WO2025088088A1 - Rna composition for improving cell therapy - Google Patents
Rna composition for improving cell therapyDownload PDFInfo
- Publication number
- WO2025088088A1 WO2025088088A1PCT/EP2024/080173EP2024080173WWO2025088088A1WO 2025088088 A1WO2025088088 A1WO 2025088088A1EP 2024080173 WEP2024080173 WEP 2024080173WWO 2025088088 A1WO2025088088 A1WO 2025088088A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- cells
- antigen
- subject
- composition
- modified
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Classifications
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/0005—Vertebrate antigens
- A61K39/0011—Cancer antigens
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/66—Phosphorus compounds
- A61K31/675—Phosphorus compounds having nitrogen as a ring hetero atom, e.g. pyridoxal phosphate
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K40/00—Cellular immunotherapy
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K40/00—Cellular immunotherapy
- A61K40/10—Cellular immunotherapy characterised by the cell type used
- A61K40/11—T-cells, e.g. tumour infiltrating lymphocytes [TIL] or regulatory T [Treg] cells; Lymphokine-activated killer [LAK] cells
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K40/00—Cellular immunotherapy
- A61K40/30—Cellular immunotherapy characterised by the recombinant expression of specific molecules in the cells of the immune system
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K40/00—Cellular immunotherapy
- A61K40/40—Cellular immunotherapy characterised by antigens that are targeted or presented by cells of the immune system
- A61K40/41—Vertebrate antigens
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K40/00—Cellular immunotherapy
- A61K40/40—Cellular immunotherapy characterised by antigens that are targeted or presented by cells of the immune system
- A61K40/41—Vertebrate antigens
- A61K40/42—Cancer antigens
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K48/00—Medicinal preparations containing genetic material which is inserted into cells of the living body to treat genetic diseases; Gene therapy
- A61K48/0008—Medicinal preparations containing genetic material which is inserted into cells of the living body to treat genetic diseases; Gene therapy characterised by an aspect of the 'non-active' part of the composition delivered, e.g. wherein such 'non-active' part is not delivered simultaneously with the 'active' part of the composition
- A61K48/0016—Medicinal preparations containing genetic material which is inserted into cells of the living body to treat genetic diseases; Gene therapy characterised by an aspect of the 'non-active' part of the composition delivered, e.g. wherein such 'non-active' part is not delivered simultaneously with the 'active' part of the composition wherein the nucleic acid is delivered as a 'naked' nucleic acid, i.e. not combined with an entity such as a cationic lipid
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K48/00—Medicinal preparations containing genetic material which is inserted into cells of the living body to treat genetic diseases; Gene therapy
- A61K48/005—Medicinal preparations containing genetic material which is inserted into cells of the living body to treat genetic diseases; Gene therapy characterised by an aspect of the 'active' part of the composition delivered, i.e. the nucleic acid delivered
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/63—Introduction of foreign genetic material using vectors; Vectors; Use of hosts therefor; Regulation of expression
- C12N15/79—Vectors or expression systems specially adapted for eukaryotic hosts
- C12N15/85—Vectors or expression systems specially adapted for eukaryotic hosts for animal cells
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/87—Introduction of foreign genetic material using processes not otherwise provided for, e.g. co-transformation
- C12N15/88—Introduction of foreign genetic material using processes not otherwise provided for, e.g. co-transformation using microencapsulation, e.g. using amphiphile liposome vesicle
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/51—Medicinal preparations containing antigens or antibodies comprising whole cells, viruses or DNA/RNA
- A61K2039/53—DNA (RNA) vaccination
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/54—Medicinal preparations containing antigens or antibodies characterised by the route of administration
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/545—Medicinal preparations containing antigens or antibodies characterised by the dose, timing or administration schedule
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/555—Medicinal preparations containing antigens or antibodies characterised by a specific combination antigen/adjuvant
- A61K2039/55511—Organic adjuvants
- A61K2039/55555—Liposomes; Vesicles, e.g. nanoparticles; Spheres, e.g. nanospheres; Polymers
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/57—Medicinal preparations containing antigens or antibodies characterised by the type of response, e.g. Th1, Th2
- A61K2039/572—Medicinal preparations containing antigens or antibodies characterised by the type of response, e.g. Th1, Th2 cytotoxic response
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K2239/00—Indexing codes associated with cellular immunotherapy of group A61K40/00
- A61K2239/31—Indexing codes associated with cellular immunotherapy of group A61K40/00 characterized by the route of administration
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K2239/00—Indexing codes associated with cellular immunotherapy of group A61K40/00
- A61K2239/38—Indexing codes associated with cellular immunotherapy of group A61K40/00 characterised by the dose, timing or administration schedule
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K48/00—Medicinal preparations containing genetic material which is inserted into cells of the living body to treat genetic diseases; Gene therapy
- A61K48/0008—Medicinal preparations containing genetic material which is inserted into cells of the living body to treat genetic diseases; Gene therapy characterised by an aspect of the 'non-active' part of the composition delivered, e.g. wherein such 'non-active' part is not delivered simultaneously with the 'active' part of the composition
- A61K48/0025—Medicinal preparations containing genetic material which is inserted into cells of the living body to treat genetic diseases; Gene therapy characterised by an aspect of the 'non-active' part of the composition delivered, e.g. wherein such 'non-active' part is not delivered simultaneously with the 'active' part of the composition wherein the non-active part clearly interacts with the delivered nucleic acid
- A61K48/0041—Medicinal preparations containing genetic material which is inserted into cells of the living body to treat genetic diseases; Gene therapy characterised by an aspect of the 'non-active' part of the composition delivered, e.g. wherein such 'non-active' part is not delivered simultaneously with the 'active' part of the composition wherein the non-active part clearly interacts with the delivered nucleic acid the non-active part being polymeric
Definitions
- ACTadoptive cell transfer
- RNAhas emerged as a promising therapeutic tool for the treatment of cancer and other diseases, as it can be used to direct the production of proteins that can e.g. inhibit or otherwise reduce the growth or survival of cancer cells.
- the object of the present inventionis to overcome challenges and inefficiencies of ACT therapies by providing novel treatment options exploiting RNA as further therapeutic modality to improve ACT therapies in particular in cancer patients.
- compositions, combinations, or kit or kit of partsconfigured for intramuscular delivery of RNA encoded antigens to e.g. stimulate adoptively transferred T cells.
- the present inventionis inter alia directed to compositions comprising at least one RNA comprising at least one coding sequence encoding at least one antigen for use in a method of treatment or prophylaxis of a disease, disorder or condition, wherein the RNA is preferably formulated in lipid nanoparticles and wherein the composition is preferably administered intramuscularly to a subject that has received modified immune cells, preferably modified T cells, targeted to the at least one antigen.
- Further aspectsrelate to a combination comprising the composition, or a kit or kit of parts comprising the composition, for use in a method of treatment or prophylaxis of a disease, disorder or condition.
- the inventionfurther provides a method for treating or preventing a disease, disorder or condition and a method of boosting a modified immune cell population, preferably a modified T cell population, in a subject.
- the inventionis inter alia based on the surprising finding that intramuscular administration of a composition comprising at least one RNA comprising at least one coding sequence encoding at least one antigen can effectively increase the levels of circulating modified immune cells such as modified T cells in the subject; induce memory formation of modified immune cells such as modified T cells in the subject, preferably formation of central and effector memory cells; activate modified immune cells such as modified T cells in the subject; re-activate modified immune cells such as modified T cells in the subject; maintain the self-renewal capacity of modified immune cells such as modified T cells in the subject; induce higher influx into the solid tumor of modified immune cells such as modified T cells in the subject; and/or induce expansion of modified immune cells such as modified T cells in the subject.
- the present inventionrelates to a composition
- a compositioncomprising at least one RNA comprising at least one coding sequence encoding at least one antigen for use in a method of treatment or prophylaxis of a disease, disorder or condition, wherein the composition is administered to a subject that has received modified immune cells, preferably modified T cells, targeted to the at least one antigen.
- the present inventionrelates to a combination comprising (i) a composition comprising at least one RNA comprising at least one coding sequence encoding at least one antigen; and (ii) modified immune cells, preferably modified T cells, targeted to the at least one antigen, for use in a method of treatment or prophylaxis of a disease, disorder or condition, wherein the modified immune cells are administered intravenously to the subject, and wherein the composition is administered to the subject that has received modified immune cells.
- the present inventionrelates to a kit or kit of parts comprising (A) at least one composition as defined in the context of the first aspect; or (B) at least one combination as defined the context of the second aspect, for use in a method of treatment or prophylaxis of a disease, disorder or condition.
- the present inventionrelates to a method of treating or preventing a disease, disorder, or condition in a subject, comprising a step of applying or administering a composition comprising at least one RNA comprising at least one coding sequence encoding at least one antigen, wherein the subject has received modified immune cells, preferably modified T cells, targeted to the at least one antigen.
- the present inventionrelates to a method of boosting a modified immune cell population in a subject, wherein the method comprises applying or administering at least one composition comprising at least one RNA comprising at least one coding sequence encoding at least one antigen to the subject.
- the composition comprising at least one RNA of the first, second, third, fourth, or fifth aspectis administered by intramuscular administration.
- the RNA of the first, second, third, fourth, or fifth aspectis formulated in lipid nanoparticles (LNPs).
- LNPslipid nanoparticles
- Adoptive cell transferis the transfer of immune cells into a patient.
- the immune cellsmay have originated from the patient or from another individual.
- the cellsare most commonly derived from the immune system with the goal of improving immune functionality and characteristics.
- Different cell typeshave been used for ACT experiments and studies like lymphokine-activated killer (LAK) cells, tumor-infiltrating lymphocytes (TILs), donor lymphocytes after hematopoietic stem cell transplantation (HSCT), tumor-specific T cell lines or clones (chimeric antigen receptor (CAR) or transgenic (tg) T cells), y5T cells (CAR-yST) natural killer cells (CAR-NK), Va24-invariant natural killer T cells (CAR-NKT) and/or macrophages (CAR-M).
- the cells used in connection with the present inventionwill preferably be autologous cells, although heterologous cells or allogenic cells can be used.
- Allogeneic antigenrefers to antigens derived from genetically distinct individuals of the same species. These antigens are recognized by the immune system as foreign and can elicit an immune response, potentially leading to the rejection of transplanted tissues or organs.
- Allogenic, allogenic modified immune cellsThe term “allogeneic” is used to describe anything that is derived from different individuals of the same species. Two or more individuals are said to be allogeneic to one another when the genes at one or more loci are not identical. Accordingly, the term “autologous modified immune cell” refers to a transplant immune cells derived from different individuals of the same species (e.g. a different human species).
- Antigenic peptiderefers to a peptide derived from a (antigenic or immunogenic) protein which stimulates the body’s adaptive or cellular immune system to provide an adaptive or cellular immune response. Therefore, an antigenic/immunogenic peptide comprises at least one epitope (as defined herein) or antigen (as defined herein) selected or derived from e.g. a tumor (neo)antigen.
- Antigen receptorrefers to a specialized protein or molecular structure found on the surface of immune cells, primarily B cells and T cells, that is responsible for recognizing and binding to antigens.
- Antigen receptorscomprise an antigen recognition domain with at least one signalling domain.
- Cellsmay naturally express an antigen receptor or be modified (e.g. ex vivo, in vitro or in vivo in a subject to be treated) to express an antigen receptor. Modification to express an antigen receptor takes place ex vivo and/or in vitro.
- Modified immune cells described hereinmay express an antigen receptor such as a chimeric antigen receptor (CAR) or a T cell receptor (TCR) or a procession product thereof.
- modified immune cells expressing the at least one antigen receptormay be administered to a subject.
- the modified cells expressing the at least one antigen receptormay be endogenous cells of the subject or may have been administered to a subject.
- autoimmune diseases, disorder or conditionrefers to a group of diseases, disorders or conditions in which the immune system loses its ability to distinguish between “self’ and “nonself’, leading to an immune response that damages the body’s own tissues, cells, and organs.
- the autoimmune disease, disorder or conditioncan be rheumatoid arthritis, systemic lupus erythematosus, Type 1 diabetes, multiple sclerosis, Hashimoto’s thyroiditis, Graves’ disease, celiac disease, Crohn’s disease and ulcerative colitis, psoriasis and Sjogren's syndrome.
- Autologous, autologous modified immune cellThe term “autologous” is used to describe anything that is derived from the same subject.
- autologous modified immune cellrefers to a transplant immune cells derived from the same subject. Autologous cells are preferred because they overcome the immunological barrier which otherwise results in rejection.
- cancer or tumor disease, disorder or conditionrefer to or describe the physiological condition in a subject that is typically characterized by unregulated cell growth.
- cancersinclude, but are not limited to, carcinoma, lymphoma, blastoma, sarcoma, and leukemia.
- cancersinclude bone cancer, blood cancer, lung cancer, liver cancer, pancreatic cancer, skin cancer, cancer of the head or neck, cutaneous or intraocular melanoma, uterine cancer, ovarian cancer, rectal cancer, cancer of the anal region, stomach cancer, colon cancer, breast cancer, prostate cancer, uterine cancer, carcinoma of the sexual and reproductive organs, Hodgkin’s disease, cancer of the esophagus, cancer of the small intestine, cancer of the endocrine system, cancer of the thyroid gland, cancer of the parathyroid gland, cancer of the adrenal gland, sarcoma of soft tissue, cancer of the bladder, cancer of the kidney, renal cell carcinoma, carcinoma of the renal pelvis, neoplasms of the central nervous system, neuroectodermal cancer, spinal axis tumors, glioma, meningioma, and pituitary adenoma.
- the term cancer disease or tumor disease according to the disclosurealso comprises cancer metastases.
- Cationic, cationisablemeans that the respective structure, compound, group, or atom bears a positive charge, either permanently or not permanently, e.g. in response to certain conditions such as pH.
- cationicmeans that the respective structure, compound, group, or atom bears a positive charge, either permanently or not permanently, e.g. in response to certain conditions such as pH.
- cationicmeans that the respective structure, compound, group, or atom bears a positive charge, either permanently or not permanently, e.g. in response to certain conditions such as pH.
- cationiccationic
- cationisablepermanently cationic
- CARchimeric antigen receptor'’ or “CAR” will be recognized and understood by the person of ordinary skill in the art. CARs relate to artificial receptors comprising a single molecule or a complex of molecules which recognizes, i.e. binds to, a target structure, e.g. an antigen, on a target cell, e.g.
- an antigen binding domainto an antigen expressed on the surface of the target cell, such as a cancer cell and may confer specificity onto an immune cell such as a T cell expressing said CAR on the cell surface.
- the binding domainbinds to an extracellular domain or to an epitope in an extracellular domain of the antigen.
- CAR expressing cellsdo not necessarily require processing and presentation of an antigen for recognition of the target cell but rather may recognize preferably with specificity any antigen present on a target cell.
- CARsare molecules that combine specificity for a desired antigen, e.g., tumor antigen, which is antibody-based with a T cell receptor-activating intracellular domain to generate a chimeric protein that exhibits a specific cellular immune activity, e.g., a specific anti-tumor cellular immune activity. Recognition of the target structure by a CAR results in activation of an immune cell expressing said CAR.
- a CARmay comprise one or more protein units said protein units comprising one or more domains, an antigen binding domain, a transmembrane domain; and an intracellular domain, mostly comprising a 4-1 BB costimulatory domain, and a CD3-zeta signalling domain.
- the antigen binding domaincomprises in some non-limiting examples a variable region of a heavy chain of an immunoglobulin (VH) with a specificity for the antigen and a variable region of a light chain of an immunoglobulin (VL) with a specificity for the antigen.
- the heavy chain variable region (VH) and the corresponding light chain variable region (VL)can be connected via a peptide linker.
- the antigen binding moiety portion in the CARcan be a scFv.
- a cellcan be genetically modified to stably express a CAR on its surface, conferring novel antigen specificity that may be MHC independent. Immune cells expressing the CAR molecule are from the subject to be treated or from a different subject and administered to the subject to be treated.
- Coding sequencecoding region, cds:
- coding sequenceand the corresponding abbreviation “cds” as used herein refers to a sequence of several nucleotide triplets that can be translated into a peptide or protein.
- a cdsmay be a DNA or RNA sequence consisting of several nucleotides that may be divided by three, which typically starts with a start codon and preferably terminates with a stop codon.
- the cdspreferably encodes at least one tumor antigen as defined herein.
- Derived fromThe term “derived from” as used herein in the context of a nucleic acid, i.e.
- nucleic acid “derived from” (another) nucleic acidmeans that the nucleic acid, which is derived from (another) nucleic acid, shares at least 70%, 80%, 81%, 82%, 83%, 84%, 85%, 86%, 87%, 88%, 89%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% sequence identity or are identical with the nucleic acid from which it is derived.
- the term “derived from”means that the amino acid sequence, which is derived from (another) amino acid sequence, shares at least 70%, 75%, 80%, 81%, 82%, 83%, 84%, 85%, 86%, 87%, 88%, 89%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% sequence identity or are identical with the amino acid sequence from which it is derived.
- Disease, disorder, conditionrefers to an abnormal condition that affects the body of an individual.
- a disease, disorder or conditionis often construed as a medical condition associated with specific symptoms and signs.
- a disease, disorder or conditionmay be caused by factors originally from an external source, such as infectious disease, or it may be caused by internal dysfunctions, such as autoimmune diseases.
- disease, disorder or conditionis often used more broadly to refer to any condition that causes pain, dysfunction, distress, social problems, or death to the individual afflicted, or similar problems for those in contact with the individual. In this broader sense, it sometimes includes injuries, disabilities, disorders, syndromes, infections, isolated symptoms, deviant behaviors, and atypical variations of structure and function, while in other contexts and for other purposes these may be considered distinguishable categories.
- Disease, disorder or condition associated with stem cell transplantationrefers to certain risks and potential complication associated with stem cell transplantation.
- Some of the diseases, disorders or conditions that can occur after stem cell transplantationinclude graft- versus-host disease, infections caused by viruses, bacteria and/or fungi, cytokine release syndrome, organ toxicity, relapse of the disease after transplantation, graft failure, thrombotic microangiopathy, endocrine complications, secondary cancers (myelodysplastic syndromes or certain solid tumors) or psychosocial and emotional challenges (anxiety, depression, and post-traumatic stress disorder).
- EpitopeThe term “epitope” will be recognized and understood by the person of ordinary skill in the art. Epitopes refer to a part or fragment of a molecule such as an antigen that is recognized by the immune system. For example, the epitope may be recognized by T cells, B cells or antibodies.
- An epitope of an antigenmay include a continuous or discontinuous portion of the antigen and may be between about 5 and about 100, such as between about 5 and about 50, more preferably between about 8 and about 30, most preferably between about 10 and about 25 amino acids in length, for example, the epitope may be preferably 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21 , 22, 23, 24, or 25 amino acids in length.
- fragmentas used herein in the context of a nucleic acid sequence (e.g. RNA or DNA) or an amino acid sequence may typically be a shorter portion of a reference sequence of e.g. a nucleic acid sequence or an amino acid sequence. Accordingly, a fragment typically consists of a sequence that is identical to the corresponding stretch within the reference sequence.
- a preferred fragment of a sequence in the context of the present inventionconsists of a continuous stretch of entities, such as nucleotides or amino acids corresponding to a continuous stretch of entities in the molecule the fragment is derived from, which represents at least 50%, 60%, 65%, 70%, 75%, 80%, 85%, 90%, 95% of the total reference molecule from which the fragment is derived.
- hematologic cancers/tumorsrefers to a group of cancers that primarily affect the blood-forming tissues in the body, including the bone marrow, lymphatic system, and blood cells. Hematologic cancers can be broadly categorized into main types: leukemia (cancer of the blood and bone marrow), lymphoma (e.g. Hodgkin lymphoma and non-Hodgkin lymphoma), multiple myeloma and myeloproliferative neoplasms (e.g. thrombocythemia and myelofibrosis).
- heterologous modified immune cellsrefers to a transplant immune cells derived from a different individual.
- identityrefers to the percentage to which two sequences are identical. To determine the percentage to which two sequences are identical, the sequences can be aligned (by also introducing gaps, if necessary) to be subsequently compared to one another.
- Immune cellsinclude immune cells like macrophages, dendritic cells, neutrophiles, natural killer cells, dendritic cells, B lymphocytes, T lymphocytes (cytotoxic T cells, helper T cells, tumor infiltrating T cells) and mast cells.
- immune cellsare cells with lytic potential, in particular lymphoid cells, and are preferably T cells, in particular cytotoxic lymphocytes, preferably selected from cytotoxic T cells, natural killer (NK) cells, and lymphokine-activated killer (LAK) cells.
- Lytic potentialcould mean the elimination of target cells, i.e., cells characterized by expression of an antigen, for example, via apoptosis or perforin-mediated cell lysis, production of cytokines such as IFN-g and TNF-a, and specific cytolytic killing of antigen expressing target cells.
- T cellsupon activation T cells release cytotoxins such as perforin, granzymes, and granulysin.
- Perforin and granulysincreate pores in the target cell, and granzymes enter the cell and trigger a caspase cascade in the cytoplasm that induces apoptosis (programmed cell death) of the cell.
- apoptosiscan be induced via Fas-Fas ligand interaction between the T cells and target cells.
- immune cellscomprise CD34-hematopoietic stem cells, immature and mature T cells and immature and mature B cells.
- the cells used in connection with the present inventionwill preferably be autologous cells, although heterologous cells or allogenic cells can be used.
- infectious disease, disorder or conditionrefers to any disease, disorder or condition which can be transmitted from individual to individual or from organism to organism and is caused by a microbial agent.
- Infectious diseases, disorder or conditionare known in the art and include, for example, a viral disease, a bacterial disease, or a parasitic disease, which diseases are caused by a virus, a bacterium, and a parasite, respectively.
- the infectious disease, disorder or conditioncan be, for example, hepatitis, sexually transmitted diseases, tuberculosis, HIV, diphtheria, hepatitis B, hepatitis C, cholera, severe acute respiratory syndrome (SARS), the bird flu, and influenza.
- SARSsevere acute respiratory syndrome
- Modified immune cellsrefers to immune cells modified to express an antigen receptor such as T cell receptor or B cell receptor or may lack expression of an antigen receptor. Immune cells can be modified to express an antigen receptor on the cell surface. In the context of the invention the immune cells lack endogenous expression of a T cell receptor.
- a modified immune cellis capable of binding an antigen such as an antigen presented in the context of MHC on a cell or expressed on the surface of a cell and mediating an immune response. Said binding resulting in stimulation, priming and/or expansion of the immune cells genetically modified to express an antigen receptor.
- the termrelates to a cell which exerts effector functions during an immune reaction.
- Immune cellscan be modified by transfection to express an endogenous antigen receptor.
- transfectioncan be transient or stable. For some applications of transfection, it is sufficient if the transfected genetic material is only transiently expressed.
- RNAcan be transfected into cells to transiently express its coded protein, e.g. an antigen receptor. Since the nucleic acid introduced in the transfection process is usually not integrated into the nuclear genome, the foreign nucleic acid will be diluted through mitosis or degraded. If it is desired that the transfected nucleic acid actually remains in the genome of the cell and its daughter cells, a stable transfection must occur. Such stable transfection can be achieved by using virus-based systems or transposon-based systems for transfection.
- cells that are genetically modified to express an antigen receptorare stably transfected with nucleic acid encoding the antigen receptor, while, generally, nucleic acid encoding antigen is transiently transfected into cells.
- Modified immune cellsare suitable for adoptive cell transfer therapy.
- Preferred in the context of the inventionare modified T cells such as CAR T cells or TCR T cells.
- nucleic acidrefers to DNA (molecules) or RNA (molecules). It is preferably used synonymously with the term polynucleotide.
- a nucleic acid or a nucleic acid moleculeis a polymer comprising or consisting of nucleotide monomers, which are covalently linked to each other by phosphodiester-bonds of a sugar/phosphate-backbone.
- nucleic acid moleculealso encompasses modified nucleic acid molecules, such as base-modified, sugar-modified, or backbone-modified DNA or RNA molecules as defined herein.
- Nucleic acid sequence, DNA sequence, RNA sequenceThe terms “nucleic acid sequence”, “DNA sequence”, “RNA sequence” refers to a particular and individual order of the succession of its nucleotides.
- Optimal therapeutic dosetypically refers to the standard therapeutic amount of a pharmaceutical agent, meaning that the amount required for the desired effect is given.
- subtherapeutic amountmeans that the dosage or amount of a particular pharmaceutical agent is sufficient to achieve the desired pharmacological action in the absence of other compounds, drugs or pharmaceutical agents, e.g., in the absence of the composition of the invention. Such desired pharmacological action may include the complete or essentially complete rejection of solid tumors.
- RNA in vitro transcriptionrelates to a process wherein RNA is synthesized in a cell-free system in vitro.
- the RNAis obtained by transcribing a DNA template in the presence of a DNA-dependent RNA polymerase (e.g. T7, SP6), ribonucleotide triphosphates (NTPs, and optionally modified NTPs) and optionally, a cap analog, in an appropriate buffer (e.g. comprising MgCb).
- a DNA-dependent RNA polymerasee.g. T7, SP6
- NTPsribonucleotide triphosphates
- cap analogoptionally, a cap analog
- Self-antigenrefers to antigens naturally present in a subject. These antigens are recognized by the immune system as “self’ because they are part of the individual’s own tissues, cells, or organs. Under normal circumstances, the immune system is tolerant of self-antigens, meaning it does not mount an immune response against them.
- self-antigensrefers to antigens that elicit an immune response and leading to inflammation, tissue damage, and the development of autoimmune diseases.
- side effectsare known as adverse reactions, are unwanted undesirable effects that are possibly related to a drug. Side effects can vary from minor problems like a runny nose to life-threatening events, such as a heart attack or liver damage.
- Solid tumorsolid cancer
- solid tumorrefers to the manifestation of a cancerous mass, as is well known in the art.
- the termrefers to a cancer or carcinoma of body tissues other than blood, preferably other than blood, bone marrow, and lymphoid system.
- solid tumorsinclude cancers of the prostate, lung cancer, colorectal tissue, bladder, oropharyngeal/laryngeal tissue, kidney, breast, endometrium, ovary, cervix, stomach, pancreas, brain, and central nervous system.
- subjectrefers to a human or another mammal (e.g. mouse, rat, rabbit, dog, cat, cattle, swine, sheep, horse or primate) that can be afflicted with or is susceptible to a disease or disorder (e.g., cancer) but may or may not have the disease or disorder.
- a disease or disordere.g., cancer
- subjectdoes not denote a particular age, and thus encompass adults, elderlies, children, and newborns.
- Syngeneic, syngeneic modified immune cellsThe term “syngeneic” is used to describe anything that is derived from individuals ortissues having identical genotypes, i.e., identical twins or animals of the same inbred strain, ortheir tissues. Accordingly, the term “syngeneic modified immune cell” refers to a transplant immune cells derived from individuals or tissues having identical genotypes.
- T cell epitoperefers to a part or fragment of a protein that is recognized by a T cell when presented in the context of MHC molecules.
- major histocompatibility complexand the abbreviation “MHC” includes MHC class I and MHC class II molecules and relates to a complex of genes which is present in all vertebrates. MHC proteins or molecules are important for signaling between lymphocytes and antigen presenting cells or diseased cells in immune reactions, wherein the MHC proteins or molecules bind peptide epitopes and present them for recognition by T cell receptors on T cells.
- the proteins encoded by the MHCare expressed on the surface of cells, and display both self-antigens, peptide fragments from the cell itself, and non-self-antigens, e.g., fragments of invading microorganisms, to a T cell.
- the binding peptidesare typically about 8 to about 10 amino acids long although longer or shorter peptides may be effective.
- the binding peptidesare typically about 10 to about 25 amino acids long and are in particular about 13 to about 18 amino acids long, whereas longer and shorter peptides may be effective.
- T cell receptorThe term “T cell receptor” as used herein in the context of the invention will be recognized and understood by the person of ordinary skill in the art.
- TCRT cell receptor
- the actual T cell receptoris composed of two separate peptide chains, which are produced from the independent T cell receptor alpha and beta (TCRa and TCR£) genes and are called a- and p-TCR chains.
- TCRa and TCR£T cell receptor alpha and beta
- y 5 T cellsrepresent a small subset of T cells that possess a distinct T cell receptor (TCR) on their surface, in this cells TCR is made up of one y -chain and one 5 -chain.
- TCRsare composed of one a-chain and one p-chain or of one y-chain and one 5-chain.
- the TCR a/p chainsare composed of an N-teiminal highly polymorphic variable region involved in antigen recognition and an invariant constant region. On the genetic level, these chains are separated into several regions, a variable (V) region, a diversity (D) region (only P- and 5-chain), a joining (J) region and a constant (C) region.
- the human p-chain genescontain over 60 variable (V), 2 diversity (D), over 10 joining (J) segments, and 2 constant region segments (C).
- the human a-chain genescontain over 50 V segments, and over 60 J segments but no D segments, as well as one C segment.
- T cell receptor genesare created by rearranging one V, one D (only p- and 5-chain), one J and one C region gene.
- the diversity of the TCRsis further amplified by imprecise V-(D)-J rearrangement wherein random nucleotides are introduced and/or deleted at the recombination sites. Since the rearrangement of the TCR gene loci occurs in the genome during maturation of T cells, each mature T cell only expresses one specific a/p ICR or y/5 TCR.
- MHC and antigen bindingis mediated by the complementary determining regions 1 , 2 and 3 (CDR1 , CDR2, CDR3) of the TCR.
- the CDR3 of the p-chain which is most critical for antigen recognition and bindingis encoded by the V-D-J junction of the rearranged TCR p-chain gene.
- the TCRis a part of a complex signalling machinery, which includes the heterodimeric complex of the TCR a- and p-chains, the co-receptor CD4 or CD8 and the CD3 signal transduction. While the CD3 chains transfer the activation signal inside the cell, the TCR a/p heterodimer is solely responsible for antigen recognition. Thus, the transfer of the I CR a/p chains offers the opportunity to redirect T cells towards any antigen of interest.
- TCRsT cell receptors
- MHCmajor histocompatibility complex
- Transgenic TCR(TCRtq): The term “transgenic T cell receptor” as used herein in the context of the invention will be recognized and understood by the person of ordinary skill in the art.
- the antigenic specificity of T cellsis rested entirely on the heterodimeric complex of the TCR a- and p-chain, the transfer of cloned TCR genes into T cells offers the potential to redirect them towards any antigen of interest.
- the transgenic TCRrecognises a known antigen expressed by cancer or infected cells and presented in the context of MHC, which leads to induction of cytotoxic T cell effector functions and the killing of targeted tumor or infected cells.
- a T cellcan be genetically modified to stably express a transgenic TCR on its surface.
- An immune cell genetically modified to express a transgenic T cell receptor (TCR) targeting the cells through binding to the antigen (or a procession product thereof)are provided to a subject such as by administration of the genetically modified immune cells to the subject or generation of genetically modified immune cells in the subject.
- TCRtransgenic T cell receptor
- Advantages of TCR gene transferare the creation of therapeutic quantities of antigen-specific T cells within a few days and the possibility to introduce specificities that are not present or ineffective in the endogenous TCR repertoire of the patient.
- Tumor antigenrefers to a constituent of cancer cells which may be derived from the cytoplasm, the cell surface and the cell nucleus.
- tumor antigenrefers to antigens that are common to specific hyperproliferative disorders such as cancer. In particular, it refers to those antigens which are produced intracellularly or as surface antigens on tumor cells.
- tumor antigensThere are two major classes of tumor antigen which are typically targeted by modified immune cells in ACT therapies, tumor specific antigens and tumor associated antigens.
- T umor specific antigensfound on cancer cells only, not on healthy tissue or cells. They arise mostly from oncogenic driver mutations that generate novel peptide sequences, i.e.
- tumor antigencan be defined in further groups: The first group defines mutation antigens, neoantigens or unique antigens. Mutation antigens resulting from specific (point) mutations and expressed throughout the tumor, such as a non-limiting example B-RAF mutation in melanoma. The second group are so called tumor-specific antigens. These are shared antigens expressed in tumors, while being absent from healthy tissue, such as a non-limiting example NY-ESO-1 found in many tumor types.
- the third groupare differentiation antigens comprising shared antigens expressed by tumors, as well as the tissue the tumor originated from, such as a non-limiting example CD19 in B cell lymphoma.
- the fourth groupare overexpressed antigens comprising shared antigens which are expressed in different healthy tissues, but are overexpressed in tumors, such as a non-limiting example p53.
- the fifth groupare virus tumor antigens comprising viral antigens associated with virus-associated cancers, such as a non-limiting example HPV-16 E7.
- Another groupare microbial or bacterial tumor antigens associated with tumors i.e. glioblastoma.
- Variantrefers to a nucleic acid sequence derived from a reference nucleic acid sequence.
- a variant of a nucleic acid sequencemay exhibit one or more nucleotide deletions, insertions, additions and/or substitutions compared to the reference nucleic acid from which the variant is derived from.
- a variantmay be a functional variant in the sense that the variant has retained at least 70%, 80%, 90%, or 95% or more of the function of the sequence where it is derived from.
- a “variant’ of a nucleic acid sequencemay have at least 70%, 75%, 80%, 85%, 90%, 95%, or 99% identity over a stretch of at least 30, 50, 75 or 100 nucleotides.
- variantrefers to a proteins or peptide having an amino acid sequence which differs from the reference sequence in one or more mutation(s) substitution(s), such as one or more substituted, inserted and/or deleted amino acid(s). Insertions and substitutions are possible at those sequence positions which cause no modification to the three-dimensional structure or do not affect the binding region.
- a variant of a peptide or proteinmay be a functional variant, which means that the variant exerts essentially the same, or at least 70%, 80%, 90% of the function of the peptide or protein it is derived from (e.g. antigenic property).
- a “variant’ of a peptide or proteinmay have at least 70%, 75%, 80%, 85%, 90%, 95%, or 99% identity over a stretch of at least 30, 50, 75 or 100 amino acids.
- compositionfor use in treatment or prophylaxis of a disease, disorder or condition
- the present inventionrelates to a composition
- a compositioncomprising at least one RNA comprising at least one coding sequence (cds) encoding at least one antigen for use in a method of treatment or prophylaxis of a disease, disorder or condition, wherein the composition is administered to a subject that has received modified immune cells targeted to the at least one antigen.
- cdscoding sequence
- the term “targeted to the at least one antigen”means that the modified immune cells such as the modified T cells are capable to bind and/or detect the at least one antigen that is provided by the RNA of the invention, or a fragment or variant thereof.
- the respective antigen or a processing product thereofe.g., a fragment thereof, binds to the specific antigen receptor (such as TCR or CAR) carried by the modified immune cells. Said binding may result in stimulation, priming and/or expansion of the modified immune cells genetically modified to express an antigen receptor and provides an immune response against the antigen which may be therapeutic or partially or fully protective.
- the antigenmay be expressed by a target cell to which the modified immune cells are targeted (e.g. tumor cell) or a fragment thereof, or a variant of the antigen or the fragment.
- the compositionis administered to a subject via subcutaneous, intravenous, intramuscular, intraarticular, intra-synovial, intranasal, oral, intrasternal, intrathecal, intrahepatic, intralesional, intracranial, transdermal, intradermal, intrapulmonal, intraperitoneal, intracardial, intraarterial, intraocular, intravitreal, subretinal, intranodal, or intratumoral, preferably intramuscular, intravenous or intratumoral administration.
- the compositionis administered to a subject via subcutaneous, intramuscular, intra-articular, intra-synovial, intranasal, oral, intrasternal, intrathecal, intrahepatic, intralesional, intracranial, transdermal, intradermal, intrapulmonal, intraperitoneal, intracardial, intraarterial, intraocular, intravitreal, subretinal, intranodal, or intratumoral, preferably intramuscular or intratumoral administration.
- the compositionis administered to a subject via intramuscular or intratumoral administration.
- the compositionis administered to a subject via intramuscular administration, in particular, intramuscular injection.
- the RNA of the compositionis formulated. Suitable formulations in the context of the invention are further described under paragraph “Formulation/Complexation of the RNA of the composition”.
- the RNAis formulated in lipid nanoparticles as further defined herein.
- the compositioncomprises at least one RNA comprising at least one cds encoding at least one antigen for use in a method of treatment or prophylaxis of a disease, disorder or condition, wherein the composition is administered to a subject that has received modified immune cells, preferably modified T cells, targeted to the at least one antigen.
- the compositioncomprises at least one RNA comprising at least one cds encoding at least one antigen for use in a method of treatment or prophylaxis of a disease, disorder or condition, wherein the composition is preferably administered intramuscularly to a subject that has received modified immune cells, preferably modified T cells, targeted to the at least one antigen.
- the RNAis formulated as further defined in paragraph “Formulation/Complexation of the RNA of the composition”. More preferably, the RNA is formulated in lipid nanoparticles (LNPs).
- modified immune cellsthat have been received by the subject are defined in more detail.
- the modified immune cellsare modified T cells.
- the modified immune cells, preferably the modified T cells, that have been received by the subjectare either from a subject to be treated or from a different subject.
- the modified immune cellspreferably the modified T cells, that have been received by the subject comprise autologous immune cells taken from the subject, heterologous immune cells, allogenic immune cells or syngeneic immune cells.
- the modified immune cellspreferably the modified T cells, that have been received by the subject may be autologous, allogeneic or syngeneic to the subject to be treated.
- the immune cellshave been removed from a subject and have subsequently been re-delivery to the subject.
- the immune cells from the subjecthave not been removed before administration of the composition. In this embodiment, all steps to modify the immune cells of the subject are performed in vivo.
- the modified immune cellspreferably the modified T cells, that have been received by the subject comprise autologous immune cells taken from the subject.
- the modified immune cellspreferably the modified T cells, that have been received by the subject express an antigen receptor targeted to the antigen encoded by the RNA of the composition and/or express the antigen corresponding to the antigen that is encoded by the RNA of the composition. Accordingly, in preferred embodiments, the modified immune cells that have been received by the subject are genetically modified to express an antigen receptor targeted to the antigen and/or to express the antigen.
- the modified immune cellspreferably the modified T cells, that have been received by the subject are genetically modified, ex vivo or in vivo, to express an antigen receptor targeted to the antigen encoded by the RNA of the composition and/or to express the antigen corresponding to the antigen that is encoded by the RNA of the composition.
- the modified immune cells, preferably the modified T cells, that have been received by the subjectare genetically modified ex vivo prior to administration or genetically modified in vivo in the subject following administration to express an antigen receptor and/or the antigen described herein.
- the modified immune cellspreferably the modified T cells, that have been received by the subject are genetically modified ex vivo to express the antigen receptor targeted to the antigen encoded by the RNA of the composition and/or to express the antigen corresponding to the antigen that is encoded by the RNA.
- the modified immune cells, preferably the modified T cells, that have been received by the subjectare genetically modified to express the antigen receptor targeted to the antigen encoded by the RNA of the composition.
- “genetically modified to express an antigen receptor and/or antigen”means that the immune cells, preferably the T cells, have been transfected with a nucleic acid, in particular RNA, into an immune cell to e.g. modify the immune cell.
- the transfectioncan be transient or stable.
- the modified immune cells, preferably the modified T cells, that have been received by the subjectcan be genetically modified to stably express an antigen receptor e.g. on its surface.
- the antigen receptor(of the modified immune cells, preferably the modified T cells) is targeted to the antigen (encoded by the RNA of the composition), that is associated with the disease, disorder or condition.
- the antigen receptor(of the modified immune cells, preferably the modified T cells) is targeted to at least one antigen associated with a disease, disorder or condition. In some embodiment, the antigen receptor (of the modified immune cells, preferably the modified T cells) is targeted to at least two antigens associated with a disease, disorder or condition.
- the antigen(to which the modified immune cells, preferably the modified T cells, are targeted to) is selected from or derived from tumor antigens, viral antigens, bacterial antigens, protozoal antigens, fungal antigens, self-antigens or allogenic antigens, preferably tumor antigens.
- the subjecthas received modified immune cells, preferably modified T cells, targeted to at least one antigen selected from or derived from tumor antigens, viral antigens, bacterial antigens, protozoal antigens, fungal antigens, self-antigens or allogenic antigens, preferably tumor antigens.
- modified immune cellspreferably modified T cells, targeted to at least one antigen selected from or derived from tumor antigens, viral antigens, bacterial antigens, protozoal antigens, fungal antigens, self-antigens or allogenic antigens, preferably tumor antigens.
- the subjecthas received modified immune cells, preferably modified T cells, targeted to at least two antigens selected from or derived from tumor antigens, viral antigens, bacterial antigens, protozoal antigens, fungal antigens, self-antigens or allogenic antigens, preferably tumor antigens.
- modified immune cellspreferably modified T cells, targeted to at least two antigens selected from or derived from tumor antigens, viral antigens, bacterial antigens, protozoal antigens, fungal antigens, self-antigens or allogenic antigens, preferably tumor antigens.
- the subjecthas received modified regulatory T cells expressing at least one antigen receptor targeted to at least one self-antigen or allogenic antigen.
- the antigen receptor (of the modified immune cells)is a chimeric antigen receptor (CAR), T cell receptor (TCR) or a transgenic T cell receptor (TCRtg), preferably a TCRtg.
- the modified immune cells that have been received by the subjectexpress at least one antigen receptor, preferably at least one CAR, TCR and/or TCRtg.
- the subjecthas received modified immune cells expressing a CAR.
- the modified immune cells that have been received by the subjectare CAR-M cells expressing at least one CAR molecule targeted to at least one antigen, preferably a tumor specific or tumor associated antigen.
- the subjecthas received modified immune cells expressing a TCR or a TCRtg, preferably a TCRtg.
- the modified immune cells that have been received by the subjectare selected from tumorinfiltrating leukocytes (TILs) or modified T cells.
- Tumor-Infiltrating Leukocyte (TIL) therapyis a form of immunotherapy used in cancer treatment. It involves the extraction, in vitro expansion, and reinfusion of autologous immune cells, specifically T lymphocytes (T cells), which have infiltrated a tumor. TIL therapy is typically utilized in the treatment of certain types of cancer, such as melanoma.
- the subjecthas received autologous and ex vivo expanded tumor-infiltrating leukocytes.
- Modified T cellsare T lymphocytes that have been altered or enhanced through genetic engineering techniques in vivo, ex vivo, in vitro to express an antigen receptor, e.g. CARs or TCRtg.
- an antigen receptore.g. CARs or TCRtg.
- the modified T cell that have been received by the subjectis a CD4 positive helper T cell, CD8 positive cytotoxic T cell, regulatory T cell (Treg), and/or a memory T cell.
- the subjecthas received modified T cells, preferably modified CD4 positive helper T cells, modified CD8 positive cytotoxic T cells, modified regulatory T cells (T regs), and/or modified memory T cells before the administration of the composition.
- modified CD4 positive helper T cellrefers to T cells that play a vital role in coordinating the immune response. These cells recognize antigens presented by antigen-presenting cells (APCs) and help activate other immune cells, such as B cells and cytotoxic T cells. Helper T cells are essential for both antibody-mediated (humoral) and cell-mediated immune responses.
- CD8 positive cytotoxic T cellrefers to T cells that are responsible for directly attacking and destroying infected or cancerous cells. These cells recognize antigens on the surface of infected or cancerous cells and release toxic molecules to induce cell death.
- regulatory T cellsrefers to T cells that help maintain immune system balance and prevent excessive immune responses. These cells play a role in suppressing immune reactions to self-antigens and preventing autoimmunity.
- memory T cellrefers to T cells, like CD4 or CD8 positive T cells, that become a memory T cell after an initial encounter with an antigen. These cells "remember” the specific antigen, allowing for a faster and more efficient immune response upon subsequent exposure to the same antigen.
- the modified T cell that have been received by the subjectis a CD8 positive cytotoxic T cell. Accordingly, the subject received modified CD8 positive cytotoxic T cells before the administration of the composition.
- the modified T cells that have been received by the subjectare selected from transgenic TCR (TCRtg) and/or chimeric antigen receptor (CAR) T cells.
- TCRtgtransgenic TCR
- CARchimeric antigen receptor
- the modified immune cells that have been received by the subjectare selected from modified T cells, preferably transgenic T cells that are genetically modified to express an antigen receptor, preferably a transgenic TCR(TCRtg) targeted to the antigen.
- modified T cellspreferably transgenic T cells that are genetically modified to express an antigen receptor, preferably a transgenic TCR(TCRtg) targeted to the antigen.
- modified T cellsselected from transgenic TCR (TCRtg) and/or chimeric antigen receptor (CAR) T cells before the administration of the composition.
- TCRtgtransgenic TCR
- CARchimeric antigen receptor
- the chimeric antigen receptor (CAR) T cellscomprise at least one CAR molecule. Accordingly, in preferred embodiments the subject has received chimeric antigen receptor (CAR) T cells comprising at least one CAR molecule. In some embodiments, the CAR molecule comprises at least one an antigen binding domain. In some embodiments, the CAR molecule comprises at least two antigen binding domains. Accordingly, in some embodiments, the subject has received modified immune cells expressing a CAR molecule that comprises at least one or at least two antigen binding domains. In some embodiments, the subject has received modified immune cells expressing a CAR molecule that comprises at least one tumor antigen binding domain.
- the subjecthas received modified immune cells expressing a chimeric antigen receptor (CAR) molecule comprising a CD19, BCMA, CD22, CD20, Mesothelin, GPC3, GD2, HER2, B7-H3, Claudin18.2 (CLDN18.2), Claudin6 (CLDN6), PSMA, PRAME CEA, EGFR, MUC1 , EGFRVIII, NKG2DL antigen binding domain, preferably an Claudin6 or PRAME antigen binding domain.
- CARchimeric antigen receptor
- the subjecthas received axicabtagene ciloleucel (YescartaTM), brexucabtagene autoleucel (TecartusTM), ciltacabtagene autoleucel (CarvyktiTM), idecabtagene vicleucel (AbecmaTM), lisocabtagene maraleucel (BreyanziTM) ortisagenlecleucel (KymriahTM).
- the subjecthas received modified immune cells expressing a chimeric antigen receptor (CAR) molecule comprising a CLDN6 antigen binding domain.
- CARchimeric antigen receptor
- the CLDN6 antigen binding domaincomprises the amino acid sequence of SEQ ID NOs: 32-42 or a functional variant thereof, preferably SEQ ID NO: 32.
- a binding domain for CLDN6comprises a heavy chain variable region (VH) comprising an amino acid sequence selected from the group consisting of SEQ ID NOs: 33-36 or a functional variant thereof.
- a binding domain for CLDN6comprises a light chain variable region (VL) comprising an amino acid sequence selected from the group consisting of SEQ ID NOs: 37-42 or a functional variant thereof.
- the subjecthas received modified immune cells, preferably modified T cells such as CAR T cells, targeted to Claudin6 or a fragment thereof, or a variant of CLDN6, and the at least one RNA of the composition of the invention that is administered to said subject encodes CLDN6 or a fragment thereof, or a variant of CLDN6.
- modified T cellssuch as CAR T cells
- Claudin6 or a fragment thereofor a variant of CLDN6
- the at least one RNA of the composition of the invention that is administered to said subjectencodes CLDN6 or a fragment thereof, or a variant of CLDN6.
- the subjecthas received modified T cells expressing a transgenic TCR molecule.
- the transgenic TCR moleculecomprises at least one an antigen binding domain. In some embodiments, the transgenic TCR molecule comprises at least two antigen binding domains. In some embodiments, the transgenic TCR molecule comprises at least one tumor antigen binding domain.
- the subjecthas received modified immune cells expressing a transgenic TCR molecule comprising an W 1 , COL6A3, HA-1, NY-ESO-1, HPV, MAGE-A4, HBV, EBV, mutated KRAS; KK-LC-1, MAGE-A3, MAGE-A10, AFP, Mesothelin, EGFRVIII, MAGE-A1, MART-1, PRAME, mutated PRAME, CD19, CD33, BCMA, CD22, CD70, NKG2DL, PD-L1 , ROBO1, 5T4, PSMA, CLDN6, DLL3, HER2, MICA/B and/or MUC1 antigen binding domain.
- a transgenic TCR moleculecomprising an W 1 , COL6A3, HA-1, NY-ESO-1, HPV, MAGE-A4, HBV, EBV, mutated KRAS; KK-LC-1, MAGE-A3, MAGE-A10, AFP, Mesothelin, EGFRVIII
- the subjecthas received modified immune cells expressing a TCR molecule comprising an PRAME or CLDN6 antigen binding domain.
- the subjecthas received modified immune cells expressing a TCR molecule comprising an PRAME antigen binding domain, wherein said antigen binding domain is capable of specifically and/or selectively binding to a PRAME antigenic peptide, such as a peptide shown in SEQ ID NO: 67.
- the antigen binding domaincomprising at least one complementary determining region (CDR) 3 having at least 80% sequence identity to an amino acid sequence selected from SEQ ID NOs: 48-65.
- the transgenic TCR a or y chaincomprises a CDR3 having at least 80% sequence identity to an amino acid sequence selected from SEQ ID NOs: 48-56.
- the transgenic TCR p or 6 chaincomprises a CDR3 having at least 80% sequence identity to an amino acid sequence selected from SEQ ID NOs: 57-65.
- the subjecthas received modified immune cells expressing a TCR molecule comprising a CLDN6 antigen binding domain, wherein the antigen binding domain is capable of specifically and/or selectively binding to a CLDN6 antigenic peptide, such as a peptide shown in SEQ ID NOs: 45-47.
- the antigen binding domaincomprising at least one TCR having at least 80% sequence identity to an amino acid sequence selected from SEQ ID NOs: 8-31.
- the antigen binding domaincomprising at least one alpha chain or a T cell receptor comprising said T cell receptor alpha chain wherein the said T cell receptor a-chain is selected from the group of SEQ ID NOs: 8- 19 or a variant thereof.
- the antigen binding domaincomprising at least one beta chain or a T cell receptor comprising said T cell receptor beta chain wherein the said T cell receptor beta chain is selected from the group of SEQ ID NOs: 20-31 or a variant thereof.
- the at least one RNA of the compositionencodes at least one antigen that is selected from the antigen to which the modified immune cells that the subject has received are targeted to.
- modified immune cellspreferably modified T cells, targeted to CD19, BCMA, CD22, CD20, Mesothelin, GPC3, GD2, HER2, B7-H3, CLDN18.2, Claudin6 (CLDN6), PSMA, PRAME, mutated PRAME, CEA, EGFR, MUC1, EGFRVIII, NKG2DL, WT1, COL6A3, HA-1, NY-ESO-1, HPV, MAGE-A4, HBV, EBV, mutated KRAS; KK-LC-1, MAGE-A3, MAGE-A10, AFP, Mesothelin, EGFRVIII, MAGE-A1, MART-1 , CD19, CD33, BCMA, CD22, CD70, NKG2DL, PD-L1 , ROBO1, 5
- the at least one antigen that is encoded by the at least one RNA of the compositionis defined in more detail. Further features relating to "antigens” in the context of the invention are also provided in paragraph “coding sequences”.
- features and embodiments disclosed herein defining "the antigen that is encoded by the at least one RNA of the composition”may likewise be applicable to the at least one antigen to which the modified immune cells that the subject has received are targeted to.
- the antigencomprises at least one epitope, at least two epitopes, at least three epitopes, at least four epitopes, at least five epitopes, at least six epitopes, at least seven epitopes, at least eight epitopes, at least nine epitopes, or at least ten epitopes.
- the antigenis associated with the disease, disorder or condition.
- a disease-associated, disorder-associated, or condition-associated antigenis a molecule which contains epitopes that will stimulate a host’s immune system to make a cellular antigen-specific immune response and/or a humoral antibody response against the disease, disorder or condition.
- the disease-associated, disorder-associated or condition- associated antigen or an epitope thereofmay be associated with infection by viruses or microbes, typically viral antigens, or associated with cancer, typically tumors.
- the at least one antigenis transiently expressed in cells of the subject.
- the RNA comprising at least one cds encoding at least one antigenis not integrated into the genome of the cells.
- the at least one antigenis expressed in cells of the subject such as antigen presenting cells to facilitate binding of the expressed antigen to the modified immune cells targeted to said (expressed) antigen which preferably results in stimulation, priming and/or expansion of the modified immune cells in the subject.
- the antigenis selected from or is derived from tumor antigens, viral antigens, bacterial antigens, protozoal antigens, fungal antigens, self-antigens or allogenic antigens, preferably tumor antigens.
- the tumor antigenis a tumor-specific antigen or tumor associated antigen, preferably selected from mutation antigen, neoantigen, unique antigen, differentiation tumor antigen, overexpressed tumor antigen, virus tumor antigen or bacterial tumor antigen as described herein.
- the antigenis a tumor antigen selected or derived from List 1 comprising p53, 5T4, ART-4, AFP, BAGE, beta-eaten in/m, BCMA, Bcr-abL B7-H3, mutated BRAF, CAMEL, CAP-1, CASP-8, CDC27/m, CDK4/m, CD19, CD22, CD20, CD33, CD70, CEA, mutated CTNNB1 , the cell surface proteins of the claudin family, such as Claudin6 (CLDN6), CLDN18.2 and CLDN12, COL6A3, c-MYC, CT, Cyp-B, DAM, DLL3, EBV, ELF2M, ETV6-AML1, EGFR, EGFRvlll, EphA2, G250, GAGE, GD2, GnT-V, GPC3, Gap 100, Gp100, HAGE, HA-1, HER-2/neu, HBV, HPV-E7,
- the tumor antigenis selected from List 2 comprising CD19, BCMA, CD22, CD20, Mesothelin, GPC3, GD2, HER2, B7-H3, CLDN18.2, Claudin6 (CLDN6), PSMA, CEA, EGFR, MUC1, EGFRVIII, NKG2DL, WT1, HA-1, NY-ESO-1, HPV, MAGE-A4, HBV, EBV, mutated KRAS; KK-LC-1 , MAGE-A3, MAGE-A10, AFP, MAGE-A1, MART-1 , PRAME, mutated PRAME, CD33, CD70, NKG2DL, PD-L1 , ROBO1 , 5T4, DLL3, COL6A3 and/or MICA/B, preferably from CLDN6 or PRAME, or an immunogenic fragment or variant of any of these.
- CLDN6Claudin6
- the tumor antigenis selected from List 3 comprising CD19, BCMA, CD22, CD20, Mesothelin, GPC3, GD2, HER2, B7-H3, CLDN18.2, Claudin6 (CLDN6), PSMA, CEA, EGFR, MUC1, EGFRVIII, PRAME or NKG2DL antigen, or an immunogenic fragment or variant of any of these.
- the subjecthas received a modified T cell expressing a CAR molecule targeted to the CD19, BCMA, CD22, CD20, Mesothelin, GPC3, GD2, HER2, B7-H3, CLDN18.2, CLDN6, PSMA, CEA, EGFR, MUC1 , EGFRVIII, PRAME or NKG2DL antigen and the subject is administered with a composition comprising at least one RNA comprising at least one coding sequence encoding CD19, BCMA, CD22, CD20, Mesothelin, GPC3, GD2, HER2, B7-H3, CLDN18.2, Claudin6 (CLDN6), PSMA, CEA, EGFR, MUC1, EGFRVIII, PRAME or NKG2DL antigen, or an immunogenic fragment or variant of any of these.
- a compositioncomprising at least one RNA comprising at least one coding sequence encoding CD19, BCMA, CD22, CD20, Mesothelin, GPC3, GD2, HER2,
- the tumor antigenis selected from List 4 comprising WT 1 , HA-1 , NY-ESO-1 , HPV, MAGE-A4, HBV, EBV, mutated KRAS, KK-LC-1, MAGE-A3, MAGE-A10, AFP, Mesothelin, EGFRVIII, MAGE-A1, MART-1 , PRAME, mutated PRAME, antigen, or an immunogenic fragment or variant of any of these.
- the subjecthas received a modified T cell expressing a TCRtg targeted to the WT1 , HA-1, NY-ESO-1, HPV, MAGE-A4, HBV, EBV, mutated KRAS; KK-LC-1, MAGE-A3, MAGE-A10, AFP, Mesothelin, EGFRVIII, MAGE-A1 , MART-1 , PRAME, mutated PRAME, antigen and the subject is administered with a composition comprising at least one RNA comprising at least one cds encoding WT 1 , HA-1 , NY-ESO-1 , HPV, MAGE- A4, HBV, EBV, mutated KRAS; KK-LC-1, MAGE-A3, MAGE-A10, AFP, Mesothelin, EGFRVIII, MAGE-A1, MART-1, PRAME, mutated PRAME, antigen, or an immunogenic fragment or variant of any of these.
- the at least one encoded antigencomprises at least the epitope recognized by the antigen receptor of the modified immune cells, preferably the modified T cells.
- the epitopecomprises at least one T cell epitope.
- the at least one encoded antigencomprises at least 1 , 2, 3, 4, 5, 6, 7, 8, 9 or 10 epitopes recognized by the antigen receptor of the modified immune cells, preferably the modified T cells.
- the at least one encoded antigencomprises at least 1 , 2, 3, 4, 5, 6, 7, 8, 9 or 10 T cell epitopes recognized by the antigen receptor of the modified immune cells, preferably the modified T cells.
- the disease, disorder or conditionis selected from a cancer or tumor disease, disorder or condition, an infectious disease, disorder or condition, an autoimmune disease, disorder or condition, or a disease, disorder or condition associated with stem cell transplantation.
- the disease, disorder or conditionis selected from a disease, disorder or condition associated with stem cell transplantation, preferably infections caused by viruses.
- the disease, disorder or conditionis selected from infectious disease, disorder or condition.
- the diseases, disorder or conditionis selected from a tumor or cancer disease, disorder or condition and an infectious disease, disorder or condition.
- the disease, disorder or conditionis selected from a tumor or cancer disease, disorder or condition.
- the cancer diseaseis derived from solid tumors or hematologic cancers or blood cancers.
- the subjectis suffering from cancer disease, preferably derived from solid tumors or hematologic cancers or blood cancers.
- composition of the inventionis administered in more detail.
- the subjecthas received modified immune cells as defined herein (see in particular paragraph “ The modified immune cells that have been received by the subject').
- the subjecthas received the modified immune, preferably the modified T cells, by intravenous administration, intratumoral administration, intraperitoneal administration, intracerebral administration, intrathecal administration, intravesical administration or intraventricular administration.
- the route of administrationdepends on the specific type of modified immune cells, depending on the disease and location of the disease, e.g. intravenous administration is the most common administration route for modified immune cells (e.g. for ACT) and allows the modified immune cells to circulate throughout the body, targeting disease cells or tissue expressing the antigen.
- Intratumoral administrationmay be preferred to treat solid tumors, the modified immune cells may be directly injected into the tumor.
- Intraperitoneal administrationinvolves the delivery of modified immune cells into the peritoneal cavity (space within the abdominal cavity). This route may be preferred to treat ovarian cancer and peritoneal mesothelioma.
- Intracerebral administrationmay be preferred in embodiments where adoptive cell therapy is used to target brain tumors or central nervous system (CNS) diseases, modified immune cells may be injected directly into the cerebrospinal fluid surrounding the brain and spinal cord.
- Intrathecal Injectioninvolves injecting modified T cells into the cerebrospinal fluid within the spinal canal. This route is used for conditions affecting the CNS, such as certain types of leukemia and lymphoma that have spread to the brain and spinal cord.
- Intravesical administrationmay be preferred in cases of bladder cancer, modified immune cells are administrated directly into the bladder through a catheter.
- Intraventricular administrationmay be preferred for certain CNS disorders and involves injecting modified immune cells directly into the ventricles of the brain.
- the subjecthas received the modified immune cells, preferably the modified T cells, by systemic administration, in particular, by intravenous administration.
- the subjectis a mammal, e.g. mouse, rat, rabbit, dog, cat, cattle, swine, sheep, horse, primate or human.
- the subjectis a mammal, preferably a human.
- the subjectpreferably the human, is an adult, elderly, child or newborn.
- modified immune cellsare correlated with therapeutic responses.
- the number of modified immune cells which can be administered to a subject for ACTis limited and the generation of a large amount of modified immune cells for ACT still remains a challenge.
- a substantial increase in cell persistencecould be achieved when patients received a lymphodepleting preparative regimen before infusion of modified immune cells, particularly TILs or modified T cells, e.g. CAR or TCRtg T cells.
- Lymphodepletion treatmentcomprises the reduction of populations of circulating lymphocytes, prior to infusion of modified immune cells, including chemotherapy, radiotherapy and/or any other method specified.
- the lymphodepletion treatmentcomprises chemotherapy or radiation therapy or a combination of both.
- the lymphodepletion treatmentcomprises chemotherapy selected from the group of fludarabine, cyclophosphamide, combination of fludarabine and cyclophosphamide, combination of cyclophosphamide and total body irradiation, busulfan, melphalan, cyclosporine, methotrexate, combination of cyclosporine and methotrexate, and/or bendamustine.
- the lymphodepletion treatmentcomprises chemotherapy selected from fludarabine and/or cyclophosphamide.
- the subjecthas received lymphodepletion, preferably with fludarabine or cyclophosphamide, within 1 , 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13 or 14 days before the administration of the modified immune cells, preferably the modified T cells.
- Lymphodepletionis correlated with adverse side effects for the subject. Almost two-thirds of the subjects developed side effects following lymphodepletion. Side effects correlated with lymphodepletion are fewer, weakness, chills, loss of appetite, immune-related toxicity and cytokine-related toxicity, cytokine release syndrome, organ dysfunction, arrhythmias, renal failure, neurotoxicity, anemia, thrombocytopenia, graft failure, increased risk of infections like bacterial infections, viral infections, fungal infections, neutropenia, pancytopenia and febrile neutropenia and/or bacteremia.
- the subjecthas not received lymphodepletion within 1 , 2, 3, 4, 5, 6, 7, 8, 9, 10, 11 , 12, 13 or 14 days before the administration of the modified immune cells, preferably the modified T cells.
- the composition of the inventionis able to stimulate and/or activate the administered modified immune cells, preferably the modified T cells, and/or the endogenous immune cells herein the subject has not received a lymphodepletion before the administration of the modified immune cells.
- the compositionis able to increase levels of circulating modified immune cells in the subject; induce memory formation of modified immune cells in the subject, preferably formation of central and effector memory T cells, activate modified immune cells in the subject; boosting metabolism and polyfunctionality of the modified immune cells in the subject, recruitment of cross-presenting cells, preferably DCs, to the tumor and elicits robust and potent antigen spreading, re-activate modified immune cells in the subject, maintaining self-renewal capacity of modified immune cells in the subject, induce higher influx into the solid tumor of modified immune cells in the subject, induce expansion of modified immune cells in the subject, activate endogenous CD4 helper cells in the subject, skew differentiation of endogenous tumor-infiltrating CD4+ T cells to a Th1 phenotype, increase levels of circulating endogenous immune cells in the subject, induce memory formation of endogenous immune cells in the subject, preferably formation of central and effector memory cells, activate endogenous immune cells in the subject, boosting metabolism and polyfunctionality of the endogenous immune cells in
- the subjecthas a reduced risk of fewer, weakness, chills, loss of appetite, immune-related toxicity and cytokine-related toxicity, cytokine release syndrome, organ dysfunction, arrhythmias, renal failure, neurotoxicity, anemia, thrombocytopenia, graft failure, increased risk of infections like bacterial infections, viral infections, fungal infections, neutropenia, pancytopenia and febrile neutropenia and bacteremia [compared to a subject that received lymphodepletionj.
- the subjecthas received a lymphodepletion within 1 , 2, 3, 4, 5, 6, 7, 8, 9, 10, 11 , 12, 13 or 14 days before the administration of the modified immune cells.
- composition of the inventionis able to stimulate/activate the administered modified immune cells wherein the subject has received lymphodepletion before the administration of the modified immune cells.
- Interleukin and/or cytokine treatment after transfer of modified immune cellsis a common therapeutic approach used to enhance the effectiveness of ACT therapy, preferably modified T cell or tumor-infiltrating lymphocyte therapy.
- Interleukinsare a group of cytokines that play a crucial role in regulating the immune system's response.
- I L2 and IL7are commonly used to support, enhance and amplify the activity of transferred modified immune cells.
- IL2plays a crucial role in expansion and activation of T cells.
- IL2is often administered as a treatment to support the survival, proliferation, and activity of transferred modified immune cells within the subject’s body.
- IL2can be associated with significant side effects like fever, chills, fatigue, muscle aches, severe toxicity, edema, pulmonary edema, gastrointestinal and neurological symptoms, skin reactions, kidney and liver dysfunction, anemia, thrombocytopenia and can promote Treg proliferation, also in modified immune cells that has transferred to the subject.
- IL7helps T cells survive and can enhance their activity against cancer cells.
- the methodcomprises a step of treating the subject with additional cytokine or interleukin, preferably at least one dose of IL2 and/or IL7, performed about 2 months after the administration of the modified immune cells.
- the methoddoes not comprise a step of treating the subject with additional cytokine or interleukin performed about 2 months after the administration of the modified immune cells.
- the methoddoes not comprise a step of treating the subject with additional interleukins, preferably IL2 or IL7, performed about 2 months after the administration of the modified immune cells.
- additional interleukinspreferably IL2 or IL7
- the subjectis not treated with additional cytokine or interleukin, preferably IL2 or IL7, performed about 2 months after the administration of the modified immune cells.
- the composition of the inventionstimulate/activate transferred modified immune cells without promoting T reg proliferation of the modified cells or cytokine or interleukin related side effects.
- the subjectnot treated with additional cytokine or interleukin, preferably IL2 or IL7, performed about 2 months after the administration of the modified immune cells, wherein the subject develops no cytokine or interleukin related side effects.
- additional cytokine or interleukinpreferably IL2 or IL7
- the optimal therapeutic dose of modified immune cellscan vary depending on several factors, including the type and stage of cancer, the specific modified immune cell product used, and individual patient characteristics.
- the optimal therapeutic dose of modified immune cellsdepends on subjects weight, specific modified immune cell product, e.g. CAR T cell products, condition regime with impact on dosing and timing of the administration of modified immune cell, administration route, clinical trial protocols, response and tolerance of the subject (may influence the subsequent doses or adjustments in the treatment schedule).
- specific modified immune cell producte.g. CAR T cell products
- condition regime with impact on dosing and timing of the administration of modified immune celladministration route, clinical trial protocols, response and tolerance of the subject (may influence the subsequent doses or adjustments in the treatment schedule).
- the subjectreceived an optimal therapeutic amount/dose of modified immune cells, preferably modified T cells.
- administration of the composition to a subjectleads to an expansion of optimal therapeutic amounts of transferred modified immune cells in the subject.
- modified immune cellsIt is generally thought that the number of transferred modified immune cells is correlated with therapeutic responses.
- the number of modified immune cells which can be administered to a subject for ACTis limited and the generation of a large amount of modified immune cells for ACT still remains a challenge.
- An increase in modified immune cell persistencecould be achieved when patients received a lymphodepleting preparative regimen before infusion of modified T cells, preferably TILs or receptor-engineered T cells like CAT T cells or TCRtg T cells.
- modified T cellspreferably TILs or receptor-engineered T cells like CAT T cells or TCRtg T cells.
- the transfer of a large amount of modified T cells into a lymphodepleted subjectalso poses the risk of severe adverse events.
- the subjecthas received a limited amount of modified immune cells, preferably modified T cells.
- the subjectreceived a subtherapeutic dose or low dose of modified immune cells, preferably modified T cells.
- the number of immune effector cells administered (including in vivo generation in a subject) for CAR T cell therapy of human beingsis about 109 cells per dose (equivalent to 1.33x10 7 per kg) or higher (equivalent to 1.3x10 7 per kg). These values can vary due to the Summary of Product Characteristics (SmPC) of the drug.
- some therapeutic approachescomprise repetitive administration of CAR T cells in a short time period (e.g. less than 4 weeks) to improve safety by dose escalation and/or to maintain the number of effective T cells in the patient. This leads to even higher “accumulated doses” within such time periods.
- a “subtherapeutic dose” of immune effector cells genetically modified to express an antigen receptoris an amount of such cells per initial dose and/or accumulated dose over a time period of at least 2 days, at least 3 days, at least 4 days, at least 5 days, at least 6 days, at least 7 days, at least 14 days, at least 21 days, at least 28 days or even longer of 10 8 or less, 10 7 or less, 10 6 or less, 10 5 or less, 10 4 or less, 10 3 or less or even lower.
- a “subtherapeutic amount” of immune effector cells genetically modified to express an antigen receptorrelates to a single dose of such cells in an amount of 10 8 or less, 10 7 or less, 10 6 or less, 10 5 or less, 10 4 or less, 10 3 or less or even lower.
- single dosemeans that one dose of a therapeutic substance is administered for a prolonged time.
- prolonged timecomprises a period of at least 14 days, at least 21 days, at least 28 days, at least 3 months, at least 6 months or even longer.
- administration of the composition of the invention to a subjectleads to an expansion of transferred modified immune cells in the subject, in particular in embodiments where the subject has received subtherapeutic amounts of modified immune cells.
- administration of the composition to a subjectleads to increased levels of circulating modified immune cells in the subject, induces memory formation of modified immune cells in the subject, preferably formation of central and effector memory cells, activates modified immune cells in the subject; boosting metabolism and polyfunctionality of the modified immune cells in the subject, recruitment of cross-presenting cells, preferably DCs, to the tumor and elicits robust and potent antigen spreading, re-activates modified immune cells in the subject; maintains the self-renewal capacity of modified immune cells in the subject, induces higher influx into the solid tumor of modified immune cells in the subject, and/or induces expansion of modified immune cells in the subject where the subject has received subtherapeutic amounts of modified immune cells.
- the first dose of the compositionis administered before the subject has received the modified immune cells, preferably the modified T cells, targeted to the antigen. In some embodiments, the first dose of the composition is administered 1 , 2, 3, 4, 5, 6 or 7 days before the subject has received the modified immune cells, preferably the modified T cells, targeted to the antigen.
- the first dose of the compositionis administered at the same day when the subject has received the modified immune cells, preferably the modified T cells, targeted to the antigen.
- the first dose of the compositionis administered after the subject has received the modified immune cells, preferably the modified T cells, targeted to the antigen.
- the first dose of the compositionis administered 1 , 2, 3, 4, 5, 6, 7, 8, 9, 10, 11 , 12, 13, 14, 15, 16, 17, 18, 19, 20, 21 , 22, 23, 24, 25, 26, 27, 28, 29, 30, 31 , 32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49, 50, 51 , 52, 53, 54, 55, 56, 57, 58, 59 or 60 days after the subject has received the modified immune cells, preferably the modified T cells, targeted to the antigen.
- the first dose of the compositionis administered 1 day to 60 days, 30 to 60 days, 30 to 40, 40 to 50 or 50 to 60 days after the subject has received the modified immune cells, preferably the modified T cells, targeted to the antigen.
- the first dose of the compositionis administered after the subject has received a sub-optimal dose of the modified immune cells, preferably the modified T cells, targeted to the antigen.
- the first dose of the compositionis administered 1 , 2, 3, 4, 5, 6 or 7 days after the subject has received a sub-optimal dose of the modified immune cells, preferably modified T cells, targeted to the antigen, preferably a tumor antigen.
- the first dose of the compositionis administered after the subject has received an optimal dose of the modified immune cells, preferably modified T cells, targeted to the antigen.
- the first dose of the compositionis administered 30 to 60 days, 30 to 40, 40 to 50 or 50 to 60 days after the subject has received an optimal dose of the modified immune cells, preferably modified T cells, targeted to the antigen, preferably a tumor antigen.
- modified immune cellspreferably modified T cells
- At least one further dose of the compositionis administered to the subject.
- the at least one further doseis administered via intramuscular or intratumoral injection, preferably via intramuscular injection.
- the at least one further dose of the compositionis administered 1 , 2, 3, 4, 5, 6, 7, 8, 9, 10, 11 , 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49, 50, 51 , 52, 53, 54, 55, 56, 57, 58, 59 or 60 days after the subject has received the first dose of the composition.
- the at least one further dose of the compositionis administered 5 days to 10 days, 11 days to 15 days, 16 days to 20 days, 21 days to 25 days, 26 days to 30 days, 31 days to 35 days, 36 days to 40 days, 41 days to 45 days, 46 days to 50 days, 51 days to 55 days, 56 days to 60 days after the subject has received the first dose of the composition.
- the at least one further dose of the compositionis administered 1 day to 60 days, 30 to 60 days, 30 to 40, 40 to 50 or 50 to 60 days after the administration of the first dose.
- the at least one further dose of the compositionis administered 6 or 7 days after the administration of the first dose of the composition when the subject has received a subtherapeutic amount of modified immune cells, preferably modified T cells. In some embodiments, the at least one further dose of the composition is administered 6 or 7 days after the administration of the first dose of the composition when the subject has received a subtherapeutic amount of modified immune cells, preferably modified T cells. In some embodiments, the at least further dose of the composition is administered 7 days, 14 days, 21 days, 28 days, 35 days, 42 days and 56 days after the subject has received modified immune cells, preferably the modified T cells, preferably a subtherapeutic amount of modified immune cells, in particular modified T cells.
- At least 2, 3, 4, 5, 6, 7, 8, 9, 10 doses of the compositionare administered to the subject. In some embodiments, at least 3, 4, 5, or more doses of the composition are administered to the subject. In some embodiments, at least 2, 3, 4, 5, or more doses of the composition are administered in a one dose per week schedule.
- the compositionis intramuscularly administered 1 day, 7 days, 14 days, 21 days, 28 days, 35 days, 42 days and 56 days after the subject has received modified immune cells, preferably a subtherapeutic amount of modified immune cells, in particular modified T cells.
- one dose of the compositioncomprises 10pg, 11pg, 12pg, 13pg, 14pg, 15pg, 16pg, 17pg, 18pg, 19pg, 20pg, 25pg, 30pg, 35pg, 40pg, 45pg, 50pg, 55pg, 60pg, 65pg, 70pg, 75pg, 80pg, 85pg, 90pg, 95pg, 100pg, 110pg, 120pg, 130pg, 140pg, 150pg, 160 g, 170pg 180pg, 190pg, 200pg, 250pg, 300pg, 350pg, 400pg, 450pg, 500pg, wherein the dose refers to the amount of RNA in the composition.
- one dose of the compositioncomprises between 1 pg and 500pg, 20pg and 500pg, 50pg and 500pg, 50pg and 250pg, 100pg and 250pg, wherein the dose refers to the amount of RNA in the composition.
- the administration of the compositionpreferably the ex vivo or in vivo administration of the composition, induces activation and stimulation of immune cells.
- Activation of immune cellsrefers to the state of an immune cell that has been sufficiently stimulated to induce detectable cellular proliferation. Activation can also be associated with initiation of signaling pathways, induced cytokine production, and detectable effector functions.
- the administration (e.g. intramuscularly) of the composition to the subjectinduces activation and stimulation of immune cells, preferably transferred modified immune cells and endogenous immune cells.
- the activation and/or stimulation of immune cellsis defined by increased levels of circulating modified immune cells, preferably modified T cells, in the subject; induced memory formation of modified immune cells, preferably modified T cells, in the subject, preferably formation of central and effector memory T cells; activated modified immune cells, preferably modified T cells, in the subject; boosted metabolism and polyfunctionality of the modified immune cells, preferably modified T cells, in the subject; recruitment of cross-presenting cells, preferably DCs, to the tumor and elicits robust and potent antigen spreading, re-activated modified immune cells, preferably modified T cells, in the subject; maintained self-renewal capacity of modified immune cells, preferably modified T cells, in the subject; induced higher influx into the solid tumor of modified immune cells, preferably modified T cells, in the subject; induced expansion of modified immune cells, preferably modified T cells, in the subject; activated endogenous CD4 helper cells in the subject; skewed differentiation of endogenous tumor-infiltrating CD4+ T cells to
- the activation and/or stimulation of immune cellscan be measured by measuring initiation of signaling pathways, induced cytokine production and/or secretion, levels of antigen specific immune cells and detectable effector functions.
- the activation and/or stimulation of immune cellsmay be evaluated using any of a variety of standard techniques, for example, within a chromium release assay, proliferation assay, synthesis of lymphokines, qRT-PCR, immunohistochemical (IHC) staining, STAR RNAseq aligner (Dobin A.
- modified immune cellspreferably modified T cells
- the percentage increase in activation and/or stimulationis at least 1%, 1.5%, 2%, 2.5%, 3%, 3.5%, 4%, 4.5%, 5%, 5.5%, 6%, 6.5%, 7%, 7.5%, 8%, 8.5%, 9%, 9.5%, 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80%, 90%, 100%, 200%, 300%, 400%, 500% or even more.
- the administration (e.g. intramuscularly) of the composition to the subject that has received modified immune cells, preferably modified T cellsresults in an increased level of circulating modified immune cells in the subject, compared to the subject that has received modified immune cells without administration of the composition, wherein the percentage increase in activation and/or stimulation is at least 0.5%, 1 %, 1.5%, 2%, 2.5%, 3%, 3.5%, 4%, 4.5%, 5%, 5.5%, 6%, 6.5%, 7%, 7.5%, 8%, 8.5%, 9%, 9.5%, 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80%, 90%, 100%, 200%, 300%, 400%, 500% or even more.
- the administration (e.g. intramuscularly) of the composition to the subject that has received modified immune cells, preferably modified T cellsresults in an increased level of central and effector memory T cells, compared to the subject that has received modified immune cells without administration of the composition, wherein the percentage increase in activation and/or stimulation is at least 0.5%, 1 %, 1.5%, 2%, 2.5%, 3%, 3.5%, 4%, 4.5%, 5%, 5.5%, 6%, 6.5%, 7%, 7.5%, 8%, 8.5%, 9%, 9.5%, 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80%, 90%, 100%, 200%, 300%, 400%, 500% or even more.
- the administration (e.g. intramuscularly) of the composition to the subject that has received modified immune cells, preferably modified T cellsresults in an increased level of re-activated modified immune cells in the subject, compared to the subject that has received modified immune cells without administration of the composition, wherein the percentage increase in activation and/or stimulation is at least 0.5%, 1 %, 1.5%, 2%, 2.5%, 3%, 3.5%, 4%, 4.5%, 5%, 5.5%, 6%, 6.5%, 7%, 7.5%, 8%, 8.5%, 9%, 9.5%, 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80%, 90%, 100%, 200%, 300%, 400%, 500% or even more.
- the administration (e.g. intramuscularly) of the composition to the subject that has received modified immune cellsresults, preferably modified T cells, results in an increased level of modified immune cells with self-renewal capacity in the subject compared to the subject that has received modified immune cells without administration of the composition, wherein the percentage increase in activation and/or stimulation is at least 0.5%, 1%, 1.5%, 2%, 2.5%, 3%, 3.5%, 4%, 4.5%, 5%, 5.5%, 6%, 6.5%, 7%, 7.5%, 8%, 8.5%, 9%, 9.5%, 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80%, 90%, 100%, 200%, 300%, 400%, 500% or even more.
- the administration (e.g. intramuscularly) of the composition to the subject that has received modified immune cells, preferably modified T cellsresults in an increased level of activated endogenous CD4 helper cells in the subject, compared to the subject that has received modified immune cells without administration of the composition, wherein the percentage increase in activation and/or stimulation is at least 0.5%, 1%, 1.5%, 2%, 2.5%, 3%, 3.5%, 4%, 4.5%, 5%, 5.5%, 6%, 6.5%, 7%, 7.5%, 8%, 8.5%, 9%, 9.5%, 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80%, 90%, 100%, 200%, 300%, 400%, 500% or even more.
- modified immune cellspreferably modified T cells
- the intramuscular administration of the composition to the subject that has received modified immune cellsresults in an increased activation and/or stimulation of the immune cells in the subject, preferably modified immune cells and/or endogenous immune cells, compared to intravenous administration of the composition to the subject that has received modified immune cells, wherein the percentage increase in activation and/or stimulation is at least 0.5%, 1 %, 1.5%, 2%, 2.5%, 3%, 3.5%, 4%, 4.5%, 5%, 5.5%, 6%, 6.5%, 7%, 7.5%, 8%, 8.5%, 9%, 9.5%, 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80%, 90%, 100%, 200%, 300%, 400%, 500% or even more.
- the activation and/or stimulation of the modified and endogenous immune cells as described aboveis determined by measuring the induction of cytokines and/or other immune response related molecules expressed intracellular or on the cell surface.
- the cytokinesare associated with proliferation of immune cells, memory formation of immune cells, differentiation of immune cells, effector functions of immune cells, as non-limiting examples IL1 , ILS, IL4, IL6, IL7, IL8, IL9, IL10, IL12, IL15, IL17, IL21 , IL23, GM-CSF, TGF-beta, Rantes, MIP-1 alpha, MIP-1 beta, McP1 , TNFalpha, IFNgamma, IFNalpha and/or IFNbeta.
- activation and/or stimulation of the modified and endogenous immune cellsis characterized by an increased level of at least one cytokine wherein the increased level of at least one cytokine is an increase of at least 0,5%, 1%, 1.5%, 2%, 2.5%, 3%, 3.5%, 4%, 4.5%, 5%, 5.5%, 6%, 6.5%, 7%, 7.5%, 8%, 8.5%, 9%, 9.5%, 10%, 15%, 20%, 25%, 30%, 35%, 40%, 45%, 50%, 55%, 60%, 65%, 70%, 75%, 80%, 85%, 90%, or 95%.
- the reduced level of at least one cytokineis a reduction of at least 30%.
- the other immune response related molecules expressed intracellular or on the cell surfaceare associated with proliferation of immune cells, memory formation of immune cells, differentiation of immune cells, effector functions of immune cells, as non-limiting examples antibodies, perforins, granzymes, histamines, chemokines, interferons, nitric oxides, lysozyme, leukotrienes, prostaglandins, defensins, cluster of differentiation (CD) molecules and or major histocompatibility complex (MHC) molecules.
- CDcluster of differentiation
- MHCmajor histocompatibility complex
- activation and/or stimulation of the modified and endogenous immune cellsis characterized by an increased level of at least one immune response related molecule wherein the increased level of at least one immune response related molecule is an increase of at least 0.5%, 1%, 1.5%, 2%, 2.5%, 3%, 3.5%, 4%, 4.5%, 5%, 5.5%, 6%, 6.5%, 7%, 7.5%, 8%, 8.5%, 9%, 9.5%, 10%, 15%, 20%, 25%, 30%, 35%, 40%, 45%, 50%, 55%, 60%, 65%, 70%, 75%, 80%, 85%, 90%, or 95%.
- the reduced level of at least one cytokineis a reduction of at least 30%.
- the administration of the compositionincreases the levels of circulating modified immune cells, preferably modified T cells, in the subject; induces memory formation of modified immune cells, preferably modified T cells, in the subject, preferably formation of central and effector memory cells; activates modified immune cells, preferably modified T cells, in the subject; boosting metabolism and polyfunctionality of the modified immune cells, preferably modified T cells, in the subject; recruitment of cross-presenting cells, preferably DCs, to the tumor and elicits robust and potent antigen spreading; re-activates modified immune cells, preferably modified T cells, in the subject; maintains the self-renewal capacity of modified immune cells, preferably modified T cells, in the subject; induces higher influx into the solid tumor of modified immune cells, preferably modified T cells, in the subject; and/or induces expansion of modified immune cells, preferably modified T cells, in the subject.
- the administration of the compositionpreferably the intramuscular administration of the composition to the subject.
- Memory T cellsplay a crucial role in the adaptive immune system’s ability to provide long-lasting immunity. These cells can persist in the body for years, or even decades, ensuring that the immune system can mount a rapid and robust response if the antigen, e.g. cancer or pathogen, recurs. This rapid response is crucial in e.g. controlling the growth and spread of e.g. cancer or virus infected cells. Memory T cells persist in the body for extended periods, sometimes even for a lifetime, ensuring that the immune system can mount a rapid and robust response if the cancer recurs. There are two distinct subsets of memory T cells: central memory T cells (Tem) and effector memory T cells (T em ).
- Temcentral memory T cells
- T emeffector memory T cells
- Tonsare primarily found in lymphoid tissues, such as lymph nodes and the spleen and have the ability to recirculate through secondary lymphoid organs.
- Central memory T cellsare known fortheir capacity to proliferate and differentiate into various effector T cell subsets when reactivated and serve as a reservoir of memory T cells and are important for the long-term maintenance of immunological memory.
- Effector memory T cellsare primarily found in peripheral tissues, such as skin, mucosal surfaces, and non-lymphoid organs and are cells that can respond rapidly to antigen reexposure. Effector memory T cells have immediate cytotoxic and cytokine-secreting functions. Tem cells are crucial for providing quick and localized immune responses at the site of infection or tissue damage and are particularly important in the defense against pathogens and in immune surveillance against cancer at peripheral sites.
- the administration of the compositionpreferably the intramuscular administration of the composition, to the subject induces formation of effector memory T cells, preferably memory precursor effector T cells and short-lived effector T cells, preferably memory precursor effector T cells.
- the administration of the compositionpreferably the intramuscular administration of the composition to the subject induces formation of effector memory T cells of the transferred modified T cell population, preferably memory precursor effector T cells and short-lived effector T cells, preferably memory precursor effector T cells.
- the administration of the compositionpreferably the intramuscular administration of the composition to the subject, induces formation of effector memory T cells of the endogenous T cell population, preferably memory precursor effector T cells and short-lived effector T cells, preferably memory precursor effector T cells.
- MPECsMemory Precursor Effector T Cells
- MPECsare a subset of T cells that differentiate into long-lasting memory T cells and contributing to the establishment of long-term immunological memory. MPECs have a high proliferative potential and are more likely to survive after the resolution of the immune response.
- Short-Lived Effector T CellsSLECs
- SLECsshort-Lived Effector T Cells
- SLECshave a limited proliferative potential and are more prone to undergo apoptosis (cell death) after the antigen is cleared and do not typically contribute significantly to long-term memory.
- the subjectcomprises higher transferred modified memory T cell population levels, preferably modified T cells levels, after intramuscular administration of the composition of the invention compared to intravenous administration.
- the administration of the composition to the subjectinduces activation and stimulation of immune cells, preferably endogenous immune cells of the subject.
- administration of the compositionpreferably intramuscular administration of the composition to the subject leads to the following immunologic effects: the administration activates endogenous CD4 helper cells in the subject, skews the differentiation of endogenous tumor-infiltrating CD4+ T cells to a Th1 phenotype, increases the levels of circulating endogenous immune cells in the subject; induces memory formation of endogenous immune cells in the subject, preferably formation of central and effector memory cells; activates endogenous immune cells in the subject; boosting metabolism and polyfunctionality of the endogenous immune cells in the subject, recruitment of cross-presenting cells, preferably DCs, to the tumor and elicits robust and potent antigen spreading (antigen spreading supports modified T cell therapy to treat antigenically heterogeneous tumors), re-activates endogenous immune cells in the subject; maintains the self-renewal capacity of endogenous immune cells in the subject; induces higher influx into the solid tumor of endogenous immune cells in the subject; and/or induces expansion of endogen
- administration of the compositionpreferably intramuscular administration of the composition to the subject leads to the following immunologic effects: the administration increases the levels of circulating modified immune cells, preferably modified T cells, in the subject; induces memory formation of modified immune cells, preferably modified T cells, in the subject, preferably formation of central and effector memory cells; activates modified immune cells, preferably modified T cells, in the subject; re-activates modified immune cells, preferably modified T cells, in the subject; maintains the self-renewal capacity of modified immune cells, preferably modified T cells, in the subject; induces higher influx into the solid tumor of modified immune cells, preferably modified T cells, in the subject; and/or induces expansion of modified immune cells, preferably modified T cells, in the subject to a larger extend compared to intravenous administration of the composition to the subject.
- the administrationincreases the levels of circulating modified immune cells, preferably modified T cells, in the subject; induces memory formation of modified immune cells, preferably modified T cells, in the subject, preferably formation of central and effector memory cells; activates
- the immunologic effects defined aboveare long-term immunologic effects, preferably immunologic effects that are detectable over 10, 15, 20, 25, 30, 35, 40, 50, 60, 70, 80, 90, 100, 110, 120, 120, 130, 140, 150, 160, 180, 200, 210, 220, 230, 240, 250, 260, 280, 300, 320, 340, 360 days or even longer.
- the immunological effects defined aboveare long-term immunological effects, preferably induction of memory formation of modified immune cells, preferably modified T cells, in the subject, e.g. formation of central and effector memory cells or reactivation of modified immune cells in the subject, that are detectable over 1 year, 1.5 years, 2 years, 2.5 years, 3 years, 3.5 years, 4 years, 4.5 years, 5 years, 5.5 years, 6 years, 6.5 years, 7 years, 7.5 years, 8 years, 8.5 years, 9 years, 9.5 years, 10 years or even longer.
- the subjecthas received a lymphodepletion therapy to minimize the numbers of endogenous (unmodified) immune cells.
- the composition of the inventionis able to stimulate the minimized population of the endogenous immune cells to induce immunologic effects as defined above.
- the administration of the compositionchanges the cytokine milieu in the solid tumor of the subject and/or eliminates minimal residual disease, preferably tumors in the subject.
- minimal residual diseasealso known as measurable residual disease or minimal residual cancer, refers to the small number of cancer cells that may remain in the body after treatment for cancer,
- the subjectreceives or has received an additional treatment selected from adjuvants, radiation therapy, surgery, hyperthermia therapy, chemotherapy, anti-cancer antibodies and/or immunotherapy.
- the additional treatment as defined hereinmay be administered to the subject after the subject has received the modified immune cells, preferably modified T cells, or before the subject has received the modified immune cells, preferably modified T cells.
- the additional treatment as defined hereinmay be administered to the subject after the subject has received the composition of the invention, or before the subject as received the composition of the invention, or concomitant or at the same time as the composition of the invention.
- adjuvantsinclude, without limitation, LPS, GP96, CpG oligodeoxynucleotides, growth factors, and cytokines, such as monokines, lymphokines, interleukins, chemokines.
- the cytokinesmay be IL1 , ILS, IL3, IL4, IL5, IL6, IL7, IL8, IL9, IL10, IL12, IFNa, IFNy, GM-CSF, LT-a.
- Further known adjuvantsare aluminium hydroxide, Freund's adjuvant or oil such as Montanide® ISA51.
- Other suitable adjuvants for use in the present disclosureinclude lipopeptides, such as Pam3Cys.
- additional treatmentsmay be administered to a patient in combination with the treatments described herein.
- additional treatmentsinclude classical cancer therapy, e.g., radiation therapy, surgery, hyperthermia therapy and/or chemotherapy.
- Chemotherapeutic agentsinclude alkylating agents, antimetabolites, anti-microtubule agents, topoisomerase inhibitors, and cytotoxic antibiotics.
- Alkylating agentshave the ability to alkylate many molecules, including proteins, RNA and DNA.
- the subtypes of alkylating agentsare the nitrogen mustards, nitrosoureas, tetrazines, aziridines, cisplatins and derivatives, and non- classical alkylating agents.
- Nitrogen mustardsinclude mechlorethamine, cyclophosphamide, melphalan, chlorambucil, ifosfamide and busulfan.
- Nitrosoureasinclude N-Nitroso-N-methylurea (MNU), carmustine (BCNU), Io ustine (CCNU) and semustine (MeCCNU), fotemustine and streptozotocin.
- Tetrazinesinclude dacarbazine, mitozolomide and temozolomide.
- Aziridinesinclude thiotepa, mytomycin and diaziquone (AZQ).
- Cisplatin and derivativesinclude cisplatin, cariaoplatin and oxaliplatin. They impair cell function by forming covalent bonds with the amino, carboxyl, sulfhydryl, and phosphate groups in biologically important molecules.
- Non-classical alkylating agentsinclude procarbazine and hexamethylmelamine. In one particularly preferred embodiment, the alkylating agent is cyclophosphamide.
- Anti-metabolitesare a group of molecules that impede DNA and RNA synthesis. Many of them have a similar structure to the building blocks of DNA and RNA. Anti-metabolites resemble either nucleobases or nucleosides but have altered chemical groups. These drugs exert their effect by either blocking the enzymes required for DNA synthesis or becoming incorporated into DNA or RNA. Subtypes of the antimetabolites are the anti-folates, fluoropyrimidines, deoxynucleoside analogues and thiopurines. The antifolates include methotrexate and pemetrexed. The fluoropyrimidines include fluorouracil and capecitabine.
- the deoxynucleoside analoguesinclude cytarabine, gemcitabine, decitabine, azacitidine, fludarabine, nelarabine, cladribine, clofarabine, and pentostatin.
- the thiopurinesinclude thioguanine and mercaptopurine.
- Anti-microtubule agentsblock cell division by preventing microtubule function.
- the vinca alkaloidsprevent the formation of the microtubules, whereas the taxanes prevent the microtubule disassembly.
- Vinca alkaloidsinclude vinorelbine, vindesine, and vinflunine.
- Taxanesinclude docetaxel (Taxotere) and paditaxel (Taxol).
- Topoisomerase inhibitorsare drugs that affect the activity of two enzymes: topoisomerase I and topoisomerase II and include irinotecan, topotecan, camptothecin, etoposide, doxorubicin, mitoxantrone, teniposide, novobiocin, merbarone, and aclarubicin.
- the cytotoxic antibioticsare a varied group of drugs that have various mechanisms of action.
- the common theme that they share in their chemotherapy indicationis that they interrupt cell division.
- the most important subgroupis the anthracyclines (e.g., doxorubicin, daunorubicin, epirubicin, idarubicin pirarubicin, and aclarubicin) and the bleomycins; other prominent examples include mitomycin C, mitoxantrone, and actinomycin.
- immune checkpoint inhibitorsare used in combination with other therapeutic agents described herein.
- “immune checkpoint inhibitor”refers to a molecule that totally or partially reduces, inhibits, interferes with or modulates one or more checkpoint proteins.
- the immune checkpoint inhibitorprevents inhibitory signals associated with the immune checkpoint.
- the immune checkpoint inhibitoris an antibody, or fragment thereof that disrupts inhibitory signaling associated with the immune checkpoint.
- the immune checkpoint inhibitoris a small molecule that disrupts inhibitory signaling.
- the immune checkpoint inhibitoris an antibody, fragment thereof, or antibody mimic, that prevents the interaction between checkpoint blocker proteins, e.g., an antibody, or fragment thereof, that prevents the interaction between PD-1 and PD-L1.
- the immune checkpoint inhibitoris an antibody, or fragment thereof, that prevents the interaction between CTLA4 and CD80 or CD86.
- the immune checkpoint inhibitoris an antibody, or fragment thereof, that prevents the interaction between LAG3 and its ligands, or TIM-3 and its ligands.
- the checkpoint inhibitormay also be in the form of the soluble form of the molecules (or variants thereof) themselves, e.g., a soluble PD-L1 or PD-L1 fusion.
- the immune checkpoint inhibitor suitable for use in the methods disclosed hereinis an antagonist of inhibitory signals, e.g., an antibody which targets, for example, PD-1 , PD-L1, CTLA4, LAG3, B7-H3, B7-H4, orT!M3.
- inhibitory signalse.g., an antibody which targets, for example, PD-1 , PD-L1, CTLA4, LAG3, B7-H3, B7-H4, orT!M3.
- Non-limiting examples of anti-cancer antibodies and potential antibody targets (in brackets)which may be used in combination with the present disclosure include: Abagovomab (CA-125), Abciximab (CD41), Adecatumumab (EpCAM), Afutuzumab (CD20), Alacizumab pegol (VEGFR2), Altumomab pentetate (CEA), Amatuximab (MORAb- 009), Anatumomab mafenatox (TAG-72), Apolizumab (HLA-DR), Arcitumomab (CEA), Atezolizumab (PD-L1 ), Bavituximab (phosphatidylserine), Bectumomab (CD22), Belimumab (BAFF), Bevacizumab (VEGF-A), Bivatuzumab mertansine (CD44 v6), Blinatumomab (CD 19),
- the at least one RNA of the compositionis complexed or associated with at least one further compound to obtain a formulated composition.
- a formulation in that contextmay have the function of a transfection agent.
- a formulationmay also have the function of protecting the RNA from degradation, e.g. to allow storage, shipment, etc.
- the at least one RNA of the compositionis formulated with at least one compound, e.g. peptides, proteins, lipids, polysaccharides, and/or polymers.
- the at least one RNA of the compositionis formulated with at least one cationic (cationic or preferably ionizable) or polycationic compound (cationic or preferably ionizable).
- the at least one RNA of the compositionis complexed or associated with or at least partially complexed or partially associated with one or more cationic (cationic or preferably ionizable) or polycationic compound.
- the at least one cationic or polycationic compoundis selected from a cationic or polycationic polymer, a cationic or polycationic polysaccharide, a cationic or polycationic lipid, a cationic or polycationic protein, a cationic or polycationic peptide, or any combinations thereof.
- the at least one cationic or polycationic compoundis a cationic or polycationic lipid.
- Further cationic or polycationic compounds being suitable in the context of the inventionmay be selected from p.88, line 24 to p.89, line 10 in WO2021156267, the respective disclosure herewith incorporated by reference.
- the at least one RNA of the compositionis formulated in lipid-based carriers.
- lipid-based carrierencompasses lipid-based delivery systems for nucleic acid, preferably RNA, that comprise a lipid component.
- a lipid-based carriermay additionally comprise other components suitable for formulating a nucleic acid including a cationic or polycationic polymer, polysaccharide, protein, and/or peptide.
- the lipid-based carriers of the compositionare selected from liposomes, LNPs, lipoplexes, solid lipid nanoparticles, lipo-polyplexes, and/or nanoliposomes.
- the lipid-based carriers of the compositionare LNPs.
- LNPsare microscopic lipid particles having a solid or partially solid core.
- the RNAmay be encapsulated or incorporated in the lipid portion of the LNP enveloped by some or the entire lipid portion of the LNP.
- An LNPmay comprise any lipid capable of forming a particle to which the RNA may be attached, or in which the RNA may be encapsulated.
- the LNPs of the compositionencapsulate the at least one RNA of the invention.
- encapsulationrefers to the essentially stable combination of RNA with one or more lipids into larger complexes or assemblies such as lipid-based carriers, preferably LNPs, preferably without covalent binding of the RNA.
- the encapsulated RNAmay be completely or partially located in the interior of the lipid-based carrier (e.g. the lipid portion and/or an interior space) and/or within the lipid layer/membrane of the lipid-based carriers.
- the lipid-based carrierscomprise at least one or more lipids selected from at least one aggregation-reducing lipid, at least one cationic lipid or ionizable lipid, at least one neutral lipid or phospholipid, or at least one steroid or steroid analogue, or any combinations thereof.
- the lipid-based carrierscomprise an (i) aggregation-reducing lipid, (ii) a cationic lipid or ionizable lipid, (iii) a neutral lipid or phospholipid, (iv) and a steroid or steroid analogue.
- the lipid-based carriers, preferably the LNPscomprise an (i) aggregation-reducing lipid, (ii) a cationic lipid or ionizable lipid, (iii) two different neutral lipids or phospholipids, (iv) and a steroid or steroid analogue.
- the lipid-based carrierspreferably the LNPs, comprise at least one aggregation reducing lipid or aggregation reducing moiety.
- aggregation reducing moietyrefers to a molecule comprising a moiety suitable of reducing or preventing aggregation of the lipid-based carriers, preferably the LNPs.
- aggregation reducing lipidrefers to a molecule comprising both a lipid portion and a moiety suitable of reducing or preventing aggregation of the lipid-based carriers.
- the lipid-based carrierssuch as LNPs may undergo charge-induced aggregation, a condition which can be undesirable for the stability of the lipid-based carriers. Therefore, it can be desirable to include a compound or moiety which can reduce aggregation, for example by sterically stabilizing the lipid- based carriers.
- Such a steric stabilizationmay occur when a compound having a sterically bulky but uncharged moiety that shields or screens the charged portions of a lipid-based carriers from close approach to other lipid-based carriers in the composition.
- stabilization of the lipid-based carriersis achieved by including lipids which may comprise a lipid bearing a sterically bulky group which, after formation of the lipid-based carrier, is preferably located on the exterior of the lipid-based carrier.
- Suitable aggregation reducing groupsinclude hydrophilic groups, e.g. monosialoganglioside GM1 , polyamide oligomers (PAO), or certain polymers, such as poly(oxyalkylenes), e.g., polyethylene glycol) or polypropylene glycol).
- the aggregation reducing lipidis a polymer conjugated lipid.
- polymer conjugated lipidrefers to a molecule comprising both a lipid portion and a polymer portion, wherein the polymer is suitable of reducing or preventing aggregation of lipid-based carriers comprising the RNA.
- a polymerhas to be understood as a substance or material consisting of very large molecules, or macromolecules, composed of many repeating subunits.
- a suitable polymer in the context of the inventionmay be a hydrophilic polymer.
- An example of a polymer conjugated lipidis a PEGylated or PEG-conjugated lipid. Lipids comprising a polymer as aggregation reducing group are herein referred to as “polymer conjugated lipid”.
- the aggregation reducing lipidis a polymer conjugated lipid selected from a PEG- conjugated lipid or a PEG-free lipid.
- the polymer conjugated lipidis a PEG-conjugated lipid.
- the average molecular weight of the PEG moiety in the PEG-conjugated lipidmay range from 500 to 8,000 Daltons (e.g., from 1 ,000 to 4,000 Daltons). In one preferred embodiment, the average molecular weight of the PEG moiety is about 2,000 Daltons.
- the PEG-conjugated lipidis selected or derived from 1 ,2-dimyristoyl-rac-glycero-3-methoxypolyethylene glycol-2000 (PEG2000 DMG or DMG-PEG 2000), C10-PEG2K, or Cer8-PEG2K.
- the polymer conjugated lipidis selected or derived from formula (IV) of WO2018078053, preferably from formula (I a) of WO2018078053.
- a preferred polymer-conjugated lipidis selected from ALC-0159 (2[(polyethylene glycol)-2000]-N,N-ditetradecylacetamide).
- the aggregation reducing lipidis selected from a PEG-free lipid, e.g. a PEG-free polymer conjugated lipid.
- the aggregation reducing lipidis a PEG-free lipid that comprises a polymer different from PEG.
- a PEG-free polymer conjugated lipidmay be selected or derived from a “POZ-lipid”.
- POZ lipidsor respectively preferred polymer conjugated lipids are described in WO2023031394, the full disclosure herewith incorporated by reference.
- disclosure relating to polymer conjugated lipids as defined in any one of claims 1 to 8 of WO2023031394are incorporated by reference.
- the polymer conjugated lipidis a PEG-free lipid selected from a POZ-lipid.
- the polymer conjugated lipidis a “POZ-lipid”, which preferably is defined as a compound according to formula (POZ): [H] - [linker] - [M], wherein
- [H]is a homopolymer moiety comprising at least one polyoxazoline (POZ) monomer unit wherein R is C1-9 alkyl or C2-9 alkenyl, preferably C1 , and n has a mean value ranging from 2 to 200, preferably from 20 to 100, more preferably from 24 to 26 or 45 to 50; [linker] is an optional linker group, and [M] is a lipid moiety.
- the aggregation-reducing lipidis selected or derived from PMOZ 1 , PMOZ 2, PMOZ 3, PMOZ 4, or PMOZ 5 of W02023031394.
- the at least one aggregation-reducing lipidis selected or derived from a POZ- lipid according to or derived from a lipid of formula PMOZ4 of W02023031394.
- the at least one aggregation-reducing lipidis selected or derived from ALC-0159, DMG- PEG 2000, C10-PEG2K, Cer8-PEG2K, or a POZ-lipid such as PMOZ4.
- the at least one aggregation-reducing lipidis ALC-0159.
- the at least one aggregation-reducing lipidis a POZ-lipid as defined herein, wherein the POZ-lipid is preferably selected from a PMOZ4 lipid.
- the at least one cationic or ionizable lipidmay be cationisable or ionizable, i.e. it becomes protonated as the pH is lowered below the pK of the ionizable group of the lipid, but is progressively more neutral at higher pH values. At pH values below the pK, the lipid is then able to associate with negatively charged nucleic acids.
- the cationic lipidcomprises a zwitterionic lipid that assumes a positive charge on pH decrease.
- the cationic or ionizable lipidpreferably carries a net positive charge at physiological pH.
- the cationic or ionizable lipidcomprises a quaternary nitrogen group or tertiary nitrogen group, most preferably a tertiary nitrogen group.
- the at least one cationic or ionizable lipidmay be selected from an amino lipid.
- the at least one cationic lipid or ionizable lipidis selected from an amino lipid, preferably wherein the amino lipid comprises a tertiary amine group.
- the at least one cationic or ionizable lipidis a lipid selected or derived from formula (II 1-1) ,
- the at least one cationic or ionizable lipidsis selected from the lipids disclosed in WO2018078053 (i.e. lipids derived from formula I, II, and III, or lipids as specified in claims 1 to 12), the disclosure of WO2018078053 hereby incorporated by reference.
- lipids disclosed in Table 7 of WO2018078053e.g. lipids derived from formula 1-1 to 141
- lipids disclosed in Table 8 of WO2018078053e.g. lipids derived from formula 11-1 to II-36
- formula 1-1 to formula 1-41 and formula 11-1 to formula II-36 of WO2018078053, and the specific disclosure relating thereto,are herewith incorporated by reference.
- the at least one cationic or ionizable lipidis selected or derived from structures 111-1 to III-36 of Table 9 of WO2018078053. Accordingly, formula 111-1 to HI-36 of WO2018078053, and the specific disclosure relating thereto, are herewith incorporated by reference.
- the at least one cationic or ionizable lipidis selected or derived from formula HI-3 of WO2018078053.
- a preferred lipid of said formula HI-3has the chemical term ((4-hydroxybutyl)azanediyl)bis(hexane-6,1-diyl)bis(2- hexyldecanoate), also referred to as ALC-0315, i.e. CAS Number 2036272-554.
- suitable cationic lipidsmay be selected or derived from cationic lipids according to PCT claims 1 to 14 of published patent application WO2021123332, or Table 1 of WO2021123332, the disclosure relating to claims 1 to 14 or Table 1 of WO2021123332 herewith incorporated by reference. Accordingly, suitable cationic lipids may be selected or derived from cationic lipids according to Compound 1 to Compound 27 (C1-C27) of Table 1 of WO2021123332.
- the at least one cationic or ionizable lipidis selected or derived from SS-33/4PE-15 (see C23 in Table 1 of WO2021123332). In other preferred embodiments, the at least one cationic or ionizable lipid is selected or derived from HEXA-C5DE-PipSS (see C2 in Table 1 of WO2021123332). In particularly preferred embodiments, the at least one cationic or ionizable lipid is selected or derived from compound C26 (VitE-C4DE-Pip- thioether) as disclosed in Table 1 of WO2021123332.
- the at least one cationic or ionizable lipidis selected or derived from C26, SM-102, SS- 33/4PE-15, HEXA-C5DE-PipSS, or ALC-0315.
- the at least one cationic or ionizable lipidis ALC-0315.
- the at least one cationic or ionizable lipidis C26 (VitE-C4DE-Pip-thioether).
- the lipid-based carriers of the inventioncomprise two or more (different) cationic lipids as defined herein.
- the lipid-based carrierspreferably the LNPs, comprise at least one neutral lipid or phospholipid.
- the lipid-based carriercomprises at least one neutral lipid or phospholipid.
- neutral lipidrefers to any lipid species that exist in either an uncharged or neutral zwitterionic form at physiological pH.
- Neutral lipidsmay be selected from DHPC, DHPC, DOPC, DPPC, DOPG, DPPG, DOPE, POPC, POPE, DOPE-mal, DPPE, DMPE, DSPE, 16-O-monomethyl PE, 16-O-dimethyl PE, 18-1-trans PE, SOPE, transDOPE, 1 ,2-diphytanoyl-sn-glycero-3-phosphoethanolamine (DPhyPE), DPhyPS (1 ,2-diphytanoyl-sn-glycero-3- phospho-L-serine), or mixtures thereof.
- DPhyPEDPhyPS (1 ,2-diphytanoyl-sn-glycero-3- phospho-L-serine
- the at least one neutral lipidis selected or derived from DSPC, DHPC, DPhyPE, or DPhyPS. Preferred in that context is DSPC. In other embodiments, the at least one neutral lipid is selected or derived from DPhyPE and/or DphyPS. In other embodiments, the lipid-based carrier, preferably the LNP, comprises DPhyPE and DPhyPS. Steroids, steroid analogs or sterols
- the lipid-based carriercomprises a steroid, steroid analog, or sterol.
- the steroid, steroid analog or sterolis derived or selected from cholesterol, cholesteryl hemisuccinate (CHEMS), or any derivate of these.
- the lipid-based carriercomprises cholesterol.
- Lipid-based carrier compositionsLipid-based carrier compositions
- the lipid-based carrierspreferably the LNPs, comprising the at least one RNA comprise
- the lipid-based carrierpreferably the LNP, comprise (i) to (iv) in a molar ratio of about 20-60% cationic lipid or ionizable lipid, about 5-25% neutral lipid, about 25-55% steroid or steroid analog, and about 0.5-15% aggregation reducing lipid e.g. polymer conjugated lipid.
- the lipid-based carrierpreferably the LNP, comprise (i) to (iv) in a molar ratio of about 45- 60% cationic lipid or ionizable lipid, about 5-15% neutral lipid, about 25-45% steroid or steroid analog, and about 0.5- 2.5% aggregation reducing lipid e.g. polymer conjugated lipid.
- the lipid-based carriercomprising the nucleic acid comprise (i) a cationic lipid ALC-0315; (ii) a neutral lipid DSPC; (iii) a steroid or steroid analog cholesterol; and (iv) an aggregation reducing lipid ALC-0159; preferably wherein (i) to (iv) are in a molar ratio of about 47.4% cationic lipid, about 10% neutral lipid, about 40.9% steroid or steroid analog, and about 1.7% aggregation reducing lipid, preferably wherein the lipid-based carrier encapsulates the nucleic acid, preferably the RNA.
- the lipid-based carriercomprising the nucleic acid comprise (i) a cationic lipid C26 (VitE-C4DE-Pip-thioether); (ii) a neutral lipid DPhyPE and a neutral lipid DPhyPS; (iii) a steroid or steroid analog cholesterol; and (iv) an aggregation reducing lipid selected from a POZ-lipid, preferably from PMOZ 4; preferably wherein (i) to (iv) are in a molar ratio of about 49% cationic lipid, about 10% neutral lipid, about 40% steroid or steroid analog, and about 1% aggregation reducing lipid, preferably wherein the lipid-based carrier encapsulates the RNA.
- a cationic lipid C26VitE-C4DE-Pip-thioether
- a neutral lipid DPhyPE and a neutral lipid DPhyPSe.g., a neutral lipid DPhyPE and a neutral
- the lipid-based carriercomprising the nucleic acid comprise (i) a cationic lipid SM-102; (ii) a neutral lipid DSPC; (iii) a steroid or steroid analog cholesterol; and (iv) an aggregation reducing lipid selected from a PEG-DMG; preferably wherein (i) to (iv) are in a molar ratio of about 50% cationic lipid, about 10% neutral lipid, about 38.5% steroid or steroid analog, and about 1.5% aggregation reducing lipid, preferably wherein the lipid-based carrier encapsulates the RNA.
- the wt/wt ratio of lipid to RNA in the lipid-based carrieris from about 10:1 to about 60:1 , e.g. about 40:1. In embodiments, the wt/wt ratio of lipid to RNA is from about 20:1 to about 30:1, e.g. about 25: 1. In other preferred embodiments, the wt/wt ratio of lipid to RNA is in the range of 20 to 60, preferably from 3 to 15, 5 to 13, 4 to 8 or from 7 to 11.
- the amount of lipid comprised in the lipid-based carriers such as LNPsmay be selected taking the amount of the RNA cargo into account. In one embodiment, these amounts are selected such as to result in an N/P ratio of the lipid-based carriers encapsulating the RNA in the range of about 0.1 to about 20.
- the N/P ratiois defined as the mole ratio of the nitrogen atoms (“N”) of the basic nitrogen-containing groups of the lipid to the phosphate groups (“P”) of the RNA which is used as cargo.
- the N/P ratiomay be calculated on the basis that, for example, 1 pg RNA typically contains about 3nmol phosphate residues, provided that the RNA exhibits a statistical distribution of bases.
- the “N”-value of the lipid or lipidoidmay be calculated on the basis of its molecular weight and the relative content of permanently cationic and - if present - cationisable groups.
- the compositioncomprises lipid-based carriers such as LNPs (encapsulating the RNA as defined herein) that have a defined size (particle size, homogeneous size distribution).
- the size of a lipid-based carrier such as an LNPis typically described as Z-average size.
- Z-average sizerefers to the mean diameter of particles as measured by dynamic light scattering (DLS) with data analysis using the so- called cumulant algorithm, which provides as results the so-called Z-average with the dimension of a length, and the polydispersity index (PDI), which is dimensionless.
- DLSdynamic light scattering
- DLSrefers to a method for analyzing particles in a liquid, wherein the liquid is typically illuminated with a monochromatic light source and wherein the light scattered by particles in the liquid is detected. Suitable DLS protocols and instruments are known in the art.
- the lipid-based carrierspreferably the LNPs, have a Z-average size of less than 400nm, preferably less than 300nm, more preferably less than 200nm.
- the lipid-based carrierspreferably the LNPs, have a Z-average size ranging from about 50nm to about 200nm, preferably in a range from about 50nm to about 150nm, more preferably from about 50nm to about 120nm.
- the compositioncomprises less than about 10%, 9%, 8%, 7%, 6%, 5%, 4%, 3%, 2%, 1% lipid-based carriers that have a particle size exceeding about 500nm.
- the pharmaceutical compositioncomprises less than about 10%, 9%, 8%, 7%, 6%, 5%, 4%, 3%, 2%, 1 % LNPs that have a particle size smaller than about 20nm.
- the lipid-based carriersexhibit a zeta potential in the range of -50 mV to +50 mV, preferably in the range of -25 mV to +25 mV, more preferably in the range of -10 mV to +10 mV, most preferably in the range of -5 mV to +5 mV.
- the polydispersity index (PDI) of the lipid-based carrierspreferably the LNPs, is in the range of 0.1 to 0.5. In preferred embodiments, the polydispersity index (PDI) value is less than about 0.3, preferably of less than about 0.2. Typically, the PDI is determined by dynamic light scattering.
- RNAis encapsulated in lipid-based carriers such as LNPs.
- the percentage of encapsulationmay be determined by a RiboGreen assay as known in the art.
- the lipid-based carrierssuch as LNPs have a spherical morphology, preferably comprising a solid core or a partially solid core.
- the lipid-based carriers, preferably the LNPshave been generated by combining an aqueous RNA solution with an ethanolic lipid solution using a mixing means at total flow rates above 15ml/min.
- the lipid-based carriershave been prepared using according to the general procedures described in PCT Pub. Nos. WO2015199952, W02017004143 and WO2017075531 , the full disclosures of which are incorporated herein by reference.
- the compositioncomprises purified lipid-based carriers, preferably purified LNPs, encapsulating an RNA as defined herein.
- the surface of the lipid-based carrierspreferably the LNPs, is uncharged at about pH.7.
- the lipid-based carrierspreferably the LNPs do not comprise DOTMA and/or DOPE.
- the RNAis not formulated in a lipoplex formulation, in particular not formulated in a lipoplex formulation comprising DOTMA and/or DOPE.
- the RNA of the inventioncomprises at least one cds encoding at least one antigen, wherein the at least one antigen that is selected from or derived from tumor antigens, viral antigens, bacterial antigens, protozoal antigens, fungal antigens, self-antigens or allogenic antigens, preferably tumor antigens.
- Suitable antigens in the context of the inventionare also specified in paragraph “Antigen” provided above. Preferred embodiments in that context are herein specified in more detail.
- the at least one antigenis a tumor-specific antigen or tumor associated antigen.
- the at least one tumor specific antigen or tumor associated antigenis selected from mutation antigen, neoantigen, unique antigen, differentiation tumor antigen, overexpressed tumor antigen, virus tumor antigen or bacterial tumor antigen.
- the tumor antigen that is encoded by the cds of the RNAis selected or derived from at least one of List 1 as defined above. In preferred embodiments, the tumor antigen that is encoded by the cds of the RNA is selected or derived from at least one of List 2 as defined above. In preferred embodiments, the tumor antigen that is encoded by the cds of the RNA is selected or derived from at least one of List 3 as defined above.
- the tumor antigen that is encoded by the cds of the RNAis selected from Claudin6 (CLDN6) or PRAME.
- the RNAcomprises at least one cds encoding at least one epitope, at least two epitopes, at least three epitopes, at least four epitopes, at least five epitopes, at least six epitopes, at least seven epitopes, at least eight epitopes, at least nine epitopes, or at least ten epitopes, wherein the epitopes derived or selected from the same antigen or from different antigens.
- the RNAcomprises at least one cds encoding at least one linker.
- linkerpreferably refers to peptide linkers, i.e. typically short (i.e. comprising 1-150 amino acids, preferably 1-50 amino acids, more preferably 1 to 20 amino acids), linear amino acid sequences connecting or linking two polypeptide sequences.
- Linkers according to the inventionmay be derived from any protein of human, animal, plant, bacterial or viral origin. Linkers according to the invention may be naturally occurring or artificial (i.e. synthetic or non-naturally occurring) linkers.
- the linker(s)is/are non-immunogenic, i.e. do not trigger an immune response.
- Linkersmay be employed to connect or link at least two components, e.g. two epitopes or an epitope and a T helper cell epitope encoded by the RNA of the invention.
- the cds of the RNA according to the inventionmay encode at least one linker, or a plurality of at least 2, 3, 4, 5, 6, 7, 8, 9 or 10 identical or different linkers, as described herein.
- the linkersdiffer in their amino acid sequence and/or nucleic acid sequence encoding the respective linkers.
- Linkers of interest in the context of the present inventionare inter alia disclosed in W02002014478, W02001008636, W02013171505, W02008017517 and WO1997047648, which are incorporated by reference in their entirety as well.
- the RNAcomprises at least one cds encoding at least one T helper epitope.
- T helper epitoperefers to an epitope capable of binding to MHC molecules, preferably MHC Class II molecules, thereby being recognized by CD4+ T helper cells.
- T helper epitopesinduce or enhance CD4+ Th cell activation, differentiation, and/or proliferation.
- Activated CD4+ Th cellsare may preferably (1) (directly or indirectly) induce or enhance cytotoxic T lymphocyte differentiation, and/or proliferation, and/or (2) (directly or indirectly) induce or enhance antibody-producing plasma cell differentiation, and/or proliferation.
- activated CD4+ Th cellsmay induce or enhance the respective immune responses either via direct interaction with target cells or precursors thereof (e.g. B cells) or indirectly via interacting with other cells (e.g. dendritic cells) that in turn directly interact with target cells or precursors thereof (e.g. modified CD8+ T cells).
- target cells or precursors thereofe.g. B cells
- dendritic cellse.g. dendritic cells
- preferred T helper epitopesinclude the T helper epitopes disclosed in WO2001062284, WO2010023247, W02004058297, W02004000873 and W02006113792.
- Particularly preferred T helper cell epitopes in the context of the present inventioninclude naturally occurring or artificial T helper epitopes derived from PADRE; TpD (tetanus toxoid and diphtheria toxoid, separated by an internal cathepsin cleavage site); Hepatitis C virus (Core); Hepatitis C virus (E1); Hepatitis C virus (E2); Hepatitis C virus (NS2); Hepatitis C virus (NS3); Hepatitis C virus (NS4); Hepatitis C virus (NS4a); Hepatitis C virus (NS4b); Hepatitis C virus (NS5a); Hepatitis C virus (NS5b); Influenza A; Influenza B; Measles virus
- the RNAcomprises at least one cds encoding at least one additional amino acid sequence derived from at least one immune response activating Signal transduction protein located in the external plasma membrane, wherein said at least one immune response activating signal transduction protein located in the external plasma membrane is CTLA4, and wherein said at least one additional amino acid sequence comprises or consists of at least one transmembrane domain from CTLA4.
- RNAcomprising at least one cds encoding at least one amino acid sequence of the following formula, preferably in 5'- ⁇ 3‘ direction:
- SIGencodes a signal peptide
- Lencodes a linker sequence
- each "AN”encodes an identical or different antigenic peptide or protein, preferably a tumor antigen as defined before
- IMencodes a helper epitope
- TMD/TMCDencodes an amino acid sequence derived from an immune response signal transduction protein located in the external plasma membrane, preferably a transmembrane domain, preferably from CTLA4, and optionally a cytoplasmic domain, preferably from CTLA4;
- b, d, m, n, ois each independently an integer selected from 0, 1 , 2, 3, 4, 5, 6, 7, 8, 9, and 10;
- a, c, e, pis each independently an integer selected from 1 , 2, 3, 4, 5, 6, 7, 8, 9 and 10.
- the RNAcomprises at least one cds encoding at least one antigen, preferably the full length of the antigen.
- the RNAcomprises at least one cds encoding at least one epitope that is targeted by the antigen receptor of the transferred modified T cells.
- the RNAis a modified and/or stabilized nucleic acid.
- the RNAmay thus be provided as a “stabilized RNA” that is to say an RNA showing improved resistance to in vivo degradation and/or an RNA showing improved stability in vivo, and/or an RNA showing improved translatability in vivo.
- the RNA of the present inventionmay be provided as a “stabilized RNA”.
- the RNAcomprises at least one codon modified cds.
- the at least one cds of the RNAis a codon modified cds.
- the amino acid sequence encoded by the at least one codon modified cdsis not modified compared to the amino acid sequence encoded by the corresponding wild type or reference cds.
- codon modified cdsrelates to coding sequences that differ in at least one codon (triplets of nucleotides coding for one amino acid) compared to the corresponding wild type or reference cds.
- a codon modified cds in the context of the inventionmay show improved resistance to in vivo degradation and/or improved stability in vivo, and/or improved translatability in vivo. Codon modifications in the broadest sense make use of the degeneracy of the genetic code wherein multiple codons may encode the same amino acid and may be used interchangeably to optimize/modify the cds for in vivo applications.
- the at least one cdsis a codon modified cds selected from a C maximized cds (as further defined in WO2021239880 [p.122, lines 33 to 39] which is hereby incorporated by reference); a CAI maximized cds (as further defined in WO2021239880 [p.123, lines 33 to 44] which is hereby incorporated by reference); a human codon usage adapted cds (as further defined in WO2021239880 [p.123, lines 7 to 17] which is hereby incorporated by reference); a G/C content modified cds (as further defined in WO2021239880 [p.123, lines 19 to 31] which is hereby incorporated by reference); a G/C optimized cds (“opt1”); or any combination thereof.
- a C maximized cdsas further defined in WO2021239880 [p.122, lines 33 to 39] which is hereby incorporated by reference
- the G/C content of the at least one cdsis optimized compared to the G/C content of the corresponding wild type or reference cds (“G/C optimized”). “Optimized” in that context means that the G/C content of the cds is preferably increased to the essentially highest possible content.
- the generation of a G/C optimized cdsis suitably carried out according to W02002098443, which is hereby incorporated by reference.
- the G/C content of the at least one cdsis increased by at least 10%, 15%, or 20% compared to the G/C content of the corresponding wild type or reference cds.
- the at least one cdshas a G/C content of at least about 50%, 55%, or 60%.
- the at least one cdshas a G/C content of at least about 50%, 51%, 52%, 53%, 54%, 55%, 56%, 57%, 58%, 59%, 60%, 61%, 62%, 63%, 64%, 65%, 66%, 67%, 68%, 69%, or 70%.
- the RNAcomprises at least one untranslated region (UTR).
- UTRuntranslated region
- UTRuntranslated region
- UTR elementrefers to a part of an RNA typically located 5’ or 3' of a cds that is not translated into protein.
- An UTRmay comprise elements for controlling gene expression, also called regulatory elements.
- regulatory elementsmay be, e.g., ribosomal binding sites, miRNA binding sites, promotor elements. Regulatory elements may determine turnover, stability, and localization of the nucleic acid, in particular the RNA.
- UTRsmay also harbour sequence elements that enhance translation.
- heterologousor “heterologous UTR” as used herein refers to a nucleic acid sequence or UTR that is not from the same gene, the same genomic fusion, or the same naturally occurring transcript. Heterologous sequences do naturally (in nature) not occur in the same RNA molecule.
- the RNA of the inventioncomprises at least one 3-UTR.
- 3’-untranslated regionrefers to a part of an RNA located 3’ (i.e. downstream) of a cds and which is not translated into protein.
- a 3-UTRmay be part of an RNA located between a cds and an (optional) terminal poly(A) sequence.
- a 3-UTRmay comprise elements for controlling gene expression, also called regulatory elements. Such regulatory elements may be, e.g., ribosomal binding sites, miRNA binding sites.
- the RNAcomprises at least one 3-UTR, which may be derivable from a gene that relates to an RNA with enhanced half-life (i.e. that provides a stable RNA).
- the 3-UTRcomprises one or more of a polyadenylation signal, a binding site for proteins that affect RNA stability of location in a cell, or one or more miRNA or binding sites for miRNAs.
- the RNAcomprises at least one 3’-UTR, wherein the at least one 3-UTR comprises or consists of a nucleic acid sequence derived or selected from a 3-UTR of a gene selected from PSMB3, AES-12S, ALB7, alpha-globin (HBA1 , HBA2), ANXA4, beta-globin (HBB), CASP1 , COX6B1, FIG4, GH1 , GNAS, NDUFA1, RPS9, SLC7A3, TUBB4B, or from a homolog, a fragment, or variant of any one of these genes.
- the at least one 3-UTR that is derived or selected from PSMB3, AES-12S, ALB7, alphaglobin (HBA1, HBA2), ANXA4, beta-globin (HBB), CASP1 , COX6B1 , FIG4, GH1 , GNAS, NDUFA1, RPS9, SLC7A3, TUBB4Bcomprises or consist of a nucleic acid sequence being identical or at least 70%, 80%, 85%, 86%, 87%, 88%, 89%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identical to any one of SEQ ID NOs: 134-191, or a fragment or a variant of any of these.
- the RNAcomprises a 3-UTR derived or selected from a PSMB3 gene.
- the at least one heterologous 3 -UTR derived or selected from PSMB3comprises or consist of a nucleic acid sequence being identical or at least 70%, 80%, 85%, 86%, 87%, 88%, 89%, 90%, 91 %, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identical to any one of SEQ ID NOs: 134, 135, 180-191 , or a fragment or a variant thereof, preferably SEQ ID NO: 135, or a fragment or a variant thereof.
- the at least one 3’-UTRcomprises or consist of a nucleic acid sequence being identical or at least 70%, 80%, 85%, 86%, 87%, 88%, 89%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identical to any one of SEQ ID NOs: 164-171, or a fragment or a variant of any of these.
- the RNA of the inventioncomprises at least one 5’-UTR.
- 5’-untranslated regionrefers to a part of a nucleic acid located 5’ (i.e. “upstream”) of a cds and which is not translated into protein.
- a 5-UTRmay be part of a nucleic acid located 5’ of the cds.
- a 5-UTRstarts with the transcriptional start site and ends before the start codon of the cds.
- a 5-UTRmay comprise elements for controlling gene expression, also called regulatory elements. Such regulatory elements may be, e.g., ribosomal binding sites, miRNA binding sites.
- the RNAcomprises at least one 5-UTR, which may be derivable from a gene that relates to an RNA with enhanced half-life (i.e. that provides a stable RNA).
- the 5-UTRcomprises one or more of a binding site for proteins that affect RNA stability or RNA location in a cell, or one or more miRNA or binding sites for miRNAs.
- the RNAcomprises at least one 5'-UTR, wherein the at least one 5’-UTR comprises a nucleic acid sequence derived or selected from a 5-UTR of gene selected from HSD17B4, AIG1 , alpha-globin (HBA1 , HBA2), ASAH1, ATP5A1 , COX6C, DPYSL2, HHV5, MDR, MP68, NDUFA4, NOSIP, RPL31, RPL32, RPL35A, SLC7A3, synthetic origin, TUBB4B, UBQLN2, or from a homolog, a fragment or variant of any one of these genes,
- the at least one 5’-UTR derived or selected from HSD17B4, AIG1 , alpha-globin (HBA1 , HBA2), ASAH1 , ATP5A1 , COX6C, DPYSL2, HHV5, MDR, MP68, NDUFA4, NOSIP, RPL31, RPL32, RPL35A, SLC7A3, synthetic origin, TUBB4B, UBQLN2comprises or consist of a nucleic acid sequence being identical or at least 70%, 80%, 85%, 86%, 87%, 88%, 89%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identical to any one of SEQ ID NOs: 80-133, or a fragment or a variant of any of these.
- the RNAcomprises a 5-UTR derived or selected from a HSD17B4 gene.
- the at least one heterologous 5’-UTR derived or selected from HSD17B4comprises or consists of a nucleic acid sequence being identical or at least 70%, 80%, 85%, 86%, 87%, 88%, 89%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identical to any one of SEQ ID NOs: 80, 81 , 132, 133 or a fragment or a variant thereof, preferably SEQ ID NO: 81 , or a fragment or a variant thereof.
- the at least one 5-UTRcomprises or consist of a nucleic acid sequence being identical or at least 70%, 80%, 85%, 86%, 87%, 88%, 89%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identical to any one of SEQ ID NOs: 122-129, or a fragment or a variant of any of these.
- the RNAcomprises at least one cds as specified herein operably linked to a 3-UTR and/or a 5-UTR selected from the 5’-UTR/3’-UTR combinations (5’UTR/3’UTR) provided in WO2021239880 [p.127, line 35 to p.128, line 2], which is hereby incorporated by reference.
- the at least one 5-UTRis selected from HSD17B4 and the at least one 3 -UTR is selected from PSMB3 (a-1 (HSD17B4/PSMB3)).
- the RNAcomprises at least one cds as defined herein encoding at least one antigen as defined herein, wherein said cds is operably linked to a HSD17B45-UTR and a PSMB3 3-UTR (HSD17B4/PSMB3 (a-1)). This embodiment is particularly beneficial for effectively expressing the antigen in humans.
- the RNAis monocistronic, bicistronic, or multicistronic, preferably the RNA is monocistronic
- the RNAcomprises a ribosome binding site, also referred to as “Kozak sequence”, that is identical to or at least 80%, 85%, 90%, 95% identical to any one of SEQ ID NOs: 70, 71 or sequences GCCGCCACC (DNA), GCCGCCACC (RNA), GCCACC (DNA), GCCACC (RNA), ACC (DNA) or ACC (RNA), or fragments or variants of any of these, preferably ACC (RNA).
- Kozak sequenceribosome binding site
- the RNA of the inventionis an artificial RNA and/or an isolated RNA.
- an artificial RNAas used herein is intended to refer to an RNA that does not occur naturally.
- an artificial RNAmay be understood as a non-natural RNA molecule.
- Such an RNA moleculemay be non-natural due to its individual sequence (e.g. G/C content modified cds, UTRs) and/or due to other modifications, e.g. structural modifications of nucleotides.
- artificial RNAmay be designed and/or generated by genetic engineering to correspond to a desired artificial sequence of ribonucleotides.
- an artificial RNAis a sequence that may not occur naturally, i.e. a sequence that differs from the wild type sequence/the naturally occurring sequence by at least one nucleotide.
- the term “artificial RNA”is not restricted to mean “one single molecule” but is understood to comprise an ensemble of essentially identical RNA molecules.
- isolated RNAdoes not comprise a cell or a subject that comprises said RNA but relates to the RNA as an isolated molecule or ensemble of isolated molecules.
- the “isolated RNA”can be an RNA isolated or purified from a cell (e.g. cell culture, bacterial culture), or can be an RNA isolated from an RNA in vitro transcription.
- the RNAmay be any type of RNA that comprises a cds as defined herein including any type of single stranded RNA, any type of double stranded RNA, any type of linear RNA, and any type of circular RNA.
- the RNAis selected from mRNA, circular RNA, replicon RNA or self-replicating RNA, or viral RNA, preferably mRNA or a circular RNA.
- the RNAis a circular RNA.
- a “circular RNA”(circRNAs) is an RNA connected to form a circle and therefore does not comprise a 3’ or 5’ terminus. Said circRNA comprises at least one cds as defined herein. CircRNA construct designs can be taken from WG2023073228, claims 1 to 51 , hereby incorporated by reference.
- the RNAis a replicon RNA or self-replicating RNA.
- Such constructsmay encode replicase elements derived from e.g. alphaviruses (e.g. SFV, SIN, VEE, or RRV) and a cds as defined herein.
- the RNA of the inventionis selected from an mRNA.
- an mRNAis preferred to provide the antigen because mRNA allows for regulated dosage, transient expression, complete degradation of the mRNA after protein synthesis, and does not pose the risk of insertional mutations.
- the mRNAis non-replicative.
- the RNAcomprises about 50 to about 20000 nucleotides, or about 500 to about 10000 nucleotides, or about 1000 to about 10000 nucleotides, or preferably about 1000 to about 5000 nucleotides.
- the RNAcomprises at least one poly(N) sequence, e.g. at least one poly(A) sequence, at least one poly(U) sequence, at least one poly(C) sequence, or combinations thereof.
- poly(A) sequencerefers to a sequence of up to 1000 adenosines typically located at the 3’-end of a linear RNA. Typically, a poly(A) sequence is homopolymeric. Alternatively, a poly(A) sequence may be interrupted by at least one nucleotide different from an adenosine.
- the at least one poly(A) sequencemay comprise about 20 to about 500 adenosine nucleotides, about 40 to about 250 adenosine nucleotides, preferably about 60 to about 150 adenosine nucleotides. In embodiments, the at least one poly(A) sequence comprises about or more than 50, 64, 75, 100, 150, 200, 300, 400, or 500 adenosines. In preferred embodiments, the at least one poly(A) sequence comprises about 60 to about 150 adenosine nucleotides, preferably about 100 adenosine nucleotides (A100).
- the RNAcomprises at least one interrupted poly(A) sequence, wherein the poly(A) sequence is interrupted by non-adenosine nucleotides, preferably by about 10 non-adenosine (N10) nucleotides.
- N10non-adenosine
- the at least one poly(A) sequence as defined hereinis located directly at the 3’ terminus of the nucleic acid, preferably the RNA. Accordingly, the 3’-terminal nucleotide in the polynucleotide chain is the 3’- terminal A nucleotide of the at least one poly(A) sequence.
- the 3’ terminus of the nucleic acidconsists of a poly(A) sequence (e.g. A100 or A30-N10-A70) and therefore terminates with an A.
- having a 3’ terminus ending on an adenosinemay decrease the induction of interferons, e.g. IFNalpha, by the RNA of the invention if, e.g., administered as a medicament to a human.
- interferonse.g. IFNalpha
- the nucleic acidpreferably the RNA, comprises a poly(A) sequence of about 100 consecutive adenosines (A100) located directly at the 3’ terminus of the RNA.
- the RNAcomprises at least one poly(A) sequence obtained by enzymatic polyadenylation, wherein the majority of RNA molecules comprise about 100 (+/- 20) to about 500 (+/- 100) adenosine nucleotides, preferably about 100 (+/- 20) to about 200 (+/- 40) adenosine nucleotides.
- the RNAcomprises at least one histone stem-loop (hSL) or histone stem loop structure.
- hSLmay be located in an UTR region, preferably in the 3’-UTR.
- the termrefers to nucleic acid sequences that forms a stem-loop secondary structure.
- a hSLmay be derived from formulae (I) or (II) of WO2012019780, or preferably from the specific formulae (la) or (Ila) of WO2012019780, that are hereby incorporated by reference.
- the at least one hSL sequencecomprises or consists of a nucleic acid sequence identical or at least 70%, 80%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identical to any one of SEQ ID NOs: 71 , 72, or a fragment or variant of any of these, preferably SEQ ID NO: 72, or a fragment or variant thereof.
- the RNAcomprises a 3’-terminal sequence element.
- the 3’-terminal sequence elementrepresents the 3’ terminus of the RNA.
- a 3’-terminal sequence elementmay comprise at least one poly(N) sequence as defined herein and, optionally, at least one hSL as defined herein.
- the at least one 3’-terminal sequence elementcomprises or consists of an RNA sequence being identical or at least 70%, 80%, 90%, 91 %, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identical to any one of SEQ ID NOs: 73-79, or a fragment or variant of these sequences.
- the at least one 3’-terminal sequence elementcomprises or consists of a nucleic acid sequence being identical or at least 70%, 80%, 90%, 91 %, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identical to SEQ ID NOs: 73 or 74, or a fragment or variant thereof.
- the RNAcomprises a 5'-terminal sequence element comprising or consisting of an RNA sequence being identical or at least 70%, 80%, 90%, 91 %, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identical to sequence AGGAGA, or a fragment or variant thereof.
- a 5’-terminal sequence elementmay comprise e.g. a binding site for T7 RNA polymerase.
- the first nucleotide of said 5’-terminal start sequencemay preferably comprise a 2'0 methylation, e.g. 2’0 methylated guanosine or a 2’0 methylated adenosine.
- the RNA of the inventionis a therapeutic RNA. Accordingly, the RNA is suitably used in a therapeutic context, in particular to provide at least one antigen for use in a method of treatment or prophylaxis of a disease, disorder or condition.
- the RNAis modified, wherein the modification refers to chemical modifications comprising backbone modifications as well as sugar modifications or base modifications.
- a backbone modificationis a chemical modification in which phosphates of the backbone of the nucleotides of the RNA are modified.
- a sugar modificationis a chemical modification of the sugar of the nucleotides of the RNA.
- a base modificationis a chemical modification of the base moiety of the nucleotides of the RNA.
- nucleotide analoguesare preferably selected from nucleotide analogues which are applicable for transcription and/or translation.
- the RNA of the inventioncomprises at least one modified nucleotide.
- the RNAcomprises at least one modified nucleotide selected from WO2021239880 [p.136, line 17 to p.137, line 19], which is hereby incorporated by reference.
- the RNAmay comprise modified uridine nucleotides that preferably comprise a chemical modification in the 5-position of the uracil. Suitable modified uridine nucleotides may be selected from WO2021239880 [p.137, lines 15 to 19], which is hereby incorporated by reference.
- the at least one modified nucleotideis selected from pseudouridine, N1 -methylpseudouridine, N1 -ethylpseudouridine, 2-thiouridine, 4’-thiouridine, 5-methylcytosine, 5-methyluridine, 2-thio-1-methyl-1 -deazapseudouridine, 2-thio-1-methyl-pseudouridine, 2-thio-5-aza-uridine, 2-thio-dihydropseudouridine, 2-thio-dihydrouridine, 2-thio-pseudouridine, 4-methoxy-2-thio-pseudouridine, 4-methoxy-pseudouridine, 4-thio-1-methyl-pseudouridine, 4- thio-pseudouridine, 5-aza-uridine, dihydropseudouridine, 5-methoxyuridine and 2’-O-methyl uridine.
- pseudouridineN1 -methylpseud
- essentially all, e.g. essentially 100% of the uracil in the cds or the full RNEA sequencehave a chemical modification, preferably a chemical modification in the 5-position of the uracil (e.g. ip and/or ml ip).
- 100% of the uracil in the full RNA sequenceare substituted with N1 -methylpseudouridine (ml i ).
- the RNAdoes not comprise chemically modified nucleotides.
- a 5’-cap structure as defined belowis typically not considered to be a chemically modified nucleotide.
- the RNAcomprises a sequence that consists only of G, C, A and U nucleotides and therefore does not comprise modified nucleotides, and optionally comprises a 5’-cap structure.
- RNA molecules that do not comprise modified nucleotides to provide the antigen as defied hereinmay be beneficial as stronger T cell responses may be induced (compared to m1i or ip modified RNA).
- the RNAcomprises a 5’-cap structure.
- 5’-cap structurerefers to a 5’ modified nucleotide, particularly a guanine nucleotide, positioned at the 5’-end of an RNA.
- the 5’-cap structureis typically connected via a 5’-5’-triphosphate linkage to the RNA.
- the RNAcomprises a 5’-cap structure, preferably m7G, capO, cap1, cap2, a modified capO or a modified cap1 structure.
- the RNAcomprises a cap1 structure or a modified cap1 structure.
- a 5'-cap structuremay be formed in chemical RNA synthesis or in RNA in vitro transcription (co-transcriptional capping) using cap analog.
- the 5’-cap structuremay be added co-transcriptionally using a cap analog, preferably a cap1 analog, as defined herein, preferably in an RNA in vitro transcription reaction.
- cap analogrefers to a non-polymerizable di-nucleotide or tri-nucleotide that has cap functionality in that it facilitates translation or localization, and/or prevents degradation of an RNA molecule when incorporated at the 5’-end of the RNA molecule.
- Non-polymerizablemeans that the cap analogue will be incorporated only at the 5’-terminus because it does not have a 5’ triphosphate and therefore cannot be extended in the 3'-direction by a template-dependent polymerase, particularly, by template-dependent RNA polymerase.
- a cap1 or a modified cap1 structureis generated using a cap analog, preferably a tri-nucleotide cap analog.
- a cap analogpreferably a tri-nucleotide cap analog.
- Any cap analog derivable from the structures defined in claims 1-13 of WO2017053297 (hereby incorporated by reference) or, alternatively, any cap analog derivable from the structures defined in claims 1 -37 of W02023007019 (hereby incorporated by reference)may be suitably used to co-transcriptionally generate a cap1 or a modified cap1.
- the cap1 structureis formed via co-transcriptional capping using tri-nucleotide cap analog m7G(5’)ppp(5’)(2’OI ⁇ /leA)pG, m7G(5’)ppp(5’)(2’OMeG)pG, or m7(3’OMeG)(5’)ppp(5’)m6(2’OMeA)pG.
- a particularly preferred cap1 analog in that contextis m7G(5’)ppp(5’)(2’OMeA)pG.
- the cap1 structureis a modified cap1 structure and is formed using co-transcriptional capping using tri-nucleotide cap analogue 3’0Me-m7G(5’)ppp(5’)(2’0MeA)pG.
- the 5’-cap structuremay be formed via enzymatic capping using capping enzymes (e.g. vaccinia virus capping enzymes and/or 2’-0 methyltransferases) to generate capO, cap1 or cap2 structures.
- RNA moleculescomprise a cap structure, preferably a cap1 structure, as determined by a capping assay (e.g. via an assay as described in cl. 27 to 46 of W02015101416).
- the RNAcomprises a 5’-terminal sequence comprising or consisting of a nucleic acid sequence being identical or at least 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identical to anyone AGGAGA, GGGAGA, GGGAAA, AGAAUA, AGAUUA, GAUGGG or GGGCG, or a fragment or variant of these, preferably AGGAGA.
- a 5'-terminal sequence elementmay comprise a binding site for an RNA polymerase.
- the first nucleotide of said 5’-terminal sequencecomprises a 2’0 methylation (2'0meA or 2’OMeG).
- the RNAis preferably an in vitro transcribed RNA (e.g. an in vitro transcribed mRNA).
- the RNA of the inventionis a purified RNA, preferably a purified mRNA.
- purified RNArefers to RNA that has a higher purity after certain purification steps than the starting material. Typical impurities comprise peptides, proteins, spermidine, RNA fragments, dsRNA, free nucleotides, DNA, etc. It is desirable for the “degree of RNA purity” to be as close as possible to 100%. Preferably, a “purified RNA” has a degree of purity of more than 75%, 80%, 85%, 90%, or 95%. The degree of purity may be determined by an analytical HPLC.
- the RNAhas been purified by (RP)HPLC, AEX, size exclusion chromatography, hydroxyapatite chromatography, tangential flow filtration (TFF), filtration, precipitation, core-bead flew through chromatography, oligo(dT) purification, and/or cellulose-based purification.
- the RNAhas been purified by (RP)HPLC (suitably as described in W02008077592), TFF (suitably as described in WO2016193206) and/or oligo d(T) purification.
- the at least one step of purificationis selected from RP-HPLC and/or TFF.
- the RNAhas an integrity of at least 60%, 70%, 80%, 90%.
- RNA integritydescribes whether the complete RNA sequence is present. RNA integrity can be determined by RP-HPLC and may be based on determining the area under the peak of the expected full-length RNA in a chromatogram.
- the RNAis suitable for use in treatment or prevention of a disease, disorder or condition.
- the RNAcomprises at least the following elements:
- the RNAcomprises the following sequence elements, preferably in 5’- to 3’-direction:
- a 5-UTRpreferably selected or derived from a 5-UTR of a HSD17B4 gene
- a poly(A) sequencepreferably comprising about 100A nucleotides, e.g. A100 or A30-N10-A70.
- Goptionally, chemically modified nucleotides, suitably selected from i or ml ip , preferably ml i .
- the RNAcomprises or consists of a nucleic acid sequence which is identical or at least 70%, 80%, 85%, 86%, 87%, 88%, 89%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identical to a nucleic acid sequence selected from the group consisting of SEQ ID NOs: 1 -7, or a fragment or variant of any of these sequences, optionally wherein at least one, preferably all uracil nucleotides in said RNA sequences are replaced by pseudouridine (ip) nucleotides and/or N1 -methylpseudouridine (m1ip) nucleotides.
- ippseudouridine
- m1ipN1 -methylpseudouridine
- the RNA of the inventionis a 5’-cap1 comprising mRNA that comprises or consists of a nucleic acid sequence which is identical or at least 95% identical to a nucleic acid sequence according to any one of SEQ ID NOs: 1-7, or a fragment or variant of any of these sequences.
- the subjecthas received CAR T cells, preferably axicabtagene ciloleucel (YescartaTM), brexucabtagene autoleucel (TecartusTM), lisocabtagene maraleucel (BreyanziTM) or tisagenlecleucel (KymriahTM).
- CAR T cellspreferably axicabtagene ciloleucel (YescartaTM), brexucabtagene autoleucel (TecartusTM), lisocabtagene maraleucel (BreyanziTM) or tisagenlecleucel (KymriahTM).
- the RNA of the compositioncomprises at least one cds encoding at least one amino acid sequence comprising CD19, a functional variant thereof, or a functional fragment of CD19.
- the RNA of the compositioncomprises at least one cds encoding at least one amino acid sequence comprising at least one epitope of or derived from CD19, a functional variant thereof, or a functional fragment of CD19.
- the tumor diseaseis a hematologic tumor disease, preferably B-cell acute lymphoblastic leukemia (ALL), non-Hodgkin lymphoma, a rare and aggressive form of non-Hodgkin lymphoma diffuse large B-cell lymphoma (DLBCL), primary mediastinal large B-cell lymphoma (PMBCL), mantle cell lymphoma (MCL), relapsed or refractory large B-cell lymphoma, transformed follicular lymphoma or high-grade B-cell lymphoma.
- ALLB-cell acute lymphoblastic leukemia
- non-Hodgkin lymphomaa rare and aggressive form of non-Hodgkin lymphoma diffuse large B-cell lymphoma (DLBCL), primary mediastinal large B-cell lymphoma (PMBCL), mantle cell lymphoma (MCL), relapsed or refractory large B-cell lymphoma, transformed follicular lymphoma
- the subjecthas received CAR T cells, preferably ciltacabtagene autoleucel (CarvyktiTM) or idecabtagene vicleucel (AbecmaTM).
- CAR T cellspreferably ciltacabtagene autoleucel (CarvyktiTM) or idecabtagene vicleucel (AbecmaTM).
- the RNA of the compositioncomprises at least one cds encoding at least one amino acid sequence comprising BCMA, a functional variant thereof, or a functional fragment of BCMA.
- the RNA of the compositioncomprises at least one cds encoding at least one amino acid sequence comprising at least one epitope of or derived from BCMA, a functional variant thereof, or a functional fragment of BCMA.
- the tumor diseaseis a hematologic tumor disease, preferably of mantle cell lymphoma (MCL), a rare and aggressive form of non-Hodgkin lymphoma or multiple myeloma.
- MCLmantle cell lymphoma
- the subjecthas received CAR T cells orTCRtg cells targeted to CLDN6.
- the RNA of the compositioncomprises at least one cds encoding at least one antigen from CLDN6 for use in a method of treatment or prophylaxis of a tumour disease, disorder or condition, wherein the RNA is formulated in LNPs and wherein the composition is administered intramuscularly to a subject that has received modified immune cells targeted to CLDN6.
- the RNA of the compositioncomprises at least one cds encoding at least one amino acid sequence comprising CLDN6, a functional variant thereof, or a functional fragment of the CLDN6 or the functional variant thereof comprises the amino acid sequence of SEQ ID NO: 43 or 44, or an amino acid sequence having at least 99%, 98%, 97%, 96%, 95%, 90%, 85%, or 80% identity to the amino acid sequence of SEQ ID NO: 43 or 44.
- the RNA of the compositioncomprises at least one cds encoding at least one amino acid sequence comprising at least one epitope of or derived from CLDN6, a functional variant thereof, or a functional fragment of the CLDN6 or the functional variant thereof comprises the amino acid sequence of SEQ ID NOs: 45, 46, 47 or an amino acid sequence having at least 99%, 98%, 97%, 96%, 95%, 90%, 85%, or 80% identity to the amino acid sequence of SEQ ID NOs: 45, 6, 47.
- the at least one RNA, preferably mRNA, of the composition that is administered to said subjectencodes Claudinl 8.2 (CLDN18.2) or Claudin6 (CLDN6) or a fragment thereof, or a variant of Claudinl 8.2 or Claudin6.
- the tumor or cancer diseaseis an ovarian cancer, lung cancer, gastric cancer, breast cancer, hepatic cancer, pancreatic cancer, skin cancer, melanomas, head neck cancer, sarcomas, bile duct cancer, renal cell cancer, and urinary bladder cancer, in particular ovarian adenocarcinoma and ovarian teratocarcinoma, small cell lung cancer (SCLC) and non-small cell lung cancer (NSCLC), squamous cell lung carcinoma and adenocarcinoma, gastric cancer, breast cancer, hepatic cancer, pancreatic cancer, skin cancer, in particular basal cell carcinoma and squamous cell carcinoma, malignant melanoma, head and neck cancer, in particular malignant pleomorphic adenoma, sarcoma, in particular synovial sarcoma and carcinosarcoma, bile duct cancer, cancer of the urinary bladder, in particular transitional cell carcinoma and papillary carcinoma, kidney cancer, in particular transitional
- the subjecthas received CAR T cells or TCRtg cells targeted to PRAME.
- the RNA of the compositioncomprises at least one cds encoding at least one antigen from PRAME for use in a method of treatment or prophylaxis of a disease, disorder or condition, wherein the RNA is formulated in LNPs and wherein the composition is administered intramuscularly to a subject that has received modified immune cells targeted to PRAME.
- the RNA of the compositioncomprises at least one cds encoding at least one amino acid sequence comprising PRAME, a functional variant thereof, or a functional fragment of the PRAME or the functional variant thereof comprises the amino acid sequence of SEQ ID NO: 66, or an amino acid sequence having at least 99%, 98%, 97%, 96%, 95%, 90%, 85%, or 80% identity to the amino acid sequence of SEQ ID NO: 66.
- the RNA of the compositioncomprises at least one cds encoding at least one amino acid sequence comprising at least one epitope of or derived from PRAME, a functional variant thereof, or a functional fragment of the PRAME or the functional variant thereof comprises the amino acid sequence of SEQ ID NO: 67 or an amino acid sequence having at least 99%, 98%, 97%, 96%, 95%, 90%, 85%, or 80% identity to the amino acid sequence of SEQ ID NO: 67.
- the tumor or cancer diseaseis any of acute lymphocytic cancer, acute myeloid leukemia, alveolar rhabdomyosarcoma, bone cancer, brain cancer, breast cancer, cancer of the anus, anal canal, or anorectum, cancer of the eye, cancer of the intrahepatic bile duct, cancer of the joints, cancer of the neck, gallbladder, or pleura, cancer of the nose, nasal cavity, or middle ear, cancer of the oral cavity, cancer of the vagina, cancer of the vulva, chronic lymphocytic leukemia, chronic myeloid cancer, colon cancer, esophageal cancer, cervical cancer, gastrointestinal carcinoid tumor, glioma, Hodgkin lymphoma, hypopharynx cancer, kidney cancer, larynx cancer, liver cancer, lung cancer, malignant mesothelioma, melanoma, multiple myeloma, nasopharynx cancer, non-Hodgkin lympho
- a preferred cancer in that contextis cancer of the uterine cervix, oropharynx, anus, anal canal, anorectum, vagina, vulva, or penis.
- a particularly preferred canceris a PRAME positive cancer, for example gastric adenocarcinomas, lung adenocarcinomas, ovarian adenocarcinomas, and endometrial carcinomas.
- the present inventionrelates to a combination comprising (i) a composition comprising at least one RNA comprising at least one cds encoding at least one antigen; and (ii) modified immune cells targeted to the at least one antigen, for use in a method of treatment or prophylaxis of a disease, disorder or condition, wherein the modified immune cells are administered intravenously to the subject, and wherein the composition is administered to the subject that has received modified immune cells.
- a “combination”refers to specific dosing regimens, delivery mechanisms, or formulations that optimize the therapeutic outcome.
- a combinationcomprising at least two compounds, (i) a composition comprising at least one RNA comprising at least one cds encoding at least one antigen; and (ii) modified immune cells targeted to the at least one antigen.
- the different compounds or compositionsmay be administered simultaneously, at essentially the same time, or sequentially. Combination in this context may refer to a medical or therapeutic approach in which multiple treatments, interventions, or medications are used together simultaneously or sequentially to address a specific health condition or disease.
- the RNA of the compositionis formulated. Suitable formulations in the context of the invention are further described under paragraph “Formulation/Complexation of the RNA of the composition" (first aspect).
- the RNAis formulated in LNPs as defined herein.
- the composition comprising at least one RNA comprising at least one cds encoding at least one antigen, or host cell modified to express the antigenis administered following administration or generation of modified immune cells targeted to an antigen, e.g., at least 1 day, such as 1 to 60 days or 1 to 50 days or 1 to 30 days following administration or generation of modified immune cells.
- the composition comprising at least one RNA comprising at least one cds encoding at least one antigen, or host cell modified to express the antigenis administered following administration or generation of modified immune cells targeted to an antigen, e.g., at least 1 day, such as 1 to 10 days or 1 to 5 days or 1 to 3 days following administration or generation of modified immune cells.
- the composition comprising at least one RNA comprising at least one cds encoding at least one antigen, or host cell modified to express the antigenmay be administered several times over time in constant or different time intervals, e.g., following administration or generation of modified immune cells targeted to an antigen, e.g., in time intervals of between 7 and 40 days, wherein the first administration of the composition comprising at least one RNA comprising at least one cds encoding at least one antigen, or host cell modified to express the antigen may be at least 1 day, such as 1 to 60 days or 1 to 50 days or 1 to 30 days following administration or generation of modified immune cells.
- the composition comprising at least one RNA comprising at least one cds encoding at least one antigen, or host cell modified to express the antigenmay be administered several times overtime in constant or different time intervals, e.g., following administration or generation of modified immune cells targeted to an antigen, e.g., in time intervals of between 7 and 40 days, wherein the first administration of the composition comprising at least one RNA comprising at least one cds encoding at least one antigen, or host cell modified to express the antigen may be at least one day, such as 1 to 10 days or 1 to 5 days or 1 to 3 days following administration or generation of modified immune cells.
- the compositionis administered to a subject as defined in the context of the first aspect.
- the compositionis administered to a subject via intratumoral or intramuscular administration. More preferably, the composition is administered to a subject via intramuscular administration, e.g. intramuscular injection.
- Intramuscular administratione.g. intramuscular injection
- intramuscular injectionsprovide a relatively consistent and sustained release, avoiding peaks of delivery.
- Intramuscular injectionsprovide slower absorption compared to intravenous administration and more controlled release or longer duration of action. Additionally, there is a lower risk of infiltration.
- Intravenous injectionscan carry a risk of infiltration, where the medication leaks into surrounding tissues if the vein is not properly secured.
- Some patientsmay find intramuscular injections less uncomfortable or invasive compared to intravenous infusions, which can be particularly relevant for pediatric or needle-phobic patients.
- Intramuscular administrationoften requires fewer resources and equipment compared to intravenous infusions, making it a cost-effective option in certain situations.
- the subject that has received the combination of the second aspectreceives at least one additional treatment, preferably at least one adjuvant, “classical” cancer treatment, immune checkpoint inhibitors and/or anti-cancer antibodies as described in the first aspect.
- the combinationcomprises
- compositioncomprising at least one RNA comprising at least one cds encoding at least one antigen
- modified immune cells targeted to the at least one antigenfor use in a method of treatment or prophylaxis of a disease, disorder or condition, wherein the modified immune cells are preferably administered intravenously to the subject, and wherein the composition is preferably administered intramuscularly to the subject that has received the modified immune cells.
- the RNAis formulated as further defined in paragraph “Formulation/Complexation of the RNA of the composition" (first aspect). More preferably, the RNA is formulated in LNPs as defined herein.
- the inventionrelates to a combination that comprises (i) a composition comprising at least one RNA comprising at least one cds encoding at least one antigen, wherein the RNA is formulated in LNPs as defined herein; and (ii) modified immune cells targeted to the at least one antigen, for use in a method of treatment or prophylaxis of a disease, disorder or condition, by administering the modified immune cells intravenously to the subject, and by administering the composition intramuscularly to the subject that has received the modified immune cells.
- the method, the composition, the modified immune cells, the subject, and the disease, disorder or conditionare further characterized by any one of the features of the first aspect.
- the method of treatment or prophylaxis of disease, disorder or conditionis selected from a tumor or cancer disease, disorder or condition or infectious disease, disorder or condition, preferably selected from a tumor or cancer disease, disorder or condition, more preferably selected from solid or hematologic cancer.
- modified immune cellscomprise modified T cells, preferably CAR T cells or TCRtg T cells.
- modified T cellsis performed with or without lymphodepletion prior to administration of modified T cells, preferably CAR T cells or TCRtg T cells.
- a low dose or subtherapeutic dose of modified T cellspreferably CAR T cells or TCRtg cells is administered to the subject.
- the modified immune cellsare administered intravenously.
- the CAR T cells or TCRtg cellsare targeted to an antigen associated with the disease, disorder or condition, preferably a tumor or cancer disease, disorder or condition or infectious disease, disorder or condition.
- the antigenis a tumor antigen, more preferably tumor specific antigen or tumor associated antigen, selected from mutation antigen, neoantigen, unique antigen, differentiation tumor antigen, overexpressed tumor antigen, virus tumor antigen or bacterial tumor antigen.
- the tumor antigenis selected or derived from CD19, BCMA, CD22, CD20, Mesothelin, GPC3, GD2, HER2, B7-H3, CLDNB18.2, Claudin6 (CLDN6), PSMA, CEA, EGFR, MUC1, EGFRVIII, NKG2DL, WT1, HA-1, NY-ESO-1, HPV, MAGE-A4, HBV, EBV, mutated KRAS; KK-LC-1 , MAGE-A3, MAGE-A10, AFP, MAGE-A1 , MART-1, PRAME, mutated PRAME, CD33, CD70, NKG2DL, PD-L1 , ROBO1, 5T4, DLL3, COL6A3 and/or MICA/B, preferably from Claudin 6 or PRAME.
- Claudin6Claudin6
- the composition comprising the RNAencodes at least one tumor antigen, preferably encodes at least one epitope targeted by the antigen receptor of the modified T cells, more preferably a full-length antigen comprising the antigen targeted by the antigen receptor of the modified T cells.
- the combination of the present aspectin particular the combination of (i) the composition and (ii) the modified immune cells, has advantageous effects as disclosed in the context of the first aspect (see in particular, paragraph “observed effects”).
- kits or kit of parts for use in treatment or prophylaxis of a disease, disorder or conditionfor use in treatment or prophylaxis of a disease, disorder or condition
- the present inventionrelates to a kit or kit of parts comprising (A) at least one composition as defined in the context of the first aspect; or (B) at least one combination as defined the context of the second aspect, for use in a method of treatment or prophylaxis of a disease, disorder or condition.
- the kit or kit of partscomprises instructions for use of the kit or kit of parts in a method of treatment or prophylaxis of a disease, disorder or condition.
- the kit or kit of partscomprises technical instructions providing information on administration and dosage of the components.
- the technical instructions of said kitmay contain information about administration and dosage and patient groups.
- kits, preferably kits of partsmay be applied e.g. for any of the applications or uses mentioned herein, preferably for the use of the therapeutic agents of the kit or kit of parts for treatment of cancer or a diseases, disorder, or condition related to cancer.
- the kit or kit of partsmay suitably comprise a buffer for re-constitution of lyophilized or spray-freeze dried or spray dried composition or combination.
- the kit or kit of parts as defined hereincomprises at least one syringe or application device.
- a syringe or application deviceas described in WO2022207862, claims 1 to 69, may be part of the kit.
- the kit of the inventionmay, for example, be affixed to a container which contains the composition or combination of the invention or be shipped together with a container which contains the composition or combination.
- the kitmay be shipped separately from the container with the intention that the kit or combination may be used cooperatively by the subject.
- the subjectreceives the kit or kit of parts comprising (A) at least one composition as defined in the context of the first aspect; or (B) at least one combination as defined the context of the second aspect, for use in a method of treatment or prophylaxis of a disease, disorder or condition, preferably a diseases, disorder, or condition related to cancer. 4.
- a method or preventing a disease, disorder or conditioncomprising (A) at least one composition as defined in the context of the first aspect; or (B) at least one combination as defined the context of the second aspect, for use in a method of treatment or prophylaxis of a disease, disorder or condition, preferably a diseases, disorder, or condition related to cancer. 4.
- the present inventionrelates to a method of treating or preventing a disease, disorder, or condition in a subject, comprising a step of applying or administering a composition comprising at least one RNA comprising at least one cds encoding at least one antigen, wherein the subject has received modified immune cells targeted to the at least one antigen.
- the method of treating or preventing a disease, disorder, or condition in a subjectcomprises a step of applying or administering intramuscularly a composition comprising at least one RNA comprising at least one cds encoding at least one antigen, wherein the RNA is formulated in LNPs, wherein the subject has received modified immune cells targeted to the at least one antigen.
- the methodis further characterized by any one of the features of the first aspect.
- the present inventionrelates to a method of activating or stimulating a modified or endogenous immune cell population in a subject, wherein the method comprises applying or administering at least one composition comprising at least one RNA comprising at least one cds encoding at least one antigen to the subject.
- the method of activating or stimulating a modified immune cell population in a subjectcomprises applying or administering at least one composition comprising at least one RNA comprising at least one cds encoding at least one antigen to the subject.
- the method of activating or stimulating a modified immune cell population in a subjectcomprises applying or administering intramuscularly at least one composition comprising at least one RNA comprising at least one cds encoding at least one antigen to the subject, wherein the RNA is formulated in LNPs.
- the methodis further characterized by any one of the features of the first aspect.
- the method of activating or stimulating a modified immune cell population in a subjectis defined in increasing levels of circulating modified immune cells in the subject;, inducing memory formation of modified immune cells in the subject, preferably formation of central and effector memory cells, activating modified immune cells in the subject; boosting metabolism and polyfunctionality of the modified immune cells in the subject, recruitment of crosspresenting cells, preferably DCs, to the tumor and elicits robust and potent antigen spreading, re-activating modified immune cells in the subject; maintaining the self-renewal capacity of modified immune cells in the subject, inducing higher influx into the solid tumor of modified immune cells in the subject, and/or inducing expansion of modified immune cells in the subject.
- the method of inducing an immune response in a subjectis a T cell-mediated immune response.
- the immune responseis an immune response to a target cell population or target tissue expressing an antigen.
- the target cell population or target tissueare cancer cells or is cancer tissue.
- the cancer cells or cancer tissueis solid cancer or hematologic cancer, preferably solid cancer.
- Item 1A composition comprising at least one RNA comprising at least one coding sequence encoding at least one antigen for use in a method of treatment or prophylaxis of a disease, disorder or condition, wherein the RNA is formulated in lipid nanoparticles and wherein the composition is administered intramuscularly to a subject that has received modified immune cells, preferably modified T cells, targeted to the at least one antigen.
- modified immune cellspreferably modified T cells
- Item 2The composition for use of item 1 , wherein the modified immune cells comprise autologous immune cells taken from the subject.
- Item 3The composition for use of item 1 or 2, wherein the modified immune cells are genetically modified to express an antigen receptor targeted to the antigen and/or to express the antigen, preferably modified to express an antigen receptor targeted to the antigen.
- Item 4The composition for use of item 3, wherein the antigen receptor is targeted to the antigen associated with the disease, disorder or condition.
- Item 5The composition for use of item 3 or 4, wherein the antigen receptor is a chimeric antigen receptor (CAR) or a T cell receptor (TCR), preferably a TCR.
- CARchimeric antigen receptor
- TCRT cell receptor
- Item 6The composition for use of items 1 to 5, wherein the modified immune cells are selected from tumor-infiltrating leukocytes (TILs) or modified T cells.
- TILstumor-infiltrating leukocytes
- Item 7The composition for use of item 6, wherein the modified T cells are selected from transgenic TCR (TCRtg) and/or chimeric antigen receptors (CAR) T cells.
- TCRtgtransgenic TCR
- CARchimeric antigen receptors
- Item 8The composition for use of items 1 to 7, wherein the modified immune cells are selected from modified T cells, preferably transgenic T cells (TCRtg), that are genetically modified to express an antigen receptor targeted to the antigen.
- modified T cellspreferably transgenic T cells (TCRtg)
- TCRtgtransgenic T cells
- Item 9The composition for use of any one of the preceding items, wherein the antigen is associated with the disease, disorder or condition.
- Item 10The composition for use of item 9, wherein the antigen is selected from or derived from tumor antigens, viral antigens, bacterial antigens, protozoal antigens, fungal antigens, self-antigens or allogenic antigens, preferably tumor antigens.
- the antigenis selected from or derived from tumor antigens, viral antigens, bacterial antigens, protozoal antigens, fungal antigens, self-antigens or allogenic antigens, preferably tumor antigens.
- Item 11The composition for use of item 10, wherein the tumor antigen is tumor specific antigen or tumor associated antigen, preferably selected from mutation antigen, neoantigen, unique antigen, differentiation tumor antigen, overexpressed tumor antigen, virus tumor antigen or bacterial tumor antigen.
- the tumor antigenis tumor specific antigen or tumor associated antigen, preferably selected from mutation antigen, neoantigen, unique antigen, differentiation tumor antigen, overexpressed tumor antigen, virus tumor antigen or bacterial tumor antigen.
- Item 12The composition for use of any one of the preceding items, wherein the antigen is a tumor antigen selected or derived from p53, 5T4, ART , AFP, BAGE, beta-eaten in/m, BCMA, Bcr-abL B7-H3, mutated BRAF, CAMEL, CAP-1 , CASP-8, CDC27/m, CDK4/m, CD19, CD22, CD20, CD33, CD70, CEA, mutated CTNNB1 , the cell surface proteins of the claudin family, such as CLDN6, CLDN18.2 and CLDN12, COL6A3, c-MYC, CT, Cyp-B, DAM, DLL3, EBV, ELF2M, ETV6-AML1, EGFR, EGFRvlll, EphA2, G250, GAGE, GD2, GnT-V, GPC3, Gap 100, Gp100, HAGE, HA-1 , HER-2/neu, HBV
- Item 13The composition for use of item 12, wherein the tumor antigen is selected or derived from CD19, BCMA, CD22, CD20, Mesothelin, GPC3, GD2, HER2, B7-H3, CLDN18.2, Claudin6 (CLDN6), PSMA, CEA, EGFR, MUC1, EGFRVIII, NKG2DL, WT1, HA-1 , NY-ESO-1 , HPV, MAGE-A4, HBV, EBV, mutated KRAS; KK-LC-1, MAGE-A3, MAGE-A10, AFP, MAGE-A1, MART-1, PRAME, mutated PRAME, CD33, CD70, NKG2DL, PD- L1 , ROBO1 , 5T4, DLL3, COL6A3 and/or MICA/B, preferably from CLDN6 or PRAME.
- CLDN6Claudin6
- Item 14The composition for use of any one of the preceding items, wherein the at least one encoded antigen comprises at least the epitope recognized by the antigen receptor of the modified immune cells, preferably wherein the at least one encoded antigen comprises at least the T cell epitope.
- Item 15The composition for use of any one of the preceding items, wherein the at least one encoded antigen comprises at least 1 , 2, 3, 4, 5, 6, 7, 8, 9 or 10 epitopes recognized by the antigen receptor of the modified immune cells.
- Item 16The composition for use of any one of the preceding items, wherein the disease, disorder or condition is selected from a cancer or tumor disease, disorder or condition, an infectious disease, disorder or condition, an autoimmune disease, disorder or condition, or a disease, disorder or condition associated with stem cell transplantation.
- Item 17The composition for use of any one of the preceding items, wherein the disease, disorder or condition is selected from a tumor or cancer disease, disorder or condition.
- Item 18The composition for use of item 17, wherein the cancer disease is derived from solid or hematologic tumors.
- Item 19The composition for use of any one of the preceding items, wherein the subject has received the modified immune cells by intravenous administration.
- Item 20The composition for use of any one of the preceding items, wherein the subject is a mammal, preferably a human.
- Item 21The composition for use of any one of the preceding items, wherein the subject has not received lymphodepletion within 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11 , 12, 13 or 14 days before the administration of the modified immune cells targeted to the antigen.
- Item 22The composition for use of item 21 , wherein the subject has a reduced risk of fewer, weakness, chills, loss of appetite, immune-related toxicity and cytokine-related toxicity, cytokine release syndrome, organ dysfunction, arrhythmias, renal failure, neurotoxicity, anemia, thrombocytopenia, graft failure, increased risk of infections like bacterial infections, viral infections, fungal infections, neutropenia, pancytopenia and febrile neutropenia and/or bacteremia, preferably compared to a subject that received lymphodepletion.
- Item 23The composition for use of items 1 to 20, wherein the subject has received a lymphodepletion within 1 , 2, 3, 4, 5, 6, 7, 8, 9, 10, 11 , 12, 13 or 14 days before the administration of the modified immune cells targeted to the antigen.
- Item 24The composition for use of any one of the preceding items, wherein the method does not comprise a step of treating the subject with additional cytokine or interleukin performed about 2 months after the administration of the modified immune cells targeted to the antigen.
- Item 25The composition for use of any one of the preceding items, wherein the subject received a therapeutic amount of modified immune cells, preferably modified T cells.
- Item 26The composition for of item 1 to 24, wherein the subject received a subtherapeutic amount of modified immune cells, preferably modified T cells.
- Item 27The composition for use in a method of treatment or prophylaxis of any one of the preceding items, wherein a first dose of the composition is administered 1 day to 60 days after the subject has received the modified immune cells targeted to the antigen.
- Item 28The composition for use of item 27, wherein at least one further dose of the composition is administered 1 day to 60 days after the administration of the first dose.
- Item 29The composition for use of any one of the preceding items, wherein at least 3, , 5, or more doses of the composition are administered to the subject.
- Item 30The composition for use of items 27 to 29, wherein one dose comprises between 10 g RNA and 100pg RNA, preferably less than 100pg RNA or less than 50pg RNA.
- Item 31The composition for use of any one of the preceding items, wherein intramuscular administration of the composition to the subject increases the levels of circulating modified immune cells in the subject, induces memory formation of modified immune cells in the subject, preferably formation of central and effector memory cells, activates modified immune cells in the subject; boosting metabolism and polyfunctionality of the modified immune cells in the subject, recruitment of cross-presenting cells, preferably DCs, to the tumor and elicits robust and potent antigen spreading, re-activates modified immune cells in the subject; maintains the self-renewal capacity of modified immune cells in the subject, induces higher influx into the solid tumor of modified immune cells in the subject, and/or induces expansion of modified immune cells in the subject.
- intramuscular administration of the composition to the subjectincreases the levels of circulating modified immune cells in the subject, induces memory formation of modified immune cells in the subject, preferably formation of central and effector memory cells, activates modified immune cells in the subject; boosting metabolism and polyfunctionality of the modified immune cells in the subject, recruitment of cross-
- Item 32The composition for use of any one of the preceding items, wherein intramuscular administration of the composition to the subject activates endogenous CD4 helper cells in the subject, skews the differentiation of endogenous tumor-infiltrating CD4+ T cells to a Th1 phenotype, increases the levels of circulating endogenous immune cells in the subject; induces memory formation of endogenous immune cells in the subject, preferably formation of central and effector memory cells; activates endogenous immune cells in the subject; boosting metabolism and polyfunctionality of the endogenous immune cells in the subject, recruitment of cross-presenting cells, preferably DCs, to the tumor and elicits robust and potent antigen spreading (antigen spreading supports modified T cell therapy to treat antigenically heterogeneous tumors), re-activates endogenous immune cells in the subject, maintains the self-renewal capacity of endogenous immune cells in the subject, induces higher influx into the solid tumor of endogenous immune cells in the subject, and/or induces expansion of endogenous immune cells
- Item 33The composition for use of any one of the preceding items, wherein intramuscular administration of the composition to the subject increases levels of circulating modified immune cells in the subject; induces memory formation of modified immune cells in the subject, preferably formation of central and effector memory T cells, activates modified immune cells in the subject; boosting metabolism and polyfunctionality of the modified immune cells in the subject, recruitment of cross-presenting cells, preferably DCs, to the tumor and elicits robust and potent antigen spreading, re-activates modified immune cells in the subject, maintaining self-renewal capacity of modified immune cells in the subject, induces higher influx into the solid tumor of modified immune cells in the subject, induces expansion of modified immune cells in the subject, activates endogenous CD4 helper cells in the subject, skews differentiation of endogenous tumor-infiltrating CD4+ T cells to a Th1 phenotype, increases levels of circulating endogenous immune cells in the subject, induces memory formation of endogenous immune cells in the subject, preferably formation of central and effector memory cells
- Item 34The composition for use of any one of the preceding items, wherein the LNPs comprise at least one lipid selected from at least one aggregation-reducing lipid, at least one cationic lipid or ionizable lipid, at least one neutral lipid or phospholipid, at least one steroid or steroid analogue.
- Item 35The composition for use of any one of the preceding items, wherein the LNPs comprise at least one aggregation-reducing lipid, at least one cationic lipid or ionizable lipid, at least one neutral lipid or phospholipid, and at least one steroid or steroid analogue.
- Item 36The composition for use of items 34 or 35, wherein the at least one aggregation reducing lipid is selected from a polymer conjugated lipid.
- Item 37The composition for use of item 36, wherein the polymer conjugated lipid is selected from a PEG-conjugated lipid or a PEG-free lipid.
- Item 38The composition for of items 34 to 37, wherein the at least one aggregation reducing lipid is selected from DMG-PEG 2000, C10-PEG2K, Cer8-PEG2K, or a POZ-lipid.
- Item 39The composition for use of items 34 to 38, wherein the at least one cationic lipid or ionizable lipid is selected from an amino lipid, preferably wherein the amino lipid comprises a tertiary amine group.
- Item 41The composition for use of items 34 to 40, wherein the at least one cationic lipid or ionizable lipid is selected from SM-102, SS-33/4PE-15, HEXA-C5DE-PipSS, or compound C26.
- Item 42The composition for use of items 34 to 41 , wherein the at least one neutral lipid or phospholipid is selected from DSPC, DHPC, DPhyPE, DphyPS.
- Item 43The composition for use of items 34 to 42, wherein the LNPs comprise at least one neutral lipid or phospholipid selected from DSPC, DHPC, DPhyPE and one neutral lipid or phospholipid selected from DphyPS.
- Item 44The composition for use of items 34 to 43, wherein the steroid or steroid analogue is selected from cholesterol, cholesteryl hemisuccinate (CHEMS), preferably cholesterol.
- CHEMScholesteryl hemisuccinate
- Item 45The composition for use of any one of the preceding items, wherein the LNPs comprise
- Item 46The composition for use of any one of the preceding items, wherein the LNPs comprise
- Item 47The composition for use of any one of the preceding items, wherein the LNPs comprise about 20-60% cationic or ionizable lipid, about 5-25% neutral lipid or phospholipids, about 25-55% steroid or steroid analogue, and about 0.5-15% aggregation reducing lipid.
- Item 48The composition for use of any one of the preceding items, wherein the wt/wt ratio of lipid to RNA in the LNPs is from about 10: 1 to about 60: 1.
- Item 49The composition for use of any one of the preceding items, wherein the N/P ratio of the LNPs comprising the RNA is in a range from about 1 to about 20.
- Item 50The composition for use of any one of the preceding items, wherein the LNPs have a Z-average size of less than 400nm, preferably less than 300nm, more preferably less than 200nm.
- Item 51The composition for use of any one of the preceding items, wherein the LNPs have a Z-average size in a range of about 50nm to about 200nm, preferably about 50nm to about 150nm.
- Item 52The composition for use of any one of the preceding items, wherein the LNPs have a polydispersity index (PDI) value of less than about 0.3, preferably of less than about 0.2.
- PDIpolydispersity index
- Item 53The composition for use of any one of the preceding items, wherein at least 70%, preferably at least 80%, more preferably at least 90% of the RNA of the composition is encapsulated in the LNPs.
- Item 54The composition for use of any one of the preceding items, wherein at least about 80%, 85%, 90%, 95% of the LNPs have a spherical morphology, preferably comprising a solid core or a partially solid core, and/or a lamellar or bilayer morphology.
- Item 55The composition for use of any one of the preceding items, wherein the LNPs have been generated by combining an aqueous RNA solution with an ethanolic lipid solution using a mixing means at total flow rates above 15ml/min.
- Item 56The composition for use of any one of the preceding items, wherein the LNPs do not comprise DOTMA and/or DOPE.
- Item 57The composition for use of any one of the preceding items, wherein the LNPs exhibit a zeta potential in the range of -50 mV to +50 mV, preferably in the range of -25 mV to +25 mV, more preferably in the range of -10 mV to +10 mV, most preferably in the range of -5 mV to +5 mV.
- Item 58The composition for use of any one of the preceding items, wherein the at least one cds encodes at least one antigen that is selected from or derived from tumor antigens, viral antigens, bacterial antigens, protozoal antigens, fungal antigens, self-antigens or allogenic antigens, preferably tumor antigens.
- Item 59The composition for use of item 58, wherein the tumor antigen is a tumor-specific antigen or tumor associated antigen that is preferably selected from p53, 5T4, ART-4, AFP, BAGE, beta-eaten in/m, BCMA, Bcr-abL B7- H3, mutated BRAF, CAMEL, CAP-1, CASP-8, CDC27/m, CDK4/m, CD19, CD22, CD20, CD33, CD70, CEA, mutated CTNNB1 , the cell surface proteins of the claudin family, such as Claudin6 (CLDN6), CLDN18.2 and CLDN12, COL6A3, c-MYC, CT, Cyp-B, DAM, DLL3, EBV, ELF2M, ETV6-AML1, EGFR, EGFRvlll, EphA2, G250, GAGE, GD2, GnT-V, GPC3, Gap 100, Gp100, HAGE, HA
- Item 60The composition for use of item 58 or 59, wherein the tumor antigen or tumor associated antigen is selected from CD19, BCMA, CD22, CD20, Mesothelin, GPC3, GD2, HER2, B7-H3, CLDN18.2, CLDN6, PSMA, CEA, EGFR, MUC1, EGFRVIII, NKG2DL, WT1, HA-1, NY-ESO-1 , HPV, MAGE-A4, HBV, EBV, mutated KRAS; KK-LC-1, MAGE-A3, MAGE-A10, AFP, MAGE-A1 , MART-1, PRAME, mutated PRAME, CD33, CD70, NKG2DL, PD-L1 , ROBO1 , 5T4, DLL3, COL6A3 and/or MICA/B, preferably from CLDN6 or PRAME.
- the tumor antigen or tumor associated antigenis selected from CD19, BCMA, CD22, CD20, Mesothelin, GPC
- Item 61The composition for use of any one of the preceding items, wherein the at least one RNA comprises at least one codon modified cds, preferably wherein the codon modified cds is selected from a C maximized cds, a CAI maximized cds, human codon usage adapted cds, a G/C content modified cds, and a G/C optimized cds, or any combination thereof.
- Item 63The composition for use of any one of the preceding items, wherein the at least one RNA comprises at least one untranslated region (UTR), preferably selected from at least one heterologous 5’-UTR and/or at least one heterologous 3-UTR.
- UTRuntranslated region
- Item 64The composition for use of item 63, wherein the at least one 3-UTR comprises or consists of a nucleic acid sequence selected or from a 3-UTR of a gene selected from PSMB3, AES-12S, ALB7, alpha-globin (HBA1 , HBA2), ANXA4, beta-globin (HBB), CASP1, COX6B1 , FIG4, GH1, GNAS, NDUFA1, RPS9, SLC7A3, TUBB4B, or from a homolog, a fragment or a variant of any one of these genes, preferably wherein the at least one 3-UTR comprises or consists of a nucleic acid sequence being identical or at least 70%, 80%, 85%, 86%, 87%, 88%, 89%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identical to SEQ ID NOs: 134-191 , or a fragment or a variant of any of these.
- Item 65The composition for use of items 63 to 64, wherein the at least one 5-UTR comprises or consists of a nucleic acid sequence selected or from a 5-UTR of a gene selected from HSD17B4, AIG1 , alpha-globin (HBA1 , HBA2), ASAH1 , ATP5A1, COX6C, DPYSL2, HHV5, MDR, MP68, NDUFA4, NOSIP, RPL31 , RPL32, RPL35A, SLC7A3, synthetic origin, TUBB4B, UBQLN2, or from a homolog, a fragment or variant of any one of these genes, preferably wherein the at least one 5-UTR comprises or consists of a nucleic acid sequence being identical or at least 70%, 80%, 85%, 86%, 87%, 88%, 89%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identical to SEQ ID NOs: 80-133,
- Item 66The composition for use of items 63 to 65, wherein the at least one 5-UTR is selected from HSD17B4 and the at least one 3-UTR is selected from PSMB3.
- Item 67The composition for use of any one of the preceding items, wherein the at least one RNA is an artificial RNA and/or an isolated RNA.
- Item 68The composition for use of any one of the preceding items, wherein the RNA is selected from an mRNA, a circular RNA, a replicon RNA, or a viral RNA.
- Item 69The composition for use of any one of the preceding items, wherein the RNA is an mRNA.
- Item 70The composition for use of any one of the preceding items, wherein the RNA comprises at least one poly(A) sequence, preferably wherein the at least one poly(A) sequence comprises about 40 to about 500 adenosine nucleotides.
- Item 71The composition for use of item 70, wherein the at least one poly(A) sequence comprises about 60 to about 150 adenosine nucleotides, preferably about 100 adenosine nucleotides.
- Item 72The composition for use of item 70 or 71 , wherein the at least one poly(A) sequence is located at the 3’ terminus, optionally, wherein the 3’ terminal nucleotide is an adenosine.
- Item 73The composition for use of any one of the preceding items, wherein the RNA comprises at least one poly(C) sequence and/or at least one miRNA binding site and/or at least one histone stem-loop sequence.
- Item 74The composition for use of any one of the preceding items, wherein the at least one RNA comprises at least one modified nucleotide, preferably at least one modified nucleotide selected from pseudouridine (i ) or N1- methylpseudouridine (ml ip) , more preferably wherein the at least one modified nucleotide is N1- methylpseudouridine.
- the at least one RNAcomprises at least one modified nucleotide, preferably at least one modified nucleotide selected from pseudouridine (i ) or N1- methylpseudouridine (ml ip) , more preferably wherein the at least one modified nucleotide is N1- methylpseudouridine.
- Item 75The composition for use of any one of the preceding items, wherein the at least one RNA is a modified RNA wherein each uracil is substituted by a modified nucleotide.
- Item 76The composition for use of item 1 to 73, wherein the at least one RNA does not comprise a modified nucleotide.
- Item 77The composition for use of any one of the preceding items, wherein the RNA comprises a 5’ -cap structure.
- Item 78The composition for use of item 77, wherein the 5’-cap structure is selected from a cap1 structure or a modified cap1 structure.
- Item 79The composition for use of any one of the preceding items, wherein the RNA is an in vitro transcribed RNA.
- Item 80The composition for use of any one of the preceding items, wherein the RNA is a purified RNA that has preferably been purified by at least one step of RP-HPLC, AEX, SEC, hydroxyapatite chromatography, TFF, filtration, precipitation, core-bead flow through chromatography, oligo(dT) purification, cellulose-based purification, or any combination thereof.
- Item 81The composition for use of any one of the preceding items, wherein the RNA comprises the following sequence elements, preferably in 5’- to 3’-direction: A) a 5'-cap structure, preferably a cap1 structure;
- a 5'-UTRpreferably selected from a 5’-UTR of a HSD17B4 gene
- a 3’-UTRpreferably selected from a 3’-UTR of a PSMB3 gene
- Item 82A combination comprising:
- compositioncomprising at least one RNA comprising at least one cds encoding at least one antigen, wherein the RNA is formulated in LNRs;
- modified immune cells targeted to the at least one antigenfor use in a method of treatment or prophylaxis of a disease, disorder or condition, wherein the modified immune cells are the administered intravenously to the subject, and wherein the composition is administered intramuscularly to the subject that has received the modified immune cells.
- Item 83The combination for use of item 82, wherein the method, the composition, the modified immune cells, the subject, and the disease, disorder or condition are further characterized by any one of the features of items 1 to 81.
- Item 84A kit or kit or parts comprising (A) at least one composition as defined in any one of items 1 to 81 ; or (B) at least one combination as defined items 82 or 83, for use in a method of treatment or prophylaxis of a disease, disorder or condition, wherein the kit or kit of parts optionally comprises instructions for use of the kit or kit of parts in a method of treatment or prophylaxis of a disease, disorder or condition.
- Item 85A method of treating or preventing a disease, disorder, or condition in a subject, comprising a step of applying or administering intramuscularly a composition comprising at least one RNA comprising at least one cds encoding at least one antigen, wherein the RNA is formulated in LNPs, wherein the subject has received modified immune cells targeted to the at least one antigen and wherein the method is further characterized by any one of the features of items 1 to 81.
- Item 86A method of activating or stimulating a modified immune cell population in a subject, wherein the method comprises applying or administering intramuscularly at least one composition comprising at least one RNA comprising at least one cds encoding at least one antigen to the subject, wherein the RNA is formulated in LNPs, and wherein the method is further characterized by any one of the features of items 1 to 81.
- Figure 1shows the tumor volume in C57BL/6 CD45.1 mice after injection of MC38-Ova tumor cells over time. Solid lines represent tumor growth in mice treated with 1 pg LNP formulated mRNA vaccine (encoding full-length Ovalbumin) administered intramuscularly. Dashed lines show tumor growth of the untreated control group. Further details are provided in Example 2.1.
- Figure 2shows the tumor volume in OT-1 CD45.1 mice after injection of MC38-Ova tumor cells over time, treated with a low dose of OT-I CD8 T cells derived from OT-I CD45.2 mice and LNP formulated mRNA vaccine administered i.m. at different time points (further details see Table 3).
- Figure 2A and 2Bshow the comparison of untreated control group ( Figure 2A, dashed lines) and ACT alone ( Figure 2B, dashed lines) to group 3 that have received a low dose of ACT and an early mRNA vaccination (solid lines). The combination of early mRNA vaccination on day 1 after T cell transfer led to complete tumor regression and long-lasting anti-tumor efficacy.
- Figure 2C and Figure 2Dshow the comparison of untreated control group (Figure 2C, dashed lines) and ACT alone ( Figure 2D, dashed lines) to group 4 that have received a low dose of ACT and a late mRNA vaccination (solid lines).
- ACTuntreated control group
- Figure 2Ddashed lines
- the combination with early and late vaccinationled to 10/10 and 7/10 complete responders, respectively. Further details are provided in Example 2.3.
- Figure 3shows the tumor volume in OT-1 CD45.1 mice after injection of MC38-Ova tumor cells on day 10 ( Figure 3A) and day 41 ( Figure 3B) after ACT, treated with high and low dose of OT-I CD8 T cells of OT-I CD45.2 mice with and without combination of mRNA vaccine formulated in LNPs administered i.m. at different time points (further details see Table 4).
- the combination therapy of low dose of ACT and early LNP formulated mRNA vaccinationshowed the same anti-tumor efficacy as compared to the high dose of ACT at day 10 ( Figure 3A) and day 41 ( Figure 3E3). Complete tumor regression was observed in both groups at day 41. Further details are provided in Example 3.1.
- Figure 4shows survival data of OT -1 CD45.1 mice after injection of MC38-Ova tumor cells over time, treated with high and low dose of OT-I CD8 T cells derived from OT-I CD45.2 mice with and without combination of LNP formulated mRNA vaccine administered i.m. at different time points (further details see Table 4). All mice that received high ACT doses (group 3) and the mice that received the combination therapy of low ACT dose with early vaccination of LNP formulated mRNA vaccine (group 5) showed long-term survival over 54 days after ACT. Further details are provided in Example 3.2.
- Figure 5shows the number of transferred antigen specific OT-I CD8 T cells derived from OT-I CD45.2 donor mice in the periphery of MC38-Ova tumor bearing CD45.1 recipient mice at day 1 , 11 , 40, 54 and 59 after tumor cell injection.
- the micewere treated with high and low dose of OT-I CD8 T cells with and without combination of LNP formulated mRNA vaccine administered i.m. at different time points. All groups, except of control group 1 , received an mRNA vaccination on day 54 (further details see Table 5). In all groups mRNA vaccination resulted in prominent expansion of transferred antigen specific CD8 T cells in the periphery. Further details are provided in Ex. 4.1.
- Figure 6shows the number of circulating antigen specific OT-I CD8 memory T cells of OT-I CD45.2 mice at day 40 after ACT in OT-I CD45.1 mice injected with MC38-Ova tumor cells. The mice were treated with high and low dose of OT-I CD8 T cells derived from OT-I CD45.2 donor mice with and without combination of mRNA vaccine formulated in LNPs administered i.m. at different time points (further details see T able 6).
- Figure 6Ashows higher frequencies of antigen specific CD8 T cells in the periphery in both groups that received an mRNA vaccination.
- Figure 6Bshows the frequency of effector memory T cells (TEM) and central memory T cells (TCM) in the periphery.
- TEMeffector memory T cells
- TCMcentral memory T cells
- FIG. 6Cshows the frequency of memory precursor cells (MPECs) and short-lived effector cells (SLECs) in the periphery.
- MPECsmemory precursor cells
- SLECsshort-lived effector cells
- Figure ?shows the PD1 , TIM3, and SLAMF6 expression at day 40 after ACT of the transferred antigen specific OT-I CD8 T cells derived from OT-I CD45.2 donor mice in the periphery of CD45.1 recipient mice after injection of MC38-Ova tumor cells.
- the micewere treated with high and low dose of OT-I CD8 T cells of OT-I CD45.2 mice with and without combination of LNP formulated mRNA vaccine administered i.m. at different time points (further details see Table 7). Both groups 4 and 5 that received an mRNA vaccination showed high populations of activated (PD1 and TIM3 positive) transferred T cells with the capacity for selfrenewal (SLAMF6 positive). Further details are provided in Example 4.3.
- Figure 8shows antigen specific (TEWTSSNVMEERKIKV; SEQ ID NO: 192) endogenous CD4 T cell population at day 59. Based on 90000 viable splenocytes, counts of the aforementioned population were calculated and are presented in the graph. All mice received a lymphodepletion prior to ACT therapy. The mice were treated with high and low dose of OT-I CD8 T cells derived from OT-I CD45.2 mice with combination of LNP formulated mRNA vaccine administered i.m. at different time points. Group 1 and 2 received only one late vaccination on day 54, group 3 and 4 received the vaccination on earlier time points (further details see Table 8). Mice that have received the composition to an earlier time point after ACT showed high antigen specific and polyfunctional (double positive IFN-y and TNF-a) endogenous CD4 helper T cell population. Further details are provided in Ex. 5.1.
- Figure 9shows the tumor volume of MC38-Ova tumors in C57BL/6 CD45.1 mice over 11 days after ACT.
- Mice displaying tumors of approximately 288mm 3were treated with 100mg/kg Cyclophoshamide intraperitoneally on Day -1 , with 0.1x10® OT-I CD8 T cells derived from OT-I CD45.2 mice intravenously on Day 0 and with 1 pg LNP-formulated mRNA encoding full length ovalbumin intramuscularly on Day 1 and 8 as indicated.
- Asterisksshow statistical significance levels for comparisons of the indicated groups. Mean values and SEM are depicted.
- ACTadoptive T cell therapy
- CpCyclophosphamide
- w/with; w/o, without.
- Figure 10shows the amount of transferred antigen-specific CD8 T cells found in the peripheral blood at Day 11 after ACT in mice treated as described in the legend of Figure 9. Mean and SEM of the amount of transferred antigen-specific CD8 T cells depicted as CD8 + Thy1.2 + CD45.2 + H-2KbTetra + cells are shown. Further details are provided in Example 6 and 8. While in the ACT alone group, i.e. without vaccination, almost no transferred CD8 T cells could be detected in the peripheral blood, the indicated group including both ACT and vaccination displayed a very high amount of transferred CD8 T cells suggesting increased survival and/or proliferation of those cells. Further details are provided in Example 6 and 8. ACT, adoptive T cell therapy; Cp, Cyclophosphamide; Vac, vaccination.
- Figure 11shows the amount of transferred antigen-specific CD8 T cells found in the spleen (left) and the spleen weight (right) at Day 11 after ACT in mice treated as described in the legend of Figure 9. Mean and SEM of the amount of transferred antigen-specific CD8 T cells depicted as CD8 + Thy 1 ,2 + CD45.2 + H-2KbT etra + cells and of spleen weights are shown. Asterisks show statistical significance levels for the indicated groups. While in the ACT alone group, i.e. without vaccination, almost no transferred CD8 T cells could be detected in the spleen, the indicated group including both ACT and vaccination displayed a very high amount of transferred CD8 T cells suggesting increased survival and/or proliferation of those cells. The high amount of transferred CD8 T cells in the combination group was further reflected by increased spleen weights in this group. Further details are provided in Example 6 and 8. ACT, adoptive T cell therapy; Cp, Cyclophosphamide; Vac, vaccination
- Figure 12shows the amount of transferred antigen-specific CD8 T cells in the tumor (top) as well as their cytotoxicity (bottom, left) and activation (bottom, right) at Day 11 after ACT in in mice treated as described in the legend of Figure 9.
- Mean and SEM of the amount of transferred antigen-specific CD8 T cells depicted as CD8 + Thy1 ,2 + CD45.2 + H-2KbTetra + cells as well as their cytotoxicity (percentage of GZMB + cells among CD8 + Thy1 ,2 + CD45.2 + T cells) and activation (percentage of CD25 + cells among CD8 + Thy1 ,2 + CD45.2 + T cells)are shown.
- Asterisksshow statistical significance levels for the indicated groups.
- the present exampleprovides methods of obtaining the RNA of the invention as well as methods of generating composition of the invention comprising RNA formulated in lipid-based carriers.
- Example 1.1Preparation of DNA templates for in vitro transcription of RNA molecules
- DNA sequences encoding proteins for use in therapywere prepared and used for subsequent RNA in vitro transcription reactions.
- Some DNA sequenceswere prepared by modifying the wild type or reference encoding DNA sequences by introducing a G/C optimized cds for stabilization and expression optimization. Sequences were introduced into a pUC derived DNA vector to comprise stabilizing heterologous UTR sequences and a stretch of adenosines and optionally a histone stem-loop (hSL).
- the obtained plasmid DNA templateswere transformed and propagated in bacteria using common protocols known in the art. Eventually, the plasmid DNA templates were extracted, purified, and linearized using a restriction enzyme.
- RNA in vitro transcriptionIVT
- T7 RNA polymeraseT7 RNA polymerase in the presence of a sequence optimized nucleotide mixture (ATP/GTP/CTP/UTP) or as an alternative with N1- methylpseudouridine (m1i ) and cap analog: m7G(5’)ppp(5’)(2’OMeA)pG; TriLink (cap1), under suitable buffer conditions.
- RNA IVT reactionwas subjected to purification steps comprising RP-HPLC.
- the herein used RNA constructis provided in Table 1.
- RNA construct used in Examples Example 1.3Preparation of lipid-based carriers encapsulating the RNA construct:
- An ethanolic lipid solutionwas prepared by solubilizing a cationic lipid, a neutral lipid, cholesterol, and an aggregation reducing lipid in ethanol.
- An aqueous RNA solutionwas prepared by adjusting the purified RNA (obtained according to Example 1.2) to a target concentration in a citrate or acetate buffer.
- Lipid-based carrierswere prepared essentially according to the procedures described in WO2015199952, W02017004143 and WO2017075531.
- pumpswere used to combine the ethanolic lipid solution with a flow rate F1 and the RNA aqueous solution with a flow rate F2 at a ratio of about 1 :5 to 1 :3 (vol/vol) in a T-piece or a microfluidics system.
- F1 and/or F2were adjusted to flow rates above 15ml/min to allow the formation of LNPs encapsulating the RNA that have a Z-average size in a range from about 50nm to about 120nm.
- the ethanolwas removed. After clarifying filtration, the filtrate was adjusted to a desired concentration. Subsequently, the resulting formulation was filtered through sterilizing filters to reduce bioburden as used for in vivo studies essentially according to Example 2 to Example 10.
- Example 2Analysis of tumor growth upon single agent treatment with an LNP-formulated and i.m. administered vaccine or with ACT in comparison to combination treatment including the identification of the optimal time point for vaccination after ACT
- the goal of the experimentwas to analyse tumor growth upon single agent treatment with an LNP-formulated and i.m. administered vaccine or with ACT in comparison to combination treatment including the identification of the optimal time point for vaccination after ACT. Additionally, the experiment demonstrated the anti-tumor efficacy in vivo of the combination of LNP formulated mRNA vaccination encoding full length ovalbumin with transferred T cells. Full length antigens induce better T cell responses, cell activation and proliferation (transferred CD8 and endogenous CD4 T cell) compared to RNA encoding only the T cell epitope.
- primed CD8 T cells specific for the ovalbumin epitope SIINFEKL(SEQ ID NO: 193) were isolated from OT-I CD45.2 mice and transferred into female C57BL/6 CD45.1 mice, inoculated subcutaneously with OVA- expressing MC38 cancer cells (murine colon cancer).
- female C57BL/6 CD45.1(abbreviated to CD45.1) mice were inoculated subcutaneously in the right rear flank region with 1x10 s MC-38-OVA cells for tumor development on Day -5.
- a randomization (10 mice/group)was performed on Day 0 (at that time the mean tumor volume reached approximately 28mm 3 ). Mice were treated as indicated in the respective Examples 2-5. Cyclophosphamide (Selleck; 2mg per mouse) was administrated intraperitoneally on Day -1 for all groups receiving ACT in the course of the experiment. 2x10 6 or 0.1x10® OT-I cells were injected i.v. on Day 0 where indicated.
- micewere vaccinated intramuscularly (m. tibialis) with 1 pg LNP-formulated mRNA encoding full length ovalbumin (SEQ ID NO: 68) on the indicated days.
- the date of ACT injectionwas denoted as Day 0.
- OT-I cells in this contextwere generated as follows. 8 female OT-I C57BL/6 CD45.2 (abbreviated to OT-I CD45.2) mice were intraperitoneally injected Ovalbumin (Sigma; 0.5pg/mouse) on Day -2. On Day 0, spleens were collected and mouse CD8+ T cells isolated using the EasySepTM Mouse CD8+ T Cell Isolation Kit (StemCell Technologies; CAT#19853) according to the manufacturer’s protocol and standard procedures in the art.
- Example 2.1Analysis of tumor growth upon single agent treatment with an LNP-formulated and i.m. administered vaccine
- Example 2.1The goal of Example 2.1 was to analyse the tumor growth of Ovalbumin-expressing MC38 tumors upon single agent treatment with an LNP-formulated and intramuscularly administered vaccine.
- mice10 female C57BL/6 CD45.1 mice per group were injected subcutaneously with 1x106 MC38-OVA tumor cells for tumor development. After tumors reached a mean tumor volume of approximately 28mm 3 , one group of the mice was vaccinated i.m. once per week for 3 weeks (Day 1 , 8, 15) with 1 pg LNP formulated mRNA encoding full length ovalbumin. Tumor growth was monitored over 52 days. Further experimental details are provided above in the introduction of Example 2.
- the LNP-formulated and i.m. administered mRNA vaccineled to a strong delay and reduction of tumor growth (Figure 1 ) resulting in 5/10 complete responders displaying no detectable tumor at the end of the observation period.
- the dose of 1 pg LNP formulated mRNAwas used in all further mouse Examples.
- Example 2.2Effect of different doses of transferred cells for ACT on tumour growth
- miceFemale C57BL/6 CD45.1 mice (10 mice/group) were injected subcutaneously with 1x10 6 MC38-OVA tumor cells for tumor development. After tumors reached a mean tumor volume of approximately 28mm 3 , mice received different doses (Table 2, group 2 and 3) of OT-I CD8 T cells derived from OT-I CD45.2 donor mice, primed before with ovalbumin. T umor growth was monitored over 52 days. Further experimental details are provided above in the introduction of Example 2.
- the high dose of 2x10 6 OT-I transferred CD8 T cellsresulted in 10/10 complete responders with no re-growth of the tumor.
- Group 3 with a lower dose of 0.1x10® of transferred OT-I CD8 T cellsshowed 6/10 complete responders displaying no detectable tumor at the end of the observation period.
- Example 2.3Identification of optimal time point for vaccination after ACT
- the goal of the analysiswas to identify an optimal time point after T cell transfer for the vaccination with an LNP- formulated and i.m. administered mRNA vaccine to induce enhanced anti-tumor efficacy of low-dose transferred T cells.
- miceFemale C57BL/6 CD45.1 mice (10 per group) were injected subcutaneously with 1 x10® MC38- OVA tumor cells for tumor development. After tumors reached a mean tumor volume of approximately 28mm 3 , group 2, 3 and 4 (see Table 3) received 0.1x10® OT-I CD8 CD45.2 T cells.
- group 3 and 4received 0.1x10® OT-I CD8 CD45.2 T cells.
- group 4received the first vaccination i.m. on day 1 (group 3) and the other group on day 8 (group 4). T umor growth was observed for 52 days. Further experimental details are provided above in the introduction of Example 2.
- ACTbenefit from early i.m. administration of LNP formulated RNA vaccines.
- the low dose of the transferred T cellswas boosted to mediate long-lasting anti-tumor efficacy, to the same amount of complete responders as observed for the high dose ACT (10/10).
- the quantity of transferred T cellsis very often limited in the clinic due to increased probability of systemic toxicity, e.g. acute cytokine release syndrome, autoimmune complications and off-target toxicities.
- a mRNA vaccinationmay enable a reduction of the number of transferred T cells to reduce adverse side effects associated with ACT without affecting therapeutic effectiveness.
- one T cell transfermay be enough for therapeutic efficacy, which reduces manufacturing costs and patient suffering (avoid repeated transfers). Companies need also to produce less autologous T cells for transfer which may result in lower manufacturing costs.
- Example 3Complete tumor regression and long-term survival with combination therapy of low ACT dose and early mRNA vaccination
- the goal of the analysiswas to show that an LNP formulated and i.m. administered mRNA vaccine is able to increase the efficacy of low and potentially suboptimal doses of transferred T cells to the same level as a high dose of transferred T cells.
- miceFemale C57BL/6 CD45.1 mice (10 mice per group) were injected subcutaneously with 1 x10 6 MC38- OVA tumor cells for tumor development. After tumors reached a mean tumor volume of approximately 28mm 3 , group 4, 5 and 6 received low dose of 0.1x10 s OT-I CD8 CD45.2 T cells and group 3 received the high dose of 2x10® OT-I CD8 CD45.2 T cells derived from OT-I CD45.2 donor mice. Group 5 received an early first intramuscular mRNA vaccination on day 1 after ACT and group 6 a late first mRNA vaccination on day 8. The control group 1 remained untreated whereas group 2 received an early first mRNA vaccination without ACT. Further experimental details are provided above in the introduction of Example 2.
- Example 3.1Anti-tumor efficacy of transferred T cells boosted with mRNA vaccine
- T umor volumewas measured at day 10 and day 41.
- the late vaccinated group 6may benefit in the long-term efficacy (day 41) compared to low ACT dose alone.
- Example 3.2Long-term survival for combination therapy of low dose of transferred T cells boosted with mRNA vaccine
- mice receiving anti-tumoral T cells in low dosescould clearly benefit from early mRNA vaccination.
- the combinationled to complete tumor regression and long-term survival in all individuals.
- T cellsmay better retain their in vivo proliferation potential and antitumor efficacy.
- Example 4Analysis of transferred T cell population boosted with the mRNA vaccine
- Goal of the analysiswas to evaluate the population of transferred T cells when combined with i.m. administered LNP formulated mRNA vaccine.
- the amount of circulating transferred antigen specific T cells and their memory- and activation statusare important parameters influencing the clinical outcome for the subjects.
- Example 4.1Analysis of adoptively transferred antigen-specific T cells in the periphery
- the goal of the analysiswas to evaluate the impact of mRNA vaccination on the number of transferred antigen specific T cells in the periphery.
- Antigen specific T cells in the peripheryare circulating in the body and are able to infiltrate tumors and metastases in different body compartments.
- the experimentwas performed as described in Example 3. All groups received an mRNA vaccination on day 54 (see Table 5). Blood samples were collected at day 1 , day 11 , day 40, day 54 and day 59 and transferred T cell populations in the periphery were analyzed by FACS.
- mouse PBMCswere stained using a standard protocol including the following steps: Fc-block (BD Biosciences), surface staining, red blood cell lysis (Bio-gems) and fixation (eBiosciences). Before acquiring the samples with the flow cytometer, counting beads (123count eBeads, eBiosciences) were added to each sample, which enabled the exact calculation of the cell count. The expression of specific markers as well as live/dead (L/D) discrimination of PBMCs were determined with fluorescent coupled antibodies or respective dyes (CD45.2:Biolegend, CD8:Biolegend, L/D:eBioscience).
- T ransferred T cells of group 4 and 5were successfully boosted 39 days after the last vaccination.
- the transferred T cells of group 2 (high dose of ACT) and group 3 (low dose of ACT)were boosted successfully with one late vaccination on day 54.
- Example 4.2Analysis of circulating memory T cell population
- the goal of the analysiswas to evaluate circulating memory T cells in the transferred T cell population.
- Memory T cellshave an important role in long lasting anti-tumor response and are in this setting mainly responsible for the protection against recurrence of the cancer.
- the circulating memory T cell populationis generally divided into two subsets, effector memory T cells (TEM) and central memory T cells (TCM). Effector memory CD8 T cells (TEM) persist and can react and expand fast to antigen (re)exposure. Durable immunosurveillance is potentially due to memory precursor cells (MPECs) of transferred T cells. MPEGs are considered precursors for long-lived memory pool and SLECs are short-lived effector cells. MPECs survive during the T cell contraction phase and form memory T cells for long-term protective immunity.
- MPECsmemory precursor cells
- the experimentwas performed as described in Example 3.
- the quantity of transferred antigen specific CD8 T cells, the frequency of transferred antigen specific CD8 TE and TC , and the frequencies of MPEC and SLECs in the peripherywere analysed on day 40.
- mouse PBMCswere stained using a standard protocol including the following steps: Fc-block (BD Biosciences), surface staining, red blood cell lyses (Bio-gems) and fixation (eBiosciences). Before acquiring the samples with the flow cytometer, counting beads (123count eBeads, eBiosciences) were added to each sample, which enabled the exact calculation of the cell count.
- the goal of the experimentwas to analyse the phenotype of the transferred antigen specific OT -I CD8 T cells.
- TILstumor infiltrating lymphocytes
- This subset of T cellshas self-renewal capacity. This self-renewal capacity can be demonstrated by SLAMF6 expression (coexpressed with transcription factor TCF1). Terminally differentiated cells without self-renewal capacity are SLAMF6 negative. Strongly activated T cells are PD1 and Tim3 positive.
- PBMCswere stained using a standard protocol including the following steps: Fc-block (BD Biosciences), surface staining, red blood cell lyses (Bio-gems) and fixation (eBiosciences). Before acquiring the samples with the flow cytometer, counting beads (123count eBeads, eBiosciences) were added to each sample, which enabled the exact calculation of the cell count. The expression of specific markers as well as live/dead (L/D) discrimination of PBMCs were determined with fluorescent coupled antibodies or respective dyes (Slamf6/Ly108:Biolegend, CD45.2:Biolegend, CD8:Biolegend, PD-1:Biolegend, L7D:eBioscience, Tim3:Biolegend).
- Table 7Study groups used in Example 4.3 Both groups that received a mRNA vaccination (group 4 and 5) showed high populations of activated (PD1 and TIM3 positive) transferred T cells with the capacity for self-renewal (SLAMF6 positive) on day 11 ( Figure 7).
- This T cell population(PD-1+; TIM3+; SLAMF6+) reflects an intermediate stage between progenitor-stem-like exhausted T cells (PD-1+; TIM3-; SLAMF6+) and terminally exhausted/terminally differentiated T cells (PD-1+; TIM3+; SLAMF6-) and act as critical resource for maintaining T cell immunity in chronic viral infections and cancer.
- TEM cellsexpress in general higher levels of receptors responsible for migration to inflamed tissues and have a stronger immediate effector function than TOM cells. High frequencies of TEM cells, as seen with combination therapy, could be beneficial for the patients in the clinic. This population of T cells can react quickly against a recurrence of tumor cells and metastasis. Vaccination with LNP formulated mRNA shifted differentiation of transferred T cells to PD-1+; TIM3+; SLAMF6+ cells.
- Example 5Analysis of endogenous T cell population after RNA vaccination with lymphodepletion prior to cell transfer
- the goal of the analysiswas to evaluate the effects of LNP formulated mRNA vaccination (i.m.) on the endogenous T cell population that remained in the subject after chemotherapeutic lymphodepletion prior to the ACT therapy.
- T cells of the spleenwere isolated at day 59.
- the spleenswere harvested, dissociated and erythrocytes were lysed with a red blood cell (ACK) lysing buffer followed by washing steps and counting.
- 2.5x10® cells/wellwere seeded to a 96-well plate and stimulated with both peptides SIINFEKL (SEQ ID NO: 193) and TEWTSSNVMEERKIKV (SEQ ID NO: 192) (each 5pg/ml) and anti-CD28 antibody (2.5pg/ml) for 6 h at 37°C.
- BFA(1 Opg/ml) was added to each well to allow accumulation of cytokines intracellularly.
- Fc-Blocksurface staining, fixation, permeabilization und intracellular staining (CD45.1 :Biolegend, CD45.2:Biolegend, CD8:Biolegend, CD4:Biolegend, INFgamma:eBioscience, TNFalpha:Biolegend, L/D:eBioscience).
- Example 5.1Analysis of endogenous CD4 positive T cell population after RNA vaccination in mice lymphodepleted prior to T cell transfer
- CD4 T helper cellsplay a crucial role in the immune response.
- a positive effect of the LNP formulated RNA vaccination (i.m.) on the endogenous CD4 T helper cell population of the subject after receiving a lymphodepletion therapymay support the therapeutic effect of the ACT therapy.
- Mice vaccinated with an early or late first vaccination after ACT therapy(vaccination after day 1 or day 8, respectively) showed increased amounts of antigen specific and polyfunctional (double positive IFN-y and TNFa) endogenous CD4 helper T cells compared to the groups (group 1 and group 2) that received only one vaccination at day 54 ( Figure 8).
- Example 5.2Analysis of endogenous CD8 T cell population after RNA vaccination in mice lymphodepleted prior to T cell transfer
- Endogenous CD8 T cellsrecognizing e.g. tumor antigens can support the transferred modified immune cells in their therapeutic effect.
- the mRNA encoded full length antigenscomprise the targeted T cell epitope/antigen of the modified transferred cells. Additionally, the mRNA encoded full-length antigen includes several T cell epitopes for the endogenous T cell population.
- the RNA compositioncan stimulate and enhance endogenous T cell populations directed to antigens or epitopes other than the transferred modified cells. Accordingly, it is advantageous to encode full length antigens, or at least larger fragments of the antigen.
- mice i.m. vaccinated with the LNP formulated RNAshowed antigen specific and polyfunctional (double positive IFN-y and TNFa) endogenous CD8 T cells.
- Activated endogenous CD4 helper T cellscan be beneficial for inducing an immune response and secures a long-term memory response with a broad repertoire of antigen specificity. Moreover, it can improve the survival of transferred modified T cells and development of memory T cell pool.
- Example 6Further in vivo analyes with focus on mode-of-action
- the goal of the experimentwas to further investigate ACT in combination with an LNP-formulated and i.m. administered mRNA vaccine in vivo, in particular its mode-of-action.
- the OT-I mouse model described abovewas modified, amongst others to allow more detailed analyses of modified CD8 T cells in different compartments such as the tumor, spleen and blood. The analyses furthermore focussed on low-dose ACT.
- micewere inoculated subcutaneously in the right rear flank region with 1x10 s MC-38-OVA cells for tumor development.
- a randomizationwas performed on Day -1 when the mean tumor volume reached approximately 288mm 3 .
- 60 micewere enrolled in the study and randomly allocated to 6 study groups, with 10 mice per group. Mice were treated as indicated in Table 9.
- Cyclophosphamide(Selleck; 10Omg/kg per mouse) was administrated intraperitoneally on Day -1.
- OT-I cellswere injected i.v. on Day 0.
- Micewere vaccinated intramuscularly (m. tibialis) with 1pg LNP-formulated mRNA encoding full length ovalbumin (SEQ ID NO: 68) on Day 1 and 8. The date of ACT injection was denoted as Day 0.
- OT-I cells in this contextwere generated as follows. 3 female OT-I C57BL/6 CD45.2 (abbreviated to OT-I CD45.2) mice were intraperitoneally injected Ovalbumin (Sigma; 0.5pg/mouse) on Day -2. On Day 0, spleens were collected and mouse CD8+ T cells isolated using the EasySepTM Mouse CD8+ T Cell Isolation Kit (StemCell Technologies; CAT#19853) according to the manufacturers protocol and standard procedures in the art.
- OvalbuminSigma; 0.5pg/mouse
- the goal of the experimentwas to analyze the effect of ACT in combination with an LNP-formulated and i.m. administered mRNA vaccine, on tumor growth.
- the experimentwas performed as described in Example 6.
- Example 8Analysis of transferred T cells in the spleen and peripheral blood
- the goal of the experimentwas to evaluate the effect of the LNP-formulated and i.m administered mRNA vaccine on the transferred T cells residing in the spleen and peripheral blood.
- Example 6The experiment was performed as described in Example 6. Mice were sacrificed on Day 11 , spleens and blood collected and further analysed as follows. Spleens were weighted and subsequently dissociated using a gentleMACS Dissociator from Miltenyi Biotec according to the manufacturer’s protocol and standard procedures in the art.
- mouse cellswere stained using a standard protocol including the following steps: red blood cell lysis (Bio-gems; for splenocytes only where necessary), mouse Fc-block (BD Biosciences), Dasatinib (Sigma) treatment, surface marker staining, fixation (eBiosciences). Before acquiring the samples with the flow cytometer, counting beads (123count eBeads, eBiosciences) were added to each sample, which enabled the exact calculation of the cell count.
- T cell populations described in Example 4.3will be performed by combining the staining described in Example 8 with fluorescent coupled antibodies or respective dyes recognizing: PD-1 :Biolegend, Tim3:Biolegend, l-Ab OVA323-339 Tetramer-ISQAVHAAHAEINEAGR:MBL and TCF1:BD Biosciences.
- the latter stainingwill be performed intracellularly using a fixation/permeabilization buffer e.g. from eBioscience.
- Resultsare shown in Figure 10 and Figure 11.
- the indicated group including both ACT and vaccinationdisplayed a very high amount of transferred CD8 T cells suggesting increased survival and/or proliferation of those cells.
- the high CD8 T cell count in this combination groupwas further reflected by increased spleen weights.
- the ACT/mRNA vaccine combination without prior lymphodepletionincreased numbers of transferred CD8+ T cells in the peripheral blood and spleen as well as a corresponding increased spleen weights could be observed (data not shown).
- the latter combinationshowed increased numbers of endogenous antigen-specific CD8 T cells in the peripheral blood and spleen.
- the goal of the experimentwas to evaluate the effect of the LNP-formulated and i.m. administered mRNA vaccine on the infiltration of transferred T cells into the tumor and on their phenotype. To this end, the amount of transferred T cells in the tumor as well as their activation (marker CD25) and cytotoxicity (marker Granzyme B) were determined.
- Example 6The experiment was performed as described in Example 6. Mice were sacrificed on Day 11 , tumors collected and further analysed as follows. Spleens were weighted and subsequently dissociated using the gentleMACS Dissociator from Miltenyi Biotec according to the manufacturer’s protocol and standard procedures in the art.
- mouse cellswere stained using a standard protocol including the following steps: mouse Fc-block (BD Biosciences), Dasatinib (Sigma) treatment, surface marker staining, fixation/permeabilization and intracellular staining of Granzyme B.
- mouse Fc-blockBD Biosciences
- DasatinibSigma
- surface marker staininge.g
- Example 10Exemplary clinical study design - Phase I dose escalation study for combination of i.m. lipid nanoparticle formulated RNA vaccine with ACT
- DLTsafety, tolerability and Dose Limiting Toxicities
- Secondary endpointsFrequency and functional status of adoptively transferred T cells in the circulation, antigen specific T cells response against the at least vaccine encoded antigen, objective response rate (ORR), Disease control rate (DCR), Duration of response (DoR)
- LNP formulated RNAcomprising a cds encoding at least one antigen, wherein the at least one encoded antigen comprises at least the epitope (T cell epitope) recognized by the antigen receptor of the adoptive immune cells, preferably the nucleic acid encoding at least one full length antigen.
- Administration routeintramuscular, dose will be increased stepwise e.g. 12pg, 25pg, 50pg or 100pg
- TCRtggenetically modified TCR
- CAR T cellstargeted to the antigen, preferably tumor antigen, more preferably an epitope derived from PRAME or CLDN6
- Cohort 1Monotherapy: ACT, lymphodepletion (chemotherapy) prior to ACT
- Cohort 2Combination therapy: ACT in combination with the composition of the invention, lymphodepletion (chemotherapy) prior to ACT.
- a subgroup of the cohortreceives a high dose of ACT with dose escalation e.g. 12pg, 25pg, 50pg or 100pg of the composition on the early vaccination time point(s) (as described above).
- the other groupreceives a low dose ACT with dose escalation of e.g. 12pg, 25pg, 50pg or 100 g of the composition on the early vaccination timepoint(s).
- Cohort 3Combination therapy: ACT in combination with the composition of the invention, without lymphodepletion prior to ACT.
- One subgroup of the cohort 3receives a high dose of ACT with dose escalation of e.g. 12 g, 25 g, 50 g or 10Opg of the composition on the early vaccination timepoint(s).
- the other groupreceives a low dose ACT with dose escalation of e.g. 12pg, 25pg, 50pg or 100pg of the composition the early vaccination time point(s).
- Cohort 4Combination therapy: ACT in combination the composition of the invention, with lymphodepletion prior to ACT.
- One subgroup of the cohort 4receives a high dose ACT in combination with dose escalation of >49pg of the composition on the late vaccination time point.
- the other groupreceives a low dose ACT with dose escalation of e.g. >49 g composition on the late vaccination time point.
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Chemical & Material Sciences (AREA)
- Genetics & Genomics (AREA)
- Epidemiology (AREA)
- Engineering & Computer Science (AREA)
- Pharmacology & Pharmacy (AREA)
- Biotechnology (AREA)
- Medicinal Chemistry (AREA)
- Organic Chemistry (AREA)
- Biomedical Technology (AREA)
- Wood Science & Technology (AREA)
- Molecular Biology (AREA)
- General Engineering & Computer Science (AREA)
- Zoology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Biochemistry (AREA)
- Microbiology (AREA)
- Plant Pathology (AREA)
- Physics & Mathematics (AREA)
- Biophysics (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Oncology (AREA)
- Immunology (AREA)
- Mycology (AREA)
- Medicines Containing Material From Animals Or Micro-Organisms (AREA)
Abstract
Description
RNA composition for improving cell therapy
Introduction
The development of adoptive cell transfer (ACT) therapies e.g. for tumor immunotherapy has promoted substantial progress for cancer treatment in recent years, resulting in approved ACT therapies for treatment of hematologic cancers. Nevertheless, there is a need to overcome challenges and inefficiencies of ACT therapy like the poor in vivo persistence of transferred cells, limited quantity of transferable cells, overcome T cell exhaustion and circumvent immunosuppressive and tolerogenic tumor microenvironment in several solid cancer entities and adverse side effects correlated with the ACT and the lymphodepletion before the transfer.
RNA has emerged as a promising therapeutic tool for the treatment of cancer and other diseases, as it can be used to direct the production of proteins that can e.g. inhibit or otherwise reduce the growth or survival of cancer cells.
However, the knowledge on how to combine ACT therapies with other therapeutic modalities such as RNA is limited, and a better understanding of how to exploit the therapeutic potential of RNA to improve ACT therapies would be of great importance for various therapeutic areas.
Accordingly, the object of the present invention is to overcome challenges and inefficiencies of ACT therapies by providing novel treatment options exploiting RNA as further therapeutic modality to improve ACT therapies in particular in cancer patients.
The object mentioned above is solved by the underlying description and the accompanying claims. In particular, the object of the invention is solved by providing compositions, combinations, or kit or kit of parts configured for intramuscular delivery of RNA encoded antigens to e.g. stimulate adoptively transferred T cells.
Short description of the invention
The present invention is inter alia directed to compositions comprising at least one RNA comprising at least one coding sequence encoding at least one antigen for use in a method of treatment or prophylaxis of a disease, disorder or condition, wherein the RNA is preferably formulated in lipid nanoparticles and wherein the composition is preferably administered intramuscularly to a subject that has received modified immune cells, preferably modified T cells, targeted to the at least one antigen. Further aspects relate to a combination comprising the composition, or a kit or kit of parts comprising the composition, for use in a method of treatment or prophylaxis of a disease, disorder or condition. The invention further provides a method for treating or preventing a disease, disorder or condition and a method of boosting a modified immune cell population, preferably a modified T cell population, in a subject.
As shown herein, the invention is inter alia based on the surprising finding that intramuscular administration of a composition comprising at least one RNA comprising at least one coding sequence encoding at least one antigen can effectively increase the levels of circulating modified immune cells such as modified T cells in the subject; induce memory formation of modified immune cells such as modified T cells in the subject, preferably formation of central and effector memory cells; activate modified immune cells such as modified T cells in the subject; re-activate modified immune cells such as modified T cells in the subject; maintain the self-renewal capacity of modified immune cells such as modified T cells in the subject; induce higher influx into the solid tumor of modified immune cells such as modified T cells in the subject; and/or induce expansion of modified immune cells such as modified T cells in the subject. In a first aspect, the present invention relates to a composition comprising at least one RNA comprising at least one coding sequence encoding at least one antigen for use in a method of treatment or prophylaxis of a disease, disorder or condition, wherein the composition is administered to a subject that has received modified immune cells, preferably modified T cells, targeted to the at least one antigen.
In a second aspect, the present invention relates to a combination comprising (i) a composition comprising at least one RNA comprising at least one coding sequence encoding at least one antigen; and (ii) modified immune cells, preferably modified T cells, targeted to the at least one antigen, for use in a method of treatment or prophylaxis of a disease, disorder or condition, wherein the modified immune cells are administered intravenously to the subject, and wherein the composition is administered to the subject that has received modified immune cells.
In a third aspect, the present invention relates to a kit or kit of parts comprising (A) at least one composition as defined in the context of the first aspect; or (B) at least one combination as defined the context of the second aspect, for use in a method of treatment or prophylaxis of a disease, disorder or condition.
In a fourth aspect, the present invention relates to a method of treating or preventing a disease, disorder, or condition in a subject, comprising a step of applying or administering a composition comprising at least one RNA comprising at least one coding sequence encoding at least one antigen, wherein the subject has received modified immune cells, preferably modified T cells, targeted to the at least one antigen.
In a fifth aspect, the present invention relates to a method of boosting a modified immune cell population in a subject, wherein the method comprises applying or administering at least one composition comprising at least one RNA comprising at least one coding sequence encoding at least one antigen to the subject.
Preferably, the composition comprising at least one RNA of the first, second, third, fourth, or fifth aspect is administered by intramuscular administration.
Preferably, the RNA of the first, second, third, fourth, or fifth aspect is formulated in lipid nanoparticles (LNPs).
Preferably, the disease, disorder or condition is selected from a tumor or cancer disease, disorder or condition.
Definitions
For the sake of clarity and readability the following definitions are provided. Any technical feature mentioned for these definitions may be read on each and every embodiment of the invention. Additional definitions and explanations may be specifically provided in the context of these embodiments.
About: The term “about” is used when values or determinants do not need to be identical, i.e. 100% the same and that e.g. the respective values or determinants may diverge by 1 % to 10%. Preferably, “about” means that a value or determinant may diverge by +/-1%, +1-2%, +/-3%, +M%, +/-5%, +/-6%, +1-7%, +/-8%, +/-9%, +/-10%.
Adoptive cell transfer, ACT; Adoptive cell transfer (ACT) is the transfer of immune cells into a patient. The immune cells may have originated from the patient or from another individual. The cells are most commonly derived from the immune system with the goal of improving immune functionality and characteristics. Different cell types have been used for ACT experiments and studies like lymphokine-activated killer (LAK) cells, tumor-infiltrating lymphocytes (TILs), donor lymphocytes after hematopoietic stem cell transplantation (HSCT), tumor-specific T cell lines or clones (chimeric antigen receptor (CAR) or transgenic (tg) T cells), y5T cells (CAR-yST) natural killer cells (CAR-NK), Va24-invariant natural killer T cells (CAR-NKT) and/or macrophages (CAR-M). The cells used in connection with the present invention will preferably be autologous cells, although heterologous cells or allogenic cells can be used.
Allogeneic antigen: The term “allogeneic antigen” refers to antigens derived from genetically distinct individuals of the same species. These antigens are recognized by the immune system as foreign and can elicit an immune response, potentially leading to the rejection of transplanted tissues or organs.
Allogenic, allogenic modified immune cells: The term “allogeneic” is used to describe anything that is derived from different individuals of the same species. Two or more individuals are said to be allogeneic to one another when the genes at one or more loci are not identical. Accordingly, the term “autologous modified immune cell” refers to a transplant immune cells derived from different individuals of the same species (e.g. a different human species).
Antigenic peptide: The term “antigenic peptide” or “immunogenic peptide” refers to a peptide derived from a (antigenic or immunogenic) protein which stimulates the body’s adaptive or cellular immune system to provide an adaptive or cellular immune response. Therefore, an antigenic/immunogenic peptide comprises at least one epitope (as defined herein) or antigen (as defined herein) selected or derived from e.g. a tumor (neo)antigen.
Antigen receptor: The term “antigen receptor’ refers to a specialized protein or molecular structure found on the surface of immune cells, primarily B cells and T cells, that is responsible for recognizing and binding to antigens. Antigen receptors comprise an antigen recognition domain with at least one signalling domain. Cells may naturally express an antigen receptor or be modified (e.g. ex vivo, in vitro or in vivo in a subject to be treated) to express an antigen receptor. Modification to express an antigen receptor takes place ex vivo and/or in vitro. Modified immune cells described herein may express an antigen receptor such as a chimeric antigen receptor (CAR) or a T cell receptor (TCR) or a procession product thereof. Subsequently, modified immune cells expressing the at least one antigen receptor may be administered to a subject. The modified cells expressing the at least one antigen receptor may be endogenous cells of the subject or may have been administered to a subject.
Autoimmune diseases, disorder or condition: The term “autoimmune diseases, disorder or condition” refers to a group of diseases, disorders or conditions in which the immune system loses its ability to distinguish between “self’ and “nonself’, leading to an immune response that damages the body’s own tissues, cells, and organs. In this regard, the autoimmune disease, disorder or condition can be rheumatoid arthritis, systemic lupus erythematosus, Type 1 diabetes, multiple sclerosis, Hashimoto’s thyroiditis, Graves’ disease, celiac disease, Crohn’s disease and ulcerative colitis, psoriasis and Sjogren's syndrome.
Autologous, autologous modified immune cell: The term “autologous” is used to describe anything that is derived from the same subject. For example, “autologous modified immune cell” refers to a transplant immune cells derived from the same subject. Autologous cells are preferred because they overcome the immunological barrier which otherwise results in rejection.
Cancer or tumor disease, disorder or condition: The terms “cancer disease, disorder or condition” or “tumor disease, disorder or condition” refer to or describe the physiological condition in a subject that is typically characterized by unregulated cell growth. Examples of cancers include, but are not limited to, carcinoma, lymphoma, blastoma, sarcoma, and leukemia. More particularly, examples of such cancers include bone cancer, blood cancer, lung cancer, liver cancer, pancreatic cancer, skin cancer, cancer of the head or neck, cutaneous or intraocular melanoma, uterine cancer, ovarian cancer, rectal cancer, cancer of the anal region, stomach cancer, colon cancer, breast cancer, prostate cancer, uterine cancer, carcinoma of the sexual and reproductive organs, Hodgkin’s disease, cancer of the esophagus, cancer of the small intestine, cancer of the endocrine system, cancer of the thyroid gland, cancer of the parathyroid gland, cancer of the adrenal gland, sarcoma of soft tissue, cancer of the bladder, cancer of the kidney, renal cell carcinoma, carcinoma of the renal pelvis, neoplasms of the central nervous system, neuroectodermal cancer, spinal axis tumors, glioma, meningioma, and pituitary adenoma. The term cancer disease or tumor disease according to the disclosure also comprises cancer metastases.
Cationic, cationisable The term “cationic” means that the respective structure, compound, group, or atom bears a positive charge, either permanently or not permanently, e.g. in response to certain conditions such as pH. The terms “cationic”, “cationisable”, and “permanently cationic” as used herein must be understood as defined in
The term “cationic” means that the respective structure, compound, group, or atom bears a positive charge, either permanently or not permanently, e.g. in response to certain conditions such as pH. The terms “cationic”, “cationisable”, and “permanently cationic” as used herein must be understood as defined in

