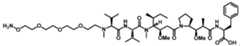
Attorney Docket No. AMB1013WOPCT1 ANTI-PSMA ADC CONJUGATE COMPOSITIONS AND METHODS OF USE THEREOF CROSS-REFERENCE [001] This Application claims the benefit of U.S. Provisional Application No. 63/578,131, filed on August 22, 2023, U.S. Provisional Application No. 63/589,823, filed on October 12, 2023, U.S. Provisional Application No. 63/591,090, filed on October 17, 2023, U.S. Provisional Application No. 63/591,665, filed on October 19, 2023, each of which are incorporated herein by reference in their entirety. FIELD OF THE INVENTION [002] The invention disclosure relates to novel anti-prostate-specific membrane antigen (anti- PSMA) antibodies and antibody drug conjugates. More particularly, the invention disclosure relates to uses of anti-PSMA antibody drug conjugates in inhibiting, preventing or treating PSMA related diseases or cancers. The invention disclosure further relates to pharmaceutical formulations containing anti-PSMA antibody drug conjugates. BACKGROUND OF THE INVENTION [003] Prostate cancer is the most commonly diagnosed non-skin related malignancy in males in developed countries. It is estimated that one in six males will be diagnosed with prostate cancer. The diagnosis of prostate cancer has improved following the use of serum-based markers such as the prostate-specific antigen (PSA). In addition, prostate tumor-associated antigens offer targets for tumor imaging, diagnosis, and targeted therapies. Prostate specific membrane antigen (PSMA), a prostate tumor associated marker, is such a target. PSMA is significantly overexpressed in androgen independent prostate cancer. Overexpression of PSMA is associated with high tumor grade, a high risk of disease progression and recurrence Attorney Docket No. AMB1013WOPCT1 (Perner S. et al., Human Pathology, 2007, 38(5):696-701). High expression of PSMA has been associated with negative clinical prognosis and significantly shorter survival. [004] High levels of PSMA expression have been found in prostate cancer, and particularly in metastatic castration-resistant prostate cancer (mCRPC). Additionally, PSMA has been found in a variety of other solid tumors. Prostate cancer represents a significant unmet need. In 2018, there were 1.3 million new cases of prostate cancer with five-year survival rates of approximately 27% and 359,000 associated deaths worldwide. Prostate cancer is the second leading cause of death in men in the United States (Siegel R.L. et al., CA: A Cancer Journal for Clinicians; 2023, 73:17–48). Treatment options include surgery, radiation, and androgen- deprivation therapy (ADT), however patients with advanced prostate cancer eventually progress to mCRPC, which has a median overall survival of approximately 2 years (Khoshkar Y. et al., BJUI Compass, 2022, 3:173–83). Although approved treatments for mCRPC patients include diverse drug classes such as taxanes, androgen receptor pathway inhibitors, pembrolizumab for microsatellite instability-high (MSI-high) or mismatch repair-deficient subpopulations, radiotherapy, and PARP inhibitors for patients with BRCA mutations (Lowrance W. et al., Journal of Urology, 2023, 209:1082–90), patients eventually develop resistance to these treatments and the disease remains incurable. [005] PSMA is a promising prostate cancer therapeutic target with its limited expression in healthy cells and high incidence of overexpression on the surface of primary prostate tumors, as well as in metastatic lesions in the lymph node and bone (Hupe M.C., et al., Front. Oncol, 2018, 8:623; Queisser A. et al., Modern Pathology, Elsevier, 2015, 28:138–45). In 2022, based on results from the Phase III VISION trial, the radiotherapy Pluvicto was approved for mCRPC (Sartor O. et al., N Engl J Med. 2021, 385:1091–103), which clinically validated PSMA as a therapeutic target for prostate cancer. PSMA also internalizes, making it an ideal target for antibody drug conjugates (ADCs) which deliver cytotoxic drugs to tumor cells via binding to Attorney Docket No. AMB1013WOPCT1 target antigens on tumor cells, internalization, and subsequent release of cytotoxic payload (i.e., free payload) to the cytoplasm (Liu H. et al., Rajasekaran AK, Moy P, Xia Y, Kim S, Navarro V, et al., Cancer Res. 1998, 58:4055–60). Multiple PSMA-targeted ADCs, using different payloads and linker technologies, were previously developed and evaluated in clinical trials (Petrylak D.P. et al., Prostate, 2020, 80:99–108; Petrylak D.P. et al., Prostate, 2019, 79:604– 13; Milowsky M.I. et al., Urol. Oncol., 2016, 34:530, e15-530.e21; de Bono J.S. et al., Clin Cancer Res. 2021, 27:3602–9). However, these ADCs appear to have discontinued their clinical development, presumably due to unacceptable toxicity and/or a narrow therapeutic index. [006] There exists a need for improved therapeutics targeting PSMA and PSMA-related cancers. To overcome this deficiency in the art, the present disclosure provides anti-PSMA ADCs and pharmaceutical compositions containing them, and methods of treating human subjects having PSMA-expressing diseases or cancers with said ADCs and compositions. SUMMARY OF THE INVENTION [007] The present invention disclosure provides anti-PSMA antibody drug conjugates for treating a PSMA-related diseases or cancer. In some embodiments, the disclosure provides anti-PSMA antibody drug conjugates for inhibiting, preventing, or treating PSMA-related conditions, disorders, diseases, or cancers. The invention disclosure further provides stable pharmaceutical formulations containing an anti-PSMA antibody drug conjugate suitable for administration to a human subject. [008] In some general aspects, the present disclosure provides a method of treating cancer, comprising: administering an effective amount of an anti-PSMA antibody drug conjugate (ADC) to a human subject in need thereof, wherein the anti-PSMA ADC comprises: a humanized anti-PSMA monoclonal antibody comprising two heavy chains and two Attorney Docket No. AMB1013WOPCT1 light chains, wherein one non-natural amino acid para-acetyl-L-phenylalanine (pAF) is incorporated into each said heavy chain at position 114 according to Kabat numbering; wherein one drug-linker is conjugated to each said pAF via an oxime linkage, wherein each said drug- linker is amberstatin269 (AS269) having the following structure: .
SEQ ID NO: 1. In some embodiments, each light chain comprises a light chain variable region of SEQ ID NO: 2. In some embodiments, each heavy chain comprises a heavy chain variable region of SEQ ID NO: 1 and each light chain comprises a light chain variable region of SEQ ID NO: 2. In some embodiments, each heavy chain amino acid sequence is SEQ ID NO: 8, comprising the pAF incorporated at Kabat position 114 (i.e., amino acid position 116 in SEQ ID NO: 8). In some embodiments, each light chain amino acid sequence is SEQ ID NO: 9. In some embodiments, each heavy chain amino acid sequence is SEQ ID NO: 8, comprising the pAF incorporated at Kabat position 114 (i.e., amino acid position 116 in SEQ ID NO: 8), and each light chain amino acid sequence is SEQ ID NO: 9. In some embodiments, the anti-PSMA ADC is ARX517. [009] In some embodiments, the effective amount of the anti-PSMA ADC is at least about 1.4 milligram per kilogram (mg/kg) of body weight of the human subject. In some embodiments, the effective amount of the anti-PSMA ADC is at most about 5 mg/kg of the body weight of the human subject. In some embodiments, the effective amount of the anti- PSMA ADC is at least about 1.4 milligram per kilogram (mg/kg) and at most about 5 mg/kg of body weight of the human subject. In some embodiments, the effective amount of the anti- Attorney Docket No. AMB1013WOPCT1 PSMA ADC is at least about 1.4 milligram per kilogram (mg/kg) and at most about 3.4 mg/kg of body weight of the human subject. [010] In some embodiments, the effective amount of the anti-PSMA ADC is about 1.4 mg/kg, about 1.7 mg/kg, about 2 mg/kg, about 2.4 mg/kg, about 2.9 mg/kg, about 3.2 mg/kg, about 3.4 mg/kg, about 3.5 mg/kg, about 4.3 mg/kg, about 4.5 mg/kg, about 4.7 mg/kg, or about 5 mg/kg of the body weight of the human subject. In some embodiments, the effective amount of the anti-PSMA ADC is about 1.4 mg/kg of the body weight of the human subject. In some embodiments, the effective amount of the anti-PSMA ADC is about 1.7 mg/kg of the body weight of the human subject. In some embodiments, the effective amount of the anti-PSMA ADC is about 2 mg/kg of the body weight of the human subject. In some embodiments, the effective amount of the anti-PSMA ADC is greater than 2.0 mg/kg of the body weight of the human subject. In some embodiments, the effective amount of the anti-PSMA ADC is about 2.4 mg/kg of the body weight of the human subject. In some embodiments, the effective amount of the anti-PSMA ADC is greater than 2.4 mg/kg of the body weight of the human subject. In some embodiments, the effective amount of the anti-PSMA ADC is at least 2.5 mg/kg of the body weight of the human subject. In some embodiments, the effective amount of the anti-PSMA ADC is about 2.9 mg/kg of the body weight of the human subject. In some embodiments, the effective amount of the anti-PSMA ADC is greater than 2.9 mg/kg of the body weight of the human subject. In some embodiments, the effective amount of the anti- PSMA ADC is at least 3.0 mg/kg of the body weight of the human subject. In some embodiments, the effective amount of the anti-PSMA ADC is about 3.1 mg/kg of the body weight of the human subject. In some embodiments, the effective amount of the anti-PSMA ADC is about 3.2 mg/kg of the body weight of the human subject. In some embodiments, the effective amount of the anti-PSMA ADC is about 3.3 mg/kg of the body weight of the human subject. In some embodiments, the effective amount of the anti-PSMA ADC is about 3.4 mg/kg Attorney Docket No. AMB1013WOPCT1 of the body weight of the human subject. In some embodiments, the effective amount of the anti-PSMA ADC is about 3.5 mg/kg of the body weight of the human subject. In some embodiments, the effective amount of the anti-PSMA ADC is about 3.6 mg/kg of the body weight of the human subject. In some embodiments, the effective amount of the anti-PSMA ADC is about 3.7 mg/kg of the body weight of the human subject. In some embodiments, the effective amount of the anti-PSMA ADC is about 3.8 mg/kg of the body weight of the human subject. In some embodiments, wherein the effective amount of the anti-PSMA ADC is about 3.9 mg/kg of the body weight of the human subject. In some embodiments, the effective amount of the anti-PSMA ADC is about 4.0 mg/kg of the body weight of the human subject. In some embodiments, the effective amount of the anti-PSMA ADC is about 4.3 mg/kg of the body weight of the human subject. In some embodiments, the effective amount of the anti- PSMA ADC is about 4.4 mg/kg of the body weight of the human subject. In some embodiments, the effective amount of the anti-PSMA ADC is about 4.5 mg/kg of the body weight of the human subject. In some embodiments, the effective amount of the anti-PSMA ADC is about 4.7 mg/kg of the body weight of the human subject. In some embodiments, the effective amount of the anti-PSMA ADC is about 5 mg/kg of the body weight of the human subject. [011] In some embodiments, the method of treating cancer comprises administering an effective amount of the anti-PSMA antibody drug conjugate (ADC) to a human subject in need thereof, wherein the effective amount of the anti-PSMA ADC is greater than 2.0 mg/kg and at most about 5.0 mg/kg of body weight of the human subject. In some embodiments, the effective amount of the anti-PSMA ADC is greater than 2.0 mg/kg and at most about 3.4 mg/kg of body weight of the human subject. In some embodiments, the effective amount of the anti- PSMA ADC is at least about 2.4 mg/kg and at most about 3.4 mg/kg of the body weight of the human subject. In some embodiments, the effective amount of the anti-PSMA ADC is at least Attorney Docket No. AMB1013WOPCT1 about 2.8 mg/kg and at most about 3.4 mg/kg of the body weight of the human subject. In some embodiments, the effective amount of the anti-PSMA ADC is at least about 3.0 mg/kg and at most about 3.4 mg/kg of the body weight of the human subject. [012] In some embodiments, an effective amount of the anti-PSMA ADC is administered to a human subject on a dosing schedule. In some embodiments, the dosing schedule is once every 1, 2, 3, 4, 5 or 6 weeks. In some embodiments, the dosing schedule is once every two weeks. In some embodiments, the dosing schedule is once every three weeks. In some embodiments, the dosing schedule is once every four weeks. In some embodiments, the dosing schedule is more than once within a three-week cycle. [013] In some embodiments, the effective amount of the anti-PSMA ADC is about 1.4 mg/kg of the body weight of the human subject, and the administering is once every three weeks. In some embodiments, the effective amount of the anti-PSMA ADC is about 1.7 mg/kg of the body weight of the human subject, and the administering is once every three weeks. In some embodiments, the effective amount of the anti-PSMA ADC is about 2.0 mg/kg of the body weight of the human subject, and the administering is once every three weeks. In some embodiments, the effective amount of the anti-PSMA ADC is about 2.4 mg/kg of the body weight of the human subject, and the administering is once every three weeks. In some embodiments, the effective amount of the anti-PSMA ADC is about 2.9 mg/kg of the body weight of the human subject, and the administering is once every three weeks. In some embodiments, the effective amount of the anti-PSMA ADC is about 3.0 mg/kg of the body weight of the human subject, and the administering is once every three weeks. In some embodiments, the effective amount of the anti-PSMA ADC is about 3.1 mg/kg of the body weight of the human subject, and the administering is once every three weeks. In some embodiments, the effective amount of the anti-PSMA ADC is about 3.2 mg/kg of the body weight of the human subject, and the administering is once every three weeks. In some Attorney Docket No. AMB1013WOPCT1 embodiments, the effective amount of the anti-PSMA ADC is about 3.3 mg/kg of the body weight of the human subject, and the administering is once every three weeks. In some embodiments, the effective amount of the anti-PSMA ADC is about 3.4 mg/kg of the body weight of the human subject, and the administering is once every three weeks. In some embodiments, the effective amount of the anti-PSMA ADC is about 3.5 mg/kg of the body weight of the human subject, and the administering is once every three weeks. [014] In some embodiments, the human subject has a PSMA-expressing prostate cancer or non-prostate cancer. In some embodiments, the PSMA-expressing cancer is a PSMA low expressing cancer. In some embodiments, the PSMA-expressing cancer is a PSMA moderate expressing cancer. In some embodiments, the PSMA-expressing cancer is a PSMA high expressing cancer. [015] In some embodiments, the human subject has prostate cancer. In some embodiments, the prostate cancer is metastatic castration-resistant prostate cancer (mCRPC). In some embodiments, the human subject has been previously administered a taxane therapy. In some embodiments, the prostate cancer has progressed after prior taxane therapy. In some embodiments, the cancer is hormone refractory prostate cancer. In some embodiments, the cancer is resistant or refractory to prior standard therapies for the treatment of prostate cancer. In some embodiments, the human subject has been previously treated with abiraterone, darolutamide, apalutamide or enzalutamide. [016] In some embodiments, the method of treatment with an anti-PSMA ADC provided herein delays or inhibits progression of cancer in the human subject. In some embodiments, the method increases the survival of the human subject as compared to the median survival of subjects who have not been previously treated with the same or a different anti-PSMA ADC. In some embodiments, the method increases the survival of the human subject, wherein the survival is increased in comparison to the median survival time of subjects with anti- Attorney Docket No. AMB1013WOPCT1 PSMA-expressing, taxane-resistant cancer not previously treated with the same or a different anti-PSMA ADC. [017] In some embodiments, the method of treatment with an anti-PSMA ADC provided herein decreases a circulating level of circulating tumor cells (CTCs) in the human subject compared to a baseline level of the CTCs in the human subject. In some embodiments, the method decreases or stabilizes a serum level of prostate specific antigen (PSA) in the human subject compared to a baseline level of PSA in the human subject. In some embodiments, the method decreases the serum level of PSA in the human subject compared to the baseline level of PSA in the human subject. In some embodiments, the decrease in the serum level of PSA in the human subject is at least about a 30% decrease in the serum level of PSA in the human subject compared to the baseline level of PSA in the human subject. In some embodiments, the decrease in the serum level of PSA in the human subject is at least about a 50% decrease in the serum level of PSA in the human subject compared to the baseline level of PSA in the human subject. In some embodiments, the decrease in the serum level of PSA in the human subject is at least about a 90% decrease in the serum level of PSA in the human subject compared to the baseline level of PSA in the human subject. [018] In some embodiments, the anti-PSMA ADC is administered intravenously. [019] In some embodiments, the method of treatment with an anti-PSMA ADC provided herein provides an anti-PSMA ADC serum terminal half-life of at least about 5 days after the administration of the effective amount of the anti-PSMA ADC to the human subject. In some embodiments, the method of treatment provides an anti-PSMA ADC serum terminal half-life within a range of about 5 to about 10 days after the administration of the effective amount of the anti-PSMA ADC to the human subject. [020] In some embodiments, the method of treatment with an anti-PSMA ADC provided herein provides a free payload time to serum maximum concentration (Tmax) of at least about Attorney Docket No. AMB1013WOPCT1 5 days after the administration of the effective amount of the anti-PSMA ADC to the human subject, wherein the free payload that is released from the anti-PSMA ADC is pAF-AS269 having the following structure (see FIG.8C): ;

Tmax of at least about 6 days after the administration of the effective amount of the anti-PSMA ADC to the human subject. In some embodiments, the method of treatment provides a free payload Tmax of about one week after the administration of the effective amount of the anti-PSMA ADC to the human subject. [021] In some embodiments, the method of treatment with an anti-PSMA ADC provided herein provides a free payload serum maximum concentration (Cmax) of at most about 1 ng/mL after the administration of the effective amount of the anti-PSMA ADC to the human subject. In some embodiments, the method of treatment provides a free payload Cmax of at most about 0.5 ng/mL, at most about 0.4 ng/mL, at most about 0.3 ng/mL or at most about 0.2 ng/mL after the administration of the effective amount of the anti-PSMA ADC to the human subject. In some embodiments, the method of treatment provides a free payload Cmax within a range of about 0.01 ng/mL to about 0.3 ng/mL after the administration of the effective amount of the anti-PSMA ADC to the human subject. [022] In some embodiments, the method of treatment with an anti-PSMA ADC provided herein provides an anti-PSMA ADC serum maximum concentration (Cmax) of at least about 20 µg/mL, at least about 30 µg/ml, at least about 40 µg/ml, at least about 50 µg/ml, or at least about 60 µg/ml after the administration of the effective amount of the anti-PSMA ADC to the human subject. In some embodiments, the method of treatment with an anti-PSMA ADC Attorney Docket No. AMB1013WOPCT1 provided herein provides an anti-PSMA ADC serum maximum concentration (Cmax) of at least about 20 µg/mL after the administration of the effective amount of the anti-PSMA ADC to the human subject. In some embodiments, the method of treatment with an anti-PSMA ADC provided herein provides an anti-PSMA ADC serum maximum concentration (Cmax) of at least about 30 µg/mL after the administration of the effective amount of the anti-PSMA ADC to the human subject. In some embodiments, the method of treatment with an anti-PSMA ADC provided herein provides an anti-PSMA ADC serum maximum concentration (Cmax) of at least about 40 µg/mL after the administration of the effective amount of the anti-PSMA ADC to the human subject. In some embodiments, the method of treatment with an anti-PSMA ADC provided herein provides an anti-PSMA ADC serum maximum concentration (Cmax) of at least about 50 µg/mL after the administration of the effective amount of the anti-PSMA ADC to the human subject. In some embodiments, the method of treatment with an anti-PSMA ADC provided herein provides an anti-PSMA ADC serum maximum concentration (Cmax) of at least about 60 µg/mL after the administration of the effective amount of the anti-PSMA ADC to the human subject [023] In some embodiments, the method of treatment with an anti-PSMA ADC provided herein provides a reduction in circulating tumor DNA (ctDNA) of at least about 50% after the administration of the effective amount of the anti-PSMA ADC to the human subject. [024] In some aspects, a method of treating cancer in a subject in need thereof comprising administering the anti-PSMA ADC of the present disclosure further comprises administering an effective amount of another therapeutic agent. In some embodiments, the therapeutic agent is a chemotherapeutic agent, a hormonal agent, an antitumor agent, an immunostimulatory agent, an immunomodulator, an immunotherapeutic agent or combination thereof. In some embodiments, the therapeutic agent is a hormonal agent. In some embodiments, the hormonal agent is enzalutamide. In some embodiments, the therapeutic agent is a checkpoint inhibitor, a Attorney Docket No. AMB1013WOPCT1 PSMA kinase inhibitor, cyclin-dependent kinase inhibitor, tyrosine kinase inhibitor, small- molecule kinase inhibitor, or a platinum-based therapeutic. [025] In some other general aspects, the present disclosure provides a pharmaceutical composition comprising an effective amount of an anti-PSMA antibody drug conjugate (ADC), wherein the anti-PSMA ADC comprises: a humanized anti-PSMA monoclonal antibody comprising two heavy chains and two light chains, wherein each heavy chain comprises a heavy chain variable region of SEQ ID NO: 1, and wherein one non-natural amino acid para-acetyl-L-phenylalanine (pAF) is incorporated into each said heavy chain at position 114 according to Kabat numbering, and each light chain comprises a light chain variable region of SEQ ID NO: 2; wherein one drug- linker is conjugated to each said pAF via an oxime linkage, wherein each said drug-linker is amberstatin269 (AS269) having the following structure: .

[026] In some embodiments, the present disclosure provides a pharmaceutical composition comprising an effective amount of an anti-PSMA antibody drug conjugate (ADC), wherein the anti-PSMA ADC comprises: a humanized anti-PSMA monoclonal antibody comprising two heavy chains and two light chains, wherein each heavy chain amino acid sequence is SEQ ID NO: 8, wherein one non-natural amino acid para-acetyl-L-phenylalanine (pAF) is incorporated into each said heavy chain at position 114 according to Kabat numbering, and each light chain amino acid sequence is SEQ ID NO: 9; wherein one drug-linker is conjugated to each said pAF via an oxime linkage, wherein each said drug-linker is amberstatin269 (AS269) having the following structure: Attorney Docket No. AMB1013WOPCT1 .
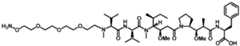
PSMA ADC is ARX517. [028] In some embodiments, the pharmaceutical composition comprises (i) the anti-PSMA ADC at a concentration within a range of about 5 mg/mL to about 25 mg/mL; and (ii) pharmaceutically acceptable components selected from the group consisting of sucrose, histidine buffer and polysorbate 80, and a combination thereof; wherein the composition pH is within a range of about 5.5 to about 6.5. In some embodiments, the pharmaceutical composition comprises the anti-PSMA ADC concentration at a concentration of about 10 mg/mL. In some embodiments, the sucrose concentration is within a range of about 5% (w/v) to about 15% (w/v); the histidine buffer concentration is within a range of about 15 mM to about 25 mM; and the polysorbate 80 concentration is within a range of about of about 0.001% (w/v) to about 0.02% (w/v). In some embodiments, the pharmaceutically acceptable components consist essentially of the sucrose, the histidine buffer and the polysorbate 80. In some embodiments, the composition pH is within a range of about 5.6 to about 6.2. In some embodiments, the composition consists essentially of the anti-PSMA ADC at a concentration of about 10 mg/mL; sucrose at a concentration of about 9% (w/v); histidine buffer at a concentration of about 20 mM; and polysorbate 80 at a concentration of about 0.01% (w/v); wherein the composition pH is about 5.9 ± 0.3. In some embodiments, the histidine buffer consists essentially of L-histidine, L-histidine hydrochloride and water. [029] In some embodiments, the pharmaceutical composition comprises (i) ARX517 at a concentration within a range of about 5 mg/mL to about 25 mg/mL; and (ii) pharmaceutically acceptable components selected from the group consisting of sucrose, histidine buffer and Attorney Docket No. AMB1013WOPCT1 polysorbate 80, and a combination thereof; wherein the composition pH is within a range of about 5.5 to about 6.5. In some embodiments, the pharmaceutical composition comprises ARX517 concentration at a concentration of about 10 mg/mL. In some embodiments, the sucrose concentration is within a range of about 5% (w/v) to about 15% (w/v); the histidine buffer concentration is within a range of about 15 mM to about 25 mM; and the polysorbate 80 concentration is within a range of about of about 0.001% (w/v) to about 0.02% (w/v). In some embodiments, the pharmaceutically acceptable components consist essentially of the sucrose, the histidine buffer and the polysorbate 80. In some embodiments, the composition pH is within a range of about 5.6 to about 6.2. In some embodiments, the composition consists essentially of ARX517 at a concentration of about 10 mg/mL; sucrose at a concentration of about 9% (w/v); histidine buffer at a concentration of about 20 mM; and polysorbate 80 at a concentration of about 0.01% (w/v); wherein the composition pH is about 5.9 ± 0.3. In some embodiments, the histidine buffer consists essentially of L-histidine, L-histidine hydrochloride and water. [030] In some embodiments, the pharmaceutical composition is a liquid formulation. In some embodiments, the composition is stored frozen. In some embodiments, the composition is stored frozen at about -20 °C. In some embodiments, the composition is stored refrigerated. In some embodiments, the composition is stored frozen at about 5 °C ± 3 °C. [031] In some embodiments, the anti-PSMA ADC of the pharmaceutical composition provided herein comprises charged variants. In some embodiments, the charged variants comprise an anti-PSMA ADC main species, an anti-PSMA ADC acidic species and an anti- PSMA ADC basic species. In some embodiments, the anti-PSMA ADC main species has an isoelectric point (pI) of about 8.3. In some embodiments, the anti-PSMA ADC acidic species has a pI of about 8.1. In some embodiments, the anti-PSMA ADC basic species has a pI of about 8.4. Attorney Docket No. AMB1013WOPCT1 [032] In some embodiments, the anti-PSMA ADC main species is present in an amount of about 40% to about 70%. In some embodiments, the anti-PSMA ADC acidic species is present in an amount of about 20% to about 40%. In some embodiments, the anti-PSMA ADC basic species is present in an amount of about 5% to about 30%. In some embodiments, the sum of the percentage of each main species, acidic species and basic species is 100%. [033] In some embodiments, the percentage of the main species, the acidic species and the basic species is a UV area % at 214 nm based on integration of the corresponding main species elution peak, acidic species elution peaks and basic species elution peaks in a cationic ion exchange chromatogram. In some embodiments, the percentage of the main species, the acidic species and the basic species is determined by a cation exchange chromatography method essentially as described in Example 17, wherein the cationic ion exchange chromatogram is obtained from the cation exchange chromatography method. In some embodiments, the cationic ion exchange chromatogram is consistent with FIG.21A and FIG.21B. [034] In some embodiments, the pharmaceutical composition has a drug-to-antibody ratio (DAR) within a range of about 1.5 to about 2.5. In some embodiments, the pharmaceutical composition has a drug-to-antibody ratio (DAR) is within a range of about 1.9 to about 2.1. [035] In some aspects, the present disclosure provides a pharmaceutical composition of the present disclosure containing an anti-PSMA ADC for use in a method of treating cancer in a human subject in need thereof. [036] It is to be understood that the methods and compositions described herein are not limited to the particular methodology, protocols, cell lines, constructs, and reagents described herein and as such may vary. It is also to be understood that the terminology used herein is for the purpose of describing particular embodiments only and is not intended to limit the scope of the methods and compositions described herein, which will be limited only by the appended claims. Attorney Docket No. AMB1013WOPCT1 BRIEF DESCRIPTION OF THE DRAWINGS [037] The novel features of the invention are set forth with particularity in the appended claims. A better understanding of the features and advantages of the present invention disclosure will be obtained by reference to the following detailed description that sets forth illustrative embodiments, in which the principles of the invention are utilized, and the accompanying drawings provided. [038] FIGS. 1A-1D depict (A) peptide maps of unconjugated ARX517 mAb and ARX517 ADC showing the site of amberstatin 269 (AS269) conjugation; (B) HIC chromatogram showing unconjugated ARX517 mAb, DAR1 and DAR2 species; (C) SEC quantification of ARX517 ADC size variants; and (D) thermal transition temperature of unconjugated ARX517 and ARX517 ADC via DSC. [039] FIGS. 2A-2E depict dose-response curves for ARX517 ADC and monomethyl auristatin E (MMAE) in vitro in various prostate cancer cell lines. (A) 22Rv1 (B) C4-2 (C) LNCaP (D) MDA-PCa-2b (E) PC3. [040] FIGS.3A-3C depict sensorgram traces from a biolayer interferometry binding assay for humanized J591 mAb (ARX517 mAb) binding to (A) human PSMA, (B) cynomolgus PSMA and (C) rat PSMA. [041] FIGS.4A-4B depict pharmacokinetics (PK) following ARX517 administration to mice (A) without tumors, and (B) with C4-2 tumors. Total Antibody (TA) and ARX517 ADC (Intact ADC) serum concentrations over time. [042] FIG. 5 depicts tumor growth inhibition (TGI) curves for mice bearing MDA-PCa-2b tumors. % TGI shown in parentheses. [043] FIGS.6A-6B depict tumor growth inhibition in mouse prostate cancer models: (A) after administration of ARX517 ± enzalutamide in a TM00298 PDX model; and (B) after Attorney Docket No. AMB1013WOPCT1 administration of ARX517 or enzalutamide in a CTG-2440 PDX model. % TGI shown in parentheses. [044] FIGS. 7A-7B depict tumor growth inhibition in an enzalutamide-resistant C4-2 CDX mouse model after administration of (A) a single dose of ARX517; and (B) a repeat dose of ARX517 ± enzalutamide. % TGI shown in parentheses. [045] FIGS. 8A-8C depict pharmacokinetics (PK) following ARX517 administration to cynomolgus monkeys: (A) Total Antibody (TA) and ARX517 ADC serum concentrations over time; (B) pAF-AS269 serum concentrations over time; and (C) pAF-AS269 structure showing drug-linker AS269 conjugated to pAF via an oxime linkage. [046] FIG. 9. depicts pK data at the highest non-severely toxic dose of ARX517 in monkey toxicology studies versus a pharmacologically active dose of ARX517 in C4-2 tumor bearing mice, showing a clear therapeutic index for ARX517. [047] FIG.10 depicts the ARX517 ADC structure, in which AS269 is conjugated via a stable oxime bond to the pAF residue incorporated into each heavy chain of the huJ591 mAb at position 114 (Kabat numbering). [048] FIG.11 depicts the amino acid sequence of the ARX517 mAb heavy chain (HC) (SEQ ID NO: 8) and light chain (LC) (SEQ ID NO: 9), respectively. Complementarity determining regions are underlined. “U” represents the non-naturally encoded amino acid (pAF) site, which is genetically encoded and biosynthetically incorporated at amino acid position 114 (Kabat numbering) of each heavy chain. [049] FIG. 12 depicts a first-in-human dose escalation and dose expansion study design for ARX517. [050] FIGS.13A-13B depict low serum concentrations of free payload observed at all doses, with the molar ratio of free payload to ADC at 0.06%. (A) Patients dosed at 2mg/KG Q3W; (B) Patients dosed at the indicated doses Q3W. Attorney Docket No. AMB1013WOPCT1 [051] FIGS.14A-14F depict virtually overlapping TA and ADC PK curves indicating strong stability at all dose levels. (A) 0.64 mg/kg; (B) 1.07 mg/kg; (C) 1.4 mg/kg; (D) 1.7 mg/kg; (E) 2 mg/kg; and (F) 2.4 mg/kg. [052] FIG.15 depicts ARX517 half-life at the indicated dosing levels Q3W. ARX517 exhibits a long half-life of ~6–10 days at doses ≥1.4 mg/kg. [053] FIG. 16A-16B depict drug exposure increases proportional to ARX517 dose. (A) ARX517 Exposure AUC; (B) ARC517 Cmax. [054] FIG. 17 depicts percentage change in PSA from baseline PSA for Cohorts 1-8 with increasing ARX517 dose. [055] FIG. 18 depicts percentage change in PSA from baseline PSA for Cohorts 6-8. 52% (12/23) of patients in Cohorts 6-8 experienced a ≥50% PSA reduction at putative therapeutic doses (≥2.0 mg kg). [056] FIG. 19 depicts percentage changes in ctDNA levels from baseline ctDNA level for Cohorts 4-8. Reductions of ≥ 50% in circulating tumor DNA (ctDNA) were observed in 81% (17/21) of patients (cohorts 4–8). [057] FIG.20 depicts the percentage change from baseline based on RECIST v1.1 criteria in cohorts 4-8. Target lesion reduction was observed in 56% (5/9) of patients (Cohorts 4–8). PD+ progressive disease; SD=stable disease; cPR = confirmed partial response. [058] FIGS.21A-21B depict a representative chromatogram (A) and expanded view thereof (B) obtained from cationic-exchange high performance liquid chromatography of ARX517 Solution for Intravenous Infusion; elution peaks corresponding to the main species ARX517, and acidic and basic charge variants thereof, are shown. Attorney Docket No. AMB1013WOPCT1 DETAILED DESCRIPTION OF THE INVENTION [059] Before describing the present invention in detail, it is to be understood that this invention is not limited to particular methodologies, or compositions, or biological systems, which can, of course, vary. It is also to be understood that the terminology used herein is for the purpose of describing particular embodiments only and is not intended to be limiting. As used in this specification and the appended claims, the singular forms "a", "an" and "the" include plural referents unless the content clearly dictates otherwise. Thus, for example, reference to "a cell" includes a combination of two or more cells and the like. [060] While preferred embodiments of the present invention have been shown and described herein, it will be obvious to those skilled in the art that such embodiments are provided by way of example only. Numerous variations, changes, and substitutions will now occur to those skilled in the art without departing from the invention. It should be understood that various alternatives to the embodiments of the invention described herein may be employed in practicing the invention. It is intended that the following claims define the scope of the invention and that methods and structures within the scope of these claims and their equivalents be covered thereby. [061] As used herein and in the appended claims, the singular forms “a,” “an,” and “the” include plural reference unless the context clearly indicates otherwise. [062] The term “about” will be understood by persons of ordinary skill in the art and will vary to some extent depending on the context in which it is used. [063] Unless defined otherwise, all technical and scientific terms used herein have the same meaning as commonly understood to one of ordinary skill in the art to which the inventions described herein belong. Various methods, materials, and the like, similar or equivalent to those described herein can be used in the practice or testing of the inventions described herein. Attorney Docket No. AMB1013WOPCT1 [064] All publications and patents mentioned herein are incorporated herein by reference in their entirety for the purpose of describing and disclosing, for example, the chemistry, chemical syntheses, compositions, and other methodologies that are described in the publications, which might be used in connection with the presently described inventions. The publications discussed herein are provided solely for their disclosure prior to the filing date of the present application. [065] The term “amino acid” refers to naturally occurring and non-natural or unnatural amino acids, as well as amino acid analogs and amino acid mimetics that function in a manner similar to the naturally occurring amino acids. Naturally encoded amino acids are the 20 common amino acids (alanine, arginine, asparagine, aspartic acid, cysteine, glutamine, glutamic acid, glycine, histidine, isoleucine, leucine, lysine, methionine, phenylalanine, proline, serine, threonine, tryptophan, tyrosine, and valine) and pyrolysine and selenocysteine. Amino acid analogs refer to compounds that have the same basic chemical structure as a naturally occurring amino acid, by way of example only, an α-carbon that is bound to a hydrogen, a carboxyl group, an amino group, and a functional R group. Such analogs may have modified R groups (by way of example, norleucine) or may have modified peptide backbones while still retaining the same basic chemical structure as a naturally occurring amino acid. Non-limiting examples of amino acid analogs include homoserine, norleucine, methionine sulfoxide, methionine methyl sulfonium. Amino acids may be referred to herein by either their name, their commonly known three letter symbols or by the one-letter symbols recommended by the IUPAC-IUB Biochemical Nomenclature Commission. Additionally, nucleotides, may be referred to by their commonly accepted single-letter codes. [066] An “amino or carboxy terminus modification group” refers to any molecule that can be attached to a terminal amine group or terminal carboxy group respectively. By way of example, such terminal amine groups or terminal carboxy groups may be at the end of polymeric Attorney Docket No. AMB1013WOPCT1 molecules, wherein such polymeric molecules include, but are not limited to, polypeptides, polynucleotides, and polysaccharides. Terminus modification groups include but are not limited to, various water soluble polymers, peptides, or proteins. By way of example only, terminus modification groups include polyethylene glycol or serum albumin. Terminus modification groups may be used to modify therapeutic characteristics of the polymeric molecule, including but not limited to increasing the serum half-life of peptides, polypeptides or proteins. [067] The term "antibody" herein refers to a protein consisting of one or more polypeptides substantially encoded by all or part of the antibody genes. The immunoglobulin genes include, but are not limited to, the kappa, lambda, alpha, gamma (IgG1, IgG2, IgG3, and IgG4), delta, epsilon and mu constant region genes, as well as the myriad immunoglobulin variable region genes. Antibody herein is also meant to include full-length antibodies and antibody fragments, and include antibodies that exist naturally in any organism, antibody variants, engineered antibodies and antibody fragments. Antibody herein is also meant to include intact antibody, monoclonal or polyclonal antibodies. Antibody herein also encompasses multispecific antibodies and/or bispecific antibodies. Antibodies of the present invention include human antibodies. Human antibodies are usually made of two light chains and two heavy chains each comprising variable regions and constant regions. The light chain variable region comprises 3 CDRs, identified herein as CDRL1, CDRL2 and CDRL3 flanked by framework regions. The heavy chain variable region comprises 3 CDRs, identified herein as CDRH1, CDRH2 and CDRH3 flanked by framework regions. [068] The term “antibody fragment” herein refers to any form of an antibody other than the full-length form. Antibody fragments herein include antibodies that are smaller components that exist within full-length antibodies, and antibodies that have been engineered, such as antibody variants. Antibody fragments include but are not limited to Fv, Fc, Fab, and (Fab')2, Attorney Docket No. AMB1013WOPCT1 single chain Fv (scFv), diabodies, triabodies, tetrabodies, bifunctional hybrid antibodies, CDR1, CDR2, CDR3, combinations of CDRs, variable regions, framework regions, constant regions, heavy chains, light chains, and variable regions, and alternative scaffold non-antibody molecules, bispecific antibodies, and the like (Maynard & Georgiou, Annu. Rev. Biomed. Eng. 2:339-76, 2000; Hudson, Curr. Opin. Biotechnol. 