WO2025045767A1 - Stabilized lipid and lipidoid nanoparticle formulations with specific surfactant properties for enhanced pharmaceutical applications - Google Patents
Stabilized lipid and lipidoid nanoparticle formulations with specific surfactant properties for enhanced pharmaceutical applicationsDownload PDFInfo
- Publication number
- WO2025045767A1 WO2025045767A1PCT/EP2024/073691EP2024073691WWO2025045767A1WO 2025045767 A1WO2025045767 A1WO 2025045767A1EP 2024073691 WEP2024073691 WEP 2024073691WWO 2025045767 A1WO2025045767 A1WO 2025045767A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- lipid
- surfactant
- lipidoid
- formulation
- nanoparticle
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Classifications
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/48—Preparations in capsules, e.g. of gelatin, of chocolate
- A61K9/50—Microcapsules having a gas, liquid or semi-solid filling; Solid microparticles or pellets surrounded by a distinct coating layer, e.g. coated microspheres, coated drug crystals
- A61K9/51—Nanocapsules; Nanoparticles
- A61K9/5107—Excipients; Inactive ingredients
- A61K9/5123—Organic compounds, e.g. fats, sugars
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
- A61K31/7088—Compounds having three or more nucleosides or nucleotides
- A61K31/7125—Nucleic acids or oligonucleotides having modified internucleoside linkage, i.e. other than 3'-5' phosphodiesters
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/06—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite
- A61K47/08—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite containing oxygen, e.g. ethers, acetals, ketones, quinones, aldehydes, peroxides
- A61K47/10—Alcohols; Phenols; Salts thereof, e.g. glycerol; Polyethylene glycols [PEG]; Poloxamers; PEG/POE alkyl ethers
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/0012—Galenical forms characterised by the site of application
- A61K9/0019—Injectable compositions; Intramuscular, intravenous, arterial, subcutaneous administration; Compositions to be administered through the skin in an invasive manner
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/10—Dispersions; Emulsions
- A61K9/127—Synthetic bilayered vehicles, e.g. liposomes or liposomes with cholesterol as the only non-phosphatidyl surfactant
- A61K9/1271—Non-conventional liposomes, e.g. PEGylated liposomes or liposomes coated or grafted with polymers
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/10—Dispersions; Emulsions
- A61K9/127—Synthetic bilayered vehicles, e.g. liposomes or liposomes with cholesterol as the only non-phosphatidyl surfactant
- A61K9/1271—Non-conventional liposomes, e.g. PEGylated liposomes or liposomes coated or grafted with polymers
- A61K9/1272—Non-conventional liposomes, e.g. PEGylated liposomes or liposomes coated or grafted with polymers comprising non-phosphatidyl surfactants as bilayer-forming substances, e.g. cationic lipids or non-phosphatidyl liposomes coated or grafted with polymers
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/87—Introduction of foreign genetic material using processes not otherwise provided for, e.g. co-transformation
- C12N15/88—Introduction of foreign genetic material using processes not otherwise provided for, e.g. co-transformation using microencapsulation, e.g. using amphiphile liposome vesicle
Definitions
- the present inventionrelates to stabilized lipid and/or lipidoid formulations comprising lipid nanoparticles or lipidoid nanoparticles and one or more surfactants with specific properties for the delivery of nucleic acids and enhanced pharmaceutical applications.
- Lipid or lipidoid nanoparticlesare frequently used for the delivery of active pharmaceutical ingredients in patients.
- lipid or lipidoid formulations of nucleic acidsare extremely useful and efficient for introducing nucleic acids into cells.
- This advantageous property of lipid or lipidoid formulations of nucleic acidshas been used for decades in biological and medical research and in therapeutic approaches to i) overexpress genes or to complement genetic defects in target cells, or ii) to downregulate or upregulate endogenous gene expression in cells, or iii) to repair genetic defects (mutations).
- mRNA formulations relying on nanoparticlesare now also established as vaccines against COVID- 19.
- the present inventionprovides novel stabilized lipid and lipidoid formulations with specific surfactants that help overcome issues arising during said purification, processing and handling.
- This inventionpresents novel lipid and lipidoid nanoparticle (LNP and LiNP) formulations enhanced with specifically characterized surfactants. These surfactants exhibit unique Langmuir isotherm properties, ensuring optimal stabilization of the nanoparticles.
- the described formulationsimprove the stability, reduce aggregation, and mitigate challenges faced during purification, such as filtration clogging or fouling.
- Integral to the innovationis a method for determining the suitability of surfactants as stabilizers based on predetermined Langmuir surface pressure/area isotherm (also referred to briefly herein as “Langmuir isotherm”) values, or filtration speeds.
- the stabilized nanoparticleswhen formulated with therapeutic agents, have demonstrated potential in medical applications, particularly in the realm of mRNA delivery, vaccination or immunization.
- the inventionprovides a lipid nanoparticle (LNP) formulation or lipidoid nanoparticle (LiNP) formulation comprising a plurality of lipid nanoparticles (LNPs) or lipidoid nanoparticles (LiNPs) each comprising a lipid mix or lipidoid mix, wherein the lipid mix or lipidoid mix comprises, as a component thereof, at least one selected from an ionizable lipid, ionizable lipidoid, and permanently cationic lipid, and wherein the formulation comprises a surfactant characterized by having a Langmuir surface pressure/area isotherm with a maximum surface pressure (Umax) that is equal to or below 4.0 mN/m at a minimum surface area established for a lipid mix or lipidoid mix as comprised by the nanoparticles.
- LNPlipid nanoparticle
- LiNPslipidoid nanoparticle
- the inventionprovides a surfactant for use in a pharmaceutical composition, the surfactant being characterized by having a Langmuir isotherm with a maximum surface pressure (n ma x) that is equal to or below 4.0 mN/m at a minimum surface area established for said pharmaceutical composition.
- the inventionprovides a method for classifying a surfactant as suitable or not suitable for use as a stabilizing agent for a pharmaceutical composition comprising a lipid or lipidoid, wherein the method comprises the steps:
- the inventionprovides a method of mitigating or avoiding clogging or fouling of a filtration system during purification of a pharmaceutical composition in the form of a lipid nanoparticle formulation (LNP) or lipidoid nanoparticle formulation (LiNP), the method comprising adding a stabilizing surfactant to a first LNP or LiNP formulation to form a second LNP or LiNP formulation, optionally before purification, wherein the LNP or LiNP formulation comprises a plurality of LNPs or LiNPs, each comprising a lipid mix or lipidoid mix and a therapeutic agent, wherein the lipid mix or lipidoid mix comprises, as a component thereof, at least one selected from an ionizable lipid, ionizable lipidoid, and permanently cationic lipid, and wherein the stabilizing surfactant is a surfactant according to the invention, e.g. a surfactant according to the third aspect discussed above or a surfactant classified as being
- the inventionprovides a method of mitigating aggregation of lipid nanoparticles or lipidoid nanoparticles in a lipid nanoparticle (LNP) formulation or lipidoid nanoparticle (LiNP) formulation, preferably a pharmaceutical composition in the form of a lipid nanoparticle (LNP) formulation or lipidoid nanoparticle (LiNP) formulation, the method comprising adding a stabilizing surfactant to a first LNP or LiNP formulation to form a second LNP or LiNP formulation optionally before purification, wherein the LNP or LiNP formulation comprises a plurality of LNPs or LiNPs, each comprising a lipid mix or lipidoid mix, wherein the lipid mix or lipidoid mix comprises, as a component thereof, at least one selected from an ionizable lipid, ionizable lipidoid, and permanently cationic lipid, and the stabilizing surfactant is a surfactant in accordance with
- the inventionprovides a lipid nanoparticle (LNP) formulation or lipidoid nanoparticle (LiNP) formulation obtained by the method in accordance with the fourth to seventh aspect.
- LNPlipid nanoparticle
- LiNPlipidoid nanoparticle
- the inventionprovides a lipid nanoparticle (LNP) formulation or lipidoid nanoparticle (LiNP) formulation in accordance with the first aspect, the second aspect, or the eighth aspect discussed above for use as a medicament.
- LNPlipid nanoparticle
- LiNPlipidoid nanoparticle
- the inventionprovides a method for classifying a surfactant as suitable or not suitable for use as a stabilizer of a pharmaceutical composition comprising a nucleic acid, optionally as a stabilizer during purification, preferably during TFF purification, the method comprising:
- the inventionprovides a use of a surfactant according to the invention, e.g. the surfactant in accordance with the above third aspect, for stabilizing a suspension of lipid nanoparticles or of lipidoid nanoparticles in an aqueous vehicle solution against particle aggregation under a physical stress condition, preferably shear stress, more preferably shear stress during purification such as TFF, wherein the lipid nanoparticles or lipidoid nanoparticles comprise the following components (a) and (b):
- a lipid nanoparticle (LNP) formulation or lipidoid nanoparticle (LiNP) formulationcomprising a plurality of lipid nanoparticles (LNPs) or lipidoid nanoparticles (LiNPs), each comprising a lipid mix or lipidoid mix, wherein the lipid mix or lipidoid mix comprises, as a component thereof, at least one selected from an ionizable lipid, ionizable lipidoid, and permanently cationic lipid, and wherein the formulation further comprises a surfactant characterized by having a Langmuir surface pressure/area isotherm (also referred to herein as “Langmuir isotherm”) with a maximum surface pressure (n ma x) that is equal to or below 4.0 mN/m at a minimum surface area established for a lipid mix or lipidoid mix as comprised by the nanoparticles.
- a surfactantcharacterized by having a Langmuir surface pressure
- a lipid nanoparticle (LNP) formulation or lipidoid nanoparticle (LiNP) formulationcomprising a plurality of lipid nanoparticles (LNPs) or lipidoid nanoparticles (LiNPs), each comprising a lipid mix or lipidoid mix, wherein the lipid mix or lipidoid mix comprises, as a component thereof, at least one selected from an ionizable lipid, ionizable lipidoid, and permanently cationic lipid, wherein the formulation comprises as a stabilizing agent a surfactant, and wherein a representative sample comprising an aqueous solution of the surfactant carrying on its surface a lipid mix or lipidoid mix as comprised by the LNPs or LiNPs is characterized by having a Langmuir isotherm An that is equal to or below 0.60, preferably equal to or below 0.45, at each area point during a Langmuir surface pressure/area isotherm cycle comprising a compression
- a surfactantselected from the list consisting of poloxamer 124 (P124), poloxamer 188 (P188), poloxamer 338 (P338), poloxamer 407 (P407), Tween-20, Tween-80, BRIJ35, tyloxapol, VitE-PEG1000, and Kolliphor EL, or from combinations thereof.
- the surfactantis a block copolymer of ethylene oxide and propylene oxide, more preferably a poloxamer, even more preferably poloxamer selected from the list consisting of poloxamer 124, poloxamer 188, poloxamer 338, and poloxamer 407, or from combinations thereof, most preferably P188.
- lipid nanoparticle (LNP) formulation or lipidoid nanoparticle (LiNP) formulationaccording to any one of items 1 to 5, wherein the lipid nanoparticles or lipidoid nanoparticles comprise the lipid mix or lipidoid mix and a therapeutic agent.
- lipid nanoparticle (LNP) formulation or lipidoid nanoparticle (LiNP) formulationaccording to any one of items 1 to 9, which is a lipidoid nanoparticle formulation comprising a plurality of lipidoid nanoparticles each comprising a lipidoid mix, wherein the lipidoid mix comprises an ionizable lipidoid of formula (L-1 ): wherein: a is 1 or 2 and b is an integer of 1 to 4 or a is an integer of 1 to 4 and b is 1 or 2, preferably a is 1 and b is an integer of 2 to 4 or a is an integer of 2 to 4 and b is 1 , p is 1 or 2, m is 1 or 2; n is 0 or 1 and m+n is > 2, and
- lipid nanoparticle (LNP) formulation or lipidoid nanoparticle (LiNP) formulationaccording to any one of items 1 to 10 which is a suspension formulation, and wherein the vehicle solution of the suspension formulation is an aqueous vehicle solution comprising the surfactant.
- a surfactantselected from the list consisting of poloxamer 124 (P124), poloxamer 188 (P188), poloxamer 338 (P338), poloxamer 407 (P407), Tween-20, Tween-80, BRIJ35, tyloxapol, VitE-PEG1000, and Kolliphor EL, or from combinations thereof.
- the surfactant for use according to item 14,which is a block copolymer of ethylene oxide and propylene oxide, more preferably a poloxamer, even more preferably poloxamer selected from the list consisting of poloxamer 124, poloxamer 188, poloxamer 338, and poloxamer 407, or from combinations thereof, most preferably P188.
- the pharmaceutical compositionis in the form of a lipid nanoparticle (LNP) suspension formulation or lipidoid nanoparticle (LiNP) suspension formulation comprising a plurality of LNPs or LiNPs, each comprising a lipid mix or lipidoid mix and a therapeutic agent, wherein the lipid mix or lipidoid mix comprises, as a component thereof, at least one selected from an ionizable lipid, ionizable lipidoid, and permanently cationic lipid, and wherein the vehicle solution of the suspension formulation is an aqueous vehicle solution comprising the surfactant
- a method for classifying a surfactant as suitable or not suitable for use as a stabilizing agent for a pharmaceutical compositioncomprising a lipid or lipidoid, optionally during purification of said composition, preferably during tangential flow filtration of said composition, wherein the method comprises the steps:
- a Langmuir pressure/area isotherm cycleincluding a compression phase and an expansion phase between a maximum surface area and a minimum surface area on a sample comprising the surfactant in the aqueous solution and carrying on its surface a lipid or lipidoid as comprised by the composition: (c) calculating a Langmuir isotherm An for each area point of the Langmuir pressure/area isotherm cycle, wherein TT is calculated as: wherein n comp is the surface pressure at said area point during the compression phase of the isotherm cycle, wherein it exp is the surface pressure at said area point during the expansion phase of the isotherm cycle, and wherein n max is the maximum surface pressure reached in the isotherm cycle, and
- a method for the preparation of a pharmaceutical compositioncomprising classifying a surfactant as suitable or not suitable for use as a stabilizing agent for a pharmaceutical composition in accordance with the method of item 17 or 18, and, if the surfactant is classified as being suitable for use as a stabilizing agent for the pharmaceutical composition, incorporating the surfactant into the pharmaceutical composition.
- the surfactantis a nonionic surfactant, preferably at least one nonionic surfactant selected from the group of fatty alcohol ethoxylates, fatty acid ethoxylates, block copolymers of ethylene oxide and propylene oxide, alkylphenol ethoxylates or oligomers of alkylphenol ethoxylates, fatty acid esters of sorbitol, ethoxylated fatty acid esters of sorbitol, fatty acid esters of glycerol, ethoxylated castor oil and ethoxylated vitamin E, e.g.
- the surfactantis a nonionic surfactant, preferably at least one nonionic surfactant selected from the group of fatty alcohol ethoxylates, fatty acid ethoxylates, block copolymers of ethylene oxide and propylene oxide, alkylphenol ethoxylates or oligomers of alkylphenol ethoxylates, fatty
- a surfactantselected from the list consisting of poloxamer 124 (P124), poloxamer 188 (P188), poloxamer 338 (P338), poloxamer 407 (P407), Tween-20, Tween-80, BRIJ35, tyloxapol, VitE-PEG1000, and Kolliphor EL, or from combinations thereof.
- the surfactantis a block copolymer of ethylene oxide and propylene oxide, more preferably a poloxamer, even more preferably poloxamer selected from the list consisting of poloxamer 124, poloxamer 188, poloxamer 338, and poloxamer 407, or from combinations thereof, most preferably P188.
- the pharmaceutical compositioncomprises a therapeutic agent comprising a nucleic acid, such as RNA, preferably mRNA.
- the pharmaceutical compositionis a lipid nanoparticle (LNP) formulation or lipidoid nanoparticle (Li NP) formulation comprising a plurality of LNPs or LiNPs, each comprising a lipid mix or lipidoid mix and a therapeutic agent, wherein the lipid mix or lipidoid mix comprises, as a component thereof, at least one selected from an ionizable lipid, ionizable lipidoid, and permanently cationic lipid.
- LNPlipid nanoparticle
- Li NPlipidoid nanoparticle
- the pharmaceutical compositionis a lipid nanoparticle (LNP) suspension formulation or lipidoid nanoparticle (LiNP) suspension formulation
- the vehicle solution of the suspension formulationis an aqueous vehicle solution comprising the surfactant.
- the pharmaceutical compositionis a lipid nanoparticle (LNP) suspension formulation or lipidoid nanoparticle (LiNP) suspension formulation comprising an aqueous vehicle solution, and wherein the stabilizing surfactant is added to the vehicle solution, optionally wherein the surfactant is essentially absent from the LNPs or LiNPs.
- LNPlipid nanoparticle
- LiNPlipidoid nanoparticle
- a method of mitigating aggregation of lipid or lipidoid nanoparticles in a lipid nanoparticle (LNP) formulation or lipidoid nanoparticle (LiNP) formulationcomprising adding a stabilizing surfactant to a first LNP or LiNP formulation to form a second LNP or LiNP formulation, optionally before purification, wherein the LNP or LiNP formulation comprises a plurality of LNPs or LiNPs, each comprising a lipid mix or lipidoid mix, wherein the lipid mix or lipidoid mix comprises, as a component thereof, at least one selected from an ionizable lipid, ionizable lipidoid, and permanently cationic lipid, and wherein the stabilizing surfactant is a surfactant as defined in any of items 12 to 16 or a surfactant classified
- the formulationis a lipid nanoparticle (LNP) suspension formulation or lipidoid nanoparticle (LiNP) suspension formulation comprising an aqueous vehicle solution, and wherein the stabilizing surfactant is added to the vehicle solution, optionally wherein the surfactant is essentially absent from the LNPs or LiNPs.
- LNPlipid nanoparticle
- LiNPlipidoid nanoparticle
- lipid nanoparticles or lipidoid nanoparticlescomprise a therapeutic agent, preferably a nucleic acid such as RNA, more preferably mRNA.
- lipid nanoparticles or lipidoid nanoparticlescomprise a nucleic acid, e.g. RNA, and preferably mRNA
- the methodcomprises the steps of: i) first, combining the nucleic acid and at least one selected from an ionizable lipid, ionizable lipidoid, and permanently cationic lipid to form LNPs or LiNPs, ii) second, purifying the LNPs or LiNPs, iii) third, adding the stabilizing surfactant before TFF purification and during TFF purification in the exchange buffer, maintaining the surfactant in a steady concentration, iv) optionally wherein the stabilizing surfactant is added to the LNP or LiNP formulation after step (i).
- the lipid mix or lipidoid mixcomprises, as a component thereof, at least one selected from an ionizable lipid, ionizable lipidoid, and a permanently cationic lipid, and further comprises one or more of the following components (c1 ) to (c6):
- the lipid mix or lipidoid mixcomprises, as a component thereof, at least one selected from an ionizable lipid, ionizable lipidoid, and a permanently cationic lipid, and further comprises the components (c1), (c2) and (c3).
- the lipid mix or lipidoid mixcomprises i) 30 to 65 mol% of at least one selected from the ionizable lipid, the ionizable lipidoid and the permanently cationic lipid, and further comprises one or more of the following components (c1 ) to (c6): ii) 10 to 50 mol% of the lipid having a sterol structure (c1 ), iii) 4 to 50 mol% of the phospholipid (c2), iv) 0.5 to 10 mol% of one of the PEG-conjugated lipid (c3), the polysarcosine-conjugated lipid (c4) and the PASylated lipid (c5), or of any combination thereof, v) 0.5 to 10 mol% of a cationic polymer (c6), such that the sum of the amounts of i) and ii) to v) is 100 mol%,
- the lipid mix or lipidoid mixcomprises i) 30 to 65 mol% of at least one selected from the ionizable lipid, the ionizable lipidoid and the permanently cationic lipid, and further comprises ii) 10 to 50 mol% of the lipid having a sterol structure (c1), iii) 4 to 50 mol% of the phospholipid (c2), iv) 0.5 to 10 mol% of a PEG-conjugated lipid (c3), such that the sum of the amounts of i) and ii) to iv) is 100%.
- the formulationis a lipidoid nanoparticle formulation comprising a plurality of lipidoid nanoparticles each comprising a lipidoid mix, wherein the lipidoid mix comprises an ionizable lipidoid of formula (L-1 ): wherein: a is 1 or 2 and b is an integer of 1 to 4 or a is an integer of 1 to 4 and b is 1 or 2, preferably a is 1 and b is an integer of 2 to 4 or a is an integer of 2 to 4 and b is 1 , p is 1 or 2, m is 1 or 2; n is 0 or 1 and m+n is 2; and
- the lipid nanoparticle formulation or lipidoid nanoparticle formulationpreferably a lipid nanoparticle suspension formulation or lipidoid nanoparticle suspension formulation, according to any one of items 1 to 11 or 40 for use as a medicament.
- the lipid nanoparticle formulation or lipidoid nanoparticle formulationpreferably a lipid nanoparticle suspension formulation or lipidoid nanoparticle suspension formulation, according to any one of items 1 to 11 or 40, for use in the treatment or prevention of a disease, preferably a disease selected from Table A, more preferably a disease selected from viral diseases, ciliopathies, autoimmune diseases, and respiratory tract diseases, even more preferably selected from a lung disease, an airway disease or a nasal disease, more preferably a lung viral disease, lung ciliopathies and a lung autoimmune diseases.
- a diseasepreferably a disease selected from Table A, more preferably a disease selected from viral diseases, ciliopathies, autoimmune diseases, and respiratory tract diseases, even more preferably selected from a lung disease, an airway disease or a nasal disease, more preferably a lung viral disease, lung ciliopathies and a lung autoimmune diseases.
- the lipid nanoparticle formulation or lipidoid nanoparticle formulation for use according to item 43wherein the lung disease or lung viral disease is at least one selected from pneumonia and asthma; the airway disease is at least one selected from bronchitis, viral induced asthma, lung fibrosis and COPD; and/or the nasal disease is at least one selected from rhinitis and sinusitis.
- the lipid nanoparticle formulation or lipidoid nanoparticle formulation according to item 41for use in vaccination or immunization.
- a method of avoiding or reducing side effects in a therapy with LNPs or LiNPs carrying at least one therapeutic agentcomprises the steps: i) determine whether LNPs or LiNPs in a pharmaceutical composition comprising LNPs or LiNPs aggregate when subjected to a mechanical stress or a temperature stress, by determining their aggregation level before and after subjecting said pharmaceutical composition is subjected to said mechanical or temperature stress ii) if the LNP or LiNP show aggregation after the test of step (i), then add to the LNP or LiNP formulation a surfactant to obtain a LNP or LiNP suspension with a final surfactant concentration of 0.01 % w/v and up to 10% w/v, preferably between 0.05% w/w surfactant and 5% surfactant, more preferably between 0.33% surfactant and 2.5% surfactant, more preferably between 0.45% and 1 .5% surfactant, most preferably between 0.5% and 1 .5% surfactant, most preferably about
- the nanoparticlescomprise a) 30 to 65 mol% of at least one selected from the ionizable lipid, an ionizable lipidoid and a permanently cationic lipid (b), and one or more of the following components: ii) 10 to 50 mol% of the lipid having a sterol structure (c1 ), iii) 4 to 50 mol% of the phospholipid (c2), iv) 0.5 to 10 mol% of one of the PEG-conjugated lipid (c3), the polysarcosine-conjugated lipid (c4) and the PASylated lipid (c5), or of any combination thereof,
- a cationic polymer (c6)0.5 to 10 mol% of a cationic polymer (c6), such that the sum of (b) and (c1 ) to (c6) amounts to 100 mol%.
- G 1 and G 2are each independently C1-C12 alkylene or C1-C12 alkenylene;
- G 3is C1-C24 alkylene, C1-C24 alkenylene, C3-C8 cycloalkylene, C3-C8 cycloalkenylene, wherein each of alkylene, alkenylene, cycloalkylene, and cyloalkenylene is optionally substituted;
- R ais H or C1-C12 alkyl wherein the alkyl is optionally substituted
- R 1 and R 2are each independently C6-C24 alkyl or C6-C24 alkenyl, wherein each of alkyl and alkenyl is optionally substituted;
- R 4is C1-C12 alkyl, wherein alkyl is optionally substituted;
- R 5is H or Ci-Ce alkyl, wherein alkyl is optionally substituted; and x is 0, 1 or 2.
- the nanoparticlescomprise an ionizable lipidoid (b) of the following formula (L-1 ), wherein: a is 1 or 2 and b is an integer of 1 to 4 or a is an integer of 1 to 4 and b is 1 or 2, preferably a is 1 and b is an integer of 2 to 4; or a is an integer of 2 to 4 and b is 1 , p is 1 or 2, m is 1 or 2; n is 0 or 1 and m+n is > 2; and
- nanoparticlescomprise, as an ionizable lipid (b), (6Z,9Z,28Z,31Z)-heptatriaconta-6,9,28,31- tetraene-19-yl 4-(dimethylamino)butanoate or a protonated form thereof wherein the nitrogen atom of the compound is protonated.
- nanoparticlescomprise, as an ionizable lipid (b), ((4-hydroxybutyl)azanediyl)bis(hexan-6,1- diyl)bis(2-hexyldecanoate) or a protonated form thereof wherein the nitrogen atom of the compound is protonated, and/or (heptadecan -9-yl 8-((2-hydroxyethyl)(6-oxo-6- (undecyloxy)hexyl)amino)octanoate, or a protonated form thereof wherein the nitrogen atom of the compound is protonated.
- nanoparticlescomprise ((4- hydroxybutyl)azanediyl)bis(hexane-6,1-diyl)bis(2-hexyldecanoate) or the protonated form thereof wherein the nitrogen atom of the compound is protonated, and optionally further comprise one or more of the following components (d1 ) to (d8):
- nanoparticlescomprise heptadecan-9-yl 8- ((2-hydroxyethyl)(6-oxo-6-(undecyloxy)hexyl)amino)octanoate or a protonated form thereof wherein the nitrogen atom of the compound is protonated, and further optionally comprise one or more of the following components (e1) to (e7):
- nanoparticlecomprises DLin-MC3-DMA ((6Z,9Z,28Z,31Z)-heptatriaconta-6,9,28,31-tetraen-19-yl-4-(dimethylamino) butanoate) or a protonated form thereof wherein the nitrogen atom of the compound is protonated, and optionally further comprise one or more of the following components (e1 ) to (e7):
- the inventionprovides in a first aspect a lipid nanoparticle (LNP) formulation or lipidoid nanoparticle (LiNP) formulation comprising a plurality of lipid nanoparticles (LNPs) or lipidoid nanoparticles (LiNPs), each comprising a lipid mix or lipidoid mix, wherein the lipid mix or lipidoid mix comprises, as a component thereof, at least one selected from an ionizable lipid, ionizable lipidoid, and permanently cationic lipid, and wherein the formulation further comprises a surfactant characterized by having a Langmuir surface pressure/area isotherm (also referred to herein as “Langmuir isotherm”) with a maximum surface pressure (IT max) that is equal to or below 4.0 mN/m at a minimum surface area established for a lipid mix or lipidoid mix as comprised by the nanoparticles.
- LNPlipid nanoparticle
- TT maxis equal to or below 3.5 mN/m, more preferably equal to or below 3.5 and equal to or above 0.5 mN/m, even more preferably equal to or below 3.0 mN/m and equal to or above 0.5 mN/m, even more preferably equal to or below 2.5 and equal to or above 1.0 mN/m, most preferably equal to or below 2.0 and equal to or above 1 .0 mN/m.
- the maximum surface pressure (TTmax) of the surfactantcan be determined by recording a Langmuir surface pressure/area isotherm of the surfactant in an aqueous solution thereof, e.g. in deionized water.
- the Langmuir isothermis recorded in a Langmuir trough.
- the recording of the Langmuir isotherm of the surfactantinvolves reducing the available surface area in the Langmuir trough at least until the minimum surface area established for the lipid mix or lipidoid mix is reached.
- the surface pressure which is observed at the minimum surface area when recording the Langmuir isotherm of the surfactantis considered as the maximum surface pressure (TT max ) of the surfactant.
- the minimum surface areacan be established, e.g., by compressing a layer, typically a monolayer, of the lipid mix or lipidoid mix (i.e. the lipid mix or lipidoid mix as comprised by the nanoparticles) in the Langmuir trough until a first phase transition is observed in a surface pressure/area isotherm of the lipid mix or lipidoid mix.
- the layercan be conveniently provided on water, e.g. deionized water.
- the surface area at the start of the phase transitionis regarded as the minimum surface area established for the lipid mix or lipidoid mix.
- the start of the phase transition as referred to herein in a pressure/area (TT-A) isotherm plotcan be objectively determined as the point where the slope of the curve (dir/dA) changes significantly, indicating a shift in the molecular organization of the monolayer.
- a step to establish the minimum surface areacan be carried out, e.g., as a calibration step in the Langmuir trough prior to determining the maximum surface pressure of the surfactant as described above.
- a Langmuir surface pressure/area isotherme.g. the maximum area provided by the Langmuir trough can be conveniently used.
- a monolayer of the lipid mix or lipidoid mix on water in a Langmuir troughcan be provided, e.g., by applying the lipid mix or lipidoid mix to the surface until a signal change in the Langmuir trough detector is generated.
- the Langmuir surface pressure/area isotherm of the surfactantcan be recorded, e.g., in the form of a Langmuir surface pressure/area isotherm cycle (also briefly referred to herein as “isotherm cycle”) which includes a compression phase and an expansion phase.
- the maximum surface pressure (n ma x)can be determined, for example, by recording a single isotherm cycle, but preferably results are obtained by recording multiple isotherm cycles in sequence, such as three isotherm cycles, e.g. with a wait time of 5 seconds or less between cycles, such as 3 seconds.
- n ma xis equal to or below 3.5 mN/m, more preferably equal to or below 3.5 and equal to or above 0.5 mN/m, even more preferably equal to or below 3.0 mN/m and equal to or above 0.5 mN/m, even more preferably equal to or below 2.5 and equal to or above 1 .0 mN/m, most preferably equal to or below 2.0 and equal to or above 1.0 mN/m, directly in this single cycle.
- the surfactantshows a Langmuir isotherm with a maximum surface pressure (Umax) as defined herein above in the last, e.g. the third isotherm cycle.
- a Langmuir surface pressure/area isotherm or isotherm cycle as referred to hereinis recorded at about room temperature, e.g. at 22.1 ⁇ 0.2°C.
- a typical concentration of the surfactant in an aqueous solution thereof for the determination of the maximum surface pressureis 1% w/v (corresponding to 1 g of surfactant in 100 ml of the total volume of the solution including the surfactant, generally measured at about room temperature, e.g. at 22.1 ⁇ 0.2°C).
- the lipid nanoparticle (LNP) formulation or lipidoid nanoparticle (LiNP) formulationis a suspension formulation comprising a liquid vehicle solution, wherein the LNPs or LiNPs can be dispersed.
- the vehicle solutionis preferably an aqueous vehicle solution. It is preferred that, in such a suspension formulation, the surfactant is comprised in the vehicle solution.
- the surfactantmay favorably act as a stabilizing agent that mitigates aggregation of the LNPs or LiNP or a subpopulation thereof which may be caused by shaking or by shear stress of the suspension during production, purification, handling or transport, preferably during production or purification, more preferably during purification, most preferably during tangential flow filtration (TFF) purification.
- a stabilizing agentthat mitigates aggregation of the LNPs or LiNP or a subpopulation thereof which may be caused by shaking or by shear stress of the suspension during production, purification, handling or transport, preferably during production or purification, more preferably during purification, most preferably during tangential flow filtration (TFF) purification.
- the lipid nanoparticle (LNP) formulation or lipidoid nanoparticle (LiNP) formulationis preferably a formulation, e.g. a suspension formulation as discussed above, wherein the LNPs or LiNPs comprise the lipid mix or lipidoid mix and a therapeutic agent, i.e. a pharmaceutical formulation.
- the first aspectalso encompasses the lipid nanoparticle (LNP) formulation or lipidoid nanoparticle (LiNP) formulation comprising the plurality of lipid nanoparticles (LNPs) or lipidoid nanoparticles (LiNPs), each comprising the lipid mix or lipidoid mix and a therapeutic agent, for use as a medicament.
- a preferred therapeutic agentis a nucleic acid, such as RNA, and particularly preferred is mRNA. Details regarding therapeutic agents which may be contained in the LNPs or LiNPs shall likewise be discussed herein below.
- the terms “pharmaceutical formulation” and “pharmaceutical composition”may be used herein as equivalents.
- the lipid nanoparticle (LNP) formulation or lipidoid nanoparticle (LiNP) formulation in accordance with the first aspectis particularly preferably a lipid nanoparticle (LNP) suspension formulation or lipidoid nanoparticle (LiNP) suspension formulation comprising a plurality of LNPs or LiNPs, each comprising a lipid mix or lipidoid mix and a therapeutic agent, wherein the lipid mix or lipidoid mix comprises, as a component thereof, at least one selected from an ionizable lipid, ionizable lipidoid, and permanently cationic lipid, and wherein the vehicle solution of the suspension formulation is an aqueous vehicle solution comprising a surfactant characterized by having a Langmuir surface pressure/area isotherm with a maximum surface pressure (n ma x) that is equal to or below 4.0 mN/m at a minimum surface area established for the lipid mix or lipidoid mix as comprised by the nano
- the inventionprovides a lipid nanoparticle (LNP) formulation or lipidoid nanoparticle (LiNP) formulation comprising a plurality of lipid nanoparticles (LNPs) or lipidoid nanoparticles (LiNPs), each comprising a lipid mix or lipidoid mix, wherein the lipid mix or lipidoid mix comprises, as a component thereof, at least one selected from an ionizable lipid, ionizable lipidoid, and permanently cationic lipid, wherein the formulation comprises as a stabilizing agent a surfactant, and wherein a representative sample comprising an aqueous solution of the surfactant carrying on its surface a lipid mix or lipidoid mix as comprised by the LNPs or LiNPs is characterized by having a Langmuir isotherm An that is equal to or below 0.60, preferably equal to or below 0.45, at each area point during a Langmuir surface pressure
- a representative sample used for determining the Langmuir isotherm An valuescomprises an aqueous solution of the surfactant, e.g. a solution in deionized water, carrying on its surface a lipid mix or lipidoid mix as comprised by the LNPs or LiNPs, typically a monolayer of the lipid mix or lipidoid mix.
- the lipid mix or lipidoid mixgenerally has the same composition as the lipid mix or lipidoid mix comprised by the nanoparticles, i.e. it comprises the same lipids and/or lipidoids in the same proportions.
- a Langmuir surface pressure/area isotherm cyclecomprising a compression phase and an expansion phase can be recorded in a Langmuir trough.
- the Langmuir isotherm ATT valuesare calculated for each area point as explained above.
- the Langmuir isotherm An valuesare equal to or below 0.60, preferably equal to or below 0.45, at each area point during a Langmuir surface pressure/area isotherm cycle.
- the Langmuir isotherm ATT values calculated as disclosed aboveare usually above 0.
- the minimum surface areacan be established, e.g., by compressing a layer, typically a monolayer, of the lipid mix or lipidoid mix (i.e.
- the layercan be conveniently provided on water, e.g. on deionized water.
- the surface area at the start of the phase transitionis regarded as the minimum surface area established for the lipid mix or lipidoid mix.
- the start of the phase transition as referred to herein in a pressure/area (TT-A) isotherm plotcan be objectively determined as the point where the slope of the curve (dn/dA) changes significantly.
- a step to establish the minimum surface areacan be carried out, e.g., as a calibration step in the Langmuir trough prior to recording a Langmuir surface pressure/area isotherm cycle on a representative sample as described above.
- the surface area before the start of the compression in a compression phasee.g., the maximum area provided by the Langmuir trough, can be conveniently used.
- a monolayer of the lipid mix or lipidoid mix on water or on an aqueous solution of the surfactant in a Langmuir troughcan be provided, e.g., by applying the lipid mix or lipidoid mix to the surface until a signal change in the Langmuir trough detector is generated.
- a Langmuir surface pressure/area isotherm or isotherm cycle as referred to hereinis recorded at about room temperature, e.g. at 22.1 ⁇ 0.2°C.
- a typical concentration of the surfactant in an aqueous solution thereof in the representative sampleis 1 % w/v (corresponding to 1 g of surfactant in 100 ml of the total volume of the solution including the surfactant, generally measured at about room temperature, e.g. at 22.1 ⁇ 0.2°C).
- a calibration stepwhich may be used to establish the minimum surface area, and the recording of the Langmuir pressure/area isotherm for the calculation of Langmuir isotherm An values are further illustrated in the “Examples” section below.
- the lipid nanoparticle (LNP) formulation or lipidoid nanoparticle (LiNP) formulationis a suspension formulation comprising a liquid vehicle solution, wherein the LNPs or LiNPs can be dispersed.
- the vehicle solutionis preferably an aqueous vehicle solution. It is preferred that, in such a suspension formulation, the surfactant is comprised in the vehicle solution.
- the surfactantmay favorably act as a stabilizing agent that mitigates aggregation of the LNPs or LiNP or a subpopulation thereof which may be caused by shaking or by shear stress of the suspension during production, purification, handling or transport, preferably during production or purification, more preferably during purification, most preferably during tangential flow filtration (TFF) purification.
- a stabilizing agentthat mitigates aggregation of the LNPs or LiNP or a subpopulation thereof which may be caused by shaking or by shear stress of the suspension during production, purification, handling or transport, preferably during production or purification, more preferably during purification, most preferably during tangential flow filtration (TFF) purification.
- the lipid nanoparticle (LNP) formulation or lipidoid nanoparticle (LiNP) formulationis preferably a formulation, e.g. a suspension formulation as discussed above, wherein the LNPs or LiNPs comprise the lipid mix or lipidoid mix and a therapeutic agent, i.e. a pharmaceutical formulation.
- the second aspectalso encompasses the lipid nanoparticle (LNP) formulation or lipidoid nanoparticle (LiNP) formulation comprising the plurality of lipid nanoparticles (LNPs) or lipidoid nanoparticles (LiNPs), each comprising the lipid mix or lipidoid mix and a therapeutic agent, for use as a medicament.
- a preferred therapeutic agentis a nucleic acid, such as RNA, and particularly preferred is mRNA. Details regarding therapeutic agents which may be contained in the LNPs or LiNPs shall likewise be discussed herein below.
- the terms “pharmaceutical formulation” and “pharmaceutical composition”may be used herein as equivalents.
- the lipid nanoparticle (LNP) formulation or lipidoid nanoparticle (LiNP) formulation in accordance with the second aspectis particularly preferably a lipid nanoparticle (LNP) suspension formulation or lipidoid nanoparticle (LiNP) suspension formulation comprising a plurality of LNPs or LiNPs, each comprising a lipid mix or lipidoid mix and a therapeutic agent, wherein the lipid mix or lipidoid mix comprises, as a component thereof, at least one selected from an ionizable lipid, ionizable lipidoid, and permanently cationic lipid, and wherein the vehicle solution of the suspension formulation is an aqueous vehicle solution comprising a surfactant as a stabilizing agent, and wherein a representative sample comprising an aqueous solution of the surfactant carrying on its surface a lipid mix or lipidoid mix as comprised by the LNPs or LiNPs is characterized by having a Langmuir is
- the inventionprovides a surfactant for use in a pharmaceutical composition, the surfactant being characterized by having a Langmuir surface pressure/area isotherm with a maximum surface pressure (IT max) that is equal to or below 4.0 mN/m at a minimum surface area established for said pharmaceutical composition.
- the aspectprovides the use of the surfactant as a component, e.g. as a stabilizing agent, in a pharmaceutical composition.
- Umaxalso in the context of this aspect is equal to or below 3.5 mN/m, more preferably equal to or below 3.5 and equal to or above 0.5 mN/m, even more preferably equal to or below 3.0 mN/m and equal to or above 0.5 mN/m, even more preferably equal to or below 2.5 and equal to or above 1 .0 mN/m, most preferably equal to or below 2.0 and equal to or above 1 .0 mN/m.
- the pharmaceutical compositioncan be a pharmaceutical composition comprising a lipid or a lipidoid, such as a lipid nanoparticle (LNP) formulation or lipidoid nanoparticle (LiNP) formulation comprising a plurality of lipid nanoparticles (LNPs) or lipidoid nanoparticles (LiNPs).
- a lipid or a lipidoidsuch as a lipid nanoparticle (LNP) formulation or lipidoid nanoparticle (LiNP) formulation comprising a plurality of lipid nanoparticles (LNPs) or lipidoid nanoparticles (LiNPs).
- the pharmaceutical compositionis a lipid nanoparticle (LNP) formulation or lipidoid nanoparticle (LiNP) formulation comprising a plurality of lipid nanoparticles (LNPs) or lipidoid nanoparticles (LiNPs), each comprising a lipid mix or lipidoid mix, wherein the lipid mix or lipidoid mix comprises, as a component thereof, at least one selected from an ionizable lipid, ionizable lipidoid, and permanently cationic lipid.
- LNPlipid nanoparticle
- LiNPslipidoid nanoparticle
- the pharmaceutical formulationis a lipid nanoparticle (LNP) formulation or lipidoid nanoparticle (LiNP) formulation
- the formulationis a suspension formulation comprising a liquid vehicle solution, wherein the LNPs or LiNPs can be dispersed.
- the vehicle solutionis preferably an aqueous vehicle solution. It is preferred that, in such a suspension formulation, the surfactant as defined in this aspect is comprised in the vehicle solution.
- the surfactantmay favorably act as a stabilizing agent that mitigates aggregation of the LNPs or LiNP or a subpopulation thereof which may be caused by shaking or by shear stress of the suspension during production, purification, handling or transport, preferably during production or purification, more preferably during purification, most preferably during tangential flow filtration (TFF) purification.
- a stabilizing agentthat mitigates aggregation of the LNPs or LiNP or a subpopulation thereof which may be caused by shaking or by shear stress of the suspension during production, purification, handling or transport, preferably during production or purification, more preferably during purification, most preferably during tangential flow filtration (TFF) purification.
- the pharmaceutical compositioncomprises a therapeutic agent, for example a nucleic acid.
- a preferred therapeutic agentis RNA, and particularly preferred is mRNA. Details regarding preferred types of therapeutic agents shall be discussed herein below.
- the pharmaceutical formulationis a lipid nanoparticle (LNP) formulation or lipidoid nanoparticle (LiNP) formulation, e.g. a suspension formulation, the therapeutic agent is typically comprised in the LNPs or LiNPs.
- the surfactant for use in accordance with the third aspectis particularly preferably a surfactant for use in a pharmaceutical composition in the form of a lipid nanoparticle (LNP) suspension formulation or lipidoid nanoparticle (LiNP) suspension formulation comprising a plurality of LNPs or LiNPs, each comprising a lipid mix or lipidoid mix and a therapeutic agent, wherein the lipid mix or lipidoid mix comprises, as a component thereof, at least one selected from an ionizable lipid, ionizable lipidoid, and permanently cationic lipid, wherein the vehicle solution of the suspension formulation is an aqueous vehicle solution and comprising the surfactant which is characterized by having a Langmuir surface pressure/area isotherm with a maximum surface pressure (n ma x) that is equal to or below 4.0 mN/m at a minimum surface area established for said pharmaceutical composition.
- LNPlipid nanoparticle
- LiNPlipidoid nanoparticle
- the aspectprovides the use of the surfactant as a component, e.g. as a stabilizing agent, in such a pharmaceutical composition.
- the maximum surface pressure (n ma x) of the surfactantcan be determined by recording a Langmuir surface pressure/area isotherm of the surfactant in an aqueous solution thereof, e.g. in deionized water.
- the Langmuir isothermis recorded in a Langmuir trough.
- the recording of the Langmuir isotherm of the surfactantinvolves reducing the available surface area in the Langmuir trough at least until the minimum surface area established for the pharmaceutical composition is reached.
- the surface pressure which is observed at the minimum surface area when recording the Langmuir isotherm of the surfactantis considered as the maximum surface pressure (TT max ) of the surfactant.
- the minimum surface areacan be established for the pharmaceutical composition, e.g., by compressing a layer, typically a monolayer, of the combination of more than one lipid and/or lipidoid as comprised by the pharmaceutical composition in a Langmuir trough until a first phase transition is observed in a surface pressure/area isotherm of the combination.
- the layercan be conveniently provided on water, e.g. deionized water.
- a Langmuir surface pressure/area isotherme.g., the maximum area provided by the Langmuir trough can be conveniently used.
- the inventionprovides a method for classifying a surfactant as suitable or not suitable for use as a stabilizing agent for a pharmaceutical composition, the method comprising:
- the methodcan be used for classifying a surfactant as suitable or not suitable for use as a stabilizing agent for a pharmaceutical composition comprising a lipid or a lipidoid, such as a lipid nanoparticle (LNP) formulation or lipidoid nanoparticle (LiNP) formulation comprising a plurality of lipid nanoparticles (LNPs) or lipidoid nanoparticles (LiNPs).
- a surfactantas suitable or not suitable for use as a stabilizing agent for a pharmaceutical composition
- a lipid or a lipidoidsuch as a lipid nanoparticle (LNP) formulation or lipidoid nanoparticle (LiNP) formulation comprising a plurality of lipid nanoparticles (LNPs) or lipidoid nanoparticles (LiNPs).
- LNPlipid nanoparticle
- LiNPlipidoid nanoparticle
- the method according to this aspectis a method for classifying a surfactant as suitable or not suitable for use as a stabilizing agent for a pharmaceutical composition
- the pharmaceutical compositionis in the form of a lipid nanoparticle (LNP) suspension formulation or lipidoid nanoparticle (LiNP) suspension formulation comprising a plurality of LNPs or LiNPs, each comprising a lipid mix or lipidoid mix and a therapeutic agent, wherein the lipid mix or lipidoid mix comprises, as a component thereof at least one selected from an ionizable lipid, ionizable lipidoid, and a permanently cationic lipid, and wherein the vehicle solution of the suspension formulation is an aqueous vehicle solution.
- LNPlipid nanoparticle
- LiNPlipidoid nanoparticle
- a surfactantis provided in an aqueous solution, e.g. as a solution in deionized water, at a concentration (C). Details regarding preferred types of surfactants subjected to the method, such as a poloxamer, shall be discussed herein below.
- the concentration of the surfactantis not particularly restricted, exemplary concentrations may be in the range of 0.1 to 10.0 % w/v (i.e. indicated as the weight of surfactant in g in 100 ml of the total volume of the solution, generally measured at about room temperature, e.g. at 22.1 ⁇ 0.2°C)., preferably in the range of 0.5 to 5.0 % w/v. Particularly preferred is a concentration of 1% w/v.
- a Langmuir surface pressure/area isotherm of the surfactant in the solution as provided in step (a)is recorded to determine a maximum surface pressure Umax of the Langmuir surface pressure/area isotherm at a predetermined minimum surface area.
- Umaxof the Langmuir surface pressure/area isotherm at a predetermined minimum surface area.
- the recording of the Langmuir isotherm of the surfactantinvolves reducing the available surface area in the Langmuir trough at least until the predetermined minimum surface area is reached.
- the surface pressure which is observed at the minimum surface area when recording the Langmuir isotherm of the surfactantis considered as the maximum surface pressure (TT ma x) of the surfactant.
- the methodcan be advantageously used, e.g., to differentiate between variants of a single surfactant, each variant nominally identified by the same name and complying with the same quality standards for medicines, to classify each variant as suitable or not suitable for use as a stabilizing agent for a pharmaceutical composition.
- a minimum surface areacan be established, e.g., by compressing a layer, typically a monolayer, of the lipid or lipidoid in a Langmuir trough until a first phase transition is observed in a surface pressure/area isotherm of the lipid or lipidoid.
- the layercan be conveniently provided on water, e.g. deionized water. If the pharmaceutical composition, e.g.
- the minimum surface areacan be established for the pharmaceutical composition, e.g., by compressing a layer, typically a monolayer, comprising at least one lipid or lipidoid of the combination in a Langmuir trough until a first phase transition is observed in a surface pressure/area isotherm of the lipid or lipidoid.
- the layercomprises at least the lipid or lipidoid which accounts for the largest proportion of the components in the combination of more than one lipid and/or lipidoid, in terms of the weight percentage based on the total weight of the combination as 100%.
- the minimum surface areacan be established for the pharmaceutical composition, e.g., by compressing a layer, typically a monolayer, comprising the same combination of more than one lipid and/or lipidoid in the same proportions in a Langmuir trough until a first phase transition is observed in a surface pressure/area isotherm of the combination.
- the layercan be conveniently provided on water, e.g. deionized water.
- a minimum surface areacan be established, e.g., by compressing a layer, typically a monolayer, comprising at least one lipid or lipidoid contained in the lipid mix or lipidoid mix (i.e. the lipid mix or lipidoid mix as comprised by the nanoparticles) in a Langmuir trough until a first phase transition is observed in a surface pressure/area isotherm of the lipid or lipidoid.
- the layercomprises at least the lipid or lipidoid which accounts for the largest proportion of the components in the lipid mix or lipidoid mix, in terms of the weight percentage based on the total weight of the lipid mix or lipidoid mix as 100%.
- a minimum surface areacan be established by compressing a layer, typically a monolayer, of the lipid mix or lipidoid mix (i.e.
- the layercan be conveniently provided on water, e.g. deionized water.
- the surface area at the start of the phase transitioncan then be relied on as the predetermined minimum surface area in step (b) of the method.
- the start of the phase transition as referred to herein in a pressure/area (TT-A) isotherm plotcan be objectively determined as the point where the slope of the curve (dn/dA) changes significantly.
- a step to establish the minimum surface areacan be carried out, e.g., as a calibration step in the Langmuir trough prior to determining the maximum surface pressure of the surfactant as described above.
- the surface area before the start of the compression in a compression phasee.g., the maximum area provided by the Langmuir trough, can be conveniently used.
- a monolayer of a lipid and/or lipidoid on water, or of a lipid mix or lipidoid mix on water, in a Langmuir troughcan be provided, e.g., by applying the lipid and/or lipidoid or the lipid mix or lipidoid mix to the surface until a signal change in the Langmuir trough detector is generated.
- the Langmuir surface pressure/area isotherm of the surfactant in the solution in step (b)can be recorded, e.g., in the form of a Langmuir surface pressure/area isotherm cycle including a compression phase and an expansion phase.
- the maximum surface pressure (n m ax)can be determined, for example, by recording a single isotherm cycle, but preferably results are obtained by recording multiple isotherm cycles in sequence, such as three isotherm cycles, e.g. with a wait time of 5 seconds or less between cycles, such as 3 seconds. If a single isotherm cycle is recorded, it is preferred that Umax is equal to or below 3.5 mN/m.
- the surfactantshows a Langmuir isotherm with a maximum surface pressure (TT ma x) equal to or below the threshold value in the last, e.g. the third isotherm cycle.
- TT ma xmaximum surface pressure
- a Langmuir surface pressure/area isotherm or isotherm cycle as referred to hereincan be recorded at about room temperature, e.g. at 22.1 ⁇ 0.2°C.
- step (c)the maximum surface pressure is subsequently compared to a threshold value. Based on this comparison, a decision can be made on whether the surfactant is suitable for use as a stabilizer.
- a threshold value of the maximum surface pressure TT ma x for comparison and classificationcan be conveniently determined e.g. based on surface pressure data prepared in preliminary tests for a stable composition comprising a surfactant.
- TTmaxcan be equal to or below 4.0 mN/m, or equal to or below 3.5 mN/m.
- a methodfor the preparation of a pharmaceutical composition, said method comprising classifying a surfactant as suitable or not suitable for use as a stabilizing agent for a pharmaceutical composition in accordance with the method as described above, and, if the surfactant is classified as being suitable for use as a stabilizing agent for a pharmaceutical composition, incorporating the surfactant into the pharmaceutical composition, e.g. into a vehicle solution of a pharmaceutical composition which is in the form of a nanoparticle suspension.
- the inventionprovides a method for classifying a surfactant as suitable or not suitable for use as a stabilizing agent for a pharmaceutical composition comprising a lipid or lipidoid, optionally during purification of said composition, preferably during tangential flow filtration of said composition, wherein the method comprises the steps:
- a method for the preparation of a pharmaceutical compositioncomprising classifying a surfactant as suitable or not suitable for use as a stabilizing agent for a pharmaceutical composition comprising a lipid or lipidoid in accordance with the method as described above, and, if the surfactant is classified as being suitable for use as a stabilizing agent, incorporating the surfactant into the pharmaceutical composition.
- the methodcan be used for classifying a surfactant as suitable or not suitable for use as a stabilizing agent in a pharmaceutical composition comprising a lipid nanoparticle (LNP) formulation or lipidoid nanoparticle (LiNP) formulation comprising a plurality of lipid nanoparticles (LNPs) or lipidoid nanoparticles (LiNPs).
- a pharmaceutical compositioncomprising a lipid nanoparticle (LNP) formulation or lipidoid nanoparticle (LiNP) formulation comprising a plurality of lipid nanoparticles (LNPs) or lipidoid nanoparticles (LiNPs).
- the pharmaceutical compositionis a lipid nanoparticle (LNP) formulation or lipidoid nanoparticle (LiNP) formulation comprising a plurality of lipid nanoparticles (LNPs) or lipidoid nanoparticles (LiNPs), each comprising a lipid mix or lipidoid mix, wherein the lipid mix or lipidoid mix comprises, as a component thereof, at least one selected from an ionizable lipid, ionizable lipidoid, and permanently cationic lipid.
- LNPlipid nanoparticle
- LiNPslipidoid nanoparticle
- the pharmaceutical formulationis a lipid nanoparticle (LNP) formulation or lipidoid nanoparticle (LiNP) formulation
- the formulationis a suspension formulation comprising a liquid vehicle solution, wherein the LNPs or LiNPs can be dispersed.
- the vehicle solutionis preferably an aqueous vehicle solution. It is preferred that, in such a suspension formulation, a surfactant classified as being suitable for use as a stabilizing agent is incorporated into the vehicle solution.
- the surfactantmay favorably act as a stabilizing agent that mitigates aggregation of the LNPs or LiNP or a subpopulation thereof which may be caused by shaking or by shear stress of the suspension during production, purification, handling or transport, preferably during production or purification, more preferably during purification, most preferably during tangential flow filtration (TFF) purification.
- a stabilizing agentthat mitigates aggregation of the LNPs or LiNP or a subpopulation thereof which may be caused by shaking or by shear stress of the suspension during production, purification, handling or transport, preferably during production or purification, more preferably during purification, most preferably during tangential flow filtration (TFF) purification.
- the pharmaceutical compositioncomprises a therapeutic agent, for example a nucleic acid.
- a preferred therapeutic agentis RNA, and particularly preferred is mRNA. Details regarding preferred types of therapeutic agents shall be discussed herein below.
- the pharmaceutical formulationis a lipid nanoparticle (LNP) formulation or lipidoid nanoparticle (LiNP) formulation, e.g. a suspension formulation, the therapeutic agent is typically comprised in the LNPs or LiNPs.
- the method according to this aspectis a method for classifying a surfactant as suitable or not suitable for use as a stabilizing agent for a pharmaceutical composition
- the pharmaceutical compositionis in the form of a lipid nanoparticle (LNP) suspension formulation or lipidoid nanoparticle (LiNP) suspension formulation comprising a plurality of LNPs or LiNPs, each comprising a lipid mix or lipidoid mix and a therapeutic agent, wherein each of the LNPs or LiNPs comprises, as a component of the lipid mix or lipidoid mix, at least one selected from an ionizable lipid, ionizable lipidoid, and permanently cationic lipid, and wherein the vehicle solution of the suspension formulation is an aqueous vehicle solution.
- LNPlipid nanoparticle
- LiNPlipidoid nanoparticle
- a surfactantis provided in an aqueous solution at a concentration (C). Details regarding preferred types of surfactants subjected to the method, such as a poloxamer, shall be discussed herein below.
- the concentration of the surfactantis not particularly restricted, exemplary concentrations may be in the range of 0.1 to 10.0 % w/v (i.e. indicated as the weight of surfactant in g in 100 ml of the combined volumes of the surfactant and aqueous solvent, generally measured at about room temperature, e.g. at 22.1 ⁇ 0.2°C), preferably in the range of 0.5 to 5.0 % w/v. Particularly preferred is a concentration of 1% w/v.
- a Langmuir pressure/area isotherm cycle including a compression phase and an expansion phaseis recorded between a maximum surface area and a minimum surface area.
- the isotherm cycleis recorded on a sample comprising the surfactant in the aqueous solution as provided in step (a), and carrying on its surface a lipid or lipidoid as comprised by the pharmaceutical composition.
- the samplecarries a monolayer of the lipid or lipidoid on its surface.
- recording of the isotherm cyclecan be accomplished using a Langmuir trough containing the sample. If the pharmaceutical composition, e.g.
- the isotherm cyclecan be recorded, e.g., on a sample carrying a layer, typically a monolayer, comprising at least one lipid or lipidoid of the combination.
- the layercomprises at least the lipid or lipidoid which accounts for the largest proportion of the components in the combination of more than one lipid and/or lipidoid in the pharmaceutical composition, in terms of the weight percentage based on the total weight of the combination as 100%.
- the isotherm cyclecan be recorded on a sample carrying a layer, typically a monolayer, comprising the same combination of more than one lipid and/or lipidoid in the same proportions as comprised in the pharmaceutical composition.
- the pharmaceutical compositionis a lipid nanoparticle (LNP) formulation or lipidoid nanoparticle (LiNP) formulation comprising a plurality of lipid nanoparticles (LNPs) or lipidoid nanoparticles (LiNPs), each comprising a lipid mix or lipidoid mix, wherein the lipid mix or lipidoid mix comprises, as a component thereof, at least one selected from an ionizable lipid, ionizable lipidoid, and permanently cationic lipid.
- LNPlipid nanoparticle
- LiNPslipidoid nanoparticle
- the isotherm cyclecan be recorded, e.g., on a sample carrying a layer, typically a monolayer, comprising at least one lipid or lipidoid contained in the lipid mix or lipidoid mix (i.e. the lipid mix or lipidoid mix as comprised by the nanoparticles).
- the layercomprises at least the lipid or lipidoid which accounts for the largest proportion of the components in the lipid mix or lipidoid mix, in terms of the weight percentage based on the total weight of the lipid mix or lipidoid mix as 100%.
- the isotherm cycleis recorded on a sample carrying a layer, typically a monolayer, of the lipid mix or lipidoid mix (i.e. the lipid mix or lipidoid mix as comprised by the nanoparticles).
- the minimum surface area to be adopted for recording the isotherm cycleis generally determined by the lipid or lipidoid, or by the combination of more than one lipid/lipidoid contained in the sample used for recording the isotherm cycle.
- the minimum surface area for recording the isotherm cycleis preferably the surface area at which the start of a phase transition can be observed when a layer, typically a monolayer, of the lipid or lipidoid or of the combination of more than one lipid/lipidoid is compressed in a Langmuir trough.
- a step to establish the minimum surface areacan be carried out, e.g., as a calibration step in the Langmuir trough prior to recording the isotherm cycle in step (b).
- a minimum surface areacan be established by compressing a layer, typically a monolayer, of the lipid or lipidoid or of the combination of more than one lipid/lipidoid as it is contained in the sample for recording the isotherm cycle in step (b) in the Langmuir trough until a first phase transition is observed in a surface pressure/area isotherm.
- the layercan be conveniently provided on water, e.g. deionized water.
- the surface area at the start of the phase transitioncan then be relied on as the minimum surface area for recording the isotherm cycle in step (b) of the method.
- the start of the phase transition as referred to herein in a pressure/area (IT -A) isotherm plotcan be objectively determined as the point where the slope of the curve (dn/dA) changes significantly.
- the surface area before the start of the compression in a compression phasee.g., the maximum area provided by the Langmuir trough, can be conveniently used.
- a monolayer of a lipid and/or lipidoid on water, or of a lipid mix or lipidoid mix on water, in a Langmuir troughcan be provided, e.g., by applying the lipid and/or lipidoid or the lipid mix or lipidoid mix to the surface until a signal change in the Langmuir trough detector is generated.
- step (c)it is sufficient if a single isotherm cycle is recorded, and the Langmuir isotherm An values are preferably calculated in step (c) for this isotherm cycle.
- Multiple isotherm cyclescan be recorded in sequence, such as three isotherm cycles, e.g. with a wait time of 5 seconds or less between cycles, such as 3 seconds.
- TT values of the first cyclefulfill the requirements set forth above, the same typically applies also for the subsequent cycles, such that it is preferred to rely on the measurement results of the first cycle for the calculation of the An values even if multiple cycles are recorded.
- a Langmuir surface pressure/area isotherm or isotherm cycle as referred to hereincan be recorded at about room temperature, e.g. at 22.1 ⁇ 0.2°C.
- step (c)a Langmuir isotherm An is calculated for each area point of the Langmuir pressure/area isotherm cycle.
- a method for the preparation of a pharmaceutical compositioncomprising classifying a surfactant as suitable or not suitable for use as a stabilizing agent for a pharmaceutical composition in accordance with the method as described above, and, if the surfactant is classified as being suitable for use as a stabilizing agent, incorporating the surfactant into the pharmaceutical composition, e.g. into a vehicle solution of a pharmaceutical composition which is in the form of a nanoparticle suspension.
- the inventionprovides a method of mitigating or avoiding clogging or fouling of a filtration system during purification of a pharmaceutical composition in the form of a lipid nanoparticle formulation (LNP) or lipidoid nanoparticle formulation (LiNP), the method comprising adding a stabilizing surfactant to a first LNP or LiNP formulation to form a second LNP or LiNP formulation, optionally before purification, wherein the LNP or LiNP formulation comprises a plurality of LNPs or LiNPs, each comprising a lipid mix or lipidoid mix and a therapeutic agent, wherein the lipid mix or lipidoid mix comprises, as a component thereof, at least one selected from an ionizable lipid, ionizable lipidoid, and permanently cationic lipid, and wherein the stabilizing surfactant is a surfactant according to the invention, e.g. a surfactant as discussed in the third aspect above, or wherein the stabilizing surfact
- the LNP formulation or LiNP formulationcomprises a therapeutic agent, for example a nucleic acid.
- a preferred therapeutic agentis RNA, and particularly preferred is mRNA. Details regarding preferred types of therapeutic agents shall be discussed herein below.
- the LNP formulation or LiNP formulationis a suspension formulation comprising a liquid vehicle solution, wherein the LNPs or LiNPs can be dispersed.
- the vehicle solutionis preferably an aqueous vehicle solution.
- the stabilizing surfactantis added in the context of the above method to the vehicle solution.
- the LNPs or LiNPs dispersed in the liquid vehicleare essentially free of the stabilizing surfactant, e.g. the surfactant is essentially not incorporated into the LNPs or LiNPs.
- the purification of the pharmaceutical compositionincludes tangential flow filtration (TFF) purification.
- the inventionprovides a method of mitigating aggregation of lipid or lipidoid nanoparticles in a lipid nanoparticle (LNP) formulation or lipidoid nanoparticle (LiNP) formulation, the method comprising adding a stabilizing surfactant to a first LNP or LiNP formulation to form a second LNP or LiNP formulation, optionally before purification, wherein the LNP or LiNP formulation comprises a plurality of LNPs or LiNPs, each comprising a lipid mix or lipidoid mix, wherein the lipid mix or lipidoid mix comprises, as a component thereof, at least one selected from an ionizable lipid, ionizable lipidoid, and permanently cationic lipid, and wherein the stabilizing surfactant is a surfactant in accordance with invention, e.g. a surfactant as discussed in the third aspect above, or wherein the stabilizing surfactant is a surfactant classified as being suitable
- the methodis a method of mitigating aggregation of lipid or lipidoid nanoparticles in a pharmaceutical formulation in the form of a lipid nanoparticle (LNP) formulation or lipidoid nanoparticle (LiNP) formulation comprising a plurality of LNPs or LiNPs, each comprising a lipid mix or lipidoid mix and a therapeutic agent, for example a nucleic acid.
- LNPlipid nanoparticle
- LiNPlipidoid nanoparticle
- a therapeutic agentfor example a nucleic acid.
- a preferred therapeutic agentis RNA, and particularly preferred is mRNA. Details regarding preferred types of therapeutic agents shall be discussed herein below.
- the LNP formulation or LiNP formulationis a suspension formulation comprising a liquid vehicle solution, wherein the LNPs or LiNPs can be dispersed.
- the vehicle solutionis preferably an aqueous vehicle solution.
- the stabilizing surfactantis added in the context of the above method to the vehicle solution.
- the LNPs or LiNPs dispersed in the liquid vehicleare essentially free of the stabilizing surfactant, e.g. the surfactant is essentially not incorporated into the LNPs or LiNPs.
- the methodis a method of mitigating aggregation of lipid or lipidoid nanoparticles in a pharmaceutical formulation in the form of a lipid nanoparticle (LNP) suspension formulation or lipidoid nanoparticle (LiNP) suspension formulation comprising a plurality of LNPs or LiNPs, each comprising a lipid mix or lipidoid mix and a therapeutic agent, wherein the lipid mix or lipidoid mix comprises, as a component thereof, at least one selected from an ionizable lipid, ionizable lipidoid, and permanently cationic lipid.
- LNPlipid nanoparticle
- LiNPlipidoid nanoparticle
- the lipid nanoparticles or lipidoid nanoparticlesmay comprise a nucleic acid, e.g. RNA, and preferably mRNA, as a therapeutic agent, and the method in accordance with this aspect may comprise, e.g., the following steps: i) first, combining the nucleic acid and at least one selected from an ionizable lipid, ionizable lipidoid, and permanently cationic lipid to form LNPs or LiNPs, ii) second, purifying the LNPs or LiNPs iii) third, adding the stabilizing surfactant before TFF purification and/or during TFF purification in an exchange buffer, maintaining the surfactant in a steady concentration, iv) optionally wherein the stabilizing surfactant is added to the LNP or LiNP formulation after step (i).
- the method in accordance with this aspectmay alternatively comprise, e.g., the following steps: i) generating an LNP or LiNP preparation by mixing of at least one selected from a permanently cationic lipid, an ionizable lipid and an ionizable lipidoid dissolved in an organic phase with a therapeutic agent dissolved in an aqueous solution, ii) diluting the LNP or LiNP preparation by dilution with a first solution, iii) concentrating the LNP or LiNP preparation by buffer exchange using ultra/diafiltration via TFF wherein a second solution is used for the ultra/diafiltration, iv) obtaining a LNP or LiNP suspension in an aqueous vehicle solution, wherein the first solution comprises between about 0.01% w/v and 10% of the stabilizing surfactant, preferably between about 0.01% w/v surfactant and 5% w/v surfactant, more preferably between about 0.01 % w/v sur
- the inventionprovides a lipid nanoparticle (LNP) formulation or lipidoid nanoparticle (LiNP) formulation obtained by the method in accordance with the fourth to seventh aspect.
- this aspectprovides a pharmaceutical composition in the form of a lipid nanoparticle (LNP) formulation or lipidoid nanoparticle (LiNP) formulation, said composition comprising a surfactant which has been classified as being suitable for use as a stabilizing agent for a pharmaceutical composition in accordance with the method of the fourth aspect.
- the eighth aspectprovides pharmaceutical composition in the form of a lipid nanoparticle (LNP) formulation or lipidoid nanoparticle (LiNP) formulation, said composition comprising a surfactant which has been classified as being suitable for use as a stabilizing agent for a pharmaceutical composition comprising a lipid or lipidoid composition in accordance with the method of the fifth aspect.
- the eighth aspectprovides a pharmaceutical composition in the form of lipid nanoparticle (LNP) formulation or lipidoid nanoparticle (LiNP) formulation obtained by the method in accordance with the sixth aspect.
- the eighth aspectprovides a lipid nanoparticle (LNP) formulation or lipidoid nanoparticle (LiNP) formulation, preferably a pharmaceutical composition in the form of a lipid nanoparticle (LNP) formulation or lipidoid nanoparticle (LiNP) formulation, obtained by the method in accordance with the sixth aspect.
- LNPlipid nanoparticle
- LiNPlipidoid nanoparticle
- the formulationis a lipid nanoparticle (LNP) suspension formulation or lipidoid nanoparticle (LiNP) suspension formulation.
- LNPlipid nanoparticle
- LiNPlipidoid nanoparticle
- the inventionprovides a lipid nanoparticle (LNP) formulation or lipidoid nanoparticle (LiNP) formulation in accordance with the first aspect, the second aspect, or the eighths aspect discussed above for use as a medicament.
- the LNPs or LiNPs of the lipid nanoparticle (LNP) formulation or lipidoid nanoparticle (LiNP) formulation for use in accordance with this aspectcomprise a therapeutic agent, e.g. a nucleic acid such as RNA, preferably mRNA. They are preferably suspension formulations as discussed in the context of the concerned aspects.
- the inventionprovides a lipid nanoparticle (LNP) formulation or lipidoid nanoparticle (LiNP) formulation in accordance with the first aspect, the second aspect, or the eighths aspect discussed above for use in the treatment or prevention of a disease.
- the LNPs or LiNPs of the lipid nanoparticle (LNP) formulation or lipidoid nanoparticle (LiNP) formulation for use in accordance with this aspectcomprise a therapeutic agent, e.g. a nucleic acid such as RNA, preferably mRNA. They are preferably suspension formulations as discussed in the context of the concerned aspects.
- the diseasemay be, e.g., a disease selected from Table A as disclosed herein below.
- the diseasemay be a disease selected from viral diseases, ciliopathies, or autoimmune diseases, and respiratory tract diseases, even more preferably selected from a lung disease, an airway disease or a nasal disease, more preferably a lung viral disease, lung ciliopathies and a lung autoimmune disease.
- the lung disease or lung viral diseaseis at least one selected from pneumonia and asthma;
- the airway diseaseis at least one selected from bronchitis, viral induced asthma, lung fibrosis and COPD;
- the nasal diseaseis at least one selected from rhinitis and sinusitis.
- the disease to be treated or preventedis a disease selected from the list consisting of pulmonary alveolar proteinosis (PAP), interstitial lung disease such as pulmonary fibrosis, e.g. idiopathic pulmonary fibrosis, viral infections such as Influenza and COVID-19, acute respiratory distress syndrome (ARDS), non-tuberculous mycobacterial (NTM) infection, lung cancer, fungal infections caused by Aspergillus sp., such as aspergillosis, fungal sinusitis, otomycosis, keratitis, and onychomycosis, preferably those caused by Aspergillus fumigatus and Aspergillus flavus, infections caused by Mycobacterium tuberculosis, Pseudomonas aeruginosa, Pneumocystis sp., Plasmodium sp., Cryptococcus sp., Nocardia sp., and combinations thereof.
- PAP
- the diseaseis selected from the list consisting of (autoimmune) pulmonary alveolar proteinosis (PAP), interstitial lung disease such as pulmonary fibrosis, e.g. idiopathic pulmonary fibrosis, viral infections such as Influenza and COVID-19, acute respiratory distress syndrome (ARDS), non-tuberculous mycobacterial (NTM) infection, lung cancer, or fungal infections caused by Aspergillus sp., such as aspergillosis, fungal sinusitis, otomycosis, keratitis, and onychomycosis, preferably those caused by Aspergillus fumigatus and Aspergillus flavus, infections caused by Mycobacterium tuberculosis, Pseudomonas aeruginosa, Pneumocystis sp., and Plasmodium sp.
- PAPpulmonary alveolar proteinosis
- interstitial lung diseasesuch as pulmonary fibrosis,
- the lipid nanoparticle (LNP) formulation or lipidoid nanoparticle (LINP) formulation for use in accordance with the tenth aspectmay be for use in vaccination or immunization.
- the inventionprovides a method for classifying a surfactant as suitable or not suitable for use as a stabilizer of a pharmaceutical composition comprising a nucleic acid, optionally as a stabilizer during purification, preferably during TFF purification, the method comprising:
- a predetermined threshold valuepreferably a time threshold value, more preferably a time threshold value of 90 minutes, wherein if the filtration speed is equal to or greater than the threshold value, the surfactant is classified as not suitable for use as a stabilizer, and if the filtration speed is less than the threshold value, the surfactant is classified as suitable for use as a stabilizer.
- the inventionprovides a method of reducing or avoiding side effects in a therapy with LNPs or LiNPs carrying at least one therapeutic agent, wherein the method comprises the steps: i) determine whether LNPs or LiNPs in a pharmaceutical composition comprising LNPs or LiNPs aggregate when subjected to a mechanical stress or a temperature stress, by determining their aggregation level before and after subjecting said pharmaceutical composition to said mechanical or temperature stress ii) if the LNP or LiNP show aggregation after the test of step (i), then add to the LNP or LiNP formulation a surfactant to obtain a LNP or LiNP suspension with a final surfactant concentration of 0.01 % w/v and up to 10% w/v, preferably between 0.05% w/w surfactant and 5% surfactant, more preferably between 0.33% surfactant and 2.5% surfactant, more preferably between 0.45% and 1 .5% surfactant, preferably between 0.5% and 1 .5% surfactant, most
- the inventionprovides a use of a surfactant according to the invention, e.g. the surfactant in accordance with the above third aspect, or a surfactant classified as being suitable as a stabilizing agent in a pharmaceutical composition in accordance with the method of the above fourth or fifth aspect, for stabilizing a suspension of lipid nanoparticles or of lipidoid nanoparticles in an aqueous vehicle solution against particle aggregation under a physical stress condition, preferably shear stress, more preferably shear stress during purification such as TFF, wherein the lipid nanoparticles or lipidoid nanoparticles comprise the following components (a) and (b):
- a therapeutic agentto a pharmaceutical composition comprising a therapeutic agent, and/or to a lipid nanoparticle formulation or lipid nanoparticle formulation comprising a therapeutic agent, one or more therapeutic agents may be used in the context of the invention.
- the therapeutic agentcomprises, essentially consists of or consists of, a nucleic acid, more preferably RNA, still more preferably single stranded RNA, and most preferably mRNA.
- a lipid nanoparticle formulation or lipid nanoparticle formulation comprising a therapeutic agentas referred to herein, e.g. a negatively charged therapeutic agent may be used.
- the therapeutic agentis typically comprised in the nanoparticles.
- the therapeutic agentcomprises, essentially consists of or consists of, a nucleic acid, more preferably RNA, still more preferably single stranded RNA, and most preferably mRNA.
- compositions in the context of the inventionare pharmaceutical compositions in the form of a lipid nanoparticle formulation or lipid nanoparticle formulation, such as a suspension formulation, wherein the lipid nanoparticles or lipid nanoparticles comprise mRNA as a therapeutic agent.
- nucleic acidis not particularly limited. In principle any type of nucleic acid can be employed in the context of the present invention. Nucleic acids are known to the skilled person and refer to biopolymers or small biomolecules composed of nucleotides which are the monomers made of three components: a 5-carbon sugar, a phosphate group and a nitrogenous base.
- the term nucleic acidis the overall name for DNA (deoxyribonucleic acid) and RNA (ribonucleic acid), i.e., the members of the above family of biopolymers. If the sugar is a compound ribose, the polymer is RNA if the sugar is derived from ribose as deoxyribose, the polymer is DNA.
- nucleic acidencompasses oligonucleotides or polynucleotides.
- a nucleic acidis a biopolymer composed of nucleotides
- nucleic acidis also often referred to as a “sequence of nucleotides” and, accordingly, as will be understood by the skilled person, the terms “nucleic acid” and “nucleic acid sequence” are often used interchangeably.
- the nanoparticlescomprise ribonucleic acid (RNA) as nucleic acid, more preferably single stranded RNA, and most preferred is mRNA.
- RNAribonucleic acid
- nucleic acidencompasses all forms of naturally occurring types of nucleic acids as well as chemically and/or enzymatically synthesized nucleic acids and also encompasses nucleic acid analogues and nucleic acid derivatives.
- the term in particularincludes any backbone-modified, sugar-modified or base-modified single-stranded or double-stranded nucleic acid, such as e.g.
- nucleic acidalso refers to any molecule that comprises nucleotides or nucleotide analogues. There are no limitations concerning sequence or size of a nucleic acid comprised in the nanoparticles of the present invention.
- the nucleic acidis predominantly defined by the biological effect that is to be achieved at the biological target the nanoparticles of the present invention are delivered to.
- the nucleic acid or nucleic acid sequencecan be defined by the gene or gene fragment that is to be expressed or by the intended substitution or repair of a defective gene or any gene target sequence or by the target sequence of a gene to be inhibited, knocked-down, down- regulated or up-regulated.
- the nanoparticlesmay comprise a nucleic acid being a DNA molecule.
- a preferred embodiment of such a DNA moleculeis a DNA molecule which can be transcribed into an mRNA molecule. Transcription is the first step of gene expression, in which a particular segment of a DNA molecule is copied into an mRNA molecule by the enzyme RNA polymerase. During transcription, a DNA sequence is read by an RNA polymerase, which produces a complementary, anti-parallel RNA strand called a primary transcript.
- a DNA moleculemay be introduced in a vector, preferably an expression vector, by standard molecular biology techniques (see, e.g. Sambrook et al., Molecular Cloning, A laboratory manual, 2nd Ed, 1989).
- vectorsuch as “expression vector” or “cloning vector” in the sense of the present invention is understood as a circular, double-stranded unit of DNA that is preferably able to replicate within a cell independently of the chromosomal DNA and which is used as a vehicle to carry genetic material into a cell, where it can be (replicated and/or) expressed (i.e., transcribed into RNA and translated into an amino acid sequence).
- a vector containing foreign DNAis termed recombinant DNA.
- the vectoritself is generally a DNA sequence that typically consists of an insert (e.g., a nucleic acid molecule/DNA molecule of the present invention) and a larger sequence that serves as the "backbone" of the vector. Plasmids in the sense of the present invention are most often found in bacteria and are used in recombinant DNA research to transfer genes between cells and are as such a subpopulation of “vectors” as used in the sense of the present invention.
- transcriptional enhancers and/or sequences which allow for induced expressionmay be employed.
- a suitable inducible systemis, for example, tetracycline-regulated gene expression as described, e.g., by Gossen and Bujard, Proc. Natl. Acad. Sci. USA 89 (1992), 5547-5551 ) and Gossen, Trends Biotech. 12 (1994), 58- 62, or a dexamethasone-inducible gene expression system as described, e.g. by Crook, EMBO J. 8 (1989), 513-519.
- the present inventionmay also use a vector, preferably an expression vector, comprising the DNA molecule.
- the vectormay be, e.g., a plasmid, cosmid, virus, bacteriophage or another vector used e.g. conventionally in genetic engineering, and may comprise further genes such as marker genes which allow for the selection of said vector in a suitable host cell and under suitable conditions.
- nucleic acid used in the context of the present inventionis a DNA molecule, it can be a plasmid DNA (pDNA) molecule.
- pDNAplasmid DNA
- the nanoparticlespreferably comprise ribonucleic acid (RNA) as nucleic acid, more preferably single stranded RNA, and most preferred is mRNA.
- RNAribonucleic acid
- RNAin principle any type of RNA can be employed in the context of the present invention.
- the RNAis a single-stranded RNA.
- the term “singlestranded RNA”means a single consecutive chain of ribonucleotides in contrast to RNA molecules in which two or more separate chains form a double-stranded molecule due to hybridization of the separate chains.
- the term “single-stranded RNA”does not exclude that the single-stranded molecule forms in itself double-stranded structures such as secondary (e.g., loops and stem-loops) or tertiary structures. Examples are tRNA and mRNA but also any other type of single-stranded RNA like antisense-RNA, siRNA, miRNA and the like.
- RNAcovers RNA which codes for an amino acid sequence as well as RNA which does not code for an amino acid sequence. It has been suggested that more than 80 % of the genome contains functional DNA elements that do not code for proteins. These noncoding sequences include regulatory DNA elements (binding sites for transcription factors, regulators and coregulators etc.) and sequences that code for transcripts that are never translated into proteins. These transcripts, which are encoded by the genome and transcribed into RNA but do not get translated into proteins, are called noncoding RNAs (ncRNAs). Thus, in one embodiment the RNA is a noncoding RNA. Preferably, the noncoding RNA is a single-stranded molecule.
- ncRNAsare critical players in gene regulation, maintenance of genomic integrity, cell differentiation, and development, and they are misregulated in various human diseases.
- ncRNAsThere are different types of ncRNAs: short (20-50 nt), medium (50-200 nt), and long (>200 nt) ncRNAs.
- Short ncRNAincludes microRNA (miRNA), small interfering RNA (siRNA), piwi-interacting RNA (piRNA), and transcription initiating RNA (tiRNA).
- medium ncRNAsare small nuclear RNAs (snRNAs), small nucleolar RNAs (snoRNAs), transfer RNAs (tRNAs), transcription start-site-associated RNAs (TSSaRNAs), promoter-associated small RNAs (PASRs), and promoter upstream transcripts (PROMPTS).
- Long noncoding RNAsinclude long-intergenic noncoding RNA (lincRNA), antisense-IncRNA, intronic IncRNA, transcribed ultra-conserved RNAs (T-UCRs), and others (Bhan A, Mandal SS, ChemMedChem. 2014 Mar 26. doi: 10.1002/cmdc.201300534).
- RNAis doublestranded.
- the noncoding RNAis single-stranded, it is preferred that the noncoding RNA is not siRNA.
- the RNAis a coding RNA, i.e. an RNA which codes for an amino acid sequence.
- RNA moleculesare also referred to as mRNA (messenger RNA) and are single-stranded RNA molecules.
- the RNAmay be made by synthetic chemical and enzymatic methodology known to one of ordinary skill in the art, or by the use of recombinant technology, or may be isolated from natural sources, or by a combination thereof.
- mRNAMessenger RNAs
- mRNAis copolymers which are built up of nucleoside phosphate building blocks mainly with adenosine, cytidine, uridine and guanosine as nucleosides, which as intermediate carriers bring the genetic information from the DNA in the cell nucleus into the cytoplasm, where it is translated into proteins. They are thus suitable as alternatives for gene expression.
- mRNAshould be understood to mean any polyribonucleotide molecule which, if it comes into the cell, is suitable for the expression of a protein or fragment thereof or is translatable to a protein or fragment thereof.
- proteinhere encompasses any kind of amino acid sequence, i.e. chains of two or more amino acids which are each linked via peptide bonds and also includes peptides and fusion proteins.
- the mRNAcontains a ribonucleotide sequence which encodes a protein or fragment thereof whose function in the cell or in the vicinity of the cell is needed or beneficial, e.g. a protein the lack or defective form of which is a trigger for a disease or an illness, the provision of which can moderate or prevent a disease or disorder, or a protein which can promote a process which is beneficial for the body, in a cell or its vicinity.
- the mRNAmay contain the sequence for the complete protein or a functional variant thereof.
- the ribonucleotide sequencecan encode a protein which acts as a factor, inducer, regulator, stimulator or enzyme, or a functional fragment thereof, where this protein is one whose function is necessary in order to remedy a disorder, in particular a metabolic disorder or in order to initiate processes in vivo such as the formation of new blood vessels, tissues, etc.
- proteins which can be encoded by mRNAinclude antibodies, cytokines or chemokines.
- functional variantis understood to mean a fragment which in the cell can undertake the function of the protein whose function in the cell is needed or the lack or defective form whereof is pathogenic.
- the mRNAmay also have further functional regions and/or 3’ or 5’ noncoding regions, in particular 3’ and/or 5’ UTRs.
- the 3’ and/or 5’ noncoding regionscan be the regions naturally flanking the protein-encoding sequence or artificial sequences, e.g. sequences which contribute to the stabilization of the RNA. Those skilled in the art can determine the sequences suitable for this in each case by routine experiments.
- the mRNAcontains a 5'-cap (five-prime-cap; cap-0) consisting of a m7GpppG connected to the mRNA via a 5' to 5' triphosphate linkage, an additional methyl group onto the penultimate nucleotide from the 5'-end of the mRNA (Cap-1 , Anti-Reverse Cap Analog (ARCA)) and/or an internal ribosome entry site (IRES) and/or a poly(A)-tail at the 3’- end, in particular, in order to improve translation.
- the mRNAcan have further regions promoting translation such as, for example, cap-2 structures or histone stem-loop structures.
- RNA which may be present in the nanoparticlesmay contain unmodified and modified nucleotides.
- unmodified nucleotideused herein refers to A, C, G and U nucleotides.
- modified nucleotideused herein refers to any naturally occurring or non -naturally occurring isomers of A, C, G and U nucleotides as well as to any naturally occurring or naturally occurring analogues, alternative or modified nucleotide or isomer thereof having for example chemical modifications or substituted residues.
- Modified nucleotidescan have a base modification and/or a sugar modification.
- Modified nucleotidescan also have phosphate group modifications, e.g., with respect to the 5’- prime cap of an mRNA molecule. Modified nucleotides also include nucleotides that are synthesized post-transcriptionally by covalent modification of the nucleotides. Further, any suitable mixture of non-modified and modified nucleotides is possible. A non-limiting number of examples of modified nucleotides can be found in the literature (e.g.
- 2-thiocytidine3-methylcytidine, N4-acetylcytidine, 5-formylcytidine, N4-methylcytidine, 5- methylcytidine, 5-hydroxymethylcytidine, 5-hydroxycytidine, lysidine, N4-acetyl-2’-O- methylcytidine, 5-formyl-2’-O-methylcytidine, 5,2’-O-dimethylcytidine, 2-O-methylcytidine, N4,2’-O-dimethylcytidine, N4,N4,2’-O-trimethylcytidine, isocytidine, pseudocytidine, pseudoisocytidine, 2-thio-cytidine, 2’-methyl-2’-deoxycytidine, 2’-amino-2’-deoxycytidine, 2’- fluoro-2’-deoxycytidine, 5-iodocytidine, 5-bromocytidine, 2
- 2-th iouridine5-methoxycarbonylmethyl-2’-O-methyluridine, 5-(isopentenylaminomethyl)-2’-O- methyluridine, 5,2’-O-dimethyluridine, 2’-O-methyluridine, 2’-O-methyl-2-thiorudine, 2-thio-2’- O-methyluridine, uridine 5-oxyacetic acid, 5-methoxycarbonylmethyluridine, uridine 5- oxyacetic acid methyl ester, 5-methoxyuridine, 5-aminomethyl-2-thiouridine, 5- carboxymethylaminomethyl-2-thiouridine, 5-methylaminomethyl-2-selenouridine, 5- methoxycarbonylmethyl-2-thiouridine, 5-taurinomethyl-2-thiouridine, pseudouridine, 1 -methyl-
- modified nucleotidecomprises nucleotides containing isotopes such as deuterium.
- isotoperefers to an element having the same number of protons but a different number of neutrons resulting in different mass numbers.
- isotopes of hydrogenfor example are not limited to deuterium but include also tritium.
- the polyribonucleotidecan also contain isotopes of other elements including for example carbon, oxygen, nitrogen and phosphor. It is also possible that modified nucleotides are deuterated or contain another isotope of hydrogen or of oxygen, carbon, nitrogen or phosphorus.
- At least one nucleotide of one nucleotide typee.g. at least one U nucleotide
- at least one nucleotide of in total two nucleotide typese.g., at least one U nucleotide and at least one C nucleotide
- at least one nucleotide of in total two nucleotide typese.g., at least one U nucleotide and at least one C nucleotide, can be a modified nucleotide.
- At least one nucleotide of in total three nucleotide typese.g., at least one G nucleotide, at least one U nucleotide and at least one C nucleotide, can be a modified nucleotide. In some embodiments, at least one nucleotide of all four nucleotide types can be a modified nucleotide.
- one or more nucleotides per nucleotide typecan be modified, the percentage of said modified nucleotides of per nucleotide type being 0%, 2.5%, 5%, 7.5%, 10%, 15%, 20%, 25%, 30%, 35%, 40%, 45%, 50%, 55%, 60%, 65%, 70%, 75%, 80%, 85%, 90% or 100%.
- the total percentage of modified nucleotides comprised in the mRNA moleculesis 0%, 2.5%, 5 %, 7.5%, 10%, 15%, 20%, 25%, 30%, 35%, 40%, 45%, 50%, 55%, 60%, 65%, 70%, 75%, 80%, 85%, 90% or 100%.
- the mRNAis an mRNA which contains a combination of modified and unmodified nucleotides.
- itis an mRNA containing a combination of modified and unmodified nucleotides as described in WO2011/012316.
- the mRNA described thereinis reported to show an increased stability and diminished immunogenicity.
- 5 to 50% of the cytidine nucleotides and 5 to 50% of the uridine nucleotidesare modified.
- 5 to 50% of the uridine nucleotidesare replaced by N1-methyl-pseudo-uridine.
- the adenosine- and guanosinecontaining nucleotidescan be unmodified.
- the adenosine and guanosine nucleotidescan be unmodified or partially modified, and they are preferably present in unmodified form.
- the percentage of analogues of a given nucleotiderefers to input percentage (e.g., the percentage of analogues in a starting reaction, such as a starting in vitro transcription reaction). In certain embodiments of any of the foregoing, the percentage of analogues of a given nucleotide refers to output (e.g., the percentage in a synthesized or transcribed compound). Both options are equally contemplated.
- the RNA, preferably the mRNA, moleculesmay be produced recombinantly in in vivo systems by methods known to a person skilled in the art.
- the modified RNApreferably the mRNA molecules may be produced in an in vitro system using, for example, an in vitro transcription system which is known to the person skilled in the art.
- An in vitro transcription system capable of producing RNA, preferably mRNArequires an input mixture of modified and unmodified nucleoside triphosphates to produce modified RNA.
- 5 to 50% of the cytidinesare analogues of cytidine in such an input mixture and 5 to 50% of the uridines are analogues of uridine in such an input mixture.
- 5 to 40% of the cytidinesare analogues of cytidine in such an input mixture and 5 to 40% of the uridines are analogues of uridine in such an input mixture.
- 5 to 30% of the cytidinesare analogues of cytidine in such a mixture and 5 to 30% of the uridines are analogues of uridine in such an input mixture.
- 5 to 30% of the cytidinesare analogues of cytidine in such mixture and 10 to 30% of the uridines are analogues of uridine in such mixture.
- 5 to 20% of the cytidinesare analogues of cytidine in such an input mixture and 5 to 20% of the uridines are analogues of uridine in such an input mixture.
- 5 to 10% of the cytidinesare analogues of cytidine in such an input mixture and 5 to 10% of the uridines are analogues of uridine in such an input mixture.
- 25% of the cytidinesare analogues of cytidine in such an input mixture and 25% of the uridines are analogues of uridine in such an input mixture.
- the input mixturedoes not comprise analogues of adenosine and/or guanosine. In other embodiments, optionally, the input mixture comprises one or more analogues of adenosine and/or guanosine (or none of either or both).
- the percentage of cytidines in an input mixture that are analogues of cytidineis not the same as the percentage of uridines in an input mixture that are analogues of uridine. In certain embodiments, the percentage of analogues of cytidine in an input mixture is lower than the percentage of analogues of uridine in an input mixture. As noted above, this may be in the presence or the absence of analogues of adenosine and guanosine in the input mixture but, in certain embodiments, is in the absence of analogues of adenosine and analogues of guanosine in the input mixture.
- an input mixture of nucleotides for an in vitro transcription system that produces a RNAcomprises analogues of cytidine and analogues of uridine, and 5 to 20% of the cytidines of the input mixture are analogues of cytidine and 25 to 45% of the uridines of the input mixture are analogues of uridine.
- the input mixturecomprises modified and unmodified cytidines and modified and unmodified uridines, and 5 to 20% of the cytidines of the input mixture comprise analogues of cytidine while 25 to 45% of the uridines of the input mixture comprise analogues of uridine.
- the input mixturecomprises 5 to 10% analogues of cytidine and 30 to 40% analogues of uridine, such as 7-9% analogues of cytidine, such as 7, 7.5 or 8% and, such as 32-38% analogues of uridine, such as 33, 34, 35, 36%.
- any of the analogues of uridine and analogues of cytidine described hereinmay be used, optionally excluding pseudouridine.
- the analogue of cytidinecomprises or consists of (e.g., it is the single C analogue type used) 5-iodocytidine and the analogue of uridine comprises or consists of (e.g., it is the single U analogue type used) 5-iodouridine.
- analoguesare described above. It should be understood that for modified polyribonucleotides encoding the desired polypeptide, the analogues and level of modification is, unless indicated otherwise, considered across the entire polyribonucleotide encoding the desired polypeptide, including 5’ and 3’ untranslated regions (e.g., the level of modification is based on input ratios of analogues in an in vitro transcription reaction such that analogues may be incorporated at positions that are transcribed).
- modified RNApreferably mRNA molecules may be chemically synthesized, e.g., by conventional chemical synthesis on an automated nucleotide sequence synthesizer using a solid-phase support and standard techniques or by chemical synthesis of the respective DNA sequences and subsequent in vitro or in vivo transcription of the same.
- the mRNAmay be combined with target binding sites, targeting sequences and/or with micro-RNA binding sites, in order to allow activity of the desired mRNA only in the relevant cells.
- the RNAcan be combined with micro-RNAs or shRNAs in the untranslated regions.
- therapeutic effectscan be achieved by the interaction of the ribonucleic acid with cellular molecules and organelles.
- Such interactionalone may for example activate the innate immune system, as is the case for certain CpG oligonucleotides and sequences designed to specifically interact with toll-like and other extra- or intracellular receptors.
- nucleic acidspreferably ribonucleic acids, more preferably mRNAs
- nucleotide sequencessuch as genes comprised in the nucleic acid (preferably ribonucleic acids, more preferably the mRNA)
- Overexpression of introduced exogenous nucleic acidsmay be intended to compensate or complement endogenous gene expression, in particular in cases where an endogenous gene is defective or silent, leading to no, insufficient or a defective or a dysfunctional product of gene expression such as is the case with many metabolic and hereditary diseases like cystic fibrosis, hemophilia or muscular dystrophy to name a few.
- Overexpression of introduced exogenous nucleic acidsmay also be intended to have the product of the expression interact or interfere with any endogenous cellular process such as the regulation of gene expression, signal transduction and other cellular processes.
- exogenous ribonucleic acidsmay also be intended to generate in vivo or ex vivo transiently genetically modified cells for cellular therapies such as modified T-cells, NK cells and other lymphocytes or precursor or stem or other cells for regenerative medicine.
- Downregulation, silencing or knockdown of endogenous gene expressioncan be exerted on the transcriptional level and on the translational level.
- Multiple mechanismsare known to the one skilled in the art and include for example epigenetic modifications, changes in chromatin structure, selective binding of transcription factors by the introduced nucleic acid, hybridization of the introduced nucleic acid to complementary sequences in genomic DNA, mRNA or other RNA species by base pairing including unconventional base pairing mechanisms such as triple helix formation.
- gene repair, base or sequence changescan be achieved at the genomic level and at the mRNA level including exon skipping.
- Base or sequence changescan for example be achieved by RNA-guided site-specific DNA cleavage, by cut and paste mechanisms exploiting trans-splicing, trans-splicing ribozymes, chimeraplasts, splicosome-mediated RNA trans- splicing, or by exploiting group II or retargeted introns, or by exploiting insertional mutagenesis mediated by viruses or exploiting targeted genomic insertion using prokaryotic, eukaryotic or viral integrase systems.
- nucleic acidsare the carriers of the building plans of living systems and as they participate in many cellular processes in a direct and indirect manner, in theory any cellular process can be influenced by the introduction of nucleic acids into cells from outside.
- this introductioncan be carried out directly in vivo and ex vivo in cell or organ culture followed by transplantation of thus modified organs or cells into a recipient.
- the particles for use in the context of the present invention with nucleic acids as therapeutically active agentmay be useful for all purposes described above.
- the RNAmay contain a ribonucleotide sequence which encodes a protein or fragment thereof whose function in the cell or in the vicinity of the cell is needed or beneficial, e.g. a protein the lack or defective form of which is a trigger for a disease or an illness, the provision of which can moderate or prevent a disease or an illness, or a protein which can promote a process which is beneficial for the body, in a cell or its vicinity.
- a proteinthe lack or defective form of which is a trigger for a disease or an illness, the provision of which can moderate or prevent a disease or an illness, or a protein which can promote a process which is beneficial for the body, in a cell or its vicinity.
- RNAin particular, mRNA
- mRNAhas become increasingly relevant as a drug entity.
- mRNAdoes not need to be transported into the nucleus but is directly translated into protein in the cytoplasm (J Control Release, 2011 , 150:238-247, and Eur J Pharm Biopharm, 2009, 71 :484-489).
- RNApreferably mRNA
- disorders caused by single-gene mutationslike cystic fibrosis, hemophilia and many others, can be dominant or recessive with respect to the likelihood that a certain trait will appear in the offspring. While a dominant allele manifests a phenotype in individuals who have only one copy of the allele, for a recessive allele the individual must have two copies, one from each parent to become manifest.
- polygenic disordersare caused by two or more genes and the manifestation of the respective disease is often fluent and associated to environmental factors. Examples for polygenic disorders are hypertension, elevated cholesterol level, cancer, neurodegenerative disorders, mental illness and others.
- therapeutic RNApreferably the mRNA, representing one or more of these genes may be beneficial to those subjects.
- a genetic disordermust not have been passed down from the parents' genes, but can also be caused by new mutations.
- therapeutic RNApreferably the mRNA, representing the correct gene sequence may be beneficial to the subjects.
- An online catalog with Human Genes and Genetic Disorders together with their respective genes and a description of their phenotypesare available at the ONIM (Online Mendelian Inheritance in Man) webpage (http://onim.org); sequences of each are available from the Uniprot database (http://www.uniprot.org).
- ONIMOnline Mendelian Inheritance in Man
- sequences of eachare available from the Uniprot database (http://www.uniprot.org).
- Table Alists some congenital diseases and disorders, and the corresponding gene(s).
- compositions as referred to hereinthus encompass compositions suitable for the treatment and prevention of a disease selected from those listed in Table A.
- a nanoparticle formulation as referred to hereinsuch as a lipid nanoparticle formulation or lipidoid nanoparticle formulation comprising RNA, preferably mRNA, may be suitable for the treatment and prevention of a disease selected from those listed in Table A.
- the therapeutic protein which is encoded by the RNApreferably the mRNA, which may be present in the suspension formulation and the aerosol of the present invention is chosen from the cellular proteins listed in Table A.
- the RNA, preferably the mRNA, moleculemay encode a therapeutic cellular protein, wherein the encoded therapeutic protein is one listed in Table A or a homolog thereof.
- the therapeutic protein which is encoded by the RNAis chosen from the secreted proteins listed in Table A.
- the RNApreferably the mRNA
- the RNAmay encode a therapeutic fusion protein, wherein the encoded therapeutic protein or a homolog thereof is one listed in Table A and the second protein is a signal peptide that allows the secretion of the therapeutic protein.
- a signal peptideis a short, typically 5-30 amino acids long sequence present at the N-terminus of said therapeutic protein and that leads the fusion protein towards the cell’s secretory pathway via certain organelles (i.e. the endoplasmic reticulum, the Golgi-apparatus or the endosomes).
- such fusion proteinis secreted from the cell or from a cellular organelle or inserted into a cellular membrane (e.g. multi-spanning trans- membrane proteins) at a cellular compartment or at the cell’s surface.
- an RNAmay encode one or more, but is not limited to, the following proteins of the genes that cause, predispose or protect from diseases.
- diseases or disordersthat may be treated (or prevented) include those wherein said polypeptide, protein or peptide is selected from the group consisting of the ones as outlined in the following Table A.
- the encoding sequence of an RNA, preferably mRNAmay be transcribed and translated into a partial or full-length protein comprising cellular activity at a level equal to or greater than that of the native protein.
- an RNApreferably the mRNA, encodes a therapeutically or pharmaceutically active polypeptide, protein or peptide having a therapeutic or preventive effect, wherein said polypeptide, protein or peptide is selected from the group consisting of the ones as outlined in the following Table A.
- the RNApreferably the mRNA, more specifically the encoding sequence thereof, may be used to express a partial or full-length protein with cellular activity at a level equal to or less than that of the native protein. This may allow the treatment of diseases for which the administration of an RNA molecule can be indicated.
- Table ANon-limiting examples of human genes and genetic diseases or disorders
- RNApreferably the mRNA
- Table Ashows examples of genes in which a defect leads to a disease which can be treated with RNA, preferably the mRNA, which may be present pharmaceutical compositions or nanoparticle formulations referred to herein wherein RNA, preferably the mRNA, comprises a ribonucleotide sequence which encodes an intact version of the protein or a functional fragment thereof of the above disclosed defective gene.
- hereditary diseasescan be addressed, which for example affect the lungs, such as SPB (surfactant protein B) deficiency, ABCA3 deficiency, cystic fibrosis and o1 -antitrypsin deficiency, or which affect plasma proteins (e.g.
- congenital hemochromatosishepcidin deficiency
- thrombotic thrombocytopenic purpuraTPP, ADAMTS 13 deficiency
- clotting defectse.g. hemophilia a and b
- complement defectse.g. protein C deficiency
- immune defectssuch as for example SCID (caused my mutations in different genes such as: RAG1 , RAG2, JAK3, IL7R, CD45, CD35, CD3E) or by deficiencies due to lack of adenosine deaminase for example (ADA-SCID), septic granulomatosis (e.g.
- RNApreferably mRNA can be useful
- disorderssuch as SMN1 -related spinal muscular atrophy (SMA); amyotrophic lateral sclerosis (ALS); GALT- related galactosemia; Cystic Fibrosis (CF); SLC3A1 -related disorders including cystinuria; COL4A5-related disorders including Alport syndrome; galactocerebrosidase deficiencies; X- linked adrenoleukodystrophy and adrenomyeloneuropathy; Friedreich's ataxia; Pelizaeus- Merzbacher disease; TSC1 and TSC2-related tuberous sclerosis; Sanfilippo B syndrome (MPS IIIB); CTNS-related cystinosis; the FMR1 -related disorders which include Fragile X syndrome, Fragile X-Associated Tremor/Ataxia Syndrome and Fragile X Premature Ovarian Failure Syndrome; Prader-Willi syndrome; hereditary hemorrhagic telan
- a proteine.g. an enzyme
- the RNApreferably the mRNA, encoding any of the above proteins, which makes the protein encoded by the defective gene or a functional fragment thereof available.
- Transcript replacement therapies/protein replacement therapiesdo not affect the underlying genetic defect, but increase the concentration of the protein in which the subject is deficient.
- the transcript replacement therapy/enzyme replacement therapyreplaces the deficient lysosomal enzyme acid alpha-glucosidase (GAA).
- proteins which can be encoded by the mRNAare erythropoietin (EPO), growth hormone (somatotropin, hGH), cystic fibrosis transmembrane conductance regulator (CFTR), growth factors such as GM-SCF, G-CSF, MPS, protein C, hepcidin, ABCA3 and surfactant protein B.
- EPOerythropoietin
- somatotropinsomatotropin
- hGHgrowth hormone
- CFTRcystic fibrosis transmembrane conductance regulator
- growth factorssuch as GM-SCF, G-CSF, MPS, protein C, hepcidin, ABCA3 and surfactant protein B.
- RNApreferably the mRNA, according to the invention contains the coding sequence for surfactant protein B (SP-B) or for erythropoietin.
- SP-Bsurfactant protein B
- proteins which can be encoded by the RNA, preferably the mRNA, of the present invention according to the inventionare growth factors such as human growth hormone hGH, BMP-2 or angiogenesis factors.
- growth factorssuch as human growth hormone hGH, BMP-2 or angiogenesis factors.
- Said DNA moleculemay encode the above RNA, preferably the above mRNA and, accordingly, harbors the genetic information for the correspondingly transcribed RNA molecule.
- RNA molecule of the present inventionas has been set forth above and below in the context of the RNA molecule, preferably the mRNA molecule, that may be in the pharmaceutical compositions or nanoparticle formulations referred to herein.
- the RNAmay contain a ribonucleotide sequence which encodes a full-length antibody or a smaller antibody (e.g., both heavy and light chains) which can be used in therapeutic settings to, e.g., confer immunity to a subject.
- a ribonucleotide sequencewhich encodes a full-length antibody or a smaller antibody (e.g., both heavy and light chains) which can be used in therapeutic settings to, e.g., confer immunity to a subject.
- a ribonucleotide sequencewhich encodes a full-length antibody or a smaller antibody (e.g., both heavy and light chains) which can be used in therapeutic settings to, e.g., confer immunity to a subject.
- a ribonucleotide sequencewhich encodes a full-length antibody or a smaller antibody (e.g., both heavy and light chains) which can be used in therapeutic settings to, e.g., confer immunity to a subject.
- the RNApreferably the mRNA may encode a functional monoclonal or polyclonal antibody, which may be useful for targeting and/or inactivating a biological target (e.g., a stimulatory cytokine such as tumor necrosis factor).
- a biological targete.g., a stimulatory cytokine such as tumor necrosis factor
- the RNA, preferably the mRNA sequencemay encode, for example, functional anti-nephrotic factor antibodies useful for the treatment of membranoproliferative glomerulonephritis type II or acute hemolytic uremic syndrome, or alternatively may encode anti-vascular endothelial growth factor (VEGF) antibodies useful for the treatment of VEGF-mediated diseases, such as cancer.
- VEGFvascular endothelial growth factor
- the RNApreferably the mRNA may encode a functional monoclonal or polyclonal antibody, which may be useful for neutralizing or otherwise inhibiting a virus or virus replication.
- the RNApreferably the mRNA, may contain a ribonucleotide sequence which encodes an antigen which preferably can be used in preventive or therapeutic settings.
- the mRNAmay encode a protein or proteins that can induce an immune modulation, such as cytokines, including chemokines, interferons (such as interferon lambda), interleukins, lymphokines, and tumor necrosis factors.
- cytokinesincluding chemokines, interferons (such as interferon lambda), interleukins, lymphokines, and tumor necrosis factors.
- the RNAmay contain a ribonucleotide sequence which encodes a polypeptide or a protein which can be used in genome editing technologies.
- Genome editingis a type of genetic engineering in which DNA is inserted, deleted, or replaced in the genome of an organism using nucleases. These nucleases create site-specific breaks at desired locations in the genome. The induced breaks are repaired by non-homologous end-joining or homologous recombination, resulting in targeted mutations in the genome, thereby “editing” the genome.
- the breaksmay either be single-strand breaks or double-strand breaks (DSBs) while double-strand breaks (DSBs) are preferred.
- the RNAmay contain a ribonucleotide sequence which encodes a polypeptide or protein of the Cas (CRISPR associated protein) protein family, preferably Cas9 (CRISPR associated protein 9).
- CasCas9
- Proteins of the Cas protein family, preferably Cas9may be used in CRISPR/Cas9 based methods and/or CRISPR/Cas9 genome editing technologies.
- CRISPR-Cas systems for genome editing, regulation and targetingare reviewed in Nat. Biotechnol., 2014, 32(4):347-355.
- the RNAmay contain a ribonucleotide sequence which encodes a meganuclease.
- Meganucleasesare endodeoxyribonucleases which, in contrast to “conventional” endodeoxyribonucleases, recognize a large recognition site (e.g., a double-stranded DNA sequence of 12 to 40 base pairs). As a result, the respective site occurs only a few times, preferably only once, in any given genome. Meganucleases are therefore considered to be the most specific naturally occurring restriction enzymes and, accordingly, are suitable tools in genome editing technologies.
- the RNApreferably the mRNA, contains a ribonucleotide sequence which encodes a zinc finger nuclease (ZFN).
- ZFNsare artificial restriction enzymes generated by fusing a zinc finger DNA-binding domain to a DNA-cleavage domain.
- Zinc finger domainscan be engineered to target specific desired DNA sequences and this enables zinc- finger nucleases to target unique sequences within complex genomes.
- ZFNscan be used to precisely alter the genome of higher organisms and are, therefore, suitable tools in genome editing technologies.
- the RNAmay contain a ribonucleotide sequence which encodes a transcription activator-like effector nuclease (TALEN).
- TALENsare restriction enzymes that can be engineered to cut specific sequences of DNA.
- TALENsare fusion proteins wherein a TAL effector DNA-binding domain is fused to a DNA cleavage domain of a nuclease.
- Transcription activator-like effectors (TALEs)can be engineered to bind practically any desired DNA sequence. Thus, when combined with a nuclease, DNA can be cut at specific desired locations.
- RNApreferably an mRNA molecule
- the present inventionis not only limited to the use of an RNA, preferably an mRNA, but may employ any nucleic acid molecule, such as a DNA molecule.
- Said DNA moleculemay encode the above RNA, preferably the above mRNA and, accordingly, harbors the genetic information for the correspondingly transcribed RNA molecule.
- RNA moleculepreferably the mRNA molecule, that may be present in the nanoparticles used in the present invention.
- Lipid or lipidoid nanoparticles referred to hereintypically comprise a lipid mix or lipidoid mix, wherein the lipid mix or lipidoid mix comprises, as a component thereof, at least one selected from an ionizable lipid, ionizable lipidoid, and permanently cationic lipid. It will be understood that this encompasses the possibility that the nanoparticles comprise a combination of different permanently cationic lipids, a combination of different ionizable lipids, a combination of different ionizable lipidoids, or a combination of one or more permanently cationic lipids, one or more ionizable lipids, and/or one or more ionizable lipidoids.
- R 4a and R 4bare, at each occurrence, independently either (a) H or C1-C12 alkyl, or (b) R 4a is H or C1-C12 alkyl, and R 4b together with the carbon atom to which it is bound is taken together with an adjacent R 4b and the carbon atom to which it is bound to form a carbon-carbon double bond;
- the ionizable lipidis selected from a lipid in Table 1 , Table 2, Table 3 or Table 4.
- Table 1Table 1 :
- the ionizable lipidhas the following structure: or a pharmaceutically acceptable salt, tautomer, prodrug or stereoisomer thereof, wherein: Ri and R2 are each independently for each occurrence optionally substituted C10-C30 alkyl, optionally substituted C10-C30 alkenyl, optionally substituted C10-C30 alkynyl or optionally substituted C10-C30 acyl; R3 is H, optionally substituted C10-C10 alkyl, optionally substituted C2-C10 alkenyl, optionally substituted C2-C10 alkynyl, alkylhetrocycle, alkylphosphate, alkylphosphorothioate, alkyl phosphorod ith ioate, alkylphosphonate, alkylamine, hydroxyalkyl, co-aminoalkyl, co-(substituted)aminoalkyl, co-phosphoalkyl, co- thiophosphoalkyl, optional
- Qis H, alkyl, co-aminoalkyl, co-(substituted)aminoalkyl, co-phosphoalkyl or co-thiophosphoalkyl.
- the ionizable lipidhas one of the following structures:
- the lipid nanoparticles referred to hereincomprise the ionizable lipid [(4-Hydroxybutyl)azanediyl]di(hexane-6,1-diyl) bis(2-hexyldecanoate), also known ALC- 0315, and as shown in the following formula:
- the lipid nanoparticlesmay further comprise a neutral lipid and/or a polymer conjugated lipid, preferably both a neutral lipid and a polymer conjugated lipid.
- the molar ratio of the above ionizable lipid to a neutral lipidpreferably ranges from about 4.1 :1.0 to about 4.9:1.0, 4.5:1.0 to about 4.8:1.0, or 4.7:1.0 to about 4.8:1.0.
- the molar ratio of ionizable lipid to the neutral lipidranges from about 2:1 to about 8:1 , preferably 5:1 to 1 :1.
- the molar ratio of ionizable lipid to the polymer conjugated lipidranges from about 35:1 to about 25:1 , or 100:1 to about 20:1.
- the ionizable lipidhas the following structure:
- a lipid nanoparticle as referred to hereincomprises: i) a first cationic lipid as an ionizable lipid (a) having a first effective pKa; ii) a second cationic lipid as an ionizable lipid (a) having a second effective pKa, the second effective pKa being greater than the first effective pKa; iii) a neutral lipid; iv) a steroid; v) a polymer conjugated lipid; vi) a therapeutic agent, or a pharmaceutically acceptable salt or prodrug thereof, encapsulated within or associated with the lipid nanoparticle, and vii) a surfactant, wherein the lipid nanoparticle has an effective pKa between the first and second effective pKa’s.
- the first effective pKais less than 5.75. In some embodiments the second effective pKa is greater than 6.25. In some embodiments, the lipid nanoparticle of any one of claims 48-50, wherein the lipid nanoparticle has an effective pKa ranging from 5.90 to 6.35. In some embodiments, the mol ratio of the first cationic lipid to the second cationic lipid ranges from 1 :20 to 1 :2.
- R ais H or C1-C12 alkyl
- R 1a and R 1bare, at each occurrence, independently either (a) H or C1-C12 alkyl, or (b) R 1a is H or C1-C12 alkyl, and R 1b together with the carbon atom to which it is bound is taken together with an adjacent R 1b and the carbon atom to which it is bound to form a carbon-carbon double bond;
- R 2a and R 2bare, at each occurrence, independently either (a) H or C1-C12 alkyl, or (b) R 2a is H or C1-C12 alkyl, and R 2b together with the carbon atom to which it is bound is taken together with an adjacent R 2b and the carbon atom to which it is bound to form a carbon-carbon double bond;
- R 3a and R 3bare, at each occurrence, independently either (a) H or C1-C12 alkyl, or (b) R 3a is H or C1-C12 alkyl, and R 3b together with the carbon atom to which it is bound is taken together with an adjacent R 3b and the carbon atom to which it is bound to form a carbon-carbon double bond;
- R 4a and R 4bare, at each occurrence, independently either (a) H or C1-C12 alkyl, or (b) R 4a is H or C1-C12 alkyl, and R 4b together with the carbon atom to which it is bound is taken together with an adjacent R 4b and the carbon atom to which it is bound to form a carbon-carbon double bond;
- R 5 and R 6are each independently methyl or cycloalkyl
- R 7is, at each occurrence, independently H or C1-C12 alkyl
- R 8 and R 9are each independently unsubstituted C1-C12 alkyl; or R 8 and R 9 , together with the nitrogen atom to which they are attached, form a 5, 6 or 7-membered heterocyclic ring comprising one nitrogen atom; a and d are each independently an integer from 0 to 24; b and c are each independently an integer from 1 to 24; e is 1 or 2; and x is 0, 1 or 2.
- the first and second cationic lipids as ionizable lipids (a)are each independently selected from a lipid of Formula a-L In some embodiments the first cationic lipid, or the second cationic lipid or both has a structure of Formula a-ll:
- G 3is Ci-Ce alkylene;
- R ais H or C1-C12 alkyl
- R 1a and R 1bare, at each occurrence, independently either: (a) H or C1-C12 alkyl; or (b) R 1a is H or C1-C12 alkyl, and R 1b together with the carbon atom to which it is bound is taken together with an adjacent R 1b and the carbon atom to which it is bound to form a carbon-carbon double bond;
- R 2a and R 2bare, at each occurrence, independently either: (a) H or C1-C12 alkyl; or (b) R 2a is H or C1-C12 alkyl, and R 2b together with the carbon atom to which it is bound is taken together with an adjacent R 2b and the carbon atom to which it is bound to form a carbon-carbon double bond;
- R 5 and R 6are each independently H or methyl
- R 7is C4-C20 alkyl
- R 8 and R 9are each independently C1-C12 alkyl; or R 8 and R 9 , together with the nitrogen atom to which they are attached, form a 5, 6 or 7-membered heterocyclic ring; a, b, c and d are each independently an integer from 1 to 24; and x is 0, 1 or 2.
- the first and second cationic lipids as ionizable lipids (a)are each, independently selected from a lipid of Formula a-ll.
- G 1 and G 2are each independently unsubstituted C1-C12 alkylene or C1-C12 alkenylene;
- G 3is C1-C24 alkylene, C1-C24 alkenylene, C3-C8 cycloalkylene, C3-C8 cycloalkenylene;
- R ais H or C1-C12 alkyl
- R 1 and R 2are each independently C6-C24 alkyl or C6-C24 alkenyl;
- R 4is C1-C12 alkyl;
- R 5is H or Ci-Ce alkyl; and x is 0, 1 or 2.
- the first and second cationic lipids as ionizable lipids (a)are each, independently, selected from a lipid of Formula a-lll.
- the first cationic lipid as an ionizable lipid (a), or the second cationic lipid as an ionizable lipid (a) or bothhas a structure of Formula a-IV:
- Xis CR a ;
- Zis alkyl, cycloalkyl or a monovalent moiety comprising at least one polar functional group when n is 1 ; or Z is alkylene, cycloalkylene or a polyvalent moiety comprising at least one polar functional group when n is greater than 1 ; R a is, at each occurrence, independently H, C1-C12 alkyl, C1-C12 hydroxylalkyl,
- Ris, at each occurrence, independently either: (a) H or C1-C12 alkyl; or (b) R together with the carbon atom to which it is bound is taken together with an adjacent R and the carbon atom to which it is bound to form a carbon-carbon double bond;
- R 1 and R 2have, at each occurrence, the following structure, respectively:
- R 1 R 2 a 1 and a 2are, at each occurrence, independently an integer from 3 to 12; b 1 and b 2 are, at each occurrence, independently 0 or 1 ; c 1 and c 2 are, at each occurrence, independently an integer from 5 to 10; d 1 and d 2 are, at each occurrence, independently an integer from 5 to 10; y is, at each occurrence, independently an integer from 0 to 2; and n is an integer from 1 to 6, wherein each alkyl, alkylene, hydroxylalkyl, aminoalkyl, alkylaminylalkyl, alkoxyalkyl, alkoxycarbonyl, alkylcarbonyloxy, alkylcarbonyloxyalkyl and alkylcarbonyl is optionally substituted with one or more substituents.
- the first and second cationic lipids as ionizable lipids (a)are each, independently, selected from a lipid of Formula a-IV.
- the first cationic lipid as an ionizable lipid (a), or the second cationic lipid as an ionizable lipid (a), or bothhas a structure of Formula a-V:
- Xis CR a ;
- Zis alkyl, cycloalkyl or a monovalent moiety comprising at least one polar functional group when n is 1 ; or Z is alkylene, cycloalkylene or a polyvalent moiety comprising at least one polar functional group when n is greater than 1 ;
- R ais, at each occurrence, independently H, C1-C12 alkyl, C1-C12 hydroxylalkyl, C1-C12 aminoalkyl, C1-C12 alkylaminylalkyl, C1-C12 alkoxyalkyl, C1-C12 alkoxycarbonyl, C1-C12 alkylcarbonyloxy, C1-C12 alkylcarbonyloxyalkyl or C1-C12 alkylcarbonyl;
- Ris, at each occurrence, independently either: (a) H or C1-C12 alkyl; or (b) R together with the carbon atom to which it is bound is taken together with an adjacent R and the carbon atom to which it is bound to form a carbon-carbon double bond;
- R 1 and R 2have at each occurrence the following structure, respectively:
- R'is, at each occurrence, independently H or C1-C12 alkyl; a 1 and a 2 are, at each occurrence, independently an integer from 3 to 12; b 1 and b 2 are, at each occurrence, independently 0 or 1; c 1 and c 2 are, at each occurrence, independently an integer from 2 to 12; d 1 and d 2 are, at each occurrence, independently an integer from 2 to 12; y is, at each occurrence, independently an integer from 0 to 2; and n is an integer from 1 to 6, wherein a 1 , a 2 , c 1 , c 2 , d 1 and d 2 are selected such that the sum of a 1 +c 1 +d 1 is an integer from 18 to 30, and the sum of a 2 +c 2 +d 2 is an integer from 18 to 30, and wherein each alkyl, alkylene, hydroxylalkyl, aminoalkyl, alkylaminylalkyl, alkoxyalkyl, alkoxycarbonyl
- the first and second cationic lipids as ionizable lipids (a)are each, independently, selected from a lipid of Formula a-V.
- the first and second cationic lipids as ionizable lipids (a)have the following structures, respectively:
- the first cationic lipid as an ionizable lipid (a), the second cationic lipid as an ionizable lipid (a), or bothhave one of the following structures:
- the total mol percent of cationic lipid as an ionizable lipid (a) in the lipid nanoparticleranges from 40 to 55 mol percent based on total lipid present in the lipid nanoparticle.
- the lipid nanoparticlesmay further comprise a neutral lipid, a steroid (such as sterol) and/or a polymer conjugated lipid, preferably all of the neutral lipid, the steroid and the polymer conjugated lipid, and the molar ratio of total cationic lipid to a neutral lipid ranges from about 2:1 to about 8:1.
- the molar ratio of total cationic lipid to a steroidranges from 5:1 to 1 :1. In some embodiments, the molar ratio of total cationic lipid to a polymer conjugated lipid ranges from about 100:1 to about 20:1 .
- the neutral lipidis distearoylphosphatidylcholine (DSPC), dioleoylphosphatidylcholine (DOPC), dipalmitoylphosphatidylcholine (DPPC), dioleoylphosphatidylglycerol (DOPG), dipalmitoylphosphatidylglycerol (DPPG), dioleoylphosphatidylethanolamine (DOPE), palmitoyloleoylphosphatidylcholine (POPO), palmitoyloleoyl-phosphatidylethanolamine (POPE) and dioleoyl-phosphatidylethanolamine 4- (N-maleimidomethyl)-cyclohexane- 1 carboxylate (DOPE-mal), dipalmitoyl phosphatidyl ethanolamine (DPPE), dimyristoylphosphoethanolamine (DMPE), distearoylphosphatidylethanolamine (DSPE), 16-O-monomethyl
- the neutral lipidis DSPC.
- the steroidis cholesterol.
- the polymer conjugated lipidis present in a concentration ranging from 1.0 to 2.5 molar percent, preferably, about 1.7 molar percent, wherein the polymer conjugated lipid is present in a concentration of about 1.5 molar percent.
- the polymer conjugated lipidis a pegylated lipid.
- the pegylated lipidis PEG-DAG, PEG-PE, PEG-S-DAG, PEG-cer or a PEG dialkyoxypropylcarbamate.
- the pegylated lipidhas the following Formula (a-VI): or a pharmaceutically acceptable salt, tautomer or stereoisomer thereof, wherein:
- R 12 and R 13are each independently a straight or branched, saturated or unsaturated alkyl chain containing from 10 to 30 carbon atoms, wherein the alkyl chain is optionally interrupted by one or more ester bonds; and w has a mean value ranging from 30 to 60.
- R 12 and R 13are each independently straight, saturated alkyl chains containing from 12 to 16 carbon atoms.
- the average wranges from 42 to 55, preferably, the average w is about 49.
- the pegylated lipidhas the following Formula (Via): wherein the average w is about 49.
- the lipid nanoparticleforms a plurality of the nanoparticles having a polydispersity of less than 0.12.
- the polydispersityis less than 0.08.
- the mean diameterranges from 50 nm to 100 nm, preferably the diameter ranges from 60 nm to 85 nm.
- the ionizable lipidoidwhich may be comprised by the nanoparticles referred to herein comprises or consists of an ionizable lipidoid of the following formula (L-1 ) or a protonated form thereof.
- the lipid nanoparticle (LNP) formulation or lipidoid nanoparticle (LiNP) formulationis a lipidoid nanoparticle formulation comprising a plurality of lipidoid nanoparticles (LiNPs), each comprising a lipidoid mix, wherein the lipidoid mix comprises the ionizable lipidoid of the following formula (L-1 ) or a protonated form thereof.
- the ionizable lipidoid of the following formula (L-1) or its protonated formswhich can be used as a preferred ionizable lipidoid in the context of the present invention are described in detail in the PCT application WO 2014/207231 A1.
- ais 1 or 2 and b is an integer of 1 to 4 or a is an integer of 1 to 4 and b is 1 or 2, preferably a is 1 and b is an integer of 2 to 4 or a is an integer of 2 to 4 and b is 1 , p is 1 or 2, m is 1 or 2; n is 0 or 1 and m+n is > 2; and
- one or more of the nitrogen atoms contained in the compound of formula (L-1 )are protonated to provide a compound carrying a positive charge.
- R 7Ais selected from C3-C18 alkyl and C3-C18 alkenyl having one C-C double bond.
- R 1A to R 6Aare independently selected from hydrogen and a group -CH2-CH(OH)-R 7A wherein R 7A is selected from C3-C18 alkyl and C3-C18 alkenyl having one C-C double bond; provided that at least two residues among R 1A to R 6A , more preferably at least three residues among R 1A to R 6A , and still more preferably at least four residues among R 1A to R 6A are a group -CH2-CH(OH)-R 7A , wherein R 7A is selected from C3-C18 alkyl and C3- C18 alkenyl having one C-C double bond.
- R 7Ais selected from C8-C18 alkyl and C8-C18 alkenyl having one C-C double bond, and more preferably from C8-C12 alkyl and C8-C12 alkenyl having one C-C double bond.
- alkyl groupsare preferred over alkenyl groups as R 7A .
- any of the groups R 1A to R 6Ais a protecting group for an amino group, such as described for example in W02006/138380, preferred embodiments thereof are t- butoxycarbonyl (Boc), 9-fluorenylmethoxycarbonyl (Fmoc), or carbobenzyloxy (Cbz).
- R 1A to R 6Aare a receptor ligand
- useful examplesare given in Philipp and Wagner in “Gene and Cell Therapy - Therapeutic Mechanisms and Strategy”, 3 rd Edition, Chapter 15. CRC Press, Taylor & Francis Group LLC, Boca Raton 2009.
- Preferred receptor ligands for lung tissueare described in Pfeifer et al. 2010, Ther Deliv. 1 (1 ):133-48.
- Preferred receptor ligandsinclude synthetic cyclic or linear peptides such as derived from screening peptide libraries for binding to a particular cell surface structure or particular cell type, cyclic or linear RGD peptides, synthetic or natural carbohydrates such as sialic acid, galactose or mannose or synthetic ligands derived from reacting a carbohydrate for example with a peptide, antibodies specifically recognizing cell surface structures, folic acid, epidermal growth factor and peptides derived thereof, transferrin, anti-transferrin receptor antibodies, nanobodies and antibody fragments, or approved drugs that bind to known cell surface molecules.
- the preferred molecular weight of the polyethylene glycol) chainis 100 - 20,000 g/mol, more preferably 1 ,000 - 10,000 g/mol and most preferred is 1 ,000 - 5,000 g/mol.
- variable p in formula (L-1 )is preferably 1 .
- nis 1 or 2; n is 0 or 1 and m+n is > 2. In other words, if m is 1 , n must also be 1 , and if m is 2, n can be 0 or 1 . If n is 0, m must be 2. If n is 1 , m can be 1 or 2.
- variable n in formula (L-1 )is preferably 1 . It is more preferred that m is 1 and n is 1 .
- ais 1 or 2 and b is an integer of 1 to 4, or a is an integer of 1 to 4 and b is 1 or 2.
- acan be 1 or 2 and b can, independently, also be 1 or 2.
- ais 1 and b is an integer of 2 to 4 or a is an integer of 2 to 4 and b is 1 , and it is more preferred that one of a and b is 1 , and the other one is 2 or 3. It is still more preferred that a is 1 and b is 2, or that a is 2 and b is 1 .
- ais 1 and b is 2.
- the compound of formula (L-1 )is a compound of formula (L-1 a) and that the ionizable lipidoid in the lipidoid nanoparticles as referred to herein comprises, or more preferably consists of, an ionizable lipidoid of the following formula (L-1 a) or a protonated form thereof:
- one or more of the nitrogen atoms contained in the compound of formula (L-1 a)are protonated to provide a compound carrying a positive charge.
- the ionizable lipidoid in the lipidoid nanoparticles as referred to hereincomprises, or more preferably consists of, an ionizable lipidoid of the following formula (L-1b) or a protonated form thereof: wherein R 1A to R 6A are defined as in formula (L-1), including preferred embodiments thereof;
- one or more of the nitrogen atoms contained in the compound of formula (L-1 b)are protonated to provide a compound carrying a positive charge.
- the ionizable lipidoid in the lipidoid nanoparticles as referred to hereincomprises, or more preferably consists of, an ionizable lipidoid of the formula (L-1b) or a protonated form thereof, wherein R 7A is selected from C8-C18 alkyl and C8-C18 alkenyl having one C-C double bond, provided that at least two residues among R 1A to R 6A are -CH2-CH(OH)-R 7A , more preferably at least three residues among R 1A to R 6A , and still more preferably at least four residues among R 1A to R 6A are -CH2- CH(OH)-R 7A , wherein R 7A is selected from C8-C18 alkyl and C8-C18 alkenyl having one C-C double bond.
- R 7Ais selected from C8-C18 alkyl and C8-C18 alkenyl having one C-C double bond.
- lipidoid dL_05(R)As an example of a suitable lipidoid compound that can be used as an ionizable lipidoid in the context of the invention, reference can be made to the lipidoid dL_05(R) with the following structure, or to a protonated form thereof wherein one or more of the nitrogen atoms contained in the compound are protonated:
- the ionizable lipidwhich may be comprised by the nanoparticles referred to herein comprises or consists of an ionizable lipid of formula (L-2)
- R 1Bis an organic group comprising one or more primary, secondary or tertiary amino groups, or a protonated form thereof, wherein one or more of the nitrogen atoms contained in the primary, secondary or tertiary amino groups comprised by R 1B are protonated to provide a compound carrying a positive charge.
- the compound of formula (L-2)has the following structure:
- the ionizable lipidwhich may be comprised by the nanoparticles referred to herein comprises or consists of an ionizable lipid of formula (L-3) wherein
- R 1C and R 2Care independently selected from a C8-C18 alkyl group and a C8-C18 alkenyl group, preferably from a C12-C18 alkyl group and a C12-C18 alkenyl group,
- R 3Cis a C1-C6 alkanediyl group, preferably a C2 or C3 alkanediyl group, and R 4C and R 5C are independently hydrogen or C1-C3 alkyl, and are preferably methyl; or a protonated form thereof, wherein one or more of the nitrogen atoms contained in the compound of formula (L-3) are protonated to provide a compound carrying a positive charge.
- lipid of formula (L-3)As an example of an ionizable lipid of formula (L-3), reference can be made to DLin-MC3-DMA (6Z,9Z,28Z,31Z)-heptatriaconta-6,9,28,31-tetraene-19-yl 4-(dimethylamino)butanoate).
- the ionizable lipidwhich may be comprised by the nanoparticles referred to herein comprises or consists of an ionizable lipid of formula (L-4) wherein
- R 1D and R 2Dare independently selected from a C8-C18 alkyl group and a C8-C18 alkenyl group, preferably from a C12-C18 alkyl group and a C12-C18 alkenyl group,
- R 3Dis a C1-C6 alkanediyl group, preferably a C2 alkanediyl group, and
- R 4D and R 5Dare independently hydrogen or C1-C3 alkyl, and are preferably methyl; or a protonated form thereof, wherein one or more of the nitrogen atoms contained in the compound of formula (L-4) are protonated to provide a compound carrying a positive charge.
- the ionizable lipidwhich may be comprised by the nanoparticles referred to herein comprises or consists of an ionizable lipidoid of formula (b-5) wherein R 1E to R 5E are independently of each other selected from hydrogen, -CH 2 -CH(OH)-R 7E , -CH(R 7E )-CH 2 -OH,
- R 1E to R 5Eare preferably independently -CH 2 -CH(OH)-R 7E , wherein R 7E is selected from C8-C18 alkyl or C8-C18 alkenyl having one C-C double bond.
- Still another exemplary ionizable lipid suitable for use in the present inventionwhich may be comprised as an ionizable lipid in the nanoparticles referred to herein, or of which the ionizable lipid may consist is the ionizable lipid disclosed as “cationic lipid of Formula I” in the PCT application WO 2012/000104 A1 , starting on page 104 of this document, and including all specific embodiments thereof also discussed in this document.
- ionizable lipidoids suitable for use in the present inventionwhich may be comprised as an ionizable lipidoid in the nanoparticles referred to herein, or of which the ionizable lipidoids may consist are the ionizable lipidoids disclosed and claimed as “aminoalcohol lipidoids” in the PCT application WO 2010/053572 A2, including the compounds of all of the general formulae shown in the summary of the invention on page 4 of the document, and further defined in the remaining application.
- Still further exemplary ionizable lipidoids suitable for use in the present inventionwhich may be comprised as an ionizable lipidoid in the nanoparticles referred to herein, or of which the ionizable lipidoids may consist are the ionizable lipidoids disclosed as amine containing lipidoids of formulae I to V in the PCT application WO 2014/028487 A1 , including specific embodiments thereof.
- an ionizable lipid suitable for use in the present inventionwhich may be comprised as an ionizable lipid in the nanoparticles referred to herein, or of which the ionizable lipid may consist is the ionizable lipid ((4-hydroxybutyl)azanediyl)bis(hexan-6,1- diyl)bis(2-hexyldecanoate) (ALC-0315) or a protonated form thereof, wherein the nitrogen atom of the compound is protonated to provide a compound carrying a positive charge.
- the ionizable lipid((4-hydroxybutyl)azanediyl)bis(hexan-6,1- diyl)bis(2-hexyldecanoate) (ALC-0315) or a protonated form thereof, wherein the nitrogen atom of the compound is protonated to provide a compound carrying a positive charge.
- an ionizable lipid suitable for use in the present inventionwhich may be comprised as an ionizable lipid in the nanoparticles referred to herein, or of which the ionizable lipid may consist is the ionizable lipid (6Z,9Z,28Z,31Z)-heptatriaconta- 6,9,28,31 -tetraene-19-yl 4-(dimethylamino)butanoate or a protonated form thereof wherein the nitrogen atom of the compound is protonated to provide a compound carrying a positive charge.
- an ionizable lipidas an ionizable lipid in the nanoparticles referred to herein, or of which the ionizable lipid may be comprised in component (b) or of which component (b) may consist is the ionizable lipid heptadecan-9-yl 8-((2-hydroxyethyl)(6- oxo-6-(undecyloxy)hexyl)amino)octanoate (SM-102) or a protonated form thereof, wherein the nitrogen atom of the compound is protonated to provide a compound carrying a positive charge.
- SM-102ionizable lipid heptadecan-9-yl 8-((2-hydroxyethyl)(6- oxo-6-(undecyloxy)hexyl)amino)octanoate
- the nanoparticlesmay comprise, typically as a component of the lipid mix or lipidoid mix, one or more of the following components (c1 ) to (c6):
- the nanoparticlescomprise one or more of the components (c1 ) to (c6) not only encompasses combinations among (c1 ) to (c6), but also combinations of different components of one type, e.g. two components (c2), or combinations of different components of one type with other components of (c1 ) to (c6).
- Component (c1)is a lipid having a sterol structure.
- suitable lipidsare compounds which have a steroid core structure with a hydroxyl group at the 3-position of the A-ring.
- An exemplary non-ionizable lipid having a sterol structurewhich may be comprised by component (c1 ) or of which component (c1 ) may consist has a structure of formula (c1 -1 )
- R 1Lis a C3-C12 alkyl group.
- non-ionizable lipids having a sterol structurewhich may be comprised by component (c1 ) or of which component (c1 ) may consist include those disclosed by S. Patel et al., Naturally-occurring cholesterol analogues in lipid nanoparticles induce polymorphic shape and enhance intracellular delivery of mRNA, Nature Communications, 2020, 11 :983, in particular those illustrated in Fig. 2 of the publication.
- component (c1 )comprises or consists of cholesterol.
- Component (c2)is a phosphoglyceride.
- component (c2)comprises or consists of a phospholipid selected from a compound of formula (c2-1 ) wherein
- R 1F and R 2Fare independently selected from a C8-C18 alkyl group and a C8-C18 alkenyl group, preferably from a C12-C18 alkyl group and a C12-C18 alkenyl group, or a pharmaceutically acceptable salt thereof; and a phospholipid of formula (c2-2) wherein
- R 1G and R 2Gare independently selected from a C8-C18 alkyl group and a C8-C18 alkenyl group, preferably from a C12-C18 alkyl group and a C12-C18 alkenyl group, or a pharmaceutically acceptable salt thereof.
- component (c2)comprises or consists of 1 ,2-dipalmitoyl-sn-glycero-3- phosphocholine (DPPC) or a pharmaceutically acceptable salt thereof or 1 ,2-Distearoyl-sn- glycero-3-phosphocholine (DSPC) or a pharmaceutically acceptable salt thereof.
- DPPC1,2-dipalmitoyl-sn-glycero-3- phosphocholine
- DSPC,2-Distearoyl-sn- glycero-3-phosphocholine
- Exemplary salt forms of the compound of formula (c2-1 )include salts formed by the acidic -OH group with a base, or salts formed by the amino group with an acid.
- salts formed with a basemention may be made of alkali metal salts such as sodium or potassium salts; alkaline- earth metal salts such as calcium or magnesium salts and ammonium salts.
- salts formed with an acidmention may be made of a salt formed with the acidic groups of the nucleic acid, but other salts are not excluded, and mineral acid salts such as chloride, bromide, or iodide, sulfate salts, nitrate salts, phosphate salts, hydrogenphosphate salts, or dihydrogenphosphate salts, carbonate salts, and hydrogencarbonate salts may be mentioned as examples.
- Exemplary salt forms of the compound of formula (c2-2)include salts formed by the acidic -OH group attached to the P atom with a base, or salts formed by the quaternary amino group with an anion.
- salts formed with a basemention may be made of alkali metal salts such as sodium or potassium salts; alkaline-earth metal salts such as calcium or magnesium salts and ammonium salts.
- Component (c3)is a PEG-conjugated lipid, i.e. a lipid which is covalently linked with a polyethylene glycol chain.
- component (c3)comprises or consists of a PEG-conjugated lipid selected from a compound of formula (c3-1) wherein
- R 1H and R 2Hare independently selected from a C8-C18 alkyl group and a C8-C18 alkenyl group, preferably from a C12-C18 alkyl group and a C12-C18 alkenyl group, and p is an integer of 5 to 200, preferably 10 to 100, and more preferably 20 to 60; wherein
- R 1J and R 2Jare independently selected from a C8-C18 alkyl group and a C8-C18 alkenyl group, preferably from a C12-C18 alkyl group and a C12-C18 alkenyl group, and q is an integer of 5 to 200, preferably 10 to 100, and more preferably 20 to 60 or a pharmaceutically acceptable salt thereof, or a compound of formula (c3-3) wherein
- R 1K and R 2Kare independently a C8-C18 alkyl group or a C8-C18 alkenyl group, preferably a C12-C18 alkyl group or a C12-C18 alkenyl group, and q is an integer of 5 to 200, preferably 10 to 100, and more preferably 20 to 60.
- Exemplary salt forms of the compound of formula (c3-2)include salts formed by the acidic -OH group attached to the P atom with a base.
- salts formed with a basemention may be made of alkali metal salts such as sodium or potassium salts; alkaline-earth metal salts such as calcium or magnesium salts and ammonium salts.
- component (c3)comprises or consists of 1 ,2-dimyristoyl-sn- glycerolmethoxy(polyethylene glycol) (DMG-PEG), and still more preferably component d) comprises or consists of 1 ,2-dimyristoyl-sn-glycerolmethoxy(polyethylene glycol)-2000 (DMG- PEG2k) or 2-[(polyethylenglycol)-2000]-N,N-ditetradecylacetamid (ALC-0159).
- Component (c4)is a polysarcosine-conjugated lipid, i.e. a lipid which is covalently linked with a polymeric moiety of the formula (c4-1 ):
- poly sarcosine based lipidis N-TETAMINE-pSar25.
- Component (c5)is a PASylated lipid, e.g. a lipid which is covalently linked with a polymeric moiety formed by proline (pro)/alanine (ala)/serine (ser) repetitive residues.
- PASylated lipidused herein, the content of WO 2017/109087 A1 and EP 3394266 B1 is incorporated herein by reference.
- the PASylated lipidcan comprise e.g. a polypeptide consisting of at least 100 amino acid residues of proline, alanine and, optionally, serine, wherein said polypeptide forms a random coil.
- Component (c6)is a cationic polymer.
- Such polymers suitable for use in the formation of nanoparticles comprising a nucleic acidare known in the art.
- Exemplary suitable cationic polymersare discussed in A.C. Silva et al., Current Drug Metabolism, 16, 2015, 3-16, and in the literature referred to therein, in J.C. Kasper et al., J. Contr. Rel. 151 (2011 ), 246-255, in WO 2014/207231 and in the literature referred to therein, and in WO 2016/097377 and in the literature referred to therein.
- Suitable cationic oligomers or polymersinclude in particular cationic polymers comprising a plurality of units wherein an amino group is contained. The amino groups may be protonated to provide the cationic charge of the polymer.
- Polymersare preferred which comprise a plurality of units independently selected from the following (1 ), (2), (3) and (4):
- Particularly preferred as cationic polymersare the following four classes of polymers comprising a plurality of units wherein an amino group is contained.
- PEIpoly(ethylene imine)
- brPEIbranched poly(ethylene imine)
- the second preferred class of cationic polymersare polymers comprising a plurality of groups of the following formula (c6-1 ) as a side chain and/or as a terminal group, as they are disclosed as groups of formula (II) in WO 2014/207231 (which is incorporated in its entirety herein, applicant Ethris GmbH):
- variables a, b, p, m, n and R 2 to R 6are defined as follows, independently for each group of formula (c6-1 ) in a plurality of such groups: a is 1 or 2 and b is an integer of 1 to 4 or a is an integer of 1 to 4 and b is 1 or 2, preferably a is 1 and b is an integer of 2 to 4 or a is an integer of 2 to 4 and b is 1 , p is 1 or 2, m is 1 or 2; n is 0 or 1 and m+n is > 2; and
- the third preferred class of cationic polymersare polymers comprising a plurality of groups of the following formula (c6-2) as repeating units, as they are disclosed as groups of formula (III) in WO 2014/207231 (which is incorporated in its entirety herein, applicant Ethris GmbH): - N— ⁇ CH 2 — (CH 2 )— N— [CH 2 — (CH 2 ) b - N] p ⁇ — [CH 2 - (CH 2 )- N]—
- R3 r5(C6-2) wherein the variables a, b, p, m, n and R 2 to R 5 are defined as follows, independently for each group of formula (c6-2) in a plurality of such groups: a is 1 or 2 and b is an integer of 1 to 4 or a is an integer of 1 to 4 and b is 1 or 2, preferably a is 1 and b is an integer of 2 to 4; or a is an integer of 2 to 4 and b is 1 , p is 1 or 2, m is 1 or 2; n is 0 or 1 and m+n is > 2; and
- the fourth preferred class of cationic polymersis provided by a statistical copolymer as it is disclosed in WO 2016/097377 (which is incorporated in its entirety herein, applicant Ethris GmbH). It comprises a plurality of repeating units (a) independently selected from repeating units of the following formulae (a1) and (a2):
- a particularly preferred copolymeris a linear copolymer which comprises repeating units (a1 ) and (b1 ), or which consists of repeating units (a1) and (b1 ).
- a polyanion component which is different from a nucleic acidmay also be comprised, especially in addition to a nucleic acid if the nanoparticles comprise a nucleic acid as the preferred therapeutic agent.
- a polyanionexamples include polyglutamic acid and chondroitin sulfate. If such a polyanion component different from the nucleic acid is used in the nanoparticles, its amount is preferably limited such that the amount of anionic charges provided by the polyanion component is not higher than the amount of the anionic charges provided by the nucleic acid.
- the nanoparticles as referred to hereincomprise, more preferably consist of: i) optionally the therapeutic agent as discussed above as a component (a), preferably a nucleic acid, more preferably RNA, still more preferably mRNA, ii) the lipid or lipidoid mix comprising or consisting of: as a component (b) at least one selected from a permanently cationic lipid, an ionizable lipid and an ionizable lipidoid, more preferably an ionizable lipid or ionizable lipidoid, and as a preferred further component of the lipid or lipidoid mix one or more of:
- Exemplary suspensions comprising nanoparticles formed from the components listed abovewhich are also suitable for use as nanoparticle formulations in the context of the present invention, include those disclosed by S. Patel et al., Naturally-occurring cholesterol analogues in lipid nanoparticles induce polymorphic shape and enhance intracellular delivery of mRNA, Nature Communications, 2020, 11 :983.
- components of the nanoparticlesand in particular components (a) and (b), and optionally one or more of (c1 ) to (c6), are typically contained as a mixture in the nanoparticles.
- the lipid or lipidoid mixcomprises the components (c1 ), (c2) and (c3).
- the nanoparticlescomprise or consist of: optionally the therapeutic agent as a component (a), and the lipid or lipidoid mix comprising or consisting of i) 30 to 65 mol% of the at least one selected from a permanently cationic lipid, an ionizable lipid and an ionizable lipidoid as a component (b), preferably of the ionizable lipid or ionizable lipidoid, and, as a further component of the lipid or lipidoid mix, one or more of the following components: ii) 10 to 50 mol% of the lipid having a sterol structure (c1 ), iii) 4 to 50 mol% of the phosphoglyceride lipid (c2), iv) 0.5 to 10 mol% of one of the PEG-conjugated lipid (c3), the polysarcosine-conjugated lipid (c4) and the PAS
- the indicated molar percentagerefers to the total amount of these constituents of the nanoparticle if two or more of the cationic lipid or lipidoid, an ionizable lipid and an ionizable lipidoid are present as component (b).
- the molar percentages for components (c1 ) to (c6)are indicated with the proviso that not all of these components need to be present in the nanoparticles.
- the cationic polymercan be present or absent in the context of this preferred embodiment, but if it is present, it is used in the amount of 0.5 to 10 mol%.
- the amount of component(s) (c1), (c2), (c3), (c4), (c5) and/or (c6) in the context of this preferred embodimentis such that the sum of (b) and (c1) to (c6) amounts to 100 mol%.
- the nanoparticlescomprise, or consist of optionally the therapeutic agent as a component (a), which is more preferably mRNA, and the lipid or lipidoid mix comprising or consisting of at least one selected from a permanently cationic lipid, an ionizable lipid and an ionizable lipidoid as a component (b), preferably the ionizable lipid or ionizable lipidoid, the non-ionizable lipid having a sterol structure (c1), the phosphoglyceride lipid (c2), and the PEG-conjugated lipid (c3).
- awhich is more preferably mRNA
- the lipid or lipidoid mixcomprising or consisting of at least one selected from a permanently cationic lipid, an ionizable lipid and an ionizable lipidoid as a component (b), preferably the ionizable lipid or ionizable lipidoid, the non
- the nanoparticlescomprise, more preferably consist of: optionally the therapeutic agent as a component (a), and the lipid or lipidoid mix comprising or consisting of
- the lipidoid nanoparticles in the context of the present inventionpreferably comprise
- R 1A to R 6Aare independently selected from hydrogen and -CH2-CH(OH)-R 7A , wherein R 7A is selected from C8-C18 alkyl and C8-C18 alkenyl having one C-C double bond, provided that at least two residues among R 1A to R 6A are -CH2-CH(OH)-R 7A , more preferably at least four residues among R 1A to R 6A are -CH2-CH(OH)-R 7A , wherein R 7A is selected from C8-C18 alkyl and C8-C18 alkenyl having one C-C double bond, or a protonated form thereof wherein one or more of the nitrogen atoms indicated in formula (L-1 b) are protonated to provide a cationic lipidoid;
- (c1)a non-ionizable lipid having a sterol structure of formula (c1 -1 ) wherein R 1L is a C3-C12 alkyl group;
- (c2)a phosphoglyceride of formula (c2-2) wherein R 1G and R 2G are independently selected from a C8-C18 alkyl group and a C8-C18 alkenyl group, preferably from a C12-C18 alkyl group and a C12-C18 alkenyl group, or a pharmaceutically acceptable salt thereof; and (c3) a PEG conjugated lipid of formula (c3-1 ) wherein R 1H and R 2H are independently selected from a C8-C18 alkyl group and a C8-C18 alkenyl group, preferably from a C12-C18 alkyl group and a C12-C18 alkenyl group, and p is an integer of 5 to 200, preferably 10 to 100, and more preferably 20 to 60.
- the lipidoid dL_05(R) with the formula shown abovewould be a particularly preferred variant of the ionizable lipid.
- composition of lipid nanoparticles suitable for use in the context of the present inventioncomprises a nucleic acid, more preferably mRNA, as a therapeutic agent, and a lipid mix comprising the ((4-hydroxybutyl)azandiyl)bis(hexan-6,1-diyl)bis(2- hexyldecanoate) or a protonated form thereof wherein the nitrogen atom of the compound is protonated to provide a compound carrying a positive charge as a ionizable lipid, and optionally further comprising one or more of the following components (d1) to (d8):
- theyfurther comprise at least (d1 ), (d2) and (d3), and still more preferably they comprise all of (d1 ) to (d8).
- composition of lipid nanoparticles suitable for use in the context of the present inventioncomprises a nucleic acid, more preferably mRNA, as a therapeutic agent, and a lipid mix comprising heptadecan-9-yl 8-((2-hydroxyethyl)(6-oxo-6- (undecyloxy)hexyl)amino)octanoate (SM-102) or a protonated form thereof wherein the nitrogen atom of the compound is protonated to provide a compound carrying a positive charge as an ionizable lipid, and optionally comprising one or more of the following components (e1) to (e7):
- theyfurther comprise at least (e1 ), (e2) and (e3), and still more preferably they comprise all of (e1 ) to (e7).
- composition of lipid nanoparticles suitable for use in the context of the present inventioncomprises a nucleic acid, more preferably mRNA, as a therapeutic agent, and a lipid mix comprising DLin-MC3-DMA ((6Z,9Z,28Z,31Z)- heptatriaconta-6,9,28,31-tetraen-19-yl-4-(dimethylamino) butanoate) or a protonated form thereof wherein the nitrogen atom of the compound is protonated, and optionally comprising one or more of the following components (e1) to (e7):
- components (e1), (e2) and (e3)are present, and still more preferably they comprise all of (e1) to (e6).
- the nanoparticlespreferably contain a therapeutic agent, preferably a nucleic acid, more preferably RNA and still more preferably mRNA, in which case the composition of the nanoparticles is preferably such that the weight ratio in the nanoparticles of the sum of the weights of components other than the therapeutic agent to the weight of the therapeutic agent is in the range of 50:1 to 1 :1 , more preferably 40:1 to 2:1 and most preferably 30:1 to 3:1 .
- the N/P ratioi.e. the ratio of the number of amine nitrogen atoms provided by the ionizable lipid, the ionizable lipidoid and/or the permanently cationic lipid to the number of phosphate groups provided by any nucleic acid of the nanoparticles, if a nucleic acid is comprised as a therapeutic agent, is preferably in the range of 0.5 to 20, more preferably in the range of 0.5 to 10.
- the lipid or lipidoid nanoparticlese.g. the lipid or lipidoid nanoparticles in a suspension formulation, preferably have a Z-average diameter in the range of 10 to 500 nm, more preferably in the range of 10 to 250 nm, still more preferably 20 to 200 nm.
- the indicated particle diameteris the hydrodynamic diameter of the particles, as determined by dynamic light scattering (DLS). Measurements are generally carried out at 25 °C.
- the polydispersity index of the nanoparticlesis preferably in the range of 0.02 to 0.4, more preferably in the range of 0.03 to 0.2.
- the polydispersity indexcan be determined by dynamic light scattering (DLS). Measurements are generally carried out at 25 °C.
- a formulatione.g. a suspension formulation, containing different lipid or lipidoid nanoparticles as defined above, i.e. particles which differ in terms of their components.
- the nanoparticles contained in the formulationare composed of the same components.
- the nanoparticles comprising an active agentcan be conveniently prepared by mixing a solution containing the active agent, e.g. in an aqueous solvent containing a buffer, such as a citrate buffer with a pH of 4.5, and optionally containing a salt such as sodium chloride, and a solution containing the ionizable lipid or ionizable lipidoid in an organic solvent, e.g. in ethanol. Further optional components can be incorporated e.g. by adding them to one of the two solutions.
- a buffersuch as a citrate buffer with a pH of 4.5
- a saltsuch as sodium chloride
- the nanoparticles generated in this mannercan be further processed by chromatography and/or dialysis and/or tangential flow filtration (TFF) in order to obtain the nanoparticles in a desired liquid composition.
- TFFtangential flow filtration
- nanoparticle suspensionIn order to provide a nanoparticle suspension, it is also possible to rely on lyophilized nanoparticles prepared following the above-mentioned procedure followed by freeze drying, which are subsequently re-suspended in an aqueous vehicle solution.
- a lipid nanoparticle formulation or lipidoid nanoparticle formulation as referred to hereinis preferably a suspension formulation wherein the nanoparticles are contained in a liquid vehicle solution.
- the liquid vehicle solutionis preferably an aqueous vehicle solution.
- an aqueous vehicle solutionis a solution wherein the main solvent, in terms of the total volume of solvent(s), is water, preferably a solution containing more than 70 % of water, more preferably more than 90 % of water, as a solvent, indicated as the volume percentage of water in the total volume of solvent(s) contained in the vehicle solution (at a temperature of 25 °C). Most preferably, water is the only solvent in the vehicle solution. Thus, the vehicle solution is a liquid at room temperature (e.g. 25 °C).
- an aqueous solvent as referred to hereinis water or a mixture of solvents wherein water represents the main solvent, in terms of the total volume of solvent(s).
- the aqueous solventcontains more than 70 % of water, more preferably more than 90 % of water, as a solvent, indicated as the volume percentage of water in the total volume of solvent(s) contained in the aqueous solvent (at a temperature of 25 °C). Most preferably, water is the only solvent in the aqueous solvent.
- the weight per volume ratio of the nanoparticles in the vehicle solution of a suspension formulationis preferably in the range 0.1 g/L to 300 g/L, more preferably 0.2 g/L to 300 g/L, still more preferably 0.5 g/L to 250 g/L and most preferably 0.5 g/L to 125 g/L (as measured at 25 °C).
- the concentration of the nucleic acid provided by the lipid or lipidoid nanoparticles in the suspensionpreferably ranges from 0.01 to 10 mg/ml, more preferably from 0.02 to 10 mg/ml, still more preferably from 0.05 to 5 mg/mL, and most preferably from 0.05 to 2.5 mg/ml, based on the total volume of the suspension (as measured at 25 °C).
- the lipid or lipidoid nanoparticles contained in the suspensionpreferably have a Z-average diameter in the range of 10 to 500 nm, more preferably in the range of 10 to 250 nm, still more preferably 20 to 200 nm.
- the indicated particle diameteris the hydrodynamic diameter of the particles, as determined by dynamic light scattering (DLS). Measurements are generally carried out at 25 °C.
- the polydispersity index of the nanoparticles contained in the suspensionis preferably in the range of 0.02 to 0.4, more preferably in the range of 0.03 to 0.2.
- the polydispersity indexcan be determined by dynamic light scattering (DLS). Measurements are generally carried out at 25 °C.
- the surfactantcomprises, more preferably is (or consists of) a nonionic surfactant.
- suitable nonionic surfactantsinclude fatty alcohol ethoxylates, fatty acid ethoxylates, block copolymers of ethylene oxide and propylene oxide, alkylphenol ethoxylates or oligomers of alkylphenol ethoxylates, fatty acid esters of sorbitol, ethoxylated fatty acid esters of sorbitol, fatty acid esters of glycerol, ethoxylated castor oil and ethoxylated vitamin E.
- the surfactant used in the context of the inventione.g. as a component of the lipid nanoparticle (LNP) formulation or lipidoid nanoparticle (LiNP) formulation, preferably comprises, still more preferably is (or consists of) at least one surfactant selected from the group of fatty alcohol ethoxylates, fatty acid ethoxylates, block copolymers of ethylene oxide and propylene oxide, alkylphenol ethoxylates or oligomers of alkylphenol ethoxylates, fatty acid esters of sorbitol, ethoxylated fatty acid esters of sorbitol, fatty acid esters of glycerol, ethoxylated castor oil and ethoxylated vitamin E.
- the block copolymer of ethylene oxide and propylene oxideis a poloxamer.
- the poloxameris preferably one which contains one polypropylene oxide) block B of formula (p-
- the surfactant used in the context of the inventioncomprises, or preferably is (or consists of), at least one selected from the group of poloxamer 124, poloxamer 188, poloxamer 338, poloxamer 407, polysorbate 20 or Tween-20, polysorbate 80 or Tween-80, polyoxyethylenelaurylether, poyloxyethylene-35 castor oil, D-a-tocopherol polyethylene glycol 1000 succinate, and tyloxapol.
- the surfactantcomprises, more preferably is (or consists of), a poloxamer, e.g. the preferred poloxamer as discussed above, still more preferably a poloxamer selected from the list of poloxamer 124, poloxamer 188, poloxamer 338, and poloxamer 407, or from combinations thereof, most preferably P188.
- a poloxamere.g. the preferred poloxamer as discussed above, still more preferably a poloxamer selected from the list of poloxamer 124, poloxamer 188, poloxamer 338, and poloxamer 407, or from combinations thereof, most preferably P188.
- a surfactant as referred to hereinis typically a surfactant which meets the requirements set forth in the aspects of the invention as defined above, e.g. in the first, second or third aspect, or which has been classified as being suitable as a stabilizing agent by the method in accordance with the fourth or fifth aspect.
- lipid nanoparticle (LNP) formulation or lipidoid nanoparticle (LiNP) formulation in the form of a suspension formulationsuch as an aqueous suspension formulation
- the vehicle solution wherein the nanoparticles are suspendedcomprises the surfactant dissolved therein.
- thisdoes not exclude the possibility that a certain amount of the surfactant molecules is adsorbed to the lipid or lipidoid nanoparticles which are contained in the suspension.
- the surfactantin the context of the invention, it has been found that a beneficial effect of the surfactant can be achieved already with relatively low concentrations of a surfactant, e.g. of 0.01 % (w/v) in the suspension.
- a surfactante.g. 0.01 % (w/v) in the suspension.
- the surfactantis contained in amounts of 0.01 % (w/v) or more in the suspension, with regard to the total volume of the suspension of the nanoparticles in the vehicle solution (typically measured at 25 °C).
- the inventioninvolves an incorporation of the surfactant into a suspension formulation, preferably into the vehicle solution, more preferably an aqueous vehicle solution, in an amount of 0.01 to 10 % (w/v), preferably 0.1 to 10 % (w/v), more preferably 0.25 to 5 % (w/v), still more preferably 0.33 to 2.5 % (w/v), even more preferably 0.45 to 1 .5 % (w/v), and most preferably 0.5 to 1 .5 % (w/v), with regard to the total volume of the suspension of the nanoparticles in the vehicle solution.
- the indication of the concentration of a substance in % (w/v) or (weight/volume)corresponds to the amount of the substance in g in a volume of 100 mL, typically measured at 25 °C, so that 1 % (w/v) corresponds to 1 g the surfactant per 100 mL of the total volume of the suspension.
- the methods in accordance with the inventionmay involve the incorporation of the surfactant into a nanoparticle suspension formulation in an amount of e.g. 0.01 to 10 % (w/v), preferably 0.1 to 10 % (w/v), more preferably 0.25 to 5 % (w/v), still more preferably 0.33 to 2.5 % (w/v), even more preferably 0.45 to 1 .5 % (w/v), and most preferably 0.5 to 1 .5 % (w/v), with regard to the total volume of the suspension of the nanoparticles in the aqueous vehicle solution (typically measured at 25 °C).
- concentrations of 0.5 to 1 .5% (w/v)are particularly preferred, as outlined above, the invention in its various aspects also provides and relates to suspension formulations wherein the concentration of the surfactant is lower, e.g. in the range of 0.01 to 0.45 % (w/v), or 0.1 to 0.40 % (w/v).
- the surfactantis essentially not attached to the nanoparticles, e.g. that it is essentially not contained in the nanoparticles and essentially not adhering to the nanoparticles.
- the vehicle solutionfurther comprises at least one of a sugar and a salt, more preferably sucrose and NaCI.
- the surfactantcan be conveniently incorporated into a nanoparticle suspension formulation, e.g. by a method including adding the surfactant to a suspension comprising an aqueous vehicle solution and the lipid or lipidoid nanoparticles, or including adding the lipid or lipidoid nanoparticles to an aqueous vehicle solution comprising the surfactant.
- a methodincluding adding the surfactant to a suspension comprising an aqueous vehicle solution and the lipid or lipidoid nanoparticles, or including adding the lipid or lipidoid nanoparticles to an aqueous vehicle solution comprising the surfactant.
- the nanoparticlesare provided in lyophilized form, they can be re-suspended in an aqueous vehicle solution containing a surfactant.
- a method for the preparation of a suspension formulation of lipid nanoparticles or lipidoid nanoparticles as defined hereinmay comprise generating a preparation of lipid nanoparticles or lipidoid nanoparticles by mixing of at least one selected from a permanently cationic lipid, an ionizable lipid and an ionizable lipidoid dissolved in an organic phase with a therapeutic agent dissolved in an aqueous solution, and combining the nanoparticles with a surfactant to obtain a suspension of the nanoparticles in an aqueous vehicle solution.
- the methodcomprises the following steps: i) generating a preparation of lipid nanoparticles or lipidoid nanoparticles by mixing of at least one selected from a permanently cationic lipid, an ionizable lipid and an ionizable lipidoid dissolved in an organic phase with a therapeutic agent dissolved in an aqueous solution, ii) diluting the preparation of lipid nanoparticles or lipidoid nanoparticles by dilution with a first solution, iii) concentrating the diluted preparation of lipid nanoparticles or lipidoid nanoparticles by buffer exchange using ultra/diafiltration by TFF, wherein a second solution is used for the ultra/diafiltration, iv) obtaining a suspension of lipid nanoparticles or lipidoid nanoparticles in an aqueous vehicle solution comprising a surfactant, wherein the first solution comprises between 0.01 % w/v and 10% w/v surfact
- the surfactantis added both with the first and the second solution.
- 30 to 70 wt% of the surfactantpreferably 40 to 60 wt%, and more preferably 45 to 55 wt% of the surfactant, based on the total weight of the surfactant in the suspension obtained in step iv
- 70 to 30 wt% of the surfactantpreferably 60 to 40 wt%, and more preferably 55 to 45 wt% of the surfactant, based on the total weight of the surfactant in the suspension obtained in step iv
- the second solutionsuch that the sum of the amount of surfactant added with the first and the second solution is 100 wt%.
- it is preferred that approximately half of the surfactantis added with the first solution and approximately half of the surfactant is added with the second solution.
- lipid nanoparticle formulations or lipidoid nanoparticle formulationsare provided which are stabilized by the use of a surfactant.
- the term “stabilized”refers to the state of a lipid nanoparticle (LNP) formulation where its physical and chemical integrity is maintained over time under specified conditions. This includes preferably preserving particle size, structural integrity, and/or homogeneity, and preventing aggregation or fusion. Stabilization may also protect encapsulated therapeutic agents from degradation, ensuring sustained efficacy and controlled release.
- Surfactantslike poloxamers, aid in stabilization by reducing surface tension and preventing agglomeration, enhancing the overall stability of the lipid nanoparticle dispersion.
- Stabilitycan be assessed, e.g., through particle size, encapsulation efficiency, mRNA integrity, and potency measurements.
- Particle sizecan be measured using Dynamic Light Scattering (DLS) with instruments such as the Malvern Zetasizer.
- Encapsulation efficiencycan be quantified using assays like the Ribogreen assay.
- the integrity of mRNAcan be assessed using a Fragment Analyzer.
- Potencycan be measured using Enzyme-Linked Immunosorbent Assay (ELISA).
- ELISAEnzyme-Linked Immunosorbent Assay
- a stabilized lipid or lipidoid formulationcan have a Z average size of 25 to 150 nm for extended periods of time such as weeks or months.
- a nanoparticle suspensioncan be stabilized in the context of the invention e.g. against particle aggregation under a physical stress condition.
- the surfactantcan be incorporated into the suspension, preferably incorporated as an excipient into the aqueous vehicle solution.
- a membrane foulingcan be avoided.
- the LNPs and/or LiNPshave not been lyophilized.
- the surfactantis added before a lyophilization process. In some embodiments, the surfactant is not present in the vehicle solution during a lyophilization process.
- the presence of the surfactantdoes not cause a change in the biological activity of the nanoparticle.
- Biological activitymeans expression level in the target cell(s) of the therapeutic nucleic acid.
- the biological activitycan e.g. be quantified by in vitro transfection of cell lines (e.g. HEK-293) with the nanoparticle followed by quantification of the produced nucleic acid by Southern/northern blot or of protein via an ELISA.
- the detected protein level calculated as average of three measurements per concentrationmust not differ more than 10%, preferably not more than 5%, more preferably is not statistically different when performing the identical assay with the same LNP or LiNP without a surfactant.
- the presence of the surfactantdoes not cause a change in the physical properties of the nanoparticle measured as the hydrodynamic diameter of the nanoparticle and as the proportion of encapsulated therapeutic agent, preferably a nucleic acid.
- the hydrodynamic diameter of the nanoparticlecan e.g. be measured via dynamic light scattering (also Photon correlation spectroscopy).
- the average of three measurements of the hydrodynamic diameter of the nanoparticle in presents of the surfactantmust not differ more than 5%, preferably not more than 1%, more preferably not statistically different of the same nanoparticle in absence of the surfactant.
- the viscosity change of the surfactantmust be taken into account during the measurement.
- the percentage of encapsulated nucleic acidcan e.g. be determined by measuring the fluorescence intensity in a RiboGreen assay. The nanoparticle is analyzed under two different conditions, untreated samples for external nucleic acid and samples treated with Triton X-100 for total mRNA.
- the percent content of encapsulated nucleic acidis calculated.
- the value calculated form an average of three measurements of the nanoparticle in absence of the surfactantshould not differ more than 5%, optionally not more than 3%, better not statistically different) from the same nanoparticle in presents of the surfactant.
- a measure which is taken for the stabilization of a nanoparticle suspension against particle aggregationmay prevent the aggregation of the nanoparticles, or may reduce the degree of aggregation of the nanoparticles compared to a situation where the concerned measure is not applied.
- the stabilization of the nanoparticle suspensionis evidenced by an increase of the Z-average particle size of the suspended particles under a physical stress condition of less than 50 %, more preferably less than 20 %, still more preferably less than 10 % and most preferably by the absence of such an increase.
- stabilization of the nanoparticle suspension against particle aggregation under a physical stress conditionmeans that an aggregation of the nanoparticles is prevented or reduced which would be observed in the absence of the stabilization when the nanoparticle suspension is exposed to a physical stress condition.
- Conditions of physical stress to which the nanoparticle suspension can be exposedare frequently physical stress conditions that are encountered during the handling or during a transport of the suspension. They include, e.g., a quick movement of a volume of the suspension which would cause a collision of nanoparticles contained in a non -stabilized suspension.
- a physical stress conditionreference may be made to shaking, stirring, vibrating, mixing, inverting, tapping, or dropping of the nanoparticle suspension, or, e.g., to a physical stress condition caused by pumping the nanoparticle suspension or by its withdrawal into a syringe.
- conditions of physical stressinclude not only conditions to which the nanoparticle suspension is exposed during its regular handling, but also conditions to which the suspension may be exposed exceptionally (such as a transport under difficult conditions) or inadvertently (such as dropping a sample of the suspension).
- compositions as referred to hereinsuch as the lipid nanoparticle (LNP) formulations or lipidoid nanoparticle (LiNP) formulations as referred to herein comprising a therapeutic agent, are particularly useful in a medical setting and in the treatment or prevention of diseases and disorders, preferably in the treatment or prevention of a disease or disorder relying on a nucleic acid, such as RNA, preferably mRNA, as an active agent.
- RNApreferably mRNA
- compositions as referred to hereinsuch as the lipid nanoparticle (LNP) formulations or lipidoid nanoparticle (LiNP) formulations as referred to herein comprising a therapeutic agent, are suitable for administration to a subject.
- the therapeutic agentpreferably the nucleic acid such as RNA, preferably the mRNA, contained in the nanoparticles can also be administered to the subject.
- the therapeutic agentpreferably the nucleic acid contained in the lipid or lipidoid nanoparticles particles
- the therapeutic agentmay be delivered to target cells.
- the term “delivered to target cells”preferably means transfer of the nucleic acid into the cell.
- the administrationcan be accomplished in various ways known to the skilled practitioner, including an administration to or via the respiratory tract, e.g. by an aerosolization of the nanoparticles, or an intramuscular or intravenous administration.
- diseases or disorderscan be treated or prevented.
- the term “disease”refers to any conceivable pathological condition that can be treated, prevented or vaccinated against by employing the suspension Said diseases may, e.g., be inherited, acquired, infectious or non-infectious, age-related, cardiovascular, metabolic, intestinal, neoplastic (in particular cancer) or genetic.
- a diseasecan be based, for example, on irregularities of physiological processes, molecular processes, biochemical reactions within an organism that in turn can be based, for instance, on the genetic equipment of an organism, on behavioral, social or environmental factors such as the exposure to chemicals or radiation.
- compositions as referred to hereinsuch as the nanoparticle (LNP) formulations or lipidoid nanoparticle (LiNP) formulations comprising a therapeutic agent as referred to herein, may generally be suitable for the treatment or prevention of a disease selected from Table A as disclosed herein above.
- the pharmaceutical compositionssuch as the nanoparticle (LNP) formulations or lipidoid nanoparticle (LiNP) formulations comprising a therapeutic agent as referred to herein, may generally be suitable for the treatment or prevention of a disease selected disease selected from viral diseases, ciliopathies, or autoimmune diseases, and respiratory tract diseases, even more preferably selected from a lung disease, an airway disease or a nasal disease, more preferably a lung viral disease, lung ciliopathies and a lung autoimmune disease.
- a disease selected diseaseselected from viral diseases, ciliopathies, or autoimmune diseases, and respiratory tract diseases, even more preferably selected from a lung disease, an airway disease or a nasal disease, more preferably a lung viral disease, lung ciliopathies and a lung autoimmune disease.
- the lung disease or lung viral diseaseis at least one selected from pneumonia and asthma; the airway disease is at least one selected from bronchitis, viral induced asthma, lung fibrosis and COPD; and/or the nasal disease is at least one selected from rhinitis and sinusitis.
- the disease to be treated or preventedis a disease selected from the list consisting of pulmonary alveolar proteinosis (PAP), interstitial lung disease such as pulmonary fibrosis, e.g. idiopathic pulmonary fibrosis, viral infections such as Influenza and COVID-19, acute respiratory distress syndrome (ARDS), non-tuberculous mycobacterial (NTM) infection, lung cancer, fungal infections caused by Aspergillus sp., such as aspergillosis, fungal sinusitis, otomycosis, keratitis, and onychomycosis, preferably those caused by Aspergillus fumigatus and Aspergillus flavus, infections caused by Mycobacterium tuberculosis, Pseudomonas aeruginosa, Pneumocystis sp., Plasmodium sp., Cryptococcus sp., Nocardia sp., and combinations thereof.
- PAP
- the diseaseis selected from the list consisting of (autoimmune) pulmonary alveolar proteinosis (PAP), interstitial lung disease such as pulmonary fibrosis, e.g. idiopathic pulmonary fibrosis, viral infections such as Influenza and COVID-19, acute respiratory distress syndrome (ARDS), non-tuberculous mycobacterial (NTM) infection, lung cancer, or fungal infections caused by Aspergillus sp., such as aspergillosis, fungal sinusitis, otomycosis, keratitis, and onychomycosis, preferably those caused by Aspergillus fumigatus and Aspergillus flavus, infections caused by Mycobacterium tuberculosis, Pseudomonas aeruginosa, Pneumocystis sp., and Plasmodium sp.
- PAPpulmonary alveolar proteinosis
- interstitial lung diseasesuch as pulmonary fibrosis,
- the inventionalso provides a formulation of lipid nanoparticles or lipidoid nanoparticles comprising a therapeutic agent as described herein for use in the treatment or prevention of a disease.
- the formulation of lipid nanoparticles or lipidoid nanoparticles according as described thereincan be used in a method for the treatment or prevention of a disease, which method includes administering the suspension or formulation to a subject in need thereof.
- the inventionalso provides the formulation of lipid nanoparticles or lipidoid nanoparticles comprising a therapeutic agent as described herein for use as a medicament.
- the inventionprovides the formulation of lipid nanoparticles or lipidoid nanoparticles comprising a therapeutic agent as described herein for use in vaccination or immunization.
- the formulation of lipid nanoparticles or lipidoid nanoparticles comprising a therapeutic agent as described hereincan be used in a method for vaccination or immunization, which method includes administering the suspension or formulation to a subject in need thereof.
- the inventionprovides a method of inducing an immune response against a target pathogen in a subject in need thereof, the method comprising administering a formulation of lipid nanoparticles or lipidoid nanoparticles comprising a therapeutic agent as described herein to the subject.
- the inventionprovides a formulation of lipid nanoparticles or lipidoid nanoparticles comprising a therapeutic agent as described therein for use in the treatment of cancer.
- the formulation of lipid nanoparticles or lipidoid nanoparticles comprising a therapeutic agent as described thereincan be used in a method for the treatment of cancer, which method includes administering the formulation to a subject in need thereof.
- the inventionprovides a method of avoiding or for alleviating side effects in a therapy with lipid nanoparticles or lipidoid nanoparticles comprising at least one therapeutic agent as they are described herein, wherein the method comprises the steps: i) determine whether lipid nanoparticles or lipidoid nanoparticles in a pharmaceutical composition comprising the lipid nanoparticles or lipidoid nanoparticles aggregate when subjected to a mechanical stress or a temperature stress, by determining their aggregation level before and after subjecting said pharmaceutical composition to said mechanical or temperature stress ii) if the lipid nanoparticles or lipidoid nanoparticles show aggregation after the test of step (i), then add to the lipid nanoparticles or lipidoid nanoparticles formulation a surfactant as defined herein to obtain a LNP or LiNP suspension with a final surfactant concentration between 0.01 % w/v and 10% w/v surfactant, preferably between
- the inventionfurther provides a method of reducing one or more side effects associated with a vaccine formulation or an anticancer formulation comprising lipid nanoparticles or lipidoid nanoparticles carrying a therapeutic agent such as nucleic acid, preferably RNA, more preferably mRNA, as they are described herein, the method comprising modifying the vaccine formulation or an anticancer formulation by adding a surfactant as described herein to a vaccine formulation or anticancer formulation comprising a suspension of the lipid nanoparticles or lipidoid nanoparticles.
- a therapeutic agentsuch as nucleic acid, preferably RNA, more preferably mRNA
- the surfactantrepresents between 0.01 % w/v and 10% w/v surfactant, preferably between 0.1 % w/v and 10% surfactant, more preferably between 0.25% w/v surfactant and 5% w/v surfactant, still more preferably between 0.33% w/v surfactant and 2.5% w/v surfactant, even more preferably between 0.45% w/v surfactant and 1 .5% w/v surfactant, most preferably between 0.5% w/v and 1 .5% w/v surfactant.
- the inventionprovides a method of reducing the occurrence or severity of one or more side effects associated with a LNP/LiNP based vaccine in a subject, the method comprising administering a vaccine formulation or an anticancer formulation comprising the formulation of lipid nanoparticles or lipidoid nanoparticles comprising a therapeutic agent as described herein to the subject.
- the reduction of the occurrence or severity of one or more side effectsmay be caused by a reduction of LNP/LiNP aggregation, as it may be measured, e.g., by determining the hydrodynamic diameter of the nanoparticle, for example by via dynamic light scattering or photon correlation spectroscopy.
- a pharmaceutical compositionsuch as the formulation of lipid or lipidoid nanoparticles comprises a therapeutic agent
- the therapeutic agentis included in an effective amount composition or formulation.
- effective amountrefers to an amount sufficient to induce a detectable therapeutic response or a preventive effect in the subject to which the pharmaceutical composition or formulation is to be administered.
- the content of the therapeutic agentis not limited as far as it is useful for treatment or prevention as described above.
- a suspension formulation wherein nanoparticles comprising the nucleic acid are containedpreferably comprises the particles in an amount so as to provide the nucleic acid contained in the particles at a concentration of 0.01 to 10 mg/ml, more preferably 0.02 to 10 mg/ml, still more preferably 0.05 to 5 mg/ml and most preferably 0.05 to 2.5 mg/ml, based on the total volume of the composition.
- a pharmaceutical compositionsuch as a formulation of lipid nanoparticles or lipidoid nanoparticles according comprising a therapeutic agent as described therein is administered to a subject, it will be administered in an effective amount.
- Exemplary subjectsinclude a mammal such as a dog, cat, pig, cow, sheep, horse, rodent, e.g., rat, mouse, and guinea pig, or a primate, e.g., gorilla, chimpanzee, and human.
- a mammalsuch as a dog, cat, pig, cow, sheep, horse, rodent, e.g., rat, mouse, and guinea pig, or a primate, e.g., gorilla, chimpanzee, and human.
- the subjectis a human.
- the Langmuir-Blodgett (LB) techniqueis a method for preparing organized molecular assemblies with precision and control over molecular orientation and layer thickness. This methodology builds upon the principle that amphiphilic molecules, when spread on an aqueous subphase, can form stable monolayers at the air-water interface. The organization and packing density of these molecules can be meticulously manipulated by adjusting the surface pressure.
- the Langmuir surface pressure/area (TT-A) isothermis a foundational concept within the realm of surface science, e.g. when it comes to studying monolayers of amphiphilic molecules at the air-water interface.
- amphiphilic moleculesmoleculess possessing both hydrophilic and hydrophobic components
- the area available to these moleculescan be altered using mobile barriers, which in turn changes the surface pressure (IT) of the monolayer.
- ITsurface pressure
- Phases of the MonolayerDifferent regions on the isotherm correspond to different molecular arrangements or phases. These can include gaseous, liquid-expanded, and liquid-condensed phases.
- Collapse PressureThe pressure at which the monolayer undergoes a phase transition and collapses can be identified on the isotherm. It might be a relevant parameter as it provides insight into the stability and strength of the monolayer.
- the area occupied by a single molecule in the monolayercan be determined from the isotherm. This is particularly useful for understanding the geometry and orientation of the molecules.
- Phase TransitionsChanges in the slope or abrupt transitions in the isotherm are indicative of phase changes within the monolayer. These can be used to deduce molecular interactions and behavior under different conditions.
- Intermolecular InteractionsThe shape and features of the isotherm can provide insights into the nature and strength of intermolecular forces in the monolayer.
- Figure 1shows a plot showing TFF processing time for different P188 solutions.
- Figure 2shows a bar diagram of the refractive index of different P188 solutions before and after TFF processing.
- Figure 3shows a Langmuir pressure-area plot for four representative poloxamer 188 samples in aqueous solutions. Considering the isotherm maximum surface pressure, two clear groups of poloxamers can be distinguished. S8 and S9 with a maximum surface pressure above 4 mN/m correspond to poloxamer P188 samples that lead to tangential flow filtration membrane fouling while samples S2 and S7 are suitable for lipid nanoparticle purification and processing.
- Figure 4shows a Langmuir pressure-area plot of three consecutive compression-expansion cycles for representative poloxamer 188 samples S1 to S11 in aqueous solutions. Considering the isotherm maximum surface pressure, samples S8 and S9 clearly stand out with a maximum surface pressure above 4 mN/m, identifying poloxamer P188 samples that lead to tangential flow filtration membrane fouling.
- Figure 5shows a Langmuir pressure-area plot for four representative poloxamer 188 samples in aqueous solution with lipid mix.
- the large hysteresis broadening observed in samples S8 and S9can be used to identify poloxamer P188 samples that lead to tangential flow filtration membrane fouling while samples S2 and S7 are suitable for lipid nanoparticle purification and processing.
- Figure 6shows a Langmuir pressure-area plot of three consecutive compression-expansion cycles for representative poloxamer 188 samples S1 to S11 in aqueous solutions and lipid mix. Considering the isotherm hysteresis, samples S8 and S9 clearly stand out with broad hysteresis identifying poloxamer P188 samples that lead to tangential flow filtration membrane fouling.
- Figure 7shows each calculated AIT datapoint obtained over three consecutive compressionexpansion Langmuir trough cycles, for a lipid mix representative of a lipid nanoparticle formulation deposited over 11 different poloxamer 188 (S1 to S11 ) aqueous subphases.
- Superimposed ATT datapointsare shown as a broader distribution to enable their visualization. Therefore, an isotherm with generally tight hysteresis and only limited regions of broader hysteresis (e.g. Figure 6, S7) will be represented here with a pear-shaped distribution. Consequently, a hysteresis broader features (e.g. Figure 6, S8 and S9) leads to higher An values and a narrower distribution of An datapoints.
- Two clear groups of poloxamerscan be distinguished and the narrow-shaped plots correspond to poloxamer P188 samples that lead to tangential flow filtration membrane fouling.
- Figure 8shows a comparison of Langmuir pressure-area plot of three consecutive compression-expansion cycles for representative poloxamer 188 sample S3 in aqueous solution with lipid mix using two different LT devices: Device 1 and Device 2.
- Device 2is characterized by a larger surface area than device 1. Plot shape and surface pressure values were reproducible in both devices.
- Figure 9shows a Langmuir pressure-area plot of three consecutive compression-expansion cycles for a poloxamer 188 test sample in aqueous solutions with lipid mix of a test sample S23. Considering the isotherm hysteresis, the sample is characterized by a low An value and thus categorized as suitable for TFF.
- Figure 10shows a plot showing TFF processing time for different P188 solutions including a test sample S23. It is shown that sample S9 shows a non-linear relationship and three of the samples (S2, S3 and test sample S23) show a linear relationship between permeate volume and time confirming the method suitability for predicting the behavior of a surfactant in during TFF purification.
- Figure 11shows a comparison of Langmuir pressure-area plot of three consecutive compression-expansion cycles for a test sample of Poloxamer 124 in aqueous solution with lipid mix using Device 1 .
- Plot shape and surface pressure valuesindicate suitability for use as stabilizer.
- Figure 12shows a plot showing TFF processing time for two different P188 solutions (S3 and S9) and a test sample of poloxamer P124. It is shown that sample S9 shows a non-linear relationship. P188 sample S2 and the P124 test sample show a linear relationship between permeate volume and time confirming the method suitability for predicting the suitability of surfactant during TFF purification.
- Poloxamer P188was obtained as pharmaceutical grade from various suppliers. A test mRNA was used at a concentration of 1mg/mL.
- Lipidoid nanoparticleswere generated using nanoprecipitation in a NanoAssemblrTM BT comprising the ionizable lipidoid (dL_05(R), Scheme 1 ), the helper lipids DPPC (1 ,2- dipalmitoyl-sn-glycero-3-phosphocholine, Avanti Polar Lipids) and cholesterol (Avanti Polar Lipids), and the PEG lipid DMG-PEG2k (1 ,2-Dimyristoyl-sn-glycerolmethoxy(polyethylene glycol)-2000, Avanti Polar lipids) at the molar ratios of 8.00/5.29/4.41/0.88 respectively.
- TFFtangential flow filtration
- a surfactantsuch as a poloxamer, in particular p188, may be qualified as suitable or not suitable for membrane purification. This method is however expensive and time consuming since a TFF column needs to be discarded in case of a poloxamer failing the test and expensively produced LiNPs or LiNP need to be discarded.
- Example 2 - TFF purification with a surfactant alonecan indicate suitability for TFF.
- the following experimentswere aimed at developing a test method for identifying P188 material suitable for LiNP formulation process, for example suitable during TFF filtration. In order to develop this test, TFF was used for purifying a solution containing only a surfactant and NaCI.
- P188 solutionswere prepared using P188 provided by different suppliers and different batches as listed in Table 2.
- the final compositionwas as follows: 0.5 % (w/v) P188, 50 mM NaCI.
- Fig 1shows that solutions containing some of the samples, in particular sample S8, had to be processed more than three times longer than all other samples. Further, for most samples the Rl did increase slightly, whereas for sample S8 the Rl of the processed solution increased to highest extent, see Fig 2.
- This experimentwas aimed at developing a test method for different poloxamer materials as incoming goods control. It provided information on whether a P188 batch or a P188 supplier is suitable for formulation and/or can be used in standard production and/or purification.
- a total of 11 aqueous solutionswere prepared by dissolving 4 g of 11 different poloxamer P188 (1 % w/v) and 584.4 mg of NaCI (25 mM) in 400 mL of purified deionized water in glass bottles.
- a lipidoid mix representative of a lipidoid nanoparticle formulationwas prepared with the ionizable lipidoid (dL_05(R), Scheme 1 ), the helper lipids DPPC (1 ,2-dipalmitoyl-sn-glycero- 3-phosphocholine, Avanti Polar Lipids) and cholesterol (Avanti Polar Lipids), and the PEG lipid DMG-PEG2k (1 ,2-Dimyristoyl-sn-glycerolmethoxy(polyethylene glycol)-2000, Avanti Polar lipids) at the molar ratios of 8.00/5.29/4.41/0.88 respectively. These were resuspended together in chloroform at 15 mg/mL and vortexed for 1 minute to reach full dissolution.
- the 15 mg/mL stockwas then transferred into a 10 mL glass vial for storage in a -20°C freezer.
- aqueous test solutionPrepare an aqueous solution of the surfactant to be tested at a concentration of 1 % w/v, thereafter, referred to as “aqueous test solution”.
- lipidoid mixa lipidoid mix solution in chloroform at a concentration of 1 mg/mL, thereafter referred to as “lipidoid mix”.
- Test samplesa) On a cleaned Langmuir trough add the aqueous test solution to the through in the volume recommended by the manufacturer so that it touches the trough sensor appropriately. b) Let the sample equilibrate for 5 minutes at 22.1 ⁇ 0.2°C. c) Start compressing the trough barriers at a speed of 20 cm 2 /min from the maximum area allowed, until the “minimum target area” is reached. d) Expand the trough back to the maximum area allowed. e) Following a three-second wait time between compression-expansion cycles, repeat steps 4.c) to d) to obtain multiple surface-pressure hysteresis cycles (e.g. three) if desired. f) Clean the trough following the manufacturer’s recommendations. Langmuir trough hardware
- a Langmuir-Blodgett trough model 112D made by Nima Technologies (U.K.)was used in all experiments of Example 4. Before use, the trough was cleaned thoroughly with a Kimwipe® tissue (type 7105, Kimtech type 75512 or EX-L WIPES 34256) soaked in chloroform wearing polythene gloves in a well-ventilated area. The chloroform was removed, and the trough was rinsed several times with purified deionized water.
- a Kimwipe® tissuetype 7105, Kimtech type 75512 or EX-L WIPES 34256
- the isotherm plots for surfactant aqueous solutionsare shown in Fig 4, the legend in the figure indicates which poloxamer P188 is present in the aqueous subphase filling the Langmuir trough and the title indicates which compression -expansion cycle is shown.
- a compression-expansion isothermcould be used to test the suitability of poloxamer P188 in lipid nanoparticle purification and processing. Particularly clear results were obtained in a third compression-expansion cycle.
- an unsuitable poloxamercan be characterized by an isotherm that reaches a maximum surface pressure above 4 mN/m.
- aqueous test solutionPrepare an aqueous solution of the surfactant to be tested at a concentration of 1 % w/v, thereafter, referred to as “aqueous test solution”.
- lipidoid mixa lipidoid mix solution in chloroform at a concentration of 1 mg/mL, thereafter, referred to as “lipidoid mix”.
- Test samplesa) On a cleaned Langmuir trough add the aqueous test solution to the through in the volume recommended by the manufacturer so that it touches the trough sensor appropriately. b) Using a glass syringe, add sufficient lipid mix on the surface of the deionized water to form a monolayer (e.g. 30 pL). c) Let the sample equilibrate for 5 minutes at 22.1 ⁇ 0.2°C. d) Start compressing the trough barriers at a speed of 20 cm 2 /min from the maximum area allowed, until the “minimum target area” is reached. e) Expand the trough back to the maximum area allowed.
- Example 4As described in Example 4, a Langmuir-Blodgett trough model 112D made by Nima Technologies (U.K.) was used in all experiments of example 5.
- the compression-expansion cyclewas repeated. This process was done a total of three to four times for each poloxamer aqueous solution and lipid mix. After each set of experiments, the trough was rinsed with five times the trough volume of deionized water.
- Fig 6The isotherm plots for surfactant aqueous solutions are shown in Fig 6, the legend in the figure indicates which stabilizing agent is present in the aqueous subphase filling the Langmuir trough and the use of the lipid mix in the experiment. The title indicates which compression-expansion cycle is shown.
- a summarizing plot with isotherms from both representative stable and unstable poloxamersis shown in Fig 5.
- TTexpbeing the surface pressure during the expansion phase
- Device 2a Langmuir-Blodgett trough model MicrotroughX from Kibron Inc was used in all experiments of Example 6. Before use, the trough was cleaned thoroughly with ethanol. The ethanol was removed, and the trough was rinsed several times with purified deionized water.
- sample S3Approximately 250 mL of poloxamer P188 aqueous solution (sample S3) was added to the Langmuir trough and left five minutes to equilibrate. 30 pL of lipid mix (1 mg/mL) was added using a 50 pL glass syringe. The sample was left five minutes equilibrate before beginning the compression-expansion cycles: starting from the maximum area of 224 cm 2 , with a compression speed of 34 m/min, and down to a minimum area of 110 cm 2 . The trough was then expanded at the same rate to reach the maximum area again. Following a wait time of three seconds, the compression-expansion cycle was repeated. This process was done a total of three times for each poloxamer aqueous solution and lipid mix. After each set of experiments, the trough was rinsed with five times the trough volume of deionized water. TFF processing
- the Langmuir isotherm from lipid mix on a poloxamer aqueous subphaseidentified this new lot as suitable for lipid nanoparticle purification and processing.
- These resultswere further confirmed by TFF processing of the new poloxamer P188 lot in aqueous solution (Fig 10) supporting that surprisingly the suitability of the test method of the invention to differentiate and determine poloxamer P188 usability for TFF purification.
- the method of the inventionprovides a method that requires significantly less time, less test material and less consumables compared to testing the surfactant in actual production settings. Multiple samples can be quickly and economically tested using the method of the invention to efficiently determine the suitability of a surfactant as stabilizer.
- this exampleexamines the performance of the method of the invention for poloxamer P124.
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Engineering & Computer Science (AREA)
- Animal Behavior & Ethology (AREA)
- Veterinary Medicine (AREA)
- Epidemiology (AREA)
- Pharmacology & Pharmacy (AREA)
- Medicinal Chemistry (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Genetics & Genomics (AREA)
- Biomedical Technology (AREA)
- Molecular Biology (AREA)
- General Engineering & Computer Science (AREA)
- Physics & Mathematics (AREA)
- Zoology (AREA)
- Organic Chemistry (AREA)
- Wood Science & Technology (AREA)
- Biochemistry (AREA)
- Dispersion Chemistry (AREA)
- Biotechnology (AREA)
- Biophysics (AREA)
- Microbiology (AREA)
- Plant Pathology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Oil, Petroleum & Natural Gas (AREA)
- Dermatology (AREA)
- Nanotechnology (AREA)
- Optics & Photonics (AREA)
- Medicinal Preparation (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
Description
Stabilized Lipid and Lipidoid Nanoparticle Formulations with Specific Surfactant Properties for Enhanced Pharmaceutical Applications
The present invention relates to stabilized lipid and/or lipidoid formulations comprising lipid nanoparticles or lipidoid nanoparticles and one or more surfactants with specific properties for the delivery of nucleic acids and enhanced pharmaceutical applications.
Lipid or lipidoid nanoparticles (LNPs or LiNPs) are frequently used for the delivery of active pharmaceutical ingredients in patients. For example, lipid or lipidoid formulations of nucleic acids are extremely useful and efficient for introducing nucleic acids into cells. This advantageous property of lipid or lipidoid formulations of nucleic acids has been used for decades in biological and medical research and in therapeutic approaches to i) overexpress genes or to complement genetic defects in target cells, or ii) to downregulate or upregulate endogenous gene expression in cells, or iii) to repair genetic defects (mutations). mRNA formulations relying on nanoparticles are now also established as vaccines against COVID- 19.
These nanoparticles, central to a range of therapeutic and vaccination applications, often face issues of aggregation and instability, especially under conditions of stress, such as purification and handling. The present invention provides novel stabilized lipid and lipidoid formulations with specific surfactants that help overcome issues arising during said purification, processing and handling.
SUMMARY OF THE INVENTION
In the context of the present invention, it was found that for pharmaceutical grade surfactants such as a poloxamer, very significant differences exist between suppliers and within a supplier’s batches. In the present invention it was also found that these differences might have significant effects, among others, in the production and purification of LNP and LiNP. This invention provides novel lipid nanoparticle (LNP) or lipidoid nanoparticle (LiNP) formulations enhanced with specifically characterized surfactants. These surfactants exhibit unique Langmuir isotherm properties, ensuring optimal stabilization of the nanoparticles and efficient membrane purification, such as tangential flow filtration (TFF). The described formulations improve the stability, reduce aggregation, and mitigate challenges faced during purification, such as filtration clogging or fouling.
This invention presents novel lipid and lipidoid nanoparticle (LNP and LiNP) formulations enhanced with specifically characterized surfactants. These surfactants exhibit unique Langmuir isotherm properties, ensuring optimal stabilization of the nanoparticles. The described formulations improve the stability, reduce aggregation, and mitigate challenges faced during purification, such as filtration clogging or fouling. Integral to the innovation is a method for determining the suitability of surfactants as stabilizers based on predetermined Langmuir surface pressure/area isotherm (also referred to briefly herein as “Langmuir isotherm”) values, or filtration speeds. The stabilized nanoparticles, when formulated with therapeutic agents, have demonstrated potential in medical applications, particularly in the realm of mRNA delivery, vaccination or immunization.
To that extent, the following aspects provided by the invention are indicated in a non-exclusive manner.
In a first aspect, the invention provides a lipid nanoparticle (LNP) formulation or lipidoid nanoparticle (LiNP) formulation comprising a plurality of lipid nanoparticles (LNPs) or lipidoid nanoparticles (LiNPs) each comprising a lipid mix or lipidoid mix, wherein the lipid mix or lipidoid mix comprises, as a component thereof, at least one selected from an ionizable lipid, ionizable lipidoid, and permanently cationic lipid, and wherein the formulation comprises a surfactant characterized by having a Langmuir surface pressure/area isotherm with a maximum surface pressure (Umax) that is equal to or below 4.0 mN/m at a minimum surface area established for a lipid mix or lipidoid mix as comprised by the nanoparticles.
In accordance with a second aspect, the invention provides a lipid nanoparticle (LNP) formulation or lipidoid nanoparticle (LiNP) formulation comprising a plurality of LNPs or LiNPs each comprising a lipid mix or lipidoid mix, wherein the lipid mix or lipidoid mix comprises, as a component thereof, at least one selected from an ionizable lipid, ionizable lipidoid, and permanently cationic lipid, wherein the formulation comprises as a stabilizing agent a surfactant, and wherein a representative sample comprising an aqueous solution of the surfactant carrying on its surface a lipid mix or lipidoid mix as comprised by the LNPs or LiNPs is characterized by having a Langmuir isotherm An that is equal to or below 0.60, preferably equal to or below 0.45, at each area point during a Langmuir surface pressure/area isotherm cycle comprising a compression phase and an expansion phase and recorded between a maximum surface area and minimum surface area established for the lipid mix or lipidoid mix , wherein TT is calculated at any area point as: wherein ncomp is the surface pressure at said area point during the compression phase of the isotherm cycle, wherein nexp is the surface pressure at said area point during the expansion phase of the isotherm cycle, and wherein nmax is the maximum surface pressure reached in the isotherm cycle.
wherein ncomp is the surface pressure at said area point during the compression phase of the isotherm cycle, wherein nexp is the surface pressure at said area point during the expansion phase of the isotherm cycle, and wherein nmax is the maximum surface pressure reached in the isotherm cycle.