W02023031394 [p.12, line 32 to p.13, line 16]. The term “polycationic” means that the respective structure, compound, group, or atom bears a plurality of positive charges. The term as used herein must be understood as defined in WO2021156267 [p.88, line 12 to p.89, line 22], The term “chimeric antigen receptor'’ or “CAR” will be recognized and understood by the person of ordinary skill in the art. CARs relate to artificial receptors comprising a single molecule or a complex of molecules which recognizes, i.e. binds to, a target structure, e.g. an antigen, on a target cell, e.g. by binding of an antigen binding domain to an antigen expressed on the surface of the target cell, such as a cancer cell and may confer specificity onto an immune cell such as a T cell expressing said CAR on the cell surface. The binding domain binds to an extracellular domain or to an epitope in an extracellular domain of the antigen. CAR expressing cells do not necessarily require processing and presentation of an antigen for recognition of the target cell but rather may recognize preferably with specificity any antigen present on a target cell. CARs are molecules that combine specificity for a desired antigen, e.g., tumor antigen, which is antibody-based with a T cell receptor-activating intracellular domain to generate a chimeric protein that exhibits a specific cellular immune activity, e.g., a specific anti-tumor cellular immune activity. Recognition of the target structure by a CAR results in activation of an immune cell expressing said CAR. A CAR may comprise one or more protein units said protein units comprising one or more domains, an antigen binding domain, a transmembrane domain; and an intracellular domain, mostly comprising a 4-1 BB costimulatory domain, and a CD3-zeta signalling domain. The antigen binding domain comprises in some non-limiting examples a variable region of a heavy chain of an immunoglobulin (VH) with a specificity for the antigen and a variable region of a light chain of an immunoglobulin (VL) with a specificity for the antigen. The heavy chain variable region (VH) and the corresponding light chain variable region (VL) can be connected via a peptide linker. In another non-limiting example, the antigen binding moiety portion in the CAR can be a scFv. A cell can be genetically modified to stably express a CAR on its surface, conferring novel antigen specificity that may be MHC independent. Immune cells expressing the CAR molecule are from the subject to be treated or from a different subject and administered to the subject to be treated.
The term “chimeric antigen receptor'’ or “CAR” will be recognized and understood by the person of ordinary skill in the art. CARs relate to artificial receptors comprising a single molecule or a complex of molecules which recognizes, i.e. binds to, a target structure, e.g. an antigen, on a target cell, e.g. by binding of an antigen binding domain to an antigen expressed on the surface of the target cell, such as a cancer cell and may confer specificity onto an immune cell such as a T cell expressing said CAR on the cell surface. The binding domain binds to an extracellular domain or to an epitope in an extracellular domain of the antigen. CAR expressing cells do not necessarily require processing and presentation of an antigen for recognition of the target cell but rather may recognize preferably with specificity any antigen present on a target cell. CARs are molecules that combine specificity for a desired antigen, e.g., tumor antigen, which is antibody-based with a T cell receptor-activating intracellular domain to generate a chimeric protein that exhibits a specific cellular immune activity, e.g., a specific anti-tumor cellular immune activity. Recognition of the target structure by a CAR results in activation of an immune cell expressing said CAR. A CAR may comprise one or more protein units said protein units comprising one or more domains, an antigen binding domain, a transmembrane domain; and an intracellular domain, mostly comprising a 4-1 BB costimulatory domain, and a CD3-zeta signalling domain. The antigen binding domain comprises in some non-limiting examples a variable region of a heavy chain of an immunoglobulin (VH) with a specificity for the antigen and a variable region of a light chain of an immunoglobulin (VL) with a specificity for the antigen. The heavy chain variable region (VH) and the corresponding light chain variable region (VL) can be connected via a peptide linker. In another non-limiting example, the antigen binding moiety portion in the CAR can be a scFv. A cell can be genetically modified to stably express a CAR on its surface, conferring novel antigen specificity that may be MHC independent. Immune cells expressing the CAR molecule are from the subject to be treated or from a different subject and administered to the subject to be treated.

