WO2025030010A1 - Compositions comprising genetically engineered hematopoietic stem cells and methods of use thereof - Google Patents
Compositions comprising genetically engineered hematopoietic stem cells and methods of use thereofDownload PDFInfo
- Publication number
- WO2025030010A1 WO2025030010A1PCT/US2024/040528US2024040528WWO2025030010A1WO 2025030010 A1WO2025030010 A1WO 2025030010A1US 2024040528 WUS2024040528 WUS 2024040528WWO 2025030010 A1WO2025030010 A1WO 2025030010A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- cell
- cell surface
- surface protein
- genetically engineered
- descendant
- Prior art date
Links
Classifications
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/87—Introduction of foreign genetic material using processes not otherwise provided for, e.g. co-transformation
- C12N15/90—Stable introduction of foreign DNA into chromosome
- C12N15/902—Stable introduction of foreign DNA into chromosome using homologous recombination
- C12N15/907—Stable introduction of foreign DNA into chromosome using homologous recombination in mammalian cells
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K35/00—Medicinal preparations containing materials or reaction products thereof with undetermined constitution
- A61K35/12—Materials from mammals; Compositions comprising non-specified tissues or cells; Compositions comprising non-embryonic stem cells; Genetically modified cells
- A61K35/28—Bone marrow; Haematopoietic stem cells; Mesenchymal stem cells of any origin, e.g. adipose-derived stem cells
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/11—DNA or RNA fragments; Modified forms thereof; Non-coding nucleic acids having a biological activity
- C12N15/113—Non-coding nucleic acids modulating the expression of genes, e.g. antisense oligonucleotides; Antisense DNA or RNA; Triplex- forming oligonucleotides; Catalytic nucleic acids, e.g. ribozymes; Nucleic acids used in co-suppression or gene silencing
- C12N15/1138—Non-coding nucleic acids modulating the expression of genes, e.g. antisense oligonucleotides; Antisense DNA or RNA; Triplex- forming oligonucleotides; Catalytic nucleic acids, e.g. ribozymes; Nucleic acids used in co-suppression or gene silencing against receptors or cell surface proteins
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N5/00—Undifferentiated human, animal or plant cells, e.g. cell lines; Tissues; Cultivation or maintenance thereof; Culture media therefor
- C12N5/06—Animal cells or tissues; Human cells or tissues
- C12N5/0602—Vertebrate cells
- C12N5/0634—Cells from the blood or the immune system
- C12N5/0647—Haematopoietic stem cells; Uncommitted or multipotent progenitors
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N9/00—Enzymes; Proenzymes; Compositions thereof; Processes for preparing, activating, inhibiting, separating or purifying enzymes
- C12N9/14—Hydrolases (3)
- C12N9/16—Hydrolases (3) acting on ester bonds (3.1)
- C12N9/22—Ribonucleases [RNase]; Deoxyribonucleases [DNase]
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N33/00—Investigating or analysing materials by specific methods not covered by groups G01N1/00 - G01N31/00
- G01N33/48—Biological material, e.g. blood, urine; Haemocytometers
- G01N33/50—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing
- G01N33/53—Immunoassay; Biospecific binding assay; Materials therefor
- G01N33/574—Immunoassay; Biospecific binding assay; Materials therefor for cancer
- G01N33/57484—Immunoassay; Biospecific binding assay; Materials therefor for cancer involving compounds serving as markers for tumor, cancer, neoplasia, e.g. cellular determinants, receptors, heat shock/stress proteins, A-protein, oligosaccharides, metabolites
- G01N33/57492—Immunoassay; Biospecific binding assay; Materials therefor for cancer involving compounds serving as markers for tumor, cancer, neoplasia, e.g. cellular determinants, receptors, heat shock/stress proteins, A-protein, oligosaccharides, metabolites involving compounds localized on the membrane of tumor or cancer cells
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2310/00—Structure or type of the nucleic acid
- C12N2310/10—Type of nucleic acid
- C12N2310/20—Type of nucleic acid involving clustered regularly interspaced short palindromic repeats [CRISPR]
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2320/00—Applications; Uses
- C12N2320/30—Special therapeutic applications
- C12N2320/33—Alteration of splicing
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2510/00—Genetically modified cells
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N2800/00—Detection or diagnosis of diseases
- G01N2800/52—Predicting or monitoring the response to treatment, e.g. for selection of therapy based on assay results in personalised medicine; Prognosis
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N2800/00—Detection or diagnosis of diseases
- G01N2800/54—Determining the risk of relapse
Definitions
- CD19-targeted chimeric antigen receptor T cellsCAR T cells
- anti-CD20 monoclonal antibodiese.g., Rituximab
- CD19 and CD20CD20
- targeting lineage-specific proteins of other cell populationsfor example, myeloid lineage cells (e.g., cancers arising from myeloid blasts, monocytes, megakaryocytes, etc.) is not feasible, as these cell populations are necessary for survival.
- aspects of the present disclosurerelate to genetically engineered hematopoietic cells, or descendants thereof, comprising a mutation in a first gene encoding a first cell surface protein and second gene encoding a second cell surface protein.
- the genetically engineered hematopoietic cells, or descendants thereofare genetically engineered such that the genetically engineered hematopoietic cells have reduced or eliminated expression of the first cell surface protein relative to a wildtype counterpart cell or express a mutant of the first cell surface protein.
- the genetically engineered hematopoietic cells, or descendants thereofare genetically engineered such that the genetically engineered hematopoietic cells have reduced or eliminated expression of the second cell surface protein relative to a wildtype counterpart cell, or express a mutant of the second cell surface protein.
- the first cell surface protein and the second cell surface proteinare of a combination as set forth in Table 3 or Table 4.
- the first cell surface protein and the second cell surface proteinare selected from the group consisting of: CD10; CD101; CD117; CD11A; CD11C; CD120B; CD123; CD13; CD133; CD152; CD200; CD205 (LY75); CD226; CD244 (SLAMF4); CD25; CD274; CD305 (LAIR1); CD32; CD33; CD34; CD366; CD38; CD41; CD42B; CD44; CD45RA; CD47; CD48; CD49D (ITGA4); CD49F; CD52; CD58; CD64; CD7; CD70; CD82; CD84; CD85; CD85K; CD86; CD9; CD96; CD99; CLL-1; EMR2 (ADGRE2); FR-B; GPR56; and IL1RAP.
- CD10CD101; CD117; CD11A; CD11C; CD120B; CD123; CD13; CD133; CD152; CD200; CD205 (LY75); CD226; CD
- the first cell surface protein and the second cell surface proteinare selected from the group consisting of: CD85K and CD64; CD45RA and IL1RAP; CD34 and CD133; IL1RAP and CD33; CD47 and CD99; CLL-1 and CD305; CD152 and CD226; CD45RA and CD96; CD45RA and CD123; CD49D and CD305; IL1RAP and CD305; CD58 and CD305; CD49D and CD47; CD33 and CD305; CD49D and CD44; CD64 and CD86; GPR56 and CD133; CD34 and CD52; EMR2 and CD82; CD64 and CD32; CD42B and CD41; CD64 and CD11C; CD99 and CD44; CD200 and CD34; CD11C and CD33; IL1RAP and CD123; CD11C and IL1RAP; CD44 and EMR2; CD205 and CD305; CD44 and CD305; CD49D and CD99; CD274 and CD226; CD49F and
- the first cell surface protein and the second cell surface proteinare selected from the group consisting of: CD33 and CD205 (LY75); CD33 and CD244 (SLAMF4); CD33 and CD305 (LAIR1); CD33 and CD49D (ITGA4); CLL-1 and CD205 (LY75); CLL-1 and CD244 (SLAMF4); CLL-1 and CD305 (LAIR1); CLL-1 and CD49D (ITGA4); EMR2 and CD205 (LY75); EMR2 and CD244 (SLAMF4); EMR2 and CD305 (LAIR1); EMR2 and CD49D (ITGA4); CD38 and CD205 (LY75); CD38 and CD244 (SLAMF4); CD38 and CD305 (LAIR1); CD38 and CD49D (ITGA4); CD305 (LAIR1) and CD49D (ITGA4); CD305 (LAIR1) and CD49D (ITGA4); CD305 (LAIR1) and CD49D (ITGA4); CD305 (LAIR1) and CD49D (ITGA
- the first cell surface proteinis CD33 and the second cell surface protein is IL1RAP. In some embodiments, the first cell surface protein is CD44 and the second cell surface protein is EMR2. In some embodiments, the first cell surface protein and the second cell surface protein are expressed in >50% of AML blasts at targetable antigen levels of >500 proteins (antigens, Ags) per cell. In some embodiments, the first cell surface protein and the second cell surface protein are expressed in >50% of AML blasts at targetable antigen levels of >1000 proteins (antigens, Ags) per cell. In some embodiments, the whole or a portion of the first gene and/or the second gene is deleted.
- the whole or a portion of the first gene and/or the second geneis deleted using genome editing.
- the genome editing carried outinvolves a zinc finger nuclease (ZFN), a transcription activator-like effector-based nuclease (TALEN), or a CRISPR-Cas system.
- ZFNzinc finger nuclease
- TALENtranscription activator-like effector-based nuclease
- the CRISPR-Cas systemcomprises a nucleic acid encoding a gRNA and an RNA-guided nuclease.
- the RNA-guided nucleaseis a base editor.
- the mutation of the first gene and/or the second genecomprises a substitution, deletion, or addition of at least one nucleotide or a combination thereof.
- the mutation of the first gene and/or the second genecomprises a substitution, deletion, or addition of at least one amino acid or a combination thereof in a non-essential epitope of the first and/or the second cell surface protein.
- the mutant of the first cell surface protein and/or the mutant of the second cell surface proteincomprises a substitution, deletion, or addition of at least one amino acid or a combination thereof.
- the mutation of the first gene and/or the second genecomprises a substitution, deletion, or addition of at least one nucleotide or a combination thereof in a sequence encoding a non-essential epitope of the first and/or the second cell surface protein.
- the mutationalters 1, 2, 3, 4, or 5 amino acid residues of the first and/or second cell surface protein. In some embodiments, the mutation alters no more than 1, no more than 2, no more than 3, no more than 4, or no more than 5 amino acid residues of the first and/or second cell surface protein. In some embodiments, expression of the first cell surface protein and/or the second cell surface protein is reduced by at least 20%, at least 30%, at least 40%, at least 50%, at least 60%, at least 70%, at least 80%, at least 90%, or at least 95% relative to the wildtype counterpart cell.
- expression of the first cell surface protein and/or the second cell surface proteinis not substantially altered, wherein the mutant of the first cell surface protein and/or the mutant of the second cell surface protein comprise a modified non-essential epitope.
- mutation in the first genedoes not affect the bioactivity of the first cell surface protein and/or the mutation in the second gene does not affect the bioactivity of the second cell surface protein.
- the cellretains the ability to differentiate normally.
- the cellis a hematopoietic stem or progenitor cell, or a descendant thereof.
- the hematopoietic cellis a hematopoietic stem cell (HSC).
- the hematopoietic cellis a CD34+ cell. In some embodiments, the hematopoietic cell is from bone marrow, blood, umbilical cord, or peripheral blood mononuclear cells (PBMCs). In some embodiments, the hematopoietic cell is a human cell. In some embodiments, the cell has reduced or eliminated binding to a first agent comprising an antigen-binding fragment that binds to the first cell surface protein and/or a second agent comprising an antigen-binding fragment that binds to the second cell surface protein. In some embodiments, the first agent and/or the second agent is a cytotoxic agent.
- the antigen-binding fragment of the cytotoxic agentis a single-chain antibody fragment (scFv) that specifically binds the cell surface protein or an epitope thereof.
- the cytotoxic agentis an antibody or an antibody-drug conjugate (ADC).
- the cytotoxic agentis an immune cell expressing a chimeric receptor that comprises the antigen-binding fragment.
- the immune cellsare T cells.
- the chimeric receptorfurther comprises: a hinge domain; a transmembrane domain; at least one co-stimulatory domain; a cytoplasmic signaling domain; or a combination thereof.
- the chimeric receptorcomprises at least one co-stimulatory signaling domain, which is derived from a co-stimulatory receptor selected from the group consisting of CD27, CD28, 4-1BB, OX40, CD30, ICOS, CD2, CD7, LIGHT, NKG2C, B7-H3, GITR, HVEM, and a combination thereof.
- the chimeric receptorcomprises a cytoplasmic signaling domain, which is from CD3 ⁇ .
- the chimeric receptorcomprises a hinge domain, which is from CD8 ⁇ or CD28.
- the immune cells, the genetically engineered hematopoietic cells, or bothare allogeneic or autologous.
- the present disclosurefurther provides a cell population comprising one or more of the genetically engineered hematopoietic cells, or descendants thereof.
- the present disclosurefurther provides a pharmaceutical composition comprising the genetically engineering hematopoietic cells, or descendants thereof.
- Other aspects of the present disclosurerelate to methods comprising administering to a subject in need thereof the genetically engineered hematopoietic cells, or descendants thereof, the cell population, or the pharmaceutical composition.
- the methodsfurther comprising administering to the subject a therapeutically effective amount of at least one agent that targets the first or second cell surface protein, wherein the agent comprises an antigen binding fragment that binds the first or second cell surface protein.
- the agentis an antibody-drug conjugate or an immune effector cell expressing a chimeric antigen receptor (CAR).
- the subjecthas blastic plasmacytoid dendritic cell neoplasm, multiple myeloma, myelodysplastic syndrome, Hodgkin’s lymphoma, non-Hodgkin’s lymphoma, leukemia, multiple myeloma (MM), myelodysplastic syndrome (MDS), B-cell acute lymphoblastic leukemia (B-ALL), chronic myelogenous leukemia, acute lymphoblastic leukemia, or chronic lymphoblastic leukemia or blastic plasmacytoid dendritic cell neoplasm (BPDCN).
- BPDCNblastic plasmacytoid dendritic cell neoplasm
- the subjecthas a hematopoietic malignancy or a hematopoietic pre-malignant disease.
- the hematopoietic malignancyis acute myeloid leukemia (AML).
- the hematopoietic pre-malignancyis myelodysplastic syndrome (MDS).
- the subjecthas or is at risk of leukemia relapse.
- Other aspects of the present disclosurerelate to methods of clinically evaluating a subject. In some embodiments, the present disclosure provides methods for determining if a subject is at risk of relapse of a disease.
- the methodcomprises: providing a first biological sample obtained from a subject at a first time point and a second biological sample obtained from the subject at a second time point; measuring a level of a first cell surface protein in the first biological sample and in the second biological sample; identifying the subject as at risk for relapse of the disease if the level of the first cell surface protein in the first biological sample deviates from the level of the first cell surface protein in the second biological sample.
- the present disclosureprovides methods for identifying a subject as a candidate for treatment with an agent.
- the methodcomprises: providing a first biological sample obtained from a subject at a first time point and a second biological sample obtained from the subject at a second time point; and measuring a level of a first cell surface protein in the first biological sample and in the second biological sample.
- the cell surface proteinis selected from Table 3 or Table 4.
- the first cell surface protein and the second cell surface proteinare selected from the group consisting of: CD10; CD101; CD117; CD11A; CD11C; CD120B; CD123; CD13; CD133; CD152; CD200; CD205 (LY75); CD226; CD244 (SLAMF4); CD25; CD274; CD305 (LAIR1); CD32; CD33; CD34; CD366; CD38; CD41; CD42B; CD44; CD45RA; CD47; CD48; CD49D (ITGA4); CD49F; CD52; CD58; CD64; CD7; CD70; CD82; CD84; CD85; CD85K; CD86; CD9; CD96; CD99; CLL-1; EMR2 (ADGRE2); FR-B; GPR56; and IL1RAP.
- CD10CD101; CD117; CD11A; CD11C; CD120B; CD123; CD13; CD133; CD152; CD200; CD205 (LY75); CD226; CD
- the first cell surface protein and the second cell surface proteinare selected from the group consisting of: CD85K and CD64; CD45RA and IL1RAP; CD34 and CD133; IL1RAP and CD33; CD47 and CD99; CLL-1 and CD305; CD152 and CD226; CD45RA and CD96; CD45RA and CD123; CD49D and CD305; IL1RAP and CD305; CD58 and CD305; CD49D and CD47; CD33 and CD305; CD49D and CD44; CD64 and CD86; GPR56 and CD133; CD34 and CD52; EMR2 and CD82; CD64 and CD32; CD42B and CD41; CD64 and CD11C; CD99 and CD44; CD200 and CD34; CD11C and CD33; IL1RAP and CD123; CD11C and IL1RAP; CD44 and EMR2; CD205 and CD305; CD44 and CD305; CD49D and CD99; CD274 and CD226; CD49F and
- the first cell surface proteinis CD33 and the second cell surface protein is IL1RAP.
- the first biological sample and/or the second biological samplecomprises blast cells.
- the first time pointis at or near the time the subject is diagnosed with the disease.
- the methodsfurther comprise administering to the subject a therapeutically effective amount of an agent comprising an antigen-binding fragment that binds to the first cell surface protein.
- the methodsfurther comprise administering to the subject a population of genetically engineered hematopoietic cells having reduced or eliminated expression of the first cell surface protein.
- FIG.1is a schematic showing a non-limiting example of a strategy to promote killing of cancer-specific cells.
- the left panelshows the traditional paradigm for drug development in which cancer antigens are targeted to kill cancer cells.
- the middle panelshows the problem with the traditional paradigm in which there are limited unique cancer antigens, so drugs target both cancer and health cells (on-target toxicity).
- FIG.2shows a cartoon representation of cell surface antigen expression heterogeneity in hematopoietic cancer cells across different subjects. This patient-to-patient heterogeneity presents an obstacle to treatment and in acute myeloid leukemia can be driven by diverse mutations and cytogenetics, manifesting distinct blast immunophenotypes that are difficult to target with a single therapy.
- FIG.3shows a cartoon representation of cell surface antigen expression changes that occur over time in hematopoietic cancer cells in individual subjects, such as over different clinical time points (e.g., from cancer diagnosis to cancer relapse).
- FIG.4Bis a heatmap showing pairwise correlation of cell surface protein marker expression in AML blast populations. Bottom and top triangles show Pearson correlation coefficients for diagnosis and relapse samples, respectively.
- FIGs.5A and 5Bshow representative results from flow cytometry analysis of cell surface antigen (CD33, CLL-1, CD123, and EMR2) expression in acute myeloid leukemia (AML) blasts.
- FIG.5Ashows the percent (%) antigen positivity on AML blasts for each of the indicated myeloid markers by flow cytometry.
- FIG.5Bshows the number (#) of antigens per cells (ABC) for each of the indicated myeloid markers by flow cytometry and quantification of the average number of antigens bound per blast cell (ABCs) by contact with QuantiBRITETM beads.
- Each pointrepresents a sample derived from a subject at the time of AML diagnosis or AML relapse.
- FIG.6shows a schematic of a strategy for identifying myeloid-specific surface antigens for analysis in samples from subjects at diagnosis and relapse.
- FIG.7shows a schematic of a strategy for characterizing cell surface antigen expression in matched samples from AML subjects at diagnosis and relapse.
- FIG.10shows cell surface antigen density of the indicated antigens (CD33, CLL-1, CD123, and EMR2) at the timepoints of diagnosis or relapse.
- the antigen derived tag (ADT) counts by single cell feature barcoding technologywas correlated to the antibodies bound per cell (ABC) as quantitated by QuantiBRITETM to estimate the antigen density in each blast.
- Each pointrepresents average cell surface antigen expression intensity for a sample derived from a subject at the time of AML diagnosis (circles) or AML relapse (triangles).
- One thousand antigenscorrespond to a normalized ADT value of 1.2.
- “R”refers to Pearson correlation coefficient.
- FIGs.11A-11Cshow estimated blast surface antigen density to identify targetable antigens across patients at the timepoints of diagnosis and relapse.
- FIG.11Ashows representative results from an unbiased analysis of cell surface antigen expression on AML blast samples, wherein 81 antigens were ranked based on average cell surface expression across all blasts.
- the two heatmaps presentedshow the estimated antigen density across blasts in each sample obtained from a subject either at AML diagnosis (left panel) or AML relapse (right panel).
- Each columncorresponds to an individual blast from a patient with the annotation differentially colored/shaded bars above each column indicating the sample of origin. Higher antigen density for a specific antigen in a single blast is denoted by the darkness of the vertical stripe in each point of the heatmap.
- FIG.11Bshows the heatmap of FIG.11A, right panel, highlighting several patients in which expression of one antigen may compensate for low expression of another antigen (e.g., CLL-1 expression compensating for low CD33 expression), suggesting these patients may benefit from a multi-targeting therapeutic approach.
- FIG.11Cshows the heatmap of FIG.11A, right panel, highlighting CD33 and CLL-1 antigen density in blasts from patient 70, demonstrating variable expression across individual blasts, which suggests that targeting one of the antigens may not completely eradicate the blast population.
- FIGs.12A and 12Bshow representative results obtained from computational analyses used to correlate pairs of cell surface antigens that exhibited co-expression on AML blast cells from patient 70.
- FIG.12Ashows a heat map representation of the correlation between expression of pairs of cell surface antigens on AML blasts.
- FIG.12Bshows a histogram representation indicating the percent of blasts exhibiting the indicated level of estimates antigen density for co-expression of CD33 and CLL-1 or expression of CD33 or CLL-1 individually, suggesting that a multi-targeting therapeutic strategy, such as with multi- specific CAR T cells, may enhance blast killing and guard against antigen escape.
- FIGs.13A-13Eshow further analyses of AML blasts from patient 26 that exhibited increased CD33 expression.
- FIG.13Ashows Uniform Manifold Approximation and Projection (UMAP) plots of blasts at AML diagnosis and AML relapse.
- FIG.13Bshows a heat map representation of differential antigen expression data obtained from analyses of AML blasts at diagnosis.
- FIG.13Bshows a heat map representation of differential antigen expression data obtained from analyses of AML blasts at relapse, indicating upregulation of CD33.
- FIG.13Dshows cell cycle module analysis indicating the cell cycle stage distribution of AML blasts at AML diagnosis and AML relapse.
- FIG.13Eshows a plot presenting differentially expressed genes (log, fold change >2) in blasts at AML diagnosis compared to AML relapse.
- FIGs.14A and 14Bshow results of pathway enrichment analysis in a subset of proliferating blasts to identify pathways that change between AML diagnosis and relapse.
- FIG.14Ashows exemplary pathways that were found to be downregulated in relapse S/G2M blasts.
- FIG.14Bshows exemplary pathways that were found to be upregulated in relapse S/G2M blasts.
- FIGs.15A-15Bshow surface density of the indicated antigens expressed on AML blasts and leukemia stem cells.
- FIG.15Ashows a heat-map representation of data from cellular indexing of transcriptomes and epitopes (CITE-seq) profiling of 81 candidate antigens across 400,000 cells from 28 paired diagnosis and relapse patient bone marrow samples.
- CITE-seqtranscriptomes and epitopes
- FIG.15Bshows percent positivity and number of antigens per antigen-positive cell in blasts and leukemia stem cells. Quantibrite PE beads were used for antigen quantitation. The dots trace expression data for select patients to demonstrate expression pattern for CD33, EMR2 (ADGRE2), CD123, and CLL-1. Data shown as median ⁇ standard error of mean. Leukemic stem cells (LSCs), Diagnosis (Dx), Relapse (Rel).
- FIGs.16A-16Dshow multiplex adenosine base editing of EMR2 (ADGRE2) and CD33 in HSPCs.
- FIG.16Ashows diagrams of the structures of the EMR2 and CD33 genes indicating the adenosine base editing (ABE) gRNA locations.
- FIG.16Bshows an example of strategy for multiple ABE of HSCs and characterizing effects of multiplex editing on CD33 and EMR2.
- FIG.16Cshows the frequency of editing types at day 2 (D2) and day 5 (D5) after electroporation of HSPCs with ABE guides.
- FIG.16Dshows percentage of the ADGRE2 and CD33 surface protein expression of ADGRE2 and CD33 edited cells, in single and multiplex ABE settings, at day 5 (D5).
- N2 donors.
- Data shown as mean ⁇ standard deviation. “dKO”refers to double knockout of EMR2 and CD33 genes.
- FIG.17shows results obtained from flow cytometry analysis of cells that were subjected to base editing conditions that utilized a non-targeting control gRNA (gCTRL) or gRNAs targeting the CD33, CD123, and EMR2 genes (triplex).
- gCTRLnon-targeting control gRNA
- FIG.18A-18Bshow estimated blast surface density of targetable antigens in patients at diagnosis (FIG.18A) and relapse (FIG.18B).
- FIGs.18A-18Bshows representative results from an unbiased analysis of cell surface antigen expression on AML blast samples, wherein 81 antigens were ranked based on average cell surface expression across all blasts.
- the two heatmaps presentedshow the estimated antigen density across blasts in each sample obtained from a subject either at AML diagnosis (FIG.18A) or AML relapse (FIG.18B).
- Each columncorresponds to an individual blast from a patient with the annotation differentially colored/shaded bars above each column indicating the sample of origin.
- Higher antigen density for a specific antigen in a single blastis denoted by the darkness of the vertical stripe in each point of the heatmap.
- the top 25 most highly expressed antigensare highlighted at the top of each heat map.
- FIG.18Bshows the heatmap of FIG.18A, highlighting four additional targets (CD305 (LAIR1), CD49d (ITGA4), CD205 (LY75), and CD244 (SLAMF4)) which are highly expressed on AML blast cells and may be able to compensate for low expression of other antigens in a multi-targeting therapeutic approach.
- CD305LAIR1
- CD49dIGA4
- CD205LY75
- SLAMF4CD244
- FIGs.19A and 19Bshow representative results from an unbiased analysis of cell surface antigen expression on 56 AML paired blast samples (28 at diagnosis and 28 at relapse), wherein 81 antigens were ranked based on average cell surface expression across all blasts.
- FIG.20Ashows CD305 (LAIR1), CD49D (ITGA4), CD205 (LY75), and CD244 (SLAMF4) antigen density in MOLM13 AML cells (shows expression levels as antigens bound per cell on MOLM13 AML cell line at complete saturation and shows a range of antigen densities on MOLM13 cells from 3800-34000 antigens bound per cell).
- FIG.20Bshows cytotoxicity analyses of cells contacted with a primary antibody targeting CD33, CD305 (LAIR1), CD49d (ITGA4), CD205 (LY75), or CD244 (SLAMF4) and then subsequently contacted with a secondary antibody-drug conjugate (Fab-aMFc-CL-MMAF).
- Fab-aMFc-CL-MMAFsecondary antibody-drug conjugate
- FIG.20Bshows the therapeutic efficacy of targeting CD33, CD305 (LAIR1), CD49d (ITGA4), CD205 (LY75), or CD244 (SLAMF4) antigens individually using in vitro cytotoxicity secondary ADC assays in MOLM-13 cells, providing a measure of sensitivity to the ADC as a function of Antigen expression. Greater than 50% cell killing was observed at ⁇ 0.1nM of primary antibody for all four Antigens (CD305, CD49d, CD205, and CD244).
- Secondary ADCsfunction by binding a primary antibody to an antigen of choice and then binding a secondary IgG1 antibody which is attached to a payload, in this case Fab-aMFc-CL-MMAF.
- FIGs.21A-21Bshow results from flow cytometry analyses of CD33, CD305 (LAIR1), CD49D (ITGA4), CD205 (LY75), and CD244 (SLAMF4) expression on four patient samples. QuantiBRITE assay was performed on four AML patient samples from the AML Atlas repository, using flow cytometry to assess percent positivity and antigen density (Antigens bound per cell).
- FIG.21Ashows overall high expression of at least 3 of 4 antigens (CD305 (LAIR1), CD49d (ITGA4), CD205 (LY75)) in all four patient samples and are shown in comparison with the validated antigen, CD33.
- FIG.21Bshows antigen density values for each patient for all five of the same antigens (CD33, CD305 (LAIR1), CD49D (ITGA4), CD205 (LY75), and CD244 (SLAMF4)). This figure tells us that each of the four antigens (CD305, CD49D, CD205, and CD244) have high expression levels and antigen density on a subset of AML patient samples.
- FIG.22shows antigen density (antigens per cell) assessed using the QuantiBRITE assay for five antigens (CD33, CD305, CD49d, CD205, and CD244) on four patient samples which had been previously run through CITE-seq.56 samples were previously sequenced using the CITE-seq method which provided ADT (Antibody derived tags) values for each of the 81 antigens in the panel, and at the same time four antigens were assessed for antigens per cell values using the flow cytometric assay, QuantiBRITE. These four antigens were used to make a model which correlated antigens per cell with ADT to predict antigens per cell values for the other 77 antigens in the panel.
- ADTAntibody derived tags
- CAR-T therapyremains a challenge in treatment of hematopoietic malignance, such as acute myeloid leukemia (AML) due to switch of cancer cell surface proteins on cancer cells, thereby escaping CAR-T therapy (see, e.g., FIG.3).
- AMLacute myeloid leukemia
- B-ALLB-cell acute lymphoblastic leukemia
- AMLacute myeloid leukemia
- the present disclosureprovides methods, cells, compositions, and kits aimed at addressing at least the above-stated problems.
- the methods, cells, compositions, and kits described hereinprovide a safe and effective treatment for hematological malignancies, allowing for targeting of one or more cell surface proteins that are present not only on cancer cells but also on cells critical for the development and/or survival of the subject (see, e.g., FIG.1, right panel). More specifically, hematopoietic cells as described herein can be used (for example) in the treatment of a subject that receives two or more different therapies for cancer. Many therapies deplete the subject’s endogenous, non-cancerous hematopoietic cells.
- Replacement or rescue hematopoietic cells described hereincan replace the subject’s depleted immune cells.
- a subjectreceives the first therapy (e.g., against IL1RAP), and then the cancer relapses, and then the subject receives the second therapy (e.g., against CD33).
- the present applicationprovides, e.g., rescue cells that are resistant to both therapies.
- the rescue cellscan be administered to the subject at or near the time of the first therapy, and if relapse occurs, the subject can then receive the second therapy without depleting the rescue cells.
- the first and second therapiestargets antigens selected from the group consisting of: CD10; CD101; CD117; CD11A; CD11C; CD120B; CD123; CD13; CD133; CD152; CD200; CD205 (LY75); CD226; CD244 (SLAMF4); CD25; CD274; CD305 (LAIR1); CD32; CD33; CD34; CD366; CD38; CD41; CD42B; CD44; CD45RA; CD47; CD48; CD49D (ITGA4); CD49F; CD52; CD58; CD64; CD7; CD70; CD82; CD84; CD85; CD85K; CD86; CD9; CD96; CD99; CLL-1; EMR2 (ADGRE2); FR-B; GPR56; and IL1RAP.
- antigensselected from the group consisting of: CD10; CD101; CD117; CD11A; CD11C; CD120B; CD123; CD13; CD133; CD152; CD200; CD205
- the first therapytargets an antigen selected from the first column in Table 3 or Table 4 and the second therapy targets an antigen selected from the second column Table 3 or Table 4 (e.g., wherein the antigens are selected from the same row in the same Table or from different tables).
- one of the therapiestargets CD33. In some embodiments, one of the therapies targets CD123. In some embodiments, one of the therapies targets CLL-1. In some embodiments, one of the therapies targets CD38. In some embodiments, one of the therapies targets CD34. In some embodiments, one of the therapies targets EMR2. In some embodiments, one of the therapies targets CD205 (LY75). In some embodiments, one of the therapies targets CD244 (SLAMF4).
- one of the therapiestargets CD49D (ITGA4). In some embodiments, one of the therapies targets CD305 (LAIR1). In some embodiments, one of the therapies targets CD305 (LAIR1) and one of therapies targets CD49D (ITGA4). In some embodiments, one of the therapies targets CD305 (LAIR1) and one of therapies targets CD205 (LY75). In some embodiments, one of the therapies targets CD305 (LAIR1) and one of therapies targets CD244 (SLAMF4). In some embodiments, one of the therapies targets CD49D (ITGA4) and one of therapies targets CD205 (LY75). In some embodiments, one of the therapies targets CD49D (ITGA4) and one of therapies targets CD244 (SLAMF4).
- one of the therapiestargets CD205 (LY75) and one of therapies targets CD244 (SLAMF4).
- a subjectmay need to receive two therapies at once, e.g., because the cancer comprises two sub-populations of cells (e.g., one expressing CD33 and the second expressing CD123 and/or CLL-1), and each therapy only attacks one of the sub- populations.
- rescue cells resistant to both therapiescan replace the subject’s depleted immune cells even in the presence of both therapies.
- the cells, which are resistant to both therapiesare genetically engineered in a gene encoding a first cell surface protein and a gene encoding a second cell surface protein.
- the first and second cell surface proteinsare selected from the group consisting of: CD10; CD101; CD117; CD11A; CD11C; CD120B; CD123; CD13; CD133; CD152; CD200; CD205 (LY75); CD226; CD244 (SLAMF4); CD25; CD274; CD305 (LAIR1); CD32; CD33; CD34; CD366; CD38; CD41; CD42B; CD44; CD45RA; CD47; CD48; CD49D (ITGA4); CD49F; CD52; CD58; CD64; CD7; CD70; CD82; CD84; CD85; CD85K; CD86; CD9; CD96; CD99; CLL-1; EMR2 (ADGRE2); FR-B; GPR56; and IL1RAP).
- CD10CD101; CD117; CD11A; CD11C; CD120B; CD123; CD13; CD133; CD152; CD200; CD205 (LY75); CD226; CD244 (S
- the cellshave reduced or eliminated expression of the first cell surface protein relative to a wildtype counterpart cell. In some embodiments, the cells have reduced or eliminated expression of the second cell surface protein relative to a wildtype counterpart cell. In some embodiments, the cells express a mutant of the first cell surface protein (e.g., a mutant that does not bind to a first therapy, such as a first cytotoxic agent). In some embodiments, the cells express a mutant of the second cell surface protein (e.g., a mutant that does not bind to a second therapy, such as a second cytotoxic agent).
- the first cell surface proteinis selected from the first column in Table 3 or Table 4 and the second column Table 3 or Table 4 (e.g., wherein the antigens are selected from the same row in the same Table or from different tables).
- one of the cell surface proteinsis CD33. In some embodiments, one of the cell surface proteins is CD123. In some embodiments, one of the cell surface proteins is CLL-1. In some embodiments, one of the cell surface proteins is CD38. In some embodiments, one of the cell surface proteins is CD34. In some embodiments, one of the cell surface proteins is EMR2.. In some embodiments, one of the cell surface proteins is CD205 (LY75). In some embodiments, one of the cell surface proteins is CD244 (SLAMF4).
- one of the cell surface proteinsis CD49D (ITGA4). In some embodiments, one of the cell surface proteins is CD305 (LAIR1). In some embodiments, one of the cell surface proteins is CD305 (LAIR1) and one of the cell surface proteins is CD49D (ITGA4). In some embodiments, one of the cell surface proteins is CD305 (LAIR1) and one of the cell surface proteins is CD205 (LY75). In some embodiments, one of the cell surface proteins is CD305 (LAIR1) and one of the cell surface proteins is CD244 (SLAMF4). In some embodiments, one of the cell surface proteins is CD49D (ITGA4) and one of the cell surface proteins is CD205 (LY75).
- one of the cell surface proteinsis CD49D (ITGA4) and one of the cell surface proteins is CD244 (SLAMF4).
- one of the cell surface proteinsis CD205 (LY75) and one of the cell surface proteins is CD244 (SLAMF4).
- the present disclosurealso provides a population of rescue hematopoietic cells that comprises a first sub-population of cells that is (and/or gives rise to) cells resistant to a first therapy and a second sub-population of cells that is (and/or gives rise to) cells resistant to a second therapy.
- the populationcan comprise cells that are (and/or give rise to) cells resistant to both therapies; however this is not required in this embodiment.
- the cell populationscan be useful, e.g., when subjects are treated with two therapies sequentially.
- the edited cell surface proteinsare proteins that are typically not expressed in normal HSCs, but become expressed in later lineages, so the transplanted HSCs are resistant to both therapies regardless of whether any HSCs are edited for both cell surface proteins.
- HSCshematopoietic stem cells
- methods of producing such, for examples, via the CRISPR approach using specific guide RNAs (gRNAs)and methods of treating a hematopoietic malignancy using the engineered hematopoietic cells, either taken alone, or in combination with one or more cytotoxic agents (e.g., CAR-T cells) that can target the wild- type cell surface proteins but not those encoded by the edited genes in the engineered hematopoietic cells.
- gRNAsspecific guide RNAs
- a diseasee.g., hematopoietic malignancy
- methods of identifying a subject as a candidate for treatment with an agentbased on the level(s) of one or more cell surface proteins in a biological sample(s).
- hematopoietic stem cellsthat carry genetically edited genes for reducing or eliminating expression of one or more cell surface proteins, or for expressing the one or more cell surface proteins in mutated form.
- the mutated cell surface proteinswould retain at least partial bioactivity of the antigens but can escape targeting by cytotoxic agents such as CAR-T cells specific to the antigens.
- the cell surface proteins of interestmay not be expressed on hematopoietic cells, such as HSCs, in nature. However, such antigens may be expressed on cells differentiated from the HSCs (e.g., descendants thereof). “Expressing a lineage-specific cell surface protein” or “expressing a lineage-specific cell surface antigen” means that at least a portion of the lineage-specific cell surface protein, or antigen thereof, can be detected on the surface of the hematopoietic cells or descendants thereof.
- hematopoietic cellsinclude any cell type or lineage of cells that arise from the hematopoietic cells.
- the descendants of the hematopoietic cellsare a cell type or lineage of cells that have differentiated from the hematopoietic cells.
- the genetically engineered hematopoietic cellsmay be used alone for treating hematopoietic malignancies, or in combination with one or more cytotoxic agents that target the wild-type cell surface proteins but not the mutant encoded by the edited genes in the genetically engineered hematopoietic cells.
- hematopoietic cellsupon differentiation, could compensate the loss of function caused by elimination of functional non-cancerous cells due to immunotherapy that targets cell surface protein(s), which may also be expressed on normal cells. This approach would also broaden the choice of target proteins for immunotherapy such as CART therapy.
- Cell Surface ProteinsAs used herein, the terms “protein,” “peptide,” and “polypeptide” may be used interchangeably and refer to a polymer of amino acid residues linked together by peptide bonds. In general, a protein may be naturally occurring, recombinant, synthetic, or any combination of these.
- lineage-specific cell surface proteinAs used herein, the terms “lineage-specific cell surface protein,” “linage-specific cell surface antigen,” and “cell surface lineage-specific protein” may be used interchangeably and refer to any protein that is sufficiently present on the surface of a cell and is associated with one or more populations of cell lineage(s). For example, the protein may be present on one or more populations of cell lineage(s) and absent (or at reduced levels) on the cell surface of other cell populations.
- the terms “lineage-specific cell surface protein” and “cell surface lineage-specific antigen”maybe used interchangeably and refer to any antigen of a lineage-specific cell surface protein.
- compositions and methods described hereinmay be used to achieve editing of a cell surface protein, which may result in making a variant lineage-specific cell surface protein.
- Such editingmay result in an amino acid substitution in an epitope of interest, such as a cell surface protein epitope.
- lineage-specific cell surface proteinscan be classified based on a number of factors, such as whether the protein and/or the populations of cells that present the protein are required for survival and/or development of the host organism.
- Table 1A summary of exemplary types of lineage-specific proteins is provide in Table 1 below.
- Table 1Classification of Lineage Specific Proteins As shown in Table 1, type 0 lineage-specific cell surface proteins are necessary for the tissue homeostasis and survival, and cell types carrying type 0 lineage-specific cell surface protein may be also necessary for survival of the subject. Thus, given the importance of type 0 lineage-specific cell surface proteins, or cells carrying type 0 lineage-specific cell surface proteins, in homeostasis and survival, targeting this category of proteins may be challenging using conventional CAR T cell immunotherapies, as the inhibition or removal of such proteins and cell carrying such proteins may be detrimental to the survival of the subject.
- lineage-specific cell surface proteinssuch as type 0 lineage-specific proteins
- the cell types that carry such proteinsmay be required for the survival, for example because it performs a vital non-redundant function in the subject, then this type of lineage specific protein may be a poor target for conventional CAR T cell-based immunotherapies.
- the selection of target cell surface proteincan be expanded to essential antigens such as type 0 lineage- specific cell surface proteins.
- the engineered hematopoietic cellshave one or more genes of type 0 antigens edited for expression of these type 0 antigens in mutated form, which retain (at least partially) bioactivity of the type 0 antigens but can escape targeting by type 0 antigen-specific cytotoxic agents such as CAR-T cells so as to remedy the loss of normal cells expressing the type 0 antigens due to the therapy.
- type 0 antigen-specific cytotoxic agentssuch as CAR-T cells so as to remedy the loss of normal cells expressing the type 0 antigens due to the therapy.
- type 1 lineage-specific cell surface proteins and cells carrying type 1 lineage-specific cell surface proteinsare not required for tissue homeostasis or survival of the subject. Targeting type 1 lineage-specific cell surface proteins is not likely to lead to acute toxicity and/or death of the subject.
- CD307a type 1 protein expressed uniquely on both normal plasma cells and multiple myeloma (MM) cells would lead to elimination of both cell types.
- CD307 and other type 1 lineage specific proteinsare proteins that are suitable for CAR T cell-based immunotherapy. Lineage specific proteins of type 1 class may be expressed in a wide variety of different tissues, including, ovaries, testes, prostate, breast, endometrium, and pancreas.
- the genetically engineered hematopoietic cellshave one or more genes of type 1 antigens for expression of the type 1 proteins in mutated forms, which retain (at least partially) bioactivity of the type 1 antigens but can escape targeting by type 1 antigen-specific cytotoxic agents such as CAR-T cells.
- Use of such engineered HSCsmay improve the longer-term survival and quality of life of the patient. For example, targeting all plasma cells, while not expected to lead to acute toxicity and/or death, could have longer-term consequences such as reduced function of the humoral immune system leading to increased risk of infection.
- Type 2 proteinsare those characterized where: (1) the protein is dispensable for the survival of an organism (i.e., is not required for the survival), and (2) the cell lineage carrying the protein is indispensable for the survival of an organism (i.e., the particular cell lineage is required for the survival).
- CD33is a type 2 protein expressed in both normal myeloid cells as well as in Acute Myeloid Leukemia (AML) cells (Dohner et al., NEJM (2015) 373:1136).
- AMLAcute Myeloid Leukemia
- the genetically engineered hematopoietic cellshave one or more genes of type 2 antigens for expression of the type 2 antigens in mutated form, which retain (at least partially) bioactivity of the type 2 antigens but can escape targeting by type 1 antigen-specific cytotoxic agents such as CAR-T cells.
- Use of such engineered HSCsmay improve the longer-term survival and quality of life of the patient. For example, targeting all plasma cells, while not expected to lead to acute toxicity and/or death, could have longer-term consequences such as reduced function of the humoral immune system leading to increased risk of infection.
- malignant hematopoietic cellsmay have altered expression or express a mutant of a cell surface protein.
- malignant hematopoietic cellsmay have altered expression or express a mutant of cell surface protein selected from a group consisting of: CD10; CD101; CD11A; CD11C; CD123; CD133; CD152; CD200; CD205; CD244; CD25; CD305; CD33; CD34; CD366; CD38; CD42B; CD44; CD45RA; CD47; CD48; CD49D; CD49F; CD52; CD58; CD64; CD7; CD84; CD85K; CD9; CD99; CLL-1; EMR2; GPR56; IL1RAP; and CD274
- malignant hematopoietic cellsmay have altered expression or express a mutant of cell surface protein selected from a group consisting of: CD117; CD11A; CD11C; CD120B; CD123; CD13; CD133; CD152;
- malignant hematopoietic cellsmay have altered expression or express of a first cell surface protein and a second cell surface protein selected from a group consisting of: CD274 and CD152; CD274 and CD226; CD152 and CD226; CD10 and CD49F; CD49F and GPR56; CD200 and CD34; CD34 and CD52; CD34 and CD133; GPR56 and CD133; CD42B and CD41; CD85K and CD64; CD85K and CD32; CD64 and CD32; CD64 and CD11C; CD64 and CD86; CD45RA and CD96; CD45RA and IL1RAP; CD45RA and CD123; CD84 and CD33; CD11C and IL1RAP; CD11C and CD33; CD11C and CD86; CD9 and IL1RAP; IL1RAP and CD33; IL1RAP and CD205; IL1RAP and CD305; IL1RAP and CD123; CD33 and CLL-1; CD33 and
- malignant hematopoietic cellsmay have altered expression or express of a first cell surface protein and a second cell surface protein selected from a group consisting of: CD85K and CD64; CD45RA and IL1RAP; CD34 and CD133; IL1RAP and CD33; CD47 and CD99; CLL-1 and CD305; CD152 and CD226; CD45RA and CD96; CD45RA and CD123; CD49D and CD305; IL1RAP and CD305; CD58 and CD305; CD49D and CD47; CD33 and CD305; CD49D and CD44; CD64 and CD86; GPR56 and CD133; CD34 and CD52; EMR2 and CD82; CD64 and CD32; CD42B and CD41; CD64 and CD11C; CD99 and CD44; CD200 and CD34; CD11C and CD33; IL1RAP and CD123; CD11C and IL1RAP; CD44 and EMR2; CD205 and CD305; CD44 and CD305;
- malignant hematopoietic cellsmay have altered expression or express of a first cell surface protein and a second cell surface protein selected from a group consisting of: CD33 and CD205 (LY75); CD33 and CD244 (SLAMF4); CD33 and CD305 (LAIR1); CD33 and CD49D (ITGA4); CLL-1 and CD205 (LY75); CLL-1 and CD244 (SLAMF4); CLL-1 and CD305 (LAIR1); CLL-1 and CD49D (ITGA4); EMR2 and CD205 (LY75); EMR2 and CD244 (SLAMF4); EMR2 and CD305 (LAIR1); EMR2 and CD49D (ITGA4); CD38 and CD205 (LY75); CD38 and CD244 (SLAMF4); CD38 and CD305 (LAIR1); CD38 and CD49D (ITGA4); CD305 (LAIR1) and CD49D (ITGA4); CD305 (LAIR1) and CD49D (ITGA4); CD305 (LAIR1) and CD49
- the hematopoietic cells (HSCs) described hereinmay contain an edited gene encoding one or more cell surface proteins of interest in mutated form (mutants or variants, which are used herein interchangeably), which has reduced binding or no binding to a cytotoxic agent as described herein.
- the variantsmay lack the epitope to which the cytotoxic agent binds.
- the mutantsmay carry one or more mutations of the epitope to which the cytotoxic agent binds, such that binding to the cytotoxic agent is reduced or abolished as compared to the natural or wild-type cell surface protein counterpart.
- Such a variantis preferred to maintain substantially similar biological activity as the wild- type counterpart.
- a cell that is “negative” for a given cell surface proteinhas a substantially reduced expression level of the cell surface protein as compared with its naturally-occurring counterpart (e.g., otherwise similar, unmodified cells), e.g., not detectable or not distinguishable from background levels, e.g., using a flow cytometry assay.
- a cell that is negative for the cell surface proteinhas a level of less than 10%, 5%, 2%, or 1% of as compared with its naturally-occurring counterpart.
- the variantmay share a sequence homology of at least 80% (e.g., 85%, 90%, 95%, 97%, 98%, 99%, or above) as the wild-type counterpart and, in some embodiments, may contain no other mutations in addition to those for mutating or deleting the epitope of interest.
- the “percent identity” of two amino acid sequencesis determined using the algorithm of Karlin and Altschul Proc. Natl. Acad. Sci. USA 87:2264-68, 1990, modified as in Karlin and Altschul Proc. Natl. Acad. Sci. USA 90:5873-77, 1993. Such an algorithm is incorporated into the NBLAST and XBLAST programs (version 2.0) of Altschul, et al. J. Mol.
- the regionis a domain of the cell surface protein of interest that encodes the epitope.
- the varianthas just the epitope deleted.
- the length of the deleted regionmay range from 3-60 amino acids, e.g., 5- 50, 5-40, 10-30, 10-20, etc.
- the mutation(s) or deletions in a mutant of a cell surface proteinmay be within or surround a non-essential epitope such that the mutation(s) or deletion(s) do not substantially affect the bioactivity of the protein.
- the term “epitope”refers to an amino acid sequence (linear or conformational) of a protein, such as a cell surface protein, that is bound by the CDRs of an antibody.
- the cytotoxic agentbinds to one or more (e.g., at least 2, 3, 4, 5 or more) epitopes of a cell surface proteins. In some embodiments, the cytotoxic agent binds to more than one epitope of the cell surface protein and the hematopoietic cells are manipulated such that each of the epitopes is absent and/or unavailable for binding by the cytotoxic agent.
- the genetically engineered HSCs described hereinhave one or more edited genes of cell surface proteins such that the edited genes express mutated cell surface proteins with mutations in one or more non-essential epitopes.
- a non-essential epitoperefers to a domain within the cell surface protein, the mutation in which (e.g., deletion) is less likely to substantially affect the bioactivity of the cell surface protein and thus the bioactivity of the cells expressing such.
- hematopoietic cellscomprising a deletion or mutation of a non-essential epitope of a cell surface protein
- Non-essential epitopes of a cell surface proteincan be identified by the methods described herein or by conventional methods relating to protein structure-function prediction.
- a non-essential epitope of a proteincan be predicted based on comparing the amino acid sequence of a protein from one species with the sequence of the protein from other species. Non-conserved domains are usually not essential to the functionality of the protein.
- non-essential epitope of a proteinis predicted using an algorithm or software, such as the PROVEAN software (see, e.g., see: provean.jcvi.org; Choi et al. PLoS ONE (2012) 7(10): e46688), to predict potential non-essential epitopes in a cell surface protein of interest (“candidate non-essential epitope”).
- genetically engineered cells of the present disclosurecomprise a mutation in a first gene encoding a first cell surface protein and a second gene encoding a second cell surface protein selected from a group consisting of: CD274 and CD152; CD274 and CD226; CD152 and CD226; CD10 and CD49F; CD49F and GPR56; CD200 and CD34; CD34 and CD52; CD34 and CD133; GPR56 and CD133; CD42B and CD41; CD85K and CD64; CD85K and CD32; CD64 and CD32; CD64 and CD11C; CD64 and CD86; CD45RA and CD96; CD45RA and IL1RAP; CD45RA and CD123; CD84 and CD33; CD11C and IL1RAP; CD11C and CD33; CD11C and CD86; CD9 and IL1RAP; IL1RAP and CD33; IL1RAP and CD205; IL1RAP and CD305; IL1RAP and CD123
- genetically engineered cells of the present disclosurecomprise a mutation in a first gene encoding a first cell surface protein and a second gene encoding a second lineage-specific cell surface antigen, wherein mutation of the first gene and second gene results in dysregulated expression of the first cell surface protein and the second cell surface protein seen in subjects diagnosed with a hematopoietic malignancy.
- the first gene and second geneencode combinations of antigens selected from a group consisting of: CD274 and CD152; CD274 and CD226; CD152 and CD226; CD10 and CD49F; CD49F and GPR56; CD200 and CD34; CD34 and CD52; CD34 and CD133; GPR56 and CD133; CD42B and CD41; CD85K and CD64; CD85K and CD32; CD64 and CD32; CD64 and CD11C; CD64 and CD86; CD45RA and CD96; CD45RA and IL1RAP; CD45RA and CD123; CD84 and CD33; CD11C and IL1RAP; CD11C and CD33; CD11C and CD86; CD9 and IL1RAP; IL1RAP and CD33; IL1RAP and CD205; IL1RAP and CD305; IL1RAP and CD123; CD33 and CLL-1; CD33 and CD305; CD11A and CD86; CLL-1 and CD305; CD
- the first gene and second geneencode combinations of antigens selected from a group consisting of: CD33 and CD205 (LY75); CD33 and CD244 (SLAMF4); CD33 and CD305 (LAIR1); CD33 and CD49D (ITGA4); CLL-1 and CD205 (LY75); CLL-1 and CD244 (SLAMF4); CLL-1 and CD305 (LAIR1); CLL-1 and CD49D (ITGA4); EMR2 and CD205 (LY75); EMR2 and CD244 (SLAMF4); EMR2 and CD305 (LAIR1); EMR2 and CD49D (ITGA4); CD38 and CD205 (LY75); CD38 and CD244 (SLAMF4); CD38 and CD305 (LAIR1); CD38 and CD49D (ITGA4); CD305 (LAIR1) and CD49D (ITGA4); CD305 (LAIR1) and CD49D (ITGA4); CD305 (LAIR1) and CD205 (LY75); CD305 (LAIR1) and CD244 (SLAMF
- the first gene and second geneencode combinations of antigens selected from a group consisting of: CD9 and CD82; CD45RA and CD123; CD133 and CD82; CD85K and CD70; CD47 and CD205; CD64 and FR-B; CD11A and CD44; CD85K and CD86; CD366 and CD96; CD101 and CD64; CLL-1 and CD49D; CD49D and CD47; EMR2 and CD82; CD38 and CD305; CD34 and CD7; CD200 and CD25; CD25 and CD45RA; CD49D and CD44; CD11A and CD244; CD133 and CD7; CD48 and CD11A; CD52 and CD133; GPR56 and CD82; CD11A and CD38; CD34 and CD13; CD58 and CD49D; CD47 and CD99; IL1RAP and CD58; CD47 and CD44; CD33 and CD305; CD64 and CD86; CD152 and CD120B; CD42B and CD41; CD34 and CD52; CD
- the first gene and second geneencode combinations of antigens selected from a group consisting of: CD33 and CD205 (LY75); CD33 and CD244 (SLAMF4); CD33 and CD305 (LAIR1); CD33 and CD49D (ITGA4); CLL-1 and CD205 (LY75); CLL-1 and CD244 (SLAMF4); CLL-1 and CD305 (LAIR1); CLL-1 and CD49D (ITGA4); EMR2 and CD205 (LY75); EMR2 and CD244 (SLAMF4); EMR2 and CD305 (LAIR1); EMR2 and CD49D (ITGA4); CD38 and CD205 (LY75); CD38 and CD244 (SLAMF4); CD38 and CD305 (LAIR1); CD38 and CD49D (ITGA4); CD305 (LAIR1) and CD49D (ITGA4); CD305 (LAIR1) and CD49D (ITGA4); CD305 (LAIR1) and CD205 (LY75); CD305 (LAIR1) and CD244 (SLAMF
- Variant Cell Surface ProteinsMethods for assessing the functionality of the cell surface protein and the hematopoietic cells or descendants thereof will be known in the art and include, for example, proliferation assays, differentiation assays, colony formation, expression analysis (e.g., gene and/or protein), protein localization, intracellular signaling, functional assays, and in vivo humanized mouse models. Any of the methods for identifying and/or verifying non-essential epitopes in cell surface proteins is also within the scope of the present disclosure.
- bindingrefers to the gRNA molecule and the target domain forming a complex.
- the complexmay comprise two strands forming a duplex structure, or three or more strands forming a multi-stranded complex.
- the bindingmay constitute a step in a more extensive process, such as the cleavage of the target domain by a Cas nuclease.
- the gRNAbinds to the target domain with perfect complementarity, and in other embodiments, the gRNA binds to the target domain with partial complementarity, e.g., with one or more mismatches.
- the full targeting domain of the gRNAbase pairs with the targeting domain.
- the interactionis sufficient to mediate a target domain-mediated cleavage event.
- the terms “binds,” “specifically binds,” “specifically recognizes” and analogous terms as used herein with reference to a protein (e.g., a cell surface protein) and an agent (e.g., a ligand or an antibody) interactionrefer to the specific binding or association between the agent (e.g., a ligand or an antibody) and protein (e.g., a cell surface protein).
- agents that specifically bind a cell surface proteinare known in the art and/or can be identified, for example, by immunoassays, BIAcoreTM, or other techniques known to those of skill in the art.
- reduced bindingrefers to binding that is reduced by at least about 5%.
- the level of bindingmay refer to the amount of binding of the agent (e.g., a ligand or an antibody) to a cell, such as a hematopoietic cell or descendant thereof, or to the amount of binding of the agent (e.g., a ligand or an antibody) to the cell surface protein.
- the level of binding of a cellsuch as a hematopoietic cell or descendant thereof, that has been modified, for example, to include an amino acid substitution in a cell surface protein epitope, may be relative to the level of binding of the agent (e.g., a ligand or an antibody) to a cell that has not been modified as determined by the same assay under the same conditions.
- the agente.g., a ligand or an antibody
- the level of binding of a cell surface protein that includes an amino acid substitution in a cell surface protein epitope to an agentmay be relative to the level of binding of the agent to a cell surface protein that does not include the amino acid substitution in the cell surface protein epitope (e.g., a wild-type protein or a variant thereof) as determined by the same assay under the same conditions.
- the level of binding of a cell surface protein that lacks an epitope to an agentmay be relative to the level of binding of the agent to a cell surface protein that contains the unmodified epitope (e.g., a wild-type protein) as determined by the same assay under the same conditions.
- the bindingis reduced by at least about 1%, at least about 2%, at least about 3%, at least about 4%, at least about 5%, at least about 6%, at least about 7%, at least about 8%, at least about 9%, at least about 10%, at least about 15%, at least about 20%, at least about 25%, at least about 30%, at least about 35%, at least about 40%, at least about 45%, at least about 50%, at least about 55%, at least about 60%, at least about 65%, at least about 70%, at least about 75%, at least about 80%, at least about 85%, at least about 90%, at least about 95%, at least about 99%, at least about 100% or more, e.g., as compared to the unmodified epitope (e.g., a wild-type protein).
- the unmodified epitopee.g., a wild-type protein
- the bindingis reduced by at least about 1-fold, at least about 2-fold, at least about 3-fold, at least about 4-fold, at least about 5-fold, at least about 6-fold, at least about 7-fold, at least about 8-fold, at least about 9-fold, at least about 10-fold, at least about 20-fold, at least about 30-fold, at least about 40-fold, at least about 50-fold, at least about 60-fold, at least about 70-fold, at least about 80- fold, at least about 90-fold, or at least about 100-fold, e.g., as compared to the unmodified epitope (e.g., a wild-type protein).
- the bindingis reduced such that there is substantially no detectable binding in a conventional assay.
- binding affinity or binding specificity for an epitope or proteincan be determined by a variety of methods including equilibrium dialysis, equilibrium binding, gel filtration, ELISA, surface plasmon resonance, or spectroscopy.
- no bindingrefers to substantially no binding, e.g., no detectable binding or only baseline binding as determined in a conventional binding assay.
- no binding of the cells, e.g., hematopoietic cells or descendant thereof, to the agentrefers to a baseline level of binding, as shown using any conventional binding assay known in the art.
- the level of binding of the cells, e.g., hematopoietic cells or descendants thereof, that have been modified and the agentis not biologically significant.
- the term “no binding”is not intended to require the absolute absence of binding.
- a subjectcan be administered the rescue cells (e.g., hematopoietic stem cells (HSCs) and/or hematopoietic progenitor cells (HPCs)) described herein comprising a modification in a cell surface protein gene, e.g., a genetic edit (i.e., mutation) that results in the rescue cells having reduced or eliminated expression of the respective gene, or a modification of an epitope of the protein encoded by the respective gene that diminishes the binding of the therapeutic agent to the protein.
- a genetic editi.e., mutation
- the bindingis reduced such that there is substantially no detectable binding in a conventional assay.
- the binding affinity or binding specificity for an epitope or proteincan be determined by a variety of methods including equilibrium dialysis, equilibrium binding, gel filtration, ELISA, surface plasmon resonance, or spectroscopy.
- the reduction in binding activitymay include an increase in K D , IC 50 , and/or EC 50 .
- the K D , IC 50 , and/or EC 50may be increased by the amino acid substitution by at least about 1%, at least about 2%, at least about 3%, at least about 4%, at least about 5%, at least about 6%, at least about 7%, at least about 8%, at least about 9%, at least about 10%, at least about 15%, at least about 20%, at least about 25%, at least about 30%, at least about 35%, at least about 40%, at least about 45%, at least about 50%, at least about 55%, at least about 60%, at least about 65%, at least about 70%, at least about 75%, at least about 80%, at least about 85%, at least about 90%, at least about 95%, at least about 99%, at least about 100% or more, e.g., as compared to the unmodified epitope (e.g., a wild- type protein).
- the unmodified epitopee.g., a wild- type protein
- the term “IC50”refers to the concentration of an agent (e.g., an inhibitor, such as a ligand or an antibody) that produces 50% of the maximal inhibition of activity or expression measurable using the same assay in the absence of the binding partner.
- the IC50can be as measured in vitro or in vivo.
- the IC50can be determined by measuring activity using a conventional in vitro assay (e.g., a protein activity assay and/or a gene expression assay).
- the term “EC 50 ”refers to the concentration of a binding partner (e.g., an activator, such as a ligand or an antibody) that produces 50% of maximal activation of measurable activity or expression using the same assay in the absence of the agent.
- KDrefers to the dissociation equilibrium constant of a particular agent- protein interaction, such as an antibody-antigen interaction or a ligand-receptor interaction, or the dissociation rate constant of an antibody or antibody-binding fragment.
- antigen Ymay be expressed as a binding ratio determined by dividing the larger KD value (lower, or weaker, affinity) by the smaller KD (higher, or stronger, affinity), for example expressed as 5-fold or 10-fold greater binding affinity, as the case may be.
- "low affinity"refers to less strong binding interaction.
- the low binding affinitycorresponds to greater than about 1 nM KD, greater than about 1 nM, about 2 nM, about 3 nM, about 4 nM, about 5 nM, about 6 nM, about 7 nM, about 8 nM, about 9 nM, about 10 nM, about 11 nM, about 12 nM, about 13 nM, about 14 nM, about 15 nM, about 16 nM, about 17 nM, about 18 nM, about 19 nM, about 20 nM, about 21 nM, about 22 nM, about 23 nM, about 24 nM, about 25 nM, about 26 nM, about 27 nM, about 28 nM, about 29 nM, about 30 nM, about 31 nM, about 32 nM, about 33 nM, about 34 nM, about 35 nM, about 36 nM, about 37 nM, about 38 nM, about 39 nM, about
- the low binding affinitycorresponds to greater than about 10 nM, about 11 nM, about 12 nM, about 13 nM, about 14 nM, about 15 nM, about 16 nM, about 17 nM, about 18 nM, about 19 nM, about 20 nM, about 21 nM, about 22 nM, about 23 nM, about 24 nM, about 25 nM, about 26 nM, about 27 nM, about 28 nM, about 29 nM, about 30 nM, about 31 nM, about 32 nM, about 33 nM, about 34 nM, about 35 nM, about 36 nM, about 37 nM, about 38 nM, about 39 nM, or about 40 nM EC50, wherein such EC50 binding affinity value is measured, e.g., in an in vitro FACS binding assay, or equivalent cell-based binding assay.
- weak affinityrefers to weak binding interaction.
- the weak binding affinitycorresponds to greater than about 100 nM K D or EC50, greater than about 200, 300, or greater than about 500 nM KD or EC50, wherein such KD binding affinity value is measured, e.g., in an in vitro surface plasmon resonance binding assay, or equivalent biomolecular interaction sensing assay, and such EC50 binding affinity value is measured, e.g., in an in vitro FACS binding assay, or equivalent cell-based interaction detecting assay to detect monovalent biding.
- genetically engineered cells of the present disclosureexpress a mutant cell surface protein selected from a group consisting of: CD10; CD101; CD117; CD11A; CD11C; CD120B; CD123; CD13; CD133; CD152; CD200; CD205 (LY75); CD226; CD244 (SLAMF4); CD25; CD274; CD305 (LAIR1); CD32; CD33; CD34; CD366; CD38; CD41; CD42B; CD44; CD45RA; CD47; CD48; CD49D (ITGA4); CD49F; CD52; CD58; CD64; CD7; CD70; CD82; CD84; CD85; CD85K; CD86; CD9; CD96; CD99; CLL- 1; EMR2 (ADGRE2); FR-B; GPR56; and
- genetically engineered cells of the present disclosureexpress a mutant cell surface protein selected from a group consisting of: CD10; CD101; CD11A; CD11C; CD123; CD133; CD152; CD200; CD205; CD244; CD25; CD305; CD33; CD34; CD366; CD38; CD42B; CD44; CD45RA; CD47; CD48; CD49D; CD49F; CD52; CD58; CD64; CD7; CD84; CD85K; CD9; CD99; CLL-1; EMR2; GPR56; IL1RAP; and CD274.
- a mutant cell surface proteinselected from a group consisting of: CD10; CD101; CD11A; CD11C; CD123; CD133; CD152; CD200; CD205; CD244; CD25; CD305; CD33; CD34; CD366; CD38; CD42B; CD44; CD45RA; CD47; CD48; CD49D; CD49F; CD52; CD58; CD64; CD7
- genetically engineered cells of the present disclosureexpress a mutant cell surface protein selected from a group consisting of: CD117; CD11A; CD11C; CD120B; CD123; CD13; CD133; CD152; CD205; CD226; CD244; CD25; CD305; CD32; CD33; CD34; CD38; CD41; CD44; CD45RA; CD47; CD49D; CD49F; CD52; CD58; CD64; CD7; CD70; CD82; CD86; CD96; CD99; CLL-1; EMR2; FR-B; GPR56; and IL1RAP.
- a mutant cell surface proteinselected from a group consisting of: CD117; CD11A; CD11C; CD120B; CD123; CD13; CD133; CD152; CD205; CD226; CD244; CD25; CD305; CD32; CD33; CD34; CD38; CD41; CD44; CD45RA; CD47; CD49D; CD49F; CD52; CD58; CD
- genetically engineered cells of the present disclosureexpress a mutant of a first cell surface protein and a mutant of a second cell surface protein selected from a group consisting of: CD10; CD101; CD117; CD11A; CD11C; CD120B; CD123; CD13; CD133; CD152; CD200; CD205 (LY75); CD226; CD244 (SLAMF4); CD25; CD274; CD305 (LAIR1); CD32; CD33; CD34; CD366; CD38; CD41; CD42B; CD44; CD45RA; CD47; CD48; CD49D (ITGA4); CD49F; CD52; CD58; CD64; CD7; CD70; CD82; CD84; CD85; CD85K; CD86; CD9; CD96; CD99; CLL-1; EMR2 (ADGRE2); FR-B; GPR56; and IL1RAP.
- CD10CD101; CD117; CD11A; CD11C; CD120B; CD123; CD13;
- genetically engineered cells of the present disclosureexpress a mutant of a first cell surface protein and a mutant of a second cell surface protein selected from a group consisting of: CD274 and CD152; CD274 and CD226; CD152 and CD226; CD10 and CD49F; CD49F and GPR56; CD200 and CD34; CD34 and CD52; CD34 and CD133; GPR56 and CD133; CD42B and CD41; CD85K and CD64; CD85K and CD32; CD64 and CD32; CD64 and CD11C; CD64 and CD86; CD45RA and CD96; CD45RA and IL1RAP; CD45RA and CD123; CD84 and CD33; CD11C and IL1RAP; CD11C and CD33; CD11C and CD86; CD9 and IL1RAP; IL1RAP and CD33; IL1RAP and CD205; IL1RAP and CD305; IL1RAP and CD123; CD33 and CLL-1; CD33
- genetically engineered cells of the present disclosureexpress a mutant of a first cell surface protein and a mutant of a second cell surface protein selected from a group consisting of: CD85K and CD64; CD45RA and IL1RAP; CD34 and CD133; IL1RAP and CD33; CD47 and CD99; CLL-1 and CD305; CD152 and CD226; CD45RA and CD96; CD45RA and CD123; CD49D and CD305; IL1RAP and CD305; CD58 and CD305; CD49D and CD47; CD33 and CD305; CD49D and CD44; CD64 and CD86; GPR56 and CD133; CD34 and CD52; EMR2 and CD82; CD64 and CD32; CD42B and CD41; CD64 and CD11C; CD99 and CD44; CD200 and CD34; CD11C and CD33; IL1RAP and CD123; CD11C and IL1RAP; CD44 and EMR2; CD205 and CD305; CD44 and CD305
- genetically engineered cells of the present disclosureexpress a mutant of a first cell surface protein and a mutant of a second cell surface protein selected from a group consisting of: CD274 and CD152; CD274 and CD226; CD152 and CD226; CD10 and CD49F; CD49F and GPR56; CD200 and CD34; CD34 and CD52; CD34 and CD133; GPR56 and CD133; CD42B and CD41; CD85K and CD64; CD85K and CD32; CD64 and CD32; CD64 and CD11C; CD64 and CD86; CD45RA and CD96; CD45RA and IL1RAP; CD45RA and CD123; CD84 and CD33; CD11C and IL1RAP; CD11C and CD33; CD11C and CD86; CD9 and IL1RAP; IL1RAP and CD33; IL1RAP and CD205; IL1RAP and CD305; IL1RAP and CD123; CD33 and CLL-1; CD33
- genetically engineered cells of the present disclosureexpress a mutant of a first cell surface protein and a mutant of a second cell surface protein selected from a group consisting of: CD9 and CD82; CD45RA and CD123; CD133 and CD82; CD85K and CD70; CD47 and CD205; CD64 and FR-B; CD11A and CD44; CD85K and CD86; CD366 and CD96; CD101 and CD64; CLL-1 and CD49D; CD49D and CD47; EMR2 and CD82; CD38 and CD305; CD34 and CD7; CD200 and CD25; CD25 and CD45RA; CD49D and CD44; CD11A and CD244; CD133 and CD7; CD48 and CD11A; CD52 and CD133; GPR56 and CD82; CD11A and CD38; CD34 and CD13; CD58 and CD49D; CD47 and CD99; IL1RAP and CD58; CD47 and CD44; CD33 and CD305; CD64 and CD86; CD152
- genetically engineered cells of the present disclosureexpress a mutant of a first cell surface protein and a mutant of a second cell surface protein selected from a group consisting of: CD33 and CD205 (LY75); CD33 and CD244 (SLAMF4); CD33 and CD305 (LAIR1); CD33 and CD49D (ITGA4); CLL-1 and CD205 (LY75); CLL-1 and CD244 (SLAMF4); CLL-1 and CD305 (LAIR1); CLL-1 and CD49D (ITGA4); EMR2 and CD205 (LY75); EMR2 and CD244 (SLAMF4); EMR2 and CD305 (LAIR1); EMR2 and CD49D (ITGA4); CD38 and CD205 (LY75); CD38 and CD244 (SLAMF4); CD38 and CD305 (LAIR1); CD38 and CD49D (ITGA4); CD305 (LAIR1) and CD49D (ITGA4); CD305 (LAIR1) and CD49D (ITGA4); CD305 (LAIR1) and CD
- a genetically engineered hematopoietic cellhas reduced or eliminated expression of a cell surface protein, or expresses a mutant of a cell surface protein, wherein the cell surface protein is CLL-1.
- C-type lectin-like molecule-1(CLL-1), which may also be referred to as CLEC12A, is a C-type lectin-like receptor family member that is frequently expressed on acute myeloid leukemia (AML) cells.
- CLL-1is expressed by hematopoietic cells, e.g., hematopoietic stem cells and/or hematopoietic progenitor cells.
- the CLL-1 genecomprises 6 exons and is located on human chromosome 12.
- An example of a CLL-1 gene sequencecan be found at NG_029426.2.
- a mutation of a gene encoding CLL-1alters one or more amino acids associated with an epitope of CLL-1.
- the epitope of CLL-1is a portion of CLL-1 bound by an agent, e.g., an immunotherapeutic agent.
- the agentis an anti-CLL-1 antibody.
- the epitope of CLL-1comprises one or more amino acids encoded by exon 2 of the gene encoding CLL-1. In some embodiments, the epitope of CLL-1 comprises one or more amino acids encoded by exon 3 of the gene encoding CLL-1. In some embodiments, the epitope of CLL-1 comprises one or more amino acids encoded by exon 4 of the gene encoding CLL-1. In some embodiments, the epitope of CLL-1 comprises one or more amino acids encoded by exon 5 of the gene encoding CLL-1. In some embodiments, the epitope of CLL-1 comprises one or more amino acids encoded by exon 6 of the gene encoding CLL-1.
- a mutationchanges the amino acid sequence in a manner corresponding to a tolerable genetic variant identified by one or more genomic sequence comparison algorithms, e.g., gnomAD (see, e.g., Gudmundsson et al. arXiv:2107.11458v3, e.g., gnomad.broadinstitute.org/) or to a position characterized by a plurality of tolerable genetic variants.
- genomic sequence comparison algorithmse.g., gnomAD (see, e.g., Gudmundsson et al. arXiv:2107.11458v3, e.g., gnomad.broadinstitute.org/) or to a position characterized by a plurality of tolerable genetic variants.
- mutations to CLL-1 corresponding to the amino acid sequence of a CLL-1 ortholog or at positions characterized by a plurality of tolerable genetic variantsdecrease or eliminate binding of an immunotherapeutic agent targeting CLL-1 while preserving some or all of CLL-1 structure, expression, and/or functionality, providing a cell expressing CLL-1 (e.g., functional CLL-1) that is targeted less or not at all by anti-CLL-1 immunotherapeutic agents.
- alterationresults in a missense variant of CLL-1.
- a genetically engineered hematopoietic cellhas reduced or eliminated expression of a cell surface protein, or of a cell surface protein, wherein the cell surface protein is CD123.
- CD123also known as interleukin-3 receptor alpha or IL3R ⁇
- IL3is a type I cytokine receptor which binds to IL3.
- IL3is a pleiotropic cytokine that regulates the function and production of hematopoietic and immune cells (see, e.g., Testa et al. Biomarker Research volume 2, Article number: 4 (2014)).
- Dysregulated expression of IL3is associated with various cancers including myeloma (see, e.g., Lee et al. Blood (2004) 103 (6): 2308- 2315).
- a hematopoietic malignancyis characterized by cells expressing (e.g., over-expressing) CD123.
- Dysregulated expression of CD123is associated with various hematopoietic malignancies including hairy cell leukemia, acute myeloid leukemia, blastic plasmacytoid dendritic cell neoplasm, and systemic mastocytosis (see, e.g., Del Giudice et al. Hematologica (2004) 89 (3): 303-308; Munoz et al.
- CD123is expressed by hematopoietic cells, e.g., hematopoietic stem cells and/or hematopoietic progenitor cells.
- a mutation of a gene encoding CD123alters one or more amino acids associated with an epitope of CD123.
- the epitope of CD123is a portion of CD123 bound by an agent, e.g., an immunotherapeutic agent.
- the agentis an anti-CD123 antibody.
- the agentcomprises an anti-CD123 antibody or portion thereof, e.g., an antibody drug conjugate (ADC), a chimeric antigen receptor (CAR), or a multispecific antibody (e.g., a bispecific T cell engager).
- the agentcan be anti-CD123 antibody 7G3 or a variant thereof (e.g., a humanized variant, e.g., antibody CSL-36).
- the agentis an anti-CD123 drug, e.g., talacotuzumab.
- the epitope of CD123is one or more amino acids of a protein domain (e.g., the extracellular domain) or the amino acids encoded by an exon or combination of exons of the gene encoding CD123.
- the epitope of CD123comprises one or more amino acids encoded by exon 2 of the gene encoding CD123.
- the epitope of CD123comprises one or more amino acids encoded by exon 3 of the gene encoding CD123. In some embodiments, the epitope of CD123 comprises one or more amino acids encoded by exon 4 of the gene encoding CD123. In some embodiments, the epitope of CD123 comprises one or more amino acids encoded by exon 5 of the gene encoding CD123. In some embodiments, the epitope of CD123 comprises one or more amino acids encoded by exon 6 of the gene encoding CD123.
- the epitope of CD123comprises one or more (e.g., two or more, three or more, four or more, or all) of the amino acids at positions 51, 59, 61, 82, or 84 of a wildtype gene encoding CD123 or at corresponding positions in a homologous CD123 gene.
- a mutation of a gene encoding CD123comprises a substitution of the amino acid at position 51 of a wildtype CD123 or at a corresponding position in a homologous CD123 gene.
- a lysineis substituted for the amino acid at position 51 of a wildtype CD123 or at a corresponding position in a homologous CD123 gene.
- a glycineis substituted for the amino acid at position 51 of a wildtype CD123 or at a corresponding position in a homologous CD123 gene.
- a mutation of a gene encoding CD123comprises a substitution of the amino acid at position 59 of a wildtype CD123 or at a corresponding position in a homologous CD123 gene.
- a phenylalanineis substituted for the amino acid at position 59 of a wildtype CD123 or at a corresponding position in a homologous CD123 gene.
- a cysteineis substituted for the amino acid at position 59 of a wildtype CD123 or at a corresponding position in a homologous CD123 gene.
- a mutation of a gene encoding CD123comprises a substitution of the amino acid at position 61 of a wildtype CD123 or at a corresponding position in a homologous CD123 gene.
- a leucineis substituted for the amino acid at position 61 of a wildtype CD123 or at a corresponding position in a homologous CD123 gene.
- a mutation of a gene encoding CD123comprises a substitution of the amino acid at position 82 of a wildtype CD123 or at a corresponding position in a homologous CD123 gene.
- an alanineis substituted for the amino acid at position 82 of a wildtype CD123 or at a corresponding position in a homologous CD123 gene.
- a mutation of a gene encoding CD123comprises a substitution of the amino acid at position 84 of a wildtype CD123 or at a corresponding position in a homologous CD123 gene.
- a glutamineis substituted for the amino acid at position 84 of a wildtype CD123 or at a corresponding position in a homologous CD123 gene.
- an alanineis substituted for the amino acid at position 84 of a wildtype CD123 or at a corresponding position in a homologous CD123 gene.
- a mutation of a gene encoding CD123makes a change in the amino acid sequence corresponding to the amino acid sequence of a CD123 ortholog.
- a mutationsubstitutes an amino acid of human CD123 for an amino acid at a corresponding position of an orthologous CD123, e.g., a non-human primate CD123.
- a mutationinserts or deletes one or more amino acids of human CD123 to correspond to the sequence of an orthologous CD123, e.g., a non-human primate CD123.
- a mutationchanges the amino acid sequence in a manner corresponding to a tolerable genetic variant identified by one or more genomic sequence comparison algorithms, e.g., gnomAD (see, e.g., Gudmundsson et al. arXiv:2107.11458v3, e.g., gnomad.broadinstitute.org/) or to a position characterized by a plurality of tolerable genetic variants.
- genomic sequence comparison algorithmse.g., gnomAD (see, e.g., Gudmundsson et al. arXiv:2107.11458v3, e.g., gnomad.broadinstitute.org/) or to a position characterized by a plurality of tolerable genetic variants.
- mutations to CD123 corresponding to the amino acid sequence of a CD123 ortholog or at positions characterized by a plurality of tolerable genetic variantsdecrease or eliminate binding of an immunotherapeutic agent targeting CD123 while preserving some or all of CD123 structure, expression, and/or functionality, providing a cell expressing CD123 (e.g., functional CD123) that is targeted less or not at all by anti-CD123 immunotherapeutic agents.
- alterationresults in a missense variant of CD123.
- CD38is expressed by hematopoietic cells, e.g., hematopoietic stem cells and/or hematopoietic progenitor cells.
- a mutation of a gene encoding CD38alters one or more amino acids associated with an epitope of CD38.
- the epitope of CD38is a portion of CD38 bound by an agent, e.g., an immunotherapeutic agent.
- the agentis an anti-CD38 antibody.
- the agentcomprises an anti-CD38 antibody or portion thereof, e.g., an antibody drug conjugate (ADC), a chimeric antigen receptor (CAR), or a multispecific antibody (e.g., a bispecific T cell engager).
- ADCantibody drug conjugate
- CARchimeric antigen receptor
- a multispecific antibodye.g., a bispecific T cell engager
- the agentcan be anti-CD38 antibody HB7 or a variant thereof (e.g., a humanized variant).
- the agentis an anti-CD38 drug, e.g., daratumumab.
- the epitope of CD38is one or more amino acids of a protein domain (e.g., the extracellular domain) or the amino acids encoded by an exon or combination of exons of the gene encoding CD38.
- an alanineis substituted for the amino acid at position 270 of a wildtype CD38 or at a corresponding position in a homologous CD38 gene.
- a mutation of a gene encoding CD38comprises a substitution of the amino acid at position 271 of a wildtype CD38 or at a corresponding position in a homologous CD38 gene.
- a mutation of a gene encoding CD38comprises a substitution of the amino acid at position 272 of a wildtype CD38 or at a corresponding position in a homologous CD38 gene.
- a mutation of a gene encoding CD38comprises a substitution of the amino acid at position 274 of a wildtype CD38 or at a corresponding position in a homologous CD38 gene.
- a phenylalanineis substituted for the amino acid at position 274 of a wildtype CD38 or at a corresponding position in a homologous CD38 gene.
- a mutation of a gene encoding CD38makes a change in the amino acid sequence corresponding to the amino acid sequence of a CD38 ortholog.
- a mutationsubstitutes an amino acid of human CD38 for an amino acid at a corresponding position of an orthologous CD38, e.g., a non-human primate CD38.
- a mutationinserts or deletes one or more amino acids of human CD38 to correspond to the sequence of an orthologous CD38, e.g., a non-human primate CD38.
- a mutationchanges the amino acid sequence in a manner corresponding to a tolerable genetic variant identified by one or more genomic sequence comparison algorithms, e.g., gnomAD, or to a position characterized by a plurality of tolerable genetic variants.
- EGF-like module-containing mucin-like hormone receptor-like 2is a 823-amino acid, ⁇ 90 kDa protein (depending on isoform) of the EGF-seven-span transmembrane (TM7) family of adhesion G protein-coupled receptors (GPCR) with a high level of homology with CD97.
- EMR2forms a heterodimer and binds to chondroitin sulfate B via its EGF-like domain 4 and mediate cell adhesion, granulocyte chemotaxis, degranulation, and the release of pro- inflammatory cytokines in macrophages. See, e.g.
- a mutation of a gene encoding EMR2alters one or more amino acids associated with an epitope of EMR2.
- the epitope of EMR2is a portion of EMR2 bound by an agent, e.g., an immunotherapeutic agent.
- the agentis an anti-EMR2 antibody.
- the agentcomprises an anti- EMR2 antibody or portion thereof, e.g., an antibody drug conjugate (ADC), a chimeric antigen receptor (CAR), or a multispecific antibody (e.g., a bispecific T cell engager).
- the agentis an anti- EMR2 drug.
- the epitope of EMR2corresponds to the amino acids of a protein domain (e.g., the extracellular domain) or the amino acids encoded by an exon or combination of exons of the gene encoding EMR2.
- the epitope of EMR2comprises amino acids encoded by exon 1, exon 2, exon 3, exon 4, exon 5, exon 6, exon 7, exon 8, exon 9, exon 10, exon 11, exon 12, exon 13, exon 14, exon 15, exon 16, exon 17, exon 18, exon 19, exon 20, or exon 21 of EMR2, or a combination thereof.
- a mutation of a gene encoding EMR2makes a change in the amino acid sequence corresponding to the amino acid sequence of a EMR2 ortholog.
- an alterationcomprises substitution of amino acid at any one or more of positions 124, 132, 146, 292, 294, 295, 296, 298, 299, 303, 304, 305, 306, 307, 308, 312, 318, 320, 328, 329, 331, 332, 335, 340, 347, 527, or 708 of a wildtype EMR2 or at a corresponding position in a homologous EMR2.
- an alterationresults in a missense variant of EMR2.
- alterationresults in a change at a splice region in EMR2.
- a mutation of a gene encoding EMR2alters one or more amino acids associated with an epitope of EMR2.
- the epitope of EMR2is a portion of EMR2 bound by an agent, e.g., an immunotherapeutic agent.
- the agentis an anti-EMR2 antibody.
- the agentcomprises an anti-EMR2 antibody or portion thereof, e.g., an antibody drug conjugate (ADC), a chimeric antigen receptor (CAR), or a multispecific antibody (e.g., a bispecific T cell engager).
- the agentcan be anti-EMR2 monoclonal antibody 2A1 (Thermo Fisher) or a variant thereof (e.g., a humanized variant), Q9UHX3, OASA01861, AB_2738756, NLS6381, ab75190, MAB4894, or A100,000. Additional anti-EMR2 antibodies will be evident to one of ordinary skill in the art. See, e.g., International Publication No. WO 2017/087800 A1; Chang et al. FEBS Letters. (2003) 547(1-3):145-150; Yona et al. FASEB J. (2008).22(3): 741-751.
- the agentcomprises an anti-EMR2 antibody or portion thereof described in International Patent Application No. PCT/US2024/029308, which is incorporated by reference in its entirety.
- mutations to EMR2 corresponding to the amino acid sequence of a EMR2 orthologdecrease or eliminate binding of an immunotherapeutic agent targeting EMR2 while preserving some or all of EMR2 expression and/or functionality, providing a cell expressing EMR2 (e.g., functional EMR2) that is targeted less or not at all by anti-EMR2 immunotherapeutic agents.
- a genetically engineered hematopoietic cellhas reduced or eliminated expression of a cell surface protein, or expresses a mutant of a cell surface protein, wherein the cell surface protein is CD47.
- CD47is a transmembrane integrin-associated protein belonging to the immunoglobulin superfamily and is involved in the increase of intracellular calcium concentration that occurs upon cell adhesion to extracellular matrix.
- CD47binds to a variety of ligands including thrombospondin-1 and signal-regulatory protein alpha and functions in processes such as apoptosis, proliferation, adhesion, and migration.
- CD47also has roles in immune and angiogenic responses including regulation of phagocytosis by macrophages (see, e.g., Brown and Frazier. Trends in Cell Biology (2001) 11 (3): 130-135). CD47 is widely expressed across various tissues in humans and also in solid tumors and hematological malignancies (see, e.g., Jiang et al. Journal of Hematology & Oncology (2021) 14: 180). Human CD47 is located on chromosome 3 and contains 13 exons. In some embodiments, a mutation of a gene encoding CD47 alters one or more amino acids associated with an epitope of CD47.
- the epitope of CD47is a portion of CD47 bound by an agent, e.g., an immunotherapeutic agent.
- the agentis an anti-CD47 antibody.
- the agentis the anti-CD47 B6H12 or 2D3 antibody.
- the agentcomprises an anti-CD47 antibody or portion thereof, e.g., an antibody drug conjugate (ADC), a chimeric antigen receptor (CAR), or a multispecific antibody (e.g., a bispecific T cell engager).
- ADCantibody drug conjugate
- CARchimeric antigen receptor
- a multispecific antibodye.g., a bispecific T cell engager
- the agentcan be anti-CD47 antibody or a variant thereof (e.g., a humanized variant).
- the agentis an anti-CD47 drug.
- one or more of amino acids 52-55 in CD47is deleted.
- amino acids 52-55 in CD47are deleted.
- amino acids 52, 53, 54, and/or 55 or any combination thereof in CD47is deleted.
- a mutation of a gene encoding CD47comprises a substitution of the amino acid at position 31 of a wildtype CD47 or at a corresponding position in a homologous CD47 gene.
- a methionineis substituted for the amino acid at position 31 of a wildtype CD47 or at a corresponding position in a homologous CD47 gene.
- a mutation of a gene encoding CD47comprises a substitution of the amino acid at position 47 of a wildtype CD47 or at a corresponding position in a homologous CD47 gene.
- a histidineis substituted for the amino acid at position 47 of a wildtype CD47 or at a corresponding position in a homologous CD47 gene.
- a glycineis substituted for the amino acid at position 47 of a wildtype CD47 or at a corresponding position in a homologous CD47 gene.
- a mutation of a gene encoding CD47comprises a substitution of the amino acid at position 49 of a wildtype CD47 or at a corresponding position in a homologous CD47 gene.
- an arginineis substituted for the amino acid at position 49 of a wildtype CD47 or at a corresponding position in a homologous CD47 gene.
- a prolineis substituted for the amino acid at position 49 of a wildtype CD47 or at a corresponding position in a homologous CD47 gene.
- a mutation of a gene encoding CD47comprises a substitution of the amino acid at position 52 of a wildtype CD47 or at a corresponding position in a homologous CD47 gene.
- a mutation of a gene encoding CD47comprises a deletion of the amino acid at position 52 of a wildtype CD47 or at a corresponding position in a homologous CD47 gene. In some embodiments, a mutation of a gene encoding CD47 comprises a substitution of the amino acid at position 53 of a wildtype CD47 or at a corresponding position in a homologous CD47 gene. In some embodiments, an alanine is substituted for the amino acid at position 53 of a wildtype CD47 or at a corresponding position in a homologous CD47 gene.
- a mutation of a gene encoding CD47comprises a deletion of the amino acid at position 53 of a wildtype CD47 or at a corresponding position in a homologous CD47 gene. In some embodiments, a mutation of a gene encoding CD47 comprises a deletion of the amino acid at position 54 of a wildtype CD47 or at a corresponding position in a homologous CD47 gene. In some embodiments, a mutation of a gene encoding CD47 comprises a deletion of the amino acid at position 55 of a wildtype CD47 or at a corresponding position in a homologous CD47 gene.
- a mutation of a gene encoding CD47comprises a substitution of the amino acid at position 120 of a wildtype CD47 or at a corresponding position in a homologous CD47 gene.
- an alanineis substituted for the amino acid at position 120 of a wildtype CD47 or at a corresponding position in a homologous CD47 gene.
- a mutation of a gene encoding CD47comprises a substitution of the amino acid at position 124 of a wildtype CD47 or at a corresponding position in a homologous CD47 gene.
- a lysineis substituted for the amino acid at position 124 of a wildtype CD47 or at a corresponding position in a homologous CD47.
- a mutation of a gene encoding CD47makes a change in the amino acid sequence corresponding to the amino acid sequence of a CD47 ortholog.
- a mutationsubstitutes an amino acid of human CD47 for an amino acid at a corresponding position of an orthologous CD47, e.g., a non-human primate CD47.
- a mutationinserts or deletes one or more amino acids of human CD47 to correspond to the sequence of an orthologous CD47, e.g., a non-human primate CD47.
- a mutationchanges the amino acid sequence in a manner corresponding to a tolerable genetic variant identified by one or more genomic sequence comparison algorithms, e.g., gnomAD (see, e.g., Gudmundsson et al. arXiv:2107.11458v3, e.g., gnomad.broadinstitute.org/), or to a position characterized by a plurality of tolerable genetic variants.
- genomic sequence comparison algorithmse.g., gnomAD (see, e.g., Gudmundsson et al. arXiv:2107.11458v3, e.g., gnomad.broadinstitute.org/)
- mutations to CD47 corresponding to the amino acid sequence of a CD47 ortholog or at positions characterized by a plurality of tolerable genetic variantsdecrease or eliminate binding of an immunotherapeutic agent targeting CD47 while preserving some or all of CD47 structure, expression, and/or functionality, providing a cell expressing CD47 (e.g., functional CD47) that is targeted less or not at all by anti-CD47 immunotherapeutic agents.
- Genetically Engineered Hematopoietic Cell Expressing CD34 MutantsIn some embodiments, a genetically engineered hematopoietic cell has reduced or eliminated expression of a cell surface protein, or expresses a mutant of a cell surface protein, wherein the cell surface protein is CD34.
- CD34is a transmembrane phosphoglycoprotein belonging to the single-pass transmembrane sialomucin protein family that functions as a cell-cell adhesion factor. Accordingly, CD34 is an important adhesion molecule required for T-cells to enter lymph nodes and for attachment of hematopoietic stem cells to bone marrow extracellular matrix or to stromal cells. CD34 is highly expressed in hematopoietic stem and progenitor cells and endothelial cells. Moreover, CD34 is commonly found expressed on the cell surface of hematopoietic cancer cells (see, e.g., Sydney et al. Stem Cells (2014) 32 (6): 1380-1389; Nielsen and McNagny.
- CD34is located on chromosome 1 and contains 8 exons.
- a mutation of a gene encoding CD34alters one or more amino acids associated with an epitope of CD34.
- the epitope of CD34is a portion of CD34 bound by an agent, e.g., an immunotherapeutic agent.
- the agentis an anti-CD34 antibody.
- the anti-CD34 antibodyis clone QBend10 or 561.
- the agentcomprises an anti-CD34 antibody or portion thereof, e.g., an antibody drug conjugate (ADC), a chimeric antigen receptor (CAR), or a multispecific antibody (e.g., a bispecific T cell engager).
- ADCantibody drug conjugate
- CARchimeric antigen receptor
- multispecific antibodye.g., a bispecific T cell engager
- the epitope of CD34is one or more amino acids of a protein domain (e.g., the extracellular domain) or the amino acids encoded by an exon or combination of exons of the gene encoding CD34.
- a mutation of a gene encoding CD34comprises a substitution of the amino acid at position 42 of a wildtype CD34 or at a corresponding position in a homologous CD34 gene.
- a glycineis substituted for the amino acid at position 42 of a wildtype CD34 or at a corresponding position in a homologous CD34 gene.
- a mutation of a gene encoding CD34comprises a substitution of the amino acid at position 46 of a wildtype CD34 or at a corresponding position in a homologous CD34 gene.
- an alanineis substituted for the amino acid at position 46 of a wildtype CD34 or at a corresponding position in a homologous CD34 gene.
- a mutation of a gene encoding CD34comprises a substitution of the amino acid at position 47 of a wildtype CD34 or at a corresponding position in a homologous CD34 gene.
- a lysineis substituted for the amino acid at position 47 of a wildtype CD34 or at a corresponding position in a homologous CD34 gene.
- a glutamateis substituted for the amino acid at position 47 of a wildtype CD34 or at a corresponding position in a homologous CD34 gene.
- a mutation of a gene encoding CD34comprises a substitution of the amino acid at position 49 of a wildtype CD34 or at a corresponding position in a homologous CD34 gene.
- a prolineis substituted for the amino acid at position 49 of a wildtype CD34 or at a corresponding position in a homologous CD34 gene.
- a serineis substituted for the amino acid at position 49 of a wildtype CD34 or at a corresponding position in a homologous CD34 gene.
- a mutation of a gene encoding CD34comprises a substitution of the amino acid at position 50 of a wildtype CD34 or at a corresponding position in a homologous CD34 gene.
- an alanineis substituted for the amino acid at position 50 of a wildtype CD34 or at a corresponding position in a homologous CD34 gene.
- a prolineis substituted for the amino acid at position 50 of a wildtype CD34 or at a corresponding position in a homologous CD34 gene.
- a mutation of a gene encoding CD34comprises a substitution of the amino acid at position 51 of a wildtype CD34 or at a corresponding position in a homologous CD34 gene.
- an alanineis substituted for the amino acid at position 51 of a wildtype CD34 or at a corresponding position in a homologous CD34 gene.
- a mutation of a gene encoding CD34comprises a substitution of the amino acid at position 54 of a wildtype CD34 or at a corresponding position in a homologous CD34 gene. In some embodiments, an alanine is substituted for the amino acid at position 54 of a wildtype CD34 or at a corresponding position in a homologous CD34 gene. In some embodiments, a mutation of a gene encoding CD34 comprises a substitution of the amino acid at position 55 of a wildtype CD34 or at a corresponding position in a homologous CD34 gene. In some embodiments, an alanine is substituted for the amino acid at position 55 of a wildtype CD34 or at a corresponding position in a homologous CD34 gene.
- a mutationchanges the amino acid sequence in a manner corresponding to a tolerable genetic variant identified by one or more genomic sequence comparison algorithms, e.g., gnomAD (see, e.g., Gudmundsson et al. arXiv:2107.11458v3, e.g., gnomad.broadinstitute.org/), or to a position characterized by a plurality of tolerable genetic variants.
- genomic sequence comparison algorithmse.g., gnomAD (see, e.g., Gudmundsson et al. arXiv:2107.11458v3, e.g., gnomad.broadinstitute.org/)
- mutations to CD34 corresponding to the amino acid sequence of a CD34 ortholog or at positions characterized by a plurality of tolerable genetic variantsdecrease or eliminate binding of an immunotherapeutic agent targeting CD34 while preserving some or all of CD34 structure, expression, and/or functionality, providing a cell expressing CD34 (e.g., functional CD34) that is targeted less or not at all by anti-CD34 immunotherapeutic agents.
- a genetically engineered hematopoietic cellhas reduced or eliminated expression of a cell surface protein, or expresses a mutant of a cell surface protein, wherein the cell surface protein is interleukin-1 receptor accessory protein (IL1RAP).
- IL1RAPis known by several aliases in the art, such as IL-1 receptor accessory protein (IL- 1RAcP), interleukin-1 receptor 3 (IL1R3), and interleukin-1 receptor accessory protein beta.
- IL1RAPis involved in initiating signaling events that regulate activation of genes that are responsive to the interleukin 1 inflammatory cytokine.
- IL1RAPalso interacts with IL1RL2 to form a heterodimer in the IL- 36 cytokine signaling system.
- IL1RAPis found at chromosomal location 3q28 and comprises 17 exons.
- Non-limiting examples of reference sequence for the gene encoding IL1RAPare found at NC_000003.12 and NC_060927.1. It will also be known to the skilled artisan that IL1RAP is encoded by gene capable of undergoing alternative splicing resulting in production of membrane-bound and soluble isoforms.
- Said IL1RAP isoformsdiffer in their C-terminus and the ratios of soluble to membrane-bound isoforms may change depending on cell signaling events (e.g., increases in the soluble isoform during acute-phase induction or stress).
- Non- limiting examples of gene products (e.g., RNAs and proteins) encoded by IL1RAPare set forth in NM_001167928.2 (encoding a protein found at NP_001161400.1), NM_001167929.1 (encoding a protein found at NP_001161401.1), NM_0011679302 (encoding a protein found at NP_001161402.1), NM_001167931.2 (encoding a protein found at NP_001161403.1), NM_001364878.1 (encoding a protein found at NP_001351808.1), NM_001364880.2 (encoding a protein found at NP_001351809.1), NM_001364881.2 (encoding a protein found at NP_001351810.1), and NM_002182.4 (encoding a protein found at NP_002173.1), NM_134470.4 (encoding a protein found at NP_608273.1).
- the IL1RAP proteincomprises an Ig-like C2-type 1 domain at amino acids 21-128, an Ig-like C2-type 2 domain at amino acids 141-230, an Ig-like C2-type 3 domain at amino acids 242-348, and a TIR domain from amino acids 403-546 which mediates NAD+ hydrolase activity.
- the membrane-bound isoformcomprises an amino acid sequence as set forth below: MTLLWCVVSLYFYGILQSDASERCDDWGLDTMRQIQVFEDEPARIKCPLFEHFLKFNYSTAHSAGLTLIWYWTRQ DRDLEEPINFRLPENRISKEKDVLWFRPTLLNDTGNYTCMLRNTTYCSKVAFPLEVVQKDSCFNSPMKLPVHKLY IEYGIQRITCPNVDGYFPSSVKPTITWYMGCYKIQNFNNVIPEGMNLSFLIALISNNGNYTCVVTYPENGRTFHL TRTLTVKVVGSPKNAVPPVIHSPNDHVVYEKEPGEELLIPCTVYFSFLMDSRNEVWWTIDGKKPDDITIDVTINE SISHSRTEDETRTQILSIKKVTSEDLKRSYVCHARSAKGEVAKAAKVKQKVPAPRYTVELACGFGATVLLVVILI VVYHVYWLEMVLFYRAHFGTDETILDG
- genetically engineered hematopoietic cellscomprise a variant gene encoding IL1RAP protein, wherein said variant gene comprises a substitution, deletion, or addition of at least one nucleotide or a combination thereof.
- a mutant IL1RAPcomprises a deletion or mutation of a fragment of the protein that is encoded by any one of the exons of the gene encoding IL1RAP.
- genetically engineered hematopoietic cellscomprise a mutant IL1RAP protein comprising a substitution, deletion, or addition of at least one amino acid or a combination thereof.
- genetically engineered hematopoietic cellscomprise a mutant IL1RAP protein comprising at least one modified non-essential epitope. In some embodiments, genetically engineered hematopoietic cells comprise IL1RAP expression that is reduced by at least 20%, at least 30%, at least 40%, at least 50%, at least 60%, at least 70%, at least 80%, at least 90%, or at least 95% relative to the wildtype counterpart cell. In some embodiments, genetically engineered hematopoietic cells express a mutant of IL1RAP that has reduced or no binding to an agent comprising an antigen-binding fragment that recognizes an epitope of IL1RAP.
- At least 40%, at least 50%, at least 60%, at least 70%, at least 75%, at least 80%, at least 85%, at least 90%, or at least 95% of copies of IL1RAP in the population of cellshave a mutation.
- a populationcan comprise a plurality of different IL1RAP mutations and each mutation of the plurality contributes to the percent of copies of IL1RAP in the population of cells that have a mutation.
- the expression of IL1RAP on the genetically engineered hematopoietic cellis compared to the expression of IL1RAP on a naturally occurring hematopoietic cell (e.g., a wild-type counterpart).
- the genetic engineeringresults in a reduction in the expression level of IL1RAP by at least50%, at least 60%, at least 70%, at least 80%, at least 90%, at least 91%, at least 92%, at least 93%, at least 94%, at least 95%, at least 96%, at least 97%, at least 98%, or at least 99% as compared to the expression of IL1RAP on a naturally occurring hematopoietic cell (e.g., a wild-type counterpart).
- a naturally occurring hematopoietic celle.g., a wild-type counterpart
- the genetically engineered hematopoietic cellexpresses less than 20%, less than 19%, less than 18%, less than 17%, less than 16%, less than 15%, less than 14%, less than 13%, less than 12%, less than 11%, less than 10%, less than 9%, less than 8%, less than 7%, less than 6%, less than 5%, less than 4%, less than 3%, less than 2%, less than or 1% of IL1RAP as compared to a naturally occurring hematopoietic cell (e.g., a wild-type counterpart).
- a naturally occurring hematopoietic celle.g., a wild-type counterpart
- the genetic engineeringresults in a reduction in the expression level of wild-type IL1RAP by at least 50%, at least 60%, at least 70%, at least 80%, at least 90%, at least 91%, at least 92%, at least 93%, at least 94%, at least 95%, at least 96%, at least 97%, at least 98%, or at least 99% as compared to the expression of the level of wild-type IL1RAP on a naturally occurring hematopoietic cell (e.g., a wild-type counterpart).
- a naturally occurring hematopoietic celle.g., a wild-type counterpart
- the genetically engineered hematopoietic cellexpresses less than 20%, less than 19%, less than 18%, less than 17%, less than 16%, less than 15%, less than 14%, less than 13%, less than 12%, less than 11%, less than 10%, less than 9%, less than 8%, less than 7%, less than 6%, less than 5%, less than 4%, less than 3%, less than 2%, or less than 1% of IL1RAP as compared to a naturally occurring hematopoietic cell (e.g., a wild-type counterpart).
- a naturally occurring hematopoietic celle.g., a wild-type counterpart
- a genetically engineered hematopoietic cellhas reduced or eliminated expression of a lineage-specific cell surface protein, or expresses a mutant of a lineage-specific cell surface protein, wherein the lineage-specific cell surface protein is CD33.
- CD33is encoded by seven exons, including the alternatively spliced exons 7A and 7B (Brinkman-Van der Linden et al. Mol Cell. Biol. (2003) 23: 4199-4206).
- CD33M-7Aamino acid sequence of human CD33 provided by SEQ ID NO: 1 (CD33M-7A).
- Table 2Exemplary deletions in CD33
- the nucleotide sequence encoding CD33are genetically manipulated to delete any epitope of the protein (of the extracellular portion of CD33), or a fragment containing such, using conventional methods of nucleic acid manipulation.
- the amino acid sequences provided beloware exemplary sequences of CD33 mutants that have been manipulated to lack each of the epitopes in Table 2.
- the amino acid sequence of the extracellular portion of CD33is provided by SEQ ID NO: 1.
- the signal peptideis shown in italics and sites for manipulation are shown in underline and boldface.
- the transmembrane domainis shown in italics with underline.
- the signal peptideis shown in italics and the transmembrane domain is shown in italics with underline.
- S248_E252insdelTARND; PROVEAN score-1.916 MPLLLLLPLL WAGALAMDPN FWLQVQESVT VQEGLCVLVP CTFFHPIPYY DKNSPVHGYW FREGAIISRD SPVATNKLDQ EVQEETQGRF RLLGDPSRNN CSLSIVDARR RDNGSYFFRM ERGSTKYSYK SPQLSVHVTD LTHRPKILIP GTLEPGHSKN LTCSVSWACE QGTPPIFSWL SAAPTSLGPR TTHSSVLIIT PRPQDHGTNL TCQVKFAGAG VTTERTIQLN VTYVPQNPTT GIFPGDGTAR NDTRAGVVHG AIGGAGVTAL LALCLCLIFF IVKTHRRKAA RTAVGRNDTH PTTGSASPKH QKKSKLHGPT ETSSCSGA
- the signal peptideis shown in italics and the transmembrane domain is shown in italics with underline.
- I47_D51insdelVPFFE; PROVEAN score-1.672 MPLLLLLPLL WAGALAMDPN FWLQVQESVT VQEGLCVLVP CTFFHPVPFF EKNSPVHGYW FREGAIISRD SPVATNKLDQ EVQEETQGRF RLLGDPSRNN CSLSIVDARR RDNGSYFFRM ERGSTKYSYK SPQLSVHVTD LTHRPKILIP GTLEPGHSKN LTCSVSWACE QGTPPIFSWL SAAPTSLGPR TTHSSVLIIT PRPQDHGTNL TCQVKFAGAG VTTERTIQLN VTYVPQNPTT GIFPGDGTAR NDTRAGVVHG AIGGAGVTAL LALCLCLIFF IVKTHRRKAA RTAVGRNDTH PTTGSASPKH QKKSKLHGPT ETSSCSGAAP TV
- the signal peptideis shown in italics and the transmembrane domain is shown in italics with underline.
- the signal peptideis shown in italics and the transmembrane domain is shown in italics with underline.
- the signal peptideis shown in italics and the transmembrane domain is shown in italics with underline.
- the predicted structure of CD33includes two immunoglobulin domains, an IgV domain and an IgC2 domain. In some embodiments, a portion of the immunoglobulin V domain of CD33 is deleted or mutated. In some embodiments, a portion of the immunoglobulin C domain of CD33 is deleted or mutated. In some embodiments, exon 2 of CD33 is deleted or mutated. In some embodiments, exon 3 of CD33 is deleted or mutated. In some embodiments, the CD33 variant lacks amino acid residues W11 to T139. In some embodiments, the CD33 variant lacks amino acid residues G13 to T139.
- the genetically engineered hematopoietic stem cellshave genetic edits in a CD33 gene, wherein exon 2 of CD33 is mutated or deleted. In some embodiments, exon 2 of CD33 is mutated or deleted (see the amino acid sequence of the exon 2-encoded fragment above). In some embodiments, the genetically engineered hematopoietic stem cells have genetic edits in a CD33 gene resulting in expression of CD33 with deleted or mutated exon 2 of CD33. In some embodiments, genetically engineered hematopoietic stem cells express CD33, in which exon 2 of CD33 is mutated or deleted.