In a third aspect, the invention provides a surfactant for use in a pharmaceutical composition, the surfactant being characterized by having a Langmuir isotherm with a maximum surface pressure (nmax) that is equal to or below 4.0 mN/m at a minimum surface area established for said pharmaceutical composition.
In a fourth aspect, the invention provides a method for classifying a surfactant as suitable or not suitable for use as a stabilizing agent for a pharmaceutical composition, the method comprising:
(a) providing a surfactant in an aqueous solution at a concentration (C),
(b) recording a Langmuir surface pressure/area isotherm of the surfactant in the solution, to determine a maximum surface pressure Umax of the Langmuir surface pressure/area isotherm at a predetermined minimum surface area;
(c) comparing the maximum surface pressure nmax to a threshold value, wherein if the maximum surface pressure TTmax is equal to or less than the threshold value, the surfactant is classified as suitable for use as a stabilizing agent, and if the maximum surface pressure TTmax is greater than the threshold value, the surfactant is classified as not suitable for use as a stabilizing agent.
In accordance with this aspect, further provided is a method for the preparation of a pharmaceutical composition, said method comprising classifying a surfactant as suitable or not suitable for use as a stabilizing agent for a pharmaceutical composition in accordance with the method for classifying as described above, and, if the surfactant is classified as being suitable for use as a stabilizing agent for a pharmaceutical composition, incorporating the surfactant into the pharmaceutical composition.
In a fifth aspect, the invention provides a method for classifying a surfactant as suitable or not suitable for use as a stabilizing agent for a pharmaceutical composition comprising a lipid or lipidoid, wherein the method comprises the steps:
(a) providing a surfactant in an aqueous solution at a concentration (C) of the surfactant in the solution;
(b) recording a Langmuir pressure/area isotherm cycle including a compression phase and an expansion phase between a maximum surface area and a minimum surface area on a sample comprising the surfactant in the aqueous solution and carrying on its surface a lipid or lipidoid as comprised by the composition:
(c) calculating a Langmuir isotherm An for each area point of the Langmuir pressure/area isotherm cycle, wherein An is calculated as: wherein ncomp is the surface pressure at said area point during the compression phase of the isotherm cycle, wherein nexp is the surface pressure at said area point during the expansion phase of the isotherm cycle, and wherein itmax is the maximum surface pressure reached in the isotherm cycle, and
wherein ncomp is the surface pressure at said area point during the compression phase of the isotherm cycle, wherein nexp is the surface pressure at said area point during the expansion phase of the isotherm cycle, and wherein itmax is the maximum surface pressure reached in the isotherm cycle, and