Coding sequence, coding region, cds: The terms “coding sequence” and the corresponding abbreviation “cds” as used herein refers to a sequence of several nucleotide triplets that can be translated into a peptide or protein. A cds may be a DNA or RNA sequence consisting of several nucleotides that may be divided by three, which typically starts with a start codon and preferably terminates with a stop codon. In the context of the invention, the cds preferably encodes at least one tumor antigen as defined herein. Derived from: The term “derived from” as used herein in the context of a nucleic acid, i.e. for a nucleic acid “derived from” (another) nucleic acid, means that the nucleic acid, which is derived from (another) nucleic acid, shares at least 70%, 80%, 81%, 82%, 83%, 84%, 85%, 86%, 87%, 88%, 89%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% sequence identity or are identical with the nucleic acid from which it is derived. In the context of amino acid sequences the term “derived from” means that the amino acid sequence, which is derived from (another) amino acid sequence, shares at least 70%, 75%, 80%, 81%, 82%, 83%, 84%, 85%, 86%, 87%, 88%, 89%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% sequence identity or are identical with the amino acid sequence from which it is derived.
Disease, disorder, condition: The term “disease”, “disorder" or “condition” refers to an abnormal condition that affects the body of an individual. A disease, disorder or condition is often construed as a medical condition associated with specific symptoms and signs. A disease, disorder or condition may be caused by factors originally from an external source, such as infectious disease, or it may be caused by internal dysfunctions, such as autoimmune diseases. In humans, disease, disorder or condition is often used more broadly to refer to any condition that causes pain, dysfunction, distress, social problems, or death to the individual afflicted, or similar problems for those in contact with the individual. In this broader sense, it sometimes includes injuries, disabilities, disorders, syndromes, infections, isolated symptoms, deviant behaviors, and atypical variations of structure and function, while in other contexts and for other purposes these may be considered distinguishable categories.
Disease, disorder or condition associated with stem cell transplantation: The term “disease, disorder or condition associated with stem cell transplantation” refers to certain risks and potential complication associated with stem cell transplantation. Some of the diseases, disorders or conditions that can occur after stem cell transplantation include graft- versus-host disease, infections caused by viruses, bacteria and/or fungi, cytokine release syndrome, organ toxicity, relapse of the disease after transplantation, graft failure, thrombotic microangiopathy, endocrine complications, secondary cancers (myelodysplastic syndromes or certain solid tumors) or psychosocial and emotional challenges (anxiety, depression, and post-traumatic stress disorder).
Epitope: The term “epitope” will be recognized and understood by the person of ordinary skill in the art. Epitopes refer to a part or fragment of a molecule such as an antigen that is recognized by the immune system. For example, the epitope may be recognized by T cells, B cells or antibodies. An epitope of an antigen may include a continuous or discontinuous portion of the antigen and may be between about 5 and about 100, such as between about 5 and about 50, more preferably between about 8 and about 30, most preferably between about 10 and about 25 amino acids in length, for example, the epitope may be preferably 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21 , 22, 23, 24, or 25 amino acids in length.
Fragment: The term “fragment” as used herein in the context of a nucleic acid sequence (e.g. RNA or DNA) or an amino acid sequence may typically be a shorter portion of a reference sequence of e.g. a nucleic acid sequence or an amino acid sequence. Accordingly, a fragment typically consists of a sequence that is identical to the corresponding stretch within the reference sequence. A preferred fragment of a sequence in the context of the present invention, consists of a continuous stretch of entities, such as nucleotides or amino acids corresponding to a continuous stretch of entities in the molecule the fragment is derived from, which represents at least 50%, 60%, 65%, 70%, 75%, 80%, 85%, 90%, 95% of the total reference molecule from which the fragment is derived. cancer or tumor, blood cancer: The term “hematologic cancers/tumors” or “blood cancers” as used herein refers to a group of cancers that primarily affect the blood-forming tissues in the body, including the bone marrow, lymphatic system, and blood cells. Hematologic cancers can be broadly categorized into main types: leukemia (cancer of the blood and bone marrow), lymphoma (e.g. Hodgkin lymphoma and non-Hodgkin lymphoma), multiple myeloma and myeloproliferative neoplasms (e.g. thrombocythemia and myelofibrosis). heterologous modified immune cells: The term “heterologous” is used to describe something consisting of multiple different elements. As an example, the transfer of one individual's bone marrow into a different individual constitutes a heterologous transplant. Accordingly, the term “heterologous modified immune cell” refers to a transplant immune cells derived from a different individual.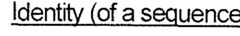 r The term “identity” as used herein in the context of a nucleic acid sequence, or an amino acid sequence, refers to the percentage to which two sequences are identical. To determine the percentage to which two sequences are identical, the sequences can be aligned (by also introducing gaps, if necessary) to be subsequently compared to one another. In the context of the invention, the substitution of a nucleotide by a modified nucleotide (e.g. U substituted by with N1 -methylpseudouridine (m1tp)) shall not be considered for calculating percent identity. The percentage to which two sequences are identical can e.g. be determined using an algorithm, e.g. an algorithm integrated in the BLAST program.
r The term “identity” as used herein in the context of a nucleic acid sequence, or an amino acid sequence, refers to the percentage to which two sequences are identical. To determine the percentage to which two sequences are identical, the sequences can be aligned (by also introducing gaps, if necessary) to be subsequently compared to one another. In the context of the invention, the substitution of a nucleotide by a modified nucleotide (e.g. U substituted by with N1 -methylpseudouridine (m1tp)) shall not be considered for calculating percent identity. The percentage to which two sequences are identical can e.g. be determined using an algorithm, e.g. an algorithm integrated in the BLAST program. lie: The terms “immunogen” or “immunogenic” will be recognized and understood by the person of ordinary skill in the art and are e.g. intended to refer to a compound that is able to stimulate/induce an immune response. Preferably, an immunogen is a peptide, polypeptide, or protein.
lie: The terms “immunogen” or “immunogenic” will be recognized and understood by the person of ordinary skill in the art and are e.g. intended to refer to a compound that is able to stimulate/induce an immune response. Preferably, an immunogen is a peptide, polypeptide, or protein.


