocyclic groups, 7- to 11-membered bicyclic groups, and 10- to 15- membered tricyclic groups, in which at least one of the rings has at least one heteroatom (O, S or N), said heteroatom containing ring preferably having 1, 2, or 3 heteroatoms selected from O, S, and N.
- Each ring of such a group containing a heteroatomcan contain one or two oxygen or sulfur atoms and/or from one to four nitrogen atoms provided that the total number of heteroatoms in each ring is four or less, and further provided that the ring contains at least one carbon atom.
- the nitrogen and sulfur atomsmay optionally be oxidized and the nitrogen atoms may optionally be quaternized.
- the fused rings completing the bicyclic and tricyclic groupsmay contain only carbon atoms and may be saturated, partially saturated, or fully unsaturated.
- the heterocyclo groupmay be attached at any available nitrogen or carbon atom.
- heterocycleAs used herein the terms “heterocycle”, “heterocycloalkyl”, “heterocyclo”, “heterocyclic”, and “heterocyclyl” include “heteroaryl” groups, as defined below.
- exemplary monocyclic heterocyclyl groupsinclude azetidinyl, pyrrolidinyl, oxetanyl, imidazolinyl, oxazolidinyl, isoxazolinyl, thiazolidinyl, isothiazolidinyl, tetrahydrofuranyl, piperidyl, piperazinyl, 2- oxopiperazinyl, 2-oxopiperidyl, 2-oxopyrrolodinyl, 2-oxoazepinyl, azepinyl, 1-pyridonyl, 4-piperidonyl, tetrahydropyranyl, morpholinyl, thiamorpholinyl
- heteroarylrefers to substituted and unsubstituted aromatic 5- or 6- membered monocyclic groups, 9- or 10-membered bicyclic groups, and 11- to 14- membered tricyclic groups which have at least one heteroatom (O, S or N) in at least one of the rings, said heteroatom-containing ring preferably having 1, 2, or 3 heteroatoms selected from O, S, and N.
- Each ring of the heteroaryl group containing a heteroatomcan contain one or two oxygen or sulfur atoms and/or from one to four nitrogen atoms provided that the total number of heteroatoms in each ring is four or less and each ring has at least one carbon atom.
- the fused rings completing the bicyclic and tricyclic groupsmay contain only carbon atoms and may be saturated, partially saturated, or unsaturated.
- the nitrogen and sulfur atomsmay optionally be oxidized and the nitrogen atoms may optionally be quaternized.
- Heteroaryl groups which are bicyclic or tricyclicmust include at least one fully aromatic ring but the other fused ring or rings may be aromatic or non- aromatic.
- Exemplary monocyclic heteroaryl groupsinclude pyrrolyl, pyrazolyl, pyrazolinyl, imidazolyl, oxazolyl, isoxazolyl, thiazolyl, thiadiazolyl, isothiazolyl, furanyl, thienyl, oxadiazolyl, pyridyl, pyrazinyl, pyrimidinyl, pyridazinyl, triazinyl and the like.
- Exemplary bicyclic heteroaryl groupsinclude indolyl, benzothiazolyl, benzodioxolyl, benzoxazolyl, benzothienyl, quinolinyl, tetrahydroisoquinolinyl, isoquinolinyl, benzimidazolyl, benzopyranyl, indolizinyl, benzofuranyl, chromonyl, coumarinyl, benzopyranyl, cinnolinyl, quinoxalinyl, indazolyl, pyrrolopyridyl, furopyridyl, dihydroisoindolyl, tetrahydroquinolinyl and the like.
- Exemplary tricyclic heteroaryl groupsinclude carbazolyl, benzindolyl, phenanthrollinyl, acridinyl, phenanthridinyl, xanthenyl and the like.
- preferred heteroaryl groupsinclude: N S O N S , , N , , , , , (e.g., cyclohexyl), heterocyclo (e.g., pyrrolidinyl, piperidinyl, and morpholinyl) or heteroaryl (e.g., tetrazolyl, imidazolyl, pyrazolyl, triazolyl, thiazolyl, and furyl) the reference is intended to include rings having 0 to 3, preferably 0 to 2, substituents selected from those recited above for the aryl, cycloalkyl, heterocyclo and/or heteroaryl groups, as appropriate.
- Carbocyclylor “carbocyclic” refers to a saturated or unsaturated monocyclic or bicyclic ring in which all atoms of all rings are carbon. Thus, the term includes cycloalkyl and aryl rings.
- Monocyclic carbocycleshave 3 to 6 ring atoms, still more typically 5 or 6 ring atoms.
- Bicyclic carbocycleshave 7 to 12 ring atoms, e.g., arranged as a bicyclo [4,5], [5,5], [5,6] or [6,6] system, or 9 or 10 ring atoms arranged as a bicyclo [5,6] or [6,6] system.
- Examples of mono- and bicyclic carbocyclesinclude cyclopropyl, cyclobutyl, cyclopentyl, 1-cyclopent-1-enyl, 1-cyclopent-2-enyl, 1- cyclopent-3-enyl, cyclohexyl, 1-cyclohex-1-enyl, 1-cyclohex-2-enyl, 1-cyclohex-3-enyl, phenyl and naphthyl.
- the carbocyclic ringmay be substituted in which case the substituents are selected from those recited above for cycloalkyl and aryl groups.
- alkylthiorefers to the group "alkyl-S-".
- acylrefers to a group -C(O)R, wherein R is hydrogen, alkyl, cycloalkyl, heterocyclyl, aryl, heteroalkyl, or heteroaryl; each of which may be optionally substituted, as defined herein.
- Ris hydrogen, alkyl, cycloalkyl, heterocyclyl, aryl, heteroalkyl, or heteroaryl; each of which may be optionally substituted, as defined herein.
- examples of acylinclude formyl, acetyl, cylcohexylcarbonyl, cyclohexylmethyl-carbonyl, and benzoyl.
- amidorefers to both a "C-amido” group which refers to the group -- C(O)NR g R h and an "N-amido” group which refers to the group –NR g C(O)R h , wherein R g and R h are independently selected from hydrogen, alkyl, aryl, haloalkyl, or heteroaryl; each of which may be optionally substituted.
- aminorefers to the group -NR g R h wherein R g and R h are independently selected from hydrogen, alkyl, haloalkyl, aryl, or heteroaryl; each of which may be optionally substituted.
- the term “azido”refers to –N 3 .
- the term “carbamoyl”refers to both an "O-carbamoyl” group which refers to the group -O-C(O)NR i R j and an "N-carbamoyl” group which refers to the group – NR i C(O)OR j , wherein R i and R j are independently selected from hydrogen, alkyl, aryl, haloalkyl, or heteroaryl; each of which may be optionally substituted.
- the term “carboxyl”refers to -C(O)OH.
- Carboxyl esterrefers to both -OC(O)R and -C(O)OR g , wherein R g is hydrogen, alkyl, cycloalkyl, heterocyclyl, aryl, heteroalkyl, or heteroaryl; each of which may be optionally substituted, as defined herein.
- cyanoor “carbonitrile” refers to the group -CN.
- cycloalkylrefers to a saturated or partially unsaturated cyclic alkyl group having a single ring or multiple rings including fused, bridged, and spiro ring systems.
- cycloalkylincludes cycloalkenyl groups (i.e.
- cycloalkylhas from 3 to 20 ring carbon atoms (i.e., C.sub.3-20 cycloalkyl), 3 to 12 ring carbon atoms (i.e., C.sub.3-12 cycloalkyl), 3 to 10 ring carbon atoms (i.e., C.sub.3-10 cycloalkyl), 3 to 8 ring carbon atoms (i.e., C.sub.3- 8 cycloalkyl), or 3 to 6 ring carbon atoms (i.e., C.sub.3-6 cycloalkyl).
- cycloalkyl groupsinclude cyclopropyl, cyclobutyl, cyclopentyl, and cyclohexyl.
- heteroatomsshall include oxygen, sulfur and nitrogen.
- unsaturatedis used herein to refer to a ring or group, the ring or group may be fully unsaturated or partially unsaturated.
- groups and substituents thereofmay be chosen by one skilled in the field to provide stable moieties and compounds and compounds useful as pharmaceutically-acceptable compounds and/or intermediate compounds useful in making pharmaceutically-acceptable compounds.
- Combinations of substituents and/or variablesare permissible only if such combinations result in stable compounds or useful synthetic intermediates.
- a stable compound or stable structureis meant to imply a compound that is sufficiently robust to survive isolation from a reaction mixture to a useful degree of purity, and subsequent formulation into an efficacious therapeutic agent.
- the presently recited compoundsdo not contain a N-halo, S(O)2H, or S(O)H group.
- the compounds described hereinmay exist in a free form (with no ionization) or can form salts which are also within the scope of this invention. Unless otherwise indicated, reference to an inventive compound is understood to include reference to the free form and to salts thereof.
- salt(s)denotes acidic and/or basic salts formed with inorganic and/or organic acids and bases.
- salt(s)may include zwitterions (inner salts), e.g., when a compound of formula I, contains both a basic moiety, such as an amine or a pyridine or imidazole ring, and an acidic moiety, such as a carboxylic acid.
- Pharmaceutically acceptable (i.e., non-toxic, physiologically acceptable) saltsare preferred, such as, for example, acceptable metal and amine salts in which the cation does not contribute significantly to the toxicity or biological activity of the salt.
- other saltsmay be useful, e.g., in isolation or purification steps which may be employed during preparation, and thus, are contemplated within the scope of the invention.
- Salts of the compounds of hereinmay be formed, for example, by reacting a compound described herein with an amount of acid or base, such as an equivalent amount, in a medium such as one in which the salt precipitates or in an aqueous medium followed by lyophilization.
- Exemplary acid addition saltsinclude acetates (such as those formed with acetic acid or trihaloacetic acid, for example, trifluoroacetic acid), adipates, alginates, ascorbates, aspartates, benzoates, benzenesulfonates, bisulfates, borates, butyrates, citrates, camphorates, camphorsulfonates, cyclopentanepropionates, digluconates, dodecylsulfates, ethanesulfonates, fumarates, glucoheptanoates, glycerophosphates, hemisulfates, heptanoates, hexanoates, hydrochlorides (formed with hydrochloric acid), hydrobromides (formed with hydrogen bromide), hydroiodides, 2- hydroxyethanesulfonates, lactates, maleates (formed with maleic acid), methanesulfonates (formed with methanesul
- Exemplary basic saltsinclude ammonium salts, alkali metal salts such as sodium, lithium, and potassium salts; alkaline earth metal salts such as calcium and magnesium salts; barium, zinc, and aluminum salts; salts with organic bases (for example, organic amines) such as trialkylamines such as triethylamine, procaine, dibenzylamine, N-benzyl- ⁇ -phenethylamine, 1-ephenamine, N,N ⁇ -dibenzylethylene-diamine, dehydroabietylamine, N-ethylpiperidine, benzylamine, dicyclohexylamine or similar pharmaceutically acceptable amines and salts with amino acids such as arginine, lysine and the like.
- organic basesfor example, organic amines
- trialkylaminessuch as triethylamine, procaine, dibenzylamine, N-benzyl- ⁇ -phenethylamine, 1-ephenamine, N,N
- Basic nitrogen-containing groupsmay be quaternized with agents such as lower alkyl halides (e.g., methyl, ethyl, propyl, and butyl chlorides, bromides and iodides), dialkyl sulfates (e.g., dimethyl, diethyl, dibutyl, and diamyl sulfates), long chain halides (e.g., decyl, lauryl, myristyl and stearyl chlorides, bromides and iodides), aralkyl halides (e.g., benzyl and phenethyl bromides), and others.
- lower alkyl halidese.g., methyl, ethyl, propyl, and butyl chlorides, bromides and iodides
- dialkyl sulfatese.g., dimethyl, diethyl, dibutyl, and diamyl sulfates
- Preferred saltsinclude monohydrochloride, hydrogensulfate, methanesulfonate, phosphate or nitrate salts.
- the compounds described hereincan be provided as amorphous solids or crystalline solids. Lyophilization can be employed to provide the compounds as a solid.
- solvatese.g., hydrates
- the term “solvate”means a physical association of a compound with one or more solvent molecules, whether organic or inorganic. This physical association includes hydrogen bonding. In certain instances, the solvate will be capable of isolation, for example when one or more solvent molecules are incorporated in the crystal lattice of the crystalline solid.
- Solidvateencompasses both solution-phase and isolable solvates.
- Exemplary solvatesinclude hydrates, ethanolates, methanolates, isopropanolates, acetonitrile solvates, and ethyl acetate solvates. Methods of solvation are known in the art.
- compounds described herein subsequent to their preparationcan be isolated and purified to obtain a composition containing an amount by weight equal to or greater than 99% of a compound (“substantially pure”), which is then used or formulated as described herein. Such “substantially pure” compounds described herein are also contemplated herein as part of the present invention.
- pharmaceutically acceptablerefers to those compounds, materials, compositions, and/or dosage forms which are, within the scope of sound medical judgment, suitable for use in contact with the tissues of human beings and animals without excessive toxicity, irritation, allergic response, or other problem or complication, commensurate with a reasonable benefit/risk ratio.
- pharmaceutically acceptable saltsrefer to derivatives of the disclosed compounds wherein the parent compound is modified by making acid or base salts thereof. Examples of pharmaceutically-acceptable salts include, but are not limited to, mineral or organic acid salts of basic groups such as amines; and alkali or organic salts of acidic groups such as carboxylic acids.
- the pharmaceutically-acceptable saltsinclude the conventional non-toxic salts or the quaternary ammonium salts of the parent compound formed, for example, from non-toxic inorganic or organic acids.
- such conventional non-toxic saltsinclude those derived from inorganic acids such as hydrochloric, hydrobromic, sulfuric, sulfamic, phosphoric, and nitric; and the salts prepared from organic acids such as acetic, propionic, succinic, glycolic, stearic, lactic, malic, tartaric, citric, ascorbic, pamoic, maleic, hydroxymaleic, phenylacetic, glutamic, benzoic, salicylic, sulfanilic, 2-acetoxybenzoic, fumaric, toluenesulfonic, methanesulfonic, ethane disulfonic, oxalic, and isethionic, and the like.
- the pharmaceutically acceptable salts of the present inventioncan be synthesized from the parent compound which contains a basic or acidic moiety by conventional chemical methods.
- such saltscan be prepared by reacting the free acid or base forms of these compounds with a stoichiometric amount of the appropriate base or acid in water or in an organic solvent, or in a mixture of the two; generally, nonaqueous media like ether, ethyl acetate, ethanol, isopropanol, or acetonitrile are preferred.
- nonaqueous medialike ether, ethyl acetate, ethanol, isopropanol, or acetonitrile are preferred.
- Lists of suitable saltsare found in Remington's Pharmaceutical Sciences, 18th Edition, Mack Publishing Company, Easton, PA (1990), the disclosure of which is hereby incorporated by reference.
- “Stable compound” and “stable structure”are meant to indicate a compound that is sufficiently robust to survive isolation to a useful degree of purity from a reaction mixture, and formulation into an efficacious therapeutic agent.
- the present inventionis intended to embody stable compounds.
- “Therapeutically effective amount”is intended to include an amount of a compound of the present invention alone or an amount of the combination of compounds claimed or an amount of a compound of the present invention in combination with other active ingredients effective to act as an inhibitor of USP1, or effective to treat or prevent proliferative disorders, such as cancer.
- treatingcover the treatment of a disease-state in a mammal, particularly in a human, and include: (a) preventing the disease-state from occurring in a mammal, in particular, when such mammal is predisposed to the disease- state but has not yet been diagnosed as having it; (b) inhibiting the disease-state, i.e., arresting its development; and/or (c) relieving the disease-state, i.e., causing regression of the disease state. All stereoisomers of the compounds of the instant invention are contemplated, either in admixture or in pure or substantially pure form.
- Stereoisomersmay include compounds which are optical isomers through possession of one or more chiral atoms, as well as compounds which are optical isomers by virtue of limited rotation about one or more bonds (atropisomers).
- the definition of compounds according to the inventionembraces all the possible stereoisomers and their mixtures. It very particularly embraces the racemic forms and the isolated optical isomers having the specified activity.
- the racemic formscan be resolved by physical methods, such as, for example, fractional crystallization, separation or crystallization of diastereomeric derivatives or separation by chiral column chromatography.
- the individual optical isomerscan be obtained from the racemates from the conventional methods, such as, for example, salt formation with an optically active acid followed by crystallization.
- the present inventionis intended to include all isotopes of atoms occurring in the present compounds.
- Isotopesinclude those atoms having the same atomic number but different mass numbers.
- isotopes of hydrogeninclude deuterium and tritium.
- Isotopes of carboninclude 13 C and 14 C.
- Isotopically-labeled compounds of the inventioncan generally be prepared by conventional techniques known to those skilled in the art or by processes analogous to those described herein, using an appropriate isotopically-labeled reagent in place of the non-labeled reagent otherwise employed. Prodrugs and solvates of the inventive compounds are also contemplated.
- prodrugdenotes a compound which, upon administration to a subject, undergoes chemical conversion by metabolic or chemical processes to yield a compound of the formula I, and/or a salt and/or solvate thereof.
- Any compound that will be converted in vivo to provide the bioactive agenti.e., the compound for formula I
- compounds containing a carboxy groupcan form physiologically hydrolyzable esters which serve as prodrugs by being hydrolyzed in the body to yield formula I compounds per se.
- prodrugsare preferably administered orally since hydrolysis in many instances occurs principally under the influence of the digestive enzymes.
- physiologically hydrolyzable esters of compounds of formula Iinclude C 1-6 alkylbenzyl, 4-methoxybenzyl, indanyl, phthalyl, methoxymethyl, C1-6alkanoyloxy-C1-6alkyl, e.g., acetoxymethyl, pivaloyloxymethyl or propionyloxymethyl, C 1-6 alkoxycarbonyloxy-C 1-6 alkyl, e.g., methoxycarbonyl-oxymethyl or ethoxycarbonyloxymethyl, glycyloxymethyl, phenylglycyloxymethyl, (5-methyl-2- oxo-1,3-dioxolen-4-yl)-methyl and other well known physiologically hydrolyzable esters used, for example, in the penicillin and cephalosporin arts.
- estersmay be prepared by conventional techniques known in the art.
- Various forms of prodrugsare well known in the art and are described in Rautio, J. et al., Nature Review Drug Discovery, 17, 559-587 (2016).
- Compounds described in the application and their saltsmay exist in their tautomeric form, in which hydrogen atoms are transposed to other parts of the molecules and the chemical bonds between the atoms of the molecules are consequently rearranged. It should be understood that the all tautomeric forms, insofar as they may exist, are included within the invention. Additionally, inventive compounds may have trans- and cis-isomers.
- the disclosure hereinfurther relates to compounds described herein, the tautomers and stereoisomeric forms thereof, and the pharmaceutically acceptable addition salts, and the solvates thereof, for use as a medicament. Furthermore, the disclosure herein relates to the use of a compound described herein, a tautomer or a stereoisomeric form thereof, or a pharmaceutically acceptable addition salt, or a solvate thereof, or a pharmaceutical composition according to the invention, for the manufacture of a medicament.
- compositionsmay contain other therapeutic agents as described above and may be formulated, for example, by employing conventional solid or liquid vehicles or diluents, as well as pharmaceutical additives of a type appropriate to the mode of desired administration (e.g., excipients, binders, preservatives, stabilizers, flavors, etc.) according to techniques such as those well known in the art of pharmaceutical formulation.
- the present inventionfurther includes compositions comprising one or more compounds described herein and a pharmaceutically acceptable carrier.
- a "pharmaceutically acceptable carrier”refers to media generally accepted in the art for the delivery of biologically active agents to animals, in particular, mammals. Pharmaceutically acceptable carriers are formulated according to a number of factors well within the purview of those of ordinary skill in the art.
- compositionsinclude without limitation the type and nature of the active agent being formulated; the subject to which the agent- containing composition is to be administered; the intended route of administration of the composition; and, the therapeutic indication being targeted.
- Pharmaceutically acceptable carriersinclude both aqueous and non-aqueous liquid media, as well as a variety of solid and semi-solid dosage forms. Such carriers can include a number of different ingredients and additives in addition to the active agent, such additional ingredients being included in the formulation for a variety of reasons, e.g., stabilization of the active agent, binders, etc., well known to those of ordinary skill in the art.
- compositions described hereinmay be administered by any means suitable for the condition to be treated, which may depend on the need for site-specific treatment or quantity of drug to be delivered.
- Topical administrationis generally preferred for skin- related diseases, and systematic treatment preferred for cancerous or pre-cancerous conditions, although other modes of delivery are contemplated.
- the compoundsmay be delivered orally, such as in the form of tablets, capsules, granules, powders, or liquid formulations including syrups; topically, such as in the form of solutions, suspensions, gels or ointments; sublingually; bucally; parenterally, such as by subcutaneous, intravenous, intramuscular or intrasternal injection or infusion techniques (e.g., as sterile injectable aq. or non-aq. solutions or suspensions); nasally such as by inhalation spray; topically, such as in the form of a cream or ointment; rectally such as in the form of suppositories; or liposomally.
- topicallysuch as in the form of solutions, suspensions, gels or ointments
- sublinguallye.g., as sterile injectable aq. or non-aq. solutions or suspensions
- nasallysuch as by inhalation spray
- topicallysuch as in the form of a cream or ointment
- Dosage unit formulations containing non-toxic, pharmaceutically acceptable vehicles or diluentsmay be administered.
- the compoundsmay be administered in a form suitable for immediate release or extended release. Immediate release or extended release may be achieved with suitable pharmaceutical compositions or, particularly in the case of extended release, with devices such as subcutaneous implants or osmotic pumps.
- compositionsmay include fast-dissolving diluents such as mannitol, lactose, sucrose, and/or cyclodextrins.
- fast-dissolving diluentssuch as mannitol, lactose, sucrose, and/or cyclodextrins.
- high molecular weight excipientssuch as celluloses (AVICEL®) or polyethylene glycols (PEG); an excipient to aid mucosal adhesion such as hydroxypropyl cellulose (HPC), hydroxypropyl methyl cellulose (HPMC), sodium carboxymethyl cellulose (SCMC), and/or maleic anhydride copolymer (e.g., GANTREZ®); and agents to control release such as polyacrylic copolymer (e.g., CARBOPOL 934®).
- HPChydroxypropyl cellulose
- HPMChydroxypropyl methyl cellulose
- SCMCsodium carboxymethyl cellulose
- Formulations for parenteral administrationmay be in the form of aqueous or non- aqueous isotonic sterile injection solutions or suspensions. These solutions and suspensions may be prepared from sterile powders or granules using one or more of the carriers or diluents mentioned for use in the formulations for oral administration or by using other suitable dispersing or wetting agents and suspending agents.
- the compoundsmay be dissolved in water, polyethylene glycol, propylene glycol, ethanol, corn oil, cottonseed oil, peanut oil, sesame oil, benzyl alcohol, sodium chloride, tragacanth gum, and/or various buffers.
- the active ingredientmay also be administered by injection as a composition with suitable carriers including saline, dextrose, or water, or with cyclodextrin (i.e. Captisol), cosolvent solubilization (i.e. propylene glycol) or micellar solubilization (i.e. Tween 80).
- suitable carriersincluding saline, dextrose, or water, or with cyclodextrin (i.e. Captisol), cosolvent solubilization (i.e. propylene glycol) or micellar solubilization (i.e. Tween 80).
- compositions for parenteral administrationinclude injectable solutions or suspensions which may contain, for example, suitable non-toxic, parenterally acceptable diluents or solvents, such as mannitol, 1,3-butanediol, water, Ringer's solution, an isotonic sodium chloride solution, or other suitable dispersing or wetting and suspending agents, including synthetic mono- or diglycerides, and fatty acids, including oleic acid.
- suitable non-toxic, parenterally acceptable diluents or solventssuch as mannitol, 1,3-butanediol, water, Ringer's solution, an isotonic sodium chloride solution, or other suitable dispersing or wetting and suspending agents, including synthetic mono- or diglycerides, and fatty acids, including oleic acid.
- the sterile injectable preparationmay also be a sterile injectable solution or suspension in a non-toxic parenterally acceptable diluent or solvent, for example as a solution in 1,
- a sterile injectable oil-in-water microemulsioncan, for example, be prepared by 1) dissolving at least one compound described herein in an oily phase, such as, for example, a mixture of soybean oil and lecithin; 2) combining the compound-containing oil phase with a water and glycerol mixture; and 3) processing the combination to form a microemulsion.
- a sterile aqueous or oleaginous suspensioncan be prepared in accordance with methods already known in the art.
- a sterile aqueous solution or suspensioncan be prepared with a non-toxic parenterally-acceptable diluent or solvent, such as, for example, 1,3-butane diol; and a sterile oleaginous suspension can be prepared with a sterile non-toxic acceptable solvent or suspending medium, such as, for example, sterile fixed oils, e.g., synthetic mono- or diglycerides; and fatty acids, such as, for example, oleic acid.
- compositions for nasal aerosol or inhalation administrationinclude solutions which may contain, for example, benzyl alcohol or other suitable preservatives, absorption promoters to enhance absorption and/or bioavailability, and/or other solubilizing or dispersing agents such as those known in the art.
- Dispersible powders and granulescan, for example, be prepared by admixing at least one compound described herein or a pharmaceutically acceptable salt thereof, with at least one dispersing and/or wetting agent; at least one suspending agent; and/or at least one preservative.
- Exemplary preservativesinclude, but are not limited to, for example, anti-oxidants, e.g., ascorbic acid.
- dispersible powders and granulescan also contain at least one excipient, including, but not limited to, for example, sweetening agents; flavoring agents; and coloring agents.
- excipientsincluding, but not limited to, for example, sweetening agents; flavoring agents; and coloring agents.
- exemplary compositions for rectal administrationinclude suppositories which may contain, for example, suitable non-irritating excipients, such as cocoa butter, synthetic glyceride esters or polyethylene glycols, which are solid at ordinary temperatures but liquefy and/or dissolve in the rectal cavity to release the drug.
- the therapeutically-effective amount of a compound of the present inventionmay be determined by one of ordinary skill in the art, and includes exemplary dosage amounts for a mammal of from about 0.05 to 1000 mg/kg; 1-1000 mg/kg; 1-50 mg/kg; 5-250 mg/kg; 250-1000 mg/kg of body weight of active compound per day, which may be administered in a single dose or in the form of individual divided doses, such as from 1 to 4 times per day.
- the specific dose level and frequency of dosage for any particular subjectmay be varied and will depend upon a variety of factors, including the activity of the specific compound employed, the metabolic stability and length of action of that compound, the species, age, body weight, general health, sex and diet of the subject, the mode and time of administration, rate of excretion, drug combination, and severity of the particular condition.
- Preferred subjects for treatmentinclude animals, most preferably mammalian species such as humans, and domestic animals such as dogs, cats, horses, and the like.
- this termis intended to include all subjects, most preferably mammalian species that are affected by modulation of USP1-mediated functions.
- the compounds described hereinare useful for the treatment of cancer.
- the present applicationprovides a combined preparation of a compound described herein and/or a pharmaceutically acceptable salt thereof, a stereoisomer thereof or a tautomer thereof, and additional therapeutic agent(s) for simultaneous, separate or sequential use in the treatment and/or prophylaxis of multiple diseases or disorders associated with USP1.
- the applicationprovides a method of treating a patient suffering from or susceptible to a medical condition that is associated with USP1. A number of medical conditions can be treated. The method comprises administering to the patient a therapeutically effective amount of a composition comprising a compound described herien and/or a pharmaceutically acceptable salt thereof, a stereoisomer thereof or a tautomer thereof.
- the compounds described hereinmay be used to treat or proliferative diseases such as cancer, immunological disorders or inflammatory disorders.
- the compounds described hereinmay be used to treat cancers that are mediated by, dependent on or associated with USP1 activity.
- the diseaseis a solid tumor.
- the solid tumoris selected from prostate cancer, pancreatic cancer, bladder cancer, colorectal cancer, breast cancer, renal cancer, hepatocellular cancer, lung cancer, ovarian cancer, cervical cancer, gastric cancer, esophageal cancer, head and neck cancer, melanoma, neuroendocrine cancers, CNS cancers, brain tumors (e.g., glioma, anaplastic oligodendroglioma, adult glioblastoma multiforme, and adult anaplastic astrocytoma), bone cancer, or soft tissue sarcoma.
- the solid tumoris from non-small cell lung cancer or small-cell lung cancer.
- the compounds hereincan be useful in the treatment of haematological malignancies.
- hematological malignancyis selected from multiple myeloma, non-Hodgkin's lymphoma, Hodgkin lymphoma, T-cell leukaemia, mucosa-associated lymphoid tissue lymphoma, diffuse large B-cell lymphoma and mantle cell lymphoma.
- solid tumoris selected from pancreatic cancer, breast cancer, melanoma and non-small cell lung cancer.
- canceris selected from a carcinoma, preferably a carcinoma of the bladder, breast, colon (including colorectal carcinomas, such as colon adenocarcinoma and colon adenoma), kidney, urothelial, uterus, epidermis, liver, lung (including adenocarcinoma, small cell lung cancer, non-small cell lung carcinomas and squamous lung cancer), oesophagus, head and neck, gall bladder, ovary, pancreas (including exocrine pancreatic carcinoma), stomach, gastrointestinal cancer (including gastrointestinal stromal tumors), cervix, endometrium, thyroid, prostate and skin.
- a carcinomapreferably a carcinoma of the bladder, breast, colon (including colorectal carcinomas, such as colon adenocarcinoma and colon adenoma), kidney, urothelial, uterus, epidermis, liver, lung (including adenocarcinoma, small cell lung cancer, non-small cell lung carcinomas and
- the canceris selected from pituitary cancer, a hematopoietic tumor of lymphoid lineage, for example leukemia, acute lymphocytic leukemia, chronic lymphocytic leukemia, B-cell lymphoma (e.g.
- B-cell lymphomadiffuse large B-cell lymphoma, mantle cell lymphoma), T-cell leukaemia/lymphoma, Hodgkin's lymphoma, non-Hodgkin's lymphoma, hairy cell lymphoma, or Burkett's lymphoma; a hematopoietic tumor of myeloid lineage, for example leukemias, acute and chronic myelogenous leukemias, chronic myelomonocytic leukemia (CMML), myeloproliferative disorder, myeloproliferative syndrome, myelodysplastic syndrome, or promyelocytic leukemia; multiple myeloma; thyroid follicular cancer; hepatocellular cancer, a tumor of mesenchymal origin (e.g.
- Ewing's sarcomafor example fibrosarcoma or rhabdomyosarcoma; a tumor of the central or peripheral nervous system, for example astrocytoma, neuroblastoma, glioma (such as glioblastoma multiforme) or schwannoma; melanoma; seminoma; teratocarcinoma; osteosarcoma; xeroderma pigmentosum; keratoctanthoma; thyroid follicular cancer; or Kaposi's sarcoma.
- the compounds described hereinmay be used to treat cancers that are mediated by, dependent on or associated with USP1 activity.
- the diseaseis a solid tumor.
- the solid tumoris from prostate cancer, pancreatic cancer, bladder cancer, colorectal cancer, breast cancer, renal cancer, hepatocellular cancer, lung cancer, ovarian cancer, cervical cancer, gastric cancer, esophageal cancer, head and neck cancer, melanoma, neuroendocrine cancers, CNS cancers, brain tumors (e.g., glioma, anaplastic oligodendroglioma, adult glioblastoma multiforme, and adult anaplastic astrocytoma), bone cancer, or soft tissue sarcoma.
- the solid tumoris from non-small cell lung cancer or small-cell lung cancer.
- the diseaseis a hematologic malignancy.
- the diseaseis lymphoma, multiple myeloma, or leukemia.
- the hematologic malignancyis leukemia or lymphoma.
- the diseaseis acute lymphocytic leukemia (ALL), acute myeloid leukemia (AML), chronic lymphocytic leukemia (CLL), small lymphocytic lymphoma (SLL), myelodysplastic syndrome (MDS), myeloproliferative disease (MPD), chronic myeloid leukemia (CML), juvenile myelomonocytic leukemia (JMML), multiple myeloma (MM), Hodgkin lymphoma, indolent non-Hodgkin's lymphoma (iNHL), refractory iNHL, non- Hodgkin's lymphoma (NHL), mantle cell lymphoma (MCL), follicular lymphoma, Waldenström’s macroglobulinemia
- the diseaseis T-cell acute lymphoblastic leukemia (T-ALL), or B-cell acute lymphoblastic leukemia (B-ALL).
- non-Hodgkin lymphomacan be indolent B-cell diseases including follicular lymphoma, lymphoplasmacytic lymphoma, Waldenström macroglobulinemia, and marginal zone lymphoma, as well as the aggressive lymphomas that include, for example, Burkitt lymphoma, diffuse large B- cell lymphoma (DLBCL) and mantle cell lymphoma (MCL).
- the canceris selected from hematological cancer, a lymphatic cancer.
- the cancercomprises cancer cells with DNA damage repair pathway deficiency. In some embodiments, the cancer is a homologous recombination deficient cancer. In some embodiments, the cancer comprises cancer cells with a mutation in a gene encoding p53. In some embodiments, the mutation in a gene encoding p53 is a germline or somatic mutation. In some embodiments, the cancer comprises with cancer cells with loss of function mutation in a gene encoding p53. In some embodiments, the cancer is a BRCA1 and/or BRCA2 deficient cancer. In some embodiments, the cancer is a somatic or germline BRCA1 and/or BRCA2 mutant cancer.
- the canceris a Poly (ADP-ribose) polymerase (“PARP”) inhibitor refractory or resistant cancer.
- PARPPoly (ADP-ribose) polymerase
- the canceris a PARP inhibitor resistant or refractory BRCA1 and/or BRCA2 deficient cancer.
- the cancer cellhas a germline or somatic mutation in a gene encoding ataxia telangiectasia mutated (ATM) protein kinase or ATM deficiency.
- ATMtelangiectasia mutated
- the cancerhas a mutation in the gene encoding more than two of p53, BRCA1, BRCA2, ATM.
- the diseaseis an autoimmune or inflammatory disease or disorder.
- autoimmune or inflammatory diseases or conditionsmay be chronic or acute and include, but are not limited to, inflammatory pelvic disease, urethritis, skin sunburn, sinusitis, pneumonitis, encephalitis, meningitis, myocarditis, pericarditis, nephritis including lupus nephritis, osteomyelitis, myositis, eczema, hepatitis, gastritis, enteritis, dermatitis, gingivitis, appendicitis, pancreatitis, primary biliary cirrhosis, cholecystitis, sclerosing cholangitis, agammaglobulinemia, psoriasis, allergy, Crohn's disease, irritable bowel syndrome, ulcerative colitis, Sjogren's disease, tissue graft rejection such as acute graft-versus-host disease, hyperacute rejection of transplanted organs, asthma, chronic obstruct
- the autoimmune and inflammatory diseases and conditionsmay also include systemic or tissue inflammation, inflammatory responses to hypoxia, cellular activation and proliferation, lipid metabolism, fibrosis, infections with bacteria, infections with viruses (e.g., herpes virus, human papilloma virus, adenovirus, poxvirus and other DNA viruses), fungi, parasites or their toxins, such as sepsis, sepsis syndrome, septic shock, endotoxaemia, systemic inflammatory response syndrome (SIRS), multi-organ dysfunction syndrome, toxic shock syndrome, acute lung injury, ARDS (adult respiratory distress syndrome), acute renal failure, fulminant hepatitis, burns, acute pancreatitis, post-surgical syndromes, sarcoidosis, Herxheimer reactions, encephalitis, myelitis, meningitis, malaria and SIRS associated with viral infections such as influenza, herpes zoster, herpes simplex and coronavirus.
- virusese.g., herpes virus
- the compounds described hereinmay be administered in conjunction with standard of care, e.g., surgery, radiation, and/or chemotherapy.
- the compoundsmay be administered in conjunction with a chemotherapeutic agent.
- the compoundsmay be administered in conjunction with one or more of carboplatin, cisplatin, paclitaxel, nab-paclitaxel, gemcitabine or FOLFOX.
- the compoundsmay be administered in conjunction with carboplatin or nab-paclitaxel.
- the compoundsmay be administered in conjunction with carboplatin and paclitaxel.
- the compoundsmay be administered in conjunction with cisplatin and pemetrexed.
- the compoundsmay be administered in conjunction with cisplatin and gemcitabine. In some embodiments, the compounds may be administered in conjunction with FOLFOX. In some embodiments, the compounds may be administered in conjunction with FOLFIRI. In one embodiment, the compounds may be administered in combination with decarbazine for the treatment of melanoma. In some embodiments, cisplatin is intravenously administered as a 100 mg/ml dose once every four weeks. In some embodiments, the compounds may be administered in conjunction with doxorubicin (adriamycin), cisplatin bleomycin sulfate, carmustine, chlorambucil, dacarbazine and/or cyclophosphamide hydroxyurea.
- doxorubicinas adriamycin
- cisplatin bleomycin sulfatecarmustine
- adriamycinis intravenously administered as a 60 mg/ml to 75 mg/ml dose once every 21 days.
- the compounds of the present applicatione.g., a compound described herein or a pharmaceutically acceptable salt, prodrug, or solvate thereof
- the one or more additional therapeutic agentmay be an inhibitor to Janus kinase (JAK) such as JAK1, JAK2 and/or JAK3, Tyroansine kinase (TYK), K-Ras, Mitogen activated protein kinases (MAPK), Bruton's tyrosine kinase (BTK), bromodomain containing protein inhibitor (BRD) such as BRD4, a lysyl oxidase protein (LOX), lysyl oxidase-like protein (LOXL) such as LOXL1-5, matrix metalloprotease (MMP) such as MMP 1-10, adenosine A2B receptor (A2B), isocitrate dehydrogenase (IDH) such as IDH1, apoptosis signal-regulating kinase (ASK) such as ASK1, serine/threonine kinase TPL2, discoidin domain receptor (DDR) such as DDR1
- the compounds of the present applicationmay be used in combination with additional chemotherapeutic agent, an immunotherapeutic agent, a radiotherapeutic agent, an anti-neoplastic agent, an anti-cancer agent, an anti- fibrotic agent, an anti-angiogenic agent, a therapeutic antibody, or any combination thereof.
- Chemotherapeutic agentsmay be categorized by their mechanism of action into, for example, the following groups: anti-metabolites/anti-cancer agents, such as pyrimidine analogs (floxuridine, capecitabine, and cytarabine); purine analogs, folate antagonists and related inhibitors antiproliferative/antimitotic agents including natural products such as vinca alkaloid (vinblastine, vincristine) and microtubule such as taxane (paclitaxel, docetaxel), vinblastin, nocodazole, epothilones and navelbine, epidipodophyllotoxins (etoposide, teniposide); DNA damaging agents (actinomycin, amsacrine, busulfan, carboplatin, chlorambucil, cisplatin, cyclophosphamide, Cytoxan, dactinomycin, daunorubicin, doxorubicin, epirubicin, iphosphamide,
- chemotherapeutic agentsinclude alkylating agents such as thiotepa and cyclophosphamide; alkyl sulfonates such as busulfan, improsulfan and piposulfan; aziridines such as benzodopa, carboquone, meturedopa, and uredopa; emylerumines and memylamelamines including alfretamine, triemylenemelamine, triethylenephosphoramide, triethylenethiophosphoramide and trimemylolomelamine; acetogenins (especially bullatacin and bullatacinone); a camptothecin (including synthetic analogue topotecan); bryostatin; callystatin; CC-1065 (including its adozelesin, carzelesin and bizelesin synthetic analogues); cryptophycins (articularly cryptophycin 1 and cryptophycin 8); dolastatin; duocarmycin (including the synthetic al
- Chemotherapeutic agentsmay also include, for example, anti-hormonal agents that act to regulate or inhibit hormone action on tumors such as anti-estrogens and selective estrogen receptor modulators (SERMs), including, for example, tamoxifen, raloxifene, droloxifene, 4-hydroxytamoxifen, trioxifene, keoxifene, onapristone, and toremifene; inhibitors of the enzyme aromatase, which regulates estrogen production in the adrenal glands, such as, for example, 4(5)-imidazoles, aminoglutethimide, megestrol acetate, exemestane, formestane, fadrozole, vorozole letrozole and anastrozole; and anti- androgens such as flutamide, nilutamide, bicalutamide, leuprohde, and goserelin; and pharmaceutically acceptable salts
- SERMsselective estrogen receptor modulators
- the anti-angiogenic agentsinclude, but are not limited to, retinoid acid and derivatives thereof, 2-methoxyestradiol, suramin, squalamine, tissue inhibitor of metalloproteinase-1, tissue inhibitor of metalloproternase-2, plasminogen activator inhibitor-1, plasminogen activator inbibitor-2, cartilage-derived inhibitor, paclitaxel (nab- paclitaxel), platelet factor 4, protamine sulphate (clupeine), sulphated chitin derivatives (prepared from queen crab shells), sulphated polysaccharide peptidoglycan complex (sp- pg), staurosporine, modulators of matrix metabolism, including for example, proline analogs ((1-azetidine-2-carboxylic acid (LACA), cishydroxyproline, d,I-3,4- dehydroproline, thiaproline, .alpha.-dipyridyl, beta-aminopropion
- anti-angiogenesis agentsinclude antibodies, preferably monoclonal antibodies against these angiogenic growth factors: beta-FGF, alpha-FGF, FGF-5, VEGF isoforms, VEGF- C, HGF/SF and Ang-1/Ang-2.
- the applicationalso provides a method for treating a subject who is undergoing one or more standard therapies, such as chemotherapy, radiotherapy, immunotherapy, surgery, or combination thereof. Accordingly, one or more therapeutic agent or inhibitors may be administered before, during, or after administration of chemotherapy, radiotherapy, immunotherapy, surgery or combination thereof.
- the subjectmay be a human who is (i) substantially refractory to at least one chemotherapy treatment, or (ii) in relapse after treatment with chemotherapy, or both (i) and (ii). In some of embodiments, the subject is refractory to at least two, at least three, or at least four chemotherapy treatments (including standard or experimental chemotherapies).
- the subjectis refractory to at least one, at least two, at least three, or at least four chemotherapy treatment (including standard or experimental chemotherapy) selected from fludarabine, rituximab, obinutuzumab, alkylating agents, alemtuzumab and other chemotherapy treatments such as CHOP (cyclophosphamide, doxorubicin, vincristine, prednisone); R-CHOP (rituximab-CHOP); hyperCVAD (hyperfractionated cyclophosphamide, vincristine, doxorubicin, dexamethasone, methotrexate, cytarabine); R-hyperCVAD (rituximab-hyperCVAD); FCM (fludarabine, cyclophosphamide, mitoxantrone); R-FCM (rituximab, fludarabine, cyclophosphamide, mitoxantrone); bortezom
- chemotherapy treatment
- immunotherapeutic agents treating lymphoma or leukemiainclude, but are not limited to, rituximab (such as Rituxan), alemtuzumab (such as Campath, MabCampath), anti-CD19 antibodies, anti-CD20 antibodies, anti-MN-14 antibodies, anti- TRAIL, Anti-TRAIL DR4 and DR5 antibodies, anti-CD74 antibodies, apolizumab, bevacizumab, CHIR-12.12, epratuzumab (hLL2-anti-CD22 humanized antibody), galiximab, ha20, ibritumomab tiuxetan, lumiliximab, milatuzumab, ofatumumab, PRO131921, SGN-40, WT-1 analog peptide vaccine, WT1126-134
- Additional immunotherapy agentsincludes using cancer vaccines based upon the genetic makeup of an individual patient's tumor, such as lymphoma vaccine GTOP-99.
- the therapeutic treatmentscan be supplemented or combined with any of the abovementioned therapies with stem cell transplantation or treatment.
- One example of modified approachis radioimmunotherapy, wherein a monoclonal antibody is combined with a radioisotope particle, such as indium In-111, yttrium Y-90, iodine I-131.
- combination therapiesinclude, but are not limited to, Iodine-131 tositumomab, Yttrium-90 ibritumomab tiuxetan with CHOP.
- the compounds of the applicationcan be used in combination with additional therapeutic procedures.
- Other therapeutic proceduresinclude peripheral blood stem cell transplantation, autologous hematopoietic stem cell transplantation, autologous bone marrow transplantation, antibody therapy, biological therapy, enzyme inhibitor therapy, total body irradiation, infusion of stem cells, bone marrow ablation with stem cell support, in vitro-treated peripheral blood stem cell transplantation, umbilical cord blood transplantation, immunoenzyme technique, pharmacological study, low-LET cobalt-60 gamma ray therapy, bleomycin, conventional surgery, radiation therapy, and nonmyeloablative allogeneic hematopoietic stem cell transplantation.
- the compounds of the applicationcan be used in combination with anti-fibrotic agents.
- the anti-fibrotic agentsinclude, but are not limited to, emylenemamine, hydrazine, phenylhydrazine, and their derivatives, semicarbazide, and urea derivatives, aminonitriles, such as beta-aminopropionitrile (BAPN), or 2-nitroethylamine, unsaturated or saturated haloamines, such as 2-bromo-ethylamine, 2-chloroethylamine, 2- trifluoroethylamine, 3-bromopropylamine, p-halobenzylamines, selenohomocysteine lactone.
- the anti-fibrotic agentsare copper chelating agents, penetrating or not penetrating the cells.
- Exemplary compoundsinclude indirect inhibitors such compounds blocking the aldehyde derivatives originating from the oxidative deamination of the lysyl and hydroxylysyl residues by the lysyl oxidases, such as the thiolamines, in particular D- penicillamine, or its analogues such as 2-amino-5-mercapto-5-methylhexanoic acid, D-2- amino-3-methyl-3-((2-acetamidoethyl)dithio)butanoic acid, p-2-amino-3-methyl-3-((2- aminoethyl)dithio)butanoic acid, sodium-4-((p-1-dimethyl-2-amino-2- carboxyethyl)dithio)butane sulphurate, 2-acetamidoethyl-2-acetamidoethanethiol sulphanate, sodium-4-mercaptobutanesulphinate trihydrate.
- indirect inhibitorssuch
- the compounds of the applicationcan be used in combination with immunotherapeutic and anti-inflammatory treatments.
- the immunotherapeutic agentsinclude and are not limited to therapeutic antibodies suitable for treating patients; such as abagovomab, adecatumumab, afutuzumab, alemtuzumab, altumomab, amatuximab, anatumomab, arcitumomab, bavituximab, bectumomab, bevacizumab, bivatuzumab, blinatumomab, brentuximab, cantuzumab, catumaxomab, cetuximab, citatuzumab, cixutumumab, clivatuzumab, conatumumab, daratumumab, drozitumab, duligotumab, dusigitumab, detumomab, dacetuzumab, da
- the exemplified therapeutic antibodiesmay be further labeled or combined with a radioisotope particle, such as indium In-111, yttrium Y-90, iodine I-131.
- the immuno-oncology agentis (i) an agonist of a stimulatory (including a co-stimulatory) receptor or (ii) an antagonist of an inhibitory (including a co- inhibitory) signal on T cells, both of which result in amplifying antigen-specific T cell responses (often referred to as immune checkpoint regulators).
- Certain of the stimulatory and inhibitory moleculesare members of the immunoglobulin super family (IgSF).
- B7 familywhich includes B7- 1, B7-2, B7-H1 (PD-L1), B7-DC (PD-L2), B7-H2 (ICOS-L), B7-H3, B7-H4, B7-H5 (VISTA), and B7-H6.
- TNF family of molecules that bind to cognate TNF receptor family memberswhich includes CD40 and CD40L, OX-40, OX-40L, CD70, CD27L, CD30, CD30L, 4-1BBL, CD137 (4-1BB), TRAIL/Apo2-L, TRAILR1/DR4, TRAILR2/DR5, TRAILR3, TRAILR4, OPG, RANK, RANKL, TWEAKR/Fn14, TWEAK, BAFFR, EDAR, XEDAR, TACI, APRIL, BCMA, LT ⁇ R, LIGHT, DcR3, HVEM, VEGI/TL1A, TRAMP/DR3, EDAR, EDA1, XEDAR, EDA2, TNFR1, Lymphotoxin ⁇ /TNF ⁇ , TNFR2, TNF ⁇ , LT ⁇ R, Lymphotoxin ⁇ 1 ⁇ 2, FAS
- T cell responsescan be stimulated by a combination of a compound of Formula (I) and one or more of (i) an antagonist of a protein that inhibits T cell activation (e.g., immune checkpoint inhibitors) such as CTLA-4, PD-1, PD-L1, PD-L2, LAG-3, TIM-3, Galectin 9, CEACAM-1, BTLA, CD69, Galectin-1, TIGIT, CD113, GPR56, VISTA, 2B4, CD48, GARP, PD1H, LAIR1, TIM-1, and TIM-4, and (ii) an agonist of a protein that stimulates T cell activation such as B7-1, B7-2, CD28, 4-1BB (CD137), 4-1BBL, ICOS, ICOS-L, OX40, OX40L, GITR, GITRL, CD70, CD27, CD40, DR3 and CD28H.
- an antagonist of a protein that inhibits T cell activatione.g., immune check
- agents that can be combined with compounds described herein for the treatment of cancerinclude antagonists of inhibitory receptors on NK cells or agonists of activating receptors on NK cells.
- compounds described heriencan be combined with antagonists of KIR, such as lirilumab.
- agents for combination therapiesinclude agents that inhibit or deplete macrophages or monocytes, including but not limited to CSF-1R antagonists such as CSF-1R antagonist antibodies including RG7155 (WO11/70024, WO11/107553,
- compounds of the present applicationcan be used with one or more of agonistic agents that ligate positive costimulatory receptors, blocking agents that attenuate signaling through inhibitory receptors, antagonists, and one or more agents that increase systemically the frequency of anti-tumor T cells, agents that overcome distinct immune suppressive pathways within the tumor microenvironment (e.g., block inhibitory receptor engagement (e.g., PD-L1/PD-1 interactions), deplete or inhibit Tregs (e.g., using an anti-CD25 monoclonal antibody (e.g., daclizumab) or by ex vivo anti-CD25 bead depletion), inhibit metabolic enzymes such as IDO, or reverse/prevent T cell anergy or exhaustion) and agents that trigger innate
- the immuno-oncology agentis a CTLA-4 antagonist, such as an antagonistic CTLA-4 antibody.
- Suitable CTLA-4 antibodiesinclude, for example, YERVOY (ipilimumab) or tremelimumab.
- the immuno-oncology agentis a PD-1 antagonist, such as an antagonistic PD-1 antibody.
- Suitable PD-1 antibodiesinclude, for example, OPDIVO (nivolumab), KEYTRUDA (pembrolizumab), or MEDI-0680 (AMP-514; WO2012/145493).
- the immuno-oncology agentmay also include pidilizumab (CT-011), though its specificity for PD-1 binding has been questioned.
- the immuno-oncology agentis a PD-L1 antagonist, such as an antagonistic PD-L1 antibody.
- Suitable PD-L1 antibodiesinclude, for example, MPDL3280A (RG7446; WO2010/077634), durvalumab (MEDI4736), BMS-936559 (WO2007/005874), and MSB0010718C (WO2013/79174).
- the immuno-oncology agentis a LAG-3 antagonist, such as an antagonistic LAG-3 antibody.
- Suitable LAG3 antibodiesinclude, for example, BMS- 986016 (WO10/19570, WO14/08218), or IMP-731 or IMP-321 (WO08/132601, WO09/44273).
- the immuno-oncology agentis a CD137 (4-1BB) agonist, such as an agonistic CD137 antibody.
- Suitable CD137 antibodiesinclude, for example, urelumab and PF-05082566 (WO12/32433).
- the immuno-oncology agentis a GITR agonist, such as an agonistic GITR antibody.
- Suitable GITR antibodiesinclude, for example, BMS-986153, BMS-986156, TRX-518 (WO06/105021, WO09/009116) and MK-4166 (WO11/028683).
- the immuno-oncology agentis an IDO antagonist.
- IDO antagonistsinclude, for example, INCB-024360 (WO2006/122150, WO07/75598, WO08/36653, WO08/36642), indoximod, BMS-986205, or NLG-919 (WO09/73620, WO09/1156652, WO11/56652, WO12/142237).
- the immuno-oncology agentis an OX40 agonist, such as an agonistic OX40 antibody.
- Suitable OX40 antibodiesinclude, for example, MEDI-6383 or MEDI-6469.
- the immuno-oncology agentis an OX40L antagonist, such as an antagonistic OX40 antibody.
- Suitable OX40L antagonistsinclude, for example, RG-7888 (WO06/029879).
- the immuno-oncology agentis a CD40 agonist, such as an agonistic CD40 antibody.
- the immuno-oncology agentis a CD40 antagonist, such as an antagonistic CD40 antibody.
- Suitable CD40 antibodiesinclude, for example, lucatumumab or dacetuzumab.
- the immuno-oncology agentis a CD47 antagonist, such as a CD47 antagonist selected from the group MIAP301, MIAP410, TTI-621, CV1, Hu5F9- G4, CC-90002, B6H12 and 2D3.
- the immuno-oncology agentis a CD27 agonist, such as an agonistic CD27 antibody. Suitable CD27 antibodies include, for example, varlilumab.
- the immuno-oncology agentis MGA271 (to B7H3) (WO11/109400).
- the combination therapyis intended to embrace administration of these therapeutic agents in a sequential manner, that is, wherein each therapeutic agent is administered at a different time, as well as administration of these therapeutic agents, or at least two of the therapeutic agents, in a substantially simultaneous manner.
- Substantially simultaneous administrationcan be accomplished, for example, by administering to the subject a single dosage form having a fixed ratio of each therapeutic agent or in multiple, single dosage forms for each of the therapeutic agents.
- Sequential or substantially simultaneous administration of each therapeutic agentcan be effected by any appropriate route including, but not limited to, oral routes, intravenous routes, intramuscular routes, and direct absorption through mucous membrane tissues.
- the therapeutic agentscan be administered by the same route or by different routes.
- a first therapeutic agent of the combination selectedmay be administered by intravenous injection while the other therapeutic agents of the combination may be administered orally.
- all therapeutic agentsmay be administered orally or all therapeutic agents may be administered by intravenous injection.
- Combination therapyalso can embrace the administration of the therapeutic agents as described above in further combination with other biologically active ingredients and non-drug therapies (e.g., surgery or radiation treatment.) Where thwe combination therapy further comprises a non drug treatment, the non drug treatment may be conducted at any suitable time so long as a beneficial effect from the co-action of the combination of the therapeutic agents and non-drug treatment is achieved.
- the beneficial effectis still achieved when the non drug treatment is temporally removed from the administration of the therapeutic agents, perhaps by days or even weeks.
- the present inventionalso provides the compounds of the present invention for use in therapy.
- compounds of Formula Iare selected from exemplified compounds or combinations of exemplified compounds or other embodiments herein.
- the present inventionmay be embodied in other specific forms without departing from the spirit or essential attributes thereof. This invention encompasses all combinations of preferred aspects and/or embodiments of the invention noted herein.
- any and all embodiments of the present inventionmay be taken in conjunction with any other embodiment or embodiments to describe additional more preferred embodiments. It is also to be understood that each individual element of the preferred embodiments is its own independent preferred embodiment. Furthermore, any element of an embodiment is meant to be combined with any and all other elements from any embodiment to describe an additional embodiment.
- METHODS OF PREPARATIONThe compounds of the present invention can be prepared in a number of ways well known to one skilled in the art of organic synthesis.
- the compounds of the present inventioncan be synthesized using the methods described below, together with synthetic methods known in the art of synthetic organic chemistry, or variations thereon as appreciated by those skilled in the art. Preferred methods include, but are not limited to, those described below.
- the compounds of this inventionmay be prepared using the reactions and techniques described in this section. The reactions are performed in solvents appropriate to the reagents and materials employed and are suitable for the transformations being effected. Also, in the description of the synthetic methods described below, it is to be understood that all proposed reaction conditions, including choice of solvent, reaction atmosphere, reaction temperature, duration of the experiment and work up procedures, are chosen to be the conditions standard for that reaction, which should be readily recognized by one skilled in the art. It is understood by one skilled in the art of organic synthesis that the functionality present on various portions of the molecule must be compatible with the reagents and reactions proposed.
- Step-24-(1-Methyl-4-(trifluoromethyl)-1H-imidazol-2-yl)benzonitrile: To a stirred solution of 4-(4- - 2-yl)benzonitrile (4.0 g, 16.86 mmol) in DMF (40 mL), sodium hydride (60% dispersion in mineral oil, 0.809 g, 33.70 mmol) was added followed by addition of iodomethane (1.26 mL, 20.24 mmol) at 0 o C and the mixture was stirred at 25 °C for 2 h. Chilled water was added and the contents extracted with ethyl acetate.
- Step-34-(1-Methyl-4-(trifluoromethyl)-1H-imidazol-2-yl)phenyl)methanamine (I- 1): To a stirred solution of 4-(1-methyl-4-(trifluoromethyl)-1H-imidazol-2-yl)benzonitrile (2.9 g, 11.54 mmol) in THF (30 mL), lithium aluminium hydride (2 M in THF, 11.54 mL, 23.09 mmol) was added at 0 o C and the mixture was stirred at 25 °C for 2 h. The reaction was quenched with saturated aqueous NH4Cl and extracted with ethyl acetate.
- Step-24-chloro-1-isopropyl-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H- a g, in tetrahydrofuran (50 mL), n-butyllithium (1.6 M in hexanes, 28.5 mL, 45.6 mmol) was added at 0 o C and the mixture was stirred at ambient temperature for 1 h. The mixture was recooled to -78 o C, 2-isopropoxy-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (8.49 g, 45.6 mmol) was added and stirred for 2 h.
- Step 22-(4-cyclopropyl-6-methoxypyrimidin-5-yl)-N-(4-(1-methyl-4- (trifluoromethyl)-1H-imidazol-2-yl)benzyl)pyrido[3,4-d]pyrimidin-4-amine (example 1): imidazol-2- mg, , 4-cyclopropyl-6- methoxy-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyrimidine (23.74 mg, 0.086 mmol), potassium phosphate tribasic (30.4 mg, 0.143 mmol) and tetrakis(triphenylphosphine)palladium(0) (4.14 mg, 3.58 ⁇ mol) in dioxane (1.0 mL) and water (0.111 mL) was flushed with nitrogen for 5 min.
- Step-2Tert-butyl 2-chloro-4-((4-(1-methyl-4-(trifluoromethyl)-1H-imidazol-2- yl)benzyl)amino)-8-oxo-5,8-dihydropyrido[3,4-d]pyrimidine-7(6H)-carboxylate(4c)
- the reaction mixturewas cooled to ambient temperature and filtered through a celite bed.
- the celite bedwas further washed with ethyl acetate, the washings and the filtrate were mixed and concentrated under reduced pressure.
- the residue obtainedwas purified by flash chromatography using a Biotage chromatography instrument, on silica gel column (100-200 mesh), eluting with 0-80% gradient of ethyl acetate in petroleum ether to obtain tert-butyl 2-(4-cyclopropyl-6-methoxypyrimidin-5- yl)-4-((4-(1-methyl-4-(trifluoromethyl)-1H-imidazol-2-yl)benzyl)amino)-8-oxo-5,8- dihydropyrido[3,4-d]pyrimidine-7(6H)-carboxylate (0.040 g, 0.047 mmol, 34.7%) as pale yellow thick gum.
- Trifluoroacetic acid(10.80 ⁇ L, 0.140 mmol) was added and the mixture was stirred for 3 h, during which time the temperature of the reaction mixture was allowed to rise to ambient temperature. After completion of the reaction (monitored by TLC and UPLC-MS), solvents were evaporated from the reaction mixture and the crude obtained was purified by preparative HPLC [Method: Diluent: WATER: THF: MeCN (30:30:40); XBridge C18 (150 x19)mm, 5micron; Temperature: Ambient; Mobile methoxypyrimidin-5-yl)-4-((4-(1-methyl-4-(trifluoromethyl)-1H-imidazol-2- yl)benzyl)amino)-6,7-dihydropyrido[3,4-d]pyrimidin-8(5H)-one (0.010 g, 0.018 mmol, 38.4%) as off white solid.
- Example 5Synthesis of 2-(4-cyclopropyl-6-methoxypyrimidin-5-yl)-7-methyl-4-((4- (1-methyl-4-(trifluoromethyl)-1H-imidazol-2-yl)benzyl)amino)-6,7- dihydropyrido[3,4-d]pyrimidin-8(5H)-one:
- Step-1Tert-butyl (2-(4-cyclopropyl-6-methoxypyrimidin-5-yl)-8-oxo-5,6,7,8- tetrahydropyrido[3,4-d]pyrimidin-4-yl)(4-(1-methyl-4-(trifluoromethyl)-1H- imidazol-2-yl)benzyl)carbamate (5a):
- Triethylamine(0.197 mL, 1.417 mmol) and di-tert-butyl carbonate (0.132 mL, 0.567 mmol) were added followed by DMAP (0.003 g, 0.028 mmol) and the mixture was stirred at ambient temperature for 16 h.
- the reaction mixturewas diluted with chilled water and extracted with dichloromethane. The combined organic extract was washed with brine, dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure.
- the crude obtainedwas purified by flash chromatography using a Biotage chromatography instrument, on silica gel column (100- 200 mesh), eluting with 0-2% gradient of methanol in dichloromethane to obtain tert- butyl (2-(4-cyclopropyl-6-methoxypyrimidin-5-yl)-8-oxo-5,6,7,8-tetrahydropyrido[3,4- d]pyrimidin-4-yl)(4-(1-methyl-4-(trifluoromethyl)-1H-imidazol-2-yl)benzyl)carbamate (0.170 g, 0.253 mmol, 89%) as pale yellow thick gum.
- Step-72-(4-cyclopropyl-6-methoxypyrimidin-5-yl)-7-methyl-4-((4-(1-methyl-4- (trifluoromethyl)-1H-imidazol-2-yl)benzyl)amino)-6,7-dihydropyrido[3,4- d]pyrimidin-8(5H)-one (example 5)
- the celite bedwas further washed with ethyl acetate, the washings and the filtrate were combined, washed with brine, dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure.
- the crude obtainedwas purified by flash chromatography using a Biotage chromatography instrument, on silica gel column (100-200 mesh), eluting with 0-30% gradient of ethyl acetate in petroleum ether to obtain tert-butyl 2,4-dichloro-5-oxo-7,8-dihydropyrido[4,3-d]pyrimidine-6(5H)- carboxylate (0.400 g, 1.157 mmol, 70.4%) as off white solid.
- 100X solutions of compounds in DMSOwere prepared by three-fold serial dilutions starting from a 10 mM stock.
- 2X solutions of His6-USP1/His6-UAF1 (200 pM, Internally Produced) and ubiquitin-rhodamine 110 (10 ⁇ M, South Bay Bio SBB-PS0001)were prepared in assay buffer (50 mM Tris pH 7.5, 100 mM NaCl, 1 mM EDTA, 1 mM TCEP, 100 ng/ ⁇ L BSA).
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Oncology (AREA)
- Hematology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
Description
UBIQUITIN SPECIFIC PROCESSING PROTEASE 1 (USP1) COMPOUNDS CROSS REFERENCE TO RELATED APPLICATIONS This application claims the benefit of U.S. Provisional Application No. 63/594,480, filed, October 31, 2023, the entire content of which is hereby incorporated herein by reference. FIELD This invention relates to compounds which are inhibitors of ubiquitin-specific- processing protease 1 (USP1) useful for treating diseases including, among others, cancer, autoimmune and inflammatory disorders. The invention further pertains to pharmaceutical compositions containing at least one compound according to the invention that are useful for the treatment of conditions related to the inhibition of USP1 in a mammal. BACKGROUND OF THE INVENTION Ubiquitination is important in the regulation of many cellular functions and cellular homeostasis. The conjugation of ubiquitin to a target protein is a multistep process involving the sequential action of a ubiquitin activating enzyme (E1), a ubiquitin- conjugating enzyme (E2), and a ubiquitin protein-ligase (E3). The ubiquitin tags can mediate non-covalent interactions of the ubiquitinated substrate with other proteins bearing different types of ubiquitin-binding motifs. A family of enzymes, termed deubiquitinases act on ubiquitinated substrates to catalyze the removal of ubiquitin moieties. One such enzyme is ubiquitin-specific protease 1 (USP1) which plays an important role in the regulation of DNA repair processes. USP1 is a regulator of several important steps in the DNA damage response, particularly in the Fanconi anemia pathway, and in the process of translesion synthesis. USP1 has also been reported to contribute to the repair of double-strand DNA breaks through homologous recombination. In addition, USP1 has been reported to deubiquitinate and stabilize members of the family of inhibitors of DNA binding (ID) proteins, ID1, ID2 and ID3. García-Santisteban, I., Peters, G.J., Giovannetti, E. et al. Mol Cancer 12, 91 (2013); US Patent Nos.7,754,463, 10,653,676, 9,518,032. SUMMARY The present disclosure provides compounds that modulate the expression or activity of USP1. The disclosure also provides compositions, including pharmaceutical compositions, kits that include the compounds, and methods of using (or administering) and making the compounds. The compounds provided herein are useful in treating diseases, disorders, or conditions that are mediated by USP1. The disclosure also provides compounds for use in therapy. The disclosure further provides compounds for use in a method of treating a disease, disorder, or condition that is mediated by USP1. Moreover, the disclosure provides uses of the compounds in the manufacture of a medicament for the treatment of a disease, disorder or condition that is mediated by (or mediated, at least in part, by) USP1. In one aspect, provided are compounds of Formula (I):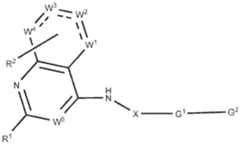 or a pharmaceutically acceptable salt, stereoisomer, mixture of stereoisomers thereof; R1 is selected from 5-6 membered heteroaryl, optionally substituted with one to four halo, hydroxy, amino, -C(O)Ra, -C(O)ORb, -C(O)NRaRb, -N(Ra)C(O)Rb, -S(O)NRaRb, - S(O)2NRaRb, -S(O)Rg, -S(O)2Rg, -NRaRb, -ORa, -SRb, C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl and C3-8 cycloalkyl; wherein each C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl and C3-8 cycloalkyl is optionally substituted with one to four R100; R2 is selected from absent, hydrogen, halo, hydroxy, amino, -CN, -C(O)Ra, -C(O)ORb, - C(O)NRaRb, -N(Ra)C(O)Rb, -N(Ra)C(O)NRaRb, -N(Ra)SO2NRaRb, -S(O)NRaRb, - S(O)2NRaRb, -N(Ra)S(O)2Rb, -S(O)Rg, -S(O)2Rg, -NRaRb, -ORa, -SRb, -OC(O)Ra, - OC(O)NRaRb, C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, C3-8 cycloalkyl and 4-10 membered heterocyclyl containing 1 to 4 heteroatoms selected from N, O, and S; wherein each C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, 4-10 membered heterocyclyl containing 1 to 4 heteroatoms selected from N, O, and S, and C3-8 cycloalkyl is optionally substituted with one to four R100; X is selected from absent and C1-6 alkyl, wherein C1-6 alkyl is optionally substituted with one to four R100; Each W1, W2, W3 and W4 is independently selected from -N(Ra)-, -C(O)- and -C(Ra)-; W5 is -N- or -C(Ra)-; wherein at least one of W1, W2, W3 and W4 is -N(Ra)-; wherein at least one of W1, W2, W3 and W4 is -C(Ra)-; G1 is selected from -C6 aryl-, 5-6 membered heteroaryl, C3-8 cycloalkyl and 5-6 membered heterocyclyl; wherein each C6 aryl, 5-6 membered heteroaryl, C3-8 cycloalkyl and 5-6 membered heterocyclyl is optionally substituted with one to four R100; G2 is a 5 or 6 membered heteroaryl or 5-6 membered heterocyclyl optionally substituted with one to four R100; each Ra and Rb is independently selected from absent, hydrogen, C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, C3-6 cycloalkyl and 4-6 membered heterocyclyl; wherein each C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl and C3-6 cycloalkyl and 4-6 membered heterocyclyl is optionally substituted with one to four R200; each R100 is independently selected from hydrogen, halo, cyano, hydroxy, amino, oxo, thioxo, vinyl, -C(O)Rc, -C(O)ORc, -C(O)NRcRd, -N(Rc)C(O)Rd, -S(O)NRcRd, - S(O)2NRcRd, -S(O)Rh, -S(O)2Rh, -NRcRd, -ORc, -SRc, C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, C3-8 cycloalkyl, C6-10 aryl, 5-10 membered heteroaryl containing 1 to 4 heteroatoms selected from N, O, and S, and 4-10 membered heterocyclyl containing 1 to 4 heteroatoms selected from N, O, and S; wherein each C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, C3-8 cycloalkyl, C6-10 aryl, 5-10 membered heteroaryl and 4-10 membered heterocyclyl is optionally substituted with one to four R201; each Rc and Rd is independently selected from hydrogen, C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, C3-8 cycloalkyl, C6-10 aryl, 5-10 membered heteroaryl containing 1 to 4 heteroatoms selected from N, O, and S, and 4-10 membered heterocyclyl containing 1 to 4 heteroatoms selected from N, O, and S; each R200 and R201 is independently selected from hydrogen, halo, cyano, hydroxy, amino, oxo, thioxo, vinyl, -C(O)Re, -C(O)ORe, -C(O)NReRf, -N(Re)C(O)Rf, -S(O)NReRf, - S(O)2NReRf, -S(O)Ri, -S(O)2Ri, -NReRf, -ORe, -SRe, C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, C3-8 cycloalkyl, C6-10 aryl, 5-10 membered heteroaryl containing 1 to 4 heteroatoms selected from N, O, and S, and 4-10 membered heterocyclyl containing 1 to 4 heteroatoms selected from N, O, and S; wherein each C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, C3-8 cycloalkyl, C6-10 aryl, 5-10 membered heteroaryl and 4-10 membered heterocyclyl is optionally substituted with one to four R300; each Rg, Rh and Ri is independently selected from C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl; wherein each C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, is optionally substituted with one to four R300; wherein each R300 is independently selected from hydrogen, halo, cyano, hydroxy, amino, oxo, thioxo, vinyl, -C(O)Re, -C(O)ORe, -C(O)NReRf, -N(Re)C(O)Rf, -S(O)NReRf, - S(O)2NReRf, -NReRf, S(O)Re, -S(O)2Re, -NReRf, -ORe, -SRe, C1-6 alkyl, C2-6 alkenyl and C2-6 alkynyl; each Re and Rf is independently selected from hydrogen, C1-6 alkyl, C2-6 alkenyl and C2-6 alkynyl; C3-8 cycloalkyl, C6-10 aryl, 5-10 membered heteroaryl containing 1 to 4 heteroatoms selected from N, O, and S, and 4-10 membered heterocyclyl containing 1 to 4 heteroatoms selected from N, O, and S; wherein each C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, C3-8 cycloalkyl, C6-10 aryl, 5-10 membered heteroaryl and 4-10 membered heterocyclyl is optionally substituted with one to four R400; each R400 is independently selected from hydrogen,
or a pharmaceutically acceptable salt, stereoisomer, mixture of stereoisomers thereof; R1 is selected from 5-6 membered heteroaryl, optionally substituted with one to four halo, hydroxy, amino, -C(O)Ra, -C(O)ORb, -C(O)NRaRb, -N(Ra)C(O)Rb, -S(O)NRaRb, - S(O)2NRaRb, -S(O)Rg, -S(O)2Rg, -NRaRb, -ORa, -SRb, C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl and C3-8 cycloalkyl; wherein each C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl and C3-8 cycloalkyl is optionally substituted with one to four R100; R2 is selected from absent, hydrogen, halo, hydroxy, amino, -CN, -C(O)Ra, -C(O)ORb, - C(O)NRaRb, -N(Ra)C(O)Rb, -N(Ra)C(O)NRaRb, -N(Ra)SO2NRaRb, -S(O)NRaRb, - S(O)2NRaRb, -N(Ra)S(O)2Rb, -S(O)Rg, -S(O)2Rg, -NRaRb, -ORa, -SRb, -OC(O)Ra, - OC(O)NRaRb, C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, C3-8 cycloalkyl and 4-10 membered heterocyclyl containing 1 to 4 heteroatoms selected from N, O, and S; wherein each C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, 4-10 membered heterocyclyl containing 1 to 4 heteroatoms selected from N, O, and S, and C3-8 cycloalkyl is optionally substituted with one to four R100; X is selected from absent and C1-6 alkyl, wherein C1-6 alkyl is optionally substituted with one to four R100; Each W1, W2, W3 and W4 is independently selected from -N(Ra)-, -C(O)- and -C(Ra)-; W5 is -N- or -C(Ra)-; wherein at least one of W1, W2, W3 and W4 is -N(Ra)-; wherein at least one of W1, W2, W3 and W4 is -C(Ra)-; G1 is selected from -C6 aryl-, 5-6 membered heteroaryl, C3-8 cycloalkyl and 5-6 membered heterocyclyl; wherein each C6 aryl, 5-6 membered heteroaryl, C3-8 cycloalkyl and 5-6 membered heterocyclyl is optionally substituted with one to four R100; G2 is a 5 or 6 membered heteroaryl or 5-6 membered heterocyclyl optionally substituted with one to four R100; each Ra and Rb is independently selected from absent, hydrogen, C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, C3-6 cycloalkyl and 4-6 membered heterocyclyl; wherein each C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl and C3-6 cycloalkyl and 4-6 membered heterocyclyl is optionally substituted with one to four R200; each R100 is independently selected from hydrogen, halo, cyano, hydroxy, amino, oxo, thioxo, vinyl, -C(O)Rc, -C(O)ORc, -C(O)NRcRd, -N(Rc)C(O)Rd, -S(O)NRcRd, - S(O)2NRcRd, -S(O)Rh, -S(O)2Rh, -NRcRd, -ORc, -SRc, C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, C3-8 cycloalkyl, C6-10 aryl, 5-10 membered heteroaryl containing 1 to 4 heteroatoms selected from N, O, and S, and 4-10 membered heterocyclyl containing 1 to 4 heteroatoms selected from N, O, and S; wherein each C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, C3-8 cycloalkyl, C6-10 aryl, 5-10 membered heteroaryl and 4-10 membered heterocyclyl is optionally substituted with one to four R201; each Rc and Rd is independently selected from hydrogen, C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, C3-8 cycloalkyl, C6-10 aryl, 5-10 membered heteroaryl containing 1 to 4 heteroatoms selected from N, O, and S, and 4-10 membered heterocyclyl containing 1 to 4 heteroatoms selected from N, O, and S; each R200 and R201 is independently selected from hydrogen, halo, cyano, hydroxy, amino, oxo, thioxo, vinyl, -C(O)Re, -C(O)ORe, -C(O)NReRf, -N(Re)C(O)Rf, -S(O)NReRf, - S(O)2NReRf, -S(O)Ri, -S(O)2Ri, -NReRf, -ORe, -SRe, C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, C3-8 cycloalkyl, C6-10 aryl, 5-10 membered heteroaryl containing 1 to 4 heteroatoms selected from N, O, and S, and 4-10 membered heterocyclyl containing 1 to 4 heteroatoms selected from N, O, and S; wherein each C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, C3-8 cycloalkyl, C6-10 aryl, 5-10 membered heteroaryl and 4-10 membered heterocyclyl is optionally substituted with one to four R300; each Rg, Rh and Ri is independently selected from C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl; wherein each C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, is optionally substituted with one to four R300; wherein each R300 is independently selected from hydrogen, halo, cyano, hydroxy, amino, oxo, thioxo, vinyl, -C(O)Re, -C(O)ORe, -C(O)NReRf, -N(Re)C(O)Rf, -S(O)NReRf, - S(O)2NReRf, -NReRf, S(O)Re, -S(O)2Re, -NReRf, -ORe, -SRe, C1-6 alkyl, C2-6 alkenyl and C2-6 alkynyl; each Re and Rf is independently selected from hydrogen, C1-6 alkyl, C2-6 alkenyl and C2-6 alkynyl; C3-8 cycloalkyl, C6-10 aryl, 5-10 membered heteroaryl containing 1 to 4 heteroatoms selected from N, O, and S, and 4-10 membered heterocyclyl containing 1 to 4 heteroatoms selected from N, O, and S; wherein each C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, C3-8 cycloalkyl, C6-10 aryl, 5-10 membered heteroaryl and 4-10 membered heterocyclyl is optionally substituted with one to four R400; each R400 is independently selected from hydrogen, cyano, hydroxy, amino, oxo, thioxo, vinyl, -C(O)Rk, -C(O)ORk, -C(O)NRkRl, -N(Rk)C(O)Rl, -S(O)NRkRl, - S(O)2NRkRl, -NRkRl, S(O)Rk, -S(O)2Rk, -NRkRl, -ORk, -SRk, C1-6 alkyl, C2-6 alkenyl and C2-6 alkynyl; each Rk and Rl is independently selected from hydrogen, C1-6 alkyl, C2-6 alkenyl and C2-6 alkynyl; C3-8 cycloalkyl, C6-10 aryl, 5-10 membered heteroaryl containing 1 to 4 heteroatoms selected from N, O, and S, and 4-10 membered heterocyclyl containing 1 to 4 heteroatoms selected from N, O, and S. In one aspect, provided are processes and intermediates for making the compounds of Formula I. In one aspect, provided are pharmaceutical compositions comprising a pharmaceutically acceptable carrier and at least one of the compounds disclosed herein. The present application also provides methods for the inhibition of USP1 comprising administering a therapeutically effective amount of at least one of Formula I. The present application also provides a method for treating proliferative, metabolic, allergic, autoimmune and inflammatory diseases, comprising administering to a host in need of such treatment a therapeutically effective amount of at least one of the compounds disclosed herein. The compounds of Formula I or a pharmaceutically acceptable salt thereof, may be used to treat cancers that are mediated by, dependent on or associated with USP1 activity. In certain embodiments, the disease is a solid tumor. DETAILED DESCRIPTION In a first aspect, provided are compounds of formula (I) that function as inhibitors of USP1:
cyano, hydroxy, amino, oxo, thioxo, vinyl, -C(O)Rk, -C(O)ORk, -C(O)NRkRl, -N(Rk)C(O)Rl, -S(O)NRkRl, - S(O)2NRkRl, -NRkRl, S(O)Rk, -S(O)2Rk, -NRkRl, -ORk, -SRk, C1-6 alkyl, C2-6 alkenyl and C2-6 alkynyl; each Rk and Rl is independently selected from hydrogen, C1-6 alkyl, C2-6 alkenyl and C2-6 alkynyl; C3-8 cycloalkyl, C6-10 aryl, 5-10 membered heteroaryl containing 1 to 4 heteroatoms selected from N, O, and S, and 4-10 membered heterocyclyl containing 1 to 4 heteroatoms selected from N, O, and S. In one aspect, provided are processes and intermediates for making the compounds of Formula I. In one aspect, provided are pharmaceutical compositions comprising a pharmaceutically acceptable carrier and at least one of the compounds disclosed herein. The present application also provides methods for the inhibition of USP1 comprising administering a therapeutically effective amount of at least one of Formula I. The present application also provides a method for treating proliferative, metabolic, allergic, autoimmune and inflammatory diseases, comprising administering to a host in need of such treatment a therapeutically effective amount of at least one of the compounds disclosed herein. The compounds of Formula I or a pharmaceutically acceptable salt thereof, may be used to treat cancers that are mediated by, dependent on or associated with USP1 activity. In certain embodiments, the disease is a solid tumor. DETAILED DESCRIPTION In a first aspect, provided are compounds of formula (I) that function as inhibitors of USP1: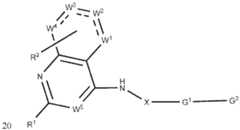 or a pharmaceutically acceptable salt, stereoisomer, mixture of stereoisomers thereof; R1 is selected from 5-6 membered heteroaryl, optionally substituted with one to four halo, hydroxy, amino, -C(O)Ra, -C(O)ORb, -C(O)NRaRb, -N(Ra)C(O)Rb, -S(O)NRaRb, - S(O)2NRaRb, -S(O)Rg, -S(O)2Rg, -NRaRb, -ORa, -SRb, C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl and C3-8 cycloalkyl; wherein each C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl and C3-8 cycloalkyl is optionally substituted with one to four R100; R2 is selected from absent, hydrogen, halo, hydroxy, amino, -CN, -C(O)Ra, -C(O)ORb, - C(O)NRaRb, -N(Ra)C(O)Rb, -N(Ra)C(O)NRaRb, -N(Ra)SO2NRaRb, -S(O)NRaRb, - S(O)2NRaRb, -N(Ra)S(O)2Rb, -S(O)Rg, -S(O)2Rg, -NRaRb, -ORa, -SRb, -OC(O)Ra, - OC(O)NRaRb, C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, C3-8 cycloalkyl and 4-10 membered heterocyclyl containing 1 to 4 heteroatoms selected from N, O, and S; wherein each C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, 4-10 membered heterocyclyl containing 1 to 4 heteroatoms selected from N, O, and S, and C3-8 cycloalkyl is optionally substituted with one to four R100; X is selected from absent and C1-6 alkyl, wherein C1-6 alkyl is optionally substituted with one to four R100; Each W1, W2, W3 and W4 is independently selected from -N(Ra)-, -C(O)- and -C(Ra)-; W5 is -N- or -C(Ra)-; wherein at least one of W1, W2, W3 and W4 is -N(Ra)-; wherein at least one of W1, W2, W3 and W4 is -C(Ra)-; G1 is selected from -C6 aryl-, 5-6 membered heteroaryl, C3-8 cycloalkyl and 5-6 membered heterocyclyl; wherein each C6 aryl, 5-6 membered heteroaryl, C3-8 cycloalkyl and 5-6 membered heterocyclyl is optionally substituted with one to four R100; G2 is a 5 or 6 membered heteroaryl or 5-6 membered heterocyclyl optionally substituted with one to four R100; each Ra and Rb is independently selected from absent, hydrogen, C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, C3-6 cycloalkyl and 4-6 membered heterocyclyl; wherein each C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl and C3-6 cycloalkyl and 4-6 membered heterocyclyl is optionally substituted with one to four R200; each R100 is independently selected from hydrogen, halo, cyano, hydroxy, amino, oxo, thioxo, vinyl, -C(O)Rc, -C(O)ORc, -C(O)NRcRd, -N(Rc)C(O)Rd, -S(O)NRcRd, - S(O)2NRcRd, -S(O)Rh, -S(O)2Rh, -NRcRd, -ORc, -SRc, C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, C3-8 cycloalkyl, C6-10 aryl, 5-10 membered heteroaryl containing 1 to 4 heteroatoms selected from N, O, and S, and 4-10 membered heterocyclyl containing 1 to 4 heteroatoms selected from N, O, and S; wherein each C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, C3-8 cycloalkyl, C6-10 aryl, 5-10 membered heteroaryl and 4-10 membered heterocyclyl is optionally substituted with one to four R201; each Rc and Rd is independently selected from hydrogen, C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, C3-8 cycloalkyl, C6-10 aryl, 5-10 membered heteroaryl containing 1 to 4 heteroatoms selected from N, O, and S, and 4-10 membered heterocyclyl containing 1 to 4 heteroatoms selected from N, O, and S; each R200 and R201 is independently selected from hydrogen, halo, cyano, hydroxy, amino, oxo, thioxo, vinyl, -C(O)Re, -C(O)ORe, -C(O)NReRf, -N(Re)C(O)Rf, -S(O)NReRf, - S(O)2NReRf, -S(O)Ri, -S(O)2Ri, -NReRf, -ORe, -SRe, C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, C3-8 cycloalkyl, C6-10 aryl, 5-10 membered heteroaryl containing 1 to 4 heteroatoms selected from N, O, and S, and 4-10 membered heterocyclyl containing 1 to 4 heteroatoms selected from N, O, and S; wherein each C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, C3-8 cycloalkyl, C6-10 aryl, 5-10 membered heteroaryl and 4-10 membered heterocyclyl is optionally substituted with one to four R300; each Rg, Rh and Ri is independently selected from C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl; wherein each C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, is optionally substituted with one to four R300; wherein each R300 is independently selected from hydrogen, halo, cyano, hydroxy, amino, oxo, thioxo, vinyl, -C(O)Re, -C(O)ORe, -C(O)NReRf, -N(Re)C(O)Rf, -S(O)NReRf, - S(O)2NReRf, -NReRf, S(O)Re, -S(O)2Re, -NReRf, -ORe, -SRe, C1-6 alkyl, C2-6 alkenyl and C2-6 alkynyl; Re and Rf is independently selected from hydrogen, C1-6 alkyl, C2-6 alkenyl and C2-6 alkynyl; C3-8 cycloalkyl, C6-10 aryl, 5-10 membered heteroaryl containing 1 to 4 heteroatoms selected from N, O, and S, and 4-10 membered heterocyclyl containing 1 to 4 heteroatoms selected from N, O, and S; wherein each C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, C3-8 cycloalkyl, C6-10 aryl, 5-10 membered heteroaryl and 4-10 membered heterocyclyl is optionally substituted with one to four R400; each R400 is independently selected from hydrogen, halo, cyano, hydroxy, amino, oxo, thioxo, vinyl, -C(O)Rk, -C(O)ORk, -C(O)NRkRl, -N(Rk)C(O)Rl, -S(O)NRkRl, - S(O)2NRkRl, -NRkRl, S(O)Rk, -S(O)2Rk, -NRkRl, -ORk, -SRk, C1-6 alkyl, C2-6 alkenyl and each Rk and Rl is independently selected from hydrogen, C1-6 alkyl, C2-6 alkenyl and C2-6 alkynyl; C3-8 cycloalkyl, C6-10 aryl, 5-10 membered heteroaryl containing 1 to 4 heteroatoms selected from N, O, and S, and 4-10 membered heterocyclyl containing 1 to 4 heteroatoms selected from N, O, and S. In one embodiment, provided are compounds of formula (II):
or a pharmaceutically acceptable salt, stereoisomer, mixture of stereoisomers thereof; R1 is selected from 5-6 membered heteroaryl, optionally substituted with one to four halo, hydroxy, amino, -C(O)Ra, -C(O)ORb, -C(O)NRaRb, -N(Ra)C(O)Rb, -S(O)NRaRb, - S(O)2NRaRb, -S(O)Rg, -S(O)2Rg, -NRaRb, -ORa, -SRb, C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl and C3-8 cycloalkyl; wherein each C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl and C3-8 cycloalkyl is optionally substituted with one to four R100; R2 is selected from absent, hydrogen, halo, hydroxy, amino, -CN, -C(O)Ra, -C(O)ORb, - C(O)NRaRb, -N(Ra)C(O)Rb, -N(Ra)C(O)NRaRb, -N(Ra)SO2NRaRb, -S(O)NRaRb, - S(O)2NRaRb, -N(Ra)S(O)2Rb, -S(O)Rg, -S(O)2Rg, -NRaRb, -ORa, -SRb, -OC(O)Ra, - OC(O)NRaRb, C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, C3-8 cycloalkyl and 4-10 membered heterocyclyl containing 1 to 4 heteroatoms selected from N, O, and S; wherein each C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, 4-10 membered heterocyclyl containing 1 to 4 heteroatoms selected from N, O, and S, and C3-8 cycloalkyl is optionally substituted with one to four R100; X is selected from absent and C1-6 alkyl, wherein C1-6 alkyl is optionally substituted with one to four R100; Each W1, W2, W3 and W4 is independently selected from -N(Ra)-, -C(O)- and -C(Ra)-; W5 is -N- or -C(Ra)-; wherein at least one of W1, W2, W3 and W4 is -N(Ra)-; wherein at least one of W1, W2, W3 and W4 is -C(Ra)-; G1 is selected from -C6 aryl-, 5-6 membered heteroaryl, C3-8 cycloalkyl and 5-6 membered heterocyclyl; wherein each C6 aryl, 5-6 membered heteroaryl, C3-8 cycloalkyl and 5-6 membered heterocyclyl is optionally substituted with one to four R100; G2 is a 5 or 6 membered heteroaryl or 5-6 membered heterocyclyl optionally substituted with one to four R100; each Ra and Rb is independently selected from absent, hydrogen, C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, C3-6 cycloalkyl and 4-6 membered heterocyclyl; wherein each C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl and C3-6 cycloalkyl and 4-6 membered heterocyclyl is optionally substituted with one to four R200; each R100 is independently selected from hydrogen, halo, cyano, hydroxy, amino, oxo, thioxo, vinyl, -C(O)Rc, -C(O)ORc, -C(O)NRcRd, -N(Rc)C(O)Rd, -S(O)NRcRd, - S(O)2NRcRd, -S(O)Rh, -S(O)2Rh, -NRcRd, -ORc, -SRc, C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, C3-8 cycloalkyl, C6-10 aryl, 5-10 membered heteroaryl containing 1 to 4 heteroatoms selected from N, O, and S, and 4-10 membered heterocyclyl containing 1 to 4 heteroatoms selected from N, O, and S; wherein each C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, C3-8 cycloalkyl, C6-10 aryl, 5-10 membered heteroaryl and 4-10 membered heterocyclyl is optionally substituted with one to four R201; each Rc and Rd is independently selected from hydrogen, C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, C3-8 cycloalkyl, C6-10 aryl, 5-10 membered heteroaryl containing 1 to 4 heteroatoms selected from N, O, and S, and 4-10 membered heterocyclyl containing 1 to 4 heteroatoms selected from N, O, and S; each R200 and R201 is independently selected from hydrogen, halo, cyano, hydroxy, amino, oxo, thioxo, vinyl, -C(O)Re, -C(O)ORe, -C(O)NReRf, -N(Re)C(O)Rf, -S(O)NReRf, - S(O)2NReRf, -S(O)Ri, -S(O)2Ri, -NReRf, -ORe, -SRe, C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, C3-8 cycloalkyl, C6-10 aryl, 5-10 membered heteroaryl containing 1 to 4 heteroatoms selected from N, O, and S, and 4-10 membered heterocyclyl containing 1 to 4 heteroatoms selected from N, O, and S; wherein each C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, C3-8 cycloalkyl, C6-10 aryl, 5-10 membered heteroaryl and 4-10 membered heterocyclyl is optionally substituted with one to four R300; each Rg, Rh and Ri is independently selected from C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl; wherein each C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, is optionally substituted with one to four R300; wherein each R300 is independently selected from hydrogen, halo, cyano, hydroxy, amino, oxo, thioxo, vinyl, -C(O)Re, -C(O)ORe, -C(O)NReRf, -N(Re)C(O)Rf, -S(O)NReRf, - S(O)2NReRf, -NReRf, S(O)Re, -S(O)2Re, -NReRf, -ORe, -SRe, C1-6 alkyl, C2-6 alkenyl and C2-6 alkynyl; Re and Rf is independently selected from hydrogen, C1-6 alkyl, C2-6 alkenyl and C2-6 alkynyl; C3-8 cycloalkyl, C6-10 aryl, 5-10 membered heteroaryl containing 1 to 4 heteroatoms selected from N, O, and S, and 4-10 membered heterocyclyl containing 1 to 4 heteroatoms selected from N, O, and S; wherein each C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, C3-8 cycloalkyl, C6-10 aryl, 5-10 membered heteroaryl and 4-10 membered heterocyclyl is optionally substituted with one to four R400; each R400 is independently selected from hydrogen, halo, cyano, hydroxy, amino, oxo, thioxo, vinyl, -C(O)Rk, -C(O)ORk, -C(O)NRkRl, -N(Rk)C(O)Rl, -S(O)NRkRl, - S(O)2NRkRl, -NRkRl, S(O)Rk, -S(O)2Rk, -NRkRl, -ORk, -SRk, C1-6 alkyl, C2-6 alkenyl and each Rk and Rl is independently selected from hydrogen, C1-6 alkyl, C2-6 alkenyl and C2-6 alkynyl; C3-8 cycloalkyl, C6-10 aryl, 5-10 membered heteroaryl containing 1 to 4 heteroatoms selected from N, O, and S, and 4-10 membered heterocyclyl containing 1 to 4 heteroatoms selected from N, O, and S. In one embodiment, provided are compounds of formula (II): or a pharmaceutically acceptable salt, stereoisomer, mixture of stereoisomers thereof. In one embodiment, provided are compounds of formula (IIIa), (IIIb), (IIIc) or (IIId):
or a pharmaceutically acceptable salt, stereoisomer, mixture of stereoisomers thereof. In one embodiment, provided are compounds of formula (IIIa), (IIIb), (IIIc) or (IIId): or a pharmaceutically acceptable salt, stereoisomer, mixture of stereoisomers thereof. In one embodiment, provided are compounds of formula (IVa) or (IVb):
or a pharmaceutically acceptable salt, stereoisomer, mixture of stereoisomers thereof. In one embodiment, provided are compounds of formula (IVa) or (IVb): In one embodiment, provided are compounds of formula (Va), (Vb) or (Vc):
In one embodiment, provided are compounds of formula (Va), (Vb) or (Vc): or a pharmaceutically acceptable salt, stereoisomer, mixture of stereoisomers thereof; wherein R5 is C1-6 alkyl. In one embodiment, provided are compounds of formula (VIa) or (VIb):
or a pharmaceutically acceptable salt, stereoisomer, mixture of stereoisomers thereof; wherein R5 is C1-6 alkyl. In one embodiment, provided are compounds of formula (VIa) or (VIb):







