WO2025059685A1 - Methods for in vivo targeted delivery of a payload - Google Patents
Methods for in vivo targeted delivery of a payloadDownload PDFInfo
- Publication number
- WO2025059685A1 WO2025059685A1PCT/US2024/046969US2024046969WWO2025059685A1WO 2025059685 A1WO2025059685 A1WO 2025059685A1US 2024046969 WUS2024046969 WUS 2024046969WWO 2025059685 A1WO2025059685 A1WO 2025059685A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- payload
- subject
- moiety
- hours
- occurrence
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Classifications
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6891—Pre-targeting systems involving an antibody for targeting specific cells
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6835—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site
- A61K47/6851—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site the antibody targeting a determinant of a tumour cell
- A61K47/6855—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site the antibody targeting a determinant of a tumour cell the tumour determinant being from breast cancer cell
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
Definitions
- the present disclosurerelates generally to methods of utilizing antibody-tetrazine conjugates for bioorthogonal delivery of a payload to a targeted location in a subject, which conjugates have applications, e.g., in the treatment of cancer, tumor growth, and immunotherapy.
- BACKGROUND[0003] Bioorthogonal conjugation or click reactions are selective and orthogonal (non-interacting with) functionalities found in biological systems, and have found use in various applications in the fields of chemistry, chemical biology, molecular diagnostics, and medicine, where they can be used to facilitate the selective manipulation of molecules, cells, particles and surfaces, and the tagging and tracking of biomolecules in vitro and in vivo.
- the present disclosureis directed to methods for delivering a payload to a target location in a subject.
- a method of forming in vivo an antibody-payload conjugate in a subject in need thereofcomprising: administering an effective amount of a targeting moiety to the subject, wherein the targeting moiety comprising at least one antibody or fragment thereof having a least one tetrazine moiety covalently linked thereto; administering a single dose of a therapeutically effective amount of a payload-TCO conjugate to the subject, wherein the payload-TCO conjugate comprises a payload having at least one trans- cyclooctene (TCO) moiety covalently linked thereto; wherein the antibody or fragment thereof has a binding affinity to a receptor on a tumor, and further wherein the amount of the antibody-payload conjugate formed in vivo is greater at a tumor site versus in plasma.
- TCOtrans- cyclooctene
- a method of administering a therapeutically effective amount of a payload to a subjectcomprising: a) administering an effective amount of a targeting moiety to the subject, wherein the targeting moiety comprises a Fab having at least one tetrazine moiety covalently linked thereto; and b) administering a single dose of a therapeutically effective amount of a payload-TCO conjugate to the subject, wherein the payload-TCO conjugate comprises a payload having at least one (e.g., 1 to 3, 1 to 2, or 1) trans-cyclooctene moiety covalently linked thereto; wherein the single dose of the payload-TCO conjugate is administered to the subject within a time frame of between about 2 hours and 48 hours, or between about 3 hours and 48 hours, or between about 4 and about 48 hours, after the targeting moiety is administered to the subject.
- the targeting moietycomprises a Fab having 1 to 5, 1 to 4, or 1 to 3, tetrazine moieties covalently linked thereto (i.e., the DAR, or ratio of Fab to tetrazine is an average of 5, 4, 3, or 2)
- the targeting moieties for use in the methods disclosed hereinare designed to, once administered to a subject, localize at a target site within the subject.
- the targeting moietiescan be administered locally or systemically.
- a prodrugcomprising a payload and one or more complimentary bioorthogonal components (i.e., a trans-cyclooctene moiety) is administered, which when in contact with the targeting moiety in vivo, allows for targeted delivery of the payload or therapeutic agent.
- a prodrugcomprising a payload and one or more complimentary bioorthogonal components (i.e., a trans-cyclooctene moiety) is administered, which when in contact with the targeting moiety in vivo, allows for targeted delivery of the payload or therapeutic agent.
- a method of treating cancer, or enhancing or eliciting an immune response, in a subject having cancercomprising: a) administering an effective amount of a targeting moiety to the subject, wherein the targeting moiety comprises a Fab having at least one (e.g., 1 to 5, 1 to 4, or 1 to 3) tetrazine moiety covalently linked thereto; and b) administering a single dose of a therapeutically effective amount of a payload-TCO conjugate to the subject, wherein the payload-TCO conjugate comprises a payload having at least one (e.g., 1 to 3, 1 to 2, or 1) trans-cyclooctene moiety covalently linked thereto; wherein the single dose of the payload-TCO conjugate is administered to the subject more than 1 hour, and less than 48 hours, after the targeting moiety is administered to the subject.
- a targeting moietycomprises a Fab having at least one (e.g., 1 to 5, 1 to 4, or 1 to 3) tetrazine
- a method of treating cancer, or enhancing or eliciting an immune response, in a subject having cancercomprising: a) administering an effective amount of a targeting moiety to the subject, wherein the targeting moiety comprises a Fab having at least one (e.g., 1 to 5, 1 to 4, or 1 to 3) tetrazine moiety covalently linked thereto; and b) administering a single dose of a therapeutically effective amount of a payload-TCO conjugate to the subject, wherein the payload-TCO conjugate comprises a payload having at least one (e.g., 1 to 3, 1 to 2, or 1) trans-cyclooctene moiety covalently linked thereto; wherein the single dose of the payload-TCO conjugate is administered to the subject more than 4 hours, and less than 48 hours, after the targeting moiety is administered to the subject.
- a targeting moietycomprises a Fab having at least one (e.g., 1 to 5, 1 to 4, or 1 to 3) tetrazine
- the canceris metastatic.
- the canceris melanoma, renal cancer, prostate cancer, ovarian cancer, endometrial carcinoma, breast cancer, glioblastoma, lung cancer, soft tissue sarcoma, fibrosarcoma, osteosarcoma, pancreatic cancer, gastric carcinoma, squamous cell carcinoma of head/neck, anal/vulvar carcinoma, esophageal carcinoma, pancreatic adenocarcinoma, cervical carcinoma, hepatocellular carcinoma, Kaposi’s sarcoma, Non-Hodgkin’s lymphoma, Hodgkin’s lymphoma Wilm’s tumor/neuroblastoma, bladder cancer, thyroid adenocarcinoma, pancreatic neuroendocrine tumors, prostatic adenocarcinoma, nasopharyngeal carcinoma, or cutaneous T-cell lymphoma.
- the canceris a melanoma, renal cancer, prostate cancer, ovarian cancer, breast cancer, glioma, lung cancer, soft tissue carcinoma, soft tissue sarcoma, osteosarcoma, or pancreatic cancer.
- the canceris a lymphoma or leukemia.
- the canceris a hematologic malignancy.
- the canceris a solid tumor.
- a targeting moiety of Formula IIFwherein p is 1 to 10; and X is an antibody fragment moiety comprising SEQ ID NO.9 and SEQ ID No.10. In some embodiments, p is 1 to 5.
- a method of reducing tumor volume in a subject having a tumorcomprising: a) administering an effective amount of a targeting moiety to the subject, wherein the targeting moiety comprises a Fab having at least one (e.g., 1 to 5, 1 to 4, or 1 to 3) tetrazine moiety covalently linked thereto; and b) administering a single dose of a therapeutically effective amount of a payload-TCO conjugate to the subject, wherein the payload-TCO conjugate comprises a payload having at least one (e.g., 1 to 3, 1 to 2, or 1) trans-cyclooctene moiety covalently linked thereto; wherein the single dose of the payload-TCO conjugate is administered to the subject more than 1 hour, and less than 48 hours, after the targeting moiety is administered to the subject.
- a method of reducing tumor volume in a subject having a tumorcomprising: a) administering an effective amount of a targeting moiety to the subject, wherein the targeting moiety comprises a Fab having at least one (e.g., 1 to 5, 1 to 4, or 1 to 3) tetrazine moiety covalently linked thereto; and b) administering a single dose of a therapeutically effective amount of a payload-TCO conjugate to the subject, wherein the payload-TCO conjugate comprises a payload having at least one (e.g., 1 to 3, 1 to 2, or 1) trans-cyclooctene moiety covalently linked thereto; wherein the single dose of the payload-TCO conjugate is administered to the subject more than 4 hours, and less than 48 hours, after the targeting moiety is administered to the subject.
- FIGs.1A-1Dshow tumor volumes of NCI-N87 tumors in SCID CB17 mice following treatment with vehicle, Compound TM-1 with Compound B, and Isotype Fab-Tz with Compound B.
- Compound Bwas dosed 4 hours (FIG.1A), 8 hours (FIG.1B), 24 hours (FIG.1C), or 48 hours (FIG.1D) after Compound TM-1 dose.
- N5 mice per group.
- FIG.2Ashows the schedule of dosing and tumor collection.
- FIG.2Bshows Compound TM-1 activates Compound B to release MMAE in tumors 15 minutes after dosing.
- FIG.3Ashows quantification of total Fab in HER2-positive tumors.
- FIG.3Bshows Compound TM-1 localizes to HER2-positive tumors.
- FIG.4Ashows the total Fab detected at the tumor site vs in the plasma.
- FIG.4Bshows the total tetrazine detected at the tumor site vs in the plasma.
- FIG.5shows Fab exposure was prolonged in plasma and at the tumor (Tz levels decreased quickly in plasma but were prolonged at the tumor).
- FIG.6shows the total tetrazine detected at the tumor site at certain time intervals.
- FIG.7shows the total Fab detected at the tumor site at certain time intervals.
- FIG.8shows the ratio of tetrazine (Fab-Biotin) at the tumor site vs tissues.
- FIG.9shows the total Fab ratio at the tumor site vs tissues.
- FIG.10 and FIG.11show preferential accumulation of Compound TM-1 at the tumor as compared to plasma DETAILED DESCRIPTION
- the following descriptionsets forth exemplary embodiments of the present technology. It should be recognized, however, that such description is not intended as a limitation on the scope of the present disclosure but is instead provided as a description of exemplary embodiments. Definitions [0029] It is appreciated that certain features of the disclosure, which are, for clarity, described in the context of separate embodiments, may also be provided in combination in a single embodiment.
- the modifier “about” used in connection with a quantityis inclusive of the stated value and has the meaning dictated by the context (for example, it includes at least the degree of error associated with the measurement of the particular quantity).
- the modifier “about”should also be considered as disclosing the range defined by the absolute values of the two endpoints.
- the expression “from about 2 to about 4”also discloses the range “from 2 to 4.”
- the term “about”may refer to plus or minus 10% of the indicated number.
- “about 10%”may indicate a range of 9% to 11%, and “about 1” may mean from 0.9-1.1.
- Other meanings of “about”may be apparent from the context, such as rounding off, so, for example “about 1” may also mean from 0.5 to 1.4.
- the conjunctive term “or”includes any and all combinations of one or more listed elements associated by the conjunctive term.
- the phrase “an apparatus comprising A or B”may refer to an apparatus including A where B is not present, an apparatus including B where A is not present, or an apparatus where both A and B are present.
- the phrases “at least one of A, B, ... and N” or “at least one of A, B, ... N, or combinations thereof”are defined in the broadest sense to mean one or more elements selected from the group comprising A, B, ... and N, that is to say, any combination of one or more of the elements A, B, ...
- alkylas used herein, means a straight or branched, saturated hydrocarbon chain containing from 1 to 30 carbon atoms.
- lower alkyl or C1-C6-alkylmeans a straight or branched chain hydrocarbon containing from 1 to 6 carbon atoms.
- C1-C3- alkylmeans a straight or branched chain hydrocarbon containing from 1 to 3 carbon atoms.
- alkylinclude, but are not limited to, methyl, ethyl, n-propyl, iso-propyl, n-butyl, sec-butyl, iso-butyl, tert- butyl, n-pentyl, isopentyl, neopentyl, n-hexyl, 3-methylhexyl, 2,2-dimethylpentyl, 2,3-dimethylpentyl, n- heptyl, n-octyl, n-nonyl, and n-decyl.
- alkoxyrefers to an alkyl group, as defined herein, appended to the parent molecular moiety through an oxygen atom. Representative examples of alkoxy include, but are not limited to, methoxy, ethoxy, propoxy, 2-propoxy, butoxy, and tert-butoxy.
- alkenylas used herein, means a hydrocarbon chain containing from 2 to 30 carbon atoms with at least one carbon-carbon double bond. The alkenyl group may be substituted or unsubstituted. For example, the alkenyl group may be substituted with an aryl group, such as a phenyl.
- alkynylrefers to straight or branched monovalent hydrocarbyl groups having from 2 to 30 carbon atoms, such as 2 to 20, or 2 to 10 carbon atoms and having at least 1 site of triple bond unsaturation.
- alkynealso includes non-aromatic cycloalkyl groups of from 5 to 20 carbon atoms, such as from 5 to 10 carbon atoms, having single or multiple rings and having at least one triple bond.
- alkynyl groupsinclude, but are not limited to acetylenyl (-C ⁇ CH), and propargyl (-CH2C ⁇ CH), and cycloalkynyl moieties, such as, but not limited to, substituted or unsubstituted cyclooctyne moieties.
- alkoxyalkylrefers to an alkoxy group, as defined herein, appended to the parent molecular moiety through an alkyl group, as defined herein.
- alkylenerefers to a divalent group derived from a straight or branched chain hydrocarbon of 1 to 30 carbon atoms, for example, of 2 to 10 carbon atoms.
- Representative examples of alkyleneinclude, but are not limited to, -CH 2 -, -CH(CH 3 )-, -C(CH 3 ) 2 -, -CH 2 CH 2 -, -CH(CH 3 )CH 2 -, -C(CH 3 ) 2 CH 2 -, -CH 2 CH 2 CH 2 -, -CH(CH 3 )CH 2 CH 2 -, -C(CH 3 ) 2 CH 2 CH 2 -, -CH 2 C(CH 3 ) 2 CH 2 -, -CH 2 CH 2 CH 2 CH 2 -, and –CH 2 CH 2 CH 2 CH 2 CH 2 -.
- amino acidrefers to both natural and unnatural amino acids, protected natural and unnatural amino acids, as well as amino acid analogs and amino acid mimetics that function in a manner similar to the naturally occurring amino acids.
- Naturally encoded amino acidsinclude 20 common amino acids (alanine, arginine, asparagine, aspartic acid, cysteine, glutamine, glutamic acid, glycine, histidine, isoleucine, leucine, lysine, methionine, phenylalanine, proline, serine, threonine, tryptophan, tyrosine and valine) and pyrrolidine and selenocysteine.
- Non-natural amino acidsrefer to amino acid analogs having the same basic chemical structure as a naturally occurring amino acid, i.e., by way of example only, an ⁇ - carbon attached to a hydrogen, carboxyl group, amino group, and R group.
- Such analogscan have a modified R group (e.g., norleucine as an example) or retain a modified peptide backbone while retaining the same basic chemical structure as a natural amino acid.
- Non-limiting examples of non-natural amino acids or amino acid analogsinclude citrulline, homoserine, norleucine, methionine sulfoxide, methionine methylsulfonium, homophenylalanine, ornithine, formyl glycine, phenyl glycine, para-azidophenyl glycine, para-azidophenylalanine, para-acetophenylalanine, 4-(3-methyl-(1,2,4,5-tetrazine))- phenylglyine, and 4-(3-methyl-(1,2,4,5-tetrazine))-phenylalanine.
- arylrefers to an aromatic carbocyclic group having a single ring (e.g. monocyclic) or multiple rings (e.g. bicyclic or tricyclic) including fused systems.
- Representative examples of arylsinclude, but are not limited to, phenyl, naphthyl, and anthracenyl.
- the monocyclic, bicyclic, and tricyclic arylsare connected to the parent molecular moiety through any carbon atom contained within the rings, and can be unsubstituted or substituted.
- the aromatic bicyclic ring system or aromatic tricyclic ring systemdoes not contain non-aromatic rings.
- a bicyclic ring system or tricyclic ring systemcontains a non-aromatic ring
- the ring systemis a cycloalkyl or heterocyclyl, depending on whether a heteroatom is present in the non-aromatic ring, regardless of the point of attachment to the remainder of the molecule.
- arylrefers to a phenyl group, or bicyclic aryl or tricyclic aryl fused ring systems.
- Bicyclic fused ring systemsare exemplified by a phenyl group appended to the parent molecular moiety and fused to a phenyl group.
- Tricyclic fused ring systemsare exemplified by a phenyl group appended to the parent molecular moiety and fused to two other phenyl groups.
- Representative examples of bicyclic arylsinclude, but are not limited to, naphthyl.
- Representative examples of tricyclic arylsinclude, but are not limited to, anthracenyl.
- the monocyclic, bicyclic, and tricyclic arylsare connected to the parent molecular moiety through any carbon atom contained within the rings, and can be unsubstituted or substituted.
- cycloalkylrefers to a non-aromatic carbocyclic ring system containing 3 to 10, or 3 to 8, or 3 to 6, or 5 to 10, carbon atoms and zero heteroatoms. Cycloalkyl ring systems may contain one or more double bonds, so long as the ring is not aromatic; and thus, the term cycloalkyl includes cycloalkenyl ring systems. Representative examples of cycloalkyl include, but are not limited to, cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl, cyclooctyl, cyclononyl, and cyclodecyl.
- Exemplary monocyclic cycloalkenyl ringsinclude cyclopentenyl, cyclohexenyl, or cycloheptenyl.
- Cycloalkylalso includes carbocyclic ring systems in which a cycloalkyl group is fused to an aryl or heteroaryl as defined herein, regardless of the point of attachment to the remainder of the molecule.
- cycloalkylinclude, but are not limited to, cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl, cyclooctyl, cyclononyl and cyclodecyl.
- Cycloalkylalso includes carbocyclic ring systems in which a cycloalkyl group is appended to the parent molecular moiety and is fused to an aryl group as defined herein, a heteroaryl group as defined herein, or a heterocycle as defined herein.
- cycloalkenylas used herein, means a non-aromatic monocyclic or multicyclic ring system containing at least one carbon-carbon double bond, e.g., having from 5-10 carbon atoms per ring.
- exemplary monocyclic cycloalkenyl ringsinclude cyclopentenyl, cyclohexenyl or cycloheptenyl.
- cyclooctenerefers to a substituted or unsubstituted non-aromatic cyclic alkyl group of 8 carbon atoms, having a single ring with a double bond.
- cyclooctene groupsinclude, but are not limited to, substituted or unsubstituted trans-cyclooctene (TCO).
- TCOtrans-cyclooctene
- fluoroalkylmeans an alkyl group, as defined herein, in which one, two, three, four, five, six, seven or eight hydrogen atoms are replaced by fluorine.
- Representative examples of fluoroalkylinclude, but are not limited to, 2-fluoroethyl, 2,2,2-trifluoroethyl, trifluoromethyl, difluoromethyl, pentafluoroethyl, and trifluoropropyl such as 3,3,3-trifluoropropyl.
- alkoxyfluoroalkylrefers to an alkoxy group, as defined herein, appended to the parent molecular moiety through a fluoroalkyl group, as defined herein.
- fluoroalkoxymeans at least one fluoroalkyl group, as defined herein, is appended to the parent molecular moiety through an oxygen atom.
- Representative examples of fluoroalkyloxyinclude, but are not limited to, difluoromethoxy, trifluoromethoxy and 2,2,2- trifluoroethoxy.
- halogenor “halo” as used herein, means Cl, Br, I, or F.
- haloalkylas used herein, means an alkyl group, as defined herein, in which one, two, three, four, five, six, seven or eight hydrogen atoms are replaced by a halogen.
- haloalkoxyas used herein, means at least one haloalkyl group, as defined herein, is appended to the parent molecular moiety through an oxygen atom.
- heteroalkylas used herein, means an alkyl group, as defined herein, in which one or more of the carbon atoms has been replaced by a heteroatom selected from S, Si, O, P and N. The heteroatom may be oxidized.
- heteroalkylsinclude, but are not limited to, alkyl ethers, secondary and tertiary alkyl amines, and alkyl sulfides.
- heteroarylrefers to an aromatic group having a single ring, multiple rings or multiple fused rings, with one or more ring heteroatoms independently selected from nitrogen, oxygen and sulfur.
- heteroarylrefers to an aromatic monocyclic ring or an aromatic bicyclic ring system or an aromatic tricyclic ring system.
- the aromatic monocyclic ringsare five or six membered rings containing at least one heteroatom independently selected from the group consisting of N, O and S (e.g.1, 2, 3, or 4 heteroatoms independently selected from O, S, and N).
- the five membered aromatic monocyclic ringshave two double bonds and the six membered aromatic monocyclic rings have three double bonds.
- monocyclic heteroarylinclude, but are not limited to, pyridinyl (including pyridin-2-yl, pyridin-3-yl, pyridin-4-yl), pyrimidinyl, pyrazinyl, thienyl, furyl, thiazolyl, thiadiazolyl, isoxazolyl, pyrazolyl, and 2-oxo-1,2- dihydropyridinyl.
- bicyclic heteroarylinclude, but are not limited to, chromenyl, benzothienyl, benzodioxolyl, benzotriazolyl, quinolinyl, thienopyrrolyl, thienothienyl, imidazothiazolyl, benzothiazolyl, benzofuranyl, indolyl, quinolinyl, imidazopyridine, benzooxadiazolyl, and benzopyrazolyl.
- tricyclic heteroarylinclude, but are not limited to, dibenzofuranyl and dibenzothienyl.
- the monocyclic, bicyclic, and tricyclic heteroarylsare connected to the parent molecular moiety through any carbon atom or any nitrogen atom contained within the rings, and can be unsubstituted or substituted.
- the aromatic bicyclic ring system or aromatic tricyclic ring systemdoes not contain non-aromatic rings.
- the ring systemis a cycloalkyl or heterocyclyl, depending on whether a heteroatom is present in the non-aromatic ring, regardless of the point of attachment to the remainder of the molecule.
- the five membered aromatic monocyclic ringshave two double bonds and the six membered aromatic monocyclic rings have three double bonds.
- exemplary bicyclic heteroaryl groupsare exemplified by a monocyclic heteroaryl ring appended to the parent molecular moiety and fused to a monocyclic cycloalkyl group, as defined herein, a monocyclic aryl group, as defined herein, a monocyclic heteroaryl group, as defined herein, or a monocyclic heterocycle, as defined herein.
- the tricyclic heteroaryl groupsare exemplified by a monocyclic heteroaryl ring appended to the parent molecular moiety and fused to two of a monocyclic cycloalkyl group, as defined herein, a monocyclic aryl group, as defined herein, a monocyclic heteroaryl group, as defined herein, or a monocyclic heterocycle, as defined herein.
- heterocyclylrefers to a non- aromatic ring system containing 3 to 10, or 3 to 8, or 3 to 6, or 5 to 10, carbon atoms and at least one (e.g., 1 to 5, 1 to 4, 1 to 3, 1 to 2, or 1) heteroatom, and optionally one or more oxo and/or double bonds.
- heterocyclylinclude monocyclic, bicyclic, tricyclic, fused, spirocyclic, or bridged ring systems, provided that at least one non-aromatic ring system containing at least one heteroatom is present.
- the monocyclic heterocycleis a three-, four-, five-, six-, seven-, or eight-membered ring containing at least one heteroatom independently selected from the group consisting of O, N, and S.
- the three- or four-membered ringcontains zero or one double bond, and one heteroatom selected from the group consisting of O, N, and S.
- the five-membered ringcontains zero or one double bond and one, two or three heteroatoms selected from the group consisting of O, N and S.
- the six-membered ringcontains zero, one or two double bonds and one, two, or three heteroatoms selected from the group consisting of O, N, and S.
- the seven- and eight-membered ringscontains zero, one, two, or three double bonds and one, two, or three heteroatoms selected from the group consisting of O, N, and S.
- monocyclic heterocyclesinclude, but are not limited to, azetidinyl, azepanyl, aziridinyl, diazepanyl, 1,3-dioxanyl, 1,3-dioxolanyl, 1,3-dithiolanyl, 1,3-dithianyl, 1,3-dimethylpyrimidine-2,4(1H,3H)-dione, imidazolinyl, imidazolidinyl, isothiazolinyl, isothiazolidinyl, isoxazolinyl, isoxazolidinyl, morpholinyl, oxadiazolinyl, oxadiazolidinyl, oxazolinyl, oxazolidinyl, morpholin
- the bicyclic heterocycleis a monocyclic heterocycle fused to a phenyl group, or a monocyclic heterocycle fused to a monocyclic cycloalkyl, or a monocyclic heterocycle fused to a monocyclic cycloalkenyl, or a monocyclic heterocycle fused to a monocyclic heterocycle, or a spiro heterocycle group, or a bridged monocyclic heterocycle ring system in which two non-adjacent atoms of the ring are linked by an alkylene bridge of 1, 2, 3, or 4 carbon atoms, or an alkenylene bridge of two, three, or four carbon atoms.
- bicyclic heterocyclesinclude, but are not limited to, benzopyranyl, benzothiopyranyl, chromanyl, 2,3- dihydrobenzofuranyl, 2,3-dihydrobenzothienyl, 2,3-dihydroisoquinoline, 2-azaspiro[3.3]heptan-2-yl, azabicyclo[2.2.1]heptyl (including 2-azabicyclo[2.2.1]hept-2-yl), 2,3-dihydro-1H-indolyl, isoindolinyl, octahydrocyclopenta[c]pyrrolyl, octahydropyrrolopyridinyl, and tetrahydroisoquinolinyl.
- Tricyclic heterocyclesare exemplified by a bicyclic heterocycle fused to a phenyl group, or a bicyclic heterocycle fused to a monocyclic cycloalkyl, or a bicyclic heterocycle fused to a monocyclic cycloalkenyl, or a bicyclic heterocycle fused to a monocyclic heterocycle, or a bicyclic heterocycle in which two non- adjacent atoms of the bicyclic ring are linked by an alkylene bridge of 1, 2, 3, or 4 carbon atoms, or an alkenylene bridge of two, three, or four carbon atoms.
- tricyclic heterocyclesinclude, but are not limited to, octahydro-2,5-epoxypentalene, hexahydro-2H-2,5-methanocyclopenta[b]furan, hexahydro-1H-1,4-methanocyclopenta[c]furan, aza-adamantane (1-azatricyclo[3.3.1.1 3,7 ]decane), and oxa-adamantane (2-oxatricyclo[3.3.1.1 3,7 ]decane).
- hydroxylas used herein, means an —OH group.
- hydroxyalkylas used herein, means an alkyl group, as defined herein, in which one, two, three, four, five, six, seven or eight hydrogen atoms are replaced by a hydroxyl group.
- the number of carbon atoms in a hydrocarbyl substituentis indicated by the prefix “C x -C y -” or “C x-y ,” wherein x is the minimum and y is the maximum number of carbon atoms in the substituent.
- C 1 -C 3 -alkyl” and “C 1-3 alkyl”refer to an alkyl substituent containing from 1 to 3 carbon atoms.
- the two conventions “C x -C y -” and “C x-y ”are used interchangeably and have the same meaning.
- substitutedrefers to a group that may be further substituted with one or more non- hydrogen substituent groups.
- tetrazinerefers to a substituted or unsubstituted aromatic cyclic group of 2 carbon atoms and 4 nitrogen atoms, having a single ring with three double bonds.
- tetrazine groupsinclude 1,2,3,4-tetrazine and 1,2,4,5-tetrazine.
- 1,2,4,5-tetrazineis referred to as a “Tz” group.
- selective deliveringrefers to delivering an agent (e.g., a payload) to an organ or tissue (or portion thereof) in need of treatment or diagnosis, without significant binding to other non- target organs or tissues (or portions thereof).
- the targeting moieties, or therapeutic targeting moiety, described hereindo not themselves have a therapeutic effect, but rather are designed to allow the selective or targeted delivery of a therapeutic agent. However, it may be that the targeting moiety does have a therapeutic effect, and thus, such constructs are not excluded by the present disclosure.
- the term “payload”refers to an agent for delivery to a target site in a subject. In some embodiments, the payloads is a therapeutic agent. In some embodiments, the payloads is a diagnostic agent.
- therapeutic agentrefers to an agent capable of treating and/or ameliorating a condition or disease, or one or more symptoms thereof, in a subject.
- Therapeutic agents of the present disclosurealso include prodrug forms of therapeutic agents, chelating agent with or without a radionuclide (e.g., diagnostic or therapeutic).
- a radionuclidee.g., diagnostic or therapeutic.
- a therapeutic “radionuclide” or “radioligand” as used hereinrefers to a radioactive substance, sometimes referred to as a radiopharmaceutical, which are used to treat medical conditions, particularly cancer.
- Radionuclides used in the trans-cyclooctene moieties described hereincomprise a chelating agent and an isotope; such as an isotope selected from the group consisting of 24 Na, 32 P, 33 P, 47 Sc, 59 Fe, 67 Cu, 76 As, 77 As, 80 Br, 82 Br, 89 Sr, 90 Nb, 90 Y, 103 Ru, 105 Rh, 109 Pd, 111 Ag, 111 In, 121 Sn, 127 Te, 131 I, 140 La, 141 Ce, 142 Pr, 143 Pr, 144 Pr, 149 Pm, 149 Tb, 151 Pm, 153 Sm, 159 Gd, 161 Tb, 165 Dy, 166 Ho, 169 Er, 172 Tm, 175 Yb, 177 Lu, 186 Re, 188 Re, 198 Au, 199 Au, 211 At, 211 Bi, 212 Bi, 212 Pb, 213 Bi, 214 Bi, 223 Ra, and
- Radionuclidescan be delivered via direct conjugation or chelation with a chelating agent. Exemplary radionuclides, chelating agents, and linkers for potential TCO-conjugate payloads are described below.
- diagnostic agentrefers to agents that assist in diagnosing conditions or diseases. Representative diagnostic agents include imaging agents such as paramagnetic agents, optical probes, radionuclides, and the like. Paramagnetic agents are imaging agents that are magnetic under an externally applied field. Examples of paramagnetic agents include, but are not limited to, iron particles including iron nanoparticles and iron microparticles. Optical probes are fluorescent compounds that can be detected by excitation at one wavelength of radiation and detection at a second, different, wavelength of radiation.
- Optical probes of the present disclosureinclude, but are not limited to, Cy5.5, Alexa 680, Cy5, DiD (1,1’-dioctadecyl-3,3,3’,3’-tetramethylindodicarbocyanine perchlorate) and DiR (1,1’- dioctadecyl-3,3,3’,3’-tetramethylindotricarbocyanine iodide).
- Other optical probesinclude quantum dots. Radionuclides are elements that undergo detectable radioactive decay.
- Radionuclidessuch as diagnostic radionuclides, useful in embodiments of the present disclosure include, but are not limited to, 3 H, 11 C, 13 N, 18 F, 19 F, 60 Co, 64 Cu, 67 Cu, 68 Ga, 82 Rb, 89 Zr, 90 Sr, 90 Y, 99 Tc, 99m Tc, 111 In, 123 I, 124 I, 125 I, 129 I, 131 I, 137 Cs, 177 Lu, 186 Re, 188 Re, 211 At, 212 Pb, 225 Ac, Rn, Ra, Th, U, Pu, and 241 Am.
- targeting agentrefers to a chemical or biological agent that specifically binds to a target (e.g., a targeted organ or tissue), thereby forming a stable association between the targeting agent and the specific target.
- stably associatedor “stable association” is meant that a moiety is bound to or otherwise associated with another moiety or structure under standard physiological conditions. Bonds may include covalent bonds and non-covalent interactions, such as, but not limited to, ionic bonds, hydrophobic interactions, hydrogen bonds, van der Waals forces (e.g., London dispersion forces), dipole- dipole interactions, and the like.
- Targeting agentsinclude ligands that specifically bind (or substantially specifically bind) a particular clinically-relevant target receptor or cell surface target.
- antibody fragment moietyrefers to one or more regions or fragments of an antibody that retain the ability to specifically bind to an antigen, or in other words, substantially retain the antigen-binding function of the antibody.
- binding fragments encompassed within the antigen-binding portion of an antibodyinclude but are not limited to a Fab fragment.
- the Fab fragmentis a monovalent fragment containing at least the V L and V H .
- the Fab fragmentis a monovalent fragment containing the V L , V H , C L , and C H 1 domains.
- Fab fragmentscan be obtained using conventional techniques known to those with skill in the art, and the fragments are screened for utility in the same manner as are intact antibodies.
- antibody fragmentscan be produced by recombinant DNA techniques, or by enzymatic or chemical cleavage of intact immunoglobulins. Additional examples are included in various embodiments disclosed herein.
- bindis meant the preferential association of an antibody or antibody fragment moiety to a target molecule (e.g., an antigen, such as a peptide, polypeptide, glycoprotein, or any other moiety with one or more antigenic determinants) or to a cell or tissue bearing the target molecule (e.g., a cell surface antigen, such as a receptor or ligand) and not to cells or tissues lacking the target molecule.
- a target moleculee.g., an antigen, such as a peptide, polypeptide, glycoprotein, or any other moiety with one or more antigenic determinants
- a cell or tissue bearing the target moleculee.g., a cell surface antigen, such as a receptor or ligand
- Specific bindingresults in a much stronger association between the targeting moiety (e.g., Fab) and e.g., cells bearing the target molecule (e.g., an antigen) than between the binding moiety and e.g., cells lacking the target molecule.
- Specific bindingtypically results in greater than 2-fold, greater than 5-fold, greater than 10-fold, or greater than 100-fold increase in amount of bound binding moiety (per unit time) to e.g., a cell or tissue bearing the target molecule or marker as compared to a cell or tissue lacking that target molecule or marker.
- binding moietiesbind to the target molecule or marker with a dissociation constant of e.g., less than 10 -5 M, less than 10 -7 M, less than 10 -8 M, less than 10 -9 M, less than 10 -10 M, less than 10 -11 M, less than 10 -12 M, less than 10 -13 M, less than 10 -14 M, or less than 10 -15 M.
- a dissociation constante.g., less than 10 -5 M, less than 10 -7 M, less than 10 -8 M, less than 10 -9 M, less than 10 -10 M, less than 10 -11 M, less than 10 -12 M, less than 10 -13 M, less than 10 -14 M, or less than 10 -15 M.
- assay formatsare appropriate for measuring binding, such as solid-phase ELISA immunoassays.
- targeted organ or tissuerefers to an organ or tissue that is being targeted for delivery of the payload.
- organs and tissues for targetinginclude those that can be targeted by chemical or biological targeting agents, as well as those organs and tissues that cannot be targeted by chemical or biological targeting agents.
- the term “contacting” or “contact”refers to the process of bringing into contact at least two distinct species such that they can interact with each other, such as in a non-covalent or covalent binding interaction or binding reaction. It should be appreciated, however, the resulting complex or reaction product can be produced directly from an interaction or a reaction between the added reagents or from an intermediate from one or more of the added reagents or moieties, which can be produced in the contacting mixture.
- administeringrefers to any suitable route of administration to a subject, such as, but not limited to, oral administration, administration as a suppository, topical contact, parenteral, intravenous, intraperitoneal, intramuscular, intralesional, intranasal or subcutaneous administration, intrathecal administration, or the implantation of a slow-release device, e.g., a mini-osmotic pump, to the subject.
- parenterallyrefers to modes of administration which include intravenous, intramuscular, intraperitoneal, intrasternal, subcutaneous and intraarticular injection, and infusion.
- a pharmaceutically or therapeutically effective amountrefers to an amount of a compound sufficient to treat a specified disorder or disease or one or more of its symptoms and/or to prevent or reduce the risk of the occurrence or reoccurrence of the disease or disorder or symptom(s) thereof.
- a pharmaceutically or therapeutically effective amountcomprises an amount sufficient to, among other things, cause the tumor to shrink or decrease the growth rate of the tumor.
- the term “subject,” “patient,” or “organism”includes humans and mammals (e.g., mice, rats, pigs, cats, dogs, and horses).
- Typical subjects to which an agent(s) of the present disclosure may be administeredmay include mammals, particularly primates, especially humans.
- suitable subjectsmay include, for example, livestock such as cattle, sheep, goats, cows, swine, and the like; poultry such as chickens, ducks, geese, turkeys, and the like; and domesticated animals particularly pets such as dogs and cats.
- suitable subjectsmay include mammals, such as rodents (e.g., mice, rats, hamsters), rabbits, primates, and swine such as inbred pigs and the like.
- treatingmeans the treating or treatment of a disease or medical condition or symptom(s) thereof in a patient, such as a mammal (e.g., a human) that includes: (a) ameliorating the disease or medical condition or symptom(s) thereof, such as, eliminating or causing regression of the disease or medical condition or symptom(s) thereof in a patient; (b) suppressing the disease or medical condition or symptom(s) thereof, for example by, slowing or arresting the development of the disease or medical condition or symptom(s) thereof in a patient; or (c) alleviating a symptom of the disease or medical condition or symptom(s) thereof in a patient.
- a mammale.g., a human
- physiological conditionsis meant to encompass those conditions compatible with living cells, e.g., predominantly aqueous conditions of a temperature, pH, salinity, etc. that are compatible with living cells.
- groups and substituents thereofmay be selected in accordance with permitted valence of the atoms and the substituents, such that the selections and substitutions result in a stable compound, e.g., which does not spontaneously undergo transformation such as by rearrangement, cyclization, elimination, etc.
- the numbers 7 and 8are contemplated in addition to 6 and 9, and for the range 6.0-7.0, the number 6.0, 6.1, 6.2, 6.3, 6.4, 6.5, 6.6, 6.7, 6.8, 6.9, and 7.0 are explicitly contemplated.
- the compoundsmay exist as stereoisomers wherein asymmetric or chiral centers are present.
- the stereoisomersare “R” or “S” depending on the configuration of substituents around the chiral carbon atom.
- the terms “R” and “S” used hereinare configurations as defined in IUPAC 1974 Recommendations for Section E, Fundamental Stereochemistry, in Pure Appl. Chem., 1976, 45: 13-30.
- Stereoisomersinclude enantiomers and diastereomers and mixtures of enantiomers or diastereomers.
- Individual stereoisomers of the compoundsmay be prepared synthetically from commercially available starting materials, which contain asymmetric or chiral centers or by preparation of racemic mixtures followed by methods of resolution well-known to those of ordinary skill in the art.
- the present disclosurealso includes isotopically-labeled compounds, which are identical to those recited herein, but for the fact that one or more atoms are replaced by an atom having an atomic mass or mass number different from the atomic mass or mass number usually found in nature.
- isotopes suitable for inclusion in the compounds of the disclosureare hydrogen, carbon, nitrogen, oxygen, phosphorus, sulfur, fluorine, and chlorine, such as, but not limited to, 2 H, 3 H, 13 C, 14 C, 15 N, 18 O, 17 O, 31 P, 32 P, 35 S, 18 F, and 36 Cl, respectively.
- the compoundmay incorporate positron-emitting isotopes for medical imaging and positron-emitting tomography (PET) studies for determining the distribution of receptors.
- positron-emitting isotopesthat can be incorporated are 11 C, 13 N, 15 O, and 18 F.
- Isotopically-labeled compounds disclosed hereincan generally be prepared by conventional techniques known to those skilled in the art or by processes analogous to those described in the accompanying Examples using appropriate isotopically-labeled reagent in place of non-isotopically-labeled reagent.
- Methods of Treatment[0086] The present disclosure is directed to methods for delivering a payload to a target location in a subject.
- a method of forming in vivo an antibody-payload conjugate in a subject in need thereofcomprising: administering an effective amount of a targeting moiety to the subject, wherein the targeting moiety comprising at least one antibody or fragment thereof having a least one tetrazine moiety covalently linked thereto; administering a single dose of a therapeutically effective amount of a payload-TCO conjugate to the subject, wherein the payload-TCO conjugate comprises a payload having at least one trans- cyclooctene (TCO) moiety covalently linked thereto; wherein the antibody or fragment thereof has a binding affinity to a receptor on a tumor, and further wherein the amount of the antibody-payload conjugate formed in vivo is greater at a tumor site versus in plasma.
- TCOtrans- cyclooctene
- a method of forming in vivo an antibody-payload conjugate in a subject in need thereofcomprising: administering an effective amount of a targeting moiety to the subject, wherein the targeting moiety comprising at least one antibody or fragment thereof having a least one tetrazine moiety covalently linked thereto; administering a single dose of a therapeutically effective amount of a payload-TCO conjugate to the subject, wherein the payload-TCO conjugate comprises a payload having at least one trans- cyclooctene (TCO) moiety covalently linked thereto; wherein the antibody or fragment thereof has a binding affinity to a receptor on a tumor, and further wherein the ratio of the antibody-payload conjugate at a tumor site versus in plasma is greater than about 2:1.
- a targeting moietycomprising at least one antibody or fragment thereof having a least one tetrazine moiety covalently linked thereto
- TCOtrans- cyclooctene
- the ratio of the antibody-payload conjugate at a tumor site versus in plasmais greater than 1:1, or about 2:1, or about 3:1, or about 4:1, or about 5:1, or about 6:1, or about 7:1, or about 8:1, or about 9:1, or about 10:1, or about 11:1, about 12:1, or about 13:1, or about 14:1, or about 15:1, or about 16:1, or about 17:1, or about 18:1, or about 19:1.
- the ratio of the antibody-payload conjugate at a tumor site versus in plasmais greater than about 2:1, or greater than about 3:1, or greater than about 4:1, or greater than about 5:1, or greater than about 6:1, or greater than about 7:1, or greater than about 8:1, or greater than about 9:1, or greater than about 10:1.
- the administeringis simultaneous.
- the administeringis sequential.
- the targeting moietyis administered prior to the payload-TCO conjugate.
- a first dose of the payload-TCO conjugateis administered to the subject less than 48 hours, or more than 4 hours and less than 48 hours, after the targeting moiety is administered to the subject.
- a single dose of the payload-TCO conjugateis administered to the subject less than 48 hours, or more than 4 hours and less than 48 hours, after the targeting moiety is administered to the subject.
- a single dose of the payload-TCO conjugateis administered to the subject less than 48 hours after the targeting moiety is administered to the subject.
- a single dose of the payload-TCO conjugateis administered to the subject more than 4 hours, and less than 48 hours, after the targeting moiety is administered to the subject.
- the targeting moietyis administered at least 8 to about 24 hours prior to the payload-TCO conjugate being administered.
- the targeting moietyis administered within 0-8 serum half-lives. It is contemplated that timing of the payload-TCO conjugate administration is calculated based on tumor disposition, which can be determined by the biology of the antigen at the tumor and the targeting moiety.
- the targeting moietycomprises a targeting format as in the table below
- the payload-TCO conjugateis administered within the serum half life range shown therein (see also Berland et al., Biomolecules 2021, 11(5), 637).
- a method of administering a payload to a subjectcomprising: a) administering an effective amount of a targeting moiety to the subject, wherein the targeting moiety comprises a Fab having at least one tetrazine moiety covalently linked thereto; and b) administering a single dose of a therapeutically effective amount of a payload-TCO conjugate to the subject, wherein the payload-TCO conjugate comprises a payload having at least one trans-cyclooctene moiety covalently linked thereto; wherein the single dose of the payload-TCO conjugate is administered to the subject at least about 2 hours, or at least about 3 hours, or at least about 4 hours, less than about 48 hours, or between about 4 and 48 hours, after the targeting moiety is administered to the subject.
- a method of administering a payload to a subjectcomprising: a) administering an effective amount of a targeting moiety to the subject, wherein the targeting moiety comprises a Fab having at least one tetrazine moiety covalently linked thereto; and b) administering a single dose of a therapeutically effective amount of a payload-TCO conjugate to the subject, wherein the payload-TCO conjugate comprises a payload having at least one trans- cyclooctene moiety covalently linked thereto; wherein the single dose of the payload-TCO conjugate is administered to the subject more than 4 hours, and less than 48 hours, after the targeting moiety is administered to the subject.
- bioorthogonal binding partnerse.g., between a tetrazine of the targeting moiety and its complementary trans-cyclooctene of a payload-TCO conjugate occurs. Due to the localized administration of the targeting moiety to a desired location in the subject as described above, the selective binding between the trans-cyclooctene of the payload-TCO conjugate and a tetrazine of the targeting moiety will localize the payload to the desired target location. [0102] The methods disclosed herein allow the payload to be selectively and safely delivered, and thus decrease side effects or toxicity associated with off-target delivery.
- a single dose of the payload-TCO conjugateis required to be therapeutically effective.
- selective delivery of a payloadmay be achieved by administering a payload- TCO conjugate within a certain therapeutic window after administering a targeting agent.
- the relatively inert, systemically administered payload-TCO conjugateis activated at the target site, e.g., a tumor, by the targeting agent via a covalent click chemistry reaction, followed by chemical rearrangement to release the active payload.
- the subjecthas cancer.
- a method of treating cancer, or enhancing or eliciting an immune response, in a subject having cancercomprising: a) administering an effective amount of a targeting moiety to the subject, wherein the targeting moiety comprises a Fab having at least one tetrazine moiety covalently linked thereto; and b) administering a single dose of a therapeutically effective amount of a payload-TCO conjugate to the subject, wherein the payload-TCO conjugate comprises a payload having at least one trans- cyclooctene moiety covalently linked thereto; wherein the single dose of the payload-TCO conjugate is administered to the subject at least about 2 hours, or at least about 3 hours, or at least about 4 hours, less than about 48 hours, or between about 4 and 48 hours, after the targeting moiety is administered to the subject.
- a method of treating cancer, or enhancing or eliciting an immune response, in a subject having cancercomprising: a) administering an effective amount of a targeting moiety to the subject, wherein the targeting moiety comprises a Fab having at least one tetrazine moiety covalently linked thereto; and b) administering a single dose of a therapeutically effective amount of a payload-TCO conjugate to the subject, wherein the payload-TCO conjugate comprises a payload having at least one trans- cyclooctene moiety covalently linked thereto; wherein the single dose of the payload-TCO conjugate is administered to the subject more than 4 hours, and less than 48 hours, after the targeting moiety is administered to the subject.
- a single dose of the payload-TCO conjugateis administered to the subject 5, or 6, or 7, or 8, or 9, or 10, or 11, or 12, or 13, or 14, or 15, or 16, or 17, or 18, or 19, or 20, or 21, or 22, or 23, or less than 24, or about 24 hours after the targeting moiety is administered to the subject.
- a single dose of the payload-TCO conjugateis administered to the subject between about 5 and about 24, or between about 6 and about 24, or between about 7 and about 24, or between about 8 and about 24, or between about 9 and about 24, or between about 10 and about 24, or between about 11 and about 24, or between about 12 and about 24, or between about 13 and about 24, or between about 14 and about 24, or between about 15 and about 24, or between about 16 and about 24, or between about 17 and about 24, or between about 18 and about 24, or between about 19 and about 24, or between about 20 and about 24, or between about 21 and about 24, or between about 22 and about 24, or between about 23 and about 24 hours after the targeting moiety is administered to the subject.
- a single dose of the payload-TCO conjugateis administered to the subject between about 5 and about 22, or between about 6 and about 22, or between about 7 and about 22, or between about 8 and about 22, or between about 9 and about 22, or between about 10 and about 22, or between about 11 and about 22, or between about 12 and about 22, or between about 13 and about 22, or between about 14 and about 22, or between about 15 and about 22, or between about 16 and about 22, or between about 17 and about 22, or between about 18 and about 22, or between about 19 and about 22, or between about 20 and about 22, or between about 21 and about 22 hours after the targeting moiety is administered to the subject.
- a single dose of the payload-TCO conjugateis administered to the subject between about 5 and about 20, or between about 6 and about 20, or between about 7 and about 20, or between about 8 and about 20, or between about 9 and about 20, or between about 10 and about 20, or between about 11 and about 20, or between about 12 and about 20, or between about 13 and about 20, or between about 14 and about 20, or between about 15 and about 20, or between about 16 and about 20, or between about 17 and about 20, or between about 18 and about 20, or between about 19 and about 20 hours after the targeting moiety is administered to the subject.
- a single dose of the payload-TCO conjugateis administered to the subject between about 8 to about 12 hours, between about 8 to about 16 hours, between about 8 and about 22 hours, between about 12 to about 16 hours, or between about 16 and about 20 hours, after the targeting moiety is administered to the subject.
- the payload-TCO conjugateis a MMAE-TCO conjugate.
- a single dose of the MMAE-TCO conjugateis administered to the subject between about 8 to about 12 hours, between about 8 to about 16 hours, between about 8 and about 22 hours, between about 12 to about 16 hours, or between about 16 and about 20 hours, after the targeting moiety is administered to the subject.
- a single dose of the payload-TCO conjugateis administered to the subject between about 16 and about 20 hours after the targeting moiety is administered to the subject.
- a method of selectively administering a payload to a tumor site in a subject in need thereofcomprising: a) administering an effective amount of a targeting moiety to the subject, wherein the targeting moiety comprises a Fab having at least one tetrazine moiety covalently linked thereto; and b) administering a single dose of a therapeutically effective amount of a payload-TCO conjugate to the subject, wherein the payload-TCO conjugate comprises a payload having at least one trans- cyclooctene moiety covalently linked thereto; wherein the single dose of the payload-TCO conjugate is administered to the subject between about 8 to about 18 hours, after the targeting moiety is administered to the subject.
- the accumulation at the tumor siteis at least about 10X greater, or up to about 200X greater, than the kidney, liver, or spleen tissue.
- the targeting moietyis of Formula I, Formula II, or Formula V:
- ring Ais aryl, cycloalkyl, heterocyclyl, or heteroaryl; the dotted lines represent additional bonds to form a tetrazine when R 3 and R 4 are both absent, or a dihydroceramide when R 3 and R 4 are both present; provided that when ring A is aryl, then R 3 and R 4 are both present;
- Xis an antibody fragment moiety;
- pis 1 to 20;
- L, at each occurrence,is independently a linker;
- the targeting moietyis of Formula IIA: wherein X, p, L, and R 20 are each independently as defined herein. [0117] In some embodiments, each R 20 is independently hydrogen or alkyl. [0118] In some embodiments, the targeting moiety is of Formula VII:
- ring Ais pyrimidinyl, triazinyl, oxazolyl, isoxazole, imidazolyl, oxadiazolyl, 6,7-dihydro-5H-pyrrolo[3,4-d]pyrimidinyl, 5,6,7,8-tetrahydropyrido[4,3-d]pyrimidinyl, or 5,6,7,8-tetrahydropyrido[3,4-d]pyrimidinyl.
- ring Ais phenyl.
- each R 1is independently hydrogen or alkyl.
- each R 2is independently halo, alkyl, or haloalkyl.
- tat each occurrence, is 0.
- Xis an antibody fragment moiety which targets TNC, FN1, CLDN4, MMP9, EpCAM, ITGAV, CEA, CEACAM5, ASPH, EGFR, EPCAM, VEGFR, PDGFR, TROP2, Nectin4, PSMA, BCMA, HER2, CD25, ANTXR1, or FAP.
- Xis an antibody fragment moiety which is “derived from” an antibody.
- the term “derived from”refers to an antibody fragment of an antibody which contains the V H and V L of the antibody, such that the antibody fragment moiety, or Fab, specifically binds to the antigen.
- Xis an antibody fragment moiety derived from daclizumab, RG6292, basiliximab, HuMax-TAC, labetuzumab, 15-1-32, PR1A3, cT84.66, tusamitiamab, CC4, PAN-622, cetuximab, necitumumab, nimotuzumab, matuzumab, AMG595, depatuxizumab, dapatuxizumab, duligotuzumab, futuximab, GC1118, imgatuzumab, panitumumab, alutumumab, tomuzotuximab, laprituximab, oportuzumab, citatuzumab, tucotuzumab, catumaxomab, edrecolomab, adecatumumab, ramucizumab, ramucirumab,
- the targeting moietyfurther comprises an imaging contrast agent.
- the imaging contrast agentis a protein.
- Lat each occurrence, is independently bonded to X via a cysteine or lysine residue on X.
- each Lcomprises one or more amino acids.
- each Lcomprises a polypeptide.
- each Lindependently comprises 1 to 100 linking atoms, from 1 to 50 linking atoms, or from 5 to 50 linking atoms, or from 10 to 50 linking atoms, or from 1 to 40 linking atoms, or from 1 to 30 linking atoms, or from 1 to 20 linking atoms, or from 1 to 10 linking atoms, or from 1 to 5 linking atoms, or from 5 to 30 linking atoms, or from 10 to 30 linking atoms, or from 5 to 40 linking atoms, or from 5 to 50 linking atoms, or from 10 to 50 linking atoms.
- each Lindependently comprises from 5 to 50 linking atoms; comprising one or more chain heteroatoms and one or more alkylene, alkenylene, alkynylene, arylene, or heteroarylene, moieties; wherein each alkylene, alkenylene, alkynylene, arylene, or heteroarylene moiety, may be independently optionally substituted with one to five substituents independently selected from oxo, halo, C1-4 alkyl, C1-4 alkoxy, and C1-4 haloalkyl.
- Xis an antibody fragment moiety that targets HER2, TROP2, Nectin-4, Claudin-18.2, MMP9, mesothelin, FN1, FAP, TNC, or ECM, EPCAM, CEA, or CEACAM5; and each L is independently selected from the group consisting of: [0135] In some embodiments, X is an antibody fragment moiety that targets HER2; p is 1 to 5; and each L is independently selected from the group consisting of: [0136] In some embodiments, the targeting moiety is of Formula IIF: and p are each independently as defined herein. [0137] In some embodiments, X is an antibody fragment moiety that targets HER2; and p is 1 to 5.
- the payload-TCO conjugateis of Formula VIII, or a pharmaceutically acceptable salt thereof:
- L 1at each occurrence, is independently a linker;
- mis an integer from 1 to 150;
- Dis a payload;
- R 1Aat each occurrence, is independently selected from the group consisting of C 1-4 alkyl, C 1-4 haloalkyl, and C 1-4 alkoxy;
- qis 0, 1, or 2;
- q1is 0 or 1;
- R 1Bat each occurrence, is independently selected from the group consisting of G 1 , -OH, -NR 1c –C 1-4 alkylene–G 1 , –NR 1c –C 1-4 alkylene–N(R 1d ) 2 , -NR 1c -C 1-6 alkylene-N(C 1-4 alkyl) 3 + , -N(R 1c )CHR 1e CO 2 H, –N(R 1c )–C 1-6 alkylene
- the payloadis selected from a therapeutic agent for treating cancer (e.g., paclitaxel, doxorubicin, daunorubicin, etoposide, irinotecan, SN-38, docetaxel, paclitaxel, gemcitabine, podophyllotoxin, Carmustine, Ixabepilone, Patupilone (epothelone class), platinum drugs, exatecan, deruxtecan, auristatin (dolastatin 10, MMAE, MMAD, MMAF) mitomycin C, bleomycin, calicheamicin, staurosporine, hemiasterlin, seco-DUBA, duocarmycin, and the like), an immunosuppressant (e.g., cyclosporin A, rapamycin, and the like), an anti-fungal agent (e.g., Amphotericin, and the like), an antibiotic (e.g., vancomycin,
- the payloadis a therapeutic agent for treating cancer (e.g., paclitaxel, doxorubicin, daunorubicin, etoposide, irinotecan, SN-38, docetaxel, paclitaxel, gemcitabine, podophyllotoxin, Carmustine, Ixabepilone, Patupilone (epothelone class), platinum drugs, exatecan, deruxtecan, auristatin (dolastatin 10, MMAE, MMAD, MMAF) mitomycin C, bleomycin, calicheamicin, staurosporine, hemiasterlin, seco-DUBA, duocarmycin, and the like), or an immunosuppressant (e.g., cyclosporin A, rapamycin, and the like).
- cancere.g., paclitaxel, doxorubicin, daunorubicin, etoposide, irinotecan,
- the payloadis paclitaxel, doxorubicin, daunorubicin, etoposide, irinotecan, SN-38, docetaxel, paclitaxel, gemcitabine, podophyllotoxin, Carmustine, Ixabepilone, Patupilone, platinum drugs, exatecan, deruxtecan, dolastatin 10, MMAE, MMAD, MMAF, mitomycin C, bleomycin, calicheamicin, staurosporine, hemiasterlin, seco-DUBA, duocarmycin, cyclosporin A, or rapamycin.
- the payload-TCO conjugateis selected from:
- a method of administering a therapeutically effective amount of monomethyl auristatin E (MMAE) to a subject having cancercomprising: a) administering an effective amount of a targeting moiety to the subject, wherein the targeting moiety is a trastuzumab Fab-tetrazine targeting moiety of formula IIF: wherein X is a trastuzumab Fab comprising (SEQ ID NO.3) and (SEQ ID NO.4); and p is 1 to 5; to the subject; and b) administering to the subject a single dose of a therapeutically effective amount of a MMAE- TCO conjugate having the structure: wherein the single dose of the MMAE-TCO conjugate is administered to the subject between about 2 and 48 hours or between about 8 and about 24 hours after the targeting moiety is administered to the subject.
- a targeting moietyis a trastuzumab Fab-tetrazine targeting moiety of formula IIF: wherein X is a trastuzumab
- a method of treating cancer, or enhancing or eliciting an immune response, in a subject having cancercomprising: a) administering an effective amount of a targeting moiety to the subject, wherein the targeting moiety is a trastuzumab Fab-tetrazine targeting moiety of formula IIF: wherein X is a trastuzumab Fab comprising (SEQ ID NO.3) and (SEQ ID NO.4); and p is 1 to 5; to the subject; and b) administering to the subject a single dose of a therapeutically effective amount of a MMAE- TCO conjugate having the structure: wherein the single dose of the MMAE-TCO conjugate is administered to the subject between about 2 and about 48 hours or between about 8 and about 24 hours after the targeting moiety is administered to the subject.
- a targeting moietyis a trastuzumab Fab-tetrazine targeting moiety of formula IIF: wherein X is a trastuzumab Fab comprising (S
- a method of reducing tumor volume in a subject having a tumorcomprising: a) administering an effective amount of a targeting moiety to the subject, wherein the targeting moiety is a trastuzumab Fab-tetrazine targeting moiety of formula IIF: wherein X is a trastuzumab Fab comprising (SEQ ID NO.3) and (SEQ ID NO.4); and p is 1 to 5; to the subject; and b) administering to the subject a single dose of a therapeutically effective amount of a MMAE- TCO conjugate having the structure: wherein the single dose of the MMAE-TCO conjugate is administered to the subject between about 2 and 48 hours or between about 8 and about 24 hours after the targeting moiety is administered to the subject.
- a targeting moietyis a trastuzumab Fab-tetrazine targeting moiety of formula IIF: wherein X is a trastuzumab Fab comprising (SEQ ID NO.3) and (SEQ ID
- a single dose of the MMAE-TCO conjugateis administered to the subject between about 8 and about 24 hours after the targeting moiety is administered to the subject.
- a single dose of the MMAE-TCO conjugateis administered to the subject between about 8 to about 12 hours, between about 8 to about 16 hours, between about 8 and about 22 hours, between about 12 to about 16 hours, or between about 16 and about 20 hours, after the targeting moiety is administered to the subject.
- a single dose of the MMAE-TCO conjugateis administered to the subject between about 8 and about 22 hours, or between about 16 and about 20 hours, after the targeting moiety is administered to the subject.
- a method of administering a therapeutically effective amount of monomethyl auristatin E (MMAE) to a subject having cancercomprising: a) administering an effective amount of a targeting moiety to the subject, wherein the targeting moiety is of formula IIF: wherein X is a trastuzumab Fab comprising (SEQ ID NO.3) and (SEQ ID NO.4); or X is a sacituzumab Fab comprising (SEQ ID NO.9) and (SEQ ID NO.10); and p is 1 to 5; to the subject; and b) administering to the subject a single dose of a therapeutically effective amount of a MMAE- TCO conjugate having the structure: wherein the single dose of the MMAE-TCO conjugate is administered to the subject between about 2 and 48 hours or between about 8 and about 24 hours after the targeting moiety is administered to the subject.
- a targeting moietyis of formula IIF: wherein X is a trastuzumab Fab comprising (SEQ
- a method of treating cancer, or enhancing or eliciting an immune response, in a subject having cancercomprising: a) administering an effective amount of a targeting moiety to the subject, wherein the targeting moiety is of formula IIF: wherein X is a trastuzumab Fab comprising (SEQ ID NO.3) and (SEQ ID NO.4); or X is a sacituzumab Fab comprising (SEQ ID NO.9) and (SEQ ID NO.10); and p is 1 to 5; to the subject; and b) administering to the subject a single dose of a therapeutically effective amount of a MMAE- TCO conjugate having the structure: wherein the single dose of the MMAE-TCO conjugate is administered to the subject between about 2 and 48 hours or between about 8 and about 24 hours after the targeting moiety is administered to the subject.
- a method of reducing tumor volume in a subject having a tumorcomprising: a) administering an effective amount of a targeting moiety to the subject, wherein the targeting moiety is of formula IIF: wherein X is a trastuzumab Fab comprising (SEQ ID NO.3) and (SEQ ID NO.4); or X is a sacituzumab Fab comprising (SEQ ID NO.9) and (SEQ ID NO.10); and p is 1 to 5; to the subject; and b) administering to the subject a single dose of a therapeutically effective amount of a MMAE- TCO conjugate having the structure: wherein the single dose of the MMAE-TCO conjugate is administered to the subject between about 2 and 48 hours or between about 8 and about 24 hours after the targeting moiety is administered to the subject.
- the targeting moietyis of formula IIF: wherein X is a trastuzumab Fab comprising (SEQ ID NO.3) and (SEQ ID NO.4); or X
- a method of administering a therapeutically effective amount of monomethyl auristatin E (MMAE) to a subject having cancercomprising: a) administering an effective amount of a targeting moiety to the subject, wherein the targeting moiety is a trastuzumab Fab-tetrazine targeting moiety of Formula IIFa: wherein X is a trastuzumab Fab comprising (SEQ ID NO.3) and (SEQ ID NO.4); and p is 2-3; to the subject; and b) administering to the subject a single dose of a therapeutically effective amount of a MMAE- TCO conjugate having the structure: wherein the single dose of the MMAE-TCO conjugate is administered to the subject between about 2 and 48 hours or between about 8 and about 24 hours after the targeting moiety is administered to the subject.
- a targeting moietyis a trastuzumab Fab-tetrazine targeting moiety of Formula IIFa: wherein X is a trastuzumab
- a method of treating cancer, or enhancing or eliciting an immune response, in a subject having cancercomprising: a) administering an effective amount of a targeting moiety to the subject, wherein the targeting moiety is a trastuzumab Fab-tetrazine targeting moiety of Formula IIFa: wherein X is a trastuzumab Fab comprising (SEQ ID NO.3) and (SEQ ID NO.4); and p is 2-3; to the subject; and b) administering to the subject a single dose of a therapeutically effective amount of a MMAE- TCO conjugate having the structure: wherein the single dose of the MMAE-TCO conjugate is administered to the subject between about 2 and 48 hours or between about 8 and about 24 hours after the targeting moiety is administered to the subject.
- a targeting moietyis a trastuzumab Fab-tetrazine targeting moiety of Formula IIFa: wherein X is a trastuzumab Fab comprising (SEQ
- a method of reducing tumor volume in a subject having a tumorcomprising: a) administering an effective amount of a targeting moiety to the subject, wherein the targeting moiety is a trastuzumab Fab-tetrazine targeting moiety of Formula IIFa: wherein X is a trastuzumab Fab comprising (SEQ ID NO.3) and (SEQ ID NO.4); and p is 2-3; to the subject; and b) administering to the subject a single dose of a therapeutically effective amount of a MMAE- TCO conjugate having the structure: wherein the single dose of the MMAE-TCO conjugate is administered to the subject between about 2 and 48 hours or between about 8 and about 24 hours after the targeting moiety is administered to the subject.
- a targeting moietyis a trastuzumab Fab-tetrazine targeting moiety of Formula IIFa: wherein X is a trastuzumab Fab comprising (SEQ ID NO.3) and (SEQ ID
- a method of administering a therapeutically effective amount of monomethyl auristatin E (MMAE) to a subject having cancercomprising: a) administering an effective amount of a targeting moiety to the subject, wherein the targeting moiety is a sacituzumab Fab-tetrazine targeting moiety of formula IIF: wherein X is a sacituzumab Fab comprising (SEQ ID NO.9) and (SEQ ID NO.10); and p is 1 to 5; to the subject; and b) administering to the subject a single dose of a therapeutically effective amount of a MMAE- TCO conjugate having the structure: wherein the single dose of the MMAE-TCO conjugate is administered to the subject between about 2 and 48 hours or between about 8 and about 24 hours after the targeting moiety is administered to the subject.
- a targeting moietyis a sacituzumab Fab-tetrazine targeting moiety of formula IIF: wherein X is a sacituzumab
- a method of treating cancer, or enhancing or eliciting an immune response, in a subject having cancercomprising: a) administering an effective amount of a targeting moiety to the subject, wherein the targeting moiety is a sacituzumab Fab-tetrazine targeting moiety of formula IIF: wherein X is a sacituzumab Fab comprising (SEQ ID NO.9) and (SEQ ID NO.10); and p is 1 to 5; to the subject; and b) administering to the subject a single dose of a therapeutically effective amount of a MMAE- TCO conjugate having the structure: wherein the single dose of the MMAE-TCO conjugate is administered to the subject between about 2 and 48 hours or between about 8 and about 24 hours after the targeting moiety is administered to the subject.
- a targeting moietyis a sacituzumab Fab-tetrazine targeting moiety of formula IIF: wherein X is a sacituzumab Fab comprising (SEQ
- a method of reducing tumor volume in a subject having a tumorcomprising: a) administering an effective amount of a targeting moiety to the subject, wherein the targeting moiety is a sacituzumab Fab-tetrazine targeting moiety of formula IIF: wherein X is a sacituzumab Fab comprising (SEQ ID NO.9) and (SEQ ID NO.10); and p is 1 to 5; to the subject; and b) administering to the subject a single dose of a therapeutically effective amount of a MMAE- TCO conjugate having the structure: wherein the single dose of the MMAE-TCO conjugate is administered to the subject between about 2 and 48 hours or between about 8 and about 24 hours after the targeting moiety is administered to the subject.
- a targeting moietyis a sacituzumab Fab-tetrazine targeting moiety of formula IIF: wherein X is a sacituzumab Fab comprising (SEQ ID NO.9) and (SEQ ID
- a single dose of the MMAE-TCO conjugateis administered to the subject between about 8 and about 24 hours after the targeting moiety is administered to the subject.
- a single dose of the MMAE-TCO conjugateis administered to the subject between about 8 to about 12 hours, between about 8 to about 16 hours, between about 8 and about 22 hours, between about 12 to about 16 hours, or between about 16 and about 20 hours, after the targeting moiety is administered to the subject.
- a single dose of the MMAE-TCO conjugateis administered to the subject between about 8 and about 22 hours, or between about 16 and about 20 hours, after the targeting moiety is administered to the subject.
- the targeting moietyis administered as a single dose in an amount ranging from 10 mg/kg to about 60 mg/kg. In some embodiments, the targeting moiety is administered as a single dose in an amount ranging from 10 mg/kg to about 50 mg/kg. In some embodiments, the targeting moiety is administered as a single dose in an amount ranging from 20 mg/kg to about 60 mg/kg. In some embodiments, the targeting moiety is administered as a single dose in an amount ranging from 20 mg/kg to about 50 mg/kg. In some embodiments, the targeting moiety is administered as a single dose in an amount ranging from 30 mg/kg to about 60 mg/kg.
- the targeting moietyis administered as a single dose in an amount ranging from 30 mg/kg to about 50 mg/kg. In some embodiments, the targeting moiety is administered as a single dose in an amount ranging from 40 mg/kg to about 60 mg/kg. In some embodiments, the targeting moiety is administered as a single dose in an amount ranging from 40 mg/kg to about 50 mg/kg. [0162] In some embodiments, the targeting moiety is administered as a single dose in an amount of about 10 mg/kg, about 15 mg/kg, about 20 mg/kg, about 25 mg/kg, about 30 mg/kg, about 35 mg/kg, about 40 mg/kg, about 45 mg/kg, about 50 mg/kg, about 55 mg/kg, or about 60 mg/kg.
- the payload-TCO conjugateis administered as a single dose in an amount ranging from 10 mg/kg to about 50 mg/kg. In some embodiments, the payload-TCO conjugate is administered as a single dose in an amount ranging from 20 mg/kg to about 50 mg/kg. In some embodiments, the payload-TCO conjugate is administered as a single dose in an amount ranging from 20 mg/kg to about 40 mg/kg. In some embodiments, the payload-TCO conjugate is administered as a single dose in an amount ranging from 25 mg/kg to about 35 mg/kg.
- the payload-TCO conjugateis administered as a single dose in an amount of about 10 mg/kg, about 15 mg/kg, about 20 mg/kg, about 25 mg/kg, about 30 mg/kg, about 35 mg/kg, about 40 mg/kg, about 45 mg/kg, or about 50 mg/kg.
- the subjecthas cancer.
- the canceris metastatic.
- the canceris melanoma, renal cancer, prostate cancer, ovarian cancer, endometrial carcinoma, breast cancer, glioblastoma, lung cancer, soft tissue sarcoma, fibrosarcoma, osteosarcoma, pancreatic cancer, gastric carcinoma, squamous cell carcinoma of head/neck, anal/vulvar carcinoma, esophageal carcinoma, pancreatic adenocarcinoma, cervical carcinoma, hepatocellular carcinoma, Kaposi's sarcoma, non-Hodgkin’s lymphoma, Hodgkin’s lymphoma, Wilm’s tumor/neuroblastoma, bladder cancer, thyroid adenocarcinoma, pancreatic neuroendocrine tumors, prostatic adenocarcinoma, nasopharyngeal carcinoma, or cutaneous T-cell lymphoma.
- the canceris a melanoma, renal cancer, prostate cancer, ovarian cancer, breast cancer, glioma, lung cancer, soft tissue carcinoma, soft tissue sarcoma, osteosarcoma, or pancreatic cancer.
- the canceris a solid tumor.
- the canceris a soft tissue sarcoma.
- the soft tissue sarcomais a fibrosarcoma, rhabdomyosarcoma, or Ewing’s sarcoma.
- the methodalso comprises enhancing or eliciting an immune response.
- the immune responseis an increase in one or more of leukocytes, lymphocytes, monocytes, and eosinophils.
- the methodfurther comprising administering a therapeutically effective amount of an additional therapeutic agent selected from the group consisting of an anticancer agent, an immunomodulatory agent, or a trans-cyclooctene prodrug thereof.
- an additional therapeutic agentselected from the group consisting of an anticancer agent, an immunomodulatory agent, or a trans-cyclooctene prodrug thereof.
- Anticancer agents, immunomodulatory agents, and their trans-cyclooctene prodrugsare known in the art.
- Indications for this approachinclude cancer, both hematological and solid cancers.
- the approachcan be used for the treatment and/or diagnosis of soft tissue sarcomas: rhabdomyosarcoma, fibrosarcoma, Ewing’s sarcoma, and all the different subtypes of soft tissue sarcoma as well as osteosarcoma.
- the compositionscan be for the treatment and/or diagnosis of pigmented villonodular synovitis.
- the approachcan be used for the treatment and/or diagnosis of hematological malignancies such as myelodysplastic syndromes, acute myeloid leukemia, myelodysplastic syndromes, chronic myelogenous leukemia, chronic myelomonocytic leukemia, primary myelofibrosis, diffuse large B-cell lymphoma, chronic lymphocytic leukemia, monoclonal gammopathy, plasma cell myeloma, follicular lymphoma, marginal zone lymphoma, classical Hodgkin’s lymphoma, monoclonal B-cell lymphocytosis, lymphoproliferative disorder NOS, T-cell lymphoma, precursor B- lymphoblastic leukemia, mantle cell lymphoma, plasmacytoma, Burkitt lymphoma, T-cell leukemia, hairy-cell leukemia, precursor T-lymphoblastic leukemia, nodular lymphocyte predominant Hod
- compositions of the present disclosurefind use in treatment and/or diagnosis of a condition or disease in a subject that is amenable to treatment or diagnosis by administration of the payload (e.g., the parent drug (i.e., the drug prior to conjugation to the composition)).
- treatmentis meant that at least an amelioration of the symptoms associated with the condition afflicting the subject is achieved, where amelioration is used in a broad sense to refer to at least a reduction in the magnitude of a parameter, e.g., symptom, associated with the condition being treated.
- treatmentalso includes situations where the pathological condition, or at least symptoms associated therewith, are completely inhibited, e.g., prevented from happening, or stopped, e.g., terminated, such that the subject no longer suffers from the condition, or at least the symptoms that characterize the condition.
- Treatmentmay include inhibition, that is, arresting the development or further development of clinical symptoms, e.g., mitigating or completely inhibiting an active disease.
- Treatmentmay include relief, that is, causing the regression of clinical symptoms.
- the term “treating”includes any or all of: reducing growth of a solid tumor, inhibiting replication of cancer cells, reducing overall tumor burden, prolonged survival and ameliorating one or more symptoms associated with a cancer.
- the subject to be treatedcan be one that is in need of therapy, where the subject to be treated is one amenable to treatment using the parent drug. Accordingly, a variety of subjects may be amenable to treatment using the compositions disclosed herein. Generally, such subjects are “mammals,” with humans being of interest. Other subjects can include domestic pets (e.g., dogs and cats), livestock (e.g., cows, pigs, goats, horses, and the like), rodents (e.g., mice, guinea pigs, and rats, e.g., as in animal models of disease), as well as non-human primates (e.g., chimpanzees, and monkeys).
- domestic petse.g., dogs and cats
- livestocke.g., cows, pigs, goats, horses, and the like
- rodentse.g., mice, guinea pigs, and rats, e.g., as in animal models of disease
- non-human primatese
- additional therapeutic agents, and methodscan be used for the treatment, prevention, and/or diagnosis of solid tumors, including but not limited to, melanoma (e.g., unresectable, metastatic melanoma), renal cancer (e.g., renal cell carcinoma), prostate cancer (e.g., metastatic castration resistant prostate cancer), ovarian cancer (e.g., epithelial ovarian cancer, such as metastatic epithelial ovarian cancer), endometrial carcinoma, breast cancer (e.g., triple negative breast cancer), glioblastoma (e.g., glioblastoma multiforme), and lung cancer (e.g., non-small cell lung cancer), soft tissue sarcoma, fibrosarcoma, osteosarcoma, pancreatic cancer, gastric carcinoma, squamous cell carcinoma of head/neck, anal/vulvar carcinoma, esophageal carcinoma, pancreatic adenocarcinoma, cervical carcinoma,
- melanomae
- the disclosed approachlends itself well as an adjuvant / neoadjuvant system.
- particles as disclosed hereincould be placed during the biopsy, once the results from the study come back, the practitioner could deliver the appropriate cocktail to the desired site in the body. This would minimize the size of the tumor particularly in the context of a surgically resectable tumor.
- the surgeoncould administer additional targeting moiety to the subject to target the surgical cavity and treat the patient with further doses of treatment (e.g. chemotherapy through the disclosed approach) to minimize the risk of any cancer cells that may have been missed in the surgical margins.
- a targeting moiety as disclosed hereincould be administered and the practitioner could deliver the appropriate cocktail to the desired site in the body.
- the disclosed methodsprovide the ability to place particles as disclosed herein at the time of the biopsy. When the results return, the practitioner can deliver through to the biopsy site immunomodulatory agents.
- the disclosed methodsprovide the ability for a practitioner to deliver immunomodulatory agents, such as TLR agonists, STING agonists, chemokines (agents that attract cancerous cells and/or immune cells) and adjuvants to enhance the immune system with fewer side effects as well as the chemotherapeutics agents combined with immunotherapy agents.
- immunomodulatory agentssuch as TLR agonists, STING agonists, chemokines (agents that attract cancerous cells and/or immune cells) and adjuvants to enhance the immune system with fewer side effects as well as the chemotherapeutics agents combined with immunotherapy agents.
- This combination approachwould be beneficial to patients.
- the chemotherapy agentwould treat the solid tumor or specific location, while the enhanced response of the immunotherapy would help with distant metastatic sites.
- the disclosed compositions and methodscould employ or be used with anthracyclines, taxanes, gemcitabine and other agents to enhance the efficacy of one or more immunomodulatory agents such as ipilimumab, nivolumab, pembrolizumab, avelumab (also known as MSB0010718C; Pfizer).
- immunomodulatory agentssuch as ipilimumab, nivolumab, pembrolizumab, avelumab (also known as MSB0010718C; Pfizer).
- Cancermay be used to treat or prevent cancer, including metastatic cancer. Cancer is a group of related diseases that may include sustained proliferative signaling, evasion of growth suppressors, resistance to cell death, enablement of replicative immortality, induction of angiogenesis, and the activation of invasion and metastasis.
- Cancer that may be treated by the disclosed methodsincludes, but is not limited to, astrocytoma, adrenocortical carcinoma, appendix cancer, basal cell carcinoma, bile duct cancer, bladder cancer, bone cancer, brain cancer, brain stem cancer, brain stem glioma, breast cancer, cervical cancer, colon cancer, colorectal cancer, cutaneous T-cell lymphoma, diffuse intrinsic pontine glioma, ductal cancer, endometrial cancer, ependymoma, Ewing’s sarcoma, esophageal cancer, eye cancer, fibrosarcoma, gallbladder cancer, gastric cancer, gastrointestinal cancer, germ cell tumor, glioma, hepatocellular cancer, histiocytosis
- the cancer that may be treated by the disclosed methodsis melanoma, renal cancer, prostate cancer, ovarian cancer, breast cancer, glioma, lung cancer, soft tissue carcinoma, soft tissue sarcoma, osteosarcoma, or pancreatic cancer.
- the canceris a solid tumor.
- the canceris a soft tissue carcinoma.
- the canceris a fibrosarcoma.
- the canceris diffuse intrinsic pontine glioma.
- the canceris a metastatic cancer.
- the cancer that may be treated by the disclosed methodsis a hematological malignancy, such as myelodysplastic syndromes, acute myeloid leukemia, myelodysplastic syndromes, chronic myelogenous leukemia, chronic myelomonocytic leukemia, primary myelofibrosis, diffuse large B-cell lymphoma, chronic lymphocytic leukemia, monoclonal gammopathy, plasma cell myeloma, follicular lymphoma, marginal zone lymphoma, classical Hodgkin’s lymphoma, monoclonal B-cell lymphocytosis, lymphoproliferative disorder NOS, T-cell lymphoma, precursor B- lymphoblastic leukemia, mantle cell lymphoma, plasmacytoma, Burkitt lymphoma, T-cell leukemia, hairy-cell leukemia, precursor T-lymphoblastic leukemia, nodular lymphocyte predominant Ho
- ICDimmunogenic cell death
- Calreticulinone of the DAMP molecules, which is normally in the lumen of endoplasmic reticulum (ER), is translocated after the induction of immunogenic apoptosis to the surface of dying cell where it functions as an "eat me” signal for professional phagocytes.
- Other important surface exposed DAMPsare heat-shock proteins (HSPs), namely HSP70 and HSP90, which under stress conditions are also translocated to the plasma membrane.
- HSPsheat-shock proteins
- HSP70 and HSP90heat-shock proteins
- APCantigen-presenting cell
- HMGB1secreted amphoterin
- ATPATP
- TLRToll-like receptor
- the targeting moietycan be used for the treatment, prevention, and/or diagnosis of solid tumors, including but not limited to, melanoma (e.g.
- unresectable, metastatic melanomarenal cancer (e.g., renal cell carcinoma), prostate cancer (e.g., metastatic castration resistant prostate cancer), ovarian cancer (e.g., epithelial ovarian cancer, such as metastatic epithelial ovarian cancer), breast cancer (e.g., triple negative breast cancer), glioblastoma (e.g., glioblastoma multiforme), and lung cancer (e.g., non-small cell lung cancer), soft tissue sarcoma, fibrosarcoma, osteosarcoma, pancreatic cancer, among others.
- the disclosed approachlends itself well as an adjuvant / neoadjuvant system.
- targeting moieties as disclosed hereincould be placed during the biopsy, once the results from the study come back, the practitioner could administer the appropriate cocktail to deliver treatment to the desired site in the body (compounds as disclosed herein and optional additional therapeutic agent(s)).
- the results of the biopsymay indicate the amount and type of treatment to deliver to the site of a tumor.
- chemokinesagents that attract cancerous cells and/or immune cells
- adjuvantsto enhance the immune system with fewer side effects as well as the chemotherapeutics agents
- the disclosed methodsmay be used to deliver a functionalized payload to these location through systemic or local administration.
- the targeting moietyis delivered systemically.
- the targeting moiety and the payload-TCO conjugateare both delivered systemically.
- the disclosed compounds and compositionsmay be administered prior to surgical resection.
- the disclosed methodsmay minimize the size of the tumor prior to surgical resection. This would minimize the size of the tumor particularly in the context of a surgically resectable tumor.
- the disclosed conjugates, compounds and compositionsmay be administered during surgical resection.
- the disclosed conjugates, compounds and compositionsmay be administered after surgical resection.
- the targeting moietymay be placed around the surgical cavity at the end of surgical resection and the subject may then be treated with further doses of a treatment to minimize the risk of any cancer cells that may have been missed in the surgical margins.
- the functionalized payloads disclosed hereinmay function as adjuvants. This combination approach would be beneficial to patients.
- the chemotherapy agentwould treat the solid tumor or specific location and may enhance or elicit an immune response, while the enhanced response of the immunotherapy of the functionalized payload and/or separate agent may help with distant metastatic sites.
- the disclosed compositions and methodscould employ or be used with anthracyclines, auristatins, vinca alkaloids, taxanes, gemcitabine, camptothecin analogues and other agents to enhance the efficacy of ipilimumab, nivolumab, pembrolizumab, avelumab (also known as MSB0010718C; Pfizer).
- the disclosed methodsmay be used to treat diffuse intrinsic pontine gliomas.
- Diffuse intrinsic pontine gliomasare pediatric brainstem tumors that may be highly malignant and may be difficult to treat.
- There is no known curative treatment for DIPGand survival odds have remained dismal over the past four decades.
- DIPG patientshave a median overall survival of just 11 months, with a two-year survival rate below 10%.
- DIPGaccount for 75–80% of brainstem tumors in children, affecting an estimated 200–300 children in the U.S. each year. The rarity of this devastating disease and previous lack of experimental model systems has impeded research, and over the past four decades survival odds have remained the same.
- Diagnosis of DIPGmay begin with clinical symptoms and may be confirmed by MRI.
- the diseasemay begin with several months of generalized symptoms, including behavioral changes and difficulties in school, double vision, abnormal or limited eye movements, an asymmetric smile, loss of balance, and weakness. Alternately, severe neurologic deterioration may happen more quickly, with symptoms present for less than a month prior to diagnosis.
- Clinical examinationmay reveal the triad of multiple cranial neuropathies, long tract signs such as hyperreflexia and clonus, as well as ataxia. Expansion of the pons section of the brainstem may cause obstructive hydrocephalus and increased intracranial pressure.
- nuclei critical for life-sustaining functionsuch as breathing and heartbeat in are located in the pons and without treatment, breathing and heartbeat may be damaged by DIPG.
- the methods disclosed hereincomprise administering both the targeting moiety and the payload-TCO conjugate parenterally.
- parenterallyrefers to modes of administration which include intravenous, intramuscular, intraperitoneal, intrasternal, subcutaneous and intraarticular injection, and infusion.
- the conjugates, compounds or compositions disclosed hereinmay be dissolved or suspended in a physiologically acceptable diluent, such as water, buffer, oils with or without solubilizers, surface-active agents, dispersants or emulsifiers.
- a physiologically acceptable diluentsuch as water, buffer, oils with or without solubilizers, surface-active agents, dispersants or emulsifiers.
- Suitable oilsmay include, for example, olive oil, peanut oil, cottonseed oil, soybean oil, castor oil and sesame oil.
- the conjugates, compounds or compositions disclosed hereinmay be administered in the form of an aqueous, lipid, oily or other kind of solution or suspension, or even administered in the form of liposomes or nano-suspensions.
- the amount of composition administered to a subjectcan be initially determined based on guidance of a dose of the parent drug.
- the compositionscan provide for targeted delivery and/or enhanced serum half-life of the bound drug, thus providing for at least one of reduced dose or reduced administrations in a dosage regimen.
- the compositionscan provide for reduced dose and/or reduced administration in a dosage regimen relative to the parent drug prior to being used in the methods of the present disclosure.
- the pharmaceutical formulationmay be provided in unit dosage form. In such form the pharmaceutical formulation may be subdivided into unit doses containing appropriate quantities of the compositions of the present disclosure.
- the unit dosage formcan be a packaged preparation, the package containing discrete quantities of the preparation, such as packeted tablets, capsules, and powders in pouches, vials or ampoules.
- a kitcomprising a targeting moiety, or a pharmaceutically acceptable salt thereof, as described herein, or the pharmaceutical composition comprising the same, and instructions for use thereof.
- the kitfurther comprising a prodrug.
- Compositions of the present disclosurecan be present in any suitable amount, and can depend on various factors including, but not limited to, weight and age of the subject, state of the disease, etc.
- Suitable dosage ranges for the composition of the present disclosureinclude from 0.1 mg to 10,000 mg, or 1 mg to 1000 mg, or 10 mg to 750 mg, or 25 mg to 500 mg, or 50 mg to 250 mg.
- suitable dosages for the composition of the present disclosureinclude 1 mg, 5 mg, 10 mg, 20 mg, 30 mg, 40 mg, 50 mg, 60 mg, 70 mg, 80 mg, 90 mg, 100 mg, 150 mg, 200 mg, 250 mg, 300 mg, 350 mg, 400 mg, 450 mg, 500 mg, 550 mg, 600 mg, 650 mg, 700 mg, 750 mg, 800 mg, 850 mg, 900 mg, 950 mg, or 1000 mg.
- the compositions of the present disclosurecan be co-administered with another active agent.
- Co-administrationincludes administering the composition of the present disclosure and active agent within 0.5 hr, 1 hr, 2 hr, 4 hr, 6 hr, 8 hr, 10 hr, 12 hr, 16 hr, 20 hr, or 24 hours of each other.
- Co- administrationalso includes administering the composition of the present disclosure and active agent simultaneously or approximately simultaneously (e.g., within about 1 min, 5 min, 10 min, 15 min, 20 min, or 30 minutes of each other), or sequentially in any order.
- the additional active agentcan be administered once a day, or two, three, or more times per day so as to provide the desired dosage level per day.
- Co-administrationcan be accomplished by coimplantation or coinjection.
- co-administrationcan be accomplished by co-formulation, e.g., preparing a single pharmaceutical formulation including both the composition of the present disclosure and the active agent.
- the composition of the present disclosure and the active agentcan be formulated separately and co-administered to the subject.
- the composition of the present disclosure and the active agentcan be present in a formulation in any suitable weight ratio, such as from 1:100 to 100:1 (w/w), or 1:50 to 50:1, or 1:25 to 25:1, or 1:10 to 10:1, or 1:5 to 5:1 (w/w).
- composition of the present disclosure and the other active agentcan be present in any suitable weight ratio, such as 1:100 (w/w), 1:75, 1:50, 1:25, 1:10, 1:5, 1:4, 1:3, 1:2, 1:1, 2:1, 3:1, 4:1, 5:1, 10:1, 25:1, 50:1, 75:1, or 100:1 (w/w).
- Other dosages and dosage ratios of the composition of the present disclosure and the active agentare suitable in the formulations and methods described herein.
- Targeting Moieties[0204] Provided herein are targeting moieties which comprise an antibody fragment moiety covalently bonded to one or more tetrazine moieties.
- the targeting moieties described hereinare designed to, once administered to a subject, localize at a target site within the subject.
- the targeting moietiescan be administered locally or systemically.
- the targeting moietyis a therapeutic targeting moiety.
- a prodrug comprising a complimentary bioorthogonal componenti.e., a trans-cyclooctene moiety
- the targeting moieties described hereincomprise a diagnostic agent such that the targeting moieties described herein can be used in diagnosing conditions or diseases, with or without administering a payload or therapeutic agent.
- the antibody fragment moietyis selected from the group consisting of a single-chain variable fragment (scFv), a divalent (or bivalent) single-chain variable fragment (di-scFvs, bi-scFvs), an antigen-binding fragment (Fab), a single-domain antibody (sdAb), a single-domain antibody (sdAb), an antigen-binding protein, a DotBody, an affibody, a DARPin, a DART, a TandAb, a diabody, a ribobody, a centyrin, a knottin, an affilin, an affimer, an alphabody, an anticalin, an atrimer, an avimer, a fynomer, a kunitz domain, an obody, a pronectin, a repebody, and a bicyclic peptide or a Humabody.
- scFvsingle-chain variable fragment
- the antibody fragment moietyis selected from the group consisting of a Fab2, Fab, scFV, minibody, diabody, VHH, V-NAR, or a fragment or polypeptide which targets a tumor by virtue of formation of a targeting moiety (e.g., peptide)-antigen complex.
- the antibody fragment moietyis selected from the group consisting of a single-chain variable fragment (scFv), a divalent (or bivalent) single-chain variable fragment (di-scFvs, bi-scFvs), an antigen-binding fragment (Fab), a single-domain antibody (sdAb), and a single-domain antibody (sdAb).
- the antibody fragment moietyis an antigen-binding protein a DotBody, affibody, DARPin, DART, TandAb, diabody, ribobody, centyrin, knottin, affilin, affimer, alphabody, anticalin, atrimer, avimer, fynomer, kunitz domain, obody, pronectin, repebody, bicyclic peptide or Humabody.
- R 22is independently a linker of 1 to 100 linking atoms, and can include ethylene-oxy groups, amines, esters, amides, carbamates, carbonates, and ketone functional groups.
- the targeting moietyis of Formula IIA: wherein L, p, X, and R 20 are each independently as defined herein.
- the targeting moietyis of Formula IIB: wherein L, p, and X are each independently as defined herein.
- the targeting moietyis of Formula IIC:
- the targeting moietyis of Formula IID: wherein X and R 20 are each independently as defined herein. In some embodiments, R 20 is methyl. In some embodiments, X is an antigen-binding protein. In some embodiments, X is an antigen- binding protein which targets HER2. [0218] In some embodiments, the targeting moiety is of Formula IIE: wherein p and X are each independently as defined herein. [0219] In some embodiments, the targeting moiety is of Formula IIF: wherein p and X are each independently as defined herein.
- the targeting moietyis of Formula IIF: wherein p is 1 to 10; and X is an antibody fragment moiety comprising SEQ ID NO.9 and SEQ ID No.10. [0221] In some embodiments, p is 1 to 5.
- a) administering an effective amount of a targeting moietyis of Formula IIF: wherein p is 1 to 10, or p is 1 to 5; and X is an antibody fragment moiety comprising SEQ ID NO.9 and SEQ ID No.10 to the subject; and b) administering a single dose of a therapeutically effective amount of a payload-TCO conjugate to the subject, wherein the payload-TCO conjugate comprises a payload having at least one trans-cyclooctene moiety covalently linked thereto.
- a method of forming in vivo an antibody-payload conjugate in a subject in need thereofcomprising: administering an effective amount of a targeting moiety is of Formula IIF: wherein p is 1 to 10, or p is 1 to 5; and X is an antibody fragment moiety comprising SEQ ID NO.9 and SEQ ID No.10 to the subject; administering a single dose of a therapeutically effective amount of a payload-TCO conjugate to the subject, wherein the payload-TCO conjugate comprises a payload having at least one trans- cyclooctene (TCO) moiety covalently linked thereto; wherein the antibody or fragment thereof has a binding affinity to a receptor on a tumor, and further wherein the amount of the antibody-payload conjugate formed in vivo is greater at a tumor site versus in plasma.
- a targeting moietyis of Formula IIF: wherein p is 1 to 10, or p is 1 to 5; and X is an antibody fragment moiety comprising SEQ ID NO.9
- the ratio of the antibody-payload conjugate at a tumor site versus in plasmais greater than 1:1, or about 2:1, or about 3:1, or about 4:1, or about 5:1, or about 6:1, or about 7:1, or about 8:1, or about 9:1, or about 10:1, or about 11:1, about 12:1, or about 13:1, or about 14:1, or about 15:1, or about 16:1, or about 17:1, or about 18:1, or about 19:1, or greater than about 2:1, or greater than about 3:1, or greater than about 4:1, or greater than about 5:1, or greater than about 6:1, or greater than about 7:1, or greater than about 8:1, or greater than about 9:1, or greater than about 10:1.
- the targeting moietyis of Formula IIG: wherein p and X are each independently as defined herein.
- pis 1 to 5; and X is an antibody fragment moiety comprising SEQ ID NO.9 and SEQ ID No.10. In some embodiments, p is 2 to 3.
- Formula IIAat least one of:
- pis 1 to 12.
- Xis an antibody.
- pis 1 to 6, or 5 to 6.
- pis 1 to 16, or 1 to 8, or 1 to 7, or 1 to 6, or 1 to 5, or 1 to 4, or 1 to 3, or 1 to 2.
- Xis an antibody fragment moiety (e.g., Fab).
- a targeting moiety of Formula Vwherein: ring A is cycloalkyl, heterocyclyl, or heteroaryl; the dotted lines represent additional bonds to form a tetrazine when R 3 and R 4 are both absent, or a dihydrotetrazine when R 3 and R 4 are both present;
- Xis an antibody fragment moiety;
- pis 1 to 20;
- Lat each occurrence, is independently a linker;
- the targeting moietyis of Formula VI: wherein each of R 1 , R 2 , R 3 , R 4 , ring A, L, p, t, and X are independently as defined herein.
- R 4is hydrogen.
- R 3is a group capable of being removed after a triggering event. In some embodiments, the triggering event occurs in vivo.
- the dihydrotetrazine moietyis oxidized to provide a tetrazine as in Formula VII: wherein each of R 1 , R 2 , ring A, L, p, t, and X are independently as defined herein.
- the triggering eventis initiated after administration of the targeting moiety to the subject, and can be initiated by any means, such as internal means (e.g., via enzymatic cleavage of a functional group, optionally followed by a decomposition) or by external means (e.g., photocleavable linkers).
- R 3comprises a targeting moiety, such as an antibody fragment as described herein.
- R 3comprises an amino acid sequence specific for cleavage by a protease or esterase. [0240] In some embodiments, R 3 comprises an amino acid sequence specific for cleavage by a protease as shown in Table 1A. Table 1A
- R 3comprises an amino acid sequence specific for cleavage by a cathepsin, matrix metalloprotease (MMP), or PSMA.
- MMPmatrix metalloprotease
- R 3comprises Val-Ala, Val-Cit, Ala-Ala, Phe-Lys, Lys-Lys, Phe-Arg, or Gly-Gly-Gly for cleavage by cathepsins.
- R 3comprises Ac- ⁇ E-PLG–S(Obn)YL, or Ac-PLG–HofOrnL, where Hof is homophenylalanine and Orn is ornithine for cleavage by MMPs.
- R 3comprises an amino acid sequence as shown Table 1B. Table 1B
- R 3is photolabile.
- the photolabile groupis labile, or decomposes, with exposure to light at a wavelength matched to the absorbance profile of the photolabile group.
- R 3is L 5 is a direct bond or linker; and X 1 is -NO2, an optionally substituted sugar moiety, or an optionally substituted peptide unit comprising one or more natural or unnatural amino acids.
- at least one of the moiety:is represented by a formula selected from:
- ring A portionmay be substituted with one or more R 2 moieties.
- at least one of the moiety:is represented by a formula selected from: X 2 is alkyl (e.g., methyl) optionally substituted with a PEG, an amino acid, ester, amide, amine, -C(O)OH, -SO2, -SO3, -PO3, -PO4, or other solubility enhancing substituent; and each of L, ring A, R 1 , R 2 , t, p, and X are independently as defined herein. [0247] In some embodiments, ring A is cycloalkyl.
- ring Ais heterocyclyl. In some embodiments, ring A is heteroaryl. In some embodiments, ring A is aryl. [0248] In some embodiments, ring A is pyrimidinyl, triazinyl, oxazolyl, isoxazole, imidazolyl, oxadiazolyl, 6,7-dihydro-5H-pyrrolo[3,4-d]pyrimidinyl, 5,6,7,8-tetrahydropyrido[4,3-d]pyrimidinyl, or 5,6,7,8-tetrahydropyrido[3,4-d]pyrimidinyl. [0249] In some embodiments, ring A is phenyl. [0250] In some embodiments, at least one of the moiety: is represented by a formula selected from:
- R 1at each occurrence, is independently hydrogen, alkyl, alkenyl, alkynyl, haloalkyl, heteroalkyl, aryl, heteroaryl, heterocyclyl, or cycloalkyl; wherein each alkyl, alkenyl, alkynyl, haloalkyl, heteroalkyl, aryl, heteroaryl, heterocyclyl, or cycloalkyl is optionally substituted with one to three Z 1 .
- R 1at each occurrence, is independently hydrogen or alkyl optionally substituted with one to three Z 1 .
- R 2at each occurrence, is independently halo, cyano, nitro, hydroxy, alkyl, haloalkyl, alkenyl, alkynyl, alkoxy, haloalkoxy, heteroalkyl, aryl, heteroaryl, heterocyclyl, or cycloalkyl.
- R 2at each occurrence, is independently halo, alkyl, or haloalkyl.
- R 2at each occurrence, is independently halo or alkyl.
- t at each occurrenceis 0.
- the targeting moietyis of Formula VA:
- the targeting moietyis of Formula VB: wherein p and X are each independently as defined herein.
- ring Ais other than pyridyl. In some embodiments, ring A is other than aryl. In some embodiments, ring A is other than phenyl.
- Xis an antibody fragment moiety which targets one or more of CD25 (NCBI Gene ID 3559), CEA (NCBI Gene ID 634), CEACAM5 (NCBI Gene ID 1048), ASPH (NCBI Gene ID 444), EGFR (NCBI Gene ID 1956), EPCAM (NCBI Gene ID 4072), VEGFR (NCBI Gene ID 3791), PDGFR (NCBI Gene ID 5159), TROP2 (NCBI Gene ID 4070), Nectin4 (NCBI Gene ID 81607), PSMA (NCBI Gene ID 2346), BCMA (NCBI Gene ID 608), CD22 (NCBI Gene ID 933), CD20 (NCBI Gene ID 920), CD19 (NCBI Gene ID 930), CD79b (NCBI Gene ID 974), CD38 (NCBI Gene ID 952), CD45 (NCBI Gene ID 5788), Endoglin (NCBI Gene ID 2022), FGFR2 (NCBI Gene ID 14183), C4.4A (NCBI Gene Gene
- Xis an antibody fragment moiety which targets CEA, CEACAM5, ASPH, EGFR, EPCAM, VEGFR, PDGFR, TROP2, Nectin4, PSMA, BCMA, HER2, CD25, CLDN4 (NCBI Gene ID 1364), TNC (NCBI Gene ID 3371), FN1 (NCBI Gene ID 2335), ITGAV (NCBI Gene ID 3685), TACSTD2 (NCBI Gene ID 4070), CD174 (NCBI Gene ID 2525), GPNMB (NCBI Gene ID 10457), GPC1 (NCBI Gene ID 2817), ITGB6 (NCBI Gene ID 3694), SEZ6 (NCBI Gene ID 124925), SLITRK6 (NCBI Gene ID 84189), NaPi-2b (NCBI Gene ID 20531), ZIP6 (NCBI Gene ID 25800), ROR1 (NCBI Gene ID 4919), or ROR2 (NCBI Gene ID 4920).
- Xis an antibody fragment moiety which targets one or more of CD25 (NCBI Gene ID 3559), CEA (NCBI Gene ID 634), CEACAM5 (NCBI Gene ID 1048), ASPH (NCBI Gene ID 444), EGFR (NCBI Gene ID 1956), EPCAM (NCBI Gene ID 4072), VEGFR (NCBI Gene ID 3791), PDGFR (NCBI Gene ID 5159), TROP2 (NCBI Gene ID 4070), Nectin4 (NCBI Gene ID 81607), PSMA (NCBI Gene ID 2346), BCMA (NCBI Gene ID 608), CD22 (NCBI Gene ID 933), CD20 (NCBI Gene ID 920), CD19 (NCBI Gene ID 930), CD79b (NCBI Gene ID 974), CD38 (NCBI Gene ID 952), CD45 (NCBI Gene ID 5788), Endoglin (NCBI Gene ID 2022), FGFR2 (NCBI Gene ID 14183), C4.4A (NCBI Gene ID 27076), Claudin
- Xis an antibody fragment moiety which targets CEA, CEACAM5, ASPH, EGFR, EPCAM, VEGFR, PDGFR, TROP2, Nectin4, PSMA, BCMA, HER2, CD25, ANTXR1, or FAP.
- Xis an antibody fragment moiety that targets HER2, TROP2, Nectin-4, Claudin-18.2, MMP9, mesothelin, FN1, FAP, TNC, or ECM, EPCAM, CEA, or CEACAM5.
- Xis an antibody fragment moiety which targets CEA, CEACAM5, ASPH, EGFR, EPCAM, VEGFR, PDGFR, TROP2, Nectin4, PSMA, BCMA, HER2, or CD25.
- Xis an antibody fragment moiety, that targets CD25, such as daclizumab, RG6292, basiliximab, or HuMax-TAC, or an antibody fragment moiety derived therefrom.
- Xis an antibody fragment moiety, that targets CEA, such as labetuzumab, 15-1-32, PR1A3, or cT84.66, or an antibody fragment moiety derived therefrom.
- Xis an antibody fragment moiety, that targets CEACAM5, such as Tusamitiamab or CC4, or an antibody fragment moiety derived therefrom.
- Xis an antibody fragment moiety, that targets ASPH, such as PAN-622, or an antibody fragment moiety derived therefrom.
- Xis an antibody fragment moiety, that targets EGFR, such as cetuximab, necitumumab, nimotuzumab, matuzumab, AMG595, depatuxizumab, dapatuxizumab, duligotuzumab, futuximab, GC1118, imgatuzumab, panitumumab, alutumumab, tomuzotuximab, or laprituximab, or an antibody fragment moiety derived therefrom.
- EGFRsuch as cetuximab, necitumumab, nimotuzumab, matuzumab, AMG595, depatuxizumab, dapatuxizumab, duligotuzumab, futuximab, GC1118, imgatuzumab, panitumumab, alutumumab, tomuzotuximab, or laprituximab, or
- Xis an antibody fragment moiety, that targets EPCAM, such as oportuzumab, citatuzumab, tucotuzumab, catumaxomab, edrecolomab, or adecatumumab, or an antibody fragment moiety derived therefrom.
- EPCAMsuch as oportuzumab, citatuzumab, tucotuzumab, catumaxomab, edrecolomab, or adecatumumab, or an antibody fragment moiety derived therefrom.
- Xis an antibody fragment moiety, that targets VEGFR, such as ramucizumab, ramucirumab, or vulinacimab, or an antibody fragment moiety derived therefrom.
- Xis an antibody fragment moiety, that targets PDGFR, such as olaratumab or ramucirumab, or an antibody fragment moiety derived therefrom.
- PDGFRsuch as olaratumab or ramucirumab
- Xis an antibody fragment moiety, that targets TROP2, such as Sacituzumab, datopotamab, or Pr1E11, or an antibody fragment moiety derived therefrom.
- Xis an antibody fragment moiety, that targets Nectin4, such as enfortumab, 15A7.5_H1L3, hNec.4.05, 14A5.2, 42D20-Hz3, 42D20-Hz10, HZD6.1C, HZD6.2C, or 74HZ or an antibody fragment moiety derived therefrom.
- Xis an antibody fragment moiety, that targets PSMA, such as J591 or MLN591, or an antibody fragment moiety derived therefrom.
- Xis an antibody fragment moiety, that targets BCMA, such as Belantamab, or an antibody fragment moiety derived therefrom.
- Xis an antibody fragment moiety, that targets CD22, such as moxetumomab, inotuzumab, epratuzumab, or pinatuzumab, or an antibody fragment moiety derived therefrom.
- CD22such as moxetumomab, inotuzumab, epratuzumab, or pinatuzumab, or an antibody fragment moiety derived therefrom.
- Xis an antibody fragment moiety, that targets CD20, such as ublituximab, ofatumumab, rituximab, obinutuzumab, tositumomab, or ibritumomab, or an antibody fragment moiety derived therefrom.
- Xis an antibody fragment moiety, that targets CD19, such as loncastuximab, XMAB-5574, MOR208, coltuximab, denintuzumab, taplitumomab, or MDX-1342, or an antibody fragment moiety derived therefrom.
- CD19such as loncastuximab, XMAB-5574, MOR208, coltuximab, denintuzumab, taplitumomab, or MDX-1342, or an antibody fragment moiety derived therefrom.
- CD79bsuch as polatuzumab
- Xis an antibody fragment moiety, that targets CD38, such as isatuximab, daratumumab, MOR202, or TAK-079, or an antibody fragment moiety derived therefrom.
- Xis an antibody fragment moiety, that targets CD45, such as I-131- BC8, or Iomab-B, or an antibody fragment moiety derived therefrom.
- Xis an antibody fragment moiety, that targets endoglin, such as carotuximab, or an antibody fragment moiety derived therefrom.
- Xis an antibody fragment moiety, that targets FGFR2, such as bemarituzumab or aprutumab, or an antibody fragment moiety derived therefrom.
- Xis an antibody fragment moiety, that targets C4.4A, such as lupartumab, or an antibody fragment moiety derived therefrom.
- Xis an antibody fragment moiety, that targets Claudin-18.2, such as zolbetuximab, or claudiximab, or an antibody fragment moiety derived therefrom.
- Xis an antibody fragment moiety, that targets MMP9, such as andecaliximab, or an antibody fragment moiety derived therefrom.
- Xis an antibody fragment moiety, that targets folate receptor, such as mirvetuximab, farletuzumab, MORAb-202, MORAb-003, or SP8166, or an antibody fragment moiety derived therefrom.
- folate receptorsuch as mirvetuximab, farletuzumab, MORAb-202, MORAb-003, or SP8166, or an antibody fragment moiety derived therefrom.
- Xis an antibody fragment moiety, that targets DLL3, such as rovalpituzumab, or an antibody fragment moiety derived therefrom.
- Xis an antibody fragment moiety, that targets CD138, such as indatuximab, or an antibody fragment moiety derived therefrom.
- Xis an antibody fragment moiety, that targets CD56, such as lorvotuzumab, promiximab, or an antibody fragment moiety derived therefrom.
- Xis an antibody fragment moiety, that targets CD37, such as BI 836826, otlertuzumab, or naratuximab, or an antibody fragment moiety derived therefrom.
- Xis an antibody fragment moiety, that targets CD74, such as milatuzumab, or an antibody fragment moiety derived therefrom.
- Xis an antibody fragment moiety, that targets mesothelin, such as anetumab, amatuximab, or MMOT-0530A, or an antibody fragment moiety derived therefrom.
- mesothelinsuch as anetumab, amatuximab, or MMOT-0530A
- Xis an antibody fragment moiety, that targets IL-6R, such as tocilizumab or sarilumab, or an antibody fragment moiety derived therefrom.
- Xis an antibody fragment moiety, that targets SLAMF7, such as elotuzumab, or an antibody fragment moiety derived therefrom.
- Xis an antibody fragment moiety, that targets BAFF, such as belimumab, or an antibody fragment moiety derived therefrom.
- Xis an antibody fragment moiety, that targets MUC1, such as KL-6, MY.1E12, hMUC1-1H7, TAB004, huC242, clivatuzumab, 8HuDS6, gatipotuzumab, AR20.5, or cantuzumab, or an antibody fragment moiety derived therefrom.
- Xis an antibody fragment moiety, that targets GPC3, such as codrituzumab, ECT204, or MDX-1414, or an antibody fragment moiety derived therefrom.
- GPC3such as codrituzumab, ECT204, or MDX-1414, or an antibody fragment moiety derived therefrom.
- Xis an antibody fragment moiety, that targets HER2, such as pertuzumab, trastuzumab, or margetuximab, or an antibody fragment moiety derived therefrom.
- Xis an antibody fragment moiety, that targets HER3, such as patritumab, seribantumab, lumretuzumab, elgemtumab, AV-203, CDX-3379, or GSK284933, or an antibody fragment moiety derived therefrom.
- HER3such as patritumab, seribantumab, lumretuzumab, elgemtumab, AV-203, CDX-3379, or GSK284933
- Xis an antibody fragment moiety, that targets CD30, such as brentuximab, or an antibody fragment moiety derived therefrom.
- Xis an antibody fragment moiety, that targets CD33, such as gemtuzumab, BI 835858, vadastuximab, or lintuzumab, or an antibody fragment moiety derived therefrom.
- CD33such as gemtuzumab, BI 835858, vadastuximab, or lintuzumab, or an antibody fragment moiety derived therefrom.
- Xis an antibody fragment moiety, that targets CD123, such as KHK2823, taclotuzumab, or G4723A, or an antibody fragment moiety derived therefrom.
- Xis an antibody fragment moiety, that targets GPNMB, such as glembatumumab, or an antibody fragment moiety derived therefrom.
- Xis an antibody fragment moiety, that targets cMET, such as telisotuzumab, onartuzumab, or SAIT301, or an antibody fragment moiety derived therefrom.
- cMETsuch as telisotuzumab, onartuzumab, or SAIT301
- Xis an antibody fragment moiety, that targets CD142, such as tisotumab, or an antibody fragment moiety derived therefrom.
- Xis an antibody fragment moiety, that targets NaPi2B, such as lifastuzumab, or an antibody fragment moiety derived therefrom.
- Xis an antibody fragment moiety, that targets GCC, such as indusatumab, or an antibody fragment moiety derived therefrom. [0310] In certain embodiments, X is an antibody fragment moiety, that targets STEAP1, such as vandortuzumab, or an antibody fragment moiety derived therefrom. [0311] In certain embodiments, X is an antibody fragment moiety, that targets MUC16, such as sofituzumab, or an antibody fragment moiety derived therefrom. [0312] In certain embodiments, X is an antibody fragment moiety, that targets CD70, such as vorsetuzumab, or an antibody fragment moiety derived therefrom.
- Xis an antibody fragment moiety, that targets CD44, such as bivatuzumab, or an antibody fragment moiety derived therefrom.
- Xis an antibody fragment moiety, that targets vWF, such as caplacizumab, or an antibody fragment moiety derived therefrom.
- Xis an antibody fragment moiety, that targets TNF, such as ozoralizumab, V565, or PF-05230905, or an antibody fragment moiety derived therefrom.
- Xis an antibody fragment moiety, that targets IL-6R, such as vobarilizumab, or an antibody fragment moiety derived therefrom.
- Xis an antibody fragment moiety, that targets BCMA, such as LCAR- B38M, or an antibody fragment moiety derived therefrom. [0318] In certain embodiments, X is an antibody fragment moiety, that targets ADAMTS5, such as M6495, or an antibody fragment moiety derived therefrom. [0319] In certain embodiments, X is an antibody fragment moiety, that targets CX3CR1, such as BI 655088, or an antibody fragment moiety derived therefrom. [0320] In certain embodiments, X is an antibody fragment moiety, that targets CXCR4, such as AD-214 or ALX-0651, or an antibody fragment moiety derived therefrom.
- Xis an antibody fragment moiety, that targets TfR1, such as TXB4, or an antibody fragment moiety derived therefrom.
- Xis an antibody fragment moiety, that targets VEGFR, such as CDP791, or an antibody fragment moiety derived therefrom.
- Xis an antibody fragment moiety, that targets PSMA, such as GY1, or an antibody fragment moiety derived therefrom.
- Xis an antibody fragment moiety, that targets FN1, such as L19 or NJB2, or an antibody fragment moiety derived therefrom.
- Xis an or antibody fragment moiety, that targets FAP, such as F19, OMTX005 or sibrotuzumab, or an antibody fragment moiety derived therefrom.
- FAPsuch as F19, OMTX005 or sibrotuzumab
- Xis an antibody fragment moiety, that targets TNC, such as F16 or R6N or an antibody fragment moiety derived therefrom.
- the antibody fragment moietyis derived from (e.g., is a Fab which comprises at least the V H and V L ) daclizumab, RG6292, basiliximab, HuMax-TAC, labetuzumab, 15-1- 32, PR1A3, cT84.66, tusamitiamab, CC4, PAN-622, cetuximab, necitumumab, nimotuzumab, matuzumab, AMG595, depatuxizumab, dapatuxizumab, duligotuzumab, Futuximab, GC1118, imgatuzumab, panitumumab, alutumumab, tomuzotuximab, laprituximab, oportuzumab, citatuzumab, tucotuzumab, catumaxomab, edrecolomab,
- Xis an antibody selected from atezolizumab, avelumab, bevacizumab, cemiplimab, cetuximab, daratumumab, dinutuximab, durvalumab, elotuzumab, ipilimumab, isatuximab, mogamulizumab, necitumumab, nivolumab, obinutuzumab, ofatumumab, olaratumab, panitumumab, pembrolizumab, pertuzumab, ramucirumab, rituximab, and trastuzumab.
- X or the antibody fragment moietyis an antigen-binding fragment (Fab).
- the Fabis a region on an antibody that binds to antigens, and is comprised of one constant and one variable domain of each of the heavy and the light chain.
- the Fabcomprises four domains: VH, CH1, VL and CL1.
- the Fabcomprises 400-500 amino acids, or 440-480 amino acids.
- the Fabhas a molecular weight of about 50 kDa, or 40-55 kDa, or 45-50 kDa, or 45-55 kDa.
- the antibody fragment moietycomprises one or more PEG units, which may enhance circulation life.
- the antibody fragment moietyis an antigen-binding protein. Antigen- binding proteins are proteins which are designed to be antibody-mimetics, exhibiting a high affinity and specificity for a given target.
- the antigen-binding proteinis a single-chain antigen-binding proteins are novel recombinant polypeptides, composed of an antibody variable light- chain amino acid sequence (VL) tethered to a variable heavy-chain sequence (VH) by a designed peptide that links the carboxyl terminus of the VL sequence to the amino terminus of the VH sequence.
- VLvariable light- chain amino acid sequence
- VHvariable heavy-chain sequence
- the antigen-binding proteinis about 5-10 kDa, or about 7 kDa. In some embodiments, the antigen-binding protein is about are about 50-80, or 60-70, or 66 amino acids in length.
- the antigen-binding proteincomprises a cysteine only at the N- or C-terminus. In some embodiments, the antigen-binding protein comprises a cysteine only at the N-terminus. In some embodiments, the antigen-binding protein comprises a cysteine only at the C-terminus.
- the antibody fragment moietyis an antigen-binding protein that targets TNC, FN1, CLDN4, MMP9, EpCAM, ITGAV, CEA, CEACAM5, ASPH, EGFR, EPCAM, VEGFR, PDGFR, TROP2, Nectin4, PSMA, BCMA, HER2, or CD25.
- the antibody fragment moietyis an antigen-binding protein that targets HER2.
- Antigen-binding proteinscan be prepared and tested according to standard methods or purchased from commercial sources (e.g., Affilogic).
- the antibody fragment moietyis derived from daclizumab, RG6292, basiliximab, HuMax-TAC, labetuzumab, 15-1-32, PR1A3, cT84.66, tusamitiamab, CC4, PAN-622, cetuximab, necitumumab, nimotuzumab, matuzumab, AMG595, depatuxizumab, dapatuxizumab, duligotuzumab, futuximab, gc1118, imgatuzumab, panitumumab, alutumumab, tomuzotuximab, laprituximab, oportuzumab, citat
- Xis an antibody fragment moiety derived from atezolizumab, avelumab, bevacizumab, cemiplimab, cetuximab, daratumumab, dinutuximab, durvalumab, elotuzumab, ipilimumab, isatuximab, mogamulizumab, necitumumab, nivolumab, obinutuzumab, ofatumumab, olaratumab, panitumumab, pembrolizumab, pertuzumab, ramucirumab, rituximab, or trastuzumab.
- Xis an antibody fragment moiety, that targets vWF, such as Caplacizumab.
- Xis an antibody fragment moiety, that targets TNF, such as Ozoralizumab, V565, or PF-05230905.
- Xis an antibody fragment moiety, that targets IL-6R, such as Vobarilizumab.
- Xis an antibody fragment moiety, that targets BCMA, such as LCAR- B38M.
- Xis an antibody fragment moiety, that targets ADAMTS5, such as M6495.
- Xis an antibody fragment moiety, that targets CX3CR1, such as BI 655088. [0343] In certain embodiments, X is an antibody fragment moiety, that targets CXCR4, such as AD-214 or ALX-0651. [0344] In certain embodiments, X is an antibody fragment moiety, that targets TfR1, such as TXB4. [0345] In certain embodiments, X is an antibody fragment moiety, that targets VEGFR, such as CDP791. [0346] In certain embodiments, X is an antibody fragment moiety, that targets PSMA, such as GY1.
- the antibody fragment moietyis caplacizumab, ozoralizumab, V565, PF- 05230905, vobarilizumab, LCAR-B38M, M6495, BI 655088, AD-214, ALX-0651, TXB4, CDP791, or GY1.
- Xfurther comprises an imaging contrast agent.
- the imaging contrast agentis a protein.
- Linker Moieties[0349] In some embodiments, L is bonded to X via a cystine or lysine residue on X. [0350] In some embodiments, L is a non-cleavable linker.
- Lis a cleavable linker.
- Lcomprises one or more amino acids.
- Lcomprises a polypeptide.
- Lcomprises one or more of a hydrazone, a hydrazide, a disulfide, a N- succinimidyl-4-(2-pyridyldithio)pentanoate (SPP), a N-succinimidyl-4-(2-pyridyldithio)butyrate (SPDB), a 4-(4’-acetylphenoxy)butanoic acid (AcBut), one or more linear or branched, natural or unnatural amino acid, a valine-citrulline (Val-Cit) moiety, or a phenylalanine-lysine (Phe-Lys) moiety.
- SPPN- succinimidyl-4-(2-pyridyldithio)pentanoate
- SPDBN-succinimidyl-4-(
- Lcomprises 1 to 100 linking atoms, from 1 to 50 linking atoms, or from 5 to 50 linking atoms, or from 10 to 50 linking atoms, or from 1 to 40 linking atoms, or from 1 to 30 linking atoms, or from 1 to 20 linking atoms, or from 1 to 10 linking atoms, or from 1 to 5 linking atoms, or from 5 to 30 linking atoms, or from 10 to 30 linking atoms, or from 5 to 40 linking atoms, or from 5 to 50 linking atoms, or from 10 to 50 linking atoms.
- Lcomprises one or more chain heteroatoms and one or more alkylene, alkenylene, alkynylene, arylene, heteroarylene, cycloalkylene or heterocycloalkylene moieties; wherein each alkylene, alkenylene, alkynylene, arylene, heteroarylene, cycloalkylene or heterocycloalkylene moiety, may be independently optionally substituted with one to five substituents independently selected from oxo, halo, C1-4 alkyl, C1-4 alkoxy, and C1-4 haloalkyl.
- Lis an alkylene linker optionally comprising one or more -O-, -S-, amine, ester, amide, carbamate, carbonate, thio-succinimide, or ketone functional groups.
- each R 110is independently hydrogen, C 1-4 alkyl, C 1-4 haloalkyl, aryl, heteroaryl, cycloalkyl or heterocyclyl; and each R 120 is independently hydrogen, C 1-4 alkyl, C 1-4 haloalkyl, aryl, heteroaryl, cycloalkyl or heterocyclyl.
- the linkeris not a bond.
- the linker Lmay comprise one or more of polyethylene glycol (e.g., PEG having an average molecular weight of from 300 g/mol to 10,000 g/mol), ethylene-1,2-diylbis(methylcarbamate, an arylene (e.g., phenylene), ethylene-oxy, amine, ester, amide, carbamate, ketone (i.e., formyl), or carbonate.
- the linkercomprises one or more of: [0363] In some embodiments, the linker comprises one or more of: [0364] In some embodiments, the linker comprises one or more of: [0365] In some embodiments, the linker comprises one or more .
- the linkercomprises one or more . [0367] In some embodiments, the linker is, or comprises one or more: [0368] In some embodiments, the linker is, or comprises one or more: [0369] In some embodiments, the linker comprises one or more natural or unnatural amino acids, which may be referred to as a peptide linker.
- the linkermay be a peptide linker made up of a carboxylic acyl unit, and one or more amino acids making up a protein or peptide sequence.
- the linkermay also contain a self-immolating spacer which spaces the drug and the protein peptide sequence.
- the linkermay be a peptide containing linker represented by “A—Y— Z—X 2 —W” in which “A” is the carboxylic acyl unit, “Y” and “Z” are each one or more natural or unnatural amino acids and together form a peptide sequence, and “X 2 ” and “W” are optional additional linkers having from 1 to 50 linking atoms, or from 5 to 10 linking atoms, or from 1 to 10 linking atoms which spaces the peptide and the payload, D, or the bioorthogonal moiety.
- one or more of the amino acids in the peptide linkeris N-methylated.
- Ymay be at least one amino acid selected from the group consisting of alanine, valine, leucine, isoleucine, methionine, phenylalanine, tryptophan, and proline. In some embodiments Y may be at least one amino acid selected from the group consisting of phenylalanine, alanine, and valine.
- Zmay be at least one amino acid selected from the group consisting of alanine, lysine, lysine protected with acetyl or formyl, arginine, arginine protected with tosyl or nitro groups, histidine, ornithine, ornithine protected with acetyl or formyl, and citrulline.
- Zmay be at least one amino acid selected from the group consisting of alanine, lysine, and citrulline.
- Exemplary Y-Z combinationsinclude Valine-Citrulline; Valine-Alanine; and Alanine-Alanine.
- Ais -OC(O)-.
- X 2is -OC(O)-.
- Wis -OC(O)-.
- X 2is absent and W is -OC(O)-. 2
- the moiety —X —Wcomprises .
- the moiety —Xis .
- —X—[0379]
- —X—[0380]
- the peptide linkeris specifically tailored so that it will be selectively cleaved (e.g., enzymatically cleaved) releasing the drug, such as by one or more of the tumor-associated proteases.
- the peptide linkerhas a chain length of two to four amino acid residues (i.e., a di-, tri-, or tetra-peptide). It will be understood, however, that peptide linkers up to five, six, seven, or eight amino acid residues may also suitably be employed.
- the peptide linkeris Phe-Lys, Val-Lys, Val-Ala, Ala-Ala, Phe-Phe-Lys, D-Phe-Phe-Lys, Gly-Phe-Lys, Ala-Lys, Val-Cit, Phe-Cit, Leu-Cit, Ile-Cit, Trp-Cit, Phe-Ala, Gly-Phe- Leu-Gly [SEQ ID NO: 1], Ala-Leu-Ala-Leu [SEQ ID NO: 2], Phe-N 9 -tosyl-Arg, or Phe-N 9 -Nitro-Arg.
- the peptide linkeris Phe-Lys, Val-Lys, Val-Ala, Ala-Ala, Val-Val, Val-Cit, or D-Phe-L-Phe-Lys. In certain embodiments, the peptide linker is Val-Cit, Val-Ala, or Ala-Ala. [0383] In some embodiments, the linker L is, or comprises one or more of:
- the linker Lcomprises one or more of:
- the linker Lcomprises one or more of: .
- the foregoing linkersmay bond to an amino acid side chain present on X, such as a lysine or [0387]
- the linker Lis –C(O)L 4 – or –C(O)C1-6alkyleneC(O)L 4 –;
- L 4is a bond, –N(R 12 )–C 2-3 alkylene–N(R 13 )C(O)–, -CH(NHC(O)R 14 )C 1-4 alkylene–S–S–C 1-4 alkylene– OC(O)–, —NHNHC(O)CH(NHC(O)R 15 )CH2C(O)–, –C1-6alkylene–CH(G x )OC(O)–,
- R 12 , R 13 , R 14 , R 15 , and R 19are each independently hydrogen or C 1-4 alkyl;
- R 16is hydrogen, C
- the linker Lcomprises a carbonyl moiety for conjugating the tetrazine moiety to the linker or X.
- the linkermay comprise a polypeptide moiety (PPM) having the lysine residue and lysine side chain and the PPM may also have additional lysines, or other amino acid side chains conjugated to the carbonyl moiety.
- PPMpolypeptide moiety
- the linker Lmay comprise [0389] In some embodiments, the linker L is, or comprises one or more of:
- the linker Lis, or comprises one or more of: . [0391] In some embodiments, the linker L is, or comprises one or more of: [0392] In some embodiments, the linker L is, or comprises one or more of:
- the linker Lis, or comprises one or more of: [0394] In some embodiments, the linker L is, or comprises one or more of: . [0395] In some embodiments, the linker L is, or comprises one or more of: [0396] In some embodiments, the linker L is, or comprises one or more of: [0397] In some embodiments, the linker L is, or comprises one or more of: [0398] In some embodiments, the linker L is, or comprises one or more of: [ [0400] In some embodiments, the linker L is, or comprises one or more of: [0401] In some embodiments, the linker L is, or comprises one or more of: [0402] In some embodiments, the linker L is, or comprises one or more of: [0403] In some embodiments, the linker L is, or comprises one or more of: .
- ring Ais phenyl.
- Xis an antibody fragment moiety, that targets HER2, TROP2, Nectin-4, FN1, FAP, TNC, or ECM.
- Xis zolbetuximab, claudiximab, andecaliximab, anetumab, amatuximab, MMOT-0530A, L19, NJB2, F19, OMTX005, sibrotuzumab, F16, or R6N, trastuzumab, enfortumab, or sacituzumab, datopotamab, or an antibody fragment moiety derived therefrom.
- Xis an antibody fragment moiety derived L19, NJB2, F19, OMTX005, sibrotuzumab, F16, or R6N, trastuzumab, enfortumab, or sacituzumab.
- pis 1 to 12. In some embodiments, p is 1 to 6, or 5 to 6.
- pis 1 to 10, or 1 to 9, or 1 to 8, or 1 to 7, or 1 to 6, or 1 to 5, or 1 to 4, or 1 to 3, or 1 to 2, or 2 to 10, or 2 to 9, or 2 to 8, or 2 to 7, or 2 to 6, or 2 to 5, or 2 to 4, or 2 to 3, or 3 to 10, or 3 to 9, or 3 to 8, or 3 to 7, or 3 to 6, or 3 to 5, or 3 to 4, or 4 to 10, or 4 to 9, or 4 to 8, or 4 to 7, or 4 to 6, or 4 to 5, or 5 to 10, or 5 to 9, or 5 to 8, or 5 to 7, or 5 to 6, or 6 to 10, or 6 to 9, or 6 to 8, or 6 to 7, or 7 to 10, or 7-9, or 7 to 8, or 8 to 10, or 8 to 9, or 9 to 10.
- pis 1 to 16, or 1 to 8, or 1 to 7, or 1 to 6, or 1 to 5, or 1 to 4, or 1 to 3, or 1 to 2.
- pis 1 to 10, or 1 to 9, or 1 to 8, or 1 to 7, or 1 to 6, or 1 to 5, or 1 to 4, or 1 to 3, or 1 to 2, or 2 to 10, or 2 to 9, or 2 to 8, or 2 to 7, or 2 to 6, or 2 to 5, or 2 to 4, or 2 to 3, or 3 to 10, or 3 to 9, or 3 to 8, or 3 to 7, or 3 to 6, or 3 to 5, or 3 to 4, or 4 to 10, or 4 to 9, or 4 to 8, or 4 to 7, or 4 to 6, or 4 to 5, or 5 to 10, or 5 to 9, or 5 to 8, or 5 to 7, or 5 to 6, or 6 to 10, or 6 to 9, or 6 to 8, or 6 to 7, or 7 to 10, or 7 to 9, or 7 to 8, or 8 to 10, or 8 to 9, or 9 to 10, and X is an antibody fragment moiety of from 15 kDa to 55 kD
- pis dependent on the size and/or number of available binding sites on X for forming a covalent bond to L.
- pis 1 to 4.
- pis 1 to 4.
- Xis an antibody fragment moiety between 45-55 kDa
- pis 1 to 4.
- Xis an antibody fragment moiety of less than 45 kDa
- pis 1 to 3, or 2 to 3, or 1 to 2, or about 1, about 2, or about 3.
- when X is an antibody fragment moiety of less than 25 kDap is 1 to 3, or 2 to 3, or 1 to 2, or about 1, about 2, or about 3.
- Cis 1 to 3, or 2 to 3, or 1 to 2, or about 1, about 2, or about 3.
- Payload-TCO Conjugates[0417] Trans-cyclooctene functionalized prodrugs (payload-TCO conjugates) are known in the art, including prodrugs of anticancer agents, as described in WO2018/187740, WO2014/205126, WO2015/139025, and WO2017/044983, which are incorporated herein by reference, and/or a chelating agent, with or without a therapeutic or diagnostic radioligand. Further exemplary embodiments follow. [0418] In some embodiments, the payload-TCO conjugate is a conjugate comprised of a payload linked to one or more trans-cyclooctene moieties.
- the payload-TCO conjugate(or trans-cyclooctene functionalized prodrug) comprises an immunomodulatory agent payload, such as for example, an immunomodulatory agent payload selected from the group consisting of a cytokine, chemokine, chemokine antagonist, therapeutic monoclonal antibody, and immune checkpoint inhibitor payload; or a pharmaceutically acceptable salt thereof.
- an immunomodulatory agent payloadselected from the group consisting of a cytokine, chemokine, chemokine antagonist, therapeutic monoclonal antibody, and immune checkpoint inhibitor payload; or a pharmaceutically acceptable salt thereof.
- the inhibitor of a cytokine payloadis an inhibitor of TNF- ⁇ , infliximab, certolizumab, TGF- ⁇ , galunisertib, fresolimumab, M7824, CSF-1, pexidartinib, or cabiralizumab.
- the payload-TCO conjugatecomprises a monoclonal antibody, or a pharmaceutically acceptable salt thereof.
- the payload-TCO conjugatecomprises a therapeutic protein payload, or a pharmaceutically acceptable salt thereof.
- the therapeutic protein payloadis an antibody-based drug, Fc fusion protein, anticoagulant, blood factor, bone morphogenetic protein, engineered protein scaffold, enzyme, growth factor, hormone, interferon, interleukin, or thrombolytic.
- the therapeutic protein payloadis a cytokine, chemokine, growth factor, hormone, antibody, or antigen.
- the therapeutic protein payloadis a payload of erythropoietin (EPO, e.g., native EPO or synthetic EPO (see, e.g., US 2003/0191291), such as, but not limited to, e.g., PROCRIT®, EPREX®, or EPOGEN® (epoetin- ⁇ ), ARANESP® (darbepoietin- ⁇ ), NEORECORMON®, EPOGIN® (epoetin- ⁇ ), and the like); a growth hormone (e.g., a somatotropin, e.g., GENOTROPIN®, NUTROPIN®, NORDITROPIN®, SAIZEN®, SEROSTIM®, HUMATROPE®, etc.); theraputic monoclonal antibody (e.g Atezolizumab, Avelumab, Bevacizumab, Cemiplimab, Cetuximab
- EPOerythrop
- the payload-TCO conjugateis of Formula VIII, or a pharmaceutically acceptable salt thereof: wherein m is an integer from 1 to 150; G, at each occurrence, is independently an optionally substituted trans-cyclooctene moiety; D is a payload; L 1 , at each occurrence, is independently a linker.
- each trans-cyclooctene moietyis independently: wherein: R 1A , at each occurrence, is independently selected from the group consisting of C 1-4 alkyl, C 1-4 haloalkyl, and C 1-4 alkoxy; q is 0, 1, or 2; q1 is 0 or 1; R 1B , at each occurrence, is independently selected from the group consisting of G 1 , -OH, –NR 1c –C 1-4 alkylene–G 1 , –NR 1c –C 1-4 alkylene–N(R 1d ) 2 , –NR 1c –C 1-6 alkylene–N(C 1-4 alkyl) 3 + , –N(R 1c )CHR 1e CO 2 H, –N(R 1c )–C 1-6 alkylene–CO 2 H, –N(R 1f )–C 2-4 alkylene–(N(C )
- the payload-TCO conjugateis of Formula VIII, or a pharmaceutically acceptable salt thereof, wherein G is the trans-cyclooctene moiety, and G, at each occurrence, is independently L 1 , at each occurrence, is independently a linker; m is an integer from 1 to 150; D is a payload; R 1A , at each occurrence, is independently selected from the group consisting of C1-4alkyl, C1-4haloalkyl, and C1-4alkoxy; q is 0, 1 or 2; q1 is 0 or 1; R 1B , at each occurrence, is independently selected from the group consisting of G 1 , OH, –NR 1c –C 1-4 alkylene–G 1 , –NR 1c –C 1-4 alkylene–N(R 1d ) 2 , –NR 1c –C 1-6 alkylene–N(C 1-4 alkyl) 3 + , –N(R 1c )CHR 1e
- q1is 1.
- the payloadis an immunomodulatory agent payload.
- the payloadis a therapeutic monoclonal antibody, cytokine, chemokine, chemokine antagonist, and immune checkpoint inhibitor payload; or a pharmaceutically acceptable salt thereof.
- the payloadis selected from a therapeutic agent for treating cancer (e.g., doxorubicin, daunorubicin, PNU-159682, etoposide, irinotecan, SN-38, docetaxel, paclitaxel, baccatin III, gemcitabine, podophyllotoxin, Carmustine, Ixabepilone, Patupilone (epothelone class), platinum drugs, exatecan, auristatin (dolastatin 10, MMAE, MMAD, MMAF), duocarmycin, pyrrolobenzodiazapene dimer, mitomycin C, bleomycin, calicheamicin, staurosporine, hemiasterlin), an immunosuppressant (e.g., cyclosporin A, rapamycin, and the like), an anti-fungal agent (e.g., Amphotericin, and the like), an antibiotic (e.g., van
- the payloadis selected from a therapeutic agent for treating cancer (e.g., paclitaxel, doxorubicin, daunorubicin, etoposide, irinotecan, SN-38, docetaxel, paclitaxel, gemcitabine, podophyllotoxin, Carmustine, Ixabepilone, Patupilone (epothelone class), platinum drugs, exatecan, auristatin (dolastatin 10, MMAE, MMAD, MMAF) mitomycin C, bleomycin, calicheamicin, staurosporine, hemiasterlin, and the like), an immunosuppressant (e.g., cyclosporin A, rapamycin, and the like), an anti-fungal agent (e.g., Amphotericin, and the like), an antibiotic (e.g., vancomycin, daptomycin, doxycycline, ceftriax
- Reference to a payloadmeans that one or more atoms, including hydrogen or non-hydrogen atoms, of the original, unmodified payload is replaced by a covalent bond to one or more linker.
- the payloadsare derived from the known nuclear payload and are modified to be covalently bonded to at least one optionally substituted trans-cyclooctene via a linker.
- the payloadseven after modification to arrive at the compounds described herein, maintain biological activity, which is comparable to that observed in the original, unmodified payload.
- the payloadsexhibit a binding activity or inhibition which is at least about 98%, about 95%, about 90%, about 85%, about 80%, about 75%, about 70%, about 65%, about 60%, about 55%, or about 50% of that observed in the original, unmodified payload.
- a hydrogen atom bound to a heteroatome.g., N, O, or S
- a halogen atom on a payloadis replaced for attachment to the remainder of the compound.
- a hydrogen atom on a payloadis replaced for attachment to the remainder of the compound.
- the hydrogen atomis on a heteroatom. In certain embodiments, the hydrogen atom is on a nitrogen. In certain embodiments, the hydrogen atom is on an oxygen. In certain embodiments, the hydrogen atom is on a carbon. [0437] In some embodiments, G, at each occurrence, is independently [0438] In some embodiments, G, at each occurrence, is independently [0439] In some embodiments, the payload is a monoclonal antibody payload.
- a monoclonal antibody for use herein as a payloadcan be an entire monoclonal antibody, or a fragment thereof (e.g., antigen- binding fragment (Fab)). In some embodiments, the antibody is an immune cell engager, and as such would induce or elicit an immune response.
- the monoclonal antibody, or fragment thereoftargets one or more of CD3 (NCBI Gene ID 916), CD28 (NCBI Gene ID 940), CD137 (4-1BB) (NCBI Gene ID 3604), CD16 (NCBI Gene ID 2214), NKG2D (NCBI Gene ID 22914), CD64 (NCBI Gene ID 2209), GITR/TNFRSF18 (NCBI Gene ID 8487), CD25 (NCBI Gene ID 3559), CD40 (NCBI Gene ID 958), CD4 (NCBI Gene ID 920), CXCR4 (NCBI Gene ID 7852), G-CSFR (NCBI Gene ID 1441), GM-CSFR (NCBI Gene ID 1438), CD122 (NCBI Gene ID 3560), PD1 (NCBI Gene ID 5133), CTLA4 (NCBI Gene ID 1493), LAG3 (NCBI Gene ID 3902), TIGIT (NCBI Gene ID 201633), NCR1 (NCBI Gene ID 9437), TIM3 (NCBI Gene ID 84868), VISTA (
- the payloadis an antibody or antibody fragment which targets CD3, such as OKT3, SP34, UCHT1, Teplizumab, Otelixizumab, Visilizumab, or Foralumab, or an antibody fragment derived therefrom.
- the payloadis an antibody or antibody fragment which targets CD28, such as Theralizumab, TGN1412, or FR104, or an antibody fragment derived therefrom.
- the payloadis an antibody or antibody fragment which targets CD137 (4-1BB), such as Utomilumab, Urelumab, LVGN6051, or AGEN2373, or an antibody fragment derived therefrom.
- the payloadis an antibody or antibody fragment which targets CD16, such as AFM13, or an antibody fragment derived therefrom.
- the payloadis an antibody or antibody fragment which targets NKG2D, such as NNC0152-0002 orJNJ-64304500, or an antibody fragment derived therefrom.
- the payloadis an antibody or antibody fragment which targets CD64, such as H22, or an antibody fragment derived therefrom.
- the payloadis an antibody or antibody fragment which targets GITR/TNFRSF18, such as MK-4166, TRX518, MS-986156, AMG-228, or INCAGN01876, or an antibody fragment derived therefrom.
- the payloadis an antibody or antibody fragment which targets CD25, such as Daclizumab, RG6292, basiliximab, or HuMax-TAC, or an antibody fragment derived therefrom.
- the payloadis an antibody or antibody fragment which targets CD40, such as Iscalimab, ABBV-323, bleselumab (ASKP-1240), BI-655064, FFP-104, BMS986090, Dacetuzumab, or Lucatumumab, or an antibody fragment derived therefrom.
- the payloadis an antibody or antibody fragment which targets CD4, such as MAX.16H5, IT1208, Zanolimumab (HuMax-CD4), UB-421, or MTRX1011A, or an antibody fragment derived therefrom.
- CD4such as MAX.16H5, IT1208, Zanolimumab (HuMax-CD4), UB-421, or MTRX1011A, or an antibody fragment derived therefrom.
- the payloadis an antibody or antibody fragment which targets CXCR4, such as F50067, or an antibody fragment derived therefrom.
- the payloadis an antibody or antibody fragment which targets G-CSFR, such as CSL324, or an antibody fragment derived therefrom.
- the payloadis an antibody or antibody fragment which targets GM- CSFR, such as Methosimumab, or an antibody fragment derived therefrom.
- the payloadis an antibody or antibody fragment which targets CD122, such as Hu-Mik(beta)1, or an antibody fragment derived therefrom.
- the payloadis an antibody or antibody fragment which targets PD-1, such as CC-90006, Cemiplimab, Camrelizumab, or TSR-042, or an antibody fragment derived therefrom.
- the payloadis an antibody or antibody fragment which targets CTLA4, such as Tremelimumab or ipilimumab, or an antibody fragment derived therefrom.
- CTLA4such as Tremelimumab or ipilimumab
- the payloadis an antibody or antibody fragment which targets LAG3, such as Relatlimab (BMS-986016), GSK2831781, Cemiplimab (REGN3767), Favezelimab, Ieramilimab, or Mavezelimab, or an antibody fragment derived therefrom.
- the payloadis an antibody or antibody fragment which targets TIGIT, such as BMS-986207, Tiragolumab, Vibostolimab, Etigilimab, Domvanalimab, ASP-8374, IBI939, BGB-A1217, COM902, or M6223, or an antibody fragment derived therefrom.
- TIGITsuch as BMS-986207, Tiragolumab, Vibostolimab, Etigilimab, Domvanalimab, ASP-8374, IBI939, BGB-A1217, COM902, or M6223, or an antibody fragment derived therefrom.
- NCR1such as hNKp46.02
- the payloadis an antibody or antibody fragment which targets TIM3, such as Cobolimab, Sym023, LY3321367, BMS-986258, SHR-1702, Sabatolimab, or INCAGN02390, or an antibody fragment derived therefrom.
- the payloadis an antibody or antibody fragment which targets VISTA, such as SG7, K01401-020, CI-8993, or JNJ-61610588, or an antibody fragment derived therefrom.
- the payloadis an antibody or antibody fragment which targets CD134, such as KHK4083 or ISB830, or an antibody fragment derived therefrom.
- the payloadis an antibody or antibody fragment which targets CD27, such as Varlilumab, MK-5890, or CDX-527, or an antibody fragment derived therefrom.
- the payloadis an antibody or antibody fragment which targets CD40L, such as Dapirolizumab, or an antibody fragment derived therefrom.
- the payloadis an antibody or antibody fragment which targets ICOS, such as MEDI-570, KY1044, JTX-2011, or GSK3359609, or an antibody fragment derived therefrom.
- the payloadis an antibody or antibody fragment which targets BAFFR, such as Ianalumab, or an antibody fragment derived therefrom.
- the payloadis an antibody or antibody fragment which targets LFA-1, such as Efalizumab, or an antibody fragment therefrom.
- the payloadis an antibody or antibody fragment which targets BTLA, such as Icatolimab, or an antibody fragment derived therefrom.
- the payloadis an anti-CD3 ( ⁇ CD3) monoclonal antibody, or a derivative, or analog thereof.
- the anti-CD3 ( ⁇ CD3) monoclonal antibodyis SP34, UCHT1, or OKT3, or a derivative, or analog thereof.
- at least one payloadis selected from an inhibitor of poly (ADP-ribose) polymerase (PARP), a duocarmycin, a pyrrolobenzodiazepine (PBD), hemiasterlin, HTI-286, an anti- CD3 ( ⁇ CD3) monoclonal antibody, lurbinectedin, MSA-2, gardiquimod, ciprofloxacin, Paclitaxel, Gemcitabine, Mitomycin C, Etoposide, exatecan, and MMAE, or a derivative, or analog thereof.
- PARPpoly (ADP-ribose) polymerase
- PPDpyrrolobenzodiazepine
- HTI-286hemiasterlin
- HTI-286hemiasterlin
- an anti- CD3 ( ⁇ CD3) monoclonal antibodylurbinectedin, MS
- Dis a payload selected from an inhibitor of poly (ADP-ribose) polymerase (PARP), a duocarmycin, a pyrrolobenzodiazepine (PBD), hemiasterlin, HTI-286, and an anti- CD3 ( ⁇ CD3) monoclonal antibody, or a derivative, or analog thereof.
- PARPpoly (ADP-ribose) polymerase
- PPDpyrrolobenzodiazepine
- hemiasterlinhemiasterlin
- HTI-286hemiasterlin
- ⁇ CD3anti- CD3
- at least one payloadis selected from lurbinectedin, MSA-2, gardiquimod, ciprofloxacin, Paclitaxel, Gemcitabine, Mitomycin C, Etoposide, exatecan, Seco-Duocarmycin SA, and MMAE, or a derivative, or analog thereof.
- a payloadis an inhibitor of poly (ADP-ribose) polymerase (PARP), or a derivative, or analog thereof.
- PARP inhibitoris niraparib, talazoparib, olaparib, pamiparib, rucaparib, veliparib, iniparib, 3- aminobenzamide, CEP-9722, E7016, or a derivative, or analog thereof.
- a payloadis:
- a payloadis a duocarmycin, or a derivative, or analog thereof.
- the duocarmycinis Duocarmycin A, Duocarmycin B1, Duocarmycin B2, Duocarmycin C1, Duocarmycin C2, Duocarmycin D, Duocarmycin SA, CC-1065, adozelesin, carzelesin, bizelesin, or a derivative, or analog thereof.
- a payloadis:
- a payloadis a pyrrolobenzodiazepine (PBD), or a derivative, or analog thereof.
- the pyrrolobenzodiazepine (PBD)is [1,2]diazepino[3,4-e]indole, or a derivative, or analog thereof.
- a payloadis an inhibitor of tubulin polymerization.
- a payloadis hemiasterlin, HTI-286, or a derivative, or analog thereof.
- a payloadis derived from: [0480] In some embodiments, a payload is: [0481] In some embodiments, the payload comprises a topoisomerase inhibitor. In some embodiments, the payload comprises camptothecin, or a derivative, or analog thereof. In some embodiments, the payload comprises topotecan, irinotecan, silatecan, cositecan, exatecan, lurtotecan, gimatecan, belotecan, or rubitecan.
- the payloadcomprises: [0483] In some embodiments, the payload comprises: [0484] In some embodiments, the payload comprises [0485] In some embodiments, the payload comprises [0486] In some embodiments, the payload comprises [0487] In some embodiments, the payload comprises . [0488] In some embodiments, the payload comprises . [0489] In some embodiments, the payload comprises a polypeptide. In some embodiments, the polypeptide comprises one or more lysine, serine, threonine, or tyrosine residues.
- the linker L 1is covalently bonded to a lysine, serine, threonine, or tyrosine residue present on the payload.
- the polypeptidecomprises one or more lysine residues.
- the linker L 1is covalently bonded to a lysine residue present on the payload.
- the payloadcomprises an N-terminal amino acid, wherein the linker L 1 is covalently bonded to a N-terminal amino acid.
- mis 1 to 20.
- the payloadis an immunomodulatory agent payload.
- the immunomodulatory agent payloadis an antibody payload.
- the immunomodulatory agent payloadis the immune checkpoint inhibitor payload.
- the immune checkpoint inhibitor payloadis a payload of pidilizumab, sintilimab, AMP-224, atezolizumab, durvalumab, BMS-936559, tremelimumab, indoximod, epacadostat, a TIGIT inhibitor (e.g., LAG-3, such as an anti-LAG-3 antibody; TIM-3, such as an anti-TIM-3 antibody), a B7 molecule, or a BTLA pathway antagonist.
- LAG-3such as an anti-LAG-3 antibody
- TIM-3such as an anti-TIM-3 antibody
- the immune checkpoint inhibitor payloadis an immune checkpoint inhibitor antibody payload.
- the immune checkpoint inhibitor antibody payloadis a PD-1 inhibitor payload.
- the PD-1 inhibitor payloadis a nivolumab, pembrolizumab, pidilizumab, sintilimab, or AMP-224 payload.
- the immune checkpoint inhibitor antibody payloadis a PD-L1 inhibitor payload.
- the PD-L1 inhibitor payloadis an atezolizumab, avelumab, durvalumab, or BMS-936559 payload.
- the immune checkpoint inhibitor antibody payloadis a CTLA4 inhibitor payload.
- the CTLA4 inhibitor payloadis an ipilimumab or tremelimumab payload.
- the immune checkpoint inhibitor payloadis an indoleamine 2,3- dioxygenase (IDO) inhibitor payload.
- the IDO inhibitor payloadis an indoximod or epacadostat payload.
- the immunomodulatory agent payloadis a cytokine payload.
- the cytokine payloadis an interferon, interleukin, tumor necrosis factor, erythropoietin, MIP3a, ICAM, macrophage colony stimulating factor, Erythropoietin (EPO), granulocyte colony stimulating factor (GCSF), or granulocyte-macrophage colony stimulating factor payload.
- the interleukin payloadis chosen from IL-1 to IL-40.
- the interleukin payloadis IL-2, IL-7, IL-12, IL-15, IL-18, or IL-21.
- the immunomodulatory agent payloadis a type 1 cytokine (IL-2, IL-12, TNF-B, IFN-g).
- the cytokine payloadis selected from the group consisting of IFN-alpha, IFN-beta, IFN-gamma, pegylated IFN- ⁇ , and apolipoprotein A-I fusion protein with IFN- ⁇ , interleukin, IL-2, IL-2 covalently bound to immunoglobulins (e.g., cergutuzumab amunaleukin, RO6874281), IL-2 covalently bound to PEG molecules (e.g., NKTR-214), IL-10, PEGylated IL-10 (e.g., pegilodecakin), IL- 7, IL-12, IL-15, recombinant aglycosylated IL-15, fusion protein of IL-15 with the binding domain of IL
- the immunomodulatory agent payloadis the chemokine payload.
- the chemokine payloadis a CCL27, CCL28, CCL2, CCL3, CCL5, CXCL1, CXCL2, CXCL3, CXCL5, CXCL6, CXCL8, CXCL9, CXCL10, CXCL11, CXCL12, or CXCL14 payload.
- the immunomodulatory agent payloadis the chemokine antagonist payload.
- the chemokine antagonist payloadis a plerixafor payload.
- the immunomodulatory agentis a monoclonal antibody specific to a cytokine or a cytokine receptor.
- the immunomodulatory agent payloadcomprises a polypeptide.
- the polypeptidecomprises one or more lysine residues.
- the polypeptidecomprises one or more lysine, serine, threonine, or tyrosine residues.
- the trans-cycloocteneis linked to one of the one or more lysine residues.
- the trans-cycloocteneis independently linked to one or more lysine, serine, threonine, or tyrosine residues.
- the polypeptidecomprises an N-terminal amino acid, wherein an occurrence of the bioorthogonal moiety is linked to the N-terminal amino acid.
- mis 1 to 20. In some embodiments, m is 1 to 10. In some embodiments, m is 1 to 5. In some embodiments, m is 20, 19, 18, 17, 16, 15, 14, 13, 12, 11, 10, 9, 8, 7, 6, 5, 4, 3, 2, or 1. In some embodiments, m is 1.
- the payload-TCO conjugateis of Formula IX: or a pharmaceutically acceptable salt thereof, wherein: R 1a , at each occurrence, is independently selected from the group consisting of hydrogen, C 1-4 alkyl, and C 1-4 haloalkyl; R 1b , at each occurrence, is independently selected from the group consisting of hydrogen, C1-4alkyl, C1-4haloalkyl, C(O)OH, C(O)OC1-4alkyl, C(O)N(R 1c )CHR 1e CO2H, C(O)N(R 1c )CHR 1e C(O)OC1-4alkyl, C(O)N(R 1c )–C1-6alkylene–CO2H, and C(O)N(R 1c )–C1-6alkylene–C(O)OC1-4alkyl; R 1c , at each occurrence, is independently hydrogen or C 1-4 alkyl; R 1e , at each occurrence, is independently –
- Dis independently selected from the group consisting of an anticancer agent payload, a toll-like receptor (TLR) agonist payload and a stimulator of interferon genes (STING) agonist payload.
- R 1ais hydrogen.
- R 1ais C 1-4 alkyl.
- R 1ais CH3.
- R 1bis selected from the group consisting of -C(O)OH, -C(O)OC 1-4 alkyl, -C(O)N(R 1c )CHR 1e CO2H, -C(O)N(R 1c )CHR 1e C(O)OC1-4alkyl, -C(O)N(R 1c )–C1-6alkylene–CO2H, and -C(O)N(R 1c )–C1-6alkylene–C(O)OC1-4alkyl.
- R 1bis selected from the group consisting of C(O)OH, C(O)N(R 1c )CHR 1e CO 2 H, and C(O)N(R 1c )CH 2 CO 2 H.
- R 1bis selected from the group consisting of –NR 1c –CH2CH2–N(CH3)3 + , –N(R 1c )–CH2CH2–SO3H, –N(R 1c )–(CH2CH2O)3–CH2CH2N((CH2CH2O)3–CH2CH2–CO2H)2, and –N(R 1c )–CH(CH2O–CH2CH2–CO2H)2.
- the trans-cyclooctene moiety (G)is: H [0524] In some embodiments, the trans-cyclooctene moiety is: H H HO [0525] In some embodiments, the trans-cyclooctene moiety is O embodiments, the trans-cyclooctene moiety is O HO [0526] In some embodiments, the trans-cyclooctene moiety is .
- the trans-cyclooctene moietyis OH O HO [0527] In some embodiments, the trans-cyclooctene moiety is O [0528] In some embodiments, the trans-cyclooctene moiety is [0529] In some embodiments, the trans-cyclooctene moiety is [0530] In some embodiments, the trans-cyclooctene moiety is . [0531] In some embodiments, the trans-cyclooctene moiety is -OH, 2-aminoethanesulfonic acid, an N-linked natural or unnatural amino acid, or an optionally substituted ethylenediamine; wherein R 2 may be optionally further substituted with a polyether.
- the trans-cyclooctene moietycomprises . [0533] In some embodiments, the trans-cyclooctene moiety comprises . [0534] In some embodiments, the trans-cyclooctene moiety of comprises [0535] In some embodiments, the trans-cyclooctene moiety of comprises [0536] In some embodiments, R 1e is –CH2CO2H, –CH2CH2CO2H, –CH2CONH2, –CH2CH2CONH2, –CH2OH, or –CH(CH3)OH. [0537] In some embodiments, R 1e is –C 1-4 alkylene–CO 2 H.
- R 1eis –CH2CO2H.
- R 1bis -C(O)N(R 1c )–C 1-6 alkylene–CO 2 H.
- R 1bis -C(O)N(R 1c )CH2CO2H.
- R 1cis hydrogen.
- R 1bis hydrogen.
- R 1bis C(O)OH.
- the methodscomprise targeted therapy. In some embodiments, this can be achieved by making use of a TCO targeting moiety and one or more pharmaceutically active agents (i.e.
- the therapeutic probecan also comprise a detectable label, such as one or more imaging agents.
- a “radionuclide”(used interchangeably herein with “radioligand”) used in the trans-cyclooctene moieties described herein, comprises a chelating agent and an isotope; such as an isotope selected from the group consisting of 24 Na, 32 P, 33 P, 47 Sc, 59 Fe, 67 Cu, 76 As, 77 As, 80 Br, 82 Br, 89 Sr, 90 Nb, 90 Y, 103 Ru, 105 Rh, 109 Pd, 111 Ag, 111 In, 121 Sn, 127 Te, 131 I, 140 La, 141 Ce, 142 Pr, 143 Pr, 144 Pr, 149 Pm, 149 Tb, 151 Pm, 153 Sm, 159 Gd,
- Radionuclidescan be delivered via direct conjugation or chelation with a chelating agent. Exemplary radionuclides, chelating agents, and linkers for potential TCO-conjugate payloads are described below.
- BFCBifunctional Chelating Agents
- Therapeutic Radionuclides[0547]
- the linkercomprises a linker as shown below, where BFC refers to a bifunctional chelating agent (e.g., as disclosed herein) and the wavy line indicates the point of attachment to the TCO.
- the payload-TCO conjugateis of Formula IX: or a pharmaceutically acceptable salt thereof, wherein R 1a , at each occurrence, is independently selected from the group consisting of hydrogen, C 1-4 alkyl, and C 1-4 haloalkyl; R 1b , at each occurrence, is independently selected from the group consisting of hydrogen, C1-4alkyl, C1-4haloalkyl, C(O)OH, C(O)OC1-4alkyl, C(O)N(R 1c )CHR 1e CO2H, C(O)N(R 1c )CHR 1e C(O)OC 1-4 alkyl, C(O)N(R 1c )–C 1-6 alkylene–CO 2 H, and C(O)N(R 1c )–C 1-6 alkylene–C(O)OC 1-4 alkyl; R 1c , at each occurrence, is independently hydrogen or C1-4alkyl; R 1e , at each occurrence, is independently
- Dis a chelating agent suitable for use in radioligand therapy.
- Dat each occurrence, is independently selected from DOTA: 1,4,7,10- tetraazacyclododecane-1,4,7,10-tetraacetic acid, DOTAGA: 1,4,7,10-tetraazacyclododececane, 1- (glutaric acid)-4,7,10-triacetic acid, DTPA: diethylenetriaminepentaacetic acid, NTA: nitrilotriacetic acid, EDTA: ethylenediaminetetraacetic acid, DO3A: 1,4,7,10-tetraazacyclododecane-1,4,7-triacetic acid, NOTA: 1,4,7-triazacyclononane-1,4,7-triacetic acid, NODAGA: 1-(1,3-carboxypropyl)-4,7- carboxymethyl-1,4,7
- the payload-TCO conjugateis of Formula IXA: or a pharmaceutically acceptable salt thereof, wherein R 1a is selected from the group consisting of hydrogen, C 1-4 alkyl, and C 1-4 haloalkyl; D is a payload; and L 1 is a linker.
- Dis a chelating agent suitable for use in radioligand therapy.
- Exemplary, non-limiting, chelating agentsinclude DOTA: 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid, DOTAGA: 1,4,7,10-tetraazacyclododececane, 1-(glutaric acid)-4,7,10-triacetic acid, DTPA: diethylenetriaminepentaacetic acid, NTA: nitrilotriacetic acid, EDTA: ethylenediaminetetraacetic acid, DO3A: 1,4,7,10-tetraazacyclododecane-1,4,7-triacetic acid, NOTA: 1,4,7-triazacyclononane-1,4,7- triacetic acid, NODAGA: 1-(1,3-carboxypropyl)-4,7-carboxymethyl-1,4,7-triazacyclononane trizoxetan, tetraxetan, macrocyclic maleimides, or a mixture thereof.
- the chelating agentis complexed to a radioligand or radionuclide.
- linker L 1may have 1 to 100 linking atoms, and may include ethylene-oxy groups, amines, esters, amides, carbamates, carbonates, and ketone functional groups.
- linkersmay have from 1 to 50 linking atoms, or from 5 to 50 linking atoms, or from 10 to 50 linking atoms, or from 1 to 40 linking atoms, or from 1 to 30 linking atoms, or from 1 to 20 linking atoms, or from 1 to 10 linking atoms, or from 1 to 5 linking atoms, or from 5 to 30 linking atoms, or from 10 to 30 linking atoms, or from 5 to 40 linking atoms, or from 5 to 50 linking atoms, or from 10 to 50 linking atoms.
- linker L 1may comprise one or more (e.g., 1-10 or 1-5) chain heteroatoms (e.g., O, N, S) and one or more (e.g., 1-10 or 1-5) alkylene, alkenylene, alkynylene, arylene, heteroarylene, cycloalkylene or heterocycloalkylene moieties; wherein each alkylene, alkenylene, alkynylene, arylene, heteroarylene, cycloalkylene or heterocycloalkylene moiety, may be independently optionally substituted with one to five substituents independently selected from oxo, halo, C1-4 alkyl, C1-4 alkoxy, and C1-4 haloalkyl.
- chain heteroatomse.g., O, N, S
- alkylene, alkenylene, alkynylene, arylene, heteroarylene, cycloalkylene or heterocycloalkylene moietieswherein each alkylene, alkenylene, alkyn
- the linkeris a bond. [0558] In certain embodiments, the linker is not a bond.
- each R 110is independently hydrogen, C 1-4 alkyl, C 1-4 haloalkyl, aryl, heteroaryl, cycloalkyl or heterocyclyl; and each R 120 is independently hydrogen, C 1-4 alkyl, C 1-4 haloalkyl, aryl, heteroaryl, cycloalkyl or heterocyclyl.
- Representative linkersinclude, but are not limited to, those shown below: .
- linker L 1may comprise one or more of polyethylene glycol (e.g., PEG having an average molecular weight of from 300 g/mol to 10,000 g/mol), ethylene-1,2- diylbis(methylcarbamate, an arylene (e.g., phenylene), ethylene-oxy, amine, ester, amide, carbamate, ketone (i.e., formyl), or carbonate.
- linker L 1may comprise [0562]
- linker L 1may comprise one or more natural or unnatural amino acids, which may be referred to as a peptide linker.
- linkermay be bound thereto using a peptide linker made up of a carboxylic acyl unit, and one or more amino acids making up a protein or peptide sequence.
- linker L 1may also contain a self- immolating spacer which spaces the drug and the protein peptide sequence.
- linker L 1may be a peptide linker represented by “A—Y—Z—X—W” in which “A” is the carboxylic acyl unit, “Y” and “Z” are each one or more natural or unnatural amino acids and together form a peptide sequence, and “X” and “W” are optional additional linkers having from 1 to 50 linking atoms, or from 5 to 10 linking atoms, or from 1 to 10 linking atoms which spaces the peptide and the drug, D, or the bioorthogonal moiety.
- one or more of the amino acids in the peptide linkeris N-methylated.
- Ymay be at least one amino acid selected from the group consisting of alanine, valine, leucine, isoleucine, methionine, phenylalanine, tryptophan and proline. In some embodiments, Y may be at least one amino acid selected from the group consisting of phenylalanine, alanine, and valine.
- Zmay be at least one amino acid selected from the group consisting of alanine, lysine, lysine protected with acetyl or formyl, arginine, arginine protected with tosyl or nitro groups, histidine, ornithine, ornithine protected with acetyl or formyl, and citrulline.
- Zmay be at least one amino acid selected from the group consisting of alanine, lysine and citrulline.
- exemplary Y-Z combinationsinclude Valine-Citrulline; Valine-Alanine; and Alanine-Alanine.
- Ais -OC(O)-.
- Xis -OC(O)-.
- Wis -OC(O)-.
- Xis absent and W is -OC(O)-.
- —X—[0571] In certain embodiments, —X— [0572]
- the peptide linkeris specifically tailored so that it will be selectively cleaved (e.g., enzymatically cleaved) releasing the drug, such as by one or more of the tumor-associated proteases.
- the peptide linkerhas a chain length of two to four amino acid residues (i.e., a di-, tri-, or tetra-peptide). It will be understood, however, that peptide linkers up to five, six, seven, or eight amino acid residues may also suitably be employed.
- the peptide linkeris Phe-Lys, Val-Lys, Val-Ala, Ala-Ala, Phe-Phe-Lys, D-Phe-Phe-Lys, Gly-Phe-Lys, Ala-Lys, Val-Cit, Phe-Cit, Leu-Cit, Ile-Cit, Trp-Cit, Phe-Ala, Gly-Phe- Leu-Gly [SEQ ID NO: ], Ala-Leu-Ala-Leu [SEQ ID NO: ], Phe-N 9 -tosyl-Arg, or Phe-N 9 -Nitro-Arg.
- the peptide linkeris Phe-Lys, Val-Lys, Val-Ala, Ala-Ala, Val-Val, Val-Cit, or D- Phe-L-Phe-Lys. In certain embodiments, the peptide linker is Val-Cit, Val-Ala, or Ala-Ala.
- linker L 1is: , , [0576]
- the foregoing linkersmay attach on the right-hand side to amino acid side chains of D such [0577]
- the payloadis covalently bonded to the linker through an amide bond; e.g., the payload may be an amine-containing payload for attachment of the payload to a carbonyl group of the linker, or, in other cases, the payload may be a carboxyl-containing payload for attachment of the payload to an amine group of the linker.
- the payload and linkertogether form a carbamate group; e.g., the payload may be an amine-containing payload for attachment of the payload to an acyloxy group of the linker.
- the payload and linkertogether form a carbonate group; e.g., the payload may be a hydroxy-containing payload for attachment of the payload to an acyloxy group of the linker.
- L 1is L 3a is a bond or C 1-6 alkylene;
- L 4ais a bond, —NHN:, –N(R 10 )–C2-6alkylene–N(R 11 )–, –N(R 12 )–C2-3alkylene–N(R 13 )C(O)–, –N(R 10 )–C1-6alkylene–C(O)NHN:, —NHNHC(O)C1-6alkylene–C(O)NHN:, –CH(NHC(O)R 14 )C 1-4 alkylene–S–S–C 1-4 alkylene–OC(O)–, —NHNHC(O)CH(NHC(O)R 15 )CH 2 C(O)–, –C 1-6 alkylene–CH(G x )OC(O)–, , R 10 , R 11 , R 12 , R 13 , R 14 , R 15 , and R
- linker L 1is -OC(O)-.
- L 1is L 3a is a bond; L 4 R 12 and R 13 are each independently hydrogen or C 1-4 alkyl.
- pis 1. In some embodiments, p’ is 1.
- R 18at each occurrence, is independently hydrogen or –CH 2 OC(O)NHD’; R D is hydrogen or C1-4alkyl on a nitrogen atom of a payload; and D and D’ are independently a payload moiety.
- D or D’is a cyclic dinucleotide payload moiety, imidazo[4,5-c]quinolin- 4-amine payload moiety, TLR agonist payload moiety, STING agonist payload moiety, or anticancer agent payload moiety.
- R 12 and R 13are each independently hydrogen or C1-4alkyl; and D and D’ are independently a payload moiety (e.g., anticancer agent payload moiety).
- p’is 0. [0586] In some embodiments, p” is 2 or 3. [0587] In some embodiments, p is 2 [0588] The person skilled in the art will recognize that a payload (D or D’) bonded to a linker does not refer to a payload molecule per se, but refers to the portion of the payload molecule bonded to the linker. Release of the payload (D or D’) from a prodrug, releases the payload per se. [0589] A payload (D or D’) may be an anticancer agent payload of any of the anticancer agents described herein. [0590] In some embodiments, the payload comprises a TLR7/8 agonist.
- the payloadcomprises a TLR7/8 agonist, and X is an antibody or antibody fragment moiety which targets HER2, TROP2, Nectin4, CEACAM5, fibronectin or extracellular matrix (ECM).
- the payloadcomprises gardiquimod, and X is an antibody or antibody fragment moiety which targets HER2, TROP2, Nectin4, CEACAM5, fibronectin or extracellular matrix (ECM).
- the payloadcomprises a camptothecin, or derivative thereof. In some embodiments, the payload comprises exatecan.
- the payloadcomprises a camptothecin, or derivative thereof, and X is an antibody or antibody fragment moiety which targets HER2, TROP2, Nectin4, or extracellular matrix (ECM).
- the payloadcomprises exatecan, and X is an antibody or antibody fragment moiety which targets HER2, TROP2, Nectin4, CEACAM5, fibronectin or extracellular matrix (ECM).
- the payloadcomprises MMAE.
- the payloadcomprises MMAE, or derivative thereof, and X is an antibody or antibody fragment moiety which targets HER2, TROP2, Nectin4, CEACAM5, fibronectin or extracellular matrix (ECM).
- the payloadcomprises paclitaxel. In some embodiments, the payload comprises paclitaxel, or derivative thereof, and X is an antibody or antibody fragment moiety which targets HER2, TROP2, Nectin4, CEACAM5, fibronectin or extracellular matrix (ECM). [0595] In some embodiments, the payload comprises docetaxel, or derivative thereof. In some embodiments, the payload comprises docetaxel, or derivative thereof, and X is an antibody or antibody fragment moiety which targets HER2, TROP2, Nectin4, CEACAM5, fibronectin or extracellular matrix (ECM). [0596] In some embodiments, the payload-TCO conjugate has the structure:
- the payload-TCO conjugateis selected from: O H
- the payload-TCO conjugateis selected from:
- a method of treating cancer or enhancing or eliciting an immune responsecomprising administering to a subject in need thereof: a therapeutically effective amount of a targeting moiety of the disclosure, or a pharmaceutically acceptable salt or composition thereof; and a prodrug, such as those as described herein; and optionally a therapeutically effective amount of an additional therapeutic agent selected from the group consisting of an anticancer agent or an immunomodulatory agent.
- the disclosurealso provides a pharmaceutical combination comprising a payload-TCO conjugate described herein, or a pharmaceutically acceptable salt, or composition thereof; and an additional therapeutic agent, such as an anticancer agent or an immunomodulatory agent, for use in the treatment or prevention of a cancer or for use in enhancing or eliciting an immune response.
- an additional therapeutic agentsuch as an anticancer agent or an immunomodulatory agent, for use in the treatment or prevention of a cancer or for use in enhancing or eliciting an immune response.
- the components of the pharmaceutical combinationsmay be administered/used simultaneously, separately, or sequentially, and in any order, and the components may be administered separately or as a fixed combination.
- the delay of progression or treatment of diseasesmay comprise administration of the first active ingredient in free or pharmaceutically acceptable salt form and administration of the second active ingredient in free or pharmaceutically acceptable salt form, simultaneously or sequentially in any order, in jointly therapeutically effective amounts or effective amounts, e.g. in daily dosages corresponding to the amounts described herein.
- the individual active ingredients of the combinationcan be administered separately at different times during the course of therapy or concurrently in divided or single dosage forms. The instant disclosure is therefore to be understood as embracing all such regimes of simultaneous or alternating treatment and the term “administering” is to be interpreted accordingly.
- a pharmaceutical combinationdefines either a fixed combination in one dosage unit form or separate dosages forms for the combined administration where the combined administration may be independently at the same time or at different times.
- the methods and uses in treating cancerinclude administering/localizing the targeting moiety at a tumor.
- Additional therapeutic agent(s)may be administered simultaneously or sequentially with the payload-TCO conjugate and compositions. Sequential administration includes administration before or after the payload-TCO conjugate and compositions. An additional therapeutic agent may be administered before the payload-TCO conjugate and compositions. An additional therapeutic agent may be administered after the payload-TCO conjugate and compositions. An additional therapeutic agent may be administered at the same time as the payload-TCO conjugate and compositions.
- the additional therapeutic agent or agentsmay be administered in the same composition as the payload-TCO conjugate. In other embodiments, there may be an interval of time between administration of the additional therapeutic agent and the payload-TCO conjugate or compositions. In some embodiments, administration of an additional therapeutic agent with a payload-TCO conjugate or composition may allow lower doses of the other therapeutic agents and/or administration at a less frequent interval.
- the payload-TCO conjugate or compositions of the present disclosure and the other active ingredientmay be used in lower doses than when each is used singly.
- the pharmaceutical compositions of the present disclosureinclude those that contain one or more other active ingredients, in addition to a payload-TCO conjugate of the present disclosure.
- Anticancer agentsinclude, but are not limited to, Abiraterone Acetate, Abitrexate (Methotrexate), Abraxane (Paclitaxel Albumin- stabilized Nanoparticle Formulation), ABVD, ABVE, ABVE-PC, AC, AC-T, Adcetris (Brentuximab Vedotin), ADE, Ado-Trastuzumab Emtansine, Adriamycin (Doxorubicin Hydrochloride), Adrucil (Fluorouracil), Afatinib Dimaleate, Afinitor (Everolimus), Aldara (Imiquimod), Aldesleukin, Alemtuzumab, Alimta (Pemetrexed Disodium), Aloxi (Palonosetron Hydrochloride), Ambochlorin (Chlorambucil), Aminolevulinic Acid, Anastrozole, Aprepitant, Aredia (Pamidron), Abidron, Abidron,
- the anticancer agentmay be a PBD dimer, calicheamicin, speromycin, tubulysin B, rhizoxin, dolastatin, didemnin B, camptothecin, CBI, temsirolimus, actinomycin D, epothilone B, taxol, cryptophycin, SN38, velcade, bruceantin, DAVLBH, DM1, Phyllanthoside, Alimta, T2 Toxin, MMC, vantalanib, vinorelbine, brefeldin, sunitinib, daunomycin, semaxanib, tarceva, iressa, irinotecan, LY- 541503, geldanomycin, gemcitabine, methotrexate, gleevec, topotecan, bleomycin, doxorubicin, cisplatin, N-mustards, etoposide, or 5-FU
- an anticancer agentis an anthracycline. In certain embodiments, anticancer agent is a taxane. In certain embodiments, anticancer agent is gemcitabine. In certain embodiments, anticancer agent is doxorubicin. In certain embodiments, anticancer agent is docetaxel. In certain embodiments, anticancer agent is SN38. In certain embodiments, anticancer agent is monomethyl auristatin E. Synthesis of the Compounds [0607] The targeting moieties may be prepared using the methods disclosed herein and routine modifications thereof, which will be apparent given the disclosure herein and methods well known in the art. Conventional and well-known synthetic methods may be used in addition to the teachings herein.
- Suitable protecting groups for various functional groups as well as suitable conditions for protecting and deprotecting particular functional groupsare well known in the art. For example, numerous protecting groups are described in Wuts, P. G. M., Greene, T. W., & Greene, T. W. (2006). Greene’s protective groups in organic synthesis. Hoboken, N.J., Wiley- Interscience, and references cited therein.
- the term “leaving group”refers to an atom (or a group of atoms) with electron withdrawing ability that can be displaced as a stable species, taking with it the bonding electrons.
- Suitable leaving groupsinclude halides (e.g., Br, Cl, I), sulfonate esters (e.g., triflate, mesylate, tosylate, and brosylate), and nitrophenols.
- R 50is a synthetic handle for bonding to L, such as a leaving group, e.g., halo, or a portion of L capable of linking to X or a further portion of L, e.g., hydroxy, amino, methylamino, etc.
- a leaving groupe.g., halo, or a portion of L capable of linking to X or a further portion of L, e.g., hydroxy, amino, methylamino, etc.
- Suitable methodscan be adapted from the literature (see, e.g., WO2020/077140, WO2018/187740, WO2017/044983, WO2015/139025, and WO2014/205126).
- Compounds of Formula I-2can be prepared by reacting compound I-1 with a precursor to L under suitable coupling reaction conditions.
- Xis an antibody fragment.
- Suitable coupling methodsinclude, but are not limited to, use of an succinimide functional group which is capable of forming an amide bond with a primary amine on the antibody fragment, or L can be functionalized with a group capable of forming a covalent bond to a cysteine residue on the antibody fragment moiety, such as a pyrrole-2,5-dione.
- Scheme II[0612] As shown in Scheme II, coupling compound II-1 with compound II-2 in the presence of N 2 H 4 provides compound II-3. Further modification of compound II-3 with compound II-4 and/or compound II-6 under standard coupling conditions provides compound II-5 and/or compound II-7. Alternatively, compound II-3 can be provided by coupling compound II-8 with compound II-9 in the presence of N 2 H 4 .
- Compound II-10can be provided by contacting compound II-3 with a suitable oxidizing agent (e.g., NaNO2). Alternatively, compound II-3 can be provided by contacting compound II-10 with thiourea dioxide.
- a suitable oxidizing agente.g., NaNO2
- compound II-3can be provided by contacting compound II-10 with thiourea dioxide.
- each of the intermediate or final compoundscan be recovered, and optionally purified, by conventional techniques such as neutralization, extraction, precipitation, chromatography, filtration and the like.
- any of the compounds or intermediates shown in Scheme I or IImay be prepared using traditional methods or purchased from commercial sources.
- any of the intermediates or any product obtained by the process outlined in Scheme I or IIcan be derivatized at any step to provide various compounds of Formula V.
- Exemplary payloads and conjugates thereofcan be prepared can be prepared according to methods adapted from the literature (see, e.g., WO2022/032191, WO2021/007160, WO2020/077140, WO2018/187740, WO2017/044983, WO2015/139025, and WO2014/205126, which methods are incorporated herein in their entirety). Exemplary procedures for certain payloads are shown in the Examples below, which procedures can be adapted to prepare other payloads such as those disclosed herein. EXAMPLES [0616] The following examples are included to demonstrate specific embodiments of the disclosure.
- Example 1Synthesis of Tetrazine-Trastuzumab Targeting Moiety
- Trastuzumab (22.1 mg/mL, 1.1 mL) in 0.01 M PBSwas mixed with 20 equivalents of methyltetrazine-PEG4-NHS (Clickchemtools #1069-10).
- the reactionwas mixed thoroughly and aged at room temperature for 1 hour, at which time the reaction was quenched by the addition of 1 volume of 0.1 M Tris buffer. The resulting solution was buffer exchanged to 0.01 M PBS to remove excess reagent and buffer salts.
- Fabwas prepared from Trastuzumab using a commercial kit (PierceTM Fab Preparation Kit #44985) according to the manufacturer’s protocol and purified by protein G resin (BioVision #6511-25).
- the purified Fab in 0.01 M PBSwas mixed with 20 equivalents of methyltetrazine-PEG4-NHS (Clickchemtools #1069-10).
- the reactionwas mixed thoroughly and aged at room temperature for 1 hour, at which time the reaction was quenched by the addition of 1 volume of 0.1 M Tris buffer.
- the resulting solutionwas buffer exchanged to 0.01 M PBS to remove excess reagent and buffer salts.
- the resulting solution of targeting moiety(1.0 mg/mL, 10.3 mL) was analyzed by SDS-Page and LCMS (data not shown) confirming the formation of the targeting moiety, and thus was used for subsequent studies.
- Fabwas prepared from Enfortumab using the following method.0.1 mg papain was pretreated with 1 mM DTT at 0.5 mg/ml concentration and 2 mM EDTA by incubating at 37 °C for 30 min. The antibody was prepared in PBS buffer, pH 7.4 (10 mg, 0.5 mg/mL). The pretreated papain was mixed with the antibody at (1 : 100) molar ratio and incubated at 37 °C for 2 hours.
- the digestion mixturewas loaded onto an anti-CH1 affinity column, washed with 25 mM Tris, 150 mM NaCl, pH 8.0, and eluted with 50 mM sodium citrate, 150 mM NaCl, pH 3.0.
- the filtrate containing the product Fabwas dialyzed into PBS.
- the purified Fabwas in PBS was concentrated to 0.2 mg/mL.
- the Fabwas mixed with Me-Tet- PEG9-NHS (prepared in DMSO at 10 mM) at 3:1 molar ratio and incubated at 37 °C for 2 hours.
- the resulting conjugatewas analyzed by LCMS and the DAR calculated to be 2.66.
- Fabwas prepared from Brentuximab using the following method.0.1 mg papain was pretreated with 1 mM DTT at 0.5 mg/ml concentration and 2 mM EDTA by incubating at 37 °C for 30 min. The antibody was prepared in PBS buffer, pH 7.4 (10 mg, 0.5 mg/mL). The pretreated papain was mixed with the antibody at (1 : 100) molar ratio and incubated at 37 °C for 2 hours.
- the digestion mixturewas loaded onto an anti-CH1 affinity column, washed with 25 mM Tris, 150 mM NaCl, pH 8.0, and eluted with 50 mM sodium citrate, 150 mM NaCl, pH 3.0.
- the filtrate containing the product Fabwas dialyzed into PBS.
- the purified Fabwas in PBS was concentrated to 0.2 mg/mL.
- the Fabwas mixed with Me-Tet- PEG9-NHS (prepared in DMSO at 10 mM) at 3:1 molar ratio and incubated at 37 °C for 2 hours.
- the resulting conjugatewas analyzed by LCMS and the DAR calculated to be 4.57.
- Example 5ATetrazine-Fab Targeting Moiety
- Fabis prepared from Sacituzumab using a commercial kit (PierceTM Fab Preparation Kit #44985) according to the manufacturers protocol and purified by protein G resin (BioVision #6511-25).
- the purified Fab 10 mM Me-Tet-PEG9-NHSis prepared in DMSO.
- the two componentsare reacted at 3:1 drug to protein molar ratio at 25 ° C for 2 hours before it is dialyzed against PBS, pH 7.4 to remove excess Me-Tet-PEG9-NHS compound from the protein component.
- the resulting solution of targeting moietyis analyzed by SDS-Page and LCMS to confirm the formation of the targeting moiety.
- Example 5BTetrazine-Fab Targeting Moiety (Compound TM-2)
- Compound TM-2was prepared and characterized as follows.
- Sacituzumab Fab amino acid sequences[0626] Heavy chain: [0627] QVQLQQSGSELKKPGASVKVSCKASGYTFTNYGMNWVKQAPGQGLKWMGWINTYTG EPTYTDDFKGRFAFSLDTSVSTAYLQISSLKADDTAVYFCARGGFGSSYWYFDVWGQGSLVTVS SASTKGPSVFPLAPSSKSTSGGTAALGCLVKDYFPEPVTVSWNSGALTSGVHTFPAVLQSSGLYS LSSVVTVPSSSLGTQTYICNVNHKPSNTKVDKKVEPKSSDKTHT (SEQ ID NO.9).
- Sacituzumab Fab-tetrazine conjugateCoding sequences of the variable region of heavy chain and light chain of sacituzumab (human anti-TROP2) antibody were used to generate TROP2 Fab-expressing constructs. Coding sequences were synthesized and subcloned into PCDN3.4 expression vector. The constructed plasmids were transformed into E. coli for propagation and scale-up. Purified plasmids were confirmed by sequencing. The constructs containing the heavy chain and light chain of the TROP2 Fab were transfected into HEK293 cells (suspension) with polymer polyethylenimine (PEI) reagent. The culture medium was harvested at 6-7 days post-transfection.
- PEIpolymer polyethylenimine
- the culture medium containing TROP2 Fabwas centrifuged, filtered, and then loaded onto KappaSelect affinity column (Mabselect Prism).
- the loading bufferwas 25 mM Tris containing 150 mM NaCl, pH 8.0, and eluted with 100 mM sodium-citrate buffer containing 150 mM NaCl, pH 2.5.
- the collected solutionwas neutralized with 1 M arginine, 400 mM succinic acid buffer, pH 9.0.
- the affinity-purified proteinwas further purified by gel filtration with Superdex S-2005/150GL column chromatography. The sample was eluted using the mobile phase 2X PBS at pH 7.4.
- the purified TROP2 Fabwas analyzed by SDS-PAGE and SEC-HPLC.
- TROP2 Fabwas buffer exchanged to PBS pH 7.4 overnight.
- the methyltetrazine-PEG9-NHS(SiChem #SC-8808) was dissolved in DMSO to make a 10 mM stock solution.
- the two componentswere reacted at 3:1 (methyltetrazine-PEG9- NHS to TROP2 Fab) molar ratio at 25 °C for 2 hours.
- the amount of Fab for tetrazine conjugationvaried from 30-100 mg but the ratio of 3:1 was always maintained for each conjugation procedure.
- HPLC-SEC analysiswas performed using a UltiMate 3000 HPLC (Thermo Scientific) fitted with a Xbridge BEH 200A SEC column (7.8 x 300 mm, Waters Corp). Proteins were eluted with PBS containing 15% isopropanol, pH 7.4). Size exclusion chromatograms showed 99% monomeric species for the Fab-tetrazine conjugate with minimal aggregation (1.0%). The tetrazine-to-antibody ratio was calculated to be 2.3.
- Example 6HER2 Fab-tetrazine Targeting Moiety (Compound TM-1) [0633] Compound TM-1 and Isotype Fab-Tz were prepared and characterized as follows.
- Trastuzumab Fab amino acid sequences[0635] Heavy chain: EVQLVESGGGLVQPGGSLRLSCAASGFNIKDTYIHWVRQAPGKGLEWVARIYPTNGYTRYADS VKGRFTISADTSKNTAYLQMNSLRAEDTAVYYCSRWGGDGFYAMDYWGQGTLVTVSSASTKG PSVFPLAPSSKSTSGGTAALGCLVKDYFPEPVTVSWNSGALTSGVHTFPAVLQSSGLYSLSSVVT VPSSSLGTQTYICNVNHKP SNTKVDKKV (SEQ ID NO.3).
- Isotype Fab-Tz amino acid sequences[0638] Heavy chain: EVQLLESGGGLVQPGGSLRLSCAASGFTFSSFSMSWVRQAPGKGLEWVSSISGSSGTTYYADSV KGRFTISRDNSKNTLYLQMNSLRAEDTAVYYCAKPFPYFDYWGQGTLVTVSSASTKGPSVFPLA PSSKSTSGGTAALGCLVKDYFPEPVTVSWNSGALTSGVHTFPAVLQSSGLYSLSSVVTVPSSSLG TQTYICNVNHKPSNTKVDKKVEPKSSDKTHT (SEQ ID NO.5).
- HER2 FabCoding sequences of the variable region of heavy chain and light chain of trastuzumab (human anti-HER2) antibody were used to generate HER2 Fab-expressing constructs. Coding sequences were synthesized and subcloned into PCDN3.4 expression vector. The constructed plasmids were transformed into E. coli for propagation and scale-up. Purified plasmids were confirmed by sequencing. The constructs containing the heavy chain and light chain of the HER2 Fab were transfected into HEK293 cells (suspension) with polymer polyethylenimine (PEI) reagent. The 6- liter culture medium was harvested at 6-7 days post-transfection.
- PEIpolymer polyethylenimine
- the culture medium containing HER2 Fabwas centrifuged, filtered, and then loaded onto KappaSelect affinity column (Mabselect Prism).
- the loading bufferwas 25 mM Tris containing 150 mM NaCl, pH 8.0, and eluted with 100 mM sodium- citrate buffer containing 150 mM NaCl, pH 2.5.
- the collected solutionwas neutralized with 1 M arginine, 400 mM succinic acid buffer, pH 9.0.
- the affinity-purified proteinwas further purified by gel filtration with Superdex S-2005/150GL column chromatography.
- the sample injectionwas 20 mL with a flow rate of 0.3 mL/min and mobile phase 2X PBS at pH 7.4.
- the purified HER2 Fabwas analyzed by SDS-PAGE and SEC-HPLC. The final yield purified yield was 742 mg and stored in -80 °C for long term storage.
- Generation of Isotype FabCoding sequences were synthesized and subcloned into pTT5 expression vector. The constructed plasmids were transformed into E. coli for propagation and scale-up. Purified plasmids were confirmed by sequencing. The constructs containing the heavy chain and light chain of the Isotype Fab were transfected into HEK293 cells (suspension) with polymer polyethylenimine (PEI) reagent. The 2-liter culture medium was harvested at 7 days post-transfection.
- PEIpolymer polyethylenimine
- the culture medium containing Isotype Fabwas centrifuged, filtered, and then loaded onto KappaSelect affinity column (Mabselect Prism).
- the loading bufferwas 25 mM Tris containing 150 mM NaCl, pH 8.0, and eluted with 50 mM sodium-citrate buffer containing 150 mM NaCl, pH 3.
- the collected solutionwas neutralized with 1 M arginine, 400 mM succinic acid buffer, pH 9.0.
- the affinity-purified proteinwas further purified by gel filtration with Superdex S-2005/150GL column chromatography.
- the sample injectionwas 3 mL with a flow rate of 0.3 mL/min and mobile phase 2X PBS at pH 7.4.
- the purified Isotype Fabwas analyzed by SDS-PAGE and SEC-HPLC. The final yield purified yield was 200 mg and stored in -80 °C for long term storage.
- Tetrazine conjugation of HER2 Fab and Isotype Fabwas buffer exchanged to PBS pH 7.4 overnight.
- the methyltetrazine-PEG9-NHS(SiChem #SC-8808) was dissolved in DMSO to make a 10 mM stock solution. For conjugation, the two components were reacted at 3:1 (methyltetrazine-PEG9-NHS to HER2 Fab) molar ratio at 25 °C for 2 hours.
- the amount of Fab for tetrazine conjugationvaried from 30-100 mg but the ratio of 3:1 was always maintained for each conjugation procedure. Then the solution was buffer exchanged to PBS pH 7.4 to remove excess methyltetrazine-PEG9-NHS and buffer salts.
- HPLC-SEC analysiswas performed using a UltiMate 3000 HPLC (Thermo Scientific) fitted with a xBridge BEH 200A SEC column (7.8 x 300 mm, Waters Corp). Proteins were eluted with PBS containing 15% isopropanol, pH 7.4). Size exclusion chromatograms for Compound TM-1 and Isotype Fab-Tz and showed > 97% monomeric species for the Fab-tetrazine conjugate with minimal aggregation ( ⁇ 3.0%). The tetrazine-to-antibody ratio was calculated to be 2.2 for Compound TM-1 and 1.8 for Isotype Fab-Tz.
- Flow cytometry analysis of Compound TM-1 and Isotype Fab-TzCell binding analysis by flow cytometry using NCI-N87 (HER2 positive) cells was tested with either unconjugated HER2 Fab, Compound TM-1, or Isotype Fab-Tz control.
- NCI-N87 (HER2-positive) human gastric cancer cellswere collected by centrifugation and resuspended with FACS buffer (PBS containing 2% FBS, pH 7.4). Cells were seeded in 96-well plates (200,000 cells per well) and centrifuged at 400 x g for 5 minutes.
- the resulting reaction mixturewas purified by Prep- HPLC (column: Welch XB-C187 ⁇ m 110 A 250*50 mm; mobile phase: [water (0.1% TFA)IN]; B%: 50-70%-40 min. number of injections: 2, Retention time: 37 min, flow rate: 60 mL/min) to give Compound A (450 mg, 99.0% purity; 64.6 mg, 99.2%, 31.2% yield).
- Example 8General procedure for preparation of Compound B [0646] To a solution of compound 2 (1.00 g, 5.43 mmol) in DCM (10 mL) was added DIEA (2.10 g, 16.3 mmol), EDCI (2.08 g, 10.9 mmol) and DMAP (1.33 g, 10.9 mmol) and compound 3 (1.61 g, 8.14 mmol). The mixture was stirred at 25 °C for 16 hrs. TLC indicated compound 2 was consumed completely and one new spot formed. The reaction mixture was partitioned between DCM (20 mL) and H2O (10 mL). The organic phase was separated, washed with sat. citric acid aq.
- Example 9Alternative route to Compound B and Synthesis of Compound C.
- the aqueous layerwas extracted with MTBE (3 ⁇ 400 mL).
- the combined MTBE layerswere dried with Na 2 SO 4 , filtered and concentrated in vacuo to provide the compound 2 (5.50 g, 35.9% yield).
- the crude productwas used into the next step without further purification.
- Example 11General procedure for preparation of Compound E [0665] To a solution of compound 13 (350 mg, 0.30 ⁇ mol) and DMAP (221 mg, 1.81 mmol) and compound 14 (249 mg, 0.39 mmol, HCl) in DMF (0.3 mL). The mixture was stirred at 25 °C for 16 hrs. LC-MS showed compound 13 was consumed completely and one main peak with desired mass was detected. The residue was purified by prep-HPLC (Water (0.1% TFA)-ACN) to give Compound E (205 mg, 41.3% yield). LCMS (m/z): 1646.5 (M+H) + .
- the aqueous layerwas extracted with MTBE (5 ⁇ 500 mL).
- the combined MTBE layerswere dried with Na2SO4, filtered and concentrated under reduced pressure to give residue and evaporated with MeCN three times to give compound 2 (9.1 g, 49.3 mmol, 47.4% yield).
- Example 21General procedure for preparation of Compound K [0696] To a solution of compound 1-2 (142 mg, 138 ⁇ mol) in DMF (2.00 mL) was added DMAP (135 mg, 1.11 mmol) and compound 5-4 (100 mg, 416 ⁇ mol) and DIEA (129 ⁇ L, 1.11 mmol). The mixture was stirred at 25 °C for 16 hrs.
- Example 22General procedure for preparation of Compound L [0699] To a solution of compound 1-2 (350 mg, 341 ⁇ mol, 1.00 equiv.) in DMF (7.00 mL) was added compound 6-1 (1.01 g, 2.73 mmol, 8.00 equiv.), DMAP (334 mg, 2.73 mmol, 8.00 equiv.) and DIEA (353 mg, 2.73 mmol, 8.00 equiv.). The mixture was stirred at 25 °C for 12 hrs. Two peaks of Cpd_6-2 and Target 287 were detected. The mixture was stirred at 25 °C for another 24 hrs. LC-MS showed one peak with desired mass detected.
- Example 23General procedure for preparation of compound 7-4 [0702] To a solution of compound 7-2 (800 mg, 5.03 mmol, 1.22 equiv.) in THF (15.0 mL) was added compound 7-3 (650 mg, 4.11 mmol, 1.00 equiv.), AcOH (376 mg, 6.25 mmol, 1.52 equiv.) and NaBH(OAc) 3 (1.33 g, 6.26 mmol, 1.52 equiv.). The mixture was stirred at 25 °C for 1 hr. LC-MS showed 99.9% of compound 7-4 was formed. The reaction mixture was filtered and purified by prep-HPLC to give compound 7-4 (1.0 g, 80.8% yield) as a colorless oil.
- Example 25General procedure for preparation of Compound M [0705] To a solution of compound 1-2 (120 mg, 117 ⁇ mol, 1.00 equiv.) in DMF (2.00 mL) was added compound 7-5 (291 mg, 936 mmol, 8.00 equiv.), DMAP (114 mg, 936 mmol, 8.00 equiv.) and DIEA (121 mg, 936 mmol, 8.00 equiv.). The mixture was stirred at 25 °C for 24 hrs. LC-MS showed Target 288 was formed.
- Example 28General procedure for preparation of Compound N [0711] To a solution of compound 1-2 (100 mg, 97.5 ⁇ mol, 1.00 equiv.) in DMF (3.00 mL) was added compound 7-5 (214 mg, 772 mmol, 7.92 equiv.), DMAP (95.3 mg, 780 mmol, 8.00 equiv.) and DIEA (101 mg, 780 mmol, 8.00 equiv.). The mixture was stirred at 25 °C for 24 hrs. LC-MS showed one peak with desired mass was formed.
- Example 29General procedure for preparation of Compound O H [0713]
- 6-(6-Methyl-1,2,4,5-tetrazin-3-yl)-3-pyridinemethanaminecan also be utilized in the compounds and methods described herein, which compound can be prepared according to the art or purchased from a commercial source (e.g., Enamine US Inc., New Jersey, USA).
- Example 33Val-Cit-PABC-dihydrotetrazine
- Example 34Dihydrotetrazine (Target 5) [0740] To a solution of N 2 H 4 .H 2 O (9.74 g, 190 mmol, 9.44 mL, 98% purity, 7.08 eq) in EtOH (35.0 mL) was added compound 1 (5.00 g, 26.9 mmol, 1.00 eq, HCl) and compound 2 (2.55 g, 26.9 mmol, 1.00 eq, HCl) at 20 °C. The mixture was stirred at 78 °C for 3 hrs. LCMS analysis of the reaction mixture showed compound 1 was consumed completely.
- Example 373-methyl-6-(1H-pyrazol-1-yl)-1,2,4,5-tetrazine (Target 13) [0748] methyl (E)-hydrazinecarbohydrazonothioate (2): To a mixture of 1,3-diaminothiourea (100 g, 942.06 mmol) in MeOH (500 mL) was added MeI (160.46 g, 1.13 mol) at 25 o C and the mixture was stirred at 80 o C for 1.5 h under N 2 . H NMR showed the reaction was completed. The resulting pale- yellow solution was cooled to room temperature until solid precipitated. And then it was diluted with MTBE (500 mL).
- MeI160.46 g, 1.13 mol
- Example 38(4-(6-methyl-1,2,4,5-tetrazin-3-yl)-2-(trifluoromethyl)phenyl)methanamine (Target 15) [0751] tert-butyl (4-cyano-2-(trifluoromethyl)benzyl)carbamate (2): To a solution of 4- (aminomethyl)-3-(trifluoromethyl)benzonitrile (300 mg, 1.50 mmol) in DCM (10 mL) was added Boc2O (360 mg, 1.65 mmol) and TEA (227 mg, 2.25 mmol) at 20 °C under N2. The mixture was stirred at 20 °C for 2 h.
- Example 39(4-(6-methyl-1,2,4,5-tetrazin-3-yl)-2-(trifluoromethyl)phenyl)methanamine (Target 17) [0755] 3-((1,3-dioxoisoindolin-2-yl)methyl)-4-(trifluoromethyl)benzonitrile (2): To a solution of 3- (hydroxymethyl)-4-(trifluoromethyl)benzonitrile (4.5 g, 22.38 mmol) in THF (50 mL) was added isoindoline-1,3-dione (3.3 g, 22.38 mmol), PPh 3 (11.7 g, 44.76 mmol) and DIAD (6.79 g, 33.57 mmol) at 0 °C under N 2 .
- Example 401-(2-(6-(4-(aminomethyl)phenyl)-1,2,4,5-tetrazin-3-yl)ethyl)-3-methylurea (Target 6) [0760] tert-butyl (2-(6-(4-iodophenyl)-1,2,4,5-tetrazin-3-yl)ethyl)carbamate: To a mixture of 4- iodobenzonitrile (15 g, 65.50 mmol) in EtOH (140 mL) was added tert-butyl N-(2-cyanoethyl)carbamate (44.59 g, 261.99 mmol), 3-sulfanylpropanoic acid (6.95 g, 65.50 mmol) and NH 2 NH 2 .H 2 O (59.02 g, 1.18 mol) at 0 °C under N 2 .
- the mixturewas stirred at 45 °C for 16 h. Then the mixture was cooled to 20 °C and added with a solution of NaNO2 in H2O (40 mL) dropwise at 20 °C. The mixture was stirred at 20 °C for 1 h. Under ice-cooling, the pH was adjusted to 3 with 1 M aqueous hydrochloride and then extracted with DCM (3 ⁇ 50 mL). The organic phase was dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure.
- tert-butyl (4-(6-(2-(3-methylureido)ethyl)-1,2,4,5-tetrazin-3-yl)benzyl)carbamateTo a mixture of 1-[2-[6-(4-iodophenyl)-1,2,4,5-tetrazin-3-yl]ethyl]-3-methyl-urea (1 g, 2.60 mmol) and (tert- butoxycarbonylamino)methyl-trifluoro-boron;potassium hydride (926 mg, 3.90 mmol) in 2-METHYL-2- BUTANOL (4 mL) and H 2 O (1 mL) was added Cs 2 CO 3 (1.70 g, 5.21 mmol) and ditert- butyl(cyclopentyl)phosphane;dichloropalladium;iron (170 mg, 0.26 mmol) at 25 °C under N 2 .
- Example 414-((S)-2-((S)-2-acetamido-3-methylbutanamido)-5-ureidopentanamido)benzyl-6- methyl-3-phenyl-1,2,4,5-tetrazine-1(4H)-carboxylate (Target 1b) [0765] 3-methyl-6-phenyl-1,4-dihydro-1,2,4,5-tetrazine (2): To a solution of benzonitrile (10 g, 96.97 mmol), ACN (31.85 g, 775.79 mmol), 3-mercaptopropanoic acid (10.29 g, 96.97 mmol) in EtOH (100 mL) was added dropwise NH 2 NH 2 .H 2 O (79.26 g, 1.55 mol) at 0 °C under N 2 .
- the mixturewas stirred for 16 h at 40 °C.
- the combined organic layerswere washed with brine (100 mL), dried over Na2SO4, filtered and concentrated under reduced pressure.
- the mixturewas slurried with MTBE:EtOAc (10:1, 100 mL), filtered and the solid was desired.
- the proteinis treated with a reducing agent, such as TCEP, at ambient temperature or on ice, followed by buffer exchange into fresh buffer.
- a reducing agentsuch as TCEP
- the proteinis exchanged into fresh buffer to remove the excess reagent to yield conjugate (Ab-Tz).
- the conjugateis analyzed by SDS- PAGE, analytical HPLC, mass spectrometry to confirm the expected properties.
- the conjugatecan also be treated with a trans-cyclooctene-functionalized fluorophore to confirm the reactivity of the tetrazine.
- An non-binding control protein with a C-terminal cysteineis expressed in E.
- the proteinis treated with a reducing agent, such as TCEP, at ambient temperature or on ice, followed by buffer exchange into fresh buffer.
- a reducing agentsuch as TCEP
- the proteinis exchanged into fresh buffer to remove the excess reagent to yield conjugate (Ab-Tz).
- the conjugateis analyzed by SDS-PAGE, analytical HPLC, mass spectrometry to confirm the expected properties.
- the conjugatecan also be treated with a trans- cyclooctene-functionalized fluorophore to confirm the reactivity of the tetrazine.
- L19-Fab HC sequence[0777] EVQLLESGGGLVQPGGSLRLSCAASGFTFSSFSMSWVRQAPGKGLEWVSSISGSSGTTYY ADSVKGRFTISRDNSKNTLYLQMNSLRAEDTAVYYCAKPFPYFDYWGQGTLVTVSSASTKGPS VFPLAPSSKSTSGGTAALGCLVKDYFPEPVTVSWNSGALTSGVHTFPAVLQSSGLYSLSSVVTVP SSSLGTQTYICNVNHKPSNTKVDKKVEPKSSDKTHT (SEQ ID NO.5).
- L19-Fab LC sequence[0779] EIVLTQSPGTLSLSPGERATLSCRASQSVSSSFLAWYQQKPGQAPRLLIYYASSRATGIPD RFSGSGSGTDFTLTISRLEPEDFAVYYCQQTGRIPPTFGQGTKVEIKRTVAAPSVFIFPPSDEQLKS GTASVVCLLNNFYPREAKVQWKVDNALQSGNSQESVTEQDSKDSTYSLSSTLTLSKADYEKHK VYACEVTHQGLSSPCTKSFNRGEC (SEQ ID NO.6).
- Protein expressionThe constructs containing heavy chain and light chain of the Fab are co- transfected into HEK293 cells with PEI.
- the culture mediumis harvested at 6-7 days post transfection.
- Conditional medium expressing target Fabis harvested by centrifugation and filtration, and can then be loaded onto KappaSelect affinity column (Mabselect Prism).
- the loading bufferis 25 mM Tris containing 150 mM NaCl, pH 8.0;
- the wash bufferis 25 mM Tris buffer containing 150 mM NaCl, 0.2% Triton X-100/114, pH 8.0;
- the elution bufferis 100 mM Sodium-citrate buffer containing 150 mM NaCl, pH 2.5.
- the collected solutionis neutralized with 1M arginine, 400 mM succinic acid buffer, pH 9.0.
- the affinity purified proteinis further purified by gel filtration with Superdex S-200 column chromatography. Purified Fab is analyzed by SDS-PAGE, SEC-HPLC and endotoxin measurement.
- Conjugate preparation120 mg Fab protein is dialyzed against PBS, pH 7.4 overnight with one buffer exchange at about 4 hours from the start.10 mM Me-Tet-PEG9-NHS is prepared in DMSO. The two components are reacted at 3:1 drug to protein molar ratio at 25 o C for 2 hours; the reaction mixture is dialyzed against PBS, pH 7.4 to remove excess Me-Tet-PEG9-NHS compound from the protein component.
- F16-Fab HC sequence[0785] EVQLLESGGGLVQPGGSLRLSCAASGFTFSRYGMSWVRQAPGKGLEWVSAISGSGGSTY YADSVKGRFTISRDNSKNTLYLQMNSLRAEDTAVYYCAKAHNAFDYWGQGTLVTVSSCSTKGP SVFPLAPSSKSTSGGTAALGCLVKDYFPEPVTVSWNSGALTSGVHTFPAVLQSSGLYSLSSVVTV PSSSLGTQTYICNVNHKPSNTKVDKKVEPKSSDKTHT (SEQ ID NO.7).
- F16-Fab LC sequence[0787] SSELTQDPAVSVALGQTVRITCQGDSLRSYYASWYQQKPGQAPVLVIYGKNNRPSGIPD RFSGSSSGNTASLTITGAQAEDEADYYCNSSVYTMPPVVFGGGTKLTVLGQPKAAPSVTLFPPSS EELQANKATLVCLISDFYPGAVTVAWKADSSPVKAGVETTTPSKQSNNKYAASSYLSLTPEQW KSHKSYSCQVTHEGSTVEKTVAPTECS (SEQ ID NO.8).
- Protein expressionThe constructs containing heavy chain and light chain of the Fab are co- transfected into HEK293 cells with PEI.
- the culture mediumis harvested at 6-7 days post transfection.
- Conditional medium expressing target Fabis harvested by centrifugation and filtration, and can then be loaded onto KappaSelect affinity column (Mabselect Prism).
- the loading bufferis 25 mM Tris containing 150 mM NaCl, pH 8.0;
- the wash bufferis 25 mM Tris buffer containing 150 mM NaCl, 0.2% Triton X-100/114, pH 8.0;
- the elution bufferis 100 mM Sodium-citrate buffer containing 150 mM NaCl, pH 2.5.
- the collected solutionis neutralized with 1M arginine, 400 mM succinic acid buffer, pH 9.0.
- the affinity purified proteinis further purified by gel filtration with Superdex S-200 column chromatography. Purified Fab is analyzed by SDS-PAGE, SEC-HPLC and endotoxin measurement.
- Conjugate preparation120 mg Fab protein is dialyzed against PBS, pH 7.4 overnight with one buffer exchange at about 4 hours from the start.10 mM Me-Tet-PEG9-NHS is prepared in DMSO. The two components are reacted at 3:1 drug to protein molar ratio at 25 o C for 2 hours; the reaction mixture is dialyzed against PBS, pH 7.4 to remove excess Me-Tet-PEG9-NHS compound from the protein component.
- Example 44Efficacy study in NCI-N87 xenograft gastric cancer model
- the following exampleshows timing of the payload dose leads to effective tumor inhibition with a single dose.
- Compound TM-1 and Isotype Fab-Tz (Iso Ctrl)were prepared according to Example 6.
- NCI-N87 (gastric carcinoma) cellswere cultured in RPMI-1640 supplemented with 10% FBS, 1% pen-strep and placed in 37 °C incubator. The cells were harvested and counted for tumor inoculation when the cells reached the exponential growth phase.
- CB-17 SCID(6-8 weeks old) female mice were inoculated subcutaneously at the right flank with 3 x 10 6 cells in a 200 mL mixture of RPMI-1640 with Matrigel (1:1) for tumor development. Animals were divided into groups of 5 for the efficacy study with mean tumor volume of ⁇ 100-150 mm 3 prior to dosing. Tumor volumes of NCI-N87 tumors in SCID CB17 mice following treatment with vehicle, Compound TM-1 with Compound B, and Isotype Fab-Tz with Compound B.
- micewere dosed with a single dose of either Isotype control (Isotype Fab-Tz) or Compound TM-1 intravenously (IV) at 50 mg/kg then mice were given a single dose of Compound B IV at 30 mg/kg after 4 hours (FIG.1A), 8 hours (FIG.1B), 24 hours (FIG.1C), or 48 hours (FIG.1D) after Iso Ctrl or Compound TM-1.
- TGI (%)[1-(T/V)] ⁇ 100; where T is the mean TV of a treatment group on a given day and V is the mean TV of the vehicle control group on the same day as T, and a two-way ANOVA with Tukey’s correction were performed among the different treatment groups. A p ⁇ 0.05 was considered statistically significant. [0793] As shown in FIGs.1A, 1B, 1C, and 1D, the time between doses can be attenuated to maximize specific and minimize non-specific activity of Compound B with Compound TM-1 on NCI-N87 tumors. Shown are means ⁇ SEM.
- NCI-N87 (gastric carcinoma) cellswere cultured in RPMI-1640 supplemented with 10% FBS, 1% pen-strep and placed in 37 oC incubator. The cells were harvested and counted for tumor inoculation when the cells reached the exponential growth phase.
- CB-17 SCID (6-8 weeks old) female micewere inoculated subcutaneously at the right flank with 5 x 10 6 cells in a 200 mL mixture of PBS with Matrigel (1:1) for tumor development. Animals were divided into groups of 3 for the PK study with mean tumor volume of ⁇ 200-250 mm 3 prior to dosing.
- FIG.2Ashows the schedule of dosing and tumor collection. Plasma and tumor tissue samples were collected after 15 minutes of dosing with Compound B. Plasma samples and tumor tissue were flash frozen and stored in -80 oC until the samples were analyzed using liquid chromatography with tandem mass spectrometry (LC-MS/MS). [0798]
- FIG.2Bshows the concentration of MMAE in tumors 15 minutes after dosing of Compound B. Shown are means ⁇ SEM. P-values were determined by two-way ANOVA with Bonferroni correction.
- CB-17 SCID(6-8 weeks old) female mice were inoculated subcutaneously at the right flank with 5 x 10 6 cells in a 200 mL mixture of RPMI- 1640 with Matrigel (1:1) for tumor development. Animals were divided into groups of 3 for the PK study with mean tumor volume of ⁇ 200-250 mm 3 prior to dosing. Animals were dosed with Compound TM-1 IV at 50 mg/kg. Then animals were dosed with TCO-biotin, 15 minutes prior to tumor collection, the animals were perfused with heparin-saline solution to remove excess/unreacted TCO-biotin, then tumors were harvested close to 1, 4, and 24 hrs.
- the tumorswere flash frozen and placed in -80 °C until the samples were analyzed using enzyme-linked immunosorbent assay (ELISA). Tumor samples were tested for total amount of Compound TM-1 and reactive tetrazine.
- ELISAenzyme-linked immunosorbent assay
- wells of 96-well platewere coated with anti-ID antibody against trastuzumab, and the plates were incubated overnight at 4 °C. The plate was washed 3 times and then samples diluted with assay diluent containing 1% BSA and 0.05 Tween-20 in PBS were added to the wells and incubated at room temperature for 2 hours with constant shaking.
- mouse anti-human IgG Fab antibody conjugated to horseradish peroxidase (HRP)detection antibody
- HRPhorseradish peroxidase
- detection antibodydetection antibody
- the concentrations from tumor sampleswere interpolated from the standard curve of Compound TM-1 using known concentration (200 ng/mL -1.563 ng/mL with 2-fold serial dilution).
- concentrations from tumor sampleswere interpolated from the standard curve of Compound TM-1 using known concentration (200 ng/mL -1.563 ng/mL with 2-fold serial dilution).
- wells of 96-well platewere coated with anti-ID antibody against trastuzumab, and the plates were incubated overnight at 4oC.
- FIG.3Ashows quantification of total Fab in the tumors by ELISA.
- FIG.3Bshows quantification of activator (tetrazine) in the tumors by ELISA. Shown are means ⁇ SEM. P-values were determined by two-way ANOVA with Bonferroni correction for multiple comparisons. ANOVA, analysis of variance; SEM, standard error of mean. [0803] As shown in FIG.3A and FIG.3B, Compound TM-1 localizes to HER2-positive tumors.
- Example 47Compound TM-2 Plasma and Tumor PK Analysis
- This studyevaluated the optimal dose and time of Compound TM-2 at the tumor site for payload-TCO conjugate dosing. [0805] Experimental Design:
- Tumor cell culture and inoculationThe HT-1376 cells (purchased from ATCC, CRL-1472 TM ) were cultured in EMEM medium supplemented with 10% FBS, 100 units/mL penicillin, and 100 ⁇ g/mL streptomycin. Cells were placed in a 37 oC incubator with an atmosphere supplemented with 5% CO 2 . The cells were harvested by trypsinization and used to inoculate female Balb/c nude mice (6 – 8 weeks old) subcutaneously at the right flank with 5x 10 6 HT-1376 cells in a 0.2 mL mixture of PBS with Matrigel (1:1) to induce tumor development.
- VehicleIsotype Control
- Compound TM-2Compound TM-2
- TCO-PEG3-Biotinintravenously at 30 mg/kg at a specific time point post-first IV treatment as follows: Groups 1, 2, and 10 were dosed at 1 hour, groups 3 and 11 at 4 hours, groups 4 – 9 at 8 hours, group 12 at 18 hours, group 13 and 24 hours, and group 14 at 36 hours.
- Sample CollectionThirty minutes after administration of the TCO-PEG3-Biotin dose, approximately 200 ⁇ L of blood was collected in EDTA-coated tubes from all animals and processed for plasma. Following blood collection, the animals were perfused with heparinized saline to remove unreacted TCO-PEG3-Biotin from circulation. Tumors were then collected and immediately frozen.
- the coated plateswere used to capture Fab-Tz in both plasma and tumor homogenates.
- streptavidin-HRPwas used, followed by a Super Signal PICO chemiluminescent substrate reaction.
- HRP-conjugated AffiniPure goat anti-human antibody F(ab’)2, fragment-specific (Jackson ImmunoResearch)was used, followed by chemiluminescent substrate reaction.
- Luminescencewas recorded using a SpectraMax i3x (Molecular Devices) plate reader. Standard curves for total Fab and intact tetrazine were prepared using eight points ranging from 100 to 0.781 ng/mL.
- FIGs.4A and 4Bthere is between 9 – 16x times more Fab detected in plasma compared to at the tumor (FIG.4A) and between 3 – 10x times more Tz detected at the tumor (FIG.4B) depending on the Compound TM-2 dose level. Shown are mean values ⁇ SD.
- the dataindicates little reaction between Compound TM-2 and the payload-TCO moiety when both molecules are in motion as seen in the plasma and Iso Ctrl at the tumor.
- the datafurther suggests that the optimal dose for Compound TM-2 is between 25 – 100 mg/kg.
- Kidney, liver, and spleen tumor datais shown in FIGs.6-9.
- ModelNCI-N87; treatment start ⁇ 150-200 mm 3
- AnimalsSpecies: Mice; Strain: CB.17 SCID; Source: Charles River (#236); Age: 7 weeks; Sex: Female; Number on Study: 81; Housing per Cage: Up to 5.
- Experimental Design:
- Tumor cell culture and inoculationThe NCI-N87 cells were cultured in EMEM medium supplemented with 10% FBS, 100 units/mL penicillin, and 100 ⁇ g/mL streptomycin. Cells were placed in a 37 oC incubator with an atmosphere supplemented with 5% CO2. The cells were harvested by trypsinization and used to inoculate female CB.17 SCID mice (6 – 8 weeks old) subcutaneously at the right flank with 10 x 10 6 NCI-N87 cells/100 microliters (0.1 mL) mixture of PBS with Matrigel (1:1) to induce tumor development.
- Animal Grouping and DosingAnimals were monitored weekly for palpable tumors, or any changes in appearance or behavior.
- Iso Controlwas administered at 50 or 200 mg/kg, while Compound TM-1was given at doses of 5,10, 25, 50, 100 or 200 mg/kg. All animals received a single dose of TCO-PEG3-Biotin intravenously at 32.73 mg/kg at a specific time point post-first IV treatment as described in above Experimental Design).
- Sample CollectionThirty minutes after administration of the TCO-PEG3-Biotin dose, approximately 200 ⁇ L of blood was collected in EDTA-coated tubes from all animals and processed for plasma.
- the standard curves and QC samples for total Fab and intact tetrazinewere prepared in PLCB using eight points ranging from 50 to 0.4 ng/mL.
- the standard curve samples, QC, and unknown sampleswere added to the plate and incubated.
- the standard curve wellswere incubated with diluted TCO-PEG3-biotin, while the unknown samples were incubated with PLCB, followed by a washing step in PBST (PBS, 0.05% Tween- 20).
- Streptavidin-HRP(Jackson ImmunoResearch, #016-030-084) was used for detection, followed by a Super Signal PICO chemiluminescent substrate reaction (ThermoFisher, #37069).
- HRP-conjugated AffiniPure goat anti-human antibody F(ab’) 2fragment-specific (Jackson ImmunoResearch, #209-035-097) was used in all wells (i.e., standard curve, QC, and unknown samples), followed by a chemiluminescent substrate reaction.
- Luminescencewas recorded using a SpectraMax i3x (Molecular Devices) plate reader.
- MSDMesoScale Discovery
- MSD QuickPlex plateswere coated with AffiniPure goat anti-human antibody F(ab’) 2 fragment specific (Jackson ImmunoResearch, #109-005-006), followed by blocking with 1% casein (ThermoFisher, #37582) and a washing step in PBST.
- Standard curves and QC samples for total Fab and intact tetrazinewere prepared in PLCB using twelve points ranging from 50,000 to 50 pg/mL. The prepared samples were added to the plate and incubated.
- FIG.10 and FIG.11show preferential accumulation of Fab-Tz at the tumor as compared to plasma.
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Chemical & Material Sciences (AREA)
- Veterinary Medicine (AREA)
- Medicinal Chemistry (AREA)
- Public Health (AREA)
- General Health & Medical Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Immunology (AREA)
- Cell Biology (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Epidemiology (AREA)
- Organic Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Oncology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
Description
METHODS FOR IN VIVO TARGETED DELIVERY OF A PAYLOAD CROSS REFERENCE TO RELATED APPLICATIONS [0001] This application claims the benefit under 35 U.S.C. §119(e) to U.S. Provisional Application Numbers 63/583,189, filed September 15, 2023, 63/660,911, filed June 17, 2024, and 63/681,755, filed August 9, 2024, both of which are incorporated by reference in their entirety. FIELD [0002] The present disclosure relates generally to methods of utilizing antibody-tetrazine conjugates for bioorthogonal delivery of a payload to a targeted location in a subject, which conjugates have applications, e.g., in the treatment of cancer, tumor growth, and immunotherapy. BACKGROUND [0003] Bioorthogonal conjugation or click reactions are selective and orthogonal (non-interacting with) functionalities found in biological systems, and have found use in various applications in the fields of chemistry, chemical biology, molecular diagnostics, and medicine, where they can be used to facilitate the selective manipulation of molecules, cells, particles and surfaces, and the tagging and tracking of biomolecules in vitro and in vivo. These reactions include the Staudinger ligation, the azide-cyclooctyne cycloaddition, and the inverse-electron-demand Diels-Alder reaction. SUMMARY [0004] The present disclosure is directed to methods for delivering a payload to a target location in a subject. In one aspect, provided is a method of forming in vivo an antibody-payload conjugate in a subject in need thereof, the method comprising: administering an effective amount of a targeting moiety to the subject, wherein the targeting moiety comprising at least one antibody or fragment thereof having a least one tetrazine moiety covalently linked thereto; administering a single dose of a therapeutically effective amount of a payload-TCO conjugate to the subject, wherein the payload-TCO conjugate comprises a payload having at least one trans- cyclooctene (TCO) moiety covalently linked thereto; wherein the antibody or fragment thereof has a binding affinity to a receptor on a tumor, and further wherein the amount of the antibody-payload conjugate formed in vivo is greater at a tumor site versus in plasma. [0005] In one aspect, provided is a method of administering a therapeutically effective amount of a payload to a subject, the method comprising: a) administering an effective amount of a targeting moiety to the subject, wherein the targeting moiety comprises a Fab having at least one tetrazine moiety covalently linked thereto; and b) administering a single dose of a therapeutically effective amount of a payload-TCO conjugate to the subject, wherein the payload-TCO conjugate comprises a payload having at least one (e.g., 1 to 3, 1 to 2, or 1) trans-cyclooctene moiety covalently linked thereto; wherein the single dose of the payload-TCO conjugate is administered to the subject within a time frame of between about 2 hours and 48 hours, or between about 3 hours and 48 hours, or between about 4 and about 48 hours, after the targeting moiety is administered to the subject. [0006] In some embodiments, the targeting moiety comprises a Fab having 1 to 5, 1 to 4, or 1 to 3, tetrazine moieties covalently linked thereto (i.e., the DAR, or ratio of Fab to tetrazine is an average of 5, 4, 3, or 2) [0007] The targeting moieties for use in the methods disclosed herein are designed to, once administered to a subject, localize at a target site within the subject. The targeting moieties can be administered locally or systemically. Once administered, a prodrug comprising a payload and one or more complimentary bioorthogonal components (i.e., a trans-cyclooctene moiety) is administered, which when in contact with the targeting moiety in vivo, allows for targeted delivery of the payload or therapeutic agent. [0008] In one aspect, provided is a method of treating cancer, or enhancing or eliciting an immune response, in a subject having cancer, the method comprising: a) administering an effective amount of a targeting moiety to the subject, wherein the targeting moiety comprises a Fab having at least one (e.g., 1 to 5, 1 to 4, or 1 to 3) tetrazine moiety covalently linked thereto; and b) administering a single dose of a therapeutically effective amount of a payload-TCO conjugate to the subject, wherein the payload-TCO conjugate comprises a payload having at least one (e.g., 1 to 3, 1 to 2, or 1) trans-cyclooctene moiety covalently linked thereto; wherein the single dose of the payload-TCO conjugate is administered to the subject more than 1 hour, and less than 48 hours, after the targeting moiety is administered to the subject. [0009] In one aspect, provided is a method of treating cancer, or enhancing or eliciting an immune response, in a subject having cancer, the method comprising: a) administering an effective amount of a targeting moiety to the subject, wherein the targeting moiety comprises a Fab having at least one (e.g., 1 to 5, 1 to 4, or 1 to 3) tetrazine moiety covalently linked thereto; and b) administering a single dose of a therapeutically effective amount of a payload-TCO conjugate to the subject, wherein the payload-TCO conjugate comprises a payload having at least one (e.g., 1 to 3, 1 to 2, or 1) trans-cyclooctene moiety covalently linked thereto; wherein the single dose of the payload-TCO conjugate is administered to the subject more than 4 hours, and less than 48 hours, after the targeting moiety is administered to the subject. [0010] In some embodiments, the cancer is metastatic. In some embodiments the cancer is melanoma, renal cancer, prostate cancer, ovarian cancer, endometrial carcinoma, breast cancer, glioblastoma, lung cancer, soft tissue sarcoma, fibrosarcoma, osteosarcoma, pancreatic cancer, gastric carcinoma, squamous cell carcinoma of head/neck, anal/vulvar carcinoma, esophageal carcinoma, pancreatic adenocarcinoma, cervical carcinoma, hepatocellular carcinoma, Kaposi’s sarcoma, Non-Hodgkin’s lymphoma, Hodgkin’s lymphoma Wilm’s tumor/neuroblastoma, bladder cancer, thyroid adenocarcinoma, pancreatic neuroendocrine tumors, prostatic adenocarcinoma, nasopharyngeal carcinoma, or cutaneous T-cell lymphoma. [0011] In some embodiments, the cancer is a melanoma, renal cancer, prostate cancer, ovarian cancer, breast cancer, glioma, lung cancer, soft tissue carcinoma, soft tissue sarcoma, osteosarcoma, or pancreatic cancer. In some embodiments, the cancer is a lymphoma or leukemia. In some embodiments, the cancer is a hematologic malignancy. In some embodiments, the cancer is a solid tumor. [0012] In some embodiments, provided is a targeting moiety of Formula IIF: wherein p is 1 to 10; and X is an antibody fragment moiety comprising SEQ ID NO.9 and SEQ ID No.10. In some embodiments, p is 1 to 5. [0013] In one aspect, provided is a method of reducing tumor volume in a subject having a tumor, the method comprising: a) administering an effective amount of a targeting moiety to the subject, wherein the targeting moiety comprises a Fab having at least one (e.g., 1 to 5, 1 to 4, or 1 to 3) tetrazine moiety covalently linked thereto; and b) administering a single dose of a therapeutically effective amount of a payload-TCO conjugate to the subject, wherein the payload-TCO conjugate comprises a payload having at least one (e.g., 1 to 3, 1 to 2, or 1) trans-cyclooctene moiety covalently linked thereto; wherein the single dose of the payload-TCO conjugate is administered to the subject more than 1 hour, and less than 48 hours, after the targeting moiety is administered to the subject. [0014] In one aspect, provided is a method of reducing tumor volume in a subject having a tumor, the method comprising: a) administering an effective amount of a targeting moiety to the subject, wherein the targeting moiety comprises a Fab having at least one (e.g., 1 to 5, 1 to 4, or 1 to 3) tetrazine moiety covalently linked thereto; and b) administering a single dose of a therapeutically effective amount of a payload-TCO conjugate to the subject, wherein the payload-TCO conjugate comprises a payload having at least one (e.g., 1 to 3, 1 to 2, or 1) trans-cyclooctene moiety covalently linked thereto; wherein the single dose of the payload-TCO conjugate is administered to the subject more than 4 hours, and less than 48 hours, after the targeting moiety is administered to the subject. BRIEF DESCRIPTION OF THE FIGURES [0015] FIGs.1A-1D show tumor volumes of NCI-N87 tumors in SCID CB17 mice following treatment with vehicle, Compound TM-1 with Compound B, and Isotype Fab-Tz with Compound B. Compound B was dosed 4 hours (FIG.1A), 8 hours (FIG.1B), 24 hours (FIG.1C), or 48 hours (FIG.1D) after Compound TM-1 dose. N = 5 mice per group. [0016] FIG.2A shows the schedule of dosing and tumor collection. [0017] FIG.2B shows Compound TM-1 activates Compound B to release MMAE in tumors 15 minutes after dosing. [0018] FIG.3A shows quantification of total Fab in HER2-positive tumors. [0019] FIG.3B shows Compound TM-1 localizes to HER2-positive tumors. [0020] FIG.4A shows the total Fab detected at the tumor site vs in the plasma. [0021] FIG.4B shows the total tetrazine detected at the tumor site vs in the plasma. [0022] FIG.5 shows Fab exposure was prolonged in plasma and at the tumor (Tz levels decreased quickly in plasma but were prolonged at the tumor). [0023] FIG.6 shows the total tetrazine detected at the tumor site at certain time intervals. [0024] FIG.7 shows the total Fab detected at the tumor site at certain time intervals. [0025] FIG.8 shows the ratio of tetrazine (Fab-Biotin) at the tumor site vs tissues. [0026] FIG.9 shows the total Fab ratio at the tumor site vs tissues. [0027] FIG.10 and FIG.11 show preferential accumulation of Compound TM-1 at the tumor as compared to plasma DETAILED DESCRIPTION [0028] The following description sets forth exemplary embodiments of the present technology. It should be recognized, however, that such description is not intended as a limitation on the scope of the present disclosure but is instead provided as a description of exemplary embodiments. Definitions [0029] It is appreciated that certain features of the disclosure, which are, for clarity, described in the context of separate embodiments, may also be provided in combination in a single embodiment. Conversely, various features of the disclosure, which are, for brevity, described in the context of a single embodiment, may also be provided separately or in any suitable sub-combination. All combinations of the embodiments pertaining to the disclosure are specifically embraced by the present disclosure and are disclosed herein just as if each and every combination was individually and explicitly disclosed, to the extent that such combinations embrace subject matter that are, for example, compounds that are stable compounds (i.e., compounds that can be made, isolated, characterized, and tested for biological activity). In addition, all sub-combinations of the various embodiments and elements thereof (e.g., elements of the chemical groups listed in the embodiments describing such variables) are also specifically embraced by the present disclosure and are disclosed herein just as if each and every such sub-combination was individually and explicitly disclosed herein. [0030] Unless otherwise defined, all technical and scientific terms used herein have the same meaning as commonly understood by one of ordinary skill in the art. In case of conflict, the present document, including definitions, will control. Exemplary methods and materials are described below, although methods and materials similar or equivalent to those described herein can be used in practice or testing of the present disclosure. All publications, patent applications, patents and other references mentioned herein are incorporated by reference in their entirety. The materials, methods, and examples disclosed herein are illustrative only and not intended to be limiting. [0031] The terms “comprise(s),” “include(s),” “having,” “has,” “can,” “contain(s),” and variants thereof, as used herein, are intended to be open-ended transitional phrases, terms, or words that do not preclude the possibility of additional acts or structures. The singular forms “a,” “an” and “the” include plural references unless the context clearly dictates otherwise. The present disclosure also contemplates other embodiments “comprising,” “consisting of” and “consisting essentially of,” the embodiments or elements presented herein, whether explicitly set forth or not. [0032] The modifier “about” used in connection with a quantity is inclusive of the stated value and has the meaning dictated by the context (for example, it includes at least the degree of error associated with the measurement of the particular quantity). The modifier “about” should also be considered as disclosing the range defined by the absolute values of the two endpoints. For example, the expression “from about 2 to about 4” also discloses the range “from 2 to 4.” The term “about” may refer to plus or minus 10% of the indicated number. For example, “about 10%” may indicate a range of 9% to 11%, and “about 1” may mean from 0.9-1.1. Other meanings of “about” may be apparent from the context, such as rounding off, so, for example “about 1” may also mean from 0.5 to 1.4. [0033] The conjunctive term “or” includes any and all combinations of one or more listed elements associated by the conjunctive term. For example, the phrase “an apparatus comprising A or B” may refer to an apparatus including A where B is not present, an apparatus including B where A is not present, or an apparatus where both A and B are present. The phrases “at least one of A, B, ... and N” or “at least one of A, B, ... N, or combinations thereof” are defined in the broadest sense to mean one or more elements selected from the group comprising A, B, ... and N, that is to say, any combination of one or more of the elements A, B, ... or N including any one element alone or in combination with one or more of the other elements which may also include, in combination, additional elements not listed. [0034] Definitions of specific functional groups and chemical terms are described in more detail below. For purposes of this disclosure, the chemical elements are identified in accordance with the Periodic Table of the Elements, CAS version, Handbook of Chemistry and Physics, 75th Ed., inside cover, and specific functional groups are generally defined as described therein. Additionally, general principles of organic chemistry, as well as specific functional moieties and reactivity, are described in Organic Chemistry, Thomas Sorrell, University Science Books, Sausalito, 1999; Smith and March March’s Advanced Organic Chemistry, 5th Edition, John Wiley & Sons, Inc., New York, 2001; Larock, Comprehensive Organic Transformations, VCH Publishers, Inc., New York, 1989; Carruthers, Some Modern Methods of Organic Synthesis, 3rd Edition, Cambridge University Press, Cambridge, 1987; the entire contents of each of which are incorporated herein by reference. [0035] The term “alkyl” as used herein, means a straight or branched, saturated hydrocarbon chain containing from 1 to 30 carbon atoms. The term “lower alkyl” or “C1-C6-alkyl” means a straight or branched chain hydrocarbon containing from 1 to 6 carbon atoms. The term “C1-C3- alkyl” means a straight or branched chain hydrocarbon containing from 1 to 3 carbon atoms. Representative examples of alkyl include, but are not limited to, methyl, ethyl, n-propyl, iso-propyl, n-butyl, sec-butyl, iso-butyl, tert- butyl, n-pentyl, isopentyl, neopentyl, n-hexyl, 3-methylhexyl, 2,2-dimethylpentyl, 2,3-dimethylpentyl, n- heptyl, n-octyl, n-nonyl, and n-decyl. [0036] The term “alkoxy” as used herein, refers to an alkyl group, as defined herein, appended to the parent molecular moiety through an oxygen atom. Representative examples of alkoxy include, but are not limited to, methoxy, ethoxy, propoxy, 2-propoxy, butoxy, and tert-butoxy. [0037] The term “alkenyl” as used herein, means a hydrocarbon chain containing from 2 to 30 carbon atoms with at least one carbon-carbon double bond. The alkenyl group may be substituted or unsubstituted. For example, the alkenyl group may be substituted with an aryl group, such as a phenyl. [0038] The term “alkynyl,” as used herein, refers to straight or branched monovalent hydrocarbyl groups having from 2 to 30 carbon atoms, such as 2 to 20, or 2 to 10 carbon atoms and having at least 1 site of triple bond unsaturation. The term “alkyne” also includes non-aromatic cycloalkyl groups of from 5 to 20 carbon atoms, such as from 5 to 10 carbon atoms, having single or multiple rings and having at least one triple bond. Examples of such alkynyl groups include, but are not limited to acetylenyl (-C≡CH), and propargyl (-CH2C≡CH), and cycloalkynyl moieties, such as, but not limited to, substituted or unsubstituted cyclooctyne moieties. [0039] The term “alkoxyalkyl” as used herein, refers to an alkoxy group, as defined herein, appended to the parent molecular moiety through an alkyl group, as defined herein. [0040] The term “alkylene” as used herein, refers to a divalent group derived from a straight or branched chain hydrocarbon of 1 to 30 carbon atoms, for example, of 2 to 10 carbon atoms. Representative examples of alkylene include, but are not limited to, -CH2-, -CH(CH3)-, -C(CH3)2-, -CH2CH2-, -CH(CH3)CH2-, -C(CH3)2CH2-, -CH2CH2CH2-, -CH(CH3)CH2CH2-, -C(CH3)2CH2CH2-, -CH2C(CH3)2CH2-, -CH2CH2CH2CH2-, and –CH2CH2CH2CH2CH2-. [0041] The term “amino acid” refers to both natural and unnatural amino acids, protected natural and unnatural amino acids, as well as amino acid analogs and amino acid mimetics that function in a manner similar to the naturally occurring amino acids. Naturally encoded amino acids include 20 common amino acids (alanine, arginine, asparagine, aspartic acid, cysteine, glutamine, glutamic acid, glycine, histidine, isoleucine, leucine, lysine, methionine, phenylalanine, proline, serine, threonine, tryptophan, tyrosine and valine) and pyrrolidine and selenocysteine. Non-natural amino acids refer to amino acid analogs having the same basic chemical structure as a naturally occurring amino acid, i.e., by way of example only, an α- carbon attached to a hydrogen, carboxyl group, amino group, and R group. Such analogs can have a modified R group (e.g., norleucine as an example) or retain a modified peptide backbone while retaining the same basic chemical structure as a natural amino acid. Non-limiting examples of non-natural amino acids or amino acid analogs include citrulline, homoserine, norleucine, methionine sulfoxide, methionine methylsulfonium, homophenylalanine, ornithine, formyl glycine, phenyl glycine, para-azidophenyl glycine, para-azidophenylalanine, para-acetophenylalanine, 4-(3-methyl-(1,2,4,5-tetrazine))- phenylglyine, and 4-(3-methyl-(1,2,4,5-tetrazine))-phenylalanine. [0042] The term “aryl” as used herein, refers to an aromatic carbocyclic group having a single ring (e.g. monocyclic) or multiple rings (e.g. bicyclic or tricyclic) including fused systems. Representative examples of aryls include, but are not limited to, phenyl, naphthyl, and anthracenyl. The monocyclic, bicyclic, and tricyclic aryls are connected to the parent molecular moiety through any carbon atom contained within the rings, and can be unsubstituted or substituted. The aromatic bicyclic ring system or aromatic tricyclic ring system does not contain non-aromatic rings. Thus, if a bicyclic ring system or tricyclic ring system contains a non-aromatic ring, the ring system is a cycloalkyl or heterocyclyl, depending on whether a heteroatom is present in the non-aromatic ring, regardless of the point of attachment to the remainder of the molecule. [0043] In some embodiments, the term “aryl” as used herein, refers to a phenyl group, or bicyclic aryl or tricyclic aryl fused ring systems. Bicyclic fused ring systems are exemplified by a phenyl group appended to the parent molecular moiety and fused to a phenyl group. Tricyclic fused ring systems are exemplified by a phenyl group appended to the parent molecular moiety and fused to two other phenyl groups. Representative examples of bicyclic aryls include, but are not limited to, naphthyl. Representative examples of tricyclic aryls include, but are not limited to, anthracenyl. The monocyclic, bicyclic, and tricyclic aryls are connected to the parent molecular moiety through any carbon atom contained within the rings, and can be unsubstituted or substituted. [0044] The term “azide” as used herein, refers to the functional group –N3. [0045] The term “cycloalkyl” as used herein, refers to a non-aromatic carbocyclic ring system containing 3 to 10, or 3 to 8, or 3 to 6, or 5 to 10, carbon atoms and zero heteroatoms. Cycloalkyl ring systems may contain one or more double bonds, so long as the ring is not aromatic; and thus, the term cycloalkyl includes cycloalkenyl ring systems. Representative examples of cycloalkyl include, but are not limited to, cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl, cyclooctyl, cyclononyl, and cyclodecyl. Exemplary monocyclic cycloalkenyl rings include cyclopentenyl, cyclohexenyl, or cycloheptenyl. “Cycloalkyl” also includes carbocyclic ring systems in which a cycloalkyl group is fused to an aryl or heteroaryl as defined herein, regardless of the point of attachment to the remainder of the molecule. [0046] In some embodiments, the term “cycloalkyl” as used herein, refers to a carbocyclic ring system containing three to ten carbon atoms, zero heteroatoms and zero double bonds. Representative examples of cycloalkyl include, but are not limited to, cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl, cyclooctyl, cyclononyl and cyclodecyl. “Cycloalkyl” also includes carbocyclic ring systems in which a cycloalkyl group is appended to the parent molecular moiety and is fused to an aryl group as defined herein, a heteroaryl group as defined herein, or a heterocycle as defined herein. [0047] The term “cycloalkenyl” as used herein, means a non-aromatic monocyclic or multicyclic ring system containing at least one carbon-carbon double bond, e.g., having from 5-10 carbon atoms per ring. Exemplary monocyclic cycloalkenyl rings include cyclopentenyl, cyclohexenyl or cycloheptenyl. [0048] The term “cyclooctene” as used herein, refers to a substituted or unsubstituted non-aromatic cyclic alkyl group of 8 carbon atoms, having a single ring with a double bond. Examples of such cyclooctene groups include, but are not limited to, substituted or unsubstituted trans-cyclooctene (TCO). [0049] The term “fluoroalkyl” as used herein, means an alkyl group, as defined herein, in which one, two, three, four, five, six, seven or eight hydrogen atoms are replaced by fluorine. Representative examples of fluoroalkyl include, but are not limited to, 2-fluoroethyl, 2,2,2-trifluoroethyl, trifluoromethyl, difluoromethyl, pentafluoroethyl, and trifluoropropyl such as 3,3,3-trifluoropropyl. [0050] The term “alkoxyfluoroalkyl” as used herein, refers to an alkoxy group, as defined herein, appended to the parent molecular moiety through a fluoroalkyl group, as defined herein. [0051] The term “fluoroalkoxy” as used herein, means at least one fluoroalkyl group, as defined herein, is appended to the parent molecular moiety through an oxygen atom. Representative examples of fluoroalkyloxy include, but are not limited to, difluoromethoxy, trifluoromethoxy and 2,2,2- trifluoroethoxy. [0052] The term “halogen” or “halo” as used herein, means Cl, Br, I, or F. [0053] The term “haloalkyl” as used herein, means an alkyl group, as defined herein, in which one, two, three, four, five, six, seven or eight hydrogen atoms are replaced by a halogen. [0054] The term “haloalkoxy” as used herein, means at least one haloalkyl group, as defined herein, is appended to the parent molecular moiety through an oxygen atom. [0055] The term “heteroalkyl” as used herein, means an alkyl group, as defined herein, in which one or more of the carbon atoms has been replaced by a heteroatom selected from S, Si, O, P and N. The heteroatom may be oxidized. Representative examples of heteroalkyls include, but are not limited to, alkyl ethers, secondary and tertiary alkyl amines, and alkyl sulfides. [0056] The term “heteroaryl” as used herein, refers to an aromatic group having a single ring, multiple rings or multiple fused rings, with one or more ring heteroatoms independently selected from nitrogen, oxygen and sulfur. In some embodiments, the term “heteroaryl” as used herein, refers to an aromatic monocyclic ring or an aromatic bicyclic ring system or an aromatic tricyclic ring system. The aromatic monocyclic rings are five or six membered rings containing at least one heteroatom independently selected from the group consisting of N, O and S (e.g.1, 2, 3, or 4 heteroatoms independently selected from O, S, and N). The five membered aromatic monocyclic rings have two double bonds and the six membered aromatic monocyclic rings have three double bonds. Representative examples of monocyclic heteroaryl include, but are not limited to, pyridinyl (including pyridin-2-yl, pyridin-3-yl, pyridin-4-yl), pyrimidinyl, pyrazinyl, thienyl, furyl, thiazolyl, thiadiazolyl, isoxazolyl, pyrazolyl, and 2-oxo-1,2- dihydropyridinyl. Representative examples of bicyclic heteroaryl include, but are not limited to, chromenyl, benzothienyl, benzodioxolyl, benzotriazolyl, quinolinyl, thienopyrrolyl, thienothienyl, imidazothiazolyl, benzothiazolyl, benzofuranyl, indolyl, quinolinyl, imidazopyridine, benzooxadiazolyl, and benzopyrazolyl. Representative examples of tricyclic heteroaryl include, but are not limited to, dibenzofuranyl and dibenzothienyl. The monocyclic, bicyclic, and tricyclic heteroaryls are connected to the parent molecular moiety through any carbon atom or any nitrogen atom contained within the rings, and can be unsubstituted or substituted. In some embodiments, the aromatic bicyclic ring system or aromatic tricyclic ring system does not contain non-aromatic rings. Thus, if a bicyclic ring system or tricyclic ring system contains a non-aromatic ring, the ring system is a cycloalkyl or heterocyclyl, depending on whether a heteroatom is present in the non-aromatic ring, regardless of the point of attachment to the remainder of the molecule. [0057] In some embodiments, the five membered aromatic monocyclic rings have two double bonds and the six membered aromatic monocyclic rings have three double bonds. In some embodiments, exemplary bicyclic heteroaryl groups are exemplified by a monocyclic heteroaryl ring appended to the parent molecular moiety and fused to a monocyclic cycloalkyl group, as defined herein, a monocyclic aryl group, as defined herein, a monocyclic heteroaryl group, as defined herein, or a monocyclic heterocycle, as defined herein. In some embodiments, the tricyclic heteroaryl groups are exemplified by a monocyclic heteroaryl ring appended to the parent molecular moiety and fused to two of a monocyclic cycloalkyl group, as defined herein, a monocyclic aryl group, as defined herein, a monocyclic heteroaryl group, as defined herein, or a monocyclic heterocycle, as defined herein. [0058] The terms “heterocyclyl,” “heterocycle,” or “heterocyclic” as used herein, refers to a non- aromatic ring system containing 3 to 10, or 3 to 8, or 3 to 6, or 5 to 10, carbon atoms and at least one (e.g., 1 to 5, 1 to 4, 1 to 3, 1 to 2, or 1) heteroatom, and optionally one or more oxo and/or double bonds. The terms “heterocyclyl”, “heterocycle” or “heterocyclic” include monocyclic, bicyclic, tricyclic, fused, spirocyclic, or bridged ring systems, provided that at least one non-aromatic ring system containing at least one heteroatom is present. In some embodiments, the monocyclic heterocycle is a three-, four-, five-, six-, seven-, or eight-membered ring containing at least one heteroatom independently selected from the group consisting of O, N, and S. In some embodiments, the three- or four-membered ring contains zero or one double bond, and one heteroatom selected from the group consisting of O, N, and S. In some embodiments, the five-membered ring contains zero or one double bond and one, two or three heteroatoms selected from the group consisting of O, N and S. In some embodiments, the six-membered ring contains zero, one or two double bonds and one, two, or three heteroatoms selected from the group consisting of O, N, and S. In some embodiments, the seven- and eight-membered rings contains zero, one, two, or three double bonds and one, two, or three heteroatoms selected from the group consisting of O, N, and S. Representative examples of monocyclic heterocycles include, but are not limited to, azetidinyl, azepanyl, aziridinyl, diazepanyl, 1,3-dioxanyl, 1,3-dioxolanyl, 1,3-dithiolanyl, 1,3-dithianyl, 1,3-dimethylpyrimidine-2,4(1H,3H)-dione, imidazolinyl, imidazolidinyl, isothiazolinyl, isothiazolidinyl, isoxazolinyl, isoxazolidinyl, morpholinyl, oxadiazolinyl, oxadiazolidinyl, oxazolinyl, oxazolidinyl, oxetanyl, piperazinyl, piperidinyl, pyranyl, pyrazolinyl, pyrazolidinyl, pyrrolinyl, pyrrolidinyl, tetrahydrofuranyl, tetrahydropyranyl, tetrahydropyridinyl, tetrahydrothienyl, thiadiazolinyl, thiadiazolidinyl, 1,2-thiazinanyl, 1,3-thiazinanyl, thiazolinyl, thiazolidinyl, thiomorpholinyl, 1,1- dioxidothiomorpholinyl (thiomorpholine sulfone), thiopyranyl, and trithianyl. The bicyclic heterocycle is a monocyclic heterocycle fused to a phenyl group, or a monocyclic heterocycle fused to a monocyclic cycloalkyl, or a monocyclic heterocycle fused to a monocyclic cycloalkenyl, or a monocyclic heterocycle fused to a monocyclic heterocycle, or a spiro heterocycle group, or a bridged monocyclic heterocycle ring system in which two non-adjacent atoms of the ring are linked by an alkylene bridge of 1, 2, 3, or 4 carbon atoms, or an alkenylene bridge of two, three, or four carbon atoms. Representative examples of bicyclic heterocycles include, but are not limited to, benzopyranyl, benzothiopyranyl, chromanyl, 2,3- dihydrobenzofuranyl, 2,3-dihydrobenzothienyl, 2,3-dihydroisoquinoline, 2-azaspiro[3.3]heptan-2-yl, azabicyclo[2.2.1]heptyl (including 2-azabicyclo[2.2.1]hept-2-yl), 2,3-dihydro-1H-indolyl, isoindolinyl, octahydrocyclopenta[c]pyrrolyl, octahydropyrrolopyridinyl, and tetrahydroisoquinolinyl. Tricyclic heterocycles are exemplified by a bicyclic heterocycle fused to a phenyl group, or a bicyclic heterocycle fused to a monocyclic cycloalkyl, or a bicyclic heterocycle fused to a monocyclic cycloalkenyl, or a bicyclic heterocycle fused to a monocyclic heterocycle, or a bicyclic heterocycle in which two non- adjacent atoms of the bicyclic ring are linked by an alkylene bridge of 1, 2, 3, or 4 carbon atoms, or an alkenylene bridge of two, three, or four carbon atoms. Examples of tricyclic heterocycles include, but are not limited to, octahydro-2,5-epoxypentalene, hexahydro-2H-2,5-methanocyclopenta[b]furan, hexahydro-1H-1,4-methanocyclopenta[c]furan, aza-adamantane (1-azatricyclo[3.3.1.13,7]decane), and oxa-adamantane (2-oxatricyclo[3.3.1.13,7]decane). The monocyclic, bicyclic, and tricyclic heterocycles are connected to the parent molecular moiety through any carbon atom or any nitrogen atom contained within the rings, and can be unsubstituted or substituted. [0059] The term “hydroxyl” as used herein, means an –OH group. [0060] The term “hydroxyalkyl” as used herein, means an alkyl group, as defined herein, in which one, two, three, four, five, six, seven or eight hydrogen atoms are replaced by a hydroxyl group. [0061] In some instances, the number of carbon atoms in a hydrocarbyl substituent (e.g., alkyl or cycloalkyl) is indicated by the prefix “Cx-Cy-” or “Cx-y,” wherein x is the minimum and y is the maximum number of carbon atoms in the substituent. Thus, for example, “C1-C3-alkyl” and “C1-3alkyl” refer to an alkyl substituent containing from 1 to 3 carbon atoms. The two conventions “Cx-Cy-” and “Cx-y” are used interchangeably and have the same meaning. [0062] The term “substituted” refers to a group that may be further substituted with one or more non- hydrogen substituent groups. Substituent groups include, but are not limited to, halogen, =O, =S, cyano, nitro, fluoroalkyl, alkoxyfluoroalkyl, fluoroalkoxy, alkyl, alkenyl, alkynyl, haloalkyl, haloalkoxy, heteroalkyl, cycloalkyl, cycloalkenyl, aryl, heteroaryl, heterocycle, cycloalkylalkyl, heteroarylalkyl, arylalkyl, hydroxy, hydroxyalkyl, alkoxy, alkoxyalkyl, alkylene, aryloxy, phenoxy, benzyloxy, amino, alkylamino, acylamino, aminoalkyl, arylamino, sulfonylamino, sulfinylamino, sulfonyl, alkylsulfonyl, arylsulfonyl, aminosulfonyl, sulfinyl, -COOH, ketone, amide, carbamate, and acyl. [0063] The term “tetrazine” refers to a substituted or unsubstituted aromatic cyclic group of 2 carbon atoms and 4 nitrogen atoms, having a single ring with three double bonds. Examples of tetrazine groups include 1,2,3,4-tetrazine and 1,2,4,5-tetrazine. As used herein, 1,2,4,5-tetrazine is referred to as a “Tz” group. [0064] The term “selectively delivering” refers to delivering an agent (e.g., a payload) to an organ or tissue (or portion thereof) in need of treatment or diagnosis, without significant binding to other non- target organs or tissues (or portions thereof). In some embodiments, the targeting moieties, or therapeutic targeting moiety, described herein do not themselves have a therapeutic effect, but rather are designed to allow the selective or targeted delivery of a therapeutic agent. However, it may be that the targeting moiety does have a therapeutic effect, and thus, such constructs are not excluded by the present disclosure. [0065] The term “payload” refers to an agent for delivery to a target site in a subject. In some embodiments, the payloads is a therapeutic agent. In some embodiments, the payloads is a diagnostic agent. [0066] The term “therapeutic agent” refers to an agent capable of treating and/or ameliorating a condition or disease, or one or more symptoms thereof, in a subject. Therapeutic agents of the present disclosure also include prodrug forms of therapeutic agents, chelating agent with or without a radionuclide (e.g., diagnostic or therapeutic). [0067] A therapeutic “radionuclide” or “radioligand” as used herein refers to a radioactive substance, sometimes referred to as a radiopharmaceutical, which are used to treat medical conditions, particularly cancer. Radionuclides used in the trans-cyclooctene moieties described herein, comprise a chelating agent and an isotope; such as an isotope selected from the group consisting of24Na,32P,33P,47Sc,59Fe,67Cu,76As,77As,80Br,82Br,89Sr,90Nb,90Y,103Ru,105Rh,109Pd,111Ag,111In,121Sn,127Te,131I,140La,141Ce,142Pr,143Pr,144Pr,149Pm,149Tb,151Pm,153Sm,159Gd,161Tb,165Dy,166Ho,169Er,172Tm,175Yb,177Lu,186Re,188Re,198Au,199Au,211At,211Bi,212Bi,212Pb,213Bi,214Bi,223Ra, and225Ac. Radionuclides can be delivered via direct conjugation or chelation with a chelating agent. Exemplary radionuclides, chelating agents, and linkers for potential TCO-conjugate payloads are described below. [0068] The term “diagnostic agent” refers to agents that assist in diagnosing conditions or diseases. Representative diagnostic agents include imaging agents such as paramagnetic agents, optical probes, radionuclides, and the like. Paramagnetic agents are imaging agents that are magnetic under an externally applied field. Examples of paramagnetic agents include, but are not limited to, iron particles including iron nanoparticles and iron microparticles. Optical probes are fluorescent compounds that can be detected by excitation at one wavelength of radiation and detection at a second, different, wavelength of radiation. Optical probes of the present disclosure include, but are not limited to, Cy5.5, Alexa 680, Cy5, DiD (1,1’-dioctadecyl-3,3,3’,3’-tetramethylindodicarbocyanine perchlorate) and DiR (1,1’- dioctadecyl-3,3,3’,3’-tetramethylindotricarbocyanine iodide). Other optical probes include quantum dots. Radionuclides are elements that undergo detectable radioactive decay. Radionuclides, such as diagnostic radionuclides, useful in embodiments of the present disclosure include, but are not limited to,3H,11C,13N,18F,19F,60Co,64Cu,67Cu,68Ga,82Rb,89Zr,90Sr,90Y,99Tc,99mTc,111In,123I,124I,125I,129I,131I,137Cs,177Lu,186Re,188Re,211At,212Pb,225Ac, Rn, Ra, Th, U, Pu, and241Am. [0069] The term “targeting agent” refers to a chemical or biological agent that specifically binds to a target (e.g., a targeted organ or tissue), thereby forming a stable association between the targeting agent and the specific target. By “stably associated” or “stable association” is meant that a moiety is bound to or otherwise associated with another moiety or structure under standard physiological conditions. Bonds may include covalent bonds and non-covalent interactions, such as, but not limited to, ionic bonds, hydrophobic interactions, hydrogen bonds, van der Waals forces (e.g., London dispersion forces), dipole- dipole interactions, and the like. Targeting agents include ligands that specifically bind (or substantially specifically bind) a particular clinically-relevant target receptor or cell surface target. [0070] The term “antibody fragment moiety” as used herein refers to one or more regions or fragments of an antibody that retain the ability to specifically bind to an antigen, or in other words, substantially retain the antigen-binding function of the antibody. Examples of binding fragments encompassed within the antigen-binding portion of an antibody include but are not limited to a Fab fragment. In some embodiments, the Fab fragment is a monovalent fragment containing at least the VL and VH. In some embodiments, the Fab fragment is a monovalent fragment containing the VL, VH, CL, and CH1 domains. Fab fragments can be obtained using conventional techniques known to those with skill in the art, and the fragments are screened for utility in the same manner as are intact antibodies. For example, antibody fragments can be produced by recombinant DNA techniques, or by enzymatic or chemical cleavage of intact immunoglobulins. Additional examples are included in various embodiments disclosed herein. [0071] By “specifically bind” is meant the preferential association of an antibody or antibody fragment moiety to a target molecule (e.g., an antigen, such as a peptide, polypeptide, glycoprotein, or any other moiety with one or more antigenic determinants) or to a cell or tissue bearing the target molecule (e.g., a cell surface antigen, such as a receptor or ligand) and not to cells or tissues lacking the target molecule. It is recognized that a certain degree of non-specific interaction may occur between a binding moiety and a non-targeted molecule (present alone or in combination with a cell or tissue). Nevertheless, specific binding may be distinguished as mediated through specific recognition of the target antigen. Specific binding results in a much stronger association between the targeting moiety (e.g., Fab) and e.g., cells bearing the target molecule (e.g., an antigen) than between the binding moiety and e.g., cells lacking the target molecule. Specific binding typically results in greater than 2-fold, greater than 5-fold, greater than 10-fold, or greater than 100-fold increase in amount of bound binding moiety (per unit time) to e.g., a cell or tissue bearing the target molecule or marker as compared to a cell or tissue lacking that target molecule or marker. In some embodiments, binding moieties bind to the target molecule or marker with a dissociation constant of e.g., less than 10-5M, less than 10-7M, less than 10-8M, less than 10-9M, less than 10-10M, less than 10-11M, less than 10-12M, less than 10-13M, less than 10-14M, or less than 10-15M. A variety of assay formats are appropriate for measuring binding, such as solid-phase ELISA immunoassays. [0072] The term “targeted organ or tissue” refers to an organ or tissue that is being targeted for delivery of the payload. Representative organs and tissues for targeting include those that can be targeted by chemical or biological targeting agents, as well as those organs and tissues that cannot be targeted by chemical or biological targeting agents. [0073] The term “contacting” or “contact” refers to the process of bringing into contact at least two distinct species such that they can interact with each other, such as in a non-covalent or covalent binding interaction or binding reaction. It should be appreciated, however, the resulting complex or reaction product can be produced directly from an interaction or a reaction between the added reagents or from an intermediate from one or more of the added reagents or moieties, which can be produced in the contacting mixture. [0074] The term “administering” refers to any suitable route of administration to a subject, such as, but not limited to, oral administration, administration as a suppository, topical contact, parenteral, intravenous, intraperitoneal, intramuscular, intralesional, intranasal or subcutaneous administration, intrathecal administration, or the implantation of a slow-release device, e.g., a mini-osmotic pump, to the subject. [0075] The term “parenterally,” as used herein, refers to modes of administration which include intravenous, intramuscular, intraperitoneal, intrasternal, subcutaneous and intraarticular injection, and infusion. [0076] The term “pharmaceutically effective amount” and “therapeutically effective amount” refer to an amount of a compound sufficient to treat a specified disorder or disease or one or more of its symptoms and/or to prevent or reduce the risk of the occurrence or reoccurrence of the disease or disorder or symptom(s) thereof. In reference to tumorigenic proliferative disorders, a pharmaceutically or therapeutically effective amount comprises an amount sufficient to, among other things, cause the tumor to shrink or decrease the growth rate of the tumor. [0077] As used herein, the term “subject,” “patient,” or “organism” includes humans and mammals (e.g., mice, rats, pigs, cats, dogs, and horses). Typical subjects to which an agent(s) of the present disclosure may be administered may include mammals, particularly primates, especially humans. For veterinary applications, suitable subjects may include, for example, livestock such as cattle, sheep, goats, cows, swine, and the like; poultry such as chickens, ducks, geese, turkeys, and the like; and domesticated animals particularly pets such as dogs and cats. For diagnostic or research applications, suitable subjects may include mammals, such as rodents (e.g., mice, rats, hamsters), rabbits, primates, and swine such as inbred pigs and the like. [0078] The term “treating” or “treatment” as used herein means the treating or treatment of a disease or medical condition or symptom(s) thereof in a patient, such as a mammal (e.g., a human) that includes: (a) ameliorating the disease or medical condition or symptom(s) thereof, such as, eliminating or causing regression of the disease or medical condition or symptom(s) thereof in a patient; (b) suppressing the disease or medical condition or symptom(s) thereof, for example by, slowing or arresting the development of the disease or medical condition or symptom(s) thereof in a patient; or (c) alleviating a symptom of the disease or medical condition or symptom(s) thereof in a patient. [0079] The term “physiological conditions” is meant to encompass those conditions compatible with living cells, e.g., predominantly aqueous conditions of a temperature, pH, salinity, etc. that are compatible with living cells. [0080] For compounds described herein, groups and substituents thereof may be selected in accordance with permitted valence of the atoms and the substituents, such that the selections and substitutions result in a stable compound, e.g., which does not spontaneously undergo transformation such as by rearrangement, cyclization, elimination, etc. [0081] Where a range of values is provided, it is understood that each intervening value, to the tenth of the unit of the lower limit unless the context clearly dictates otherwise, between the upper and lower limit of that range and any other stated or intervening value in that stated range, is encompassed within the disclosure. The upper and lower limits of these smaller ranges may independently be included in the smaller ranges, and are also encompassed within the disclosure, subject to any specifically excluded limit in the stated range. Where the stated range includes one or both of the limits, ranges excluding either or both of those included limits are also included in the disclosure. [0082] For the recitation of numeric ranges herein, each intervening number there between with the same degree of precision is explicitly contemplated. For example, for the range of 6-9, the numbers 7 and 8 are contemplated in addition to 6 and 9, and for the range 6.0-7.0, the number 6.0, 6.1, 6.2, 6.3, 6.4, 6.5, 6.6, 6.7, 6.8, 6.9, and 7.0 are explicitly contemplated. [0083] The compounds may exist as stereoisomers wherein asymmetric or chiral centers are present. The stereoisomers are “R” or “S” depending on the configuration of substituents around the chiral carbon atom. The terms “R” and “S” used herein are configurations as defined in IUPAC 1974 Recommendations for Section E, Fundamental Stereochemistry, in Pure Appl. Chem., 1976, 45: 13-30. The disclosure contemplates various stereoisomers and mixtures thereof, and these are specifically included within the scope of this disclosure. Stereoisomers include enantiomers and diastereomers and mixtures of enantiomers or diastereomers. Individual stereoisomers of the compounds may be prepared synthetically from commercially available starting materials, which contain asymmetric or chiral centers or by preparation of racemic mixtures followed by methods of resolution well-known to those of ordinary skill in the art. These methods of resolution are exemplified by (1) attachment of a mixture of enantiomers to a chiral auxiliary, separation of the resulting mixture of diastereomers by recrystallization or chromatography, and optional liberation of the optically pure product from the auxiliary as described in Furniss, Hannaford, Smith, and Tatchell, “Vogel’s Textbook of Practical Organic Chemistry,” 5th edition (1989), Longman Scientific & Technical, Essex CM202JE, England, or (2) direct separation of the mixture of optical enantiomers on chiral chromatographic columns, or (3) fractional recrystallization methods. [0084] It should be understood that the compounds may possess tautomeric forms as well as geometric isomers, and that these also constitute an aspect of the disclosure. [0085] The present disclosure also includes isotopically-labeled compounds, which are identical to those recited herein, but for the fact that one or more atoms are replaced by an atom having an atomic mass or mass number different from the atomic mass or mass number usually found in nature. Examples of isotopes suitable for inclusion in the compounds of the disclosure are hydrogen, carbon, nitrogen, oxygen, phosphorus, sulfur, fluorine, and chlorine, such as, but not limited to,2H,3H,13C,14C,15N,18O,17O,31P,32P,35S,18F, and36Cl, respectively. Substitution with heavier isotopes such as deuterium, i.e.,2H, can afford certain therapeutic advantages resulting from greater metabolic stability, for example increased in vivo half-life or reduced dosage requirements, and, hence, may be preferred in some circumstances. The compound may incorporate positron-emitting isotopes for medical imaging and positron-emitting tomography (PET) studies for determining the distribution of receptors. Suitable positron-emitting isotopes that can be incorporated are11C,13N,15O, and18F. Isotopically-labeled compounds disclosed herein can generally be prepared by conventional techniques known to those skilled in the art or by processes analogous to those described in the accompanying Examples using appropriate isotopically-labeled reagent in place of non-isotopically-labeled reagent. Methods of Treatment [0086] The present disclosure is directed to methods for delivering a payload to a target location in a subject. [0087] In one aspect, provided is a method of forming in vivo an antibody-payload conjugate in a subject in need thereof, the method comprising: administering an effective amount of a targeting moiety to the subject, wherein the targeting moiety comprising at least one antibody or fragment thereof having a least one tetrazine moiety covalently linked thereto; administering a single dose of a therapeutically effective amount of a payload-TCO conjugate to the subject, wherein the payload-TCO conjugate comprises a payload having at least one trans- cyclooctene (TCO) moiety covalently linked thereto; wherein the antibody or fragment thereof has a binding affinity to a receptor on a tumor, and further wherein the amount of the antibody-payload conjugate formed in vivo is greater at a tumor site versus in plasma. [0088] In one aspect, provided is a method of forming in vivo an antibody-payload conjugate in a subject in need thereof, the method comprising: administering an effective amount of a targeting moiety to the subject, wherein the targeting moiety comprising at least one antibody or fragment thereof having a least one tetrazine moiety covalently linked thereto; administering a single dose of a therapeutically effective amount of a payload-TCO conjugate to the subject, wherein the payload-TCO conjugate comprises a payload having at least one trans- cyclooctene (TCO) moiety covalently linked thereto; wherein the antibody or fragment thereof has a binding affinity to a receptor on a tumor, and further wherein the ratio of the antibody-payload conjugate at a tumor site versus in plasma is greater than about 2:1. [0089] In some embodiments, the ratio of the antibody-payload conjugate at a tumor site versus in plasma is greater than 1:1, or about 2:1, or about 3:1, or about 4:1, or about 5:1, or about 6:1, or about 7:1, or about 8:1, or about 9:1, or about 10:1, or about 11:1, about 12:1, or about 13:1, or about 14:1, or about 15:1, or about 16:1, or about 17:1, or about 18:1, or about 19:1. [0090] In some embodiments, the ratio of the antibody-payload conjugate at a tumor site versus in plasma is greater than about 2:1, or greater than about 3:1, or greater than about 4:1, or greater than about 5:1, or greater than about 6:1, or greater than about 7:1, or greater than about 8:1, or greater than about 9:1, or greater than about 10:1. [0091] In some embodiments, the administering is simultaneous. [0092] In some embodiments, the administering is sequential. In some embodiments, the targeting moiety is administered prior to the payload-TCO conjugate. [0093] In some embodiments, a first dose of the payload-TCO conjugate is administered to the subject less than 48 hours, or more than 4 hours and less than 48 hours, after the targeting moiety is administered to the subject. [0094] In some embodiments, a single dose of the payload-TCO conjugate is administered to the subject less than 48 hours, or more than 4 hours and less than 48 hours, after the targeting moiety is administered to the subject. [0095] In some embodiments, a single dose of the payload-TCO conjugate is administered to the subject less than 48 hours after the targeting moiety is administered to the subject. [0096] In some embodiments, a single dose of the payload-TCO conjugate is administered to the subject more than 4 hours, and less than 48 hours, after the targeting moiety is administered to the subject. [0097] In some embodiments, the targeting moiety is administered at least 8 to about 24 hours prior to the payload-TCO conjugate being administered. [0098] In some embodiments, the targeting moiety is administered within 0-8 serum half-lives. It is contemplated that timing of the payload-TCO conjugate administration is calculated based on tumor disposition, which can be determined by the biology of the antigen at the tumor and the targeting moiety. For example, in some embodiments, when the targeting moiety comprises a targeting format as in the table below, the payload-TCO conjugate is administered within the serum half life range shown therein (see also Berland et al., Biomolecules 2021, 11(5), 637).
wherein p is 1 to 10; and X is an antibody fragment moiety comprising SEQ ID NO.9 and SEQ ID No.10. In some embodiments, p is 1 to 5. [0013] In one aspect, provided is a method of reducing tumor volume in a subject having a tumor, the method comprising: a) administering an effective amount of a targeting moiety to the subject, wherein the targeting moiety comprises a Fab having at least one (e.g., 1 to 5, 1 to 4, or 1 to 3) tetrazine moiety covalently linked thereto; and b) administering a single dose of a therapeutically effective amount of a payload-TCO conjugate to the subject, wherein the payload-TCO conjugate comprises a payload having at least one (e.g., 1 to 3, 1 to 2, or 1) trans-cyclooctene moiety covalently linked thereto; wherein the single dose of the payload-TCO conjugate is administered to the subject more than 1 hour, and less than 48 hours, after the targeting moiety is administered to the subject. [0014] In one aspect, provided is a method of reducing tumor volume in a subject having a tumor, the method comprising: a) administering an effective amount of a targeting moiety to the subject, wherein the targeting moiety comprises a Fab having at least one (e.g., 1 to 5, 1 to 4, or 1 to 3) tetrazine moiety covalently linked thereto; and b) administering a single dose of a therapeutically effective amount of a payload-TCO conjugate to the subject, wherein the payload-TCO conjugate comprises a payload having at least one (e.g., 1 to 3, 1 to 2, or 1) trans-cyclooctene moiety covalently linked thereto; wherein the single dose of the payload-TCO conjugate is administered to the subject more than 4 hours, and less than 48 hours, after the targeting moiety is administered to the subject. BRIEF DESCRIPTION OF THE FIGURES [0015] FIGs.1A-1D show tumor volumes of NCI-N87 tumors in SCID CB17 mice following treatment with vehicle, Compound TM-1 with Compound B, and Isotype Fab-Tz with Compound B. Compound B was dosed 4 hours (FIG.1A), 8 hours (FIG.1B), 24 hours (FIG.1C), or 48 hours (FIG.1D) after Compound TM-1 dose. N = 5 mice per group. [0016] FIG.2A shows the schedule of dosing and tumor collection. [0017] FIG.2B shows Compound TM-1 activates Compound B to release MMAE in tumors 15 minutes after dosing. [0018] FIG.3A shows quantification of total Fab in HER2-positive tumors. [0019] FIG.3B shows Compound TM-1 localizes to HER2-positive tumors. [0020] FIG.4A shows the total Fab detected at the tumor site vs in the plasma. [0021] FIG.4B shows the total tetrazine detected at the tumor site vs in the plasma. [0022] FIG.5 shows Fab exposure was prolonged in plasma and at the tumor (Tz levels decreased quickly in plasma but were prolonged at the tumor). [0023] FIG.6 shows the total tetrazine detected at the tumor site at certain time intervals. [0024] FIG.7 shows the total Fab detected at the tumor site at certain time intervals. [0025] FIG.8 shows the ratio of tetrazine (Fab-Biotin) at the tumor site vs tissues. [0026] FIG.9 shows the total Fab ratio at the tumor site vs tissues. [0027] FIG.10 and FIG.11 show preferential accumulation of Compound TM-1 at the tumor as compared to plasma DETAILED DESCRIPTION [0028] The following description sets forth exemplary embodiments of the present technology. It should be recognized, however, that such description is not intended as a limitation on the scope of the present disclosure but is instead provided as a description of exemplary embodiments. Definitions [0029] It is appreciated that certain features of the disclosure, which are, for clarity, described in the context of separate embodiments, may also be provided in combination in a single embodiment. Conversely, various features of the disclosure, which are, for brevity, described in the context of a single embodiment, may also be provided separately or in any suitable sub-combination. All combinations of the embodiments pertaining to the disclosure are specifically embraced by the present disclosure and are disclosed herein just as if each and every combination was individually and explicitly disclosed, to the extent that such combinations embrace subject matter that are, for example, compounds that are stable compounds (i.e., compounds that can be made, isolated, characterized, and tested for biological activity). In addition, all sub-combinations of the various embodiments and elements thereof (e.g., elements of the chemical groups listed in the embodiments describing such variables) are also specifically embraced by the present disclosure and are disclosed herein just as if each and every such sub-combination was individually and explicitly disclosed herein. [0030] Unless otherwise defined, all technical and scientific terms used herein have the same meaning as commonly understood by one of ordinary skill in the art. In case of conflict, the present document, including definitions, will control. Exemplary methods and materials are described below, although methods and materials similar or equivalent to those described herein can be used in practice or testing of the present disclosure. All publications, patent applications, patents and other references mentioned herein are incorporated by reference in their entirety. The materials, methods, and examples disclosed herein are illustrative only and not intended to be limiting. [0031] The terms “comprise(s),” “include(s),” “having,” “has,” “can,” “contain(s),” and variants thereof, as used herein, are intended to be open-ended transitional phrases, terms, or words that do not preclude the possibility of additional acts or structures. The singular forms “a,” “an” and “the” include plural references unless the context clearly dictates otherwise. The present disclosure also contemplates other embodiments “comprising,” “consisting of” and “consisting essentially of,” the embodiments or elements presented herein, whether explicitly set forth or not. [0032] The modifier “about” used in connection with a quantity is inclusive of the stated value and has the meaning dictated by the context (for example, it includes at least the degree of error associated with the measurement of the particular quantity). The modifier “about” should also be considered as disclosing the range defined by the absolute values of the two endpoints. For example, the expression “from about 2 to about 4” also discloses the range “from 2 to 4.” The term “about” may refer to plus or minus 10% of the indicated number. For example, “about 10%” may indicate a range of 9% to 11%, and “about 1” may mean from 0.9-1.1. Other meanings of “about” may be apparent from the context, such as rounding off, so, for example “about 1” may also mean from 0.5 to 1.4. [0033] The conjunctive term “or” includes any and all combinations of one or more listed elements associated by the conjunctive term. For example, the phrase “an apparatus comprising A or B” may refer to an apparatus including A where B is not present, an apparatus including B where A is not present, or an apparatus where both A and B are present. The phrases “at least one of A, B, ... and N” or “at least one of A, B, ... N, or combinations thereof” are defined in the broadest sense to mean one or more elements selected from the group comprising A, B, ... and N, that is to say, any combination of one or more of the elements A, B, ... or N including any one element alone or in combination with one or more of the other elements which may also include, in combination, additional elements not listed. [0034] Definitions of specific functional groups and chemical terms are described in more detail below. For purposes of this disclosure, the chemical elements are identified in accordance with the Periodic Table of the Elements, CAS version, Handbook of Chemistry and Physics, 75th Ed., inside cover, and specific functional groups are generally defined as described therein. Additionally, general principles of organic chemistry, as well as specific functional moieties and reactivity, are described in Organic Chemistry, Thomas Sorrell, University Science Books, Sausalito, 1999; Smith and March March’s Advanced Organic Chemistry, 5th Edition, John Wiley & Sons, Inc., New York, 2001; Larock, Comprehensive Organic Transformations, VCH Publishers, Inc., New York, 1989; Carruthers, Some Modern Methods of Organic Synthesis, 3rd Edition, Cambridge University Press, Cambridge, 1987; the entire contents of each of which are incorporated herein by reference. [0035] The term “alkyl” as used herein, means a straight or branched, saturated hydrocarbon chain containing from 1 to 30 carbon atoms. The term “lower alkyl” or “C1-C6-alkyl” means a straight or branched chain hydrocarbon containing from 1 to 6 carbon atoms. The term “C1-C3- alkyl” means a straight or branched chain hydrocarbon containing from 1 to 3 carbon atoms. Representative examples of alkyl include, but are not limited to, methyl, ethyl, n-propyl, iso-propyl, n-butyl, sec-butyl, iso-butyl, tert- butyl, n-pentyl, isopentyl, neopentyl, n-hexyl, 3-methylhexyl, 2,2-dimethylpentyl, 2,3-dimethylpentyl, n- heptyl, n-octyl, n-nonyl, and n-decyl. [0036] The term “alkoxy” as used herein, refers to an alkyl group, as defined herein, appended to the parent molecular moiety through an oxygen atom. Representative examples of alkoxy include, but are not limited to, methoxy, ethoxy, propoxy, 2-propoxy, butoxy, and tert-butoxy. [0037] The term “alkenyl” as used herein, means a hydrocarbon chain containing from 2 to 30 carbon atoms with at least one carbon-carbon double bond. The alkenyl group may be substituted or unsubstituted. For example, the alkenyl group may be substituted with an aryl group, such as a phenyl. [0038] The term “alkynyl,” as used herein, refers to straight or branched monovalent hydrocarbyl groups having from 2 to 30 carbon atoms, such as 2 to 20, or 2 to 10 carbon atoms and having at least 1 site of triple bond unsaturation. The term “alkyne” also includes non-aromatic cycloalkyl groups of from 5 to 20 carbon atoms, such as from 5 to 10 carbon atoms, having single or multiple rings and having at least one triple bond. Examples of such alkynyl groups include, but are not limited to acetylenyl (-C≡CH), and propargyl (-CH2C≡CH), and cycloalkynyl moieties, such as, but not limited to, substituted or unsubstituted cyclooctyne moieties. [0039] The term “alkoxyalkyl” as used herein, refers to an alkoxy group, as defined herein, appended to the parent molecular moiety through an alkyl group, as defined herein. [0040] The term “alkylene” as used herein, refers to a divalent group derived from a straight or branched chain hydrocarbon of 1 to 30 carbon atoms, for example, of 2 to 10 carbon atoms. Representative examples of alkylene include, but are not limited to, -CH2-, -CH(CH3)-, -C(CH3)2-, -CH2CH2-, -CH(CH3)CH2-, -C(CH3)2CH2-, -CH2CH2CH2-, -CH(CH3)CH2CH2-, -C(CH3)2CH2CH2-, -CH2C(CH3)2CH2-, -CH2CH2CH2CH2-, and –CH2CH2CH2CH2CH2-. [0041] The term “amino acid” refers to both natural and unnatural amino acids, protected natural and unnatural amino acids, as well as amino acid analogs and amino acid mimetics that function in a manner similar to the naturally occurring amino acids. Naturally encoded amino acids include 20 common amino acids (alanine, arginine, asparagine, aspartic acid, cysteine, glutamine, glutamic acid, glycine, histidine, isoleucine, leucine, lysine, methionine, phenylalanine, proline, serine, threonine, tryptophan, tyrosine and valine) and pyrrolidine and selenocysteine. Non-natural amino acids refer to amino acid analogs having the same basic chemical structure as a naturally occurring amino acid, i.e., by way of example only, an α- carbon attached to a hydrogen, carboxyl group, amino group, and R group. Such analogs can have a modified R group (e.g., norleucine as an example) or retain a modified peptide backbone while retaining the same basic chemical structure as a natural amino acid. Non-limiting examples of non-natural amino acids or amino acid analogs include citrulline, homoserine, norleucine, methionine sulfoxide, methionine methylsulfonium, homophenylalanine, ornithine, formyl glycine, phenyl glycine, para-azidophenyl glycine, para-azidophenylalanine, para-acetophenylalanine, 4-(3-methyl-(1,2,4,5-tetrazine))- phenylglyine, and 4-(3-methyl-(1,2,4,5-tetrazine))-phenylalanine. [0042] The term “aryl” as used herein, refers to an aromatic carbocyclic group having a single ring (e.g. monocyclic) or multiple rings (e.g. bicyclic or tricyclic) including fused systems. Representative examples of aryls include, but are not limited to, phenyl, naphthyl, and anthracenyl. The monocyclic, bicyclic, and tricyclic aryls are connected to the parent molecular moiety through any carbon atom contained within the rings, and can be unsubstituted or substituted. The aromatic bicyclic ring system or aromatic tricyclic ring system does not contain non-aromatic rings. Thus, if a bicyclic ring system or tricyclic ring system contains a non-aromatic ring, the ring system is a cycloalkyl or heterocyclyl, depending on whether a heteroatom is present in the non-aromatic ring, regardless of the point of attachment to the remainder of the molecule. [0043] In some embodiments, the term “aryl” as used herein, refers to a phenyl group, or bicyclic aryl or tricyclic aryl fused ring systems. Bicyclic fused ring systems are exemplified by a phenyl group appended to the parent molecular moiety and fused to a phenyl group. Tricyclic fused ring systems are exemplified by a phenyl group appended to the parent molecular moiety and fused to two other phenyl groups. Representative examples of bicyclic aryls include, but are not limited to, naphthyl. Representative examples of tricyclic aryls include, but are not limited to, anthracenyl. The monocyclic, bicyclic, and tricyclic aryls are connected to the parent molecular moiety through any carbon atom contained within the rings, and can be unsubstituted or substituted. [0044] The term “azide” as used herein, refers to the functional group –N3. [0045] The term “cycloalkyl” as used herein, refers to a non-aromatic carbocyclic ring system containing 3 to 10, or 3 to 8, or 3 to 6, or 5 to 10, carbon atoms and zero heteroatoms. Cycloalkyl ring systems may contain one or more double bonds, so long as the ring is not aromatic; and thus, the term cycloalkyl includes cycloalkenyl ring systems. Representative examples of cycloalkyl include, but are not limited to, cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl, cyclooctyl, cyclononyl, and cyclodecyl. Exemplary monocyclic cycloalkenyl rings include cyclopentenyl, cyclohexenyl, or cycloheptenyl. “Cycloalkyl” also includes carbocyclic ring systems in which a cycloalkyl group is fused to an aryl or heteroaryl as defined herein, regardless of the point of attachment to the remainder of the molecule. [0046] In some embodiments, the term “cycloalkyl” as used herein, refers to a carbocyclic ring system containing three to ten carbon atoms, zero heteroatoms and zero double bonds. Representative examples of cycloalkyl include, but are not limited to, cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl, cyclooctyl, cyclononyl and cyclodecyl. “Cycloalkyl” also includes carbocyclic ring systems in which a cycloalkyl group is appended to the parent molecular moiety and is fused to an aryl group as defined herein, a heteroaryl group as defined herein, or a heterocycle as defined herein. [0047] The term “cycloalkenyl” as used herein, means a non-aromatic monocyclic or multicyclic ring system containing at least one carbon-carbon double bond, e.g., having from 5-10 carbon atoms per ring. Exemplary monocyclic cycloalkenyl rings include cyclopentenyl, cyclohexenyl or cycloheptenyl. [0048] The term “cyclooctene” as used herein, refers to a substituted or unsubstituted non-aromatic cyclic alkyl group of 8 carbon atoms, having a single ring with a double bond. Examples of such cyclooctene groups include, but are not limited to, substituted or unsubstituted trans-cyclooctene (TCO). [0049] The term “fluoroalkyl” as used herein, means an alkyl group, as defined herein, in which one, two, three, four, five, six, seven or eight hydrogen atoms are replaced by fluorine. Representative examples of fluoroalkyl include, but are not limited to, 2-fluoroethyl, 2,2,2-trifluoroethyl, trifluoromethyl, difluoromethyl, pentafluoroethyl, and trifluoropropyl such as 3,3,3-trifluoropropyl. [0050] The term “alkoxyfluoroalkyl” as used herein, refers to an alkoxy group, as defined herein, appended to the parent molecular moiety through a fluoroalkyl group, as defined herein. [0051] The term “fluoroalkoxy” as used herein, means at least one fluoroalkyl group, as defined herein, is appended to the parent molecular moiety through an oxygen atom. Representative examples of fluoroalkyloxy include, but are not limited to, difluoromethoxy, trifluoromethoxy and 2,2,2- trifluoroethoxy. [0052] The term “halogen” or “halo” as used herein, means Cl, Br, I, or F. [0053] The term “haloalkyl” as used herein, means an alkyl group, as defined herein, in which one, two, three, four, five, six, seven or eight hydrogen atoms are replaced by a halogen. [0054] The term “haloalkoxy” as used herein, means at least one haloalkyl group, as defined herein, is appended to the parent molecular moiety through an oxygen atom. [0055] The term “heteroalkyl” as used herein, means an alkyl group, as defined herein, in which one or more of the carbon atoms has been replaced by a heteroatom selected from S, Si, O, P and N. The heteroatom may be oxidized. Representative examples of heteroalkyls include, but are not limited to, alkyl ethers, secondary and tertiary alkyl amines, and alkyl sulfides. [0056] The term “heteroaryl” as used herein, refers to an aromatic group having a single ring, multiple rings or multiple fused rings, with one or more ring heteroatoms independently selected from nitrogen, oxygen and sulfur. In some embodiments, the term “heteroaryl” as used herein, refers to an aromatic monocyclic ring or an aromatic bicyclic ring system or an aromatic tricyclic ring system. The aromatic monocyclic rings are five or six membered rings containing at least one heteroatom independently selected from the group consisting of N, O and S (e.g.1, 2, 3, or 4 heteroatoms independently selected from O, S, and N). The five membered aromatic monocyclic rings have two double bonds and the six membered aromatic monocyclic rings have three double bonds. Representative examples of monocyclic heteroaryl include, but are not limited to, pyridinyl (including pyridin-2-yl, pyridin-3-yl, pyridin-4-yl), pyrimidinyl, pyrazinyl, thienyl, furyl, thiazolyl, thiadiazolyl, isoxazolyl, pyrazolyl, and 2-oxo-1,2- dihydropyridinyl. Representative examples of bicyclic heteroaryl include, but are not limited to, chromenyl, benzothienyl, benzodioxolyl, benzotriazolyl, quinolinyl, thienopyrrolyl, thienothienyl, imidazothiazolyl, benzothiazolyl, benzofuranyl, indolyl, quinolinyl, imidazopyridine, benzooxadiazolyl, and benzopyrazolyl. Representative examples of tricyclic heteroaryl include, but are not limited to, dibenzofuranyl and dibenzothienyl. The monocyclic, bicyclic, and tricyclic heteroaryls are connected to the parent molecular moiety through any carbon atom or any nitrogen atom contained within the rings, and can be unsubstituted or substituted. In some embodiments, the aromatic bicyclic ring system or aromatic tricyclic ring system does not contain non-aromatic rings. Thus, if a bicyclic ring system or tricyclic ring system contains a non-aromatic ring, the ring system is a cycloalkyl or heterocyclyl, depending on whether a heteroatom is present in the non-aromatic ring, regardless of the point of attachment to the remainder of the molecule. [0057] In some embodiments, the five membered aromatic monocyclic rings have two double bonds and the six membered aromatic monocyclic rings have three double bonds. In some embodiments, exemplary bicyclic heteroaryl groups are exemplified by a monocyclic heteroaryl ring appended to the parent molecular moiety and fused to a monocyclic cycloalkyl group, as defined herein, a monocyclic aryl group, as defined herein, a monocyclic heteroaryl group, as defined herein, or a monocyclic heterocycle, as defined herein. In some embodiments, the tricyclic heteroaryl groups are exemplified by a monocyclic heteroaryl ring appended to the parent molecular moiety and fused to two of a monocyclic cycloalkyl group, as defined herein, a monocyclic aryl group, as defined herein, a monocyclic heteroaryl group, as defined herein, or a monocyclic heterocycle, as defined herein. [0058] The terms “heterocyclyl,” “heterocycle,” or “heterocyclic” as used herein, refers to a non- aromatic ring system containing 3 to 10, or 3 to 8, or 3 to 6, or 5 to 10, carbon atoms and at least one (e.g., 1 to 5, 1 to 4, 1 to 3, 1 to 2, or 1) heteroatom, and optionally one or more oxo and/or double bonds. The terms “heterocyclyl”, “heterocycle” or “heterocyclic” include monocyclic, bicyclic, tricyclic, fused, spirocyclic, or bridged ring systems, provided that at least one non-aromatic ring system containing at least one heteroatom is present. In some embodiments, the monocyclic heterocycle is a three-, four-, five-, six-, seven-, or eight-membered ring containing at least one heteroatom independently selected from the group consisting of O, N, and S. In some embodiments, the three- or four-membered ring contains zero or one double bond, and one heteroatom selected from the group consisting of O, N, and S. In some embodiments, the five-membered ring contains zero or one double bond and one, two or three heteroatoms selected from the group consisting of O, N and S. In some embodiments, the six-membered ring contains zero, one or two double bonds and one, two, or three heteroatoms selected from the group consisting of O, N, and S. In some embodiments, the seven- and eight-membered rings contains zero, one, two, or three double bonds and one, two, or three heteroatoms selected from the group consisting of O, N, and S. Representative examples of monocyclic heterocycles include, but are not limited to, azetidinyl, azepanyl, aziridinyl, diazepanyl, 1,3-dioxanyl, 1,3-dioxolanyl, 1,3-dithiolanyl, 1,3-dithianyl, 1,3-dimethylpyrimidine-2,4(1H,3H)-dione, imidazolinyl, imidazolidinyl, isothiazolinyl, isothiazolidinyl, isoxazolinyl, isoxazolidinyl, morpholinyl, oxadiazolinyl, oxadiazolidinyl, oxazolinyl, oxazolidinyl, oxetanyl, piperazinyl, piperidinyl, pyranyl, pyrazolinyl, pyrazolidinyl, pyrrolinyl, pyrrolidinyl, tetrahydrofuranyl, tetrahydropyranyl, tetrahydropyridinyl, tetrahydrothienyl, thiadiazolinyl, thiadiazolidinyl, 1,2-thiazinanyl, 1,3-thiazinanyl, thiazolinyl, thiazolidinyl, thiomorpholinyl, 1,1- dioxidothiomorpholinyl (thiomorpholine sulfone), thiopyranyl, and trithianyl. The bicyclic heterocycle is a monocyclic heterocycle fused to a phenyl group, or a monocyclic heterocycle fused to a monocyclic cycloalkyl, or a monocyclic heterocycle fused to a monocyclic cycloalkenyl, or a monocyclic heterocycle fused to a monocyclic heterocycle, or a spiro heterocycle group, or a bridged monocyclic heterocycle ring system in which two non-adjacent atoms of the ring are linked by an alkylene bridge of 1, 2, 3, or 4 carbon atoms, or an alkenylene bridge of two, three, or four carbon atoms. Representative examples of bicyclic heterocycles include, but are not limited to, benzopyranyl, benzothiopyranyl, chromanyl, 2,3- dihydrobenzofuranyl, 2,3-dihydrobenzothienyl, 2,3-dihydroisoquinoline, 2-azaspiro[3.3]heptan-2-yl, azabicyclo[2.2.1]heptyl (including 2-azabicyclo[2.2.1]hept-2-yl), 2,3-dihydro-1H-indolyl, isoindolinyl, octahydrocyclopenta[c]pyrrolyl, octahydropyrrolopyridinyl, and tetrahydroisoquinolinyl. Tricyclic heterocycles are exemplified by a bicyclic heterocycle fused to a phenyl group, or a bicyclic heterocycle fused to a monocyclic cycloalkyl, or a bicyclic heterocycle fused to a monocyclic cycloalkenyl, or a bicyclic heterocycle fused to a monocyclic heterocycle, or a bicyclic heterocycle in which two non- adjacent atoms of the bicyclic ring are linked by an alkylene bridge of 1, 2, 3, or 4 carbon atoms, or an alkenylene bridge of two, three, or four carbon atoms. Examples of tricyclic heterocycles include, but are not limited to, octahydro-2,5-epoxypentalene, hexahydro-2H-2,5-methanocyclopenta[b]furan, hexahydro-1H-1,4-methanocyclopenta[c]furan, aza-adamantane (1-azatricyclo[3.3.1.13,7]decane), and oxa-adamantane (2-oxatricyclo[3.3.1.13,7]decane). The monocyclic, bicyclic, and tricyclic heterocycles are connected to the parent molecular moiety through any carbon atom or any nitrogen atom contained within the rings, and can be unsubstituted or substituted. [0059] The term “hydroxyl” as used herein, means an –OH group. [0060] The term “hydroxyalkyl” as used herein, means an alkyl group, as defined herein, in which one, two, three, four, five, six, seven or eight hydrogen atoms are replaced by a hydroxyl group. [0061] In some instances, the number of carbon atoms in a hydrocarbyl substituent (e.g., alkyl or cycloalkyl) is indicated by the prefix “Cx-Cy-” or “Cx-y,” wherein x is the minimum and y is the maximum number of carbon atoms in the substituent. Thus, for example, “C1-C3-alkyl” and “C1-3alkyl” refer to an alkyl substituent containing from 1 to 3 carbon atoms. The two conventions “Cx-Cy-” and “Cx-y” are used interchangeably and have the same meaning. [0062] The term “substituted” refers to a group that may be further substituted with one or more non- hydrogen substituent groups. Substituent groups include, but are not limited to, halogen, =O, =S, cyano, nitro, fluoroalkyl, alkoxyfluoroalkyl, fluoroalkoxy, alkyl, alkenyl, alkynyl, haloalkyl, haloalkoxy, heteroalkyl, cycloalkyl, cycloalkenyl, aryl, heteroaryl, heterocycle, cycloalkylalkyl, heteroarylalkyl, arylalkyl, hydroxy, hydroxyalkyl, alkoxy, alkoxyalkyl, alkylene, aryloxy, phenoxy, benzyloxy, amino, alkylamino, acylamino, aminoalkyl, arylamino, sulfonylamino, sulfinylamino, sulfonyl, alkylsulfonyl, arylsulfonyl, aminosulfonyl, sulfinyl, -COOH, ketone, amide, carbamate, and acyl. [0063] The term “tetrazine” refers to a substituted or unsubstituted aromatic cyclic group of 2 carbon atoms and 4 nitrogen atoms, having a single ring with three double bonds. Examples of tetrazine groups include 1,2,3,4-tetrazine and 1,2,4,5-tetrazine. As used herein, 1,2,4,5-tetrazine is referred to as a “Tz” group. [0064] The term “selectively delivering” refers to delivering an agent (e.g., a payload) to an organ or tissue (or portion thereof) in need of treatment or diagnosis, without significant binding to other non- target organs or tissues (or portions thereof). In some embodiments, the targeting moieties, or therapeutic targeting moiety, described herein do not themselves have a therapeutic effect, but rather are designed to allow the selective or targeted delivery of a therapeutic agent. However, it may be that the targeting moiety does have a therapeutic effect, and thus, such constructs are not excluded by the present disclosure. [0065] The term “payload” refers to an agent for delivery to a target site in a subject. In some embodiments, the payloads is a therapeutic agent. In some embodiments, the payloads is a diagnostic agent. [0066] The term “therapeutic agent” refers to an agent capable of treating and/or ameliorating a condition or disease, or one or more symptoms thereof, in a subject. Therapeutic agents of the present disclosure also include prodrug forms of therapeutic agents, chelating agent with or without a radionuclide (e.g., diagnostic or therapeutic). [0067] A therapeutic “radionuclide” or “radioligand” as used herein refers to a radioactive substance, sometimes referred to as a radiopharmaceutical, which are used to treat medical conditions, particularly cancer. Radionuclides used in the trans-cyclooctene moieties described herein, comprise a chelating agent and an isotope; such as an isotope selected from the group consisting of24Na,32P,33P,47Sc,59Fe,67Cu,76As,77As,80Br,82Br,89Sr,90Nb,90Y,103Ru,105Rh,109Pd,111Ag,111In,121Sn,127Te,131I,140La,141Ce,142Pr,143Pr,144Pr,149Pm,149Tb,151Pm,153Sm,159Gd,161Tb,165Dy,166Ho,169Er,172Tm,175Yb,177Lu,186Re,188Re,198Au,199Au,211At,211Bi,212Bi,212Pb,213Bi,214Bi,223Ra, and225Ac. Radionuclides can be delivered via direct conjugation or chelation with a chelating agent. Exemplary radionuclides, chelating agents, and linkers for potential TCO-conjugate payloads are described below. [0068] The term “diagnostic agent” refers to agents that assist in diagnosing conditions or diseases. Representative diagnostic agents include imaging agents such as paramagnetic agents, optical probes, radionuclides, and the like. Paramagnetic agents are imaging agents that are magnetic under an externally applied field. Examples of paramagnetic agents include, but are not limited to, iron particles including iron nanoparticles and iron microparticles. Optical probes are fluorescent compounds that can be detected by excitation at one wavelength of radiation and detection at a second, different, wavelength of radiation. Optical probes of the present disclosure include, but are not limited to, Cy5.5, Alexa 680, Cy5, DiD (1,1’-dioctadecyl-3,3,3’,3’-tetramethylindodicarbocyanine perchlorate) and DiR (1,1’- dioctadecyl-3,3,3’,3’-tetramethylindotricarbocyanine iodide). Other optical probes include quantum dots. Radionuclides are elements that undergo detectable radioactive decay. Radionuclides, such as diagnostic radionuclides, useful in embodiments of the present disclosure include, but are not limited to,3H,11C,13N,18F,19F,60Co,64Cu,67Cu,68Ga,82Rb,89Zr,90Sr,90Y,99Tc,99mTc,111In,123I,124I,125I,129I,131I,137Cs,177Lu,186Re,188Re,211At,212Pb,225Ac, Rn, Ra, Th, U, Pu, and241Am. [0069] The term “targeting agent” refers to a chemical or biological agent that specifically binds to a target (e.g., a targeted organ or tissue), thereby forming a stable association between the targeting agent and the specific target. By “stably associated” or “stable association” is meant that a moiety is bound to or otherwise associated with another moiety or structure under standard physiological conditions. Bonds may include covalent bonds and non-covalent interactions, such as, but not limited to, ionic bonds, hydrophobic interactions, hydrogen bonds, van der Waals forces (e.g., London dispersion forces), dipole- dipole interactions, and the like. Targeting agents include ligands that specifically bind (or substantially specifically bind) a particular clinically-relevant target receptor or cell surface target. [0070] The term “antibody fragment moiety” as used herein refers to one or more regions or fragments of an antibody that retain the ability to specifically bind to an antigen, or in other words, substantially retain the antigen-binding function of the antibody. Examples of binding fragments encompassed within the antigen-binding portion of an antibody include but are not limited to a Fab fragment. In some embodiments, the Fab fragment is a monovalent fragment containing at least the VL and VH. In some embodiments, the Fab fragment is a monovalent fragment containing the VL, VH, CL, and CH1 domains. Fab fragments can be obtained using conventional techniques known to those with skill in the art, and the fragments are screened for utility in the same manner as are intact antibodies. For example, antibody fragments can be produced by recombinant DNA techniques, or by enzymatic or chemical cleavage of intact immunoglobulins. Additional examples are included in various embodiments disclosed herein. [0071] By “specifically bind” is meant the preferential association of an antibody or antibody fragment moiety to a target molecule (e.g., an antigen, such as a peptide, polypeptide, glycoprotein, or any other moiety with one or more antigenic determinants) or to a cell or tissue bearing the target molecule (e.g., a cell surface antigen, such as a receptor or ligand) and not to cells or tissues lacking the target molecule. It is recognized that a certain degree of non-specific interaction may occur between a binding moiety and a non-targeted molecule (present alone or in combination with a cell or tissue). Nevertheless, specific binding may be distinguished as mediated through specific recognition of the target antigen. Specific binding results in a much stronger association between the targeting moiety (e.g., Fab) and e.g., cells bearing the target molecule (e.g., an antigen) than between the binding moiety and e.g., cells lacking the target molecule. Specific binding typically results in greater than 2-fold, greater than 5-fold, greater than 10-fold, or greater than 100-fold increase in amount of bound binding moiety (per unit time) to e.g., a cell or tissue bearing the target molecule or marker as compared to a cell or tissue lacking that target molecule or marker. In some embodiments, binding moieties bind to the target molecule or marker with a dissociation constant of e.g., less than 10-5M, less than 10-7M, less than 10-8M, less than 10-9M, less than 10-10M, less than 10-11M, less than 10-12M, less than 10-13M, less than 10-14M, or less than 10-15M. A variety of assay formats are appropriate for measuring binding, such as solid-phase ELISA immunoassays. [0072] The term “targeted organ or tissue” refers to an organ or tissue that is being targeted for delivery of the payload. Representative organs and tissues for targeting include those that can be targeted by chemical or biological targeting agents, as well as those organs and tissues that cannot be targeted by chemical or biological targeting agents. [0073] The term “contacting” or “contact” refers to the process of bringing into contact at least two distinct species such that they can interact with each other, such as in a non-covalent or covalent binding interaction or binding reaction. It should be appreciated, however, the resulting complex or reaction product can be produced directly from an interaction or a reaction between the added reagents or from an intermediate from one or more of the added reagents or moieties, which can be produced in the contacting mixture. [0074] The term “administering” refers to any suitable route of administration to a subject, such as, but not limited to, oral administration, administration as a suppository, topical contact, parenteral, intravenous, intraperitoneal, intramuscular, intralesional, intranasal or subcutaneous administration, intrathecal administration, or the implantation of a slow-release device, e.g., a mini-osmotic pump, to the subject. [0075] The term “parenterally,” as used herein, refers to modes of administration which include intravenous, intramuscular, intraperitoneal, intrasternal, subcutaneous and intraarticular injection, and infusion. [0076] The term “pharmaceutically effective amount” and “therapeutically effective amount” refer to an amount of a compound sufficient to treat a specified disorder or disease or one or more of its symptoms and/or to prevent or reduce the risk of the occurrence or reoccurrence of the disease or disorder or symptom(s) thereof. In reference to tumorigenic proliferative disorders, a pharmaceutically or therapeutically effective amount comprises an amount sufficient to, among other things, cause the tumor to shrink or decrease the growth rate of the tumor. [0077] As used herein, the term “subject,” “patient,” or “organism” includes humans and mammals (e.g., mice, rats, pigs, cats, dogs, and horses). Typical subjects to which an agent(s) of the present disclosure may be administered may include mammals, particularly primates, especially humans. For veterinary applications, suitable subjects may include, for example, livestock such as cattle, sheep, goats, cows, swine, and the like; poultry such as chickens, ducks, geese, turkeys, and the like; and domesticated animals particularly pets such as dogs and cats. For diagnostic or research applications, suitable subjects may include mammals, such as rodents (e.g., mice, rats, hamsters), rabbits, primates, and swine such as inbred pigs and the like. [0078] The term “treating” or “treatment” as used herein means the treating or treatment of a disease or medical condition or symptom(s) thereof in a patient, such as a mammal (e.g., a human) that includes: (a) ameliorating the disease or medical condition or symptom(s) thereof, such as, eliminating or causing regression of the disease or medical condition or symptom(s) thereof in a patient; (b) suppressing the disease or medical condition or symptom(s) thereof, for example by, slowing or arresting the development of the disease or medical condition or symptom(s) thereof in a patient; or (c) alleviating a symptom of the disease or medical condition or symptom(s) thereof in a patient. [0079] The term “physiological conditions” is meant to encompass those conditions compatible with living cells, e.g., predominantly aqueous conditions of a temperature, pH, salinity, etc. that are compatible with living cells. [0080] For compounds described herein, groups and substituents thereof may be selected in accordance with permitted valence of the atoms and the substituents, such that the selections and substitutions result in a stable compound, e.g., which does not spontaneously undergo transformation such as by rearrangement, cyclization, elimination, etc. [0081] Where a range of values is provided, it is understood that each intervening value, to the tenth of the unit of the lower limit unless the context clearly dictates otherwise, between the upper and lower limit of that range and any other stated or intervening value in that stated range, is encompassed within the disclosure. The upper and lower limits of these smaller ranges may independently be included in the smaller ranges, and are also encompassed within the disclosure, subject to any specifically excluded limit in the stated range. Where the stated range includes one or both of the limits, ranges excluding either or both of those included limits are also included in the disclosure. [0082] For the recitation of numeric ranges herein, each intervening number there between with the same degree of precision is explicitly contemplated. For example, for the range of 6-9, the numbers 7 and 8 are contemplated in addition to 6 and 9, and for the range 6.0-7.0, the number 6.0, 6.1, 6.2, 6.3, 6.4, 6.5, 6.6, 6.7, 6.8, 6.9, and 7.0 are explicitly contemplated. [0083] The compounds may exist as stereoisomers wherein asymmetric or chiral centers are present. The stereoisomers are “R” or “S” depending on the configuration of substituents around the chiral carbon atom. The terms “R” and “S” used herein are configurations as defined in IUPAC 1974 Recommendations for Section E, Fundamental Stereochemistry, in Pure Appl. Chem., 1976, 45: 13-30. The disclosure contemplates various stereoisomers and mixtures thereof, and these are specifically included within the scope of this disclosure. Stereoisomers include enantiomers and diastereomers and mixtures of enantiomers or diastereomers. Individual stereoisomers of the compounds may be prepared synthetically from commercially available starting materials, which contain asymmetric or chiral centers or by preparation of racemic mixtures followed by methods of resolution well-known to those of ordinary skill in the art. These methods of resolution are exemplified by (1) attachment of a mixture of enantiomers to a chiral auxiliary, separation of the resulting mixture of diastereomers by recrystallization or chromatography, and optional liberation of the optically pure product from the auxiliary as described in Furniss, Hannaford, Smith, and Tatchell, “Vogel’s Textbook of Practical Organic Chemistry,” 5th edition (1989), Longman Scientific & Technical, Essex CM202JE, England, or (2) direct separation of the mixture of optical enantiomers on chiral chromatographic columns, or (3) fractional recrystallization methods. [0084] It should be understood that the compounds may possess tautomeric forms as well as geometric isomers, and that these also constitute an aspect of the disclosure. [0085] The present disclosure also includes isotopically-labeled compounds, which are identical to those recited herein, but for the fact that one or more atoms are replaced by an atom having an atomic mass or mass number different from the atomic mass or mass number usually found in nature. Examples of isotopes suitable for inclusion in the compounds of the disclosure are hydrogen, carbon, nitrogen, oxygen, phosphorus, sulfur, fluorine, and chlorine, such as, but not limited to,2H,3H,13C,14C,15N,18O,17O,31P,32P,35S,18F, and36Cl, respectively. Substitution with heavier isotopes such as deuterium, i.e.,2H, can afford certain therapeutic advantages resulting from greater metabolic stability, for example increased in vivo half-life or reduced dosage requirements, and, hence, may be preferred in some circumstances. The compound may incorporate positron-emitting isotopes for medical imaging and positron-emitting tomography (PET) studies for determining the distribution of receptors. Suitable positron-emitting isotopes that can be incorporated are11C,13N,15O, and18F. Isotopically-labeled compounds disclosed herein can generally be prepared by conventional techniques known to those skilled in the art or by processes analogous to those described in the accompanying Examples using appropriate isotopically-labeled reagent in place of non-isotopically-labeled reagent. Methods of Treatment [0086] The present disclosure is directed to methods for delivering a payload to a target location in a subject. [0087] In one aspect, provided is a method of forming in vivo an antibody-payload conjugate in a subject in need thereof, the method comprising: administering an effective amount of a targeting moiety to the subject, wherein the targeting moiety comprising at least one antibody or fragment thereof having a least one tetrazine moiety covalently linked thereto; administering a single dose of a therapeutically effective amount of a payload-TCO conjugate to the subject, wherein the payload-TCO conjugate comprises a payload having at least one trans- cyclooctene (TCO) moiety covalently linked thereto; wherein the antibody or fragment thereof has a binding affinity to a receptor on a tumor, and further wherein the amount of the antibody-payload conjugate formed in vivo is greater at a tumor site versus in plasma. [0088] In one aspect, provided is a method of forming in vivo an antibody-payload conjugate in a subject in need thereof, the method comprising: administering an effective amount of a targeting moiety to the subject, wherein the targeting moiety comprising at least one antibody or fragment thereof having a least one tetrazine moiety covalently linked thereto; administering a single dose of a therapeutically effective amount of a payload-TCO conjugate to the subject, wherein the payload-TCO conjugate comprises a payload having at least one trans- cyclooctene (TCO) moiety covalently linked thereto; wherein the antibody or fragment thereof has a binding affinity to a receptor on a tumor, and further wherein the ratio of the antibody-payload conjugate at a tumor site versus in plasma is greater than about 2:1. [0089] In some embodiments, the ratio of the antibody-payload conjugate at a tumor site versus in plasma is greater than 1:1, or about 2:1, or about 3:1, or about 4:1, or about 5:1, or about 6:1, or about 7:1, or about 8:1, or about 9:1, or about 10:1, or about 11:1, about 12:1, or about 13:1, or about 14:1, or about 15:1, or about 16:1, or about 17:1, or about 18:1, or about 19:1. [0090] In some embodiments, the ratio of the antibody-payload conjugate at a tumor site versus in plasma is greater than about 2:1, or greater than about 3:1, or greater than about 4:1, or greater than about 5:1, or greater than about 6:1, or greater than about 7:1, or greater than about 8:1, or greater than about 9:1, or greater than about 10:1. [0091] In some embodiments, the administering is simultaneous. [0092] In some embodiments, the administering is sequential. In some embodiments, the targeting moiety is administered prior to the payload-TCO conjugate. [0093] In some embodiments, a first dose of the payload-TCO conjugate is administered to the subject less than 48 hours, or more than 4 hours and less than 48 hours, after the targeting moiety is administered to the subject. [0094] In some embodiments, a single dose of the payload-TCO conjugate is administered to the subject less than 48 hours, or more than 4 hours and less than 48 hours, after the targeting moiety is administered to the subject. [0095] In some embodiments, a single dose of the payload-TCO conjugate is administered to the subject less than 48 hours after the targeting moiety is administered to the subject. [0096] In some embodiments, a single dose of the payload-TCO conjugate is administered to the subject more than 4 hours, and less than 48 hours, after the targeting moiety is administered to the subject. [0097] In some embodiments, the targeting moiety is administered at least 8 to about 24 hours prior to the payload-TCO conjugate being administered. [0098] In some embodiments, the targeting moiety is administered within 0-8 serum half-lives. It is contemplated that timing of the payload-TCO conjugate administration is calculated based on tumor disposition, which can be determined by the biology of the antigen at the tumor and the targeting moiety. For example, in some embodiments, when the targeting moiety comprises a targeting format as in the table below, the payload-TCO conjugate is administered within the serum half life range shown therein (see also Berland et al., Biomolecules 2021, 11(5), 637). [0099] In one aspect, provided is a method of administering a payload to a subject, the method comprising: a) administering an effective amount of a targeting moiety to the subject, wherein the targeting moiety comprises a Fab having at least one tetrazine moiety covalently linked thereto; and b) administering a single dose of a therapeutically effective amount of a payload-TCO conjugate to the subject, wherein the payload-TCO conjugate comprises a payload having at least one trans-cyclooctene moiety covalently linked thereto; wherein the single dose of the payload-TCO conjugate is administered to the subject at least about 2 hours, or at least about 3 hours, or at least about 4 hours, less than about 48 hours, or between about 4 and 48 hours, after the targeting moiety is administered to the subject. [0100] In one aspect, provided is a method of administering a payload to a subject, the method comprising: a) administering an effective amount of a targeting moiety to the subject, wherein the targeting moiety comprises a Fab having at least one tetrazine moiety covalently linked thereto; and b) administering a single dose of a therapeutically effective amount of a payload-TCO conjugate to the subject, wherein the payload-TCO conjugate comprises a payload having at least one trans- cyclooctene moiety covalently linked thereto; wherein the single dose of the payload-TCO conjugate is administered to the subject more than 4 hours, and less than 48 hours, after the targeting moiety is administered to the subject. [0101] Selective binding between bioorthogonal binding partners (e.g., between a tetrazine of the targeting moiety and its complementary trans-cyclooctene of a payload-TCO conjugate occurs. Due to the localized administration of the targeting moiety to a desired location in the subject as described above, the selective binding between the trans-cyclooctene of the payload-TCO conjugate and a tetrazine of the targeting moiety will localize the payload to the desired target location. [0102] The methods disclosed herein allow the payload to be selectively and safely delivered, and thus decrease side effects or toxicity associated with off-target delivery. Furthermore, by administering the two agents as described herein, only a single dose of the payload-TCO conjugate is required to be therapeutically effective. [0103] As described herein, selective delivery of a payload may be achieved by administering a payload- TCO conjugate within a certain therapeutic window after administering a targeting agent. The relatively inert, systemically administered payload-TCO conjugate is activated at the target site, e.g., a tumor, by the targeting agent via a covalent click chemistry reaction, followed by chemical rearrangement to release the active payload. [0104] In some embodiments, the subject has cancer. In one aspect, provided is a method of treating cancer, or enhancing or eliciting an immune response, in a subject having cancer, the method comprising: a) administering an effective amount of a targeting moiety to the subject, wherein the targeting moiety comprises a Fab having at least one tetrazine moiety covalently linked thereto; and b) administering a single dose of a therapeutically effective amount of a payload-TCO conjugate to the subject, wherein the payload-TCO conjugate comprises a payload having at least one trans- cyclooctene moiety covalently linked thereto; wherein the single dose of the payload-TCO conjugate is administered to the subject at least about 2 hours, or at least about 3 hours, or at least about 4 hours, less than about 48 hours, or between about 4 and 48 hours, after the targeting moiety is administered to the subject. [0105] In one aspect, provided is a method of treating cancer, or enhancing or eliciting an immune response, in a subject having cancer, the method comprising: a) administering an effective amount of a targeting moiety to the subject, wherein the targeting moiety comprises a Fab having at least one tetrazine moiety covalently linked thereto; and b) administering a single dose of a therapeutically effective amount of a payload-TCO conjugate to the subject, wherein the payload-TCO conjugate comprises a payload having at least one trans- cyclooctene moiety covalently linked thereto; wherein the single dose of the payload-TCO conjugate is administered to the subject more than 4 hours, and less than 48 hours, after the targeting moiety is administered to the subject. [0106] In some embodiments, a single dose of the payload-TCO conjugate is administered to the subject 5, or 6, or 7, or 8, or 9, or 10, or 11, or 12, or 13, or 14, or 15, or 16, or 17, or 18, or 19, or 20, or 21, or 22, or 23, or less than 24, or about 24 hours after the targeting moiety is administered to the subject. [0107] In some embodiments, a single dose of the payload-TCO conjugate is administered to the subject between about 5 and about 24, or between about 6 and about 24, or between about 7 and about 24, or between about 8 and about 24, or between about 9 and about 24, or between about 10 and about 24, or between about 11 and about 24, or between about 12 and about 24, or between about 13 and about 24, or between about 14 and about 24, or between about 15 and about 24, or between about 16 and about 24, or between about 17 and about 24, or between about 18 and about 24, or between about 19 and about 24, or between about 20 and about 24, or between about 21 and about 24, or between about 22 and about 24, or between about 23 and about 24 hours after the targeting moiety is administered to the subject. [0108] In some embodiments, a single dose of the payload-TCO conjugate is administered to the subject between about 5 and about 22, or between about 6 and about 22, or between about 7 and about 22, or between about 8 and about 22, or between about 9 and about 22, or between about 10 and about 22, or between about 11 and about 22, or between about 12 and about 22, or between about 13 and about 22, or between about 14 and about 22, or between about 15 and about 22, or between about 16 and about 22, or between about 17 and about 22, or between about 18 and about 22, or between about 19 and about 22, or between about 20 and about 22, or between about 21 and about 22 hours after the targeting moiety is administered to the subject. [0109] In some embodiments, a single dose of the payload-TCO conjugate is administered to the subject between about 5 and about 20, or between about 6 and about 20, or between about 7 and about 20, or between about 8 and about 20, or between about 9 and about 20, or between about 10 and about 20, or between about 11 and about 20, or between about 12 and about 20, or between about 13 and about 20, or between about 14 and about 20, or between about 15 and about 20, or between about 16 and about 20, or between about 17 and about 20, or between about 18 and about 20, or between about 19 and about 20 hours after the targeting moiety is administered to the subject. [0110] In some embodiments, a single dose of the payload-TCO conjugate is administered to the subject between about 8 to about 12 hours, between about 8 to about 16 hours, between about 8 and about 22 hours, between about 12 to about 16 hours, or between about 16 and about 20 hours, after the targeting moiety is administered to the subject. [0111] In some embodiments, the payload-TCO conjugate is a MMAE-TCO conjugate. In some embodiments, a single dose of the MMAE-TCO conjugate is administered to the subject between about 8 to about 12 hours, between about 8 to about 16 hours, between about 8 and about 22 hours, between about 12 to about 16 hours, or between about 16 and about 20 hours, after the targeting moiety is administered to the subject. [0112] In some embodiments, a single dose of the payload-TCO conjugate is administered to the subject between about 16 and about 20 hours after the targeting moiety is administered to the subject. [0113] In one aspect, provided is a method of selectively administering a payload to a tumor site in a subject in need thereof, the method comprising: a) administering an effective amount of a targeting moiety to the subject, wherein the targeting moiety comprises a Fab having at least one tetrazine moiety covalently linked thereto; and b) administering a single dose of a therapeutically effective amount of a payload-TCO conjugate to the subject, wherein the payload-TCO conjugate comprises a payload having at least one trans- cyclooctene moiety covalently linked thereto; wherein the single dose of the payload-TCO conjugate is administered to the subject between about 8 to about 18 hours, after the targeting moiety is administered to the subject. [0114] In some embodiments, the accumulation at the tumor site is at least about 10X greater, or up to about 200X greater, than the kidney, liver, or spleen tissue. In some embodiments, the targeting moiety is of Formula I, Formula II, or Formula V:
[0099] In one aspect, provided is a method of administering a payload to a subject, the method comprising: a) administering an effective amount of a targeting moiety to the subject, wherein the targeting moiety comprises a Fab having at least one tetrazine moiety covalently linked thereto; and b) administering a single dose of a therapeutically effective amount of a payload-TCO conjugate to the subject, wherein the payload-TCO conjugate comprises a payload having at least one trans-cyclooctene moiety covalently linked thereto; wherein the single dose of the payload-TCO conjugate is administered to the subject at least about 2 hours, or at least about 3 hours, or at least about 4 hours, less than about 48 hours, or between about 4 and 48 hours, after the targeting moiety is administered to the subject. [0100] In one aspect, provided is a method of administering a payload to a subject, the method comprising: a) administering an effective amount of a targeting moiety to the subject, wherein the targeting moiety comprises a Fab having at least one tetrazine moiety covalently linked thereto; and b) administering a single dose of a therapeutically effective amount of a payload-TCO conjugate to the subject, wherein the payload-TCO conjugate comprises a payload having at least one trans- cyclooctene moiety covalently linked thereto; wherein the single dose of the payload-TCO conjugate is administered to the subject more than 4 hours, and less than 48 hours, after the targeting moiety is administered to the subject. [0101] Selective binding between bioorthogonal binding partners (e.g., between a tetrazine of the targeting moiety and its complementary trans-cyclooctene of a payload-TCO conjugate occurs. Due to the localized administration of the targeting moiety to a desired location in the subject as described above, the selective binding between the trans-cyclooctene of the payload-TCO conjugate and a tetrazine of the targeting moiety will localize the payload to the desired target location. [0102] The methods disclosed herein allow the payload to be selectively and safely delivered, and thus decrease side effects or toxicity associated with off-target delivery. Furthermore, by administering the two agents as described herein, only a single dose of the payload-TCO conjugate is required to be therapeutically effective. [0103] As described herein, selective delivery of a payload may be achieved by administering a payload- TCO conjugate within a certain therapeutic window after administering a targeting agent. The relatively inert, systemically administered payload-TCO conjugate is activated at the target site, e.g., a tumor, by the targeting agent via a covalent click chemistry reaction, followed by chemical rearrangement to release the active payload. [0104] In some embodiments, the subject has cancer. In one aspect, provided is a method of treating cancer, or enhancing or eliciting an immune response, in a subject having cancer, the method comprising: a) administering an effective amount of a targeting moiety to the subject, wherein the targeting moiety comprises a Fab having at least one tetrazine moiety covalently linked thereto; and b) administering a single dose of a therapeutically effective amount of a payload-TCO conjugate to the subject, wherein the payload-TCO conjugate comprises a payload having at least one trans- cyclooctene moiety covalently linked thereto; wherein the single dose of the payload-TCO conjugate is administered to the subject at least about 2 hours, or at least about 3 hours, or at least about 4 hours, less than about 48 hours, or between about 4 and 48 hours, after the targeting moiety is administered to the subject. [0105] In one aspect, provided is a method of treating cancer, or enhancing or eliciting an immune response, in a subject having cancer, the method comprising: a) administering an effective amount of a targeting moiety to the subject, wherein the targeting moiety comprises a Fab having at least one tetrazine moiety covalently linked thereto; and b) administering a single dose of a therapeutically effective amount of a payload-TCO conjugate to the subject, wherein the payload-TCO conjugate comprises a payload having at least one trans- cyclooctene moiety covalently linked thereto; wherein the single dose of the payload-TCO conjugate is administered to the subject more than 4 hours, and less than 48 hours, after the targeting moiety is administered to the subject. [0106] In some embodiments, a single dose of the payload-TCO conjugate is administered to the subject 5, or 6, or 7, or 8, or 9, or 10, or 11, or 12, or 13, or 14, or 15, or 16, or 17, or 18, or 19, or 20, or 21, or 22, or 23, or less than 24, or about 24 hours after the targeting moiety is administered to the subject. [0107] In some embodiments, a single dose of the payload-TCO conjugate is administered to the subject between about 5 and about 24, or between about 6 and about 24, or between about 7 and about 24, or between about 8 and about 24, or between about 9 and about 24, or between about 10 and about 24, or between about 11 and about 24, or between about 12 and about 24, or between about 13 and about 24, or between about 14 and about 24, or between about 15 and about 24, or between about 16 and about 24, or between about 17 and about 24, or between about 18 and about 24, or between about 19 and about 24, or between about 20 and about 24, or between about 21 and about 24, or between about 22 and about 24, or between about 23 and about 24 hours after the targeting moiety is administered to the subject. [0108] In some embodiments, a single dose of the payload-TCO conjugate is administered to the subject between about 5 and about 22, or between about 6 and about 22, or between about 7 and about 22, or between about 8 and about 22, or between about 9 and about 22, or between about 10 and about 22, or between about 11 and about 22, or between about 12 and about 22, or between about 13 and about 22, or between about 14 and about 22, or between about 15 and about 22, or between about 16 and about 22, or between about 17 and about 22, or between about 18 and about 22, or between about 19 and about 22, or between about 20 and about 22, or between about 21 and about 22 hours after the targeting moiety is administered to the subject. [0109] In some embodiments, a single dose of the payload-TCO conjugate is administered to the subject between about 5 and about 20, or between about 6 and about 20, or between about 7 and about 20, or between about 8 and about 20, or between about 9 and about 20, or between about 10 and about 20, or between about 11 and about 20, or between about 12 and about 20, or between about 13 and about 20, or between about 14 and about 20, or between about 15 and about 20, or between about 16 and about 20, or between about 17 and about 20, or between about 18 and about 20, or between about 19 and about 20 hours after the targeting moiety is administered to the subject. [0110] In some embodiments, a single dose of the payload-TCO conjugate is administered to the subject between about 8 to about 12 hours, between about 8 to about 16 hours, between about 8 and about 22 hours, between about 12 to about 16 hours, or between about 16 and about 20 hours, after the targeting moiety is administered to the subject. [0111] In some embodiments, the payload-TCO conjugate is a MMAE-TCO conjugate. In some embodiments, a single dose of the MMAE-TCO conjugate is administered to the subject between about 8 to about 12 hours, between about 8 to about 16 hours, between about 8 and about 22 hours, between about 12 to about 16 hours, or between about 16 and about 20 hours, after the targeting moiety is administered to the subject. [0112] In some embodiments, a single dose of the payload-TCO conjugate is administered to the subject between about 16 and about 20 hours after the targeting moiety is administered to the subject. [0113] In one aspect, provided is a method of selectively administering a payload to a tumor site in a subject in need thereof, the method comprising: a) administering an effective amount of a targeting moiety to the subject, wherein the targeting moiety comprises a Fab having at least one tetrazine moiety covalently linked thereto; and b) administering a single dose of a therapeutically effective amount of a payload-TCO conjugate to the subject, wherein the payload-TCO conjugate comprises a payload having at least one trans- cyclooctene moiety covalently linked thereto; wherein the single dose of the payload-TCO conjugate is administered to the subject between about 8 to about 18 hours, after the targeting moiety is administered to the subject. [0114] In some embodiments, the accumulation at the tumor site is at least about 10X greater, or up to about 200X greater, than the kidney, liver, or spleen tissue. In some embodiments, the targeting moiety is of Formula I, Formula II, or Formula V:

















































