Immune cells: The term “immune cells” including immune cells like macrophages, dendritic cells, neutrophiles, natural killer cells, dendritic cells, B lymphocytes, T lymphocytes (cytotoxic T cells, helper T cells, tumor infiltrating T cells) and mast cells. In the context of the invention immune cells are cells with lytic potential, in particular lymphoid cells, and are preferably T cells, in particular cytotoxic lymphocytes, preferably selected from cytotoxic T cells, natural killer (NK) cells, and lymphokine-activated killer (LAK) cells. Lytic potential could mean the elimination of target cells, i.e., cells characterized by expression of an antigen, for example, via apoptosis or perforin-mediated cell lysis, production of cytokines such as IFN-g and TNF-a, and specific cytolytic killing of antigen expressing target cells. As a non-limiting example first, upon activation T cells release cytotoxins such as perforin, granzymes, and granulysin. Perforin and granulysin create pores in the target cell, and granzymes enter the cell and trigger a caspase cascade in the cytoplasm that induces apoptosis (programmed cell death) of the cell. Second, apoptosis can be induced via Fas-Fas ligand interaction between the T cells and target cells. Additionally, the term immune cells comprise CD34-hematopoietic stem cells, immature and mature T cells and immature and mature B cells. The cells used in connection with the present invention will preferably be autologous cells, although heterologous cells or allogenic cells can be used.
Infectious disease, disorder or condition: The term “infectious disease, disorder or condition” refers to any disease, disorder or condition which can be transmitted from individual to individual or from organism to organism and is caused by a microbial agent. Infectious diseases, disorder or condition are known in the art and include, for example, a viral disease, a bacterial disease, or a parasitic disease, which diseases are caused by a virus, a bacterium, and a parasite, respectively. In this regard, the infectious disease, disorder or condition can be, for example, hepatitis, sexually transmitted diseases, tuberculosis, HIV, diphtheria, hepatitis B, hepatitis C, cholera, severe acute respiratory syndrome (SARS), the bird flu, and influenza. Modified immune cells: The term “modified immune cells” refers to immune cells modified to express an antigen receptor such as T cell receptor or B cell receptor or may lack expression of an antigen receptor. Immune cells can be modified to express an antigen receptor on the cell surface. In the context of the invention the immune cells lack endogenous expression of a T cell receptor. A modified immune cell is capable of binding an antigen such as an antigen presented in the context of MHC on a cell or expressed on the surface of a cell and mediating an immune response. Said binding resulting in stimulation, priming and/or expansion of the immune cells genetically modified to express an antigen receptor. In the context of the present invention, the term relates to a cell which exerts effector functions during an immune reaction. Immune cells can be modified by transfection to express an endogenous antigen receptor. According to the invention, transfection can be transient or stable. For some applications of transfection, it is sufficient if the transfected genetic material is only transiently expressed. RNA can be transfected into cells to transiently express its coded protein, e.g. an antigen receptor. Since the nucleic acid introduced in the transfection process is usually not integrated into the nuclear genome, the foreign nucleic acid will be diluted through mitosis or degraded. If it is desired that the transfected nucleic acid actually remains in the genome of the cell and its daughter cells, a stable transfection must occur. Such stable transfection can be achieved by using virus-based systems or transposon-based systems for transfection. Generally, cells that are genetically modified to express an antigen receptor are stably transfected with nucleic acid encoding the antigen receptor, while, generally, nucleic acid encoding antigen is transiently transfected into cells. Modified immune cells are suitable for adoptive cell transfer therapy. Preferred in the context of the invention are modified T cells such as CAR T cells or TCR T cells.
Nucleic acid, nucleic acid molecule: The terms “nucleic acid” or “nucleic acid molecule” refers to DNA (molecules) or RNA (molecules). It is preferably used synonymously with the term polynucleotide. Preferably, a nucleic acid or a nucleic acid molecule is a polymer comprising or consisting of nucleotide monomers, which are covalently linked to each other by phosphodiester-bonds of a sugar/phosphate-backbone. The term “nucleic acid molecule” also encompasses modified nucleic acid molecules, such as base-modified, sugar-modified, or backbone-modified DNA or RNA molecules as defined herein.
Nucleic acid sequence, DNA sequence, RNA sequence: The terms “nucleic acid sequence”, “DNA sequence”, “RNA sequence” refers to a particular and individual order of the succession of its nucleotides.
Optimal therapeutic dose: The term “optimal therapeutic amount’ or “optimal therapeutic dose" typically refers to the standard therapeutic amount of a pharmaceutical agent, meaning that the amount required for the desired effect is given. As used herein, the term “subtherapeutic amount" means that the dosage or amount of a particular pharmaceutical agent is sufficient to achieve the desired pharmacological action in the absence of other compounds, drugs or pharmaceutical agents, e.g., in the absence of the composition of the invention. Such desired pharmacological action may include the complete or essentially complete rejection of solid tumors.
RNA in vitro transcription: The term “RNA in vitro transcription" (IVT) relates to a process wherein RNA is synthesized in a cell-free system in vitro. In IVT, the RNA is obtained by transcribing a DNA template in the presence of a DNA- dependent RNA polymerase (e.g. T7, SP6), ribonucleotide triphosphates (NTPs, and optionally modified NTPs) and optionally, a cap analog, in an appropriate buffer (e.g. comprising MgCb).
Self-antigen: The term “self-antigens”, also known as autoantigens, refers to antigens naturally present in a subject. These antigens are recognized by the immune system as “self’ because they are part of the individual’s own tissues, cells, or organs. Under normal circumstances, the immune system is tolerant of self-antigens, meaning it does not mount an immune response against them. In the context of the invention self-antigens refers to antigens that elicit an immune response and leading to inflammation, tissue damage, and the development of autoimmune diseases.
Side effects: The term “side effects” as used herein in the context of the invention will be recognized and understood by the person of ordinary skill in the art. Side effects are known as adverse reactions, are unwanted undesirable effects that are possibly related to a drug. Side effects can vary from minor problems like a runny nose to life-threatening events, such as a heart attack or liver damage.
Solid tumor, solid cancer: The term “solid tumor” or “solid cancer" as used herein refers to the manifestation of a cancerous mass, as is well known in the art. Preferably, the term refers to a cancer or carcinoma of body tissues other than blood, preferably other than blood, bone marrow, and lymphoid system. For example, but not by way of limitation, solid tumors include cancers of the prostate, lung cancer, colorectal tissue, bladder, oropharyngeal/laryngeal tissue, kidney, breast, endometrium, ovary, cervix, stomach, pancreas, brain, and central nervous system.
Subject: The term “subject” refers to a human or another mammal (e.g. mouse, rat, rabbit, dog, cat, cattle, swine, sheep, horse or primate) that can be afflicted with or is susceptible to a disease or disorder (e.g., cancer) but may or may not have the disease or disorder. The term subject does not denote a particular age, and thus encompass adults, elderlies, children, and newborns.
Subtherapeutic dose, low dose, sub-optimal dose: The term “subtherapeutic dose” or “low dose” or “sub-optimal dose” typically refers to a less than standard or optimal therapeutic amount of a pharmaceutical agent, meaning that the amount required for the desired effect is lower than when the pharmaceutical agent is used alone. As used herein, the term “subtherapeutic dose" means that the dosage or amount of a particular pharmaceutical agent is insufficient to achieve the desired pharmacological action in the absence of other compounds, drugs or pharmaceutical agents, e.g., in the absence of the composition of the invention. Such desired pharmacological action may include the complete or essentially complete rejection of solid tumors. Subtherapeutic amounts and doses will usually not be less than about 5%, typically not less than about 10%, and typically not greater than about 75%, more typically not greater than about 60% of the therapeutic dosage or amount.
Syngeneic, syngeneic modified immune cells: The term “syngeneic” is used to describe anything that is derived from individuals ortissues having identical genotypes, i.e., identical twins or animals of the same inbred strain, ortheir tissues. Accordingly, the term "syngeneic modified immune cell" refers to a transplant immune cells derived from individuals or tissues having identical genotypes.
T cell epitope: The term “T cell epitope” refers to a part or fragment of a protein that is recognized by a T cell when presented in the context of MHC molecules. The term “major histocompatibility complex” and the abbreviation “MHC” includes MHC class I and MHC class II molecules and relates to a complex of genes which is present in all vertebrates. MHC proteins or molecules are important for signaling between lymphocytes and antigen presenting cells or diseased cells in immune reactions, wherein the MHC proteins or molecules bind peptide epitopes and present them for recognition by T cell receptors on T cells. The proteins encoded by the MHC are expressed on the surface of cells, and display both self-antigens, peptide fragments from the cell itself, and non-self-antigens, e.g., fragments of invading microorganisms, to a T cell. In the case of class I MHC/peptide complexes, the binding peptides are typically about 8 to about 10 amino acids long although longer or shorter peptides may be effective. In the case of class II MHC/peptide complexes, the binding peptides are typically about 10 to about 25 amino acids long and are in particular about 13 to about 18 amino acids long, whereas longer and shorter peptides may be effective.
T cell receptor (TCR): The term “T cell receptor” as used herein in the context of the invention will be recognized and understood by the person of ordinary skill in the art. A majority of T cells have a T cell receptor (TCR) existing as a complex of several proteins. The actual T cell receptor is composed of two separate peptide chains, which are produced from the independent T cell receptor alpha and beta (TCRa and TCR£) genes and are called a- and p-TCR chains. Further y 5 T cells (gamma delta T cells) represent a small subset of T cells that possess a distinct T cell receptor (TCR) on their surface, in this cells TCR is made up of one y -chain and one 5 -chain. TCRs are composed of one a-chain and one p-chain or of one y-chain and one 5-chain. The TCR a/p chains are composed of an N-teiminal highly polymorphic variable region involved in antigen recognition and an invariant constant region. On the genetic level, these chains are separated into several regions, a variable (V) region, a diversity (D) region (only P- and 5-chain), a joining (J) region and a constant (C) region. The human p-chain genes contain over 60 variable (V), 2 diversity (D), over 10 joining (J) segments, and 2 constant region segments (C). The human a-chain genes contain over 50 V segments, and over 60 J segments but no D segments, as well as one C segment. During the differentiation of T cells, specific T cell receptor genes are created by rearranging one V, one D (only p- and 5-chain), one J and one C region gene. The diversity of the TCRs is further amplified by imprecise V-(D)-J rearrangement wherein random nucleotides are introduced and/or deleted at the recombination sites. Since the rearrangement of the TCR gene loci occurs in the genome during maturation of T cells, each mature T cell only expresses one specific a/p ICR or y/5 TCR. MHC and antigen binding is mediated by the complementary determining regions 1 , 2 and 3 (CDR1 , CDR2, CDR3) of the TCR. The CDR3 of the p-chain which is most critical for antigen recognition and binding is encoded by the V-D-J junction of the rearranged TCR p-chain gene. The TCR is a part of a complex signalling machinery, which includes the heterodimeric complex of the TCR a- and p-chains, the co-receptor CD4 or CD8 and the CD3 signal transduction. While the CD3 chains transfer the activation signal inside the cell, the TCR a/p heterodimer is solely responsible for antigen recognition. Thus, the transfer of the I CR a/p chains offers the opportunity to redirect T cells towards any antigen of interest. The recognition and binding of a particular antigen is mediated by the T cell receptors (TCRs) expressed on the surface of T cells. The TCR of a T cell is able to interact with immunogenic peptides (epitopes) bound to major histocompatibility complex (MHC) molecules and presented on the surface of target cells. Specific binding of the TCR triggers a signal cascade inside the T cell leading to proliferation and differentiation into a maturated effector T cell. T cell receptor (TCR) having a binding specificity for a procession product of vaccine antigen and disease- associated antigen when presented on antigen presenting cells and diseased cells, respectively.
Transgenic TCR (TCRtq): The term “transgenic T cell receptor" as used herein in the context of the invention will be recognized and understood by the person of ordinary skill in the art. The antigenic specificity of T cells is rested entirely on the heterodimeric complex of the TCR a- and p-chain, the transfer of cloned TCR genes into T cells offers the potential to redirect them towards any antigen of interest. The transgenic TCR recognises a known antigen expressed by cancer or infected cells and presented in the context of MHC, which leads to induction of cytotoxic T cell effector functions and the killing of targeted tumor or infected cells. A T cell can be genetically modified to stably express a transgenic TCR on its surface. An immune cell genetically modified to express a transgenic T cell receptor (TCR) targeting the cells through binding to the antigen (or a procession product thereof) are provided to a subject such as by administration of the genetically modified immune cells to the subject or generation of genetically modified immune cells in the subject. Advantages of TCR gene transfer are the creation of therapeutic quantities of antigen-specific T cells within a few days and the possibility to introduce specificities that are not present or ineffective in the endogenous TCR repertoire of the patient.
Tumor antigen: The term "tumor antigen” refers to a constituent of cancer cells which may be derived from the cytoplasm, the cell surface and the cell nucleus. In the context of the present invention, the term "tumor antigen” refers to antigens that are common to specific hyperproliferative disorders such as cancer. In particular, it refers to those antigens which are produced intracellularly or as surface antigens on tumor cells. There are two major classes of tumor antigen which are typically targeted by modified immune cells in ACT therapies, tumor specific antigens and tumor associated antigens. T umor specific antigens found on cancer cells only, not on healthy tissue or cells. They arise mostly from oncogenic driver mutations that generate novel peptide sequences, i.e. neoantigens, but can also be generated by oncoviruses. T umor associated antigens are present in a subset of normal host cells as well. The tendency for expression that is higher and preferential for tumor cells. In a non-limiting example tumor antigen can be defined in further groups: The first group defines mutation antigens, neoantigens or unique antigens. Mutation antigens resulting from specific (point) mutations and expressed throughout the tumor, such as a non-limiting example B-RAF mutation in melanoma. The second group are so called tumor-specific antigens. These are shared antigens expressed in tumors, while being absent from healthy tissue, such as a non-limiting example NY-ESO-1 found in many tumor types. The third group are differentiation antigens comprising shared antigens expressed by tumors, as well as the tissue the tumor originated from, such as a non-limiting example CD19 in B cell lymphoma. The fourth group are overexpressed antigens comprising shared antigens which are expressed in different healthy tissues, but are overexpressed in tumors, such as a non-limiting example p53. The fifth group are virus tumor antigens comprising viral antigens associated with virus-associated cancers, such as a non-limiting example HPV-16 E7. Another group are microbial or bacterial tumor antigens associated with tumors i.e. glioblastoma.
Variant (of a sequence): The term “variant” as used herein in the context of a nucleic acid sequence refers to a nucleic acid sequence derived from a reference nucleic acid sequence. A variant of a nucleic acid sequence may exhibit one or more nucleotide deletions, insertions, additions and/or substitutions compared to the reference nucleic acid from which the variant is derived from. A variant may be a functional variant in the sense that the variant has retained at least 70%, 80%, 90%, or 95% or more of the function of the sequence where it is derived from. A “variant’ of a nucleic acid sequence may have at least 70%, 75%, 80%, 85%, 90%, 95%, or 99% identity over a stretch of at least 30, 50, 75 or 100 nucleotides.
The term “variant” as used herein in the context of proteins or peptides refers to a proteins or peptide having an amino acid sequence which differs from the reference sequence in one or more mutation(s) substitution(s), such as one or more substituted, inserted and/or deleted amino acid(s). Insertions and substitutions are possible at those sequence positions which cause no modification to the three-dimensional structure or do not affect the binding region. A variant of a peptide or protein may be a functional variant, which means that the variant exerts essentially the same, or at least 70%, 80%, 90% of the function of the peptide or protein it is derived from (e.g. antigenic property). A “variant’ of a peptide or protein may have at least 70%, 75%, 80%, 85%, 90%, 95%, or 99% identity over a stretch of at least 30, 50, 75 or 100 amino acids. Detailed description of the invention
1. Composition for use in treatment or prophylaxis of a disease, disorder or condition
In a first aspect, the present invention relates to a composition comprising at least one RNA comprising at least one coding sequence (cds) encoding at least one antigen for use in a method of treatment or prophylaxis of a disease, disorder or condition, wherein the composition is administered to a subject that has received modified immune cells targeted to the at least one antigen.
It has to be noted that specific features and embodiments that are described in the context of the first aspect of the invention, that is the composition for use of the invention, are likewise applicable to any other aspect of the invention.
The term “targeted to the at least one antigen” means that the modified immune cells such as the modified T cells are capable to bind and/or detect the at least one antigen that is provided by the RNA of the invention, or a fragment or variant thereof. Typically, the respective antigen or a processing product thereof, e.g., a fragment thereof, binds to the specific antigen receptor (such as TCR or CAR) carried by the modified immune cells. Said binding may result in stimulation, priming and/or expansion of the modified immune cells genetically modified to express an antigen receptor and provides an immune response against the antigen which may be therapeutic or partially or fully protective. The antigen may be expressed by a target cell to which the modified immune cells are targeted (e.g. tumor cell) or a fragment thereof, or a variant of the antigen or the fragment.
In embodiments, the composition is administered to a subject via subcutaneous, intravenous, intramuscular, intraarticular, intra-synovial, intranasal, oral, intrasternal, intrathecal, intrahepatic, intralesional, intracranial, transdermal, intradermal, intrapulmonal, intraperitoneal, intracardial, intraarterial, intraocular, intravitreal, subretinal, intranodal, or intratumoral, preferably intramuscular, intravenous or intratumoral administration. In embodiments, the composition is administered to a subject via subcutaneous, intramuscular, intra-articular, intra-synovial, intranasal, oral, intrasternal, intrathecal, intrahepatic, intralesional, intracranial, transdermal, intradermal, intrapulmonal, intraperitoneal, intracardial, intraarterial, intraocular, intravitreal, subretinal, intranodal, or intratumoral, preferably intramuscular or intratumoral administration. In preferred embodiments, the composition is administered to a subject via intramuscular or intratumoral administration. In particularly preferred embodiments, the composition is administered to a subject via intramuscular administration, in particular, intramuscular injection.
In embodiments, the RNA of the composition is formulated. Suitable formulations in the context of the invention are further described under paragraph “Formulation/Complexation of the RNA of the composition". In particularly preferred embodiments, the RNA is formulated in lipid nanoparticles as further defined herein.
In some embodiments, the composition comprises at least one RNA comprising at least one cds encoding at least one antigen for use in a method of treatment or prophylaxis of a disease, disorder or condition, wherein the composition is administered to a subject that has received modified immune cells, preferably modified T cells, targeted to the at least one antigen. In preferred embodiments, the composition comprises at least one RNA comprising at least one cds encoding at least one antigen for use in a method of treatment or prophylaxis of a disease, disorder or condition, wherein the composition is preferably administered intramuscularly to a subject that has received modified immune cells, preferably modified T cells, targeted to the at least one antigen. In preferred embodiments, the RNA is formulated as further defined in paragraph “Formulation/Complexation of the RNA of the composition". More preferably, the RNA is formulated in lipid nanoparticles (LNPs).