or a pharmaceutically acceptable salt, stereoisomer, mixture of stereoisomers thereof. In one embodiment, provided are compounds wherein R1 is selected from: ; salt, stereoisomer, mixture of stereoisomers thereof. -OCH3, -H, -SCH3, -S(O)2CH3, -S(O)2CH3, -C(O)OCH3, -C(O)OCH2CH3, and -C(O)NH2. or a pharmaceutically acceptable salt, stereoisomer, mixture of stereoisomers, or deuterated analog thereof. In one embodiment, provided are compounds wherein G2 is selected from:
-OCH3, -H, -SCH3, -S(O)2CH3, -S(O)2CH3, -C(O)OCH3, -C(O)OCH2CH3, and -C(O)NH2. or a pharmaceutically acceptable salt, stereoisomer, mixture of stereoisomers, or deuterated analog thereof. In one embodiment, provided are compounds wherein G2 is selected from: or deuterated analog thereof. In one embodiment, provided are compounds of Table A or a pharmaceutically acceptable salt thereof; TABLE A The compounds described herein (e.g., a compound of Formula I, IIa, IIIa, IIIb, IIIc, IVa, IVb, Va, Vb, VIa, VIb or Table A) or a pharmaceutically acceptable salt thereof, may be used to treat cancers that are mediated by, dependent on or associated with USP1 activity. In certain embodiments, the disease is a solid tumor. In particular embodiments, the solid tumor is selected from prostate cancer, pancreatic cancer, bladder cancer, colorectal cancer, breast cancer, renal cancer, hepatocellular cancer, lung cancer, ovarian cancer, cervical cancer, gastric cancer, esophageal cancer, head and neck cancer, melanoma, neuroendocrine cancers, CNS cancers, brain tumors (e.g., glioma, anaplastic oligodendroglioma, adult glioblastoma multiforme, and adult anaplastic astrocytoma), bone cancer, or soft tissue sarcoma. In some embodiments, the solid tumor is from non- small cell lung cancer or small-cell lung cancer. The following are definitions of terms used in this specification and appended claims. The initial definition provided for a group or term herein applies to that group or term throughout the specification and claims, individually or as part of another group, unless otherwise indicated. As used in the present specification, the following words, phrases and symbols are generally intended to have the meanings as set forth below, except to the extent that the context in which they are used indicates otherwise. Compounds of this invention may have one or more asymmetric centers. Unless otherwise indicated, all chiral (enantiomeric and diastereomeric) and racemic forms of compounds of the present invention are included in the present invention. Many geometric isomers of olefins, C=N double bonds, and the like can also be present in the compounds, and all such stable isomers are contemplated in the present invention. Cis- and trans-geometric isomers of the compounds of the present invention are described and may be isolated as a mixture of isomers or as separated isomeric forms. The present compounds can be isolated in optically active or racemic forms. It is well known in the art how to prepare optically active forms, such as by resolution of racemic forms or by synthesis from optically active starting materials. All chiral, (enantiomeric and diastereomeric) and racemic forms and all geometric isomeric forms of a structure are intended, unless the specific stereochemistry or isomer form is specifically indicated. When any variable (e.g., R3) occurs more than one time in any constituent or formula for a compound, its definition at each occurrence is independent of its definition at everyother occurrence. Thus, for example, if a group is shown to be substituted with 0-2 R3, then said group may optionally be substituted with up to two R3 groups and R3 at each occurrence is selected independently from the definition of R3. Also, combinations of substituents and/or variables are permissible only if such combinations result in stable compounds. When a bond to a substituent is shown to cross a bond connecting two atoms in a ring, then such substituent may be bonded to any atom on the ring. When a substituent is listed without indicating the atom via which such substituent is bonded to the rest of the compound of a given formula, then such substituent may be bonded via any atom in such substituent. Combinations of substituents and/or variables are permissible only if such combinations result in stable compounds. In cases wherein there are nitrogen atoms (e.g., amines) on compounds of the present invention, these can be converted to N-oxides by treatment with an oxidizing agent (e.g., MCPBA and/or hydrogen peroxides) to afford other compounds of this invention. Thus, all shown and claimed nitrogen atoms are considered to cover both the shown nitrogen and its N-oxide (NoO) derivative. In accordance with a convention used in the is used in structural formulas herein to depict the bond that is the point of
or deuterated analog thereof. In one embodiment, provided are compounds of Table A or a pharmaceutically acceptable salt thereof; TABLE A The compounds described herein (e.g., a compound of Formula I, IIa, IIIa, IIIb, IIIc, IVa, IVb, Va, Vb, VIa, VIb or Table A) or a pharmaceutically acceptable salt thereof, may be used to treat cancers that are mediated by, dependent on or associated with USP1 activity. In certain embodiments, the disease is a solid tumor. In particular embodiments, the solid tumor is selected from prostate cancer, pancreatic cancer, bladder cancer, colorectal cancer, breast cancer, renal cancer, hepatocellular cancer, lung cancer, ovarian cancer, cervical cancer, gastric cancer, esophageal cancer, head and neck cancer, melanoma, neuroendocrine cancers, CNS cancers, brain tumors (e.g., glioma, anaplastic oligodendroglioma, adult glioblastoma multiforme, and adult anaplastic astrocytoma), bone cancer, or soft tissue sarcoma. In some embodiments, the solid tumor is from non- small cell lung cancer or small-cell lung cancer. The following are definitions of terms used in this specification and appended claims. The initial definition provided for a group or term herein applies to that group or term throughout the specification and claims, individually or as part of another group, unless otherwise indicated. As used in the present specification, the following words, phrases and symbols are generally intended to have the meanings as set forth below, except to the extent that the context in which they are used indicates otherwise. Compounds of this invention may have one or more asymmetric centers. Unless otherwise indicated, all chiral (enantiomeric and diastereomeric) and racemic forms of compounds of the present invention are included in the present invention. Many geometric isomers of olefins, C=N double bonds, and the like can also be present in the compounds, and all such stable isomers are contemplated in the present invention. Cis- and trans-geometric isomers of the compounds of the present invention are described and may be isolated as a mixture of isomers or as separated isomeric forms. The present compounds can be isolated in optically active or racemic forms. It is well known in the art how to prepare optically active forms, such as by resolution of racemic forms or by synthesis from optically active starting materials. All chiral, (enantiomeric and diastereomeric) and racemic forms and all geometric isomeric forms of a structure are intended, unless the specific stereochemistry or isomer form is specifically indicated. When any variable (e.g., R3) occurs more than one time in any constituent or formula for a compound, its definition at each occurrence is independent of its definition at everyother occurrence. Thus, for example, if a group is shown to be substituted with 0-2 R3, then said group may optionally be substituted with up to two R3 groups and R3 at each occurrence is selected independently from the definition of R3. Also, combinations of substituents and/or variables are permissible only if such combinations result in stable compounds. When a bond to a substituent is shown to cross a bond connecting two atoms in a ring, then such substituent may be bonded to any atom on the ring. When a substituent is listed without indicating the atom via which such substituent is bonded to the rest of the compound of a given formula, then such substituent may be bonded via any atom in such substituent. Combinations of substituents and/or variables are permissible only if such combinations result in stable compounds. In cases wherein there are nitrogen atoms (e.g., amines) on compounds of the present invention, these can be converted to N-oxides by treatment with an oxidizing agent (e.g., MCPBA and/or hydrogen peroxides) to afford other compounds of this invention. Thus, all shown and claimed nitrogen atoms are considered to cover both the shown nitrogen and its N-oxide (NoO) derivative. In accordance with a convention used in the is used in structural formulas herein to depict the bond that is the point of the moiety or substituent to the core or backbone structure. A dash "-" that is not between two letters or symbols is used to indicate a point of attachment for a substituent. For example, -CONH2 is attached through the carbon atom. A dash at the front or end of a chemical group is a matter of convenience; chemical groups may be depicted with or without one or more dashes without losing their ordinary meaning. A wavy line drawn through a line in a structure indicates a point of attachment of a group. Unless chemically or structurally required, no directionality is indicated or implied by the order in which a chemical group is written or named. The term "optionally substituted" in reference to a particular moiety of the compound of Formula I (e.g., an optionally substituted heteroaryl group) refers to a moiety having 0, 1, 2, or more substituents. For example, "optionally substituted alkyl" encompasses both "alkyl" and "substituted alkyl" as defined below. It will be understood by those skilled in the art, with respect to any group containing one or more substituents, that such groups are not intended to introduce any substitution or substitution patterns that are sterically impractical, synthetically non-feasible and/or inherently unstable. As used herein, the term "at least one chemical entity" is interchangeable with the term "a compound". The prefix "Cu-v" indicates that the following group has from u to v carbon atoms. For example, "C1-6 alkyl" indicates that the alkyl group has from 1 to 6 carbon atoms. As used herein, the term "alkyl" or "alkylene" is intended to include both branched and straight-chain saturated aliphatic hydrocarbon groups having the specified number of carbon atoms. For example, "C1-10 alkyl" (or alkylene), is intended to include C1, C2, C3, C4, C5, C6, C7, C8, C9, and C10 alkyl groups. Additionally, for example, "C1-C6 alkyl" denotes alkyl having 1 to 6 carbon atoms. Alkyl groups can be unsubstituted or substituted so that one or more of its hydrogens are replaced by another chemical group. Example alkyl groups include, but are not limited to, methyl (Me), ethyl (Et), propyl (e.g., n-propyl and isopropyl), butyl (e.g., n-butyl, isobutyl, t-butyl), pentyl (e.g., n-pentyl, isopentyl, neopentyl), and the like. "Alkenyl" or "alkenylene" is intended to include hydrocarbon chains of either straight or branched configuration and having one or more double carbon-carbon bonds that may occur in any stable point along the chain. For example, "C2-6 alkenyl" (or alkenylene), is intended to include C2, C3, C4, C5, and C6 alkenyl groups. Examples of alkenyl include, but are not limited to, ethenyl, 1-propenyl, 2-propenyl, 2-butenyl, 3-butenyl, 2-pentenyl, 3- pentenyl, 4-pentenyl, 2-hexenyl, 3-hexenyl, 4-hexenyl, 5-hexenyl, 2-methyl-2-propenyl, 4- methyl-3-pentenyl, and the like. "Alkynyl" or "alkynylene" is intended to include hydrocarbon chains of either straight or branched configuration and having one or more triple carbon-carbon bonds that may occur in any stable point along the chain. For example, "C2-6 alkynyl" (or alkynylene), is intended to include C2, C3, C4, C5, and C6 alkynyl groups; such as ethynyl, propynyl, butynyl, pentynyl, hexynyl and the like. One skilled in the field will understand that, when the designation "CO2" is used O herein, this is intended to refer to the groupC O . When the term "alkyl" is used together with another group, such as in "arylalkyl", this conjunction defines with more specificity at least one of the substituents that the substituted alkyl will contain. For example, "arylalkyl" refers to a substituted alkyl group as defined above where at least one of the substituents is an aryl, such as benzyl. Thus, the term aryl(C0-4)alkyl includes a substituted lower alkyl having at least one aryl substituent and also includes an aryl directly bonded to another group, i.e., aryl(C0)alkyl. The term "heteroarylalkyl" refers to a substituted alkyl group as defined above where at least one of the substituents is a heteroaryl. When reference is made to a substituted alkenyl, alkynyl, alkylene, alkenylene, or alkynylene group, these groups are substituted with one to three substituents as defined above for substituted alkyl groups. The term "alkoxy" refers to an oxygen atom substituted by alkyl or substituted alkyl, as defined herein. For example, the term "alkoxy" includes the group -O-C1-6alkyl such as methoxy, ethoxy, propoxy, isopropoxy, n-butoxy, sec-butoxy, tert-butoxy, pentoxy, 2-pentyloxy, isopentoxy, neopentoxy, hexoxy, 2-hexoxy, 3-hexoxy, 3- methylpentoxy, and the like. "Lower alkoxy" refers to alkoxy groups having one to four carbons. The term "cycloalkyl" refers to cyclized alkyl groups, including mono-, bi- or poly- cyclic ring systems. C3-7 cycloalkyl is intended to include C3, C4, C5, C6, and C7 cycloalkyl groups. Example cycloalkyl groups include, but are not limited to, cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, norbornyl, and the like. As used herein, "carbocycle" or "carbocyclic residue" is intended to mean any stable 3-, 4-, 5-, 6-, or 7-membered monocyclic or bicyclic or 7-, 8-, 9-, 10-, 11-, 12-, or 13-membered bicyclic or tricyclic ring, any of which may be saturated, partially unsaturated, unsaturated or aromatic. Examples of such carbocycles include, but are not limited to, cyclopropyl, cyclobutyl, cyclobutenyl, cyclopentyl, cyclopentenyl, cyclohexyl, cycloheptenyl, cycloheptyl, cycloheptenyl, adamantyl, cyclooctyl, cyclooctenyl, cyclooctadienyl, [3.3.0]bicyclooctane, [4.3.0]bicyclononane, [4.4.0]bicyclodecane, [2.2.2]bicyclooctane, fluorenyl, phenyl, naphthyl, indanyl, adamantyl, anthracenyl, and tetrahydronaphthyl (tetralin). As shown above, bridged rings are also included in the definition of carbocycle (e.g., [2.2.2]bicyclooctane). Preferred carbocycles, unless otherwise specified, are cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, and phenyl. When the term "carbocycle" is used, it is intended to include "aryl". A bridged ring occurs when one or more carbon atoms link two non-adjacent carbon atoms. Preferred bridges are one or two carbon atoms. It is noted that a bridge always converts a monocyclic ring into a bicyclic ring. When a ring is bridged, the substituents recited for the ring may also be present on the bridge. The term "aryl" refers to monocyclic or bicyclic aromatic hydrocarbon groups having 6 to 12 carbon atoms in the ring portion, such as phenyl, and naphthyl groups, each of which may be substituted. Accordingly, in compounds of formula I, the term "cycloalkyl" includes cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl, bicyclooctyl, etc., as well as the following ring systems: , .
the moiety or substituent to the core or backbone structure. A dash "-" that is not between two letters or symbols is used to indicate a point of attachment for a substituent. For example, -CONH2 is attached through the carbon atom. A dash at the front or end of a chemical group is a matter of convenience; chemical groups may be depicted with or without one or more dashes without losing their ordinary meaning. A wavy line drawn through a line in a structure indicates a point of attachment of a group. Unless chemically or structurally required, no directionality is indicated or implied by the order in which a chemical group is written or named. The term "optionally substituted" in reference to a particular moiety of the compound of Formula I (e.g., an optionally substituted heteroaryl group) refers to a moiety having 0, 1, 2, or more substituents. For example, "optionally substituted alkyl" encompasses both "alkyl" and "substituted alkyl" as defined below. It will be understood by those skilled in the art, with respect to any group containing one or more substituents, that such groups are not intended to introduce any substitution or substitution patterns that are sterically impractical, synthetically non-feasible and/or inherently unstable. As used herein, the term "at least one chemical entity" is interchangeable with the term "a compound". The prefix "Cu-v" indicates that the following group has from u to v carbon atoms. For example, "C1-6 alkyl" indicates that the alkyl group has from 1 to 6 carbon atoms. As used herein, the term "alkyl" or "alkylene" is intended to include both branched and straight-chain saturated aliphatic hydrocarbon groups having the specified number of carbon atoms. For example, "C1-10 alkyl" (or alkylene), is intended to include C1, C2, C3, C4, C5, C6, C7, C8, C9, and C10 alkyl groups. Additionally, for example, "C1-C6 alkyl" denotes alkyl having 1 to 6 carbon atoms. Alkyl groups can be unsubstituted or substituted so that one or more of its hydrogens are replaced by another chemical group. Example alkyl groups include, but are not limited to, methyl (Me), ethyl (Et), propyl (e.g., n-propyl and isopropyl), butyl (e.g., n-butyl, isobutyl, t-butyl), pentyl (e.g., n-pentyl, isopentyl, neopentyl), and the like. "Alkenyl" or "alkenylene" is intended to include hydrocarbon chains of either straight or branched configuration and having one or more double carbon-carbon bonds that may occur in any stable point along the chain. For example, "C2-6 alkenyl" (or alkenylene), is intended to include C2, C3, C4, C5, and C6 alkenyl groups. Examples of alkenyl include, but are not limited to, ethenyl, 1-propenyl, 2-propenyl, 2-butenyl, 3-butenyl, 2-pentenyl, 3- pentenyl, 4-pentenyl, 2-hexenyl, 3-hexenyl, 4-hexenyl, 5-hexenyl, 2-methyl-2-propenyl, 4- methyl-3-pentenyl, and the like. "Alkynyl" or "alkynylene" is intended to include hydrocarbon chains of either straight or branched configuration and having one or more triple carbon-carbon bonds that may occur in any stable point along the chain. For example, "C2-6 alkynyl" (or alkynylene), is intended to include C2, C3, C4, C5, and C6 alkynyl groups; such as ethynyl, propynyl, butynyl, pentynyl, hexynyl and the like. One skilled in the field will understand that, when the designation "CO2" is used O herein, this is intended to refer to the groupC O . When the term "alkyl" is used together with another group, such as in "arylalkyl", this conjunction defines with more specificity at least one of the substituents that the substituted alkyl will contain. For example, "arylalkyl" refers to a substituted alkyl group as defined above where at least one of the substituents is an aryl, such as benzyl. Thus, the term aryl(C0-4)alkyl includes a substituted lower alkyl having at least one aryl substituent and also includes an aryl directly bonded to another group, i.e., aryl(C0)alkyl. The term "heteroarylalkyl" refers to a substituted alkyl group as defined above where at least one of the substituents is a heteroaryl. When reference is made to a substituted alkenyl, alkynyl, alkylene, alkenylene, or alkynylene group, these groups are substituted with one to three substituents as defined above for substituted alkyl groups. The term "alkoxy" refers to an oxygen atom substituted by alkyl or substituted alkyl, as defined herein. For example, the term "alkoxy" includes the group -O-C1-6alkyl such as methoxy, ethoxy, propoxy, isopropoxy, n-butoxy, sec-butoxy, tert-butoxy, pentoxy, 2-pentyloxy, isopentoxy, neopentoxy, hexoxy, 2-hexoxy, 3-hexoxy, 3- methylpentoxy, and the like. "Lower alkoxy" refers to alkoxy groups having one to four carbons. The term "cycloalkyl" refers to cyclized alkyl groups, including mono-, bi- or poly- cyclic ring systems. C3-7 cycloalkyl is intended to include C3, C4, C5, C6, and C7 cycloalkyl groups. Example cycloalkyl groups include, but are not limited to, cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, norbornyl, and the like. As used herein, "carbocycle" or "carbocyclic residue" is intended to mean any stable 3-, 4-, 5-, 6-, or 7-membered monocyclic or bicyclic or 7-, 8-, 9-, 10-, 11-, 12-, or 13-membered bicyclic or tricyclic ring, any of which may be saturated, partially unsaturated, unsaturated or aromatic. Examples of such carbocycles include, but are not limited to, cyclopropyl, cyclobutyl, cyclobutenyl, cyclopentyl, cyclopentenyl, cyclohexyl, cycloheptenyl, cycloheptyl, cycloheptenyl, adamantyl, cyclooctyl, cyclooctenyl, cyclooctadienyl, [3.3.0]bicyclooctane, [4.3.0]bicyclononane, [4.4.0]bicyclodecane, [2.2.2]bicyclooctane, fluorenyl, phenyl, naphthyl, indanyl, adamantyl, anthracenyl, and tetrahydronaphthyl (tetralin). As shown above, bridged rings are also included in the definition of carbocycle (e.g., [2.2.2]bicyclooctane). Preferred carbocycles, unless otherwise specified, are cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, and phenyl. When the term "carbocycle" is used, it is intended to include "aryl". A bridged ring occurs when one or more carbon atoms link two non-adjacent carbon atoms. Preferred bridges are one or two carbon atoms. It is noted that a bridge always converts a monocyclic ring into a bicyclic ring. When a ring is bridged, the substituents recited for the ring may also be present on the bridge. The term "aryl" refers to monocyclic or bicyclic aromatic hydrocarbon groups having 6 to 12 carbon atoms in the ring portion, such as phenyl, and naphthyl groups, each of which may be substituted. Accordingly, in compounds of formula I, the term "cycloalkyl" includes cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl, bicyclooctyl, etc., as well as the following ring systems: , . or The term "haloalkyl" means a substituted alkyl having one or more halo substituents. For example, "haloalkyl" includes mono, bi, and trifluoromethyl. The term "haloalkoxy" means an alkoxy group having one or more halo substituents. For example, "haloalkoxy" includes OCF3. The terms "heterocycle", "heterocycloalkyl", "heterocyclo", "heterocyclic", or "heterocyclyl" may be used interchangeably and refer to substituted and unsubstituted 3- to 7-membered monocyclic groups, 7- to 11-membered bicyclic groups, and 10- to 15- membered tricyclic groups, in which at least one of the rings has at least one heteroatom (O, S or N), said heteroatom containing ring preferably having 1, 2, or 3 heteroatoms selected from O, S, and N. Each ring of such a group containing a heteroatom can contain one or two oxygen or sulfur atoms and/or from one to four nitrogen atoms provided that the total number of heteroatoms in each ring is four or less, and further provided that the ring contains at least one carbon atom. The nitrogen and sulfur atoms may optionally be oxidized and the nitrogen atoms may optionally be quaternized. The fused rings completing the bicyclic and tricyclic groups may contain only carbon atoms and may be saturated, partially saturated, or fully unsaturated. The heterocyclo group may be attached at any available nitrogen or carbon atom. As used herein the terms "heterocycle", "heterocycloalkyl", "heterocyclo", "heterocyclic", and "heterocyclyl" include "heteroaryl" groups, as defined below. In addition to the heteroaryl groups described below, exemplary monocyclic heterocyclyl groups include azetidinyl, pyrrolidinyl, oxetanyl, imidazolinyl, oxazolidinyl, isoxazolinyl, thiazolidinyl, isothiazolidinyl, tetrahydrofuranyl, piperidyl, piperazinyl, 2- oxopiperazinyl, 2-oxopiperidyl, 2-oxopyrrolodinyl, 2-oxoazepinyl, azepinyl, 1-pyridonyl, 4-piperidonyl, tetrahydropyranyl, morpholinyl, thiamorpholinyl, thiamorpholinyl sulfoxide, thiamorpholinyl sulfone, 1,3-dioxolane and tetrahydro-1,1-dioxothienyl and the like. Exemplary bicyclic heterocyclo groups include quinuclidinyl. The term "heteroaryl" refers to substituted and unsubstituted aromatic 5- or 6- membered monocyclic groups, 9- or 10-membered bicyclic groups, and 11- to 14- membered tricyclic groups which have at least one heteroatom (O, S or N) in at least one of the rings, said heteroatom-containing ring preferably having 1, 2, or 3 heteroatoms selected from O, S, and N. Each ring of the heteroaryl group containing a heteroatom can contain one or two oxygen or sulfur atoms and/or from one to four nitrogen atoms provided that the total number of heteroatoms in each ring is four or less and each ring has at least one carbon atom. The fused rings completing the bicyclic and tricyclic groups may contain only carbon atoms and may be saturated, partially saturated, or unsaturated. The nitrogen and sulfur atoms may optionally be oxidized and the nitrogen atoms may optionally be quaternized. Heteroaryl groups which are bicyclic or tricyclic must include at least one fully aromatic ring but the other fused ring or rings may be aromatic or non- aromatic. The heteroaryl group may be attached at any available nitrogen or carbon atom of any ring. As valence allows, if said further ring is cycloalkyl or heterocyclo it is additionally optionally substituted with =O (oxo). Exemplary monocyclic heteroaryl groups include pyrrolyl, pyrazolyl, pyrazolinyl, imidazolyl, oxazolyl, isoxazolyl, thiazolyl, thiadiazolyl, isothiazolyl, furanyl, thienyl, oxadiazolyl, pyridyl, pyrazinyl, pyrimidinyl, pyridazinyl, triazinyl and the like. Exemplary bicyclic heteroaryl groups include indolyl, benzothiazolyl, benzodioxolyl, benzoxazolyl, benzothienyl, quinolinyl, tetrahydroisoquinolinyl, isoquinolinyl, benzimidazolyl, benzopyranyl, indolizinyl, benzofuranyl, chromonyl, coumarinyl, benzopyranyl, cinnolinyl, quinoxalinyl, indazolyl, pyrrolopyridyl, furopyridyl, dihydroisoindolyl, tetrahydroquinolinyl and the like. Exemplary tricyclic heteroaryl groups include carbazolyl, benzindolyl, phenanthrollinyl, acridinyl, phenanthridinyl, xanthenyl and the like. In compounds of formula I, preferred heteroaryl groups include: N SO N S , , N , ,
or The term "haloalkyl" means a substituted alkyl having one or more halo substituents. For example, "haloalkyl" includes mono, bi, and trifluoromethyl. The term "haloalkoxy" means an alkoxy group having one or more halo substituents. For example, "haloalkoxy" includes OCF3. The terms "heterocycle", "heterocycloalkyl", "heterocyclo", "heterocyclic", or "heterocyclyl" may be used interchangeably and refer to substituted and unsubstituted 3- to 7-membered monocyclic groups, 7- to 11-membered bicyclic groups, and 10- to 15- membered tricyclic groups, in which at least one of the rings has at least one heteroatom (O, S or N), said heteroatom containing ring preferably having 1, 2, or 3 heteroatoms selected from O, S, and N. Each ring of such a group containing a heteroatom can contain one or two oxygen or sulfur atoms and/or from one to four nitrogen atoms provided that the total number of heteroatoms in each ring is four or less, and further provided that the ring contains at least one carbon atom. The nitrogen and sulfur atoms may optionally be oxidized and the nitrogen atoms may optionally be quaternized. The fused rings completing the bicyclic and tricyclic groups may contain only carbon atoms and may be saturated, partially saturated, or fully unsaturated. The heterocyclo group may be attached at any available nitrogen or carbon atom. As used herein the terms "heterocycle", "heterocycloalkyl", "heterocyclo", "heterocyclic", and "heterocyclyl" include "heteroaryl" groups, as defined below. In addition to the heteroaryl groups described below, exemplary monocyclic heterocyclyl groups include azetidinyl, pyrrolidinyl, oxetanyl, imidazolinyl, oxazolidinyl, isoxazolinyl, thiazolidinyl, isothiazolidinyl, tetrahydrofuranyl, piperidyl, piperazinyl, 2- oxopiperazinyl, 2-oxopiperidyl, 2-oxopyrrolodinyl, 2-oxoazepinyl, azepinyl, 1-pyridonyl, 4-piperidonyl, tetrahydropyranyl, morpholinyl, thiamorpholinyl, thiamorpholinyl sulfoxide, thiamorpholinyl sulfone, 1,3-dioxolane and tetrahydro-1,1-dioxothienyl and the like. Exemplary bicyclic heterocyclo groups include quinuclidinyl. The term "heteroaryl" refers to substituted and unsubstituted aromatic 5- or 6- membered monocyclic groups, 9- or 10-membered bicyclic groups, and 11- to 14- membered tricyclic groups which have at least one heteroatom (O, S or N) in at least one of the rings, said heteroatom-containing ring preferably having 1, 2, or 3 heteroatoms selected from O, S, and N. Each ring of the heteroaryl group containing a heteroatom can contain one or two oxygen or sulfur atoms and/or from one to four nitrogen atoms provided that the total number of heteroatoms in each ring is four or less and each ring has at least one carbon atom. The fused rings completing the bicyclic and tricyclic groups may contain only carbon atoms and may be saturated, partially saturated, or unsaturated. The nitrogen and sulfur atoms may optionally be oxidized and the nitrogen atoms may optionally be quaternized. Heteroaryl groups which are bicyclic or tricyclic must include at least one fully aromatic ring but the other fused ring or rings may be aromatic or non- aromatic. The heteroaryl group may be attached at any available nitrogen or carbon atom of any ring. As valence allows, if said further ring is cycloalkyl or heterocyclo it is additionally optionally substituted with =O (oxo). Exemplary monocyclic heteroaryl groups include pyrrolyl, pyrazolyl, pyrazolinyl, imidazolyl, oxazolyl, isoxazolyl, thiazolyl, thiadiazolyl, isothiazolyl, furanyl, thienyl, oxadiazolyl, pyridyl, pyrazinyl, pyrimidinyl, pyridazinyl, triazinyl and the like. Exemplary bicyclic heteroaryl groups include indolyl, benzothiazolyl, benzodioxolyl, benzoxazolyl, benzothienyl, quinolinyl, tetrahydroisoquinolinyl, isoquinolinyl, benzimidazolyl, benzopyranyl, indolizinyl, benzofuranyl, chromonyl, coumarinyl, benzopyranyl, cinnolinyl, quinoxalinyl, indazolyl, pyrrolopyridyl, furopyridyl, dihydroisoindolyl, tetrahydroquinolinyl and the like. Exemplary tricyclic heteroaryl groups include carbazolyl, benzindolyl, phenanthrollinyl, acridinyl, phenanthridinyl, xanthenyl and the like. In compounds of formula I, preferred heteroaryl groups include: N SO N S , , N , , , ,
, ,
 , (e.g., cyclohexyl), heterocyclo (e.g., pyrrolidinyl, piperidinyl, and morpholinyl) or heteroaryl (e.g., tetrazolyl, imidazolyl, pyrazolyl, triazolyl, thiazolyl, and furyl) the reference is intended to include rings having 0 to 3, preferably 0 to 2, substituents selected from those recited above for the aryl, cycloalkyl, heterocyclo and/or heteroaryl groups, as appropriate. The term "carbocyclyl" or "carbocyclic" refers to a saturated or unsaturated monocyclic or bicyclic ring in which all atoms of all rings are carbon. Thus, the term includes cycloalkyl and aryl rings. Monocyclic carbocycles have 3 to 6 ring atoms, still more typically 5 or 6 ring atoms. Bicyclic carbocycles have 7 to 12 ring atoms, e.g., arranged as a bicyclo [4,5], [5,5], [5,6] or [6,6] system, or 9 or 10 ring atoms arranged as a bicyclo [5,6] or [6,6] system. Examples of mono- and bicyclic carbocycles include cyclopropyl, cyclobutyl, cyclopentyl, 1-cyclopent-1-enyl, 1-cyclopent-2-enyl, 1- cyclopent-3-enyl, cyclohexyl, 1-cyclohex-1-enyl, 1-cyclohex-2-enyl, 1-cyclohex-3-enyl, phenyl and naphthyl. The carbocyclic ring may be substituted in which case the substituents are selected from those recited above for cycloalkyl and aryl groups. The term "alkylthio" refers to the group "alkyl-S-". The term "acyl" refers to a group -C(O)R, wherein R is hydrogen, alkyl, cycloalkyl, heterocyclyl, aryl, heteroalkyl, or heteroaryl; each of which may be optionally substituted, as defined herein. Examples of acyl include formyl, acetyl, cylcohexylcarbonyl, cyclohexylmethyl-carbonyl, and benzoyl. The term "amido" refers to both a "C-amido" group which refers to the group -- C(O)NRgRh and an "N-amido" group which refers to the group –NRgC(O)Rh, wherein Rg and Rh are independently selected from hydrogen, alkyl, aryl, haloalkyl, or heteroaryl; each of which may be optionally substituted. The term "amino" refers to the group -NRg Rh wherein Rg and Rh are independently selected from hydrogen, alkyl, haloalkyl, aryl, or heteroaryl; each of which may be optionally substituted. The term "azido" refers to –N3. The term "carbamoyl" refers to both an "O-carbamoyl" group which refers to the group -O-C(O)NRiRj and an "N-carbamoyl" group which refers to the group – NRiC(O)ORj, wherein Ri and Rj are independently selected from hydrogen, alkyl, aryl, haloalkyl, or heteroaryl; each of which may be optionally substituted. The term "carboxyl" refers to -C(O)OH. The term "carboxyl ester" refers to both -OC(O)R and -C(O)ORg, wherein Rg is hydrogen, alkyl, cycloalkyl, heterocyclyl, aryl, heteroalkyl, or heteroaryl; each of which may be optionally substituted, as defined herein. The term "cyano" or "carbonitrile" refers to the group -CN. The term "cycloalkyl" refers to a saturated or partially unsaturated cyclic alkyl group having a single ring or multiple rings including fused, bridged, and spiro ring systems. The term "cycloalkyl" includes cycloalkenyl groups (i.e. the cyclic group having at least one double bond). As used herein, cycloalkyl has from 3 to 20 ring carbon atoms (i.e., C.sub.3-20 cycloalkyl), 3 to 12 ring carbon atoms (i.e., C.sub.3-12 cycloalkyl), 3 to 10 ring carbon atoms (i.e., C.sub.3-10 cycloalkyl), 3 to 8 ring carbon atoms (i.e., C.sub.3- 8 cycloalkyl), or 3 to 6 ring carbon atoms (i.e., C.sub.3-6 cycloalkyl). Examples of cycloalkyl groups include cyclopropyl, cyclobutyl, cyclopentyl, and cyclohexyl. The term "heteroatoms" shall include oxygen, sulfur and nitrogen. When the term "unsaturated" is used herein to refer to a ring or group, the ring or group may be fully unsaturated or partially unsaturated. Throughout the specification, groups and substituents thereof may be chosen by one skilled in the field to provide stable moieties and compounds and compounds useful as pharmaceutically-acceptable compounds and/or intermediate compounds useful in making pharmaceutically-acceptable compounds. It should be understood that the selections for all groups, including for example, alkoxy, thioalkyl, and aminoalkyl, will be made by one skilled in the field to provide stable compounds. The term "substituted", as used herein, means that any one or more hydrogens on the designated atom or group is replaced with a selection from the indicated group, provided that the designated atom's normal valence is not exceeded. When a substituent is oxo, or keto, (i.e., =O) then 2 hydrogens on the atom are replaced. Keto substituents are not present on aromatic moieties. Unless otherwise specified, substituents are named into the core structure. For example, it is to be understood that when (cycloalkyl)alkyl is listed as a possible substituent, the point of attachment of this substituent to the core structure is in the alkyl portion. Ring double bonds, as used herein, are double bonds that are formed between two adjacent ring atoms (e.g., C=C, C=N, or N=N). Combinations of substituents and/or variables are permissible only if such combinations result in stable compounds or useful synthetic intermediates. A stable compound or stable structure is meant to imply a compound that is sufficiently robust to survive isolation from a reaction mixture to a useful degree of purity, and subsequent formulation into an efficacious therapeutic agent. It is preferred that the presently recited compounds do not contain a N-halo, S(O)2H, or S(O)H group. The compounds described herein may exist in a free form (with no ionization) or can form salts which are also within the scope of this invention. Unless otherwise indicated, reference to an inventive compound is understood to include reference to the free form and to salts thereof. The term "salt(s)" denotes acidic and/or basic salts formed with inorganic and/or organic acids and bases. In addition, the term "salt(s)" may include zwitterions (inner salts), e.g., when a compound of formula I, contains both a basic moiety, such as an amine or a pyridine or imidazole ring, and an acidic moiety, such as a carboxylic acid. Pharmaceutically acceptable (i.e., non-toxic, physiologically acceptable) salts are preferred, such as, for example, acceptable metal and amine salts in which the cation does not contribute significantly to the toxicity or biological activity of the salt. However, other salts may be useful, e.g., in isolation or purification steps which may be employed during preparation, and thus, are contemplated within the scope of the invention. Salts of the compounds of herein may be formed, for example, by reacting a compound described herein with an amount of acid or base, such as an equivalent amount, in a medium such as one in which the salt precipitates or in an aqueous medium followed by lyophilization. Exemplary acid addition salts include acetates (such as those formed with acetic acid or trihaloacetic acid, for example, trifluoroacetic acid), adipates, alginates, ascorbates, aspartates, benzoates, benzenesulfonates, bisulfates, borates, butyrates, citrates, camphorates, camphorsulfonates, cyclopentanepropionates, digluconates, dodecylsulfates, ethanesulfonates, fumarates, glucoheptanoates, glycerophosphates, hemisulfates, heptanoates, hexanoates, hydrochlorides (formed with hydrochloric acid), hydrobromides (formed with hydrogen bromide), hydroiodides, 2- hydroxyethanesulfonates, lactates, maleates (formed with maleic acid), methanesulfonates (formed with methanesulfonic acid), 2-naphthalenesulfonates, nicotinates, nitrates, oxalates, pectinates, persulfates, 3-phenylpropionates, phosphates, picrates, pivalates, propionates, salicylates, succinates, sulfates (such as those formed with sulfuric acid), sulfonates (such as those mentioned herein), tartrates, thiocyanates, toluenesulfonates such as tosylates, undecanoates, and the like. Exemplary basic salts include ammonium salts, alkali metal salts such as sodium, lithium, and potassium salts; alkaline earth metal salts such as calcium and magnesium salts; barium, zinc, and aluminum salts; salts with organic bases (for example, organic amines) such as trialkylamines such as triethylamine, procaine, dibenzylamine, N-benzyl- ȕ-phenethylamine, 1-ephenamine, N,Nƍ-dibenzylethylene-diamine, dehydroabietylamine, N-ethylpiperidine, benzylamine, dicyclohexylamine or similar pharmaceutically acceptable amines and salts with amino acids such as arginine, lysine and the like. Basic nitrogen-containing groups may be quaternized with agents such as lower alkyl halides (e.g., methyl, ethyl, propyl, and butyl chlorides, bromides and iodides), dialkyl sulfates (e.g., dimethyl, diethyl, dibutyl, and diamyl sulfates), long chain halides (e.g., decyl, lauryl, myristyl and stearyl chlorides, bromides and iodides), aralkyl halides (e.g., benzyl and phenethyl bromides), and others. Preferred salts include monohydrochloride, hydrogensulfate, methanesulfonate, phosphate or nitrate salts. The compounds described herein can be provided as amorphous solids or crystalline solids. Lyophilization can be employed to provide the compounds as a solid. It should further be understood that solvates (e.g., hydrates) of the compounds described herein are also within the scope of the present invention. The term “solvate” means a physical association of a compound with one or more solvent molecules, whether organic or inorganic. This physical association includes hydrogen bonding. In certain instances, the solvate will be capable of isolation, for example when one or more solvent molecules are incorporated in the crystal lattice of the crystalline solid. “Solvate” encompasses both solution-phase and isolable solvates. Exemplary solvates include hydrates, ethanolates, methanolates, isopropanolates, acetonitrile solvates, and ethyl acetate solvates. Methods of solvation are known in the art. In addition, compounds described herein subsequent to their preparation, can be isolated and purified to obtain a composition containing an amount by weight equal to or greater than 99% of a compound (“substantially pure”), which is then used or formulated as described herein. Such “substantially pure” compounds described herein are also contemplated herein as part of the present invention. The phrase "pharmaceutically acceptable" is employed herein to refer to those compounds, materials, compositions, and/or dosage forms which are, within the scope of sound medical judgment, suitable for use in contact with the tissues of human beings and animals without excessive toxicity, irritation, allergic response, or other problem or complication, commensurate with a reasonable benefit/risk ratio. As used herein, "pharmaceutically acceptable salts" refer to derivatives of the disclosed compounds wherein the parent compound is modified by making acid or base salts thereof. Examples of pharmaceutically-acceptable salts include, but are not limited to, mineral or organic acid salts of basic groups such as amines; and alkali or organic salts of acidic groups such as carboxylic acids. The pharmaceutically-acceptable salts include the conventional non-toxic salts or the quaternary ammonium salts of the parent compound formed, for example, from non-toxic inorganic or organic acids. For example, such conventional non-toxic salts include those derived from inorganic acids such as hydrochloric, hydrobromic, sulfuric, sulfamic, phosphoric, and nitric; and the salts prepared from organic acids such as acetic, propionic, succinic, glycolic, stearic, lactic, malic, tartaric, citric, ascorbic, pamoic, maleic, hydroxymaleic, phenylacetic, glutamic, benzoic, salicylic, sulfanilic, 2-acetoxybenzoic, fumaric, toluenesulfonic, methanesulfonic, ethane disulfonic, oxalic, and isethionic, and the like. The pharmaceutically acceptable salts of the present invention can be synthesized from the parent compound which contains a basic or acidic moiety by conventional chemical methods. Generally, such salts can be prepared by reacting the free acid or base forms of these compounds with a stoichiometric amount of the appropriate base or acid in water or in an organic solvent, or in a mixture of the two; generally, nonaqueous media like ether, ethyl acetate, ethanol, isopropanol, or acetonitrile are preferred. Lists of suitable salts are found in Remington's Pharmaceutical Sciences, 18th Edition, Mack Publishing Company, Easton, PA (1990), the disclosure of which is hereby incorporated by reference. “Stable compound” and “stable structure” are meant to indicate a compound that is sufficiently robust to survive isolation to a useful degree of purity from a reaction mixture, and formulation into an efficacious therapeutic agent. The present invention is intended to embody stable compounds. “Therapeutically effective amount” is intended to include an amount of a compound of the present invention alone or an amount of the combination of compounds claimed or an amount of a compound of the present invention in combination with other active ingredients effective to act as an inhibitor of USP1, or effective to treat or prevent proliferative disorders, such as cancer. As used herein, “treating” or “treatment” cover the treatment of a disease-state in a mammal, particularly in a human, and include: (a) preventing the disease-state from occurring in a mammal, in particular, when such mammal is predisposed to the disease- state but has not yet been diagnosed as having it; (b) inhibiting the disease-state, i.e., arresting its development; and/or (c) relieving the disease-state, i.e., causing regression of the disease state. All stereoisomers of the compounds of the instant invention are contemplated, either in admixture or in pure or substantially pure form. Stereoisomers may include compounds which are optical isomers through possession of one or more chiral atoms, as well as compounds which are optical isomers by virtue of limited rotation about one or more bonds (atropisomers). The definition of compounds according to the invention embraces all the possible stereoisomers and their mixtures. It very particularly embraces the racemic forms and the isolated optical isomers having the specified activity. The racemic forms can be resolved by physical methods, such as, for example, fractional crystallization, separation or crystallization of diastereomeric derivatives or separation by chiral column chromatography. The individual optical isomers can be obtained from the racemates from the conventional methods, such as, for example, salt formation with an optically active acid followed by crystallization. The present invention is intended to include all isotopes of atoms occurring in the present compounds. Isotopes include those atoms having the same atomic number but different mass numbers. By way of general example and without limitation, isotopes of hydrogen include deuterium and tritium. Isotopes of carbon include13C and14C. Isotopically-labeled compounds of the invention can generally be prepared by conventional techniques known to those skilled in the art or by processes analogous to those described herein, using an appropriate isotopically-labeled reagent in place of the non-labeled reagent otherwise employed. Prodrugs and solvates of the inventive compounds are also contemplated. The term "prodrug" denotes a compound which, upon administration to a subject, undergoes chemical conversion by metabolic or chemical processes to yield a compound of the formula I, and/or a salt and/or solvate thereof. Any compound that will be converted in vivo to provide the bioactive agent (i.e., the compound for formula I) is a prodrug within the scope and spirit of the invention. For example, compounds containing a carboxy group can form physiologically hydrolyzable esters which serve as prodrugs by being hydrolyzed in the body to yield formula I compounds per se. Such prodrugs are preferably administered orally since hydrolysis in many instances occurs principally under the influence of the digestive enzymes. Parenteral administration may be used where the ester per se is active, or in those instances where hydrolysis occurs in the blood. Examples of physiologically hydrolyzable esters of compounds of formula I include C1-6alkylbenzyl, 4-methoxybenzyl, indanyl, phthalyl, methoxymethyl, C1-6alkanoyloxy-C1-6alkyl, e.g., acetoxymethyl, pivaloyloxymethyl or propionyloxymethyl, C1-6alkoxycarbonyloxy-C1-6alkyl, e.g., methoxycarbonyl-oxymethyl or ethoxycarbonyloxymethyl, glycyloxymethyl, phenylglycyloxymethyl, (5-methyl-2- oxo-1,3-dioxolen-4-yl)-methyl and other well known physiologically hydrolyzable esters used, for example, in the penicillin and cephalosporin arts. Such esters may be prepared by conventional techniques known in the art. Various forms of prodrugs are well known in the art and are described in Rautio, J. et al., Nature Review Drug Discovery, 17, 559-587 (2018). Compounds described in the application and their salts may exist in their tautomeric form, in which hydrogen atoms are transposed to other parts of the molecules and the chemical bonds between the atoms of the molecules are consequently rearranged. It should be understood that the all tautomeric forms, insofar as they may exist, are included within the invention. Additionally, inventive compounds may have trans- and cis-isomers. The disclosure herein further relates to compounds described herein, the tautomers and stereoisomeric forms thereof, and the pharmaceutically acceptable addition salts, and the solvates thereof, for use as a medicament. Furthermore, the disclosure herein relates to the use of a compound described herein, a tautomer or a stereoisomeric form thereof, or a pharmaceutically acceptable addition salt, or a solvate thereof, or a pharmaceutical composition according to the invention, for the manufacture of a medicament. The inventive compositions may contain other therapeutic agents as described above and may be formulated, for example, by employing conventional solid or liquid vehicles or diluents, as well as pharmaceutical additives of a type appropriate to the mode of desired administration (e.g., excipients, binders, preservatives, stabilizers, flavors, etc.) according to techniques such as those well known in the art of pharmaceutical formulation. Accordingly, the present invention further includes compositions comprising one or more compounds described herein and a pharmaceutically acceptable carrier. A "pharmaceutically acceptable carrier" refers to media generally accepted in the art for the delivery of biologically active agents to animals, in particular, mammals. Pharmaceutically acceptable carriers are formulated according to a number of factors well within the purview of those of ordinary skill in the art. These include without limitation the type and nature of the active agent being formulated; the subject to which the agent- containing composition is to be administered; the intended route of administration of the composition; and, the therapeutic indication being targeted. Pharmaceutically acceptable carriers include both aqueous and non-aqueous liquid media, as well as a variety of solid and semi-solid dosage forms. Such carriers can include a number of different ingredients and additives in addition to the active agent, such additional ingredients being included in the formulation for a variety of reasons, e.g., stabilization of the active agent, binders, etc., well known to those of ordinary skill in the art. Descriptions of suitable pharmaceutically acceptable carriers, and factors involved in their selection, are found in a variety of readily available sources such as, for example, Remington's Pharmaceutical Sciences, 17th Edition (1985), which is incorporated herein by reference in its entirety. The compounds described herein may be administered by any means suitable for the condition to be treated, which may depend on the need for site-specific treatment or quantity of drug to be delivered. Topical administration is generally preferred for skin- related diseases, and systematic treatment preferred for cancerous or pre-cancerous conditions, although other modes of delivery are contemplated. For example, the compounds may be delivered orally, such as in the form of tablets, capsules, granules, powders, or liquid formulations including syrups; topically, such as in the form of solutions, suspensions, gels or ointments; sublingually; bucally; parenterally, such as by subcutaneous, intravenous, intramuscular or intrasternal injection or infusion techniques (e.g., as sterile injectable aq. or non-aq. solutions or suspensions); nasally such as by inhalation spray; topically, such as in the form of a cream or ointment; rectally such as in the form of suppositories; or liposomally. Dosage unit formulations containing non-toxic, pharmaceutically acceptable vehicles or diluents may be administered. The compounds may be administered in a form suitable for immediate release or extended release. Immediate release or extended release may be achieved with suitable pharmaceutical compositions or, particularly in the case of extended release, with devices such as subcutaneous implants or osmotic pumps. Exemplary compositions for oral administration include suspensions which may contain, for example, microcrystalline cellulose for imparting bulk, alginic acid or sodium alginate as a suspending agent, methylcellulose as a viscosity enhancer, and sweeteners or flavoring agents such as those known in the art; and immediate release tablets which may contain, for example, microcrystalline cellulose, dicalcium phosphate, starch, magnesium stearate and/or lactose and/or other excipients, binders, extenders, disintegrants, diluents and lubricants such as those known in the art. The inventive compounds may also be orally delivered by sublingual and/or buccal administration, e.g., with molded, compressed, or freeze-dried tablets. Exemplary compositions may include fast-dissolving diluents such as mannitol, lactose, sucrose, and/or cyclodextrins. Also included in such formulations may be high molecular weight excipients such as celluloses (AVICEL®) or polyethylene glycols (PEG); an excipient to aid mucosal adhesion such as hydroxypropyl cellulose (HPC), hydroxypropyl methyl cellulose (HPMC), sodium carboxymethyl cellulose (SCMC), and/or maleic anhydride copolymer (e.g., GANTREZ®); and agents to control release such as polyacrylic copolymer (e.g., CARBOPOL 934®). Lubricants, glidants, flavors, coloring agents and stabilizers may also be added for ease of fabrication and use. Formulations for parenteral administration may be in the form of aqueous or non- aqueous isotonic sterile injection solutions or suspensions. These solutions and suspensions may be prepared from sterile powders or granules using one or more of the carriers or diluents mentioned for use in the formulations for oral administration or by using other suitable dispersing or wetting agents and suspending agents. The compounds may be dissolved in water, polyethylene glycol, propylene glycol, ethanol, corn oil, cottonseed oil, peanut oil, sesame oil, benzyl alcohol, sodium chloride, tragacanth gum, and/or various buffers. Other adjuvants and modes of administration are well and widely known in the pharmaceutical art. The active ingredient may also be administered by injection as a composition with suitable carriers including saline, dextrose, or water, or with cyclodextrin (i.e. Captisol), cosolvent solubilization (i.e. propylene glycol) or micellar solubilization (i.e. Tween 80). Exemplary compositions for parenteral administration include injectable solutions or suspensions which may contain, for example, suitable non-toxic, parenterally acceptable diluents or solvents, such as mannitol, 1,3-butanediol, water, Ringer's solution, an isotonic sodium chloride solution, or other suitable dispersing or wetting and suspending agents, including synthetic mono- or diglycerides, and fatty acids, including oleic acid. The sterile injectable preparation may also be a sterile injectable solution or suspension in a non-toxic parenterally acceptable diluent or solvent, for example as a solution in 1,3-butanediol. Among the acceptable vehicles and solvents that may be employed are water, Ringer’s solution, and isotonic sodium chloride solution. In addition, sterile, fixed oils are conventionally employed as a solvent or suspending medium. For this purpose any bland fixed oil may be employed, including synthetic mono- or diglycerides. In addition, fatty acids such as oleic acid find use in the preparation of injectables. A sterile injectable oil-in-water microemulsion can, for example, be prepared by 1) dissolving at least one compound described herein in an oily phase, such as, for example, a mixture of soybean oil and lecithin; 2) combining the compound-containing oil phase with a water and glycerol mixture; and 3) processing the combination to form a microemulsion. A sterile aqueous or oleaginous suspension can be prepared in accordance with methods already known in the art. For example, a sterile aqueous solution or suspension can be prepared with a non-toxic parenterally-acceptable diluent or solvent, such as, for example, 1,3-butane diol; and a sterile oleaginous suspension can be prepared with a sterile non-toxic acceptable solvent or suspending medium, such as, for example, sterile fixed oils, e.g., synthetic mono- or diglycerides; and fatty acids, such as, for example, oleic acid. Exemplary compositions for nasal aerosol or inhalation administration include solutions which may contain, for example, benzyl alcohol or other suitable preservatives, absorption promoters to enhance absorption and/or bioavailability, and/or other solubilizing or dispersing agents such as those known in the art. Dispersible powders and granules can, for example, be prepared by admixing at least one compound described herein or a pharmaceutically acceptable salt thereof, with at least one dispersing and/or wetting agent; at least one suspending agent; and/or at least one preservative. Exemplary preservatives include, but are not limited to, for example, anti-oxidants, e.g., ascorbic acid. In addition, dispersible powders and granules can also contain at least one excipient, including, but not limited to, for example, sweetening agents; flavoring agents; and coloring agents. Exemplary compositions for rectal administration include suppositories which may contain, for example, suitable non-irritating excipients, such as cocoa butter, synthetic glyceride esters or polyethylene glycols, which are solid at ordinary temperatures but liquefy and/or dissolve in the rectal cavity to release the drug. The therapeutically-effective amount of a compound of the present invention may be determined by one of ordinary skill in the art, and includes exemplary dosage amounts for a mammal of from about 0.05 to 1000 mg/kg; 1-1000 mg/kg; 1-50 mg/kg; 5-250 mg/kg; 250-1000 mg/kg of body weight of active compound per day, which may be administered in a single dose or in the form of individual divided doses, such as from 1 to 4 times per day. It will be understood that the specific dose level and frequency of dosage for any particular subject may be varied and will depend upon a variety of factors, including the activity of the specific compound employed, the metabolic stability and length of action of that compound, the species, age, body weight, general health, sex and diet of the subject, the mode and time of administration, rate of excretion, drug combination, and severity of the particular condition. Preferred subjects for treatment include animals, most preferably mammalian species such as humans, and domestic animals such as dogs, cats, horses, and the like. Thus, when the term "patient" is used herein, this term is intended to include all subjects, most preferably mammalian species that are affected by modulation of USP1-mediated functions. The compounds described herein are useful for the treatment of cancer. In one embodiment, the present application provides a combined preparation of a compound described herein and/or a pharmaceutically acceptable salt thereof, a stereoisomer thereof or a tautomer thereof, and additional therapeutic agent(s) for simultaneous, separate or sequential use in the treatment and/or prophylaxis of multiple diseases or disorders associated with USP1. In another aspect, the application provides a method of treating a patient suffering from or susceptible to a medical condition that is associated with USP1. A number of medical conditions can be treated. The method comprises administering to the patient a therapeutically effective amount of a composition comprising a compound described herien and/or a pharmaceutically acceptable salt thereof, a stereoisomer thereof or a tautomer thereof. For example, the compounds described herein may be used to treat or proliferative diseases such as cancer, immunological disorders or inflammatory disorders. In other embodiments, the compounds described herein may be used to treat cancers that are mediated by, dependent on or associated with USP1 activity. In certain embodiments, the disease is a solid tumor. In particular embodiments, the solid tumor is selected from prostate cancer, pancreatic cancer, bladder cancer, colorectal cancer, breast cancer, renal cancer, hepatocellular cancer, lung cancer, ovarian cancer, cervical cancer, gastric cancer, esophageal cancer, head and neck cancer, melanoma, neuroendocrine cancers, CNS cancers, brain tumors (e.g., glioma, anaplastic oligodendroglioma, adult glioblastoma multiforme, and adult anaplastic astrocytoma), bone cancer, or soft tissue sarcoma. In some embodiments, the solid tumor is from non-small cell lung cancer or small-cell lung cancer. In one embodiment, the compounds herein can be useful in the treatment of haematological malignancies. In one embodiment, hematological malignancy is selected from multiple myeloma, non-Hodgkin's lymphoma, Hodgkin lymphoma, T-cell leukaemia, mucosa-associated lymphoid tissue lymphoma, diffuse large B-cell lymphoma and mantle cell lymphoma. In one embodiment solid tumor is selected from pancreatic cancer, breast cancer, melanoma and non-small cell lung cancer. In one embodiment, cancer is selected from a carcinoma, preferably a carcinoma of the bladder, breast, colon (including colorectal carcinomas, such as colon adenocarcinoma and colon adenoma), kidney, urothelial, uterus, epidermis, liver, lung (including adenocarcinoma, small cell lung cancer, non-small cell lung carcinomas and squamous lung cancer), oesophagus, head and neck, gall bladder, ovary, pancreas (including exocrine pancreatic carcinoma), stomach, gastrointestinal cancer (including gastrointestinal stromal tumors), cervix, endometrium, thyroid, prostate and skin. In one embodiment, the cancer is selected from pituitary cancer, a hematopoietic tumor of lymphoid lineage, for example leukemia, acute lymphocytic leukemia, chronic lymphocytic leukemia, B-cell lymphoma (e.g. diffuse large B-cell lymphoma, mantle cell lymphoma), T-cell leukaemia/lymphoma, Hodgkin's lymphoma, non-Hodgkin's lymphoma, hairy cell lymphoma, or Burkett's lymphoma; a hematopoietic tumor of myeloid lineage, for example leukemias, acute and chronic myelogenous leukemias, chronic myelomonocytic leukemia (CMML), myeloproliferative disorder, myeloproliferative syndrome, myelodysplastic syndrome, or promyelocytic leukemia; multiple myeloma; thyroid follicular cancer; hepatocellular cancer, a tumor of mesenchymal origin (e.g. Ewing's sarcoma), for example fibrosarcoma or rhabdomyosarcoma; a tumor of the central or peripheral nervous system, for example astrocytoma, neuroblastoma, glioma (such as glioblastoma multiforme) or schwannoma; melanoma; seminoma; teratocarcinoma; osteosarcoma; xeroderma pigmentosum; keratoctanthoma; thyroid follicular cancer; or Kaposi's sarcoma. In other embodiments, the compounds described herein may be used to treat cancers that are mediated by, dependent on or associated with USP1 activity. In certain embodiments, the disease is a solid tumor. In particular embodiments, the solid tumor is from prostate cancer, pancreatic cancer, bladder cancer, colorectal cancer, breast cancer, renal cancer, hepatocellular cancer, lung cancer, ovarian cancer, cervical cancer, gastric cancer, esophageal cancer, head and neck cancer, melanoma, neuroendocrine cancers, CNS cancers, brain tumors (e.g., glioma, anaplastic oligodendroglioma, adult glioblastoma multiforme, and adult anaplastic astrocytoma), bone cancer, or soft tissue sarcoma. In some embodiments, the solid tumor is from non-small cell lung cancer or small-cell lung cancer. In other embodiments, the disease is a hematologic malignancy. In certain embodiments, the disease is lymphoma, multiple myeloma, or leukemia. In certain embodiments, the hematologic malignancy is leukemia or lymphoma. In specific embodiments, the disease is acute lymphocytic leukemia (ALL), acute myeloid leukemia (AML), chronic lymphocytic leukemia (CLL), small lymphocytic lymphoma (SLL), myelodysplastic syndrome (MDS), myeloproliferative disease (MPD), chronic myeloid leukemia (CML), juvenile myelomonocytic leukemia (JMML), multiple myeloma (MM), Hodgkin lymphoma, indolent non-Hodgkin's lymphoma (iNHL), refractory iNHL, non- Hodgkin's lymphoma (NHL), mantle cell lymphoma (MCL), follicular lymphoma, Waldenström’s macroglobulinemia (WM), minimal residual disease (MRD), T-cell lymphoma, B-cell lymphoma, diffuse large B-cell lymphoma (DLBCL), T-cell acute lymphoblastic leukemia (T-ALL), B-cell acute lymphoblastic leukemia (B-ALL), lymphoplasmacytic lymphoma, marginal zone lymphoma, or Burkitt lymphoma. In one embodiment, the disease is T-cell acute lymphoblastic leukemia (T-ALL), or B-cell acute lymphoblastic leukemia (B-ALL). In some embodiments, non-Hodgkin lymphoma can be indolent B-cell diseases including follicular lymphoma, lymphoplasmacytic lymphoma, Waldenström macroglobulinemia, and marginal zone lymphoma, as well as the aggressive lymphomas that include, for example, Burkitt lymphoma, diffuse large B- cell lymphoma (DLBCL) and mantle cell lymphoma (MCL). In some embodiments, the cancer is selected from hematological cancer, a lymphatic cancer. In some embodiments, the cancer comprises cancer cells with DNA damage repair pathway deficiency. In some embodiments, the cancer is a homologous recombination deficient cancer. In some embodiments, the cancer comprises cancer cells with a mutation in a gene encoding p53. In some embodiments, the mutation in a gene encoding p53 is a germline or somatic mutation. In some embodiments, the cancer comprises with cancer cells with loss of function mutation in a gene encoding p53. In some embodiments, the cancer is a BRCA1 and/or BRCA2 deficient cancer. In some embodiments, the cancer is a somatic or germline BRCA1 and/or BRCA2 mutant cancer. In some embodiments, the cancer is a Poly (ADP-ribose) polymerase (“PARP”) inhibitor refractory or resistant cancer. In some embodiments, the cancer is a PARP inhibitor resistant or refractory BRCA1 and/or BRCA2 deficient cancer. In some embodiments, the cancer cell has a germline or somatic mutation in a gene encoding ataxia telangiectasia mutated (ATM) protein kinase or ATM deficiency. In some embodiments, the cancer has a mutation in the gene encoding more than two of p53, BRCA1, BRCA2, ATM. In some embodiments, the disease is an autoimmune or inflammatory disease or disorder. These autoimmune or inflammatory diseases or conditions may be chronic or acute and include, but are not limited to, inflammatory pelvic disease, urethritis, skin sunburn, sinusitis, pneumonitis, encephalitis, meningitis, myocarditis, pericarditis, nephritis including lupus nephritis, osteomyelitis, myositis, eczema, hepatitis, gastritis, enteritis, dermatitis, gingivitis, appendicitis, pancreatitis, primary biliary cirrhosis, cholecystitis, sclerosing cholangitis, agammaglobulinemia, psoriasis, allergy, Crohn's disease, irritable bowel syndrome, ulcerative colitis, Sjogren's disease, tissue graft rejection such as acute graft-versus-host disease, hyperacute rejection of transplanted organs, asthma, chronic obstructive airways disease, allergic rhinitis, chronic obstructive pulmonary disease (COPD), autoimmune polyglandular disease (also known as autoimmune polyglandular syndrome), autoimmune alopecia, pernicious anemia, vasculitis, glomerulonephritis, giant cell arteritis, Wegener's granulomatosis, Polyarteritis nodosa, dermatomyositis, multiple sclerosis, scleroderma, autoimmune hemolytic and thrombocytopenic states, Goodpasture's syndrome, atherosclerosis, Addison's disease, hypophysitis, Parkinson's disease, Alzheimer's disease, Kawasaki disease, Takayasu's Arteritis, depression, retinitis, uveitis, scleritis, Type I diabetes, septic shock, systemic lupus erythematosus (SLE), rheumatoid arthritis, psoriatic arthritis, juvenile arthritis, osteoarthritis, gout, chronic idiopathic thrombocytopenic purpura, Waldenström macroglobulinemia, myasthenia gravis, Hashimoto's thyroiditis, atopic dermatitis, degenerative joint disease, vitiligo, bullous skin diseases, autoimmune hypopituitarism, Guillain-Barre syndrome, Behcet's disease, scleracierma, mycosis fungoides, acute inflammatory responses (such as acute respiratory distress syndrome and ischemia/reperfusion injury), and Graves' disease. In some embodiments, the autoimmune and inflammatory diseases and conditions may also include systemic or tissue inflammation, inflammatory responses to hypoxia, cellular activation and proliferation, lipid metabolism, fibrosis, infections with bacteria, infections with viruses (e.g., herpes virus, human papilloma virus, adenovirus, poxvirus and other DNA viruses), fungi, parasites or their toxins, such as sepsis, sepsis syndrome, septic shock, endotoxaemia, systemic inflammatory response syndrome (SIRS), multi-organ dysfunction syndrome, toxic shock syndrome, acute lung injury, ARDS (adult respiratory distress syndrome), acute renal failure, fulminant hepatitis, burns, acute pancreatitis, post-surgical syndromes, sarcoidosis, Herxheimer reactions, encephalitis, myelitis, meningitis, malaria and SIRS associated with viral infections such as influenza, herpes zoster, herpes simplex and coronavirus. Combination Therapies In some embodiments, the compounds described herein may be administered in conjunction with standard of care, e.g., surgery, radiation, and/or chemotherapy. In some embodiments, the compounds may be administered in conjunction with a chemotherapeutic agent. In some embodiments, the compounds may be administered in conjunction with one or more of carboplatin, cisplatin, paclitaxel, nab-paclitaxel, gemcitabine or FOLFOX. In some embodiment, the compounds may be administered in conjunction with carboplatin or nab-paclitaxel. In some embodiments, the compounds may be administered in conjunction with carboplatin and paclitaxel. In some embodiments, the compounds may be administered in conjunction with cisplatin and pemetrexed. In some embodiments, the compounds may be administered in conjunction with cisplatin and gemcitabine. In some embodiments, the compounds may be administered in conjunction with FOLFOX. In some embodiments, the compounds may be administered in conjunction with FOLFIRI. In one embodiment, the compounds may be administered in combination with decarbazine for the treatment of melanoma. In some embodiments, cisplatin is intravenously administered as a 100 mg/ml dose once every four weeks. In some embodiments, the compounds may be administered in conjunction with doxorubicin (adriamycin), cisplatin bleomycin sulfate, carmustine, chlorambucil, dacarbazine and/or cyclophosphamide hydroxyurea. In some embodiments, adriamycin is intravenously administered as a 60 mg/ml to 75 mg/ml dose once every 21 days. In one embodiment, the compounds of the present application (e.g., a compound described herein or a pharmaceutically acceptable salt, prodrug, or solvate thereof) may be used in combination with one or more additional therapeutic agent that are being used and/or developed to treat cancers or inflammatory disorders. The one or more additional therapeutic agent may be an inhibitor to Janus kinase (JAK) such as JAK1, JAK2 and/or JAK3, Tyroansine kinase (TYK), K-Ras, Mitogen activated protein kinases (MAPK), Bruton's tyrosine kinase (BTK), bromodomain containing protein inhibitor (BRD) such as BRD4, a lysyl oxidase protein (LOX), lysyl oxidase-like protein (LOXL) such as LOXL1-5, matrix metalloprotease (MMP) such as MMP 1-10, adenosine A2B receptor (A2B), isocitrate dehydrogenase (IDH) such as IDH1, apoptosis signal-regulating kinase (ASK) such as ASK1, serine/threonine kinase TPL2, discoidin domain receptor (DDR) such as DDR1 and DDR2, histone deacetylase (HDAC), protein kinase C (PKC), or any combination thereof. In one embodiment, the compounds of the present application (e.g., a compound described herein or a pharmaceutically acceptable salt, prodrug, or solvate thereof) may be used in combination with additional chemotherapeutic agent, an immunotherapeutic agent, a radiotherapeutic agent, an anti-neoplastic agent, an anti-cancer agent, an anti- fibrotic agent, an anti-angiogenic agent, a therapeutic antibody, or any combination thereof. Chemotherapeutic agents may be categorized by their mechanism of action into, for example, the following groups: anti-metabolites/anti-cancer agents, such as pyrimidine analogs (floxuridine, capecitabine, and cytarabine); purine analogs, folate antagonists and related inhibitors antiproliferative/antimitotic agents including natural products such as vinca alkaloid (vinblastine, vincristine) and microtubule such as taxane (paclitaxel, docetaxel), vinblastin, nocodazole, epothilones and navelbine, epidipodophyllotoxins (etoposide, teniposide); DNA damaging agents (actinomycin, amsacrine, busulfan, carboplatin, chlorambucil, cisplatin, cyclophosphamide, Cytoxan, dactinomycin, daunorubicin, doxorubicin, epirubicin, iphosphamide, melphalan, merchlorehtamine, mitomycin, mitoxantrone, nitrosourea, procarbazine, taxol, taxotere, teniposide, etoposide, triethylenethiophosphoramide); antibiotics such as dactinomycin (actinomycin D), daunorubicin, doxorubicin (adriamycin), idarubicin, anthracyclines, mitoxantrone, bleomycins, plicamycin (mithramycin) and mitomycin; enzymes (L- asparaginase which systemically metabolizes L-asparagine and deprives cells which do not have the capacity to synthesize their own asparagine); antiplatelet agents; antiproliferative/antimitotic alkylating agents such as nitrogen mustards cyclophosphamide and analogs, melphalan, chlorambucil), and (hexamethylmelamine and thiotepa), alkyl nitrosoureas (BCNU) and analogs, streptozocin), trazenes-dacarbazinine (DTIC); antiproliferative/antimitotic antimetabolites such as folic acid analogs (methotrexate); platinum coordination complexes (cisplatin, oxiloplatinim, carboplatin), procarbazine, hydroxyurea, mitotane, aminoglutethimide; hormones, hormone analogs (estrogen, tamoxifen, goserelin, bicalutamide, nilutamide) and aromatase inhibitors (letrozole, anastrozole); anticoagulants (heparin, synthetic heparin salts and other inhibitors of thrombin); fibrinolytic agents (such as tissue plasminogen activator, streptokinase and urokinase), aspirin, dipyridamole, ticlopidine, clopidogrel; antimigratory agents; antisecretory agents (breveldin); immunosuppressives tacrolimus sirolimus azathioprine, mycophenolate; compounds (TNP-470, genistein) and growth factor inhibitors (vascular endothelial growth factor inhibitors, fibroblast growth factor inhibitors); angiotensin receptor blocker, nitric oxide donors; anti-sense oligonucleotides; antibodies (trastuzumab, rituximab); cell cycle inhibitors and differentiation inducers (tretinoin); inhibitors, topoisomerase inhibitors (doxorubicin (adriamycin), daunorubicin, dactinomycin, eniposide, epirubicin, etoposide, idarubicin, irinotecan and mitoxantrone, topotecan, irinotecan, camptothesin), corticosteroids (cortisone, dexamethasone, hydrocortisone, methylpednisolone, prednisone, and prenisolone); growth factor signal transduction kinase inhibitors; dysfunction inducers, toxins such as Cholera toxin, ricin, Pseudomonas exotoxin, Bordetella pertussis adenylate cyclase toxin, or diphtheria toxin, and caspase activators; and chromatin. Examples of chemotherapeutic agents include alkylating agents such as thiotepa and cyclophosphamide; alkyl sulfonates such as busulfan, improsulfan and piposulfan; aziridines such as benzodopa, carboquone, meturedopa, and uredopa; emylerumines and memylamelamines including alfretamine, triemylenemelamine, triethylenephosphoramide, triethylenethiophosphoramide and trimemylolomelamine; acetogenins (especially bullatacin and bullatacinone); a camptothecin (including synthetic analogue topotecan); bryostatin; callystatin; CC-1065 (including its adozelesin, carzelesin and bizelesin synthetic analogues); cryptophycins (articularly cryptophycin 1 and cryptophycin 8); dolastatin; duocarmycin (including the synthetic analogues, KW-2189 and CBI-TMI); eleutherobin; pancratistatin; a sarcodictyin; spongistatin; nitrogen mustards such as chlorambucil, chlornaphazine, cholophosphamide, estramustine, ifosfamide, mechlorethamine, mechlorethamine oxide hydrochloride, melphalan, novembichin, phenesterine, prednimustine, trofosfamide, uracil mustard; nitrosoureas such as carmustine, chlorozotocin, foremustine, lomustine, nimustine, ranimustine; antibiotics such as the enediyne antibiotics (e.g., calicheamicin, calicheamicin gammall, dynemicin, dynemicin A; bisphosphonates, such as clodronate; an esperamicin; as well as neocarzinostatin chromophore and related chromoprotein enediyne antibiotic chromomophores), aclacinomysins, actinomycin, authramycin, azaserine, bleomycins, cactinomycin, carabicin, carrninomycin, carzinophilin, chromomycins, dactinomycin, daunorubicin, detorubicin, 6-diazo-5-oxo-L-norleucine, doxorubicin (including morpholino-doxorubicin, cyanomorpholino-doxorubicin, 2-pyrrolino-doxorubicin and deoxydoxorubicin), epirubicin, esorubicin, idarubicin, marcellomycin, mitomycins such as mitomycin C, mycophenolic acid, nogalamycin, olivomycins, peplomycin, potfiromycin, puromycin, quelamycin, rodorubicin, streptonigrin, streptozocin, tubercidin, ubenimex, zinostatin, zorubicin; anti-metabolites such as methotrexate and 5- fluorouracil (5-FU); folic acid analogues such as demopterin, methotrexate, pteropterin, trimetrexate; purine analogs such as fludarabine, 6-mercaptopurine, thiamiprine, thioguanine; pyrimidine analogues such as ancitabine, azacitidine, 6-azauridine, carmofur, cytarabine, dideoxyuridine, doxifluridine, enocitabine, floxuridine; androgens such as calusterone, dromostanolone propionate, epitiostanol, mepitiostane, testolactone; anti-adrenals such as aminoglutethimide, mitotane, trilostane; folic acid replinisher such as frolinic acid; aceglatone; aldophosphamide glycoside; aminolevulinic acid; eniluracil; amsacrine; hestrabucil; bisantrene; edatraxate; defofamine; demecolcine; diaziquone; elformthine; elliptinium acetate; an epothilone; etoglucid; gallium nitrate; hydroxyurea; lentinan; leucovorin; lonidamine; maytansinoids such as maytansine and ansamitocins; mitoguazone; mitoxantrone; mopidamol; nitracrine; pentostatin; phenamet; pirarubicin; losoxantrone; fluoropyrimidine; folinic acid; podophyllinic acid; 2-ethylhydrazide; procarbazine; razoxane; rhizoxin; sizofiran; spirogermanium; tenuazonic acid; triaziquone; 2,2',2''-tricUorotriemylamine; trichothecenes (especially T-2 toxin, verracurin A, roridin A and anguidine); urethane; vindesine; dacarbazine; mannomustine; mitobronitol; mitolactol; pipobroman; gacytosine; arabinoside ("Ara-C"); cyclophosphamide; thiopeta; taxoids, e.g., paclitaxel and docetaxel, chlorambucil; gemcitabine; 6-thioguanine; mercaptopurine; methotrexate; platinum analogs such as cisplatin and carboplatin; vinblastine; platinum; etoposide; ifosfamide; mitroxantrone; vancristine; vinorelbine; novantrone; teniposide; edatrexate; daunomycin; aminopterin; xeoloda; ibandronate; CPT-11; topoisomerase inhibitor RFS 2000; difluoromethylornithine (DMFO); retinoids such as retinoic acid; capecitabine; FOLFIRI (fluorouracil, leucovorin, and irinotecan) and pharmaceutically acceptable salts, acids or derivatives of any of the above. One or more chemotherapeutic agent is used or included in the present application. Chemotherapeutic agents may also include, for example, anti-hormonal agents that act to regulate or inhibit hormone action on tumors such as anti-estrogens and selective estrogen receptor modulators (SERMs), including, for example, tamoxifen, raloxifene, droloxifene, 4-hydroxytamoxifen, trioxifene, keoxifene, onapristone, and toremifene; inhibitors of the enzyme aromatase, which regulates estrogen production in the adrenal glands, such as, for example, 4(5)-imidazoles, aminoglutethimide, megestrol acetate, exemestane, formestane, fadrozole, vorozole letrozole and anastrozole; and anti- androgens such as flutamide, nilutamide, bicalutamide, leuprohde, and goserelin; and pharmaceutically acceptable salts thereof. The anti-angiogenic agents include, but are not limited to, retinoid acid and derivatives thereof, 2-methoxyestradiol, suramin, squalamine, tissue inhibitor of metalloproteinase-1, tissue inhibitor of metalloproternase-2, plasminogen activator inhibitor-1, plasminogen activator inbibitor-2, cartilage-derived inhibitor, paclitaxel (nab- paclitaxel), platelet factor 4, protamine sulphate (clupeine), sulphated chitin derivatives (prepared from queen crab shells), sulphated polysaccharide peptidoglycan complex (sp- pg), staurosporine, modulators of matrix metabolism, including for example, proline analogs ((1-azetidine-2-carboxylic acid (LACA), cishydroxyproline, d,I-3,4- dehydroproline, thiaproline, .alpha.-dipyridyl, beta-aminopropionitrile fumarate, 4- propyl-5-(4-pyridinyl)-2(3h)-oxazolone; methotrexate, mitoxantrone, heparin, interferons, 2 macroglobulin-serum, chimp-3, chymostatin, beta-cyclodextrin tetradecasulfate, eponemycin; fumagillin, gold sodium thiomalate, d-penicillamine (CDPT), beta-1- anticollagenase-serum, alpba-2-antiplasmin, bisantrene, lobenzarit disodium, n-2- carboxyphenyl-4-chloroanthronilic acid disodium or "CCA", thalidomide; angiostatic steroid, cargboxynaminolmidazole; metalloproteinase inhibitors such as BB94. Other anti-angiogenesis agents include antibodies, preferably monoclonal antibodies against these angiogenic growth factors: beta-FGF, alpha-FGF, FGF-5, VEGF isoforms, VEGF- C, HGF/SF and Ang-1/Ang-2. The application also provides a method for treating a subject who is undergoing one or more standard therapies, such as chemotherapy, radiotherapy, immunotherapy, surgery, or combination thereof. Accordingly, one or more therapeutic agent or inhibitors may be administered before, during, or after administration of chemotherapy, radiotherapy, immunotherapy, surgery or combination thereof. In certain embodiments, the subject may be a human who is (i) substantially refractory to at least one chemotherapy treatment, or (ii) in relapse after treatment with chemotherapy, or both (i) and (ii). In some of embodiments, the subject is refractory to at least two, at least three, or at least four chemotherapy treatments (including standard or experimental chemotherapies). In certain embodiments, the subject is refractory to at least one, at least two, at least three, or at least four chemotherapy treatment (including standard or experimental chemotherapy) selected from fludarabine, rituximab, obinutuzumab, alkylating agents, alemtuzumab and other chemotherapy treatments such as CHOP (cyclophosphamide, doxorubicin, vincristine, prednisone); R-CHOP (rituximab-CHOP); hyperCVAD (hyperfractionated cyclophosphamide, vincristine, doxorubicin, dexamethasone, methotrexate, cytarabine); R-hyperCVAD (rituximab-hyperCVAD); FCM (fludarabine, cyclophosphamide, mitoxantrone); R-FCM (rituximab, fludarabine, cyclophosphamide, mitoxantrone); bortezomib and rituximab; temsirolimus and rituximab; temsirolimus and Velcade.RTM.; Iodine-131 tositumomab (Bexxar.RTM.) and CHOP; CVP (cyclophosphamide, vincristine, prednisone); R-CVP (rituximab-CVP); ICE (iphosphamide, carboplatin, etoposide); R-ICE (rituximab-ICE); FCR (fludarabine, cyclophosphamide, rituximab); FR (fludarabine, rituximab); and D.T. PACE (dexamethasone, thalidomide, cisplatin, Adriamycin.RTM., cyclophosphamide, etoposide). Examples of immunotherapeutic agents treating lymphoma or leukemia include, but are not limited to, rituximab (such as Rituxan), alemtuzumab (such as Campath, MabCampath), anti-CD19 antibodies, anti-CD20 antibodies, anti-MN-14 antibodies, anti- TRAIL, Anti-TRAIL DR4 and DR5 antibodies, anti-CD74 antibodies, apolizumab, bevacizumab, CHIR-12.12, epratuzumab (hLL2-anti-CD22 humanized antibody), galiximab, ha20, ibritumomab tiuxetan, lumiliximab, milatuzumab, ofatumumab, PRO131921, SGN-40, WT-1 analog peptide vaccine, WT1126-134 peptide vaccine, tositumomab, autologous human tumor-derived HSPPC-96, and veltuzumab. Additional immunotherapy agents includes using cancer vaccines based upon the genetic makeup of an individual patient's tumor, such as lymphoma vaccine GTOP-99. The therapeutic treatments can be supplemented or combined with any of the abovementioned therapies with stem cell transplantation or treatment. One example of modified approach is radioimmunotherapy, wherein a monoclonal antibody is combined with a radioisotope particle, such as indium In-111, yttrium Y-90, iodine I-131. Examples of combination therapies include, but are not limited to, Iodine-131 tositumomab, Yttrium-90 ibritumomab tiuxetan with CHOP. The compounds of the application can be used in combination with additional therapeutic procedures. Other therapeutic procedures include peripheral blood stem cell transplantation, autologous hematopoietic stem cell transplantation, autologous bone marrow transplantation, antibody therapy, biological therapy, enzyme inhibitor therapy, total body irradiation, infusion of stem cells, bone marrow ablation with stem cell support, in vitro-treated peripheral blood stem cell transplantation, umbilical cord blood transplantation, immunoenzyme technique, pharmacological study, low-LET cobalt-60 gamma ray therapy, bleomycin, conventional surgery, radiation therapy, and nonmyeloablative allogeneic hematopoietic stem cell transplantation. The compounds of the application can be used in combination with anti-fibrotic agents. The anti-fibrotic agents include, but are not limited to, emylenemamine, hydrazine, phenylhydrazine, and their derivatives, semicarbazide, and urea derivatives, aminonitriles, such as beta-aminopropionitrile (BAPN), or 2-nitroethylamine, unsaturated or saturated haloamines, such as 2-bromo-ethylamine, 2-chloroethylamine, 2- trifluoroethylamine, 3-bromopropylamine, p-halobenzylamines, selenohomocysteine lactone. Also, the anti-fibrotic agents are copper chelating agents, penetrating or not penetrating the cells. Exemplary compounds include indirect inhibitors such compounds blocking the aldehyde derivatives originating from the oxidative deamination of the lysyl and hydroxylysyl residues by the lysyl oxidases, such as the thiolamines, in particular D- penicillamine, or its analogues such as 2-amino-5-mercapto-5-methylhexanoic acid, D-2- amino-3-methyl-3-((2-acetamidoethyl)dithio)butanoic acid, p-2-amino-3-methyl-3-((2- aminoethyl)dithio)butanoic acid, sodium-4-((p-1-dimethyl-2-amino-2- carboxyethyl)dithio)butane sulphurate, 2-acetamidoethyl-2-acetamidoethanethiol sulphanate, sodium-4-mercaptobutanesulphinate trihydrate. The compounds of the application can be used in combination with immunotherapeutic and anti-inflammatory treatments. The immunotherapeutic agents include and are not limited to therapeutic antibodies suitable for treating patients; such as abagovomab, adecatumumab, afutuzumab, alemtuzumab, altumomab, amatuximab, anatumomab, arcitumomab, bavituximab, bectumomab, bevacizumab, bivatuzumab, blinatumomab, brentuximab, cantuzumab, catumaxomab, cetuximab, citatuzumab, cixutumumab, clivatuzumab, conatumumab, daratumumab, drozitumab, duligotumab, dusigitumab, detumomab, dacetuzumab, dalotuzumab, ecromeximab, elotuzumab, ensituximab, ertumaxomab, etaracizumab, farietuzumab, ficlatuzumab, figitumumab, flanvotumab, futuximab, ganitumab, gemtuzumab, girentuximab, glembatumumab, ibritumomab, igovomab, imgatuzumab, indatuximab, inotuzumab, intetumumab, ipilimumab, iratumumab, labetuzumab, lexatumumab, lintuzumab, lorvotuzumab, lucatumumab, mapatumumab, matuzumab, milatuzumab, minretumomab, mitumomab, moxetumomab, narnatumab, naptumomab, necitumumab, nimotuzumab, nofetumomabn, ocaratuzumab, ofatumumab, olaratumab, onartuzumab, oportuzumab, oregovomab, panitumumab, parsatuzumab, patritumab, pemtumomab, pertuzumab, pintumomab, pritumumab, racotumomab, radretumab, rilotumumab, rituximab, robatumumab, satumomab, sibrotuzumab, siltuximab, simtuzumab, solitomab, tacatuzumab, taplitumomab, tenatumomab, teprotumumab, tigatuzumab, tositumomab, trastuzumab, tucotuzumab, ublituximab, veltuzumab, vorsetuzumab, votumumab, zalutumumab, CC49 and 3F8. The exemplified therapeutic antibodies may be further labeled or combined with a radioisotope particle, such as indium In-111, yttrium Y-90, iodine I-131. In one aspect, the immuno-oncology agent is (i) an agonist of a stimulatory (including a co-stimulatory) receptor or (ii) an antagonist of an inhibitory (including a co- inhibitory) signal on T cells, both of which result in amplifying antigen-specific T cell responses (often referred to as immune checkpoint regulators). Certain of the stimulatory and inhibitory molecules are members of the immunoglobulin super family (IgSF). One important family of membrane-bound ligands that bind to co-stimulatory or co-inhibitory receptors is the B7 family, which includes B7- 1, B7-2, B7-H1 (PD-L1), B7-DC (PD-L2), B7-H2 (ICOS-L), B7-H3, B7-H4, B7-H5 (VISTA), and B7-H6. Another family of membrane bound ligands that bind to co- stimulatory or co-inhibitory receptors is the TNF family of molecules that bind to cognate TNF receptor family members, which includes CD40 and CD40L, OX-40, OX-40L, CD70, CD27L, CD30, CD30L, 4-1BBL, CD137 (4-1BB), TRAIL/Apo2-L, TRAILR1/DR4, TRAILR2/DR5, TRAILR3, TRAILR4, OPG, RANK, RANKL, TWEAKR/Fn14, TWEAK, BAFFR, EDAR, XEDAR, TACI, APRIL, BCMA, LTȕR, LIGHT, DcR3, HVEM, VEGI/TL1A, TRAMP/DR3, EDAR, EDA1, XEDAR, EDA2, TNFR1, Lymphotoxin Į/TNFȕ, TNFR2, TNFĮ, LTȕR, Lymphotoxin Į 1ȕ2, FAS, FASL, RELT, DR6, TROY, NGFR. In one aspect, T cell responses can be stimulated by a combination of a compound of Formula (I) and one or more of (i) an antagonist of a protein that inhibits T cell activation (e.g., immune checkpoint inhibitors) such as CTLA-4, PD-1, PD-L1, PD-L2, LAG-3, TIM-3, Galectin 9, CEACAM-1, BTLA, CD69, Galectin-1, TIGIT, CD113, GPR56, VISTA, 2B4, CD48, GARP, PD1H, LAIR1, TIM-1, and TIM-4, and (ii) an agonist of a protein that stimulates T cell activation such as B7-1, B7-2, CD28, 4-1BB (CD137), 4-1BBL, ICOS, ICOS-L, OX40, OX40L, GITR, GITRL, CD70, CD27, CD40, DR3 and CD28H. Other agents that can be combined with compounds described herein for the treatment of cancer include antagonists of inhibitory receptors on NK cells or agonists of activating receptors on NK cells. For example, compounds described herien can be combined with antagonists of KIR, such as lirilumab. Yet other agents for combination therapies include agents that inhibit or deplete macrophages or monocytes, including but not limited to CSF-1R antagonists such as CSF-1R antagonist antibodies including RG7155 (WO11/70024, WO11/107553, In another aspect, compounds of the present application can be used with one or more of agonistic agents that ligate positive costimulatory receptors, blocking agents that attenuate signaling through inhibitory receptors, antagonists, and one or more agents that increase systemically the frequency of anti-tumor T cells, agents that overcome distinct immune suppressive pathways within the tumor microenvironment (e.g., block inhibitory receptor engagement (e.g., PD-L1/PD-1 interactions), deplete or inhibit Tregs (e.g., using an anti-CD25 monoclonal antibody (e.g., daclizumab) or by ex vivo anti-CD25 bead depletion), inhibit metabolic enzymes such as IDO, or reverse/prevent T cell anergy or exhaustion) and agents that trigger innate immune activation and/or inflammation at tumor sites. In one aspect, the immuno-oncology agent is a CTLA-4 antagonist, such as an antagonistic CTLA-4 antibody. Suitable CTLA-4 antibodies include, for example, YERVOY (ipilimumab) or tremelimumab. In another aspect, the immuno-oncology agent is a PD-1 antagonist, such as an antagonistic PD-1 antibody. Suitable PD-1 antibodies include, for example, OPDIVO (nivolumab), KEYTRUDA (pembrolizumab), or MEDI-0680 (AMP-514; WO2012/145493). The immuno-oncology agent may also include pidilizumab (CT-011), though its specificity for PD-1 binding has been questioned. Another approach to target the PD-1 receptor is the recombinant protein composed of the extracellular domain of PD-L2 (B7-DC) fused to the Fc portion of IgG1, called AMP-224 In another aspect, the immuno-oncology agent is a PD-L1 antagonist, such as an antagonistic PD-L1 antibody. Suitable PD-L1 antibodies include, for example, MPDL3280A (RG7446; WO2010/077634), durvalumab (MEDI4736), BMS-936559 (WO2007/005874), and MSB0010718C (WO2013/79174). In another aspect, the immuno-oncology agent is a LAG-3 antagonist, such as an antagonistic LAG-3 antibody. Suitable LAG3 antibodies include, for example, BMS- 986016 (WO10/19570, WO14/08218), or IMP-731 or IMP-321 (WO08/132601, WO09/44273). In another aspect, the immuno-oncology agent is a CD137 (4-1BB) agonist, such as an agonistic CD137 antibody. Suitable CD137 antibodies include, for example, urelumab and PF-05082566 (WO12/32433). In another aspect, the immuno-oncology agent is a GITR agonist, such as an agonistic GITR antibody. Suitable GITR antibodies include, for example, BMS-986153, BMS-986156, TRX-518 (WO06/105021, WO09/009116) and MK-4166 (WO11/028683). In another aspect, the immuno-oncology agent is an IDO antagonist. Suitable IDO antagonists include, for example, INCB-024360 (WO2006/122150, WO07/75598, WO08/36653, WO08/36642), indoximod, BMS-986205, or NLG-919 (WO09/73620, WO09/1156652, WO11/56652, WO12/142237). In another aspect, the immuno-oncology agent is an OX40 agonist, such as an agonistic OX40 antibody. Suitable OX40 antibodies include, for example, MEDI-6383 or MEDI-6469. In another aspect, the immuno-oncology agent is an OX40L antagonist, such as an antagonistic OX40 antibody. Suitable OX40L antagonists include, for example, RG-7888 (WO06/029879). In another aspect, the immuno-oncology agent is a CD40 agonist, such as an agonistic CD40 antibody. In yet another embodiment, the immuno-oncology agent is a CD40 antagonist, such as an antagonistic CD40 antibody. Suitable CD40 antibodies include, for example, lucatumumab or dacetuzumab. In another aspect, the immuno-oncology agent is a CD47 antagonist, such as a CD47 antagonist selected from the group MIAP301, MIAP410, TTI-621, CV1, Hu5F9- G4, CC-90002, B6H12 and 2D3. In another aspect, the immuno-oncology agent is a CD27 agonist, such as an agonistic CD27 antibody. Suitable CD27 antibodies include, for example, varlilumab. In another aspect, the immuno-oncology agent is MGA271 (to B7H3) (WO11/109400). The combination therapy is intended to embrace administration of these therapeutic agents in a sequential manner, that is, wherein each therapeutic agent is administered at a different time, as well as administration of these therapeutic agents, or at least two of the therapeutic agents, in a substantially simultaneous manner. Substantially simultaneous administration can be accomplished, for example, by administering to the subject a single dosage form having a fixed ratio of each therapeutic agent or in multiple, single dosage forms for each of the therapeutic agents. Sequential or substantially simultaneous administration of each therapeutic agent can be effected by any appropriate route including, but not limited to, oral routes, intravenous routes, intramuscular routes, and direct absorption through mucous membrane tissues. The therapeutic agents can be administered by the same route or by different routes. For example, a first therapeutic agent of the combination selected may be administered by intravenous injection while the other therapeutic agents of the combination may be administered orally. Alternatively, for example, all therapeutic agents may be administered orally or all therapeutic agents may be administered by intravenous injection. Combination therapy also can embrace the administration of the therapeutic agents as described above in further combination with other biologically active ingredients and non-drug therapies (e.g., surgery or radiation treatment.) Where thwe combination therapy further comprises a non drug treatment, the non drug treatment may be conducted at any suitable time so long as a beneficial effect from the co-action of the combination of the therapeutic agents and non-drug treatment is achieved. For example, in appropriate cases, the beneficial effect is still achieved when the non drug treatment is temporally removed from the administration of the therapeutic agents, perhaps by days or even weeks. The present invention also provides the compounds of the present invention for use in therapy. In another embodiment, compounds of Formula I are selected from exemplified compounds or combinations of exemplified compounds or other embodiments herein. In another embodiment are compounds having an IC50 < 1000 nM in at least one of the assays described below. The present invention may be embodied in other specific forms without departing from the spirit or essential attributes thereof. This invention encompasses all combinations of preferred aspects and/or embodiments of the invention noted herein. It is understood that any and all embodiments of the present invention may be taken in conjunction with any other embodiment or embodiments to describe additional more preferred embodiments. It is also to be understood that each individual element of the preferred embodiments is its own independent preferred embodiment. Furthermore, any element of an embodiment is meant to be combined with any and all other elements from any embodiment to describe an additional embodiment. METHODS OF PREPARATION The compounds of the present invention can be prepared in a number of ways well known to one skilled in the art of organic synthesis. The compounds of the present invention can be synthesized using the methods described below, together with synthetic methods known in the art of synthetic organic chemistry, or variations thereon as appreciated by those skilled in the art. Preferred methods include, but are not limited to, those described below. All references cited herein are hereby incorporated in their entirety by reference. The compounds of this invention may be prepared using the reactions and techniques described in this section. The reactions are performed in solvents appropriate to the reagents and materials employed and are suitable for the transformations being effected. Also, in the description of the synthetic methods described below, it is to be understood that all proposed reaction conditions, including choice of solvent, reaction atmosphere, reaction temperature, duration of the experiment and work up procedures, are chosen to be the conditions standard for that reaction, which should be readily recognized by one skilled in the art. It is understood by one skilled in the art of organic synthesis that the functionality present on various portions of the molecule must be compatible with the reagents and reactions proposed. Such restrictions to the substituents that are compatible with the reaction conditions will be readily apparent to one skilled in the art and alternate methods must then be used. This will sometimes require a judgment to modify the order of the synthetic steps or to select one particular process scheme over another in order to obtain a desired compound of the invention. It will also be recognized that another major consideration in the planning of any synthetic route in this field is the judicious choice of the protecting group used for protection of the reactive functional groups present in the compounds described in this invention. An authoritative account describing the many alternatives to the trained practitioner is Greene and Wuts (Protective Groups In Organic Synthesis, Third Edition, Wiley and Sons, 1999). EXAMPLES Preparation of compounds of Formula I and intermediates used in the preparation of compounds of Formula I can be prepared using procedures shown in the following Examples and related procedures. The methods and conditions used in these examples, and the actual compounds prepared in these Examples, are not meant to be limiting, but are meant to demonstrate how the compounds of Formula I can be prepared. Starting materials and reagents used in these examples, when not prepared by a procedure described herein, are generally either commercially available, or are reported in the chemical literature, or may be prepared by using procedures described in the chemical literature. EXAMPLES The following examples illustrate the particular and preferred embodiments of the present invention and do not limit the scope of the present invention. Chemical abbreviations and symbols as well as scientific abbreviations and symbols have their usual and customary meanings unless otherwise specified. Common intermediates are generally useful for the preparation of more than one Example as shown in the Tables. Chemical names were determined using ChemBioDraw Ultra, version 14.0.0.126 (CambridgeSoft). The following abbreviations are used: ABBREVIATIONNAME Boctert-butyloxycarbonyl CDCl3deuterated chloroform DASTdiethylaminosulfur trifluoride DCMdichloromethane DIBAL-H diisobutylaluminium hydride DMF N,N-dimethylformamide DMSO dimethyl sulfoxide DMSO-d6 deuterated dimethyl sulfoxide Et3N triethylamine EtOAc ethyl acetate H hours HATU O-(7-azabenzotriazol- l-yl)-1, 1,3,3-tetramethyluronium hexafluorophosphateABBREVIATIONNAME HPLC high performance liquid chromatography LC liquid chromatography LCMS liquid chromatography – mass spectrometry MeCN acetonitrile MeOH methanol MeOH-d4 deuterated methanol mCPBA meta-chloroperoxybenzoic acid Min minutes MsCl methanesulfonyl chloride NH4OAc ammonium acetate PyBOP benzotriazol-1-yl- oxytripyrrolidinophosphonium hexafluorophosphate Rt room temperature SFC super-critical fluid chromatography TFA trifluoroacetic acid THF tetrahydrofuran TLC thin layer chromatography UPLC ultra performance liquid chromatography LCMS-Method 1: COLUMN: Kinetex XB-C18(75X3.0) mm, 2.6um Mobile phase A: 5mm Ammonium formate pH 3.3: Acetonitrile (98:2) Mobile phase B: ACN :5mm Ammonium formate pH 3.3 (98:2) Flow rate: 1 ml/min Temperature: 25° C GRADIENT: 20%B to 100%B over 4min, 1.5 ml/min flow at 0.5 min hold at 100%B MS and UV: 220 & 254 LCMS-Method 2: COLUMN: Kinetex -C18(75X3.0) mm, 2.6um Mobile phase A: 0.1% TFA in water Mobile phase B: 0.1% TFA in Acetonitrile Flow rate: 1 ml/min Temperature: 25° C GRADIENT: 05%B to 95%B over 2.5min, 1.5 ml\min flow at 1.49min hold at 95%B MS and UV: 220 & 254 nm LCMS-Method 3: COLUMN: Acquity BEH C18(50X3.0) mm, 1.7um Mobile phase A: 5mm Ammonium formate pH 3.3: Acetonitrile (98:2) Mobile phase B: ACN :5mm Ammonium formate pH 3.3 (98:2) Flow rate: 0.7 ml/min Temperature: 25° C GRADIENT: 20%B to 98%B over 1.5min, 0.5 min hold at 98%B MS and UV: 220 nm LCMS-Method 4: COLUMN: Acquity Uplc BEH C18(50X3.0) mm, 1.7um Mobile phase A: 5mm Ammonium formate pH 3.3: Acetonitrile (98:2) Mobile phase B: ACN :5mm Ammonium formate pH 3.3 (98:2) Flow rate: 0.7 ml/min Temperature: 25° C GRADIENT: 20%B to 98%B over 1.5min, 0.5 min hold at 98%B MS and UV: 220 nm
, (e.g., cyclohexyl), heterocyclo (e.g., pyrrolidinyl, piperidinyl, and morpholinyl) or heteroaryl (e.g., tetrazolyl, imidazolyl, pyrazolyl, triazolyl, thiazolyl, and furyl) the reference is intended to include rings having 0 to 3, preferably 0 to 2, substituents selected from those recited above for the aryl, cycloalkyl, heterocyclo and/or heteroaryl groups, as appropriate. The term "carbocyclyl" or "carbocyclic" refers to a saturated or unsaturated monocyclic or bicyclic ring in which all atoms of all rings are carbon. Thus, the term includes cycloalkyl and aryl rings. Monocyclic carbocycles have 3 to 6 ring atoms, still more typically 5 or 6 ring atoms. Bicyclic carbocycles have 7 to 12 ring atoms, e.g., arranged as a bicyclo [4,5], [5,5], [5,6] or [6,6] system, or 9 or 10 ring atoms arranged as a bicyclo [5,6] or [6,6] system. Examples of mono- and bicyclic carbocycles include cyclopropyl, cyclobutyl, cyclopentyl, 1-cyclopent-1-enyl, 1-cyclopent-2-enyl, 1- cyclopent-3-enyl, cyclohexyl, 1-cyclohex-1-enyl, 1-cyclohex-2-enyl, 1-cyclohex-3-enyl, phenyl and naphthyl. The carbocyclic ring may be substituted in which case the substituents are selected from those recited above for cycloalkyl and aryl groups. The term "alkylthio" refers to the group "alkyl-S-". The term "acyl" refers to a group -C(O)R, wherein R is hydrogen, alkyl, cycloalkyl, heterocyclyl, aryl, heteroalkyl, or heteroaryl; each of which may be optionally substituted, as defined herein. Examples of acyl include formyl, acetyl, cylcohexylcarbonyl, cyclohexylmethyl-carbonyl, and benzoyl. The term "amido" refers to both a "C-amido" group which refers to the group -- C(O)NRgRh and an "N-amido" group which refers to the group –NRgC(O)Rh, wherein Rg and Rh are independently selected from hydrogen, alkyl, aryl, haloalkyl, or heteroaryl; each of which may be optionally substituted. The term "amino" refers to the group -NRg Rh wherein Rg and Rh are independently selected from hydrogen, alkyl, haloalkyl, aryl, or heteroaryl; each of which may be optionally substituted. The term "azido" refers to –N3. The term "carbamoyl" refers to both an "O-carbamoyl" group which refers to the group -O-C(O)NRiRj and an "N-carbamoyl" group which refers to the group – NRiC(O)ORj, wherein Ri and Rj are independently selected from hydrogen, alkyl, aryl, haloalkyl, or heteroaryl; each of which may be optionally substituted. The term "carboxyl" refers to -C(O)OH. The term "carboxyl ester" refers to both -OC(O)R and -C(O)ORg, wherein Rg is hydrogen, alkyl, cycloalkyl, heterocyclyl, aryl, heteroalkyl, or heteroaryl; each of which may be optionally substituted, as defined herein. The term "cyano" or "carbonitrile" refers to the group -CN. The term "cycloalkyl" refers to a saturated or partially unsaturated cyclic alkyl group having a single ring or multiple rings including fused, bridged, and spiro ring systems. The term "cycloalkyl" includes cycloalkenyl groups (i.e. the cyclic group having at least one double bond). As used herein, cycloalkyl has from 3 to 20 ring carbon atoms (i.e., C.sub.3-20 cycloalkyl), 3 to 12 ring carbon atoms (i.e., C.sub.3-12 cycloalkyl), 3 to 10 ring carbon atoms (i.e., C.sub.3-10 cycloalkyl), 3 to 8 ring carbon atoms (i.e., C.sub.3- 8 cycloalkyl), or 3 to 6 ring carbon atoms (i.e., C.sub.3-6 cycloalkyl). Examples of cycloalkyl groups include cyclopropyl, cyclobutyl, cyclopentyl, and cyclohexyl. The term "heteroatoms" shall include oxygen, sulfur and nitrogen. When the term "unsaturated" is used herein to refer to a ring or group, the ring or group may be fully unsaturated or partially unsaturated. Throughout the specification, groups and substituents thereof may be chosen by one skilled in the field to provide stable moieties and compounds and compounds useful as pharmaceutically-acceptable compounds and/or intermediate compounds useful in making pharmaceutically-acceptable compounds. It should be understood that the selections for all groups, including for example, alkoxy, thioalkyl, and aminoalkyl, will be made by one skilled in the field to provide stable compounds. The term "substituted", as used herein, means that any one or more hydrogens on the designated atom or group is replaced with a selection from the indicated group, provided that the designated atom's normal valence is not exceeded. When a substituent is oxo, or keto, (i.e., =O) then 2 hydrogens on the atom are replaced. Keto substituents are not present on aromatic moieties. Unless otherwise specified, substituents are named into the core structure. For example, it is to be understood that when (cycloalkyl)alkyl is listed as a possible substituent, the point of attachment of this substituent to the core structure is in the alkyl portion. Ring double bonds, as used herein, are double bonds that are formed between two adjacent ring atoms (e.g., C=C, C=N, or N=N). Combinations of substituents and/or variables are permissible only if such combinations result in stable compounds or useful synthetic intermediates. A stable compound or stable structure is meant to imply a compound that is sufficiently robust to survive isolation from a reaction mixture to a useful degree of purity, and subsequent formulation into an efficacious therapeutic agent. It is preferred that the presently recited compounds do not contain a N-halo, S(O)2H, or S(O)H group. The compounds described herein may exist in a free form (with no ionization) or can form salts which are also within the scope of this invention. Unless otherwise indicated, reference to an inventive compound is understood to include reference to the free form and to salts thereof. The term "salt(s)" denotes acidic and/or basic salts formed with inorganic and/or organic acids and bases. In addition, the term "salt(s)" may include zwitterions (inner salts), e.g., when a compound of formula I, contains both a basic moiety, such as an amine or a pyridine or imidazole ring, and an acidic moiety, such as a carboxylic acid. Pharmaceutically acceptable (i.e., non-toxic, physiologically acceptable) salts are preferred, such as, for example, acceptable metal and amine salts in which the cation does not contribute significantly to the toxicity or biological activity of the salt. However, other salts may be useful, e.g., in isolation or purification steps which may be employed during preparation, and thus, are contemplated within the scope of the invention. Salts of the compounds of herein may be formed, for example, by reacting a compound described herein with an amount of acid or base, such as an equivalent amount, in a medium such as one in which the salt precipitates or in an aqueous medium followed by lyophilization. Exemplary acid addition salts include acetates (such as those formed with acetic acid or trihaloacetic acid, for example, trifluoroacetic acid), adipates, alginates, ascorbates, aspartates, benzoates, benzenesulfonates, bisulfates, borates, butyrates, citrates, camphorates, camphorsulfonates, cyclopentanepropionates, digluconates, dodecylsulfates, ethanesulfonates, fumarates, glucoheptanoates, glycerophosphates, hemisulfates, heptanoates, hexanoates, hydrochlorides (formed with hydrochloric acid), hydrobromides (formed with hydrogen bromide), hydroiodides, 2- hydroxyethanesulfonates, lactates, maleates (formed with maleic acid), methanesulfonates (formed with methanesulfonic acid), 2-naphthalenesulfonates, nicotinates, nitrates, oxalates, pectinates, persulfates, 3-phenylpropionates, phosphates, picrates, pivalates, propionates, salicylates, succinates, sulfates (such as those formed with sulfuric acid), sulfonates (such as those mentioned herein), tartrates, thiocyanates, toluenesulfonates such as tosylates, undecanoates, and the like. Exemplary basic salts include ammonium salts, alkali metal salts such as sodium, lithium, and potassium salts; alkaline earth metal salts such as calcium and magnesium salts; barium, zinc, and aluminum salts; salts with organic bases (for example, organic amines) such as trialkylamines such as triethylamine, procaine, dibenzylamine, N-benzyl- ȕ-phenethylamine, 1-ephenamine, N,Nƍ-dibenzylethylene-diamine, dehydroabietylamine, N-ethylpiperidine, benzylamine, dicyclohexylamine or similar pharmaceutically acceptable amines and salts with amino acids such as arginine, lysine and the like. Basic nitrogen-containing groups may be quaternized with agents such as lower alkyl halides (e.g., methyl, ethyl, propyl, and butyl chlorides, bromides and iodides), dialkyl sulfates (e.g., dimethyl, diethyl, dibutyl, and diamyl sulfates), long chain halides (e.g., decyl, lauryl, myristyl and stearyl chlorides, bromides and iodides), aralkyl halides (e.g., benzyl and phenethyl bromides), and others. Preferred salts include monohydrochloride, hydrogensulfate, methanesulfonate, phosphate or nitrate salts. The compounds described herein can be provided as amorphous solids or crystalline solids. Lyophilization can be employed to provide the compounds as a solid. It should further be understood that solvates (e.g., hydrates) of the compounds described herein are also within the scope of the present invention. The term “solvate” means a physical association of a compound with one or more solvent molecules, whether organic or inorganic. This physical association includes hydrogen bonding. In certain instances, the solvate will be capable of isolation, for example when one or more solvent molecules are incorporated in the crystal lattice of the crystalline solid. “Solvate” encompasses both solution-phase and isolable solvates. Exemplary solvates include hydrates, ethanolates, methanolates, isopropanolates, acetonitrile solvates, and ethyl acetate solvates. Methods of solvation are known in the art. In addition, compounds described herein subsequent to their preparation, can be isolated and purified to obtain a composition containing an amount by weight equal to or greater than 99% of a compound (“substantially pure”), which is then used or formulated as described herein. Such “substantially pure” compounds described herein are also contemplated herein as part of the present invention. The phrase "pharmaceutically acceptable" is employed herein to refer to those compounds, materials, compositions, and/or dosage forms which are, within the scope of sound medical judgment, suitable for use in contact with the tissues of human beings and animals without excessive toxicity, irritation, allergic response, or other problem or complication, commensurate with a reasonable benefit/risk ratio. As used herein, "pharmaceutically acceptable salts" refer to derivatives of the disclosed compounds wherein the parent compound is modified by making acid or base salts thereof. Examples of pharmaceutically-acceptable salts include, but are not limited to, mineral or organic acid salts of basic groups such as amines; and alkali or organic salts of acidic groups such as carboxylic acids. The pharmaceutically-acceptable salts include the conventional non-toxic salts or the quaternary ammonium salts of the parent compound formed, for example, from non-toxic inorganic or organic acids. For example, such conventional non-toxic salts include those derived from inorganic acids such as hydrochloric, hydrobromic, sulfuric, sulfamic, phosphoric, and nitric; and the salts prepared from organic acids such as acetic, propionic, succinic, glycolic, stearic, lactic, malic, tartaric, citric, ascorbic, pamoic, maleic, hydroxymaleic, phenylacetic, glutamic, benzoic, salicylic, sulfanilic, 2-acetoxybenzoic, fumaric, toluenesulfonic, methanesulfonic, ethane disulfonic, oxalic, and isethionic, and the like. The pharmaceutically acceptable salts of the present invention can be synthesized from the parent compound which contains a basic or acidic moiety by conventional chemical methods. Generally, such salts can be prepared by reacting the free acid or base forms of these compounds with a stoichiometric amount of the appropriate base or acid in water or in an organic solvent, or in a mixture of the two; generally, nonaqueous media like ether, ethyl acetate, ethanol, isopropanol, or acetonitrile are preferred. Lists of suitable salts are found in Remington's Pharmaceutical Sciences, 18th Edition, Mack Publishing Company, Easton, PA (1990), the disclosure of which is hereby incorporated by reference. “Stable compound” and “stable structure” are meant to indicate a compound that is sufficiently robust to survive isolation to a useful degree of purity from a reaction mixture, and formulation into an efficacious therapeutic agent. The present invention is intended to embody stable compounds. “Therapeutically effective amount” is intended to include an amount of a compound of the present invention alone or an amount of the combination of compounds claimed or an amount of a compound of the present invention in combination with other active ingredients effective to act as an inhibitor of USP1, or effective to treat or prevent proliferative disorders, such as cancer. As used herein, “treating” or “treatment” cover the treatment of a disease-state in a mammal, particularly in a human, and include: (a) preventing the disease-state from occurring in a mammal, in particular, when such mammal is predisposed to the disease- state but has not yet been diagnosed as having it; (b) inhibiting the disease-state, i.e., arresting its development; and/or (c) relieving the disease-state, i.e., causing regression of the disease state. All stereoisomers of the compounds of the instant invention are contemplated, either in admixture or in pure or substantially pure form. Stereoisomers may include compounds which are optical isomers through possession of one or more chiral atoms, as well as compounds which are optical isomers by virtue of limited rotation about one or more bonds (atropisomers). The definition of compounds according to the invention embraces all the possible stereoisomers and their mixtures. It very particularly embraces the racemic forms and the isolated optical isomers having the specified activity. The racemic forms can be resolved by physical methods, such as, for example, fractional crystallization, separation or crystallization of diastereomeric derivatives or separation by chiral column chromatography. The individual optical isomers can be obtained from the racemates from the conventional methods, such as, for example, salt formation with an optically active acid followed by crystallization. The present invention is intended to include all isotopes of atoms occurring in the present compounds. Isotopes include those atoms having the same atomic number but different mass numbers. By way of general example and without limitation, isotopes of hydrogen include deuterium and tritium. Isotopes of carbon include13C and14C. Isotopically-labeled compounds of the invention can generally be prepared by conventional techniques known to those skilled in the art or by processes analogous to those described herein, using an appropriate isotopically-labeled reagent in place of the non-labeled reagent otherwise employed. Prodrugs and solvates of the inventive compounds are also contemplated. The term "prodrug" denotes a compound which, upon administration to a subject, undergoes chemical conversion by metabolic or chemical processes to yield a compound of the formula I, and/or a salt and/or solvate thereof. Any compound that will be converted in vivo to provide the bioactive agent (i.e., the compound for formula I) is a prodrug within the scope and spirit of the invention. For example, compounds containing a carboxy group can form physiologically hydrolyzable esters which serve as prodrugs by being hydrolyzed in the body to yield formula I compounds per se. Such prodrugs are preferably administered orally since hydrolysis in many instances occurs principally under the influence of the digestive enzymes. Parenteral administration may be used where the ester per se is active, or in those instances where hydrolysis occurs in the blood. Examples of physiologically hydrolyzable esters of compounds of formula I include C1-6alkylbenzyl, 4-methoxybenzyl, indanyl, phthalyl, methoxymethyl, C1-6alkanoyloxy-C1-6alkyl, e.g., acetoxymethyl, pivaloyloxymethyl or propionyloxymethyl, C1-6alkoxycarbonyloxy-C1-6alkyl, e.g., methoxycarbonyl-oxymethyl or ethoxycarbonyloxymethyl, glycyloxymethyl, phenylglycyloxymethyl, (5-methyl-2- oxo-1,3-dioxolen-4-yl)-methyl and other well known physiologically hydrolyzable esters used, for example, in the penicillin and cephalosporin arts. Such esters may be prepared by conventional techniques known in the art. Various forms of prodrugs are well known in the art and are described in Rautio, J. et al., Nature Review Drug Discovery, 17, 559-587 (2018). Compounds described in the application and their salts may exist in their tautomeric form, in which hydrogen atoms are transposed to other parts of the molecules and the chemical bonds between the atoms of the molecules are consequently rearranged. It should be understood that the all tautomeric forms, insofar as they may exist, are included within the invention. Additionally, inventive compounds may have trans- and cis-isomers. The disclosure herein further relates to compounds described herein, the tautomers and stereoisomeric forms thereof, and the pharmaceutically acceptable addition salts, and the solvates thereof, for use as a medicament. Furthermore, the disclosure herein relates to the use of a compound described herein, a tautomer or a stereoisomeric form thereof, or a pharmaceutically acceptable addition salt, or a solvate thereof, or a pharmaceutical composition according to the invention, for the manufacture of a medicament. The inventive compositions may contain other therapeutic agents as described above and may be formulated, for example, by employing conventional solid or liquid vehicles or diluents, as well as pharmaceutical additives of a type appropriate to the mode of desired administration (e.g., excipients, binders, preservatives, stabilizers, flavors, etc.) according to techniques such as those well known in the art of pharmaceutical formulation. Accordingly, the present invention further includes compositions comprising one or more compounds described herein and a pharmaceutically acceptable carrier. A "pharmaceutically acceptable carrier" refers to media generally accepted in the art for the delivery of biologically active agents to animals, in particular, mammals. Pharmaceutically acceptable carriers are formulated according to a number of factors well within the purview of those of ordinary skill in the art. These include without limitation the type and nature of the active agent being formulated; the subject to which the agent- containing composition is to be administered; the intended route of administration of the composition; and, the therapeutic indication being targeted. Pharmaceutically acceptable carriers include both aqueous and non-aqueous liquid media, as well as a variety of solid and semi-solid dosage forms. Such carriers can include a number of different ingredients and additives in addition to the active agent, such additional ingredients being included in the formulation for a variety of reasons, e.g., stabilization of the active agent, binders, etc., well known to those of ordinary skill in the art. Descriptions of suitable pharmaceutically acceptable carriers, and factors involved in their selection, are found in a variety of readily available sources such as, for example, Remington's Pharmaceutical Sciences, 17th Edition (1985), which is incorporated herein by reference in its entirety. The compounds described herein may be administered by any means suitable for the condition to be treated, which may depend on the need for site-specific treatment or quantity of drug to be delivered. Topical administration is generally preferred for skin- related diseases, and systematic treatment preferred for cancerous or pre-cancerous conditions, although other modes of delivery are contemplated. For example, the compounds may be delivered orally, such as in the form of tablets, capsules, granules, powders, or liquid formulations including syrups; topically, such as in the form of solutions, suspensions, gels or ointments; sublingually; bucally; parenterally, such as by subcutaneous, intravenous, intramuscular or intrasternal injection or infusion techniques (e.g., as sterile injectable aq. or non-aq. solutions or suspensions); nasally such as by inhalation spray; topically, such as in the form of a cream or ointment; rectally such as in the form of suppositories; or liposomally. Dosage unit formulations containing non-toxic, pharmaceutically acceptable vehicles or diluents may be administered. The compounds may be administered in a form suitable for immediate release or extended release. Immediate release or extended release may be achieved with suitable pharmaceutical compositions or, particularly in the case of extended release, with devices such as subcutaneous implants or osmotic pumps. Exemplary compositions for oral administration include suspensions which may contain, for example, microcrystalline cellulose for imparting bulk, alginic acid or sodium alginate as a suspending agent, methylcellulose as a viscosity enhancer, and sweeteners or flavoring agents such as those known in the art; and immediate release tablets which may contain, for example, microcrystalline cellulose, dicalcium phosphate, starch, magnesium stearate and/or lactose and/or other excipients, binders, extenders, disintegrants, diluents and lubricants such as those known in the art. The inventive compounds may also be orally delivered by sublingual and/or buccal administration, e.g., with molded, compressed, or freeze-dried tablets. Exemplary compositions may include fast-dissolving diluents such as mannitol, lactose, sucrose, and/or cyclodextrins. Also included in such formulations may be high molecular weight excipients such as celluloses (AVICEL®) or polyethylene glycols (PEG); an excipient to aid mucosal adhesion such as hydroxypropyl cellulose (HPC), hydroxypropyl methyl cellulose (HPMC), sodium carboxymethyl cellulose (SCMC), and/or maleic anhydride copolymer (e.g., GANTREZ®); and agents to control release such as polyacrylic copolymer (e.g., CARBOPOL 934®). Lubricants, glidants, flavors, coloring agents and stabilizers may also be added for ease of fabrication and use. Formulations for parenteral administration may be in the form of aqueous or non- aqueous isotonic sterile injection solutions or suspensions. These solutions and suspensions may be prepared from sterile powders or granules using one or more of the carriers or diluents mentioned for use in the formulations for oral administration or by using other suitable dispersing or wetting agents and suspending agents. The compounds may be dissolved in water, polyethylene glycol, propylene glycol, ethanol, corn oil, cottonseed oil, peanut oil, sesame oil, benzyl alcohol, sodium chloride, tragacanth gum, and/or various buffers. Other adjuvants and modes of administration are well and widely known in the pharmaceutical art. The active ingredient may also be administered by injection as a composition with suitable carriers including saline, dextrose, or water, or with cyclodextrin (i.e. Captisol), cosolvent solubilization (i.e. propylene glycol) or micellar solubilization (i.e. Tween 80). Exemplary compositions for parenteral administration include injectable solutions or suspensions which may contain, for example, suitable non-toxic, parenterally acceptable diluents or solvents, such as mannitol, 1,3-butanediol, water, Ringer's solution, an isotonic sodium chloride solution, or other suitable dispersing or wetting and suspending agents, including synthetic mono- or diglycerides, and fatty acids, including oleic acid. The sterile injectable preparation may also be a sterile injectable solution or suspension in a non-toxic parenterally acceptable diluent or solvent, for example as a solution in 1,3-butanediol. Among the acceptable vehicles and solvents that may be employed are water, Ringer’s solution, and isotonic sodium chloride solution. In addition, sterile, fixed oils are conventionally employed as a solvent or suspending medium. For this purpose any bland fixed oil may be employed, including synthetic mono- or diglycerides. In addition, fatty acids such as oleic acid find use in the preparation of injectables. A sterile injectable oil-in-water microemulsion can, for example, be prepared by 1) dissolving at least one compound described herein in an oily phase, such as, for example, a mixture of soybean oil and lecithin; 2) combining the compound-containing oil phase with a water and glycerol mixture; and 3) processing the combination to form a microemulsion. A sterile aqueous or oleaginous suspension can be prepared in accordance with methods already known in the art. For example, a sterile aqueous solution or suspension can be prepared with a non-toxic parenterally-acceptable diluent or solvent, such as, for example, 1,3-butane diol; and a sterile oleaginous suspension can be prepared with a sterile non-toxic acceptable solvent or suspending medium, such as, for example, sterile fixed oils, e.g., synthetic mono- or diglycerides; and fatty acids, such as, for example, oleic acid. Exemplary compositions for nasal aerosol or inhalation administration include solutions which may contain, for example, benzyl alcohol or other suitable preservatives, absorption promoters to enhance absorption and/or bioavailability, and/or other solubilizing or dispersing agents such as those known in the art. Dispersible powders and granules can, for example, be prepared by admixing at least one compound described herein or a pharmaceutically acceptable salt thereof, with at least one dispersing and/or wetting agent; at least one suspending agent; and/or at least one preservative. Exemplary preservatives include, but are not limited to, for example, anti-oxidants, e.g., ascorbic acid. In addition, dispersible powders and granules can also contain at least one excipient, including, but not limited to, for example, sweetening agents; flavoring agents; and coloring agents. Exemplary compositions for rectal administration include suppositories which may contain, for example, suitable non-irritating excipients, such as cocoa butter, synthetic glyceride esters or polyethylene glycols, which are solid at ordinary temperatures but liquefy and/or dissolve in the rectal cavity to release the drug. The therapeutically-effective amount of a compound of the present invention may be determined by one of ordinary skill in the art, and includes exemplary dosage amounts for a mammal of from about 0.05 to 1000 mg/kg; 1-1000 mg/kg; 1-50 mg/kg; 5-250 mg/kg; 250-1000 mg/kg of body weight of active compound per day, which may be administered in a single dose or in the form of individual divided doses, such as from 1 to 4 times per day. It will be understood that the specific dose level and frequency of dosage for any particular subject may be varied and will depend upon a variety of factors, including the activity of the specific compound employed, the metabolic stability and length of action of that compound, the species, age, body weight, general health, sex and diet of the subject, the mode and time of administration, rate of excretion, drug combination, and severity of the particular condition. Preferred subjects for treatment include animals, most preferably mammalian species such as humans, and domestic animals such as dogs, cats, horses, and the like. Thus, when the term "patient" is used herein, this term is intended to include all subjects, most preferably mammalian species that are affected by modulation of USP1-mediated functions. The compounds described herein are useful for the treatment of cancer. In one embodiment, the present application provides a combined preparation of a compound described herein and/or a pharmaceutically acceptable salt thereof, a stereoisomer thereof or a tautomer thereof, and additional therapeutic agent(s) for simultaneous, separate or sequential use in the treatment and/or prophylaxis of multiple diseases or disorders associated with USP1. In another aspect, the application provides a method of treating a patient suffering from or susceptible to a medical condition that is associated with USP1. A number of medical conditions can be treated. The method comprises administering to the patient a therapeutically effective amount of a composition comprising a compound described herien and/or a pharmaceutically acceptable salt thereof, a stereoisomer thereof or a tautomer thereof. For example, the compounds described herein may be used to treat or proliferative diseases such as cancer, immunological disorders or inflammatory disorders. In other embodiments, the compounds described herein may be used to treat cancers that are mediated by, dependent on or associated with USP1 activity. In certain embodiments, the disease is a solid tumor. In particular embodiments, the solid tumor is selected from prostate cancer, pancreatic cancer, bladder cancer, colorectal cancer, breast cancer, renal cancer, hepatocellular cancer, lung cancer, ovarian cancer, cervical cancer, gastric cancer, esophageal cancer, head and neck cancer, melanoma, neuroendocrine cancers, CNS cancers, brain tumors (e.g., glioma, anaplastic oligodendroglioma, adult glioblastoma multiforme, and adult anaplastic astrocytoma), bone cancer, or soft tissue sarcoma. In some embodiments, the solid tumor is from non-small cell lung cancer or small-cell lung cancer. In one embodiment, the compounds herein can be useful in the treatment of haematological malignancies. In one embodiment, hematological malignancy is selected from multiple myeloma, non-Hodgkin's lymphoma, Hodgkin lymphoma, T-cell leukaemia, mucosa-associated lymphoid tissue lymphoma, diffuse large B-cell lymphoma and mantle cell lymphoma. In one embodiment solid tumor is selected from pancreatic cancer, breast cancer, melanoma and non-small cell lung cancer. In one embodiment, cancer is selected from a carcinoma, preferably a carcinoma of the bladder, breast, colon (including colorectal carcinomas, such as colon adenocarcinoma and colon adenoma), kidney, urothelial, uterus, epidermis, liver, lung (including adenocarcinoma, small cell lung cancer, non-small cell lung carcinomas and squamous lung cancer), oesophagus, head and neck, gall bladder, ovary, pancreas (including exocrine pancreatic carcinoma), stomach, gastrointestinal cancer (including gastrointestinal stromal tumors), cervix, endometrium, thyroid, prostate and skin. In one embodiment, the cancer is selected from pituitary cancer, a hematopoietic tumor of lymphoid lineage, for example leukemia, acute lymphocytic leukemia, chronic lymphocytic leukemia, B-cell lymphoma (e.g. diffuse large B-cell lymphoma, mantle cell lymphoma), T-cell leukaemia/lymphoma, Hodgkin's lymphoma, non-Hodgkin's lymphoma, hairy cell lymphoma, or Burkett's lymphoma; a hematopoietic tumor of myeloid lineage, for example leukemias, acute and chronic myelogenous leukemias, chronic myelomonocytic leukemia (CMML), myeloproliferative disorder, myeloproliferative syndrome, myelodysplastic syndrome, or promyelocytic leukemia; multiple myeloma; thyroid follicular cancer; hepatocellular cancer, a tumor of mesenchymal origin (e.g. Ewing's sarcoma), for example fibrosarcoma or rhabdomyosarcoma; a tumor of the central or peripheral nervous system, for example astrocytoma, neuroblastoma, glioma (such as glioblastoma multiforme) or schwannoma; melanoma; seminoma; teratocarcinoma; osteosarcoma; xeroderma pigmentosum; keratoctanthoma; thyroid follicular cancer; or Kaposi's sarcoma. In other embodiments, the compounds described herein may be used to treat cancers that are mediated by, dependent on or associated with USP1 activity. In certain embodiments, the disease is a solid tumor. In particular embodiments, the solid tumor is from prostate cancer, pancreatic cancer, bladder cancer, colorectal cancer, breast cancer, renal cancer, hepatocellular cancer, lung cancer, ovarian cancer, cervical cancer, gastric cancer, esophageal cancer, head and neck cancer, melanoma, neuroendocrine cancers, CNS cancers, brain tumors (e.g., glioma, anaplastic oligodendroglioma, adult glioblastoma multiforme, and adult anaplastic astrocytoma), bone cancer, or soft tissue sarcoma. In some embodiments, the solid tumor is from non-small cell lung cancer or small-cell lung cancer. In other embodiments, the disease is a hematologic malignancy. In certain embodiments, the disease is lymphoma, multiple myeloma, or leukemia. In certain embodiments, the hematologic malignancy is leukemia or lymphoma. In specific embodiments, the disease is acute lymphocytic leukemia (ALL), acute myeloid leukemia (AML), chronic lymphocytic leukemia (CLL), small lymphocytic lymphoma (SLL), myelodysplastic syndrome (MDS), myeloproliferative disease (MPD), chronic myeloid leukemia (CML), juvenile myelomonocytic leukemia (JMML), multiple myeloma (MM), Hodgkin lymphoma, indolent non-Hodgkin's lymphoma (iNHL), refractory iNHL, non- Hodgkin's lymphoma (NHL), mantle cell lymphoma (MCL), follicular lymphoma, Waldenström’s macroglobulinemia (WM), minimal residual disease (MRD), T-cell lymphoma, B-cell lymphoma, diffuse large B-cell lymphoma (DLBCL), T-cell acute lymphoblastic leukemia (T-ALL), B-cell acute lymphoblastic leukemia (B-ALL), lymphoplasmacytic lymphoma, marginal zone lymphoma, or Burkitt lymphoma. In one embodiment, the disease is T-cell acute lymphoblastic leukemia (T-ALL), or B-cell acute lymphoblastic leukemia (B-ALL). In some embodiments, non-Hodgkin lymphoma can be indolent B-cell diseases including follicular lymphoma, lymphoplasmacytic lymphoma, Waldenström macroglobulinemia, and marginal zone lymphoma, as well as the aggressive lymphomas that include, for example, Burkitt lymphoma, diffuse large B- cell lymphoma (DLBCL) and mantle cell lymphoma (MCL). In some embodiments, the cancer is selected from hematological cancer, a lymphatic cancer. In some embodiments, the cancer comprises cancer cells with DNA damage repair pathway deficiency. In some embodiments, the cancer is a homologous recombination deficient cancer. In some embodiments, the cancer comprises cancer cells with a mutation in a gene encoding p53. In some embodiments, the mutation in a gene encoding p53 is a germline or somatic mutation. In some embodiments, the cancer comprises with cancer cells with loss of function mutation in a gene encoding p53. In some embodiments, the cancer is a BRCA1 and/or BRCA2 deficient cancer. In some embodiments, the cancer is a somatic or germline BRCA1 and/or BRCA2 mutant cancer. In some embodiments, the cancer is a Poly (ADP-ribose) polymerase (“PARP”) inhibitor refractory or resistant cancer. In some embodiments, the cancer is a PARP inhibitor resistant or refractory BRCA1 and/or BRCA2 deficient cancer. In some embodiments, the cancer cell has a germline or somatic mutation in a gene encoding ataxia telangiectasia mutated (ATM) protein kinase or ATM deficiency. In some embodiments, the cancer has a mutation in the gene encoding more than two of p53, BRCA1, BRCA2, ATM. In some embodiments, the disease is an autoimmune or inflammatory disease or disorder. These autoimmune or inflammatory diseases or conditions may be chronic or acute and include, but are not limited to, inflammatory pelvic disease, urethritis, skin sunburn, sinusitis, pneumonitis, encephalitis, meningitis, myocarditis, pericarditis, nephritis including lupus nephritis, osteomyelitis, myositis, eczema, hepatitis, gastritis, enteritis, dermatitis, gingivitis, appendicitis, pancreatitis, primary biliary cirrhosis, cholecystitis, sclerosing cholangitis, agammaglobulinemia, psoriasis, allergy, Crohn's disease, irritable bowel syndrome, ulcerative colitis, Sjogren's disease, tissue graft rejection such as acute graft-versus-host disease, hyperacute rejection of transplanted organs, asthma, chronic obstructive airways disease, allergic rhinitis, chronic obstructive pulmonary disease (COPD), autoimmune polyglandular disease (also known as autoimmune polyglandular syndrome), autoimmune alopecia, pernicious anemia, vasculitis, glomerulonephritis, giant cell arteritis, Wegener's granulomatosis, Polyarteritis nodosa, dermatomyositis, multiple sclerosis, scleroderma, autoimmune hemolytic and thrombocytopenic states, Goodpasture's syndrome, atherosclerosis, Addison's disease, hypophysitis, Parkinson's disease, Alzheimer's disease, Kawasaki disease, Takayasu's Arteritis, depression, retinitis, uveitis, scleritis, Type I diabetes, septic shock, systemic lupus erythematosus (SLE), rheumatoid arthritis, psoriatic arthritis, juvenile arthritis, osteoarthritis, gout, chronic idiopathic thrombocytopenic purpura, Waldenström macroglobulinemia, myasthenia gravis, Hashimoto's thyroiditis, atopic dermatitis, degenerative joint disease, vitiligo, bullous skin diseases, autoimmune hypopituitarism, Guillain-Barre syndrome, Behcet's disease, scleracierma, mycosis fungoides, acute inflammatory responses (such as acute respiratory distress syndrome and ischemia/reperfusion injury), and Graves' disease. In some embodiments, the autoimmune and inflammatory diseases and conditions may also include systemic or tissue inflammation, inflammatory responses to hypoxia, cellular activation and proliferation, lipid metabolism, fibrosis, infections with bacteria, infections with viruses (e.g., herpes virus, human papilloma virus, adenovirus, poxvirus and other DNA viruses), fungi, parasites or their toxins, such as sepsis, sepsis syndrome, septic shock, endotoxaemia, systemic inflammatory response syndrome (SIRS), multi-organ dysfunction syndrome, toxic shock syndrome, acute lung injury, ARDS (adult respiratory distress syndrome), acute renal failure, fulminant hepatitis, burns, acute pancreatitis, post-surgical syndromes, sarcoidosis, Herxheimer reactions, encephalitis, myelitis, meningitis, malaria and SIRS associated with viral infections such as influenza, herpes zoster, herpes simplex and coronavirus. Combination Therapies In some embodiments, the compounds described herein may be administered in conjunction with standard of care, e.g., surgery, radiation, and/or chemotherapy. In some embodiments, the compounds may be administered in conjunction with a chemotherapeutic agent. In some embodiments, the compounds may be administered in conjunction with one or more of carboplatin, cisplatin, paclitaxel, nab-paclitaxel, gemcitabine or FOLFOX. In some embodiment, the compounds may be administered in conjunction with carboplatin or nab-paclitaxel. In some embodiments, the compounds may be administered in conjunction with carboplatin and paclitaxel. In some embodiments, the compounds may be administered in conjunction with cisplatin and pemetrexed. In some embodiments, the compounds may be administered in conjunction with cisplatin and gemcitabine. In some embodiments, the compounds may be administered in conjunction with FOLFOX. In some embodiments, the compounds may be administered in conjunction with FOLFIRI. In one embodiment, the compounds may be administered in combination with decarbazine for the treatment of melanoma. In some embodiments, cisplatin is intravenously administered as a 100 mg/ml dose once every four weeks. In some embodiments, the compounds may be administered in conjunction with doxorubicin (adriamycin), cisplatin bleomycin sulfate, carmustine, chlorambucil, dacarbazine and/or cyclophosphamide hydroxyurea. In some embodiments, adriamycin is intravenously administered as a 60 mg/ml to 75 mg/ml dose once every 21 days. In one embodiment, the compounds of the present application (e.g., a compound described herein or a pharmaceutically acceptable salt, prodrug, or solvate thereof) may be used in combination with one or more additional therapeutic agent that are being used and/or developed to treat cancers or inflammatory disorders. The one or more additional therapeutic agent may be an inhibitor to Janus kinase (JAK) such as JAK1, JAK2 and/or JAK3, Tyroansine kinase (TYK), K-Ras, Mitogen activated protein kinases (MAPK), Bruton's tyrosine kinase (BTK), bromodomain containing protein inhibitor (BRD) such as BRD4, a lysyl oxidase protein (LOX), lysyl oxidase-like protein (LOXL) such as LOXL1-5, matrix metalloprotease (MMP) such as MMP 1-10, adenosine A2B receptor (A2B), isocitrate dehydrogenase (IDH) such as IDH1, apoptosis signal-regulating kinase (ASK) such as ASK1, serine/threonine kinase TPL2, discoidin domain receptor (DDR) such as DDR1 and DDR2, histone deacetylase (HDAC), protein kinase C (PKC), or any combination thereof. In one embodiment, the compounds of the present application (e.g., a compound described herein or a pharmaceutically acceptable salt, prodrug, or solvate thereof) may be used in combination with additional chemotherapeutic agent, an immunotherapeutic agent, a radiotherapeutic agent, an anti-neoplastic agent, an anti-cancer agent, an anti- fibrotic agent, an anti-angiogenic agent, a therapeutic antibody, or any combination thereof. Chemotherapeutic agents may be categorized by their mechanism of action into, for example, the following groups: anti-metabolites/anti-cancer agents, such as pyrimidine analogs (floxuridine, capecitabine, and cytarabine); purine analogs, folate antagonists and related inhibitors antiproliferative/antimitotic agents including natural products such as vinca alkaloid (vinblastine, vincristine) and microtubule such as taxane (paclitaxel, docetaxel), vinblastin, nocodazole, epothilones and navelbine, epidipodophyllotoxins (etoposide, teniposide); DNA damaging agents (actinomycin, amsacrine, busulfan, carboplatin, chlorambucil, cisplatin, cyclophosphamide, Cytoxan, dactinomycin, daunorubicin, doxorubicin, epirubicin, iphosphamide, melphalan, merchlorehtamine, mitomycin, mitoxantrone, nitrosourea, procarbazine, taxol, taxotere, teniposide, etoposide, triethylenethiophosphoramide); antibiotics such as dactinomycin (actinomycin D), daunorubicin, doxorubicin (adriamycin), idarubicin, anthracyclines, mitoxantrone, bleomycins, plicamycin (mithramycin) and mitomycin; enzymes (L- asparaginase which systemically metabolizes L-asparagine and deprives cells which do not have the capacity to synthesize their own asparagine); antiplatelet agents; antiproliferative/antimitotic alkylating agents such as nitrogen mustards cyclophosphamide and analogs, melphalan, chlorambucil), and (hexamethylmelamine and thiotepa), alkyl nitrosoureas (BCNU) and analogs, streptozocin), trazenes-dacarbazinine (DTIC); antiproliferative/antimitotic antimetabolites such as folic acid analogs (methotrexate); platinum coordination complexes (cisplatin, oxiloplatinim, carboplatin), procarbazine, hydroxyurea, mitotane, aminoglutethimide; hormones, hormone analogs (estrogen, tamoxifen, goserelin, bicalutamide, nilutamide) and aromatase inhibitors (letrozole, anastrozole); anticoagulants (heparin, synthetic heparin salts and other inhibitors of thrombin); fibrinolytic agents (such as tissue plasminogen activator, streptokinase and urokinase), aspirin, dipyridamole, ticlopidine, clopidogrel; antimigratory agents; antisecretory agents (breveldin); immunosuppressives tacrolimus sirolimus azathioprine, mycophenolate; compounds (TNP-470, genistein) and growth factor inhibitors (vascular endothelial growth factor inhibitors, fibroblast growth factor inhibitors); angiotensin receptor blocker, nitric oxide donors; anti-sense oligonucleotides; antibodies (trastuzumab, rituximab); cell cycle inhibitors and differentiation inducers (tretinoin); inhibitors, topoisomerase inhibitors (doxorubicin (adriamycin), daunorubicin, dactinomycin, eniposide, epirubicin, etoposide, idarubicin, irinotecan and mitoxantrone, topotecan, irinotecan, camptothesin), corticosteroids (cortisone, dexamethasone, hydrocortisone, methylpednisolone, prednisone, and prenisolone); growth factor signal transduction kinase inhibitors; dysfunction inducers, toxins such as Cholera toxin, ricin, Pseudomonas exotoxin, Bordetella pertussis adenylate cyclase toxin, or diphtheria toxin, and caspase activators; and chromatin. Examples of chemotherapeutic agents include alkylating agents such as thiotepa and cyclophosphamide; alkyl sulfonates such as busulfan, improsulfan and piposulfan; aziridines such as benzodopa, carboquone, meturedopa, and uredopa; emylerumines and memylamelamines including alfretamine, triemylenemelamine, triethylenephosphoramide, triethylenethiophosphoramide and trimemylolomelamine; acetogenins (especially bullatacin and bullatacinone); a camptothecin (including synthetic analogue topotecan); bryostatin; callystatin; CC-1065 (including its adozelesin, carzelesin and bizelesin synthetic analogues); cryptophycins (articularly cryptophycin 1 and cryptophycin 8); dolastatin; duocarmycin (including the synthetic analogues, KW-2189 and CBI-TMI); eleutherobin; pancratistatin; a sarcodictyin; spongistatin; nitrogen mustards such as chlorambucil, chlornaphazine, cholophosphamide, estramustine, ifosfamide, mechlorethamine, mechlorethamine oxide hydrochloride, melphalan, novembichin, phenesterine, prednimustine, trofosfamide, uracil mustard; nitrosoureas such as carmustine, chlorozotocin, foremustine, lomustine, nimustine, ranimustine; antibiotics such as the enediyne antibiotics (e.g., calicheamicin, calicheamicin gammall, dynemicin, dynemicin A; bisphosphonates, such as clodronate; an esperamicin; as well as neocarzinostatin chromophore and related chromoprotein enediyne antibiotic chromomophores), aclacinomysins, actinomycin, authramycin, azaserine, bleomycins, cactinomycin, carabicin, carrninomycin, carzinophilin, chromomycins, dactinomycin, daunorubicin, detorubicin, 6-diazo-5-oxo-L-norleucine, doxorubicin (including morpholino-doxorubicin, cyanomorpholino-doxorubicin, 2-pyrrolino-doxorubicin and deoxydoxorubicin), epirubicin, esorubicin, idarubicin, marcellomycin, mitomycins such as mitomycin C, mycophenolic acid, nogalamycin, olivomycins, peplomycin, potfiromycin, puromycin, quelamycin, rodorubicin, streptonigrin, streptozocin, tubercidin, ubenimex, zinostatin, zorubicin; anti-metabolites such as methotrexate and 5- fluorouracil (5-FU); folic acid analogues such as demopterin, methotrexate, pteropterin, trimetrexate; purine analogs such as fludarabine, 6-mercaptopurine, thiamiprine, thioguanine; pyrimidine analogues such as ancitabine, azacitidine, 6-azauridine, carmofur, cytarabine, dideoxyuridine, doxifluridine, enocitabine, floxuridine; androgens such as calusterone, dromostanolone propionate, epitiostanol, mepitiostane, testolactone; anti-adrenals such as aminoglutethimide, mitotane, trilostane; folic acid replinisher such as frolinic acid; aceglatone; aldophosphamide glycoside; aminolevulinic acid; eniluracil; amsacrine; hestrabucil; bisantrene; edatraxate; defofamine; demecolcine; diaziquone; elformthine; elliptinium acetate; an epothilone; etoglucid; gallium nitrate; hydroxyurea; lentinan; leucovorin; lonidamine; maytansinoids such as maytansine and ansamitocins; mitoguazone; mitoxantrone; mopidamol; nitracrine; pentostatin; phenamet; pirarubicin; losoxantrone; fluoropyrimidine; folinic acid; podophyllinic acid; 2-ethylhydrazide; procarbazine; razoxane; rhizoxin; sizofiran; spirogermanium; tenuazonic acid; triaziquone; 2,2',2''-tricUorotriemylamine; trichothecenes (especially T-2 toxin, verracurin A, roridin A and anguidine); urethane; vindesine; dacarbazine; mannomustine; mitobronitol; mitolactol; pipobroman; gacytosine; arabinoside ("Ara-C"); cyclophosphamide; thiopeta; taxoids, e.g., paclitaxel and docetaxel, chlorambucil; gemcitabine; 6-thioguanine; mercaptopurine; methotrexate; platinum analogs such as cisplatin and carboplatin; vinblastine; platinum; etoposide; ifosfamide; mitroxantrone; vancristine; vinorelbine; novantrone; teniposide; edatrexate; daunomycin; aminopterin; xeoloda; ibandronate; CPT-11; topoisomerase inhibitor RFS 2000; difluoromethylornithine (DMFO); retinoids such as retinoic acid; capecitabine; FOLFIRI (fluorouracil, leucovorin, and irinotecan) and pharmaceutically acceptable salts, acids or derivatives of any of the above. One or more chemotherapeutic agent is used or included in the present application. Chemotherapeutic agents may also include, for example, anti-hormonal agents that act to regulate or inhibit hormone action on tumors such as anti-estrogens and selective estrogen receptor modulators (SERMs), including, for example, tamoxifen, raloxifene, droloxifene, 4-hydroxytamoxifen, trioxifene, keoxifene, onapristone, and toremifene; inhibitors of the enzyme aromatase, which regulates estrogen production in the adrenal glands, such as, for example, 4(5)-imidazoles, aminoglutethimide, megestrol acetate, exemestane, formestane, fadrozole, vorozole letrozole and anastrozole; and anti- androgens such as flutamide, nilutamide, bicalutamide, leuprohde, and goserelin; and pharmaceutically acceptable salts thereof. The anti-angiogenic agents include, but are not limited to, retinoid acid and derivatives thereof, 2-methoxyestradiol, suramin, squalamine, tissue inhibitor of metalloproteinase-1, tissue inhibitor of metalloproternase-2, plasminogen activator inhibitor-1, plasminogen activator inbibitor-2, cartilage-derived inhibitor, paclitaxel (nab- paclitaxel), platelet factor 4, protamine sulphate (clupeine), sulphated chitin derivatives (prepared from queen crab shells), sulphated polysaccharide peptidoglycan complex (sp- pg), staurosporine, modulators of matrix metabolism, including for example, proline analogs ((1-azetidine-2-carboxylic acid (LACA), cishydroxyproline, d,I-3,4- dehydroproline, thiaproline, .alpha.-dipyridyl, beta-aminopropionitrile fumarate, 4- propyl-5-(4-pyridinyl)-2(3h)-oxazolone; methotrexate, mitoxantrone, heparin, interferons, 2 macroglobulin-serum, chimp-3, chymostatin, beta-cyclodextrin tetradecasulfate, eponemycin; fumagillin, gold sodium thiomalate, d-penicillamine (CDPT), beta-1- anticollagenase-serum, alpba-2-antiplasmin, bisantrene, lobenzarit disodium, n-2- carboxyphenyl-4-chloroanthronilic acid disodium or "CCA", thalidomide; angiostatic steroid, cargboxynaminolmidazole; metalloproteinase inhibitors such as BB94. Other anti-angiogenesis agents include antibodies, preferably monoclonal antibodies against these angiogenic growth factors: beta-FGF, alpha-FGF, FGF-5, VEGF isoforms, VEGF- C, HGF/SF and Ang-1/Ang-2. The application also provides a method for treating a subject who is undergoing one or more standard therapies, such as chemotherapy, radiotherapy, immunotherapy, surgery, or combination thereof. Accordingly, one or more therapeutic agent or inhibitors may be administered before, during, or after administration of chemotherapy, radiotherapy, immunotherapy, surgery or combination thereof. In certain embodiments, the subject may be a human who is (i) substantially refractory to at least one chemotherapy treatment, or (ii) in relapse after treatment with chemotherapy, or both (i) and (ii). In some of embodiments, the subject is refractory to at least two, at least three, or at least four chemotherapy treatments (including standard or experimental chemotherapies). In certain embodiments, the subject is refractory to at least one, at least two, at least three, or at least four chemotherapy treatment (including standard or experimental chemotherapy) selected from fludarabine, rituximab, obinutuzumab, alkylating agents, alemtuzumab and other chemotherapy treatments such as CHOP (cyclophosphamide, doxorubicin, vincristine, prednisone); R-CHOP (rituximab-CHOP); hyperCVAD (hyperfractionated cyclophosphamide, vincristine, doxorubicin, dexamethasone, methotrexate, cytarabine); R-hyperCVAD (rituximab-hyperCVAD); FCM (fludarabine, cyclophosphamide, mitoxantrone); R-FCM (rituximab, fludarabine, cyclophosphamide, mitoxantrone); bortezomib and rituximab; temsirolimus and rituximab; temsirolimus and Velcade.RTM.; Iodine-131 tositumomab (Bexxar.RTM.) and CHOP; CVP (cyclophosphamide, vincristine, prednisone); R-CVP (rituximab-CVP); ICE (iphosphamide, carboplatin, etoposide); R-ICE (rituximab-ICE); FCR (fludarabine, cyclophosphamide, rituximab); FR (fludarabine, rituximab); and D.T. PACE (dexamethasone, thalidomide, cisplatin, Adriamycin.RTM., cyclophosphamide, etoposide). Examples of immunotherapeutic agents treating lymphoma or leukemia include, but are not limited to, rituximab (such as Rituxan), alemtuzumab (such as Campath, MabCampath), anti-CD19 antibodies, anti-CD20 antibodies, anti-MN-14 antibodies, anti- TRAIL, Anti-TRAIL DR4 and DR5 antibodies, anti-CD74 antibodies, apolizumab, bevacizumab, CHIR-12.12, epratuzumab (hLL2-anti-CD22 humanized antibody), galiximab, ha20, ibritumomab tiuxetan, lumiliximab, milatuzumab, ofatumumab, PRO131921, SGN-40, WT-1 analog peptide vaccine, WT1126-134 peptide vaccine, tositumomab, autologous human tumor-derived HSPPC-96, and veltuzumab. Additional immunotherapy agents includes using cancer vaccines based upon the genetic makeup of an individual patient's tumor, such as lymphoma vaccine GTOP-99. The therapeutic treatments can be supplemented or combined with any of the abovementioned therapies with stem cell transplantation or treatment. One example of modified approach is radioimmunotherapy, wherein a monoclonal antibody is combined with a radioisotope particle, such as indium In-111, yttrium Y-90, iodine I-131. Examples of combination therapies include, but are not limited to, Iodine-131 tositumomab, Yttrium-90 ibritumomab tiuxetan with CHOP. The compounds of the application can be used in combination with additional therapeutic procedures. Other therapeutic procedures include peripheral blood stem cell transplantation, autologous hematopoietic stem cell transplantation, autologous bone marrow transplantation, antibody therapy, biological therapy, enzyme inhibitor therapy, total body irradiation, infusion of stem cells, bone marrow ablation with stem cell support, in vitro-treated peripheral blood stem cell transplantation, umbilical cord blood transplantation, immunoenzyme technique, pharmacological study, low-LET cobalt-60 gamma ray therapy, bleomycin, conventional surgery, radiation therapy, and nonmyeloablative allogeneic hematopoietic stem cell transplantation. The compounds of the application can be used in combination with anti-fibrotic agents. The anti-fibrotic agents include, but are not limited to, emylenemamine, hydrazine, phenylhydrazine, and their derivatives, semicarbazide, and urea derivatives, aminonitriles, such as beta-aminopropionitrile (BAPN), or 2-nitroethylamine, unsaturated or saturated haloamines, such as 2-bromo-ethylamine, 2-chloroethylamine, 2- trifluoroethylamine, 3-bromopropylamine, p-halobenzylamines, selenohomocysteine lactone. Also, the anti-fibrotic agents are copper chelating agents, penetrating or not penetrating the cells. Exemplary compounds include indirect inhibitors such compounds blocking the aldehyde derivatives originating from the oxidative deamination of the lysyl and hydroxylysyl residues by the lysyl oxidases, such as the thiolamines, in particular D- penicillamine, or its analogues such as 2-amino-5-mercapto-5-methylhexanoic acid, D-2- amino-3-methyl-3-((2-acetamidoethyl)dithio)butanoic acid, p-2-amino-3-methyl-3-((2- aminoethyl)dithio)butanoic acid, sodium-4-((p-1-dimethyl-2-amino-2- carboxyethyl)dithio)butane sulphurate, 2-acetamidoethyl-2-acetamidoethanethiol sulphanate, sodium-4-mercaptobutanesulphinate trihydrate. The compounds of the application can be used in combination with immunotherapeutic and anti-inflammatory treatments. The immunotherapeutic agents include and are not limited to therapeutic antibodies suitable for treating patients; such as abagovomab, adecatumumab, afutuzumab, alemtuzumab, altumomab, amatuximab, anatumomab, arcitumomab, bavituximab, bectumomab, bevacizumab, bivatuzumab, blinatumomab, brentuximab, cantuzumab, catumaxomab, cetuximab, citatuzumab, cixutumumab, clivatuzumab, conatumumab, daratumumab, drozitumab, duligotumab, dusigitumab, detumomab, dacetuzumab, dalotuzumab, ecromeximab, elotuzumab, ensituximab, ertumaxomab, etaracizumab, farietuzumab, ficlatuzumab, figitumumab, flanvotumab, futuximab, ganitumab, gemtuzumab, girentuximab, glembatumumab, ibritumomab, igovomab, imgatuzumab, indatuximab, inotuzumab, intetumumab, ipilimumab, iratumumab, labetuzumab, lexatumumab, lintuzumab, lorvotuzumab, lucatumumab, mapatumumab, matuzumab, milatuzumab, minretumomab, mitumomab, moxetumomab, narnatumab, naptumomab, necitumumab, nimotuzumab, nofetumomabn, ocaratuzumab, ofatumumab, olaratumab, onartuzumab, oportuzumab, oregovomab, panitumumab, parsatuzumab, patritumab, pemtumomab, pertuzumab, pintumomab, pritumumab, racotumomab, radretumab, rilotumumab, rituximab, robatumumab, satumomab, sibrotuzumab, siltuximab, simtuzumab, solitomab, tacatuzumab, taplitumomab, tenatumomab, teprotumumab, tigatuzumab, tositumomab, trastuzumab, tucotuzumab, ublituximab, veltuzumab, vorsetuzumab, votumumab, zalutumumab, CC49 and 3F8. The exemplified therapeutic antibodies may be further labeled or combined with a radioisotope particle, such as indium In-111, yttrium Y-90, iodine I-131. In one aspect, the immuno-oncology agent is (i) an agonist of a stimulatory (including a co-stimulatory) receptor or (ii) an antagonist of an inhibitory (including a co- inhibitory) signal on T cells, both of which result in amplifying antigen-specific T cell responses (often referred to as immune checkpoint regulators). Certain of the stimulatory and inhibitory molecules are members of the immunoglobulin super family (IgSF). One important family of membrane-bound ligands that bind to co-stimulatory or co-inhibitory receptors is the B7 family, which includes B7- 1, B7-2, B7-H1 (PD-L1), B7-DC (PD-L2), B7-H2 (ICOS-L), B7-H3, B7-H4, B7-H5 (VISTA), and B7-H6. Another family of membrane bound ligands that bind to co- stimulatory or co-inhibitory receptors is the TNF family of molecules that bind to cognate TNF receptor family members, which includes CD40 and CD40L, OX-40, OX-40L, CD70, CD27L, CD30, CD30L, 4-1BBL, CD137 (4-1BB), TRAIL/Apo2-L, TRAILR1/DR4, TRAILR2/DR5, TRAILR3, TRAILR4, OPG, RANK, RANKL, TWEAKR/Fn14, TWEAK, BAFFR, EDAR, XEDAR, TACI, APRIL, BCMA, LTȕR, LIGHT, DcR3, HVEM, VEGI/TL1A, TRAMP/DR3, EDAR, EDA1, XEDAR, EDA2, TNFR1, Lymphotoxin Į/TNFȕ, TNFR2, TNFĮ, LTȕR, Lymphotoxin Į 1ȕ2, FAS, FASL, RELT, DR6, TROY, NGFR. In one aspect, T cell responses can be stimulated by a combination of a compound of Formula (I) and one or more of (i) an antagonist of a protein that inhibits T cell activation (e.g., immune checkpoint inhibitors) such as CTLA-4, PD-1, PD-L1, PD-L2, LAG-3, TIM-3, Galectin 9, CEACAM-1, BTLA, CD69, Galectin-1, TIGIT, CD113, GPR56, VISTA, 2B4, CD48, GARP, PD1H, LAIR1, TIM-1, and TIM-4, and (ii) an agonist of a protein that stimulates T cell activation such as B7-1, B7-2, CD28, 4-1BB (CD137), 4-1BBL, ICOS, ICOS-L, OX40, OX40L, GITR, GITRL, CD70, CD27, CD40, DR3 and CD28H. Other agents that can be combined with compounds described herein for the treatment of cancer include antagonists of inhibitory receptors on NK cells or agonists of activating receptors on NK cells. For example, compounds described herien can be combined with antagonists of KIR, such as lirilumab. Yet other agents for combination therapies include agents that inhibit or deplete macrophages or monocytes, including but not limited to CSF-1R antagonists such as CSF-1R antagonist antibodies including RG7155 (WO11/70024, WO11/107553, In another aspect, compounds of the present application can be used with one or more of agonistic agents that ligate positive costimulatory receptors, blocking agents that attenuate signaling through inhibitory receptors, antagonists, and one or more agents that increase systemically the frequency of anti-tumor T cells, agents that overcome distinct immune suppressive pathways within the tumor microenvironment (e.g., block inhibitory receptor engagement (e.g., PD-L1/PD-1 interactions), deplete or inhibit Tregs (e.g., using an anti-CD25 monoclonal antibody (e.g., daclizumab) or by ex vivo anti-CD25 bead depletion), inhibit metabolic enzymes such as IDO, or reverse/prevent T cell anergy or exhaustion) and agents that trigger innate immune activation and/or inflammation at tumor sites. In one aspect, the immuno-oncology agent is a CTLA-4 antagonist, such as an antagonistic CTLA-4 antibody. Suitable CTLA-4 antibodies include, for example, YERVOY (ipilimumab) or tremelimumab. In another aspect, the immuno-oncology agent is a PD-1 antagonist, such as an antagonistic PD-1 antibody. Suitable PD-1 antibodies include, for example, OPDIVO (nivolumab), KEYTRUDA (pembrolizumab), or MEDI-0680 (AMP-514; WO2012/145493). The immuno-oncology agent may also include pidilizumab (CT-011), though its specificity for PD-1 binding has been questioned. Another approach to target the PD-1 receptor is the recombinant protein composed of the extracellular domain of PD-L2 (B7-DC) fused to the Fc portion of IgG1, called AMP-224 In another aspect, the immuno-oncology agent is a PD-L1 antagonist, such as an antagonistic PD-L1 antibody. Suitable PD-L1 antibodies include, for example, MPDL3280A (RG7446; WO2010/077634), durvalumab (MEDI4736), BMS-936559 (WO2007/005874), and MSB0010718C (WO2013/79174). In another aspect, the immuno-oncology agent is a LAG-3 antagonist, such as an antagonistic LAG-3 antibody. Suitable LAG3 antibodies include, for example, BMS- 986016 (WO10/19570, WO14/08218), or IMP-731 or IMP-321 (WO08/132601, WO09/44273). In another aspect, the immuno-oncology agent is a CD137 (4-1BB) agonist, such as an agonistic CD137 antibody. Suitable CD137 antibodies include, for example, urelumab and PF-05082566 (WO12/32433). In another aspect, the immuno-oncology agent is a GITR agonist, such as an agonistic GITR antibody. Suitable GITR antibodies include, for example, BMS-986153, BMS-986156, TRX-518 (WO06/105021, WO09/009116) and MK-4166 (WO11/028683). In another aspect, the immuno-oncology agent is an IDO antagonist. Suitable IDO antagonists include, for example, INCB-024360 (WO2006/122150, WO07/75598, WO08/36653, WO08/36642), indoximod, BMS-986205, or NLG-919 (WO09/73620, WO09/1156652, WO11/56652, WO12/142237). In another aspect, the immuno-oncology agent is an OX40 agonist, such as an agonistic OX40 antibody. Suitable OX40 antibodies include, for example, MEDI-6383 or MEDI-6469. In another aspect, the immuno-oncology agent is an OX40L antagonist, such as an antagonistic OX40 antibody. Suitable OX40L antagonists include, for example, RG-7888 (WO06/029879). In another aspect, the immuno-oncology agent is a CD40 agonist, such as an agonistic CD40 antibody. In yet another embodiment, the immuno-oncology agent is a CD40 antagonist, such as an antagonistic CD40 antibody. Suitable CD40 antibodies include, for example, lucatumumab or dacetuzumab. In another aspect, the immuno-oncology agent is a CD47 antagonist, such as a CD47 antagonist selected from the group MIAP301, MIAP410, TTI-621, CV1, Hu5F9- G4, CC-90002, B6H12 and 2D3. In another aspect, the immuno-oncology agent is a CD27 agonist, such as an agonistic CD27 antibody. Suitable CD27 antibodies include, for example, varlilumab. In another aspect, the immuno-oncology agent is MGA271 (to B7H3) (WO11/109400). The combination therapy is intended to embrace administration of these therapeutic agents in a sequential manner, that is, wherein each therapeutic agent is administered at a different time, as well as administration of these therapeutic agents, or at least two of the therapeutic agents, in a substantially simultaneous manner. Substantially simultaneous administration can be accomplished, for example, by administering to the subject a single dosage form having a fixed ratio of each therapeutic agent or in multiple, single dosage forms for each of the therapeutic agents. Sequential or substantially simultaneous administration of each therapeutic agent can be effected by any appropriate route including, but not limited to, oral routes, intravenous routes, intramuscular routes, and direct absorption through mucous membrane tissues. The therapeutic agents can be administered by the same route or by different routes. For example, a first therapeutic agent of the combination selected may be administered by intravenous injection while the other therapeutic agents of the combination may be administered orally. Alternatively, for example, all therapeutic agents may be administered orally or all therapeutic agents may be administered by intravenous injection. Combination therapy also can embrace the administration of the therapeutic agents as described above in further combination with other biologically active ingredients and non-drug therapies (e.g., surgery or radiation treatment.) Where thwe combination therapy further comprises a non drug treatment, the non drug treatment may be conducted at any suitable time so long as a beneficial effect from the co-action of the combination of the therapeutic agents and non-drug treatment is achieved. For example, in appropriate cases, the beneficial effect is still achieved when the non drug treatment is temporally removed from the administration of the therapeutic agents, perhaps by days or even weeks. The present invention also provides the compounds of the present invention for use in therapy. In another embodiment, compounds of Formula I are selected from exemplified compounds or combinations of exemplified compounds or other embodiments herein. In another embodiment are compounds having an IC50 < 1000 nM in at least one of the assays described below. The present invention may be embodied in other specific forms without departing from the spirit or essential attributes thereof. This invention encompasses all combinations of preferred aspects and/or embodiments of the invention noted herein. It is understood that any and all embodiments of the present invention may be taken in conjunction with any other embodiment or embodiments to describe additional more preferred embodiments. It is also to be understood that each individual element of the preferred embodiments is its own independent preferred embodiment. Furthermore, any element of an embodiment is meant to be combined with any and all other elements from any embodiment to describe an additional embodiment. METHODS OF PREPARATION The compounds of the present invention can be prepared in a number of ways well known to one skilled in the art of organic synthesis. The compounds of the present invention can be synthesized using the methods described below, together with synthetic methods known in the art of synthetic organic chemistry, or variations thereon as appreciated by those skilled in the art. Preferred methods include, but are not limited to, those described below. All references cited herein are hereby incorporated in their entirety by reference. The compounds of this invention may be prepared using the reactions and techniques described in this section. The reactions are performed in solvents appropriate to the reagents and materials employed and are suitable for the transformations being effected. Also, in the description of the synthetic methods described below, it is to be understood that all proposed reaction conditions, including choice of solvent, reaction atmosphere, reaction temperature, duration of the experiment and work up procedures, are chosen to be the conditions standard for that reaction, which should be readily recognized by one skilled in the art. It is understood by one skilled in the art of organic synthesis that the functionality present on various portions of the molecule must be compatible with the reagents and reactions proposed. Such restrictions to the substituents that are compatible with the reaction conditions will be readily apparent to one skilled in the art and alternate methods must then be used. This will sometimes require a judgment to modify the order of the synthetic steps or to select one particular process scheme over another in order to obtain a desired compound of the invention. It will also be recognized that another major consideration in the planning of any synthetic route in this field is the judicious choice of the protecting group used for protection of the reactive functional groups present in the compounds described in this invention. An authoritative account describing the many alternatives to the trained practitioner is Greene and Wuts (Protective Groups In Organic Synthesis, Third Edition, Wiley and Sons, 1999). EXAMPLES Preparation of compounds of Formula I and intermediates used in the preparation of compounds of Formula I can be prepared using procedures shown in the following Examples and related procedures. The methods and conditions used in these examples, and the actual compounds prepared in these Examples, are not meant to be limiting, but are meant to demonstrate how the compounds of Formula I can be prepared. Starting materials and reagents used in these examples, when not prepared by a procedure described herein, are generally either commercially available, or are reported in the chemical literature, or may be prepared by using procedures described in the chemical literature. EXAMPLES The following examples illustrate the particular and preferred embodiments of the present invention and do not limit the scope of the present invention. Chemical abbreviations and symbols as well as scientific abbreviations and symbols have their usual and customary meanings unless otherwise specified. Common intermediates are generally useful for the preparation of more than one Example as shown in the Tables. Chemical names were determined using ChemBioDraw Ultra, version 14.0.0.126 (CambridgeSoft). The following abbreviations are used: ABBREVIATIONNAME Boctert-butyloxycarbonyl CDCl3deuterated chloroform DASTdiethylaminosulfur trifluoride DCMdichloromethane DIBAL-H diisobutylaluminium hydride DMF N,N-dimethylformamide DMSO dimethyl sulfoxide DMSO-d6 deuterated dimethyl sulfoxide Et3N triethylamine EtOAc ethyl acetate H hours HATU O-(7-azabenzotriazol- l-yl)-1, 1,3,3-tetramethyluronium hexafluorophosphateABBREVIATIONNAME HPLC high performance liquid chromatography LC liquid chromatography LCMS liquid chromatography – mass spectrometry MeCN acetonitrile MeOH methanol MeOH-d4 deuterated methanol mCPBA meta-chloroperoxybenzoic acid Min minutes MsCl methanesulfonyl chloride NH4OAc ammonium acetate PyBOP benzotriazol-1-yl- oxytripyrrolidinophosphonium hexafluorophosphate Rt room temperature SFC super-critical fluid chromatography TFA trifluoroacetic acid THF tetrahydrofuran TLC thin layer chromatography UPLC ultra performance liquid chromatography LCMS-Method 1: COLUMN: Kinetex XB-C18(75X3.0) mm, 2.6um Mobile phase A: 5mm Ammonium formate pH 3.3: Acetonitrile (98:2) Mobile phase B: ACN :5mm Ammonium formate pH 3.3 (98:2) Flow rate: 1 ml/min Temperature: 25° C GRADIENT: 20%B to 100%B over 4min, 1.5 ml/min flow at 0.5 min hold at 100%B MS and UV: 220 & 254 LCMS-Method 2: COLUMN: Kinetex -C18(75X3.0) mm, 2.6um Mobile phase A: 0.1% TFA in water Mobile phase B: 0.1% TFA in Acetonitrile Flow rate: 1 ml/min Temperature: 25° C GRADIENT: 05%B to 95%B over 2.5min, 1.5 ml\min flow at 1.49min hold at 95%B MS and UV: 220 & 254 nm LCMS-Method 3: COLUMN: Acquity BEH C18(50X3.0) mm, 1.7um Mobile phase A: 5mm Ammonium formate pH 3.3: Acetonitrile (98:2) Mobile phase B: ACN :5mm Ammonium formate pH 3.3 (98:2) Flow rate: 0.7 ml/min Temperature: 25° C GRADIENT: 20%B to 98%B over 1.5min, 0.5 min hold at 98%B MS and UV: 220 nm LCMS-Method 4: COLUMN: Acquity Uplc BEH C18(50X3.0) mm, 1.7um Mobile phase A: 5mm Ammonium formate pH 3.3: Acetonitrile (98:2) Mobile phase B: ACN :5mm Ammonium formate pH 3.3 (98:2) Flow rate: 0.7 ml/min Temperature: 25° C GRADIENT: 20%B to 98%B over 1.5min, 0.5 min hold at 98%B MS and UV: 220 nm







MS and UV: 220 & 254 nm LCMS-Method 6: COLUMN: Kinetex -C18(75X3.0) mm, 2.6um Mobile phase A: 5mm Ammonium formate pH 3.3: Acetonitrile (98:2) Mobile phase B: ACN :5mm Ammonium formate pH 3.3 (98:2) Flow rate: 0.7 ml/min Temperature: 25° C GRADIENT: 20%B to 100%B over 4min, 1.5 ml/min flow at 0.5 min hold at 100%B MS and UV: 220 & 254 LCMS-Method 7: COLUMN: Acquity BEH C18(50X3.0) mm, 1.7um Mobile phase A: 0.1% TFA in water Mobile phase B: 0.1% TFA in Acetonitrile Flow rate: 0.7 ml/min Temperature: 25° C GRADIENT: 20%B to 98%B over 1.5min, 0.5 min hold at 98%B MS and UV: 220 nm LCMS-Method 8: COLUMN: X-bridge C8(50X4.5) mm, 5um Mobile phase A: 0.1% TFA in water Mobile phase B: 0.1% TFA in Acetonitrile Flow rate: 1 ml/min Temperature: Ambient GRADIENT: 5%B to 95%B over 2.5min, 1.5ml/min flow at 1.5 min hold at 95%B MS and UV: 220 LCMS-Method 9: COLUMN: Waters Acquity BEH C181.7um 2.1 x 50 mm Mobile phase A: 0.05% TFA in CH3CN:Water (5:95) Mobile phase B: 0.05% TFA in CH3CN:Water (95:5) Flow rate: 1 ml/min Temperature: 50° C GRADIENT: 0%B to 100%B over 1 min, stop time 1.5 min. MS and UV: 220, 254 LCMS-Method 10: COLUMN: XBridge C18, 2.1 mm x 50 mm, 1.7 ^m particles Mobile phase A: 0.05% TFA in CH3CN:Water (5:95) Mobile phase B: 0.05% TFA in CH3CN:Water (95:5) Flow rate: 1 ml/min Temperature: 50° C GRADIENT: 0-100 %B (0-3 min), 100 %B (3-3.5 min). MS and UV: 220 Scheme 1: Synthesis of Intermediates I-1 and I-2: Synthesis of I-1 Step-1: 4-(4-(Trifluoromethyl)-1H-imidazol-2-yl)benzonitrile: NC A mixture of 3,3-dibromo-1,1,1-trifluoropropan-2-one (22.64 g, 84 mmol) and sodium acetate (12.51 g, 153 mmol) in water (30 mL) was stirred at 95 °C for 30 min. The reaction mixture was cooled to 0 °C, a cold solution of 4-formylbenzonitrile (10.00 g, 76 mmol) in a mixture of NH4OH (28% aq, 35 mL) and MeOH (100 mL) was added. The resulting mixture was stirred at 25oC for 16 h. Volatiles were evaporated, the residue obtained was diluted with ethyl acetate and washed with water. The organic extract was dried over anhydrous Na2SO4, filtered and concentrated. The crude obtained was purified by flash chromatography on silica gel, 230-400 mesh, to obtain 4-(4-(trifluoromethyl)- 1H-imidazol-2-yl)benzonitrile (10.00 g, 37.6 mmol, 49.3%) as pale yellow solid. LCMS (ESI) m/z: 238.0 [M+H]+, LC retention time: 1.150 min. (LCMS Method 4). Step-2: 4-(1-Methyl-4-(trifluoromethyl)-1H-imidazol-2-yl)benzonitrile:
A mixture of 3,3-dibromo-1,1,1-trifluoropropan-2-one (22.64 g, 84 mmol) and sodium acetate (12.51 g, 153 mmol) in water (30 mL) was stirred at 95 °C for 30 min. The reaction mixture was cooled to 0 °C, a cold solution of 4-formylbenzonitrile (10.00 g, 76 mmol) in a mixture of NH4OH (28% aq, 35 mL) and MeOH (100 mL) was added. The resulting mixture was stirred at 25oC for 16 h. Volatiles were evaporated, the residue obtained was diluted with ethyl acetate and washed with water. The organic extract was dried over anhydrous Na2SO4, filtered and concentrated. The crude obtained was purified by flash chromatography on silica gel, 230-400 mesh, to obtain 4-(4-(trifluoromethyl)- 1H-imidazol-2-yl)benzonitrile (10.00 g, 37.6 mmol, 49.3%) as pale yellow solid. LCMS (ESI) m/z: 238.0 [M+H]+, LC retention time: 1.150 min. (LCMS Method 4). Step-2: 4-(1-Methyl-4-(trifluoromethyl)-1H-imidazol-2-yl)benzonitrile: To a stirred solution of 4-(4- - 2-yl)benzonitrile (4.0 g, 16.86 mmol) in DMF (40 mL), sodium hydride (60% dispersion in mineral oil, 0.809 g, 33.70 mmol) was added followed by addition of iodomethane (1.26 mL, 20.24 mmol) at 0oC and the mixture was stirred at 25 °C for 2 h. Chilled water was added and the contents extracted with ethyl acetate. The organic extract was dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The crude obtained was purified by flash chromatography on silica gel, 230-400 mesh to obtain 4-(1-methyl-4- (trifluoromethyl)-1H-imidazol-2-yl)benzonitrile 14a (3.0 g, 11.83 mmol, 70.1 %) as pale yellow solid. LCMS (ESI) m/z: 252.0 [M+H]+, LC retention time: 1.191 min. (LCMS Method 4). Step-3: 4-(1-Methyl-4-(trifluoromethyl)-1H-imidazol-2-yl)phenyl)methanamine (I- 1):
To a stirred solution of 4-(4- - 2-yl)benzonitrile (4.0 g, 16.86 mmol) in DMF (40 mL), sodium hydride (60% dispersion in mineral oil, 0.809 g, 33.70 mmol) was added followed by addition of iodomethane (1.26 mL, 20.24 mmol) at 0oC and the mixture was stirred at 25 °C for 2 h. Chilled water was added and the contents extracted with ethyl acetate. The organic extract was dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The crude obtained was purified by flash chromatography on silica gel, 230-400 mesh to obtain 4-(1-methyl-4- (trifluoromethyl)-1H-imidazol-2-yl)benzonitrile 14a (3.0 g, 11.83 mmol, 70.1 %) as pale yellow solid. LCMS (ESI) m/z: 252.0 [M+H]+, LC retention time: 1.191 min. (LCMS Method 4). Step-3: 4-(1-Methyl-4-(trifluoromethyl)-1H-imidazol-2-yl)phenyl)methanamine (I- 1): To a stirred solution of 4-(1-methyl-4-(trifluoromethyl)-1H-imidazol-2-yl)benzonitrile (2.9 g, 11.54 mmol) in THF (30 mL), lithium aluminium hydride (2 M in THF, 11.54 mL, 23.09 mmol) was added at 0oC and the mixture was stirred at 25 °C for 2 h. The reaction was quenched with saturated aqueous NH4Cl and extracted with ethyl acetate. The organic extract was dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The residue was purified by flash chromatography on silica gel, 230- 400 mesh, to obtain (4-(1-methyl-4-(trifluoromethyl)-1H-imidazol-2- yl)phenyl)methanamine (2.2 g, 7.7 mmol, 66.7%) as pale yellow gum. LCMS (ESI) m/z: 256.1 [M+H]+, LC retention time: 1.952 min. (LCMS Method 8). Synthesis of I-2 Step-1: 4-(1-Isopropyl-4-(trifluoromethyl)-1H-imidazol-2-yl)benzonitrile:
To a stirred solution of 4-(1-methyl-4-(trifluoromethyl)-1H-imidazol-2-yl)benzonitrile (2.9 g, 11.54 mmol) in THF (30 mL), lithium aluminium hydride (2 M in THF, 11.54 mL, 23.09 mmol) was added at 0oC and the mixture was stirred at 25 °C for 2 h. The reaction was quenched with saturated aqueous NH4Cl and extracted with ethyl acetate. The organic extract was dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The residue was purified by flash chromatography on silica gel, 230- 400 mesh, to obtain (4-(1-methyl-4-(trifluoromethyl)-1H-imidazol-2- yl)phenyl)methanamine (2.2 g, 7.7 mmol, 66.7%) as pale yellow gum. LCMS (ESI) m/z: 256.1 [M+H]+, LC retention time: 1.952 min. (LCMS Method 8). Synthesis of I-2 Step-1: 4-(1-Isopropyl-4-(trifluoromethyl)-1H-imidazol-2-yl)benzonitrile: a - 2-yl)benzonitrile (4.0 g, 16.86 mmol) in acetonitrile (60 mL), Cs2CO3 (11.0 g, 33.7 mmol) was added followed by addition of 2-iodopropane (2.46 mL, 25.3 mmol) at 0 oC and the mixture was stirred at 80 °C for 16 h. Volatiles were removed under reduced pressure, extracted with ethyl acetate and washed with water. The organic extract was dried over anhydrous Na2SO4, filtered and solvents evaporated under reduced pressure. The crude product obtained was purified by flash chromatography on silica gel, 230-400 mesh, to obtain 4-(1-isopropyl-4- (trifluoromethyl)-1H-imidazol-2-yl)benzonitrile (2.0 g, 7.08 mmol, 42%) as pale yellow solid. LCMS (ESI) m/z: 280.0 [M+H]+, LC retention time: 1.350 min (LCMS Method 4). Step-2: 4-(1-Isopropyl-4-(trifluoromethyl)-1H-imidazol-2-yl)phenyl)methanamine (I- 2):
a - 2-yl)benzonitrile (4.0 g, 16.86 mmol) in acetonitrile (60 mL), Cs2CO3 (11.0 g, 33.7 mmol) was added followed by addition of 2-iodopropane (2.46 mL, 25.3 mmol) at 0 oC and the mixture was stirred at 80 °C for 16 h. Volatiles were removed under reduced pressure, extracted with ethyl acetate and washed with water. The organic extract was dried over anhydrous Na2SO4, filtered and solvents evaporated under reduced pressure. The crude product obtained was purified by flash chromatography on silica gel, 230-400 mesh, to obtain 4-(1-isopropyl-4- (trifluoromethyl)-1H-imidazol-2-yl)benzonitrile (2.0 g, 7.08 mmol, 42%) as pale yellow solid. LCMS (ESI) m/z: 280.0 [M+H]+, LC retention time: 1.350 min (LCMS Method 4). Step-2: 4-(1-Isopropyl-4-(trifluoromethyl)-1H-imidazol-2-yl)phenyl)methanamine (I- 2):