- the genetically engineered hematopoietic cellshave a genetically engineered CD33 gene (e.g., a genetically engineered endogenous CD33 gene), wherein the engineering results in expression of a CD33 variant having the fragment encoded by exon 2 deleted (CD33ex2).
- CD33 genes edited HSCs expressing CD33 variant having the fragment encoded by exon 2 deletedalso comprise a partial or complete deletion in the adjacent introns (intron 1 and intron 2) in addition to the deletion of exon 2.
- the parent hematopoietic stem cells for producing the genetically engineered HSCscarry a CD33 gene that produces exon 2- containing transcripts.
- Genetically engineered hematopoietic stem cells carrying an edited CD33 gene that expresses this CD33 mutantare also within the scope of the present disclosure.
- Such cellsmay be a homogenous population containing cells expressing the same CD33 mutant (e.g., CD33ex2).
- the cellsmay be a heterogeneous population containing cells expressing different CD33 mutants (which may be due to heterogeneous editing/repairing events inside cells) or cells that do not express CD33 (CD33KO).
- the genetically engineered HSCsmay be a heterogeneous population containing cells expressing CD33ex2 and cells that do not express CD33 (CD33KO).
- Genetically engineered hematopoietic stem cells having edited a CD33 genecan be prepared by a suitable genome editing method, such as those known in the art or disclosed herein.
- the genetically engineered hematopoietic stem cells described hereincan be generated using the CRISPR approach. See discussions herein.
- specific guide RNAs targeting a fragment of the CD33 genecan be used in the CRISPR method.
- a CD33 pseudogeneknown as SIGLEC22P (Gene ID 114195), is located upstream of the CD33 gene and shares a certain degree of sequence homology with the CD33 gene. gRNAs that cross-target regions in the pseudogene and regions in the CD33 gene may lead to production of aberrant gene products.
- the gRNAs used in methods of editing CD33 via CRISPRpreferably have low or no cross-reactivity with regions inside the pseudogene.
- the gRNAs used in methods of editing CD33 via CRISPRpreferably have low or no cross-reactivity with region(s) in Exon 1, intron 1 or Exon 2 of CD33 that are homologous to the pseudogene.
- Such gRNAscan be designed by comparing the sequences of the pseudogene and the CD33 gene to choose targeting sites inside the CD33 gene that have less or no homology to regions of the pseudogene.
- the pair of gRNA 18(TTCATGGGTACTGCAGGGCA)(SEQ ID NO: 24) and gRNA 24 (GTGAGTGGCTGTGGGGAGAG)(SEQ ID NO: 25) are used for editing CD33 via CRISPR.
- methods of genetically editing CD33 in hematopoietic cellse.g., HSCs
- the pair of gRNA18 + gRNA24are also provided herein.
- the length of a gRNA sequencemay be modified (increased or decreased), for example, to enhance editing specificity and/or efficiency.
- the length of gRNA 24is 20 base pairs.
- reducing the length of gRNA 24may reduce the editing efficiency of gRNA 24 for CD33.
- at least 40%, at least 50%, at least 60%, at least 70%, at least 75%, at least 80%, at least 85%, at least 90%, or at least 95% of copies of CD33 in the population of cellshave a mutation.
- a populationcan comprise a plurality of different CD33 mutations and each mutation of the plurality contributes to the percent of copies of CD33 in the population of cells that have a mutation.
- the expression of CD33 on the genetically engineered hematopoietic cellis compared to the expression of CD33 on a naturally occurring hematopoietic cell (e.g., a wild-type counterpart).
- the genetic engineeringresults in a reduction in the expression level of CD33 by at least50%, at least 60%, at least 70%, at least 80%, at least 90%, at least 91%, at least 92%, at least 93%, at least 94%, at least 95%, at least 96%, at least 97%, at least 98%, or at least 99% as compared to the expression of CD33 on a naturally occurring hematopoietic cell (e.g., a wild-type counterpart).
- the genetically engineered hematopoietic cellexpresses less than 20%, less than 19%, less than 18%, less than 17%, less than 16%, less than 15%, less than 14%, less than 13%, less than 12%, less than 11%, less than 10%, less than 9%, less than 8%, less than 7%, less than 6%, less than 5%, less than 4%, less than 3%, less than 2%, less than or 1% of CD33 as compared to a naturally occurring hematopoietic cell (e.g., a wild-type counterpart).
- a naturally occurring hematopoietic celle.g., a wild-type counterpart
- the genetic engineeringresults in a reduction in the expression level of wild-type CD33 by at least 50%, at least 60%, at least 70%, at least 80%, at least 90%, at least 91%, at least 92%, at least 93%, at least 94%, at least 95%, at least 96%, at least 97%, at least 98%, or at least 99% as compared to the expression of the level of wild-type CD33 on a naturally occurring hematopoietic cell (e.g., a wild-type counterpart).
- a naturally occurring hematopoietic celle.g., a wild-type counterpart
- the genetically engineered hematopoietic cellexpresses less than 20%, less than 19%, less than 18%, less than 17%, less than 16%, less than 15%, less than 14%, less than 13%, less than 12%, less than 11%, less than 10%, less than 9%, less than 8%, less than 7%, less than 6%, less than 5%, less than 4%, less than 3%, less than 2%, or less than 1% of CD33 as compared to a naturally occurring hematopoietic cell (e.g., a wild-type counterpart).
- a naturally occurring hematopoietic celle.g., a wild-type counterpart
- a genetically engineered hematopoietic cellssuch as HSCs, that have both the IL1RAP and CD33 genes edited.
- HSCsgenetically engineered hematopoietic cells
- a population of genetically engineered hematopoietic HSCsin which at least 50% of the cells carry genetically edited IL1RAP and CD33 cells in at least on chromosome.
- the edited IL1RAP geneis capable of expressing an IL1RAP mutant having at least one fragment encoded by one or more exons deleted.
- the edited CD33 geneis capable of expressing a CD33 mutant having at least one fragment encoded by one or more exons deleted.
- the genetically engineered hematopoietic cells having both IL1RAP and CD33 genes editedcan be prepared by conventional methods. In some embodiments, such cells are prepared by CRISPR using a pair of gRNAs, one targeting IL1RAP and the other targeting CD33. In one example, the pair of gRNAs can be introduced into parent HSCs simultaneously and cells having genetic edits in IL1RAP and/or CD33 can be harvested for further use.
- HSCsGenetically Engineered Hematopoietic Cells Expressing both CD44 and EMR2 Mutants
- a genetically engineered hematopoietic cellssuch as HSCs, that have both the EMR2 and CD44 genes edited.
- HSCsgenetically engineered hematopoietic cells
- the edited EMR2 geneis capable of expressing a EMR2 mutant having at least one fragment encoded by a one or more exons deleted.
- the edited CD44 geneis capable of expressing a CD44 mutant having at least one fragment encoded by one or more exons deleted.
- the genetically engineered hematopoietic cells having both EMR2 and CD44 genes editedcan be prepared by conventional methods. In some embodiments, such cells are prepared by CRISPR using a pair of gRNAs, one targeting EMR2 and the other targeting CD44. In one example, the pair of gRNAs can be introduced into parent HSCs simultaneously and cells having genetic edits in EMR2 and/or CD44 can be harvested for further use.
- hematopoietic cellssuch as HSCs
- HSCsgenetically engineered hematopoietic cells, such as HSCs, that have genetic editing in a first gene encoding a first cell surface protein and a second gene encoding a second cell surface protein, wherein at least one of the cell surface proteins is selected from the group consisting of: CD305 (LAIR1); CD49D (ITGA4); CD205 (LY75); and CD244 (SLAMF4).
- one of the cell surface proteinsis selected from the group consisting of: CD305 (LAIR1); CD49D (ITGA4); CD205 (LY75); and CD244 (SLAMF4), and one of the cell surface proteins is selected from the group consisting of: CD10; CD101; CD117; CD11A; CD11C; CD120B; CD123; CD13; CD133; CD152; CD200; CD205 (LY75); CD226; CD244 (SLAMF4); CD25; CD274; CD305 (LAIR1); CD32; CD33; CD34; CD366; CD38; CD41; CD42B; CD44; CD45RA; CD47; CD48; CD49D (ITGA4); CD49F; CD52; CD58; CD64; CD7; CD70; CD82; CD84; CD85; CD85K; CD86; CD9; CD96; CD99; CLL-1; EMR2 (ADGRE2); FR-B; GPR56; and IL1RAP.
- CD10CD101
- the edited first geneis capable of expressing a mutant of the first cell surface protein having at least one fragment encoded by a one or more exons deleted.
- the edited second geneis capable of expressing a mutant of the second cell surface protein having at least one fragment encoded by one or more exons deleted.
- a mutant of the first cell surface protein and/or a mutant of the second cell surface proteincomprises a substitution, deletion, or insertion of an amino acid at one or more amino acid positions (e.g., 1-10, 10-20, 20-30, 30-40, 40-50, or more than 50 amino acid positions).
- the mutated amino acid positionsare located in an epitope (e.g., wherein the epitope is deleted in the mutant cell surface protein).
- a mutant of the first cell surface protein and/or a mutant of the second cell surface proteincomprises a deletion of one or more domains.
- the first gene and/or the second geneis deleted.
- the genetically engineered hematopoietic cells having both first and second genes editedcan be prepared by conventional methods. In some embodiments, such cells are prepared by CRISPR using a pair of gRNAs, one targeting the first gene and the other targeting the second gene.
- the lineage-specific cell surface proteinis CD45.
- maskingis performed on a cell type described herein, e.g., an HSC or HPC. In some embodiments, masking is accomplished by expressing a masking protein in the cell of interest (e.g., by stably expressing DNA encoding the masking protein in the cell).
- the masking proteincomprises a binding domain (e.g., an antibody or antigen-binding fragment, e.g., an scFv) that binds the lineage-specific cell surface protein, e.g., in a way that reduces binding of an immunotherapeutic agent to the lineage-specific cell surface protein, e.g., by competing for binding at the same epitope.
- the binding domainbinds CD45.
- the making proteinfurther comprises one or more sequences that direct its localization to the surface of the cell.
- the masking proteincomprises a transmembrane domain fused to the binding domain.
- the masking proteinmay comprise a linker disposed between the transmembrane domain and the binding domain.
- the masking proteinmay be expressed at a level that binds to a sufficient amount of the cell surface protein that an immunotherapeutic agent displays reduced binding to and/or reduced killing of a cell expressing the masking protein compared to an otherwise similar cell that does not express the masking protein.
- a cell described hereinhas reduced binding to (and/or reduced killing by) two different immunotherapeutic agents that recognize two different lineage- specific cell surface antigens.
- the cellmay have a mutation at a gene encoding a first lineage-specific cell surface antigen, and may comprise a masking protein that masks a second lineage-specific cell surface antigen.
- the cellmay comprise a first masking protein that masks a first cell surface protein and a second masking protein that masks a second lineage-specific cell surface antigen.
- Cells Altered at One or More Lineage-Specific Cell Surface AntigensWhile many of the embodiments described herein involve two or more lineage-specific cell surface antigens, the application also discloses various cells altered with respect to a single lineage-specific cell surface antigen. For instance, the disclosure describes cells mutated at any one of the lineage-specific cell surface antigens described herein (e.g., mutated at one or both alleles of the lineage-specific cell surface antigen). The disclosure also describes cells expressing a single masking protein for a single lineage-specific cell surface antigen.
- the hematopoietic cells described hereinare hematopoietic stem cells.
- Hematopoietic stem cellsare capable of giving rise to both myeloid and lymphoid progenitor cells that further give rise to myeloid cells (e.g., monocytes, macrophages, neutrophils, basophils, dendritic cells, erythrocytes, platelets, etc) and lymphoid cells (e.g., T cells, B cells, NK cells), respectively.
- myeloid cellse.g., monocytes, macrophages, neutrophils, basophils, dendritic cells, erythrocytes, platelets, etc
- lymphoid cellse.g., T cells, B cells, NK cells
- the HSCsare characterized by the expression of the cell surface marker CD34 (e.g., CD34 + ), which can be used for the identification and/or isolation of HSCs, and absence of cell surface markers associated with commitment to a cell lineage.
- the HSCsare obtained from a subject, such as a mammalian subject.
- the mammalian subjectis a non-human primate, a rodent (e.g., mouse or rat), a bovine, a porcine, an equine, or a domestic animal.
- the HSCsare obtained from a human patient, such as a human patient having a hematopoietic malignancy.
- the HSCsare obtained from a healthy donor. In some embodiments, the HSCs are obtained from the subject to whom the immune cells expressing the chimeric receptors will be subsequently administered. HSCs that are administered to the same subject from which the cells were obtained are referred to as autologous cells, whereas HSCs that are obtained from a subject who is not the subject to whom the cells will be administered are referred to as allogeneic cells. In some embodiments, the HSCs that are administered to the subject are allogeneic cells. In some embodiments, the HSCs are obtained from a donor having a HLA haplotype that is matched with the HLA haplotype of the subject.
- HLAHuman Leukocyte Antigen encodes major histocompatibility complex (MHC) proteins in humans.
- MHC moleculesare present on the surface of antigen-presenting cells as well as many other cell types and present peptides of self and non-self (e.g., foreign) antigens for immunosurveillance.
- HLAare highly polymorphic, which results in many distinct alleles. Different (foreign, non-self) alleles may be antigenic and stimulate robust adverse immune responses, particularly in organ and cell transplantation.
- HLA molecules that are recognized as foreign (non-self)can result in transplant rejection.
- the HLA loci of a donor subjectmay be typed to identify an individual as a HLA- matched donor for the subject.
- Methods for typing the HLA lociwill be evident to one of ordinary skill in the art and include, for example, serology (serotyping), cellular typing, gene sequencing, phenotyping, and PCR methods.
- An HLA from a donoris considered “matched” with the HLA of the subject if the HLA loci of the donor and the subject are identical or sufficiently similar such that an adverse immune response is not expected.
- the HLA from the donoris not matched with the HLA of the subject.
- the subjectis administered HSCs that are not HLA matched with the HLA of the subject.
- the subjectis further administered one or more immunosuppressive agents to reduce or prevent rejection of the donor HSC cells.
- the HSCsdo not comprise a CART.
- HSCsmay be obtained from any suitable source using convention means known in the art.
- HSCsare obtained from a sample from a subject (or donor), such as bone marrow sample or from a blood sample.
- HSCsmay be obtained from an umbilical cord.
- the HSCsare from bone marrow, cord blood cells, or peripheral blood mononuclear cells (PBMCs).
- PBMCsperipheral blood mononuclear cells
- bone marrow cellsmay be obtained from iliac crest, femora, tibiae, spine, rib or other medullary spaces of a subject (or donor). Bone marrow may be taken out of the patient and isolated through various separations and washing procedures known in the art.
- An exemplary procedure for isolation of bone marrow cellscomprises the following steps: a) extraction of a bone marrow sample; b) centrifugal separation of bone marrow suspension in three fractions and collecting the intermediate fraction, or buffycoat; c) the buffycoat fraction from step (b) is centrifuged one more time in a separation fluid, commonly Ficoll TM , and an intermediate fraction which contains the bone marrow cells is collected; and d) washing of the collected fraction from step (c) for recovery of re-transfusable bone marrow cells.
- HSCstypically reside in the bone marrow but can be mobilized into the circulating blood by administering a mobilizing agent in order to harvest HSCs from the peripheral blood.
- the subject (or donor) from which the HSCs are obtainedis administered a mobilizing agent, such as granulocyte colony-stimulating factor (G-CSF).
- G-CSFgranulocyte colony-stimulating factor
- the number of the HSCs collected following mobilization using a mobilizing agentis typically greater than the number of cells obtained without use of a mobilizing agent.
- the HSCs for use in the methods described hereinmay express the cell surface protein of interest. Upon any of the modifications described herein (e.g., genetic modification or incubation with a blocking agent), the HSCs would not be targeted by the cytotoxicity agent also described herein.
- the HSCs for use in the methods described hereinmay not express the lineage-specific cell surface protein of interest (e.g., CD33); however, descendant cells differentiated from the HSCs (e.g., B cells) express the lineage-specific cell surface protein.
- the HSCsmay not express the lineage-specific cell surface protein of interest (e.g., CD33); however, descendant cells differentiated from the HSCs (e.g., B cells) express the lineage-specific cell surface protein.
- an endogenous gene of the HSCs coding for the lineage-specific cell surface proteinmay be disrupted at a region encoding a non-essential epitope of the lineage-specific cell surface protein.
- Descendant cells differentiated from such modified HSCswould express a modified lineage-specific cell surface protein having the non-essential epitope mutated such that they would not be targeted by the cytotoxicity agent capable of binding the non-essential epitope.
- a sampleis obtained from a subject (or donor) and is then enriched for a desired cell type (e.g. CD34 + /CD33- cells).
- a desired cell typee.g. CD34 + /CD33- cells.
- PBMCs and/or CD34 + hematopoietic cellscan be isolated from blood as described herein. Cells can also be isolated from other cells, for example by isolation and/or activation with an antibody binding to an epitope on the cell surface of the desired cell type.
- HSChuman immunodeficiency virus
- the cellsmay be cultured under conditions that comprise an expansion medium comprising one or more cytokines, such as stem cell factor (SCF), Flt-3 ligand (Flt3L), thrombopoietin (TPO), Interleukin 3 (IL-3), or Interleukin 6 (IL-6).
- SCFstem cell factor
- Flt3LFlt-3 ligand
- TPOthrombopoietin
- IL-3Interleukin 3
- IL-6Interleukin 6
- the cellmay be expanded for about 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25 days or any range necessary.
- the HSCare expanded after isolation of a desired cell population (e.g., CD34 + /CD33-) from a sample obtained from a subject (or donor) and prior to manipulation (e.g., genetic engineering, contact with a blocking agent).
- a desired cell populatione.g., CD34 + /CD33-
- the HSCare expanded after genetic engineering, thereby selectively expanding cells that have undergone the genetic modification and lack the epitope (e.g., have a deletion or substitution of at least a portion of the epitope) of the cell surface protein to which the cytotoxic agent binds.
- a cell (“a clone”) or several cells having a desired characteristic (e.g., phenotype or genotype) following genetic modificationmay be selected and independently expanded.
- the HSCare expanded prior to contacting the HSC with a blocking agent that binds the epitope of the cell surface proteins, thereby providing a population of HSC expressing the cell surface proteins that cannot be bound by the cytotoxic agent due to blocking of the corresponding epitope by the blocking agent.
- a blocking agentthat binds the epitope of the cell surface proteins
- genome editingrefers to a method of modifying the genome, including any protein-coding or non-coding nucleotide sequence, of an organism to knock out the expression of a target gene.
- genome editing methodsinvolve use of an endonuclease that is capable of cleaving the nucleic acid of the genome, for example at a targeted nucleotide sequence. Repair of the double-stranded breaks in the genome may be repaired introducing mutations and/or exogenous nucleic acid may be inserted into the targeted site.
- Genome editing methodsare generally classified based on the type of endonuclease that is involved in generating double stranded breaks in the target nucleic acid.
- TALENTranscription Activator-Like-Effector-Based Nuclease
- the modified cellsare manipulated as described herein using the TALEN technology known in the art.
- TALENsare engineered restriction enzymes that can specifically bind and cleave a desired target DNA molecule.
- a TALENtypically contains a Transcriptional Activator-Like Effector (TALE) DNA-binding domain fused to a DNA cleavage domain.
- TALETranscriptional Activator-Like Effector
- the DNA binding domainmay contain a highly conserved 33-34 amino acid sequence with a divergent 2 amino acid RVD (repeat variable dipeptide motif) at positions 12 and 13.
- RVDrepeat variable dipeptide motif
- the RVD motifdetermines binding specificity to a nucleic acid sequence and can be engineered according to methods known to those of skill in the art to specifically bind a desired DNA sequence.
- the DNA cleavage domainmay be derived from the FokI endonuclease.
- the FokI domainfunctions as a dimer, requiring two constructs with unique DNA binding domains for sites in the target genome with proper orientation and spacing.
- TALENs specific to sequences in a target gene of intereste.g., IL1RAP, CD33
- a TALEN specific to a target gene of interestcan be used inside a cell to produce a double-stranded break (DSB).
- a mutationcan be introduced at the break site if the repair mechanisms improperly repair the break via non-homologous end joining. For example, improper repair may introduce a frame shift mutation.
- a foreign DNA molecule having a desired sequencecan be introduced into the cell along with the TALEN. Depending on the sequence of the foreign DNA and chromosomal sequence, this process can be used to correct a defect or introduce a DNA fragment into a target gene of interest, or introduce such a defect into the endogenous gene, thus decreasing expression of the target gene.
- one or more population of hematopoietic cellsis generated by genetic engineering of a cell surface protein (e.g., those described herein) using a TALEN.
- the genetically engineered hematopoietic cellsmay not express the lineage-specific cell surface antigen.
- the hematopoietic cellsmay be engineered to express an altered version of the lineage-specific cell surface antigen, e.g., having a deletion or mutation relative to the wild-type counterpart.
- Such a mutated cell surface proteinmay preserve a certain level of the bioactivity as the wild-type counterpart.
- a population of hematopoietic cells containing a mutated CD33is generated by genetic engineering using a TALEN.
- exon 2 or exon 3 of CD33is mutated using a TALEN.
- Zinc Finger NucleaseZFN
- the cellscan be genetically manipulated using zinc finger (ZFN) technology known in the art.
- ZFNzinc finger
- zinc finger mediated genomic editinginvolves use of a zinc finger nuclease, which typically comprises a DNA binding domain (i.e., zinc finger) and a cleavage domain (i.e., nuclease).
- the zinc finger binding domainmay be engineered to recognize and bind to any target gene of interest (e.g., IL1RAP, CD33) using methods known in the art and in particular, may be designed to recognize a DNA sequence ranging from about 3 nucleotides to about 21 nucleotides in length, or from about 8 to about 19 nucleotides in length.
- Zinc finger binding domainstypically comprise at least three zinc finger recognition regions (e.g., zinc fingers).
- Restriction endonucleasescapable of sequence-specific binding to DNA (at a recognition site) and cleaving DNA at or near the site of binding are known in the art and may be used to form ZFN for use in genomic editing.
- Type IIS restriction endonucleasescleave DNA at sites removed from the recognition site and have separable binding and cleavage domains.
- the DNA cleavage domainmay be derived from the FokI endonuclease.
- one or more population of hematopoietic cellsis generated by genetic engineering of a cell surface protein (e.g., those described herein) using a ZFN.
- the genetically engineered hematopoietic cellsmay not express the cell surface protein.
- the hematopoietic cellsmay be engineered to express an altered version of the cell surface protein, e.g., having a deletion or mutation relative to the wild-type counterpart.
- Such a mutated cell surface proteinmay preserve a certain level of the bioactivity as the wild- type counterpart.
- a population of hematopoietic cells containing a mutated CD33is generated by genetic engineering using a ZFN.
- exon 2 or exon 3 of CD33is mutated using a ZFN.
- CRISPR/Cas SystemsOne exemplary suitable genome editing technology comprises the use of a CRISPR/Cas nuclease to introduce targeted single- or double-stranded DNA breaks in the genome of a cell, which trigger cellular repair mechanisms, such as, for example, nonhomologous end joining (NHEJ), microhomology-mediated end joining (MMEJ, also sometimes referred to as “alternative NHEJ” or “alt-NHEJ”), or homology-directed repair (HDR) that typically result in an altered nucleic acid sequence (e.g., via nucleotide or nucleotide sequence insertion, deletion, inversion, or substitution) at or immediately proximal to the site of the nuclease cut.
- NHEJnonhomologous end joining
- MMEJmicrohomology-mediated end joining
- HDRhomology-directed repair
- base editingincludes the use of a base editor, e.g., a nuclease-impaired or partially nuclease impaired enzyme (e.g., RNA-guided CRISPR/Cas protein) fused to a deaminase that targets and deaminates a specific nucleobase, e.g., a cytosine or adenosine nucleobase of a C or A nucleotide, which, via cellular mismatch repair mechanisms, results in a change from a C to a T nucleotide, or a change from an A to a G nucleotide.
- a base editore.g., a nuclease-impaired or partially nuclease impaired enzyme (e.g., RNA-guided CRISPR/Cas protein) fused to a deaminase that targets and deaminates a specific nucleobase, e.g., a cytosine or
- Yet another exemplary suitable genome editing technologyincludes “prime editing,” which includes the introduction of new genetic information, e.g., an altered nucleotide sequence, into a specifically targeted genomic site using a catalytically impaired or partially catalytically impaired nuclease (e.g., a CRISPR/Cas nuclease), fused to an engineered reverse transcriptase (RT) domain.
- primary editingincludes the introduction of new genetic information, e.g., an altered nucleotide sequence, into a specifically targeted genomic site using a catalytically impaired or partially catalytically impaired nuclease (e.g., a CRISPR/Cas nuclease), fused to an engineered reverse transcriptase (RT) domain.
- a catalytically impaired or partially catalytically impaired nucleasee.g., a CRISPR/Cas nuclease
- the Cas/RT fusionis targeted to a target site within the genome by a guide RNA that also comprises a nucleic acid sequence encoding the desired edit, and that can serve as a primer for the RT.
- a genetically engineered cell described hereinis made using gene editing technology, also referred to herein as DNA-editing technology, e.g., CRISPR/Cas-based DNA editing as described herein.
- Exemplary CRISPR/Cas systemsare described herein, and additional suitable CRISPR/Cas systems will be apparent to the skilled artisan based on the present disclosure and the knowledge in the art.
- a gRNA targeting a gene of intereste.g. a cell surface protein
- a Cas9 nucleaseVarious Cas9 nuclease orthologs or variants can be used.
- a Cas9 nucleaseis selected that has the desired PAM specificity to target the gRNA/Cas9 nuclease complex to the target domain in the gene of interest (e.g., cell surface antigen).
- genetically engineering a cellalso comprises introducing one or more (e.g., 1, 2, 3 or more) Cas9 nucleases into the cell.
- Cas nucleaserefers to a CRISPR/Cas nuclease (e.g., Cas9) that can interact with a gRNA and, in concert with the gRNA, home or localize to a site which comprises a target domain.
- Cas nucleasesinclude naturally occurring Cas nucleases and engineered, altered, or modified Cas nucleases that differ, e.g., by at least one amino acid residue, from a naturally occurring Cas nuclease.
- Cas9 nucleases of a variety of speciescan be used in the methods and compositions described herein.
- the Cas9 nucleaseis of, or derived from, Streptococcus pyogenes (SpCas9), Staphylococcus aureus (SaCas9), or Streptococcus thermophilus (StCas9).
- Cas9 nucleasesinclude those of, or derived from, Staphylococcus aureus, Neisseria meningitidis (NmCas9), Acidovorax avenae, Actinobacillus pleuropneumoniae, Actinobacillus succinogenes, Actinobacillus suis, Actinomyces sp., Cycliphilus denitrificans, Aminomonas paucivorans, Bacillus cereus, Bacillus smithii, Bacillus thuringiensis, Bacteroides sp., Blastopirellula marina, Bradyrhizobium sp., Brevibacillus laterosporus, Campylobacter coli, Campylobacter jejuni (CjCas9), Campylobacter lari, Candidatus puniceispirillum, Clostridium cellulolyticum, Clostridium perfringens, Corynebacterium acco
- the Cas9 nucleaseis a naturally occurring Cas9 nuclease.
- the Cas9 nucleaseis an engineered, altered, or modified Cas9 nuclease that differs, e.g., by at least one amino acid residue, from a reference sequence, e.g., the most similar naturally occurring Cas9 nuclease or a sequence of Table 50 of International Publication No.
- the Cas nucleasecomprises Cpf1 or a fragment or variant thereof.
- a naturally occurring Cas9 nucleasetypically comprises two lobes: a recognition (REC) lobe and a nuclease (NUC) lobe; each of which further comprises domains described, e.g., in International Publication No. WO 2015/157070, e.g., in Figs.9A-9B therein (which application is incorporated herein by reference in its entirety).
- the REC lobecomprises the arginine-rich bridge helix (BH), the REC1 domain, and the REC2 domain.
- the REC lobeappears to be a Cas9-specific functional domain.
- the BH domainis a long alpha helix and arginine rich region and comprises amino acids 60-93 of the sequence of S. pyogenes Cas9.
- the REC1 domainis involved in recognition of the repeat:anti-repeat duplex, e.g., of a gRNA or a tracrRNA.
- the REC1 domaincomprises two REC1 motifs at amino acids 94 to 179 and 308 to 717 of the sequence of S. pyogenes Cas9. These two REC1 domains, though separated by the REC2 domain in the linear primary structure, assemble in the tertiary structure to form the REC1 domain.
- the REC2 domainmay also play a role in the recognition of the repeat: anti-repeat duplex.
- the REC2 domaincomprises amino acids 180-307 of the sequence of S. pyogenes Cas9.
- the NUC lobecomprises the RuvC domain (also referred to herein as RuvC-like domain), the HNH domain (also referred to herein as HNH-like domain), and the PAM- interacting (PI) domain.
- the RuvC domainshares structural similarity to retroviral integrase superfamily members and cleaves a single strand, e.g., the non-complementary strand of the target nucleic acid molecule.
- the RuvC domainis assembled from the three split RuvC motifs (RuvC I, RuvCII, and RuvCIII, which are often commonly referred to in the art as RuvCI domain, or N-terminal RuvC domain, RuvCII domain, and RuvCIII domain) at amino acids 1-59, 718-769, and 909-1098, respectively, of the sequence of S. pyogenes Cas9. Similar to the REC1 domain, the three RuvC motifs are linearly separated by other domains in the primary structure, however in the tertiary structure, the three RuvC motifs assemble and form the RuvC domain.
- the HNH domainshares structural similarity with HNH endonucleases, and cleaves a single strand, e.g., the complementary strand of the target nucleic acid molecule.
- the HNH domainlies between the RuvC II-III motifs and comprises amino acids 775-908 of the sequence of S. pyogenes Cas9.
- the PI domaininteracts with the PAM of the target nucleic acid molecule, and comprises amino acids 1099-1368 of the sequence of S. pyogenes Cas9. Crystal structures have been determined for naturally occurring bacterial Cas9 nucleases (Jinek et al., Science, 343(6176): 1247997, 2014) and for S.
- pyogenes Cas9 with a guide RNAe.g., a synthetic fusion of crRNA and tracrRNA
- a guide RNAe.g., a synthetic fusion of crRNA and tracrRNA
- nuclease activitye.g., double strand break activity in or directly proximal to a target site.
- the Cas9 nucleasehas been modified to inactivate one of the catalytic residues of the nuclease.
- the Cas9 nucleaseis a nickase and produces a single stranded break. See, e.g., Dabrowska et al. Frontiers in Neuroscience (2016) 12(75). It has been shown that one or more mutations in the RuvC and HNH catalytic domains of the enzyme may improve Cas9 efficiency. See, e.g., Sarai et al. Currently Pharma. Biotechnol. (2017) 18(13).
- the Cas9 nucleaseis fused to a second domain, e.g., a domain that modifies DNA or chromatin, e.g., a deaminase or demethylase domain.
- the Cas9 nucleaseis modified to eliminate its nuclease activity. In some embodiments, the Cas nuclease is modified to enhance specificity of the enzyme (e.g., reduce off-target effects, maintain robust on-target cleavage). In some embodiments, the Cas9 nuclease is an enhanced specificity Cas9 variant (e.g., eSPCas9). See, e.g., Slaymaker et al. Science (2016) 351 (6268): 84-88. In some embodiments, the Cas9 nuclease is a high fidelity Cas9 variant (e.g., SpCas9-HF1). See, e.g., Kleinstiver et al.
- PAMprotospacer adjacent motif
- the PAM specificitymay be a function of the DNA-binding specificity of the nuclease, e.g., Cas9 protein, e.g., a “protospacer adjacent motif recognition domain” at the C-terminus of Cas9.
- the PAMcomprises a 2 to 6 base pair DNA sequence immediately downstream or upstream of the target site in the genome, which may be recognized directly by a Cas nuclease, e.g., a Cas9, to promote cleavage of the target site by the Cas nuclease, or in the case of nuclease-deficient Cas9 allows binding to the DNA at that locus.
- a Cas nucleasee.g., a Cas9
- the PAMcan be a 5' PAM (i.e., located upstream of the 5' end of the protospacer).
- the PAMcan be a 3' PAM (i.e., located downstream of the 5' end of the protospacer).
- Cas nuclease PAMrefers to a DNA sequence that may be required to home or localize a CRISPR/Cas nuclease (e.g., Cas9 or other Cas nucleases, such as Cas12a (Cpf1) or Cas12b) interacting with a gRNA to sequence comprising a target domain.
- CRISPR/Cas nucleasese.g., Cas9 or other Cas nucleases, such as Cas12a (Cpf1) or Cas12b
- Non-limiting examples of CRISPR/Cas nucleases and their corresponding consensus PAMare described herein.
- SpyCas9can also weakly recognize NGA, NNGG, and a selection of other sequences.
- the MAD7 endonuclease from Eubacterium rectalecan recognize 5’- TTTV on the 5’ end of the gRNA sequence, and to some extent 5’-YTTV and YTTN.
- the Cpf1 endonuclease from Acidaminococcus sp. and Lachnospiraceae sp.recognize 5’-TTTN and the Cpf1 endonuclease from Francisella novicide can recognize 5’-TTN-3’ on the 5’ end of the gRNA.
- Cas9 nucleasesare known in the art and may be obtained from various sources and/or engineered/modified to modulate one or more activities or specificities of the enzymes.
- the Cas9 nucleasehas been engineered/modified to recognize one or more PAM sequence.
- the Cas9 nucleasehas been engineered/modified to recognize one or more PAM sequence that is different than the PAM sequence the Cas9 nuclease recognizes without engineering/modification.
- the Cas9 nucleasehas been engineered/modified to reduce off-target activity of the enzyme.
- the nucleotide sequence encoding the Cas9 nucleaseis modified further to alter the specificity of the nuclease activity (e.g., reduce off-target cleavage, decrease the nuclease activity or lifetime in cells, increase homology-directed recombination and reduce non-homologous end joining). See, e.g., Komor et al. Cell (2017) 168: 20-36.
- the nucleotide sequence encoding the Cas9 nucleaseis modified to alter the PAM recognition of the nuclease.
- the Cas9 nuclease SpCas9recognizes PAM sequence NGG
- relaxed variants of the SpCas9 comprising one or more modifications of the nucleasee.g., VQR SpCas9, EQR SpCas9, VRER SpCas9
- PAM recognition of a modified Cas9 nucleaseis considered “relaxed” if the Cas9 nuclease recognizes more potential PAM sequences as compared to the Cas9 nuclease that has not been modified.
- the Cas9 nuclease SaCas9recognizes PAM sequence NNGRRT, whereas a relaxed variant of the SaCas9 comprising one or more modifications (e.g., KKH SaCas9) may recognize the PAM sequence NNNRRT.
- the Cas9 nuclease FnCas9recognizes PAM sequence NNG, whereas a relaxed variant of the FnCas9 comprising one or more modifications of the nuclease (e.g., RHA FnCas9) may recognize the PAM sequence YG.
- the Cas nucleaseis a Cpf1 nuclease comprising substitution mutations S542R and K607R and recognize the PAM sequence TYCV. In one example, the Cas nuclease is a Cpf1 nuclease comprising substitution mutations S542R, K607R, and N552R and recognize the PAM sequence TATV. See, e.g., Gao et al. Nat. Biotechnol. (2017) 35(8): 789-792.
- a Cas nucleaserecognizes a PAM selected from: a SpyCas9 PAM, a SpGCas9 PAM, a SpRYCas9 PAM, a Sth1Cas9 PAM, a SauCas9 PAM, a NmeCas9 PAM, a RspCas9 PAM, a Cca1Cas9 PAM, a PspCas9 PAM, a OrhCas9 PAM, a ScCas9 PAM, a SmacCas9 PAM, a TdeCas9 PAM, a Nme2Cas9 PAM, a CjeCas9 PAM, a SmuCas9 PAM, a Smu2Cas9 PAM, a PmuCas9 PAM, a SpaCas9 PAM, a NciCas9 PAM, a ClaCas9
- a Cas nucleaserecognizes a PAM comprising, and/or consisting of a nucleotide sequence selected from: NGG; NNAGAAW; NNGRRT; NNNNGATT; NVNDCCY; BRTTTTT; NR(A or G)TTTT; NNAAAR(G or A); N(N or A)G; NAAN; NAAAAY; NHDTCCA; NNNVRYM; NNNNRYAC; NAA; GNNNNCNNA; NNGTGA; NNNNGTA; NNGGG; NNNCAT; NNRHHHY; NRRNAT; NNNNCNAA; NNNNCMCA; NNNNCRAA; NNNNGMAA; NNNCC; NGGNG; NNNNCNDD; NYAAA; NRGNN; N(C or D)GGN(T or A or G or C)NN; NRTAW; N(C or K or A)AARC; NAAAG; NV(A or G or C)R(A or G)
- more than one (e.g., 2, 3, or more) Cas nucleasesare used.
- at least one of the Cas nucleaseis a Cas9.
- at least one of the Cas nucleasesis a Cpf1 enzyme.
- at least one of the Cas9 nucleaseis derived from Streptococcus pyogenes.
- at least one of the Cas9 nucleaseis derived from Streptococcus pyogenes and at least one Cas9 nuclease is derived from an organism that is not Streptococcus pyogenes.
- gRNAand “guide RNA” are used interchangeably throughout and refer to a nucleic acid that promotes the specific targeting or homing of a CRISPR/Cas system to a target nucleic acid sequence.
- a gRNAcan be unimolecular (having a single RNA molecule), sometimes referred to herein as sgRNAs, or modular (comprising more than one, and typically two, separate RNA molecules).
- a gRNAmay bind to a target domain in the genome of a host cell.
- the gRNAmay comprise a targeting domain that may be partially or completely complementary to the target domain.
- the gRNAmay also comprise a “scaffold sequence,” (e.g., a tracrRNA sequence), that recruits a Cas nuclease to a target domain bound to a gRNA sequence (e.g., by the targeting domain of the gRNA sequence).
- the scaffold sequencemay comprise at least one stem loop structure and recruits an endonuclease. Exemplary scaffold sequences can be found, for example, in Jinek, et al. Science (2012) 337(6096):816-821, Ran, et al. Nature Protocols (2013) 8:2281-2308, International Publication No. WO2014/093694, and International Publication No. WO2013/176772.
- proximityrefers to a first, second or subsequent target site within distance of a PAM that can be bound by a CRISPR/Cas system, according to any method described herein.
- the mutation of the genomic DNA sequence at the first target siteis within 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, or 25 nucleotides from a PAM sequence.
- sgRNArefers to single guide RNA used in conjunction with CRISPR associated systems (Cas).
- sgRNAsare a fusion of crRNA and tracrRNA and contain nucleotides of sequence complementary to the desired target site (Jinek et al., Science, 343(6176): 1247997, 2014). Watson-Crick pairing of the sgRNA with the target site permits R-loop formation, which in conjunction with a functional PAM permits DNA cleavage or in the case of nuclease-deficient Cas9 allows binding to the DNA at that locus.
- Some exemplary suitable Cas9 gRNA scaffold sequencesare provided herein, and additional suitable gRNA scaffold sequences will be apparent to the skilled artisan based on the present disclosure. Such additional suitable scaffold sequences include, without limitation, those recited in Jinek, et al.
- the binding domains of naturally occurring SpCas9 gRNAtypically comprise two RNA molecules, the crRNA (partially) and the tracrRNA.
- SpCas9 gRNAsthat comprise only a single RNA molecule including both crRNA and tracrRNA sequences, covalently bound to each other, e.g., via a tetraloop or via click chemistry type covalent linkage, have been engineered and are commonly referred to as “single guide RNA” or “sgRNA.”
- sgRNAsfor use with other Cas nucleases, for example, with Cas12a nucleases, typically comprise only a single RNA molecule, as the naturally occurring Cas12a guide RNA comprises a single RNA molecule.
- a suitable gRNAmay thus be unimolecular (having a single RNA molecule), sometimes referred to herein as sgRNAs, or modular (comprising more than one, and typically two, separate RNA molecules).
- a gRNA as provided hereintypically comprises a targeting domain that binds to a target site in the genome of a cell.
- the “targeting domain” of the gRNAis complementary to the “target domain” on the target nucleic acid.
- the strand of the target nucleic acid comprising the nucleotide sequence complementary to the core domain of the gRNAis referred to herein as the “complementary strand” of the target nucleic acid.
- the targeting domainmediates targeting of the gRNA-bound Cas nuclease to a target site.
- Guidance on the selection of targeting domainscan be found, e.g., in Fu Y et al, Nat Biotechnol 2014 (doi: 10.1038/nbt.2808) and Sternberg SH et al., Nature 2014 (doi: 10.1038/naturel3011).
- the targeting domain of the gRNAthus base-pairs (in full or partial complementarity) with the sequence of the double-stranded genomic target site that is complementary to the sequence of the target domain, and thus with the strand complementary to the strand that comprises the PAM sequence. It will be understood that the targeting domain of the gRNA typically does not include the PAM sequence.
- the location of the PAMmay be 5’ or 3’ of the target domain sequence, depending on the nuclease employed.
- the PAMis typically 3’ of the target domain sequences for Cas9 nucleases, and 5’ of the target domain sequence for Cas12a nucleases.
- the length and complementarity of the targeting domain with the target sequencecontributes to specificity of the interaction of the gRNA/Cas9 nuclease complex with a target nucleic acid.
- the targeting domain of a gRNA provided hereinis 5 to 50 nucleotides in length. In some embodiments, the targeting domain is 15 to 25 nucleotides in length. In some embodiments, the targeting domain is 18 to 22 nucleotides in length. In some embodiments, the targeting domain is 19-21 nucleotides in length. In some embodiments, the targeting domain is 15 nucleotides in length. In some embodiments, the targeting domain is 16 nucleotides in length.
- the targeting domainis 17 nucleotides in length. In some embodiments, the targeting domain is 18 nucleotides in length. In some embodiments, the targeting domain is 19 nucleotides in length. In some embodiments, the targeting domain is 20 nucleotides in length. In some embodiments, the targeting domain is 21 nucleotides in length. In some embodiments, the targeting domain is 22 nucleotides in length. In some embodiments, the targeting domain is 23 nucleotides in length. In some embodiments, the targeting domain is 24 nucleotides in length. In some embodiments, the targeting domain is 25 nucleotides in length.
- the targeting domainfully corresponds, without mismatch, to a target domain sequence provided herein, or a part thereof.
- the targeting domain of a gRNA provided hereincomprises 1 mismatch relative to a target domain sequence provided herein.
- the targeting domaincomprises 2 mismatches relative to the target domain sequence.
- the target domaincomprises 3 mismatches relative to the target domain sequence.
- a targeting domaincomprises a core domain and a secondary targeting domain, e.g., as described in International Publication No. WO 2015/157070, which is incorporated by reference in its entirety.
- the core domaincomprises about 8 to about 13 nucleotides from the 3' end of the targeting domain (e.g., the most 3' 8 to 13 nucleotides of the targeting domain).
- the secondary domainis positioned 5' to the core domain.
- the core domainhas exact complementarity (corresponds fully) with the corresponding region of the target sequence, or part thereof.
- the core domaincan have 1 or more nucleotides that are not complementary (mismatched) with the corresponding nucleotide of the target domain sequence.
- the first complementarity domainis complementary with the second complementarity domain, and in an embodiment, has sufficient complementarity to the second complementarity domain to form a duplexed region under at least some physiological conditions.
- the first complementarity domainis 5 to 30 nucleotides in length.
- the first complementarity domaincomprises 3 subdomains, which, in the 5' to 3' direction are: a 5' subdomain, a central subdomain, and a 3' subdomain.
- the 5' subdomainis 4 to 9, e.g., 4, 5, 6, 7, 8 or 9 nucleotides in length.
- the central subdomainis 1, 2, or 3, e.g., 1, nucleotide in length.
- the 3' subdomainis 3 to 25, e.g., 4 to 22, 4 to 18, or 4 to 10, or 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, or 25 nucleotides in length.
- the first complementarity domaincan share homology with, or be derived from, a naturally occurring first complementarity domain. In an embodiment, it has at least 50% homology with a S. pyogenes, S. aureus or S. thermophilus, first complementarity domain.
- S. pyogenesS. aureus or S. thermophilus
- a linking domainserves to link the first complementarity domain with the second complementarity domain of a unimolecular gRNA.
- the linking domaincan link the first and second complementarity domains covalently or non-covalently.
- the linkageis covalent.
- the linking domainis, or comprises, a covalent bond interposed between the first complementarity domain and the second complementarity domain.
- the linking domaincomprises one or more, e.g., 2, 3, 4, 5, 6, 7, 8, 9, or 10 nucleotides.
- the linking domaincomprises at least one non-nucleotide bond, e.g., as disclosed in International Publication No. WO2018/126176, the entire contents of which are incorporated herein by reference.
- the second complementarity domainis complementary, at least in part, with the first complementarity domain, and in an embodiment, has sufficient complementarity to the second complementarity domain to form a duplexed region under at least some physiological conditions.
- the second complementarity domaincan include a sequence that lacks complementarity with the first complementarity domain, e.g., a sequence that loops out from the duplexed region.
- the second complementarity domainis 5 to 27 nucleotides in length. In an embodiment, the second complementarity domain is longer than the first complementarity region.
- the complementary domainis 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24 or 25 nucleotides in length.
- the second complementarity domaincomprises 3 subdomains, which, in the 5' to 3' direction are: a 5' subdomain, a central subdomain, and a 3' subdomain.
- the 5' subdomainis 3 to 25, e.g., 4 to 22, 4 to 18, or 4 to 10, or 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, or 25 nucleotides in length.
- the central subdomainis 1, 2, 3, 4 or 5, e.g., 3, nucleotides in length.
- the 3' subdomainis 4 to 9, e.g., 4, 5, 6, 7, 8 or 9 nucleotides in length.
- the 5' subdomain and the 3' subdomain of the first complementarity domainare respectively, complementary, e.g., fully complementary, with the 3' subdomain and the 5' subdomain of the second complementarity domain.
- the proximal domainis 5 to 20 nucleotides in length.
- the proximal domaincan share homology with or be derived from a naturally occurring proximal domain. In an embodiment, it has at least 50% homology with an S. pyogenes, S. aureus or S. thermophilus, proximal domain.
- a broad spectrum of tail domainsare suitable for use in gRNAs.
- the tail domainis 0 (absent), 1, 2, 3, 4, 5, 6, 7, 8, 9, or 10 nucleotides in length.
- the tail domain nucleotidesare from or share homology with a sequence from the 5' end of a naturally occurring tail domain.
- the tail domainincludes sequences that are complementary to each other and which, under at least some physiological conditions, form a duplexed region.
- the tail domainis absent or is 1 to 50 nucleotides in length.
- the tail domaincan share homology with or be derived from a naturally occurring proximal tail domain. In some embodiments, it has at least 50% homology with an S. pyogenes, S. aureus or S.
- thermophilus, tail domainincludes nucleotides at the 3' end that are related to the method of in vitro or in vivo transcription.
- the gRNAis chemically modified.
- any of the gRNAs provided hereincomprise one or more nucleotides that are chemically modified. Chemical modifications of gRNAs have previously been described, and suitable chemical modifications include any modifications that are beneficial for gRNA function and do not measurably increase any undesired characteristics, e.g., off-target effects, of a given gRNA.
- Suitable chemical modificationsinclude, for example, those that make a gRNA less susceptible to endo- or exonuclease catalytic activity, and include, without limitation, that the gRNA may comprise one or more modification chosen from phosphorothioate backbone modification, 2′-O-Me–modified sugars (e.g., at one or both of the 3’ and 5’ termini), 2’F- modified sugar, replacement of the ribose sugar with the bicyclic nucleotide-cEt, 3′thioPACE (MSP), or any combination thereof.
- MSP3′thioPACE
- gRNA modificationswill be apparent to the skilled artisan based on this disclosure, and such suitable gRNA modification include, without limitation, those described, e.g., in Rahdar et al. PNAS December 22, 2015 112 (51) E7110-E7117 and Hendel et al., Nat Biotechnol.2015 Sep; 33(9): 985–989, each of which is incorporated herein by reference in its entirety.
- a gRNA described hereincomprises one or more 2′-O-methyl-3′-phosphorothioate nucleotides, e.g., at least 2, 3, 4, 5, or 62′-O-methyl-3′-phosphorothioate nucleotides.
- a gRNA described hereincomprises modified nucleotides (e.g., 2′-O-methyl-3′- phosphorothioate nucleotides) at the three terminal positions and the 5’ end and/or at the three terminal positions and the 3’ end.
- the gRNAmay comprise one or more modified nucleotides, e.g., as described in International Publication Nos. WO2017/214460, WO2016/089433, and WO2016/164356, which are incorporated by reference their entirety.
- a gRNA described hereinis chemically modified.
- the gRNAmay comprise one or more 2’-O modified nucleotides, e.g., 2’-O-methyl nucleotides.
- the gRNAcomprises a 2’-O modified nucleotide, e.g., 2’-O-methyl nucleotide at the 5’ end of the gRNA.
- the gRNAcomprises a 2’-O modified nucleotide, e.g., 2’-O-methyl nucleotide at the 3’ end of the gRNA.
- the gRNAcomprises a 2’-O-modified nucleotide, e.g., 2’-O- methyl nucleotide at both the 5’ and 3’ ends of the gRNA.
- the gRNAis 2’-O-modified, e.g.2’-O-methyl-modified at the nucleotide at the 5’ end of the gRNA, the second nucleotide from the 5’ end of the gRNA, and the third nucleotide from the 5’ end of the gRNA.
- the gRNAis 2’-O-modified, e.g.2’-O-methyl-modified at the nucleotide at the 3’ end of the gRNA, the second nucleotide from the 3’ end of the gRNA, and the third nucleotide from the 3’ end of the gRNA.
- the gRNAis 2’-O-modified, e.g.2’-O-methyl-modified at the nucleotide at the 5’ end of the gRNA, the second nucleotide from the 5’ end of the gRNA, the third nucleotide from the 5’ end of the gRNA, the nucleotide at the 3’ end of the gRNA, the second nucleotide from the 3’ end of the gRNA, and the third nucleotide from the 3’ end of the gRNA.
- the gRNAis 2’-O-modified, e.g.2’-O-methyl-modified at the second nucleotide from the 3’ end of the gRNA, the third nucleotide from the 3’ end of the gRNA, and at the fourth nucleotide from the 3’ end of the gRNA.
- the nucleotide at the 3’ end of the gRNAis not chemically modified. In some embodiments, the nucleotide at the 3’ end of the gRNA does not have a chemically modified sugar.
- the gRNAis 2’-O- modified, e.g.2’-O-methyl-modified, at the nucleotide at the 5’ end of the gRNA, the second nucleotide from the 5’ end of the gRNA, the third nucleotide from the 5’ end of the gRNA, the second nucleotide from the 3’ end of the gRNA, the third nucleotide from the 3’ end of the gRNA, and the fourth nucleotide from the 3’ end of the gRNA.
- the 2’-O-methyl nucleotidecomprises a phosphate linkage to an adjacent nucleotide.
- the 2’-O-methyl nucleotidecomprises a phosphorothioate linkage to an adjacent nucleotide. In some embodiments, the 2’-O-methyl nucleotide comprises a thioPACE linkage to an adjacent nucleotide. In some embodiments, the gRNA may comprise one or more 2’-O-modified and 3’phosphorous-modified nucleotide, e.g., a 2’-O-methyl 3’phosphorothioate nucleotide.
- the gRNAcomprises a 2’-O-modified and 3’phosphorous-modified, e.g., 2’-O-methyl 3’phosphorothioate nucleotide at the 5’ and 3’ ends of the gRNA.
- the gRNAcomprises a backbone in which one or more non-bridging oxygen atoms has been replaced with a sulfur atom.
- the gRNAis 2’-O-modified and 3’phosphorous-modified, e.g.2’-O-methyl 3’phosphorothioate-modified at the nucleotide at the 3’ end of the gRNA, the second nucleotide from the 3’ end of the gRNA, and the third nucleotide from the 3’ end of the gRNA.
- nucleotide at the 3’ end of the gRNAis not chemically modified. In some embodiments, the nucleotide at the 3’ end of the gRNA does not have a chemically modified sugar.
- the gRNAmay comprise one or more 2’-O-modified and 3’- phosphorous-modified, e.g., 2’-O-methyl 3’thioPACE nucleotide.
- the gRNAcomprises a 2’-O-modified and 3’phosphorous-modified, e.g., 2’-O-methyl 3’thioPACE nucleotide at the 5’ end of the gRNA.
- the gRNAcomprises a 2’-O-modified and 3’phosphorous-modified, e.g., 2’-O-methyl 3’thioPACE nucleotide at the 3’ end of the gRNA.
- the gRNAcomprises a 2’-O- modified and 3’phosphorous-modified, e.g., 2’-O-methyl 3’thioPACE nucleotide at the 5’ and 3’ ends of the gRNA.
- the gRNAcomprises a backbone in which one or more non-bridging oxygen atoms have been replaced with a sulfur atom and one or more non-bridging oxygen atoms have been replaced with an acetate group.
- the gRNAis 2’-O-modified and 3’phosphorous-modified, e.g.2’-O-methyl 3’ thioPACE-modified at the nucleotide at the 5’ end of the gRNA, the second nucleotide from the 5’ end of the gRNA, and the third nucleotide from the 5’ end of the gRNA.
- the gRNAis 2’-O-modified and 3’phosphorous-modified, e.g.2’-O-methyl 3’thioPACE-modified at the nucleotide at the 5’ end of the gRNA, the second nucleotide from the 5’ end of the gRNA, the third nucleotide from the 5’ end of the gRNA, the nucleotide at the 3’ end of the gRNA, the second nucleotide from the 3’ end of the gRNA, and the third nucleotide from the 3’ end of the gRNA.
- the gRNAis 2’-O-modified and 3’phosphorous-modified, e.g.2’-O-methyl 3’thioPACE-modified at the second nucleotide from the 3’ end of the gRNA, the third nucleotide from the 3’ end of the gRNA, and the fourth nucleotide from the 3’ end of the gRNA.
- the nucleotide at the 3’ end of the gRNAis not chemically modified. In some embodiments, the nucleotide at the 3’ end of the gRNA does not have a chemically modified sugar.
- the gRNAis 2’-O-modified and 3’phosphorous-modified, e.g.2’-O-methyl 3’thioPACE-modified at the nucleotide at the 5’ end of the gRNA, the second nucleotide from the 5’ end of the gRNA, the third nucleotide from the 5’ end of the gRNA, the second nucleotide from the 3’ end of the gRNA, the third nucleotide from the 3’ end of the gRNA, and the fourth nucleotide from the 3’ end of the gRNA.
- the gRNAcomprises a chemically modified backbone.
- the gRNAcomprises a phosphorothioate linkage. In some embodiments, one or more non-bridging oxygen atoms have been replaced with a sulfur atom.
- the nucleotide at the 5’ end of the gRNA, the second nucleotide from the 5’ end of the gRNA, and the third nucleotide from the 5’ end of the gRNAeach comprise a phosphorothioate linkage. In some embodiments, the nucleotide at the 3’ end of the gRNA, the second nucleotide from the 3’ end of the gRNA, and the third nucleotide from the 3’ end of the gRNA each comprise a phosphorothioate linkage.
- the nucleotide at the 5’ end of the gRNA, the second nucleotide from the 5’ end of the gRNA, the third nucleotide from the 5’ end of the gRNA, the nucleotide at the 3’ end of the gRNA, the second nucleotide from the 3’ end of the gRNA, and the third nucleotide from the 3’ end of the gRNAeach comprise a phosphorothioate linkage.
- the second nucleotide from the 3’ end of the gRNA, the third nucleotide from the 3’ end of the gRNA, and at the fourth nucleotide from the 3’ end of the gRNAeach comprise a phosphorothioate linkage.
- the nucleotide at the 5’ end of the gRNA, the second nucleotide from the 5’ end of the gRNA, the third nucleotide from the 5’ end, the second nucleotide from the 3’ end of the gRNA, the third nucleotide from the 3’ end of the gRNA, and the fourth nucleotide from the 3’ end of the gRNAeach comprise a phosphorothioate linkage.
- the gRNAcomprises a thioPACE linkage.
- the nucleotide at the 3’ end of the gRNA, the second nucleotide from the 3’ end of the gRNA, and the third nucleotide from the 3’ end of the gRNAeach comprise a thioPACE linkage.
- the nucleotide at the 5’ end of the gRNA, the second nucleotide from the 5’ end of the gRNA, the third nucleotide from the 5’ end of the gRNA, the nucleotide at the 3’ end of the gRNA, the second nucleotide from the 3’ end of the gRNA, and the third nucleotide from the 3’ end of the gRNAeach comprise a thioPACE linkage.
- the second nucleotide from the 3’ end of the gRNA, the third nucleotide from the 3’ end of the gRNA, and at the fourth nucleotide from the 3’ end of the gRNAeach comprise a thioPACE linkage.
- the nucleotide at the 5’ end of the gRNA, the second nucleotide from the 5’ end of the gRNA, the third nucleotide from the 5’ end, the second nucleotide from the 3’ end of the gRNA, the third nucleotide from the 3’ end of the gRNA, and the fourth nucleotide from the 3’ end of the gRNAeach comprise a thioPACE linkage.
- modificationse.g., chemical modifications
- modificationssuitable for use in connection with the guide RNAs and genetic engineering methods provided herein have been described above. Additional suitable modifications, e.g., chemical modifications, will be apparent to those of skill in the art based on the present disclosure and the knowledge in the art, including, but not limited to those described in Hendel, A. et al., Nature Biotech., 2015, Vol 33, No.9; in International Publication No. WO2017/214460; WO2016/089433; and in WO2016/164356; each one of which is herein incorporated by reference in its entirety.
- a gRNA described hereincan be used in combination with one or more gRNA, e.g., for directing nucleases to one or more sites in a genome.
- multiple gRNA described hereincan be used in combination, e.g., for directing nucleases to multiple sites in a genome.
- a gRNA described hereincan be used in combination with a second gRNA, e.g., for directing nucleases to two sites in a genome.
- a gRNA described hereincan be used in combination with a third gRNA, e.g., for directing nucleases to three sites in a genome.
- a gRNA described hereincan be used in combination with a fourth gRNA, e.g., for directing nucleases to four sites in a genome.
- a gRNA described hereincan be used in combination with a fifth gRNA, e.g., for directing nucleases to five sites in a genome.
- a gRNA described hereincan be used in combination with a sixth gRNA, e.g., for directing nucleases to six sites in a genome.
- a gRNA described hereincan be used in combination with a seventh gRNA, e.g., for directing nucleases to seven sites in a genome.
- a gRNA described hereincan be used in combination with an eight gRNA, e.g., for directing nucleases to eight sites in a genome.
- a gRNA described hereincan be used in combination with a ninth gRNA, e.g., for directing nucleases to nine sites in a genome.
- a gRNA described hereincan be used in combination with a tenth gRNA, e.g., for directing nucleases to ten sites in a genome.
- a gRNA described hereincan be used in combination with more than tenth gRNA, e.g., for directing nucleases to more than ten sites in a genome.
- Exemplary Cell Surface Protein Editing StrategiesFurther examples of editing strategies targeting cell surface proteins of the present disclosure are provided herein.
- genetically engineered cells of the present disclosureare produced by contacting a hematopoietic cell with a gRNA comprising a targeting domain that is capable of binding to a target gene encoding a cell surface protein selected from a group consisting of: CD10; CD101; CD117; CD11A; CD11C; CD120B; CD123; CD13; CD133; CD152; CD200; CD205 (LY75); CD226; CD244 (SLAMF4); CD25; CD274; CD305 (LAIR1); CD32; CD33; CD34; CD366; CD38; CD41; CD42B; CD44; CD45RA; CD47; CD48; CD49D (ITGA4); CD49F; CD52; CD58; CD64; CD7; CD70; CD82; CD84; CD85; CD85K; CD86; CD9; CD96; CD99; CLL-1; EMR2 (ADGRE2); FR-B; GPR56
- genetically engineered cells of the present disclosureare produced by contacting a hematopoietic cell with a gRNA comprising a targeting domain that is capable of binding to a target gene encoding a cell surface protein selected from a group consisting of: CD10; CD11A; CD11C; CD152; CD200; CD274; CD33; CD34; CD42B; CD45RA; CD47; CD49D; CD49F; CD58; CD64; CD84; CD85K; CD9; CLL-1; GPR56; and IL1RAP.
- a target geneselected from a group consisting of: CD10; CD11A; CD11C; CD152; CD200; CD274; CD33; CD34; CD42B; CD45RA; CD47; CD49D; CD49F; CD58; CD64; CD84; CD85K; CD9; CLL-1; GPR56; and IL1RAP.
- genetically engineered cells of the present disclosureare produced by contacting a hematopoietic cell with a gRNA comprising a targeting domain that is capable of binding to a target gene encoding a cell surface protein selected from a group consisting of: CD10; CD117; CD11A; CD11C; CD120B; CD123; CD13; CD133; CD152; CD205; CD22; CD226; CD244; CD25; CD305; CD32; CD33; CD34; CD38; CD41; CD44; CD45RA; CD47; CD48; CD49D; CD49F; CD52; CD56; CD58; CD64; CD7; CD70; CD71; CD82; CD84; CD85K; CD86; CD96; CD99; CLL-1; EMR2; FR-B; GPR56; HLA-DR; and IL1RAP.
- a targeting domainthat is capable of binding to a target gene encoding a cell surface protein selected from a group consisting of: CD10;
- genetically engineered cells of the present disclosureare produced by contacting a hematopoietic cell with a first gRNA comprising a targeting domain that is capable of binding to a target gene encoding a first cell surface protein and a second gRNA comprising a targeting domain that is capable of binding to a target gene encoding a second cell surface protein selected from a group consisting of: CD274 and CD152; CD274 and CD226; CD152 and CD226; CD10 and CD49F; CD49F and GPR56; CD200 and CD34; CD34 and CD52; CD34 and CD133; GPR56 and CD133; CD42B and CD41; CD85K and CD64; CD85K and CD32; CD64 and CD32; CD64 and CD11C; CD64 and CD86; CD45RA and CD96; CD45RA and IL1RAP; CD45RA and CD123; CD84 and CD33; CD11C and IL1RAP; CD11C and CD33; CD11C and
- genetically engineered cells of the present disclosureare produced by contacting a hematopoietic cell with a first gRNA comprising a targeting domain that is capable of binding to a target gene encoding a first cell surface protein and a second gRNA comprising a targeting domain that is capable of binding to a target gene encoding a second cell surface protein selected from a group consisting of: CD85K and CD64; CD45RA and IL1RAP; CD34 and CD133; IL1RAP and CD33; CD47 and CD99; CLL-1 and CD305; CD152 and CD226; CD45RA and CD96; CD45RA and CD123; CD49D and CD305; IL1RAP and CD305; CD58 and CD305; CD49D and CD47; CD33 and CD305; CD49D and CD44; CD64 and CD86; GPR56 and CD133; CD34 and CD52; EMR2 and CD82; CD64 and CD32; CD42B and CD41; CD64 and CD11C
- genetically engineered cells of the present disclosureare produced by contacting a hematopoietic cell with a first gRNA comprising a targeting domain that is capable of binding to a target gene encoding a first cell surface protein and a second gRNA comprising a targeting domain that is capable of binding to a target gene encoding a second cell surface protein selected from a group consisting of: CD33 and CD205 (LY75); CD33 and CD244 (SLAMF4); CD33 and CD305 (LAIR1); CD33 and CD49D (ITGA4); CLL-1 and CD205 (LY75); CLL-1 and CD244 (SLAMF4); CLL-1 and CD305 (LAIR1); CLL-1 and CD49D (ITGA4); EMR2 and CD205 (LY75); EMR2 and CD244 (SLAMF4); EMR2 and CD305 (LAIR1); EMR2 and CD49D (ITGA4); CD38 and CD205 (LY75); CD38 and CD244 (SLAMF4); EMR
- a targeting domain which is complementary to a target nucleic acid in a cell surface proteinis complementary to a target nucleic acid encoding human CD33 or a portion thereof, e.g., having a sequence at least 70%, 75%, 80%, 85%, 90%, 95%, 96%, 97%, 98%, or 99% identical thereto to the CD33 amino acid sequence set forth below: MPLLLLLPLLWAGALAMDPNFWLQVQESVTVQEGLCVLVPCTFFHPIPYYDKNSPVHGYW FREGAIISRDSPVATNKLDQEVQEETQGRFRLLGDPSRNNCSLSIVDARRRDNGSYFFRM ERGSTKYSYKSPQLSVHVTDLTHRPKILIPGTLEPGHSKNLTCSVSWACEQGTPPIFSWL GIFPGDGSGKQETRAGVVHGAIGGAGVTALLALCLCLIFFIVKTHRRKAARTAVGRNDTH PTTGSASPKHQKKSKLHGPTETSSCSGA
- Non-limiting examples of CD33 gRNAsmay be found in International Patent Application Publication Nos. WO 2017/066760, WO 2020/150478, WO 2020/237217, WO 2018/160768, WO 2019/046285, WO2023/015182, WO2024/015925, WO2023/196816, WO2023/043858, and WO2022/217086, and WO2020/047164 each of which are incorporated by reference herein in their entirety.
- a targeting domain which is complementary to a target nucleic acid in a cell surface proteinis complementary to a target nucleic acid encoding human CLL- 1 or a portion thereof, e.g., having a sequence at least 70%, 75%, 80%, 85%, 90%, 95%, 96%, 97%, 98%, or 99% identical thereto to the CLL-1 amino acid sequence set forth below: MSEEVTYADLQFQNSSEMEKIPEIGKFGEKAPPAPSHVWRPAALFLTLLCLLLLIGLGVL A SMFHVTLKIEMKKMNKLQNISEELQRNISLQLMSNMNISNKIRNLSTTLQTIATKLCRE LYSKEQEHKCKPCPRRWIWHKDSCYFLSDDVQTWQESKMACAAQNASLLKINNKNALEFI KSQSRSYDYWLGLSPEEDSTRGMRVDNIINSSAWVIRNAPDLNNMYCGYINRLYVQYYHC TYKKRMICEKMAN
- Non-limiting examples of CLL-1 gRNAsmay be found in PCT Publication Nos. WO 2021/041971 WO2020/047164, WO2023/015182, WO2024/015925, WO2023/196816, WO2023/043858, and WO2022/047168, each of which are incorporated by reference herein in their entirety.
- a targeting domain which is complementary to a target nucleic acid in a cell surface proteinis complementary to a target nucleic acid encoding human CD123 or a portion thereof, e.g., having a sequence at least 70%, 75%, 80%, 85%, 90%, 95%, 96%, 97%, 98%, or 99% identical thereto to the CD123 amino acid sequence set forth below: M VLLWLTLLLIALPCLLQTKEDPNPPITNLRMKAKAQQLTWDLNRNVTDIECVKDADYSM PAVNNSYCQFGAISLCEVTNYTVRVANPPFSTWILFPENSGKPWAGAENLTCWIHDVDFL SCSWAVGPGAPADVQYDLYLNVANRRQQYECLHYKTDAQGTRIGCRFDDISRLSSGSQSS H ILVRGRSAAFGIPCTDKFVVFSQIEILTPPNMTAKCNKTHSFMHWKMRSHFNRKFRYEL QIQKRMQPVITEQVRDRTSFQLL
- Non-limiting examples of CD123 gRNAsmay be found in PCT Publication Nos. WO 2021/041977, WO2023/015182, WO2024015925, WO2023196816, WO2023043858, and WO2022/047165, each of which are incorporated by reference herein in their entirety.
- a targeting domain which is complementary to a target nucleic acid in a cell surface proteinis complementary to a target nucleic acid encoding human CD38 or a portion thereof, e.g., having a sequence at least 70%, 75%, 80%, 85%, 90%, 95%, 96%, 97%, 98%, or 99% identical thereto to the CD38 amino acid sequence set forth below: MANCEFSPVSGDKPCCRLSRRAQLCLGVSILVLILVVVLAVVVPRWRQQWSGPGTTKRFP ETVLARCVKYTEIHPEMRHVDCQSVWDAFKGAFISKHPCNITEEDYQPLMKLGTQTVPCN KILLWSRIKDLAHQFTQVQRDMFTLEDTLLGYLADDLTWCGEFNTSKINYQSCPDWRKDC SNNPVSVFWKTVSRRFAEAACDVVHVMLNGSRSKIFDKNSTFGSVEVHNLQPEKVQTLEA WVIHGGREDSRDLC
- Non-limiting examples of CD38 gRNAsmay be found in PCT Publication Nos. WO 2022/056489, WO2023/015182, and WO2023/196816, which are incorporated by reference herein in their entirety.
- a targeting domain which is complementary to a target nucleic acid in a cell surface proteinis complementary to a target nucleic acid encoding human EMR2 or a portion thereof, e.g., having a sequence at least 70%, 75%, 80%, 85%, 90%, 95%, 96%, 97%, 98%, or 99% identical thereto to the EMR2 amino acid sequence set forth below: MGGRVFLVFLAFCVWLTLPGAETQDSRGCARWCPQDSSCVNATACRCNPGFSSFSEIITT PMETCDDINECATLSKVSCGKFSDCWNTEGSYDCVCSPGYEPVSGAKTFKNESENTCQDV DECQQNPRLCKSYGTCVNTLGSYTCQCLPGFKLKPEDPKLCTDVNECTSGQNPCHSSTHC LNNVGSYQCRCRPGWQPIPGSPNGPNNTVCEDVDECSSGQHQCDSSTVCFNTVGSYSCRC RPGWKPRHGIPNNQKDTVCEDMTF
- EMR2 gRNAsmay be found in PCT Publication Nos. WO 2023/086422, WO2024/015925, WO2023/196816, and WO2023/043858, which are incorporated by reference herein in their entirety.
- a targeting domain which is complementary to a target nucleic acid in a cell surface proteinis complementary to a target nucleic acid encoding human CD117 or a portion thereof, e.g., having a sequence at least 70%, 75%, 80%, 85%, 90%, 95%, 96%, 97%, 98%, or 99% identical thereto to the CD117 amino acid sequence set forth below: MRGARGAWDFLCVLLLLLRVQTGSSQPSVSPGEPSPPSIHPGKSDLIVRVGDEIRLLCTD PGFVKWTFEILDETNENKQNEWITEKAEATNTGKYTCTNKHGLSNSIYVFVRDPAKLFLV DRSLYGKEDNDTLVRCPLTDPEVTNYSLKGCQGKPLPKDLRFIPDPKAGIMIKSVKRAYH RLCLHCSVDQEGKSVLSEKFILKVRPAFKAVPVVSVSKASYLLREGEEFTVTCTIKDVSS SVYSTWKRENSQTKLQE
- a targeting domain which is complementary to a target nucleic acid in a cell surface proteinis complementary to a target nucleic acid encoding human IL1RAP or a portion thereof, e.g., having a sequence at least 70%, 75%, 80%, 85%, 90%, 95%, 96%, 97%, 98%, or 99% identical thereto to the IL1RAP amino acid sequence set forth below: MTLLWCVVSLYFYGILQSDASERCDDWGLDTMRQIQVFEDEPARIKCPLFEHFLKFNYSTAHSAGLTLIWYWTRQ DRDLEEPINFRLPENRISKEKDVLWFRPTLLNDTGNYTCMLRNTTYCSKVAFPLEVVQKDSCFNSPMKLPVHKLY IEYGIQRITCPNVDGYFPSSVKPTITWYMGCYKIQNFNNVIPEGMNLSFLIALISNNGNYTCVVTYPENGRTFHL TRTLTVKVVGSPKNAVPPVIHSPN
- a genetically engineered hematopoietic cellcomprises genetic engineering in a first gene encoding a first cell surface protein as described herein and genetic engineering in a second gene encoding a second cell surface protein as described herein, wherein the first gene and the second gene each comprise a mutation (e.g., a substitution, a deletion, or an insertion at one or more nucleotide positions) which is located in a sequence (e.g., an exon and/or an intron) that hybridizes with a gRNA, or hybridizes with a portion of a gRNA (e.g., a sequence of at least 10 nucleotides in the gRNA), described in any of the foregoing references (e.g., see: disclosures related to CD47 editing in WO2023/196816; disclosures related to CD7 editing in WO2022/061115 and WO2023/015182; disclosures related to CD34 editing in WO2022/147347; disclosures
- HDRHomology-Directed Repair
- CRISPR/Cas-based HDR-mediated gene editinge.g., CRISPR/Cas-based HDR-mediated gene editing.
- HDRis a process wherein damage to DNA (e.g., a break in the DNA) is repaired using a donor sequence with flanking sequences comprising homology to the site of DNA damage.
- a CRISPR/Cas systemis used to introduce a break in the DNA (e.g., a double-stranded break (DSB)).
- DSBdouble-stranded break
- HDRcan be promoted (e.g., relative to other DNA repair pathways, e.g., NHEJ).
- HDRcan result in substitution or insertion mutations that replace endogenous or naturally occurring sequences with those of the donor sequence.
- methods described hereincan be used to introduce a mutation into a target DNA, e.g., to correct a disease-associated genetic mutation.
- HDRmay be induced by a DNA damage event that is capable of being mutagenic if left unrepaired or unprocessed, e.g., a double-stranded break.
- the DNA damage eventis induced by a CRISPR/Cas system, e.g., comprising a Cas nuclease, e.g., Cas9.
- DNA damage capable of producing a mutationexamples include, but are not limited to, DNA alkylation, base deamination, base depurination, incidence of abasic sites, single-stranded breaks, and double-stranded breaks.
- sequence “proximal” to the sites of damageis defined as a sequence that is found anywhere 10, 20, 30, 40, 50, 60, 70, 80, 90, 100, 150, 200, 250, 300, 350, 400, 450, or 500 nucleotides in the 5’ or 3’ direction of site of damage.
- Processing by nucleasesin turn, generates single- stranded overhangs comprised of a stretch of nucleotides that are not participating in base pairing interactions with nucleotides on the cognate strand to which the strand bearing the overhang is hybridized.
- Strand invasionfollows, wherein the overhangs transiently base pair with a donor sequence that is located in close physical proximity to the damaged DNA molecule.
- template polynucleotide homology to a target site provided by the flanking sequencesdirects template polynucleotide participation in HDR.
- Strand invasionis followed by cellular polymerase-dependent recombination wherein the donor sequence serves as the template to direct the repair of the damaged DNA.
- Recombination between the donor sequence and the damaged DNAcan incorporate the sequence of the donor sequence into the damaged DNA molecule. Following recombination, the repair is completed by a cellular ligase enzyme.
- the donor sequenceis provided by, for example, a template polynucleotide. When the donor sequence differs at one or more positions relative to a target DNA, integration of the donor sequence by HDR results in a mutation.
- the target DNAcomprises a mutation relative to a reference sequence (e.g., a wild-type sequence, or a sequence dominant in a population of subjects, or a sequence not characteristic of, or causally associated with, a disease or disorder); such a mutation may be referred to herein as a prior mutation (as distinguished from a mutation introduced by a method described herein).
- a prior mutationis characteristic of, or causally associated with, a disease, e.g., a mutation that is known to cause a genetic disease, or is a mutation that is known to convey increased risk of a genetic disease.
- a method described hereinalters a genomic sequence comprising a mutation characteristic for and/or causally associated with a disease or disorder, changing the genomic sequence to a sequence that is not characteristic for and/or causally associated with that disease or disorder.
- such alterationcomprises correcting a prior mutation.
- such alterationcomprises introducing a silent mutation, a restriction site, or a tag sequence.
- a donor sequencediffers from a sequence in the target DNA in one or more nucleotides, and integration of the donor sequence into the target DNA produces a genetic modification in the target DNA.
- the donor sequencediffers from a target DNA in a manner that integration of the donor sequence corrects a prior mutation in the target DNA, e.g., integration of the donor sequence into a target DNA comprising a prior mutation results in a modification of the mutation within the target DNA, e.g., in a modification of the target DNA sequence to the wild-type sequence, to the dominant sequence in a population of subjects, or, where the prior mutation is characteristic of, or causally associated with, a disease or disorder, in a modification to a sequence that is not characteristic of, or causally associated with a disease or disorder.
- the prior mutationis characteristic for, or causally associated with, a disease or disorder.
- the prior mutationis not characteristic for, or not causally associated with, a disease or disorder.
- a template polynucleotide comprising the donor sequenceis referred to as a template for HDR of the mutation.
- the donor sequencecomprises a gene or a portion thereof, e.g., a gene, or portion thereof, that (e.g., prior to any genetic modification described herein) is mutated or non-functional in the target DNA or the genome of a cell.
- a template polynucleotide comprising the donor sequenceis referred to as a template for HDR insertion of a gene, or portion thereof, in the target DNA.
- a template polynucleotideis single-stranded, e.g., a single- strand donor oligonucleotide (ssODN).
- a template polynucleotideis double-stranded, e.g., a plasmid or a double-stranded donor oligonucleotide (dsODN).
- dsODNdouble-stranded donor oligonucleotide
- a template polynucleotideis a minicircle plasmid.
- a template polynucleotideis a nanoplasmid.
- minicircle plasmidis a plasmid that contains double stranded donor template without conventional plasmid backbones. Minicircle plasmids may be processed via a single DSB leading to linearization, whereas larger plasmids might require two DSBs which flank the template polynucleotide to excise the donor. Those of ordinary skill in the art will also recognize that a nanoplasmid comprises a circular DNA molecule of 500 base pairs or less and can be generated using services provided by Aldevron®.
- minicircle plasmids and nanoplasmidsare, in some embodiments, cut in a host cell prior to HDR via, for example, an exogenous nuclease (e.g., Cas9) targeting gRNA cut sites engineered into the plasmid sequence.
- minicircle plasmids and nanoplasmids comprising a template polynucleotideneed not be cut by an exogenous prior to HDR.
- a template polynucleotiderefers to a nucleic acid that is a template for HDR, e.g., HDR of a mutation in the target DNA.
- a template polynucleotideis approximately 10, 20, 30, 40, 50, 60, 70, 80, 90, 100, 110, 120, 130, 140, 150, 160, 170, 180, 190, or 200 nucleotides long +/- 1, 2, 3, 4, 5, 6, 7, 8, 9, or 10 nucleotides long.
- a template polynucleotideis approximately 300, 400, 500, 600, 700, 800, 900, 1000, 1100, 1200, 1300, 1400, 1500, 1600, 1700, 1800, 1900, 2000, 2100, 2200, 2300, 2400, 2500, 2600, 2700, 2800, 2900, 3000, 3100, 3200, 3300, 3400, or 3500 nucleotides +/- 10, 25, 50, or 75 nucleotides long.
- the donor sequencecomprises a modification as compared to the target DNA, for example, a mutation, e.g., an insertion, deletion, or substitution as compared to the target DNA nucleotide sequence.
- the donor sequencecomprises a substitution of a single nucleotide as compared to the target DNA.
- Such donor sequencesare useful, for example, to effect genetic modifications that correct a single nucleotide mutation in a target DNA sequence that is characteristic for, or causally associated with, a disease or disorder.
- the donor sequencecomprises a substitution of two or more nucleotides as compared to the target DNA.
- Such donor sequencesare useful, for example, to effect genetic modifications that correct more complex mutations, e.g., affecting two or more nucleotides, in a target DNA sequence that are characteristic for, or causally associated with, a disease or disorder.
- the donor sequencecomprises one or more insertions (e.g., of one or more nucleotides) as compared to the target DNA. Such donor sequences are useful, for example, to effect genetic modifications that create insertion mutations in a target DNA sequence.
- the donor sequencecomprises one or more deletions (e.g., of one or more nucleotides) as compared to the target DNA. Such donor sequences are useful, for example, to effect genetic modifications that create deletion mutations in a target DNA sequence.
- the donor sequencecomprises two or more substitutions as compared to the target DNA, wherein, if integrated into the target DNA, at least one such substitution results in the correction of a mutation that is characteristic of, or causally associated with, a disease or mutation, and wherein at least one such substitution results in a silent mutation in the target DNA, e.g., a substitution of a wobble base within an amino acid-encoding codon of a target DNA.
- donor sequencesare useful, for example, to effect genetic modifications that correct disease-associated mutations in a target DNA sequence, while at the same time creating a sequence tag, e.g., a non-naturally occurring sequence or a sequence that did was not previously present in the target DNA, which is useful for identification and/or tracking of the modified cells.
- the donor sequencecomprises a restriction site or a unique sequence tag, for example, a unique primer binding site.
- the sequence comprising the restriction site or a unique sequence tagis an insertion relative to the target DNA, e.g., the target DNA does not comprise a restriction site or a unique sequence tag where the donor sequence comprises one.
- the sequence comprising the restriction site or a unique sequence tagis not an insertion relative to the target DNA.
- the sequence comprising the restriction site or a unique sequence tagcomprises a mutation (e.g., a substitution) as compared to the target DNA that, upon integration into the target DNA, produces a restriction site or a unique sequence tag.
- the sequence comprising the restriction site or a unique sequence tagdoes not alter an amino acid sequence encoded by the target DNA.
- a restriction site or a unique sequence tag thus introducedmay be used, e.g., to confirm the success of integration of the donor sequence (e.g., in an experiment where the modified target DNA is cleaved and fragments or sequences thereof are analyzed).
- the restriction endonuclease sitecomprises a Pvu1 site, e.g., 5’-CGATCG-3’.
- the target DNAcomprises a prior mutation and the donor sequence differs from the target DNA in a manner such that integration of the donor sequence corrects the prior mutation and produces one or more additional mutations (e.g., a second, third, fourth, or fifth mutation relative to the correction of the prior mutation (the first mutation)).
- the one or more additional mutationscomprise one or more silent mutations that do not alter the amino acid encoded by the nucleic acid sequence of the target DNA.
- the one or more silent mutationsare contiguous (i.e., directly adjacent) to the prior mutation or the codon containing the prior mutation.
- silent mutationsare used, e.g., as identifiers (e.g., “tags” or “bar codes”) of a given correction of a prior mutation or to facilitate confirmation of integration of the donor sequence (e.g., in an experiment where the modified target DNA sequences are analyzed).
- methods and compositions provided by the present disclosureare applied to a target DNA, e.g., in order to modify the target DNA.
- the target DNAcomprises a nucleotide sequence that is characteristic for, or causally associated with, a disease or disorder.
- a template polynucleotidecomprises a first flanking sequence and a second flanking sequence, also referred to herein as a first homology sequence and a second homology sequence.
- the first flanking sequence and second flanking sequencedirect the binding of the template polynucleotide to a target DNA sequence in the cell.
- a first flanking sequenceis at least 50, at least 60, at least 70, at least 80, at least 90, at least 100, at least 110, at least 120, at least 130, at least 140, at least 150, at least 160, at least 170, at least 180, at least 190, at least 200, at least 210, at least 220, at least 230, at least 240, or at least 250 nucleotides long (and optionally no more than 1000, no more than 750, no more than 500, no more than 400, no more than 300, or no more than 250 nucleotides long).
- a first flanking sequencecomprises 25- 300, 50-300, 75-300, 100-300, 125-300, 150-300, 175-300, 200-300, 225-300, 250-300, 275- 300, 100-400, 125-400, 150-400, 175-400, 200-400, 225-400, 250-400, 275-400, 300-400, 325, 400, 350-400, 375-400, 200-500, 225-500, 250-500, 275-500, 300-500, 325-500, 350- 500, 375-500, 400-500, 425-500, 450-500, or 475-500 nucleotides in length.
- a first flanking sequencecomprises 500-2000, 600-2000, 700-2000, 800-2000, 900-2000, 1000-2000, 1100-2000, 1200-2000, 1300-2000, 1400-2000, 1500-2000, 1600- 2000, 1700-2000, 1800-2000, or 1900-2000 nucleotides in length.
- the first flanking sequencehas at least 50%, at least 60%, at least 70%, at least at least 80%, at least 90%, at least 91%, at least 92%, at least 93%, at least 94%, at least 95%, at least 96%, at least 97%, at least 98%, at least 99%, or at least 100% identity to a sequence upstream of a DSB in the target DNA (e.g., upstream of a site where a DSB is produced by a CRISPR/Cas system described herein), or a sequence complementary thereto.
- the first flanking sequencehas 100% identity to a sequence upstream of a DSB in the target DNA (e.g., upstream of a site where a DSB is produced by a CRISPR/Cas system described herein), or a sequence complementary thereto.
- sequence “upstream” and “downstream”refer to a region within 10, within 20, within 30, within 40, within 50, within 60, within 70, within 80, within 90, or within 100 nucleotides of a feature in the DNA (e.g., a DSB), with each term referring to a different direction from the target site, and, in the case where the target DNA is a gene or portion thereof upstream is toward the transcription start site for the gene and downstream is away from the transcription start site for the gene.
- a feature in the DNAe.g., a DSB
- the first flanking sequenceis a 5’ homology arm of a template polynucleotide and is 5’ of a donor sequence, e.g., in an ssODN, dsODN, minicircle plasmid, or nanoplasmid.
- a second flanking sequenceis at least 50, at least 60, at least 70, at least 80, at least 90, at least 100, at least 110, at least 120, at least 130, at least 140, at least 150, at least 160, at least 170, at least 180, at least 190, at least 200, at least 210, at least 220, at least 230, at least 240, or at least 250 nucleotides long (and optionally no more than 1000, no more than 750, no more than 500, no more than 400, no more than 300, or no more than 250 nucleotides long).
- a second flanking sequencecomprises 25-300, 50-300, 75-300, 100-300, 125-300, 150-300, 175-300, 200-300, 225-300, 250-300, 275-300, 100-400, 125-400, 150-400, 175-400, 200-400, 225-400, 250-400, 275- 400, 300-400, 325, 400, 350-400, 375-400, 200-500, 225-500, 250-500, 275-500, 300-500, 325-500, 350-500, 375-500, 400-500, 425-500, 450-500, or 475-500 nucleotides in length.
- a second flanking sequencecomprises 500-2000, 600-2000, 700-2000, 800-2000, 900-2000, 1000-2000, 1100-2000, 1200-2000, 1300-2000, 1400-2000, 1500-2000, 1600-2000, 1700-2000, 1800-2000, or 1900-2000 nucleotides in length.
- the second flanking sequencehas at least 50%, at least 60%, at least 70%, at least 80%, at least 90%, at least 91%, at least 92%, at least 93%, at least 94%, at least 95%, at least 96%, at least 97%, at least 98%, at least 99%, or at least 100% identity to a sequence downstream of a target site (e.g., downstream of a DSB produced by a CRISPR/Cas system in the target site), or a sequence complementary thereto.
- a target sitee.g., downstream of a DSB produced by a CRISPR/Cas system in the target site
- the second flanking sequencehas 100% identity to a sequence downstream of a DSB in the target DNA (e.g., downstream of a site where a DSB is produced by a CRISPR/Cas system described herein), or a sequence complementary thereto.
- the second flanking sequenceis a 3’ homology arm of a template polynucleotide and is 3’ of a donor sequence, e.g., in an ssODN, dsODN, minicircle plasmid, or nanoplasmid.
- the first flanking sequence and the second flanking sequencehave identity or complementarity to different sequences within or proximal to the target DNA.
- the first flanking sequencehas identity or complementarity to a first target sequence within or proximal to a target DNA and the second flanking sequence has identity or complementarity to a second target sequence within or proximal to the target DNA.
- the first target sequence and second target sequenceare no more than 5, no more than 10, no more than 20, no more than 30, no more than 40, no more than 50, no more than 100, no more than 150, no more than 200, no more than 250, no more than 300, no more than 500, or no more than 1000 bases apart in the nucleic acid molecule comprising the target DNA.
- the first flanking sequencehas 100% identity to a sequence upstream of a DSB in the target DNA, or a sequence complementary thereto
- the second flanking sequencehas 100% identity to a sequence downstream of a DSB in the target DNA, or a sequence complementary thereto.
- a flanking sequencecomprises 500-2000, 600-2000, 700-2000, 800-2000, 900-2000, 1000-2000, 1100-2000, 1200-2000, 1300-2000, 1400-2000, 1500-2000, 1600-2000, 1700-2000, 1800-2000, or 1900-2000 consecutive nucleotides that are 100% identical to a target sequence within a target DNA.
- a second flanking sequencecomprises 500-2000, 600-2000, 700-2000, 800-2000, 900-2000, 1000-2000, 1100- 2000, 1200-2000, 1300-2000, 1400-2000, 1500-2000, 1600-2000, 1700-2000, 1800-2000, or 1900-2000 nucleotides in length.
- a flanking sequence(e.g., a 3’ homology arm or 5’ homology arm) comprises 25-300, 50-300, 75-300, 100-300, 125-300, 150-300, 175-300, 200-300, 225-300, 250-300, 275-300, 100-400, 125-400, 150-400, 175- 400, 200-400, 225-400, 250-400, 275-400, 300-400, 325, 400, 350-400, 375-400, 200-500, 225-500, 250-500, 275-500, 300-500, 325-500, 350-500, 375-500, 400-500, 425-500, 450- 500, or 475-500 consecutive nucleotides that are 100% identical to a target sequence within a target DNA.
- a flanking sequence(e.g., a 3’ homology arm or 5’ homology arm) comprises 2-100, 10-100, 20-100, 30-100, 40-100, 50-100, 60-100, 70-100, 80-100, 90-100, 2-150, 2-200, 2-250, 10-150, 10-200, 10-250, 50-150, 50-200, 50-250, 100- 150, 100-200, 100-250, 150-200, 150-200, or 200-250 consecutive nucleotides that are 100% identical to a target sequence within a target DNA.
- a flanking sequence(e.g., a 3’ homology arm or 5’ homology arm) comprises at least 2, at least 10, at least 20, at least 30, at least 40, at least 50, at least 60, at least 70, at least 80, at least 90, or at least 100 consecutive nucleotides that are 100% identical to a target sequence within a target DNA (and optionally no more than 200, no more than 180, no more than 160, no more than 140, no more than 120, or no more than 100 consecutive nucleotides that are 100% identical to a target sequence within a target DNA).
- a flanking sequence(e.g., a 3’ homology arm or a 5’ homology arm) comprises a nucleotide sequence that is 100% identical to a PAM sequence in the target DNA.
- the nucleotide sequence identical to the PAM sequenceis 2-3, 2-4, 2-5, 2-6, 3-4, 3-5, 3-6, 4-5, 4-6, or 5-6 nucleotides in length (e.g., 2, 3, 4, 5, or 6 nucleotides in length).
- a template polynucleotidecomprises a donor sequence.
- the donor sequenceis integrated into a target DNA at the site of a DSB.
- the donor sequenceis homologous to the target DNA or a portion thereof, e.g., the sequence of the target DNA surrounding or adjacent to the DSB. In some embodiments, the donor sequence is contiguous with the first and second flanking sequences in a template polynucleotide.
- a target DNAcomprises a gene or a portion thereof, and the donor sequence is homologous to the target DNA or a portion thereof (e.g., in proximity to a DSB or a site targeted for a DSB by a CRISPR/Cas system as described herein).
- the first and second flanking sequencesguide binding of the template polynucleotide to a target DNA, facilitating interaction of the donor sequence with its homologous sequence in the target DNA and/or with cellular DNA repair (e.g., HDR) pathway components.
- cellular DNA repaire.g., HDR
- the donor sequencediffers from a homologous sequence of the target DNA at 1, 2, 3, 4, 5, 6, 7, 8, 9, or 10 bases (e.g., 1- 10, 1-9, 1-8, 1-7, 1-6, 1-5, 1-4, 1-3, 1-2, 2-10, 2-9, 2-8, 2-7, 2-6, 2-5, 2-4, 2-3, 3-10, 3-9, 3-8, 3-7, 3-6, 3-5, 3-4, 4-10, 4-9, 4-8, 4-7, 4-6, 4-5, 5-10, 5-9, 5-8, 5-7, 5-6, 6-10, 6-9, 6-8, 6-7, 7- 10, 7-9, 7-8, 8-10, 8-9, or 9-10 bases), or at a number of positions corresponding to up to 1, 5, 10, 15, or 20% of the length of the donor sequence.
- basese.g., 1- 10, 1-9, 1-8, 1-7, 1-6, 1-5, 1-4, 1-3, 1-2, 2-10, 2-9, 2-8, 2-7, 2-6, 2-5, 2-4, 2-3,
- a donor sequenceis 1-100, 1-80, 1-60, 1-40, 1-20, 1-15, 1-10, 1-9, 1-8, 1-7, 1-6, 1-5, 1-4, 1-3, 1-2, 5-100, 5-80, 5-60, 5-40, 5-20, 5-15, 5-10, 5-9, 5-8, 5-7, 5-6, 10-100, 10-80, 10-60, 10-40, 10-20, 10-15, 20-100, 20-80, 20-60, 20-40, 60-100, or 60-80 nucleotides in length.
- a donor sequenceis no more than 100, no more than 90, no more than 80, no more than 70, no more than 60, no more than 50, no more than 45, no more than 40, no more than 35, no more than 30, no more than 25, no more than 20, no more than 15, no more than 14, no more than 13, no more than 12, no more than 11, no more than 10, no more than 9, no more than 8, no more than 7, no more than 6, no more than 5, no more than 4, no more than 3, no more than 2, or no more than 1 bases long.
- the donor sequencecomprises sequence corresponding to the wild-type, functional, and/or naturally-occurring sequence at a position or positions corresponding to a prior mutation in the target DNA.
- the donor sequencecomprises an artificial or heterologous sequence.
- a donor sequenceis 200-2000, 200-1900, 200-1800, 200-1700, 200-1600, 200-1500, 200-1400, 200-1300, 200-1200, 200- 1100, 100-1000, 100-900, 100-800, 100-700, 100-600, 100-500, 100-400, 100-300, or 100- 200 nucleotides in length.
- a donor sequenceis no more than 2000, no more than 1900, no more than 1800, no more than 1700, no more than 1600, no more than 1500, no more than 1400, no more than 1300, no more than 1200, no more than 1100, no more than 1000, no more than 900, no more than 800, no more than 700, no more than 600, no more than 500, no more than 400, no more than 300, or no more than 200 nucleotides in length.
- a CRISPR/Cas system for use in a method of the disclosurecomprises a Cas nuclease that recognizes a PAM sequence in the target DNA and cuts the target DNA at a position near to the PAM sequence (e.g., 5’ or 3’ of the PAM sequence).
- a PAM homologous sequenceis present in a 3’ homology arm or a 5’ homology arm of a template polynucleotide.
- the PAM homologous sequenceis positioned such that HDR of a DSB produced by a Cas nuclease promotes integration of a donor sequence.
- the DSBis positioned in a target DNA sequence homologous to the donor sequence.
- a schematic of an exemplary 3’ homology arme.g., where a CRISPR/Cas system (e.g., comprising Cas9) cuts a target DNA 5’ of a PAM sequence
- [N]x – [PAM] – [N]yFor example, an exemplary Cas nuclease, Cas9, cuts a target DNA 3-4 nucleotides 5’ of a PAM sequence.
- xis 3-4
- yis the number of nucleotides in the remaining length of the homology arm (e.g., wherein the length of the homology arm is described herein).
- xis 2 and the homology arm is 160-170 nucleotides long. In some embodiments, x is 2 and the homology arm is 170-180 nucleotides long. In some embodiments, x is 2 and the homology arm is 180-190 nucleotides long. In some embodiments, x is 2 and the homology arm is 190-200 nucleotides long. In some embodiments, x is 2 and the homology arm is 210-220 nucleotides long. In some embodiments, x is 2 and the homology arm is 220-230 nucleotides long. In some embodiments, x is 2 and the homology arm is 230-240 nucleotides long.
- xis 2 and the homology arm is 240-250 nucleotides long. In some embodiments, x is 3 and the homology arm is 50-60 nucleotides long. In some embodiments, x is 3 and the homology arm is 60-70 nucleotides long. In some embodiments, x is 3 and the homology arm is 70-80 nucleotides long. In some embodiments, x is 3 and the homology arm is 80-90 nucleotides long. In some embodiments, x is 3 and the homology arm is 90-100 nucleotides long. In some embodiments, x is 3 and the homology arm is 100-110 nucleotides long.
- xis 3 and the homology arm is 110-120 nucleotides long. In some embodiments, x is 3 and the homology arm is 120-130 nucleotides long. In some embodiments, x is 3 and the homology arm is 130-140 nucleotides long. In some embodiments, x is 3 and the homology arm is 140-150 nucleotides long. In some embodiments, x is 3 and the homology arm is 150-160 nucleotides long. In some embodiments, x is 3 and the homology arm is 160-170 nucleotides long. In some embodiments, x is 3 and the homology arm is 170-180 nucleotides long.
- xis 3 and the homology arm is 180-190 nucleotides long. In some embodiments, x is 3 and the homology arm is 190-200 nucleotides long. In some embodiments, x is 3 and the homology arm is 210-220 nucleotides long. In some embodiments, x is 3 and the homology arm is 220-230 nucleotides long. In some embodiments, x is 3 and the homology arm is 230-240 nucleotides long. In some embodiments, x is 3 and the homology arm is 240-250 nucleotides long. In some embodiments, x is 4 and the homology arm is 50-60 nucleotides long.
- xis 4 and the homology arm is 60-70 nucleotides long. In some embodiments, x is 4 and the homology arm is 70-80 nucleotides long. In some embodiments, x is 4 and the homology arm is 80-90 nucleotides long. In some embodiments, x is 4 and the homology arm is 90-100 nucleotides long. In some embodiments, x is 4 and the homology arm is 100-110 nucleotides long. In some embodiments, x is 4 and the homology arm is 110-120 nucleotides long. In some embodiments, x is 4 and the homology arm is 120-130 nucleotides long.
- xis 4 and the homology arm is 130-140 nucleotides long. In some embodiments, x is 4 and the homology arm is 140-150 nucleotides long. In some embodiments, x is 4 and the homology arm is 150-160 nucleotides long. In some embodiments, x is 4 and the homology arm is 160-170 nucleotides long. In some embodiments, x is 4 and the homology arm is 170-180 nucleotides long. In some embodiments, x is 4 and the homology arm is 180-190 nucleotides long. In some embodiments, x is 4 and the homology arm is 190-200 nucleotides long.
- xis 4 and the homology arm is 210-220 nucleotides long. In some embodiments, x is 4 and the homology arm is 220-230 nucleotides long. In some embodiments, x is 4 and the homology arm is 230-240 nucleotides long. In some embodiments, x is 4 and the homology arm is 240-250 nucleotides long.
- a schematic of an exemplary 5’ homology arm(e.g., where a CRISPR/Cas system (e.g., comprising Cas12a) cuts a target DNA 3’ of a PAM sequence) is provided below: [N]a – [PAM] – [N]b
- another exemplary Cas nuclease, Cas12acuts a target DNA 18-19 nucleotides 3’ of a PAM sequence.
- bis 18-19
- ais the number of nucleotides in the remaining length of the homology arm (e.g., wherein the length of the homology arm is described herein).
- b18 and a homology arm length of 100 nucleotides, a would be 100 minus 18 and minus the length of the PAM homologous sequence (e.g., where the PAM sequence is 3 nucleotides long, a would be 79 (100-18-3).
- bis 17 and the homology arm is 50-60 nucleotides long.
- bis 17 and the homology arm is 60-70 nucleotides long.
- bis 17 and the homology arm is 70-80 nucleotides long.
- bis 17 and the homology arm is 80-90 nucleotides long.
- bis 17 and the homology arm is 90-100 nucleotides long. In some embodiments, b is 17 and the homology arm is 100-110 nucleotides long. In some embodiments, b is 17 and the homology arm is 110-120 nucleotides long. In some embodiments, b is 17 and the homology arm is 120-130 nucleotides long. In some embodiments, b is 17 and the homology arm is 130-140 nucleotides long. In some embodiments, b is 17 and the homology arm is 140-150 nucleotides long. In some embodiments, b is 17 and the homology arm is 150-160 nucleotides long.
- bis 17 and the homology arm is 160-170 nucleotides long. In some embodiments, b is 17 and the homology arm is 170-180 nucleotides long. In some embodiments, b is 17 and the homology arm is 180-190 nucleotides long. In some embodiments, b is 17 and the homology arm is 190-200 nucleotides long. In some embodiments, b is 17 and the homology arm is 210-220 nucleotides long. In some embodiments, b is 17 and the homology arm is 220-230 nucleotides long. In some embodiments, b is 17 and the homology arm is 230-240 nucleotides long.
- bis 17 and the homology arm is 240-250 nucleotides long. In some embodiments, b is 18 and the homology arm is 50-60 nucleotides long. In some embodiments, b is 18 and the homology arm is 60-70 nucleotides long. In some embodiments, b is 18 and the homology arm is 70-80 nucleotides long. In some embodiments, b is 18 and the homology arm is 80-90 nucleotides long. In some embodiments, b is 18 and the homology arm is 90-100 nucleotides long. In some embodiments, b is 18 and the homology arm is 100-110 nucleotides long.
- bis 18 and the homology arm is 110-120 nucleotides long. In some embodiments, b is 18 and the homology arm is 120-130 nucleotides long. In some embodiments, b is 18 and the homology arm is 130-140 nucleotides long. In some embodiments, b is 18 and the homology arm is 140-150 nucleotides long. In some embodiments, x is 3 and the homology arm is 150-160 nucleotides long. In some embodiments, b is 18 and the homology arm is 160-170 nucleotides long. In some embodiments, b is 18 and the homology arm is 170-180 nucleotides long.
- bis 18 and the homology arm is 180-190 nucleotides long. In some embodiments, b is 18 and the homology arm is 190-200 nucleotides long. In some embodiments, b is 18 and the homology arm is 210-220 nucleotides long. In some embodiments, b is 18 and the homology arm is 220-230 nucleotides long. In some embodiments, b is 18 and the homology arm is 230-240 nucleotides long. In some embodiments, b is 18 and the homology arm is 240-250 nucleotides long. In some embodiments, b is 19 and the homology arm is 50-60 nucleotides long.
- bis 19 and the homology arm is 60-70 nucleotides long. In some embodiments, b is 19 and the homology arm is 70-80 nucleotides long. In some embodiments, b is 19 and the homology arm is 80-90 nucleotides long. In some embodiments, b is 19 and the homology arm is 90-100 nucleotides long. In some embodiments, b is 19 and the homology arm is 100-110 nucleotides long. In some embodiments, b is 19 and the homology arm is 110-120 nucleotides long. In some embodiments, b is 19 and the homology arm is 120-130 nucleotides long.
- bis 19 and the homology arm is 130-140 nucleotides long. In some embodiments, b is 19 and the homology arm is 140-150 nucleotides long. In some embodiments, b is 19 and the homology arm is 150-160 nucleotides long. In some embodiments, b is 19 and the homology arm is 160-170 nucleotides long. In some embodiments, b is 19 and the homology arm is 170-180 nucleotides long. In some embodiments, b is 19 and the homology arm is 180-190 nucleotides long. In some embodiments, b is 19 and the homology arm is 190-200 nucleotides long.
- a genetically engineered hematopoietic cellcomprises a genetic modification corresponding to integration of a donor sequence (e.g., from a template polynucleotide described herein) into a target DNA in the hematopoietic cell.
- the genetic modificationcorresponds to a position or positions where the donor sequence differs from the sequence of the target DNA.
- integration of the donor sequenceresults in modification at 1, 2, 3, 4, 5, 6, 7, 8, 9, or 10 bases (e.g., 1-10, 1- 9, 1-8, 1-7, 1-6, 1-5, 1-4, 1-3, 1-2, 2-10, 2-9, 2-8, 2-7, 2-6, 2-5, 2-4, 2-3, 3-10, 3-9, 3-8, 3-7, 3-6, 3-5, 3-4, 4-10, 4-9, 4-8, 4-7, 4-6, 4-5, 5-10, 5-9, 5-8, 5-7, 5-6, 6-10, 6-9, 6-8, 6-7, 7-10, 7-9, 7-8, 8-10, 8-9, or 9-10 bases) in the target DNA.
- 1, 2, 3, 4, 5, 6, 7, 8, 9, or 10 basese.g., 1-10, 1- 9, 1-8, 1-7, 1-6, 1-5, 1-4, 1-3, 1-2, 2-10, 2-9, 2-8, 2-7, 2-6
- integration of the donor sequenceresults in an insertion of 1, 2, 3, 4, 5, 6, 7, 8, 9, or 10 bases (e.g., 1-10, 1- 9, 1-8, 1-7, 1-6, 1-5, 1-4, 1-3, 1-2, 2-10, 2-9, 2-8, 2-7, 2-6, 2-5, 2-4, 2-3, 3-10, 3-9, 3-8, 3-7, 3-6, 3-5, 3-4, 4-10, 4-9, 4-8, 4-7, 4-6, 4-5, 5-10, 5-9, 5-8, 5-7, 5-6, 6-10, 6-9, 6-8, 6-7, 7-10, 7-9, 7-8, 8-10, 8-9, or 9-10 bases) in the target DNA.
- basese.g., 1-10, 1- 9, 1-8, 1-7, 1-6, 1-5, 1-4, 1-3, 1-2, 2-10, 2-9, 2-8, 2-7, 2-6, 2-5, 2-4, 2-3, 3-10, 3-9, 3-8, 3-7, 3-6, 3-5, 3-4, 4-10, 4-9,
- integration of the donor sequenceresults in substitution of 1, 2, 3, 4, 5, 6, 7, 8, 9, or 10 bases (e.g., 1-10, 1- 9, 1-8, 1-7, 1-6, 1-5, 1-4, 1-3, 1-2, 2-10, 2-9, 2-8, 2-7, 2-6, 2-5, 2-4, 2-3, 3-10, 3-9, 3-8, 3-7, 3-6, 3-5, 3-4, 4-10, 4-9, 4-8, 4-7, 4-6, 4-5, 5-10, 5-9, 5-8, 5-7, 5-6, 6-10, 6-9, 6-8, 6-7, 7-10, 7-9, 7-8, 8-10, 8-9, or 9-10 bases) in the target DNA.
- basese.g., 1-10, 1- 9, 1-8, 1-7, 1-6, 1-5, 1-4, 1-3, 1-2, 2-10, 2-9, 2-8, 2-7, 2-6, 2-5, 2-4, 2-3, 3-10, 3-9, 3-8, 3-7, 3-6, 3-5, 3-4, 4-10, 4-9, 4-8
- integration of the donor sequenceresults in modification at a number of positions in the target DNA corresponding to up to 1, 5, 10, 15, 20, 25, 30, 35, 40, 45, 50, 55, 60, 65, 70, 75, 80, 85, 90, 95, or 100% of the length of the donor sequence.