In the following, the modified immune cells that have been received by the subject are defined in more detail.
In particularly preferred embodiments, the modified immune cells are modified T cells.
In some embodiments, the modified immune cells, preferably the modified T cells, that have been received by the subject are either from a subject to be treated or from a different subject.
In some embodiments, the modified immune cells, preferably the modified T cells, that have been received by the subject comprise autologous immune cells taken from the subject, heterologous immune cells, allogenic immune cells or syngeneic immune cells.
In some embodiments, the modified immune cells, preferably the modified T cells, that have been received by the subject may be autologous, allogeneic or syngeneic to the subject to be treated. In one embodiment, the immune cells have been removed from a subject and have subsequently been re-delivery to the subject.
In one embodiment, the immune cells from the subject have not been removed before administration of the composition. In this embodiment, all steps to modify the immune cells of the subject are performed in vivo.
In preferred embodiments, the modified immune cells, preferably the modified T cells, that have been received by the subject comprise autologous immune cells taken from the subject.
In some embodiments, the modified immune cells, preferably the modified T cells, that have been received by the subject express an antigen receptor targeted to the antigen encoded by the RNA of the composition and/or express the antigen corresponding to the antigen that is encoded by the RNA of the composition. Accordingly, in preferred embodiments, the modified immune cells that have been received by the subject are genetically modified to express an antigen receptor targeted to the antigen and/or to express the antigen. In some embodiments, the modified immune cells, preferably the modified T cells, that have been received by the subject are genetically modified, ex vivo or in vivo, to express an antigen receptor targeted to the antigen encoded by the RNA of the composition and/or to express the antigen corresponding to the antigen that is encoded by the RNA of the composition. In preferred embodiments, the modified immune cells, preferably the modified T cells, that have been received by the subject are genetically modified ex vivo prior to administration or genetically modified in vivo in the subject following administration to express an antigen receptor and/or the antigen described herein. In preferred embodiments, the modified immune cells, preferably the modified T cells, that have been received by the subject are genetically modified ex vivo to express the antigen receptor targeted to the antigen encoded by the RNA of the composition and/or to express the antigen corresponding to the antigen that is encoded by the RNA. In particularly preferred embodiments, the modified immune cells, preferably the modified T cells, that have been received by the subject are genetically modified to express the antigen receptor targeted to the antigen encoded by the RNA of the composition.
In some embodiments, “genetically modified to express an antigen receptor and/or antigen" means that the immune cells, preferably the T cells, have been transfected with a nucleic acid, in particular RNA, into an immune cell to e.g. modify the immune cell. According to the invention the transfection can be transient or stable. In a preferred embodiment, the modified immune cells, preferably the modified T cells, that have been received by the subject can be genetically modified to stably express an antigen receptor e.g. on its surface. In preferred embodiments in that context, the antigen receptor (of the modified immune cells, preferably the modified T cells) is targeted to the antigen (encoded by the RNA of the composition), that is associated with the disease, disorder or condition. In some embodiments, the antigen receptor (of the modified immune cells, preferably the modified T cells) is targeted to at least one antigen associated with a disease, disorder or condition. In some embodiment, the antigen receptor (of the modified immune cells, preferably the modified T cells) is targeted to at least two antigens associated with a disease, disorder or condition.
In embodiments, the antigen (to which the modified immune cells, preferably the modified T cells, are targeted to) is selected from or derived from tumor antigens, viral antigens, bacterial antigens, protozoal antigens, fungal antigens, self-antigens or allogenic antigens, preferably tumor antigens.
In some embodiments, the subject has received modified immune cells, preferably modified T cells, targeted to at least one antigen selected from or derived from tumor antigens, viral antigens, bacterial antigens, protozoal antigens, fungal antigens, self-antigens or allogenic antigens, preferably tumor antigens.
In some embodiments the subject has received modified immune cells, preferably modified T cells, targeted to at least two antigens selected from or derived from tumor antigens, viral antigens, bacterial antigens, protozoal antigens, fungal antigens, self-antigens or allogenic antigens, preferably tumor antigens.
In some embodiments, the subject has received modified regulatory T cells expressing at least one antigen receptor targeted to at least one self-antigen or allogenic antigen.
In preferred embodiments in that context, the antigen receptor (of the modified immune cells) is a chimeric antigen receptor (CAR), T cell receptor (TCR) or a transgenic T cell receptor (TCRtg), preferably a TCRtg. Accordingly, in some embodiments, the modified immune cells that have been received by the subject express at least one antigen receptor, preferably at least one CAR, TCR and/or TCRtg. Accordingly, in some embodiments, the subject has received modified immune cells expressing a CAR. In some embodiments, the modified immune cells that have been received by the subject are CAR-M cells expressing at least one CAR molecule targeted to at least one antigen, preferably a tumor specific or tumor associated antigen. In preferred embodiments, the subject has received modified immune cells expressing a TCR or a TCRtg, preferably a TCRtg.
In preferred embodiments, the modified immune cells that have been received by the subject are selected from tumorinfiltrating leukocytes (TILs) or modified T cells. Tumor-Infiltrating Leukocyte (TIL) therapy is a form of immunotherapy used in cancer treatment. It involves the extraction, in vitro expansion, and reinfusion of autologous immune cells, specifically T lymphocytes (T cells), which have infiltrated a tumor. TIL therapy is typically utilized in the treatment of certain types of cancer, such as melanoma. In preferred embodiments, the TILs expressing at least one TCR. In some embodiments, the subject has received autologous and ex vivo expanded tumor-infiltrating leukocytes.
Modified T cells, often referred to as engineered or genetically modified T cells, are T lymphocytes that have been altered or enhanced through genetic engineering techniques in vivo, ex vivo, in vitro to express an antigen receptor, e.g. CARs or TCRtg.
In some embodiments, the modified T cell that have been received by the subject is a CD4 positive helper T cell, CD8 positive cytotoxic T cell, regulatory T cell (Treg), and/or a memory T cell. Accordingly, the subject has received modified T cells, preferably modified CD4 positive helper T cells, modified CD8 positive cytotoxic T cells, modified regulatory T cells (T regs), and/or modified memory T cells before the administration of the composition. The term “CD4 positive helper T cell” refers to T cells that play a vital role in coordinating the immune response. These cells recognize antigens presented by antigen-presenting cells (APCs) and help activate other immune cells, such as B cells and cytotoxic T cells. Helper T cells are essential for both antibody-mediated (humoral) and cell-mediated immune responses.
The term “CD8 positive cytotoxic T cell” refers to T cells that are responsible for directly attacking and destroying infected or cancerous cells. These cells recognize antigens on the surface of infected or cancerous cells and release toxic molecules to induce cell death.
The term “regulatory T cells” or“Treg” refers to T cells that help maintain immune system balance and prevent excessive immune responses. These cells play a role in suppressing immune reactions to self-antigens and preventing autoimmunity.
The term “memory T cell” refers to T cells, like CD4 or CD8 positive T cells, that become a memory T cell after an initial encounter with an antigen. These cells "remember” the specific antigen, allowing for a faster and more efficient immune response upon subsequent exposure to the same antigen.
In preferred embodiments, the modified T cell that have been received by the subject is a CD8 positive cytotoxic T cell. Accordingly, the subject received modified CD8 positive cytotoxic T cells before the administration of the composition.
In preferred embodiments, the modified T cells that have been received by the subject are selected from transgenic TCR (TCRtg) and/or chimeric antigen receptor (CAR) T cells.
In preferred embodiments, the modified immune cells that have been received by the subject are selected from modified T cells, preferably transgenic T cells that are genetically modified to express an antigen receptor, preferably a transgenic TCR(TCRtg) targeted to the antigen.
Accordingly, the subject has received modified T cells selected from transgenic TCR (TCRtg) and/or chimeric antigen receptor (CAR) T cells before the administration of the composition.
In some embodiments, the chimeric antigen receptor (CAR) T cells comprise at least one CAR molecule. Accordingly, in preferred embodiments the subject has received chimeric antigen receptor (CAR) T cells comprising at least one CAR molecule. In some embodiments, the CAR molecule comprises at least one an antigen binding domain. In some embodiments, the CAR molecule comprises at least two antigen binding domains. Accordingly, in some embodiments, the subject has received modified immune cells expressing a CAR molecule that comprises at least one or at least two antigen binding domains. In some embodiments, the subject has received modified immune cells expressing a CAR molecule that comprises at least one tumor antigen binding domain.
In preferred embodiments, the subject has received modified immune cells expressing a chimeric antigen receptor (CAR) molecule comprising a CD19, BCMA, CD22, CD20, Mesothelin, GPC3, GD2, HER2, B7-H3, Claudin18.2 (CLDN18.2), Claudin6 (CLDN6), PSMA, PRAME CEA, EGFR, MUC1 , EGFRVIII, NKG2DL antigen binding domain, preferably an Claudin6 or PRAME antigen binding domain.
In preferred embodiments, the subject has received axicabtagene ciloleucel (Yescarta™), brexucabtagene autoleucel (Tecartus™), ciltacabtagene autoleucel (Carvykti™), idecabtagene vicleucel (Abecma™), lisocabtagene maraleucel (Breyanzi™) ortisagenlecleucel (Kymriah™). In preferred embodiments, the subject has received modified immune cells expressing a chimeric antigen receptor (CAR) molecule comprising a CLDN6 antigen binding domain. In embodiments, the CLDN6 antigen binding domain comprises the amino acid sequence of SEQ ID NOs: 32-42 or a functional variant thereof, preferably SEQ ID NO: 32. In embodiments, a binding domain for CLDN6 comprises a heavy chain variable region (VH) comprising an amino acid sequence selected from the group consisting of SEQ ID NOs: 33-36 or a functional variant thereof. In embodiments, a binding domain for CLDN6 comprises a light chain variable region (VL) comprising an amino acid sequence selected from the group consisting of SEQ ID NOs: 37-42 or a functional variant thereof.
In embodiments, the subject has received modified immune cells, preferably modified T cells such as CAR T cells, targeted to Claudin6 or a fragment thereof, or a variant of CLDN6, and the at least one RNA of the composition of the invention that is administered to said subject encodes CLDN6 or a fragment thereof, or a variant of CLDN6.
In some embodiments, the subject has received modified T cells expressing a transgenic TCR molecule. In some embodiments, the transgenic TCR molecule comprises at least one an antigen binding domain. In some embodiments, the transgenic TCR molecule comprises at least two antigen binding domains. In some embodiments, the transgenic TCR molecule comprises at least one tumor antigen binding domain. In preferred embodiments, the subject has received modified immune cells expressing a transgenic TCR molecule comprising an W 1 , COL6A3, HA-1, NY-ESO-1, HPV, MAGE-A4, HBV, EBV, mutated KRAS; KK-LC-1, MAGE-A3, MAGE-A10, AFP, Mesothelin, EGFRVIII, MAGE-A1, MART-1, PRAME, mutated PRAME, CD19, CD33, BCMA, CD22, CD70, NKG2DL, PD-L1 , ROBO1, 5T4, PSMA, CLDN6, DLL3, HER2, MICA/B and/or MUC1 antigen binding domain. In embodiments, the subject has received modified immune cells expressing a TCR molecule comprising an PRAME or CLDN6 antigen binding domain. In embodiments, the subject has received modified immune cells expressing a TCR molecule comprising an PRAME antigen binding domain, wherein said antigen binding domain is capable of specifically and/or selectively binding to a PRAME antigenic peptide, such as a peptide shown in SEQ ID NO: 67. In preferred embodiments, the antigen binding domain comprising at least one complementary determining region (CDR) 3 having at least 80% sequence identity to an amino acid sequence selected from SEQ ID NOs: 48-65. In some embodiments, the transgenic TCR a or y chain comprises a CDR3 having at least 80% sequence identity to an amino acid sequence selected from SEQ ID NOs: 48-56. In some embodiments, the transgenic TCR p or 6 chain comprises a CDR3 having at least 80% sequence identity to an amino acid sequence selected from SEQ ID NOs: 57-65. In embodiments, the subject has received modified immune cells expressing a TCR molecule comprising a CLDN6 antigen binding domain, wherein the antigen binding domain is capable of specifically and/or selectively binding to a CLDN6 antigenic peptide, such as a peptide shown in SEQ ID NOs: 45-47. In some embodiments, the antigen binding domain comprising at least one TCR having at least 80% sequence identity to an amino acid sequence selected from SEQ ID NOs: 8-31. In preferred embodiments, the antigen binding domain comprising at least one alpha chain or a T cell receptor comprising said T cell receptor alpha chain wherein the said T cell receptor a-chain is selected from the group of SEQ ID NOs: 8- 19 or a variant thereof. In preferred embodiments, the antigen binding domain comprising at least one beta chain or a T cell receptor comprising said T cell receptor beta chain wherein the said T cell receptor beta chain is selected from the group of SEQ ID NOs: 20-31 or a variant thereof. As specified herein, the at least one RNA of the composition encodes at least one antigen that is selected from the antigen to which the modified immune cells that the subject has received are targeted to. For example, in case the subject has received modified immune cells, preferably modified T cells, targeted to CD19, BCMA, CD22, CD20, Mesothelin, GPC3, GD2, HER2, B7-H3, CLDN18.2, Claudin6 (CLDN6), PSMA, PRAME, mutated PRAME, CEA, EGFR, MUC1, EGFRVIII, NKG2DL, WT1, COL6A3, HA-1, NY-ESO-1, HPV, MAGE-A4, HBV, EBV, mutated KRAS; KK-LC-1, MAGE-A3, MAGE-A10, AFP, Mesothelin, EGFRVIII, MAGE-A1, MART-1 , CD19, CD33, BCMA, CD22, CD70, NKG2DL, PD-L1 , ROBO1, 5T4, DLL3, HER2, MICA/B and/or MUC1 or a fragment thereof, the at least one RNA of the composition that is administered to said subject encodes the respective antigen (or a fragment thereof) selected from CD19, BCMA, CD22, CD20, Mesothelin, GPC3, GD2, HER2, B7-H3, CLDN18.2, Claudin6 (CLDN6), PSMA, PRAME, mutated PRAME, CEA, EGFR, MUC1 , EGFRVIII, NKG2DL, WT1, COL6A3, HA-1, NY-ESO-1, HPV, MAGE-A4, HBV, EBV, mutated KRAS; KK-LC-1, MAGE-A3, MAGE-A10, AFP, Mesothelin, EGFRVIII, MAGE-A1, MART-1, CD19, CD33, BCMA, CD22, CD70, NKG2DL, PD-L1, ROBO1, 5T4, PSMA, DLL3, HER2, MICA/B.
As an example, in case the subject has received modified immune cells, preferably modified T cells such as TCR T cells, targeted to PRAME or a fragment thereof, or a variant of PRAME, the at least one RNA of the composition that is administered to said subject encodes PRAME or a fragment thereof, or a variant of PRAME.
Antigen
In the following, the at least one antigen that is encoded by the at least one RNA of the composition is defined in more detail. Further features relating to "antigens” in the context of the invention are also provided in paragraph “coding sequences”.
Notably, features and embodiments disclosed herein defining "the antigen that is encoded by the at least one RNA of the composition” may likewise be applicable to the at least one antigen to which the modified immune cells that the subject has received are targeted to.
In referred embodiments, the antigen comprises at least one epitope, at least two epitopes, at least three epitopes, at least four epitopes, at least five epitopes, at least six epitopes, at least seven epitopes, at least eight epitopes, at least nine epitopes, or at least ten epitopes.
In preferred embodiments, the antigen is associated with the disease, disorder or condition.
A disease-associated, disorder-associated, or condition-associated antigen is a molecule which contains epitopes that will stimulate a host’s immune system to make a cellular antigen-specific immune response and/or a humoral antibody response against the disease, disorder or condition. The disease-associated, disorder-associated or condition- associated antigen or an epitope thereof may be associated with infection by viruses or microbes, typically viral antigens, or associated with cancer, typically tumors.
In preferred embodiments, the at least one antigen is transiently expressed in cells of the subject. Thus, in preferred embodiments, the RNA comprising at least one cds encoding at least one antigen is not integrated into the genome of the cells.
In preferred embodiments, upon administration of the composition to the subject, the at least one antigen is expressed in cells of the subject such as antigen presenting cells to facilitate binding of the expressed antigen to the modified immune cells targeted to said (expressed) antigen which preferably results in stimulation, priming and/or expansion of the modified immune cells in the subject. In preferred embodiments, the antigen is selected from or is derived from tumor antigens, viral antigens, bacterial antigens, protozoal antigens, fungal antigens, self-antigens or allogenic antigens, preferably tumor antigens.
In preferred embodiments, the tumor antigen is a tumor-specific antigen or tumor associated antigen, preferably selected from mutation antigen, neoantigen, unique antigen, differentiation tumor antigen, overexpressed tumor antigen, virus tumor antigen or bacterial tumor antigen as described herein.
In embodiments, the antigen is a tumor antigen selected or derived from List 1 comprising p53, 5T4, ART-4, AFP, BAGE, beta-eaten in/m, BCMA, Bcr-abL B7-H3, mutated BRAF, CAMEL, CAP-1, CASP-8, CDC27/m, CDK4/m, CD19, CD22, CD20, CD33, CD70, CEA, mutated CTNNB1 , the cell surface proteins of the claudin family, such as Claudin6 (CLDN6), CLDN18.2 and CLDN12, COL6A3, c-MYC, CT, Cyp-B, DAM, DLL3, EBV, ELF2M, ETV6-AML1, EGFR, EGFRvlll, EphA2, G250, GAGE, GD2, GnT-V, GPC3, Gap 100, Gp100, HAGE, HA-1, HER-2/neu, HBV, HPV-E7, HPV-E6, HAST-2, hTERT (or hTRT), KRAS, mutated KRAS, KK-LC-1, LAGE, LDLR/FUT, MAGE-A, preferably MAGE-A1 , MAGE-A2, MAGE-A3, MAGE-A4, MAGE-A5, MAGE-A6, MAGE-A7, MAGE-A8, MAGE-A9, MAGE-A10, MAGE-A11, or MAGE-A12, MAGE-B, MAGE-C, MART-1 /Melan-A, MICA/B, MC1R, Mesothelin, Myosin/m, MUC1 , MUM-1, MUM-2, MUM-3, NA88-A, NF1 , NY-ESO-1, NY-BR-1, NKG2DL, PAP, pl90 minor BCR- abL, Pml/RARa, PRAME, mutated PRAME, proteinase 3, PD-L1 , PSA, PSM, PSMA, PSCA, RAGE, ROBO1 , RU1 or RU2, SAGE, SART-1 or SART-3, SCGB3A2, SCP1 , SCP2, SCP3, SSX, STEAP, SURVIVIN, TEL/AML1, mutated TP53, TPI/m, TRP-1 , TRP-2, TRP-2/INT2, TPTE, WT, and WT-1 , or an immunogenic fragment or variant of these.
In preferred embodiments, the tumor antigen is selected from List 2 comprising CD19, BCMA, CD22, CD20, Mesothelin, GPC3, GD2, HER2, B7-H3, CLDN18.2, Claudin6 (CLDN6), PSMA, CEA, EGFR, MUC1, EGFRVIII, NKG2DL, WT1, HA-1, NY-ESO-1, HPV, MAGE-A4, HBV, EBV, mutated KRAS; KK-LC-1 , MAGE-A3, MAGE-A10, AFP, MAGE-A1, MART-1 , PRAME, mutated PRAME, CD33, CD70, NKG2DL, PD-L1 , ROBO1 , 5T4, DLL3, COL6A3 and/or MICA/B, preferably from CLDN6 or PRAME, or an immunogenic fragment or variant of any of these.
In preferred embodiments, the tumor antigen is selected from List 3 comprising CD19, BCMA, CD22, CD20, Mesothelin, GPC3, GD2, HER2, B7-H3, CLDN18.2, Claudin6 (CLDN6), PSMA, CEA, EGFR, MUC1, EGFRVIII, PRAME or NKG2DL antigen, or an immunogenic fragment or variant of any of these. Accordingly, in preferred embodiments in that context, the subject has received a modified T cell expressing a CAR molecule targeted to the CD19, BCMA, CD22, CD20, Mesothelin, GPC3, GD2, HER2, B7-H3, CLDN18.2, CLDN6, PSMA, CEA, EGFR, MUC1 , EGFRVIII, PRAME or NKG2DL antigen and the subject is administered with a composition comprising at least one RNA comprising at least one coding sequence encoding CD19, BCMA, CD22, CD20, Mesothelin, GPC3, GD2, HER2, B7-H3, CLDN18.2, Claudin6 (CLDN6), PSMA, CEA, EGFR, MUC1, EGFRVIII, PRAME or NKG2DL antigen, or an immunogenic fragment or variant of any of these.
In other preferred embodiments, the tumor antigen is selected from List 4 comprising WT 1 , HA-1 , NY-ESO-1 , HPV, MAGE-A4, HBV, EBV, mutated KRAS, KK-LC-1, MAGE-A3, MAGE-A10, AFP, Mesothelin, EGFRVIII, MAGE-A1, MART-1 , PRAME, mutated PRAME, antigen, or an immunogenic fragment or variant of any of these. Accordingly, in preferred embodiments in that context, the subject has received a modified T cell expressing a TCRtg targeted to the WT1 , HA-1, NY-ESO-1, HPV, MAGE-A4, HBV, EBV, mutated KRAS; KK-LC-1, MAGE-A3, MAGE-A10, AFP, Mesothelin, EGFRVIII, MAGE-A1 , MART-1 , PRAME, mutated PRAME, antigen and the subject is administered with a composition comprising at least one RNA comprising at least one cds encoding WT 1 , HA-1 , NY-ESO-1 , HPV, MAGE- A4, HBV, EBV, mutated KRAS; KK-LC-1, MAGE-A3, MAGE-A10, AFP, Mesothelin, EGFRVIII, MAGE-A1, MART-1, PRAME, mutated PRAME, antigen, or an immunogenic fragment or variant of any of these.
In preferred embodiments, the at least one encoded antigen comprises at least the epitope recognized by the antigen receptor of the modified immune cells, preferably the modified T cells. In some preferred embodiments, the epitope comprises at least one T cell epitope. In preferred embodiments, the at least one encoded antigen comprises at least 1 , 2, 3, 4, 5, 6, 7, 8, 9 or 10 epitopes recognized by the antigen receptor of the modified immune cells, preferably the modified T cells. In some embodiments the at least one encoded antigen comprises at least 1 , 2, 3, 4, 5, 6, 7, 8, 9 or 10 T cell epitopes recognized by the antigen receptor of the modified immune cells, preferably the modified T cells.
Disease, disorder or conditions
In preferred embodiments, the disease, disorder or condition is selected from a cancer or tumor disease, disorder or condition, an infectious disease, disorder or condition, an autoimmune disease, disorder or condition, or a disease, disorder or condition associated with stem cell transplantation.
In some embodiments, the disease, disorder or condition is selected from a disease, disorder or condition associated with stem cell transplantation, preferably infections caused by viruses. In some embodiments, the disease, disorder or condition is selected from infectious disease, disorder or condition. In some embodiments, the diseases, disorder or condition is selected from a tumor or cancer disease, disorder or condition and an infectious disease, disorder or condition.
In preferred embodiments, the disease, disorder or condition is selected from a tumor or cancer disease, disorder or condition. In preferred embodiments in that context, the cancer disease is derived from solid tumors or hematologic cancers or blood cancers. In some embodiments the subject is suffering from cancer disease, preferably derived from solid tumors or hematologic cancers or blood cancers.
The subject
In the following, the subject to which the composition of the invention is administered is defined in more detail.
In particularly preferred embodiments of the invention, the subject has received modified immune cells as defined herein (see in particular paragraph “ The modified immune cells that have been received by the subject').
In some embodiments, the subject has received the modified immune, preferably the modified T cells, by intravenous administration, intratumoral administration, intraperitoneal administration, intracerebral administration, intrathecal administration, intravesical administration or intraventricular administration.
In some embodiments, the route of administration depends on the specific type of modified immune cells, depending on the disease and location of the disease, e.g. intravenous administration is the most common administration route for modified immune cells (e.g. for ACT) and allows the modified immune cells to circulate throughout the body, targeting disease cells or tissue expressing the antigen. Intratumoral administration may be preferred to treat solid tumors, the modified immune cells may be directly injected into the tumor. Intraperitoneal administration involves the delivery of modified immune cells into the peritoneal cavity (space within the abdominal cavity). This route may be preferred to treat ovarian cancer and peritoneal mesothelioma. Intracerebral administration may be preferred in embodiments where adoptive cell therapy is used to target brain tumors or central nervous system (CNS) diseases, modified immune cells may be injected directly into the cerebrospinal fluid surrounding the brain and spinal cord. Intrathecal Injection: Intrathecal administration involves injecting modified T cells into the cerebrospinal fluid within the spinal canal. This route is used for conditions affecting the CNS, such as certain types of leukemia and lymphoma that have spread to the brain and spinal cord. Intravesical administration may be preferred in cases of bladder cancer, modified immune cells are administrated directly into the bladder through a catheter. Intraventricular administration may be preferred for certain CNS disorders and involves injecting modified immune cells directly into the ventricles of the brain.
In preferred embodiments, the subject has received the modified immune cells, preferably the modified T cells, by systemic administration, in particular, by intravenous administration.
In some embodiments, the subject is a mammal, e.g. mouse, rat, rabbit, dog, cat, cattle, swine, sheep, horse, primate or human. In preferred embodiments, the subject is a mammal, preferably a human.
In some embodiments the subject, preferably the human, is an adult, elderly, child or newborn.
It is generally thought that the number of transferred modified immune cells is correlated with therapeutic responses. The number of modified immune cells which can be administered to a subject for ACT is limited and the generation of a large amount of modified immune cells for ACT still remains a challenge. A substantial increase in cell persistence could be achieved when patients received a lymphodepleting preparative regimen before infusion of modified immune cells, particularly TILs or modified T cells, e.g. CAR or TCRtg T cells.
Lymphodepletion treatment comprises the reduction of populations of circulating lymphocytes, prior to infusion of modified immune cells, including chemotherapy, radiotherapy and/or any other method specified.
In some embodiments, the lymphodepletion treatment comprises chemotherapy or radiation therapy or a combination of both. In some embodiments, the lymphodepletion treatment comprises chemotherapy selected from the group of fludarabine, cyclophosphamide, combination of fludarabine and cyclophosphamide, combination of cyclophosphamide and total body irradiation, busulfan, melphalan, cyclosporine, methotrexate, combination of cyclosporine and methotrexate, and/or bendamustine. In preferred embodiments, the lymphodepletion treatment comprises chemotherapy selected from fludarabine and/or cyclophosphamide.
In embodiments, the subject has received lymphodepletion, preferably with fludarabine or cyclophosphamide, within 1 , 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13 or 14 days before the administration of the modified immune cells, preferably the modified T cells.
Lymphodepletion is correlated with adverse side effects for the subject. Almost two-thirds of the subjects developed side effects following lymphodepletion. Side effects correlated with lymphodepletion are fewer, weakness, chills, loss of appetite, immune-related toxicity and cytokine-related toxicity, cytokine release syndrome, organ dysfunction, arrhythmias, renal failure, neurotoxicity, anemia, thrombocytopenia, graft failure, increased risk of infections like bacterial infections, viral infections, fungal infections, neutropenia, pancytopenia and febrile neutropenia and/or bacteremia.
In preferred embodiments, the subject has not received lymphodepletion within 1 , 2, 3, 4, 5, 6, 7, 8, 9, 10, 11 , 12, 13 or 14 days before the administration of the modified immune cells, preferably the modified T cells.
In preferred embodiments, the composition of the invention is able to stimulate and/or activate the administered modified immune cells, preferably the modified T cells, and/or the endogenous immune cells herein the subject has not received a lymphodepletion before the administration of the modified immune cells. In this embodiment, the composition is able to increase levels of circulating modified immune cells in the subject; induce memory formation of modified immune cells in the subject, preferably formation of central and effector memory T cells, activate modified immune cells in the subject; boosting metabolism and polyfunctionality of the modified immune cells in the subject, recruitment of cross-presenting cells, preferably DCs, to the tumor and elicits robust and potent antigen spreading, re-activate modified immune cells in the subject, maintaining self-renewal capacity of modified immune cells in the subject, induce higher influx into the solid tumor of modified immune cells in the subject, induce expansion of modified immune cells in the subject, activate endogenous CD4 helper cells in the subject, skew differentiation of endogenous tumor-infiltrating CD4+ T cells to a Th1 phenotype, increase levels of circulating endogenous immune cells in the subject, induce memory formation of endogenous immune cells in the subject, preferably formation of central and effector memory cells, activate endogenous immune cells in the subject, boosting metabolism and polyfunctionality of the endogenous immune cells in the subject, recruitment of cross-presenting cells, preferably DCs, to the tumor and elicits robust and potent antigen spreading (antigen spreading supports modified T cell therapy to treat antigenically heterogeneous tumors), re-activate endogenous immune cells in the subject, maintaining self-renewal capacity of endogenous immune cells in the subject, induce higher influx into the solid tumor of endogenous immune cells in the subject, induce expansion of endogenous immune cells in the subject and/or increase anti-tumor potential of endogenous tumor-infiltrating CD8+ T cells in the subject to a larger extend compared to intravenous administration of the composition to the subject.
In preferred embodiments in that context, the subject has a reduced risk of fewer, weakness, chills, loss of appetite, immune-related toxicity and cytokine-related toxicity, cytokine release syndrome, organ dysfunction, arrhythmias, renal failure, neurotoxicity, anemia, thrombocytopenia, graft failure, increased risk of infections like bacterial infections, viral infections, fungal infections, neutropenia, pancytopenia and febrile neutropenia and bacteremia [compared to a subject that received lymphodepletionj.
In alternative embodiments, the subject has received a lymphodepletion within 1 , 2, 3, 4, 5, 6, 7, 8, 9, 10, 11 , 12, 13 or 14 days before the administration of the modified immune cells.
In preferred embodiments, the composition of the invention is able to stimulate/activate the administered modified immune cells wherein the subject has received lymphodepletion before the administration of the modified immune cells.
Interleukin and/or cytokine treatment after transfer of modified immune cells is a common therapeutic approach used to enhance the effectiveness of ACT therapy, preferably modified T cell or tumor-infiltrating lymphocyte therapy. Interleukins are a group of cytokines that play a crucial role in regulating the immune system's response.
In the context of ACT therapy, I L2 and IL7 are commonly used to support, enhance and amplify the activity of transferred modified immune cells. IL2 plays a crucial role in expansion and activation of T cells. IL2 is often administered as a treatment to support the survival, proliferation, and activity of transferred modified immune cells within the subject’s body. IL2 can be associated with significant side effects like fever, chills, fatigue, muscle aches, severe toxicity, edema, pulmonary edema, gastrointestinal and neurological symptoms, skin reactions, kidney and liver dysfunction, anemia, thrombocytopenia and can promote Treg proliferation, also in modified immune cells that has transferred to the subject. IL7 helps T cells survive and can enhance their activity against cancer cells. IL7 can be associated with significant side effects like fever, chills, fatigue, muscle aches, gastrointestinal symptoms, lymph node enlargement, elevated liver enzymes, leukocytosis, anemia and skin reactions. In some embodiments, the method comprises a step of treating the subject with additional cytokine or interleukin, preferably at least one dose of IL2 and/or IL7, performed about 2 months after the administration of the modified immune cells. In preferred embodiments, the method does not comprise a step of treating the subject with additional cytokine or interleukin performed about 2 months after the administration of the modified immune cells. In particularly preferred embodiments, the method does not comprise a step of treating the subject with additional interleukins, preferably IL2 or IL7, performed about 2 months after the administration of the modified immune cells. In some embodiments, the subject is not treated with additional cytokine or interleukin, preferably IL2 or IL7, performed about 2 months after the administration of the modified immune cells.
In some embodiments, the composition of the invention stimulate/activate transferred modified immune cells without promoting T reg proliferation of the modified cells or cytokine or interleukin related side effects.
In some embodiments, the subject not treated with additional cytokine or interleukin, preferably IL2 or IL7, performed about 2 months after the administration of the modified immune cells, wherein the subject develops no cytokine or interleukin related side effects.
In some embodiments, the optimal therapeutic dose of modified immune cells can vary depending on several factors, including the type and stage of cancer, the specific modified immune cell product used, and individual patient characteristics.
In some embodiments, the optimal therapeutic dose of modified immune cells depends on subjects weight, specific modified immune cell product, e.g. CAR T cell products, condition regime with impact on dosing and timing of the administration of modified immune cell, administration route, clinical trial protocols, response and tolerance of the subject (may influence the subsequent doses or adjustments in the treatment schedule).
In preferred embodiments, the subject received an optimal therapeutic amount/dose of modified immune cells, preferably modified T cells.
In some embodiments, administration of the composition to a subject leads to an expansion of optimal therapeutic amounts of transferred modified immune cells in the subject.
It is generally thought that the number of transferred modified immune cells is correlated with therapeutic responses. The number of modified immune cells which can be administered to a subject for ACT is limited and the generation of a large amount of modified immune cells for ACT still remains a challenge. An increase in modified immune cell persistence could be achieved when patients received a lymphodepleting preparative regimen before infusion of modified T cells, preferably TILs or receptor-engineered T cells like CAT T cells or TCRtg T cells. The transfer of a large amount of modified T cells into a lymphodepleted subject also poses the risk of severe adverse events.
In some embodiments, the subject has received a limited amount of modified immune cells, preferably modified T cells.
In alterative embodiments, the subject received a subtherapeutic dose or low dose of modified immune cells, preferably modified T cells.
As a non-limiting example, the number of immune effector cells administered (including in vivo generation in a subject) for CAR T cell therapy of human beings is about 109 cells per dose (equivalent to 1.33x107 per kg) or higher (equivalent to 1.3x107 per kg). These values can vary due to the Summary of Product Characteristics (SmPC) of the drug. Furthermore, some therapeutic approaches comprise repetitive administration of CAR T cells in a short time period (e.g. less than 4 weeks) to improve safety by dose escalation and/or to maintain the number of effective T cells in the patient. This leads to even higher “accumulated doses” within such time periods. Thus, a “subtherapeutic dose” of immune effector cells genetically modified to express an antigen receptor is an amount of such cells per initial dose and/or accumulated dose over a time period of at least 2 days, at least 3 days, at least 4 days, at least 5 days, at least 6 days, at least 7 days, at least 14 days, at least 21 days, at least 28 days or even longer of 108 or less, 107 or less, 106 or less, 105 or less, 104 or less, 103 or less or even lower. In one embodiment, a “subtherapeutic amount” of immune effector cells genetically modified to express an antigen receptor relates to a single dose of such cells in an amount of 108 or less, 107 or less, 106 or less, 105 or less, 104 or less, 103 or less or even lower. The term “single dose” means that one dose of a therapeutic substance is administered for a prolonged time. The term “prolonged time” comprises a period of at least 14 days, at least 21 days, at least 28 days, at least 3 months, at least 6 months or even longer.
In some embodiments, administration of the composition of the invention to a subject leads to an expansion of transferred modified immune cells in the subject, in particular in embodiments where the subject has received subtherapeutic amounts of modified immune cells.
In preferred embodiments, administration of the composition to a subject leads to increased levels of circulating modified immune cells in the subject, induces memory formation of modified immune cells in the subject, preferably formation of central and effector memory cells, activates modified immune cells in the subject; boosting metabolism and polyfunctionality of the modified immune cells in the subject, recruitment of cross-presenting cells, preferably DCs, to the tumor and elicits robust and potent antigen spreading, re-activates modified immune cells in the subject; maintains the self-renewal capacity of modified immune cells in the subject, induces higher influx into the solid tumor of modified immune cells in the subject, and/or induces expansion of modified immune cells in the subject where the subject has received subtherapeutic amounts of modified immune cells.
Application regimen and dosing
In some embodiments, the first dose of the composition is administered before the subject has received the modified immune cells, preferably the modified T cells, targeted to the antigen. In some embodiments, the first dose of the composition is administered 1 , 2, 3, 4, 5, 6 or 7 days before the subject has received the modified immune cells, preferably the modified T cells, targeted to the antigen.
In some embodiments, the first dose of the composition is administered at the same day when the subject has received the modified immune cells, preferably the modified T cells, targeted to the antigen.
In preferred embodiments, the first dose of the composition is administered after the subject has received the modified immune cells, preferably the modified T cells, targeted to the antigen.
Accordingly, in some embodiments, the first dose of the composition is administered 1 , 2, 3, 4, 5, 6, 7, 8, 9, 10, 11 , 12, 13, 14, 15, 16, 17, 18, 19, 20, 21 , 22, 23, 24, 25, 26, 27, 28, 29, 30, 31 , 32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49, 50, 51 , 52, 53, 54, 55, 56, 57, 58, 59 or 60 days after the subject has received the modified immune cells, preferably the modified T cells, targeted to the antigen. In preferred embodiments, the first dose of the composition is administered 1 day to 60 days, 30 to 60 days, 30 to 40, 40 to 50 or 50 to 60 days after the subject has received the modified immune cells, preferably the modified T cells, targeted to the antigen.
In preferred embodiments, the first dose of the composition is administered after the subject has received a sub-optimal dose of the modified immune cells, preferably the modified T cells, targeted to the antigen. In some embodiments, the first dose of the composition is administered 1 , 2, 3, 4, 5, 6 or 7 days after the subject has received a sub-optimal dose of the modified immune cells, preferably modified T cells, targeted to the antigen, preferably a tumor antigen. In some embodiments, the first dose of the composition is administered after the subject has received an optimal dose of the modified immune cells, preferably modified T cells, targeted to the antigen. In preferred embodiments, the first dose of the composition is administered 30 to 60 days, 30 to 40, 40 to 50 or 50 to 60 days after the subject has received an optimal dose of the modified immune cells, preferably modified T cells, targeted to the antigen, preferably a tumor antigen.
In some embodiments, at least one further dose of the composition is administered to the subject. Suitably, the at least one further dose is administered via intramuscular or intratumoral injection, preferably via intramuscular injection. In some embodiments, the at least one further dose of the composition is administered 1 , 2, 3, 4, 5, 6, 7, 8, 9, 10, 11 , 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49, 50, 51 , 52, 53, 54, 55, 56, 57, 58, 59 or 60 days after the subject has received the first dose of the composition. In some embodiments, the at least one further dose of the composition is administered 5 days to 10 days, 11 days to 15 days, 16 days to 20 days, 21 days to 25 days, 26 days to 30 days, 31 days to 35 days, 36 days to 40 days, 41 days to 45 days, 46 days to 50 days, 51 days to 55 days, 56 days to 60 days after the subject has received the first dose of the composition. In preferred embodiments, the at least one further dose of the composition is administered 1 day to 60 days, 30 to 60 days, 30 to 40, 40 to 50 or 50 to 60 days after the administration of the first dose.
In some embodiments, the at least one further dose of the composition is administered 6 or 7 days after the administration of the first dose of the composition when the subject has received a subtherapeutic amount of modified immune cells, preferably modified T cells. In some embodiments, the at least one further dose of the composition is administered 6 or 7 days after the administration of the first dose of the composition when the subject has received a subtherapeutic amount of modified immune cells, preferably modified T cells. In some embodiments, the at least further dose of the composition is administered 7 days, 14 days, 21 days, 28 days, 35 days, 42 days and 56 days after the subject has received modified immune cells, preferably the modified T cells, preferably a subtherapeutic amount of modified immune cells, in particular modified T cells.
In some embodiments, at least 2, 3, 4, 5, 6, 7, 8, 9, 10 doses of the composition are administered to the subject. In some embodiments, at least 3, 4, 5, or more doses of the composition are administered to the subject. In some embodiments, at least 2, 3, 4, 5, or more doses of the composition are administered in a one dose per week schedule.
In preferred embodiments, the composition is intramuscularly administered 1 day, 7 days, 14 days, 21 days, 28 days, 35 days, 42 days and 56 days after the subject has received modified immune cells, preferably a subtherapeutic amount of modified immune cells, in particular modified T cells.
In embodiments, one dose of the composition comprises 10pg, 11pg, 12pg, 13pg, 14pg, 15pg, 16pg, 17pg, 18pg, 19pg, 20pg, 25pg, 30pg, 35pg, 40pg, 45pg, 50pg, 55pg, 60pg, 65pg, 70pg, 75pg, 80pg, 85pg, 90pg, 95pg, 100pg, 110pg, 120pg, 130pg, 140pg, 150pg, 160 g, 170pg 180pg, 190pg, 200pg, 250pg, 300pg, 350pg, 400pg, 450pg, 500pg, wherein the dose refers to the amount of RNA in the composition. In embodiments, one dose of the composition comprises between 1 pg and 500pg, 20pg and 500pg, 50pg and 500pg, 50pg and 250pg, 100pg and 250pg, wherein the dose refers to the amount of RNA in the composition. Observed effects
In some embodiments, the administration of the composition, preferably the ex vivo or in vivo administration of the composition, induces activation and stimulation of immune cells.
“Activation of immune cells” or “stimulation of immune cells”, as used herein, refers to the state of an immune cell that has been sufficiently stimulated to induce detectable cellular proliferation. Activation can also be associated with initiation of signaling pathways, induced cytokine production, and detectable effector functions.
In preferred embodiments, the administration (e.g. intramuscularly) of the composition to the subject induces activation and stimulation of immune cells, preferably transferred modified immune cells and endogenous immune cells.
In the context of the invention, the activation and/or stimulation of immune cells is defined by increased levels of circulating modified immune cells, preferably modified T cells, in the subject; induced memory formation of modified immune cells, preferably modified T cells, in the subject, preferably formation of central and effector memory T cells; activated modified immune cells, preferably modified T cells, in the subject; boosted metabolism and polyfunctionality of the modified immune cells, preferably modified T cells, in the subject; recruitment of cross-presenting cells, preferably DCs, to the tumor and elicits robust and potent antigen spreading, re-activated modified immune cells, preferably modified T cells, in the subject; maintained self-renewal capacity of modified immune cells, preferably modified T cells, in the subject; induced higher influx into the solid tumor of modified immune cells, preferably modified T cells, in the subject; induced expansion of modified immune cells, preferably modified T cells, in the subject; activated endogenous CD4 helper cells in the subject; skewed differentiation of endogenous tumor-infiltrating CD4+ T cells to a Th1 phenotype; increased levels of circulating endogenous immune cells in the subject; induced memory formation of endogenous immune cells in the subject; preferably formation of central and effector memory cells; activated endogenous immune cells in the subject; boosting metabolism and polyfunctionality of the endogenous immune cells in the subject; recruitment of cross-presenting cells, preferably DCs, to the tumor and elicits robust and potent antigen spreading (antigen spreading supports modified T cell therapy to treat antigenically heterogeneous tumors); reactivated endogenous immune cells in the subject; maintained self-renewal capacity of endogenous immune cells in the subject; induced higher influx into the solid tumor of endogenous immune cells in the subject; induced expansion of endogenous immune cells in the subject and/or increased anti-tumor potential of endogenous tumor-infiltrating CD8+ T cells, in the subject.
The activation and/or stimulation of immune cells, preferably modified immune cells or endogenous immune cells, can be measured by measuring initiation of signaling pathways, induced cytokine production and/or secretion, levels of antigen specific immune cells and detectable effector functions.
The activation and/or stimulation of immune cells, preferably modified immune cells or endogenous immune cells may be evaluated using any of a variety of standard techniques, for example, within a chromium release assay, proliferation assay, synthesis of lymphokines, qRT-PCR, immunohistochemical (IHC) staining, STAR RNAseq aligner (Dobin A. et al., Bioinformatics (Oxford, England) 29, 15-21 (2013)), tissue micro arrays (TMAs), quantitative real-time PCR, flow cytometric measurements (surface and intracellular staining), cytokine multiplex analysis, bead based cytokine assays, cytometric bead array, quantitative mass spectrometry, western blot, ELISA and cytotoxicity assays (spheroid based cytotoxicity or xCELLigence system). Accordingly, administration (e.g. intramuscularly) of the composition to the subject that has received modified immune cells, preferably modified T cells, results in an increased activation and/or stimulation of the immune cells in the subject, preferably modified immune cells and/or endogenous immune cells, compared to the subject that has received modified immune cells without administration of the composition, wherein the percentage increase in activation and/or stimulation is at least 1%, 1.5%, 2%, 2.5%, 3%, 3.5%, 4%, 4.5%, 5%, 5.5%, 6%, 6.5%, 7%, 7.5%, 8%, 8.5%, 9%, 9.5%, 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80%, 90%, 100%, 200%, 300%, 400%, 500% or even more.
In preferred embodiments, the administration (e.g. intramuscularly) of the composition to the subject that has received modified immune cells, preferably modified T cells, results in an increased level of circulating modified immune cells in the subject, compared to the subject that has received modified immune cells without administration of the composition, wherein the percentage increase in activation and/or stimulation is at least 0.5%, 1 %, 1.5%, 2%, 2.5%, 3%, 3.5%, 4%, 4.5%, 5%, 5.5%, 6%, 6.5%, 7%, 7.5%, 8%, 8.5%, 9%, 9.5%, 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80%, 90%, 100%, 200%, 300%, 400%, 500% or even more.
In preferred embodiments, the administration (e.g. intramuscularly) of the composition to the subject that has received modified immune cells, preferably modified T cells, results in an increased level of central and effector memory T cells, compared to the subject that has received modified immune cells without administration of the composition, wherein the percentage increase in activation and/or stimulation is at least 0.5%, 1 %, 1.5%, 2%, 2.5%, 3%, 3.5%, 4%, 4.5%, 5%, 5.5%, 6%, 6.5%, 7%, 7.5%, 8%, 8.5%, 9%, 9.5%, 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80%, 90%, 100%, 200%, 300%, 400%, 500% or even more.
In preferred embodiments, the administration (e.g. intramuscularly) of the composition to the subject that has received modified immune cells, preferably modified T cells, results in an increased level of re-activated modified immune cells in the subject, compared to the subject that has received modified immune cells without administration of the composition, wherein the percentage increase in activation and/or stimulation is at least 0.5%, 1 %, 1.5%, 2%, 2.5%, 3%, 3.5%, 4%, 4.5%, 5%, 5.5%, 6%, 6.5%, 7%, 7.5%, 8%, 8.5%, 9%, 9.5%, 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80%, 90%, 100%, 200%, 300%, 400%, 500% or even more.
In preferred embodiments, the administration (e.g. intramuscularly) of the composition to the subject that has received modified immune cells results, preferably modified T cells, results in an increased level of modified immune cells with self-renewal capacity in the subject compared to the subject that has received modified immune cells without administration of the composition, wherein the percentage increase in activation and/or stimulation is at least 0.5%, 1%, 1.5%, 2%, 2.5%, 3%, 3.5%, 4%, 4.5%, 5%, 5.5%, 6%, 6.5%, 7%, 7.5%, 8%, 8.5%, 9%, 9.5%, 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80%, 90%, 100%, 200%, 300%, 400%, 500% or even more.
In preferred embodiments, the administration (e.g. intramuscularly) of the composition to the subject that has received modified immune cells, preferably modified T cells, results in an increased level of activated endogenous CD4 helper cells in the subject, compared to the subject that has received modified immune cells without administration of the composition, wherein the percentage increase in activation and/or stimulation is at least 0.5%, 1%, 1.5%, 2%, 2.5%, 3%, 3.5%, 4%, 4.5%, 5%, 5.5%, 6%, 6.5%, 7%, 7.5%, 8%, 8.5%, 9%, 9.5%, 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80%, 90%, 100%, 200%, 300%, 400%, 500% or even more.
In some embodiments, the intramuscular administration of the composition to the subject that has received modified immune cells, preferably modified T cells, results in an increased activation and/or stimulation of the immune cells in the subject, preferably modified immune cells and/or endogenous immune cells, compared to intravenous administration of the composition to the subject that has received modified immune cells, wherein the percentage increase in activation and/or stimulation is at least 0.5%, 1 %, 1.5%, 2%, 2.5%, 3%, 3.5%, 4%, 4.5%, 5%, 5.5%, 6%, 6.5%, 7%, 7.5%, 8%, 8.5%, 9%, 9.5%, 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80%, 90%, 100%, 200%, 300%, 400%, 500% or even more.
In some embodiments, the activation and/or stimulation of the modified and endogenous immune cells as described above is determined by measuring the induction of cytokines and/or other immune response related molecules expressed intracellular or on the cell surface.
In some embodiments, the cytokines are associated with proliferation of immune cells, memory formation of immune cells, differentiation of immune cells, effector functions of immune cells, as non-limiting examples IL1 , ILS, IL4, IL6, IL7, IL8, IL9, IL10, IL12, IL15, IL17, IL21 , IL23, GM-CSF, TGF-beta, Rantes, MIP-1 alpha, MIP-1 beta, McP1 , TNFalpha, IFNgamma, IFNalpha and/or IFNbeta.
Accordingly, activation and/or stimulation of the modified and endogenous immune cells is characterized by an increased level of at least one cytokine wherein the increased level of at least one cytokine is an increase of at least 0,5%, 1%, 1.5%, 2%, 2.5%, 3%, 3.5%, 4%, 4.5%, 5%, 5.5%, 6%, 6.5%, 7%, 7.5%, 8%, 8.5%, 9%, 9.5%, 10%, 15%, 20%, 25%, 30%, 35%, 40%, 45%, 50%, 55%, 60%, 65%, 70%, 75%, 80%, 85%, 90%, or 95%. Preferably, the reduced level of at least one cytokine is a reduction of at least 30%.
In some embodiments, the other immune response related molecules expressed intracellular or on the cell surface are associated with proliferation of immune cells, memory formation of immune cells, differentiation of immune cells, effector functions of immune cells, as non-limiting examples antibodies, perforins, granzymes, histamines, chemokines, interferons, nitric oxides, lysozyme, leukotrienes, prostaglandins, defensins, cluster of differentiation (CD) molecules and or major histocompatibility complex (MHC) molecules.
Accordingly, activation and/or stimulation of the modified and endogenous immune cells is characterized by an increased level of at least one immune response related molecule wherein the increased level of at least one immune response related molecule is an increase of at least 0.5%, 1%, 1.5%, 2%, 2.5%, 3%, 3.5%, 4%, 4.5%, 5%, 5.5%, 6%, 6.5%, 7%, 7.5%, 8%, 8.5%, 9%, 9.5%, 10%, 15%, 20%, 25%, 30%, 35%, 40%, 45%, 50%, 55%, 60%, 65%, 70%, 75%, 80%, 85%, 90%, or 95%. Preferably, the reduced level of at least one cytokine is a reduction of at least 30%.
In preferred embodiments, the administration of the composition, preferably the intramuscular administration of the composition to the subject, increases the levels of circulating modified immune cells, preferably modified T cells, in the subject; induces memory formation of modified immune cells, preferably modified T cells, in the subject, preferably formation of central and effector memory cells; activates modified immune cells, preferably modified T cells, in the subject; boosting metabolism and polyfunctionality of the modified immune cells, preferably modified T cells, in the subject; recruitment of cross-presenting cells, preferably DCs, to the tumor and elicits robust and potent antigen spreading; re-activates modified immune cells, preferably modified T cells, in the subject; maintains the self-renewal capacity of modified immune cells, preferably modified T cells, in the subject; induces higher influx into the solid tumor of modified immune cells, preferably modified T cells, in the subject; and/or induces expansion of modified immune cells, preferably modified T cells, in the subject. In particularly preferred embodiments, the administration of the composition, preferably the intramuscular administration of the composition to the subject induces formation of memory T cells, preferably central memory T cells or effector memory T cells, more preferably effector memory T cells.
Memory T cells play a crucial role in the adaptive immune system’s ability to provide long-lasting immunity. These cells can persist in the body for years, or even decades, ensuring that the immune system can mount a rapid and robust response if the antigen, e.g. cancer or pathogen, recurs. This rapid response is crucial in e.g. controlling the growth and spread of e.g. cancer or virus infected cells. Memory T cells persist in the body for extended periods, sometimes even for a lifetime, ensuring that the immune system can mount a rapid and robust response if the cancer recurs. There are two distinct subsets of memory T cells: central memory T cells (Tem) and effector memory T cells (Tem). Tons are primarily found in lymphoid tissues, such as lymph nodes and the spleen and have the ability to recirculate through secondary lymphoid organs. Central memory T cells are known fortheir capacity to proliferate and differentiate into various effector T cell subsets when reactivated and serve as a reservoir of memory T cells and are important for the long-term maintenance of immunological memory. Effector memory T cells are primarily found in peripheral tissues, such as skin, mucosal surfaces, and non-lymphoid organs and are cells that can respond rapidly to antigen reexposure. Effector memory T cells have immediate cytotoxic and cytokine-secreting functions. Tem cells are crucial for providing quick and localized immune responses at the site of infection or tissue damage and are particularly important in the defense against pathogens and in immune surveillance against cancer at peripheral sites.
In preferred embodiments, the administration of the composition, preferably the intramuscular administration of the composition, to the subject induces formation of effector memory T cells, preferably memory precursor effector T cells and short-lived effector T cells, preferably memory precursor effector T cells.
In preferred embodiments, the administration of the composition, preferably the intramuscular administration of the composition to the subject induces formation of effector memory T cells of the transferred modified T cell population, preferably memory precursor effector T cells and short-lived effector T cells, preferably memory precursor effector T cells.
In preferred embodiments, the administration of the composition, preferably the intramuscular administration of the composition to the subject, induces formation of effector memory T cells of the endogenous T cell population, preferably memory precursor effector T cells and short-lived effector T cells, preferably memory precursor effector T cells.
Memory Precursor Effector T Cells (MPECs) are a subset of T cells that differentiate into long-lasting memory T cells and contributing to the establishment of long-term immunological memory. MPECs have a high proliferative potential and are more likely to survive after the resolution of the immune response. Short-Lived Effector T Cells (SLECs) play a crucial role in the acute phase of the immune response, contributing to the rapid control of infections or the destruction of infected or cancerous cells via cytokine production and cytotoxic activity. SLECs have a limited proliferative potential and are more prone to undergo apoptosis (cell death) after the antigen is cleared and do not typically contribute significantly to long-term memory.
In some embodiments, the subject (that has received modified immune cells) comprises higher transferred modified memory T cell population levels, preferably modified T cells levels, after intramuscular administration of the composition of the invention compared to intravenous administration. In preferred embodiments, the administration of the composition to the subject induces activation and stimulation of immune cells, preferably endogenous immune cells of the subject.
In preferred embodiments, administration of the composition, preferably intramuscular administration of the composition to the subject leads to the following immunologic effects: the administration activates endogenous CD4 helper cells in the subject, skews the differentiation of endogenous tumor-infiltrating CD4+ T cells to a Th1 phenotype, increases the levels of circulating endogenous immune cells in the subject; induces memory formation of endogenous immune cells in the subject, preferably formation of central and effector memory cells; activates endogenous immune cells in the subject; boosting metabolism and polyfunctionality of the endogenous immune cells in the subject, recruitment of cross-presenting cells, preferably DCs, to the tumor and elicits robust and potent antigen spreading (antigen spreading supports modified T cell therapy to treat antigenically heterogeneous tumors), re-activates endogenous immune cells in the subject; maintains the self-renewal capacity of endogenous immune cells in the subject; induces higher influx into the solid tumor of endogenous immune cells in the subject; and/or induces expansion of endogenous immune cells in the subject and increases the anti-tumor potential of endogenous tumor-infiltrating CD8+ T cells, in the subject.
In preferred embodiments, administration of the composition, preferably intramuscular administration of the composition to the subject leads to the following immunologic effects: the administration increases the levels of circulating modified immune cells, preferably modified T cells, in the subject; induces memory formation of modified immune cells, preferably modified T cells, in the subject, preferably formation of central and effector memory cells; activates modified immune cells, preferably modified T cells, in the subject; re-activates modified immune cells, preferably modified T cells, in the subject; maintains the self-renewal capacity of modified immune cells, preferably modified T cells, in the subject; induces higher influx into the solid tumor of modified immune cells, preferably modified T cells, in the subject; and/or induces expansion of modified immune cells, preferably modified T cells, in the subject to a larger extend compared to intravenous administration of the composition to the subject.
In some embodiments, the immunologic effects defined above are long-term immunologic effects, preferably immunologic effects that are detectable over 10, 15, 20, 25, 30, 35, 40, 50, 60, 70, 80, 90, 100, 110, 120, 120, 130, 140, 150, 160, 180, 200, 210, 220, 230, 240, 250, 260, 280, 300, 320, 340, 360 days or even longer.
In preferred embodiments, the immunological effects defined above are long-term immunological effects, preferably induction of memory formation of modified immune cells, preferably modified T cells, in the subject, e.g. formation of central and effector memory cells or reactivation of modified immune cells in the subject, that are detectable over 1 year, 1.5 years, 2 years, 2.5 years, 3 years, 3.5 years, 4 years, 4.5 years, 5 years, 5.5 years, 6 years, 6.5 years, 7 years, 7.5 years, 8 years, 8.5 years, 9 years, 9.5 years, 10 years or even longer.
In preferred embodiments, the subject has received a lymphodepletion therapy to minimize the numbers of endogenous (unmodified) immune cells. Suitably, the composition of the invention is able to stimulate the minimized population of the endogenous immune cells to induce immunologic effects as defined above.
In some embodiments, the administration of the composition, preferably intramuscular administration of the composition to the subject changes the cytokine milieu in the solid tumor of the subject and/or eliminates minimal residual disease, preferably tumors in the subject. The term “minimal residual disease”, also known as measurable residual disease or minimal residual cancer, refers to the small number of cancer cells that may remain in the body after treatment for cancer,
Additional therapeutic modalities
In some embodiments, the subject receives or has received an additional treatment selected from adjuvants, radiation therapy, surgery, hyperthermia therapy, chemotherapy, anti-cancer antibodies and/or immunotherapy.
The additional treatment as defined herein may be administered to the subject after the subject has received the modified immune cells, preferably modified T cells, or before the subject has received the modified immune cells, preferably modified T cells.
The additional treatment as defined herein may be administered to the subject after the subject has received the composition of the invention, or before the subject as received the composition of the invention, or concomitant or at the same time as the composition of the invention.
Examples of adjuvants include, without limitation, LPS, GP96, CpG oligodeoxynucleotides, growth factors, and cytokines, such as monokines, lymphokines, interleukins, chemokines. The cytokines may be IL1 , ILS, IL3, IL4, IL5, IL6, IL7, IL8, IL9, IL10, IL12, IFNa, IFNy, GM-CSF, LT-a. Further known adjuvants are aluminium hydroxide, Freund's adjuvant or oil such as Montanide® ISA51. Other suitable adjuvants for use in the present disclosure include lipopeptides, such as Pam3Cys.
In certain embodiments, additional treatments may be administered to a patient in combination with the treatments described herein. Such additional treatments include classical cancer therapy, e.g., radiation therapy, surgery, hyperthermia therapy and/or chemotherapy.
Chemotherapeutic agents include alkylating agents, antimetabolites, anti-microtubule agents, topoisomerase inhibitors, and cytotoxic antibiotics.
Alkylating agents have the ability to alkylate many molecules, including proteins, RNA and DNA. The subtypes of alkylating agents are the nitrogen mustards, nitrosoureas, tetrazines, aziridines, cisplatins and derivatives, and non- classical alkylating agents. Nitrogen mustards include mechlorethamine, cyclophosphamide, melphalan, chlorambucil, ifosfamide and busulfan. Nitrosoureas include N-Nitroso-N-methylurea (MNU), carmustine (BCNU), Io ustine (CCNU) and semustine (MeCCNU), fotemustine and streptozotocin. Tetrazines include dacarbazine, mitozolomide and temozolomide. Aziridines include thiotepa, mytomycin and diaziquone (AZQ). Cisplatin and derivatives include cisplatin, cariaoplatin and oxaliplatin. They impair cell function by forming covalent bonds with the amino, carboxyl, sulfhydryl, and phosphate groups in biologically important molecules. Non-classical alkylating agents include procarbazine and hexamethylmelamine. In one particularly preferred embodiment, the alkylating agent is cyclophosphamide.
Anti-metabolites are a group of molecules that impede DNA and RNA synthesis. Many of them have a similar structure to the building blocks of DNA and RNA. Anti-metabolites resemble either nucleobases or nucleosides but have altered chemical groups. These drugs exert their effect by either blocking the enzymes required for DNA synthesis or becoming incorporated into DNA or RNA. Subtypes of the antimetabolites are the anti-folates, fluoropyrimidines, deoxynucleoside analogues and thiopurines. The antifolates include methotrexate and pemetrexed. The fluoropyrimidines include fluorouracil and capecitabine. The deoxynucleoside analogues include cytarabine, gemcitabine, decitabine, azacitidine, fludarabine, nelarabine, cladribine, clofarabine, and pentostatin. The thiopurines include thioguanine and mercaptopurine.
Anti-microtubule agents block cell division by preventing microtubule function. The vinca alkaloids prevent the formation of the microtubules, whereas the taxanes prevent the microtubule disassembly. Vinca alkaloids include vinorelbine, vindesine, and vinflunine. Taxanes include docetaxel (Taxotere) and paditaxel (Taxol).
Topoisomerase inhibitors are drugs that affect the activity of two enzymes: topoisomerase I and topoisomerase II and include irinotecan, topotecan, camptothecin, etoposide, doxorubicin, mitoxantrone, teniposide, novobiocin, merbarone, and aclarubicin.
The cytotoxic antibiotics are a varied group of drugs that have various mechanisms of action. The common theme that they share in their chemotherapy indication is that they interrupt cell division. The most important subgroup is the anthracyclines (e.g., doxorubicin, daunorubicin, epirubicin, idarubicin pirarubicin, and aclarubicin) and the bleomycins; other prominent examples include mitomycin C, mitoxantrone, and actinomycin.
In certain embodiments, immune checkpoint inhibitors are used in combination with other therapeutic agents described herein. As used herein, “immune checkpoint inhibitor” refers to a molecule that totally or partially reduces, inhibits, interferes with or modulates one or more checkpoint proteins. In certain embodiments, the immune checkpoint inhibitor prevents inhibitory signals associated with the immune checkpoint. In certain embodiments, the immune checkpoint inhibitor is an antibody, or fragment thereof that disrupts inhibitory signaling associated with the immune checkpoint. In certain embodiments, the immune checkpoint inhibitor is a small molecule that disrupts inhibitory signaling. In certain embodiments, the immune checkpoint inhibitor is an antibody, fragment thereof, or antibody mimic, that prevents the interaction between checkpoint blocker proteins, e.g., an antibody, or fragment thereof, that prevents the interaction between PD-1 and PD-L1. In certain embodiments, the immune checkpoint inhibitor is an antibody, or fragment thereof, that prevents the interaction between CTLA4 and CD80 or CD86. In certain embodiments, the immune checkpoint inhibitor is an antibody, or fragment thereof, that prevents the interaction between LAG3 and its ligands, or TIM-3 and its ligands. The checkpoint inhibitor may also be in the form of the soluble form of the molecules (or variants thereof) themselves, e.g., a soluble PD-L1 or PD-L1 fusion. In certain embodiments, the immune checkpoint inhibitor suitable for use in the methods disclosed herein, is an antagonist of inhibitory signals, e.g., an antibody which targets, for example, PD-1 , PD-L1, CTLA4, LAG3, B7-H3, B7-H4, orT!M3.
Non-limiting examples of anti-cancer antibodies and potential antibody targets (in brackets) which may be used in combination with the present disclosure include: Abagovomab (CA-125), Abciximab (CD41), Adecatumumab (EpCAM), Afutuzumab (CD20), Alacizumab pegol (VEGFR2), Altumomab pentetate (CEA), Amatuximab (MORAb- 009), Anatumomab mafenatox (TAG-72), Apolizumab (HLA-DR), Arcitumomab (CEA), Atezolizumab (PD-L1 ), Bavituximab (phosphatidylserine), Bectumomab (CD22), Belimumab (BAFF), Bevacizumab (VEGF-A), Bivatuzumab mertansine (CD44 v6), Blinatumomab (CD 19), Brentuximab vedotin (CD30 TNFRSF8), Cantuzumab mertansin (mucin CanAg), Cantuzumab ravtansine (MUC1), Capromab pendetide (prostatic carcinoma cells), Carlumab (CNT0888), Catumaxomab (EpCAM, CD3), Cetuximab (EGFR), Citatuzumab bogatox (EpCAM), Cixutumumab (IGF- 1 receptor), Claudiximab (Claudin), Clivatuzumab tetraxetan (MUC1), Conatumumab (TRAIL-R2), Dacetuzumab (CD40), Dalotuzumab (insulin like growth factor I receptor), Denosumab (RANKL), Detumomab (B-lymphoma cell), Drozitumab (DR5), Ecromeximab (GD3 ganglioside), Edrecolomab (EpCAM), Elotuzumab (SLAMF7), Enavatuzumab (PDL192), Ensituximab (NPC-1C), Epratuzumab (CD22), Ertumaxomab (FIER2/neu, CD3), Etaracizumab (integrin anb3), Farletuzumab (folate receptor 1), FBTA05 (CD20), Ficlatuzumab (SCH 900105), Figitumumab (IGF-1 receptor), Flanvotumab (glycoprotein 75), Fresolimumab (TGF-b), Galiximab (CD80), Ganitumab (IGF-I), Gemtuzumab ozogamicin (CD33), Gevokizumab (lilb), Girentuximab (carbonic anhydrase 9 (CA-IX)), Glembatumumab vedotin (GPNMB), Ibritumomab tiuxetan (CD20), Icrucumab (VEGFR-1 ), Igovoma (CA-125), Indatuximab ravtansine (SDC1), Intetumumab (CD51), Inotuzumab ozogamicin (CD22), Ipilimumab (CD 152), Iratumumab (CD30), Labetuzumab (CEA), Lexatumumab (TRAIL-R2), Libivirumab (hepatitis B surface antigen), Lintuzumab (CD33), Lorvotuzumab mertansine (CD56), Lucatumumab (CD40), Lumiliximab (CD23), Mapatumumab (TRAIL-R1), Matuzumab (EGFR), Mepolizumab (IL5), Milatuzumab (CD74), Mitumomab (GD3 ganglioside), Mogamulizumab (CCR4), Moxetumomab pasudotox (CD22), Nacolomab tafenatox (C242 antigen), Naptumomab estafenatox (5T4), Namatumab (RON), Necitumumab (EGFR), Nimotuzumab (EGFR), Nivolumab (lgG4), Ofatumumab (CD20), Olaratumab (PDGF-R a), Onartuzumab (human scatter factor receptor kinase), Oportuzumab monatox (EpCAM), Oregovomab (CA-125), Oxelumab (OX-40), Panitumumab (EGFR), Patritumab (HER3), Pemtumoma (MUC1), Pertuzuma (HER2/neu), Pintumomab (adenocarcinoma antigen), Pritumumab (vimentin), Racotumomab (N- glycolylneuraminic acid), Radretumab (fibronectin extra domain-B), Rafivirumab (rabies virus glycoprotein), Ramucirumab (VEGFR2), Rilotumumab (HGF), Rituximab (CD20), Robatumumab (IGF-1 receptor), Samalizumab (CD200), Sibrotuzumab (FAP), Siltuximab (IL6), Tabalumab (BAFF), Tacatuzumab tetraxetan (alpha-fetoprotein), Taplitumomab paptox (CD 19), Tenatumomab (tenascin C), Teprotumumab (CD221), Ticilimumab (CTLA4), Tigatuzumab (TRAIL-R2), TNX-650 (IL13), Tositumomab (CD20), Trastuzumab (HER2/neu), TRBS07 (GD2), Tremelimumab (CTLA4), Tucotuzumab celmoleukin (EpCAM), Ublituximab (MS4A1), Urelumab (4-1 BB), Volociximab (integrin a5b1), Votumumab (tumor antigen CTAA 16.88), Zalutumumab (EGFR), and Zanolimumab (CD4).
"Formulation/Complexation of the RNA of the composition"
In preferred embodiments, the at least one RNA of the composition is complexed or associated with at least one further compound to obtain a formulated composition. A formulation in that context may have the function of a transfection agent. A formulation may also have the function of protecting the RNA from degradation, e.g. to allow storage, shipment, etc.
In embodiments, the at least one RNA of the composition is formulated with at least one compound, e.g. peptides, proteins, lipids, polysaccharides, and/or polymers.
In embodiments, the at least one RNA of the composition is formulated with at least one cationic (cationic or preferably ionizable) or polycationic compound (cationic or preferably ionizable). In preferred embodiments, the at least one RNA of the composition is complexed or associated with or at least partially complexed or partially associated with one or more cationic (cationic or preferably ionizable) or polycationic compound.
In preferred embodiments, the at least one cationic or polycationic compound is selected from a cationic or polycationic polymer, a cationic or polycationic polysaccharide, a cationic or polycationic lipid, a cationic or polycationic protein, a cationic or polycationic peptide, or any combinations thereof. Preferably, the at least one cationic or polycationic compound is a cationic or polycationic lipid. Further cationic or polycationic compounds being suitable in the context of the invention may be selected from p.88, line 24 to p.89, line 10 in WO2021156267, the respective disclosure herewith incorporated by reference. In particularly preferred embodiments, the at least one RNA of the composition is formulated in lipid-based carriers.
The term “lipid-based carrier” as used herein encompasses lipid-based delivery systems for nucleic acid, preferably RNA, that comprise a lipid component. A lipid-based carrier may additionally comprise other components suitable for formulating a nucleic acid including a cationic or polycationic polymer, polysaccharide, protein, and/or peptide.
In embodiments, the lipid-based carriers of the composition are selected from liposomes, LNPs, lipoplexes, solid lipid nanoparticles, lipo-polyplexes, and/or nanoliposomes.
In particularly preferred embodiments, the lipid-based carriers of the composition are LNPs.
LNPs are microscopic lipid particles having a solid or partially solid core. In an LNP, the RNA may be encapsulated or incorporated in the lipid portion of the LNP enveloped by some or the entire lipid portion of the LNP. An LNP may comprise any lipid capable of forming a particle to which the RNA may be attached, or in which the RNA may be encapsulated.
In particularly preferred embodiments, the LNPs of the composition encapsulate the at least one RNA of the invention.
The term “encapsulation” as used herein refers to the essentially stable combination of RNA with one or more lipids into larger complexes or assemblies such as lipid-based carriers, preferably LNPs, preferably without covalent binding of the RNA. The encapsulated RNA may be completely or partially located in the interior of the lipid-based carrier (e.g. the lipid portion and/or an interior space) and/or within the lipid layer/membrane of the lipid-based carriers.
In embodiments, the lipid-based carriers, preferably the LNPs, comprise at least one or more lipids selected from at least one aggregation-reducing lipid, at least one cationic lipid or ionizable lipid, at least one neutral lipid or phospholipid, or at least one steroid or steroid analogue, or any combinations thereof.
In preferred embodiments, the lipid-based carriers, preferably the LNPs, comprise an (i) aggregation-reducing lipid, (ii) a cationic lipid or ionizable lipid, (iii) a neutral lipid or phospholipid, (iv) and a steroid or steroid analogue. In other preferred embodiments, the lipid-based carriers, preferably the LNPs, comprise an (i) aggregation-reducing lipid, (ii) a cationic lipid or ionizable lipid, (iii) two different neutral lipids or phospholipids, (iv) and a steroid or steroid analogue.
Aggregation reducing lipids / polymer conjugated lipids
In preferred embodiments, the lipid-based carriers, preferably the LNPs, comprise at least one aggregation reducing lipid or aggregation reducing moiety.
The term “aggregation reducing moiety” refers to a molecule comprising a moiety suitable of reducing or preventing aggregation of the lipid-based carriers, preferably the LNPs. The term “aggregation reducing lipid” refers to a molecule comprising both a lipid portion and a moiety suitable of reducing or preventing aggregation of the lipid-based carriers. Under storage conditions or during formulation, the lipid-based carriers such as LNPs may undergo charge-induced aggregation, a condition which can be undesirable for the stability of the lipid-based carriers. Therefore, it can be desirable to include a compound or moiety which can reduce aggregation, for example by sterically stabilizing the lipid- based carriers. Such a steric stabilization may occur when a compound having a sterically bulky but uncharged moiety that shields or screens the charged portions of a lipid-based carriers from close approach to other lipid-based carriers in the composition. In the context of the invention, stabilization of the lipid-based carriers, preferably the LNPs, is achieved by including lipids which may comprise a lipid bearing a sterically bulky group which, after formation of the lipid-based carrier, is preferably located on the exterior of the lipid-based carrier. Suitable aggregation reducing groups include hydrophilic groups, e.g. monosialoganglioside GM1 , polyamide oligomers (PAO), or certain polymers, such as poly(oxyalkylenes), e.g., polyethylene glycol) or polypropylene glycol).
In preferred embodiments, the aggregation reducing lipid is a polymer conjugated lipid.
The term “polymer conjugated lipid” refers to a molecule comprising both a lipid portion and a polymer portion, wherein the polymer is suitable of reducing or preventing aggregation of lipid-based carriers comprising the RNA. A polymer has to be understood as a substance or material consisting of very large molecules, or macromolecules, composed of many repeating subunits. A suitable polymer in the context of the invention may be a hydrophilic polymer. An example of a polymer conjugated lipid is a PEGylated or PEG-conjugated lipid. Lipids comprising a polymer as aggregation reducing group are herein referred to as “polymer conjugated lipid".
In preferred embodiments, the aggregation reducing lipid is a polymer conjugated lipid selected from a PEG- conjugated lipid or a PEG-free lipid.
In preferred embodiments, the polymer conjugated lipid is a PEG-conjugated lipid. The average molecular weight of the PEG moiety in the PEG-conjugated lipid may range from 500 to 8,000 Daltons (e.g., from 1 ,000 to 4,000 Daltons). In one preferred embodiment, the average molecular weight of the PEG moiety is about 2,000 Daltons. In embodiments, the PEG-conjugated lipid is selected or derived from 1 ,2-dimyristoyl-rac-glycero-3-methoxypolyethylene glycol-2000 (PEG2000 DMG or DMG-PEG 2000), C10-PEG2K, or Cer8-PEG2K.
In preferred embodiments, the polymer conjugated lipid is selected or derived from formula (IV) of WO2018078053, preferably from formula (I a) of WO2018078053. In that context, a preferred polymer-conjugated lipid is selected from ALC-0159 (2[(polyethylene glycol)-2000]-N,N-ditetradecylacetamide).
In embodiments, the aggregation reducing lipid is selected from a PEG-free lipid, e.g. a PEG-free polymer conjugated lipid. Preferably, the aggregation reducing lipid is a PEG-free lipid that comprises a polymer different from PEG.
A PEG-free polymer conjugated lipid may be selected or derived from a “POZ-lipid”. In embodiments, the “POZ lipids" or respectively preferred polymer conjugated lipids are described in WO2023031394, the full disclosure herewith incorporated by reference. In particular, the disclosure relating to polymer conjugated lipids as defined in any one of claims 1 to 8 of WO2023031394 are incorporated by reference.
In embodiments, the polymer conjugated lipid is a PEG-free lipid selected from a POZ-lipid.
Accordingly, in embodiments, the polymer conjugated lipid is a “POZ-lipid”, which preferably is defined as a compound according to formula (POZ): [H] - [linker] - [M], wherein
[H] is a homopolymer moiety comprising at least one polyoxazoline (POZ) monomer unit wherein R is C1-9 alkyl or C2-9 alkenyl, preferably C1 , and n has a mean value ranging from 2 to 200, preferably from 20 to 100, more preferably from 24 to 26 or 45 to 50; [linker] is an optional linker group, and [M] is a lipid moiety. In preferred embodiment in the context of POZ-lipids, the aggregation-reducing lipid is selected or derived from PMOZ 1 , PMOZ 2, PMOZ 3, PMOZ 4, or PMOZ 5 of W02023031394.
wherein R is C1-9 alkyl or C2-9 alkenyl, preferably C1 , and n has a mean value ranging from 2 to 200, preferably from 20 to 100, more preferably from 24 to 26 or 45 to 50; [linker] is an optional linker group, and [M] is a lipid moiety. In preferred embodiment in the context of POZ-lipids, the aggregation-reducing lipid is selected or derived from PMOZ 1 , PMOZ 2, PMOZ 3, PMOZ 4, or PMOZ 5 of W02023031394.