9:395-402, 1998). Another functional substructure is a single chain Fv (scFv), comprised of the variable regions of the immunoglobulin heavy and light chain, covalently connected by a peptide linker (Hu et al., Cancer Research, 56, 3055-3061, 1996). These small (Mr 25,000) proteins generally retain specificity and affinity for antigen in a single polypeptide and can provide a convenient building block for larger, antigen-specific molecules. Unless specifically noted otherwise, statements and claims that use the term “antibody” or “antibodies” specifically includes “antibody fragment” and “antibody fragments.” [069] The term “antibody-drug conjugate, or “ADC”, as used herein, refers to an antibody molecule, or fragment thereof, that is covalently bonded to one or more biologically active molecule(s). The biologically active molecule may be conjugated to the antibody through a linker, polymer, or other covalent bond. ADCs are a potent class of therapeutic constructs that allow targeted delivery of cytotoxic agents to target cells, such as cancer cells. Because of the targeting function, these compounds show a much higher therapeutic index compared to the same systemically delivered agents. ADCs have been developed as intact antibodies or antibody fragments, such as scFvs. The antibody or fragment is linked to one or more copies of drug (e.g., a toxic moiety) via a linker that is stable under physiological conditions, but that may be cleaved once inside the target cell. [070] The term "antigen-binding fragment", as used herein, refers to one or more fragments of an antibody that retain the ability to bind to an antigen. It has been shown that the antigen- binding function of an antibody can be performed by fragments of an intact antibody. Examples Attorney Docket No. AMB1013WOPCT1 of binding fragments encompassed within the term "antigen-binding fragment" of an antibody include (i) a Fab fragment, a monovalent fragment consisting of the VL, VH, CL and CH1 domains; (ii) a F(ab')2 fragment, a bivalent fragment comprising two Fab fragments linked by a disulfide bridge at the hinge region; (iii) a Fd fragment consisting of the VH and CH1 domains; (iv) a Fv fragment consisting of the VL and VH domains of a single arm of an antibody, (v) a dAb fragment (Ward et al., Nature 341:544-546, 1989), which consists of a VH domain; (vi) an isolated complementarity determining region (CDR), e.g., VH CDR3 comprising or not additional sequence (linker, framework region(s) etc.) and (v) a combination of two to six isolated CDRs comprising or not additional sequence (linker, framework region(s) etc.). Furthermore, although the two domains of the Fv fragment, VL and VH, are coded for by separate genes, they can be joined, using recombinant methods, by a synthetic linker that enables them to be made as a single polypeptide chain in which the VL and VH regions pair to form monovalent molecules (known as single chain Fv (scFv); see e.g., Bird et al., Science 242:423-426, 1988); and (Huston et al., Proc. Natl. Acad. Sci. USA 85:5879-5883, 1988). Such single chain antibodies are also intended to be encompassed within the term "antigen-binding fragment" of an antibody. Furthermore, the antigen-binding fragments include binding-domain immunoglobulin fusion proteins comprising (i) a binding domain polypeptide (such as a heavy chain variable region, a light chain variable region, or a heavy chain variable region fused to a light chain variable region via a linker peptide) that is fused to an immunoglobulin hinge region polypeptide, (ii) an immunoglobulin heavy chain CH2 constant region fused to the hinge region, and (iii) an immunoglobulin heavy chain CH3 constant region fused to the CH2 constant region. The hinge region may be modified by replacing one or more cysteine residues with serine residues to prevent dimerization. Such binding-domain immunoglobulin fusion proteins are further disclosed in US 2003/0118592 and US 2003/0133939. These antibody Attorney Docket No. AMB1013WOPCT1 fragments are obtained using conventional techniques known to those with skill in the art, and the fragments are screened for utility in the same manner as are intact antibodies. [071] A typical antigen binding site is comprised of the variable regions formed by the pairing of a light chain immunoglobulin and a heavy chain immunoglobulin. The structure of the antibody variable regions is very consistent and exhibits very similar structures. These variable regions are typically comprised of relatively homologous framework regions (FR) interspaced with three hypervariable regions termed Complementarity Determining Regions (CDRs). The overall binding activity of the antigen binding fragment is often dictated by the sequence of the CDRs. The FRs often play a role in the proper positioning and alignment in three dimensions of the CDRs for optimal antigen binding. In fact, because CDR sequences are responsible for most antibody-antigen interactions, it is possible to express recombinant antibodies that shows the properties of specific naturally occurring antibodies by constructing expression vectors that include CDR sequences from the specific naturally occurring antibody grafted onto framework sequences from a different antibody with different properties (see, e.g., Riechmann, L. et al., Nature 332:323-327, 1998; Jones, P. et al., Nature 321:522-525, 1986; and Queen, C. et al., Proc. Natl. Acad. USA 86:10029-10033, 1989). Such framework sequences can be obtained from public DNA databases that include germline antibody gene sequences. These germline sequences will differ from mature antibody gene sequences because they will not include completely assembled variable genes, which are formed by V(D)J joining during B cell maturation. Germline gene sequences will also differ from the sequences of a high affinity secondary repertoire antibody which contains mutations throughout the variable gene but typically clustered in the CDRs. For example, somatic mutations are relatively infrequent in the amino terminal portion of framework region 1 and in the carboxy-terminal portion of framework region 4. Furthermore, many somatic mutations do not significantly alter the binding properties of the antibody. For this reason, it is not necessary to obtain the entire DNA Attorney Docket No. AMB1013WOPCT1 sequence of a particular antibody in order to recreate an intact recombinant antibody having binding properties similar to those of the original antibody. Partial heavy and light chain sequence spanning the CDR regions is typically sufficient for this purpose. The partial sequence is used to determine which germline variable and joining gene segments contributed to the recombined antibody variable genes. The germline sequence is then used to fill in missing portions of the variable regions. Heavy and light chain leader sequences are cleaved during protein maturation and do not contribute to the properties of the final antibody. To add missing sequences, cloned cDNA sequences can be combined with synthetic oligonucleotides by ligation or PCR amplification. Alternatively, the entire variable region can be synthesized to create an entirely synthetic variable region clone. This process has certain advantages such as elimination or inclusion of particular restriction sites, or optimization of particular codons. Of course, the totality or portions of the framework region of the antibody described herein may be used in conjunction with the CDRs in order to optimize the affinity, specificity or any other desired properties of the antibody. [072] As used herein, “ARX517” refers to an anti-PSMA antibody drug conjugate (ADC) comprising a humanized anti-PSMA monoclonal antibody comprising two heavy chains and two light chains, wherein each heavy chain has the amino acid sequence of SEQ ID NO: 8, wherein one non-natural amino acid para-acetyl-L-phenylalanine (pAF) is incorporated into each said heavy chain at position 114 according to Kabat numbering, and each light chain has the amino acid sequence of SEQ ID NO: 9; wherein one drug-linker is conjugated to each said pAF via an oxime linkage, wherein each said drug-linker is amberstatin269 (AS269) having the following structure: . Attorney Docket No. AMB1013WOPCT1 The structure of ARX517 is shown at FIG.10, and SEQ ID NOs: 8 and 9 are shown at FIG. 11 and at Table 3, which shows SEQ ID NOs: 8 and 9 in connection with Variant 1 and the pAF incorporation site at Kabat amino acid position 114 (i.e., amino acid position 116 in SEQ ID NO:8). [073] In some embodiments the invention concerns polymers such as a bifunctional polymer. A “bifunctional polymer”, also referred to as a “bifunctional linker”, refers to a polymer comprising two functional groups that are capable of reacting specifically with other moieties to form covalent or non-covalent linkages. Such moieties may include, but are not limited to, the side groups on natural or non-natural amino acids or peptides which contain such natural or non-natural amino acids. The other moieties that may be linked to the bifunctional linker or bifunctional polymer may be the same or different moieties. By way of example only, a bifunctional linker may have a functional group reactive with a group on a first peptide, and another functional group which is reactive with a group on a second peptide, whereby forming a conjugate that includes the first peptide, the bifunctional linker and the second peptide. Many procedures and linker molecules for attachment of various compounds to peptides are known. See, for example, European Patent Application No. 0188256; U.S. Patent Nos. 4,659,839; 4,414,148; 4,699,784; 4,680,338; and 4,569,789 incorporated herein by reference in their entirety. A “multi-functional polymer” also referred to as a “multi-functional linker”, refers to a polymer comprising two or more functional groups that are capable of reacting with other moieties. Such moieties may include, but are not limited to, the side groups on natural or non- natural amino acids or peptides which contain such natural or non-natural amino acids. (including but not limited to, amino acid side groups) to form covalent or non-covalent linkages. A bi-functional polymer or multi-functional polymer may be any desired length or molecular weight and may be selected to provide a particular desired spacing or conformation Attorney Docket No. AMB1013WOPCT1 between one or more molecules linked to a compound and molecules it binds to, or to the compound. [074] The term “bioavailability,” as used herein, refers to the rate and extent to which a substance or its active moiety is delivered from a pharmaceutical dosage form and becomes available at the site of action or in the general circulation. Increases in bioavailability refers to increasing the rate and extent a substance or its active moiety is delivered from a pharmaceutical dosage form and becomes available at the site of action or in the general circulation. By way of example, an increase in bioavailability may be indicated as an increase in concentration of the substance or its active moiety in the blood when compared to other substances or active moieties. [075] The term “biologically active molecule”, “biologically active moiety” or “biologically active agent” when used herein means any substance which can affect any physical or biochemical properties of a biological system, pathway, molecule, or interaction relating to an organism, including but not limited to, viruses, bacteria, bacteriophage, transposon, prion, insects, fungi, plants, animals, and humans. In particular, as used herein, biologically active molecules include but are not limited to any substance intended for diagnosis, cure, mitigation, treatment, or prevention of disease in humans or other animals, or to otherwise enhance physical or mental well-being of humans or animals. Examples of biologically active molecules include, but are not limited to, peptides, proteins, enzymes, small molecule drugs, hard drugs, soft drugs, prodrugs, carbohydrates, inorganic atoms or molecules, dyes, lipids, nucleosides, radionuclides, oligonucleotides, toxins, cells, viruses, liposomes, microparticles and micelles. Classes of biologically active agents that are suitable for use with the methods and compositions described herein include, but are not limited to, drugs, prodrugs, radionuclides, imaging agents, polymers, antibiotics, fungicides, anti-viral agents, anti-inflammatory agents, Attorney Docket No. AMB1013WOPCT1 anti-tumor agents, cardiovascular agents, anti-anxiety agents, hormones, growth factors, steroidal and nonsteroidal agents, microbially derived toxins, and the like. [076] By “modulating biological activity” is meant increasing or decreasing the reactivity of a polypeptide, altering the selectivity of the polypeptide, enhancing or decreasing the substrate selectivity of the polypeptide. Analysis of modified biological activity can be performed by comparing the biological activity of the non-natural polypeptide to that of the natural polypeptide. [077] In some embodiments, the disclosure concerns amino acids that have been biosynthetically incorporated in the antibody. The term “biosynthetically,” as used herein, refers to any method utilizing a translation system (cellular or non-cellular), including use of at least one of the following components: a polynucleotide, a codon, a tRNA, and a ribosome. By way of example, non-natural amino acids may be “biosynthetically incorporated” into non- natural amino acid polypeptides using the methods and techniques described herein and as is well known in the art. See for example, WO2010/011735 and WO2005/074650. [078] The term “conservatively modified variants” applies to both natural and non-natural amino acid and natural and non-natural nucleic acid sequences, and combinations thereof. With respect to particular nucleic acid sequences, “conservatively modified variants” refers to those natural and non-natural nucleic acids which encode identical or essentially identical natural and non-natural amino acid sequences, or where the natural and non-natural nucleic acid does not encode a natural and non-natural amino acid sequence, to essentially identical sequences. By way of example, because of the degeneracy of the genetic code, a large number of functionally identical nucleic acids encode any given protein. For instance, the codons GCA, GCC, GCG and GCU all encode the amino acid alanine. Thus, at every position where an alanine is specified by a codon, the codon can be altered to any of the corresponding codons described without altering the encoded polypeptide. Such nucleic acid variations are “silent Attorney Docket No. AMB1013WOPCT1 variations,” which are one species of conservatively modified variations. Thus, by way of example every natural or non-natural nucleic acid sequence herein which encodes a natural or non-natural polypeptide also describes every possible silent variation of the natural or non- natural nucleic acid. One of ordinary skill in the art will recognize that each codon in a natural or non-natural nucleic acid (except AUG, which is ordinarily the only codon for methionine, and TGG, which is ordinarily the only codon for tryptophan) can be modified to yield a functionally identical molecule. Accordingly, each silent variation of a natural and non-natural nucleic acid which encodes a natural and non-natural polypeptide is implicit in each described sequence. As to amino acid sequences, individual substitutions, deletions or additions to a nucleic acid, peptide, polypeptide, or protein sequence which alters, adds or deletes a single natural and non-natural amino acid or a small percentage of natural and non-natural amino acids in the encoded sequence is a “conservatively modified variant” where the alteration results in the deletion of an amino acid, addition of an amino acid, or substitution of a natural and non-natural amino acid with a chemically similar amino acid. Conservative substitution tables providing functionally similar natural amino acids are well known in the art. Conservative substitution tables providing functionally similar amino acids are known to those of ordinary skill in the art. The following eight groups each contain amino acids that are conservative substitutions for one another: 1) Alanine (A), Glycine (G); 2) Aspartic acid (D), Glutamic acid (E); 3) Asparagine (N), Glutamine (Q); 4) Arginine (R), Lysine (K); 5) Isoleucine (I), Leucine (L), Methionine (M), Valine (V); 6) Phenylalanine (F), Tyrosine (Y), Tryptophan (W); 7) Serine (S), Threonine (T); and 8) Cysteine (C), Methionine (M) (see, e.g., Creighton, Proteins: Structures and Molecular Properties (W H Freeman & Co.; 2nd edition, 1993). Such conservatively modified variants are in addition to and do not exclude polymorphic variants, interspecies homologs, and alleles of the compositions described herein. Attorney Docket No. AMB1013WOPCT1 [079] The term “drug,” as used herein, refers to any substance used in the prevention, diagnosis, alleviation, treatment, or cure of a disease or condition such as cancer, including prostate cancer. [080] The term “effective amount,” as used herein, refers to a sufficient amount of an agent, compound or composition being administered which will relieve to some extent one or more of the symptoms of the disease or condition being treated. The result can be reduction and/or alleviation of the signs, symptoms, or causes of a disease, or any other desired alteration of a biological system. By way of example, an agent, compound or composition being administered includes, but is not limited to, a natural amino acid polypeptide, non-natural amino acid polypeptide, modified natural amino acid polypeptide, modified non-amino acid polypeptide, or an antibody or variant thereof. Compositions containing such natural amino acid polypeptides, non-natural amino acid polypeptides, modified natural amino acid polypeptides, modified non-natural amino acid polypeptides, or an antibody or variant thereof can be administered for prophylactic, enhancing, and/or therapeutic treatments. An appropriate “effective” amount in any individual case may be determined using techniques, such as a dose escalation study. [081] The terms “enhance” or “enhancing” means to increase or prolong either in potency or duration a desired effect. By way of example, “enhancing” the effect of therapeutic agents refers to the ability to increase or prolong, either in potency or duration, the effect of therapeutic agents on during treatment of a disease, disorder or condition. An “enhancing-effective amount,” as used herein, refers to an amount adequate to enhance the effect of a therapeutic agent in the treatment of a disease, disorder or condition. When used in a patient, amounts effective for this use will depend on the severity and course of the disease, disorder or condition, previous therapy, the patient's health status and response to the drugs, and the judgment of the treating physician. Attorney Docket No. AMB1013WOPCT1 [082] The term “half-life” as used herein refers to the time required for any specified property to decrease by half. Typically, the specified property is the concentration of a substance in the body, or a compartment thereof, wherein the substance is a conjugated protein of the present disclosure, or the corresponding protein or a comparable protein in its unconjugated form. The term “half-life” can be referred to herein as “t1/2” or “T1/2”. [083] The term “elimination half-life” as used herein refers to a pharmacokinetic parameter that is defined as the period of time that it takes for the concentration of a biotherapeutic in the plasma or serum of a subject, or the total amount in the whole body of a subject, to be reduced by about 50%. Thus, after one half-life, the concentration of the biotherapeutic in the plasma or serum, or in the whole body, of the subject will be half of the starting concentration. Typically, the period of time that it takes for the concentration of the biotherapeutic to be reduced by about 50% (e.g., in the blood, plasma or serum, or in the whole body) commences at or about the time of administration of the biotherapeutic to the subject. [084] The term “terminal half-life” as used herein refers to the time required to divide the serum or plasma concentration by two after reaching pseudo-equilibrium, and not the time required to eliminate half the administered dose. See, e.g., Toutain P.L. and Bousquet-Melou A. (2004) J. Vetinary Pharmacology and Therapeutics, 27(6):427-439. [085] The term "humanized or chimeric antibody" refer to a molecule, generally prepared using recombinant techniques, having an antigen binding site derived from an immunoglobulin from a non-human species, (e.g., murine), and the remaining immunoglobulin structure of the molecule based upon the structure and/or sequence of a human immunoglobulin. In general, the humanized antibody will comprise substantially all of at least one, and typically two, variable domains, in which all or substantially all of the hypervariable loops correspond to those of a non-human immunoglobulin and all or substantially all of the framework residues/regions (FR) are those of a human immunoglobulin sequence. The humanized Attorney Docket No. AMB1013WOPCT1 antibody optionally also will comprise at least a portion of an immunoglobulin constant region (Fc), typically that of a human immunoglobulin. The humanized forms of rodent antibodies will essentially comprise the same CDR sequences of the parental rodent antibodies, although certain amino acid substitutions may be included to increase affinity, increase stability of the humanized antibody, or for other reasons. However, as CDR loop exchanges do not uniformly result in an antibody with the same binding properties as the antibody of origin, changes in framework residues (FR), residues involved in CDR loop support, might also be introduced in humanized antibodies to preserve antigen binding affinity. The antigen-binding site may comprise either complete variable domains fused onto constant domains or only the complementarity determining regions (CDRs) grafted onto appropriate framework regions in the variable domains. Antigen binding sites may be wild type or modified by one or more amino acid substitutions. This eliminates the constant region as an immunogen in human individuals, but the possibility of an immune response to the foreign variable region remains (LoBuglio, A. F. et al., "Mouse/Human Chimeric Monoclonal Antibody In Man: Kinetics And Immune Response," Proc. Natl. Acad. Sci. (USA) 86:4220-4224, 1989). Another approach focuses not only on providing human-derived constant regions but modifying the variable regions as well so as to reshape them as closely as possible to human form. It is known that the variable regions of both heavy and light chains contain three complementarity-determining regions (CDRs) which vary in response to the antigens in question and determine binding capability, flanked by four framework regions (FRs) which are relatively conserved in a given species and which putatively provide a scaffolding for the CDRs. When nonhuman antibodies are prepared with respect to a particular antigen, the variable regions can be "humanized" by grafting CDRs derived from nonhuman antibody on the FRs present in the human antibody to be modified. Application of this approach to various antibodies has been reported by Kettleborough, C. A. et al., "Humanization Of A Mouse Monoclonal Antibody By CDR- Attorney Docket No. AMB1013WOPCT1 Grafting: The Importance Of Framework Residues On Loop Conformation," Protein Engineering 4:773-3783,1991; Co, M. S. et al., "Humanized Antibodies For Antiviral Therapy," Proc. Natl. Acad. Sci. (USA) 88:2869-2873,1991; Carter, P. et al., "Humanization Of An Anti-p185her2 Antibody For Human Cancer Therapy," Proc. Natl. Acad. Sci. (USA) 89:4285-4289,1992; and Co, M. S. et al., "Chimeric And Humanized Antibodies With Specificity For The CD33 Antigen," J. Immunol.148:1149-1154,1992. In some embodiments, humanized antibodies preserve all CDR sequences (for example, a humanized mouse antibody which contains all six CDRs from the mouse antibodies). In other embodiments, humanized antibodies have one or more CDRs (one, two, three, four, five, six) which are altered with respect to the original antibody, which are also termed one or more CDRs "derived from" one or more CDRs from the original antibody. [086] The term “identical,” as used herein, refers to two or more sequences or subsequences which are the same. In addition, the term “substantially identical,” as used herein, refers to two or more sequences which have a percentage of sequential units which are the same when compared and aligned for maximum correspondence over a comparison window, or designated region as measured using comparison algorithms or by manual alignment and visual inspection. By way of example only, two or more sequences may be “substantially identical” if the sequential units are about 60% identical, about 65% identical, about 70% identical, about 75% identical, about 80% identical, about 85% identical, about 90% identical, or about 95% identical over a specified region. Such percentages describe the “percent identity” of two or more sequences. The identity of a sequence can exist over a region that is at least about 75-100 sequential units in length, over a region that is about 50 sequential units in length, or, where not specified, across the entire sequence. This definition also refers to the complement of a test sequence. By way of example only, two or more polypeptide sequences are identical when the amino acid residues are the same, while two or more polypeptide sequences are “substantially Attorney Docket No. AMB1013WOPCT1 identical” if the amino acid residues are about 60% identical, about 65% identical, about 70% identical, about 75% identical, about 80% identical, about 85% identical, about 90% identical, or about 95% identical over a specified region. The identity can exist over a region that is at least about 75 to about 100 amino acids in length, over a region that is about 50 amino acids in length, or, where not specified, across the entire sequence of a polypeptide sequence. In addition, by way of example only, two or more polynucleotide sequences are identical when the nucleic acid residues are the same, while two or more polynucleotide sequences are “substantially identical” if the nucleic acid residues are about 60% identical, about 65% identical, about 70% identical, about 75% identical, about 80% identical, about 85% identical, about 90% identical, or about 95% identical over a specified region. The identity can exist over a region that is at least about 75 to about 100 nucleic acids in length, over a region that is about 50 nucleic acids in length, or, where not specified, across the entire sequence of a polynucleotide sequence. [087] The term “immunogenicity,” as used herein, refers to an antibody response to administration of a therapeutic drug. The immunogenicity toward therapeutic non-natural amino acid polypeptides can be obtained using quantitative and qualitative assays for detection of anti-non-natural amino acid polypeptides antibodies in biological fluids. Such assays include, but are not limited to, Radioimmunoassay (RIA), Enzyme-linked immunosorbent assay (ELISA), luminescent immunoassay (LIA), and fluorescent immunoassay (FIA). Analysis of immunogenicity toward therapeutic non-natural amino acid polypeptides involves comparing the antibody response upon administration of therapeutic non-natural amino acid polypeptides to the antibody response upon administration of therapeutic natural amino acid polypeptides. [088] The term “isolated,” as used herein, refers to separating and removing a component of interest from components not of interest. Isolated substances can be in either a dry or semi-dry Attorney Docket No. AMB1013WOPCT1 state, or in solution, including but not limited to an aqueous solution. The isolated component can be in a homogeneous state or the isolated component can be a part of a pharmaceutical composition that comprises additional pharmaceutically acceptable carriers and/or excipients. Purity and homogeneity may be determined using analytical chemistry techniques including, but not limited to, polyacrylamide gel electrophoresis or high-performance liquid chromatography. In addition, when a component of interest is isolated and is the predominant species present in a preparation, the component is described herein as substantially purified. The term “purified,” as used herein, may refer to a component of interest which is at least 85% pure, at least 90% pure, at least 95% pure, at least 99% or greater pure. By way of example only, nucleic acids or proteins are “isolated” when such nucleic acids or proteins are free of at least some of the cellular components with which it is associated in the natural state, or that the nucleic acid or protein has been concentrated to a level greater than the concentration of its in vivo or in vitro production. Also, by way of example, a gene is isolated when separated from open reading frames which flank the gene and encode a protein other than the gene of interest. [089] The term “linkage,” as used herein to refer to bonds or chemical moiety formed from a chemical reaction between the functional group of a linker and another molecule. Such bonds may include, but are not limited to, covalent linkages and non-covalent bonds, while such chemical moieties may include, but are not limited to, esters, carbonates, imines phosphate esters, hydrazones, acetals, orthoesters, peptide linkages, and oligonucleotide linkages. Hydrolytically stable linkages mean that the linkages are substantially stable in water and do not react with water at useful pH values, including but not limited to, under physiological conditions for an extended period of time, perhaps even indefinitely. Hydrolytically unstable or degradable linkages mean that the linkages are degradable in water or in aqueous solutions, including for example, blood. Enzymatically unstable or degradable linkages mean that the linkage can be degraded by one or more enzymes. By way of example only, PEG and related Attorney Docket No. AMB1013WOPCT1 polymers may include degradable linkages in the polymer backbone or in the linker group between the polymer backbone and one or more of the terminal functional groups of the polymer molecule. Such degradable linkages include but are not limited to ester linkages formed by the reaction of PEG carboxylic acids or activated PEG carboxylic acids with alcohol groups on a biologically active agent, wherein such ester groups generally hydrolyze under physiological conditions to release the biologically active agent. Other hydrolytically degradable linkages include but are not limited to carbonate linkages; imine linkages resulted from reaction of an amine and an aldehyde; phosphate ester linkages formed by reacting an alcohol with a phosphate group; hydrazone linkages which are reaction product of a hydrazide and an aldehyde; acetal linkages that are the reaction product of an aldehyde and an alcohol; orthoester linkages that are the reaction product of a formate and an alcohol; peptide linkages formed by an amine group, including but not limited to, at an end of a polymer such as PEG, and a carboxyl group of a peptide; and oligonucleotide linkages formed by a phosphoramidite group, including but not limited to, at the end of a polymer, and a 5' hydroxyl group of an oligonucleotide. [090] The term “metabolite,” as used herein, refers to a derivative of a compound, by way of example natural amino acid polypeptide, a non-natural amino acid polypeptide, a modified natural amino acid polypeptide, or a modified non-natural amino acid polypeptide, that is formed when the compound, by way of example natural amino acid polypeptide, non-natural amino acid polypeptide, modified natural amino acid polypeptide, or modified non-natural amino acid polypeptide, is metabolized. The term “pharmaceutically active metabolite” or “active metabolite” refers to a biologically active derivative of a compound, by way of example natural amino acid polypeptide, a non-natural amino acid polypeptide, a modified natural amino acid polypeptide, or a modified non-natural amino acid polypeptide, that is formed when such a compound, by way of example a natural amino acid polypeptide, non-natural amino acid Attorney Docket No. AMB1013WOPCT1 polypeptide, modified natural amino acid polypeptide, or modified non-natural amino acid polypeptide, is metabolized. [091] The term “metabolized,” as used herein, refers to the sum of the processes by which a particular substance is changed by an organism. Such processes include, but are not limited to, hydrolysis reactions and reactions catalyzed by enzymes. Further information on metabolism may be obtained from The Pharmacological Basis of Therapeutics, 9th Edition, McGraw-Hill (1996). By way of example only, metabolites of natural amino acid polypeptides, non-natural amino acid polypeptides, modified natural amino acid polypeptides, or modified non-natural amino acid polypeptides may be identified either by administration of the natural amino acid polypeptides, non-natural amino acid polypeptides, modified natural amino acid polypeptides, or modified non-natural amino acid polypeptides to a host and analysis of tissue samples from the host, or by incubation of natural amino acid polypeptides, non-natural amino acid polypeptides, modified natural amino acid polypeptides, or modified non-natural amino acid polypeptides with hepatic cells in vitro and analysis of the resulting compounds. [092] The term “modified,” as used herein refers to the presence of a change to a natural amino acid, a non-natural amino acid, a natural amino acid polypeptide or a non-natural amino acid polypeptide. Such changes, or modifications, may be obtained by post synthesis modifications of natural amino acids, non-natural amino acids, natural amino acid polypeptides or non-natural amino acid polypeptides, or by co-translational, or by post-translational modification of natural amino acids, non-natural amino acids, natural amino acid polypeptides or non-natural amino acid polypeptides. [093] A “non-natural amino acid” refers to an amino acid that is not one of the 20 common amino acids or pyrolysine or selenocysteine. Other terms that may be used synonymously with the term “non-natural amino acid” is “non-naturally encoded amino acid,” “unnatural amino acid,” “non-naturally-occurring amino acid,” and variously hyphenated and non-hyphenated Attorney Docket No. AMB1013WOPCT1 versions thereof. The term “non-natural amino acid” includes, but is not limited to, amino acids which occur naturally by modification of a naturally encoded amino acid (including but not limited to, the 20 common amino acids or pyrrolysine and selenocysteine) but are not themselves incorporated into a growing polypeptide chain by the translation complex. Examples of naturally-occurring amino acids that are not naturally-encoded include, but are not limited to, N-acetylglucosaminyl-L-serine, N-acetylglucosaminyl-L-threonine, and O- phosphotyrosine. Additionally, the term “non-natural amino acid” includes, but is not limited to, amino acids which do not occur naturally and may be obtained synthetically or may be obtained by modification of non-natural amino acids. [094] The term “nucleic acid,” as used herein, refers to deoxyribonucleotides, deoxyribonucleosides, ribonucleosides or ribonucleotides and polymers thereof in either single- or double-stranded form. By way of example only, such nucleic acids and nucleic acid polymers include, but are not limited to, (i) analogues of natural nucleotides which have similar binding properties as a reference nucleic acid and are metabolized in a manner similar to naturally occurring nucleotides; (ii) oligonucleotide analogs including, but are not limited to, PNA (peptidonucleic acid), analogs of DNA used in antisense technology (phosphorothioates, phosphoroamidates, and the like); (iii) conservatively modified variants thereof (including but not limited to, degenerate codon substitutions) and complementary sequences and sequence explicitly indicated. By way of example, degenerate codon substitutions may be achieved by generating sequences in which the third position of one or more selected (or all) codons is substituted with mixed-base and/or deoxyinosine residues (Batzer et al., Nucleic Acid Res. 19:5081, 1991; Ohtsuka et al., J. Biol. Chem.260:2605-2608, 1985; and Rossolini et al., Mol. Cell. Probes 8:91-98, 1994). [095] The term “pharmaceutically acceptable”, as used herein, refers to a material, including but not limited, to a salt, binder, adjuvant, excipient, carrier or diluent, which does not abrogate Attorney Docket No. AMB1013WOPCT1 the biological activity or properties of the compound, and is relatively nontoxic, i.e., the material may be administered to an individual without causing undesirable biological effects or interacting in a deleterious manner with any of the components of the composition in which it is contained. [096] In some embodiments the invention concerns polymers. The term “polymer,” as used herein, refers to a molecule composed of repeated subunits. Such molecules include, but are not limited to, polypeptides, polynucleotides, or polysaccharides or polyalkylene glycols. Polymers of the invention can be linear or branched polymeric polyether polyols including, but are not limited to, polyethylene glycol, polypropylene glycol, polybutylene glycol, and derivatives thereof. Other exemplary embodiments are listed, for example, in commercial supplier catalogs, such as Shearwater Corporation's catalog “Polyethylene Glycol and Derivatives for Biomedical Applications” (2001). By way of example only, such polymers have average molecular weights between about 0.1 kDa to about 100 kDa. Such polymers include, but are not limited to, between about 100 Da and about 100,000 Da or more. The molecular weight of the polymer may be between about 100 Da and about 100,000 Da, including but not limited to, about 100,000 Da, about 95,000 Da, about 90,000 Da, about 85,000 Da, about 80,000 Da, about 75,000 Da, about 70,000 Da, about 65,000 Da, about 60,000 Da, about 55,000 Da, about 50,000 Da, about 45,000 Da, about 40,000 Da, about 35,000 Da, about 30,000 Da, about 25,000 Da, about 20,000 Da, about 15,000 Da, about 10,000 Da, about 9,000 Da, about 8,000 Da, about 7,000 Da, about 6,000 Da, about 5,000 Da, about 4,000 Da, about 3,000 Da, about 2,000 Da, about 1,000 Da, about 900 Da, about 800 Da, about 700 Da, about 600 Da, about 500 Da, 400 Da, about 300 Da, about 200 Da, and about 100 Da. In some embodiments molecular weight of the polymer is between about 100 Da and about 50,000 Da. In some embodiments, the molecular weight of the polymer is between about 100 Da and about 40,000 Da. In some embodiments, the molecular weight of the polymer is between about 1,000 Attorney Docket No. AMB1013WOPCT1 Da and about 40,000 Da. In some embodiments, the molecular weight of the polymer is between about 2,000 to about 50,000 Da. In some embodiments, the molecular weight of the polymer is between about 5,000 Da and about 40,000 Da. In some embodiments, the molecular weight of the polymer is between about 10,000 Da and about 40,000 Da. In some embodiments, the poly(ethylene glycol) molecule is a branched polymer. The molecular weight of the branched chain PEG may be between about 1,000 Da and about 100,000 Da, including but not limited to, about 100,000 Da, about 95,000 Da, about 90,000 Da, about 85,000 Da, about 80,000 Da, about 75,000 Da, about 70,000 Da, about 65,000 Da, about 60,000 Da, about 55,000 Da, about 50,000 Da, about 45,000 Da, about 40,000 Da, about 35,000 Da, about 30,000 Da, about 25,000 Da, about 20,000 Da, about 15,000 Da, about 10,000 Da, about 9,000 Da, about 8,000 Da, about 7,000 Da, about 6,000 Da, about 5,000 Da, about 4,000 Da, about 3,000 Da, about 2,000 Da, and about 1,000 Da. In some embodiments, the molecular weight of the branched chain PEG is between about 1,000 Da and about 50,000 Da. In some embodiments, the molecular weight of the branched chain PEG is between about 1,000 Da and about 40,000 Da. In some embodiments, the molecular weight of the branched chain PEG is between about 5,000 Da and about 40,000 Da. In some embodiments, the molecular weight of the branched chain PEG is between about 5,000 Da and about 20,000 Da. In other embodiments, the molecular weight of the branched chain PEG is between about 2,000 to about 50,000 Da. The term “PEGylating” or “PEGylated” is meant to refer to the covalent bonding of the specified synthetic amino acid to a polyethylene glycol (PEG) molecule. The method can comprise contacting an isolated α-PSMA ADCs polypeptide comprising a synthetic amino acid with a water soluble polymer comprising a moiety that reacts with the synthetic amino acid. [097] The terms “polypeptide,” “peptide” and “protein” are used interchangeably herein to refer to a polymer of amino acid residues. That is, a description directed to a polypeptide applies equally to a description of a peptide and a description of a protein, and vice versa. The terms Attorney Docket No. AMB1013WOPCT1 apply to naturally occurring amino acid polymers as well as amino acid polymers in which one or more amino acid residues is a non-natural amino acid. Additionally, such “polypeptides,” “peptides” and “proteins” include amino acid chains of any length, including full length proteins, wherein the amino acid residues are linked by covalent peptide bonds. [098] The term “post-translationally modified” refers to any modification of a natural or non- natural amino acid which occurs after such an amino acid has been translationally incorporated into a polypeptide chain. Such modifications include, but are not limited to, co-translational in vivo modifications, co-translational in vitro modifications (such as in a cell-free translation system), post-translational in vivo modifications, and post-translational in vitro modifications. [099] The terms “prodrug” or “pharmaceutically acceptable prodrug,” as used herein, refers to an agent that is converted into the parent drug in vivo or in vitro, which does not abrogate the biological activity or properties of the drug, and is relatively nontoxic, i.e., the material may be administered to an individual without causing undesirable biological effects or interacting in a deleterious manner with any of the components of the composition in which it is contained. Prodrugs are generally drug precursors that, following administration to a subject and subsequent absorption, are converted to an active, or a more active species via some process, such as conversion by a metabolic pathway. Some prodrugs have a chemical group present on the prodrug that renders it less active and/or confers solubility or some other property to the drug. Once the chemical group has been cleaved and/or modified from the prodrug the active drug is generated. Prodrugs are converted into active drug within the body through enzymatic or non-enzymatic reactions. Prodrugs may provide improved physiochemical properties such as better solubility, enhanced delivery characteristics, such as specifically targeting a particular cell, tissue, organ or ligand, and improved therapeutic value of the drug. The benefits of such prodrugs include, but are not limited to, (i) ease of administration compared with the parent drug; (ii) the prodrug may be bioavailable by oral administration whereas the parent is not; and Attorney Docket No. AMB1013WOPCT1 (iii) the prodrug may also have improved solubility in pharmaceutical compositions compared with the parent drug. A prodrug includes a pharmacologically inactive, or reduced activity, derivative of an active drug. Prodrugs may be designed to modulate the amount of a drug or biologically active molecule that reaches a desired site of action through the manipulation of the properties of a drug, such as physiochemical, biopharmaceutical, or pharmacokinetic properties. An example, without limitation, of a prodrug would be a non-natural amino acid polypeptide which is administered as an ester (the “prodrug”) to facilitate transmittal across a cell membrane where water solubility is detrimental to mobility and that is then metabolically hydrolyzed to the carboxylic acid, the active entity, once inside the cell where water solubility is beneficial. Prodrugs may be designed as reversible drug derivatives, for use as modifiers to enhance drug transport to site-specific tissues. [100] The term “prophylactically effective amount,” as used herein, refers to an amount of a composition containing at least one non-natural amino acid polypeptide or at least one modified non-natural amino acid polypeptide prophylactically applied to a patient which will relieve to some extent one or more of the symptoms of a disease, condition or disorder being treated. In such prophylactic applications, such amounts may depend on the patient's state of health, weight, and the like. It is considered well within the skill of the art for one to determine such prophylactically effective amounts by routine experimentation, including, but not limited to, a dose escalation clinical trial. [101] The term “recombinant host cell,” also referred to as “host cell,” refers to a cell which includes an exogenous polynucleotide, wherein the methods used to insert the exogenous polynucleotide into a cell include, but are not limited to, direct uptake, transduction, f-mating, or other methods known in the art to create recombinant host cells. By way of example only, such exogenous polynucleotide may be a nonintegrated vector, including but not limited to a plasmid, or may be integrated into the host genome. Attorney Docket No. AMB1013WOPCT1 [102] The term “subject” as used herein, refers to an animal which is the object of treatment, observation or experiment. By way of example only, a subject may be, but is not limited to, a mammal including, but not limited to, a human. The terms, "subject" and "patient" are used interchangeably herein. [103] The term “substantially purified,” as used herein, refers to a component of interest that may be substantially or essentially free of other components which normally accompany or interact with the component of interest prior to purification. By way of example only, a component of interest may be “substantially purified” when the preparation of the component of interest contains less than about 30%, less than about 25%, less than about 20%, less than about 15%, less than about 10%, less than about 5%, less than about 4%, less than about 3%, less than about 2%, or less than about l% (by dry weight) of contaminating components. Thus, a “substantially purified” component of interest may have a purity level of about 70%, about 75%, about 80%, about 85%, about 90%, about 95%, about 96%, about 97%, about 98%, about 99% or greater. By way of example only, a natural amino acid polypeptide or a non-natural amino acid polypeptide may be purified from a native cell, or host cell in the case of recombinantly produced natural amino acid polypeptides or non-natural amino acid polypeptides. By way of example a preparation of a natural amino acid polypeptide or a non- natural amino acid polypeptide may be “substantially purified” when the preparation contains less than about 30%, less than about 25%, less than about 20%, less than about 15%, less than about 10%, less than about 5%, less than about 4%, less than about 3%, less than about 2%, or less than about l% (by dry weight) of contaminating material. By way of example when a natural amino acid polypeptide or a non-natural amino acid polypeptide is recombinantly produced by host cells, the natural amino acid polypeptide or non-natural amino acid polypeptide may be present at about 30%, about 25%, about 20%, about 15%, about 10%, about 5%, about 4%, about 3%, about 2%, or about 1% or less of the dry weight of the cells. Attorney Docket No. AMB1013WOPCT1 By way of example when a natural amino acid polypeptide or a non-natural amino acid polypeptide is recombinantly produced by host cells, the natural amino acid polypeptide or non-natural amino acid polypeptide may be present in the culture medium at about 5g/L, about 4g/L, about 3g/L, about 2g/L, about 1g/L, about 750mg/L, about 500mg/L, about 250mg/L, about 100mg/L, about 50mg/L, about 10mg/L, or about 1mg/L or less of the dry weight of the cells. By way of example, “substantially purified” natural amino acid polypeptides or non- natural amino acid polypeptides may have a purity level of about 30%, about 35%, about 40%, about 45%, about 50%, about 55%, about 60%, about 65%, about 70%, about 75%, about 80%, about 85%, about 90%, about 95%, about 99% or greater as determined by appropriate methods, including, but not limited to, SDS/PAGE analysis, RP-HPLC, SEC, and capillary electrophoresis. [104] The term “therapeutically effective amount,” as used herein, refers to the amount of a composition containing at least one non-natural amino acid polypeptide and/or at least one modified non-natural amino acid polypeptide administered to a patient already suffering from a disease, condition or disorder, sufficient to cure or at least partially arrest, or relieve to some extent one or more of the symptoms of the disease, disorder or condition being treated. The effectiveness of such compositions depends on conditions including, but not limited to, the severity and course of the disease, disorder or condition, previous therapy, the patient's health status and response to the drugs, and the judgment of the treating physician. By way of example only, therapeutically effective amounts may be determined by routine experimentation, including but not limited to a dose escalation clinical trial. [105] The term “toxic”, or “toxic moiety” or “toxic group” or “cytotoxic” or “cytotoxic payload” or “payload” or “free payload” as used herein, refers to a compound which can cause harm, disturbances, or death. Toxic moieties and like terms, including payload and free payload, include, but are not limited to, auristatin, amberstatin269 (AS269), DNA minor Attorney Docket No. AMB1013WOPCT1 groove binding agent, DNA minor groove alkylating agent, enediyne, lexitropsin, duocarmycin, taxane, puromycin, dolastatin, maytansinoid, vinca alkaloid, AFP, monomethyl auristatin F (MMAF), MMAE, AEB, AEVB, auristatin E, paclitaxel, docetaxel, CC-1065, SN- 38, topotecan, morpholino-doxorubicin, rhizoxin, cyanomorpholino-doxorubicin, dolastatin- 10, echinomycin, combretatstatin, chalicheamicin, maytansine, DM-1, netropsin, podophyllotoxin (e.g. etoposide, teniposide, etc.), baccatin and its derivatives, anti-tubulin agents, cryptophysin, combretastatin, vincristine, vinblastine, vindesine, vinorelbine, VP-16, camptothecin, epothilone A, epothilone B, nocodazole, colchicines, colcimid, estramustine, cemadotin, discodermolide, maytansine, eleutherobin, mechlorethamine, cyclophosphamide, melphalan, carmustine, lomustine, semustine, streptozocin, chlorozotocin, uracil mustard, chlormethine, ifosfamide, chlorambucil, pipobroman, triethylenemelamine, triethylenethiophosphoramine, busulfan, dacarbazine, and temozolomide, ytarabine, cytosine arabinoside, fluorouracil, floxuridine, 6-thioguanine, 6-mercaptopurine, pentostatin, 5- fluorouracil, methotrexate, 10-propargyl-5,8-dideazafolate, 5,8-dideazatetrahydrofolic acid, leucovorin, fludarabine phosphate, pentostatine, gemcitabine, Ara-C, deoxycoformycin, mitomycin-C, L-asparaginase, azathioprine, brequinar, antibiotics (e.g., anthracycline, gentamicin, cefalotin, vancomycin, telavancin, daptomycin, azithromycin, erythromycin, rocithromycin, furazolidone, amoxicillin, ampicillin, carbenicillin, flucloxacillin, methicillin, penicillin, ciprofloxacin, moxifloxacin, ofloxacin, doxycycline, minocycline, oxytetracycline, tetracycline, streptomycin, rifabutin, ethambutol, rifaximin, etc.), antiviral drugs (e.g., abacavir, acyclovir, ampligen, cidofovir, delavirdine, didanosine, efavirenz, entecavir, fosfonet, ganciclovir, ibacitabine, imunovir, idoxuridine, inosine, lopinavir, methisazone, nexavir, nevirapine, oseltamivir, penciclovir, stavudine, trifluridine, truvada, valaciclovir, zanamivir, etc.), daunorubicin hydrochloride, daunomycin, rubidomycin, cerubidine, idarubicin, doxorubicin, epirubicin and morpholino derivatives, phenoxizone biscyclopeptides Attorney Docket No. AMB1013WOPCT1 (e.g., dactinomycin), basic glycopeptides (e.g., bleomycin), anthraquinone glycosides (e.g., plicamycin, mithramycin), anthracenediones (e.g., mitoxantrone), azirinopyrrolo indolediones (e.g., mitomycin), macrocyclic immunosuppressants (e.g., cyclosporine, FK-506, tacrolimus, prograf, rapamycin etc.), navelbene, CPT-11, anastrazole, letrazole, capecitabine, reloxafine, cyclophosphamide, ifosamide, droloxafine, allocolchicine, Halichondrin B, colchicine, colchicine derivatives , maytansine, rhizoxin, paclitaxel, paclitaxel derivatives, docetaxel, thiocolchicine, trityl cysterin, vinblastine sulfate, vincristine sulfate, cisplatin, carboplatin, hydroxyurea, N-methylhydrazine, epidophyllotoxin, procarbazine, mitoxantrone, leucovorin, and tegafur; and any one of the foregoing further conjugated to a linker and/or an amino acid, including but not limited to a non-natural amino acid. For example, the terms “payload” or “free payload” can refer to a compound which can cause harm, disturbances, or death, wherein the compound comprises, or consists of, a toxic moiety further conjugated to linker and/or a non-natural amino acid. As described herein, a free payload can be released in vivo from an anti-PSMA ADC of the present disclosure following administration of the anti-PSMA ADC to a subject. For example, as disclosed herein, the free payload pAF-AS269 is released from the anti-PSMA ADC ARX517. As disclosed at Example 12, once ARX517 is internalized by a cancer cell, ARX517 undergoes proteolytic degradation and releases free payload pAF-AS269, which has the structure depicted in FIG.8C comprising the cytotoxic moiety AS269 conjugated to non-natural amino acid para-acetyl phenylalanine, where the non-natural amino acid is derived from the ARX517 antibody heavy chain amino acid sequence. Thus, in some embodiments, a “free payload” of the present disclosure is pAF-AS269. [106] The term “taxanes” include paclitaxel, as well as any active taxane derivative or pro- drug. [107] The terms “treat,” “treating” or “treatment”, as used herein, include alleviating, preventing, abating or ameliorating a disease or condition symptoms, preventing additional Attorney Docket No. AMB1013WOPCT1 symptoms, ameliorating or preventing the underlying metabolic causes of symptoms, inhibiting the disease or condition, e.g., arresting the development of the disease or condition, relieving the disease or condition, causing regression of the disease or condition, relieving a condition caused by the disease or condition, or stopping the symptoms of the disease or condition. The terms “treat,” “treating” or “treatment”, include, but are not limited to, prophylactic and/or therapeutic treatments. The term “treat”, “treating”, or “treatment” can refers to the decrease, reduction or amelioration of one or more symptoms associated with prostate cancer. [108] As used herein, the term “water soluble polymer” refers to any polymer that is soluble in aqueous solvents. Such water soluble polymers include, but are not limited to, polyethylene glycol, polyethylene glycol propionaldehyde, mono C1-C10 alkoxy or aryloxy derivatives thereof (described in U.S. Patent No. 5,252,714 which is incorporated by reference herein), monomethoxy-polyethylene glycol, polyvinyl pyrrolidone, polyvinyl alcohol, polyamino acids, divinylether maleic anhydride, N-(2-Hydroxypropyl)-methacrylamide, dextran, dextran derivatives including dextran sulfate, polypropylene glycol, polypropylene oxide/ethylene oxide copolymer, polyoxyethylated polyol, heparin, heparin fragments, polysaccharides, oligosaccharides, glycans, cellulose and cellulose derivatives, including but not limited to methylcellulose and carboxymethyl cellulose, serum albumin, starch and starch derivatives, polypeptides, polyalkylene glycol and derivatives thereof, copolymers of polyalkylene glycols and derivatives thereof, polyvinyl ethyl ethers, and alpha-beta-poly[(2-hydroxyethyl)-DL- aspartamide, and the like, or mixtures thereof. By way of example only, coupling of such water soluble polymers to natural amino acid polypeptides or non-natural polypeptides may result in changes including, but not limited to, increased water solubility, increased or modulated serum half-life, increased or modulated therapeutic half-life relative to the unmodified form, increased bioavailability, modulated biological activity, extended circulation time, modulated immunogenicity, modulated physical association characteristics including, but not limited to, Attorney Docket No. AMB1013WOPCT1 aggregation and multimer formation, altered receptor binding, altered binding to one or more binding partners, and altered receptor dimerization or multimerization. In addition, such water soluble polymers may or may not have their own biological activity. [109] As used herein, the term “modulated serum half-life” refers to positive or negative changes in the circulating half-life of a modified biologically active molecule relative to its non-modified form. By way of example, the modified biologically active molecules include, but are not limited to, natural amino acid, non-natural amino acid, natural amino acid polypeptide or non-natural amino acid polypeptide. By way of example, serum half-life is measured by taking blood samples at various time points after administration of the biologically active molecule or modified biologically active molecule and determining the concentration of that molecule in each sample. Correlation of the serum concentration with time allows calculation of the serum half-life. By way of example, modulated serum half-life may be an increased in serum half-life, which may enable an improved dosing regimen or avoid toxic effects. Such increases in serum may be at least about two-fold, at least about three-fold, at least about five-fold, or at least about ten-fold. Methods for evaluating serum half-life are known in the art and may be used for evaluating the serum half-life of antibodies and antibody drug conjugates of the present invention. [110] As used herein, "taxanes" are anti-cancer agents that interfere with or disrupt microtubule stability, formation and/or function. Such agents include paclitaxel, docetaxel and cabazitaxel, as well as any prodrugs or active derivatives of any of the foregoing, wherein the derivatives function against microtubules by the same mode of action as the taxanes from which they are derived. In some embodiments, “taxanes” refers to one or more of the following: paclitaxel, docetaxel, cabazitaxel. [111] The term “modulated therapeutic half-life,” as used herein, refers to positive or negative change in the half-life of the therapeutically effective amount of a modified biologically active Attorney Docket No. AMB1013WOPCT1 molecule, relative to its non-modified form. By way of example, the modified biologically active molecules include, but are not limited to, natural amino acid, non-natural amino acid, natural amino acid polypeptide or non-natural amino acid polypeptide. By way of example, therapeutic half-life is measured by measuring pharmacokinetic and/or pharmacodynamic properties of the molecule at various time points after administration. Increased therapeutic half-life may enable a particular beneficial dosing regimen, a particular beneficial total dose, or avoids an undesired effect. By way of example, the increased therapeutic half-life may result from increased potency, increased or decreased binding of the modified molecule to its target, an increase or decrease in another parameter or mechanism of action of the non-modified molecule, or an increased or decreased breakdown of the molecules by enzymes such as, by way of example only, proteases. Methods for evaluating therapeutic half-life are known in the art and may be used for evaluating the therapeutic half-life of antibodies and antibody drug conjugates of the present invention. [112] Antibody-based therapeutics have emerged as important components of therapies for an increasing number of human malignancies in such fields as oncology, inflammatory and infectious diseases. In most cases, the basis of the therapeutic function is the high degree of specificity and affinity the antibody-based drug has for its target antigen. Arming monoclonal antibodies with drugs, toxins, or radionuclides is yet another strategy by which monoclonal antibodies may induce therapeutic effect. By combining the exquisite targeting specificity of antibody with the tumor killing power of toxic effector molecules, immunoconjugates permit sensitive discrimination between target and normal tissue thereby resulting in fewer side effects than most conventional chemotherapeutic drugs. The toxins utilized can specifically, stably and irreversibly conjugate to unique sites in the antibody. This unique process of conjugation allows for the precise control of the location of the toxin on the antibody, and also the number of toxins conjugated to each antibody. Both of these features are critical for controlling Attorney Docket No. AMB1013WOPCT1 biophysical characteristics and toxicities associated with ADCs. (See for example Jackson et al., 2014, Tian et al., 2014). [113] Anti-PSMA antibody drug conjugates provided in the present disclosure include humanized or chimeric monoclonal antibodies and variants that bind to the extracellular domain of prostate specific membrane antigen (PSMA). Prostate specific membrane antigen is a type II membrane protein that is highly expressed, for example, in prostatic intraepithelial neoplasia (PIN), primary prostate cancers, and metastatic prostate cancers Anti-PSMA antibody disclosed herein can be any known PSMA antibody with at least one non-naturally or unnaturally encoded amino acid. [114] In one embodiment, the invention provides anti-PSMA antibodies, antibody fragments and variants thereof having a non-naturally encoded amino acid that facilitate antibody conjugation to a drug (e.g., a drug, toxin molecule). In one embodiment, the ADC comprises an anti-PSMA antibody conjugated to a drug wherein the conjugation occurs via a non- naturally encoded amino acid in the antibody. In one embodiment, the ADC comprises an anti- PSMA antibody conjugated to a drug wherein the conjugation occurs via a non-naturally encoded amino acid in the heavy chain of the antibody. In one embodiment, the ADC comprises an anti-PSMA antibody conjugated to a drug wherein the conjugation occurs via a non- naturally encoded amino acid in the light chain of the antibody. In one embodiment, the ADC comprises a full-length antibody conjugated to a drug wherein the conjugation occurs via a non-naturally encoded amino acid in the antibody. In one embodiment, the ADC comprises a full-length antibody conjugated to a drug wherein the conjugation occurs via a non-naturally encoded amino acid in the heavy chain of the antibody. In one embodiment, the ADC comprises a full-length antibody conjugated to a drug wherein the conjugation occurs via a non-naturally encoded amino acid in the light chain of the antibody. Attorney Docket No. AMB1013WOPCT1 [115] In some embodiments, the drug of the ADC is a cytotoxic drug or agent. In some aspects of the invention, the cytotoxic drug is selected from the group consisting of an auristatin, a DNA minor groove binding agent, a DNA minor groove alkylating agent, an enediyne, a lexitropsin, a duocarmycin, a taxane, a puromycin, a dolastatin, a maytansinoid, and a vinca alkaloid. In some aspects of the invention, the cytotoxic drug is AFP, MMAF, MMAE, AEB, AEVB, auristatin E, paclitaxel, docetaxel, CC-1065, SN-38, topotecan, morpholino- doxorubicin, rhizoxin, cyanomorpholino-doxorubicin, dolastatin-10, echinomycin, combretatstatin, chalicheamicin, maytansine, DM-1, or netropsin, but not limiting to such. [116] In some aspects of the invention, the cytotoxic drug is an anti-tubulin agent. In some embodiments, the anti-tubulin agent is an auristatin, a vinca alkaloid, a podophyllotoxin, a taxane, a baccatin derivative, a cryptophysin, a maytansinoid, a combretastatin, or a dolastatin. In other aspects of the invention, the antitubulin agent is AFP, MMAF, MMAE, AEB, AEVB, auristatin E, vincristine, vinblastine, vindesine, vinorelbine, VP-16, camptothecin, paclitaxel, docetaxel, epothilone A, epothilone B, nocodazole, colchicines, colcimid, estramustine, cemadotin, discodennolide, maytansine, DM-1, or eleutherobin but not limiting to such. [117] In other aspects of the invention, the cytotoxic drug of the ADC is gancyclovir, etanercept, cyclosporine, tacrolimus, rapamycin, cyclophosphamide, azathioprine, mycophenolate mofetil, methotrexate, cortisol, aldosterone, dexamethasone, a cyclooxygenase inhibitor, a 5-ipoxygenase inhibitor, or a leukotriene receptor antagonist. [118] In some embodiments of the invention, the antibody of the ADC comprises a full length antibody or fragment thereof that binds to PSMA, and is conjugated to a cytotoxic agent or an immunosuppressive agent, wherein the antibody-drug conjugate exerts: (a) a cytotoxic or cytostatic effect on a PSMA-expressing cancer cell line, or (b) a cytotoxic, cytostatic, or immunosuppressive effect on a PSMA-expressing immune cell, wherein the conjugation occurs at a non-naturally encoded amino acid in the antibody. Attorney Docket No. AMB1013WOPCT1 [119] In some embodiments, the antibody, variant, or composition of the present disclosure may be an antibody, variant, or composition that binds to a PSMA receptor. In other embodiments of the present invention the antibody, variant, or composition may be an antibody, variant, or composition that binds to extracellular surface of PSMA receptor. In another embodiment of the present invention the antibody, variant, or composition disclosed may be an antibody, variant, or composition that bind to a PSMA dimer. In some embodiments the antibody, variant, or composition of the present disclosure may be an antibody, variant, or composition that has CDRs from J591 grafted onto the framework region of the variable region. In other embodiments the antibody, variant, or composition of the present invention disclosure may be an antibody, variant, or composition that has a non-naturally encoded amino acid. In some embodiments the antibody, variant, or composition may be an antibody, variant, or composition that is described by more than one of the embodiments elsewhere herein the present invention disclosure. In some embodiments the antibody, antibody variant or antibody composition(s) disclosed herein may be fully humanized. In other embodiments the antibody, antibody variant or antibody composition(s) disclosed herein may be chimeric. In some embodiments of the present invention the antibody may be an antibody that is full length antibody (Variable + Fc regions), Fab, bispecific, Fab-dimers, Fab-bispecific, Fab-trispecific, bispecific T-cell engagers, dual-affinity re-targeting antibody, IgG1/IgG3 bispecific antibody, diabody, bispecific diabody, scFv-Fc, minibody. [120] Methods, compositions, and techniques for creating and using dolastatin linker derivatives or analogs comprising at least one carbonyl, dicarbonyl, oxime, hydroxylamine, aldehyde, protected aldehyde, ketone, protected ketone, thioester, ester, dicarbonyl, hydrazine, azide, amidine, imine, diamine, keto-amine, keto-alkyne, alkyne, cycloalkyne, or ene-dione are well known to one of ordinary skill in the art, (see, for example, WO2013/185117, incorporated herein by reference in its entirety). Methods, compositions, and techniques for creating and Attorney Docket No. AMB1013WOPCT1 using dolastatin linker derivatives or analogs comprising at least one non-natural amino acid or modified non-natural amino acid with an oxime, aromatic amine, heterocycle (e.g., indole, quinoxaline, phenazine, pyrazole, triazole, etc.) are also well known to the skilled artisan and described in, for example, WO2013/185117, incorporated herein by reference in its entirety. Such dolastatin linker derivatives comprising non-natural amino acids may contain further functionality, including but not limited to, a polymer; a water-soluble polymer; a derivative of polyethylene glycol; a second protein or polypeptide or polypeptide analog; an antibody or antibody fragment; and any combination thereof. Note that the various aforementioned functionalities are not meant to imply that the members of one functionality cannot be classified as members of another functionality. Indeed, there will be overlap depending upon the particular circumstances. By way of example only, a water-soluble polymer overlaps in scope with a derivative of polyethylene glycol, however the overlap is not complete and thus both functionalities are cited above. [121] Provided herein in some embodiments, is a toxic group linker derivative comprising a carbonyl, dicarbonyl, oxime, hydroxylamine, amino-oxy, aldehyde, protected aldehyde, ketone, protected ketone, thioester, ester, dicarbonyl, hydrazine, azide, amidine, imine, diamine, keto-amine, keto-alkyne, alkyne, cycloalkyne, or ene-dione. In some embodiments, the toxic group derivative comprises any of the linkers disclosed herein. Methods, compositions, and techniques for creating and using toxic group derivatives or analogs comprising at least one non-natural amino acid or modified non-natural amino acid with an oxime, aromatic amine, heterocycle (e.g., indole, quinoxaline, phenazine, pyrazole, triazole, etc.) are described in WO2013/185117 (incorporated herein by reference in its entirety). In some embodiments, such toxic derivatives comprising non-natural amino acids may contain further functionality, including but not limited to, a polymer; a water-soluble polymer; a derivative of polyethylene glycol; a second protein or polypeptide or polypeptide analog; an Attorney Docket No. AMB1013WOPCT1 antibody or antibody fragment; and any combination thereof. In specific embodiments, the toxic group is a tubulin inhibitor. In certain specific embodiments, the toxic group is dolastatin or auristatin. In other specific embodiments, the toxic group is dolastatin or auristatin derivative. Note that the various aforementioned functionalities are not meant to imply that the members of one functionality cannot be classified as members of another functionality. Indeed, there will be overlap depending upon the particular circumstances. By way of example only, a water-soluble polymer overlaps in scope with a derivative of polyethylene glycol, however the overlap is not complete and thus both functionalities are cited above. [122] Certain embodiments of the present invention describe preparations of certain toxic moieties with linkers that reduce the toxicity of the moiety in vivo while the toxic moiety retains pharmacological activity. In some embodiments, the toxicity of the linked toxic group, when administered to an animal or human, is reduced or eliminated compared to the free toxic group or toxic group derivatives comprising labile linkages, while retaining pharmacological activity. In some embodiments, increased doses of the linked toxic group (e.g., dolastatin linker derivatives, non-natural amino acid linked dolastatin derivatives) may be administered to animals or humans with greater safety. In certain embodiments, the non-natural amino acid polypeptides linked to a toxic moiety (e.g., dolastatin derivative) provides in vitro and in vivo stability. In some embodiments, the non-natural amino acid polypeptides linked to a toxic moiety (e.g., tubulin inhibitor, dolastatin-10 derivative) are efficacious and less toxic compared to the free toxic moiety (e.g., tubulin inhibitor, dolastatin-10). [123] The present invention provides anti-PSMA antibody drug conjugates (ADCs) and pharmaceutical formulations containing them. More particularly, the present disclosure relates to methods and uses of anti-PSMA ADCs and pharmaceutical compositions containing them in inhibiting, preventing or treating PSMA related diseases or cancers. In some embodiments, the cancer is prostate cancer or metastatic castration-resistant prostate cancer. In some Attorney Docket No. AMB1013WOPCT1 embodiments, the method comprises administering to the subject an anti-PSMA ADC of the present disclosure such as ARX517. Pharmaceutical formulations containing effective amounts of anti-PSMA ADCs and that are stable in storage are also provided. Methodology and Techniques [124] The present disclosure encompasses methodologies and technologies well known in the art. These include conventional methods of mass spectroscopy, NMR, HPLC, protein chemistry, biochemistry, recombinant DNA techniques and pharmacology, within the skill of the art. Compounds of the present invention disclosure can be synthesized using several processes or schemes employed in the art. See for example, Dubowchik et al., Bioconjugate Chem. 13: 855-869, 2002; Doronina et al., Nature Biotechnology 21(7): 778-784, 2003; WO2012/166560; WO2013/185117, each incorporated herein by reference. Many methodologies and techniques for synthesis of pharmaceutical, diagnostic or therapeutic compounds are well known to one of ordinary skill in the art. [125] The present invention, unless otherwise indicated, also encompass conventional techniques of molecular biology (including recombinant techniques), cell biology, biochemistry and immunology, all within the skill of the art. Such techniques are explained fully in the literature, such as, Molecular Cloning: A Laboratory Manual, Third Edition, Cold Spring Harbor Press, Cold Spring Harbor, NY (Sambrook et al. Eds., 2001); Oligonucleotide Synthesis: Methods And Applications (Methods in Molecular Biology), Herdewijn, P., Ed., Humana Press, Totowa, NJ; Oligonucleotide Synthesis (Gait, M. J., Ed., 1984); Methods In Molecular Biology, Humana Press, Totowa, NJ; Cell Biology: A Laboratory Notebook ,Academic Press, New York, NY (Cellis, J. E., Ed., 1998); Animal Cell Culture (Freshney, R. I., Ed., 1987); Introduction To Cell And Tissue Culture Plenum Press, New York, NY, (Mather, J. P. and Roberts, P. E., Eds., 1998); Cell And Tissue Culture: Laboratory Procedures John Wiley and Sons, Hoboken, NJ, (Doyle, A. et al., Eds., 1993-8); Methods In Enzymology Attorney Docket No. AMB1013WOPCT1 (Academic Press, Inc.) New York, NY; Weir's Handbook Of Experimental Immunology Wiley-Blackwell Publishers, New York, NY, (Herzenberg, L. A. et al. Eds., 1997); Gene Transfer Vectors For Mammalian Cells Cold Spring Harbor Press, Cold Spring Harbor, NY, (Miller, J. M. et al. Eds., 1987); Current Protocols In Molecular Biology, Greene Pub. Associates, New York, NY, (Ausubel, F. M. et al., Eds., 1987); PCR: The Polymerase Chain Reaction, Birkhauser, Boston, MA, (Mullis, K. et al., Eds., 1994); Current Protocols In Immunology, John Wiley and Sons, Hoboken, NJ, (Coligan, J. E. et al., eds., 1991); Short Protocols In Molecular Biology, Hoboken, NJ, (John Wiley and Sons, 1999); Immunobiology 7 Garland Science, London, UK, (Janeway, C. A. et al., 2007); Antibodies. Stride Publications, Devoran, UK, (P. Finch, 1997); Antibodies: A Practical Approach Oxford University Press, USA, New York, NY, (D. Catty., ed., 1989); Monoclonal Antibodies: A Practical Approach Oxford University Press, USA, New York NY, (Shepherd, P. et al. Eds., 2000); Using Antibodies: A Laboratory Manual Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, (Harlow, E. et al. Eds., 1998); The Antibodies Harwood Academic Publishers, London, UK, (Zanetti, M. et al. Eds.1995). Dolastatin Linker Derivatives [126] In embodiments of the present invention disclosure dolastatin linker derivatives or analogs comprising at least one non-natural amino acid or modified non-natural amino acid with a carbonyl, dicarbonyl, oxime or hydroxylamine group may be utilized. Methods for selecting and designing a dolastatin linker derivative to be modified using the methods, compositions and techniques are well known in the art, see for example WO2013/185117, incorporated herein by reference in its entirety. Dolastatin linker derivative may be designed de novo, including by way of example only, as part of high-throughput screening process (in which case numerous polypeptides may be designed, synthesized, characterized and/or tested) or based on the interests of the researcher. The new dolastatin linker derivative may also be Attorney Docket No. AMB1013WOPCT1 designed based on the structure of a known or partially characterized polypeptide. The principles for selecting which amino acid(s) to substitute and/or modify and the choice of which modification to employ are described in WO2013/185117, for example. Dolastatin linker derivative may be designed to meet the needs of the experimenter or end user. Such needs may include, but are not limited to, manipulating the therapeutic effectiveness of the polypeptide, improving the safety profile of the polypeptide, adjusting the pharmacokinetics, pharmacologics and/or pharmacodynamics of the polypeptide, such as, by way of example only, increasing water solubility, bioavailability, increasing serum half-life, increasing therapeutic half-life, modulating immunogenicity, modulating biological activity, or extending the circulation time. In addition, such modifications include, by way of example only, providing additional functionality to the polypeptide, incorporating an antibody, and any combination of the aforementioned modifications. Such dolastatin linker derivatives can be modified to contain an oxime, carbonyl, dicarbonyl, or hydroxylamine group. The dolastatin linker derivative may contain at least one, at least two, at least three, at least four, at least five, at least six, at least seven, at least eight, at least nine, or ten or more of a carbonyl or dicarbonyl group, oxime group, hydroxylamine group, or protected forms thereof. The dolastatin linker derivative can be the same or different, for example, there can be 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, or more different sites in the derivative that comprise 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, or more different reactive groups. [127] For example, dolastatin derivatives with linkers containing a hydroxylamine (also called an aminooxy) group allow for reaction with a variety of electrophilic groups to form conjugates, including but not limited to, with PEG or other water soluble polymers. Like hydrazines, hydrazides and semicarbazides, the enhanced nucleophilicity of the aminooxy group permits it to react efficiently and selectively with a variety of molecules that contain carbonyl- or dicarbonyl-groups, including but not limited to, ketones, aldehydes or other Attorney Docket No. AMB1013WOPCT1 functional groups with similar chemical reactivity. (See, for example, Shao, J. and Tam, J., J. Am. Chem. Soc.117:3893-3899, 1995; H. Hang and C. Bertozzi, Acc. Chem. Res.34(9): 727- 736, 2001). Whereas the result of reaction with a hydrazine group is the corresponding hydrazone, an oxime, however, results generally from the reaction of an aminooxy group with a carbonyl- or dicarbonyl-containing group such as, by way of example, a ketones, aldehydes or other functional groups with similar chemical reactivity. In some embodiments, dolastatin derivatives with linkers comprising an azide, alkyne or cycloalkyne allow for linking of molecules via cycloaddition reactions (e.g., 1,3-dipolar cycloadditions, azide-alkyne Huisgen cycloaddition, etc., described in U.S. Patent No.7,807,619 incorporated by reference herein to the extent relative to the reaction). [128] Thus, in certain embodiments described herein are dolastatin derivatives with linkers comprising a hydroxylamine, amino-oxy, aldehyde, protected aldehyde, ketone, protected ketone, thioester, ester, dicarbonyl, hydrazine, amidine, imine, diamine, keto-amine, keto- alkyne, and ene-dione hydroxylamine group, a hydroxylamine-like group (which has reactivity similar to a hydroxylamine group and is structurally similar to a hydroxylamine group), a masked hydroxylamine group (which can be readily converted into a hydroxylamine group), or a protected hydroxylamine group (which has reactivity similar to a hydroxylamine group upon deprotection). In some embodiments, the dolastatin derivatives with linkers comprise azides, alkynes or cycloalkynes. Examples of such dolastatin linker derivatives are included elsewhere herein and in WO2013/185117 and WO2005/074650 (each incorporated herein by reference in its entirety). Non-Natural Amino Acids [129] Non-naturally encoded amino acid site selection was based on surface exposure/site accessibility within the antibody and hydrophobic or neutral amino acid sites were selected to maintain the charge on the antibody. Methods for introducing non-natural amino acids inserted Attorney Docket No. AMB1013WOPCT1 into sites in a protein are described for example in WO2010/011735 and in WO2005/074650. The present invention employs such methodologies and techniques. The non-natural amino acids used in the methods and compositions described herein have at least one of the following four properties: (1) at least one functional group on the sidechain of the non-natural amino acid has at least one characteristics and/or activity and/or reactivity orthogonal to the chemical reactivity of the 20 common, genetically-encoded amino acids (i.e., alanine, arginine, asparagine, aspartic acid, cysteine, glutamine, glutamic acid, glycine, histidine, isoleucine, leucine, lysine, methionine, phenylalanine, proline, serine, threonine, tryptophan, tyrosine, and valine), or at least orthogonal to the chemical reactivity of the naturally occurring amino acids present in the polypeptide that includes the non-natural amino acid; (2) the introduced non- natural amino acids are substantially chemically inert toward the 20 common, genetically- encoded amino acids; (3) the non-natural amino acid can be stably incorporated into a polypeptide, preferably with the stability commensurate with the naturally-occurring amino acids or under typical physiological conditions, and further preferably such incorporation can occur via an in vivo system; and (4) the non-natural amino acid includes an oxime functional group or a functional group that can be transformed into an oxime group by reacting with a reagent, preferably under conditions that do not destroy the biological properties of the polypeptide that includes the non-natural amino acid (unless of course such a destruction of biological properties is the purpose of the modification/transformation), or where the transformation can occur under aqueous conditions at a pH between about 4 and about 8, or where the reactive site on the non-natural amino acid is an electrophilic site. Any number of non-natural amino acids can be introduced into the polypeptide. Non-natural amino acids may also include protected or masked oximes or protected or masked groups that can be transformed into an oxime group after deprotection of the protected group or unmasking of the masked group. Non-natural amino acids may also include protected or masked carbonyl or dicarbonyl Attorney Docket No. AMB1013WOPCT1 groups, which can be transformed into a carbonyl or dicarbonyl group after deprotection of the protected group or unmasking of the masked group and thereby are available to react with hydroxylamines or oximes to form oxime groups. Oxime-based non-natural amino acids may be synthesized by methods well known in the art, (see for example WO2013/185117 and WO2005/074650), including: (a) reaction of a hydroxylamine-containing non-natural amino acid with a carbonyl- or dicarbonyl-containing reagent; (b) reaction of a carbonyl- or dicarbonyl-containing non-natural amino acid with a hydroxylamine-containing reagent; or (c) reaction of an oxime-containing non-natural amino acid with certain carbonyl- or dicarbonyl- containing reagents. [130] Non-natural amino acids that may be used in the methods and compositions described herein include, but are not limited to, amino acids comprising amino acids with novel functional groups, amino acids that covalently or noncovalently interact with other molecules, glycosylated amino acids such as a sugar substituted serine, other carbohydrate modified amino acids, keto-containing amino acids, aldehyde-containing amino acids, amino acids comprising polyethylene glycol or other polyethers, heavy atom substituted amino acids, chemically cleavable and/or photocleavable amino acids, amino acids with an elongated side chains as compared to natural amino acids, including but not limited to, polyethers or long chain hydrocarbons, including but not limited to, greater than about 5 or greater than about 10 carbons, carbon-linked sugar-containing amino acids, redox-active amino acids, amino thioacid containing amino acids, and amino acids comprising one or more toxic moiety. [131] In some embodiments disclosed herein are anti-PSMA antibodies comprising one or more non-natural amino acids. The one or more non-natural amino acids may be encoded by a codon that does not code for one of the twenty natural amino acids. The one or more non- natural amino acids may be encoded by a nonsense codon (stop codon). The stop codon may be an amber codon. The amber codon may comprise a UAG sequence. The stop codon may be Attorney Docket No. AMB1013WOPCT1 an ochre codon. The ochre codon may comprise a UAA sequence. The stop codon may be an opal or umber codon. The opal or umber codon may comprise a UGA sequence. The one or more non-natural amino acids may be encoded by a four-base codon. [132] Non-natural amino acids of the present disclosure include, but are not limited to, 1) substituted phenylalanine and tyrosine analogues, such as 4-amino-L-phenylalanine, 4-acetyl- L-phenylalanine, 4-azido-L-phenylalanine, 4-nitro-L-phenylalanine, 3-methoxy-L- phenylalanine, 4-isopropyl-L-phenylalanine, 3-nitro-L-tyrosine, O-methyl-L-tyrosine and O- phosphotyrosine; 2) amino acids that can be photo-cross-linked, e.g., amino acids with aryl azide or benzophenone groups, such as 4-azidophenylalanine or 4-benzoylphenylalanine; 3) amino acids that have unique chemical reactivity, such as 4-acetyl-L-phenylalanine, 3-acetyl- L-phenylalanine, O-allyl-L-tyrosine, O-2-propyn-1-yl-L-tyrosine, N-(ethylthio)thiocarbonyl- L-phenylalanine and p-(3-oxobutanoyl)-L-phenylalanine; 4) heavy-atom-containing amino acids, e.g., for phasing in X-ray crystallography, such as 4-iodo-L-phenylalanine or 4-bromo- L-phenylalanine; 5) a redox-active amino acid, such as 3,4-dihydroxy-L-phenylalanine; 6) a fluorinated amino acid, such as a 2-fluorophenylalanine (e.g., 2-fluoro-L-phenylalanine), a 3- fluorophenylalanine (e.g., 3-fluoro-L-phenylalanine) or a 4-fluorophenylalanine (e.g., 4- fluoro-L-phenylalanine; 7) a fluorescent amino acid, such as an amino acid containing a naphthyl, dansyl or 7-aminocoumarin side chain; 8) a photocleavable or photoisomerizable amino acid, such as an amino acid comprising an azobenzyl or nitrobenzyl, e.g., cysteine, serine or tyrosine comprising azobenzyl or nitrobenzyl; 9) a β-amino acid (e.g., a β