(d) comparing the calculated Langmuir isotherm An to a predetermined threshold value, wherein if the calculated Langmuir isotherm An is at every isotherm area point equal to or less than the threshold value the surfactant is classified as suitable for use as a stabilizer, and if the calculated Langmuir An at any area point is greater than the threshold value the surfactant is classified as not suitable for use as a stabilizer.
Further provided in the context of this aspect is a method for the preparation of a pharmaceutical composition, said method comprising classifying a surfactant as suitable or not suitable for use as a stabilizing agent for a pharmaceutical composition comprising a lipid or lipidoid in accordance with the method as described above, and, if the surfactant is classified as being suitable for use as a stabilizing agent, incorporating the surfactant into the pharmaceutical composition.
In a related sixth aspect, the invention provides a method of mitigating or avoiding clogging or fouling of a filtration system during purification of a pharmaceutical composition in the form of a lipid nanoparticle formulation (LNP) or lipidoid nanoparticle formulation (LiNP), the method comprising adding a stabilizing surfactant to a first LNP or LiNP formulation to form a second LNP or LiNP formulation, optionally before purification, wherein the LNP or LiNP formulation comprises a plurality of LNPs or LiNPs, each comprising a lipid mix or lipidoid mix and a therapeutic agent, wherein the lipid mix or lipidoid mix comprises, as a component thereof, at least one selected from an ionizable lipid, ionizable lipidoid, and permanently cationic lipid, and wherein the stabilizing surfactant is a surfactant according to the invention, e.g. a surfactant according to the third aspect discussed above or a surfactant classified as being suitable as a stabilizing agent by the method according to the fourth or fifth aspect.
In a further related seventh aspect, the invention provides a method of mitigating aggregation of lipid nanoparticles or lipidoid nanoparticles in a lipid nanoparticle (LNP) formulation or lipidoid nanoparticle (LiNP) formulation, preferably a pharmaceutical composition in the form of a lipid nanoparticle (LNP) formulation or lipidoid nanoparticle (LiNP) formulation, the method comprising adding a stabilizing surfactant to a first LNP or LiNP formulation to form a second LNP or LiNP formulation optionally before purification, wherein the LNP or LiNP formulation comprises a plurality of LNPs or LiNPs, each comprising a lipid mix or lipidoid mix, wherein the lipid mix or lipidoid mix comprises, as a component thereof, at least one selected from an ionizable lipid, ionizable lipidoid, and permanently cationic lipid, and the stabilizing surfactant is a surfactant in accordance with the invention, e.g. a surfactant according to the third aspect discussed above or a surfactant classified as being suitable as a stabilizing agent by the method according to the fourth or fifth aspect.
As a related eighth aspect, the invention provides a lipid nanoparticle (LNP) formulation or lipidoid nanoparticle (LiNP) formulation obtained by the method in accordance with the fourth to seventh aspect.
Furthermore, in a ninth aspect, the invention provides a lipid nanoparticle (LNP) formulation or lipidoid nanoparticle (LiNP) formulation in accordance with the first aspect, the second aspect, or the eighth aspect discussed above for use as a medicament.
Likewise, in a tenth aspect, the invention provides a lipid nanoparticle (LNP) formulation or lipidoid nanoparticle (LiNP) formulation in accordance with the first aspect, the second aspect, or the eighth aspect discussed above for use in the treatment or prevention of a disease, preferably a disease selected from Table A as disclosed herein below, more preferably a disease selected from viral diseases, ciliopathies, or autoimmune diseases, even more preferably a lung disease, a lung viral disease, lung ciliopathies or a lung autoimmune diseases.
In a further aspect, the invention provides a method for classifying a surfactant as suitable or not suitable for use as a stabilizer of a pharmaceutical composition comprising a nucleic acid, optionally as a stabilizer during purification, preferably during TFF purification, the method comprising:
(a) providing a surfactant in an aqueous solution;
(b) optionally combining the surfactant with a LNP or LiNP formulation;
(c) purifying the aqueous solution comprising the surfactant or optionally the LNP or LiNP formulation comprising the surfactant using a membrane purification system, preferably TFF or ultrafiltration, most preferably TFF;
(d) measuring the filtration time of the aqueous solution or the LNP formulation during diafiltration or ultrafiltration;
(e) comparing the measured filtration speed to a predetermined threshold value, preferably a time threshold value, more preferably a time threshold value of 90 minutes, wherein if the filtration speed is equal to or greater than the threshold value, the surfactant is classified as not suitable for use as a stabilizer, and if the filtration speed is less than the threshold value, the surfactant is classified as suitable for use as a stabilizer.
In a further aspect, the invention provides a method of reducing or avoiding side effects in a therapy with LNPs or LiNPs carrying at least one therapeutic agent, wherein the method comprises the steps: i) determine whether LNPs or LiNPs in a pharmaceutical composition comprising LNPs or LiNPs aggregate when subjected to a mechanical stress or a temperature stress, by determining their aggregation level before and after subjecting said pharmaceutical composition to said mechanical or temperature stress ii) if the LNP or LiNP show aggregation after the test of step (i), then add to the LNP or LiNP formulation a surfactant, preferably a surfactant according to the third aspect above, to obtain a LNP or LiNP suspension with a final surfactant concentration of 0.01% w/v and up to 10% w/v, preferably between 0.05% w/w surfactant and 5% surfactant, more preferably between 0.33% surfactant and 2.5% surfactant, more preferably between 0.45% and 1 .5% surfactant, preferably between 0.5% and 1.5% surfactant, most preferably 1 %. ill) reconstitute with mixing to generate a stable LNP or LiNP suspension.
In a related aspect, the invention provides a use of a surfactant according to the invention, e.g. the surfactant in accordance with the above third aspect, for stabilizing a suspension of lipid nanoparticles or of lipidoid nanoparticles in an aqueous vehicle solution against particle aggregation under a physical stress condition, preferably shear stress, more preferably shear stress during purification such as TFF, wherein the lipid nanoparticles or lipidoid nanoparticles comprise the following components (a) and (b):
(a) a therapeutic agent and
(b) at least one selected from a permanently cationic lipid, an ionizable lipid and an ionizable lipidoid.
Without wanting to be bound by theory, the present invention provides stabilized LNP or LiNP formulations, such as LNP or LiNP suspension formulations, uses thereof and uses in method of treatment based on the findings that the differences at a molecular level or differences in the quality within pharmaceutical grade surfactants compositions are responsible for their suitability or unsuitability for use in during processing and purification of LNPs and LiNPs, and further uses to avoid aggregation. The surfactant selected with the method of the invention further allows a surprisingly long shelf life and extended stability to shaking. Said reduction of aggregation according to the invention results in a reduction of side effects of the formulations and suspension of the invention, such as a reduction of side effects caused by vaccines formulations or anticancer formulations comprising LNPs or LiNPs.
An overview over the various aspects of the invention is provided in the following set of items.
Set of Items
1. A lipid nanoparticle (LNP) formulation or lipidoid nanoparticle (LiNP) formulation comprising a plurality of lipid nanoparticles (LNPs) or lipidoid nanoparticles (LiNPs), each comprising a lipid mix or lipidoid mix, wherein the lipid mix or lipidoid mix comprises, as a component thereof, at least one selected from an ionizable lipid, ionizable lipidoid, and permanently cationic lipid, and wherein the formulation further comprises a surfactant characterized by having a Langmuir surface pressure/area isotherm (also referred to herein as “Langmuir isotherm”) with a maximum surface pressure (nmax) that is equal to or below 4.0 mN/m at a minimum surface area established for a lipid mix or lipidoid mix as comprised by the nanoparticles.
2. The lipid nanoparticle (LNP) formulation or lipidoid nanoparticle (LiNP) formulation according to item 1 , wherein Umax is equal to or below 3.5 mN/m, more preferably equal to or below 3.5 and equal to or above 0.5 mN/m, even more preferably equal to or below 3.0 mN/m and equal to or above 0.5 mN/m, even more preferably equal to or below 2.5 and equal to or above 1 .0 mN/m, most preferably equal to or below 2.0 and equal to or above 1 .0 mN/m.
3. A lipid nanoparticle (LNP) formulation or lipidoid nanoparticle (LiNP) formulation comprising a plurality of lipid nanoparticles (LNPs) or lipidoid nanoparticles (LiNPs), each comprising a lipid mix or lipidoid mix, wherein the lipid mix or lipidoid mix comprises, as a component thereof, at least one selected from an ionizable lipid, ionizable lipidoid, and permanently cationic lipid, wherein the formulation comprises as a stabilizing agent a surfactant, and wherein a representative sample comprising an aqueous solution of the surfactant carrying on its surface a lipid mix or lipidoid mix as comprised by the LNPs or LiNPs is characterized by having a Langmuir isotherm An that is equal to or below 0.60, preferably equal to or below 0.45, at each area point during a Langmuir surface pressure/area isotherm cycle comprising a compression phase and an expansion phase and recorded between a maximum surface area and minimum surface area established for the lipid mix or lipidoid mix , wherein TT is calculated at any area point as: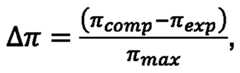 wherein itcomp is the surface pressure at said area point during the compression phase of the isotherm cycle, wherein nexp is the surface pressure at said area point during the expansion phase of the isotherm cycle, and wherein nmax is the maximum surface pressure reached in the isotherm cycle.
wherein itcomp is the surface pressure at said area point during the compression phase of the isotherm cycle, wherein nexp is the surface pressure at said area point during the expansion phase of the isotherm cycle, and wherein nmax is the maximum surface pressure reached in the isotherm cycle.