- integration of the donor sequenceresults in insertion of a number of bases in the target DNA corresponding to up to 1, 5, 10, 15, 20, 25, 30, 35, 40, 45, 50, 55, 60, 65, 70, 75, 80, 85, 90, 95, or 100% of the length of the donor sequence.
- a donor sequenceis 200-2000, 200- 1900, 200-1800, 200-1700, 200-1600, 200-1500, 200-1400, 200-1300, 200-1200, 200-1100, 100-1000, 100-900, 100-800, 100-700, 100-600, 100-500, 100-400, 100-300, or 100-200 nucleotides in length.
- a donor sequenceis no more than 2000, no more than 1900, no more than 1800, no more than 1700, no more than 1600, no more than 1500, no more than 1400, no more than 1300, no more than 1200, no more than 1100, no more than 1000, no more than 900, no more than 800, no more than 700, no more than 600, no more than 500, no more than 400, no more than 300, or no more than 200 nucleotides in length.
- the donor sequenceis 1-100, 1-80, 1-60, 1-40, 1-20, 1-15, 1-10, 1-9, 1-8, 1-7, 1-6, 1-5, 1-4, 1-3, 1-2, 5-100, 5-80, 5-60, 5-40, 5-20, 5-15, 5-10, 5-9, 5-8, 5-7, 5-6, 10- 100, 10-80, 10-60, 10-40, 10-20, 10-15, 20-100, 20-80, 20-60, 20-40, 60-100, or 60-80 nucleotides in length.
- a donor sequenceis no more than 100, no more than 90, no more than 80, no more than 70, no more than 60, no more than 50, no more than 45, no more than 40, no more than 35, no more than 30, no more than 25, no more than 20, no more than 15, no more than 14, no more than 13, no more than 12, no more than 11, no more than 10, no more than 9, no more than 8, no more than 7, no more than 6, no more than 5, no more than 4, no more than 3, no more than 2, or no more than 1 bases long.
- a donor sequenceis 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49, 50, 55, 60, 65, 70, 75, 80, 85, 90, 95, or 100 bases long.
- the donor sequence integratedcomprises a sequence corresponding to the wild-type, functional, and/or naturally-occurring sequence at a position or positions corresponding to a prior mutation in the target DNA.
- the donor sequencecomprises an artificial or heterologous sequence.
- integration of the donor sequenceproduces a restriction nuclease site or a unique sequence tag in the target DNA of the genetically engineered hematopoietic cell.
- integration of the donor sequence into the target DNA of the genetically engineered hematopoietic cellproduces one or more silent mutations along with a non-silent mutation (e.g., correction of a prior mutation, e.g., in a coding sequence).
- the one or more silent mutationsare contiguous with another mutation described herein (e.g., contiguous with correction of a prior mutation).
- a genetically engineered hematopoietic cellcomprises a genetic modification corresponding to correction of a prior mutation which substitutes a single prior mutation, e.g., a single nucleotide point mutation, for the corresponding base(s) present in a wild-type cell, and one or more silent mutations contiguous with the prior mutation.
- the disclosureprovides a genetically engineered hematopoietic cell comprising a genetic modification corresponding to integration of a donor sequence as described herein, e.g., a donor sequence described herein.
- integration of the donor sequence into a genetically engineered T cellresults in the expression of a chimeric antigen receptor (CAR).
- CARchimeric antigen receptor
- the CARbinds to at least one antigen present on a cell surface protein.
- the cell surface proteinis CD33. It will be understood that, upon engrafting donor cells into a recipient host organism, the relative levels of the engrafted donor cells (and descendants thereof) and the host cells, e.g., in a given niche (e.g., bone marrow), are important for physiological and/or therapeutic outcomes for the host organism. The level of engrafted donor cells, or descendants thereof, relative to host cells in a given tissue or niche is referred to herein as chimerism.
- a cell described hereine.g., an HSC or HPC
- a cell described hereinis capable of engrafting in a human subject and does not exhibit any difference in chimerism as compared to a hematopoietic cell of the same cell type, but not comprising a genomic modification that results in expression of a variant form (e.g., a wild-type form or having wild-type functionality) of a gene or a loss of expression of a gene.
- a variant forme.g., a wild-type form or having wild-type functionality
- a cell described hereinis capable of engrafting in a human subject exhibits no more than a 1, no more than a 2, no more than a 5, no more than a 10, no more than a 15, no more than a 20, no more than a 25, no more than a 30, no more than a 35, no more than a 40, no more than a 45, or no more than a 50% difference in chimerism as compared to a hematopoietic cell of the same cell type, but not comprising a genomic modification that results in expression of a variant form (e.g., a wild-type form or having wild-type functionality) of a gene or a loss of expression of a gene.
- a variant forme.g., a wild-type form or having wild-type functionality
- a genetic modificationis produced using HDR.
- producing a genetic modification using HDRcomprises contacting cells with a template polynucleotide, a CRISPR/Cas system, and one or more other agents (e.g., one or more HDR-promoting agents or expansion agents), e.g., contacting cells with a genetic modification mixture described herein.
- the disclosureprovides, in part, methods and compositions that achieve unexpectedly high editing efficiencies utilizing HDR.
- efficiency of HDR-mediated editing and/or efficiency of total/overall editingis determined by a method described herein.
- the efficiency of HDRis at least 20, 30, 40, 50, 60, 70, 80, 90, 95, or 99% (e.g., 50%, 60%, 70%, 80%, 90% or higher).
- contacting cells to produce a genetic modification using HDRcomprises contacting cells with one or more HDR-promoting agents as described herein.
- the disclosureis directed, in part, to the discovery that the presence of one or more HDR- promoting agents may result in unexpectedly and advantageously high efficiency of HDR. Accordingly, methods describing contacting a cell herein also contemplate contacting a population of cells to produce a population of genetically modified cells, e.g., an editing efficiency, percent viability, and/or HDR efficiency described herein.
- producing a genetic modification using HDRcomprises contacting a cell with a genetic modification mixture.
- a genetic modification mixturerefers to a mixture comprising a plurality of components that may be used to genetically modify a target DNA, e.g., in a cell.
- a genetic modification mixturecomprises one, two, three, or all of a CRISPR/Cas system, a template polynucleotide, one or more HDR-promoting agents, and one or more expansion agents.
- a genetic modification mixturepromotes HDR and HDR-mediated genetic modification (e.g., relative to another DNA repair pathway or genetic modifications utilizing another DNA repair pathway).
- contacting a cell with the genetic modification mixturecomprises adding the genetic modification mixture directly to media comprising the cell. In some embodiments, contacting a cell with the genetic modification mixture comprises adding media comprising the genetic modification mixture to the cell or adding the cell to media comprising the genetic modification mixture.
- the mediais a growth media, e.g., a growth media suited to hematopoietic cells (e.g., hematopoietic stem cells (HSCs)). Examples of growth media include, but are not limited to, a Stromal cell Growth Media (SCGM TM , e.g., as available from Lonza Bioscience) or serum- and feeder-free media (SFFM).
- SCGM TMStromal cell Growth Media
- SFFMserum- and feeder-free media
- contacting a cell with the genetic modification mixturecomprises electroporating the genetic modification mixture or one or more components of the mixture into the cell. In some embodiments, contacting a cell with the genetic modification mixture comprises solvating the mixture in a lipid-permeable buffer, e.g., to serve as a carrier for movement of mixture components across the cell membrane.
- lipid-permeable buffersinclude, but are not limited to, DMSO and lipofectamine.
- the genetic modification mixturecomprises a template polynucleotide, e.g., a single-strand donor oligonucleotide (ssODN), a double-stranded donor oligonucleotide (dsODN), a minicircle plasmid, or a nanoplasmid, comprising a donor sequence, a first flanking sequence and a second flanking sequence.
- the genetic modification mixturecomprises a dsODN comprising “capped” or “closed-ends” in order to minimize the chances that the NHEJ pathway is used in the genetic modification process.
- the dsODN comprising “capped” or “closed-ends”is a GenWand® double-stranded DNA.
- the genetic modification mixturecomprises a CRISPR/Cas system capable of producing a break, e.g., a double-stranded break, at a target site in the genome of the cell.
- the genetic modification mixturecomprises one or more other agents (e.g., an expansion agent and/or HDR-promoting agent) that promote genetic modification.
- generating a genetically engineered hematopoietic cellcomprises contacting a hematopoietic cell with a CRISPR/Cas system comprising a Cas nuclease, wherein the Cas nuclease comprises a base editor.
- a base editoris used to a create a genomic modification resulting in an artificial PAM.
- a base editoris used to a create a genomic modification resulting in a loss of expression of a protein antigen of interest (e.g., a cell surface protein antigen ), or in expression of a protein antigen variant not targeted by an immunotherapy.
- a base editor nucleasegenerally comprises a catalytically inactive Cas9 nuclease fused to a functional domain, e.g., a deaminase domain. See, e.g., Eid et al. Biochem. J. (2016) 475(11): 1955-1964; Rees et al. Nature Reviews Genetics (2016) 19:770-788.
- the catalytically inactive Cas9 nucleaseis referred to as “dead Cas” or “dCas9.”
- the catalytically inactive Cas nucleasehas reduced nuclease activity and is, e.g., a nickase (referred to as “nCas”).
- the nucleasecomprises a dCas9 fused to one or more uracil glycosylase inhibitor (UGI) domains.
- UBIuracil glycosylase inhibitor
- the nucleasecomprises a dCas9 fused to an adenine base editor (ABE), for example an ABE evolved from the RNA adenine deaminase TadA.
- ABEadenine base editor
- the nucleasecomprises a dCas9 fused to cytidine deaminase enzyme (e.g., APOBEC deaminase, pmCDA1, activation-induced cytidine deaminase (AID)).
- the catalytically inactive Cas9 nucleasehas reduced activity and is nCas9.
- the catalytically inactive Cas9 nucleaseis fused to one or more uracil glycosylase inhibitor (UGI) domains.
- the Cas9 nucleasecomprises an inactive Cas9 nuclease (dCas9) fused to an adenine base editor (ABE), for example an ABE evolved from the RNA adenine deaminase TadA.
- the Cas9 nucleasecomprises a nCas9 fused to an adenine base editor (ABE), for example an ABE evolved from the RNA adenine deaminase TadA.
- the Cas9 nucleasecomprises a dCas9 fused to cytidine deaminase enzyme (e.g., APOBEC deaminase, pmCDA1, activation- induced cytidine deaminase (AID)).
- the Cas9 nucleasecomprises a nCas9 fused to cytidine deaminase enzyme (e.g., APOBEC deaminase, pmCDA1, activation- induced cytidine deaminase (AID)).
- base editorsinclude, without limitation, BE1, BE2, BE3, HF-BE3, BE4, BE4max, BE4-Gam, YE1-BE3, EE-BE3, YE2-BE3, YEE-CE3, VQR-BE3, VRER-BE3, SaBE3, SaBE4, SaBE4-Gam, Sa(KKH)-BE3, Target-AID, Target-AID-NG, xBE3, eA3A- BE3, BE-PLUS, TAM, CRISPR-X, ABE7.9, ABE7.10, ABE7.10*, ABE8, ABE8e, ABE8.20-m, xABE, ABESa, VQR-ABE, VRER-ABE, Sa(KKH)-ABE, and CRISPR-SKIP.
- the base editoris a cytosine base editor (CBE).
- the CBEis CBE1, CBE2, CBE3, or CBE4.
- the CBEis nCas9-2xUGI; BE4-rAPOBEC1; BE4-rAPOBEC1 K34A H122A; BE4-PpAPOBEC1; BE4- PpAPOBEC1 R33A; BE4-PpAPOBEC1 H122A; BE4-RrA3F; BE4-AmAPOBEC1; or BE4- SsAPOBEC3B.
- the base editoris an adenine base editor (ABE).
- the ABEis ABE1, ABE2, ABE3, ABE4, ABE5, ABE6, ABE7, or ABE8.
- the ABEis ABE7.10-m; ABE7.10-d; ABE8e; ABE8.8-m; ABE8.8-d; ABE8.13-m; ABE8.13-d; ABE8.17-m; ABE8.17-d; ABE8.20-m; or ABE8.20-d.
- the base editorsincludes, without limitation, BE1, BE2, BE3, HF-BE3, BE4, BE4max, BE4-Gam, YE1-BE3, EE-BE3, YE2-BE3, YEE-CE3, VQR-BE3, VRER-BE3, SaBE3, SaBE4, SaBE4-Gam, Sa(KKH)-BE3, Target-AID, Target-AID-NG, xBE3, eA3A-BE3, BE-PLUS, TAM, CRISPR-X, ABE7.9, ABE7.10, ABE7.10*, xABE, ABESa, VQR-ABE, VRER-ABE, Sa(KKH)-ABE, and CRISPR-SKIP.
- the base editoris ABE8e or ABE8.20-m. Additional examples of base editors can be found, for example, in US Publication No. 2018/0312825A1, US Publication No.2018/0312828A1, International Publication No. WO 2018/165629A1, Yu et al. Nat Commun. (2020) 11(1):2052, and Gaudelli et al. Nat Biotechnol. (2020) 38(7):892-900. which are incorporated by reference herein in their entireties.
- abe8_20m sequenceis provided below: atgtccgaagtcgagttttcccatgagtactggatgagacacgcattgactctcgcaaagagggctcgagatgaa cgcgaggtgcccgtgggggcagtactcgtgctcaacaatcgcgtaatcggcgaaggttggaatagggcaatcgga ctccacgaccccactgcacatgcggaaatcatggcccttcgacagggagggcttgtgatgcagaattatcgactt tatgatgcgacgctgtactcgacgtttgaaccttgcgtaatgtgcgcgggagctatgattcactcccgcattgga cgagtttgaaccttgcgtaatgtg
- any of the Cas9 nucleases described hereinmay be fused to a Gam domain (bacteriophage Mu protein) to protect the Cas9 nuclease from degradation and exonuclease activity. See, e.g., Eid et al. Biochem. J. (2016) 475(11): 1955-1964.
- the Cas nucleasebelongs to class 2 type V of Cas endonuclease.
- Class 2 type V Cas endonucleasescan be further categorized as type V-A, type V-B, type V-C, and type V-U. See, e.g., Stella et al. Nature Structural & Molecular Biology (2017) 24: 882-892.
- the Cas nucleaseis a type V-A Cas endonuclease, such as a Cpf1 (Cas12a) nuclease.
- the Cas9 nucleaseis a type V-B Cas endonuclease, such as a C2c1 endonuclease. See, e.g., Shmakov et al. Mol Cell (2015) 60: 385-397.
- the Cas nucleaseis MAD7 TM .
- the Cas9 nucleaseis a Cpf1 nuclease or a variant thereof.
- the Cpf1 nucleasemay also be referred to as Cas12a. See, e.g., Strohkendl et al. Mol. Cell (2016) 71: 1-9.
- a composition or method described hereininvolves, or a host cell expresses a Cpf1 nuclease derived from Provetella spp. or Francisella spp., Acidaminococcus sp. (AsCpf1), Lachnospiraceae bacterium (LpCpf1), or Eubacterium rectale.
- the nucleotide sequence encoding the Cpf1 nucleasemay be codon optimized for expression in a host cell. In some embodiments, the nucleotide sequence encoding the Cpf1 endonuclease is further modified to alter the activity of the protein.
- CRISPR/Cas nucleasesare suitable for use according to aspects of this disclosure. For example, dCas or nickase variants, Cas variants having altered PAM specificities, and Cas variants having improved nuclease activities are embraced by some embodiments of this disclosure.
- catalytically inactive variants of Cas nucleasesare used according to the methods described herein.
- a catalytically inactive variant of Cpf1 (Cas12a)may be referred to dCas12a.
- catalytically inactive variants of Cpf1maybe fused to a function domain to form a base editor. See, e.g., Rees et al. Nature Reviews Genetics (2016) 19:770-788.
- the catalytically inactive Cas9 nucleaseis dCas9.
- the nucleasecomprises a dCas12a fused to one or more uracil glycosylase inhibitor (UGI) domains.
- the Cas9 nucleasecomprises a dCas12a fused to an adenine base editor (ABE), for example an ABE evolved from the RNA adenine deaminase TadA.
- the Cas nucleasecomprises a dCas12a fused to cytidine deaminase enzyme (e.g., APOBEC deaminase, pmCDA1, activation- induced cytidine deaminase (AID)).
- the Cas9 nucleasemay be a Cas14 nuclease or variant thereof.
- Cas14 nucleasesare derived from archaea and tend to be smaller in size (e.g., 400– 700 amino acids). Additionally Cas14 nucleases do not require a PAM sequence. See, e.g., Harrington et al. Science (2016). Any of the Cas9 nucleases described herein may be modulated to regulate levels of expression and/or activity of the Cas9 nuclease at a desired time. For example, it may be advantageous to increase levels of expression and/or activity of the Cas9 nuclease during particular phase(s) of the cell cycle.
- levels of homology- directed repairare reduced during the G1 phase of the cell cycle, therefore increasing levels of expression and/or activity of the Cas9 nuclease during the S phase, G2 phase, and/or M phase may increase homology-directed repair following the Cas endonuclease editing.
- levels of expression and/or activity of the Cas9 nucleaseare increased during the S phase, G2 phase, and/or M phase of the cell cycle.
- the Cas9 nucleasefused to the N-terminal region of human Geminin. See, e.g., Gutschner et al. Cell Rep. (2016) 14(6): 1555-1566.
- levels of expression and/or activity of the Cas9 nucleaseare reduced during the G1 phase.
- the Cas9 nucleaseis modified such that it has reduced activity during the G1 phase.
- an epigenetic modifiere.g., a chromatin-modifying enzyme, e.g., DNA methylase, histone deacetylase. See, e.g., Kungulovski et al. Trends Genet. (2016) 32(2):101-113.
- Cas9 nuclease fused to an epigenetic modifiermay be referred to as “epieffectors” and may allow for temporal and/or transient endonuclease activity.
- the Cas9 nucleaseis a dCas9 fused to a chromatin-modifying enzyme.
- Cytotoxic AgentsCytotoxic agents targeting cells (e.g., cancer cells) expressing a cell surface protein can be co-used with the genetically engineered hematopoietic cells as described herein.
- the term “cytotoxic agent”refers to any agent that can directly or indirectly induce cytotoxicity of a target cell, which expresses the cell surface protein (e.g., a target cancer cell).
- Such a cytotoxic agentmay comprise a protein-binding fragment that binds and targets an epitope of the lineage-specific cell surface antigen.
- the cytotoxic agentmay comprise an antibody, which may be conjugated to a drug (e.g., an anti- cancer drug) to form an antibody-drug conjugate (ADC).
- ADCantibody-drug conjugate
- the cytotoxic agent for use in the methods described hereinmay directly cause cell death of a target cell.
- the cytotoxic agentcan be an immune cell (e.g., a cytotoxic T cell) expressing a chimeric receptor.
- a signal(e.g., activation signal) may be transduced to the immune cell resulting in release of cytotoxic molecules, such as perforins and granzymes, as well as activation of effector functions, leading to death of the target cell.
- the cytotoxic agentmay be an ADC molecule.
- the drug moiety in the ADCwould exert cytotoxic activity, leading to target cell death.
- the cytotoxic agentmay indirectly induce cell death of the target cell.
- the cytotoxic agentmay be an antibody, which, upon binding to the target cell, would trigger effector activities (e.g., ADCC) and/or recruit other factors (e.g., complements), resulting in target cell death.
- Any of the cytotoxic agents described hereintarget a lineage-specific cell surface antigen, e.g., comprising a protein-binding fragment that specifically binds an epitope in the cell surface protein.
- Non-limiting examples of cytotoxic agents that can be utilized in the compositions and methods described hereininclude those described in PCT Publication Nos. WO2022/056497, WO2022/056490, and WO2019/178382, WO2023/201238, and International Patent Application No. PCT/US2024/029308.
- cytotoxic agentis used to target more than one (e.g., 1, 2, 3, 4, 5 or more) epitopes of a lineage-specific cell surface antigen.
- more than one (e.g., 1, 2, 3, 4, 5 or more) cytotoxic agentis used to target an epitope(s) of one or more lineage-specific cell surface antigen(s) (e.g., additional/alternative antigens).
- targeting of more than one cell surface proteinreduces relapse of a hematopoietic malignancy.
- two or more cytotoxic agentsare used in the methods described herein.
- the two or more cytotoxic agentsare administered concurrently.
- the two or more cytotoxic agentsare administered sequentially. Examples of additional cell surface proteins that may be targeted are known in the art (see, e.g., Tasian, Ther. Adv.
- the methods described hereininvolve targeting a cell surface protein and one or more additional cell surface proteins.
- a genetically engineered cell of the present disclosuremay be contacted with a cytotoxic agent comprising an antigen-binding fragment that recognizes a cell surface protein selected from a group consisting of: CD10; CD101; CD117; CD11A; CD11C; CD120B; CD123; CD13; CD133; CD152; CD200; CD205 (LY75); CD226; CD244 (SLAMF4); CD25; CD274; CD305 (LAIR1); CD32; CD33; CD34; CD366; CD38; CD41; CD42B; CD44; CD45RA; CD47; CD48; CD49D (ITGA4); CD49F; CD52; CD58; CD64; CD7; CD70; CD82; CD84; CD85; CD85K; CD86; CD9; CD96; CD99; CLL-1; EMR2 (ADGRE2); FR-B; GPR56; and IL1RAP.
- a cytotoxic agentcomprising an antigen-binding fragment that recognizes
- a genetically engineered cell of the present disclosuremay be contacted with a cytotoxic agent comprising an antigen-binding fragment that recognizes a cell surface protein selected from a group consisting of: CD10; CD101; CD11A; CD11C; CD123; CD133; CD152; CD200; CD205; CD244; CD25; CD305; CD33; CD34; CD366; CD38; CD42B; CD44; CD45RA; CD47; CD48; CD49D; CD49F; CD52; CD58; CD64; CD7; CD84; CD85K; CD9; CD99; CLL-1; EMR2; GPR56; IL1RAP; and CD274.
- a cytotoxic agentcomprising an antigen-binding fragment that recognizes a cell surface protein selected from a group consisting of: CD10; CD101; CD11A; CD11C; CD123; CD133; CD152; CD200; CD205; CD244; CD25; CD305; CD33; CD34; CD366
- a genetically engineered cell of the present disclosuremay be contacted with a cytotoxic agent comprising an antigen-binding fragment that recognizes a cell surface protein selected from a group consisting of: CD10; CD117; CD11A; CD11C; CD120B; CD123; CD13; CD133; CD152; CD205; CD22; CD226; CD244; CD25; CD305; CD32; CD33; CD34; CD38; CD41; CD44; CD45RA; CD47; CD48; CD49D; CD49F; CD52; CD56; CD58; CD64; CD7; CD70; CD71; CD82; CD84; CD85K; CD86; CD96; CD99; CLL- 1; EMR2; FR-B; GPR56; HLA-DR; and IL1RAP.
- a cytotoxic agentcomprising an antigen-binding fragment that recognizes a cell surface protein selected from a group consisting of: CD10; CD117; CD11A; CD11C; CD120
- a genetically engineered cell of the present disclosuremay be contacted with a first cytotoxic agent comprising an antigen-binding fragment that recognizes a first cell surface protein and a second cytotoxic agent comprising an antigen-binding fragment that recognizes a second cell surface protein selected from a group consisting of: CD274 and CD152; CD274 and CD226; CD152 and CD226; CD10 and CD49F; CD49F and GPR56; CD200 and CD34; CD34 and CD52; CD34 and CD133; GPR56 and CD133; CD42B and CD41; CD85K and CD64; CD85K and CD32; CD64 and CD32; CD64 and CD11C; CD64 and CD86; CD45RA and CD96; CD45RA and IL1RAP; CD45RA and CD123; CD84 and CD33; CD11C and IL1RAP; CD11C and CD33; CD11C and CD86; CD9 and IL1RAP; IL1RAP and CD33;
- a genetically engineered cell of the present disclosuremay be contacted with a first cytotoxic agent comprising an antigen-binding fragment that recognizes a first cell surface protein and a second cytotoxic agent comprising an antigen-binding fragment that recognizes a second cell surface protein selected from a group consisting of: CD33 and CD205 (LY75); CD33 and CD244 (SLAMF4); CD33 and CD305 (LAIR1); CD33 and CD49D (ITGA4); CLL-1 and CD205 (LY75); CLL-1 and CD244 (SLAMF4); CLL-1 and CD305 (LAIR1); CLL-1 and CD49D (ITGA4); EMR2 and CD205 (LY75); EMR2 and CD244 (SLAMF4); EMR2 and CD305 (LAIR1); EMR2 and CD49D (ITGA4); CD38 and CD205 (LY75); CD38 and CD244 (SLAMF4); CD38 and CD305 (LAIR1); CD38 and CD49D (ITGA), CD38 and
- a genetically engineered cell of the present disclosuremay be contacted with a first cytotoxic agent comprising an antigen-binding fragment that recognizes a first cell surface protein and a second cytotoxic agent comprising an antigen-binding fragment that recognizes a second cell surface protein selected from a group consisting of: CD85K and CD64; CD45RA and IL1RAP; CD34 and CD133; IL1RAP and CD33; CD47 and CD99; CLL-1 and CD305; CD152 and CD226; CD45RA and CD96; CD45RA and CD123; CD49D and CD305; IL1RAP and CD305; CD58 and CD305; CD49D and CD47; CD33 and CD305; CD49D and CD44; CD64 and CD86; GPR56 and CD133; CD34 and CD52; EMR2 and CD82; CD64 and CD32; CD42B and CD41; CD64 and CD11C; CD99 and CD44; CD200 and CD34; CD11C and CD33;
- a genetically engineered cell of the present disclosuremay be contacted with a first cytotoxic agent comprising an antigen-binding fragment that recognizes a first cell surface protein and a second cytotoxic agent comprising an antigen-binding fragment that recognizes a second cell surface protein selected from a group consisting of: CD274 and CD152; CD274 and CD226; CD152 and CD226; CD10 and CD49F; CD49F and GPR56; CD200 and CD34; CD34 and CD52; CD34 and CD133; GPR56 and CD133; CD42B and CD41; CD85K and CD64; CD85K and CD32; CD64 and CD32; CD64 and CD11C; CD64 and CD86; CD45RA and CD96; CD45RA and IL1RAP; CD45RA and CD123; CD84 and CD33; CD11C and IL1RAP; CD11C and CD33; CD11C and CD86; CD9 and IL1RAP; IL1RAP and CD33;
- a genetically engineered cell of the present disclosuremay be contacted with a first cytotoxic agent comprising an antigen-binding fragment that recognizes a first cell surface protein and a second cytotoxic agent comprising an antigen-binding fragment that recognizes a second cell surface protein selected from a group consisting of: CD9 and CD82; CD45RA and CD123; CD133 and CD82; CD85K and CD70; CD47 and CD205; CD64 and FR-B; CD11A and CD44; CD85K and CD86; CD366 and CD96; CD101 and CD64; CLL-1 and CD49D; CD49D and CD47; EMR2 and CD82; CD38 and CD305; CD34 and CD7; CD200 and CD25; CD25 and CD45RA; CD49D and CD44; CD11A and CD244; CD133 and CD7; CD48 and CD11A; CD52 and CD133; GPR56 and CD82; CD11A and CD38; CD34 and CD13; CD58 and CD49D
- agentscomprising antigen-binding fragments that may be used to target malignant hematopoietic cells in a subject are further provided herein.
- Therapeutic AntibodiesAny antibody or an antigen-binding fragment thereof can be used as a cytotoxic agent or for constructing a cytotoxic agent that targets an epitope of a lineage-specific cell surface antigen, as described herein.
- Such an antibody or antigen-binding fragmentcan be prepared by a conventional method, for example, the hybridoma technology or recombinant technology.
- the term "antibody”refers to a glycoprotein comprising at least two heavy (H) chains and two light (L) chains inter-connected by disulfide bonds, i.e., covalent heterotetramers comprised of two identical Ig H chains and two identical L chains that are encoded by different genes.
- Each heavy chainis comprised of a heavy chain variable region (abbreviated herein as HCVR or VH) and a heavy chain constant region.
- the heavy chain constant regionis comprised of three domains, CH1, CH2 and CH3.
- Each light chainis comprised of a light chain variable region (abbreviated herein as LCVR or VL) and a light chain constant region.
- the light chain constant regionis comprised of one domain, CL.
- VH and VL regionscan be further subdivided into regions of hypervariability, termed complementarity determining regions (CDR), interspersed with regions that are more conserved, termed framework regions (FR).
- CDRcomplementarity determining regions
- FRframework regions
- Each VH and VLis composed of three CDRs and four FRs, arranged from amino-terminus to carboxy-terminus in the following order: FR1, CDR1, FR2, CDR2, FR3, CDR3, FR4.
- the variable regions of the heavy and light chainscontain a binding domain that interacts with an antigen.
- the constant regions of the antibodiesmay mediate the binding of the immunoglobulin to host tissues or factors, including various cells of the immune system (e.g., effector cells) and the first component (Clq) of the classical complement system.
- the antigen-binding fragmentis a single-chain antibody fragment (scFv) that specifically binds the epitope of the lineage-specific cell surface antigen.
- the antigen-binding fragmentis a full-length antibody that specifically binds the epitope of the lineage-specific cell surface antigen.
- the CDRs of an antibodyspecifically bind to the epitope of a target protein/antigen (the cell surface protein/antigen).
- the antibodiesare full-length antibodies, meaning the antibodies comprise a fragment crystallizable (Fc) portion and a fragment antigen-binding (Fab) portion.
- the antibodiesare of the isotype IgG, IgA, IgM, IgA, or IgD.
- a population of antibodiescomprises one isotype of antibody.
- the antibodiesare IgG antibodies.
- the antibodiesare IgM antibodies.
- a population of antibodiescomprises more than one isotype of antibody.
- a population of antibodiesis comprised of a majority of one isotype of antibodies but also contains one or more other isotypes of antibodies.
- the antibodiesare selected from the group consisting of IgGl, IgG2, IgG3, IgG4, IgM, IgAl, IgA2, IgAsec, IgD, and IgE.
- the antibodies described hereinmay specifically bind to a target protein.
- specific bindingrefers to antibody binding to a predetermined protein, such as a cancer antigen. “Specific binding” involves more frequent, more rapid, greater duration of interaction, and/or greater affinity to a target protein relative to alternative proteins.
- a population of antibodiesspecifically binds to a particular epitope of a target protein, meaning the antibodies bind to the particular protein with more frequently, more rapidly, for greater duration of interaction, and/or with greater affinity to the epitope relative to alternative epitopes of the same target protein or to epitopes of another protein.
- the antibodies that specifically bind to a particular epitope of a target proteinmay not bind to other epitopes of the same protein.
- Antibodiesmay be selected based on the binding affinity of the antibody to the target protein or epitope. Alternatively or in additional, the antibodies may be mutated to introduce one or more mutations to modify (e.g., enhance or reduce) the binding affinity of the antibody to the target protein or epitope.
- the present antibodies or antigen-binding portionscan specifically bind with a dissociation constant (KD) of less than about 10 -7 M, less than about 10 -8 M, less than about 10 -9 M, less than about 10 -10 M, less than about 10 -11 M, or less than about 10 -12 M.
- KDdissociation constant
- Affinities of the antibodies according to the present disclosurecan be readily determined using conventional techniques (see, e.g., Scatchard et al., Ann. N.Y. Acad. Sci. (1949) 51:660; and U.S. Patent Nos.5,283,173, 5,468,614, or the equivalent).
- the binding affinity or binding specificity for an epitope or proteincan be determined by a variety of methods including equilibrium dialysis, equilibrium binding, gel filtration, ELISA, surface plasmon resonance, or spectroscopy.
- antibodies (of antigen-binding fragments thereof) specific to an epitope of a cell surface protein of interestcan be made by the conventional hybridoma technology.
- the cell surface proteinwhich may be coupled to a carrier protein such as KLH, can be used to immunize a host animal for generating antibodies binding to that complex.
- the route and schedule of immunization of the host animalare generally in keeping with established and conventional techniques for antibody stimulation and production, as further described herein. General techniques for production of mouse, humanized, and human antibodies are known in the art and are described herein.
- any mammalian subject including humans or antibody producing cells therefromcan be manipulated to serve as the basis for production of mammalian, including human hybridoma cell lines.
- the host animalis inoculated intraperitoneally, intramuscularly, orally, subcutaneously, intraplantar, and/or intradermally with an amount of immunogen, including as described herein.
- Hybridomascan be prepared from the lymphocytes and immortalized myeloma cells using the general somatic cell hybridization technique of Kohler, B. and Milstein, C. (1975) Nature 256:495-497 or as modified by Buck, D. W., et al., In Vitro, 18:377-381 (1982).
- myeloma linesincluding but not limited to X63-Ag8.653 and those from the Salk Institute, Cell Distribution Center, San Diego, Calif., USA, may be used in the hybridization.
- the techniqueinvolves fusing myeloma cells and lymphoid cells using a fusogen such as polyethylene glycol, or by electrical means well known to those skilled in the art.
- the cellsare separated from the fusion medium and grown in a selective growth medium, such as hypoxanthine-aminopterin-thymidine (HAT) medium, to eliminate unhybridized parent cells.
- HAThypoxanthine-aminopterin-thymidine
- EBV immortalized B cellsmay be used to produce the TCR-like monoclonal antibodies described herein.
- the hybridomasare expanded and subcloned, if desired, and supernatants are assayed for anti-immunogen activity by conventional immunoassay procedures (e.g., radioimmunoassay, enzyme immunoassay, or fluorescence immunoassay).
- immunoassay procedurese.g., radioimmunoassay, enzyme immunoassay, or fluorescence immunoassay.
- Hybridomas that may be used as source of antibodiesencompass all derivatives, progeny cells of the parent hybridomas that produce monoclonal antibodies capable of binding to a cell surface protein.
- Hybridomas that produce such antibodiesmay be grown in vitro or in vivo using known procedures.
- the monoclonal antibodiesmay be isolated from the culture media or body fluids, by conventional immunoglobulin purification procedures such as ammonium sulfate precipitation, gel electrophoresis, dialysis, chromatography, and ultrafiltration, if desired.
- Undesired activity if present,can be removed, for example, by running the preparation over adsorbents made of the immunogen attached to a solid phase and eluting or releasing the desired antibodies off the immunogen.
- a protein that is immunogenic in the species to be immunizede.g., keyhole limpet hemocyanin, serum albumin, bovine thyroglobulin, or soybean trypsin inhibitor
- an antibody of interest(e.g., produced by a hybridoma) may be sequenced and the polynucleotide sequence may then be cloned into a vector for expression or propagation.
- the sequence encoding the antibody of interestmay be maintained in vector in a host cell and the host cell can then be expanded and frozen for future use.
- the polynucleotide sequencemay be used for genetic manipulation to "humanize" the antibody or to improve the affinity (affinity maturation), or other characteristics of the antibody.
- the constant regionmay be engineered to more resemble human constant regions to avoid immune response if the antibody is used in clinical trials and treatments in humans. It may be desirable to genetically manipulate the antibody sequence to obtain greater affinity to the cell surface protein.
- the antibody sequenceis manipulated to increase binding affinity of the antibody to the cell surface protein such that lower levels of the cell surface protein are detected by the antibody.
- antibodies that have increased binding to the cell surface proteinmay be used to reduce or prevent relapse of a hematopoietic malignancy. It will be apparent to one of skill in the art that one or more polynucleotide changes can be made to the antibody and still maintain its binding specificity to the target protein.
- fully human antibodiescan be obtained by using commercially available mice that have been engineered to express specific human immunoglobulin proteins. Transgenic animals that are designed to produce a more desirable (e.g., fully human antibodies) or more robust immune response may also be used for generation of humanized or human antibodies.
- antibodiesmay be made recombinantly by phage display or yeast technology. See, for example, U.S. Pat. Nos.5,565,332; 5,580,717; 5,733,743; and 6,265,150; and Winter et al., (1994) Annu. Rev. Immunol.12:433-455.
- the phage display technology(McCafferty et al., (1990) Nature 348:552-553) can be used to produce human antibodies and antibody fragments in vitro, from immunoglobulin variable (V) domain gene repertoires from unimmunized donors.
- Antigen-binding fragments of an intact antibodycan be prepared via routine methods. For example, F(ab')2 fragments can be produced by pepsin digestion of an antibody molecule, and Fab fragments that can be generated by reducing the disulfide bridges of F(ab')2 fragments.
- DNA encoding a monoclonal antibodies specific to a target proteincan be readily isolated and sequenced using conventional procedures (e.g., by using oligonucleotide probes that are capable of binding specifically to genes encoding the heavy and light chains of the monoclonal antibodies).
- the hybridoma cellsserve as a preferred source of such DNA. Once isolated, the DNA may be placed into one or more expression vectors, which are then transfected into host cells such as E.
- the DNAcan then be modified, for example, by substituting the coding sequence for human heavy and light chain constant domains in place of the homologous murine sequences, Morrison et al., (1984) Proc. Nat. Acad. Sci.81:6851, or by covalently joining to the immunoglobulin coding sequence all or part of the coding sequence for a non- immunoglobulin polypeptide.
- chimeric antibodiessuch as “chimeric” or “hybrid” antibodies
- Techniques developed for the production of “chimeric antibodies”are well known in the art. See, e.g., Morrison et al. (1984) Proc. Natl. Acad. Sci. USA 81, 6851; Neuberger et al. (1984) Nature 312, 604; and Takeda et al. (1984) Nature 314:452. Methods for constructing humanized antibodies are also well known in the art. See, e.g., Queen et al., Proc. Natl. Acad. Sci. USA, 86:10029-10033 (1989).
- variable regions of VH and VL of a parent non-human antibodyare subjected to three- dimensional molecular modeling analysis following methods known in the art.
- framework amino acid residues predicted to be important for the formation of the correct CDR structuresare identified using the same molecular modeling analysis.
- human VH and VL chains having amino acid sequences that are homologous to those of the parent non-human antibodyare identified from any antibody gene database using the parent VH and VL sequences as search queries.
- Human VH and VL acceptor genesare then selected.
- the CDR regions within the selected human acceptor genescan be replaced with the CDR regions from the parent non-human antibody or functional variants thereof.
- a single-chain antibodycan be prepared via recombinant technology by linking a nucleotide sequence coding for a heavy chain variable region and a nucleotide sequence coding for a light chain variable region.
- a flexible linkeris incorporated between the two variable regions.
- Patent Nos.4,946,778 and 4,704,692can be adapted to produce a phage or yeast scFv library and scFv clones specific to a cell surface protein can be identified from the library following routine procedures. Positive clones can be subjected to further screening to identify those that bind cell surface protein.
- the cytotoxic agent for use in the methods described hereincomprises an antigen-binding fragment that targets CD33.
- the cytotoxic agent for use in the methods described hereincomprises an antigen-binding fragment that targets IL1RAP.
- two or more cytotoxic agentsare used in the methods described herein. In some embodiments, the two or more cytotoxic agents are administered concurrently.
- the two or more cytotoxic agentsare administered sequentially.
- antibodies or antigen-binding fragments thereof targeting CD33 and IL1RAP in combinationare used in the methods described herein.
- antibodies or antigen-binding fragments thereof targeting CD44 and EMR2 in combinationare used in the methods described herein.
- antibodies or antigen-binding fragments thereof targeting CD33are used in combination with a cytotoxic agent (e.g., antibodies, immune cells expressing chimeric antigen receptors, antibody-drug conjugates) that targets a second cell surface protein or an additional cell surface protein.
- a cytotoxic agente.g., antibodies, immune cells expressing chimeric antigen receptors, antibody-drug conjugates
- antibodies or antigen-binding fragments thereof targeting IL1RAPare used in combination with a cytotoxic agent (e.g., antibodies, immune cells expressing chimeric antigen receptors, antibody-drug conjugates) that targets a second cell surface protein or an additional cell surface protein.
- a cytotoxic agente.g., antibodies, immune cells expressing chimeric antigen receptors, antibody-drug conjugates
- bispecific or multi-specific antibodiesmay be used to target more than one epitope (e.g., more than one epitope of a lineage-specific cell surface antigen, epitopes of more than one lineage-specific cell surface antigen, an epitope of cell surface protein and an epitope of an additional cell surface antigen). See, e.g., Hoseini et al. Blood Cancer Journal (2017) 7, e552.
- bispecific antibodiesinclude tandem double scFv (e.g., single-chain bispecific tandem fragment variable (scBsTaFv), bispecific T-cell engager (BiTE), bispecific single-chain Fv (bsscFv), bispecific killer-cell engager (BiKE), dual-affinity re-targeting (DART), diabody, tandem diabodies (TandAb), single-chain Fv triplebody (sctb), bispecific scFv immunofusion (Blf), Fabsc, dual-variable- domain immunoglobulin (DVD-Ig), CrossMab (CH1-CL), modular bispecific antibody (IgG- scFv).
- scBsTaFvsingle-chain bispecific tandem fragment variable
- biTEbispecific T-cell engager
- bsscFvbispecific single-chain Fv
- BiKEbispecific killer-cell engager
- DARTdual-affinity re-targeting
- diabodytandem diabo
- the antibodyis a bispecific T-cell engager (BiTE) comprising two linked scFv molecules.
- the linked scFv of the BiTEbinds an epitope of a lineage-specific cell surface protein (e.g., CD33 or IL1RAP).
- the BiTEis blinatumomab. See, e.g.
- an antibody that targets both CD33 and IL1RAPmay be used in the methods described herein.
- Antibodies and antigen-binding fragments targeting CD33 or IL1RAP or a combination thereofcan be prepared by routine practice.
- an antibody that targets both CD44 and EMR2may be used in the methods described herein.
- Antibodies and antigen-binding fragments targeting CD44 or EMR2 or a combination thereofcan be prepared by routine practice.
- a bispecific antibodymay be used in which one molecule targets an epitope of a lineage-specific cell surface protein on a target cell and the other molecule targets a surface antigen on an effector cell (e.g., T cell, NK cell) such that the target cell is brought into proximity with the effector cell.
- an effector celle.g., T cell, NK cell
- two or more epitopes of a cell surface proteinhave been modified, enabling two or more (e.g., 2, 3, 4, 5 or more) different cytotoxic agents (e.g., two antibodies) to be targeted to the two or more epitopes.
- the antibodiescould work synergistically to enhance efficacy.
- epitopes of two or more (e.g., 2, 3, 4, 5 or more) cell surface proteinhave been modified, enabling two or more (e.g., 2, 3, 4, 5 or more) different cytotoxic agents (e.g., two antibodies) to be targeted to epitopes of the two or more cell surface proteins.
- one or more (e.g., 1, 2, 3, 4, 5 or more) epitopes of a cell surface proteinhave been modified and one or more (e.g., 1, 2, 3, 4, 5 or more) epitopes of an additional cell surface protein have been modified, enabling two or more (e.g., 2, 3, 4, 5 or more) different cytotoxic agents (e.g., two antibodies) to be targeted to epitopes of the cell surface protein and epitopes of the additional cell surface protein.
- targeting of two or more cell surface proteinmay reduce relapse of a hematopoietic malignancy.
- the methods described hereininvolve administering a cytotoxic agent that targets an epitope of a cell surface protein that is mutated in the population of genetically engineered hematopoietic cells. In some embodiments, the methods described herein involve administering a cytotoxic agent that targets an epitope of a cell surface protein that is mutated in the population of genetically engineered hematopoietic cells and one or more additional cytotoxic agents that target one or more additional cell surface proteins. In some embodiments, the antibodies work synergistically to enhance efficacy by targeting more than one cell surface protein.
- the methods described hereininvolve administering to the subject a population of genetically engineered cells lacking a non-essential epitope in a cell surface protein and one or more immunotherapeutic agents (e.g., antibodies) that target cells expressing the lineage-specific cell surface antigen. In some embodiments, the methods described herein involve administering to the subject a population of genetically engineered cells lacking a non-essential epitope in a type 1 cell surface protein and one or more immunotherapeutic agents (e.g., antibodies) that target cells expressing the lineage-specific cell surface antigen.
- immunotherapeutic agentse.g., antibodies
- the methods described hereininvolve administering to the subject a population of genetically engineered cells lacking a non- essential epitope in a type 2 cell surface protein and one or more immunotherapeutic agents (e.g., antibodies) that target cells expressing the lineage-specific cell surface antigen.
- one or more additional immunotherapeutic agentsmay be further administered to the subject (e.g., targeting one or more additional epitopes and/or antigens), for example if the hematopoietic malignancy relapses.
- Antibody-Drug ConjugatesIn some embodiments, the cytotoxic agent targeting an epitope of a cell surface protein is an antibody-drug conjugate (ADC).
- ADCantibody-drug conjugate
- antibody-drug conjugatecan be used interchangeably with “immunotoxin” and refers to a fusion molecule comprising an antibody (or antigen-binding fragment thereof) conjugated to a toxin or drug molecule. Binding of the antibody to the corresponding epitope of the target protein allows for delivery of the toxin or drug molecule to a cell that presents the protein (and epitope thereof) on the cell surface (e.g., target cell), thereby resulting in death of the target cell.
- the antibody-drug conjugate(or antigen- binding fragment thereof) binds to its corresponding epitope of a cell surface protein but does not bind to a cell surface protein that lacks the epitope or in which the epitope has been mutated.
- the agentis an antibody-drug conjugate.
- the antibody-drug conjugatecomprises an antigen-binding fragment and a toxin or drug that induces cytotoxicity in a target cell.
- the antibody- drug conjugatebinds to an epitope in an exon of IL1RAP.
- the antibody-drug conjugatetargets a type 2 protein.
- the antibody-drug conjugatetargets CD33.
- the antibody-drug conjugatebinds to an epitope in exon 2 or exon 3 of CD33. In some embodiments, the antibody-drug conjugate targets a type 1 protein. In some embodiments, the antibody-drug conjugate targets IL1RAP. In some embodiments, the antibody-drug conjugate targets a type 0 protein.
- Toxins or drugs compatible for use in antibody-drug conjugatesare well known in the art and will be evident to one of ordinary skill in the art. See, e.g., Peters et al. Biosci. Rep.(2015) 35(4): e00225, Beck et al. Nature Reviews Drug Discovery (2017) 16:315-337; Marin-Acevedo et al. J.
- the antibody-drug conjugatemay further comprise a linker (e.g., a peptide linker, such as a cleavable linker or a non-cleavable linker) attaching the antibody and drug molecule.
- a linkere.g., a peptide linker, such as a cleavable linker or a non-cleavable linker
- antibody-drug conjugatesinclude, without limitation, brentuximab vedotin, glembatumumab vedotin/CDX-011, depatuxizumab mafodotin/ABT-414, PSMA ADC, polatuzumab vedotin/RG7596/DCDS4501A, denintuzumab mafodotin/SGN-CD19A, AGS-16C3F, CDX-014, RG7841/DLYE5953A, RG7882/DMUC406A, RG7986/DCDS0780A, SGN-LIV1A, enfortumab vedotin/ASG- 22ME, AG-15ME, AGS67E, telisotuzumab vedotin/ABBV-399, ABBV-221, ABBV-085, GSK-2857916, tisotumab vedotin/HuMax-
- the antibody- drug conjugateis gemtuzumab ozogamicin.
- binding of the antibody-drug conjugate to the epitope of the cell surface proteininduces internalization of the antibody-drug conjugate, and the drug (or toxin) may be released intracellularly.
- binding of the antibody-drug conjugate to the epitope of a cell surface proteininduces internalization of the toxin or drug, which allows the toxin or drug to kill the cells expressing the cell surface protein (target cells).
- binding of the antibody-drug conjugate to the epitope of a cell surface proteininduces internalization of the toxin or drug, which may regulate the activity of the cell expressing the cell surface protein (target cells).
- toxin or drug used in the antibody-drug conjugates described hereinis not limited to any specific type.
- two or more (e.g., 2, 3, 4, 5 or more) epitopes of a cell surface proteinhave been modified, enabling two or more (e.g., 2, 3, 4, 5 or more) different cytotoxic agents (e.g., two ADCs) to be targeted to the two or more epitopes.
- the toxins carried by the ADCscould work synergistically to enhance efficacy (e.g., death of the target cells).
- epitopes of two or more (e.g., 2, 3, 4, 5 or more) lineage-specific cell surface proteinhave been modified, enabling two or more (e.g., 2, 3, 4, 5 or more) different cytotoxic agents (e.g., two ADCs) to be targeted to epitopes of the two or more cell surface proteins.
- two or more (e.g., 2, 3, 4, 5 or more) different cytotoxic agentse.g., two ADCs
- one or more (e.g., 1, 2, 3, 4, 5 or more) epitopes of a cell surface proteinhave been modified and one or more (e.g., 1, 2, 3, 4, 5 or more) epitopes of an additional cell surface protein have been modified, enabling two or more (e.g., 2, 3, 4, 5 or more) different cytotoxic agents (e.g., two ADCs) to be targeted to epitopes of the cell surface protein and epitopes of additional cell surface antigen.
- two or more (e.g., 2, 3, 4, 5 or more) different cytotoxic agentse.g., two ADCs
- targeting of more than one cell surface protein or a cell surface protein and one or more additional cell surface protein/antigenmay reduce relapse of a hematopoietic malignancy.
- the methods described hereininvolve administering ADCs that target an epitope of a cell surface protein that is mutated in the population of genetically engineered hematopoietic cells. In some embodiments, the methods described herein involve administering ADCs that target an epitope of a cell surface protein that is mutated in the population of genetically engineered hematopoietic cells and one or more additional cytotoxic agents that may target one or more additional cell surface proteins. In some embodiments, the agents could work synergistically to enhance efficacy by targeting more than one cell surface protein.
- An ADC described hereinmay be used as a follow-on treatment to subjects who have been undergone the combined therapy as described herein.
- the methods described hereininvolve administering to the subject a population of genetically engineered cells lacking a non-essential epitope in a cell surface protein and one or more immunotherapeutic agents (e.g., ADCs) that target cells expressing the lineage-specific cell surface antigen. In some embodiments, the methods described herein involve administering to the subject a population of genetically engineered cells lacking a non-essential epitope in a type 1 cell surface protein and one or more immunotherapeutic agents (e.g., ADCs) that target cells expressing the lineage-specific cell surface antigen.
- ADCsimmunotherapeutic agents
- the methods described hereininvolve administering to the subject a population of genetically engineered cells lacking a non-essential epitope in a type 2 cell surface protein and one or more immunotherapeutic agents (e.g., ADCs) that target cells expressing the lineage-specific cell surface antigen.
- one or more additional immunotherapeutic agentsmay be further administered to the subject (e.g., targeting one or more additional epitopes and/or antigens), for example if the hematopoietic malignancy relapses.
- the methods described hereininvolve administering to the subject a population of genetically engineered cells lacking a non-essential epitope of CD33 and one or more antibody-drug conjugates that target cells expressing CD33. In some examples, the methods described herein involve administering to the subject a population of genetically engineered cells lacking an epitope in exon 2 or exon 3 of CD33 and one or more antibodies antibody-drug conjugates that target cells expressing CD33. In some examples, the methods described herein involve administering to the subject a population of genetically engineered cells expressing a mutated CD33 comprising the amino acid sequence of SEQ ID NO: 56 or SEQ ID NO: 58 and one or more antibody-drug conjugates that target cells expressing CD33.
- the methods described hereininvolve administering to the subject a population of genetically engineered cells lacking a non-essential epitope of IL1RAP and one or more antibody-drug conjugates that target cells expressing IL1RAP. In some examples, the methods described herein involve administering to the subject a population of genetically engineered cells lacking an epitope in and of IL1RAP and one or more antibody-drug conjugates that target cells expressing IL1RAP.
- the cytotoxic agent that targets an epitope of a cell surface protein as described hereinis an immune cell that expresses a chimeric receptor, which comprises an antigen-binding fragment (e.g., a single-chain antibody) capable of binding to the epitope of the cell surface protein (e.g., CD33 or IL1RAP).
- a chimeric receptorwhich comprises an antigen-binding fragment (e.g., a single-chain antibody) capable of binding to the epitope of the cell surface protein (e.g., CD33 or IL1RAP).
- a target celle.g., a cancer cell
- the antigen-binding fragment of the chimeric receptortransduces an activation signal to the signaling domain(s) (e.g., co-stimulatory signaling domain and/or the cytoplasmic signaling domain) of the chimeric receptor, which may activate an effector function in the immune cell expressing the chimeric receptor.
- the immune cellexpresses more than one chimeric receptor (e.g., 2, 3, 4, 5 or more), referred to as a bispecific or multi- specific immune cell.
- the immune cellexpresses more than one chimeric receptor, at least one of which targets an epitope of a lineage-specific cell surface antigen. In some embodiments, the immune cell expresses more than one chimeric receptor, each of which targets an epitope of a lineage-specific cell surface antigen. In some embodiments, the immune cell expresses more than one chimeric receptor, at least one of which targets an epitope of a cell surface protein and at least one of which targets an epitope of an additional cell surface antigen. In some embodiments, targeting of more than one lineage-specific cell surface protein or a lineage-specific cell surface protein and one or more additional cell surface protein may reduce relapse of a hematopoietic malignancy.
- the immune cellexpresses a chimeric receptor that targets more than one epitopes (e.g., more than one epitopes of one cell surface protein or epitopes of more than one cell surface protein), referred to as a bispecific chimeric receptor.
- epitopes of two or more lineage-specific cell surface proteinsare targeted by cytotoxic agents.
- two or more chimeric receptorsare expressed in the same immune cell, e.g., bispecific chimeric receptors.
- Such cellscan be used in any of the methods described herein.
- cells expressing a chimeric receptorare “pooled,, i.e., two or more groups of cells express two or more different chimeric receptors.
- two or more cells expressing different chimeric antigen receptorsare administered concurrently. In some embodiments, two or more cells expressing different chimeric antigen receptors are administered sequentially.
- epitopes of CD33 and IL1RAPare targeted by cytotoxic agents.
- the chimeric receptors targeting CD33 and IL1RAPare expressed in the same immune cell (i.e., a bispecific immune cell). Such cells can be used in any of the methods described herein.
- cells expressing chimeric receptors targeting CD33 and IL1RAP “pooled,” i.e., two or more groups of cellsexpress two or more different chimeric receptors.
- two or more groups of cells expressing chimeric receptors targeting CD33 and IL1RAPare administered concurrently. In some embodiments, two or more groups of cells expressing chimeric receptors targeting CD33 and IL1RAP are administered sequentially.
- epitopes of CD44 and EMR2are targeted by cytotoxic agents. In some embodiments, the chimeric receptors targeting CD44 and EMR2 are expressed in the same immune cell (i.e., a bispecific immune cell). Such cells can be used in any of the methods described herein. In some embodiments, cells expressing chimeric receptors targeting CD44 and EMR2 “pooled,” i.e., two or more groups of cells express two or more different chimeric receptors.
- two or more groups of cells expressing chimeric receptors targeting CD44 and EMR2are administered concurrently. In some embodiments, two or more groups of cells expressing chimeric receptors targeting CD44 and EMR2 are administered sequentially.
- a chimeric receptorrefers to a non-naturally occurring molecule that can be expressed on the surface of a host cell and comprises binding domain that provides specificity of the chimeric receptor (e.g., an antigen-binding fragment that binds to an epitope of a cell surface protein).
- binding domainthat provides specificity of the chimeric receptor (e.g., an antigen-binding fragment that binds to an epitope of a cell surface protein).
- chimeric receptorscomprise at least two domains that are derived from different molecules.
- the chimeric receptormay further comprise one or more of the following: a hinge domain, a transmembrane domain, a co-stimulatory domain, a cytoplasmic signaling domain, and combinations thereof.
- the chimeric receptorcomprises from N terminus to C terminus, an antigen-binding fragment that binds to a cell surface protein, a hinge domain, a transmembrane domain, and a cytoplasmic signaling domain.
- the chimeric receptorfurther comprises at least one co-stimulatory domain. See, e.g., Marin-Acevedo et al. J. Hematol. Oncol.(2018)11: 8.
- the chimeric receptormay be a switchable chimeric receptor.
- a switchable chimeric receptorcomprises a binding domain that binds a soluble antigen-binding fragment, which has antigen binding specificity and may be administered concomitantly with the immune cells.
- the chimeric receptormay be a masked chimeric receptor, which is maintained in an “off” state until the immune cell expressing the chimeric receptor is localized to a desired location in the subject.
- the binding domain of the chimeric receptormay be blocked by an inhibitory peptide that is cleaved by a protease present at a desired location in the subject.
- relapse of hematopoietic malignanciesresults due to the reduced expression of the targeted cell surface protein on the surface of target cells (e.g., antigen escape) and the lower levels of cell surface protein any be inefficient or less efficient in stimulating cytotoxicity of the target cells. See, e.g., Majzner et al. Cancer Discovery (2016) 8(10).
- the binding affinity of the binding domainmay be enhanced, for example by mutating one or more amino acid residues of the binding domain. Binding domains having enhanced binding affinity to a cell surface protein may result in immune cells that response to lower levels of cell surface protein (lower cell surface protein density) and reduce or prevent relapse.
- Hinge Domains of Chimeric Antigen ReceptorsIn some embodiments, the chimeric receptors described herein comprise one or more hinge domain(s). In some embodiments, the hinge domain may be located between the antigen-binding fragment and a transmembrane domain.
- a hinge domainis an amino acid segment that is generally found between two domains of a protein and may allow for flexibility of the protein and movement of one or both of the domains relative to one another. Any amino acid sequence that provides such flexibility and movement of the antigen-binding fragment relative to another domain of the chimeric receptor can be used.
- the hinge domainmay contain about 10-200 amino acids, e.g., 15-150 amino acids, 20-100 amino acids, or 30-60 amino acids.
- the hinge domainmay be of about 10, 11, 12, 13, 14,15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 35, 40, 45, 50, 55, 60, 65, 70, 75, 80, 85, 90, 95, 100, 110, 120, 130, 140, 150, 160, 170, 180, 190, or 200 amino acids in length.
- the hinge domainis a hinge domain of a naturally occurring protein. Hinge domains of any protein known in the art to comprise a hinge domain are compatible for use in the chimeric receptors described herein. In some embodiments, the hinge domain is at least a portion of a hinge domain of a naturally occurring protein and confers flexibility to the chimeric receptor.
- the hinge domainis of CD8 ⁇ or CD28. In some embodiments, the hinge domain is a portion of the hinge domain of CD8 ⁇ , e.g., a fragment containing at least 15 (e.g., 20, 25, 30, 35, or 40) consecutive amino acids of the hinge domain of CD8 ⁇ or CD28. Hinge domains of antibodies, such as an IgG, IgA, IgM, IgE, or IgD antibody, are also compatible for use in the chimeric receptors described herein. In some embodiments, the hinge domain is the hinge domain that joins the constant domains CH1 and CH2 of an antibody. In some embodiments, the hinge domain is of an antibody and comprises the hinge domain of the antibody and one or more constant regions of the antibody.
- the hinge domaincomprises the hinge domain of an antibody and the CH3 constant region of the antibody. In some embodiments, the hinge domain comprises the hinge domain of an antibody and the CH2 and CH3 constant regions of the antibody.
- the antibodyis an IgG, IgA, IgM, IgE, or IgD antibody. In some embodiments, the antibody is an IgG antibody. In some embodiments, the antibody is an IgG1, IgG2, IgG3, or IgG4 antibody.
- the hinge regioncomprises the hinge region and the CH2 and CH3 constant regions of an IgG1 antibody. In some embodiments, the hinge region comprises the hinge region and the CH3 constant region of an IgG1 antibody.
- chimeric receptorscomprising a hinge domain that is a non-naturally occurring peptide.
- the hinge domain between the C-terminus of the extracellular ligand-binding domain of an Fc receptor and the N-terminus of the transmembrane domainis a peptide linker, such as a (Gly x Ser)n linker, wherein x and n, independently can be an integer between 3 and 12, including 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, or more.
- Additional peptide linkersthat may be used in a hinge domain of the chimeric receptors described herein are known in the art. See, e.g., Wriggers et al.
- the chimeric receptors described hereinmay comprise one or more transmembrane domain(s).
- the transmembrane domain for use in the chimeric receptorscan be in any form known in the art.
- a “transmembrane domain”refers to any protein structure that is thermodynamically stable in a cell membrane, preferably a eukaryotic cell membrane.
- Transmembrane domains compatible for use in the chimeric receptors used hereinmay be obtained from a naturally occurring protein.
- the transmembrane domainmay be a synthetic, non-naturally occurring protein segment, e.g., a hydrophobic protein segment that is thermodynamically stable in a cell membrane.
- Transmembrane domainsare classified based on the transmembrane domain topology, including the number of passes that the transmembrane domain makes across the membrane and the orientation of the protein. For example, single-pass membrane proteins cross the cell membrane once, and multi-pass membrane proteins cross the cell membrane at least twice (e.g., 2, 3, 4, 5, 6, 7 or more times).
- the transmembrane domainis a single-pass transmembrane domain.
- the transmembrane domainis a single-pass transmembrane domain that orients the N terminus of the chimeric receptor to the extracellular side of the cell and the C terminus of the chimeric receptor to the intracellular side of the cell.
- the transmembrane domainis obtained from a single pass transmembrane protein.
- the transmembrane domainis of CD8 ⁇ .
- the transmembrane domainis of CD28.
- the transmembrane domainis of ICOS. Costimulatory and Signaling Domains of Chimeric Antigen Receptors
- the chimeric receptors described hereincomprise one or more costimulatory signaling domains.
- co-stimulatory signaling domainrefers to at least a portion of a protein that mediates signal transduction within a cell to induce an immune response, such as an effector function.
- the co-stimulatory signaling domain of the chimeric receptor described hereincan be a cytoplasmic signaling domain from a co-stimulatory protein, which transduces a signal and modulates responses mediated by immune cells, such as T cells, NK cells, macrophages, neutrophils, or eosinophils.
- the chimeric receptorcomprises more than one (at least 2, 3, 4, or more) co-stimulatory signaling domains.
- the chimeric receptorcomprises more than one co-stimulatory signaling domains obtained from different costimulatory proteins. In some embodiments, the chimeric receptor does not comprise a co- stimulatory signaling domain.
- many immune cellsrequire co-stimulation, in addition to stimulation of an antigen-specific signal, to promote cell proliferation, differentiation and survival, and to activate effector functions of the cell.
- Activation of a co-stimulatory signaling domain in a host celle.g., an immune cell
- co-stimulatory signaling domain of any co-stimulatory proteinmay be compatible for use in the chimeric receptors described herein.
- the type(s) of co-stimulatory signaling domainis selected based on factors such as the type of the immune cells in which the chimeric receptors would be expressed (e.g., primary T cells, T cell lines, NK cell lines) and the desired immune effector function (e.g., cytotoxicity).
- co-stimulatory signaling domains for use in the chimeric receptorscan be the cytoplasmic signaling domain of co-stimulatory proteins, including, without limitation, CD27, CD28, 4-1BB, OX40, CD30, ICOS, CD2, CD7, LIGHT, NKG2C, B7-H3.
- the co-stimulatory domainis derived from 4-1BB, CD28, or ICOS.
- the costimulatory domainis derived from CD28 and chimeric receptor comprises a second co-stimulatory domain from 4-1BB or ICOS.
- the costimulatory domainis a fusion domain comprising more than one costimulatory domain or portions of more than one costimulatory domains.
- the costimulatory domainis a fusion of costimulatory domains from CD28 and ICOS.
- the chimeric receptors described hereincomprise one or more cytoplasmic signaling domain(s). Any cytoplasmic signaling domain can be used in the chimeric receptors described herein.
- a cytoplasmic signaling domainrelays a signal, such as interaction of an extracellular ligand-binding domain with its ligand, to stimulate a cellular response, such as inducing an effector function of the cell (e.g., cytotoxicity).
- a factor involved in T cell activationis the phosphorylation of immunoreceptor tyrosine-based activation motif (ITAM) of a cytoplasmic signaling domain.
- ITAMimmunoreceptor tyrosine-based activation motif
- an ITAM motifmay comprise two repeats of the amino acid sequence YxxL/I separated by 6-8 amino acids, wherein each x is independently any amino acid, producing the conserved motif YxxL/Ix(6- 8)YxxL/I.
- the cytoplasmic signaling domainis from CD3 ⁇ .
- Exemplary Uses and Methods Related to Chimeric Antigen Receptorsthe chimeric receptor described herein targets a type 2 protein. In some embodiments, the chimeric receptor targets CD33. In some embodiments, the chimeric receptor described herein targets a type 1 protein. In some embodiments, the chimeric receptor targets IL1RAP. In some embodiments, the chimeric receptor targets a type 0 protein.
- Such a chimeric receptormay comprise an antigen-binding fragment (e.g., an scFv) comprising a heavy chain variable region and a light chain variable region that bind to IL1RAP.
- the chimeric receptormay comprise an antigen-binding fragment (e.g., scFv) comprising a heavy chain variable region and a light chain variable region that bind to CD33.
- the immune cells described hereinexpress a chimeric receptor (e.g., bispecific chimeric receptor) that targets a type 2 protein and a chimeric receptor that targets a type 1 protein.
- the immune cells described hereinexpress a chimeric receptor (e.g., bispecific chimeric receptor) that targets a type 2 protein and at least one additional cell surface protein, such as any of those described herein.
- the immune cells described hereinexpress a chimeric receptor that targets CD33 and at least one additional cell surface protein, such as any of those described herein.
- the immune cells described hereinexpress a chimeric receptor that targets a type 1 protein and at least one additional cell surface protein, such as any of those described herein.
- the immune cells described hereinexpress a chimeric receptor that targets IL1RAP and at least one additional cell surface protein, such as any of those described herein.
- the chimeric receptor described hereintargets CD33 and IL1RAP.
- the immune cells described hereinexpress a chimeric receptor that targets EMR2 and at least one additional cell surface protein, such as any of those described herein.
- the chimeric receptor described hereintargets CD44 and EMR2.
- the immune cells described hereinexpress a chimeric receptor that targets a type 2 protein and at least one additional chimeric receptor that targets an additional cell surface protein, such as any of those described herein.
- the immune cells described hereinexpress a chimeric receptor that targets CD33 and at least one additional chimeric receptor that targets another cell surface protein, such as any of those described herein.
- the immune cells described hereinexpress a chimeric receptor that targets a type 1 protein and at least one additional chimeric receptor that targets another cell surface protein, such as any of those described herein. In some embodiments, the immune cells described herein express a chimeric receptor that targets IL1RAP and at least one additional chimeric receptor that targets another cell surface protein, such as any of those described herein. In some embodiments, the immune cells express a chimeric receptor that targets CD33 and a chimeric receptor that targets IL1RAP.
- a chimeric receptor construct targeting CD33 and/or IL1RAPmay further comprise at least a hinge domain (e.g., from CD28, CD8 ⁇ , or an antibody), a transmembrane domain (e.g., from CD8 ⁇ , CD28 or ICOS), one or more co-stimulatory domains (from one or more of CD28, ICOS, or 4-1BB) and a cytoplasmic signaling domain (e.g., from CD3 ⁇ ), or a combination thereof.
- a hinge domaine.g., from CD28, CD8 ⁇ , or an antibody
- a transmembrane domaine.g., from CD8 ⁇ , CD28 or ICOS
- co-stimulatory domainsfrom one or more of CD28, ICOS, or 4-1BB
- cytoplasmic signaling domaine.g., from CD3 ⁇
- the methods described hereininvolve administering to a subject a population of genetically engineered hematopoietic cells and an immune cell expressing a chimeric receptor that targets CD33 and/or IL1RAP, which may further comprise at least a hinge domain (e.g., from CD28, CD8 ⁇ , or an antibody), a transmembrane domain (e.g., from CD8 ⁇ , CD28 or ICOS), one or more co-stimulatory domains (from one or more of CD28, ICOS, or 4-1BB) and a cytoplasmic signaling domain (e.g., from CD3 ⁇ ), or a combination thereof
- a hinge domaine.g., from CD28, CD8 ⁇ , or an antibody
- a transmembrane domaine.g., from CD8 ⁇ , CD28 or ICOS
- co-stimulatory domainsfrom one or more of CD28, ICOS, or 4-1BB
- cytoplasmic signaling domaine.g., from CD3
- Methods for preparing the chimeric receptors hereininvolve generation of a nucleic acid that encodes a polypeptide comprising each of the domains of the chimeric receptors, including the antigen-binding fragment and optionally, the hinge domain, the transmembrane domain, at least one co-stimulatory signaling domain, and the cytoplasmic signaling domain.
- nucleic acids encoding the components of a chimeric receptorare joined together using recombinant technology. Sequences of each of the components of the chimeric receptors may be obtained via routine technology, e.g., PCR amplification from any one of a variety of sources known in the art.
- sequences of one or more of the components of the chimeric receptorsare obtained from a human cell.
- the sequences of one or more components of the chimeric receptorscan be synthesized. Sequences of each of the components (e.g., domains) can be joined directly or indirectly (e.g., using a nucleic acid sequence encoding a peptide linker) to form a nucleic acid sequence encoding the chimeric receptor, using methods such as PCR amplification or ligation.
- the nucleic acid encoding the chimeric receptormay be synthesized.
- the nucleic acidis DNA. In other embodiments, the nucleic acid is RNA.
- Mutation of one or more residues within one or more of the components of the chimeric receptormay be performed prior to or after joining the sequences of each of the components.
- one or more mutations in a component of the chimeric receptormay be made to modulate (increase or decrease) the affinity of the component for an epitope (e.g., the antigen-binding fragment for the target protein) and/or modulate the activity of the component.
- Any of the chimeric receptors described hereincan be introduced into a suitable immune cell for expression via conventional technology.
- the immune cellsare T cells, such as primary T cells or T cell lines.
- the immune cellscan be NK cells, such as established NK cell lines (e.g., NK-92 cells).
- the immune cellsare T cells that express CD8 (CD8 + ) or CD8 and CD4 (CD8 + /CD4 + ).
- the T cellsare T cells of an established T cell line, for example, 293T cells or Jurkat cells.
- Primary T cellsmay be obtained from any source, such as peripheral blood mononuclear cells (PBMCs), bone marrow, tissues such as spleen, lymph node, thymus, or tumor tissue.
- PBMCsperipheral blood mononuclear cells
- marrowtissues
- tissuessuch as spleen, lymph node, thymus, or tumor tissue.
- a source suitable for obtaining the type of immune cells desiredwould be evident to one of skill in the art.
- the population of immune cellsis derived from a human patient having a hematopoietic malignancy, such as from the bone marrow or from PBMCs obtained from the patient. In some embodiments, the population of immune cells is derived from a healthy donor. In some embodiments, the immune cells are obtained from the subject to whom the immune cells expressing the chimeric receptors will be subsequently administered. Immune cells that are administered to the same subject from which the cells were obtained are referred to as autologous cells, whereas immune cells that are obtained from a subject who is not the subject to whom the cells will be administered are referred to as allogeneic cells.
- the type of host cells desiredmay be expanded within the population of cells obtained by co-incubating the cells with stimulatory molecules, for example, anti-CD3 and anti-CD28 antibodies may be used for expansion of T cells.
- stimulatory moleculesfor example, anti-CD3 and anti-CD28 antibodies may be used for expansion of T cells.
- expression vectors for stable or transient expression of the chimeric receptor constructmay be constructed via conventional methods as described herein and introduced into immune host cells.
- nucleic acids encoding the chimeric receptorsmay be cloned into a suitable expression vector, such as a viral vector in operable linkage to a suitable promoter.
- the nucleic acids and the vectormay be contacted, under suitable conditions, with a restriction enzyme to create complementary ends on each molecule that can pair with each other and be joined with a ligase.
- synthetic nucleic acid linkerscan be ligated to the termini of the nucleic acid encoding the chimeric receptors.
- the synthetic linkersmay contain nucleic acid sequences that correspond to a particular restriction site in the vector.
- the selection of expression vectors/plasmids/viral vectorswould depend on the type of host cells for expression of the chimeric receptors, but should be suitable for integration and replication in eukaryotic cells.
- the chimeric receptorsare expressed using a non-integrating transient expression system.
- the chimeric receptorsare integrated into the genome of the immune cell. In some embodiments, the chimeric receptors are integrated into a specific loci of the genome of immune cell using gene editing (e.g., zinc-finger nucleases, meganucleases, TALENs, CRISPR systems).
- gene editinge.g., zinc-finger nucleases, meganucleases, TALENs, CRISPR systems.
- promoterscan be used for expression of the chimeric receptors described herein, including, without limitation, cytomegalovirus (CMV) intermediate early promoter, a viral LTR such as the Rous sarcoma virus LTR, HIV-LTR, HTLV-1 LTR, Maloney murine leukemia virus (MMLV) LTR, myeloproliferative sarcoma virus (MPSV) LTR, spleen focus- forming virus (SFFV) LTR, the simian virus 40 (SV40) early promoter, herpes simplex tk virus promoter, elongation factor 1-alpha (EF1- ⁇ ) promoter with or without the EF1- ⁇ intron.
- CMVcytomegalovirus
- a viral LTRsuch as the Rous sarcoma virus LTR, HIV-LTR, HTLV-1 LTR, Maloney murine leukemia virus (MMLV) LTR, myeloproliferative sarcoma virus (MPSV)
- Additional promoters for expression of the chimeric receptorsinclude any constitutively active promoter in an immune cell.
- any regulatable promotermay be used, such that its expression can be modulated within an immune cell.
- the promoter regulating expression of a chimeric receptoris an inducible promoter.
- the activity of “inducible promoters”may be regulated based on the presence (or absence) of a signal, such as an endogenous signal or an exogenous signal, for example a small molecule or drug that is administered to the subject.
- the vectormay contain, for example, some or all of the following: a selectable marker gene, such as the neomycin gene for selection of stable or transient transfectants in host cells; enhancer/promoter sequences from the immediate early gene of human CMV for high levels of transcription; transcription termination and RNA processing signals from SV40 for mRNA stability; 5’-and 3’-untranslated regions for mRNA stability and translation efficiency from highly-expressed genes like ⁇ -globin or ⁇ -globin; SV40 polyoma origins of replication and ColE1 for proper episomal replication; internal ribosome binding sites (IRESes), versatile multiple cloning sites; T7 and SP6 RNA promoters for in vitro transcription of sense and antisense RNA; a “suicide switch” or “suicide gene” which when triggered causes cells carrying the vector to die (e.g., HSV thymidine kinase, an inducible caspase such as iCasp9, drug
- Suitable vectors and methods for producing vectors containing transgenesare well known and available in the art. Examples of the preparation of vectors for expression of chimeric receptors can be found, for example, in US2014/0106449, herein incorporated by reference in its entirety. As will be appreciated by one of ordinary skill in the art, in some embodiments, it may be advantageous to quickly and efficiently reduce or eliminate the immune cells expressing chimeric receptors, for example at a time point following administration to a subject. Mechanisms of killing immune cells expressing chimeric receptors, or inducing cytotoxicity of such cells, are known in the art, see, e.g., Labanieh et al. Nature Biomedical Engineering (2016)2: 337-391; Slaney et al.
- the immune cell expressing the chimeric receptormay also express a suicide switch” or “suicide gene,” which may or may not be encoded by the same vector as the chimeric receptor, as described above.
- the immune cell expressing chimeric receptorsfurther express an epitope tag such that upon engagement of the epitope tag, the immune cell is killed through antibody-dependent cell-mediated cytotoxicity and/or complement-mediated cytotoxicity. See, e.g., Paszkiewicz et al. J. Clin. Invest. (2016) 126: 4262-4272; Wang et al. Blood (2011) 118: 1255-1263; Tasian et al.
- the chimeric receptor construct or the nucleic acid encoding said chimeric receptoris a DNA molecule.
- chimeric receptor construct or the nucleic acid encoding said chimeric receptoris a DNA vector and may be electroporated to immune cells (see, e.g., Till, et al. Blood (2012) 119(17): 3940-3950).
- the nucleic acid encoding the chimeric receptoris an RNA molecule, which may be electroporated to immune cells.
- any of the vectors comprising a nucleic acid sequence that encodes a chimeric receptor construct described hereinis also within the scope of the present disclosure.
- a vectormay be delivered into host cells such as host immune cells by a suitable method.
- Methods of delivering vectors to immune cellsare well known in the art and may include DNA, RNA, or transposon electroporation, transfection reagents such as liposomes or nanoparticles to delivery DNA, RNA, or transposons; delivery of DNA, RNA, or transposons or protein by mechanical deformation (see, e.g., Sharei et al. Proc. Natl. Acad. Sci. USA (2013) 110(6): 2082-2087); or viral transduction.
- the vectors for expression of the chimeric receptorsare delivered to host cells by viral transduction.
- viral methods for deliveryinclude, but are not limited to, recombinant retroviruses (see, e.g., PCT Publication Nos. WO 90/07936; WO 94/03622; WO 93/25698; WO 93/25234; WO 93/11230; WO 93/10218; WO 91/02805; U.S. Pat.
- the vectors for expression of the chimeric receptorsare retroviruses. In some embodiments, the vectors for expression of the chimeric receptors are lentiviruses.
- the vectors for expression of the chimeric receptorsare adeno-associated viruses.
- viral particles that are capable of infecting the immune cells and carry the vectormay be produced by any method known in the art and can be found, for example in PCT Application No. WO 1991/002805A2, WO 1998/009271 A1, and U.S. Patent 6,194,191.
- the viral particlesare harvested from the cell culture supernatant and may be isolated and/or purified prior to contacting the viral particles with the immune cells.
- the methods described hereininvolve use of immune cells that express more than one chimeric receptor (e.g., chimeric receptors that target first epitope and chimeric receptors that target a second epitope).
- more than one chimeric receptore.g., chimeric receptors that target first epitope and chimeric receptors that target a second epitope
- more than one chimeric receptorare expressed from a single vector.
- more than one chimeric receptore.g., chimeric receptors that target first epitope and chimeric receptors that target a second epitope
- Such immune cellsmay be prepared using methods known in the art, for example by delivering a vector that encodes a first chimeric receptor simultaneously or sequentially with a second vector that encodes a second chimeric receptor.
- the resulting immune cell populationis a mixed population comprising a subset of immune cells that express one chimeric receptor and a subset of immune cells that express both chimeric receptors.
- the domains of the chimeric receptorare encoded by and expressed from a single vector.
- the domains of the chimeric receptormay be encoded by and expressed from more than one vector.
- activity of immune cells expressing the chimeric receptorsmay be regulated by controlling assembly of the chimeric receptor.
- the domains of a chimeric receptormay be expressed as two or more non-functional segments of the chimeric receptor and induced to assemble at a desired time and/or location, for example through use of a dimerization agent (e.g., dimerizing drug).
- the methods of preparing host cells expressing any of the chimeric receptors described hereinmay comprise activating and/or expanding the immune cells ex vivo.
- Activating a host cellmeans stimulating a host cell into an activate state in which the cell may be able to perform effector functions (e.g., cytotoxicity). Methods of activating a host cell will depend on the type of host cell used for expression of the chimeric receptors.
- Expanding host cellsmay involve any method that results in an increase in the number of cells expressing chimeric receptors, for example, allowing the host cells to proliferate or stimulating the host cells to proliferate. Methods for stimulating expansion of host cells will depend on the type of host cell used for expression of the chimeric receptors and will be evident to one of skill in the art.
- the host cells expressing any of the chimeric receptors described hereinare activated and/or expanded ex vivo prior to administration to a subject. It has been appreciated that administration of immunostimulatory cytokines may enhance proliferation of immune cells expressing chimeric receptors following administration to a subject and/or promote engraftment of the immune cells. See, e.g., Pegram et al.
- Any of the methods described hereinmay further involve administering one or more immunostimulatory cytokines concurrently with any of the immune cells expressing chimeric receptors.
- immunostimulatory cytokinesinclude IL-12, IL-15, IL-1, IL-21, and combinations thereof.
- anti-IL1RAP CAR and anti-CD33 CAR moleculesknown in the art can be used together with the genetically engineered HSCs described herein.
- Exemplary anti- IL1RAP CARsinclude axicabtagene and tisagenlecleucel.
- Exemplary anti-CD33 CARsinclude those disclosed in PCT Publication Nos. WO2017/066760 and WO2017/079400.
- primary human CD8 + T cellsare isolated from patients’ peripheral blood by immunomagnetic separation (Miltenyi Biotec). T cells are cultured and stimulated with anti-CD3 and anti-CD28 mAbs–coated beads (Invitrogen) as previously described (Levine et al., J. Immunol.
- Chimeric receptors that target a lineage-specific cell surface proteinsare generated using conventional recombinant DNA technologies and inserted into a lentiviral vector.
- the vectors containing the chimeric receptorsare used to generate lentiviral particles, which are used to transduce primary CD8 + T cells.
- Human recombinant IL-2may be added every other day (50 IU/mL). T cells are cultured for ⁇ 14 days after stimulation. Expression of the chimeric receptors can be confirmed using methods, such as Western blotting and flow cytometry.
- T cells expressing the chimeric receptorsare selected and assessed for their ability to bind to the lineage-specific cell surface protein such as IL1RAP or CD33 and to induce cytotoxicity of cells expressing the cell surface protein.
- Immune cells expressing the chimeric receptorare also evaluated for their ability to induce cytotoxicity of cells expressing the cell surface protein in mutated form.
- immune cells expressing chimeric receptors that binds to the wild-type cell surface protein but not the mutated formare selected and assessed for their ability to bind to the lineage-specific cell surface protein such as IL1RAP or CD33 and to induce cytotoxicity of cells expressing the cell surface protein.
- Immune cells expressing the chimeric receptorare also evaluated for their ability to induce cytotoxicity of cells expressing the cell surface protein in mutated form.
- immune cells expressing chimeric receptors that binds to the wild-type cell surface protein but not the mutated formare selected and assessed for their ability to bind to the lineage-specific cell surface protein such as
- the methods described hereininvolve administering to the subject a population of genetically engineered cells lacking a non-essential epitope in a cell surface protein and one or more immunotherapeutic agents (e.g., immune cells expressing chimeric receptor(s)) that target cells expressing the lineage-specific cell surface antigen.
- the methods described hereininvolve administering to the subject a population of genetically engineered cells lacking a non-essential epitope in a type 1 cell surface protein and one or more immunotherapeutic agents (e.g., immune cells expressing chimeric receptor(s)) that target cells expressing the lineage-specific cell surface antigen.
- the methods described hereininvolve administering to the subject a population of genetically engineered cells lacking a non-essential epitope in a type 2 cell surface protein and one or more immunotherapeutic agents (e.g., immune cells expressing chimeric receptor(s)) that target cells expressing the lineage-specific cell surface antigen.
- one or more additional immunotherapeutic agentsmay be further administered to the subject (e.g., targeting one or more additional epitopes and/or antigens), for example if the hematopoietic malignancy relapses.
- the methods described hereininvolve administering immune cells expressing chimeric receptors that target an epitope of a lineage-specific cell surface protein that is mutated in the population of genetically engineered hematopoietic cells. In some embodiments, the methods described herein involve administering immune cells expressing chimeric receptors that target an epitope of a lineage-specific cell surface protein that is mutated in the population of genetically engineered hematopoietic cells and one or more additional cytotoxic agents that may target one or more additional cell surface proteins. In some embodiments, the agents could work synergistically to enhance efficacy by targeting more than one cell surface protein.
- the methods described hereininvolve administering to the subject a population of genetically engineered cells lacking a non-essential epitope of CD33 and one or more immune cells expressing chimeric receptor(s) that target cells expressing CD33. In some examples, the methods described herein involve administering to the subject a population of genetically engineered cells lacking an epitope in exon 2 or exon 3 of CD33 and one or more immune cells expressing chimeric receptor(s) that target cells expressing CD33.
- the methods described hereininvolve administering to the subject a population of genetically engineered cells expressing a mutated CD33 comprising the amino acid sequence of SEQ ID NO: 56 or SEQ ID NO: 58 and one or more immune cells expressing chimeric receptor(s) that target cells expressing CD33. In some examples, the methods described herein involve administering to the subject a population of genetically engineered cells lacking a non-essential epitope of IL1RAP and one or more immune cells expressing chimeric receptor(s) that target cells expressing IL1RAP.
- the methods described hereininvolve administering to the subject a population of genetically engineered cells lacking an epitope in an exon of IL1RAP and one or more immune cells expressing chimeric receptor(s) that target cells expressing IL1RAP.
- the genetically engineered hematopoietic cellssuch as HSCs may be administered to a subject in need of the treatment, either taken alone or in combination of one or more cytotoxic agents that target one or more antigens as described herein.
- the hematopoietic cellsare genetically edited in the genes of the one or more antigens, the hematopoietic cells and/or descendant cells thereof would express the one or more antigens in mutated form (e.g., but functional) such that they can escape being targeted by the cytotoxic agents, for example, CAR-T cells.
- the cytotoxic agentsfor example, CAR-T cells.
- the present disclosureprovides methods for treating a hematopoietic malignancy, the method comprising administering to a subject in need thereof (i) a population of the genetically engineered hematopoietic cells described herein, and optionally (ii) a cytotoxic agent such as CAR-T cells that target an antigen, the gene of which is genetically edited in the hematopoietic cells such that the cytotoxic agent does not target hematopoietic cells or descendant cells thereof.
- a cytotoxic agentsuch as CAR-T cells that target an antigen, the gene of which is genetically edited in the hematopoietic cells such that the cytotoxic agent does not target hematopoietic cells or descendant cells thereof.
- the cytotoxic agents and/or the hematopoietic cellsmay be mixed with a pharmaceutically acceptable carrier to form a pharmaceutical composition, which is also within the scope of the present disclosure.
- the genetically engineered hematopoietic stem cellshave genetic editing in genes of at least two antigens A and B.
- Such hematopoietic stem cellscan be administered to a subject, who has been or will be treated with a first cytotoxic agent specific to A (e.g., anti-protein A CAR-T cells), or who is at risk of a hematopoietic malignancy that would need treatment of the cytotoxic agent.
- a second cytotoxic agent specific to Bmay be administered to the subject.
- Be.g., anti-protein B CAR-T cells
- the genetically engineered hematopoietic cellshave genetic editing in both A and B genes, those cells and descendant cells thereof would also be resistant to cytotoxicity induced by the second cytotoxic agent.
- administering once the genetically engineered hematopoietic cellswould be sufficient to compensate loss of normal cells expressing at least lineage-specific cell surface proteins/antigens A and B due to multiple treatment by cytotoxic agents specific to at least proteins/antigens A and B.
- subjectAs used herein, “subject,” “individual,” and “patient” are used interchangeably, and refer to a vertebrate, preferably a mammal such as a human. Mammals include, but are not limited to, human primates, non-human primates or murine, bovine, equine, canine or feline species. In some embodiments, the subject is a human patient having a hematopoietic malignancy. To perform the methods described herein, an effective amount of the genetically engineered hematopoietic cells can be administered to a subject in need of the treatment. Optionally, the hematopoietic cells can be co-used with a cytotoxic agent as described herein.
- the term “effective amount”may be used interchangeably with the term “therapeutically effective amount” and refers to that quantity of a cytotoxic agent, hematopoietic cell population, or pharmaceutical composition (e.g., a composition comprising cytotoxic agents and/or hematopoietic cells) that is sufficient to result in a desired activity upon administration to a subject in need thereof.
- the term “effective amount”refers to that quantity of a compound, cell population, or pharmaceutical composition that is sufficient to delay the manifestation, arrest the progression, relieve or alleviate at least one symptom of a disorder treated by the methods of the present disclosure.
- the effective amount of the combinationmay or may not include amounts of each ingredient that would have been effective if administered individually.
- Effective amountsvary, as recognized by those skilled in the art, depending on the particular condition being treated, the severity of the condition, the individual patient parameters including age, physical condition, size, gender and weight, the duration of the treatment, the nature of concurrent therapy (if any), the specific route of administration and like factors within the knowledge and expertise of the health practitioner.
- the effective amountalleviates, relieves, ameliorates, improves, reduces the symptoms, or delays the progression of any disease or disorder in the subject.
- the subjectis a human.
- the subjectis a human patient having a hematopoietic malignancy.
- the hematopoietic cells and/or immune cells expressing chimeric receptorsmay be autologous to the subject, i.e., the cells are obtained from the subject in need of the treatment, manipulated such that the cells do not bind the cytotoxic agents, and then administered to the same subject.
- Administration of autologous cells to a subjectmay result in reduced rejection of the host cells as compared to administration of non-autologous cells.
- the host cellsare allogeneic cells, i.e., the cells are obtained from a first subject, manipulated such that the cells do not bind the cytotoxic agents, and then administered to a second subject that is different from the first subject but of the same species.
- allogeneic immune cellsmay be derived from a human donor and administered to a human recipient who is different from the donor.
- engineered hematopoietic cellscomprising one or more genetically engineered gene(s) encoding lineage-specific cell surface protein(s) (e.g., CD33 and IL1RAP or CD44 and EMR2) and the immune cells engineered to target an epitope of the antigen(s) are both allogeneic to the subject.
- engineered hematopoietic cellscomprising one or more genetically engineered gene(s) encoding antigen(s) (e.g., CD33 and IL1RAP or CD44 and EMR2 or any other combination of antigens described herein) and the immune cells engineered to target an epitope of the antigen(s) are from the same allogeneic donor.
- antigen(s)e.g., CD33 and IL1RAP or CD44 and EMR2 or any other combination of antigens described herein
- the immune cells engineered to target an epitope of the antigen(s)are from the same allogeneic donor.
- engineered hematopoietic cellscomprising one or more genetically engineered gene(s) encoding antigen(s) (e.g., CD33 and IL1RAP or CD44 and EMR2 or any other combination of antigens described herein) and the immune cells engineered to target an epitope of the antigen(s) are from two different allogeneic donors.
- antigen(s)e.g., CD33 and IL1RAP or CD44 and EMR2 or any other combination of antigens described herein
- the immune cells engineered to target an epitope of the antigen(s)are from two different allogeneic donors.
- engineered hematopoietic cellscomprising one or more genetically engineered gene(s) encoding antigen(s) (e.g., CD33 and IL1RAP or CD44 and EMR2 or any other combination of antigens described herein) and the immune cells engineered to target an epitope of the antigen(s) are both autologous to the subject.
- antigen(s)e.g., CD33 and IL1RAP or CD44 and EMR2 or any other combination of antigens described herein
- engineered hematopoietic cellscomprising one or more genetically engineered gene(s) encoding antigen(s) (e.g., CD33 and IL1RAP or CD44 and EMR2 or any other combination of antigens described herein) are autologous to the subject and the immune cells engineered to target an epitope of the antigen(s) are from an allogeneic donor.
- antigen(s)e.g., CD33 and IL1RAP or CD44 and EMR2 or any other combination of antigens described herein
- engineered hematopoietic cellscomprising one or more genetically engineered gene(s) encoding antigen(s) (e.g., CD33 and IL1RAP or CD44 and EMR2 or any other combination of antigens described herein) are from an allogeneic donor and the immune cells engineered to target an epitope of the lineage-specific cell surface protein(s) are autologous to the subject.
- the immune cells and/or hematopoietic cellsare allogeneic cells and have been further genetically engineered to reduce graft-versus-host disease.
- the hematopoietic stem cellsmay be genetically engineered (e.g., using genome editing) to have reduced expression of CD45RA.
- Methods for reducing graft-versus-host diseaseare known in the art, see, e.g., Yang et al. Curr. Opin. Hematol. (2015) 22(6): 509-515.
- the immune cellse.g., T cells
- the gene encoding the TCRis knocked out or silenced (e.g., using gene editing methods, or shRNAs).
- the TCRis silenced using peptide inhibitors of the TCR.
- the immune cellse.g., T cells
- the immune cellsare subjected to a selection process to select for immune cells or a population of immune cells that do not contain an alloreactive TCR.
- immune cells that naturally do not express TCRse.g., NK cells
- the immune cells and/or hematopoietic cellshave been further genetically engineered to reduce host-versus-graft effects.
- immune cells and/or hematopoietic cellsmay be subjected to gene editing or silencing methods to reduce or eliminate expression of one or more proteins involved in inducing host immune responses, e.g., CD52, MHC molecules, and/or MHC beta-2 microglobulin.
- the immune cells expressing any of the chimeric receptors described hereinare administered to a subject in an amount effective in to reduce the number of target cells (e.g., cancer cells) by least 20%, e.g., 50%, 80%, 100%, 2-fold, 5-fold, 10-fold, 20-fold, 50-fold, 100-fold or more.
- a typical amount of cells, i.e., immune cells or hematopoietic cells, administered to a mammalcan be, for example, in the range of about 10 6 to 10 11 cells. In some embodiments it may be desirable to administer fewer than 10 6 cells to the subject. In some embodiments, it may be desirable to administer more than 10 11 cells to the subject.
- one or more doses of cellsincludes about 10 6 cells to about 10 11 cells, about 10 7 cells to about 10 10 cells, about 10 8 cells to about 10 9 cells, about 10 6 cells to about 10 8 cells, about 10 7 cells to about 10 9 cells, about 10 7 cells to about 10 10 cells, about 10 7 cells to about 10 11 cells, about 10 8 cells to about 10 10 cells, about 10 8 cells to about 10 11 cells, about 10 9 cells to about 10 10 cells, about 10 9 cells to about 10 11 cells, or about 10 10 cells to about 10 11 cells.
- the subjectis preconditioned prior to administration of the cytotoxic agent and/or hematopoietic cells.
- the methodfurther comprises pre-conditioning the subject.
- preconditioning a subjectinvolves subjecting the patient to one or more therapy, such as a chemotherapy or other type of therapy, such as irradiation.
- the preconditioningmay induce or enhance the patient’s tolerance of one or more subsequent therapy (e.g., a targeted therapy), as described herein.
- the pre-conditioninginvolves administering one or more chemotherapeutic agents to the subject.
- Non-limiting examples of chemotherapeutic agentsinclude actinomycin, azacitidine, azathioprine, bleomycin, bortezomib, carboplatin, capecitabine, cisplatin, chlorambucil, cyclophosphamide, cytarabine, daunorubicin, docetaxel, doxifluridine, doxorubicin, epirubicin, epothilone, etoposide, fludarabine, fluorouracil, gemcitabine, hydroxyurea, idarubicin, imatinib, irinotecan, mechlorethamine, mercaptopurine, methotrexate, mitoxantrone, oxaliplatin, paclitaxel, pemetrexed, teniposide, tioguanine, topotecan, valrubicin, vinblastine, vincristine, vindesine, and vinore
- the subjectis preconditioned at least one day, two days, three days, four days, 5 days, 6 days, 7 days, 8 days, 9 days, 10 days, 11 days, 12 days, 13 days, two weeks, three weeks, four weeks, five weeks, six weeks, seven weeks, eight weeks, nine weeks, ten weeks, two months, three months, four months, five months, or at least six months prior to administering the cytotoxic agent and/or hematopoietic cells.
- the chemotherapy(ies) or other therapy(ies)are administered concurrently with the cytotoxic agent and manipulated hematopoietic cells.
- the chemotherapy(ies) or other therapy(ies)are administered after the cytotoxic agent and manipulated hematopoietic cells.
- the chimeric receptor(e.g., a nucleic acid encoding the chimeric receptor) is introduced into an immune cell, and the subject (e.g., human patient) receives an initial administration or dose of the immune cells expressing the chimeric receptor.
- One or more subsequent administrations of the cytotoxic agente.g., immune cells expressing the chimeric receptor
- the subjectmay receive more than one doses of the cytotoxic agent (e.g., an immune cell expressing a chimeric receptor) per week, followed by a week of no administration of the agent, and finally followed by one or more additional doses of the cytotoxic agent (e.g., more than one administration of immune cells expressing a chimeric receptor per week).
- the immune cells expressing a chimeric receptormay be administered every other day for 3 administrations per week for two, three, four, five, six, seven, eight or more weeks. Any of the methods described herein may be for the treatment of a hematological malignancy in a subject.
- the terms “treat,” “treating,” and “treatment”mean to relieve or alleviate at least one symptom associated with the disease or disorder, or to slow or reverse the progression of the disease or disorder.
- the term “treat”also denotes to arrest, delay the onset (i.e., the period prior to clinical manifestation of a disease) and/or reduce the risk of developing or worsening a disease.
- the term “treat”may mean eliminate or reduce the number or replication of cancer cells, and/or prevent, delay or inhibit metastasis, etc.
- a population of genetically engineered hematopoietic cellse.g., carrying genetic edits in genes encoding antigens or expressing those proteins in mutated form
- a cytotoxic agent(s) specific to the cell surface proteinare co-administered to a subject via a suitable route (e.g., infusion).
- the cytotoxic agentrecognizes (binds) a target cell expressing the cell surface protein for targeted killing.
- the hematopoietic cells that express the protein in mutated form, which has reduced binding activity or do not bind the cytotoxic acid (e.g., because of lacking binding epitope)allow for repopulation of a cell type that is targeted by the cytotoxic agent.
- the methods described hereininvolve administering a population of genetically engineered hematopoietic cells to a subject and administering one or more immunotherapeutic agents (e.g., cytotoxic agents).
- the immunotherapeutic agentsmay be of the same or different type (e.g., therapeutic antibodies, populations of immune cells expressing chimeric antigen receptor(s), and/or antibody-drug conjugates).
- the treatment of the patientcan involve the following steps: (1) administering a therapeutically effective amount of the cytotoxic agent to the patient and (2) infusing or reinfusing the patient with hematopoietic stem cells, either autologous or allogenic.
- the treatment of the patientcan involve the following steps: (1) administering a therapeutically effective amount of an immune cell expressing a chimeric receptor to the patient, wherein the immune cell comprises a nucleic acid sequence encoding a chimeric receptor that binds an epitope of a cell surface lineage-specific, disease-associated protein; and (2) infusing or reinfusing the patient with hematopoietic cells (e.g., hematopoietic stem cells), either autologous or allogenic.
- the cytotoxic agente.g., CAR-T cells
- the genetically engineered hematopoietic cellscan be administered to the subject in any order.
- the hematopoietic cellsare given to the subject prior to the cytotoxic agent.
- a second cytotoxic agentcan be administered to the subject after treatment with the first cytotoxic agent, e.g., when the patient develops resistance or disease relapse.
- the hematopoietic cells given the same subjectmay have multiple edited genes expressing lineage-specific cell surface proteins in mutated form such that the cytotoxic agents can target wild-type proteins but not the mutated form.
- the efficacy of the therapeutic methods using a population of genetically engineered hematopoietic cells described herein, optionally in combination with a cytotoxic agentmay be assessed by any method known in the art and would be evident to a skilled medical professional.
- the efficacy of the therapymay be assessed by survival of the subject or cancer burden in the subject or tissue or sample thereof.
- the efficacy of the therapyis assessed by quantifying the absolute number of a particular cell surface protein on a cell or population of cells.
- the efficacy of the therapyis assessed by quantifying the number of cells presenting the antigen.
- the cytotoxic agent comprising an antigen-binding fragment that binds to the epitope of the cell surface protein and the population of hematopoietic cellsis administered concomitantly.
- the cytotoxic agent comprising an antigen-binding fragment that binds an epitope of the cell surface proteine.g., immune cells expressing a chimeric receptor as described herein
- the agent comprising an antigen-binding fragment that binds an epitope of the cell surface proteinis administered at least about 1 day, 2 days, 3 days, 4 days, 5 days, 6 days, 1 week, 2 weeks, 3 weeks, 4 weeks, 5 weeks, 6 weeks, 7 weeks, 8 weeks, 9 weeks, 10 weeks, 11 weeks, 12 weeks, 3 months, 4 months, 5 months, 6 months or more prior to administration of the hematopoietic cells.
- the hematopoietic cellsare administered prior to the cytotoxic agent comprising an antigen-binding fragment that binds an epitope of the cell surface protein (e.g., immune cells expressing a chimeric receptor as described herein).
- the population of hematopoietic cellsis administered at least about 1 day, 2 days, 3 days, 4 days, 5 days, 6 days, 1 week, 2 weeks, 3 weeks, 4 weeks, 5 weeks, 6 weeks, 7 weeks, 8 weeks, 9 weeks, 10 weeks, 11 weeks, 12 weeks, 3 months, 4 months, 5 months, 6 months or more prior to administration of the cytotoxic agent comprising an antigen-binding fragment that binds to an epitope of the antigen.
- the cytotoxic agent targeting the cell surface protein and the population of hematopoietic cellsare administered at substantially the same time. In some embodiments, the cytotoxic agent targeting the cell surface protein is administered and the patient is assessed for a period of time, after which the population of hematopoietic cells is administered. In some embodiments, the population of hematopoietic cells is administered and the patient is assessed for a period of time, after which the cytotoxic agent targeting the cell surface protein is administered. Also within the scope of the present disclosure are multiple administrations (e.g., doses) of the cytotoxic agents and/or populations of hematopoietic cells.
- the cytotoxic agents and/or populations of hematopoietic cellsare administered to the subject once. In some embodiments, cytotoxic agents and/or populations of hematopoietic cells are administered to the subject more than once (e.g., at least 2, 3, 4, 5, or more times). In some embodiments, the cytotoxic agents and/or populations of hematopoietic cells are administered to the subject at a regular interval, e.g., every six months. In some embodiments, the subject is a human subject having a hematopoietic malignancy.
- hematopoietic malignancyrefers to a malignant abnormality involving hematopoietic cells (e.g., blood cells, including progenitor and stem cells).
- hematopoietic malignanciesinclude, without limitation, Hodgkin’s lymphoma, non-Hodgkin’s lymphoma, leukemia, or multiple myeloma.
- leukemiasinclude, without limitation, acute myeloid leukemia, acute lymphoid leukemia, chronic myelogenous leukemia, acute lymphoblastic leukemia or chronic lymphoblastic leukemia, and chronic lymphoid leukemia.
- cells involved in the hematopoietic malignancyare resistant to convention or standard therapeutics used to treat the malignancy.
- the cellse.g., cancer cells
- the cellsmay be resistant to a chemotherapeutic agent and/or CAR T cells used to treat the malignancy.
- hematopoietic malignanciesinclude, but are not limited to, B cell malignancies such as non-Hodgkin’s lymphoma, Hodgkin’s lymphoma, leukemia, multiple myeloma, acute lymphoblastic leukemia, acute lymphoid leukemia, acute lymphocytic leukemia, chronic lymphoblastic leukemia, chronic lymphoid leukemia, and chronic lymphocytic leukemia.
- the hematopoietic malignancyis a relapsing hematopoietic malignancy.
- the leukemiais acute myeloid leukemia (AML).
- AMLis characterized as a heterogeneous, clonal, neoplastic disease that originates from transformed cells that have progressively acquired critical genetic changes that disrupt key differentiation and growth-regulatory pathways.
- CD33 glycoproteinis expressed on the majority of myeloid leukemia cells as well as on normal myeloid and monocytic precursors and has been considered to be an attractive target for AML therapy (Laszlo et al., Blood Rev. (2014) 28(4):143-53). While clinical trials using anti-CD33 monoclonal antibody based therapy have shown improved survival in a subset of AML patients when combined with standard chemotherapy, these effects were also accompanied by safety and efficacy concerns.
- a subjectmay initially respond to a therapy (e.g., for a hematopoietic malignancy) and subsequently experience relapse.
- a therapye.g., for a hematopoietic malignancy
- Any of the methods or populations of genetically engineered hematopoietic cells described hereinmay be used to reduce or prevent relapse of a hematopoietic malignancy.
- any of the methods described hereinmay involve administering any of the populations of genetically engineered hematopoietic cells described herein and an immunotherapeutic agent (e.g., cytotoxic agent) that targets cells associated with the hematopoietic malignancy and further administering one or more additional immunotherapeutic agents when the hematopoietic malignancy relapses.
- an immunotherapeutic agente.g., cytotoxic agent
- the term “relapse”refers to the reemergence or reappearance of cells associated with a hematopoietic malignancy following a period of responsiveness to a therapy. Methods of determining whether a hematopoietic malignancy has relapsed in a subject will be appreciated by one of ordinary skill in the art.
- the period of responsiveness to a therapyinvolves the level or quantity of cells associated with the hematopoietic malignancy the falling below a threshold, e.g., below 20%, 15%, 10%, 5%, 4%, 3%, 2%, or 1% of the level or quantity of cells prior to administration of the therapy.
- a relapseis characterized by the level or quantity of cells associated with the hematopoietic malignancy above a threshold, e.g., above 20%, 15%, 10%, 5%, 4%, 3%, 2%, or 1% higher than the level or quantity of cells during the period of responsiveness.
- a thresholde.g., above 20%, 15%, 10%, 5%, 4%, 3%, 2%, or 1% higher than the level or quantity of cells during the period of responsiveness.
- the subjecthas or is susceptible to relapse of a hematopoietic malignancy (e.g., AML) following administration of one or more previous therapies.
- a hematopoietic malignancye.g., AML
- the methods described hereinreduce the subject’s risk of relapse or the severity of relapse.
- some cancers, including hematopoietic malignanciesare thought to relapse after an initial period of responsiveness to a therapy due to mechanisms such as antigen loss/antigen escape or lineage switch.
- antigen loss/antigen escaperesults in relapse with a phenotypically similar hematopoietic malignancy characterized by cells that lack surface expression of the antigen targeted by the previous therapy (e.g., immunotherapeutic agent) such that the cells are no longer targeted by the previous therapy.
- lineage switchpresents as a genetically related but phenotypically different malignancy in which the cells lack surface expression of the antigen targeted by the previous therapy (e.g., immunotherapeutic agent) such that the cells are no longer targeted by the previous therapy. See, e.g., Brown et al. Nature Reviews Immunology (2019) 19:73-74; Majzner et al. Cancer Discovery (2016) 8(10).
- the hematopoietic malignancyhas relapsed due to antigen loss/antigen escape.
- the target cellshave lost the targeted epitope (e.g., of a lineage-specific cell surface antigen) or have reduced expression of the antigen (e.g., lineage-specific cell surface antigen) such that the targeted epitope is not recognized by the immunotherapeutic agent or is not sufficient to induce cytotoxicity.
- the hematopoietic malignancyhas relapsed due to lineage switch.
- the methods described hereinreduce or avoid relapse of a hematopoietic malignancy by targeting more than one antigen.
- the populations of genetically engineered hematopoietic cellsexpress mutants of more than one antigens such that the mutated antigens are not targeted by an immunotherapeutic agent(s).
- a cancer treated with the methods hereincomprises a first sub- population of cancer cells and a second sub-population of cancer cells.
- One of the sub- populationsmay be cancer stem cells.
- One of the sub-populationsmay be cancer bulk cells.
- One of the sub-populationsmay have one or more (e.g., at least 2, 3, 4, 5, or all) markers of differentiated hematopoietic cells.