2 or β
3 amino acid); 10) a homo-amino acid, such as homoglutamine (e.g., beta-homoglutamine) or homophenylalanine (e.g., beta-homophenylalanine); 11) a proline or pyruvic acid derivative; 12) a 3-substituted alanine derivative; 14) a glycine derivative; 15) a linear core amino acid; 16) a diamino acid; 17) a D-amino acid; 18) an N-methyl amino acid; 19) a phosphotyrosine mimetic, such as a carboxymethylphenylalanine (pCmF) (e.g., 4-carboxymethyl-L- Attorney Docket No. AMB1013WOPCT1 phenylalanine); 20) 2-aminooctanoic acid; and 21) an amino acid comprising a saccharide moiety, such as N-acetyl-L-glucosaminyl-L-serine, beta-N-acetylglucosamine-O-serine, N- acetyl-L-galactosaminyl-L-serine, alpha-N-acetylgalactosamine-O-serine, O-(3-O-D- galactosyl-N-acetyl-beta-D-galactosaminyl)-L-serine, N-acetyl-L-glucosaminyl-L-threonine, alpha-N-acetylgalactosamine-O-threonine, 3-O-(N-acetyl-beta-D-glucosaminyl)-L-threonine, N-acetyl-L-glucosaminyl-L-asparagine, N4-(β-N-Acetyl-D-glucosaminyl)-L-asparagine and O-(mannosyl)-L-serine; an amino acid wherein the naturally-occurring N- or O- linkage between the amino acid and the saccharide is replaced by a covalent linkage not commonly found in nature, including but not limited to, an alkene, an oxime, a thioether, an amide and the like; or an amino acid containing saccharides that are not commonly found in naturally- occurring polypeptides, such as 2-deoxy-glucose, 2-deoxy-galactose and the like. Specific examples of non-natural amino acids include, but are not limited to, a p-acetylphenylalanine (4-acetyl phenylalanine) (including 4-acetyl-L-phenylalanine, also referred to herein as p- acetyl-L-phenylalanine (pAF)), a 4-boronophenylalanine (pBoF) (e.g., 4-borono-L- phenylalanine, a 4-propargyloxyphenylalanine (pPrF) (e.g., 4-propargyloxy-L-phenylalanine), an O-methyltyrosine (e.g., O-methyl-L-tyrosine), a 3-(2-naphthyl)alanine (NapA) (e.g., 3-(2- naphthyl)-L-alanine), a 3-methylphenylalanine (e.g., 3-methyl-L-phenylalanine), an O- allyltyrosine (e.g., O-allyl-L-tyrosine), an O-isopropyltyrosine (e.g., O-isopropyl-L-tyrosine), a dopamine (e.g., L-Dopa), a 4-isopropylphenylalanine (e.g., 4-isopropyl-L-phenylalanine), a 4-azidophenylalanine (pAz) (e.g., 4-azido-L-phenylalanine), a 4-benzoylphenylalanine (pBpF) (e.g., 4-benzoyl-L-phenylalanine), an O-phosphoserine (e.g., O-phospho-L-serine), an O- phosphotyrosine (e.g., O-phospho-L-tyrosine), a 4-iodophenylalanine (pIF) (e.g., 4-iodo-L- phenylalanine, a 4-bromophenylalanine (e.g., 4-bromo-L-phenylalanine), a 4- aminophenylalanine (e.g., 4-amino-L-phenylalanine), a 4-cyanophenylalanine (pCNF) (e.g., 4- cyano-L-phenylalanine, a (8-hydroxyquinolin-3-yl)alanine (HQA) (e.g., (8-hydroxyquinolin- Attorney Docket No. AMB1013WOPCT1 3-yl)-L-alanine), a (2,2-bipyridin-5-yl)alanine (BipyA) (e.g., (2,2-bipyridin-5-yl)-L-alanine), and the like. Additional non-natural amino acids are disclosed in Liu et al. (2010) Annu Rev Biochem, 79:413-44; Wang et al. (2005) Angew Chem Int Ed, 44:34-66; and Published International Application Nos.: WO 2012/166560, WO 2012/166559, WO 2011/028195, WO 2010/037062, WO 2008/083346, WO 2008/077079, WO 2007/094916, WO 2007/079130, WO 2007/070659 and WO 2007/059312, the entire contents of each of which are hereby incorporated by reference herein in their entirety. In some embodiments, the one or more non-natural amino acids can be p-acetylphenylalanine. In some more particular embodiments, the one or more non-natural amino acids can be p-acetyl-L-phenylalanine (pAF). [133] In some embodiments, one or more non-natural amino acids is selected from the group consisting of 4-acetyl phenylalanine, 3-O-(N-acetyl-beta-D-glucosaminyl)threonine, N4-(β-N- Acetyl-D-glucosaminyl)asparagine, O-allyltyrosine, alpha-N-acetylgalactosamine-O-serine, alpha-N-acetylgalactosamine-O-threonine, 2-aminooctanoic acid, 2-aminophenylalanine, 3- aminophenylalanine, 4-aminophenylalanine, 2-aminotyrosine, 3-aminotyrosine, 4- azidophenylalanine, 4-benzoylphenylalanine, (2,2-bipyridin-5yl)alanine, 3- boronophenylalanine, 4-boronophenylalanine, 4-bromophenylalanine, p- carboxymethylphenylalanine, 4-carboxyphenylalanine, p-cyanophenylalanine, 3,4- dihydroxyphenylalanine, 4-ethynylphenylalanine, 2-fluorophenylalanine, 3- fluorophenylalanine, 4-fluorophenylalanine, O-(3-O-D-galactosyl-N-acetyl-beta-D- galactosaminyl)serine, homoglutamine, (8-hydroxyquinolin-3-yl)alanine, 4- iodophenylalanine, 4-isopropylphenylalanine, O-i-propyltyrosine, 3-isopropyltyrosine, O- mannopyranosylserine, 2-methoxyphenylalanine, 3-methoxyphenylalanine, 4- methoxyphenylalanine, 3-methylphenylalanine, O-methyltyrosine, 3-(2-naphthyl)alanine, 5- nitrohistidine, 4-nitrohistidine, 4-nitroleucine, 2-nitrophenylalanine, 3-nitrophenylalanine, 4- nitrophenylalanine, 4-nitrotryptophan, 5-nitrotryptophan, 6-nitrotryptophan, 7- Attorney Docket No. AMB1013WOPCT1 nitrotryptophan, 2-nitrotyrosine, 3-nitrotyrosine, O-phosphoserine, O-phosphotyrosine, 4- propargyloxyphenylalanine, O-2-propyn-1-yltyrosine, 4-sulfophenylalanine and O- sulfotyrosine. [134] In some further embodiments, one or more non-natural amino acids is selected from the group consisting of 4-acetyl-L-phenylalanine (para-acetyl-L-phenylalanine (pAF)), 3-O-(N- acetyl-beta-D-glucosaminyl)-L-threonine, N4-(β-N-Acetyl-D-glucosaminyl)-L-asparagine, O- allyl-L-tyrosine, alpha-N-acetylgalactosamine-O-L-serine, alpha-N-acetylgalactosamine-O-L- threonine, 2-aminooctanoic acid, 2-amino-L-phenylalanine, 3-amino-L-phenylalanine, 4- amino-L-phenylalanine, 2-amino-L-tyrosine, 3-amino-L-tyrosine, 4-azido-L-phenylalanine, 4- benzoyl-L-phenylalanine, (2,2-bipyridin-5yl)-L-alanine, 3-borono-L-phenylalanine, 4-borono- L-phenylalanine, 4-bromo-L-phenylalanine, p-carboxymethyl-L-phenylalanine, 4-carboxy-L- phenylalanine, p-cyano-L-phenylalanine, 3,4-dihydroxy-L-phenylalanine (L-DOPA), 4- ethynyl-L-phenylalanine, 2-fluoro-L-phenylalanine, 3-fluoro-L-phenylalanine, 4-fluoro-L- phenylalanine, O-(3-O-D-galactosyl-N-acetyl-beta-D-galactosaminyl)-L-serine, L- homoglutamine, (8-hydroxyquinolin-3-yl)-L-alanine, 4-iodo-L-phenylalanine, 4-isopropyl-L- phenylalanine, O-i-propyl-L-tyrosine, 3-isopropyl-L-tyrosine, O-mannopyranosyl-L-serine, 2- methoxy-L-phenylalanine, 3-methoxy-L-phenylalanine, 4-methoxy-L-phenylalanine, 3- methyl-L-phenylalanine, O-methyl-L-tyrosine, 3-(2-naphthyl)-L-alanine, 5-nitro-L-histidine, 4-nitro-L-histidine, 4-nitro-L-leucine, 2-nitro-L-phenylalanine, 3-nitro-L-phenylalanine, 4- nitro-L-phenylalanine, 4-nitro-L-tryptophan, 5-nitro-L-tryptophan, 6-nitro-L-tryptophan, 7- nitro-L-tryptophan, 2-nitro-L-tyrosine, 3-nitro-L-tyrosine, O-phospho-L-serine, O-phospho-L- tyrosine, 4-propargyloxy-L-phenylalanine, O-2-propyn-1-yl-L-tyrosine, 4-sulfo-L- phenylalanine and O-sulfo-L-tyrosine. In some embodiments, the one or more non-natural amino acids can be p-acetyl-L-phenylalanine (pAF). Thus, in some embodiments, each and every one of the one or more non-natural amino acids is pAF. Attorney Docket No. AMB1013WOPCT1 [135] In certain embodiments of the disclosure, an antibody with at least one non-natural amino acid includes at least one post-translational modification. In one embodiment, the at least one post-translational modification comprises attachment of a molecule including but not limited to, a water soluble polymer, a derivative of polyethylene glycol, a drug, a second protein or polypeptide or polypeptide analog, an antibody or antibody fragment, a biologically active agent, a small molecule, or any combination of the above or any other desirable compound or substance, comprising a second reactive group to at least one non-natural amino acid comprising a first reactive group utilizing chemistry methodology that is known to one of ordinary skill in the art to be suitable for the particular reactive groups. For example, the first reactive group is an alkynyl moiety (including but not limited to, the non-natural amino acid p-propargyloxyphenylalanine, where the propargyl group is also sometimes referred to as an acetylene moiety) and the second reactive group is an azido moiety, and [3+2] cycloaddition chemistry methodologies are utilized. In another example, the first reactive group is the azido moiety (including but not limited to, the non-natural amino acid p-azido-L-phenylalanine) and the second reactive group is the alkynyl moiety. In certain embodiments of the modified antibody polypeptide of the present disclosure at least one non-natural amino acid, (including but not limited to, non-natural amino acid containing a keto functional group), comprising at least one post-translational modification is used where the at least one post-translational modification comprises a saccharide moiety. In certain embodiments, the post-translational modification is made in vivo in a eukaryotic cell or in a non-eukaryotic cell. In other embodiments the post-translational modification is made in vitro. In another embodiment, the post-translational modification is made in vitro and in vivo. [136] In some embodiments, the non-natural amino acid may be modified to incorporate a chemical group. In some embodiments the non-natural amino acid may be modified to incorporate a ketone group. The one or more non-natural amino acids may comprise at least Attorney Docket No. AMB1013WOPCT1 one oxime, carbonyl, dicarbonyl, hydroxylamine group or a combination thereof. The one or more non-natural amino acids may comprise at least one carbonyl, dicarbonyl, alkoxy-amine, hydrazine, acyclic alkene, acyclic alkyne, cyclooctyne, aryl/alkyl azide, norbornene, cyclopropene, trans-cyclooctene, or tetrazine functional group or a combination thereof. [137] In some embodiments disclosed herein the non-natural amino acid is site-specifically incorporated into the antibody, antibody fragment or variant. In some embodiments the non- natural amino acid is site-specifically incorporated into an antibody, antibody fragment or variant. Methods for incorporating a non-natural amino acid into a molecule, for example, proteins, polypeptides or peptides, are disclosed in U.S. Patent Nos.: 7,332,571; 7,928,163; 7,696,312; 8,008,456; 8,048,988; 8,809,511; 8,859,802; 8,791,231; 8,476,411; or 9,637,411, (each of which is incorporated herein by reference in its entirety), and in the Examples herein. The one or more non-natural amino acids may be incorporated by methods known in the art. For example, cell-based or cell-free systems may be used, and auxotrophic strains may also be used in place of engineered tRNA and synthetase. In certain embodiments, orthogonal tRNA synthetase are used as disclosed in for example, WO2002085923A2; WO2002086075A2; WO2004035743A2; WO2007021297A1; WO2006068802A2; and WO2006069246A2; the contents of each of which are incorporated herein by reference in their entirety. Incorporating one or more non-natural amino acids into the antibody or antibody fragment or variant may comprise modifying one or more amino acid residues in the antibody or antibody fragment or variant. Modifying the one or more amino acid residues in the antibody or antibody fragment or variant may comprise mutating one or more nucleotides in the nucleotide sequence encoding the antibody or antibody fragment or variant. Mutating the one or more nucleotides in the nucleotide sequence encoding the antibody or antibody fragment or variant may comprise altering a codon encoding an amino acid to a nonsense codon. Incorporating one or more non- natural amino acids into the antibody or antibody fragment or variant may comprise modifying Attorney Docket No. AMB1013WOPCT1 one or more amino acid residues in the antibody or antibody fragment or variant to produce one or more amber codons in the antibody or antibody fragment or variant. The one or more non-natural amino acids may be incorporated into the antibody or antibody fragment or variant in response to an amber codon. The one or more non-natural amino acids may be site- specifically incorporated into the antibody or antibody fragment or variant. Incorporating one or more non-natural amino acids into the antibody or antibody fragment or variant may comprise one or more genetically encoded non-natural amino acids with orthogonal chemical reactivity relative to the canonical twenty amino acids to site-specifically modify the biologically active molecule or targeting agent. Incorporating the one or more non-natural amino acids may comprise use of a tRNA/aminoacyl-tRNA synthetase pair to site-specifically incorporate one or more non-natural amino acids at defined sites in the biologically active molecule or targeting agent in response to one or more amber nonsense codon. Additional methods for incorporating non-natural amino acids include, but are not limited to, methods disclosed in Chatterjee et al., A Versatile Platform for Single- and Multiple-Unnatural Amino Acid Mutagenesis in Escherichia coli, Biochemistry, 2013; Kazane et al., J Am Chem Soc, 135(1):340-6, 2013; Kim et al., J Am Chem Soc, 134(24):9918-21, 2012; Johnson et al., Nat Chem Biol, 7(11):779-86, 2011; and Hutchins et al., J Mol Biol, 406(4):595-603, 2011. The one or more non-natural amino acids may be produced through selective reaction of one or more natural amino acids. The selective reaction may be mediated by one or more enzymes. In non-limiting examples, the selective reaction of one or more cysteines with formylglycine generating enzyme (FGE) may produce one or more formylglycines as described in Rabuka et al., Nature Protocols 7: 1052-1067, 2012. The one or more non-natural amino acids may involve a chemical reaction to form a linker. The chemical reaction to form the linker may include a bioorthogonal reaction. The chemical reaction to form the linker may include click chemistry. See for example WO2006/050262 incorporated herein by reference in its entirety. Attorney Docket No. AMB1013WOPCT1 [138] In some embodiments, non-natural amino acids comprise a saccharide moiety. Examples of such amino acids include N-acetyl-L-glucosaminyl-L-serine, N-acetyl-L- galactosaminyl-L-serine, N-acetyl-L-glucosaminyl-L-threonine, N-acetyl-L-glucosaminyl-L- asparagine and O-mannosaminyl-L-serine. Examples of such amino acids also include examples where the naturally-occurring N- or O- linkage between the amino acid and the saccharide is replaced by a covalent linkage not commonly found in nature – including but not limited to, an alkene, an oxime, a thioether, an amide and the like. Examples of such amino acids also include saccharides that are not commonly found in naturally-occurring proteins such as 2-deoxy-glucose, 2-deoxygalactose and the like. [139] The chemical moieties incorporated into polypeptides via incorporation of non-natural amino acids into such polypeptides offer a variety of advantages and manipulations of polypeptides. For example, the unique reactivity of a carbonyl or dicarbonyl functional group (including a keto- or aldehyde- functional group) allows selective modification of proteins with any of a number of hydrazine- or hydroxylamine-containing reagents in vivo and in vitro. A heavy atom non-natural amino acid, for example, can be useful for phasing x-ray structure data. The site-specific introduction of heavy atoms using non-natural amino acids also provides selectivity and flexibility in choosing positions for heavy atoms. Photoreactive non-natural amino acids (including but not limited to, amino acids with benzophenone and arylazides (including but not limited to, phenylazide side chains), for example, allow for efficient in vivo and in vitro photocrosslinking of polypeptides. Examples of photoreactive non-natural amino acids include, but are not limited to, p-azido-phenylalanine and p-benzoyl-phenylalanine. The polypeptide with the photoreactive non-natural amino acids may then be crosslinked at will by excitation of the photoreactive group-providing temporal control. In a non-limiting example, the methyl group of a non-natural amino can be substituted with an isotopically labeled, including but not limited to, with a methyl group, as a probe of local structure and dynamics, Attorney Docket No. AMB1013WOPCT1 including but not limited to, with the use of nuclear magnetic resonance and vibrational spectroscopy. Non-Natural Amino Acid Linked Dolastatin Derivatives [140] In other embodiments of the present invention described herein are methods, strategies and techniques for incorporating at least one dolastatin linker derivatives into a non-natural amino acid. The present invention described herein includes methods for producing, purifying, characterizing and using dolastatin linker derivatives containing at least one such non-natural amino acid. Also included with this aspect are compositions of and methods for producing, purifying, characterizing and using oligonucleotides (including DNA and RNA) that can be used to produce, at least in part, a dolastatin linker derivative containing at least one non-natural amino acid. Also included with this aspect are compositions of and methods for producing, purifying, characterizing and using cells that can express such oligonucleotides that can be used to produce, at least in part, a dolastatin linker derivative containing at least one non-natural amino acid. [141] Thus, dolastatin linker derivatives comprising at least one non-natural amino acid or modified non-natural amino acid with a carbonyl, dicarbonyl, alkyne, cycloalkyne, azide, oxime or hydroxylamine group are provided and described herein. In certain embodiments, dolastatin linker derivatives with at least one non-natural amino acid or modified non-natural amino acid with a carbonyl, dicarbonyl, alkyne, cycloalkyne, azide, oxime or hydroxylamine group include at least one post-translational modification at some position on the polypeptide. In some embodiments the co-translational or post-translational modification occurs via the cellular machinery (e.g., glycosylation, acetylation, acylation, lipid-modification, palmitoylation, palmitate addition, phosphorylation, glycolipid-linkage modification, and the like), in many instances, such cellular-machinery-based co-translational or post-translational modifications occur at the naturally occurring amino acid sites on the polypeptide, however, in Attorney Docket No. AMB1013WOPCT1 certain embodiments, the cellular-machinery-based co-translational or post-translational modifications occur on the non-natural amino acid site(s) on the polypeptide. [142] In other embodiments, the post-translational modification does not utilize the cellular machinery, but the functionality is instead provided by attachment of a molecule (a polymer; a water soluble polymer; a derivative of polyethylene glycol; a second protein or polypeptide or polypeptide analog; an antibody or antibody fragment; and any combination thereof) comprising a second reactive group to the at least one non-natural amino acid comprising a first reactive group (including but not limited to, non-natural amino acid containing a ketone, aldehyde, acetal, hemiacetal, alkyne, cycloalkyne, azide, oxime, or hydroxylamine functional group) utilizing chemistry methodology described herein, or others suitable for the particular reactive groups. In certain embodiments, the co-translational or post-translational modification is made in vivo in a eukaryotic cell or in a non-eukaryotic cell. In certain embodiments, the post-translational modification is made in vitro not utilizing the cellular machinery. Also included with this aspect are methods for producing, purifying, characterizing and using such dolastatin linker derivatives containing at least one such co-translationally or post- translationally modified non-natural amino acids. [143] Also included within the scope of the methods, compositions, strategies and techniques described herein are reagents capable of reacting with a dolastatin linker derivative (containing a carbonyl or dicarbonyl group, oxime group, alkyne, cycloalkyne, azide, hydroxylamine group, or masked or protected forms thereof) that is part of a polypeptide so as to produce any of the aforementioned post-translational modifications. In certain embodiments, the resulting post-translationally modified dolastatin linker derivative will contain at least one oxime group; the resulting modified oxime-containing dolastatin linker derivative may undergo subsequent modification reactions. Also included with this aspect are methods for producing, purifying, Attorney Docket No. AMB1013WOPCT1 characterizing and using such reagents that are capable of any such post-translational modifications of such dolastatin linker derivative(s). [144] In certain embodiments, the polypeptide or non-natural amino acid linked dolastatin derivative includes at least one co-translational or post-translational modification that is made in vivo by one host cell, where the post-translational modification is not normally made by another host cell type. In certain embodiments, the polypeptide includes at least one co- translational or post-translational modification that is made in vivo by a eukaryotic cell, where the co-translational or post-translational modification is not normally made by a non-eukaryotic cell. Examples of such co-translational or post-translational modifications include, but are not limited to, glycosylation, acetylation, acylation, lipid-modification, palmitoylation, palmitate addition, phosphorylation, glycolipid-linkage modification, and the like. In one embodiment, the co-translational or post-translational modification comprises attachment of an oligosaccharide to an asparagine by a GlcNAc-asparagine linkage (including but not limited to, where the oligosaccharide comprises (GlcNAc-Man)
2-Man-GlcNAc-GlcNAc, and the like). In another embodiment, the co-translational or post-translational modification comprises attachment of an oligosaccharide (including but not limited to, Gal-GalNAc, Gal-GlcNAc, etc.) to a serine or threonine by a GalNAc-serine, a GalNAc-threonine, a GlcNAc-serine, or a GlcNAc-threonine linkage. In certain embodiments, a protein or polypeptide can comprise a secretion or localization sequence, an epitope tag, a FLAG tag, a polyhistidine tag, a GST fusion, and/or the like. Also included with this aspect are methods for producing, purifying, characterizing and using such polypeptides containing at least one such co-translational or post- translational modification. In other embodiments, the glycosylated non-natural amino acid polypeptide is produced in a non-glycosylated form. Such a non-glycosylated form of a glycosylated non-natural amino acid may be produced by methods that include chemical or enzymatic removal of oligosaccharide groups from an isolated or substantially purified or Attorney Docket No. AMB1013WOPCT1 unpurified glycosylated non-natural amino acid polypeptide; production of the non-natural amino acid in a host that does not glycosylate such a non-natural amino acid polypeptide (such a host including, prokaryotes or eukaryotes engineered or mutated to not glycosylate such a polypeptide), the introduction of a glycosylation inhibitor into the cell culture medium in which such a non-natural amino acid polypeptide is being produced by a eukaryote that normally would glycosylate such a polypeptide, or a combination of any such methods. Also described herein are such non-glycosylated forms of normally-glycosylated non-natural amino acid polypeptides (by normally-glycosylated is meant a polypeptide that would be glycosylated when produced under conditions in which naturally-occurring polypeptides are glycosylated). Of course, such non-glycosylated forms of normally-glycosylated non-natural amino acid polypeptides (or indeed any polypeptide described herein) may be in an unpurified form, a substantially purified form, or in an isolated form. Oxime-Containing Linked Dolastatin Derivatives [145] Non-natural amino acid dolastatin linked derivatives containing an oxime group allow for reaction with a variety of reagents that contain certain reactive carbonyl- or dicarbonyl- groups (including but not limited to, ketones, aldehydes, or other groups with similar reactivity) to form new non-natural amino acids comprising a new oxime group. Such an oxime exchange reaction allows for the further functionalization of dolastatin linked derivatives. Further, the original dolastatin linked derivative containing an oxime group may be useful in their own right as long as the oxime linkage is stable under conditions necessary to incorporate the amino acid into a polypeptide (e.g., the in vivo, in vitro and chemical synthetic methods described herein and in WO2013/185117 and WO2005/074650, each incorporated herein by reference). [146] Thus, in certain embodiments described herein are non-natural amino acid dolastatin linked derivatives with sidechains comprising an oxime group, an oxime-like group (which has reactivity similar to an oxime group and is structurally similar to an oxime group), a masked Attorney Docket No. AMB1013WOPCT1 oxime group (which can be readily converted into an oxime group), or a protected oxime group (which has reactivity similar to an oxime group upon deprotection). [147] The methods and compositions for incorporation of one or more non-natural amino acids into a dolastatin linker derivative are well known in the art, (see for example WO2013/185117 and WO2005/074650, each incorporated herein by reference in its entirety). One or more non-natural amino acids may be incorporated at one or more particular positions which do not disrupt activity of the dolastatin linker derivative. This can be achieved by making “conservative” substitutions, including but not limited to, substituting hydrophobic amino acids with non-natural or natural hydrophobic amino acids, bulky amino acids with non-natural or natural bulky amino acids, hydrophilic amino acids with non-natural or natural hydrophilic amino acids) and/or inserting the non-natural amino acid in a location that is not required for activity. [148] A variety of biochemical and structural approaches can be employed to select the desired sites for substitution with a non-natural amino acid within the dolastatin linker derivative. In some embodiments, the non-natural amino acid is linked at the C-terminus of the dolastatin derivative. In other embodiments, the non-natural amino acid is linked at the N- terminus of the dolastatin derivative. Any position of the dolastatin linker derivative is suitable for selection to incorporate a non-natural amino acid, and selection may be based on rational design or by random selection for any or no particular desired purpose. Selection of desired sites may be based on producing a non-natural amino acid polypeptide (which may be further modified or remain unmodified) having any desired property or activity, including but not limited to a receptor binding modulators, receptor activity modulators, modulators of binding to binder partners, binding partner activity modulators, binding partner conformation modulators, dimer or multimer formation, no change to activity or property compared to the native molecule, or manipulating any physical or chemical property of the polypeptide such as Attorney Docket No. AMB1013WOPCT1 solubility, aggregation, or stability. Alternatively, the sites identified as critical to biological activity may also be good candidates for substitution with a non-natural amino acid, again depending on the desired activity sought for the polypeptide. Another alternative would be to simply make serial substitutions in each position on the polypeptide chain with a non-natural amino acid and observe the effect on the activities of the polypeptide. Any means, technique, or method for selecting a position for substitution with a non-natural amino acid into any polypeptide is suitable for use in the methods, techniques and compositions described herein. [149] The structure and activity of naturally-occurring mutants of a polypeptide that contain deletions can also be examined to determine regions of the protein that are likely to be tolerant of substitution with a non-natural amino acid. Once residues that are likely to be intolerant to substitution with non-natural amino acids have been eliminated, the impact of proposed substitutions at each of the remaining positions can be examined using methods including, but not limited to, the three-dimensional structure of the relevant polypeptide, and any associated ligands or binding proteins. X-ray crystallographic and NMR structures of many polypeptides are available in the Protein Data Bank (PDB, see world wide web for rcsb.org), a centralized database containing three-dimensional structural data of large molecules of proteins and nucleic acids, one can be used to identify amino acid positions that can be substituted with non- natural amino acids. In addition, models may be made investigating the secondary and tertiary structure of polypeptides, if three-dimensional structural data is not available. Thus, the identity of amino acid positions that can be substituted with non-natural amino acids can be readily obtained. [150] Exemplary sites of incorporation of a non-natural amino acid include, but are not limited to, those that are excluded from potential receptor binding regions, or regions for binding to binding proteins or ligands may be fully or partially solvent exposed, have minimal or no hydrogen-bonding interactions with nearby residues, may be minimally exposed to Attorney Docket No. AMB1013WOPCT1 nearby reactive residues, and/or may be in regions that are highly flexible as predicted by the three-dimensional crystal structure of a particular polypeptide with its associated receptor, ligand or binding proteins. [151] A wide variety of non-natural amino acids can be substituted for, or incorporated into, a given position in a polypeptide. By way of example, a particular non-natural amino acid may be selected for incorporation based on an examination of the three-dimensional crystal structure of a polypeptide with its associated ligand, receptor and/or binding proteins, a preference for conservative substitutions. [152] In one embodiment, the methods described herein include incorporating into the dolastatin linker derivative, where the dolastatin linker derivative comprises a first reactive group; and contacting the dolastatin linker derivative with a molecule (including but not limited to a second protein or polypeptide or polypeptide analog; an antibody or antibody fragment; and any combination thereof) that comprises a second reactive group. In certain embodiments, the first reactive group is a hydroxylamine moiety and the second reactive group is a carbonyl or dicarbonyl moiety, whereby an oxime linkage is formed. In certain embodiments, the first reactive group is a carbonyl or dicarbonyl moiety and the second reactive group is a hydroxylamine moiety, whereby an oxime linkage is formed. In certain embodiments, the first reactive group is a carbonyl or dicarbonyl moiety and the second reactive group is an oxime moiety, whereby an oxime exchange reaction occurs. In certain embodiments, the first reactive group is an oxime moiety and the second reactive group is carbonyl or dicarbonyl moiety, whereby an oxime exchange reaction occurs. [153] In some cases, the dolastatin linker derivative incorporation(s) will be combined with other additions, substitutions, or deletions within the polypeptide to affect other chemical, physical, pharmacologic and/or biological traits. In some cases, the other additions, substitutions or deletions may increase the stability (including but not limited to, resistance to Attorney Docket No. AMB1013WOPCT1 proteolytic degradation) of the polypeptide or increase affinity of the polypeptide for its appropriate receptor, ligand and/or binding proteins. In some cases, the other additions, substitutions or deletions may increase the solubility (including but not limited to, when expressed in E. coli or other host cells) of the polypeptide. In some embodiments sites are selected for substitution with a naturally encoded or non-natural amino acid in addition to another site for incorporation of a non-natural amino acid for the purpose of increasing the polypeptide solubility following expression in E. coli, or other recombinant host cells. In some embodiments, the polypeptides comprise another addition, substitution, or deletion that modulates affinity for the associated ligand, binding proteins, and/or receptor, modulates (including but not limited to, increases or decreases) receptor dimerization, stabilizes receptor dimers, modulates circulating half-life, modulates release or bio-availability, facilitates purification, or improves or alters a particular route of administration. Similarly, the non- natural amino acid polypeptide can comprise chemical or enzyme cleavage sequences, protease cleavage sequences, reactive groups, antibody-binding domains (including but not limited to, FLAG or poly-His) or other affinity based sequences (including but not limited to, FLAG, poly-His, GST, etc.) or linked molecules (including but not limited to, biotin) that improve detection (including but not limited to, GFP), purification, transport thru tissues or cell membranes, prodrug release or activation, size reduction, or other traits of the polypeptide. [154] In some embodiments a payload or toxin moiety is employed in the anti PSMA antibody drug conjugates of the present invention disclosure. Examples of payloads can include in a non-limiting manner: inhibitors of DNA replication, inhibitors of DNA transcription, inhibitors of RNA translation, inhibitors of cell division, inhibitors of cell signaling, kinase inhibitors, tubulin polymerase inhibitors, tubulin depolymerizing agents, DNA cleavage agents, DNA binding agents, RNA polymerase inhibitors, auristatins, dolastatins, MMAF, MMAE, MMAD, duocarmycin analogs, pyrrolobenzodiazepine (PBD) analogs, tubulysin analogs, maytansine Attorney Docket No. AMB1013WOPCT1 analogs, amanitin analogs, cryptophycin analogs, epothilone analogs, calicheamicin analogs, doxorubicin analogs, camptothecin analogs. [155] In other embodiments of the present invention are provided drug payload linkers. Anti- tubulin inhibitors were chosen as the payload in combination with cleavable, short cleavable and non-cleavable linkers. In an exemplary manner, the payload linker combination of the present invention disclosure can include, but is not limited to, cleavable, non-cleavable, short cleavable payload linkers. Cleavable payload linkers can include cleavable dipeptides, (including, but not limited to Val-Cti, Val-Ala, Val-Lys and Ala- Ala), hydrazine linkage, disulfide linkage or pyrophosphate linkage. Example of payload linkers employed in anti- PSMA antibody drug conjugate of the present disclosure includes non-cleavable MMAE, non- cleavable MMAF, Val-Citruline-Acetyl MMAF, short Val-Citruline-Acetyl MMAF, or short Val-Citruline-Acetyl MMAE. Both MMAE and MMAF were utilized in the study. Non- limiting examples of dolastatin linker derivatives include the following: Non-cleavable MMAF (AS269) having the structure: .

having the structure: .
Short-cleavable or short Val-Citruline-Acetyl (Val-Cit) MMAF having the structure: Attorney Docket No. AMB1013WOPCT1 .
Cit) MMAE having the structure: .
be employed with the anti PSMA antibody or antibody drug conjugates of the present invention. Synthesis of such payload linkers are well known to the skilled artisan. See for example, Dubowchik et al., Bioconjugate Chem. 13: 855-869, (2002); Doronina et al., Nature Biotechnology 21(7): 778- 784, (2003); WO2012/166560; WO2013/185117, the entire contents of each of which are hereby incorporated by reference in their entirety. Table 1. Drug-linker Compounds – Single Payload Compound Structure
Attorney Docket No. AMB1013WOPCT1
Attorney Docket No. AMB1013WOPCT1
Attorney Docket No. AMB1013WOPCT1 17 18 Table 2. Drug-linker Compounds – Dual Payload Compound Structure 19 20
Attorney Docket No. AMB1013WOPCT1 21 22 23 [157] In some aspects of the disclosure, anti-PSMA antibody, variant or drug conjugate with increase serum half-life, water solubility, bioavailability, therapeutic half-life, or circulation time, or to modulate immunogenicity, or biological activity is desired. One method of achieving such desired features of the anti-PSMA composition disclosed herein, is by covalent attachment of the polymer polyethylene glycol, (PEG). To maximize the desired properties of Attorney Docket No. AMB1013WOPCT1 polyethylene glycol, the total molecular weight and hydration state of the polymer or polymers attached to the biologically active molecule must be sufficiently high to impart the advantageous characteristics typically associated with such polymer attachment, such as increased water solubility and circulating half-life, while not adversely impacting the bioactivity of the molecule to which the polyethylene glycol is attached. [158] Polyethylene glycol derivatives are frequently linked to biologically active molecules through reactive chemical functionalities, such as amino acid residues, the N-terminus, and/or carbohydrate moieties. WO99/67291 discloses a process for conjugating a protein with polyethylene glycol, wherein at least one amino acid residue on the protein is substituted with a synthetic amino acid and the protein is contacted with polyethylene glycol under conditions sufficient to achieve conjugation to the protein. [159] Proteins and other molecules often have a limited number of reactive sites available for polymer attachment. The sites most suitable for modification via polymer attachment may play a significant role in receptor binding, and such sites may be necessary for retention of the biological activity of the molecule therefore making them inappropriate for polymer attachment. As a result, indiscriminate attachment of polymer chains to such reactive sites on a biologically active molecule often leads to a significant reduction or even total loss of biological activity of the polymer-modified molecule, polyethylene glycol attachment can be directed to a particular position within a protein such that the polyethylene glycol moiety does not interfere with the function of that protein. One method of directing polyethylene glycol attachment is to introduce a synthetic amino acid into the protein sequence. The protein biosynthetic machinery of the prokaryote Escherichia coli (E. coli) can be altered in order to incorporate synthetic amino acids efficiently and with high fidelity into proteins in response to the amber codon, UAG. See, e.g., J. W. Chin et al., J. Amer. Chem. Soc.124: 9026-9027, 2002; J. W. Chin, & P. G. Schultz, ChemBioChem 3(11): 1135-1137, 2002; J. W. Chin, et al., PNAS Attorney Docket No. AMB1013WOPCT1 USA 99: 11020-11024, 2002; and, L. Wang, & P. G. Schultz, Chem. Comm., 1: 1-11, 2002. A similar method can be accomplished with the eukaryote, Saccharomyces cerevisiae (S. cerevisiae) (e.g., J. Chin et al., Science 301: 964-7, 2003). Using this method, a non-naturally encoded amino acid can be incorporated into anti-PSMA antibody, variant or drug conjugate of the present invention, providing an attachment site for polyethylene glycol. See, for example WO2010/011735 and WO2005/074650. Pharmaceutical Compositions [160] In other aspects, the present disclosure provides pharmaceutical compositions or formulations containing the anti-PSMA antibody, antibody fragments, variants or anti-PSMA ADCs of the present disclosure. Such pharmaceutical compositions can employ various pharmaceutically acceptable excipients, stabilizers, buffers, and other components for administration to animals. See, for example, Remington, The Science and Practice of Pharmacy, 19th ed., Gennaro, ed., Mack Publishing Co., Easton, PA, 1995. Identifying suitable composition or formulations for stability, administration to a subject, and activity varies with each compound as a number of components, (for example, purifying, stabilizing components), need to be considered. Suitable salts for inclusion into the composition or formulation can include, but not limited to, sodium chloride, potassium chloride or calcium chloride. Buffering and/or stabilizing agents such as sodium acetate can be used. Suitable buffers can include acetate buffer, phosphate-citrate buffer, phosphate buffer, citrate buffer, histidine buffer, L- histidine, L-arginine hydrochloride, bicarbonate buffer, succinate buffer, citrate buffer, and TRIS buffer, either alone or in combination. Surfactants can also be employed, including polysorbates (e.g., polysorbate 80), dodecyl sulfate (SDS), lecithin either alone or in combination. Attorney Docket No. AMB1013WOPCT1 [161] In some embodiments, a pharmaceutical composition can be a formulation, comprising an anti-PSMA ADC of the present disclosure and one or more pharmaceutically acceptable excipients, stabilizers or buffers. [162] In some embodiments, the anti-PSMA ADC comprises: a humanized anti-PSMA monoclonal antibody comprising two heavy chains and two light chains, wherein one non-natural amino acid para-acetyl-L-phenylalanine (pAF) is incorporated into each said heavy chain at position 114 according to Kabat numbering, wherein one drug-linker is conjugated to each said pAF via an oxime linkage, wherein each said drug- linker is amberstatin269 (AS269) having the following structure: .