4. The lipid nanoparticle (LNP) formulation or lipidoid nanoparticle (LiNP) formulation according to any one of items 1 to 3, wherein the surfactant is a non-ionic surfactant, preferably at least one nonionic surfactant selected from the group of fatty alcohol ethoxylates, fatty acid ethoxylates, block copolymers of ethylene oxide and propylene oxide, alkylphenol ethoxylates or oligomers of alkylphenol ethoxylates, fatty acid esters of sorbitol, ethoxylated fatty acid esters of sorbitol, fatty acid esters of glycerol, ethoxylated castor oil and ethoxylated vitamin E, e.g. a surfactant selected from the list consisting of poloxamer 124 (P124), poloxamer 188 (P188), poloxamer 338 (P338), poloxamer 407 (P407), Tween-20, Tween-80, BRIJ35, tyloxapol, VitE-PEG1000, and Kolliphor EL, or from combinations thereof.
5. The lipid nanoparticle (LNP) formulation or lipidoid nanoparticle (LiNP) formulation according to item 4, wherein the surfactant is a block copolymer of ethylene oxide and propylene oxide, more preferably a poloxamer, even more preferably poloxamer selected from the list consisting of poloxamer 124, poloxamer 188, poloxamer 338, and poloxamer 407, or from combinations thereof, most preferably P188.
6. The lipid nanoparticle (LNP) formulation or lipidoid nanoparticle (LiNP) formulation according to any one of items 1 to 5, wherein the lipid nanoparticles or lipidoid nanoparticles comprise the lipid mix or lipidoid mix and a therapeutic agent.
7. The lipid nanoparticle (LNP) formulation or lipidoid nanoparticle (LiNP) formulation according to item 6, wherein the therapeutic agent comprises a nucleic acid, such as RNA, preferably mRNA.
8. The lipid nanoparticle (LNP) formulation or lipidoid nanoparticle (LiNP) formulation according to any one of items 1 to 7, wherein the lipid mix or lipidoid mix comprises, as a component thereof, at least one selected from an ionizable lipid, ionizable lipidoid, and a permanently cationic lipid, and further comprises one or more of the following components (c1 ) to (c6), preferably further comprises the components (c1 ), (c2) and (c3):
(c1) a non-ionizable lipid having a sterol structure;
(c2) a phospholipid;
(c3) a PEG-conjugated lipid;
(c4) a polysarcosine-conjugated lipid
(c5) a PASylated lipid;
(c6) an ionizable or a cationic polymer.
9. The lipid nanoparticle (LNP) formulation or lipidoid nanoparticle (LiNP) formulation according to item 8, wherein the lipid mix or lipidoid mix comprises i) 30 to 65 mol% of at least one selected from the ionizable lipid, the ionizable lipidoid, and the permanently cationic lipid and further comprises one or more of the following components (c1 ) to (c6): ii) 10 to 50 mol% of the lipid having a sterol structure (c1 ), iii) 4 to 50 mol% of the phospholipid (c2), iv) 0.5 to 10 mol% of one of the PEG-conjugated lipid (c3), the polysarcosine-conjugated lipid (c4) and the PASylated lipid (c5), or of any combination thereof, v) 0.5 to 10 mol% of a cationic polymer (c6), such that the sum of the amounts of i) and ii) to v) is 100 mol%, and more preferably further comprises the components (c1), (c2) and (c3), such that the sum of the amounts of i) and ii) to iv) is 100%.
10. The lipid nanoparticle (LNP) formulation or lipidoid nanoparticle (LiNP) formulation according to any one of items 1 to 9, which is a lipidoid nanoparticle formulation comprising a plurality of lipidoid nanoparticles each comprising a lipidoid mix, wherein the lipidoid mix comprises an ionizable lipidoid of formula (L-1 ): wherein: a is 1 or 2 and b is an integer of 1 to 4 or a is an integer of 1 to 4 and b is 1 or 2, preferably a is 1 and b is an integer of 2 to 4 or a is an integer of 2 to 4 and b is 1 , p is 1 or 2, m is 1 or 2; n is 0 or 1 and m+n is > 2, and
wherein: a is 1 or 2 and b is an integer of 1 to 4 or a is an integer of 1 to 4 and b is 1 or 2, preferably a is 1 and b is an integer of 2 to 4 or a is an integer of 2 to 4 and b is 1 , p is 1 or 2, m is 1 or 2; n is 0 or 1 and m+n is > 2, and

R1A to R6A are independently of each other selected from: hydrogen, -CH2-CH(OH)-R7A, -CH(R7A)-CH2-OH, -CH2-CH2-(C=O)-O-R7A, -CH2-CH2-(C=O)-NH-R7A, -CH2-R7A, -C(NH)-NH2, a polyethylene glycol) chain, and a receptor ligand; wherein R7A is selected from C3-C18 alkyl and C3-C18 alkenyl having one C-C double bond; provided that at least two residues among R1A to R6A are selected from -CH2-CH(OH)-R7A, -CH(R7A)-CH2-OH, -CH2-CH2-(C=O)-O-R7A, -CH2-CH2-(C=O)-NH-R7A and -CH2-R7A, wherein R7A is selected from C3-C18 alkyl or C3-C18 alkenyl having one C-C double bond, or a protonated form of the ionizable lipid of formula (L-1 ) wherein one or more of the nitrogen atoms contained in the compound of formula (L-1 ) are protonated to provide a compound carrying a positive charge.
11. The lipid nanoparticle (LNP) formulation or lipidoid nanoparticle (LiNP) formulation according to any one of items 1 to 10 which is a suspension formulation, and wherein the vehicle solution of the suspension formulation is an aqueous vehicle solution comprising the surfactant.
12. A surfactant for use in a pharmaceutical composition, the surfactant being characterized by having a Langmuir surface pressure/area isotherm with a maximum surface pressure (nmax) that is equal to or below 4.0 mN/m at a minimum surface area established for said pharmaceutical composition.
13. The surfactant for use according to item 12, wherein the maximum surface pressure is equal to or below 3.5 mN/m, more preferably equal to or below 3.5 and equal to or above 0.5 mN/m, even more preferably equal to or below 3.0 mN/m and equal to or above 0.5 mN/m, even more preferably equal to or below 2.5 and equal to or above 1 .0 mN/m, most preferably equal to or below 2.0 and equal to or above 1.0 mN/m.
14. The surfactant for use according to item 12 or 13, which is a non-ionic surfactant, preferably at least one nonionic surfactant selected from the group of fatty alcohol ethoxylates, fatty acid ethoxylates, block copolymers of ethylene oxide and propylene oxide, alkylphenol ethoxylates or oligomers of alkylphenol ethoxylates, fatty acid esters of sorbitol, ethoxylated fatty acid esters of sorbitol, fatty acid esters of glycerol, ethoxylated castor oil and ethoxylated vitamin E, e.g. a surfactant selected from the list consisting of poloxamer 124 (P124), poloxamer 188 (P188), poloxamer 338 (P338), poloxamer 407 (P407), Tween-20, Tween-80, BRIJ35, tyloxapol, VitE-PEG1000, and Kolliphor EL, or from combinations thereof.
15. The surfactant for use according to item 14, which is a block copolymer of ethylene oxide and propylene oxide, more preferably a poloxamer, even more preferably poloxamer selected from the list consisting of poloxamer 124, poloxamer 188, poloxamer 338, and poloxamer 407, or from combinations thereof, most preferably P188.
16. The surfactant for use according to any of items 12 to 15, wherein the pharmaceutical composition is in the form of a lipid nanoparticle (LNP) suspension formulation or lipidoid nanoparticle (LiNP) suspension formulation comprising a plurality of LNPs or LiNPs, each comprising a lipid mix or lipidoid mix and a therapeutic agent, wherein the lipid mix or lipidoid mix comprises, as a component thereof, at least one selected from an ionizable lipid, ionizable lipidoid, and permanently cationic lipid, and wherein the vehicle solution of the suspension formulation is an aqueous vehicle solution comprising the surfactant
17. A method for classifying a surfactant as suitable or not suitable for use as a stabilizing agent for a pharmaceutical composition, the method comprising:
(a) providing a surfactant in an aqueous solution at a concentration (C),
(b) recording a Langmuir surface pressure/area isotherm of the surfactant in the solution to determine a maximum surface pressure Umax of the Langmuir isotherm at a predetermined minimum surface area;
(c) comparing the maximum surface pressure nmax to a threshold value, wherein if the maximum surface pressure TTmax is equal to or less than the threshold value, the surfactant is classified as suitable for use as a stabilizing agent, and if the maximum surface pressure TTmax is greater than the threshold value, the surfactant is classified as not suitable for use as a stabilizing agent.
18. A method for classifying a surfactant as suitable or not suitable for use as a stabilizing agent for a pharmaceutical composition comprising a lipid or lipidoid, optionally during purification of said composition, preferably during tangential flow filtration of said composition, wherein the method comprises the steps:
(a) providing a surfactant in an aqueous solution at a concentration (C) of the surfactant in the solution;
(b) recording a Langmuir pressure/area isotherm cycle including a compression phase and an expansion phase between a maximum surface area and a minimum surface area on a sample comprising the surfactant in the aqueous solution and carrying on its surface a lipid or lipidoid as comprised by the composition: (c) calculating a Langmuir isotherm An for each area point of the Langmuir pressure/area isotherm cycle, wherein TT is calculated as: wherein ncomp is the surface pressure at said area point during the compression phase of the isotherm cycle, wherein itexp is the surface pressure at said area point during the expansion phase of the isotherm cycle, and wherein nmax is the maximum surface pressure reached in the isotherm cycle, and
wherein ncomp is the surface pressure at said area point during the compression phase of the isotherm cycle, wherein itexp is the surface pressure at said area point during the expansion phase of the isotherm cycle, and wherein nmax is the maximum surface pressure reached in the isotherm cycle, and

(d) comparing the calculated Langmuir isotherm TT to a threshold value, wherein, if the calculated Langmuir isotherm An is at every isotherm area point equal to or less than the threshold value, the surfactant is classified as suitable for use as a stabilizing agent, and if the calculated Langmuir An at any area point is greater than the threshold value the surfactant is classified as not suitable for use as a stabilizing agent.
19. A method for the preparation of a pharmaceutical composition, said method comprising classifying a surfactant as suitable or not suitable for use as a stabilizing agent for a pharmaceutical composition in accordance with the method of item 17 or 18, and, if the surfactant is classified as being suitable for use as a stabilizing agent for the pharmaceutical composition, incorporating the surfactant into the pharmaceutical composition.
20. The method according to any one of items 17 to 19, wherein the surfactant is a nonionic surfactant, preferably at least one nonionic surfactant selected from the group of fatty alcohol ethoxylates, fatty acid ethoxylates, block copolymers of ethylene oxide and propylene oxide, alkylphenol ethoxylates or oligomers of alkylphenol ethoxylates, fatty acid esters of sorbitol, ethoxylated fatty acid esters of sorbitol, fatty acid esters of glycerol, ethoxylated castor oil and ethoxylated vitamin E, e.g. a surfactant selected from the list consisting of poloxamer 124 (P124), poloxamer 188 (P188), poloxamer 338 (P338), poloxamer 407 (P407), Tween-20, Tween-80, BRIJ35, tyloxapol, VitE-PEG1000, and Kolliphor EL, or from combinations thereof.
21. The method according to item 20, wherein the surfactant is a block copolymer of ethylene oxide and propylene oxide, more preferably a poloxamer, even more preferably poloxamer selected from the list consisting of poloxamer 124, poloxamer 188, poloxamer 338, and poloxamer 407, or from combinations thereof, most preferably P188. 22. The method according to any one of items 17 to 21 , wherein the pharmaceutical composition comprises a therapeutic agent comprising a nucleic acid, such as RNA, preferably mRNA.
23. The method according to any one of items 17 to 22, wherein the pharmaceutical composition is a lipid nanoparticle (LNP) formulation or lipidoid nanoparticle (Li NP) formulation comprising a plurality of LNPs or LiNPs, each comprising a lipid mix or lipidoid mix and a therapeutic agent, wherein the lipid mix or lipidoid mix comprises, as a component thereof, at least one selected from an ionizable lipid, ionizable lipidoid, and permanently cationic lipid.
24. The method according to item 23, wherein the pharmaceutical composition is a lipid nanoparticle (LNP) suspension formulation or lipidoid nanoparticle (LiNP) suspension formulation, and wherein the vehicle solution of the suspension formulation is an aqueous vehicle solution comprising the surfactant.
25. A method of mitigating or avoiding clogging or fouling of a filtration system during purification of a pharmaceutical composition in the form of a lipid nanoparticle formulation (LNP) or lipidoid nanoparticle formulation (LiNP), the method comprising adding a stabilizing surfactant to a first LNP or LiNP formulation to form a second LNP or LiNP formulation, optionally before purification, wherein the LNP or LiNP formulation comprises a plurality of LNPs or LiNPs, each comprising a lipid mix or lipidoid mix and a therapeutic agent, wherein the lipid mix or lipidoid mix comprises, as a component thereof, at least one selected from an ionizable lipid, ionizable lipidoid, and permanently cationic lipid, and wherein the stabilizing surfactant is a surfactant as defined in any of items 12 to 16 or a surfactant classified as being suitable as a stabilizing agent by the method according to any of items 17 to 27.
26. The method according to item 25, wherein the purification includes tangential flow filtration.
27. The method according to item 25 or 26, wherein the pharmaceutical composition is a lipid nanoparticle (LNP) suspension formulation or lipidoid nanoparticle (LiNP) suspension formulation comprising an aqueous vehicle solution, and wherein the stabilizing surfactant is added to the vehicle solution, optionally wherein the surfactant is essentially absent from the LNPs or LiNPs.
28. The method according to any one of items 25 to 27, wherein therapeutic agent, is a nucleic acid such as RNA, more preferably mRNA. 29. A method of mitigating aggregation of lipid or lipidoid nanoparticles in a lipid nanoparticle (LNP) formulation or lipidoid nanoparticle (LiNP) formulation, the method comprising adding a stabilizing surfactant to a first LNP or LiNP formulation to form a second LNP or LiNP formulation, optionally before purification, wherein the LNP or LiNP formulation comprises a plurality of LNPs or LiNPs, each comprising a lipid mix or lipidoid mix, wherein the lipid mix or lipidoid mix comprises, as a component thereof, at least one selected from an ionizable lipid, ionizable lipidoid, and permanently cationic lipid, and wherein the stabilizing surfactant is a surfactant as defined in any of items 12 to 16 or a surfactant classified as being suitable as a stabilizing agent by the method according to any of items 17 to 27.
30. The method according to item 29, wherein the formulation is a lipid nanoparticle (LNP) suspension formulation or lipidoid nanoparticle (LiNP) suspension formulation comprising an aqueous vehicle solution, and wherein the stabilizing surfactant is added to the vehicle solution, optionally wherein the surfactant is essentially absent from the LNPs or LiNPs.
31. The method according to item 29 or 30, wherein the lipid nanoparticles or lipidoid nanoparticles comprise a therapeutic agent, preferably a nucleic acid such as RNA, more preferably mRNA.
32. The method according to item 31 , wherein the lipid nanoparticles or lipidoid nanoparticles comprise a nucleic acid, e.g. RNA, and preferably mRNA, and wherein the method comprises the steps of: i) first, combining the nucleic acid and at least one selected from an ionizable lipid, ionizable lipidoid, and permanently cationic lipid to form LNPs or LiNPs, ii) second, purifying the LNPs or LiNPs, iii) third, adding the stabilizing surfactant before TFF purification and during TFF purification in the exchange buffer, maintaining the surfactant in a steady concentration, iv) optionally wherein the stabilizing surfactant is added to the LNP or LiNP formulation after step (i).
33. The method according to items 31 or 32, wherein the method comprises the following steps: i) generating a LNP or LiNP preparation by mixing of at least one selected from a permanently cationic lipid, an ionizable lipid and an ionizable lipidoid dissolved in an organic phase with a therapeutic agent dissolved in an aqueous solution, ii) diluting the LNP or LiNP preparation by dilution with a first solution, iii) concentrating the LNP or LiNP preparation by buffer exchange using ultra/diafiltration via TFF wherein a second solution is used for the ultra/diafiltration, iv) obtaining a LNP or LiNP suspension in an aqueous vehicle solution, wherein the first solution comprises between about 0.01% w/v and 10% of stabilizing surfactant, preferably between about 0.01 % w/v surfactant and 5% w/v surfactant, more preferably between about 0.01% w/v surfactant and 2.5% w/v surfactant, more preferably between about 0.05% w/v and 1.5% w/v surfactant, even more preferably between about 0.05% w/v and 1.5% w/v surfactant, most preferably about 1% w/v surfactant, and/or wherein the second solution comprises between about 0.01 % w/v and about 10% of stabilizing surfactant, preferably between about 0.01% w/v surfactant and about 5% w/v surfactant, more preferably between about 0.01 % w/v surfactant and about 2.5% w/v surfactant, even more preferably between about 0.05% w/v and 1.5% w/v surfactant, most preferably about 1 % w/v; and wherein the final concentration of stabilizing surfactant from combined first and second solution is between 0.01% and 10% surfactant, preferably between 0.01% w/v surfactant and 5% w/v surfactant, more preferably between 0.01% w/v surfactant and 2.5% w/v surfactant, even more preferably between 0.05% w/v and 1.5 % w/v surfactant, most preferably about 1 % w/v surfactant with regard to the total volume of the suspension of the nanoparticles in the aqueous vehicle solution.
34. The method of item 33, wherein: a) the incorporation of the stabilizing surfactant into the suspension does not occur before or during step i), b) the stabilizing surfactant is added in the first and the second solution, and/or c) approximately half of the stabilizing surfactant is added to the first solution and approximately half of the surfactant is added to the second solution.
35. The method according to any one of items 23 to 34, wherein the lipid mix or lipidoid mix comprises, as a component thereof, at least one selected from an ionizable lipid, ionizable lipidoid, and a permanently cationic lipid, and further comprises one or more of the following components (c1 ) to (c6):
(c1) a non-ionizable lipid having a sterol structure;
(c2) a phospholipid;
(c3) a PEG-conjugated lipid;
(c4) a polysarcosine-conjugated lipid
(c5) a PASylated lipid;
(c6) an ionizable or a cationic polymer.
36. The method according to item 35, wherein the lipid mix or lipidoid mix comprises, as a component thereof, at least one selected from an ionizable lipid, ionizable lipidoid, and a permanently cationic lipid, and further comprises the components (c1), (c2) and (c3).
37. The method according to item 35, wherein the lipid mix or lipidoid mix comprises i) 30 to 65 mol% of at least one selected from the ionizable lipid, the ionizable lipidoid and the permanently cationic lipid, and further comprises one or more of the following components (c1 ) to (c6): ii) 10 to 50 mol% of the lipid having a sterol structure (c1 ), iii) 4 to 50 mol% of the phospholipid (c2), iv) 0.5 to 10 mol% of one of the PEG-conjugated lipid (c3), the polysarcosine-conjugated lipid (c4) and the PASylated lipid (c5), or of any combination thereof, v) 0.5 to 10 mol% of a cationic polymer (c6), such that the sum of the amounts of i) and ii) to v) is 100 mol%,
38. The method according to item 37, wherein the lipid mix or lipidoid mix comprises i) 30 to 65 mol% of at least one selected from the ionizable lipid, the ionizable lipidoid and the permanently cationic lipid, and further comprises ii) 10 to 50 mol% of the lipid having a sterol structure (c1), iii) 4 to 50 mol% of the phospholipid (c2), iv) 0.5 to 10 mol% of a PEG-conjugated lipid (c3), such that the sum of the amounts of i) and ii) to iv) is 100%.
39. The method according to any one of items 23 to 38, wherein the formulation is a lipidoid nanoparticle formulation comprising a plurality of lipidoid nanoparticles each comprising a lipidoid mix, wherein the lipidoid mix comprises an ionizable lipidoid of formula (L-1 ): wherein: a is 1 or 2 and b is an integer of 1 to 4 or a is an integer of 1 to 4 and b is 1 or 2, preferably a is 1 and b is an integer of 2 to 4 or a is an integer of 2 to 4 and b is 1 , p is 1 or 2, m is 1 or 2; n is 0 or 1 and m+n is 2; and
wherein: a is 1 or 2 and b is an integer of 1 to 4 or a is an integer of 1 to 4 and b is 1 or 2, preferably a is 1 and b is an integer of 2 to 4 or a is an integer of 2 to 4 and b is 1 , p is 1 or 2, m is 1 or 2; n is 0 or 1 and m+n is 2; and

R1A to R6A are independently of each other selected from: hydrogen; -CH2-CH(OH)-R7A, -CH(R7A)-CH2-OH, -CH2-CH2-(C=O)-O-R7A, -CH2-CH2-(C=O)-NH-R7A, -CH2-R7A, -C(NH)-NH2, a polyethylene glycol) chain, and a receptor ligand; wherein R7A is selected from C3-C18 alkyl and C3-C18 alkenyl having one C-C double bond; provided that at least two residues among R1A to R6A are selected from -CH2-CH(OH)-R7A, -CH(R7A)-CH2-OH, -CH2-CH2-(C=O)-O-R7A, -CH2-CH2-(C=O)-NH-R7A and -CH2-R7A, wherein R7A is selected from C3-C18 alkyl or C3-C18 alkenyl having one C-C double bond, or a protonated form of the ionizable lipid of formula (L-1 ) wherein one or more of the nitrogen atoms contained in the compound of formula (L-1 ) are protonated to provide a compound carrying a positive charge. 40. A lipid nanoparticle formulation or lipidoid nanoparticle formulation, preferably a lipid nanoparticle suspension formulation or lipidoid nanoparticle suspension formulation, obtained by the method according to any one of items 23 to 39.
41 . The lipid nanoparticle formulation or lipidoid nanoparticle formulation, preferably a lipid nanoparticle suspension formulation or lipidoid nanoparticle suspension formulation, according to any one of items 1 to 11 or 40 for use as a medicament.
42. The lipid nanoparticle formulation or lipidoid nanoparticle formulation, preferably a lipid nanoparticle suspension formulation or lipidoid nanoparticle suspension formulation, according to any one of items 1 to 11 or 40, for use in the treatment or prevention of a disease, preferably a disease selected from Table A, more preferably a disease selected from viral diseases, ciliopathies, autoimmune diseases, and respiratory tract diseases, even more preferably selected from a lung disease, an airway disease or a nasal disease, more preferably a lung viral disease, lung ciliopathies and a lung autoimmune diseases.
43. The lipid nanoparticle formulation or lipidoid nanoparticle formulation for use according to item 43, wherein the lung disease or lung viral disease is at least one selected from pneumonia and asthma; the airway disease is at least one selected from bronchitis, viral induced asthma, lung fibrosis and COPD; and/or the nasal disease is at least one selected from rhinitis and sinusitis.
44. The lipid nanoparticle formulation or lipidoid nanoparticle formulation according to item 41 , for use in vaccination or immunization.
45. A method of avoiding or reducing side effects in a therapy with LNPs or LiNPs carrying at least one therapeutic agent, wherein the method comprises the steps: i) determine whether LNPs or LiNPs in a pharmaceutical composition comprising LNPs or LiNPs aggregate when subjected to a mechanical stress or a temperature stress, by determining their aggregation level before and after subjecting said pharmaceutical composition is subjected to said mechanical or temperature stress ii) if the LNP or LiNP show aggregation after the test of step (i), then add to the LNP or LiNP formulation a surfactant to obtain a LNP or LiNP suspension with a final surfactant concentration of 0.01 % w/v and up to 10% w/v, preferably between 0.05% w/w surfactant and 5% surfactant, more preferably between 0.33% surfactant and 2.5% surfactant, more preferably between 0.45% and 1 .5% surfactant, most preferably between 0.5% and 1 .5% surfactant, most preferably about 1% w/v. iii) reconstitute with mixing to generate a stable LNP or LiNP suspension.
46. The method in accordance with item 45, wherein the surfactant is a surfactant according to any one of items 12 to 16, or a surfactant classified as being suitable as a stabilizing agent by the method according to any one of items 17 to 24
47. Use of a surfactant according to any one of items 12 to 16, or of a surfactant classified as being suitable as a stabilizing agent by the method according to any one of items 17 to 24, for stabilizing a suspension of lipid nanoparticles or of lipidoid nanoparticles in an aqueous vehicle solution against particle aggregation under a physical stress condition, preferably shear stress, more preferably shear stress during purification such as TFF, wherein the lipid nanoparticles or lipidoid nanoparticles comprise the following components (a) and (b):
(a) a therapeutic agent and
(b) at least one selected from an ionizable lipid, an ionizable lipidoid and a permanently cationic lipid.
48. The use of the surfactant in accordance with item 47, wherein the physical stress condition is selected from shaking, stirring, vibrating, mixing, inverting, tapping, or dropping of the suspension, or a combination thereof, or wherein the physical stress condition is caused by pumping the suspension or by its withdrawal into a syringe.
49. The use of the surfactant in accordance with item 47 or 48, wherein the surfactant is incorporated as an excipient into the aqueous vehicle solution.
50. The use of the surfactant in accordance with any one of items 47 to 49, wherein the nanoparticle formulation is not lyophilized.
51. The use of the surfactant in accordance with any one of items 47 to 50, wherein the surfactant is added before a lyophilization process.
52. The use of the surfactant in accordance with any one of items 47 to 51 , wherein the presence of the surfactant does not cause a change in the biological activity of the nanoparticle.
53. The use of the surfactant in accordance with any one of items 47 to 52, wherein the presence of the surfactant does not cause a change in the physical properties of the nanoparticle measured as the hydrodynamic diameter of the nanoparticle and as the proportion of therapeutic agent comprised by the nanoparticle. 54. The use of the surfactant in accordance with any of items 47 to 53, wherein the suspension of lipid nanoparticles or lipidoid nanoparticles in an aqueous vehicle solution comprises the surfactant at a concentration of 0.01 to 10 % (w/v).
55. The use of the surfactant in accordance with any of items 47 to 54, wherein the therapeutic agent is a nucleic acid.
56. The use of the surfactant in accordance with Item 55, wherein the nucleic acid is mRNA.
57. The use of the surfactant in accordance with any of items 47 to 56, wherein the concentration of the nucleic acid in the suspension formulation ranges from 0.01 to 10 mg/mL, based on the total volume of the suspension formulation.
58. The use of the surfactant in accordance with any of items 47 to 57, wherein the nanoparticles have a Z-average diameter, as determined by dynamic light scattering, in the range of 10 to 500 nm, preferably around 30 to 100 nm.
59. The use of the surfactant in accordance with any of items 47 to 58, wherein the nanoparticles further comprise one or more of the following components (c1 ) to (c6):
(c1) a non-ionizable lipid having a sterol structure;
(c2) a phospholipid lipid;
(c3) a PEG-conjugated lipid;
(c4) a polysarcosine-conjugated lipid
(c5) a PASylated lipid;
(c6) an ionizable or a cationic polymer or lipidoid.
60. The use of the surfactant in accordance with any of items 47 to 59, wherein the nanoparticles comprise a) 30 to 65 mol% of at least one selected from the ionizable lipid, an ionizable lipidoid and a permanently cationic lipid (b), and one or more of the following components: ii) 10 to 50 mol% of the lipid having a sterol structure (c1 ), iii) 4 to 50 mol% of the phospholipid (c2), iv) 0.5 to 10 mol% of one of the PEG-conjugated lipid (c3), the polysarcosine-conjugated lipid (c4) and the PASylated lipid (c5), or of any combination thereof,
0.5 to 10 mol% of a cationic polymer (c6), such that the sum of (b) and (c1 ) to (c6) amounts to 100 mol%.
61. The use of the surfactant in accordance with any of items 47 to 60, wherein the nanoparticles comprise an ionizable lipid (b) of the following formula (a-lll):

III a-lll or a pharmaceutically acceptable salt, prodrug or stereoisomer thereof, wherein: one of L1 or L2 is -O(C=O)-, -(C=O)O-, -C(=O)-, -O-, -S(O)X-, -S-S-, - C(=O)S-, SC(=O)-, - NRaC(=O)-, -C(=O)NRa-, NRaC(=O)NRa-, -OC(=O)NRa- or - NRaC(=O)O-, and the other of L1 or L2 is-O(C=O)-, -(C=O)O-, -C(=O)-, -O-, -S(O)X-, -S- S-, -C(=O)S-, SC(=O)-,
-NRaC(=O)-, -C(=O)NRa-, -NRaC(=O)NRa-, -OC(=O)NRa- or -NRaC(=O)O- or a direct bond;
G1 and G2 are each independently C1-C12 alkylene or C1-C12 alkenylene;
G3 is C1-C24 alkylene, C1-C24 alkenylene, C3-C8 cycloalkylene, C3-C8 cycloalkenylene, wherein each of alkylene, alkenylene, cycloalkylene, and cyloalkenylene is optionally substituted;
Ra is H or C1-C12 alkyl wherein the alkyl is optionally substituted;
R1 and R2 are each independently C6-C24 alkyl or C6-C24 alkenyl, wherein each of alkyl and alkenyl is optionally substituted;
R3 is H, OR5, ON, -C(=O)OR4, -OC(=O)R4 or-NR5C(=O)R4; R4 is C1-C12 alkyl, wherein alkyl is optionally substituted;
R5 is H or Ci-Ce alkyl, wherein alkyl is optionally substituted; and x is 0, 1 or 2. 62. The use of the surfactant in accordance with any of items 47 to 61 , wherein the nanoparticles comprise an ionizable lipidoid (b) of the following formula (L-1 ), wherein: a is 1 or 2 and b is an integer of 1 to 4 or a is an integer of 1 to 4 and b is 1 or 2, preferably a is 1 and b is an integer of 2 to 4; or a is an integer of 2 to 4 and b is 1 , p is 1 or 2, m is 1 or 2; n is 0 or 1 and m+n is > 2; and
wherein: a is 1 or 2 and b is an integer of 1 to 4 or a is an integer of 1 to 4 and b is 1 or 2, preferably a is 1 and b is an integer of 2 to 4; or a is an integer of 2 to 4 and b is 1 , p is 1 or 2, m is 1 or 2; n is 0 or 1 and m+n is > 2; and

R1A to R6A are independently of each other selected from: hydrogen; -CH2-CH(OH)-R7A, -CH(R7A)-CH2-OH, -CH2-CH2-(C=O)-O-R7A, -CH2-CH2-(C=O)-NH-R7A, -CH2-R7A, -C(NH)-NH2I a polyethylene glycol) chain; and a receptor ligand, wherein R7A is selected from C3-C18 alkyl and C3-C18 alkenyl having one C-C double bond, provided that at least two residues among R1A to R6A are selected from -CH2-CH(OH)-R7A, -CH(R7A)-CH2-OH, -CH2-CH2-(C=O)-O-R7A, -CH2-CH2-(C=O)-NH-R7A and -CH2-R7A, wherein R7A is selected from C3-C18 alkyl or C3-C18 alkenyl having one C-C double bond, or a protonated form of the ionizable lipid of formula (L-1 ), wherein one or more of the nitrogen atoms contained in the compound of formula (L-1 ) are protonated to provide a compound carrying a positive charge.
63. Use of the surfactant in accordance with any of items 47 to 60, wherein the nanoparticles comprise, as an ionizable lipid (b), (6Z,9Z,28Z,31Z)-heptatriaconta-6,9,28,31- tetraene-19-yl 4-(dimethylamino)butanoate or a protonated form thereof wherein the nitrogen atom of the compound is protonated.
64. Use of the surfactant in accordance with any of items 47 to 60, wherein the nanoparticles comprise, as an ionizable lipid (b), ((4-hydroxybutyl)azanediyl)bis(hexan-6,1- diyl)bis(2-hexyldecanoate) or a protonated form thereof wherein the nitrogen atom of the compound is protonated, and/or (heptadecan -9-yl 8-((2-hydroxyethyl)(6-oxo-6- (undecyloxy)hexyl)amino)octanoate, or a protonated form thereof wherein the nitrogen atom of the compound is protonated.
65. The use according to item 64, wherein the nanoparticles comprise ((4- hydroxybutyl)azanediyl)bis(hexane-6,1-diyl)bis(2-hexyldecanoate) or the protonated form thereof wherein the nitrogen atom of the compound is protonated, and optionally further comprise one or more of the following components (d1 ) to (d8):
(d1) 2-[(polyethylene glycol)-2000]-N,N-ditetradecylacetamide (ALC-0159)
(d2) 1 ,2-distearoyl-sn-glycero-3-phosphocholine (DSPC)
(d3) cholesterol
(d4) potassium chloride
(d5) potassium dihydrogen phosphate
(d6) sodium chloride
(d7) disodium phosphate dihydrate
(d8) sucrose.
66. The use according to item 64, wherein the nanoparticles comprise heptadecan-9-yl 8- ((2-hydroxyethyl)(6-oxo-6-(undecyloxy)hexyl)amino)octanoate or a protonated form thereof wherein the nitrogen atom of the compound is protonated, and further optionally comprise one or more of the following components (e1) to (e7):
(e1) 1 ,2-distearoyl-sn-glycero-3-phosphocholine (DSPC),
(e2) cholesterol,
(e3) 1 ,2-dimyristoyl-rac-glycero-3-methoxypolyethylene glycol-2000 (PEG2000 DMG),
(e4) 2-amino-2-(hydroxymethyl)propan-1 ,3-diol (tromethamol) hydrochloride
(e5) sodium acetate trihydrate
(e6) acetic acid
(e7) sucrose. 67. The use according to item 64, wherein the nanoparticle comprises DLin-MC3-DMA ((6Z,9Z,28Z,31Z)-heptatriaconta-6,9,28,31-tetraen-19-yl-4-(dimethylamino) butanoate) or a protonated form thereof wherein the nitrogen atom of the compound is protonated, and optionally further comprise one or more of the following components (e1 ) to (e7):
(e1) 1 ,2-distearoyl-sn-glycero-3-phosphocholine (DSPC),
(e2) cholesterol,
(e3) PEG2000-C-DMG (a-(3’-{[1 ,2-di(myristyloxy)propanoxy]carbonylamino}propyl)-(jj- methoxy, polyoxyethylene),
(e4) 2-amino-2-(hydroxymethyl)propan-1 ,3-diol (trometamol) hydrochloride
(e5) Disodium hydrogen phosphate, heptahydrate
(e6) Potassium dihydrogen phosphate, anhydrous
(e7) Sodium chloride.
It will be understood that the summary in the above items forms a part of the general disclosure of the present invention, such that the information provided in the following detailed description, e.g. with regard to further preferred embodiments or optional features, also applies for the above items and vice versa.
DETAILED DESCRIPTION
Unless indicated to the contrary in any specific context, the following explanation, e.g. with respect to therapeutic agents, lipid nanoparticles or lipidoid nanoparticles or surfactants, applies for all aspects of the invention which contain or make use of any of these components.
As noted above, the invention provides in a first aspect a lipid nanoparticle (LNP) formulation or lipidoid nanoparticle (LiNP) formulation comprising a plurality of lipid nanoparticles (LNPs) or lipidoid nanoparticles (LiNPs), each comprising a lipid mix or lipidoid mix, wherein the lipid mix or lipidoid mix comprises, as a component thereof, at least one selected from an ionizable lipid, ionizable lipidoid, and permanently cationic lipid, and wherein the formulation further comprises a surfactant characterized by having a Langmuir surface pressure/area isotherm (also referred to herein as “Langmuir isotherm”) with a maximum surface pressure (IT max) that is equal to or below 4.0 mN/m at a minimum surface area established for a lipid mix or lipidoid mix as comprised by the nanoparticles.
Preferably, TTmax is equal to or below 3.5 mN/m, more preferably equal to or below 3.5 and equal to or above 0.5 mN/m, even more preferably equal to or below 3.0 mN/m and equal to or above 0.5 mN/m, even more preferably equal to or below 2.5 and equal to or above 1.0 mN/m, most preferably equal to or below 2.0 and equal to or above 1 .0 mN/m.
The maximum surface pressure (TTmax) of the surfactant can be determined by recording a Langmuir surface pressure/area isotherm of the surfactant in an aqueous solution thereof, e.g. in deionized water. The Langmuir isotherm is recorded in a Langmuir trough. The recording of the Langmuir isotherm of the surfactant involves reducing the available surface area in the Langmuir trough at least until the minimum surface area established for the lipid mix or lipidoid mix is reached. The surface pressure which is observed at the minimum surface area when recording the Langmuir isotherm of the surfactant is considered as the maximum surface pressure (TT max ) of the surfactant.
The minimum surface area can be established, e.g., by compressing a layer, typically a monolayer, of the lipid mix or lipidoid mix (i.e. the lipid mix or lipidoid mix as comprised by the nanoparticles) in the Langmuir trough until a first phase transition is observed in a surface pressure/area isotherm of the lipid mix or lipidoid mix. In order to establish the minimum surface area, the layer can be conveniently provided on water, e.g. deionized water. The surface area at the start of the phase transition is regarded as the minimum surface area established for the lipid mix or lipidoid mix. As will be understood by the skilled reader, the start of the phase transition as referred to herein in a pressure/area (TT-A) isotherm plot can be objectively determined as the point where the slope of the curve (dir/dA) changes significantly, indicating a shift in the molecular organization of the monolayer. A step to establish the minimum surface area can be carried out, e.g., as a calibration step in the Langmuir trough prior to determining the maximum surface pressure of the surfactant as described above.
As a maximum surface area during recording a Langmuir surface pressure/area isotherm e.g. the maximum area provided by the Langmuir trough can be conveniently used.
A monolayer of the lipid mix or lipidoid mix on water in a Langmuir trough can be provided, e.g., by applying the lipid mix or lipidoid mix to the surface until a signal change in the Langmuir trough detector is generated. The Langmuir surface pressure/area isotherm of the surfactant can be recorded, e.g., in the form of a Langmuir surface pressure/area isotherm cycle (also briefly referred to herein as “isotherm cycle”) which includes a compression phase and an expansion phase. The maximum surface pressure (nmax) can be determined, for example, by recording a single isotherm cycle, but preferably results are obtained by recording multiple isotherm cycles in sequence, such as three isotherm cycles, e.g. with a wait time of 5 seconds or less between cycles, such as 3 seconds. If a single isotherm cycle is recorded, it is preferred that nmax is equal to or below 3.5 mN/m, more preferably equal to or below 3.5 and equal to or above 0.5 mN/m, even more preferably equal to or below 3.0 mN/m and equal to or above 0.5 mN/m, even more preferably equal to or below 2.5 and equal to or above 1 .0 mN/m, most preferably equal to or below 2.0 and equal to or above 1.0 mN/m, directly in this single cycle. If multiple cycles are recorded, such as three cycles, it is preferred that the surfactant shows a Langmuir isotherm with a maximum surface pressure (Umax) as defined herein above in the last, e.g. the third isotherm cycle.
Typically, a Langmuir surface pressure/area isotherm or isotherm cycle as referred to herein is recorded at about room temperature, e.g. at 22.1 ± 0.2°C. A typical concentration of the surfactant in an aqueous solution thereof for the determination of the maximum surface pressure is 1% w/v (corresponding to 1 g of surfactant in 100 ml of the total volume of the solution including the surfactant, generally measured at about room temperature, e.g. at 22.1 ± 0.2°C).
A calibration step, which may be used to establish the minimum surface area, and the measurement of the maximum surface pressure of the surfactant are further illustrated in the “Examples” section below.
Details regarding preferred compositions of the lipid nanoparticles, the lipidoid nanoparticles, the lipid mix and the lipidoid mix shall be discussed herein below.
Details regarding preferred types of surfactants, such as a poloxamer, shall also be discussed herein below.
Preferably, the lipid nanoparticle (LNP) formulation or lipidoid nanoparticle (LiNP) formulation is a suspension formulation comprising a liquid vehicle solution, wherein the LNPs or LiNPs can be dispersed. The vehicle solution is preferably an aqueous vehicle solution. It is preferred that, in such a suspension formulation, the surfactant is comprised in the vehicle solution. For example, in such a suspension formulation, the surfactant may favorably act as a stabilizing agent that mitigates aggregation of the LNPs or LiNP or a subpopulation thereof which may be caused by shaking or by shear stress of the suspension during production, purification, handling or transport, preferably during production or purification, more preferably during purification, most preferably during tangential flow filtration (TFF) purification.
The lipid nanoparticle (LNP) formulation or lipidoid nanoparticle (LiNP) formulation is preferably a formulation, e.g. a suspension formulation as discussed above, wherein the LNPs or LiNPs comprise the lipid mix or lipidoid mix and a therapeutic agent, i.e. a pharmaceutical formulation. Thus, the first aspect also encompasses the lipid nanoparticle (LNP) formulation or lipidoid nanoparticle (LiNP) formulation comprising the plurality of lipid nanoparticles (LNPs) or lipidoid nanoparticles (LiNPs), each comprising the lipid mix or lipidoid mix and a therapeutic agent, for use as a medicament. A preferred therapeutic agent is a nucleic acid, such as RNA, and particularly preferred is mRNA. Details regarding therapeutic agents which may be contained in the LNPs or LiNPs shall likewise be discussed herein below. The terms “pharmaceutical formulation” and “pharmaceutical composition” may be used herein as equivalents.
In line with the above, the lipid nanoparticle (LNP) formulation or lipidoid nanoparticle (LiNP) formulation in accordance with the first aspect is particularly preferably a lipid nanoparticle (LNP) suspension formulation or lipidoid nanoparticle (LiNP) suspension formulation comprising a plurality of LNPs or LiNPs, each comprising a lipid mix or lipidoid mix and a therapeutic agent, wherein the lipid mix or lipidoid mix comprises, as a component thereof, at least one selected from an ionizable lipid, ionizable lipidoid, and permanently cationic lipid, and wherein the vehicle solution of the suspension formulation is an aqueous vehicle solution comprising a surfactant characterized by having a Langmuir surface pressure/area isotherm with a maximum surface pressure (nmax) that is equal to or below 4.0 mN/m at a minimum surface area established for the lipid mix or lipidoid mix as comprised by the nanoparticles. As will be understood by the skilled person, the preferred values discussed above for the maximum surface pressure continue to apply for this preferred embodiment.
In accordance with the second aspect, the invention provides a lipid nanoparticle (LNP) formulation or lipidoid nanoparticle (LiNP) formulation comprising a plurality of lipid nanoparticles (LNPs) or lipidoid nanoparticles (LiNPs), each comprising a lipid mix or lipidoid mix, wherein the lipid mix or lipidoid mix comprises, as a component thereof, at least one selected from an ionizable lipid, ionizable lipidoid, and permanently cationic lipid, wherein the formulation comprises as a stabilizing agent a surfactant, and wherein a representative sample comprising an aqueous solution of the surfactant carrying on its surface a lipid mix or lipidoid mix as comprised by the LNPs or LiNPs is characterized by having a Langmuir isotherm An that is equal to or below 0.60, preferably equal to or below 0.45, at each area point during a Langmuir surface pressure/area isotherm cycle comprising a compression phase and an expansion phase and recorded between a maximum surface area and minimum surface area established for the lipid mix or lipidoid mix , wherein TT is calculated at any area point as: wherein ncomp is the surface pressure at said area point during the compression phase of the isotherm cycle, wherein itexp is the surface pressure at said area point during the expansion phase of the isotherm cycle, and wherein nmax is the maximum surface pressure reached in the isotherm cycle.
wherein ncomp is the surface pressure at said area point during the compression phase of the isotherm cycle, wherein itexp is the surface pressure at said area point during the expansion phase of the isotherm cycle, and wherein nmax is the maximum surface pressure reached in the isotherm cycle.

A representative sample used for determining the Langmuir isotherm An values comprises an aqueous solution of the surfactant, e.g. a solution in deionized water, carrying on its surface a lipid mix or lipidoid mix as comprised by the LNPs or LiNPs, typically a monolayer of the lipid mix or lipidoid mix. The lipid mix or lipidoid mix generally has the same composition as the lipid mix or lipidoid mix comprised by the nanoparticles, i.e. it comprises the same lipids and/or lipidoids in the same proportions. Using this representative sample, a Langmuir surface pressure/area isotherm cycle comprising a compression phase and an expansion phase can be recorded in a Langmuir trough. From the surface pressure determined during compression and the surface pressure determined during expansion at each area point of the isotherms, the Langmuir isotherm ATT values are calculated for each area point as explained above. As noted above, the Langmuir isotherm An values are equal to or below 0.60, preferably equal to or below 0.45, at each area point during a Langmuir surface pressure/area isotherm cycle. The Langmuir isotherm ATT values calculated as disclosed above are usually above 0.
It is sufficient if a single isotherm cycle is recorded, and the Langmuir isotherm An values are preferably calculated for this isotherm cycle. Multiple isotherm cycles can be recorded in sequence, such as three isotherm cycles, e.g. with a wait time of 5 seconds or less between cycles, such as 3 seconds. However, if An values of the first cycle fulfill the requirements set forth above, the same typically applies also for the subsequent cycles, such that it is preferred to rely on the measurement results of the first cycle for the calculation of the An values even if multiple cycles are recorded. The minimum surface area can be established, e.g., by compressing a layer, typically a monolayer, of the lipid mix or lipidoid mix (i.e. the lipid mix or lipidoid mix as comprised by the nanoparticles) in the Langmuir trough until a first phase transition is observed in a surface pressure/area isotherm of the lipid mix or lipidoid mix. In order to establish the minimum surface area, the layer can be conveniently provided on water, e.g. on deionized water. The surface area at the start of the phase transition is regarded as the minimum surface area established for the lipid mix or lipidoid mix. As noted above, the start of the phase transition as referred to herein in a pressure/area (TT-A) isotherm plot can be objectively determined as the point where the slope of the curve (dn/dA) changes significantly. A step to establish the minimum surface area can be carried out, e.g., as a calibration step in the Langmuir trough prior to recording a Langmuir surface pressure/area isotherm cycle on a representative sample as described above.
As a maximum surface area during recording a Langmuir surface pressure/area isotherm, the surface area before the start of the compression in a compression phase, e.g., the maximum area provided by the Langmuir trough, can be conveniently used.
A monolayer of the lipid mix or lipidoid mix on water or on an aqueous solution of the surfactant in a Langmuir trough can be provided, e.g., by applying the lipid mix or lipidoid mix to the surface until a signal change in the Langmuir trough detector is generated.
Typically, a Langmuir surface pressure/area isotherm or isotherm cycle as referred to herein is recorded at about room temperature, e.g. at 22.1 ± 0.2°C. A typical concentration of the surfactant in an aqueous solution thereof in the representative sample is 1 % w/v (corresponding to 1 g of surfactant in 100 ml of the total volume of the solution including the surfactant, generally measured at about room temperature, e.g. at 22.1 ± 0.2°C).
A calibration step, which may be used to establish the minimum surface area, and the recording of the Langmuir pressure/area isotherm for the calculation of Langmuir isotherm An values are further illustrated in the “Examples” section below.
Details regarding preferred types of surfactants, such as a poloxamer, shall be discussed herein below.
Details regarding preferred compositions of the lipid mix and the lipidoid mix shall also be discussed herein below. Preferably, the lipid nanoparticle (LNP) formulation or lipidoid nanoparticle (LiNP) formulation is a suspension formulation comprising a liquid vehicle solution, wherein the LNPs or LiNPs can be dispersed. The vehicle solution is preferably an aqueous vehicle solution. It is preferred that, in such a suspension formulation, the surfactant is comprised in the vehicle solution.
For example, in such a suspension formulation, the surfactant may favorably act as a stabilizing agent that mitigates aggregation of the LNPs or LiNP or a subpopulation thereof which may be caused by shaking or by shear stress of the suspension during production, purification, handling or transport, preferably during production or purification, more preferably during purification, most preferably during tangential flow filtration (TFF) purification.
The lipid nanoparticle (LNP) formulation or lipidoid nanoparticle (LiNP) formulation is preferably a formulation, e.g. a suspension formulation as discussed above, wherein the LNPs or LiNPs comprise the lipid mix or lipidoid mix and a therapeutic agent, i.e. a pharmaceutical formulation. Thus, the second aspect also encompasses the lipid nanoparticle (LNP) formulation or lipidoid nanoparticle (LiNP) formulation comprising the plurality of lipid nanoparticles (LNPs) or lipidoid nanoparticles (LiNPs), each comprising the lipid mix or lipidoid mix and a therapeutic agent, for use as a medicament. A preferred therapeutic agent is a nucleic acid, such as RNA, and particularly preferred is mRNA. Details regarding therapeutic agents which may be contained in the LNPs or LiNPs shall likewise be discussed herein below. The terms “pharmaceutical formulation” and "pharmaceutical composition” may be used herein as equivalents.
In line with the above, the lipid nanoparticle (LNP) formulation or lipidoid nanoparticle (LiNP) formulation in accordance with the second aspect is particularly preferably a lipid nanoparticle (LNP) suspension formulation or lipidoid nanoparticle (LiNP) suspension formulation comprising a plurality of LNPs or LiNPs, each comprising a lipid mix or lipidoid mix and a therapeutic agent, wherein the lipid mix or lipidoid mix comprises, as a component thereof, at least one selected from an ionizable lipid, ionizable lipidoid, and permanently cationic lipid, and wherein the vehicle solution of the suspension formulation is an aqueous vehicle solution comprising a surfactant as a stabilizing agent, and wherein a representative sample comprising an aqueous solution of the surfactant carrying on its surface a lipid mix or lipidoid mix as comprised by the LNPs or LiNPs is characterized by having a Langmuir isotherm An that is equal to or below 0.60, preferably equal to or below 0.45, at each area point during a Langmuir surface pressure/area isotherm cycle comprising a compression phase and an expansion phase and recorded between a maximum surface area and minimum surface area established for the lipid mix or lipidoid mix , wherein TT is calculated at any area point as defined above. In accordance with the third aspect, the invention provides a surfactant for use in a pharmaceutical composition, the surfactant being characterized by having a Langmuir surface pressure/area isotherm with a maximum surface pressure (IT max) that is equal to or below 4.0 mN/m at a minimum surface area established for said pharmaceutical composition. Likewise, the aspect provides the use of the surfactant as a component, e.g. as a stabilizing agent, in a pharmaceutical composition.
Preferably, Umax also in the context of this aspect is equal to or below 3.5 mN/m, more preferably equal to or below 3.5 and equal to or above 0.5 mN/m, even more preferably equal to or below 3.0 mN/m and equal to or above 0.5 mN/m, even more preferably equal to or below 2.5 and equal to or above 1 .0 mN/m, most preferably equal to or below 2.0 and equal to or above 1 .0 mN/m.
For example, the pharmaceutical composition can be a pharmaceutical composition comprising a lipid or a lipidoid, such as a lipid nanoparticle (LNP) formulation or lipidoid nanoparticle (LiNP) formulation comprising a plurality of lipid nanoparticles (LNPs) or lipidoid nanoparticles (LiNPs). More preferably, the pharmaceutical composition is a lipid nanoparticle (LNP) formulation or lipidoid nanoparticle (LiNP) formulation comprising a plurality of lipid nanoparticles (LNPs) or lipidoid nanoparticles (LiNPs), each comprising a lipid mix or lipidoid mix, wherein the lipid mix or lipidoid mix comprises, as a component thereof, at least one selected from an ionizable lipid, ionizable lipidoid, and permanently cationic lipid.
If the pharmaceutical formulation is a lipid nanoparticle (LNP) formulation or lipidoid nanoparticle (LiNP) formulation, it is preferred that the formulation is a suspension formulation comprising a liquid vehicle solution, wherein the LNPs or LiNPs can be dispersed. The vehicle solution is preferably an aqueous vehicle solution. It is preferred that, in such a suspension formulation, the surfactant as defined in this aspect is comprised in the vehicle solution.
For example, in such a suspension formulation, the surfactant may favorably act as a stabilizing agent that mitigates aggregation of the LNPs or LiNP or a subpopulation thereof which may be caused by shaking or by shear stress of the suspension during production, purification, handling or transport, preferably during production or purification, more preferably during purification, most preferably during tangential flow filtration (TFF) purification.
As will be understood by the skilled person, the pharmaceutical composition comprises a therapeutic agent, for example a nucleic acid. A preferred therapeutic agent is RNA, and particularly preferred is mRNA. Details regarding preferred types of therapeutic agents shall be discussed herein below. If the pharmaceutical formulation is a lipid nanoparticle (LNP) formulation or lipidoid nanoparticle (LiNP) formulation, e.g. a suspension formulation, the therapeutic agent is typically comprised in the LNPs or LiNPs.
In line with the above, the surfactant for use in accordance with the third aspect is particularly preferably a surfactant for use in a pharmaceutical composition in the form of a lipid nanoparticle (LNP) suspension formulation or lipidoid nanoparticle (LiNP) suspension formulation comprising a plurality of LNPs or LiNPs, each comprising a lipid mix or lipidoid mix and a therapeutic agent, wherein the lipid mix or lipidoid mix comprises, as a component thereof, at least one selected from an ionizable lipid, ionizable lipidoid, and permanently cationic lipid, wherein the vehicle solution of the suspension formulation is an aqueous vehicle solution and comprising the surfactant which is characterized by having a Langmuir surface pressure/area isotherm with a maximum surface pressure (nmax) that is equal to or below 4.0 mN/m at a minimum surface area established for said pharmaceutical composition. As will be understood by the skilled person, the preferred values discussed above for the maximum surface pressure continue to apply for this preferred embodiment. Likewise, the aspect provides the use of the surfactant as a component, e.g. as a stabilizing agent, in such a pharmaceutical composition.
The maximum surface pressure (nmax) of the surfactant can be determined by recording a Langmuir surface pressure/area isotherm of the surfactant in an aqueous solution thereof, e.g. in deionized water. The Langmuir isotherm is recorded in a Langmuir trough. The recording of the Langmuir isotherm of the surfactant involves reducing the available surface area in the Langmuir trough at least until the minimum surface area established for the pharmaceutical composition is reached. The surface pressure which is observed at the minimum surface area when recording the Langmuir isotherm of the surfactant is considered as the maximum surface pressure (TT max ) of the surfactant.
The Langmuir surface pressure/area isotherm of the surfactant can be recorded, e.g., in the form of a Langmuir surface pressure/area isotherm cycle (also briefly referred to herein as “isotherm cycle”) which includes a compression phase and an expansion phase. The maximum surface pressure (nmax) can be determined, for example, by recording a single isotherm cycle, but preferably results are obtained by recording multiple isotherm cycles in sequence, such as three isotherm cycles, e.g. with a wait time of 5 seconds or less between cycles, such as 3 seconds. If a single isotherm cycle is recorded, it is preferred that TTmax is equal to or below 3.5 mN/m, more preferably equal to or below 3.5 and equal to or above 0.5 mN/m, even more preferably equal to or below 3.0 mN/m and equal to or above 0.5 mN/m, even more preferably equal to or below 2.5 and equal to or above 1 .0 mN/m, most preferably equal to or below 2.0 and equal to or above 1.0 mN/m, directly in this single cycle. If multiple cycles are recorded, such as three cycles, it is preferred that the surfactant shows a Langmuir isotherm with a maximum surface pressure (Umax) as defined hereinabove in the last, e.g. the third isotherm cycle.
Typically, a Langmuir surface pressure/area isotherm or isotherm cycle as referred to herein is recorded at about room temperature, e.g. at 22.1 ± 0.2°C. A typical concentration of the surfactant in an aqueous solution thereof for the determination of the maximum surface pressure is 1 % w/v (corresponding to 1 g of surfactant in 100 ml of the total volume of the solution including the surfactant, generally measured at about room temperature, e.g. at 22.1 ± 0.2°C).
Details regarding preferred types of surfactants, such as a poloxamer, shall also be discussed herein below.
For example, if the pharmaceutical composition is a pharmaceutical composition comprising a lipid or a lipidoid, such as a lipid nanoparticle (LNP) formulation or lipidoid nanoparticle (LiNP) formulation comprising a plurality of lipid nanoparticles (LNPs) or lipidoid nanoparticles (LiNPs), the minimum surface area when recording the Langmuir isotherm of the surfactant can be established for the pharmaceutical composition, e.g., by compressing a layer, typically a monolayer, of the lipid or lipidoid as comprised by the pharmaceutical composition in a Langmuir trough until a first phase transition is observed in a surface pressure/area isotherm of the lipid or lipidoid. If the pharmaceutical composition, e.g. as a nanoparticle formulation, comprises a combination of more than one lipid and/or lipidoid, the minimum surface area can be established for the pharmaceutical composition, e.g., by compressing a layer, typically a monolayer, of the combination of more than one lipid and/or lipidoid as comprised by the pharmaceutical composition in a Langmuir trough until a first phase transition is observed in a surface pressure/area isotherm of the combination. In order to establish the minimum surface area, the layer can be conveniently provided on water, e.g. deionized water.
If, as set out above, the pharmaceutical composition is the lipid nanoparticle (LNP) formulation or lipidoid nanoparticle (LiNP) formulation comprising a plurality of lipid nanoparticles (LNPs) or lipidoid nanoparticles (LiNPs), each comprising a lipid mix or lipidoid mix, wherein the lipid mix or lipidoid mix comprises, as a component thereof, at least one selected from an ionizable lipid, ionizable lipidoid, and permanently cationic lipid, the minimum surface area when recording the Langmuir isotherm of the surfactant can be established for the pharmaceutical composition, e.g., by compressing a layer, typically a monolayer, of the lipid mix or lipidoid mix as comprised by the LNPs or LiNPs in a Langmuir trough until a first phase transition is observed in a surface pressure/area isotherm of the lipid mix or lipidoid mix. In order to establish the minimum surface area, the layer can be conveniently provided on water, e.g. deionized water.
In either case, the surface area at the start of the phase transition is regarded as the minimum surface area established for the pharmaceutical composition. As noted above, the start of the phase transition as referred to herein in a pressure/area (TT-A) isotherm plot can be objectively determined as the point where the slope of the curve (dn/dA) changes significantly.
A step to establish the minimum surface area can be carried out, e.g., as a calibration step in the Langmuir trough prior to determining the maximum surface pressure of the surfactant as described above.
As a maximum surface area during recording a Langmuir surface pressure/area isotherm, e.g., the maximum area provided by the Langmuir trough can be conveniently used.
A monolayer of a lipid and/or lipidoid on water, or of a lipid mix or lipidoid mix on water, in a Langmuir trough can be provided, e.g., by applying the lipid and/or lipidoid or the lipid mix or lipidoid mix to the surface until a signal change in the Langmuir trough detector is generated.
A calibration step which may be used to establish the minimum surface area and the measurement of the maximum surface pressure are further illustrated in the “Examples” section below.
In a fourth aspect, the invention provides a method for classifying a surfactant as suitable or not suitable for use as a stabilizing agent for a pharmaceutical composition, the method comprising:
(a) providing a surfactant in an aqueous solution at a concentration (C),
(b) recording a Langmuir surface pressure/area isotherm of the surfactant in the solution to determine a maximum surface pressure nmax of the Langmuir isotherm at a predetermined minimum surface area;
(c) comparing the maximum surface pressure nmax to a threshold value, wherein if the maximum surface pressure Umax is equal to or less than the threshold value, the surfactant is classified as suitable for use as a stabilizing agent, and if the maximum surface pressure nmax is greater than the threshold value, the surfactant is classified as not suitable for use as a stabilizing agent. Further provided in the context of this aspect is a method for the preparation of a pharmaceutical composition, said method comprising classifying a surfactant as suitable or not suitable for use as a stabilizing agent for a pharmaceutical composition in accordance with the method as described above, and, if the surfactant is classified as being suitable for use as a stabilizing agent for a pharmaceutical composition, incorporating the surfactant into the pharmaceutical composition.
For example, the method can be used for classifying a surfactant as suitable or not suitable for use as a stabilizing agent for a pharmaceutical composition comprising a lipid or a lipidoid, such as a lipid nanoparticle (LNP) formulation or lipidoid nanoparticle (LiNP) formulation comprising a plurality of lipid nanoparticles (LNPs) or lipidoid nanoparticles (LiNPs). More preferably, the pharmaceutical composition is a lipid nanoparticle (LNP) formulation or lipidoid nanoparticle (LiNP) formulation comprising a plurality of lipid nanoparticles (LNPs) or lipidoid nanoparticles (LiNPs), each comprising a lipid mix or lipidoid mix, wherein the lipid mix or lipidoid mix comprises, as a component thereof, at least one selected from an ionizable lipid, ionizable lipidoid, and permanently cationic lipid.
If the pharmaceutical formulation is a lipid nanoparticle (LNP) formulation or lipidoid nanoparticle (LiNP) formulation, it is preferred that the formulation is a suspension formulation comprising a liquid vehicle solution, wherein the LNPs or LiNPs can be dispersed. The vehicle solution is preferably an aqueous vehicle solution. It is preferred that, in such a suspension formulation, a surfactant classified as being suitable for use as a stabilizing agent is incorporated into the vehicle solution.
For example, in such a suspension formulation, the surfactant may favorably act as a stabilizing agent that mitigates aggregation of the LNPs or LiNP or a subpopulation thereof which may be caused by shaking or by shear stress of the suspension during production, purification, handling or transport, preferably during production or purification, more preferably during purification, most preferably during tangential flow filtration (TFF) purification.
As will be understood by the skilled person, the pharmaceutical composition comprises a therapeutic agent, for example a nucleic acid. A preferred therapeutic agent is RNA, and particularly preferred is mRNA. Details regarding preferred types of therapeutic agents shall be discussed herein below. If the pharmaceutical formulation is a lipid nanoparticle (LNP) formulation or lipidoid nanoparticle (LiNP) formulation, e.g. a suspension formulation, the therapeutic agent is typically comprised in the LNPs or LiNPs. In line with the above, it is particularly preferred if the method according to this aspect is a method for classifying a surfactant as suitable or not suitable for use as a stabilizing agent for a pharmaceutical composition wherein the pharmaceutical composition is in the form of a lipid nanoparticle (LNP) suspension formulation or lipidoid nanoparticle (LiNP) suspension formulation comprising a plurality of LNPs or LiNPs, each comprising a lipid mix or lipidoid mix and a therapeutic agent, wherein the lipid mix or lipidoid mix comprises, as a component thereof at least one selected from an ionizable lipid, ionizable lipidoid, and a permanently cationic lipid, and wherein the vehicle solution of the suspension formulation is an aqueous vehicle solution.
In step (a) of the method, a surfactant is provided in an aqueous solution, e.g. as a solution in deionized water, at a concentration (C). Details regarding preferred types of surfactants subjected to the method, such as a poloxamer, shall be discussed herein below. The concentration of the surfactant is not particularly restricted, exemplary concentrations may be in the range of 0.1 to 10.0 % w/v (i.e. indicated as the weight of surfactant in g in 100 ml of the total volume of the solution, generally measured at about room temperature, e.g. at 22.1 ± 0.2°C)., preferably in the range of 0.5 to 5.0 % w/v. Particularly preferred is a concentration of 1% w/v.
In step (b), a Langmuir surface pressure/area isotherm of the surfactant in the solution as provided in step (a) is recorded to determine a maximum surface pressure Umax of the Langmuir surface pressure/area isotherm at a predetermined minimum surface area. As will be understood by the skilled reader, this can be accomplished using a Langmuir trough. The recording of the Langmuir isotherm of the surfactant involves reducing the available surface area in the Langmuir trough at least until the predetermined minimum surface area is reached. The surface pressure which is observed at the minimum surface area when recording the Langmuir isotherm of the surfactant is considered as the maximum surface pressure (TTmax) of the surfactant.
As explained herein, it has been surprisingly found by the present inventors that different Langmuir surface pressure/area isotherms may be observed not only for different types of surfactants, but even for surfactants of the same type, e.g. obtained from different sources or production batches, and that the Langmuir isotherms reflect the suitability of the surfactants to act as a stabilizing agent. A skilled person can conveniently determine a minimum surface area for the Langmuir isotherm as a reference point. This reference point can be used to assess the suitability and determine the threshold value that must be observed. The determination can be based on preliminary stability tests of a desired composition containing a surfactant as a stabilizing agent and by selecting a minimum surface area and a threshold value based on a composition with favorable stability. On this basis, a selection and/or quality control of the surfactant during the actual production of the composition can be conveniently carried out by obtaining the Langmuir isotherm data as discussed above and comparing them to the data previously established using a stable composition as a template.
The method can be advantageously used, e.g., to differentiate between variants of a single surfactant, each variant nominally identified by the same name and complying with the same quality standards for medicines, to classify each variant as suitable or not suitable for use as a stabilizing agent for a pharmaceutical composition.
For example, in order to classify a surfactant as suitable or not suitable for use as a stabilizing agent for a pharmaceutical composition comprising a lipid or a lipidoid, a minimum surface area can be established, e.g., by compressing a layer, typically a monolayer, of the lipid or lipidoid in a Langmuir trough until a first phase transition is observed in a surface pressure/area isotherm of the lipid or lipidoid. In order to establish the minimum surface area, the layer can be conveniently provided on water, e.g. deionized water. If the pharmaceutical composition, e.g. as a nanoparticle formulation, comprises a combination of more than one lipid and/or lipidoid, the minimum surface area can be established for the pharmaceutical composition, e.g., by compressing a layer, typically a monolayer, comprising at least one lipid or lipidoid of the combination in a Langmuir trough until a first phase transition is observed in a surface pressure/area isotherm of the lipid or lipidoid. Preferably, the layer comprises at least the lipid or lipidoid which accounts for the largest proportion of the components in the combination of more than one lipid and/or lipidoid, in terms of the weight percentage based on the total weight of the combination as 100%. More preferably, for a pharmaceutical composition comprising a combination of more than one lipid and/or lipidoid, the minimum surface area can be established for the pharmaceutical composition, e.g., by compressing a layer, typically a monolayer, comprising the same combination of more than one lipid and/or lipidoid in the same proportions in a Langmuir trough until a first phase transition is observed in a surface pressure/area isotherm of the combination. In order to establish the minimum surface area, the layer can be conveniently provided on water, e.g. deionized water.
Similarly, in order to classify a surfactant as suitable or not suitable for use as a stabilizing agent for a pharmaceutical composition in the form of a lipid nanoparticle (LNP) formulation or lipidoid nanoparticle (LiNP) formulation as discussed above, a minimum surface area can be established, e.g., by compressing a layer, typically a monolayer, comprising at least one lipid or lipidoid contained in the lipid mix or lipidoid mix (i.e. the lipid mix or lipidoid mix as comprised by the nanoparticles) in a Langmuir trough until a first phase transition is observed in a surface pressure/area isotherm of the lipid or lipidoid. Preferably, the layer comprises at least the lipid or lipidoid which accounts for the largest proportion of the components in the lipid mix or lipidoid mix, in terms of the weight percentage based on the total weight of the lipid mix or lipidoid mix as 100%. More preferably, for a pharmaceutical composition in the form of a lipid nanoparticle (LNP) formulation or lipidoid nanoparticle (LiNP) formulation as discussed above, a minimum surface area can be established by compressing a layer, typically a monolayer, of the lipid mix or lipidoid mix (i.e. the lipid mix or lipidoid mix as comprised by the nanoparticles) in a Langmuir trough until a first phase transition is observed in a surface pressure/area isotherm of the lipid mix or lipidoid mix. In order to establish the minimum surface area, the layer can be conveniently provided on water, e.g. deionized water.
In either case, the surface area at the start of the phase transition can then be relied on as the predetermined minimum surface area in step (b) of the method. As noted above, the start of the phase transition as referred to herein in a pressure/area (TT-A) isotherm plot can be objectively determined as the point where the slope of the curve (dn/dA) changes significantly.
Optionally, a step to establish the minimum surface area can be carried out, e.g., as a calibration step in the Langmuir trough prior to determining the maximum surface pressure of the surfactant as described above.
As a maximum surface area during recording a Langmuir surface pressure/area isotherm, the surface area before the start of the compression in a compression phase, e.g., the maximum area provided by the Langmuir trough, can be conveniently used.
A monolayer of a lipid and/or lipidoid on water, or of a lipid mix or lipidoid mix on water, in a Langmuir trough can be provided, e.g., by applying the lipid and/or lipidoid or the lipid mix or lipidoid mix to the surface until a signal change in the Langmuir trough detector is generated.
The Langmuir surface pressure/area isotherm of the surfactant in the solution in step (b) can be recorded, e.g., in the form of a Langmuir surface pressure/area isotherm cycle including a compression phase and an expansion phase. The maximum surface pressure (nmax) can be determined, for example, by recording a single isotherm cycle, but preferably results are obtained by recording multiple isotherm cycles in sequence, such as three isotherm cycles, e.g. with a wait time of 5 seconds or less between cycles, such as 3 seconds. If a single isotherm cycle is recorded, it is preferred that Umax is equal to or below 3.5 mN/m. If multiple cycles are recorded, such as three cycles, it is preferred that the surfactant shows a Langmuir isotherm with a maximum surface pressure (TTmax) equal to or below the threshold value in the last, e.g. the third isotherm cycle. For example, a Langmuir surface pressure/area isotherm or isotherm cycle as referred to herein can be recorded at about room temperature, e.g. at 22.1 ± 0.2°C.
In step (c), the maximum surface pressure is subsequently compared to a threshold value. Based on this comparison, a decision can be made on whether the surfactant is suitable for use as a stabilizer.
As noted above, a threshold value of the maximum surface pressure TTmax for comparison and classification can be conveniently determined e.g. based on surface pressure data prepared in preliminary tests for a stable composition comprising a surfactant. For example, TTmax can be equal to or below 4.0 mN/m, or equal to or below 3.5 mN/m.
As noted above, in the context of this aspect, a method is further provided for the preparation of a pharmaceutical composition, said method comprising classifying a surfactant as suitable or not suitable for use as a stabilizing agent for a pharmaceutical composition in accordance with the method as described above, and, if the surfactant is classified as being suitable for use as a stabilizing agent for a pharmaceutical composition, incorporating the surfactant into the pharmaceutical composition, e.g. into a vehicle solution of a pharmaceutical composition which is in the form of a nanoparticle suspension.
In a fifth aspect, the invention provides a method for classifying a surfactant as suitable or not suitable for use as a stabilizing agent for a pharmaceutical composition comprising a lipid or lipidoid, optionally during purification of said composition, preferably during tangential flow filtration of said composition, wherein the method comprises the steps:
(a) providing a surfactant in an aqueous solution at a concentration (C) of the surfactant in the solution;
(b) recording a Langmuir pressure/area isotherm cycle including a compression phase and an expansion phase between a maximum surface area and a minimum surface area on a sample comprising the surfactant in the aqueous solution and carrying on its surface a lipid or lipidoid as comprised by the composition:
(c) calculating a Langmuir isotherm An for each area point of the Langmuir pressure/area isotherm cycle, wherein TT is calculated as: wherein itcomp is the surface pressure at said area point during the compression phase of the isotherm cycle, wherein nexp is the surface pressure at said area point during the expansion phase of the isotherm cycle, and wherein nmax is the maximum surface pressure reached in the isotherm cycle, and
wherein itcomp is the surface pressure at said area point during the compression phase of the isotherm cycle, wherein nexp is the surface pressure at said area point during the expansion phase of the isotherm cycle, and wherein nmax is the maximum surface pressure reached in the isotherm cycle, and