To a stirred solution of 4-(1-isopropyl-4-(trifluoromethyl)-1H-imidazol-2-yl)benzonitrile (1.9 g, 6.80 mmol) in THF (20 mL), lithium aluminium hydride (2 M in THF, 6.80 mL, 13.61 mmol) at 0oC and the mixture was stirred at 25 °C for 2 h. The reaction was quenched with saturated aqueous NH4Cl and extracted with ethyl acetate. The organic extract was dried over anhydrous Na2SO4, filtered and concentrated. The crude residue was purified by flash chromatography on silica gel, 230-400 mesh, to obtain (4-(1- isopropyl-4-(trifluoromethyl)-1H-imidazol-2-yl)phenyl)methanamine (1.2 g, 4.14 mmol, 60.9%) as pale yellow gummy solid. LCMS (ESI) m/z: 284.0 [M+H]+, LC retention time: 2.759 min (LCMS Method 1). Scheme 2: Synthesis of Intermediate 3 Step-1: 4-chloro-1-isopropyl-1H-pyrazole: To a stirred solution of 4-chloro-1H-pyrazole (5.0 g, 48.8 mmol) in acetonitrile (60 mL), Cs2CO3 (31.8 g, 98 mmol) was added followed by addition of 2-iodopropane (5.69 mL, 58.5 mmol) and the mixture was heated at 80 °C for 2 h. The reaction mixture was cooled to ambient temperature, diluted with water and extracted with diethyl ether. The organic extract was dried over anhydrous Na2SO4, filtered and solvents evaporated from the filtrate under reduced pressure, at ~35oC, to obtain crude 4-chloro-1-isopropyl-1H-pyrazole (6.0 g, 39.4 mmol, 81%) as pale yellow oil, which was used as such for the next step. LCMS (ESI) m/z: 145.0 [M+H]+, LC retention time: 1.187 min. (LCMS Method 7). Step-2: 4-chloro-1-isopropyl-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-
To a stirred solution of 4-chloro-1H-pyrazole (5.0 g, 48.8 mmol) in acetonitrile (60 mL), Cs2CO3 (31.8 g, 98 mmol) was added followed by addition of 2-iodopropane (5.69 mL, 58.5 mmol) and the mixture was heated at 80 °C for 2 h. The reaction mixture was cooled to ambient temperature, diluted with water and extracted with diethyl ether. The organic extract was dried over anhydrous Na2SO4, filtered and solvents evaporated from the filtrate under reduced pressure, at ~35oC, to obtain crude 4-chloro-1-isopropyl-1H-pyrazole (6.0 g, 39.4 mmol, 81%) as pale yellow oil, which was used as such for the next step. LCMS (ESI) m/z: 145.0 [M+H]+, LC retention time: 1.187 min. (LCMS Method 7). Step-2: 4-chloro-1-isopropyl-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H- a g, in tetrahydrofuran (50 mL), n-butyllithium (1.6 M in hexanes, 28.5 mL, 45.6 mmol) was added at 0oC and the mixture was stirred at ambient temperature for 1 h. The mixture was recooled to -78oC, 2-isopropoxy-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (8.49 g, 45.6 mmol) was added and stirred for 2 h. The reaction was quenched with saturated aqueous NH4Cl and extracted with ethyl acetate. The organic extract was dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure to obtained crude 4-chloro-1- isopropyl-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole (6.00 g, 14.41 mmol, 37.9%) as dark brown oil, which was used as such in the subsequent steps. LCMS (ESI) m/z: 271.0 [M+H]+, LC retention time: 1.327 min. (LCMS Method 4).
a g, in tetrahydrofuran (50 mL), n-butyllithium (1.6 M in hexanes, 28.5 mL, 45.6 mmol) was added at 0oC and the mixture was stirred at ambient temperature for 1 h. The mixture was recooled to -78oC, 2-isopropoxy-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (8.49 g, 45.6 mmol) was added and stirred for 2 h. The reaction was quenched with saturated aqueous NH4Cl and extracted with ethyl acetate. The organic extract was dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure to obtained crude 4-chloro-1- isopropyl-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole (6.00 g, 14.41 mmol, 37.9%) as dark brown oil, which was used as such in the subsequent steps. LCMS (ESI) m/z: 271.0 [M+H]+, LC retention time: 1.327 min. (LCMS Method 4).