- One of the sub-populationsmay have one or more (e.g., at least 2, 3, 4, 5, or all) markers of earlier lineage cells, e.g., HSCs or HPCs.
- Any of the immune cells expressing chimeric receptors described hereinmay be administered in a pharmaceutically acceptable carrier or excipient as a pharmaceutical composition.
- pharmaceutically acceptablerefers to molecular entities and other ingredients of such compositions that are physiologically tolerable and do not typically produce untoward reactions when administered to a mammal (e.g., a human).
- the term “pharmaceutically acceptable”means approved by a regulatory agency of the Federal or a state government or listed in the U.S. Pharmacopeia or other generally recognized pharmacopeia for use in mammals, and more particularly in humans. “Acceptable” means that the carrier is compatible with the active ingredient of the composition (e.g., the nucleic acids, vectors, cells, or therapeutic antibodies) and does not negatively affect the subject to which the composition(s) are administered.
- Any of the pharmaceutical compositions and/or cells to be used in the present methodscan comprise pharmaceutically acceptable carriers, excipients, or stabilizers in the form of lyophilized formations or aqueous solutions.
- Pharmaceutically acceptable carriersincluding buffers, are well known in the art, and may comprise phosphate, citrate, and other organic acids; antioxidants including ascorbic acid and methionine; preservatives; low molecular weight polypeptides; proteins, such as serum albumin, gelatin, or immunoglobulins; amino acids; hydrophobic polymers; monosaccharides; disaccharides; and other carbohydrates; metal complexes; and/or non-ionic surfactants. See, e.g. Remington: The Science and Practice of Pharmacy 20th Ed. (2000) Lippincott Williams and Wilkins, Ed. K. E. Hoover. V.
- aspects of the present disclosurerelate to methods for determining if a subject is at risk of relapse of a disease.
- such methodscomprise providing a first biological sample obtained from a subject at a first time point and a second biological sample obtained from the subject at a second time point, measuring a level of a first cell surface protein in the first biological sample and in the second biological sample, identifying the subject as at risk for relapse of the disease if the level of the first cell surface protein in the first biological sample deviates from the level of the first cell surface protein in the second biological sample.
- the cell surface proteinis one selected from Table 3 or Table 4.
- aspects of the present disclosurerelate to methods for identifying a subject as a candidate for treatment with an agent, such as a cytotoxic agent described herein.
- such methodscomprise providing a first biological sample obtained from a subject at a first time point and a second biological sample obtained from the subject at a second time point, and measuring a level of a first cell surface protein in the first biological sample and in the second biological sample.
- the cell surface proteinis one selected from Table 3 or Table 4.
- methods of identifying subjects as being at risk of relapse of a disease and/or as a candidate for treatment with an agentfurther comprises administering to the subject a therapeutically effective amount of an agent comprising an antigen-binding fragment that binds to the first cell surface protein, such as a cytotoxic agent described herein.
- methods of identifying subjects as being at risk of relapse of a disease and/or as a candidate for treatment with an agentfurther comprises administering to the subject a population of genetically engineered hematopoietic cells having reduced or eliminated expression of the first cell surface protein, such as a genetically engineered hematopoietic cell described herein, or a cell population thereof.
- first biological sample and/or the second biological samplecomprises AML blast cells.
- first time pointis at or near the time the subject is diagnosed with the disease.
- the subjectis a human subject having a hematopoietic malignancy.
- a subjecthas, is suspected of having, or at risk of developing Hodgkin’s lymphoma, non-Hodgkin’s lymphoma, leukemia, or multiple myeloma.
- Exemplary leukemiasinclude, without limitation, acute myeloid leukemia, acute lymphoid leukemia, chronic myelogenous leukemia, acute lymphoblastic leukemia or chronic lymphoblastic leukemia, or chronic lymphoid leukemia.
- a subjectmay initially respond to a therapy (e.g., for a hematopoietic malignancy) and subsequently experience relapse.
- the subjecthas or is susceptible to relapse of a hematopoietic malignancy (e.g., AML) following administration of one or more previous therapies.
- a subjectexhibits an initial period of responsiveness to a therapy due to mechanisms such as antigen loss/antigen escape or lineage switch.
- a subjectmay have, be suspect of having, or at risk of developing antigen loss/antigen escape which results in relapse with a phenotypically similar hematopoietic malignancy characterized by cells that lack surface expression of the cell surface protein targeted by the previous therapy (e.g., immunotherapeutic agent) such that the cells are no longer targeted by the previous therapy.
- a subjectmay have, be suspect of having, or at risk of developing lineage switch wherein a genetically related but phenotypically different malignancy in which the cells lack surface expression of the cell surface protein targeted by the previous therapy (e.g., immunotherapeutic agent) such that the cells are no longer targeted by the previous therapy. See, e.g., Brown et al.
- antigen loss/antigen escapecomprises the target antigen no longer being present on the target cells (e.g., cells of the hematopoietic malignancy) and may occur as a result of genetic mutation and/or enrichment of cells that express a variant of the cell surface protein (e.g., lineage-specific cell surface antigen) that is not targeted by the immunotherapeutic agent (e.g., cytotoxic agent).
- the hematopoietic malignancyhas relapsed due to antigen loss/antigen escape.
- the target cellshave lost the targeted epitope (e.g., of a lineage-specific cell surface antigen) or have reduced expression of the cell surface protein (e.g., lineage-specific cell surface antigen) such that the targeted epitope is not recognized by the immunotherapeutic agent or is not sufficient to induce cytotoxicity.
- the hematopoietic malignancyhas relapsed due to lineage switch.
- the methods described hereinreduce or avoid relapse of a hematopoietic malignancy by targeting more than one antigen.
- the populations of genetically engineered hematopoietic cellsexpress mutants of more than one antigens such that the mutated antigens are not targeted by an immunotherapeutic agent(s).
- the methods described hereinreduce the subject’s risk of relapse or the severity of relapse.
- the period of responsiveness to a therapyinvolves the level or quantity of cells associated with the hematopoietic malignancy the falling below a threshold, e.g., below 20%, 15%, 10%, 5%, 4%, 3%, 2%, or 1% of the level or quantity of cells prior to administration of the therapy.
- a relapseis characterized by the level or quantity of cells associated with the hematopoietic malignancy above a threshold, e.g., above 20%, 15%, 10%, 5%, 4%, 3%, 2%, or 1% higher than the level or quantity of cells during the period of responsiveness.
- a thresholde.g., above 20%, 15%, 10%, 5%, 4%, 3%, 2%, or 1% higher than the level or quantity of cells during the period of responsiveness.
- a subjecthas, is suspected of having, or is at risk of developing a mutation in a gene encoding a lineage-specific cell surface antigen.
- the mutationis associated with altered function a lineage-specific cell surface antigen.
- the mutationis associated with upregulating a lineage-specific cell surface antigen.
- the mutationis associated with downregulating a lineage-specific cell surface antigen.
- the subjecthas, is suspected of having, or is at risk of developing more than one (e.g., two, three, four, or more) mutated gene encoding cell surface proteins.
- the subjectmay comprise a first mutated gene that results in downregulation of a first cell surface protein and second mutated gene that results in downregulation of a second lineage-specific cell surface antigen. In some embodiments, the subject may comprise a first mutated gene that results in upregulation of a first cell surface protein and second mutated gene that results in downregulation of a second lineage-specific cell surface antigen. In some embodiments, the subject may comprise a first mutated gene that results in upregulation of a first cell surface protein and second mutated gene that results in upregulation of a second lineage-specific cell surface antigen.
- a subjecthas, is suspected of having, or is at risk of developing a mutation in a gene encoding a lineage-specific cell surface antigen, such as CD1a, CD1b, CD1c, CD1d, CD1e, CD2, CD3, CD3d, CD3e, CD3g, CD4, CD5, CD6, CD7, CD8a, CD8b, CD9, CD10, CD11a, CD11b, CD11c, CD11d, CDw12, CD13, CD14, CD15, CD16, CD16b, CD17, CD18, CD19, CD20, CD21, CD22, CD23, CD24, CD25, CD26, CD27, CD28, CD29, CD30, CD31, CD32a, CD32b, CD32c, CD34, CD35, CD36, CD37, CD38, CD39, CD40, CD41, CD42a, CD42b, CD42c, CD42d, CD43, CD44, CD45, CD45RA, CD45RB, CD45
- a subjecthas, is suspected of having, or is at risk of developing a mutation in a gene encoding a cell surface protein selected from the group consisting of: CD19; CD123; CD22; CD30; CD171; CS-1 (also referred to as CD2 subset 1, CRACC, SLAMF7, CD319, and 19A24); C-type lectin-like molecule-1 (CLECL1); epidermal growth factor receptor variant III (EGFRvIII); ganglioside G2 (CD2); ganglioside GD3 (aNeu5Ac(2- 8)aNeu5Ac(2-3)bDGalp(1-4)bDGlep(1-1)Cer); TNF receptor family member B cell maturation (BCMA), Tn antigen ((Tn Ag) or (GalNAc ⁇ -Ser/Thr)); prostate-specific membrane antigen (PSMA); Receptor tyrosine kinase-like orphan receptor 1 (ROR1); Fms- Like t
- a subjecthas, is suspected of having, or is at risk of developing a mutation in a gene encoding a cell surface protein known to be associated with a cancer, such as CD20, CD22 (Non-Hodgkin's lymphoma, B-cell lymphoma, chronic lymphocytic leukemia (CLL)), CD52 (B-cell CLL), CD33 (Acute myelogenous leukemia (AML)), CD10 (gp100) (Common (pre-B) acute lymphocytic leukemia and malignant melanoma), CD3/T- cell receptor (TCR) (T-cell lymphoma and leukemia), CD79/B-cell receptor (BCR) (B-cell lymphoma and leukemia), CD26 (epithelial and lymphoid malignancies), human leukocyte antigen (HLA)-DR, HLA-DP, and HLA-DQ (lymphoid malignancies), RCAS1 (gynec
- a subjecthas, is suspected of having, or is at risk of developing a mutation in a gene encoding a cell surface protein selected from a group consisting of: CD10; CD101; CD117; CD11A; CD11C; CD120B; CD123; CD13; CD133; CD152; CD200; CD205 (LY75); CD226; CD244 (SLAMF4); CD25; CD274; CD305 (LAIR1); CD32; CD33; CD34; CD366; CD38; CD41; CD42B; CD44; CD45RA; CD47; CD48; CD49D (ITGA4); CD49F; CD52; CD58; CD64; CD7; CD70; CD82; CD84; CD85; CD85K; CD86; CD9; CD96; CD99; CLL-1; EMR2 (ADGRE2); FR-B; GPR56; and IL1RAP.
- a cell surface proteinselected from a group consisting of: CD10; CD101; CD117; CD11A
- a subjecthas, is suspected of having, or is at risk of developing a mutation in a gene encoding a cell surface protein selected from a group consisting of: CD10; CD101; CD11A; CD11C; CD123; CD133; CD152; CD200; CD205; CD244; CD25; CD305; CD33; CD34; CD366; CD38; CD42B; CD44; CD45RA; CD47; CD48; CD49D; CD49F; CD52; CD58; CD64; CD7; CD84; CD85K; CD9; CD99; CLL-1; EMR2; GPR56; IL1RAP; and CD274.
- a cell surface proteinselected from a group consisting of: CD10; CD101; CD11A; CD11C; CD123; CD133; CD152; CD200; CD205; CD244; CD25; CD305; CD33; CD34; CD366; CD38; CD42B; CD44; CD45RA; CD47; CD48; CD49D; CD
- a subjecthas, is suspected of having, or is at risk of developing a mutation in a gene encoding a cell surface protein selected from a group consisting of: CD10; CD117; CD11A; CD11C; CD120B; CD123; CD13; CD133; CD152; CD205; CD22; CD226; CD244; CD25; CD305; CD32; CD33; CD34; CD38; CD41; CD44; CD45RA; CD47; CD48; CD49D; CD49F; CD52; CD56; CD58; CD64; CD7; CD70; CD71; CD82; CD84; CD85K; CD86; CD96; CD99; CLL-1; EMR2; FR-B; GPR56; HLA-DR; and IL1RAP.
- a cell surface proteinselected from a group consisting of: CD10; CD117; CD11A; CD11C; CD120B; CD123; CD13; CD133; CD152; CD205; CD22; CD226; CD244
- a subjecthas, is suspected of having, or is at risk of developing a mutation in a first gene encoding a first cell surface protein and a second gene encoding a second cell surface protein selected from a group consisting of: CD274 and CD152; CD274 and CD226; CD152 and CD226; CD10 and CD49F; CD49F and GPR56; CD200 and CD34; CD34 and CD52; CD34 and CD133; GPR56 and CD133; CD42B and CD41; CD85K and CD64; CD85K and CD32; CD64 and CD32; CD64 and CD11C; CD64 and CD86; CD45RA and CD96; CD45RA and IL1RAP; CD45RA and CD123; CD84 and CD33; CD11C and IL1RAP; CD11C and CD33; CD11C and CD86; CD9 and IL1RAP; IL1RAP and CD33; IL1RAP and CD205; IL1RAP and CD305; IL
- a subjecthas, is suspected of having, or is at risk of developing a mutation in a first gene encoding a first cell surface protein and a second gene encoding a second cell surface protein selected from a group consisting of: CD85K and CD64; CD45RA and IL1RAP; CD34 and CD133; IL1RAP and CD33; CD47 and CD99; CLL-1 and CD305; CD152 and CD226; CD45RA and CD96; CD45RA and CD123; CD49D and CD305; IL1RAP and CD305; CD58 and CD305; CD49D and CD47; CD33 and CD305; CD49D and CD44; CD64 and CD86; GPR56 and CD133; CD34 and CD52; EMR2 and CD82; CD64 and CD32; CD42B and CD41; CD64 and CD11C; CD99 and CD44; CD200 and CD34; CD11C and CD33; IL1RAP and CD123; CD11C and IL1RAP; CD44
- a subjecthas, is suspected of having, or is at risk of developing a mutation in a first gene encoding a first cell surface protein and a second gene encoding a second cell surface protein selected from a group consisting of: CD33 and CD205 (LY75); CD33 and CD244 (SLAMF4); CD33 and CD305 (LAIR1); CD33 and CD49D (ITGA4); CLL-1 and CD205 (LY75); CLL-1 and CD244 (SLAMF4); CLL-1 and CD305 (LAIR1); CLL-1 and CD49D (ITGA4); EMR2 and CD205 (LY75); EMR2 and CD244 (SLAMF4); EMR2 and CD305 (LAIR1); EMR2 and CD49D (ITGA4); CD38 and CD205 (LY75); CD38 and CD244 (SLAMF4); CD38 and CD305 (LAIR1); CD38 and CD49D (ITGA4); CD305 (LAIR1) and CD49D (ITGA4); CD305 (LAIR1) and
- a subjecthas, is suspected of having, or is at risk of developing a mutation in a first gene encoding a first cell surface protein and a second gene encoding a second lineage-specific cell surface antigen, wherein mutation of the first gene and second gene results in dysregulated expression of the first cell surface protein and the second cell surface protein which is indicative (e.g., diagnostic) of the subject having a hematopoietic malignancy.
- the first gene and second geneencode combinations of antigens selected from a group consisting of: CD274 and CD152; CD274 and CD226; CD152 and CD226; CD10 and CD49F; CD49F and GPR56; CD200 and CD34; CD34 and CD52; CD34 and CD133; GPR56 and CD133; CD42B and CD41; CD85K and CD64; CD85K and CD32; CD64 and CD32; CD64 and CD11C; CD64 and CD86; CD45RA and CD96; CD45RA and IL1RAP; CD45RA and CD123; CD84 and CD33; CD11C and IL1RAP; CD11C and CD33; CD11C and CD86; CD9 and IL1RAP; IL1RAP and CD33; IL1RAP and CD205; IL1RAP and CD305; IL1RAP and CD123; CD33 and CLL-1; CD33 and CD305; CD11A and CD86; CLL-1 and CD305; CD
- the first gene and second geneencode combinations of antigens selected from a group consisting of: CD33 and CD205 (LY75); CD33 and CD244 (SLAMF4); CD33 and CD305 (LAIR1); CD33 and CD49D (ITGA4); CLL-1 and CD205 (LY75); CLL-1 and CD244 (SLAMF4); CLL-1 and CD305 (LAIR1); CLL-1 and CD49D (ITGA4); EMR2 and CD205 (LY75); EMR2 and CD244 (SLAMF4); EMR2 and CD305 (LAIR1); EMR2 and CD49D (ITGA4); CD38 and CD205 (LY75); CD38 and CD244 (SLAMF4); CD38 and CD305 (LAIR1); CD38 and CD49D (ITGA4); CD305 (LAIR1) and CD49D (ITGA4); CD305 (LAIR1) and CD49D (ITGA4); CD305 (LAIR1) and CD205 (LY75); CD305 (LAIR1) and CD244 (SLAMF
- a subjecthas, is suspected of having, or is at risk of developing a mutation in a first gene encoding a first cell surface protein and a second gene encoding a second lineage-specific cell surface antigen, wherein mutation of the first gene and second gene results in dysregulated expression of the first cell surface protein and the second cell surface protein which is indicative (e.g., diagnostic) of the subject having experienced a relapse of a hematopoietic malignancy.
- the first gene and second geneencode combinations of antigens selected from a group consisting of: CD9 and CD82; CD45RA and CD123; CD133 and CD82; CD85K and CD70; CD47 and CD205; CD64 and FR-B; CD11A and CD44; CD85K and CD86; CD366 and CD96; CD101 and CD64; CLL-1 and CD49D; CD49D and CD47; EMR2 and CD82; CD38 and CD305; CD34 and CD7; CD200 and CD25; CD25 and CD45RA; CD49D and CD44; CD11A and CD244; CD133 and CD7; CD48 and CD11A; CD52 and CD133; GPR56 and CD82; CD11A and CD38; CD34 and CD13; CD58 and CD49D; CD47 and CD99; IL1RAP and CD58; CD47 and CD44; CD33 and CD305; CD64 and CD86; CD152 and CD120B; CD42B and CD41; CD34 and CD52; CD
- the first gene and second geneencode combinations of antigens selected from a group consisting of: CD33 and CD205 (LY75); CD33 and CD244 (SLAMF4); CD33 and CD305 (LAIR1); CD33 and CD49D (ITGA4); CLL-1 and CD205 (LY75); CLL-1 and CD244 (SLAMF4); CLL-1 and CD305 (LAIR1); CLL-1 and CD49D (ITGA4); EMR2 and CD205 (LY75); EMR2 and CD244 (SLAMF4); EMR2 and CD305 (LAIR1); EMR2 and CD49D (ITGA4); CD38 and CD205 (LY75); CD38 and CD244 (SLAMF4); CD38 and CD305 (LAIR1); CD38 and CD49D (ITGA4); CD305 (LAIR1) and CD49D (ITGA4); CD305 (LAIR1) and CD49D (ITGA4); CD305 (LAIR1) and CD205 (LY75); CD305 (LAIR1) and CD244 (SLAMF
- tissue samplessuch as tissue sections and needle biopsies of a tissue
- cell samplese.g., cytological smears (such as Pap or blood smears) or samples of cells obtained by microdissection); samples of whole organisms (such as samples of yeasts or bacteria); or cell fractions, fragments or organelles (such as obtained by lysing cells and separating the components thereof by centrifugation or otherwise).
- biological samplesinclude blood, serum, urine, semen, fecal matter, cerebrospinal fluid, interstitial fluid, mucous, tears, sweat, pus, biopsied tissue (e.g., obtained by a surgical biopsy or needle biopsy), nipple aspirates, milk, vaginal fluid, saliva, swabs (such as buccal swabs), or any material containing biomolecules that is derived from a first biological sample.
- a biological sampleis derived from a subject who has, is suspected of having, or is at risk of developing a hematopoietic malignancy.
- a biological sampleis obtained at a time during or near the time the subject is diagnosed with a hematopoietic malignancy. In some embodiments, a biological sample is obtained at a time during or near when the subject has or is suspected to have experienced relapse of the hematopoietic malignancy. In some embodiments, a biological sample is obtained after the subject has undergone has received at least one treatment for a hematopoietic malignancy. In some embodiments, a biological sample comprises malignant hematopoietic cells. In some embodiments, a biological sample comprises AML blast cells.
- methods of the present disclosurefurther comprise determining the expression level of a first cell surface protein and the expression level of a second cell surface protein in a first biological sample by obtaining or having obtained the first biological sample from a subject and performing or having performed an assay comprising detecting the presence or absence of expression of the first cell surface protein and the second cell surface protein in the first biological sample.
- methods of the present disclosurefurther comprise determining the expression level of the first cell surface protein and the expression level of the second cell surface protein in a second biological sample by obtaining or having obtained the second biological sample from the subject and performing or having performed an assay comprising detecting the presence or absence of expression of the first cell surface protein and the second cell surface protein in the second biological sample.
- methods of the present disclosurefurther comprise identifying or having identified the second biological sample as comprising altered expression of the first cell surface protein and the second cell surface protein when the expression levels of the first cell surface protein and the second cell surface protein are different in the second biological sample relative to the first biological sample.
- identifying or having identified the second biological sample as comprising altered expression of the first cell surface protein and the second cell surface proteinalso identifies the subject as having experienced relapse of the hematopoietic malignancy.
- such methodsfurther comprise administering to the subject a at least one of a genetically engineered hematopoietic, or cell population thereof, a cytotoxic agent described herein, or both.
- identifying or having identified the second biological sample as comprising altered expression of the first cell surface protein and the second cell surface proteinalso identifies subject as being in need of administration to the subject a at least one of a genetically engineered hematopoietic, or cell population thereof, a cytotoxic agent described herein, or both,
- a biological sampleis derived from a subject who has, is suspected of having, or is at risk of developing a mutation in a gene encoding a cell surface protein selected from a group consisting of: CD10; CD101; CD117; CD11A; CD11C; CD120B; CD123; CD13; CD133; CD152; CD200; CD205 (LY75); CD226; CD244 (SLAMF4); CD25; CD274; CD305 (LAIR1); CD32; CD33; CD34; CD366; CD38; CD41; CD42B; CD44; CD45RA; CD47; CD48; CD49D (ITGA4); CD49F; CD
- a biological sampleis derived from a subject who has, is suspected of having, or is at risk of developing a mutation in a gene encoding a cell surface protein selected from a group consisting of: CD10; CD101; CD11A; CD11C; CD123; CD133; CD152; CD200; CD205; CD244; CD25; CD305; CD33; CD34; CD366; CD38; CD42B; CD44; CD45RA; CD47; CD48; CD49D; CD49F; CD52; CD58; CD64; CD7; CD84; CD85K; CD9; CD99; CLL-1; EMR2; GPR56; IL1RAP; and CD274.
- a cell surface proteinselected from a group consisting of: CD10; CD101; CD11A; CD11C; CD123; CD133; CD152; CD200; CD205; CD244; CD25; CD305; CD33; CD34; CD366; CD38; CD42B; CD44; CD45RA; CD47
- a biological sampleis derived from a subject who has, is suspected of having, or is at risk of developing a mutation in a gene encoding a cell surface protein selected from a group consisting of: CD10; CD117; CD11A; CD11C; CD120B; CD123; CD13; CD133; CD152; CD205; CD22; CD226; CD244; CD25; CD305; CD32; CD33; CD34; CD38; CD41; CD44; CD45RA; CD47; CD48; CD49D; CD49F; CD52; CD56; CD58; CD64; CD7; CD70; CD71; CD82; CD84; CD85K; CD86; CD96; CD99; CLL-1; EMR2; FR-B; GPR56; HLA-DR; and IL1RAP.
- a cell surface proteinselected from a group consisting of: CD10; CD117; CD11A; CD11C; CD120B; CD123; CD13; CD133; CD152; CD205; CD
- a biological sampleis derived from a subject who has, is suspected of having, or is at risk of developing a mutation in a first gene encoding a first cell surface protein and a second gene encoding a second cell surface protein selected from a group consisting of: CD274 and CD152; CD274 and CD226; CD152 and CD226; CD10 and CD49F; CD49F and GPR56; CD200 and CD34; CD34 and CD52; CD34 and CD133; GPR56 and CD133; CD42B and CD41; CD85K and CD64; CD85K and CD32; CD64 and CD32; CD64 and CD11C; CD64 and CD86; CD45RA and CD96; CD45RA and IL1RAP; CD45RA and CD123; CD84 and CD33; CD11C and IL1RAP; CD11C and CD33; CD11C and CD86; CD9 and IL1RAP; IL1RAP and CD33; IL1RAP and CD205; IL
- a biological sampleis derived from a subject who has, is suspected of having, or is at risk of developing a mutation in a first gene encoding a first cell surface protein and a second gene encoding a second cell surface protein selected from a group consisting of: CD33 and CD205 (LY75); CD33 and CD244 (SLAMF4); CD33 and CD305 (LAIR1); CD33 and CD49D (ITGA4); CLL-1 and CD205 (LY75); CLL-1 and CD244 (SLAMF4); CLL-1 and CD305 (LAIR1); CLL-1 and CD49D (ITGA4); EMR2 and CD205 (LY75); EMR2 and CD244 (SLAMF4); EMR2 and CD305 (LAIR1); EMR2 and CD49D (ITGA4); CD38 and CD205 (LY75); CD38 and CD244 (SLAMF4); CD38 and CD305 (LAIR1); CD38 and CD49D (ITGA4); CD305 (LAIR1) and CD49D (ITGA
- kits for use in treating hematopoietic malignancymay comprise the genetically engineered hematopoietic cells such as HSCs, and optionally one or more cytotoxic agents targeting cell surface proteins, the genes of which are edited in the hematopoietic cells.
- kitsmay include a container comprising a first pharmaceutical composition that comprises any of the genetically engineered hematopoietic cells as described herein, and optionally one or more additional containers comprising one or more cytotoxic agents (e.g., immune cells expressing chimeric receptors described herein) targeting the cell surface proteins as also described herein.
- the kitcan comprise instructions for use in any of the methods described herein.
- the included instructionscan comprise a description of administration of the genetically engineered hematopoietic cells and optionally descriptions of administration of the one or more cytotoxic agents to a subject to achieve the intended activity in a subject.
- the kitmay further comprise a description of selecting a subject suitable for treatment based on identifying whether the subject is in need of the treatment.
- the instructionscomprise a description of administering the genetically engineered hematopoietic cells and optionally the one or more cytotoxic agents to a subject who is in need of the treatment.
- the instructions relating to the use of the genetically engineered hematopoietic cells and optionally the cytotoxic agents described hereingenerally include information as to dosage, dosing schedule, and route of administration for the intended treatment.
- the containersmay be unit doses, bulk packages (e.g., multi-dose packages) or sub-unit doses.
- Instructions supplied in the kits of the disclosureare typically written instructions on a label or package insert.
- the label or package insertindicates that the pharmaceutical compositions are used for treating, delaying the onset, and/or alleviating a disease or disorder in a subject.
- the kits provided hereinare in suitable packaging. Suitable packaging includes, but is not limited to, vials, bottles, jars, flexible packaging, and the like.
- kits for use in combination with a specific devicesuch as an inhaler, nasal administration device, or an infusion device.
- a kitmay have a sterile access port (for example, the container may be an intravenous solution bag or a vial having a stopper pierceable by a hypodermic injection needle).
- the containermay also have a sterile access port.
- Kitsoptionally may provide additional components such as buffers and interpretive information.
- the kitcomprises a container and a label or package insert(s) on or associated with the container.
- the disclosureprovides articles of manufacture comprising contents of the kits described above.
- Acute myeloid leukemiais an aggressive clonal malignancy characterized by combinations of chromosomal abnormalities, gene mutations, and cell surface antigen (Ag) expression profiles. This heterogeneity contributes to refractory or relapsed disease that presents a major challenge when treating AML.
- Previous single cell sequencing studies of primary AML sampleshave provided remarkable insight into the clonal architecture of AML cell populations and how their mutational, transcriptomic, and surface Ag profiles vary between patients. However, information addressing clonal shifts during progression from diagnosis to relapse in large cohorts is lacking.
- single cell RNA sequencingwas adapted with surface Ag feature barcoding to analyze more than 450,000 cells from 28 paired AML patient bone marrow mononuclear cell samples collected at diagnosis and relapse (56 samples) to generate the largest and most comprehensive single cell AML atlas to date.
- This atlascontains rich clinical metadata including cytogenetics, mutation status of canonical AML genes, treatment history, and survival information.
- the feature barcoding panelconsisted of a comprehensive list of 81 surface Ags reported in clinicaltrials.gov or mined from literature, including large proteomics mass spectrometry datasets of AML patient bone marrow samples (DeBoer et. al., Cancer Cell 2018; Jayavelu et al, Cancer Cell 2022).
- Feature barcodingproduced antibody derived tag (ADT) counts which are interpreted as relative values but alone does not indicate absolute number of Ags per cell. This limitation was addressed by measuring absolute Ag density of AML Ags, CD33, CLL-1, CD123, and EMR2 in patient samples using the flow cytometric QuantiBRITE assay.
- Flow cytometrywas further employed to characterize surface expression of four key myeloid antigens including CD33, CLL-1, CD123, and EMR2. More than 80% of AML blasts expressed CD33 at both diagnosis and relapse. A similar trend was observed for CLL- 1, CD123, and EMR2 (FIG.5A).
- Antigen densitywhich refers to the number of molecules of an antigen present on a cell surface, can inform the development of effective targeted therapies.
- AML blastswere contacted with QuantiBRITE beads for quantification of the average number of antibodies bound per blast cell (ABCs). The results suggested that CD33- positive blasts had on average more than 1000 antigens per cell, with the same trend observed for the other 3 antigens (FIG.5B).
- 81 antigenswere selected based on whether oligo-tagged antibodies for these antigens were commercially available.
- scRNAseqwas performed on matched samples from diagnosis and relapse with feature barcoding on all 56 samples to construct the atlas which was used to characterize heterogeneity across patients and within patients across clinical timepoints (FIGs.6-7).
- Single-cell RNA sequencing analyses of samples derived from AML subjectswere used to correlate cell surface antigen expression profiles to hematopoietic cell lineage (FIGs. 8A-9).
- a UMAP plotwas generated to represent the entire atlas consisting of about 400,00 cells across all patients and timepoints (FIGs.8 and 10).
- the datawas harmonized, clustered, and annotated into broad cell type categories using a combination of cell surface protein and RNA information.
- Blast cell populationswere identified that were characterized by high CD33, CLL-1, CD34 surface expression and diminished CD45 expression (FIG.8B).
- Monocyte, dendritic cell, T cell, B cell, and erythroid cell populationswere also identified in each sample (FIGs.8A and 9).
- ADTraw antigen-derived counts
- QuantiBRITETM data setwas used to analyze absolute antigen counts. Then, the relationship between ADTs and QuantiBRITE data for CD33, CLL-1, CD123, and EMR2 was analyzed.
- FIG.10A strong linear correlation between the two modalities was observed (FIG.10). This relationship was used to estimate antigen density on each blast based on its ADT expression. An unbiased analysis of the data described above was performed to rank the 81 antigens based on the average surface expression across all blasts. Heatmaps were generated to illustrate estimated antigen density across blasts in each patient at diagnosis and relapse. CD33 and CLL-1 were among the top antigens expressed in blasts at both time points. High expression of both CD99 and CD44 was also observed (FIG.11A). Antigen expression changes between timepoints within individual patients were also assessed which identified antigens with increased expression at relapse (FIG.11A).
- the proliferating blastswere further analyzed using differential gene expression analysis between the two timepoints (AML diagnosis and relapse). This identified 256 upregulated genes and 772 down-regulated genes (FIG.13E). Pathway enrichment analysis using the Hallmark gene sets showed a significant downregulation of inflammatory response pathways including interferon gamma response, TNF alpha signaling, and interferon alpha response (FIG.14A). There was also significant upregulation of oxidative phosphorylation and an increased expression of MYC and E2F target genes (FIG.14B).
- CITE-seq profilingwas performed to assess expression of 81 candidate antigens across 400,000 cells from 28 paired diagnosis and relapse patient bone marrow samples (FIG.15A).
- One AML target, EMR2is among the top 30 highest surface expressors in both diagnosis and relapsed samples (FIG. 15A and Table 4). This multimodal profiling was paired with flow cytometry surface expression analysis.
- Single cell fluorescent intensities of CD33, CLL-1, CD123, and EMR2 from the flow cytometry datawere converted to antigen density based on a standard curve produced from the QuantiBRITE assay.5% quantiles of this antigen density distribution were calculated for each antigen for each sample.5% quantiles were then calculated for the Antigen Derived Tag (ADT) readout for same antigens and samples and matched with the former quantiles produced from QuantiBRITE.
- ADTAntigen Derived Tag
- Example 2Multiplex Editing of Cell Surface Antigens Showing High Expression at AML Diagnosis and Relapse This Example relates to a strategy for multiplexed genetic editing of cell surface antigens (e.g., two or more of EMR2, CD33, CD123, and CLL-1) in healthy HSCs to enable combinatorial targeting of AML cells.
- cell surface antigense.g., two or more of EMR2, CD33, CD123, and CLL-1
- a “protected” HSC transplantcan be generated which enables selective eradication of AML blast and leukemic stem cells by administration of combinatorial immunotherapy.
- CD34+ cellswere thawed and two days later electroporated with adenosine base editor mRNA and either a gRNA targeting splicing regulatory sequences in exon 19 of EMR2, a gRNA targeting splicing regulatory sequences in exon 5 of CD33, or both gRNAs simultaneously (FIG.16A and Table 5).
- EMR2 and CD33 gRNAswere electroporated.
- multiplex base editingwas utilized to genetically engineer the EMR2, CD33, CD123, and CLL-1 genes.
- flow cytometrywas used to analyze the number of cells in the population having cell surface expression of one, two, three, or four of the antigens selected from CD33, CD123, EMR2, and CLL-1 (FIG. 17).
- Example 3Multimodal Atlas of Paired Diagnosis and Relapse AML Reveals Surface Antigens for Multi-specific Immunotherapy
- This Examplerelates to an expanded cell surface antigen atlas that included additional samples from healthy donors and categorizations of blasts into hematopoietic cell lineages.
- the dataset of blast heterogeneity across 68 sampleswas used to identify novel cell surface antigens (Ags) that can be targeted with multi-specific immunotherapies.
- all samples in the atlasincluded both feature barcoding with antigen derived tags (ADT) on 81 Ags and QuantiBRITE flow cytometric data, measured as antibodies bound per cell (ABC), on four well-characterized AML Ags (CD33, CLL-1, CD123, and EMR2).
- the Ags in this expanded cell surface antigen atlaswere ranked which, when combined together, or with CD33 and CLL-1, label the maximum number of blasts at a sufficient therapeutic threshold for immunotherapy (Ag count >1000/cell) (Sharma and Kranz, 2016).
- Four additional Ag candidateswere identified from the atlas: LAIR1, ITGA4, LY75, and CD244 that were expressed in 23, 26, 18, and 15 of 28 diagnosis samples, and 24, 25, 18, and 17 of 28 relapse samples, respectively, on >80% of blasts in the sample at >500 predicted Ags/blast (FIGs.18A-18B and FIG.19A).
- At least one of A and Bcan refer, in one embodiment, to at least one, optionally including more than one, A, with no B present (and optionally including elements other than B); in another embodiment, to at least one, optionally including more than one, B, with no A present (and optionally including elements other than A); in yet another embodiment, to at least one, optionally including more than one, A, and at least one, optionally including more than one, B (and optionally including other elements); etc.
- the order of the steps or acts of the methodis not necessarily limited to the order in which the steps or
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Engineering & Computer Science (AREA)
- Genetics & Genomics (AREA)
- Chemical & Material Sciences (AREA)
- Biomedical Technology (AREA)
- Biotechnology (AREA)
- Zoology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Organic Chemistry (AREA)
- Molecular Biology (AREA)
- Wood Science & Technology (AREA)
- Cell Biology (AREA)
- General Health & Medical Sciences (AREA)
- General Engineering & Computer Science (AREA)
- Immunology (AREA)
- Biochemistry (AREA)
- Microbiology (AREA)
- Hematology (AREA)
- Physics & Mathematics (AREA)
- Developmental Biology & Embryology (AREA)
- Medicinal Chemistry (AREA)
- Biophysics (AREA)
- Plant Pathology (AREA)
- Oncology (AREA)
- Urology & Nephrology (AREA)
- Virology (AREA)
- Analytical Chemistry (AREA)
- General Physics & Mathematics (AREA)
- Pathology (AREA)
- Food Science & Technology (AREA)
- Hospice & Palliative Care (AREA)
- Pharmacology & Pharmacy (AREA)
- Epidemiology (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Mycology (AREA)
- Micro-Organisms Or Cultivation Processes Thereof (AREA)
Abstract
Description
COMPOSITIONS COMPRISING GENETICALLY ENGINEERED HEMATOPOIETIC STEM CELLS AND METHODS OF USE THEREOF RELATED APPLICATIONS The application claims the benefit under 35 U.S.C.119(e) of U.S. Provisional Application number 63/517,109 filed August 1, 2023, U.S. Provisional Application number 63/607,780 filed December 8, 2023, and U.S. Provisional Application number 63/645,618 filed May 10, 2024, each of which is incorporated by reference in its entirety. REFERENCE TO AN ELECTRONIC SEQUENCE LISTING The contents of the electronic sequence listing (V029170046WO00-SEQ-MFM.xml; Size: 71,279 bytes; and Date of Creation: July 25, 2024) is herein incorporated by reference in its entirety. BACKGROUND OF THE INVENTION A major challenge in designing targeted therapies is the successful identification of proteins that are uniquely expressed on cells that would be therapeutically relevant to eliminate (e.g., abnormal, malignant, or other target cells) but not present on cells that one does not wish to eliminate (e.g., normal, healthy, or other non-target cells). For example, many cancer therapeutics struggle to effectively target cancer cells while leaving normal cells unharmed. An alternative strategy that has emerged involves targeting an entire cell lineage, which includes targeting normal cells, cancer cells, and pre-cancerous cells. For example, CD19-targeted chimeric antigen receptor T cells (CAR T cells) and anti-CD20 monoclonal antibodies (e.g., Rituximab) each target B cell lineage proteins (CD19 and CD20, respectively). While potentially effective in treating B cell malignancies, use of such therapies is limited as elimination of B cells is detrimental. Similarly, targeting lineage- specific proteins of other cell populations, for example, myeloid lineage cells (e.g., cancers arising from myeloid blasts, monocytes, megakaryocytes, etc.) is not feasible, as these cell populations are necessary for survival. Thus, there remains an unmet need to effectively target cells of interest, e.g., cancer cells, without targeting or harming normal cell populations. SUMMARY OF THE INVENTION Aspects of the present disclosure relate to genetically engineered hematopoietic cells, or descendants thereof, comprising a mutation in a first gene encoding a first cell surface protein and second gene encoding a second cell surface protein. In some embodiments, the genetically engineered hematopoietic cells, or descendants thereof, are genetically engineered such that the genetically engineered hematopoietic cells have reduced or eliminated expression of the first cell surface protein relative to a wildtype counterpart cell or express a mutant of the first cell surface protein. In some embodiments, the genetically engineered hematopoietic cells, or descendants thereof, are genetically engineered such that the genetically engineered hematopoietic cells have reduced or eliminated expression of the second cell surface protein relative to a wildtype counterpart cell, or express a mutant of the second cell surface protein. In some embodiments, the first cell surface protein and the second cell surface protein are of a combination as set forth in Table 3 or Table 4. In some embodiments, the first cell surface protein and the second cell surface protein are selected from the group consisting of: CD10; CD101; CD117; CD11A; CD11C; CD120B; CD123; CD13; CD133; CD152; CD200; CD205 (LY75); CD226; CD244 (SLAMF4); CD25; CD274; CD305 (LAIR1); CD32; CD33; CD34; CD366; CD38; CD41; CD42B; CD44; CD45RA; CD47; CD48; CD49D (ITGA4); CD49F; CD52; CD58; CD64; CD7; CD70; CD82; CD84; CD85; CD85K; CD86; CD9; CD96; CD99; CLL-1; EMR2 (ADGRE2); FR-B; GPR56; and IL1RAP. In some embodiments, the first cell surface protein and the second cell surface protein are selected from the group consisting of: CD85K and CD64; CD45RA and IL1RAP; CD34 and CD133; IL1RAP and CD33; CD47 and CD99; CLL-1 and CD305; CD152 and CD226; CD45RA and CD96; CD45RA and CD123; CD49D and CD305; IL1RAP and CD305; CD58 and CD305; CD49D and CD47; CD33 and CD305; CD49D and CD44; CD64 and CD86; GPR56 and CD133; CD34 and CD52; EMR2 and CD82; CD64 and CD32; CD42B and CD41; CD64 and CD11C; CD99 and CD44; CD200 and CD34; CD11C and CD33; IL1RAP and CD123; CD11C and IL1RAP; CD44 and EMR2; CD205 and CD305; CD44 and CD305; CD49D and CD99; CD274 and CD226; CD49F and GPR56; CD9 and IL1RAP; CD99 and CD305; CD10 and CD49F; CD11A and CD86; CD33 and CLL-1; CD84 and CD33; CD244 and CD117; CD85K and CD32; CD47 and EMR2; CD274 and CD152; IL1RAP and CD205; CD11C and CD86; CD99 and EMR2; CD205 and CD123; and CD123 and EMR2. In some embodiments, the first cell surface protein and the second cell surface protein are selected from the group consisting of: CD33 and CD205 (LY75); CD33 and CD244 (SLAMF4); CD33 and CD305 (LAIR1); CD33 and CD49D (ITGA4); CLL-1 and CD205 (LY75); CLL-1 and CD244 (SLAMF4); CLL-1 and CD305 (LAIR1); CLL-1 and CD49D (ITGA4); EMR2 and CD205 (LY75); EMR2 and CD244 (SLAMF4); EMR2 and CD305 (LAIR1); EMR2 and CD49D (ITGA4); CD38 and CD205 (LY75); CD38 and CD244 (SLAMF4); CD38 and CD305 (LAIR1); CD38 and CD49D (ITGA4); CD305 (LAIR1) and CD49D (ITGA4); CD305 (LAIR1) and CD205 (LY75); CD305 (LAIR1) and CD244 (SLAMF4); CD49D (ITGA4) and CD205 (LY75); CD49D (ITGA4) and CD244 (SLAMF4); and CD205 (LY75) and CD244 (SLAMF4). In some embodiments, the first cell surface protein is CD33 and the second cell surface protein is IL1RAP. In some embodiments, the first cell surface protein is CD44 and the second cell surface protein is EMR2. In some embodiments, the first cell surface protein and the second cell surface protein are expressed in >50% of AML blasts at targetable antigen levels of >500 proteins (antigens, Ags) per cell. In some embodiments, the first cell surface protein and the second cell surface protein are expressed in >50% of AML blasts at targetable antigen levels of >1000 proteins (antigens, Ags) per cell. In some embodiments, the whole or a portion of the first gene and/or the second gene is deleted. In some embodiments, the whole or a portion of the first gene and/or the second gene is deleted using genome editing. In some embodiments, the genome editing carried out involves a zinc finger nuclease (ZFN), a transcription activator-like effector-based nuclease (TALEN), or a CRISPR-Cas system. In some embodiments, the CRISPR-Cas system comprises a nucleic acid encoding a gRNA and an RNA-guided nuclease. In some embodiments, the RNA-guided nuclease is a base editor. In some embodiments, the mutation of the first gene and/or the second gene comprises a substitution, deletion, or addition of at least one nucleotide or a combination thereof. In some embodiments, the mutation of the first gene and/or the second gene comprises a substitution, deletion, or addition of at least one amino acid or a combination thereof in a non-essential epitope of the first and/or the second cell surface protein. In some embodiments, the mutant of the first cell surface protein and/or the mutant of the second cell surface protein comprises a substitution, deletion, or addition of at least one amino acid or a combination thereof. In some embodiments, the mutation of the first gene and/or the second gene comprises a substitution, deletion, or addition of at least one nucleotide or a combination thereof in a sequence encoding a non-essential epitope of the first and/or the second cell surface protein. In some embodiments, the mutation alters 1, 2, 3, 4, or 5 amino acid residues of the first and/or second cell surface protein. In some embodiments, the mutation alters no more than 1, no more than 2, no more than 3, no more than 4, or no more than 5 amino acid residues of the first and/or second cell surface protein. In some embodiments, expression of the first cell surface protein and/or the second cell surface protein is reduced by at least 20%, at least 30%, at least 40%, at least 50%, at least 60%, at least 70%, at least 80%, at least 90%, or at least 95% relative to the wildtype counterpart cell. However, in other embodiments, expression of the first cell surface protein and/or the second cell surface protein is not substantially altered, wherein the mutant of the first cell surface protein and/or the mutant of the second cell surface protein comprise a modified non-essential epitope. In some embodiments, mutation in the first gene does not affect the bioactivity of the first cell surface protein and/or the mutation in the second gene does not affect the bioactivity of the second cell surface protein. In some embodiments, the cell retains the ability to differentiate normally. In some embodiments, the cell is a hematopoietic stem or progenitor cell, or a descendant thereof. In some embodiments, the hematopoietic cell is a hematopoietic stem cell (HSC). In some embodiments, the hematopoietic cell is a CD34+ cell. In some embodiments, the hematopoietic cell is from bone marrow, blood, umbilical cord, or peripheral blood mononuclear cells (PBMCs). In some embodiments, the hematopoietic cell is a human cell. In some embodiments, the cell has reduced or eliminated binding to a first agent comprising an antigen-binding fragment that binds to the first cell surface protein and/or a second agent comprising an antigen-binding fragment that binds to the second cell surface protein. In some embodiments, the first agent and/or the second agent is a cytotoxic agent. In some embodiments, the antigen-binding fragment of the cytotoxic agent is a single-chain antibody fragment (scFv) that specifically binds the cell surface protein or an epitope thereof. In some embodiments, the cytotoxic agent is an antibody or an antibody-drug conjugate (ADC). In some embodiments, the cytotoxic agent is an immune cell expressing a chimeric receptor that comprises the antigen-binding fragment. In some embodiments, the immune cells are T cells. In some embodiments, the chimeric receptor further comprises: a hinge domain; a transmembrane domain; at least one co-stimulatory domain; a cytoplasmic signaling domain; or a combination thereof. In some embodiments, the chimeric receptor comprises at least one co-stimulatory signaling domain, which is derived from a co-stimulatory receptor selected from the group consisting of CD27, CD28, 4-1BB, OX40, CD30, ICOS, CD2, CD7, LIGHT, NKG2C, B7-H3, GITR, HVEM, and a combination thereof. In some embodiments, the chimeric receptor comprises a cytoplasmic signaling domain, which is from CD3ζ. In some embodiments, the chimeric receptor comprises a hinge domain, which is from CD8α or CD28. In some embodiments, the immune cells, the genetically engineered hematopoietic cells, or both, are allogeneic or autologous. In some aspects, the present disclosure further provides a cell population comprising one or more of the genetically engineered hematopoietic cells, or descendants thereof. In some aspects, the present disclosure further provides a pharmaceutical composition comprising the genetically engineering hematopoietic cells, or descendants thereof. Other aspects of the present disclosure relate to methods comprising administering to a subject in need thereof the genetically engineered hematopoietic cells, or descendants thereof, the cell population, or the pharmaceutical composition. In some embodiments, the methods further comprising administering to the subject a therapeutically effective amount of at least one agent that targets the first or second cell surface protein, wherein the agent comprises an antigen binding fragment that binds the first or second cell surface protein. In some embodiments, the agent is an antibody-drug conjugate or an immune effector cell expressing a chimeric antigen receptor (CAR). In some embodiments, the subject has blastic plasmacytoid dendritic cell neoplasm, multiple myeloma, myelodysplastic syndrome, Hodgkin’s lymphoma, non-Hodgkin’s lymphoma, leukemia, multiple myeloma (MM), myelodysplastic syndrome (MDS), B-cell acute lymphoblastic leukemia (B-ALL), chronic myelogenous leukemia, acute lymphoblastic leukemia, or chronic lymphoblastic leukemia or blastic plasmacytoid dendritic cell neoplasm (BPDCN). In some embodiments, the subject has a hematopoietic malignancy or a hematopoietic pre-malignant disease. In some embodiments, the hematopoietic malignancy is acute myeloid leukemia (AML). In some embodiments, the hematopoietic pre-malignancy is myelodysplastic syndrome (MDS). In some embodiments, the subject has or is at risk of leukemia relapse. Other aspects of the present disclosure relate to methods of clinically evaluating a subject. In some embodiments, the present disclosure provides methods for determining if a subject is at risk of relapse of a disease. In some embodiments, the method comprises: providing a first biological sample obtained from a subject at a first time point and a second biological sample obtained from the subject at a second time point; measuring a level of a first cell surface protein in the first biological sample and in the second biological sample; identifying the subject as at risk for relapse of the disease if the level of the first cell surface protein in the first biological sample deviates from the level of the first cell surface protein in the second biological sample. In some embodiments, the present disclosure provides methods for identifying a subject as a candidate for treatment with an agent. In some embodiments, the method comprises: providing a first biological sample obtained from a subject at a first time point and a second biological sample obtained from the subject at a second time point; and measuring a level of a first cell surface protein in the first biological sample and in the second biological sample. In some embodiments, the cell surface protein is selected from Table 3 or Table 4. In some embodiments, the first cell surface protein and the second cell surface protein are selected from the group consisting of: CD10; CD101; CD117; CD11A; CD11C; CD120B; CD123; CD13; CD133; CD152; CD200; CD205 (LY75); CD226; CD244 (SLAMF4); CD25; CD274; CD305 (LAIR1); CD32; CD33; CD34; CD366; CD38; CD41; CD42B; CD44; CD45RA; CD47; CD48; CD49D (ITGA4); CD49F; CD52; CD58; CD64; CD7; CD70; CD82; CD84; CD85; CD85K; CD86; CD9; CD96; CD99; CLL-1; EMR2 (ADGRE2); FR-B; GPR56; and IL1RAP. In some embodiments, the first cell surface protein and the second cell surface protein are selected from the group consisting of: CD85K and CD64; CD45RA and IL1RAP; CD34 and CD133; IL1RAP and CD33; CD47 and CD99; CLL-1 and CD305; CD152 and CD226; CD45RA and CD96; CD45RA and CD123; CD49D and CD305; IL1RAP and CD305; CD58 and CD305; CD49D and CD47; CD33 and CD305; CD49D and CD44; CD64 and CD86; GPR56 and CD133; CD34 and CD52; EMR2 and CD82; CD64 and CD32; CD42B and CD41; CD64 and CD11C; CD99 and CD44; CD200 and CD34; CD11C and CD33; IL1RAP and CD123; CD11C and IL1RAP; CD44 and EMR2; CD205 and CD305; CD44 and CD305; CD49D and CD99; CD274 and CD226; CD49F and GPR56; CD9 and IL1RAP; CD99 and CD305; CD10 and CD49F; CD11A and CD86; CD33 and CLL-1; CD84 and CD33; CD244 and CD117; CD85K and CD32; CD47 and EMR2; CD274 and CD152; IL1RAP and CD205; CD11C and CD86; CD99 and EMR2; CD205 and CD123; and CD123 and EMR2. In some embodiments, the first cell surface protein is CD33 and the second cell surface protein is IL1RAP. In some embodiments, the first biological sample and/or the second biological sample comprises blast cells. In some embodiments, the first time point is at or near the time the subject is diagnosed with the disease. In some embodiments, the methods further comprise administering to the subject a therapeutically effective amount of an agent comprising an antigen-binding fragment that binds to the first cell surface protein. In some embodiments, the methods further comprise administering to the subject a population of genetically engineered hematopoietic cells having reduced or eliminated expression of the first cell surface protein. The details of one or more embodiments of the invention are set forth in the description below. Other features or advantages of the present invention will be apparent from the following drawings and detailed description of several embodiments, and also from the appended claims. BRIEF DESCRIPTION OF THE DRAWINGS The following drawings form part of the present specification and are included to further demonstrate certain aspects of the present disclosure, which can be better understood by reference to one or more of these drawings in combination with the detailed description of specific embodiments presented herein. FIG.1 is a schematic showing a non-limiting example of a strategy to promote killing of cancer-specific cells. The left panel shows the traditional paradigm for drug development in which cancer antigens are targeted to kill cancer cells. The middle panel shows the problem with the traditional paradigm in which there are limited unique cancer antigens, so drugs target both cancer and health cells (on-target toxicity). The right panel shows a non- limited strategy as described herein, in which target expression is removed from healthy cells so that the killing is cancer-specific. FIG.2 shows a cartoon representation of cell surface antigen expression heterogeneity in hematopoietic cancer cells across different subjects. This patient-to-patient heterogeneity presents an obstacle to treatment and in acute myeloid leukemia can be driven by diverse mutations and cytogenetics, manifesting distinct blast immunophenotypes that are difficult to target with a single therapy. FIG.3 shows a cartoon representation of cell surface antigen expression changes that occur over time in hematopoietic cancer cells in individual subjects, such as over different clinical time points (e.g., from cancer diagnosis to cancer relapse). The plastic and evolving nature of leukemic cells within a patient after diagnosis through the growth of new clones or the rejuvenation of existing clones that had not been completely eradicated represents a further challenge in treatment. FIGs.4A and 4B show cell surface protein expression in AML patient myeloblasts across clinical timepoints. FIG.4A is a heatmap showing cell surface protein log2-fold changes between relapse and diagnosis blasts for all AML bone marrow samples. Positive values indicate the marker is upregulated in bone marrow samples from relapsed patients. Clinical annotation for each patient is displayed above the heatmap. Cell surface proteins (antigens, Ag) are categorized into three groups based on whether they are differentially expressed between timepoints in multiple, one, or no patients. FIG.4B is a heatmap showing pairwise correlation of cell surface protein marker expression in AML blast populations. Bottom and top triangles show Pearson correlation coefficients for diagnosis and relapse samples, respectively. FIGs.5A and 5B show representative results from flow cytometry analysis of cell surface antigen (CD33, CLL-1, CD123, and EMR2) expression in acute myeloid leukemia (AML) blasts. FIG.5A shows the percent (%) antigen positivity on AML blasts for each of the indicated myeloid markers by flow cytometry. FIG.5B shows the number (#) of antigens per cells (ABC) for each of the indicated myeloid markers by flow cytometry and quantification of the average number of antigens bound per blast cell (ABCs) by contact with QuantiBRITE™ beads. Each point represents a sample derived from a subject at the time of AML diagnosis or AML relapse. FIG.6 shows a schematic of a strategy for identifying myeloid-specific surface antigens for analysis in samples from subjects at diagnosis and relapse. FIG.7 shows a schematic of a strategy for characterizing cell surface antigen expression in matched samples from AML subjects at diagnosis and relapse. FIGs.8A and 8B show a representative Uniform Manifold Approximation and Projection (UMAP) plot consisting of about 400,000 cells across all acute myeloid leukemia patient samples and timepoints assessed. FIG.8A shows specific cells populations including blasts, monocytes, dendritic cells, T cells, B cells, and erythroid cell populations. FIG.8B shows surface antigen expression of the cell populations, including the blast population having high CD33, CLL-1 and CD34 surface expression and reduced CD45 expression. FIG.9 shows representative Uniform Manifold Approximation and Projection (UMAP) plot further analyzing the blast population of cells, revealing distinct cell populations across patients and between timepoints (e.g., diagnosis, at relapse). FIG.10 shows cell surface antigen density of the indicated antigens (CD33, CLL-1, CD123, and EMR2) at the timepoints of diagnosis or relapse. The antigen derived tag (ADT) counts by single cell feature barcoding technology was correlated to the antibodies bound per cell (ABC) as quantitated by QuantiBRITE™ to estimate the antigen density in each blast. Each point represents average cell surface antigen expression intensity for a sample derived from a subject at the time of AML diagnosis (circles) or AML relapse (triangles). One thousand antigens correspond to a normalized ADT value of 1.2. “R” refers to Pearson correlation coefficient. FIGs.11A-11C show estimated blast surface antigen density to identify targetable antigens across patients at the timepoints of diagnosis and relapse. FIG.11A shows representative results from an unbiased analysis of cell surface antigen expression on AML blast samples, wherein 81 antigens were ranked based on average cell surface expression across all blasts. The two heatmaps presented show the estimated antigen density across blasts in each sample obtained from a subject either at AML diagnosis (left panel) or AML relapse (right panel). Each column corresponds to an individual blast from a patient with the annotation differentially colored/shaded bars above each column indicating the sample of origin. Higher antigen density for a specific antigen in a single blast is denoted by the darkness of the vertical stripe in each point of the heatmap. The top 25 most highly expressed antigens are highlighted at the top of each heat map. The black horizontal line corresponds to an average antigen density threshold of 2000. FIG.11B shows the heatmap of FIG.11A, right panel, highlighting several patients in which expression of one antigen may compensate for low expression of another antigen (e.g., CLL-1 expression compensating for low CD33 expression), suggesting these patients may benefit from a multi-targeting therapeutic approach. FIG.11C shows the heatmap of FIG.11A, right panel, highlighting CD33 and CLL-1 antigen density in blasts from patient 70, demonstrating variable expression across individual blasts, which suggests that targeting one of the antigens may not completely eradicate the blast population. FIGs.12A and 12B show representative results obtained from computational analyses used to correlate pairs of cell surface antigens that exhibited co-expression on AML blast cells from patient 70. FIG.12A shows a heat map representation of the correlation between expression of pairs of cell surface antigens on AML blasts. FIG.12B shows a histogram representation indicating the percent of blasts exhibiting the indicated level of estimates antigen density for co-expression of CD33 and CLL-1 or expression of CD33 or CLL-1 individually, suggesting that a multi-targeting therapeutic strategy, such as with multi- specific CAR T cells, may enhance blast killing and guard against antigen escape. FIGs.13A-13E show further analyses of AML blasts from patient 26 that exhibited increased CD33 expression. FIG.13A shows Uniform Manifold Approximation and Projection (UMAP) plots of blasts at AML diagnosis and AML relapse. FIG.13B shows a heat map representation of differential antigen expression data obtained from analyses of AML blasts at diagnosis. FIG.13B shows a heat map representation of differential antigen expression data obtained from analyses of AML blasts at relapse, indicating upregulation of CD33. FIG.13D shows cell cycle module analysis indicating the cell cycle stage distribution of AML blasts at AML diagnosis and AML relapse. FIG.13E shows a plot presenting differentially expressed genes (log, fold change >2) in blasts at AML diagnosis compared to AML relapse. FIGs.14A and 14B show results of pathway enrichment analysis in a subset of proliferating blasts to identify pathways that change between AML diagnosis and relapse. FIG.14A shows exemplary pathways that were found to be downregulated in relapse S/G2M blasts. FIG.14B shows exemplary pathways that were found to be upregulated in relapse S/G2M blasts. FIGs.15A-15B show surface density of the indicated antigens expressed on AML blasts and leukemia stem cells. FIG.15A shows a heat-map representation of data from cellular indexing of transcriptomes and epitopes (CITE-seq) profiling of 81 candidate antigens across 400,000 cells from 28 paired diagnosis and relapse patient bone marrow samples. FIG.15B shows percent positivity and number of antigens per antigen-positive cell in blasts and leukemia stem cells. Quantibrite PE beads were used for antigen quantitation. The dots trace expression data for select patients to demonstrate expression pattern for CD33, EMR2 (ADGRE2), CD123, and CLL-1. Data shown as median ± standard error of mean. Leukemic stem cells (LSCs), Diagnosis (Dx), Relapse (Rel). FIGs.16A-16D show multiplex adenosine base editing of EMR2 (ADGRE2) and CD33 in HSPCs. FIG.16A shows diagrams of the structures of the EMR2 and CD33 genes indicating the adenosine base editing (ABE) gRNA locations. FIG.16B shows an example of strategy for multiple ABE of HSCs and characterizing effects of multiplex editing on CD33 and EMR2. FIG.16C shows the frequency of editing types at day 2 (D2) and day 5 (D5) after electroporation of HSPCs with ABE guides. FIG.16D shows percentage of the ADGRE2 and CD33 surface protein expression of ADGRE2 and CD33 edited cells, in single and multiplex ABE settings, at day 5 (D5). N=2 donors. Data shown as mean ± standard deviation. “dKO” refers to double knockout of EMR2 and CD33 genes. FIG.17 shows results obtained from flow cytometry analysis of cells that were subjected to base editing conditions that utilized a non-targeting control gRNA (gCTRL) or gRNAs targeting the CD33, CD123, and EMR2 genes (triplex). At 16 days (D16) post- electroporation under base editing conditions, cell surface expression of CD33, CD123, EMR2, and CLL-1 was analyzed. Each fraction of the left (gCTRL) and right (triplex) bars represent the number of cells in the population expressing the corresponding combination of antigens shown in the key below the histogram. FIGs.18A-18B show estimated blast surface density of targetable antigens in patients at diagnosis (FIG.18A) and relapse (FIG.18B). FIGs.18A-18B shows representative results from an unbiased analysis of cell surface antigen expression on AML blast samples, wherein 81 antigens were ranked based on average cell surface expression across all blasts. The two heatmaps presented show the estimated antigen density across blasts in each sample obtained from a subject either at AML diagnosis (FIG.18A) or AML relapse (FIG.18B). Each column corresponds to an individual blast from a patient with the annotation differentially colored/shaded bars above each column indicating the sample of origin. Higher antigen density for a specific antigen in a single blast is denoted by the darkness of the vertical stripe in each point of the heatmap. The top 25 most highly expressed antigens are highlighted at the top of each heat map. The black horizontal line corresponds to an average antigen density threshold of 2000. FIG.18B shows the heatmap of FIG.18A, highlighting four additional targets (CD305 (LAIR1), CD49d (ITGA4), CD205 (LY75), and CD244 (SLAMF4)) which are highly expressed on AML blast cells and may be able to compensate for low expression of other antigens in a multi-targeting therapeutic approach. These four antigens were selected out of the top ranked antigens due to their high expression on AML blasts in addition to minimal expression on non-heme healthy tissues according to public databases Gtex and Tabula Sapiens (https://gtexportal.org/home/) (https://tabula-sapiens- portal.ds.czbiohub.org/) which would enable these antigens to be targets in an edited HSC transplant paired with single or multi-targeting therapeutics. FIGs.19A and 19B show representative results from an unbiased analysis of cell surface antigen expression on 56 AML paired blast samples (28 at diagnosis and 28 at relapse), wherein 81 antigens were ranked based on average cell surface expression across all blasts. For FIG 19A, once ranked, each antigen was evaluated for how many patients would be covered at different thresholds. FIG.19A shows a threshold of greater than or equal to 80% positive expression on AML blast cells (coverage) at 500 plus antigens per cell. The number of patients at diagnosis (out of 28 samples), relapse (out of 28 samples), and total combined (Out of 56 samples) are shown. FIG.19A shows that the four antigens (CD305 (LAIR1), CD49d (ITGA4), CD205 (LY75), and CD244 (SLAMF4)) represent targets for therapeutics since they are highly expressed in a large number of patient samples. For FIG 19B, once ranked, each antigen was evaluated in pairs in addition to single antigens for the number of patient samples that have blast coverage at different antigen thresholds when 2 antigens are combined. FIG 19B shows a threshold of greater than or equal to 90% positive expression on AML blast cells (coverage), greater than or equal to 50% co-expression (Two antigens expressed on the same cell), and at 500 plus antigens per cell. The number of patients at diagnosis (out of 28 samples), relapse (out of 28 samples), and total combined (out of 56 samples) are shown for each pair. FIG 19B shows evidence that these six antigen pairs (CD305 & CD49d; CD305 & CD205; CD305 & CD244; CD49d & CD205; CD49d & CD244; CD205 & CD244) would be effective for multi-targeted therapeutics because when combined, they are highly expressed in a large number of patient samples. FIGs.20A-20B show results from analyses of antigen density-dependent cytotoxicity effects of an antibody-drug conjugate. FIG.20A shows CD305 (LAIR1), CD49D (ITGA4), CD205 (LY75), and CD244 (SLAMF4) antigen density in MOLM13 AML cells (shows expression levels as antigens bound per cell on MOLM13 AML cell line at complete saturation and shows a range of antigen densities on MOLM13 cells from 3800-34000 antigens bound per cell). FIG.20B shows cytotoxicity analyses of cells contacted with a primary antibody targeting CD33, CD305 (LAIR1), CD49d (ITGA4), CD205 (LY75), or CD244 (SLAMF4) and then subsequently contacted with a secondary antibody-drug conjugate (Fab-aMFc-CL-MMAF). FIG.20B shows the therapeutic efficacy of targeting CD33, CD305 (LAIR1), CD49d (ITGA4), CD205 (LY75), or CD244 (SLAMF4) antigens individually using in vitro cytotoxicity secondary ADC assays in MOLM-13 cells, providing a measure of sensitivity to the ADC as a function of Antigen expression. Greater than 50% cell killing was observed at <0.1nM of primary antibody for all four Antigens (CD305, CD49d, CD205, and CD244). Secondary ADCs function by binding a primary antibody to an antigen of choice and then binding a secondary IgG1 antibody which is attached to a payload, in this case Fab-aMFc-CL-MMAF. Once the antibody is internalized by the cell, the payload kills the cell by inhibiting cell division by blocking the polymerization of tubulin (https://www.moradec.com/product_info/Fab-aMFc-CL-MMAF.pdf). Antigen density dependent cytotoxicity was observed which further provides evidence that higher antigen densities allow for more effective targeted cytotoxicity in vitro. FIGs.21A-21B show results from flow cytometry analyses of CD33, CD305 (LAIR1), CD49D (ITGA4), CD205 (LY75), and CD244 (SLAMF4) expression on four patient samples. QuantiBRITE assay was performed on four AML patient samples from the AML Atlas repository, using flow cytometry to assess percent positivity and antigen density (Antigens bound per cell). FIG.21A shows overall high expression of at least 3 of 4 antigens (CD305 (LAIR1), CD49d (ITGA4), CD205 (LY75)) in all four patient samples and are shown in comparison with the validated antigen, CD33. FIG.21B shows antigen density values for each patient for all five of the same antigens (CD33, CD305 (LAIR1), CD49D (ITGA4), CD205 (LY75), and CD244 (SLAMF4)). This figure tells us that each of the four antigens (CD305, CD49D, CD205, and CD244) have high expression levels and antigen density on a subset of AML patient samples. FIG.22 shows antigen density (antigens per cell) assessed using the QuantiBRITE assay for five antigens (CD33, CD305, CD49d, CD205, and CD244) on four patient samples which had been previously run through CITE-seq.56 samples were previously sequenced using the CITE-seq method which provided ADT (Antibody derived tags) values for each of the 81 antigens in the panel, and at the same time four antigens were assessed for antigens per cell values using the flow cytometric assay, QuantiBRITE. These four antigens were used to make a model which correlated antigens per cell with ADT to predict antigens per cell values for the other 77 antigens in the panel. Four of the top ranked antigens (CD305, CD49d, CD205, and CD244) were evaluated along with a previously validated antigen (CD33) using the QuantiBRITE assay on four patient samples which had been previously run in the AML Atlas. From this assay, antigens per cell values were acquired for these 5 antigens (light gray) and compared these with the predicted CITE-seq antigens per cell values (dark gray). A high consistency was observed between the measured antigens per cell readout and the predicted antigens per cell for all four antigens and samples (Pearson r = 0.87, p = 9.9e-6). This provides evidence for the accuracy of the CITE-seq prediction model. DETAILED DESCRIPTION OF THE INVENTION Successfully identifying suitable proteins for targeted cancer therapies presents a significant challenge. Many potential target proteins are present on both the cell surface of a cancer cell and on the cell surface of normal, non-cancer cells, which may be required or critically involved in the development and/or survival of the subject. Many of the target proteins contribute to the functionality of such essential cells. Thus, therapies targeting these proteins may lead to deleterious effects in the subject, such as significant toxicity and/or other side effects (see, e.g., FIG.1, left panel and center panel). The profiles of cell surface antigen expression can also vary from subject to subject (see, FIG.2). Further, resistance to CAR-T therapy remains a challenge in treatment of hematopoietic malignance, such as acute myeloid leukemia (AML) due to switch of cancer cell surface proteins on cancer cells, thereby escaping CAR-T therapy (see, e.g., FIG.3). For example, patients having B-cell acute lymphoblastic leukemia (B-ALL) were found to develop acute myeloid leukemia (AML) with CD19- cancer cells after CAR-T therapy. Thus, characterizing shifts in antigen expression during cancer progression (e.g., from cancer diagnosis to cancer relapse) can be used to identify correlations in cell surface antigen expression for targeting cancer cells and enhancing eradication of blasts. The present disclosure provides methods, cells, compositions, and kits aimed at addressing at least the above-stated problems. The methods, cells, compositions, and kits described herein provide a safe and effective treatment for hematological malignancies, allowing for targeting of one or more cell surface proteins that are present not only on cancer cells but also on cells critical for the development and/or survival of the subject (see, e.g., FIG.1, right panel). More specifically, hematopoietic cells as described herein can be used (for example) in the treatment of a subject that receives two or more different therapies for cancer. Many therapies deplete the subject’s endogenous, non-cancerous hematopoietic cells. Replacement or rescue hematopoietic cells described herein can replace the subject’s depleted immune cells. First, in some cases, a subject receives the first therapy (e.g., against IL1RAP), and then the cancer relapses, and then the subject receives the second therapy (e.g., against CD33). The present application provides, e.g., rescue cells that are resistant to both therapies. Thus, the rescue cells can be administered to the subject at or near the time of the first therapy, and if relapse occurs, the subject can then receive the second therapy without depleting the rescue cells. In some embodiments, the first and second therapies targets antigens selected from the group consisting of: CD10; CD101; CD117; CD11A; CD11C; CD120B; CD123; CD13; CD133; CD152; CD200; CD205 (LY75); CD226; CD244 (SLAMF4); CD25; CD274; CD305 (LAIR1); CD32; CD33; CD34; CD366; CD38; CD41; CD42B; CD44; CD45RA; CD47; CD48; CD49D (ITGA4); CD49F; CD52; CD58; CD64; CD7; CD70; CD82; CD84; CD85; CD85K; CD86; CD9; CD96; CD99; CLL-1; EMR2 (ADGRE2); FR-B; GPR56; and IL1RAP. In some embodiments, the first therapy targets an antigen selected from the first column in Table 3 or Table 4 and the second therapy targets an antigen selected from the second column Table 3 or Table 4 (e.g., wherein the antigens are selected from the same row in the same Table or from different tables). In some embodiments, one of the therapies targets CD33. In some embodiments, one of the therapies targets CD123. In some embodiments, one of the therapies targets CLL-1. In some embodiments, one of the therapies targets CD38. In some embodiments, one of the therapies targets CD34. In some embodiments, one of the therapies targets EMR2. In some embodiments, one of the therapies targets CD205 (LY75). In some embodiments, one of the therapies targets CD244 (SLAMF4). In some embodiments, one of the therapies targets CD49D (ITGA4). In some embodiments, one of the therapies targets CD305 (LAIR1). In some embodiments, one of the therapies targets CD305 (LAIR1) and one of therapies targets CD49D (ITGA4). In some embodiments, one of the therapies targets CD305 (LAIR1) and one of therapies targets CD205 (LY75). In some embodiments, one of the therapies targets CD305 (LAIR1) and one of therapies targets CD244 (SLAMF4). In some embodiments, one of the therapies targets CD49D (ITGA4) and one of therapies targets CD205 (LY75). In some embodiments, one of the therapies targets CD49D (ITGA4) and one of therapies targets CD244 (SLAMF4). In some embodiments, one of the therapies targets CD205 (LY75) and one of therapies targets CD244 (SLAMF4). Second, in some cases, a subject may need to receive two therapies at once, e.g., because the cancer comprises two sub-populations of cells (e.g., one expressing CD33 and the second expressing CD123 and/or CLL-1), and each therapy only attacks one of the sub- populations. As described herein, rescue cells resistant to both therapies can replace the subject’s depleted immune cells even in the presence of both therapies. In some embodiments, the cells, which are resistant to both therapies, are genetically engineered in a gene encoding a first cell surface protein and a gene encoding a second cell surface protein. In some embodiments, the first and second cell surface proteins are selected from the group consisting of: CD10; CD101; CD117; CD11A; CD11C; CD120B; CD123; CD13; CD133; CD152; CD200; CD205 (LY75); CD226; CD244 (SLAMF4); CD25; CD274; CD305 (LAIR1); CD32; CD33; CD34; CD366; CD38; CD41; CD42B; CD44; CD45RA; CD47; CD48; CD49D (ITGA4); CD49F; CD52; CD58; CD64; CD7; CD70; CD82; CD84; CD85; CD85K; CD86; CD9; CD96; CD99; CLL-1; EMR2 (ADGRE2); FR-B; GPR56; and IL1RAP). In some embodiments, the cells have reduced or eliminated expression of the first cell surface protein relative to a wildtype counterpart cell. In some embodiments, the cells have reduced or eliminated expression of the second cell surface protein relative to a wildtype counterpart cell. In some embodiments, the cells express a mutant of the first cell surface protein (e.g., a mutant that does not bind to a first therapy, such as a first cytotoxic agent). In some embodiments, the cells express a mutant of the second cell surface protein (e.g., a mutant that does not bind to a second therapy, such as a second cytotoxic agent). In some embodiments, the first cell surface protein is selected from the first column in Table 3 or Table 4 and the second column Table 3 or Table 4 (e.g., wherein the antigens are selected from the same row in the same Table or from different tables). In some embodiments, one of the cell surface proteins is CD33. In some embodiments, one of the cell surface proteins is CD123. In some embodiments, one of the cell surface proteins is CLL-1. In some embodiments, one of the cell surface proteins is CD38. In some embodiments, one of the cell surface proteins is CD34. In some embodiments, one of the cell surface proteins is EMR2.. In some embodiments, one of the cell surface proteins is CD205 (LY75). In some embodiments, one of the cell surface proteins is CD244 (SLAMF4). In some embodiments, one of the cell surface proteins is CD49D (ITGA4). In some embodiments, one of the cell surface proteins is CD305 (LAIR1). In some embodiments, one of the cell surface proteins is CD305 (LAIR1) and one of the cell surface proteins is CD49D (ITGA4). In some embodiments, one of the cell surface proteins is CD305 (LAIR1) and one of the cell surface proteins is CD205 (LY75). In some embodiments, one of the cell surface proteins is CD305 (LAIR1) and one of the cell surface proteins is CD244 (SLAMF4). In some embodiments, one of the cell surface proteins is CD49D (ITGA4) and one of the cell surface proteins is CD205 (LY75). In some embodiments, one of the cell surface proteins is CD49D (ITGA4) and one of the cell surface proteins is CD244 (SLAMF4). In some embodiments, one of the cell surface proteins is CD205 (LY75) and one of the cell surface proteins is CD244 (SLAMF4).The present disclosure also provides a population of rescue hematopoietic cells that comprises a first sub-population of cells that is (and/or gives rise to) cells resistant to a first therapy and a second sub-population of cells that is (and/or gives rise to) cells resistant to a second therapy. Optionally, the population can comprise cells that are (and/or give rise to) cells resistant to both therapies; however this is not required in this embodiment. The cell populations can be useful, e.g., when subjects are treated with two therapies sequentially. For instance, in some embodiments, the edited cell surface proteins are proteins that are typically not expressed in normal HSCs, but become expressed in later lineages, so the transplanted HSCs are resistant to both therapies regardless of whether any HSCs are edited for both cell surface proteins. Accordingly, described herein are genetically engineered hematopoietic cells such as hematopoietic stem cells (HSCs) having genetic editing in genes coding for cell surface proteins (e.g., CD33 and IL1RAP); methods of producing such, for examples, via the CRISPR approach using specific guide RNAs (gRNAs); and methods of treating a hematopoietic malignancy using the engineered hematopoietic cells, either taken alone, or in combination with one or more cytotoxic agents (e.g., CAR-T cells) that can target the wild- type cell surface proteins but not those encoded by the edited genes in the engineered hematopoietic cells. Also described herein are methods of determining whether a subject is at risk for relapse of a disease (e.g., hematopoietic malignancy) and methods of identifying a subject as a candidate for treatment with an agent, based on the level(s) of one or more cell surface proteins in a biological sample(s). I. Genetically Engineered Hematopoietic Cells The present disclosure provides genetically engineered hematopoietic cells, such as hematopoietic stem cells that carry genetically edited genes for reducing or eliminating expression of one or more cell surface proteins, or for expressing the one or more cell surface proteins in mutated form. The mutated cell surface proteins would retain at least partial bioactivity of the antigens but can escape targeting by cytotoxic agents such as CAR-T cells specific to the antigens. In some embodiments, the cell surface proteins of interest may not be expressed on hematopoietic cells, such as HSCs, in nature. However, such antigens may be expressed on cells differentiated from the HSCs (e.g., descendants thereof). “Expressing a lineage-specific cell surface protein” or “expressing a lineage-specific cell surface antigen” means that at least a portion of the lineage-specific cell surface protein, or antigen thereof, can be detected on the surface of the hematopoietic cells or descendants thereof. As used herein, “descendants” of hematopoietic cells include any cell type or lineage of cells that arise from the hematopoietic cells. In some embodiments, the descendants of the hematopoietic cells are a cell type or lineage of cells that have differentiated from the hematopoietic cells. The genetically engineered hematopoietic cells may be used alone for treating hematopoietic malignancies, or in combination with one or more cytotoxic agents that target the wild-type cell surface proteins but not the mutant encoded by the edited genes in the genetically engineered hematopoietic cells. Such hematopoietic cells, upon differentiation, could compensate the loss of function caused by elimination of functional non-cancerous cells due to immunotherapy that targets cell surface protein(s), which may also be expressed on normal cells. This approach would also broaden the choice of target proteins for immunotherapy such as CART therapy. Cell Surface Proteins As used herein, the terms “protein,” “peptide,” and “polypeptide” may be used interchangeably and refer to a polymer of amino acid residues linked together by peptide bonds. In general, a protein may be naturally occurring, recombinant, synthetic, or any combination of these. Also within the scope of the term are variant proteins, which comprise a mutation (e.g., substitution, insertion, or deletion) of one or more amino acid residues relative to the unmodified, e.g., wild-type, counterpart. As used herein, the term “cell surface protein” refers to a protein, at least a portion of which is present on the extracellular surface of a cell. In some embodiments, a “cell surface protein” may be a lineage-specific cell surface protein. A cell surface protein may be displayed on the surface of a cell such that it is capable of being bound by another agent, such as a ligand or an antibody. As used herein, the terms “lineage-specific cell surface protein,” “linage-specific cell surface antigen,” and “cell surface lineage-specific protein” may be used interchangeably and refer to any protein that is sufficiently present on the surface of a cell and is associated with one or more populations of cell lineage(s). For example, the protein may be present on one or more populations of cell lineage(s) and absent (or at reduced levels) on the cell surface of other cell populations. In some embodiments, the terms “lineage-specific cell surface protein” and “cell surface lineage-specific antigen” maybe used interchangeably and refer to any antigen of a lineage-specific cell surface protein. The compositions and methods described herein may be used to achieve editing of a cell surface protein, which may result in making a variant lineage-specific cell surface protein. Such editing may result in an amino acid substitution in an epitope of interest, such as a cell surface protein epitope. In general, lineage-specific cell surface proteins can be classified based on a number of factors, such as whether the protein and/or the populations of cells that present the protein are required for survival and/or development of the host organism. A summary of exemplary types of lineage-specific proteins is provide in Table 1 below. Table 1: Classification of Lineage Specific Proteins
 As shown in Table 1, type 0 lineage-specific cell surface proteins are necessary for the tissue homeostasis and survival, and cell types carrying type 0 lineage-specific cell surface protein may be also necessary for survival of the subject. Thus, given the importance of type 0 lineage-specific cell surface proteins, or cells carrying type 0 lineage-specific cell surface proteins, in homeostasis and survival, targeting this category of proteins may be challenging using conventional CAR T cell immunotherapies, as the inhibition or removal of such proteins and cell carrying such proteins may be detrimental to the survival of the subject. Consequently, lineage-specific cell surface proteins (such as type 0 lineage-specific proteins) and/or the cell types that carry such proteins may be required for the survival, for example because it performs a vital non-redundant function in the subject, then this type of lineage specific protein may be a poor target for conventional CAR T cell-based immunotherapies. However, by combining the genetically engineered hematopoietic stem cells described herein and a cytotoxic agent such as CAR-T cell-based therapy, the selection of target cell surface protein can be expanded to essential antigens such as type 0 lineage- specific cell surface proteins. In some embodiments, the engineered hematopoietic cells (e.g., HSCs) have one or more genes of type 0 antigens edited for expression of these type 0 antigens in mutated form, which retain (at least partially) bioactivity of the type 0 antigens but can escape targeting by type 0 antigen-specific cytotoxic agents such as CAR-T cells so as to remedy the loss of normal cells expressing the type 0 antigens due to the therapy. In contrast to type 0 proteins, type 1 lineage-specific cell surface proteins and cells carrying type 1 lineage-specific cell surface proteins are not required for tissue homeostasis or survival of the subject. Targeting type 1 lineage-specific cell surface proteins is not likely to lead to acute toxicity and/or death of the subject. For example, as described in Elkins et al. (Mol. Cancer Ther. (2012) 10:2222-32) a CAR T cell engineered to target CD307, a type 1 protein expressed uniquely on both normal plasma cells and multiple myeloma (MM) cells would lead to elimination of both cell types. However, since the plasma cell lineage is expendable for the survival of the organism, CD307 and other type 1 lineage specific proteins are proteins that are suitable for CAR T cell-based immunotherapy. Lineage specific proteins of type 1 class may be expressed in a wide variety of different tissues, including, ovaries, testes, prostate, breast, endometrium, and pancreas. In some embodiments, the genetically engineered hematopoietic cells (e.g., HSCs) have one or more genes of type 1 antigens for expression of the type 1 proteins in mutated forms, which retain (at least partially) bioactivity of the type 1 antigens but can escape targeting by type 1 antigen-specific cytotoxic agents such as CAR-T cells. Use of such engineered HSCs (either alone or in combination with cytotoxic agents such as CAR-T cells targeting the type 1 antigens) may improve the longer-term survival and quality of life of the patient. For example, targeting all plasma cells, while not expected to lead to acute toxicity and/or death, could have longer-term consequences such as reduced function of the humoral immune system leading to increased risk of infection. Targeting type 2 proteins presents a significant difficulty as compared to type 1 proteins. Type 2 proteins are those characterized where: (1) the protein is dispensable for the survival of an organism (i.e., is not required for the survival), and (2) the cell lineage carrying the protein is indispensable for the survival of an organism (i.e., the particular cell lineage is required for the survival). For example, CD33 is a type 2 protein expressed in both normal myeloid cells as well as in Acute Myeloid Leukemia (AML) cells (Dohner et al., NEJM (2015) 373:1136). As a result, a CAR T cell engineered to target CD33 protein could lead to the killing of both normal myeloid cells as well as AML cells, which may be incompatible with survival of the subject. In some embodiments, the genetically engineered hematopoietic cells (e.g., HSCs) have one or more genes of type 2 antigens for expression of the type 2 antigens in mutated form, which retain (at least partially) bioactivity of the type 2 antigens but can escape targeting by type 1 antigen-specific cytotoxic agents such as CAR-T cells. Use of such engineered HSCs (either alone or in combination with cytotoxic agents, such as CAR-T cells targeting the type 2 antigens) may improve the longer-term survival and quality of life of the patient. For example, targeting all plasma cells, while not expected to lead to acute toxicity and/or death, could have longer-term consequences such as reduced function of the humoral immune system leading to increased risk of infection. In some embodiments, malignant hematopoietic cells may have altered expression or express a mutant of a cell surface protein. In some embodiments malignant hematopoietic cells may have altered expression or express a mutant of cell surface protein selected from a group consisting of: CD10; CD101; CD11A; CD11C; CD123; CD133; CD152; CD200; CD205; CD244; CD25; CD305; CD33; CD34; CD366; CD38; CD42B; CD44; CD45RA; CD47; CD48; CD49D; CD49F; CD52; CD58; CD64; CD7; CD84; CD85K; CD9; CD99; CLL-1; EMR2; GPR56; IL1RAP; and CD274 In some embodiments, malignant hematopoietic cells may have altered expression or express a mutant of cell surface protein selected from a group consisting of: CD117; CD11A; CD11C; CD120B; CD123; CD13; CD133; CD152; CD205; CD226; CD244; CD25; CD305; CD32; CD33; CD34; CD38; CD41; CD44; CD45RA; CD47; CD49D; CD49F; CD52; CD58; CD64; CD7; CD70; CD82; CD86; CD96; CD99; CLL-1; EMR2; FR-B; GPR56; and IL1RAP. In some embodiments, malignant hematopoietic cells may have altered expression or express of a first cell surface protein and a second cell surface protein selected from a group consisting of: CD274 and CD152; CD274 and CD226; CD152 and CD226; CD10 and CD49F; CD49F and GPR56; CD200 and CD34; CD34 and CD52; CD34 and CD133; GPR56 and CD133; CD42B and CD41; CD85K and CD64; CD85K and CD32; CD64 and CD32; CD64 and CD11C; CD64 and CD86; CD45RA and CD96; CD45RA and IL1RAP; CD45RA and CD123; CD84 and CD33; CD11C and IL1RAP; CD11C and CD33; CD11C and CD86; CD9 and IL1RAP; IL1RAP and CD33; IL1RAP and CD205; IL1RAP and CD305; IL1RAP and CD123; CD33 and CLL-1; CD33 and CD305; CD11A and CD86; CLL-1 and CD305; CD58 and CD305; CD49D and CD47; CD49D and CD99; CD49D and CD44; CD49D and CD305; CD47 and CD99; CD47 and EMR2; CD99 and CD44; CD99 and CD305; CD99 and EMR2; CD205 and CD305; CD205 and CD123; CD44 and CD305; CD44 and EMR2; CD244 and CD117; CD123 and EMR2; EMR2 and CD82; CD152 and CD120B; CD48 and CD11A; CD200 and CD25; CD34 and CD7; CD34 and CD13; CD52 and GPR56; CD52and CD133; CD52 and CD7; GPR56 and CD7; GPR56 and CD82; CD133 and CD7; CD133 and CD82; CD7 and CD13; CD101 and CD64; CD85 and CD70; CD85K and CD86; CD64 and FR-B; CD366 and CD96; CD25 and CD45RA; CD25 and CD96; CD9 and CD82; IL1RAP and CD58; CD11A and CD38; CD11A and CD44; CD11A and CD244; CD38 and CD305; CD38 and EMR2; CLL-1 and CD49D; CD58 and CD49D; CD47 and CD205; and CD47 and CD44. In some embodiments, malignant hematopoietic cells may have altered expression or express of a first cell surface protein and a second cell surface protein selected from a group consisting of: CD85K and CD64; CD45RA and IL1RAP; CD34 and CD133; IL1RAP and CD33; CD47 and CD99; CLL-1 and CD305; CD152 and CD226; CD45RA and CD96; CD45RA and CD123; CD49D and CD305; IL1RAP and CD305; CD58 and CD305; CD49D and CD47; CD33 and CD305; CD49D and CD44; CD64 and CD86; GPR56 and CD133; CD34 and CD52; EMR2 and CD82; CD64 and CD32; CD42B and CD41; CD64 and CD11C; CD99 and CD44; CD200 and CD34; CD11C and CD33; IL1RAP and CD123; CD11C and IL1RAP; CD44 and EMR2; CD205 and CD305; CD44 and CD305; CD49D and CD99; CD274 and CD226; CD49F and GPR56; CD9 and IL1RAP; CD99 and CD305; CD10 and CD49F; CD11A and CD86; CD33 and CLL-1; CD84 and CD33; CD244 and CD117; CD85K and CD32; CD47 and EMR2; CD274 and CD152; IL1RAP and CD205; CD11C and CD86; CD99 and EMR2; CD205 and CD123; and CD123 and EMR2. In some embodiments, malignant hematopoietic cells may have altered expression or express of a first cell surface protein and a second cell surface protein selected from a group consisting of: CD33 and CD205 (LY75); CD33 and CD244 (SLAMF4); CD33 and CD305 (LAIR1); CD33 and CD49D (ITGA4); CLL-1 and CD205 (LY75); CLL-1 and CD244 (SLAMF4); CLL-1 and CD305 (LAIR1); CLL-1 and CD49D (ITGA4); EMR2 and CD205 (LY75); EMR2 and CD244 (SLAMF4); EMR2 and CD305 (LAIR1); EMR2 and CD49D (ITGA4); CD38 and CD205 (LY75); CD38 and CD244 (SLAMF4); CD38 and CD305 (LAIR1); CD38 and CD49D (ITGA4); CD305 (LAIR1) and CD49D (ITGA4); CD305 (LAIR1) and CD205 (LY75); CD305 (LAIR1) and CD244 (SLAMF4); CD49D (ITGA4) and CD205 (LY75); CD49D (ITGA4) and CD244 (SLAMF4); and CD205 (LY75) and CD244 (SLAMF4). Mutated Genes Encoding Cell Surface Proteins In some embodiments, the hematopoietic cells (HSCs) described herein may contain an edited gene encoding one or more cell surface proteins of interest in mutated form (mutants or variants, which are used herein interchangeably), which has reduced binding or no binding to a cytotoxic agent as described herein. The variants may lack the epitope to which the cytotoxic agent binds. Alternatively, the mutants may carry one or more mutations of the epitope to which the cytotoxic agent binds, such that binding to the cytotoxic agent is reduced or abolished as compared to the natural or wild-type cell surface protein counterpart. Such a variant is preferred to maintain substantially similar biological activity as the wild- type counterpart. A cell that is “negative” for a given cell surface protein has a substantially reduced expression level of the cell surface protein as compared with its naturally-occurring counterpart (e.g., otherwise similar, unmodified cells), e.g., not detectable or not distinguishable from background levels, e.g., using a flow cytometry assay. In some instances, a cell that is negative for the cell surface protein has a level of less than 10%, 5%, 2%, or 1% of as compared with its naturally-occurring counterpart. The variant may share a sequence homology of at least 80% (e.g., 85%, 90%, 95%, 97%, 98%, 99%, or above) as the wild-type counterpart and, in some embodiments, may contain no other mutations in addition to those for mutating or deleting the epitope of interest. The “percent identity” of two amino acid sequences is determined using the algorithm of Karlin and Altschul Proc. Natl. Acad. Sci. USA 87:2264-68, 1990, modified as in Karlin and Altschul Proc. Natl. Acad. Sci. USA 90:5873-77, 1993. Such an algorithm is incorporated into the NBLAST and XBLAST programs (version 2.0) of Altschul, et al. J. Mol. Biol.215:403-10, 1990. BLAST protein searches can be performed with the XBLAST program, score=50, wordlength=3 to obtain amino acid sequences homologous to the protein molecules of the invention. Where gaps exist between two sequences, Gapped BLAST can be utilized as described in Altschul et al., Nucleic Acids Res.25(17):3389-3402, 1997. When utilizing BLAST and Gapped BLAST programs, the default parameters of the respective programs (e.g., XBLAST and NBLAST) can be used. In some instances, the variant contains one or more amino acid residue substitutions (e.g., 2, 3, 4, 5, or more) within the epitope of interest such that the cytotoxic agent does not bind or has reduced binding to the mutated epitope. Such a variant may have substantially reduced binding affinity to the cytotoxic agent (e.g., having a binding affinity that is at least 40%, 50%, 60%, 70%, 80% or 90% lower than its wild-type counterpart). In some examples, such a variant may have abolished binding activity to the cytotoxic agent. In other instances, the variant contains a deletion of a region that comprises the epitope of interest. Such a region may be encoded by an exon. In some embodiments, the region is a domain of the cell surface protein of interest that encodes the epitope. In one example, the variant has just the epitope deleted. The length of the deleted region may range from 3-60 amino acids, e.g., 5- 50, 5-40, 10-30, 10-20, etc. The mutation(s) or deletions in a mutant of a cell surface protein may be within or surround a non-essential epitope such that the mutation(s) or deletion(s) do not substantially affect the bioactivity of the protein. As used herein, the term “epitope” refers to an amino acid sequence (linear or conformational) of a protein, such as a cell surface protein, that is bound by the CDRs of an antibody. In some embodiments, the cytotoxic agent binds to one or more (e.g., at least 2, 3, 4, 5 or more) epitopes of a cell surface proteins. In some embodiments, the cytotoxic agent binds to more than one epitope of the cell surface protein and the hematopoietic cells are manipulated such that each of the epitopes is absent and/or unavailable for binding by the cytotoxic agent. In some embodiments, the genetically engineered HSCs described herein have one or more edited genes of cell surface proteins such that the edited genes express mutated cell surface proteins with mutations in one or more non-essential epitopes. A non-essential epitope (or a fragment comprising such) refers to a domain within the cell surface protein, the mutation in which (e.g., deletion) is less likely to substantially affect the bioactivity of the cell surface protein and thus the bioactivity of the cells expressing such. For example, when hematopoietic cells comprising a deletion or mutation of a non-essential epitope of a cell surface protein, such hematopoietic cells are able to proliferate and/or undergo erythropoietic differentiation to a similar level as hematopoietic cells that express a wild-type cell surface protein. Non-essential epitopes of a cell surface protein can be identified by the methods described herein or by conventional methods relating to protein structure-function prediction. For example, a non-essential epitope of a protein can be predicted based on comparing the amino acid sequence of a protein from one species with the sequence of the protein from other species. Non-conserved domains are usually not essential to the functionality of the protein. As will be evident to one of ordinary skill in the art, non-essential epitope of a protein is predicted using an algorithm or software, such as the PROVEAN software (see, e.g., see: provean.jcvi.org; Choi et al. PLoS ONE (2012) 7(10): e46688), to predict potential non-essential epitopes in a cell surface protein of interest (“candidate non-essential epitope”). Mutations, including substitution and/or deletion, many be made in any one or more amino acid residues of a candidate non-essential epitope using convention nucleic acid modification technologies. The protein variants thus prepared may be introduced into a suitable type of cells, such as hematopoietic cells, and the functionality of the protein variant can be investigated to confirm that the candidate non-essential epitope is indeed a non-essential epitope. Alternatively, a non-essential epitope of a cell surface protein may be identified by introducing a mutation into a candidate region in a cell surface protein of interest in a suitable type of host cells (e.g., hematopoietic cells) and examining the functionality of the mutated cell surface protein in the host cells. If the mutated cell surface protein maintains substantially the biological activity of the native counterpart, this indicates that the region where the mutation is introduced is non-essential to the function of the cell surface protein. In some embodiments, genetically engineered cells of the present disclosure comprise a mutation in a gene encoding a cell surface protein selected from a group consisting of: CD10; CD101; CD117; CD11A; CD11C; CD120B; CD123; CD13; CD133; CD152; CD200; CD205 (LY75); CD226; CD244 (SLAMF4); CD25; CD274; CD305 (LAIR1); CD32; CD33; CD34; CD366; CD38; CD41; CD42B; CD44; CD45RA; CD47; CD48; CD49D (ITGA4); CD49F; CD52; CD58; CD64; CD7; CD70; CD82; CD84; CD85; CD85K; CD86; CD9; CD96; CD99; CLL-1; EMR2 (ADGRE2); FR-B; GPR56; and IL1RAP). In some embodiments, genetically engineered cells of the present disclosure comprise a mutation in a gene encoding a cell surface protein selected from a group consisting of: CD10; CD101; CD11A; CD11C; CD123; CD133; CD152; CD200; CD205; CD244; CD25; CD305; CD33; CD34; CD366; CD38; CD42B; CD44; CD45RA; CD47; CD48; CD49D; CD49F; CD52; CD58; CD64; CD7; CD84; CD85K; CD9; CD99; CLL-1; EMR2; GPR56; IL1RAP; and CD274. In some embodiments, genetically engineered cells of the present disclosure comprise a mutation in a gene encoding a cell surface protein selected from a group consisting of: CD117; CD11A; CD11C; CD120B; CD123; CD13; CD133; CD152; CD205; CD226; CD244; CD25; CD305; CD32; CD33; CD34; CD38; CD41; CD44; CD45RA; CD47; CD49D; CD49F; CD52; CD58; CD64; CD7; CD70; CD82; CD86; CD96; CD99; CLL-1; EMR2; FR-B; GPR56; and IL1RAP. In some embodiments, genetically engineered cells of the present disclosure comprise a mutation in a first gene encoding a first cell surface protein and a second gene encoding a second cell surface protein selected from a group consisting of: CD274 and CD152; CD274 and CD226; CD152 and CD226; CD10 and CD49F; CD49F and GPR56; CD200 and CD34; CD34 and CD52; CD34 and CD133; GPR56 and CD133; CD42B and CD41; CD85K and CD64; CD85K and CD32; CD64 and CD32; CD64 and CD11C; CD64 and CD86; CD45RA and CD96; CD45RA and IL1RAP; CD45RA and CD123; CD84 and CD33; CD11C and IL1RAP; CD11C and CD33; CD11C and CD86; CD9 and IL1RAP; IL1RAP and CD33; IL1RAP and CD205; IL1RAP and CD305; IL1RAP and CD123; CD33 