N N N are independently as defined herein. [0251] In some embodiments, R1, at each occurrence, is independently hydrogen, alkyl, alkenyl, alkynyl, haloalkyl, heteroalkyl, aryl, heteroaryl, heterocyclyl, or cycloalkyl; wherein each alkyl, alkenyl, alkynyl, haloalkyl, heteroalkyl, aryl, heteroaryl, heterocyclyl, or cycloalkyl is optionally substituted with one to three Z1. [0252] In some embodiments, R1, at each occurrence, is independently hydrogen or alkyl optionally substituted with one to three Z1. [0253] In some embodiments, Z1, at each occurrence, is independently selected from halo, hydroxy, alkoxy, and OC(=O)OR’. [0254] In some embodiments, R2, at each occurrence, is independently halo, cyano, nitro, hydroxy, alkyl, haloalkyl, alkenyl, alkynyl, alkoxy, haloalkoxy, heteroalkyl, aryl, heteroaryl, heterocyclyl, or cycloalkyl. In some embodiments, R2, at each occurrence, is independently halo, alkyl, or haloalkyl. In some embodiments, R2, at each occurrence, is independently halo or alkyl. [0255] In some embodiments, t at each occurrence, is 0. [0256] In some embodiments, the targeting moiety is of Formula VA:
are independently as defined herein. [0251] In some embodiments, R1, at each occurrence, is independently hydrogen, alkyl, alkenyl, alkynyl, haloalkyl, heteroalkyl, aryl, heteroaryl, heterocyclyl, or cycloalkyl; wherein each alkyl, alkenyl, alkynyl, haloalkyl, heteroalkyl, aryl, heteroaryl, heterocyclyl, or cycloalkyl is optionally substituted with one to three Z1. [0252] In some embodiments, R1, at each occurrence, is independently hydrogen or alkyl optionally substituted with one to three Z1. [0253] In some embodiments, Z1, at each occurrence, is independently selected from halo, hydroxy, alkoxy, and OC(=O)OR’. [0254] In some embodiments, R2, at each occurrence, is independently halo, cyano, nitro, hydroxy, alkyl, haloalkyl, alkenyl, alkynyl, alkoxy, haloalkoxy, heteroalkyl, aryl, heteroaryl, heterocyclyl, or cycloalkyl. In some embodiments, R2, at each occurrence, is independently halo, alkyl, or haloalkyl. In some embodiments, R2, at each occurrence, is independently halo or alkyl. [0255] In some embodiments, t at each occurrence, is 0. [0256] In some embodiments, the targeting moiety is of Formula VA:


























