[163] some a humanized anti-PSMA monoclonal antibody comprising two heavy chains and two light chains, wherein each heavy chain comprises a heavy chain variable region of SEQ ID NO: 1, wherein one non-natural amino acid para-acetyl-L-phenylalanine (pAF) is incorporated into each said heavy chain at position 114 according to Kabat numbering; wherein one drug- linker is conjugated to each said pAF via an oxime linkage, wherein each said drug-linker is amberstatin269 (AS269) having the following structure: .

[164] In some embodiments, the anti-PSMA ADC comprises: Attorney Docket No. AMB1013WOPCT1 a humanized anti-PSMA monoclonal antibody comprising two heavy chains and two light chains, wherein each heavy chain comprises a heavy chain variable region of SEQ ID NO: 1, wherein one non-natural amino acid para-acetyl-L-phenylalanine (pAF) is incorporated into each said heavy chain at position 114 according to Kabat numbering, wherein each light chain comprises a light chain variable region of SEQ ID NO: 2; wherein one drug-linker is conjugated to each said pAF via an oxime linkage, wherein each said drug-linker is amberstatin269 (AS269) having the following structure: .

[165] some a amino acid sequence of SEQ ID NO: 8. In some embodiments, the anti-PSMA ADC comprises a light chain having the amino acid sequence of SEQ ID NO: 9. In some embodiments, the anti-PSMA ADC comprises a heavy chain having the amino acid sequence of SEQ ID NO: 8 and a light chain having the amino acid sequence of SEQ ID NO: 9. [166] In some embodiments, the anti-PSMA ADC is ARX517. Thus, in some embodiments, the anti-PSMA ADC comprises: a humanized anti-PSMA monoclonal antibody comprising two heavy chains and two light chains, wherein each heavy chain has the amino acid sequence of SEQ ID NO: 8, wherein one non-natural amino acid para-acetyl-L-phenylalanine (pAF) is incorporated into each said heavy chain at position 114 according to Kabat numbering, and each light chain has the amino acid sequence of SEQ ID NO: 9; wherein one drug-linker is conjugated to each said pAF via an oxime linkage, wherein each said drug-linker is amberstatin269 (AS269) having the following structure: Attorney Docket No. AMB1013WOPCT1 . [167]

aqueous composition or in the form of a reconstituted liquid composition or as a powder. The composition or formulation can have a pH range from about 4.0 to about 7.0 or from about 4.5 to about 6.5 when the formulation is in a liquid form. However, the pH can be adjusted to provide acceptable stability and administration by the skilled medical practitioner. The composition can further be stored in a vial or cartridge, a pen delivery device, a syringe, intravenous administration tubing or an intravenous administration bag but is not limited to such. [168] Formulation selection criteria for an anti-PSMA ADCs of the present disclosure can include solubility, chemical stability and physical stability. [169] In some embodiments, the formulation can contain an anti-PSMA ADC of the present disclosure and a buffer, sugar or surfactant; or any combination thereof. [170] In some embodiments, the formulation can contain the anti-PSMA ADC of the present disclosure at a concentration within a range of about 5 mg/mL to about 25 mg/mL. In some embodiments, the formulation can contain the anti-PSMA ADC at a concentration of about 5 mg/mL, about 10 mg/mL, about 15 mg/mL, about 20 mg/mL or about 25 mg/mL. In some embodiments, the formulation contains the anti-PSMA ADC at a concentration of about 9 mg/mL, about 10 mg/mL or about 11 mg/mL. In some embodiments, the formulation contains the anti-PSMA ADC at a concentration of about 10 mg/mL. In some embodiments, the formulation contains the anti-PSMA ADC at a concentration of about 10 mg/mL ± 1 mg/mL. [171] In some embodiments, the anti-PSMA ADC formulation can contain a buffer. In some embodiments, the buffer is acetate buffer, succinate buffer, histidine buffer or phosphate buffer. Attorney Docket No. AMB1013WOPCT1 In some embodiments, the buffer is a histidine buffer. In some embodiments, the formulation can have a histidine buffer concentration within a range of about 10 mM to about 50 mM. In some embodiments, the formulation can have a histidine buffer concentration within a range of about 10 mM to about 30 mM. In some embodiments, the formulation can have a histidine buffer concentration within a range of about 15 mM to about 25 mM. In some embodiments, the formulation can have a histidine buffer concentration of about 5 mM, about 10 mM, about 15 mM, about 20 mM, about 25 mM, about 30 mM, about 35 mM, about 40 mM, about 45 mM or about 50 mM. In some embodiments, the formulation the formulation can have a histidine buffer concentration of about 5 mM, about 10 mM, about 15 mM, about 20 mM or about 25 mM. In some embodiments, the formulation the formulation can have a histidine buffer concentration of about 20 mM. In some embodiments, the histidine is L-histidine. In some embodiments, the histidine buffer comprises L-histidine and a salt of L-histidine, such as L- histidine hydrochloride, and can be provided in water. Various combinations of L-histidine and L-histidine hydrochloride concentrations can be used by the person of ordinary skill in the art to achieve a target pH of the histidine buffer. [172] In some embodiments, the anti-PSMA ADC formulation is characterized as having a pH value. In some embodiments, the formulation can have a pH within a range of about 5 to about 7.4. In some embodiments, the formulation can have a pH of at most about 7, at most about 6.5, at most about 6.2 or at most about 6. In some embodiments, the formulation can have a pH within a range of about 5.5 to about 6.5. In some embodiments, the formulation can have a pH within a range of about 5.4 to about 6.4. In some embodiments, the formulation can have a pH of about 5.0, about 5.1, about 5.2, about 5.3, about 5.4, about 5.5, about 5.6, about 5.7, about 5.8, about 5.9, about 6.0, about 6.1, about 6.3, about 6.4 or about 6.5. In some embodiments, the formulation pH is less than 6. In some embodiments, the formulation can have a pH within a range of about 5.5 to about 6.3. In some embodiments, the formulation can Attorney Docket No. AMB1013WOPCT1 have a pH within a range of about 5.6 to about 6.2. In some embodiments, the formulation can have a pH of about 5.6, about 5.7, about 5.8, about 5.9, about 6.0, about 6.1 or about 6.2. In some embodiments, the formulation can have a pH of about 5.9 ± 0.3. [173] In some embodiments, the anti-PSMA ADC formulation contains a water soluble polymer. In some embodiments, the water-soluble polymer is polyvinyl pyrrolidone, glycerol, trehalose, fructose, sucrose, glucose or mannose; or a combination thereof. In some embodiments, the water-soluble polymer is a sugar. In some embodiments, the sugar is trehalose, fructose, sucrose, glucose or mannose. In some embodiments, the formulation has a sugar concentration within a range of about 1% (w/v) to about 20% (w/v). In some embodiments, the formulation has a sugar concentration of at most about 15% (w/v). In some other embodiments, the formulation has a sugar concentration within a range of about 5% (w/v) and about 15% (w/v). In some embodiments, the formulation has a sugar concentration of at most about 10% (w/v). In some embodiments, the formulation has a sugar concentration of about 1%, about 2%, about 3%, about 4%, about 5%, about 6%, about 7%, about 8%, about 9%, about 10%, about 11%, about 12%, about 13%, about 14% or about 15% (w/v). In some embodiments, the sugar is sucrose. In some embodiments, the formulation has a sucrose concentration within a range of about 1% (w/v) to about 20% (w/v). In some embodiments, the formulation has a sucrose concentration of at most about 15% (w/v). In some embodiments, the formulation has a sucrose concentration of about 1%, about 2%, about 3%, about 4%, about 5%, about 6%, about 7%, about 8%, about 9%, about 10%, about 11%, about 12%, about 13%, about 14% or about 15% (w/v). In some other embodiments, the formulation has a sucrose concentration within a range of about 5% (w/v) and about 15% (w/v). In some embodiments, the formulation has a sucrose concentration of about 5%, about 6%, about 7%, about 8%, about 9%, about 10%, about 11%, about 12%, about 13%, about 14% or about 15% (w/v). In some embodiments, the formulation has a sucrose concentration of at most about 10% (w/v). In some Attorney Docket No. AMB1013WOPCT1 embodiments, the formulation has a sucrose concentration within a range of about 5% (w/v) and about 10% (w/v). In some embodiments, the formulation has a sucrose concentration of about 5%, about 6%, about 7%, about 8%, about 9% or about 10% (w/v). In some embodiments, the formulation has a sucrose concentration of about 9% (w/v). [174] In some embodiments, the anti-PSMA ADC formulation can contain a surfactant. In some embodiments, the surfactant is a polysorbate. In some embodiments, the surfactant is polysorbate 20. In some other embodiments, the surfactant is polysorbate 80. In some embodiments the formulation has a surfactant concentration of at most about 1% (w/v). In some embodiments, the formulation has a surfactant concentration of at most about 0.1% (w/v). In some embodiments, the formulation has a surfactant concentration within a range of about 0.01% (w/v) to about 0.1% (w/v). In some embodiments, the formulation has a surfactant concentration of about 0.01% (w/v), about 0.02% (w/v), about 0.03% (w/v), about 0.04% (w/v), about 0.05% (w/v), about 0.06% (w/v), about 0.07% (w/v), about 0.08% (w/v), about 0.09% (w/v) or about 0.10% (w/v). In some embodiments the formulation has a polysorbate 80 concentration of at most about 1% (w/v). In some embodiments, the formulation has a polysorbate 80 concentration of at most about 0.1% (w/v). In some embodiments, the formulation has a polysorbate 80 concentration within a range of about 0.01% (w/v) to about 0.1% (w/v). In some embodiments, the formulation has a polysorbate 80 concentration of about 0.01% (w/v), about 0.02% (w/v), about 0.03% (w/v), about 0.04% (w/v), about 0.05% (w/v), about 0.06% (w/v), about 0.07% (w/v), about 0.08% (w/v), about 0.09% (w/v) or about 0.10% (w/v). In some embodiments, the formulation has a polysorbate 80 concentration within a range of about 0.001% (w/v) to about 0.02% (w/v). In some embodiments, the formulation has a polysorbate 80 concentration of about 0.001% (w/v), about 0.002% (w/v), about 0.003% (w/v), about 0.004% (w/v), about 0.005% (w/v), about 0.006% (w/v), about 0.007% (w/v), about 0.008% (w/v), about 0.009% (w/v), about 0.01% (w/v), about 0.015% (w/v) or about 0.02% Attorney Docket No. AMB1013WOPCT1 (w/v). In some embodiments, the formulation has a polysorbate 80 concentration of about 0.01% (w/v). [175] In some embodiments, there is provided an aqueous formulation comprising an anti- PSMA ADC of the present disclosure, histidine buffer, sucrose and polysorbate 80. It is to be understood that “aqueous formulation” as used herein refers to a formulation containing water. In some embodiments, the aqueous formulation comprises the anti-PSMA ADC at a concentration within a range of about 5 mg/mL to about 25 mg/mL; histidine buffer at a concentration within a range of about 10 mM to about 50 mM; sucrose at a concentration within a range of about 1% (w/v) to about 20% (w/v); and polysorbate 80 at a concentration within a range of about 0.005% (w/v) to about 0.1% (w/v). In some embodiments, the aqueous formulation can have a pH within a range of about 5.6 to about 6.2. In some embodiments, the aqueous formulation can have a pH of about 5.6, about 5.7, about 5.8, about 5.9, about 6.0, about 6.1 or about 6.2. In some embodiments, the anti-PSMA ADC is ARX517. In some embodiments, the histidine is L-histidine. [176] In some more particular embodiments, there is provided an aqueous formulation comprising an anti-PSMA ADC at a concentration of about 10 mg/mL; histidine buffer at a concentration of about 20 mM; sucrose at a concentration of about 9% (w/v); and polysorbate 80 at a concentration of about 0.01% (w/v); wherein the anti-PSMA ADC is ARX517, and the aqueous formulation pH is about 5.9 ± 0.3. In some embodiments, the histidine is L-histidine. [177] In yet some more particular embodiments, there is provided an aqueous formulation consisting essentially of, or consisting of, an anti-PSMA ADC at a concentration of about 10 mg/mL; histidine at a concentration of about 20 mM; sucrose at a concentration of about 9% (w/v); polysorbate 80 at a concentration of about 0.01% (w/v); and water; wherein the anti- PSMA ADC is ARX517, and the aqueous formulation pH is about 5.9 ± 0.3. In some embodiments, the histidine is L-histidine. Attorney Docket No. AMB1013WOPCT1 [178] In some embodiments, a pharmaceutical composition such as a liquid formulation containing the anti-PSMA ADC of the present disclosure does not contain preservatives. [179] In some embodiments, the formulation is a liquid formulation. In some embodiments, liquid formulation can be stored at room temperature. In some other embodiments, the liquid formulation can be stored frozen. In some embodiments, the liquid formulation can be stored frozen at -20 °C. In yet some other embodiments, the liquid formulation can be stored refrigerated. In some embodiments, the liquid formulation can be stored refrigerated at 5 °C ± 3 °C. The formulation can be contained in glass vials. For example, glass vials can contain 5.0 mL ARX517 at 10 mg/mL in frozen solution. In another example, glass vials can contain 5.0 mL ARX517 at 10 mg/mL in refrigerated solution. [180] The composition can be stored in a vial or cartridge, a pen delivery device, a syringe, intravenous administration tubing or an intravenous administration bag but is not limited to such. A composition can be diluted into a diluent, such as water, pH-adjusted water, water for injections and the like. A composition can be diluted into an IV bag, for example, in 0.9% saline for injection. For example, a liquid formulation containing an anti-PSMA ADC at a concentration of about 10 mg/mL; histidine at a concentration of about 20 mM; sucrose at a concentration of about 9% (w/v); polysorbate 80 at a concentration of about 0.01% (w/v); and water; wherein the anti-PSMA ADC is ARX517, and the aqueous formulation pH is about 5.9 ± 0.3, can be added to an infusion bag containing 0.9% saline for injection. [181] Biotherapeutic proteins can contain charged variants, as understood by a person of ordinary skill in the art. Accordingly, a composition comprising an ADC of the present disclosure, such as ARX517, can comprise one or more charged variants, as further discussed herein. In some embodiments, the composition is a liquid formulation of the present disclosure. In some embodiments, the composition is ARX517 Solution for Intravenous Infusion. Attorney Docket No. AMB1013WOPCT1 [182] The analysis of charged variants is a regulatory requirement for bio-therapeutic proteins. Methods of analyzing biotherapeutic proteins, such as mAbs or ADCs, for charge variants are known in the art, and include ion exchange chromatography, such as cationic exchange chromatography (CEX) or anionic exchange chromatography, which are typically performed with salt gradients. CEX is a type of ion exchange chromatography that separates molecules, such as proteins, based upon their net surface charge. [183] As is further understood by a person of ordinary skill in the art, the isoelectric point (pI) of a protein is the pH value at which the protein carries no net electrical charge. The net surface charge of a protein changes with pH, as follows: i) a protein will have no net charge under conditions wherein the pH (e.g., the pH of a solution containing the protein) is the same as the pI of the protein; ii) a protein will have a net positive charge under conditions wherein the pH (e.g., the pH of a solution containing the protein) is less than the protein pI, and iii) a protein will carry a net negative charge when the pH (e.g., the pH of a solution containing the protein) is greater than the protein pI. By using a negatively charged ion exchange stationary phase, such as a resin, that has affinity for molecules having net positive surface charges, CEX can be used to separate a mixture of proteins having different pI values. [184] Because the pI of a protein of interest can be predicted based on the protein’s primary amino acid sequence, or can be determined by methods known by persons of ordinary skill in the art, such as isoelectric focusing, a suitable buffer and buffer pH can be chosen to ensure a known net charge for a protein of interest. A negatively charged cation exchange resin can be chosen when the protein of interest carries a net positive charge at the working buffer pH, such that the negatively charged resin will attract the positive surface charges of proteins in a test sample. Proteins in the sample with different pI values will have varying degrees of charge at a given pH and will thus have different affinities for the positively charged surface groups on the particles of the anion exchange media. Consequently, different proteins will bind to the Attorney Docket No. AMB1013WOPCT1 resin with different strengths, facilitating their separation. In a non-limiting example, if a cation exchange resin is used at a pH of 7.5, in general, all proteins that have a pI >7.5 will carry a net positive charge and will bind the negatively charged resin. A salt gradient can then be used to separate the protein of interest from other bound proteins. Proteins will be eluted in an order that depends on their net surface charge. Proteins with pI values closer to 7.5 will elute at a lower ionic strength (e.g., a lower salt concentration), while proteins with very high pI values will elute at a higher ionic strength (e.g., a higher salt concentration). [185] Compositions comprising monoclonal antibodies (mAbs) or ADCs can contain mAb or ADC variants thereof, respectively, that result from post-translational modifications and/or degradation events. Typical post-translational modifications can include cyclization of the N- terminal glutamine (Gln) to pyroglutamic acid (pyroGlu); removal of a heavy chain C-terminal lysine (Lys); and/or glycosylation of a conserved asparagine (Asn) in the CH2 domain with neutral oligosaccharides. Such variants can be observed when compositions containing the mAbs or ADCs are analyzed using CEX. These variants are generally referred to as acidic or basic species as compared with the main species. When CEX chromatography is used, acidic species are the variants that elute earlier than the main species, and basic species are the variants that elute later than the main species. The corresponding peaks in the CEX chromatogram can be referred to as the acidic peak(s), which corresponds to the acidic species; the main peak, which corresponds to the main species; and the basic peak(s), which corresponds to the elution of the basic species. (See, e.g., Du, Y. et al., Chromatographic analysis of the acidic and basic species of recombinant monoclonal antibodies. MAbs. 2012 Sep-Oct;4(5):578-85. doi: 10.4161/mabs.21328. Epub 2012 Jul 23. PMID: 22820257; PMCID: PMC3499298.) [186] In some aspects, the present disclosure provides a composition comprising an anti- PSMA ADC, wherein the composition comprises a main species of the anti-PSMA ADC. In some embodiments, the anti-PSMA ADC main species has a pI of about 8.3. For example, Attorney Docket No. AMB1013WOPCT1 ARX517 was determined to have a pI of 8.3 via Imaged Capillary Isoelectric Focusing (iCIEF; see, e.g., Example 18). In some embodiments, the composition comprises the anti-PSMA ADC main species and one or more variants thereof. In some embodiments, the one or more variants comprise an anti-PSMA ADC acidic species, anti-PSMA ADC basic species, or a combination thereof. In some embodiments, the anti-PSMA ADC acidic species has a pI of about 8.1, and the anti-PSMA ADC basic species has a pI of about 8.4. For example, ARX517 acidic and basic species were determined to have a pI of 8.1 and 8.4 respectively via iCIEF. In some embodiments, the anti-PSMA ADC is ARX517. In some embodiments, the composition is ARX517 Solution for Intravenous Infusion, as disclosed herein. [187] In some embodiments, an anti-PSMA ADC of the present disclosure, or a composition comprising an anti-PSMA ADC of the present disclosure, is characterized by the ADC charge variant profile. In some embodiments, the composition is characterized as having, or the composition comprises, an anti-PSMA ADC main species and one or more variants thereof, wherein the one or more variants comprises an anti-PSMA ADC acidic species and an anti- PSMA ADC basic species. In some embodiments, the anti-PSMA ADC main species has a pI of about 8.3, the anti-PSMA ADC acidic species has a pI of about 8.1 and the anti-PSMA ADC basic species has a pI of about 8.4, respectively. In some embodiments, the anti-PSMA ADC is ARX517. In some embodiments, the composition is ARX517 Solution for Intravenous Infusion, as disclosed herein. [188] In some embodiments, the composition is characterized as having, or the composition comprises, an anti-PSMA ADC main species, wherein the anti-PSMA ADC main species is present in an amount of at least about 35%. In some further embodiments, the composition is characterized as having, or the composition comprises, an anti-PSMA ADC main species, an anti-PSMA ADC acidic species and an anti-PSMA ADC basic species, wherein the anti-PSMA ADC main species is present in an amount of at least about 35%, the anti-PSMA ADC acidic Attorney Docket No. AMB1013WOPCT1 species is present in an amount of at most about 45%, and the anti-PSMA ADC basic species is present in an amount of at most about 35%; wherein the sum of the percentage of the main species, acidic species and basic species is 100%. In some embodiments, the composition comprises the anti-PSMA ADC main species, the anti-PSMA ADC acidic species and the anti- PSMA ADC basic species, wherein the anti-PSMA ADC main species is present in an amount of about 35% to about 70%, the anti-PSMA ADC acidic species is present in an amount of about 20% to about 45%, and the anti-PSMA ADC basic species is present in an amount of about 5% to about 35%; wherein the sum of the percentage of the main species, acidic species and basic species is 100%. In some embodiments, the composition comprises the anti-PSMA ADC main species, the anti-PSMA ADC acidic species and the anti-PSMA ADC basic species, wherein the anti-PSMA ADC main species is present in an amount of about 35% to about 60%, the anti-PSMA ADC acidic species is present in an amount of about 20% to about 45%, and the anti-PSMA ADC basic species is present in an amount of about 5% to about 30%; wherein the sum of the percentage of the main species, acidic species and basic species is 100%. In some embodiments, the composition comprises the anti-PSMA ADC main species, the anti- PSMA ADC acidic species and the anti-PSMA ADC basic species, wherein the anti-PSMA ADC main species is present in an amount of about 35% to about 55%, the anti-PSMA ADC acidic species is present in an amount of about 25% to about 45%, and the anti-PSMA ADC basic species is present in an amount of about 5% to about 25%; wherein the sum of the percentage of the main species, acidic species and basic species is 100%. In some other embodiments, the composition is characterized as having, or the composition comprises, the anti-PSMA ADC main species, wherein the anti-PSMA ADC main species is present in an amount of at least about 40%. In some embodiments, the composition is characterized as having, or the composition comprises, the anti-PSMA ADC main species, the anti-PSMA ADC acidic species and the anti-PSMA ADC basic species, wherein the anti-PSMA ADC main Attorney Docket No. AMB1013WOPCT1 species is present in an amount of at least about 40%, the anti-PSMA ADC acidic species is present in an amount of at most about 40%, and the anti-PSMA ADC basic species is present in an amount of at most about 30%; wherein the sum of the percentage of the main species, acidic species and basic species is 100%. In some other embodiments, the composition comprises the anti-PSMA ADC main species, the anti-PSMA ADC acidic species and the anti- PSMA ADC basic species, wherein the anti-PSMA ADC main species is present in an amount of about 40% to about 70%, the anti-PSMA ADC acidic species is present in an amount of about 20% to about 40%, and the anti-PSMA ADC basic species is present in an amount of about 5% to about 30%; wherein the sum of the percentage of the main species, acidic species and basic species is 100%. In yet some other embodiments, the composition comprises the anti- PSMA ADC main species, the anti-PSMA ADC acidic species and the anti-PSMA ADC basic species, wherein the anti-PSMA ADC main species is present in an amount of about 40% to about 60%, the anti-PSMA ADC acidic species is present in an amount of about 25% to about 40%, and the anti-PSMA ADC basic species is present in an amount of about 5% to about 20%; wherein the sum of the percentage of the main species, acidic species and basic species is 100%. In some more particular embodiments, the composition comprises the anti-PSMA ADC main species, the anti-PSMA ADC acidic species and the anti-PSMA ADC basic species, wherein the anti-PSMA ADC main species is present in an amount of about 45% to about 60%, the anti-PSMA ADC acidic species is present in an amount of about 25% to about 35%, and the anti-PSMA ADC basic species is present in an amount of about 10% to about 20%; wherein the sum of the percentage of the main species, acidic species and basic species is 100%. In some other embodiments, the composition comprises the anti-PSMA ADC main species, the anti-PSMA ADC acidic species and the anti-PSMA ADC basic species, wherein the anti- PSMA ADC main species is present in an amount of at least about 50%, the anti-PSMA ADC acidic species is present in an amount of at most about 35%, and the anti-PSMA ADC basic Attorney Docket No. AMB1013WOPCT1 species is present in an amount of at most about 20%; wherein the sum of the percentage of the main species, acidic species and basic species is 100%. In yet some other embodiments, the composition comprises the anti-PSMA ADC main species, the anti-PSMA ADC acidic species and the anti-PSMA ADC basic species, wherein the anti-PSMA ADC main species is present in an amount of about 50% to about 55%, the anti-PSMA ADC acidic species is present in an amount of about 30% to about 35%, and the anti-PSMA ADC basic species is present in an amount of about 12% to about 17%; wherein the sum of the percentage of the main species, acidic species and basic species is 100%. In some embodiments, the percentage of the main species, the acidic species and the basic species is a UV area % at 214 nm based on integration of the corresponding main species elution peak, acidic species elution peaks and basic species elution peaks in a cationic ion exchange chromatogram. In some embodiments, the acidic species elution peaks comprise, or consist of, all of the peaks in the cationic exchange chromatogram that elute before the main peak, and the basic species elution peaks comprise, or consist of, all of the peaks in the cationic exchange chromatogram that elute after the main peak. In some embodiments, the cation exchange chromatogram is obtained from a CEX method as disclosed at Example 17. In some embodiments, a charge variant profile of an anti- PSMA ADC, or a composition comprising an anti-PSMA ADC, is determined by a CEX method as disclosed at Example 17. In some embodiments, the anti-PSMA ADC is ARX517. In some embodiments, the composition is ARX517 Solution for Intravenous Infusion, as disclosed at Example 14. [189] A pharmaceutical composition of the invention can be administered as a single dose or followed by one or more subsequent dose(s) minutes, days, or weeks after the first dose. Further administrations may be contemplated as needed to treat, reduce or prevent a cancer, condition, disorder or disease, including prostate cancer. Therapeutic Uses of Anti-PSMA Antibodies and ADCs Attorney Docket No. AMB1013WOPCT1 [190] The present disclosure provides anti-PSMA ADCs for use in inhibiting, preventing or treating PSMA related diseases or cancers, and to methods of treating subjects having, or at risk of having, a PSMA related disease or cancer. The uses and methods can include dosing regimens for inhibiting, preventing or treating PSMA related diseases or cancers. In some embodiments, the use or method of treatment comprises administering an anti-PSMA ADC of the present disclosure to a subject in need thereof. The use or method of treatment can further include treating the subject with additional therapy, as further disclosed herein. [191] The anti-PSMA antibodies or anti-PSMA ADCs of the disclosure are useful for treating a wide range of diseases, disorders, conditions or cancers. The present disclosure includes compositions for use in, and methods of treating, a mammal (e.g., a human) that is at risk for, is having, and/or has had a disease or condition, such as a cancer, that is responsive to PSMA overexpression, amplification, mutation and/or targeted therapies. In some embodiments, a composition comprising an effective amount of an ADC of the present disclosure can be used for reducing or inhibiting tumor growth or progression in an antigen-expressing cancer or cancer cell, such as a prostate-specific antigen-expressing cancer or cancer cell. [192] Compositions disclosed herein can be used to modulate an immune response. Modulation of an immune response can comprise stimulating, activating, increasing, enhancing, or up-regulating an immune response. Modulation of an immune response can comprise suppressing, inhibiting, preventing, reducing, or downregulating an immune response. [193] Administration of ADCs can result in a short-term effect, i.e., an immediate beneficial effect on several clinical parameters observed, and this can occur 12 or 24 hours from the time of administration, and/or can result in a long-term effect, such as a beneficial slowing of progression of tumor growth or reduction in tumor size. The ADCs of the present disclosure can be administered by any means known to those skilled in the art, and can beneficially be Attorney Docket No. AMB1013WOPCT1 administered via infusion, e.g., by arterial, intraperitoneal or intravenous injection and/or infusion in a dosage which is sufficient to obtain the desired pharmacological effect. In some embodiments, the ADC, or the composition or formulation comprising the ADC, is administered orally, intradermally, intratumorally, intravenously or subcutaneously. In some embodiments, the ADC, or the composition or formulation comprising the ADC, is administered intravenously. [194] The present disclosure provides a method of treating a disease or condition, such as a tumor or cancer, by administering to a patient (e.g., a human subject) in need thereof a therapeutically effective amount of an anti-PSMA ADC of the present disclosure, or a pharmaceutical composition comprising an anti-PSMA ADC of the present disclosure. [195] In some embodiments, an anti-PSMA ADC of the present disclosure can be used to inhibit the growth of cancer cells. In some embodiments, the cancer cells are prostate cancer cells, bladder cancer cells, pancreatic cancer cells, liver cancer cells, lung cancer cells, kidney cancer cells, sarcoma cells, breast cancer cells, brain cancer cells, neuroendocrine carcinoma cells, colon cancer cells, testicular cancer cells or melanoma cells. In some more particular embodiments, the cancer cells are prostate cancer cells. [196] In some embodiments, an anti-PSMA ADC of the present disclosure can be used to treat cancer in a subject, wherein the cancer is prostate cancer. In some embodiments, the cancer is metastatic castration resistant prostate cancer. [197] In some other embodiments, an anti-PSMA ADC of the present disclosure can be used to treat cancer in a subject, wherein the cancer is a non-prostate cancer. In some embodiments, the non-prostate cancer is bladder cancer, pancreatic cancer, liver cancer, lung cancer, kidney cancer, sarcoma, breast cancer, brain cancer, neuroendocrine carcinoma, colon cancer, testicular cancer or melanoma. Attorney Docket No. AMB1013WOPCT1 [198] A dose of an anti-PSMA ADC of the present disclosure for administration to a subject can be sufficient to cause a beneficial response in the subject over time. The anti-PSMA ADC dosage can be given as a bolus injection and/or as an infusion for a clinically necessary period of time, e.g., for a period ranging from a few minutes to several hours, e.g., up to 24 hours. In some embodiments, the anti-PSMA ADC is administered by intravenous infusion from about 30 minutes to about 120 minutes. In some embodiments, the anti-PSMA ADC is administered by intravenous infusion for about 90 minutes. In some embodiments, the anti-PSMA ADC is administered by intravenous infusion for about 60 minutes. If necessary, the anti-PSMA ADC administration can be repeated one or several times. In some embodiments, the infusion time of the anti-PSMA ADC is reduced over subsequent administrations. In some embodiments, the subject is administered a first anti-PSMA ADC dose by intravenous infusion for about 90 minutes and subsequently administered a second anti-PSMA ADC dose by intravenous infusion for about 60 minutes. The anti-PSMA ADC dosage can be an effective amount or dose. In some embodiments, the dose is administered at more than once in an hour interval. In some embodiments, the dose is administered at more than once in a day interval. In another embodiment, the dose is administered at more than once in a week intervals. In another embodiment, the dose is administered at 1-week intervals. In another embodiment, the dose is administered at 2-week intervals. In another embodiment, the dose is administered at 3-week intervals. In another embodiment, the dose is administered at 4-week intervals. In another embodiment, the dose is administered at 5-week intervals. In another embodiment, the dose is administered at 6-week intervals. [199] In some embodiments, an effective amount of anti-PSMA ADC is sufficient to 1) delay or inhibit progression of cancer in a subject, 2) increase survival of a subject as compared to the median survival of subjects who have not been treated with an anti-PSMA ADC and who have metastatic castration-resistant prostate cancer that has progressed after Attorney Docket No. AMB1013WOPCT1 prior taxane therapy, 3) decrease a circulating level of circulating tumor cells (CTCs) in a subject compared to a baseline level, 4) decrease or stabilize a serum level of prostate specific antigen (PSA) in a subject compared to a baseline level of PSA. In another embodiment, treatment of a subject with an anti-PSMA ADC of the present disclosure results in an improvement in the subject's quality of life as compared to the quality of life of the subject prior to treatment with the anti-PSMA ADC. When assessing any one or more of the foregoing changes, a first time point can, in some embodiments, be at baseline, or before the administration of any PSMA ADC, while the second time point is any time after the administration of an anti-PSMA ADC. In other embodiments, the first time point occurs after the administration of an anti-PSMA ADC, and the second time point occurs any time subsequent to the first time point. In some embodiments, the first time point occurs before a particular dose of anti-PSMA ADC, and the second time point occurs subsequent to that particular dose of anti-PSMA ADC. [200] In some embodiments, treatment of a subject with an anti-PSMA ADC of the present disclosure results in a delay or inhibition of progression of the cancer in the subject. As used herein, "delay or inhibition of progression of the cancer" is intended to refer to any slowing or halting of the progression of the cancer in the subject. A slowing or halting of the progression of the cancer includes a reduction or stabilization in the number of cancer cells, the number of tumors, and/or the number of metastases in a subject. A slowing or halting is also intended to include a reduction or stabilization in the size (e.g., length or volume) of tumors and/or size of metastases in a subject. In further embodiments, a delay or inhibition of progression of the cancer is demonstrated by radiographic image changes in tumor burden compared to a baseline radiographic image in the subject prior to anti- PSMA ADC treatment. Methods for assessing radiographic changes, include, for example, computerized axial tomography (CT) scan and magnetic resonance imaging (MRI). Attorney Docket No. AMB1013WOPCT1 Methods for assessing the progression of cancer in a subject will be apparent to one of ordinary skill in the art. [201] In another embodiment, treatment with an anti-PSMA ADC results in increased survival of the subject, wherein the survival is increased in comparison to the median survival time of subjects with anti-PSMA-expressing, taxane-resistant cancer not treated with an anti-PSMA ADC. In some embodiments, survival in the subject is increased by four weeks. In another embodiment, survival in the subject is increased by six weeks. In another embodiment, survival in the subject is increased by two months. In another embodiment, survival in the subject is increased by four months. In another embodiment, survival in the subject is increased by six months. In another embodiment, survival in the subject is increased by eight months. In another embodiment, survival in the subject is increased by ten months. In another embodiment, survival in the subject is increased by twelve months. In another embodiment, survival in the subject is increased by fourteen months. In some embodiments, the survival of the a subject is increased by 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49, 50, 51, 52, 56, 58 or 60 weeks or more as compared to the median survival of subjects who have not been treated with an anti-PSMA ADC and has metastatic castration-resistant prostate cancer that has progressed after prior taxane therapy. In some other embodiments, the survival is increased by 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49, 50, 51, 52, 56, 58 or 60 weeks or more as compared to the expected survival time for the subject prior to treatment with the anti-PSMA ADC. In other embodiments, the survival of the subject is increased by 6, 10, 12, 14, 16, 18, 20, 22, 24, 26, 28, 30, 32, 34, 36, 38, 40, 42, 44, 46, 48, 50, 52, 54, 56, 58 or 60 months or more according to either of the foregoing comparisons. Attorney Docket No. AMB1013WOPCT1 [202] In some embodiments, the subject treated with an anti-PSMA ADC has metastatic castration-resistant prostate cancer that has progressed after prior taxane therapy, and the increase in survival of the subject is as compared to the median survival of subjects having metastatic castration-resistant prostate cancer that has progressed after prior taxane therapy and not treated with the ADC. In another embodiment, the subject treated with an anti- PSMA ADC is one that has metastatic castration-resistant prostate cancer that has progressed after prior taxane therapy, and the increase in survival of the subject is as compared to the expected survival time for the subject prior to treatment with the ADC. [203] In some embodiments, treatment with an anti-PSMA ADC can result in a decrease in a circulating level of circulating tumor cells (CTCs) compared to a baseline level. In further embodiments, treatment with an ADC can result in a decrease or stabilization (no significant increase or decrease) in a serum level of prostate-specific antigen (PSA) compared to a baseline level of PSA. Methods for assessing the circulating level of CTCs or the serum level of PSA are well known to those of ordinary skill in the art. [204] In some embodiments, treatment of a subject with an anti-PSMA ADC of the present disclosure results in a reduction of tumor volume. In some embodiments, the tumor volume is reduced by at least about 1%, 2%, 3%, 4%, 5%, 6%, 7%, 8%, 9%, 10%, 11%, 12%, 13%, 14%, 15%, 16%, 17%, 18%, 19%, 20%, 21%, 22%, 23%, 24%, 25%, 26%, 27%, 28%, 29%, 30%, 31%, 32%, 33%, 34%, 35%, 36%, 37%, 38%, 39%, 40%, 41%, 42%, 43%, 44%, 45%, 46%, 47%, 48%, 49%, 50%, 51%, 52%, 53%, 54%, 55%, 56%, 57%, 58%, 59%, 60%, 61%, 62%, 63%, 64%, 65%, 66%, 67%, 68%, 69%, 70%, 71%, 72%, 73%, 74%, 75%, 76%, 77%, 78%, 79%, 80%, 81%, 82%, 83%, 84%, 85%, 86%, 87%, 88%, 89%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97% or 98%. In another embodiment, the tumor volume is reduced by at least about 99%. Attorney Docket No. AMB1013WOPCT1 [205] In a further embodiment, administration of an anti-PSMA ADC to a subject results in a gain in body weight in the subject (as compared to the weight of the subject prior to administration of the PSMA ADC). The gain in body weight can result from a single administration of the PSMA ADC, or from a course of treatment with the anti-PSMA ADC (i.e., more than one administration). In some embodiments, the body weight gain is an increase in body weight of about 5% to about 10%. In another embodiment, the body weight gain is an increase in body weight of about 10% to about 20%. In still another embodiment, the body weight gain is an increase in body weight of about 15%. In still a further embodiment, the body weight gain is an increase in body weight of about 25%. In yet another embodiment, the body weight gain is an increase in body weight of about 30%, about 35% or more. [206] In some embodiments, the dose of anti-PSMA ADC administered to a subject can be chosen in accordance with different parameters, in particular, in accordance with the mode of administration used and the state of the subject. Other factors include the desired period of treatment. In the event that a response in a subject is insufficient at the initial doses applied, higher doses (or effectively higher doses by a different, more localized delivery route) can be employed to the extent that patient tolerance permits. [207] In some embodiments, the anti-PSMA ADC comprises: a humanized anti-PSMA monoclonal antibody comprising two heavy chains and two light chains, wherein one non-natural amino acid para-acetyl-L-phenylalanine (pAF) is incorporated into each said heavy chain at position 114 according to Kabat numbering, wherein one drug-linker is conjugated to each said pAF via an oxime linkage, wherein each said drug- linker is amberstatin269 (AS269) having the following structure: . Attorney Docket No. AMB1013WOPCT1 [208] In some embodiments, each heavy chain comprises a heavy chain variable region of SEQ ID NO: 1. In some embodiments, each light chain comprises a light chain variable region of SEQ ID NO: 2. In some embodiments, each heavy chain comprises a heavy chain variable region of SEQ ID NO: 1 and each light chain comprises a light chain variable region of SEQ ID NO: 2 [209] In some embodiments, the anti-PSMA ADC comprises: a humanized anti-PSMA monoclonal antibody comprising two heavy chains and two light chains, wherein each heavy chain comprises a heavy chain variable region of SEQ ID NO: 1, wherein one non-natural amino acid para-acetyl-L-phenylalanine (pAF) is incorporated into each said heavy chain at position 114 according to Kabat numbering, wherein each light chain comprises a light chain variable region of SEQ ID NO: 2; wherein one drug-linker is conjugated to each said pAF via an oxime linkage, wherein each said drug-linker is amberstatin269 (AS269) having the following structure: .