In particularly preferred embodiments, the at least one aggregation-reducing lipid is selected or derived from a POZ- lipid according to or derived from a lipid of formula PMOZ4 of W02023031394.
Accordingly, in embodiments, the at least one aggregation-reducing lipid is selected or derived from ALC-0159, DMG- PEG 2000, C10-PEG2K, Cer8-PEG2K, or a POZ-lipid such as PMOZ4. In preferred embodiments, the at least one aggregation-reducing lipid is ALC-0159. In other preferred embodiments, the at least one aggregation-reducing lipid is a POZ-lipid as defined herein, wherein the POZ-lipid is preferably selected from a PMOZ4 lipid.
Cationic lipids
In embodiments, the lipid-based carriers, preferably the LNPs, comprise at least one cationic or ionizable lipid.
The at least one cationic or ionizable lipid may be cationisable or ionizable, i.e. it becomes protonated as the pH is lowered below the pK of the ionizable group of the lipid, but is progressively more neutral at higher pH values. At pH values below the pK, the lipid is then able to associate with negatively charged nucleic acids. In certain embodiments, the cationic lipid comprises a zwitterionic lipid that assumes a positive charge on pH decrease.
In preferred embodiments, the cationic or ionizable lipid preferably carries a net positive charge at physiological pH. Preferably the cationic or ionizable lipid comprises a quaternary nitrogen group or tertiary nitrogen group, most preferably a tertiary nitrogen group. Accordingly, in preferred embodiments, the at least one cationic or ionizable lipid may be selected from an amino lipid.
Preferably, the at least one cationic lipid or ionizable lipid is selected from an amino lipid, preferably wherein the amino lipid comprises a tertiary amine group.
In preferred embodiments, the at least one cationic or ionizable lipid is a lipid selected or derived from formula (II 1-1) ,
,

In embodiments, the at least one cationic or ionizable lipids is selected from the lipids disclosed in WO2018078053 (i.e. lipids derived from formula I, II, and III, or lipids as specified in claims 1 to 12), the disclosure of WO2018078053 hereby incorporated by reference. In that context, lipids disclosed in Table 7 of WO2018078053 (e.g. lipids derived from formula 1-1 to 141) and lipids disclosed in Table 8 of WO2018078053 (e.g. lipids derived from formula 11-1 to II-36) may be suitably used in the context of the invention. Accordingly, formula 1-1 to formula 1-41 and formula 11-1 to formula II-36 of WO2018078053, and the specific disclosure relating thereto, are herewith incorporated by reference.
In embodiments, the at least one cationic or ionizable lipid is selected or derived from structures 111-1 to III-36 of Table 9 of WO2018078053. Accordingly, formula 111-1 to HI-36 of WO2018078053, and the specific disclosure relating thereto, are herewith incorporated by reference.
In embodiments, the at least one cationic or ionizable lipid is selected or derived from formula HI-3 of WO2018078053. A preferred lipid of said formula HI-3 has the chemical term ((4-hydroxybutyl)azanediyl)bis(hexane-6,1-diyl)bis(2- hexyldecanoate), also referred to as ALC-0315, i.e. CAS Number 2036272-554.
Further suitable cationic lipids may be selected or derived from cationic lipids according to PCT claims 1 to 14 of published patent application WO2021123332, or Table 1 of WO2021123332, the disclosure relating to claims 1 to 14 or Table 1 of WO2021123332 herewith incorporated by reference. Accordingly, suitable cationic lipids may be selected or derived from cationic lipids according to Compound 1 to Compound 27 (C1-C27) of Table 1 of WO2021123332.
In preferred embodiments, the at least one cationic or ionizable lipid is selected or derived from SS-33/4PE-15 (see C23 in Table 1 of WO2021123332). In other preferred embodiments, the at least one cationic or ionizable lipid is selected or derived from HEXA-C5DE-PipSS (see C2 in Table 1 of WO2021123332). In particularly preferred embodiments, the at least one cationic or ionizable lipid is selected or derived from compound C26 (VitE-C4DE-Pip- thioether) as disclosed in Table 1 of WO2021123332.
Accordingly, in embodiments, the at least one cationic or ionizable lipid is selected or derived from C26, SM-102, SS- 33/4PE-15, HEXA-C5DE-PipSS, or ALC-0315. In preferred embodiments, the at least one cationic or ionizable lipid is ALC-0315. In other preferred embodiments, the at least one cationic or ionizable lipid is C26 (VitE-C4DE-Pip-thioether).
In some embodiments, the lipid-based carriers of the invention comprise two or more (different) cationic lipids as defined herein.
Neutral Lipids
In preferred embodiments, the lipid-based carriers, preferably the LNPs, comprise at least one neutral lipid or phospholipid.
In preferred embodiments, the lipid-based carrier comprises at least one neutral lipid or phospholipid.
The term “neutral lipid” refers to any lipid species that exist in either an uncharged or neutral zwitterionic form at physiological pH. Neutral lipids may be selected from DHPC, DHPC, DOPC, DPPC, DOPG, DPPG, DOPE, POPC, POPE, DOPE-mal, DPPE, DMPE, DSPE, 16-O-monomethyl PE, 16-O-dimethyl PE, 18-1-trans PE, SOPE, transDOPE, 1 ,2-diphytanoyl-sn-glycero-3-phosphoethanolamine (DPhyPE), DPhyPS (1 ,2-diphytanoyl-sn-glycero-3- phospho-L-serine), or mixtures thereof.
In embodiments, the at least one neutral lipid is selected or derived from DSPC, DHPC, DPhyPE, or DPhyPS. Preferred in that context is DSPC. In other embodiments, the at least one neutral lipid is selected or derived from DPhyPE and/or DphyPS. In other embodiments, the lipid-based carrier, preferably the LNP, comprises DPhyPE and DPhyPS. Steroids, steroid analogs or sterols
In preferred embodiments, the lipid-based carrier comprises a steroid, steroid analog, or sterol.
In embodiments, the steroid, steroid analog or sterol is derived or selected from cholesterol, cholesteryl hemisuccinate (CHEMS), or any derivate of these. In preferred embodiments, the lipid-based carrier comprises cholesterol.
Lipid-based carrier compositions
In embodiments, the lipid-based carriers, preferably the LNPs, comprising the at least one RNA comprise
(i) at least one cationic lipid or ionizable lipid, preferably as defined herein;
(ii) at least one or two (e.g. two different) neutral lipids or phospholipids, preferably as defined herein;
(iii) at least one steroid or steroid analogue, preferably as defined herein; and
(iv) at least one aggregation reducing lipid, preferably as defined herein.
In embodiments, the lipid-based carrier, preferably the LNP, comprise (i) to (iv) in a molar ratio of about 20-60% cationic lipid or ionizable lipid, about 5-25% neutral lipid, about 25-55% steroid or steroid analog, and about 0.5-15% aggregation reducing lipid e.g. polymer conjugated lipid.
In preferred embodiments, the lipid-based carrier, preferably the LNP, comprise (i) to (iv) in a molar ratio of about 45- 60% cationic lipid or ionizable lipid, about 5-15% neutral lipid, about 25-45% steroid or steroid analog, and about 0.5- 2.5% aggregation reducing lipid e.g. polymer conjugated lipid.
In embodiments, the lipid-based carrier, preferably the LNP, comprising the nucleic acid comprise (i) a cationic lipid ALC-0315; (ii) a neutral lipid DSPC; (iii) a steroid or steroid analog cholesterol; and (iv) an aggregation reducing lipid ALC-0159; preferably wherein (i) to (iv) are in a molar ratio of about 47.4% cationic lipid, about 10% neutral lipid, about 40.9% steroid or steroid analog, and about 1.7% aggregation reducing lipid, preferably wherein the lipid-based carrier encapsulates the nucleic acid, preferably the RNA.
In embodiments, the lipid-based carrier, preferably the LNP, comprising the nucleic acid comprise (i) a cationic lipid C26 (VitE-C4DE-Pip-thioether); (ii) a neutral lipid DPhyPE and a neutral lipid DPhyPS; (iii) a steroid or steroid analog cholesterol; and (iv) an aggregation reducing lipid selected from a POZ-lipid, preferably from PMOZ 4; preferably wherein (i) to (iv) are in a molar ratio of about 49% cationic lipid, about 10% neutral lipid, about 40% steroid or steroid analog, and about 1% aggregation reducing lipid, preferably wherein the lipid-based carrier encapsulates the RNA.
In embodiments, the lipid-based carrier, preferably the LNP, comprising the nucleic acid comprise (i) a cationic lipid SM-102; (ii) a neutral lipid DSPC; (iii) a steroid or steroid analog cholesterol; and (iv) an aggregation reducing lipid selected from a PEG-DMG; preferably wherein (i) to (iv) are in a molar ratio of about 50% cationic lipid, about 10% neutral lipid, about 38.5% steroid or steroid analog, and about 1.5% aggregation reducing lipid, preferably wherein the lipid-based carrier encapsulates the RNA.
In preferred embodiments, the wt/wt ratio of lipid to RNA in the lipid-based carrier, preferably the LNPs, is from about 10:1 to about 60:1 , e.g. about 40:1. In embodiments, the wt/wt ratio of lipid to RNA is from about 20:1 to about 30:1, e.g. about 25: 1. In other preferred embodiments, the wt/wt ratio of lipid to RNA is in the range of 20 to 60, preferably from 3 to 15, 5 to 13, 4 to 8 or from 7 to 11.
The amount of lipid comprised in the lipid-based carriers such as LNPs may be selected taking the amount of the RNA cargo into account. In one embodiment, these amounts are selected such as to result in an N/P ratio of the lipid-based carriers encapsulating the RNA in the range of about 0.1 to about 20. The N/P ratio is defined as the mole ratio of the nitrogen atoms (“N”) of the basic nitrogen-containing groups of the lipid to the phosphate groups (“P”) of the RNA which is used as cargo. The N/P ratio may be calculated on the basis that, for example, 1 pg RNA typically contains about 3nmol phosphate residues, provided that the RNA exhibits a statistical distribution of bases. The “N”-value of the lipid or lipidoid may be calculated on the basis of its molecular weight and the relative content of permanently cationic and - if present - cationisable groups.
In embodiments, the N/P ratio can be in the range of about 1 to about 50. In preferred embodiments, the range is from about 5 to about 20. In some embodiments, the N/P ratio is at about 17. In some embodiments, the N/P ratio is at about 14. In some embodiments, the N/P ratio is at about 6.
In various embodiments, the composition comprises lipid-based carriers such as LNPs (encapsulating the RNA as defined herein) that have a defined size (particle size, homogeneous size distribution).
The size of a lipid-based carrier such as an LNP is typically described as Z-average size. The term “Z-average size” refers to the mean diameter of particles as measured by dynamic light scattering (DLS) with data analysis using the so- called cumulant algorithm, which provides as results the so-called Z-average with the dimension of a length, and the polydispersity index (PDI), which is dimensionless. The term “dynamic light scattering” or “DLS” refers to a method for analyzing particles in a liquid, wherein the liquid is typically illuminated with a monochromatic light source and wherein the light scattered by particles in the liquid is detected. Suitable DLS protocols and instruments are known in the art.
In preferred embodiments, the lipid-based carriers, preferably the LNPs, have a Z-average size of less than 400nm, preferably less than 300nm, more preferably less than 200nm.
In preferred embodiments, the lipid-based carriers, preferably the LNPs, have a Z-average size ranging from about 50nm to about 200nm, preferably in a range from about 50nm to about 150nm, more preferably from about 50nm to about 120nm.
Preferably, the composition comprises less than about 10%, 9%, 8%, 7%, 6%, 5%, 4%, 3%, 2%, 1% lipid-based carriers that have a particle size exceeding about 500nm. Preferably, the pharmaceutical composition comprises less than about 10%, 9%, 8%, 7%, 6%, 5%, 4%, 3%, 2%, 1 % LNPs that have a particle size smaller than about 20nm.
In some embodiments the lipid-based carriers exhibit a zeta potential in the range of -50 mV to +50 mV, preferably in the range of -25 mV to +25 mV, more preferably in the range of -10 mV to +10 mV, most preferably in the range of -5 mV to +5 mV.
In preferred embodiments, the polydispersity index (PDI) of the lipid-based carriers, preferably the LNPs, is in the range of 0.1 to 0.5. In preferred embodiments, the polydispersity index (PDI) value is less than about 0.3, preferably of less than about 0.2. Typically, the PDI is determined by dynamic light scattering.
In embodiments, at least 70%, 80%, 90%, 95% of the RNA is encapsulated in lipid-based carriers such as LNPs. The percentage of encapsulation may be determined by a RiboGreen assay as known in the art.
In preferred embodiments, at least about 80%, 85%, 90%, 95% of the lipid-based carriers such as LNPs have a spherical morphology, preferably comprising a solid core or a partially solid core. In preferred embodiments, the lipid-based carriers, preferably the LNPs have been generated by combining an aqueous RNA solution with an ethanolic lipid solution using a mixing means at total flow rates above 15ml/min.
Suitably, the lipid-based carriers have been prepared using according to the general procedures described in PCT Pub. Nos. WO2015199952, W02017004143 and WO2017075531 , the full disclosures of which are incorporated herein by reference. Accordingly, in preferred embodiments, the composition comprises purified lipid-based carriers, preferably purified LNPs, encapsulating an RNA as defined herein.
In preferred embodiments, the surface of the lipid-based carriers, preferably the LNPs, is uncharged at about pH.7.
In preferred embodiments, the lipid-based carriers, preferably the LNPs do not comprise DOTMA and/or DOPE. In preferred embodiments, the RNA is not formulated in a lipoplex formulation, in particular not formulated in a lipoplex formulation comprising DOTMA and/or DOPE.

According to preferred embodiments, the RNA of the invention comprises at least one cds encoding at least one antigen, wherein the at least one antigen that is selected from or derived from tumor antigens, viral antigens, bacterial antigens, protozoal antigens, fungal antigens, self-antigens or allogenic antigens, preferably tumor antigens.
Suitable antigens in the context of the invention are also specified in paragraph “Antigen” provided above. Preferred embodiments in that context are herein specified in more detail.
In preferred embodiments in that context, the at least one antigen is a tumor-specific antigen or tumor associated antigen.
In preferred embodiments, the at least one tumor specific antigen or tumor associated antigen is selected from mutation antigen, neoantigen, unique antigen, differentiation tumor antigen, overexpressed tumor antigen, virus tumor antigen or bacterial tumor antigen.
In embodiments, the tumor antigen that is encoded by the cds of the RNA is selected or derived from at least one of List 1 as defined above. In preferred embodiments, the tumor antigen that is encoded by the cds of the RNA is selected or derived from at least one of List 2 as defined above. In preferred embodiments, the tumor antigen that is encoded by the cds of the RNA is selected or derived from at least one of List 3 as defined above.
In particularly preferred embodiments, the tumor antigen that is encoded by the cds of the RNA is selected from Claudin6 (CLDN6) or PRAME.
In some embodiments, the RNA comprises at least one cds encoding at least one epitope, at least two epitopes, at least three epitopes, at least four epitopes, at least five epitopes, at least six epitopes, at least seven epitopes, at least eight epitopes, at least nine epitopes, or at least ten epitopes, wherein the epitopes derived or selected from the same antigen or from different antigens.
In some embodiments, the RNA comprises at least one cds encoding at least one linker. The term “linker” preferably refers to peptide linkers, i.e. typically short (i.e. comprising 1-150 amino acids, preferably 1-50 amino acids, more preferably 1 to 20 amino acids), linear amino acid sequences connecting or linking two polypeptide sequences. Linkers according to the invention may be derived from any protein of human, animal, plant, bacterial or viral origin. Linkers according to the invention may be naturally occurring or artificial (i.e. synthetic or non-naturally occurring) linkers.
Preferably, the linker(s) is/are non-immunogenic, i.e. do not trigger an immune response. Linkers may be employed to connect or link at least two components, e.g. two epitopes or an epitope and a T helper cell epitope encoded by the RNA of the invention. The cds of the RNA according to the invention may encode at least one linker, or a plurality of at least 2, 3, 4, 5, 6, 7, 8, 9 or 10 identical or different linkers, as described herein. In case a plurality of linkers is encoded by the RNA it is particularly preferred that the linkers differ in their amino acid sequence and/or nucleic acid sequence encoding the respective linkers. Linkers of interest in the context of the present invention are inter alia disclosed in W02002014478, W02001008636, W02013171505, W02008017517 and WO1997047648, which are incorporated by reference in their entirety as well.
In some embodiments, the RNA comprises at least one cds encoding at least one T helper epitope. The term “T helper epitope” refers to an epitope capable of binding to MHC molecules, preferably MHC Class II molecules, thereby being recognized by CD4+ T helper cells. Preferably, such T helper epitopes induce or enhance CD4+ Th cell activation, differentiation, and/or proliferation. Activated CD4+ Th cells are may preferably (1) (directly or indirectly) induce or enhance cytotoxic T lymphocyte differentiation, and/or proliferation, and/or (2) (directly or indirectly) induce or enhance antibody-producing plasma cell differentiation, and/or proliferation. In this respect, ‘‘directly or indirectly” means that activated CD4+ Th cells may induce or enhance the respective immune responses either via direct interaction with target cells or precursors thereof (e.g. B cells) or indirectly via interacting with other cells (e.g. dendritic cells) that in turn directly interact with target cells or precursors thereof (e.g. modified CD8+ T cells).
In the context of the present invention, preferred T helper epitopes include the T helper epitopes disclosed in WO2001062284, WO2010023247, W02004058297, W02004000873 and W02006113792. Particularly preferred T helper cell epitopes in the context of the present invention include naturally occurring or artificial T helper epitopes derived from PADRE; TpD (tetanus toxoid and diphtheria toxoid, separated by an internal cathepsin cleavage site); Hepatitis C virus (Core); Hepatitis C virus (E1); Hepatitis C virus (E2); Hepatitis C virus (NS2); Hepatitis C virus (NS3); Hepatitis C virus (NS4); Hepatitis C virus (NS4a); Hepatitis C virus (NS4b); Hepatitis C virus (NS5a); Hepatitis C virus (NS5b); Influenza A; Influenza B; Measles virus (F protein); Canine distemper virus (Fusion protein); Mucin-1 ; Foot- and-mouth disease virus (VP3); Foot-and-mouth disease virus; Clostridium tetani; Human immunodeficiency virus 1 (gp120 protein); Human immunodeficiency virus 1 (gag protein); Human immunodeficiency virus 1 (envelope glycoprotein); Tetanus toxoid; Human papilloma virus 16 (E17); Diphtheria toxoid; Plasmodium falciparum (CS) or a functional fragment, variant or derivative thereof.
In some embodiments, the RNA comprises at least one cds encoding at least one additional amino acid sequence derived from at least one immune response activating Signal transduction protein located in the external plasma membrane, wherein said at least one immune response activating signal transduction protein located in the external plasma membrane is CTLA4, and wherein said at least one additional amino acid sequence comprises or consists of at least one transmembrane domain from CTLA4.
In some embodiments the RNA comprising at least one cds encoding at least one amino acid sequence of the following formula, preferably in 5'-^3‘ direction:
-(SIG) a -(L) b -[(AN) c -(L) d ] e -[(IM) m -(L) n ] o -(TMD/TMCD) p - , wherein "SIG" encodes a signal peptide; "L" encodes a linker sequence; each "AN" encodes an identical or different antigenic peptide or protein, preferably a tumor antigen as defined before, "IM" encodes a helper epitope;
"TMD/TMCD" encodes an amino acid sequence derived from an immune response signal transduction protein located in the external plasma membrane, preferably a transmembrane domain, preferably from CTLA4, and optionally a cytoplasmic domain, preferably from CTLA4; b, d, m, n, o is each independently an integer selected from 0, 1 , 2, 3, 4, 5, 6, 7, 8, 9, and 10; a, c, e, p is each independently an integer selected from 1 , 2, 3, 4, 5, 6, 7, 8, 9 and 10.
In preferred embodiments, the RNA comprises at least one cds encoding at least one antigen, preferably the full length of the antigen.
In preferred embodiments, the RNA comprises at least one cds encoding at least one epitope that is targeted by the antigen receptor of the transferred modified T cells.
In preferred embodiments, the RNA is a modified and/or stabilized nucleic acid.
According to preferred embodiments, the RNA may thus be provided as a “stabilized RNA” that is to say an RNA showing improved resistance to in vivo degradation and/or an RNA showing improved stability in vivo, and/or an RNA showing improved translatability in vivo.
Preferably, the RNA of the present invention may be provided as a “stabilized RNA”.
In the following, suitable modifications/adaptations are described that are capable of “stabilizing” the RNA.
In particularly preferred embodiments, the RNA comprises at least one codon modified cds.
In preferred embodiments, the at least one cds of the RNA is a codon modified cds. Suitably, the amino acid sequence encoded by the at least one codon modified cds is not modified compared to the amino acid sequence encoded by the corresponding wild type or reference cds.
The term “codon modified cds” relates to coding sequences that differ in at least one codon (triplets of nucleotides coding for one amino acid) compared to the corresponding wild type or reference cds. Suitably, a codon modified cds in the context of the invention may show improved resistance to in vivo degradation and/or improved stability in vivo, and/or improved translatability in vivo. Codon modifications in the broadest sense make use of the degeneracy of the genetic code wherein multiple codons may encode the same amino acid and may be used interchangeably to optimize/modify the cds for in vivo applications.
In embodiments, the at least one cds is a codon modified cds selected from a C maximized cds (as further defined in WO2021239880 [p.122, lines 33 to 39] which is hereby incorporated by reference); a CAI maximized cds (as further defined in WO2021239880 [p.123, lines 33 to 44] which is hereby incorporated by reference); a human codon usage adapted cds (as further defined in WO2021239880 [p.123, lines 7 to 17] which is hereby incorporated by reference); a G/C content modified cds (as further defined in WO2021239880 [p.123, lines 19 to 31] which is hereby incorporated by reference); a G/C optimized cds (“opt1”); or any combination thereof.
In preferred embodiments, the G/C content of the at least one cds is optimized compared to the G/C content of the corresponding wild type or reference cds (“G/C optimized”). “Optimized” in that context means that the G/C content of the cds is preferably increased to the essentially highest possible content. The generation of a G/C optimized cds is suitably carried out according to W02002098443, which is hereby incorporated by reference.
In preferred embodiments, the G/C content of the at least one cds is increased by at least 10%, 15%, or 20% compared to the G/C content of the corresponding wild type or reference cds. In preferred embodiments, the at least one cds has a G/C content of at least about 50%, 55%, or 60%. In particular embodiments, the at least one cds has a G/C content of at least about 50%, 51%, 52%, 53%, 54%, 55%, 56%, 57%, 58%, 59%, 60%, 61%, 62%, 63%, 64%, 65%, 66%, 67%, 68%, 69%, or 70%.
UTR
In preferred embodiments, the RNA comprises at least one untranslated region (UTR).
The term “untranslated region”, “UTR” or “UTR element” refers to a part of an RNA typically located 5’ or 3' of a cds that is not translated into protein. An UTR may comprise elements for controlling gene expression, also called regulatory elements. Such regulatory elements may be, e.g., ribosomal binding sites, miRNA binding sites, promotor elements. Regulatory elements may determine turnover, stability, and localization of the nucleic acid, in particular the RNA. UTRs may also harbour sequence elements that enhance translation.
Preferably, the at least one UTR is selected from at least one 5’-UTR and/or at least one 3’-UTR, preferably selected from at least one heterologous 5-UTR and/or at least one heterologous 3-UTR.
The term “heterologous” or “heterologous UTR” as used herein refers to a nucleic acid sequence or UTR that is not from the same gene, the same genomic fusion, or the same naturally occurring transcript. Heterologous sequences do naturally (in nature) not occur in the same RNA molecule.
In preferred embodiments, the RNA of the invention comprises at least one 3-UTR.
The term “3’-untranslated region” or “3-UTR” refers to a part of an RNA located 3’ (i.e. downstream) of a cds and which is not translated into protein. A 3-UTR may be part of an RNA located between a cds and an (optional) terminal poly(A) sequence. A 3-UTR may comprise elements for controlling gene expression, also called regulatory elements. Such regulatory elements may be, e.g., ribosomal binding sites, miRNA binding sites.
Preferably, the RNA comprises at least one 3-UTR, which may be derivable from a gene that relates to an RNA with enhanced half-life (i.e. that provides a stable RNA).
In some embodiments, the 3-UTR comprises one or more of a polyadenylation signal, a binding site for proteins that affect RNA stability of location in a cell, or one or more miRNA or binding sites for miRNAs.
In preferred embodiments, the RNA comprises at least one 3’-UTR, wherein the at least one 3-UTR comprises or consists of a nucleic acid sequence derived or selected from a 3-UTR of a gene selected from PSMB3, AES-12S, ALB7, alpha-globin (HBA1 , HBA2), ANXA4, beta-globin (HBB), CASP1 , COX6B1, FIG4, GH1 , GNAS, NDUFA1, RPS9, SLC7A3, TUBB4B, or from a homolog, a fragment, or variant of any one of these genes.
In preferred embodiments, the at least one 3-UTR that is derived or selected from PSMB3, AES-12S, ALB7, alphaglobin (HBA1, HBA2), ANXA4, beta-globin (HBB), CASP1 , COX6B1 , FIG4, GH1 , GNAS, NDUFA1, RPS9, SLC7A3, TUBB4B comprises or consist of a nucleic acid sequence being identical or at least 70%, 80%, 85%, 86%, 87%, 88%, 89%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identical to any one of SEQ ID NOs: 134-191, or a fragment or a variant of any of these.
In particularly preferred embodiments, the RNA comprises a 3-UTR derived or selected from a PSMB3 gene. Preferably, the at least one heterologous 3 -UTR derived or selected from PSMB3, comprises or consist of a nucleic acid sequence being identical or at least 70%, 80%, 85%, 86%, 87%, 88%, 89%, 90%, 91 %, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identical to any one of SEQ ID NOs: 134, 135, 180-191 , or a fragment or a variant thereof, preferably SEQ ID NO: 135, or a fragment or a variant thereof.
In other embodiments, the at least one 3’-UTR comprises or consist of a nucleic acid sequence being identical or at least 70%, 80%, 85%, 86%, 87%, 88%, 89%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identical to any one of SEQ ID NOs: 164-171, or a fragment or a variant of any of these.
In preferred embodiments, the RNA of the invention comprises at least one 5’-UTR.
The terms “5’-untranslated region” or “5’-UTR" refers to a part of a nucleic acid located 5’ (i.e. “upstream”) of a cds and which is not translated into protein. A 5-UTR may be part of a nucleic acid located 5’ of the cds. Typically, a 5-UTR starts with the transcriptional start site and ends before the start codon of the cds. A 5-UTR may comprise elements for controlling gene expression, also called regulatory elements. Such regulatory elements may be, e.g., ribosomal binding sites, miRNA binding sites.
Preferably, the RNA comprises at least one 5-UTR, which may be derivable from a gene that relates to an RNA with enhanced half-life (i.e. that provides a stable RNA).
In some embodiments, the 5-UTR comprises one or more of a binding site for proteins that affect RNA stability or RNA location in a cell, or one or more miRNA or binding sites for miRNAs.
In preferred embodiments, the RNA comprises at least one 5'-UTR, wherein the at least one 5’-UTR comprises a nucleic acid sequence derived or selected from a 5-UTR of gene selected from HSD17B4, AIG1 , alpha-globin (HBA1 , HBA2), ASAH1, ATP5A1 , COX6C, DPYSL2, HHV5, MDR, MP68, NDUFA4, NOSIP, RPL31, RPL32, RPL35A, SLC7A3, synthetic origin, TUBB4B, UBQLN2, or from a homolog, a fragment or variant of any one of these genes,
In preferred embodiments, the at least one 5’-UTR derived or selected from HSD17B4, AIG1 , alpha-globin (HBA1 , HBA2), ASAH1 , ATP5A1 , COX6C, DPYSL2, HHV5, MDR, MP68, NDUFA4, NOSIP, RPL31, RPL32, RPL35A, SLC7A3, synthetic origin, TUBB4B, UBQLN2 comprises or consist of a nucleic acid sequence being identical or at least 70%, 80%, 85%, 86%, 87%, 88%, 89%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identical to any one of SEQ ID NOs: 80-133, or a fragment or a variant of any of these.
In particularly preferred embodiments, the RNA comprises a 5-UTR derived or selected from a HSD17B4 gene. Preferably, the at least one heterologous 5’-UTR derived or selected from HSD17B4, comprises or consists of a nucleic acid sequence being identical or at least 70%, 80%, 85%, 86%, 87%, 88%, 89%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identical to any one of SEQ ID NOs: 80, 81 , 132, 133 or a fragment or a variant thereof, preferably SEQ ID NO: 81 , or a fragment or a variant thereof.
In other embodiments, the at least one 5-UTR comprises or consist of a nucleic acid sequence being identical or at least 70%, 80%, 85%, 86%, 87%, 88%, 89%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identical to any one of SEQ ID NOs: 122-129, or a fragment or a variant of any of these.
In embodiments, the RNA comprises at least one cds as specified herein operably linked to a 3-UTR and/or a 5-UTR selected from the 5’-UTR/3’-UTR combinations (5’UTR/3’UTR) provided in WO2021239880 [p.127, line 35 to p.128, line 2], which is hereby incorporated by reference.
In preferred embodiments, the at least one 5-UTR is selected from HSD17B4 and the at least one 3 -UTR is selected from PSMB3 (a-1 (HSD17B4/PSMB3)). Accordingly, in particularly preferred embodiments, the RNA comprises at least one cds as defined herein encoding at least one antigen as defined herein, wherein said cds is operably linked to a HSD17B45-UTR and a PSMB3 3-UTR (HSD17B4/PSMB3 (a-1)). This embodiment is particularly beneficial for effectively expressing the antigen in humans.
In embodiments, the RNA is monocistronic, bicistronic, or multicistronic, preferably the RNA is monocistronic
In preferred embodiments, the RNA comprises a ribosome binding site, also referred to as “Kozak sequence”, that is identical to or at least 80%, 85%, 90%, 95% identical to any one of SEQ ID NOs: 70, 71 or sequences GCCGCCACC (DNA), GCCGCCACC (RNA), GCCACC (DNA), GCCACC (RNA), ACC (DNA) or ACC (RNA), or fragments or variants of any of these, preferably ACC (RNA).
In preferred embodiments, the RNA of the invention is an artificial RNA and/or an isolated RNA.
The term “artificial RNA” as used herein is intended to refer to an RNA that does not occur naturally. In other words, an artificial RNA may be understood as a non-natural RNA molecule. Such an RNA molecule may be non-natural due to its individual sequence (e.g. G/C content modified cds, UTRs) and/or due to other modifications, e.g. structural modifications of nucleotides. Typically, artificial RNA may be designed and/or generated by genetic engineering to correspond to a desired artificial sequence of ribonucleotides. In this context, an artificial RNA is a sequence that may not occur naturally, i.e. a sequence that differs from the wild type sequence/the naturally occurring sequence by at least one nucleotide. The term “artificial RNA” is not restricted to mean “one single molecule” but is understood to comprise an ensemble of essentially identical RNA molecules.
The term “isolated RNA” does not comprise a cell or a subject that comprises said RNA but relates to the RNA as an isolated molecule or ensemble of isolated molecules. For example, the “isolated RNA” can be an RNA isolated or purified from a cell (e.g. cell culture, bacterial culture), or can be an RNA isolated from an RNA in vitro transcription.
The RNA may be any type of RNA that comprises a cds as defined herein including any type of single stranded RNA, any type of double stranded RNA, any type of linear RNA, and any type of circular RNA.
In preferred embodiments, the RNA is selected from mRNA, circular RNA, replicon RNA or self-replicating RNA, or viral RNA, preferably mRNA or a circular RNA.
In embodiments, the RNA is a circular RNA. A “circular RNA” (circRNAs) is an RNA connected to form a circle and therefore does not comprise a 3’ or 5’ terminus. Said circRNA comprises at least one cds as defined herein. CircRNA construct designs can be taken from WG2023073228, claims 1 to 51 , hereby incorporated by reference.
In other embodiments, the RNA is a replicon RNA or self-replicating RNA. Such constructs may encode replicase elements derived from e.g. alphaviruses (e.g. SFV, SIN, VEE, or RRV) and a cds as defined herein.
In particularly preferred embodiments, the RNA of the invention is selected from an mRNA.
In the context of the invention, an mRNA is preferred to provide the antigen because mRNA allows for regulated dosage, transient expression, complete degradation of the mRNA after protein synthesis, and does not pose the risk of insertional mutations. Preferably, the mRNA is non-replicative.
Preferably, the RNA comprises about 50 to about 20000 nucleotides, or about 500 to about 10000 nucleotides, or about 1000 to about 10000 nucleotides, or preferably about 1000 to about 5000 nucleotides. PoMNjsequences, histone stem loops:
In preferred embodiments, the RNA comprises at least one poly(N) sequence, e.g. at least one poly(A) sequence, at least one poly(U) sequence, at least one poly(C) sequence, or combinations thereof.
In preferred embodiments, the RNA comprises at least one poly(A) sequence. In some embodiments, the RNA comprises at least two, three, or more poly(A) sequences.
The term “poly(A) sequence” refers to a sequence of up to 1000 adenosines typically located at the 3’-end of a linear RNA. Typically, a poly(A) sequence is homopolymeric. Alternatively, a poly(A) sequence may be interrupted by at least one nucleotide different from an adenosine.
In preferred embodiments, the at least one poly(A) sequence may comprise about 20 to about 500 adenosine nucleotides, about 40 to about 250 adenosine nucleotides, preferably about 60 to about 150 adenosine nucleotides. In embodiments, the at least one poly(A) sequence comprises about or more than 50, 64, 75, 100, 150, 200, 300, 400, or 500 adenosines. In preferred embodiments, the at least one poly(A) sequence comprises about 60 to about 150 adenosine nucleotides, preferably about 100 adenosine nucleotides (A100).
In alternative embodiments, the RNA comprises at least one interrupted poly(A) sequence, wherein the poly(A) sequence is interrupted by non-adenosine nucleotides, preferably by about 10 non-adenosine (N10) nucleotides. In that context, a poly(A) sequence A30-N10-A70 is preferred.
In preferred embodiments, the at least one poly(A) sequence as defined herein is located directly at the 3’ terminus of the nucleic acid, preferably the RNA. Accordingly, the 3’-terminal nucleotide in the polynucleotide chain is the 3’- terminal A nucleotide of the at least one poly(A) sequence. In other words, the 3’ terminus of the nucleic acid consists of a poly(A) sequence (e.g. A100 or A30-N10-A70) and therefore terminates with an A.
Advantageously, having a 3’ terminus ending on an adenosine may decrease the induction of interferons, e.g. IFNalpha, by the RNA of the invention if, e.g., administered as a medicament to a human. This is important as the induction of interferons, e.g. IFNalpha, is thought to be one main factor for induction of side effects.
In particularly preferred embodiments, the nucleic acid, preferably the RNA, comprises a poly(A) sequence of about 100 consecutive adenosines (A100) located directly at the 3’ terminus of the RNA.
In alternative embodiments, the RNA comprises at least one poly(A) sequence obtained by enzymatic polyadenylation, wherein the majority of RNA molecules comprise about 100 (+/- 20) to about 500 (+/- 100) adenosine nucleotides, preferably about 100 (+/- 20) to about 200 (+/- 40) adenosine nucleotides.
In preferred embodiments, the RNA comprises at least one histone stem-loop (hSL) or histone stem loop structure. A hSL may be located in an UTR region, preferably in the 3’-UTR. The term refers to nucleic acid sequences that forms a stem-loop secondary structure. A hSL may be derived from formulae (I) or (II) of WO2012019780, or preferably from the specific formulae (la) or (Ila) of WO2012019780, that are hereby incorporated by reference.
In preferred embodiments, the at least one hSL sequence comprises or consists of a nucleic acid sequence identical or at least 70%, 80%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identical to any one of SEQ ID NOs: 71 , 72, or a fragment or variant of any of these, preferably SEQ ID NO: 72, or a fragment or variant thereof. In preferred embodiments, the RNA comprises a 3’-terminal sequence element. The 3’-terminal sequence element represents the 3’ terminus of the RNA. A 3’-terminal sequence element may comprise at least one poly(N) sequence as defined herein and, optionally, at least one hSL as defined herein.
In embodiments, the at least one 3’-terminal sequence element comprises or consists of an RNA sequence being identical or at least 70%, 80%, 90%, 91 %, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identical to any one of SEQ ID NOs: 73-79, or a fragment or variant of these sequences. In preferred embodiments, the at least one 3’-terminal sequence element comprises or consists of a nucleic acid sequence being identical or at least 70%, 80%, 90%, 91 %, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identical to SEQ ID NOs: 73 or 74, or a fragment or variant thereof.
In preferred embodiments, the RNA comprises a 5'-terminal sequence element comprising or consisting of an RNA sequence being identical or at least 70%, 80%, 90%, 91 %, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identical to sequence AGGAGA, or a fragment or variant thereof. Such a 5’-terminal sequence element may comprise e.g. a binding site for T7 RNA polymerase. Further, the first nucleotide of said 5’-terminal start sequence may preferably comprise a 2'0 methylation, e.g. 2’0 methylated guanosine or a 2’0 methylated adenosine.
In particularly preferred embodiments, the RNA of the invention is a therapeutic RNA. Accordingly, the RNA is suitably used in a therapeutic context, in particular to provide at least one antigen for use in a method of treatment or prophylaxis of a disease, disorder or condition.
Modified nucleotides
According to various embodiments, the RNA is modified, wherein the modification refers to chemical modifications comprising backbone modifications as well as sugar modifications or base modifications.
A backbone modification is a chemical modification in which phosphates of the backbone of the nucleotides of the RNA are modified. A sugar modification is a chemical modification of the sugar of the nucleotides of the RNA. A base modification is a chemical modification of the base moiety of the nucleotides of the RNA. In this context, nucleotide analogues are preferably selected from nucleotide analogues which are applicable for transcription and/or translation.
Accordingly, in preferred embodiments, the RNA of the invention comprises at least one modified nucleotide. In embodiments, the RNA comprises at least one modified nucleotide selected from WO2021239880 [p.136, line 17 to p.137, line 19], which is hereby incorporated by reference. In embodiments, the RNA may comprise modified uridine nucleotides that preferably comprise a chemical modification in the 5-position of the uracil. Suitable modified uridine nucleotides may be selected from WO2021239880 [p.137, lines 15 to 19], which is hereby incorporated by reference.
In some embodiments, the at least one modified nucleotide is selected from pseudouridine, N1 -methylpseudouridine, N1 -ethylpseudouridine, 2-thiouridine, 4’-thiouridine, 5-methylcytosine, 5-methyluridine, 2-thio-1-methyl-1 -deazapseudouridine, 2-thio-1-methyl-pseudouridine, 2-thio-5-aza-uridine, 2-thio-dihydropseudouridine, 2-thio-dihydrouridine, 2-thio-pseudouridine, 4-methoxy-2-thio-pseudouridine, 4-methoxy-pseudouridine, 4-thio-1-methyl-pseudouridine, 4- thio-pseudouridine, 5-aza-uridine, dihydropseudouridine, 5-methoxyuridine and 2’-O-methyl uridine. Particularly preferred in that context are pseudouridine (ip) and N1 -methylpseudouridine (ml ip).
In some embodiments, essentially all, e.g. essentially 100% of the uracil in the cds or the full RNEA sequence have a chemical modification, preferably a chemical modification in the 5-position of the uracil (e.g. ip and/or ml ip). In preferred embodiments, 100% of the uracil in the full RNA sequence are substituted with N1 -methylpseudouridine (ml i ). Alternatively, 100% of the uracil in the full nucleic acid sequence, preferably the RNA sequence is substituted with pseudouridine (i ).
Incorporating modified nucleotides such as e.g. pseudouridine (qj) or N1 -methylpseudouridine (m1qi) into the cds (or the full RNA sequence) may be advantageous as unwanted innate immune responses (upon administration of the RNA) may be adjusted or reduced (if required).
In alternative embodiments, the RNA does not comprise chemically modified nucleotides. Notably, a 5’-cap structure as defined below is typically not considered to be a chemically modified nucleotide. Accordingly, the RNA comprises a sequence that consists only of G, C, A and U nucleotides and therefore does not comprise modified nucleotides, and optionally comprises a 5’-cap structure.
Using RNA molecules that do not comprise modified nucleotides to provide the antigen as defied herein may be beneficial as stronger T cell responses may be induced (compared to m1i or ip modified RNA).
Cap structures
In preferred embodiments, the RNA comprises a 5’-cap structure.
The term “5’-cap structure” refers to a 5’ modified nucleotide, particularly a guanine nucleotide, positioned at the 5’-end of an RNA. The 5’-cap structure is typically connected via a 5’-5’-triphosphate linkage to the RNA.
Accordingly, in preferred embodiments, the RNA comprises a 5’-cap structure, preferably m7G, capO, cap1, cap2, a modified capO or a modified cap1 structure. In particularly preferred embodiments, the RNA comprises a cap1 structure or a modified cap1 structure.
Suitably, a 5'-cap structure may be formed in chemical RNA synthesis or in RNA in vitro transcription (co-transcriptional capping) using cap analog. In preferred embodiments, the 5’-cap structure may be added co-transcriptionally using a cap analog, preferably a cap1 analog, as defined herein, preferably in an RNA in vitro transcription reaction.
The term “cap analog” as used herein refers to a non-polymerizable di-nucleotide or tri-nucleotide that has cap functionality in that it facilitates translation or localization, and/or prevents degradation of an RNA molecule when incorporated at the 5’-end of the RNA molecule. Non-polymerizable means that the cap analogue will be incorporated only at the 5’-terminus because it does not have a 5’ triphosphate and therefore cannot be extended in the 3'-direction by a template-dependent polymerase, particularly, by template-dependent RNA polymerase.
In embodiments, a cap1 or a modified cap1 structure is generated using a cap analog, preferably a tri-nucleotide cap analog. Any cap analog derivable from the structures defined in claims 1-13 of WO2017053297 (hereby incorporated by reference) or, alternatively, any cap analog derivable from the structures defined in claims 1 -37 of W02023007019 (hereby incorporated by reference) may be suitably used to co-transcriptionally generate a cap1 or a modified cap1.
In preferred embodiments, the cap1 structure is formed via co-transcriptional capping using tri-nucleotide cap analog m7G(5’)ppp(5’)(2’OI\/leA)pG, m7G(5’)ppp(5’)(2’OMeG)pG, or m7(3’OMeG)(5’)ppp(5’)m6(2’OMeA)pG. A particularly preferred cap1 analog in that context is m7G(5’)ppp(5’)(2’OMeA)pG. In other preferred embodiments, the cap1 structure is a modified cap1 structure and is formed using co-transcriptional capping using tri-nucleotide cap analogue 3’0Me-m7G(5’)ppp(5’)(2’0MeA)pG. Alternatively, the 5’-cap structure may be formed via enzymatic capping using capping enzymes (e.g. vaccinia virus capping enzymes and/or 2’-0 methyltransferases) to generate capO, cap1 or cap2 structures.
It is preferred that at least 80%, 85%, 90%, 95% of the RNA molecules comprise a cap structure, preferably a cap1 structure, as determined by a capping assay (e.g. via an assay as described in cl. 27 to 46 of W02015101416).
In preferred embodiments, the RNA comprises a 5’-terminal sequence comprising or consisting of a nucleic acid sequence being identical or at least 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identical to anyone AGGAGA, GGGAGA, GGGAAA, AGAAUA, AGAUUA, GAUGGG or GGGCG, or a fragment or variant of these, preferably AGGAGA. Such a 5'-terminal sequence element may comprise a binding site for an RNA polymerase. Preferably, the first nucleotide of said 5’-terminal sequence comprises a 2’0 methylation (2'0meA or 2’OMeG).
Further RNA features
In preferred embodiments, the RNA is preferably an in vitro transcribed RNA (e.g. an in vitro transcribed mRNA).
In preferred embodiments, the RNA of the invention is a purified RNA, preferably a purified mRNA.
The term “purified RNA” refers to RNA that has a higher purity after certain purification steps than the starting material. Typical impurities comprise peptides, proteins, spermidine, RNA fragments, dsRNA, free nucleotides, DNA, etc. It is desirable for the “degree of RNA purity” to be as close as possible to 100%. Preferably, a “purified RNA” has a degree of purity of more than 75%, 80%, 85%, 90%, or 95%. The degree of purity may be determined by an analytical HPLC.
In embodiments, the RNA has been purified by (RP)HPLC, AEX, size exclusion chromatography, hydroxyapatite chromatography, tangential flow filtration (TFF), filtration, precipitation, core-bead flew through chromatography, oligo(dT) purification, and/or cellulose-based purification. Preferably, the RNA has been purified by (RP)HPLC (suitably as described in W02008077592), TFF (suitably as described in WO2016193206) and/or oligo d(T) purification.
In preferred embodiments, the at least one step of purification is selected from RP-HPLC and/or TFF.
In embodiments, the RNA has an integrity of at least 60%, 70%, 80%, 90%. The term “RNA integrity” describes whether the complete RNA sequence is present. RNA integrity can be determined by RP-HPLC and may be based on determining the area under the peak of the expected full-length RNA in a chromatogram.
In preferred embodiments, the RNA is suitable for use in treatment or prevention of a disease, disorder or condition.
Preferred RNA construct designs
In various embodiments, the RNA comprises at least the following elements:
A) a 5’-cap structure, preferably as specified herein;
B) a 5’-UTR, preferably as specified herein;
C) at least one cds encoding at least one antigen as defined herein;
D) a 3-UTR, preferably as specified herein;
E) optionally, a histone stem-loop as specified herein; and
F) at least one poly(A) sequence, preferably as specified herein.
In preferred embodiments, the RNA comprises the following sequence elements, preferably in 5’- to 3’-direction:
A) a 5’-cap structure, preferably a cap1 structure.
B) a 5-UTR, preferably selected or derived from a 5-UTR of a HSD17B4 gene;
C) at least one cds encoding at least one antigen as defined herein; D) a 3-UTR, preferably selected or derived from a 3-UTR of a PSMB3 gene;
E) optionally, a histone stem-loop; and
F) a poly(A) sequence, preferably comprising about 100A nucleotides, e.g. A100 or A30-N10-A70.
G) optionally, chemically modified nucleotides, suitably selected from i or ml ip , preferably ml i .
In preferred embodiments, the RNA comprises or consists of a nucleic acid sequence which is identical or at least 70%, 80%, 85%, 86%, 87%, 88%, 89%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identical to a nucleic acid sequence selected from the group consisting of SEQ ID NOs: 1 -7, or a fragment or variant of any of these sequences, optionally wherein at least one, preferably all uracil nucleotides in said RNA sequences are replaced by pseudouridine (ip) nucleotides and/or N1 -methylpseudouridine (m1ip) nucleotides.
In preferred embodiments, the RNA of the invention is a 5’-cap1 comprising mRNA that comprises or consists of a nucleic acid sequence which is identical or at least 95% identical to a nucleic acid sequence according to any one of SEQ ID NOs: 1-7, or a fragment or variant of any of these sequences.