o [0476] In some embodiments, a payload is a pyrrolobenzodiazepine (PBD), or a derivative, or analog thereof. In some embodiments, the pyrrolobenzodiazepine (PBD) is [1,2]diazepino[3,4-e]indole, or a derivative, or analog thereof. [
[0476] In some embodiments, a payload is a pyrrolobenzodiazepine (PBD), or a derivative, or analog thereof. In some embodiments, the pyrrolobenzodiazepine (PBD) is [1,2]diazepino[3,4-e]indole, or a derivative, or analog thereof. [
 . [0478] In some embodiments, a payload is an inhibitor of tubulin polymerization. In some embodiments, a payload is hemiasterlin, HTI-286, or a derivative, or analog thereof. [0479] In some embodiments, a payload is derived from:
. [0478] In some embodiments, a payload is an inhibitor of tubulin polymerization. In some embodiments, a payload is hemiasterlin, HTI-286, or a derivative, or analog thereof. [0479] In some embodiments, a payload is derived from: [0480] In some embodiments, a payload is:
[0480] In some embodiments, a payload is: [0481] In some embodiments, the payload comprises a topoisomerase inhibitor. In some embodiments, the payload comprises camptothecin, or a derivative, or analog thereof. In some embodiments, the payload comprises topotecan, irinotecan, silatecan, cositecan, exatecan, lurtotecan, gimatecan, belotecan, or rubitecan. [0482] In some embodiments, the payload comprises:
[0481] In some embodiments, the payload comprises a topoisomerase inhibitor. In some embodiments, the payload comprises camptothecin, or a derivative, or analog thereof. In some embodiments, the payload comprises topotecan, irinotecan, silatecan, cositecan, exatecan, lurtotecan, gimatecan, belotecan, or rubitecan. [0482] In some embodiments, the payload comprises: [0483] In some embodiments, the payload comprises:
[0483] In some embodiments, the payload comprises: [0484] In some embodiments, the payload comprises
[0484] In some embodiments, the payload comprises [0485] In some embodiments, the payload comprises
[0485] In some embodiments, the payload comprises [0486] In some embodiments, the payload comprises
[0486] In some embodiments, the payload comprises [0487] In some embodiments, the payload comprises
[0487] In some embodiments, the payload comprises . [0488] In some embodiments, the payload comprises
. [0488] In some embodiments, the payload comprises
 . [0489] In some embodiments, the payload comprises a polypeptide. In some embodiments, the polypeptide comprises one or more lysine, serine, threonine, or tyrosine residues. In some embodiments, the linker L1 is covalently bonded to a lysine, serine, threonine, or tyrosine residue present on the payload. In some embodiments, the polypeptide comprises one or more lysine residues. In some embodiments, the linker L1 is covalently bonded to a lysine residue present on the payload. [0490] In some embodiments, the payload comprises an N-terminal amino acid, wherein the linker L1 is covalently bonded to a N-terminal amino acid. [0491] In some embodiments, m is 1 to 20. [0492] In some embodiments, the payload is an immunomodulatory agent payload. [0493] In some embodiments, the immunomodulatory agent payload is an antibody payload. [0494] In some embodiments, the immunomodulatory agent payload is the immune checkpoint inhibitor payload. In some embodiments, the immune checkpoint inhibitor payload is a payload of pidilizumab, sintilimab, AMP-224, atezolizumab, durvalumab, BMS-936559, tremelimumab, indoximod, epacadostat, a TIGIT inhibitor (e.g., LAG-3, such as an anti-LAG-3 antibody; TIM-3, such as an anti-TIM-3 antibody), a B7 molecule, or a BTLA pathway antagonist. [0495] In some embodiments, the immune checkpoint inhibitor payload is an immune checkpoint inhibitor antibody payload. In some embodiments, the immune checkpoint inhibitor antibody payload is a PD-1 inhibitor payload. In some embodiments, the PD-1 inhibitor payload is a nivolumab, pembrolizumab, pidilizumab, sintilimab, or AMP-224 payload. [0496] In some embodiments, the immune checkpoint inhibitor antibody payload is a PD-L1 inhibitor payload. In some embodiments, the PD-L1 inhibitor payload is an atezolizumab, avelumab, durvalumab, or BMS-936559 payload. [0497] In some embodiments, the immune checkpoint inhibitor antibody payload is a CTLA4 inhibitor payload. In some embodiments, the CTLA4 inhibitor payload is an ipilimumab or tremelimumab payload. [0498] In some embodiments, the immune checkpoint inhibitor payload is an indoleamine 2,3- dioxygenase (IDO) inhibitor payload. In some embodiments, the IDO inhibitor payload is an indoximod or epacadostat payload. [0499] In some embodiments, the immunomodulatory agent payload is a cytokine payload. [0500] In some embodiments, the cytokine payload is an interferon, interleukin, tumor necrosis factor, erythropoietin, MIP3a, ICAM, macrophage colony stimulating factor, Erythropoietin (EPO), granulocyte colony stimulating factor (GCSF), or granulocyte-macrophage colony stimulating factor payload. [0501] In some embodiments, the interleukin payload is chosen from IL-1 to IL-40. In some embodiments, the interleukin payload is IL-2, IL-7, IL-12, IL-15, IL-18, or IL-21. [0502] In some embodiments, the immunomodulatory agent payload is a type 1 cytokine (IL-2, IL-12, TNF-B, IFN-g). [0503] In some embodiments, the cytokine payload is selected from the group consisting of IFN-alpha, IFN-beta, IFN-gamma, pegylated IFN-α, and apolipoprotein A-I fusion protein with IFN-α, interleukin, IL-2, IL-2 covalently bound to immunoglobulins (e.g., cergutuzumab amunaleukin, RO6874281), IL-2 covalently bound to PEG molecules (e.g., NKTR-214), IL-10, PEGylated IL-10 (e.g., pegilodecakin), IL- 7, IL-12, IL-15, recombinant aglycosylated IL-15, fusion protein of IL-15 with the binding domain of IL- 15Rα (e.g., RLI), triple fusion protein comprising human IL-15, the binding domain of IL-15Rα and apolipoprotein A-I, ALT-803 (IL-15 fused to IgG1 Fc domain), IL-18, IL-21, tumor necrosis factor, (TNF-alpha, TNF-beta), erythropoietin (EPO), MIP3a, ICAM, macrophage colony stimulating factor (M- CSF), granulocyte colony stimulating factor (GCSF), granulocyte-macrophage colony stimulating factor (GM-CSF), GM-CSF, and talimogene laherparepvec. [0504] In some embodiments, the immunomodulatory agent payload is the chemokine payload. [0505] In some embodiments, the chemokine payload is a CCL27, CCL28, CCL2, CCL3, CCL5, CXCL1, CXCL2, CXCL3, CXCL5, CXCL6, CXCL8, CXCL9, CXCL10, CXCL11, CXCL12, or CXCL14 payload. [0506] In some embodiments, the immunomodulatory agent payload is the chemokine antagonist payload. In some embodiments, the chemokine antagonist payload is a plerixafor payload. [0507] In some embodiments, the immunomodulatory agent is a monoclonal antibody specific to a cytokine or a cytokine receptor. [0508] In some embodiments, the immunomodulatory agent payload comprises a polypeptide. [0509] In some embodiments, the polypeptide comprises one or more lysine residues. [0510] In some embodiments, the polypeptide comprises one or more lysine, serine, threonine, or tyrosine residues. [0511] In some embodiments, the trans-cyclooctene is linked to one of the one or more lysine residues. [0512] In some embodiments, the trans-cyclooctene is independently linked to one or more lysine, serine, threonine, or tyrosine residues. [0513] In some embodiments, the polypeptide comprises an N-terminal amino acid, wherein an occurrence of the bioorthogonal moiety is linked to the N-terminal amino acid. [0514] In some embodiments, m is 1 to 20. In some embodiments, m is 1 to 10. In some embodiments, m is 1 to 5. In some embodiments, m is 20, 19, 18, 17, 16, 15, 14, 13, 12, 11, 10, 9, 8, 7, 6, 5, 4, 3, 2, or 1. In some embodiments, m is 1. [0515] In some embodiments, the payload-TCO conjugate is of Formula IX:
. [0489] In some embodiments, the payload comprises a polypeptide. In some embodiments, the polypeptide comprises one or more lysine, serine, threonine, or tyrosine residues. In some embodiments, the linker L1 is covalently bonded to a lysine, serine, threonine, or tyrosine residue present on the payload. In some embodiments, the polypeptide comprises one or more lysine residues. In some embodiments, the linker L1 is covalently bonded to a lysine residue present on the payload. [0490] In some embodiments, the payload comprises an N-terminal amino acid, wherein the linker L1 is covalently bonded to a N-terminal amino acid. [0491] In some embodiments, m is 1 to 20. [0492] In some embodiments, the payload is an immunomodulatory agent payload. [0493] In some embodiments, the immunomodulatory agent payload is an antibody payload. [0494] In some embodiments, the immunomodulatory agent payload is the immune checkpoint inhibitor payload. In some embodiments, the immune checkpoint inhibitor payload is a payload of pidilizumab, sintilimab, AMP-224, atezolizumab, durvalumab, BMS-936559, tremelimumab, indoximod, epacadostat, a TIGIT inhibitor (e.g., LAG-3, such as an anti-LAG-3 antibody; TIM-3, such as an anti-TIM-3 antibody), a B7 molecule, or a BTLA pathway antagonist. [0495] In some embodiments, the immune checkpoint inhibitor payload is an immune checkpoint inhibitor antibody payload. In some embodiments, the immune checkpoint inhibitor antibody payload is a PD-1 inhibitor payload. In some embodiments, the PD-1 inhibitor payload is a nivolumab, pembrolizumab, pidilizumab, sintilimab, or AMP-224 payload. [0496] In some embodiments, the immune checkpoint inhibitor antibody payload is a PD-L1 inhibitor payload. In some embodiments, the PD-L1 inhibitor payload is an atezolizumab, avelumab, durvalumab, or BMS-936559 payload. [0497] In some embodiments, the immune checkpoint inhibitor antibody payload is a CTLA4 inhibitor payload. In some embodiments, the CTLA4 inhibitor payload is an ipilimumab or tremelimumab payload. [0498] In some embodiments, the immune checkpoint inhibitor payload is an indoleamine 2,3- dioxygenase (IDO) inhibitor payload. In some embodiments, the IDO inhibitor payload is an indoximod or epacadostat payload. [0499] In some embodiments, the immunomodulatory agent payload is a cytokine payload. [0500] In some embodiments, the cytokine payload is an interferon, interleukin, tumor necrosis factor, erythropoietin, MIP3a, ICAM, macrophage colony stimulating factor, Erythropoietin (EPO), granulocyte colony stimulating factor (GCSF), or granulocyte-macrophage colony stimulating factor payload. [0501] In some embodiments, the interleukin payload is chosen from IL-1 to IL-40. In some embodiments, the interleukin payload is IL-2, IL-7, IL-12, IL-15, IL-18, or IL-21. [0502] In some embodiments, the immunomodulatory agent payload is a type 1 cytokine (IL-2, IL-12, TNF-B, IFN-g). [0503] In some embodiments, the cytokine payload is selected from the group consisting of IFN-alpha, IFN-beta, IFN-gamma, pegylated IFN-α, and apolipoprotein A-I fusion protein with IFN-α, interleukin, IL-2, IL-2 covalently bound to immunoglobulins (e.g., cergutuzumab amunaleukin, RO6874281), IL-2 covalently bound to PEG molecules (e.g., NKTR-214), IL-10, PEGylated IL-10 (e.g., pegilodecakin), IL- 7, IL-12, IL-15, recombinant aglycosylated IL-15, fusion protein of IL-15 with the binding domain of IL- 15Rα (e.g., RLI), triple fusion protein comprising human IL-15, the binding domain of IL-15Rα and apolipoprotein A-I, ALT-803 (IL-15 fused to IgG1 Fc domain), IL-18, IL-21, tumor necrosis factor, (TNF-alpha, TNF-beta), erythropoietin (EPO), MIP3a, ICAM, macrophage colony stimulating factor (M- CSF), granulocyte colony stimulating factor (GCSF), granulocyte-macrophage colony stimulating factor (GM-CSF), GM-CSF, and talimogene laherparepvec. [0504] In some embodiments, the immunomodulatory agent payload is the chemokine payload. [0505] In some embodiments, the chemokine payload is a CCL27, CCL28, CCL2, CCL3, CCL5, CXCL1, CXCL2, CXCL3, CXCL5, CXCL6, CXCL8, CXCL9, CXCL10, CXCL11, CXCL12, or CXCL14 payload. [0506] In some embodiments, the immunomodulatory agent payload is the chemokine antagonist payload. In some embodiments, the chemokine antagonist payload is a plerixafor payload. [0507] In some embodiments, the immunomodulatory agent is a monoclonal antibody specific to a cytokine or a cytokine receptor. [0508] In some embodiments, the immunomodulatory agent payload comprises a polypeptide. [0509] In some embodiments, the polypeptide comprises one or more lysine residues. [0510] In some embodiments, the polypeptide comprises one or more lysine, serine, threonine, or tyrosine residues. [0511] In some embodiments, the trans-cyclooctene is linked to one of the one or more lysine residues. [0512] In some embodiments, the trans-cyclooctene is independently linked to one or more lysine, serine, threonine, or tyrosine residues. [0513] In some embodiments, the polypeptide comprises an N-terminal amino acid, wherein an occurrence of the bioorthogonal moiety is linked to the N-terminal amino acid. [0514] In some embodiments, m is 1 to 20. In some embodiments, m is 1 to 10. In some embodiments, m is 1 to 5. In some embodiments, m is 20, 19, 18, 17, 16, 15, 14, 13, 12, 11, 10, 9, 8, 7, 6, 5, 4, 3, 2, or 1. In some embodiments, m is 1. [0515] In some embodiments, the payload-TCO conjugate is of Formula IX: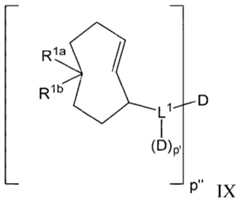 or a pharmaceutically acceptable salt thereof, wherein: R1a, at each occurrence, is independently selected from the group consisting of hydrogen, C1-4alkyl, and C1-4haloalkyl; R1b, at each occurrence, is independently selected from the group consisting of hydrogen, C1-4alkyl, C1-4haloalkyl, C(O)OH, C(O)OC1-4alkyl, C(O)N(R1c)CHR1eCO2H, C(O)N(R1c)CHR1eC(O)OC1-4alkyl, C(O)N(R1c)–C1-6alkylene–CO2H, and C(O)N(R1c)–C1-6alkylene–C(O)OC1-4alkyl; R1c, at each occurrence, is independently hydrogen or C1-4alkyl; R1e, at each occurrence, is independently –C1-4alkylene–CO2H, –C1-4alkylene–CONH2, or –C1-4alkylene–OH; D, at each occurrence, is independently a payload; L1, at each occurrence, is independently a linker; p’, at each occurrence, is independently 0, 1, or 2; and p’’, at each occurrence, is independently 1, 2, or 3. [0516] In some embodiments, D, at each occurrence, is independently selected from the group consisting of an anticancer agent payload, a toll-like receptor (TLR) agonist payload and a stimulator of interferon genes (STING) agonist payload. [0517] In some embodiments, R1a is hydrogen. [0518] In some embodiments, R1a is C1-4alkyl. [0519] In some embodiments, R1a is CH3. [0520] In some embodiments, R1b is selected from the group consisting of -C(O)OH, -C(O)OC1-4alkyl, -C(O)N(R1c)CHR1eCO2H, -C(O)N(R1c)CHR1eC(O)OC1-4alkyl, -C(O)N(R1c)–C1-6alkylene–CO2H, and -C(O)N(R1c)–C1-6alkylene–C(O)OC1-4alkyl. [0521] In some embodiments, R1b is selected from the group consisting of C(O)OH, C(O)N(R1c)CHR1eCO2H, and C(O)N(R1c)CH2CO2H. [0522] In some embodiments, R1b is selected from the group consisting of –NR1c–CH2CH2–N(CH3)3+, –N(R1c)–CH2CH2–SO3H, –N(R1c)–(CH2CH2O)3–CH2CH2N((CH2CH2O)3–CH2CH2–CO2H)2, and –N(R1c)–CH(CH2O–CH2CH2–CO2H)2. [0523] In some embodiments, the trans-cyclooctene moiety (G) is: H
or a pharmaceutically acceptable salt thereof, wherein: R1a, at each occurrence, is independently selected from the group consisting of hydrogen, C1-4alkyl, and C1-4haloalkyl; R1b, at each occurrence, is independently selected from the group consisting of hydrogen, C1-4alkyl, C1-4haloalkyl, C(O)OH, C(O)OC1-4alkyl, C(O)N(R1c)CHR1eCO2H, C(O)N(R1c)CHR1eC(O)OC1-4alkyl, C(O)N(R1c)–C1-6alkylene–CO2H, and C(O)N(R1c)–C1-6alkylene–C(O)OC1-4alkyl; R1c, at each occurrence, is independently hydrogen or C1-4alkyl; R1e, at each occurrence, is independently –C1-4alkylene–CO2H, –C1-4alkylene–CONH2, or –C1-4alkylene–OH; D, at each occurrence, is independently a payload; L1, at each occurrence, is independently a linker; p’, at each occurrence, is independently 0, 1, or 2; and p’’, at each occurrence, is independently 1, 2, or 3. [0516] In some embodiments, D, at each occurrence, is independently selected from the group consisting of an anticancer agent payload, a toll-like receptor (TLR) agonist payload and a stimulator of interferon genes (STING) agonist payload. [0517] In some embodiments, R1a is hydrogen. [0518] In some embodiments, R1a is C1-4alkyl. [0519] In some embodiments, R1a is CH3. [0520] In some embodiments, R1b is selected from the group consisting of -C(O)OH, -C(O)OC1-4alkyl, -C(O)N(R1c)CHR1eCO2H, -C(O)N(R1c)CHR1eC(O)OC1-4alkyl, -C(O)N(R1c)–C1-6alkylene–CO2H, and -C(O)N(R1c)–C1-6alkylene–C(O)OC1-4alkyl. [0521] In some embodiments, R1b is selected from the group consisting of C(O)OH, C(O)N(R1c)CHR1eCO2H, and C(O)N(R1c)CH2CO2H. [0522] In some embodiments, R1b is selected from the group consisting of –NR1c–CH2CH2–N(CH3)3+, –N(R1c)–CH2CH2–SO3H, –N(R1c)–(CH2CH2O)3–CH2CH2N((CH2CH2O)3–CH2CH2–CO2H)2, and –N(R1c)–CH(CH2O–CH2CH2–CO2H)2. [0523] In some embodiments, the trans-cyclooctene moiety (G) is: H [0524] In some embodiments, the trans-cyclooctene moiety is: H H
[0524] In some embodiments, the trans-cyclooctene moiety is: H H HO [0525] In some embodiments, the trans-cyclooctene moiety is O
HO [0525] In some embodiments, the trans-cyclooctene moiety is O embodiments, the trans-cyclooctene moiety is
embodiments, the trans-cyclooctene moiety is O HO [0526] In some embodiments, the trans-cyclooctene moiety is
O HO [0526] In some embodiments, the trans-cyclooctene moiety is . In some embodiments, the trans-cyclooctene moiety is
. In some embodiments, the trans-cyclooctene moiety is OH O HO [0527] In some embodiments, the trans-cyclooctene moiety is O
OH O HO [0527] In some embodiments, the trans-cyclooctene moiety is O [0528] In some embodiments, the trans-cyclooctene moiety is
[0528] In some embodiments, the trans-cyclooctene moiety is [0529] In some embodiments, the trans-cyclooctene moiety is
[0529] In some embodiments, the trans-cyclooctene moiety is [0530] In some embodiments, the trans-cyclooctene moiety is .
[0530] In some embodiments, the trans-cyclooctene moiety is . [0531] In some embodiments, the trans-cyclooctene moiety is
[0531] In some embodiments, the trans-cyclooctene moiety is -OH, 2-aminoethanesulfonic acid, an N-linked natural or unnatural amino acid, or an optionally substituted ethylenediamine; wherein R2 may be optionally further substituted with a polyether. [0532] In some embodiments, the trans-cyclooctene moiety comprises
-OH, 2-aminoethanesulfonic acid, an N-linked natural or unnatural amino acid, or an optionally substituted ethylenediamine; wherein R2 may be optionally further substituted with a polyether. [0532] In some embodiments, the trans-cyclooctene moiety comprises . [0533] In some embodiments, the trans-cyclooctene moiety comprises
. [0533] In some embodiments, the trans-cyclooctene moiety comprises . [0534] In some embodiments, the trans-cyclooctene moiety of comprises
. [0534] In some embodiments, the trans-cyclooctene moiety of comprises [0535] In some embodiments, the trans-cyclooctene moiety of comprises
[0535] In some embodiments, the trans-cyclooctene moiety of comprises [0536] In some embodiments, R1e is –CH2CO2H, –CH2CH2CO2H, –CH2CONH2, –CH2CH2CONH2, –CH2OH, or –CH(CH3)OH. [0537] In some embodiments, R1e is –C1-4alkylene–CO2H. [0538] In some embodiments, R1e is –CH2CO2H. [0539] In some embodiments, R1b is -C(O)N(R1c)–C1-6alkylene–CO2H. [0540] In some embodiments, R1b is -C(O)N(R1c)CH2CO2H. [0541] In some embodiments, R1c is hydrogen. [0542] In some embodiments, R1b is hydrogen. [0543] In some embodiments, R1b is C(O)OH. [0544] In some embodiments, the methods comprise targeted therapy. In some embodiments, this can be achieved by making use of a TCO targeting moiety and one or more pharmaceutically active agents (i.e. a drug or a radioactive isotope for radiation therapy or radioligand therapy). Suitable drugs for use in the context of targeted drug delivery are known in the art. Optionally, the therapeutic probe can also comprise a detectable label, such as one or more imaging agents. A “radionuclide” (used interchangeably herein with “radioligand”) used in the trans-cyclooctene moieties described herein, comprises a chelating agent and an isotope; such as an isotope selected from the group consisting of24Na,32P,33P,47Sc,59Fe,67Cu,76As,77As,80Br,82Br,89Sr,90Nb,90Y,103Ru,105Rh,109Pd,111Ag,111In,121Sn,127Te,131I,140La,141Ce,142Pr,143Pr,144Pr,149Pm,149Tb,151Pm,153Sm,159Gd,161Tb,165Dy,166Ho,169Er,172Tm,175Yb,177Lu,186Re,188Re,198Au,199Au,211At,211Bi,212Bi,212Pb,213Bi,214Bi,223Ra, and225Ac. Radionuclides can be delivered via direct conjugation or chelation with a chelating agent. Exemplary radionuclides, chelating agents, and linkers for potential TCO-conjugate payloads are described below. [0545] Bifunctional Chelating Agents (BFC):
[0536] In some embodiments, R1e is –CH2CO2H, –CH2CH2CO2H, –CH2CONH2, –CH2CH2CONH2, –CH2OH, or –CH(CH3)OH. [0537] In some embodiments, R1e is –C1-4alkylene–CO2H. [0538] In some embodiments, R1e is –CH2CO2H. [0539] In some embodiments, R1b is -C(O)N(R1c)–C1-6alkylene–CO2H. [0540] In some embodiments, R1b is -C(O)N(R1c)CH2CO2H. [0541] In some embodiments, R1c is hydrogen. [0542] In some embodiments, R1b is hydrogen. [0543] In some embodiments, R1b is C(O)OH. [0544] In some embodiments, the methods comprise targeted therapy. In some embodiments, this can be achieved by making use of a TCO targeting moiety and one or more pharmaceutically active agents (i.e. a drug or a radioactive isotope for radiation therapy or radioligand therapy). Suitable drugs for use in the context of targeted drug delivery are known in the art. Optionally, the therapeutic probe can also comprise a detectable label, such as one or more imaging agents. A “radionuclide” (used interchangeably herein with “radioligand”) used in the trans-cyclooctene moieties described herein, comprises a chelating agent and an isotope; such as an isotope selected from the group consisting of24Na,32P,33P,47Sc,59Fe,67Cu,76As,77As,80Br,82Br,89Sr,90Nb,90Y,103Ru,105Rh,109Pd,111Ag,111In,121Sn,127Te,131I,140La,141Ce,142Pr,143Pr,144Pr,149Pm,149Tb,151Pm,153Sm,159Gd,161Tb,165Dy,166Ho,169Er,172Tm,175Yb,177Lu,186Re,188Re,198Au,199Au,211At,211Bi,212Bi,212Pb,213Bi,214Bi,223Ra, and225Ac. Radionuclides can be delivered via direct conjugation or chelation with a chelating agent. Exemplary radionuclides, chelating agents, and linkers for potential TCO-conjugate payloads are described below. [0545] Bifunctional Chelating Agents (BFC): [0546] Therapeutic Radionuclides:
[0546] Therapeutic Radionuclides: [0547] In some embodiments, the linker comprises a linker as shown below, where BFC refers to a bifunctional chelating agent (e.g., as disclosed herein) and the wavy line indicates the point of attachment to the TCO.
[0547] In some embodiments, the linker comprises a linker as shown below, where BFC refers to a bifunctional chelating agent (e.g., as disclosed herein) and the wavy line indicates the point of attachment to the TCO.
 [0548] In some embodiments, the payload-TCO conjugate is of Formula IX:
[0548] In some embodiments, the payload-TCO conjugate is of Formula IX: or a pharmaceutically acceptable salt thereof, wherein R1a, at each occurrence, is independently selected from the group consisting of hydrogen, C1-4alkyl, and C1-4haloalkyl; R1b, at each occurrence, is independently selected from the group consisting of hydrogen, C1-4alkyl, C1-4haloalkyl, C(O)OH, C(O)OC1-4alkyl, C(O)N(R1c)CHR1eCO2H, C(O)N(R1c)CHR1eC(O)OC1-4alkyl, C(O)N(R1c)–C1-6alkylene–CO2H, and C(O)N(R1c)–C1-6alkylene–C(O)OC1-4alkyl; R1c, at each occurrence, is independently hydrogen or C1-4alkyl; R1e, at each occurrence, is independently –C1-4alkylene–CO2H, –C1-4alkylene–CONH2, or –C1-4alkylene–OH; D, at each occurrence, is independently a payload; L1, at each occurrence, is independently a linker; p’, at each occurrence, is independently 0, 1, or 2; and p’’, at each occurrence, is independently 1, 2, or 3. [0549] In some embodiments, D, at each occurrence, is a chelating agent suitable for use in radioligand therapy. [0550] In some embodiments, D, at each occurrence, is independently selected from DOTA: 1,4,7,10- tetraazacyclododecane-1,4,7,10-tetraacetic acid, DOTAGA: 1,4,7,10-tetraazacyclododececane, 1- (glutaric acid)-4,7,10-triacetic acid, DTPA: diethylenetriaminepentaacetic acid, NTA: nitrilotriacetic acid, EDTA: ethylenediaminetetraacetic acid, DO3A: 1,4,7,10-tetraazacyclododecane-1,4,7-triacetic acid, NOTA: 1,4,7-triazacyclononane-1,4,7-triacetic acid, NODAGA: 1-(1,3-carboxypropyl)-4,7- carboxymethyl-1,4,7-triazacyclononane trizoxetan, tetraxetan, macrocyclic maleimides, or a mixture thereof. [0551] In some embodiments, the payload-TCO conjugate is of Formula IXA:
or a pharmaceutically acceptable salt thereof, wherein R1a, at each occurrence, is independently selected from the group consisting of hydrogen, C1-4alkyl, and C1-4haloalkyl; R1b, at each occurrence, is independently selected from the group consisting of hydrogen, C1-4alkyl, C1-4haloalkyl, C(O)OH, C(O)OC1-4alkyl, C(O)N(R1c)CHR1eCO2H, C(O)N(R1c)CHR1eC(O)OC1-4alkyl, C(O)N(R1c)–C1-6alkylene–CO2H, and C(O)N(R1c)–C1-6alkylene–C(O)OC1-4alkyl; R1c, at each occurrence, is independently hydrogen or C1-4alkyl; R1e, at each occurrence, is independently –C1-4alkylene–CO2H, –C1-4alkylene–CONH2, or –C1-4alkylene–OH; D, at each occurrence, is independently a payload; L1, at each occurrence, is independently a linker; p’, at each occurrence, is independently 0, 1, or 2; and p’’, at each occurrence, is independently 1, 2, or 3. [0549] In some embodiments, D, at each occurrence, is a chelating agent suitable for use in radioligand therapy. [0550] In some embodiments, D, at each occurrence, is independently selected from DOTA: 1,4,7,10- tetraazacyclododecane-1,4,7,10-tetraacetic acid, DOTAGA: 1,4,7,10-tetraazacyclododececane, 1- (glutaric acid)-4,7,10-triacetic acid, DTPA: diethylenetriaminepentaacetic acid, NTA: nitrilotriacetic acid, EDTA: ethylenediaminetetraacetic acid, DO3A: 1,4,7,10-tetraazacyclododecane-1,4,7-triacetic acid, NOTA: 1,4,7-triazacyclononane-1,4,7-triacetic acid, NODAGA: 1-(1,3-carboxypropyl)-4,7- carboxymethyl-1,4,7-triazacyclononane trizoxetan, tetraxetan, macrocyclic maleimides, or a mixture thereof. [0551] In some embodiments, the payload-TCO conjugate is of Formula IXA: or a pharmaceutically acceptable salt thereof, wherein R1a is selected from the group consisting of hydrogen, C1-4alkyl, and C1-4haloalkyl; D is a payload; and L1 is a linker. [0552] In some embodiments, D is a chelating agent suitable for use in radioligand therapy. Exemplary, non-limiting, chelating agents include DOTA: 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid, DOTAGA: 1,4,7,10-tetraazacyclododececane, 1-(glutaric acid)-4,7,10-triacetic acid, DTPA: diethylenetriaminepentaacetic acid, NTA: nitrilotriacetic acid, EDTA: ethylenediaminetetraacetic acid, DO3A: 1,4,7,10-tetraazacyclododecane-1,4,7-triacetic acid, NOTA: 1,4,7-triazacyclononane-1,4,7- triacetic acid, NODAGA: 1-(1,3-carboxypropyl)-4,7-carboxymethyl-1,4,7-triazacyclononane trizoxetan, tetraxetan, macrocyclic maleimides, or a mixture thereof. [0553] In some embodiments, the chelating agent is complexed to a radioligand or radionuclide. [0554] In some embodiments, linker L1 may have 1 to 100 linking atoms, and may include ethylene-oxy groups, amines, esters, amides, carbamates, carbonates, and ketone functional groups. For example, linkers may have from 1 to 50 linking atoms, or from 5 to 50 linking atoms, or from 10 to 50 linking atoms, or from 1 to 40 linking atoms, or from 1 to 30 linking atoms, or from 1 to 20 linking atoms, or from 1 to 10 linking atoms, or from 1 to 5 linking atoms, or from 5 to 30 linking atoms, or from 10 to 30 linking atoms, or from 5 to 40 linking atoms, or from 5 to 50 linking atoms, or from 10 to 50 linking atoms. [0555] In some embodiments, linker L1 may comprise one or more (e.g., 1-10 or 1-5) chain heteroatoms (e.g., O, N, S) and one or more (e.g., 1-10 or 1-5) alkylene, alkenylene, alkynylene, arylene, heteroarylene, cycloalkylene or heterocycloalkylene moieties; wherein each alkylene, alkenylene, alkynylene, arylene, heteroarylene, cycloalkylene or heterocycloalkylene moiety, may be independently optionally substituted with one to five substituents independently selected from oxo, halo, C1-4 alkyl, C1-4 alkoxy, and C1-4 haloalkyl. [0556] In some embodiments, linker L1 may be of the formula: -Y10-(CH2)n’-Y20-(CH2)m’’-Y30- wherein: each of Y10, Y20, and Y30 are independently a bond, -NR110-, -O-, -S(O)0-2-, -NR110C(O)-, -C(O)NR110-, -NR110S(O)2-, -S(O)2NR110-, -CR120=N-NR110-, -NR110-N=CR120-, -C(O)-, -OC(O)-, -OC(O)O-, alkylene, alkenylene, alkynylene, arylene, heteroarylene, cycloalkylene or heterocycloalkylene; wherein each alkylene, alkenylene, alkynylene, arylene, heteroarylene, cycloalkylene or heterocycloalkylene is independently optionally substituted with one to five substituents independently selected from oxo, halo, C1-4 alkyl, C1-4 alkoxy, and C1-4 haloalkyl; each R110 is independently hydrogen, C1-4 alkyl, C1-4 haloalkyl, aryl, heteroaryl, cycloalkyl or heterocyclyl; each R120 is independently hydrogen, C1-4 alkyl, C1-4 haloalkyl, aryl, heteroaryl, cycloalkyl or heterocyclyl; and n’ and m’’ are each independently 0, 1, 2, 3, 4, 5, 6, 7, or 8. [0557] In certain embodiments, the linker is a bond. [0558] In certain embodiments, the linker is not a bond. In certain embodiments, each R110 is independently hydrogen, C1-4 alkyl, C1-4 haloalkyl, aryl, heteroaryl, cycloalkyl or heterocyclyl; and each R120 is independently hydrogen, C1-4 alkyl, C1-4 haloalkyl, aryl, heteroaryl, cycloalkyl or heterocyclyl. [0559] Representative linkers include, but are not limited to, those shown below:
or a pharmaceutically acceptable salt thereof, wherein R1a is selected from the group consisting of hydrogen, C1-4alkyl, and C1-4haloalkyl; D is a payload; and L1 is a linker. [0552] In some embodiments, D is a chelating agent suitable for use in radioligand therapy. Exemplary, non-limiting, chelating agents include DOTA: 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid, DOTAGA: 1,4,7,10-tetraazacyclododececane, 1-(glutaric acid)-4,7,10-triacetic acid, DTPA: diethylenetriaminepentaacetic acid, NTA: nitrilotriacetic acid, EDTA: ethylenediaminetetraacetic acid, DO3A: 1,4,7,10-tetraazacyclododecane-1,4,7-triacetic acid, NOTA: 1,4,7-triazacyclononane-1,4,7- triacetic acid, NODAGA: 1-(1,3-carboxypropyl)-4,7-carboxymethyl-1,4,7-triazacyclononane trizoxetan, tetraxetan, macrocyclic maleimides, or a mixture thereof. [0553] In some embodiments, the chelating agent is complexed to a radioligand or radionuclide. [0554] In some embodiments, linker L1 may have 1 to 100 linking atoms, and may include ethylene-oxy groups, amines, esters, amides, carbamates, carbonates, and ketone functional groups. For example, linkers may have from 1 to 50 linking atoms, or from 5 to 50 linking atoms, or from 10 to 50 linking atoms, or from 1 to 40 linking atoms, or from 1 to 30 linking atoms, or from 1 to 20 linking atoms, or from 1 to 10 linking atoms, or from 1 to 5 linking atoms, or from 5 to 30 linking atoms, or from 10 to 30 linking atoms, or from 5 to 40 linking atoms, or from 5 to 50 linking atoms, or from 10 to 50 linking atoms. [0555] In some embodiments, linker L1 may comprise one or more (e.g., 1-10 or 1-5) chain heteroatoms (e.g., O, N, S) and one or more (e.g., 1-10 or 1-5) alkylene, alkenylene, alkynylene, arylene, heteroarylene, cycloalkylene or heterocycloalkylene moieties; wherein each alkylene, alkenylene, alkynylene, arylene, heteroarylene, cycloalkylene or heterocycloalkylene moiety, may be independently optionally substituted with one to five substituents independently selected from oxo, halo, C1-4 alkyl, C1-4 alkoxy, and C1-4 haloalkyl. [0556] In some embodiments, linker L1 may be of the formula: -Y10-(CH2)n’-Y20-(CH2)m’’-Y30- wherein: each of Y10, Y20, and Y30 are independently a bond, -NR110-, -O-, -S(O)0-2-, -NR110C(O)-, -C(O)NR110-, -NR110S(O)2-, -S(O)2NR110-, -CR120=N-NR110-, -NR110-N=CR120-, -C(O)-, -OC(O)-, -OC(O)O-, alkylene, alkenylene, alkynylene, arylene, heteroarylene, cycloalkylene or heterocycloalkylene; wherein each alkylene, alkenylene, alkynylene, arylene, heteroarylene, cycloalkylene or heterocycloalkylene is independently optionally substituted with one to five substituents independently selected from oxo, halo, C1-4 alkyl, C1-4 alkoxy, and C1-4 haloalkyl; each R110 is independently hydrogen, C1-4 alkyl, C1-4 haloalkyl, aryl, heteroaryl, cycloalkyl or heterocyclyl; each R120 is independently hydrogen, C1-4 alkyl, C1-4 haloalkyl, aryl, heteroaryl, cycloalkyl or heterocyclyl; and n’ and m’’ are each independently 0, 1, 2, 3, 4, 5, 6, 7, or 8. [0557] In certain embodiments, the linker is a bond. [0558] In certain embodiments, the linker is not a bond. In certain embodiments, each R110 is independently hydrogen, C1-4 alkyl, C1-4 haloalkyl, aryl, heteroaryl, cycloalkyl or heterocyclyl; and each R120 is independently hydrogen, C1-4 alkyl, C1-4 haloalkyl, aryl, heteroaryl, cycloalkyl or heterocyclyl. [0559] Representative linkers include, but are not limited to, those shown below:
 . [0560] Representative linkers include, but are not limited to, those shown below:
. [0560] Representative linkers include, but are not limited to, those shown below: [0561] In some embodiments, linker L1 may comprise one or more of polyethylene glycol (e.g., PEG having an average molecular weight of from 300 g/mol to 10,000 g/mol), ethylene-1,2- diylbis(methylcarbamate, an arylene (e.g., phenylene), ethylene-oxy, amine, ester, amide, carbamate, ketone (i.e., formyl), or carbonate. In some embodiments, linker L1 may comprise
[0561] In some embodiments, linker L1 may comprise one or more of polyethylene glycol (e.g., PEG having an average molecular weight of from 300 g/mol to 10,000 g/mol), ethylene-1,2- diylbis(methylcarbamate, an arylene (e.g., phenylene), ethylene-oxy, amine, ester, amide, carbamate, ketone (i.e., formyl), or carbonate. In some embodiments, linker L1 may comprise [0562] In some embodiments, linker L1 may comprise one or more natural or unnatural amino acids, which may be referred to as a peptide linker. Where the drug (D) comprises an amino moiety, the linker may be bound thereto using a peptide linker made up of a carboxylic acyl unit, and one or more amino acids making up a protein or peptide sequence. In some embodiments, linker L1 may also contain a self- immolating spacer which spaces the drug and the protein peptide sequence. [0563] In some embodiments, linker L1 may be a peptide linker represented by “A—Y—Z—X—W” in which “A” is the carboxylic acyl unit, “Y” and “Z” are each one or more natural or unnatural amino acids and together form a peptide sequence, and “X” and “W” are optional additional linkers having from 1 to 50 linking atoms, or from 5 to 10 linking atoms, or from 1 to 10 linking atoms which spaces the peptide and the drug, D, or the bioorthogonal moiety. In certain embodiments, one or more of the amino acids in the peptide linker is N-methylated. [0564] In some embodiments, Y may be at least one amino acid selected from the group consisting of alanine, valine, leucine, isoleucine, methionine, phenylalanine, tryptophan and proline. In some embodiments, Y may be at least one amino acid selected from the group consisting of phenylalanine, alanine, and valine. [0565] In some embodiments, Z may be at least one amino acid selected from the group consisting of alanine, lysine, lysine protected with acetyl or formyl, arginine, arginine protected with tosyl or nitro groups, histidine, ornithine, ornithine protected with acetyl or formyl, and citrulline. In some embodiments, Z may be at least one amino acid selected from the group consisting of alanine, lysine and citrulline. [0566] In some embodiments, exemplary Y-Z combinations include Valine-Citrulline; Valine-Alanine; and Alanine-Alanine. [0567] In certain embodiments, A is -OC(O)-. [0568] In certain embodiments, X is -OC(O)-. [0569] In certain embodiments, W is -OC(O)-. In certain embodiments, X is absent and W is -OC(O)-. [0570] In certain embodiments, —X—
[0562] In some embodiments, linker L1 may comprise one or more natural or unnatural amino acids, which may be referred to as a peptide linker. Where the drug (D) comprises an amino moiety, the linker may be bound thereto using a peptide linker made up of a carboxylic acyl unit, and one or more amino acids making up a protein or peptide sequence. In some embodiments, linker L1 may also contain a self- immolating spacer which spaces the drug and the protein peptide sequence. [0563] In some embodiments, linker L1 may be a peptide linker represented by “A—Y—Z—X—W” in which “A” is the carboxylic acyl unit, “Y” and “Z” are each one or more natural or unnatural amino acids and together form a peptide sequence, and “X” and “W” are optional additional linkers having from 1 to 50 linking atoms, or from 5 to 10 linking atoms, or from 1 to 10 linking atoms which spaces the peptide and the drug, D, or the bioorthogonal moiety. In certain embodiments, one or more of the amino acids in the peptide linker is N-methylated. [0564] In some embodiments, Y may be at least one amino acid selected from the group consisting of alanine, valine, leucine, isoleucine, methionine, phenylalanine, tryptophan and proline. In some embodiments, Y may be at least one amino acid selected from the group consisting of phenylalanine, alanine, and valine. [0565] In some embodiments, Z may be at least one amino acid selected from the group consisting of alanine, lysine, lysine protected with acetyl or formyl, arginine, arginine protected with tosyl or nitro groups, histidine, ornithine, ornithine protected with acetyl or formyl, and citrulline. In some embodiments, Z may be at least one amino acid selected from the group consisting of alanine, lysine and citrulline. [0566] In some embodiments, exemplary Y-Z combinations include Valine-Citrulline; Valine-Alanine; and Alanine-Alanine. [0567] In certain embodiments, A is -OC(O)-. [0568] In certain embodiments, X is -OC(O)-. [0569] In certain embodiments, W is -OC(O)-. In certain embodiments, X is absent and W is -OC(O)-. [0570] In certain embodiments, —X— [0571] In certain embodiments, —X—
[0571] In certain embodiments, —X— [0572] In certain embodiments, the peptide linker is specifically tailored so that it will be selectively cleaved (e.g., enzymatically cleaved) releasing the drug, such as by one or more of the tumor-associated proteases. [0573] In certain embodiments, the peptide linker has a chain length of two to four amino acid residues (i.e., a di-, tri-, or tetra-peptide). It will be understood, however, that peptide linkers up to five, six, seven, or eight amino acid residues may also suitably be employed. [0574] In certain embodiments, the peptide linker is Phe-Lys, Val-Lys, Val-Ala, Ala-Ala, Phe-Phe-Lys, D-Phe-Phe-Lys, Gly-Phe-Lys, Ala-Lys, Val-Cit, Phe-Cit, Leu-Cit, Ile-Cit, Trp-Cit, Phe-Ala, Gly-Phe- Leu-Gly [SEQ ID NO: ], Ala-Leu-Ala-Leu [SEQ ID NO: ], Phe-N9-tosyl-Arg, or Phe-N9-Nitro-Arg. In certain embodiments, the peptide linker is Phe-Lys, Val-Lys, Val-Ala, Ala-Ala, Val-Val, Val-Cit, or D- Phe-L-Phe-Lys. In certain embodiments, the peptide linker is Val-Cit, Val-Ala, or Ala-Ala. [0575] In certain embodiments, linker L1 is:
[0572] In certain embodiments, the peptide linker is specifically tailored so that it will be selectively cleaved (e.g., enzymatically cleaved) releasing the drug, such as by one or more of the tumor-associated proteases. [0573] In certain embodiments, the peptide linker has a chain length of two to four amino acid residues (i.e., a di-, tri-, or tetra-peptide). It will be understood, however, that peptide linkers up to five, six, seven, or eight amino acid residues may also suitably be employed. [0574] In certain embodiments, the peptide linker is Phe-Lys, Val-Lys, Val-Ala, Ala-Ala, Phe-Phe-Lys, D-Phe-Phe-Lys, Gly-Phe-Lys, Ala-Lys, Val-Cit, Phe-Cit, Leu-Cit, Ile-Cit, Trp-Cit, Phe-Ala, Gly-Phe- Leu-Gly [SEQ ID NO: ], Ala-Leu-Ala-Leu [SEQ ID NO: ], Phe-N9-tosyl-Arg, or Phe-N9-Nitro-Arg. In certain embodiments, the peptide linker is Phe-Lys, Val-Lys, Val-Ala, Ala-Ala, Val-Val, Val-Cit, or D- Phe-L-Phe-Lys. In certain embodiments, the peptide linker is Val-Cit, Val-Ala, or Ala-Ala. [0575] In certain embodiments, linker L1 is: , ,
, , [0576] The foregoing linkers may attach on the right-hand side to amino acid side chains of D such
[0576] The foregoing linkers may attach on the right-hand side to amino acid side chains of D such
 [0577] In some embodiments, the payload is covalently bonded to the linker through an amide bond; e.g., the payload may be an amine-containing payload for attachment of the payload to a carbonyl group of the linker, or, in other cases, the payload may be a carboxyl-containing payload for attachment of the payload to an amine group of the linker. In some instances, the payload and linker, together form a carbamate group; e.g., the payload may be an amine-containing payload for attachment of the payload to an acyloxy group of the linker. In some instances, the payload and linker, together form a carbonate group; e.g., the payload may be a hydroxy-containing payload for attachment of the payload to an acyloxy group of the linker. O [0578] In some embodiments, L1 is L3a is a bond or C1-6alkylene;
[0577] In some embodiments, the payload is covalently bonded to the linker through an amide bond; e.g., the payload may be an amine-containing payload for attachment of the payload to a carbonyl group of the linker, or, in other cases, the payload may be a carboxyl-containing payload for attachment of the payload to an amine group of the linker. In some instances, the payload and linker, together form a carbamate group; e.g., the payload may be an amine-containing payload for attachment of the payload to an acyloxy group of the linker. In some instances, the payload and linker, together form a carbonate group; e.g., the payload may be a hydroxy-containing payload for attachment of the payload to an acyloxy group of the linker. O [0578] In some embodiments, L1 is L3a is a bond or C1-6alkylene; L4a is a bond, –NHN:, –N(R10)–C2-6alkylene–N(R11)–, –N(R12)–C2-3alkylene–N(R13)C(O)–, –N(R10)–C1-6alkylene–C(O)NHN:, –NHNHC(O)C1-6alkylene–C(O)NHN:, –CH(NHC(O)R14)C1-4alkylene–S–S–C1-4alkylene–OC(O)–, –NHNHC(O)CH(NHC(O)R15)CH2C(O)–, –C1-6alkylene–CH(Gx)OC(O)–,
L4a is a bond, –NHN:, –N(R10)–C2-6alkylene–N(R11)–, –N(R12)–C2-3alkylene–N(R13)C(O)–, –N(R10)–C1-6alkylene–C(O)NHN:, –NHNHC(O)C1-6alkylene–C(O)NHN:, –CH(NHC(O)R14)C1-4alkylene–S–S–C1-4alkylene–OC(O)–, –NHNHC(O)CH(NHC(O)R15)CH2C(O)–, –C1-6alkylene–CH(Gx)OC(O)–,
 ,
, ,
, R10, R11, R12, R13, R14, R15, and R19 are each independently hydrogen or C1-4alkyl; R16 is hydrogen, C1-4alkyl, –C1-4alkylene–OH, –C1-4alkylene–OC1-4alkyl, –C1-4alkylene–CO2H, or –C1-4alkylene–CONH2; R17, at each occurrence, is independently hydrogen or –CH2OC(O)–; and Gx is phenyl optionally substituted with 1 to 5 substituents independently selected from the group consisting of halogen, C1-4alkyl, C1-4haloalkyl, C1-4alkoxy, cyano, and nitro. [0579] In certain embodiments, linker L1 is -OC(O)-. [0580] In some embodiments, L1 is
R10, R11, R12, R13, R14, R15, and R19 are each independently hydrogen or C1-4alkyl; R16 is hydrogen, C1-4alkyl, –C1-4alkylene–OH, –C1-4alkylene–OC1-4alkyl, –C1-4alkylene–CO2H, or –C1-4alkylene–CONH2; R17, at each occurrence, is independently hydrogen or –CH2OC(O)–; and Gx is phenyl optionally substituted with 1 to 5 substituents independently selected from the group consisting of halogen, C1-4alkyl, C1-4haloalkyl, C1-4alkoxy, cyano, and nitro. [0579] In certain embodiments, linker L1 is -OC(O)-. [0580] In some embodiments, L1 is L3a is a bond; L4
L3a is a bond; L4 R12 and R13 are each independently hydrogen or C1-4alkyl. [0581] In some embodiments, p” is 1. In some embodiments, p’ is 1. [0582] In some embodiments,
R12 and R13 are each independently hydrogen or C1-4alkyl. [0581] In some embodiments, p” is 1. In some embodiments, p’ is 1. [0582] In some embodiments,








































