Scheme 3. Synthesis of example 1. Step 1: 2-chloro-N-(4-(1-methyl-4-(trifluoromethyl)-1H-imidazol-2- yl)benzyl)pyrido[3,4-d]pyrimidin-4-amine (4) A mixture of 2,4-
Step 1: 2-chloro-N-(4-(1-methyl-4-(trifluoromethyl)-1H-imidazol-2- yl)benzyl)pyrido[3,4-d]pyrimidin-4-amine (4) A mixture of 2,4- 0.300 mmol), (4-(1-methyl-4- (trifluoromethyl)-1H-imidazol-2-yl)phenyl)methanamine (I-1, 77 mg, 0.300 mmol) and TEA (41.8 μl, 0.300 mmol) in IPA (3.0 mL) was stirred at 80 °C for 18 hours. The reaction mixture was cooled to RT and concentrated. The crude product was subjected to ISCO flash chromatography (silica gel/hexane-EtOAc 100:0 to 0:100 gradient). Yield of 2- chloro-N-(4-(1-methyl-4-(trifluoromethyl)-1H-imidazol-2-yl)benzyl)pyrido[3,4- d]pyrimidin-4-amine (113 mg, 0.270 mmol, 90 % yield) as light brown solid. LCMS (ESI) m/z: 419.0 [M+H]+, LC retention time: 0.88 min (LCMS method 9). Examples in Table 1 were prepared according to procedure described in step-1 above. Table 1 Intermediate Structure Name LCMS (ESI) Number 2-chloro-N-(4-(1- m/z: 447.0 isopropyl-4- [M+H]+ (trifluoromethyl)-1H- LC retention 5 imidazol-2- time: 0.95 min yl)benzyl)pyrido[3,4- (LCMS Method d]pyrimidin-4-amine 9) m/z: 447.0 2-chloro-N-(4-(1- [M+H]+, LC isopropyl-4- retention time: (trifluoromethyl)-1H- 6 1.00 min imidazol-2- (LCMS Method yl)benzyl)pyrido[2,3- 9). d]pyrimidin-4-amine
0.300 mmol), (4-(1-methyl-4- (trifluoromethyl)-1H-imidazol-2-yl)phenyl)methanamine (I-1, 77 mg, 0.300 mmol) and TEA (41.8 μl, 0.300 mmol) in IPA (3.0 mL) was stirred at 80 °C for 18 hours. The reaction mixture was cooled to RT and concentrated. The crude product was subjected to ISCO flash chromatography (silica gel/hexane-EtOAc 100:0 to 0:100 gradient). Yield of 2- chloro-N-(4-(1-methyl-4-(trifluoromethyl)-1H-imidazol-2-yl)benzyl)pyrido[3,4- d]pyrimidin-4-amine (113 mg, 0.270 mmol, 90 % yield) as light brown solid. LCMS (ESI) m/z: 419.0 [M+H]+, LC retention time: 0.88 min (LCMS method 9). Examples in Table 1 were prepared according to procedure described in step-1 above. Table 1 Intermediate Structure Name LCMS (ESI) Number 2-chloro-N-(4-(1- m/z: 447.0 isopropyl-4- [M+H]+ (trifluoromethyl)-1H- LC retention 5 imidazol-2- time: 0.95 min yl)benzyl)pyrido[3,4- (LCMS Method d]pyrimidin-4-amine 9) m/z: 447.0 2-chloro-N-(4-(1- [M+H]+, LC isopropyl-4- retention time: (trifluoromethyl)-1H- 6 1.00 min imidazol-2- (LCMS Method yl)benzyl)pyrido[2,3- 9). d]pyrimidin-4-amine