In preferred embodiments, the subject has received CAR T cells, preferably axicabtagene ciloleucel (Yescarta™), brexucabtagene autoleucel (Tecartus™), lisocabtagene maraleucel (Breyanzi™) or tisagenlecleucel (Kymriah™).
In this embodiment, the RNA of the composition comprises at least one cds encoding at least one amino acid sequence comprising CD19, a functional variant thereof, or a functional fragment of CD19.
In particularly preferred embodiment, the RNA of the composition comprises at least one cds encoding at least one amino acid sequence comprising at least one epitope of or derived from CD19, a functional variant thereof, or a functional fragment of CD19.
In that context, the tumor disease is a hematologic tumor disease, preferably B-cell acute lymphoblastic leukemia (ALL), non-Hodgkin lymphoma, a rare and aggressive form of non-Hodgkin lymphoma diffuse large B-cell lymphoma (DLBCL), primary mediastinal large B-cell lymphoma (PMBCL), mantle cell lymphoma (MCL), relapsed or refractory large B-cell lymphoma, transformed follicular lymphoma or high-grade B-cell lymphoma.
In other preferred embodiments, the subject has received CAR T cells, preferably ciltacabtagene autoleucel (Carvykti™) or idecabtagene vicleucel (Abecma™).
In this embodiment, the RNA of the composition comprises at least one cds encoding at least one amino acid sequence comprising BCMA, a functional variant thereof, or a functional fragment of BCMA.
In particularly preferred embodiment, the RNA of the composition comprises at least one cds encoding at least one amino acid sequence comprising at least one epitope of or derived from BCMA, a functional variant thereof, or a functional fragment of BCMA.
In that context, the tumor disease is a hematologic tumor disease, preferably of mantle cell lymphoma (MCL), a rare and aggressive form of non-Hodgkin lymphoma or multiple myeloma.
In other embodiments, the subject has received CAR T cells orTCRtg cells targeted to CLDN6.
In this embodiment, the RNA of the composition comprises at least one cds encoding at least one antigen from CLDN6 for use in a method of treatment or prophylaxis of a tumour disease, disorder or condition, wherein the RNA is formulated in LNPs and wherein the composition is administered intramuscularly to a subject that has received modified immune cells targeted to CLDN6.
In this embodiment, the RNA of the composition comprises at least one cds encoding at least one amino acid sequence comprising CLDN6, a functional variant thereof, or a functional fragment of the CLDN6 or the functional variant thereof comprises the amino acid sequence of SEQ ID NO: 43 or 44, or an amino acid sequence having at least 99%, 98%, 97%, 96%, 95%, 90%, 85%, or 80% identity to the amino acid sequence of SEQ ID NO: 43 or 44.
In particularly preferred embodiment, the RNA of the composition comprises at least one cds encoding at least one amino acid sequence comprising at least one epitope of or derived from CLDN6, a functional variant thereof, or a functional fragment of the CLDN6 or the functional variant thereof comprises the amino acid sequence of SEQ ID NOs: 45, 46, 47 or an amino acid sequence having at least 99%, 98%, 97%, 96%, 95%, 90%, 85%, or 80% identity to the amino acid sequence of SEQ ID NOs: 45, 6, 47.
In another example, in case the subject has received modified immune cells targeted to Claudinl 8.2 or Claudin6 or a fragment thereof, or a variant of Claudinl 8.2 or Claudin6, the at least one RNA, preferably mRNA, of the composition that is administered to said subject encodes Claudinl 8.2 (CLDN18.2) or Claudin6 (CLDN6) or a fragment thereof, or a variant of Claudinl 8.2 or Claudin6.
In that context, the tumor or cancer disease is an ovarian cancer, lung cancer, gastric cancer, breast cancer, hepatic cancer, pancreatic cancer, skin cancer, melanomas, head neck cancer, sarcomas, bile duct cancer, renal cell cancer, and urinary bladder cancer, in particular ovarian adenocarcinoma and ovarian teratocarcinoma, small cell lung cancer (SCLC) and non-small cell lung cancer (NSCLC), squamous cell lung carcinoma and adenocarcinoma, gastric cancer, breast cancer, hepatic cancer, pancreatic cancer, skin cancer, in particular basal cell carcinoma and squamous cell carcinoma, malignant melanoma, head and neck cancer, in particular malignant pleomorphic adenoma, sarcoma, in particular synovial sarcoma and carcinosarcoma, bile duct cancer, cancer of the urinary bladder, in particular transitional cell carcinoma and papillary carcinoma, kidney cancer, in particular renal cell carcinoma including clear cell renal cell carcinoma and papillary renal cell carcinoma, colon cancer, small bowel cancer, including cancer of the ileum, in particular small bowel adenocarcinoma and adenocarcinoma of the ileum, testicular embryonal carcinoma, placental choriocarcinoma, cervical cancer, testicular cancer, in particular testicular seminoma, testicular teratoma and embryonic testicular cancer, uterine cancer, germ cell tumors such as a teratocarcinoma or an embryonal carcinoma, in particular germ cell tumors of the testis, and the metastatic forms thereof. A particularly preferred cancer is a CLDN6 and/or a CLDN18.2 positive cancer, for example gastric adenocarcinomas, lung adenocarcinomas, ovarian adenocarcinomas, and endometrial carcinomas.
In other preferred embodiments, the subject has received CAR T cells or TCRtg cells targeted to PRAME.
In this embodiment, the RNA of the composition comprises at least one cds encoding at least one antigen from PRAME for use in a method of treatment or prophylaxis of a disease, disorder or condition, wherein the RNA is formulated in LNPs and wherein the composition is administered intramuscularly to a subject that has received modified immune cells targeted to PRAME.
In this embodiment, the RNA of the composition comprises at least one cds encoding at least one amino acid sequence comprising PRAME, a functional variant thereof, or a functional fragment of the PRAME or the functional variant thereof comprises the amino acid sequence of SEQ ID NO: 66, or an amino acid sequence having at least 99%, 98%, 97%, 96%, 95%, 90%, 85%, or 80% identity to the amino acid sequence of SEQ ID NO: 66.
In particularly preferred embodiment, the RNA of the composition comprises at least one cds encoding at least one amino acid sequence comprising at least one epitope of or derived from PRAME, a functional variant thereof, or a functional fragment of the PRAME or the functional variant thereof comprises the amino acid sequence of SEQ ID NO: 67 or an amino acid sequence having at least 99%, 98%, 97%, 96%, 95%, 90%, 85%, or 80% identity to the amino acid sequence of SEQ ID NO: 67.
In that context, the tumor or cancer disease is any of acute lymphocytic cancer, acute myeloid leukemia, alveolar rhabdomyosarcoma, bone cancer, brain cancer, breast cancer, cancer of the anus, anal canal, or anorectum, cancer of the eye, cancer of the intrahepatic bile duct, cancer of the joints, cancer of the neck, gallbladder, or pleura, cancer of the nose, nasal cavity, or middle ear, cancer of the oral cavity, cancer of the vagina, cancer of the vulva, chronic lymphocytic leukemia, chronic myeloid cancer, colon cancer, esophageal cancer, cervical cancer, gastrointestinal carcinoid tumor, glioma, Hodgkin lymphoma, hypopharynx cancer, kidney cancer, larynx cancer, liver cancer, lung cancer, malignant mesothelioma, melanoma, multiple myeloma, nasopharynx cancer, non-Hodgkin lymphoma, cancer of the oropharynx, ovarian cancer, cancer of the penis, pancreatic cancer, peritoneum, omentum, and mesentery cancer, pharynx cancer, prostate cancer, rectal cancer, renal cancer, skin cancer, small intestine cancer, soft tissue cancer, stomach cancer, testicular cancer, thyroid cancer, cancer of the uterus, ureter cancer, and urinary bladder cancer. A preferred cancer in that context is cancer of the uterine cervix, oropharynx, anus, anal canal, anorectum, vagina, vulva, or penis. A particularly preferred cancer is a PRAME positive cancer, for example gastric adenocarcinomas, lung adenocarcinomas, ovarian adenocarcinomas, and endometrial carcinomas.
2. Combination for use in treatment or prophylaxis of a disease, disorder or condition
In a second aspect, the present invention relates to a combination comprising (i) a composition comprising at least one RNA comprising at least one cds encoding at least one antigen; and (ii) modified immune cells targeted to the at least one antigen, for use in a method of treatment or prophylaxis of a disease, disorder or condition, wherein the modified immune cells are administered intravenously to the subject, and wherein the composition is administered to the subject that has received modified immune cells.
It has to be noted that specific features and embodiments that are described in the context of the first aspect of the invention, that is the composition for use of the invention, are likewise applicable to the second aspect of the invention. Moreover, specific features and embodiments that are described in the context of the second aspect of the invention, that is the combination for use of the invention, are likewise applicable to any other aspect of the invention.
In the context of the invention, a “combination” refers to specific dosing regimens, delivery mechanisms, or formulations that optimize the therapeutic outcome. In this context a combination comprising at least two compounds, (i) a composition comprising at least one RNA comprising at least one cds encoding at least one antigen; and (ii) modified immune cells targeted to the at least one antigen. The different compounds or compositions may be administered simultaneously, at essentially the same time, or sequentially. Combination in this context may refer to a medical or therapeutic approach in which multiple treatments, interventions, or medications are used together simultaneously or sequentially to address a specific health condition or disease. In embodiments, the RNA of the composition is formulated. Suitable formulations in the context of the invention are further described under paragraph “Formulation/Complexation of the RNA of the composition" (first aspect). In particularly preferred embodiments in the context of formulations, the RNA is formulated in LNPs as defined herein.
In some embodiment, the composition comprising at least one RNA comprising at least one cds encoding at least one antigen, or host cell modified to express the antigen, is administered following administration or generation of modified immune cells targeted to an antigen, e.g., at least 1 day, such as 1 to 60 days or 1 to 50 days or 1 to 30 days following administration or generation of modified immune cells.
In preferred embodiments, the composition comprising at least one RNA comprising at least one cds encoding at least one antigen, or host cell modified to express the antigen, is administered following administration or generation of modified immune cells targeted to an antigen, e.g., at least 1 day, such as 1 to 10 days or 1 to 5 days or 1 to 3 days following administration or generation of modified immune cells.
In some embodiments, the composition comprising at least one RNA comprising at least one cds encoding at least one antigen, or host cell modified to express the antigen, may be administered several times over time in constant or different time intervals, e.g., following administration or generation of modified immune cells targeted to an antigen, e.g., in time intervals of between 7 and 40 days, wherein the first administration of the composition comprising at least one RNA comprising at least one cds encoding at least one antigen, or host cell modified to express the antigen may be at least 1 day, such as 1 to 60 days or 1 to 50 days or 1 to 30 days following administration or generation of modified immune cells.
In preferred embodiments, the composition comprising at least one RNA comprising at least one cds encoding at least one antigen, or host cell modified to express the antigen, may be administered several times overtime in constant or different time intervals, e.g., following administration or generation of modified immune cells targeted to an antigen, e.g., in time intervals of between 7 and 40 days, wherein the first administration of the composition comprising at least one RNA comprising at least one cds encoding at least one antigen, or host cell modified to express the antigen may be at least one day, such as 1 to 10 days or 1 to 5 days or 1 to 3 days following administration or generation of modified immune cells.
In preferred embodiments, the composition is administered to a subject as defined in the context of the first aspect. Preferably, the composition is administered to a subject via intratumoral or intramuscular administration. More preferably, the composition is administered to a subject via intramuscular administration, e.g. intramuscular injection.
Intramuscular administration, e.g. intramuscular injection, has many advantages, non-limiting examples are i.m. injections provide a relatively consistent and sustained release, avoiding peaks of delivery. Intramuscular injections provide slower absorption compared to intravenous administration and more controlled release or longer duration of action. Additionally, there is a lower risk of infiltration. Intravenous injections can carry a risk of infiltration, where the medication leaks into surrounding tissues if the vein is not properly secured. Some patients may find intramuscular injections less uncomfortable or invasive compared to intravenous infusions, which can be particularly relevant for pediatric or needle-phobic patients. Intramuscular administration often requires fewer resources and equipment compared to intravenous infusions, making it a cost-effective option in certain situations. In some embodiments, the subject that has received the combination of the second aspect receives at least one additional treatment, preferably at least one adjuvant, “classical” cancer treatment, immune checkpoint inhibitors and/or anti-cancer antibodies as described in the first aspect.
In preferred embodiments, the combination comprises
(i) a composition comprising at least one RNA comprising at least one cds encoding at least one antigen; and
(ii) modified immune cells targeted to the at least one antigen, for use in a method of treatment or prophylaxis of a disease, disorder or condition, wherein the modified immune cells are preferably administered intravenously to the subject, and wherein the composition is preferably administered intramuscularly to the subject that has received the modified immune cells. In preferred embodiments, the RNA is formulated as further defined in paragraph “Formulation/Complexation of the RNA of the composition" (first aspect). More preferably, the RNA is formulated in LNPs as defined herein.
Accordingly, the invention relates to a combination that comprises (i) a composition comprising at least one RNA comprising at least one cds encoding at least one antigen, wherein the RNA is formulated in LNPs as defined herein; and (ii) modified immune cells targeted to the at least one antigen, for use in a method of treatment or prophylaxis of a disease, disorder or condition, by administering the modified immune cells intravenously to the subject, and by administering the composition intramuscularly to the subject that has received the modified immune cells.
In particularly preferred embodiments of the combination, the method, the composition, the modified immune cells, the subject, and the disease, disorder or condition are further characterized by any one of the features of the first aspect.
In this embodiment, the method of treatment or prophylaxis of disease, disorder or condition is selected from a tumor or cancer disease, disorder or condition or infectious disease, disorder or condition, preferably selected from a tumor or cancer disease, disorder or condition, more preferably selected from solid or hematologic cancer.
Further the modified immune cells comprise modified T cells, preferably CAR T cells or TCRtg T cells.
In preferred embodiments, the administration of modified T cells is performed with or without lymphodepletion prior to administration of modified T cells, preferably CAR T cells or TCRtg T cells.
In preferred embodiments, a low dose or subtherapeutic dose of modified T cells, preferably CAR T cells or TCRtg cells is administered to the subject.
In preferred embodiments of the combination, the modified immune cells are administered intravenously.
In preferred embodiments, the CAR T cells or TCRtg cells are targeted to an antigen associated with the disease, disorder or condition, preferably a tumor or cancer disease, disorder or condition or infectious disease, disorder or condition.
In some embodiments, the antigen is a tumor antigen, more preferably tumor specific antigen or tumor associated antigen, selected from mutation antigen, neoantigen, unique antigen, differentiation tumor antigen, overexpressed tumor antigen, virus tumor antigen or bacterial tumor antigen.
In preferred embodiments, the tumor antigen is selected or derived from CD19, BCMA, CD22, CD20, Mesothelin, GPC3, GD2, HER2, B7-H3, CLDNB18.2, Claudin6 (CLDN6), PSMA, CEA, EGFR, MUC1, EGFRVIII, NKG2DL, WT1, HA-1, NY-ESO-1, HPV, MAGE-A4, HBV, EBV, mutated KRAS; KK-LC-1 , MAGE-A3, MAGE-A10, AFP, MAGE-A1 , MART-1, PRAME, mutated PRAME, CD33, CD70, NKG2DL, PD-L1 , ROBO1, 5T4, DLL3, COL6A3 and/or MICA/B, preferably from Claudin 6 or PRAME.
In this embodiment, the composition comprising the RNA encodes at least one tumor antigen, preferably encodes at least one epitope targeted by the antigen receptor of the modified T cells, more preferably a full-length antigen comprising the antigen targeted by the antigen receptor of the modified T cells.
The combination of the present aspect, in particular the combination of (i) the composition and (ii) the modified immune cells, has advantageous effects as disclosed in the context of the first aspect (see in particular, paragraph “observed effects”).
3. A kit or kit of parts for use in treatment or prophylaxis of a disease, disorder or condition
In a third aspect, the present invention relates to a kit or kit of parts comprising (A) at least one composition as defined in the context of the first aspect; or (B) at least one combination as defined the context of the second aspect, for use in a method of treatment or prophylaxis of a disease, disorder or condition.
It has to be noted that specific features and embodiments that are described in the context of the first aspect of the invention and the second aspect of the invention are likewise applicable to the third aspect of the invention. Moreover, specific features and embodiments that are described in the context of the third aspect of the invention, that is the kit or kit of parts of the invention, are likewise applicable to any other aspect of the invention.
In embodiments, the kit or kit of parts comprises instructions for use of the kit or kit of parts in a method of treatment or prophylaxis of a disease, disorder or condition. In embodiments, the kit or kit of parts comprises technical instructions providing information on administration and dosage of the components. The technical instructions of said kit may contain information about administration and dosage and patient groups. Such kits, preferably kits of parts, may be applied e.g. for any of the applications or uses mentioned herein, preferably for the use of the therapeutic agents of the kit or kit of parts for treatment of cancer or a diseases, disorder, or condition related to cancer.
In embodiments where the composition or combination is provided as a lyophilized or spray-freeze dried or spray dried composition, the kit or kit of parts may suitably comprise a buffer for re-constitution of lyophilized or spray-freeze dried or spray dried composition or combination.
In preferred embodiments, the kit or kit of parts as defined herein comprises at least one syringe or application device. Suitably, in embodiments where lipid-based carrier formulated RNA is to be administered, a syringe or application device as described in WO2022207862, claims 1 to 69, may be part of the kit.
In some embodiments, the kit of the invention may, for example, be affixed to a container which contains the composition or combination of the invention or be shipped together with a container which contains the composition or combination. Alternatively, the kit may be shipped separately from the container with the intention that the kit or combination may be used cooperatively by the subject.
In a preferred embodiment, the subject receives the kit or kit of parts comprising (A) at least one composition as defined in the context of the first aspect; or (B) at least one combination as defined the context of the second aspect, for use in a method of treatment or prophylaxis of a disease, disorder or condition, preferably a diseases, disorder, or condition related to cancer. 4. A method or preventing a disease, disorder or condition
In a fourth aspect, the present invention relates to a method of treating or preventing a disease, disorder, or condition in a subject, comprising a step of applying or administering a composition comprising at least one RNA comprising at least one cds encoding at least one antigen, wherein the subject has received modified immune cells targeted to the at least one antigen.
It has to be noted that specific features and embodiments that are described in the context of the first aspect of the invention and the second aspect of the invention are likewise applicable to the fourth aspect of the invention. Moreover, specific features and embodiments that are described in the context of the fourth aspect of the invention are likewise applicable to any other aspect of the invention.
In preferred embodiments, the method of treating or preventing a disease, disorder, or condition in a subject, comprises a step of applying or administering intramuscularly a composition comprising at least one RNA comprising at least one cds encoding at least one antigen, wherein the RNA is formulated in LNPs, wherein the subject has received modified immune cells targeted to the at least one antigen. Preferably, the method is further characterized by any one of the features of the first aspect.
5. A method a modified immune cell
a modified immune cell


In a fifth aspect, the present invention relates to a method of activating or stimulating a modified or endogenous immune cell population in a subject, wherein the method comprises applying or administering at least one composition comprising at least one RNA comprising at least one cds encoding at least one antigen to the subject.
In preferred embodiments, the method of activating or stimulating a modified immune cell population in a subject, wherein the method comprises applying or administering at least one composition comprising at least one RNA comprising at least one cds encoding at least one antigen to the subject.
It has to be noted that specific features and embodiments that are described in the context of the first aspect of the invention and the second aspect of the invention are likewise applicable to the fifth aspect of the invention. Moreover, specific features and embodiments that are described in the context of the fifth aspect of the invention are likewise applicable to any other aspect of the invention.
In preferred embodiments, the method of activating or stimulating a modified immune cell population in a subject comprises applying or administering intramuscularly at least one composition comprising at least one RNA comprising at least one cds encoding at least one antigen to the subject, wherein the RNA is formulated in LNPs. Preferably, the method is further characterized by any one of the features of the first aspect.
In some embodiments, the method of activating or stimulating a modified immune cell population in a subject is defined in increasing levels of circulating modified immune cells in the subject;, inducing memory formation of modified immune cells in the subject, preferably formation of central and effector memory cells, activating modified immune cells in the subject; boosting metabolism and polyfunctionality of the modified immune cells in the subject, recruitment of crosspresenting cells, preferably DCs, to the tumor and elicits robust and potent antigen spreading, re-activating modified immune cells in the subject; maintaining the self-renewal capacity of modified immune cells in the subject, inducing higher influx into the solid tumor of modified immune cells in the subject, and/or inducing expansion of modified immune cells in the subject. In one embodiment, the method of inducing an immune response in a subject. In one embodiment, the immune response is a T cell-mediated immune response. In one embodiment, the immune response is an immune response to a target cell population or target tissue expressing an antigen. In one embodiment, the target cell population or target tissue are cancer cells or is cancer tissue. In one embodiment, the cancer cells or cancer tissue is solid cancer or hematologic cancer, preferably solid cancer.
Item list
Preferred embodiments of the present invention are provided in the following item list:
Item 1 : A composition comprising at least one RNA comprising at least one coding sequence encoding at least one antigen for use in a method of treatment or prophylaxis of a disease, disorder or condition, wherein the RNA is formulated in lipid nanoparticles and wherein the composition is administered intramuscularly to a subject that has received modified immune cells, preferably modified T cells, targeted to the at least one antigen.
Item 2: The composition for use of item 1 , wherein the modified immune cells comprise autologous immune cells taken from the subject.
Item 3: The composition for use of item 1 or 2, wherein the modified immune cells are genetically modified to express an antigen receptor targeted to the antigen and/or to express the antigen, preferably modified to express an antigen receptor targeted to the antigen.
Item 4: The composition for use of item 3, wherein the antigen receptor is targeted to the antigen associated with the disease, disorder or condition.
Item 5: The composition for use of item 3 or 4, wherein the antigen receptor is a chimeric antigen receptor (CAR) or a T cell receptor (TCR), preferably a TCR.
Item 6: The composition for use of items 1 to 5, wherein the modified immune cells are selected from tumor-infiltrating leukocytes (TILs) or modified T cells.
Item 7: The composition for use of item 6, wherein the modified T cells are selected from transgenic TCR (TCRtg) and/or chimeric antigen receptors (CAR) T cells.
Item 8: The composition for use of items 1 to 7, wherein the modified immune cells are selected from modified T cells, preferably transgenic T cells (TCRtg), that are genetically modified to express an antigen receptor targeted to the antigen.
Item 9: The composition for use of any one of the preceding items, wherein the antigen is associated with the disease, disorder or condition.
Item 10: The composition for use of item 9, wherein the antigen is selected from or derived from tumor antigens, viral antigens, bacterial antigens, protozoal antigens, fungal antigens, self-antigens or allogenic antigens, preferably tumor antigens.
Item 11 : The composition for use of item 10, wherein the tumor antigen is tumor specific antigen or tumor associated antigen, preferably selected from mutation antigen, neoantigen, unique antigen, differentiation tumor antigen, overexpressed tumor antigen, virus tumor antigen or bacterial tumor antigen.
Item 12: The composition for use of any one of the preceding items, wherein the antigen is a tumor antigen selected or derived from p53, 5T4, ART , AFP, BAGE, beta-eaten in/m, BCMA, Bcr-abL B7-H3, mutated BRAF, CAMEL, CAP-1 , CASP-8, CDC27/m, CDK4/m, CD19, CD22, CD20, CD33, CD70, CEA, mutated CTNNB1 , the cell surface proteins of the claudin family, such as CLDN6, CLDN18.2 and CLDN12, COL6A3, c-MYC, CT, Cyp-B, DAM, DLL3, EBV, ELF2M, ETV6-AML1, EGFR, EGFRvlll, EphA2, G250, GAGE, GD2, GnT-V, GPC3, Gap 100, Gp100, HAGE, HA-1 , HER-2/neu, HBV, HPV-E7, HPV-E6, HAST-2, hTERT (or hTRT), KRAS, mutated KRAS, KK-LC-1, LAGE, LDLR/FUT, MAGE-A, preferably MAGE-A1 , MAGE-A2, MAGE-A3, MAGE-A4, MAGE-A5, MAGE-A6, MAGE-A7, MAGE-A8, MAGE-A9, MAGE-A10, MAGE-A11 , or MAGE- A12, MAGE-B, MAGE-C, MART-1 /Melan-A, MICA/B, MC1R, Mesothelin, Myosin/m, MUC1, MUM-1, MUM- 2, MUM-3, NA88-A, NF1, NY-ESO-1 , NY-BR-1, NKG2DL, PAP, pl90 minor BCR-abL, Pml/RARa, PRAME, mutated PRAME, proteinase 3, PD-L1 , PSA, PSM, PSMA, PSCA, RAGE, ROBO1 , RU1 or RU2, SAGE, SART-1 or SART-3, SCGB3A2, SCP1 , SCP2, SCP3, SSX, STEAP, SURVIVIN, TEL/AML1, mutated TP53, TPI/m, TRP-1 , TRP-2, TRP-2/INT2, TPTE, WT, and WT1.
Item 13: The composition for use of item 12, wherein the tumor antigen is selected or derived from CD19, BCMA, CD22, CD20, Mesothelin, GPC3, GD2, HER2, B7-H3, CLDN18.2, Claudin6 (CLDN6), PSMA, CEA, EGFR, MUC1, EGFRVIII, NKG2DL, WT1, HA-1 , NY-ESO-1 , HPV, MAGE-A4, HBV, EBV, mutated KRAS; KK-LC-1, MAGE-A3, MAGE-A10, AFP, MAGE-A1, MART-1, PRAME, mutated PRAME, CD33, CD70, NKG2DL, PD- L1 , ROBO1 , 5T4, DLL3, COL6A3 and/or MICA/B, preferably from CLDN6 or PRAME.
Item 14: The composition for use of any one of the preceding items, wherein the at least one encoded antigen comprises at least the epitope recognized by the antigen receptor of the modified immune cells, preferably wherein the at least one encoded antigen comprises at least the T cell epitope.
Item 15: The composition for use of any one of the preceding items, wherein the at least one encoded antigen comprises at least 1 , 2, 3, 4, 5, 6, 7, 8, 9 or 10 epitopes recognized by the antigen receptor of the modified immune cells.
Item 16: The composition for use of any one of the preceding items, wherein the disease, disorder or condition is selected from a cancer or tumor disease, disorder or condition, an infectious disease, disorder or condition, an autoimmune disease, disorder or condition, or a disease, disorder or condition associated with stem cell transplantation.
Item 17: The composition for use of any one of the preceding items, wherein the disease, disorder or condition is selected from a tumor or cancer disease, disorder or condition.
Item 18: The composition for use of item 17, wherein the cancer disease is derived from solid or hematologic tumors. Item 19: The composition for use of any one of the preceding items, wherein the subject has received the modified immune cells by intravenous administration.
Item 20: The composition for use of any one of the preceding items, wherein the subject is a mammal, preferably a human.
Item 21 : The composition for use of any one of the preceding items, wherein the subject has not received lymphodepletion within 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11 , 12, 13 or 14 days before the administration of the modified immune cells targeted to the antigen.
Item 22: The composition for use of item 21 , wherein the subject has a reduced risk of fewer, weakness, chills, loss of appetite, immune-related toxicity and cytokine-related toxicity, cytokine release syndrome, organ dysfunction, arrhythmias, renal failure, neurotoxicity, anemia, thrombocytopenia, graft failure, increased risk of infections like bacterial infections, viral infections, fungal infections, neutropenia, pancytopenia and febrile neutropenia and/or bacteremia, preferably compared to a subject that received lymphodepletion. Item 23: The composition for use of items 1 to 20, wherein the subject has received a lymphodepletion within 1 , 2, 3, 4, 5, 6, 7, 8, 9, 10, 11 , 12, 13 or 14 days before the administration of the modified immune cells targeted to the antigen.
Item 24: The composition for use of any one of the preceding items, wherein the method does not comprise a step of treating the subject with additional cytokine or interleukin performed about 2 months after the administration of the modified immune cells targeted to the antigen.
Item 25: The composition for use of any one of the preceding items, wherein the subject received a therapeutic amount of modified immune cells, preferably modified T cells.
Item 26: The composition for of item 1 to 24, wherein the subject received a subtherapeutic amount of modified immune cells, preferably modified T cells.
Item 27: The composition for use in a method of treatment or prophylaxis of any one of the preceding items, wherein a first dose of the composition is administered 1 day to 60 days after the subject has received the modified immune cells targeted to the antigen.
Item 28: The composition for use of item 27, wherein at least one further dose of the composition is administered 1 day to 60 days after the administration of the first dose.
Item 29: The composition for use of any one of the preceding items, wherein at least 3, , 5, or more doses of the composition are administered to the subject.
Item 30: The composition for use of items 27 to 29, wherein one dose comprises between 10 g RNA and 100pg RNA, preferably less than 100pg RNA or less than 50pg RNA.
Item 31 : The composition for use of any one of the preceding items, wherein intramuscular administration of the composition to the subject increases the levels of circulating modified immune cells in the subject, induces memory formation of modified immune cells in the subject, preferably formation of central and effector memory cells, activates modified immune cells in the subject; boosting metabolism and polyfunctionality of the modified immune cells in the subject, recruitment of cross-presenting cells, preferably DCs, to the tumor and elicits robust and potent antigen spreading, re-activates modified immune cells in the subject; maintains the self-renewal capacity of modified immune cells in the subject, induces higher influx into the solid tumor of modified immune cells in the subject, and/or induces expansion of modified immune cells in the subject.
Item 32: The composition for use of any one of the preceding items, wherein intramuscular administration of the composition to the subject activates endogenous CD4 helper cells in the subject, skews the differentiation of endogenous tumor-infiltrating CD4+ T cells to a Th1 phenotype, increases the levels of circulating endogenous immune cells in the subject; induces memory formation of endogenous immune cells in the subject, preferably formation of central and effector memory cells; activates endogenous immune cells in the subject; boosting metabolism and polyfunctionality of the endogenous immune cells in the subject, recruitment of cross-presenting cells, preferably DCs, to the tumor and elicits robust and potent antigen spreading (antigen spreading supports modified T cell therapy to treat antigenically heterogeneous tumors), re-activates endogenous immune cells in the subject, maintains the self-renewal capacity of endogenous immune cells in the subject, induces higher influx into the solid tumor of endogenous immune cells in the subject, and/or induces expansion of endogenous immune cells in the subject and increases the anti-tumor potential of endogenous tumor-infiltrating CD8+ T cells in the subject.
Item 33: The composition for use of any one of the preceding items, wherein intramuscular administration of the composition to the subject increases levels of circulating modified immune cells in the subject; induces memory formation of modified immune cells in the subject, preferably formation of central and effector memory T cells, activates modified immune cells in the subject; boosting metabolism and polyfunctionality of the modified immune cells in the subject, recruitment of cross-presenting cells, preferably DCs, to the tumor and elicits robust and potent antigen spreading, re-activates modified immune cells in the subject, maintaining self-renewal capacity of modified immune cells in the subject, induces higher influx into the solid tumor of modified immune cells in the subject, induces expansion of modified immune cells in the subject, activates endogenous CD4 helper cells in the subject, skews differentiation of endogenous tumor-infiltrating CD4+ T cells to a Th1 phenotype, increases levels of circulating endogenous immune cells in the subject, induces memory formation of endogenous immune cells in the subject, preferably formation of central and effector memory cells, activates endogenous immune cells in the subject, boosting metabolism and polyfunctionality of the endogenous immune cells in the subject, recruitment of cross-presenting cells, preferably DCs, to the tumor and elicits robust and potent antigen spreading (antigen spreading supports modified T cell therapy to treat antigenically heterogeneous tumors), re-activates endogenous immune cells in the subject, maintaining self-renewal capacity of endogenous immune cells in the subject, induces higher influx into the solid tumor of endogenous immune cells in the subject, induces expansion of endogenous immune cells in the subject and/or increases anti-tumor potential of endogenous tumor-infiltrating CD8+ T cells in the subject to a larger extend compared to intravenous administration of the composition to the subject.
Item 34: The composition for use of any one of the preceding items, wherein the LNPs comprise at least one lipid selected from at least one aggregation-reducing lipid, at least one cationic lipid or ionizable lipid, at least one neutral lipid or phospholipid, at least one steroid or steroid analogue.
Item 35: The composition for use of any one of the preceding items, wherein the LNPs comprise at least one aggregation-reducing lipid, at least one cationic lipid or ionizable lipid, at least one neutral lipid or phospholipid, and at least one steroid or steroid analogue.
Item 36: The composition for use of items 34 or 35, wherein the at least one aggregation reducing lipid is selected from a polymer conjugated lipid.
Item 37: The composition for use of item 36, wherein the polymer conjugated lipid is selected from a PEG-conjugated lipid or a PEG-free lipid.
Item 38: The composition for of items 34 to 37, wherein the at least one aggregation reducing lipid is selected from DMG-PEG 2000, C10-PEG2K, Cer8-PEG2K, or a POZ-lipid.
Item 39: The composition for use of items 34 to 38, wherein the at least one cationic lipid or ionizable lipid is selected from an amino lipid, preferably wherein the amino lipid comprises a tertiary amine group.
Item 40: The composition for use of items 34 to 39, wherein the at least one cationic or ionizable lipid is a lipid selected or derived from formula (111-1 ), preferably, wherein one of L1 or L2 is -O(C=O)-, -(C=O)O-, -C(=O)-, -O-, - S(O)x-, -S-S-, -C(=O)S-, SC(=O)-, -NRaC(=O)-, -C(=O)NRa- -NRaC(=O)NRa-, -OC(=O)NRa- or - NRaC(=O)O-, and the other of L1 or L2 is -O(C=O)-, -(C=O)O-, -C(=O)-, -O-, -S(O)x-, -S S-, -C(=O)S- SC(=O)-, -NRaC(=O)-, -C(=O)NRa-, -NRaC(=0)NRa-, -OC(=O)NRa- or -NRaC(=O)O- or a direct bond; G1 and G2 are each independently unsubstituted C1-C12 alkylene or C1-C12 alkenylene; G3 is C1-C24 alkylene, C1-C24 alkenylene, C3-C8 cycloalkylene, C3-C8 cycloalkenylene; Ra is H or C1-C12 alkyl; R1 and R2 are each independently C6-C24 alkyl or C6-C24 alkenyl; R3 is H, OR5, CN, C(=O)OR4, 0C(=0)R4 or- NR5C(=O)R4; R4 is C1-C12 alkyl; R5 is H or C1-C6 alkyl; and x is 0, 1 or 2. Item 41 : The composition for use of items 34 to 40, wherein the at least one cationic lipid or ionizable lipid is selected from SM-102, SS-33/4PE-15, HEXA-C5DE-PipSS, or compound C26.
Item 42: The composition for use of items 34 to 41 , wherein the at least one neutral lipid or phospholipid is selected from DSPC, DHPC, DPhyPE, DphyPS.
Item 43: The composition for use of items 34 to 42, wherein the LNPs comprise at least one neutral lipid or phospholipid selected from DSPC, DHPC, DPhyPE and one neutral lipid or phospholipid selected from DphyPS.
Item 44: The composition for use of items 34 to 43, wherein the steroid or steroid analogue is selected from cholesterol, cholesteryl hemisuccinate (CHEMS), preferably cholesterol.
Item 45: The composition for use of any one of the preceding items, wherein the LNPs comprise
(i) at least one cationic or ionizable lipid, preferably as defined in items 39 to 41 ;
(ii) at least one or two neutral lipids or phospholipids, preferably as defined in item 42 or 43;
(iii) at least one steroid or steroid analogue, preferably as defined in item 44; and
(iv) at least one aggregation reducing lipid, preferably as defined in items 36 to 38.
Item 46: The composition for use of any one of the preceding items, wherein the LNPs comprise
(i) a cationic or ionizable lipid selected from compound C26;
(ii) one neutral lipid selected from DphyPE and one neutral lipid selected from DphyPS;
(iii) a steroid or steroid analogue selected from cholesterol; and
(iv) an aggregation reducing lipid selected from a POZ-lipid.
Item 47: The composition for use of any one of the preceding items, wherein the LNPs comprise about 20-60% cationic or ionizable lipid, about 5-25% neutral lipid or phospholipids, about 25-55% steroid or steroid analogue, and about 0.5-15% aggregation reducing lipid.
Item 48: The composition for use of any one of the preceding items, wherein the wt/wt ratio of lipid to RNA in the LNPs is from about 10: 1 to about 60: 1.
Item 49: The composition for use of any one of the preceding items, wherein the N/P ratio of the LNPs comprising the RNA is in a range from about 1 to about 20.
Item 50: The composition for use of any one of the preceding items, wherein the LNPs have a Z-average size of less than 400nm, preferably less than 300nm, more preferably less than 200nm.
Item 51 : The composition for use of any one of the preceding items, wherein the LNPs have a Z-average size in a range of about 50nm to about 200nm, preferably about 50nm to about 150nm.
Item 52: The composition for use of any one of the preceding items, wherein the LNPs have a polydispersity index (PDI) value of less than about 0.3, preferably of less than about 0.2.
Item 53: The composition for use of any one of the preceding items, wherein at least 70%, preferably at least 80%, more preferably at least 90% of the RNA of the composition is encapsulated in the LNPs.
Item 54: The composition for use of any one of the preceding items, wherein at least about 80%, 85%, 90%, 95% of the LNPs have a spherical morphology, preferably comprising a solid core or a partially solid core, and/or a lamellar or bilayer morphology.
Item 55: The composition for use of any one of the preceding items, wherein the LNPs have been generated by combining an aqueous RNA solution with an ethanolic lipid solution using a mixing means at total flow rates above 15ml/min. Item 56: The composition for use of any one of the preceding items, wherein the LNPs do not comprise DOTMA and/or DOPE.
Item 57: The composition for use of any one of the preceding items, wherein the LNPs exhibit a zeta potential in the range of -50 mV to +50 mV, preferably in the range of -25 mV to +25 mV, more preferably in the range of -10 mV to +10 mV, most preferably in the range of -5 mV to +5 mV.
Item 58: The composition for use of any one of the preceding items, wherein the at least one cds encodes at least one antigen that is selected from or derived from tumor antigens, viral antigens, bacterial antigens, protozoal antigens, fungal antigens, self-antigens or allogenic antigens, preferably tumor antigens.
Item 59: The composition for use of item 58, wherein the tumor antigen is a tumor-specific antigen or tumor associated antigen that is preferably selected from p53, 5T4, ART-4, AFP, BAGE, beta-eaten in/m, BCMA, Bcr-abL B7- H3, mutated BRAF, CAMEL, CAP-1, CASP-8, CDC27/m, CDK4/m, CD19, CD22, CD20, CD33, CD70, CEA, mutated CTNNB1 , the cell surface proteins of the claudin family, such as Claudin6 (CLDN6), CLDN18.2 and CLDN12, COL6A3, c-MYC, CT, Cyp-B, DAM, DLL3, EBV, ELF2M, ETV6-AML1, EGFR, EGFRvlll, EphA2, G250, GAGE, GD2, GnT-V, GPC3, Gap 100, Gp100, HAGE, HA-1, HER-2/neu, HBV, HPV-E7, HPV-E6, HAST-2, hTERT (or hTRT), KRAS, mutated KRAS, KK-LC-1, LAGE, LDLR/FUT, MAGE-A, preferably MAGE-A1, MAGE-A2, MAGE-A3, MAGE-A4, MAGE-A5, MAGE-A6, MAGE-A7, MAGE-A8, MAGE-A9, MAGE-A10, MAGE-A11, or MAGE-A12, MAGE-B, MAGE-C, MART-1 /Melan-A, MICA/B, MC1R, Mesothelin, Myosin/m, MUC1, MUM-1, MUM-2, MUM-3, NA88-A, NF1, NY-ESO-1 , NY-BR-1, NKG2DL, PAP, pl90 minor BCR-abL, Pml/RARa, PRAME, mutated PRAME, proteinase 3, PD-L1 , PSA, PSM, PSMA, PSCA, RAGE, ROBO1, RU1 or RU2, SAGE, SART-1 or SART-3, SCGB3A2, SCP1 , SCP2, SCP3, SSX, STEAP, SURVIVIN, TEL/AML1 , mutated TP53, TPI/m, TRP-1 , TRP-2, TRP-2/INT2, TPTE, WT, and WT1.
Item 60: The composition for use of item 58 or 59, wherein the tumor antigen or tumor associated antigen is selected from CD19, BCMA, CD22, CD20, Mesothelin, GPC3, GD2, HER2, B7-H3, CLDN18.2, CLDN6, PSMA, CEA, EGFR, MUC1, EGFRVIII, NKG2DL, WT1, HA-1, NY-ESO-1 , HPV, MAGE-A4, HBV, EBV, mutated KRAS; KK-LC-1, MAGE-A3, MAGE-A10, AFP, MAGE-A1 , MART-1, PRAME, mutated PRAME, CD33, CD70, NKG2DL, PD-L1 , ROBO1 , 5T4, DLL3, COL6A3 and/or MICA/B, preferably from CLDN6 or PRAME.
Item 61 : The composition for use of any one of the preceding items, wherein the at least one RNA comprises at least one codon modified cds, preferably wherein the codon modified cds is selected from a C maximized cds, a CAI maximized cds, human codon usage adapted cds, a G/C content modified cds, and a G/C optimized cds, or any combination thereof.
Item 62: The composition for use of item 61 , wherein the at least one codon modified cds is a G/C optimized cds.
Item 63: The composition for use of any one of the preceding items, wherein the at least one RNA comprises at least one untranslated region (UTR), preferably selected from at least one heterologous 5’-UTR and/or at least one heterologous 3-UTR.
Item 64: The composition for use of item 63, wherein the at least one 3-UTR comprises or consists of a nucleic acid sequence selected or from a 3-UTR of a gene selected from PSMB3, AES-12S, ALB7, alpha-globin (HBA1 , HBA2), ANXA4, beta-globin (HBB), CASP1, COX6B1 , FIG4, GH1, GNAS, NDUFA1, RPS9, SLC7A3, TUBB4B, or from a homolog, a fragment or a variant of any one of these genes, preferably wherein the at least one 3-UTR comprises or consists of a nucleic acid sequence being identical or at least 70%, 80%, 85%, 86%, 87%, 88%, 89%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identical to SEQ ID NOs: 134-191 , or a fragment or a variant of any of these. Item 65: The composition for use of items 63 to 64, wherein the at least one 5-UTR comprises or consists of a nucleic acid sequence selected or from a 5-UTR of a gene selected from HSD17B4, AIG1 , alpha-globin (HBA1 , HBA2), ASAH1 , ATP5A1, COX6C, DPYSL2, HHV5, MDR, MP68, NDUFA4, NOSIP, RPL31 , RPL32, RPL35A, SLC7A3, synthetic origin, TUBB4B, UBQLN2, or from a homolog, a fragment or variant of any one of these genes, preferably wherein the at least one 5-UTR comprises or consists of a nucleic acid sequence being identical or at least 70%, 80%, 85%, 86%, 87%, 88%, 89%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identical to SEQ ID NOs: 80-133, or a fragment or a variant of any of these.
Item 66: The composition for use of items 63 to 65, wherein the at least one 5-UTR is selected from HSD17B4 and the at least one 3-UTR is selected from PSMB3.
Item 67: The composition for use of any one of the preceding items, wherein the at least one RNA is an artificial RNA and/or an isolated RNA.
Item 68: The composition for use of any one of the preceding items, wherein the RNA is selected from an mRNA, a circular RNA, a replicon RNA, or a viral RNA.
Item 69: The composition for use of any one of the preceding items, wherein the RNA is an mRNA.
Item 70: The composition for use of any one of the preceding items, wherein the RNA comprises at least one poly(A) sequence, preferably wherein the at least one poly(A) sequence comprises about 40 to about 500 adenosine nucleotides.
Item 71 : The composition for use of item 70, wherein the at least one poly(A) sequence comprises about 60 to about 150 adenosine nucleotides, preferably about 100 adenosine nucleotides.
Item 72: The composition for use of item 70 or 71 , wherein the at least one poly(A) sequence is located at the 3’ terminus, optionally, wherein the 3’ terminal nucleotide is an adenosine.
Item 73: The composition for use of any one of the preceding items, wherein the RNA comprises at least one poly(C) sequence and/or at least one miRNA binding site and/or at least one histone stem-loop sequence.
Item 74: The composition for use of any one of the preceding items, wherein the at least one RNA comprises at least one modified nucleotide, preferably at least one modified nucleotide selected from pseudouridine (i ) or N1- methylpseudouridine (ml ip) , more preferably wherein the at least one modified nucleotide is N1- methylpseudouridine.
Item 75: The composition for use of any one of the preceding items, wherein the at least one RNA is a modified RNA wherein each uracil is substituted by a modified nucleotide.
Item 76: The composition for use of item 1 to 73, wherein the at least one RNA does not comprise a modified nucleotide.
Item 77: The composition for use of any one of the preceding items, wherein the RNA comprises a 5’ -cap structure. Item 78: The composition for use of item 77, wherein the 5’-cap structure is selected from a cap1 structure or a modified cap1 structure.
Item 79: The composition for use of any one of the preceding items, wherein the RNA is an in vitro transcribed RNA. Item 80: The composition for use of any one of the preceding items, wherein the RNA is a purified RNA that has preferably been purified by at least one step of RP-HPLC, AEX, SEC, hydroxyapatite chromatography, TFF, filtration, precipitation, core-bead flow through chromatography, oligo(dT) purification, cellulose-based purification, or any combination thereof.
Item 81 : The composition for use of any one of the preceding items, wherein the RNA comprises the following sequence elements, preferably in 5’- to 3’-direction: A) a 5'-cap structure, preferably a cap1 structure;
B) a 5'-UTR, preferably selected from a 5’-UTR of a HSD17B4 gene;
C) a cds encoding at least one tumor antigen;
D) a 3’-UTR, preferably selected from a 3’-UTR of a PSMB3 gene;
E) optionally, a histone stem-loop; and
F) a poly(A) sequence, preferably comprising about 100 A nucleotides.
Item 82: A combination comprising:
(i) a composition comprising at least one RNA comprising at least one cds encoding at least one antigen, wherein the RNA is formulated in LNRs; and
(ii) modified immune cells targeted to the at least one antigen, for use in a method of treatment or prophylaxis of a disease, disorder or condition, wherein the modified immune cells are the administered intravenously to the subject, and wherein the composition is administered intramuscularly to the subject that has received the modified immune cells.
Item 83: The combination for use of item 82, wherein the method, the composition, the modified immune cells, the subject, and the disease, disorder or condition are further characterized by any one of the features of items 1 to 81.
Item 84: A kit or kit or parts comprising (A) at least one composition as defined in any one of items 1 to 81 ; or (B) at least one combination as defined items 82 or 83, for use in a method of treatment or prophylaxis of a disease, disorder or condition, wherein the kit or kit of parts optionally comprises instructions for use of the kit or kit of parts in a method of treatment or prophylaxis of a disease, disorder or condition.
Item 85: A method of treating or preventing a disease, disorder, or condition in a subject, comprising a step of applying or administering intramuscularly a composition comprising at least one RNA comprising at least one cds encoding at least one antigen, wherein the RNA is formulated in LNPs, wherein the subject has received modified immune cells targeted to the at least one antigen and wherein the method is further characterized by any one of the features of items 1 to 81.
Item 86: A method of activating or stimulating a modified immune cell population in a subject, wherein the method comprises applying or administering intramuscularly at least one composition comprising at least one RNA comprising at least one cds encoding at least one antigen to the subject, wherein the RNA is formulated in LNPs, and wherein the method is further characterized by any one of the features of items 1 to 81.