H D. Combination Therapies [0599] In one aspect, provided is a method of treating cancer or enhancing or eliciting an immune response comprising administering to a subject in need thereof: a therapeutically effective amount of a targeting moiety of the disclosure, or a pharmaceutically acceptable salt or composition thereof; and a prodrug, such as those as described herein; and optionally a therapeutically effective amount of an additional therapeutic agent selected from the group consisting of an anticancer agent or an immunomodulatory agent. [0600] The disclosure also provides a pharmaceutical combination comprising a payload-TCO conjugate described herein, or a pharmaceutically acceptable salt, or composition thereof; and an additional therapeutic agent, such as an anticancer agent or an immunomodulatory agent, for use in the treatment or prevention of a cancer or for use in enhancing or eliciting an immune response. [0601] In the methods and uses described herein, the components of the pharmaceutical combinations may be administered/used simultaneously, separately, or sequentially, and in any order, and the components may be administered separately or as a fixed combination. For example, the delay of progression or treatment of diseases according to the disclosure may comprise administration of the first active ingredient in free or pharmaceutically acceptable salt form and administration of the second active ingredient in free or pharmaceutically acceptable salt form, simultaneously or sequentially in any order, in jointly therapeutically effective amounts or effective amounts, e.g. in daily dosages corresponding to the amounts described herein. The individual active ingredients of the combination can be administered separately at different times during the course of therapy or concurrently in divided or single dosage forms. The instant disclosure is therefore to be understood as embracing all such regimes of simultaneous or alternating treatment and the term “administering” is to be interpreted accordingly. Thus, a pharmaceutical combination, as used herein, defines either a fixed combination in one dosage unit form or separate dosages forms for the combined administration where the combined administration may be independently at the same time or at different times. [0602] The methods and uses in treating cancer include administering/localizing the targeting moiety at a tumor. [0603] Additional therapeutic agent(s) may be administered simultaneously or sequentially with the payload-TCO conjugate and compositions. Sequential administration includes administration before or after the payload-TCO conjugate and compositions. An additional therapeutic agent may be administered before the payload-TCO conjugate and compositions. An additional therapeutic agent may be administered after the payload-TCO conjugate and compositions. An additional therapeutic agent may be administered at the same time as the payload-TCO conjugate and compositions. In some embodiments, the additional therapeutic agent or agents may be administered in the same composition as the payload-TCO conjugate. In other embodiments, there may be an interval of time between administration of the additional therapeutic agent and the payload-TCO conjugate or compositions. In some embodiments, administration of an additional therapeutic agent with a payload-TCO conjugate or composition may allow lower doses of the other therapeutic agents and/or administration at a less frequent interval. When used in combination with one or more other active ingredients, the payload-TCO conjugate or compositions of the present disclosure and the other active ingredient may be used in lower doses than when each is used singly. Accordingly, the pharmaceutical compositions of the present disclosure include those that contain one or more other active ingredients, in addition to a payload-TCO conjugate of the present disclosure. Anticancer agents [0604] Exemplary anti-cancer agents include, but are not limited to, Abiraterone Acetate, Abitrexate (Methotrexate), Abraxane (Paclitaxel Albumin- stabilized Nanoparticle Formulation), ABVD, ABVE, ABVE-PC, AC, AC-T, Adcetris (Brentuximab Vedotin), ADE, Ado-Trastuzumab Emtansine, Adriamycin (Doxorubicin Hydrochloride), Adrucil (Fluorouracil), Afatinib Dimaleate, Afinitor (Everolimus), Aldara (Imiquimod), Aldesleukin, Alemtuzumab, Alimta (Pemetrexed Disodium), Aloxi (Palonosetron Hydrochloride), Ambochlorin (Chlorambucil), Aminolevulinic Acid, Anastrozole, Aprepitant, Aredia (Pamidronate Disodium), Arimidex (Anastrozole), Aromasin (Exemestane), Arranon (Nelarabine), Arsenic Trioxide, Arzerra (Ofatumumab), Asparaginase Erwinia chrysanthemi, Avastin (Bevacizumab), Axitinib, Azacitidine, BEACOPP, Bendamustine Hydrochloride, BEP, Bevacizumab, Bexarotene, Bexxar (Tositumomab and I 131 Iodine Tositumomab), Bicalutamide, Bleomycin, Bortezomib, Bosulif (Bosutinib), Bosutinib, Brentuximab Vedotin, Busulfan, Busulfex (Busulfan), Cabazitaxel, Cabozantinib- S-Malate, CAF, Campath (Alemtuzumab), Camptosar (Irinotecan Hydrochloride), Capecitabine, CAPOX, Carboplatin, Carboplatin-Taxol, Carfilzomib, Casodex (Bicalutamide), CeeNU (Lomustine), Cerubidine (Daunorubicin Hydrochloride), Cervarix (Recombinant HPV Bivalent Vaccine), Cetuximab, Chlorambucil, Chlorambucil-Prednisone, CHOP, Cisplatin, Clafen (Cyclophosphamide), Clofarabine, Clofarex (Clofarabine), Clolar (Clofarabine), CMF, Cometriq (Cabozantinib-S-Malate), COPP, COPP-ABV, Cosmegen (Dactinomycin), Crizotinib, CVP, Cyclophosphamide, Cyfos (Ifosfamide), Cytarabine, Cytarabine liposomal, Cytosar-U (Cytarabine), Cytoxan (Cyclophosphamide), Dabrafenib, Dacarbazine, Dacogen (Decitabine), Dactinomycin, Dasatinib, Daunorubicin Hydrochloride, Decitabine, Degarelix, Denileukin Diftitox, Denosumab, DepoCyt (Liposomal Cytarabine), DepoFoam (Liposomal Cytarabine), Dexrazoxane Hydrochloride, Docetaxel, Doxil (Doxorubicin Hydrochloride Liposome), Doxorubicin Hydrochloride, Doxorubicin Hydrochloride Liposome, Dox-SL (Doxorubicin Hydrochloride Liposome), DTIC-Dome (Dacarbazine), Efudex (Fluorouracil), Elitek (Rasburicase), Ellence (Epirubicin Hydrochloride), Eloxatin (Oxaliplatin), Eltrombopag Olamine, Emend (Aprepitant), Enzalutamide, Epirubicin Hydrochloride, EPOCH, Erbitux (Cetuximab), Eribulin Mesylate, Erivedge (Vismodegib), Erlotinib Hydrochloride, Erwinaze (Asparaginase Erwinia chrysanthemi), Etopophos (Etoposide Phosphate), Etoposide, Etoposide Phosphate, Evacet (Doxorubicin Hydrochloride Liposome), Everolimus, Evista (Raloxifene Hydrochloride), Exemestane, Fareston (Toremifene), Faslodex (Fulvestrant), FEC, Femara (Letrozole), Filgrastim, Fludara (Fludarabine Phosphate), Fludarabine Phosphate, Fluoroplex (Fluorouracil), Fluorouracil, Folex (Methotrexate), Folex PFS (Methotrexate), Folfiri, Folfiri- Bevacizumab, Folfiri- Cetuximab, Folfirinox, Folfox (Leucovorin, Fluorouracil, Oxaliplatin), Folotyn (Pralatrexate), FU-LV, Fulvestrant, Gardasil (Recombinant HPV Quadrivalent Vaccine), Gazyva (Obinutuzumab), Gefitinib, Gemcitabine Hydrochloride, Gemcitabine-Cisplatin, Gemcitabine-Oxaliplatin, Gemtuzumab Ozogamicin, Gemzar (Gemcitabine Hydrochloride), Gilotrif (Afatinib Dimaleate), Gleevec (Imatinib Mesylate), Glucarpidase, Goserelin Acetate, Halaven (Eribulin Mesylate), Herceptin (Trastuzumab), HPV Bivalent Vaccine, Recombinant, HPV Quadrivalent Vaccine, Recombinant, Hycamtin (Topotecan Hydrochloride), Hyper-CVAD, Ibritumomab Tiuxetan, Ibrutinib, ICE, Iclusig (Ponatinib Hydrochloride), Ifex (Ifosfamide), Ifosf amide, Ifosfamidum (Ifosfamide), Imatinib Mesylate, Imbruvica (Ibrutinib), Imiquimod, Inlyta (Axitinib), Intron A (Recombinant Interferon Alfa- 2b), Iodine 131 Tositumomab and Tositumomab, Ipilimumab, Iressa (Gefitinib), Irinotecan Hydrochloride, Istodax (Romidepsin), Ixabepilone, Ixempra (Ixabepilone), Jakafi (Ruxolitinib Phosphate), Jevtana (Cabazitaxel), Kadcyla (Ado-Trastuzumab Emtansine), Keoxifene (Raloxifene Hydrochloride), Kepivance (Palifermin), Kyprolis (Carfilzomib), Lapatinib Ditosylate, Lenalidomide, Letrozole, Leucovorin Calcium, Leukeran (Chlorambucil), Leuprolide Acetate, Levulan (Aminolevulinic Acid), Linfolizin (Chlorambucil), LipoDox (Doxorubicin Hydrochloride Liposome), Liposomal Cytarabine, Lomustine, Lupron (Leuprolide Acetate), Lupron Depot (Leuprolide Acetate), Lupron Depot-Ped (Leuprolide Acetate), Lupron Depot- 3 Month (Leuprolide Acetate), Lupron Depot-4 Month (Leuprolide Acetate), Marqibo (Vincristine Sulfate Liposome), Matulane (Procarbazine Hydrochloride), Mechlorethamine Hydrochloride, Megace (Megestrol Acetate), Megestrol Acetate, Mekinist (Trametinib), Mercaptopurine, Mesna, Mesnex (Mesna), Methazolastone (Temozolomide), Methotrexate, Methotrexate LPF (Methotrexate), Mexate (Methotrexate), Mexate-AQ (Methotrexate), Mitomycin C, Mitozytrex (Mitomycin C), MOPP, Mozobil (Plerixafor), Mustargen (Mechlorethamine Hydrochloride), Mutamycin (Mitomycin C), Myleran (Busulfan), Mylosar (Azacitidine), Mylotarg (Gemtuzumab Ozogamicin), Nanoparticle Paclitaxel (Paclitaxel Albumin- stabilized Nanoparticle Formulation), Navelbine (Vinorelbine Tartrate), Nelarabine, Neosar (Cyclophosphamide), Neupogen (Filgrastim), Nexavar (Sorafenib Tosylate), Nilotinib, Nolvadex (Tamoxifen Citrate), Nplate (Romiplostim), Obinutuzumab, Ofatumumab, Omacetaxine Mepesuccinate, Oncaspar (Pegaspargase), Ontak (Denileukin Diftitox), OEPA, OPPA, Oxaliplatin, Paclitaxel, Paclitaxel Albumin- stabilized Nanoparticle Formulation, Palifermin, Palonosetron Hydrochloride, Pamidronate Disodium, Panitumumab, Paraplat (Carboplatin), Paraplatin (Carboplatin), Pazopanib Hydrochloride, Pegaspargase, Peginterferon Alfa-2b, PEG-Intron (Peginterferon Alfa-2b), Pemetrexed Disodium, Perjeta (Pertuzumab), Pertuzumab, Platinol (Cisplatin), Platinol-AQ (Cisplatin), Plerixafor, Pomalidomide, Pomalyst (Pomalidomide), Ponatinib Hydrochloride, Pralatrexate, Prednisone, Procarbazine Hydrochloride, Proleukin (Aldesleukin), Prolia (Denosumab), Promacta (Eltrombopag Olamine), Provenge (Sipuleucel-T), Purinethol (Mercaptopurine), Radium 223 Dichloride, Raloxifene Hydrochloride, Rasburicase, R-CHOP, R-CVP, Recombinant HPV Bivalent Vaccine, Recombinant HPV Quadrivalent Vaccine, Recombinant Interferon Alfa- 2b, Regorafenib, Revlimid (Lenalidomide), Rheumatrex (Methotrexate), Rituxan (Rituximab), Rituximab, Romidepsin, Romiplostim, Rubidomycin (Daunorubicin Hydrochloride), Ruxolitinib Phosphate, Sclerosol Intrapleural Aerosol (Talc), Sipuleucel-T, Sorafenib Tosylate, Sprycel (Dasatinib), Stanford V, Sterile Talc Powder (Talc), Steritalc (Talc), Stivarga (Regorafenib), Sunitinib Malate, Sutent (Sunitinib Malate), Sylatron (Peginterferon Alfa- 2b), Synovir (Thalidomide), Synribo (Omacetaxine Mepesuccinate), Tafinlar (Dabrafenib), Talc, Tamoxifen Citrate, Tarabine PFS (Cytarabine), Tarceva (Erlotinib Hydrochloride), Targretin (Bexarotene), Tasigna (Nilotinib), Taxol (Paclitaxel), Taxotere (Docetaxel), Temodar (Temozolomide), Temozolomide, Temsirolimus, Thalidomide, Thalomid (Thalidomide), Toposar (Etoposide), Topotecan Hydrochloride, Toremifene, Torisel (Temsirolimus), Tositumomab and 1131 Iodine Tositumomab, Totect (Dexrazoxane Hydrochloride), Trametinib, Trastuzumab, Treanda (Bendamustine Hydrochloride), Trisenox (Arsenic Trioxide), Tykerb (Lapatinib Ditosylate), Vandetanib, VAMP, Vectibix (Panitumumab), VelP, Velban (Vinblastine Sulfate), Velcade (Bortezomib), Velsar (Vinblastine Sulfate), Vemurafenib, VePesid (Etoposide), Viadur (Leuprolide Acetate), Vidaza (Azacitidine), Vinblastine Sulfate, Vincasar PFS (Vincristine Sulfate), Vincristine Sulfate, Vincristine Sulfate Liposome, Vinorelbine Tartrate, Vismodegib, Voraxaze (Glucarpidase), Vorinostat, Votrient (Pazopanib Hydrochloride), Wellcovorin (Leucovorin Calcium), Xalkori (Crizotinib), Xeloda (Capecitabine), Xelox, Xgeva (Denosumab), Xofigo (Radium 223 Dichloride), Xtandi (Enzalutamide), Yervoy (Ipilimumab), Zaltrap (Ziv-Aflibercept), Zelboraf (Vemurafenib), Zevalin (Ibritumomab Tiuxetan), Zinecard (Dexrazoxane Hydrochloride), Ziv-Aflibercept, Zoladex (Goserelin Acetate), Zoledronic Acid, Zolinza (Vorinostat), Zometa (Zoledronic Acid), and Zytiga (Abiraterone Acetate). [0605] The anticancer agent may be a PBD dimer, calicheamicin, speromycin, tubulysin B, rhizoxin, dolastatin, didemnin B, camptothecin, CBI, temsirolimus, actinomycin D, epothilone B, taxol, cryptophycin, SN38, velcade, bruceantin, DAVLBH, DM1, Phyllanthoside, Alimta, T2 Toxin, MMC, vantalanib, vinorelbine, brefeldin, sunitinib, daunomycin, semaxanib, tarceva, iressa, irinotecan, LY- 541503, geldanomycin, gemcitabine, methotrexate, gleevec, topotecan, bleomycin, doxorubicin, cisplatin, N-mustards, etoposide, or 5-FU. [0606] In certain embodiments, an anticancer agent is an anthracycline. In certain embodiments, anticancer agent is a taxane. In certain embodiments, anticancer agent is gemcitabine. In certain embodiments, anticancer agent is doxorubicin. In certain embodiments, anticancer agent is docetaxel. In certain embodiments, anticancer agent is SN38. In certain embodiments, anticancer agent is monomethyl auristatin E. Synthesis of the Compounds [0607] The targeting moieties may be prepared using the methods disclosed herein and routine modifications thereof, which will be apparent given the disclosure herein and methods well known in the art. Conventional and well-known synthetic methods may be used in addition to the teachings herein. The synthesis of typical targeting moieties described herein may be accomplished as described in the following examples. If available, reagents and starting materials may be purchased commercially, e.g., from Sigma Aldrich or other chemical suppliers. [0608] It will be appreciated that where typical or preferred process conditions (i.e., reaction temperatures, times, mole ratios of reactants, solvents, pressures, etc.) are given, other process conditions can also be used unless otherwise stated. Optimum reaction conditions may vary with the particular reactants or solvent used, but such conditions can be determined by one skilled in the art by routine optimization procedures. [0609] Additionally, conventional protecting groups may be necessary to prevent certain functional groups from undergoing undesired reactions. Suitable protecting groups for various functional groups as well as suitable conditions for protecting and deprotecting particular functional groups are well known in the art. For example, numerous protecting groups are described in Wuts, P. G. M., Greene, T. W., & Greene, T. W. (2006). Greene’s protective groups in organic synthesis. Hoboken, N.J., Wiley- Interscience, and references cited therein. In some embodiments, the term “leaving group” refers to an atom (or a group of atoms) with electron withdrawing ability that can be displaced as a stable species, taking with it the bonding electrons. Examples of suitable leaving groups include halides (e.g., Br, Cl, I), sulfonate esters (e.g., triflate, mesylate, tosylate, and brosylate), and nitrophenols. [0610] As shown in Scheme I, compounds of Formula V (wherein each of the dotted lines, R1, R2, R3, R4, ring A, t, L, p, and X are independently defined herein, and R50 is a synthetic handle for bonding to L, such as a leaving group, e.g., halo, or a portion of L capable of linking to X or a further portion of L, e.g., hydroxy, amino, methylamino, etc.) can be prepared by coupling a compound of Formula I-2 with a suitably functionalized antibody fragment moiety X. Suitable methods can be adapted from the literature (see, e.g., WO2020/077140, WO2018/187740, WO2017/044983, WO2015/139025, and WO2014/205126). Compounds of Formula I-2 can be prepared by reacting compound I-1 with a precursor to L under suitable coupling reaction conditions. In some embodiments, X is an antibody fragment. Suitable coupling methods include, but are not limited to, use of an succinimide functional group which is capable of forming an amide bond with a primary amine on the antibody fragment, or L can be functionalized with a group capable of forming a covalent bond to a cysteine residue on the antibody fragment moiety, such as a pyrrole-2,5-dione. Scheme I
D. Combination Therapies [0599] In one aspect, provided is a method of treating cancer or enhancing or eliciting an immune response comprising administering to a subject in need thereof: a therapeutically effective amount of a targeting moiety of the disclosure, or a pharmaceutically acceptable salt or composition thereof; and a prodrug, such as those as described herein; and optionally a therapeutically effective amount of an additional therapeutic agent selected from the group consisting of an anticancer agent or an immunomodulatory agent. [0600] The disclosure also provides a pharmaceutical combination comprising a payload-TCO conjugate described herein, or a pharmaceutically acceptable salt, or composition thereof; and an additional therapeutic agent, such as an anticancer agent or an immunomodulatory agent, for use in the treatment or prevention of a cancer or for use in enhancing or eliciting an immune response. [0601] In the methods and uses described herein, the components of the pharmaceutical combinations may be administered/used simultaneously, separately, or sequentially, and in any order, and the components may be administered separately or as a fixed combination. For example, the delay of progression or treatment of diseases according to the disclosure may comprise administration of the first active ingredient in free or pharmaceutically acceptable salt form and administration of the second active ingredient in free or pharmaceutically acceptable salt form, simultaneously or sequentially in any order, in jointly therapeutically effective amounts or effective amounts, e.g. in daily dosages corresponding to the amounts described herein. The individual active ingredients of the combination can be administered separately at different times during the course of therapy or concurrently in divided or single dosage forms. The instant disclosure is therefore to be understood as embracing all such regimes of simultaneous or alternating treatment and the term “administering” is to be interpreted accordingly. Thus, a pharmaceutical combination, as used herein, defines either a fixed combination in one dosage unit form or separate dosages forms for the combined administration where the combined administration may be independently at the same time or at different times. [0602] The methods and uses in treating cancer include administering/localizing the targeting moiety at a tumor. [0603] Additional therapeutic agent(s) may be administered simultaneously or sequentially with the payload-TCO conjugate and compositions. Sequential administration includes administration before or after the payload-TCO conjugate and compositions. An additional therapeutic agent may be administered before the payload-TCO conjugate and compositions. An additional therapeutic agent may be administered after the payload-TCO conjugate and compositions. An additional therapeutic agent may be administered at the same time as the payload-TCO conjugate and compositions. In some embodiments, the additional therapeutic agent or agents may be administered in the same composition as the payload-TCO conjugate. In other embodiments, there may be an interval of time between administration of the additional therapeutic agent and the payload-TCO conjugate or compositions. In some embodiments, administration of an additional therapeutic agent with a payload-TCO conjugate or composition may allow lower doses of the other therapeutic agents and/or administration at a less frequent interval. When used in combination with one or more other active ingredients, the payload-TCO conjugate or compositions of the present disclosure and the other active ingredient may be used in lower doses than when each is used singly. Accordingly, the pharmaceutical compositions of the present disclosure include those that contain one or more other active ingredients, in addition to a payload-TCO conjugate of the present disclosure. Anticancer agents [0604] Exemplary anti-cancer agents include, but are not limited to, Abiraterone Acetate, Abitrexate (Methotrexate), Abraxane (Paclitaxel Albumin- stabilized Nanoparticle Formulation), ABVD, ABVE, ABVE-PC, AC, AC-T, Adcetris (Brentuximab Vedotin), ADE, Ado-Trastuzumab Emtansine, Adriamycin (Doxorubicin Hydrochloride), Adrucil (Fluorouracil), Afatinib Dimaleate, Afinitor (Everolimus), Aldara (Imiquimod), Aldesleukin, Alemtuzumab, Alimta (Pemetrexed Disodium), Aloxi (Palonosetron Hydrochloride), Ambochlorin (Chlorambucil), Aminolevulinic Acid, Anastrozole, Aprepitant, Aredia (Pamidronate Disodium), Arimidex (Anastrozole), Aromasin (Exemestane), Arranon (Nelarabine), Arsenic Trioxide, Arzerra (Ofatumumab), Asparaginase Erwinia chrysanthemi, Avastin (Bevacizumab), Axitinib, Azacitidine, BEACOPP, Bendamustine Hydrochloride, BEP, Bevacizumab, Bexarotene, Bexxar (Tositumomab and I 131 Iodine Tositumomab), Bicalutamide, Bleomycin, Bortezomib, Bosulif (Bosutinib), Bosutinib, Brentuximab Vedotin, Busulfan, Busulfex (Busulfan), Cabazitaxel, Cabozantinib- S-Malate, CAF, Campath (Alemtuzumab), Camptosar (Irinotecan Hydrochloride), Capecitabine, CAPOX, Carboplatin, Carboplatin-Taxol, Carfilzomib, Casodex (Bicalutamide), CeeNU (Lomustine), Cerubidine (Daunorubicin Hydrochloride), Cervarix (Recombinant HPV Bivalent Vaccine), Cetuximab, Chlorambucil, Chlorambucil-Prednisone, CHOP, Cisplatin, Clafen (Cyclophosphamide), Clofarabine, Clofarex (Clofarabine), Clolar (Clofarabine), CMF, Cometriq (Cabozantinib-S-Malate), COPP, COPP-ABV, Cosmegen (Dactinomycin), Crizotinib, CVP, Cyclophosphamide, Cyfos (Ifosfamide), Cytarabine, Cytarabine liposomal, Cytosar-U (Cytarabine), Cytoxan (Cyclophosphamide), Dabrafenib, Dacarbazine, Dacogen (Decitabine), Dactinomycin, Dasatinib, Daunorubicin Hydrochloride, Decitabine, Degarelix, Denileukin Diftitox, Denosumab, DepoCyt (Liposomal Cytarabine), DepoFoam (Liposomal Cytarabine), Dexrazoxane Hydrochloride, Docetaxel, Doxil (Doxorubicin Hydrochloride Liposome), Doxorubicin Hydrochloride, Doxorubicin Hydrochloride Liposome, Dox-SL (Doxorubicin Hydrochloride Liposome), DTIC-Dome (Dacarbazine), Efudex (Fluorouracil), Elitek (Rasburicase), Ellence (Epirubicin Hydrochloride), Eloxatin (Oxaliplatin), Eltrombopag Olamine, Emend (Aprepitant), Enzalutamide, Epirubicin Hydrochloride, EPOCH, Erbitux (Cetuximab), Eribulin Mesylate, Erivedge (Vismodegib), Erlotinib Hydrochloride, Erwinaze (Asparaginase Erwinia chrysanthemi), Etopophos (Etoposide Phosphate), Etoposide, Etoposide Phosphate, Evacet (Doxorubicin Hydrochloride Liposome), Everolimus, Evista (Raloxifene Hydrochloride), Exemestane, Fareston (Toremifene), Faslodex (Fulvestrant), FEC, Femara (Letrozole), Filgrastim, Fludara (Fludarabine Phosphate), Fludarabine Phosphate, Fluoroplex (Fluorouracil), Fluorouracil, Folex (Methotrexate), Folex PFS (Methotrexate), Folfiri, Folfiri- Bevacizumab, Folfiri- Cetuximab, Folfirinox, Folfox (Leucovorin, Fluorouracil, Oxaliplatin), Folotyn (Pralatrexate), FU-LV, Fulvestrant, Gardasil (Recombinant HPV Quadrivalent Vaccine), Gazyva (Obinutuzumab), Gefitinib, Gemcitabine Hydrochloride, Gemcitabine-Cisplatin, Gemcitabine-Oxaliplatin, Gemtuzumab Ozogamicin, Gemzar (Gemcitabine Hydrochloride), Gilotrif (Afatinib Dimaleate), Gleevec (Imatinib Mesylate), Glucarpidase, Goserelin Acetate, Halaven (Eribulin Mesylate), Herceptin (Trastuzumab), HPV Bivalent Vaccine, Recombinant, HPV Quadrivalent Vaccine, Recombinant, Hycamtin (Topotecan Hydrochloride), Hyper-CVAD, Ibritumomab Tiuxetan, Ibrutinib, ICE, Iclusig (Ponatinib Hydrochloride), Ifex (Ifosfamide), Ifosf amide, Ifosfamidum (Ifosfamide), Imatinib Mesylate, Imbruvica (Ibrutinib), Imiquimod, Inlyta (Axitinib), Intron A (Recombinant Interferon Alfa- 2b), Iodine 131 Tositumomab and Tositumomab, Ipilimumab, Iressa (Gefitinib), Irinotecan Hydrochloride, Istodax (Romidepsin), Ixabepilone, Ixempra (Ixabepilone), Jakafi (Ruxolitinib Phosphate), Jevtana (Cabazitaxel), Kadcyla (Ado-Trastuzumab Emtansine), Keoxifene (Raloxifene Hydrochloride), Kepivance (Palifermin), Kyprolis (Carfilzomib), Lapatinib Ditosylate, Lenalidomide, Letrozole, Leucovorin Calcium, Leukeran (Chlorambucil), Leuprolide Acetate, Levulan (Aminolevulinic Acid), Linfolizin (Chlorambucil), LipoDox (Doxorubicin Hydrochloride Liposome), Liposomal Cytarabine, Lomustine, Lupron (Leuprolide Acetate), Lupron Depot (Leuprolide Acetate), Lupron Depot-Ped (Leuprolide Acetate), Lupron Depot- 3 Month (Leuprolide Acetate), Lupron Depot-4 Month (Leuprolide Acetate), Marqibo (Vincristine Sulfate Liposome), Matulane (Procarbazine Hydrochloride), Mechlorethamine Hydrochloride, Megace (Megestrol Acetate), Megestrol Acetate, Mekinist (Trametinib), Mercaptopurine, Mesna, Mesnex (Mesna), Methazolastone (Temozolomide), Methotrexate, Methotrexate LPF (Methotrexate), Mexate (Methotrexate), Mexate-AQ (Methotrexate), Mitomycin C, Mitozytrex (Mitomycin C), MOPP, Mozobil (Plerixafor), Mustargen (Mechlorethamine Hydrochloride), Mutamycin (Mitomycin C), Myleran (Busulfan), Mylosar (Azacitidine), Mylotarg (Gemtuzumab Ozogamicin), Nanoparticle Paclitaxel (Paclitaxel Albumin- stabilized Nanoparticle Formulation), Navelbine (Vinorelbine Tartrate), Nelarabine, Neosar (Cyclophosphamide), Neupogen (Filgrastim), Nexavar (Sorafenib Tosylate), Nilotinib, Nolvadex (Tamoxifen Citrate), Nplate (Romiplostim), Obinutuzumab, Ofatumumab, Omacetaxine Mepesuccinate, Oncaspar (Pegaspargase), Ontak (Denileukin Diftitox), OEPA, OPPA, Oxaliplatin, Paclitaxel, Paclitaxel Albumin- stabilized Nanoparticle Formulation, Palifermin, Palonosetron Hydrochloride, Pamidronate Disodium, Panitumumab, Paraplat (Carboplatin), Paraplatin (Carboplatin), Pazopanib Hydrochloride, Pegaspargase, Peginterferon Alfa-2b, PEG-Intron (Peginterferon Alfa-2b), Pemetrexed Disodium, Perjeta (Pertuzumab), Pertuzumab, Platinol (Cisplatin), Platinol-AQ (Cisplatin), Plerixafor, Pomalidomide, Pomalyst (Pomalidomide), Ponatinib Hydrochloride, Pralatrexate, Prednisone, Procarbazine Hydrochloride, Proleukin (Aldesleukin), Prolia (Denosumab), Promacta (Eltrombopag Olamine), Provenge (Sipuleucel-T), Purinethol (Mercaptopurine), Radium 223 Dichloride, Raloxifene Hydrochloride, Rasburicase, R-CHOP, R-CVP, Recombinant HPV Bivalent Vaccine, Recombinant HPV Quadrivalent Vaccine, Recombinant Interferon Alfa- 2b, Regorafenib, Revlimid (Lenalidomide), Rheumatrex (Methotrexate), Rituxan (Rituximab), Rituximab, Romidepsin, Romiplostim, Rubidomycin (Daunorubicin Hydrochloride), Ruxolitinib Phosphate, Sclerosol Intrapleural Aerosol (Talc), Sipuleucel-T, Sorafenib Tosylate, Sprycel (Dasatinib), Stanford V, Sterile Talc Powder (Talc), Steritalc (Talc), Stivarga (Regorafenib), Sunitinib Malate, Sutent (Sunitinib Malate), Sylatron (Peginterferon Alfa- 2b), Synovir (Thalidomide), Synribo (Omacetaxine Mepesuccinate), Tafinlar (Dabrafenib), Talc, Tamoxifen Citrate, Tarabine PFS (Cytarabine), Tarceva (Erlotinib Hydrochloride), Targretin (Bexarotene), Tasigna (Nilotinib), Taxol (Paclitaxel), Taxotere (Docetaxel), Temodar (Temozolomide), Temozolomide, Temsirolimus, Thalidomide, Thalomid (Thalidomide), Toposar (Etoposide), Topotecan Hydrochloride, Toremifene, Torisel (Temsirolimus), Tositumomab and 1131 Iodine Tositumomab, Totect (Dexrazoxane Hydrochloride), Trametinib, Trastuzumab, Treanda (Bendamustine Hydrochloride), Trisenox (Arsenic Trioxide), Tykerb (Lapatinib Ditosylate), Vandetanib, VAMP, Vectibix (Panitumumab), VelP, Velban (Vinblastine Sulfate), Velcade (Bortezomib), Velsar (Vinblastine Sulfate), Vemurafenib, VePesid (Etoposide), Viadur (Leuprolide Acetate), Vidaza (Azacitidine), Vinblastine Sulfate, Vincasar PFS (Vincristine Sulfate), Vincristine Sulfate, Vincristine Sulfate Liposome, Vinorelbine Tartrate, Vismodegib, Voraxaze (Glucarpidase), Vorinostat, Votrient (Pazopanib Hydrochloride), Wellcovorin (Leucovorin Calcium), Xalkori (Crizotinib), Xeloda (Capecitabine), Xelox, Xgeva (Denosumab), Xofigo (Radium 223 Dichloride), Xtandi (Enzalutamide), Yervoy (Ipilimumab), Zaltrap (Ziv-Aflibercept), Zelboraf (Vemurafenib), Zevalin (Ibritumomab Tiuxetan), Zinecard (Dexrazoxane Hydrochloride), Ziv-Aflibercept, Zoladex (Goserelin Acetate), Zoledronic Acid, Zolinza (Vorinostat), Zometa (Zoledronic Acid), and Zytiga (Abiraterone Acetate). [0605] The anticancer agent may be a PBD dimer, calicheamicin, speromycin, tubulysin B, rhizoxin, dolastatin, didemnin B, camptothecin, CBI, temsirolimus, actinomycin D, epothilone B, taxol, cryptophycin, SN38, velcade, bruceantin, DAVLBH, DM1, Phyllanthoside, Alimta, T2 Toxin, MMC, vantalanib, vinorelbine, brefeldin, sunitinib, daunomycin, semaxanib, tarceva, iressa, irinotecan, LY- 541503, geldanomycin, gemcitabine, methotrexate, gleevec, topotecan, bleomycin, doxorubicin, cisplatin, N-mustards, etoposide, or 5-FU. [0606] In certain embodiments, an anticancer agent is an anthracycline. In certain embodiments, anticancer agent is a taxane. In certain embodiments, anticancer agent is gemcitabine. In certain embodiments, anticancer agent is doxorubicin. In certain embodiments, anticancer agent is docetaxel. In certain embodiments, anticancer agent is SN38. In certain embodiments, anticancer agent is monomethyl auristatin E. Synthesis of the Compounds [0607] The targeting moieties may be prepared using the methods disclosed herein and routine modifications thereof, which will be apparent given the disclosure herein and methods well known in the art. Conventional and well-known synthetic methods may be used in addition to the teachings herein. The synthesis of typical targeting moieties described herein may be accomplished as described in the following examples. If available, reagents and starting materials may be purchased commercially, e.g., from Sigma Aldrich or other chemical suppliers. [0608] It will be appreciated that where typical or preferred process conditions (i.e., reaction temperatures, times, mole ratios of reactants, solvents, pressures, etc.) are given, other process conditions can also be used unless otherwise stated. Optimum reaction conditions may vary with the particular reactants or solvent used, but such conditions can be determined by one skilled in the art by routine optimization procedures. [0609] Additionally, conventional protecting groups may be necessary to prevent certain functional groups from undergoing undesired reactions. Suitable protecting groups for various functional groups as well as suitable conditions for protecting and deprotecting particular functional groups are well known in the art. For example, numerous protecting groups are described in Wuts, P. G. M., Greene, T. W., & Greene, T. W. (2006). Greene’s protective groups in organic synthesis. Hoboken, N.J., Wiley- Interscience, and references cited therein. In some embodiments, the term “leaving group” refers to an atom (or a group of atoms) with electron withdrawing ability that can be displaced as a stable species, taking with it the bonding electrons. Examples of suitable leaving groups include halides (e.g., Br, Cl, I), sulfonate esters (e.g., triflate, mesylate, tosylate, and brosylate), and nitrophenols. [0610] As shown in Scheme I, compounds of Formula V (wherein each of the dotted lines, R1, R2, R3, R4, ring A, t, L, p, and X are independently defined herein, and R50 is a synthetic handle for bonding to L, such as a leaving group, e.g., halo, or a portion of L capable of linking to X or a further portion of L, e.g., hydroxy, amino, methylamino, etc.) can be prepared by coupling a compound of Formula I-2 with a suitably functionalized antibody fragment moiety X. Suitable methods can be adapted from the literature (see, e.g., WO2020/077140, WO2018/187740, WO2017/044983, WO2015/139025, and WO2014/205126). Compounds of Formula I-2 can be prepared by reacting compound I-1 with a precursor to L under suitable coupling reaction conditions. In some embodiments, X is an antibody fragment. Suitable coupling methods include, but are not limited to, use of an succinimide functional group which is capable of forming an amide bond with a primary amine on the antibody fragment, or L can be functionalized with a group capable of forming a covalent bond to a cysteine residue on the antibody fragment moiety, such as a pyrrole-2,5-dione. Scheme I [0611] Compounds of Formula I-1 can be prepared according to Scheme II, wherein each of the dotted lines, R1, R2, R3, R4, ring A, t, and L are independently defined herein, and R50 is a synthetic handle for bonding to L, such as a leaving group, e.g., halo or a thioether, or a portion of L capable of linking to X or a further portion of L, e.g., hydroxy, amino, methylamino, etc., and each LG is independently a leaving group, e.g., halo. Scheme II
[0611] Compounds of Formula I-1 can be prepared according to Scheme II, wherein each of the dotted lines, R1, R2, R3, R4, ring A, t, and L are independently defined herein, and R50 is a synthetic handle for bonding to L, such as a leaving group, e.g., halo or a thioether, or a portion of L capable of linking to X or a further portion of L, e.g., hydroxy, amino, methylamino, etc., and each LG is independently a leaving group, e.g., halo. Scheme II [0612] As shown in Scheme II, coupling compound II-1 with compound II-2 in the presence of N2H4 provides compound II-3. Further modification of compound II-3 with compound II-4 and/or compound II-6 under standard coupling conditions provides compound II-5 and/or compound II-7. Alternatively, compound II-3 can be provided by coupling compound II-8 with compound II-9 in the presence of N2H4. Compound II-10 can be provided by contacting compound II-3 with a suitable oxidizing agent (e.g., NaNO2). Alternatively, compound II-3 can be provided by contacting compound II-10 with thiourea dioxide. [0613] Upon each reaction completion, each of the intermediate or final compounds can be recovered, and optionally purified, by conventional techniques such as neutralization, extraction, precipitation, chromatography, filtration and the like. [0614] It should be understood that any of the compounds or intermediates shown in Scheme I or II may be prepared using traditional methods or purchased from commercial sources. In addition, any of the intermediates or any product obtained by the process outlined in Scheme I or II can be derivatized at any step to provide various compounds of Formula V. [0615] Exemplary payloads and conjugates thereof can be prepared can be prepared according to methods adapted from the literature (see, e.g., WO2022/032191, WO2021/007160, WO2020/077140, WO2018/187740, WO2017/044983, WO2015/139025, and WO2014/205126, which methods are incorporated herein in their entirety). Exemplary procedures for certain payloads are shown in the Examples below, which procedures can be adapted to prepare other payloads such as those disclosed herein. EXAMPLES [0616] The following examples are included to demonstrate specific embodiments of the disclosure. It should be appreciated by those of skill in the art that the techniques disclosed in the examples which follow represent techniques to function well in the practice of the disclosure, and thus can be considered to constitute specific modes for its practice. However, those of skill in the art should, in light of the present disclosure, appreciate that many changes can be made in the specific embodiments which are disclosed and still obtain a like or similar result without departing from the spirit and scope of the disclosure. [0617] LCMS Analysis Method: Test articles were subjected to PNGaseF (IgG) and DTT or DTT alone (Fab) in RapiGest according to the manufacturer’s protocol. Samples were diluted to 100 µg/mL with water and centrifuged at 16.1k RCF for 10 min at 4 °C. The samples were then analyzed by LCMS (LC-Q-TOF) and the mass spectra reconstructed from the charge ladder. Example 1: Synthesis of Tetrazine-Trastuzumab Targeting Moiety [0618] Trastuzumab (22.1 mg/mL, 1.1 mL) in 0.01 M PBS was mixed with 20 equivalents of methyltetrazine-PEG4-NHS (Clickchemtools #1069-10).
[0612] As shown in Scheme II, coupling compound II-1 with compound II-2 in the presence of N2H4 provides compound II-3. Further modification of compound II-3 with compound II-4 and/or compound II-6 under standard coupling conditions provides compound II-5 and/or compound II-7. Alternatively, compound II-3 can be provided by coupling compound II-8 with compound II-9 in the presence of N2H4. Compound II-10 can be provided by contacting compound II-3 with a suitable oxidizing agent (e.g., NaNO2). Alternatively, compound II-3 can be provided by contacting compound II-10 with thiourea dioxide. [0613] Upon each reaction completion, each of the intermediate or final compounds can be recovered, and optionally purified, by conventional techniques such as neutralization, extraction, precipitation, chromatography, filtration and the like. [0614] It should be understood that any of the compounds or intermediates shown in Scheme I or II may be prepared using traditional methods or purchased from commercial sources. In addition, any of the intermediates or any product obtained by the process outlined in Scheme I or II can be derivatized at any step to provide various compounds of Formula V. [0615] Exemplary payloads and conjugates thereof can be prepared can be prepared according to methods adapted from the literature (see, e.g., WO2022/032191, WO2021/007160, WO2020/077140, WO2018/187740, WO2017/044983, WO2015/139025, and WO2014/205126, which methods are incorporated herein in their entirety). Exemplary procedures for certain payloads are shown in the Examples below, which procedures can be adapted to prepare other payloads such as those disclosed herein. EXAMPLES [0616] The following examples are included to demonstrate specific embodiments of the disclosure. It should be appreciated by those of skill in the art that the techniques disclosed in the examples which follow represent techniques to function well in the practice of the disclosure, and thus can be considered to constitute specific modes for its practice. However, those of skill in the art should, in light of the present disclosure, appreciate that many changes can be made in the specific embodiments which are disclosed and still obtain a like or similar result without departing from the spirit and scope of the disclosure. [0617] LCMS Analysis Method: Test articles were subjected to PNGaseF (IgG) and DTT or DTT alone (Fab) in RapiGest according to the manufacturer’s protocol. Samples were diluted to 100 µg/mL with water and centrifuged at 16.1k RCF for 10 min at 4 °C. The samples were then analyzed by LCMS (LC-Q-TOF) and the mass spectra reconstructed from the charge ladder. Example 1: Synthesis of Tetrazine-Trastuzumab Targeting Moiety [0618] Trastuzumab (22.1 mg/mL, 1.1 mL) in 0.01 M PBS was mixed with 20 equivalents of methyltetrazine-PEG4-NHS (Clickchemtools #1069-10). [0619] The reaction was mixed thoroughly and aged at room temperature for 1 hour, at which time the reaction was quenched by the addition of 1 volume of 0.1 M Tris buffer. The resulting solution was buffer exchanged to 0.01 M PBS to remove excess reagent and buffer salts. The resulting solution of targeting moiety (6.3 mg/mL, 1.6 mL) was analyzed by SDS-Page and LCMS (data not shown) confirming the formation of the targeting moiety, and thus was used for subsequent studies. Based on the analysis, it is contemplated that up to 12 methyltetrazine-PEG4 units are covalently bonded to the antibody. Example 2: Tetrazine-Fab Targeting Moiety [0620] Fab was prepared from Trastuzumab using a commercial kit (Pierce™ Fab Preparation Kit #44985) according to the manufacturer’s protocol and purified by protein G resin (BioVision #6511-25). The purified Fab in 0.01 M PBS was mixed with 20 equivalents of methyltetrazine-PEG4-NHS (Clickchemtools #1069-10). The reaction was mixed thoroughly and aged at room temperature for 1 hour, at which time the reaction was quenched by the addition of 1 volume of 0.1 M Tris buffer. The resulting solution was buffer exchanged to 0.01 M PBS to remove excess reagent and buffer salts. The resulting solution of targeting moiety (1.0 mg/mL, 10.3 mL) was analyzed by SDS-Page and LCMS (data not shown) confirming the formation of the targeting moiety, and thus was used for subsequent studies. Based on the analysis, it is contemplated that up to about 6 methyltetrazine-PEG4 units are covalently bonded to the Fab. Example 3: Tetrazine-Fab Targeting Moiety [0621] Fab was prepared from Enfortumab using the following method.0.1 mg papain was pretreated with 1 mM DTT at 0.5 mg/ml concentration and 2 mM EDTA by incubating at 37 ℃ for 30 min. The antibody was prepared in PBS buffer, pH 7.4 (10 mg, 0.5 mg/mL). The pretreated papain was mixed with the antibody at (1 : 100) molar ratio and incubated at 37 ℃ for 2 hours. The digestion mixture was loaded onto an anti-CH1 affinity column, washed with 25 mM Tris, 150 mM NaCl, pH 8.0, and eluted with 50 mM sodium citrate, 150 mM NaCl, pH 3.0. The filtrate containing the product Fab was dialyzed into PBS. The purified Fab was in PBS was concentrated to 0.2 mg/mL. The Fab was mixed with Me-Tet- PEG9-NHS (prepared in DMSO at 10 mM) at 3:1 molar ratio and incubated at 37 °C for 2 hours. The resulting conjugate was analyzed by LCMS and the DAR calculated to be 2.66. Example 4: Tetrazine-Fab Targeting Moiety [0622] Fab was prepared from Brentuximab using the following method.0.1 mg papain was pretreated with 1 mM DTT at 0.5 mg/ml concentration and 2 mM EDTA by incubating at 37 ℃ for 30 min. The antibody was prepared in PBS buffer, pH 7.4 (10 mg, 0.5 mg/mL). The pretreated papain was mixed with the antibody at (1 : 100) molar ratio and incubated at 37 ℃ for 2 hours. The digestion mixture was loaded onto an anti-CH1 affinity column, washed with 25 mM Tris, 150 mM NaCl, pH 8.0, and eluted with 50 mM sodium citrate, 150 mM NaCl, pH 3.0. The filtrate containing the product Fab was dialyzed into PBS. The purified Fab was in PBS was concentrated to 0.2 mg/mL. The Fab was mixed with Me-Tet- PEG9-NHS (prepared in DMSO at 10 mM) at 3:1 molar ratio and incubated at 37 °C for 2 hours. The resulting conjugate was analyzed by LCMS and the DAR calculated to be 4.57. Example 5A: Tetrazine-Fab Targeting Moiety [0623] Fab is prepared from Sacituzumab using a commercial kit (Pierce™ Fab Preparation Kit #44985) according to the manufacturers protocol and purified by protein G resin (BioVision #6511-25). The purified Fab 10 mM Me-Tet-PEG9-NHS is prepared in DMSO. The two components are reacted at 3:1 drug to protein molar ratio at 25°C for 2 hours before it is dialyzed against PBS, pH 7.4 to remove excess Me-Tet-PEG9-NHS compound from the protein component. The resulting solution of targeting moiety is analyzed by SDS-Page and LCMS to confirm the formation of the targeting moiety. It is contemplated that approximately two methyltetrazines will be covalently bonded to each Fab, on average, as confirmed by LCMS. Example 5B: Tetrazine-Fab Targeting Moiety (Compound TM-2)
[0619] The reaction was mixed thoroughly and aged at room temperature for 1 hour, at which time the reaction was quenched by the addition of 1 volume of 0.1 M Tris buffer. The resulting solution was buffer exchanged to 0.01 M PBS to remove excess reagent and buffer salts. The resulting solution of targeting moiety (6.3 mg/mL, 1.6 mL) was analyzed by SDS-Page and LCMS (data not shown) confirming the formation of the targeting moiety, and thus was used for subsequent studies. Based on the analysis, it is contemplated that up to 12 methyltetrazine-PEG4 units are covalently bonded to the antibody. Example 2: Tetrazine-Fab Targeting Moiety [0620] Fab was prepared from Trastuzumab using a commercial kit (Pierce™ Fab Preparation Kit #44985) according to the manufacturer’s protocol and purified by protein G resin (BioVision #6511-25). The purified Fab in 0.01 M PBS was mixed with 20 equivalents of methyltetrazine-PEG4-NHS (Clickchemtools #1069-10). The reaction was mixed thoroughly and aged at room temperature for 1 hour, at which time the reaction was quenched by the addition of 1 volume of 0.1 M Tris buffer. The resulting solution was buffer exchanged to 0.01 M PBS to remove excess reagent and buffer salts. The resulting solution of targeting moiety (1.0 mg/mL, 10.3 mL) was analyzed by SDS-Page and LCMS (data not shown) confirming the formation of the targeting moiety, and thus was used for subsequent studies. Based on the analysis, it is contemplated that up to about 6 methyltetrazine-PEG4 units are covalently bonded to the Fab. Example 3: Tetrazine-Fab Targeting Moiety [0621] Fab was prepared from Enfortumab using the following method.0.1 mg papain was pretreated with 1 mM DTT at 0.5 mg/ml concentration and 2 mM EDTA by incubating at 37 ℃ for 30 min. The antibody was prepared in PBS buffer, pH 7.4 (10 mg, 0.5 mg/mL). The pretreated papain was mixed with the antibody at (1 : 100) molar ratio and incubated at 37 ℃ for 2 hours. The digestion mixture was loaded onto an anti-CH1 affinity column, washed with 25 mM Tris, 150 mM NaCl, pH 8.0, and eluted with 50 mM sodium citrate, 150 mM NaCl, pH 3.0. The filtrate containing the product Fab was dialyzed into PBS. The purified Fab was in PBS was concentrated to 0.2 mg/mL. The Fab was mixed with Me-Tet- PEG9-NHS (prepared in DMSO at 10 mM) at 3:1 molar ratio and incubated at 37 °C for 2 hours. The resulting conjugate was analyzed by LCMS and the DAR calculated to be 2.66. Example 4: Tetrazine-Fab Targeting Moiety [0622] Fab was prepared from Brentuximab using the following method.0.1 mg papain was pretreated with 1 mM DTT at 0.5 mg/ml concentration and 2 mM EDTA by incubating at 37 ℃ for 30 min. The antibody was prepared in PBS buffer, pH 7.4 (10 mg, 0.5 mg/mL). The pretreated papain was mixed with the antibody at (1 : 100) molar ratio and incubated at 37 ℃ for 2 hours. The digestion mixture was loaded onto an anti-CH1 affinity column, washed with 25 mM Tris, 150 mM NaCl, pH 8.0, and eluted with 50 mM sodium citrate, 150 mM NaCl, pH 3.0. The filtrate containing the product Fab was dialyzed into PBS. The purified Fab was in PBS was concentrated to 0.2 mg/mL. The Fab was mixed with Me-Tet- PEG9-NHS (prepared in DMSO at 10 mM) at 3:1 molar ratio and incubated at 37 °C for 2 hours. The resulting conjugate was analyzed by LCMS and the DAR calculated to be 4.57. Example 5A: Tetrazine-Fab Targeting Moiety [0623] Fab is prepared from Sacituzumab using a commercial kit (Pierce™ Fab Preparation Kit #44985) according to the manufacturers protocol and purified by protein G resin (BioVision #6511-25). The purified Fab 10 mM Me-Tet-PEG9-NHS is prepared in DMSO. The two components are reacted at 3:1 drug to protein molar ratio at 25°C for 2 hours before it is dialyzed against PBS, pH 7.4 to remove excess Me-Tet-PEG9-NHS compound from the protein component. The resulting solution of targeting moiety is analyzed by SDS-Page and LCMS to confirm the formation of the targeting moiety. It is contemplated that approximately two methyltetrazines will be covalently bonded to each Fab, on average, as confirmed by LCMS. Example 5B: Tetrazine-Fab Targeting Moiety (Compound TM-2)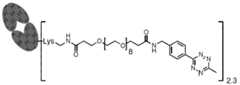 [0624] Compound TM-2 was prepared and characterized as follows. [0625] Sacituzumab Fab amino acid sequences: [0626] Heavy chain: [0627] QVQLQQSGSELKKPGASVKVSCKASGYTFTNYGMNWVKQAPGQGLKWMGWINTYTG EPTYTDDFKGRFAFSLDTSVSTAYLQISSLKADDTAVYFCARGGFGSSYWYFDVWGQGSLVTVS SASTKGPSVFPLAPSSKSTSGGTAALGCLVKDYFPEPVTVSWNSGALTSGVHTFPAVLQSSGLYS LSSVVTVPSSSLGTQTYICNVNHKPSNTKVDKKVEPKSSDKTHT (SEQ ID NO.9). [0628] Light chain: [0629] DIQLTQSPSSLSASVGDRVSITCKASQDVSIAVAWYQQKPGKAPKLLIYSASYRYTGVPD RFSGSGSGTDFTLTISSLQPEDFAVYYCQQHYITPLTFGAGTKVEIKRTVAAPSVFIFPPSDEQLKS GTASVVCLLNNFYPREAKVQWKVDNALQSGNSQESVTEQDSKDSTYSLSSTLTLSKADYEKHK VYACEVTHQGLSSPCTKSFNRGEC (SEQ ID NO.10). [0630] Generation of Sacituzumab Fab-tetrazine conjugate: Coding sequences of the variable region of heavy chain and light chain of sacituzumab (human anti-TROP2) antibody were used to generate TROP2 Fab-expressing constructs. Coding sequences were synthesized and subcloned into PCDN3.4 expression vector. The constructed plasmids were transformed into E. coli for propagation and scale-up. Purified plasmids were confirmed by sequencing. The constructs containing the heavy chain and light chain of the TROP2 Fab were transfected into HEK293 cells (suspension) with polymer polyethylenimine (PEI) reagent. The culture medium was harvested at 6-7 days post-transfection. The culture medium containing TROP2 Fab was centrifuged, filtered, and then loaded onto KappaSelect affinity column (Mabselect Prism). The loading buffer was 25 mM Tris containing 150 mM NaCl, pH 8.0, and eluted with 100 mM sodium-citrate buffer containing 150 mM NaCl, pH 2.5. The collected solution was neutralized with 1 M arginine, 400 mM succinic acid buffer, pH 9.0. The affinity-purified protein was further purified by gel filtration with Superdex S-2005/150GL column chromatography. The sample was eluted using the mobile phase 2X PBS at pH 7.4. The purified TROP2 Fab was analyzed by SDS-PAGE and SEC-HPLC. [0631] Tetrazine conjugation of TROP2 Fab: TROP2 Fab was buffer exchanged to PBS pH 7.4 overnight. The methyltetrazine-PEG9-NHS (SiChem #SC-8808) was dissolved in DMSO to make a 10 mM stock solution. For conjugation, the two components were reacted at 3:1 (methyltetrazine-PEG9- NHS to TROP2 Fab) molar ratio at 25 °C for 2 hours. The amount of Fab for tetrazine conjugation varied from 30-100 mg but the ratio of 3:1 was always maintained for each conjugation procedure. Then the solution was buffer exchanged to PBS pH 7.4 to remove excess methyltetrazine-PEG9-NHS and buffer salts. [0632] Characterization of Sacituzumab Fab-Tz conjugate (Compound TM-2): The sample of prepared Fab-Tz conjugate was analyzed by SDS-PAGE and Q Exactive HF-X LC-MS (Thermo Scientific) fitted with an Acquity UPLC protein BEH C4 column (300A, 1.7 µm, 2.1 x 50 mm) to confirm the formation of the conjugate and to determine the tetrazine-to-antibody ratio. HPLC-SEC analysis was performed using a UltiMate 3000 HPLC (Thermo Scientific) fitted with a Xbridge BEH 200A SEC column (7.8 x 300 mm, Waters Corp). Proteins were eluted with PBS containing 15% isopropanol, pH 7.4). Size exclusion chromatograms showed 99% monomeric species for the Fab-tetrazine conjugate with minimal aggregation (1.0%). The tetrazine-to-antibody ratio was calculated to be 2.3.
[0624] Compound TM-2 was prepared and characterized as follows. [0625] Sacituzumab Fab amino acid sequences: [0626] Heavy chain: [0627] QVQLQQSGSELKKPGASVKVSCKASGYTFTNYGMNWVKQAPGQGLKWMGWINTYTG EPTYTDDFKGRFAFSLDTSVSTAYLQISSLKADDTAVYFCARGGFGSSYWYFDVWGQGSLVTVS SASTKGPSVFPLAPSSKSTSGGTAALGCLVKDYFPEPVTVSWNSGALTSGVHTFPAVLQSSGLYS LSSVVTVPSSSLGTQTYICNVNHKPSNTKVDKKVEPKSSDKTHT (SEQ ID NO.9). [0628] Light chain: [0629] DIQLTQSPSSLSASVGDRVSITCKASQDVSIAVAWYQQKPGKAPKLLIYSASYRYTGVPD RFSGSGSGTDFTLTISSLQPEDFAVYYCQQHYITPLTFGAGTKVEIKRTVAAPSVFIFPPSDEQLKS GTASVVCLLNNFYPREAKVQWKVDNALQSGNSQESVTEQDSKDSTYSLSSTLTLSKADYEKHK VYACEVTHQGLSSPCTKSFNRGEC (SEQ ID NO.10). [0630] Generation of Sacituzumab Fab-tetrazine conjugate: Coding sequences of the variable region of heavy chain and light chain of sacituzumab (human anti-TROP2) antibody were used to generate TROP2 Fab-expressing constructs. Coding sequences were synthesized and subcloned into PCDN3.4 expression vector. The constructed plasmids were transformed into E. coli for propagation and scale-up. Purified plasmids were confirmed by sequencing. The constructs containing the heavy chain and light chain of the TROP2 Fab were transfected into HEK293 cells (suspension) with polymer polyethylenimine (PEI) reagent. The culture medium was harvested at 6-7 days post-transfection. The culture medium containing TROP2 Fab was centrifuged, filtered, and then loaded onto KappaSelect affinity column (Mabselect Prism). The loading buffer was 25 mM Tris containing 150 mM NaCl, pH 8.0, and eluted with 100 mM sodium-citrate buffer containing 150 mM NaCl, pH 2.5. The collected solution was neutralized with 1 M arginine, 400 mM succinic acid buffer, pH 9.0. The affinity-purified protein was further purified by gel filtration with Superdex S-2005/150GL column chromatography. The sample was eluted using the mobile phase 2X PBS at pH 7.4. The purified TROP2 Fab was analyzed by SDS-PAGE and SEC-HPLC. [0631] Tetrazine conjugation of TROP2 Fab: TROP2 Fab was buffer exchanged to PBS pH 7.4 overnight. The methyltetrazine-PEG9-NHS (SiChem #SC-8808) was dissolved in DMSO to make a 10 mM stock solution. For conjugation, the two components were reacted at 3:1 (methyltetrazine-PEG9- NHS to TROP2 Fab) molar ratio at 25 °C for 2 hours. The amount of Fab for tetrazine conjugation varied from 30-100 mg but the ratio of 3:1 was always maintained for each conjugation procedure. Then the solution was buffer exchanged to PBS pH 7.4 to remove excess methyltetrazine-PEG9-NHS and buffer salts. [0632] Characterization of Sacituzumab Fab-Tz conjugate (Compound TM-2): The sample of prepared Fab-Tz conjugate was analyzed by SDS-PAGE and Q Exactive HF-X LC-MS (Thermo Scientific) fitted with an Acquity UPLC protein BEH C4 column (300A, 1.7 µm, 2.1 x 50 mm) to confirm the formation of the conjugate and to determine the tetrazine-to-antibody ratio. HPLC-SEC analysis was performed using a UltiMate 3000 HPLC (Thermo Scientific) fitted with a Xbridge BEH 200A SEC column (7.8 x 300 mm, Waters Corp). Proteins were eluted with PBS containing 15% isopropanol, pH 7.4). Size exclusion chromatograms showed 99% monomeric species for the Fab-tetrazine conjugate with minimal aggregation (1.0%). The tetrazine-to-antibody ratio was calculated to be 2.3. Example 6: HER2 Fab-tetrazine Targeting Moiety (Compound TM-1) [0633] Compound TM-1 and Isotype Fab-Tz were prepared and characterized as follows. [0634] Trastuzumab Fab amino acid sequences: [0635] Heavy chain: EVQLVESGGGLVQPGGSLRLSCAASGFNIKDTYIHWVRQAPGKGLEWVARIYPTNGYTRYADS VKGRFTISADTSKNTAYLQMNSLRAEDTAVYYCSRWGGDGFYAMDYWGQGTLVTVSSASTKG PSVFPLAPSSKSTSGGTAALGCLVKDYFPEPVTVSWNSGALTSGVHTFPAVLQSSGLYSLSSVVT VPSSSLGTQTYICNVNHKP SNTKVDKKV (SEQ ID NO.3). [0636] Light chain: DIQMTQSPSSLSASVGDRVTITCRASQDVNTAVAWYQQKPGKAPKLLIYSASFLYSGVPSRFSGS RSGTDFTLTISSLQPEDFATYYCQQHYTTPPTFGQGTKVEIKRTVAAPSVFIFPPSDEQLKSGTASV VCLLNNFYPREAKVQWKVDNALQSGNSQESVTEQDSKDSTYSLSSTLTLSKADYEKHKVYACE VTHQGLSSPVTKSFNRGEC (SEQ ID NO.4). [0637] Isotype Fab-Tz amino acid sequences: [0638] Heavy chain: EVQLLESGGGLVQPGGSLRLSCAASGFTFSSFSMSWVRQAPGKGLEWVSSISGSSGTTYYADSV KGRFTISRDNSKNTLYLQMNSLRAEDTAVYYCAKPFPYFDYWGQGTLVTVSSASTKGPSVFPLA PSSKSTSGGTAALGCLVKDYFPEPVTVSWNSGALTSGVHTFPAVLQSSGLYSLSSVVTVPSSSLG TQTYICNVNHKPSNTKVDKKVEPKSSDKTHT (SEQ ID NO.5). [0639] Light chain: EIVLTQSPGTLSLSPGERATLSCRASQSVSSSFLAWYQQKPGQAPRLLIYYASSRATGIPDRFSGSG SGTDFTLTISRLEPEDFAVYYCQQTGRIPPTFGQGTKVEIKRTVAAPSVFIFPPSDEQLKSGTASVV CLLNNFYPREAKVQWKVDNALQSGNSQESVTEQDSKDSTYSLSSTLTLSKADYEKHKVYACEV THQGLSSPCTKSFNRGEC (SEQ ID NO.6). [0640] Generation of HER2 Fab: Coding sequences of the variable region of heavy chain and light chain of trastuzumab (human anti-HER2) antibody were used to generate HER2 Fab-expressing constructs. Coding sequences were synthesized and subcloned into PCDN3.4 expression vector. The constructed plasmids were transformed into E. coli for propagation and scale-up. Purified plasmids were confirmed by sequencing. The constructs containing the heavy chain and light chain of the HER2 Fab were transfected into HEK293 cells (suspension) with polymer polyethylenimine (PEI) reagent. The 6- liter culture medium was harvested at 6-7 days post-transfection. The culture medium containing HER2 Fab was centrifuged, filtered, and then loaded onto KappaSelect affinity column (Mabselect Prism). The loading buffer was 25 mM Tris containing 150 mM NaCl, pH 8.0, and eluted with 100 mM sodium- citrate buffer containing 150 mM NaCl, pH 2.5. The collected solution was neutralized with 1 M arginine, 400 mM succinic acid buffer, pH 9.0. The affinity-purified protein was further purified by gel filtration with Superdex S-2005/150GL column chromatography. The sample injection was 20 mL with a flow rate of 0.3 mL/min and mobile phase 2X PBS at pH 7.4. The purified HER2 Fab was analyzed by SDS-PAGE and SEC-HPLC. The final yield purified yield was 742 mg and stored in -80 °C for long term storage. [0641] Generation of Isotype Fab: Coding sequences were synthesized and subcloned into pTT5 expression vector. The constructed plasmids were transformed into E. coli for propagation and scale-up. Purified plasmids were confirmed by sequencing. The constructs containing the heavy chain and light chain of the Isotype Fab were transfected into HEK293 cells (suspension) with polymer polyethylenimine (PEI) reagent. The 2-liter culture medium was harvested at 7 days post-transfection. The culture medium containing Isotype Fab was centrifuged, filtered, and then loaded onto KappaSelect affinity column (Mabselect Prism). The loading buffer was 25 mM Tris containing 150 mM NaCl, pH 8.0, and eluted with 50 mM sodium-citrate buffer containing 150 mM NaCl, pH 3. The collected solution was neutralized with 1 M arginine, 400 mM succinic acid buffer, pH 9.0. The affinity-purified protein was further purified by gel filtration with Superdex S-2005/150GL column chromatography. The sample injection was 3 mL with a flow rate of 0.3 mL/min and mobile phase 2X PBS at pH 7.4. The purified Isotype Fab was analyzed by SDS-PAGE and SEC-HPLC. The final yield purified yield was 200 mg and stored in -80 °C for long term storage. [0642] Tetrazine conjugation of HER2 Fab and Isotype Fab: HER2 Fab and Isotype Fab was buffer exchanged to PBS pH 7.4 overnight. The methyltetrazine-PEG9-NHS (SiChem #SC-8808) was dissolved in DMSO to make a 10 mM stock solution. For conjugation, the two components were reacted at 3:1 (methyltetrazine-PEG9-NHS to HER2 Fab) molar ratio at 25 °C for 2 hours. The amount of Fab for tetrazine conjugation varied from 30-100 mg but the ratio of 3:1 was always maintained for each conjugation procedure. Then the solution was buffer exchanged to PBS pH 7.4 to remove excess methyltetrazine-PEG9-NHS and buffer salts.
Example 6: HER2 Fab-tetrazine Targeting Moiety (Compound TM-1) [0633] Compound TM-1 and Isotype Fab-Tz were prepared and characterized as follows. [0634] Trastuzumab Fab amino acid sequences: [0635] Heavy chain: EVQLVESGGGLVQPGGSLRLSCAASGFNIKDTYIHWVRQAPGKGLEWVARIYPTNGYTRYADS VKGRFTISADTSKNTAYLQMNSLRAEDTAVYYCSRWGGDGFYAMDYWGQGTLVTVSSASTKG PSVFPLAPSSKSTSGGTAALGCLVKDYFPEPVTVSWNSGALTSGVHTFPAVLQSSGLYSLSSVVT VPSSSLGTQTYICNVNHKP SNTKVDKKV (SEQ ID NO.3). [0636] Light chain: DIQMTQSPSSLSASVGDRVTITCRASQDVNTAVAWYQQKPGKAPKLLIYSASFLYSGVPSRFSGS RSGTDFTLTISSLQPEDFATYYCQQHYTTPPTFGQGTKVEIKRTVAAPSVFIFPPSDEQLKSGTASV VCLLNNFYPREAKVQWKVDNALQSGNSQESVTEQDSKDSTYSLSSTLTLSKADYEKHKVYACE VTHQGLSSPVTKSFNRGEC (SEQ ID NO.4). [0637] Isotype Fab-Tz amino acid sequences: [0638] Heavy chain: EVQLLESGGGLVQPGGSLRLSCAASGFTFSSFSMSWVRQAPGKGLEWVSSISGSSGTTYYADSV KGRFTISRDNSKNTLYLQMNSLRAEDTAVYYCAKPFPYFDYWGQGTLVTVSSASTKGPSVFPLA PSSKSTSGGTAALGCLVKDYFPEPVTVSWNSGALTSGVHTFPAVLQSSGLYSLSSVVTVPSSSLG TQTYICNVNHKPSNTKVDKKVEPKSSDKTHT (SEQ ID NO.5). [0639] Light chain: EIVLTQSPGTLSLSPGERATLSCRASQSVSSSFLAWYQQKPGQAPRLLIYYASSRATGIPDRFSGSG SGTDFTLTISRLEPEDFAVYYCQQTGRIPPTFGQGTKVEIKRTVAAPSVFIFPPSDEQLKSGTASVV CLLNNFYPREAKVQWKVDNALQSGNSQESVTEQDSKDSTYSLSSTLTLSKADYEKHKVYACEV THQGLSSPCTKSFNRGEC (SEQ ID NO.6). [0640] Generation of HER2 Fab: Coding sequences of the variable region of heavy chain and light chain of trastuzumab (human anti-HER2) antibody were used to generate HER2 Fab-expressing constructs. Coding sequences were synthesized and subcloned into PCDN3.4 expression vector. The constructed plasmids were transformed into E. coli for propagation and scale-up. Purified plasmids were confirmed by sequencing. The constructs containing the heavy chain and light chain of the HER2 Fab were transfected into HEK293 cells (suspension) with polymer polyethylenimine (PEI) reagent. The 6- liter culture medium was harvested at 6-7 days post-transfection. The culture medium containing HER2 Fab was centrifuged, filtered, and then loaded onto KappaSelect affinity column (Mabselect Prism). The loading buffer was 25 mM Tris containing 150 mM NaCl, pH 8.0, and eluted with 100 mM sodium- citrate buffer containing 150 mM NaCl, pH 2.5. The collected solution was neutralized with 1 M arginine, 400 mM succinic acid buffer, pH 9.0. The affinity-purified protein was further purified by gel filtration with Superdex S-2005/150GL column chromatography. The sample injection was 20 mL with a flow rate of 0.3 mL/min and mobile phase 2X PBS at pH 7.4. The purified HER2 Fab was analyzed by SDS-PAGE and SEC-HPLC. The final yield purified yield was 742 mg and stored in -80 °C for long term storage. [0641] Generation of Isotype Fab: Coding sequences were synthesized and subcloned into pTT5 expression vector. The constructed plasmids were transformed into E. coli for propagation and scale-up. Purified plasmids were confirmed by sequencing. The constructs containing the heavy chain and light chain of the Isotype Fab were transfected into HEK293 cells (suspension) with polymer polyethylenimine (PEI) reagent. The 2-liter culture medium was harvested at 7 days post-transfection. The culture medium containing Isotype Fab was centrifuged, filtered, and then loaded onto KappaSelect affinity column (Mabselect Prism). The loading buffer was 25 mM Tris containing 150 mM NaCl, pH 8.0, and eluted with 50 mM sodium-citrate buffer containing 150 mM NaCl, pH 3. The collected solution was neutralized with 1 M arginine, 400 mM succinic acid buffer, pH 9.0. The affinity-purified protein was further purified by gel filtration with Superdex S-2005/150GL column chromatography. The sample injection was 3 mL with a flow rate of 0.3 mL/min and mobile phase 2X PBS at pH 7.4. The purified Isotype Fab was analyzed by SDS-PAGE and SEC-HPLC. The final yield purified yield was 200 mg and stored in -80 °C for long term storage. [0642] Tetrazine conjugation of HER2 Fab and Isotype Fab: HER2 Fab and Isotype Fab was buffer exchanged to PBS pH 7.4 overnight. The methyltetrazine-PEG9-NHS (SiChem #SC-8808) was dissolved in DMSO to make a 10 mM stock solution. For conjugation, the two components were reacted at 3:1 (methyltetrazine-PEG9-NHS to HER2 Fab) molar ratio at 25 °C for 2 hours. The amount of Fab for tetrazine conjugation varied from 30-100 mg but the ratio of 3:1 was always maintained for each conjugation procedure. Then the solution was buffer exchanged to PBS pH 7.4 to remove excess methyltetrazine-PEG9-NHS and buffer salts.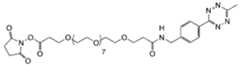 met y tetrazine-PEG9-NHS [0643] Characterization of Compound TM-1 and Isotype Fab-Tz: The sample of prepared Compound TM-1 or Isotype Fab-Tz conjugate was analyzed by SDS-PAGE and Q Exactive HF-X LC-MS (Thermo Scientific) fitted with an Acquity UPLC protein BEH C4 column (300A, 1.7 µm, 2.1 x 50 mm) to confirm the formation of Compound TM-1 and to determine the tetrazine-to-antibody ratio. HPLC-SEC analysis was performed using a UltiMate 3000 HPLC (Thermo Scientific) fitted with a xBridge BEH 200A SEC column (7.8 x 300 mm, Waters Corp). Proteins were eluted with PBS containing 15% isopropanol, pH 7.4). Size exclusion chromatograms for Compound TM-1 and Isotype Fab-Tz and showed > 97% monomeric species for the Fab-tetrazine conjugate with minimal aggregation (≤ 3.0%). The tetrazine-to-antibody ratio was calculated to be 2.2 for Compound TM-1 and 1.8 for Isotype Fab-Tz. [0644] Flow cytometry analysis of Compound TM-1 and Isotype Fab-Tz: Cell binding analysis by flow cytometry using NCI-N87 (HER2 positive) cells was tested with either unconjugated HER2 Fab, Compound TM-1, or Isotype Fab-Tz control. NCI-N87 (HER2-positive) human gastric cancer cells were collected by centrifugation and resuspended with FACS buffer (PBS containing 2% FBS, pH 7.4). Cells were seeded in 96-well plates (200,000 cells per well) and centrifuged at 400 x g for 5 minutes. Supernatants were removed, and cells were incubated at 4 °C for 1 hour with 10-fold titration starting at 400 nM Compound TM-1, HER2 Fab unconjugated, or isotype control (IgG) and for Isotype Fab-Tz cell binding experiment the titration was 3-fold starting at 300 nM. Plates were centrifuged, washed 3 times with FACS buffer and resuspended in 100 mL of secondary antibody of goat anti-human IgG Alexa 488 (catalog #A-11013, Thermo Fisher Scientific 1:500 dilution) or Goat anti-human (Fab’) 2 fragment- specific (109-116-097, Jackson Immunoresearch, PA, USA, 1:200 dilution), and incubated in the dark at 4 °C for 1 hour. Supernatants were removed, cells washed twice with PBS and analyzed by CytoFLEX flow cytometer (Beckman Coulter, Brea, CA, USA) or Attune NxT (Thermo Scientific, MA, USA). Mean fluorescent intensity (MFI) values were calculated and plotted against antibody concentrations. Compound TM-1 binding to HER2-positive cells was observed to be comparable to unconjugated HER2 Fab and no binding was detected with Isotype Fab-Tz. Example 7: General procedure for preparation of Compound A
met y tetrazine-PEG9-NHS [0643] Characterization of Compound TM-1 and Isotype Fab-Tz: The sample of prepared Compound TM-1 or Isotype Fab-Tz conjugate was analyzed by SDS-PAGE and Q Exactive HF-X LC-MS (Thermo Scientific) fitted with an Acquity UPLC protein BEH C4 column (300A, 1.7 µm, 2.1 x 50 mm) to confirm the formation of Compound TM-1 and to determine the tetrazine-to-antibody ratio. HPLC-SEC analysis was performed using a UltiMate 3000 HPLC (Thermo Scientific) fitted with a xBridge BEH 200A SEC column (7.8 x 300 mm, Waters Corp). Proteins were eluted with PBS containing 15% isopropanol, pH 7.4). Size exclusion chromatograms for Compound TM-1 and Isotype Fab-Tz and showed > 97% monomeric species for the Fab-tetrazine conjugate with minimal aggregation (≤ 3.0%). The tetrazine-to-antibody ratio was calculated to be 2.2 for Compound TM-1 and 1.8 for Isotype Fab-Tz. [0644] Flow cytometry analysis of Compound TM-1 and Isotype Fab-Tz: Cell binding analysis by flow cytometry using NCI-N87 (HER2 positive) cells was tested with either unconjugated HER2 Fab, Compound TM-1, or Isotype Fab-Tz control. NCI-N87 (HER2-positive) human gastric cancer cells were collected by centrifugation and resuspended with FACS buffer (PBS containing 2% FBS, pH 7.4). Cells were seeded in 96-well plates (200,000 cells per well) and centrifuged at 400 x g for 5 minutes. Supernatants were removed, and cells were incubated at 4 °C for 1 hour with 10-fold titration starting at 400 nM Compound TM-1, HER2 Fab unconjugated, or isotype control (IgG) and for Isotype Fab-Tz cell binding experiment the titration was 3-fold starting at 300 nM. Plates were centrifuged, washed 3 times with FACS buffer and resuspended in 100 mL of secondary antibody of goat anti-human IgG Alexa 488 (catalog #A-11013, Thermo Fisher Scientific 1:500 dilution) or Goat anti-human (Fab’) 2 fragment- specific (109-116-097, Jackson Immunoresearch, PA, USA, 1:200 dilution), and incubated in the dark at 4 °C for 1 hour. Supernatants were removed, cells washed twice with PBS and analyzed by CytoFLEX flow cytometer (Beckman Coulter, Brea, CA, USA) or Attune NxT (Thermo Scientific, MA, USA). Mean fluorescent intensity (MFI) values were calculated and plotted against antibody concentrations. Compound TM-1 binding to HER2-positive cells was observed to be comparable to unconjugated HER2 Fab and no binding was detected with Isotype Fab-Tz. Example 7: General procedure for preparation of Compound A [0645] To a solution of MMAE (1.40 g, 1.95 mmol) and DIEA (690 mg, 5.34 mmol) in DMF (4.00 mL) was added compound 2 (800 mg, 1.79 mmol) in DMF (4.00 mL) at 0 °C, the mixture was stirred at 25 °C for 16 hrs. Then HOBt (480 mg, 3.56 mmol) in DMF (0.50 mL) was added to the above reaction mixture at 0 °C, after the reaction mixture was stirred at 25 °C for 1.0 hr, the reaction mixture was cooled to 0 °C, and TBAF (1 M in THF, 4.45 mL) was added. After the mixture was stirred for 2.0 hrs at 25 °C, another batch of TBAF (1 M in THF, 4.45 mL) was added at 0 °C, the reaction mixture was continued to stirring for 12.0 hrs at 25 °C. LC-MS showed one main peak was desired mass. The resulting reaction mixture was purified by Prep- HPLC (column: Welch XB-C187 µm 110 A 250*50 mm; mobile phase: [water (0.1% TFA)IN]; B%: 50-70%-40 min. number of injections: 2, Retention time: 37 min, flow rate: 60 mL/min) to give Compound A (450 mg, 99.0% purity; 64.6 mg, 99.2%, 31.2% yield). LCMS (m/z): 928.6 [M+H]+.1HNMR: (400 MHz, DMSO-d6): δ 8–46 - 8.28 (m, 1H), 8–03 - 7.84 (m, 1H), 7.64 (d, J = 8.8 Hz, 1H), 7–35 - 7.23 (m, 4H), 7–21 - 7.13 (m, 1H), 6–05 - 5.57 (m, 2H), 5.10 (s, 1H), 4–81 - 4.39 (m, 3H), 4–35 - 4.19 (m, 1H), 4–05 - 3.92 (m, 2H), 3–40 - 3.09 (m, 11H), 3–08 - 2.83 (m, 5H), 2–48 - 2.37 (m, 2H), 2–31 - 2.09 (m, 5H), 2–07 - 1.89 (m, 3H), 1–88 - 1.64 (m, 6H), 1–63 - 1.33 (m, 4H), 1–32 - 1.16 (m, 1H), 1–08 - 0.97 (m, 9H), 0–90 - 0.68 (m, 18H). Example 8: General procedure for preparation of Compound B
[0645] To a solution of MMAE (1.40 g, 1.95 mmol) and DIEA (690 mg, 5.34 mmol) in DMF (4.00 mL) was added compound 2 (800 mg, 1.79 mmol) in DMF (4.00 mL) at 0 °C, the mixture was stirred at 25 °C for 16 hrs. Then HOBt (480 mg, 3.56 mmol) in DMF (0.50 mL) was added to the above reaction mixture at 0 °C, after the reaction mixture was stirred at 25 °C for 1.0 hr, the reaction mixture was cooled to 0 °C, and TBAF (1 M in THF, 4.45 mL) was added. After the mixture was stirred for 2.0 hrs at 25 °C, another batch of TBAF (1 M in THF, 4.45 mL) was added at 0 °C, the reaction mixture was continued to stirring for 12.0 hrs at 25 °C. LC-MS showed one main peak was desired mass. The resulting reaction mixture was purified by Prep- HPLC (column: Welch XB-C187 µm 110 A 250*50 mm; mobile phase: [water (0.1% TFA)IN]; B%: 50-70%-40 min. number of injections: 2, Retention time: 37 min, flow rate: 60 mL/min) to give Compound A (450 mg, 99.0% purity; 64.6 mg, 99.2%, 31.2% yield). LCMS (m/z): 928.6 [M+H]+.1HNMR: (400 MHz, DMSO-d6): δ 8–46 - 8.28 (m, 1H), 8–03 - 7.84 (m, 1H), 7.64 (d, J = 8.8 Hz, 1H), 7–35 - 7.23 (m, 4H), 7–21 - 7.13 (m, 1H), 6–05 - 5.57 (m, 2H), 5.10 (s, 1H), 4–81 - 4.39 (m, 3H), 4–35 - 4.19 (m, 1H), 4–05 - 3.92 (m, 2H), 3–40 - 3.09 (m, 11H), 3–08 - 2.83 (m, 5H), 2–48 - 2.37 (m, 2H), 2–31 - 2.09 (m, 5H), 2–07 - 1.89 (m, 3H), 1–88 - 1.64 (m, 6H), 1–63 - 1.33 (m, 4H), 1–32 - 1.16 (m, 1H), 1–08 - 0.97 (m, 9H), 0–90 - 0.68 (m, 18H). Example 8: General procedure for preparation of Compound B [0646] To a solution of compound 2 (1.00 g, 5.43 mmol) in DCM (10 mL) was added DIEA (2.10 g, 16.3 mmol), EDCI (2.08 g, 10.9 mmol) and DMAP (1.33 g, 10.9 mmol) and compound 3 (1.61 g, 8.14 mmol). The mixture was stirred at 25 °C for 16 hrs. TLC indicated compound 2 was consumed completely and one new spot formed. The reaction mixture was partitioned between DCM (20 mL) and H2O (10 mL). The organic phase was separated, washed with sat. citric acid aq. (3 mL) and brine (20 mL), then dried over Na2SO4, filtered and concentrated under reduced pressure to give a residue. The residue was purified by column chromatography (Petroleum ether/Ethyl acetate = 3/1 to 1/1) to give compound 4 (700 mg, 39.4% yield).1HNMR (400MHz, CDCl3): δ ppm 1.12 (s, 3 H), 1.60 (dd, J =15.45, 6.19 Hz, 1 H), 1–79 - 1.87 (m, 2 H), 1.92 (br d, J = 5.88 Hz, 1 H), 1.95 (s, 1 H), 1–98 - 2.00 (m, 1 H), 2.02 (br d, J = 4.13 Hz, 1 H), 2.26 (dd, J = 11.63, 3.88 Hz, 1 H), 2–30 - 2.36 (m, 1 H), 2–77 - 2.89 (m, 1 H), 2–88 - 2.88 (m, 1 H), 3.00 (dd, J = 16.95, 4.57 Hz, 1 H), 3.70 (s, 4 H), 3.75 (s, 3 H), 4.80 (dt, J = 8.00, 4.50 Hz, 1 H), 5.66 (dd, J = 16.63, 2.38 Hz, 1 H), 6–02 - 6.12 (m, 1 H), 6.54 (br d, J = 7.88 Hz, 2 H). General procedure for preparation of Compound 6
[0646] To a solution of compound 2 (1.00 g, 5.43 mmol) in DCM (10 mL) was added DIEA (2.10 g, 16.3 mmol), EDCI (2.08 g, 10.9 mmol) and DMAP (1.33 g, 10.9 mmol) and compound 3 (1.61 g, 8.14 mmol). The mixture was stirred at 25 °C for 16 hrs. TLC indicated compound 2 was consumed completely and one new spot formed. The reaction mixture was partitioned between DCM (20 mL) and H2O (10 mL). The organic phase was separated, washed with sat. citric acid aq. (3 mL) and brine (20 mL), then dried over Na2SO4, filtered and concentrated under reduced pressure to give a residue. The residue was purified by column chromatography (Petroleum ether/Ethyl acetate = 3/1 to 1/1) to give compound 4 (700 mg, 39.4% yield).1HNMR (400MHz, CDCl3): δ ppm 1.12 (s, 3 H), 1.60 (dd, J =15.45, 6.19 Hz, 1 H), 1–79 - 1.87 (m, 2 H), 1.92 (br d, J = 5.88 Hz, 1 H), 1.95 (s, 1 H), 1–98 - 2.00 (m, 1 H), 2.02 (br d, J = 4.13 Hz, 1 H), 2.26 (dd, J = 11.63, 3.88 Hz, 1 H), 2–30 - 2.36 (m, 1 H), 2–77 - 2.89 (m, 1 H), 2–88 - 2.88 (m, 1 H), 3.00 (dd, J = 16.95, 4.57 Hz, 1 H), 3.70 (s, 4 H), 3.75 (s, 3 H), 4.80 (dt, J = 8.00, 4.50 Hz, 1 H), 5.66 (dd, J = 16.63, 2.38 Hz, 1 H), 6–02 - 6.12 (m, 1 H), 6.54 (br d, J = 7.88 Hz, 2 H). General procedure for preparation of Compound 6 [0647] To a solution of compound 4 (700 mg, 2.14 mmol) in DCM (5 mL) was added Py (846 mg, 10.7 mmol) and compound 5 (1.72 g, 8.55 mmol) in DCM (5 mL). The mixture was stirred at 25 °C for 1 hrs. TLC indicated compound 4 was consumed completely and one new spot formed. The reaction mixture was partitioned between DCM (20 mL) and H2O (10 mL). The organic phase was separated, washed with sat. citric acid aq. (3 mL) and brine (20 mL), dried over Na2SO4, filtered and concentrated under reduced pressure to give a residue. The residue was purified by column chromatography (Petroleum ether/Ethyl acetate = 3/1 to 1/1) to give compound 6 (490 mg, 46.5% yield).1HNMR (400MHz, CDCl3): δ ppm 1.17 (s, 3 H), 1–55 - 1.61 (m, 1 H), 1.58 (br s, 1 H), 1.76 (dd, J = 14.76, 6.25 Hz, 1 H), 1–87 - 2.03 (m, 3 H), 2–06 - 2.15 (m, 1 H), 2–19 - 2.41 (m, 3 H), 2.82 (dd, J = 17.13, 4.50 Hz, 1 H), 3.03 (dd, J = 17.07, 4.44 Hz, 1 H), 3.72 (s, 3 H), 3.77 (s, 3 H), 4–78 - 4.86 (m, 1 H), 5.67 (dd, J = 16.70, 2.44 Hz, 1 H), 6–03 - 6.14 (m, 1 H), 6.58 (br d, J = 7.88 Hz, 1 H), 7–40 - 7.45 (m, 2 H), 8–27 - 8.33 (m, 2 H). General procedure for preparation of compound 7
[0647] To a solution of compound 4 (700 mg, 2.14 mmol) in DCM (5 mL) was added Py (846 mg, 10.7 mmol) and compound 5 (1.72 g, 8.55 mmol) in DCM (5 mL). The mixture was stirred at 25 °C for 1 hrs. TLC indicated compound 4 was consumed completely and one new spot formed. The reaction mixture was partitioned between DCM (20 mL) and H2O (10 mL). The organic phase was separated, washed with sat. citric acid aq. (3 mL) and brine (20 mL), dried over Na2SO4, filtered and concentrated under reduced pressure to give a residue. The residue was purified by column chromatography (Petroleum ether/Ethyl acetate = 3/1 to 1/1) to give compound 6 (490 mg, 46.5% yield).1HNMR (400MHz, CDCl3): δ ppm 1.17 (s, 3 H), 1–55 - 1.61 (m, 1 H), 1.58 (br s, 1 H), 1.76 (dd, J = 14.76, 6.25 Hz, 1 H), 1–87 - 2.03 (m, 3 H), 2–06 - 2.15 (m, 1 H), 2–19 - 2.41 (m, 3 H), 2.82 (dd, J = 17.13, 4.50 Hz, 1 H), 3.03 (dd, J = 17.07, 4.44 Hz, 1 H), 3.72 (s, 3 H), 3.77 (s, 3 H), 4–78 - 4.86 (m, 1 H), 5.67 (dd, J = 16.70, 2.44 Hz, 1 H), 6–03 - 6.14 (m, 1 H), 6.58 (br d, J = 7.88 Hz, 1 H), 7–40 - 7.45 (m, 2 H), 8–27 - 8.33 (m, 2 H). General procedure for preparation of compound 7 [0648] To a solution of compound 6 (490 mg, 995 µmol) and MMAE (714 mg, 995 µmol) in DMF (4 mL) was added DIEA (64.3 mg, 497 µmol) and HOBt (202 mg, 1.49 mmol). The mixture was stirred at 25 °C for 16 hrs. LC-MS showed compound 6 was consumed completely and one main peak with desired mass was detected. The residue was purified by prep-HPLC (0.1% TFA conditions) to give compound 7 (500 mg, 46.9% yield).1HNMR (400MHz, CDCl3): δ ppm 0.84 (br d, J = 6.75 Hz, 4 H), 0.89 (br d, J = 4.50 Hz, 5 H), 0.92 (br d, J = 6.63 Hz, 4 H), 0.98 (br d, J = 6.25 Hz, 3 H), 1.04 (br d, J = 6.88 Hz, 3 H), 1.16 (s, 3 H), 1–25 - 1.27 (m, 3 H), 1–59 - 1.74 (m, 3 H), 1.88 (br d, J = 9.38 Hz, 4 H), 2.07 (br d, J = 8.38 Hz, 5 H), 2.27 (br s, 4 H), 2–36 - 2.43 (m, 2 H), 2–45 - 2.53 (m, 1 H), 2.89 (br s, 6 H), 2–95 - 3.01 (m, 4 H), 3.04 (br s, 2 H), 3–29 - 3.34 (m, 3 H), 3–36 - 3.47 (m, 5 H), 3–67 - 3.73 (m, 4 H), 3.76 (s, 3 H), 3–82 - 3.89 (m, 1 H), 4–05 - 4.19 (m, 3 H), 4.28 (br s, 1 H), 4–63 - 4.86 (m, 3 H), 4.96 (d, J = 2.50 Hz, 1 H), 5.24 (br s, 1 H), 5.63 (br d, J = 18.14 Hz, 1 H), 5.82 (br s, 1 H), 6–53 - 6.74 (m, 3 H), 7–30 - 7.41 (m, 5 H). General procedure for preparation of Compound B
[0648] To a solution of compound 6 (490 mg, 995 µmol) and MMAE (714 mg, 995 µmol) in DMF (4 mL) was added DIEA (64.3 mg, 497 µmol) and HOBt (202 mg, 1.49 mmol). The mixture was stirred at 25 °C for 16 hrs. LC-MS showed compound 6 was consumed completely and one main peak with desired mass was detected. The residue was purified by prep-HPLC (0.1% TFA conditions) to give compound 7 (500 mg, 46.9% yield).1HNMR (400MHz, CDCl3): δ ppm 0.84 (br d, J = 6.75 Hz, 4 H), 0.89 (br d, J = 4.50 Hz, 5 H), 0.92 (br d, J = 6.63 Hz, 4 H), 0.98 (br d, J = 6.25 Hz, 3 H), 1.04 (br d, J = 6.88 Hz, 3 H), 1.16 (s, 3 H), 1–25 - 1.27 (m, 3 H), 1–59 - 1.74 (m, 3 H), 1.88 (br d, J = 9.38 Hz, 4 H), 2.07 (br d, J = 8.38 Hz, 5 H), 2.27 (br s, 4 H), 2–36 - 2.43 (m, 2 H), 2–45 - 2.53 (m, 1 H), 2.89 (br s, 6 H), 2–95 - 3.01 (m, 4 H), 3.04 (br s, 2 H), 3–29 - 3.34 (m, 3 H), 3–36 - 3.47 (m, 5 H), 3–67 - 3.73 (m, 4 H), 3.76 (s, 3 H), 3–82 - 3.89 (m, 1 H), 4–05 - 4.19 (m, 3 H), 4.28 (br s, 1 H), 4–63 - 4.86 (m, 3 H), 4.96 (d, J = 2.50 Hz, 1 H), 5.24 (br s, 1 H), 5.63 (br d, J = 18.14 Hz, 1 H), 5.82 (br s, 1 H), 6–53 - 6.74 (m, 3 H), 7–30 - 7.41 (m, 5 H). General procedure for preparation of Compound B [0649] To a solution of compound 7 (500 mg, 467 µmol) in MeOH (5 mL) was added LiOH·H2O (196 mg, 4.67 mmol) in H2O (2 mL). The mixture was stirred at 25 °C for 16 hrs. LC-MS showed compound 7 was consumed completely and one main peak with desired mass was detected. The residue was adjusted pH ~ 2 with sat. citric acid aq., then purified by prep-HPLC (0.1% TFA condition) to give Compound B (265 mg 53.4% yield).1HNMR (400MHz, CDCl3): δ ppm 0–80 - 1.04 (m, 25 H), 1.09 (s, 3 H), 1.24 (d, J = 6.88 Hz, 3 H), 1–69 - 1.82 (m, 2 H), 1–88 - 1.94 (m, 3 H), 2–02 - 2.11 (m, 4 H), 2.16 (br d, J = 18.64 Hz, 1 H), 2–11 - 2.24 (m, 2 H), 2.32 (br d, J = 5.25 Hz, 2 H), 2–40 - 2.45 (m, 1 H), 2.52 (br d, J = 5.50 Hz, 2 H), 2.81 (br dd, J = 14.01, 4.88 Hz, 1 H), 2–94 - 3.04 (m, 2 H), 3.08 (s, 2 H), 3–14 - 3.27 (m, 6 H), 3.33 (s, 1 H), 3.39 (s, 3 H), 3–48 - 3.57 (m, 2 H), 3.94 (br d, J = 1.25 Hz, 1 H), 4–05 - 4.18 (m, 4 H), 4.30 (br dd, J = 6.19, 4.82 Hz, 2 H), 4–54 - 4.67 (m, 4 H), 4.91 (br d, J = 2.00 Hz, 2 H), 5.30 (br s, 1 H), 5–64 - 5.73 (m, 1 H), 5–79 - 5.89 (m, 1 H), 6.61 (br d, J = 7.38 Hz, 1 H), 7–30 - 7.42 (m, 5 H), 7–55 - 7.64 (m, 1 H).
[0649] To a solution of compound 7 (500 mg, 467 µmol) in MeOH (5 mL) was added LiOH·H2O (196 mg, 4.67 mmol) in H2O (2 mL). The mixture was stirred at 25 °C for 16 hrs. LC-MS showed compound 7 was consumed completely and one main peak with desired mass was detected. The residue was adjusted pH ~ 2 with sat. citric acid aq., then purified by prep-HPLC (0.1% TFA condition) to give Compound B (265 mg 53.4% yield).1HNMR (400MHz, CDCl3): δ ppm 0–80 - 1.04 (m, 25 H), 1.09 (s, 3 H), 1.24 (d, J = 6.88 Hz, 3 H), 1–69 - 1.82 (m, 2 H), 1–88 - 1.94 (m, 3 H), 2–02 - 2.11 (m, 4 H), 2.16 (br d, J = 18.64 Hz, 1 H), 2–11 - 2.24 (m, 2 H), 2.32 (br d, J = 5.25 Hz, 2 H), 2–40 - 2.45 (m, 1 H), 2.52 (br d, J = 5.50 Hz, 2 H), 2.81 (br dd, J = 14.01, 4.88 Hz, 1 H), 2–94 - 3.04 (m, 2 H), 3.08 (s, 2 H), 3–14 - 3.27 (m, 6 H), 3.33 (s, 1 H), 3.39 (s, 3 H), 3–48 - 3.57 (m, 2 H), 3.94 (br d, J = 1.25 Hz, 1 H), 4–05 - 4.18 (m, 4 H), 4.30 (br dd, J = 6.19, 4.82 Hz, 2 H), 4–54 - 4.67 (m, 4 H), 4.91 (br d, J = 2.00 Hz, 2 H), 5.30 (br s, 1 H), 5–64 - 5.73 (m, 1 H), 5–79 - 5.89 (m, 1 H), 6.61 (br d, J = 7.38 Hz, 1 H), 7–30 - 7.42 (m, 5 H), 7–55 - 7.64 (m, 1 H).