and CLL-1; CD33 and CD305; CD11A and CD86; CLL-1 and CD305; CD58 and CD305; CD49D and CD47; CD49D and CD99; CD49D and CD44; CD49D and CD305; CD47 and CD99; CD47 and EMR2; CD99 and CD44; CD99 and CD305; CD99 and EMR2; CD205 and CD305; CD205 and CD123; CD44 and CD305; CD44 and EMR2; CD244 and CD117; CD123 and EMR2; EMR2 and CD82; CD152 and CD120B; CD48 and CD11A; CD200 and CD25; CD34 and CD7; CD34 and CD13; CD52 and GPR56; CD52and CD133; CD52 and CD7; GPR56 and CD7; GPR56 and CD82; CD133 and CD7; CD133 and CD82; CD7 and CD13; CD101 and CD64; CD85 and CD70; CD85K and CD86; CD64 and FR-B; CD366 and CD96; CD25 and CD45RA; CD25 and CD96; CD9 and CD82; IL1RAP and CD58; CD11A and CD38; CD11A and CD44; CD11A and CD244; CD38 and CD305; CD38 and EMR2; CLL-1 and CD49D; CD58 and CD49D; CD47 and CD205; and CD47 and CD44. In some embodiments, genetically engineered cells of the present disclosure comprise a mutation in a first gene encoding a first cell surface protein and a second gene encoding a second cell surface protein selected from a group consisting of: CD85K and CD64; CD45RA and IL1RAP; CD34 and CD133; IL1RAP and CD33; CD47 and CD99; CLL-1 and CD305; CD152 and CD226; CD45RA and CD96; CD45RA and CD123; CD49D and CD305; IL1RAP and CD305; CD58 and CD305; CD49D and CD47; CD33 and CD305; CD49D and CD44; CD64 and CD86; GPR56 and CD133; CD34 and CD52; EMR2 and CD82; CD64 and CD32; CD42B and CD41; CD64 and CD11C; CD99 and CD44; CD200 and CD34; CD11C and CD33; IL1RAP and CD123; CD11C and IL1RAP; CD44 and EMR2; CD205 and CD305; CD44 and CD305; CD49D and CD99; CD274 and CD226; CD49F and GPR56; CD9 and IL1RAP; CD99 and CD305; CD10 and CD49F; CD11A and CD86; CD33 and CLL-1; CD84 and CD33; CD244 and CD117; CD85K and CD32; CD47 and EMR2; CD274 and CD152; IL1RAP and CD205; CD11C and CD86; CD99 and EMR2; CD205 and CD123; and CD123 and EMR2. In some embodiments, genetically engineered cells of the present disclosure comprise a mutation in a first gene encoding a first cell surface protein and a second gene encoding a second cell surface protein selected from a group consisting of: CD33 and CD205 (LY75); CD33 and CD244 (SLAMF4); CD33 and CD305 (LAIR1); CD33 and CD49D (ITGA4); CLL-1 and CD205 (LY75); CLL-1 and CD244 (SLAMF4); CLL-1 and CD305 (LAIR1); CLL-1 and CD49D (ITGA4); EMR2 and CD205 (LY75); EMR2 and CD244 (SLAMF4); EMR2 and CD305 (LAIR1); EMR2 and CD49D (ITGA4); CD38 and CD205 (LY75); CD38 and CD244 (SLAMF4); CD38 and CD305 (LAIR1); CD38 and CD49D (ITGA4); CD305 (LAIR1) and CD49D (ITGA4); CD305 (LAIR1) and CD205 (LY75); CD305 (LAIR1) and CD244 (SLAMF4); CD49D (ITGA4) and CD205 (LY75); CD49D (ITGA4) and CD244 (SLAMF4); and CD205 (LY75) and CD244 (SLAMF4). In some embodiments, genetically engineered cells of the present disclosure comprise a mutation in a first gene encoding a first cell surface protein and a second gene encoding a second lineage-specific cell surface antigen, wherein mutation of the first gene and second gene results in dysregulated expression of the first cell surface protein and the second cell surface protein seen in subjects diagnosed with a hematopoietic malignancy. In some embodiments, the first gene and second gene encode combinations of antigens selected from a group consisting of: CD274 and CD152; CD274 and CD226; CD152 and CD226; CD10 and CD49F; CD49F and GPR56; CD200 and CD34; CD34 and CD52; CD34 and CD133; GPR56 and CD133; CD42B and CD41; CD85K and CD64; CD85K and CD32; CD64 and CD32; CD64 and CD11C; CD64 and CD86; CD45RA and CD96; CD45RA and IL1RAP; CD45RA and CD123; CD84 and CD33; CD11C and IL1RAP; CD11C and CD33; CD11C and CD86; CD9 and IL1RAP; IL1RAP and CD33; IL1RAP and CD205; IL1RAP and CD305; IL1RAP and CD123; CD33 and CLL-1; CD33 and CD305; CD11A and CD86; CLL-1 and CD305; CD58 and CD305; CD49D and CD47; CD49D and CD99; CD49D and CD44; CD49D and CD305; CD47 and CD99; CD47 and EMR2; CD99 and CD44; CD99 and CD305; CD99 and EMR2; CD205 and CD305; CD205 and CD123; CD44 and CD305; CD44 and EMR2; CD244 and CD117; CD123 and EMR2; and EMR2 and CD82. In some embodiments, the first gene and second gene encode combinations of antigens selected from a group consisting of: CD33 and CD205 (LY75); CD33 and CD244 (SLAMF4); CD33 and CD305 (LAIR1); CD33 and CD49D (ITGA4); CLL-1 and CD205 (LY75); CLL-1 and CD244 (SLAMF4); CLL-1 and CD305 (LAIR1); CLL-1 and CD49D (ITGA4); EMR2 and CD205 (LY75); EMR2 and CD244 (SLAMF4); EMR2 and CD305 (LAIR1); EMR2 and CD49D (ITGA4); CD38 and CD205 (LY75); CD38 and CD244 (SLAMF4); CD38 and CD305 (LAIR1); CD38 and CD49D (ITGA4); CD305 (LAIR1) and CD49D (ITGA4); CD305 (LAIR1) and CD205 (LY75); CD305 (LAIR1) and CD244 (SLAMF4); CD49D (ITGA4) and CD205 (LY75); CD49D (ITGA4) and CD244 (SLAMF4); and CD205 (LY75) and CD244 (SLAMF4). In some embodiments, genetically engineered cells of the present disclosure comprise a mutation in a first gene encoding a first cell surface protein and a second gene encoding a second lineage-specific cell surface antigen, wherein mutation of the first gene and second gene results in dysregulated expression of the first cell surface protein and the second cell surface protein seen in subjects who have experienced relapse of a hematopoietic malignancy. In some embodiments, the first gene and second gene encode combinations of antigens selected from a group consisting of: CD9 and CD82; CD45RA and CD123; CD133 and CD82; CD85K and CD70; CD47 and CD205; CD64 and FR-B; CD11A and CD44; CD85K and CD86; CD366 and CD96; CD101 and CD64; CLL-1 and CD49D; CD49D and CD47; EMR2 and CD82; CD38 and CD305; CD34 and CD7; CD200 and CD25; CD25 and CD45RA; CD49D and CD44; CD11A and CD244; CD133 and CD7; CD48 and CD11A; CD52 and CD133; GPR56 and CD82; CD11A and CD38; CD34 and CD13; CD58 and CD49D; CD47 and CD99; IL1RAP and CD58; CD47 and CD44; CD33 and CD305; CD64 and CD86; CD152 and CD120B; CD42B and CD41; CD34 and CD52; CD152 and CD226; CD7 and CD13; CD38 and EMR2; IL1RAP and CD33; CLL-1 and CD305; CD45RA and IL1RAP; CD34 and CD133; GPR56 and CD133; CD49D and CD305; CD25 and CD96; CD52 and CD7; CD64 and CD32; CD58 and CD305; CD45RA and CD96; IL1RAP and CD305; CD52 and GPR56; GPR56 and CD7; and CD85K and CD64. In some embodiments, the first gene and second gene encode combinations of antigens selected from a group consisting of: CD33 and CD205 (LY75); CD33 and CD244 (SLAMF4); CD33 and CD305 (LAIR1); CD33 and CD49D (ITGA4); CLL-1 and CD205 (LY75); CLL-1 and CD244 (SLAMF4); CLL-1 and CD305 (LAIR1); CLL-1 and CD49D (ITGA4); EMR2 and CD205 (LY75); EMR2 and CD244 (SLAMF4); EMR2 and CD305 (LAIR1); EMR2 and CD49D (ITGA4); CD38 and CD205 (LY75); CD38 and CD244 (SLAMF4); CD38 and CD305 (LAIR1); CD38 and CD49D (ITGA4); CD305 (LAIR1) and CD49D (ITGA4); CD305 (LAIR1) and CD205 (LY75); CD305 (LAIR1) and CD244 (SLAMF4); CD49D (ITGA4) and CD205 (LY75); CD49D (ITGA4) and CD244 (SLAMF4); and CD205 (LY75) and CD244 (SLAMF4). Variant Cell Surface Proteins Methods for assessing the functionality of the cell surface protein and the hematopoietic cells or descendants thereof will be known in the art and include, for example, proliferation assays, differentiation assays, colony formation, expression analysis (e.g., gene and/or protein), protein localization, intracellular signaling, functional assays, and in vivo humanized mouse models. Any of the methods for identifying and/or verifying non-essential epitopes in cell surface proteins is also within the scope of the present disclosure. The term “binds,” as used herein with reference to a gRNA interaction with a target domain (e.g., a first target domain and/or a second or subsequent target domain), refers to the gRNA molecule and the target domain forming a complex. The complex may comprise two strands forming a duplex structure, or three or more strands forming a multi-stranded complex. The binding may constitute a step in a more extensive process, such as the cleavage of the target domain by a Cas nuclease. In some embodiments, the gRNA binds to the target domain with perfect complementarity, and in other embodiments, the gRNA binds to the target domain with partial complementarity, e.g., with one or more mismatches. In some embodiments, when a gRNA binds to a target domain, the full targeting domain of the gRNA base pairs with the targeting domain. In other embodiments, only a portion of the target domain and/or only a portion of the targeting domain base pairs with the other. In an embodiment, the interaction is sufficient to mediate a target domain-mediated cleavage event. As used herein, the terms “binds,” “specifically binds,” “specifically recognizes” and analogous terms as used herein with reference to a protein (e.g., a cell surface protein) and an agent (e.g., a ligand or an antibody) interaction refer to the specific binding or association between the agent (e.g., a ligand or an antibody) and protein (e.g., a cell surface protein). Agents that specifically bind a cell surface protein are known in the art and/or can be identified, for example, by immunoassays, BIAcore™, or other techniques known to those of skill in the art. The term “reduced binding,” as used herein with reference to the binding activity of an agent (e.g., a ligand or an antibody) to a cell surface protein epitope, refers to binding that is reduced by at least about 5%. The level of binding may refer to the amount of binding of the agent (e.g., a ligand or an antibody) to a cell, such as a hematopoietic cell or descendant thereof, or to the amount of binding of the agent (e.g., a ligand or an antibody) to the cell surface protein. The level of binding of a cell, such as a hematopoietic cell or descendant thereof, that has been modified, for example, to include an amino acid substitution in a cell surface protein epitope, may be relative to the level of binding of the agent (e.g., a ligand or an antibody) to a cell that has not been modified as determined by the same assay under the same conditions. In exemplary embodiments, the level of binding of a cell surface protein that includes an amino acid substitution in a cell surface protein epitope to an agent (e.g., a ligand or an antibody) may be relative to the level of binding of the agent to a cell surface protein that does not include the amino acid substitution in the cell surface protein epitope (e.g., a wild-type protein or a variant thereof) as determined by the same assay under the same conditions. Alternatively, the level of binding of a cell surface protein that lacks an epitope to an agent (e.g., a ligand or an antibody) may be relative to the level of binding of the agent to a cell surface protein that contains the unmodified epitope (e.g., a wild-type protein) as determined by the same assay under the same conditions. In some embodiments, the binding is reduced by at least about 1%, at least about 2%, at least about 3%, at least about 4%, at least about 5%, at least about 6%, at least about 7%, at least about 8%, at least about 9%, at least about 10%, at least about 15%, at least about 20%, at least about 25%, at least about 30%, at least about 35%, at least about 40%, at least about 45%, at least about 50%, at least about 55%, at least about 60%, at least about 65%, at least about 70%, at least about 75%, at least about 80%, at least about 85%, at least about 90%, at least about 95%, at least about 99%, at least about 100% or more, e.g., as compared to the unmodified epitope (e.g., a wild-type protein). In some embodiments, the binding is reduced by at least about 1-fold, at least about 2-fold, at least about 3-fold, at least about 4-fold, at least about 5-fold, at least about 6-fold, at least about 7-fold, at least about 8-fold, at least about 9-fold, at least about 10-fold, at least about 20-fold, at least about 30-fold, at least about 40-fold, at least about 50-fold, at least about 60-fold, at least about 70-fold, at least about 80- fold, at least about 90-fold, or at least about 100-fold, e.g., as compared to the unmodified epitope (e.g., a wild-type protein). In some embodiments, the binding is reduced such that there is substantially no detectable binding in a conventional assay. The binding affinity or binding specificity for an epitope or protein can be determined by a variety of methods including equilibrium dialysis, equilibrium binding, gel filtration, ELISA, surface plasmon resonance, or spectroscopy. As used herein, “no binding” refers to substantially no binding, e.g., no detectable binding or only baseline binding as determined in a conventional binding assay. In some embodiments, there is no binding between the cells, e.g., hematopoietic cells or descendants thereof, that have been modified and the agent. In some embodiments, there is no detectable binding between the cells, e.g., hematopoietic cells or descendants thereof, that have been modified and the agent. In some embodiments, no binding of the cells, e.g., hematopoietic cells or descendant thereof, to the agent refers to a baseline level of binding, as shown using any conventional binding assay known in the art. In some embodiments, the level of binding of the cells, e.g., hematopoietic cells or descendants thereof, that have been modified and the agent is not biologically significant. The term “no binding” is not intended to require the absolute absence of binding. Without wishing to be bound by theory, a subject can be administered the rescue cells (e.g., hematopoietic stem cells (HSCs) and/or hematopoietic progenitor cells (HPCs)) described herein comprising a modification in a cell surface protein gene, e.g., a genetic edit (i.e., mutation) that results in the rescue cells having reduced or eliminated expression of the respective gene, or a modification of an epitope of the protein encoded by the respective gene that diminishes the binding of the therapeutic agent to the protein. These genetically engineered (modified) cells can thus be resistant to an agent, such as an anti-cancer therapy, and can therefore repopulate the hematopoietic system during or after anti-cancer therapy. The binding activity of the agent (e.g., a ligand or an antibody) to the epitope may be reduced by the amino acid substitution by at least about 1%, at least about 2%, at least about 3%, at least about 4%, at least about 5%, at least about 6%, at least about 7%, at least about 8%, at least about 9%, at least about 10%, at least about 15%, at least about 20%, at least about 25%, at least about 30%, at least about 35%, at least about 40%, at least about 45%, at least about 50%, at least about 55%, at least about 60%, at least about 65%, at least about 70%, at least about 75%, at least about 80%, at least about 85%, at least about 90%, at least about 95%, at least about 99%, at least about 100% or more, e.g., as compared to the unmodified epitope (e.g., a wild-type protein). In some embodiments, the binding activity of the agent (e.g., a ligand or an antibody) to the epitope may be reduced by the amino acid substitution by at least about at least about 1-fold, at least about 2-fold, at least about 3-fold, at least about 4-fold, at least about 5-fold, at least about 6-fold, at least about 7-fold, at least about 8-fold, at least about 9-fold, at least about 10-fold, at least about 20-fold, at least about 30-fold, at least about 40-fold, at least about 50-fold, at least about 60-fold, at least about 70- fold, at least about 80-fold, at least about 90-fold, or at least about 100-fold, e.g., as compared to the unmodified epitope (e.g., a wild-type protein). In some embodiments, the binding is reduced such that there is substantially no detectable binding in a conventional assay. The binding affinity or binding specificity for an epitope or protein can be determined by a variety of methods including equilibrium dialysis, equilibrium binding, gel filtration, ELISA, surface plasmon resonance, or spectroscopy. In some embodiments, the reduction in binding activity may include an increase in KD, IC50, and/or EC50. For example, the KD, IC50, and/or EC50 may be increased by the amino acid substitution by at least about 1%, at least about 2%, at least about 3%, at least about 4%, at least about 5%, at least about 6%, at least about 7%, at least about 8%, at least about 9%, at least about 10%, at least about 15%, at least about 20%, at least about 25%, at least about 30%, at least about 35%, at least about 40%, at least about 45%, at least about 50%, at least about 55%, at least about 60%, at least about 65%, at least about 70%, at least about 75%, at least about 80%, at least about 85%, at least about 90%, at least about 95%, at least about 99%, at least about 100% or more, e.g., as compared to the unmodified epitope (e.g., a wild- type protein). In some embodiments, the KD, IC50, and/or EC50 may be increased by the amino acid substitution by at least about 1-fold, at least about 2-fold, at least about 3-fold, at least about 4-fold, at least about 5-fold, at least about 6-fold, at least about 7-fold, at least about 8-fold, at least about 9-fold, at least about 10-fold, at least about 20-fold, at least about 30-fold, at least about 40-fold, at least about 50-fold, at least about 60-fold, at least about 70- fold, at least about 80-fold, at least about 90-fold, or at least about 100-fold, e.g., as compared to the unmodified epitope (e.g., a wild-type protein). As used herein, the term "IC50" refers to the concentration of an agent (e.g., an inhibitor, such as a ligand or an antibody) that produces 50% of the maximal inhibition of activity or expression measurable using the same assay in the absence of the binding partner. The IC50 can be as measured in vitro or in vivo. The IC50 can be determined by measuring activity using a conventional in vitro assay (e.g., a protein activity assay and/or a gene expression assay). As used herein, the term "EC50," refers to the concentration of a binding partner (e.g., an activator, such as a ligand or an antibody) that produces 50% of maximal activation of measurable activity or expression using the same assay in the absence of the agent. Stated differently, the "EC50" is the concentration of agent that gives 50% activation, when 100% activation is set at the amount of activity that does not increase with the addition of more agent. The EC50 may refer to the half maximal effective concentration, which includes the concentration of an antibody which induces a response halfway between the baseline and maximum after a specified exposure time. The EC50 may represent the concentration of an antibody where 50% of its maximal effect is observed. In certain embodiments, the EC50 value may equal the concentration of an agent (e.g., a ligand or an antibody) that gives half- maximal binding to cells expressing a cell surface protein, such as a cell surface protein, as determined by, e.g. a FACS binding assay. Thus, reduced or weaker binding is observed with an increased EC50 value, or half maximal effective concentration value such that 500 nM EC50 is indicative of a weaker binding affinity than 50 nM EC50. The EC50 can be as measured in vitro or in vivo. The term "KD" refers to the dissociation equilibrium constant of a particular agent- protein interaction, such as an antibody-antigen interaction or a ligand-receptor interaction, or the dissociation rate constant of an antibody or antibody-binding fragment. There is an inverse relationship between KD and binding affinity, therefore the smaller the KD value, the higher, i.e. stronger, the affinity. Thus, the terms "higher affinity" or "stronger affinity" relate to a higher ability to form an interaction and therefore a smaller KD value, and conversely the terms "lower affinity" or "weaker affinity" relate to a lower ability to form an interaction and therefore a larger KD value. In some circumstances, a higher binding affinity (or KD) of a particular molecule (e.g., antibody) to its interactive partner molecule (e.g., antigen X) compared to the binding affinity of the molecule (e.g., antibody) to another interactive partner molecule (e.g. antigen Y) may be expressed as a binding ratio determined by dividing the larger KD value (lower, or weaker, affinity) by the smaller KD (higher, or stronger, affinity), for example expressed as 5-fold or 10-fold greater binding affinity, as the case may be. In some embodiments, "low affinity" refers to less strong binding interaction. In some embodiments, the low binding affinity corresponds to greater than about 1 nM KD, greater than about 1 nM, about 2 nM, about 3 nM, about 4 nM, about 5 nM, about 6 nM, about 7 nM, about 8 nM, about 9 nM, about 10 nM, about 11 nM, about 12 nM, about 13 nM, about 14 nM, about 15 nM, about 16 nM, about 17 nM, about 18 nM, about 19 nM, about 20 nM, about 21 nM, about 22 nM, about 23 nM, about 24 nM, about 25 nM, about 26 nM, about 27 nM, about 28 nM, about 29 nM, about 30 nM, about 31 nM, about 32 nM, about 33 nM, about 34 nM, about 35 nM, about 36 nM, about 37 nM, about 38 nM, about 39 nM, or about 40 nM KD, wherein such KD binding affinity value is measured, e.g., in an in vitro surface plasmon resonance binding assay, or equivalent biomolecular interaction sensing assay. In some embodiments, the low binding affinity corresponds to greater than about 10 nM, about 11 nM, about 12 nM, about 13 nM, about 14 nM, about 15 nM, about 16 nM, about 17 nM, about 18 nM, about 19 nM, about 20 nM, about 21 nM, about 22 nM, about 23 nM, about 24 nM, about 25 nM, about 26 nM, about 27 nM, about 28 nM, about 29 nM, about 30 nM, about 31 nM, about 32 nM, about 33 nM, about 34 nM, about 35 nM, about 36 nM, about 37 nM, about 38 nM, about 39 nM, or about 40 nM EC50, wherein such EC50 binding affinity value is measured, e.g., in an in vitro FACS binding assay, or equivalent cell-based binding assay. In some embodiments, "weak affinity" refers to weak binding interaction. In some embodiments, the weak binding affinity corresponds to greater than about 100 nM KD or EC50, greater than about 200, 300, or greater than about 500 nM KD or EC50, wherein such KD binding affinity value is measured, e.g., in an in vitro surface plasmon resonance binding assay, or equivalent biomolecular interaction sensing assay, and such EC50 binding affinity value is measured, e.g., in an in vitro FACS binding assay, or equivalent cell-based interaction detecting assay to detect monovalent biding. No detectable binding means that the affinity between the two biomolecules, for example, especially between the monovalent antibody binding arm and its target antigen, is beyond the detection limit of the assay being used. In some embodiments, genetically engineered cells of the present disclosure express a mutant cell surface protein selected from a group consisting of: CD10; CD101; CD117; CD11A; CD11C; CD120B; CD123; CD13; CD133; CD152; CD200; CD205 (LY75); CD226; CD244 (SLAMF4); CD25; CD274; CD305 (LAIR1); CD32; CD33; CD34; CD366; CD38; CD41; CD42B; CD44; CD45RA; CD47; CD48; CD49D (ITGA4); CD49F; CD52; CD58; CD64; CD7; CD70; CD82; CD84; CD85; CD85K; CD86; CD9; CD96; CD99; CLL- 1; EMR2 (ADGRE2); FR-B; GPR56; and IL1RAP. In some embodiments, genetically engineered cells of the present disclosure express a mutant cell surface protein selected from a group consisting of: CD10; CD101; CD11A; CD11C; CD123; CD133; CD152; CD200; CD205; CD244; CD25; CD305; CD33; CD34; CD366; CD38; CD42B; CD44; CD45RA; CD47; CD48; CD49D; CD49F; CD52; CD58; CD64; CD7; CD84; CD85K; CD9; CD99; CLL-1; EMR2; GPR56; IL1RAP; and CD274. In some embodiments, genetically engineered cells of the present disclosure express a mutant cell surface protein selected from a group consisting of: CD117; CD11A; CD11C; CD120B; CD123; CD13; CD133; CD152; CD205; CD226; CD244; CD25; CD305; CD32; CD33; CD34; CD38; CD41; CD44; CD45RA; CD47; CD49D; CD49F; CD52; CD58; CD64; CD7; CD70; CD82; CD86; CD96; CD99; CLL-1; EMR2; FR-B; GPR56; and IL1RAP. In some embodiments, genetically engineered cells of the present disclosure express a mutant of a first cell surface protein and a mutant of a second cell surface protein selected from a group consisting of: CD10; CD101; CD117; CD11A; CD11C; CD120B; CD123; CD13; CD133; CD152; CD200; CD205 (LY75); CD226; CD244 (SLAMF4); CD25; CD274; CD305 (LAIR1); CD32; CD33; CD34; CD366; CD38; CD41; CD42B; CD44; CD45RA; CD47; CD48; CD49D (ITGA4); CD49F; CD52; CD58; CD64; CD7; CD70; CD82; CD84; CD85; CD85K; CD86; CD9; CD96; CD99; CLL-1; EMR2 (ADGRE2); FR-B; GPR56; and IL1RAP. In some embodiments, genetically engineered cells of the present disclosure express a mutant of a first cell surface protein and a mutant of a second cell surface protein selected from a group consisting of: CD274 and CD152; CD274 and CD226; CD152 and CD226; CD10 and CD49F; CD49F and GPR56; CD200 and CD34; CD34 and CD52; CD34 and CD133; GPR56 and CD133; CD42B and CD41; CD85K and CD64; CD85K and CD32; CD64 and CD32; CD64 and CD11C; CD64 and CD86; CD45RA and CD96; CD45RA and IL1RAP; CD45RA and CD123; CD84 and CD33; CD11C and IL1RAP; CD11C and CD33; CD11C and CD86; CD9 and IL1RAP; IL1RAP and CD33; IL1RAP and CD205; IL1RAP and CD305; IL1RAP and CD123; CD33 and CLL-1; CD33 and CD305; CD11A and CD86; CLL-1 and CD305; CD58 and CD305; CD49D and CD47; CD49D and CD99; CD49D and CD44; CD49D and CD305; CD47 and CD99; CD47 and EMR2; CD99 and CD44; CD99 and CD305; CD99 and EMR2; CD205 and CD305; CD205 and CD123; CD44 and CD305; CD44 and EMR2; CD244 and CD117; CD123 and EMR2; EMR2 and CD82; CD152 and CD120B; CD48 and CD11A; CD200 and CD25; CD34 and CD7; CD34 and CD13; CD52 and GPR56; CD52and CD133; CD52 and CD7; GPR56 and CD7; GPR56 and CD82; CD133 and CD7; CD133 and CD82; CD7 and CD13; CD101 and CD64; CD85 and CD70; CD85K and CD86; CD64 and FR-B; CD366 and CD96; CD25 and CD45RA; CD25 and CD96; CD9 and CD82; IL1RAP and CD58; CD11A and CD38; CD11A and CD44; CD11A and CD244; CD38 and CD305; CD38 and EMR2; CLL-1 and CD49D; CD58 and CD49D; CD47 and CD205; and CD47 and CD44. In some embodiments, genetically engineered cells of the present disclosure express a mutant of a first cell surface protein and a mutant of a second cell surface protein selected from a group consisting of: CD85K and CD64; CD45RA and IL1RAP; CD34 and CD133; IL1RAP and CD33; CD47 and CD99; CLL-1 and CD305; CD152 and CD226; CD45RA and CD96; CD45RA and CD123; CD49D and CD305; IL1RAP and CD305; CD58 and CD305; CD49D and CD47; CD33 and CD305; CD49D and CD44; CD64 and CD86; GPR56 and CD133; CD34 and CD52; EMR2 and CD82; CD64 and CD32; CD42B and CD41; CD64 and CD11C; CD99 and CD44; CD200 and CD34; CD11C and CD33; IL1RAP and CD123; CD11C and IL1RAP; CD44 and EMR2; CD205 and CD305; CD44 and CD305; CD49D and CD99; CD274 and CD226; CD49F and GPR56; CD9 and IL1RAP; CD99 and CD305; CD10 and CD49F; CD11A and CD86; CD33 and CLL-1; CD84 and CD33; CD244 and CD117; CD85K and CD32; CD47 and EMR2; CD274 and CD152; IL1RAP and CD205; CD11C and CD86; CD99 and EMR2; CD205 and CD123; and CD123 and EMR2. In some embodiments, genetically engineered cells of the present disclosure express a mutant of a first cell surface protein and a mutant of a second cell surface protein selected from a group consisting of: CD274 and CD152; CD274 and CD226; CD152 and CD226; CD10 and CD49F; CD49F and GPR56; CD200 and CD34; CD34 and CD52; CD34 and CD133; GPR56 and CD133; CD42B and CD41; CD85K and CD64; CD85K and CD32; CD64 and CD32; CD64 and CD11C; CD64 and CD86; CD45RA and CD96; CD45RA and IL1RAP; CD45RA and CD123; CD84 and CD33; CD11C and IL1RAP; CD11C and CD33; CD11C and CD86; CD9 and IL1RAP; IL1RAP and CD33; IL1RAP and CD205; IL1RAP and CD305; IL1RAP and CD123; CD33 and CLL-1; CD33 and CD305; CD11A and CD86; CLL-1 and CD305; CD58 and CD305; CD49D and CD47; CD49D and CD99; CD49D and CD44; CD49D and CD305; CD47 and CD99; CD47 and EMR2; CD99 and CD44; CD99 and CD305; CD99 and EMR2; CD205 and CD305; CD205 and CD123; CD44 and CD305; CD44 and EMR2; CD244 and CD117; CD123 and EMR2; and EMR2 and CD82. In some embodiments, genetically engineered cells of the present disclosure express a mutant of a first cell surface protein and a mutant of a second cell surface protein selected from a group consisting of: CD9 and CD82; CD45RA and CD123; CD133 and CD82; CD85K and CD70; CD47 and CD205; CD64 and FR-B; CD11A and CD44; CD85K and CD86; CD366 and CD96; CD101 and CD64; CLL-1 and CD49D; CD49D and CD47; EMR2 and CD82; CD38 and CD305; CD34 and CD7; CD200 and CD25; CD25 and CD45RA; CD49D and CD44; CD11A and CD244; CD133 and CD7; CD48 and CD11A; CD52 and CD133; GPR56 and CD82; CD11A and CD38; CD34 and CD13; CD58 and CD49D; CD47 and CD99; IL1RAP and CD58; CD47 and CD44; CD33 and CD305; CD64 and CD86; CD152 and CD120B; CD42B and CD41; CD34 and CD52; CD152 and CD226; CD7 and CD13; CD38 and EMR2; IL1RAP and CD33; CLL-1 and CD305; CD45RA and IL1RAP; CD34 and CD133; GPR56 and CD133; CD49D and CD305; CD25 and CD96; CD52 and CD7; CD64 and CD32; CD58 and CD305; CD45RA and CD96; IL1RAP and CD305; CD52 and GPR56; GPR56 and CD7; and CD85K and CD64. In some embodiments, genetically engineered cells of the present disclosure express a mutant of a first cell surface protein and a mutant of a second cell surface protein selected from a group consisting of: CD33 and CD205 (LY75); CD33 and CD244 (SLAMF4); CD33 and CD305 (LAIR1); CD33 and CD49D (ITGA4); CLL-1 and CD205 (LY75); CLL-1 and CD244 (SLAMF4); CLL-1 and CD305 (LAIR1); CLL-1 and CD49D (ITGA4); EMR2 and CD205 (LY75); EMR2 and CD244 (SLAMF4); EMR2 and CD305 (LAIR1); EMR2 and CD49D (ITGA4); CD38 and CD205 (LY75); CD38 and CD244 (SLAMF4); CD38 and CD305 (LAIR1); CD38 and CD49D (ITGA4); CD305 (LAIR1) and CD49D (ITGA4); CD305 (LAIR1) and CD205 (LY75); CD305 (LAIR1) and CD244 (SLAMF4); CD49D (ITGA4) and CD205 (LY75); CD49D (ITGA4) and CD244 (SLAMF4); and CD205 (LY75) and CD244 (SLAMF4). Further examples of genetically engineered hematopoietic cells are further described herein. Genetically Engineered Hematopoietic Cell Expressing CLL-1 Mutants In some embodiments, a genetically engineered hematopoietic cell has reduced or eliminated expression of a cell surface protein, or expresses a mutant of a cell surface protein, wherein the cell surface protein is CLL-1. C-type lectin-like molecule-1 (CLL-1), which may also be referred to as CLEC12A, is a C-type lectin-like receptor family member that is frequently expressed on acute myeloid leukemia (AML) cells. In some embodiments, CLL-1 is expressed by hematopoietic cells, e.g., hematopoietic stem cells and/or hematopoietic progenitor cells. The CLL-1 gene comprises 6 exons and is located on human chromosome 12. An example of a CLL-1 gene sequence can be found at NG_029426.2. In some embodiments, a mutation of a gene encoding CLL-1 alters one or more amino acids associated with an epitope of CLL-1. In some embodiments, the epitope of CLL-1 is a portion of CLL-1 bound by an agent, e.g., an immunotherapeutic agent. In some embodiments, the agent is an anti-CLL-1 antibody. In some embodiments, the agent comprises an anti-CLL-1 antibody or portion thereof, e.g., an antibody drug conjugate (ADC), a chimeric antigen receptor (CAR), or a multispecific antibody (e.g., a bispecific T cell engager). In some embodiments, the agent comprises an antibody or antigen-binding fragment thereof described in PCT Publication No. WO2023/201238, which is incorporated by reference herein in entirety. In some embodiments, the epitope of CLL-1 is one or more amino acids of a protein domain (e.g., the extracellular domain) or the amino acids encoded by an exon or combination of exons of the gene encoding CLL-1. In some embodiments, the epitope of CLL-1 comprises one or more amino acids encoded by exon 2 of the gene encoding CLL-1. In some embodiments, the epitope of CLL-1 comprises one or more amino acids encoded by exon 3 of the gene encoding CLL-1. In some embodiments, the epitope of CLL-1 comprises one or more amino acids encoded by exon 4 of the gene encoding CLL-1. In some embodiments, the epitope of CLL-1 comprises one or more amino acids encoded by exon 5 of the gene encoding CLL-1. In some embodiments, the epitope of CLL-1 comprises one or more amino acids encoded by exon 6 of the gene encoding CLL-1. In some embodiments, a mutation of a gene encoding CLL-1 makes a change in the amino acid sequence corresponding to the amino acid sequence of a CLL-1 ortholog. In some embodiments, a mutation substitutes an amino acid of human CLL-1 for an amino acid at a corresponding position of an orthologous CLL-1, e.g., a non-human primate CLL-1. In some embodiments, a mutation inserts or deletes one or more amino acids of human CLL-1 to correspond to the sequence of an orthologous CLL-1, e.g., a non-human primate CLL-1. In some embodiments, a mutation changes the amino acid sequence in a manner corresponding to a tolerable genetic variant identified by one or more genomic sequence comparison algorithms, e.g., gnomAD (see, e.g., Gudmundsson et al. arXiv:2107.11458v3, e.g., gnomad.broadinstitute.org/) or to a position characterized by a plurality of tolerable genetic variants. In some embodiments, mutations to CLL-1 corresponding to the amino acid sequence of a CLL-1 ortholog or at positions characterized by a plurality of tolerable genetic variants decrease or eliminate binding of an immunotherapeutic agent targeting CLL-1 while preserving some or all of CLL-1 structure, expression, and/or functionality, providing a cell expressing CLL-1 (e.g., functional CLL-1) that is targeted less or not at all by anti-CLL-1 immunotherapeutic agents. In some embodiments, alteration results in a missense variant of CLL-1. Genetically Engineered Hematopoietic Cell Expressing CD123 Mutants In some embodiments, a genetically engineered hematopoietic cell has reduced or eliminated expression of a cell surface protein, or of a cell surface protein, wherein the cell surface protein is CD123. CD123 (also known as interleukin-3 receptor alpha or IL3Rα) is a type I cytokine receptor which binds to IL3. IL3 is a pleiotropic cytokine that regulates the function and production of hematopoietic and immune cells (see, e.g., Testa et al. Biomarker Research volume 2, Article number: 4 (2014)). Dysregulated expression of IL3 is associated with various cancers including myeloma (see, e.g., Lee et al. Blood (2004) 103 (6): 2308- 2315). In some embodiments, a hematopoietic malignancy is characterized by cells expressing (e.g., over-expressing) CD123. Dysregulated expression of CD123 is associated with various hematopoietic malignancies including hairy cell leukemia, acute myeloid leukemia, blastic plasmacytoid dendritic cell neoplasm, and systemic mastocytosis (see, e.g., Del Giudice et al. Hematologica (2004) 89 (3): 303-308; Munoz et al. Hematologica (2001) 86 (12): 1261-1269; Angelot-Delettre et al. Hematologica (2015) 100 (2): 223-230; Alayed et al. American Journal of Hematology (2013) 88 (12): 1055-1061; Paradanani et al. Leukemia (2016) 30 (4): 914-918; Testa et al. Biomarker Research (2014) 2: 4; and Lamble et al. Journal of Clinical Oncology (2022) 40 (3): 252-261). In some embodiments, CD123 is expressed by hematopoietic cells, e.g., hematopoietic stem cells and/or hematopoietic progenitor cells. In some embodiments, a mutation of a gene encoding CD123 alters one or more amino acids associated with an epitope of CD123. In some embodiments, the epitope of CD123 is a portion of CD123 bound by an agent, e.g., an immunotherapeutic agent. In some embodiments, the agent is an anti-CD123 antibody. In some embodiments, the agent comprises an anti-CD123 antibody or portion thereof, e.g., an antibody drug conjugate (ADC), a chimeric antigen receptor (CAR), or a multispecific antibody (e.g., a bispecific T cell engager). For example, the agent can be anti-CD123 antibody 7G3 or a variant thereof (e.g., a humanized variant, e.g., antibody CSL-36). In some embodiments, the agent is an anti-CD123 drug, e.g., talacotuzumab. In some embodiments, the epitope of CD123 is one or more amino acids of a protein domain (e.g., the extracellular domain) or the amino acids encoded by an exon or combination of exons of the gene encoding CD123. In some embodiments, the epitope of CD123 comprises one or more amino acids encoded by exon 2 of the gene encoding CD123. In some embodiments, the epitope of CD123 comprises one or more amino acids encoded by exon 3 of the gene encoding CD123. In some embodiments, the epitope of CD123 comprises one or more amino acids encoded by exon 4 of the gene encoding CD123. In some embodiments, the epitope of CD123 comprises one or more amino acids encoded by exon 5 of the gene encoding CD123. In some embodiments, the epitope of CD123 comprises one or more amino acids encoded by exon 6 of the gene encoding CD123. In some embodiments, the epitope of CD123 comprises one or more (e.g., two or more, three or more, four or more, or all) of the amino acids at positions 51, 59, 61, 82, or 84 of a wildtype gene encoding CD123 or at corresponding positions in a homologous CD123 gene. In some embodiments, a mutation of a gene encoding CD123 comprises a substitution of the amino acid at position 51 of a wildtype CD123 or at a corresponding position in a homologous CD123 gene. In some embodiments, a lysine is substituted for the amino acid at position 51 of a wildtype CD123 or at a corresponding position in a homologous CD123 gene. In some embodiments, a glycine is substituted for the amino acid at position 51 of a wildtype CD123 or at a corresponding position in a homologous CD123 gene. In some embodiments, a mutation of a gene encoding CD123 comprises a substitution of the amino acid at position 59 of a wildtype CD123 or at a corresponding position in a homologous CD123 gene. In some embodiments, a phenylalanine is substituted for the amino acid at position 59 of a wildtype CD123 or at a corresponding position in a homologous CD123 gene. In some embodiments, a cysteine is substituted for the amino acid at position 59 of a wildtype CD123 or at a corresponding position in a homologous CD123 gene. In some embodiments, a mutation of a gene encoding CD123 comprises a substitution of the amino acid at position 61 of a wildtype CD123 or at a corresponding position in a homologous CD123 gene. In some embodiments, a leucine is substituted for the amino acid at position 61 of a wildtype CD123 or at a corresponding position in a homologous CD123 gene. In some embodiments, a mutation of a gene encoding CD123 comprises a substitution of the amino acid at position 82 of a wildtype CD123 or at a corresponding position in a homologous CD123 gene. In some embodiments, an alanine is substituted for the amino acid at position 82 of a wildtype CD123 or at a corresponding position in a homologous CD123 gene. In some embodiments, a mutation of a gene encoding CD123 comprises a substitution of the amino acid at position 84 of a wildtype CD123 or at a corresponding position in a homologous CD123 gene. In some embodiments, a glutamine is substituted for the amino acid at position 84 of a wildtype CD123 or at a corresponding position in a homologous CD123 gene. In some embodiments, an alanine is substituted for the amino acid at position 84 of a wildtype CD123 or at a corresponding position in a homologous CD123 gene. In some embodiments, a mutation of a gene encoding CD123 makes a change in the amino acid sequence corresponding to the amino acid sequence of a CD123 ortholog. In some embodiments, a mutation substitutes an amino acid of human CD123 for an amino acid at a corresponding position of an orthologous CD123, e.g., a non-human primate CD123. In some embodiments, a mutation inserts or deletes one or more amino acids of human CD123 to correspond to the sequence of an orthologous CD123, e.g., a non-human primate CD123. In some embodiments, a mutation changes the amino acid sequence in a manner corresponding to a tolerable genetic variant identified by one or more genomic sequence comparison algorithms, e.g., gnomAD (see, e.g., Gudmundsson et al. arXiv:2107.11458v3, e.g., gnomad.broadinstitute.org/) or to a position characterized by a plurality of tolerable genetic variants. In some embodiments, mutations to CD123 corresponding to the amino acid sequence of a CD123 ortholog or at positions characterized by a plurality of tolerable genetic variants decrease or eliminate binding of an immunotherapeutic agent targeting CD123 while preserving some or all of CD123 structure, expression, and/or functionality, providing a cell expressing CD123 (e.g., functional CD123) that is targeted less or not at all by anti-CD123 immunotherapeutic agents. In some embodiments, alteration results in a missense variant of CD123. Genetically Engineered Hematopoietic Cell Expressing CD38 Mutants In some embodiments, a genetically engineered hematopoietic cell has reduced or eliminated expression of a cell surface protein, or expresses a mutant of a cell surface protein, wherein the cell surface protein is CD38. CD38 (also known as cyclic ADP ribose hydrolase) is a transmembrane ectoenzymatic glycoprotein involved in cell adhesion, signal transduction, and calcium signaling (see, e.g., van de Donk et al. Blood (2018) 131 (1): 13– 29). In some embodiments, a hematopoietic malignancy is characterized by cells expressing (e.g., over-expressing) CD38. In some embodiments, CD38 is expressed by hematopoietic cells, e.g., hematopoietic stem cells and/or hematopoietic progenitor cells. In some embodiments, a mutation of a gene encoding CD38 alters one or more amino acids associated with an epitope of CD38. In some embodiments, the epitope of CD38 is a portion of CD38 bound by an agent, e.g., an immunotherapeutic agent. In some embodiments, the agent is an anti-CD38 antibody. In some embodiments, the agent comprises an anti-CD38 antibody or portion thereof, e.g., an antibody drug conjugate (ADC), a chimeric antigen receptor (CAR), or a multispecific antibody (e.g., a bispecific T cell engager). For example, the agent can be anti-CD38 antibody HB7 or a variant thereof (e.g., a humanized variant). In some embodiments, the agent is an anti-CD38 drug, e.g., daratumumab. In some embodiments, the epitope of CD38 is one or more amino acids of a protein domain (e.g., the extracellular domain) or the amino acids encoded by an exon or combination of exons of the gene encoding CD38. In some embodiments, the epitope of CD38 comprises one or more amino acids encoded by exon 7 of the gene encoding CD38. In some embodiments, the epitope of CD38 comprises one or more (e.g., two or more, three or more, four or more, or all) of the amino acids at positions 270-274 of a wildtype gene encoding CD38 or at corresponding positions in a homologous CD38 gene. In some embodiments, a mutation of a gene encoding CD38 comprises a substitution of the amino acid at position 270 of a wildtype CD38 or at a corresponding position in a homologous CD38 gene. In some embodiments, an alanine is substituted for the amino acid at position 270 of a wildtype CD38 or at a corresponding position in a homologous CD38 gene. In some embodiments, a mutation of a gene encoding CD38 comprises a substitution of the amino acid at position 271 of a wildtype CD38 or at a corresponding position in a homologous CD38 gene. In some embodiments, a mutation of a gene encoding CD38 comprises a substitution of the amino acid at position 272 of a wildtype CD38 or at a corresponding position in a homologous CD38 gene. In some embodiments, a histidine is substituted for the amino acid at position 272 of a wildtype CD38 or at a corresponding position in a homologous CD38 gene. In some embodiments, an arginine is substituted for the amino acid at position 272 of a wildtype CD38 or at a corresponding position in a homologous CD38 gene. In some embodiments, an alanine is substituted for the amino acid at position 272 of a wildtype CD38 or at a corresponding position in a homologous CD38 gene. In some embodiments, a mutation of a gene encoding CD38 comprises a substitution of the amino acid at position 273 of a wildtype CD38 or at a corresponding position in a homologous CD38 gene. In some embodiments, a mutation of a gene encoding CD38 comprises a substitution of the amino acid at position 274 of a wildtype CD38 or at a corresponding position in a homologous CD38 gene. In some embodiments, a phenylalanine is substituted for the amino acid at position 274 of a wildtype CD38 or at a corresponding position in a homologous CD38 gene. In some embodiments, a mutation of a gene encoding CD38 makes a change in the amino acid sequence corresponding to the amino acid sequence of a CD38 ortholog. In some embodiments, a mutation substitutes an amino acid of human CD38 for an amino acid at a corresponding position of an orthologous CD38, e.g., a non-human primate CD38. In some embodiments, a mutation inserts or deletes one or more amino acids of human CD38 to correspond to the sequence of an orthologous CD38, e.g., a non-human primate CD38. In some embodiments, a mutation changes the amino acid sequence in a manner corresponding to a tolerable genetic variant identified by one or more genomic sequence comparison algorithms, e.g., gnomAD, or to a position characterized by a plurality of tolerable genetic variants. In some embodiments, mutations to CD38 corresponding to the amino acid sequence of a CD38 ortholog or at positions characterized by a plurality of tolerable genetic variants decrease or eliminate binding of an immunotherapeutic agent targeting CD38 while preserving some or all of CD38 structure, expression, and/or functionality, providing a cell expressing CD38 (e.g., functional CD38) that is targeted less or not at all by anti-CD38 immunotherapeutic agents. Genetically Engineered Hematopoietic Cell Expressing EMR2 Mutants In some embodiments, a genetically engineered hematopoietic cell has reduced or eliminated expression of a cell surface protein, or expresses a mutant of a cell surface protein, wherein the cell surface protein is EMR2. EGF-like module-containing mucin-like hormone receptor-like 2 (EMR2), also referred to as CD312 and ADGRE2, is a 823-amino acid, ~90 kDa protein (depending on isoform) of the EGF-seven-span transmembrane (TM7) family of adhesion G protein-coupled receptors (GPCR) with a high level of homology with CD97. EMR2 forms a heterodimer and binds to chondroitin sulfate B via its EGF-like domain 4 and mediate cell adhesion, granulocyte chemotaxis, degranulation, and the release of pro- inflammatory cytokines in macrophages. See, e.g. Kuan-Yu et al. Front. Immunol. (2017) 8:373. Without wishing to be bound by any particular theory, EMR2 is expressed on myeloid cells with highest expression in granulocytes, macrophages, and Kupffer cells. The ADGRE2 gene located on human chromosome 19 encodes human EMR2 and canonically contains 19 exons, although a number of isoforms exist with varying number EGF domains due to alternative RNA splicing. The dominant isoform in whole blood contains 17 exons. See, e.g. Safaee et al. Onc. Rev. (2014).8(242):20-24. In some embodiments, a mutation of a gene encoding EMR2 alters one or more amino acids associated with an epitope of EMR2. In some embodiments, the epitope of EMR2 is a portion of EMR2 bound by an agent, e.g., an immunotherapeutic agent. In some embodiments, the agent is an anti-EMR2 antibody. In some embodiments, the agent comprises an anti- EMR2 antibody or portion thereof, e.g., an antibody drug conjugate (ADC), a chimeric antigen receptor (CAR), or a multispecific antibody (e.g., a bispecific T cell engager). In some embodiments, the agent is an anti- EMR2 drug. In some embodiments, the epitope of EMR2 corresponds to the amino acids of a protein domain (e.g., the extracellular domain) or the amino acids encoded by an exon or combination of exons of the gene encoding EMR2. In some embodiments, the epitope of EMR2 comprises amino acids encoded by exon 1, exon 2, exon 3, exon 4, exon 5, exon 6, exon 7, exon 8, exon 9, exon 10, exon 11, exon 12, exon 13, exon 14, exon 15, exon 16, exon 17, exon 18, exon 19, exon 20, or exon 21 of EMR2, or a combination thereof. In some embodiments, the epitope of EMR2 comprises amino acids encoded by one, two, three, four, or all of exons 6, 10, 11, 14, and 18 of EMR2. In some embodiments, the epitope of EMR2 comprises the amino acids encoded by exon 6 of EMR2. In some embodiments, the epitope of EMR2 comprises the amino acids encoded by exon 10 of EMR2. In some embodiments, the epitope of EMR2 comprises the amino acids encoded by exon 11 of EMR2. In some embodiments, the epitope of EMR2 comprises the amino acids encoded by exon 14 of EMR2. In some embodiments, the epitope of EMR2 comprises the amino acids encoded by exon 18 of EMR2. In some embodiments, a mutation of a gene encoding EMR2 makes a change in the amino acid sequence corresponding to the amino acid sequence of a EMR2 ortholog. In some embodiments, an alteration comprises substitution of amino acid at any one or more of positions 124, 132, 146, 292, 294, 295, 296, 298, 299, 303, 304, 305, 306, 307, 308, 312, 318, 320, 328, 329, 331, 332, 335, 340, 347, 527, or 708 of a wildtype EMR2 or at a corresponding position in a homologous EMR2. In some embodiments, an alteration results in a missense variant of EMR2. In some embodiments, alteration results in a change at a splice region in EMR2. In some embodiments, a mutation of a gene encoding EMR2 alters one or more amino acids associated with an epitope of EMR2. In some embodiments, the epitope of EMR2 is a portion of EMR2 bound by an agent, e.g., an immunotherapeutic agent. In some embodiments, the agent is an anti-EMR2 antibody. In some embodiments, the agent comprises an anti-EMR2 antibody or portion thereof, e.g., an antibody drug conjugate (ADC), a chimeric antigen receptor (CAR), or a multispecific antibody (e.g., a bispecific T cell engager). For example, the agent can be anti-EMR2 monoclonal antibody 2A1 (Thermo Fisher) or a variant thereof (e.g., a humanized variant), Q9UHX3, OASA01861, AB_2738756, NLS6381, ab75190, MAB4894, or A100,000. Additional anti-EMR2 antibodies will be evident to one of ordinary skill in the art. See, e.g., International Publication No. WO 2017/087800 A1; Chang et al. FEBS Letters. (2003) 547(1-3):145-150; Yona et al. FASEB J. (2008).22(3): 741-751. In some embodiments, the agent comprises an anti-EMR2 antibody or portion thereof described in International Patent Application No. PCT/US2024/029308, which is incorporated by reference in its entirety. In some embodiments, mutations to EMR2 corresponding to the amino acid sequence of a EMR2 ortholog decrease or eliminate binding of an immunotherapeutic agent targeting EMR2 while preserving some or all of EMR2 expression and/or functionality, providing a cell expressing EMR2 (e.g., functional EMR2) that is targeted less or not at all by anti-EMR2 immunotherapeutic agents. Genetically Engineered Hematopoietic Cell Expressing CD47 Mutants In some embodiments, a genetically engineered hematopoietic cell has reduced or eliminated expression of a cell surface protein, or expresses a mutant of a cell surface protein, wherein the cell surface protein is CD47. CD47 is a transmembrane integrin-associated protein belonging to the immunoglobulin superfamily and is involved in the increase of intracellular calcium concentration that occurs upon cell adhesion to extracellular matrix. CD47 binds to a variety of ligands including thrombospondin-1 and signal-regulatory protein alpha and functions in processes such as apoptosis, proliferation, adhesion, and migration. CD47 also has roles in immune and angiogenic responses including regulation of phagocytosis by macrophages (see, e.g., Brown and Frazier. Trends in Cell Biology (2001) 11 (3): 130-135). CD47 is widely expressed across various tissues in humans and also in solid tumors and hematological malignancies (see, e.g., Jiang et al. Journal of Hematology & Oncology (2021) 14: 180). Human CD47 is located on chromosome 3 and contains 13 exons. In some embodiments, a mutation of a gene encoding CD47 alters one or more amino acids associated with an epitope of CD47. In some embodiments, the epitope of CD47 is a portion of CD47 bound by an agent, e.g., an immunotherapeutic agent. In some embodiments, the agent is an anti-CD47 antibody. In some embodiments, the agent is the anti-CD47 B6H12 or 2D3 antibody. In some embodiments, the agent comprises an anti-CD47 antibody or portion thereof, e.g., an antibody drug conjugate (ADC), a chimeric antigen receptor (CAR), or a multispecific antibody (e.g., a bispecific T cell engager). For example, the agent can be anti-CD47 antibody or a variant thereof (e.g., a humanized variant). In some embodiments, the agent is an anti-CD47 drug. In some embodiments, the epitope of CD47 is one or more amino acids of a protein domain (e.g., the extracellular domain) or the amino acids encoded by an exon or combination of exons of the gene encoding CD47. In some embodiments, the epitope of CD47 comprises one or more of amino acids 117-122 in CD47. In some embodiments, one or more of amino acids 117-122 in CD47 is deleted. In some embodiments, amino acids 117-122 in CD47 are deleted. In some embodiments, amino acids 117, 118, 119, 120, 121, and/or 122 or any combination thereof in CD47 is deleted. In some embodiments, the epitope of CD47 comprises one or more of amino acids 52-55 in CD47. In some embodiments, one or more of amino acids 52-55 in CD47 is deleted. In some embodiments, amino acids 52-55 in CD47 are deleted. In some embodiments, amino acids 52, 53, 54, and/or 55 or any combination thereof in CD47 is deleted. In some embodiments, a mutation of a gene encoding CD47 comprises a substitution of the amino acid at position 31 of a wildtype CD47 or at a corresponding position in a homologous CD47 gene. In some embodiments, a methionine is substituted for the amino acid at position 31 of a wildtype CD47 or at a corresponding position in a homologous CD47 gene. In some embodiments, a mutation of a gene encoding CD47 comprises a substitution of the amino acid at position 47 of a wildtype CD47 or at a corresponding position in a homologous CD47 gene. In some embodiments, a histidine is substituted for the amino acid at position 47 of a wildtype CD47 or at a corresponding position in a homologous CD47 gene. In some embodiments, a glycine is substituted for the amino acid at position 47 of a wildtype CD47 or at a corresponding position in a homologous CD47 gene. In some embodiments, a mutation of a gene encoding CD47 comprises a substitution of the amino acid at position 49 of a wildtype CD47 or at a corresponding position in a homologous CD47 gene. In some embodiments, an arginine is substituted for the amino acid at position 49 of a wildtype CD47 or at a corresponding position in a homologous CD47 gene. In some embodiments, a proline is substituted for the amino acid at position 49 of a wildtype CD47 or at a corresponding position in a homologous CD47 gene. In some embodiments, a mutation of a gene encoding CD47 comprises a substitution of the amino acid at position 52 of a wildtype CD47 or at a corresponding position in a homologous CD47 gene. In some embodiments, a mutation of a gene encoding CD47 comprises a deletion of the amino acid at position 52 of a wildtype CD47 or at a corresponding position in a homologous CD47 gene. In some embodiments, a mutation of a gene encoding CD47 comprises a substitution of the amino acid at position 53 of a wildtype CD47 or at a corresponding position in a homologous CD47 gene. In some embodiments, an alanine is substituted for the amino acid at position 53 of a wildtype CD47 or at a corresponding position in a homologous CD47 gene. In some embodiments, a mutation of a gene encoding CD47 comprises a deletion of the amino acid at position 53 of a wildtype CD47 or at a corresponding position in a homologous CD47 gene. In some embodiments, a mutation of a gene encoding CD47 comprises a deletion of the amino acid at position 54 of a wildtype CD47 or at a corresponding position in a homologous CD47 gene. In some embodiments, a mutation of a gene encoding CD47 comprises a deletion of the amino acid at position 55 of a wildtype CD47 or at a corresponding position in a homologous CD47 gene. In some embodiments, a mutation of a gene encoding CD47 comprises a substitution of the amino acid at position 120 of a wildtype CD47 or at a corresponding position in a homologous CD47 gene. In some embodiments, an alanine is substituted for the amino acid at position 120 of a wildtype CD47 or at a corresponding position in a homologous CD47 gene. In some embodiments, a mutation of a gene encoding CD47 comprises a substitution of the amino acid at position 124 of a wildtype CD47 or at a corresponding position in a homologous CD47 gene. In some embodiments, a lysine is substituted for the amino acid at position 124 of a wildtype CD47 or at a corresponding position in a homologous CD47. In some embodiments, a mutation of a gene encoding CD47 makes a change in the amino acid sequence corresponding to the amino acid sequence of a CD47 ortholog. In some embodiments, a mutation substitutes an amino acid of human CD47 for an amino acid at a corresponding position of an orthologous CD47, e.g., a non-human primate CD47. In some embodiments, a mutation inserts or deletes one or more amino acids of human CD47 to correspond to the sequence of an orthologous CD47, e.g., a non-human primate CD47. In some embodiments, a mutation changes the amino acid sequence in a manner corresponding to a tolerable genetic variant identified by one or more genomic sequence comparison algorithms, e.g., gnomAD (see, e.g., Gudmundsson et al. arXiv:2107.11458v3, e.g., gnomad.broadinstitute.org/), or to a position characterized by a plurality of tolerable genetic variants. In some embodiments, mutations to CD47 corresponding to the amino acid sequence of a CD47 ortholog or at positions characterized by a plurality of tolerable genetic variants decrease or eliminate binding of an immunotherapeutic agent targeting CD47 while preserving some or all of CD47 structure, expression, and/or functionality, providing a cell expressing CD47 (e.g., functional CD47) that is targeted less or not at all by anti-CD47 immunotherapeutic agents. Genetically Engineered Hematopoietic Cell Expressing CD34 Mutants In some embodiments, a genetically engineered hematopoietic cell has reduced or eliminated expression of a cell surface protein, or expresses a mutant of a cell surface protein, wherein the cell surface protein is CD34. CD34 is a transmembrane phosphoglycoprotein belonging to the single-pass transmembrane sialomucin protein family that functions as a cell-cell adhesion factor. Accordingly, CD34 is an important adhesion molecule required for T-cells to enter lymph nodes and for attachment of hematopoietic stem cells to bone marrow extracellular matrix or to stromal cells. CD34 is highly expressed in hematopoietic stem and progenitor cells and endothelial cells. Moreover, CD34 is commonly found expressed on the cell surface of hematopoietic cancer cells (see, e.g., Sydney et al. Stem Cells (2014) 32 (6): 1380-1389; Nielsen and McNagny. Journal of Cell Science (2008) 121 (22): 3683-3692; Lanze et al. Journal of Biological Regulators and Homeostatic Agents (2001) 15 (1): 1-13; Sutherland and Keating. Journal of Hematotherapy (2009) 1 (2): 115-129). CD34 is located on chromosome 1 and contains 8 exons. In some embodiments, a mutation of a gene encoding CD34 alters one or more amino acids associated with an epitope of CD34. In some embodiments, the epitope of CD34 is a portion of CD34 bound by an agent, e.g., an immunotherapeutic agent. In some embodiments, the agent is an anti-CD34 antibody. In some embodiments, the anti-CD34 antibody is clone QBend10 or 561. In some embodiments, the agent comprises an anti-CD34 antibody or portion thereof, e.g., an antibody drug conjugate (ADC), a chimeric antigen receptor (CAR), or a multispecific antibody (e.g., a bispecific T cell engager). In some embodiments, the epitope of CD34 is one or more amino acids of a protein domain (e.g., the extracellular domain) or the amino acids encoded by an exon or combination of exons of the gene encoding CD34. In some embodiments, a mutation of a gene encoding CD34 comprises a substitution of the amino acid at position 42 of a wildtype CD34 or at a corresponding position in a homologous CD34 gene. In some embodiments, a glycine is substituted for the amino acid at position 42 of a wildtype CD34 or at a corresponding position in a homologous CD34 gene. In some embodiments, a mutation of a gene encoding CD34 comprises a substitution of the amino acid at position 46 of a wildtype CD34 or at a corresponding position in a homologous CD34 gene. In some embodiments, an alanine is substituted for the amino acid at position 46 of a wildtype CD34 or at a corresponding position in a homologous CD34 gene. In some embodiments, a mutation of a gene encoding CD34 comprises a substitution of the amino acid at position 47 of a wildtype CD34 or at a corresponding position in a homologous CD34 gene. In some embodiments, a lysine is substituted for the amino acid at position 47 of a wildtype CD34 or at a corresponding position in a homologous CD34 gene. In some embodiments, a glutamate is substituted for the amino acid at position 47 of a wildtype CD34 or at a corresponding position in a homologous CD34 gene. In some embodiments, a mutation of a gene encoding CD34 comprises a substitution of the amino acid at position 49 of a wildtype CD34 or at a corresponding position in a homologous CD34 gene. In some embodiments, a proline is substituted for the amino acid at position 49 of a wildtype CD34 or at a corresponding position in a homologous CD34 gene. In some embodiments, a serine is substituted for the amino acid at position 49 of a wildtype CD34 or at a corresponding position in a homologous CD34 gene. In some embodiments, a mutation of a gene encoding CD34 comprises a substitution of the amino acid at position 50 of a wildtype CD34 or at a corresponding position in a homologous CD34 gene. In some embodiments, an alanine is substituted for the amino acid at position 50 of a wildtype CD34 or at a corresponding position in a homologous CD34 gene. In some embodiments, a proline is substituted for the amino acid at position 50 of a wildtype CD34 or at a corresponding position in a homologous CD34 gene. In some embodiments, a mutation of a gene encoding CD34 comprises a substitution of the amino acid at position 51 of a wildtype CD34 or at a corresponding position in a homologous CD34 gene. In some embodiments, an alanine is substituted for the amino acid at position 51 of a wildtype CD34 or at a corresponding position in a homologous CD34 gene. In some embodiments, a mutation of a gene encoding CD34 comprises a substitution of the amino acid at position 54 of a wildtype CD34 or at a corresponding position in a homologous CD34 gene. In some embodiments, an alanine is substituted for the amino acid at position 54 of a wildtype CD34 or at a corresponding position in a homologous CD34 gene. In some embodiments, a mutation of a gene encoding CD34 comprises a substitution of the amino acid at position 55 of a wildtype CD34 or at a corresponding position in a homologous CD34 gene. In some embodiments, an alanine is substituted for the amino acid at position 55 of a wildtype CD34 or at a corresponding position in a homologous CD34 gene. In some embodiments, a mutation of a gene encoding CD34 makes a change in the amino acid sequence corresponding to the amino acid sequence of a CD34 ortholog. In some embodiments, a mutation substitutes an amino acid of human CD34 for an amino acid at a corresponding position of an orthologous CD34, e.g., a non-human primate CD34. In some embodiments, a mutation inserts or deletes one or more amino acids of human CD34 to correspond to the sequence of an orthologous CD34, e.g., a non-human primate CD34. In some embodiments, a mutation changes the amino acid sequence in a manner corresponding to a tolerable genetic variant identified by one or more genomic sequence comparison algorithms, e.g., gnomAD (see, e.g., Gudmundsson et al. arXiv:2107.11458v3, e.g., gnomad.broadinstitute.org/), or to a position characterized by a plurality of tolerable genetic variants. In some embodiments, mutations to CD34 corresponding to the amino acid sequence of a CD34 ortholog or at positions characterized by a plurality of tolerable genetic variants decrease or eliminate binding of an immunotherapeutic agent targeting CD34 while preserving some or all of CD34 structure, expression, and/or functionality, providing a cell expressing CD34 (e.g., functional CD34) that is targeted less or not at all by anti-CD34 immunotherapeutic agents. Genetically Engineered Hematopoietic Cell Expressing IL1RAP Mutants In some embodiments, a genetically engineered hematopoietic cell has reduced or eliminated expression of a cell surface protein, or expresses a mutant of a cell surface protein, wherein the cell surface protein is interleukin-1 receptor accessory protein (IL1RAP). IL1RAP is known by several aliases in the art, such as IL-1 receptor accessory protein (IL- 1RAcP), interleukin-1 receptor 3 (IL1R3), and interleukin-1 receptor accessory protein beta. As will be known to one of ordinary skill in the art, IL1RAP is involved in initiating signaling events that regulate activation of genes that are responsive to the interleukin 1 inflammatory cytokine. IL1RAP also interacts with IL1RL2 to form a heterodimer in the IL- 36 cytokine signaling system. In humans, IL1RAP is found at chromosomal location 3q28 and comprises 17 exons. Non-limiting examples of reference sequence for the gene encoding IL1RAP are found at NC_000003.12 and NC_060927.1. It will also be known to the skilled artisan that IL1RAP is encoded by gene capable of undergoing alternative splicing resulting in production of membrane-bound and soluble isoforms. Said IL1RAP isoforms differ in their C-terminus and the ratios of soluble to membrane-bound isoforms may change depending on cell signaling events (e.g., increases in the soluble isoform during acute-phase induction or stress). Non- limiting examples of gene products (e.g., RNAs and proteins) encoded by IL1RAP are set forth in NM_001167928.2 (encoding a protein found at NP_001161400.1), NM_001167929.1 (encoding a protein found at NP_001161401.1), NM_0011679302 (encoding a protein found at NP_001161402.1), NM_001167931.2 (encoding a protein found at NP_001161403.1), NM_001364878.1 (encoding a protein found at NP_001351808.1), NM_001364880.2 (encoding a protein found at NP_001351809.1), NM_001364881.2 (encoding a protein found at NP_001351810.1), and NM_002182.4 (encoding a protein found at NP_002173.1), NM_134470.4 (encoding a protein found at NP_608273.1). In some embodiments, the IL1RAP protein comprises an Ig-like C2-type 1 domain at amino acids 21-128, an Ig-like C2-type 2 domain at amino acids 141-230, an Ig-like C2-type 3 domain at amino acids 242-348, and a TIR domain from amino acids 403-546 which mediates NAD+ hydrolase activity. In some embodiments, the membrane-bound isoform comprises an amino acid sequence as set forth below: MTLLWCVVSLYFYGILQSDASERCDDWGLDTMRQIQVFEDEPARIKCPLFEHFLKFNYSTAHSAGLTLIWYWTRQ DRDLEEPINFRLPENRISKEKDVLWFRPTLLNDTGNYTCMLRNTTYCSKVAFPLEVVQKDSCFNSPMKLPVHKLY IEYGIQRITCPNVDGYFPSSVKPTITWYMGCYKIQNFNNVIPEGMNLSFLIALISNNGNYTCVVTYPENGRTFHL TRTLTVKVVGSPKNAVPPVIHSPNDHVVYEKEPGEELLIPCTVYFSFLMDSRNEVWWTIDGKKPDDITIDVTINE SISHSRTEDETRTQILSIKKVTSEDLKRSYVCHARSAKGEVAKAAKVKQKVPAPRYTVELACGFGATVLLVVILI VVYHVYWLEMVLFYRAHFGTDETILDGKEYDIYVSYARNAEEEEFVLLTLRGVLENEFGYKLCIFDRDSLPGGIV TDETLSFIQKSRRLLVVLSPNYVLQGTQALLELKAGLENMASRGNINVILVQYKAVKETKVKELKRAKTVLTVIK WKGEKSKYPQGRFWKQLQVAMPVKKSPRRSSSDEQGLSYSSLKNV (SEQ ID NO: 8)(UniPort ID: Q9NPH3-1) In some embodiments, the soluble-bound isoform comprises an amino acid sequence as set forth below: MTLLWCVVSLYFYGILQSDASERCDDWGLDTMRQIQVFEDEPARIKCPLFEHFLKFNYSTAHSAGLTLIWYWTRQ DRDLEEPINFRLPENRISKEKDVLWFRPTLLNDTGNYTCMLRNTTYCSKVAFPLEVVQKDSCFNSPMKLPVHKLY IEYGIQRITCPNVDGYFPSSVKPTITWYMGCYKIQNFNNVIPEGMNLSFLIALISNNGNYTCVVTYPENGRTFHL TRTLTVKVVGSPKNAVPPVIHSPNDHVVYEKEPGEELLIPCTVYFSFLMDSRNEVWWTIDGKKPDDITIDVTINE SISHSRTEDETRTQILSIKKVTSEDLKRSYVCHARSAKGEVAKAAKVKQKGNRCGQ (SEQ ID NO: 9) (UniPort ID: Q9NPH3-2) MTLLWCVVSLYFYGILQSDASERCDDWGLDTMRQIQVFEDEPARIKCPLFEHFLKFNYSTAHSAGLTLIWYWTRQ DRDLEEPINFRLPENRISKEKDVLWFRPTLLNDTGNYTCMLRNTTYCSKVAFPLEVVQKDSCFNSPMKLPVHKLY IEYGIQRITCPNVDGYFPSSVKPTITWYMGCYKIQNFNNVIPEGMNLSFLIALISNNGNYTCVVTYPENGRTFHL TRTLTVKVVGSPKNAVPPVIHSPNDHVVYEKEPGEELLIPCTVYFSFLMDSRNEVWWTIDGKKPDDITIDVTINE SASSKIHSGTGLWFWSHSPASGDSHCCLPCLLARDGPILPGSFWNR (SEQ ID NO: 10)(UniPort ID: Q9NPH3-3) In some embodiments, the present disclosure relates to variants of IL1RAP. In some embodiments, genetically engineered hematopoietic cells comprise a variant gene encoding IL1RAP protein, wherein said variant gene comprises a substitution, deletion, or addition of at least one nucleotide or a combination thereof. In some embodiments, a mutant IL1RAP comprises a deletion or mutation of a fragment of the protein that is encoded by any one of the exons of the gene encoding IL1RAP. In some embodiments, genetically engineered hematopoietic cells comprise a mutant IL1RAP protein comprising a substitution, deletion, or addition of at least one amino acid or a combination thereof. In some embodiments, genetically engineered hematopoietic cells comprise a mutant IL1RAP protein comprising at least one modified non-essential epitope. In some embodiments, genetically engineered hematopoietic cells comprise IL1RAP expression that is reduced by at least 20%, at least 30%, at least 40%, at least 50%, at least 60%, at least 70%, at least 80%, at least 90%, or at least 95% relative to the wildtype counterpart cell. In some embodiments, genetically engineered hematopoietic cells express a mutant of IL1RAP that has reduced or no binding to an agent comprising an antigen-binding fragment that recognizes an epitope of IL1RAP. In some embodiments, at least 40%, at least 50%, at least 60%, at least 70%, at least 75%, at least 80%, at least 85%, at least 90%, or at least 95% of copies of IL1RAP in the population of cells have a mutation. By way of example, a population can comprise a plurality of different IL1RAP mutations and each mutation of the plurality contributes to the percent of copies of IL1RAP in the population of cells that have a mutation. In some embodiments, the expression of IL1RAP on the genetically engineered hematopoietic cell is compared to the expression of IL1RAP on a naturally occurring hematopoietic cell (e.g., a wild-type counterpart). In some embodiments, the genetic engineering results in a reduction in the expression level of IL1RAP by at least50%, at least 60%, at least 70%, at least 80%, at least 90%, at least 91%, at least 92%, at least 93%, at least 94%, at least 95%, at least 96%, at least 97%, at least 98%, or at least 99% as compared to the expression of IL1RAP on a naturally occurring hematopoietic cell (e.g., a wild-type counterpart). For example, in some embodiments, the genetically engineered hematopoietic cell expresses less than 20%, less than 19%, less than 18%, less than 17%, less than 16%, less than 15%, less than 14%, less than 13%, less than 12%, less than 11%, less than 10%, less than 9%, less than 8%, less than 7%, less than 6%, less than 5%, less than 4%, less than 3%, less than 2%, less than or 1% of IL1RAP as compared to a naturally occurring hematopoietic cell (e.g., a wild-type counterpart). In some embodiments, the genetic engineering results in a reduction in the expression level of wild-type IL1RAP by at least 50%, at least 60%, at least 70%, at least 80%, at least 90%, at least 91%, at least 92%, at least 93%, at least 94%, at least 95%, at least 96%, at least 97%, at least 98%, or at least 99% as compared to the expression of the level of wild-type IL1RAP on a naturally occurring hematopoietic cell (e.g., a wild-type counterpart). That is, in some embodiments, the genetically engineered hematopoietic cell expresses less than 20%, less than 19%, less than 18%, less than 17%, less than 16%, less than 15%, less than 14%, less than 13%, less than 12%, less than 11%, less than 10%, less than 9%, less than 8%, less than 7%, less than 6%, less than 5%, less than 4%, less than 3%, less than 2%, or less than 1% of IL1RAP as compared to a naturally occurring hematopoietic cell (e.g., a wild-type counterpart). Genetically Engineered Hematopoietic Cell Expressing CD33 Mutants In some embodiments, a genetically engineered hematopoietic cell has reduced or eliminated expression of a lineage-specific cell surface protein, or expresses a mutant of a lineage-specific cell surface protein, wherein the lineage-specific cell surface protein is CD33. As will be known to one of ordinary skill in the art, CD33 is encoded by seven exons, including the alternatively spliced exons 7A and 7B (Brinkman-Van der Linden et al. Mol Cell. Biol. (2003) 23: 4199-4206). Further, the CD33 gene encodes two isoforms, one of which retains exon 2, referred to as CD33M, and one that excludes exon 2, referred to as CD33m. Exemplary amino acid sequences of the 7A and 7B splicing isoforms are provided below: CD33M-7A: Amino Acid (underlined=exon 2; italicized= exon 7A) MPLLLLLPLLWAGALAMDPNFWLQVQESVTVQEGLCVLVPCTFFHPIPYYDKNSPVHGYWFREGAIIS RDSPVATNKLDQEVQEETQGRFRLLGDPSRNNCSLSIVDARRRDNGSYFFRMERGSTKYSYKSPQLSV HVTDLTHRPKILIPGTLEPGHSKNLTCSVSWACEQGTPPIFSWLSAAPTSLGPRTTHSSVLIITPRPQ DHGTNLTCQVKFAGAGVTTERTIQLNVTYVPQNPTTGIFPGDGSGKQETRAGVVHGAIGGAGVTALLA LCLCLIFFIVKTHRRKAARTAVGRNDTHPTTGSASPKHQKKSKLHGPTETSSCSGAAPTVEMDEELHY ASLNFHGMNPSKDTSTEYSEVRTQ (SEQ ID NO: 1) CD33-M; transcript variant 1 mRNA. NM_001772 (underlined=exon 2; italicized= exon 7A) AUGCCGCUGCUGCUACUGCUGCCCCUGCUGUGGGCAGGGGCCCUGGCUAUGGAUCCAAAUUUCUGGCU GCAAGUGCAGGAGUCAGUGACGGUACAGGAGGGUUUGUGCGUCCUCGUGCCCUGCACUUUCUUCCAUC CCAUACCCUACUACGACAAGAACUCCCCAGUUCAUGGUUACUGGUUCCGGGAAGGAGCCAUUAUAUCC AGGGACUCUCCAGUGGCCACAAACAAGCUAGAUCAAGAAGUACAGGAGGAGACUCAGGGCAGAUUCCG CCUCCUUGGGGAUCCCAGUAGGAACAACUGCUCCCUGAGCAUCGUAGACGCCAGGAGGAGGGAUAAUG GUUCAUACUUCUUUCGGAUGGAGAGAGGAAGUACCAAAUACAGUUACAAAUCUCCCCAGCUCUCUGUG CAUGUGACAGACUUGACCCACAGGCCCAAAAUCCUCAUCCCUGGCACUCUAGAACCCGGCCACUCCAA AAACCUGACCUGCUCUGUGUCCUGGGCCUGUGAGCAGGGAACACCCCCGAUCUUCUCCUGGUUGUCAG CUGCCCCCACCUCCCUGGGCCCCAGGACUACUCACUCCUCGGUGCUCAUAAUCACCCCACGGCCCCAG GACCACGGCACCAACCUGACCUGUCAGGUGAAGUUCGCUGGAGCUGGUGUGACUACGGAGAGAACCAU CCAGCUCAACGUCACCUAUGUUCCACAGAACCCAACAACUGGUAUCUUUCCAGGAGAUGGCUCAGGGA AACAAGAGACCAGAGCAGGAGUGGUUCAUGGGGCCAUUGGAGGAGCUGGUGUUACAGCCCUGCUCGCU CUUUGUCUCUGCCUCAUCUUCUUCAUAGUGAAGACCCACAGGAGGAAAGCAGCCAGGACAGCAGUGGG CAGGAAUGACACCCACCCUACCACAGGGUCAGCCUCCCCGAAACACCAGAAGAAGUCCAAGUUACAUG GCCCCACUGAAACCUCAAGCUGUUCAGGUGCCGCCCCUACUGUGGAGAUGGAUGAGGAGCUGCAUUAU GCUUCCCUCAACUUUCAUGGGAUGAAUCCUUCCAAGGACACCUCCACCGAAUACUCAGAGGUCAGGAC CCAGUGA (SEQ ID NO: 11) CD33m-7A: Amino Acid (italicized= exon 7A) MPLLLLLPLLWADLTHRPKILIPGTLEPGHSKNLTCSVSWACEQGTPPIFSWLSAAPTSLGPRTTHSS VLIITPRPQDHGTNLTCQVKFAGAGVTTERTIQLNVTYVPQNPTTGIFPGDGSGKQETRAGVVHGAIG GAGVTALLALCLCLIFFIVKTHRRKAARTAVGRNDTHPTTGSASPKHQKKSKLHGPTETSSCSGAAPT VEMDEELHYASLNFHGMNPSKDTSTEYSEVRTQ(SEQ ID NO: 12) CD33-m transcript variant 2; no exon 2, exon 7A mRNA. NM_001082618 (italicized= exon 7A)) AUGCCGCUGCUGCUACUGCUGCCCCUGCUGUGGGCAGACUUGACCCACAGGCCCAAAAUCCUCAUCCC UGGCACUCUAGAACCCGGCCACUCCAAAAACCUGACCUGCUCUGUGUCCUGGGCCUGUGAGCAGGGAA CACCCCCGAUCUUCUCCUGGUUGUCAGCUGCCCCCACCUCCCUGGGCCCCAGGACUACUCACUCCUCG GUGCUCAUAAUCACCCCACGGCCCCAGGACCACGGCACCAACCUGACCUGUCAGGUGAAGUUCGCUGG AGCUGGUGUGACUACGGAGAGAACCAUCCAGCUCAACGUCACCUAUGUUCCACAGAACCCAACAACUG GUAUCUUUCCAGGAGAUGGCUCAGGGAAACAAGAGACCAGAGCAGGAGUGGUUCAUGGGGCCAUUGGA GGAGCUGGUGUUACAGCCCUGCUCGCUCUUUGUCUCUGCCUCAUCUUCUUCAUAGUGAAGACCCACAG GAGGAAAGCAGCCAGGACAGCAGUGGGCAGGAAUGACACCCACCCUACCACAGGGUCAGCCUCCCCGA AACACCAGAAGAAGUCCAAGUUACAUGGCCCCACUGAAACCUCAAGCUGUUCAGGUGCCGCCCCUACU GUGGAGAUGGAUGAGGAGCUGCAUUAUGCUUCCCUCAACUUUCAUGGGAUGAAUCCUUCCAAGGACAC CUCCACCGAAUACUCAGAGGUCAGGACCCAGUGA (SEQ ID NO: 13) CD33M-7B: Amino Acid (underlined=exon 2; italicized= exon 7B) MPLLLLLPLLWAGALAMDPNFWLQVQESVTVQEGLCVLVPCTFFHPIPYYDKNSPVHGYWFREGAIIS RDSPVATNKLDQEVQEETQGRFRLLGDPSRNNCSLSIVDARRRDNGSYFFRMERGSTKYSYKSPQLSV HVTDLTHRPKILIPGTLEPGHSKNLTCSVSWACEQGTPPIFSWLSAAPTSLGPRTTHSSVLIITPRPQ DHGTNLTCQVKFAGAGVTTERTIQLNVTYVPQNPTTGIFPGDGSGKQETRAGVVHGAIGGAGVTALLA LCLCLIFFIVKTHRRKAARTAVGRNDTHPTTGSASPVR (SEQ ID NO: 14) CD33 transcript variant 3; exon 7B mRNA. NM_001177608 (underlined= exon 2; italicized= exon 7B)) AUGCCGCUGCUGCUACUGCUGCCCCUGCUGUGGGCAGGGGCCCUGGCUAUGGAUCCAAAUUUCUGGCUGCAAGUG CAGGAGUCAGUGACGGUACAGGAGGGUUUGUGCGUCCUCGUGCCCUGCACUUUCUUCCAUCCCAUACCCUACUAC GACAAGAACUCCCCAGUUCAUGGUUACUGGUUCCGGGAAGGAGCCAUUAUAUCCAGGGACUCUCCAGUGGCCACA AACAAGCUAGAUCAAGAAGUACAGGAGGAGACUCAGGGCAGAUUCCGCCUCCUUGGGGAUCCCAGUAGGAACAAC UGCUCCCUGAGCAUCGUAGACGCCAGGAGGAGGGAUAAUGGUUCAUACUUCUUUCGGAUGGAGAGAGGAAGUACC AAAUACAGUUACAAAUCUCCCCAGCUCUCUGUGCAUGUGACAGACUUGACCCACAGGCCCAAAAUCCUCAUCCCU GGCACUCUAGAACCCGGCCACUCCAAAAACCUGACCUGCUCUGUGUCCUGGCCUGUGAGCAGGGAACACCCCCGA UCUUCUCCGGUUGUCAGCUGCCCCCACCUCCCUGGGCCCCAGGACUACUCACUCCUCGGUGCUCAUAAUCACCCC ACGGCCCCAGGACCACGGCACCAACCUGACCUGUCAGGUGAAGUUCGCUGGAGCUGGUGUGACUACGGAGAGAAC CAUCCAGCUCAACGUCACCUAUGUUCCACAGAACCCAACAACUGGUAUCUUUCCGGAGAUGGCUCAGGGAAACAA GAGACCAGAGCAGGAGUGGUUCAUGGGGCCAUUGGAGGAGCUGGUGUUACAGCCCUGCUCGCUCUUUGUCUCUGC CUCAUCUUCUUCAUAGUGAAGACCCACAGGAGGAAAGCAGCCAGGACAGCAGUGGGCAGGAAUGACACCCACCCU ACCACAGGGUCAGCCUCCCCGGUACGUUGA (SEQ ID NO: 15) CD33m-7B Amino Acid (italicized= exon 7B) MPLLLLLPLLWADLTHRPKILIPGTLEPGHSKNLTCSVSWACEQGTPPIFSWLSAAPTSLGPRTTHSS VLIITPRPQDHGTNLTCQVKFAGAGVTTERTIQLNVTYVPQNPTTGIFPGDGSGKQETRAGVVHGAIG GAGVTALLALCLCLIFFIVKTHRRKAARTAVGRNDTHPTTGSASPVR (SEQ ID NO: 16) CD33 transcript variant 4; no exon 2, exon 7B. mRNA (italicized= exon 7B) AUGCCGCUGCUGCUACUGCUGCCCCUGCUGUGGGCAAGGAACAACUGCUCCCUGAGCAUCGUAGACGCCAGGAGG AGGGAUAAUGGUUCAUACUUCUUUCGGAUGGAGAGAGGAAGUACCAAAUACAGUUACAAAUCUCCCCAGCUCUCU GUGCAUGUGACAGACUUGACCCACAGGCCCAAAAUCCUCAUCCCUGGCACUCUAGAACCCGGCCACUCCAAAAAC CUGACCUGCUCUGUGUCCUGGCCUGUGAGCAGGGAACACCCCCGAUCUUCUCCGGUUGUCAGCUGCCCCCACCUC CCUGGGCCCCAGGACUACUCACUCCUCGGUGCUCAUAAUCACCCCACGGCCCCAGGACCACGGCACCAACCUGAC CUGUCAGGUGAAGUUCGCUGGAGCUGGUGUGACUACGGAGAGAACCAUCCAGCUCAACGUCACCUAUGUUCCACA GAACCCAACAACUGGUAUCUUUCCGGAGAUGGCUCAGGGAAACAAGAGACCAGAGCAGGAGUGGUUCAUGGGGCC AUUGGAGGAGCUGGUGUUACAGCCCUGCUCGCUCUUUGUCUCUGCCUCAUCUUCUUCAUAGUGAAGACCCACAGG AGGAAAGCAGCCAGGACAGCAGUGGGCAGGAAUGACACCCACCCUACCACAGGGUCAGCCUCCCCGGUACGUUGA (SEQ ID NO: 17) Using human CD33 as an exemplary lineage-specific cell surface protein, regions of the protein in which mutation and/or deletion of amino acids are less likely to result in deleterious effects (e.g., a reduction or abrogation of function) were predicted using PROVEAN software (see: provean.jcvi.org; Choi et al. PLoS ONE (2012) 7(10): e46688). Examples of the predicted regions are shown in boxes in FIGURE 2 and exemplary deletions in the predicated regions are presented in Table 2. Numbering of the amino acid residues is based on the amino acid sequence of human CD33 provided by SEQ ID NO: 1 (CD33M-7A). Table 2: Exemplary deletions in CD33
As shown in Table 1, type 0 lineage-specific cell surface proteins are necessary for the tissue homeostasis and survival, and cell types carrying type 0 lineage-specific cell surface protein may be also necessary for survival of the subject. Thus, given the importance of type 0 lineage-specific cell surface proteins, or cells carrying type 0 lineage-specific cell surface proteins, in homeostasis and survival, targeting this category of proteins may be challenging using conventional CAR T cell immunotherapies, as the inhibition or removal of such proteins and cell carrying such proteins may be detrimental to the survival of the subject. Consequently, lineage-specific cell surface proteins (such as type 0 lineage-specific proteins) and/or the cell types that carry such proteins may be required for the survival, for example because it performs a vital non-redundant function in the subject, then this type of lineage specific protein may be a poor target for conventional CAR T cell-based immunotherapies. However, by combining the genetically engineered hematopoietic stem cells described herein and a cytotoxic agent such as CAR-T cell-based therapy, the selection of target cell surface protein can be expanded to essential antigens such as type 0 lineage- specific cell surface proteins. In some embodiments, the engineered hematopoietic cells (e.g., HSCs) have one or more genes of type 0 antigens edited for expression of these type 0 antigens in mutated form, which retain (at least partially) bioactivity of the type 0 antigens but can escape targeting by type 0 antigen-specific cytotoxic agents such as CAR-T cells so as to remedy the loss of normal cells expressing the type 0 antigens due to the therapy. In contrast to type 0 proteins, type 1 lineage-specific cell surface proteins and cells carrying type 1 lineage-specific cell surface proteins are not required for tissue homeostasis or survival of the subject. Targeting type 1 lineage-specific cell surface proteins is not likely to lead to acute toxicity and/or death of the subject. For example, as described in Elkins et al. (Mol. Cancer Ther. (2012) 10:2222-32) a CAR T cell engineered to target CD307, a type 1 protein expressed uniquely on both normal plasma cells and multiple myeloma (MM) cells would lead to elimination of both cell types. However, since the plasma cell lineage is expendable for the survival of the organism, CD307 and other type 1 lineage specific proteins are proteins that are suitable for CAR T cell-based immunotherapy. Lineage specific proteins of type 1 class may be expressed in a wide variety of different tissues, including, ovaries, testes, prostate, breast, endometrium, and pancreas. In some embodiments, the genetically engineered hematopoietic cells (e.g., HSCs) have one or more genes of type 1 antigens for expression of the type 1 proteins in mutated forms, which retain (at least partially) bioactivity of the type 1 antigens but can escape targeting by type 1 antigen-specific cytotoxic agents such as CAR-T cells. Use of such engineered HSCs (either alone or in combination with cytotoxic agents such as CAR-T cells targeting the type 1 antigens) may improve the longer-term survival and quality of life of the patient. For example, targeting all plasma cells, while not expected to lead to acute toxicity and/or death, could have longer-term consequences such as reduced function of the humoral immune system leading to increased risk of infection. Targeting type 2 proteins presents a significant difficulty as compared to type 1 proteins. Type 2 proteins are those characterized where: (1) the protein is dispensable for the survival of an organism (i.e., is not required for the survival), and (2) the cell lineage carrying the protein is indispensable for the survival of an organism (i.e., the particular cell lineage is required for the survival). For example, CD33 is a type 2 protein expressed in both normal myeloid cells as well as in Acute Myeloid Leukemia (AML) cells (Dohner et al., NEJM (2015) 373:1136). As a result, a CAR T cell engineered to target CD33 protein could lead to the killing of both normal myeloid cells as well as AML cells, which may be incompatible with survival of the subject. In some embodiments, the genetically engineered hematopoietic cells (e.g., HSCs) have one or more genes of type 2 antigens for expression of the type 2 antigens in mutated form, which retain (at least partially) bioactivity of the type 2 antigens but can escape targeting by type 1 antigen-specific cytotoxic agents such as CAR-T cells. Use of such engineered HSCs (either alone or in combination with cytotoxic agents, such as CAR-T cells targeting the type 2 antigens) may improve the longer-term survival and quality of life of the patient. For example, targeting all plasma cells, while not expected to lead to acute toxicity and/or death, could have longer-term consequences such as reduced function of the humoral immune system leading to increased risk of infection. In some embodiments, malignant hematopoietic cells may have altered expression or express a mutant of a cell surface protein. In some embodiments malignant hematopoietic cells may have altered expression or express a mutant of cell surface protein selected from a group consisting of: CD10; CD101; CD11A; CD11C; CD123; CD133; CD152; CD200; CD205; CD244; CD25; CD305; CD33; CD34; CD366; CD38; CD42B; CD44; CD45RA; CD47; CD48; CD49D; CD49F; CD52; CD58; CD64; CD7; CD84; CD85K; CD9; CD99; CLL-1; EMR2; GPR56; IL1RAP; and CD274 In some embodiments, malignant hematopoietic cells may have altered expression or express a mutant of cell surface protein selected from a group consisting of: CD117; CD11A; CD11C; CD120B; CD123; CD13; CD133; CD152; CD205; CD226; CD244; CD25; CD305; CD32; CD33; CD34; CD38; CD41; CD44; CD45RA; CD47; CD49D; CD49F; CD52; CD58; CD64; CD7; CD70; CD82; CD86; CD96; CD99; CLL-1; EMR2; FR-B; GPR56; and IL1RAP. In some embodiments, malignant hematopoietic cells may have altered expression or express of a first cell surface protein and a second cell surface protein selected from a group consisting of: CD274 and CD152; CD274 and CD226; CD152 and CD226; CD10 and CD49F; CD49F and GPR56; CD200 and CD34; CD34 and CD52; CD34 and CD133; GPR56 and CD133; CD42B and CD41; CD85K and CD64; CD85K and CD32; CD64 and CD32; CD64 and CD11C; CD64 and CD86; CD45RA and CD96; CD45RA and IL1RAP; CD45RA and CD123; CD84 and CD33; CD11C and IL1RAP; CD11C and CD33; CD11C and CD86; CD9 and IL1RAP; IL1RAP and CD33; IL1RAP and CD205; IL1RAP and CD305; IL1RAP and CD123; CD33 and CLL-1; CD33 and CD305; CD11A and CD86; CLL-1 and CD305; CD58 and CD305; CD49D and CD47; CD49D and CD99; CD49D and CD44; CD49D and CD305; CD47 and CD99; CD47 and EMR2; CD99 and CD44; CD99 and CD305; CD99 and EMR2; CD205 and CD305; CD205 and CD123; CD44 and CD305; CD44 and EMR2; CD244 and CD117; CD123 and EMR2; EMR2 and CD82; CD152 and CD120B; CD48 and CD11A; CD200 and CD25; CD34 and CD7; CD34 and CD13; CD52 and GPR56; CD52and CD133; CD52 and CD7; GPR56 and CD7; GPR56 and CD82; CD133 and CD7; CD133 and CD82; CD7 and CD13; CD101 and CD64; CD85 and CD70; CD85K and CD86; CD64 and FR-B; CD366 and CD96; CD25 and CD45RA; CD25 and CD96; CD9 and CD82; IL1RAP and CD58; CD11A and CD38; CD11A and CD44; CD11A and CD244; CD38 and CD305; CD38 and EMR2; CLL-1 and CD49D; CD58 and CD49D; CD47 and CD205; and CD47 and CD44. In some embodiments, malignant hematopoietic cells may have altered expression or express of a first cell surface protein and a second cell surface protein selected from a group consisting of: CD85K and CD64; CD45RA and IL1RAP; CD34 and CD133; IL1RAP and CD33; CD47 and CD99; CLL-1 and CD305; CD152 and CD226; CD45RA and CD96; CD45RA and CD123; CD49D and CD305; IL1RAP and CD305; CD58 and CD305; CD49D and CD47; CD33 and CD305; CD49D and CD44; CD64 and CD86; GPR56 and CD133; CD34 and CD52; EMR2 and CD82; CD64 and CD32; CD42B and CD41; CD64 and CD11C; CD99 and CD44; CD200 and CD34; CD11C and CD33; IL1RAP and CD123; CD11C and IL1RAP; CD44 and EMR2; CD205 and CD305; CD44 and CD305; CD49D and CD99; CD274 and CD226; CD49F and GPR56; CD9 and IL1RAP; CD99 and CD305; CD10 and CD49F; CD11A and CD86; CD33 and CLL-1; CD84 and CD33; CD244 and CD117; CD85K and CD32; CD47 and EMR2; CD274 and CD152; IL1RAP and CD205; CD11C and CD86; CD99 and EMR2; CD205 and CD123; and CD123 and EMR2. In some embodiments, malignant hematopoietic cells may have altered expression or express of a first cell surface protein and a second cell surface protein selected from a group consisting of: CD33 and CD205 (LY75); CD33 and CD244 (SLAMF4); CD33 and CD305 (LAIR1); CD33 and CD49D (ITGA4); CLL-1 and CD205 (LY75); CLL-1 and CD244 (SLAMF4); CLL-1 and CD305 (LAIR1); CLL-1 and CD49D (ITGA4); EMR2 and CD205 (LY75); EMR2 and CD244 (SLAMF4); EMR2 and CD305 (LAIR1); EMR2 and CD49D (ITGA4); CD38 and CD205 (LY75); CD38 and CD244 (SLAMF4); CD38 and CD305 (LAIR1); CD38 and CD49D (ITGA4); CD305 (LAIR1) and CD49D (ITGA4); CD305 (LAIR1) and CD205 (LY75); CD305 (LAIR1) and CD244 (SLAMF4); CD49D (ITGA4) and CD205 (LY75); CD49D (ITGA4) and CD244 (SLAMF4); and CD205 (LY75) and CD244 (SLAMF4). Mutated Genes Encoding Cell Surface Proteins In some embodiments, the hematopoietic cells (HSCs) described herein may contain an edited gene encoding one or more cell surface proteins of interest in mutated form (mutants or variants, which are used herein interchangeably), which has reduced binding or no binding to a cytotoxic agent as described herein. The variants may lack the epitope to which the cytotoxic agent binds. Alternatively, the mutants may carry one or more mutations of the epitope to which the cytotoxic agent binds, such that binding to the cytotoxic agent is reduced or abolished as compared to the natural or wild-type cell surface protein counterpart. Such a variant is preferred to maintain substantially similar biological activity as the wild- type counterpart. A cell that is “negative” for a given cell surface protein has a substantially reduced expression level of the cell surface protein as compared with its naturally-occurring counterpart (e.g., otherwise similar, unmodified cells), e.g., not detectable or not distinguishable from background levels, e.g., using a flow cytometry assay. In some instances, a cell that is negative for the cell surface protein has a level of less than 10%, 5%, 2%, or 1% of as compared with its naturally-occurring counterpart. The variant may share a sequence homology of at least 80% (e.g., 85%, 90%, 95%, 97%, 98%, 99%, or above) as the wild-type counterpart and, in some embodiments, may contain no other mutations in addition to those for mutating or deleting the epitope of interest. The “percent identity” of two amino acid sequences is determined using the algorithm of Karlin and Altschul Proc. Natl. Acad. Sci. USA 87:2264-68, 1990, modified as in Karlin and Altschul Proc. Natl. Acad. Sci. USA 90:5873-77, 1993. Such an algorithm is incorporated into the NBLAST and XBLAST programs (version 2.0) of Altschul, et al. J. Mol. Biol.215:403-10, 1990. BLAST protein searches can be performed with the XBLAST program, score=50, wordlength=3 to obtain amino acid sequences homologous to the protein molecules of the invention. Where gaps exist between two sequences, Gapped BLAST can be utilized as described in Altschul et al., Nucleic Acids Res.25(17):3389-3402, 1997. When utilizing BLAST and Gapped BLAST programs, the default parameters of the respective programs (e.g., XBLAST and NBLAST) can be used. In some instances, the variant contains one or more amino acid residue substitutions (e.g., 2, 3, 4, 5, or more) within the epitope of interest such that the cytotoxic agent does not bind or has reduced binding to the mutated epitope. Such a variant may have substantially reduced binding affinity to the cytotoxic agent (e.g., having a binding affinity that is at least 40%, 50%, 60%, 70%, 80% or 90% lower than its wild-type counterpart). In some examples, such a variant may have abolished binding activity to the cytotoxic agent. In other instances, the variant contains a deletion of a region that comprises the epitope of interest. Such a region may be encoded by an exon. In some embodiments, the region is a domain of the cell surface protein of interest that encodes the epitope. In one example, the variant has just the epitope deleted. The length of the deleted region may range from 3-60 amino acids, e.g., 5- 50, 5-40, 10-30, 10-20, etc. The mutation(s) or deletions in a mutant of a cell surface protein may be within or surround a non-essential epitope such that the mutation(s) or deletion(s) do not substantially affect the bioactivity of the protein. As used herein, the term “epitope” refers to an amino acid sequence (linear or conformational) of a protein, such as a cell surface protein, that is bound by the CDRs of an antibody. In some embodiments, the cytotoxic agent binds to one or more (e.g., at least 2, 3, 4, 5 or more) epitopes of a cell surface proteins. In some embodiments, the cytotoxic agent binds to more than one epitope of the cell surface protein and the hematopoietic cells are manipulated such that each of the epitopes is absent and/or unavailable for binding by the cytotoxic agent. In some embodiments, the genetically engineered HSCs described herein have one or more edited genes of cell surface proteins such that the edited genes express mutated cell surface proteins with mutations in one or more non-essential epitopes. A non-essential epitope (or a fragment comprising such) refers to a domain within the cell surface protein, the mutation in which (e.g., deletion) is less likely to substantially affect the bioactivity of the cell surface protein and thus the bioactivity of the cells expressing such. For example, when hematopoietic cells comprising a deletion or mutation of a non-essential epitope of a cell surface protein, such hematopoietic cells are able to proliferate and/or undergo erythropoietic differentiation to a similar level as hematopoietic cells that express a wild-type cell surface protein. Non-essential epitopes of a cell surface protein can be identified by the methods described herein or by conventional methods relating to protein structure-function prediction. For example, a non-essential epitope of a protein can be predicted based on comparing the amino acid sequence of a protein from one species with the sequence of the protein from other species. Non-conserved domains are usually not essential to the functionality of the protein. As will be evident to one of ordinary skill in the art, non-essential epitope of a protein is predicted using an algorithm or software, such as the PROVEAN software (see, e.g., see: provean.jcvi.org; Choi et al. PLoS ONE (2012) 7(10): e46688), to predict potential non-essential epitopes in a cell surface protein of interest (“candidate non-essential epitope”). Mutations, including substitution and/or deletion, many be made in any one or more amino acid residues of a candidate non-essential epitope using convention nucleic acid modification technologies. The protein variants thus prepared may be introduced into a suitable type of cells, such as hematopoietic cells, and the functionality of the protein variant can be investigated to confirm that the candidate non-essential epitope is indeed a non-essential epitope. Alternatively, a non-essential epitope of a cell surface protein may be identified by introducing a mutation into a candidate region in a cell surface protein of interest in a suitable type of host cells (e.g., hematopoietic cells) and examining the functionality of the mutated cell surface protein in the host cells. If the mutated cell surface protein maintains substantially the biological activity of the native counterpart, this indicates that the region where the mutation is introduced is non-essential to the function of the cell surface protein. In some embodiments, genetically engineered cells of the present disclosure comprise a mutation in a gene encoding a cell surface protein selected from a group consisting of: CD10; CD101; CD117; CD11A; CD11C; CD120B; CD123; CD13; CD133; CD152; CD200; CD205 (LY75); CD226; CD244 (SLAMF4); CD25; CD274; CD305 (LAIR1); CD32; CD33; CD34; CD366; CD38; CD41; CD42B; CD44; CD45RA; CD47; CD48; CD49D (ITGA4); CD49F; CD52; CD58; CD64; CD7; CD70; CD82; CD84; CD85; CD85K; CD86; CD9; CD96; CD99; CLL-1; EMR2 (ADGRE2); FR-B; GPR56; and IL1RAP). In some embodiments, genetically engineered cells of the present disclosure comprise a mutation in a gene encoding a cell surface protein selected from a group consisting of: CD10; CD101; CD11A; CD11C; CD123; CD133; CD152; CD200; CD205; CD244; CD25; CD305; CD33; CD34; CD366; CD38; CD42B; CD44; CD45RA; CD47; CD48; CD49D; CD49F; CD52; CD58; CD64; CD7; CD84; CD85K; CD9; CD99; CLL-1; EMR2; GPR56; IL1RAP; and CD274. In some embodiments, genetically engineered cells of the present disclosure comprise a mutation in a gene encoding a cell surface protein selected from a group consisting of: CD117; CD11A; CD11C; CD120B; CD123; CD13; CD133; CD152; CD205; CD226; CD244; CD25; CD305; CD32; CD33; CD34; CD38; CD41; CD44; CD45RA; CD47; CD49D; CD49F; CD52; CD58; CD64; CD7; CD70; CD82; CD86; CD96; CD99; CLL-1; EMR2; FR-B; GPR56; and IL1RAP. In some embodiments, genetically engineered cells of the present disclosure comprise a mutation in a first gene encoding a first cell surface protein and a second gene encoding a second cell surface protein selected from a group consisting of: CD274 and CD152; CD274 and CD226; CD152 and CD226; CD10 and CD49F; CD49F and GPR56; CD200 and CD34; CD34 and CD52; CD34 and CD133; GPR56 and CD133; CD42B and CD41; CD85K and CD64; CD85K and CD32; CD64 and CD32; CD64 and CD11C; CD64 and CD86; CD45RA and CD96; CD45RA and IL1RAP; CD45RA and CD123; CD84 and CD33; CD11C and IL1RAP; CD11C and CD33; CD11C and CD86; CD9 and IL1RAP; IL1RAP and CD33; IL1RAP and CD205; IL1RAP and CD305; IL1RAP and CD123; CD33 and CLL-1; CD33 and CD305; CD11A and CD86; CLL-1 and CD305; CD58 and CD305; CD49D and CD47; CD49D and CD99; CD49D and CD44; CD49D and CD305; CD47 and CD99; CD47 and EMR2; CD99 and CD44; CD99 and CD305; CD99 and EMR2; CD205 and CD305; CD205 and CD123; CD44 and CD305; CD44 and EMR2; CD244 and CD117; CD123 and EMR2; EMR2 and CD82; CD152 and CD120B; CD48 and CD11A; CD200 and CD25; CD34 and CD7; CD34 and CD13; CD52 and GPR56; CD52and CD133; CD52 and CD7; GPR56 and CD7; GPR56 and CD82; CD133 and CD7; CD133 and CD82; CD7 and CD13; CD101 and CD64; CD85 and CD70; CD85K and CD86; CD64 and FR-B; CD366 and CD96; CD25 and CD45RA; CD25 and CD96; CD9 and CD82; IL1RAP and CD58; CD11A and CD38; CD11A and CD44; CD11A and CD244; CD38 and CD305; CD38 and EMR2; CLL-1 and CD49D; CD58 and CD49D; CD47 and CD205; and CD47 and CD44. In some embodiments, genetically engineered cells of the present disclosure comprise a mutation in a first gene encoding a first cell surface protein and a second gene encoding a second cell surface protein selected from a group consisting of: CD85K and CD64; CD45RA and IL1RAP; CD34 and CD133; IL1RAP and CD33; CD47 and CD99; CLL-1 and CD305; CD152 and CD226; CD45RA and CD96; CD45RA and CD123; CD49D and CD305; IL1RAP and CD305; CD58 and CD305; CD49D and CD47; CD33 and CD305; CD49D and CD44; CD64 and CD86; GPR56 and CD133; CD34 and CD52; EMR2 and CD82; CD64 and CD32; CD42B and CD41; CD64 and CD11C; CD99 and CD44; CD200 and CD34; CD11C and CD33; IL1RAP and CD123; CD11C and IL1RAP; CD44 and EMR2; CD205 and CD305; CD44 and CD305; CD49D and CD99; CD274 and CD226; CD49F and GPR56; CD9 and IL1RAP; CD99 and CD305; CD10 and CD49F; CD11A and CD86; CD33 and CLL-1; CD84 and CD33; CD244 and CD117; CD85K and CD32; CD47 and EMR2; CD274 and CD152; IL1RAP and CD205; CD11C and CD86; CD99 and EMR2; CD205 and CD123; and CD123 and EMR2. In some embodiments, genetically engineered cells of the present disclosure comprise a mutation in a first gene encoding a first cell surface protein and a second gene encoding a second cell surface protein selected from a group consisting of: CD33 and CD205 (LY75); CD33 and CD244 (SLAMF4); CD33 and CD305 (LAIR1); CD33 and CD49D (ITGA4); CLL-1 and CD205 (LY75); CLL-1 and CD244 (SLAMF4); CLL-1 and CD305 (LAIR1); CLL-1 and CD49D (ITGA4); EMR2 and CD205 (LY75); EMR2 and CD244 (SLAMF4); EMR2 and CD305 (LAIR1); EMR2 and CD49D (ITGA4); CD38 and CD205 (LY75); CD38 and CD244 (SLAMF4); CD38 and CD305 (LAIR1); CD38 and CD49D (ITGA4); CD305 (LAIR1) and CD49D (ITGA4); CD305 (LAIR1) and CD205 (LY75); CD305 (LAIR1) and CD244 (SLAMF4); CD49D (ITGA4) and CD205 (LY75); CD49D (ITGA4) and CD244 (SLAMF4); and CD205 (LY75) and CD244 (SLAMF4). In some embodiments, genetically engineered cells of the present disclosure comprise a mutation in a first gene encoding a first cell surface protein and a second gene encoding a second lineage-specific cell surface antigen, wherein mutation of the first gene and second gene results in dysregulated expression of the first cell surface protein and the second cell surface protein seen in subjects diagnosed with a hematopoietic malignancy. In some embodiments, the first gene and second gene encode combinations of antigens selected from a group consisting of: CD274 and CD152; CD274 and CD226; CD152 and CD226; CD10 and CD49F; CD49F and GPR56; CD200 and CD34; CD34 and CD52; CD34 and CD133; GPR56 and CD133; CD42B and CD41; CD85K and CD64; CD85K and CD32; CD64 and CD32; CD64 and CD11C; CD64 and CD86; CD45RA and CD96; CD45RA and IL1RAP; CD45RA and CD123; CD84 and CD33; CD11C and IL1RAP; CD11C and CD33; CD11C and CD86; CD9 and IL1RAP; IL1RAP and CD33; IL1RAP and CD205; IL1RAP and CD305; IL1RAP and CD123; CD33 and CLL-1; CD33 and CD305; CD11A and CD86; CLL-1 and CD305; CD58 and CD305; CD49D and CD47; CD49D and CD99; CD49D and CD44; CD49D and CD305; CD47 and CD99; CD47 and EMR2; CD99 and CD44; CD99 and CD305; CD99 and EMR2; CD205 and CD305; CD205 and CD123; CD44 and CD305; CD44 and EMR2; CD244 and CD117; CD123 and EMR2; and EMR2 and CD82. In some embodiments, the first gene and second gene encode combinations of antigens selected from a group consisting of: CD33 and CD205 (LY75); CD33 and CD244 (SLAMF4); CD33 and CD305 (LAIR1); CD33 and CD49D (ITGA4); CLL-1 and CD205 (LY75); CLL-1 and CD244 (SLAMF4); CLL-1 and CD305 (LAIR1); CLL-1 and CD49D (ITGA4); EMR2 and CD205 (LY75); EMR2 and CD244 (SLAMF4); EMR2 and CD305 (LAIR1); EMR2 and CD49D (ITGA4); CD38 and CD205 (LY75); CD38 and CD244 (SLAMF4); CD38 and CD305 (LAIR1); CD38 and CD49D (ITGA4); CD305 (LAIR1) and CD49D (ITGA4); CD305 (LAIR1) and CD205 (LY75); CD305 (LAIR1) and CD244 (SLAMF4); CD49D (ITGA4) and CD205 (LY75); CD49D (ITGA4) and CD244 (SLAMF4); and CD205 (LY75) and CD244 (SLAMF4). In some embodiments, genetically engineered cells of the present disclosure comprise a mutation in a first gene encoding a first cell surface protein and a second gene encoding a second lineage-specific cell surface antigen, wherein mutation of the first gene and second gene results in dysregulated expression of the first cell surface protein and the second cell surface protein seen in subjects who have experienced relapse of a hematopoietic malignancy. In some embodiments, the first gene and second gene encode combinations of antigens selected from a group consisting of: CD9 and CD82; CD45RA and CD123; CD133 and CD82; CD85K and CD70; CD47 and CD205; CD64 and FR-B; CD11A and CD44; CD85K and CD86; CD366 and CD96; CD101 and CD64; CLL-1 and CD49D; CD49D and CD47; EMR2 and CD82; CD38 and CD305; CD34 and CD7; CD200 and CD25; CD25 and CD45RA; CD49D and CD44; CD11A and CD244; CD133 and CD7; CD48 and CD11A; CD52 and CD133; GPR56 and CD82; CD11A and CD38; CD34 and CD13; CD58 and CD49D; CD47 and CD99; IL1RAP and CD58; CD47 and CD44; CD33 and CD305; CD64 and CD86; CD152 and CD120B; CD42B and CD41; CD34 and CD52; CD152 and CD226; CD7 and CD13; CD38 and EMR2; IL1RAP and CD33; CLL-1 and CD305; CD45RA and IL1RAP; CD34 and CD133; GPR56 and CD133; CD49D and CD305; CD25 and CD96; CD52 and CD7; CD64 and CD32; CD58 and CD305; CD45RA and CD96; IL1RAP and CD305; CD52 and GPR56; GPR56 and CD7; and CD85K and CD64. In some embodiments, the first gene and second gene encode combinations of antigens selected from a group consisting of: CD33 and CD205 (LY75); CD33 and CD244 (SLAMF4); CD33 and CD305 (LAIR1); CD33 and CD49D (ITGA4); CLL-1 and CD205 (LY75); CLL-1 and CD244 (SLAMF4); CLL-1 and CD305 (LAIR1); CLL-1 and CD49D (ITGA4); EMR2 and CD205 (LY75); EMR2 and CD244 (SLAMF4); EMR2 and CD305 (LAIR1); EMR2 and CD49D (ITGA4); CD38 and CD205 (LY75); CD38 and CD244 (SLAMF4); CD38 and CD305 (LAIR1); CD38 and CD49D (ITGA4); CD305 (LAIR1) and CD49D (ITGA4); CD305 (LAIR1) and CD205 (LY75); CD305 (LAIR1) and CD244 (SLAMF4); CD49D (ITGA4) and CD205 (LY75); CD49D (ITGA4) and CD244 (SLAMF4); and CD205 (LY75) and CD244 (SLAMF4). Variant Cell Surface Proteins Methods for assessing the functionality of the cell surface protein and the hematopoietic cells or descendants thereof will be known in the art and include, for example, proliferation assays, differentiation assays, colony formation, expression analysis (e.g., gene and/or protein), protein localization, intracellular signaling, functional assays, and in vivo humanized mouse models. Any of the methods for identifying and/or verifying non-essential epitopes in cell surface proteins is also within the scope of the present disclosure. The term “binds,” as used herein with reference to a gRNA interaction with a target domain (e.g., a first target domain and/or a second or subsequent target domain), refers to the gRNA molecule and the target domain forming a complex. The complex may comprise two strands forming a duplex structure, or three or more strands forming a multi-stranded complex. The binding may constitute a step in a more extensive process, such as the cleavage of the target domain by a Cas nuclease. In some embodiments, the gRNA binds to the target domain with perfect complementarity, and in other embodiments, the gRNA binds to the target domain with partial complementarity, e.g., with one or more mismatches. In some embodiments, when a gRNA binds to a target domain, the full targeting domain of the gRNA base pairs with the targeting domain. In other embodiments, only a portion of the target domain and/or only a portion of the targeting domain base pairs with the other. In an embodiment, the interaction is sufficient to mediate a target domain-mediated cleavage event. As used herein, the terms “binds,” “specifically binds,” “specifically recognizes” and analogous terms as used herein with reference to a protein (e.g., a cell surface protein) and an agent (e.g., a ligand or an antibody) interaction refer to the specific binding or association between the agent (e.g., a ligand or an antibody) and protein (e.g., a cell surface protein). Agents that specifically bind a cell surface protein are known in the art and/or can be identified, for example, by immunoassays, BIAcore™, or other techniques known to those of skill in the art. The term “reduced binding,” as used herein with reference to the binding activity of an agent (e.g., a ligand or an antibody) to a cell surface protein epitope, refers to binding that is reduced by at least about 5%. The level of binding may refer to the amount of binding of the agent (e.g., a ligand or an antibody) to a cell, such as a hematopoietic cell or descendant thereof, or to the amount of binding of the agent (e.g., a ligand or an antibody) to the cell surface protein. The level of binding of a cell, such as a hematopoietic cell or descendant thereof, that has been modified, for example, to include an amino acid substitution in a cell surface protein epitope, may be relative to the level of binding of the agent (e.g., a ligand or an antibody) to a cell that has not been modified as determined by the same assay under the same conditions. In exemplary embodiments, the level of binding of a cell surface protein that includes an amino acid substitution in a cell surface protein epitope to an agent (e.g., a ligand or an antibody) may be relative to the level of binding of the agent to a cell surface protein that does not include the amino acid substitution in the cell surface protein epitope (e.g., a wild-type protein or a variant thereof) as determined by the same assay under the same conditions. Alternatively, the level of binding of a cell surface protein that lacks an epitope to an agent (e.g., a ligand or an antibody) may be relative to the level of binding of the agent to a cell surface protein that contains the unmodified epitope (e.g., a wild-type protein) as determined by the same assay under the same conditions. In some embodiments, the binding is reduced by at least about 1%, at least about 2%, at least about 3%, at least about 4%, at least about 5%, at least about 6%, at least about 7%, at least about 8%, at least about 9%, at least about 10%, at least about 15%, at least about 20%, at least about 25%, at least about 30%, at least about 35%, at least about 40%, at least about 45%, at least about 50%, at least about 55%, at least about 60%, at least about 65%, at least about 70%, at least about 75%, at least about 80%, at least about 85%, at least about 90%, at least about 95%, at least about 99%, at least about 100% or more, e.g., as compared to the unmodified epitope (e.g., a wild-type protein). In some embodiments, the binding is reduced by at least about 1-fold, at least about 2-fold, at least about 3-fold, at least about 4-fold, at least about 5-fold, at least about 6-fold, at least about 7-fold, at least about 8-fold, at least about 9-fold, at least about 10-fold, at least about 20-fold, at least about 30-fold, at least about 40-fold, at least about 50-fold, at least about 60-fold, at least about 70-fold, at least about 80- fold, at least about 90-fold, or at least about 100-fold, e.g., as compared to the unmodified epitope (e.g., a wild-type protein). In some embodiments, the binding is reduced such that there is substantially no detectable binding in a conventional assay. The binding affinity or binding specificity for an epitope or protein can be determined by a variety of methods including equilibrium dialysis, equilibrium binding, gel filtration, ELISA, surface plasmon resonance, or spectroscopy. As used herein, “no binding” refers to substantially no binding, e.g., no detectable binding or only baseline binding as determined in a conventional binding assay. In some embodiments, there is no binding between the cells, e.g., hematopoietic cells or descendants thereof, that have been modified and the agent. In some embodiments, there is no detectable binding between the cells, e.g., hematopoietic cells or descendants thereof, that have been modified and the agent. In some embodiments, no binding of the cells, e.g., hematopoietic cells or descendant thereof, to the agent refers to a baseline level of binding, as shown using any conventional binding assay known in the art. In some embodiments, the level of binding of the cells, e.g., hematopoietic cells or descendants thereof, that have been modified and the agent is not biologically significant. The term “no binding” is not intended to require the absolute absence of binding. Without wishing to be bound by theory, a subject can be administered the rescue cells (e.g., hematopoietic stem cells (HSCs) and/or hematopoietic progenitor cells (HPCs)) described herein comprising a modification in a cell surface protein gene, e.g., a genetic edit (i.e., mutation) that results in the rescue cells having reduced or eliminated expression of the respective gene, or a modification of an epitope of the protein encoded by the respective gene that diminishes the binding of the therapeutic agent to the protein. These genetically engineered (modified) cells can thus be resistant to an agent, such as an anti-cancer therapy, and can therefore repopulate the hematopoietic system during or after anti-cancer therapy. The binding activity of the agent (e.g., a ligand or an antibody) to the epitope may be reduced by the amino acid substitution by at least about 1%, at least about 2%, at least about 3%, at least about 4%, at least about 5%, at least about 6%, at least about 7%, at least about 8%, at least about 9%, at least about 10%, at least about 15%, at least about 20%, at least about 25%, at least about 30%, at least about 35%, at least about 40%, at least about 45%, at least about 50%, at least about 55%, at least about 60%, at least about 65%, at least about 70%, at least about 75%, at least about 80%, at least about 85%, at least about 90%, at least about 95%, at least about 99%, at least about 100% or more, e.g., as compared to the unmodified epitope (e.g., a wild-type protein). In some embodiments, the binding activity of the agent (e.g., a ligand or an antibody) to the epitope may be reduced by the amino acid substitution by at least about at least about 1-fold, at least about 2-fold, at least about 3-fold, at least about 4-fold, at least about 5-fold, at least about 6-fold, at least about 7-fold, at least about 8-fold, at least about 9-fold, at least about 10-fold, at least about 20-fold, at least about 30-fold, at least about 40-fold, at least about 50-fold, at least about 60-fold, at least about 70- fold, at least about 80-fold, at least about 90-fold, or at least about 100-fold, e.g., as compared to the unmodified epitope (e.g., a wild-type protein). In some embodiments, the binding is reduced such that there is substantially no detectable binding in a conventional assay. The binding affinity or binding specificity for an epitope or protein can be determined by a variety of methods including equilibrium dialysis, equilibrium binding, gel filtration, ELISA, surface plasmon resonance, or spectroscopy. In some embodiments, the reduction in binding activity may include an increase in KD, IC50, and/or EC50. For example, the KD, IC50, and/or EC50 may be increased by the amino acid substitution by at least about 1%, at least about 2%, at least about 3%, at least about 4%, at least about 5%, at least about 6%, at least about 7%, at least about 8%, at least about 9%, at least about 10%, at least about 15%, at least about 20%, at least about 25%, at least about 30%, at least about 35%, at least about 40%, at least about 45%, at least about 50%, at least about 55%, at least about 60%, at least about 65%, at least about 70%, at least about 75%, at least about 80%, at least about 85%, at least about 90%, at least about 95%, at least about 99%, at least about 100% or more, e.g., as compared to the unmodified epitope (e.g., a wild- type protein). In some embodiments, the KD, IC50, and/or EC50 may be increased by the amino acid substitution by at least about 1-fold, at least about 2-fold, at least about 3-fold, at least about 4-fold, at least about 5-fold, at least about 6-fold, at least about 7-fold, at least about 8-fold, at least about 9-fold, at least about 10-fold, at least about 20-fold, at least about 30-fold, at least about 40-fold, at least about 50-fold, at least about 60-fold, at least about 70- fold, at least about 80-fold, at least about 90-fold, or at least about 100-fold, e.g., as compared to the unmodified epitope (e.g., a wild-type protein). As used herein, the term "IC50" refers to the concentration of an agent (e.g., an inhibitor, such as a ligand or an antibody) that produces 50% of the maximal inhibition of activity or expression measurable using the same assay in the absence of the binding partner. The IC50 can be as measured in vitro or in vivo. The IC50 can be determined by measuring activity using a conventional in vitro assay (e.g., a protein activity assay and/or a gene expression assay). As used herein, the term "EC50," refers to the concentration of a binding partner (e.g., an activator, such as a ligand or an antibody) that produces 50% of maximal activation of measurable activity or expression using the same assay in the absence of the agent. Stated differently, the "EC50" is the concentration of agent that gives 50% activation, when 100% activation is set at the amount of activity that does not increase with the addition of more agent. The EC50 may refer to the half maximal effective concentration, which includes the concentration of an antibody which induces a response halfway between the baseline and maximum after a specified exposure time. The EC50 may represent the concentration of an antibody where 50% of its maximal effect is observed. In certain embodiments, the EC50 value may equal the concentration of an agent (e.g., a ligand or an antibody) that gives half- maximal binding to cells expressing a cell surface protein, such as a cell surface protein, as determined by, e.g. a FACS binding assay. Thus, reduced or weaker binding is observed with an increased EC50 value, or half maximal effective concentration value such that 500 nM EC50 is indicative of a weaker binding affinity than 50 nM EC50. The EC50 can be as measured in vitro or in vivo. The term "KD" refers to the dissociation equilibrium constant of a particular agent- protein interaction, such as an antibody-antigen interaction or a ligand-receptor interaction, or the dissociation rate constant of an antibody or antibody-binding fragment. There is an inverse relationship between KD and binding affinity, therefore the smaller the KD value, the higher, i.e. stronger, the affinity. Thus, the terms "higher affinity" or "stronger affinity" relate to a higher ability to form an interaction and therefore a smaller KD value, and conversely the terms "lower affinity" or "weaker affinity" relate to a lower ability to form an interaction and therefore a larger KD value. In some circumstances, a higher binding affinity (or KD) of a particular molecule (e.g., antibody) to its interactive partner molecule (e.g., antigen X) compared to the binding affinity of the molecule (e.g., antibody) to another interactive partner molecule (e.g. antigen Y) may be expressed as a binding ratio determined by dividing the larger KD value (lower, or weaker, affinity) by the smaller KD (higher, or stronger, affinity), for example expressed as 5-fold or 10-fold greater binding affinity, as the case may be. In some embodiments, "low affinity" refers to less strong binding interaction. In some embodiments, the low binding affinity corresponds to greater than about 1 nM KD, greater than about 1 nM, about 2 nM, about 3 nM, about 4 nM, about 5 nM, about 6 nM, about 7 nM, about 8 nM, about 9 nM, about 10 nM, about 11 nM, about 12 nM, about 13 nM, about 14 nM, about 15 nM, about 16 nM, about 17 nM, about 18 nM, about 19 nM, about 20 nM, about 21 nM, about 22 nM, about 23 nM, about 24 nM, about 25 nM, about 26 nM, about 27 nM, about 28 nM, about 29 nM, about 30 nM, about 31 nM, about 32 nM, about 33 nM, about 34 nM, about 35 nM, about 36 nM, about 37 nM, about 38 nM, about 39 nM, or about 40 nM KD, wherein such KD binding affinity value is measured, e.g., in an in vitro surface plasmon resonance binding assay, or equivalent biomolecular interaction sensing assay. In some embodiments, the low binding affinity corresponds to greater than about 10 nM, about 11 nM, about 12 nM, about 13 nM, about 14 nM, about 15 nM, about 16 nM, about 17 nM, about 18 nM, about 19 nM, about 20 nM, about 21 nM, about 22 nM, about 23 nM, about 24 nM, about 25 nM, about 26 nM, about 27 nM, about 28 nM, about 29 nM, about 30 nM, about 31 nM, about 32 nM, about 33 nM, about 34 nM, about 35 nM, about 36 nM, about 37 nM, about 38 nM, about 39 nM, or about 40 nM EC50, wherein such EC50 binding affinity value is measured, e.g., in an in vitro FACS binding assay, or equivalent cell-based binding assay. In some embodiments, "weak affinity" refers to weak binding interaction. In some embodiments, the weak binding affinity corresponds to greater than about 100 nM KD or EC50, greater than about 200, 300, or greater than about 500 nM KD or EC50, wherein such KD binding affinity value is measured, e.g., in an in vitro surface plasmon resonance binding assay, or equivalent biomolecular interaction sensing assay, and such EC50 binding affinity value is measured, e.g., in an in vitro FACS binding assay, or equivalent cell-based interaction detecting assay to detect monovalent biding. No detectable binding means that the affinity between the two biomolecules, for example, especially between the monovalent antibody binding arm and its target antigen, is beyond the detection limit of the assay being used. In some embodiments, genetically engineered cells of the present disclosure express a mutant cell surface protein selected from a group consisting of: CD10; CD101; CD117; CD11A; CD11C; CD120B; CD123; CD13; CD133; CD152; CD200; CD205 (LY75); CD226; CD244 (SLAMF4); CD25; CD274; CD305 (LAIR1); CD32; CD33; CD34; CD366; CD38; CD41; CD42B; CD44; CD45RA; CD47; CD48; CD49D (ITGA4); CD49F; CD52; CD58; CD64; CD7; CD70; CD82; CD84; CD85; CD85K; CD86; CD9; CD96; CD99; CLL- 1; EMR2 (ADGRE2); FR-B; GPR56; and IL1RAP. In some embodiments, genetically engineered cells of the present disclosure express a mutant cell surface protein selected from a group consisting of: CD10; CD101; CD11A; CD11C; CD123; CD133; CD152; CD200; CD205; CD244; CD25; CD305; CD33; CD34; CD366; CD38; CD42B; CD44; CD45RA; CD47; CD48; CD49D; CD49F; CD52; CD58; CD64; CD7; CD84; CD85K; CD9; CD99; CLL-1; EMR2; GPR56; IL1RAP; and CD274. In some embodiments, genetically engineered cells of the present disclosure express a mutant cell surface protein selected from a group consisting of: CD117; CD11A; CD11C; CD120B; CD123; CD13; CD133; CD152; CD205; CD226; CD244; CD25; CD305; CD32; CD33; CD34; CD38; CD41; CD44; CD45RA; CD47; CD49D; CD49F; CD52; CD58; CD64; CD7; CD70; CD82; CD86; CD96; CD99; CLL-1; EMR2; FR-B; GPR56; and IL1RAP. In some embodiments, genetically engineered cells of the present disclosure express a mutant of a first cell surface protein and a mutant of a second cell surface protein selected from a group consisting of: CD10; CD101; CD117; CD11A; CD11C; CD120B; CD123; CD13; CD133; CD152; CD200; CD205 (LY75); CD226; CD244 (SLAMF4); CD25; CD274; CD305 (LAIR1); CD32; CD33; CD34; CD366; CD38; CD41; CD42B; CD44; CD45RA; CD47; CD48; CD49D (ITGA4); CD49F; CD52; CD58; CD64; CD7; CD70; CD82; CD84; CD85; CD85K; CD86; CD9; CD96; CD99; CLL-1; EMR2 (ADGRE2); FR-B; GPR56; and IL1RAP. In some embodiments, genetically engineered cells of the present disclosure express a mutant of a first cell surface protein and a mutant of a second cell surface protein selected from a group consisting of: CD274 and CD152; CD274 and CD226; CD152 and CD226; CD10 and CD49F; CD49F and GPR56; CD200 and CD34; CD34 and CD52; CD34 and CD133; GPR56 and CD133; CD42B and CD41; CD85K and CD64; CD85K and CD32; CD64 and CD32; CD64 and CD11C; CD64 and CD86; CD45RA and CD96; CD45RA and IL1RAP; CD45RA and CD123; CD84 and CD33; CD11C and IL1RAP; CD11C and CD33; CD11C and CD86; CD9 and IL1RAP; IL1RAP and CD33; IL1RAP and CD205; IL1RAP and CD305; IL1RAP and CD123; CD33 and CLL-1; CD33 and CD305; CD11A and CD86; CLL-1 and CD305; CD58 and CD305; CD49D and CD47; CD49D and CD99; CD49D and CD44; CD49D and CD305; CD47 and CD99; CD47 and EMR2; CD99 and CD44; CD99 and CD305; CD99 and EMR2; CD205 and CD305; CD205 and CD123; CD44 and CD305; CD44 and EMR2; CD244 and CD117; CD123 and EMR2; EMR2 and CD82; CD152 and CD120B; CD48 and CD11A; CD200 and CD25; CD34 and CD7; CD34 and CD13; CD52 and GPR56; CD52and CD133; CD52 and CD7; GPR56 and CD7; GPR56 and CD82; CD133 and CD7; CD133 and CD82; CD7 and CD13; CD101 and CD64; CD85 and CD70; CD85K and CD86; CD64 and FR-B; CD366 and CD96; CD25 and CD45RA; CD25 and CD96; CD9 and CD82; IL1RAP and CD58; CD11A and CD38; CD11A and CD44; CD11A and CD244; CD38 and CD305; CD38 and EMR2; CLL-1 and CD49D; CD58 and CD49D; CD47 and CD205; and CD47 and CD44. In some embodiments, genetically engineered cells of the present disclosure express a mutant of a first cell surface protein and a mutant of a second cell surface protein selected from a group consisting of: CD85K and CD64; CD45RA and IL1RAP; CD34 and CD133; IL1RAP and CD33; CD47 and CD99; CLL-1 and CD305; CD152 and CD226; CD45RA and CD96; CD45RA and CD123; CD49D and CD305; IL1RAP and CD305; CD58 and CD305; CD49D and CD47; CD33 and CD305; CD49D and CD44; CD64 and CD86; GPR56 and CD133; CD34 and CD52; EMR2 and CD82; CD64 and CD32; CD42B and CD41; CD64 and CD11C; CD99 and CD44; CD200 and CD34; CD11C and CD33; IL1RAP and CD123; CD11C and IL1RAP; CD44 and EMR2; CD205 and CD305; CD44 and CD305; CD49D and CD99; CD274 and CD226; CD49F and GPR56; CD9 and IL1RAP; CD99 and CD305; CD10 and CD49F; CD11A and CD86; CD33 and CLL-1; CD84 and CD33; CD244 and CD117; CD85K and CD32; CD47 and EMR2; CD274 and CD152; IL1RAP and CD205; CD11C and CD86; CD99 and EMR2; CD205 and CD123; and CD123 and EMR2. In some embodiments, genetically engineered cells of the present disclosure express a mutant of a first cell surface protein and a mutant of a second cell surface protein selected from a group consisting of: CD274 and CD152; CD274 and CD226; CD152 and CD226; CD10 and CD49F; CD49F and GPR56; CD200 and CD34; CD34 and CD52; CD34 and CD133; GPR56 and CD133; CD42B and CD41; CD85K and CD64; CD85K and CD32; CD64 and CD32; CD64 and CD11C; CD64 and CD86; CD45RA and CD96; CD45RA and IL1RAP; CD45RA and CD123; CD84 and CD33; CD11C and IL1RAP; CD11C and CD33; CD11C and CD86; CD9 and IL1RAP; IL1RAP and CD33; IL1RAP and CD205; IL1RAP and CD305; IL1RAP and CD123; CD33 and CLL-1; CD33 and CD305; CD11A and CD86; CLL-1 and CD305; CD58 and CD305; CD49D and CD47; CD49D and CD99; CD49D and CD44; CD49D and CD305; CD47 and CD99; CD47 and EMR2; CD99 and CD44; CD99 and CD305; CD99 and EMR2; CD205 and CD305; CD205 and CD123; CD44 and CD305; CD44 and EMR2; CD244 and CD117; CD123 and EMR2; and EMR2 and CD82. In some embodiments, genetically engineered cells of the present disclosure express a mutant of a first cell surface protein and a mutant of a second cell surface protein selected from a group consisting of: CD9 and CD82; CD45RA and CD123; CD133 and CD82; CD85K and CD70; CD47 and CD205; CD64 and FR-B; CD11A and CD44; CD85K and CD86; CD366 and CD96; CD101 and CD64; CLL-1 and CD49D; CD49D and CD47; EMR2 and CD82; CD38 and CD305; CD34 and CD7; CD200 and CD25; CD25 and CD45RA; CD49D and CD44; CD11A and CD244; CD133 and CD7; CD48 and CD11A; CD52 and CD133; GPR56 and CD82; CD11A and CD38; CD34 and CD13; CD58 and CD49D; CD47 and CD99; IL1RAP and CD58; CD47 and CD44; CD33 and CD305; CD64 and CD86; CD152 and CD120B; CD42B and CD41; CD34 and CD52; CD152 and CD226; CD7 and CD13; CD38 and EMR2; IL1RAP and CD33; CLL-1 and CD305; CD45RA and IL1RAP; CD34 and CD133; GPR56 and CD133; CD49D and CD305; CD25 and CD96; CD52 and CD7; CD64 and CD32; CD58 and CD305; CD45RA and CD96; IL1RAP and CD305; CD52 and GPR56; GPR56 and CD7; and CD85K and CD64. In some embodiments, genetically engineered cells of the present disclosure express a mutant of a first cell surface protein and a mutant of a second cell surface protein selected from a group consisting of: CD33 and CD205 (LY75); CD33 and CD244 (SLAMF4); CD33 and CD305 (LAIR1); CD33 and CD49D (ITGA4); CLL-1 and CD205 (LY75); CLL-1 and CD244 (SLAMF4); CLL-1 and CD305 (LAIR1); CLL-1 and CD49D (ITGA4); EMR2 and CD205 (LY75); EMR2 and CD244 (SLAMF4); EMR2 and CD305 (LAIR1); EMR2 and CD49D (ITGA4); CD38 and CD205 (LY75); CD38 and CD244 (SLAMF4); CD38 and CD305 (LAIR1); CD38 and CD49D (ITGA4); CD305 (LAIR1) and CD49D (ITGA4); CD305 (LAIR1) and CD205 (LY75); CD305 (LAIR1) and CD244 (SLAMF4); CD49D (ITGA4) and CD205 (LY75); CD49D (ITGA4) and CD244 (SLAMF4); and CD205 (LY75) and CD244 (SLAMF4). Further examples of genetically engineered hematopoietic cells are further described herein. Genetically Engineered Hematopoietic Cell Expressing CLL-1 Mutants In some embodiments, a genetically engineered hematopoietic cell has reduced or eliminated expression of a cell surface protein, or expresses a mutant of a cell surface protein, wherein the cell surface protein is CLL-1. C-type lectin-like molecule-1 (CLL-1), which may also be referred to as CLEC12A, is a C-type lectin-like receptor family member that is frequently expressed on acute myeloid leukemia (AML) cells. In some embodiments, CLL-1 is expressed by hematopoietic cells, e.g., hematopoietic stem cells and/or hematopoietic progenitor cells. The CLL-1 gene comprises 6 exons and is located on human chromosome 12. An example of a CLL-1 gene sequence can be found at NG_029426.2. In some embodiments, a mutation of a gene encoding CLL-1 alters one or more amino acids associated with an epitope of CLL-1. In some embodiments, the epitope of CLL-1 is a portion of CLL-1 bound by an agent, e.g., an immunotherapeutic agent. In some embodiments, the agent is an anti-CLL-1 antibody. In some embodiments, the agent comprises an anti-CLL-1 antibody or portion thereof, e.g., an antibody drug conjugate (ADC), a chimeric antigen receptor (CAR), or a multispecific antibody (e.g., a bispecific T cell engager). In some embodiments, the agent comprises an antibody or antigen-binding fragment thereof described in PCT Publication No. WO2023/201238, which is incorporated by reference herein in entirety. In some embodiments, the epitope of CLL-1 is one or more amino acids of a protein domain (e.g., the extracellular domain) or the amino acids encoded by an exon or combination of exons of the gene encoding CLL-1. In some embodiments, the epitope of CLL-1 comprises one or more amino acids encoded by exon 2 of the gene encoding CLL-1. In some embodiments, the epitope of CLL-1 comprises one or more amino acids encoded by exon 3 of the gene encoding CLL-1. In some embodiments, the epitope of CLL-1 comprises one or more amino acids encoded by exon 4 of the gene encoding CLL-1. In some embodiments, the epitope of CLL-1 comprises one or more amino acids encoded by exon 5 of the gene encoding CLL-1. In some embodiments, the epitope of CLL-1 comprises one or more amino acids encoded by exon 6 of the gene encoding CLL-1. In some embodiments, a mutation of a gene encoding CLL-1 makes a change in the amino acid sequence corresponding to the amino acid sequence of a CLL-1 ortholog. In some embodiments, a mutation substitutes an amino acid of human CLL-1 for an amino acid at a corresponding position of an orthologous CLL-1, e.g., a non-human primate CLL-1. In some embodiments, a mutation inserts or deletes one or more amino acids of human CLL-1 to correspond to the sequence of an orthologous CLL-1, e.g., a non-human primate CLL-1. In some embodiments, a mutation changes the amino acid sequence in a manner corresponding to a tolerable genetic variant identified by one or more genomic sequence comparison algorithms, e.g., gnomAD (see, e.g., Gudmundsson et al. arXiv:2107.11458v3, e.g., gnomad.broadinstitute.org/) or to a position characterized by a plurality of tolerable genetic variants. In some embodiments, mutations to CLL-1 corresponding to the amino acid sequence of a CLL-1 ortholog or at positions characterized by a plurality of tolerable genetic variants decrease or eliminate binding of an immunotherapeutic agent targeting CLL-1 while preserving some or all of CLL-1 structure, expression, and/or functionality, providing a cell expressing CLL-1 (e.g., functional CLL-1) that is targeted less or not at all by anti-CLL-1 immunotherapeutic agents. In some embodiments, alteration results in a missense variant of CLL-1. Genetically Engineered Hematopoietic Cell Expressing CD123 Mutants In some embodiments, a genetically engineered hematopoietic cell has reduced or eliminated expression of a cell surface protein, or of a cell surface protein, wherein the cell surface protein is CD123. CD123 (also known as interleukin-3 receptor alpha or IL3Rα) is a type I cytokine receptor which binds to IL3. IL3 is a pleiotropic cytokine that regulates the function and production of hematopoietic and immune cells (see, e.g., Testa et al. Biomarker Research volume 2, Article number: 4 (2014)). Dysregulated expression of IL3 is associated with various cancers including myeloma (see, e.g., Lee et al. Blood (2004) 103 (6): 2308- 2315). In some embodiments, a hematopoietic malignancy is characterized by cells expressing (e.g., over-expressing) CD123. Dysregulated expression of CD123 is associated with various hematopoietic malignancies including hairy cell leukemia, acute myeloid leukemia, blastic plasmacytoid dendritic cell neoplasm, and systemic mastocytosis (see, e.g., Del Giudice et al. Hematologica (2004) 89 (3): 303-308; Munoz et al. Hematologica (2001) 86 (12): 1261-1269; Angelot-Delettre et al. Hematologica (2015) 100 (2): 223-230; Alayed et al. American Journal of Hematology (2013) 88 (12): 1055-1061; Paradanani et al. Leukemia (2016) 30 (4): 914-918; Testa et al. Biomarker Research (2014) 2: 4; and Lamble et al. Journal of Clinical Oncology (2022) 40 (3): 252-261). In some embodiments, CD123 is expressed by hematopoietic cells, e.g., hematopoietic stem cells and/or hematopoietic progenitor cells. In some embodiments, a mutation of a gene encoding CD123 alters one or more amino acids associated with an epitope of CD123. In some embodiments, the epitope of CD123 is a portion of CD123 bound by an agent, e.g., an immunotherapeutic agent. In some embodiments, the agent is an anti-CD123 antibody. In some embodiments, the agent comprises an anti-CD123 antibody or portion thereof, e.g., an antibody drug conjugate (ADC), a chimeric antigen receptor (CAR), or a multispecific antibody (e.g., a bispecific T cell engager). For example, the agent can be anti-CD123 antibody 7G3 or a variant thereof (e.g., a humanized variant, e.g., antibody CSL-36). In some embodiments, the agent is an anti-CD123 drug, e.g., talacotuzumab. In some embodiments, the epitope of CD123 is one or more amino acids of a protein domain (e.g., the extracellular domain) or the amino acids encoded by an exon or combination of exons of the gene encoding CD123. In some embodiments, the epitope of CD123 comprises one or more amino acids encoded by exon 2 of the gene encoding CD123. In some embodiments, the epitope of CD123 comprises one or more amino acids encoded by exon 3 of the gene encoding CD123. In some embodiments, the epitope of CD123 comprises one or more amino acids encoded by exon 4 of the gene encoding CD123. In some embodiments, the epitope of CD123 comprises one or more amino acids encoded by exon 5 of the gene encoding CD123. In some embodiments, the epitope of CD123 comprises one or more amino acids encoded by exon 6 of the gene encoding CD123. In some embodiments, the epitope of CD123 comprises one or more (e.g., two or more, three or more, four or more, or all) of the amino acids at positions 51, 59, 61, 82, or 84 of a wildtype gene encoding CD123 or at corresponding positions in a homologous CD123 gene. In some embodiments, a mutation of a gene encoding CD123 comprises a substitution of the amino acid at position 51 of a wildtype CD123 or at a corresponding position in a homologous CD123 gene. In some embodiments, a lysine is substituted for the amino acid at position 51 of a wildtype CD123 or at a corresponding position in a homologous CD123 gene. In some embodiments, a glycine is substituted for the amino acid at position 51 of a wildtype CD123 or at a corresponding position in a homologous CD123 gene. In some embodiments, a mutation of a gene encoding CD123 comprises a substitution of the amino acid at position 59 of a wildtype CD123 or at a corresponding position in a homologous CD123 gene. In some embodiments, a phenylalanine is substituted for the amino acid at position 59 of a wildtype CD123 or at a corresponding position in a homologous CD123 gene. In some embodiments, a cysteine is substituted for the amino acid at position 59 of a wildtype CD123 or at a corresponding position in a homologous CD123 gene. In some embodiments, a mutation of a gene encoding CD123 comprises a substitution of the amino acid at position 61 of a wildtype CD123 or at a corresponding position in a homologous CD123 gene. In some embodiments, a leucine is substituted for the amino acid at position 61 of a wildtype CD123 or at a corresponding position in a homologous CD123 gene. In some embodiments, a mutation of a gene encoding CD123 comprises a substitution of the amino acid at position 82 of a wildtype CD123 or at a corresponding position in a homologous CD123 gene. In some embodiments, an alanine is substituted for the amino acid at position 82 of a wildtype CD123 or at a corresponding position in a homologous CD123 gene. In some embodiments, a mutation of a gene encoding CD123 comprises a substitution of the amino acid at position 84 of a wildtype CD123 or at a corresponding position in a homologous CD123 gene. In some embodiments, a glutamine is substituted for the amino acid at position 84 of a wildtype CD123 or at a corresponding position in a homologous CD123 gene. In some embodiments, an alanine is substituted for the amino acid at position 84 of a wildtype CD123 or at a corresponding position in a homologous CD123 gene. In some embodiments, a mutation of a gene encoding CD123 makes a change in the amino acid sequence corresponding to the amino acid sequence of a CD123 ortholog. In some embodiments, a mutation substitutes an amino acid of human CD123 for an amino acid at a corresponding position of an orthologous CD123, e.g., a non-human primate CD123. In some embodiments, a mutation inserts or deletes one or more amino acids of human CD123 to correspond to the sequence of an orthologous CD123, e.g., a non-human primate CD123. In some embodiments, a mutation changes the amino acid sequence in a manner corresponding to a tolerable genetic variant identified by one or more genomic sequence comparison algorithms, e.g., gnomAD (see, e.g., Gudmundsson et al. arXiv:2107.11458v3, e.g., gnomad.broadinstitute.org/) or to a position characterized by a plurality of tolerable genetic variants. In some embodiments, mutations to CD123 corresponding to the amino acid sequence of a CD123 ortholog or at positions characterized by a plurality of tolerable genetic variants decrease or eliminate binding of an immunotherapeutic agent targeting CD123 while preserving some or all of CD123 structure, expression, and/or functionality, providing a cell expressing CD123 (e.g., functional CD123) that is targeted less or not at all by anti-CD123 immunotherapeutic agents. In some embodiments, alteration results in a missense variant of CD123. Genetically Engineered Hematopoietic Cell Expressing CD38 Mutants In some embodiments, a genetically engineered hematopoietic cell has reduced or eliminated expression of a cell surface protein, or expresses a mutant of a cell surface protein, wherein the cell surface protein is CD38. CD38 (also known as cyclic ADP ribose hydrolase) is a transmembrane ectoenzymatic glycoprotein involved in cell adhesion, signal transduction, and calcium signaling (see, e.g., van de Donk et al. Blood (2018) 131 (1): 13– 29). In some embodiments, a hematopoietic malignancy is characterized by cells expressing (e.g., over-expressing) CD38. In some embodiments, CD38 is expressed by hematopoietic cells, e.g., hematopoietic stem cells and/or hematopoietic progenitor cells. In some embodiments, a mutation of a gene encoding CD38 alters one or more amino acids associated with an epitope of CD38. In some embodiments, the epitope of CD38 is a portion of CD38 bound by an agent, e.g., an immunotherapeutic agent. In some embodiments, the agent is an anti-CD38 antibody. In some embodiments, the agent comprises an anti-CD38 antibody or portion thereof, e.g., an antibody drug conjugate (ADC), a chimeric antigen receptor (CAR), or a multispecific antibody (e.g., a bispecific T cell engager). For example, the agent can be anti-CD38 antibody HB7 or a variant thereof (e.g., a humanized variant). In some embodiments, the agent is an anti-CD38 drug, e.g., daratumumab. In some embodiments, the epitope of CD38 is one or more amino acids of a protein domain (e.g., the extracellular domain) or the amino acids encoded by an exon or combination of exons of the gene encoding CD38. In some embodiments, the epitope of CD38 comprises one or more amino acids encoded by exon 7 of the gene encoding CD38. In some embodiments, the epitope of CD38 comprises one or more (e.g., two or more, three or more, four or more, or all) of the amino acids at positions 270-274 of a wildtype gene encoding CD38 or at corresponding positions in a homologous CD38 gene. In some embodiments, a mutation of a gene encoding CD38 comprises a substitution of the amino acid at position 270 of a wildtype CD38 or at a corresponding position in a homologous CD38 gene. In some embodiments, an alanine is substituted for the amino acid at position 270 of a wildtype CD38 or at a corresponding position in a homologous CD38 gene. In some embodiments, a mutation of a gene encoding CD38 comprises a substitution of the amino acid at position 271 of a wildtype CD38 or at a corresponding position in a homologous CD38 gene. In some embodiments, a mutation of a gene encoding CD38 comprises a substitution of the amino acid at position 272 of a wildtype CD38 or at a corresponding position in a homologous CD38 gene. In some embodiments, a histidine is substituted for the amino acid at position 272 of a wildtype CD38 or at a corresponding position in a homologous CD38 gene. In some embodiments, an arginine is substituted for the amino acid at position 272 of a wildtype CD38 or at a corresponding position in a homologous CD38 gene. In some embodiments, an alanine is substituted for the amino acid at position 272 of a wildtype CD38 or at a corresponding position in a homologous CD38 gene. In some embodiments, a mutation of a gene encoding CD38 comprises a substitution of the amino acid at position 273 of a wildtype CD38 or at a corresponding position in a homologous CD38 gene. In some embodiments, a mutation of a gene encoding CD38 comprises a substitution of the amino acid at position 274 of a wildtype CD38 or at a corresponding position in a homologous CD38 gene. In some embodiments, a phenylalanine is substituted for the amino acid at position 274 of a wildtype CD38 or at a corresponding position in a homologous CD38 gene. In some embodiments, a mutation of a gene encoding CD38 makes a change in the amino acid sequence corresponding to the amino acid sequence of a CD38 ortholog. In some embodiments, a mutation substitutes an amino acid of human CD38 for an amino acid at a corresponding position of an orthologous CD38, e.g., a non-human primate CD38. In some embodiments, a mutation inserts or deletes one or more amino acids of human CD38 to correspond to the sequence of an orthologous CD38, e.g., a non-human primate CD38. In some embodiments, a mutation changes the amino acid sequence in a manner corresponding to a tolerable genetic variant identified by one or more genomic sequence comparison algorithms, e.g., gnomAD, or to a position characterized by a plurality of tolerable genetic variants. In some embodiments, mutations to CD38 corresponding to the amino acid sequence of a CD38 ortholog or at positions characterized by a plurality of tolerable genetic variants decrease or eliminate binding of an immunotherapeutic agent targeting CD38 while preserving some or all of CD38 structure, expression, and/or functionality, providing a cell expressing CD38 (e.g., functional CD38) that is targeted less or not at all by anti-CD38 immunotherapeutic agents. Genetically Engineered Hematopoietic Cell Expressing EMR2 Mutants In some embodiments, a genetically engineered hematopoietic cell has reduced or eliminated expression of a cell surface protein, or expresses a mutant of a cell surface protein, wherein the cell surface protein is EMR2. EGF-like module-containing mucin-like hormone receptor-like 2 (EMR2), also referred to as CD312 and ADGRE2, is a 823-amino acid, ~90 kDa protein (depending on isoform) of the EGF-seven-span transmembrane (TM7) family of adhesion G protein-coupled receptors (GPCR) with a high level of homology with CD97. EMR2 forms a heterodimer and binds to chondroitin sulfate B via its EGF-like domain 4 and mediate cell adhesion, granulocyte chemotaxis, degranulation, and the release of pro- inflammatory cytokines in macrophages. See, e.g. Kuan-Yu et al. Front. Immunol. (2017) 8:373. Without wishing to be bound by any particular theory, EMR2 is expressed on myeloid cells with highest expression in granulocytes, macrophages, and Kupffer cells. The ADGRE2 gene located on human chromosome 19 encodes human EMR2 and canonically contains 19 exons, although a number of isoforms exist with varying number EGF domains due to alternative RNA splicing. The dominant isoform in whole blood contains 17 exons. See, e.g. Safaee et al. Onc. Rev. (2014).8(242):20-24. In some embodiments, a mutation of a gene encoding EMR2 alters one or more amino acids associated with an epitope of EMR2. In some embodiments, the epitope of EMR2 is a portion of EMR2 bound by an agent, e.g., an immunotherapeutic agent. In some embodiments, the agent is an anti-EMR2 antibody. In some embodiments, the agent comprises an anti- EMR2 antibody or portion thereof, e.g., an antibody drug conjugate (ADC), a chimeric antigen receptor (CAR), or a multispecific antibody (e.g., a bispecific T cell engager). In some embodiments, the agent is an anti- EMR2 drug. In some embodiments, the epitope of EMR2 corresponds to the amino acids of a protein domain (e.g., the extracellular domain) or the amino acids encoded by an exon or combination of exons of the gene encoding EMR2. In some embodiments, the epitope of EMR2 comprises amino acids encoded by exon 1, exon 2, exon 3, exon 4, exon 5, exon 6, exon 7, exon 8, exon 9, exon 10, exon 11, exon 12, exon 13, exon 14, exon 15, exon 16, exon 17, exon 18, exon 19, exon 20, or exon 21 of EMR2, or a combination thereof. In some embodiments, the epitope of EMR2 comprises amino acids encoded by one, two, three, four, or all of exons 6, 10, 11, 14, and 18 of EMR2. In some embodiments, the epitope of EMR2 comprises the amino acids encoded by exon 6 of EMR2. In some embodiments, the epitope of EMR2 comprises the amino acids encoded by exon 10 of EMR2. In some embodiments, the epitope of EMR2 comprises the amino acids encoded by exon 11 of EMR2. In some embodiments, the epitope of EMR2 comprises the amino acids encoded by exon 14 of EMR2. In some embodiments, the epitope of EMR2 comprises the amino acids encoded by exon 18 of EMR2. In some embodiments, a mutation of a gene encoding EMR2 makes a change in the amino acid sequence corresponding to the amino acid sequence of a EMR2 ortholog. In some embodiments, an alteration comprises substitution of amino acid at any one or more of positions 124, 132, 146, 292, 294, 295, 296, 298, 299, 303, 304, 305, 306, 307, 308, 312, 318, 320, 328, 329, 331, 332, 335, 340, 347, 527, or 708 of a wildtype EMR2 or at a corresponding position in a homologous EMR2. In some embodiments, an alteration results in a missense variant of EMR2. In some embodiments, alteration results in a change at a splice region in EMR2. In some embodiments, a mutation of a gene encoding EMR2 alters one or more amino acids associated with an epitope of EMR2. In some embodiments, the epitope of EMR2 is a portion of EMR2 bound by an agent, e.g., an immunotherapeutic agent. In some embodiments, the agent is an anti-EMR2 antibody. In some embodiments, the agent comprises an anti-EMR2 antibody or portion thereof, e.g., an antibody drug conjugate (ADC), a chimeric antigen receptor (CAR), or a multispecific antibody (e.g., a bispecific T cell engager). For example, the agent can be anti-EMR2 monoclonal antibody 2A1 (Thermo Fisher) or a variant thereof (e.g., a humanized variant), Q9UHX3, OASA01861, AB_2738756, NLS6381, ab75190, MAB4894, or A100,000. Additional anti-EMR2 antibodies will be evident to one of ordinary skill in the art. See, e.g., International Publication No. WO 2017/087800 A1; Chang et al. FEBS Letters. (2003) 547(1-3):145-150; Yona et al. FASEB J. (2008).22(3): 741-751. In some embodiments, the agent comprises an anti-EMR2 antibody or portion thereof described in International Patent Application No. PCT/US2024/029308, which is incorporated by reference in its entirety. In some embodiments, mutations to EMR2 corresponding to the amino acid sequence of a EMR2 ortholog decrease or eliminate binding of an immunotherapeutic agent targeting EMR2 while preserving some or all of EMR2 expression and/or functionality, providing a cell expressing EMR2 (e.g., functional EMR2) that is targeted less or not at all by anti-EMR2 immunotherapeutic agents. Genetically Engineered Hematopoietic Cell Expressing CD47 Mutants In some embodiments, a genetically engineered hematopoietic cell has reduced or eliminated expression of a cell surface protein, or expresses a mutant of a cell surface protein, wherein the cell surface protein is CD47. CD47 is a transmembrane integrin-associated protein belonging to the immunoglobulin superfamily and is involved in the increase of intracellular calcium concentration that occurs upon cell adhesion to extracellular matrix. CD47 binds to a variety of ligands including thrombospondin-1 and signal-regulatory protein alpha and functions in processes such as apoptosis, proliferation, adhesion, and migration. CD47 also has roles in immune and angiogenic responses including regulation of phagocytosis by macrophages (see, e.g., Brown and Frazier. Trends in Cell Biology (2001) 11 (3): 130-135). CD47 is widely expressed across various tissues in humans and also in solid tumors and hematological malignancies (see, e.g., Jiang et al. Journal of Hematology & Oncology (2021) 14: 180). Human CD47 is located on chromosome 3 and contains 13 exons. In some embodiments, a mutation of a gene encoding CD47 alters one or more amino acids associated with an epitope of CD47. In some embodiments, the epitope of CD47 is a portion of CD47 bound by an agent, e.g., an immunotherapeutic agent. In some embodiments, the agent is an anti-CD47 antibody. In some embodiments, the agent is the anti-CD47 B6H12 or 2D3 antibody. In some embodiments, the agent comprises an anti-CD47 antibody or portion thereof, e.g., an antibody drug conjugate (ADC), a chimeric antigen receptor (CAR), or a multispecific antibody (e.g., a bispecific T cell engager). For example, the agent can be anti-CD47 antibody or a variant thereof (e.g., a humanized variant). In some embodiments, the agent is an anti-CD47 drug. In some embodiments, the epitope of CD47 is one or more amino acids of a protein domain (e.g., the extracellular domain) or the amino acids encoded by an exon or combination of exons of the gene encoding CD47. In some embodiments, the epitope of CD47 comprises one or more of amino acids 117-122 in CD47. In some embodiments, one or more of amino acids 117-122 in CD47 is deleted. In some embodiments, amino acids 117-122 in CD47 are deleted. In some embodiments, amino acids 117, 118, 119, 120, 121, and/or 122 or any combination thereof in CD47 is deleted. In some embodiments, the epitope of CD47 comprises one or more of amino acids 52-55 in CD47. In some embodiments, one or more of amino acids 52-55 in CD47 is deleted. In some embodiments, amino acids 52-55 in CD47 are deleted. In some embodiments, amino acids 52, 53, 54, and/or 55 or any combination thereof in CD47 is deleted. In some embodiments, a mutation of a gene encoding CD47 comprises a substitution of the amino acid at position 31 of a wildtype CD47 or at a corresponding position in a homologous CD47 gene. In some embodiments, a methionine is substituted for the amino acid at position 31 of a wildtype CD47 or at a corresponding position in a homologous CD47 gene. In some embodiments, a mutation of a gene encoding CD47 comprises a substitution of the amino acid at position 47 of a wildtype CD47 or at a corresponding position in a homologous CD47 gene. In some embodiments, a histidine is substituted for the amino acid at position 47 of a wildtype CD47 or at a corresponding position in a homologous CD47 gene. In some embodiments, a glycine is substituted for the amino acid at position 47 of a wildtype CD47 or at a corresponding position in a homologous CD47 gene. In some embodiments, a mutation of a gene encoding CD47 comprises a substitution of the amino acid at position 49 of a wildtype CD47 or at a corresponding position in a homologous CD47 gene. In some embodiments, an arginine is substituted for the amino acid at position 49 of a wildtype CD47 or at a corresponding position in a homologous CD47 gene. In some embodiments, a proline is substituted for the amino acid at position 49 of a wildtype CD47 or at a corresponding position in a homologous CD47 gene. In some embodiments, a mutation of a gene encoding CD47 comprises a substitution of the amino acid at position 52 of a wildtype CD47 or at a corresponding position in a homologous CD47 gene. In some embodiments, a mutation of a gene encoding CD47 comprises a deletion of the amino acid at position 52 of a wildtype CD47 or at a corresponding position in a homologous CD47 gene. In some embodiments, a mutation of a gene encoding CD47 comprises a substitution of the amino acid at position 53 of a wildtype CD47 or at a corresponding position in a homologous CD47 gene. In some embodiments, an alanine is substituted for the amino acid at position 53 of a wildtype CD47 or at a corresponding position in a homologous CD47 gene. In some embodiments, a mutation of a gene encoding CD47 comprises a deletion of the amino acid at position 53 of a wildtype CD47 or at a corresponding position in a homologous CD47 gene. In some embodiments, a mutation of a gene encoding CD47 comprises a deletion of the amino acid at position 54 of a wildtype CD47 or at a corresponding position in a homologous CD47 gene. In some embodiments, a mutation of a gene encoding CD47 comprises a deletion of the amino acid at position 55 of a wildtype CD47 or at a corresponding position in a homologous CD47 gene. In some embodiments, a mutation of a gene encoding CD47 comprises a substitution of the amino acid at position 120 of a wildtype CD47 or at a corresponding position in a homologous CD47 gene. In some embodiments, an alanine is substituted for the amino acid at position 120 of a wildtype CD47 or at a corresponding position in a homologous CD47 gene. In some embodiments, a mutation of a gene encoding CD47 comprises a substitution of the amino acid at position 124 of a wildtype CD47 or at a corresponding position in a homologous CD47 gene. In some embodiments, a lysine is substituted for the amino acid at position 124 of a wildtype CD47 or at a corresponding position in a homologous CD47. In some embodiments, a mutation of a gene encoding CD47 makes a change in the amino acid sequence corresponding to the amino acid sequence of a CD47 ortholog. In some embodiments, a mutation substitutes an amino acid of human CD47 for an amino acid at a corresponding position of an orthologous CD47, e.g., a non-human primate CD47. In some embodiments, a mutation inserts or deletes one or more amino acids of human CD47 to correspond to the sequence of an orthologous CD47, e.g., a non-human primate CD47. In some embodiments, a mutation changes the amino acid sequence in a manner corresponding to a tolerable genetic variant identified by one or more genomic sequence comparison algorithms, e.g., gnomAD (see, e.g., Gudmundsson et al. arXiv:2107.11458v3, e.g., gnomad.broadinstitute.org/), or to a position characterized by a plurality of tolerable genetic variants. In some embodiments, mutations to CD47 corresponding to the amino acid sequence of a CD47 ortholog or at positions characterized by a plurality of tolerable genetic variants decrease or eliminate binding of an immunotherapeutic agent targeting CD47 while preserving some or all of CD47 structure, expression, and/or functionality, providing a cell expressing CD47 (e.g., functional CD47) that is targeted less or not at all by anti-CD47 immunotherapeutic agents. Genetically Engineered Hematopoietic Cell Expressing CD34 Mutants In some embodiments, a genetically engineered hematopoietic cell has reduced or eliminated expression of a cell surface protein, or expresses a mutant of a cell surface protein, wherein the cell surface protein is CD34. CD34 is a transmembrane phosphoglycoprotein belonging to the single-pass transmembrane sialomucin protein family that functions as a cell-cell adhesion factor. Accordingly, CD34 is an important adhesion molecule required for T-cells to enter lymph nodes and for attachment of hematopoietic stem cells to bone marrow extracellular matrix or to stromal cells. CD34 is highly expressed in hematopoietic stem and progenitor cells and endothelial cells. Moreover, CD34 is commonly found expressed on the cell surface of hematopoietic cancer cells (see, e.g., Sydney et al. Stem Cells (2014) 32 (6): 1380-1389; Nielsen and McNagny. Journal of Cell Science (2008) 121 (22): 3683-3692; Lanze et al. Journal of Biological Regulators and Homeostatic Agents (2001) 15 (1): 1-13; Sutherland and Keating. Journal of Hematotherapy (2009) 1 (2): 115-129). CD34 is located on chromosome 1 and contains 8 exons. In some embodiments, a mutation of a gene encoding CD34 alters one or more amino acids associated with an epitope of CD34. In some embodiments, the epitope of CD34 is a portion of CD34 bound by an agent, e.g., an immunotherapeutic agent. In some embodiments, the agent is an anti-CD34 antibody. In some embodiments, the anti-CD34 antibody is clone QBend10 or 561. In some embodiments, the agent comprises an anti-CD34 antibody or portion thereof, e.g., an antibody drug conjugate (ADC), a chimeric antigen receptor (CAR), or a multispecific antibody (e.g., a bispecific T cell engager). In some embodiments, the epitope of CD34 is one or more amino acids of a protein domain (e.g., the extracellular domain) or the amino acids encoded by an exon or combination of exons of the gene encoding CD34. In some embodiments, a mutation of a gene encoding CD34 comprises a substitution of the amino acid at position 42 of a wildtype CD34 or at a corresponding position in a homologous CD34 gene. In some embodiments, a glycine is substituted for the amino acid at position 42 of a wildtype CD34 or at a corresponding position in a homologous CD34 gene. In some embodiments, a mutation of a gene encoding CD34 comprises a substitution of the amino acid at position 46 of a wildtype CD34 or at a corresponding position in a homologous CD34 gene. In some embodiments, an alanine is substituted for the amino acid at position 46 of a wildtype CD34 or at a corresponding position in a homologous CD34 gene. In some embodiments, a mutation of a gene encoding CD34 comprises a substitution of the amino acid at position 47 of a wildtype CD34 or at a corresponding position in a homologous CD34 gene. In some embodiments, a lysine is substituted for the amino acid at position 47 of a wildtype CD34 or at a corresponding position in a homologous CD34 gene. In some embodiments, a glutamate is substituted for the amino acid at position 47 of a wildtype CD34 or at a corresponding position in a homologous CD34 gene. In some embodiments, a mutation of a gene encoding CD34 comprises a substitution of the amino acid at position 49 of a wildtype CD34 or at a corresponding position in a homologous CD34 gene. In some embodiments, a proline is substituted for the amino acid at position 49 of a wildtype CD34 or at a corresponding position in a homologous CD34 gene. In some embodiments, a serine is substituted for the amino acid at position 49 of a wildtype CD34 or at a corresponding position in a homologous CD34 gene. In some embodiments, a mutation of a gene encoding CD34 comprises a substitution of the amino acid at position 50 of a wildtype CD34 or at a corresponding position in a homologous CD34 gene. In some embodiments, an alanine is substituted for the amino acid at position 50 of a wildtype CD34 or at a corresponding position in a homologous CD34 gene. In some embodiments, a proline is substituted for the amino acid at position 50 of a wildtype CD34 or at a corresponding position in a homologous CD34 gene. In some embodiments, a mutation of a gene encoding CD34 comprises a substitution of the amino acid at position 51 of a wildtype CD34 or at a corresponding position in a homologous CD34 gene. In some embodiments, an alanine is substituted for the amino acid at position 51 of a wildtype CD34 or at a corresponding position in a homologous CD34 gene. In some embodiments, a mutation of a gene encoding CD34 comprises a substitution of the amino acid at position 54 of a wildtype CD34 or at a corresponding position in a homologous CD34 gene. In some embodiments, an alanine is substituted for the amino acid at position 54 of a wildtype CD34 or at a corresponding position in a homologous CD34 gene. In some embodiments, a mutation of a gene encoding CD34 comprises a substitution of the amino acid at position 55 of a wildtype CD34 or at a corresponding position in a homologous CD34 gene. In some embodiments, an alanine is substituted for the amino acid at position 55 of a wildtype CD34 or at a corresponding position in a homologous CD34 gene. In some embodiments, a mutation of a gene encoding CD34 makes a change in the amino acid sequence corresponding to the amino acid sequence of a CD34 ortholog. In some embodiments, a mutation substitutes an amino acid of human CD34 for an amino acid at a corresponding position of an orthologous CD34, e.g., a non-human primate CD34. In some embodiments, a mutation inserts or deletes one or more amino acids of human CD34 to correspond to the sequence of an orthologous CD34, e.g., a non-human primate CD34. In some embodiments, a mutation changes the amino acid sequence in a manner corresponding to a tolerable genetic variant identified by one or more genomic sequence comparison algorithms, e.g., gnomAD (see, e.g., Gudmundsson et al. arXiv:2107.11458v3, e.g., gnomad.broadinstitute.org/), or to a position characterized by a plurality of tolerable genetic variants. In some embodiments, mutations to CD34 corresponding to the amino acid sequence of a CD34 ortholog or at positions characterized by a plurality of tolerable genetic variants decrease or eliminate binding of an immunotherapeutic agent targeting CD34 while preserving some or all of CD34 structure, expression, and/or functionality, providing a cell expressing CD34 (e.g., functional CD34) that is targeted less or not at all by anti-CD34 immunotherapeutic agents. Genetically Engineered Hematopoietic Cell Expressing IL1RAP Mutants In some embodiments, a genetically engineered hematopoietic cell has reduced or eliminated expression of a cell surface protein, or expresses a mutant of a cell surface protein, wherein the cell surface protein is interleukin-1 receptor accessory protein (IL1RAP). IL1RAP is known by several aliases in the art, such as IL-1 receptor accessory protein (IL- 1RAcP), interleukin-1 receptor 3 (IL1R3), and interleukin-1 receptor accessory protein beta. As will be known to one of ordinary skill in the art, IL1RAP is involved in initiating signaling events that regulate activation of genes that are responsive to the interleukin 1 inflammatory cytokine. IL1RAP also interacts with IL1RL2 to form a heterodimer in the IL- 36 cytokine signaling system. In humans, IL1RAP is found at chromosomal location 3q28 and comprises 17 exons. Non-limiting examples of reference sequence for the gene encoding IL1RAP are found at NC_000003.12 and NC_060927.1. It will also be known to the skilled artisan that IL1RAP is encoded by gene capable of undergoing alternative splicing resulting in production of membrane-bound and soluble isoforms. Said IL1RAP isoforms differ in their C-terminus and the ratios of soluble to membrane-bound isoforms may change depending on cell signaling events (e.g., increases in the soluble isoform during acute-phase induction or stress). Non- limiting examples of gene products (e.g., RNAs and proteins) encoded by IL1RAP are set forth in NM_001167928.2 (encoding a protein found at NP_001161400.1), NM_001167929.1 (encoding a protein found at NP_001161401.1), NM_0011679302 (encoding a protein found at NP_001161402.1), NM_001167931.2 (encoding a protein found at NP_001161403.1), NM_001364878.1 (encoding a protein found at NP_001351808.1), NM_001364880.2 (encoding a protein found at NP_001351809.1), NM_001364881.2 (encoding a protein found at NP_001351810.1), and NM_002182.4 (encoding a protein found at NP_002173.1), NM_134470.4 (encoding a protein found at NP_608273.1). In some embodiments, the IL1RAP protein comprises an Ig-like C2-type 1 domain at amino acids 21-128, an Ig-like C2-type 2 domain at amino acids 141-230, an Ig-like C2-type 3 domain at amino acids 242-348, and a TIR domain from amino acids 403-546 which mediates NAD+ hydrolase activity. In some embodiments, the membrane-bound isoform comprises an amino acid sequence as set forth below: MTLLWCVVSLYFYGILQSDASERCDDWGLDTMRQIQVFEDEPARIKCPLFEHFLKFNYSTAHSAGLTLIWYWTRQ DRDLEEPINFRLPENRISKEKDVLWFRPTLLNDTGNYTCMLRNTTYCSKVAFPLEVVQKDSCFNSPMKLPVHKLY IEYGIQRITCPNVDGYFPSSVKPTITWYMGCYKIQNFNNVIPEGMNLSFLIALISNNGNYTCVVTYPENGRTFHL TRTLTVKVVGSPKNAVPPVIHSPNDHVVYEKEPGEELLIPCTVYFSFLMDSRNEVWWTIDGKKPDDITIDVTINE SISHSRTEDETRTQILSIKKVTSEDLKRSYVCHARSAKGEVAKAAKVKQKVPAPRYTVELACGFGATVLLVVILI VVYHVYWLEMVLFYRAHFGTDETILDGKEYDIYVSYARNAEEEEFVLLTLRGVLENEFGYKLCIFDRDSLPGGIV TDETLSFIQKSRRLLVVLSPNYVLQGTQALLELKAGLENMASRGNINVILVQYKAVKETKVKELKRAKTVLTVIK WKGEKSKYPQGRFWKQLQVAMPVKKSPRRSSSDEQGLSYSSLKNV (SEQ ID NO: 8)(UniPort ID: Q9NPH3-1) In some embodiments, the soluble-bound isoform comprises an amino acid sequence as set forth below: MTLLWCVVSLYFYGILQSDASERCDDWGLDTMRQIQVFEDEPARIKCPLFEHFLKFNYSTAHSAGLTLIWYWTRQ DRDLEEPINFRLPENRISKEKDVLWFRPTLLNDTGNYTCMLRNTTYCSKVAFPLEVVQKDSCFNSPMKLPVHKLY IEYGIQRITCPNVDGYFPSSVKPTITWYMGCYKIQNFNNVIPEGMNLSFLIALISNNGNYTCVVTYPENGRTFHL TRTLTVKVVGSPKNAVPPVIHSPNDHVVYEKEPGEELLIPCTVYFSFLMDSRNEVWWTIDGKKPDDITIDVTINE SISHSRTEDETRTQILSIKKVTSEDLKRSYVCHARSAKGEVAKAAKVKQKGNRCGQ (SEQ ID NO: 9) (UniPort ID: Q9NPH3-2) MTLLWCVVSLYFYGILQSDASERCDDWGLDTMRQIQVFEDEPARIKCPLFEHFLKFNYSTAHSAGLTLIWYWTRQ DRDLEEPINFRLPENRISKEKDVLWFRPTLLNDTGNYTCMLRNTTYCSKVAFPLEVVQKDSCFNSPMKLPVHKLY IEYGIQRITCPNVDGYFPSSVKPTITWYMGCYKIQNFNNVIPEGMNLSFLIALISNNGNYTCVVTYPENGRTFHL TRTLTVKVVGSPKNAVPPVIHSPNDHVVYEKEPGEELLIPCTVYFSFLMDSRNEVWWTIDGKKPDDITIDVTINE SASSKIHSGTGLWFWSHSPASGDSHCCLPCLLARDGPILPGSFWNR (SEQ ID NO: 10)(UniPort ID: Q9NPH3-3) In some embodiments, the present disclosure relates to variants of IL1RAP. In some embodiments, genetically engineered hematopoietic cells comprise a variant gene encoding IL1RAP protein, wherein said variant gene comprises a substitution, deletion, or addition of at least one nucleotide or a combination thereof. In some embodiments, a mutant IL1RAP comprises a deletion or mutation of a fragment of the protein that is encoded by any one of the exons of the gene encoding IL1RAP. In some embodiments, genetically engineered hematopoietic cells comprise a mutant IL1RAP protein comprising a substitution, deletion, or addition of at least one amino acid or a combination thereof. In some embodiments, genetically engineered hematopoietic cells comprise a mutant IL1RAP protein comprising at least one modified non-essential epitope. In some embodiments, genetically engineered hematopoietic cells comprise IL1RAP expression that is reduced by at least 20%, at least 30%, at least 40%, at least 50%, at least 60%, at least 70%, at least 80%, at least 90%, or at least 95% relative to the wildtype counterpart cell. In some embodiments, genetically engineered hematopoietic cells express a mutant of IL1RAP that has reduced or no binding to an agent comprising an antigen-binding fragment that recognizes an epitope of IL1RAP. In some embodiments, at least 40%, at least 50%, at least 60%, at least 70%, at least 75%, at least 80%, at least 85%, at least 90%, or at least 95% of copies of IL1RAP in the population of cells have a mutation. By way of example, a population can comprise a plurality of different IL1RAP mutations and each mutation of the plurality contributes to the percent of copies of IL1RAP in the population of cells that have a mutation. In some embodiments, the expression of IL1RAP on the genetically engineered hematopoietic cell is compared to the expression of IL1RAP on a naturally occurring hematopoietic cell (e.g., a wild-type counterpart). In some embodiments, the genetic engineering results in a reduction in the expression level of IL1RAP by at least50%, at least 60%, at least 70%, at least 80%, at least 90%, at least 91%, at least 92%, at least 93%, at least 94%, at least 95%, at least 96%, at least 97%, at least 98%, or at least 99% as compared to the expression of IL1RAP on a naturally occurring hematopoietic cell (e.g., a wild-type counterpart). For example, in some embodiments, the genetically engineered hematopoietic cell expresses less than 20%, less than 19%, less than 18%, less than 17%, less than 16%, less than 15%, less than 14%, less than 13%, less than 12%, less than 11%, less than 10%, less than 9%, less than 8%, less than 7%, less than 6%, less than 5%, less than 4%, less than 3%, less than 2%, less than or 1% of IL1RAP as compared to a naturally occurring hematopoietic cell (e.g., a wild-type counterpart). In some embodiments, the genetic engineering results in a reduction in the expression level of wild-type IL1RAP by at least 50%, at least 60%, at least 70%, at least 80%, at least 90%, at least 91%, at least 92%, at least 93%, at least 94%, at least 95%, at least 96%, at least 97%, at least 98%, or at least 99% as compared to the expression of the level of wild-type IL1RAP on a naturally occurring hematopoietic cell (e.g., a wild-type counterpart). That is, in some embodiments, the genetically engineered hematopoietic cell expresses less than 20%, less than 19%, less than 18%, less than 17%, less than 16%, less than 15%, less than 14%, less than 13%, less than 12%, less than 11%, less than 10%, less than 9%, less than 8%, less than 7%, less than 6%, less than 5%, less than 4%, less than 3%, less than 2%, or less than 1% of IL1RAP as compared to a naturally occurring hematopoietic cell (e.g., a wild-type counterpart). Genetically Engineered Hematopoietic Cell Expressing CD33 Mutants In some embodiments, a genetically engineered hematopoietic cell has reduced or eliminated expression of a lineage-specific cell surface protein, or expresses a mutant of a lineage-specific cell surface protein, wherein the lineage-specific cell surface protein is CD33. As will be known to one of ordinary skill in the art, CD33 is encoded by seven exons, including the alternatively spliced exons 7A and 7B (Brinkman-Van der Linden et al. Mol Cell. Biol. (2003) 23: 4199-4206). Further, the CD33 gene encodes two isoforms, one of which retains exon 2, referred to as CD33M, and one that excludes exon 2, referred to as CD33m. Exemplary amino acid sequences of the 7A and 7B splicing isoforms are provided below: CD33M-7A: Amino Acid (underlined=exon 2; italicized= exon 7A) MPLLLLLPLLWAGALAMDPNFWLQVQESVTVQEGLCVLVPCTFFHPIPYYDKNSPVHGYWFREGAIIS RDSPVATNKLDQEVQEETQGRFRLLGDPSRNNCSLSIVDARRRDNGSYFFRMERGSTKYSYKSPQLSV HVTDLTHRPKILIPGTLEPGHSKNLTCSVSWACEQGTPPIFSWLSAAPTSLGPRTTHSSVLIITPRPQ DHGTNLTCQVKFAGAGVTTERTIQLNVTYVPQNPTTGIFPGDGSGKQETRAGVVHGAIGGAGVTALLA LCLCLIFFIVKTHRRKAARTAVGRNDTHPTTGSASPKHQKKSKLHGPTETSSCSGAAPTVEMDEELHY ASLNFHGMNPSKDTSTEYSEVRTQ (SEQ ID NO: 1) CD33-M; transcript variant 1 mRNA. NM_001772 (underlined=exon 2; italicized= exon 7A) AUGCCGCUGCUGCUACUGCUGCCCCUGCUGUGGGCAGGGGCCCUGGCUAUGGAUCCAAAUUUCUGGCU GCAAGUGCAGGAGUCAGUGACGGUACAGGAGGGUUUGUGCGUCCUCGUGCCCUGCACUUUCUUCCAUC CCAUACCCUACUACGACAAGAACUCCCCAGUUCAUGGUUACUGGUUCCGGGAAGGAGCCAUUAUAUCC AGGGACUCUCCAGUGGCCACAAACAAGCUAGAUCAAGAAGUACAGGAGGAGACUCAGGGCAGAUUCCG CCUCCUUGGGGAUCCCAGUAGGAACAACUGCUCCCUGAGCAUCGUAGACGCCAGGAGGAGGGAUAAUG GUUCAUACUUCUUUCGGAUGGAGAGAGGAAGUACCAAAUACAGUUACAAAUCUCCCCAGCUCUCUGUG CAUGUGACAGACUUGACCCACAGGCCCAAAAUCCUCAUCCCUGGCACUCUAGAACCCGGCCACUCCAA AAACCUGACCUGCUCUGUGUCCUGGGCCUGUGAGCAGGGAACACCCCCGAUCUUCUCCUGGUUGUCAG CUGCCCCCACCUCCCUGGGCCCCAGGACUACUCACUCCUCGGUGCUCAUAAUCACCCCACGGCCCCAG GACCACGGCACCAACCUGACCUGUCAGGUGAAGUUCGCUGGAGCUGGUGUGACUACGGAGAGAACCAU CCAGCUCAACGUCACCUAUGUUCCACAGAACCCAACAACUGGUAUCUUUCCAGGAGAUGGCUCAGGGA AACAAGAGACCAGAGCAGGAGUGGUUCAUGGGGCCAUUGGAGGAGCUGGUGUUACAGCCCUGCUCGCU CUUUGUCUCUGCCUCAUCUUCUUCAUAGUGAAGACCCACAGGAGGAAAGCAGCCAGGACAGCAGUGGG CAGGAAUGACACCCACCCUACCACAGGGUCAGCCUCCCCGAAACACCAGAAGAAGUCCAAGUUACAUG GCCCCACUGAAACCUCAAGCUGUUCAGGUGCCGCCCCUACUGUGGAGAUGGAUGAGGAGCUGCAUUAU GCUUCCCUCAACUUUCAUGGGAUGAAUCCUUCCAAGGACACCUCCACCGAAUACUCAGAGGUCAGGAC CCAGUGA (SEQ ID NO: 11) CD33m-7A: Amino Acid (italicized= exon 7A) MPLLLLLPLLWADLTHRPKILIPGTLEPGHSKNLTCSVSWACEQGTPPIFSWLSAAPTSLGPRTTHSS VLIITPRPQDHGTNLTCQVKFAGAGVTTERTIQLNVTYVPQNPTTGIFPGDGSGKQETRAGVVHGAIG GAGVTALLALCLCLIFFIVKTHRRKAARTAVGRNDTHPTTGSASPKHQKKSKLHGPTETSSCSGAAPT VEMDEELHYASLNFHGMNPSKDTSTEYSEVRTQ(SEQ ID NO: 12) CD33-m transcript variant 2; no exon 2, exon 7A mRNA. NM_001082618 (italicized= exon 7A)) AUGCCGCUGCUGCUACUGCUGCCCCUGCUGUGGGCAGACUUGACCCACAGGCCCAAAAUCCUCAUCCC UGGCACUCUAGAACCCGGCCACUCCAAAAACCUGACCUGCUCUGUGUCCUGGGCCUGUGAGCAGGGAA CACCCCCGAUCUUCUCCUGGUUGUCAGCUGCCCCCACCUCCCUGGGCCCCAGGACUACUCACUCCUCG GUGCUCAUAAUCACCCCACGGCCCCAGGACCACGGCACCAACCUGACCUGUCAGGUGAAGUUCGCUGG AGCUGGUGUGACUACGGAGAGAACCAUCCAGCUCAACGUCACCUAUGUUCCACAGAACCCAACAACUG GUAUCUUUCCAGGAGAUGGCUCAGGGAAACAAGAGACCAGAGCAGGAGUGGUUCAUGGGGCCAUUGGA GGAGCUGGUGUUACAGCCCUGCUCGCUCUUUGUCUCUGCCUCAUCUUCUUCAUAGUGAAGACCCACAG GAGGAAAGCAGCCAGGACAGCAGUGGGCAGGAAUGACACCCACCCUACCACAGGGUCAGCCUCCCCGA AACACCAGAAGAAGUCCAAGUUACAUGGCCCCACUGAAACCUCAAGCUGUUCAGGUGCCGCCCCUACU GUGGAGAUGGAUGAGGAGCUGCAUUAUGCUUCCCUCAACUUUCAUGGGAUGAAUCCUUCCAAGGACAC CUCCACCGAAUACUCAGAGGUCAGGACCCAGUGA (SEQ ID NO: 13) CD33M-7B: Amino Acid (underlined=exon 2; italicized= exon 7B) MPLLLLLPLLWAGALAMDPNFWLQVQESVTVQEGLCVLVPCTFFHPIPYYDKNSPVHGYWFREGAIIS RDSPVATNKLDQEVQEETQGRFRLLGDPSRNNCSLSIVDARRRDNGSYFFRMERGSTKYSYKSPQLSV HVTDLTHRPKILIPGTLEPGHSKNLTCSVSWACEQGTPPIFSWLSAAPTSLGPRTTHSSVLIITPRPQ DHGTNLTCQVKFAGAGVTTERTIQLNVTYVPQNPTTGIFPGDGSGKQETRAGVVHGAIGGAGVTALLA LCLCLIFFIVKTHRRKAARTAVGRNDTHPTTGSASPVR (SEQ ID NO: 14) CD33 transcript variant 3; exon 7B mRNA. NM_001177608 (underlined= exon 2; italicized= exon 7B)) AUGCCGCUGCUGCUACUGCUGCCCCUGCUGUGGGCAGGGGCCCUGGCUAUGGAUCCAAAUUUCUGGCUGCAAGUG CAGGAGUCAGUGACGGUACAGGAGGGUUUGUGCGUCCUCGUGCCCUGCACUUUCUUCCAUCCCAUACCCUACUAC GACAAGAACUCCCCAGUUCAUGGUUACUGGUUCCGGGAAGGAGCCAUUAUAUCCAGGGACUCUCCAGUGGCCACA AACAAGCUAGAUCAAGAAGUACAGGAGGAGACUCAGGGCAGAUUCCGCCUCCUUGGGGAUCCCAGUAGGAACAAC UGCUCCCUGAGCAUCGUAGACGCCAGGAGGAGGGAUAAUGGUUCAUACUUCUUUCGGAUGGAGAGAGGAAGUACC AAAUACAGUUACAAAUCUCCCCAGCUCUCUGUGCAUGUGACAGACUUGACCCACAGGCCCAAAAUCCUCAUCCCU GGCACUCUAGAACCCGGCCACUCCAAAAACCUGACCUGCUCUGUGUCCUGGCCUGUGAGCAGGGAACACCCCCGA UCUUCUCCGGUUGUCAGCUGCCCCCACCUCCCUGGGCCCCAGGACUACUCACUCCUCGGUGCUCAUAAUCACCCC ACGGCCCCAGGACCACGGCACCAACCUGACCUGUCAGGUGAAGUUCGCUGGAGCUGGUGUGACUACGGAGAGAAC CAUCCAGCUCAACGUCACCUAUGUUCCACAGAACCCAACAACUGGUAUCUUUCCGGAGAUGGCUCAGGGAAACAA GAGACCAGAGCAGGAGUGGUUCAUGGGGCCAUUGGAGGAGCUGGUGUUACAGCCCUGCUCGCUCUUUGUCUCUGC CUCAUCUUCUUCAUAGUGAAGACCCACAGGAGGAAAGCAGCCAGGACAGCAGUGGGCAGGAAUGACACCCACCCU ACCACAGGGUCAGCCUCCCCGGUACGUUGA (SEQ ID NO: 15) CD33m-7B Amino Acid (italicized= exon 7B) MPLLLLLPLLWADLTHRPKILIPGTLEPGHSKNLTCSVSWACEQGTPPIFSWLSAAPTSLGPRTTHSS VLIITPRPQDHGTNLTCQVKFAGAGVTTERTIQLNVTYVPQNPTTGIFPGDGSGKQETRAGVVHGAIG GAGVTALLALCLCLIFFIVKTHRRKAARTAVGRNDTHPTTGSASPVR (SEQ ID NO: 16) CD33 transcript variant 4; no exon 2, exon 7B. mRNA (italicized= exon 7B) AUGCCGCUGCUGCUACUGCUGCCCCUGCUGUGGGCAAGGAACAACUGCUCCCUGAGCAUCGUAGACGCCAGGAGG AGGGAUAAUGGUUCAUACUUCUUUCGGAUGGAGAGAGGAAGUACCAAAUACAGUUACAAAUCUCCCCAGCUCUCU GUGCAUGUGACAGACUUGACCCACAGGCCCAAAAUCCUCAUCCCUGGCACUCUAGAACCCGGCCACUCCAAAAAC CUGACCUGCUCUGUGUCCUGGCCUGUGAGCAGGGAACACCCCCGAUCUUCUCCGGUUGUCAGCUGCCCCCACCUC CCUGGGCCCCAGGACUACUCACUCCUCGGUGCUCAUAAUCACCCCACGGCCCCAGGACCACGGCACCAACCUGAC CUGUCAGGUGAAGUUCGCUGGAGCUGGUGUGACUACGGAGAGAACCAUCCAGCUCAACGUCACCUAUGUUCCACA GAACCCAACAACUGGUAUCUUUCCGGAGAUGGCUCAGGGAAACAAGAGACCAGAGCAGGAGUGGUUCAUGGGGCC AUUGGAGGAGCUGGUGUUACAGCCCUGCUCGCUCUUUGUCUCUGCCUCAUCUUCUUCAUAGUGAAGACCCACAGG AGGAAAGCAGCCAGGACAGCAGUGGGCAGGAAUGACACCCACCCUACCACAGGGUCAGCCUCCCCGGUACGUUGA (SEQ ID NO: 17) Using human CD33 as an exemplary lineage-specific cell surface protein, regions of the protein in which mutation and/or deletion of amino acids are less likely to result in deleterious effects (e.g., a reduction or abrogation of function) were predicted using PROVEAN software (see: provean.jcvi.org; Choi et al. PLoS ONE (2012) 7(10): e46688). Examples of the predicted regions are shown in boxes in FIGURE 2 and exemplary deletions in the predicated regions are presented in Table 2. Numbering of the amino acid residues is based on the amino acid sequence of human CD33 provided by SEQ ID NO: 1 (CD33M-7A). Table 2: Exemplary deletions in CD33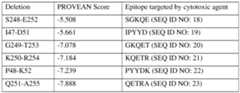 The nucleotide sequence encoding CD33 are genetically manipulated to delete any epitope of the protein (of the extracellular portion of CD33), or a fragment containing such, using conventional methods of nucleic acid manipulation. The amino acid sequences provided below are exemplary sequences of CD33 mutants that have been manipulated to lack each of the epitopes in Table 2. The amino acid sequence of the extracellular portion of CD33 is provided by SEQ ID NO: 1. The signal peptide is shown in italics and sites for manipulation are shown in underline and boldface. The transmembrane domain is shown in italics with underline. MPLLLLLPLL WAGALAMDPN FWLQVQESVT VQEGLCVLVP CTFFHPIPYY DKNSPVHGYW FREGAIISRD SPVATNKLDQ EVQEETQGRF RLLGDPSRNN CSLSIVDARR RDNGSYFFRM ERGSTKYSYK SPQLSVHVTD LTHRPKILIP GTLEPGHSKN LTCSVSWACE QGTPPIFSWL SAAPTSLGPR TTHSSVLIIT PRPQDHGTNL TCQVKFAGAG VTTERTIQLN VTYVPQNPTT GIFPGDGSGK QETRAGVVHG AIGGAGVTAL LALCLCLIFF IVKTHRRKAA RTAVGRNDTH PTTGSASPKH QKKSKLHGPT ETSSCSGAAP TVEMDEELHY ASLNFHGMNP SKDTSTEYSE VRTQ (SEQ ID NO: 1) The amino acid sequence of the extracellular portion of CD33 comprising a deletion of residues S248 through E252 is provided by SEQ ID NO: 2. The signal peptide is shown in italics and the transmembrane domain is shown in italics with underline. S248_E252insdelTARND; PROVEAN score = -1.916 MPLLLLLPLL WAGALAMDPN FWLQVQESVT VQEGLCVLVP CTFFHPIPYY DKNSPVHGYW FREGAIISRD SPVATNKLDQ EVQEETQGRF RLLGDPSRNN CSLSIVDARR RDNGSYFFRM ERGSTKYSYK SPQLSVHVTD LTHRPKILIP GTLEPGHSKN LTCSVSWACE QGTPPIFSWL SAAPTSLGPR TTHSSVLIIT PRPQDHGTNL TCQVKFAGAG VTTERTIQLN VTYVPQNPTT GIFPGDGTAR NDTRAGVVHG AIGGAGVTAL LALCLCLIFF IVKTHRRKAA RTAVGRNDTH PTTGSASPKH QKKSKLHGPT ETSSCSGAAP TVEMDEELHY ASLNFHGMNP SKDTSTEYSE VRTQ (SEQ ID NO: 2) The amino acid sequence of the extracellular portion of CD33 comprising a deletion of residues I47 through D51 is provided by SEQ ID NO: 3. The signal peptide is shown in italics and the transmembrane domain is shown in italics with underline. I47_D51insdelVPFFE; PROVEAN score = -1.672 MPLLLLLPLL WAGALAMDPN FWLQVQESVT VQEGLCVLVP CTFFHPVPFF EKNSPVHGYW FREGAIISRD SPVATNKLDQ EVQEETQGRF RLLGDPSRNN CSLSIVDARR RDNGSYFFRM ERGSTKYSYK SPQLSVHVTD LTHRPKILIP GTLEPGHSKN LTCSVSWACE QGTPPIFSWL SAAPTSLGPR TTHSSVLIIT PRPQDHGTNL TCQVKFAGAG VTTERTIQLN VTYVPQNPTT GIFPGDGTAR NDTRAGVVHG AIGGAGVTAL LALCLCLIFF IVKTHRRKAA RTAVGRNDTH PTTGSASPKH QKKSKLHGPT ETSSCSGAAP TVEMDEELHY ASLNFHGMNP SKDTSTEYSE VRTQ (SEQ ID NO: 3) The amino acid sequence of the extracellular portion of CD33 comprising a deletion of residues G249 through T253 is provided by SEQ ID NO: 4. The signal peptide is shown in italics and the transmembrane domain is shown in italics with underline. MPLLLLLPLL WAGALAMDPN FWLQVQESVT VQEGLCVLVP CTFFHPIPYY DKNSPVHGYW FREGAIISRD SPVATNKLDQ EVQEETQGRF RLLGDPSRNN CSLSIVDARR RDNGSYFFRM ERGSTKYSYK SPQLSVHVTD LTHRPKILIP GTLEPGHSKN LTCSVSWACE QGTPPIFSWL SAAPTSLGPR TTHSSVLIIT PRPQDHGTNL TCQVKFAGAG VTTERTIQLN VTYVPQNPTT GIFPGDGSRA GVVHGAIGGA GVTALLALCL CLIFFIVKTH RRKAARTAVG RNDTHPTTGS ASPKHQKKSK LHGPTETSSC SGAAPTVEMD EELHYASLNF HGMNPSKDTS TEYSEVRTQ (SEQ ID NO: 4) The amino acid sequence of the extracellular portion of CD33 comprising a deletion of residues K250 through R254 is provided by SEQ ID NO: 5. The signal peptide is shown in italics and the transmembrane domain is shown in italics with underline. MPLLLLLPLL WAGALAMDPN FWLQVQESVT VQEGLCVLVP CTFFHPIPYY DKNSPVHGYW FREGAIISRD SPVATNKLDQ EVQEETQGRF RLLGDPSRNN CSLSIVDARR RDNGSYFFRM ERGSTKYSYK SPQLSVHVTD LTHRPKILIP GTLEPGHSKN LTCSVSWACE QGTPPIFSWL SAAPTSLGPR TTHSSVLIIT PRPQDHGTNL TCQVKFAGAG VTTERTIQLN VTYVPQNPTT GIFPGDGSGA GVVHGAIGGA GVTALLALCL CLIFFIVKTH RRKAARTAVG RNDTHPTTGS ASPKHQKKSK LHGPTETSSC SGAAPTVEMD EELHYASLNF HGMNPSKDTS TEYSEVRTQ (SEQ ID NO: 5) The amino acid sequence of the extracellular portion of CD33 comprising a deletion of residues P48 through K52 is provided by SEQ ID NO: 6. The signal peptide is shown in italics and the transmembrane domain is shown in italics with underline. MPLLLLLPLL WAGALAMDPN FWLQVQESVT VQEGLCVLVP CTFFHPINSP VHGYWFREGA IISRDSPVAT NKLDQEVQEE TQGRFRLLGD PSRNNCSLSI VDARRRDNGS YFFRMERGST KYSYKSPQLS VHVTDLTHRP KILIPGTLEP GHSKNLTCSV SWACEQGTPP IFSWLSAAPT SLGPRTTHSS VLIITPRPQD HGTNLTCQVK FAGAGVTTER TIQLNVTYVP QNPTTGIFPG DGSGKQETRA GVVHGAIGGA GVTALLALCL CLIFFIVKTH RRKAARTAVG RNDTHPTTGS ASPKHQKKSK LHGPTETSSC SGAAPTVEMD EELHYASLNF HGMNPSKDTS TEYSEVRTQ (SEQ ID NO: 6) The amino acid sequence of the extracellular portion of CD33 comprising a deletion of residues Q251 through A255 is provided by SEQ ID NO: 7. The signal peptide is shown in italics and the transmembrane domain is shown in italics with underline. MPLLLLLPLL WAGALAMDPN FWLQVQESVT VQEGLCVLVP CTFFHPIPYY DKNSPVHGYW FREGAIISRD SPVATNKLDQ EVQEETQGRF RLLGDPSRNN CSLSIVDARR RDNGSYFFRM ERGSTKYSYK SPQLSVHVTD LTHRPKILIP GTLEPGHSKN LTCSVSWACE QGTPPIFSWL SAAPTSLGPR TTHSSVLIIT PRPQDHGTNL TCQVKFAGAG VTTERTIQLN VTYVPQNPTT GIFPGDGSGK GVVHGAIGGA GVTALLALCL CLIFFIVKTH RRKAARTAVG RNDTHPTTGS ASPKHQKKSK LHGPTETSSC SGAAPTVEMD EELHYASLNF HGMNPSKDTS TEYSEVRTQ (SEQ ID NO: 7) In some examples, provided herein are variants of CD33, which may comprise a deletion or mutation of a fragment of the protein that is encoded by any one of the exons of CD33, or a deletion or mutation in a non-essential epitope. The predicted structure of CD33 includes two immunoglobulin domains, an IgV domain and an IgC2 domain. In some embodiments, a portion of the immunoglobulin V domain of CD33 is deleted or mutated. In some embodiments, a portion of the immunoglobulin C domain of CD33 is deleted or mutated. In some embodiments, exon 2 of CD33 is deleted or mutated. In some embodiments, exon 3 of CD33 is deleted or mutated. In some embodiments, the CD33 variant lacks amino acid residues W11 to T139. In some embodiments, the CD33 variant lacks amino acid residues G13 to T139. In some embodiments, the deleted or mutated fragment overlaps or encompasses the epitope to which a cytotoxic agent binds, such as a cytotoxic agent described in PCT Publication No. WO2022/056497, WO2022/056490, or WO2019/178382, which are incorporated by reference in their entirety. In some embodiments, the epitope comprises amino acids 47-51 or 248-252 of CD33. In some embodiments, the epitope comprises amino acids 248-252, 47-51, 249-253, 250-254, 48-52, or 251-255 of the extracellular portion of CD33. In some embodiments, the genetically engineered hematopoietic stem cells have genetic edits in a CD33 gene, wherein exon 2 of CD33 is mutated or deleted. In some embodiments, exon 2 of CD33 is mutated or deleted (see the amino acid sequence of the exon 2-encoded fragment above). In some embodiments, the genetically engineered hematopoietic stem cells have genetic edits in a CD33 gene resulting in expression of CD33 with deleted or mutated exon 2 of CD33. In some embodiments, genetically engineered hematopoietic stem cells express CD33, in which exon 2 of CD33 is mutated or deleted. In some examples, the genetically engineered hematopoietic cells have a genetically engineered CD33 gene (e.g., a genetically engineered endogenous CD33 gene), wherein the engineering results in expression of a CD33 variant having the fragment encoded by exon 2 deleted (CD33ex2). In some embodiments, the CD33 genes edited HSCs expressing CD33 variant having the fragment encoded by exon 2 deleted also comprise a partial or complete deletion in the adjacent introns (intron 1 and intron 2) in addition to the deletion of exon 2. Exemplary amino acid sequence of CD33 mutants, with fragments G13-T139 deleted, is provided below (the junction of exon 1-encoded fragment and exon-3 encoded fragment is shown in boldface): MPLLLLLPLLWADLTHRPKILIPGTLEPGHSKNLTCSVSWACEQGTPPIFSWLSAAPTSLGPRTTHSS VLIITPRPQDHGTNLTCQVKFAGAGVTTERTIQLNVTYVPQNPTTGIFPGDGSGKQETRAGVVHGAIG GAGVTALLALCLCLIFFIVKTHRRKAARTAVGRNDTHPTTGSASPKHQKKSKLHGPTETSSCSGAAPT VEMDEELHYASLNFHGMNPSKDTSTEYSEVRTQ (SEQ ID NO: 12) MPLLLLLPLLWADLTHRPKILIPGTLEPGHSKNLTCSVSWACEQGTPPIFSWLSAAPTSLGPRTTHSS VLIITPRPQDHGTNLTCQVKFAGAGVTTERTIQLNVTYVPQNPTTGIFPGDGSGKQETRAGVVHGAIG GAGVTALLALCLCLIFFIVKTHRRKAARTAVGRNDTHPTTGSASPVR (SEQ ID NO: 16) While certain cells may express CD33 proteins lacking the fragment encoded by exon 2, the genetically engineered hematopoietic cells are different from such native cells in at least the aspect that these cells have undergone genome editing to modify a CD33 gene, such as an endogenous CD33 gene. In other words, the parent hematopoietic stem cells for producing the genetically engineered HSCs carry a CD33 gene that produces exon 2- containing transcripts. Genetically engineered hematopoietic stem cells carrying an edited CD33 gene that expresses this CD33 mutant are also within the scope of the present disclosure. Such cells may be a homogenous population containing cells expressing the same CD33 mutant (e.g., CD33ex2). Alternatively, the cells may be a heterogeneous population containing cells expressing different CD33 mutants (which may be due to heterogeneous editing/repairing events inside cells) or cells that do not express CD33 (CD33KO). In specific examples, the genetically engineered HSCs may be a heterogeneous population containing cells expressing CD33ex2 and cells that do not express CD33 (CD33KO). Genetically engineered hematopoietic stem cells having edited a CD33 gene can be prepared by a suitable genome editing method, such as those known in the art or disclosed herein. In some embodiments, the genetically engineered hematopoietic stem cells described herein can be generated using the CRISPR approach. See discussions herein. In certain examples, specific guide RNAs targeting a fragment of the CD33 gene (an exon sequence or an intron sequence) can be used in the CRISPR method. A CD33 pseudogene, known as SIGLEC22P (Gene ID 114195), is located upstream of the CD33 gene and shares a certain degree of sequence homology with the CD33 gene. gRNAs that cross-target regions in the pseudogene and regions in the CD33 gene may lead to production of aberrant gene products. Thus, in some embodiments, the gRNAs used in methods of editing CD33 via CRISPR preferably have low or no cross-reactivity with regions inside the pseudogene. In some instances, the gRNAs used in methods of editing CD33 via CRISPR preferably have low or no cross-reactivity with region(s) in Exon 1, intron 1 or Exon 2 of CD33 that are homologous to the pseudogene. Such gRNAs can be designed by comparing the sequences of the pseudogene and the CD33 gene to choose targeting sites inside the CD33 gene that have less or no homology to regions of the pseudogene. In one example, the pair of gRNA 18 (TTCATGGGTACTGCAGGGCA)(SEQ ID NO: 24) and gRNA 24 (GTGAGTGGCTGTGGGGAGAG)(SEQ ID NO: 25) are used for editing CD33 via CRISPR. Also provided herein are methods of genetically editing CD33 in hematopoietic cells (e.g., HSCs) via CRISPR, using one or more of the gRNAs described herein, for example, the pair of gRNA18 + gRNA24. As described herein, the length of a gRNA sequence may be modified (increased or decreased), for example, to enhance editing specificity and/or efficiency. In some embodiments, the length of gRNA 24 is 20 base pairs. In some embodiments, reducing the length of gRNA 24 may reduce the editing efficiency of gRNA 24 for CD33. In some embodiments, at least 40%, at least 50%, at least 60%, at least 70%, at least 75%, at least 80%, at least 85%, at least 90%, or at least 95% of copies of CD33 in the population of cells have a mutation. By way of example, a population can comprise a plurality of different CD33 mutations and each mutation of the plurality contributes to the percent of copies of CD33 in the population of cells that have a mutation. In some embodiments, the expression of CD33 on the genetically engineered hematopoietic cell is compared to the expression of CD33 on a naturally occurring hematopoietic cell (e.g., a wild-type counterpart). In some embodiments, the genetic engineering results in a reduction in the expression level of CD33 by at least50%, at least 60%, at least 70%, at least 80%, at least 90%, at least 91%, at least 92%, at least 93%, at least 94%, at least 95%, at least 96%, at least 97%, at least 98%, or at least 99% as compared to the expression of CD33 on a naturally occurring hematopoietic cell (e.g., a wild-type counterpart). For example, in some embodiments, the genetically engineered hematopoietic cell expresses less than 20%, less than 19%, less than 18%, less than 17%, less than 16%, less than 15%, less than 14%, less than 13%, less than 12%, less than 11%, less than 10%, less than 9%, less than 8%, less than 7%, less than 6%, less than 5%, less than 4%, less than 3%, less than 2%, less than or 1% of CD33 as compared to a naturally occurring hematopoietic cell (e.g., a wild-type counterpart). In some embodiments, the genetic engineering results in a reduction in the expression level of wild-type CD33 by at least 50%, at least 60%, at least 70%, at least 80%, at least 90%, at least 91%, at least 92%, at least 93%, at least 94%, at least 95%, at least 96%, at least 97%, at least 98%, or at least 99% as compared to the expression of the level of wild-type CD33 on a naturally occurring hematopoietic cell (e.g., a wild-type counterpart). That is, in some embodiments, the genetically engineered hematopoietic cell expresses less than 20%, less than 19%, less than 18%, less than 17%, less than 16%, less than 15%, less than 14%, less than 13%, less than 12%, less than 11%, less than 10%, less than 9%, less than 8%, less than 7%, less than 6%, less than 5%, less than 4%, less than 3%, less than 2%, or less than 1% of CD33 as compared to a naturally occurring hematopoietic cell (e.g., a wild-type counterpart). Genetically Engineered Hematopoietic Cells Expressing both CD33 and IL1RAP Mutants Also provided herein are a genetically engineered hematopoietic cells, such as HSCs, that have both the IL1RAP and CD33 genes edited. In some embodiments, provided herein is a population of genetically engineered hematopoietic HSCs in which at least 50% of the cells carry genetically edited IL1RAP and CD33 cells in at least on chromosome. In some embodiments, the edited IL1RAP gene is capable of expressing an IL1RAP mutant having at least one fragment encoded by one or more exons deleted. Alternatively or in addition, the edited CD33 gene is capable of expressing a CD33 mutant having at least one fragment encoded by one or more exons deleted. The genetically engineered hematopoietic cells having both IL1RAP and CD33 genes edited can be prepared by conventional methods. In some embodiments, such cells are prepared by CRISPR using a pair of gRNAs, one targeting IL1RAP and the other targeting CD33. In one example, the pair of gRNAs can be introduced into parent HSCs simultaneously and cells having genetic edits in IL1RAP and/or CD33 can be harvested for further use. Genetically Engineered Hematopoietic Cells Expressing both CD44 and EMR2 Mutants Also provided herein are a genetically engineered hematopoietic cells, such as HSCs, that have both the EMR2 and CD44 genes edited. In some embodiments, provide herein is a population of genetically engineered hematopoietic HSCs in which at least 50% of the cells carry genetically edited EMR2 and CD44 cells in at least one chromosome. In some embodiments, the edited EMR2 gene is capable of expressing a EMR2 mutant having at least one fragment encoded by a one or more exons deleted. Alternatively or in addition, the edited CD44 gene is capable of expressing a CD44 mutant having at least one fragment encoded by one or more exons deleted. The genetically engineered hematopoietic cells having both EMR2 and CD44 genes edited can be prepared by conventional methods. In some embodiments, such cells are prepared by CRISPR using a pair of gRNAs, one targeting EMR2 and the other targeting CD44. In one example, the pair of gRNAs can be introduced into parent HSCs simultaneously and cells having genetic edits in EMR2 and/or CD44 can be harvested for further use. Genetically Engineered Hematopoietic Cells Expressing CD305, CD49D, CD205, and CD244 Mutants Also provided herein are a genetically engineered hematopoietic cells, such as HSCs, that have genetic editing in a first gene encoding a first cell surface protein and a second gene encoding a second cell surface protein, wherein at least one of the cell surface proteins is selected from the group consisting of: CD305 (LAIR1); CD49D (ITGA4); CD205 (LY75); and CD244 (SLAMF4). In some embodiments, one of the cell surface proteins is selected from the group consisting of: CD305 (LAIR1); CD49D (ITGA4); CD205 (LY75); and CD244 (SLAMF4), and one of the cell surface proteins is selected from the group consisting of: CD10; CD101; CD117; CD11A; CD11C; CD120B; CD123; CD13; CD133; CD152; CD200; CD205 (LY75); CD226; CD244 (SLAMF4); CD25; CD274; CD305 (LAIR1); CD32; CD33; CD34; CD366; CD38; CD41; CD42B; CD44; CD45RA; CD47; CD48; CD49D (ITGA4); CD49F; CD52; CD58; CD64; CD7; CD70; CD82; CD84; CD85; CD85K; CD86; CD9; CD96; CD99; CLL-1; EMR2 (ADGRE2); FR-B; GPR56; and IL1RAP. In some embodiments, the first cell surface protein and the second cell surface protein is a combination selected from the group consisting of: CD33 and CD205 (LY75); CD33 and CD244 (SLAMF4); CD33 and CD305 (LAIR1); CD33 and CD49D (ITGA4); CLL-1 and CD205 (LY75); CLL-1 and CD244 (SLAMF4); CLL-1 and CD305 (LAIR1); CLL-1 and CD49D (ITGA4); EMR2 and CD205 (LY75); EMR2 and CD244 (SLAMF4); EMR2 and CD305 (LAIR1); EMR2 and CD49D (ITGA4); CD38 and CD205 (LY75); CD38 and CD244 (SLAMF4); CD38 and CD305 (LAIR1); CD38 and CD49D (ITGA4); CD305 (LAIR1) and CD49D (ITGA4); CD305 (LAIR1) and CD205 (LY75); CD305 (LAIR1) and CD244 (SLAMF4); CD49D (ITGA4) and CD205 (LY75); CD49D (ITGA4) and CD244 (SLAMF4); and CD205 (LY75) and CD244 (SLAMF4) In some embodiments, a population of genetically engineered hematopoietic HSCs in which at least 50% of the cells carry the genetic editing of the first gene in at least one chromosome and genetic editing of the second gene in at least one chromosome. In some embodiments, the edited first gene is capable of expressing a mutant of the first cell surface protein having at least one fragment encoded by a one or more exons deleted. Alternatively or in addition, the edited second gene is capable of expressing a mutant of the second cell surface protein having at least one fragment encoded by one or more exons deleted. For example, in some embodiments, a mutant of the first cell surface protein and/or a mutant of the second cell surface protein comprises a substitution, deletion, or insertion of an amino acid at one or more amino acid positions (e.g., 1-10, 10-20, 20-30, 30-40, 40-50, or more than 50 amino acid positions). In some embodiments, the mutated amino acid positions are located in an epitope (e.g., wherein the epitope is deleted in the mutant cell surface protein). In some embodiments, a mutant of the first cell surface protein and/or a mutant of the second cell surface protein comprises a deletion of one or more domains. In some embodiments, the first gene and/or the second gene is deleted. The genetically engineered hematopoietic cells having both first and second genes edited can be prepared by conventional methods. In some embodiments, such cells are prepared by CRISPR using a pair of gRNAs, one targeting the first gene and the other targeting the second gene. In one example, the pair of gRNAs can be introduced into parent HSCs simultaneously and cells having genetic edits in the first gene and/or the second gene can be harvested for further use. Masked lineage-specific cell surface antigens While many of the embodiments described herein involve mutations to the endogenous genes encoding lineage-specific cell surface antigens, it is understood that other approaches may be used instead of or in addition to mutation. For instance, a cell surface protein can be masked, e.g., to prevent or reduce its recognition by an immunotherapeutic agent. In some embodiments, masking is used on a cell surface protein that is difficult to mutate, e.g., because mutation of the gene is inefficient or is deleterious to cells expressing the mutant. In some embodiments, the lineage-specific cell surface protein is CD45. In some embodiments, masking is performed on a cell type described herein, e.g., an HSC or HPC. In some embodiments, masking is accomplished by expressing a masking protein in the cell of interest (e.g., by stably expressing DNA encoding the masking protein in the cell). In some embodiments, the masking protein comprises a binding domain (e.g., an antibody or antigen-binding fragment, e.g., an scFv) that binds the lineage-specific cell surface protein, e.g., in a way that reduces binding of an immunotherapeutic agent to the lineage-specific cell surface protein, e.g., by competing for binding at the same epitope. In some embodiments, the binding domain binds CD45. In some embodiments, the making protein further comprises one or more sequences that direct its localization to the surface of the cell. In some embodiments, the masking protein comprises a transmembrane domain fused to the binding domain. The masking protein may comprise a linker disposed between the transmembrane domain and the binding domain. The masking protein may be expressed at a level that binds to a sufficient amount of the cell surface protein that an immunotherapeutic agent displays reduced binding to and/or reduced killing of a cell expressing the masking protein compared to an otherwise similar cell that does not express the masking protein. In some embodiments, a cell described herein has reduced binding to (and/or reduced killing by) two different immunotherapeutic agents that recognize two different lineage- specific cell surface antigens. For instance, the cell may have a mutation at a gene encoding a first lineage-specific cell surface antigen, and may comprise a masking protein that masks a second lineage-specific cell surface antigen. In some embodiments, the cell may comprise a first masking protein that masks a first cell surface protein and a second masking protein that masks a second lineage-specific cell surface antigen. Cells Altered at One or More Lineage-Specific Cell Surface Antigens While many of the embodiments described herein involve two or more lineage- specific cell surface antigens, the application also discloses various cells altered with respect to a single lineage-specific cell surface antigen. For instance, the disclosure describes cells mutated at any one of the lineage-specific cell surface antigens described herein (e.g., mutated at one or both alleles of the lineage-specific cell surface antigen). The disclosure also describes cells expressing a single masking protein for a single lineage-specific cell surface antigen. Hematopoietic Stem Cells In some embodiments, the hematopoietic cells described herein are hematopoietic stem cells. Hematopoietic stem cells (HSCs) are capable of giving rise to both myeloid and lymphoid progenitor cells that further give rise to myeloid cells (e.g., monocytes, macrophages, neutrophils, basophils, dendritic cells, erythrocytes, platelets, etc) and lymphoid cells (e.g., T cells, B cells, NK cells), respectively. HSCs are characterized by the expression of the cell surface marker CD34 (e.g., CD34+), which can be used for the identification and/or isolation of HSCs, and absence of cell surface markers associated with commitment to a cell lineage. In some embodiments, the HSCs are obtained from a subject, such as a mammalian subject. In some embodiments, the mammalian subject is a non-human primate, a rodent (e.g., mouse or rat), a bovine, a porcine, an equine, or a domestic animal. In some embodiments, the HSCs are obtained from a human patient, such as a human patient having a hematopoietic malignancy. In some embodiments, the HSCs are obtained from a healthy donor. In some embodiments, the HSCs are obtained from the subject to whom the immune cells expressing the chimeric receptors will be subsequently administered. HSCs that are administered to the same subject from which the cells were obtained are referred to as autologous cells, whereas HSCs that are obtained from a subject who is not the subject to whom the cells will be administered are referred to as allogeneic cells. In some embodiments, the HSCs that are administered to the subject are allogeneic cells. In some embodiments, the HSCs are obtained from a donor having a HLA haplotype that is matched with the HLA haplotype of the subject. Human Leukocyte Antigen (HLA) encodes major histocompatibility complex (MHC) proteins in humans. MHC molecules are present on the surface of antigen-presenting cells as well as many other cell types and present peptides of self and non-self (e.g., foreign) antigens for immunosurveillance. However, HLA are highly polymorphic, which results in many distinct alleles. Different (foreign, non-self) alleles may be antigenic and stimulate robust adverse immune responses, particularly in organ and cell transplantation. HLA molecules that are recognized as foreign (non-self) can result in transplant rejection. In some embodiments, it is desirable to administer HSCs from donor that has the same HLA type as the patient to reduce the incidence of rejection. The HLA loci of a donor subject may be typed to identify an individual as a HLA- matched donor for the subject. Methods for typing the HLA loci will be evident to one of ordinary skill in the art and include, for example, serology (serotyping), cellular typing, gene sequencing, phenotyping, and PCR methods. An HLA from a donor is considered “matched” with the HLA of the subject if the HLA loci of the donor and the subject are identical or sufficiently similar such that an adverse immune response is not expected. In some embodiments, the HLA from the donor is not matched with the HLA of the subject. In some embodiments, the subject is administered HSCs that are not HLA matched with the HLA of the subject. In some embodiments, the subject is further administered one or more immunosuppressive agents to reduce or prevent rejection of the donor HSC cells. In some embodiments, the HSCs do not comprise a CART. HSCs may be obtained from any suitable source using convention means known in the art. In some embodiments, HSCs are obtained from a sample from a subject (or donor), such as bone marrow sample or from a blood sample. Alternatively or in addition, HSCs may be obtained from an umbilical cord. In some embodiments, the HSCs are from bone marrow, cord blood cells, or peripheral blood mononuclear cells (PBMCs). In general, bone marrow cells may be obtained from iliac crest, femora, tibiae, spine, rib or other medullary spaces of a subject (or donor). Bone marrow may be taken out of the patient and isolated through various separations and washing procedures known in the art. An exemplary procedure for isolation of bone marrow cells comprises the following steps: a) extraction of a bone marrow sample; b) centrifugal separation of bone marrow suspension in three fractions and collecting the intermediate fraction, or buffycoat; c) the buffycoat fraction from step (b) is centrifuged one more time in a separation fluid, commonly FicollTM, and an intermediate fraction which contains the bone marrow cells is collected; and d) washing of the collected fraction from step (c) for recovery of re-transfusable bone marrow cells. HSCs typically reside in the bone marrow but can be mobilized into the circulating blood by administering a mobilizing agent in order to harvest HSCs from the peripheral blood. In some embodiments, the subject (or donor) from which the HSCs are obtained is administered a mobilizing agent, such as granulocyte colony-stimulating factor (G-CSF). The number of the HSCs collected following mobilization using a mobilizing agent is typically greater than the number of cells obtained without use of a mobilizing agent. The HSCs for use in the methods described herein may express the cell surface protein of interest. Upon any of the modifications described herein (e.g., genetic modification or incubation with a blocking agent), the HSCs would not be targeted by the cytotoxicity agent also described herein. Alternatively, the HSCs for use in the methods described herein may not express the lineage-specific cell surface protein of interest (e.g., CD33); however, descendant cells differentiated from the HSCs (e.g., B cells) express the lineage-specific cell surface protein. Upon genetic modification, an endogenous gene of the HSCs coding for the lineage-specific cell surface protein may be disrupted at a region encoding a non-essential epitope of the lineage-specific cell surface protein. Descendant cells differentiated from such modified HSCs (e.g., in vivo) would express a modified lineage-specific cell surface protein having the non-essential epitope mutated such that they would not be targeted by the cytotoxicity agent capable of binding the non-essential epitope. In some embodiments, a sample is obtained from a subject (or donor) and is then enriched for a desired cell type (e.g. CD34+/CD33- cells). For example, PBMCs and/or CD34+ hematopoietic cells can be isolated from blood as described herein. Cells can also be isolated from other cells, for example by isolation and/or activation with an antibody binding to an epitope on the cell surface of the desired cell type. Another method that can be used includes negative selection using antibodies to cell surface markers to selectively enrich for a specific cell type without activating the cell by receptor engagement. Populations of HSC can be expanded prior to or after manipulating the HSC such that they don’t bind the cytotoxic agent or have reduced binding to the cytotoxic agent. The cells may be cultured under conditions that comprise an expansion medium comprising one or more cytokines, such as stem cell factor (SCF), Flt-3 ligand (Flt3L), thrombopoietin (TPO), Interleukin 3 (IL-3), or Interleukin 6 (IL-6). The cell may be expanded for about 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25 days or any range necessary. In some embodiments, the HSC are expanded after isolation of a desired cell population (e.g., CD34+/CD33-) from a sample obtained from a subject (or donor) and prior to manipulation (e.g., genetic engineering, contact with a blocking agent). In some embodiments, the HSC are expanded after genetic engineering, thereby selectively expanding cells that have undergone the genetic modification and lack the epitope (e.g., have a deletion or substitution of at least a portion of the epitope) of the cell surface protein to which the cytotoxic agent binds. In some embodiments, a cell (“a clone”) or several cells having a desired characteristic (e.g., phenotype or genotype) following genetic modification may be selected and independently expanded. In some embodiments, the HSC are expanded prior to contacting the HSC with a blocking agent that binds the epitope of the cell surface proteins, thereby providing a population of HSC expressing the cell surface proteins that cannot be bound by the cytotoxic agent due to blocking of the corresponding epitope by the blocking agent. II. Preparation of Genetically Engineered Hematopoietic Cells Any of the genetically engineering hematopoietic cells, such as HSCs, that carry edited genes of one or