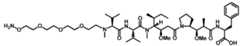
ID NO: 8. In some embodiments, each light chain has the amino acid sequence of SEQ ID NO: 9. In some embodiments, each heavy chain has the amino acid sequence of SEQ ID NO: 8, comprising the pAF at Kabat amino acid position 114 (i.e., amino acid position 116 of SEQ ID NO:8), and each light chain has the amino acid sequence of SEQ ID NO: 9 [211] In some embodiments, the anti-PSMA ADC is ARX517. Thus, in some embodiments, the anti-PSMA ADC comprises: a humanized anti-PSMA monoclonal antibody comprising two heavy chains and two light chains, wherein each heavy chain has the amino acid sequence of SEQ ID NO: 8, wherein Attorney Docket No. AMB1013WOPCT1 one non-natural amino acid para-acetyl-L-phenylalanine (pAF) is incorporated into each said heavy chain at position 114 according to Kabat numbering, and each light chain has the amino acid sequence of SEQ ID NO: 9; wherein one drug-linker is conjugated to each said pAF via an oxime linkage, wherein each said drug-linker is amberstatin269 (AS269) having the following structure: . [212]
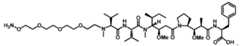
present disclosure for administration to a human subject is a dose within a range of about 0.32 mg/kg to about 10 mg/kg of the body weight of the human subject. In still other embodiments, the effective dose is a dose within a range of about 0.32 mg/kg to about 5 mg/kg, about 1.4 mg/kg to about 5 mg/kg, about 1.4 mg/kg to about 3.4 mg/kg, about 2 mg/kg to about 10 mg/kg, about 2 mg/kg to about 5 mg/kg, about 2 mg/kg to about 3.4 mg/kg of the body weight of the human subject. In further embodiments, the dose is about 0.32 mg/kg, about 0.5 mg/kg, about 1 mg/kg, about 2 mg/kg, about 3 mg/kg, about 4 mg/kg, about 5 mg/kg, about 7 mg/kg, or about 10 mg/kg of the body weight of the human subject. In further embodiments, the dose is about 0.32 mg/kg, about 0.64 mg/kg. about 1.07 mg/kg, about 1.4 mg/kg, about 1.7 mg/kg, about 2.0 mg/kg, about 2.4 mg/kg, about 2.88 mg/kg, about 3.4 mg/kg, or about 4.5mg/kg of the body weight of the human subject. In some embodiments, the dose is greater than 2 mg/kg of the body weight of the human subject. In some embodiments, the dose is about 2.1 mg/kg, about 2.2 mg/kg, about 2.3 mg/kg, about 2.4 mg/kg, about 2.5 mg/kg, about 2.6 mg/kg, about 2.7 mg/kg, about 2.8 mg/kg, about 2.9 mg/kg, or about 3.0 mg/kg of the body weight of the human subject. In some embodiments, the dose is greater than 3 mg/kg of the body weight of the human subject. In some embodiments, the dose is about 3.1 mg/kg, about 3.2 mg/kg, about Attorney Docket No. AMB1013WOPCT1 3.3 mg/kg or about 3.4 mg/kg of the body weight of the human subject. In some embodiments, the dose is about 3.5 mg/kg, about 3.6 mg/kg, about 3.7 mg/kg, about 3.8 mg/kg, about 3.9 mg/kg, about 4.0 mg/kg, about 4.1 mg/kg, about 4.2 mg/kg, about 4.3 mg/kg, about 4.4 mg/kg or about 4.5 mg/kg of the body weight of the human subject. In some embodiments, the dose is administered to the human subject on a dosing schedule of about once per week. In some embodiments, the dose is administered to the human subject on a dosing schedule of about once every two weeks. In some embodiments, the dose is administered to the human subject on a dosing schedule of about once every three weeks. In some embodiments, the dose is administered to the human subject on a dosing schedule of about once every four weeks. In some embodiments, the anti-PSMA ADC is ARX517. [213] In some embodiments, an effective amount of anti-PSMA ADC of the present disclosure for administration to a human subject is a dose within a range of about 0.05 mg/kg to about 10 mg/kg of the body weight of the human subject, or any value in between. In some other embodiments, the effective amount of the anti-PSMA ADC for administration to a human subject is a dose is within a range of about 2 mg/kg to about 10 mg/kg of the body weight of the human subject, or any value in between. In some embodiments, the effective amount of the anti-PSMA ADC for administration to a human subject is a dose is within a range of about 1.4 mg/kg to about 5 mg/kg of the body weight of the human subject, or any value in between. In some embodiments, the effective amount of the anti-PSMA ADC for administration to a human subject is a dose is within a range of about 1.4 mg/kg to about 5 mg/kg of the body weight of the human subject, or any value in between. In some embodiments, the effective amount of the anti-PSMA ADC for administration to a human subject is a dose is within a range of about 1.4 mg/kg to about 3.4 mg/kg of the body weight of the human subject, or any value in between. In some embodiments, the effective amount of the anti-PSMA ADC for administration to a human subject is a dose is within a range of about 2.0 mg/kg to about 5 Attorney Docket No. AMB1013WOPCT1 mg/kg of the body weight of the human subject, or any value in between. In some embodiments, the effective amount of the anti-PSMA ADC for administration to a human subject is a dose within a range of 2.1 mg/kg and about 4.5 mg/kg of the body weight of the human subject, or any value in between. In some embodiments, the effective amount of the anti-PSMA ADC for administration to a human subject is a dose within a range of 3.0 mg/kg to about 4.5 mg/kg of the body weight of the human subject, or any value in between. In some embodiments, the effective amount of the anti-PSMA ADC is a dose of about 1.0 mg/kg, about 1.2 mg/kg, about 1.3 mg/kg, about 1.4 mg/kg, about 1.5 mg/kg, about 1.6 mg/kg, about 1.7 mg/kg, about 1.8 mg/kg, about 1.9 mg/kg, about 2.0 mg/kg, about 2.1 mg/kg, about 2.2 mg/kg, about 2.3 mg/kg, about 2.4 mg/kg, about 2.5 mg/kg, about 2.6 mg/kg, about 2.7 mg/kg, about 2.8 mg/kg, about 2.9 mg/kg, about 3.0 mg/kg, about 3.1 mg/kg, about 3.2 mg/kg, about 3.3 mg/kg, about 3.4 mg/kg, about 3.5 mg/kg, about 3.6 mg/kg, about 3.7 mg/kg, about 3.8 mg/kg, about 3.9 mg/kg, about 4 mg/kg, about 4.1 mg/kg, about 4.2 mg/kg, about 4.3 mg/kg, about 4.4 mg/kg, about 4.5 mg/kg, about 4.6 mg/kg, about 4.7 mg/kg, about 4.8 mg/kg, about 4.9 mg/kg or about 5 mg/kg of the body weight of the human subject. In some embodiments, the dose is administered to the human subject on a dosing schedule of about once per week. In some embodiments, the dose is administered to the human subject on a dosing schedule of about once every two weeks. In some embodiments, the dose is administered to the human subject on a dosing schedule of about once every three weeks. In some embodiments, the dose is administered to the human subject on a dosing schedule of about once every four weeks. In some embodiments, the anti-PSMA ADC is ARX517. [214] In some embodiments, an effective amount of anti-PSMA ADC of the present disclosure for administration to a human subject is a dose that is greater than 2.0 mg/kg and at most about 10.0 mg/kg of the body weight of the human subject. In some embodiments, the effective amount is a dose of at least about 2.4 mg/kg of the body weight of the human subject. Attorney Docket No. AMB1013WOPCT1 In some embodiments, the effective amount is a dose of at least about 2.4 mg/kg and at most about 5.0 mg/kg of the body weight of the human subject. In some embodiments, the effective amount is a dose of at least about 2.4 mg/kg and at most about 4.0 mg/kg of the body weight of the human subject. In some embodiments, the effective amount is a dose of about 2.4 mg/kg, about 2.9 mg/kg, about 3.2 mg/kg, about 3.4 mg/kg, about 3.5 mg/kg or about 3.9 mg/kg of the body weight of the human subject. In some embodiments, the effective amount is a dose of at least about 2.4 mg/kg and at most about 3.5 mg/kg of the body weight of the human subject. In some embodiments, the effective amount is a dose of at least about 2.4 mg/kg and at most about 3.4 mg/kg of the body weight of the human subject. In some embodiments, the effective amount is a dose of at least about 2.4 mg/kg and at most about 3.2 mg/kg of the body weight of the human subject. In some embodiments, the effective amount is a dose of at least about 2.4 mg/kg and at most about 3.0 mg/kg of the body weight of the human subject. In some embodiments, the effective amount is a dose of at least about 2.8 mg/kg and at most about 3.4 mg/kg of the body weight of the human subject. In some embodiments, the effective amount is a dose of at least about 2.8 mg/kg and at most about 3.2 mg/kg of the body weight of the human subject. In some embodiments, the effective amount is administered to the human subject on a dosing schedule of about once per week. In some embodiments, the effective amount is administered to the human subject on a dosing schedule of about once every two weeks. In some embodiments, the effective amount is administered to the human subject on a dosing schedule of about once every three weeks. In some embodiments, the effective amount is administered to the human subject on a dosing schedule of about once every four weeks. [215] In some embodiments, an effective amount of anti-PSMA ADC of the present disclosure for administration to a human subject is a dose within a range of about 2.0 mg/kg to about 4.0 mg/kg of the body weight of the human subject, wherein the dose is administered Attorney Docket No. AMB1013WOPCT1 to the human subject once every three weeks, and wherein the anti-PSMA ADC is ARX517. In some embodiments, the dose is about 2.0 mg/kg of the body weight of the human subject, wherein the dose is administered to the human subject once every three weeks, and wherein the anti-PSMA ADC is ARX517. In some embodiments, the dose is greater than 2.0 mg/kg of the body weight of the human subject wherein the dose is administered to the human subject once every three weeks, and wherein the anti-PSMA ADC is ARX517. In some embodiments, the dose is about 2.1 mg/kg of the body weight of the human subject, wherein the dose is administered to the human subject once every three weeks, and wherein the anti-PSMA ADC is ARX517. In some embodiments, the dose is about 2.2 mg/kg of the body weight of the human subject, wherein the dose is administered to the human subject once every three weeks, and wherein the anti-PSMA ADC is ARX517. In some embodiments, the dose is about 2.3 mg/kg of the body weight of the human subject, wherein the dose is administered to the human subject once every three weeks, and wherein the anti-PSMA ADC is ARX517. In some embodiments, the dose is about 2.4 mg/kg of the body weight of the human subject, wherein the dose is administered to the human subject once every three weeks, and wherein the anti- PSMA ADC is ARX517. In some embodiments, the dose is about 2.5 mg/kg of the body weight of the human subject, wherein the dose is administered to the human subject once every three weeks, and wherein the anti-PSMA ADC is ARX517. In some embodiments, the dose is greater than 2.5 mg/kg of the body weight of the human subject. In some embodiments, the dose is about 2.6 mg/kg of the body weight of the human subject, wherein the dose is administered to the human subject once every three weeks, and wherein the anti- PSMA ADC is ARX517. In some embodiments, the dose is about 2.7 mg/kg of the body weight of the human subject, wherein the dose is administered to the human subject once every three weeks, and wherein the anti-PSMA ADC is ARX517. In some embodiments, the dose is about 2.8 mg/kg of the body weight of the human subject, wherein the dose is Attorney Docket No. AMB1013WOPCT1 administered to the human subject once every three weeks, and wherein the anti-PSMA ADC is ARX517. In some embodiments, the dose is about 2.9 mg/kg of the body weight of the human subject, wherein the dose is administered to the human subject once every three weeks, and wherein the anti-PSMA ADC is ARX517. In some embodiments, the dose is about 3.0 mg/kg of the body weight of the human subject, wherein the dose is administered to the human subject once every three weeks, and wherein the anti-PSMA ADC is ARX517. In some embodiments, the dose is about 3.1 mg/kg of the body weight of the human subject, wherein the dose is administered to the human subject once every three weeks, and wherein the anti- PSMA ADC is ARX517. In some embodiments, the dose is about 3.2 mg/kg of the body weight of the human subject, wherein the dose is administered to the human subject once every three weeks, and wherein the anti-PSMA ADC is ARX517. In some embodiments, the dose is about 3.3 mg/kg of the body weight of the human subject, wherein the dose is administered to the human subject once every three weeks, and wherein the anti-PSMA ADC is ARX517. In some embodiments, the dose is about 3.4 mg/kg of the body weight of the human subject, wherein the dose is administered to the human subject once every three weeks, and wherein the anti-PSMA ADC is ARX517. In some embodiments, the dose is about 3.5 mg/kg of the body weight of the human subject, wherein the dose is administered to the human subject once every three weeks, and wherein the anti-PSMA ADC is ARX517. In some embodiments, the dose is about 3.6 mg/kg of the body weight of the human subject, wherein the dose is administered to the human subject once every three weeks, and wherein the anti- PSMA ADC is ARX517. In some embodiments, the dose is about 3.7 mg/kg of the body weight of the human subject, wherein the dose is administered to the human subject once every three weeks, and wherein the anti-PSMA ADC is ARX517. In some embodiments, the dose is about 3.8 mg/kg of the body weight of the human subject, wherein the dose is administered to the human subject once every three weeks, and wherein the anti-PSMA ADC Attorney Docket No. AMB1013WOPCT1 is ARX517. In some embodiments, the dose is about 3.9 mg/kg of the body weight of the human subject, wherein the dose is administered to the human subject once every three weeks, and wherein the anti-PSMA ADC is ARX517. In some embodiments, the dose is about 4.0 mg/kg of the body weight of the human subject, wherein the dose is administered to the human subject once every three weeks, and wherein the anti-PSMA ADC is ARX517. In some embodiments, the dose is about 4.1 mg/kg of the body weight of the human subject, wherein the dose is administered to the human subject once every three weeks, and wherein the anti- PSMA ADC is ARX517. In some embodiments, the dose is about 4.2 mg/kg of the body weight of the human subject, wherein the dose is administered to the human subject once every three weeks, and wherein the anti-PSMA ADC is ARX517. In some embodiments, the dose is about 4.3 mg/kg of the body weight of the human subject, wherein the dose is administered to the human subject once every three weeks, and wherein the anti-PSMA ADC is ARX517. In some embodiments, the dose is about 4.4 mg/kg of the body weight of the human subject, wherein the dose is administered to the human subject once every three weeks, and wherein the anti-PSMA ADC is ARX517. In some embodiments, the dose is about 4.5 mg/kg of the body weight of the human subject, wherein the dose is administered to the human subject once every three weeks, and wherein the anti-PSMA ADC is ARX517. [216] In other embodiments, based upon the composition, the dose can be delivered continuously, such as by continuous pump, or at periodic intervals. Desired time intervals of multiple doses of a particular composition can be determined without undue experimentation by one skilled in the art. Other protocols for the administration of the compositions provided will be known to one of ordinary skill in the art, in which the dose amount, schedule of administration, site(s) of administration, mode of administration and the like vary from the foregoing. In some embodiments, the dose regimen is a single intravenous dose. In some other embodiments, the dose regimen begins with a single Attorney Docket No. AMB1013WOPCT1 intravenous dose and is followed by a series of additional single intravenous doses, with each dose administered about 3 weeks apart. [217] In some embodiments, the method of treatment provides an anti-PSMA ADC serum terminal half-life of at least about 5 days after the administration of the effective amount of the anti-PSMA ADC to the human subject. In some embodiments, the method of treatment provides an anti-PSMA ADC serum terminal half-life within a range of about 5 to about 10 days after the administration of the effective amount of the anti-PSMA ADC to the human subject. In some embodiments, the method of treatment provides an anti-PSMA ADC serum terminal half-life of at least about 6 days after the administration of the effective amount of the anti-PSMA ADC to the human subject. In some embodiments, the method of treatment provides an anti-PSMA ADC serum terminal half-life of at least about one week after the administration of the effective amount of the anti-PSMA ADC to the human subject. In some embodiments, the method of treatment provides an anti-PSMA ADC serum terminal half-life within a range of about 6 to about 10 days after the administration of the effective amount of the anti-PSMA ADC to the human subject. [218] In some embodiments, the method of treatment provides a free payload time to serum maximum concentration (Tmax) of at least about 5 days after the administration of the effective amount of the anti-PSMA ADC to the human subject, wherein the free payload has the following structure: ;

or a some a Tmax of at least about 6 days after the administration of the effective amount of the anti-PSMA ADC to the human subject. In some embodiments, the method of treatment provides a free payload Attorney Docket No. AMB1013WOPCT1 Tmax of about one week after the administration of the effective amount of the anti-PSMA ADC to the human subject. [219] In some embodiments, the method of treatment provides a free payload serum maximum concentration (Cmax) of at most about 1 ng/mL after the administration of the effective amount of the anti-PSMA ADC to the human subject. In some embodiments, the method of treatment provides a free payload Cmax of at most about 0.5 ng/mL, at most about 0.4 ng/mL, at most about 0.3 ng/mL or at most about 0.2 ng/mL after the administration of the effective amount of the anti-PSMA ADC to the human subject. In some embodiments, the method of treatment provides a free payload Cmax within a range of about 0.01 ng/mL to about 0.3 ng/mL after the administration of the effective amount of the anti-PSMA ADC to the human subject. [220] In some embodiments, the method of treatment provides an anti-PSMA ADC serum maximum concentration (Cmax) of at least about 20 µg/ml (20 micrograms per milliliter), at least about 25 µg/ml (20 micrograms per milliliter), at least about 30 µg/ml, at least 35 µg/ml, at least about 40 µg/ml, at least about 45 µg/ml, at least about 50 µg/ml, at least about 55 µg/ml, or at least about 60 µ/ml after the administration of the effective amount of the anti-PSMA ADC to the human subject. (See, e.g., FIG.16B.) [221] In some instances, the PSMA antibodies, variants or anti-PSMA ADC compositions of the present disclosure can be used in conjunction with an additional therapy or treatment including but not limited to surgery, radiation, cryosurgery, thermotherapy, hormone treatment, chemotherapy, vaccines and other immunotherapies. In some embodiments such additional treatment can include a therapeutic agent such as chemotherapeutic agent, hormonal agent, antitumor agent, immunostimulatory agent, immunomodulator, corticosteroid or combination thereof. In some embodiments, the hormonal agent is enzalutamide. Attorney Docket No. AMB1013WOPCT1 [222] In other embodiments, the anti-PSMA ADCs of the present disclosure can be administered with one or more immunostimulatory agents to induce or enhance an immune response. Immunostimulatory agents that can stimulate specific arms of the immune system, such as natural killer (NK) cells that mediate antibody-dependent cell cytotoxicity (ADCC). Such immunostimulatory agents include, but are not limited to, IL-2, immunostimulatory oligonucleotides (for example, CpG motifs), α-interferon, γ-interferon, tumor necrosis factor alpha (TNFα). In other embodiments, the anti-PSMA ADCs of the present disclosure can be administered with one or more immunomodulators including, but not limited to, cytokines, chemokines (including, but are not limited to, SLC5 ELC, MIP3α, MIP3β, IP-IO, MIG, and combinations thereof). Other therapeutic agents can be a vaccine that immunizes a subject against PSMA. Such vaccines, in some embodiments, include antigens, such as PSMA dimers, with, optionally, one or more adjuvants to induce or enhance an immune response. Adjuvants of many kinds are well known in the art. [223] The chemotherapeutic agent or any agent involved in treating, reducing or preventing a disease, condition or cancer in a subject in need thereof can also be administered in combination with an anti-PSMA ADC of the present disclosure. Chemotherapeutic agents can include, but are not limited to, erlotinib (TARCEVA®, Genentech/OSI Pharm.), bortezomib (VELCADE®, Millennium Pharm.), fulvestrant (FASLODEX®, AstraZeneca), sutent (SU11248, Pfizer), letrozole (FEMARA®, Novartis), imatinib mesylate (GLEEVEC®, Novartis), PTK787/ZK 222584 (Novartis), oxaliplatin (Eloxatin®, Sanofi), 5-FU (5- fluorouracil), leucovorin, Rapamycin (Sirolimus, RAPAMUNE®, Wyeth), lapatinib (TYKERB®, GSK572016, GlaxoSmithKline), lonafarnib (SCH 66336), sorafenib (BAY43- 9006, Bayer Labs.), and gefitinib (IRESSA®, AstraZeneca), AG1478, AG1571 (SU 5271; Sugen), alkylating agents such as thiotepa and CYTOXAN® cyclosphosphamide; alkyl sulfonates such as busulfan, improsulfan and piposulfan; antifolate antineoplastic such as Attorney Docket No. AMB1013WOPCT1 pemetrexed (ALIMTA®, Eli Lilly), aziridines such as benzodopa, carboquone, meturedopa, and uredopa; ethylenimines and methylamelamines including altretamine, triethylenemelamine, triethylenephosphoramide, triethylenethiophosphoramide and trimethylomelamine; acetogenins (especially bullatacin and bullatacinone); a camptothecin (including the synthetic analogue topotecan); bryostatin; callystatin; CC-1065 (including its adozelesin, carzelesin and bizelesin synthetic analogues); cryptophycins (particularly cryptophycin 1 and cryptophycin 8); dolastatin; duocarmycin (including the synthetic analogues, KW-2189 and CB1-TM1); eleutherobin; pancratistatin; a sarcodictyin; spongistatin; nitrogen mustards such as chlorambucil, chlornaphazine, cholophosphamide, estramustine, ifosfamide, mechlorethamine, mechlorethamine oxide hydrochloride, melphalan, novembichin, phenesterine, prednimustine, trofosfamide, uracil mustard; nitrosoureas such as carmustine, chlorozotocin, fotemustine, lomustine, nimustine, and ranimnustine; antibiotics such as the enediyne antibiotics, calicheamicin, calicheamicin gamma1I and calicheamicin omegaI1; dynemicin, including dynemicin A; bisphosphonates, such as clodronate; an esperamicin; as well as neocarzinostatin chromophore and related chromoprotein enediyne antibiotic chromophores, aclacinomysins, actinomycin, anthramycin, azaserine, bleomycins, cactinomycin, carabicin, caminomycin, carzinophilin, chromomycinis, dactinomycin, daunorubicin, detorubicin, 6-diazo-5-oxo-L-norleucine, ADRIAMYCIN® doxorubicin (including morpholino-doxorubicin, cyanomorpholino-doxorubicin, 2-pyrrolino-doxorubicin and deoxydoxorubicin), epirubicin, esorubicin, idarubicin, marcellomycin, mitomycins such as mitomycin C, mycophenolic acid, nogalamycin, olivomycins, peplomycin, potfiromycin, puromycin, quelamycin, rodorubicin, streptonigrin, streptozocin, tubercidin, ubenimex, zinostatin, zorubicin; anti-metabolites such as methotrexate and 5-fluorouracil (5-FU); folic acid analogues such as denopterin, methotrexate, pteropterin, trimetrexate; purine analogs such as fludarabine, 6-mercaptopurine, thiamiprine, thioguanine; pyrimidine analogs such as Attorney Docket No. AMB1013WOPCT1 ancitabine, azacitidine, 6-azauridine, carmofur, cytarabine, dideoxyuridine, doxifluridine, enocitabine, floxuridine; androgens such as calusterone, dromostanolone propionate, epitiostanol, mepitiostane, testolactone; antiandrogens (for example, enzalutamide) or androgen deprivation therapy; anti-adrenals such as aminoglutethimide, mitotane, trilostane; folic acid replenisher such as frolinic acid; aceglatone; aldophosphamide glycoside; aminolevulinic acid; eniluracil; amsacrine; bestrabucil; bisantrene; edatraxate; defofamine; demecolcine; diaziquone; elformithine; elliptinium acetate; an epothilone; etoglucid; gallium nitrate; hydroxyurea; lentinan; lonidainine; maytansinoids such as maytansine and ansamitocins; mitoguazone; mitoxantrone; mopidanmol; nitraerine; pentostatin; phenamet; pirarubicin; losoxantrone; podophyllinic acid; 2-ethylhydrazide; procarbazine; PSK® polysaccharide complex (JHS Natural Products, Eugene, OR); razoxane; rhizoxin; sizofuran; spirogermanium; tenuazonic acid; triaziquone; 2,2',2''-trichlorotriethylamine; trichothecenes (especially T-2 toxin, verracurin A, roridin A and anguidine); urethan; vindesine; dacarbazine; mannomustine; mitobronitol; mitolactol; pipobroman; gacytosine; arabinoside ("Ara-C"); cyclophosphamide; thiotepa; taxoids, e.g., paclitaxel (TAXOL®, Bristol-Myers Squibb Oncology, Princeton, N.J.), ABRAXANE™ Cremophor-free, albumin, nanoparticle formulation of paclitaxel (American Pharmaceutical Partners, Schaumberg, Ill.), and TAXOTERE® doxetaxel (Rhone-Poulenc Rorer, Antony, France); chloranbucil; GEMZAR® gemcitabine; 6-thioguanine; mercaptopurine; methotrexate; platinum analogs such as cisplatin and carboplatin; vinblastine; platinum; etoposide (VP-16); ifosfamide; mitoxantrone; vincristine; NAVELBINE® vinorelbine; novantrone; teniposide; edatrexate; daunomycin; aminopterin; xeloda; ibandronate; CPT-11; topoisomerase inhibitor RFS 2000; difluoromethylornithine (DMFO); retinoids such as retinoic acid; capecitabine; and pharmaceutically acceptable salts, acids or derivatives of any of the above. Attorney Docket No. AMB1013WOPCT1 [224] The anti-PSMA ADCs of the present invention can be used in combination with any agent involved in treating, reducing, ameliorating, or preventing prostate cancer in a subject in need of such, including but not limited to hormone therapy or chemotherapy. For example, the hormone therapy enzalutamide has been demonstrated, in vitro, to increased expression of cell surface PSMA expression approximately 3-fold (Murga et al., Prostate 15;75(3):242-54, 2015). Additionally, enzalutamide treated mice bearing human PDX prostate tumors showed increased inhibition of tumor growth when combined with a PSMA ADC with the auristatin payload, MMAE (DiPippo et al., Prostate 15;76(3):325-34, 2016). [225] Most prostate cancer patients eventually progress to androgen deprivation therapy (ADT)-resistant disease. Anti-PSMA ADCs of the present disclosure, such as ARX517, can inhibit tumor growth in enzalutamide-sensitive and -resistant CDX and PDX prostate cancer models and may therefore be useful in treating subjects having ADT-resistant disease. ARX517 inhibited tumor growth in multiple enzalutamide-sensitive and -resistant xenograft models and produced additive efficacy with enzalutamide in an enzalutamide-sensitive tumor model. EXAMPLES [226] It is understood that the examples and embodiments described herein are for illustrative purposes only and that various modifications or changes in light thereof will be suggested to persons skilled in the art and are to be included within the spirit and purview of this application and scope of the appended claims. [227] Example 1: Transient Transfection. [228] CHO-S cultures were seeded at 0.75x10
6/mL approximately 16 hours pre-transfection in FreeStyle CHO medium. Cells were transfected the following day when the cell count reached 1.4 – 1.6 x 10
6/mL. At the target cell count, 400 mM para-acetyl phenylalanine stock was added to a 1.4 mM final culture concentration. Attorney Docket No. AMB1013WOPCT1 [229] Polyethylenimine/DNA (PEI/DNA) complex was prepared as described: DNA (1.42 ug/1x10
6 cells) was dissolved in RPMI media (5% (v/v) of total culture volume), DNA/RPMI mixture was incubated at room temperature for 2 minutes, PEI stock (1 mg/mL) was added to DNA solution at a 3:1 ratio (PEI/DNA (ug/ug)), and the mixture incubated at room temperature for 5 min. [230] The mixture was gently added to the cell culture and swirled. The flasks were transferred to a 32 °C incubator. At day 6 post-transfection, a western blot analysis was performed. At day 7 post-transfection, the supernatant was harvested. [231] Example 2: Antibody Humanization. [232] Humanization of parental mouse anti-PSMA J591 monoclonal antibody (mAb) (Liu et al., Cancer Research, 57, 3629-36354, 1997) was performed by grafting mouse heavy chain and light chain CDRs (HC-CDR1:31-35, HC-CDR2:50-66, HC-CDR3:99-104 and LC- CDR1:24-34, LC-CDR2:50-56, LC-CDR3:89-97) onto different human frameworks selected for highest homology to the mouse framework sequences and cloning into human IgG1/kappa constant regions as a backbone. Multiple humanized mAb variants were generated by pairing four human heavy chain (HC) variants with six light chain (LC) variants, using transient expression of in HEK293 cells. Key back mutations were added to the LC framework region to retain binding activity of the antibody. To select the lead clone, mAb variant supernatants were tested for binding to PSMA-positive LNCaP cells by flow cytometry. Table 3 describes a selected heavy chain variable region sequence and four light chain variable region sequences used for further studies. Binding analysis showed that four humanized full length variants, shown as humanized anti-PSMA variant 1, 2, 3 and 4 (Table 3), retained comparable binding affinity as the chimera (Table 3), exhibiting nanomolar range binding affinity. As disclosed in Table 3, humanized anti-PSMA variant 1 comprises the heavy chain sequence of SEQ ID NO: Attorney Docket No. AMB1013WOPCT1 8 and the light chain sequence of SEQ ID NO: 9; humanized anti-PSMA variant 2 comprises the heavy chain sequence of SEQ ID NO: 10 and the light chain sequence of SEQ ID NO: 11; humanized anti-PSMA variant 3 comprises the heavy chain sequence of SEQ ID NO: 12 and the light chain sequence of SEQ ID NO: 13; and humanized anti-PSMA variant 4 comprises the heavy chain sequence of SEQ ID NO: 14 and the light chain sequence of SEQ ID NO: 15. Underlined Alanine amino acids (“A”) in the heavy chain sequences of SEQ ID NOs: 8, 10, 12, 14, and 16 in Table 3 indicate amino acid position 114 according to the Kabat numbering scheme (see Kabat et al., NIH Publication No.369–847, 1993) at which the non-natural amino acid para-acetyl-L-phenylalanine (pAF) is incorporated (see Example 3). Table 3. Anti-PSMA Antibody Amino Acid Sequences Sequences of Humanized and Chimeric anti-PSMA antibodies Humanized Heavy Chain Variable Region Sequence SEQ ID NO: 1 QVQLVQSGAEVKKPGASVKVSCKASGYTFTEYTIHWVRQAPGQRLEWMGNINPN NGGTTYNQKFEDRVTITRDTSASTAYMELSSLRSEDTAVYYCAAGWNFDYWGQG TTLTVSS Humanized Light Chain Variable Region Sequence SEQ ID NO: 2 DIQLTQSPSFLSASVGDRVTITCKASQDVGTAVDWYQQKPGKAPKLLIYWASTRH TGVPSRFSGSGSGTEFTLTISSLQPEDFATYYCQQYNSYPLTFGGGTKVEIKRTV SEQ ID NO: 3 DIQLTQSPSFLSASVGDRVTITCKASQDVGTAVDWYQQKPGKAPKLLIYWASTRH TGVPDRFSGSGSGTEFTLTISSLQPEDFATYYCQQYNSYPLTFGGGTKVEIKRTV SEQ ID NO: 4 DIQLTQSPSFLSASVGDRVTITCKASQDVGTAVDWYQQKPGKAPKLLIYWASTRH TGVPSRFSGSGSGTEFTLTISSLQSEDFATYYCQQYNSYPLTFGGGTKVEIKRTV SEQ ID NO: 5 DIQLTQSPSFLSASVGDRVTITCKASQDVGTAVDWYQQKPGKAPKLLIYWASTRH TGVPSRFSGSGSGTEFTLTISNLQPEDFATYYCQQYNSYPLTFGGGTKVEIKRTV Chimeric Heavy Chain Variable Region Sequence SEQ ID NO: 6 EVQLQQSGPELVKPGTSVRISCKTSGYTFTEYTIHWVKQSHGKSLEWIGNINPNNG GTTYNQKFEDKATLTVDKSSSTAYMELRSLTSEDSAVYYCAAGWNFDYWGQGTT LTVSS Attorney Docket No. AMB1013WOPCT1 Chimeric Light Chain Variable Region Sequence SEQ ID NO: 7 DIVMTQSHKFMSTSVGDRVSIICKASQDVGTAVDWYQQKPGQSPKLLIYWASTRH TGVPDRFTGSGSGTDFTLTITNVQSEDLADYFCQQYNSYPLTFGAGTKVEIKRTV Humanized Anti- PSMA (full length) Variant 1 SEQ ID NO: 8 Heavy Chain QVQLVQSGAEVKKPGASVKVSCKASGYTFTEYTIHWVRQAPGQRLEWMGNINPN NGGTTYNQKFEDRVTITRDTSASTAYMELSSLRSEDTAVYYCAAGWNFDYWGQG TTLTVSSASTKGPSVFPLAPSSKSTSGGTAALGCLVKDYFPEPVTVSWNSGALTSG VHTFPAVLQSSGLYSLSSVVTVPSSSLGTQTYICNVNHKPSNTKVDKKVEPKSCDK THTCPPCPAPELLGGPSVFLFPPKPKDTLMISRTPEVTCVVVDVSHEDPEVKFNWY VDGVEVHNAKTKPREEQYNSTYRVVSVLTVLHQDWLNGKEYKCKVSNKALPAPI EKTISKAKGQPREPQVYTLPPSRDELTKNQVSLTCLVKGFYPSDIAVEWESNGQPE NNYKTTPPVLDSDGSFFLYSKLTVDKSRWQQGNVFSCSVMHEALHNHYTQKSLSL SPG SEQ ID NO: 9 Light Chain DIQLTQSPSFLSASVGDRVTITCKASQDVGTAVDWYQQKPGKAPKLLIYWASTRH TGVPSRFSGSGSGTEFTLTISSLQPEDFATYYCQQYNSYPLTFGGGTKVEIKRTVAA PSVFIFPPSDEQLKSGTASVVCLLNNFYPREAKVQWKVDNALQSGNSQESVTEQDS KDSTYSLSSTLTLSKADYEKHKVYACEVTHQGLSSPVTKSFNRGEC Humanized Anti- PSMA (full length) Variant 2 SEQ ID NO: 10 Heavy Chain QVQLVQSGAEVKKPGASVKVSCKASGYTFTEYTIHWVRQAPGQRLEWMGNINPN NGGTTYNQKFEDRVTITRDTSASTAYMELSSLRSEDTAVYYCAAGWNFDYWGQG TTLTVSSASTKGPSVFPLAPSSKSTSGGTAALGCLVKDYFPEPVTVSWNSGALTSG VHTFPAVLQSSGLYSLSSVVTVPSSSLGTQTYICNVNHKPSNTKVDKKVEPKSCDK THTCPPCPAPELLGGPSVFLFPPKPKDTLMISRTPEVTCVVVDVSHEDPEVKFNWY VDGVEVHNAKTKPREEQYNSTYRVVSVLTVLHQDWLNGKEYKCKVSNKALPAPI EKTISKAKGQPREPQVYTLPPSRDELTKNQVSLTCLVKGFYPSDIAVEWESNGQPE NNYKTTPPVLDSDGSFFLYSKLTVDKSRWQQGNVFSCSVMHEALHNHYTQKSLSL SPG SEQ ID NO: 11 Light Chain DIQLTQSPSFLSASVGDRVTITCKASQDVGTAVDWYQQKPGKAPKLLIYWASTRH TGVPSRFSGSGSGTEFTLTISSLQSEDFATYYCQQYNSYPLTFGGGTKVEIKRTVAA PSVFIFPPSDEQLKSGTASVVCLLNNFYPREAKVQWKVDNALQSGNSQESVTEQDS KDSTYSLSSTLTLSKADYEKHKVYACEVTHQGLSSPVTKSFNRGEC Humanized Anti- PSMA (full length) Variant 3 SEQ ID NO: 12 Heavy Chain Attorney Docket No. AMB1013WOPCT1 QVQLVQSGAEVKKPGASVKVSCKASGYTFTEYTIHWVRQAPGQRLEWMGNINPN NGGTTYNQKFEDRVTITRDTSASTAYMELSSLRSEDTAVYYCAAGWNFDYWGQG TTLTVSSASTKGPSVFPLAPSSKSTSGGTAALGCLVKDYFPEPVTVSWNSGALTSG VHTFPAVLQSSGLYSLSSVVTVPSSSLGTQTYICNVNHKPSNTKVDKKVEPKSCDK THTCPPCPAPELLGGPSVFLFPPKPKDTLMISRTPEVTCVVVDVSHEDPEVKFNWY VDGVEVHNAKTKPREEQYNSTYRVVSVLTVLHQDWLNGKEYKCKVSNKALPAPI EKTISKAKGQPREPQVYTLPPSRDELTKNQVSLTCLVKGFYPSDIAVEWESNGQPE NNYKTTPPVLDSDGSFFLYSKLTVDKSRWQQGNVFSCSVMHEALHNHYTQKSLSL SPG SEQ ID NO: 13 Light Chain DIQLTQSPSFLSASVGDRVTITCKASQDVGTAVDWYQQKPGKAPKLLIYWASTRH TGVPDRFSGSGSGTEFTLTISSLQPEDFATYYCQQYNSYPLTFGGGTKVEIKRTVAA PSVFIFPPSDEQLKSGTASVVCLLNNFYPREAKVQWKVDNALQSGNSQESVTEQDS KDSTYSLSSTLTLSKADYEKHKVYACEVTHQGLSSPVTKSFNRGEC Humanized Anti- PSMA (full length) Variant 4 SEQ ID NO: 14 Heavy Chain QVQLVQSGAEVKKPGASVKVSCKASGYTFTEYTIHWVRQAPGQRLEWMGNINPN NGGTTYNQKFEDRVTITRDTSASTAYMELSSLRSEDTAVYYCAAGWNFDYWGQG TTLTVSSASTKGPSVFPLAPSSKSTSGGTAALGCLVKDYFPEPVTVSWNSGALTSG VHTFPAVLQSSGLYSLSSVVTVPSSSLGTQTYICNVNHKPSNTKVDKKVEPKSCDK THTCPPCPAPELLGGPSVFLFPPKPKDTLMISRTPEVTCVVVDVSHEDPEVKFNWY VDGVEVHNAKTKPREEQYNSTYRVVSVLTVLHQDWLNGKEYKCKVSNKALPAPI EKTISKAKGQPREPQVYTLPPSRDELTKNQVSLTCLVKGFYPSDIAVEWESNGQPE NNYKTTPPVLDSDGSFFLYSKLTVDKSRWQQGNVFSCSVMHEALHNHYTQKSLSL SPG SEQ ID NO: 15 Light Chain DIQLTQSPSFLSASVGDRVTITCKASQDVGTAVDWYQQKPGKAPKLLIYWASTRH TGVPSRFSGSGSGTEFTLTISNLQPEDFATYYCQQYNSYPLTFGGGTKVEIKRTVAA PSVFIFPPSDEQLKSGTASVVCLLNNFYPREAKVQWKVDNALQSGNSQESVTEQDS KDSTYSLSSTLTLSKADYEKHKVYACEVTHQGLSSPVTKSFNRGEC Chimeric Anti-PSMA Antibody (full length) Variant SEQ ID NO: 16 Heavy Chain EVQLQQSGPELVKPGTSVRISCKTSGYTFTEYTIHWVKQSHGKSLEWIGNINPNNG GTTYNQKFEDKATLTVDKSSSTAYMELRSLTSEDSAVYYCAAGWNFDYWGQGTT LTVSSASTKGPSVFPLAPSSKSTSGGTAALGCLVKDYFPEPVTVSWNSGALTSGVH TFPAVLQSSGLYSLSSVVTVPSSSLGTQTYICNVNHKPSNTKVDKKVEPKSCDKTH TCPPCPAPELLGGPSVFLFPPKPKDTLMISRTPEVTCVVVDVSHEDPEVKFNWYVD GVEVHNAKTKPREEQYNSTYRVVSVLTVLHQDWLNGKEYKCKVSNKALPAPIEK TISKAKGQPREPQVYTLPPSRDELTKNQVSLTCLVKGFYPSDIAVEWESNGQPENN YKTTPPVLDSDGSFFLYSKLTVDKSRWQQGNVFSCSVMHEALHNHYTQKSLSLSP GSEQ ID NO: 17 Light Chain Attorney Docket No. AMB1013WOPCT1 DIVMTQSHKFMSTSVGDRVSIICKASQDVGTAVDWYQQKPGQSPKLLIYWASTRH TGVPDRFTGSGSGTDFTLTITNVQSEDLADYFCQQYNSYPLTFGAGTKVEIKRTVA APSVFIFPPSDEQLKSGTASVVCLLNNFYPREAKVQWKVDNALQSGNSQESVTEQ DSKDSTYSLSSTLTLSKADYEKHK [233] Example 3: Anti-PSMA Antibody Selection and Recombinant Expression. [234] Lead humanized anti-PSMA antibodies (see Example 2) were selected and recombinantly expressed in CHO cells by both transient transfection (described in Example 1) and stable bulk pool method to examine expression of the humanized sequence. In transient transfection and stable bulk pool approaches, proprietary technology was built into the expression vector and CHO host cells, respectively (see for example WO2018/223108). The non-natural amino acid para-acetyl-L-phenylalanine (pAF) was incorporated at position A114, (Kabat numbering scheme (see Kabat et al., NIH Publication No. 369–847, 1993); position indicated in Table 3 by A), in the PSMA antibody, which is the first amino acid residue of the constant region of the heavy chain. [235] Candidate humanized J591 antibodies were evaluated for binding to human PSMA expressed on LNCaP cells. Selection of the final humanized J591 (huJ591) mAb candidate was based on successful retention of human PSMA binding activity and the lowest number of back- mutations required to maintain binding affinity (data not shown). [236] Example 4: Purification of Humanized anti-PSMA Antibody. [237] Humanized mAb variant 1 (Table 3) containing non-natural amino acid pAF in each heavy chain at Kabat position 114 (hereinafter, “ARX517 mAb” or “unconjugated mAb”) in clarified cell culture fluid (HCCF) was loaded over protein A column (MabSelect SuRe, Cytiva) equilibrated in 25 mM sodium phosphate, 100 mM sodium chloride, pH 7.3. After loading, the column was washed with wash buffer I (20 mM sodium phosphate, 100 mM sodium chloride, pH 7.5) followed by wash buffer II (50 mM sodium acetate, pH 5.5) to remove Attorney Docket No. AMB1013WOPCT1 host cell contaminants. ARX517 mAb was eluted from the column with elution buffer (50 mM sodium acetate, pH 3.4), pooled, and pH adjusted to pH 5.5 with 1.0 M tris base. ARX517 mAb was further purified by loading over a cation exchange column (Capto SP Impres, Cytiva) equilibrated in 50 mM sodium acetate, pH 5.5. After loading, the column was washed with wash buffer (50 mM sodium acetate, pH 5.5), and the mAb was eluted from the column using a linear gradient over 20 column volumes to 100% elution buffer (50 mM sodium acetate, 0.5 M sodium chloride, pH 5.5) and pooled. The cation exchange pool was buffer exchanged into formulation buffer (20 mM L-histidine, 2.5% trehalose, pH 6.0), concentrated to 40 mg/mL, 0.22 µM filtered, and stored at ≤ -60 °C. [238] Example 5: Production of Antibody Drug Conjugates (ADCs) [239] ARX517 mAb, purified as described in Example 4, was conjugated to drug-linker compound 6 (AS269) as follows. Acetohydrazide (Sigma) and AS269 were added to the ARX517 mAb at a molar excess of 300:1 and 10:1, respectively. The reaction mixture pH was adjusted to 4.0 with 2.0 M acetic acid followed by a water dilution to a final ARX517 mAb concentration of 30 mg/mL. The reaction was incubated at 28 °C for 24 hours and then buffer exchanged by ultrafiltration and diafiltration (UF/DF) into formulation buffer to remove excess reagents. The resulting antibody-drug conjugate (hereinafter, “ARX517 ADC” or simply “ARX517”) was 0.22 micron filtered and stored at -60 °C. [240] Example 6: Analytical Characterization of ARX517. [241] Peptide Mapping [242] Peptide maps of ARX517 mAb and ARX517 ADC were obtained using a Reversed Phase High Performance Liquid Chromatography (RP-HPLC) method with mass spectrometric (MS) detection. Approximately 200 µg of samples were denatured in Tris buffered 6M GdHCl Attorney Docket No. AMB1013WOPCT1 (pH 8) and reduced in 20 mM DTT by heating at 37 °C for 30 min. The samples were allowed to cool to room temperature, and the reduced thiols were alkylated by incubation in 45 mM iodoacetamide in the dark for 30 min. The samples were then buffer exchanged using 0.5 mL Amicon Ultra 10 KDa MWCO spin-filters into trypsin digestion buffer containing 50mM Tris- HCl pH 8. Sequencing grade modified Trypsin (Promega) was then added, and the samples were incubated at 37 °C for 4 hours. To stop the reaction, 15 µL of 20% TFA was added to each sample. Thereafter, 20 µL volume of each sample were injected into a 2 x 250 mm Phenomenex Jupiter Proteo 90Å (4 µm) column and the peptides were separated using a gradient of TFA and acetonitrile at a flow rate of 0.3 mL/min. A Thermo Qexactive Plus mass spectrometer was used to acquire HCD tandem MS data on top 10 peaks per MS scan. The raw data were analyzed using Thermo PepFinder software and the identities of the peptides were verified manually thereafter. The mass spectrometry data confirmed AS269 conjugation only occurred at the heavy chain alanine 114 (“HA114”) site (Kabat numbering), with the heavy chain peptide 101-119 shifting to the theoretical conjugate retention time in the ADC chromatogram (FIG. 1A shows peptide maps of unconjugated mAb and ARX517 ADC using reversed phase-HPLC method with mass spectrometric detection). [243] Hydrophobic interaction chromatography (HIC) [244] HIC was applied to determine the drug-to-antibody ratio (DAR) of ARX517 batches. About 50 µg of each sample was loaded on a MAbPac HIC-butyl HPLC column (Thermo) and eluted using a gradient of ammonium sulphate in a phosphate buffered mobile phase at 0.5 mL/min. The HIC method with UV detection at 280 nm resolved ARX517 unconjugated mAb, DAR1, and DAR2 species (FIG.1B). The DAR was calculated using the following equation: ∑
^ ^^^ =
^ ×^ ^^^ ; where d
i is the % peak area of the ADC with a drug load of i.