Example 9: Alternative route to Compound B and Synthesis of Compound C. ` General procedure for preparation of compound 2
General procedure for preparation of compound 2 [0650] To a solution of compound 1 (20.0 g, 83.2 mmol) in MeOH (80 mL) was added KOH (8.19 g, 124 mmol) in H2O (80 mL). The mixture was stirred at 25 °C for 24 hrs. The reaction was monitored by TLC (compound 1, PE/EtOAc = 5/1, Rf = 0.5). The reaction mixture was extracted with MTBE (3 × 400 mL). The combined organic layers were washed with water (100 mL), dried with Na2SO4, filtered and concentrated in vacuo to provide the undesired ester. The aqueous layer was acidified with 1 M HCl until pH = 4 while cooling in an ice-water bath (T < 7 °C). The aqueous layer was extracted with MTBE (3 × 400 mL). The combined MTBE layers were dried with Na2SO4, filtered and concentrated in vacuo to provide the compound 2 (5.50 g, 35.9% yield). The crude product was used into the next step without further purification.1H NMR: (400 MHz, DMSO-d6): δ ppm 11.9 (br s, 1 H), 5–81 - 5.94 (m, 1 H), 5.58 (dd, J = 16.45, 2.31 Hz, 1 H), 4.65 (br s, 1 H), 4.24 (br s, 1 H), 2–04 - 2.24 (m, 2 H), 1–87 - 2.03 (m, 1 H), 1–61 - 1.86 (m, 4 H), 1–36 - 1.46 (m, 1 H), 0.97 (s, 3 H). General procedure for preparation of compound 3
[0650] To a solution of compound 1 (20.0 g, 83.2 mmol) in MeOH (80 mL) was added KOH (8.19 g, 124 mmol) in H2O (80 mL). The mixture was stirred at 25 °C for 24 hrs. The reaction was monitored by TLC (compound 1, PE/EtOAc = 5/1, Rf = 0.5). The reaction mixture was extracted with MTBE (3 × 400 mL). The combined organic layers were washed with water (100 mL), dried with Na2SO4, filtered and concentrated in vacuo to provide the undesired ester. The aqueous layer was acidified with 1 M HCl until pH = 4 while cooling in an ice-water bath (T < 7 °C). The aqueous layer was extracted with MTBE (3 × 400 mL). The combined MTBE layers were dried with Na2SO4, filtered and concentrated in vacuo to provide the compound 2 (5.50 g, 35.9% yield). The crude product was used into the next step without further purification.1H NMR: (400 MHz, DMSO-d6): δ ppm 11.9 (br s, 1 H), 5–81 - 5.94 (m, 1 H), 5.58 (dd, J = 16.45, 2.31 Hz, 1 H), 4.65 (br s, 1 H), 4.24 (br s, 1 H), 2–04 - 2.24 (m, 2 H), 1–87 - 2.03 (m, 1 H), 1–61 - 1.86 (m, 4 H), 1–36 - 1.46 (m, 1 H), 0.97 (s, 3 H). General procedure for preparation of compound 3 [0651] To a solution of compound 2 (8.00 g, 43.4 mmol) in MeCN (160 mL) was added DIEA (39.3 g, 304 mmol) and DSC (47.8 g, 186.4 mmol). The mixture was stirred at 40 °C for 14 hrs. The completion of the reaction was confirmed by TLC (compound 2, DCM/MeOH = 10/1, Rf = 0.5). The reaction mixture was poured in water (400 mL), then the temperature rose from 20 °C to 27 °C. After 15 mins, the mixture was cooled to 17 °C in an ice-water bath and stirred for 15 mins. The solid was filtered, washed with water (3 × 20 mL) and was dried under vacuum at 35 °C for 4 hrs to give crude compound 3 (9.60 g). Acetonitrile (20 mL) was added to the crude and the mixture was heated at 40 °C for 1 hrs using mechanical stirring. The heating was stopped and the mixture was cooled to 8 °C in an ice-water bath for 15 mins. The solid was filtered, washed with acetonitrile (2 ×10 mL) and dried under vacuum at 35 °C for 3 hrs to give compound 3 (6.65 g, 36.3% yield).1H NMR: (400 MHz, CDCl3): δ 6–03 - 6.14 (m, 1 H), 5–60 - 5.67 (m, 1 H), 5.29 (br s, 1 H), 2–80 - 2.88 (m, 8 H), 2–25 - 2.47 (m, 4 H), 1–94 - 2.18 (m, 4 H), 1.29 (s, 3 H). General procedure for preparation of compound 4
[0651] To a solution of compound 2 (8.00 g, 43.4 mmol) in MeCN (160 mL) was added DIEA (39.3 g, 304 mmol) and DSC (47.8 g, 186.4 mmol). The mixture was stirred at 40 °C for 14 hrs. The completion of the reaction was confirmed by TLC (compound 2, DCM/MeOH = 10/1, Rf = 0.5). The reaction mixture was poured in water (400 mL), then the temperature rose from 20 °C to 27 °C. After 15 mins, the mixture was cooled to 17 °C in an ice-water bath and stirred for 15 mins. The solid was filtered, washed with water (3 × 20 mL) and was dried under vacuum at 35 °C for 4 hrs to give crude compound 3 (9.60 g). Acetonitrile (20 mL) was added to the crude and the mixture was heated at 40 °C for 1 hrs using mechanical stirring. The heating was stopped and the mixture was cooled to 8 °C in an ice-water bath for 15 mins. The solid was filtered, washed with acetonitrile (2 ×10 mL) and dried under vacuum at 35 °C for 3 hrs to give compound 3 (6.65 g, 36.3% yield).1H NMR: (400 MHz, CDCl3): δ 6–03 - 6.14 (m, 1 H), 5–60 - 5.67 (m, 1 H), 5.29 (br s, 1 H), 2–80 - 2.88 (m, 8 H), 2–25 - 2.47 (m, 4 H), 1–94 - 2.18 (m, 4 H), 1.29 (s, 3 H). General procedure for preparation of compound 4 [0652] To a solution of compound 3 (100 mg, 0.24 mmol) in DMF (20 mL) was added MMAE (136 mg, 0.19 mmol) and DIEA (61.2 mg, 0.47 mmol). The mixture was stirred at 25 °C for 16 hrs. LC-MS showed one main peak with desired mass was detected. The residue was purified by prep-HPLC (Water (0.1% I-ACN) to give compound 4 (41.0 mg, 16.9% yield). LCMS (m/z): 1025.6 (M+H)+. General procedure for preparation of Compound B
[0652] To a solution of compound 3 (100 mg, 0.24 mmol) in DMF (20 mL) was added MMAE (136 mg, 0.19 mmol) and DIEA (61.2 mg, 0.47 mmol). The mixture was stirred at 25 °C for 16 hrs. LC-MS showed one main peak with desired mass was detected. The residue was purified by prep-HPLC (Water (0.1% I-ACN) to give compound 4 (41.0 mg, 16.9% yield). LCMS (m/z): 1025.6 (M+H)+. General procedure for preparation of Compound B [0653] To a solution of compound 4 (500 mg, 0.49 mmol) and compound 4-1 (519 mg, 3.90 mmol) in DMF (10 mL) was added DIEA (378 mg, 2.93 mmol) and DMAP (119 mg, 0.97 mmol). The mixture was stirred at 25 °C for 12 hrs. LC-MS showed one main peak with desired mass was detected. The residue was purified by prep-HPLC (Water (0.1IFA)-ACN) to give Compound B (161 mg, 31.6% yield). 1H NMR: (400 MHz, MeOD–: δ 7.73 - 8.00 (m, 1–H), 7.20 - 7.39 (m, 4–H), 5.78 - 5.96 (m, 1 H), 5.73 (br s, 1–H), 5.20 - 5.28 (m, 1–H), 5.13 - 5.20 (m, 1–H), 4.49 - 4.74 (m, 3–H), 4.17 - 4.28 (m, 2–H), 4.04 - 4.10 (m, 1–H), 3.85 - 3.90 (m, 1–H), 3.50 - 3.80 (m, 2–H), 3.31 - 3.50 (m, 9–H), 3.28 - 3.30 (m, 3–H), 2.77 - 3.12 (m, 6–H), 2.44 - 2.58 (m, 2–H), 1.78 - 2.36 (m, 13–H), 1.56 - 1.72 (m, 2–H), 1.22 - 1.48 (m, 3–H), 1.08 - 1.23 (m, 9–H), 0.80 - 1.07 (m, 18 H). LCMS (m/z): 1043.62 (M+H)+; 1065.61 (M+Na)+. General procedure for preparation of Compound C
[0653] To a solution of compound 4 (500 mg, 0.49 mmol) and compound 4-1 (519 mg, 3.90 mmol) in DMF (10 mL) was added DIEA (378 mg, 2.93 mmol) and DMAP (119 mg, 0.97 mmol). The mixture was stirred at 25 °C for 12 hrs. LC-MS showed one main peak with desired mass was detected. The residue was purified by prep-HPLC (Water (0.1IFA)-ACN) to give Compound B (161 mg, 31.6% yield). 1H NMR: (400 MHz, MeOD–: δ 7.73 - 8.00 (m, 1–H), 7.20 - 7.39 (m, 4–H), 5.78 - 5.96 (m, 1 H), 5.73 (br s, 1–H), 5.20 - 5.28 (m, 1–H), 5.13 - 5.20 (m, 1–H), 4.49 - 4.74 (m, 3–H), 4.17 - 4.28 (m, 2–H), 4.04 - 4.10 (m, 1–H), 3.85 - 3.90 (m, 1–H), 3.50 - 3.80 (m, 2–H), 3.31 - 3.50 (m, 9–H), 3.28 - 3.30 (m, 3–H), 2.77 - 3.12 (m, 6–H), 2.44 - 2.58 (m, 2–H), 1.78 - 2.36 (m, 13–H), 1.56 - 1.72 (m, 2–H), 1.22 - 1.48 (m, 3–H), 1.08 - 1.23 (m, 9–H), 0.80 - 1.07 (m, 18 H). LCMS (m/z): 1043.62 (M+H)+; 1065.61 (M+Na)+. General procedure for preparation of Compound C [0654] To a solution of compound 4 (320 mg, 0.31 mmol) and compound 4-2 (187 mg, 2.50 mmol) in DMF (3.2 mL) was added DIEA (242 mg, 1.87 mmol) and DMAP (76.3 mg, 0.62 mmol). The mixture was stirred at 25 °C for 12 hrs. LC-MS showed compound 4 was consumed completely and one main peak with desired mass was detected. The residue was purified by prep-HPLC (Water (0.1% TFA)- ACN) to give Compound C (92.0 mg, 29.9% yield).1H NMR (400 MHz, MeOD–: δ 7.86 - 8.00 (m, 1– H), 7.15 - 7.45 (m, 5–H), 5.67 - 5.98 (m, 2 H), 5.17 (br s, 1–H), 4.50 - 4.74 (m, 2–H), 4.03 - 4.29 (m, 3– H), 3.81 - 3.89 (m, 2–H), 3.51 - 3.77 (m, 2–H), 3.46 - 3.50 (m, 1–H), 3.33 - 3.45 (m, 5 H), 3.30 (br s, 4 H), 3.20 (dt, J = 11.57, 7.47 Hz, 1–H), 3.02 - 3.15 (m, 3–H), 2.90 - 3.01 (m, 1–H), 2.41 - 2.57 (m, 2–H), 1.65 - 2.39 (m, 15–H), 1.52 - 1.64 (m, 1–H), 1.26 - 1.51 (m, 2–H), 1.08 - 1.23 (m, 9–H), 0.82 - 1.07 (m, 18 H). LCMS (m/z): 985.6 (M+H)+. Example 10: Synthesis of Compound D
[0654] To a solution of compound 4 (320 mg, 0.31 mmol) and compound 4-2 (187 mg, 2.50 mmol) in DMF (3.2 mL) was added DIEA (242 mg, 1.87 mmol) and DMAP (76.3 mg, 0.62 mmol). The mixture was stirred at 25 °C for 12 hrs. LC-MS showed compound 4 was consumed completely and one main peak with desired mass was detected. The residue was purified by prep-HPLC (Water (0.1% TFA)- ACN) to give Compound C (92.0 mg, 29.9% yield).1H NMR (400 MHz, MeOD–: δ 7.86 - 8.00 (m, 1– H), 7.15 - 7.45 (m, 5–H), 5.67 - 5.98 (m, 2 H), 5.17 (br s, 1–H), 4.50 - 4.74 (m, 2–H), 4.03 - 4.29 (m, 3– H), 3.81 - 3.89 (m, 2–H), 3.51 - 3.77 (m, 2–H), 3.46 - 3.50 (m, 1–H), 3.33 - 3.45 (m, 5 H), 3.30 (br s, 4 H), 3.20 (dt, J = 11.57, 7.47 Hz, 1–H), 3.02 - 3.15 (m, 3–H), 2.90 - 3.01 (m, 1–H), 2.41 - 2.57 (m, 2–H), 1.65 - 2.39 (m, 15–H), 1.52 - 1.64 (m, 1–H), 1.26 - 1.51 (m, 2–H), 1.08 - 1.23 (m, 9–H), 0.82 - 1.07 (m, 18 H). LCMS (m/z): 985.6 (M+H)+. Example 10: Synthesis of Compound D



