more cell surface proteins can be prepared by a routine method or by a method described herein. In some embodiments, the genetic engineering is performed using genome editing. As used herein, “genome editing” refers to a method of modifying the genome, including any protein-coding or non-coding nucleotide sequence, of an organism to knock out the expression of a target gene. In general, genome editing methods involve use of an endonuclease that is capable of cleaving the nucleic acid of the genome, for example at a targeted nucleotide sequence. Repair of the double-stranded breaks in the genome may be repaired introducing mutations and/or exogenous nucleic acid may be inserted into the targeted site. Genome editing methods are generally classified based on the type of endonuclease that is involved in generating double stranded breaks in the target nucleic acid. These methods include use of zinc finger nucleases (ZFN), transcription activator-like effector- based nuclease (TALEN), meganucleases, and CRISPR/Cas systems. Transcription Activator-Like-Effector-Based Nuclease (TALEN) In some embodiments, the modified cells are manipulated as described herein using the TALEN technology known in the art. In general, TALENs are engineered restriction enzymes that can specifically bind and cleave a desired target DNA molecule. A TALEN typically contains a Transcriptional Activator-Like Effector (TALE) DNA-binding domain fused to a DNA cleavage domain. The DNA binding domain may contain a highly conserved 33-34 amino acid sequence with a divergent 2 amino acid RVD (repeat variable dipeptide motif) at positions 12 and 13. The RVD motif determines binding specificity to a nucleic acid sequence and can be engineered according to methods known to those of skill in the art to specifically bind a desired DNA sequence. In one example, the DNA cleavage domain may be derived from the FokI endonuclease. The FokI domain functions as a dimer, requiring two constructs with unique DNA binding domains for sites in the target genome with proper orientation and spacing. TALENs specific to sequences in a target gene of interest (e.g., IL1RAP, CD33) can be constructed using any method known in the art. A TALEN specific to a target gene of interest can be used inside a cell to produce a double-stranded break (DSB). A mutation can be introduced at the break site if the repair mechanisms improperly repair the break via non-homologous end joining. For example, improper repair may introduce a frame shift mutation. Alternatively, a foreign DNA molecule having a desired sequence can be introduced into the cell along with the TALEN. Depending on the sequence of the foreign DNA and chromosomal sequence, this process can be used to correct a defect or introduce a DNA fragment into a target gene of interest, or introduce such a defect into the endogenous gene, thus decreasing expression of the target gene. In some embodiments, one or more population of hematopoietic cells is generated by genetic engineering of a cell surface protein (e.g., those described herein) using a TALEN. The genetically engineered hematopoietic cells may not express the lineage-specific cell surface antigen. Alternatively, the hematopoietic cells may be engineered to express an altered version of the lineage-specific cell surface antigen, e.g., having a deletion or mutation relative to the wild-type counterpart. Such a mutated cell surface protein may preserve a certain level of the bioactivity as the wild-type counterpart. In some embodiments, a population of hematopoietic cells containing a mutated CD33 is generated by genetic engineering using a TALEN. In some embodiments, exon 2 or exon 3 of CD33 is mutated using a TALEN. Zinc Finger Nuclease (ZFN) In some embodiments, the cells can be genetically manipulated using zinc finger (ZFN) technology known in the art. In general, zinc finger mediated genomic editing involves use of a zinc finger nuclease, which typically comprises a DNA binding domain (i.e., zinc finger) and a cleavage domain (i.e., nuclease). The zinc finger binding domain may be engineered to recognize and bind to any target gene of interest (e.g., IL1RAP, CD33) using methods known in the art and in particular, may be designed to recognize a DNA sequence ranging from about 3 nucleotides to about 21 nucleotides in length, or from about 8 to about 19 nucleotides in length. Zinc finger binding domains typically comprise at least three zinc finger recognition regions (e.g., zinc fingers). Restriction endonucleases (restriction enzymes) capable of sequence-specific binding to DNA (at a recognition site) and cleaving DNA at or near the site of binding are known in the art and may be used to form ZFN for use in genomic editing. For example, Type IIS restriction endonucleases cleave DNA at sites removed from the recognition site and have separable binding and cleavage domains. In one example, the DNA cleavage domain may be derived from the FokI endonuclease. In some embodiments, one or more population of hematopoietic cells is generated by genetic engineering of a cell surface protein (e.g., those described herein) using a ZFN. The genetically engineered hematopoietic cells may not express the cell surface protein. Alternatively, the hematopoietic cells may be engineered to express an altered version of the cell surface protein, e.g., having a deletion or mutation relative to the wild-type counterpart. Such a mutated cell surface protein may preserve a certain level of the bioactivity as the wild- type counterpart. In some examples, a population of hematopoietic cells containing a mutated CD33 is generated by genetic engineering using a ZFN. In some embodiments, exon 2 or exon 3 of CD33 is mutated using a ZFN. CRISPR/Cas Systems One exemplary suitable genome editing technology comprises the use of a CRISPR/Cas nuclease to introduce targeted single- or double-stranded DNA breaks in the genome of a cell, which trigger cellular repair mechanisms, such as, for example, nonhomologous end joining (NHEJ), microhomology-mediated end joining (MMEJ, also sometimes referred to as “alternative NHEJ” or “alt-NHEJ”), or homology-directed repair (HDR) that typically result in an altered nucleic acid sequence (e.g., via nucleotide or nucleotide sequence insertion, deletion, inversion, or substitution) at or immediately proximal to the site of the nuclease cut. See, Yeh et al. Nat. Cell. Biol. (2019) 21: 1468-1478; e.g., Hsu et al. Cell (2014) 157: 1262-1278; Jasin et al. DNA Repair (2016) 44: 6-16; Sfeir et al. Trends Biochem. Sci. (2015) 40: 701-714. Another exemplary suitable genome editing technology is “base editing,” which includes the use of a base editor, e.g., a nuclease-impaired or partially nuclease impaired enzyme (e.g., RNA-guided CRISPR/Cas protein) fused to a deaminase that targets and deaminates a specific nucleobase, e.g., a cytosine or adenosine nucleobase of a C or A nucleotide, which, via cellular mismatch repair mechanisms, results in a change from a C to a T nucleotide, or a change from an A to a G nucleotide. See, e.g., Komor et al. Nature (2016) 533: 420-424; Rees et al. Nat. Rev. Genet. (2018) 19(12): 770-788; Anzalone et al. Nat. Biotechnol. (2020) 38: 824-844. Yet another exemplary suitable genome editing technology includes “prime editing,” which includes the introduction of new genetic information, e.g., an altered nucleotide sequence, into a specifically targeted genomic site using a catalytically impaired or partially catalytically impaired nuclease (e.g., a CRISPR/Cas nuclease), fused to an engineered reverse transcriptase (RT) domain. The Cas/RT fusion is targeted to a target site within the genome by a guide RNA that also comprises a nucleic acid sequence encoding the desired edit, and that can serve as a primer for the RT. See, e.g., Anzalone et al. Nature (2019) 576 (7785): 149-157. In some embodiments, a genetically engineered cell described herein is made using gene editing technology, also referred to herein as DNA-editing technology, e.g., CRISPR/Cas-based DNA editing as described herein. Exemplary CRISPR/Cas systems are described herein, and additional suitable CRISPR/Cas systems will be apparent to the skilled artisan based on the present disclosure and the knowledge in the art. In some embodiments, a gRNA targeting a gene of interest (e.g. a cell surface protein) described herein is complexed with a Cas9 nuclease. Various Cas9 nuclease orthologs or variants can be used. In some embodiments, a Cas9 nuclease is selected that has the desired PAM specificity to target the gRNA/Cas9 nuclease complex to the target domain in the gene of interest (e.g., cell surface antigen). In some embodiments, genetically engineering a cell also comprises introducing one or more (e.g., 1, 2, 3 or more) Cas9 nucleases into the cell. Cas Nucleases A “Cas nuclease” as that term is used herein, refers to a CRISPR/Cas nuclease (e.g., Cas9) that can interact with a gRNA and, in concert with the gRNA, home or localize to a site which comprises a target domain. Cas nucleases include naturally occurring Cas nucleases and engineered, altered, or modified Cas nucleases that differ, e.g., by at least one amino acid residue, from a naturally occurring Cas nuclease. Cas9 nucleases of a variety of species can be used in the methods and compositions described herein. In various embodiments, the Cas9 nuclease is of, or derived from, Streptococcus pyogenes (SpCas9), Staphylococcus aureus (SaCas9), or Streptococcus thermophilus (StCas9). Additional suitable Cas9 nucleases include those of, or derived from, Staphylococcus aureus, Neisseria meningitidis (NmCas9), Acidovorax avenae, Actinobacillus pleuropneumoniae, Actinobacillus succinogenes, Actinobacillus suis, Actinomyces sp., Cycliphilus denitrificans, Aminomonas paucivorans, Bacillus cereus, Bacillus smithii, Bacillus thuringiensis, Bacteroides sp., Blastopirellula marina, Bradyrhizobium sp., Brevibacillus laterosporus, Campylobacter coli, Campylobacter jejuni (CjCas9), Campylobacter lari, Candidatus puniceispirillum, Clostridium cellulolyticum, Clostridium perfringens, Corynebacterium accolens, Corynebacterium diphtheria, Corynebacterium matruchotii, Dinoroseobacter shibae, Eubacterium dolichum, gamma proteobacterium, Gluconacetobacter diazotrophicus, Haemophilus parainfluenzae, Haemophilus sputorum, Helicobacter canadensis, Helicobacter cinaedi, Helicobacter mustelae, Ilyobacter polytropus, Kingella kingae, Lactobacillus crispatus, Listeria ivanovii, Listeria monocytogenes, Listeriaceae bacterium, Methylocystis sp., Methylosinus trichosporium, Mobiluncus mulieris, Neisseria bacilliformis, Neisseria cinerea, Neisseria flavescens, Neisseria lactamica, Neisseria sp., Neisseria wadsworthii, Nitrosomonas sp., Parvibaculum lavamentivorans, Pasteurella multocida, Phascolarctobacterium succinatutens, Ralstonia syzygii, Rhodopseudomonas palustris, Rhodovulum sp., Simonsiella muelleri, Sphingomonas sp., Sporolactobacillus vineae, Staphylococcus lugdunensis, Streptococcus sp., Subdoligranulum sp., Tistrella mobilis, Treponema sp., or Verminephrobacter eiseniae. In some embodiments, catalytically impaired, or partially impaired, variants of such Cas9 nucleases may be used. Additional suitable Cas9 nucleases, and nuclease variants, will be apparent to those of skill in the art based on the present disclosure. The disclosure is not limited in this respect. In some embodiments, the Cas9 nuclease is a naturally occurring Cas9 nuclease. In some embodiments, the Cas9 nuclease is an engineered, altered, or modified Cas9 nuclease that differs, e.g., by at least one amino acid residue, from a reference sequence, e.g., the most similar naturally occurring Cas9 nuclease or a sequence of Table 50 of International Publication No. WO 2015/157070, which is herein incorporated by reference in its entirety. In some embodiments, the Cas nuclease comprises Cpf1 or a fragment or variant thereof. A naturally occurring Cas9 nuclease typically comprises two lobes: a recognition (REC) lobe and a nuclease (NUC) lobe; each of which further comprises domains described, e.g., in International Publication No. WO 2015/157070, e.g., in Figs.9A-9B therein (which application is incorporated herein by reference in its entirety). The REC lobe comprises the arginine-rich bridge helix (BH), the REC1 domain, and the REC2 domain. The REC lobe appears to be a Cas9-specific functional domain. The BH domain is a long alpha helix and arginine rich region and comprises amino acids 60-93 of the sequence of S. pyogenes Cas9. The REC1 domain is involved in recognition of the repeat:anti-repeat duplex, e.g., of a gRNA or a tracrRNA. The REC1 domain comprises two REC1 motifs at amino acids 94 to 179 and 308 to 717 of the sequence of S. pyogenes Cas9. These two REC1 domains, though separated by the REC2 domain in the linear primary structure, assemble in the tertiary structure to form the REC1 domain. The REC2 domain, or parts thereof, may also play a role in the recognition of the repeat: anti-repeat duplex. The REC2 domain comprises amino acids 180-307 of the sequence of S. pyogenes Cas9. The NUC lobe comprises the RuvC domain (also referred to herein as RuvC-like domain), the HNH domain (also referred to herein as HNH-like domain), and the PAM- interacting (PI) domain. The RuvC domain shares structural similarity to retroviral integrase superfamily members and cleaves a single strand, e.g., the non-complementary strand of the target nucleic acid molecule. The RuvC domain is assembled from the three split RuvC motifs (RuvC I, RuvCII, and RuvCIII, which are often commonly referred to in the art as RuvCI domain, or N-terminal RuvC domain, RuvCII domain, and RuvCIII domain) at amino acids 1-59, 718-769, and 909-1098, respectively, of the sequence of S. pyogenes Cas9. Similar to the REC1 domain, the three RuvC motifs are linearly separated by other domains in the primary structure, however in the tertiary structure, the three RuvC motifs assemble and form the RuvC domain. The HNH domain shares structural similarity with HNH endonucleases, and cleaves a single strand, e.g., the complementary strand of the target nucleic acid molecule. The HNH domain lies between the RuvC II-III motifs and comprises amino acids 775-908 of the sequence of S. pyogenes Cas9. The PI domain interacts with the PAM of the target nucleic acid molecule, and comprises amino acids 1099-1368 of the sequence of S. pyogenes Cas9. Crystal structures have been determined for naturally occurring bacterial Cas9 nucleases (Jinek et al., Science, 343(6176): 1247997, 2014) and for S. pyogenes Cas9 with a guide RNA (e.g., a synthetic fusion of crRNA and tracrRNA) (Nishimasu et al., Cell, 156:935-949, 2014; and Anders et al., Nature, 2014, doi: 10.1038/naturel3579). In some embodiments, a Cas nuclease described herein has nuclease activity, e.g., double strand break activity in or directly proximal to a target site. In some embodiments, the Cas9 nuclease has been modified to inactivate one of the catalytic residues of the nuclease. In some embodiments, the Cas9 nuclease is a nickase and produces a single stranded break. See, e.g., Dabrowska et al. Frontiers in Neuroscience (2018) 12(75). It has been shown that one or more mutations in the RuvC and HNH catalytic domains of the enzyme may improve Cas9 efficiency. See, e.g., Sarai et al. Currently Pharma. Biotechnol. (2017) 18(13). In some embodiments, the Cas9 nuclease is fused to a second domain, e.g., a domain that modifies DNA or chromatin, e.g., a deaminase or demethylase domain. In some such embodiments, the Cas9 nuclease is modified to eliminate its nuclease activity. In some embodiments, the Cas nuclease is modified to enhance specificity of the enzyme (e.g., reduce off-target effects, maintain robust on-target cleavage). In some embodiments, the Cas9 nuclease is an enhanced specificity Cas9 variant (e.g., eSPCas9). See, e.g., Slaymaker et al. Science (2016) 351 (6268): 84-88. In some embodiments, the Cas9 nuclease is a high fidelity Cas9 variant (e.g., SpCas9-HF1). See, e.g., Kleinstiver et al. Nature (2016) 529: 490-495. As used herein, the term “protospacer adjacent motif” or “PAM” refers to a DNA sequence that may be required for a nuclease and a gRNA (e.g., Cas9/sgRNA) to form an R- loop to interrogate a specific DNA sequence through Watson-Crick pairing of its guide RNA with the genome. The PAM specificity may be a function of the DNA-binding specificity of the nuclease, e.g., Cas9 protein, e.g., a “protospacer adjacent motif recognition domain” at the C-terminus of Cas9. In some embodiments, the PAM comprises a 2 to 6 base pair DNA sequence immediately downstream or upstream of the target site in the genome, which may be recognized directly by a Cas nuclease, e.g., a Cas9, to promote cleavage of the target site by the Cas nuclease, or in the case of nuclease-deficient Cas9 allows binding to the DNA at that locus. In some embodiments, the PAM can be a 5' PAM (i.e., located upstream of the 5' end of the protospacer). In other embodiments, the PAM can be a 3' PAM (i.e., located downstream of the 5' end of the protospacer). As used herein, the term “Cas nuclease PAM” refers to a DNA sequence that may be required to home or localize a CRISPR/Cas nuclease (e.g., Cas9 or other Cas nucleases, such as Cas12a (Cpf1) or Cas12b) interacting with a gRNA to sequence comprising a target domain. Non-limiting examples of CRISPR/Cas nucleases and their corresponding consensus PAM are described herein. For example, the Cas9 endonuclease from Streptococcus pyogenes (SpyCas9) can recognize a NGG consensus PAM (e.g., a 5’-NGG on the 3’ end of the gRNA sequence) (N = any base). SpyCas9 can also weakly recognize NGA, NNGG, and a selection of other sequences. The SpGCas9 (a variant of SpyCas9) can recognize a NGN (N= any base) consensus PAM. The SpRYCas9 (a variant of SpGCas9) can recognize a NRN (N= any base; R= A,G) consensus PAM. SpRYCas9 can also weakly some NYN (N= any base; Y= C or T) sequences. The Cas9 nuclease from the CRISPR1 locus (Sth1Cas9) in the model lactic-acid bacterium Streptococcus thermophilus can recognize a NNATAAW (W = A, T; N= any base) consensus PAM. The Cas9 nuclease from the pathogen Staphylococcus aureus (SauCas9) can recognize an NNGRRT consensus PAM (R = A, G; N= any base). The Cas9 nuclease from the pathogen Neisseria meningitidis (NmeCas9) can recognize a NNNNGATT consensus PAM (N= any base). The MAD7 endonuclease from Eubacterium rectale can recognize 5’- TTTV on the 5’ end of the gRNA sequence, and to some extent 5’-YTTV and YTTN. The Cpf1 endonuclease from Acidaminococcus sp. and Lachnospiraceae sp. recognize 5’-TTTN and the Cpf1 endonuclease from Francisella novicide can recognize 5’-TTN-3’ on the 5’ end of the gRNA. Various Cas9 nucleases are known in the art and may be obtained from various sources and/or engineered/modified to modulate one or more activities or specificities of the enzymes. In some embodiments, the Cas9 nuclease has been engineered/modified to recognize one or more PAM sequence. In some embodiments, the Cas9 nuclease has been engineered/modified to recognize one or more PAM sequence that is different than the PAM sequence the Cas9 nuclease recognizes without engineering/modification. In some embodiments, the Cas9 nuclease has been engineered/modified to reduce off-target activity of the enzyme. In some embodiments, the nucleotide sequence encoding the Cas9 nuclease is modified further to alter the specificity of the nuclease activity (e.g., reduce off-target cleavage, decrease the nuclease activity or lifetime in cells, increase homology-directed recombination and reduce non-homologous end joining). See, e.g., Komor et al. Cell (2017) 168: 20-36. In some embodiments, the nucleotide sequence encoding the Cas9 nuclease is modified to alter the PAM recognition of the nuclease. For example, the Cas9 nuclease SpCas9 recognizes PAM sequence NGG, whereas relaxed variants of the SpCas9 comprising one or more modifications of the nuclease (e.g., VQR SpCas9, EQR SpCas9, VRER SpCas9) may recognize the PAM sequences NGA, NGAG, NGCG. PAM recognition of a modified Cas9 nuclease is considered “relaxed” if the Cas9 nuclease recognizes more potential PAM sequences as compared to the Cas9 nuclease that has not been modified. For example, the Cas9 nuclease SaCas9 recognizes PAM sequence NNGRRT, whereas a relaxed variant of the SaCas9 comprising one or more modifications (e.g., KKH SaCas9) may recognize the PAM sequence NNNRRT. In one example, the Cas9 nuclease FnCas9 recognizes PAM sequence NNG, whereas a relaxed variant of the FnCas9 comprising one or more modifications of the nuclease (e.g., RHA FnCas9) may recognize the PAM sequence YG. In one example, the Cas nuclease is a Cpf1 nuclease comprising substitution mutations S542R and K607R and recognize the PAM sequence TYCV. In one example, the Cas nuclease is a Cpf1 nuclease comprising substitution mutations S542R, K607R, and N552R and recognize the PAM sequence TATV. See, e.g., Gao et al. Nat. Biotechnol. (2017) 35(8): 789-792. In some embodiments, a Cas nuclease recognizes a PAM selected from: a SpyCas9 PAM, a SpGCas9 PAM, a SpRYCas9 PAM, a Sth1Cas9 PAM, a SauCas9 PAM, a NmeCas9 PAM, a RspCas9 PAM, a Cca1Cas9 PAM, a PspCas9 PAM, a OrhCas9 PAM, a ScCas9 PAM, a SmacCas9 PAM, a TdeCas9 PAM, a Nme2Cas9 PAM, a CjeCas9 PAM, a SmuCas9 PAM, a Smu2Cas9 PAM, a PmuCas9 PAM, a SpaCas9 PAM, a NciCas9 PAM, a ClaCas9 PAM, a PlaCas9 PAM, a CdCas9 PAM, a IgnaviCas9 PAM, a ThermoCas9 PAM, a GeoCas9 PAM, a G. LC300 Cas9 PAM, a AceCas9 PAM, a Sth3Cas9 PAM, a BlatCas9 PAM, a FnCas9 PAM, a LfeCas9 PAM, a LpnCas9 PAM, a KhuCas9 PAM, a AinCas9 PAM, a CglCas9 PAM, a Esp1Cas9 PAM, a Esp2Cas9 PAM, a FmaCas9 PAM, a LceCas9 PAM, a LrhCas9 PAM, a Lsp1Cas9 PAM, a Lsp2Cas9 PAM, a PacCas9 PAM, a TbaCas9 PAM, a TpuCas9 PAM, a VpaCas9 PAM, a EfaCas9 PAM, a EitCas9 PAM, a LanCas9 PAM, a LmoCas9 PAM, a Sag1Cas9 PAM, a Sag2Cas9 PAM, a SdyCas9 PAM, a Seq1Cas9 PAM, a Seq2Cas9 PAM, a SgaCas9 PAM, a Smu3Cas9 PAM, a SraCas9 PAM, a BniCas9 PAM, a EceCas9 PAM, a EdoCas9 PAM, a FhoCas9 PAM, a MgaCas9 PAM, a MseCas9 PAM, a SgoCas9 PAM, a SmaCas9 PAM, a SsaCas9 PAM, a SsiCas9 PAM, a SsuCas9 PAM, a Sth1ACas9 PAM, a TspCas9 PAM, a BokCas9 PAM, a CcoCas9 PAM, a CpeCas9 PAM, a DdeCas9 PAM, a Ghc2Cas9 PAM, a Ghy3Cas9 PAM, a Ghy4Cas9 PAM, a GspCas9 PAM, a KkiCas9 PAM, a NspCas9 PAM, a TmoCas9 PAM, a NsaCas9 PAM, a JpaCas9 PAM, a BboCas9 PAM, a Cca2Cas9 PAM, a Cme2Cas9 PAM, a Cme3Cas9 PAM, a Cme4Cas9 PAM, a CsaCas9 PAM, a Ghc1Cas9 PAM, a GheCas9 PAM, a Ghh1Cas9 PAM, a Ghh2Cas9 PAM, a Ghy1Cas9 PAM, a MscCas9 PAM, a SdoCas9 PAM, a SpacCas9 PAM, a CgaCas9 PAM, a Cme1Cas9 PAM, a FfrCas9 PAM, a Ghy2Cas9 PAM, a PhiCas9 PAM, a WviCas9 PAM, a CcCas9 PAM, a FnCas12a PAM, a AsCas12a PAM, a HkCas12a PAM, a PiCas12a PAM, a PdCas12a PAM, a LbCas12a PAM, a Lb2Cas12a or Lb5Cas12a PAM, a CmtCas12a PAM, a MbCas12a PAM, a TsCas12a PAM, a Pb2Cas12a PAM, a MlCas12a PAM, a Mb2Cas12a PAM, a Mb3Cas12a PAM, a CmaCas12a PAM, a BsCas12a PAM, a BfCas12a PAM, a BoCas12a PAM, a Adurb193Cas12a PAM, a Adurb336Cas12a PAM, a Fn3Cas12a PAM, a Lb6Cas12a PAM, a EcCas12a PAM, a PsCas12a PAM, a McCas12a PAM, a AacCas12b PAM, a BthCas12b PAM, a AkCas12b PAM, a Ebcas12b PAM, a bvCas12b PAM, a BhCas12b PAM, a LsCas12b PAM, a BrCas12b PAM, a Cas12c1 PAM, a Cas12c2 PAM, a OspCas12c PAM, a Cas12d.15 PAM, a Cas12d.1 (CasY.1) PAM, a DpbCas12e (DpbCasX) PAM, a PlmCas12e (PlmCasX) PAM, a Mi1Cas12f2 PAM, a Un1Cas12f1 PAM, a Un2Cas12f1 PAM, a Mi2Cas12f2 PAM, a AuCas12f2 PAM, a PtCas12f1 PAM, a AsCas12f1 PAM, a RuCas12f1 PAM, a SpCas12f1 PAM, a CnCas12f1 PAM, a Cas12h1 PAM, a Cas12i1 PAM, a Cas12i2 PAM, a Cas12j-1 (CasPhi-1) PAM, a Cas12j-2 (CasPhi-2) PAM, a Cas12j-3 (CasPhi-3) PAM, a ShCas12k PAM, or a AcCas12k PAM. In some embodiments, a Cas nuclease recognizes a PAM comprising, and/or consisting of a nucleotide sequence selected from: NGG; NNAGAAW; NNGRRT; NNNNGATT; NVNDCCY; BRTTTTT; NR(A or G)TTTT; NNAAAR(G or A); N(N or A)G; NAAN; NAAAAY; NHDTCCA; NNNVRYM; NNNNRYAC; NAA; GNNNNCNNA; NNGTGA; NNNNGTA; NNGGG; NNNCAT; NNRHHHY; NRRNAT; NNNNCNAA; NNNNCMCA; NNNNCRAA; NNNNGMAA; NNNCC; NGGNG; NNNNCNDD; NYAAA; NRGNN; N(C or D)GGN(T or A or G or C)NN; NRTAW; N(C or K or A)AARC; NAAAG; NV(A or G or C)R(A or G)ACCN; NNGAC; NATGNT; N(T or V)NTAAW(A or T); NNGW(A or T)AY(T or C); NCAA(H(Y or A)B(Y or G); NH(T or C or A)AAAA; NNNATTT; NATAWN(A or T or S); NATARCH; B(T or G or C)GGD(A or T or G)TNN; N(G or T or M)GGAH(T or A or C)N(A or C or K)N; NRG; N(B or A)GG; NGGD(A or K)W(T or A); N(T or C or R)AGAN(A or K or C)NN; NGGD(A or T or G)H(T or M); NGGDT; NGGD(A or T or G)GNN; NNGTAM(A or C)Y; NNGH(W or C)AAA; NTGAR(G or A)N(A or Y or G)N(Y or R); NNGAAAN; NNGAD; NHARMC; NNAAAG; NHGYNAN(A or B); NNAGAAA; NHAAAAA; NH(T or M)AAAAA; NHGYRAA; NNAAACN; NN(H or G)D(A or K)GGDN(A or B); NNNNCTA; NNNNCVGAA; NNNNGYAA; NNNNATN(W or S)ANN; NNWHR(G or A)TA(not G)AA; YHHNGTH; NNNNCDAANN; NNNNCTAA; N(C or D)NNTCCN; NNNNCCAA; NAGRGN(T or V)N(T or C); NNAH(T or M)I; CN(C or W or G)AV(A or S)GAC; NAR(G or A)H(W or C)H(A or T or C)GN(C or T or R); NAGNGC; NATCCTN; NGTGANN; HGCNGCR; NAR(A or G)W(T or A)AC; N(C or D)M(A or C)RN(A or B)AY(C or T); NNNCAC; BGGGTCD; NNRRCC; NRRNTT; KARDAT; BRRTTTW; NARNCCN; NAR(A or G)TC; NAAN(A or T or S)RCN; HHAAATD; NNNNGNA; TTV; TTTV; YYV; KKYV; TTTM; TTYV; TTTN; TTTTA; TTN; BTTV; YTV; YTN; NYTV; DTTD; ATTN; RTTNT; HATT; ATTW; RTTN; TVT; TG; TN; TR; TA; TTCN; TTAT; TTTR; TTR; YTTR; YTTN; CTT; TTC; CCD; RTR; VTTR; TBN; VTTN; NGTT; CGTT; AGG; CGG; GTT, or RGTG, wherein “N” is any nucleotide or base, “W” is adenine (A) or thymine (T), “R” is A or guanine (G), “V” is A, cytosine (C), or G, “Y” is C or T, and “H” is A, C, or T. In some embodiments, more than one (e.g., 2, 3, or more) Cas nucleases are used. In some embodiments, at least one of the Cas nuclease is a Cas9. In some embodiments, at least one of the Cas nucleases is a Cpf1 enzyme. In some embodiments, at least one of the Cas9 nuclease is derived from Streptococcus pyogenes. In some embodiments, at least one of the Cas9 nuclease is derived from Streptococcus pyogenes and at least one Cas9 nuclease is derived from an organism that is not Streptococcus pyogenes. Guide Nucleic Acids The terms “gRNA” and “guide RNA” are used interchangeably throughout and refer to a nucleic acid that promotes the specific targeting or homing of a CRISPR/Cas system to a target nucleic acid sequence. A gRNA can be unimolecular (having a single RNA molecule), sometimes referred to herein as sgRNAs, or modular (comprising more than one, and typically two, separate RNA molecules). A gRNA may bind to a target domain in the genome of a host cell. The gRNA may comprise a targeting domain that may be partially or completely complementary to the target domain. The gRNA may also comprise a “scaffold sequence,” (e.g., a tracrRNA sequence), that recruits a Cas nuclease to a target domain bound to a gRNA sequence (e.g., by the targeting domain of the gRNA sequence). The scaffold sequence may comprise at least one stem loop structure and recruits an endonuclease. Exemplary scaffold sequences can be found, for example, in Jinek, et al. Science (2012) 337(6096):816-821, Ran, et al. Nature Protocols (2013) 8:2281-2308, International Publication No. WO2014/093694, and International Publication No. WO2013/176772. As used herein, the terms “proximity,” “in proximity,” or “within proximity,” refers to a first, second or subsequent target site within distance of a PAM that can be bound by a CRISPR/Cas system, according to any method described herein. In various embodiments, the mutation of the genomic DNA sequence at the first target site is within 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, or 25 nucleotides from a PAM sequence. As used herein, the term “sgRNA,” refers to single guide RNA used in conjunction with CRISPR associated systems (Cas). sgRNAs are a fusion of crRNA and tracrRNA and contain nucleotides of sequence complementary to the desired target site (Jinek et al., Science, 343(6176): 1247997, 2014). Watson-Crick pairing of the sgRNA with the target site permits R-loop formation, which in conjunction with a functional PAM permits DNA cleavage or in the case of nuclease-deficient Cas9 allows binding to the DNA at that locus. Some exemplary suitable Cas9 gRNA scaffold sequences are provided herein, and additional suitable gRNA scaffold sequences will be apparent to the skilled artisan based on the present disclosure. Such additional suitable scaffold sequences include, without limitation, those recited in Jinek, et al. Science (2012) 337(6096):816-821, Ran, et al. Nature Protocols (2013) 8:2281-2308, PCT Publication No. WO2014/093694, and PCT Publication No. WO2013/176772. For example, the binding domains of naturally occurring SpCas9 gRNA typically comprise two RNA molecules, the crRNA (partially) and the tracrRNA. Variants of SpCas9 gRNAs that comprise only a single RNA molecule including both crRNA and tracrRNA sequences, covalently bound to each other, e.g., via a tetraloop or via click chemistry type covalent linkage, have been engineered and are commonly referred to as “single guide RNA” or “sgRNA.” Suitable gRNAs for use with other Cas nucleases, for example, with Cas12a nucleases, typically comprise only a single RNA molecule, as the naturally occurring Cas12a guide RNA comprises a single RNA molecule. A suitable gRNA may thus be unimolecular (having a single RNA molecule), sometimes referred to herein as sgRNAs, or modular (comprising more than one, and typically two, separate RNA molecules). A gRNA as provided herein typically comprises a targeting domain that binds to a target site in the genome of a cell. The “targeting domain” of the gRNA is complementary to the “target domain” on the target nucleic acid. The strand of the target nucleic acid comprising the nucleotide sequence complementary to the core domain of the gRNA is referred to herein as the “complementary strand” of the target nucleic acid. The targeting domain mediates targeting of the gRNA-bound Cas nuclease to a target site. Guidance on the selection of targeting domains can be found, e.g., in Fu Y et al, Nat Biotechnol 2014 (doi: 10.1038/nbt.2808) and Sternberg SH et al., Nature 2014 (doi: 10.1038/naturel3011). The targeting domain of the gRNA thus base-pairs (in full or partial complementarity) with the sequence of the double-stranded genomic target site that is complementary to the sequence of the target domain, and thus with the strand complementary to the strand that comprises the PAM sequence. It will be understood that the targeting domain of the gRNA typically does not include the PAM sequence. It will further be understood that the location of the PAM may be 5’ or 3’ of the target domain sequence, depending on the nuclease employed. For example, the PAM is typically 3’ of the target domain sequences for Cas9 nucleases, and 5’ of the target domain sequence for Cas12a nucleases. For an illustration of the location of the PAM and the mechanism of gRNA binding a target site, see, e.g., Figure 1 of Vanegas et al., Fungal Biol Biotechnol.2019; 6: 6, which is incorporated by reference herein. For additional illustration and description of the mechanism of gRNA targeting an CRISPR/Cas nuclease to a target site, see Fu Y et al, Nat Biotechnol 2014 (doi: 10.1038/nbt.2808) and Sternberg SH et al., Nature 2014 (doi: 10.1038/naturel3011), both incorporated herein by reference. An exemplary illustration of a Cas9 target site, comprising a 22 nucleotide target domain, and an NGG PAM sequence, as well as of a gRNA comprising a targeting domain that fully corresponds to the target domain (and thus base-pairs with full complementarity with the DNA strand complementary to the strand comprising the target domain and PAM) is provided below: [ target domain (DNA) ][ PAM ] 5’-N-N-N-N-N-N-N-N-N-N-N-N-N-N-N-N-N-N-N-N-N-N-N-G-G-3’ (DNA) 3’-N-N-N-N-N-N-N-N-N-N-N-N-N-N-N-N-N-N-N-N-N-N-N-C-C-5’ (DNA) | | | | | | | | | | | | | | | | | | | | | | 5’-N-N-N-N-N-N-N-N-N-N-N-N-N-N-N-N-N-N-N-N-N-N-[gRNA scaffold]-3’ (RNA) [ targeting domain (RNA) ][binding domain] An exemplary illustration of a Cas12a target site, comprising a 22 nucleotide target domain, and a TTN PAM sequence, as well as of a gRNA comprising a targeting domain that fully corresponds to the target domain (and thus base-pairs with full complementarity with the DNA strand complementary to the strand comprising the target domain and PAM) is provided below: [ PAM ][ target domain (DNA) ] 5’-T-T-N-N-N-N-N-N-N-N-N-N-N-N-N-N-N-N-N-N-N-N-N-N-N-3’ (DNA) 3’-A-A-N-N-N-N-N-N-N-N-N-N-N-N-N-N-N-N-N-N-N-N-N-N-N-5’ (DNA) | | | | | | | | | | | | | | | | | | | | | |5’-[gRNA scaffold]-N-N-N-N-N-N-N-N-N-N-N-N-N-N-N-N-N-N-N-N-N-N-3’ (RNA) [binding domain][ targeting domain (RNA) ] In some embodiments, the Cas12a PAM sequence is 5’-T-T-T-V-3’. While not wishing to be bound by theory, at least in some embodiments, it is believed that the length and complementarity of the targeting domain with the target sequence contributes to specificity of the interaction of the gRNA/Cas9 nuclease complex with a target nucleic acid. In some embodiments, the targeting domain of a gRNA provided herein is 5 to 50 nucleotides in length. In some embodiments, the targeting domain is 15 to 25 nucleotides in length. In some embodiments, the targeting domain is 18 to 22 nucleotides in length. In some embodiments, the targeting domain is 19-21 nucleotides in length. In some embodiments, the targeting domain is 15 nucleotides in length. In some embodiments, the targeting domain is 16 nucleotides in length. In some embodiments, the targeting domain is 17 nucleotides in length. In some embodiments, the targeting domain is 18 nucleotides in length. In some embodiments, the targeting domain is 19 nucleotides in length. In some embodiments, the targeting domain is 20 nucleotides in length. In some embodiments, the targeting domain is 21 nucleotides in length. In some embodiments, the targeting domain is 22 nucleotides in length. In some embodiments, the targeting domain is 23 nucleotides in length. In some embodiments, the targeting domain is 24 nucleotides in length. In some embodiments, the targeting domain is 25 nucleotides in length. In some embodiments, the targeting domain fully corresponds, without mismatch, to a target domain sequence provided herein, or a part thereof. In some embodiments, the targeting domain of a gRNA provided herein comprises 1 mismatch relative to a target domain sequence provided herein. In some embodiments, the targeting domain comprises 2 mismatches relative to the target domain sequence. In some embodiments, the target domain comprises 3 mismatches relative to the target domain sequence. In some embodiments, a targeting domain comprises a core domain and a secondary targeting domain, e.g., as described in International Publication No. WO 2015/157070, which is incorporated by reference in its entirety. In some embodiments, the core domain comprises about 8 to about 13 nucleotides from the 3' end of the targeting domain (e.g., the most 3' 8 to 13 nucleotides of the targeting domain). In an embodiment, the secondary domain is positioned 5' to the core domain. In many embodiments, the core domain has exact complementarity (corresponds fully) with the corresponding region of the target sequence, or part thereof. In other embodiments, the core domain can have 1 or more nucleotides that are not complementary (mismatched) with the corresponding nucleotide of the target domain sequence. The first complementarity domain is complementary with the second complementarity domain, and in an embodiment, has sufficient complementarity to the second complementarity domain to form a duplexed region under at least some physiological conditions. In an embodiment, the first complementarity domain is 5 to 30 nucleotides in length. In an embodiment, the first complementarity domain comprises 3 subdomains, which, in the 5' to 3' direction are: a 5' subdomain, a central subdomain, and a 3' subdomain. In an embodiment, the 5' subdomain is 4 to 9, e.g., 4, 5, 6, 7, 8 or 9 nucleotides in length. In an embodiment, the central subdomain is 1, 2, or 3, e.g., 1, nucleotide in length. In an embodiment, the 3' subdomain is 3 to 25, e.g., 4 to 22, 4 to 18, or 4 to 10, or 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, or 25 nucleotides in length. The first complementarity domain can share homology with, or be derived from, a naturally occurring first complementarity domain. In an embodiment, it has at least 50% homology with a S. pyogenes, S. aureus or S. thermophilus, first complementarity domain. The sequence and placement of the above-mentioned domains are described in more detail in International Publication No. WO2015/157070, which is herein incorporated by reference in its entirety, including p.88-112 therein. A linking domain serves to link the first complementarity domain with the second complementarity domain of a unimolecular gRNA. The linking domain can link the first and second complementarity domains covalently or non-covalently. In an embodiment, the linkage is covalent. In an embodiment, the linking domain is, or comprises, a covalent bond interposed between the first complementarity domain and the second complementarity domain. In some embodiments, the linking domain comprises one or more, e.g., 2, 3, 4, 5, 6, 7, 8, 9, or 10 nucleotides. In some embodiments, the linking domain comprises at least one non-nucleotide bond, e.g., as disclosed in International Publication No. WO2018/126176, the entire contents of which are incorporated herein by reference. The second complementarity domain is complementary, at least in part, with the first complementarity domain, and in an embodiment, has sufficient complementarity to the second complementarity domain to form a duplexed region under at least some physiological conditions. In an embodiment, the second complementarity domain can include a sequence that lacks complementarity with the first complementarity domain, e.g., a sequence that loops out from the duplexed region. In an embodiment, the second complementarity domain is 5 to 27 nucleotides in length. In an embodiment, the second complementarity domain is longer than the first complementarity region. In an embodiment, the complementary domain is 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24 or 25 nucleotides in length. In an embodiment, the second complementarity domain comprises 3 subdomains, which, in the 5' to 3' direction are: a 5' subdomain, a central subdomain, and a 3' subdomain. In an embodiment, the 5' subdomain is 3 to 25, e.g., 4 to 22, 4 to 18, or 4 to 10, or 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, or 25 nucleotides in length. In an embodiment, the central subdomain is 1, 2, 3, 4 or 5, e.g., 3, nucleotides in length. In an embodiment, the 3' subdomain is 4 to 9, e.g., 4, 5, 6, 7, 8 or 9 nucleotides in length. In an embodiment, the 5' subdomain and the 3' subdomain of the first complementarity domain, are respectively, complementary, e.g., fully complementary, with the 3' subdomain and the 5' subdomain of the second complementarity domain. In an embodiment, the proximal domain is 5 to 20 nucleotides in length. In an embodiment, the proximal domain can share homology with or be derived from a naturally occurring proximal domain. In an embodiment, it has at least 50% homology with an S. pyogenes, S. aureus or S. thermophilus, proximal domain. A broad spectrum of tail domains are suitable for use in gRNAs. In an embodiment, the tail domain is 0 (absent), 1, 2, 3, 4, 5, 6, 7, 8, 9, or 10 nucleotides in length. In some embodiments, the tail domain nucleotides are from or share homology with a sequence from the 5' end of a naturally occurring tail domain. In some embodiments, the tail domain includes sequences that are complementary to each other and which, under at least some physiological conditions, form a duplexed region. In some embodiments, the tail domain is absent or is 1 to 50 nucleotides in length. In some embodiments, the tail domain can share homology with or be derived from a naturally occurring proximal tail domain. In some embodiments, it has at least 50% homology with an S. pyogenes, S. aureus or S. thermophilus, tail domain. In an embodiment, the tail domain includes nucleotides at the 3' end that are related to the method of in vitro or in vivo transcription. In some embodiments, the gRNA is chemically modified. In some embodiments, any of the gRNAs provided herein comprise one or more nucleotides that are chemically modified. Chemical modifications of gRNAs have previously been described, and suitable chemical modifications include any modifications that are beneficial for gRNA function and do not measurably increase any undesired characteristics, e.g., off-target effects, of a given gRNA. Suitable chemical modifications include, for example, those that make a gRNA less susceptible to endo- or exonuclease catalytic activity, and include, without limitation, that the gRNA may comprise one or more modification chosen from phosphorothioate backbone modification, 2′-O-Me–modified sugars (e.g., at one or both of the 3’ and 5’ termini), 2’F- modified sugar, replacement of the ribose sugar with the bicyclic nucleotide-cEt, 3′thioPACE (MSP), or any combination thereof. Additional suitable gRNA modifications will be apparent to the skilled artisan based on this disclosure, and such suitable gRNA modification include, without limitation, those described, e.g., in Rahdar et al. PNAS December 22, 2015 112 (51) E7110-E7117 and Hendel et al., Nat Biotechnol.2015 Sep; 33(9): 985–989, each of which is incorporated herein by reference in its entirety. In some embodiments, a gRNA described herein comprises one or more 2′-O-methyl-3′-phosphorothioate nucleotides, e.g., at least 2, 3, 4, 5, or 62′-O-methyl-3′-phosphorothioate nucleotides. In some embodiments, a gRNA described herein comprises modified nucleotides (e.g., 2′-O-methyl-3′- phosphorothioate nucleotides) at the three terminal positions and the 5’ end and/or at the three terminal positions and the 3’ end. In some embodiments, the gRNA may comprise one or more modified nucleotides, e.g., as described in International Publication Nos. WO2017/214460, WO2016/089433, and WO2016/164356, which are incorporated by reference their entirety. In some embodiments, a gRNA described herein is chemically modified. For example, the gRNA may comprise one or more 2’-O modified nucleotides, e.g., 2’-O-methyl nucleotides. In some embodiments, the gRNA comprises a 2’-O modified nucleotide, e.g., 2’-O-methyl nucleotide at the 5’ end of the gRNA. In some embodiments, the gRNA comprises a 2’-O modified nucleotide, e.g., 2’-O-methyl nucleotide at the 3’ end of the gRNA. In some embodiments, the gRNA comprises a 2’-O-modified nucleotide, e.g., 2’-O- methyl nucleotide at both the 5’ and 3’ ends of the gRNA. In some embodiments, the gRNA is 2’-O-modified, e.g.2’-O-methyl-modified at the nucleotide at the 5’ end of the gRNA, the second nucleotide from the 5’ end of the gRNA, and the third nucleotide from the 5’ end of the gRNA. In some embodiments, the gRNA is 2’-O-modified, e.g.2’-O-methyl-modified at the nucleotide at the 3’ end of the gRNA, the second nucleotide from the 3’ end of the gRNA, and the third nucleotide from the 3’ end of the gRNA. In some embodiments, the gRNA is 2’-O-modified, e.g.2’-O-methyl-modified at the nucleotide at the 5’ end of the gRNA, the second nucleotide from the 5’ end of the gRNA, the third nucleotide from the 5’ end of the gRNA, the nucleotide at the 3’ end of the gRNA, the second nucleotide from the 3’ end of the gRNA, and the third nucleotide from the 3’ end of the gRNA. In some embodiments, the gRNA is 2’-O-modified, e.g.2’-O-methyl-modified at the second nucleotide from the 3’ end of the gRNA, the third nucleotide from the 3’ end of the gRNA, and at the fourth nucleotide from the 3’ end of the gRNA. In some embodiments, the nucleotide at the 3’ end of the gRNA is not chemically modified. In some embodiments, the nucleotide at the 3’ end of the gRNA does not have a chemically modified sugar. In some embodiments, the gRNA is 2’-O- modified, e.g.2’-O-methyl-modified, at the nucleotide at the 5’ end of the gRNA, the second nucleotide from the 5’ end of the gRNA, the third nucleotide from the 5’ end of the gRNA, the second nucleotide from the 3’ end of the gRNA, the third nucleotide from the 3’ end of the gRNA, and the fourth nucleotide from the 3’ end of the gRNA. In some embodiments, the 2’-O-methyl nucleotide comprises a phosphate linkage to an adjacent nucleotide. In some embodiments, the 2’-O-methyl nucleotide comprises a phosphorothioate linkage to an adjacent nucleotide. In some embodiments, the 2’-O-methyl nucleotide comprises a thioPACE linkage to an adjacent nucleotide. In some embodiments, the gRNA may comprise one or more 2’-O-modified and 3’phosphorous-modified nucleotide, e.g., a 2’-O-methyl 3’phosphorothioate nucleotide. In some embodiments, the gRNA comprises a 2’-O-modified and 3’phosphorous-modified, e.g., 2’-O-methyl 3’phosphorothioate nucleotide at the 5’ end of the gRNA. In some embodiments, the gRNA comprises a 2’-O-modified and 3’phosphorous-modified, e.g., 2’-O- methyl 3’phosphorothioate nucleotide at the 3’ end of the gRNA. In some embodiments, the gRNA comprises a 2’-O-modified and 3’phosphorous-modified, e.g., 2’-O-methyl 3’phosphorothioate nucleotide at the 5’ and 3’ ends of the gRNA. In some embodiments, the gRNA comprises a backbone in which one or more non-bridging oxygen atoms has been replaced with a sulfur atom. In some embodiments, the gRNA is 2’-O-modified and 3’phosphorous-modified, e.g.2’-O-methyl 3’phosphorothioate-modified at the nucleotide at the 5’ end of the gRNA, the second nucleotide from the 5’ end of the gRNA, and the third nucleotide from the 5’ end of the gRNA. In some embodiments, the gRNA is 2’-O-modified and 3’phosphorous-modified, e.g.2’-O-methyl 3’phosphorothioate-modified at the nucleotide at the 3’ end of the gRNA, the second nucleotide from the 3’ end of the gRNA, and the third nucleotide from the 3’ end of the gRNA. In some embodiments, the gRNA is 2’-O-modified and 3’phosphorous-modified, e.g.2’-O-methyl 3’phosphorothioate-modified at the nucleotide at the 5’ end of the gRNA, the second nucleotide from the 5’ end of the gRNA, the third nucleotide from the 5’ end of the gRNA, the nucleotide at the 3’ end of the gRNA, the second nucleotide from the 3’ end of the gRNA, and the third nucleotide from the 3’ end of the gRNA. In some embodiments, the gRNA is 2’-O-modified and 3’phosphorous-modified, e.g. 2’-O-methyl 3’phosphorothioate-modified at the second nucleotide from the 3’ end of the gRNA, the third nucleotide from the 3’ end of the gRNA, and the fourth nucleotide from the 3’ end of the gRNA. In some embodiments, the nucleotide at the 3’ end of the gRNA is not chemically modified. In some embodiments, the nucleotide at the 3’ end of the gRNA does not have a chemically modified sugar. In some embodiments, the gRNA is 2’-O-modified and 3’phosphorous-modified, e.g.2’-O-methyl 3’phosphorothioate-modified at the nucleotide at the 5’ end of the gRNA, the second nucleotide from the 5’ end of the gRNA, the third nucleotide from the 5’ end of the gRNA, the second nucleotide from the 3’ end of the gRNA, the third nucleotide from the 3’ end of the gRNA, and the fourth nucleotide from the 3’ end of the gRNA. In some embodiments, the gRNA may comprise one or more 2’-O-modified and 3’- phosphorous-modified, e.g., 2’-O-methyl 3’thioPACE nucleotide. In some embodiments, the gRNA comprises a 2’-O-modified and 3’phosphorous-modified, e.g., 2’-O-methyl 3’thioPACE nucleotide at the 5’ end of the gRNA. In some embodiments, the gRNA comprises a 2’-O-modified and 3’phosphorous-modified, e.g., 2’-O-methyl 3’thioPACE nucleotide at the 3’ end of the gRNA. In some embodiments, the gRNA comprises a 2’-O- modified and 3’phosphorous-modified, e.g., 2’-O-methyl 3’thioPACE nucleotide at the 5’ and 3’ ends of the gRNA. In some embodiments, the gRNA comprises a backbone in which one or more non-bridging oxygen atoms have been replaced with a sulfur atom and one or more non-bridging oxygen atoms have been replaced with an acetate group. In some embodiments, the gRNA is 2’-O-modified and 3’phosphorous-modified, e.g.2’-O-methyl 3’ thioPACE-modified at the nucleotide at the 5’ end of the gRNA, the second nucleotide from the 5’ end of the gRNA, and the third nucleotide from the 5’ end of the gRNA. In some embodiments, the gRNA is 2’-O-modified and 3’phosphorous-modified, e.g.2’-O-methyl 3’thioPACE-modified at the nucleotide at the 3’ end of the gRNA, the second nucleotide from the 3’ end of the gRNA, and the third nucleotide from the 3’ end of the gRNA. In some embodiments, the gRNA is 2’-O-modified and 3’phosphorous-modified, e.g.2’-O-methyl 3’thioPACE-modified at the nucleotide at the 5’ end of the gRNA, the second nucleotide from the 5’ end of the gRNA, the third nucleotide from the 5’ end of the gRNA, the nucleotide at the 3’ end of the gRNA, the second nucleotide from the 3’ end of the gRNA, and the third nucleotide from the 3’ end of the gRNA. In some embodiments, the gRNA is 2’-O-modified and 3’phosphorous-modified, e.g.2’-O-methyl 3’thioPACE-modified at the second nucleotide from the 3’ end of the gRNA, the third nucleotide from the 3’ end of the gRNA, and the fourth nucleotide from the 3’ end of the gRNA. In some embodiments, the nucleotide at the 3’ end of the gRNA is not chemically modified. In some embodiments, the nucleotide at the 3’ end of the gRNA does not have a chemically modified sugar. In some embodiments, the gRNA is 2’-O-modified and 3’phosphorous-modified, e.g.2’-O-methyl 3’thioPACE-modified at the nucleotide at the 5’ end of the gRNA, the second nucleotide from the 5’ end of the gRNA, the third nucleotide from the 5’ end of the gRNA, the second nucleotide from the 3’ end of the gRNA, the third nucleotide from the 3’ end of the gRNA, and the fourth nucleotide from the 3’ end of the gRNA. In some embodiments, the gRNA comprises a chemically modified backbone. In some embodiments, the gRNA comprises a phosphorothioate linkage. In some embodiments, one or more non-bridging oxygen atoms have been replaced with a sulfur atom. In some embodiments, the nucleotide at the 5’ end of the gRNA, the second nucleotide from the 5’ end of the gRNA, and the third nucleotide from the 5’ end of the gRNA each comprise a phosphorothioate linkage. In some embodiments, the nucleotide at the 3’ end of the gRNA, the second nucleotide from the 3’ end of the gRNA, and the third nucleotide from the 3’ end of the gRNA each comprise a phosphorothioate linkage. In some embodiments, the nucleotide at the 5’ end of the gRNA, the second nucleotide from the 5’ end of the gRNA, the third nucleotide from the 5’ end of the gRNA, the nucleotide at the 3’ end of the gRNA, the second nucleotide from the 3’ end of the gRNA, and the third nucleotide from the 3’ end of the gRNA each comprise a phosphorothioate linkage. In some embodiments, the second nucleotide from the 3’ end of the gRNA, the third nucleotide from the 3’ end of the gRNA, and at the fourth nucleotide from the 3’ end of the gRNA each comprise a phosphorothioate linkage. In some embodiments, the nucleotide at the 5’ end of the gRNA, the second nucleotide from the 5’ end of the gRNA, the third nucleotide from the 5’ end, the second nucleotide from the 3’ end of the gRNA, the third nucleotide from the 3’ end of the gRNA, and the fourth nucleotide from the 3’ end of the gRNA each comprise a phosphorothioate linkage. In some embodiments, the gRNA comprises a thioPACE linkage. In some embodiments, the gRNA comprises a backbone in which one or more non-bridging oxygen atoms have been replaced with a sulfur atom and one or more non-bridging oxygen atoms have been replaced with an acetate group. In some embodiments, the nucleotide at the 5’ end of the gRNA, the second nucleotide from the 5’ end of the gRNA, and the third nucleotide from the 5’ end of the gRNA each comprise a thioPACE linkage. In some embodiments, the nucleotide at the 3’ end of the gRNA, the second nucleotide from the 3’ end of the gRNA, and the third nucleotide from the 3’ end of the gRNA each comprise a thioPACE linkage. In some embodiments, the nucleotide at the 5’ end of the gRNA, the second nucleotide from the 5’ end of the gRNA, the third nucleotide from the 5’ end of the gRNA, the nucleotide at the 3’ end of the gRNA, the second nucleotide from the 3’ end of the gRNA, and the third nucleotide from the 3’ end of the gRNA each comprise a thioPACE linkage. In some embodiments, the second nucleotide from the 3’ end of the gRNA, the third nucleotide from the 3’ end of the gRNA, and at the fourth nucleotide from the 3’ end of the gRNA each comprise a thioPACE linkage. In some embodiments, the nucleotide at the 5’ end of the gRNA, the second nucleotide from the 5’ end of the gRNA, the third nucleotide from the 5’ end, the second nucleotide from the 3’ end of the gRNA, the third nucleotide from the 3’ end of the gRNA, and the fourth nucleotide from the 3’ end of the gRNA each comprise a thioPACE linkage. Some exemplary, non-limiting embodiments of modifications, e.g., chemical modifications, suitable for use in connection with the guide RNAs and genetic engineering methods provided herein have been described above. Additional suitable modifications, e.g., chemical modifications, will be apparent to those of skill in the art based on the present disclosure and the knowledge in the art, including, but not limited to those described in Hendel, A. et al., Nature Biotech., 2015, Vol 33, No.9; in International Publication No. WO2017/214460; WO2016/089433; and in WO2016/164356; each one of which is herein incorporated by reference in its entirety. Multiple gRNA Compositions In some embodiments, a gRNA described herein can be used in combination with one or more gRNA, e.g., for directing nucleases to one or more sites in a genome. In some embodiments, multiple gRNA described herein can be used in combination, e.g., for directing nucleases to multiple sites in a genome. In some embodiments, a gRNA described herein can be used in combination with a second gRNA, e.g., for directing nucleases to two sites in a genome. In some embodiments, a gRNA described herein can be used in combination with a third gRNA, e.g., for directing nucleases to three sites in a genome. In some embodiments, a gRNA described herein can be used in combination with a fourth gRNA, e.g., for directing nucleases to four sites in a genome. In some embodiments, a gRNA described herein can be used in combination with a fifth gRNA, e.g., for directing nucleases to five sites in a genome. In some embodiments, a gRNA described herein can be used in combination with a sixth gRNA, e.g., for directing nucleases to six sites in a genome. In some embodiments, a gRNA described herein can be used in combination with a seventh gRNA, e.g., for directing nucleases to seven sites in a genome. In some embodiments, a gRNA described herein can be used in combination with an eight gRNA, e.g., for directing nucleases to eight sites in a genome. In some embodiments, a gRNA described herein can be used in combination with a ninth gRNA, e.g., for directing nucleases to nine sites in a genome. In some embodiments, a gRNA described herein can be used in combination with a tenth gRNA, e.g., for directing nucleases to ten sites in a genome. In some embodiments, a gRNA described herein can be used in combination with more than tenth gRNA, e.g., for directing nucleases to more than ten sites in a genome. Exemplary Cell Surface Protein Editing Strategies Further examples of editing strategies targeting cell surface proteins of the present disclosure are provided herein. Exemplary Guide Nucleic Acid Targets In some embodiments, genetically engineered cells of the present disclosure are produced by contacting a hematopoietic cell with a gRNA comprising a targeting domain that is capable of binding to a target gene encoding a cell surface protein selected from a group consisting of: CD10; CD101; CD117; CD11A; CD11C; CD120B; CD123; CD13; CD133; CD152; CD200; CD205 (LY75); CD226; CD244 (SLAMF4); CD25; CD274; CD305 (LAIR1); CD32; CD33; CD34; CD366; CD38; CD41; CD42B; CD44; CD45RA; CD47; CD48; CD49D (ITGA4); CD49F; CD52; CD58; CD64; CD7; CD70; CD82; CD84; CD85; CD85K; CD86; CD9; CD96; CD99; CLL-1; EMR2 (ADGRE2); FR-B; GPR56; and IL1RAP. In some embodiments, genetically engineered cells of the present disclosure are produced by contacting a hematopoietic cell with a gRNA comprising a targeting domain that is capable of binding to a target gene encoding a cell surface protein selected from a group consisting of: CD10; CD11A; CD11C; CD152; CD200; CD274; CD33; CD34; CD42B; CD45RA; CD47; CD49D; CD49F; CD58; CD64; CD84; CD85K; CD9; CLL-1; GPR56; and IL1RAP. In some embodiments, genetically engineered cells of the present disclosure are produced by contacting a hematopoietic cell with a gRNA comprising a targeting domain that is capable of binding to a target gene encoding a cell surface protein selected from a group consisting of: CD10; CD117; CD11A; CD11C; CD120B; CD123; CD13; CD133; CD152; CD205; CD22; CD226; CD244; CD25; CD305; CD32; CD33; CD34; CD38; CD41; CD44; CD45RA; CD47; CD48; CD49D; CD49F; CD52; CD56; CD58; CD64; CD7; CD70; CD71; CD82; CD84; CD85K; CD86; CD96; CD99; CLL-1; EMR2; FR-B; GPR56; HLA-DR; and IL1RAP. In some embodiments, genetically engineered cells of the present disclosure are produced by contacting a hematopoietic cell with a first gRNA comprising a targeting domain that is capable of binding to a target gene encoding a first cell surface protein and a second gRNA comprising a targeting domain that is capable of binding to a target gene encoding a second cell surface protein selected from a group consisting of: CD274 and CD152; CD274 and CD226; CD152 and CD226; CD10 and CD49F; CD49F and GPR56; CD200 and CD34; CD34 and CD52; CD34 and CD133; GPR56 and CD133; CD42B and CD41; CD85K and CD64; CD85K and CD32; CD64 and CD32; CD64 and CD11C; CD64 and CD86; CD45RA and CD96; CD45RA and IL1RAP; CD45RA and CD123; CD84 and CD33; CD11C and IL1RAP; CD11C and CD33; CD11C and CD86; CD9 and IL1RAP; IL1RAP and CD33; IL1RAP and CD205; IL1RAP and CD305; IL1RAP and CD123; CD33 and CLL-1; CD33 and CD305; CD11A and CD86; CLL-1 and CD305; CD58 and CD305; CD49D and CD47; CD49D and CD99; CD49D and CD44; CD49D and CD305; CD47 and CD99; CD47 and EMR2; CD99 and CD44; CD99 and CD305; CD99 and EMR2; CD205 and CD305; CD205 and CD123; CD44 and CD305; CD44 and EMR2; CD244 and CD117; CD123 and EMR2; EMR2 and CD82; CD152 and CD120B; CD48 and CD11A; CD200 and CD25; CD34 and CD7; CD34 and CD13; CD52 and GPR56; CD52and CD133; CD52 and CD7; GPR56 and CD7; GPR56 and CD82; CD133 and CD7; CD133 and CD82; CD7 and CD13; CD101 and CD64; CD85 and CD70; CD85K and CD86; CD64 and FR-B; CD366 and CD96; CD25 and CD45RA; CD25 and CD96; CD9 and CD82; IL1RAP and CD58; CD11A and CD38; CD11A and CD44; CD11A and CD244; CD38 and CD305; CD38 and EMR2; CLL-1 and CD49D; CD58 and CD49D; CD47 and CD205; and CD47 and CD44. In some embodiments, genetically engineered cells of the present disclosure are produced by contacting a hematopoietic cell with a first gRNA comprising a targeting domain that is capable of binding to a target gene encoding a first cell surface protein and a second gRNA comprising a targeting domain that is capable of binding to a target gene encoding a second cell surface protein selected from a group consisting of: CD85K and CD64; CD45RA and IL1RAP; CD34 and CD133; IL1RAP and CD33; CD47 and CD99; CLL-1 and CD305; CD152 and CD226; CD45RA and CD96; CD45RA and CD123; CD49D and CD305; IL1RAP and CD305; CD58 and CD305; CD49D and CD47; CD33 and CD305; CD49D and CD44; CD64 and CD86; GPR56 and CD133; CD34 and CD52; EMR2 and CD82; CD64 and CD32; CD42B and CD41; CD64 and CD11C; CD99 and CD44; CD200 and CD34; CD11C and CD33; IL1RAP and CD123; CD11C and IL1RAP; CD44 and EMR2; CD205 and CD305; CD44 and CD305; CD49D and CD99; CD274 and CD226; CD49F and GPR56; CD9 and IL1RAP; CD99 and CD305; CD10 and CD49F; CD11A and CD86; CD33 and CLL-1; CD84 and CD33; CD244 and CD117; CD85K and CD32; CD47 and EMR2; CD274 and CD152; IL1RAP and CD205; CD11C and CD86; CD99 and EMR2; CD205 and CD123; and CD123 and EMR2. In some embodiments, genetically engineered cells of the present disclosure are produced by contacting a hematopoietic cell with a first gRNA comprising a targeting domain that is capable of binding to a target gene encoding a first cell surface protein and a second gRNA comprising a targeting domain that is capable of binding to a target gene encoding a second cell surface protein selected from a group consisting of: CD33 and CD205 (LY75); CD33 and CD244 (SLAMF4); CD33 and CD305 (LAIR1); CD33 and CD49D (ITGA4); CLL-1 and CD205 (LY75); CLL-1 and CD244 (SLAMF4); CLL-1 and CD305 (LAIR1); CLL-1 and CD49D (ITGA4); EMR2 and CD205 (LY75); EMR2 and CD244 (SLAMF4); EMR2 and CD305 (LAIR1); EMR2 and CD49D (ITGA4); CD38 and CD205 (LY75); CD38 and CD244 (SLAMF4); CD38 and CD305 (LAIR1); CD38 and CD49D (ITGA4); CD305 (LAIR1) and CD49D (ITGA4); CD305 (LAIR1) and CD205 (LY75); CD305 (LAIR1) and CD244 (SLAMF4); CD49D (ITGA4) and CD205 (LY75); CD49D (ITGA4) and CD244 (SLAMF4); and CD205 (LY75) and CD244 (SLAMF4). In some embodiments, a targeting domain which is complementary to a target nucleic acid in a cell surface protein is complementary to a target nucleic acid encoding human CD33 or a portion thereof, e.g., having a sequence at least 70%, 75%, 80%, 85%, 90%, 95%, 96%, 97%, 98%, or 99% identical thereto to the CD33 amino acid sequence set forth below: MPLLLLLPLLWAGALAMDPNFWLQVQESVTVQEGLCVLVPCTFFHPIPYYDKNSPVHGYW FREGAIISRDSPVATNKLDQEVQEETQGRFRLLGDPSRNNCSLSIVDARRRDNGSYFFRM ERGSTKYSYKSPQLSVHVTDLTHRPKILIPGTLEPGHSKNLTCSVSWACEQGTPPIFSWL
The nucleotide sequence encoding CD33 are genetically manipulated to delete any epitope of the protein (of the extracellular portion of CD33), or a fragment containing such, using conventional methods of nucleic acid manipulation. The amino acid sequences provided below are exemplary sequences of CD33 mutants that have been manipulated to lack each of the epitopes in Table 2. The amino acid sequence of the extracellular portion of CD33 is provided by SEQ ID NO: 1. The signal peptide is shown in italics and sites for manipulation are shown in underline and boldface. The transmembrane domain is shown in italics with underline. MPLLLLLPLL WAGALAMDPN FWLQVQESVT VQEGLCVLVP CTFFHPIPYY DKNSPVHGYW FREGAIISRD SPVATNKLDQ EVQEETQGRF RLLGDPSRNN CSLSIVDARR RDNGSYFFRM ERGSTKYSYK SPQLSVHVTD LTHRPKILIP GTLEPGHSKN LTCSVSWACE QGTPPIFSWL SAAPTSLGPR TTHSSVLIIT PRPQDHGTNL TCQVKFAGAG VTTERTIQLN VTYVPQNPTT GIFPGDGSGK QETRAGVVHG AIGGAGVTAL LALCLCLIFF IVKTHRRKAA RTAVGRNDTH PTTGSASPKH QKKSKLHGPT ETSSCSGAAP TVEMDEELHY ASLNFHGMNP SKDTSTEYSE VRTQ (SEQ ID NO: 1) The amino acid sequence of the extracellular portion of CD33 comprising a deletion of residues S248 through E252 is provided by SEQ ID NO: 2. The signal peptide is shown in italics and the transmembrane domain is shown in italics with underline. S248_E252insdelTARND; PROVEAN score = -1.916 MPLLLLLPLL WAGALAMDPN FWLQVQESVT VQEGLCVLVP CTFFHPIPYY DKNSPVHGYW FREGAIISRD SPVATNKLDQ EVQEETQGRF RLLGDPSRNN CSLSIVDARR RDNGSYFFRM ERGSTKYSYK SPQLSVHVTD LTHRPKILIP GTLEPGHSKN LTCSVSWACE QGTPPIFSWL SAAPTSLGPR TTHSSVLIIT PRPQDHGTNL TCQVKFAGAG VTTERTIQLN VTYVPQNPTT GIFPGDGTAR NDTRAGVVHG AIGGAGVTAL LALCLCLIFF IVKTHRRKAA RTAVGRNDTH PTTGSASPKH QKKSKLHGPT ETSSCSGAAP TVEMDEELHY ASLNFHGMNP SKDTSTEYSE VRTQ (SEQ ID NO: 2) The amino acid sequence of the extracellular portion of CD33 comprising a deletion of residues I47 through D51 is provided by SEQ ID NO: 3. The signal peptide is shown in italics and the transmembrane domain is shown in italics with underline. I47_D51insdelVPFFE; PROVEAN score = -1.672 MPLLLLLPLL WAGALAMDPN FWLQVQESVT VQEGLCVLVP CTFFHPVPFF EKNSPVHGYW FREGAIISRD SPVATNKLDQ EVQEETQGRF RLLGDPSRNN CSLSIVDARR RDNGSYFFRM ERGSTKYSYK SPQLSVHVTD LTHRPKILIP GTLEPGHSKN LTCSVSWACE QGTPPIFSWL SAAPTSLGPR TTHSSVLIIT PRPQDHGTNL TCQVKFAGAG VTTERTIQLN VTYVPQNPTT GIFPGDGTAR NDTRAGVVHG AIGGAGVTAL LALCLCLIFF IVKTHRRKAA RTAVGRNDTH PTTGSASPKH QKKSKLHGPT ETSSCSGAAP TVEMDEELHY ASLNFHGMNP SKDTSTEYSE VRTQ (SEQ ID NO: 3) The amino acid sequence of the extracellular portion of CD33 comprising a deletion of residues G249 through T253 is provided by SEQ ID NO: 4. The signal peptide is shown in italics and the transmembrane domain is shown in italics with underline. MPLLLLLPLL WAGALAMDPN FWLQVQESVT VQEGLCVLVP CTFFHPIPYY DKNSPVHGYW FREGAIISRD SPVATNKLDQ EVQEETQGRF RLLGDPSRNN CSLSIVDARR RDNGSYFFRM ERGSTKYSYK SPQLSVHVTD LTHRPKILIP GTLEPGHSKN LTCSVSWACE QGTPPIFSWL SAAPTSLGPR TTHSSVLIIT PRPQDHGTNL TCQVKFAGAG VTTERTIQLN VTYVPQNPTT GIFPGDGSRA GVVHGAIGGA GVTALLALCL CLIFFIVKTH RRKAARTAVG RNDTHPTTGS ASPKHQKKSK LHGPTETSSC SGAAPTVEMD EELHYASLNF HGMNPSKDTS TEYSEVRTQ (SEQ ID NO: 4) The amino acid sequence of the extracellular portion of CD33 comprising a deletion of residues K250 through R254 is provided by SEQ ID NO: 5. The signal peptide is shown in italics and the transmembrane domain is shown in italics with underline. MPLLLLLPLL WAGALAMDPN FWLQVQESVT VQEGLCVLVP CTFFHPIPYY DKNSPVHGYW FREGAIISRD SPVATNKLDQ EVQEETQGRF RLLGDPSRNN CSLSIVDARR RDNGSYFFRM ERGSTKYSYK SPQLSVHVTD LTHRPKILIP GTLEPGHSKN LTCSVSWACE QGTPPIFSWL SAAPTSLGPR TTHSSVLIIT PRPQDHGTNL TCQVKFAGAG VTTERTIQLN VTYVPQNPTT GIFPGDGSGA GVVHGAIGGA GVTALLALCL CLIFFIVKTH RRKAARTAVG RNDTHPTTGS ASPKHQKKSK LHGPTETSSC SGAAPTVEMD EELHYASLNF HGMNPSKDTS TEYSEVRTQ (SEQ ID NO: 5) The amino acid sequence of the extracellular portion of CD33 comprising a deletion of residues P48 through K52 is provided by SEQ ID NO: 6. The signal peptide is shown in italics and the transmembrane domain is shown in italics with underline. MPLLLLLPLL WAGALAMDPN FWLQVQESVT VQEGLCVLVP CTFFHPINSP VHGYWFREGA IISRDSPVAT NKLDQEVQEE TQGRFRLLGD PSRNNCSLSI VDARRRDNGS YFFRMERGST KYSYKSPQLS VHVTDLTHRP KILIPGTLEP GHSKNLTCSV SWACEQGTPP IFSWLSAAPT SLGPRTTHSS VLIITPRPQD HGTNLTCQVK FAGAGVTTER TIQLNVTYVP QNPTTGIFPG DGSGKQETRA GVVHGAIGGA GVTALLALCL CLIFFIVKTH RRKAARTAVG RNDTHPTTGS ASPKHQKKSK LHGPTETSSC SGAAPTVEMD EELHYASLNF HGMNPSKDTS TEYSEVRTQ (SEQ ID NO: 6) The amino acid sequence of the extracellular portion of CD33 comprising a deletion of residues Q251 through A255 is provided by SEQ ID NO: 7. The signal peptide is shown in italics and the transmembrane domain is shown in italics with underline. MPLLLLLPLL WAGALAMDPN FWLQVQESVT VQEGLCVLVP CTFFHPIPYY DKNSPVHGYW FREGAIISRD SPVATNKLDQ EVQEETQGRF RLLGDPSRNN CSLSIVDARR RDNGSYFFRM ERGSTKYSYK SPQLSVHVTD LTHRPKILIP GTLEPGHSKN LTCSVSWACE QGTPPIFSWL SAAPTSLGPR TTHSSVLIIT PRPQDHGTNL TCQVKFAGAG VTTERTIQLN VTYVPQNPTT GIFPGDGSGK GVVHGAIGGA GVTALLALCL CLIFFIVKTH RRKAARTAVG RNDTHPTTGS ASPKHQKKSK LHGPTETSSC SGAAPTVEMD EELHYASLNF HGMNPSKDTS TEYSEVRTQ (SEQ ID NO: 7) In some examples, provided herein are variants of CD33, which may comprise a deletion or mutation of a fragment of the protein that is encoded by any one of the exons of CD33, or a deletion or mutation in a non-essential epitope. The predicted structure of CD33 includes two immunoglobulin domains, an IgV domain and an IgC2 domain. In some embodiments, a portion of the immunoglobulin V domain of CD33 is deleted or mutated. In some embodiments, a portion of the immunoglobulin C domain of CD33 is deleted or mutated. In some embodiments, exon 2 of CD33 is deleted or mutated. In some embodiments, exon 3 of CD33 is deleted or mutated. In some embodiments, the CD33 variant lacks amino acid residues W11 to T139. In some embodiments, the CD33 variant lacks amino acid residues G13 to T139. In some embodiments, the deleted or mutated fragment overlaps or encompasses the epitope to which a cytotoxic agent binds, such as a cytotoxic agent described in PCT Publication No. WO2022/056497, WO2022/056490, or WO2019/178382, which are incorporated by reference in their entirety. In some embodiments, the epitope comprises amino acids 47-51 or 248-252 of CD33. In some embodiments, the epitope comprises amino acids 248-252, 47-51, 249-253, 250-254, 48-52, or 251-255 of the extracellular portion of CD33. In some embodiments, the genetically engineered hematopoietic stem cells have genetic edits in a CD33 gene, wherein exon 2 of CD33 is mutated or deleted. In some embodiments, exon 2 of CD33 is mutated or deleted (see the amino acid sequence of the exon 2-encoded fragment above). In some embodiments, the genetically engineered hematopoietic stem cells have genetic edits in a CD33 gene resulting in expression of CD33 with deleted or mutated exon 2 of CD33. In some embodiments, genetically engineered hematopoietic stem cells express CD33, in which exon 2 of CD33 is mutated or deleted. In some examples, the genetically engineered hematopoietic cells have a genetically engineered CD33 gene (e.g., a genetically engineered endogenous CD33 gene), wherein the engineering results in expression of a CD33 variant having the fragment encoded by exon 2 deleted (CD33ex2). In some embodiments, the CD33 genes edited HSCs expressing CD33 variant having the fragment encoded by exon 2 deleted also comprise a partial or complete deletion in the adjacent introns (intron 1 and intron 2) in addition to the deletion of exon 2. Exemplary amino acid sequence of CD33 mutants, with fragments G13-T139 deleted, is provided below (the junction of exon 1-encoded fragment and exon-3 encoded fragment is shown in boldface): MPLLLLLPLLWADLTHRPKILIPGTLEPGHSKNLTCSVSWACEQGTPPIFSWLSAAPTSLGPRTTHSS VLIITPRPQDHGTNLTCQVKFAGAGVTTERTIQLNVTYVPQNPTTGIFPGDGSGKQETRAGVVHGAIG GAGVTALLALCLCLIFFIVKTHRRKAARTAVGRNDTHPTTGSASPKHQKKSKLHGPTETSSCSGAAPT VEMDEELHYASLNFHGMNPSKDTSTEYSEVRTQ (SEQ ID NO: 12) MPLLLLLPLLWADLTHRPKILIPGTLEPGHSKNLTCSVSWACEQGTPPIFSWLSAAPTSLGPRTTHSS VLIITPRPQDHGTNLTCQVKFAGAGVTTERTIQLNVTYVPQNPTTGIFPGDGSGKQETRAGVVHGAIG GAGVTALLALCLCLIFFIVKTHRRKAARTAVGRNDTHPTTGSASPVR (SEQ ID NO: 16) While certain cells may express CD33 proteins lacking the fragment encoded by exon 2, the genetically engineered hematopoietic cells are different from such native cells in at least the aspect that these cells have undergone genome editing to modify a CD33 gene, such as an endogenous CD33 gene. In other words, the parent hematopoietic stem cells for producing the genetically engineered HSCs carry a CD33 gene that produces exon 2- containing transcripts. Genetically engineered hematopoietic stem cells carrying an edited CD33 gene that expresses this CD33 mutant are also within the scope of the present disclosure. Such cells may be a homogenous population containing cells expressing the same CD33 mutant (e.g., CD33ex2). Alternatively, the cells may be a heterogeneous population containing cells expressing different CD33 mutants (which may be due to heterogeneous editing/repairing events inside cells) or cells that do not express CD33 (CD33KO). In specific examples, the genetically engineered HSCs may be a heterogeneous population containing cells expressing CD33ex2 and cells that do not express CD33 (CD33KO). Genetically engineered hematopoietic stem cells having edited a CD33 gene can be prepared by a suitable genome editing method, such as those known in the art or disclosed herein. In some embodiments, the genetically engineered hematopoietic stem cells described herein can be generated using the CRISPR approach. See discussions herein. In certain examples, specific guide RNAs targeting a fragment of the CD33 gene (an exon sequence or an intron sequence) can be used in the CRISPR method. A CD33 pseudogene, known as SIGLEC22P (Gene ID 114195), is located upstream of the CD33 gene and shares a certain degree of sequence homology with the CD33 gene. gRNAs that cross-target regions in the pseudogene and regions in the CD33 gene may lead to production of aberrant gene products. Thus, in some embodiments, the gRNAs used in methods of editing CD33 via CRISPR preferably have low or no cross-reactivity with regions inside the pseudogene. In some instances, the gRNAs used in methods of editing CD33 via CRISPR preferably have low or no cross-reactivity with region(s) in Exon 1, intron 1 or Exon 2 of CD33 that are homologous to the pseudogene. Such gRNAs can be designed by comparing the sequences of the pseudogene and the CD33 gene to choose targeting sites inside the CD33 gene that have less or no homology to regions of the pseudogene. In one example, the pair of gRNA 18 (TTCATGGGTACTGCAGGGCA)(SEQ ID NO: 24) and gRNA 24 (GTGAGTGGCTGTGGGGAGAG)(SEQ ID NO: 25) are used for editing CD33 via CRISPR. Also provided herein are methods of genetically editing CD33 in hematopoietic cells (e.g., HSCs) via CRISPR, using one or more of the gRNAs described herein, for example, the pair of gRNA18 + gRNA24. As described herein, the length of a gRNA sequence may be modified (increased or decreased), for example, to enhance editing specificity and/or efficiency. In some embodiments, the length of gRNA 24 is 20 base pairs. In some embodiments, reducing the length of gRNA 24 may reduce the editing efficiency of gRNA 24 for CD33. In some embodiments, at least 40%, at least 50%, at least 60%, at least 70%, at least 75%, at least 80%, at least 85%, at least 90%, or at least 95% of copies of CD33 in the population of cells have a mutation. By way of example, a population can comprise a plurality of different CD33 mutations and each mutation of the plurality contributes to the percent of copies of CD33 in the population of cells that have a mutation. In some embodiments, the expression of CD33 on the genetically engineered hematopoietic cell is compared to the expression of CD33 on a naturally occurring hematopoietic cell (e.g., a wild-type counterpart). In some embodiments, the genetic engineering results in a reduction in the expression level of CD33 by at least50%, at least 60%, at least 70%, at least 80%, at least 90%, at least 91%, at least 92%, at least 93%, at least 94%, at least 95%, at least 96%, at least 97%, at least 98%, or at least 99% as compared to the expression of CD33 on a naturally occurring hematopoietic cell (e.g., a wild-type counterpart). For example, in some embodiments, the genetically engineered hematopoietic cell expresses less than 20%, less than 19%, less than 18%, less than 17%, less than 16%, less than 15%, less than 14%, less than 13%, less than 12%, less than 11%, less than 10%, less than 9%, less than 8%, less than 7%, less than 6%, less than 5%, less than 4%, less than 3%, less than 2%, less than or 1% of CD33 as compared to a naturally occurring hematopoietic cell (e.g., a wild-type counterpart). In some embodiments, the genetic engineering results in a reduction in the expression level of wild-type CD33 by at least 50%, at least 60%, at least 70%, at least 80%, at least 90%, at least 91%, at least 92%, at least 93%, at least 94%, at least 95%, at least 96%, at least 97%, at least 98%, or at least 99% as compared to the expression of the level of wild-type CD33 on a naturally occurring hematopoietic cell (e.g., a wild-type counterpart). That is, in some embodiments, the genetically engineered hematopoietic cell expresses less than 20%, less than 19%, less than 18%, less than 17%, less than 16%, less than 15%, less than 14%, less than 13%, less than 12%, less than 11%, less than 10%, less than 9%, less than 8%, less than 7%, less than 6%, less than 5%, less than 4%, less than 3%, less than 2%, or less than 1% of CD33 as compared to a naturally occurring hematopoietic cell (e.g., a wild-type counterpart). Genetically Engineered Hematopoietic Cells Expressing both CD33 and IL1RAP Mutants Also provided herein are a genetically engineered hematopoietic cells, such as HSCs, that have both the IL1RAP and CD33 genes edited. In some embodiments, provided herein is a population of genetically engineered hematopoietic HSCs in which at least 50% of the cells carry genetically edited IL1RAP and CD33 cells in at least on chromosome. In some embodiments, the edited IL1RAP gene is capable of expressing an IL1RAP mutant having at least one fragment encoded by one or more exons deleted. Alternatively or in addition, the edited CD33 gene is capable of expressing a CD33 mutant having at least one fragment encoded by one or more exons deleted. The genetically engineered hematopoietic cells having both IL1RAP and CD33 genes edited can be prepared by conventional methods. In some embodiments, such cells are prepared by CRISPR using a pair of gRNAs, one targeting IL1RAP and the other targeting CD33. In one example, the pair of gRNAs can be introduced into parent HSCs simultaneously and cells having genetic edits in IL1RAP and/or CD33 can be harvested for further use. Genetically Engineered Hematopoietic Cells Expressing both CD44 and EMR2 Mutants Also provided herein are a genetically engineered hematopoietic cells, such as HSCs, that have both the EMR2 and CD44 genes edited. In some embodiments, provide herein is a population of genetically engineered hematopoietic HSCs in which at least 50% of the cells carry genetically edited EMR2 and CD44 cells in at least one chromosome. In some embodiments, the edited EMR2 gene is capable of expressing a EMR2 mutant having at least one fragment encoded by a one or more exons deleted. Alternatively or in addition, the edited CD44 gene is capable of expressing a CD44 mutant having at least one fragment encoded by one or more exons deleted. The genetically engineered hematopoietic cells having both EMR2 and CD44 genes edited can be prepared by conventional methods. In some embodiments, such cells are prepared by CRISPR using a pair of gRNAs, one targeting EMR2 and the other targeting CD44. In one example, the pair of gRNAs can be introduced into parent HSCs simultaneously and cells having genetic edits in EMR2 and/or CD44 can be harvested for further use. Genetically Engineered Hematopoietic Cells Expressing CD305, CD49D, CD205, and CD244 Mutants Also provided herein are a genetically engineered hematopoietic cells, such as HSCs, that have genetic editing in a first gene encoding a first cell surface protein and a second gene encoding a second cell surface protein, wherein at least one of the cell surface proteins is selected from the group consisting of: CD305 (LAIR1); CD49D (ITGA4); CD205 (LY75); and CD244 (SLAMF4). In some embodiments, one of the cell surface proteins is selected from the group consisting of: CD305 (LAIR1); CD49D (ITGA4); CD205 (LY75); and CD244 (SLAMF4), and one of the cell surface proteins is selected from the group consisting of: CD10; CD101; CD117; CD11A; CD11C; CD120B; CD123; CD13; CD133; CD152; CD200; CD205 (LY75); CD226; CD244 (SLAMF4); CD25; CD274; CD305 (LAIR1); CD32; CD33; CD34; CD366; CD38; CD41; CD42B; CD44; CD45RA; CD47; CD48; CD49D (ITGA4); CD49F; CD52; CD58; CD64; CD7; CD70; CD82; CD84; CD85; CD85K; CD86; CD9; CD96; CD99; CLL-1; EMR2 (ADGRE2); FR-B; GPR56; and IL1RAP. In some embodiments, the first cell surface protein and the second cell surface protein is a combination selected from the group consisting of: CD33 and CD205 (LY75); CD33 and CD244 (SLAMF4); CD33 and CD305 (LAIR1); CD33 and CD49D (ITGA4); CLL-1 and CD205 (LY75); CLL-1 and CD244 (SLAMF4); CLL-1 and CD305 (LAIR1); CLL-1 and CD49D (ITGA4); EMR2 and CD205 (LY75); EMR2 and CD244 (SLAMF4); EMR2 and CD305 (LAIR1); EMR2 and CD49D (ITGA4); CD38 and CD205 (LY75); CD38 and CD244 (SLAMF4); CD38 and CD305 (LAIR1); CD38 and CD49D (ITGA4); CD305 (LAIR1) and CD49D (ITGA4); CD305 (LAIR1) and CD205 (LY75); CD305 (LAIR1) and CD244 (SLAMF4); CD49D (ITGA4) and CD205 (LY75); CD49D (ITGA4) and CD244 (SLAMF4); and CD205 (LY75) and CD244 (SLAMF4) In some embodiments, a population of genetically engineered hematopoietic HSCs in which at least 50% of the cells carry the genetic editing of the first gene in at least one chromosome and genetic editing of the second gene in at least one chromosome. In some embodiments, the edited first gene is capable of expressing a mutant of the first cell surface protein having at least one fragment encoded by a one or more exons deleted. Alternatively or in addition, the edited second gene is capable of expressing a mutant of the second cell surface protein having at least one fragment encoded by one or more exons deleted. For example, in some embodiments, a mutant of the first cell surface protein and/or a mutant of the second cell surface protein comprises a substitution, deletion, or insertion of an amino acid at one or more amino acid positions (e.g., 1-10, 10-20, 20-30, 30-40, 40-50, or more than 50 amino acid positions). In some embodiments, the mutated amino acid positions are located in an epitope (e.g., wherein the epitope is deleted in the mutant cell surface protein). In some embodiments, a mutant of the first cell surface protein and/or a mutant of the second cell surface protein comprises a deletion of one or more domains. In some embodiments, the first gene and/or the second gene is deleted. The genetically engineered hematopoietic cells having both first and second genes edited can be prepared by conventional methods. In some embodiments, such cells are prepared by CRISPR using a pair of gRNAs, one targeting the first gene and the other targeting the second gene. In one example, the pair of gRNAs can be introduced into parent HSCs simultaneously and cells having genetic edits in the first gene and/or the second gene can be harvested for further use. Masked lineage-specific cell surface antigens While many of the embodiments described herein involve mutations to the endogenous genes encoding lineage-specific cell surface antigens, it is understood that other approaches may be used instead of or in addition to mutation. For instance, a cell surface protein can be masked, e.g., to prevent or reduce its recognition by an immunotherapeutic agent. In some embodiments, masking is used on a cell surface protein that is difficult to mutate, e.g., because mutation of the gene is inefficient or is deleterious to cells expressing the mutant. In some embodiments, the lineage-specific cell surface protein is CD45. In some embodiments, masking is performed on a cell type described herein, e.g., an HSC or HPC. In some embodiments, masking is accomplished by expressing a masking protein in the cell of interest (e.g., by stably expressing DNA encoding the masking protein in the cell). In some embodiments, the masking protein comprises a binding domain (e.g., an antibody or antigen-binding fragment, e.g., an scFv) that binds the lineage-specific cell surface protein, e.g., in a way that reduces binding of an immunotherapeutic agent to the lineage-specific cell surface protein, e.g., by competing for binding at the same epitope. In some embodiments, the binding domain binds CD45. In some embodiments, the making protein further comprises one or more sequences that direct its localization to the surface of the cell. In some embodiments, the masking protein comprises a transmembrane domain fused to the binding domain. The masking protein may comprise a linker disposed between the transmembrane domain and the binding domain. The masking protein may be expressed at a level that binds to a sufficient amount of the cell surface protein that an immunotherapeutic agent displays reduced binding to and/or reduced killing of a cell expressing the masking protein compared to an otherwise similar cell that does not express the masking protein. In some embodiments, a cell described herein has reduced binding to (and/or reduced killing by) two different immunotherapeutic agents that recognize two different lineage- specific cell surface antigens. For instance, the cell may have a mutation at a gene encoding a first lineage-specific cell surface antigen, and may comprise a masking protein that masks a second lineage-specific cell surface antigen. In some embodiments, the cell may comprise a first masking protein that masks a first cell surface protein and a second masking protein that masks a second lineage-specific cell surface antigen. Cells Altered at One or More Lineage-Specific Cell Surface Antigens While many of the embodiments described herein involve two or more lineage- specific cell surface antigens, the application also discloses various cells altered with respect to a single lineage-specific cell surface antigen. For instance, the disclosure describes cells mutated at any one of the lineage-specific cell surface antigens described herein (e.g., mutated at one or both alleles of the lineage-specific cell surface antigen). The disclosure also describes cells expressing a single masking protein for a single lineage-specific cell surface antigen. Hematopoietic Stem Cells In some embodiments, the hematopoietic cells described herein are hematopoietic stem cells. Hematopoietic stem cells (HSCs) are capable of giving rise to both myeloid and lymphoid progenitor cells that further give rise to myeloid cells (e.g., monocytes, macrophages, neutrophils, basophils, dendritic cells, erythrocytes, platelets, etc) and lymphoid cells (e.g., T cells, B cells, NK cells), respectively. HSCs are characterized by the expression of the cell surface marker CD34 (e.g., CD34+), which can be used for the identification and/or isolation of HSCs, and absence of cell surface markers associated with commitment to a cell lineage. In some embodiments, the HSCs are obtained from a subject, such as a mammalian subject. In some embodiments, the mammalian subject is a non-human primate, a rodent (e.g., mouse or rat), a bovine, a porcine, an equine, or a domestic animal. In some embodiments, the HSCs are obtained from a human patient, such as a human patient having a hematopoietic malignancy. In some embodiments, the HSCs are obtained from a healthy donor. In some embodiments, the HSCs are obtained from the subject to whom the immune cells expressing the chimeric receptors will be subsequently administered. HSCs that are administered to the same subject from which the cells were obtained are referred to as autologous cells, whereas HSCs that are obtained from a subject who is not the subject to whom the cells will be administered are referred to as allogeneic cells. In some embodiments, the HSCs that are administered to the subject are allogeneic cells. In some embodiments, the HSCs are obtained from a donor having a HLA haplotype that is matched with the HLA haplotype of the subject. Human Leukocyte Antigen (HLA) encodes major histocompatibility complex (MHC) proteins in humans. MHC molecules are present on the surface of antigen-presenting cells as well as many other cell types and present peptides of self and non-self (e.g., foreign) antigens for immunosurveillance. However, HLA are highly polymorphic, which results in many distinct alleles. Different (foreign, non-self) alleles may be antigenic and stimulate robust adverse immune responses, particularly in organ and cell transplantation. HLA molecules that are recognized as foreign (non-self) can result in transplant rejection. In some embodiments, it is desirable to administer HSCs from donor that has the same HLA type as the patient to reduce the incidence of rejection. The HLA loci of a donor subject may be typed to identify an individual as a HLA- matched donor for the subject. Methods for typing the HLA loci will be evident to one of ordinary skill in the art and include, for example, serology (serotyping), cellular typing, gene sequencing, phenotyping, and PCR methods. An HLA from a donor is considered “matched” with the HLA of the subject if the HLA loci of the donor and the subject are identical or sufficiently similar such that an adverse immune response is not expected. In some embodiments, the HLA from the donor is not matched with the HLA of the subject. In some embodiments, the subject is administered HSCs that are not HLA matched with the HLA of the subject. In some embodiments, the subject is further administered one or more immunosuppressive agents to reduce or prevent rejection of the donor HSC cells. In some embodiments, the HSCs do not comprise a CART. HSCs may be obtained from any suitable source using convention means known in the art. In some embodiments, HSCs are obtained from a sample from a subject (or donor), such as bone marrow sample or from a blood sample. Alternatively or in addition, HSCs may be obtained from an umbilical cord. In some embodiments, the HSCs are from bone marrow, cord blood cells, or peripheral blood mononuclear cells (PBMCs). In general, bone marrow cells may be obtained from iliac crest, femora, tibiae, spine, rib or other medullary spaces of a subject (or donor). Bone marrow may be taken out of the patient and isolated through various separations and washing procedures known in the art. An exemplary procedure for isolation of bone marrow cells comprises the following steps: a) extraction of a bone marrow sample; b) centrifugal separation of bone marrow suspension in three fractions and collecting the intermediate fraction, or buffycoat; c) the buffycoat fraction from step (b) is centrifuged one more time in a separation fluid, commonly FicollTM, and an intermediate fraction which contains the bone marrow cells is collected; and d) washing of the collected fraction from step (c) for recovery of re-transfusable bone marrow cells. HSCs typically reside in the bone marrow but can be mobilized into the circulating blood by administering a mobilizing agent in order to harvest HSCs from the peripheral blood. In some embodiments, the subject (or donor) from which the HSCs are obtained is administered a mobilizing agent, such as granulocyte colony-stimulating factor (G-CSF). The number of the HSCs collected following mobilization using a mobilizing agent is typically greater than the number of cells obtained without use of a mobilizing agent. The HSCs for use in the methods described herein may express the cell surface protein of interest. Upon any of the modifications described herein (e.g., genetic modification or incubation with a blocking agent), the HSCs would not be targeted by the cytotoxicity agent also described herein. Alternatively, the HSCs for use in the methods described herein may not express the lineage-specific cell surface protein of interest (e.g., CD33); however, descendant cells differentiated from the HSCs (e.g., B cells) express the lineage-specific cell surface protein. Upon genetic modification, an endogenous gene of the HSCs coding for the lineage-specific cell surface protein may be disrupted at a region encoding a non-essential epitope of the lineage-specific cell surface protein. Descendant cells differentiated from such modified HSCs (e.g., in vivo) would express a modified lineage-specific cell surface protein having the non-essential epitope mutated such that they would not be targeted by the cytotoxicity agent capable of binding the non-essential epitope. In some embodiments, a sample is obtained from a subject (or donor) and is then enriched for a desired cell type (e.g. CD34+/CD33- cells). For example, PBMCs and/or CD34+ hematopoietic cells can be isolated from blood as described herein. Cells can also be isolated from other cells, for example by isolation and/or activation with an antibody binding to an epitope on the cell surface of the desired cell type. Another method that can be used includes negative selection using antibodies to cell surface markers to selectively enrich for a specific cell type without activating the cell by receptor engagement. Populations of HSC can be expanded prior to or after manipulating the HSC such that they don’t bind the cytotoxic agent or have reduced binding to the cytotoxic agent. The cells may be cultured under conditions that comprise an expansion medium comprising one or more cytokines, such as stem cell factor (SCF), Flt-3 ligand (Flt3L), thrombopoietin (TPO), Interleukin 3 (IL-3), or Interleukin 6 (IL-6). The cell may be expanded for about 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25 days or any range necessary. In some embodiments, the HSC are expanded after isolation of a desired cell population (e.g., CD34+/CD33-) from a sample obtained from a subject (or donor) and prior to manipulation (e.g., genetic engineering, contact with a blocking agent). In some embodiments, the HSC are expanded after genetic engineering, thereby selectively expanding cells that have undergone the genetic modification and lack the epitope (e.g., have a deletion or substitution of at least a portion of the epitope) of the cell surface protein to which the cytotoxic agent binds. In some embodiments, a cell (“a clone”) or several cells having a desired characteristic (e.g., phenotype or genotype) following genetic modification may be selected and independently expanded. In some embodiments, the HSC are expanded prior to contacting the HSC with a blocking agent that binds the epitope of the cell surface proteins, thereby providing a population of HSC expressing the cell surface proteins that cannot be bound by the cytotoxic agent due to blocking of the corresponding epitope by the blocking agent. II. Preparation of Genetically Engineered Hematopoietic Cells Any of the genetically engineering hematopoietic cells, such as HSCs, that carry edited genes of one or more cell surface proteins can be prepared by a routine method or by a method described herein. In some embodiments, the genetic engineering is performed using genome editing. As used herein, “genome editing” refers to a method of modifying the genome, including any protein-coding or non-coding nucleotide sequence, of an organism to knock out the expression of a target gene. In general, genome editing methods involve use of an endonuclease that is capable of cleaving the nucleic acid of the genome, for example at a targeted nucleotide sequence. Repair of the double-stranded breaks in the genome may be repaired introducing mutations and/or exogenous nucleic acid may be inserted into the targeted site. Genome editing methods are generally classified based on the type of endonuclease that is involved in generating double stranded breaks in the target nucleic acid. These methods include use of zinc finger nucleases (ZFN), transcription activator-like effector- based nuclease (TALEN), meganucleases, and CRISPR/Cas systems. Transcription Activator-Like-Effector-Based Nuclease (TALEN) In some embodiments, the modified cells are manipulated as described herein using the TALEN technology known in the art. In general, TALENs are engineered restriction enzymes that can specifically bind and cleave a desired target DNA molecule. A TALEN typically contains a Transcriptional Activator-Like Effector (TALE) DNA-binding domain fused to a DNA cleavage domain. The DNA binding domain may contain a highly conserved 33-34 amino acid sequence with a divergent 2 amino acid RVD (repeat variable dipeptide motif) at positions 12 and 13. The RVD motif determines binding specificity to a nucleic acid sequence and can be engineered according to methods known to those of skill in the art to specifically bind a desired DNA sequence. In one example, the DNA cleavage domain may be derived from the FokI endonuclease. The FokI domain functions as a dimer, requiring two constructs with unique DNA binding domains for sites in the target genome with proper orientation and spacing. TALENs specific to sequences in a target gene of interest (e.g., IL1RAP, CD33) can be constructed using any method known in the art. A TALEN specific to a target gene of interest can be used inside a cell to produce a double-stranded break (DSB). A mutation can be introduced at the break site if the repair mechanisms improperly repair the break via non-homologous end joining. For example, improper repair may introduce a frame shift mutation. Alternatively, a foreign DNA molecule having a desired sequence can be introduced into the cell along with the TALEN. Depending on the sequence of the foreign DNA and chromosomal sequence, this process can be used to correct a defect or introduce a DNA fragment into a target gene of interest, or introduce such a defect into the endogenous gene, thus decreasing expression of the target gene. In some embodiments, one or more population of hematopoietic cells is generated by genetic engineering of a cell surface protein (e.g., those described herein) using a TALEN. The genetically engineered hematopoietic cells may not express the lineage-specific cell surface antigen. Alternatively, the hematopoietic cells may be engineered to express an altered version of the lineage-specific cell surface antigen, e.g., having a deletion or mutation relative to the wild-type counterpart. Such a mutated cell surface protein may preserve a certain level of the bioactivity as the wild-type counterpart. In some embodiments, a population of hematopoietic cells containing a mutated CD33 is generated by genetic engineering using a TALEN. In some embodiments, exon 2 or exon 3 of CD33 is mutated using a TALEN. Zinc Finger Nuclease (ZFN) In some embodiments, the cells can be genetically manipulated using zinc finger (ZFN) technology known in the art. In general, zinc finger mediated genomic editing involves use of a zinc finger nuclease, which typically comprises a DNA binding domain (i.e., zinc finger) and a cleavage domain (i.e., nuclease). The zinc finger binding domain may be engineered to recognize and bind to any target gene of interest (e.g., IL1RAP, CD33) using methods known in the art and in particular, may be designed to recognize a DNA sequence ranging from about 3 nucleotides to about 21 nucleotides in length, or from about 8 to about 19 nucleotides in length. Zinc finger binding domains typically comprise at least three zinc finger recognition regions (e.g., zinc fingers). Restriction endonucleases (restriction enzymes) capable of sequence-specific binding to DNA (at a recognition site) and cleaving DNA at or near the site of binding are known in the art and may be used to form ZFN for use in genomic editing. For example, Type IIS restriction endonucleases cleave DNA at sites removed from the recognition site and have separable binding and cleavage domains. In one example, the DNA cleavage domain may be derived from the FokI endonuclease. In some embodiments, one or more population of hematopoietic cells is generated by genetic engineering of a cell surface protein (e.g., those described herein) using a ZFN. The genetically engineered hematopoietic cells may not express the cell surface protein. Alternatively, the hematopoietic cells may be engineered to express an altered version of the cell surface protein, e.g., having a deletion or mutation relative to the wild-type counterpart. Such a mutated cell surface protein may preserve a certain level of the bioactivity as the wild- type counterpart. In some examples, a population of hematopoietic cells containing a mutated CD33 is generated by genetic engineering using a ZFN. In some embodiments, exon 2 or exon 3 of CD33 is mutated using a ZFN. CRISPR/Cas Systems One exemplary suitable genome editing technology comprises the use of a CRISPR/Cas nuclease to introduce targeted single- or double-stranded DNA breaks in the genome of a cell, which trigger cellular repair mechanisms, such as, for example, nonhomologous end joining (NHEJ), microhomology-mediated end joining (MMEJ, also sometimes referred to as “alternative NHEJ” or “alt-NHEJ”), or homology-directed repair (HDR) that typically result in an altered nucleic acid sequence (e.g., via nucleotide or nucleotide sequence insertion, deletion, inversion, or substitution) at or immediately proximal to the site of the nuclease cut. See, Yeh et al. Nat. Cell. Biol. (2019) 21: 1468-1478; e.g., Hsu et al. Cell (2014) 157: 1262-1278; Jasin et al. DNA Repair (2016) 44: 6-16; Sfeir et al. Trends Biochem. Sci. (2015) 40: 701-714. Another exemplary suitable genome editing technology is “base editing,” which includes the use of a base editor, e.g., a nuclease-impaired or partially nuclease impaired enzyme (e.g., RNA-guided CRISPR/Cas protein) fused to a deaminase that targets and deaminates a specific nucleobase, e.g., a cytosine or adenosine nucleobase of a C or A nucleotide, which, via cellular mismatch repair mechanisms, results in a change from a C to a T nucleotide, or a change from an A to a G nucleotide. See, e.g., Komor et al. Nature (2016) 533: 420-424; Rees et al. Nat. Rev. Genet. (2018) 19(12): 770-788; Anzalone et al. Nat. Biotechnol. (2020) 38: 824-844. Yet another exemplary suitable genome editing technology includes “prime editing,” which includes the introduction of new genetic information, e.g., an altered nucleotide sequence, into a specifically targeted genomic site using a catalytically impaired or partially catalytically impaired nuclease (e.g., a CRISPR/Cas nuclease), fused to an engineered reverse transcriptase (RT) domain. The Cas/RT fusion is targeted to a target site within the genome by a guide RNA that also comprises a nucleic acid sequence encoding the desired edit, and that can serve as a primer for the RT. See, e.g., Anzalone et al. Nature (2019) 576 (7785): 149-157. In some embodiments, a genetically engineered cell described herein is made using gene editing technology, also referred to herein as DNA-editing technology, e.g., CRISPR/Cas-based DNA editing as described herein. Exemplary CRISPR/Cas systems are described herein, and additional suitable CRISPR/Cas systems will be apparent to the skilled artisan based on the present disclosure and the knowledge in the art. In some embodiments, a gRNA targeting a gene of interest (e.g. a cell surface protein) described herein is complexed with a Cas9 nuclease. Various Cas9 nuclease orthologs or variants can be used. In some embodiments, a Cas9 nuclease is selected that has the desired PAM specificity to target the gRNA/Cas9 nuclease complex to the target domain in the gene of interest (e.g., cell surface antigen). In some embodiments, genetically engineering a cell also comprises introducing one or more (e.g., 1, 2, 3 or more) Cas9 nucleases into the cell. Cas Nucleases A “Cas nuclease” as that term is used herein, refers to a CRISPR/Cas nuclease (e.g., Cas9) that can interact with a gRNA and, in concert with the gRNA, home or localize to a site which comprises a target domain. Cas nucleases include naturally occurring Cas nucleases and engineered, altered, or modified Cas nucleases that differ, e.g., by at least one amino acid residue, from a naturally occurring Cas nuclease. Cas9 nucleases of a variety of species can be used in the methods and compositions described herein. In various embodiments, the Cas9 nuclease is of, or derived from, Streptococcus pyogenes (SpCas9), Staphylococcus aureus (SaCas9), or Streptococcus thermophilus (StCas9). Additional suitable Cas9 nucleases include those of, or derived from, Staphylococcus aureus, Neisseria meningitidis (NmCas9), Acidovorax avenae, Actinobacillus pleuropneumoniae, Actinobacillus succinogenes, Actinobacillus suis, Actinomyces sp., Cycliphilus denitrificans, Aminomonas paucivorans, Bacillus cereus, Bacillus smithii, Bacillus thuringiensis, Bacteroides sp., Blastopirellula marina, Bradyrhizobium sp., Brevibacillus laterosporus, Campylobacter coli, Campylobacter jejuni (CjCas9), Campylobacter lari, Candidatus puniceispirillum, Clostridium cellulolyticum, Clostridium perfringens, Corynebacterium accolens, Corynebacterium diphtheria, Corynebacterium matruchotii, Dinoroseobacter shibae, Eubacterium dolichum, gamma proteobacterium, Gluconacetobacter diazotrophicus, Haemophilus parainfluenzae, Haemophilus sputorum, Helicobacter