Step 2: 2-(4-cyclopropyl-6-methoxypyrimidin-5-yl)-N-(4-(1-methyl-4- (trifluoromethyl)-1H-imidazol-2-yl)benzyl)pyrido[3,4-d]pyrimidin-4-amine (example 1): imidazol-2- mg, , 4-cyclopropyl-6- methoxy-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyrimidine (23.74 mg, 0.086 mmol), potassium phosphate tribasic (30.4 mg, 0.143 mmol) and tetrakis(triphenylphosphine)palladium(0) (4.14 mg, 3.58 μmol) in dioxane (1.0 mL) and water (0.111 mL) was flushed with nitrogen for 5 min. and stirred at 100 °C for 18 hours. The reaction mixture was cooled to RT and partitioned between EtOAc (5 mL) and water. The ethyl acetate layer was dried over sodium sulphate and concentrated. The crude product was purified with XBridge C18, 19 mm x 200 mm, 5 ^m particles; Flow Rate: 20 mL/min; Column Temperature: 25 °C. Mobile Phase A (ACN/H2O (5:95) with10 mM AA), Mobile Phase B (ACN/H2O (95:5) with 10 mM AA). Gradient: 0 B to 100% B in 10 min. Yield 2-(4-cyclopropyl-6-methoxypyrimidin-5-yl)-N-(4-(1-methyl-4- (trifluoromethyl)-1H-imidazol-2-yl)benzyl)pyrido[3,4-d]pyrimidin-4-amine (25 mg, 0.045 mmol, 62.7 % yield). LCMS (ESI) m/z: 533.1 [M+H]+, LC retention time: 1.53 min. (LCMS Method 10); 1H NMR (500 MHz, DMSO-d6) į 9.42 (br t, J=5.4 Hz, 1H), 9.16 (s, 1H), 8.70 (d, J=5.9 Hz, 1H), 8.64 (s, 1H), 8.24 (d, J=5.6 Hz, 1H), 7.91 (s, 1H), 7.66 (d, J=7.4 Hz, 2H), 7.50 (br d, J=8.2 Hz, 2H), 4.86 (br d, J=5.2 Hz, 2H), 3.83 (s, 3H), 3.75 (s, 3H), 1.78 - 1.75 (m, 1H), 1.01 - 0.98 (m, 2H), 0.79 (br dd, J=7.8, 3.1 Hz, 2H). Examples in Table 2 were prepared according to procedure described in step-2 above Table 2 Example Structure Name LCMS (ESI)1H NMR Number 1H NMR (500 MHz, DMSO-d6) į 9.43 (br t, J=5.4 Hz, 1H), 9.16 (s, 1H), 8.70 (d, J=5.6 2-(4-cyclopropyl-6- Hz, 1H), 8.64 (s, methoxypyrimidin- m/z: 561.2 + 1H), 8.24 (d, J=5.3 5-yl)-N-(4-(1- [M+H] , LC Hz, 1H), 8.15 (s, isopropyl-4- retention 1H), 7.51 (s, 4H), 2. (trifluoromethyl)- time: 1.91 4.87 (br d, J=5.4 1H-imidazol-2- min. Hz, 2H), 4.46 - yl)benzyl)pyrido[3,4 (LCMS 4.41 (m, 1H), 3.83 -d]pyrimidin-4- Method 10). (s, 3H), 1.75 (br dd, amine J=8.3, 3.9 Hz, 1H), 1.39 (br d, J=6.6 Hz, 6H), 0.98 (br s, 2H), 0.77 (br dd, J=7.7, 3.1 Hz, 2H).
mg, , 4-cyclopropyl-6- methoxy-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyrimidine (23.74 mg, 0.086 mmol), potassium phosphate tribasic (30.4 mg, 0.143 mmol) and tetrakis(triphenylphosphine)palladium(0) (4.14 mg, 3.58 μmol) in dioxane (1.0 mL) and water (0.111 mL) was flushed with nitrogen for 5 min. and stirred at 100 °C for 18 hours. The reaction mixture was cooled to RT and partitioned between EtOAc (5 mL) and water. The ethyl acetate layer was dried over sodium sulphate and concentrated. The crude product was purified with XBridge C18, 19 mm x 200 mm, 5 ^m particles; Flow Rate: 20 mL/min; Column Temperature: 25 °C. Mobile Phase A (ACN/H2O (5:95) with10 mM AA), Mobile Phase B (ACN/H2O (95:5) with 10 mM AA). Gradient: 0 B to 100% B in 10 min. Yield 2-(4-cyclopropyl-6-methoxypyrimidin-5-yl)-N-(4-(1-methyl-4- (trifluoromethyl)-1H-imidazol-2-yl)benzyl)pyrido[3,4-d]pyrimidin-4-amine (25 mg, 0.045 mmol, 62.7 % yield). LCMS (ESI) m/z: 533.1 [M+H]+, LC retention time: 1.53 min. (LCMS Method 10); 1H NMR (500 MHz, DMSO-d6) į 9.42 (br t, J=5.4 Hz, 1H), 9.16 (s, 1H), 8.70 (d, J=5.9 Hz, 1H), 8.64 (s, 1H), 8.24 (d, J=5.6 Hz, 1H), 7.91 (s, 1H), 7.66 (d, J=7.4 Hz, 2H), 7.50 (br d, J=8.2 Hz, 2H), 4.86 (br d, J=5.2 Hz, 2H), 3.83 (s, 3H), 3.75 (s, 3H), 1.78 - 1.75 (m, 1H), 1.01 - 0.98 (m, 2H), 0.79 (br dd, J=7.8, 3.1 Hz, 2H). Examples in Table 2 were prepared according to procedure described in step-2 above Table 2 Example Structure Name LCMS (ESI)1H NMR Number 1H NMR (500 MHz, DMSO-d6) į 9.43 (br t, J=5.4 Hz, 1H), 9.16 (s, 1H), 8.70 (d, J=5.6 2-(4-cyclopropyl-6- Hz, 1H), 8.64 (s, methoxypyrimidin- m/z: 561.2 + 1H), 8.24 (d, J=5.3 5-yl)-N-(4-(1- [M+H] , LC Hz, 1H), 8.15 (s, isopropyl-4- retention 1H), 7.51 (s, 4H), 2. (trifluoromethyl)- time: 1.91 4.87 (br d, J=5.4 1H-imidazol-2- min. Hz, 2H), 4.46 - yl)benzyl)pyrido[3,4 (LCMS 4.41 (m, 1H), 3.83 -d]pyrimidin-4- Method 10). (s, 3H), 1.75 (br dd, amine J=8.3, 3.9 Hz, 1H), 1.39 (br d, J=6.6 Hz, 6H), 0.98 (br s, 2H), 0.77 (br dd, J=7.7, 3.1 Hz, 2H).