Figure 1 : shows the tumor volume in C57BL/6 CD45.1 mice after injection of MC38-Ova tumor cells over time. Solid lines represent tumor growth in mice treated with 1 pg LNP formulated mRNA vaccine (encoding full-length Ovalbumin) administered intramuscularly. Dashed lines show tumor growth of the untreated control group. Further details are provided in Example 2.1.
Figure 2: shows the tumor volume in OT-1 CD45.1 mice after injection of MC38-Ova tumor cells over time, treated with a low dose of OT-I CD8 T cells derived from OT-I CD45.2 mice and LNP formulated mRNA vaccine administered i.m. at different time points (further details see Table 3). Figure 2A and 2B show the comparison of untreated control group (Figure 2A, dashed lines) and ACT alone (Figure 2B, dashed lines) to group 3 that have received a low dose of ACT and an early mRNA vaccination (solid lines). The combination of early mRNA vaccination on day 1 after T cell transfer led to complete tumor regression and long-lasting anti-tumor efficacy. Figure 2C and Figure 2D show the comparison of untreated control group (Figure 2C, dashed lines) and ACT alone (Figure 2D, dashed lines) to group 4 that have received a low dose of ACT and a late mRNA vaccination (solid lines). In summary, the combination with early and late vaccination led to 10/10 and 7/10 complete responders, respectively. Further details are provided in Example 2.3.
Figure 3: shows the tumor volume in OT-1 CD45.1 mice after injection of MC38-Ova tumor cells on day 10 (Figure 3A) and day 41 (Figure 3B) after ACT, treated with high and low dose of OT-I CD8 T cells of OT-I CD45.2 mice with and without combination of mRNA vaccine formulated in LNPs administered i.m. at different time points (further details see Table 4). The combination therapy of low dose of ACT and early LNP formulated mRNA vaccination showed the same anti-tumor efficacy as compared to the high dose of ACT at day 10 (Figure 3A) and day 41 (Figure 3E3). Complete tumor regression was observed in both groups at day 41. Further details are provided in Example 3.1.
Figure 4: shows survival data of OT -1 CD45.1 mice after injection of MC38-Ova tumor cells over time, treated with high and low dose of OT-I CD8 T cells derived from OT-I CD45.2 mice with and without combination of LNP formulated mRNA vaccine administered i.m. at different time points (further details see Table 4). All mice that received high ACT doses (group 3) and the mice that received the combination therapy of low ACT dose with early vaccination of LNP formulated mRNA vaccine (group 5) showed long-term survival over 54 days after ACT. Further details are provided in Example 3.2.
Figure 5: shows the number of transferred antigen specific OT-I CD8 T cells derived from OT-I CD45.2 donor mice in the periphery of MC38-Ova tumor bearing CD45.1 recipient mice at day 1 , 11 , 40, 54 and 59 after tumor cell injection. The mice were treated with high and low dose of OT-I CD8 T cells with and without combination of LNP formulated mRNA vaccine administered i.m. at different time points. All groups, except of control group 1 , received an mRNA vaccination on day 54 (further details see Table 5). In all groups mRNA vaccination resulted in prominent expansion of transferred antigen specific CD8 T cells in the periphery. Further details are provided in Ex. 4.1.
Figure 6: shows the number of circulating antigen specific OT-I CD8 memory T cells of OT-I CD45.2 mice at day 40 after ACT in OT-I CD45.1 mice injected with MC38-Ova tumor cells. The mice were treated with high and low dose of OT-I CD8 T cells derived from OT-I CD45.2 donor mice with and without combination of mRNA vaccine formulated in LNPs administered i.m. at different time points (further details see T able 6). Figure 6A shows higher frequencies of antigen specific CD8 T cells in the periphery in both groups that received an mRNA vaccination. Figure 6B shows the frequency of effector memory T cells (TEM) and central memory T cells (TCM) in the periphery. The combination with mRNA vaccination shifted the memory differentiation to effector memory T cells. Figure 6C shows the frequency of memory precursor cells (MPECs) and short-lived effector cells (SLECs) in the periphery. The combination with mRNA vaccination induced high MPECs and SLECs levels in group 4 and 5. Further details are provided in Example 4.2.
Figure ?: shows the PD1 , TIM3, and SLAMF6 expression at day 40 after ACT of the transferred antigen specific OT-I CD8 T cells derived from OT-I CD45.2 donor mice in the periphery of CD45.1 recipient mice after injection of MC38-Ova tumor cells. The mice were treated with high and low dose of OT-I CD8 T cells of OT-I CD45.2 mice with and without combination of LNP formulated mRNA vaccine administered i.m. at different time points (further details see Table 7). Both groups 4 and 5 that received an mRNA vaccination showed high populations of activated (PD1 and TIM3 positive) transferred T cells with the capacity for selfrenewal (SLAMF6 positive). Further details are provided in Example 4.3.
Figure 8: shows antigen specific (TEWTSSNVMEERKIKV; SEQ ID NO: 192) endogenous CD4 T cell population at day 59. Based on 90000 viable splenocytes, counts of the aforementioned population were calculated and are presented in the graph. All mice received a lymphodepletion prior to ACT therapy. The mice were treated with high and low dose of OT-I CD8 T cells derived from OT-I CD45.2 mice with combination of LNP formulated mRNA vaccine administered i.m. at different time points. Group 1 and 2 received only one late vaccination on day 54, group 3 and 4 received the vaccination on earlier time points (further details see Table 8). Mice that have received the composition to an earlier time point after ACT showed high antigen specific and polyfunctional (double positive IFN-y and TNF-a) endogenous CD4 helper T cell population. Further details are provided in Ex. 5.1.
Figure 9: shows the tumor volume of MC38-Ova tumors in C57BL/6 CD45.1 mice over 11 days after ACT. Mice displaying tumors of approximately 288mm3 were treated with 100mg/kg Cyclophoshamide intraperitoneally on Day -1 , with 0.1x10® OT-I CD8 T cells derived from OT-I CD45.2 mice intravenously on Day 0 and with 1 pg LNP-formulated mRNA encoding full length ovalbumin intramuscularly on Day 1 and 8 as indicated. Asterisks show statistical significance levels for comparisons of the indicated groups. Mean values and SEM are depicted. The indicated combination of ACT and mRNA vaccine significantly slowed tumor growth and even reduced or eradicated the very large tumors that were present at the start of the treatment. Further details are provided in Example 6 and 7. ACT, adoptive T cell therapy; Cp, Cyclophosphamide; w/, with; w/o, without.
Figure 10: shows the amount of transferred antigen-specific CD8 T cells found in the peripheral blood at Day 11 after ACT in mice treated as described in the legend of Figure 9. Mean and SEM of the amount of transferred antigen-specific CD8 T cells depicted as CD8+Thy1.2+CD45.2+H-2KbTetra+ cells are shown. Further details are provided in Example 6 and 8. While in the ACT alone group, i.e. without vaccination, almost no transferred CD8 T cells could be detected in the peripheral blood, the indicated group including both ACT and vaccination displayed a very high amount of transferred CD8 T cells suggesting increased survival and/or proliferation of those cells. Further details are provided in Example 6 and 8. ACT, adoptive T cell therapy; Cp, Cyclophosphamide; Vac, vaccination.
Figure 11 : shows the amount of transferred antigen-specific CD8 T cells found in the spleen (left) and the spleen weight (right) at Day 11 after ACT in mice treated as described in the legend of Figure 9. Mean and SEM of the amount of transferred antigen-specific CD8 T cells depicted as CD8+Thy 1 ,2+CD45.2+H-2KbT etra+ cells and of spleen weights are shown. Asterisks show statistical significance levels for the indicated groups. While in the ACT alone group, i.e. without vaccination, almost no transferred CD8 T cells could be detected in the spleen, the indicated group including both ACT and vaccination displayed a very high amount of transferred CD8 T cells suggesting increased survival and/or proliferation of those cells. The high amount of transferred CD8 T cells in the combination group was further reflected by increased spleen weights in this group. Further details are provided in Example 6 and 8. ACT, adoptive T cell therapy; Cp, Cyclophosphamide; Vac, vaccination
Figure 12: shows the amount of transferred antigen-specific CD8 T cells in the tumor (top) as well as their cytotoxicity (bottom, left) and activation (bottom, right) at Day 11 after ACT in in mice treated as described in the legend of Figure 9. Mean and SEM of the amount of transferred antigen-specific CD8 T cells depicted as CD8+Thy1 ,2+CD45.2+ H-2KbTetra+ cells as well as their cytotoxicity (percentage of GZMB+ cells among CD8+Thy1 ,2+CD45.2+ T cells) and activation (percentage of CD25+ cells among CD8+Thy1 ,2+CD45.2+ T cells) are shown. Asterisks show statistical significance levels for the indicated groups. A starkly increased amount of transferred antigen-specific CD8 T cells in the tumor could be observed for the indicated ACT/mRNA vaccine combination. Furthermore, the transferred CD8 T cells in this combination group also showed a higher cytotoxicity and activation as reflected by upregulation of Granzyme B and CD25, respectively. Further details are provided in Example 6 and 9. ACT, adoptive T cell therapy; Cp, Cyclophosphamide; GZMB, Granzyme B; Vac, vaccination.
Examples
In the following, examples illustrating various embodiments and aspects of the invention are presented. However, the present invention shall not to be limited in scope by the specific embodiments presented herein and should rather be understood as being applicable to other compositions or uses as e.g. defined in the specification. Accordingly, the following preparations and examples are given to enable those skilled in the art to more clearly understand and to practice the invention. Various modifications of the invention in addition to those described herein will become readily apparent to those skilled in the art from the foregoing description, accompanying figures and the examples below.
Example 1 : Preparation of RNA formulation of the invention
The present example provides methods of obtaining the RNA of the invention as well as methods of generating composition of the invention comprising RNA formulated in lipid-based carriers.
Example 1.1 : Preparation of DNA templates for in vitro transcription of RNA molecules
DNA sequences encoding proteins for use in therapy, e.g. Ovalbumin, were prepared and used for subsequent RNA in vitro transcription reactions. Some DNA sequences were prepared by modifying the wild type or reference encoding DNA sequences by introducing a G/C optimized cds for stabilization and expression optimization. Sequences were introduced into a pUC derived DNA vector to comprise stabilizing heterologous UTR sequences and a stretch of adenosines and optionally a histone stem-loop (hSL). The obtained plasmid DNA templates were transformed and propagated in bacteria using common protocols known in the art. Eventually, the plasmid DNA templates were extracted, purified, and linearized using a restriction enzyme.
Example 1.2. RNA in vitro transcription from plasmid DNA templates:
Linearized DNA templates were used for DNA dependent RNA in vitro transcription (IVT) using T7 RNA polymerase in the presence of a sequence optimized nucleotide mixture (ATP/GTP/CTP/UTP) or as an alternative with N1- methylpseudouridine (m1i ) and cap analog: m7G(5’)ppp(5’)(2’OMeA)pG; TriLink (cap1), under suitable buffer conditions. After RNA in vitro transcription, the obtained RNA IVT reaction was subjected to purification steps comprising RP-HPLC. The herein used RNA construct is provided in Table 1.
Table 1: RNA construct used in Examples Example 1.3. Preparation of lipid-based carriers encapsulating the RNA construct:
Example 1.3. Preparation of lipid-based carriers encapsulating the RNA construct:

An ethanolic lipid solution was prepared by solubilizing a cationic lipid, a neutral lipid, cholesterol, and an aggregation reducing lipid in ethanol. An aqueous RNA solution was prepared by adjusting the purified RNA (obtained according to Example 1.2) to a target concentration in a citrate or acetate buffer. Lipid-based carriers were prepared essentially according to the procedures described in WO2015199952, W02017004143 and WO2017075531. In short, pumps were used to combine the ethanolic lipid solution with a flow rate F1 and the RNA aqueous solution with a flow rate F2 at a ratio of about 1 :5 to 1 :3 (vol/vol) in a T-piece or a microfluidics system. F1 and/or F2 were adjusted to flow rates above 15ml/min to allow the formation of LNPs encapsulating the RNA that have a Z-average size in a range from about 50nm to about 120nm. After formulation, the ethanol was removed. After clarifying filtration, the filtrate was adjusted to a desired concentration. Subsequently, the resulting formulation was filtered through sterilizing filters to reduce bioburden as used for in vivo studies essentially according to Example 2 to Example 10.
Example 2: Analysis of tumor growth upon single agent treatment with an LNP-formulated and i.m. administered vaccine or with ACT in comparison to combination treatment including the identification of the optimal time point for vaccination after ACT
The goal of the experiment was to analyse tumor growth upon single agent treatment with an LNP-formulated and i.m. administered vaccine or with ACT in comparison to combination treatment including the identification of the optimal time point for vaccination after ACT. Additionally, the experiment demonstrated the anti-tumor efficacy in vivo of the combination of LNP formulated mRNA vaccination encoding full length ovalbumin with transferred T cells. Full length antigens induce better T cell responses, cell activation and proliferation (transferred CD8 and endogenous CD4 T cell) compared to RNA encoding only the T cell epitope.
To this end, primed CD8 T cells specific for the ovalbumin epitope SIINFEKL (SEQ ID NO: 193) were isolated from OT-I CD45.2 mice and transferred into female C57BL/6 CD45.1 mice, inoculated subcutaneously with OVA- expressing MC38 cancer cells (murine colon cancer). In brief, female C57BL/6 CD45.1 (abbreviated to CD45.1) mice were inoculated subcutaneously in the right rear flank region with 1x10s MC-38-OVA cells for tumor development on Day -5. T umor volumes were measured in two dimensions using a calliper, and the volume was expressed in mm3 using the formula: V= (L x W * W) /2, where V is tumor volume, L is tumor length (the longest tumor dimension) and W is tumor width (the longest tumor dimension perpendicular to L). A randomization (10 mice/group) was performed on Day 0 (at that time the mean tumor volume reached approximately 28mm3). Mice were treated as indicated in the respective Examples 2-5. Cyclophosphamide (Selleck; 2mg per mouse) was administrated intraperitoneally on Day -1 for all groups receiving ACT in the course of the experiment. 2x106 or 0.1x10® OT-I cells were injected i.v. on Day 0 where indicated. Mice were vaccinated intramuscularly (m. tibialis) with 1 pg LNP-formulated mRNA encoding full length ovalbumin (SEQ ID NO: 68) on the indicated days. The date of ACT injection was denoted as Day 0.
OT-I cells in this context were generated as follows. 8 female OT-I C57BL/6 CD45.2 (abbreviated to OT-I CD45.2) mice were intraperitoneally injected Ovalbumin (Sigma; 0.5pg/mouse) on Day -2. On Day 0, spleens were collected and mouse CD8+ T cells isolated using the EasySep™ Mouse CD8+ T Cell Isolation Kit (StemCell Technologies; CAT#19853) according to the manufacturer’s protocol and standard procedures in the art. Example 2.1 : Analysis of tumor growth upon single agent treatment with an LNP-formulated and i.m. administered vaccine
The goal of Example 2.1 was to analyse the tumor growth of Ovalbumin-expressing MC38 tumors upon single agent treatment with an LNP-formulated and intramuscularly administered vaccine.
For the experiment, 10 female C57BL/6 CD45.1 mice per group were injected subcutaneously with 1x106 MC38-OVA tumor cells for tumor development. After tumors reached a mean tumor volume of approximately 28mm3, one group of the mice was vaccinated i.m. once per week for 3 weeks (Day 1 , 8, 15) with 1 pg LNP formulated mRNA encoding full length ovalbumin. Tumor growth was monitored over 52 days. Further experimental details are provided above in the introduction of Example 2.
The LNP-formulated and i.m. administered mRNA vaccine led to a strong delay and reduction of tumor growth (Figure 1 ) resulting in 5/10 complete responders displaying no detectable tumor at the end of the observation period. The dose of 1 pg LNP formulated mRNA was used in all further mouse Examples.
Example 2.2: Effect of different doses of transferred cells for ACT on tumour growth
The goal of the analysis was to identify the effects of different numbers of transferred CD8 T cells on tumor growth. For the experiment female C57BL/6 CD45.1 mice (10 mice/group) were injected subcutaneously with 1x106 MC38-OVA tumor cells for tumor development. After tumors reached a mean tumor volume of approximately 28mm3, mice received different doses (Table 2, group 2 and 3) of OT-I CD8 T cells derived from OT-I CD45.2 donor mice, primed before with ovalbumin. T umor growth was monitored over 52 days. Further experimental details are provided above in the introduction of Example 2.
Table 2: Study groups used in Example 2.2

The high dose of 2x106 OT-I transferred CD8 T cells resulted in 10/10 complete responders with no re-growth of the tumor. Group 3 with a lower dose of 0.1x10® of transferred OT-I CD8 T cells showed 6/10 complete responders displaying no detectable tumor at the end of the observation period.
The results demonstrated that anti-tumoral efficacy can be modulated by the quantity of transferred T cells. Notably, however, as stated in the next Example, the quantity of transferred T cells is very often limited in the clinic.
Example 2.3: Identification of optimal time point for vaccination after ACT
The goal of the analysis was to identify an optimal time point after T cell transfer for the vaccination with an LNP- formulated and i.m. administered mRNA vaccine to induce enhanced anti-tumor efficacy of low-dose transferred T cells.
For the experiment, female C57BL/6 CD45.1 mice (10 per group) were injected subcutaneously with 1 x10® MC38- OVA tumor cells for tumor development. After tumors reached a mean tumor volume of approximately 28mm3, group 2, 3 and 4 (see Table 3) received 0.1x10® OT-I CD8 CD45.2 T cells. For the identification of the optimal time point for mRNA vaccination, two groups received the first vaccination i.m. on day 1 (group 3) and the other group on day 8 (group 4). T umor growth was observed for 52 days. Further experimental details are provided above in the introduction of Example 2.
Table 3: Study groups used in Example 2.3

The combination of early mRNA vaccination on day 1 after T cell transfer led to tumor regression and long-lasting antitumor efficacy of the transferred T cells compared to the untreated control group 1 (Figure 2A) and the T cell transfer alone (Figure 2B), with 10/10 complete responders displaying no detectable tumor at the end of the observation period. The combination of late vaccination on day 8 after T cell transfer resulted in a less effective anti-tumoral response, with only 7/10 complete responders displaying no detectable tumor at the end of the observation period.
(Figure 2C and Figure 2D).
Results of Example 2
The experiments demonstrated that ACT benefit from early i.m. administration of LNP formulated RNA vaccines. With the early vaccination the low dose of the transferred T cells was boosted to mediate long-lasting anti-tumor efficacy, to the same amount of complete responders as observed for the high dose ACT (10/10). Notably, the quantity of transferred T cells is very often limited in the clinic due to increased probability of systemic toxicity, e.g. acute cytokine release syndrome, autoimmune complications and off-target toxicities. Accordingly, a mRNA vaccination may enable a reduction of the number of transferred T cells to reduce adverse side effects associated with ACT without affecting therapeutic effectiveness. When combined with the mRNA vaccine of the invention, one T cell transfer may be enough for therapeutic efficacy, which reduces manufacturing costs and patient suffering (avoid repeated transfers). Companies need also to produce less autologous T cells for transfer which may result in lower manufacturing costs.
Example 3: Complete tumor regression and long-term survival with combination therapy of low ACT dose and early mRNA vaccination
The goal of the analysis was to show that an LNP formulated and i.m. administered mRNA vaccine is able to increase the efficacy of low and potentially suboptimal doses of transferred T cells to the same level as a high dose of transferred T cells.
For the study, female C57BL/6 CD45.1 mice (10 mice per group) were injected subcutaneously with 1 x106 MC38- OVA tumor cells for tumor development. After tumors reached a mean tumor volume of approximately 28mm3, group 4, 5 and 6 received low dose of 0.1x10s OT-I CD8 CD45.2 T cells and group 3 received the high dose of 2x10® OT-I CD8 CD45.2 T cells derived from OT-I CD45.2 donor mice. Group 5 received an early first intramuscular mRNA vaccination on day 1 after ACT and group 6 a late first mRNA vaccination on day 8. The control group 1 remained untreated whereas group 2 received an early first mRNA vaccination without ACT. Further experimental details are provided above in the introduction of Example 2.
Table 4: Study groups used in Example 3

Example 3.1 : Anti-tumor efficacy of transferred T cells boosted with mRNA vaccine
Experiment was performed as described in Example 3. T umor volume was measured at day 10 and day 41.
The combination therapy of low dose of ACT and early LNP formulated mRNA vaccination showed the same antitumor efficacy as compared to the high dose of ACT at day 10 (Figure 3A) and day 41 (Figure 3B). Complete tumor regression was obsen/ed in both groups at day 41. The mice of the untreated control group 1 had to be sacrificed during the study due to high tumor volume and could not be measured again on day 41.
The late vaccinated group 6 may benefit in the long-term efficacy (day 41) compared to low ACT dose alone.
Example 3.2: Long-term survival for combination therapy of low dose of transferred T cells boosted with mRNA vaccine
Experiment was performed as described in Example 3. Survival of the animals remaining in the study was observed over 54 days. All mice that received high ACT doses (group 3) and the mice that received the combination therapy of low ACT dose with early vaccination of LNP formulated mRNA vaccine (group 5) showed long-term survival (Figure 4) over 54 days.
Results of Example 3
Mice receiving anti-tumoral T cells in low doses could clearly benefit from early mRNA vaccination. The combination led to complete tumor regression and long-term survival in all individuals.
Patients might benefit from the combination therapy of low ACT dose and mRNA vaccine as the adverse side effects correlated with high numbers of transferred T cells may be avoided. When T cells do not need to be amplified during the life cycle (manufacturing process) of cell therapy to maintain high cell numbers, the T cells may better retain their in vivo proliferation potential and antitumor efficacy.
Example 4: Analysis of transferred T cell population boosted with the mRNA vaccine
Goal of the analysis was to evaluate the population of transferred T cells when combined with i.m. administered LNP formulated mRNA vaccine. The amount of circulating transferred antigen specific T cells and their memory- and activation status are important parameters influencing the clinical outcome for the subjects.
Example 4.1 : Analysis of adoptively transferred antigen-specific T cells in the periphery
The goal of the analysis was to evaluate the impact of mRNA vaccination on the number of transferred antigen specific T cells in the periphery. Antigen specific T cells in the periphery are circulating in the body and are able to infiltrate tumors and metastases in different body compartments. The experiment was performed as described in Example 3. All groups received an mRNA vaccination on day 54 (see Table 5). Blood samples were collected at day 1 , day 11 , day 40, day 54 and day 59 and transferred T cell populations in the periphery were analyzed by FACS.
For FACS analysis, mouse PBMCs were stained using a standard protocol including the following steps: Fc-block (BD Biosciences), surface staining, red blood cell lysis (Bio-gems) and fixation (eBiosciences). Before acquiring the samples with the flow cytometer, counting beads (123count eBeads, eBiosciences) were added to each sample, which enabled the exact calculation of the cell count. The expression of specific markers as well as live/dead (L/D) discrimination of PBMCs were determined with fluorescent coupled antibodies or respective dyes (CD45.2:Biolegend, CD8:Biolegend, L/D:eBioscience).
Table 5: Study groups used in Example 4. 1

In all groups, mRNA vaccination resulted in prominent expansion of transferred antigen specific CD8 T cells in the periphery (Figure 5). T ransferred T cells of group 4 and 5 were successfully boosted 39 days after the last vaccination. Of note, the transferred T cells of group 2 (high dose of ACT) and group 3 (low dose of ACT) were boosted successfully with one late vaccination on day 54.
Example 4.2: Analysis of circulating memory T cell population
The goal of the analysis was to evaluate circulating memory T cells in the transferred T cell population. Memory T cells have an important role in long lasting anti-tumor response and are in this setting mainly responsible for the protection against recurrence of the cancer.
The circulating memory T cell population is generally divided into two subsets, effector memory T cells (TEM) and central memory T cells (TCM). Effector memory CD8 T cells (TEM) persist and can react and expand fast to antigen (re)exposure. Durable immunosurveillance is potentially due to memory precursor cells (MPECs) of transferred T cells. MPEGs are considered precursors for long-lived memory pool and SLECs are short-lived effector cells. MPECs survive during the T cell contraction phase and form memory T cells for long-term protective immunity.
The experiment was performed as described in Example 3. The quantity of transferred antigen specific CD8 T cells, the frequency of transferred antigen specific CD8 TE and TC , and the frequencies of MPEC and SLECs in the periphery were analysed on day 40.
For FACS analysis, mouse PBMCs were stained using a standard protocol including the following steps: Fc-block (BD Biosciences), surface staining, red blood cell lyses (Bio-gems) and fixation (eBiosciences). Before acquiring the samples with the flow cytometer, counting beads (123count eBeads, eBiosciences) were added to each sample, which enabled the exact calculation of the cell count. The expression of specific markers as well as live/dead (L/D) discrimination of PBMCs were determined with fluorescent coupled antibodies or respective dyes (CD45.2:Biolegend, CD8: Biolegend, CD44:Biolegend, CD62L:Biolegend, CD127: Biolegend, KLRG1:Biolegend, H-2Kb bound to SlINFEKLBiolegend, L/D:eBioscience).
Table 6: Study groups used in Example 4.2

Both groups that received an mRNA vaccination showed higher frequencies of antigen specific CD8 T cells in the periphery at day 40 (Figure 6A). The mRNA vaccination shifted memory differentiation to effector memory status of transferred CD8 T cells in the periphery (Figure 6B) and increased the numbers of SLECs and MPECs T cells significantly (Figure 6C) at day 40. Mice of untreated control group 1 had to be sacrificed during the study due to high tumor volume and could not be measured on day 40.
Example 4.3: Analysis of phenotype of activated transferred T cells
The goal of the experiment was to analyse the phenotype of the transferred antigen specific OT -I CD8 T cells. Recent studies have shown that the pool of exhausted CD8 T cells is maintained within the tumor infiltrating lymphocytes (TILs) by a precursor subset of cells called progenitor- or stem-like exhausted T cells for recall responses. This subset of T cells has self-renewal capacity. This self-renewal capacity can be demonstrated by SLAMF6 expression (coexpressed with transcription factor TCF1). Terminally differentiated cells without self-renewal capacity are SLAMF6 negative. Strongly activated T cells are PD1 and Tim3 positive.
Experiment was performed as described in Example 3. Blood samples were collected at day 11 and analyzed with FACS.
For FACS analysis mouse PBMCs were stained using a standard protocol including the following steps: Fc-block (BD Biosciences), surface staining, red blood cell lyses (Bio-gems) and fixation (eBiosciences). Before acquiring the samples with the flow cytometer, counting beads (123count eBeads, eBiosciences) were added to each sample, which enabled the exact calculation of the cell count. The expression of specific markers as well as live/dead (L/D) discrimination of PBMCs were determined with fluorescent coupled antibodies or respective dyes (Slamf6/Ly108:Biolegend, CD45.2:Biolegend, CD8:Biolegend, PD-1:Biolegend, L7D:eBioscience, Tim3:Biolegend).
Table 7 Study groups used in Example 4.3 Both groups that received a mRNA vaccination (group 4 and 5) showed high populations of activated (PD1 and TIM3 positive) transferred T cells with the capacity for self-renewal (SLAMF6 positive) on day 11 (Figure 7). This T cell population (PD-1+; TIM3+; SLAMF6+) reflects an intermediate stage between progenitor-stem-like exhausted T cells (PD-1+; TIM3-; SLAMF6+) and terminally exhausted/terminally differentiated T cells (PD-1+; TIM3+; SLAMF6-) and act as critical resource for maintaining T cell immunity in chronic viral infections and cancer.
Both groups that received a mRNA vaccination (group 4 and 5) showed high populations of activated (PD1 and TIM3 positive) transferred T cells with the capacity for self-renewal (SLAMF6 positive) on day 11 (Figure 7). This T cell population (PD-1+; TIM3+; SLAMF6+) reflects an intermediate stage between progenitor-stem-like exhausted T cells (PD-1+; TIM3-; SLAMF6+) and terminally exhausted/terminally differentiated T cells (PD-1+; TIM3+; SLAMF6-) and act as critical resource for maintaining T cell immunity in chronic viral infections and cancer.

Results of Example 4
In summary, the data demonstrated that vaccination with an LNP-formulated and i.m. administered mRNA vaccine could boost transferred CD8 T cells for different ACT doses and vaccination schedules (e.g. Figure 5). Such vaccines could furthermore induce CD8 T cell subsets particular important for strong and durable anti-tumor responses. TEM cells express in general higher levels of receptors responsible for migration to inflamed tissues and have a stronger immediate effector function than TOM cells. High frequencies of TEM cells, as seen with combination therapy, could be beneficial for the patients in the clinic. This population of T cells can react quickly against a recurrence of tumor cells and metastasis. Vaccination with LNP formulated mRNA shifted differentiation of transferred T cells to PD-1+; TIM3+; SLAMF6+ cells.
Example 5: Analysis of endogenous T cell population after RNA vaccination with lymphodepletion prior to cell transfer
The goal of the analysis was to evaluate the effects of LNP formulated mRNA vaccination (i.m.) on the endogenous T cell population that remained in the subject after chemotherapeutic lymphodepletion prior to the ACT therapy.
Table 8: Study groups used in Example 5

The experiment was performed as described in Example 4.1. T cells of the spleen were isolated at day 59. The spleens were harvested, dissociated and erythrocytes were lysed with a red blood cell (ACK) lysing buffer followed by washing steps and counting. 2.5x10® cells/well were seeded to a 96-well plate and stimulated with both peptides SIINFEKL (SEQ ID NO: 193) and TEWTSSNVMEERKIKV (SEQ ID NO: 192) (each 5pg/ml) and anti-CD28 antibody (2.5pg/ml) for 6 h at 37°C. 5h prior endpoint, BFA (1 Opg/ml)) was added to each well to allow accumulation of cytokines intracellularly. For FACS staining the samples were prepared following standard procedures as Fc-Block, surface staining, fixation, permeabilization und intracellular staining (CD45.1 :Biolegend, CD45.2:Biolegend, CD8:Biolegend, CD4:Biolegend, INFgamma:eBioscience, TNFalpha:Biolegend, L/D:eBioscience).
Example 5.1 : Analysis of endogenous CD4 positive T cell population after RNA vaccination in mice lymphodepleted prior to T cell transfer
CD4 T helper cells play a crucial role in the immune response. A positive effect of the LNP formulated RNA vaccination (i.m.) on the endogenous CD4 T helper cell population of the subject after receiving a lymphodepletion therapy may support the therapeutic effect of the ACT therapy. Mice vaccinated with an early or late first vaccination after ACT therapy (vaccination after day 1 or day 8, respectively) showed increased amounts of antigen specific and polyfunctional (double positive IFN-y and TNFa) endogenous CD4 helper T cells compared to the groups (group 1 and group 2) that received only one vaccination at day 54 (Figure 8).
Example 5.2: Analysis of endogenous CD8 T cell population after RNA vaccination in mice lymphodepleted prior to T cell transfer
Endogenous CD8 T cells recognizing e.g. tumor antigens can support the transferred modified immune cells in their therapeutic effect. The mRNA encoded full length antigens comprise the targeted T cell epitope/antigen of the modified transferred cells. Additionally, the mRNA encoded full-length antigen includes several T cell epitopes for the endogenous T cell population. The RNA composition can stimulate and enhance endogenous T cell populations directed to antigens or epitopes other than the transferred modified cells. Accordingly, it is advantageous to encode full length antigens, or at least larger fragments of the antigen.
Mice i.m. vaccinated with the LNP formulated RNA showed antigen specific and polyfunctional (double positive IFN-y and TNFa) endogenous CD8 T cells.
Results of Example 5
The combination with the LNP-formulated mRNA vaccine induced antigen specific and polyfunctional endogenous CD4 and CD8 T cell populations. Activated endogenous CD4 helper T cells can be beneficial for inducing an immune response and secures a long-term memory response with a broad repertoire of antigen specificity. Moreover, it can improve the survival of transferred modified T cells and development of memory T cell pool.
Example 6: Further in vivo analyes with focus on mode-of-action
The goal of the experiment was to further investigate ACT in combination with an LNP-formulated and i.m. administered mRNA vaccine in vivo, in particular its mode-of-action. To this end, the OT-I mouse model described above was modified, amongst others to allow more detailed analyses of modified CD8 T cells in different compartments such as the tumor, spleen and blood. The analyses furthermore focussed on low-dose ACT.
In brief, female C57BL/6 CD45.1 (abbreviated to CD45.1) mice were inoculated subcutaneously in the right rear flank region with 1x10s MC-38-OVA cells for tumor development. T umor volumes were measured in two dimensions using a calliper, and the volume was expressed in mm3 using the formula: V= (L x W* W) /2, where V is tumor volume, L is tumor length (the longest tumor dimension) and W is tumor width (the longest tumor dimension perpendicular to L). A randomization was performed on Day -1 when the mean tumor volume reached approximately 288mm3. 60 mice were enrolled in the study and randomly allocated to 6 study groups, with 10 mice per group. Mice were treated as indicated in Table 9. Cyclophosphamide (Selleck; 10Omg/kg per mouse) was administrated intraperitoneally on Day -1. OT-I cells were injected i.v. on Day 0. Mice were vaccinated intramuscularly (m. tibialis) with 1pg LNP-formulated mRNA encoding full length ovalbumin (SEQ ID NO: 68) on Day 1 and 8. The date of ACT injection was denoted as Day 0.
OT-I cells in this context were generated as follows. 3 female OT-I C57BL/6 CD45.2 (abbreviated to OT-I CD45.2) mice were intraperitoneally injected Ovalbumin (Sigma; 0.5pg/mouse) on Day -2. On Day 0, spleens were collected and mouse CD8+ T cells isolated using the EasySep™ Mouse CD8+ T Cell Isolation Kit (StemCell Technologies; CAT#19853) according to the manufacturers protocol and standard procedures in the art.
Tumor volumes were measured on Day 2, 5, 9 and 11. Mice were sacrificed on Day 11 and tumors, spleens and blood collected for further analysis. Table 9: Study groups used in Example 6-9

Example 7: Analysis of tumor growth
The goal of the experiment was to analyze the effect of ACT in combination with an LNP-formulated and i.m. administered mRNA vaccine, on tumor growth. The experiment was performed as described in Example 6.
Results of Example 7
The results are shown in Figure 9. Combination of ACT with an LNP-formulated and i.m. administered mRNA vaccine was able to significantly slow tumor growth and reduce or even eradicate tumor burden of the very large tumors that were present at the start of the treatment. In contrast, ACT alone could only slow tumor growth, but not reduce tumor burden compared to the start of the treatment, while vaccination alone did not show any effects on tumor growth. This highlights the synergistic effect of the ACT/mRNA vaccine combination. The experiment furthermore shows that this combination enables a significant reduction in tumor burden even in cases where mice were not lymphodepleted (“ACT + vaccination w/o Cp”). This might help to overcome the higher risk for infections and cytopenia correlated with lymphodepleting regimens in the clinic by applying this combination without prior lymphodepleting regimens, e.g. in patients particularly vulnerable or prone to those side effects.
Example 8: Analysis of transferred T cells in the spleen and peripheral blood
The goal of the experiment was to evaluate the effect of the LNP-formulated and i.m administered mRNA vaccine on the transferred T cells residing in the spleen and peripheral blood.
The experiment was performed as described in Example 6. Mice were sacrificed on Day 11 , spleens and blood collected and further analysed as follows. Spleens were weighted and subsequently dissociated using a gentleMACS Dissociator from Miltenyi Biotec according to the manufacturer’s protocol and standard procedures in the art.
For FACS analysis, mouse cells were stained using a standard protocol including the following steps: red blood cell lysis (Bio-gems; for splenocytes only where necessary), mouse Fc-block (BD Biosciences), Dasatinib (Sigma) treatment, surface marker staining, fixation (eBiosciences). Before acquiring the samples with the flow cytometer, counting beads (123count eBeads, eBiosciences) were added to each sample, which enabled the exact calculation of the cell count. The expression of specific markers as well as live/dead (L/D) discrimination of cells were determined with fluorescent coupled antibodies or respective dyes including CD45.1 : BD, CD45.2:Biolegend, CD8:MBL, Thy1.2: Biolegend, H-2Kb tetramer bound to SIINFEKL:MBL.
Further analyses of the T cell populations described in Example 4.3 will be performed by combining the staining described in Example 8 with fluorescent coupled antibodies or respective dyes recognizing: PD-1 :Biolegend, Tim3:Biolegend, l-Ab OVA323-339 Tetramer-ISQAVHAAHAEINEAGR:MBL and TCF1:BD Biosciences. The latter staining will be performed intracellularly using a fixation/permeabilization buffer e.g. from eBioscience.
Results of Example 8
Results are shown in Figure 10 and Figure 11. As can be seen herein, at day 11 almost no transferred CD8 T cells could be detected in the spleen and peripheral blood for the ACT alone group (i.e. without vaccination). In stark contrast, the indicated group including both ACT and vaccination displayed a very high amount of transferred CD8 T cells suggesting increased survival and/or proliferation of those cells. The high CD8 T cell count in this combination group was further reflected by increased spleen weights. Notably, also for the ACT/mRNA vaccine combination without prior lymphodepletion increased numbers of transferred CD8+ T cells in the peripheral blood and spleen as well as a corresponding increased spleen weights could be observed (data not shown). Moreover, the latter combination showed increased numbers of endogenous antigen-specific CD8 T cells in the peripheral blood and spleen.
Example 9: Analysis of transferred T cells in the tumor
The goal of the experiment was to evaluate the effect of the LNP-formulated and i.m. administered mRNA vaccine on the infiltration of transferred T cells into the tumor and on their phenotype. To this end, the amount of transferred T cells in the tumor as well as their activation (marker CD25) and cytotoxicity (marker Granzyme B) were determined.
The experiment was performed as described in Example 6. Mice were sacrificed on Day 11 , tumors collected and further analysed as follows. Spleens were weighted and subsequently dissociated using the gentleMACS Dissociator from Miltenyi Biotec according to the manufacturer’s protocol and standard procedures in the art.
For FACS analysis, mouse cells were stained using a standard protocol including the following steps: mouse Fc-block (BD Biosciences), Dasatinib (Sigma) treatment, surface marker staining, fixation/permeabilization and intracellular staining of Granzyme B. Before acquiring the samples with the flow cytometer, counting beads (123count eBeads, eBiosciences) were added to each sample, which enabled the exact calculation of the cell count. The expression of specific markers as well as live/dead (L/D) discrimination of cells were determined with fluorescent coupled antibodies or respective dyes including CD45.1 : BD, CD45.2:Biolegend, CD8:MBL, Thy1.2: Biolegend, H-2Kb tetramer bound to SIINFEKLMBL, CD25: Biolegend, Granzyme B: Biolegend, L/D:eBioscience.
Results of Example 9
The results are shown in Figure 12. As can be seen, a starkly increased amount of transferred CD8 T cells in the tumor could be observed for the indicated ACT/mRNA vaccine combination. Furthermore, the transferred CD8 T cells in this combination group also showed a higher cytotoxicity and activation as reflected by upregulation of Granzyme B and CD25, respectively. Notably, also for the ACT/mRNA vaccine combination without prior lymphodepletion strong effects could be observed (data not shown).
Example 10: Exemplary clinical study design - Phase I dose escalation study for combination of i.m. lipid nanoparticle formulated RNA vaccine with ACT
Primary endpoints: safety, tolerability and Dose Limiting Toxicities (DLT) Secondary endpoints: Frequency and functional status of adoptively transferred T cells in the circulation, antigen specific T cells response against the at least vaccine encoded antigen, objective response rate (ORR), Disease control rate (DCR), Duration of response (DoR)
Key inclusion criteria:
• Patients with advanced solid tumors or hematologic malignancies without curative treatment options, refractory to standard treatments
• measurable disease or elevated tumor marker
• some tumor cells of patient are tested positive for the target antigen of the adoptive T cells
Further description:
• Composition:
LNP formulated RNA comprising a cds encoding at least one antigen, wherein the at least one encoded antigen comprises at least the epitope (T cell epitope) recognized by the antigen receptor of the adoptive immune cells, preferably the nucleic acid encoding at least one full length antigen.
• Administration route: intramuscular, dose will be increased stepwise e.g. 12pg, 25pg, 50pg or 100pg
• Early vaccination time point: 1 day after low dose ACT, optionally on day 7, day 14, day 21 , day 28,
• Late vaccination time point: 6 weeks after high dose of ACT
ACT: genetically modified TCR (TCRtg) or CAR T cells, targeted to the antigen, preferably tumor antigen, more preferably an epitope derived from PRAME or CLDN6
• ACT high dose
• ACT low dose
• Administration time point: on day 1
Cohort 1: Monotherapy: ACT, lymphodepletion (chemotherapy) prior to ACT
Cohort 2: Combination therapy: ACT in combination with the composition of the invention, lymphodepletion (chemotherapy) prior to ACT. A subgroup of the cohort receives a high dose of ACT with dose escalation e.g. 12pg, 25pg, 50pg or 100pg of the composition on the early vaccination time point(s) (as described above). The other group receives a low dose ACT with dose escalation of e.g. 12pg, 25pg, 50pg or 100 g of the composition on the early vaccination timepoint(s).
Cohort 3: Combination therapy: ACT in combination with the composition of the invention, without lymphodepletion prior to ACT. One subgroup of the cohort 3 receives a high dose of ACT with dose escalation of e.g. 12 g, 25 g, 50 g or 10Opg of the composition on the early vaccination timepoint(s). The other group receives a low dose ACT with dose escalation of e.g. 12pg, 25pg, 50pg or 100pg of the composition the early vaccination time point(s).
Cohort 4: Combination therapy: ACT in combination the composition of the invention, with lymphodepletion prior to ACT. One subgroup of the cohort 4 receives a high dose ACT in combination with dose escalation of >49pg of the composition on the late vaccination time point. The other group receives a low dose ACT with dose escalation of e.g. >49 g composition on the late vaccination time point.
Claims
1. A composition comprising at least one RNA comprising at least one coding sequence encoding at least one antigen for use in a method of treatment or prophylaxis of a disease, disorder or condition, wherein the RNA is formulated in lipid nanoparticles (LNPs) and wherein the composition is administered intramuscularly to a subject that has received modified T cells targeted to the at least one antigen.
2. The composition for use of claim 1 , wherein the modified T cells are genetically modified to express an antigen receptor targeted to the antigen, preferably wherein the antigen receptor is a chimeric antigen receptor (CAR) or a T cell receptor (TCR), preferably a TCR.
3. The composition for use of claim 1 or 2, wherein the modified T cells are selected from TCR T cells and/or CAR T cells.
4. The composition for use of any one of the preceding claims, wherein the antigen is associated with the disease, disorder or condition, preferably wherein the antigen is selected from or derived from tumor antigens, viral antigens, bacterial antigens, protozoal antigens, fungal antigens, self-antigens or allogenic antigens, preferably tumor antigens.
5. The composition for use of claim 4, wherein the tumor antigen is tumor specific antigen or tumor associated antigen, preferably selected from mutation antigen, neoantigen, unique antigen, differentiation tumor antigen, overexpressed tumor antigen, virus tumor antigen or bacterial tumor antigen.
6. The composition for use of any one of the preceding claims, wherein the antigen is a tumor antigen selected or derived from p53, 5T4, ART-4, AFP, BAGE, beta-eaten in/m, BCMA, Bcr-abL B7-H3, mutated BRAF, CAMEL, CAP-1, CASP-8, CDC27/m, CDK4/m, CD19, CD22, CD20, CD33, CD70, CEA, mutated CTNNB1 , the cell surface proteins of the claudin family, such as Claudin6 (CLDN6), CLDN18.2 and CLDN12, COL6A3, c-MYC, CT, Cyp-B, DAM, DLL3, EBV, ELF2M, ETV6-AML1 , EGFR, EGFRvlll, EphA2, G250, GAGE, GD2, GnT-V, GPC3, Gap 100, Gp100, HAGE, HA-1, HER-2/neu, HBV, HPV-E7, HPV-E6, HAST-2, hTERT (or hTRT), KRAS, mutated KRAS, KK-LC-1 , LAGE, LDLR/FUT, MAGE-A, preferably MAGE-A1 , MAGE-A2, MAGE- A3, MAGE-A4, MAGE-A5, MAGE-A6, MAGE-A7, MAGE-A8, MAGE-A9, MAGE-A10, MAGE-A11, or MAGE-A12, MAGE-B, MAGE-C, MART-1/Melan-A, MICA/B, MC1R, Mesothelin, Myosin/m, MUC1, MUM-1, MUM-2, MUM-3, NA88-A, NF1 , NY-ESO-1 , NY-BR-1, NKG2DL, PAP, pl90 minor BCR-abL, Pml/RARa, PRAME, mutated PRAME, proteinase 3, PD-L1 , PSA, PSM, PSMA, PSCA, RAGE, ROBO1 , RU1 or RU2, SAGE, SART-1 or SART-3, SCGB3A2, SCP1 , SCP2, SCP3, SSX, STEAP, SURVIVIN, TEL/AML1 , mutated TP53, TPI/m, TRP-1 , TRP-2, TRP-2/INT2, TPTE, WT, and WT1.
7. The composition for use of any one of the preceding claims, wherein the at least one encoded antigen comprises at least the T cell epitope, optionally, wherein the at least one encoded antigen comprises at least 1 , 2, 3, 4, 5, 6, 7, 8, 9 or 10 epitopes recognized by the antigen receptor of the modified T cells.
8. The composition for use of any one of the preceding claims, wherein the disease, disorder or condition is selected from a cancer or tumor disease, disorder or condition, an infectious disease, disorder or condition, an autoimmune disease, disorder or condition, or a disease, disorder or condition associated with stem cell transplantation, preferably wherein the disease, disorder or condition is selected from a tumor or cancer disease, disorder or condition, optionally
9. The composition for use of any one of the preceding claims, wherein the subject has received the modified T cells by intravenous administration.
10. The composition for use of any one of claims 1 to 9, wherein the subject has not received lymphodepletion within 1 , 2, 3, 4, 5, 6, 7, 8, 9, 10, 11 , 12, 13 or 14 days before the administration of the modified T cells targeted to the antigen.
11. The composition for use of any one of claims 1 to 9, wherein the subject has received a lymphodepletion within 1 , 2, 3, 4, 5, 6, 7, 8, 9, 10, 11 , 12, 13 or 14 days before the administration of the modified T cells targeted to the antigen.
12. The composition for use of any one of the preceding claims, wherein at least 3, 4, 5, or more doses of the composition are administered to the subject.
13. The composition for use of any one of the preceding claims, wherein intramuscular administration of the composition to the subject increases levels of circulating modified T cells in the subject; induces memory formation of modified T cells in the subject, preferably formation of central and effector memory T cells; activates modified T cells in the subject; re-activates modified T cells in the subject; maintains self-renewal capacity of modified T cells in the subject; induces higher influx into the solid tumor of modified T cells in the subject; induces expansion of modified T cells in the subject; activates endogenous CD4 helper cells in the subject; skews differentiation of endogenous CD4+ T cells to a Th1 phenotype; increases levels of circulating endogenous T cells in the subject; activates endogenous T cells in the subject; boosts polyfunctionality of the endogenous T cells in the subject; preferably to a larger extend compared to intravenous administration of the composition to the subject.
14. The composition for use of any one of the preceding claims, wherein the LNPs comprise at least one aggregation-reducing lipid, at least one cationic lipid or ionizable lipid, at least one neutral lipid or phospholipid, and at least one steroid or steroid analogue.
15. The composition for use of any one of the preceding claims, wherein the LNPs have a Z-average size in a range of about 50nm to about 200nm, preferably about 50nm to about 150nm.
16. The composition for use of any one of the preceding claims, wherein the LNPs have a polydispersity index (PDI) value of less than about 0.3, preferably of less than about 0.2.
17. The composition for use of any one of the preceding claims, wherein at least 70%, preferably at least 80%, more preferably at least 90% of the RNA of the composition is encapsulated in the lipid nanoparticles.
18. The composition for use of any one of the preceding claims, wherein at least about 80%, 85%, 90%, 95% of the LNPs have a spherical morphology.
19. The composition for use of any one of the preceding claims, wherein the LNPs exhibit a zeta potential in the range of -50 mV to +50 mV, preferably in the range of -25 mV to +25 mV, more preferably in the range of -10 mV to +10 mV, most preferably in the range of -5 mV to +5 mV.
20. The composition for use of any one of the preceding claims, wherein the at least one coding sequence encodes at least one antigen that is selected from or derived from tumor antigens, viral antigens, bacterial antigens, protozoal antigens, fungal antigens, self-antigens or allogenic antigens, preferably tumor antigens, preferably wherein the tumor antigen is selected from any one of the antigens as defined in Claim 5 or, preferably, Claim 6.
21. The composition for use of any one of the preceding claims, wherein the at least one RNA comprises at least one untranslated region (UTR), preferably selected from at least one heterologous 5'-UTR and/or at least one heterologous 3’-UTR.
22. The composition for use of claim 21 , wherein the at least one 5 -UTR is selected from HSD17B4 and the at least one 3-UTR is selected from PSMB3.
23. The composition for use of any one of the preceding claims, wherein the at least one RNA is selected from an mRNA, a circular RNA, a replicon RNA, or a viral RNA, preferably wherein the RNA is an mRNA.
24. The composition for use of any one of the preceding claims, wherein the RNA comprises at least one poly(A) sequence, preferably wherein the at least one poly(A) sequence comprises about 40 to about 500 adenosine nucleotides, preferably about 60 to about 150 adenosine nucleotides, more preferably about
100 adenosine nucleotides.
25. The composition for use of any one of the preceding claims, wherein the at least one RNA comprises at least one modified nucleotide selected from N1 -methylpseudouridine (m1yj) or wherein the at least one RNA does not comprise a modified nucleotide.
26. The composition for use of any one of the preceding claims, wherein the least one RNA comprises a 5’- cap structure, preferably a 5’-cap structure is selected from a cap1 structure or a modified cap1 structure.
27. A combination comprising: (i) a composition comprising at least one RNA comprising at least one coding sequence encoding at least one antigen, wherein the RNA is formulated in lipid nanoparticles; and (ii) modified T cells targeted to the at least one antigen; for use in a method of treatment or prophylaxis of a disease, disorder or condition, wherein the modified T cells are administered intravenously to a subject, and wherein the composition is administered intramuscularly to the subject that received the modified T cells.
28. The combination for use of claim 27, wherein the method, the composition, the modified T cells, the subject, and the disease, disorder or condition are further characterized by any one of the features of claims 1 to 26.
29. A kit or kit or parts comprising (A) at least one composition as defined in any one of claims 1 to 26, or
(B) at least one combination as defined in claims 27 or 28, for use in a method of treatment or prophylaxis of a disease, disorder or condition, wherein the kit or kit of parts optionally comprises instructions for use of the kit or kit of parts in a method of treatment or prophylaxis of a disease, disorder or condition.
30. A method of treating or preventing a disease, disorder, or condition in a subject, comprising a step of applying or administering intramuscularly a composition comprising at least one RNA comprising at least one coding sequence encoding at least one antigen, wherein the RNA is formulated in lipid nanoparticles, wherein the subject has received modified T cells targeted to the at least one antigen, and wherein the method is optionally further characterized by any one of the features of any one of claims 1 to 26.
31. A method of activating or stimulating a modified T cell population in a subject, wherein the method comprises applying or administering intramuscularly at least one composition comprising at least one RNA comprising at least one coding sequence encoding at least one antigen to the subject, wherein the RNA is formulated in lipid nanoparticles, and wherein the method is optionally further characterized by any one of the features of any one of claims 1 to 26.
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EPPCT/EP2023/080127 | 2023-10-27 | ||
| EP2023080127 | 2023-10-27 | ||
| EP2024072756 | 2024-08-12 | ||
| EPPCT/EP2024/072756 | 2024-08-12 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2025088088A1true WO2025088088A1 (en) | 2025-05-01 |
Family
ID=93257927
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2024/080173PendingWO2025088088A1 (en) | 2023-10-27 | 2024-10-25 | Rna composition for improving cell therapy |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2025088088A1 (en) |
Citations (28)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1997047648A1 (en) | 1996-06-14 | 1997-12-18 | Meiji Milk Products Co., Ltd. | T cell epitope peptide |
| WO2001008636A2 (en) | 1999-08-03 | 2001-02-08 | The Ohio State University | Polypeptides and polynucleotides for enhancing immune reactivity to her-2 protein |
| WO2001062284A2 (en) | 2000-02-21 | 2001-08-30 | Pharmexa A/S | Novel method for down-regulation of amyloid |
| WO2002014478A2 (en) | 2000-08-16 | 2002-02-21 | Apovia, Inc. | IMMUNOGENIC HBc CHIMER PARTICLES HAVING ENHANCED STABILITY |
| WO2002098443A2 (en) | 2001-06-05 | 2002-12-12 | Curevac Gmbh | Stabilised mrna with an increased g/c content and optimised codon for use in gene therapy |
| WO2004000873A2 (en) | 2002-06-25 | 2003-12-31 | City Of Hope | Adjuvant-free peptide vaccine |
| WO2004058297A1 (en) | 2002-12-27 | 2004-07-15 | Centro De Ingeniería Genética Y Biotecnología | Combinations of growth- and hormone-regulating factors for the treatment of neoplasia |
| WO2006113792A2 (en) | 2005-04-19 | 2006-10-26 | Eli Lilly And Company | Monovalent and polyvalent synthetic polysaccharide antigens for immunological intervention in disease |
| WO2008017517A1 (en) | 2006-08-11 | 2008-02-14 | Life Sciences Research Partners Vzw | Immunogenic peptides and their use in immune disorders |
| WO2008077592A1 (en) | 2006-12-22 | 2008-07-03 | Curevac Gmbh | Method for purifying rna on a preparative scale by means of hplc |
| WO2010023247A1 (en) | 2008-08-29 | 2010-03-04 | Centre National De La Recherche Scientifique | Synthetic peptides corresponding to overlapping neutralizing determinants in the cbd1 epitope induce broadly neutralizing antibodies. |
| WO2012019780A1 (en) | 2010-08-13 | 2012-02-16 | Curevac Gmbh | Nucleic acid comprising or coding for a histone stem-loop and a poly(a) sequence or a polyadenylation signal for increasing the expression of an encoded protein |
| WO2013171505A2 (en) | 2012-05-17 | 2013-11-21 | Kymab Limited | In vivo guided selection & antibodies |
| WO2015101416A1 (en) | 2013-12-30 | 2015-07-09 | Curevac Gmbh | Methods for rna analysis |
| WO2015199952A1 (en) | 2014-06-25 | 2015-12-30 | Acuitas Therapeutics Inc. | Novel lipids and lipid nanoparticle formulations for delivery of nucleic acids |
| WO2016193206A1 (en) | 2015-05-29 | 2016-12-08 | Curevac Ag | A method for producing and purifying rna, comprising at least one step of tangential flow filtration |
| WO2017004143A1 (en) | 2015-06-29 | 2017-01-05 | Acuitas Therapeutics Inc. | Lipids and lipid nanoparticle formulations for delivery of nucleic acids |
| WO2017053297A1 (en) | 2015-09-21 | 2017-03-30 | Trilink Biotechnologies, Inc. | Compositions and methods for synthesizing 5'-capped rnas |
| WO2017075531A1 (en) | 2015-10-28 | 2017-05-04 | Acuitas Therapeutics, Inc. | Novel lipids and lipid nanoparticle formulations for delivery of nucleic acids |
| WO2018078053A1 (en) | 2016-10-26 | 2018-05-03 | Curevac Ag | Lipid nanoparticle mrna vaccines |
| WO2021123332A1 (en) | 2019-12-20 | 2021-06-24 | Curevac Ag | Lipid nanoparticles for delivery of nucleic acids |
| WO2021156267A1 (en) | 2020-02-04 | 2021-08-12 | Curevac Ag | Coronavirus vaccine |
| WO2021239880A1 (en) | 2020-05-29 | 2021-12-02 | Curevac Ag | Nucleic acid based combination vaccines |
| US20220249557A1 (en)* | 2021-02-05 | 2022-08-11 | Innovative Cellular Therapeutics Holdings, Ltd. | Vaccine and Uses thereof in Cell Therapy |
| WO2022207862A2 (en) | 2021-03-31 | 2022-10-06 | Curevac Ag | Syringes containing pharmaceutical compositions comprising rna |
| WO2023007019A1 (en) | 2021-07-30 | 2023-02-02 | CureVac SE | Cap analogs having an acyclic linker to the guanine derivative nucleobase |
| WO2023031394A1 (en) | 2021-09-03 | 2023-03-09 | CureVac SE | Novel lipid nanoparticles for delivery of nucleic acids |
| WO2023073228A1 (en) | 2021-10-29 | 2023-05-04 | CureVac SE | Improved circular rna for expressing therapeutic proteins |
- 2024
- 2024-10-25WOPCT/EP2024/080173patent/WO2025088088A1/enactivePending
Patent Citations (28)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1997047648A1 (en) | 1996-06-14 | 1997-12-18 | Meiji Milk Products Co., Ltd. | T cell epitope peptide |
| WO2001008636A2 (en) | 1999-08-03 | 2001-02-08 | The Ohio State University | Polypeptides and polynucleotides for enhancing immune reactivity to her-2 protein |
| WO2001062284A2 (en) | 2000-02-21 | 2001-08-30 | Pharmexa A/S | Novel method for down-regulation of amyloid |
| WO2002014478A2 (en) | 2000-08-16 | 2002-02-21 | Apovia, Inc. | IMMUNOGENIC HBc CHIMER PARTICLES HAVING ENHANCED STABILITY |
| WO2002098443A2 (en) | 2001-06-05 | 2002-12-12 | Curevac Gmbh | Stabilised mrna with an increased g/c content and optimised codon for use in gene therapy |
| WO2004000873A2 (en) | 2002-06-25 | 2003-12-31 | City Of Hope | Adjuvant-free peptide vaccine |
| WO2004058297A1 (en) | 2002-12-27 | 2004-07-15 | Centro De Ingeniería Genética Y Biotecnología | Combinations of growth- and hormone-regulating factors for the treatment of neoplasia |
| WO2006113792A2 (en) | 2005-04-19 | 2006-10-26 | Eli Lilly And Company | Monovalent and polyvalent synthetic polysaccharide antigens for immunological intervention in disease |
| WO2008017517A1 (en) | 2006-08-11 | 2008-02-14 | Life Sciences Research Partners Vzw | Immunogenic peptides and their use in immune disorders |
| WO2008077592A1 (en) | 2006-12-22 | 2008-07-03 | Curevac Gmbh | Method for purifying rna on a preparative scale by means of hplc |
| WO2010023247A1 (en) | 2008-08-29 | 2010-03-04 | Centre National De La Recherche Scientifique | Synthetic peptides corresponding to overlapping neutralizing determinants in the cbd1 epitope induce broadly neutralizing antibodies. |
| WO2012019780A1 (en) | 2010-08-13 | 2012-02-16 | Curevac Gmbh | Nucleic acid comprising or coding for a histone stem-loop and a poly(a) sequence or a polyadenylation signal for increasing the expression of an encoded protein |
| WO2013171505A2 (en) | 2012-05-17 | 2013-11-21 | Kymab Limited | In vivo guided selection & antibodies |
| WO2015101416A1 (en) | 2013-12-30 | 2015-07-09 | Curevac Gmbh | Methods for rna analysis |
| WO2015199952A1 (en) | 2014-06-25 | 2015-12-30 | Acuitas Therapeutics Inc. | Novel lipids and lipid nanoparticle formulations for delivery of nucleic acids |
| WO2016193206A1 (en) | 2015-05-29 | 2016-12-08 | Curevac Ag | A method for producing and purifying rna, comprising at least one step of tangential flow filtration |
| WO2017004143A1 (en) | 2015-06-29 | 2017-01-05 | Acuitas Therapeutics Inc. | Lipids and lipid nanoparticle formulations for delivery of nucleic acids |
| WO2017053297A1 (en) | 2015-09-21 | 2017-03-30 | Trilink Biotechnologies, Inc. | Compositions and methods for synthesizing 5'-capped rnas |
| WO2017075531A1 (en) | 2015-10-28 | 2017-05-04 | Acuitas Therapeutics, Inc. | Novel lipids and lipid nanoparticle formulations for delivery of nucleic acids |
| WO2018078053A1 (en) | 2016-10-26 | 2018-05-03 | Curevac Ag | Lipid nanoparticle mrna vaccines |
| WO2021123332A1 (en) | 2019-12-20 | 2021-06-24 | Curevac Ag | Lipid nanoparticles for delivery of nucleic acids |
| WO2021156267A1 (en) | 2020-02-04 | 2021-08-12 | Curevac Ag | Coronavirus vaccine |
| WO2021239880A1 (en) | 2020-05-29 | 2021-12-02 | Curevac Ag | Nucleic acid based combination vaccines |
| US20220249557A1 (en)* | 2021-02-05 | 2022-08-11 | Innovative Cellular Therapeutics Holdings, Ltd. | Vaccine and Uses thereof in Cell Therapy |
| WO2022207862A2 (en) | 2021-03-31 | 2022-10-06 | Curevac Ag | Syringes containing pharmaceutical compositions comprising rna |
| WO2023007019A1 (en) | 2021-07-30 | 2023-02-02 | CureVac SE | Cap analogs having an acyclic linker to the guanine derivative nucleobase |
| WO2023031394A1 (en) | 2021-09-03 | 2023-03-09 | CureVac SE | Novel lipid nanoparticles for delivery of nucleic acids |
| WO2023073228A1 (en) | 2021-10-29 | 2023-05-04 | CureVac SE | Improved circular rna for expressing therapeutic proteins |
Non-Patent Citations (9)
| Title |
|---|
| ANDREAS MACKENSEN: "CLDN6-specific CAR-T cells plus amplifying RNA vaccine in relapsed or refractory solid tumors: the phase 1 BNT211-01 trial", vol. 29, no. 11, 23 October 2023 (2023-10-23), New York, pages 2844 - 2853, XP093162046, ISSN: 1078-8956, Retrieved from the Internet <URL:https://www.nature.com/articles/s41591-023-02612-0> DOI: 10.1038/s41591-023-02612-0* |
| BOLIN WANG: "Recent advances in mRNA cancer vaccines: meeting challenges and embracing opportunities", FRONTIERS IN IMMUNOLOGY, vol. 14, 6 September 2023 (2023-09-06), Lausanne, CH, XP093162270, ISSN: 1664-3224, DOI: 10.3389/fimmu.2023.1246682* |
| DOBIN A. ET AL., BIOINFORMATICS, vol. 29, 2013, pages 15 - 21 |
| EMMANUEL OWUSU ANSAH: "Vaccine Boosting CAR-T Cell Therapy: Current and Future Strategies", vol. 2023, 31 January 2023 (2023-01-31), pages 1 - 9, XP093162119, ISSN: 2573-8461, Retrieved from the Internet <URL:https://downloads.hindawi.com/journals/acgt/2023/8030440.pdf?_gl=1*1qndjt9*_ga*NzI0MTM5NTQ3LjE3MTU2Nzc1MjQ.*_ga_NF5QFMJT5V*MTcxNTY3NzUyNC4xLjAuMTcxNTY3NzUyNC42MC4wLjA.&_ga=2.218596441.1128931839.1715677524-724139547.1715677524> DOI: 10.1155/2023/8030440* |
| GREG SLABODKIN: "Moderna, CARsgen Join Forces Combining mRNA Cancer Vaccine with CAR T | BioSpace", 21 August 2023 (2023-08-21), pages 1 - 4, XP093162175, Retrieved from the Internet <URL:https://www.biospace.com/article/moderna-carsgen-join-forces-combining-mrna-cancer-vaccine-with-car-t/>* |
| HAN SUN: "mRNA-Based Therapeutics in Cancer Treatment", PHARMACEUTICS, vol. 15, no. 2, 13 February 2023 (2023-02-13), Switzerland, pages 622, XP093162824, ISSN: 1999-4923, DOI: 10.3390/pharmaceutics15020622* |
| KATHARINA REINHARD ET AL: "An RNA vaccine drives expansion and efficacy of claudin-CAR-T cells against solid tumors", SCIENCE, vol. 367, no. 6476, 24 January 2020 (2020-01-24), US, pages 446 - 453, XP055681271, ISSN: 0036-8075, DOI: 10.1126/science.aay5967* |
| LEYUAN MA: "Vaccine-boosted CAR T crosstalk with host immunity to reject tumors with antigen heterogeneity", vol. 186, no. 15, 1 July 2023 (2023-07-01), Amsterdam NL, pages 3148 - 3165.e20, XP093162109, ISSN: 0092-8674, Retrieved from the Internet <URL:https://www.cell.com/cell/pdf/S0092-8674(23)00642-6.pdf> DOI: 10.1016/j.cell.2023.06.002* |
| no. 2036272-55-4 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP7096282B2 (en) | RNA preparation for immunotherapy | |
| EP4081239A1 (en) | In vitro and in vivo gene delivery to immune effector cells using nanoparticles functionalized with designed ankyrin repeat proteins (darpins) | |
| JP2025131724A (en) | Treatments including interleukin 2 (IL2) and interferon (IFN) | |
| US20230145774A1 (en) | Treatment involving non-immunogenic rna for antigen vaccination | |
| US20230000962A1 (en) | Treatment involving immune effector cells genetically modified to express antigen receptors | |
| US20220356223A1 (en) | IL2 Agonists | |
| WO2025088088A1 (en) | Rna composition for improving cell therapy | |
| US20220177544A1 (en) | Interleukin-2 receptor (IL2R) and interleukin-2 (IL2) variants for specific activation of immune effector cells | |
| US20230405046A1 (en) | Antigen-specific t cell receptors and t cell epitopes |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | Ref document number:24794806 Country of ref document:EP Kind code of ref document:A1 |