(d) comparing the calculated Langmuir isotherm ATT to a threshold value, wherein, if the calculated Langmuir isotherm An is at every isotherm area point equal to or less than the threshold value, the surfactant is classified as suitable for use as a stabilizing agent, and if the calculated Langmuir ATT at any area point is greater than the threshold value the surfactant is classified as not suitable for use as a stabilizing agent.
Further provided in the context of this aspect is a method for the preparation of a pharmaceutical composition, said method comprising classifying a surfactant as suitable or not suitable for use as a stabilizing agent for a pharmaceutical composition comprising a lipid or lipidoid in accordance with the method as described above, and, if the surfactant is classified as being suitable for use as a stabilizing agent, incorporating the surfactant into the pharmaceutical composition.
For example, the method can be used for classifying a surfactant as suitable or not suitable for use as a stabilizing agent in a pharmaceutical composition comprising a lipid nanoparticle (LNP) formulation or lipidoid nanoparticle (LiNP) formulation comprising a plurality of lipid nanoparticles (LNPs) or lipidoid nanoparticles (LiNPs). Preferably, the pharmaceutical composition is a lipid nanoparticle (LNP) formulation or lipidoid nanoparticle (LiNP) formulation comprising a plurality of lipid nanoparticles (LNPs) or lipidoid nanoparticles (LiNPs), each comprising a lipid mix or lipidoid mix, wherein the lipid mix or lipidoid mix comprises, as a component thereof, at least one selected from an ionizable lipid, ionizable lipidoid, and permanently cationic lipid.
If the pharmaceutical formulation is a lipid nanoparticle (LNP) formulation or lipidoid nanoparticle (LiNP) formulation, it is preferred that the formulation is a suspension formulation comprising a liquid vehicle solution, wherein the LNPs or LiNPs can be dispersed. The vehicle solution is preferably an aqueous vehicle solution. It is preferred that, in such a suspension formulation, a surfactant classified as being suitable for use as a stabilizing agent is incorporated into the vehicle solution. For example, in such a suspension formulation, the surfactant may favorably act as a stabilizing agent that mitigates aggregation of the LNPs or LiNP or a subpopulation thereof which may be caused by shaking or by shear stress of the suspension during production, purification, handling or transport, preferably during production or purification, more preferably during purification, most preferably during tangential flow filtration (TFF) purification.
As will be understood by the skilled person, the pharmaceutical composition comprises a therapeutic agent, for example a nucleic acid. A preferred therapeutic agent is RNA, and particularly preferred is mRNA. Details regarding preferred types of therapeutic agents shall be discussed herein below. If the pharmaceutical formulation is a lipid nanoparticle (LNP) formulation or lipidoid nanoparticle (LiNP) formulation, e.g. a suspension formulation, the therapeutic agent is typically comprised in the LNPs or LiNPs.
In line with the above, it is particularly preferred if the method according to this aspect is a method for classifying a surfactant as suitable or not suitable for use as a stabilizing agent for a pharmaceutical composition, wherein the pharmaceutical composition is in the form of a lipid nanoparticle (LNP) suspension formulation or lipidoid nanoparticle (LiNP) suspension formulation comprising a plurality of LNPs or LiNPs, each comprising a lipid mix or lipidoid mix and a therapeutic agent, wherein each of the LNPs or LiNPs comprises, as a component of the lipid mix or lipidoid mix, at least one selected from an ionizable lipid, ionizable lipidoid, and permanently cationic lipid, and wherein the vehicle solution of the suspension formulation is an aqueous vehicle solution.
In step (a) of the method, a surfactant is provided in an aqueous solution at a concentration (C). Details regarding preferred types of surfactants subjected to the method, such as a poloxamer, shall be discussed herein below. The concentration of the surfactant is not particularly restricted, exemplary concentrations may be in the range of 0.1 to 10.0 % w/v (i.e. indicated as the weight of surfactant in g in 100 ml of the combined volumes of the surfactant and aqueous solvent, generally measured at about room temperature, e.g. at 22.1 ± 0.2°C), preferably in the range of 0.5 to 5.0 % w/v. Particularly preferred is a concentration of 1% w/v.
In step (b), a Langmuir pressure/area isotherm cycle including a compression phase and an expansion phase is recorded between a maximum surface area and a minimum surface area. The isotherm cycle is recorded on a sample comprising the surfactant in the aqueous solution as provided in step (a), and carrying on its surface a lipid or lipidoid as comprised by the pharmaceutical composition. Typically, the sample carries a monolayer of the lipid or lipidoid on its surface. As will be understood by the skilled reader, recording of the isotherm cycle can be accomplished using a Langmuir trough containing the sample. If the pharmaceutical composition, e.g. as a nanoparticle formulation, comprises a combination of more than one lipid and/or lipidoid, the isotherm cycle can be recorded, e.g., on a sample carrying a layer, typically a monolayer, comprising at least one lipid or lipidoid of the combination. Preferably, the layer comprises at least the lipid or lipidoid which accounts for the largest proportion of the components in the combination of more than one lipid and/or lipidoid in the pharmaceutical composition, in terms of the weight percentage based on the total weight of the combination as 100%. More preferably, for a pharmaceutical composition comprising a combination of more than one lipid and/or lipidoid, the isotherm cycle can be recorded on a sample carrying a layer, typically a monolayer, comprising the same combination of more than one lipid and/or lipidoid in the same proportions as comprised in the pharmaceutical composition.
In line with a preferred embodiment discussed above, the pharmaceutical composition is a lipid nanoparticle (LNP) formulation or lipidoid nanoparticle (LiNP) formulation comprising a plurality of lipid nanoparticles (LNPs) or lipidoid nanoparticles (LiNPs), each comprising a lipid mix or lipidoid mix, wherein the lipid mix or lipidoid mix comprises, as a component thereof, at least one selected from an ionizable lipid, ionizable lipidoid, and permanently cationic lipid. In this case, the isotherm cycle can be recorded, e.g., on a sample carrying a layer, typically a monolayer, comprising at least one lipid or lipidoid contained in the lipid mix or lipidoid mix (i.e. the lipid mix or lipidoid mix as comprised by the nanoparticles). Preferably, the layer comprises at least the lipid or lipidoid which accounts for the largest proportion of the components in the lipid mix or lipidoid mix, in terms of the weight percentage based on the total weight of the lipid mix or lipidoid mix as 100%. More preferably, for a pharmaceutical composition in the form of a lipid nanoparticle (LNP) formulation or lipidoid nanoparticle (LiNP) formulation as discussed above, the isotherm cycle is recorded on a sample carrying a layer, typically a monolayer, of the lipid mix or lipidoid mix (i.e. the lipid mix or lipidoid mix as comprised by the nanoparticles).
The minimum surface area to be adopted for recording the isotherm cycle is generally determined by the lipid or lipidoid, or by the combination of more than one lipid/lipidoid contained in the sample used for recording the isotherm cycle. Specifically, the minimum surface area for recording the isotherm cycle is preferably the surface area at which the start of a phase transition can be observed when a layer, typically a monolayer, of the lipid or lipidoid or of the combination of more than one lipid/lipidoid is compressed in a Langmuir trough.
Thus, optionally, a step to establish the minimum surface area can be carried out, e.g., as a calibration step in the Langmuir trough prior to recording the isotherm cycle in step (b). For example, a minimum surface area can be established by compressing a layer, typically a monolayer, of the lipid or lipidoid or of the combination of more than one lipid/lipidoid as it is contained in the sample for recording the isotherm cycle in step (b) in the Langmuir trough until a first phase transition is observed in a surface pressure/area isotherm. In order to establish the minimum surface area, the layer can be conveniently provided on water, e.g. deionized water. The surface area at the start of the phase transition can then be relied on as the minimum surface area for recording the isotherm cycle in step (b) of the method. As noted above, the start of the phase transition as referred to herein in a pressure/area (IT -A) isotherm plot can be objectively determined as the point where the slope of the curve (dn/dA) changes significantly.
As a maximum surface area during recording a Langmuir surface pressure/area isotherm, the surface area before the start of the compression in a compression phase, e.g., the maximum area provided by the Langmuir trough, can be conveniently used.
A monolayer of a lipid and/or lipidoid on water, or of a lipid mix or lipidoid mix on water, in a Langmuir trough can be provided, e.g., by applying the lipid and/or lipidoid or the lipid mix or lipidoid mix to the surface until a signal change in the Langmuir trough detector is generated.
It is sufficient if a single isotherm cycle is recorded, and the Langmuir isotherm An values are preferably calculated in step (c) for this isotherm cycle. Multiple isotherm cycles can be recorded in sequence, such as three isotherm cycles, e.g. with a wait time of 5 seconds or less between cycles, such as 3 seconds. However, if TT values of the first cycle fulfill the requirements set forth above, the same typically applies also for the subsequent cycles, such that it is preferred to rely on the measurement results of the first cycle for the calculation of the An values even if multiple cycles are recorded.
For example, a Langmuir surface pressure/area isotherm or isotherm cycle as referred to herein can be recorded at about room temperature, e.g. at 22.1 ± 0.2°C.
In step (c), a Langmuir isotherm An is calculated for each area point of the Langmuir pressure/area isotherm cycle.
As an aspect closely related to the method for classifying a surfactant described above, a method is provided for the preparation of a pharmaceutical composition, said method comprising classifying a surfactant as suitable or not suitable for use as a stabilizing agent for a pharmaceutical composition in accordance with the method as described above, and, if the surfactant is classified as being suitable for use as a stabilizing agent, incorporating the surfactant into the pharmaceutical composition, e.g. into a vehicle solution of a pharmaceutical composition which is in the form of a nanoparticle suspension. In a related sixth aspect, the invention provides a method of mitigating or avoiding clogging or fouling of a filtration system during purification of a pharmaceutical composition in the form of a lipid nanoparticle formulation (LNP) or lipidoid nanoparticle formulation (LiNP), the method comprising adding a stabilizing surfactant to a first LNP or LiNP formulation to form a second LNP or LiNP formulation, optionally before purification, wherein the LNP or LiNP formulation comprises a plurality of LNPs or LiNPs, each comprising a lipid mix or lipidoid mix and a therapeutic agent, wherein the lipid mix or lipidoid mix comprises, as a component thereof, at least one selected from an ionizable lipid, ionizable lipidoid, and permanently cationic lipid, and wherein the stabilizing surfactant is a surfactant according to the invention, e.g. a surfactant as discussed in the third aspect above, or wherein the stabilizing surfactant is a surfactant classified as being suitable as a stabilizing agent by the method according to the fourth or the fifth aspect discussed above.
The LNP formulation or LiNP formulation comprises a therapeutic agent, for example a nucleic acid. A preferred therapeutic agent is RNA, and particularly preferred is mRNA. Details regarding preferred types of therapeutic agents shall be discussed herein below.
It is preferred that the LNP formulation or LiNP formulation is a suspension formulation comprising a liquid vehicle solution, wherein the LNPs or LiNPs can be dispersed. The vehicle solution is preferably an aqueous vehicle solution. It is preferred that, in such a suspension formulation, the stabilizing surfactant is added in the context of the above method to the vehicle solution. Optionally, the LNPs or LiNPs dispersed in the liquid vehicle are essentially free of the stabilizing surfactant, e.g. the surfactant is essentially not incorporated into the LNPs or LiNPs.
Preferably, the purification of the pharmaceutical composition includes tangential flow filtration (TFF) purification.
In a further related seventh aspect, the invention provides a method of mitigating aggregation of lipid or lipidoid nanoparticles in a lipid nanoparticle (LNP) formulation or lipidoid nanoparticle (LiNP) formulation, the method comprising adding a stabilizing surfactant to a first LNP or LiNP formulation to form a second LNP or LiNP formulation, optionally before purification, wherein the LNP or LiNP formulation comprises a plurality of LNPs or LiNPs, each comprising a lipid mix or lipidoid mix, wherein the lipid mix or lipidoid mix comprises, as a component thereof, at least one selected from an ionizable lipid, ionizable lipidoid, and permanently cationic lipid, and wherein the stabilizing surfactant is a surfactant in accordance with invention, e.g. a surfactant as discussed in the third aspect above, or wherein the stabilizing surfactant is a surfactant classified as being suitable as a stabilizing agent by the method according to the fourth or the fifth aspect discussed above.
Preferably, the method is a method of mitigating aggregation of lipid or lipidoid nanoparticles in a pharmaceutical formulation in the form of a lipid nanoparticle (LNP) formulation or lipidoid nanoparticle (LiNP) formulation comprising a plurality of LNPs or LiNPs, each comprising a lipid mix or lipidoid mix and a therapeutic agent, for example a nucleic acid. A preferred therapeutic agent is RNA, and particularly preferred is mRNA. Details regarding preferred types of therapeutic agents shall be discussed herein below.
Also in the context of this method, it is preferred that the LNP formulation or LiNP formulation is a suspension formulation comprising a liquid vehicle solution, wherein the LNPs or LiNPs can be dispersed. The vehicle solution is preferably an aqueous vehicle solution. It is preferred that, in such a suspension formulation, the stabilizing surfactant is added in the context of the above method to the vehicle solution. Optionally, the LNPs or LiNPs dispersed in the liquid vehicle are essentially free of the stabilizing surfactant, e.g. the surfactant is essentially not incorporated into the LNPs or LiNPs.
Thus, it is particularly preferred that the method is a method of mitigating aggregation of lipid or lipidoid nanoparticles in a pharmaceutical formulation in the form of a lipid nanoparticle (LNP) suspension formulation or lipidoid nanoparticle (LiNP) suspension formulation comprising a plurality of LNPs or LiNPs, each comprising a lipid mix or lipidoid mix and a therapeutic agent, wherein the lipid mix or lipidoid mix comprises, as a component thereof, at least one selected from an ionizable lipid, ionizable lipidoid, and permanently cationic lipid.
The lipid nanoparticles or lipidoid nanoparticles may comprise a nucleic acid, e.g. RNA, and preferably mRNA, as a therapeutic agent, and the method in accordance with this aspect may comprise, e.g., the following steps: i) first, combining the nucleic acid and at least one selected from an ionizable lipid, ionizable lipidoid, and permanently cationic lipid to form LNPs or LiNPs, ii) second, purifying the LNPs or LiNPs iii) third, adding the stabilizing surfactant before TFF purification and/or during TFF purification in an exchange buffer, maintaining the surfactant in a steady concentration, iv) optionally wherein the stabilizing surfactant is added to the LNP or LiNP formulation after step (i). In a preferred approach, the method in accordance with this aspect may alternatively comprise, e.g., the following steps: i) generating an LNP or LiNP preparation by mixing of at least one selected from a permanently cationic lipid, an ionizable lipid and an ionizable lipidoid dissolved in an organic phase with a therapeutic agent dissolved in an aqueous solution, ii) diluting the LNP or LiNP preparation by dilution with a first solution, iii) concentrating the LNP or LiNP preparation by buffer exchange using ultra/diafiltration via TFF wherein a second solution is used for the ultra/diafiltration, iv) obtaining a LNP or LiNP suspension in an aqueous vehicle solution, wherein the first solution comprises between about 0.01% w/v and 10% of the stabilizing surfactant, preferably between about 0.01% w/v surfactant and 5% w/v surfactant, more preferably between about 0.01 % w/v surfactant and 2.5% w/v surfactant, more preferably between about 0.05% w/v and 1 .5% w/v surfactant, even more preferably between about 0.05% w/v and 1.5% w/v surfactant, most preferably about 1 % w/v surfactant, and/or wherein the second solution comprises between about 0.01% w/v and about 10% of the stabilizing surfactant, preferably between about 0.01% w/v surfactant and about 5% w/v surfactant, more preferably between about 0.01 % w/v surfactant and about 2.5% w/v surfactant, even more preferably between about 0.05% w/v and 1.5% w/v surfactant, most preferably about 1 % w/v; and wherein the final concentration of the stabilizing surfactant from combined first and second solution is between 0.01% and 10% surfactant, preferably between 0.01% w/v surfactant and 5% w/v surfactant, more preferably between 0.01% w/v surfactant and 2.5% w/v surfactant, even more preferably between 0.05% w/v and 1.5 % w/v surfactant, most preferably about 1% w/v surfactant with regard to the total volume of the suspension of the nanoparticles in the aqueous vehicle solution.
In the context of this preferred approach, these further optional features may be observed: a) the incorporation of the stabilizing surfactant into the suspension does not occur before or during step i), b) the stabilizing surfactant is added in the first and the second solution, and/or c) approximately half of the stabilizing surfactant is added to the first solution and approximately half of the surfactant is added to the second solution.
As a related eighth aspect, the invention provides a lipid nanoparticle (LNP) formulation or lipidoid nanoparticle (LiNP) formulation obtained by the method in accordance with the fourth to seventh aspect. For example, this aspect provides a pharmaceutical composition in the form of a lipid nanoparticle (LNP) formulation or lipidoid nanoparticle (LiNP) formulation, said composition comprising a surfactant which has been classified as being suitable for use as a stabilizing agent for a pharmaceutical composition in accordance with the method of the fourth aspect. As another example, the eighth aspect provides pharmaceutical composition in the form of a lipid nanoparticle (LNP) formulation or lipidoid nanoparticle (LiNP) formulation, said composition comprising a surfactant which has been classified as being suitable for use as a stabilizing agent for a pharmaceutical composition comprising a lipid or lipidoid composition in accordance with the method of the fifth aspect. As a further example, the eighth aspect provides a pharmaceutical composition in the form of lipid nanoparticle (LNP) formulation or lipidoid nanoparticle (LiNP) formulation obtained by the method in accordance with the sixth aspect. As still a further example, the eighth aspect provides a lipid nanoparticle (LNP) formulation or lipidoid nanoparticle (LiNP) formulation, preferably a pharmaceutical composition in the form of a lipid nanoparticle (LNP) formulation or lipidoid nanoparticle (LiNP) formulation, obtained by the method in accordance with the sixth aspect.
Preferably, the formulation is a lipid nanoparticle (LNP) suspension formulation or lipidoid nanoparticle (LiNP) suspension formulation.
Furthermore, in a ninth aspect, the invention provides a lipid nanoparticle (LNP) formulation or lipidoid nanoparticle (LiNP) formulation in accordance with the first aspect, the second aspect, or the eighths aspect discussed above for use as a medicament. As will be understood, the LNPs or LiNPs of the lipid nanoparticle (LNP) formulation or lipidoid nanoparticle (LiNP) formulation for use in accordance with this aspect comprise a therapeutic agent, e.g. a nucleic acid such as RNA, preferably mRNA. They are preferably suspension formulations as discussed in the context of the concerned aspects.
In a related tenth aspect, the invention provides a lipid nanoparticle (LNP) formulation or lipidoid nanoparticle (LiNP) formulation in accordance with the first aspect, the second aspect, or the eighths aspect discussed above for use in the treatment or prevention of a disease. As will be understood, the LNPs or LiNPs of the lipid nanoparticle (LNP) formulation or lipidoid nanoparticle (LiNP) formulation for use in accordance with this aspect comprise a therapeutic agent, e.g. a nucleic acid such as RNA, preferably mRNA. They are preferably suspension formulations as discussed in the context of the concerned aspects. The disease may be, e.g., a disease selected from Table A as disclosed herein below.
Likewise, and preferably, the disease may be a disease selected from viral diseases, ciliopathies, or autoimmune diseases, and respiratory tract diseases, even more preferably selected from a lung disease, an airway disease or a nasal disease, more preferably a lung viral disease, lung ciliopathies and a lung autoimmune disease. Preferably, the lung disease or lung viral disease is at least one selected from pneumonia and asthma; the airway disease is at least one selected from bronchitis, viral induced asthma, lung fibrosis and COPD; and/or the nasal disease is at least one selected from rhinitis and sinusitis.
In some embodiments, the disease to be treated or prevented is a disease selected from the list consisting of pulmonary alveolar proteinosis (PAP), interstitial lung disease such as pulmonary fibrosis, e.g. idiopathic pulmonary fibrosis, viral infections such as Influenza and COVID-19, acute respiratory distress syndrome (ARDS), non-tuberculous mycobacterial (NTM) infection, lung cancer, fungal infections caused by Aspergillus sp., such as aspergillosis, fungal sinusitis, otomycosis, keratitis, and onychomycosis, preferably those caused by Aspergillus fumigatus and Aspergillus flavus, infections caused by Mycobacterium tuberculosis, Pseudomonas aeruginosa, Pneumocystis sp., Plasmodium sp., Cryptococcus sp., Nocardia sp., and combinations thereof.
In some embodiments the disease is selected from the list consisting of (autoimmune) pulmonary alveolar proteinosis (PAP), interstitial lung disease such as pulmonary fibrosis, e.g. idiopathic pulmonary fibrosis, viral infections such as Influenza and COVID-19, acute respiratory distress syndrome (ARDS), non-tuberculous mycobacterial (NTM) infection, lung cancer, or fungal infections caused by Aspergillus sp., such as aspergillosis, fungal sinusitis, otomycosis, keratitis, and onychomycosis, preferably those caused by Aspergillus fumigatus and Aspergillus flavus, infections caused by Mycobacterium tuberculosis, Pseudomonas aeruginosa, Pneumocystis sp., and Plasmodium sp.
The lipid nanoparticle (LNP) formulation or lipidoid nanoparticle (LINP) formulation for use in accordance with the tenth aspect may be for use in vaccination or immunization.
In a further aspect, the invention provides a method for classifying a surfactant as suitable or not suitable for use as a stabilizer of a pharmaceutical composition comprising a nucleic acid, optionally as a stabilizer during purification, preferably during TFF purification, the method comprising:
(a) providing a surfactant in an aqueous solution; (b) optionally combining the surfactant with a LNP or LiNP formulation;
(c) purifying the aqueous solution comprising the surfactant or optionally the LNP or LiNP formulation comprising the surfactant using a membrane purification system, preferably TFF or ultrafiltration, most preferably TFF;
(d) measuring the filtration time of the aqueous solution or the LNP formulation during diafiltration or ultrafiltration;
(e) comparing the measured filtration speed to a predetermined threshold value, preferably a time threshold value, more preferably a time threshold value of 90 minutes, wherein if the filtration speed is equal to or greater than the threshold value, the surfactant is classified as not suitable for use as a stabilizer, and if the filtration speed is less than the threshold value, the surfactant is classified as suitable for use as a stabilizer.
In a further aspect, the invention provides a method of reducing or avoiding side effects in a therapy with LNPs or LiNPs carrying at least one therapeutic agent, wherein the method comprises the steps: i) determine whether LNPs or LiNPs in a pharmaceutical composition comprising LNPs or LiNPs aggregate when subjected to a mechanical stress or a temperature stress, by determining their aggregation level before and after subjecting said pharmaceutical composition to said mechanical or temperature stress ii) if the LNP or LiNP show aggregation after the test of step (i), then add to the LNP or LiNP formulation a surfactant to obtain a LNP or LiNP suspension with a final surfactant concentration of 0.01 % w/v and up to 10% w/v, preferably between 0.05% w/w surfactant and 5% surfactant, more preferably between 0.33% surfactant and 2.5% surfactant, more preferably between 0.45% and 1 .5% surfactant, preferably between 0.5% and 1 .5% surfactant, most preferably 1%. iii) reconstitute with mixing to generate a stable LNP or LiNP suspension.
In a related aspect, the invention provides a use of a surfactant according to the invention, e.g. the surfactant in accordance with the above third aspect, or a surfactant classified as being suitable as a stabilizing agent in a pharmaceutical composition in accordance with the method of the above fourth or fifth aspect, for stabilizing a suspension of lipid nanoparticles or of lipidoid nanoparticles in an aqueous vehicle solution against particle aggregation under a physical stress condition, preferably shear stress, more preferably shear stress during purification such as TFF, wherein the lipid nanoparticles or lipidoid nanoparticles comprise the following components (a) and (b):
(a) a therapeutic agent and
(b) at least one selected from a permanently cationic lipid, an ionizable lipid and an ionizable lipidoid.
THERAPEUTIC AGENTS
When reference is made herein to a therapeutic agent, to a pharmaceutical composition comprising a therapeutic agent, and/or to a lipid nanoparticle formulation or lipid nanoparticle formulation comprising a therapeutic agent, one or more therapeutic agents may be used in the context of the invention.
Preferably, the therapeutic agent comprises, essentially consists of or consists of, a nucleic acid, more preferably RNA, still more preferably single stranded RNA, and most preferably mRNA.
In a lipid nanoparticle formulation or lipid nanoparticle formulation comprising a therapeutic agent as referred to herein, e.g. a negatively charged therapeutic agent may be used. As will be understood by the skilled reader, in such a lipid nanoparticle formulation or lipid nanoparticle formulation comprising a therapeutic agent, the therapeutic agent is typically comprised in the nanoparticles. Also in such a formulation, it is preferred that the therapeutic agent comprises, essentially consists of or consists of, a nucleic acid, more preferably RNA, still more preferably single stranded RNA, and most preferably mRNA. Thus, particularly preferred pharmaceutical compositions in the context of the invention are pharmaceutical compositions in the form of a lipid nanoparticle formulation or lipid nanoparticle formulation, such as a suspension formulation, wherein the lipid nanoparticles or lipid nanoparticles comprise mRNA as a therapeutic agent.
The nature of a nucleic acid is not particularly limited. In principle any type of nucleic acid can be employed in the context of the present invention. Nucleic acids are known to the skilled person and refer to biopolymers or small biomolecules composed of nucleotides which are the monomers made of three components: a 5-carbon sugar, a phosphate group and a nitrogenous base. The term nucleic acid is the overall name for DNA (deoxyribonucleic acid) and RNA (ribonucleic acid), i.e., the members of the above family of biopolymers. If the sugar is a compound ribose, the polymer is RNA if the sugar is derived from ribose as deoxyribose, the polymer is DNA. The term “nucleic acid” encompasses oligonucleotides or polynucleotides. As a nucleic acid is a biopolymer composed of nucleotides, the term “nucleic acid” is also often referred to as a “sequence of nucleotides” and, accordingly, as will be understood by the skilled person, the terms “nucleic acid” and “nucleic acid sequence” are often used interchangeably.
In a preferred embodiment, the nanoparticles comprise ribonucleic acid (RNA) as nucleic acid, more preferably single stranded RNA, and most preferred is mRNA.
The term "nucleic acid" encompasses all forms of naturally occurring types of nucleic acids as well as chemically and/or enzymatically synthesized nucleic acids and also encompasses nucleic acid analogues and nucleic acid derivatives. The term in particular includes any backbone-modified, sugar-modified or base-modified single-stranded or double-stranded nucleic acid, such as e.g. locked nucleic acids (LNA), peptide nucleic acids (PNA), oligonucleoside thiophosphates and phosphotriesters, morpholino oligonucleotides, cationic oligonucleotides (US6017700 A, WO/2007/069092), substituted ribo-oligonucleotides or phosphorothioates. Furthermore, the term "nucleic acid" also refers to any molecule that comprises nucleotides or nucleotide analogues. There are no limitations concerning sequence or size of a nucleic acid comprised in the nanoparticles of the present invention. The nucleic acid is predominantly defined by the biological effect that is to be achieved at the biological target the nanoparticles of the present invention are delivered to. For instance, as will be outlined in more detail further below, in the case of an application in gene or nucleic acid therapy, the nucleic acid or nucleic acid sequence can be defined by the gene or gene fragment that is to be expressed or by the intended substitution or repair of a defective gene or any gene target sequence or by the target sequence of a gene to be inhibited, knocked-down, down- regulated or up-regulated.
The nanoparticles may comprise a nucleic acid being a DNA molecule. A preferred embodiment of such a DNA molecule is a DNA molecule which can be transcribed into an mRNA molecule. Transcription is the first step of gene expression, in which a particular segment of a DNA molecule is copied into an mRNA molecule by the enzyme RNA polymerase. During transcription, a DNA sequence is read by an RNA polymerase, which produces a complementary, anti-parallel RNA strand called a primary transcript. A DNA molecule may be introduced in a vector, preferably an expression vector, by standard molecular biology techniques (see, e.g. Sambrook et al., Molecular Cloning, A laboratory manual, 2nd Ed, 1989). The term “vector” such as “expression vector” or “cloning vector” in the sense of the present invention is understood as a circular, double-stranded unit of DNA that is preferably able to replicate within a cell independently of the chromosomal DNA and which is used as a vehicle to carry genetic material into a cell, where it can be (replicated and/or) expressed (i.e., transcribed into RNA and translated into an amino acid sequence). A vector containing foreign DNA is termed recombinant DNA. The vector itself is generally a DNA sequence that typically consists of an insert (e.g., a nucleic acid molecule/DNA molecule of the present invention) and a larger sequence that serves as the "backbone" of the vector. Plasmids in the sense of the present invention are most often found in bacteria and are used in recombinant DNA research to transfer genes between cells and are as such a subpopulation of “vectors” as used in the sense of the present invention.
It is evident to the person skilled in the art that further regulatory sequences may be added to the DNA molecule of the invention. For example, transcriptional enhancers and/or sequences which allow for induced expression may be employed. A suitable inducible system is, for example, tetracycline-regulated gene expression as described, e.g., by Gossen and Bujard, Proc. Natl. Acad. Sci. USA 89 (1992), 5547-5551 ) and Gossen, Trends Biotech. 12 (1994), 58- 62, or a dexamethasone-inducible gene expression system as described, e.g. by Crook, EMBO J. 8 (1989), 513-519. The present invention may also use a vector, preferably an expression vector, comprising the DNA molecule. The vector may be, e.g., a plasmid, cosmid, virus, bacteriophage or another vector used e.g. conventionally in genetic engineering, and may comprise further genes such as marker genes which allow for the selection of said vector in a suitable host cell and under suitable conditions.
If the nucleic acid used in the context of the present invention is a DNA molecule, it can be a plasmid DNA (pDNA) molecule.
As noted above, the nanoparticles preferably comprise ribonucleic acid (RNA) as nucleic acid, more preferably single stranded RNA, and most preferred is mRNA.
As regards RNA, in principle any type of RNA can be employed in the context of the present invention. In a preferred embodiment the RNA is a single-stranded RNA. The term “singlestranded RNA” means a single consecutive chain of ribonucleotides in contrast to RNA molecules in which two or more separate chains form a double-stranded molecule due to hybridization of the separate chains. The term “single-stranded RNA” does not exclude that the single-stranded molecule forms in itself double-stranded structures such as secondary (e.g., loops and stem-loops) or tertiary structures. Examples are tRNA and mRNA but also any other type of single-stranded RNA like antisense-RNA, siRNA, miRNA and the like.
The term “RNA” covers RNA which codes for an amino acid sequence as well as RNA which does not code for an amino acid sequence. It has been suggested that more than 80 % of the genome contains functional DNA elements that do not code for proteins. These noncoding sequences include regulatory DNA elements (binding sites for transcription factors, regulators and coregulators etc.) and sequences that code for transcripts that are never translated into proteins. These transcripts, which are encoded by the genome and transcribed into RNA but do not get translated into proteins, are called noncoding RNAs (ncRNAs). Thus, in one embodiment the RNA is a noncoding RNA. Preferably, the noncoding RNA is a single-stranded molecule. Studies demonstrate that ncRNAs are critical players in gene regulation, maintenance of genomic integrity, cell differentiation, and development, and they are misregulated in various human diseases. There are different types of ncRNAs: short (20-50 nt), medium (50-200 nt), and long (>200 nt) ncRNAs. Short ncRNA includes microRNA (miRNA), small interfering RNA (siRNA), piwi-interacting RNA (piRNA), and transcription initiating RNA (tiRNA). Examples of medium ncRNAs are small nuclear RNAs (snRNAs), small nucleolar RNAs (snoRNAs), transfer RNAs (tRNAs), transcription start-site-associated RNAs (TSSaRNAs), promoter-associated small RNAs (PASRs), and promoter upstream transcripts (PROMPTS). Long noncoding RNAs (IncRNA) include long-intergenic noncoding RNA (lincRNA), antisense-IncRNA, intronic IncRNA, transcribed ultra-conserved RNAs (T-UCRs), and others (Bhan A, Mandal SS, ChemMedChem. 2014 Mar 26. doi: 10.1002/cmdc.201300534). Of the above-mentioned non-coding RNAs only siRNA is doublestranded. Thus, since in a preferred embodiment the noncoding RNA is single-stranded, it is preferred that the noncoding RNA is not siRNA. In another embodiment the RNA is a coding RNA, i.e. an RNA which codes for an amino acid sequence. Such RNA molecules are also referred to as mRNA (messenger RNA) and are single-stranded RNA molecules. The RNA may be made by synthetic chemical and enzymatic methodology known to one of ordinary skill in the art, or by the use of recombinant technology, or may be isolated from natural sources, or by a combination thereof.
Messenger RNAs (mRNA) are copolymers which are built up of nucleoside phosphate building blocks mainly with adenosine, cytidine, uridine and guanosine as nucleosides, which as intermediate carriers bring the genetic information from the DNA in the cell nucleus into the cytoplasm, where it is translated into proteins. They are thus suitable as alternatives for gene expression. In the context of the present invention, mRNA should be understood to mean any polyribonucleotide molecule which, if it comes into the cell, is suitable for the expression of a protein or fragment thereof or is translatable to a protein or fragment thereof. The term “protein” here encompasses any kind of amino acid sequence, i.e. chains of two or more amino acids which are each linked via peptide bonds and also includes peptides and fusion proteins.
The mRNA contains a ribonucleotide sequence which encodes a protein or fragment thereof whose function in the cell or in the vicinity of the cell is needed or beneficial, e.g. a protein the lack or defective form of which is a trigger for a disease or an illness, the provision of which can moderate or prevent a disease or disorder, or a protein which can promote a process which is beneficial for the body, in a cell or its vicinity. The mRNA may contain the sequence for the complete protein or a functional variant thereof. Further, the ribonucleotide sequence can encode a protein which acts as a factor, inducer, regulator, stimulator or enzyme, or a functional fragment thereof, where this protein is one whose function is necessary in order to remedy a disorder, in particular a metabolic disorder or in order to initiate processes in vivo such as the formation of new blood vessels, tissues, etc. Examples of proteins which can be encoded by mRNA include antibodies, cytokines or chemokines. Here, functional variant is understood to mean a fragment which in the cell can undertake the function of the protein whose function in the cell is needed or the lack or defective form whereof is pathogenic. In addition, the mRNA may also have further functional regions and/or 3’ or 5’ noncoding regions, in particular 3’ and/or 5’ UTRs. The 3’ and/or 5’ noncoding regions can be the regions naturally flanking the protein-encoding sequence or artificial sequences, e.g. sequences which contribute to the stabilization of the RNA. Those skilled in the art can determine the sequences suitable for this in each case by routine experiments.
In a preferred embodiment, the mRNA contains a 5'-cap (five-prime-cap; cap-0) consisting of a m7GpppG connected to the mRNA via a 5' to 5' triphosphate linkage, an additional methyl group onto the penultimate nucleotide from the 5'-end of the mRNA (Cap-1 , Anti-Reverse Cap Analog (ARCA)) and/or an internal ribosome entry site (IRES) and/or a poly(A)-tail at the 3’- end, in particular, in order to improve translation. The mRNA can have further regions promoting translation such as, for example, cap-2 structures or histone stem-loop structures.
The RNA which may be present in the nanoparticles may contain unmodified and modified nucleotides. The term “unmodified nucleotide” used herein refers to A, C, G and U nucleotides. The term “modified nucleotide” used herein refers to any naturally occurring or non -naturally occurring isomers of A, C, G and U nucleotides as well as to any naturally occurring or naturally occurring analogues, alternative or modified nucleotide or isomer thereof having for example chemical modifications or substituted residues. Modified nucleotides can have a base modification and/or a sugar modification. Modified nucleotides can also have phosphate group modifications, e.g., with respect to the 5’- prime cap of an mRNA molecule. Modified nucleotides also include nucleotides that are synthesized post-transcriptionally by covalent modification of the nucleotides. Further, any suitable mixture of non-modified and modified nucleotides is possible. A non-limiting number of examples of modified nucleotides can be found in the literature (e.g. US 2013/0123481 A1; Cantara et al., Nucleic Acids Res, 2011, 39(lssue suppl_1):D195-D201; Helm and Alfonzo, Chem Biol, 2014, 21 (2):174-185; or Carell et al., Angew Chem Int Ed Engl, 2012, 51 (29):7110-31) and some preferable modified nucleotides are mentioned exemplarily in the following based on their respective nucleoside residue:
1 -methyladenosine, 2-methylthio-N6-hydroxynorvalyl carbamoyladenosine, 2- methyladenosine, 2’-O-ribosylphosphate adenosine, N6-methyl-N6- threonylcarbamoyladenosine, N6-acetyladenosine, N6-glycinylcarbamoyladenosine, N6- isopentenyladenosine, N6-methyladenosine, N6-threonylcarbamoyladenosine, N6,N6- dimethyladenosine, N6-(cis-hydroxyisopentenyl)adenosine, N6- hydroxynorvalylcarbamoyladenosine, 1 ,2’-O-dimethyladenosine, N6,2’-O-dimethyladenosine, 2’-O-methyladenosine, N6,N6,2’-O-trimethyladenosine, 2-methylthio-N6-(cis- hydroxyisopentenyl)adenosine, 2-methylthio-N6-methyladenosine, 2-methylthio-N6- isopentenyladenosine, 2-methylthio-N6-threonyl carbamoyladenosine, N6-2-methylthio-N6- threonyl carbamoyladenosine, 2-methylthio-N6-(cis-hydroxyisopentenyl)adenosine, 7- methyladenosine, 2-methylthio-adenosine, 2-methoxy-adenosine, 2’-amino-2’- deoxyadenosine, 2’-azido-2’-deoxyadenosine, 2’-fluoro-2’-deoxyadenosine, 2-aminopurine, 2,6-diaminopurine, 7-deaza-adenosine, 7-deaza-8-aza-adenosine, 7-deaza-2-aminopurine, 7- deaza-8-aza-2-aminopurine, 7-deaza-2,6-diaminopurine, 7-deaza-8-aza-2,6-diaminopurine;
2-thiocytidine, 3-methylcytidine, N4-acetylcytidine, 5-formylcytidine, N4-methylcytidine, 5- methylcytidine, 5-hydroxymethylcytidine, 5-hydroxycytidine, lysidine, N4-acetyl-2’-O- methylcytidine, 5-formyl-2’-O-methylcytidine, 5,2’-O-dimethylcytidine, 2-O-methylcytidine, N4,2’-O-dimethylcytidine, N4,N4,2’-O-trimethylcytidine, isocytidine, pseudocytidine, pseudoisocytidine, 2-thio-cytidine, 2’-methyl-2’-deoxycytidine, 2’-amino-2’-deoxycytidine, 2’- fluoro-2’-deoxycytidine, 5-iodocytidine, 5-bromocytidine, 2’-azido-2’-deoxycytidine, 2’-amino- 2’-deoxycytidine, 2’-fluor-2’-deoxycytidine, 5-aza-cytidine, 3-methyl-cytidine, 1-methyl- pseudoisocytidine, pyrrolo-cytidine, pyrrolo-pseudoisocytidine, 2-thio-5-methyl-cytidine, 4- thio-pseudoisocytidine, 4-thio-l-methyl-pseudoisocytidine, 4-thio-l-methyl-1 -deaza- pseudoisocytidine, 1-methyl-l-deaza-pseudoisocytidine, 2-methoxy-cytidine, 2-methoxy-5- methyl-cytidine, 4-methoxy-pseudoisocytidine, 4-methoxy-l-methyl-pseudoisocytidine, zebularine,5-aza-zebularine, 5-methyl-zebularine, 5-aza-2-thio-zebularine, 2-thio-zebularine; 1 -methylguanosine, N2,7-dimethylguanosine, N2-methylguanosine, 2’-O-ribosylphosphate guanosine, 7-methylguanosine, hydroxywybutosine, 7-aminomethyl-7-deazaguanosine, 7- cyano-7-deazaguanosine, N2,N2-dimethylguanosine, N2,7,2’-O-trimethylguanosine, N2,2’-O- dimethylguanosine, 1 ,2’-O-dimethylguanosine, 2’-O-methylguanosine, N2,N2,2’-O- trimethylguanosine, N2,N2J-trimethylguanosine, Isoguanosine, 4-demethylwyosine, epoxyqueuosine, undermodified hydroxywybutosine, methylated undermodified hydroxywybutosine, isowyosine, peroxywybutosine, galactosyl-queuosine, mannosyl- queuosine, queuosine, archaeosine, wybutosine, methylwyosine, wyosine, 7- aminocarboxypropyldemethylwyosine, 7-aminocarboxypropylwyosine, 7- aminocarboxypropylwyosinemethylester, 7-deaza-guanosine, 7-deaza-8-aza-guanosine, 6- thio-guanosine, 6-thio-7-deaza-guanosine, 6-thio-7-deaza-8-aza-guanosine, 7-methyl- guanosine, 6-thio-7-methyl-guanosine, 7-methylinosine, 6-methoxy-guanosine, 1- methylguanosine, 8-oxo-guanosine, 7-methyl-8-oxo-guanosine, 1 -methyl-6-thio-guanosine, N2-methyl-6-thio-guanosine, N2,N2-dimethyl-6-thio-guanosine, N1 -methylguanosine, 2’- amino-3’-deoxyguanosine, 2’-azido-2’-deoxyguanosine, 2’-fluoro-2’-deoxyguanosine, 2- thiouridine, 3-(3-amino-3-carboxypropyl)uridine, 3-methyluridine, 4-thiouridine, 5-methyl-2- thiouridine, 5-methylaminomethyluridine, 5-carboxymethyluridine, 5- carboxymethylaminomethyluridine, 5-hydroxyuridine, 5-methyluridine, 5-taurinomethyluridine, 5-carbamoylmethyluridine, 5-(carboxyhydroxymethyl)uridine methyl ester, dihydrouridine, 5- methyldihydrouridine, 5-methylaminomethyl-2-thiouridine, 5-(carboxyhydroxymethyl)uridine, 5-(carboxyhydroxymethyl)-2'-O-methyluridine methyl ester, 5-
(isopentenylaminomethyl)uridine, 5-(isopentenylaminomethyl)-2-thiouridine, 3,2’-O- dimethyluridine, 5-carboxymethylaminomethyl-2’-O-methyluridine, 5- carbamoylhydroxymethyluridine, 5-carbamoylmethyl-2’-O-methyluridine, 5-carbamoylmethyl-
2-th iouridine, 5-methoxycarbonylmethyl-2’-O-methyluridine, 5-(isopentenylaminomethyl)-2’-O- methyluridine, 5,2’-O-dimethyluridine, 2’-O-methyluridine, 2’-O-methyl-2-thiorudine, 2-thio-2’- O-methyluridine, uridine 5-oxyacetic acid, 5-methoxycarbonylmethyluridine, uridine 5- oxyacetic acid methyl ester, 5-methoxyuridine, 5-aminomethyl-2-thiouridine, 5- carboxymethylaminomethyl-2-thiouridine, 5-methylaminomethyl-2-selenouridine, 5- methoxycarbonylmethyl-2-thiouridine, 5-taurinomethyl-2-thiouridine, pseudouridine, 1 -methyl-
3-(3-amino-3-carboxypropyl)pseudouridine, 1 -methylpseudouridine, 3-methylpseudouridine, 2’-O-methylpseudouridine, 5-formyluridine, 5-aminomethyl-2-geranyluridine, 5- taurinomethyluridine, 5-iodouridine, 5-bromouridine, 2’-methyl-2’-deoxyuridine, 2’-amino-2’- deoxyuridine, 2’-azido-2’-deoxyuridine, 2’-fluoro-2’-deoxyuridine, inosine, 1 -methylinosine, 1 ,2’-O-dimethylinosine, 2’-O-methylinosine, 5-aza-uridine, 2-thio-5-aza-uridine, 4-thio- pseudouridine, 2-thio-pseudouridine, 5-carboxymethyl-uridine, 1 -carboxymethylpseudouridine, 5-propynyl-uridine, 1-propynyl-pseudouridine, 1-taurinomethyl-pseudouridine, 5-taurinomethyl-2-thio-uridine, 1-taurinomethyl-4-thio-uridine, 5-methyl-uridine, 1-methyl- pseudouridine, 4-thio-l-methyl-pseudouridine, 2-thio-l-methyl-pseudouridine, 1-methyl-l- deaza-pseudouridine, 2-thio-1-methyl-l-deaza-pseudouridine, dihydropseudouridine, 2-thio- dihydrouridine, 2-thio-dihydropseudouridine, 2-methoxyuridine, 2-methoxy-4-thio-uridine, 4- methoxy-pseudouridine, 4-methoxy-2-thio-pseudouridine, 1 ,2'-O-dimethyladenosine, 1 ,2'-O- dimethylguanosine, 1 ,2'-O-dimethylinosine, 2,8-dimethyladenosine, 2- methylthiomethylenethio-N6-isopentenyl-adenosine, 2-geranylthiouridine, 2-lysidine, 2- methylthio cyclic N6-threonylcarbamoyladenosine, 2-methylthio-N6-(cis-hydroxyisopentenyl) adenosine, 2-methylthio-N6-hydroxynorvalylcarbamoyladenosine, 2-methylthio-N6- threonylcarbamoyladenosine, 2-selenouridine, 2-thio-2'-O-methyluridine, 2'-O- methyladenosine, 2'-O-methylcytidine, 2'-O-methylguanosine, 2'-O-methylinosine, 2’-O- methylpseudouridine, 2'-O-methyluridine, 2'-O-methyluridine 5-oxyacetic acid methyl ester, 2'- O-ribosyladenosinephosphate, 2'-O-ribosylguanosinephosphate, 3,2'-O-dimethyluridine, 3-(3- amino-3-carboxypropyl)-5,6-dihydrouridine, 3-(3-amino-3-carboxypropyl)pseudouridine, 5,2’- O-dimethylcytidine, 5,2'-O-dimethyluridine, 5-(carboxyhydroxymethyl)-2'-O-methyluridine methyl ester, 55-(isopentenylaminomethyl)-2'-O-methyluridine, 5-aminomethyl-2- geranylthiouridine, 5-aminomethyl-2-selenouridine, 5-aminomethyluridine, 5- carbamoylmethyl-2'-O-methyluridine, 5-carboxyhydroxymethyluridine, 5-carboxymethyl-2- thiouridine, 5-carboxymethylaminomethyl-2-geranylthiouridine, 5-carboxymethylaminomethyl- 2-selenouridine, 5-carboxymethylaminomethyl-2'-O-methyluridine, 5-cyanomethyluridine, 5- formyl-2'-O-methylcytidine, 5-methoxycarbonylmethyl-2'-O-methyluridine, 5- methylaminomethyl-2-geranylthiouridine, 7-aminocarboxypropyl-demethylwyosine, 7- methylguanosine, 8-methyladenosine, N2,2'-O-dimethylguanosine, N2,7,2'-O- trimethylguanosine, N2,7-dimethylguanosine, N2,N2,2'-O-trimethylguanosine, N2,N2,7- trimethylguanosine, N2,N2,7-trimethylguanosine , N4,2'-O-dimethylcytidine, N4,N4,2'-O- trimethylcytidine, N4,N4-dimethylcytidine, N4-acetyl-2'-O-methylcytidine, N6,2'-O- dimethyladenosine, N6,N6,2'-O-trimethyladenosine, N6-formyladenosine, N6- hydroxymethyladenosine, agmatidine, 2-methylthio cyclic N6-threonylcarbamoyladenosine, glutamyl-queuosine, guanosine added to any nucleotide, guanylylated 5' end , hydroxy-N6- threonylcarbamoyladenosine; most preferably pseudo-uridine, N1-methyl-pseudo-uridine, 2 - fluoro-2'-deoxycytidine, 5-iodocytidine, 5-methylcytidine, 2-thiouridine, 5-iodouridine and/or 5- methyl-uridine.
Furthermore, the term “modified nucleotide” comprises nucleotides containing isotopes such as deuterium. The term "isotope" refers to an element having the same number of protons but a different number of neutrons resulting in different mass numbers. Thus, isotopes of hydrogen for example are not limited to deuterium but include also tritium. Furthermore, the polyribonucleotide can also contain isotopes of other elements including for example carbon, oxygen, nitrogen and phosphor. It is also possible that modified nucleotides are deuterated or contain another isotope of hydrogen or of oxygen, carbon, nitrogen or phosphorus. Among the U, C, A and G nucleotides either none, one, two, three or all of them can be modified. Hence, in some embodiments, at least one nucleotide of one nucleotide type, e.g. at least one U nucleotide, can be a modified nucleotide. In some embodiments, at least one nucleotide of in total two nucleotide types, e.g., at least one U nucleotide and at least one C nucleotide, can be a modified nucleotide. In some embodiments, at least one nucleotide of in total three nucleotide types, e.g., at least one G nucleotide, at least one U nucleotide and at least one C nucleotide, can be a modified nucleotide. In some embodiments, at least one nucleotide of all four nucleotide types can be a modified nucleotide. In all these embodiments one or more nucleotides per nucleotide type can be modified, the percentage of said modified nucleotides of per nucleotide type being 0%, 2.5%, 5%, 7.5%, 10%, 15%, 20%, 25%, 30%, 35%, 40%, 45%, 50%, 55%, 60%, 65%, 70%, 75%, 80%, 85%, 90% or 100%.
In some embodiments, the total percentage of modified nucleotides comprised in the mRNA molecules is 0%, 2.5%, 5 %, 7.5%, 10%, 15%, 20%, 25%, 30%, 35%, 40%, 45%, 50%, 55%, 60%, 65%, 70%, 75%, 80%, 85%, 90% or 100%.
In a preferred embodiment the mRNA is an mRNA which contains a combination of modified and unmodified nucleotides. Preferably, it is an mRNA containing a combination of modified and unmodified nucleotides as described in WO2011/012316. The mRNA described therein is reported to show an increased stability and diminished immunogenicity. In a preferred embodiment, in such a modified mRNA 5 to 50% of the cytidine nucleotides and 5 to 50% of the uridine nucleotides are modified. In another preferred embodiment, 5 to 50% of the uridine nucleotides are replaced by N1-methyl-pseudo-uridine. The adenosine- and guanosinecontaining nucleotides can be unmodified. The adenosine and guanosine nucleotides can be unmodified or partially modified, and they are preferably present in unmodified form.
In certain embodiments of any of the foregoing, the percentage of analogues of a given nucleotide refers to input percentage (e.g., the percentage of analogues in a starting reaction, such as a starting in vitro transcription reaction). In certain embodiments of any of the foregoing, the percentage of analogues of a given nucleotide refers to output (e.g., the percentage in a synthesized or transcribed compound). Both options are equally contemplated. The RNA, preferably the mRNA, molecules may be produced recombinantly in in vivo systems by methods known to a person skilled in the art.
Alternatively, the modified RNA, preferably the mRNA molecules may be produced in an in vitro system using, for example, an in vitro transcription system which is known to the person skilled in the art. An in vitro transcription system capable of producing RNA, preferably mRNA requires an input mixture of modified and unmodified nucleoside triphosphates to produce modified RNA. In certain embodiments, 5 to 50% of the cytidines are analogues of cytidine in such an input mixture and 5 to 50% of the uridines are analogues of uridine in such an input mixture. In certain embodiments, 5 to 40% of the cytidines are analogues of cytidine in such an input mixture and 5 to 40% of the uridines are analogues of uridine in such an input mixture. In certain embodiments, 5 to 30% of the cytidines are analogues of cytidine in such a mixture and 5 to 30% of the uridines are analogues of uridine in such an input mixture. In certain embodiments, 5 to 30% of the cytidines are analogues of cytidine in such mixture and 10 to 30% of the uridines are analogues of uridine in such mixture. In certain embodiments, 5 to 20% of the cytidines are analogues of cytidine in such an input mixture and 5 to 20% of the uridines are analogues of uridine in such an input mixture. In certain embodiments, 5 to 10% of the cytidines are analogues of cytidine in such an input mixture and 5 to 10% of the uridines are analogues of uridine in such an input mixture. In certain embodiments, 25% of the cytidines are analogues of cytidine in such an input mixture and 25% of the uridines are analogues of uridine in such an input mixture. In certain embodiments, the input mixture does not comprise analogues of adenosine and/or guanosine. In other embodiments, optionally, the input mixture comprises one or more analogues of adenosine and/or guanosine (or none of either or both).
In certain embodiments, the percentage of cytidines in an input mixture that are analogues of cytidine is not the same as the percentage of uridines in an input mixture that are analogues of uridine. In certain embodiments, the percentage of analogues of cytidine in an input mixture is lower than the percentage of analogues of uridine in an input mixture. As noted above, this may be in the presence or the absence of analogues of adenosine and guanosine in the input mixture but, in certain embodiments, is in the absence of analogues of adenosine and analogues of guanosine in the input mixture.
In certain embodiments, an input mixture of nucleotides for an in vitro transcription system that produces a RNA, preferably mRNA of the present invention comprises analogues of cytidine and analogues of uridine, and 5 to 20% of the cytidines of the input mixture are analogues of cytidine and 25 to 45% of the uridines of the input mixture are analogues of uridine. In other words, the input mixture comprises modified and unmodified cytidines and modified and unmodified uridines, and 5 to 20% of the cytidines of the input mixture comprise analogues of cytidine while 25 to 45% of the uridines of the input mixture comprise analogues of uridine. In other embodiments, the input mixture comprises 5 to 10% analogues of cytidine and 30 to 40% analogues of uridine, such as 7-9% analogues of cytidine, such as 7, 7.5 or 8% and, such as 32-38% analogues of uridine, such as 33, 34, 35, 36%.
In certain embodiments, any of the analogues of uridine and analogues of cytidine described herein may be used, optionally excluding pseudouridine. In certain embodiments, the analogue of cytidine comprises or consists of (e.g., it is the single C analogue type used) 5-iodocytidine and the analogue of uridine comprises or consists of (e.g., it is the single U analogue type used) 5-iodouridine.
Exemplary analogues are described above. It should be understood that for modified polyribonucleotides encoding the desired polypeptide, the analogues and level of modification is, unless indicated otherwise, considered across the entire polyribonucleotide encoding the desired polypeptide, including 5’ and 3’ untranslated regions (e.g., the level of modification is based on input ratios of analogues in an in vitro transcription reaction such that analogues may be incorporated at positions that are transcribed).
Furthermore, the modified RNA, preferably mRNA molecules may be chemically synthesized, e.g., by conventional chemical synthesis on an automated nucleotide sequence synthesizer using a solid-phase support and standard techniques or by chemical synthesis of the respective DNA sequences and subsequent in vitro or in vivo transcription of the same.
In another preferred embodiment, the mRNA may be combined with target binding sites, targeting sequences and/or with micro-RNA binding sites, in order to allow activity of the desired mRNA only in the relevant cells. In a further preferred embodiment, the RNA can be combined with micro-RNAs or shRNAs in the untranslated regions.
In general, therapeutic effects can be achieved by the interaction of the ribonucleic acid with cellular molecules and organelles. Such interaction alone may for example activate the innate immune system, as is the case for certain CpG oligonucleotides and sequences designed to specifically interact with toll-like and other extra- or intracellular receptors. Furthermore, the uptake or introduction of nucleic acids (preferably ribonucleic acids, more preferably mRNAs) in cells can be intended to lead to the expression of nucleotide sequences such as genes comprised in the nucleic acid (preferably ribonucleic acids, more preferably the mRNA), can be intended for the downregulation, silencing or knockdown of endogenous gene expression as a consequence of the intracellular presence of an introduced exogenous nucleic acid, or can be intended for the modification of endogenous nucleic acid sequences such as repair, excision, insertion or exchange of selected bases or of whole stretches of endogenous nucleic acid sequences, or can be intended for interference with virtually any cellular process as a consequence of the intracellular presence and interaction of an introduced exogenous ribonucleic acid (preferably an mRNA). Overexpression of introduced exogenous nucleic acids (preferably ribonucleic acids, more preferably mRNAs) may be intended to compensate or complement endogenous gene expression, in particular in cases where an endogenous gene is defective or silent, leading to no, insufficient or a defective or a dysfunctional product of gene expression such as is the case with many metabolic and hereditary diseases like cystic fibrosis, hemophilia or muscular dystrophy to name a few. Overexpression of introduced exogenous nucleic acids (preferably ribonucleic acids, more preferably mRNAs) may also be intended to have the product of the expression interact or interfere with any endogenous cellular process such as the regulation of gene expression, signal transduction and other cellular processes. The overexpression of introduced exogenous nucleic acids (preferably ribonucleic acids, more preferably mRNAs) may also be intended to give rise to an immune response in context of the organism in which a transfected or transduced cell resides or is made to reside. Examples are the genetic modification of antigen-presenting cells such as dendritic cells in order to have them present an antigen for vaccination purposes. Other examples are the overexpression of cytokines in tumors in order to elicit a tumor-specific immune response. Furthermore, the overexpression of introduced exogenous ribonucleic acids (preferably mRNAs) may also be intended to generate in vivo or ex vivo transiently genetically modified cells for cellular therapies such as modified T-cells, NK cells and other lymphocytes or precursor or stem or other cells for regenerative medicine.
Downregulation, silencing or knockdown of endogenous gene expression for therapeutic purposes can for example be achieved by RNA interference (RNAi), with ribozymes, antisense oligonucleotides, tRNAs, long double-stranded RNA where such down regulation can be sequence-specific or unspecific and can also lead to cell death as is the case when long double-stranded RNAs are introduced into cells. Downregulation, silencing or knockdown of endogenous or pre-existing gene expression can be useful in the treatment of acquired, hereditary or spontaneously incurring diseases including viral infections and cancer. It can also be envisaged that the introduction of nucleic acids into cells can be practiced as a preventive measure in order to prevent, for example, viral infection or neoplasias. Downregulation, silencing or knockdown of endogenous gene expression can be exerted on the transcriptional level and on the translational level. Multiple mechanisms are known to the one skilled in the art and include for example epigenetic modifications, changes in chromatin structure, selective binding of transcription factors by the introduced nucleic acid, hybridization of the introduced nucleic acid to complementary sequences in genomic DNA, mRNA or other RNA species by base pairing including unconventional base pairing mechanisms such as triple helix formation. Similarly, gene repair, base or sequence changes can be achieved at the genomic level and at the mRNA level including exon skipping. Base or sequence changes can for example be achieved by RNA-guided site-specific DNA cleavage, by cut and paste mechanisms exploiting trans-splicing, trans-splicing ribozymes, chimeraplasts, splicosome-mediated RNA trans- splicing, or by exploiting group II or retargeted introns, or by exploiting insertional mutagenesis mediated by viruses or exploiting targeted genomic insertion using prokaryotic, eukaryotic or viral integrase systems. As nucleic acids are the carriers of the building plans of living systems and as they participate in many cellular processes in a direct and indirect manner, in theory any cellular process can be influenced by the introduction of nucleic acids into cells from outside. Notably, this introduction can be carried out directly in vivo and ex vivo in cell or organ culture followed by transplantation of thus modified organs or cells into a recipient. The particles for use in the context of the present invention with nucleic acids as therapeutically active agent may be useful for all purposes described above.
As mentioned above, the RNA, preferably the mRNA, may contain a ribonucleotide sequence which encodes a protein or fragment thereof whose function in the cell or in the vicinity of the cell is needed or beneficial, e.g. a protein the lack or defective form of which is a trigger for a disease or an illness, the provision of which can moderate or prevent a disease or an illness, or a protein which can promote a process which is beneficial for the body, in a cell or its vicinity.
Indeed, in recent years, RNA (in particular, mRNA) has become increasingly relevant as a drug entity. As opposed to DNA-based gene therapeutics, mRNA does not need to be transported into the nucleus but is directly translated into protein in the cytoplasm (J Control Release, 2011 , 150:238-247, and Eur J Pharm Biopharm, 2009, 71 :484-489).
Moreover, numerous genetic disorders, caused by the mutation of a single gene are known and candidates for RNA, preferably mRNA, therapeutic approaches. Disorders caused by single-gene mutations, like cystic fibrosis, hemophilia and many others, can be dominant or recessive with respect to the likelihood that a certain trait will appear in the offspring. While a dominant allele manifests a phenotype in individuals who have only one copy of the allele, for a recessive allele the individual must have two copies, one from each parent to become manifest. In contrast, polygenic disorders are caused by two or more genes and the manifestation of the respective disease is often fluent and associated to environmental factors. Examples for polygenic disorders are hypertension, elevated cholesterol level, cancer, neurodegenerative disorders, mental illness and others. Also in these cases therapeutic RNA, preferably the mRNA, representing one or more of these genes may be beneficial to those subjects. Furthermore, a genetic disorder must not have been passed down from the parents' genes, but can also be caused by new mutations. Also in these cases therapeutic RNA, preferably the mRNA, representing the correct gene sequence may be beneficial to the subjects. An online catalog with Human Genes and Genetic Disorders together with their respective genes and a description of their phenotypes are available at the ONIM (Online Mendelian Inheritance in Man) webpage (http://onim.org); sequences of each are available from the Uniprot database (http://www.uniprot.org). As non-limiting examples, the following Table A lists some congenital diseases and disorders, and the corresponding gene(s). Due to the high degree of interaction of cellular signaling pathways, the mutation of a certain gene causes a multiply of pathogenic symptoms, of which only a characteristic one is listed in Table A. Pharmaceutical compositions as referred to herein thus encompass compositions suitable for the treatment and prevention of a disease selected from those listed in Table A. Likewise, a nanoparticle formulation as referred to herein, such as a lipid nanoparticle formulation or lipidoid nanoparticle formulation comprising RNA, preferably mRNA, may be suitable for the treatment and prevention of a disease selected from those listed in Table A.
In some embodiments of the present invention, the therapeutic protein which is encoded by the RNA, preferably the mRNA, which may be present in the suspension formulation and the aerosol of the present invention is chosen from the cellular proteins listed in Table A. Thus, the RNA, preferably the mRNA, molecule may encode a therapeutic cellular protein, wherein the encoded therapeutic protein is one listed in Table A or a homolog thereof.
In another embodiment of the present invention, the therapeutic protein which is encoded by the RNA, preferably the mRNA, is chosen from the secreted proteins listed in Table A. Thus, the RNA, preferably the mRNA, may encode a therapeutic fusion protein, wherein the encoded therapeutic protein or a homolog thereof is one listed in Table A and the second protein is a signal peptide that allows the secretion of the therapeutic protein. A signal peptide is a short, typically 5-30 amino acids long sequence present at the N-terminus of said therapeutic protein and that leads the fusion protein towards the cell’s secretory pathway via certain organelles (i.e. the endoplasmic reticulum, the Golgi-apparatus or the endosomes). Thus, such fusion protein is secreted from the cell or from a cellular organelle or inserted into a cellular membrane (e.g. multi-spanning trans- membrane proteins) at a cellular compartment or at the cell’s surface.
Thus, in embodiments of the present invention an RNA, preferably mRNA, may encode one or more, but is not limited to, the following proteins of the genes that cause, predispose or protect from diseases. Non-limiting examples of such diseases or disorders that may be treated (or prevented) include those wherein said polypeptide, protein or peptide is selected from the group consisting of the ones as outlined in the following Table A. In some embodiments, the encoding sequence of an RNA, preferably mRNA, may be transcribed and translated into a partial or full-length protein comprising cellular activity at a level equal to or greater than that of the native protein. In some embodiments, an RNA, preferably the mRNA, encodes a therapeutically or pharmaceutically active polypeptide, protein or peptide having a therapeutic or preventive effect, wherein said polypeptide, protein or peptide is selected from the group consisting of the ones as outlined in the following Table A. The RNA, preferably the mRNA, more specifically the encoding sequence thereof, may be used to express a partial or full-length protein with cellular activity at a level equal to or less than that of the native protein. This may allow the treatment of diseases for which the administration of an RNA molecule can be indicated.
Table A: Non-limiting examples of human genes and genetic diseases or disorders