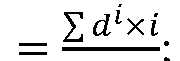

[245] The assays confirmed an average drug-to-antibody ratio (DAR) of 1.9 for ARX517 ADC. Attorney Docket No. AMB1013WOPCT1 [246] Size Exclusion Chromatography (SEC) [247] Size variants present in ARX517 were analyzed by SEC on a TSKgel G3000SWXL column (Tosoh) using an Agilent 1200 series HPLC system with UV detection at 280 nm. Samples were injected at 50 µg load and separation was achieved by an isocratic elution using a mobile phase consisting of 200 mM potassium phosphate and 250 mM potassium chloride, pH 6.0 at a flow rate of 0.5 mL/min and column temperature set at 25 °C. Percent area of HMW, monomer, and LWM species were calculated using Agilent ChemStation Software. ARX517 ADC exhibited high purity (98% main peak) and minimal aggregation (1.7% high molecular weight species) from the SE-HPLC data (FIG.1C). [248] Differential Scanning Calorimetry (DSC) [249] The thermal transition temperature of ARX517 mAb and ARX517 ADC (diluted to 1 mg/mL using formulation buffer) was determined using a MicroCal capillary VP DSC. The sample and reference cells were loaded with sample and formulation buffer, respectively. The instrument was programmed to scan from 10 to 110°C at a rate of 1 °C/min, with a 10 second data averaging period and 15 min equilibration time. Buffer vs. buffer scans were recorded throughout the experiment sequence to obtain a baseline scan to subtract from the experimental data and to ensure the shapes of the baseline scans were reproducible over the course of the experiment. The raw data was processed using MicroCal PEAQ DSC software to calculate T
m values. This DSC characterization showed minimal impact of conjugation on the thermodynamic stability, with no detectable differences in overall conformation between mAb and ADC lots (FIG.1D). [250] Example 7: In Vitro PSMA Expression and ARX517 Activity [251] PSMA cell surface expression levels were quantified in multiple prostate cancer cell lines using QiFiKit (Agilent Dako, Part number K007811-8) according to manufacturer Attorney Docket No. AMB1013WOPCT1 recommendations. Each cell line was harvested using StemPro Accutase Cell dissociation Reagent (Gibco, Catalog number A1110501) and incubated with mouse anti-human PSMA antibody (BioLegend, Catalog #342502, clone LNI-17), isotype control antibody (BioLegend, Catalog number 401401, Clone MGI-45) or FACS buffer only for 1 hour at 4 °C. After washing, the cells were incubated with FITC-conjugated anti-mouse secondary antibody, along with set-up beads and calibration beads, for 45 minutes at 4 °C in the dark. Following washing and resuspension of the cells and beads, FITC fluorescence was analyzed using FACSCanto II (BD Biosciences). PSMA receptor numbers were calculated using lot-specific standard curve obtained from calibration beads. PSMA expression ranged from <1,000 to 136,000 receptors per cell. [252] Prostate cancer cell lines including LNCaP (clone FGC, CRL-1740), MDA-PCa-2b (CRL-2422), 22Rv1 (CRL-2505), and PC-3 (CRL-1435) were purchased from ATCC and cultured in the recommended media. C4-2 cells were purchased directly from MD Anderson Cancer Center and cultured in RPMI-1640 media supplemented with 10% FBS and 100 U/mL penicillin/streptomycin. MDA-PCa-2b cells were initially propagated in male nu/nu mice (Charles River Laboratories), isolated, and then maintained in F-12K medium supplemented with 20% FBS, 100 U/mL penicillin/streptomycin, 25 ng/mL cholera toxin, 10 ng/mL mouse epidermal growth factor, 5 µM phosphoethanolamine, 100 pg/mL hydrocortisone, 45 nM sodium selenite, and 5 µg/mL human recombinant insulin as recommended by ATCC. All cell lines were used in the in vitro assays within 1 month after thawing the frozen vial. Cells were seeded at 3,000 cells/well in 96-well white plates using their respective media, and incubated overnight in a 37°C, 5% CO2 incubator. The next day, serially diluted ARX517 or MMAE were added to the cells and incubated for 4 days in a 37°C, 5% CO2 incubator. At the end of incubation, cell viability was measured using CellTiter-Glo2.0 reagent (Promega) in a SpectraMax M5E luminometer. IC50 values were determined by a 4PL curve fitting using Attorney Docket No. AMB1013WOPCT1 GraphPad Prism software (Version 8.2.1) and the % E
max for the dose of 30 nM was calculated by subtracting the % viability from 100%. [253] Treatment of cell lines with ARX517 ADC in 4-day cellular proliferation assays resulted in potent sub-nanomolar activity (IC50 values ≤ 0.5 nM) and high maximum efficacy of ARX517 in cells expressing moderate to high PSMA levels (MDA-PCa-2b, LNCaP, and C4-2). Minimal ARX517 activity was observed in cell lines with low or no PSMA expression (22Rv1 and PC-3), demonstrating the selectivity of ARX517 which requires expression of PSMA for activity. All tested cell lines were sensitive to the auristatin class of drugs as the positive control MMAE strongly inhibited proliferation in a similar manner across all tested cell lines with IC50 values ranging from 0.18-0.78 nM (see FIGs. 2A-2E for dose-response curves, showing relative cell viability (% of untreated control samples) and Table 4, which summarizes PSMA expression, and potency (IC50) and maximum efficacy (Emax) for ARX517 ADC and MMAE across the prostate cancer cell lines). Table 4. PSMA Expression and In Vitro Cytotoxicity Activity of ARX517 and MMAE In Human Prostate Cancer Cell Lines Cell Line PSMA ARX517 ARX517 MMAE MMAE Surface IC50 (nM) Emax (%) IC50 (nM) Emax (%) Number (QiFiKit) C4-2 87,000 0.228 92 0.178 96 LNCaP 136,000 0.449 63 0.566 89 MDA-PCa-2b 46,000 0.451 89 0.641 94 22Rv1 6,000 >3.366 ≥24 0.771 91 PC-3 <1000 >30 3 0.778 79 [254] The dependency of cell killing activity of the invented anti-PSMA ADCs on receptor copy number of PSMA is indicative of therapeutic benefit for treating prostate cancer. Receptor copy number of PSMA is usually high in cancerous prostate tissue compared to expression of PSMA in tissues with no cancer. This differential expression in PSMA receptor copy number Attorney Docket No. AMB1013WOPCT1 between cancerous prostate tissue and normal tissues can facilitate reducing and/or eliminating target-related non-prostate tissue related toxicity. This analysis suggests that ARX517 ADC can exhibit low activity in tissues expressing low PSMA receptor copy numbers than in cancerous prostate tissue with high PSMA receptor copy number. Hence, the background toxicity of ARX517 ADC can be significantly lowered. [255] Example 8: PSMA Species Cross Reactivity Binding by Bio-Layer Interferometry (BLI) [256] The binding of ARX517 mAb or ARX517 ADC to human, cynomolgus and rat PSMA/FOLH1 was measured in a biolayer interferometry assay on an Octet RED96 system (Sartorius). Anti-human IgG Fc capture biosensors (Sartorius) were loaded with purified ARX517 mAb or ARX517 ADC in HBS-P+ buffer (Cytiva). After washing biosensors with HBS-P+ buffer to remove unbound protein, serially diluted PSMA/FOLH1 (human, cynomolgus or rat) in HBS-P+ buffer was monitored for association and dissociation kinetics with ARX517 mAb- or ARX517 ADC-loaded biosensors. Data were referenced using a parallel buffer blank subtraction. The processed binding curves were globally fitted using the Langmuir model describing a 1:1 binding stoichiometry to calculate affinities. [257] The results are shown in FIGs. 3A-3C and Table 5. ARX517 mAb (unconjugated) showed similar high-affinity binding to human and cynomolgus monkey PSMA (apparent KD value was 0.62 nM and 0.79 nM, respectively; FIGs 3A and 3B) and no binding to rat PSMA (FIG. 3C) in the cross-species PSMA biolayer interferometry binding assay. ARX517 ADC bound human, cynomolgus monkey, and rat PSMA with similar affinities as the unconjugated ARX517 mAb, confirming the conjugation of AS269 at site HA114 did not impact binding activity of the ADC (FIGA.3A-3C). Table 5. Dissociation kinetics for huJ591 mAb and ARX517 to human, rat or cynomolgus PSMA. Attorney Docket No. AMB1013WOPCT1 Analyte Human KD (M) Rat KD (M) Cynomolgus KD (M) HuJ591 mAb 6.23 E-10 No binding 7.9 E-10 ARX517 5.04 E-10 No binding 6.81 E-10 [258] Example 9: Pharmacokinetic and Stability Studies Following ARX517 Dosing in Mice with and without C4-2 tumors. [259] To assess the in vivo stability and pharmacokinetics of ARX517, male nude (nu/nu) mice bearing C4-2 prostate tumors or no tumors were administered a single intravenous dose of ARX517 at 1 mg/kg or 5 mg/kg (n=5/group). Blood samples were collected from all animals at pre-dose, and up to 28 days post-dose (0.5, 2, 6, 24, 48, 72, 168, 240, 336, 504, and 672 hours post-dose). Blood samples were immediately diluted 10-fold into Casein-PBS (Thermo Scientific catalog# 37528) and frozen in tubes at -60°C to -80°C. The samples were analyzed in qualified ligand-binding assays designed to measure Total Antibody (“TA”; unconjugated and conjugated antibody species) and Intact ADC (ARX517 ADC DAR2 species only, i.e., 2- drug ADC species only), as described below. [260] Bioanalytical Assays in Mouse Matrix: An ARX517 TA assay was developed to detect unconjugated and all conjugated antibody species in mouse nude (nu/nu) serum. Meso Scale Discovery (MSD) High Bind plates were coated overnight at 4◦C with recombinant human PSMA (rhPSMA, R&D Systems catalog# 4234-ZN-010), then blocked with Casein-PBS (Thermo Scientific catalog# 37528) for at least 1 hour at room temperature the next day. The plates were washed 3 times in 1x Wash Buffer (20x KPL Wash Solution, KPL catalog# 50-63- 04) and ARX517 Standard (STD), Quality Controls (QCs), and samples pre-diluted 1:50 in Casein-PBS were added in duplicate to the plates. After incubation for 2 hours at room temperature, MSD plates were washed 3 times as before, and biotinylated goat anti-human kappa detection antibody (Southern Biotech catalog# 2061-08) was incubated for 1 hour at Attorney Docket No. AMB1013WOPCT1 room temperature. Following a wash step to remove unbound antibodies, Streptavidin-SULFO tag (MSD catalog# R32AD-1) reagent was added for 1 hour at room temperature. After a final wash step, 1x Read Buffer T (4x Read Buffer T, MSD catalog# R92TC-2) was added and plates were read in an MSD QuickPlex SQ120 unit. The LLOQ of the assay in nu/nu mouse serum was 39.1 ng/mL. The ARX517 Intact ADC assay in mouse nude (nu/nu) serum was designed to specifically only detect ADC with two drug-linkers (not ADC with one drug-linker, unconjugated mAb or free pAF-AS269) to enable determination of any loss of AS269 drug- linker when comparing intact ARX517 ADC curves with TA curves. The assay procedure was similar to the TA assay except MSD High Bind plates were coated with AMB-20, an anti- AS269 specific rabbit monoclonal Ab (mAb), and detection antibody was biotinylated-AMB- 20. The LLOQ of the intact ADC assay in nu/nu mouse serum was 125 ng/mL. Pharmacokinetic (PK) parameters for TA and Intact ADC data were analyzed in Phoenix
WinNonlin software, using non-compartmental analysis and a linear up/log down trapezoidal method. A minimum of 3 time points were required for determination of the elimination rate constant, upon which disposition parameters were calculated. Concentrations that were below the limit of quantitation (BLQ) were set to zero for PK analysis. [261] Comparison of the TA and intact ADC concentration-time curves enables stringent detection of any loss of AS269. ARX517 exhibited overlapping TA and intact ADC concentration-time curves at both dose levels, indicating that deconjugation of drug-linker is not observed, and ARX517 was stable in circulation for the duration of the 28-day study (FIG. 4A). [262] Linear pharmacokinetics (pK) and prolonged stability were observed following ARX517 dosing in mice with and without C4-2 tumors (Table 6). Mean PK parameters (n=5 mice per group) were analyzed using non-compartmental analysis. The ARX517 intact ADC versus TA exposures were comparable at both doses with mean area under the curve from time Attorney Docket No. AMB1013WOPCT1 zero to last quantifiable
concentration (AUC0-last) ratios from 1.07 to 1.23. The resulting ADC terminal half-life (t1/2) was long in mice with no tumors (mean t1/2 ranged from 261 to 350 hours) and in mice bearing tumors (mean t
1/2 ranged from 210 to 237 hours). The t
1/2 appeared slightly shorter for tumor-bearing mice, but the difference may fall within variability of the study. The ADC mean AUC
0-last in tumor-bearing versus non-tumor-bearing mice was comparable, with the ratio ranging from 0.86 (at 1 mg/kg) to 1.03 (at 5 mg/kg), suggesting no significant target-mediated drug disposition in the C4-2 xenograft model (FIG.4B). Table 6. PK parameters after ARX517 dosing in mice with and without CR-2 tumors. [263] Example 10: ARX517 Stability in Human Serum [264] ARX517 was diluted to 200 µg/mL in pooled human serum (Bioreclamation Cat# HMNSRM). Aliquots (100 µL) in sterile 0.6 mL Eppendorf tubes were incubated upright in an incubator set to 37 °C, 5% CO2 for 21 days. Individual tubes were removed after 0, 1, 4, 6, 8, 11, 14, 21 days incubation and frozen in a -80 °C freezer until all timepoints were collected. All samples were diluted to 500 ng/mL in human serum to fall within the quantitative range of the PK assays and analyzed in duplicate in Total Antibody (TA) and Intact ADC assays using the MSD platform to assess stability. The TA and Intact ADC assays in human serum used the Attorney Docket No. AMB1013WOPCT1 same reagents for capture and detection as the mouse TA and intact ADC assays (see Example 9). The LLOQ of the TA and intact ADC assays in human serum was 65.3 ng/mL and 32.7 ng/mL, respectively. The TA and Intact ADC concentrations at all time points were highly similar, with ADC versus TA ratios ranging from 0.91 to 1.16 (Table 7). The percent difference from theoretical (PDT) values for TA and intact ADC were all within 30% difference, which was considered within assay variability. Overall, the data demonstrated ARX517 is stable in human serum under the tested assay conditions for 21 days. Table 7. ARX517 stability in human serum. [265] Example 11: ARX517 In Vivo Efficacy in CDX and PDX Models in Mouse [266] ARX517 was evaluated in a series of in vivo cell line-derived xenograft (CDX) and patient-derived xenograft (PDX) models. [267] Cell line-Derived Xenograft (CDX) and Patient-Derived Xenograft (PDX) Models: C4- 2 and MDA-PCa-2b cells were purchased from ATCC (CRL-1595 and CRL-2422, respectively) and grown to near confluency. Freshly harvested cells were suspended in PBS and mixed 1:1 with matrigel. NCG or nu/nu mice were anesthetized with isoflurane (2-3%, 2 L/min oxygen) and implanted subcutaneously in the right flank with 5 x 10
6 cells/mouse in 0.2 mL cell suspension. NSG mice containing subcutaneously implanted TM00298 PDX cells were obtained from Jackson Laboratories. For CTG-2440 studies (Champions Oncology), Attorney Docket No. AMB1013WOPCT1 NOG mice were subcutaneously implanted with fragments derived from 1000-1500mm
3 donor mouse tumors. [268] Twice weekly, body weight was recorded, and both tumor length and width were measured by electronic calipers. Tumor volume (TV) was calculated as LxWxWx0.5 (wherein L is tumor length and W is tumor width). When tumor volumes equaled 150-500 mm
3, mice were randomized into approximately equal TV average groups, and dosed intravenously at 10 mL/kg with vehicle or the indicated test articles. Enzalutamide was administered orally at 10 mg/kg in 1% carboxymethyl cellulose and 0.1% Tween-80. Percent tumor growth Inhibition (%TGI) was calculated as follows: 100 x (1 – [tumor volume or wet weight of treatment group / tumor volume or wet weight of control group]). [269] In these studies, ARX517 did not induce severe body weight loss, mortality or morbidity. [270] Enzalutamide-sensitive MDA-PCa-2b tumor model. [271] MDA-Pca-2b cells were implanted subcutaneously into the flank of male nude mice (n=10/group). When tumors reached 100-200mm
3, mice were given a single intravenous dose of test article at the dose indicated in FIG.5 (dotted line, day 14). %TGI was calculated based on tumor wet weights at day 45 after cell implantation. Statistical analysis: * = p < 0.05 and ** = p < 0.01 for tumor wet weights on day 45, calculated using non-parametric Mann Whitney ttest. The single intravenous ARX517 administration demonstrated robustTGI up to 83% (at the 5 and 10 mg/kg doses), whereas 10 mg/kg unconjugated mAb or isotype control ADC (containing AS269) did not inhibit tumor growth (FIG.5; graph shows mean tumor volumes ± SEM over time). [272] Enzalutamide-sensitive TM00298 prostate cancer PDX model. [273] NSG mice were subcutaneously implanted with TM00298 patient-derived tumor cells. When tumors reached 100-200mm
3, mice were dosed intravenously once weekly with the test Attorney Docket No. AMB1013WOPCT1 articles indicated in FIG. 6A; dotted lines show ARX517 doses). %TGI was calculated based on tumor volume measurements at study end. Statistical analysis: * = p < 0.05, ** = p < 0.01, and *** = p < 0.001; calculated using non-parametric Mann Whitney t-test and final tumor volumes. The once weekly ARX517 administration inhibited tumor growth in a dose- dependent manner (up to 66% in the 3 mg/kg group, p<0.0001 vs. vehicle), and daily enzalutamide administration (10 mg/kg) induced 41% TGI (p<0.01). In this TM00298 model, ARX517 and enzalutamide combination resulted in a TGI of 85% (p<0.0001) (FIG.6A; graph shows mean tumor volumes ± SEM over time). [274] CTG-2440 PDX prostate cancer model. [275] NOG mice (n=10/group) were subcutaneously implanted with CTG-2440 tumor fragments derived from 1000-1500 mm
3 stock mouse tumors. Mice were dosed intravenously once weekly with the test articles indicated in FIG. 6B; dotted lines show ARX517 doses). %TGI was calculated based on tumor volume measurements at study end. Statistical analysis: * = p<0.05, ** = p< 0.01, and *** = p<0.001; calculated using one-way Analysis of Variance (ANOVA) followed by Tukey’s multiple comparisons test. The once weekly administration of ARX517 as a single agent inhibited tumor growth in a dose dependent manner up to 92%. At 3 mg/kg in this model, ARX517 induced more robust TGI than 10 mg/kg enzalutamide (92% vs.38%, respectively) (FIG.6B; graph shows mean tumor volumes ± SEM over time). [276] Most prostate cancer patients eventually progress to androgen deprivation therapy (ADT)-resistant disease. ARX517 was evaluated in the enzalutamide-resistant C4-2 CDX model. Nude mice (n=5-10/group) were subcutaneously implanted with C4-2 tumor cells in matrigel. When tumors reached 200-500 mm
3, mice were dosed intravenously with test articles at doses indicated in FIGS. 7A and 7B (dotted lines show ARX517 doses; graphs show mean tumor volumes ± SEM over time). %TGI was calculated based on tumor volume measurements at study end. Statistical analysis: ** = p< 0.01 and *** = p<0.001 calculated using a two-way Attorney Docket No. AMB1013WOPCT1 ANOVA with repeated measures and Tukey’s post-test. ARX517 promoted C4-2 TGI after a single dose as low as 1 mg/kg, and progression free disease was observed after a single dose of 5.0 mg/kg (FIG. 7A). Once weekly ARX517 administration resulted in 37% (1mg/kg) to 79% (3 mg/kg) TGI in the C4-2 model. Consistent with C4-2 enzalutamide-resistance, combination of ARX517 and enzalutamide (10 mg/kg) did not lead to additive TGI (FIG.7B). [277] Example 12: Pharmacokinetic and Toxicology Studies of ARX517 in Rat and Cynomolgus Monkey. [278] Rat Studies. ARX517 was evaluated in a single-dose Good Laboratory Practice (GLP) toxicity study in Sprague-Dawley rats. Male and female Sprague Dawley rats (7-10 weeks old) were dosed with vehicle or ARX517 (20, 40, or 60 mg/kg) via intravenous infusion over approximately 20 minutes at a dose volume of 15 mL/kg. Following test article administration, rats were observed for 28 days and necropsied on Day 29. Toxicological observations/evaluations were all performed using standard methodology, and included: mortality, clinical observations, injection site observation, body weight, food consumption, safety pharmacology examinations (functional observational battery test and respiration examination), clinical pathology (hematology, coagulation, serum chemistry, and urinalysis), toxicokinetics, gross observations, terminal necropsy organ weights, and histopathological evaluation. At various timepoints after dosing, jugular vein blood samples were collected for clinical pathology and toxicokinetic analysis. For histopathology, all major tissues were trimmed and fixed in 10% neutral buffered formalin except for eyes with optic nerves, testes, and epididymides, which were fixed in Modified Davidson’s solution for 24 to 72 hours. Preserved tissues were embedded in paraffin, sectioned, stained with hematoxylin and eosin, and examined microscopically by a licensed pathologist. [279] Because ARX517 does not bind rodent PSMA (see Example 8, Table 5), the rat study findings reveal ARX517’s acute target-independent toxicologic profile. Administration of a Attorney Docket No. AMB1013WOPCT1 single ARX517 dose to rats over a 20-minute intravenous infusion at 20, 40, or 60 mg/kg did not result in treatment-related mortality, moribundity, abnormalities in behavioral testing, or respiratory findings. At day 4 post-infusion, clinical pathology findings included increased neutrophil and monocyte counts, reduced lymphocyte counts, decreased red blood cell parameters (RBC/HGB/HCT), and changes in hepatic function (increased AST/ALT/ALP/TBIL) and renal function (increased CRE) at doses ≥ 40 mg/kg. Both sexes had significant test article-related weight increases in liver, spleen, and lung. At the end of a 28-day observation period, ARX517-related histopathologic adverse findings were observed in male reproductive organs and the lungs of both sexes at doses ≥ 20 mg/kg, and the ARX517 maximum tolerated dose (MTD) was 60 mg/kg. [280] Cynomolgus Monkey Studies. ARX517 was evaluated in a GLP repeat-dose toxicity study in cynomolgus monkeys. Male and female cynomolgus monkeys (2.5-3.5 years old, n=6/sex/group) were dosed twice, 3 weeks apart, with vehicle or ARX517 (1, 6, or 9 mg/kg) via intravenous infusion over approximately 20 minutes at a dose volume of 5 mL/kg. Four monkeys/sex in each group were euthanized and necropsied one week after the final ARX517 dose, and the remaining two animals/sex (recovery group) were observed for another six weeks, followed by terminal necropsy. Toxicological observations/evaluations were all performed using standard methodology, and included: mortality, clinical observations, injection site observation, body weight, food consumption, safety pharmacology examinations (functional observational battery test and respiration examination), clinical pathology (hematology, coagulation, serum chemistry, and urinalysis), toxicokinetics, gross observations, terminal necropsy organ weights, and histopathological evaluation. At various timepoints after dosing, jugular vein blood samples were collected for clinical pathology and toxicokinetic analysis. For histopathology, all major tissues were trimmed and fixed in 10% neutral buffered formalin except for eyes with optic nerves, testes, and epididymides were fixed in Modified Davidson’s Attorney Docket No. AMB1013WOPCT1 solution for 24 to 72 hours. Preserved tissues were embedded in paraffin, sectioned, stained with hematoxylin and eosin, and examined microscopically by a licensed pathologist. [281] To determine serum concentrations of ARX517 Total Antibody (TA), ARX517 ADC and free payload pAF-AS269, blood samples (n=6/sex/group) were collected following ARX517 dosing from all available study animals at 0 (pre-dose), 0.5, 4, 8, 24, 72, 120, 168, 336 and 504 hours after the first ARX517 dose, and at 0.5, 4, 8, 24, 72, 120 and 168 hours (dosing phase), and 336, 504, 672, 840 and 1176 hours (recovery phase) after the second ARX517 dose. Blood samples were collected into tubes without anticoagulant, allowed to clot at room temperature for at least 30 minutes, and centrifuged at 3200 x g for 15 minutes at 4°C. Serum was transferred into aliquots and stored in a freezer set to ≤ -65 °C. [282] The samples were analyzed in validated PK assays. The TA PK assay in cynomolgus monkey serum used the same capture and detection reagents as the TA assay in mouse matrix (see Example 9). The ARX517 ADC PK assay in cynomolgus serum was similar to the Intact ADC assay in mouse matrix (Example 9), except that MSD High Bind plates were coated with rhPSMA. The lower limit of quantification (LLOQ) of the TA and ADC assays in cynomolgus serum was 78.1 ng/mL and 19.5 ng/mL, respectively. [283] To extract pAF-AS269 from 30 µL of study samples, STDs, and QCs, a protein precipitation step was performed with 200 ng/mL Internal Standard (Diclofenac) and 190 µL of 0.1% Formic Acid in Acetonitrile in a 96-well deep-well plate. Samples, STDs, and QCs were vortexed for 10 minutes, centrifuged at 3220 x g for 15 minutes at 4 °C, and 100 µL supernatant was transferred to a new 96-well deep-well plate. 100 µL deionized water was added to the transferred supernatants, vortexed for 5 minutes, and centrifuged at 3220g for 5 minutes at 4 °C. 10 µL of the extracted samples was injected into an LC-MS/MS for chromatographic separation and detection with the positive ion ESI mode. The LLOQ of the pAF-AS269 assay in cynomolgus serum was 0.2 ng/mL. Attorney Docket No. AMB1013WOPCT1 [284] Because ARX517 binds to cynomolgus monkey and human PSMA with similar affinity (see Example 8 and Table 5), we evaluated ARX517 on-target and off-target toxicologic profiles in cynomolgus monkeys following two intravenous administrations of 1, 6, or 9 mg/kg given 3 weeks apart. In this study, the ARX517 no-observed-adverse-effect level (NOAEL) was 1 mg/kg/dose, and the highest non-severely toxic dose (HNSTD) was 6 mg/kg/dose for both male and female monkeys. At day 6 following the second ARX517 dose, clinical pathology findings were all considered non-adverse, were only observed at doses ≥ 6 mg/kg, and included: increased monocyte counts, reduced platelet (PLT) counts, increased coagulation time (APTT), and increased hepatocellular enzymes (AST/ALP/GGT). At the HNSTD, although histopathologic target organs included liver, spleen, and thymus, all adverse findings had normalized and were reduced in incidence/severity at the end of the 6-week recovery period. At the 9 mg/kg dose (above the HNSTD), additional target organs were kidney and lung. No ophthalmologic, respiratory, urinary, cardiovascular, or neurologic findings were observed at any dose. [285] The ARX517 toxicokinetic profile in the cynomolgus repeat-dose study showed stability of ARX517 in the systemic circulation, with overlapping TA and ARX517 ADC concentration-time curves at all doses (FIG. 8A; shown are mean ± SD from n=12 monkeys/group). TA versus ADC exposure was similar with mean maximum observed concentration (C
max) and AUC
last ratios within 0.77 to 1.07 (Table 8). Table 8: ARX517 Toxicokinetic Parameters in Cynomolgus Monkey Repeat-Dose Tox Study ADC ADC/TA C /TA Dose
max Tmax AUC0-last
t1/2 AUC0-168h Cmax AUC (mg/kg)
Sex Analyte last (
ng/mL) (h)* (h*ng/mL) (h)
(h*ng/mL) Ratio Ratio TA 29,300 0.5 3,110,000 108 2,480,000 NA NA Male ADC 24,900 0.5 2,870,000 151 2,130,000 0.85 0.92 1 TA 27,500 0.5 3,300,000 132 2,570,000 NA NA Female ADC 23,800 0.5 3,300,000 135 2,350,000 0.87 1.00 TA 177,000 0.5 34,600,000 265 19,800,000 NA NA Male ADC 175,000 0.5 36,500,000 324 18,800,000 0.99 1.05 6 TA 168,000 0.5 33,500,000 378 18,000,000 NA NA Female ADC 152,000 0.5 35,100,000 315 16,500,000 0.90 1.05 Attorney Docket No. AMB1013WOPCT1 TA 257,000 0.5 41,400,000 250 27,100,000 NA NA Male ADC 266,000 0.5 44,100,000 284 29,600,000 1.04 1.07 9 TA 316,000 0.5 54,100,000 320 33,700,000 NA NA Female ADC 243,000 0.5 47,000,000 308 26,500,000 0.77 0.87 *Median value for Tmax [286] After administration of the second dose of ARX517, free payload pAF-AS269 that is released from ARX517 was not quantifiable for all animals in the 1 mg/kg dose group and for 3 animals in the 6 mg/kg dose group. The remaining animals in the 6 mg/kg or 9 mg/kg dose groups showed pAF-AS269 appeared slowly in circulation with a small number of quantifiable time points at very low concentrations (< 0.5 ng/mL) near the assay LLOQ (FIG. 8B; shown are mean ± SD for pAF-AS269). These low concentrations of pAF-AS269 are > 200-fold lower than the observed in vitro activity of pAF-AS269 (IC50 >100 nM, or 111 ng/mL; data not shown). Internalization and intracellular proteolytic degradation of ARX517 is required for release of pAF-AS269 from ARX517 as a free payload (or metabolite; see structure at FIG. 8C), which correlates with the slow appearance of free pAF-AS269 (Cmax observed 96 to 120 hours after dosing ARX517) in circulation. [287] At the HNSTD of 6 mg/kg in male monkeys, the ARX517 AUC
last, C
max, and t
1/2 after the second dose were 36,500,000 h*ng/mL, 175,000 ng/mL and 324 hours (about 13.5 days), respectively (Table 8). At the pharmacologically active dose of 5 mg/kg in C4-2 tumor bearing mice (see Example 9), ARX517 mean AUC
last, C
max, and t
1/2 were 7,570,000 h*ng/mL, 57,100 ng/mL, and 237 hours, respectively (Table 6 and FIG.4B). Comparison of ARX517 exposure at the 5 mg/kg dose evaluated in PK studies, which is higher than the pharmacologically active dose of 3 mg/kg observed in multiple models, versus exposure at the HNSTD in monkey toxicology studies shows a clear therapeutic index of more than 5-fold for AUClast and 3-fold for Cmax (FIG.9). Attorney Docket No. AMB1013WOPCT1 [288] Taken together, the pharmacologic, toxicologic, and pharmacokinetic profiles suggest that ARX517 is well-tolerated at exposures much higher than those required for efficacy, providing rationale for clinical investigation as a potential mCRPC treatment option. [289] Example 13: A Phase 1/2, Multicenter, Open-label, Dose-escalation, and Dose- expansion Study to Evaluate the Safety, Pharmacokinetics, and Anti-tumor Activity of ARX517. [290] With Randomized Comparison to Investigator's Choice of Treatment, in Subjects With Metastatic Castration-resistant Prostate Cancer Who Are Resistant or Refractory to Prior Standard Therapies. [291] ARX517 (FIG. 10) is currently being evaluated in a multi-center Phase1 clinical trial in the US (ARX517-2011 (APEX-01); NCT04662580 (see, on the World Wide Web, clinicaltrials.gov/study/NCT04662580?term=NCT04662580&rank=1). Briefly, the APEX-01 trial is a Phase 1/2, multicenter, open-label, dose-escalation, and dose-expansion study to evaluate the safety, pharmacokinetics (PK) and anti-tumor activity of ARX517, with randomized comparison to investigator's choice of treatment (ICT), in subjects with metastatic castration-resistant prostate cancer (mCRPC) who are resistant or refractory to prior standard therapies. [292] This first-in-human, Phase 1/2, multicenter, open-label study is to evaluate the safety, PK, pharmacodynamics (PDy) and preliminary anti-tumor activity of ARX517 in subjects with mCRPC who are resistant or refractory to standard therapies. Phase 1a and 1b dose-escalation and dose expansion stages were designed to identify the MTD and/or RP2D(s). Phase 2 will randomize subjects to receive ARX517 at the RP2D(s) or ICT as comparator. The ICT to be used in Phase 2 will be determined after reviewing all available clinical data in Phase 1. [293] Primary Objectives: Safety, tolerability, maximum tolerated dose (MTD), recommended Phase 2 Dose(s) (RP2D(s)). Attorney Docket No. AMB1013WOPCT1 [294] Secondary Objectives: PK, immunogenicity, preliminary anti-tumor activity (RECIST 1.1, prostate specific antigen (PSA) response [PSA30, PSA50, PSA90], PCWG3. [295] Key Eligibility Criteria. Key inclusion criteria include: male subjects ≥ 18 years at the first time of providing written informed consent; histologically confirmed prostate adenocarcinoma; documented metastatic disease; castration-resistant prostate cancer, per the Prostate Cancer Working Group 3 (PCWG3); ongoing therapy with (and willing to continue with) a gonadotropin-releasing hormone agonist or antagonist (unless prior orchiectomy) and serum testosterone level < 50 ng/dL at Screening; prior treatment with at least two FDA- approved therapies for metastatic prostate cancer with at least one being an androgen receptor signaling inhibitor; adequate blood counts; disease progression by PSA, RECIST 1.1 or new bone lesions; Eastern Cooperative Oncology Group (ECOG) performance status ≤ 1. [296] Key Exclusion Criteria. Key exclusion criteria include: Subjects having central nervous system (CNS) metastasis, unless the CNS metastasis was treated with local therapy and has proven to be stable over the last 2 months prior to the enrollment date, and not currently requiring ongoing systemic steroid treatment; history of any invasive malignancy (other than primary) within previous 2 years prior to the enrollment date that requires active therapy; marked baseline prolongation of QT/QT interval corrected for heart rate (QTc), e.g., a triplicate average QTc interval > 480 milliseconds (CTCAE Grade 1) using Fridericia's QT correction formula; prior history of interstitial lung disease, pneumonitis, or other clinically significant lung disease within 12 months prior to enrollment date; clinically significant ocular findings by a qualified ophthalmologist or optometrist including active ocular infections or chronic corneal disorders;, ongoing use of bisphosphonate or denosumab therapy, estrogen therapy, systemic glucocorticoids (> 10 mg prednisone), finasteride/dutasteride. [297] Design Details. Primary Purpose: Treatment. Interventional Model: Single Group Assignment. Interventional Model Description: This is a first-in-human, Phase 1/2, Attorney Docket No. AMB1013WOPCT1 multicenter, open-label study to evaluate the safety, PK, PDy, and preliminary anti-tumor activity of ARX517 in subjects with mCRPC who are resistant or refractory to standard therapies. Phase 1a and 1b dose-escalation and dose expansion stages will identify the MTD and/or RP2D(s). Phase 2 will randomize subjects to receive ARX517 at the RP2D(s) or ICT as comparator. The ICT to be used in Phase 2 will be determined after reviewing all available clinical data in Phase 1. Masking: None (Open Label). [298] Participant Group/Arm: Experimental: ARX517 (Phase 1 Dose Escalation and Expansion). During the Dose Escalation period of the study, subjects are enrolled in cohorts administered ascending dose levels of ARX517 via intravenous (IV) infusion every 3, 4 or 6 weeks. During the Dose Expansion period of the study, subjects receive dose levels and dose intervals at or below the MTD, identified as putative RP2D(s). [299] Intervention/Treatment: Drug: ARX517. ARX517 is an ADC consisting of a humanized anti-PSMA monoclonal antibody (mAb) (IgG1κ) covalently conjugated to two (2) proprietary microtubule-disrupting toxins referred to as AS269. [300] Primary Outcome Measures. Phase 1: Assess incidence of adverse events. Incidence and severity of adverse events or serious adverse events of ARX517 are assessed to determine the safety and tolerability of the treatment using National Cancer Institute (NCI) Common Terminology Criteria for Adverse Events version 5 (CTCAE) (Time Frame: 1.5 years). [301] Secondary Outcome Measures. (A) Phase 1: Area under the serum concentration-time curve (AUC) for ARX517. Pharmacokinetic parameter area under the serum concentration- time curve (AUC) is analyzed through different analytes such as ADC, total antibody and pAF- AS269 (Time Frame: 3 years). (B) Phase 1: Maximum serum concentration (Cmax) for ARX517. Pharmacokinetic parameter maximum serum concentration (Cmax) will be analyzed through different analytes such as, ADC, total antibody and pAF-AS269 (Time Frame: 3 years). (C) Phase 1: Trough concentration (Ctrough) for ARX517. Pharmacokinetic parameter Attorney Docket No. AMB1013WOPCT1 trough concentration (Ctrough) will be analyzed through different analytes such as, ADC, total antibody and pAF-AS269 (Time Frame: 3 years). (D) Phase 1: Incidence of ADA against ARX517. Assess the incidence of anti-drug antibodies (ADA) against ARX517 at selected timepoints (Time Frame: 3 years). (E) Overall survival. Overall survival (OS) is defined as the time from first dose of study therapy to the date of death (any cause). Subjects who are alive are censored at the last known time that the subject was alive (Time Frame: 3 years). (F) Assess changes in serum prostate specific antigen (PSA) levels. Proportion of subjects who show a confirmed reduction of 30% and 50% from baseline in serum prostate specific antigen (PSA) levels (PSA30, PSA50; Time Frame: 3 years). (G) Progression-free survival (PFS). PFS, defined as the time between date of first dose of study therapy and date of progression or death, whichever occurs first, is computed for response evaluable subjects. Subjects are censored at time of subsequent therapy (Time Frame: 3 years). [302] Other Outcome Measures. (A) Phase 1: Evaluate biomarkers. Evaluate exploratory blood- and tissue-based biomarkers related to study drug response (Time Frame: 3 years). (B) Phase 1: Evaluate PSMA expression. Evaluate baseline PSMA expression (Time Frame: 3 years). (C) Phase 1: Assess changes in Brief Pain Inventory. A Brief Pain Inventory questionnaire is utilized to assess subject quality of life. Higher scores mean a worse outcome (Time Frame: 3 years). (D) Phase 1: Assess changes in Functional Assessment of Cancer Therapy for Patients with Prostate Cancer (FACIT-P). A FACIT-P questionnaire is utilized to assess subject quality of life. Higher scores mean a worse outcome (Time Frame: 3 years). [303] Study Design. This study includes two parts: a dose-escalation part and a dose- expansion part. [304] Dose escalation. The dose-escalation part of the study consists of ascending dose levels of ARX517 administered as a single agent to determine MTD and RP2D. The subject will be Attorney Docket No. AMB1013WOPCT1 enrolled using the i3+3 design (Liu et al., 2019, J Biopharm Stat. 2019:1-11) with a starting dose of 0.32 mg/kg. [305] Dose expansion. The dose expansion part of the study will evaluate the putative RP2D(s). Patients in dose escalation enroll using the i3+3 design. If no DLTs are observed within the 21-day DLT period, the subsequent dose cohort may enroll upon Safety Monitoring Committee’s (SMC) review of available safety and tolerability data. This study began by evaluating 6 dose levels of ARX517 Q3W following a modified Fibonacci scheme as shown in Table 9. Table 9. Dose levels of ARX517 Q3W following a modified Fibonacci scheme. [306] Additional cohorts were added following the start of the clinical trial with a dose level increment ≤ 20% of the highest dose level in the subsequent cohort upon SMC’s recommendation. The study enrolled patients in Cohort 7 at a dose level of 2.4 mg/kg, Q3W, in Cohort 8 at a dose level of 2.88 mg/kg, Q3W, in Cohort 9 at a dose level of 3.4 mg/kg, Q3W, and in Cohort 10 at a dose level of 4.5 mg/kg, Q3W. (FIG. 12). Data presented herein was available at the time of the present filing through cohort 8. [307] Preliminary Results for Patients in Cohorts 1 to 6 of APEX-01 Clinical Trial. In the Phase 1 dose-escalation portion of the study, ascending dose levels of ARX517 is administered as a single agent every 3 weeks. The primary endpoints are safety, tolerability and pharmacokinetics. The key secondary endpoint is objective decline of prostate-specific antigen (PSA) from baseline and/or tumor shrinkage. PSA is a protein produced by the prostate gland Attorney Docket No. AMB1013WOPCT1 and is commonly used as a biomarker to diagnose and follow prostate cancer. A ≥ 50% reduction in PSA levels from baseline is considered clinically relevant and has been shown to correlate with improved overall survival in prostate cancer. A greater than 50% reduction in PSA levels in three of three patients at the 2.0 mg/kg dose level in patients with prostate cancer was observed. Two patients in this cohort had a reduction in PSA > 90%. Additionally, one of these three patients had soft tissue measurable disease and experienced a partial response at the first on-treatment scan. [308] Results for Patients in Cohorts 1 to 8 of APEX-01 Clinical Trial. The i3+3 dose escalation design was used. Eligible patients had ≥ 2 FDA-approved treatments for mCRPC with progression by Prostate Cancer Working Group criteria. Key objectives include safety, PK and clinical efficacy. Baseline PSMA PET expression was not required for eligibility but was evaluated as a biomarker. [309] Twenty-four (24) patients received ARX517 Q3W via intravenous infusion at escalating doses (Table 10). Patients had a median of 4 prior lines of therapy; 100% received ≥ 1 androgen pathway inhibitor, 50% received taxane, and 12.5% received PSMA-targeted radionuclide. Grade 1/2 treatment-related adverse events (TRAEs) were dry mouth (41.7%), fatigue (33.3%), diarrhea and platelet count decrease (20.8% each). Four Grade 3 TRAEs were reported at 1.7, 2.4, and 2.88 mg/kg (two lymphopenia, two platelet count decrease). No dose limiting toxicities DLTs, treatment-related serious adverse events (SAEs), or ≥ Grade 4 adverse events were reported. At higher doses (Cohorts 4-8), median duration on-treatment was 6.3 months (range 0.7+,11.8+). Additionally, > 50% PSA and circulating tumor DNA (ctDNA) decline were observed (Table 10); 2/7 pts had confirmed RECIST v1.1 responses. PK profiles for total antibody and ADC were similar, suggesting strong stability of ARX517 with minimal premature free payload release. Attorney Docket No. AMB1013WOPCT1 Table 10. PSA and ctDNA decline, confirmed responses, DLTs and Grade 3/4/5 TRAEs by ARX517 dose level. Best % Cohort Cohort Cohort Cohort Cohort Cohort Cohort Cohort change 1 (0.32 2 (0.64 3 (1.07 4 (1.4 5 (1.7 6 (2.0 7 (2.4 8 (2.88 from mg/kg) mg/kg) mg/kg) mg/kg) mg/kg) mg/kg) mg/kg) mg/kg baseline* PSA > 0/1 1/3 1/3 2/3 2/5 3/3 2/3 2/3 30% PSA > 0/1 0/3 0/3 1/3 0/5 3/3 2/3 2/3 50% PSA > 0/1 0/3 0/3 0/3 0/5 2/3 0/3 0/3 90% ctDNA > 0/1 1/2 2/3 1/3 3/5 3/3 2/2 n/d 50% Confirmed RECIST 0/1 0/1 0/0 1/2 0/3 1/1 0/1 n/d v1.1 response DLT 0/3 0/3 0/3 0/3 0/5 0/3 0/3 0/3 Grade 3 0/3 0/3 0/3 0/3 1/5 0/3 1/3 2/3 TRAE Grade 4/5 0/3 0/3 0/3 0/3 0/5 0/3 0/3 0/3 TRAE *n/d = no data available. [310] On treatment changes in ctDNA have been shown to predict time to progression and survival (see Tolmeijer SH et al. Clin Cancer Res. 2023 Aug 1;29(15):2835-2844, doi: 10.1158/1078-0432.CCR-22-2998; Sartor O. Clin Cancer Res.2023 Aug 1;29(15):2745-2747, doi: 10.1158/1078-0432.CCR-23-1043). Serial plasma samples were collected at baseline, C3D1, C4D1 and EOT, best percent change from baseline is shown. ctDNA was measured using GuardantINFINITY test (Guardant Health) with a specificity of 96.9%, a sensitivity of 91.3% and a reported lower limit of detection 0.06%. Samples were processed after passing multiple quality control measurements encompassing DNA yield, GC bias, methylation bias, diversity, and contamination checks. ctDNA changes compared with its baseline level were measured based on aggregated tumor-specific methylation signal scores. Reductions of greater than 50% ctDNA were observed in 81% (17/21) of patients (cohorts 4 to 8); see FIG.19. Attorney Docket No. AMB1013WOPCT1 [311] ARX517 treatment resulted in PSA declines and RECIST v1.1 responses without treatment-related SAEs. Dose expansion to 3.4 mg/kg (cohort 9) and 4.5 mg/kg (cohort 10) has begun (FIG.12, cohort 10 not shown in figure). [312] Pharmacokinetics Results for Patients in Cohorts 1 to 7 of APEX-01 Clinical Trial. [313] As disclosed herein, ARX517 is an anti-PSMA ADC containing anti-PSMA mAb conjugated to AS269 at a drug-to-antibody ratio (DAR) of two (2). The ARX517 design addresses instability issues encountered by previous PSMA-targeted ADCs via three key components: a non-cleavable PEG linker, a non-cell permeable payload, and stable oxime conjugation chemistry. The site-specific conjugation of AS269 to the mAb is enabled using synthetic amino acid (pAF) incorporation. This ADC design minimizes premature free payload release and off-target toxicity. [314] Twenty-one (21) patients in Cohorts 1 to 7 received ARX517 Q3W via intravenous infusion at doses ranging from 0.32 to 2.4 mg/kg. Patient serum samples were collected at fixed time points and evaluated in validated Total Antibody (TA; sum of deconjugated antibody and conjugated antibody), ADC (conjugated antibody with a DAR of 1 or 2), and free payload pAF- AS269 assays. The ARX517 TA, ADC and pAF-AS269 PK assays in patient serum samples were similar to the TA and Intact ADC in mouse serum (see Example 9), and the pAF-AS269 assay in cyno serum (see Example 12), except for the following parameters: (i) for TA assay, STD/QC/sample predilution step was 1:100; (ii) for ADC assay, MSD High Bind plates were coated with rhPSMA and STD/QC/sample predilution step was 1:20; (iii) for pAF-AS269 assay, Dexamethasone-d4 was used as the internal standard and 100 µL STD/QC/sample were transferred for the protein precipitation step. The lower limit of quantitation (LLOQ) for the TA, ADC, and pAF-AS269 assays in human serum were 62.5 ng/mL, 7.8 ng/mL, and 0.02 ng/mL, respectively. PK parameters were determined using noncompartmental analysis based on serum concentrations of TA, ADC, and pAF-AS269. Attorney Docket No. AMB1013WOPCT1 [315] ARX517 exhibited virtually overlapping TA and ADC PK concentration-time curves at all dose levels tested, indicating strong stability of the ADC with minimal premature free payload release. A long ADC terminal half-life of ~6–10 days was observed at doses ≥ 1.4 mg/kg, thereby maximizing drug exposure over a dosing cycle of 3 weeks. Low concentrations of pAF-AS269 (approximately 0.02–0.2 ng/mL) were observed at all dose levels and appeared slowly in the circulation, with Cmax observed approximately 7 days after administration. This contrasts with other ADCs that typically exhibit free payload Cmax hours to days after dosing. [316] ARX517 is the first anti-PSMA ADC tested in the clinic to demonstrate notable stability and a long terminal half-life. These attributes enable continuous drug delivery throughout the dosing cycle to potentially improve efficacy and minimize toxicity due to premature free payload release, indicating a clear and favorable therapeutic index. [317] The U.S. Food and Drug Administration (FDA) has granted Fast Track designation to anti-PSMA antibody-drug conjugate (ADC) investigational therapy, ARX517, for the treatment of patients with metastatic castration-resistant prostate cancer (mCRPC) upon progression on an androgen receptor pathway inhibitor. [318] Example 14: ARX517 Solution for Intravenous Infusion. [319] ARX517 ADC formulation development included chemical and physical characterization of ARX517 (e.g., amino acid sequence, charge, isoelectric