Example 12: Synthesis of Exatecan Prodrug Compound F [0666] Procedure for preparation of Compound 2
[0666] Procedure for preparation of Compound 2 [0667] To a solution of methyl compound 1 (25.0 g, 104 mmol, 1.0 eq.) in MeOH (125 mL) was added NaOMe (5.40 M, 105 mL, 5.5 eq.) in H2O (125 mL). The mixture was stirred at 25 °C for 24 hrs. The reaction was monitored by TLC, TLC (PE: EA = 2:1, product Rf = 0.20) indicated the reactant (Rf = 0.60) was consumed. The reaction mixture was diluted with H2O (50 mL) and extracted with MTBE (4 × 500 mL). The aqueous layer was acidified with 1 M HCl until pH = 4 while cooling in an ice-water bath (T < 7 °C). The aqueous layer was extracted with MTBE (5 × 500 mL). The combined MTBE layers were dried with Na2SO4, filtered and concentrated under reduced pressure to give residue and evaporated with MeCN three times to give compound 2 (9.1 g, 49.3 mmol, 47.4% yield).1H NMR: 400 MHz, CDCl3 δ ppm 1.11 (s, 3 H), 1.65 (br dd, J = 15.82, 6.19 Hz, 1–H), 1.78 - 2.02 (m, 4–H), 2.05 - 2.42 (m, 4 H), 4.49 (br s, 1 H), 5.64 (dd, J = 16.63, 2.00 Hz, 1–H), 6.00 - 6.14 (m, 1 H). [0668] Procedure for preparation of Compound 3
[0667] To a solution of methyl compound 1 (25.0 g, 104 mmol, 1.0 eq.) in MeOH (125 mL) was added NaOMe (5.40 M, 105 mL, 5.5 eq.) in H2O (125 mL). The mixture was stirred at 25 °C for 24 hrs. The reaction was monitored by TLC, TLC (PE: EA = 2:1, product Rf = 0.20) indicated the reactant (Rf = 0.60) was consumed. The reaction mixture was diluted with H2O (50 mL) and extracted with MTBE (4 × 500 mL). The aqueous layer was acidified with 1 M HCl until pH = 4 while cooling in an ice-water bath (T < 7 °C). The aqueous layer was extracted with MTBE (5 × 500 mL). The combined MTBE layers were dried with Na2SO4, filtered and concentrated under reduced pressure to give residue and evaporated with MeCN three times to give compound 2 (9.1 g, 49.3 mmol, 47.4% yield).1H NMR: 400 MHz, CDCl3 δ ppm 1.11 (s, 3 H), 1.65 (br dd, J = 15.82, 6.19 Hz, 1–H), 1.78 - 2.02 (m, 4–H), 2.05 - 2.42 (m, 4 H), 4.49 (br s, 1 H), 5.64 (dd, J = 16.63, 2.00 Hz, 1–H), 6.00 - 6.14 (m, 1 H). [0668] Procedure for preparation of Compound 3 [0669] To a solution of compound 2 (6.6 g, 35.8 mmol, 1.0 eq.) in MeCN (130 mL) was added DIEA (32.4 g, 250 mmol, 43.6 mL, 7.0 eq.) and DSC (39.4 g, 154 mmol, 4.3 eq.). The mixture was stirred at 25 °C for 16 hrs. The reaction was monitored by TLC, TLC (DCM: MeOH = 10:1, product Rf = 0.60) indicated the reactant (Rf = 0.20) was consumed. The reaction mixture was purified by re-crystallization from H2O (350 mL) at 25 °C, filtered and concentrated under reduced pressure to give a residue. The crude product was evaporated with MeCN for three times to give compound 3 (10.0 g, 21.6 mmol, 60.4% yield, 91.4% purity). HPLC: Rt = 2.14 min, purity: 91.4%.1H NMR: 400 MHz, CDCl3 δ ppm 1.28 (s, 3– H), 1.97 - 2.17 (m, 4–H), 2.26 - 2.49 (m, 4–H), 2.82 - 2.86 (m, 8 H), 5.29 (br s, 1 H), 5.63 (dd, J = 16.70, 2.19 Hz, 1–H), 6.02 - 6.15 (m, 1 H). [0670] Procedure for preparation of Compound 5
[0669] To a solution of compound 2 (6.6 g, 35.8 mmol, 1.0 eq.) in MeCN (130 mL) was added DIEA (32.4 g, 250 mmol, 43.6 mL, 7.0 eq.) and DSC (39.4 g, 154 mmol, 4.3 eq.). The mixture was stirred at 25 °C for 16 hrs. The reaction was monitored by TLC, TLC (DCM: MeOH = 10:1, product Rf = 0.60) indicated the reactant (Rf = 0.20) was consumed. The reaction mixture was purified by re-crystallization from H2O (350 mL) at 25 °C, filtered and concentrated under reduced pressure to give a residue. The crude product was evaporated with MeCN for three times to give compound 3 (10.0 g, 21.6 mmol, 60.4% yield, 91.4% purity). HPLC: Rt = 2.14 min, purity: 91.4%.1H NMR: 400 MHz, CDCl3 δ ppm 1.28 (s, 3– H), 1.97 - 2.17 (m, 4–H), 2.26 - 2.49 (m, 4–H), 2.82 - 2.86 (m, 8 H), 5.29 (br s, 1 H), 5.63 (dd, J = 16.70, 2.19 Hz, 1–H), 6.02 - 6.15 (m, 1 H). [0670] Procedure for preparation of Compound 5 [0671] To a solution of compound 3 (6.6 g, 15.6 mmol, 1.0 eq.) in DMF (70 mL) was added DIEA (4.04 g, 31.2 mmol, 5.44 mL, 2.0 eq.) and Exatecan (6.23 g, 11.7 mmol, 0.75 eq.). The mixture was stirred at 25 °C for 1 hr. LC-MS (EC17183-7-P1A1) showed one main peak with desired mass (RT =0.44 min) was detected. The reaction mixture was filtered and purified by prep-HPLC (column: Phenomenex luna C18 (250*70mm, 10 um); mobile phase: [wIr (FA)-ACN]; gradient: 40%-70% B over 15 min) to give compound 5 (6.0 g, 7.76 mmol, 49.6% yield, 96.1% purity). [0672] LCMS (monitor): Rt = 0.44 min, MS cal.: 742.2, MS observed: [M+H]+ = 743.1. [0673] LCMS: Rt = 0.44 min, MS cal.: 742.2, MS observed: [M+H]+ = 743.3. [0674] HPLC: Rt = 3.04 min, purity: 96.1%. [0675] Procedure for preparation of Compound F
[0671] To a solution of compound 3 (6.6 g, 15.6 mmol, 1.0 eq.) in DMF (70 mL) was added DIEA (4.04 g, 31.2 mmol, 5.44 mL, 2.0 eq.) and Exatecan (6.23 g, 11.7 mmol, 0.75 eq.). The mixture was stirred at 25 °C for 1 hr. LC-MS (EC17183-7-P1A1) showed one main peak with desired mass (RT =0.44 min) was detected. The reaction mixture was filtered and purified by prep-HPLC (column: Phenomenex luna C18 (250*70mm, 10 um); mobile phase: [wIr (FA)-ACN]; gradient: 40%-70% B over 15 min) to give compound 5 (6.0 g, 7.76 mmol, 49.6% yield, 96.1% purity). [0672] LCMS (monitor): Rt = 0.44 min, MS cal.: 742.2, MS observed: [M+H]+ = 743.1. [0673] LCMS: Rt = 0.44 min, MS cal.: 742.2, MS observed: [M+H]+ = 743.3. [0674] HPLC: Rt = 3.04 min, purity: 96.1%. [0675] Procedure for preparation of Compound F [0676] To a solution of compound 5-1 (9.95 g, 16.5 mmol, 1.5 eq.) in DMSO (40 mL) was added DIEA (2.85 g, 22.0 mmol, 3.85 mL, 2.0 eq.) and DMAP (2.70 g, 22.08 mmol, 2.0 eq.) and compound 5 (8.2 g, 11.04 mmol, 1 eq). The mixture was stirred at 25 °C for 16 hrs. LC-MS (EC17183-21-P1A11) showed one main peak with desired mass (Rt = 0.39 min) was detected. The reaction mixture was filtered, purified by prep-HPLC (FA condition) and exchange to AcOH salt to give Compound F (7.5 g, 5.89 mmol, 53.3% yield, 96.5% purity). [0677] LCMS (monitor): Rt = 0.39 min, MS cal.: 1227.5, MS observed: [M+H]+ = 1228.8. [0678] LCMS: Rt = 0.38 min, MS cal.: 1227.5, MS observed: [M+H]+ = 1228.7. [0679] HPLC: Rt = 2.28 min, purity: 96.5%. Example 13: Synthesis of MMAE-TCO Compound G
[0676] To a solution of compound 5-1 (9.95 g, 16.5 mmol, 1.5 eq.) in DMSO (40 mL) was added DIEA (2.85 g, 22.0 mmol, 3.85 mL, 2.0 eq.) and DMAP (2.70 g, 22.08 mmol, 2.0 eq.) and compound 5 (8.2 g, 11.04 mmol, 1 eq). The mixture was stirred at 25 °C for 16 hrs. LC-MS (EC17183-21-P1A11) showed one main peak with desired mass (Rt = 0.39 min) was detected. The reaction mixture was filtered, purified by prep-HPLC (FA condition) and exchange to AcOH salt to give Compound F (7.5 g, 5.89 mmol, 53.3% yield, 96.5% purity). [0677] LCMS (monitor): Rt = 0.39 min, MS cal.: 1227.5, MS observed: [M+H]+ = 1228.8. [0678] LCMS: Rt = 0.38 min, MS cal.: 1227.5, MS observed: [M+H]+ = 1228.7. [0679] HPLC: Rt = 2.28 min, purity: 96.5%. Example 13: Synthesis of MMAE-TCO Compound G [0680] To a solution of compound 1-2 (100 mg, 97.5 μmol) in DMF (1.00 mL) was added DMAP (95.3 mg, 780 μmol) and compound 1-3 (175 mg, 292 μmol) and DIEA (129 μL, 780 μmol). The mixture was stirred at 25 °C for 24 hrs. LC-MS showed one main peak with desired mass (MS cal.: 1510.91, MS observed: [M+H]+ = 1512.1) was detected. The mixture was filtered and purified by prep-HPLC (AcOH condition) to give Compound G (44 mg, 29.8% yield) as a white solid. [0681] LCMS: MS cal.: 1510.91, MS observed: [M+H]+ = 1511.1. [0682] HPLC: Rt = 6.981 min, purity: 95.3%
[0680] To a solution of compound 1-2 (100 mg, 97.5 μmol) in DMF (1.00 mL) was added DMAP (95.3 mg, 780 μmol) and compound 1-3 (175 mg, 292 μmol) and DIEA (129 μL, 780 μmol). The mixture was stirred at 25 °C for 24 hrs. LC-MS showed one main peak with desired mass (MS cal.: 1510.91, MS observed: [M+H]+ = 1512.1) was detected. The mixture was filtered and purified by prep-HPLC (AcOH condition) to give Compound G (44 mg, 29.8% yield) as a white solid. [0681] LCMS: MS cal.: 1510.91, MS observed: [M+H]+ = 1511.1. [0682] HPLC: Rt = 6.981 min, purity: 95.3%