The above Table A shows examples of genes in which a defect leads to a disease which can be treated with RNA, preferably the mRNA, which may be present pharmaceutical compositions or nanoparticle formulations referred to herein wherein RNA, preferably the mRNA, comprises a ribonucleotide sequence which encodes an intact version of the protein or a functional fragment thereof of the above disclosed defective gene. In particularly preferred embodiments, hereditary diseases can be addressed, which for example affect the lungs, such as SPB (surfactant protein B) deficiency, ABCA3 deficiency, cystic fibrosis and o1 -antitrypsin deficiency, or which affect plasma proteins (e.g. congenital hemochromatosis (hepcidin deficiency), thrombotic thrombocytopenic purpura (TPP, ADAMTS 13 deficiency) and cause clotting defects (e.g. hemophilia a and b) and complement defects (e.g. protein C deficiency), immune defects such as for example SCID (caused my mutations in different genes such as: RAG1 , RAG2, JAK3, IL7R, CD45, CD35, CD3E) or by deficiencies due to lack of adenosine deaminase for example (ADA-SCID), septic granulomatosis (e.g. caused by mutations of the gp-91-phox gene, the p47-phox gene, the p67-phox gene or the p33-phox gene) and storage diseases like Gaucher’s disease, Fabry’s disease, Krabbe’s disease, MPS I, MPS II (Hunter syndrome), MPS VI, Glycogen storage disease type II or mucopolysaccharidoses.
Other disorders for which RNA, preferably mRNA can be useful include disorders such as SMN1 -related spinal muscular atrophy (SMA); amyotrophic lateral sclerosis (ALS); GALT- related galactosemia; Cystic Fibrosis (CF); SLC3A1 -related disorders including cystinuria; COL4A5-related disorders including Alport syndrome; galactocerebrosidase deficiencies; X- linked adrenoleukodystrophy and adrenomyeloneuropathy; Friedreich's ataxia; Pelizaeus- Merzbacher disease; TSC1 and TSC2-related tuberous sclerosis; Sanfilippo B syndrome (MPS IIIB); CTNS-related cystinosis; the FMR1 -related disorders which include Fragile X syndrome, Fragile X-Associated Tremor/Ataxia Syndrome and Fragile X Premature Ovarian Failure Syndrome; Prader-Willi syndrome; hereditary hemorrhagic telangiectasia (AT); Niemann-Pick disease Type C1 ; the neuronal ceroid lipofuscinoses-related diseases including Juvenile Neuronal Ceroid Lipofuscinosis (JNCL), Juvenile Batten disease, Santavuori-Haltia disease, Jansky-Bielschowsky disease, and PTT-1 and TPP1 deficiencies; EIF2B1 , EIF2B2, EIF2B3, EIF2B4 and EIF2B5-related childhood ataxia with central nervous system hypomyelination/vanishing white matter; CACNA1A and CACNB4 -related Episodic Ataxia Type 2; the MECP2-related disorders including Classic Rett Syndrome, MECP2-related Severe Neonatal Encephalopathy and PPM-X Syndrome; CDKL5-related Atypical Rett Syndrome; Kennedy's disease (SBMA); Notch-3 related cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL); SCN1A and SCN1 B-related seizure disorders; the Polymerase G-related disorders which include Alpers- Huttenlocher syndrome, POLG-related sensory ataxic neuropathy, dysarthria, and ophthalmoparesis, and autosomal dominant and recessive progressive external ophthalmoplegia with mitochondrial DNA deletions; X-Linked adrenal hypoplasia; X-linked agammaglobulinemia; Fabry disease; and Wilson's disease.
In all these diseases, a protein, e.g. an enzyme, is defective, which can be treated with the RNA, preferably the mRNA, encoding any of the above proteins, which makes the protein encoded by the defective gene or a functional fragment thereof available. Transcript replacement therapies/protein replacement therapies do not affect the underlying genetic defect, but increase the concentration of the protein in which the subject is deficient. As an example, in Pompe's disease, the transcript replacement therapy/enzyme replacement therapy replaces the deficient lysosomal enzyme acid alpha-glucosidase (GAA).
Thus, non-limiting examples of proteins which can be encoded by the mRNA are erythropoietin (EPO), growth hormone (somatotropin, hGH), cystic fibrosis transmembrane conductance regulator (CFTR), growth factors such as GM-SCF, G-CSF, MPS, protein C, hepcidin, ABCA3 and surfactant protein B. Further examples of diseases which can be treated with the RNA according to the invention are hemophilia A/B, Fabry’s disease, CGD, ADAMTS13, Hurler’s disease, X chromosome-mediated A-y-globulinemia, adenosine deaminase-related immunodeficiency and respiratory distress syndrome in the newborn, which is linked with SP- B. Particularly preferably, the RNA, preferably the mRNA, according to the invention contains the coding sequence for surfactant protein B (SP-B) or for erythropoietin. Further examples of proteins which can be encoded by the RNA, preferably the mRNA, of the present invention according to the invention are growth factors such as human growth hormone hGH, BMP-2 or angiogenesis factors. Although the above embodiments are described in the context of the RNA, preferably an mRNA molecule, that may be present in the pharmaceutical compositions or nanoparticle formulations referred to herein, as mentioned above, is not limited to the use of an RNA, preferably an mRNA but may employ other therapeutic agents, e.g., nucleic acid molecules, such as DNA molecules.
Said DNA molecule may encode the above RNA, preferably the above mRNA and, accordingly, harbors the genetic information for the correspondingly transcribed RNA molecule.
Hence, as regards preferred embodiments the same applies, mutatis mutandis, to the DNA molecule of the present invention as has been set forth above and below in the context of the RNA molecule, preferably the mRNA molecule, that may be in the pharmaceutical compositions or nanoparticle formulations referred to herein.
Alternatively, the RNA, preferably the mRNA, may contain a ribonucleotide sequence which encodes a full-length antibody or a smaller antibody (e.g., both heavy and light chains) which can be used in therapeutic settings to, e.g., confer immunity to a subject. Corresponding antibodies and their therapeutic application(s) are known in the art. The antibody may be encoded by a single mRNA strand or by more than one mRNA strand.
In another embodiment, the RNA, preferably the mRNA may encode a functional monoclonal or polyclonal antibody, which may be useful for targeting and/or inactivating a biological target (e.g., a stimulatory cytokine such as tumor necrosis factor). Similarly, the RNA, preferably the mRNA sequence may encode, for example, functional anti-nephrotic factor antibodies useful for the treatment of membranoproliferative glomerulonephritis type II or acute hemolytic uremic syndrome, or alternatively may encode anti-vascular endothelial growth factor (VEGF) antibodies useful for the treatment of VEGF-mediated diseases, such as cancer.
In another embodiment, the RNA, preferably the mRNA may encode a functional monoclonal or polyclonal antibody, which may be useful for neutralizing or otherwise inhibiting a virus or virus replication.
Alternatively, the RNA, preferably the mRNA, may contain a ribonucleotide sequence which encodes an antigen which preferably can be used in preventive or therapeutic settings.
In another embodiment, the mRNA may encode a protein or proteins that can induce an immune modulation, such as cytokines, including chemokines, interferons (such as interferon lambda), interleukins, lymphokines, and tumor necrosis factors.
In another embodiment, the RNA, preferably the mRNA, may contain a ribonucleotide sequence which encodes a polypeptide or a protein which can be used in genome editing technologies. Genome editing is a type of genetic engineering in which DNA is inserted, deleted, or replaced in the genome of an organism using nucleases. These nucleases create site-specific breaks at desired locations in the genome. The induced breaks are repaired by non-homologous end-joining or homologous recombination, resulting in targeted mutations in the genome, thereby “editing” the genome. The breaks may either be single-strand breaks or double-strand breaks (DSBs) while double-strand breaks (DSBs) are preferred. Numerous genome editing systems utilizing different polypeptides or proteins are known in the art, i.e., e.g., the CRISPR-Cas system, meganucleases, zinc finger nucleases (ZFNs) and transcription activator-like effector-based nucleases (TALEN). Methods for genome engineering are reviewed in Trends in Biotechnology, 2013, 31 (7), 397-405.
Thus, in a preferred embodiment, the RNA, preferably the mRNA, may contain a ribonucleotide sequence which encodes a polypeptide or protein of the Cas (CRISPR associated protein) protein family, preferably Cas9 (CRISPR associated protein 9). Proteins of the Cas protein family, preferably Cas9, may be used in CRISPR/Cas9 based methods and/or CRISPR/Cas9 genome editing technologies. CRISPR-Cas systems for genome editing, regulation and targeting are reviewed in Nat. Biotechnol., 2014, 32(4):347-355.
In another preferred embodiment, the RNA, preferably the mRNA, may contain a ribonucleotide sequence which encodes a meganuclease. Meganucleases are endodeoxyribonucleases which, in contrast to “conventional” endodeoxyribonucleases, recognize a large recognition site (e.g., a double-stranded DNA sequence of 12 to 40 base pairs). As a result, the respective site occurs only a few times, preferably only once, in any given genome. Meganucleases are therefore considered to be the most specific naturally occurring restriction enzymes and, accordingly, are suitable tools in genome editing technologies.
In another preferred embodiment, the RNA, preferably the mRNA, contains a ribonucleotide sequence which encodes a zinc finger nuclease (ZFN). ZFNs are artificial restriction enzymes generated by fusing a zinc finger DNA-binding domain to a DNA-cleavage domain. Zinc finger domains can be engineered to target specific desired DNA sequences and this enables zinc- finger nucleases to target unique sequences within complex genomes. By taking advantage of the endogenous DNA repair machinery, ZFNs can be used to precisely alter the genome of higher organisms and are, therefore, suitable tools in genome editing technologies.
In another preferred embodiment, the RNA, preferably the mRNA, may contain a ribonucleotide sequence which encodes a transcription activator-like effector nuclease (TALEN). TALENs are restriction enzymes that can be engineered to cut specific sequences of DNA. TALENs are fusion proteins wherein a TAL effector DNA-binding domain is fused to a DNA cleavage domain of a nuclease. Transcription activator-like effectors (TALEs) can be engineered to bind practically any desired DNA sequence. Thus, when combined with a nuclease, DNA can be cut at specific desired locations.
Although the above embodiments are described in the context of the RNA, preferably an mRNA molecule, the present invention, as mentioned above, is not only limited to the use of an RNA, preferably an mRNA, but may employ any nucleic acid molecule, such as a DNA molecule.
Said DNA molecule may encode the above RNA, preferably the above mRNA and, accordingly, harbors the genetic information for the correspondingly transcribed RNA molecule.
Hence, as regards preferred embodiments the same applies, mutatis mutandis, to the DNA molecule as has been set forth above and below in the context of the RNA molecule, preferably the mRNA molecule, that may be present in the nanoparticles used in the present invention.
Alternatively to the above, the RNA contains a ribonucleotide sequence which is not to be expressed as a protein or a polypeptide. Thus, the term RNA should not only be understood to mean any polynucleotide molecule which, if introduced into a cell, is translatable to a polypeptide/protein or fragment thereof. Rather, it is also contemplated that the RNA contains a ribonucleotide sequence which is not translated into a protein. In this context, it is envisaged that the RNA contains a ribonucleotide sequence which preferably provides the genetic information for an antisense RNA, an siRNA or a miRNA sequence or another desired noncoding ribonucleotide sequence.
Thus, the RNA may also be an antisense RNA, an siRNA, or a miRNA sequence. Antisense RNA, siRNA or miRNA sequences can be used to silence the effect of a certain RNA molecule at some stage. This may, in particular, be desirable and useful in certain medical settings and in the treatment of a certain disease and, in particular, in RNA-based therapies as described herein above and below. Silencing the effect of an RNA molecule can be achieved by making use of an RNAi (RNA interference) mechanism by using the nucleic acid strand which is complementary to a certain RNA sequence. The term "RNA interference" or "inhibiting RNA" (RNAi/iRNA) describes the use of double-stranded RNA to target specific mRNAs for degradation, thereby silencing their translation. Preferred inhibiting RNA molecules may be selected from the group consisting of double-stranded RNA (dsRNA), siRNA, shRNA and stRNA. dsRNA matching a gene sequence may be synthesized in vitro and introduced into a cell. The dsRNA may also be introduced into a cell in form of a vector expressing a target gene sequence in sense and antisense orientation, for example in form of a hairpin mRNA. The sense and antisense sequences may also be expressed from separate vectors, whereby the individual antisense and sense molecules form double-stranded RNA upon their expression. It is known in the art that in some occasions the expression of a sequence in sense orientation or even of a promoter sequence suffices to give rise to dsRNA and subsequently to siRNA due to internal amplification mechanisms in a cell. Accordingly, all means and methods which result in a decrease in activity of the polypeptide or protein encoded by the coding region are to be used in accordance with the present invention. For example sense constructs, antisense constructs, hairpin constructs, sense and antisense molecules and combinations thereof can be used to generate/introduce these siRNAs. The dsRNA feeds into a natural process including the highly conserved nuclease dicer which cleaves dsRNA precursor molecules into short interfering RNAs (siRNAs). The generation and preparation of siRNA(s) as well as the method for inhibiting the expression of a target gene is, inter alia, described in WO 02/055693, Wei (2000) Dev. Biol. 15:239-255; La Count (2000) Biochem. Paras. 111 :67-76; Baker (2000) Curr. Biol. 10:1071 -1074; Svoboda (2000) Development 127:4147-4156 or Marie (2000) Curr. Biol. 10:289-292. These siRNAs build then the sequence specific part of an RNA-induced silencing complex (RISC), a multicomplex nuclease that destroys messenger RNAs homologous to the silencing trigger). Elbashir (2001 ) EMBO J. 20:6877-6888 showed that duplexes of 21 nucleotide RNAs may be used in cell culture to interfere with gene expression in mammalian cells.
Methods to deduce and construct siRNAs are known in the art and are described in Elbashir (2002) Methods 26:199-213, at the internet web sites of commercial vendors of siRNA, e.g., Qiagen GmbH; Dharmacon; Xeragon Inc., and Ambion, or at the web site of the research group of Tom TuschL In addition, programs are available online to deduce siRNAs from a given mRNA sequence. Uridine residues in the 2-nt 3’ overhang can be replaced by 2’deoxythymidine without loss of activity, which significantly reduces costs of RNA synthesis and may also enhance resistance of siRNA duplexes when applied to mammalian cells (Elbashir et al., nature 411.6836 (2001 ): 494-498). The siRNAs may also be synthesized enzymatically using T7 or other RNA polymerases (Donze (2002) Nucleic Acids Res 30:e46). Short RNA duplexes that mediate effective RNA interference (esiRNA) may also be produced by hydrolysis with Escherichia coli RNase III (Yang (2002) PNAS 99:9942-9947). Furthermore, expression vectors have been developed to express double stranded siRNAs connected by small hairpin RNA loops in eukaryotic cells (e.g. (Brummelkamp (2002) Science 296:550-553). All of these constructs may be developed with the help of the programs named above. In addition, commercially available sequence prediction tools incorporated in sequence analysis programs or sold separately, e.g., the siRNA Design Tool offered by www.oligoEngine.com (Seattle, WA) may be used for siRNA sequence prediction. microRNA (miRNA) resembles small interfering RNAs (siRNAs) described above. microRNA (miRNA) is a small non-coding RNA molecule (containing about 22 nucleotides) found in plants, animals and some viruses, that functions in RNA silencing and post-transcriptional regulation of gene expression. miRNAs function via base-pairing with complementary sequences within mRNA molecules. As a result, these mRNA molecules are silenced, by one or more of the following processes: (1) cleavage of the mRNA strand into two pieces, (2) destabilization of the mRNA through shortening of its poly(A) tail, and (3) less efficient translation of the mRNA into proteins by ribosomes. As mentioned, miRNAs resemble the small interfering RNAs (siRNAs) of the RNA interference (RNAi) pathway, except miRNAs derive from regions of RNA transcripts that fold back on themselves to form short hairpins, whereas siRNAs derive from longer regions of double-stranded RNA.
A DNA molecule which may be present in the pharmaceutical compositions or nanoparticle formulations referred to herein may also be one which encodes the above RNA, e.g., the above siRNA or miRNA, accordingly, harbors the genetic information for the correspondingly transcribed RNA molecule. Hence, as regards preferred embodiments the same applies, mutatis mutandis, to the DNA molecule as has been set forth above in the context of the RNA molecule, preferably the mRNA molecule, that may be present in the nanoparticles used in the present invention.
It will be understood that pharmaceutical compositions, in particular the nanoparticles in the context of the present invention can comprise a single type of nucleic acid, preferably an RNA such as mRNA, but may alternatively comprise a combination of two or more types of nucleic acids, preferably RNAs, e.g. in the form of particles comprising two or more types of nucleic acids, preferably RNAs, in single particles, or in the form of a blend of particles which differ in the type of nucleic acid, preferably RNA such as mRNA, contained therein. NANOPARTICLES, LIPIDS and LIPIDOIDS
In various aspects of the invention, reference is made to a lipid nanoparticle formulation or lipidoid nanoparticle formulation comprising a plurality of lipid nanoparticles (LNPs) or lipidoid nanoparticles (LiNPs). In order to facilitate the discussion, the lipid nanoparticles and lipidoid nanoparticles may be collectively referred to as nanoparticles herein. As will be understood by the skilled reader, a nanoparticle comprising at least one lipidoid, e.g., an ionizable lipidoid, is referred to as a lipidoid nanoparticle (LiNP), notwithstanding the fact that, in addition, the lipidoid nanoparticle may comprise one or more lipids. Likewise, a lipidoid mix as it may be comprised by the lipidoid particles as referred to herein, comprises at least one lipidoid, e.g., an ionizable lipidoid, while one or more further components of the lipidoid mix may be a lipid(s). Moreover, unless indicated to the contrary in a specific context, reference to a lipid nanoparticle formulation or lipidoid nanoparticle (LiNP) formulation comprising a plurality of lipid nanoparticles (LNPs) or lipidoid nanoparticles (LiNPs) is intended to encompass formulations comprising only lipid or lipidoid nanoparticles and combinations thereof as nanoparticles.
For example, a preferred component of a lipid nanoparticle or a lipidoid particle, or of a lipid mix or a lipidoid mix may be a structural lipid. The term “structural lipid” refers herein to a lipid component that provides structural integrity and shape to the nanoparticle. These lipids may play a foundational role in forming the primary lipid bilayer or multi-lamellar structures of LNPs or LiNPs, and may provide a scaffold of the particle.
LNPs or LiNPs often comprise, in a lipid or lipidoid mix together with the at least one selected from an ionizable lipid, ionizable lipidoid, and permanently cationic lipid, a combination of different lipid components, each serving specific roles: i) Phospholipids: These are commonly used as structural lipids due to their ability to form bilayers, mimicking natural cell membranes, ii) Cholesterol: Helps in modulating the rigidity and permeability of the lipid bilayers, iii) PEGylated lipids or poly sarcosine based lipids: May be added to increase the circulation time of LNPs or LiNPs in the bloodstream by making them more stealthy and less recognizable to the immune system.
Among these, the phospholipids and cholesterol are often considered as the main "structural lipids" as they contribute to the stability to the LNP or LiNP structure. They may be used for maintaining the integrity, size, and overall morphology of the LNPs or LiNPs. The LNPs and LiNPs referred to in the context of the invention preferably comprise at least one selected from an ionizable lipid, ionizable lipidoid, and permanently cationic lipid. These may be included to facilitate encapsulation of negatively charged molecules, e.g., a nucleic acid such as RNA, preferably mRNA.
Lipid or lipidoid nanoparticles referred to herein typically comprise a lipid mix or lipidoid mix, wherein the lipid mix or lipidoid mix comprises, as a component thereof, at least one selected from an ionizable lipid, ionizable lipidoid, and permanently cationic lipid. It will be understood that this encompasses the possibility that the nanoparticles comprise a combination of different permanently cationic lipids, a combination of different ionizable lipids, a combination of different ionizable lipidoids, or a combination of one or more permanently cationic lipids, one or more ionizable lipids, and/or one or more ionizable lipidoids. Preferred as a component are an ionizable lipid and an ionizable lipidoid, i.e., it is preferred that the nanoparticles comprise at least one selected from an ionizable lipid and an ionizable lipidoid. If the lipid nanoparticles or lipidoid nanoparticles comprise the lipid mix or lipidoid mix and a therapeutic agent, the lipid mix or lipidoid mix and the therapeutic agent are typically comprised as a mixture in the nanoparticles.
The term "permanently cationic lipid” is used in the field of lipid nanoparticles to refer to a lipid or lipidoid which contains a permanent cationic charge, e.g., in the form of a quaternary nitrogen atom.
The terms “ionizable lipid” and “ionizable lipidoid”, are used in the field of lipid nanoparticles and lipidoid nanoparticles to refer to a lipid or a lipidoid which is protonated to carry a cationic charge, or which can be protonated to carry a cationic charge. Thus, ionizable lipids and lipidoids, respectively, are also referred to as “protonatable lipids” and “protonatable lipidoids”, as “ionizable cationic lipids” and “ionizable cationic lipidoids”, or as “titratable lipids” or “titratable lipidoids”, respectively. As will be understood by the skilled reader, the reference to an “ionizable lipid” or an “ionizable lipidoid” encompasses the ionizable lipid or lipidoid in its protonated or non-protonated form. As will further be understood, the protonated or nonprotonated state of the lipid or lipidoid is generally determined by the pH value of a medium surrounding the lipid or lipidoid, e.g. by the pH value of the aqueous vehicle solution wherein the nanoparticles are suspended. Thus, the terms “ionizable lipid” and “ionizable lipidoid” also include lipids or lipidoids which are positively charged at neutral pH.
Counterions (anions) for the positive charges of positively charged permanently cationic lipids ionizable lipids or ionizable lipidoids in the context of the invention are typically provided by anionic moieties contained in the nucleic acid. If positively charged groups are present in excess compared to the anionic moieties in the nucleic acid, positive charges may be balanced by other pharmaceutically acceptable anions, such as chloride, bromide, or iodide, sulfate, nitrate, phosphate, hydrogenphosphate, dihydrogenphosphate, carbonate, or hydrogencarbonate, or by a polyanion component different from the nucleic acid, which may be present as an optional component in the nanoparticles.
Permanently cationic lipids, ionizable lipids and ionizable lipidoids are well known as components of lipid nanoparticles or lipidoid nanoparticles. In the context of the present invention, there are no particular restrictions imposed on the type of permanently cationic lipid, ionizable lipid or ionizable lipidoid contained in the nanoparticles.
Generally, an ionizable lipid or lipidoid, respectively, comprises a primary, secondary or tertiary amino group which can act as proton acceptor, and which may thus be protonated or nonprotonated. An ionizable lipidoid generally comprises a plurality of such amino groups, such as two or more, preferably three or more.
Preferably, an ionizable lipid which may be comprised by the nanoparticles is a lipid which comprises a protonatable head group which contains one or more, preferably one, primary, secondary or tertiary amino group(s) as a protonatable or protonated group, and one or more, preferably one or two, hydrophobic moieties, linked to the head group.
Examples of these preferred ionizable lipids are i) a lipid which comprises a protonatable head group which contains one or more, preferably one, primary, secondary, or tertiary amino group(s) as a protonatable or protonated group, and one hydrophobic moiety linked to the head group; ii) a lipid which comprises one secondary or tertiary amino group as a protonatable or protonated head group, and two hydrophobic moieties linked to the head group.
A hydrophobic moiety comprised in these preferred lipids preferably contains one or more of a linear chain aliphatic residue, e.g. a linear chain residue comprising 8 to 18 carbon atoms, a branched chain aliphatic residue, e.g. a branched chain residue comprising 8 to 18 carbon atoms, or an alicyclic ring structure which may be a condensed ring structure, e.g. an alicyclic ring structure comprising 10 to 18 carbon atoms. In addition, the hydrophobic moiety may comprise one or more linking groups which facilitate the linking of the moiety to the head group, or which allow two or more of the above aliphatic residues to be combined with each other. Furthermore, it may comprise one or more substituents, to the extent that the hydrophobic characteristics of the moiety are maintained. Preferably, an ionizable lipidoid which may be comprised in the nanoparticles is an oligoamine, more preferably an oligoalkylamine, which comprises at least two, preferably at least three, amino groups selected from a protonatable or protonated secondary and a tertiary amino group, each of which may carry a hydrophobic moiety attached to it. In addition to the amino groups carrying a hydrophobic residue, the lipidoid may comprise further protonatable or protonated amino groups selected from a primary, a secondary and a tertiary amino group. Preferably, the total number of the amino groups is 2 to 10, more preferably 3 to 6. Preferably, the total number of hydrophobic moieties attached to the amino groups is 2 to 6, more preferably 3 to 6. Preferably, the ratio of the number of hydrophobic moieties attached to amino groups to the total number of amino groups in the oligoalkylamine is 0.5 to 2, more preferably 0.75 to 1 .5.
A hydrophobic moiety comprised in such a preferred lipidoid preferably contains one or more of a linear chain aliphatic residue, e.g., a linear chain residue comprising 8 to 18 carbon atoms and a branched chain aliphatic residue, e.g. a branched chain residue comprising 8 to 18 carbon atoms. In addition, the hydrophobic moiety may comprise one or more linking groups which facilitate the linking of the moiety to an amino group, or which allow two or more of the above aliphatic residues to be combined with each other. Furthermore, it may comprise one or more substituents, to the extent that the hydrophobic characteristics of the moiety are maintained.
Suitable exemplary ionizable lipids or ionizable lipidoids which can be comprised as component (b) in the in the nanoparticles used in the context of the present invention are disclosed, e.g., in WO 2006/138380 A2, EP2476756 A1 , US 2016/0114042 A1, US 8,058,069 B2, US 8,492,359 B2, US 8,822,668 B2, US 8,969,535, US 9,006,417 B2, US 9,018,187 B2, US 9,345,780 B2, US 9,352,042 B2, US 9,364,435 B2, US 9,394,234 B2, US 9,492,386 B2, US 9,504,651 B2, US 9,518,272 B2, DE 19834683 A1, WO 2010/053572 A2, US 9,227,917 B2, US 9,556,110 B2, US 8,969,353 B2, US 10,189,802 B2, WO 2012/000104 A1 , WO 2010/053572, WO 2014/028487, WO 2015/095351 , US 2013/0156849 A1 (e.g. claims 13, 33, 34), US 9254311 B2 (e.g. Item 14), US 10501512 B2 (e.g. claims 1 , 6, 9), US 2014/0010861 A1 (e.g. claims 44 and 78-82), US 2013/0115272 A1 (e.g. Item 12) or by Akinc, A., et aL, Nature Biotechnology, 26(5), 2008, 561-569; Sabnis, S. et al., Molecular Therapy, 26(6), 2018, Vol. 26 No 6 June 2018, 1509-1519; Kowalski, P.S., et aL, Molecular Therapy, 27(4), 2019, 710-728; Kulkami, J. A. et al, Nucleic Acid Therapeutics, 28(3), 2018, 146-157; and Li, B. et aL, Nano Letters, 15, 2015, 8099-8107.
Preferably, a permanently cationic lipid which may be comprised by the nanoparticles is a lipid which comprises a head group containing one quaternary nitrogen atom and one or more, preferably one or two, hydrophobic moieties, linked to the head group. Preferably, the quaternary nitrogen atom is provided by a group of the formula -N(Me)3+, wherein Me is a methyl group.
A hydrophobic moiety comprised in these preferred lipids preferably contains one or more of a linear chain aliphatic residue, e.g. a linear chain residue comprising 8 to 18 carbon atoms, or a branched chain aliphatic residue, e.g. a branched chain residue comprising 8 to 18 carbon atoms, In addition, the hydrophobic moiety may comprise one or more linking groups which facilitate the linking of the moiety to the head group, or which allow two or more of the above aliphatic residues to be combined with each other. Furthermore, it may comprise one or more substituents, to the extent that the hydrophobic characteristics of the moiety are maintained. As examples of a permanently cationic lipid, reference can be made to DOTMA (Dioleoyl-3- trimethylammonium propane and DOTAP (Dioleoyl-3-trimethylammonium propane).
In as one exemplary embodiment, the nanoparticles may comprise an ionizable lipid or lipidoid of Formula a-l as disclosed in WO 2016176330 A1 which is incorporated in its entirety herein: a-l or a pharmaceutically acceptable salt, tautomer, prodrug, or stereoisomer thereof, wherein: one of L1 or L2 is-O(C=O)-, -(C=O)O-, -C(=O)-, -O-, -S(O)X-, -S-S-, - C(=O)S-, SC(=O)-, - NRaC(=O)-, -C(=O)NRa-, NRaC(=O)NRa-, -OC(=O)NRa- or - NRaC(=O)O-, and the other of L1 or L2 is-O(C=O)-, -(C=O)O-, -C(=O)-, -O-, -S(O)X-, -S- S-, -C(=O)S-, SC(=O)-,
a-l or a pharmaceutically acceptable salt, tautomer, prodrug, or stereoisomer thereof, wherein: one of L1 or L2 is-O(C=O)-, -(C=O)O-, -C(=O)-, -O-, -S(O)X-, -S-S-, - C(=O)S-, SC(=O)-, - NRaC(=O)-, -C(=O)NRa-, NRaC(=O)NRa-, -OC(=O)NRa- or - NRaC(=O)O-, and the other of L1 or L2 is-O(C=O)-, -(C=O)O-, -C(=O)-, -O-, -S(O)X-, -S- S-, -C(=O)S-, SC(=O)-,

-NRaC(=O)-, -C(=O)NRa-, -NRaC(=O)NRa-, -OC(=O)NRa- or -NRaC(=O)O- or a direct bond;
Ra is H or C1-C12 alkyl;
R1a and R1b are, at each occurrence, independently either (a) H or C1-C12 alkyl, or (b) R1a is H or C1-C12 alkyl, and R1b together with the carbon atom to which it is bound is taken together with an adjacent R1b and the carbon atom to which it is bound to form a carbon-carbon double bond;
R2a and R2b are, at each occurrence, independently either (a) H or C1-C12 alkyl, or (b) R2a is H or C1-C12 alkyl, and R2b together with the carbon atom to which it is bound is taken together with an adjacent R2b and the carbon atom to which it is bound to form a carbon-carbon double bond;
R3a and R3b are, at each occurrence, independently either (a) H or C1-C12 alkyl, or (b) R3a is H or C1-C12 alkyl, and R3b together with the carbon atom to which it is bound is taken together with an adjacent R3b and the carbon atom to which it is bound to form a carbon-carbon double bond;
R4a and R4b are, at each occurrence, independently either (a) H or C1-C12 alkyl, or (b) R4a is H or C1-C12 alkyl, and R4b together with the carbon atom to which it is bound is taken together with an adjacent R4b and the carbon atom to which it is bound to form a carbon-carbon double bond;
R5 and R6 are each independently methyl or cycloalkyl;
R7 is, at each occurrence, independently H or C1-C12 alkyl;
R8 and R9 are each independently unsubstituted C1-C12 alkyl; or R8 and R9, together with the nitrogen atom to which they are attached, form a 5, 6 or 7-membered heterocyclic ring comprising one nitrogen atom; a and d are each independently an integer from 0 to 24; b and c are each independently an integer from 1 to 24; e is 1 or 2; and x is 0, 1 or 2.
In some embodiments, the ionizable lipid has a structure of Formula a-ll:

-NRaC(=O)-, -C(=O)NRa-, ,NRaC(=O)NRa-, -OC(=O)NRa- or -NRaC(=O)O- or a direct bond;
G1 is C1-C2 alkylene, -(C=O)-, -O(C=O)-, -SC(=O)-, -NRaC(=O)- or a direct bond;
G2 is-C(=0)-, -(0=0)0-, -C(=0)S-, -C(=0)NRa- or a direct bond;
G3 is Ci-Os alkylene;
Ra is H or C1-C12 alkyl;
R1a and R1b are, at each occurrence, independently either: (a) H or C1-C12 alkyl; or (b) R1a is H or C1-C12 alkyl, and R1b together with the carbon atom to which it is bound is taken together with an adjacent R1b and the carbon atom to which it is bound to form a carbon-carbon double bond;
R2a and R2b are, at each occurrence, independently either: (a) H or C1-C12 alkyl; or (b) R2a is H or C1-C12 alkyl, and R2b together with the carbon atom to which it is bound is taken together with an adjacent R2b and the carbon atom to which it is bound to form a carbon-carbon double bond;
R3a and R3b are, at each occurrence, independently either (a): H or C1-C12 alkyl; or (b) R3a is H or C1-C12 alkyl, and R3b together with the carbon atom to which it is bound is taken together with an adjacent R3b and the carbon atom to which it is bound to form a carbon-carbon double bond; R4a and R4b are, at each occurrence, independently either: (a) H or C1-C12 alkyl; or (b) R4a is H or C1-C12 alkyl, and R4b together with the carbon atom to which it is bound is taken together with an adjacent R4b and the carbon atom to which it is bound to form a carbon-carbon double bond;
R5 and R6 are each independently H or methyl;
R7 is C4-C20 alkyl;
R8 and R9 are each independently C1-C12 alkyl; or R8 and R9, together with the nitrogen atom to which they are attached, form a 5, 6 or 7-membered heterocyclic ring; a, b, c, and d are each independently an integer from 1 to 24; and x is 0, 1 or 2.
In some embodiments, the ionizable lipid has a structure of Formula a-lll:

III a-lll or a pharmaceutically acceptable salt, prodrug, or stereoisomer thereof, wherein: one of L1 or L2 is-O(C=O)-, -(C=O)O-, -C(=O)-, -O-, -S(O)X-, -S-S-, - C(=O)S-, SC(=O)-, - NRaC(=O)-, -C(=O)NRa-, NRaC(=O)NRa-, -OC(=O)NRa- or - NRaC(=O)O-, and the other of L1 or L2 is-O(C=O)-, -(C=O)O-, -C(=O)-, -O-, -S(O)X-, -S- S-, -C(=O)S-, SC(=O)-,
-NRaC(=O)-, -C(=O)NRa-, ,NRaC(=O)NRa-, -OC(=O)NRa- or -NRaC(=O)O- or a direct bond;
G1 and G2 are each independently unsubstituted C1-C12 alkylene or C1-C12 alkenylene;
G3 is C1-C24 alkylene, C1-C24 alkenylene, C3-C8 cycloalkylene, C3-C8 cycloalkenylene;
Ra is H or C1-C12 alkyl;
R1 and R2 are each independently C6-C24 alkyl or C6-C24 alkenyl;
R3 is H, OR5, ON, -C(=O)OR4, -OC(=O)R4 or-NR5C(=O)R4; R4 is C1-C12 alkyl; R5 is H or Ci-Ce alkyl; and x is 0, 1 or 2.
In some embodiments, the ionizable lipid has the following Formula a-IV :

(a-IV) or a pharmaceutically acceptable salt, prodrug or stereoisomer thereof, wherein: one of G1 or G2 is, at each occurrence, -O(C=O)-, -(C=0)0-, -C(=0)-, -0-, -S(0)y-, -S-S-, -C(=O)S-, SC(=O)-, -N(Ra)C(=O)-, -C(=O)N(Ra)- , -N(Ra)C(=O)N(Ra)-,
-OC(=O)N(Ra)- or -N(Ra)C(=O)O-, and the other of G1 or G2 is, at each occurrence, -0(0=0)-, -(0=0)0-, -C(=0)-, -O-, -S(O)y-, -S-S-, -C(=0)S-, -SC(=O)-, -N(Ra)C(=O)-, -C(=0)N(Ra)-, -N(Ra)C(=0)N(Ra)-, -OC(=O)N(Ra)- or
-N(Ra)C(=0)0- or a direct bond;
L is, at each occurrence, ~0(C=0)-, wherein ~ represents a covalent bond to X;
X is CRa;
Z is alkyl, cycloalkyl or a monovalent moiety comprising at least one polar functional group when n is 1 ; or Z is alkylene, cycloalkylene or a polyvalent moiety comprising at least one polar functional group when n is greater than 1 ;
Ra is, at each occurrence, independently H, C1-C12 alkyl, C1-C12 hydroxylalkyl, Ci- 012 aminoalkyl, C1-C12 alkylaminylalkyl, C1-C12 alkoxyalkyl, C1-C12 alkoxycarbonyl, C1-C12 alkylcarbonyloxy, C1-C12 alkylcarbonyloxyalkyl or C1-C12 alkylcarbonyl;
R is, at each occurrence, independently either: (a) H or C1-C12 alkyl; or (b) R together with the carbon atom to which it is bound is taken together with an adjacent R and the carbon atom to which it is bound to form a carbon-carbon double bond; R1 and R2 have at each occurrence the following structure, respectively:

R1 R2 a1 and a2 are, at each occurrence, independently an integer from 3 to 12; b1 and b2 are, at each occurrence, independently 0 or 1 ; c1 and c2 are, at each occurrence, independently an integer from 5 to 10; d1 and d2 are, at each occurrence, independently an integer from 5 to 10; y is, at each occurrence, independently an integer from 0 to 2; and n is an integer from 1 to 6, wherein each alkyl, alkylene, hydroxylalkyl, aminoalkyl, alkylaminylalkyl, alkoxyalkyl, alkoxycarbonyl, alkylcarbonyloxy, alkylcarbonyloxyalkyl and alkylcarbonyl is optionally substituted with one or more substituents.
In some embodiments, the ionizable lipid has the following Formula (a-V):

(a-V) or a pharmaceutically acceptable salt, prodrug or stereoisomer thereof, wherein: one of G1 or G2 is, at each occurrence, -O(C=O)-, -(0=0)0-, -C(=O)-, -O-, -S(O)y-, -S-S-, -C(=O)S-, SC(=O)-, -N(Ra)C(=O)-, -C(=O)N(Ra)- , -N(Ra)C(=O)N(Ra)-,
-OC(=O)N(Ra)- or -N(Ra)C(=O)O-, and the other of G1 or G2 is, at each occurrence, -0(0=0)-, -(C=0)0-, -C(=0)-, -0-, -S(O)y-, -S-S-, -C(=O)S-, -SC(=O)-, -N(Ra)C(=O)-, - C(=O)N(Ra)-, -N(Ra)C(=O)N(Ra)-, -OC(=O)N(Ra)- or
-N(Ra)C(=O)O- or a direct bond;
L is, at each occurrence, ~O(C=O)-, wherein ~ represents a covalent bond to X;
X is CRa;
Z is alkyl, cycloalkyl or a monovalent moiety comprising at least one polar functional group when n is 1 ; or Z is alkylene, cycloalkylene or a polyvalent moiety comprising at least one polar functional group when n is greater than 1 ;
Ra is, at each occurrence, independently H, C1-C12 alkyl, C1-C12 hydroxylalkyl, C1-C12 aminoalkyl, C1-C12 alkylaminylalkyl, C1-C12 alkoxyalkyl, C1-C12 alkoxycarbonyl, C1-C12 alkylcarbonyloxy, C1-C12 alkylcarbonyloxyalkyl or C1-C12 alkylcarbonyl;
R is, at each occurrence, independently either: (a) H or C1-C12 alkyl; or (b) R together with the carbon atom to which it is bound is taken together with an adjacent R and the carbon atom to which it is bound to form a carbon-carbon double bond;
R1 and R2 have at each occurrence the following structure, respectively:

R' is, at each occurrence, independently H or C1-C12 alkyl; a1 and a2 are, at each occurrence, independently an integer from 3 to 12; b1 and b2 are, at each occurrence, independently 0 or 1; c1 and c2 are, at each occurrence, independently an integer from 2 to 12; d1 and d2 are, at each occurrence, independently an integer from 2 to 12; y is, at each occurrence, independently an integer from 0 to 2; and n is an integer from 1 to 6, wherein a1, a2, c1, c2, d1 and d2 are selected such that the sum of a1+c1+d1 is an integer from 18 to 30, and the sum of a2+c2+d2 is an integer from 18 to 30, and wherein each alkyl, alkylene, hydroxylalkyl, aminoalkyl, alkylaminylalkyl, alkoxyalkyl, alkoxycarbonyl, alkylcarbonyloxy, alkylcarbonyloxyalkyl and alkylcarbonyl is optionally substituted with one or more substituent.
In some embodiments, the ionizable lipid is selected from a lipid in Table 1 , Table 2, Table 3 or Table 4. Table 1 :



41





Table 2:








Table 3:








Table 4:

In some embodiments the ionizable lipid has one of the following structures:

In a further exemplary embodiment, the ionizable lipid has the following structure: or a pharmaceutically acceptable salt, tautomer, prodrug or stereoisomer thereof, wherein: Ri and R2 are each independently for each occurrence optionally substituted C10-C30 alkyl, optionally substituted C10-C30 alkenyl, optionally substituted C10-C30 alkynyl or optionally substituted C10-C30 acyl; R3 is H, optionally substituted C10-C10 alkyl, optionally substituted C2-C10 alkenyl, optionally substituted C2-C10 alkynyl, alkylhetrocycle, alkylphosphate, alkylphosphorothioate, alkyl phosphorod ith ioate, alkylphosphonate, alkylamine, hydroxyalkyl, co-aminoalkyl, co-(substituted)aminoalkyl, co-phosphoalkyl, co- thiophosphoalkyl, optionally substituted polyethylene glycol (PEG, mw 100-40K), optionally substituted mPEG (mw 120-40K), heteroaryl, or heterocycle, or linker-ligand; and
or a pharmaceutically acceptable salt, tautomer, prodrug or stereoisomer thereof, wherein: Ri and R2 are each independently for each occurrence optionally substituted C10-C30 alkyl, optionally substituted C10-C30 alkenyl, optionally substituted C10-C30 alkynyl or optionally substituted C10-C30 acyl; R3 is H, optionally substituted C10-C10 alkyl, optionally substituted C2-C10 alkenyl, optionally substituted C2-C10 alkynyl, alkylhetrocycle, alkylphosphate, alkylphosphorothioate, alkyl phosphorod ith ioate, alkylphosphonate, alkylamine, hydroxyalkyl, co-aminoalkyl, co-(substituted)aminoalkyl, co-phosphoalkyl, co- thiophosphoalkyl, optionally substituted polyethylene glycol (PEG, mw 100-40K), optionally substituted mPEG (mw 120-40K), heteroaryl, or heterocycle, or linker-ligand; and

E is O, S, N(Q), C(O), N(Q)C(O), C(O)N(Q), (Q)N(CO)O, O(CO)N(Q), S(O), NS(O)2N(Q), S(O)2, N(Q)S(O)2, SS, O=N, aryl, heteroaryl, cyclic or heterocycle; and
Q is H, alkyl, co-aminoalkyl, co-(substituted)aminoalkyl, co-phosphoalkyl or co-thiophosphoalkyl. In some embodiments the ionizable lipid has one of the following structures:

In a preferred embodiment, the lipid nanoparticles referred to herein comprise the ionizable lipid [(4-Hydroxybutyl)azanediyl]di(hexane-6,1-diyl) bis(2-hexyldecanoate), also known ALC- 0315, and as shown in the following formula:

In some embodiments, the lipid nanoparticles may further comprise a neutral lipid and/or a polymer conjugated lipid, preferably both a neutral lipid and a polymer conjugated lipid. The molar ratio of the above ionizable lipid to a neutral lipid preferably ranges from about 4.1 :1.0 to about 4.9:1.0, 4.5:1.0 to about 4.8:1.0, or 4.7:1.0 to about 4.8:1.0. In some embodiment the molar ratio of ionizable lipid to the neutral lipid ranges from about 2:1 to about 8:1 , preferably 5:1 to 1 :1.
The molar ratio of ionizable lipid to the polymer conjugated lipid ranges from about 35:1 to about 25:1 , or 100:1 to about 20:1.
In another exemplary embodiment, the ionizable lipid has the following structure:

In accordance with a further exemplary embodiment, a lipid nanoparticle as referred to herein comprises: i) a first cationic lipid as an ionizable lipid (a) having a first effective pKa; ii) a second cationic lipid as an ionizable lipid (a) having a second effective pKa, the second effective pKa being greater than the first effective pKa; iii) a neutral lipid; iv) a steroid; v) a polymer conjugated lipid; vi) a therapeutic agent, or a pharmaceutically acceptable salt or prodrug thereof, encapsulated within or associated with the lipid nanoparticle, and vii) a surfactant, wherein the lipid nanoparticle has an effective pKa between the first and second effective pKa’s.
In some embodiments the first effective pKa is less than 5.75. In some embodiments the second effective pKa is greater than 6.25. In some embodiments, the lipid nanoparticle of any one of claims 48-50, wherein the lipid nanoparticle has an effective pKa ranging from 5.90 to 6.35. In some embodiments, the mol ratio of the first cationic lipid to the second cationic lipid ranges from 1 :20 to 1 :2.
In some embodiments, a LNP or LiNP of the invention comprises a first cationic lipid as an ionizable lipid (a), or a second cationic lipid as an ionizable lipid (a), and one or both has a structure of Formula a-l: a-l or a pharmaceutically acceptable salt, tautomer, prodrug or stereoisomer thereof, wherein: one of L1 or L2 is-O(C=O)-, -(C=O)O-, -C(=O)-, -O-, -S(O)X-, -S-S-, - C(=O)S-, SC(=O)-, -NRaC(=O)-, -C(=O)NRa-, NRaC(=O)NRa-, -OC(=O)NRa- or - NRaC(=O)O-, and the other of L1 or L2 is-O(C=O)-, -(C=O)O-, -C(=O)-, -O-, -S(O)X-, -S- S-, -C(=O)S-, SC(=O)-,
a-l or a pharmaceutically acceptable salt, tautomer, prodrug or stereoisomer thereof, wherein: one of L1 or L2 is-O(C=O)-, -(C=O)O-, -C(=O)-, -O-, -S(O)X-, -S-S-, - C(=O)S-, SC(=O)-, -NRaC(=O)-, -C(=O)NRa-, NRaC(=O)NRa-, -OC(=O)NRa- or - NRaC(=O)O-, and the other of L1 or L2 is-O(C=O)-, -(C=O)O-, -C(=O)-, -O-, -S(O)X-, -S- S-, -C(=O)S-, SC(=O)-,

-NRaC(=O)-, -C(=O)NRa-, ,NRaC(=O)NRa-, -OC(=O)NRa- or -NRaC(=O)O- or a direct bond;
Ra is H or C1-C12 alkyl;
R1a and R1b are, at each occurrence, independently either (a) H or C1-C12 alkyl, or (b) R1a is H or C1-C12 alkyl, and R1b together with the carbon atom to which it is bound is taken together with an adjacent R1b and the carbon atom to which it is bound to form a carbon-carbon double bond; R2a and R2b are, at each occurrence, independently either (a) H or C1-C12 alkyl, or (b) R2a is H or C1-C12 alkyl, and R2b together with the carbon atom to which it is bound is taken together with an adjacent R2b and the carbon atom to which it is bound to form a carbon-carbon double bond;
R3a and R3b are, at each occurrence, independently either (a) H or C1-C12 alkyl, or (b) R3a is H or C1-C12 alkyl, and R3b together with the carbon atom to which it is bound is taken together with an adjacent R3b and the carbon atom to which it is bound to form a carbon-carbon double bond; R4a and R4b are, at each occurrence, independently either (a) H or C1-C12 alkyl, or (b) R4a is H or C1-C12 alkyl, and R4b together with the carbon atom to which it is bound is taken together with an adjacent R4b and the carbon atom to which it is bound to form a carbon-carbon double bond;
R5 and R6 are each independently methyl or cycloalkyl;
R7 is, at each occurrence, independently H or C1-C12 alkyl;
R8 and R9 are each independently unsubstituted C1-C12 alkyl; or R8 and R9, together with the nitrogen atom to which they are attached, form a 5, 6 or 7-membered heterocyclic ring comprising one nitrogen atom; a and d are each independently an integer from 0 to 24; b and c are each independently an integer from 1 to 24; e is 1 or 2; and x is 0, 1 or 2.
In some embodiments, the first and second cationic lipids as ionizable lipids (a) are each independently selected from a lipid of Formula a-L In some embodiments the first cationic lipid, or the second cationic lipid or both has a structure of Formula a-ll:

-NRaC(=O)-, -C(=O)NRa-, NRaC(=O)NRa-, -OC(=O)NRa- or -NRaC(=O)O- or a direct bond;
G1 is C1-C2 alkylene, -(C=O)-, -O(C=O)-, -SC(=0)-, -NRaC(=O)- or a direct bond;
G2 is-C(=0)-, -(0=0)0-, -C(=0)S-, -C(=0)NRa- or a direct bond; G3 is Ci-Ce alkylene;
Ra is H or C1-C12 alkyl;
R1a and R1b are, at each occurrence, independently either: (a) H or C1-C12 alkyl; or (b) R1a is H or C1-C12 alkyl, and R1b together with the carbon atom to which it is bound is taken together with an adjacent R1b and the carbon atom to which it is bound to form a carbon-carbon double bond;
R2a and R2b are, at each occurrence, independently either: (a) H or C1-C12 alkyl; or (b) R2a is H or C1-C12 alkyl, and R2b together with the carbon atom to which it is bound is taken together with an adjacent R2b and the carbon atom to which it is bound to form a carbon-carbon double bond;
R3a and R3b are, at each occurrence, independently either (a): H or C1-C12 alkyl; or (b) R3a is H or C1-C12 alkyl, and R3b together with the carbon atom to which it is bound is taken together with an adjacent R3b and the carbon atom to which it is bound to form a carbon-carbon double bond; R4a and R4b are, at each occurrence, independently either: (a) H or C1-C12 alkyl; or (b) R4a is H or C1-C12 alkyl, and R4b together with the carbon atom to which it is bound is taken together with an adjacent R4b and the carbon atom to which it is bound to form a carbon-carbon double bond;
R5 and R6 are each independently H or methyl;
R7 is C4-C20 alkyl;
R8 and R9 are each independently C1-C12 alkyl; or R8 and R9, together with the nitrogen atom to which they are attached, form a 5, 6 or 7-membered heterocyclic ring; a, b, c and d are each independently an integer from 1 to 24; and x is 0, 1 or 2.
In some embodiments, the first and second cationic lipids as ionizable lipids (a) are each, independently selected from a lipid of Formula a-ll.
In some embodiments, the first cationic lipid as an ionizable lipid (a), or the second cationic lipid as an ionizable lipid (a) or both has a structure of Formula a-lll: a-lll or a pharmaceutically acceptable salt, prodrug or stereoisomer thereof, wherein: one of L1 or L2 is-O(C=O)-, -(C=O)O-, -C(=0)-, -0-, -S(0)x-, -S-S-, - C(=O)S-, SC(=O)-, -NRaC(=0)-, -C(=0)NRa-, NRaC(=0)NRa-, -OC(=O)NRa- or - NRaC(=O)O-, and the other of L1 or L2 is-0(C=0)-, -(C=0)0-, -C(=0)-, -0-, -S(0)x-, -S- S-, -C(=O)S-, SC(=O)-,
a-lll or a pharmaceutically acceptable salt, prodrug or stereoisomer thereof, wherein: one of L1 or L2 is-O(C=O)-, -(C=O)O-, -C(=0)-, -0-, -S(0)x-, -S-S-, - C(=O)S-, SC(=O)-, -NRaC(=0)-, -C(=0)NRa-, NRaC(=0)NRa-, -OC(=O)NRa- or - NRaC(=O)O-, and the other of L1 or L2 is-0(C=0)-, -(C=0)0-, -C(=0)-, -0-, -S(0)x-, -S- S-, -C(=O)S-, SC(=O)-,

-NRaC(=0)-, -C(=0)NRa-, NRaC(=0)NRa-, -OC(=O)NRa- or -NRaC(=O)O- or a direct bond;
G1 and G2 are each independently unsubstituted C1-C12 alkylene or C1-C12 alkenylene;
G3 is C1-C24 alkylene, C1-C24 alkenylene, C3-C8 cycloalkylene, C3-C8 cycloalkenylene;
Ra is H or C1-C12 alkyl;
R1 and R2 are each independently C6-C24 alkyl or C6-C24 alkenyl; R3 is H, OR5, CN, -C(=O)OR4, -OC(=O)R4 or-NR5C(=O)R4; R4 is C1-C12 alkyl;
R5 is H or Ci-Ce alkyl; and x is 0, 1 or 2.
In some embodiments, the first and second cationic lipids as ionizable lipids (a) are each, independently, selected from a lipid of Formula a-lll.
In some embodiments, the first cationic lipid as an ionizable lipid (a), or the second cationic lipid as an ionizable lipid (a) or both has a structure of Formula a-IV:

(a-IV) or a pharmaceutically acceptable salt, prodrug or stereoisomer thereof, wherein: one of G1 or G2 is, at each occurrence, -O(C=O)-, -(C=O)O-, -C(=O)-, -O-, -S(O)y-, -S-S-, -C(=O)S-, SC(=O)-, -N(Ra)C(=O)-, -C(=O)N(Ra)- , -N(Ra)C(=O)N(Ra)-,
-OC(=O)N(Ra)- or -N(Ra)C(=O)O-, and the other of G1 or G2 is, at each occurrence, -O(C=O)-,
-(C=O)O-, -C(=O)-, -O-, -S(O)y-, -S-S-, -C(=O)S-, -SC(=O)-, -N(Ra)C(=O)-, - C(=O)N(Ra)-,
-N(Ra)C(=O)N(Ra)-, -OC(=O)N(Ra)- or-N(Ra)C(=O)O- or a direct bond; L is, at each occurrence, ~O(C=O)-, wherein ~ represents a covalent bond to X;
X is CRa;
Z is alkyl, cycloalkyl or a monovalent moiety comprising at least one polar functional group when n is 1 ; or Z is alkylene, cycloalkylene or a polyvalent moiety comprising at least one polar functional group when n is greater than 1 ; Ra is, at each occurrence, independently H, C1-C12 alkyl, C1-C12 hydroxylalkyl,
C1-C12 aminoalkyl, C1-C12 alkylaminylalkyl, C1-C12 alkoxyalkyl, C1-C12 alkoxycarbonyl, C1-C12 alkylcarbonyloxy, C1-C12 alkylcarbonyloxyalkyl or C1-C12 alkylcarbonyl;
R is, at each occurrence, independently either: (a) H or C1-C12 alkyl; or (b) R together with the carbon atom to which it is bound is taken together with an adjacent R and the carbon atom to which it is bound to form a carbon-carbon double bond;
R1 and R2 have, at each occurrence, the following structure, respectively:

R1 R2 a1 and a2 are, at each occurrence, independently an integer from 3 to 12; b1 and b2 are, at each occurrence, independently 0 or 1 ; c1 and c2 are, at each occurrence, independently an integer from 5 to 10; d1 and d2 are, at each occurrence, independently an integer from 5 to 10; y is, at each occurrence, independently an integer from 0 to 2; and n is an integer from 1 to 6, wherein each alkyl, alkylene, hydroxylalkyl, aminoalkyl, alkylaminylalkyl, alkoxyalkyl, alkoxycarbonyl, alkylcarbonyloxy, alkylcarbonyloxyalkyl and alkylcarbonyl is optionally substituted with one or more substituents.
In some embodiments, the first and second cationic lipids as ionizable lipids (a) are each, independently, selected from a lipid of Formula a-IV.
In some embodiments, the first cationic lipid as an ionizable lipid (a), or the second cationic lipid as an ionizable lipid (a), or both has a structure of Formula a-V:

(a-V) or a pharmaceutically acceptable salt, prodrug or stereoisomer thereof, wherein: one of G1 or G2 is, at each occurrence, -O(C=O)-, -(0=0)0-, -C(=0)-, -0-, -S(0)y-, -S-S-, -C(=O)S-, SC(=O)-, -N(Ra)C(=0)-, -C(=0)N(Ra)- , -N(Ra)C(=0)N(Ra)-,
-0C(=0)N(Ra)- or -N(Ra)C(=0)0-, and the other of G1 or G2 is, at each occurrence, -0(C=0)-,
-(0=0)0-, -C(=0)-, -0-, -S(0)y-, -S-S-, -C(=0)S-, -SC(=0)-, -N(Ra)C(=0)-, - C(=0)N(Ra)-,
-N(Ra)C(=0)N(Ra)-, -0C(=0)N(Ra)- or-N(Ra)C(=O)O- or a direct bond;
L is, at each occurrence, ~0(C=0)-, wherein ~ represents a covalent bond to X;
X is CRa;
Z is alkyl, cycloalkyl or a monovalent moiety comprising at least one polar functional group when n is 1 ; or Z is alkylene, cycloalkylene or a polyvalent moiety comprising at least one polar functional group when n is greater than 1 ;
Ra is, at each occurrence, independently H, C1-C12 alkyl, C1-C12 hydroxylalkyl, C1-C12 aminoalkyl, C1-C12 alkylaminylalkyl, C1-C12 alkoxyalkyl, C1-C12 alkoxycarbonyl, C1-C12 alkylcarbonyloxy, C1-C12 alkylcarbonyloxyalkyl or C1-C12 alkylcarbonyl;
R is, at each occurrence, independently either: (a) H or C1-C12 alkyl; or (b) R together with the carbon atom to which it is bound is taken together with an adjacent R and the carbon atom to which it is bound to form a carbon-carbon double bond;
R1 and R2 have at each occurrence the following structure, respectively:

R' is, at each occurrence, independently H or C1-C12 alkyl; a1 and a2 are, at each occurrence, independently an integer from 3 to 12; b1 and b2 are, at each occurrence, independently 0 or 1; c1 and c2 are, at each occurrence, independently an integer from 2 to 12; d1 and d2 are, at each occurrence, independently an integer from 2 to 12; y is, at each occurrence, independently an integer from 0 to 2; and n is an integer from 1 to 6, wherein a1, a2, c1, c2, d1 and d2 are selected such that the sum of a1+c1+d1 is an integer from 18 to 30, and the sum of a2+c2+d2 is an integer from 18 to 30, and wherein each alkyl, alkylene, hydroxylalkyl, aminoalkyl, alkylaminylalkyl, alkoxyalkyl, alkoxycarbonyl, alkylcarbonyloxy, alkylcarbonyloxyalkyl and alkylcarbonyl is optionally substituted with one or more substituent.
In some embodiments, the first and second cationic lipids as ionizable lipids (a) are each, independently, selected from a lipid of Formula a-V.
In some embodiments, the first and second cationic lipids as ionizable lipids (a) have the following structures, respectively:

In some embodiments, the first cationic lipid as an ionizable lipid (a), the second cationic lipid as an ionizable lipid (a), or both have one of the following structures:

In some embodiments, when a compound of formula a-ll is used, the total mol percent of cationic lipid as an ionizable lipid (a) in the lipid nanoparticle ranges from 40 to 55 mol percent based on total lipid present in the lipid nanoparticle. In some embodiments, the lipid nanoparticles may further comprise a neutral lipid, a steroid (such as sterol) and/or a polymer conjugated lipid, preferably all of the neutral lipid, the steroid and the polymer conjugated lipid, and the molar ratio of total cationic lipid to a neutral lipid ranges from about 2:1 to about 8:1. In some embodiments, the molar ratio of total cationic lipid to a steroid ranges from 5:1 to 1 :1. In some embodiments, the molar ratio of total cationic lipid to a polymer conjugated lipid ranges from about 100:1 to about 20:1 .
In some embodiments, the neutral lipid is distearoylphosphatidylcholine (DSPC), dioleoylphosphatidylcholine (DOPC), dipalmitoylphosphatidylcholine (DPPC), dioleoylphosphatidylglycerol (DOPG), dipalmitoylphosphatidylglycerol (DPPG), dioleoylphosphatidylethanolamine (DOPE), palmitoyloleoylphosphatidylcholine (POPO), palmitoyloleoyl-phosphatidylethanolamine (POPE) and dioleoyl-phosphatidylethanolamine 4- (N-maleimidomethyl)-cyclohexane- 1 carboxylate (DOPE-mal), dipalmitoyl phosphatidyl ethanolamine (DPPE), dimyristoylphosphoethanolamine (DMPE), distearoylphosphatidylethanolamine (DSPE), 16-O-monomethyl PE, 16-O-dimethyl PE, 18-1 -trans PE, 1 -stearioyl-2- oleoylphosphatidyethanol amine (SOPE) or 1,2-dielaidoyl-sn-glycero-3- phophoethanolamine (transDOPE), preferably the neutral lipid is DSPC, DPPC, DMPC, DOPC, POPC, DOPE or SM. In some embodiments, the neutral lipid is DSPC. In some embodiments, the steroid is cholesterol. In some embodiments, the polymer conjugated lipid is present in a concentration ranging from 1.0 to 2.5 molar percent, preferably, about 1.7 molar percent, wherein the polymer conjugated lipid is present in a concentration of about 1.5 molar percent.
In some embodiments, the polymer conjugated lipid is a pegylated lipid. In some embodiments, the pegylated lipid is PEG-DAG, PEG-PE, PEG-S-DAG, PEG-cer or a PEG dialkyoxypropylcarbamate. In some embodiments, the pegylated lipid has the following Formula (a-VI): or a pharmaceutically acceptable salt, tautomer or stereoisomer thereof, wherein:
or a pharmaceutically acceptable salt, tautomer or stereoisomer thereof, wherein:

R12 and R13 are each independently a straight or branched, saturated or unsaturated alkyl chain containing from 10 to 30 carbon atoms, wherein the alkyl chain is optionally interrupted by one or more ester bonds; and w has a mean value ranging from 30 to 60.
Optionally, R12 and R13 are each independently straight, saturated alkyl chains containing from 12 to 16 carbon atoms. Optionally, the average w ranges from 42 to 55, preferably, the average w is about 49. In some embodiments, the pegylated lipid has the following Formula (Via): wherein the average w is about 49.
wherein the average w is about 49.