point (pI), molecular weight, drug-to-antibody ratio (DAR), size and charge variants), solubility, excipient screening (e.g., excipient identities and concentrations, buffer pH, identity and concentration) and stability studies, and consideration of the ability to minimize manufacturing processing steps between bulk drug substance (ARX517 ADC) and formulated drug product. The lead clinical formulation was selected based on outcomes of evaluations of the foregoing, including solubility (protein recovery), chemical and physical stability criteria using high performance liquid chromatography (HPLC) analysis and capillary electrophoresis. Attorney Docket No. AMB1013WOPCT1 [320] For example, stability testing of ARX517 formulations containing succinate buffers (pH 5.5 or 5.0) versus histidine buffers (pH 5.5, 6.0 and 6.5) at 40 °C after four weeks, and sample analysis via size exclusion chromatography (SEC) for % monomer (% main peak), % high molecular weight (HMW; e.g., containing aggregates) and % low molecular weight (LMW) species, showed that formulations containing succinate buffer had lower % monomer species when compared to formulations containing histidine buffer. Notably, formulations containing succinate buffer had markedly increased high molecular weight species (0.91% to 1.69%) compared to formulations containing histidine buffer (0.52% to 0.55%), indicating that formulations containing succinate buffer had a higher tendency towards aggregation. Based on this analysis, the formulation containing succinate buffer at pH 5.5 was the least stable, followed by the formulation containing the succinate buffer at pH 5.0, while formulation containing histidine buffer at pH 5.5 contained higher % monomer species than formulation containing succinate buffer at the same pH (Table 11). Table 11. ARX517 Formulation Main Peak Purity and Impurities by SEC-HPLC after 4 weeks at 40°C [321] Samples stored at 40 °C and analyzed via reduced capillary electrophoresis sodium dodecyl sulfate (CE-SDS), which measures sample purity as the sum of % heavy chain (HC) and % light chain (LC) under reducing conditions, also indicated that the histidine formulations showed better stability profiles (e.g., higher % intact species and lower % other impurities) Attorney Docket No. AMB1013WOPCT1 than succinate formulations; however, among the histidine formulations, the pH 6.5 histidine buffer formulation showed increased amounts of % other impurities (data not shown). [322] Imaged capillary isoelectric focusing (icIEF) analysis of the formulations held at 40°C for 4 weeks showed an increase in acidic charged species, with a corresponding decrease in main peak purity. Based on this analysis, ARX517 formulations contained acidic species in the following amounts: 52.5% and 48.2% (succinate buffer, pH 5.5 and 5.0, respectively); 48.5% acidic (histidine buffer pH 6.5); and 39.4% and 41.6% (histidine buffer, pH 5.5 and 6.0, respectively). [323] Formulation samples stored at 40°C also were analyzed via hydrophobic interaction chromatography (HIC) for drug-to-antibody ratio (DAR); the formulation containing succinate buffer pH 5.0 showed a decrease in DAR while higher pH formulations were more stable under these conditions (Table 12). Table 12. ARX517 formulation DAR value by HIC-HPLC after 4 weeks at 40°C [324] The final formulation selection was based on balancing the impurity profiles from SEC- HPLC, reduced capillary electrophoresis sodium dodecyl sulfate CE-SDS, imaged capillary isoelectric focusing (icIEF) and DAR evaluation by hydrophobic interaction chromatography (HIC)-HPLC. A confirmatory stability study was conducted in Type I borosilicate glass vials with chlorobutyl rubber stopper and aluminum seal in histidine buffer at pH 5.6, 5.9, and 6.2 to determine the range and robustness of the selected clinical formulation. Attorney Docket No. AMB1013WOPCT1 [325] The clinical formulation for IV administration, ARX517 Solution for IV Injection (hereinafter, ARX517 Drug Product), contains ARX517 ADC formulated at a concentration of 10 mg/mL in a solution of histidine buffer (L-histidine, L-histidine hydrochloride) at a concentration of 20 mM, sucrose at a concentration of 9% (w/v) and polysorbate 80 at a concentration of 0.01% (w/v); the final solution pH is 5.9 ± 0.3. The excipients comply with the requirements of the applicable compendial monographs (Ph. Eur., USP/NF, JP, ChP). [326] ARX517 Drug Product, manufactured under current Good Manufacturing Practices (cGMP), was evaluated in accordance with International Conference on Harmonisation (ICH) guidelines for long term stability at -20 °C ± 5 °C, accelerated stability at 5 °C ± 3 °C and under stressed conditions of 25 °C/60% relative humidity (RH). Stability acceptance criteria included appearance, pH, content (protein concentration), potency (binding ELISA and cell-based assays) and purity (including unconjugated mAb, DAR, CE-SDS, size exclusion chromatography (SEC), and cation ion exchange chromatography (CEX; see Example 17)) measures, and safety measures (endotoxin, sterility and particulates). [327] Long-Term Stability. ARX517 Drug Product exhibited the following characteristics under the long-term stability conditions of -20 °C ± 5 °C (stored upright) over a period of 36 months, with all tests performed at zero and 36 months, and most tests additionally performed at 1, 3, 6, 9, 12, 18 and 24 months: the appearance was a colorless to light yellow and clear to opalescent liquid, essentially free of visible particles; the pH was within a range of 5.6 to 6.2 (specifically, 5.8 to 5.9); particulates ≥ 25 µm were less than 20/vial, and ≥ 10 µm were less than 100/vial; content (protein concentration) was within a range of 9.0 to 11.0 mg/mL (specifically, 10.1 to 10.7 mg/mL); cell-based and binding ELISA assay results were each > 90% of reference standard at all time points; purity based on CEX that measured % main peak, % acidic peak and % basic peak indicated ≥ 40.0% main peak, ≤ 40.0% acidic peak and ≤ 30.0% basic peak over the duration of the study (specifically, 51.9% to 53.8% main peak, Attorney Docket No. AMB1013WOPCT1 31.8% to 34.0% acidic species peak, and 13.8% to 14.3% basic species peak); purity based on SEC that measured % monomer (% main peak), % high molecular weight (HMW) and % low molecular weight (LMW) species indicated high purity and minimal aggregation (98.0% to 98.3% main peak, 1.7% to 1.9% HMW species and 0.1% to 0.2% LMW species) over the duration of the study; endotoxins were < 0.05 endotoxin units (EU)/mg, and no growth was observed, indicating that the drug product remained sterile; drug-to-antibody ratio (DAR) as measured by hydrophobic interaction chromatography (HIC) was within the range of 1.9 to 2.1 (specifically, 2.1 over all measured time points, including zero and 36 months), and unconjugated mAb (also measured by HIC) was 0.2% at each measured time point, including zero and 36 months; and free drug related impurities were less than 0.10% (w/w). Based on these and other test results, ARX517 Drug Product was found to be stable for at least 36 months at the long-term storage condition of -20 °C ± 5 °C. [328] Additional tests were performed at a 48 month timepoint: the appearance was a clear and colorless liquid, free of visible particles; the pH was within a range of 5.6 to 6.2 (specifically, 5.9); particulates ≥ 25 µm or ≥ 10 µm were not more than 6,000/vial (specifically, 31/vial or 558/vial, respectively); content (protein concentration) was within a range of 9.0 to 11.0 mg/mL (specifically, 10.3 mg/mL); purity based on CEX that measured % main peak, % acidic peak and % basic peak indicated ≥ 40.0% main peak, ≤ 40.0% acidic peak and ≤ 30.0% basic peak (specifically, 52.2% main peak, 33.7% acidic species peak and 14.1% basic species peak); purity based on SEC indicated high purity and minimal aggregation (98.2% main peak, 1.7% HMW species and 0.1% LMW species); DAR was within the range of 1.9 to 2.1 (specifically, 2.1); unconjugated mAb was 0.2%; and free drug related impurities were less than 0.10% (w/w) (specifically, less than the lower limit of quantification). Based on these and other test results, ARX517 Drug Product was found to be stable for at least 48 months at the long-term storage condition of -20 °C ± 5 °C. Attorney Docket No. AMB1013WOPCT1 [329] Accelerated Stability. ARX517 Drug Product exhibited the following characteristics under accelerated stability conditions at 5 °C ± 3 °C (stored upright) over a period of 6 months, with all tests performed at zero and 6 months, and most tests additionally performed at 1 and 3 months: the appearance was a colorless to light yellow and clear to opalescent liquid, essentially free of visible particles; the pH was within a range of 5.6 to 6.2 (specifically, 5.8 to 5.9); particulates ≥ 25 µm were less than 5, and ≥ 10 µm were less than 100; content (protein concentration) was within a range of 9.0 to 11.0 mg/mL (specifically, 10.1 to 10.4 mg/mL); cell-based and binding ELISA assay results were each > 85% of reference standard at all time points; purity based on CEX that measured % main peak, % acidic peak and % basic peak indicated ≥ 40.0% main peak, ≤ 40.0% acidic peak and ≤ 30.0% basic peak over the duration of the study (specifically, 51.2% to 54.1% main peak, 31.8% to 34.5% acidic species peak, and 14.1% to 14.3% basic species peak); purity based on SEC that measured % monomer (% main peak), % high molecular weight (HMW) and % low molecular weight (LMW) species was 97.5% to 98.2% main peak, 1.7% to 2.4% HMW species and 0.1% LMW species over the duration of the study; endotoxins were < 0.05 EU/mg, and no growth was observed, indicating that the drug product remained sterile; drug-to-antibody ratio (DAR) as measured by hydrophobic interaction chromatography (HIC) was within the range of 1.9 to 2.1 (specifically, 2.1 over all measured time points, including zero and 6 months), and unconjugated mAb (also measured by HIC) was 0.2% at each measured time point, including zero and 6 months; and free drug related impurities were less than 0.10% (w/w). [330] Additional tests were performed at the accelerated condition at the 12-month timepoint: purity based on CEX indicated 53.3% main peak, 31.9% acidic species peak, and 14.8% basic species peak. Analysis of Covariance (ANCOVA) of the zero to 12-month CEX data indicate that ARX517 Drug Product is stable for at least 24 months when stored at 5 °C ± 3 °C. Attorney Docket No. AMB1013WOPCT1 [331] Stressed conditions of 25 °C/60% RH. ARX517 Drug Product exhibited the following characteristics under stressed conditions of 25 °C/60% relative humidity (RH) (stored upright) over a period of 3 months, with all tests performed at zero and 3 months, and most tests additionally performed at 1 month: the appearance was a colorless and clear liquid, essentially free of visible particles; the pH was within a range of 5.6 to 6.2 (specifically, 5.8 to 5.9); particulates ≥ 25 µm were less than 15, and ≥ 10 µm were less than 100; content (protein concentration) was within a range of 9.0 to 11.0 mg/mL (specifically, 10.1 to 10.3 mg/mL); cell-based and binding ELISA assay results were each ≥ 75% of reference standard at all time points; purity based on size exclusion chromatography (SEC) that measured % monomer (% main peak), % high molecular weight (HMW) and % low molecular weight (LMW) species was 96.3% to 98.2% main peak, 1.7% to 3.2% HMW species and 0.1% to 0.5% LMW species over the duration of the study; endotoxins were < 0.05 EU/mg, and no growth was observed, indicating that the drug product remained sterile; drug-to-antibody ratio (DAR) as measured by hydrophobic interaction chromatography (HIC) was within the range of 2.0 to 2.1, and unconjugated mAb (also measured by HIC) was 0.2% to 0.3%; and free drug related impurities were less than 0.10% (w/w). [332] In summary, the stability results showed that ARX517 Drug Product is stable for at least 48 months at -20 °C ± 5 °C, at least 12 months at accelerated condition of 5 °C ± 3 °C, and at least 3 months at stressed conditions of 25 °C/60% RH. [333] Example 15: ARX517 Demonstrates Stability, Dose-Dependent Exposure, and Long Half-Life. [334] As discussed in Example 13, a Phase 1/2 first-in-human trial evaluating ARX517 in patients with mCRPC resistant or refractory to prior therapies, (APEX-01), was conducted. The pharmacokinetics population consisted of 32 patients having received ARX517 at doses ranging from 0.32 to 2.4 mg/kg as an intravenous infusion Q3W. Table 13 shows the baseline Attorney Docket No. AMB1013WOPCT1 demographics of the pharmacokinetic population consisting of 32 patients. The data shows the mean and median body weight, creatinine clearance, hepatic function and PSA for patients administered ARX517 at a dose of 0.32 mg/kg, 0.64 mg/kg, 1.07 mg/kg, 1.4 mg/kg, 1.7 mg/kg, 2.0 mg/kg, and 2.4 mg/kg. Table 13 – Baseline demographics of the pharmacokinetic population (n=32) Dose (mg/kg) 0.32 0.64 1.07 1.4 1.7 2 2.4 Number of 1 3 3 5 5 9 6 Patients Age (yr) Mean (SD) 57.0 67.3 73.0 77.8 73.6 67.9 67.5 (NA) (11.5) (7.00) (12.9) (9.84) (9.33) (10.7) Median 57.0 67.0 70.0 72.0 75.0 68.0 64.5 [Min, [57.0, [56.0, [68.0, [69.0, [62.0, [50.0, [56.0, Max] 57.0] 79.0] 81.0] 100] 83.0] 83.0] 82.0] Body Weight (kg) Mean 69.3 102 83.4 78.6 80.7 88.0 85.3 (NA) (20.3) (29.3) (17.5) (15.9) (21.4) (18.0) Median 69.3 108 71.3 80.7 78.7 92.1 81.7 [Min, [69.3, [80.0, [62.0, [54.4, [60.1, [61.9, [64.1, Max] 69.3] 120] 117] 95.0] 98.6] 127] 108] Creatinine Clearance (mL/min) Mean 125 (NA) 142 92.4 76.6 71.0 105 98.1 (49.4) (36.0) (37.7) (26.5) (27.7) (37.1) Median 125 123 112 79.3 63.6 105 107 [Min, [125, [105, [50.8, [32.0, [48.2, [66.2, [41.1, Max] 125] 198] 115] 132] 117] 153] 144] Hepatic Function Normal 1 (100%) 3 3 3 5 8 5 (100%) (100%) (60.0%) (100%) (88.9%) (83.3%) Mild 0 (0%) 0 (0%) 0 (0%) 2 0 (0%) 1 1 impairment (40.0%) (11.1%) (16.7%) PSA (ng/mL) Attorney Docket No. AMB1013WOPCT1 Mean 33.8 1310 51.1 195 462 51.4 273 (NA) (2190) (58.7) (277) (786) (43.7) (410) Median 33.8 63.5 26.8 101 51.7 57.0 125 [Min, [33.8, [26.0, [8.50, [8.30, [10.6, [2.33, [0.520, Max] 33.8] 3850] 118] 685] 1840] 121] 1080] [335] Patient serum samples were collected at fixed time points and evaluated in validated Total Antibody (TA; sum of deconjugated antibody and conjugated antibody), ADC (conjugated antibody with a DAR of 1 or 2), and free payload (pAF-AS269) assays. The lower limit of quantitation for the TA, ADC, and free payload assays were 62.5 ng/mL, 7.8 ng/mL, and 0.02 ng/mL, respectively. [336] Cycle 1 and Cycle 3 pharmacokinetic parameters were determined by noncompartmental analysis using Phoenix WINNONLIN (version 8.4) using serum concentrations of TA, ADC, and free payload. [337] Low serum concentrations of free payload were observed at all doses, with the molar ratio of free payload to ADC at 0.06%, FIGs. 13A and 13B. Pharmacokinetic parameter maximum serum concentration (Cmax) was analyzed through different analytes such as ADC, total antibody and pAF-AS269. FIG. 13A shows the pAF-AS269 serum concentrations from serum samples collected from patients receiving 2mg/kg ARX517 Q3W. FIG. 13B shows the pAF-AS269 serum concentrations from serum samples collected from patients receiving 0.32 mg/kg, 0.64 mg/kg, 1.07 mg/kg, 1.4 mg/kg, 1.7 mg/kg, 2.0 mg/kg, and 2.4 mg/kg ARX517 Q3W. [338] Serum samples collected from patients receiving 0.64 mg/kg, 1.07 mg/kg, 1.4 mg/kg, 1.7 mg/kg, 2.0 mg/kg, or 2.4 mg/kg ARX517 Q3W via intravenous infusion, were also evaluated in validated Total Antibody (TA; sum of deconjugated antibody and conjugated antibody), and ADC (conjugated antibody with a DAR of 1 or 2) assays. FIGs.14A-14F show Attorney Docket No. AMB1013WOPCT1 virtually overlapping total antibody (TA) and ADC PK concentration-time curves demonstrating strong stability of the ADC ARX517 at all dose levels. [339] Serum samples collected from patients receiving ARX517 at doses ranging from 0.32 mg/kg to 2.4 mg/kg exhibited a long half-life of about 6-10 days at doses >1.4mg/kg, (FIG.15 and Table 14). Further, drug exposure was shown to increase proportional to ARX517 dose, as indicated in FIGs.16A (AUC) and 16B (Cmax). Long half-life indicated maximum drug exposure over the dosing cycle. Table 14 – ARX517 exhibits a long half-life of about 6-10 days at doses > 1.4 mg/kg Dose (mg/kg) 0.32 0.64 1.07 1.4 1.7 2 2.4 Number of P
atients1 3 3 3 5 7 6 Minimum 1.6 2.2 4.5 3.2 4.9 6.2 5.0 (days) Maximum 1.6 4.1 5.3 8.5 8.2 10.4 11.8 (days) Range 0.0 1.9 0.8 5.4 3.3 4.1 6.7 Geometric 1.6 2.9 4.9 5.6 6.0 7.9 8.3 Mean Geometric SD 1.0 1.4 1.0 1.7 1.2 1.2 1.4 [340] Example 16: Safety and efficacy of ARX517 in heavily pre-treated patients with Metastatic Castration-Resistant Prostate Cancer (mCRPC) [341] Overall, the clinical data showed no ARX517 treatment related adverse events and no DLTs in patients. [342] Deep PSA reductions with increasing ARX517 dose were observed as shown in FIG. 17. The PSA waterfall plot includes patients with at least two on-treatment PSA assessments or discontinued before the second assessment. Prior to reaching MTD, two dose cohorts (4 and Attorney Docket No. AMB1013WOPCT1 6) were expanded based on three criteria 1) PSA decline of ≥50%; 2) no treatment-related SAEs; and 3) target lesion reduction or RECIST v1.1 response. [343] Prior PSMA-TRT, ≥50% PSA reductions were achieved in patients who had prior PSMA-TRT: 37% (4/11) and 50% (3/6) of patients at doses ≥1.4 mg/kg and ≥2.0 mg/kg, respectively. 52% (12/23) of patients experienced a ≥50% PSA reduction at putative therapeutic doses (≥2.0 mg/kg), FIG.18. Greater frequency and depth of PSA response at putative therapeutic doses (≥2.0 mg kg) are illustrated in Table 15. Table 15. Frequency and Depth of PSA Response at Putative Therapeutic Doses (≥2.0 mg kg). C1–3 C4 C5 C6 C7 C8 C6–8 n = 7 n = 16 n = 5 n = 14 n = 6 n = 3 n = 23 ≥30% 29% 38% 40%, 64% 50% 67% 61% PSA ≥50% 0 25% 0 50% 50% 67% 52% PSA ≥90% 0 6% 0 36% 0 33% 26% PSA [344] On treatment changes in ctDNA have been shown to predict time to progression and survival, (Tolmeijer SH et al. Clin Cancer Res. 2023 Aug 1;29(15):2835-2844. doi: 10.1158/1078-0432.CCR-22-2998; Sartor O. Clin Cancer Res.2023 Aug 1;29(15):2745-2747. doi: 10.1158/1078-0432.CCR-23-1043). Serial plasma samples were collected at baseline, C3D1, C4D1 and EOT, best percent change from baseline is shown. ctDNA was measured using GuardantINFINITY test (Guardant Health) with a specificity of 96.9%, a sensitivity of 91.3% and a reported lower limit of detection 0.06%. Samples were processed after passing multiple quality control measurements encompassing DNA yield, GC bias, methylation bias, Attorney Docket No. AMB1013WOPCT1 diversity, and contamination checks. ctDNA changes compared with its baseline level were measured based on aggregated tumor-specific methylation signal scores. Reductions of ≥50% in circulating tumor DNA (ctDNA) in 81% (17/21) of patients (cohorts 4–8) was also observed as shown in FIG.19 for ARX517. [345] Duration on treatment from first dose was observed in Cohorts 4-8 administered 1.4 mg/kg, 1.7 mg/kg, 2.0 mg/kg, 2.4 mg/kg, and 2.88 mg/kg of ARX517 Q3W. ARX517 has a strong safety profile at all doses tested up to 2.88mg/kg Q3W. Data from later cohorts not yet reported. [346] RECIST v1.1 Target lesion reduction was observed with ARX517. Evaluable population included all patients with measurable target lesion(s) at baseline per RECIST v1.1 who had at least two post baseline tumor assessments or progressed or discontinued treatment prior to the second assessment. [347] Note, cPR, confirmed partial response per RECIST v1.1; *Patients with lung/liver target lesions;
†Patients with prior PSMA-TRT; Patient had PR in target lesions; 1 liver lesion reduced in size from 38 to 14 mm and 1 lung lesion from 18 to 9 mm, but growth in non-target lesion resulted in PD by RECIST v1.1. In cohorts 4–8 patients 56% (5/9) of RECIST v1.1 target lesion reduction was observed, FIG.20. [348] Overall, without PSMA imaging selection, ARX517 monotherapy achieved favorable safety and demonstrated early efficacy, with deep PSA and ctDNA reductions and confirmed RECIST v1.1 tumor response in patients with mCRPC who progressed on multiple FDA- approved treatments. [349] Example 17: Cationic Exchange High Performance Liquid Chromatography (CEX-HPLC) Analysis of ARX517 Solution for Intravenous Infusion [350] CEX-HPLC was used to quantify ARX517 ADC main species, acidic species and basic species in samples of ARX517 Solution for Intravenous Infusion manufactured under cGMP Attorney Docket No. AMB1013WOPCT1 that were evaluated in accordance with ICH guidelines for long term stability at -20 °C ± 5 °C, accelerated stability at 5 °C ± 3 °C and under stressed conditions of 25 °C/60% relative humidity (RH), as described in Example 14. The CEX method employed sodium phosphate buffer pH 6.6 and an increasing gradient of sodium chloride. [351] Method. Briefly, CEX was performed on an Agilent analytical HPLC system configured with a UV detector and autosampler. A CEX column (ProPac WCX-10 (Thermo Fisher Scientific): nonporous polymer packing material; 4 × 250 mm dimensions, particle size 10 µm) was equilibrated with mobile phase A containing 20 mM sodium phosphate, pH 6.6. Mobile phase B contained 20 mM sodium phosphate, 200 mM sodium chloride, pH 6.6. The column temperature was 40°C and the flow rate was 1.0 mL/min. Samples of ARX517 Solution for Intravenous Infusion (10 mg/mL) were diluted to 1 mg/mL with mobile phase A and mixed to provide test samples for analysis, which were maintained at 5°C on a temperature controlled autosampler. The target sample column load was 50 µg (5 µL injection volume). [352] The gradient program was as follows: 100% mobile phase A for 5 minutes; zero (0)% to 30% mobile phase B from 5 minutes to 35 minutes; 30% to 80% mobile phase B from 35 to 36 minutes; hold at 80% mobile phase B from 36 to 40 minutes; 80% to 0% mobile phase B from 40 to 40.1 minutes. [353] The detection wavelengths were 214 nm (bandwidth: 4 nm) for quantification and 280 nm (bandwidth: 4 nm) for column recovery calculation. The main peak (corresponding to the ARX517 ADC main species), the acidic peaks (corresponding to the ARX517 ADC acidic species, which included all peaks eluting prior to the main peak), and the basic peaks (corresponding to the ARX517 ADC basic species, which included all peaks eluting after the main peak) were integrated to quantify the relative UV area % at 214 nm of the main species, the acidic species and the basic species present in samples of ARX517 Solution for Intravenous Infusion. Attorney Docket No. AMB1013WOPCT1 [354] Results. The relative UV area % at 214 nm of the main species, the acidic species and the basic species in samples of ARX517 Solution for Intravenous Infusion that were evaluated for long term stability at -20 °C ± 5 °C and accelerated stability at 5 °C ± 3 °C was at least 40% main species, not more than 40% acidic species, and not more than 30% basic species, as described in Example 14. Exemplary cation exchange chromatograms of ARX517 Solution for Intravenous Infusion are shown in FIGs.21A-21B. [355] Example 18: Isoelectric Point Analysis by Imaged Capillary Isoelectric Focusing (iCIEF) [356] The isoelectric point (pI) of ARX517 main species and of charged variants thereof (i.e., acidic and basic species) were determined via iCIEF. [357] The iCIEF method employed a capillary isoelectric focusing electrophoresis system (ProteinSimple iCE3 (Bio-Techne; or equivalent, e.g., Maurice iCE platform) equipped with fluorocarbon (FC) coated capillary cartridges (e.g., iCE3101701 for single FC coated capillary cartridge; iCE3101700 for two FC capillary coated cartridge) and a PrinCE Next MicroInjector (or equivalent). [358] In brief, iCIEF was used to separate ARX517 main species from its acidic and basic species based on their charge differences in a pH gradient. Under an applied external electric field, ARX517 main species and its charged variants move along a continuous pH gradient formed by ampholytes, and each species stops where the pH equals its pI. The method is suitable for, and was used to evaluate, in-process samples, drug substance release samples, drug product release samples, and stability samples containing ARX517. [359] Samples containing ARX517 were prepared in L-arginine (Sigma), 500 mM; and urea (Sigma), 8 M. Reference standard isoelectric point markers (marker pI 7.05 and marker pI 9.50) were added to samples for calibration purposes. Test samples were centrifuged (13,000 rpm, 1 to 5 minutes) prior to analysis. First and second focusing conditions were used, as follows. Attorney Docket No. AMB1013WOPCT1 Focus period 1: 2 (two) minutes at 1500 V. Focus period 2: 8 (eight) minutes at 3000 V. An electrogram was obtained at 280 nm. The pI values and relative abundance of the resolved peaks were quantified using chromatographic software (Empower 3 Chromatograph Data System (CDS); Waters). All peaks within the two pI markers were integrated. The average pI of main species and charged variants thereof were reported, rounded to one decimal place, as follows: main species, pI 8.3; major acidic species eluting closes to main species, pI 8.1; major basic species eluting closes to main species, pI 8.4. [360] While preferred embodiments of the present disclosures have been shown and described herein, it will be obvious to those skilled in the art that such embodiments are provided by way of example only. Numerous variations, changes, and substitutions will now occur to those skilled in the art without departing from the disclosures. It should be understood that various alternatives to the embodiments of the disclosures described herein may be employed in practicing the disclosures. It is intended that the following claims define the scope of the disclosures and that methods and structures within the scope of these claims and their equivalents be covered thereby.
 Short-cleavable or short Val-Citruline-Acetyl (Val-Cit) MMAF having the structure: Attorney Docket No. AMB1013WOPCT1 .
Short-cleavable or short Val-Citruline-Acetyl (Val-Cit) MMAF having the structure: Attorney Docket No. AMB1013WOPCT1 . Cit) MMAE having the structure: .
Cit) MMAE having the structure: . be employed with the anti PSMA antibody or antibody drug conjugates of the present invention. Synthesis of such payload linkers are well known to the skilled artisan. See for example, Dubowchik et al., Bioconjugate Chem. 13: 855-869, (2002); Doronina et al., Nature Biotechnology 21(7): 778- 784, (2003); WO2012/166560; WO2013/185117, the entire contents of each of which are hereby incorporated by reference in their entirety. Table 1. Drug-linker Compounds – Single Payload Compound Structure
be employed with the anti PSMA antibody or antibody drug conjugates of the present invention. Synthesis of such payload linkers are well known to the skilled artisan. See for example, Dubowchik et al., Bioconjugate Chem. 13: 855-869, (2002); Doronina et al., Nature Biotechnology 21(7): 778- 784, (2003); WO2012/166560; WO2013/185117, the entire contents of each of which are hereby incorporated by reference in their entirety. Table 1. Drug-linker Compounds – Single Payload Compound Structure
















 milligram per kilogram (mg/kg) and at most about 5.0 mg/kg of the body weight of the human subject. 2. The method of claim 1, wherein each light chain comprises a light chain variable region of SEQ ID NO: 2. 3. The method of claim 1 or claim 2, wherein each heavy chain amino acid sequence is SEQ ID NO: 8, comprising the pAF at Kabat position 114. 4. The method of any one of claims 1 to 3, wherein each light chain amino acid sequence is SEQ ID NO: 9. Attorney Docket No. AMB1013WOPCT1 5. The method of any one of claims 1 to 4, wherein the effective amount of the anti- PSMA ADC is at least about 1.4 mg/kg and at most about 3.4 mg/kg of the body weight of the human subject. 6. The method of any one of claims 1 to 4, wherein the effective amount of the anti- PSMA ADC is about 1.4 mg/kg, about 1.7 mg/kg, about 2 mg/kg, about 2.4 mg/kg, about 2.9 mg/kg, about 3.2 mg/kg, about 3.4 mg/kg, about 3.5 mg/kg, about 4.3 mg/kg, about 4.5 mg/kg, about 4.7 mg/kg or about 5 mg/kg of the body weight of the human subject. 7. The method of any one of clams 1 to 6, wherein the effective amount of the anti- PSMA ADC is greater than 2.0 mg/kg of the body weight of the human subject. 8. The method of any one of claims 1 to 7, wherein the anti-PSMA ADC is administered once every three weeks or once every four weeks. 9. The method of any one of claims 1 to 8, wherein the human subject has a PSMA- expressing prostate cancer or a PSMA-expressing non-prostate cancer. 10. The method of any one of claims 1 to 9, wherein the human subject has prostate cancer. 11. The method of claim 10, wherein the prostate cancer is metastatic castration-resistant prostate cancer (mCRPC). 12. The method of any one of claims 9 to 11, wherein the prostate cancer has progressed after prior taxane therapy. Attorney Docket No. AMB1013WOPCT1 13. The method of any one of claims 1 to 12, wherein the cancer is hormone refractory prostate cancer. 14. The method of any one of claims 1 to 13, wherein the method delays or inhibits progression of the cancer in the human subject. 15. The method of any one of claims 1 to 14, wherein the method increases the survival of the human subject as compared to the median survival of subjects who have not been previously treated with the same or a different anti-PSMA ADC. 16. The method of any one of claims 1 to 15, wherein the method decreases a circulating level of circulating tumor cells (CTCs) in the human subject compared to a baseline level of the CTCs in the human subject. 17. The method of any one of claims 1 to 16, wherein the method decreases or stabilizes a serum level of prostate specific antigen (PSA) in the human subject compared to a baseline level of PSA in the human subject. 18. The method of claim 17, wherein the decrease in the serum level of PSA in the human subject is at least about a 30% decrease in the serum level of PSA in the human subject compared to the baseline level of PSA in the human subject. 19. The method of claim 17, wherein the decrease in the serum level of PSA in the human subject is at least about a 50% decrease in the serum level of PSA in the human subject compared to the baseline level of PSA in the human subject. 20. The method of any one of claims 1 to 19, wherein the administering is intravenous. Attorney Docket No. AMB1013WOPCT1 21. The method of any one of claims 1 to 20, wherein the cancer is resistant or refractory to prior standard therapies. 22. The method of any one of claims 1 to 21, wherein the human subject has been previously treated with abiraterone, darolutamide, apalutamide or enzalutamide. 23. The method of any one of claims 1 to 22, further comprising administering an effective amount of an additional therapeutic agent. 24. The method of claims 23, wherein the additional therapeutic agent is a chemotherapeutic agent, a hormonal agent, an antitumor agent, an immunostimulatory agent, an immunomodulator, an immunotherapeutic agent or combination thereof. 25. The method of claim 24, wherein the hormonal agent is enzalutamide. 26. The method of any one of claims 1 to 25, wherein the method increases the survival of the human subject, wherein the survival is increased in comparison to the median survival time of subjects with anti-PSMA-expressing, taxane-resistant cancer not previously treated with the same or a different anti-PSMA ADC. 27. The method of any one of claims 1 to 26, wherein the method provides an anti-PSMA ADC serum terminal half-life of at least about 5 days after the administration of the effective amount of the anti-PSMA ADC to the human subject. 28. The method of any one of claims 1 to 27, wherein the method of treatment provides a time to free payload serum maximum concentration (Tmax) of at least about 5 days after the administration of the effective amount of the anti-PSMA ADC to the human subject, wherein the free payload has the following structure: Attorney Docket No. AMB1013WOPCT1 ;
milligram per kilogram (mg/kg) and at most about 5.0 mg/kg of the body weight of the human subject. 2. The method of claim 1, wherein each light chain comprises a light chain variable region of SEQ ID NO: 2. 3. The method of claim 1 or claim 2, wherein each heavy chain amino acid sequence is SEQ ID NO: 8, comprising the pAF at Kabat position 114. 4. The method of any one of claims 1 to 3, wherein each light chain amino acid sequence is SEQ ID NO: 9. Attorney Docket No. AMB1013WOPCT1 5. The method of any one of claims 1 to 4, wherein the effective amount of the anti- PSMA ADC is at least about 1.4 mg/kg and at most about 3.4 mg/kg of the body weight of the human subject. 6. The method of any one of claims 1 to 4, wherein the effective amount of the anti- PSMA ADC is about 1.4 mg/kg, about 1.7 mg/kg, about 2 mg/kg, about 2.4 mg/kg, about 2.9 mg/kg, about 3.2 mg/kg, about 3.4 mg/kg, about 3.5 mg/kg, about 4.3 mg/kg, about 4.5 mg/kg, about 4.7 mg/kg or about 5 mg/kg of the body weight of the human subject. 7. The method of any one of clams 1 to 6, wherein the effective amount of the anti- PSMA ADC is greater than 2.0 mg/kg of the body weight of the human subject. 8. The method of any one of claims 1 to 7, wherein the anti-PSMA ADC is administered once every three weeks or once every four weeks. 9. The method of any one of claims 1 to 8, wherein the human subject has a PSMA- expressing prostate cancer or a PSMA-expressing non-prostate cancer. 10. The method of any one of claims 1 to 9, wherein the human subject has prostate cancer. 11. The method of claim 10, wherein the prostate cancer is metastatic castration-resistant prostate cancer (mCRPC). 12. The method of any one of claims 9 to 11, wherein the prostate cancer has progressed after prior taxane therapy. Attorney Docket No. AMB1013WOPCT1 13. The method of any one of claims 1 to 12, wherein the cancer is hormone refractory prostate cancer. 14. The method of any one of claims 1 to 13, wherein the method delays or inhibits progression of the cancer in the human subject. 15. The method of any one of claims 1 to 14, wherein the method increases the survival of the human subject as compared to the median survival of subjects who have not been previously treated with the same or a different anti-PSMA ADC. 16. The method of any one of claims 1 to 15, wherein the method decreases a circulating level of circulating tumor cells (CTCs) in the human subject compared to a baseline level of the CTCs in the human subject. 17. The method of any one of claims 1 to 16, wherein the method decreases or stabilizes a serum level of prostate specific antigen (PSA) in the human subject compared to a baseline level of PSA in the human subject. 18. The method of claim 17, wherein the decrease in the serum level of PSA in the human subject is at least about a 30% decrease in the serum level of PSA in the human subject compared to the baseline level of PSA in the human subject. 19. The method of claim 17, wherein the decrease in the serum level of PSA in the human subject is at least about a 50% decrease in the serum level of PSA in the human subject compared to the baseline level of PSA in the human subject. 20. The method of any one of claims 1 to 19, wherein the administering is intravenous. Attorney Docket No. AMB1013WOPCT1 21. The method of any one of claims 1 to 20, wherein the cancer is resistant or refractory to prior standard therapies. 22. The method of any one of claims 1 to 21, wherein the human subject has been previously treated with abiraterone, darolutamide, apalutamide or enzalutamide. 23. The method of any one of claims 1 to 22, further comprising administering an effective amount of an additional therapeutic agent. 24. The method of claims 23, wherein the additional therapeutic agent is a chemotherapeutic agent, a hormonal agent, an antitumor agent, an immunostimulatory agent, an immunomodulator, an immunotherapeutic agent or combination thereof. 25. The method of claim 24, wherein the hormonal agent is enzalutamide. 26. The method of any one of claims 1 to 25, wherein the method increases the survival of the human subject, wherein the survival is increased in comparison to the median survival time of subjects with anti-PSMA-expressing, taxane-resistant cancer not previously treated with the same or a different anti-PSMA ADC. 27. The method of any one of claims 1 to 26, wherein the method provides an anti-PSMA ADC serum terminal half-life of at least about 5 days after the administration of the effective amount of the anti-PSMA ADC to the human subject. 28. The method of any one of claims 1 to 27, wherein the method of treatment provides a time to free payload serum maximum concentration (Tmax) of at least about 5 days after the administration of the effective amount of the anti-PSMA ADC to the human subject, wherein the free payload has the following structure: Attorney Docket No. AMB1013WOPCT1 ; 29. The method of any one of claims 1 to 28, wherein the method of treatment provides a free payload serum maximum concentration (Cmax) of at most about 1 ng/mL after the administration of the effective amount of the anti-PSMA ADC to the human subject, wherein the free payload has the following structure: ;
29. The method of any one of claims 1 to 28, wherein the method of treatment provides a free payload serum maximum concentration (Cmax) of at most about 1 ng/mL after the administration of the effective amount of the anti-PSMA ADC to the human subject, wherein the free payload has the following structure: ; or a salt thereof. 30. The method of claim 29, wherein the serum payload Cmax is at most about 0.5 ng/mL, at most about 0.4 ng/mL, at most about 0.3 ng/mL or at most about 0.2 ng/mL after the administration of the effective amount of the anti-PSMA ADC to the human subject. 31. The method of any one of claims 1 to 30, wherein the method of treatment provides an anti-PSMA ADC serum maximum concentration (Cmax) of at least about 20 µg/mL, at least about 30 µg/ml, at least about 40 µg/ml, at least about 50 µg/ml, or at least about 60 µg/ml after the administration of the effective amount of the anti-PSMA ADC to the human subject. Attorney Docket No. AMB1013WOPCT1 32. The method of any one of claims 1 to 31, wherein the method of treatment provides a reduction in circulating tumor DNA (ctDNA) of at least about 50% after the administration of the effective amount of the anti-PSMA ADC to the human subject. 33. The method of any one of claims 1 to 32, wherein the anti-PSMA ADC is ARX517. 34. A pharmaceutical composition comprising an effective amount of an anti-PSMA antibody drug conjugate (ADC), wherein the anti-PSMA ADC comprises: a humanized anti-PSMA monoclonal antibody comprising two heavy chains and two light chains, wherein each heavy chain comprises a heavy chain variable region of SEQ ID NO: 1, wherein one non-natural amino acid para-acetyl-L-phenylalanine (pAF) is incorporated into each said heavy chain at position 114 according to Kabat numbering, and each light chain comprises a light chain variable region of SEQ ID NO: 2; wherein one drug-linker is conjugated to each said pAF via an oxime linkage, wherein each said drug-linker is amberstatin269 (AS269) having the following structure: the
or a salt thereof. 30. The method of claim 29, wherein the serum payload Cmax is at most about 0.5 ng/mL, at most about 0.4 ng/mL, at most about 0.3 ng/mL or at most about 0.2 ng/mL after the administration of the effective amount of the anti-PSMA ADC to the human subject. 31. The method of any one of claims 1 to 30, wherein the method of treatment provides an anti-PSMA ADC serum maximum concentration (Cmax) of at least about 20 µg/mL, at least about 30 µg/ml, at least about 40 µg/ml, at least about 50 µg/ml, or at least about 60 µg/ml after the administration of the effective amount of the anti-PSMA ADC to the human subject. Attorney Docket No. AMB1013WOPCT1 32. The method of any one of claims 1 to 31, wherein the method of treatment provides a reduction in circulating tumor DNA (ctDNA) of at least about 50% after the administration of the effective amount of the anti-PSMA ADC to the human subject. 33. The method of any one of claims 1 to 32, wherein the anti-PSMA ADC is ARX517. 34. A pharmaceutical composition comprising an effective amount of an anti-PSMA antibody drug conjugate (ADC), wherein the anti-PSMA ADC comprises: a humanized anti-PSMA monoclonal antibody comprising two heavy chains and two light chains, wherein each heavy chain comprises a heavy chain variable region of SEQ ID NO: 1, wherein one non-natural amino acid para-acetyl-L-phenylalanine (pAF) is incorporated into each said heavy chain at position 114 according to Kabat numbering, and each light chain comprises a light chain variable region of SEQ ID NO: 2; wherein one drug-linker is conjugated to each said pAF via an oxime linkage, wherein each said drug-linker is amberstatin269 (AS269) having the following structure: the and one or more pharmaceutically acceptable components selected from the group consisting of sucrose, histidine buffer and polysorbate 80, and a combination thereof, and wherein the composition pH is within a range of about 5.5 to about 6.5. 35. The pharmaceutical composition of claim 34, wherein each heavy chain amino acid sequence is SEQ ID NO: 8, comprising the pAF at Kabat position 114, and/or each light chain amino acid sequence is SEQ ID NO: 9. Attorney Docket No. AMB1013WOPCT1 36. The pharmaceutical composition of claim 34 or claim 35, wherein the anti-PSMA ADC concentration is about 10 mg/mL. 37. The pharmaceutical composition of any one of claims 34 to 36, wherein the sucrose concentration is within a range of about 5% (w/v) to about 15% (w/v); the histidine buffer concentration is within a range of about 15 mM to about 25 mM; and the polysorbate 80 concentration is within a range of about of about 0.001% (w/v) to about 0.02% (w/v). 38. The pharmaceutical composition of any one of claims 34 to 37, wherein the composition consists essentially of the anti-PSMA ADC at a concentration of about 10 mg/mL; sucrose at a concentration of about 9% (w/v); histidine buffer at a concentration of about 20 mM; and polysorbate 80 at a concentration of about 0.01% (w/v); wherein the composition pH is about 5.9 ± 0.3. 39. The pharmaceutical composition of any one of claims 34 to 38, wherein the composition is a liquid formulation. 40. The pharmaceutical composition of any one of claims 34 to 39, wherein the anti- PSMA ADC comprises charged variants, wherein the charged variants comprise an anti- PSMA ADC main species, an anti-PSMA ADC acidic species and an anti-PSMA ADC basic species. 41. The pharmaceutical composition of claim 40, wherein the anti-PSMA ADC main species has an isoelectric point (pI) of about 8.3, the anti-PSMA ADC acidic species has a pI of about 8.1, and the anti-PSMA ADC basic species has a pI of about 8.4. Attorney Docket No. AMB1013WOPCT1 42. The pharmaceutical composition of claim 40 or 41, wherein the anti-PSMA ADC main species is present in an amount of about 40% to about 70%, the anti-PSMA ADC acidic species is present in an amount of about 20% to about 40%, and anti-PSMA ADC basic species is present in an amount of about 5% to about 30%; wherein the sum of the percentage of each said main species, acidic species and basic species is 100%. 43. The pharmaceutical composition of any one of claims 34 to 42, wherein the drug-to- antibody ratio (DAR) is within a range of about 1.5 to about 2.5. 44. The pharmaceutical composition of claim 43, wherein the drug-to-antibody ratio (DAR) is within a range of about 1.9 to about 2.1. 45. The pharmaceutical composition of any one of claims 34 to 44, wherein the anti- PSMA ADC is ARX517. 46. The pharmaceutical composition of any one of claims 34 to 45 for use in a method of any one of claims 1 to 33.
and one or more pharmaceutically acceptable components selected from the group consisting of sucrose, histidine buffer and polysorbate 80, and a combination thereof, and wherein the composition pH is within a range of about 5.5 to about 6.5. 35. The pharmaceutical composition of claim 34, wherein each heavy chain amino acid sequence is SEQ ID NO: 8, comprising the pAF at Kabat position 114, and/or each light chain amino acid sequence is SEQ ID NO: 9. Attorney Docket No. AMB1013WOPCT1 36. The pharmaceutical composition of claim 34 or claim 35, wherein the anti-PSMA ADC concentration is about 10 mg/mL. 37. The pharmaceutical composition of any one of claims 34 to 36, wherein the sucrose concentration is within a range of about 5% (w/v) to about 15% (w/v); the histidine buffer concentration is within a range of about 15 mM to about 25 mM; and the polysorbate 80 concentration is within a range of about of about 0.001% (w/v) to about 0.02% (w/v). 38. The pharmaceutical composition of any one of claims 34 to 37, wherein the composition consists essentially of the anti-PSMA ADC at a concentration of about 10 mg/mL; sucrose at a concentration of about 9% (w/v); histidine buffer at a concentration of about 20 mM; and polysorbate 80 at a concentration of about 0.01% (w/v); wherein the composition pH is about 5.9 ± 0.3. 39. The pharmaceutical composition of any one of claims 34 to 38, wherein the composition is a liquid formulation. 40. The pharmaceutical composition of any one of claims 34 to 39, wherein the anti- PSMA ADC comprises charged variants, wherein the charged variants comprise an anti- PSMA ADC main species, an anti-PSMA ADC acidic species and an anti-PSMA ADC basic species. 41. The pharmaceutical composition of claim 40, wherein the anti-PSMA ADC main species has an isoelectric point (pI) of about 8.3, the anti-PSMA ADC acidic species has a pI of about 8.1, and the anti-PSMA ADC basic species has a pI of about 8.4. Attorney Docket No. AMB1013WOPCT1 42. The pharmaceutical composition of claim 40 or 41, wherein the anti-PSMA ADC main species is present in an amount of about 40% to about 70%, the anti-PSMA ADC acidic species is present in an amount of about 20% to about 40%, and anti-PSMA ADC basic species is present in an amount of about 5% to about 30%; wherein the sum of the percentage of each said main species, acidic species and basic species is 100%. 43. The pharmaceutical composition of any one of claims 34 to 42, wherein the drug-to- antibody ratio (DAR) is within a range of about 1.5 to about 2.5. 44. The pharmaceutical composition of claim 43, wherein the drug-to-antibody ratio (DAR) is within a range of about 1.9 to about 2.1. 45. The pharmaceutical composition of any one of claims 34 to 44, wherein the anti- PSMA ADC is ARX517. 46. The pharmaceutical composition of any one of claims 34 to 45 for use in a method of any one of claims 1 to 33.