Example Structure Name LCMS (ESI)1H NMR Number 1H NMR (500 MHz, DMSO-d6) į 9.47 (br t, J=6.0 Hz, 1H), 8.93 - 2-(4-chloro-1- 8.90 (m, 1H), 8.20 isopropyl-1H- m/z: 555.2 + (br d, J=8.5 Hz, pyrazol-5-yl)-N-(4- [M+H] , 1H), 8.13 (s, 1H), (1-isopropyl-4- LC retention 7.95 - 7.90 (m, 1H), 3. (trifluoromethyl)- time: 2.15 7.64 (s, 1H), 7.51 1H-imidazol-2- min. (s, 4H), 5.22 - 5.15 yl)benzyl)pyrido[2,3 (LCMS (m, 1H), 4.91 (br d, -d]pyrimidin-4- Method 10). J=6.3 Hz, 2H), 4.47 amine - 4.41 (m, 1H), 1.38 (br d, J=6.6 Hz, 6H), 1.25 (br d, J=6.6 Hz, 6H).
Scheme 4: Synthesis of examples 4 and 5 Example 4: 2-(4-cyclopropyl-6-methoxypyrimidin-5-yl)-4-((4-(1-methyl-4- (trifluoromethyl)-1H-imidazol-2-yl)benzyl)amino)-6,7-dihydropyrido[3,4- d]pyrimidin-8(5H)-one
Example 4: 2-(4-cyclopropyl-6-methoxypyrimidin-5-yl)-4-((4-(1-methyl-4- (trifluoromethyl)-1H-imidazol-2-yl)benzyl)amino)-6,7-dihydropyrido[3,4- d]pyrimidin-8(5H)-one Step-1: Tert-butyl 2,4-dichloro-8-oxo-5,8-dihydropyrido[3,4-d]pyrimidine-7(6H)- carboxylate (4b) was added to sodium periodate
Step-1: Tert-butyl 2,4-dichloro-8-oxo-5,8-dihydropyrido[3,4-d]pyrimidine-7(6H)- carboxylate (4b) was added to sodium periodate g, a mL) and water (2 mL) at 0oC. To the above mixture, a solution of tert-butyl 2,4-dichloro-5,8-dihydropyrido[3,4- d]pyrimidine-7(6H)-carboxylate (0.100 g, 0.329 mmol) in ethyl acetate (1 ml) was added at 0oC and the mixture was stirred at ambient temperature for 16 h. The reaction mixture was filtered through a celite bed and the celite bed was further washed with ethyl acetate. The filtrate was concentrated under reduced pressure. The residue was purified by flash chromatography using a Biotage chromatography instrument, on silica gel column (100- 200 mesh), eluting with 0-40% gradient of ethyl acetate in petroleum ether to obtain tert- butyl 2,4-dichloro-8-oxo-5,8-dihydropyrido[3,4-d]pyrimidine-7(6H)-carboxylate (0.060 g, 0.186 mmol, 56.6%) as off white solid. LCMS (ESI) m/z: 218.0 [M-Boc+H]+, LC retention time: 2.33 min. (LCMS Method 1). Step-2 : Tert-butyl 2-chloro-4-((4-(1-methyl-4-(trifluoromethyl)-1H-imidazol-2- yl)benzyl)amino)-8-oxo-5,8-dihydropyrido[3,4-d]pyrimidine-7(6H)-carboxylate(4c)
g, a mL) and water (2 mL) at 0oC. To the above mixture, a solution of tert-butyl 2,4-dichloro-5,8-dihydropyrido[3,4- d]pyrimidine-7(6H)-carboxylate (0.100 g, 0.329 mmol) in ethyl acetate (1 ml) was added at 0oC and the mixture was stirred at ambient temperature for 16 h. The reaction mixture was filtered through a celite bed and the celite bed was further washed with ethyl acetate. The filtrate was concentrated under reduced pressure. The residue was purified by flash chromatography using a Biotage chromatography instrument, on silica gel column (100- 200 mesh), eluting with 0-40% gradient of ethyl acetate in petroleum ether to obtain tert- butyl 2,4-dichloro-8-oxo-5,8-dihydropyrido[3,4-d]pyrimidine-7(6H)-carboxylate (0.060 g, 0.186 mmol, 56.6%) as off white solid. LCMS (ESI) m/z: 218.0 [M-Boc+H]+, LC retention time: 2.33 min. (LCMS Method 1). Step-2 : Tert-butyl 2-chloro-4-((4-(1-methyl-4-(trifluoromethyl)-1H-imidazol-2- yl)benzyl)amino)-8-oxo-5,8-dihydropyrido[3,4-d]pyrimidine-7(6H)-carboxylate(4c)



To a stirred solution of tert-butyl 2,4-dichloro-8-oxo-5,8-dihydropyrido[3,4-d]pyrimidine- 7(6H)-carboxylate (0.060 g, 0.186 mmol) and (4-(1-methyl-4-(trifluoromethyl)-1H- imidazol-2-yl)phenyl)methanamine (I-1, 0.0523 g, 0.205 mmol) in acetonitrile (2 mL), Cs2CO3 (0.091 g, 0.28 mmol) was added at ambient temperature and the mixture was heated at 80 °C for 3 h. The reaction mixture was cooled to ambient temperature, and filtered through a celite bed. The celite bed was further washed with ethyl acetate, the washings and the filtrate were concentrated under reduced pressure. The residue was purified by flash chromatography using a Biotage chromatography instrument, on silica gel column (100-200 mesh), eluting with 0-60% gradient of ethyl acetate in petroleum ether to obtain tert-butyl 2-chloro-4-((4-(1-methyl-4-(trifluoromethyl)-1H-imidazol-2- yl)benzyl)amino)-8-oxo-5,8-dihydropyrido[3,4-d]pyrimidine-7(6H)-carboxylate (0.080 g, 0.134 mmol, 71.9%) as liquid. LCMS (ESI) m/z: 537.0 [M+H]+, LC retention time: 2.88 min. (LCMS Method 1). Step 3: Tert-butyl 2-(4-cyclopropyl-6-methoxypyrimidin-5-yl)-4-((4-(1-methyl-4- (trifluoromethyl)-1H-imidazol-2-yl)benzyl)amino)-8-oxo-5,8-dihydropyrido[3,4- d]pyrimidine-7(6H)-carboxylate (4d) (trifluoromethyl)-1H- imidazol-2-yl)benzyl)amino)-8-oxo-5,8-dihydropyrido[3,4-d]pyrimidine-7(6H)- carboxylate (0.080 g, 0.134 mmol) and (4-cyclopropyl-6-methoxypyrimidin-5-yl)boronic acid (0.0325 g, 0.168 mmol) in 1,4-dioxane (2 mL), Tetrakis(triphenylphosphine)palladium(0) (0.015 mg, 0.13 mmol) and tripotassium phosphate (0.057 g, 0.268 mmol) were added under nitrogen atmosphere and the mixture was heated at 90 °C for 16 h. The reaction mixture was cooled to ambient temperature and filtered through a celite bed. The celite bed was further washed with ethyl acetate, the washings and the filtrate were mixed and concentrated under reduced pressure. The residue obtained was purified by flash chromatography using a Biotage chromatography instrument, on silica gel column (100-200 mesh), eluting with 0-80% gradient of ethyl acetate in petroleum ether to obtain tert-butyl 2-(4-cyclopropyl-6-methoxypyrimidin-5- yl)-4-((4-(1-methyl-4-(trifluoromethyl)-1H-imidazol-2-yl)benzyl)amino)-8-oxo-5,8- dihydropyrido[3,4-d]pyrimidine-7(6H)-carboxylate (0.040 g, 0.047 mmol, 34.7%) as pale yellow thick gum. LCMS (ESI) m/z: 651.0 [M+H]+, LC retention time: 1.20 min. (LCMS Method 7). Step 4: 2-(4-cyclopropyl-6-methoxypyrimidin-5-yl)-4-((4-(1-methyl-4- (trifluoromethyl)-1H-imidazol-2-yl)benzyl)amino)-6,7-dihydropyrido[3,4- d]pyrimidin-8(5H)-one (example 4)
(trifluoromethyl)-1H- imidazol-2-yl)benzyl)amino)-8-oxo-5,8-dihydropyrido[3,4-d]pyrimidine-7(6H)- carboxylate (0.080 g, 0.134 mmol) and (4-cyclopropyl-6-methoxypyrimidin-5-yl)boronic acid (0.0325 g, 0.168 mmol) in 1,4-dioxane (2 mL), Tetrakis(triphenylphosphine)palladium(0) (0.015 mg, 0.13 mmol) and tripotassium phosphate (0.057 g, 0.268 mmol) were added under nitrogen atmosphere and the mixture was heated at 90 °C for 16 h. The reaction mixture was cooled to ambient temperature and filtered through a celite bed. The celite bed was further washed with ethyl acetate, the washings and the filtrate were mixed and concentrated under reduced pressure. The residue obtained was purified by flash chromatography using a Biotage chromatography instrument, on silica gel column (100-200 mesh), eluting with 0-80% gradient of ethyl acetate in petroleum ether to obtain tert-butyl 2-(4-cyclopropyl-6-methoxypyrimidin-5- yl)-4-((4-(1-methyl-4-(trifluoromethyl)-1H-imidazol-2-yl)benzyl)amino)-8-oxo-5,8- dihydropyrido[3,4-d]pyrimidine-7(6H)-carboxylate (0.040 g, 0.047 mmol, 34.7%) as pale yellow thick gum. LCMS (ESI) m/z: 651.0 [M+H]+, LC retention time: 1.20 min. (LCMS Method 7). Step 4: 2-(4-cyclopropyl-6-methoxypyrimidin-5-yl)-4-((4-(1-methyl-4- (trifluoromethyl)-1H-imidazol-2-yl)benzyl)amino)-6,7-dihydropyrido[3,4- d]pyrimidin-8(5H)-one (example 4) A solution of tert-butyl 2-(4-cyclopropyl-6-methoxypyrimidin-5-yl)-4-((4-(1-methyl-4- (trifluoromethyl)-1H-imidazol-2-yl)benzyl)amino)-8-oxo-5,8-dihydropyrido[3,4- d]pyrimidine-7(6H)-carboxylate (0.040 g, 0.047 mmol) in dichloromethane (2 mL) was cooled to 0oC (ice bath). Trifluoroacetic acid (10.80 μL, 0.140 mmol) was added and the mixture was stirred for 3 h, during which time the temperature of the reaction mixture was allowed to rise to ambient temperature. After completion of the reaction (monitored by TLC and UPLC-MS), solvents were evaporated from the reaction mixture and the crude obtained was purified by preparative HPLC [Method: Diluent: WATER: THF: MeCN (30:30:40); XBridge C18 (150 x19)mm, 5micron; Temperature: Ambient; Mobile methoxypyrimidin-5-yl)-4-((4-(1-methyl-4-(trifluoromethyl)-1H-imidazol-2- yl)benzyl)amino)-6,7-dihydropyrido[3,4-d]pyrimidin-8(5H)-one (0.010 g, 0.018 mmol, 38.4%) as off white solid. LCMS (ESI) m/z: 551.2 [M+H]+, LC retention time: 2.61 min. (LCMS Method 1);1H-NMR: (400 MHz,DMSO-d6) į = 8.61 (s, 1H), 8.19 (s, 1H), 8.04 (t, J = 6.0 Hz, 1H), 7.91 (d, J = 1.2 Hz, 1H), 7.64 (d, J = 8.4, Hz, 2H), 7.43 (d, J = 8.40 Hz, 2H), 4.68 (d, J = 5.6 Hz, 2H), 3.81 (s, 3H), 3.75 (s, 3H), 3.45 (t, J = 2.8 Hz, 2H), 2.79 (t, J = 6.8 Hz, 2H), 1.61-1.65 (m, 1H), 0.94-0.97 (m, 2H), 0.75-0.78 (m, 2H). Example 5: Synthesis of 2-(4-cyclopropyl-6-methoxypyrimidin-5-yl)-7-methyl-4-((4- (1-methyl-4-(trifluoromethyl)-1H-imidazol-2-yl)benzyl)amino)-6,7- dihydropyrido[3,4-d]pyrimidin-8(5H)-one:
A solution of tert-butyl 2-(4-cyclopropyl-6-methoxypyrimidin-5-yl)-4-((4-(1-methyl-4- (trifluoromethyl)-1H-imidazol-2-yl)benzyl)amino)-8-oxo-5,8-dihydropyrido[3,4- d]pyrimidine-7(6H)-carboxylate (0.040 g, 0.047 mmol) in dichloromethane (2 mL) was cooled to 0oC (ice bath). Trifluoroacetic acid (10.80 μL, 0.140 mmol) was added and the mixture was stirred for 3 h, during which time the temperature of the reaction mixture was allowed to rise to ambient temperature. After completion of the reaction (monitored by TLC and UPLC-MS), solvents were evaporated from the reaction mixture and the crude obtained was purified by preparative HPLC [Method: Diluent: WATER: THF: MeCN (30:30:40); XBridge C18 (150 x19)mm, 5micron; Temperature: Ambient; Mobile methoxypyrimidin-5-yl)-4-((4-(1-methyl-4-(trifluoromethyl)-1H-imidazol-2- yl)benzyl)amino)-6,7-dihydropyrido[3,4-d]pyrimidin-8(5H)-one (0.010 g, 0.018 mmol, 38.4%) as off white solid. LCMS (ESI) m/z: 551.2 [M+H]+, LC retention time: 2.61 min. (LCMS Method 1);1H-NMR: (400 MHz,DMSO-d6) į = 8.61 (s, 1H), 8.19 (s, 1H), 8.04 (t, J = 6.0 Hz, 1H), 7.91 (d, J = 1.2 Hz, 1H), 7.64 (d, J = 8.4, Hz, 2H), 7.43 (d, J = 8.40 Hz, 2H), 4.68 (d, J = 5.6 Hz, 2H), 3.81 (s, 3H), 3.75 (s, 3H), 3.45 (t, J = 2.8 Hz, 2H), 2.79 (t, J = 6.8 Hz, 2H), 1.61-1.65 (m, 1H), 0.94-0.97 (m, 2H), 0.75-0.78 (m, 2H). Example 5: Synthesis of 2-(4-cyclopropyl-6-methoxypyrimidin-5-yl)-7-methyl-4-((4- (1-methyl-4-(trifluoromethyl)-1H-imidazol-2-yl)benzyl)amino)-6,7- dihydropyrido[3,4-d]pyrimidin-8(5H)-one: Step-1: Tert-butyl (2-(4-cyclopropyl-6-methoxypyrimidin-5-yl)-8-oxo-5,6,7,8- tetrahydropyrido[3,4-d]pyrimidin-4-yl)(4-(1-methyl-4-(trifluoromethyl)-1H- imidazol-2-yl)benzyl)carbamate (5a):
Step-1: Tert-butyl (2-(4-cyclopropyl-6-methoxypyrimidin-5-yl)-8-oxo-5,6,7,8- tetrahydropyrido[3,4-d]pyrimidin-4-yl)(4-(1-methyl-4-(trifluoromethyl)-1H- imidazol-2-yl)benzyl)carbamate (5a):



A solution of 2-(4-cyclopropyl-6-methoxypyrimidin-5-yl)-4-((4-(1-methyl-4- (trifluoromethyl)-1H-imidazol-2-yl)benzyl)amino)-6,7-dihydropyrido[3,4-d]pyrimidin- 8(5H)-one trifluoroacetate salt (0.214 g, 0.283 mmol) in dichloromethane (8 mL) was cooled to 0oC. (ice bath), Triethylamine (0.197 mL, 1.417 mmol) and di-tert-butyl carbonate (0.132 mL, 0.567 mmol) were added followed by DMAP (0.003 g, 0.028 mmol) and the mixture was stirred at ambient temperature for 16 h. The reaction mixture was diluted with chilled water and extracted with dichloromethane. The combined organic extract was washed with brine, dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure. The crude obtained was purified by flash chromatography using a Biotage chromatography instrument, on silica gel column (100- 200 mesh), eluting with 0-2% gradient of methanol in dichloromethane to obtain tert- butyl (2-(4-cyclopropyl-6-methoxypyrimidin-5-yl)-8-oxo-5,6,7,8-tetrahydropyrido[3,4- d]pyrimidin-4-yl)(4-(1-methyl-4-(trifluoromethyl)-1H-imidazol-2-yl)benzyl)carbamate (0.170 g, 0.253 mmol, 89%) as pale yellow thick gum. LCMS (ESI) m/z: 651.2 [M+H]+, Step-6: tert-butyl (2-(4-cyclopropyl-6-methoxypyrimidin-5-yl)-7-methyl-8-oxo- 5,6,7,8-tetrahydropyrido[3,4-d]pyrimidin-4-yl)(4-(1-methyl-4-(trifluoromethyl)-1H- imidazol-2-yl)benzyl)carbamate (5b)
Step-6: tert-butyl (2-(4-cyclopropyl-6-methoxypyrimidin-5-yl)-7-methyl-8-oxo- 5,6,7,8-tetrahydropyrido[3,4-d]pyrimidin-4-yl)(4-(1-methyl-4-(trifluoromethyl)-1H- imidazol-2-yl)benzyl)carbamate (5b) To a stirred solution of tert-butyl (2-(4-cyclopropyl-6-methoxypyrimidin-5-yl)-8-oxo- 5,6,7,8-tetrahydropyrido[3,4-d]pyrimidin-4-yl)(4-(1-methyl-4-(trifluoromethyl)-1H- imidazol-2-yl)benzyl)carbamate (0.150 g, 0.223 mmol) in DMF (2 mL), Cs2CO3 (0.145 g, 0.446 mmol) and iodomethane (0.028 mL, 0.446 mmol) was added and the mixture was stirred at ambient temperature for 16 hour. The reaction mixture was poured into chilled water, the precipitated solid was filtered and dried to obtain tert-butyl (2-(4-cyclopropyl- 6-methoxypyrimidin-5-yl)-7-methyl-8-oxo-5,6,7,8-tetrahydropyrido[3,4-d]pyrimidin-4- yl)(4-(1-methyl-4-(trifluoromethyl)-1H-imidazol-2-yl)benzyl)carbamate (0.120 g, 0.119 mmol, 53.4%) as light brown solid. LCMS (ESI) m/z: 665.2 [M+H]+, LC retention time: 1.45 min. (LCMS Method 3). Step-7: 2-(4-cyclopropyl-6-methoxypyrimidin-5-yl)-7-methyl-4-((4-(1-methyl-4- (trifluoromethyl)-1H-imidazol-2-yl)benzyl)amino)-6,7-dihydropyrido[3,4- d]pyrimidin-8(5H)-one (example 5)
To a stirred solution of tert-butyl (2-(4-cyclopropyl-6-methoxypyrimidin-5-yl)-8-oxo- 5,6,7,8-tetrahydropyrido[3,4-d]pyrimidin-4-yl)(4-(1-methyl-4-(trifluoromethyl)-1H- imidazol-2-yl)benzyl)carbamate (0.150 g, 0.223 mmol) in DMF (2 mL), Cs2CO3 (0.145 g, 0.446 mmol) and iodomethane (0.028 mL, 0.446 mmol) was added and the mixture was stirred at ambient temperature for 16 hour. The reaction mixture was poured into chilled water, the precipitated solid was filtered and dried to obtain tert-butyl (2-(4-cyclopropyl- 6-methoxypyrimidin-5-yl)-7-methyl-8-oxo-5,6,7,8-tetrahydropyrido[3,4-d]pyrimidin-4- yl)(4-(1-methyl-4-(trifluoromethyl)-1H-imidazol-2-yl)benzyl)carbamate (0.120 g, 0.119 mmol, 53.4%) as light brown solid. LCMS (ESI) m/z: 665.2 [M+H]+, LC retention time: 1.45 min. (LCMS Method 3). Step-7: 2-(4-cyclopropyl-6-methoxypyrimidin-5-yl)-7-methyl-4-((4-(1-methyl-4- (trifluoromethyl)-1H-imidazol-2-yl)benzyl)amino)-6,7-dihydropyrido[3,4- d]pyrimidin-8(5H)-one (example 5) A solution of tert-butyl (2-(4-cyclopropyl-6-methoxypyrimidin-5-yl)-7-methyl-8-oxo- 5,6,7,8-tetrahydropyrido[3,4-d]pyrimidin-4-yl)(4-(1-methyl-4-(trifluoromethyl)-1H- imidazol-2-yl)benzyl)carbamate (0.115 g, 0.118 mmol) in dichloromethane (5 mL) was cooled to 0oC ( ice bath) and trifluoroacetic acid (0.027 mL, 0.353 mmol) was added. The mixture was stirred for 4 h, during which time temperature of the solution was allowed to rise to ambient temperature. After completion of the reaction (monitored by TLC and UPLC-MS), 10% (w/v) aqueous sodium bicarbonate was added to the reaction mixture until pH of the reaction mixture reached 7~8. The contents were extracted with 10% methanol in dichloromethane. The combined organic extract was washed with brine, dried over anhydrous sodium sulfate, filtered and solvents concentrated under reduced pressure. The crude obtained was purified by reverse phase flash chromatography on a Teledyne Isco-Combi flash instrument [Method: Diluent: THF: MeCN (30:70); Column: RediSep 40g C18, 20-40 micron; Temperature: Ambient; Mobile phase A: 5mM Ammonium format in Water, Mobile phase B: Acetonitrile; Flow :20mL/min; Compound elution: 43%Acetonitrile/ 5mM Ammonium formate in water] to obtain 2-(4-cyclopropyl- 6-methoxypyrimidin-5-yl)-7-methyl-4-((4-(1-methyl-4-(trifluoromethyl)-1H-imidazol-2- yl)benzyl)amino)-6,7-dihydropyrido[3,4-d]pyrimidin-8(5H)-one (0.038 g, 0.065 mmol, 55.4%) as off white solid. LCMS (ESI) m/z: 565.2 [M+H]+, LC retention time: 2.79 min. (LCMS Method 1);1H-NMR: (400 MHz,DMSO-d6) į = 8.63 (s, 1H), 8.32 (s, 1H), 7.93 (q, J = 1.2 Hz, 1H), 7.68 (d, J = 8.4 Hz, 2H), 7.44 (d, J = 8.0 Hz, 2H), 4.81 (s, 2H), 3.85 (s, 3H), 3.78 (s, 3H), 3.30-3.32 (m, 2H), 3.09 (s, 3H), 3.04 (t, J = 6.4 Hz, 2H), 1.66-1.70 To
A solution of tert-butyl (2-(4-cyclopropyl-6-methoxypyrimidin-5-yl)-7-methyl-8-oxo- 5,6,7,8-tetrahydropyrido[3,4-d]pyrimidin-4-yl)(4-(1-methyl-4-(trifluoromethyl)-1H- imidazol-2-yl)benzyl)carbamate (0.115 g, 0.118 mmol) in dichloromethane (5 mL) was cooled to 0oC ( ice bath) and trifluoroacetic acid (0.027 mL, 0.353 mmol) was added. The mixture was stirred for 4 h, during which time temperature of the solution was allowed to rise to ambient temperature. After completion of the reaction (monitored by TLC and UPLC-MS), 10% (w/v) aqueous sodium bicarbonate was added to the reaction mixture until pH of the reaction mixture reached 7~8. The contents were extracted with 10% methanol in dichloromethane. The combined organic extract was washed with brine, dried over anhydrous sodium sulfate, filtered and solvents concentrated under reduced pressure. The crude obtained was purified by reverse phase flash chromatography on a Teledyne Isco-Combi flash instrument [Method: Diluent: THF: MeCN (30:70); Column: RediSep 40g C18, 20-40 micron; Temperature: Ambient; Mobile phase A: 5mM Ammonium format in Water, Mobile phase B: Acetonitrile; Flow :20mL/min; Compound elution: 43%Acetonitrile/ 5mM Ammonium formate in water] to obtain 2-(4-cyclopropyl- 6-methoxypyrimidin-5-yl)-7-methyl-4-((4-(1-methyl-4-(trifluoromethyl)-1H-imidazol-2- yl)benzyl)amino)-6,7-dihydropyrido[3,4-d]pyrimidin-8(5H)-one (0.038 g, 0.065 mmol, 55.4%) as off white solid. LCMS (ESI) m/z: 565.2 [M+H]+, LC retention time: 2.79 min. (LCMS Method 1);1H-NMR: (400 MHz,DMSO-d6) į = 8.63 (s, 1H), 8.32 (s, 1H), 7.93 (q, J = 1.2 Hz, 1H), 7.68 (d, J = 8.4 Hz, 2H), 7.44 (d, J = 8.0 Hz, 2H), 4.81 (s, 2H), 3.85 (s, 3H), 3.78 (s, 3H), 3.30-3.32 (m, 2H), 3.09 (s, 3H), 3.04 (t, J = 6.4 Hz, 2H), 1.66-1.70 To was filtered through a celite bed. The celite bed was further washed with ethyl acetate, the washings and the filtrate were combined, washed with brine, dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure. The crude obtained was purified by flash chromatography using a Biotage chromatography instrument, on silica gel column (100-200 mesh), eluting with 0-30% gradient of ethyl acetate in petroleum ether to obtain tert-butyl 2,4-dichloro-5-oxo-7,8-dihydropyrido[4,3-d]pyrimidine-6(5H)- carboxylate (0.400 g, 1.157 mmol, 70.4%) as off white solid. LCMS (ESI) m/z: 217.9 [M-Boc+H]+, LC retention time: 1.35 min. (LCMS Method 7). Step-2 Tert-butyl 2-chloro-4-((4-(1-methyl-4-(trifluoromethyl)-1H-imidazol-2- yl)benzyl)amino)-5-oxo-7,8-dihydropyrido[4,3-d]pyrimidine-6(5H)-carboxylate (6c):
was filtered through a celite bed. The celite bed was further washed with ethyl acetate, the washings and the filtrate were combined, washed with brine, dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure. The crude obtained was purified by flash chromatography using a Biotage chromatography instrument, on silica gel column (100-200 mesh), eluting with 0-30% gradient of ethyl acetate in petroleum ether to obtain tert-butyl 2,4-dichloro-5-oxo-7,8-dihydropyrido[4,3-d]pyrimidine-6(5H)- carboxylate (0.400 g, 1.157 mmol, 70.4%) as off white solid. LCMS (ESI) m/z: 217.9 [M-Boc+H]+, LC retention time: 1.35 min. (LCMS Method 7). Step-2 Tert-butyl 2-chloro-4-((4-(1-methyl-4-(trifluoromethyl)-1H-imidazol-2- yl)benzyl)amino)-5-oxo-7,8-dihydropyrido[4,3-d]pyrimidine-6(5H)-carboxylate (6c): To a stirred solution of tert-butyl 2,4-dichloro-5-oxo-7,8-dihydropyrido[4,3-d]pyrimidine- 6(5H)-carboxylate (0.250 g, 0.723 mmol) and (4-(1-methyl-4-(trifluoromethyl)-1H- imidazol-2-yl)phenyl)methanamine (I-1, 0.203 g, 0.795 mmol) in acetonitrile (5 mL), Cs2CO3 (0.353 g, 1.084 mmol) was added at ambient temperature and the mixture was heated at 80 °C for 5 h. The reaction mixture was cooled to ambient temperature, diluted with water and extracted with ethyl acetate. The organic layer was washed with brine, dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure. The residue was purified by flash chromatography using a Biotage chromatography instrument, on silica gel column (100-200 mesh), eluting with 0-40% gradient of ethyl acetate in petroleum ether to obtain tert-butyl 2-chloro-4-((4-(1-methyl-4- (trifluoromethyl)-1H-imidazol-2-yl)benzyl)amino)-5-oxo-7,8-dihydropyrido[4,3- d]pyrimidine-6(5H)-carboxylate (270 mg, 0.402 mmol, 55.6 % yield) as a pale yellow thick gum. LCMS (ESI) m/z: 537.2 [M+H]+, LC retention time: 3.135 min. (LCMS Method 6). Example in Table 3 was prepared according to procedure described in step-2 above. Table 3 Example Structure Name LCMS (ESI) Number tert-butyl 2-chloro-4-((4- m/z: 565.2 [M (1-isopropyl-4- +H]+ (trifluoromethyl)-1H- LC retention imidazol-2- 7c time: 2.91 min yl)benzyl)amino)-5-oxo- (LCMS-Method 7,8-dihydropyrido[4,3- 1) d]pyrimidine-6(5H)- carboxylate Step 3: Tert-butyl 2-(4-cyclopropyl-6-methoxypyrimidin-5-yl)-4-((4-(1-methyl-4- (trifluoromethyl)-1H-imidazol-2-yl)benzyl)amino)-5-oxo-7,8-dihydropyrido[4,3- d]pyrimidine-6(5H)-carboxylate (6d):
To a stirred solution of tert-butyl 2,4-dichloro-5-oxo-7,8-dihydropyrido[4,3-d]pyrimidine- 6(5H)-carboxylate (0.250 g, 0.723 mmol) and (4-(1-methyl-4-(trifluoromethyl)-1H- imidazol-2-yl)phenyl)methanamine (I-1, 0.203 g, 0.795 mmol) in acetonitrile (5 mL), Cs2CO3 (0.353 g, 1.084 mmol) was added at ambient temperature and the mixture was heated at 80 °C for 5 h. The reaction mixture was cooled to ambient temperature, diluted with water and extracted with ethyl acetate. The organic layer was washed with brine, dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure. The residue was purified by flash chromatography using a Biotage chromatography instrument, on silica gel column (100-200 mesh), eluting with 0-40% gradient of ethyl acetate in petroleum ether to obtain tert-butyl 2-chloro-4-((4-(1-methyl-4- (trifluoromethyl)-1H-imidazol-2-yl)benzyl)amino)-5-oxo-7,8-dihydropyrido[4,3- d]pyrimidine-6(5H)-carboxylate (270 mg, 0.402 mmol, 55.6 % yield) as a pale yellow thick gum. LCMS (ESI) m/z: 537.2 [M+H]+, LC retention time: 3.135 min. (LCMS Method 6). Example in Table 3 was prepared according to procedure described in step-2 above. Table 3 Example Structure Name LCMS (ESI) Number tert-butyl 2-chloro-4-((4- m/z: 565.2 [M (1-isopropyl-4- +H]+ (trifluoromethyl)-1H- LC retention imidazol-2- 7c time: 2.91 min yl)benzyl)amino)-5-oxo- (LCMS-Method 7,8-dihydropyrido[4,3- 1) d]pyrimidine-6(5H)- carboxylate Step 3: Tert-butyl 2-(4-cyclopropyl-6-methoxypyrimidin-5-yl)-4-((4-(1-methyl-4- (trifluoromethyl)-1H-imidazol-2-yl)benzyl)amino)-5-oxo-7,8-dihydropyrido[4,3- d]pyrimidine-6(5H)-carboxylate (6d): To a stirred solution of tert-butyl 2-chloro-4-((4-(1-methyl-4-(trifluoromethyl)-1H- imidazol-2-yl)benzyl)amino)-5-oxo-7,8-dihydropyrido[4,3-d]pyrimidine-6(5H)- carboxylate (0.250 g, 0.372 mmol) and (4-cyclopropyl-6-methoxypyrimidin-5-yl)boronic acid (4-cyclopropyl-6-methoxypyrimidin-5-yl)boronic acid (0.108 g, 0.559 mmol) in 1,4- dioxane (4 mL), tetrakis(triphenylphosphine)palladium(0) (0.043 g, 0.037 mmol)) and tripotassium phosphate (0.158 g, 0.745 mmol) were added under nitrogen atmosphere and the mixture was heated at 95 °C for 16 h. After 16 h, the reaction mixture was cooled to ambient temperature and filtered through a celite bed. The celite bed was further washed with ethyl acetate, the washings and the filtrate were mixed and concentrated under reduced pressure. The crude obtained was purified by flash chromatography on silica gel, 230-400 mesh, eluting with 0-60% gradient of ethyl acetate in petroleum ether to obtain tert-butyl 2-(4-cyclopropyl-6-methoxypyrimidin-5-yl)-4-((4-(1-methyl-4- (trifluoromethyl)-1H-imidazol-2-yl)benzyl)amino)-5-oxo-7,8-dihydropyrido[4,3- d]pyrimidine-6(5H)-carboxylate (0.230 g, 0.191 mmol, 51.2%) as pale yellow thick gum. LCMS (ESI) m/z: 651.0 [M+H]+, LC retention time: 1.343 min. (LCMS Method 7). Example in Table 4 was prepared according to procedure described in step-3 above.
To a stirred solution of tert-butyl 2-chloro-4-((4-(1-methyl-4-(trifluoromethyl)-1H- imidazol-2-yl)benzyl)amino)-5-oxo-7,8-dihydropyrido[4,3-d]pyrimidine-6(5H)- carboxylate (0.250 g, 0.372 mmol) and (4-cyclopropyl-6-methoxypyrimidin-5-yl)boronic acid (4-cyclopropyl-6-methoxypyrimidin-5-yl)boronic acid (0.108 g, 0.559 mmol) in 1,4- dioxane (4 mL), tetrakis(triphenylphosphine)palladium(0) (0.043 g, 0.037 mmol)) and tripotassium phosphate (0.158 g, 0.745 mmol) were added under nitrogen atmosphere and the mixture was heated at 95 °C for 16 h. After 16 h, the reaction mixture was cooled to ambient temperature and filtered through a celite bed. The celite bed was further washed with ethyl acetate, the washings and the filtrate were mixed and concentrated under reduced pressure. The crude obtained was purified by flash chromatography on silica gel, 230-400 mesh, eluting with 0-60% gradient of ethyl acetate in petroleum ether to obtain tert-butyl 2-(4-cyclopropyl-6-methoxypyrimidin-5-yl)-4-((4-(1-methyl-4- (trifluoromethyl)-1H-imidazol-2-yl)benzyl)amino)-5-oxo-7,8-dihydropyrido[4,3- d]pyrimidine-6(5H)-carboxylate (0.230 g, 0.191 mmol, 51.2%) as pale yellow thick gum. LCMS (ESI) m/z: 651.0 [M+H]+, LC retention time: 1.343 min. (LCMS Method 7). Example in Table 4 was prepared according to procedure described in step-3 above.