Example 14: Synthesis of MMAE-TCO Compound H [0683] To a solution of compound 1-2 (100 mg, 97.5 μmol) in DMF (1.00 mL) was added DMAP (95.3 mg, 780 μmol) and compound 2-1 (97.6 mg, 780 μmol) and DIEA (129 μL, 780 μmol). The mixture was stirred at 25 °C for 16 hrs. LC-MS showed one main peak with desired mass (MS cal.: 1035.3, MS observed: [M+H]+ = 1036.6) was detected. The mixture was filtered and purified by prep-HPLC to give Compound H (20 mg, 19.8% yield) as a white solid. [0684] LCMS: MS cal.: 1034.6, MS observed: [M+H]+ = 1035.8. [0685] HPLC: Rt = 4.566 min, purity: 95.06% Example 15: Synthesis of MMAE-TCO Compound I
[0683] To a solution of compound 1-2 (100 mg, 97.5 μmol) in DMF (1.00 mL) was added DMAP (95.3 mg, 780 μmol) and compound 2-1 (97.6 mg, 780 μmol) and DIEA (129 μL, 780 μmol). The mixture was stirred at 25 °C for 16 hrs. LC-MS showed one main peak with desired mass (MS cal.: 1035.3, MS observed: [M+H]+ = 1036.6) was detected. The mixture was filtered and purified by prep-HPLC to give Compound H (20 mg, 19.8% yield) as a white solid. [0684] LCMS: MS cal.: 1034.6, MS observed: [M+H]+ = 1035.8. [0685] HPLC: Rt = 4.566 min, purity: 95.06% Example 15: Synthesis of MMAE-TCO Compound I [0686] To a solution of compound 1-2 (50.0 mg, 48.7 μmol) in DMF (0.50 mL) was added DMAP (47.7 mg, 390 μmol) and compound 3-1 (48.4 mg, 390 μmol) and DIEA (129 μL, 390 μmol). The mixture was stirred at 25 °C for 24 hrs. LC-MS showed one main peak with desired mass (MS cal.: 1033.6, MS observed: [M+H]+ = 1035.1) was detected. The mixture was filtered and purified by prep-HPLC to give Compound I (19 mg, 37.7% yield) as a white solid. [0687] LCMS: MS cal.: 1033.6, MS observed: [M+H]+ = 1034.8. [0688] HPLC: Rt = 5.863 min, purity: 96.39% Example 16: Synthesis of MMAE-TCO Compound J
[0686] To a solution of compound 1-2 (50.0 mg, 48.7 μmol) in DMF (0.50 mL) was added DMAP (47.7 mg, 390 μmol) and compound 3-1 (48.4 mg, 390 μmol) and DIEA (129 μL, 390 μmol). The mixture was stirred at 25 °C for 24 hrs. LC-MS showed one main peak with desired mass (MS cal.: 1033.6, MS observed: [M+H]+ = 1035.1) was detected. The mixture was filtered and purified by prep-HPLC to give Compound I (19 mg, 37.7% yield) as a white solid. [0687] LCMS: MS cal.: 1033.6, MS observed: [M+H]+ = 1034.8. [0688] HPLC: Rt = 5.863 min, purity: 96.39% Example 16: Synthesis of MMAE-TCO Compound J [0689] To a solution of compound 1-2 (50.0 mg, 48.7 μmol) in DMF (0.50 mL) was added DMAP (47.7 mg, 390 μmol) and compound 4-1 (48.8 mg, 390 μmol) and DIEA (129 μL, 390 μmol). The mixture was stirred at 25 °C for 24 hrs. LC-MS showed one main peak with desired mass (MS cal.: 1034.6, MS observed: [M+H]+ = 1036.2) was detected. The mixture was filtered and purified by prep-HPLC to give Compound J (37 mg, 73.3% yield) as a white solid. [0690] LCMS: MS cal.: 1034.6, MS observed: [M+H]+ = 1035.8. [0691] HPLC: Rt = 7.609 min, purity: 95.48% Example 17: General procedure for preparation of Intermediate 5-1B
[0689] To a solution of compound 1-2 (50.0 mg, 48.7 μmol) in DMF (0.50 mL) was added DMAP (47.7 mg, 390 μmol) and compound 4-1 (48.8 mg, 390 μmol) and DIEA (129 μL, 390 μmol). The mixture was stirred at 25 °C for 24 hrs. LC-MS showed one main peak with desired mass (MS cal.: 1034.6, MS observed: [M+H]+ = 1036.2) was detected. The mixture was filtered and purified by prep-HPLC to give Compound J (37 mg, 73.3% yield) as a white solid. [0690] LCMS: MS cal.: 1034.6, MS observed: [M+H]+ = 1035.8. [0691] HPLC: Rt = 7.609 min, purity: 95.48% Example 17: General procedure for preparation of Intermediate 5-1B [0692] To a solution of tert-butyl 3-hydroxypropanoate (0.50 g, 3.42 mmol,) and pyridine (552 μL, 6.84 mmol) in THF (10.0 mL) was added triphosgene (405 mg, 1.36 mmol) portion-wise over 5 min at 0 °C under N2 atmosphere. The mixture was stirred at 0 °C for 1 hrs. TLC indicated Reactant 1 was consumed completely and one new spot formed. An aliquot of the reaction mixture was quenched with benzylamine and the desired mass was detected via LCMS (MS cal.: 279.15, MS observed: [M+H-56]+ = 224.1). The reaction mixture was diluted with DCM (20 mL) and washed with 0.5 M HCL (20ml), brine (20 mL), dried over Na2SO4, filtered and concentrated under reduced pressure to give a residue. Intermediate 5-1B (520 mg, 2.49 mmol, 72.8% yield) was obtained as colorless oil. The residue was directly used in the next step without purification. Example 18: General procedure for preparation of Intermediate 5-3B
[0692] To a solution of tert-butyl 3-hydroxypropanoate (0.50 g, 3.42 mmol,) and pyridine (552 μL, 6.84 mmol) in THF (10.0 mL) was added triphosgene (405 mg, 1.36 mmol) portion-wise over 5 min at 0 °C under N2 atmosphere. The mixture was stirred at 0 °C for 1 hrs. TLC indicated Reactant 1 was consumed completely and one new spot formed. An aliquot of the reaction mixture was quenched with benzylamine and the desired mass was detected via LCMS (MS cal.: 279.15, MS observed: [M+H-56]+ = 224.1). The reaction mixture was diluted with DCM (20 mL) and washed with 0.5 M HCL (20ml), brine (20 mL), dried over Na2SO4, filtered and concentrated under reduced pressure to give a residue. Intermediate 5-1B (520 mg, 2.49 mmol, 72.8% yield) was obtained as colorless oil. The residue was directly used in the next step without purification. Example 18: General procedure for preparation of Intermediate 5-3B [0693] To a solution of 5-1B (321 mg, 1.24 mmol) in THF (10 mL) was added DMAP (304 mg, 2.49 mmol) and 5-2 C (520 mg, 2.49 mmol,) at 0 °C. The mixture was stirred at 25 °C for 2 hrs. LC-MS showed 5-2 C was consumed completely and one main peak with desired mass (MS cal.: 430.1, MS observed: [M+Na]+ = 453.1) was detected. The reaction mixture was filtered and concentrated under reduced pressure to give a residue. The residue was purified by column chromatography (silica gel 12 g, PE/EA = 0% to 100%) to give the product. Intermediate 5-3B (502 mg, 1.16 mmol, 93.5% yield) was obtained as colorless oil. LCMS: MS cal.: 430.1, MS observed: [M+Na]+ = 453.1.1H NMR: (400 MHz, CDC–3) δ: 7.35 - 7.22 (m, 5H), 5.38 (br d, J = 6.3 Hz, 1H), 5.04 (s, 2H), 4.35 (t, J = 6.1 H–, 2H), 3.71 - 3.60 (–, 2H), 3.59 - 3.51 (m, 2H), 2.52 (t, J = 6.1 Hz, 2H), 1.38 (s, 9H). Example 19: General procedure for preparation of Intermediate 5-3C O
[0693] To a solution of 5-1B (321 mg, 1.24 mmol) in THF (10 mL) was added DMAP (304 mg, 2.49 mmol) and 5-2 C (520 mg, 2.49 mmol,) at 0 °C. The mixture was stirred at 25 °C for 2 hrs. LC-MS showed 5-2 C was consumed completely and one main peak with desired mass (MS cal.: 430.1, MS observed: [M+Na]+ = 453.1) was detected. The reaction mixture was filtered and concentrated under reduced pressure to give a residue. The residue was purified by column chromatography (silica gel 12 g, PE/EA = 0% to 100%) to give the product. Intermediate 5-3B (502 mg, 1.16 mmol, 93.5% yield) was obtained as colorless oil. LCMS: MS cal.: 430.1, MS observed: [M+Na]+ = 453.1.1H NMR: (400 MHz, CDC–3) δ: 7.35 - 7.22 (m, 5H), 5.38 (br d, J = 6.3 Hz, 1H), 5.04 (s, 2H), 4.35 (t, J = 6.1 H–, 2H), 3.71 - 3.60 (–, 2H), 3.59 - 3.51 (m, 2H), 2.52 (t, J = 6.1 Hz, 2H), 1.38 (s, 9H). Example 19: General procedure for preparation of Intermediate 5-3C O [0694] To mixture of 5-3B (500 mg, 1.16 mmol) was added HCl/dioxane (5 mL, 2M) at 0 °C, the mixture was stirred at 25 °C for 48 hrs. LC-MS showed 5-3 B was consumed completely and one main peak with desired mass (MS cal.: 374.1, MS observed: [M+Na]+ = 397.0) was detected. The reaction mixture was filtered and concentrated under reduced pressure to give a colorless oil. Intermediate 5-3C (400 mg, 1.06 mmol, 91.9% yield) was obtained as colorless oil. LCMS: MS cal.: 374.1, MS observed: [M+Na]+ = 397.0 Example 20: General procedure for preparation of Intermediate 5-4
[0694] To mixture of 5-3B (500 mg, 1.16 mmol) was added HCl/dioxane (5 mL, 2M) at 0 °C, the mixture was stirred at 25 °C for 48 hrs. LC-MS showed 5-3 B was consumed completely and one main peak with desired mass (MS cal.: 374.1, MS observed: [M+Na]+ = 397.0) was detected. The reaction mixture was filtered and concentrated under reduced pressure to give a colorless oil. Intermediate 5-3C (400 mg, 1.06 mmol, 91.9% yield) was obtained as colorless oil. LCMS: MS cal.: 374.1, MS observed: [M+Na]+ = 397.0 Example 20: General procedure for preparation of Intermediate 5-4 [0695] To a dry hydrogenated bottle was added Pd(OH)2 (68.9 mg, 245 μmol) under Ar atmosphere, then the MeOH (5 mL) was added to infiltrate the Pd(OH)2 completely, followed by the solution of 5-3C (400 mg, 1.06 mmol) in MeOH (5 mL) slowly under Ar atmosphere. The mixture was degassed and purged with H2 for 3 times, and then the mixture was s–irred at 20 - 25 °C for 12 hrs under H2 atmosphere (15 psi). LC-MS showed 5-3 C was consumed completely. The reaction mixture was filtered under reduced pressure carefully under Ar atmosphere and the organic layer concentrated under reduced pressure to give a crude compound. Intermediate 5-4 (324 mg, crude) was obtained as colorless oil. The residue was directly used in the next step without purification. Example 21: General procedure for preparation of Compound K
[0695] To a dry hydrogenated bottle was added Pd(OH)2 (68.9 mg, 245 μmol) under Ar atmosphere, then the MeOH (5 mL) was added to infiltrate the Pd(OH)2 completely, followed by the solution of 5-3C (400 mg, 1.06 mmol) in MeOH (5 mL) slowly under Ar atmosphere. The mixture was degassed and purged with H2 for 3 times, and then the mixture was s–irred at 20 - 25 °C for 12 hrs under H2 atmosphere (15 psi). LC-MS showed 5-3 C was consumed completely. The reaction mixture was filtered under reduced pressure carefully under Ar atmosphere and the organic layer concentrated under reduced pressure to give a crude compound. Intermediate 5-4 (324 mg, crude) was obtained as colorless oil. The residue was directly used in the next step without purification. Example 21: General procedure for preparation of Compound K [0696] To a solution of compound 1-2 (142 mg, 138 μmol) in DMF (2.00 mL) was added DMAP (135 mg, 1.11 mmol) and compound 5-4 (100 mg, 416 μmol) and DIEA (129 μL, 1.11 mmol). The mixture was stirred at 25 °C for 16 hrs. LC-MS showed one main peak with desired mass (MS cal.: 1149.6, MS observed: [M+H]+ = 1151.5) was detected. The mixture was filtered and purified by prep-HPLC (TFA condition) to give Compound K (22 mg, 13.8% yield) as a white solid. [0697] LCMS: MS cal.: 1149.6, MS observed: [M+H]+ = 1150.7. [0698] HPLC: Rt = 6.137 min, purity: 98.35%
[0696] To a solution of compound 1-2 (142 mg, 138 μmol) in DMF (2.00 mL) was added DMAP (135 mg, 1.11 mmol) and compound 5-4 (100 mg, 416 μmol) and DIEA (129 μL, 1.11 mmol). The mixture was stirred at 25 °C for 16 hrs. LC-MS showed one main peak with desired mass (MS cal.: 1149.6, MS observed: [M+H]+ = 1151.5) was detected. The mixture was filtered and purified by prep-HPLC (TFA condition) to give Compound K (22 mg, 13.8% yield) as a white solid. [0697] LCMS: MS cal.: 1149.6, MS observed: [M+H]+ = 1150.7. [0698] HPLC: Rt = 6.137 min, purity: 98.35%








Example 22: General procedure for preparation of Compound L [0699] To a solution of compound 1-2 (350 mg, 341 μmol, 1.00 equiv.) in DMF (7.00 mL) was added compound 6-1 (1.01 g, 2.73 mmol, 8.00 equiv.), DMAP (334 mg, 2.73 mmol, 8.00 equiv.) and DIEA (353 mg, 2.73 mmol, 8.00 equiv.). The mixture was stirred at 25 °C for 12 hrs. Two peaks of Cpd_6-2 and Target 287 were detected. The mixture was stirred at 25 °C for another 24 hrs. LC-MS showed one peak with desired mass detected. The reaction mixture was filtered and purified by prep-HPLC (TFA condition) and transfer to AcOH Salt to give Compound L (49 mg, 13.6% yield, 98.6% purity) as a white solid. [0700] LCMS: MS cal.: 1055.7, MS observed: [M+H]+ = 1056.9. [0701] HPLC: Rt = 5.715 min, purity: 98.6%. Example 23: General procedure for preparation of compound 7-4
[0699] To a solution of compound 1-2 (350 mg, 341 μmol, 1.00 equiv.) in DMF (7.00 mL) was added compound 6-1 (1.01 g, 2.73 mmol, 8.00 equiv.), DMAP (334 mg, 2.73 mmol, 8.00 equiv.) and DIEA (353 mg, 2.73 mmol, 8.00 equiv.). The mixture was stirred at 25 °C for 12 hrs. Two peaks of Cpd_6-2 and Target 287 were detected. The mixture was stirred at 25 °C for another 24 hrs. LC-MS showed one peak with desired mass detected. The reaction mixture was filtered and purified by prep-HPLC (TFA condition) and transfer to AcOH Salt to give Compound L (49 mg, 13.6% yield, 98.6% purity) as a white solid. [0700] LCMS: MS cal.: 1055.7, MS observed: [M+H]+ = 1056.9. [0701] HPLC: Rt = 5.715 min, purity: 98.6%. Example 23: General procedure for preparation of compound 7-4 [0702] To a solution of compound 7-2 (800 mg, 5.03 mmol, 1.22 equiv.) in THF (15.0 mL) was added compound 7-3 (650 mg, 4.11 mmol, 1.00 equiv.), AcOH (376 mg, 6.25 mmol, 1.52 equiv.) and NaBH(OAc)3 (1.33 g, 6.26 mmol, 1.52 equiv.). The mixture was stirred at 25 °C for 1 hr. LC-MS showed 99.9% of compound 7-4 was formed. The reaction mixture was filtered and purified by prep-HPLC to give compound 7-4 (1.0 g, 80.8% yield) as a colorless oil. [0703] LCMS: MS cal.: 301.2, MS observed: [M+H]+ = 302.2. Example 24: General procedure for preparation of compound 7-5
[0702] To a solution of compound 7-2 (800 mg, 5.03 mmol, 1.22 equiv.) in THF (15.0 mL) was added compound 7-3 (650 mg, 4.11 mmol, 1.00 equiv.), AcOH (376 mg, 6.25 mmol, 1.52 equiv.) and NaBH(OAc)3 (1.33 g, 6.26 mmol, 1.52 equiv.). The mixture was stirred at 25 °C for 1 hr. LC-MS showed 99.9% of compound 7-4 was formed. The reaction mixture was filtered and purified by prep-HPLC to give compound 7-4 (1.0 g, 80.8% yield) as a colorless oil. [0703] LCMS: MS cal.: 301.2, MS observed: [M+H]+ = 302.2. Example 24: General procedure for preparation of compound 7-5 [0704] To the compound 7-4 (1.00 g, 3.32 mmol, 1.00 equiv.) was added HCl in 1,4-dioxane (4 mL, 8 mmol, 2 M). The mixture was stirred at 25 °C for 1 hr. TLC shows 7-4 was consumed completely. The reaction mixture was concentrated under vacuum to give compound 7-5 (890 mg, 86.3% yield, crud–) as an off - white solid, which is used in next without purification Example 25: General procedure for preparation of Compound M
[0704] To the compound 7-4 (1.00 g, 3.32 mmol, 1.00 equiv.) was added HCl in 1,4-dioxane (4 mL, 8 mmol, 2 M). The mixture was stirred at 25 °C for 1 hr. TLC shows 7-4 was consumed completely. The reaction mixture was concentrated under vacuum to give compound 7-5 (890 mg, 86.3% yield, crud–) as an off - white solid, which is used in next without purification Example 25: General procedure for preparation of Compound M [0705] To a solution of compound 1-2 (120 mg, 117 μmol, 1.00 equiv.) in DMF (2.00 mL) was added compound 7-5 (291 mg, 936 mmol, 8.00 equiv.), DMAP (114 mg, 936 mmol, 8.00 equiv.) and DIEA (121 mg, 936 mmol, 8.00 equiv.). The mixture was stirred at 25 °C for 24 hrs. LC-MS showed Target 288 was formed. The reaction mixture was filtered, purified by prep-HPLC (TFA condition) and transfer to AcOH salt to give Compound M (55 mg, 42.3% yield, 99.0% purity) as a white solid. [0706] LCMS: MS cal.: 1110, MS observed: [M+H]+ = 1111.9. [0707] HPLC: Rt = 5.105 min, purity: 99.0%. Example 26: General procedure for preparation of compound 8-5
[0705] To a solution of compound 1-2 (120 mg, 117 μmol, 1.00 equiv.) in DMF (2.00 mL) was added compound 7-5 (291 mg, 936 mmol, 8.00 equiv.), DMAP (114 mg, 936 mmol, 8.00 equiv.) and DIEA (121 mg, 936 mmol, 8.00 equiv.). The mixture was stirred at 25 °C for 24 hrs. LC-MS showed Target 288 was formed. The reaction mixture was filtered, purified by prep-HPLC (TFA condition) and transfer to AcOH salt to give Compound M (55 mg, 42.3% yield, 99.0% purity) as a white solid. [0706] LCMS: MS cal.: 1110, MS observed: [M+H]+ = 1111.9. [0707] HPLC: Rt = 5.105 min, purity: 99.0%. Example 26: General procedure for preparation of compound 8-5 [0708] To a solution of compound 8-2 (500 mg, 3.12 mmol, 1.00 equiv.) in MeOH (10.0 mL) was added compound 8-4 (1.20 g, 9.36 mmol, 3.00 equiv.). The mixture was stirred at 25 °C for 12 hrs. LC-MS showed compound 8-5 was formed. The reaction mixture was concentrated under vacuum and purified by chromatography (silica gel column, EA/PE=0/1- 1/10) to give compound 8-5 (390 mg, 30.0% yield), which was obtained as a colorless oil. LCMS: MS cal.: 416.3, MS observed: [M+H]+ = 417.6. HPLC: Rt = 4.181 min, purity: 83.59%. Example 27: General procedure for preparation of compound 8-6
[0708] To a solution of compound 8-2 (500 mg, 3.12 mmol, 1.00 equiv.) in MeOH (10.0 mL) was added compound 8-4 (1.20 g, 9.36 mmol, 3.00 equiv.). The mixture was stirred at 25 °C for 12 hrs. LC-MS showed compound 8-5 was formed. The reaction mixture was concentrated under vacuum and purified by chromatography (silica gel column, EA/PE=0/1- 1/10) to give compound 8-5 (390 mg, 30.0% yield), which was obtained as a colorless oil. LCMS: MS cal.: 416.3, MS observed: [M+H]+ = 417.6. HPLC: Rt = 4.181 min, purity: 83.59%. Example 27: General procedure for preparation of compound 8-6 [0709] To the compound 8-5 (380 mg, 912 µmol, 1.00 equiv.) was added HCl in 1,4-dioxane (2 mL, 2 M). The mixture was stirred at 25 °C for 20 hrs. LC-MS showed one peak with desired mass (MS cal.: 204.1, MS observed: [M+H]+ = 204.9) was detected. The reaction mixture was concentrated under vacuum to give compound 8-5 (214 mg, crude– as a light - yellow oil. [0710] LCMS: MS cal.: 204.1, MS observed: [M+H]+ = 204.9. Example 28: General procedure for preparation of Compound N
[0709] To the compound 8-5 (380 mg, 912 µmol, 1.00 equiv.) was added HCl in 1,4-dioxane (2 mL, 2 M). The mixture was stirred at 25 °C for 20 hrs. LC-MS showed one peak with desired mass (MS cal.: 204.1, MS observed: [M+H]+ = 204.9) was detected. The reaction mixture was concentrated under vacuum to give compound 8-5 (214 mg, crude– as a light - yellow oil. [0710] LCMS: MS cal.: 204.1, MS observed: [M+H]+ = 204.9. Example 28: General procedure for preparation of Compound N [0711] To a solution of compound 1-2 (100 mg, 97.5 μmol, 1.00 equiv.) in DMF (3.00 mL) was added compound 7-5 (214 mg, 772 mmol, 7.92 equiv.), DMAP (95.3 mg, 780 mmol, 8.00 equiv.) and DIEA (101 mg, 780 mmol, 8.00 equiv.). The mixture was stirred at 25 °C for 24 hrs. LC-MS showed one peak with desired mass was formed. The reaction mixture was filtered and purified by prep-HPLC (TFA condition) and transfer to AcOH salt to give Compound N (39 mg, 35.9% yield, 98.6% purity) as a white solid. [0712] LCMS: MS cal.: 1113.7, MS observed: [M+H]+ = 1114.9. HPLC: Rt = 5.861 min, purity: 98.6%. Example 29: General procedure for preparation of Compound O H
[0711] To a solution of compound 1-2 (100 mg, 97.5 μmol, 1.00 equiv.) in DMF (3.00 mL) was added compound 7-5 (214 mg, 772 mmol, 7.92 equiv.), DMAP (95.3 mg, 780 mmol, 8.00 equiv.) and DIEA (101 mg, 780 mmol, 8.00 equiv.). The mixture was stirred at 25 °C for 24 hrs. LC-MS showed one peak with desired mass was formed. The reaction mixture was filtered and purified by prep-HPLC (TFA condition) and transfer to AcOH salt to give Compound N (39 mg, 35.9% yield, 98.6% purity) as a white solid. [0712] LCMS: MS cal.: 1113.7, MS observed: [M+H]+ = 1114.9. HPLC: Rt = 5.861 min, purity: 98.6%. Example 29: General procedure for preparation of Compound O H [0713] General procedure for preparation of compound
[0713] General procedure for preparation of compound [0714] To a solution of compound 5 (4.00 g, 16.3 mmol, 1.00 eq.) in ACN (15.0 mL) was added DIEA (8.42 g, 65.1 mmol, 11.3 mL, 3.00 eq.) and DSC (5.56 g, 21.7 mmol, 1.00 eq.) in ACN (25.0 mL). The mixture was stirred at 25 °C for 1 hr. The reaction was monitored by TLC, TLC (Petroleum ether/Ethyl acetate = 1:1, product Rf = 0.41). The reaction mixture was concentrated under reduced pressure to give a residue. The residue was purified by column chromatography (SiO2, Petroleum ether/Ethyl acetate=50/1 to 1/1). LCMS showed compound 5 (4.7 g, 17.6 mmol, 81.1% yield) as a colorless oil. LCMS: Rt = 0.316 min, MS cal.: 281.1, MS observed: [M+Na]+ = 304.0.1HNMR (400 MHz, DMSO-d6) δ: 5.86 - 5.97 (m, 1 H), 5.55 - 5.72 (m, 1 H), 4.77 (d, J=3.0 Hz, 1 H), 4.21 - 4.33 (m, 1 H), 2.79 (s, 4 H), 2.09 - 2.27 (m, 3 H), 1.89 - 2.02 (m, 2 H), 1.65 - 1.81 (m, 3 H), 1.12 - 1.21 (m, 3 H). [0715] General procedure for preparation of compound 3
[0714] To a solution of compound 5 (4.00 g, 16.3 mmol, 1.00 eq.) in ACN (15.0 mL) was added DIEA (8.42 g, 65.1 mmol, 11.3 mL, 3.00 eq.) and DSC (5.56 g, 21.7 mmol, 1.00 eq.) in ACN (25.0 mL). The mixture was stirred at 25 °C for 1 hr. The reaction was monitored by TLC, TLC (Petroleum ether/Ethyl acetate = 1:1, product Rf = 0.41). The reaction mixture was concentrated under reduced pressure to give a residue. The residue was purified by column chromatography (SiO2, Petroleum ether/Ethyl acetate=50/1 to 1/1). LCMS showed compound 5 (4.7 g, 17.6 mmol, 81.1% yield) as a colorless oil. LCMS: Rt = 0.316 min, MS cal.: 281.1, MS observed: [M+Na]+ = 304.0.1HNMR (400 MHz, DMSO-d6) δ: 5.86 - 5.97 (m, 1 H), 5.55 - 5.72 (m, 1 H), 4.77 (d, J=3.0 Hz, 1 H), 4.21 - 4.33 (m, 1 H), 2.79 (s, 4 H), 2.09 - 2.27 (m, 3 H), 1.89 - 2.02 (m, 2 H), 1.65 - 1.81 (m, 3 H), 1.12 - 1.21 (m, 3 H). [0715] General procedure for preparation of compound 3 [0716] To a solution of compound 1 (3.15 g, 4.49 mmol, 1.00 eq.) and in DCM (31.5 mL) was added HBTU (2.05 g, 5.39 mmol, 1.20 eq.) and DIEA (1.16 g, 8.99 mmol, 1.57 mL, 2.00 eq.), then was added compound 2 (2.63 g, 8.99 mmol, 2.00 eq.). The mixture was stirred at 25 °C for 2 hrs. LCMS showed one peak (Rt = 0.428 min) with desired mass (MS cal.: 974.6, MS observed: [M+H]+ = 975.5 ) detected. The reaction mixture was diluted with H2O (60.0 mL) and extracted with DCM (60.0 mL *2). The combined organic layers were washed with Brine (60.0 mL * 2), dried over Na2SO4, filtered and concentrated under reduced pressure to give a residue. The residue was purified by column chromatography (SiO2, DCM: MeOH =100/1 to 50/1) (TLC: DCM:MeOH=10:1, Rf = 0.62). LCMS, HNMR, and HPLC showed compound 3 (4.05 g, 4.04 mmol, 90.0% yield, 97.4% purity) as a yellow oil. LCMS: Rt = 0.429 min, MS cal.: 974.6, MS observed: [M+H]+ = 975.5. HPLC: Rt = 2.326 min, purity: 97.4%. HNMR (400 MHz, DMSO-d6) δ: 7.89 (br t, J = 5.6 Hz, 1 H), 6.74 (br s, 1 H), 3.56 - 3.67 (m, 1 H), 3.50 (br d, J = 2.4 Hz, 8 H), 3.35 - 3.45 (m, 8 H), 3.02 - 3.23 (m, 8 H), 2.73 - 2.93 (m, 4 H), 2.52 - 2.66 (m, 4 H), 2.34 (br d, J = 5.3 Hz, 2 H), 2.04 - 2.20 (m, 4 H), 1.84 - 2.04 (m, 4 H), 1.41 - 1.46 (m, 36 H), 1.37 (s, 9 H) [0717] General procedure for preparation of compound 4
[0716] To a solution of compound 1 (3.15 g, 4.49 mmol, 1.00 eq.) and in DCM (31.5 mL) was added HBTU (2.05 g, 5.39 mmol, 1.20 eq.) and DIEA (1.16 g, 8.99 mmol, 1.57 mL, 2.00 eq.), then was added compound 2 (2.63 g, 8.99 mmol, 2.00 eq.). The mixture was stirred at 25 °C for 2 hrs. LCMS showed one peak (Rt = 0.428 min) with desired mass (MS cal.: 974.6, MS observed: [M+H]+ = 975.5 ) detected. The reaction mixture was diluted with H2O (60.0 mL) and extracted with DCM (60.0 mL *2). The combined organic layers were washed with Brine (60.0 mL * 2), dried over Na2SO4, filtered and concentrated under reduced pressure to give a residue. The residue was purified by column chromatography (SiO2, DCM: MeOH =100/1 to 50/1) (TLC: DCM:MeOH=10:1, Rf = 0.62). LCMS, HNMR, and HPLC showed compound 3 (4.05 g, 4.04 mmol, 90.0% yield, 97.4% purity) as a yellow oil. LCMS: Rt = 0.429 min, MS cal.: 974.6, MS observed: [M+H]+ = 975.5. HPLC: Rt = 2.326 min, purity: 97.4%. HNMR (400 MHz, DMSO-d6) δ: 7.89 (br t, J = 5.6 Hz, 1 H), 6.74 (br s, 1 H), 3.56 - 3.67 (m, 1 H), 3.50 (br d, J = 2.4 Hz, 8 H), 3.35 - 3.45 (m, 8 H), 3.02 - 3.23 (m, 8 H), 2.73 - 2.93 (m, 4 H), 2.52 - 2.66 (m, 4 H), 2.34 (br d, J = 5.3 Hz, 2 H), 2.04 - 2.20 (m, 4 H), 1.84 - 2.04 (m, 4 H), 1.41 - 1.46 (m, 36 H), 1.37 (s, 9 H) [0717] General procedure for preparation of compound 4 [0718] To a solution of compound 3 (4.50 g, 4.61 mmol, 1.00 eq.) and in HCOOH (42.0 mL) .The mixture was stirred at 40 °C for 16 hrs. LCMS showed one peak (Rt = 0.424 min) with desired mass (MS cal.: 650.3, MS observed: [M+H]+ = 651.2 ) detected. The reaction mixture concentrated with PhMe/MeCN = 1:1 under reduced pressure to give a residue. LCMS and HPLC showed compound 4 (4.2 g, crude, HCOOH salt) as a yellow oil. LCMS: Rt = 0.423 min, MS cal.: 650.3, MS observed: [M+H]+ = 651.2. HPLC: Rt = 0.202, purity: 86.1% [0719] General procedure for preparation of Compound O
[0718] To a solution of compound 3 (4.50 g, 4.61 mmol, 1.00 eq.) and in HCOOH (42.0 mL) .The mixture was stirred at 40 °C for 16 hrs. LCMS showed one peak (Rt = 0.424 min) with desired mass (MS cal.: 650.3, MS observed: [M+H]+ = 651.2 ) detected. The reaction mixture concentrated with PhMe/MeCN = 1:1 under reduced pressure to give a residue. LCMS and HPLC showed compound 4 (4.2 g, crude, HCOOH salt) as a yellow oil. LCMS: Rt = 0.423 min, MS cal.: 650.3, MS observed: [M+H]+ = 651.2. HPLC: Rt = 0.202, purity: 86.1% [0719] General procedure for preparation of Compound O [0720] To a solution of compound 4 (2.80 g, 4.30 mmol, 1.00 eq.) in DMSO (28.0 mL) was added DIEA (2.78 g, 21.5 mmol, 3.75 mL, 5.00 eq.) and compound 5 (1.45 g, 5.16 mmol, 1.20 eq.). The mixture was stirred at 25 °C for 16 hrs. LCMS showed one peak (Rt = 1.281 min) with desired mass (MS cal.: 816.4, MS observed: [M+H]+ = 817.4) was detected. The reaction liquid is filtered for high efficiency mass spectrometry separation. The residue was purified by prep-HPLC (TFA condition) to afford Compound O (peak 1, 610 mg, 698.9 μmol, 16.2% yield, 93.6% purity) and Compound O (peak 2, 500 mg, 606.5 μmol, 14.1% yield, 99.1% purity) as a white solid. LCMS (Peak 1): Rt = 0.300 min, MS cal.: 816.4, MS observed: [M+H]+ = 817.4. LCMS (Peak 2): Rt = 0.337 min, MS cal.: 816.4, MS observed: [M+H]+ = 817.4. HPLC (Peak 1): Rt =1.603 min, purity: 93.6%. HPLC (Peak 2): Rt =1.634 min, purity: 99.1%. Example 30: General procedure for preparation of Compound P
[0720] To a solution of compound 4 (2.80 g, 4.30 mmol, 1.00 eq.) in DMSO (28.0 mL) was added DIEA (2.78 g, 21.5 mmol, 3.75 mL, 5.00 eq.) and compound 5 (1.45 g, 5.16 mmol, 1.20 eq.). The mixture was stirred at 25 °C for 16 hrs. LCMS showed one peak (Rt = 1.281 min) with desired mass (MS cal.: 816.4, MS observed: [M+H]+ = 817.4) was detected. The reaction liquid is filtered for high efficiency mass spectrometry separation. The residue was purified by prep-HPLC (TFA condition) to afford Compound O (peak 1, 610 mg, 698.9 μmol, 16.2% yield, 93.6% purity) and Compound O (peak 2, 500 mg, 606.5 μmol, 14.1% yield, 99.1% purity) as a white solid. LCMS (Peak 1): Rt = 0.300 min, MS cal.: 816.4, MS observed: [M+H]+ = 817.4. LCMS (Peak 2): Rt = 0.337 min, MS cal.: 816.4, MS observed: [M+H]+ = 817.4. HPLC (Peak 1): Rt =1.603 min, purity: 93.6%. HPLC (Peak 2): Rt =1.634 min, purity: 99.1%. Example 30: General procedure for preparation of Compound P [0721] General procedure for preparation of compound 7
[0721] General procedure for preparation of compound 7 [0722] To a solution of compound 6 (10.0 g, 17.4 mmol, 1.00 eq.) in ACN (250.0 mL) was added HOSu (2.21 g, 19.2 mmol, 1.10 eq.) and HBTU (7.28 g, 19.2 mmol, 1.10 eq.). The mixture was stirred at 25 °C for 24 hrs. LCMS showed one main peak (Rt = 1.35 min) with desired mass (MS cal.: 669.3, MS observed: [M+H]+ = 670.3) was detected. The reaction mixture was concentrated under reduced pressure to give a residue. The crude product was purified by re-crystallization from MTBE (2500 mL). HNMR, LCMS, and HPLC showed compound 7 (12.0 g, 17.1 mmol, 97.6% yield, 95.2% purity) as a white solid. [0723] LCMS: Rt = 1.318 min, MS cal.: 669.3, MS observed: [M+H]+ = 670.2. HPLC: Rt = 1.836 min, purity: 95.2%.1HNMR (400 MHz, DMSO-d6) δ : 4.10 - 4.02 (m, 2 H), 3.77 - 3.72 (m, 2 H), 3.72 - 3.66 (m, 4 H), 3.72 - 3.64 (m, 4 H), 3.03 (br s, 8 H), 3.01 – 2.96 (m, 4 H), 2.88 - 2.76 (m, 8 H), 1.49 - 1.40 (m, 27 H). [0724] General procedure for preparation of compound 9
[0722] To a solution of compound 6 (10.0 g, 17.4 mmol, 1.00 eq.) in ACN (250.0 mL) was added HOSu (2.21 g, 19.2 mmol, 1.10 eq.) and HBTU (7.28 g, 19.2 mmol, 1.10 eq.). The mixture was stirred at 25 °C for 24 hrs. LCMS showed one main peak (Rt = 1.35 min) with desired mass (MS cal.: 669.3, MS observed: [M+H]+ = 670.3) was detected. The reaction mixture was concentrated under reduced pressure to give a residue. The crude product was purified by re-crystallization from MTBE (2500 mL). HNMR, LCMS, and HPLC showed compound 7 (12.0 g, 17.1 mmol, 97.6% yield, 95.2% purity) as a white solid. [0723] LCMS: Rt = 1.318 min, MS cal.: 669.3, MS observed: [M+H]+ = 670.2. HPLC: Rt = 1.836 min, purity: 95.2%.1HNMR (400 MHz, DMSO-d6) δ : 4.10 - 4.02 (m, 2 H), 3.77 - 3.72 (m, 2 H), 3.72 - 3.66 (m, 4 H), 3.72 - 3.64 (m, 4 H), 3.03 (br s, 8 H), 3.01 – 2.96 (m, 4 H), 2.88 - 2.76 (m, 8 H), 1.49 - 1.40 (m, 27 H). [0724] General procedure for preparation of compound 9 [0725] To a solution of compound 8 (5.74 g, 29.8 mmol, 2.00 eq.) in DCM (100 mL) was added DIEA (5.79 g, 44.8 mmol, 7.80 mL, 3.00 eq.) and compound 7 (10.0 g, 14.9 mmol, 1.00 eq.). LCMS showed one main peak (Rt = 1.205 min) with desired mass (MS cal.: 746.5, MS observed: [M+H]+ = 747.2) was detected. The reaction mixture was washed with H2O (50.0 mL * 3), dried over Na2SO4, filtered and concentrated under reduced. The residue was purified by prep-HPLC (HCOOH condition). HNMR, LCMS, and HPLC showed compound 3 (5.50 g, 7.33 mmol, 49.0% yield, 99.5% purity) as a yellow oil. LCMS: Rt = 1.209 min, (MS cal.: 746.5, MS observed: [M+H]+ = 747.2). HPLC: Rt = 1.632 min, 99.5% purity.1HNMR 400 MHz, DMSO-d6) δ : 8.49 (br t, J = 5.7 Hz, 1 H), 3.63- 3.46 (m, 11 H), 3.45 - 3.40 (m, 2 H), 3.36 - 3.32 (m, 2 H), 3.30 - 3.22 (m, 6 H), 3.12 - 3.04 (m, 2 H), 2.96 - 2.90 (m, 2 H), 2.84 - 2.65 (m, 12 H), 2.59 (br s, 4 H), 1.46 - 1.35 (m, 27 H). [0726] General procedure for preparation of compound 10
[0725] To a solution of compound 8 (5.74 g, 29.8 mmol, 2.00 eq.) in DCM (100 mL) was added DIEA (5.79 g, 44.8 mmol, 7.80 mL, 3.00 eq.) and compound 7 (10.0 g, 14.9 mmol, 1.00 eq.). LCMS showed one main peak (Rt = 1.205 min) with desired mass (MS cal.: 746.5, MS observed: [M+H]+ = 747.2) was detected. The reaction mixture was washed with H2O (50.0 mL * 3), dried over Na2SO4, filtered and concentrated under reduced. The residue was purified by prep-HPLC (HCOOH condition). HNMR, LCMS, and HPLC showed compound 3 (5.50 g, 7.33 mmol, 49.0% yield, 99.5% purity) as a yellow oil. LCMS: Rt = 1.209 min, (MS cal.: 746.5, MS observed: [M+H]+ = 747.2). HPLC: Rt = 1.632 min, 99.5% purity.1HNMR 400 MHz, DMSO-d6) δ : 8.49 (br t, J = 5.7 Hz, 1 H), 3.63- 3.46 (m, 11 H), 3.45 - 3.40 (m, 2 H), 3.36 - 3.32 (m, 2 H), 3.30 - 3.22 (m, 6 H), 3.12 - 3.04 (m, 2 H), 2.96 - 2.90 (m, 2 H), 2.84 - 2.65 (m, 12 H), 2.59 (br s, 4 H), 1.46 - 1.35 (m, 27 H). [0726] General procedure for preparation of compound 10 [0727] To a solution of compound 9 (1.50 g, 2.01 mmol, 1.00 eq.) in HCOOH (15.0 mL). The mixture was stirred at 40 °C for 16 hrs. LCMS showed one main peak (Rt = 0.191 min) with desired mass (MS cal.: 578.3, MS observed: [M+H]+ = 579.0) was detected. The reaction mixture was concentrated with PhMe/MeCN = 1:1 under reduced pressure to give a residue. LCMS showed compound 10 (1.40 g, crude) as a yellow oil. LCMS: Rt = 0.167 min, MS cal.: 578.3, MS observed: [(M+H)]+=579.0. [0728] General procedure for preparation of Compound P
[0727] To a solution of compound 9 (1.50 g, 2.01 mmol, 1.00 eq.) in HCOOH (15.0 mL). The mixture was stirred at 40 °C for 16 hrs. LCMS showed one main peak (Rt = 0.191 min) with desired mass (MS cal.: 578.3, MS observed: [M+H]+ = 579.0) was detected. The reaction mixture was concentrated with PhMe/MeCN = 1:1 under reduced pressure to give a residue. LCMS showed compound 10 (1.40 g, crude) as a yellow oil. LCMS: Rt = 0.167 min, MS cal.: 578.3, MS observed: [(M+H)]+=579.0. [0728] General procedure for preparation of Compound P [0729] To a solution of compound 10 (1.30 g, 4.62 mmol, 1.00 eq.) in DMSO (13.0 mL) was added DIEA (1.79 g, 13.8 mmol, 2.41 mL, 3.00 eq.) and compound 5 (3.21 g, 5.55 mmol, 1.20 eq.). The mixture was stirred at 25 °C for 16 hrs. LCMS showed one main peak (Rt =1.403 min) with desired mass (MS cal.: 744.4, MS observed: [M+H]+ = 745.2) was detected. The reaction liquid is filtered for high efficiency mass spectrometry separation. The residue was purified by prep-HPLC (TFA condition). LCMS and HPLC showed compound Compound P (590 mg, 754.8 μmol, 16.3% yield, 95.3% purity) as a white solid. LCMS: Rt = 0.361 min, (MS cal.: 744.4, MS observed: [M+H]+= 745.2). HPLC: Rt = 1.549 min, purity: 95.3% Example 31: General procedure for preparation of Compound Q
[0729] To a solution of compound 10 (1.30 g, 4.62 mmol, 1.00 eq.) in DMSO (13.0 mL) was added DIEA (1.79 g, 13.8 mmol, 2.41 mL, 3.00 eq.) and compound 5 (3.21 g, 5.55 mmol, 1.20 eq.). The mixture was stirred at 25 °C for 16 hrs. LCMS showed one main peak (Rt =1.403 min) with desired mass (MS cal.: 744.4, MS observed: [M+H]+ = 745.2) was detected. The reaction liquid is filtered for high efficiency mass spectrometry separation. The residue was purified by prep-HPLC (TFA condition). LCMS and HPLC showed compound Compound P (590 mg, 754.8 μmol, 16.3% yield, 95.3% purity) as a white solid. LCMS: Rt = 0.361 min, (MS cal.: 744.4, MS observed: [M+H]+= 745.2). HPLC: Rt = 1.549 min, purity: 95.3% Example 31: General procedure for preparation of Compound Q [0730] General procedure for preparation of compound 11
[0730] General procedure for preparation of compound 11 [0731] To a solution of compound 10 (1.09 g, 2.59 mmol, 1.00 eq.) in DMF (12.0 mL) was added DIEA (1.01 g, 7.78 mmol, 1.35 mL, 3.00 eq.) and compound TCO-NHS ester (1.50 g, 2.59 mmol, 1.00 eq.) The mixture was stirred at 25 °C for 0.5 hr. LCMS showed one main peak (Rt = 1.09 min) with desired mass (MS cal.: 885.4, MS observed: [M+H]+ = 886.2) was detected. The crude product was not work-up and used for next step directly. LCMS: Rt = 1.087 min, MS cal.: 885.4, MS observed: [M+H]+ = 886.2. [0732] General procedure for preparation of Compound Q
[0731] To a solution of compound 10 (1.09 g, 2.59 mmol, 1.00 eq.) in DMF (12.0 mL) was added DIEA (1.01 g, 7.78 mmol, 1.35 mL, 3.00 eq.) and compound TCO-NHS ester (1.50 g, 2.59 mmol, 1.00 eq.) The mixture was stirred at 25 °C for 0.5 hr. LCMS showed one main peak (Rt = 1.09 min) with desired mass (MS cal.: 885.4, MS observed: [M+H]+ = 886.2) was detected. The crude product was not work-up and used for next step directly. LCMS: Rt = 1.087 min, MS cal.: 885.4, MS observed: [M+H]+ = 886.2. [0732] General procedure for preparation of Compound Q




























Example 39: (4-(6-methyl-1,2,4,5-tetrazin-3-yl)-2-(trifluoromethyl)phenyl)methanamine (Target 17) [0755] 3-((1,3-dioxoisoindolin-2-yl)methyl)-4-(trifluoromethyl)benzonitrile (2): To a solution of 3- (hydroxymethyl)-4-(trifluoromethyl)benzonitrile (4.5 g, 22.38 mmol) in THF (50 mL) was added isoindoline-1,3-dione (3.3 g, 22.38 mmol), PPh3 (11.7 g, 44.76 mmol) and DIAD (6.79 g, 33.57 mmol) at 0 °C under N2. Then the resulting mixture was stirred at 20 °C for 16 h. The reaction mixture was quenched by addition of water (15 mL) and extracted with EtOAc (3 × 10 mL). The combined organic phase was washed with brine (15 mL), dried over sodium sulfate, filtered and the filtrate was concentrated under reduced pressure to give a residue, which was purified by column to give 3-((1,3- dioxoisoindolin-2-yl)methyl)-4-(trifluoromethyl)benzonitrile (2) (5.1 g, 69%).1H NMR (400 MHz, CDCl3) δ = 7.91-8.03 (m, 2H), 7.84 (dd, J = 5.6, 2.8 Hz, 3H), 7.70 (d, J = 8.0 Hz, 1H), 7.46 (s, 1H), 5.13 (s, 2H). [0756] 3-(aminomethyl)-4-(trifluoromethyl)benzonitrile (3): To a solution of 3-((1,3-dioxoisoindolin- 2-yl)methyl)-4-(trifluoromethyl)benzonitrile (1 g, 3.03 mmol) in EtOH (20 mL) was added NH2NH2.H2O (3.79 g, 60.56 mmol, 80% purity) at 20 °C, then the mixture was stirred at 20 °C for 12 h.6 M hydrochloric acid was added until pH = 1, and the reaction mixture was stirred at 20 °C for 2 h. After the completion of the reaction, the mixture was filtered and the filtrate was concentrated under reduced pressure. The residue was poured into water (50 mL), adjusting pH to 11 with 6 M NaOH aqueous solution (20 mL). Then the mixture was extracted with EtOAc (3 × 50 mL) and organic layer was washed with brine (200 mL), dried over Na2SO4, filtered and concentrated under reduced pressure to give 3- (aminomethyl)-4-(trifluoromethyl)benzonitrile (3) (600 mg, 99.0%).1H NMR (400 MHz, CDCl3): δ = 8.09 (s, 1H), 7.72-7.80 (m, 1H), 7.62-7.71 (m, 1H), 4.12 ppm (s, 2H). [0757] tert-butyl (5-cyano-2-(trifluoromethyl)benzyl)carbamate (4): To a solution of 3- (aminomethyl)-4-(trifluoromethyl)benzonitrile (0.3 g, 1.50 mmol) and NaOH (180 mg, 4.50 mmol) in dioxane (6 mL) and H2O (3 mL) was added Boc2O (654 mg, 3.00 mmol) at 20 °C, then the mixture was stirred at 20 °C for 2 h. The reaction mixture was quenched with H2O (10 mL) and extracted with EtOAc (3 × 10 mL). The combined organic layers were washed with brine (10 mL), dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure to give a residue. The residue was purified by silica gel column chromatography (PE:EtOAc = 1:0 to 0:1) to give tert-butyl (5-cyano-2- (trifluoromethyl)benzyl)carbamate (4) (300 mg, 66.6%).1H NMR (400 MHz, CDCl3): δ = 7.89 (s, 1H), 7.75 (s, 1H), 7.65-7.71 (m, 1H), 4.88-5.19 (m, 1H), 4.54 (br d, J = 6.0 Hz, 2H), 1.48 ppm (s, 9H). [0758] tert-butyl (5-(6-methyl-1,2,4,5-tetrazin-3-yl)-2-(trifluoromethyl)benzyl)carbamate (5): To a solution of tert-butyl (5-cyano-2-(trifluoromethyl)benzyl)carbamate (500 mg, 1.67 mmol), MeCN (273 mg, 6.66 mmol) and 3-sulfanylpropanoic acid (177 mg, 1.67 mmol) in EtOH (3 mL) was added N2H4.H2O (1.33 g, 26.64 mmol) at 25oC and the mixture was stirred at 65oC for 16 h under N2. Then the mixture was cooled to r.t and added with a solution of NaNO2 (345 mg, 5.00 mmol) in H2O (2.5 mL) dropwise at 25 °C. The mixture was stirred at 25oC for 3 h. The mixture was cooled to room temperature and adjusted to pH = 3 with 1 M aqueous hydrochloric acid. The mixture was extracted with DCM (20 mL). The organic phase was dried over anhydrous sodium sulfate and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (PE:EtOAc = 20:1 to 1:1) to give tert-butyl (5-(6-methyl-1,2,4,5-tetrazin-3-yl)-2-(trifluoromethyl)benzyl)carbamate (5) (140 mg, 22.8%).1H NMR (400 MHz, CDCl3): δ = 8.78 (s, 1H), 8.60 (d, J = 8.0 Hz, 1H), 7.88 (d, J = 8.0 Hz, 1H), 5.08 (s, 1H), 4.67 (s, 1H), 3.15 (s, 3H), 1.49 (s, 9H). [0759] (5-(6-methyl-1,2,4,5-tetrazin-3-yl)-2-(trifluoromethyl)phenyl)methanamine (Target 15): To a solution of tert-butyl N-[[5-(6-methyl-1,2,4,5-tetrazin-3-yl)-2- (trifluoromethyl)phenyl]methyl]carbamate (5) (140 mg, 0.38 mmol) in MeOH (2 mL) was added HCl/MeOH (2 mL, 4 M) at 20 °C under N2. The mixture was stirred at 20 °C for 2 h. The reaction mixture was concentrated under reduced pressure. The crude product was purified by prep-TLC (SiO2, DCM:MeOH = 10:1) to give [5-(6-methyl-1,2,4,5-tetrazin-3-yl)-2- (trifluoromethyl)phenyl]methanamine (18.0 mg, 17.7%). LCMS (ESI+): m/z = 270.0 [M+H]+.1H NMR (400MHz, MeOD): δ 8.88 (s, 1H), 8.60 (d, J = 8.4 Hz, 1H), 7.94 (d, J = 8.4 Hz, 1H), 4.11 (s, 2H), 3.08 (s, 3H)
[0755] 3-((1,3-dioxoisoindolin-2-yl)methyl)-4-(trifluoromethyl)benzonitrile (2): To a solution of 3- (hydroxymethyl)-4-(trifluoromethyl)benzonitrile (4.5 g, 22.38 mmol) in THF (50 mL) was added isoindoline-1,3-dione (3.3 g, 22.38 mmol), PPh3 (11.7 g, 44.76 mmol) and DIAD (6.79 g, 33.57 mmol) at 0 °C under N2. Then the resulting mixture was stirred at 20 °C for 16 h. The reaction mixture was quenched by addition of water (15 mL) and extracted with EtOAc (3 × 10 mL). The combined organic phase was washed with brine (15 mL), dried over sodium sulfate, filtered and the filtrate was concentrated under reduced pressure to give a residue, which was purified by column to give 3-((1,3- dioxoisoindolin-2-yl)methyl)-4-(trifluoromethyl)benzonitrile (2) (5.1 g, 69%).1H NMR (400 MHz, CDCl3) δ = 7.91-8.03 (m, 2H), 7.84 (dd, J = 5.6, 2.8 Hz, 3H), 7.70 (d, J = 8.0 Hz, 1H), 7.46 (s, 1H), 5.13 (s, 2H). [0756] 3-(aminomethyl)-4-(trifluoromethyl)benzonitrile (3): To a solution of 3-((1,3-dioxoisoindolin- 2-yl)methyl)-4-(trifluoromethyl)benzonitrile (1 g, 3.03 mmol) in EtOH (20 mL) was added NH2NH2.H2O (3.79 g, 60.56 mmol, 80% purity) at 20 °C, then the mixture was stirred at 20 °C for 12 h.6 M hydrochloric acid was added until pH = 1, and the reaction mixture was stirred at 20 °C for 2 h. After the completion of the reaction, the mixture was filtered and the filtrate was concentrated under reduced pressure. The residue was poured into water (50 mL), adjusting pH to 11 with 6 M NaOH aqueous solution (20 mL). Then the mixture was extracted with EtOAc (3 × 50 mL) and organic layer was washed with brine (200 mL), dried over Na2SO4, filtered and concentrated under reduced pressure to give 3- (aminomethyl)-4-(trifluoromethyl)benzonitrile (3) (600 mg, 99.0%).1H NMR (400 MHz, CDCl3): δ = 8.09 (s, 1H), 7.72-7.80 (m, 1H), 7.62-7.71 (m, 1H), 4.12 ppm (s, 2H). [0757] tert-butyl (5-cyano-2-(trifluoromethyl)benzyl)carbamate (4): To a solution of 3- (aminomethyl)-4-(trifluoromethyl)benzonitrile (0.3 g, 1.50 mmol) and NaOH (180 mg, 4.50 mmol) in dioxane (6 mL) and H2O (3 mL) was added Boc2O (654 mg, 3.00 mmol) at 20 °C, then the mixture was stirred at 20 °C for 2 h. The reaction mixture was quenched with H2O (10 mL) and extracted with EtOAc (3 × 10 mL). The combined organic layers were washed with brine (10 mL), dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure to give a residue. The residue was purified by silica gel column chromatography (PE:EtOAc = 1:0 to 0:1) to give tert-butyl (5-cyano-2- (trifluoromethyl)benzyl)carbamate (4) (300 mg, 66.6%).1H NMR (400 MHz, CDCl3): δ = 7.89 (s, 1H), 7.75 (s, 1H), 7.65-7.71 (m, 1H), 4.88-5.19 (m, 1H), 4.54 (br d, J = 6.0 Hz, 2H), 1.48 ppm (s, 9H). [0758] tert-butyl (5-(6-methyl-1,2,4,5-tetrazin-3-yl)-2-(trifluoromethyl)benzyl)carbamate (5): To a solution of tert-butyl (5-cyano-2-(trifluoromethyl)benzyl)carbamate (500 mg, 1.67 mmol), MeCN (273 mg, 6.66 mmol) and 3-sulfanylpropanoic acid (177 mg, 1.67 mmol) in EtOH (3 mL) was added N2H4.H2O (1.33 g, 26.64 mmol) at 25oC and the mixture was stirred at 65oC for 16 h under N2. Then the mixture was cooled to r.t and added with a solution of NaNO2 (345 mg, 5.00 mmol) in H2O (2.5 mL) dropwise at 25 °C. The mixture was stirred at 25oC for 3 h. The mixture was cooled to room temperature and adjusted to pH = 3 with 1 M aqueous hydrochloric acid. The mixture was extracted with DCM (20 mL). The organic phase was dried over anhydrous sodium sulfate and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (PE:EtOAc = 20:1 to 1:1) to give tert-butyl (5-(6-methyl-1,2,4,5-tetrazin-3-yl)-2-(trifluoromethyl)benzyl)carbamate (5) (140 mg, 22.8%).1H NMR (400 MHz, CDCl3): δ = 8.78 (s, 1H), 8.60 (d, J = 8.0 Hz, 1H), 7.88 (d, J = 8.0 Hz, 1H), 5.08 (s, 1H), 4.67 (s, 1H), 3.15 (s, 3H), 1.49 (s, 9H). [0759] (5-(6-methyl-1,2,4,5-tetrazin-3-yl)-2-(trifluoromethyl)phenyl)methanamine (Target 15): To a solution of tert-butyl N-[[5-(6-methyl-1,2,4,5-tetrazin-3-yl)-2- (trifluoromethyl)phenyl]methyl]carbamate (5) (140 mg, 0.38 mmol) in MeOH (2 mL) was added HCl/MeOH (2 mL, 4 M) at 20 °C under N2. The mixture was stirred at 20 °C for 2 h. The reaction mixture was concentrated under reduced pressure. The crude product was purified by prep-TLC (SiO2, DCM:MeOH = 10:1) to give [5-(6-methyl-1,2,4,5-tetrazin-3-yl)-2- (trifluoromethyl)phenyl]methanamine (18.0 mg, 17.7%). LCMS (ESI+): m/z = 270.0 [M+H]+.1H NMR (400MHz, MeOD): δ 8.88 (s, 1H), 8.60 (d, J = 8.4 Hz, 1H), 7.94 (d, J = 8.4 Hz, 1H), 4.11 (s, 2H), 3.08 (s, 3H)

Example 40: 1-(2-(6-(4-(aminomethyl)phenyl)-1,2,4,5-tetrazin-3-yl)ethyl)-3-methylurea (Target 6) [0760] tert-butyl (2-(6-(4-iodophenyl)-1,2,4,5-tetrazin-3-yl)ethyl)carbamate: To a mixture of 4- iodobenzonitrile (15 g, 65.50 mmol) in EtOH (140 mL) was added tert-butyl N-(2-cyanoethyl)carbamate (44.59 g, 261.99 mmol), 3-sulfanylpropanoic acid (6.95 g, 65.50 mmol) and NH2NH2.H2O (59.02 g, 1.18 mol) at 0 °C under N2. The mixture was stirred at 45 °C for 16 h. Then the mixture was cooled to 20 °C and added with a solution of NaNO2 in H2O (40 mL) dropwise at 20 °C. The mixture was stirred at 20 °C for 1 h. Under ice-cooling, the pH was adjusted to 3 with 1 M aqueous hydrochloride and then extracted with DCM (3 × 50 mL). The organic phase was dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (PE:EtOAc = 10:1 to 1:1) to give tert-butyl (2-(6-(4-iodophenyl)-1,2,4,5-tetrazin-3-yl)ethyl)carbamate (14 g, 50%). LCMS: m/z = 372.0 [M-BuOH+H]+. [0761] 2-(6-(4-iodophenyl)-1,2,4,5-tetrazin-3-yl)ethan-1-amine: A mixture of tert-butyl (2-(6-(4- iodophenyl)-1,2,4,5-tetrazin-3-yl)ethyl)carbamate (6.5 g, 5.85 mmol) in HCl/EtOAc (4M, 100 mL) was stirred at 25 °C for 1 h. The reaction was concentrated under reduced pressure to give 2-(6-(4- iodophenyl)-1,2,4,5-tetrazin-3-yl)ethan-1-amine; hydrochloride (5.8 g, 80%). LCMS: m/z = 328.2 [M+H]+. [0762] 1-(2-(6-(4-iodophenyl)-1,2,4,5-tetrazin-3-yl)ethyl)-3-methylurea: To a mixture of CDI (3.35 g, 20.63 mmol) in DCM (45 mL) was added TEA (4.18 g, 41.27 mmol) and 2-(6-(4-iodophenyl)-1,2,4,5- tetrazin-3-yl)ethan-1-amine (5.8 g, 13.76 mmol) at -40 °C under N2. The mixture was stirred at -40 °C for 1 h. Then MeNH2 (2 M, 17 mL) and TEA (4.17 g, 41.24 mmol) were added at 0 °C under N2. The mixture was stirred at 25 °C for 12 h. The reaction was concentrated under reduced pressure to give a residue. The residue was triturated with DCM. The resulting solid was collected by filtration, washed with DCM (40 mL) and dried under reduced pressure to give 1-[2-[6-(4-iodophenyl)-1,2,4,5-tetrazin-3- yl]ethyl]-3-methyl-urea (3 g, 57%). LCMS: m/z = 385.0 [M+H]+.1H NMR (400 MHz, DMSO): δ = 8.24 (d, J = 8.4 Hz, 2 H) 8.06 (d, J = 8.4 Hz, 2 H) 6.09 (br t, J = 5.6 Hz, 1 H) 5.77 (br d, J = 4.8 H–, 1 H) 3.53 - 3.57 (–, 2 H) 3.36 - 3.40 (m, 2 H) 2.45 (d, J = 4.4 Hz, 3 H). [0763] tert-butyl (4-(6-(2-(3-methylureido)ethyl)-1,2,4,5-tetrazin-3-yl)benzyl)carbamate: To a mixture of 1-[2-[6-(4-iodophenyl)-1,2,4,5-tetrazin-3-yl]ethyl]-3-methyl-urea (1 g, 2.60 mmol) and (tert- butoxycarbonylamino)methyl-trifluoro-boron;potassium hydride (926 mg, 3.90 mmol) in 2-METHYL-2- BUTANOL (4 mL) and H2O (1 mL) was added Cs2CO3 (1.70 g, 5.21 mmol) and ditert- butyl(cyclopentyl)phosphane;dichloropalladium;iron (170 mg, 0.26 mmol) at 25 °C under N2. The mixture was stirred at 80 °C for 16 h. The mixture was diluted with H2O (15 mL) and extracted with EtOAc (3 × 10 mL). The combined organics were dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (PE:EtOAc = 5:1 to 0:1) to give tert-butyl (4-(6-(2-(3-methylureido)ethyl)-1,2,4,5-tetrazin-3- yl)benzyl)carbamate (200 mg, 20%). LCMS: m/z = 388.3 [M+H]+.1H NMR (400 MHz, DMSO): δ = 8.44 (br d, J = 8.0 Hz, 2 H), 7.52 (br d, J = 7.6 Hz, 3 H), 6.08 (br t, J = 5.6 Hz, 1 H), 5.78 (br s, 1 H), 4.26 (br d, J = 5.6 Hz– 2 H), 3.54 - 3.58 (m, 2 H), 3.38 (br d, J = 6.4 Hz, 2 H), 2.45 (d, J = 4.4 Hz, 3 H), 1.41 (s, 9 H). [0764] 1-(2-(6-(4-(aminomethyl)phenyl)-1,2,4,5-tetrazin-3-yl)ethyl)-3-methylurea (Target 6): To a mixture of tert-butyl (4-(6-(2-(3-methylureido)ethyl)-1,2,4,5-tetrazin-3-yl)benzyl)carbamate (150 mg, 0.39 mmol) was added HCl/EtOAc (4 M, 3 mL) at 25°C under N2. The mixture was stirred at 25 °C for 1 h. The reaction was concentrated under reduced pressure. The crude product was purified by prep-HPLC with the following conditions: column: Phenomenex C18100 × 30 mm × 5 μm; mobile phase: A: 10 mM NH4HCO3 in water, B: MeCN; B% in A: 20%-50%, 10 min to give 1-(2-(6-(4-(aminomethyl)phenyl)- 1,2,4,5-tetrazin-3-yl)ethyl)-3-methylurea (63 mg, 29%). LCMS: m/z = 288.2 [M+H]+.1H NMR (400 MHz, DMSO): δ = 8.52 (d, J = 8.0 Hz, 2 H), 8.45 (br s, 2 H), 7.77 (d, J = 8.2 Hz, 2 H), 6.14 (br t, J = 5.6 Hz– 1 H), 5.76 - 5.83 (m, 1 H), 4.18 (br s, 2 H), 3.56 (q, J = 6.4 Hz– 2 H), 3.38 - 3.41 (m, 2 H), 2.44 (d, J = 4.4 Hz, 3 H).
[0760] tert-butyl (2-(6-(4-iodophenyl)-1,2,4,5-tetrazin-3-yl)ethyl)carbamate: To a mixture of 4- iodobenzonitrile (15 g, 65.50 mmol) in EtOH (140 mL) was added tert-butyl N-(2-cyanoethyl)carbamate (44.59 g, 261.99 mmol), 3-sulfanylpropanoic acid (6.95 g, 65.50 mmol) and NH2NH2.H2O (59.02 g, 1.18 mol) at 0 °C under N2. The mixture was stirred at 45 °C for 16 h. Then the mixture was cooled to 20 °C and added with a solution of NaNO2 in H2O (40 mL) dropwise at 20 °C. The mixture was stirred at 20 °C for 1 h. Under ice-cooling, the pH was adjusted to 3 with 1 M aqueous hydrochloride and then extracted with DCM (3 × 50 mL). The organic phase was dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (PE:EtOAc = 10:1 to 1:1) to give tert-butyl (2-(6-(4-iodophenyl)-1,2,4,5-tetrazin-3-yl)ethyl)carbamate (14 g, 50%). LCMS: m/z = 372.0 [M-BuOH+H]+. [0761] 2-(6-(4-iodophenyl)-1,2,4,5-tetrazin-3-yl)ethan-1-amine: A mixture of tert-butyl (2-(6-(4- iodophenyl)-1,2,4,5-tetrazin-3-yl)ethyl)carbamate (6.5 g, 5.85 mmol) in HCl/EtOAc (4M, 100 mL) was stirred at 25 °C for 1 h. The reaction was concentrated under reduced pressure to give 2-(6-(4- iodophenyl)-1,2,4,5-tetrazin-3-yl)ethan-1-amine; hydrochloride (5.8 g, 80%). LCMS: m/z = 328.2 [M+H]+. [0762] 1-(2-(6-(4-iodophenyl)-1,2,4,5-tetrazin-3-yl)ethyl)-3-methylurea: To a mixture of CDI (3.35 g, 20.63 mmol) in DCM (45 mL) was added TEA (4.18 g, 41.27 mmol) and 2-(6-(4-iodophenyl)-1,2,4,5- tetrazin-3-yl)ethan-1-amine (5.8 g, 13.76 mmol) at -40 °C under N2. The mixture was stirred at -40 °C for 1 h. Then MeNH2 (2 M, 17 mL) and TEA (4.17 g, 41.24 mmol) were added at 0 °C under N2. The mixture was stirred at 25 °C for 12 h. The reaction was concentrated under reduced pressure to give a residue. The residue was triturated with DCM. The resulting solid was collected by filtration, washed with DCM (40 mL) and dried under reduced pressure to give 1-[2-[6-(4-iodophenyl)-1,2,4,5-tetrazin-3- yl]ethyl]-3-methyl-urea (3 g, 57%). LCMS: m/z = 385.0 [M+H]+.1H NMR (400 MHz, DMSO): δ = 8.24 (d, J = 8.4 Hz, 2 H) 8.06 (d, J = 8.4 Hz, 2 H) 6.09 (br t, J = 5.6 Hz, 1 H) 5.77 (br d, J = 4.8 H–, 1 H) 3.53 - 3.57 (–, 2 H) 3.36 - 3.40 (m, 2 H) 2.45 (d, J = 4.4 Hz, 3 H). [0763] tert-butyl (4-(6-(2-(3-methylureido)ethyl)-1,2,4,5-tetrazin-3-yl)benzyl)carbamate: To a mixture of 1-[2-[6-(4-iodophenyl)-1,2,4,5-tetrazin-3-yl]ethyl]-3-methyl-urea (1 g, 2.60 mmol) and (tert- butoxycarbonylamino)methyl-trifluoro-boron;potassium hydride (926 mg, 3.90 mmol) in 2-METHYL-2- BUTANOL (4 mL) and H2O (1 mL) was added Cs2CO3 (1.70 g, 5.21 mmol) and ditert- butyl(cyclopentyl)phosphane;dichloropalladium;iron (170 mg, 0.26 mmol) at 25 °C under N2. The mixture was stirred at 80 °C for 16 h. The mixture was diluted with H2O (15 mL) and extracted with EtOAc (3 × 10 mL). The combined organics were dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (PE:EtOAc = 5:1 to 0:1) to give tert-butyl (4-(6-(2-(3-methylureido)ethyl)-1,2,4,5-tetrazin-3- yl)benzyl)carbamate (200 mg, 20%). LCMS: m/z = 388.3 [M+H]+.1H NMR (400 MHz, DMSO): δ = 8.44 (br d, J = 8.0 Hz, 2 H), 7.52 (br d, J = 7.6 Hz, 3 H), 6.08 (br t, J = 5.6 Hz, 1 H), 5.78 (br s, 1 H), 4.26 (br d, J = 5.6 Hz– 2 H), 3.54 - 3.58 (m, 2 H), 3.38 (br d, J = 6.4 Hz, 2 H), 2.45 (d, J = 4.4 Hz, 3 H), 1.41 (s, 9 H). [0764] 1-(2-(6-(4-(aminomethyl)phenyl)-1,2,4,5-tetrazin-3-yl)ethyl)-3-methylurea (Target 6): To a mixture of tert-butyl (4-(6-(2-(3-methylureido)ethyl)-1,2,4,5-tetrazin-3-yl)benzyl)carbamate (150 mg, 0.39 mmol) was added HCl/EtOAc (4 M, 3 mL) at 25°C under N2. The mixture was stirred at 25 °C for 1 h. The reaction was concentrated under reduced pressure. The crude product was purified by prep-HPLC with the following conditions: column: Phenomenex C18100 × 30 mm × 5 μm; mobile phase: A: 10 mM NH4HCO3 in water, B: MeCN; B% in A: 20%-50%, 10 min to give 1-(2-(6-(4-(aminomethyl)phenyl)- 1,2,4,5-tetrazin-3-yl)ethyl)-3-methylurea (63 mg, 29%). LCMS: m/z = 288.2 [M+H]+.1H NMR (400 MHz, DMSO): δ = 8.52 (d, J = 8.0 Hz, 2 H), 8.45 (br s, 2 H), 7.77 (d, J = 8.2 Hz, 2 H), 6.14 (br t, J = 5.6 Hz– 1 H), 5.76 - 5.83 (m, 1 H), 4.18 (br s, 2 H), 3.56 (q, J = 6.4 Hz– 2 H), 3.38 - 3.41 (m, 2 H), 2.44 (d, J = 4.4 Hz, 3 H).