In some embodiments, the lipid nanoparticle forms a plurality of the nanoparticles having a polydispersity of less than 0.12. Preferably, the polydispersity is less than 0.08.
In some embodiments, the mean diameter ranges from 50 nm to 100 nm, preferably the diameter ranges from 60 nm to 85 nm. In accordance with a preferred embodiment, the ionizable lipidoid which may be comprised by the nanoparticles referred to herein comprises or consists of an ionizable lipidoid of the following formula (L-1 ) or a protonated form thereof. It is particularly preferred that the lipid nanoparticle (LNP) formulation or lipidoid nanoparticle (LiNP) formulation is a lipidoid nanoparticle formulation comprising a plurality of lipidoid nanoparticles (LiNPs), each comprising a lipidoid mix, wherein the lipidoid mix comprises the ionizable lipidoid of the following formula (L-1 ) or a protonated form thereof. The ionizable lipidoid of the following formula (L-1) or its protonated forms which can be used as a preferred ionizable lipidoid in the context of the present invention are described in detail in the PCT application WO 2014/207231 A1.
R2A R4A
R1 A— N— {CH2- (CH2)— N— [CH2— (CH2)b— N]p}— [CH2- (CH2)~ N]- R6A 3A 5A wherein the variables a, b, p, m, n and R1A to R6A are defined as follows: a is 1 or 2 and b is an integer of 1 to 4 or a is an integer of 1 to 4 and b is 1 or 2, preferably a is 1 and b is an integer of 2 to 4 or a is an integer of 2 to 4 and b is 1 , p is 1 or 2, m is 1 or 2; n is 0 or 1 and m+n is > 2; and
wherein the variables a, b, p, m, n and R1A to R6A are defined as follows: a is 1 or 2 and b is an integer of 1 to 4 or a is an integer of 1 to 4 and b is 1 or 2, preferably a is 1 and b is an integer of 2 to 4 or a is an integer of 2 to 4 and b is 1 , p is 1 or 2, m is 1 or 2; n is 0 or 1 and m+n is > 2; and

R1A to R6A are independently of each other selected from hydrogen, -CH2-CH(OH)-R7A, -CH(R7A)-CH2-OH, -CH2-CH2-(C=O)-O-R7A, -CH2-CH2-(C=O)-NH-R7A, -CH2-R7A, -C(NH)-NH2I a polyethylene glycol) chain, and a receptor ligand; wherein R7A is selected from C3-C18 alkyl and C3-C18 alkenyl having one C-C double bond, provided that at least two residues among R1A to R6A are selected from -CH2-CH(OH)-R7A, -CH(R7A)-CH2-OH, -CH2-CH2-(C=O)-O-R7A, -CH2-CH2-(C=O)-NH-R7A and -CH2-R7A wherein R7A is selected from C3-C18 alkyl or C3-C18 alkenyl having one C-C double bond.
In the protonated form of the compound of formula (L-1 ), one or more of the nitrogen atoms contained in the compound of formula (L-1 ) are protonated to provide a compound carrying a positive charge.
Preferably, R1A to R6A are independently selected from hydrogen, a group -CH2-CH(OH)-R7A, -CH(R7A)-CH2-OH, -CH2-CH2-(C=O)-O-R7A, -CH2-CH2-(C=O)-NH-R7A; and -CH2-R7A wherein R7A is selected from C3-C18 alkyl and C3- C18 alkenyl having one C-C double bond, provided that at least two residues among R1A to R6A, more preferably at least three residues among R1A to R6A, and still more preferably at least four residues among R1A to R6A are a group selected from -CH2-CH(OH)-R7A, -CH(R7A)-CH2-OH,
-CH2-CH2-(C=O)-O-R7A, -CH2-CH2-(C=O)-NH-R7A and -CH2-R7A wherein R7A is selected from C3-C18 alkyl and C3-C18 alkenyl having one C-C double bond.
More preferably, R1A to R6A are independently selected from hydrogen and a group -CH2-CH(OH)-R7A wherein R7A is selected from C3-C18 alkyl and C3-C18 alkenyl having one C-C double bond; provided that at least two residues among R1A to R6A, more preferably at least three residues among R1A to R6A, and still more preferably at least four residues among R1A to R6A are a group -CH2-CH(OH)-R7A, wherein R7A is selected from C3-C18 alkyl and C3- C18 alkenyl having one C-C double bond.
Preferably, R7A is selected from C8-C18 alkyl and C8-C18 alkenyl having one C-C double bond, and more preferably from C8-C12 alkyl and C8-C12 alkenyl having one C-C double bond. Generally, alkyl groups are preferred over alkenyl groups as R7A.
As far as any of the groups R1A to R6A is a protecting group for an amino group, such as described for example in W02006/138380, preferred embodiments thereof are t- butoxycarbonyl (Boc), 9-fluorenylmethoxycarbonyl (Fmoc), or carbobenzyloxy (Cbz).
As far as any of the groups R1A to R6A are a receptor ligand, useful examples are given in Philipp and Wagner in “Gene and Cell Therapy - Therapeutic Mechanisms and Strategy”, 3rd Edition, Chapter 15. CRC Press, Taylor & Francis Group LLC, Boca Raton 2009. Preferred receptor ligands for lung tissue are described in Pfeifer et al. 2010, Ther Deliv. 1 (1 ):133-48. Preferred receptor ligands include synthetic cyclic or linear peptides such as derived from screening peptide libraries for binding to a particular cell surface structure or particular cell type, cyclic or linear RGD peptides, synthetic or natural carbohydrates such as sialic acid, galactose or mannose or synthetic ligands derived from reacting a carbohydrate for example with a peptide, antibodies specifically recognizing cell surface structures, folic acid, epidermal growth factor and peptides derived thereof, transferrin, anti-transferrin receptor antibodies, nanobodies and antibody fragments, or approved drugs that bind to known cell surface molecules. As far as any of the groups R1A to R6A are a polyethylene glycol) chain, the preferred molecular weight of the polyethylene glycol) chain is 100 - 20,000 g/mol, more preferably 1 ,000 - 10,000 g/mol and most preferred is 1 ,000 - 5,000 g/mol.
The variable p in formula (L-1 ) is preferably 1 .
In formula (L-1), m is 1 or 2; n is 0 or 1 and m+n is > 2. In other words, if m is 1 , n must also be 1 , and if m is 2, n can be 0 or 1 . If n is 0, m must be 2. If n is 1 , m can be 1 or 2.
The variable n in formula (L-1 ) is preferably 1 . It is more preferred that m is 1 and n is 1 .
Thus, the combination of p = 1 , m = 1 and n = 1 is likewise preferred.
As for the variables a and b in formula (L-1 ), a is 1 or 2 and b is an integer of 1 to 4, or a is an integer of 1 to 4 and b is 1 or 2. Thus, for example, a can be 1 or 2 and b can, independently, also be 1 or 2. Preferably, a is 1 and b is an integer of 2 to 4 or a is an integer of 2 to 4 and b is 1 , and it is more preferred that one of a and b is 1 , and the other one is 2 or 3. It is still more preferred that a is 1 and b is 2, or that a is 2 and b is 1 . Most preferably, a is 1 and b is 2.
In view of the above, it is further preferred that the compound of formula (L-1 ) is a compound of formula (L-1 a) and that the ionizable lipidoid in the lipidoid nanoparticles as referred to herein comprises, or more preferably consists of, an ionizable lipidoid of the following formula (L-1 a) or a protonated form thereof:
R1A-NR2A-CH2-(CH2)a-NR3A-CH2-(CH2)b-NR4A-CH2-(CH2)a-NR5A-R6A (L-1a), wherein a, b, and R1A to R6A are defined as in formula (L-1 ), including exemplary and preferred embodiments thereof.
In the protonated form of the compound of formula (L-1 a), one or more of the nitrogen atoms contained in the compound of formula (L-1 a) are protonated to provide a compound carrying a positive charge.
In accordance with a still further preferred embodiment, the ionizable lipidoid in the lipidoid nanoparticles as referred to herein comprises, or more preferably consists of, an ionizable lipidoid of the following formula (L-1b) or a protonated form thereof: wherein R1A to R6A are defined as in formula (L-1), including preferred embodiments thereof;
wherein R1A to R6A are defined as in formula (L-1), including preferred embodiments thereof;

In the protonated form of the compound of formula (L-1 b), one or more of the nitrogen atoms contained in the compound of formula (L-1 b) are protonated to provide a compound carrying a positive charge.
Thus, in a accordance with a particularly preferred embodiment, the ionizable lipidoid in the lipidoid nanoparticles as referred to herein comprises, or more preferably consists of, an ionizable lipidoid of the formula (L-1b) or a protonated form thereof, wherein R7A is selected from C8-C18 alkyl and C8-C18 alkenyl having one C-C double bond, provided that at least two residues among R1A to R6A are -CH2-CH(OH)-R7A, more preferably at least three residues among R1A to R6A, and still more preferably at least four residues among R1A to R6A are -CH2- CH(OH)-R7A, wherein R7A is selected from C8-C18 alkyl and C8-C18 alkenyl having one C-C double bond.
As an example of a suitable lipidoid compound that can be used as an ionizable lipidoid in the context of the invention, reference can be made to the lipidoid dL_05(R) with the following structure, or to a protonated form thereof wherein one or more of the nitrogen atoms contained in the compound are protonated:

In accordance with a further exemplary embodiment, the ionizable lipid which may be comprised by the nanoparticles referred to herein comprises or consists of an ionizable lipid of formula (L-2)

Preferably, the compound of formula (L-2) has the following structure:

In accordance with another exemplary embodiment, the ionizable lipid which may be comprised by the nanoparticles referred to herein comprises or consists of an ionizable lipid of formula (L-3) wherein
wherein

R1C and R2C are independently selected from a C8-C18 alkyl group and a C8-C18 alkenyl group, preferably from a C12-C18 alkyl group and a C12-C18 alkenyl group,
R3C is a C1-C6 alkanediyl group, preferably a C2 or C3 alkanediyl group, and R4C and R5C are independently hydrogen or C1-C3 alkyl, and are preferably methyl; or a protonated form thereof, wherein one or more of the nitrogen atoms contained in the compound of formula (L-3) are protonated to provide a compound carrying a positive charge.
As an example of an ionizable lipid of formula (L-3), reference can be made to DLin-MC3-DMA (6Z,9Z,28Z,31Z)-heptatriaconta-6,9,28,31-tetraene-19-yl 4-(dimethylamino)butanoate).
In accordance with still another exemplary embodiment, the ionizable lipid which may be comprised by the nanoparticles referred to herein comprises or consists of an ionizable lipid of formula (L-4) wherein
wherein

R1D and R2D are independently selected from a C8-C18 alkyl group and a C8-C18 alkenyl group, preferably from a C12-C18 alkyl group and a C12-C18 alkenyl group,
R3D is a C1-C6 alkanediyl group, preferably a C2 alkanediyl group, and
R4D and R5D are independently hydrogen or C1-C3 alkyl, and are preferably methyl; or a protonated form thereof, wherein one or more of the nitrogen atoms contained in the compound of formula (L-4) are protonated to provide a compound carrying a positive charge.
In accordance with still another exemplary embodiment, the ionizable lipid which may be comprised by the nanoparticles referred to herein comprises or consists of an ionizable lipidoid of formula (b-5) wherein R1E to R5E are independently of each other selected from hydrogen, -CH2-CH(OH)-R7E, -CH(R7E)-CH2-OH,
wherein R1E to R5E are independently of each other selected from hydrogen, -CH2-CH(OH)-R7E, -CH(R7E)-CH2-OH,

-CH2-CH2-(C=O)-O-R7E, -CH2-CH2-(C=O)-NH-R7E and -CH2-R7Ewherein R7E is selected from C3-C18 alkyl or C3-C18 alkenyl having one C-C double bond, provided that at least two residues among R1E to R5E are selected from -CH2-CH(OH)-R7E, -CH(R7E)- CH2-OH, -CH2-CH2-(C=O)-O-R7E, -CH2-CH2-(C=O)-NH-R7E and -CH2-R7E wherein R7E is selected from C3-C18 alkyl or C3-C18 alkenyl having one C-C double bond; or a protonated form thereof, wherein one or more of the nitrogen atoms contained in the compound of formula (L-5) are protonated to provide a compound carrying a positive charge.
In formula (L-5), R1E to R5E are preferably independently -CH2-CH(OH)-R7E, wherein R7E is selected from C8-C18 alkyl or C8-C18 alkenyl having one C-C double bond.
Still another exemplary ionizable lipid suitable for use in the present invention which may be comprised as an ionizable lipid in the nanoparticles referred to herein, or of which the ionizable lipid may consist is the ionizable lipid disclosed as “cationic lipid of Formula I” in the PCT application WO 2012/000104 A1 , starting on page 104 of this document, and including all specific embodiments thereof also discussed in this document.
Further exemplary ionizable lipidoids suitable for use in the present invention which may be comprised as an ionizable lipidoid in the nanoparticles referred to herein, or of which the ionizable lipidoids may consist are the ionizable lipidoids disclosed and claimed as “aminoalcohol lipidoids” in the PCT application WO 2010/053572 A2, including the compounds of all of the general formulae shown in the summary of the invention on page 4 of the document, and further defined in the remaining application.
Still further exemplary ionizable lipidoids suitable for use in the present invention which may be comprised as an ionizable lipidoid in the nanoparticles referred to herein, or of which the ionizable lipidoids may consist are the ionizable lipidoids disclosed as amine containing lipidoids of formulae I to V in the PCT application WO 2014/028487 A1 , including specific embodiments thereof.
A further preferred example of an ionizable lipid suitable for use in the present invention which may be comprised as an ionizable lipid in the nanoparticles referred to herein, or of which the ionizable lipid may consist is the ionizable lipid ((4-hydroxybutyl)azanediyl)bis(hexan-6,1- diyl)bis(2-hexyldecanoate) (ALC-0315) or a protonated form thereof, wherein the nitrogen atom of the compound is protonated to provide a compound carrying a positive charge.
Still a further preferred example of an ionizable lipid suitable for use in the present invention which may be comprised as an ionizable lipid in the nanoparticles referred to herein, or of which the ionizable lipid may consist is the ionizable lipid (6Z,9Z,28Z,31Z)-heptatriaconta- 6,9,28,31 -tetraene-19-yl 4-(dimethylamino)butanoate or a protonated form thereof wherein the nitrogen atom of the compound is protonated to provide a compound carrying a positive charge.
Yet a further preferred example of an ionizable lipid as an ionizable lipid in the nanoparticles referred to herein, or of which the ionizable lipid may be comprised in component (b) or of which component (b) may consist is the ionizable lipid heptadecan-9-yl 8-((2-hydroxyethyl)(6- oxo-6-(undecyloxy)hexyl)amino)octanoate (SM-102) or a protonated form thereof, wherein the nitrogen atom of the compound is protonated to provide a compound carrying a positive charge.
As preferred optional components in to at least one selected from a permanently cationic lipid, an ionizable lipid and an ionizable lipidoid, the nanoparticles may comprise, typically as a component of the lipid mix or lipidoid mix, one or more of the following components (c1 ) to (c6):
(c1) a non-ionizable lipid having a sterol structure;
(c2) a phosphoglyceride lipid;
(c3) a PEG-conjugated lipid;
(c4) a polysarcosine-conjugated lipid;
(c5) a PASylated lipid; and
(c6) a cationic polymer.
As will be understood by the skilled reader, the possibility that the nanoparticles comprise one or more of the components (c1 ) to (c6) not only encompasses combinations among (c1 ) to (c6), but also combinations of different components of one type, e.g. two components (c2), or combinations of different components of one type with other components of (c1 ) to (c6).
Component (c1) is a lipid having a sterol structure. As such, suitable lipids are compounds which have a steroid core structure with a hydroxyl group at the 3-position of the A-ring.
An exemplary non-ionizable lipid having a sterol structure which may be comprised by component (c1 ) or of which component (c1 ) may consist has a structure of formula (c1 -1 )

Further exemplary non-ionizable lipids having a sterol structure which may be comprised by component (c1 ) or of which component (c1 ) may consist include those disclosed by S. Patel et al., Naturally-occurring cholesterol analogues in lipid nanoparticles induce polymorphic shape and enhance intracellular delivery of mRNA, Nature Communications, 2020, 11 :983, in particular those illustrated in Fig. 2 of the publication.
Preferably, component (c1 ) comprises or consists of cholesterol.
Component (c2) is a phosphoglyceride.
Preferably, component (c2) comprises or consists of a phospholipid selected from a compound of formula (c2-1 ) wherein
wherein

R1F and R2F are independently selected from a C8-C18 alkyl group and a C8-C18 alkenyl group, preferably from a C12-C18 alkyl group and a C12-C18 alkenyl group, or a pharmaceutically acceptable salt thereof; and a phospholipid of formula (c2-2) wherein
wherein

R1G and R2G are independently selected from a C8-C18 alkyl group and a C8-C18 alkenyl group, preferably from a C12-C18 alkyl group and a C12-C18 alkenyl group, or a pharmaceutically acceptable salt thereof.
More preferably, component (c2) comprises or consists of 1 ,2-dipalmitoyl-sn-glycero-3- phosphocholine (DPPC) or a pharmaceutically acceptable salt thereof or 1 ,2-Distearoyl-sn- glycero-3-phosphocholine (DSPC) or a pharmaceutically acceptable salt thereof.
Exemplary salt forms of the compound of formula (c2-1 ) include salts formed by the acidic -OH group with a base, or salts formed by the amino group with an acid. As salts formed with a base, mention may be made of alkali metal salts such as sodium or potassium salts; alkaline- earth metal salts such as calcium or magnesium salts and ammonium salts. As exemplary salts formed with an acid, mention may be made of a salt formed with the acidic groups of the nucleic acid, but other salts are not excluded, and mineral acid salts such as chloride, bromide, or iodide, sulfate salts, nitrate salts, phosphate salts, hydrogenphosphate salts, or dihydrogenphosphate salts, carbonate salts, and hydrogencarbonate salts may be mentioned as examples.
Exemplary salt forms of the compound of formula (c2-2) include salts formed by the acidic -OH group attached to the P atom with a base, or salts formed by the quaternary amino group with an anion. As salts formed with a base, mention may be made of alkali metal salts such as sodium or potassium salts; alkaline-earth metal salts such as calcium or magnesium salts and ammonium salts. As exemplary salts formed with anion, mention may be made of a salt formed with the acidic groups of the nucleic acid, but other salts are not excluded, and mineral acid salts such as chloride, bromide, or iodide, sulfate salts, nitrate salts, phosphate salts, hydrogenphosphate salts, or dihydrogenphosphate salts, carbonate salts, and hydrogencarbonate salts may be mentioned as examples. Component (c3) is a PEG-conjugated lipid, i.e. a lipid which is covalently linked with a polyethylene glycol chain.
Preferably, component (c3) comprises or consists of a PEG-conjugated lipid selected from a compound of formula (c3-1) wherein
wherein

R1H and R2H are independently selected from a C8-C18 alkyl group and a C8-C18 alkenyl group, preferably from a C12-C18 alkyl group and a C12-C18 alkenyl group, and p is an integer of 5 to 200, preferably 10 to 100, and more preferably 20 to 60; wherein
wherein

R1J and R2J are independently selected from a C8-C18 alkyl group and a C8-C18 alkenyl group, preferably from a C12-C18 alkyl group and a C12-C18 alkenyl group, and q is an integer of 5 to 200, preferably 10 to 100, and more preferably 20 to 60 or a pharmaceutically acceptable salt thereof, or a compound of formula (c3-3) wherein
wherein

R1K and R2K are independently a C8-C18 alkyl group or a C8-C18 alkenyl group, preferably a C12-C18 alkyl group or a C12-C18 alkenyl group, and q is an integer of 5 to 200, preferably 10 to 100, and more preferably 20 to 60.
Exemplary salt forms of the compound of formula (c3-2) include salts formed by the acidic -OH group attached to the P atom with a base. As salts formed with a base, mention may be made of alkali metal salts such as sodium or potassium salts; alkaline-earth metal salts such as calcium or magnesium salts and ammonium salts.
More preferably, component (c3) comprises or consists of 1 ,2-dimyristoyl-sn- glycerolmethoxy(polyethylene glycol) (DMG-PEG), and still more preferably component d) comprises or consists of 1 ,2-dimyristoyl-sn-glycerolmethoxy(polyethylene glycol)-2000 (DMG- PEG2k) or 2-[(polyethylenglycol)-2000]-N,N-ditetradecylacetamid (ALC-0159).
Component (c4) is a polysarcosine-conjugated lipid, i.e. a lipid which is covalently linked with a polymeric moiety of the formula (c4-1 ):
-[C(O)-CH2-N(CH3)]r (c4-1 ) wherein r denotes the number of repeating units, and is preferably 10 to 100. An example of poly sarcosine based lipid is N-TETAMINE-pSar25.
Component (c5) is a PASylated lipid, e.g. a lipid which is covalently linked with a polymeric moiety formed by proline (pro)/alanine (ala)/serine (ser) repetitive residues.
With regards to the PASylated lipid used herein, the content of WO 2017/109087 A1 and EP 3394266 B1 is incorporated herein by reference. In particular the definitions and embodiments as recited below as incorporated herein by reference, specifically the embodiments reciting a nucleic acid coding for a PAS polypeptide. The PASylated lipid can comprise e.g. a polypeptide consisting of at least 100 amino acid residues of proline, alanine and, optionally, serine, wherein said polypeptide forms a random coil. Component (c6) is a cationic polymer. Such polymers suitable for use in the formation of nanoparticles comprising a nucleic acid are known in the art. Exemplary suitable cationic polymers are discussed in A.C. Silva et al., Current Drug Metabolism, 16, 2015, 3-16, and in the literature referred to therein, in J.C. Kasper et al., J. Contr. Rel. 151 (2011 ), 246-255, in WO 2014/207231 and in the literature referred to therein, and in WO 2016/097377 and in the literature referred to therein.
Suitable cationic oligomers or polymers include in particular cationic polymers comprising a plurality of units wherein an amino group is contained. The amino groups may be protonated to provide the cationic charge of the polymer.
Polymers are preferred which comprise a plurality of units independently selected from the following (1 ), (2), (3) and (4):
-CH2-CH2-NH- (1 ) wherein one or more of the nitrogen atoms of the repeating units (1 ), (2), (3) and/or (4) may be protonated to provide the cationic charge of the polymer.
wherein one or more of the nitrogen atoms of the repeating units (1 ), (2), (3) and/or (4) may be protonated to provide the cationic charge of the polymer.

Particularly preferred as cationic polymers are the following four classes of polymers comprising a plurality of units wherein an amino group is contained.
As the first preferred class, poly(ethylene imine) (“PEI”) is mentioned, including branched poly(ethylene imine) (“brPEI”).
The second preferred class of cationic polymers are polymers comprising a plurality of groups of the following formula (c6-1 ) as a side chain and/or as a terminal group, as they are disclosed as groups of formula (II) in WO 2014/207231 (which is incorporated in its entirety herein, applicant Ethris GmbH):
R2 R4
- N— {CH2— (CH2)— N— [CH2— (CH2)b- N]p}— [CH2- (CH2)- N]— R6 R3 R5 (C6-1 ) wherein the variables a, b, p, m, n and R2 to R6 are defined as follows, independently for each group of formula (c6-1 ) in a plurality of such groups: a is 1 or 2 and b is an integer of 1 to 4 or a is an integer of 1 to 4 and b is 1 or 2, preferably a is 1 and b is an integer of 2 to 4 or a is an integer of 2 to 4 and b is 1 , p is 1 or 2, m is 1 or 2; n is 0 or 1 and m+n is > 2; and
R2 to R5 are, independently of each other, selected from hydrogen, a group -CH2-CH(OH)-R7,-CH(R7)-CH2-OH, -CH2-CH2-(C=O)-O-R7, -CH2-CH2-(C=O)-NH-R7, -CH2-R7,a protecting group for an amino group, and a poly(ethylene glycol) chain wherein R7 is selected from C3-C18 alkyl or 03-018 alkenyl having one C-C double bond;
R6 is selected from hydrogen, a group -CH2-CH(OH)-R7, -CH(R7)-CH-OH, -CH2-CH2-(C=O)-O-R7, -CH2-CH2-(C=O)-NH-R7,-CH2-R7a protecting group for an amino group, -C(NH)-NH2, a poly(ethylene glycol) chain, and a receptor ligand, wherein R7 is selected from C3-C18 alkyl or C3-C18 alkenyl having one C-C double bond; and wherein one or more of the nitrogen atoms indicated in formula (c6-1 ) may be protonated to provide a cationic group of formula (c6-1 ).
As regards further preferred definitions of these polymers, and of the variables contained in formula (c6-1) above, the respective disclosure in WO 2014/207231 with regard to the groups of formula (II) as disclosed in this document also applies for the invention described herein.
The third preferred class of cationic polymers are polymers comprising a plurality of groups of the following formula (c6-2) as repeating units, as they are disclosed as groups of formula (III) in WO 2014/207231 (which is incorporated in its entirety herein, applicant Ethris GmbH): - N— {CH2— (CH2)— N— [CH2— (CH2)b- N]p}— [CH2- (CH2)- N]—
R3 r5 (C6-2) wherein the variables a, b, p, m, n and R2 to R5 are defined as follows, independently for each group of formula (c6-2) in a plurality of such groups: a is 1 or 2 and b is an integer of 1 to 4 or a is an integer of 1 to 4 and b is 1 or 2, preferably a is 1 and b is an integer of 2 to 4; or a is an integer of 2 to 4 and b is 1 , p is 1 or 2, m is 1 or 2; n is 0 or 1 and m+n is > 2; and
R2to R5 are, independently of each other, selected from hydrogen, a group -CH2-CH(OH)-R7, -CH(R7)-CH2-OH, -CH2-CH2-(C=O)-O-R7, -CH2-CH2-(C=O)-NH-R7, -CH2-R7, a protecting group for an amino group, -C(NH)-NH2, and a poly(ethylene glycol) chain; and wherein one or more of the nitrogen atoms indicated in formula (c6-2) may be protonated to provide a cationic group of formula (c6-2).
As regards further preferred definitions of these polymers, and of the variables contained in formula (c6-2) above, the respective disclosure in WO 2014/207231 with regard to the repeating units of formula (III) as disclosed in this document also applies for the invention described herein.
The fourth preferred class of cationic polymers is provided by a statistical copolymer as it is disclosed in WO 2016/097377 (which is incorporated in its entirety herein, applicant Ethris GmbH). It comprises a plurality of repeating units (a) independently selected from repeating units of the following formulae (a1) and (a2):
-CH2-CH2-NH- (a1) and a plurality of repeating units (b) independently selected from repeating units of the following formulae (b1 ) to (b4): -CH2-CH2-CH2-NH- (b1 )
and a plurality of repeating units (b) independently selected from repeating units of the following formulae (b1 ) to (b4): -CH2-CH2-CH2-NH- (b1 ) and the molar ratio of the sum of the repeating units (a) to the sum of the repeating units (b) lies within the range of 0.7/1 .0 to 1.0/0.7, and one or more of the nitrogen atoms of the repeating units (a) and/or (b) contained in the copolymer may be protonated to provide a cationic copolymer.
and the molar ratio of the sum of the repeating units (a) to the sum of the repeating units (b) lies within the range of 0.7/1 .0 to 1.0/0.7, and one or more of the nitrogen atoms of the repeating units (a) and/or (b) contained in the copolymer may be protonated to provide a cationic copolymer.


As regards further preferred definitions of this copolymer, the respective disclosure in WO 2016/097377 also applies for the invention described herein. As noted therein, a particularly preferred copolymer is a linear copolymer which comprises repeating units (a1 ) and (b1 ), or which consists of repeating units (a1) and (b1 ).
As an optional component of the nanoparticles, a polyanion component which is different from a nucleic acid may also be comprised, especially in addition to a nucleic acid if the nanoparticles comprise a nucleic acid as the preferred therapeutic agent. Examples of such a polyanion are polyglutamic acid and chondroitin sulfate. If such a polyanion component different from the nucleic acid is used in the nanoparticles, its amount is preferably limited such that the amount of anionic charges provided by the polyanion component is not higher than the amount of the anionic charges provided by the nucleic acid.
Preferably, the nanoparticles as referred to herein comprise, more preferably consist of: i) optionally the therapeutic agent as discussed above as a component (a), preferably a nucleic acid, more preferably RNA, still more preferably mRNA, ii) the lipid or lipidoid mix comprising or consisting of: as a component (b) at least one selected from a permanently cationic lipid, an ionizable lipid and an ionizable lipidoid, more preferably an ionizable lipid or ionizable lipidoid, and as a preferred further component of the lipid or lipidoid mix one or more of:
-the non-ionizable lipid having a sterol structure (c1 );
-the phosphoglyceride lipid (c2);
-the PEG-conjugated lipid (c3); the polysarcosine-conjugated lipid (c4); the PASylated lipid (c5); the cationic polymer (c6).
Exemplary suspensions comprising nanoparticles formed from the components listed above, which are also suitable for use as nanoparticle formulations in the context of the present invention, include those disclosed by S. Patel et al., Naturally-occurring cholesterol analogues in lipid nanoparticles induce polymorphic shape and enhance intracellular delivery of mRNA, Nature Communications, 2020, 11 :983.
It will be understood that components of the nanoparticles, and in particular components (a) and (b), and optionally one or more of (c1 ) to (c6), are typically contained as a mixture in the nanoparticles.
It is particularly preferred that the lipid or lipidoid mix comprises the components (c1 ), (c2) and (c3).
In terms of the amounts of these components, it is further preferred that the nanoparticles comprise or consist of: optionally the therapeutic agent as a component (a), and the lipid or lipidoid mix comprising or consisting of i) 30 to 65 mol% of the at least one selected from a permanently cationic lipid, an ionizable lipid and an ionizable lipidoid as a component (b), preferably of the ionizable lipid or ionizable lipidoid, and, as a further component of the lipid or lipidoid mix, one or more of the following components: ii) 10 to 50 mol% of the lipid having a sterol structure (c1 ), iii) 4 to 50 mol% of the phosphoglyceride lipid (c2), iv) 0.5 to 10 mol% of one of the PEG-conjugated lipid (c3), the polysarcosine-conjugated lipid (c4) and the PASylated lipid (c5), or of any combination thereof, v) 0.5 to 10 mol% of the cationic polymer (c6), such that the sum of i) and ii) to v) amounts to 100 mol%.
As regards the 30 to 65 mol% of the at least one selected from a permanently cationic lipid, an ionizable lipid and an ionizable lipidoid as component (b), it will be understood that the indicated molar percentage refers to the total amount of these constituents of the nanoparticle if two or more of the cationic lipid or lipidoid, an ionizable lipid and an ionizable lipidoid are present as component (b). Likewise, it will be understood that the molar percentages for components (c1 ) to (c6) are indicated with the proviso that not all of these components need to be present in the nanoparticles. Thus, for example, the cationic polymer can be present or absent in the context of this preferred embodiment, but if it is present, it is used in the amount of 0.5 to 10 mol%. As further indicated above, the amount of component(s) (c1), (c2), (c3), (c4), (c5) and/or (c6) in the context of this preferred embodiment is such that the sum of (b) and (c1) to (c6) amounts to 100 mol%.
It is still further preferred that the nanoparticles comprise, or consist of optionally the therapeutic agent as a component (a), which is more preferably mRNA, and the lipid or lipidoid mix comprising or consisting of at least one selected from a permanently cationic lipid, an ionizable lipid and an ionizable lipidoid as a component (b), preferably the ionizable lipid or ionizable lipidoid, the non-ionizable lipid having a sterol structure (c1), the phosphoglyceride lipid (c2), and the PEG-conjugated lipid (c3).
In terms of the amounts of these components, it is still further preferred that the nanoparticles comprise, more preferably consist of: optionally the therapeutic agent as a component (a), and the lipid or lipidoid mix comprising or consisting of
30 to 65 mol% of the at least one selected from a permanently cationic lipid, an ionizable lipid and an ionizable lipidoid (b), preferably of the ionizable lipid or ionizable lipidoid,
10 to 50 mol% of the lipid having a sterol structure (c1),
4 to 50 mol% of the phosphoglyceride lipid (c2), and
0.5 to 10 mol% of the PEG-conjugated lipid (c3), such that the sum of (b) and (c1 ) to (c3) amounts to 100 mol%.
In line with the above information related to preferred therapeutic agents, in particular nucleic acids and related to the preferred components of the lipid composition other than the therapeutic agent, the lipidoid nanoparticles in the context of the present invention preferably comprise
(a) mRNA as a therapeutic agent, and a lipidoid mix comprising
(b) an ionizable lipidoid of formula (L-1 b)

(L-1b) wherein R1A to R6A are independently selected from hydrogen and -CH2-CH(OH)-R7A, wherein R7A is selected from C8-C18 alkyl and C8-C18 alkenyl having one C-C double bond, provided that at least two residues among R1A to R6A are -CH2-CH(OH)-R7A, more preferably at least four residues among R1A to R6A are -CH2-CH(OH)-R7A, wherein R7A is selected from C8-C18 alkyl and C8-C18 alkenyl having one C-C double bond, or a protonated form thereof wherein one or more of the nitrogen atoms indicated in formula (L-1 b) are protonated to provide a cationic lipidoid;
(c1) a non-ionizable lipid having a sterol structure of formula (c1 -1 ) wherein R1L is a C3-C12 alkyl group;
wherein R1L is a C3-C12 alkyl group;

(c2) a phosphoglyceride of formula (c2-2) wherein R1G and R2G are independently selected from a C8-C18 alkyl group and a C8-C18 alkenyl group, preferably from a C12-C18 alkyl group and a C12-C18 alkenyl group, or a pharmaceutically acceptable salt thereof; and (c3) a PEG conjugated lipid of formula (c3-1 )
wherein R1G and R2G are independently selected from a C8-C18 alkyl group and a C8-C18 alkenyl group, preferably from a C12-C18 alkyl group and a C12-C18 alkenyl group, or a pharmaceutically acceptable salt thereof; and (c3) a PEG conjugated lipid of formula (c3-1 ) wherein R1H and R2H are independently selected from a C8-C18 alkyl group and a C8-C18 alkenyl group, preferably from a C12-C18 alkyl group and a C12-C18 alkenyl group, and p is an integer of 5 to 200, preferably 10 to 100, and more preferably 20 to 60. In such a lipidoid particle composition, the lipidoid dL_05(R) with the formula shown above would be a particularly preferred variant of the ionizable lipid.
wherein R1H and R2H are independently selected from a C8-C18 alkyl group and a C8-C18 alkenyl group, preferably from a C12-C18 alkyl group and a C12-C18 alkenyl group, and p is an integer of 5 to 200, preferably 10 to 100, and more preferably 20 to 60. In such a lipidoid particle composition, the lipidoid dL_05(R) with the formula shown above would be a particularly preferred variant of the ionizable lipid.


Another preferred exemplary composition of lipid nanoparticles suitable for use in the context of the present invention comprises a nucleic acid, more preferably mRNA, as a therapeutic agent, and a lipid mix comprising the ((4-hydroxybutyl)azandiyl)bis(hexan-6,1-diyl)bis(2- hexyldecanoate) or a protonated form thereof wherein the nitrogen atom of the compound is protonated to provide a compound carrying a positive charge as a ionizable lipid, and optionally further comprising one or more of the following components (d1) to (d8):
(d1) 2-[(polyethylene glycol)-2000]-N,N-ditetradecylacetamide (ALC-0159)
(d2) 1 ,2-distearoyl-sn-glycero-3-phosphocholine (DSPC)
(d3) cholesterol
(d4) potassium chloride
(d5) potassium dihydrogen phosphate
(d6) sodium chloride
(d7) disodium phosphate dihydrate
(d8) sucrose.
More preferably, they further comprise at least (d1 ), (d2) and (d3), and still more preferably they comprise all of (d1 ) to (d8).
Still another preferred exemplary composition of lipid nanoparticles suitable for use in the context of the present invention comprises a nucleic acid, more preferably mRNA, as a therapeutic agent, and a lipid mix comprising heptadecan-9-yl 8-((2-hydroxyethyl)(6-oxo-6- (undecyloxy)hexyl)amino)octanoate (SM-102) or a protonated form thereof wherein the nitrogen atom of the compound is protonated to provide a compound carrying a positive charge as an ionizable lipid, and optionally comprising one or more of the following components (e1) to (e7):
(e1) 1 ,2-distearoyl-sn-glycero-3-phosphocholine (DSPC),
(e2) cholesterol,
(e3) 1 ,2-dimyristoyl-rac-glycero-3-methoxypolyethylene glycol-2000 (PEG2000 DMG),
(e4) trometamol hydrochloride
(e5) sodium acetate trihydrate
(e6) acetic acid (e7) sucrose.
More preferably, they further comprise at least (e1 ), (e2) and (e3), and still more preferably they comprise all of (e1 ) to (e7).
Still another preferred exemplary composition of lipid nanoparticles suitable for use in the context of the present invention comprises a nucleic acid, more preferably mRNA, as a therapeutic agent, and a lipid mix comprising DLin-MC3-DMA ((6Z,9Z,28Z,31Z)- heptatriaconta-6,9,28,31-tetraen-19-yl-4-(dimethylamino) butanoate) or a protonated form thereof wherein the nitrogen atom of the compound is protonated, and optionally comprising one or more of the following components (e1) to (e7):
(e1) 1 ,2-distearoyl-sn-glycero-3-phosphocholine (DSPC),
(e2) cholesterol,
(e3) PEG2000-C-DMG (a-(3’-{[1 ,2-di(myristyloxy)propanoxy]carbonylamino}propyl)-(jj- methoxy, polyoxyethylene),
(e4) 2-amino-2-(hydroxymethyl)propan-1 ,3-diol (trometamol) hydrochloride
(e5) Disodium hydrogen phosphate, heptahydrate
(e6) Potassium dihydrogen phosphate, anhydrous
(e7) Sodium chloride
More preferably, components (e1), (e2) and (e3) are present, and still more preferably they comprise all of (e1) to (e6).
The nanoparticles preferably contain a therapeutic agent, preferably a nucleic acid, more preferably RNA and still more preferably mRNA, in which case the composition of the nanoparticles is preferably such that the weight ratio in the nanoparticles of the sum of the weights of components other than the therapeutic agent to the weight of the therapeutic agent is in the range of 50:1 to 1 :1 , more preferably 40:1 to 2:1 and most preferably 30:1 to 3:1 .
The N/P ratio, i.e. the ratio of the number of amine nitrogen atoms provided by the ionizable lipid, the ionizable lipidoid and/or the permanently cationic lipid to the number of phosphate groups provided by any nucleic acid of the nanoparticles, if a nucleic acid is comprised as a therapeutic agent, is preferably in the range of 0.5 to 20, more preferably in the range of 0.5 to 10.
The lipid or lipidoid nanoparticles, e.g. the lipid or lipidoid nanoparticles in a suspension formulation, preferably have a Z-average diameter in the range of 10 to 500 nm, more preferably in the range of 10 to 250 nm, still more preferably 20 to 200 nm. The indicated particle diameter is the hydrodynamic diameter of the particles, as determined by dynamic light scattering (DLS). Measurements are generally carried out at 25 °C.
The polydispersity index of the nanoparticles, e.g. of the lipid or lipidoid nanoparticles in a suspension formulation, is preferably in the range of 0.02 to 0.4, more preferably in the range of 0.03 to 0.2. The polydispersity index can be determined by dynamic light scattering (DLS). Measurements are generally carried out at 25 °C.
It is possible to provide a formulation, e.g. a suspension formulation, containing different lipid or lipidoid nanoparticles as defined above, i.e. particles which differ in terms of their components. However, preferably the nanoparticles contained in the formulation are composed of the same components.
The nanoparticles comprising an active agent, such as a nucleic acid, can be conveniently prepared by mixing a solution containing the active agent, e.g. in an aqueous solvent containing a buffer, such as a citrate buffer with a pH of 4.5, and optionally containing a salt such as sodium chloride, and a solution containing the ionizable lipid or ionizable lipidoid in an organic solvent, e.g. in ethanol. Further optional components can be incorporated e.g. by adding them to one of the two solutions. The nanoparticles generated in this manner can be further processed by chromatography and/or dialysis and/or tangential flow filtration (TFF) in order to obtain the nanoparticles in a desired liquid composition. Preferably, they are further processed using TFF.
In order to provide a nanoparticle suspension, it is also possible to rely on lyophilized nanoparticles prepared following the above-mentioned procedure followed by freeze drying, which are subsequently re-suspended in an aqueous vehicle solution.
A lipid nanoparticle formulation or lipidoid nanoparticle formulation as referred to herein is preferably a suspension formulation wherein the nanoparticles are contained in a liquid vehicle solution. The liquid vehicle solution is preferably an aqueous vehicle solution.
As will be understood by the skilled reader, an aqueous vehicle solution is a solution wherein the main solvent, in terms of the total volume of solvent(s), is water, preferably a solution containing more than 70 % of water, more preferably more than 90 % of water, as a solvent, indicated as the volume percentage of water in the total volume of solvent(s) contained in the vehicle solution (at a temperature of 25 °C). Most preferably, water is the only solvent in the vehicle solution. Thus, the vehicle solution is a liquid at room temperature (e.g. 25 °C). Likewise, an aqueous solvent as referred to herein is water or a mixture of solvents wherein water represents the main solvent, in terms of the total volume of solvent(s). Typically, the aqueous solvent contains more than 70 % of water, more preferably more than 90 % of water, as a solvent, indicated as the volume percentage of water in the total volume of solvent(s) contained in the aqueous solvent (at a temperature of 25 °C). Most preferably, water is the only solvent in the aqueous solvent.
The weight per volume ratio of the nanoparticles in the vehicle solution of a suspension formulation is preferably in the range 0.1 g/L to 300 g/L, more preferably 0.2 g/L to 300 g/L, still more preferably 0.5 g/L to 250 g/L and most preferably 0.5 g/L to 125 g/L (as measured at 25 °C).
If the nanoparticles comprise a nucleic acid as a therapeutic agent and are provided as a suspension formulation, the concentration of the nucleic acid provided by the lipid or lipidoid nanoparticles in the suspension preferably ranges from 0.01 to 10 mg/ml, more preferably from 0.02 to 10 mg/ml, still more preferably from 0.05 to 5 mg/mL, and most preferably from 0.05 to 2.5 mg/ml, based on the total volume of the suspension (as measured at 25 °C).
As noted above, the lipid or lipidoid nanoparticles contained in the suspension preferably have a Z-average diameter in the range of 10 to 500 nm, more preferably in the range of 10 to 250 nm, still more preferably 20 to 200 nm. The indicated particle diameter is the hydrodynamic diameter of the particles, as determined by dynamic light scattering (DLS). Measurements are generally carried out at 25 °C.
The polydispersity index of the nanoparticles contained in the suspension is preferably in the range of 0.02 to 0.4, more preferably in the range of 0.03 to 0.2. The polydispersity index can be determined by dynamic light scattering (DLS). Measurements are generally carried out at 25 °C.
SURFACTANTS
Diverse types of surfactants can be used in the context of the present invention. It is preferred that the surfactant comprises, more preferably is (or consists of) a nonionic surfactant. Examples of suitable nonionic surfactants include fatty alcohol ethoxylates, fatty acid ethoxylates, block copolymers of ethylene oxide and propylene oxide, alkylphenol ethoxylates or oligomers of alkylphenol ethoxylates, fatty acid esters of sorbitol, ethoxylated fatty acid esters of sorbitol, fatty acid esters of glycerol, ethoxylated castor oil and ethoxylated vitamin E.
Thus, the surfactant used in the context of the invention, e.g. as a component of the lipid nanoparticle (LNP) formulation or lipidoid nanoparticle (LiNP) formulation, preferably comprises, still more preferably is (or consists of) at least one surfactant selected from the group of fatty alcohol ethoxylates, fatty acid ethoxylates, block copolymers of ethylene oxide and propylene oxide, alkylphenol ethoxylates or oligomers of alkylphenol ethoxylates, fatty acid esters of sorbitol, ethoxylated fatty acid esters of sorbitol, fatty acid esters of glycerol, ethoxylated castor oil and ethoxylated vitamin E.
Preferably, the block copolymer of ethylene oxide and propylene oxide is a poloxamer. The poloxamer is preferably one which contains one polypropylene oxide) block B of formula (p-
1): wherein s is an integer of 15 to 60, and two poly(ethylene oxides) blocks A of formula (p-2):
wherein s is an integer of 15 to 60, and two poly(ethylene oxides) blocks A of formula (p-2): (P-2) wherein r is, independently for each block, an integer of 8 to 150, preferably 10 to 150.
(P-2) wherein r is, independently for each block, an integer of 8 to 150, preferably 10 to 150.


It is preferred that the surfactant used in the context of the invention comprises, or preferably is (or consists of), at least one selected from the group of poloxamer 124, poloxamer 188, poloxamer 338, poloxamer 407, polysorbate 20 or Tween-20, polysorbate 80 or Tween-80, polyoxyethylenelaurylether, poyloxyethylene-35 castor oil, D-a-tocopherol polyethylene glycol 1000 succinate, and tyloxapol.
It is particularly preferred that the surfactant comprises, more preferably is (or consists of), a poloxamer, e.g. the preferred poloxamer as discussed above, still more preferably a poloxamer selected from the list of poloxamer 124, poloxamer 188, poloxamer 338, and poloxamer 407, or from combinations thereof, most preferably P188.
It will be understood that a surfactant as referred to herein is typically a surfactant which meets the requirements set forth in the aspects of the invention as defined above, e.g. in the first, second or third aspect, or which has been classified as being suitable as a stabilizing agent by the method in accordance with the fourth or fifth aspect.
In the case of a lipid nanoparticle (LNP) formulation or lipidoid nanoparticle (LiNP) formulation in the form of a suspension formulation, such as an aqueous suspension formulation, it is preferred that the vehicle solution wherein the nanoparticles are suspended comprises the surfactant dissolved therein. As will be appreciated by the skilled reader, this does not exclude the possibility that a certain amount of the surfactant molecules is adsorbed to the lipid or lipidoid nanoparticles which are contained in the suspension.
In the context of the invention, it has been found that a beneficial effect of the surfactant can be achieved already with relatively low concentrations of a surfactant, e.g. of 0.01 % (w/v) in the suspension. Thus, typically, the surfactant is contained in amounts of 0.01 % (w/v) or more in the suspension, with regard to the total volume of the suspension of the nanoparticles in the vehicle solution (typically measured at 25 °C).
For example, the invention involves an incorporation of the surfactant into a suspension formulation, preferably into the vehicle solution, more preferably an aqueous vehicle solution, in an amount of 0.01 to 10 % (w/v), preferably 0.1 to 10 % (w/v), more preferably 0.25 to 5 % (w/v), still more preferably 0.33 to 2.5 % (w/v), even more preferably 0.45 to 1 .5 % (w/v), and most preferably 0.5 to 1 .5 % (w/v), with regard to the total volume of the suspension of the nanoparticles in the vehicle solution. As will be appreciated, the indication of the concentration of a substance in % (w/v) or (weight/volume) corresponds to the amount of the substance in g in a volume of 100 mL, typically measured at 25 °C, so that 1 % (w/v) corresponds to 1 g the surfactant per 100 mL of the total volume of the suspension.
Likewise, it is the methods in accordance with the invention may involve the incorporation of the surfactant into a nanoparticle suspension formulation in an amount of e.g. 0.01 to 10 % (w/v), preferably 0.1 to 10 % (w/v), more preferably 0.25 to 5 % (w/v), still more preferably 0.33 to 2.5 % (w/v), even more preferably 0.45 to 1 .5 % (w/v), and most preferably 0.5 to 1 .5 % (w/v), with regard to the total volume of the suspension of the nanoparticles in the aqueous vehicle solution (typically measured at 25 °C). While concentrations of 0.5 to 1 .5% (w/v) are particularly preferred, as outlined above, the invention in its various aspects also provides and relates to suspension formulations wherein the concentration of the surfactant is lower, e.g. in the range of 0.01 to 0.45 % (w/v), or 0.1 to 0.40 % (w/v).
In a suspension formulation of lipid nanoparticles or of lipidoid nanoparticles in a vehicle solution, preferably an aqueous vehicle solution in the context of the various aspects of the invention, it is generally preferred that the surfactant is essentially not attached to the nanoparticles, e.g. that it is essentially not contained in the nanoparticles and essentially not adhering to the nanoparticles. For example, more than 90 wt%, preferably more than 95 wt% of the total amount of the surfactant which is contained in or incorporated into the suspension of lipid nanoparticles or of lipidoid nanoparticles in an aqueous vehicle solution in the context of the various aspects of the invention, is present in the aqueous vehicle solution without adhering to the nanoparticles.
In addition to the surfactant, other excipients may be present in the vehicle solution. Preferably, the vehicle solution further comprises at least one of a sugar and a salt, more preferably sucrose and NaCI.
The surfactant can be conveniently incorporated into a nanoparticle suspension formulation, e.g. by a method including adding the surfactant to a suspension comprising an aqueous vehicle solution and the lipid or lipidoid nanoparticles, or including adding the lipid or lipidoid nanoparticles to an aqueous vehicle solution comprising the surfactant. For example, as noted above, if the nanoparticles are provided in lyophilized form, they can be re-suspended in an aqueous vehicle solution containing a surfactant.
To that extent, a method for the preparation of a suspension formulation of lipid nanoparticles or lipidoid nanoparticles as defined herein may comprise generating a preparation of lipid nanoparticles or lipidoid nanoparticles by mixing of at least one selected from a permanently cationic lipid, an ionizable lipid and an ionizable lipidoid dissolved in an organic phase with a therapeutic agent dissolved in an aqueous solution, and combining the nanoparticles with a surfactant to obtain a suspension of the nanoparticles in an aqueous vehicle solution.
Preferably, the method comprises the following steps: i) generating a preparation of lipid nanoparticles or lipidoid nanoparticles by mixing of at least one selected from a permanently cationic lipid, an ionizable lipid and an ionizable lipidoid dissolved in an organic phase with a therapeutic agent dissolved in an aqueous solution, ii) diluting the preparation of lipid nanoparticles or lipidoid nanoparticles by dilution with a first solution, iii) concentrating the diluted preparation of lipid nanoparticles or lipidoid nanoparticles by buffer exchange using ultra/diafiltration by TFF, wherein a second solution is used for the ultra/diafiltration, iv) obtaining a suspension of lipid nanoparticles or lipidoid nanoparticles in an aqueous vehicle solution comprising a surfactant, wherein the first solution comprises between 0.01 % w/v and 10% w/v surfactant, preferably between 0.1% w/v and 10% surfactant, more preferably between 0.25% w/v surfactant and 5% w/v surfactant, still more preferably between 0.33% w/v surfactant and 2.5% w/v surfactant, even more preferably between 0.45% w/v surfactant and 1 .5% w/v surfactant, most preferably between 0.5% w/v and 1.5% w/v surfactant, and/or wherein the second solution comprises between 0.01 % w/v and 10% w/v surfactant, preferably between 0.1 % w/v and 10% surfactant, more preferably between 0.25% w/v surfactant and 5% w/v surfactant, still more preferably between 0.33% w/v surfactant and 2.5% w/v surfactant, even more preferably between 0.45% w/v surfactant and 1 .5% w/v surfactant, most preferably between 0.5% w/v and 1.5% w/v surfactant, and wherein the final concentration of surfactant from combined first and second solution is between 0.01 % w/v and 10% w/v surfactant, preferably between 0.1 % w/v and 10% surfactant, more preferably between 0.25% w/v surfactant and 5% w/v surfactant, still more preferably between 0.33% w/v surfactant and 2.5% w/v surfactant, even more preferably between 0.45% w/v surfactant and 1 .5% w/v surfactant, most preferably between 0.5% w/v and 1 .5% w/v surfactant, with regard to the total volume of the suspension of the nanoparticles in the aqueous vehicle solution.
In the above method, it is preferred that an incorporation of a surfactant into the suspension does not occur before or during step i).
Moreover, it is preferred that the surfactant is added both with the first and the second solution. For example, wherein 30 to 70 wt% of the surfactant, preferably 40 to 60 wt%, and more preferably 45 to 55 wt% of the surfactant, based on the total weight of the surfactant in the suspension obtained in step iv), can be added with the first solution, and 70 to 30 wt% of the surfactant, preferably 60 to 40 wt%, and more preferably 55 to 45 wt% of the surfactant, based on the total weight of the surfactant in the suspension obtained in step iv), can be added with the second solution, such that the sum of the amount of surfactant added with the first and the second solution is 100 wt%. Generally, it is preferred that approximately half of the surfactant is added with the first solution and approximately half of the surfactant is added with the second solution.
In the context of the invention, lipid nanoparticle formulations or lipidoid nanoparticle formulations are provided which are stabilized by the use of a surfactant. As used herein, the term “stabilized” refers to the state of a lipid nanoparticle (LNP) formulation where its physical and chemical integrity is maintained over time under specified conditions. This includes preferably preserving particle size, structural integrity, and/or homogeneity, and preventing aggregation or fusion. Stabilization may also protect encapsulated therapeutic agents from degradation, ensuring sustained efficacy and controlled release. Surfactants, like poloxamers, aid in stabilization by reducing surface tension and preventing agglomeration, enhancing the overall stability of the lipid nanoparticle dispersion. Stability can be assessed, e.g., through particle size, encapsulation efficiency, mRNA integrity, and potency measurements. Particle size can be measured using Dynamic Light Scattering (DLS) with instruments such as the Malvern Zetasizer. Encapsulation efficiency can be quantified using assays like the Ribogreen assay. The integrity of mRNA can be assessed using a Fragment Analyzer. Potency can be measured using Enzyme-Linked Immunosorbent Assay (ELISA). For example, a stabilized lipid or lipidoid formulation can have a Z average size of 25 to 150 nm for extended periods of time such as weeks or months.
For example, by using a surfactant as described herein, preferably a nonionic surfactant, a nanoparticle suspension can be stabilized in the context of the invention e.g. against particle aggregation under a physical stress condition. To achieve this effect, the surfactant can be incorporated into the suspension, preferably incorporated as an excipient into the aqueous vehicle solution. In preferred embodiments, by using the surfactant as described herein, a membrane fouling can be avoided.
In some embodiments, the LNPs and/or LiNPs have not been lyophilized. In some embodiments, the surfactant is added before a lyophilization process. In some embodiments, the surfactant is not present in the vehicle solution during a lyophilization process.
Typically, the presence of the surfactant does not cause a change in the biological activity of the nanoparticle. Biological activity means expression level in the target cell(s) of the therapeutic nucleic acid. The biological activity can e.g. be quantified by in vitro transfection of cell lines (e.g. HEK-293) with the nanoparticle followed by quantification of the produced nucleic acid by Southern/northern blot or of protein via an ELISA. The detected protein level calculated as average of three measurements per concentration must not differ more than 10%, preferably not more than 5%, more preferably is not statistically different when performing the identical assay with the same LNP or LiNP without a surfactant.
Typically, the presence of the surfactant does not cause a change in the physical properties of the nanoparticle measured as the hydrodynamic diameter of the nanoparticle and as the proportion of encapsulated therapeutic agent, preferably a nucleic acid.
The hydrodynamic diameter of the nanoparticle can e.g. be measured via dynamic light scattering (also Photon correlation spectroscopy). Optionally, the average of three measurements of the hydrodynamic diameter of the nanoparticle in presents of the surfactant must not differ more than 5%, preferably not more than 1%, more preferably not statistically different of the same nanoparticle in absence of the surfactant. The viscosity change of the surfactant must be taken into account during the measurement. The percentage of encapsulated nucleic acid can e.g. be determined by measuring the fluorescence intensity in a RiboGreen assay. The nanoparticle is analyzed under two different conditions, untreated samples for external nucleic acid and samples treated with Triton X-100 for total mRNA. The percent content of encapsulated nucleic acid is calculated. Optionally, the value calculated form an average of three measurements of the nanoparticle in absence of the surfactant should not differ more than 5%, optionally not more than 3%, better not statistically different) from the same nanoparticle in presents of the surfactant.
As will be understood by the skilled reader, a measure which is taken for the stabilization of a nanoparticle suspension against particle aggregation may prevent the aggregation of the nanoparticles, or may reduce the degree of aggregation of the nanoparticles compared to a situation where the concerned measure is not applied. Preferably, the stabilization of the nanoparticle suspension is evidenced by an increase of the Z-average particle size of the suspended particles under a physical stress condition of less than 50 %, more preferably less than 20 %, still more preferably less than 10 % and most preferably by the absence of such an increase.
Similarly, it will be understood that stabilization of the nanoparticle suspension against particle aggregation under a physical stress condition means that an aggregation of the nanoparticles is prevented or reduced which would be observed in the absence of the stabilization when the nanoparticle suspension is exposed to a physical stress condition. Conditions of physical stress to which the nanoparticle suspension can be exposed are frequently physical stress conditions that are encountered during the handling or during a transport of the suspension. They include, e.g., a quick movement of a volume of the suspension which would cause a collision of nanoparticles contained in a non -stabilized suspension. As examples of a physical stress condition, reference may be made to shaking, stirring, vibrating, mixing, inverting, tapping, or dropping of the nanoparticle suspension, or, e.g., to a physical stress condition caused by pumping the nanoparticle suspension or by its withdrawal into a syringe. As will be understood by the skilled reader, conditions of physical stress include not only conditions to which the nanoparticle suspension is exposed during its regular handling, but also conditions to which the suspension may be exposed exceptionally (such as a transport under difficult conditions) or inadvertently (such as dropping a sample of the suspension).
FURTHER PHARMACEUTICAL ASPECTS
Pharmaceutical compositions as referred to herein, such as the lipid nanoparticle (LNP) formulations or lipidoid nanoparticle (LiNP) formulations as referred to herein comprising a therapeutic agent, are particularly useful in a medical setting and in the treatment or prevention of diseases and disorders, preferably in the treatment or prevention of a disease or disorder relying on a nucleic acid, such as RNA, preferably mRNA, as an active agent. Thus, such compositions are generally provided as or used as a medicament or as a pharmaceutical composition.
In particular, pharmaceutical compositions as referred to herein, such as the lipid nanoparticle (LNP) formulations or lipidoid nanoparticle (LiNP) formulations as referred to herein comprising a therapeutic agent, are suitable for administration to a subject. In this manner, the therapeutic agent, preferably the nucleic acid such as RNA, preferably the mRNA, contained in the nanoparticles can also be administered to the subject.
Via administration to a subject, the therapeutic agent, preferably the nucleic acid contained in the lipid or lipidoid nanoparticles particles, may be delivered to target cells. The term “delivered to target cells” preferably means transfer of the nucleic acid into the cell. The administration can be accomplished in various ways known to the skilled practitioner, including an administration to or via the respiratory tract, e.g. by an aerosolization of the nanoparticles, or an intramuscular or intravenous administration.
By administering the pharmaceutical composition, such as the lipid nanoparticle (LNP) formulations or lipidoid nanoparticle (LiNP) formulations as referred to herein comprising a therapeutic agent, to a subject, diseases or disorders can be treated or prevented. The term “disease” refers to any conceivable pathological condition that can be treated, prevented or vaccinated against by employing the suspension Said diseases may, e.g., be inherited, acquired, infectious or non-infectious, age-related, cardiovascular, metabolic, intestinal, neoplastic (in particular cancer) or genetic. A disease can be based, for example, on irregularities of physiological processes, molecular processes, biochemical reactions within an organism that in turn can be based, for instance, on the genetic equipment of an organism, on behavioral, social or environmental factors such as the exposure to chemicals or radiation.
Pharmaceutical compositions as referred to herein, such as the nanoparticle (LNP) formulations or lipidoid nanoparticle (LiNP) formulations comprising a therapeutic agent as referred to herein, may generally be suitable for the treatment or prevention of a disease selected from Table A as disclosed herein above.
Likewise, the pharmaceutical compositions, such as the nanoparticle (LNP) formulations or lipidoid nanoparticle (LiNP) formulations comprising a therapeutic agent as referred to herein,, may generally be suitable for the treatment or prevention of a disease selected disease selected from viral diseases, ciliopathies, or autoimmune diseases, and respiratory tract diseases, even more preferably selected from a lung disease, an airway disease or a nasal disease, more preferably a lung viral disease, lung ciliopathies and a lung autoimmune disease. Preferably, the lung disease or lung viral disease is at least one selected from pneumonia and asthma; the airway disease is at least one selected from bronchitis, viral induced asthma, lung fibrosis and COPD; and/or the nasal disease is at least one selected from rhinitis and sinusitis.
In some embodiments, the disease to be treated or prevented is a disease selected from the list consisting of pulmonary alveolar proteinosis (PAP), interstitial lung disease such as pulmonary fibrosis, e.g. idiopathic pulmonary fibrosis, viral infections such as Influenza and COVID-19, acute respiratory distress syndrome (ARDS), non-tuberculous mycobacterial (NTM) infection, lung cancer, fungal infections caused by Aspergillus sp., such as aspergillosis, fungal sinusitis, otomycosis, keratitis, and onychomycosis, preferably those caused by Aspergillus fumigatus and Aspergillus flavus, infections caused by Mycobacterium tuberculosis, Pseudomonas aeruginosa, Pneumocystis sp., Plasmodium sp., Cryptococcus sp., Nocardia sp., and combinations thereof. In some embodiments the disease is selected from the list consisting of (autoimmune) pulmonary alveolar proteinosis (PAP), interstitial lung disease such as pulmonary fibrosis, e.g. idiopathic pulmonary fibrosis, viral infections such as Influenza and COVID-19, acute respiratory distress syndrome (ARDS), non-tuberculous mycobacterial (NTM) infection, lung cancer, or fungal infections caused by Aspergillus sp., such as aspergillosis, fungal sinusitis, otomycosis, keratitis, and onychomycosis, preferably those caused by Aspergillus fumigatus and Aspergillus flavus, infections caused by Mycobacterium tuberculosis, Pseudomonas aeruginosa, Pneumocystis sp., and Plasmodium sp.
To that extent, the invention also provides a formulation of lipid nanoparticles or lipidoid nanoparticles comprising a therapeutic agent as described herein for use in the treatment or prevention of a disease. Likewise, the formulation of lipid nanoparticles or lipidoid nanoparticles according as described therein can be used in a method for the treatment or prevention of a disease, which method includes administering the suspension or formulation to a subject in need thereof.
In a related aspect, the invention also provides the formulation of lipid nanoparticles or lipidoid nanoparticles comprising a therapeutic agent as described herein for use as a medicament.
For example, the invention provides the formulation of lipid nanoparticles or lipidoid nanoparticles comprising a therapeutic agent as described herein for use in vaccination or immunization. Likewise, the formulation of lipid nanoparticles or lipidoid nanoparticles comprising a therapeutic agent as described herein can be used in a method for vaccination or immunization, which method includes administering the suspension or formulation to a subject in need thereof.
In line with a further aspect, the invention provides a method of inducing an immune response against a target pathogen in a subject in need thereof, the method comprising administering a formulation of lipid nanoparticles or lipidoid nanoparticles comprising a therapeutic agent as described herein to the subject.
In another example, the invention provides a formulation of lipid nanoparticles or lipidoid nanoparticles comprising a therapeutic agent as described therein for use in the treatment of cancer. Likewise, the formulation of lipid nanoparticles or lipidoid nanoparticles comprising a therapeutic agent as described therein can be used in a method for the treatment of cancer, which method includes administering the formulation to a subject in need thereof. In a further aspect, the invention provides a method of avoiding or for alleviating side effects in a therapy with lipid nanoparticles or lipidoid nanoparticles comprising at least one therapeutic agent as they are described herein, wherein the method comprises the steps: i) determine whether lipid nanoparticles or lipidoid nanoparticles in a pharmaceutical composition comprising the lipid nanoparticles or lipidoid nanoparticles aggregate when subjected to a mechanical stress or a temperature stress, by determining their aggregation level before and after subjecting said pharmaceutical composition to said mechanical or temperature stress ii) if the lipid nanoparticles or lipidoid nanoparticles show aggregation after the test of step (i), then add to the lipid nanoparticles or lipidoid nanoparticles formulation a surfactant as defined herein to obtain a LNP or LiNP suspension with a final surfactant concentration between 0.01 % w/v and 10% w/v surfactant, preferably between 0.1 % w/v and 10% surfactant, more preferably between 0.25% w/v surfactant and 5% w/v surfactant, still more preferably between 0.33% w/v surfactant and 2.5% w/v surfactant, even more preferably between 0.45% w/v surfactant and 1 .5% w/v surfactant, most preferably between 0.5% w/v and 1.5% w/v surfactant, iii) reconstitute with mixing to generate a stable suspension of lipid nanoparticles or lipidoid nanoparticles.
In a related aspect, the invention further provides a method of reducing one or more side effects associated with a vaccine formulation or an anticancer formulation comprising lipid nanoparticles or lipidoid nanoparticles carrying a therapeutic agent such as nucleic acid, preferably RNA, more preferably mRNA, as they are described herein, the method comprising modifying the vaccine formulation or an anticancer formulation by adding a surfactant as described herein to a vaccine formulation or anticancer formulation comprising a suspension of the lipid nanoparticles or lipidoid nanoparticles. Preferably, the surfactant represents between 0.01 % w/v and 10% w/v surfactant, preferably between 0.1 % w/v and 10% surfactant, more preferably between 0.25% w/v surfactant and 5% w/v surfactant, still more preferably between 0.33% w/v surfactant and 2.5% w/v surfactant, even more preferably between 0.45% w/v surfactant and 1 .5% w/v surfactant, most preferably between 0.5% w/v and 1 .5% w/v surfactant.
In a related aspect, the invention provides a method of reducing the occurrence or severity of one or more side effects associated with a LNP/LiNP based vaccine in a subject, the method comprising administering a vaccine formulation or an anticancer formulation comprising the formulation of lipid nanoparticles or lipidoid nanoparticles comprising a therapeutic agent as described herein to the subject. The reduction of the occurrence or severity of one or more side effects may be caused by a reduction of LNP/LiNP aggregation, as it may be measured, e.g., by determining the hydrodynamic diameter of the nanoparticle, for example by via dynamic light scattering or photon correlation spectroscopy.
It will be understood that, if a pharmaceutical composition, such as the formulation of lipid or lipidoid nanoparticles comprises a therapeutic agent, the therapeutic agent is included in an effective amount composition or formulation. The term "effective amount" refers to an amount sufficient to induce a detectable therapeutic response or a preventive effect in the subject to which the pharmaceutical composition or formulation is to be administered. In accordance with the above, the content of the therapeutic agent is not limited as far as it is useful for treatment or prevention as described above. For example, as noted above, a suspension formulation wherein nanoparticles comprising the nucleic acid are contained, preferably comprises the particles in an amount so as to provide the nucleic acid contained in the particles at a concentration of 0.01 to 10 mg/ml, more preferably 0.02 to 10 mg/ml, still more preferably 0.05 to 5 mg/ml and most preferably 0.05 to 2.5 mg/ml, based on the total volume of the composition. Likewise, it will be understood that, in cases where a pharmaceutical composition, such as a formulation of lipid nanoparticles or lipidoid nanoparticles according comprising a therapeutic agent as described therein is administered to a subject, it will be administered in an effective amount.
Exemplary subjects include a mammal such as a dog, cat, pig, cow, sheep, horse, rodent, e.g., rat, mouse, and guinea pig, or a primate, e.g., gorilla, chimpanzee, and human. In a most preferable embodiment, the subject is a human.
LANGMUIR BLODGETT TROUGH
The Langmuir-Blodgett (LB) technique is a method for preparing organized molecular assemblies with precision and control over molecular orientation and layer thickness. This methodology builds upon the principle that amphiphilic molecules, when spread on an aqueous subphase, can form stable monolayers at the air-water interface. The organization and packing density of these molecules can be meticulously manipulated by adjusting the surface pressure.
The Langmuir surface pressure/area (TT-A) isotherm is a foundational concept within the realm of surface science, e.g. when it comes to studying monolayers of amphiphilic molecules at the air-water interface. In a typical Langmuir setup, amphiphilic molecules (molecules possessing both hydrophilic and hydrophobic components) are spread onto an aqueous subphase, creating a monolayer at the air-water interface. The area available to these molecules can be altered using mobile barriers, which in turn changes the surface pressure (IT) of the monolayer. By plotting the changes in surface pressure (IT) as a function of the area (A) per molecule in the monolayer, one derives the TT-A isotherm.
Key Features of TT-A Isotherms are:
Phases of the Monolayer. Different regions on the isotherm correspond to different molecular arrangements or phases. These can include gaseous, liquid-expanded, and liquid-condensed phases.
Collapse Pressure: The pressure at which the monolayer undergoes a phase transition and collapses can be identified on the isotherm. It might be a relevant parameter as it provides insight into the stability and strength of the monolayer.
Molecular Area: The area occupied by a single molecule in the monolayer can be determined from the isotherm. This is particularly useful for understanding the geometry and orientation of the molecules.
Phase Transitions: Changes in the slope or abrupt transitions in the isotherm are indicative of phase changes within the monolayer. These can be used to deduce molecular interactions and behavior under different conditions.
Intermolecular Interactions: The shape and features of the isotherm can provide insights into the nature and strength of intermolecular forces in the monolayer.
In the context of the invention, it was surprisingly found that a compression -expansion isotherm can be used to test the suitability of a surfactant in lipid or lipidoid nanoparticle purification and processing.
* * *
In this specification, a number of documents including patent applications and manufacturer’s manuals are cited. The disclosure of these documents, while not considered relevant for the patentability of this invention, is herewith incorporated by reference in its entirety. More specifically, all referenced documents are incorporated by reference to the same extent as if each individual document was specifically and individually indicated to be incorporated by reference.
Brief description of the drawings:
Figure 1 shows a plot showing TFF processing time for different P188 solutions.
Figure 2 shows a bar diagram of the refractive index of different P188 solutions before and after TFF processing.
Figure 3 shows a Langmuir pressure-area plot for four representative poloxamer 188 samples in aqueous solutions. Considering the isotherm maximum surface pressure, two clear groups of poloxamers can be distinguished. S8 and S9 with a maximum surface pressure above 4 mN/m correspond to poloxamer P188 samples that lead to tangential flow filtration membrane fouling while samples S2 and S7 are suitable for lipid nanoparticle purification and processing.
Figure 4 shows a Langmuir pressure-area plot of three consecutive compression-expansion cycles for representative poloxamer 188 samples S1 to S11 in aqueous solutions. Considering the isotherm maximum surface pressure, samples S8 and S9 clearly stand out with a maximum surface pressure above 4 mN/m, identifying poloxamer P188 samples that lead to tangential flow filtration membrane fouling.
Figure 5 shows a Langmuir pressure-area plot for four representative poloxamer 188 samples in aqueous solution with lipid mix. The large hysteresis broadening observed in samples S8 and S9 can be used to identify poloxamer P188 samples that lead to tangential flow filtration membrane fouling while samples S2 and S7 are suitable for lipid nanoparticle purification and processing.
Figure 6 shows a Langmuir pressure-area plot of three consecutive compression-expansion cycles for representative poloxamer 188 samples S1 to S11 in aqueous solutions and lipid mix. Considering the isotherm hysteresis, samples S8 and S9 clearly stand out with broad hysteresis identifying poloxamer P188 samples that lead to tangential flow filtration membrane fouling.
Figure 7 shows each calculated AIT datapoint obtained over three consecutive compressionexpansion Langmuir trough cycles, for a lipid mix representative of a lipid nanoparticle formulation deposited over 11 different poloxamer 188 (S1 to S11 ) aqueous subphases. Superimposed ATT datapoints are shown as a broader distribution to enable their visualization. Therefore, an isotherm with generally tight hysteresis and only limited regions of broader hysteresis (e.g. Figure 6, S7) will be represented here with a pear-shaped distribution. Consequently, a hysteresis broader features (e.g. Figure 6, S8 and S9) leads to higher An values and a narrower distribution of An datapoints. Two clear groups of poloxamers can be distinguished and the narrow-shaped plots correspond to poloxamer P188 samples that lead to tangential flow filtration membrane fouling.
Figure 8 shows a comparison of Langmuir pressure-area plot of three consecutive compression-expansion cycles for representative poloxamer 188 sample S3 in aqueous solution with lipid mix using two different LT devices: Device 1 and Device 2. Device 2 is characterized by a larger surface area than device 1. Plot shape and surface pressure values were reproducible in both devices.
Figure 9 shows a Langmuir pressure-area plot of three consecutive compression-expansion cycles for a poloxamer 188 test sample in aqueous solutions with lipid mix of a test sample S23. Considering the isotherm hysteresis, the sample is characterized by a low An value and thus categorized as suitable for TFF.
Figure 10 shows a plot showing TFF processing time for different P188 solutions including a test sample S23. It is shown that sample S9 shows a non-linear relationship and three of the samples (S2, S3 and test sample S23) show a linear relationship between permeate volume and time confirming the method suitability for predicting the behavior of a surfactant in during TFF purification.
Figure 11 shows a comparison of Langmuir pressure-area plot of three consecutive compression-expansion cycles for a test sample of Poloxamer 124 in aqueous solution with lipid mix using Device 1 . Plot shape and surface pressure values indicate suitability for use as stabilizer.
Figure 12 shows a plot showing TFF processing time for two different P188 solutions (S3 and S9) and a test sample of poloxamer P124. It is shown that sample S9 shows a non-linear relationship. P188 sample S2 and the P124 test sample show a linear relationship between permeate volume and time confirming the method suitability for predicting the suitability of surfactant during TFF purification. EXAMPLES
Example 1 - Some P188 causes membrane fouling during TFF purification
During LiNP purification it was observed that some components of the nanoparticle preparation on certain occasions cause TFF membrane clogging or fouling causing considerable delays in filtration time or complete loss of the LiNP preparation.
In order to test the origin of the substance causing fouling different batches of poloxamer P188 were tested during LiNP purification using TFF.
Materials & Methods
Materials Poloxamer P188 was obtained as pharmaceutical grade from various suppliers. A test mRNA was used at a concentration of 1mg/mL.
Table 1

Methods LiNP formulation and TFF
Lipidoid nanoparticles were generated using nanoprecipitation in a NanoAssemblr™ BT comprising the ionizable lipidoid (dL_05(R), Scheme 1 ), the helper lipids DPPC (1 ,2- dipalmitoyl-sn-glycero-3-phosphocholine, Avanti Polar Lipids) and cholesterol (Avanti Polar Lipids), and the PEG lipid DMG-PEG2k (1 ,2-Dimyristoyl-sn-glycerolmethoxy(polyethylene glycol)-2000, Avanti Polar lipids) at the molar ratios of 8.00/5.29/4.41/0.88 respectively.. Before purification with tangential flow filtration (TFF), poloxamer P188 to a final concentration of 1% w/v was added. TFF used a C02-E100-05-N filter unit from Repligen. The devices were prepared according to the manual of the manufacturer. A flow rate of 54 mL/min (main pump) was selected for the system to reach the desired maximal sheer rate of 12000 s’1. TFF was performed in diafiltration mode for 10 volumes, followed by ultrafiltration to reduce volume twofold.
Quality Control (QC)
Time measurement for TFF were noted. Samples wherein TFF duration extended over 75 minutes were considered as failing the test and as not suitable for TFF purification.
Results
Fouling or clogging of the TFF column could be observed for some LINP/P188 samples. Summary is shown in Table 2.
Table 2 - Summary of TFF clogging obtained for some pharmaceutical grade poloxamer 188 samples. Conclusion
Conclusion

Fouling or clogging could be observed for different Poloxamer P188 used at the same concentration. By using TFF for purifying a composition comprising nanoparticles and a poloxamer, a surfactant such as a poloxamer, in particular p188, may be qualified as suitable or not suitable for membrane purification. This method is however expensive and time consuming since a TFF column needs to be discarded in case of a poloxamer failing the test and expensively produced LiNPs or LiNP need to be discarded.
Example 2 - TFF purification with a surfactant alone can indicate suitability for TFF. The following experiments were aimed at developing a test method for identifying P188 material suitable for LiNP formulation process, for example suitable during TFF filtration. In order to develop this test, TFF was used for purifying a solution containing only a surfactant and NaCI.
Samples S1-S11 of poloxamers as discussed in Example 1 , were tested measuring the duration of TFF purification or measuring a refractive index (Rl) of the poloxamer solutions:
Material and Method
Materials
Table 3 - Materials Table 4 - Devices
Table 4 - Devices


Sample preparation
P188 solutions were prepared using P188 provided by different suppliers and different batches as listed in Table 2.
The final composition was as follows: 0.5 % (w/v) P188, 50 mM NaCI.
Solutions were visually inspected for aggregates. For the described solutions, no aggregates or cloudiness occurred unless stated otherwise.
TFF processing
All poloxamer 188 solutions were tested at a concentration of 0.5 % (w/v). The TFF system was flushed with the respective poloxamer solution and 100 mL were diafiltrated with the automatic backpressure valve fully opened. The feed flow rate was set to 54 mL/min with a max TMP of 690 mbar.
Before and after TFF processing, a 100 pL sample of each solution was drawn for refractive index (Rl) measurements.
Refractive Index Measurements
A refractometer (DR201-95, Kriiss Optronic™, Hamburg, Germany) was calibrated with 100 L HPLC grade H2O. A sample volume of 100 pL was used for determination of Rl before and after processing the poloxamer solutions by TFF. Table 5: tabulated data for Fig 2

The experiment was aimed at evaluating the processing time of P188 materials of different suppliers. Fig 1 shows that solutions containing some of the samples, in particular sample S8, had to be processed more than three times longer than all other samples. Further, for most samples the Rl did increase slightly, whereas for sample S8 the Rl of the processed solution increased to highest extent, see Fig 2.
Discussion and Conclusion
This experiment was aimed at developing a test method for different poloxamer materials as incoming goods control. It provided information on whether a P188 batch or a P188 supplier is suitable for formulation and/or can be used in standard production and/or purification.
The experiment showed that surprisingly, the clogging observed during nanoparticle purification in the presence of poloxamer can be reproduced in the absence of said nanoparticles. It was thus found that there are differences between the processability of the tested poloxamers when tested without nanoparticles. According to the generated results, a limit of 75 minutes for the processing time during this material test is defined. Materials with longer processing times than 75 minutes were considered as not suitable for formulation of LiNP. Poloxamers P188 characterized by shorter processing times were assigned a better suitability score.
In summary, it was found that when running a TFF with a poloxamer dissolved in an aqueous phase, the poloxamer quality could be determined. From 5 samples of pharmaceutical grade poloxamer tested, one sample contained a pharmaceutical grade poloxamer that caused filtration delays or clogging resulting in samples with increased reflective index.
Example 3 - Preparation of surfactant solution and lipidoid mix
Surfactant aqueous solution preparation
A total of 11 aqueous solutions were prepared by dissolving 4 g of 11 different poloxamer P188 (1 % w/v) and 584.4 mg of NaCI (25 mM) in 400 mL of purified deionized water in glass bottles.
Lipidoid mix preparation
A lipidoid mix representative of a lipidoid nanoparticle formulation was prepared with the ionizable lipidoid (dL_05(R), Scheme 1 ), the helper lipids DPPC (1 ,2-dipalmitoyl-sn-glycero- 3-phosphocholine, Avanti Polar Lipids) and cholesterol (Avanti Polar Lipids), and the PEG lipid DMG-PEG2k (1 ,2-Dimyristoyl-sn-glycerolmethoxy(polyethylene glycol)-2000, Avanti Polar lipids) at the molar ratios of 8.00/5.29/4.41/0.88 respectively. These were resuspended together in chloroform at 15 mg/mL and vortexed for 1 minute to reach full dissolution.