Table 4 Example Structure Name LCMS (ESI) Number tert-butyl 2-(4- cyclopropyl-6- methoxypyrimidin-5- yl)-4-((4-(1-isopropyl- 4-(trifluoromethyl)-1H- m/z: 679.5 imidazol-2- [M+H]+ yl)benzyl)amino)-5- LC retention oxo-7,8- 7d time: 3.37 min dihydropyrido[4,3- (LCMS- d]pyrimidine-6(5H)- Method 4): carboxylate [Note: For this intermediate, Suzuki coupling reaction was carried out under MW at 80oC for 30 min.] Step 4: 2-(4-cyclopropyl-6-methoxypyrimidin-5-yl)-4-((4-(1-methyl-4- (trifluoromethyl)-1H-imidazol-2-yl)benzyl)amino)-7,8-dihydropyrido[4,3- d]pyrimidin-5(6H)-one (example 6):
A solution of tert-butyl 2-(4-cyclopropyl-6-methoxypyrimidin-5-yl)-4-((4-(1-methyl-4- (trifluoromethyl)-1H-imidazol-2-yl)benzyl)amino)-5-oxo-7,8-dihydropyrido[4,3- d]pyrimidine-6(5H)-carboxylate (0.230 g, 0.191 mmol) in dichloromethane (5 mL) was cooled to 0oC (ice bath), trifluoroacetic acid (0.074 mL, 0.954 mmol) was added and the mixture was stirred at ambient temperature for 3 h. After completion of the reaction (monitored by TLC and UPLC-MS), solvents were evaporated from the reaction mixture under reduced pressure and 10% aqueous sodium bicarbonate was slowly added to at 0oC (ice bath) until pH of the system reached 7~8. The contents wereextracted with a mixture of 10% methanol in dichloromethane and the combined organic extract was washed with brine, dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure. The crude obtained was purified by reverse phase flash chromatography [Method: Diluent: WATER: THF: MeCN (50:20:30); Column: RediSep 80 g C18, 20-40 micron; Temperature: Ambient; Mobile phase A: 5mM Ammonium formate in water, Mobile phase B: Acetonitrile; Flow :40mL/min; 42%Acetonitrile/ 5mM Ammonium formate in water] to obtain 2-(4-cyclopropyl-6-methoxypyrimidin-5-yl)-4-((4-(1-methyl- 4-(trifluoromethyl)-1H-imidazol-2-yl)benzyl)amino)-7,8-dihydropyrido[4,3-d]pyrimidin- 5(6H)-one (0.040 g, 0.073 mmol, 38.0%) as off white solid. LCMS (ESI) m/z: 551.2 [M+H]+, LC retention time: 2.43 min. (LCMS Method 1);1H-NMR: (400 MHz,DMSO- d6) į = 9.63 (t, J = 6.4 Hz, 1H), 8.62 (s, 1H), 8.11 (s, 1H), 7.91 (d, J = 1.2 Hz, 1H), 7.65 (dd, J = 6.8 Hz & 1.6 Hz, 2H), 7.43 (d, J = 8.4 Hz, 2H), 4.74 (d, J = 6.4Hz, 2H), 3.84 (s, 3H), 3.76 (s, 3H), 3.46-3.42 (m, 2H), 2.89 (t, J = 7.2 Hz, 2H), 1.76-1.74 (m, 1H), 1.01- 0.98 (m, 2H), 0.85-0.81 (m, 2H). Example in Table 5 was prepared according to procedure described in step-4 above.
Table 5 Example LCMS Structure1H NMR Number (ESI) (400 MHz, DMSO- d6) į = 9.64 (t, J = 6.4 Hz, 1H), 8.62 2-(4- (s, 1H), 8.15 (d, J Cyclopropyl-6- =1.2, Hz, 1H), 8.11 methoxypyrimidi m/z: 579.2 + (s, 1H), 7.50 (d, J = n-5-yl)-4-((4-(1- [M+H] , 8.4 Hz, 2H), 7.44 isopropyl-4- LC (d, J = 8.4, Hz, (trifluoromethyl)- retention 7 2H), 4.75 (d, J = 6.4 1H-imidazol-2- time: 2.80 Hz, 2H), 4.50-4.41 yl)benzyl)amino)- min. (m, 1H), 3.84 (s, 7,8- (LCMS 3H), 3.48-3.41 (m, dihydropyrido[4, Method 1). 2H), 2.89 (t, J = 7.2 3-d]pyrimidin- Hz, 2H), 1.78-1.70 5(6H)-one (m, 1H), 1.00-0.95 (m, 2H), 0.82-0.76 (m, 2H). USP1-UAF1 Rhodamine assay USP1/UAF1 ubiquitin-rhodamine 110 hydrolysis assays were performed at room 5 temperature in black, low-volume 384 well plates (Corning 3821). 100X solutions of compounds in DMSO were prepared by three-fold serial dilutions starting from a 10 mM stock. 2X solutions of His6-USP1/His6-UAF1 (200 pM, Internally Produced) and ubiquitin-rhodamine 110 (10 ^M, South Bay Bio SBB-PS0001) were prepared in assay buffer (50 mM Tris pH 7.5, 100 mM NaCl, 1 mM EDTA, 1 mM TCEP, 100 ng/^L BSA). 10 Serially diluted compounds in DMSO were transferred to the assay plate by acoustic dispensing (100 nL per well).5 ^L of assay buffer was added to column 1 of the plates, and 5 ^L of 2X USP1/UAF1 solution was added to columns 2-24 and incubated with compounds for 3 hrs. Reactions were initiated by addition of 5 ^L of 2X ubiquitin- rhodamine 110 solution to each well for final concentrations of 100 pM USP1/UAF1 and 5 ^M ubiquitin-rhodamine 110. Fluorescence was read at the minimum kinetic interval for 1 h using a BioTek Synergy HTX plate reader (Agilent Technologies) with excitation at 485 nm and emission at 528 nm. Initial rates were calculated by fitting the linear range of the plot of fluorescence vs time to a linear equation. IC50 values were calculated from dose- response curves. IC50 values for compounds of the invention in the USP1-UAF1 Rhodamine assay are shown below. Table 1: USP1-UAF1 Rhodamine Assay 3-hour Incubation Results Compound # USP1-UAF1 IC50 PM 1 0.058 2 0.028 3 0.021 4 0.030 5 >10 6 0.020 7 0.015
Claims
WE CLAIM: 1. A compound having the structure of formula (I): W3 W2 or a of stereoisomers thereof; R1 is selected from 5-6 membered heteroaryl, optionally substituted with one to four halo, hydroxy, amino, -C(O)Ra, -C(O)ORb, -C(O)NRaRb, -N(Ra)C(O)Rb, -S(O)NRaRb, - S(O)2NRaRb, -S(O)Rg, -S(O)2Rg, -NRaRb, -ORa, -SRb, C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl and C3-8 cycloalkyl; wherein each C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl and C3-8 cycloalkyl is optionally substituted with one to four R100; R2 is selected from absent, hydrogen, halo, hydroxy, amino, -CN, -C(O)Ra, -C(O)ORb, - C(O)NRaRb, -N(Ra)C(O)Rb, -N(Ra)C(O)NRaRb, -N(Ra)SO2NRaRb, -S(O)NRaRb, - -
or a of stereoisomers thereof; R1 is selected from 5-6 membered heteroaryl, optionally substituted with one to four halo, hydroxy, amino, -C(O)Ra, -C(O)ORb, -C(O)NRaRb, -N(Ra)C(O)Rb, -S(O)NRaRb, - S(O)2NRaRb, -S(O)Rg, -S(O)2Rg, -NRaRb, -ORa, -SRb, C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl and C3-8 cycloalkyl; wherein each C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl and C3-8 cycloalkyl is optionally substituted with one to four R100; R2 is selected from absent, hydrogen, halo, hydroxy, amino, -CN, -C(O)Ra, -C(O)ORb, - C(O)NRaRb, -N(Ra)C(O)Rb, -N(Ra)C(O)NRaRb, -N(Ra)SO2NRaRb, -S(O)NRaRb, - - heterocyclyl containing 1 to 4 heteroatoms selected from N, O, and S; wherein each C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, 4-10 membered heterocyclyl containing 1 to 4 heteroatoms selected from N, O, and S, and C3-8 cycloalkyl is optionally substituted with one to four R100; X is selected from absent and C1-6 alkyl, wherein C1-6 alkyl is optionally substituted with one to four R100; Each W1, W2, W3 and W4 is independently selected from -N(Ra)-, -C(O)- and -C(Ra)-; W5 is -N- or -C(Ra)-; wherein at least one of W1, W2, W3 and W4 is -N(Ra)-; wherein at least one of W1, W2, W3 and W4 is -C(Ra)-; G1 is selected from -C6 aryl-, 5-6 membered heteroaryl, C3-8 cycloalkyl and 5-6 membered heterocyclyl; wherein each C6 aryl, 5-6 membered heteroaryl, C3-8 cycloalkyl and 5-6 membered heterocyclyl is optionally substituted with one to four R100; G2 is a 5 or 6 membered heteroaryl or 5-6 membered heterocyclyl optionally substituted with one to four R100; each Ra and Rb is independently selected from absent, hydrogen, C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, C3-6 cycloalkyl and 4-6 membered heterocyclyl; wherein each C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, C3-6 cycloalkyl and 4-6 membered heterocyclyl is optionally substituted with one to four R200; each R100 is independently selected from hydrogen, halo, cyano, hydroxy, amino, oxo, thioxo, vinyl, -C(O)Rc, -C(O)ORc, -C(O)NRcRd, -N(Rc)C(O)Rd, -S(O)NRcRd, - S(O)2NRcRd, -S(O)Rh, -S(O)2Rh, -NRcRd, -ORc, -SRc, C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, C3-8 cycloalkyl, C6-10 aryl, 5-10 membered heteroaryl containing 1 to 4 heteroatoms selected from N, O, and S, and 4-10 membered heterocyclyl containing 1 to 4 heteroatoms selected from N, O, and S; wherein each C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, C3-8 cycloalkyl, C6-10 aryl, 5-10 membered heteroaryl and 4-10 membered heterocyclyl is optionally substituted with one to four R201; each Rc and Rd is independently selected from hydrogen, C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, C3-8 cycloalkyl, C6-10 aryl, 5-10 membered heteroaryl containing 1 to 4 heteroatoms selected from N, O, and S, and 4-10 membered heterocyclyl containing 1 to 4 heteroatoms selected from N, O, and S; each R200 and R201 is independently selected from hydrogen, halo, cyano, hydroxy, amino, oxo, thioxo, vinyl, -C(O)Re, -C(O)ORe, -C(O)NReRf, -N(Re)C(O)Rf, -S(O)NReRf, - S(O)2NReRf, -S(O)Ri, -S(O)2Ri, -NReRf, -ORe, -SRe, C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, C3-8 cycloalkyl, C6-10 aryl, 5-10 membered heteroaryl containing 1 to 4 heteroatoms selected from N, O, and S, and 4-10 membered heterocyclyl containing 1 to 4 heteroatoms selected from N, O, and S; wherein each C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, C3-8 cycloalkyl, C6-10 aryl, 5-10 membered heteroaryl and 4-10 membered heterocyclyl is optionally substituted with one to four R300; each Rg, Rh and Ri is independently
heterocyclyl containing 1 to 4 heteroatoms selected from N, O, and S; wherein each C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, 4-10 membered heterocyclyl containing 1 to 4 heteroatoms selected from N, O, and S, and C3-8 cycloalkyl is optionally substituted with one to four R100; X is selected from absent and C1-6 alkyl, wherein C1-6 alkyl is optionally substituted with one to four R100; Each W1, W2, W3 and W4 is independently selected from -N(Ra)-, -C(O)- and -C(Ra)-; W5 is -N- or -C(Ra)-; wherein at least one of W1, W2, W3 and W4 is -N(Ra)-; wherein at least one of W1, W2, W3 and W4 is -C(Ra)-; G1 is selected from -C6 aryl-, 5-6 membered heteroaryl, C3-8 cycloalkyl and 5-6 membered heterocyclyl; wherein each C6 aryl, 5-6 membered heteroaryl, C3-8 cycloalkyl and 5-6 membered heterocyclyl is optionally substituted with one to four R100; G2 is a 5 or 6 membered heteroaryl or 5-6 membered heterocyclyl optionally substituted with one to four R100; each Ra and Rb is independently selected from absent, hydrogen, C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, C3-6 cycloalkyl and 4-6 membered heterocyclyl; wherein each C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, C3-6 cycloalkyl and 4-6 membered heterocyclyl is optionally substituted with one to four R200; each R100 is independently selected from hydrogen, halo, cyano, hydroxy, amino, oxo, thioxo, vinyl, -C(O)Rc, -C(O)ORc, -C(O)NRcRd, -N(Rc)C(O)Rd, -S(O)NRcRd, - S(O)2NRcRd, -S(O)Rh, -S(O)2Rh, -NRcRd, -ORc, -SRc, C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, C3-8 cycloalkyl, C6-10 aryl, 5-10 membered heteroaryl containing 1 to 4 heteroatoms selected from N, O, and S, and 4-10 membered heterocyclyl containing 1 to 4 heteroatoms selected from N, O, and S; wherein each C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, C3-8 cycloalkyl, C6-10 aryl, 5-10 membered heteroaryl and 4-10 membered heterocyclyl is optionally substituted with one to four R201; each Rc and Rd is independently selected from hydrogen, C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, C3-8 cycloalkyl, C6-10 aryl, 5-10 membered heteroaryl containing 1 to 4 heteroatoms selected from N, O, and S, and 4-10 membered heterocyclyl containing 1 to 4 heteroatoms selected from N, O, and S; each R200 and R201 is independently selected from hydrogen, halo, cyano, hydroxy, amino, oxo, thioxo, vinyl, -C(O)Re, -C(O)ORe, -C(O)NReRf, -N(Re)C(O)Rf, -S(O)NReRf, - S(O)2NReRf, -S(O)Ri, -S(O)2Ri, -NReRf, -ORe, -SRe, C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, C3-8 cycloalkyl, C6-10 aryl, 5-10 membered heteroaryl containing 1 to 4 heteroatoms selected from N, O, and S, and 4-10 membered heterocyclyl containing 1 to 4 heteroatoms selected from N, O, and S; wherein each C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, C3-8 cycloalkyl, C6-10 aryl, 5-10 membered heteroaryl and 4-10 membered heterocyclyl is optionally substituted with one to four R300; each Rg, Rh and Ri is independently from C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl; wherein each C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, is optionally substituted with one to four R300; wherein each R300 is independently selected from hydrogen, halo, cyano, hydroxy, amino, oxo, thioxo, vinyl, -C(O)Re, -C(O)ORe, -C(O)NReRf, -N(Re)C(O)Rf, -S(O)NReRf, - S(O)2NReRf, -NReRf, S(O)Re, -S(O)2Re, -NReRf, -ORe, -SRe, C1-6 alkyl, C2-6 alkenyl and C2-6 alkynyl; each Re and Rf is independently selected from hydrogen, C1-6 alkyl, C2-6 alkenyl and C2-6 alkynyl; C3-8 cycloalkyl, C6-10 aryl, 5-10 membered heteroaryl containing 1 to 4 heteroatoms selected from N, O, and S, and 4-10 membered heterocyclyl containing 1 to 4 heteroatoms selected from N, O, and S; wherein each C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, C3-8 cycloalkyl, C6-10 aryl, 5-10 membered heteroaryl and 4-10 membered heterocyclyl is optionally substituted with one to four R400; each R400 is independently selected from hydrogen, halo, cyano, hydroxy, amino, oxo, thioxo, vinyl, -C(O)Rk, -C(O)ORk, -C(O)NRkRl, -N(Rk)C(O)Rl, -S(O)NRkRl, - S(O)2NRkRl, -NRkRl, S(O)Rk, -S(O)2Rk, -NRkRl, -ORk, -SRk, C1-6 alkyl, C2-6 alkenyl and C2-6 alkynyl; each Rk and Rl is independently selected from hydrogen, C1-6 alkyl, C2-6 alkenyl and C2-6 alkynyl; C3-8 cycloalkyl, C6-10 aryl, 5-10 membered heteroaryl containing 1 to 4 heteroatoms selected from N, O, and S, and 4-10 membered heterocyclyl containing 1 to 4 heteroatoms selected from N, O, and S.
from C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl; wherein each C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, is optionally substituted with one to four R300; wherein each R300 is independently selected from hydrogen, halo, cyano, hydroxy, amino, oxo, thioxo, vinyl, -C(O)Re, -C(O)ORe, -C(O)NReRf, -N(Re)C(O)Rf, -S(O)NReRf, - S(O)2NReRf, -NReRf, S(O)Re, -S(O)2Re, -NReRf, -ORe, -SRe, C1-6 alkyl, C2-6 alkenyl and C2-6 alkynyl; each Re and Rf is independently selected from hydrogen, C1-6 alkyl, C2-6 alkenyl and C2-6 alkynyl; C3-8 cycloalkyl, C6-10 aryl, 5-10 membered heteroaryl containing 1 to 4 heteroatoms selected from N, O, and S, and 4-10 membered heterocyclyl containing 1 to 4 heteroatoms selected from N, O, and S; wherein each C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, C3-8 cycloalkyl, C6-10 aryl, 5-10 membered heteroaryl and 4-10 membered heterocyclyl is optionally substituted with one to four R400; each R400 is independently selected from hydrogen, halo, cyano, hydroxy, amino, oxo, thioxo, vinyl, -C(O)Rk, -C(O)ORk, -C(O)NRkRl, -N(Rk)C(O)Rl, -S(O)NRkRl, - S(O)2NRkRl, -NRkRl, S(O)Rk, -S(O)2Rk, -NRkRl, -ORk, -SRk, C1-6 alkyl, C2-6 alkenyl and C2-6 alkynyl; each Rk and Rl is independently selected from hydrogen, C1-6 alkyl, C2-6 alkenyl and C2-6 alkynyl; C3-8 cycloalkyl, C6-10 aryl, 5-10 membered heteroaryl containing 1 to 4 heteroatoms selected from N, O, and S, and 4-10 membered heterocyclyl containing 1 to 4 heteroatoms selected from N, O, and S.



2. A compound of claim 1, having the structure of Formula (II): or a pharmaceutically acceptable salt, stereoisomer, mixture of stereoisomers thereof.
or a pharmaceutically acceptable salt, stereoisomer, mixture of stereoisomers thereof.

3. A compound of claim 1, having the structure of Formula (IIIa), (IIIb), (IIIc) or (IIId): or a pharmaceutically acceptable salt, stereoisomer, mixture of stereoisomers thereof.
4. A compound of claim 1, having the structure of Formula (IVa) or (IVb): or a pharmaceutically acceptable salt, stereoisomer, mixture of stereoisomers thereof; wherein R5 is C1-6 alkyl.
or a pharmaceutically acceptable salt, stereoisomer, mixture of stereoisomers thereof; wherein R5 is C1-6 alkyl.

5. A compound of claim 1, having the structure of Formula (Va) or (Vb): N R5 or a pharmaceutically acceptable salt, stereoisomer, mixture of stereoisomers thereof; wherein R5 is C1-6 alkyl.
or a pharmaceutically acceptable salt, stereoisomer, mixture of stereoisomers thereof; wherein R5 is C1-6 alkyl.

6. A compound of claim 1, having the structure of Formula (VIa) or (VIb): or a pharmaceutically acceptable salt, stereoisomer, mixture of stereoisomers thereof; wherein R5 is C1-6 alkyl.
or a pharmaceutically acceptable salt, stereoisomer, mixture of stereoisomers thereof; wherein R5 is C1-6 alkyl.

7. A compound according to any of claims 1-6 wherein R1 is selected from: ; salt, stereoisomer, mixture of stereoisomers thereof.
salt, stereoisomer, mixture of stereoisomers thereof.

8. A compound according to any of claims 1-7 wherein R2 is selected from: -H, -OCH3, -SCH3, -S(O)2CH3, -S(O)2CH3, -C(O)OCH3, -C(O)OCH2CH3, and -C(O)NH2. or a pharmaceutically acceptable salt, stereoisomer, mixture of stereoisomers, or deuterated analog thereof.
9. A compound according to any of claims 1-3 and 7-8 wherein G2 is selected from: or deuterated analog thereof.
or deuterated analog thereof.

10. A compound of claim 1, selected from the Table A or a pharmaceutically acceptable salt thereof; TABLE A
11. A pharmaceutical composition comprising one or more compounds according to any of the above claims and a pharmaceutically acceptable carrier or diluent.
12. A method of treating or preventing a disease or disorder associated with the inhibition of ubiquitin specific protease 1 (USP1) comprising, administering to a patient in need thereof an effective amount of a compound account to any of the above claims.
13. A method for treating cancer comprising administering a therapeutically effective amount of a compound according to claims 1-10 or a pharmaceutically acceptable salt thereof, to a patient in need thereof.
14. The method according to claim 13 wherein said disease or condition is a solid tumor selected from pancreatic cancer, bladder cancer, colorectal cancer, breast cancer, prostate cancer, renal cancer, hepatocellular cancer, lung cancer, ovarian cancer, cervical cancer, gastric cancer, esophageal cancer, head and neck cancer, melanoma, neuroendocrine cancers, CNS cancers, brain tumors (e.g., glioma, anaplastic oligodendroglioma, adult glioblastoma multiforme, and adult anaplastic astrocytoma), bone cancer, and soft tissue sarcoma.
15. The method according to claim 13, wherein the cancer is pancreatic cancer, bladder cancer, colorectal cancer, breast cancer, prostate cancer, renal cancer, hepatocellular cancer, lung cancer, ovarian cancer, cervical cancer, gastric cancer, esophageal cancer, head and neck cancer, melanoma, neuroendocrine cancer, CNS cancer, brain cancer, bone cancer, soft tissue sarcoma, non-small cell lung cancer, small-cell lung cancer or colon cancer.
16. The method according to claim 13, wherein the cancer is acute lymphocytic leukemia (ALL), acute myeloid leukemia (AML), chronic lymphocytic leukemia (CLL), small lymphocytic lymphoma (SLL), myelodysplastic syndrome (MDS), myeloproliferative disease (MPD), chronic myeloid leukemia (CML), multiple myeloma (MM), non-Hodgkin's lymphoma (NHL), mantle cell lymphoma (MCL), follicular lymphoma, Waldenström’s macroglobulinemia (WM), T-cell lymphoma, B-cell lymphoma or diffuse large B-cell lymphoma (DLBCL).
17. The method according to any of claims 12-16, further comprising administering at least one additional anticancer agent or therapy.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US202363594480P | 2023-10-31 | 2023-10-31 | |
| US63/594,480 | 2023-10-31 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2025096489A1true WO2025096489A1 (en) | 2025-05-08 |
Family
ID=93566439
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2024/053492PendingWO2025096489A1 (en) | 2023-10-31 | 2024-10-30 | Ubiquitin specific processing protease 1 (usp1) compounds |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2025096489A1 (en) |
Citations (32)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2006029879A2 (en) | 2004-09-17 | 2006-03-23 | F.Hoffmann-La Roche Ag | Anti-ox40l antibodies |
| WO2006105021A2 (en) | 2005-03-25 | 2006-10-05 | Tolerrx, Inc. | Gitr binding molecules and uses therefor |
| WO2006122150A1 (en) | 2005-05-10 | 2006-11-16 | Incyte Corporation | Modulators of indoleamine 2,3-dioxygenase and methods of using the same |
| WO2007005874A2 (en) | 2005-07-01 | 2007-01-11 | Medarex, Inc. | Human monoclonal antibodies to programmed death ligand 1 (pd-l1) |
| WO2007075598A2 (en) | 2005-12-20 | 2007-07-05 | Incyte Corporation | N-hydroxyamidinoheterocycles as modulators of indoleamine 2,3-dioxygenase |
| WO2008036642A2 (en) | 2006-09-19 | 2008-03-27 | Incyte Corporation | N-hydroxyamidinoheterocycles as modulators of indoleamine 2,3-dioxygenase |
| WO2008132601A1 (en) | 2007-04-30 | 2008-11-06 | Immutep | Cytotoxic anti-lag-3 monoclonal antibody and its use in the treatment or prevention of organ transplant rejection and autoimmune disease |
| WO2009009116A2 (en) | 2007-07-12 | 2009-01-15 | Tolerx, Inc. | Combination therapies employing gitr binding molecules |
| WO2009044273A2 (en) | 2007-10-05 | 2009-04-09 | Immutep | Use of recombinant lag-3 or the derivatives thereof for eliciting monocyte immune response |
| WO2009073620A2 (en) | 2007-11-30 | 2009-06-11 | Newlink Genetics | Ido inhibitors |
| US7754463B2 (en) | 2006-06-20 | 2010-07-13 | Dana-Farber Cancer Institute | Inhibitors of USP1 Deubiquitinating Enzyme Complex |
| WO2011028683A1 (en) | 2009-09-03 | 2011-03-10 | Schering Corporation | Anti-gitr antibodies |
| WO2011070024A1 (en) | 2009-12-10 | 2011-06-16 | F. Hoffmann-La Roche Ag | Antibodies binding preferentially human csf1r extracellular domain 4 and their use |
| WO2011107553A1 (en) | 2010-03-05 | 2011-09-09 | F. Hoffmann-La Roche Ag | Antibodies against human csf-1r and uses thereof |
| WO2011109400A2 (en) | 2010-03-04 | 2011-09-09 | Macrogenics,Inc. | Antibodies reactive with b7-h3, immunologically active fragments thereof and uses thereof |
| WO2011131407A1 (en) | 2010-03-05 | 2011-10-27 | F. Hoffmann-La Roche Ag | Antibodies against human csf-1r and uses thereof |
| WO2012032433A1 (en) | 2010-09-09 | 2012-03-15 | Pfizer Inc. | 4-1bb binding molecules |
| WO2012142237A1 (en) | 2011-04-15 | 2012-10-18 | Newlink Geneticks Corporation | Fused imidazole derivatives useful as ido inhibitors |
| WO2012145493A1 (en) | 2011-04-20 | 2012-10-26 | Amplimmune, Inc. | Antibodies and other molecules that bind b7-h1 and pd-1 |
| WO2013079174A1 (en) | 2011-11-28 | 2013-06-06 | Merck Patent Gmbh | Anti-pd-l1 antibodies and uses thereof |
| WO2013087699A1 (en) | 2011-12-15 | 2013-06-20 | F. Hoffmann-La Roche Ag | Antibodies against human csf-1r and uses thereof |
| WO2013119716A1 (en) | 2012-02-06 | 2013-08-15 | Genentech, Inc. | Compositions and methods for using csf1r inhibitors |
| WO2013132044A1 (en) | 2012-03-08 | 2013-09-12 | F. Hoffmann-La Roche Ag | Combination therapy of antibodies against human csf-1r and uses thereof |
| WO2013169264A1 (en) | 2012-05-11 | 2013-11-14 | Five Prime Therapeutics, Inc. | Methods of treating conditions with antibodies that bind colony stimulating factor 1 receptor (csf1r) |
| WO2014036357A1 (en) | 2012-08-31 | 2014-03-06 | Five Prime Therapeutics, Inc. | Methods of treating conditions with antibodies that bind colony stimulating factor 1 receptor (csf1r) |
| US9518032B2 (en) | 2010-04-30 | 2016-12-13 | Dana-Farber Cancer Institute, Inc. | Small molecule inhibitors of USP1 deubiquitinating enzyme activity |
| WO2018229139A1 (en)* | 2017-06-14 | 2018-12-20 | Fundación Para La Investigación Médica Aplicada | Novel compounds for use in cancer |
| WO2023148643A1 (en)* | 2022-02-03 | 2023-08-10 | Aurigene Oncology Limited | Fused bicyclic heterocyclyl compounds as usp1 inhibitors |
| WO2024006879A1 (en)* | 2022-06-29 | 2024-01-04 | Zentaur Therapeutics Usa Inc. | Usp1 inhibitors and uses thereof |
| WO2024094170A1 (en)* | 2022-11-04 | 2024-05-10 | 深圳晶泰科技有限公司 | Inhibitor of ubiquitin-specific protease 1 and use thereof |
| WO2024208292A1 (en)* | 2023-04-04 | 2024-10-10 | 江苏亚虹医药科技股份有限公司 | Ubiquitin-specific protease 1 inhibitor, preparation method therefor, and medical use thereof |
| WO2024233605A1 (en)* | 2023-05-08 | 2024-11-14 | Tango Therapeutics, Inc. | Compounds and their use against cancer |
- 2024
- 2024-10-30WOPCT/US2024/053492patent/WO2025096489A1/enactivePending
Patent Citations (33)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2006029879A2 (en) | 2004-09-17 | 2006-03-23 | F.Hoffmann-La Roche Ag | Anti-ox40l antibodies |
| WO2006105021A2 (en) | 2005-03-25 | 2006-10-05 | Tolerrx, Inc. | Gitr binding molecules and uses therefor |
| WO2006122150A1 (en) | 2005-05-10 | 2006-11-16 | Incyte Corporation | Modulators of indoleamine 2,3-dioxygenase and methods of using the same |
| WO2007005874A2 (en) | 2005-07-01 | 2007-01-11 | Medarex, Inc. | Human monoclonal antibodies to programmed death ligand 1 (pd-l1) |
| WO2007075598A2 (en) | 2005-12-20 | 2007-07-05 | Incyte Corporation | N-hydroxyamidinoheterocycles as modulators of indoleamine 2,3-dioxygenase |
| US7754463B2 (en) | 2006-06-20 | 2010-07-13 | Dana-Farber Cancer Institute | Inhibitors of USP1 Deubiquitinating Enzyme Complex |
| WO2008036642A2 (en) | 2006-09-19 | 2008-03-27 | Incyte Corporation | N-hydroxyamidinoheterocycles as modulators of indoleamine 2,3-dioxygenase |
| WO2008132601A1 (en) | 2007-04-30 | 2008-11-06 | Immutep | Cytotoxic anti-lag-3 monoclonal antibody and its use in the treatment or prevention of organ transplant rejection and autoimmune disease |
| WO2009009116A2 (en) | 2007-07-12 | 2009-01-15 | Tolerx, Inc. | Combination therapies employing gitr binding molecules |
| WO2009044273A2 (en) | 2007-10-05 | 2009-04-09 | Immutep | Use of recombinant lag-3 or the derivatives thereof for eliciting monocyte immune response |
| WO2009073620A2 (en) | 2007-11-30 | 2009-06-11 | Newlink Genetics | Ido inhibitors |
| WO2011028683A1 (en) | 2009-09-03 | 2011-03-10 | Schering Corporation | Anti-gitr antibodies |
| WO2011070024A1 (en) | 2009-12-10 | 2011-06-16 | F. Hoffmann-La Roche Ag | Antibodies binding preferentially human csf1r extracellular domain 4 and their use |
| WO2011109400A2 (en) | 2010-03-04 | 2011-09-09 | Macrogenics,Inc. | Antibodies reactive with b7-h3, immunologically active fragments thereof and uses thereof |
| WO2011107553A1 (en) | 2010-03-05 | 2011-09-09 | F. Hoffmann-La Roche Ag | Antibodies against human csf-1r and uses thereof |
| WO2011131407A1 (en) | 2010-03-05 | 2011-10-27 | F. Hoffmann-La Roche Ag | Antibodies against human csf-1r and uses thereof |
| US9518032B2 (en) | 2010-04-30 | 2016-12-13 | Dana-Farber Cancer Institute, Inc. | Small molecule inhibitors of USP1 deubiquitinating enzyme activity |
| US10653676B2 (en) | 2010-04-30 | 2020-05-19 | Dana-Farber Cancer Institute, Inc. | Small molecule inhibitors of USP1 deubiquitinating enzyme activity |
| WO2012032433A1 (en) | 2010-09-09 | 2012-03-15 | Pfizer Inc. | 4-1bb binding molecules |
| WO2012142237A1 (en) | 2011-04-15 | 2012-10-18 | Newlink Geneticks Corporation | Fused imidazole derivatives useful as ido inhibitors |
| WO2012145493A1 (en) | 2011-04-20 | 2012-10-26 | Amplimmune, Inc. | Antibodies and other molecules that bind b7-h1 and pd-1 |
| WO2013079174A1 (en) | 2011-11-28 | 2013-06-06 | Merck Patent Gmbh | Anti-pd-l1 antibodies and uses thereof |
| WO2013087699A1 (en) | 2011-12-15 | 2013-06-20 | F. Hoffmann-La Roche Ag | Antibodies against human csf-1r and uses thereof |
| WO2013119716A1 (en) | 2012-02-06 | 2013-08-15 | Genentech, Inc. | Compositions and methods for using csf1r inhibitors |
| WO2013132044A1 (en) | 2012-03-08 | 2013-09-12 | F. Hoffmann-La Roche Ag | Combination therapy of antibodies against human csf-1r and uses thereof |
| WO2013169264A1 (en) | 2012-05-11 | 2013-11-14 | Five Prime Therapeutics, Inc. | Methods of treating conditions with antibodies that bind colony stimulating factor 1 receptor (csf1r) |
| WO2014036357A1 (en) | 2012-08-31 | 2014-03-06 | Five Prime Therapeutics, Inc. | Methods of treating conditions with antibodies that bind colony stimulating factor 1 receptor (csf1r) |
| WO2018229139A1 (en)* | 2017-06-14 | 2018-12-20 | Fundación Para La Investigación Médica Aplicada | Novel compounds for use in cancer |
| WO2023148643A1 (en)* | 2022-02-03 | 2023-08-10 | Aurigene Oncology Limited | Fused bicyclic heterocyclyl compounds as usp1 inhibitors |
| WO2024006879A1 (en)* | 2022-06-29 | 2024-01-04 | Zentaur Therapeutics Usa Inc. | Usp1 inhibitors and uses thereof |
| WO2024094170A1 (en)* | 2022-11-04 | 2024-05-10 | 深圳晶泰科技有限公司 | Inhibitor of ubiquitin-specific protease 1 and use thereof |
| WO2024208292A1 (en)* | 2023-04-04 | 2024-10-10 | 江苏亚虹医药科技股份有限公司 | Ubiquitin-specific protease 1 inhibitor, preparation method therefor, and medical use thereof |
| WO2024233605A1 (en)* | 2023-05-08 | 2024-11-14 | Tango Therapeutics, Inc. | Compounds and their use against cancer |
Non-Patent Citations (5)
| Title |
|---|
| "Remington's Pharmaceutical Sciences", 1985, MACK PUBLISHING COMPANY |
| GREENEWUTS: "Protective Groups In Organic Synthesis", 1999, WILEY AND SONS |
| GΑRCIA-SCRNTISTEBAN. I.PETERS, G.JGIOVANNETTI. L: ET AL., MOL CANCER, vol. 12, 2013, pages 91 |
| RAUTIO., J. ET AL., NATURE REVIEW DRUG DISCOVERY, vol. 17, 2018, pages 559 - 587 |
| THOMAS S. DEXHEIMER ET AL: "Synthesis and Structure-Activity Relationship Studies of N -Benzyl-2-phenylpyrimidin-4-amine Derivatives as Potent USP1/UAF1 Deubiquitinase Inhibitors with Anticancer Activity against Nonsmall Cell Lung Cancer", JOURNAL OF MEDICINAL CHEMISTRY, vol. 57, no. 19, 9 October 2014 (2014-10-09), US, pages 8099 - 8110, XP055330334, ISSN: 0022-2623, DOI: 10.1021/jm5010495* |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP7723743B2 (en) | Compounds and their uses | |
| JP6165977B2 (en) | Heteroarylpyridone and aza-pyridone amide compounds | |
| EP3154958B1 (en) | Phosphatidylinositol 3-kinase inhibitors | |
| US11021467B2 (en) | Phosphatidylinositol 3-kinase inhibitors | |
| EP3154961B1 (en) | Quinazolinone derivatives as phosphatidylinositol 3-kinase inhibitors | |
| JP7693688B2 (en) | Compounds and their uses | |
| TW201737913A (en) | TANK-binding kinase inhibitor compounds | |
| EP3154962B1 (en) | Phosphatidylinositol 3-kinase inhibitors | |
| EP4247815A1 (en) | Compounds and uses thereof | |
| US20230150974A1 (en) | Compounds and uses thereof | |
| AU2013364070A1 (en) | Isoquinolinone or quinazolinone phosphatidylinositol 3-kinase inhibitors | |
| AU2015280138A1 (en) | Phosphatidylinositol 3-kinase inhibitors | |
| WO2018156901A1 (en) | Inhibitors of bruton's tyrosine kinase | |
| JP2021529806A (en) | Therapeutic heterocyclic compounds | |
| AU2022272181B9 (en) | Compounds and uses thereof | |
| WO2025096489A1 (en) | Ubiquitin specific processing protease 1 (usp1) compounds | |
| WO2025096490A1 (en) | Ubiquitin specific processing protease 1 (usp1) compounds | |
| WO2025096487A1 (en) | Ubiquitin specific processing protease 1 (usp1) compounds | |
| WO2025096488A1 (en) | Ubiquitin specific processing protease 1 (usp1) compounds | |
| WO2025096539A1 (en) | Ubiquitin specific processing protease 1 (usp1) compounds | |
| WO2025096494A1 (en) | Ubiquitin specific processing protease 1 (usp1) compounds | |
| WO2025096505A1 (en) | Ubiquitin specific processing protease 1 (usp1) compounds | |
| EP4522279A1 (en) | Pyrazine derivatives and uses thereof | |
| HK40021417A (en) | Heteroaryl pyridone and aza-pyridone amide compounds |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | Ref document number:24809085 Country of ref document:EP Kind code of ref document:A1 |