Example 41: 4-((S)-2-((S)-2-acetamido-3-methylbutanamido)-5-ureidopentanamido)benzyl-6- methyl-3-phenyl-1,2,4,5-tetrazine-1(4H)-carboxylate (Target 1b) [0765] 3-methyl-6-phenyl-1,4-dihydro-1,2,4,5-tetrazine (2): To a solution of benzonitrile (10 g, 96.97 mmol), ACN (31.85 g, 775.79 mmol), 3-mercaptopropanoic acid (10.29 g, 96.97 mmol) in EtOH (100 mL) was added dropwise NH2NH2.H2O (79.26 g, 1.55 mol) at 0 °C under N2. The mixture was stirred for 16 h at 40 °C. The mixture was quenched by water (100 mL), adjusted to pH = 4 by addition of HCl (1 M), extracted with EtOAc (3 × 100 mL). The combined organic layers were washed with brine (100 mL), dried over Na2SO4, filtered and concentrated under reduced pressure. The mixture was slurried with MTBE:EtOAc (10:1, 100 mL), filtered and the solid was desired. The solid was purified by p-HPLC (FA) under the following condition: column: Phenomenex luna C18 (250 × 70 mm, 15 um); mobile phase: [water(FA)-ACN]; B%: 8%-35%, 22 min to give 3-methyl-6-phenyl-1,4-dihydro-1,2,4,5-tetrazine (10 g, 29%). LCMS, m/z = 175.1 [M+H]+.1H NMR (400MHz, DMSO): δ 8.50 (br s, 1H), 8.29 (br s, 1H), 7.74 (dd, J = 1.6, 7.6 Hz, 2H), 7.47-7.35 (m, 3H), 1.78 (s, 3H). [0766] 4-nitrophenyl 6-methyl-3-phenyl-1,2,4,5-tetrazine-1(4H)-carboxylate (4): To a solution of 3- methyl-6-phenyl-1,4-dihydro-1,2,4,5-tetrazine (3 g, 17.22 mmol) in DCM (30 mL) was added DIEA (6.68 g, 51.66 mmol) and then (4-nitrophenyl) carbonochloridate (3.64 g, 18.08 mmol) at 0 °C under N2. The mixture was stirred for 2 h at 25 °C. The mixture was poured into water (45 mL), extracted with DCM (3 × 15 mL). The combined organic layers were washed with brine (15 mL), dried over Na2SO4, filtered and concentrated under reduced pressure. The crude was purified by silica gel column chromatography (PE:EtOAc = 1:0 to 0:1) to give 4-nitrophenyl 6-methyl-3-phenyl-1,2,4,5-tetrazine- 1(4H)-carboxylate (1 g, 17%). LCMS, m/z = 340.0 [M+H]+.1H NMR (400MHz, CDCl3): δ 8.36-8.28 (m, 2H), 7.76 (d, J = 7.6 Hz, 2H), 7.56-7.42 (m, 5H), 6.30-6.02 (m, 2H), 2.44 (s, 3H). [0767] 4-((S)-2-((S)-2-amino-3-methylbutanamido)-5-ureidopentanamido)benzyl-6-methyl-3- phenyl-1,2,4,5-tetrazine-1(4H)-carboxylate (6): To a solution of 4-nitrophenyl 6-methyl-3-phenyl- 1,2,4,5-tetrazine-1(4H)-carboxylate (705 mg, 1.66 mmol) and (9H-fluoren-9-yl)methyl ((S)-1-(((S)-1-((4- (hydroxymethyl)phenyl)amino)-1-oxo-5-ureidopentan-2-yl)amino)-3-methyl-1-oxobutan-2-yl)carbamate (1 g, 1.66 mmol) in DMF (10 mL) was added DIEA (644 mg, 4.99 mmol) at 25 °C under N2. The mixture was heated to 80 °C and stirred for 2 h. The crude was purified by p-HPLC (neutral) under the following condition: column: Phenomenex C18250 × 50 mm × 10 um; mobile phase: [watI NH4HCO3)- ACN]; B%: 14%-44%, 8 min to give 4-((S)-2-((S)-2-amino-3-methylbutanamido)-5- ureidopentanamido)benzyl-6-methyl-3-phenyl-1,2,4,5-tetrazine-1(4H)-carboxylate (240 mg, 25%). LCMS, m/z = 580.2 [M+H]+.1H NMR (400MHz, DMSO): δ 9.97 (s, 1H), 9.84 (s, 1H), 8.42 (d, J = 7.6 Hz, 1H), 7.88-7.79 (m, 2H), 7.60-7.47 (m, 5H), 7.23 (d, J = 8.4 Hz, 2H), 7.14 (d, J = 8.4 Hz, 1H), 5.98 (br t, J = 6.0 Hz, 1H), 5.47-5.35 (m, 2H), 5.12-5.04 (m, 1H), 4.43 (d, J = 5.6 Hz, 3H), 4.26 (dd, J = 5.6, 8.8 Hz, 1H), 3.07-2.94 (m, 2H), 2.18 (s, 3H), 2.14-2.04 (m, 1H), 1.75-1.55 (m, 2H), 1.51-1.30 (m, 2H), 0.92 (d, J = 6.8 Hz, 3H), 0.85 (d, J = 6.8 Hz, 3H). [0768] 4-((S)-2-((S)-2-acetamido-3-methylbutanamido)-5-ureidopentanamido)benzyl-6-methyl-3- phenyl-1,2,4,5-tetrazine-1(4H)-carboxylate (Target 1b): To a solution of 4-((S)-2-((S)-2-amino-3- methylbutanamido)-5-ureidopentanamido)benzyl 6-methyl-3-phenyl-1,2,4,5-tetrazine-1(4H)-carboxylate (100 mg, 0.17 mmol) and DIEA (45 mg, 0.35 mmol) in THF (0.5 mL) was added DMAP (4 mg, 0.03 mmol) and then Ac2O (23 mg, 0.22 mmol) at 0 °C under N2. The mixture was stirred for 2 h at 25 °C. The mixture was concentrated under reduced pressure. The crude was purified by p-HPLC (neutral) under the following condition: column: Waters xbridge 150 × 25 mm × 10 um; mobile phase: [Ier( NH4HCO3)-ACN]; B%: 24%-54%, 8 min to give 4-((S)-2-((S)-2-acetamido-3-methylbutanamido)-5- ureidopentanamido)benzyl 6-methyl-3-phenyl-1,2,4,5-tetrazine-1(4H)-carboxylate (58.9 mg, 55%). LCMS, m/z = 622.2 [M+H]+.1H NMR (400MHz, DMSO): δ 10.09 (s, 1H), 9.85 (s, 1H), 8.44 (d, J = 7.2 Hz, 1H), 7.87-7.77 (m, 2H), 7.64-7.47 (m, 5H), 7.30 (d, J = 8.4 Hz, 2H), 7.14 (d, J = 8.4 Hz, 1H), 5.99 (br t, J = 5.6 Hz, 1H), 5.41 (s, 2H), 5.00 (s, 2H), 4.47-4.36 (m, 1H), 4.27 (dd, J = 5.6, 8.7 Hz, 1H), 3.10- 2.88 (m, 2H), 2.18 (s, 3H), 2.14-2.06 (m, 1H), 2.04 (s, 3H), 1.77-1.57 (m, 2H), 1.52-1.32 (m, 2H), 0.92 (d, J = 6.8 Hz, 3H), 0.85 (d, J = 6.8 Hz, 3H). Example 42: LCMS analysis of drug release by tetrazines [0769] A solution of each tetrazine (20 µM final concentration) in PBS is mixed with TCO-MMAE (10 µM final concentration). The solution is mixed thoroughly and an aliquot removed at the appropriate time for analysis by LCMS. The results are reported in the table below.
[0765] 3-methyl-6-phenyl-1,4-dihydro-1,2,4,5-tetrazine (2): To a solution of benzonitrile (10 g, 96.97 mmol), ACN (31.85 g, 775.79 mmol), 3-mercaptopropanoic acid (10.29 g, 96.97 mmol) in EtOH (100 mL) was added dropwise NH2NH2.H2O (79.26 g, 1.55 mol) at 0 °C under N2. The mixture was stirred for 16 h at 40 °C. The mixture was quenched by water (100 mL), adjusted to pH = 4 by addition of HCl (1 M), extracted with EtOAc (3 × 100 mL). The combined organic layers were washed with brine (100 mL), dried over Na2SO4, filtered and concentrated under reduced pressure. The mixture was slurried with MTBE:EtOAc (10:1, 100 mL), filtered and the solid was desired. The solid was purified by p-HPLC (FA) under the following condition: column: Phenomenex luna C18 (250 × 70 mm, 15 um); mobile phase: [water(FA)-ACN]; B%: 8%-35%, 22 min to give 3-methyl-6-phenyl-1,4-dihydro-1,2,4,5-tetrazine (10 g, 29%). LCMS, m/z = 175.1 [M+H]+.1H NMR (400MHz, DMSO): δ 8.50 (br s, 1H), 8.29 (br s, 1H), 7.74 (dd, J = 1.6, 7.6 Hz, 2H), 7.47-7.35 (m, 3H), 1.78 (s, 3H). [0766] 4-nitrophenyl 6-methyl-3-phenyl-1,2,4,5-tetrazine-1(4H)-carboxylate (4): To a solution of 3- methyl-6-phenyl-1,4-dihydro-1,2,4,5-tetrazine (3 g, 17.22 mmol) in DCM (30 mL) was added DIEA (6.68 g, 51.66 mmol) and then (4-nitrophenyl) carbonochloridate (3.64 g, 18.08 mmol) at 0 °C under N2. The mixture was stirred for 2 h at 25 °C. The mixture was poured into water (45 mL), extracted with DCM (3 × 15 mL). The combined organic layers were washed with brine (15 mL), dried over Na2SO4, filtered and concentrated under reduced pressure. The crude was purified by silica gel column chromatography (PE:EtOAc = 1:0 to 0:1) to give 4-nitrophenyl 6-methyl-3-phenyl-1,2,4,5-tetrazine- 1(4H)-carboxylate (1 g, 17%). LCMS, m/z = 340.0 [M+H]+.1H NMR (400MHz, CDCl3): δ 8.36-8.28 (m, 2H), 7.76 (d, J = 7.6 Hz, 2H), 7.56-7.42 (m, 5H), 6.30-6.02 (m, 2H), 2.44 (s, 3H). [0767] 4-((S)-2-((S)-2-amino-3-methylbutanamido)-5-ureidopentanamido)benzyl-6-methyl-3- phenyl-1,2,4,5-tetrazine-1(4H)-carboxylate (6): To a solution of 4-nitrophenyl 6-methyl-3-phenyl- 1,2,4,5-tetrazine-1(4H)-carboxylate (705 mg, 1.66 mmol) and (9H-fluoren-9-yl)methyl ((S)-1-(((S)-1-((4- (hydroxymethyl)phenyl)amino)-1-oxo-5-ureidopentan-2-yl)amino)-3-methyl-1-oxobutan-2-yl)carbamate (1 g, 1.66 mmol) in DMF (10 mL) was added DIEA (644 mg, 4.99 mmol) at 25 °C under N2. The mixture was heated to 80 °C and stirred for 2 h. The crude was purified by p-HPLC (neutral) under the following condition: column: Phenomenex C18250 × 50 mm × 10 um; mobile phase: [watI NH4HCO3)- ACN]; B%: 14%-44%, 8 min to give 4-((S)-2-((S)-2-amino-3-methylbutanamido)-5- ureidopentanamido)benzyl-6-methyl-3-phenyl-1,2,4,5-tetrazine-1(4H)-carboxylate (240 mg, 25%). LCMS, m/z = 580.2 [M+H]+.1H NMR (400MHz, DMSO): δ 9.97 (s, 1H), 9.84 (s, 1H), 8.42 (d, J = 7.6 Hz, 1H), 7.88-7.79 (m, 2H), 7.60-7.47 (m, 5H), 7.23 (d, J = 8.4 Hz, 2H), 7.14 (d, J = 8.4 Hz, 1H), 5.98 (br t, J = 6.0 Hz, 1H), 5.47-5.35 (m, 2H), 5.12-5.04 (m, 1H), 4.43 (d, J = 5.6 Hz, 3H), 4.26 (dd, J = 5.6, 8.8 Hz, 1H), 3.07-2.94 (m, 2H), 2.18 (s, 3H), 2.14-2.04 (m, 1H), 1.75-1.55 (m, 2H), 1.51-1.30 (m, 2H), 0.92 (d, J = 6.8 Hz, 3H), 0.85 (d, J = 6.8 Hz, 3H). [0768] 4-((S)-2-((S)-2-acetamido-3-methylbutanamido)-5-ureidopentanamido)benzyl-6-methyl-3- phenyl-1,2,4,5-tetrazine-1(4H)-carboxylate (Target 1b): To a solution of 4-((S)-2-((S)-2-amino-3- methylbutanamido)-5-ureidopentanamido)benzyl 6-methyl-3-phenyl-1,2,4,5-tetrazine-1(4H)-carboxylate (100 mg, 0.17 mmol) and DIEA (45 mg, 0.35 mmol) in THF (0.5 mL) was added DMAP (4 mg, 0.03 mmol) and then Ac2O (23 mg, 0.22 mmol) at 0 °C under N2. The mixture was stirred for 2 h at 25 °C. The mixture was concentrated under reduced pressure. The crude was purified by p-HPLC (neutral) under the following condition: column: Waters xbridge 150 × 25 mm × 10 um; mobile phase: [Ier( NH4HCO3)-ACN]; B%: 24%-54%, 8 min to give 4-((S)-2-((S)-2-acetamido-3-methylbutanamido)-5- ureidopentanamido)benzyl 6-methyl-3-phenyl-1,2,4,5-tetrazine-1(4H)-carboxylate (58.9 mg, 55%). LCMS, m/z = 622.2 [M+H]+.1H NMR (400MHz, DMSO): δ 10.09 (s, 1H), 9.85 (s, 1H), 8.44 (d, J = 7.2 Hz, 1H), 7.87-7.77 (m, 2H), 7.64-7.47 (m, 5H), 7.30 (d, J = 8.4 Hz, 2H), 7.14 (d, J = 8.4 Hz, 1H), 5.99 (br t, J = 5.6 Hz, 1H), 5.41 (s, 2H), 5.00 (s, 2H), 4.47-4.36 (m, 1H), 4.27 (dd, J = 5.6, 8.7 Hz, 1H), 3.10- 2.88 (m, 2H), 2.18 (s, 3H), 2.14-2.06 (m, 1H), 2.04 (s, 3H), 1.77-1.57 (m, 2H), 1.52-1.32 (m, 2H), 0.92 (d, J = 6.8 Hz, 3H), 0.85 (d, J = 6.8 Hz, 3H). Example 42: LCMS analysis of drug release by tetrazines [0769] A solution of each tetrazine (20 µM final concentration) in PBS is mixed with TCO-MMAE (10 µM final concentration). The solution is mixed thoroughly and an aliquot removed at the appropriate time for analysis by LCMS. The results are reported in the table below. [0770] Based on the above data, it is contemplated that the drug release profile can be modified, enhanced, or attenuated by adjusting the structure of the bicyclic tetrazine or dihydrotetrazine portion targeting moiety. Example 43: Tetrazine-Antigen-Binding Protein Targeting Moiety and Antibody Fragment Moieties [0771] Antigen-binding proteins are engineered proteins that can bind an antigen. The proteins are approximately 66 amino acids in length with a molecular weight of 7 kDa. The protein can be expressed in E. coli and a cysteine residue can be included at the N- or C-terminus of the sequence that can be conjugated with a cysteine-reactive group and can be purchased from commercial sources (e.g., Nanofitins® from Affilogic). [0772] An antigen-binding protein targeting HER2 with a C-terminal cysteine is expressed in E. coli and purified to homogeneity. The protein is treated with a reducing agent, such as TCEP, at ambient temperature or on ice, followed by buffer exchange into fresh buffer. The protein is then treated with maleimide-PEG-tetrazine (n = 3). After the reaction is complete, the protein is exchanged into fresh buffer to remove the excess reagent to yield conjugate (Ab-Tz). The conjugate is analyzed by SDS- PAGE, analytical HPLC, mass spectrometry to confirm the expected properties. The conjugate can also be treated with a trans-cyclooctene-functionalized fluorophore to confirm the reactivity of the tetrazine. [0773] An non-binding control protein with a C-terminal cysteine is expressed in E. coli and purified to homogeneity. The protein is treated with a reducing agent, such as TCEP, at ambient temperature or on ice, followed by buffer exchange into fresh buffer. The protein is then treated with maleimide-PEG- tetrazine (n = 3). After the reaction is complete, the protein is exchanged into fresh buffer to remove the excess reagent to yield conjugate (Ab-Tz). The conjugate is analyzed by SDS-PAGE, analytical HPLC, mass spectrometry to confirm the expected properties. The conjugate can also be treated with a trans- cyclooctene-functionalized fluorophore to confirm the reactivity of the tetrazine. [0774] Further targeting moieties can be prepared, as described above, using the seques shown below. 32) a) Synthesis of Fab of L19 -binding to FN-1 (Gene ID 2335) [0775] Vector construction: Coding sequences (listed below) are synthesized and subcloned into expression vector. Constructed plasmids are transformed to E.coli for propagation. NucleoBond Xtra Maxi Plus EF kit are used for large scale plasmid generation. Purified plasmids are checked by agarose gel and confirmed by sequencing. [0776] L19-Fab HC sequence: [0777] EVQLLESGGGLVQPGGSLRLSCAASGFTFSSFSMSWVRQAPGKGLEWVSSISGSSGTTYY ADSVKGRFTISRDNSKNTLYLQMNSLRAEDTAVYYCAKPFPYFDYWGQGTLVTVSSASTKGPS VFPLAPSSKSTSGGTAALGCLVKDYFPEPVTVSWNSGALTSGVHTFPAVLQSSGLYSLSSVVTVP SSSLGTQTYICNVNHKPSNTKVDKKVEPKSSDKTHT (SEQ ID NO.5). [0778] L19-Fab LC sequence: [0779] EIVLTQSPGTLSLSPGERATLSCRASQSVSSSFLAWYQQKPGQAPRLLIYYASSRATGIPD RFSGSGSGTDFTLTISRLEPEDFAVYYCQQTGRIPPTFGQGTKVEIKRTVAAPSVFIFPPSDEQLKS GTASVVCLLNNFYPREAKVQWKVDNALQSGNSQESVTEQDSKDSTYSLSSTLTLSKADYEKHK VYACEVTHQGLSSPCTKSFNRGEC (SEQ ID NO.6). [0780] Protein expression: The constructs containing heavy chain and light chain of the Fab are co- transfected into HEK293 cells with PEI. The culture medium is harvested at 6-7 days post transfection. [0781] Protein purification: Conditional medium expressing target Fab is harvested by centrifugation and filtration, and can then be loaded onto KappaSelect affinity column (Mabselect Prism). The loading buffer is 25 mM Tris containing 150 mM NaCl, pH 8.0; the wash buffer is 25 mM Tris buffer containing 150 mM NaCl, 0.2% Triton X-100/114, pH 8.0; the elution buffer is 100 mM Sodium-citrate buffer containing 150 mM NaCl, pH 2.5. The collected solution is neutralized with 1M arginine, 400 mM succinic acid buffer, pH 9.0. The affinity purified protein is further purified by gel filtration with Superdex S-200 column chromatography. Purified Fab is analyzed by SDS-PAGE, SEC-HPLC and endotoxin measurement. [0782] Conjugate preparation: 120 mg Fab protein is dialyzed against PBS, pH 7.4 overnight with one buffer exchange at about 4 hours from the start.10 mM Me-Tet-PEG9-NHS is prepared in DMSO. The two components are reacted at 3:1 drug to protein molar ratio at 25oC for 2 hours; the reaction mixture is dialyzed against PBS, pH 7.4 to remove excess Me-Tet-PEG9-NHS compound from the protein component. b) Synth–sis of Fab of F16 - binding to TNC (Gene ID 3371) [0783] Vector construction: Coding sequences (listed below) are synthesized and subcloned into expression vector. Constructed plasmids are transformed to E.coli for propagation. NucleoBond Xtra Maxi Plus EF kit are used for large scale plasmid generation. Purified plasmids are checked by agarose gel and confirmed by sequencing. [0784] F16-Fab HC sequence: [0785] EVQLLESGGGLVQPGGSLRLSCAASGFTFSRYGMSWVRQAPGKGLEWVSAISGSGGSTY YADSVKGRFTISRDNSKNTLYLQMNSLRAEDTAVYYCAKAHNAFDYWGQGTLVTVSSCSTKGP SVFPLAPSSKSTSGGTAALGCLVKDYFPEPVTVSWNSGALTSGVHTFPAVLQSSGLYSLSSVVTV PSSSLGTQTYICNVNHKPSNTKVDKKVEPKSSDKTHT (SEQ ID NO.7). [0786] F16-Fab LC sequence: [0787] SSELTQDPAVSVALGQTVRITCQGDSLRSYYASWYQQKPGQAPVLVIYGKNNRPSGIPD RFSGSSSGNTASLTITGAQAEDEADYYCNSSVYTMPPVVFGGGTKLTVLGQPKAAPSVTLFPPSS EELQANKATLVCLISDFYPGAVTVAWKADSSPVKAGVETTTPSKQSNNKYAASSYLSLTPEQW KSHKSYSCQVTHEGSTVEKTVAPTECS (SEQ ID NO.8). [0788] Protein expression: The constructs containing heavy chain and light chain of the Fab are co- transfected into HEK293 cells with PEI. The culture medium is harvested at 6-7 days post transfection. [0789] Protein purification: Conditional medium expressing target Fab is harvested by centrifugation and filtration, and can then be loaded onto KappaSelect affinity column (Mabselect Prism). The loading buffer is 25 mM Tris containing 150 mM NaCl, pH 8.0; the wash buffer is 25 mM Tris buffer containing 150 mM NaCl, 0.2% Triton X-100/114, pH 8.0; the elution buffer is 100 mM Sodium-citrate buffer containing 150 mM NaCl, pH 2.5. The collected solution is neutralized with 1M arginine, 400 mM succinic acid buffer, pH 9.0. The affinity purified protein is further purified by gel filtration with Superdex S-200 column chromatography. Purified Fab is analyzed by SDS-PAGE, SEC-HPLC and endotoxin measurement. [0790] Conjugate preparation: 120 mg Fab protein is dialyzed against PBS, pH 7.4 overnight with one buffer exchange at about 4 hours from the start.10 mM Me-Tet-PEG9-NHS is prepared in DMSO. The two components are reacted at 3:1 drug to protein molar ratio at 25oC for 2 hours; the reaction mixture is dialyzed against PBS, pH 7.4 to remove excess Me-Tet-PEG9-NHS compound from the protein component. Example 44: Efficacy study in NCI-N87 xenograft gastric cancer model [0791] The following example shows timing of the payload dose leads to effective tumor inhibition with a single dose. Compound TM-1 and Isotype Fab-Tz (Iso Ctrl) were prepared according to Example 6. [0792] NCI-N87 (gastric carcinoma) cells were cultured in RPMI-1640 supplemented with 10% FBS, 1% pen-strep and placed in 37 °C incubator. The cells were harvested and counted for tumor inoculation when the cells reached the exponential growth phase. CB-17 SCID (6-8 weeks old) female mice were inoculated subcutaneously at the right flank with 3 x 106 cells in a 200 mL mixture of RPMI-1640 with Matrigel (1:1) for tumor development. Animals were divided into groups of 5 for the efficacy study with mean tumor volume of ~100-150 mm3 prior to dosing. Tumor volumes of NCI-N87 tumors in SCID CB17 mice following treatment with vehicle, Compound TM-1 with Compound B, and Isotype Fab-Tz with Compound B. Animals were dosed with a single dose of either Isotype control (Isotype Fab-Tz) or Compound TM-1 intravenously (IV) at 50 mg/kg then mice were given a single dose of Compound B IV at 30 mg/kg after 4 hours (FIG.1A), 8 hours (FIG.1B), 24 hours (FIG.1C), or 48 hours (FIG.1D) after Iso Ctrl or Compound TM-1. Tumor volume was measured two times weekly in two dimensions using a caliper and the volume was expressed in mm3 using the formula: V = 0.5 a x b2 where a and b are the long and short diameters of the tumor. For tumor growth inhibition (Δ%TGI) was calculated to compare between Iso Ctrl and Compound TM-1 using the formula: TGI (%) = [1-(T/V)] ×100; where T is the mean TV of a treatment group on a given day and V is the mean TV of the vehicle control group on the same day as T, and a two-way ANOVA with Tukey’s correction were performed among the different treatment groups. A p < 0.05 was considered statistically significant. [0793] As shown in FIGs.1A, 1B, 1C, and 1D, the time between doses can be attenuated to maximize specific and minimize non-specific activity of Compound B with Compound TM-1 on NCI-N87 tumors. Shown are means ± SEM. P-values were determined by two-way ANOVA with Dunnet’s correction (* p < 0.05, ** p < 0.01). ANOVA, analysis of variance; SEM, standard error of mean. [0794] As shown in FIG.1A, a portion of the 50 mg/kg of Compound TM-1 is systemically circulating around 4 hours after dose, leading to higher non-specific activity. [0795] As shown in FIG.1D, administering Compound B 48 hours after 50 mg/kg of Compound TM-1 leads to diminished activity. [0796] Accordingly, a single dose of the MMAE prodrug, Compound B, achieves prolonged tumor growth inhibition when administered within about 8 to 24 hours after administering Compound TM-1. Example 45: Plasma and Tumor PK Analysis [0797] NCI-N87 (gastric carcinoma) cells were cultured in RPMI-1640 supplemented with 10% FBS, 1% pen-strep and placed in 37 ºC incubator. The cells were harvested and counted for tumor inoculation when the cells reached the exponential growth phase. CB-17 SCID (6-8 weeks old) female mice were inoculated subcutaneously at the right flank with 5 x 106 cells in a 200 mL mixture of PBS with Matrigel (1:1) for tumor development. Animals were divided into groups of 3 for the PK study with mean tumor volume of ~200-250 mm3 prior to dosing. Animals were dosed with a single dose of Compound TM-1 intravenously (IV) at 50 or 5 mg/kg and 4 hours later dosed with Compound B intravenously (IV) at 10 mg/kg. FIG.2A shows the schedule of dosing and tumor collection. Plasma and tumor tissue samples were collected after 15 minutes of dosing with Compound B. Plasma samples and tumor tissue were flash frozen and stored in -80 ºC until the samples were analyzed using liquid chromatography with tandem mass spectrometry (LC-MS/MS). [0798] FIG.2B shows the concentration of MMAE in tumors 15 minutes after dosing of Compound B. Shown are means ± SEM. P-values were determined by two-way ANOVA with Bonferroni correction. ANOVA, analysis of variance; MMAE, monomethyl auristatin E; SEM, standard error of mean. [0799] As shown in the FIG.2B, Compound TM-1 is converted to active payload, i.e., MMAE, within 15 minutes. Example 46: PK measurement of total amount of Compound TM-1 and reactive tetrazine (Tz) in plasma and tumor samples [0800] The NCI-N87 (gastric carcinoma) cells were cultured in RPMI-1640 supplemented with 10% FBS and 1% pen-strep and placed in 37ºC incubator. The cells were harvested and counted for tumor inoculation when the cells reached the exponential growth phase. CB-17 SCID (6-8 weeks old) female mice were inoculated subcutaneously at the right flank with 5 x 106 cells in a 200 mL mixture of RPMI- 1640 with Matrigel (1:1) for tumor development. Animals were divided into groups of 3 for the PK study with mean tumor volume of ~200-250 mm3 prior to dosing. Animals were dosed with Compound TM-1 IV at 50 mg/kg. Then animals were dosed with TCO-biotin, 15 minutes prior to tumor collection, the animals were perfused with heparin-saline solution to remove excess/unreacted TCO-biotin, then tumors were harvested close to 1, 4, and 24 hrs. The tumors were flash frozen and placed in -80 °C until the samples were analyzed using enzyme-linked immunosorbent assay (ELISA). Tumor samples were tested for total amount of Compound TM-1 and reactive tetrazine. For measuring the total amount of Compound TM-1, wells of 96-well plate were coated with anti-ID antibody against trastuzumab, and the plates were incubated overnight at 4 °C. The plate was washed 3 times and then samples diluted with assay diluent containing 1% BSA and 0.05 Tween-20 in PBS were added to the wells and incubated at room temperature for 2 hours with constant shaking. After washing, mouse anti-human IgG Fab antibody conjugated to horseradish peroxidase (HRP) (detection antibody) was addeI to the wells and incubated for 1 hr. Then the plates were again washed for 3 times, then mixed with detection reagent, and read using SepctraMax i3x. The concentrations from tumor samples were interpolated from the standard curve of Compound TM-1 using known concentration (200 ng/mL -1.563 ng/mL with 2-fold serial dilution). [0801] For measuring reactive tetrazine of Compound TM-1, wells of 96-well plate were coated with anti-ID antibody against trastuzumab, and the plates were incubated overnight at 4ºC. The plates were washed 3X times and then samples diluted with assay diluent containing 1% BSA and 0.05 Tween-20 in PBS were added to the wells and incubated at room temperature for 2 hours with constant shaking. After washing, streptavidin-HRP were added to the wells and incubated for 1 hour. Then the plates were mixed with detection reagent and read using SepctraMax i3x. The concentrations from tumor samples were interpolated from the standard curve of Compound TM-1 using known conce–tration (100 ng/mL - 0.781 ng/mL with 2-fold serial dilution). [0802] FIG.3A shows quantification of total Fab in the tumors by ELISA. FIG.3B shows quantification of activator (tetrazine) in the tumors by ELISA. Shown are means ± SEM. P-values were determined by two-way ANOVA with Bonferroni correction for multiple comparisons. ANOVA, analysis of variance; SEM, standard error of mean. [0803] As shown in FIG.3A and FIG.3B, Compound TM-1 localizes to HER2-positive tumors. Example 47: Compound TM-2 Plasma and Tumor PK Analysis [0804] This study evaluated the optimal dose and time of Compound TM-2 at the tumor site for payload-TCO conjugate dosing. [0805] Experimental Design:
[0770] Based on the above data, it is contemplated that the drug release profile can be modified, enhanced, or attenuated by adjusting the structure of the bicyclic tetrazine or dihydrotetrazine portion targeting moiety. Example 43: Tetrazine-Antigen-Binding Protein Targeting Moiety and Antibody Fragment Moieties [0771] Antigen-binding proteins are engineered proteins that can bind an antigen. The proteins are approximately 66 amino acids in length with a molecular weight of 7 kDa. The protein can be expressed in E. coli and a cysteine residue can be included at the N- or C-terminus of the sequence that can be conjugated with a cysteine-reactive group and can be purchased from commercial sources (e.g., Nanofitins® from Affilogic). [0772] An antigen-binding protein targeting HER2 with a C-terminal cysteine is expressed in E. coli and purified to homogeneity. The protein is treated with a reducing agent, such as TCEP, at ambient temperature or on ice, followed by buffer exchange into fresh buffer. The protein is then treated with maleimide-PEG-tetrazine (n = 3). After the reaction is complete, the protein is exchanged into fresh buffer to remove the excess reagent to yield conjugate (Ab-Tz). The conjugate is analyzed by SDS- PAGE, analytical HPLC, mass spectrometry to confirm the expected properties. The conjugate can also be treated with a trans-cyclooctene-functionalized fluorophore to confirm the reactivity of the tetrazine. [0773] An non-binding control protein with a C-terminal cysteine is expressed in E. coli and purified to homogeneity. The protein is treated with a reducing agent, such as TCEP, at ambient temperature or on ice, followed by buffer exchange into fresh buffer. The protein is then treated with maleimide-PEG- tetrazine (n = 3). After the reaction is complete, the protein is exchanged into fresh buffer to remove the excess reagent to yield conjugate (Ab-Tz). The conjugate is analyzed by SDS-PAGE, analytical HPLC, mass spectrometry to confirm the expected properties. The conjugate can also be treated with a trans- cyclooctene-functionalized fluorophore to confirm the reactivity of the tetrazine. [0774] Further targeting moieties can be prepared, as described above, using the seques shown below. 32) a) Synthesis of Fab of L19 -binding to FN-1 (Gene ID 2335) [0775] Vector construction: Coding sequences (listed below) are synthesized and subcloned into expression vector. Constructed plasmids are transformed to E.coli for propagation. NucleoBond Xtra Maxi Plus EF kit are used for large scale plasmid generation. Purified plasmids are checked by agarose gel and confirmed by sequencing. [0776] L19-Fab HC sequence: [0777] EVQLLESGGGLVQPGGSLRLSCAASGFTFSSFSMSWVRQAPGKGLEWVSSISGSSGTTYY ADSVKGRFTISRDNSKNTLYLQMNSLRAEDTAVYYCAKPFPYFDYWGQGTLVTVSSASTKGPS VFPLAPSSKSTSGGTAALGCLVKDYFPEPVTVSWNSGALTSGVHTFPAVLQSSGLYSLSSVVTVP SSSLGTQTYICNVNHKPSNTKVDKKVEPKSSDKTHT (SEQ ID NO.5). [0778] L19-Fab LC sequence: [0779] EIVLTQSPGTLSLSPGERATLSCRASQSVSSSFLAWYQQKPGQAPRLLIYYASSRATGIPD RFSGSGSGTDFTLTISRLEPEDFAVYYCQQTGRIPPTFGQGTKVEIKRTVAAPSVFIFPPSDEQLKS GTASVVCLLNNFYPREAKVQWKVDNALQSGNSQESVTEQDSKDSTYSLSSTLTLSKADYEKHK VYACEVTHQGLSSPCTKSFNRGEC (SEQ ID NO.6). [0780] Protein expression: The constructs containing heavy chain and light chain of the Fab are co- transfected into HEK293 cells with PEI. The culture medium is harvested at 6-7 days post transfection. [0781] Protein purification: Conditional medium expressing target Fab is harvested by centrifugation and filtration, and can then be loaded onto KappaSelect affinity column (Mabselect Prism). The loading buffer is 25 mM Tris containing 150 mM NaCl, pH 8.0; the wash buffer is 25 mM Tris buffer containing 150 mM NaCl, 0.2% Triton X-100/114, pH 8.0; the elution buffer is 100 mM Sodium-citrate buffer containing 150 mM NaCl, pH 2.5. The collected solution is neutralized with 1M arginine, 400 mM succinic acid buffer, pH 9.0. The affinity purified protein is further purified by gel filtration with Superdex S-200 column chromatography. Purified Fab is analyzed by SDS-PAGE, SEC-HPLC and endotoxin measurement. [0782] Conjugate preparation: 120 mg Fab protein is dialyzed against PBS, pH 7.4 overnight with one buffer exchange at about 4 hours from the start.10 mM Me-Tet-PEG9-NHS is prepared in DMSO. The two components are reacted at 3:1 drug to protein molar ratio at 25oC for 2 hours; the reaction mixture is dialyzed against PBS, pH 7.4 to remove excess Me-Tet-PEG9-NHS compound from the protein component. b) Synth–sis of Fab of F16 - binding to TNC (Gene ID 3371) [0783] Vector construction: Coding sequences (listed below) are synthesized and subcloned into expression vector. Constructed plasmids are transformed to E.coli for propagation. NucleoBond Xtra Maxi Plus EF kit are used for large scale plasmid generation. Purified plasmids are checked by agarose gel and confirmed by sequencing. [0784] F16-Fab HC sequence: [0785] EVQLLESGGGLVQPGGSLRLSCAASGFTFSRYGMSWVRQAPGKGLEWVSAISGSGGSTY YADSVKGRFTISRDNSKNTLYLQMNSLRAEDTAVYYCAKAHNAFDYWGQGTLVTVSSCSTKGP SVFPLAPSSKSTSGGTAALGCLVKDYFPEPVTVSWNSGALTSGVHTFPAVLQSSGLYSLSSVVTV PSSSLGTQTYICNVNHKPSNTKVDKKVEPKSSDKTHT (SEQ ID NO.7). [0786] F16-Fab LC sequence: [0787] SSELTQDPAVSVALGQTVRITCQGDSLRSYYASWYQQKPGQAPVLVIYGKNNRPSGIPD RFSGSSSGNTASLTITGAQAEDEADYYCNSSVYTMPPVVFGGGTKLTVLGQPKAAPSVTLFPPSS EELQANKATLVCLISDFYPGAVTVAWKADSSPVKAGVETTTPSKQSNNKYAASSYLSLTPEQW KSHKSYSCQVTHEGSTVEKTVAPTECS (SEQ ID NO.8). [0788] Protein expression: The constructs containing heavy chain and light chain of the Fab are co- transfected into HEK293 cells with PEI. The culture medium is harvested at 6-7 days post transfection. [0789] Protein purification: Conditional medium expressing target Fab is harvested by centrifugation and filtration, and can then be loaded onto KappaSelect affinity column (Mabselect Prism). The loading buffer is 25 mM Tris containing 150 mM NaCl, pH 8.0; the wash buffer is 25 mM Tris buffer containing 150 mM NaCl, 0.2% Triton X-100/114, pH 8.0; the elution buffer is 100 mM Sodium-citrate buffer containing 150 mM NaCl, pH 2.5. The collected solution is neutralized with 1M arginine, 400 mM succinic acid buffer, pH 9.0. The affinity purified protein is further purified by gel filtration with Superdex S-200 column chromatography. Purified Fab is analyzed by SDS-PAGE, SEC-HPLC and endotoxin measurement. [0790] Conjugate preparation: 120 mg Fab protein is dialyzed against PBS, pH 7.4 overnight with one buffer exchange at about 4 hours from the start.10 mM Me-Tet-PEG9-NHS is prepared in DMSO. The two components are reacted at 3:1 drug to protein molar ratio at 25oC for 2 hours; the reaction mixture is dialyzed against PBS, pH 7.4 to remove excess Me-Tet-PEG9-NHS compound from the protein component. Example 44: Efficacy study in NCI-N87 xenograft gastric cancer model [0791] The following example shows timing of the payload dose leads to effective tumor inhibition with a single dose. Compound TM-1 and Isotype Fab-Tz (Iso Ctrl) were prepared according to Example 6. [0792] NCI-N87 (gastric carcinoma) cells were cultured in RPMI-1640 supplemented with 10% FBS, 1% pen-strep and placed in 37 °C incubator. The cells were harvested and counted for tumor inoculation when the cells reached the exponential growth phase. CB-17 SCID (6-8 weeks old) female mice were inoculated subcutaneously at the right flank with 3 x 106 cells in a 200 mL mixture of RPMI-1640 with Matrigel (1:1) for tumor development. Animals were divided into groups of 5 for the efficacy study with mean tumor volume of ~100-150 mm3 prior to dosing. Tumor volumes of NCI-N87 tumors in SCID CB17 mice following treatment with vehicle, Compound TM-1 with Compound B, and Isotype Fab-Tz with Compound B. Animals were dosed with a single dose of either Isotype control (Isotype Fab-Tz) or Compound TM-1 intravenously (IV) at 50 mg/kg then mice were given a single dose of Compound B IV at 30 mg/kg after 4 hours (FIG.1A), 8 hours (FIG.1B), 24 hours (FIG.1C), or 48 hours (FIG.1D) after Iso Ctrl or Compound TM-1. Tumor volume was measured two times weekly in two dimensions using a caliper and the volume was expressed in mm3 using the formula: V = 0.5 a x b2 where a and b are the long and short diameters of the tumor. For tumor growth inhibition (Δ%TGI) was calculated to compare between Iso Ctrl and Compound TM-1 using the formula: TGI (%) = [1-(T/V)] ×100; where T is the mean TV of a treatment group on a given day and V is the mean TV of the vehicle control group on the same day as T, and a two-way ANOVA with Tukey’s correction were performed among the different treatment groups. A p < 0.05 was considered statistically significant. [0793] As shown in FIGs.1A, 1B, 1C, and 1D, the time between doses can be attenuated to maximize specific and minimize non-specific activity of Compound B with Compound TM-1 on NCI-N87 tumors. Shown are means ± SEM. P-values were determined by two-way ANOVA with Dunnet’s correction (* p < 0.05, ** p < 0.01). ANOVA, analysis of variance; SEM, standard error of mean. [0794] As shown in FIG.1A, a portion of the 50 mg/kg of Compound TM-1 is systemically circulating around 4 hours after dose, leading to higher non-specific activity. [0795] As shown in FIG.1D, administering Compound B 48 hours after 50 mg/kg of Compound TM-1 leads to diminished activity. [0796] Accordingly, a single dose of the MMAE prodrug, Compound B, achieves prolonged tumor growth inhibition when administered within about 8 to 24 hours after administering Compound TM-1. Example 45: Plasma and Tumor PK Analysis [0797] NCI-N87 (gastric carcinoma) cells were cultured in RPMI-1640 supplemented with 10% FBS, 1% pen-strep and placed in 37 ºC incubator. The cells were harvested and counted for tumor inoculation when the cells reached the exponential growth phase. CB-17 SCID (6-8 weeks old) female mice were inoculated subcutaneously at the right flank with 5 x 106 cells in a 200 mL mixture of PBS with Matrigel (1:1) for tumor development. Animals were divided into groups of 3 for the PK study with mean tumor volume of ~200-250 mm3 prior to dosing. Animals were dosed with a single dose of Compound TM-1 intravenously (IV) at 50 or 5 mg/kg and 4 hours later dosed with Compound B intravenously (IV) at 10 mg/kg. FIG.2A shows the schedule of dosing and tumor collection. Plasma and tumor tissue samples were collected after 15 minutes of dosing with Compound B. Plasma samples and tumor tissue were flash frozen and stored in -80 ºC until the samples were analyzed using liquid chromatography with tandem mass spectrometry (LC-MS/MS). [0798] FIG.2B shows the concentration of MMAE in tumors 15 minutes after dosing of Compound B. Shown are means ± SEM. P-values were determined by two-way ANOVA with Bonferroni correction. ANOVA, analysis of variance; MMAE, monomethyl auristatin E; SEM, standard error of mean. [0799] As shown in the FIG.2B, Compound TM-1 is converted to active payload, i.e., MMAE, within 15 minutes. Example 46: PK measurement of total amount of Compound TM-1 and reactive tetrazine (Tz) in plasma and tumor samples [0800] The NCI-N87 (gastric carcinoma) cells were cultured in RPMI-1640 supplemented with 10% FBS and 1% pen-strep and placed in 37ºC incubator. The cells were harvested and counted for tumor inoculation when the cells reached the exponential growth phase. CB-17 SCID (6-8 weeks old) female mice were inoculated subcutaneously at the right flank with 5 x 106 cells in a 200 mL mixture of RPMI- 1640 with Matrigel (1:1) for tumor development. Animals were divided into groups of 3 for the PK study with mean tumor volume of ~200-250 mm3 prior to dosing. Animals were dosed with Compound TM-1 IV at 50 mg/kg. Then animals were dosed with TCO-biotin, 15 minutes prior to tumor collection, the animals were perfused with heparin-saline solution to remove excess/unreacted TCO-biotin, then tumors were harvested close to 1, 4, and 24 hrs. The tumors were flash frozen and placed in -80 °C until the samples were analyzed using enzyme-linked immunosorbent assay (ELISA). Tumor samples were tested for total amount of Compound TM-1 and reactive tetrazine. For measuring the total amount of Compound TM-1, wells of 96-well plate were coated with anti-ID antibody against trastuzumab, and the plates were incubated overnight at 4 °C. The plate was washed 3 times and then samples diluted with assay diluent containing 1% BSA and 0.05 Tween-20 in PBS were added to the wells and incubated at room temperature for 2 hours with constant shaking. After washing, mouse anti-human IgG Fab antibody conjugated to horseradish peroxidase (HRP) (detection antibody) was addeI to the wells and incubated for 1 hr. Then the plates were again washed for 3 times, then mixed with detection reagent, and read using SepctraMax i3x. The concentrations from tumor samples were interpolated from the standard curve of Compound TM-1 using known concentration (200 ng/mL -1.563 ng/mL with 2-fold serial dilution). [0801] For measuring reactive tetrazine of Compound TM-1, wells of 96-well plate were coated with anti-ID antibody against trastuzumab, and the plates were incubated overnight at 4ºC. The plates were washed 3X times and then samples diluted with assay diluent containing 1% BSA and 0.05 Tween-20 in PBS were added to the wells and incubated at room temperature for 2 hours with constant shaking. After washing, streptavidin-HRP were added to the wells and incubated for 1 hour. Then the plates were mixed with detection reagent and read using SepctraMax i3x. The concentrations from tumor samples were interpolated from the standard curve of Compound TM-1 using known conce–tration (100 ng/mL - 0.781 ng/mL with 2-fold serial dilution). [0802] FIG.3A shows quantification of total Fab in the tumors by ELISA. FIG.3B shows quantification of activator (tetrazine) in the tumors by ELISA. Shown are means ± SEM. P-values were determined by two-way ANOVA with Bonferroni correction for multiple comparisons. ANOVA, analysis of variance; SEM, standard error of mean. [0803] As shown in FIG.3A and FIG.3B, Compound TM-1 localizes to HER2-positive tumors. Example 47: Compound TM-2 Plasma and Tumor PK Analysis [0804] This study evaluated the optimal dose and time of Compound TM-2 at the tumor site for payload-TCO conjugate dosing. [0805] Experimental Design:






Claims
What is claimed is: 1. A method of forming in vivo an antibody-payload conjugate in a subject in need thereof, the method comprising: administering an effective amount of a targeting moiety to the subject, wherein the targeting moiety comprising at least one antibody or fragment thereof having a least one tetrazine moiety covalently linked thereto; administering a single dose of a therapeutically effective amount of a payload-TCO conjugate to the subject, wherein the payload-TCO conjugate comprises a payload having at least one trans- cyclooctene (TCO) moiety covalently linked thereto; wherein the antibody or fragment thereof has a binding affinity to a receptor on a tumor, and further wherein the ratio of the antibody-payload conjugate at a tumor site versus in plasma is greater than about 2:1.
2. The method of claim 1, wherein the ratio of the antibody-payload conjugate at a tumor site versus in plasma is greater than about 5:1, about 8:1, or about 10:1.
3. The method of claim 1 or 2, wherein the administering is sequential.
4. The method of any preceding claim, wherein a single dose of the payload-TCO conjugate is administered to the subject between about 2 hours and about 48 hours, or between about 3 hours and about 48 hours, or between about 4 and about 48 hours, after the targeting moiety is administered to the subject.
5. The method any preceding claim, wherein the targeting moiety is administered at least about 8 to about 24 hours prior to the payload-TCO conjugate being administered.
6. A method of administering a payload to a subject, the method comprising: a) administering an effective amount of a targeting moiety to the subject, wherein the targeting moiety comprises a Fab having at least one tetrazine moiety covalently linked thereto; and b) administering a single dose of a therapeutically effective amount of a payload-TCO conjugate to the subject, wherein the payload-TCO conjugate comprises a payload having at least one trans-cyclooctene moiety covalently linked thereto; wherein the single dose of the payload-TCO conjugate is administered to the subject between about 2 hours and about 48 hours, or between about 3 hours and about 48 hours, or between about 4 and about 48 hours, after the targeting moiety is administered to the subject.
7. A method of treating cancer, or enhancing or eliciting an immune response, in a subject having cancer, the method comprising: a) administering an effective amount of a targeting moiety to the subject, wherein the targeting moiety comprises a Fab having at least one tetrazine moiety covalently linked thereto; and b) administering a single dose of a therapeutically effective amount of a payload-TCO conjugate to the subject, wherein the payload-TCO conjugate comprises a payload having at least one trans-cyclooctene moiety covalently linked thereto; wherein the single dose of the payload-TCO conjugate is administered to the subject between about 2 hours and 48 hours, or between about 3 hours and 48 hours, or between about 4 and about 48 hours, after the targeting moiety is administered to the subject.
8. The method of claim 7 or 8, wherein a single dose of the payload-TCO conjugate is administered to the subject between about 8 and about 24 hours after the targeting moiety is administered to the subject.
9. The method of claim 7 or 8, wherein a single dose of the payload-TCO conjugate is administered to the subject between about 8 and about 22 hours, between about 8 and about 16 hours, between about 8 and about 22 hours, between about 12 to about 16 hours, or between about 16 and about 20 hours, after the targeting moiety is administered to the subject.
10. The method of any preceding claim, wherein the targeting moiety is of Formula I, Formula II, or Formula V: wherein: ring A is aryl, cycloalkyl, heterocyclyl, or heteroaryl; the dotted lines represent additional bonds to form a tetrazine when R3 and R4 are both absent, or a dihydrotetrazine when R3 and R4 are both present; provided that when ring A is aryl, then R3 and R4 are both present; X is an antibody fragment moiety; p is 1 to 20; L, at each occurrence, is independently a linker; R1, at each occurrence, is independently selected from the group consisting of hydrogen, halo, cyano, nitro, alkyl, alkenyl, alkynyl, haloalkyl, heteroalkyl, aryl, heteroaryl, heterocyclyl, cycloalkyl, -OR', -SR', -C(=O)R', -C(=S)R', -OC(=O)R"', -SC(=O)R'", -OC(=S)R"', -SC(=S)R"', -S(=O)R', -S(=O)2R"', -S(=O)2NR'R", -C(=O)O-R', -C(=O)S-R', -C(=S)OR', -C(=S)SR', -C(=O)NR'R", -C(=S)NR'R'', -NR'R", -NR'C(=O)R", -NR'C(=S)R'', -NR'C(=O)OR'', -NR'C(=S)OR'', -NR'C(=O)SR", -NR'C(=S)SR", -OC(=O)NR'R", -SC(=O)NR'R", -OC(=S)R'R''', -SC(=S)R'R'', -NR'C(=O)NR"R", and -NR'C(=S)NR"R''; wherein each alkyl, alkenyl, alkynyl, haloalkyl, heteroalkyl, aryl, heteroaryl, heterocyclyl, or cycloalkyl is optionally substituted with one to three Z1; R2, at each occurrence, is independently halo, cyano, nitro, hydroxy, alkyl, haloalkyl, alkenyl, alkynyl, alkoxy, haloalkoxy, heteroalkyl, aryl, heteroaryl, heterocyclyl, cycloalkyl, -C(=O)-alkyl, -C(=O)-haloalkyl, -C(=O)-alkenyl, -C(=O)-alkynyl, -C(=O)-alkoxy, -C(=O)-haloalkoxy, -C(=O)-heteroalkyl, -C(=O)-aryl, -C(=O)-heteroaryl, -C(=O)-heterocyclyl, or -C(=O)-cycloalkyl; wherein each alkyl, haloalkyl, alkenyl, alkynyl, alkoxy, haloalkoxy, heteroalkyl, aryl, heteroaryl, heterocyclyl, or cycloalkyl is optionally substituted with one to three Z1; R3 and R4 are both absent; or R3 and R4 are each independently hydrogen or a group capable of being removed after a triggering event; R20, at each occurrence, is independently selected from the group consisting of hydrogen, halogen, cyano, nitro, alkyl, alkenyl, alkynyl, heteroalkyl, aryl, heteroaryl, heterocyclyl, cycloalkyl, cycloalkenyl, -CF3, -CF2R', -NO2, -OR', -SR', -C(=O)R', -C(=S)R', -OC(=O)R"', -SC(=O)R'", -OC(=S)R"', -SC(=S)R"', -S(=O)R', -S(=O)2R"', -S(=O)2NR'R", -C(=O)O-R', -C(=O)S-R', -C(=S)O-R', -C(=S)S-R', -C(=O)NR'R", -C(=S)NR'R'', -NR'R", -NR'C(=O)R", -NR'C(=S)R'', -NR'C(=O)OR'', -NR'C(=S)OR'', -NR'C(=O)SR", -NR'C(=S)SR", -OC(=O)NR'R", -SC(=O)NR'R", -OC(=S)R'R''', -SC(=S)R'R'', -NR'C(=O)NR"R", and -NR'C(=S)NR"R''; R22, at each occurrence, is independently a linker of 1 to 100 linking atoms optionally comprising one or more ethylene-oxy, amine, ester, amide, carbamate, carbonate, or ketone functional group; R30, at each occurrence, is independently halogen, cyano, nitro, hydroxy, alkyl, haloalkyl, alkenyl, alkynyl, alkoxy, haloalkoxy, heteroalkyl, aryl, heteroaryl, heterocyclyl, cycloalkyl, or cycloalkenyl; Ra, R31a, and R31b are each independently hydrogen, C1-C6-alkyl, or C1-C6-haloalkyl; each Z1 is independently selected from halo, oxo, cyano, nitro, hydroxy, alkyl, haloalkyl, alkenyl, alkynyl, alkoxy, haloalkoxy, heteroalkyl, aryl, heteroaryl, heterocyclyl, cycloalkyl, -OR', -SR', -C(=O)R', -C(=S)R', -OC(=O)R"', -SC(=O)R'", -OC(=S)R"', -SC(=S)R"', -S(=O)R', -S(=O)2R"', -S(=O)2NR'R", -C(=O)O-R', -C(=O)S-R', -C(=S)O-R', -C(=S)S-R', -C(=O)NR'R", -C(=S)NR'R'', NR'R", -NR'C(=O)R", -NR'C(=S)R'', -NR'C(=O)OR'', -NR'C(=S)OR'', -NR'C(=O)SR", -NR'C(=S)SR", -OC(=O)NR'R", -SC(=O)NR'R", -OC(=S)R'R''', -SC(=S)R'R'', -NR'C(=O)NR"R", and -NR'C(=S)NR"R''; R' and R", at each occurrence, are independently selected from hydrogen, aryl, and alkyl; R''', at each occurrence, is independently selected from aryl and alkyl; and t, at each occurrence, is independently 0, 1, 2, 3, or 4.
wherein: ring A is aryl, cycloalkyl, heterocyclyl, or heteroaryl; the dotted lines represent additional bonds to form a tetrazine when R3 and R4 are both absent, or a dihydrotetrazine when R3 and R4 are both present; provided that when ring A is aryl, then R3 and R4 are both present; X is an antibody fragment moiety; p is 1 to 20; L, at each occurrence, is independently a linker; R1, at each occurrence, is independently selected from the group consisting of hydrogen, halo, cyano, nitro, alkyl, alkenyl, alkynyl, haloalkyl, heteroalkyl, aryl, heteroaryl, heterocyclyl, cycloalkyl, -OR', -SR', -C(=O)R', -C(=S)R', -OC(=O)R"', -SC(=O)R'", -OC(=S)R"', -SC(=S)R"', -S(=O)R', -S(=O)2R"', -S(=O)2NR'R", -C(=O)O-R', -C(=O)S-R', -C(=S)OR', -C(=S)SR', -C(=O)NR'R", -C(=S)NR'R'', -NR'R", -NR'C(=O)R", -NR'C(=S)R'', -NR'C(=O)OR'', -NR'C(=S)OR'', -NR'C(=O)SR", -NR'C(=S)SR", -OC(=O)NR'R", -SC(=O)NR'R", -OC(=S)R'R''', -SC(=S)R'R'', -NR'C(=O)NR"R", and -NR'C(=S)NR"R''; wherein each alkyl, alkenyl, alkynyl, haloalkyl, heteroalkyl, aryl, heteroaryl, heterocyclyl, or cycloalkyl is optionally substituted with one to three Z1; R2, at each occurrence, is independently halo, cyano, nitro, hydroxy, alkyl, haloalkyl, alkenyl, alkynyl, alkoxy, haloalkoxy, heteroalkyl, aryl, heteroaryl, heterocyclyl, cycloalkyl, -C(=O)-alkyl, -C(=O)-haloalkyl, -C(=O)-alkenyl, -C(=O)-alkynyl, -C(=O)-alkoxy, -C(=O)-haloalkoxy, -C(=O)-heteroalkyl, -C(=O)-aryl, -C(=O)-heteroaryl, -C(=O)-heterocyclyl, or -C(=O)-cycloalkyl; wherein each alkyl, haloalkyl, alkenyl, alkynyl, alkoxy, haloalkoxy, heteroalkyl, aryl, heteroaryl, heterocyclyl, or cycloalkyl is optionally substituted with one to three Z1; R3 and R4 are both absent; or R3 and R4 are each independently hydrogen or a group capable of being removed after a triggering event; R20, at each occurrence, is independently selected from the group consisting of hydrogen, halogen, cyano, nitro, alkyl, alkenyl, alkynyl, heteroalkyl, aryl, heteroaryl, heterocyclyl, cycloalkyl, cycloalkenyl, -CF3, -CF2R', -NO2, -OR', -SR', -C(=O)R', -C(=S)R', -OC(=O)R"', -SC(=O)R'", -OC(=S)R"', -SC(=S)R"', -S(=O)R', -S(=O)2R"', -S(=O)2NR'R", -C(=O)O-R', -C(=O)S-R', -C(=S)O-R', -C(=S)S-R', -C(=O)NR'R", -C(=S)NR'R'', -NR'R", -NR'C(=O)R", -NR'C(=S)R'', -NR'C(=O)OR'', -NR'C(=S)OR'', -NR'C(=O)SR", -NR'C(=S)SR", -OC(=O)NR'R", -SC(=O)NR'R", -OC(=S)R'R''', -SC(=S)R'R'', -NR'C(=O)NR"R", and -NR'C(=S)NR"R''; R22, at each occurrence, is independently a linker of 1 to 100 linking atoms optionally comprising one or more ethylene-oxy, amine, ester, amide, carbamate, carbonate, or ketone functional group; R30, at each occurrence, is independently halogen, cyano, nitro, hydroxy, alkyl, haloalkyl, alkenyl, alkynyl, alkoxy, haloalkoxy, heteroalkyl, aryl, heteroaryl, heterocyclyl, cycloalkyl, or cycloalkenyl; Ra, R31a, and R31b are each independently hydrogen, C1-C6-alkyl, or C1-C6-haloalkyl; each Z1 is independently selected from halo, oxo, cyano, nitro, hydroxy, alkyl, haloalkyl, alkenyl, alkynyl, alkoxy, haloalkoxy, heteroalkyl, aryl, heteroaryl, heterocyclyl, cycloalkyl, -OR', -SR', -C(=O)R', -C(=S)R', -OC(=O)R"', -SC(=O)R'", -OC(=S)R"', -SC(=S)R"', -S(=O)R', -S(=O)2R"', -S(=O)2NR'R", -C(=O)O-R', -C(=O)S-R', -C(=S)O-R', -C(=S)S-R', -C(=O)NR'R", -C(=S)NR'R'', NR'R", -NR'C(=O)R", -NR'C(=S)R'', -NR'C(=O)OR'', -NR'C(=S)OR'', -NR'C(=O)SR", -NR'C(=S)SR", -OC(=O)NR'R", -SC(=O)NR'R", -OC(=S)R'R''', -SC(=S)R'R'', -NR'C(=O)NR"R", and -NR'C(=S)NR"R''; R' and R", at each occurrence, are independently selected from hydrogen, aryl, and alkyl; R''', at each occurrence, is independently selected from aryl and alkyl; and t, at each occurrence, is independently 0, 1, 2, 3, or 4.

11. The method of any preceding claim, wherein the targeting moiety is of Formula I or II: wherein: X is an antibody fragment moiety; p is 1 to 16; L, at each occurrence, is independently a linker; R20, at each occurrence, is independently selected from the group consisting of hydrogen, halogen, cyano, nitro, alkyl, alkenyl, alkynyl, heteroalkyl, aryl, heteroaryl, heterocyclyl, cycloalkyl, cycloalkenyl, CF3, CF2-R', NO2, OR', SR', C(=O)R', C(=S)R', OC(=O)R"', SC(=O)R'", OC(=S)R"', SC(=S)R"', S(=O)R', S(=O)2R"', S(=O)2NR' R", C(=O)O-R', C(=O)S-R', C(=S)O-R', C(=S)S-R', C(=O)NR'R", C(=S)NR' R'', NR'R", NR'C(=O)R", NR'C(=S)R'', NR'C(=O)OR'', NR'C(=S)OR'', NR'C(=O)SR", NR'C(=S)SR", OC(=O)NR'R", SC(=O)NR'R", OC(=S) R'R''', SC(=S)R'R'', NR'C(=O)NR"R", and NR'C(=S)NR"R''; R22, at each occurrence, is independently a linker of 1 to 100 linking atoms optionally comprising one or more ethylene-oxy, amine, ester, amide, carbamate, carbonate, or ketone functional group; R30, at each occurrence, is independently halogen, cyano, nitro, hydroxy, alkyl, haloalkyl, alkenyl, alkynyl, alkoxy, haloalkoxy, heteroalkyl, aryl, heteroaryl, heterocyclyl, cycloalkyl, or cycloalkenyl; Ra, R31a and R31b are each independently hydrogen, C1-C6-alkyl, or C1-C6-haloalkyl; R' and R", at each occurrence, are independently selected from hydrogen, aryl, and alkyl; R''' at each occurrence is independently selected from aryl and alkyl; and t, at each occurrence, is independently 0, 1, 2, 3, or 4.
wherein: X is an antibody fragment moiety; p is 1 to 16; L, at each occurrence, is independently a linker; R20, at each occurrence, is independently selected from the group consisting of hydrogen, halogen, cyano, nitro, alkyl, alkenyl, alkynyl, heteroalkyl, aryl, heteroaryl, heterocyclyl, cycloalkyl, cycloalkenyl, CF3, CF2-R', NO2, OR', SR', C(=O)R', C(=S)R', OC(=O)R"', SC(=O)R'", OC(=S)R"', SC(=S)R"', S(=O)R', S(=O)2R"', S(=O)2NR' R", C(=O)O-R', C(=O)S-R', C(=S)O-R', C(=S)S-R', C(=O)NR'R", C(=S)NR' R'', NR'R", NR'C(=O)R", NR'C(=S)R'', NR'C(=O)OR'', NR'C(=S)OR'', NR'C(=O)SR", NR'C(=S)SR", OC(=O)NR'R", SC(=O)NR'R", OC(=S) R'R''', SC(=S)R'R'', NR'C(=O)NR"R", and NR'C(=S)NR"R''; R22, at each occurrence, is independently a linker of 1 to 100 linking atoms optionally comprising one or more ethylene-oxy, amine, ester, amide, carbamate, carbonate, or ketone functional group; R30, at each occurrence, is independently halogen, cyano, nitro, hydroxy, alkyl, haloalkyl, alkenyl, alkynyl, alkoxy, haloalkoxy, heteroalkyl, aryl, heteroaryl, heterocyclyl, cycloalkyl, or cycloalkenyl; Ra, R31a and R31b are each independently hydrogen, C1-C6-alkyl, or C1-C6-haloalkyl; R' and R", at each occurrence, are independently selected from hydrogen, aryl, and alkyl; R''' at each occurrence is independently selected from aryl and alkyl; and t, at each occurrence, is independently 0, 1, 2, 3, or 4.

12. The method of any preceding claim, wherein the targeting moiety is of Formula IIA:

13. The method of any one of claims 10-12, wherein each R20 is independently hydrogen or alkyl.
14. The method of claim 10, wherein the targeting moiety is of Formula VII:

15. The method of claim 10 or 11, wherein ring A is pyrimidinyl, triazinyl, oxazolyl, isoxazole, imidazolyl, oxadiazolyl, 6,7-dihydro-5H-pyrrolo[3,4-d]pyrimidinyl, 5,6,7,8-tetrahydropyrido[4,3- d]pyrimidinyl, or 5,6,7,8-tetrahydropyrido[3,4-d]pyrimidinyl.
16. The method of claim 10 or 11, wherein ring A is phenyl.
17. The method of any one of claims 10-16, wherein p is 1 to 16, or 1 to 8, or 1 to 7, or 1 to 6, or 1 to 5, or 1 to 4, or 1 to 3, or 1 to 2.
18. The method of any one of claims 10 or 14-17, wherein each R1 is independently hydrogen or alkyl.
19. The method of any one of claims 14-18, wherein each R2 is independently halo, alkyl, or haloalkyl.
20. The method of any one of claims 14-18, wherein t, at each occurrence, is 0.
21. The method of any one of claims 5-16, wherein X is an antibody fragment moiety which targets TNC, FN1, CLDN4, MMP9, EpCAM, ITGAV, CEA, CEACAM5, ASPH, EGFR, EPCAM, VEGFR, PDGFR, TROP2, Nectin4, PSMA, BCMA, HER2, CD25, ANTXR1, or FAP.
22. The method of any one of claims 10-21, wherein X is an antibody fragment moiety derived from daclizumab, RG6292, basiliximab, HuMax-TAC, labetuzumab, 15-1-32, PR1A3, cT84.66, tusamitiamab, CC4, PAN-622, cetuximab, necitumumab, nimotuzumab, matuzumab, AMG595, depatuxizumab, dapatuxizumab, duligotuzumab, futuximab, GC1118, imgatuzumab, panitumumab, alutumumab, tomuzotuximab, laprituximab, oportuzumab, citatuzumab, tucotuzumab, catumaxomab, edrecolomab, adecatumumab, ramucizumab, ramucirumab, vulinacimab, olaratumab, ramucirumab, sacituzumab, Pr1E11, Enfortumab, J591, MLN591, belantamab, moxetumomab, inotuzumab, epratuzumab, pinatuzumab, ublituximab, ofatumumab, rituximab, obinutuzumab, tositumomab, ibritumomab, loncastuximab, XMAB-5574, MOR208, coltuximab, denintuzumab, taplitumomab, MDX- 1342, polatuzumab, isatuximab, daratumumab, MOR202, TAK-079, I-131-BC8, Iomab-B, carotuximab, bemarituzumab, aprutumab, lupartumab, zolbetuximab, claudiximab, andecaliximab, mirvetuximab, farletuzumab, MORAb-202, MORAb-003, SP8166, rovalpituzumab, indatuximab, lorvotuzumab, promiximab, BI 836826, otlertuzumab, naratuximab, milatuzumab, anetumab, amatuximab, MMOT- 0530A, sarilumab, elotuzumab, belimumab, KL-6, MY.1E12, hMUC1-1H7, TAB004, huC242, clivatuzumab, 8HuDS6, gatipotuzumab, AR20.5, cantuzumab, codrituzumab, ECT204, MDX-1414, pertuzumab, trastuzumab, margetuximab, patritumab, seribantumab, lumretuzumab, elgemtumab, AV- 203, CDX-3379, GSK284933, brentuximab, gemtuzumab, BI 835858, vadastuximab, lintuzumab, KHK2823, taclotuzumab, G4723A, glembatumumab, telisotuzumab, onartuzumab, SAIT301, tisotumab, lifastuzumab, indusatumab, vandortuzumab, sofituzumab, vorsetuzumab, bivatuzumab, caplacizumab, ozoralizumab, V565, PF-05230905, vobarilizumab, LCAR-B38M, BI 655088, AD-214, ALX-0651, TXB4, CDP791, GY1, L19, NJB2, F19, OMTX005, sibrotuzumab, F16, R6N, datopotamab, 15A7.5_H1L3, hNec.4.05, 14A5.2, 42D20-Hz3, 42D20-Hz10, HZD6.1C, HZD6.2C, 74HZ.
23. The method of any one of claims 1-22, wherein the targeting moiety further comprises an imaging contrast agent.
24. The method of claim 23, wherein the imaging contrast agent is a protein.
25. The method of any one of claims 10-24, wherein L, at each occurrence, is independently bonded to X via a cystine or lysine residue on X.
26. The method of any one of claims 10-25, wherein each L comprises one or more amino acids.
27. The method of any one of claims 10-26, wherein each L comprises a polypeptide.
28. The method of any one of claims 10-27, wherein each L independently comprises 1 to 100 linking atoms, from 1 to 50 linking atoms, or from 5 to 50 linking atoms, or from 10 to 50 linking atoms, or from 1 to 40 linking atoms, or from 1 to 30 linking atoms, or from 1 to 20 linking atoms, or from 1 to 10 linking atoms, or from 1 to 5 linking atoms, or from 5 to 30 linking atoms, or from 10 to 30 linking atoms, or from 5 to 40 linking atoms, or from 5 to 50 linking atoms, or from 10 to 50 linking atoms.
29. The method of any one of claims 10-28, wherein each L independently comprises from 5 to 50 linking atoms; comprising one or more chain heteroatoms and one or more alkylene, alkenylene, alkynylene, arylene, or heteroarylene; wherein each alkylene, alkenylene, alkynylene, arylene, or heteroarylene, may be independently optionally substituted with one to five substituents independently selected from oxo, halo, C1-4 alkyl, C1-4 alkoxy, and C1-4 haloalkyl.
30. The method of any one of claims 10-29, wherein X is an antibody fragment moiety that targets HER2, TROP2, Nectin-4, Claudin-18.2, MMP9, mesothelin, FN1, FAP, TNC, or ECM, EPCAM, CEA, or CEACAM5; and each L is independently selected from the group consisting of: 31. The method of any one of claims 10-14, wherein X is an antibody fragment moiety that targets HER2; p is 1 to 5; and each L is independently selected from the group consisting of:
31. The method of any one of claims 10-14, wherein X is an antibody fragment moiety that targets HER2; p is 1 to 5; and each L is independently selected from the group consisting of: 32. The method of any preceding claim, wherein the targeting moiety is of Formula IIF:
32. The method of any preceding claim, wherein the targeting moiety is of Formula IIF: 33. The method of claim 32, wherein X is an antibody fragment moiety that targets HER2; and p is 1 to 5. 34. The method of any preceding claim, wherein the payload-TCO conjugate is of Formula VIII, or a pharmaceutically acceptable salt thereof:
33. The method of claim 32, wherein X is an antibody fragment moiety that targets HER2; and p is 1 to 5. 34. The method of any preceding claim, wherein the payload-TCO conjugate is of Formula VIII, or a pharmaceutically acceptable salt thereof: wherein:
wherein: L1, at each occurrence, is independently a linker; m is an integer from 1 to 150; D is a payload; R1A, at each occurrence, is independently selected from the group consisting of C1-4alkyl, C1-4haloalkyl, and C1-4alkoxy; q is 0, 1, or 2; q1 is 0 or 1; R1B, at each occurrence, is independently selected from the group consisting of G1, -OH, -NR1c–C1-4alkylene–G1, –NR1c–C1-4alkylene–N(R1d)2, -NR1c-C1-6alkylene-N(C1-4alkyl)3+, -N(R1c)CHR1eCO2H, –N(R1c)–C1-6alkylene–CO2H, –N(R1c)CHR1eC(O)OC1-6alkyl, -N(R1f)-C2-4alkylene-(N(C1-4alkylene-CO2H)-C2-4alkylene)n–N(C1-4alkylene–CO2H)2, -N(R1f)-C2-4alkylene-(N(C1-4alkylene-C(O)OC1-6alkyl)-C2-4alkylene)n-N(C1-4alkylene-C(O)OC1-6alkyl)2, -N(R1c)–C1-6alkylene–SO3H, –N(R1c)–(CH2CH2O)1-3–CH2CH2N((CH2CH2O)1-3–C1-6alkylene–CO2H)2, -N(R1c)-C1-6alkylene-C(O)OC1-6alkyl, and –N(R1c)–CH(CH2O–(CH2CH2O)0-2–C1-6alkylene–CO2H)2; R1c and R1d, at each occurrence, are independently hydrogen or C1-4alkyl; R1e, at each occurrence, is independently –C1-4alkylene–CO2H, –C1-4alkylene–CONH2, or –C1-4alkylene–OH; R1f, at each occurrence, is independently hydrogen, C1-6alkyl, or C1-4alkylene–CO2H; n, at each occurrence, is independently 0, 1, 2, or 3; L2, at each occurrence, is independently selected from the group consisting of –C(O)– and C1-3alkylene; and G1, at each occurrence, is independently an optionally substituted heterocyclyl. 35. The method of any preceding claim, wherein the payload is paclitaxel, doxorubicin, daunorubicin, etoposide, irinotecan, SN-38, docetaxel, paclitaxel, gemcitabine, podophyllotoxin, Carmustine, Ixabepilone, Patupilone, platinum drugs, exatecan, deruxtecan, dolastatin 10, MMAE, MMAD, MMAF, mitomycin C, bleomycin, calicheamicin, staurosporine, hemiasterlin, seco-DUBA, duocarmycin, cyclosporin A, or rapamycin. 36. The method of any preceding claim, wherein the payload-TCO conjugate is selected from:
L1, at each occurrence, is independently a linker; m is an integer from 1 to 150; D is a payload; R1A, at each occurrence, is independently selected from the group consisting of C1-4alkyl, C1-4haloalkyl, and C1-4alkoxy; q is 0, 1, or 2; q1 is 0 or 1; R1B, at each occurrence, is independently selected from the group consisting of G1, -OH, -NR1c–C1-4alkylene–G1, –NR1c–C1-4alkylene–N(R1d)2, -NR1c-C1-6alkylene-N(C1-4alkyl)3+, -N(R1c)CHR1eCO2H, –N(R1c)–C1-6alkylene–CO2H, –N(R1c)CHR1eC(O)OC1-6alkyl, -N(R1f)-C2-4alkylene-(N(C1-4alkylene-CO2H)-C2-4alkylene)n–N(C1-4alkylene–CO2H)2, -N(R1f)-C2-4alkylene-(N(C1-4alkylene-C(O)OC1-6alkyl)-C2-4alkylene)n-N(C1-4alkylene-C(O)OC1-6alkyl)2, -N(R1c)–C1-6alkylene–SO3H, –N(R1c)–(CH2CH2O)1-3–CH2CH2N((CH2CH2O)1-3–C1-6alkylene–CO2H)2, -N(R1c)-C1-6alkylene-C(O)OC1-6alkyl, and –N(R1c)–CH(CH2O–(CH2CH2O)0-2–C1-6alkylene–CO2H)2; R1c and R1d, at each occurrence, are independently hydrogen or C1-4alkyl; R1e, at each occurrence, is independently –C1-4alkylene–CO2H, –C1-4alkylene–CONH2, or –C1-4alkylene–OH; R1f, at each occurrence, is independently hydrogen, C1-6alkyl, or C1-4alkylene–CO2H; n, at each occurrence, is independently 0, 1, 2, or 3; L2, at each occurrence, is independently selected from the group consisting of –C(O)– and C1-3alkylene; and G1, at each occurrence, is independently an optionally substituted heterocyclyl. 35. The method of any preceding claim, wherein the payload is paclitaxel, doxorubicin, daunorubicin, etoposide, irinotecan, SN-38, docetaxel, paclitaxel, gemcitabine, podophyllotoxin, Carmustine, Ixabepilone, Patupilone, platinum drugs, exatecan, deruxtecan, dolastatin 10, MMAE, MMAD, MMAF, mitomycin C, bleomycin, calicheamicin, staurosporine, hemiasterlin, seco-DUBA, duocarmycin, cyclosporin A, or rapamycin. 36. The method of any preceding claim, wherein the payload-TCO conjugate is selected from:















Applications Claiming Priority (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US202363583189P | 2023-09-15 | 2023-09-15 | |
| US63/583,189 | 2023-09-15 | ||
| US202463660911P | 2024-06-17 | 2024-06-17 | |
| US63/660,911 | 2024-06-17 | ||
| US202463681755P | 2024-08-09 | 2024-08-09 | |
| US63/681,755 | 2024-08-09 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2025059685A1true WO2025059685A1 (en) | 2025-03-20 |
Family
ID=92966746
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2024/046969PendingWO2025059685A1 (en) | 2023-09-15 | 2024-09-16 | Methods for in vivo targeted delivery of a payload |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2025059685A1 (en) |
Citations (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20030191291A1 (en) | 2000-09-08 | 2003-10-09 | Kochendoerfer Gerd G. | Synthetic erythropoiesis stimulating proteins |
| US20130189184A1 (en)* | 2010-10-14 | 2013-07-25 | Koninklijke Philips Electronics N.V. | Pretargeting kit, method and agents used therein |
| WO2014138186A1 (en)* | 2013-03-05 | 2014-09-12 | The Johns Hopkins University | Bioorthogonal two-component delivery systems for enhanced internalization of nanotherapeutics |
| WO2014205126A1 (en) | 2013-06-19 | 2014-12-24 | The Regents Of The University Of California | Chemical structures for localized delivery of therapeutic agents |
| WO2015139025A1 (en) | 2014-03-14 | 2015-09-17 | The Regents Of The University Of California | Tco conjugates and methods for delivery of therapeutic agents |
| WO2017044983A1 (en) | 2015-09-10 | 2017-03-16 | Shasqi, Inc. | Bioorthogonal compositions |
| WO2018187740A1 (en) | 2017-04-07 | 2018-10-11 | Shasqi, Inc. | Bioorthogonal compositions |
| US20190091351A1 (en)* | 2015-05-11 | 2019-03-28 | Memorial Sloan Kettering Cancer Center | Radioligands for pretargeted pet imaging and methods of their therapeutic use |
| WO2020077140A1 (en) | 2018-10-10 | 2020-04-16 | Tambo, Inc. | Processes for preparing functionalized cyclooctenes |
| WO2021007160A1 (en) | 2019-07-05 | 2021-01-14 | Tambo, Inc. | Trans-cyclooctene bioorthogonal agents and uses in cancer and immunotherapy |
| WO2022032191A1 (en) | 2020-08-07 | 2022-02-10 | Tambo, Inc. | Trans-cyclooctene bioorthogonal agents and uses in cancer and immunotherapy |
| WO2022189304A2 (en)* | 2021-03-11 | 2022-09-15 | University Of Copenhagen | Aliphatic 18f-radiolabeling of a tetrazine precursor |
| WO2023077129A1 (en)* | 2021-10-29 | 2023-05-04 | Tambo, Inc. | Tetrazine conjugates for in vivo targeted delivery of a payload |
- 2024
- 2024-09-16WOPCT/US2024/046969patent/WO2025059685A1/enactivePending
Patent Citations (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20030191291A1 (en) | 2000-09-08 | 2003-10-09 | Kochendoerfer Gerd G. | Synthetic erythropoiesis stimulating proteins |
| US20130189184A1 (en)* | 2010-10-14 | 2013-07-25 | Koninklijke Philips Electronics N.V. | Pretargeting kit, method and agents used therein |
| WO2014138186A1 (en)* | 2013-03-05 | 2014-09-12 | The Johns Hopkins University | Bioorthogonal two-component delivery systems for enhanced internalization of nanotherapeutics |
| WO2014205126A1 (en) | 2013-06-19 | 2014-12-24 | The Regents Of The University Of California | Chemical structures for localized delivery of therapeutic agents |
| WO2015139025A1 (en) | 2014-03-14 | 2015-09-17 | The Regents Of The University Of California | Tco conjugates and methods for delivery of therapeutic agents |
| US20190091351A1 (en)* | 2015-05-11 | 2019-03-28 | Memorial Sloan Kettering Cancer Center | Radioligands for pretargeted pet imaging and methods of their therapeutic use |
| WO2017044983A1 (en) | 2015-09-10 | 2017-03-16 | Shasqi, Inc. | Bioorthogonal compositions |
| WO2018187740A1 (en) | 2017-04-07 | 2018-10-11 | Shasqi, Inc. | Bioorthogonal compositions |
| WO2020077140A1 (en) | 2018-10-10 | 2020-04-16 | Tambo, Inc. | Processes for preparing functionalized cyclooctenes |
| WO2021007160A1 (en) | 2019-07-05 | 2021-01-14 | Tambo, Inc. | Trans-cyclooctene bioorthogonal agents and uses in cancer and immunotherapy |
| WO2022032191A1 (en) | 2020-08-07 | 2022-02-10 | Tambo, Inc. | Trans-cyclooctene bioorthogonal agents and uses in cancer and immunotherapy |
| WO2022189304A2 (en)* | 2021-03-11 | 2022-09-15 | University Of Copenhagen | Aliphatic 18f-radiolabeling of a tetrazine precursor |
| WO2023077129A1 (en)* | 2021-10-29 | 2023-05-04 | Tambo, Inc. | Tetrazine conjugates for in vivo targeted delivery of a payload |
Non-Patent Citations (17)
| Title |
|---|
| "Periodic Table of the Elements", 1999, HANDBOOK OF CHEMISTRY AND PHYSICS |
| AGNESE MAGGI ET AL: "Development of a novel antibody?tetrazine conjugate for bioorthogonal pretargeting", ORGANIC & BIOMOLECULAR CHEMISTRY, vol. 14, no. 31, 1 January 2016 (2016-01-01), pages 7544 - 7551, XP055385205, ISSN: 1477-0520, DOI: 10.1039/C6OB01411A* |
| BERLAND ET AL., BIOMOLECULES, vol. 11, no. 5, 2021, pages 637 |
| BILLAUD EMILIE M. F. ET AL: "Pretargeted PET Imaging Using a Bioorthogonal 18 F-Labeled trans -Cyclooctene in an Ovarian Carcinoma Model", BIOCONJUGATE CHEMISTRY, vol. 28, no. 12, 11 December 2017 (2017-12-11), US, pages 2915 - 2920, XP093151560, ISSN: 1043-1802, DOI: 10.1021/acs.bioconjchem.7b00635* |
| CARRUTHERS: "Some Modern Methods of Organic Synthesis", 1987, CAMBRIDGE UNIVERSITY PRESS |
| CHOI ET AL., THERANOSTICS, vol. 2, no. 2, 2012, pages 156 - 178 |
| GARCÍA MARÍA FERNANDA ET AL: "99mTc-bioorthogonal click chemistry reagent for in vivo pretargeted imaging", BIOORGANIC & MEDICINAL CHEMISTRY, ELSEVIER, AMSTERDAM, NL, vol. 24, no. 6, 29 January 2016 (2016-01-29), pages 1209 - 1215, XP029439440, ISSN: 0968-0896, DOI: 10.1016/J.BMC.2016.01.046* |
| KROEMER ET AL., ANNU. REV. IMMUNOL., no. 31, 2013, pages 51 - 72 |
| KROEMER, CURR. OP. IMMUNOL., vol. 20, 2008, pages 504 - 511 |
| LAROCK: "Comprehensive Organic Transformations", 1989, VCH PUBLISHERS, INC. |
| PURE APPL. CHEM., vol. 45, 1976, pages 13 - 30 |
| RAFFAELLA ROSSIN ET AL: "In Vivo Chemistry for Pretargeted Tumor Imaging in Live Mice", ANGEWANDTE CHEMIE INTERNATIONAL EDITION, VERLAG CHEMIE, HOBOKEN, USA, vol. 49, no. 19, 12 April 2010 (2010-04-12), pages 3375 - 3378, XP072067029, ISSN: 1433-7851, DOI: 10.1002/ANIE.200906294* |
| ROSSIN RAFFAELLA ET AL: "Pretargeted imaging using bioorthogonal chemistry in mice", CURRENT OPINION IN CHEMICAL BIOLOGY, vol. 21, 19 August 2014 (2014-08-19), GB, pages 161 - 169, XP093230984, ISSN: 1367-5931, Retrieved from the Internet <URL:https://www.sciencedirect.com/science/article/pii/S1367593114000969> DOI: 10.1016/j.cbpa.2014.07.023* |
| RUIVO EDUARDO ET AL: "Preclinical Evaluation of a Novel 18 F-Labeled dTCO-Amide Derivative for Bioorthogonal Pretargeted Positron Emission Tomography Imaging", ACS OMEGA, vol. 5, no. 9, 26 February 2020 (2020-02-26), US, pages 4449 - 4456, XP093151342, ISSN: 2470-1343, Retrieved from the Internet <URL:http://pubs.acs.org/doi/pdf/10.1021/acsomega.9b03584> DOI: 10.1021/acsomega.9b03584* |
| S. M. J. VAN DUIJNHOVEN ET AL: "Diabody Pretargeting with Click Chemistry In Vivo", THE JOURNAL OF NUCLEAR MEDICINE, vol. 56, no. 9, 1 September 2015 (2015-09-01), US, pages 1422 - 1428, XP055648065, ISSN: 0161-5505, DOI: 10.2967/jnumed.115.159145* |
| SMITHMARCH MARCH 'S: "Advanced Organic Chemistry,", 2001, JOHN WILEY & SONS, INC. |
| WUTS, P. G. M.GREENE, T. W.GREENE, T. W.: "Greene's protective groups in organic synthesis.", 2006, WILEY-INTERSCIENCE |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP2024096335A (en) | Hydrophilic antibody-drug conjugates | |
| JP6751165B2 (en) | Anti-B7-H3 antibody and antibody drug conjugate | |
| JP2021185176A (en) | Antibody-drug conjugates based on eribulin and methods of use | |
| US11999748B2 (en) | Exatecan derivatives, linker-payloads, and conjugates and thereof | |
| US20250186592A1 (en) | Tetrazine conjugates for in vivo targeted delivery of a payload | |
| JP2018502839A (en) | Bcl-xL inhibitory compound and antibody drug conjugate containing the same | |
| EP4422693A1 (en) | Trans-cyclooctene conjugates | |
| US20240058467A1 (en) | Anti-ror1 antibody conjugates, compositions comprising anti ror1 antibody conjugates, and methods of making and using anti-ror1 antibody conjugates | |
| TW202519271A (en) | Herboxidiene splicing modulator antibody-drug conjugates and methods of use | |
| TW201920192A (en) | THAILANSTATIN analogs | |
| US20250144229A1 (en) | Conjugate of antibody-eribulin or derivative thereof, intermediate thereof, preparation method therefor, pharmaceutical composition thereof and use thereof | |
| US20250009890A1 (en) | Trans-cyclooctene-modified bispecific antibodies | |
| KR20250060248A (en) | Tissue factor antibody-drug conjugates and uses thereof | |
| WO2025059685A1 (en) | Methods for in vivo targeted delivery of a payload | |
| WO2024229427A1 (en) | Tetrazine-based targeting agents for in vivo delivery of a payload | |
| CN118369123A (en) | Tetrazine conjugates for targeted delivery of payloads in vivo | |
| US12419964B2 (en) | Antibody drug conjugate (ADC) targeting Nectin 4 and comprising an exatecan payload | |
| WO2024238969A1 (en) | Trans-cyclooctene-modified targeted protein degrader conjugates | |
| WO2024178155A1 (en) | Trans-cyclooctene conjugates | |
| WO2025166216A1 (en) | Trop2-targeting antibodies and uses thereof | |
| US20250000991A1 (en) | Nectin-4 antibodies and antibody-drug conjugates | |
| WO2025064427A1 (en) | Dual-payload antibody-drug conjugates and uses thereof |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | Ref document number:24783438 Country of ref document:EP Kind code of ref document:A1 |