Scheme 1 : Chemical structure of dL_05(R)
The 15 mg/mL stock was then transferred into a 10 mL glass vial for storage in a -20°C freezer.
The lipids were then further diluted in chloroform to 1 mg/mL before use on the Langmuir trough. Example 4 - Langmuir trough analysis of poloxamer P188
Exemplary Protocol of Langmuir surface pressure/area isotherm measurement on aqueous solution of test surfactant
1. Prepare an aqueous solution of the surfactant to be tested at a concentration of 1 % w/v, thereafter, referred to as “aqueous test solution”.
2. Prepare a lipidoid mix solution in chloroform at a concentration of 1 mg/mL, thereafter referred to as “lipidoid mix”.
3. Ensure the Langmuir trough (e.g. model MicrotroughX from Kibron Inc, or model 112D from Nima Technologies) is ready for use, according to the manufacturer’s recommendations.
4. Calibration control run: a) On a cleaned Langmuir trough add deionized water to the through in the volume recommended by the manufacturer so that it touches the trough sensor appropriately. b) Using a glass syringe, add sufficient lipidoid mix on the surface of the deionized water to form a monolayer (e.g. 30 pL). c) Let the sample equilibrate for 5 minutes at 22.1 ± 0.2°C. d) Start compressing the trough barriers at a speed of 20 cm2/min from the maximum area allowed, until the first phase transition is observed on the surfacepressure plot. Stop the compression when it is just past the first phase transition and note down the area reached at the beginning of the first phase transition, thereafter referred to as “minimum target area”. e) Expand the trough back to the maximum area allowed.
4. Test samples: a) On a cleaned Langmuir trough add the aqueous test solution to the through in the volume recommended by the manufacturer so that it touches the trough sensor appropriately. b) Let the sample equilibrate for 5 minutes at 22.1 ± 0.2°C. c) Start compressing the trough barriers at a speed of 20 cm2/min from the maximum area allowed, until the “minimum target area” is reached. d) Expand the trough back to the maximum area allowed. e) Following a three-second wait time between compression-expansion cycles, repeat steps 4.c) to d) to obtain multiple surface-pressure hysteresis cycles (e.g. three) if desired. f) Clean the trough following the manufacturer’s recommendations. Langmuir trough hardware
A Langmuir-Blodgett trough model 112D made by Nima Technologies (U.K.) was used in all experiments of Example 4. Before use, the trough was cleaned thoroughly with a Kimwipe® tissue (type 7105, Kimtech type 75512 or EX-L WIPES 34256) soaked in chloroform wearing polythene gloves in a well-ventilated area. The chloroform was removed, and the trough was rinsed several times with purified deionized water.
Langmuir trough settings
Isotherm cycle settings:
- Compression/expansion speed: 20 cm/min
- Total cycles: 3 to 4 cycles
Max area: 79 cm2
Min area: 20 cm2
- Wait time of 3 seconds when fully opened.
During all experiments the trough was covered, and the temperature maintained at 22.1 ± 0.2°C.
Langmuir trough experiment
Approximately 80 mL of poloxamer P188 aqueous solution was added to the Langmuir trough and left five minutes to equilibrate. A com pression -expansion isotherm was run, beginning at a maximum area of 79 cm2 and with a compression speed of 20 cm/min to a minimum area of 20 cm2. The trough was then expanded at the same rate to reach the maximum area again. Following a wait time of three seconds, the com pression -expansion cycle was repeated. This process was done a total of three to four times for each poloxamer aqueous solution. After each set of experiments, the trough was rinsed with five times the trough volume of deionized water.
Results
The isotherm plots for surfactant aqueous solutions are shown in Fig 4, the legend in the figure indicates which poloxamer P188 is present in the aqueous subphase filling the Langmuir trough and the title indicates which compression -expansion cycle is shown. Surprisingly, it was found that a compression-expansion isotherm could be used to test the suitability of poloxamer P188 in lipid nanoparticle purification and processing. Particularly clear results were obtained in a third compression-expansion cycle. It was further found that an unsuitable poloxamer can be characterized by an isotherm that reaches a maximum surface pressure above 4 mN/m.
A summarizing plot with isotherms from both representative stable and unstable poloxamers is shown in Fig 3.
Example 5 - Langmuir trough analysis of poloxamer P188 with lipidoid mix
Exemplary Protocol of Langmuir surface pressure/area isotherm measurement on aqueous solution of test surfactant with lipidoid mix layer added on surface
1. Prepare an aqueous solution of the surfactant to be tested at a concentration of 1 % w/v, thereafter, referred to as “aqueous test solution”.
2. Prepare a lipidoid mix solution in chloroform at a concentration of 1 mg/mL, thereafter, referred to as “lipidoid mix”.
3. Ensure the Langmuir trough (e.g. model MicrotroughX from Kibron Inc, or model 112D from Nima Technologies) is ready for use, according to the manufacturer’s recommendations.
4. Calibration control run: a) On a cleaned Langmuir trough add deionized water to the through in the volume recommended by the manufacturer so that it touches the trough sensor appropriately. b) Using a glass syringe, add sufficient lipid mix on the surface of the deionized water to form a monolayer (e.g. 30 pL). c) Let the sample equilibrate for 5 minutes at 22.1 ± 0.2°C. d) Start compressing the trough barriers at a speed of 20 cm2/min from the maximum area allowed, until the first phase transition is observed on the surfacepressure plot. Stop the compression when it is just past the first phase transition and note down the area reached at the beginning of the first phase transition, thereafter referred to as “minimum target area”. e) Expand the trough back to the maximum area allowed.
5. Test samples: a) On a cleaned Langmuir trough add the aqueous test solution to the through in the volume recommended by the manufacturer so that it touches the trough sensor appropriately. b) Using a glass syringe, add sufficient lipid mix on the surface of the deionized water to form a monolayer (e.g. 30 pL). c) Let the sample equilibrate for 5 minutes at 22.1 ± 0.2°C. d) Start compressing the trough barriers at a speed of 20 cm2/min from the maximum area allowed, until the “minimum target area” is reached. e) Expand the trough back to the maximum area allowed. f) Following a three-second wait time between com pression -expansion cycles, repeat steps 5.d-e to obtain multiple surface-pressure hysteresis cycles (e.g. three) if desired. g) Clean the trough following the manufacturer’s recommendations.
Langmuir trough hardware
As described in Example 4, a Langmuir-Blodgett trough model 112D made by Nima Technologies (U.K.) was used in all experiments of example 5.
Langmuir trough settings
As described in Example 4, the following Langmuir trough settings were used:
Compression/expansion speed: 20 cm/min
- Total cycles: 3 to 4 cycles
Max area: 79 cm2
Min area: 20 cm2
- Wait time of 3 seconds when fully opened.
During all experiments the trough was covered, and the temperature maintained at 22.1 ± 0.2°C.
Langmuir trough experiment
Approximately 80 mL of poloxamer P188 aqueous solution was added to the Langmuir trough and left five minutes to equilibrate. Control compression-expansion isotherms were recorded for the poloxamer aqueous solution alone. After the control run, 4 pL of lipid mix (1 mg/mL) was added using a 10 pL glass syringe. The sample was left five minutes equilibrate before beginning the compression-expansion cycles: starting from the maximum area of 79 cm2, with a compression speed of 20 cm/min, and down to a minimum area of 20 cm2. The trough was then expanded at the same rate to reach the maximum area again. Following a wait time of three seconds, the compression-expansion cycle was repeated. This process was done a total of three to four times for each poloxamer aqueous solution and lipid mix. After each set of experiments, the trough was rinsed with five times the trough volume of deionized water.
Results
The isotherm plots for surfactant aqueous solutions are shown in Fig 6, the legend in the figure indicates which stabilizing agent is present in the aqueous subphase filling the Langmuir trough and the use of the lipid mix in the experiment. The title indicates which compression-expansion cycle is shown. A summarizing plot with isotherms from both representative stable and unstable poloxamers is shown in Fig 5.
Surprisingly, it was found out that an analysis of the Langmuir isotherm from lipid mix on a poloxamer aqueous subphase, could be used to test the suitability of poloxamer P188 in lipid nanoparticle purification and processing.
To define a threshold between what makes a suitable and unsuitable poloxamer P188, a parameter An was defined for the isotherm, calculated for any area point within three compression-expansion cycles.
For each cycle, the TT of the hysteresis was calculated as Arc =ncomp nexp with:
^max
TTcomp being the surface pressure during the compression phase,
TTexp being the surface pressure during the expansion phase,
Umax being the maximum surface pressure reached in that isotherm cycle
This calculation was done fornCOmp and neXp matching in area. An was calculated for any area point of the Langmuir trough experiment and plotted in Fig 7. Poloxamers P188 unsuitable for lipid nanoparticle purification and processing were identified if at any Au was above 0.6 considering up to three isotherm cycles. As shown in Fig 7, consistently low Au values were observed for during the first isotherm cycle. Example 6 - Langmuir trough analysis of poloxamer P188 with lipid mix in LT Hardware with larger test area.
Langmuir trough hardware
To confirm the reproducibility of the method of the invention, Device 2, a Langmuir-Blodgett trough model MicrotroughX from Kibron Inc was used in all experiments of Example 6. Before use, the trough was cleaned thoroughly with ethanol. The ethanol was removed, and the trough was rinsed several times with purified deionized water.
Langmuir trough settings
The following Langmuir trough settings were used:
Compression/expansion speed: 34 mm/min
- Total cycles: 3 cycles
Max area: 224 cm2
Min area: 110 cm2
- Wait time of 3 seconds when fully opened.
During all experiments the trough was covered, and the temperature maintained at 22.1 ± 0.2°C.
Langmuir trough experiment
Approximately 250 mL of poloxamer P188 aqueous solution (sample S3) was added to the Langmuir trough and left five minutes to equilibrate. 30 pL of lipid mix (1 mg/mL) was added using a 50 pL glass syringe. The sample was left five minutes equilibrate before beginning the compression-expansion cycles: starting from the maximum area of 224 cm2, with a compression speed of 34 m/min, and down to a minimum area of 110 cm2. The trough was then expanded at the same rate to reach the maximum area again. Following a wait time of three seconds, the compression-expansion cycle was repeated. This process was done a total of three times for each poloxamer aqueous solution and lipid mix. After each set of experiments, the trough was rinsed with five times the trough volume of deionized water. TFF processing
The same tangential flow filtration processing as described in Example 2 was used here as well.
Results
The isotherm plots for surfactant aqueous solutions are shown in Fig 8 and Fig 9, the legend in the figure indicates which stabilizing agent is present in the aqueous subphase filling the Langmuir trough and the use of the lipid mix in the experiment. The title indicates which compression-expansion cycle is shown.
It was found that the Langmuir isotherm for lipid mix on a poloxamer aqueous subphase were reproducible between Device 1 (112D, Nima Technologies) and Device 2 (MicrotroughX, Kibron Inc) (Fig 8). This illustrates the characteristics of a suitable poloxamer P188 showing a thin hysteresis and a TT < 0.6. The suitability of the poloxamer sample S3 and test sample S23 was further confirmed by reprocessing of the same sample via TFF (Fig 10).
Considering a new lot of poloxamer P188 and the method described herein, the Langmuir isotherm from lipid mix on a poloxamer aqueous subphase (Fig. 9) identified this new lot as suitable for lipid nanoparticle purification and processing. These results were further confirmed by TFF processing of the new poloxamer P188 lot in aqueous solution (Fig 10) supporting that surprisingly the suitability of the test method of the invention to differentiate and determine poloxamer P188 usability for TFF purification. The method of the invention provides a method that requires significantly less time, less test material and less consumables compared to testing the surfactant in actual production settings. Multiple samples can be quickly and economically tested using the method of the invention to efficiently determine the suitability of a surfactant as stabilizer.
Example 7 - Langmuir trough analysis and TFF processing of poloxamer P124.
Expanding the analysis to a different poloxamer, this example examines the performance of the method of the invention for poloxamer P124.
Langmuir trough hardware, settings, and experimental procedure
The same Langmuir trough hardware, settings, experimental set up and procedure was used, as described in Example 5. TFF processing
The same experimental set up and procedure was followed, as described in Example 2.
Results
The isotherm plots for a surfactant aqueous solution comprising P124 with the addition of lipid mix on the surface are shown in Fig 11 over three compression-expansion cycles. The thin hysteresis (An < 0.6) observed in these samples identifies this excipient as suitable for lipid nanoparticle purification and processing. These results were further confirmed by TFF processing of this poloxamer P124 in aqueous solution shown as a permeate /time plot in Fig 12. The sample of P124 showed a linear relationship between permeate and time, confirming the suitability of poloxamer P124 for TFF purification and of the sample in particular. The results show that the method of the invention requires significantly less time, less test material and less consumables compared to testing the surfactant in actual production setting.
Claims
1. A lipid nanoparticle (LNP) formulation or lipidoid nanoparticle (LiNP) formulation comprising a plurality of lipid nanoparticles (LNPs) or lipidoid nanoparticles (LiNPs), each comprising a lipid mix or lipidoid mix, wherein the lipid mix or lipidoid mix comprises, as a component thereof, at least one selected from an ionizable lipid, ionizable lipidoid, and permanently cationic lipid, and wherein the formulation further comprises a surfactant characterized by having a Langmuir surface pressure/area isotherm with a maximum surface pressure (nmax) that is equal to or below 4.0 mN/m at a minimum surface area established for a lipid mix or lipidoid mix as comprised by the nanoparticles.
2. The lipid nanoparticle (LNP) formulation or lipidoid nanoparticle (LiNP) formulation according to claim 1 , wherein Umax is equal to or below 3.5 mN/m, more preferably equal to or below 3.5 and equal to or above 0.5 mN/m, even more preferably equal to or below 3.0 mN/m and equal to or above 0.5 mN/m, even more preferably equal to or below 2.5 and equal to or above 1 .0 mN/m, most preferably equal to or below 2.0 and equal to or above 1 .0 mN/m.
3. A lipid nanoparticle (LNP) formulation or lipidoid nanoparticle (LiNP) formulation comprising a plurality of lipid nanoparticles (LNPs) or lipidoid nanoparticles (LiNPs), each comprising a lipid mix or lipidoid mix, wherein the lipid mix or lipidoid mix comprises, as a component thereof, at least one selected from an ionizable lipid, ionizable lipidoid, and permanently cationic lipid, wherein the formulation comprises as a stabilizing agent a surfactant, and wherein a representative sample comprising an aqueous solution of the surfactant carrying on its surface a lipid mix or lipidoid mix as comprised by the LNPs or LiNPs is characterized by having a Langmuir isotherm An that is equal to or below 0.60, preferably equal to or below 0.45, at each area point during a Langmuir surface pressure/area isotherm cycle comprising a compression phase and an expansion phase and recorded between a maximum surface area and minimum surface area established for the lipid mix or lipidoid mix , wherein TT is calculated at any area point as: ft _ (,n comp ~n exp) nmax ’ wherein ncomp is the surface pressure at said area point during the compression phase of the isotherm cycle, wherein itexp is the surface pressure at said area point during the expansion phase of the isotherm cycle, and wherein nmax is the maximum surface pressure reached in the isotherm cycle.
4. The lipid nanoparticle (LNP) formulation or lipidoid nanoparticle (LiNP) formulation according to any one of claims 1 to 3, wherein the surfactant is a non-ionic surfactant, preferably at least one nonionic surfactant selected from the group of fatty alcohol ethoxylates, fatty acid ethoxylates, block copolymers of ethylene oxide and propylene oxide, alkylphenol ethoxylates or oligomers of alkylphenol ethoxylates, fatty acid esters of sorbitol, ethoxylated fatty acid esters of sorbitol, fatty acid esters of glycerol, ethoxylated castor oil and ethoxylated vitamin E.
5. The lipid nanoparticle (LNP) formulation or lipidoid nanoparticle (LiNP) formulation according to claim 4, wherein the surfactant is a block copolymer of ethylene oxide and propylene oxide.
6. The lipid nanoparticle (LNP) formulation or lipidoid nanoparticle (LiNP) formulation according to any one of claims 1 to 5, wherein the lipid nanoparticles or lipidoid nanoparticles comprise the lipid mix or lipidoid mix and a therapeutic agent.
7. The lipid nanoparticle (LNP) formulation or lipidoid nanoparticle (LiNP) formulation according to claim 6, wherein the therapeutic agent comprises a nucleic acid, preferably mRNA.
8. The lipid nanoparticle (LNP) formulation or lipidoid nanoparticle (LiNP) formulation according to any one of claims 1 to 7, wherein the lipid mix or lipidoid mix comprises, as a component thereof, at least one selected from an ionizable lipid, ionizable lipidoid, and a permanently cationic lipid, and further comprises one or more of the following components (c1) to (c6), preferably further comprises the components (c1 ), (c2) and (c3):
(c1) a non-ionizable lipid having a sterol structure;
(c2) a phospholipid;
(c3) a PEG-conjugated lipid;
(c4) a polysarcosine-conjugated lipid
(c5) a PASylated lipid; (c6) an ionizable or a cationic polymer.
9. The lipid nanoparticle (LNP) formulation or lipidoid nanoparticle (LiNP) formulation according to claim 8, wherein the lipid mix or lipidoid mix comprises i) 30 to 65 mol% of at least one selected from the ionizable lipid, the ionizable lipidoid, and the permanently cationic lipid and further comprises one or more of the following components (c1 ) to (c6): ii) 10 to 50 mol% of the lipid having a sterol structure (c1 ), iii) 4 to 50 mol% of the phospholipid (c2), iv) 0.5 to 10 mol% of one of the PEG-conjugated lipid (c3), the polysarcosine-conjugated lipid (c4) and the PASylated lipid (c5), or of any combination thereof, v) 0.5 to 10 mol% of a cationic polymer (c6), such that the sum of the amounts of i) and ii) to v) is 100 mol%, and more preferably further comprises the components (c1 ), (c2) and (c3), such that the sum of the amounts of i) and ii) to iv) is 100%.
10. The lipid nanoparticle (LNP) formulation or lipidoid nanoparticle (LiNP) formulation according to any one of claims 1 to 9, which is a lipidoid nanoparticle formulation comprising a plurality of lipidoid nanoparticles each comprising a lipidoid mix, wherein the lipidoid mix comprises an ionizable lipidoid of formula (L-1 ): wherein: a is 1 or 2 and b is an integer of 1 to 4 or a is an integer of 1 to 4 and b is 1 or 2, p is 1 or 2, m is 1 or 2; n is 0 or 1 and m+n is 2, and
wherein: a is 1 or 2 and b is an integer of 1 to 4 or a is an integer of 1 to 4 and b is 1 or 2, p is 1 or 2, m is 1 or 2; n is 0 or 1 and m+n is 2, and

R1A to R6A are independently of each other selected from: hydrogen, -CH2-CH(OH)-R7A, -CH(R7A)-CH2-OH, -CH2-CH2-(C=O)-O-R7A, -CH2-CH2-(C=O)-NH-R7A, -CH2-R7A, -C(NH)-NH2J a polyethylene glycol) chain, and a receptor ligand; wherein R7A is selected from C3-C18 alkyl and C3-C18 alkenyl having one C-C double bond; provided that at least two residues among R1A to R6A are selected from -CH2-CH(OH)-R7A, -CH(R7A)-CH2-OH, -CH2-CH2-(C=O)-O-R7A, -CH2-CH2-(C=O)-NH-R7A and -CH2-R7A, wherein R7A is selected from C3-C18 alkyl or C3-C18 alkenyl having one C-C double bond, or a protonated form of the ionizable lipid of formula (L-1 ) wherein one or more of the nitrogen atoms contained in the compound of formula (L-1 ) are protonated to provide a compound carrying a positive charge.
11. The lipid nanoparticle (LNP) formulation or lipidoid nanoparticle (LiNP) formulation according to any one of claims 1 to 10 which is a suspension formulation, and wherein the vehicle solution of the suspension formulation is an aqueous vehicle solution comprising the surfactant.
12. A surfactant for use in a pharmaceutical composition, the surfactant being characterized by having a Langmuir surface pressure/area isotherm with a maximum surface pressure (nmax) that is equal to or below 4.0 mN/m at a minimum surface area established for said pharmaceutical composition.
13. The surfactant for use according to claim 12, wherein the maximum surface pressure is equal to or below 3.5 mN/m, more preferably equal to or below 3.5 and equal to or above 0.5 mN/m, even more preferably equal to or below 3.0 mN/m and equal to or above 0.5 mN/m, even more preferably equal to or below 2.5 and equal to or above 1 .0 mN/m, most preferably equal to or below 2.0 and equal to or above 1.0 mN/m.
14. The surfactant for use according to claim 12 or 13, which is a non-ionic surfactant, preferably at least one nonionic surfactant selected from the group of fatty alcohol ethoxylates, fatty acid ethoxylates, block copolymers of ethylene oxide and propylene oxide, alkylphenol ethoxylates or oligomers of alkylphenol ethoxylates, fatty acid esters of sorbitol, ethoxylated fatty acid esters of sorbitol, fatty acid esters of glycerol, ethoxylated castor oil and ethoxylated vitamin E.
15. The surfactant for use according to claim 14, which is a block copolymer of ethylene oxide and propylene oxide, more preferably a.
16. The surfactant for use according to any of claims 12 to 15, wherein the pharmaceutical composition is in the form of a lipid nanoparticle (LNP) suspension formulation or lipidoid nanoparticle (LiNP) suspension formulation comprising a plurality of LNPs or LiNPs, each comprising a lipid mix or lipidoid mix and a therapeutic agent, wherein the lipid mix or lipidoid mix comprises, as a component thereof, at least one selected from an ionizable lipid, ionizable lipidoid, and permanently cationic lipid, and wherein the vehicle solution of the suspension formulation is an aqueous vehicle solution comprising the surfactant
17. A method for classifying a surfactant as suitable or not suitable for use as a stabilizing agent for a pharmaceutical composition, the method comprising:
(a) providing a surfactant in an aqueous solution at a concentration (C),
(b) recording a Langmuir surface pressure/area isotherm of the surfactant in the solution to determine a maximum surface pressure nmax of the Langmuir isotherm at a predetermined minimum surface area;
(c) comparing the maximum surface pressure nmax to a threshold value, wherein if the maximum surface pressure TTmax is equal to or less than the threshold value, the surfactant is classified as suitable for use as a stabilizing agent, and if the maximum surface pressure TTmax is greater than the threshold value, the surfactant is classified as not suitable for use as a stabilizing agent.
18. A method for classifying a surfactant as suitable or not suitable for use as a stabilizing agent for a pharmaceutical composition comprising a lipid or lipidoid, optionally during purification of said composition, preferably during tangential flow filtration of said composition, wherein the method comprises the steps:
(a) providing a surfactant in an aqueous solution at a concentration (C) of the surfactant in the solution;
(b) recording a Langmuir pressure/area isotherm cycle including a compression phase and an expansion phase between a maximum surface area and a minimum surface area on a sample comprising the surfactant in the aqueous solution and carrying on its surface a lipid or lipidoid as comprised by the composition: (c) calculating a Langmuir isotherm An for each area point of the Langmuir pressure/area isotherm cycle, wherein TT is calculated as: wherein ncomp is the surface pressure at said area point during the compression phase of the isotherm cycle, wherein itexp is the surface pressure at said area point during the expansion phase of the isotherm cycle, and wherein nmax is the maximum surface pressure reached in the isotherm cycle, and
wherein ncomp is the surface pressure at said area point during the compression phase of the isotherm cycle, wherein itexp is the surface pressure at said area point during the expansion phase of the isotherm cycle, and wherein nmax is the maximum surface pressure reached in the isotherm cycle, and

(d) comparing the calculated Langmuir isotherm TT to a threshold value, wherein, if the calculated Langmuir isotherm An is at every isotherm area point equal to or less than the threshold value, the surfactant is classified as suitable for use as a stabilizing agent, and if the calculated Langmuir An at any area point is greater than the threshold value the surfactant is classified as not suitable for use as a stabilizing agent.
19. A method for the preparation of a pharmaceutical composition, said method comprising classifying a surfactant as suitable or not suitable for use as a stabilizing agent for a pharmaceutical composition in accordance with the method of claim 17 or 18, and, if the surfactant is classified as being suitable for use as a stabilizing agent for the pharmaceutical composition, incorporating the surfactant into the pharmaceutical composition.
20. The method according to any one of claims 17 to 19, wherein the surfactant is a nonionic surfactant, preferably at least one nonionic surfactant selected from the group of fatty alcohol ethoxylates, fatty acid ethoxylates, block copolymers of ethylene oxide and propylene oxide, alkylphenol ethoxylates or oligomers of alkylphenol ethoxylates, fatty acid esters of sorbitol, ethoxylated fatty acid esters of sorbitol, fatty acid esters of glycerol, ethoxylated castor oil and ethoxylated vitamin E.
21. The method according to claim 20, wherein the surfactant is a block copolymer of ethylene oxide and propylene oxide, more preferably a poloxamer, even more preferably poloxamer selected from the list consisting of poloxamer 124, poloxamer 188, poloxamer 338, and poloxamer 407, or from combinations thereof, most preferably P188.
22. The method according to any one of claims 17 to 21, wherein the pharmaceutical composition comprises a therapeutic agent comprising a nucleic acid, such as RNA, preferably mRNA.
23. The method according to any one of claims 17 to 22, wherein the pharmaceutical composition is a lipid nanoparticle (LNP) formulation or lipidoid nanoparticle (LiNP) formulation comprising a plurality of LNPs or LiNPs, each comprising a lipid mix or lipidoid mix and a therapeutic agent, wherein the lipid mix or lipidoid mix comprises, as a component thereof, at least one selected from an ionizable lipid, ionizable lipidoid, and permanently cationic lipid.
24. The method according to claim 23, wherein the pharmaceutical composition is a lipid nanoparticle (LNP) suspension formulation or lipidoid nanoparticle (LiNP) suspension formulation, and wherein the vehicle solution of the suspension formulation is an aqueous vehicle solution comprising the surfactant.
25. A method of mitigating or avoiding clogging or fouling of a filtration system during purification of a pharmaceutical composition in the form of a lipid nanoparticle formulation (LNP) or lipidoid nanoparticle formulation (LiNP), the method comprising adding a stabilizing surfactant to a first LNP or LiNP formulation to form a second LNP or LiNP formulation, optionally before purification, wherein the LNP or LiNP formulation comprises a plurality of LNPs or LiNPs, each comprising a lipid mix or lipidoid mix and a therapeutic agent, wherein the lipid mix or lipidoid mix comprises, as a component thereof, at least one selected from an ionizable lipid, ionizable lipidoid, and permanently cationic lipid, and wherein the stabilizing surfactant is a surfactant as defined in any of claims 12 to 16 or a surfactant classified as being suitable as a stabilizing agent by the method according to any of claims 17 to 27.
26. The method according to claim 25, wherein the purification includes tangential flow filtration.
27. The method according to claim 25 or 26, wherein the pharmaceutical composition is a lipid nanoparticle (LNP) suspension formulation or lipidoid nanoparticle (LiNP) suspension formulation comprising an aqueous vehicle solution, and wherein the stabilizing surfactant is added to the vehicle solution, optionally wherein the surfactant is essentially absent from the LNPs or LiNPs.
28. The method according to any one of claims 25 to 27, wherein therapeutic agent, is a nucleic acid such as RNA, more preferably mRNA.
29. A method of mitigating aggregation of lipid or lipidoid nanoparticles in a lipid nanoparticle (LNP) formulation or lipidoid nanoparticle (LiNP) formulation, the method comprising adding a stabilizing surfactant to a first LNP or LiNP formulation to form a second LNP or LiNP formulation, optionally before purification, wherein the LNP or LiNP formulation comprises a plurality of LNPs or LiNPs, each comprising a lipid mix or lipidoid mix, wherein the lipid mix or lipidoid mix comprises, as a component thereof, at least one selected from an ionizable lipid, ionizable lipidoid, and permanently cationic lipid, and wherein the stabilizing surfactant is a surfactant as defined in any of claims 12 to 16 or a surfactant classified as being suitable as a stabilizing agent by the method according to any of claims 17 to 27.
30. The method according to claim 29, wherein the formulation is a lipid nanoparticle (LNP) suspension formulation or lipidoid nanoparticle (LiNP) suspension formulation comprising an aqueous vehicle solution, and wherein the stabilizing surfactant is added to the vehicle solution, optionally wherein the surfactant is essentially absent from the LNPs or LiNPs.
31. The method according to claim 29 or 30, wherein the lipid nanoparticles or lipidoid nanoparticles comprise a therapeutic agent, preferably a nucleic acid such as RNA, more preferably mRNA.
32. The method according to claim 31, wherein the lipid nanoparticles or lipidoid nanoparticles comprise a nucleic acid, e.g. RNA, and preferably mRNA, and wherein the method comprises the steps of: i) first, combining the nucleic acid and at least one selected from an ionizable lipid, ionizable lipidoid, and permanently cationic lipid to form LNPs or LiNPs, ii) second, purifying the LNPs or LiNPs, iii) third, adding the stabilizing surfactant before TFF purification and during TFF purification in the exchange buffer, maintaining the surfactant in a steady concentration, iv) optionally wherein the stabilizing surfactant is added to the LNP or LiNP formulation after step (i).
33. The method according to claims 31 or 32, wherein the method comprises the following steps: i) generating a LNP or LiNP preparation by mixing of at least one selected from a permanently cationic lipid, an ionizable lipid and an ionizable lipidoid dissolved in an organic phase with a therapeutic agent dissolved in an aqueous solution, ii) diluting the LNP or LiNP preparation by dilution with a first solution, iii) concentrating the LNP or LiNP preparation by buffer exchange using ultra/diafiltration via TFF wherein a second solution is used for the ultra/diafiltration, iv) obtaining a LNP or LiNP suspension in an aqueous vehicle solution, wherein the first solution comprises between about 0.01% w/v and 10% of stabilizing surfactant, preferably between about 0.01 % w/v surfactant and 5% w/v surfactant, more preferably between about 0.01% w/v surfactant and 2.5% w/v surfactant, more preferably between about 0.05% w/v and 1.5% w/v surfactant, even more preferably between about 0.05% w/v and 1 .5% w/v surfactant, most preferably about 1 % w/v surfactant, and/or wherein the second solution comprises between about 0.01 % w/v and about 10% of stabilizing surfactant, preferably between about 0.01% w/v surfactant and about 5% w/v surfactant, more preferably between about 0.01 % w/v surfactant and about 2.5% w/v surfactant, even more preferably between about 0.05% w/v and 1.5% w/v surfactant, most preferably about 1% w/v; and wherein the final concentration of stabilizing surfactant from combined first and second solution is between 0.01% and 10% surfactant, preferably between 0.01% w/v surfactant and 5% w/v surfactant, more preferably between 0.01 % w/v surfactant and 2.5% w/v surfactant, even more preferably between 0.05% w/v and 1.5 % w/v surfactant, most preferably about 1% w/v surfactant with regard to the total volume of the suspension of the nanoparticles in the aqueous vehicle solution.
34. The method of claim 33, wherein: a) the incorporation of the stabilizing surfactant into the suspension does not occur before or during step i), b) the stabilizing surfactant is added in the first and the second solution, and/or c) approximately half of the stabilizing surfactant is added to the first solution and approximately half of the surfactant is added to the second solution.
35. The method according to any one of claims 23 to 34, wherein the lipid mix or lipidoid mix comprises, as a component thereof, at least one selected from an ionizable lipid, ionizable lipidoid, and a permanently cationic lipid, and further comprises one or more of the following components (c1 ) to (c6):
(c1) a non-ionizable lipid having a sterol structure;
(c2) a phospholipid;
(c3) a PEG-conjugated lipid;
(c4) a polysarcosine-conjugated lipid
(c5) a PASylated lipid;
(c6) an ionizable or a cationic polymer.
36. The method according to claim 35, wherein the lipid mix or lipidoid mix comprises, as a component thereof, at least one selected from an ionizable lipid, ionizable lipidoid, and a permanently cationic lipid, and further comprises the components (c1), (c2) and (c3).
37. The method according to claim 35, wherein the lipid mix or lipidoid mix comprises i) 30 to 65 mol% of at least one selected from the ionizable lipid, the ionizable lipidoid and the permanently cationic lipid, and further comprises one or more of the following components (c1 ) to (c6): ii) 10 to 50 mol% of the lipid having a sterol structure (c1 ), iii) 4 to 50 mol% of the phospholipid (c2), iv) 0.5 to 10 mol% of one of the PEG-conjugated lipid (c3), the polysarcosine-conjugated lipid (c4) and the PASylated lipid (c5), or of any combination thereof, v) 0.5 to 10 mol% of a cationic polymer (c6), such that the sum of the amounts of i) and ii) to v) is 100 mol%,
38. The method according to claim 37, wherein the lipid mix or lipidoid mix comprises i) 30 to 65 mol% of at least one selected from the ionizable lipid, the ionizable lipidoid and the permanently cationic lipid, and further comprises ii) 10 to 50 mol% of the lipid having a sterol structure (c1 ), iii) 4 to 50 mol% of the phospholipid (c2), iv) 0.5 to 10 mol% of a PEG-conjugated lipid (c3), such that the sum of the amounts of i) and ii) to iv) is 100%.
39. The method according to any one of claims 23 to 38, wherein the formulation is a lipidoid nanoparticle formulation comprising a plurality of lipidoid nanoparticles each comprising a lipidoid mix, wherein the lipidoid mix comprises an ionizable lipidoid of formula (L-1 ):
R2A R4A
R1 A— N— {CH2- (CH2)— N— [CH2— (CH2)b- N]p}— [CH2- (CH2)- NJ- R6A 3A 5A
(L-1 ) wherein: a is 1 or 2 and b is an integer of 1 to 4 or a is an integer of 1 to 4 and b is 1 or 2, p is 1 or 2, m is 1 or 2; n is 0 or 1 and m+n is 2; and
R1A to R6A are independently of each other selected from: hydrogen; -CH2-CH(OH)-R7A, -CH(R7A)-CH2-OH, -CH2-CH2-(C=O)-O-R7A, -CH2-CH2-(C=O)-NH-R7A, -CH2-R7A, -C(NH)-NH2I a polyethylene glycol) chain, and a receptor ligand; wherein R7A is selected from C3-C18 alkyl and C3-C18 alkenyl having one C-C double bond; provided that at least two residues among R1A to R6A are selected from -CH2-CH(OH)-R7A, -CH(R7A)-CH2-OH, -CH2-CH2-(C=O)-O-R7A, -CH2-CH2-(C=O)-NH-R7A and -CH2-R7A, wherein R7A is selected from C3-C18 alkyl or C3-C18 alkenyl having one C-C double bond, or a protonated form of the ionizable lipid of formula (L-1 ) wherein one or more of the nitrogen atoms contained in the compound of formula (L-1 ) are protonated to provide a compound carrying a positive charge.
40. A lipid nanoparticle formulation or lipidoid nanoparticle formulation, preferably a lipid nanoparticle suspension formulation or lipidoid nanoparticle suspension formulation, obtained by the method according to any one of claims 23 to 39.
41 . The lipid nanoparticle formulation or lipidoid nanoparticle formulation, preferably a lipid nanoparticle suspension formulation or lipidoid nanoparticle suspension formulation, according to any one of claims 1 to 11 or 40 for use as a medicament.
42. The lipid nanoparticle formulation or lipidoid nanoparticle formulation, preferably a lipid nanoparticle suspension formulation or lipidoid nanoparticle suspension formulation, according to any one of claims 1 to 11 or 40, for use in the treatment or prevention of a disease, preferably a disease selected from Table A, more preferably a disease selected from viral diseases, ciliopathies, autoimmune diseases, and respiratory tract diseases, even more preferably selected from a lung disease, an airway disease or a nasal disease, more preferably a lung viral disease, lung ciliopathies and a lung autoimmune diseases.
43. The lipid nanoparticle formulation or lipidoid nanoparticle formulation for use according to claim 43, wherein the lung disease or lung viral disease is at least one selected from pneumonia and asthma; the airway disease is at least one selected from bronchitis, viral induced asthma, lung fibrosis and COPD; and/or the nasal disease is at least one selected from rhinitis and sinusitis.
44. The lipid nanoparticle formulation or lipidoid nanoparticle formulation according to claim 41 , for use in vaccination or immunization.
45. A method of avoiding or reducing side effects in a therapy with LNPs or LiNPs carrying at least one therapeutic agent, wherein the method comprises the steps: i) determine whether LNPs or LiNPs in a pharmaceutical composition comprising LNPs or LiNPs aggregate when subjected to a mechanical stress or a temperature stress, by determining their aggregation level before and after subjecting said pharmaceutical composition is subjected to said mechanical or temperature stress ii) if the LNP or LiNP show aggregation after the test of step (i), then add to the LNP or LiNP formulation a surfactant to obtain a LNP or LiNP suspension with a final surfactant concentration of 0.01 % w/v and up to 10% w/v, preferably between 0.05% w/w surfactant and 5% surfactant, more preferably between 0.33% surfactant and 2.5% surfactant, more preferably between 0.45% and 1 .5% surfactant, most preferably between 0.5% and 1 .5% surfactant, most preferably about 1% w/v. iii) reconstitute with mixing to generate a stable LNP or LiNP suspension.
46. The method in accordance with claim 45, wherein the surfactant is a surfactant according to any one of claims 12 to 16, or a surfactant classified as being suitable as a stabilizing agent by the method according to any one of claims 17 to 24
47. Use of a surfactant according to any one of claims 12 to 16, or of a surfactant classified as being suitable as a stabilizing agent by the method according to any one of claims 17 to 24, for stabilizing a suspension of lipid nanoparticles or of lipidoid nanoparticles in an aqueous vehicle solution against particle aggregation under a physical stress condition, preferably shear stress, more preferably shear stress during purification such as TFF, wherein the lipid nanoparticles or lipidoid nanoparticles comprise the following components (a) and (b):
(a) a therapeutic agent and
(b) at least one selected from an ionizable lipid, an ionizable lipidoid and a permanently cationic lipid.
48. The use of the surfactant in accordance with claim 47, wherein the physical stress condition is selected from shaking, stirring, vibrating, mixing, inverting, tapping, or dropping of the suspension, or a combination thereof, or wherein the physical stress condition is caused by pumping the suspension or by its withdrawal into a syringe.
49. The use of the surfactant in accordance with claim 47 or 48, wherein the surfactant is incorporated as an excipient into the aqueous vehicle solution.
50. The use of the surfactant in accordance with any one of claims 47 to 49, wherein the nanoparticle formulation is not lyophilized.
51 . The use of the surfactant in accordance with any one of claims 47 to 50, wherein the surfactant is added before a lyophilization process.
52. The use of the surfactant in accordance with any one of claims 47 to 51 , wherein the presence of the surfactant does not cause a change in the biological activity of the nanoparticle.
53. The use of the surfactant in accordance with any one of claims 47 to 52, wherein the presence of the surfactant does not cause a change in the physical properties of the nanoparticle measured as the hydrodynamic diameter of the nanoparticle and as the proportion of therapeutic agent comprised by the nanoparticle.
54. The use of the surfactant in accordance with any of claims 47 to 53, wherein the suspension of lipid nanoparticles or lipidoid nanoparticles in an aqueous vehicle solution comprises the surfactant at a concentration of 0.01 to 10 % (w/v).
55. The use of the surfactant in accordance with any of claims 47 to 54, wherein the therapeutic agent is a nucleic acid.
56. The use of the surfactant in accordance with Claim 55, wherein the nucleic acid is mRNA.
57. The use of the surfactant in accordance with any of claims 47 to 56, wherein the concentration of the nucleic acid in the suspension formulation ranges from 0.01 to 10 mg/mL, based on the total volume of the suspension formulation.
58. The use of the surfactant in accordance with any of claims 47 to 57, wherein the nanoparticles have a Z-average diameter, as determined by dynamic light scattering, in the range of 10 to 500 nm, preferably around 30 to 100 nm.
59. The use of the surfactant in accordance with any of claims 47 to 58, wherein the nanoparticles further comprise one or more of the following components (c1 ) to (c6):
(c1) a non-ionizable lipid having a sterol structure;
(c2) a phospholipid lipid;
(c3) a PEG-conjugated lipid;
(c4) a polysarcosine-conjugated lipid
(c5) a PASylated lipid;
(c6) an ionizable or a cationic polymer or lipidoid.
60. The use of the surfactant in accordance with any of claims 47 to 59, wherein the nanoparticles comprise a) 30 to 65 mol% of at least one selected from the ionizable lipid, an ionizable lipidoid and a permanently cationic lipid (b), and one or more of the following components: ii) 10 to 50 mol% of the lipid having a sterol structure (c1 ), iii) 4 to 50 mol% of the phospholipid (c2), iv) 0.5 to 10 mol% of one of the PEG-conjugated lipid (c3), the polysarcosine-conjugated lipid (c4) and the PASylated lipid (c5), or of any combination thereof,
0.5 to 10 mol% of a cationic polymer (c6), such that the sum of (b) and (c1 ) to (c6) amounts to 100 mol%.
61. The use of the surfactant in accordance with any of claims 47 to 60, wherein the nanoparticles comprise an ionizable lipid (b) of the following formula (a-lll):

III a-lll or a pharmaceutically acceptable salt, prodrug or stereoisomer thereof, wherein: one of L1 or L2 is -O(C=O)-, -(C=O)O-, -C(=O)-, -O-, -S(O)X-, -S-S-, - C(=O)S-, SC(=O)-, - NRaC(=O)-, -C(=O)NRa-, NRaC(=O)NRa-, -OC(=O)NRa- or - NRaC(=O)O-, and the other of L1 or L2 is-O(C=O)-, -(C=O)O-, -C(=O)-, -O-, -S(O)X-, -S- S-, -C(=O)S-, SC(=O)-,
-NRaC(=O)-, -C(=O)NRa-, ,NRaC(=O)NRa-, -OC(=O)NRa- or -NRaC(=O)O- or a direct bond;
G1 and G2 are each independently C1-C12 alkylene or C1-C12 alkenylene;
G3 is C1-C24 alkylene, C1-C24 alkenylene, C3-C8 cycloalkylene, C3-C8 cycloalkenylene, wherein each of alkylene, alkenylene, cycloalkylene, and cyloalkenylene is optionally substituted;
Ra is H or C1-C12 alkyl wherein the alkyl is optionally substituted;
R1 and R2 are each independently C6-C24 alkyl or C6-C24 alkenyl, wherein each of alkyl and alkenyl is optionally substituted;
R3 is H, OR5, ON, -C(=O)OR4, -OC(=O)R4 or-NR5C(=O)R4; R4 is C1-C12 alkyl, wherein alkyl is optionally substituted;
R5 is H or Ci-Ce alkyl, wherein alkyl is optionally substituted; and x is 0, 1 or 2.
62. The use of the surfactant in accordance with any of claims 47 to 61 , wherein the nanoparticles comprise an ionizable lipidoid (b) of the following formula (L-1 ), wherein: a is 1 or 2 and b is an integer of 1 to 4 or a is an integer of 1 to 4 and b is 1 or 2, p is 1 or 2, m is 1 or 2; n is 0 or 1 and m+n is > 2; and
wherein: a is 1 or 2 and b is an integer of 1 to 4 or a is an integer of 1 to 4 and b is 1 or 2, p is 1 or 2, m is 1 or 2; n is 0 or 1 and m+n is > 2; and

R1A to R6A are independently of each other selected from: hydrogen; -CH2-CH(OH)-R7A, -CH(R7A)-CH2-OH, -CH2-CH2-(C=O)-O-R7A, -CH2-CH2-(C=O)-NH-R7A, -CH2-R7A, -C(NH)-NH2I a polyethylene glycol) chain; and a receptor ligand, wherein R7A is selected from C3-C18 alkyl and C3-C18 alkenyl having one C-C double bond, provided that at least two residues among R1A to R6A are selected from -CH2-CH(OH)-R7A, -CH(R7A)-CH2-OH, -CH2-CH2-(C=O)-O-R7A, -CH2-CH2-(C=O)-NH-R7A and -CH2-R7A, wherein R7A is selected from C3-C18 alkyl or C3-C18 alkenyl having one C-C double bond, or a protonated form of the ionizable lipid of formula (L-1 ), wherein one or more of the nitrogen atoms contained in the compound of formula (L-1 ) are protonated to provide a compound carrying a positive charge.
63. Use of the surfactant in accordance with any of claims 47 to 60, wherein the nanoparticles comprise, as an ionizable lipid (b), (6Z,9Z,28Z,31Z)-heptatriaconta-6,9,28,31- tetraene-19-yl 4-(dimethylamino)butanoate or a protonated form thereof wherein the nitrogen atom of the compound is protonated.
64. Use of the surfactant in accordance with any of claims 47 to 60, wherein the nanoparticles comprise, as an ionizable lipid (b), ((4-hydroxybutyl)azanediyl)bis(hexan-6,1- diyl)bis(2-hexyldecanoate) or a protonated form thereof wherein the nitrogen atom of the compound is protonated, and/or (heptadecan -9-yl 8-((2-hydroxyethyl)(6-oxo-6- (undecyloxy)hexyl)amino)octanoate, or a protonated form thereof wherein the nitrogen atom of the compound is protonated.
65. The use according to Claim 64, wherein the nanoparticles comprise
((4-hydroxybutyl)azanediyl)bis(hexane-6,1-diyl)bis(2-hexyldecanoate) or the protonated form thereof wherein the nitrogen atom of the compound is protonated, and optionally further comprise one or more of the following components (d1 ) to (d8):
(d1) 2-[(polyethylene glycol)-2000]-N,N-ditetradecylacetamide (ALC-0159)
(d2) 1 ,2-distearoyl-sn-glycero-3-phosphocholine (DSPC)
(d3) cholesterol
(d4) potassium chloride
(d5) potassium dihydrogen phosphate
(d6) sodium chloride
(d7) disodium phosphate dihydrate
(d8) sucrose.
66. The use according to claim 64, wherein the nanoparticles comprise heptadecan-9-yl 8- ((2-hydroxyethyl)(6-oxo-6-(undecyloxy)hexyl)amino)octanoate or a protonated form thereof wherein the nitrogen atom of the compound is protonated, and further optionally comprise one or more of the following components (e1) to (e7):
(e1) 1 ,2-distearoyl-sn-glycero-3-phosphocholine (DSPC),
(e2) cholesterol,
(e3) 1 ,2-dimyristoyl-rac-glycero-3-methoxypolyethylene glycol-2000 (PEG2000 DMG),
(e4) 2-amino-2-(hydroxymethyl)propan-1 ,3-diol (trometamol) hydrochloride
(e5) sodium acetate trihydrate (e6) acetic acid
(e7) sucrose.
67. The use according to Claim 64, wherein the nanoparticle comprises DLin-MC3-DMA ((6Z,9Z,28Z,31Z)-heptatriaconta-6,9,28,31-tetraen-19-yl-4-(dimethylamino) butanoate) or a protonated form thereof wherein the nitrogen atom of the compound is protonated, and optionally further comprise one or more of the following components (e1 ) to (e7):
(e1) 1,2-distearoyl-sn-glycero-3-phosphocholine (DSPC),
(e2) cholesterol,
(e3) PEG2000-C-DMG (a-(3’-{[1 ,2-di(myristyloxy)propanoxy]carbonylamino}propyl)-(jj- methoxy, polyoxyethylene),
(e4) 2-amino-2-(hydroxymethyl)propan-1 ,3-diol (trometamol) hydrochloride
(e5) Disodium hydrogen phosphate, heptahydrate
(e6) Potassium dihydrogen phosphate, anhydrous
(e7) Sodium chloride.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP23020399.4 | 2023-08-25 | ||
| EP23020399 | 2023-08-25 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2025045767A1true WO2025045767A1 (en) | 2025-03-06 |
Family
ID=87847907
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2024/073691PendingWO2025045767A1 (en) | 2023-08-25 | 2024-08-23 | Stabilized lipid and lipidoid nanoparticle formulations with specific surfactant properties for enhanced pharmaceutical applications |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2025045767A1 (en) |
Citations (31)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4765987A (en)* | 1985-08-30 | 1988-08-23 | Adir & Cie | Artificial surfactants, pharmaceutical compositions containing them and use thereof |
| DE19834683A1 (en) | 1997-08-13 | 1999-04-01 | Biontex Lab Gmbh | New lipopolyamines, their presentation and application |
| US6017700A (en) | 1995-08-04 | 2000-01-25 | Bayer Corporation | Cationic oligonucleotides, and related methods of synthesis and use |
| WO2002055693A2 (en) | 2001-01-09 | 2002-07-18 | Ribopharma Ag | Method for inhibiting the expression of a target gene |
| WO2006138380A2 (en) | 2005-06-15 | 2006-12-28 | Massachusetts Institute Of Technology | Amine-containing lipids and uses thereof |
| WO2007069092A2 (en) | 2005-12-15 | 2007-06-21 | Centre National De La Recherche Scientifique (Cnrs) | Cationic oligonucleotides, automated methods for preparing same and their uses |
| WO2010053572A2 (en) | 2008-11-07 | 2010-05-14 | Massachusetts Institute Of Technology | Aminoalcohol lipidoids and uses thereof |
| WO2011012316A2 (en) | 2009-07-31 | 2011-02-03 | Ludwig-Maximilians-Universität | Rna with a combination of unmodified and modified nucleotides for protein expression |
| US8058069B2 (en) | 2008-04-15 | 2011-11-15 | Protiva Biotherapeutics, Inc. | Lipid formulations for nucleic acid delivery |
| WO2012000104A1 (en) | 2010-06-30 | 2012-01-05 | Protiva Biotherapeutics, Inc. | Non-liposomal systems for nucleic acid delivery |
| US20130115272A1 (en) | 2011-10-03 | 2013-05-09 | modeRNA Therapeutics | Modified nucleosides, nucleotides, and nucleic acids, and uses thereof |
| US20130156849A1 (en) | 2011-12-16 | 2013-06-20 | modeRNA Therapeutics | Modified nucleoside, nucleotide, and nucleic acid compositions |
| US20140010861A1 (en) | 2012-04-02 | 2014-01-09 | modeRNA Therapeutics | Modified polynucleotides for the production of proteins associated with human disease |
| WO2014028487A1 (en) | 2012-08-13 | 2014-02-20 | Massachusetts Institute Of Technology | Amine-containing lipidoids and uses thereof |
| WO2014207231A1 (en) | 2013-06-28 | 2014-12-31 | Ethris Gmbh | Compositions for introducing rna into cells |
| US8969535B2 (en) | 2006-12-05 | 2015-03-03 | Lasergen, Inc. | Photocleavable labeled nucleotides and nucleosides and methods for their use in DNA sequencing |
| US9018187B2 (en) | 2009-07-01 | 2015-04-28 | Protiva Biotherapeutics, Inc. | Cationic lipids and methods for the delivery of therapeutic agents |
| WO2015095351A1 (en) | 2013-12-19 | 2015-06-25 | Novartis Ag | LEPTIN mRNA COMPOSITIONS AND FORMULATIONS |
| US9254311B2 (en) | 2012-04-02 | 2016-02-09 | Moderna Therapeutics, Inc. | Modified polynucleotides for the production of proteins |
| US9345780B2 (en) | 2008-04-11 | 2016-05-24 | Tekmira Pharmaceuticals Corporation | Site specific delivery of nucleic acids by combining targeting ligands with endosomolytic components |
| US9352042B2 (en) | 2012-02-24 | 2016-05-31 | Protiva Biotherapeutics, Inc. | Trialkyl cationic lipids and methods of use thereof |
| WO2016097377A1 (en) | 2014-12-19 | 2016-06-23 | Ethris Gmbh | Compositions for introducing nucleic acid into cells |
| US9394234B2 (en) | 2009-06-10 | 2016-07-19 | Arbutus Biopharma Corporation | Lipid formulations |
| WO2016176330A1 (en) | 2015-04-27 | 2016-11-03 | The Trustees Of The University Of Pennsylvania | Nucleoside-modified rna for inducing an adaptive immune response |
| US9492386B2 (en) | 2002-06-28 | 2016-11-15 | Protiva Biotherapeutics, Inc. | Liposomal apparatus and manufacturing methods |
| WO2017109087A1 (en) | 2015-12-22 | 2017-06-29 | Xl-Protein Gmbh | Nucleic acids encoding repetitive amino acid sequences rich in proline and alanine residues that have low repetitive nucleotide sequences |
| AU2017286606A1 (en)* | 2016-06-14 | 2018-12-13 | Modernatx, Inc. | Stabilized formulations of lipid nanoparticles |
| US20200306191A1 (en)* | 2017-08-31 | 2020-10-01 | Modernatx, Inc. | Methods of making lipid nanoparticles |
| WO2022180213A1 (en)* | 2021-02-26 | 2022-09-01 | Ethris Gmbh | Formulations for aerosol formation and aerosols for the delivery of nucleic acid |
| EP4327829A1 (en)* | 2022-08-26 | 2024-02-28 | Ethris GmbH | Stabilization of lipid or lipidoid nanoparticle suspensions |
| WO2024042236A1 (en)* | 2022-08-26 | 2024-02-29 | Ethris Gmbh | Stable lipid or lipidoid nanoparticle suspensions |
- 2024
- 2024-08-23WOPCT/EP2024/073691patent/WO2025045767A1/enactivePending
Patent Citations (46)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4765987A (en)* | 1985-08-30 | 1988-08-23 | Adir & Cie | Artificial surfactants, pharmaceutical compositions containing them and use thereof |
| US6017700A (en) | 1995-08-04 | 2000-01-25 | Bayer Corporation | Cationic oligonucleotides, and related methods of synthesis and use |
| DE19834683A1 (en) | 1997-08-13 | 1999-04-01 | Biontex Lab Gmbh | New lipopolyamines, their presentation and application |
| WO2002055693A2 (en) | 2001-01-09 | 2002-07-18 | Ribopharma Ag | Method for inhibiting the expression of a target gene |
| US9492386B2 (en) | 2002-06-28 | 2016-11-15 | Protiva Biotherapeutics, Inc. | Liposomal apparatus and manufacturing methods |
| US9504651B2 (en) | 2002-06-28 | 2016-11-29 | Protiva Biotherapeutics, Inc. | Lipid compositions for nucleic acid delivery |
| EP2476756A1 (en) | 2005-06-15 | 2012-07-18 | Massachusetts Institute of Technology | Amine-containing lipids and uses thereof |
| WO2006138380A2 (en) | 2005-06-15 | 2006-12-28 | Massachusetts Institute Of Technology | Amine-containing lipids and uses thereof |
| WO2007069092A2 (en) | 2005-12-15 | 2007-06-21 | Centre National De La Recherche Scientifique (Cnrs) | Cationic oligonucleotides, automated methods for preparing same and their uses |
| US8969535B2 (en) | 2006-12-05 | 2015-03-03 | Lasergen, Inc. | Photocleavable labeled nucleotides and nucleosides and methods for their use in DNA sequencing |
| US9345780B2 (en) | 2008-04-11 | 2016-05-24 | Tekmira Pharmaceuticals Corporation | Site specific delivery of nucleic acids by combining targeting ligands with endosomolytic components |
| US8058069B2 (en) | 2008-04-15 | 2011-11-15 | Protiva Biotherapeutics, Inc. | Lipid formulations for nucleic acid delivery |
| US8492359B2 (en) | 2008-04-15 | 2013-07-23 | Protiva Biotherapeutics, Inc. | Lipid formulations for nucleic acid delivery |
| US9364435B2 (en) | 2008-04-15 | 2016-06-14 | Protiva Biotherapeutics, Inc. | Lipid formulations for nucleic acid delivery |
| US8822668B2 (en) | 2008-04-15 | 2014-09-02 | Protiva Biotherapeutics, Inc. | Lipid formulations for nucleic acid delivery |
| US9556110B2 (en) | 2008-11-07 | 2017-01-31 | Massachusetts Institute Of Technology | Aminoalcohol lipidoids and uses thereof |
| US10189802B2 (en) | 2008-11-07 | 2019-01-29 | Massachusetts Institute Of Technology | Aminoalcohol lipidoids and uses thereof |
| WO2010053572A2 (en) | 2008-11-07 | 2010-05-14 | Massachusetts Institute Of Technology | Aminoalcohol lipidoids and uses thereof |
| US8969353B2 (en) | 2008-11-07 | 2015-03-03 | Massachusetts Institute Of Technology | Aminoalcohol lipidoids and uses thereof |
| US9394234B2 (en) | 2009-06-10 | 2016-07-19 | Arbutus Biopharma Corporation | Lipid formulations |
| US9018187B2 (en) | 2009-07-01 | 2015-04-28 | Protiva Biotherapeutics, Inc. | Cationic lipids and methods for the delivery of therapeutic agents |
| WO2011012316A2 (en) | 2009-07-31 | 2011-02-03 | Ludwig-Maximilians-Universität | Rna with a combination of unmodified and modified nucleotides for protein expression |
| US9006417B2 (en) | 2010-06-30 | 2015-04-14 | Protiva Biotherapeutics, Inc. | Non-liposomal systems for nucleic acid delivery |
| WO2012000104A1 (en) | 2010-06-30 | 2012-01-05 | Protiva Biotherapeutics, Inc. | Non-liposomal systems for nucleic acid delivery |
| US9518272B2 (en) | 2010-06-30 | 2016-12-13 | Protiva Biotherapeutics, Inc. | Non-liposomal systems for nucleic acid delivery |
| US20130123481A1 (en) | 2011-10-03 | 2013-05-16 | modeRNA Therapeutics | Modified nucleosides, nucleotides, and nucleic acids, and uses thereof |
| US20130115272A1 (en) | 2011-10-03 | 2013-05-09 | modeRNA Therapeutics | Modified nucleosides, nucleotides, and nucleic acids, and uses thereof |
| US20130156849A1 (en) | 2011-12-16 | 2013-06-20 | modeRNA Therapeutics | Modified nucleoside, nucleotide, and nucleic acid compositions |
| US9352042B2 (en) | 2012-02-24 | 2016-05-31 | Protiva Biotherapeutics, Inc. | Trialkyl cationic lipids and methods of use thereof |
| US20140010861A1 (en) | 2012-04-02 | 2014-01-09 | modeRNA Therapeutics | Modified polynucleotides for the production of proteins associated with human disease |
| US10501512B2 (en) | 2012-04-02 | 2019-12-10 | Modernatx, Inc. | Modified polynucleotides |
| US9254311B2 (en) | 2012-04-02 | 2016-02-09 | Moderna Therapeutics, Inc. | Modified polynucleotides for the production of proteins |
| US20160114042A1 (en) | 2012-08-13 | 2016-04-28 | Massachusetts Institute Of Technology | Amine-containing lipidoids and uses thereof |
| WO2014028487A1 (en) | 2012-08-13 | 2014-02-20 | Massachusetts Institute Of Technology | Amine-containing lipidoids and uses thereof |
| US9227917B2 (en) | 2012-08-13 | 2016-01-05 | Massachusetts Institute Of Technology | Amine-containing lipidoids and uses thereof |
| WO2014207231A1 (en) | 2013-06-28 | 2014-12-31 | Ethris Gmbh | Compositions for introducing rna into cells |
| WO2015095351A1 (en) | 2013-12-19 | 2015-06-25 | Novartis Ag | LEPTIN mRNA COMPOSITIONS AND FORMULATIONS |
| WO2016097377A1 (en) | 2014-12-19 | 2016-06-23 | Ethris Gmbh | Compositions for introducing nucleic acid into cells |
| WO2016176330A1 (en) | 2015-04-27 | 2016-11-03 | The Trustees Of The University Of Pennsylvania | Nucleoside-modified rna for inducing an adaptive immune response |
| WO2017109087A1 (en) | 2015-12-22 | 2017-06-29 | Xl-Protein Gmbh | Nucleic acids encoding repetitive amino acid sequences rich in proline and alanine residues that have low repetitive nucleotide sequences |
| EP3394266B1 (en) | 2015-12-22 | 2021-04-14 | XL-protein GmbH | Nucleic acids encoding repetitive amino acid sequences rich in proline and alanine residues that have low repetitive nucleotide sequences |
| AU2017286606A1 (en)* | 2016-06-14 | 2018-12-13 | Modernatx, Inc. | Stabilized formulations of lipid nanoparticles |
| US20200306191A1 (en)* | 2017-08-31 | 2020-10-01 | Modernatx, Inc. | Methods of making lipid nanoparticles |
| WO2022180213A1 (en)* | 2021-02-26 | 2022-09-01 | Ethris Gmbh | Formulations for aerosol formation and aerosols for the delivery of nucleic acid |
| EP4327829A1 (en)* | 2022-08-26 | 2024-02-28 | Ethris GmbH | Stabilization of lipid or lipidoid nanoparticle suspensions |
| WO2024042236A1 (en)* | 2022-08-26 | 2024-02-29 | Ethris Gmbh | Stable lipid or lipidoid nanoparticle suspensions |
Non-Patent Citations (32)
| Title |
|---|
| A.C. SILVA ET AL., CURRENT DRUG METABOLISM, vol. 16, 2015, pages 3 - 16 |
| AKINC, A. ET AL., NATURE BIOTECHNOLOGY, vol. 26, no. 5, 2008, pages 561 - 569 |
| BHAN AMANDAL SS, CHEMMEDCHEM, 26 March 2014 (2014-03-26) |
| BRUMMELKAMP, SCIENCE, vol. 296, 2002, pages 550 - 553 |
| CANTARA ET AL., NUCLEIC ACIDS RES, vol. 39, 2011, pages D195 - D201 |
| CARELL ET AL., ANGEW CHEM INT ED ENGL, vol. 51, no. 29, 2012, pages 7110 - 31 |
| CROOK, EMBO J., vol. 8, 1989, pages 513 - 519 |
| DONZE, NUCLEIC ACIDS RES, vol. 30, 2002, pages e46 |
| ELBASHIR ET AL., NATURE, vol. 411, no. 6836, 2001, pages 494 - 498 |
| ELBASHIR, EMBO J., vol. 20, 2001, pages 6877 - 6888 |
| ELBASHIR, METHODS, vol. 26, 2002, pages 199 - 213 |
| EUR J PHARM BIOPHARM, vol. 71, 2009, pages 484 - 489 |
| GOSSEN, TRENDS BIOTECH, vol. 12, 1994, pages 58 - 62 |
| GOSSENBUJARD, PROC. NATL. ACAD. SCI. USA, vol. 89, 1992, pages 5547 - 5551 |
| HELMALFONZO, CHEM BIOL, vol. 21, no. 2, 2014, pages 174 - 185 |
| J CONTROL RELEASE, vol. 150, 2011, pages 238 - 247 |
| J.C. KASPER ET AL., J. CONTR. REL., vol. 151, 2011, pages 246 - 255 |
| KOWALSKI, P.S. ET AL., MOLECULAR THERAPY, vol. 27, no. 4, 2019, pages 710 - 728 |
| KULKAMI, J. A. ET AL., NUCLEIC ACID THERAPEUTICS, vol. 28, no. 3, 2018, pages 146 - 157 |
| LA COUNT, BIOCHEM. PARAS., vol. 111, 2000, pages 67 - 76 |
| LI, B. ET AL., NANO LETTERS, vol. 15, 2015, pages 8099 - 8107 |
| MARIE, CURR. BIOL., vol. 10, 2000, pages 1071 - 1074 |
| METHODS FOR GENOME ENGINEERING ARE REVIEWED IN TRENDS IN BIOTECHNOLOGY, vol. 31, no. 7, 2013, pages 397 - 405 |
| NAT. BIOTECHNOL., vol. 32, no. 4, 2014, pages 347 - 355 |
| PFEIFER ET AL., THER DELIV., vol. 1, no. 1, 2010, pages 133 - 48 |
| S. PATEL ET AL.: "Naturally-occurring cholesterol analogues in lipid nanoparticles induce polymorphic shape and enhance intracellular delivery of mRNA", NATURE COMMUNICATIONS, vol. 11, 2020, pages 983, XP055877888, DOI: 10.1038/s41467-020-14527-2 |
| SABNIS, S. ET AL., MOLECULAR THERAPY, vol. 26, no. 6, June 2018 (2018-06-01), pages 1509 - 1519 |
| SAMBROOK ET AL., MOLECULAR CLONING, A LABORATORY MANUAL, 1989 |
| SVOBODA, DEVELOPMENT, vol. 127, 2000, pages 4147 - 4156 |
| TAYLORFRANCIS: "Gene and Cell Therapy - Therapeutic Mechanisms and Strategy", 2009, CRC PRESS |
| WEI, DEV. BIOL., vol. 15, 2000, pages 239 - 255 |
| YANG, PNAS, vol. 99, 2002, pages 9942 - 9947 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP7482028B2 (en) | Compositions and methods for gene editing for hemophilia A | |
| US11692205B2 (en) | Systems and methods for one-shot guide RNA (ogRNA) targeting of endogenous and source DNA | |
| AU2018224326B2 (en) | Novel codon-optimized CFTR mRNA | |
| WO2022180213A1 (en) | Formulations for aerosol formation and aerosols for the delivery of nucleic acid | |
| KR102312903B1 (en) | New minimal UTR sequence | |
| EP4577243A1 (en) | Stable lipid or lipidoid nanoparticle suspensions | |
| CN112153986A (en) | Lipid-based formulations for delivery of RNA | |
| CN115398546A (en) | Improved method for in vitro transcription of messenger RNA | |
| EP4367228A1 (en) | Modular prime editor systems for genome engineering | |
| EP4083227A1 (en) | Linear dna with enhanced resistance against exonucleases | |
| WO2023235501A1 (en) | High fidelity nucleotide polymerase chimeric prime editor systems | |
| CN116438298A (en) | Large-Scale Synthesis of Messenger RNA | |
| CN118234855A (en) | RNA-guided kilobase-scale genome recombination engineering | |
| CN118166004A (en) | MRNA for encoding human PCCA or PCCB protein and use thereof | |
| WO2021055609A1 (en) | Mrna encoding engineered cftr | |
| EP4327829A1 (en) | Stabilization of lipid or lipidoid nanoparticle suspensions | |
| TW202513796A (en) | Engineered rna molecules with adjustable expression and uses thereof | |
| WO2025045767A1 (en) | Stabilized lipid and lipidoid nanoparticle formulations with specific surfactant properties for enhanced pharmaceutical applications | |
| EP4311559A1 (en) | Nanoparticles and peptides for the delivery of cargos to chondrocytes | |
| WO2025039972A1 (en) | Tls-based gene editing systems | |
| EP4606900A1 (en) | Modified rna for the treatment of cfdna-associated diseases | |
| WO2025140249A1 (en) | A circular rna and relevant constructs and a method using the same | |
| WO2025061842A1 (en) | Gene editing tgm1 mutations for treating autosomal recessive congenital ichthyosis (arci) | |
| WO2024263919A2 (en) | Methods and compositions for treating epithelial diseases | |
| WO2019144064A2 (en) | Nanoparticles comprising protein-polynucleotide complexes and for delivering protein based complexes |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | Ref document number:24761220 Country of ref document:EP Kind code of ref document:A1 |