canadensis, Helicobacter cinaedi, Helicobacter mustelae, Ilyobacter polytropus, Kingella kingae, Lactobacillus crispatus, Listeria ivanovii, Listeria monocytogenes, Listeriaceae bacterium, Methylocystis sp., Methylosinus trichosporium, Mobiluncus mulieris, Neisseria bacilliformis, Neisseria cinerea, Neisseria flavescens, Neisseria lactamica, Neisseria sp., Neisseria wadsworthii, Nitrosomonas sp., Parvibaculum lavamentivorans, Pasteurella multocida, Phascolarctobacterium succinatutens, Ralstonia syzygii, Rhodopseudomonas palustris, Rhodovulum sp., Simonsiella muelleri, Sphingomonas sp., Sporolactobacillus vineae, Staphylococcus lugdunensis, Streptococcus sp., Subdoligranulum sp., Tistrella mobilis, Treponema sp., or Verminephrobacter eiseniae. In some embodiments, catalytically impaired, or partially impaired, variants of such Cas9 nucleases may be used. Additional suitable Cas9 nucleases, and nuclease variants, will be apparent to those of skill in the art based on the present disclosure. The disclosure is not limited in this respect. In some embodiments, the Cas9 nuclease is a naturally occurring Cas9 nuclease. In some embodiments, the Cas9 nuclease is an engineered, altered, or modified Cas9 nuclease that differs, e.g., by at least one amino acid residue, from a reference sequence, e.g., the most similar naturally occurring Cas9 nuclease or a sequence of Table 50 of International Publication No. WO 2015/157070, which is herein incorporated by reference in its entirety. In some embodiments, the Cas nuclease comprises Cpf1 or a fragment or variant thereof. A naturally occurring Cas9 nuclease typically comprises two lobes: a recognition (REC) lobe and a nuclease (NUC) lobe; each of which further comprises domains described, e.g., in International Publication No. WO 2015/157070, e.g., in Figs.9A-9B therein (which application is incorporated herein by reference in its entirety). The REC lobe comprises the arginine-rich bridge helix (BH), the REC1 domain, and the REC2 domain. The REC lobe appears to be a Cas9-specific functional domain. The BH domain is a long alpha helix and arginine rich region and comprises amino acids 60-93 of the sequence of S. pyogenes Cas9. The REC1 domain is involved in recognition of the repeat:anti-repeat duplex, e.g., of a gRNA or a tracrRNA. The REC1 domain comprises two REC1 motifs at amino acids 94 to 179 and 308 to 717 of the sequence of S. pyogenes Cas9. These two REC1 domains, though separated by the REC2 domain in the linear primary structure, assemble in the tertiary structure to form the REC1 domain. The REC2 domain, or parts thereof, may also play a role in the recognition of the repeat: anti-repeat duplex. The REC2 domain comprises amino acids 180-307 of the sequence of S. pyogenes Cas9. The NUC lobe comprises the RuvC domain (also referred to herein as RuvC-like domain), the HNH domain (also referred to herein as HNH-like domain), and the PAM- interacting (PI) domain. The RuvC domain shares structural similarity to retroviral integrase superfamily members and cleaves a single strand, e.g., the non-complementary strand of the target nucleic acid molecule. The RuvC domain is assembled from the three split RuvC motifs (RuvC I, RuvCII, and RuvCIII, which are often commonly referred to in the art as RuvCI domain, or N-terminal RuvC domain, RuvCII domain, and RuvCIII domain) at amino acids 1-59, 718-769, and 909-1098, respectively, of the sequence of S. pyogenes Cas9. Similar to the REC1 domain, the three RuvC motifs are linearly separated by other domains in the primary structure, however in the tertiary structure, the three RuvC motifs assemble and form the RuvC domain. The HNH domain shares structural similarity with HNH endonucleases, and cleaves a single strand, e.g., the complementary strand of the target nucleic acid molecule. The HNH domain lies between the RuvC II-III motifs and comprises amino acids 775-908 of the sequence of S. pyogenes Cas9. The PI domain interacts with the PAM of the target nucleic acid molecule, and comprises amino acids 1099-1368 of the sequence of S. pyogenes Cas9. Crystal structures have been determined for naturally occurring bacterial Cas9 nucleases (Jinek et al., Science, 343(6176): 1247997, 2014) and for S. pyogenes Cas9 with a guide RNA (e.g., a synthetic fusion of crRNA and tracrRNA) (Nishimasu et al., Cell, 156:935-949, 2014; and Anders et al., Nature, 2014, doi: 10.1038/naturel3579). In some embodiments, a Cas nuclease described herein has nuclease activity, e.g., double strand break activity in or directly proximal to a target site. In some embodiments, the Cas9 nuclease has been modified to inactivate one of the catalytic residues of the nuclease. In some embodiments, the Cas9 nuclease is a nickase and produces a single stranded break. See, e.g., Dabrowska et al. Frontiers in Neuroscience (2018) 12(75). It has been shown that one or more mutations in the RuvC and HNH catalytic domains of the enzyme may improve Cas9 efficiency. See, e.g., Sarai et al. Currently Pharma. Biotechnol. (2017) 18(13). In some embodiments, the Cas9 nuclease is fused to a second domain, e.g., a domain that modifies DNA or chromatin, e.g., a deaminase or demethylase domain. In some such embodiments, the Cas9 nuclease is modified to eliminate its nuclease activity. In some embodiments, the Cas nuclease is modified to enhance specificity of the enzyme (e.g., reduce off-target effects, maintain robust on-target cleavage). In some embodiments, the Cas9 nuclease is an enhanced specificity Cas9 variant (e.g., eSPCas9). See, e.g., Slaymaker et al. Science (2016) 351 (6268): 84-88. In some embodiments, the Cas9 nuclease is a high fidelity Cas9 variant (e.g., SpCas9-HF1). See, e.g., Kleinstiver et al. Nature (2016) 529: 490-495. As used herein, the term “protospacer adjacent motif” or “PAM” refers to a DNA sequence that may be required for a nuclease and a gRNA (e.g., Cas9/sgRNA) to form an R- loop to interrogate a specific DNA sequence through Watson-Crick pairing of its guide RNA with the genome. The PAM specificity may be a function of the DNA-binding specificity of the nuclease, e.g., Cas9 protein, e.g., a “protospacer adjacent motif recognition domain” at the C-terminus of Cas9. In some embodiments, the PAM comprises a 2 to 6 base pair DNA sequence immediately downstream or upstream of the target site in the genome, which may be recognized directly by a Cas nuclease, e.g., a Cas9, to promote cleavage of the target site by the Cas nuclease, or in the case of nuclease-deficient Cas9 allows binding to the DNA at that locus. In some embodiments, the PAM can be a 5' PAM (i.e., located upstream of the 5' end of the protospacer). In other embodiments, the PAM can be a 3' PAM (i.e., located downstream of the 5' end of the protospacer). As used herein, the term “Cas nuclease PAM” refers to a DNA sequence that may be required to home or localize a CRISPR/Cas nuclease (e.g., Cas9 or other Cas nucleases, such as Cas12a (Cpf1) or Cas12b) interacting with a gRNA to sequence comprising a target domain. Non-limiting examples of CRISPR/Cas nucleases and their corresponding consensus PAM are described herein. For example, the Cas9 endonuclease from Streptococcus pyogenes (SpyCas9) can recognize a NGG consensus PAM (e.g., a 5’-NGG on the 3’ end of the gRNA sequence) (N = any base). SpyCas9 can also weakly recognize NGA, NNGG, and a selection of other sequences. The SpGCas9 (a variant of SpyCas9) can recognize a NGN (N= any base) consensus PAM. The SpRYCas9 (a variant of SpGCas9) can recognize a NRN (N= any base; R= A,G) consensus PAM. SpRYCas9 can also weakly some NYN (N= any base; Y= C or T) sequences. The Cas9 nuclease from the CRISPR1 locus (Sth1Cas9) in the model lactic-acid bacterium Streptococcus thermophilus can recognize a NNATAAW (W = A, T; N= any base) consensus PAM. The Cas9 nuclease from the pathogen Staphylococcus aureus (SauCas9) can recognize an NNGRRT consensus PAM (R = A, G; N= any base). The Cas9 nuclease from the pathogen Neisseria meningitidis (NmeCas9) can recognize a NNNNGATT consensus PAM (N= any base). The MAD7 endonuclease from Eubacterium rectale can recognize 5’- TTTV on the 5’ end of the gRNA sequence, and to some extent 5’-YTTV and YTTN. The Cpf1 endonuclease from Acidaminococcus sp. and Lachnospiraceae sp. recognize 5’-TTTN and the Cpf1 endonuclease from Francisella novicide can recognize 5’-TTN-3’ on the 5’ end of the gRNA. Various Cas9 nucleases are known in the art and may be obtained from various sources and/or engineered/modified to modulate one or more activities or specificities of the enzymes. In some embodiments, the Cas9 nuclease has been engineered/modified to recognize one or more PAM sequence. In some embodiments, the Cas9 nuclease has been engineered/modified to recognize one or more PAM sequence that is different than the PAM sequence the Cas9 nuclease recognizes without engineering/modification. In some embodiments, the Cas9 nuclease has been engineered/modified to reduce off-target activity of the enzyme. In some embodiments, the nucleotide sequence encoding the Cas9 nuclease is modified further to alter the specificity of the nuclease activity (e.g., reduce off-target cleavage, decrease the nuclease activity or lifetime in cells, increase homology-directed recombination and reduce non-homologous end joining). See, e.g., Komor et al. Cell (2017) 168: 20-36. In some embodiments, the nucleotide sequence encoding the Cas9 nuclease is modified to alter the PAM recognition of the nuclease. For example, the Cas9 nuclease SpCas9 recognizes PAM sequence NGG, whereas relaxed variants of the SpCas9 comprising one or more modifications of the nuclease (e.g., VQR SpCas9, EQR SpCas9, VRER SpCas9) may recognize the PAM sequences NGA, NGAG, NGCG. PAM recognition of a modified Cas9 nuclease is considered “relaxed” if the Cas9 nuclease recognizes more potential PAM sequences as compared to the Cas9 nuclease that has not been modified. For example, the Cas9 nuclease SaCas9 recognizes PAM sequence NNGRRT, whereas a relaxed variant of the SaCas9 comprising one or more modifications (e.g., KKH SaCas9) may recognize the PAM sequence NNNRRT. In one example, the Cas9 nuclease FnCas9 recognizes PAM sequence NNG, whereas a relaxed variant of the FnCas9 comprising one or more modifications of the nuclease (e.g., RHA FnCas9) may recognize the PAM sequence YG. In one example, the Cas nuclease is a Cpf1 nuclease comprising substitution mutations S542R and K607R and recognize the PAM sequence TYCV. In one example, the Cas nuclease is a Cpf1 nuclease comprising substitution mutations S542R, K607R, and N552R and recognize the PAM sequence TATV. See, e.g., Gao et al. Nat. Biotechnol. (2017) 35(8): 789-792. In some embodiments, a Cas nuclease recognizes a PAM selected from: a SpyCas9 PAM, a SpGCas9 PAM, a SpRYCas9 PAM, a Sth1Cas9 PAM, a SauCas9 PAM, a NmeCas9 PAM, a RspCas9 PAM, a Cca1Cas9 PAM, a PspCas9 PAM, a OrhCas9 PAM, a ScCas9 PAM, a SmacCas9 PAM, a TdeCas9 PAM, a Nme2Cas9 PAM, a CjeCas9 PAM, a SmuCas9 PAM, a Smu2Cas9 PAM, a PmuCas9 PAM, a SpaCas9 PAM, a NciCas9 PAM, a ClaCas9 PAM, a PlaCas9 PAM, a CdCas9 PAM, a IgnaviCas9 PAM, a ThermoCas9 PAM, a GeoCas9 PAM, a G. LC300 Cas9 PAM, a AceCas9 PAM, a Sth3Cas9 PAM, a BlatCas9 PAM, a FnCas9 PAM, a LfeCas9 PAM, a LpnCas9 PAM, a KhuCas9 PAM, a AinCas9 PAM, a CglCas9 PAM, a Esp1Cas9 PAM, a Esp2Cas9 PAM, a FmaCas9 PAM, a LceCas9 PAM, a LrhCas9 PAM, a Lsp1Cas9 PAM, a Lsp2Cas9 PAM, a PacCas9 PAM, a TbaCas9 PAM, a TpuCas9 PAM, a VpaCas9 PAM, a EfaCas9 PAM, a EitCas9 PAM, a LanCas9 PAM, a LmoCas9 PAM, a Sag1Cas9 PAM, a Sag2Cas9 PAM, a SdyCas9 PAM, a Seq1Cas9 PAM, a Seq2Cas9 PAM, a SgaCas9 PAM, a Smu3Cas9 PAM, a SraCas9 PAM, a BniCas9 PAM, a EceCas9 PAM, a EdoCas9 PAM, a FhoCas9 PAM, a MgaCas9 PAM, a MseCas9 PAM, a SgoCas9 PAM, a SmaCas9 PAM, a SsaCas9 PAM, a SsiCas9 PAM, a SsuCas9 PAM, a Sth1ACas9 PAM, a TspCas9 PAM, a BokCas9 PAM, a CcoCas9 PAM, a CpeCas9 PAM, a DdeCas9 PAM, a Ghc2Cas9 PAM, a Ghy3Cas9 PAM, a Ghy4Cas9 PAM, a GspCas9 PAM, a KkiCas9 PAM, a NspCas9 PAM, a TmoCas9 PAM, a NsaCas9 PAM, a JpaCas9 PAM, a BboCas9 PAM, a Cca2Cas9 PAM, a Cme2Cas9 PAM, a Cme3Cas9 PAM, a Cme4Cas9 PAM, a CsaCas9 PAM, a Ghc1Cas9 PAM, a GheCas9 PAM, a Ghh1Cas9 PAM, a Ghh2Cas9 PAM, a Ghy1Cas9 PAM, a MscCas9 PAM, a SdoCas9 PAM, a SpacCas9 PAM, a CgaCas9 PAM, a Cme1Cas9 PAM, a FfrCas9 PAM, a Ghy2Cas9 PAM, a PhiCas9 PAM, a WviCas9 PAM, a CcCas9 PAM, a FnCas12a PAM, a AsCas12a PAM, a HkCas12a PAM, a PiCas12a PAM, a PdCas12a PAM, a LbCas12a PAM, a Lb2Cas12a or Lb5Cas12a PAM, a CmtCas12a PAM, a MbCas12a PAM, a TsCas12a PAM, a Pb2Cas12a PAM, a MlCas12a PAM, a Mb2Cas12a PAM, a Mb3Cas12a PAM, a CmaCas12a PAM, a BsCas12a PAM, a BfCas12a PAM, a BoCas12a PAM, a Adurb193Cas12a PAM, a Adurb336Cas12a PAM, a Fn3Cas12a PAM, a Lb6Cas12a PAM, a EcCas12a PAM, a PsCas12a PAM, a McCas12a PAM, a AacCas12b PAM, a BthCas12b PAM, a AkCas12b PAM, a Ebcas12b PAM, a bvCas12b PAM, a BhCas12b PAM, a LsCas12b PAM, a BrCas12b PAM, a Cas12c1 PAM, a Cas12c2 PAM, a OspCas12c PAM, a Cas12d.15 PAM, a Cas12d.1 (CasY.1) PAM, a DpbCas12e (DpbCasX) PAM, a PlmCas12e (PlmCasX) PAM, a Mi1Cas12f2 PAM, a Un1Cas12f1 PAM, a Un2Cas12f1 PAM, a Mi2Cas12f2 PAM, a AuCas12f2 PAM, a PtCas12f1 PAM, a AsCas12f1 PAM, a RuCas12f1 PAM, a SpCas12f1 PAM, a CnCas12f1 PAM, a Cas12h1 PAM, a Cas12i1 PAM, a Cas12i2 PAM, a Cas12j-1 (CasPhi-1) PAM, a Cas12j-2 (CasPhi-2) PAM, a Cas12j-3 (CasPhi-3) PAM, a ShCas12k PAM, or a AcCas12k PAM. In some embodiments, a Cas nuclease recognizes a PAM comprising, and/or consisting of a nucleotide sequence selected from: NGG; NNAGAAW; NNGRRT; NNNNGATT; NVNDCCY; BRTTTTT; NR(A or G)TTTT; NNAAAR(G or A); N(N or A)G; NAAN; NAAAAY; NHDTCCA; NNNVRYM; NNNNRYAC; NAA; GNNNNCNNA; NNGTGA; NNNNGTA; NNGGG; NNNCAT; NNRHHHY; NRRNAT; NNNNCNAA; NNNNCMCA; NNNNCRAA; NNNNGMAA; NNNCC; NGGNG; NNNNCNDD; NYAAA; NRGNN; N(C or D)GGN(T or A or G or C)NN; NRTAW; N(C or K or A)AARC; NAAAG; NV(A or G or C)R(A or G)ACCN; NNGAC; NATGNT; N(T or V)NTAAW(A or T); NNGW(A or T)AY(T or C); NCAA(H(Y or A)B(Y or G); NH(T or C or A)AAAA; NNNATTT; NATAWN(A or T or S); NATARCH; B(T or G or C)GGD(A or T or G)TNN; N(G or T or M)GGAH(T or A or C)N(A or C or K)N; NRG; N(B or A)GG; NGGD(A or K)W(T or A); N(T or C or R)AGAN(A or K or C)NN; NGGD(A or T or G)H(T or M); NGGDT; NGGD(A or T or G)GNN; NNGTAM(A or C)Y; NNGH(W or C)AAA; NTGAR(G or A)N(A or Y or G)N(Y or R); NNGAAAN; NNGAD; NHARMC; NNAAAG; NHGYNAN(A or B); NNAGAAA; NHAAAAA; NH(T or M)AAAAA; NHGYRAA; NNAAACN; NN(H or G)D(A or K)GGDN(A or B); NNNNCTA; NNNNCVGAA; NNNNGYAA; NNNNATN(W or S)ANN; NNWHR(G or A)TA(not G)AA; YHHNGTH; NNNNCDAANN; NNNNCTAA; N(C or D)NNTCCN; NNNNCCAA; NAGRGN(T or V)N(T or C); NNAH(T or M)I; CN(C or W or G)AV(A or S)GAC; NAR(G or A)H(W or C)H(A or T or C)GN(C or T or R); NAGNGC; NATCCTN; NGTGANN; HGCNGCR; NAR(A or G)W(T or A)AC; N(C or D)M(A or C)RN(A or B)AY(C or T); NNNCAC; BGGGTCD; NNRRCC; NRRNTT; KARDAT; BRRTTTW; NARNCCN; NAR(A or G)TC; NAAN(A or T or S)RCN; HHAAATD; NNNNGNA; TTV; TTTV; YYV; KKYV; TTTM; TTYV; TTTN; TTTTA; TTN; BTTV; YTV; YTN; NYTV; DTTD; ATTN; RTTNT; HATT; ATTW; RTTN; TVT; TG; TN; TR; TA; TTCN; TTAT; TTTR; TTR; YTTR; YTTN; CTT; TTC; CCD; RTR; VTTR; TBN; VTTN; NGTT; CGTT; AGG; CGG; GTT, or RGTG, wherein “N” is any nucleotide or base, “W” is adenine (A) or thymine (T), “R” is A or guanine (G), “V” is A, cytosine (C), or G, “Y” is C or T, and “H” is A, C, or T. In some embodiments, more than one (e.g., 2, 3, or more) Cas nucleases are used. In some embodiments, at least one of the Cas nuclease is a Cas9. In some embodiments, at least one of the Cas nucleases is a Cpf1 enzyme. In some embodiments, at least one of the Cas9 nuclease is derived from Streptococcus pyogenes. In some embodiments, at least one of the Cas9 nuclease is derived from Streptococcus pyogenes and at least one Cas9 nuclease is derived from an organism that is not Streptococcus pyogenes. Guide Nucleic Acids The terms “gRNA” and “guide RNA” are used interchangeably throughout and refer to a nucleic acid that promotes the specific targeting or homing of a CRISPR/Cas system to a target nucleic acid sequence. A gRNA can be unimolecular (having a single RNA molecule), sometimes referred to herein as sgRNAs, or modular (comprising more than one, and typically two, separate RNA molecules). A gRNA may bind to a target domain in the genome of a host cell. The gRNA may comprise a targeting domain that may be partially or completely complementary to the target domain. The gRNA may also comprise a “scaffold sequence,” (e.g., a tracrRNA sequence), that recruits a Cas nuclease to a target domain bound to a gRNA sequence (e.g., by the targeting domain of the gRNA sequence). The scaffold sequence may comprise at least one stem loop structure and recruits an endonuclease. Exemplary scaffold sequences can be found, for example, in Jinek, et al. Science (2012) 337(6096):816-821, Ran, et al. Nature Protocols (2013) 8:2281-2308, International Publication No. WO2014/093694, and International Publication No. WO2013/176772. As used herein, the terms “proximity,” “in proximity,” or “within proximity,” refers to a first, second or subsequent target site within distance of a PAM that can be bound by a CRISPR/Cas system, according to any method described herein. In various embodiments, the mutation of the genomic DNA sequence at the first target site is within 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, or 25 nucleotides from a PAM sequence. As used herein, the term “sgRNA,” refers to single guide RNA used in conjunction with CRISPR associated systems (Cas). sgRNAs are a fusion of crRNA and tracrRNA and contain nucleotides of sequence complementary to the desired target site (Jinek et al., Science, 343(6176): 1247997, 2014). Watson-Crick pairing of the sgRNA with the target site permits R-loop formation, which in conjunction with a functional PAM permits DNA cleavage or in the case of nuclease-deficient Cas9 allows binding to the DNA at that locus. Some exemplary suitable Cas9 gRNA scaffold sequences are provided herein, and additional suitable gRNA scaffold sequences will be apparent to the skilled artisan based on the present disclosure. Such additional suitable scaffold sequences include, without limitation, those recited in Jinek, et al. Science (2012) 337(6096):816-821, Ran, et al. Nature Protocols (2013) 8:2281-2308, PCT Publication No. WO2014/093694, and PCT Publication No. WO2013/176772. For example, the binding domains of naturally occurring SpCas9 gRNA typically comprise two RNA molecules, the crRNA (partially) and the tracrRNA. Variants of SpCas9 gRNAs that comprise only a single RNA molecule including both crRNA and tracrRNA sequences, covalently bound to each other, e.g., via a tetraloop or via click chemistry type covalent linkage, have been engineered and are commonly referred to as “single guide RNA” or “sgRNA.” Suitable gRNAs for use with other Cas nucleases, for example, with Cas12a nucleases, typically comprise only a single RNA molecule, as the naturally occurring Cas12a guide RNA comprises a single RNA molecule. A suitable gRNA may thus be unimolecular (having a single RNA molecule), sometimes referred to herein as sgRNAs, or modular (comprising more than one, and typically two, separate RNA molecules). A gRNA as provided herein typically comprises a targeting domain that binds to a target site in the genome of a cell. The “targeting domain” of the gRNA is complementary to the “target domain” on the target nucleic acid. The strand of the target nucleic acid comprising the nucleotide sequence complementary to the core domain of the gRNA is referred to herein as the “complementary strand” of the target nucleic acid. The targeting domain mediates targeting of the gRNA-bound Cas nuclease to a target site. Guidance on the selection of targeting domains can be found, e.g., in Fu Y et al, Nat Biotechnol 2014 (doi: 10.1038/nbt.2808) and Sternberg SH et al., Nature 2014 (doi: 10.1038/naturel3011). The targeting domain of the gRNA thus base-pairs (in full or partial complementarity) with the sequence of the double-stranded genomic target site that is complementary to the sequence of the target domain, and thus with the strand complementary to the strand that comprises the PAM sequence. It will be understood that the targeting domain of the gRNA typically does not include the PAM sequence. It will further be understood that the location of the PAM may be 5’ or 3’ of the target domain sequence, depending on the nuclease employed. For example, the PAM is typically 3’ of the target domain sequences for Cas9 nucleases, and 5’ of the target domain sequence for Cas12a nucleases. For an illustration of the location of the PAM and the mechanism of gRNA binding a target site, see, e.g., Figure 1 of Vanegas et al., Fungal Biol Biotechnol.2019; 6: 6, which is incorporated by reference herein. For additional illustration and description of the mechanism of gRNA targeting an CRISPR/Cas nuclease to a target site, see Fu Y et al, Nat Biotechnol 2014 (doi: 10.1038/nbt.2808) and Sternberg SH et al., Nature 2014 (doi: 10.1038/naturel3011), both incorporated herein by reference. An exemplary illustration of a Cas9 target site, comprising a 22 nucleotide target domain, and an NGG PAM sequence, as well as of a gRNA comprising a targeting domain that fully corresponds to the target domain (and thus base-pairs with full complementarity with the DNA strand complementary to the strand comprising the target domain and PAM) is provided below: [ target domain (DNA) ][ PAM ] 5’-N-N-N-N-N-N-N-N-N-N-N-N-N-N-N-N-N-N-N-N-N-N-N-G-G-3’ (DNA) 3’-N-N-N-N-N-N-N-N-N-N-N-N-N-N-N-N-N-N-N-N-N-N-N-C-C-5’ (DNA) | | | | | | | | | | | | | | | | | | | | | | 5’-N-N-N-N-N-N-N-N-N-N-N-N-N-N-N-N-N-N-N-N-N-N-[gRNA scaffold]-3’ (RNA) [ targeting domain (RNA) ][binding domain] An exemplary illustration of a Cas12a target site, comprising a 22 nucleotide target domain, and a TTN PAM sequence, as well as of a gRNA comprising a targeting domain that fully corresponds to the target domain (and thus base-pairs with full complementarity with the DNA strand complementary to the strand comprising the target domain and PAM) is provided below: [ PAM ][ target domain (DNA) ] 5’-T-T-N-N-N-N-N-N-N-N-N-N-N-N-N-N-N-N-N-N-N-N-N-N-N-3’ (DNA) 3’-A-A-N-N-N-N-N-N-N-N-N-N-N-N-N-N-N-N-N-N-N-N-N-N-N-5’ (DNA) | | | | | | | | | | | | | | | | | | | | | |5’-[gRNA scaffold]-N-N-N-N-N-N-N-N-N-N-N-N-N-N-N-N-N-N-N-N-N-N-3’ (RNA) [binding domain][ targeting domain (RNA) ] In some embodiments, the Cas12a PAM sequence is 5’-T-T-T-V-3’. While not wishing to be bound by theory, at least in some embodiments, it is believed that the length and complementarity of the targeting domain with the target sequence contributes to specificity of the interaction of the gRNA/Cas9 nuclease complex with a target nucleic acid. In some embodiments, the targeting domain of a gRNA provided herein is 5 to 50 nucleotides in length. In some embodiments, the targeting domain is 15 to 25 nucleotides in length. In some embodiments, the targeting domain is 18 to 22 nucleotides in length. In some embodiments, the targeting domain is 19-21 nucleotides in length. In some embodiments, the targeting domain is 15 nucleotides in length. In some embodiments, the targeting domain is 16 nucleotides in length. In some embodiments, the targeting domain is 17 nucleotides in length. In some embodiments, the targeting domain is 18 nucleotides in length. In some embodiments, the targeting domain is 19 nucleotides in length. In some embodiments, the targeting domain is 20 nucleotides in length. In some embodiments, the targeting domain is 21 nucleotides in length. In some embodiments, the targeting domain is 22 nucleotides in length. In some embodiments, the targeting domain is 23 nucleotides in length. In some embodiments, the targeting domain is 24 nucleotides in length. In some embodiments, the targeting domain is 25 nucleotides in length. In some embodiments, the targeting domain fully corresponds, without mismatch, to a target domain sequence provided herein, or a part thereof. In some embodiments, the targeting domain of a gRNA provided herein comprises 1 mismatch relative to a target domain sequence provided herein. In some embodiments, the targeting domain comprises 2 mismatches relative to the target domain sequence. In some embodiments, the target domain comprises 3 mismatches relative to the target domain sequence. In some embodiments, a targeting domain comprises a core domain and a secondary targeting domain, e.g., as described in International Publication No. WO 2015/157070, which is incorporated by reference in its entirety. In some embodiments, the core domain comprises about 8 to about 13 nucleotides from the 3' end of the targeting domain (e.g., the most 3' 8 to 13 nucleotides of the targeting domain). In an embodiment, the secondary domain is positioned 5' to the core domain. In many embodiments, the core domain has exact complementarity (corresponds fully) with the corresponding region of the target sequence, or part thereof. In other embodiments, the core domain can have 1 or more nucleotides that are not complementary (mismatched) with the corresponding nucleotide of the target domain sequence. The first complementarity domain is complementary with the second complementarity domain, and in an embodiment, has sufficient complementarity to the second complementarity domain to form a duplexed region under at least some physiological conditions. In an embodiment, the first complementarity domain is 5 to 30 nucleotides in length. In an embodiment, the first complementarity domain comprises 3 subdomains, which, in the 5' to 3' direction are: a 5' subdomain, a central subdomain, and a 3' subdomain. In an embodiment, the 5' subdomain is 4 to 9, e.g., 4, 5, 6, 7, 8 or 9 nucleotides in length. In an embodiment, the central subdomain is 1, 2, or 3, e.g., 1, nucleotide in length. In an embodiment, the 3' subdomain is 3 to 25, e.g., 4 to 22, 4 to 18, or 4 to 10, or 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, or 25 nucleotides in length. The first complementarity domain can share homology with, or be derived from, a naturally occurring first complementarity domain. In an embodiment, it has at least 50% homology with a S. pyogenes, S. aureus or S. thermophilus, first complementarity domain. The sequence and placement of the above-mentioned domains are described in more detail in International Publication No. WO2015/157070, which is herein incorporated by reference in its entirety, including p.88-112 therein. A linking domain serves to link the first complementarity domain with the second complementarity domain of a unimolecular gRNA. The linking domain can link the first and second complementarity domains covalently or non-covalently. In an embodiment, the linkage is covalent. In an embodiment, the linking domain is, or comprises, a covalent bond interposed between the first complementarity domain and the second complementarity domain. In some embodiments, the linking domain comprises one or more, e.g., 2, 3, 4, 5, 6, 7, 8, 9, or 10 nucleotides. In some embodiments, the linking domain comprises at least one non-nucleotide bond, e.g., as disclosed in International Publication No. WO2018/126176, the entire contents of which are incorporated herein by reference. The second complementarity domain is complementary, at least in part, with the first complementarity domain, and in an embodiment, has sufficient complementarity to the second complementarity domain to form a duplexed region under at least some physiological conditions. In an embodiment, the second complementarity domain can include a sequence that lacks complementarity with the first complementarity domain, e.g., a sequence that loops out from the duplexed region. In an embodiment, the second complementarity domain is 5 to 27 nucleotides in length. In an embodiment, the second complementarity domain is longer than the first complementarity region. In an embodiment, the complementary domain is 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24 or 25 nucleotides in length. In an embodiment, the second complementarity domain comprises 3 subdomains, which, in the 5' to 3' direction are: a 5' subdomain, a central subdomain, and a 3' subdomain. In an embodiment, the 5' subdomain is 3 to 25, e.g., 4 to 22, 4 to 18, or 4 to 10, or 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, or 25 nucleotides in length. In an embodiment, the central subdomain is 1, 2, 3, 4 or 5, e.g., 3, nucleotides in length. In an embodiment, the 3' subdomain is 4 to 9, e.g., 4, 5, 6, 7, 8 or 9 nucleotides in length. In an embodiment, the 5' subdomain and the 3' subdomain of the first complementarity domain, are respectively, complementary, e.g., fully complementary, with the 3' subdomain and the 5' subdomain of the second complementarity domain. In an embodiment, the proximal domain is 5 to 20 nucleotides in length. In an embodiment, the proximal domain can share homology with or be derived from a naturally occurring proximal domain. In an embodiment, it has at least 50% homology with an S. pyogenes, S. aureus or S. thermophilus, proximal domain. A broad spectrum of tail domains are suitable for use in gRNAs. In an embodiment, the tail domain is 0 (absent), 1, 2, 3, 4, 5, 6, 7, 8, 9, or 10 nucleotides in length. In some embodiments, the tail domain nucleotides are from or share homology with a sequence from the 5' end of a naturally occurring tail domain. In some embodiments, the tail domain includes sequences that are complementary to each other and which, under at least some physiological conditions, form a duplexed region. In some embodiments, the tail domain is absent or is 1 to 50 nucleotides in length. In some embodiments, the tail domain can share homology with or be derived from a naturally occurring proximal tail domain. In some embodiments, it has at least 50% homology with an S. pyogenes, S. aureus or S. thermophilus, tail domain. In an embodiment, the tail domain includes nucleotides at the 3' end that are related to the method of in vitro or in vivo transcription. In some embodiments, the gRNA is chemically modified. In some embodiments, any of the gRNAs provided herein comprise one or more nucleotides that are chemically modified. Chemical modifications of gRNAs have previously been described, and suitable chemical modifications include any modifications that are beneficial for gRNA function and do not measurably increase any undesired characteristics, e.g., off-target effects, of a given gRNA. Suitable chemical modifications include, for example, those that make a gRNA less susceptible to endo- or exonuclease catalytic activity, and include, without limitation, that the gRNA may comprise one or more modification chosen from phosphorothioate backbone modification, 2′-O-Me–modified sugars (e.g., at one or both of the 3’ and 5’ termini), 2’F- modified sugar, replacement of the ribose sugar with the bicyclic nucleotide-cEt, 3′thioPACE (MSP), or any combination thereof. Additional suitable gRNA modifications will be apparent to the skilled artisan based on this disclosure, and such suitable gRNA modification include, without limitation, those described, e.g., in Rahdar et al. PNAS December 22, 2015 112 (51) E7110-E7117 and Hendel et al., Nat Biotechnol.2015 Sep; 33(9): 985–989, each of which is incorporated herein by reference in its entirety. In some embodiments, a gRNA described herein comprises one or more 2′-O-methyl-3′-phosphorothioate nucleotides, e.g., at least 2, 3, 4, 5, or 62′-O-methyl-3′-phosphorothioate nucleotides. In some embodiments, a gRNA described herein comprises modified nucleotides (e.g., 2′-O-methyl-3′- phosphorothioate nucleotides) at the three terminal positions and the 5’ end and/or at the three terminal positions and the 3’ end. In some embodiments, the gRNA may comprise one or more modified nucleotides, e.g., as described in International Publication Nos. WO2017/214460, WO2016/089433, and WO2016/164356, which are incorporated by reference their entirety. In some embodiments, a gRNA described herein is chemically modified. For example, the gRNA may comprise one or more 2’-O modified nucleotides, e.g., 2’-O-methyl nucleotides. In some embodiments, the gRNA comprises a 2’-O modified nucleotide, e.g., 2’-O-methyl nucleotide at the 5’ end of the gRNA. In some embodiments, the gRNA comprises a 2’-O modified nucleotide, e.g., 2’-O-methyl nucleotide at the 3’ end of the gRNA. In some embodiments, the gRNA comprises a 2’-O-modified nucleotide, e.g., 2’-O- methyl nucleotide at both the 5’ and 3’ ends of the gRNA. In some embodiments, the gRNA is 2’-O-modified, e.g.2’-O-methyl-modified at the nucleotide at the 5’ end of the gRNA, the second nucleotide from the 5’ end of the gRNA, and the third nucleotide from the 5’ end of the gRNA. In some embodiments, the gRNA is 2’-O-modified, e.g.2’-O-methyl-modified at the nucleotide at the 3’ end of the gRNA, the second nucleotide from the 3’ end of the gRNA, and the third nucleotide from the 3’ end of the gRNA. In some embodiments, the gRNA is 2’-O-modified, e.g.2’-O-methyl-modified at the nucleotide at the 5’ end of the gRNA, the second nucleotide from the 5’ end of the gRNA, the third nucleotide from the 5’ end of the gRNA, the nucleotide at the 3’ end of the gRNA, the second nucleotide from the 3’ end of the gRNA, and the third nucleotide from the 3’ end of the gRNA. In some embodiments, the gRNA is 2’-O-modified, e.g.2’-O-methyl-modified at the second nucleotide from the 3’ end of the gRNA, the third nucleotide from the 3’ end of the gRNA, and at the fourth nucleotide from the 3’ end of the gRNA. In some embodiments, the nucleotide at the 3’ end of the gRNA is not chemically modified. In some embodiments, the nucleotide at the 3’ end of the gRNA does not have a chemically modified sugar. In some embodiments, the gRNA is 2’-O- modified, e.g.2’-O-methyl-modified, at the nucleotide at the 5’ end of the gRNA, the second nucleotide from the 5’ end of the gRNA, the third nucleotide from the 5’ end of the gRNA, the second nucleotide from the 3’ end of the gRNA, the third nucleotide from the 3’ end of the gRNA, and the fourth nucleotide from the 3’ end of the gRNA. In some embodiments, the 2’-O-methyl nucleotide comprises a phosphate linkage to an adjacent nucleotide. In some embodiments, the 2’-O-methyl nucleotide comprises a phosphorothioate linkage to an adjacent nucleotide. In some embodiments, the 2’-O-methyl nucleotide comprises a thioPACE linkage to an adjacent nucleotide. In some embodiments, the gRNA may comprise one or more 2’-O-modified and 3’phosphorous-modified nucleotide, e.g., a 2’-O-methyl 3’phosphorothioate nucleotide. In some embodiments, the gRNA comprises a 2’-O-modified and 3’phosphorous-modified, e.g., 2’-O-methyl 3’phosphorothioate nucleotide at the 5’ end of the gRNA. In some embodiments, the gRNA comprises a 2’-O-modified and 3’phosphorous-modified, e.g., 2’-O- methyl 3’phosphorothioate nucleotide at the 3’ end of the gRNA. In some embodiments, the gRNA comprises a 2’-O-modified and 3’phosphorous-modified, e.g., 2’-O-methyl 3’phosphorothioate nucleotide at the 5’ and 3’ ends of the gRNA. In some embodiments, the gRNA comprises a backbone in which one or more non-bridging oxygen atoms has been replaced with a sulfur atom. In some embodiments, the gRNA is 2’-O-modified and 3’phosphorous-modified, e.g.2’-O-methyl 3’phosphorothioate-modified at the nucleotide at the 5’ end of the gRNA, the second nucleotide from the 5’ end of the gRNA, and the third nucleotide from the 5’ end of the gRNA. In some embodiments, the gRNA is 2’-O-modified and 3’phosphorous-modified, e.g.2’-O-methyl 3’phosphorothioate-modified at the nucleotide at the 3’ end of the gRNA, the second nucleotide from the 3’ end of the gRNA, and the third nucleotide from the 3’ end of the gRNA. In some embodiments, the gRNA is 2’-O-modified and 3’phosphorous-modified, e.g.2’-O-methyl 3’phosphorothioate-modified at the nucleotide at the 5’ end of the gRNA, the second nucleotide from the 5’ end of the gRNA, the third nucleotide from the 5’ end of the gRNA, the nucleotide at the 3’ end of the gRNA, the second nucleotide from the 3’ end of the gRNA, and the third nucleotide from the 3’ end of the gRNA. In some embodiments, the gRNA is 2’-O-modified and 3’phosphorous-modified, e.g. 2’-O-methyl 3’phosphorothioate-modified at the second nucleotide from the 3’ end of the gRNA, the third nucleotide from the 3’ end of the gRNA, and the fourth nucleotide from the 3’ end of the gRNA. In some embodiments, the nucleotide at the 3’ end of the gRNA is not chemically modified. In some embodiments, the nucleotide at the 3’ end of the gRNA does not have a chemically modified sugar. In some embodiments, the gRNA is 2’-O-modified and 3’phosphorous-modified, e.g.2’-O-methyl 3’phosphorothioate-modified at the nucleotide at the 5’ end of the gRNA, the second nucleotide from the 5’ end of the gRNA, the third nucleotide from the 5’ end of the gRNA, the second nucleotide from the 3’ end of the gRNA, the third nucleotide from the 3’ end of the gRNA, and the fourth nucleotide from the 3’ end of the gRNA. In some embodiments, the gRNA may comprise one or more 2’-O-modified and 3’- phosphorous-modified, e.g., 2’-O-methyl 3’thioPACE nucleotide. In some embodiments, the gRNA comprises a 2’-O-modified and 3’phosphorous-modified, e.g., 2’-O-methyl 3’thioPACE nucleotide at the 5’ end of the gRNA. In some embodiments, the gRNA comprises a 2’-O-modified and 3’phosphorous-modified, e.g., 2’-O-methyl 3’thioPACE nucleotide at the 3’ end of the gRNA. In some embodiments, the gRNA comprises a 2’-O- modified and 3’phosphorous-modified, e.g., 2’-O-methyl 3’thioPACE nucleotide at the 5’ and 3’ ends of the gRNA. In some embodiments, the gRNA comprises a backbone in which one or more non-bridging oxygen atoms have been replaced with a sulfur atom and one or more non-bridging oxygen atoms have been replaced with an acetate group. In some embodiments, the gRNA is 2’-O-modified and 3’phosphorous-modified, e.g.2’-O-methyl 3’ thioPACE-modified at the nucleotide at the 5’ end of the gRNA, the second nucleotide from the 5’ end of the gRNA, and the third nucleotide from the 5’ end of the gRNA. In some embodiments, the gRNA is 2’-O-modified and 3’phosphorous-modified, e.g.2’-O-methyl 3’thioPACE-modified at the nucleotide at the 3’ end of the gRNA, the second nucleotide from the 3’ end of the gRNA, and the third nucleotide from the 3’ end of the gRNA. In some embodiments, the gRNA is 2’-O-modified and 3’phosphorous-modified, e.g.2’-O-methyl 3’thioPACE-modified at the nucleotide at the 5’ end of the gRNA, the second nucleotide from the 5’ end of the gRNA, the third nucleotide from the 5’ end of the gRNA, the nucleotide at the 3’ end of the gRNA, the second nucleotide from the 3’ end of the gRNA, and the third nucleotide from the 3’ end of the gRNA. In some embodiments, the gRNA is 2’-O-modified and 3’phosphorous-modified, e.g.2’-O-methyl 3’thioPACE-modified at the second nucleotide from the 3’ end of the gRNA, the third nucleotide from the 3’ end of the gRNA, and the fourth nucleotide from the 3’ end of the gRNA. In some embodiments, the nucleotide at the 3’ end of the gRNA is not chemically modified. In some embodiments, the nucleotide at the 3’ end of the gRNA does not have a chemically modified sugar. In some embodiments, the gRNA is 2’-O-modified and 3’phosphorous-modified, e.g.2’-O-methyl 3’thioPACE-modified at the nucleotide at the 5’ end of the gRNA, the second nucleotide from the 5’ end of the gRNA, the third nucleotide from the 5’ end of the gRNA, the second nucleotide from the 3’ end of the gRNA, the third nucleotide from the 3’ end of the gRNA, and the fourth nucleotide from the 3’ end of the gRNA. In some embodiments, the gRNA comprises a chemically modified backbone. In some embodiments, the gRNA comprises a phosphorothioate linkage. In some embodiments, one or more non-bridging oxygen atoms have been replaced with a sulfur atom. In some embodiments, the nucleotide at the 5’ end of the gRNA, the second nucleotide from the 5’ end of the gRNA, and the third nucleotide from the 5’ end of the gRNA each comprise a phosphorothioate linkage. In some embodiments, the nucleotide at the 3’ end of the gRNA, the second nucleotide from the 3’ end of the gRNA, and the third nucleotide from the 3’ end of the gRNA each comprise a phosphorothioate linkage. In some embodiments, the nucleotide at the 5’ end of the gRNA, the second nucleotide from the 5’ end of the gRNA, the third nucleotide from the 5’ end of the gRNA, the nucleotide at the 3’ end of the gRNA, the second nucleotide from the 3’ end of the gRNA, and the third nucleotide from the 3’ end of the gRNA each comprise a phosphorothioate linkage. In some embodiments, the second nucleotide from the 3’ end of the gRNA, the third nucleotide from the 3’ end of the gRNA, and at the fourth nucleotide from the 3’ end of the gRNA each comprise a phosphorothioate linkage. In some embodiments, the nucleotide at the 5’ end of the gRNA, the second nucleotide from the 5’ end of the gRNA, the third nucleotide from the 5’ end, the second nucleotide from the 3’ end of the gRNA, the third nucleotide from the 3’ end of the gRNA, and the fourth nucleotide from the 3’ end of the gRNA each comprise a phosphorothioate linkage. In some embodiments, the gRNA comprises a thioPACE linkage. In some embodiments, the gRNA comprises a backbone in which one or more non-bridging oxygen atoms have been replaced with a sulfur atom and one or more non-bridging oxygen atoms have been replaced with an acetate group. In some embodiments, the nucleotide at the 5’ end of the gRNA, the second nucleotide from the 5’ end of the gRNA, and the third nucleotide from the 5’ end of the gRNA each comprise a thioPACE linkage. In some embodiments, the nucleotide at the 3’ end of the gRNA, the second nucleotide from the 3’ end of the gRNA, and the third nucleotide from the 3’ end of the gRNA each comprise a thioPACE linkage. In some embodiments, the nucleotide at the 5’ end of the gRNA, the second nucleotide from the 5’ end of the gRNA, the third nucleotide from the 5’ end of the gRNA, the nucleotide at the 3’ end of the gRNA, the second nucleotide from the 3’ end of the gRNA, and the third nucleotide from the 3’ end of the gRNA each comprise a thioPACE linkage. In some embodiments, the second nucleotide from the 3’ end of the gRNA, the third nucleotide from the 3’ end of the gRNA, and at the fourth nucleotide from the 3’ end of the gRNA each comprise a thioPACE linkage. In some embodiments, the nucleotide at the 5’ end of the gRNA, the second nucleotide from the 5’ end of the gRNA, the third nucleotide from the 5’ end, the second nucleotide from the 3’ end of the gRNA, the third nucleotide from the 3’ end of the gRNA, and the fourth nucleotide from the 3’ end of the gRNA each comprise a thioPACE linkage. Some exemplary, non-limiting embodiments of modifications, e.g., chemical modifications, suitable for use in connection with the guide RNAs and genetic engineering methods provided herein have been described above. Additional suitable modifications, e.g., chemical modifications, will be apparent to those of skill in the art based on the present disclosure and the knowledge in the art, including, but not limited to those described in Hendel, A. et al., Nature Biotech., 2015, Vol 33, No.9; in International Publication No. WO2017/214460; WO2016/089433; and in WO2016/164356; each one of which is herein incorporated by reference in its entirety. Multiple gRNA Compositions In some embodiments, a gRNA described herein can be used in combination with one or more gRNA, e.g., for directing nucleases to one or more sites in a genome. In some embodiments, multiple gRNA described herein can be used in combination, e.g., for directing nucleases to multiple sites in a genome. In some embodiments, a gRNA described herein can be used in combination with a second gRNA, e.g., for directing nucleases to two sites in a genome. In some embodiments, a gRNA described herein can be used in combination with a third gRNA, e.g., for directing nucleases to three sites in a genome. In some embodiments, a gRNA described herein can be used in combination with a fourth gRNA, e.g., for directing nucleases to four sites in a genome. In some embodiments, a gRNA described herein can be used in combination with a fifth gRNA, e.g., for directing nucleases to five sites in a genome. In some embodiments, a gRNA described herein can be used in combination with a sixth gRNA, e.g., for directing nucleases to six sites in a genome. In some embodiments, a gRNA described herein can be used in combination with a seventh gRNA, e.g., for directing nucleases to seven sites in a genome. In some embodiments, a gRNA described herein can be used in combination with an eight gRNA, e.g., for directing nucleases to eight sites in a genome. In some embodiments, a gRNA described herein can be used in combination with a ninth gRNA, e.g., for directing nucleases to nine sites in a genome. In some embodiments, a gRNA described herein can be used in combination with a tenth gRNA, e.g., for directing nucleases to ten sites in a genome. In some embodiments, a gRNA described herein can be used in combination with more than tenth gRNA, e.g., for directing nucleases to more than ten sites in a genome. Exemplary Cell Surface Protein Editing Strategies Further examples of editing strategies targeting cell surface proteins of the present disclosure are provided herein. Exemplary Guide Nucleic Acid Targets In some embodiments, genetically engineered cells of the present disclosure are produced by contacting a hematopoietic cell with a gRNA comprising a targeting domain that is capable of binding to a target gene encoding a cell surface protein selected from a group consisting of: CD10; CD101; CD117; CD11A; CD11C; CD120B; CD123; CD13; CD133; CD152; CD200; CD205 (LY75); CD226; CD244 (SLAMF4); CD25; CD274; CD305 (LAIR1); CD32; CD33; CD34; CD366; CD38; CD41; CD42B; CD44; CD45RA; CD47; CD48; CD49D (ITGA4); CD49F; CD52; CD58; CD64; CD7; CD70; CD82; CD84; CD85; CD85K; CD86; CD9; CD96; CD99; CLL-1; EMR2 (ADGRE2); FR-B; GPR56; and IL1RAP. In some embodiments, genetically engineered cells of the present disclosure are produced by contacting a hematopoietic cell with a gRNA comprising a targeting domain that is capable of binding to a target gene encoding a cell surface protein selected from a group consisting of: CD10; CD11A; CD11C; CD152; CD200; CD274; CD33; CD34; CD42B; CD45RA; CD47; CD49D; CD49F; CD58; CD64; CD84; CD85K; CD9; CLL-1; GPR56; and IL1RAP. In some embodiments, genetically engineered cells of the present disclosure are produced by contacting a hematopoietic cell with a gRNA comprising a targeting domain that is capable of binding to a target gene encoding a cell surface protein selected from a group consisting of: CD10; CD117; CD11A; CD11C; CD120B; CD123; CD13; CD133; CD152; CD205; CD22; CD226; CD244; CD25; CD305; CD32; CD33; CD34; CD38; CD41; CD44; CD45RA; CD47; CD48; CD49D; CD49F; CD52; CD56; CD58; CD64; CD7; CD70; CD71; CD82; CD84; CD85K; CD86; CD96; CD99; CLL-1; EMR2; FR-B; GPR56; HLA-DR; and IL1RAP. In some embodiments, genetically engineered cells of the present disclosure are produced by contacting a hematopoietic cell with a first gRNA comprising a targeting domain that is capable of binding to a target gene encoding a first cell surface protein and a second gRNA comprising a targeting domain that is capable of binding to a target gene encoding a second cell surface protein selected from a group consisting of: CD274 and CD152; CD274 and CD226; CD152 and CD226; CD10 and CD49F; CD49F and GPR56; CD200 and CD34; CD34 and CD52; CD34 and CD133; GPR56 and CD133; CD42B and CD41; CD85K and CD64; CD85K and CD32; CD64 and CD32; CD64 and CD11C; CD64 and CD86; CD45RA and CD96; CD45RA and IL1RAP; CD45RA and CD123; CD84 and CD33; CD11C and IL1RAP; CD11C and CD33; CD11C and CD86; CD9 and IL1RAP; IL1RAP and CD33; IL1RAP and CD205; IL1RAP and CD305; IL1RAP and CD123; CD33 and CLL-1; CD33 and CD305; CD11A and CD86; CLL-1 and CD305; CD58 and CD305; CD49D and CD47; CD49D and CD99; CD49D and CD44; CD49D and CD305; CD47 and CD99; CD47 and EMR2; CD99 and CD44; CD99 and CD305; CD99 and EMR2; CD205 and CD305; CD205 and CD123; CD44 and CD305; CD44 and EMR2; CD244 and CD117; CD123 and EMR2; EMR2 and CD82; CD152 and CD120B; CD48 and CD11A; CD200 and CD25; CD34 and CD7; CD34 and CD13; CD52 and GPR56; CD52and CD133; CD52 and CD7; GPR56 and CD7; GPR56 and CD82; CD133 and CD7; CD133 and CD82; CD7 and CD13; CD101 and CD64; CD85 and CD70; CD85K and CD86; CD64 and FR-B; CD366 and CD96; CD25 and CD45RA; CD25 and CD96; CD9 and CD82; IL1RAP and CD58; CD11A and CD38; CD11A and CD44; CD11A and CD244; CD38 and CD305; CD38 and EMR2; CLL-1 and CD49D; CD58 and CD49D; CD47 and CD205; and CD47 and CD44. In some embodiments, genetically engineered cells of the present disclosure are produced by contacting a hematopoietic cell with a first gRNA comprising a targeting domain that is capable of binding to a target gene encoding a first cell surface protein and a second gRNA comprising a targeting domain that is capable of binding to a target gene encoding a second cell surface protein selected from a group consisting of: CD85K and CD64; CD45RA and IL1RAP; CD34 and CD133; IL1RAP and CD33; CD47 and CD99; CLL-1 and CD305; CD152 and CD226; CD45RA and CD96; CD45RA and CD123; CD49D and CD305; IL1RAP and CD305; CD58 and CD305; CD49D and CD47; CD33 and CD305; CD49D and CD44; CD64 and CD86; GPR56 and CD133; CD34 and CD52; EMR2 and CD82; CD64 and CD32; CD42B and CD41; CD64 and CD11C; CD99 and CD44; CD200 and CD34; CD11C and CD33; IL1RAP and CD123; CD11C and IL1RAP; CD44 and EMR2; CD205 and CD305; CD44 and CD305; CD49D and CD99; CD274 and CD226; CD49F and GPR56; CD9 and IL1RAP; CD99 and CD305; CD10 and CD49F; CD11A and CD86; CD33 and CLL-1; CD84 and CD33; CD244 and CD117; CD85K and CD32; CD47 and EMR2; CD274 and CD152; IL1RAP and CD205; CD11C and CD86; CD99 and EMR2; CD205 and CD123; and CD123 and EMR2. In some embodiments, genetically engineered cells of the present disclosure are produced by contacting a hematopoietic cell with a first gRNA comprising a targeting domain that is capable of binding to a target gene encoding a first cell surface protein and a second gRNA comprising a targeting domain that is capable of binding to a target gene encoding a second cell surface protein selected from a group consisting of: CD33 and CD205 (LY75); CD33 and CD244 (SLAMF4); CD33 and CD305 (LAIR1); CD33 and CD49D (ITGA4); CLL-1 and CD205 (LY75); CLL-1 and CD244 (SLAMF4); CLL-1 and CD305 (LAIR1); CLL-1 and CD49D (ITGA4); EMR2 and CD205 (LY75); EMR2 and CD244 (SLAMF4); EMR2 and CD305 (LAIR1); EMR2 and CD49D (ITGA4); CD38 and CD205 (LY75); CD38 and CD244 (SLAMF4); CD38 and CD305 (LAIR1); CD38 and CD49D (ITGA4); CD305 (LAIR1) and CD49D (ITGA4); CD305 (LAIR1) and CD205 (LY75); CD305 (LAIR1) and CD244 (SLAMF4); CD49D (ITGA4) and CD205 (LY75); CD49D (ITGA4) and CD244 (SLAMF4); and CD205 (LY75) and CD244 (SLAMF4). In some embodiments, a targeting domain which is complementary to a target nucleic acid in a cell surface protein is complementary to a target nucleic acid encoding human CD33 or a portion thereof, e.g., having a sequence at least 70%, 75%, 80%, 85%, 90%, 95%, 96%, 97%, 98%, or 99% identical thereto to the CD33 amino acid sequence set forth below: MPLLLLLPLLWAGALAMDPNFWLQVQESVTVQEGLCVLVPCTFFHPIPYYDKNSPVHGYW FREGAIISRDSPVATNKLDQEVQEETQGRFRLLGDPSRNNCSLSIVDARRRDNGSYFFRM ERGSTKYSYKSPQLSVHVTDLTHRPKILIPGTLEPGHSKNLTCSVSWACEQGTPPIFSWL GIFPGDGSGKQETRAGVVHGAIGGAGVTALLALCLCLIFFIVKTHRRKAARTAVGRNDTH PTTGSASPKHQKKSKLHGPTETSSCSGAAPTVEMDEELHYASLNFHGMNPSKDTSTEYSE VRTQ (SEQ ID NO: 1) (Uniprot No. P20138) Non-limiting examples of CD33 gRNAs may be found in International Patent Application Publication Nos. WO 2017/066760, WO 2020/150478, WO 2020/237217, WO 2018/160768, WO 2019/046285, WO2023/015182, WO2024/015925, WO2023/196816, WO2023/043858, and WO2022/217086, and WO2020/047164 each of which are incorporated by reference herein in their entirety. In some embodiments, a targeting domain which is complementary to a target nucleic acid in a cell surface protein is complementary to a target nucleic acid encoding human CLL- 1 or a portion thereof, e.g., having a sequence at least 70%, 75%, 80%, 85%, 90%, 95%, 96%, 97%, 98%, or 99% identical thereto to the CLL-1 amino acid sequence set forth below: MSEEVTYADLQFQNSSEMEKIPEIGKFGEKAPPAPSHVWRPAALFLTLLCLLLLIGLGVL ASMFHVTLKIEMKKMNKLQNISEELQRNISLQLMSNMNISNKIRNLSTTLQTIATKLCRE LYSKEQEHKCKPCPRRWIWHKDSCYFLSDDVQTWQESKMACAAQNASLLKINNKNALEFI KSQSRSYDYWLGLSPEEDSTRGMRVDNIINSSAWVIRNAPDLNNMYCGYINRLYVQYYHC TYKKRMICEKMANPVQLGSTYFREA(SEQ ID NO: 26)(Uniprot No. Q5QGZ9) Non-limiting examples of CLL-1 gRNAs may be found in PCT Publication Nos. WO 2021/041971 WO2020/047164, WO2023/015182, WO2024/015925, WO2023/196816, WO2023/043858, and WO2022/047168, each of which are incorporated by reference herein in their entirety. In some embodiments, a targeting domain which is complementary to a target nucleic acid in a cell surface protein is complementary to a target nucleic acid encoding human CD123 or a portion thereof, e.g., having a sequence at least 70%, 75%, 80%, 85%, 90%, 95%, 96%, 97%, 98%, or 99% identical thereto to the CD123 amino acid sequence set forth below: MVLLWLTLLLIALPCLLQTKEDPNPPITNLRMKAKAQQLTWDLNRNVTDIECVKDADYSM PAVNNSYCQFGAISLCEVTNYTVRVANPPFSTWILFPENSGKPWAGAENLTCWIHDVDFL SCSWAVGPGAPADVQYDLYLNVANRRQQYECLHYKTDAQGTRIGCRFDDISRLSSGSQSS HILVRGRSAAFGIPCTDKFVVFSQIEILTPPNMTAKCNKTHSFMHWKMRSHFNRKFRYEL QIQKRMQPVITEQVRDRTSFQLLNPGTYTVQIRARERVYEFLSAWSTPQRFECDQEEGAN TRAWRTSLLIALGTLLALVCVFVICRRYLVMQRLFPRIPHMKDPIGDSFQNDKLVVWEAG KAGLEECLVTEVQVVQKT (SEQ ID NO: 27)(Uniport No. P26951) Non-limiting examples of CD123 gRNAs may be found in PCT Publication Nos. WO 2021/041977, WO2023/015182, WO2024015925, WO2023196816, WO2023043858, and WO2022/047165, each of which are incorporated by reference herein in their entirety. In some embodiments, a targeting domain which is complementary to a target nucleic acid in a cell surface protein is complementary to a target nucleic acid encoding human CD38 or a portion thereof, e.g., having a sequence at least 70%, 75%, 80%, 85%, 90%, 95%, 96%, 97%, 98%, or 99% identical thereto to the CD38 amino acid sequence set forth below: MANCEFSPVSGDKPCCRLSRRAQLCLGVSILVLILVVVLAVVVPRWRQQWSGPGTTKRFP ETVLARCVKYTEIHPEMRHVDCQSVWDAFKGAFISKHPCNITEEDYQPLMKLGTQTVPCN KILLWSRIKDLAHQFTQVQRDMFTLEDTLLGYLADDLTWCGEFNTSKINYQSCPDWRKDC SNNPVSVFWKTVSRRFAEAACDVVHVMLNGSRSKIFDKNSTFGSVEVHNLQPEKVQTLEA WVIHGGREDSRDLCQDPTIKELESIISKRNIQFSCKNIYRPDKFLQCVKNPEDSSCTSEI (SEQ ID NO: 28) (Uniprot No. P28907) Non-limiting examples of CD38 gRNAs may be found in PCT Publication Nos. WO 2022/056489, WO2023/015182, and WO2023/196816, which are incorporated by reference herein in their entirety. In some embodiments, a targeting domain which is complementary to a target nucleic acid in a cell surface protein is complementary to a target nucleic acid encoding human EMR2 or a portion thereof, e.g., having a sequence at least 70%, 75%, 80%, 85%, 90%, 95%, 96%, 97%, 98%, or 99% identical thereto to the EMR2 amino acid sequence set forth below: MGGRVFLVFLAFCVWLTLPGAETQDSRGCARWCPQDSSCVNATACRCNPGFSSFSEIITT PMETCDDINECATLSKVSCGKFSDCWNTEGSYDCVCSPGYEPVSGAKTFKNESENTCQDV DECQQNPRLCKSYGTCVNTLGSYTCQCLPGFKLKPEDPKLCTDVNECTSGQNPCHSSTHC LNNVGSYQCRCRPGWQPIPGSPNGPNNTVCEDVDECSSGQHQCDSSTVCFNTVGSYSCRC RPGWKPRHGIPNNQKDTVCEDMTFSTWTPPPGVHSQTLSRFFDKVQDLGRDYKPGLANNT IQSILQALDELLEAPGDLETLPRLQQHCVASHLLDGLEDVLRGLSKNLSNGLLNFSYPAG TELSLEVQKQVDRSVTLRQNQAVMQLDWNQAQKSGDPGPSVVGLVSIPGMGKLLAEAPLV LEPEKQMLLHETHQGLLQDGSPILLSDVISAFLSNNDTQNLSSPVTFTFSHRSVIPRQKV LCVFWEHGQNGCGHWATTGCSTIGTRDTSTICRCTHLSSFAVLMAHYDVQEEDPVLTVIT YMGLSVSLLCLLLAALTFLLCKAIQNTSTSLHLQLSLCLFLAHLLFLVAIDQTGHKVLCS IIAGTLHYLYLATLTWMLLEALYLFLTARNLTVVNYSSINRFMKKLMFPVGYGVPAVTVA ISAASRPHLYGTPSRCWLQPEKGFIWGFLGPVCAIFSVNLVLFLVTLWILKNRLSSLNSE VSTLRNTRMLAFKATAQLFILGCTWCLGILQVGPAARVMAYLFTIINSLQGVFIFLVYCL LSQQVREQYGKWSKGIRKLKTESEMHTLSSSAKADTSKPSTVN (SEQ ID NO: 29)(Uniprot No. Q9UHX3) Non-limiting examples of EMR2 gRNAs may be found in PCT Publication Nos. WO 2023/086422, WO2024/015925, WO2023/196816, and WO2023/043858, which are incorporated by reference herein in their entirety. In some embodiments, a targeting domain which is complementary to a target nucleic acid in a cell surface protein is complementary to a target nucleic acid encoding human CD117 or a portion thereof, e.g., having a sequence at least 70%, 75%, 80%, 85%, 90%, 95%, 96%, 97%, 98%, or 99% identical thereto to the CD117 amino acid sequence set forth below: MRGARGAWDFLCVLLLLLRVQTGSSQPSVSPGEPSPPSIHPGKSDLIVRVGDEIRLLCTD PGFVKWTFEILDETNENKQNEWITEKAEATNTGKYTCTNKHGLSNSIYVFVRDPAKLFLV DRSLYGKEDNDTLVRCPLTDPEVTNYSLKGCQGKPLPKDLRFIPDPKAGIMIKSVKRAYH RLCLHCSVDQEGKSVLSEKFILKVRPAFKAVPVVSVSKASYLLREGEEFTVTCTIKDVSS SVYSTWKRENSQTKLQEKYNSWHHGDFNYERQATLTISSARVNDSGVFMCYANNTFGSAN VTTTLEVVDKGFINIFPMINTTVFVNDGENVDLIVEYEAFPKPEHQQWIYMNRTFTDKWE DYPKSENESNIRYVSELHLTRLKGTEGGTYTFLVSNSDVNAAIAFNVYVNTKPEILTYDR LVNGMLQCVAAGFPEPTIDWYFCPGTEQRCSASVLPVDVQTLNSSGPPFGKLVVQSSIDS SAFKHNGTVECKAYNDVGKTSAYFNFAFKGNNKEQIHPHTLFTPLLIGFVIVAGMMCIIV MILTYKYLQKPMYEVQWKVVEEINGNNYVYIDPTQLPYDHKWEFPRNRLSFGKTLGAGAF GKVVEATAYGLIKSDAAMTVAVKMLKPSAHLTEREALMSELKVLSYLGNHMNIVNLLGAC TIGGPTLVITEYCCYGDLLNFLRRKRDSFICSKQEDHAEAALYKNLLHSKESSCSDSTNE YMDMKPGVSYVVPTKADKRRSVRIGSYIERDVTPAIMEDDELALDLEDLLSFSYQVAKGM AFLASKNCIHRDLAARNILLTHGRITKICDFGLARDIKNDSNYVVKGNARLPVKWMAPES IFNCVYTFESDVWSYGIFLWELFSLGSSPYPGMPVDSKFYKMIKEGFRMLSPEHAPAEMY DIMKTCWDADPLKRPTFKQIVQLIEKQISESTNHIYSNLANCSPNRQKPVVDHSVRINSV GSTASSSQPLLVHDDV (SEQ ID NO: 30) (Uniprot No. P10721) In some embodiments, a targeting domain which is complementary to a target nucleic acid in a cell surface protein is complementary to a target nucleic acid encoding human IL1RAP or a portion thereof, e.g., having a sequence at least 70%, 75%, 80%, 85%, 90%, 95%, 96%, 97%, 98%, or 99% identical thereto to the IL1RAP amino acid sequence set forth below: MTLLWCVVSLYFYGILQSDASERCDDWGLDTMRQIQVFEDEPARIKCPLFEHFLKFNYSTAHSAGLTLIWYWTRQ DRDLEEPINFRLPENRISKEKDVLWFRPTLLNDTGNYTCMLRNTTYCSKVAFPLEVVQKDSCFNSPMKLPVHKLYIEYGIQRITCPNVDGYFPSSVKPTITWYMGCYKIQNFNNVIPEGMNLSFLIALISNNGNYTCVVTYPENGRTFHL TRTLTVKVVGSPKNAVPPVIHSPNDHVVYEKEPGEELLIPCTVYFSFLMDSRNEVWWTIDGKKPDDITIDVTINE SISHSRTEDETRTQILSIKKVTSEDLKRSYVCHARSAKGEVAKAAKVKQKVPAPRYTVELACGFGATVLLVVILI VVYHVYWLEMVLFYRAHFGTDETILDGKEYDIYVSYARNAEEEEFVLLTLRGVLENEFGYKLCIFDRDSLPGGIV TDETLSFIQKSRRLLVVLSPNYVLQGTQALLELKAGLENMASRGNINVILVQYKAVKETKVKELKRAKTVLTVIK WKGEKSKYPQGRFWKQLQVAMPVKKSPRRSSSDEQGLSYSSLKNV (SEQ ID NO: 8)(UniPort ID: Q9NPH3-1) MTLLWCVVSLYFYGILQSDASERCDDWGLDTMRQIQVFEDEPARIKCPLFEHFLKFNYSTAHSAGLTLIWYWTRQ DRDLEEPINFRLPENRISKEKDVLWFRPTLLNDTGNYTCMLRNTTYCSKVAFPLEVVQKDSCFNSPMKLPVHKLY IEYGIQRITCPNVDGYFPSSVKPTITWYMGCYKIQNFNNVIPEGMNLSFLIALISNNGNYTCVVTYPENGRTFHL TRTLTVKVVGSPKNAVPPVIHSPNDHVVYEKEPGEELLIPCTVYFSFLMDSRNEVWWTIDGKKPDDITIDVTINE SISHSRTEDETRTQILSIKKVTSEDLKRSYVCHARSAKGEVAKAAKVKQKGNRCGQ (SEQ ID NO: 9) (UniPort ID: Q9NPH3-2) MTLLWCVVSLYFYGILQSDASERCDDWGLDTMRQIQVFEDEPARIKCPLFEHFLKFNYSTAHSAGLTLIWYWTRQ DRDLEEPINFRLPENRISKEKDVLWFRPTLLNDTGNYTCMLRNTTYCSKVAFPLEVVQKDSCFNSPMKLPVHKLY IEYGIQRITCPNVDGYFPSSVKPTITWYMGCYKIQNFNNVIPEGMNLSFLIALISNNGNYTCVVTYPENGRTFHL TRTLTVKVVGSPKNAVPPVIHSPNDHVVYEKEPGEELLIPCTVYFSFLMDSRNEVWWTIDGKKPDDITIDVTINE SASSKIHSGTGLWFWSHSPASGDSHCCLPCLLARDGPILPGSFWNR(SEQ ID NO: 10) ((UniPort ID: Q9NPH3-3) Additional non-limiting examples of gRNAs which, in some embodiments, may be useful for editing genes encoding cell surface proteins are found in PCT Publication Nos. WO2022/061115, WO2022/067240, WO2022/072643, WO2022/094245, WO2022/147347, WO2018/160768, WO2019/046285, WO2020/047164, WO2020/150478, WO2021/041971, WO2021/041977, WO2022/056489, WO2023/015182, WO2024/015925, WO2023/196816, WO2023/086422, WO2023/043858, and WO2022/217086, which are incorporated by reference herein in their entirety. For example, in some embodiments, a genetically engineered hematopoietic cell comprises genetic engineering in a first gene encoding a first cell surface protein as described herein and genetic engineering in a second gene encoding a second cell surface protein as described herein, wherein the first gene and the second gene each comprise a mutation (e.g., a substitution, a deletion, or an insertion at one or more nucleotide positions) which is located in a sequence (e.g., an exon and/or an intron) that hybridizes with a gRNA, or hybridizes with a portion of a gRNA (e.g., a sequence of at least 10 nucleotides in the gRNA), described in any of the foregoing references (e.g., see: disclosures related to CD47 editing in WO2023/196816; disclosures related to CD7 editing in WO2022/061115 and WO2023/015182; disclosures related to CD34 editing in WO2022/147347; disclosures related to CD33 editing in WO 2020/237217, WO2018/160768, WO2020/047164, WO2019/046285, WO2020/150478, WO2023/015182, WO2024/015925, WO2023/196816, WO2023/043858, and WO2022/217086; disclosures related to CD123 editing in WO2020/047164, WO2021/041977, WO2023/015182, WO2024/015925, WO2023/196816, WO2023/043858, and WO2022/047165; disclosures related to CLL-1 editing in WO2020/047164, WO2021/041971, WO2023/015182, WO2024/015925, WO2023/196816, WO2023/043858, and WO2022/047168; disclosures related to CD38 editing in WO2022/056489, WO2023/196816, WO2023/015182; disclosures related to CD30 editing in WO2023/015182; and disclosures related to EMR2 editing in WO2024/015925, WO2023/086422, WO2023/043858, and WO2023/196816). Homology-Directed Repair (HDR) Using Template Polynucleotides In some embodiments, the present disclosure provides genetically engineered cells and cell populations, and methods of producing genetically engineered cells and cell populations using HDR-mediated gene editing, e.g., CRISPR/Cas-based HDR-mediated gene editing. Without being bound by any particular theory, HDR is a process wherein damage to DNA (e.g., a break in the DNA) is repaired using a donor sequence with flanking sequences comprising homology to the site of DNA damage. In some embodiments, a CRISPR/Cas system is used to introduce a break in the DNA (e.g., a double-stranded break (DSB)). Without wishing to be bound by theory, by providing a donor sequence (e.g., via a template polynucleotide) in the presence of a DSB, it is thought that HDR can be promoted (e.g., relative to other DNA repair pathways, e.g., NHEJ). HDR can result in substitution or insertion mutations that replace endogenous or naturally occurring sequences with those of the donor sequence. For example, methods described herein can be used to introduce a mutation into a target DNA, e.g., to correct a disease-associated genetic mutation. As a further example, methods described herein can be used to introduce a mutation, e.g., to insert a nucleotide sequence (e.g., a nucleotide sequence correcting a mutation in a genomic sequence). HDR may be induced by a DNA damage event that is capable of being mutagenic if left unrepaired or unprocessed, e.g., a double-stranded break. In some embodiments, the DNA damage event is induced by a CRISPR/Cas system, e.g., comprising a Cas nuclease, e.g., Cas9. Examples of DNA damage capable of producing a mutation include, but are not limited to, DNA alkylation, base deamination, base depurination, incidence of abasic sites, single-stranded breaks, and double-stranded breaks. Once DNA is damaged, the damage is repaired in multiple steps wherein cellular nucleases degrade nucleotide sequences at and proximal to the sites of the damage on one strand of the DNA. As used in this context, sequence “proximal” to the sites of damage is defined as a sequence that is found anywhere 10, 20, 30, 40, 50, 60, 70, 80, 90, 100, 150, 200, 250, 300, 350, 400, 450, or 500 nucleotides in the 5’ or 3’ direction of site of damage. Processing by nucleases, in turn, generates single- stranded overhangs comprised of a stretch of nucleotides that are not participating in base pairing interactions with nucleotides on the cognate strand to which the strand bearing the overhang is hybridized. Strand invasion follows, wherein the overhangs transiently base pair with a donor sequence that is located in close physical proximity to the damaged DNA molecule. In this way, template polynucleotide homology to a target site provided by the flanking sequences directs template polynucleotide participation in HDR. Strand invasion is followed by cellular polymerase-dependent recombination wherein the donor sequence serves as the template to direct the repair of the damaged DNA. Recombination between the donor sequence and the damaged DNA can incorporate the sequence of the donor sequence into the damaged DNA molecule. Following recombination, the repair is completed by a cellular ligase enzyme. In some embodiments, the donor sequence is provided by, for example, a template polynucleotide. When the donor sequence differs at one or more positions relative to a target DNA, integration of the donor sequence by HDR results in a mutation. In some embodiments, the target DNA comprises a mutation relative to a reference sequence (e.g., a wild-type sequence, or a sequence dominant in a population of subjects, or a sequence not characteristic of, or causally associated with, a disease or disorder); such a mutation may be referred to herein as a prior mutation (as distinguished from a mutation introduced by a method described herein). In some embodiments, the prior mutation is characteristic of, or causally associated with, a disease, e.g., a mutation that is known to cause a genetic disease, or is a mutation that is known to convey increased risk of a genetic disease. In some embodiments, a method described herein alters a genomic sequence comprising a mutation characteristic for and/or causally associated with a disease or disorder, changing the genomic sequence to a sequence that is not characteristic for and/or causally associated with that disease or disorder. In some embodiments, such alteration comprises correcting a prior mutation. In some embodiments, such alteration comprises introducing a silent mutation, a restriction site, or a tag sequence. In some embodiments, a donor sequence differs from a sequence in the target DNA in one or more nucleotides, and integration of the donor sequence into the target DNA produces a genetic modification in the target DNA. In some embodiments, the donor sequence differs from a target DNA in a manner that integration of the donor sequence corrects a prior mutation in the target DNA, e.g., integration of the donor sequence into a target DNA comprising a prior mutation results in a modification of the mutation within the target DNA, e.g., in a modification of the target DNA sequence to the wild-type sequence, to the dominant sequence in a population of subjects, or, where the prior mutation is characteristic of, or causally associated with, a disease or disorder, in a modification to a sequence that is not characteristic of, or causally associated with a disease or disorder. In some embodiments, the prior mutation is characteristic for, or causally associated with, a disease or disorder. In some embodiments, the prior mutation is not characteristic for, or not causally associated with, a disease or disorder. In some such embodiments, a template polynucleotide comprising the donor sequence is referred to as a template for HDR of the mutation. In some embodiments, the donor sequence comprises a gene or a portion thereof, e.g., a gene, or portion thereof, that (e.g., prior to any genetic modification described herein) is mutated or non-functional in the target DNA or the genome of a cell. In some such embodiments, a template polynucleotide comprising the donor sequence is referred to as a template for HDR insertion of a gene, or portion thereof, in the target DNA. In some embodiments, a template polynucleotide is single-stranded, e.g., a single- strand donor oligonucleotide (ssODN). In some embodiments, a template polynucleotide is double-stranded, e.g., a plasmid or a double-stranded donor oligonucleotide (dsODN). In some embodiments, a template polynucleotide is a minicircle plasmid. In some embodiments, a template polynucleotide is a nanoplasmid. Those of ordinary skill in the art will recognize that a minicircle plasmid is a plasmid that contains double stranded donor template without conventional plasmid backbones. Minicircle plasmids may be processed via a single DSB leading to linearization, whereas larger plasmids might require two DSBs which flank the template polynucleotide to excise the donor. Those of ordinary skill in the art will also recognize that a nanoplasmid comprises a circular DNA molecule of 500 base pairs or less and can be generated using services provided by Aldevron®. As such, minicircle plasmids and nanoplasmids are, in some embodiments, cut in a host cell prior to HDR via, for example, an exogenous nuclease (e.g., Cas9) targeting gRNA cut sites engineered into the plasmid sequence. However, in some embodiments, minicircle plasmids and nanoplasmids comprising a template polynucleotide need not be cut by an exogenous prior to HDR. As used herein, a template polynucleotide refers to a nucleic acid that is a template for HDR, e.g., HDR of a mutation in the target DNA. In some embodiments, a template polynucleotide is approximately 10, 20, 30, 40, 50, 60, 70, 80, 90, 100, 110, 120, 130, 140, 150, 160, 170, 180, 190, or 200 nucleotides long +/- 1, 2, 3, 4, 5, 6, 7, 8, 9, or 10 nucleotides long. In some embodiments, a template polynucleotide is approximately 300, 400, 500, 600, 700, 800, 900, 1000, 1100, 1200, 1300, 1400, 1500, 1600, 1700, 1800, 1900, 2000, 2100, 2200, 2300, 2400, 2500, 2600, 2700, 2800, 2900, 3000, 3100, 3200, 3300, 3400, or 3500 nucleotides +/- 10, 25, 50, or 75 nucleotides long. In some embodiments, the donor sequence comprises a modification as compared to the target DNA, for example, a mutation, e.g., an insertion, deletion, or substitution as compared to the target DNA nucleotide sequence. In some embodiments, the donor sequence comprises a substitution of a single nucleotide as compared to the target DNA. Such donor sequences are useful, for example, to effect genetic modifications that correct a single nucleotide mutation in a target DNA sequence that is characteristic for, or causally associated with, a disease or disorder. In some embodiments, the donor sequence comprises a substitution of two or more nucleotides as compared to the target DNA. Such donor sequences are useful, for example, to effect genetic modifications that correct more complex mutations, e.g., affecting two or more nucleotides, in a target DNA sequence that are characteristic for, or causally associated with, a disease or disorder. In some embodiments, the donor sequence comprises one or more insertions (e.g., of one or more nucleotides) as compared to the target DNA. Such donor sequences are useful, for example, to effect genetic modifications that create insertion mutations in a target DNA sequence. In some embodiments, the donor sequence comprises one or more deletions (e.g., of one or more nucleotides) as compared to the target DNA. Such donor sequences are useful, for example, to effect genetic modifications that create deletion mutations in a target DNA sequence. In some embodiments, the donor sequence comprises two or more substitutions as compared to the target DNA, wherein, if integrated into the target DNA, at least one such substitution results in the correction of a mutation that is characteristic of, or causally associated with, a disease or mutation, and wherein at least one such substitution results in a silent mutation in the target DNA, e.g., a substitution of a wobble base within an amino acid-encoding codon of a target DNA. Such donor sequences are useful, for example, to effect genetic modifications that correct disease-associated mutations in a target DNA sequence, while at the same time creating a sequence tag, e.g., a non-naturally occurring sequence or a sequence that did was not previously present in the target DNA, which is useful for identification and/or tracking of the modified cells. In some embodiments, the donor sequence comprises a restriction site or a unique sequence tag, for example, a unique primer binding site. In some embodiments, the sequence comprising the restriction site or a unique sequence tag is an insertion relative to the target DNA, e.g., the target DNA does not comprise a restriction site or a unique sequence tag where the donor sequence comprises one. In some embodiments, the sequence comprising the restriction site or a unique sequence tag is not an insertion relative to the target DNA. For example, in some embodiments, the sequence comprising the restriction site or a unique sequence tag comprises a mutation (e.g., a substitution) as compared to the target DNA that, upon integration into the target DNA, produces a restriction site or a unique sequence tag. In some embodiments, the sequence comprising the restriction site or a unique sequence tag does not alter an amino acid sequence encoded by the target DNA. A restriction site or a unique sequence tag thus introduced may be used, e.g., to confirm the success of integration of the donor sequence (e.g., in an experiment where the modified target DNA is cleaved and fragments or sequences thereof are analyzed). In some embodiments, the restriction endonuclease site comprises a Pvu1 site, e.g., 5’-CGATCG-3’. In some embodiments, the target DNA comprises a prior mutation and the donor sequence differs from the target DNA in a manner such that integration of the donor sequence corrects the prior mutation and produces one or more additional mutations (e.g., a second, third, fourth, or fifth mutation relative to the correction of the prior mutation (the first mutation)). In some embodiments, the one or more additional mutations comprise one or more silent mutations that do not alter the amino acid encoded by the nucleic acid sequence of the target DNA. In some embodiments, the one or more silent mutations are contiguous (i.e., directly adjacent) to the prior mutation or the codon containing the prior mutation. In some embodiments, silent mutations are used, e.g., as identifiers (e.g., “tags” or “bar codes”) of a given correction of a prior mutation or to facilitate confirmation of integration of the donor sequence (e.g., in an experiment where the modified target DNA sequences are analyzed). In some embodiments, methods and compositions provided by the present disclosure are applied to a target DNA, e.g., in order to modify the target DNA. For example, in some embodiments, the target DNA comprises a nucleotide sequence that is characteristic for, or causally associated with, a disease or disorder. Where such a target DNA sequence is different from the wild-type DNA sequence, or from a dominant DNA sequence at this locus within a population of subjects not affected by the disease or disorder, the divergence between the target DNA and the wild-type or dominant DNA sequence is also sometimes referred to herein as a prior mutation. In some embodiments, a template polynucleotide comprises a first flanking sequence and a second flanking sequence, also referred to herein as a first homology sequence and a second homology sequence. In some embodiments, the first flanking sequence and second flanking sequence direct the binding of the template polynucleotide to a target DNA sequence in the cell. In some embodiments, a first flanking sequence is at least 50, at least 60, at least 70, at least 80, at least 90, at least 100, at least 110, at least 120, at least 130, at least 140, at least 150, at least 160, at least 170, at least 180, at least 190, at least 200, at least 210, at least 220, at least 230, at least 240, or at least 250 nucleotides long (and optionally no more than 1000, no more than 750, no more than 500, no more than 400, no more than 300, or no more than 250 nucleotides long). In some embodiments, a first flanking sequence comprises 25- 300, 50-300, 75-300, 100-300, 125-300, 150-300, 175-300, 200-300, 225-300, 250-300, 275- 300, 100-400, 125-400, 150-400, 175-400, 200-400, 225-400, 250-400, 275-400, 300-400, 325, 400, 350-400, 375-400, 200-500, 225-500, 250-500, 275-500, 300-500, 325-500, 350- 500, 375-500, 400-500, 425-500, 450-500, or 475-500 nucleotides in length. In some embodiments, a first flanking sequence comprises 500-2000, 600-2000, 700-2000, 800-2000, 900-2000, 1000-2000, 1100-2000, 1200-2000, 1300-2000, 1400-2000, 1500-2000, 1600- 2000, 1700-2000, 1800-2000, or 1900-2000 nucleotides in length. In some embodiments, the first flanking sequence has at least 50%, at least 60%, at least 70%, at least at least 80%, at least 90%, at least 91%, at least 92%, at least 93%, at least 94%, at least 95%, at least 96%, at least 97%, at least 98%, at least 99%, or at least 100% identity to a sequence upstream of a DSB in the target DNA (e.g., upstream of a site where a DSB is produced by a CRISPR/Cas system described herein), or a sequence complementary thereto. In some embodiments, the first flanking sequence has 100% identity to a sequence upstream of a DSB in the target DNA (e.g., upstream of a site where a DSB is produced by a CRISPR/Cas system described herein), or a sequence complementary thereto. As used in this context, sequence “upstream” and “downstream” refer to a region within 10, within 20, within 30, within 40, within 50, within 60, within 70, within 80, within 90, or within 100 nucleotides of a feature in the DNA (e.g., a DSB), with each term referring to a different direction from the target site, and, in the case where the target DNA is a gene or portion thereof upstream is toward the transcription start site for the gene and downstream is away from the transcription start site for the gene. In some embodiments, the first flanking sequence is a 5’ homology arm of a template polynucleotide and is 5’ of a donor sequence, e.g., in an ssODN, dsODN, minicircle plasmid, or nanoplasmid. In some embodiments, a second flanking sequence is at least 50, at least 60, at least 70, at least 80, at least 90, at least 100, at least 110, at least 120, at least 130, at least 140, at least 150, at least 160, at least 170, at least 180, at least 190, at least 200, at least 210, at least 220, at least 230, at least 240, or at least 250 nucleotides long (and optionally no more than 1000, no more than 750, no more than 500, no more than 400, no more than 300, or no more than 250 nucleotides long). In some embodiments, a second flanking sequence comprises 25-300, 50-300, 75-300, 100-300, 125-300, 150-300, 175-300, 200-300, 225-300, 250-300, 275-300, 100-400, 125-400, 150-400, 175-400, 200-400, 225-400, 250-400, 275- 400, 300-400, 325, 400, 350-400, 375-400, 200-500, 225-500, 250-500, 275-500, 300-500, 325-500, 350-500, 375-500, 400-500, 425-500, 450-500, or 475-500 nucleotides in length. In some embodiments, a second flanking sequence comprises 500-2000, 600-2000, 700-2000, 800-2000, 900-2000, 1000-2000, 1100-2000, 1200-2000, 1300-2000, 1400-2000, 1500-2000, 1600-2000, 1700-2000, 1800-2000, or 1900-2000 nucleotides in length. In some embodiments, the second flanking sequence has at least 50%, at least 60%, at least 70%, at least 80%, at least 90%, at least 91%, at least 92%, at least 93%, at least 94%, at least 95%, at least 96%, at least 97%, at least 98%, at least 99%, or at least 100% identity to a sequence downstream of a target site (e.g., downstream of a DSB produced by a CRISPR/Cas system in the target site), or a sequence complementary thereto. In some embodiments, the second flanking sequence has 100% identity to a sequence downstream of a DSB in the target DNA (e.g., downstream of a site where a DSB is produced by a CRISPR/Cas system described herein), or a sequence complementary thereto. In some embodiments, the second flanking sequence is a 3’ homology arm of a template polynucleotide and is 3’ of a donor sequence, e.g., in an ssODN, dsODN, minicircle plasmid, or nanoplasmid. In some embodiments, the first flanking sequence and the second flanking sequence have identity or complementarity to different sequences within or proximal to the target DNA. For example, in some embodiments the first flanking sequence has identity or complementarity to a first target sequence within or proximal to a target DNA and the second flanking sequence has identity or complementarity to a second target sequence within or proximal to the target DNA. In some embodiments, the first target sequence and second target sequence are no more than 5, no more than 10, no more than 20, no more than 30, no more than 40, no more than 50, no more than 100, no more than 150, no more than 200, no more than 250, no more than 300, no more than 500, or no more than 1000 bases apart in the nucleic acid molecule comprising the target DNA. In some embodiments, the first flanking sequence has 100% identity to a sequence upstream of a DSB in the target DNA, or a sequence complementary thereto, and the second flanking sequence has 100% identity to a sequence downstream of a DSB in the target DNA, or a sequence complementary thereto. In some embodiments, a flanking sequence comprises 500-2000, 600-2000, 700-2000, 800-2000, 900-2000, 1000-2000, 1100-2000, 1200-2000, 1300-2000, 1400-2000, 1500-2000, 1600-2000, 1700-2000, 1800-2000, or 1900-2000 consecutive nucleotides that are 100% identical to a target sequence within a target DNA. In some embodiments, a second flanking sequence comprises 500-2000, 600-2000, 700-2000, 800-2000, 900-2000, 1000-2000, 1100- 2000, 1200-2000, 1300-2000, 1400-2000, 1500-2000, 1600-2000, 1700-2000, 1800-2000, or 1900-2000 nucleotides in length. In some embodiments, a flanking sequence (e.g., a 3’ homology arm or 5’ homology arm) comprises 25-300, 50-300, 75-300, 100-300, 125-300, 150-300, 175-300, 200-300, 225-300, 250-300, 275-300, 100-400, 125-400, 150-400, 175- 400, 200-400, 225-400, 250-400, 275-400, 300-400, 325, 400, 350-400, 375-400, 200-500, 225-500, 250-500, 275-500, 300-500, 325-500, 350-500, 375-500, 400-500, 425-500, 450- 500, or 475-500 consecutive nucleotides that are 100% identical to a target sequence within a target DNA. In some embodiments, a flanking sequence (e.g., a 3’ homology arm or 5’ homology arm) comprises 2-100, 10-100, 20-100, 30-100, 40-100, 50-100, 60-100, 70-100, 80-100, 90-100, 2-150, 2-200, 2-250, 10-150, 10-200, 10-250, 50-150, 50-200, 50-250, 100- 150, 100-200, 100-250, 150-200, 150-200, or 200-250 consecutive nucleotides that are 100% identical to a target sequence within a target DNA. In some embodiments, a flanking sequence (e.g., a 3’ homology arm or 5’ homology arm) comprises at least 2, at least 10, at least 20, at least 30, at least 40, at least 50, at least 60, at least 70, at least 80, at least 90, or at least 100 consecutive nucleotides that are 100% identical to a target sequence within a target DNA (and optionally no more than 200, no more than 180, no more than 160, no more than 140, no more than 120, or no more than 100 consecutive nucleotides that are 100% identical to a target sequence within a target DNA). In some embodiments, a flanking sequence (e.g., a 3’ homology arm or a 5’ homology arm) comprises a nucleotide sequence that is 100% identical to a PAM sequence in the target DNA. In some embodiments, the nucleotide sequence identical to the PAM sequence is 2-3, 2-4, 2-5, 2-6, 3-4, 3-5, 3-6, 4-5, 4-6, or 5-6 nucleotides in length (e.g., 2, 3, 4, 5, or 6 nucleotides in length). In some embodiments, a template polynucleotide comprises a donor sequence. In some embodiments, the donor sequence is integrated into a target DNA at the site of a DSB. In some embodiments, the donor sequence is homologous to the target DNA or a portion thereof, e.g., the sequence of the target DNA surrounding or adjacent to the DSB. In some embodiments, the donor sequence is contiguous with the first and second flanking sequences in a template polynucleotide. For example, in some embodiments a target DNA comprises a gene or a portion thereof, and the donor sequence is homologous to the target DNA or a portion thereof (e.g., in proximity to a DSB or a site targeted for a DSB by a CRISPR/Cas system as described herein). In some embodiments, the first and second flanking sequences guide binding of the template polynucleotide to a target DNA, facilitating interaction of the donor sequence with its homologous sequence in the target DNA and/or with cellular DNA repair (e.g., HDR) pathway components. In some embodiments, the donor sequence differs from a homologous sequence of the target DNA at 1, 2, 3, 4, 5, 6, 7, 8, 9, or 10 bases (e.g., 1- 10, 1-9, 1-8, 1-7, 1-6, 1-5, 1-4, 1-3, 1-2, 2-10, 2-9, 2-8, 2-7, 2-6, 2-5, 2-4, 2-3, 3-10, 3-9, 3-8, 3-7, 3-6, 3-5, 3-4, 4-10, 4-9, 4-8, 4-7, 4-6, 4-5, 5-10, 5-9, 5-8, 5-7, 5-6, 6-10, 6-9, 6-8, 6-7, 7- 10, 7-9, 7-8, 8-10, 8-9, or 9-10 bases), or at a number of positions corresponding to up to 1, 5, 10, 15, or 20% of the length of the donor sequence. In some embodiments, a donor sequence is 1-100, 1-80, 1-60, 1-40, 1-20, 1-15, 1-10, 1-9, 1-8, 1-7, 1-6, 1-5, 1-4, 1-3, 1-2, 5-100, 5-80, 5-60, 5-40, 5-20, 5-15, 5-10, 5-9, 5-8, 5-7, 5-6, 10-100, 10-80, 10-60, 10-40, 10-20, 10-15, 20-100, 20-80, 20-60, 20-40, 60-100, or 60-80 nucleotides in length. In some embodiments, a donor sequence is no more than 100, no more than 90, no more than 80, no more than 70, no more than 60, no more than 50, no more than 45, no more than 40, no more than 35, no more than 30, no more than 25, no more than 20, no more than 15, no more than 14, no more than 13, no more than 12, no more than 11, no more than 10, no more than 9, no more than 8, no more than 7, no more than 6, no more than 5, no more than 4, no more than 3, no more than 2, or no more than 1 bases long. In some embodiments, a donor sequence is 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49, 50, 55, 60, 65, 70, 75, 80, 85, 90, 95, or 100 bases long. In some embodiments, a donor sequence differs from a homologous sequence of the target DNA at a position or positions corresponding to a prior mutation in the target DNA (e.g., characteristic of, or causally associated with, a disease or disorder, or risk of developing a disease or disorder), e.g., a prior point mutation. In some embodiments, the donor sequence comprises sequence corresponding to the wild-type, functional, and/or naturally-occurring sequence at a position or positions corresponding to a prior mutation in the target DNA. In some embodiments, the donor sequence comprises an artificial or heterologous sequence. In some embodiments, a donor sequence is 200-2000, 200-1900, 200-1800, 200-1700, 200-1600, 200-1500, 200-1400, 200-1300, 200-1200, 200- 1100, 100-1000, 100-900, 100-800, 100-700, 100-600, 100-500, 100-400, 100-300, or 100- 200 nucleotides in length. In some embodiments, a donor sequence is no more than 2000, no more than 1900, no more than 1800, no more than 1700, no more than 1600, no more than 1500, no more than 1400, no more than 1300, no more than 1200, no more than 1100, no more than 1000, no more than 900, no more than 800, no more than 700, no more than 600, no more than 500, no more than 400, no more than 300, or no more than 200 nucleotides in length. A schematic of an exemplary template polynucleotide, such as an ssODN, a double- stranded dsODN, minicircle plasmid, or nanoplasmid is provided below: [5’-homology arm] – [donor sequence] – [3’ homology arm] Each homology arm (e.g., a flanking sequence described herein) has homology to a sequence in the target DNA proximal to the sequence homologous to the donor sequence. In some embodiments, a homology arm comprises a sequence homologous to a PAM sequence in the target DNA. In some embodiments, a CRISPR/Cas system for use in a method of the disclosure comprises a Cas nuclease that recognizes a PAM sequence in the target DNA and cuts the target DNA at a position near to the PAM sequence (e.g., 5’ or 3’ of the PAM sequence). Accordingly, in some embodiments a PAM homologous sequence is present in a 3’ homology arm or a 5’ homology arm of a template polynucleotide. In some embodiments, the PAM homologous sequence is positioned such that HDR of a DSB produced by a Cas nuclease promotes integration of a donor sequence. In some embodiments, the DSB is positioned in a target DNA sequence homologous to the donor sequence. A schematic of an exemplary 3’ homology arm (e.g., where a CRISPR/Cas system (e.g., comprising Cas9) cuts a target DNA 5’ of a PAM sequence) is provided below: [N]x – [PAM] – [N]y For example, an exemplary Cas nuclease, Cas9, cuts a target DNA 3-4 nucleotides 5’ of a PAM sequence. In some embodiments, x is 3-4, and y is the number of nucleotides in the remaining length of the homology arm (e.g., wherein the length of the homology arm is described herein). For example, for x = 3, and a homology arm length of 100 nucleotides, y would be 100 minus 3 and minus the length of the PAM homologous sequence (e.g., where the PAM sequence is 3 nucleotides long, y would be 94 (100-3-3). In some embodiments, x is 2 and the homology arm is 50-60 nucleotides long. In some embodiments, x is 2 and the homology arm is 60-70 nucleotides long. In some embodiments, x is 2 and the homology arm is 70-80 nucleotides long. In some embodiments, x is 2 and the homology arm is 80-90 nucleotides long. In some embodiments, x is 2 and the homology arm is 90-100 nucleotides long. In some embodiments, x is 2 and the homology arm is 100-110 nucleotides long. In some embodiments, x is 2 and the homology arm is 110-120 nucleotides long. In some embodiments, x is 2 and the homology arm is 120-130 nucleotides long. In some embodiments, x is 2 and the homology arm is 130-140 nucleotides long. In some embodiments, x is 2 and the homology arm is 140-150 nucleotides long. In some embodiments, x is 2 and the homology arm is 150-160 nucleotides long. In some embodiments, x is 2 and the homology arm is 160-170 nucleotides long. In some embodiments, x is 2 and the homology arm is 170-180 nucleotides long. In some embodiments, x is 2 and the homology arm is 180-190 nucleotides long. In some embodiments, x is 2 and the homology arm is 190-200 nucleotides long. In some embodiments, x is 2 and the homology arm is 210-220 nucleotides long. In some embodiments, x is 2 and the homology arm is 220-230 nucleotides long. In some embodiments, x is 2 and the homology arm is 230-240 nucleotides long. In some embodiments, x is 2 and the homology arm is 240-250 nucleotides long. In some embodiments, x is 3 and the homology arm is 50-60 nucleotides long. In some embodiments, x is 3 and the homology arm is 60-70 nucleotides long. In some embodiments, x is 3 and the homology arm is 70-80 nucleotides long. In some embodiments, x is 3 and the homology arm is 80-90 nucleotides long. In some embodiments, x is 3 and the homology arm is 90-100 nucleotides long. In some embodiments, x is 3 and the homology arm is 100-110 nucleotides long. In some embodiments, x is 3 and the homology arm is 110-120 nucleotides long. In some embodiments, x is 3 and the homology arm is 120-130 nucleotides long. In some embodiments, x is 3 and the homology arm is 130-140 nucleotides long. In some embodiments, x is 3 and the homology arm is 140-150 nucleotides long. In some embodiments, x is 3 and the homology arm is 150-160 nucleotides long. In some embodiments, x is 3 and the homology arm is 160-170 nucleotides long. In some embodiments, x is 3 and the homology arm is 170-180 nucleotides long. In some embodiments, x is 3 and the homology arm is 180-190 nucleotides long. In some embodiments, x is 3 and the homology arm is 190-200 nucleotides long. In some embodiments, x is 3 and the homology arm is 210-220 nucleotides long. In some embodiments, x is 3 and the homology arm is 220-230 nucleotides long. In some embodiments, x is 3 and the homology arm is 230-240 nucleotides long. In some embodiments, x is 3 and the homology arm is 240-250 nucleotides long. In some embodiments, x is 4 and the homology arm is 50-60 nucleotides long. In some embodiments, x is 4 and the homology arm is 60-70 nucleotides long. In some embodiments, x is 4 and the homology arm is 70-80 nucleotides long. In some embodiments, x is 4 and the homology arm is 80-90 nucleotides long. In some embodiments, x is 4 and the homology arm is 90-100 nucleotides long. In some embodiments, x is 4 and the homology arm is 100-110 nucleotides long. In some embodiments, x is 4 and the homology arm is 110-120 nucleotides long. In some embodiments, x is 4 and the homology arm is 120-130 nucleotides long. In some embodiments, x is 4 and the homology arm is 130-140 nucleotides long. In some embodiments, x is 4 and the homology arm is 140-150 nucleotides long. In some embodiments, x is 4 and the homology arm is 150-160 nucleotides long. In some embodiments, x is 4 and the homology arm is 160-170 nucleotides long. In some embodiments, x is 4 and the homology arm is 170-180 nucleotides long. In some embodiments, x is 4 and the homology arm is 180-190 nucleotides long. In some embodiments, x is 4 and the homology arm is 190-200 nucleotides long. In some embodiments, x is 4 and the homology arm is 210-220 nucleotides long. In some embodiments, x is 4 and the homology arm is 220-230 nucleotides long. In some embodiments, x is 4 and the homology arm is 230-240 nucleotides long. In some embodiments, x is 4 and the homology arm is 240-250 nucleotides long. A schematic of an exemplary 5’ homology arm (e.g., where a CRISPR/Cas system (e.g., comprising Cas12a) cuts a target DNA 3’ of a PAM sequence) is provided below: [N]a – [PAM] – [N]b As a further example, another exemplary Cas nuclease, Cas12a, cuts a target DNA 18-19 nucleotides 3’ of a PAM sequence. In some embodiments, b is 18-19, and a is the number of nucleotides in the remaining length of the homology arm (e.g., wherein the length of the homology arm is described herein). For example, for b = 18, and a homology arm length of 100 nucleotides, a would be 100 minus 18 and minus the length of the PAM homologous sequence (e.g., where the PAM sequence is 3 nucleotides long, a would be 79 (100-18-3). In some embodiments, b is 17 and the homology arm is 50-60 nucleotides long. In some embodiments, b is 17 and the homology arm is 60-70 nucleotides long. In some embodiments, b is 17 and the homology arm is 70-80 nucleotides long. In some embodiments, b is 17 and the homology arm is 80-90 nucleotides long. In some embodiments, b is 17 and the homology arm is 90-100 nucleotides long. In some embodiments, b is 17 and the homology arm is 100-110 nucleotides long. In some embodiments, b is 17 and the homology arm is 110-120 nucleotides long. In some embodiments, b is 17 and the homology arm is 120-130 nucleotides long. In some embodiments, b is 17 and the homology arm is 130-140 nucleotides long. In some embodiments, b is 17 and the homology arm is 140-150 nucleotides long. In some embodiments, b is 17 and the homology arm is 150-160 nucleotides long. In some embodiments, b is 17 and the homology arm is 160-170 nucleotides long. In some embodiments, b is 17 and the homology arm is 170-180 nucleotides long. In some embodiments, b is 17 and the homology arm is 180-190 nucleotides long. In some embodiments, b is 17 and the homology arm is 190-200 nucleotides long. In some embodiments, b is 17 and the homology arm is 210-220 nucleotides long. In some embodiments, b is 17 and the homology arm is 220-230 nucleotides long. In some embodiments, b is 17 and the homology arm is 230-240 nucleotides long. In some embodiments, b is 17 and the homology arm is 240-250 nucleotides long. In some embodiments, b is 18 and the homology arm is 50-60 nucleotides long. In some embodiments, b is 18 and the homology arm is 60-70 nucleotides long. In some embodiments, b is 18 and the homology arm is 70-80 nucleotides long. In some embodiments, b is 18 and the homology arm is 80-90 nucleotides long. In some embodiments, b is 18 and the homology arm is 90-100 nucleotides long. In some embodiments, b is 18 and the homology arm is 100-110 nucleotides long. In some embodiments, b is 18 and the homology arm is 110-120 nucleotides long. In some embodiments, b is 18 and the homology arm is 120-130 nucleotides long. In some embodiments, b is 18 and the homology arm is 130-140 nucleotides long. In some embodiments, b is 18 and the homology arm is 140-150 nucleotides long. In some embodiments, x is 3 and the homology arm is 150-160 nucleotides long. In some embodiments, b is 18 and the homology arm is 160-170 nucleotides long. In some embodiments, b is 18 and the homology arm is 170-180 nucleotides long. In some embodiments, b is 18 and the homology arm is 180-190 nucleotides long. In some embodiments, b is 18 and the homology arm is 190-200 nucleotides long. In some embodiments, b is 18 and the homology arm is 210-220 nucleotides long. In some embodiments, b is 18 and the homology arm is 220-230 nucleotides long. In some embodiments, b is 18 and the homology arm is 230-240 nucleotides long. In some embodiments, b is 18 and the homology arm is 240-250 nucleotides long. In some embodiments, b is 19 and the homology arm is 50-60 nucleotides long. In some embodiments, b is 19 and the homology arm is 60-70 nucleotides long. In some embodiments, b is 19 and the homology arm is 70-80 nucleotides long. In some embodiments, b is 19 and the homology arm is 80-90 nucleotides long. In some embodiments, b is 19 and the homology arm is 90-100 nucleotides long. In some embodiments, b is 19 and the homology arm is 100-110 nucleotides long. In some embodiments, b is 19 and the homology arm is 110-120 nucleotides long. In some embodiments, b is 19 and the homology arm is 120-130 nucleotides long. In some embodiments, b is 19 and the homology arm is 130-140 nucleotides long. In some embodiments, b is 19 and the homology arm is 140-150 nucleotides long. In some embodiments, b is 19 and the homology arm is 150-160 nucleotides long. In some embodiments, b is 19 and the homology arm is 160-170 nucleotides long. In some embodiments, b is 19 and the homology arm is 170-180 nucleotides long. In some embodiments, b is 19 and the homology arm is 180-190 nucleotides long. In some embodiments, b is 19 and the homology arm is 190-200 nucleotides long. In some embodiments, b is 19 and the homology arm is 210-220 nucleotides long. In some embodiments, b is 19 and the homology arm is 220-230 nucleotides long. In some embodiments, b is 19 and the homology arm is 230-240 nucleotides long. In some embodiments, b is 19 and the homology arm is 240-250 nucleotides long. In some embodiments, a genetically engineered hematopoietic cell comprises a genetic modification corresponding to integration of a donor sequence (e.g., from a template polynucleotide described herein) into a target DNA in the hematopoietic cell. In some embodiments, the genetic modification corresponds to a position or positions where the donor sequence differs from the sequence of the target DNA. In some embodiments, integration of the donor sequence results in modification at 1, 2, 3, 4, 5, 6, 7, 8, 9, or 10 bases (e.g., 1-10, 1- 9, 1-8, 1-7, 1-6, 1-5, 1-4, 1-3, 1-2, 2-10, 2-9, 2-8, 2-7, 2-6, 2-5, 2-4, 2-3, 3-10, 3-9, 3-8, 3-7, 3-6, 3-5, 3-4, 4-10, 4-9, 4-8, 4-7, 4-6, 4-5, 5-10, 5-9, 5-8, 5-7, 5-6, 6-10, 6-9, 6-8, 6-7, 7-10, 7-9, 7-8, 8-10, 8-9, or 9-10 bases) in the target DNA. In some embodiments, integration of the donor sequence results in an insertion of 1, 2, 3, 4, 5, 6, 7, 8, 9, or 10 bases (e.g., 1-10, 1- 9, 1-8, 1-7, 1-6, 1-5, 1-4, 1-3, 1-2, 2-10, 2-9, 2-8, 2-7, 2-6, 2-5, 2-4, 2-3, 3-10, 3-9, 3-8, 3-7, 3-6, 3-5, 3-4, 4-10, 4-9, 4-8, 4-7, 4-6, 4-5, 5-10, 5-9, 5-8, 5-7, 5-6, 6-10, 6-9, 6-8, 6-7, 7-10, 7-9, 7-8, 8-10, 8-9, or 9-10 bases) in the target DNA. In some embodiments, integration of the donor sequence results in substitution of 1, 2, 3, 4, 5, 6, 7, 8, 9, or 10 bases (e.g., 1-10, 1- 9, 1-8, 1-7, 1-6, 1-5, 1-4, 1-3, 1-2, 2-10, 2-9, 2-8, 2-7, 2-6, 2-5, 2-4, 2-3, 3-10, 3-9, 3-8, 3-7, 3-6, 3-5, 3-4, 4-10, 4-9, 4-8, 4-7, 4-6, 4-5, 5-10, 5-9, 5-8, 5-7, 5-6, 6-10, 6-9, 6-8, 6-7, 7-10, 7-9, 7-8, 8-10, 8-9, or 9-10 bases) in the target DNA. In some embodiments, integration of the donor sequence results in modification at a number of positions in the target DNA corresponding to up to 1, 5, 10, 15, 20, 25, 30, 35, 40, 45, 50, 55, 60, 65, 70, 75, 80, 85, 90, 95, or 100% of the length of the donor sequence. In some embodiments, integration of the donor sequence results in insertion of a number of bases in the target DNA corresponding to up to 1, 5, 10, 15, 20, 25, 30, 35, 40, 45, 50, 55, 60, 65, 70, 75, 80, 85, 90, 95, or 100% of the length of the donor sequence. In some embodiments, a donor sequence is 200-2000, 200- 1900, 200-1800, 200-1700, 200-1600, 200-1500, 200-1400, 200-1300, 200-1200, 200-1100, 100-1000, 100-900, 100-800, 100-700, 100-600, 100-500, 100-400, 100-300, or 100-200 nucleotides in length. In some embodiments, a donor sequence is no more than 2000, no more than 1900, no more than 1800, no more than 1700, no more than 1600, no more than 1500, no more than 1400, no more than 1300, no more than 1200, no more than 1100, no more than 1000, no more than 900, no more than 800, no more than 700, no more than 600, no more than 500, no more than 400, no more than 300, or no more than 200 nucleotides in length. In some embodiments, the donor sequence is 1-100, 1-80, 1-60, 1-40, 1-20, 1-15, 1-10, 1-9, 1-8, 1-7, 1-6, 1-5, 1-4, 1-3, 1-2, 5-100, 5-80, 5-60, 5-40, 5-20, 5-15, 5-10, 5-9, 5-8, 5-7, 5-6, 10- 100, 10-80, 10-60, 10-40, 10-20, 10-15, 20-100, 20-80, 20-60, 20-40, 60-100, or 60-80 nucleotides in length. In some embodiments, a donor sequence is no more than 100, no more than 90, no more than 80, no more than 70, no more than 60, no more than 50, no more than 45, no more than 40, no more than 35, no more than 30, no more than 25, no more than 20, no more than 15, no more than 14, no more than 13, no more than 12, no more than 11, no more than 10, no more than 9, no more than 8, no more than 7, no more than 6, no more than 5, no more than 4, no more than 3, no more than 2, or no more than 1 bases long. In some embodiments, a donor sequence is 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49, 50, 55, 60, 65, 70, 75, 80, 85, 90, 95, or 100 bases long. In some embodiments, integration of the donor sequence into the genetically engineered hematopoietic cell corrected a prior mutation in the target DNA (e.g., a disease-associated mutation (a mutation characteristic of, or causally associated with, a disease or disorder, or risk of developing a disease or disorder) or a non-disease-associated mutation), e.g., as described herein. In some embodiments, the donor sequence integrated comprises a sequence corresponding to the wild-type, functional, and/or naturally-occurring sequence at a position or positions corresponding to a prior mutation in the target DNA. In some embodiments, the donor sequence comprises an artificial or heterologous sequence. In some embodiments, integration of the donor sequence produces a restriction nuclease site or a unique sequence tag in the target DNA of the genetically engineered hematopoietic cell. In some embodiments, integration of the donor sequence into the target DNA of the genetically engineered hematopoietic cell produces one or more silent mutations along with a non-silent mutation (e.g., correction of a prior mutation, e.g., in a coding sequence). In some embodiments, the one or more silent mutations are contiguous with another mutation described herein (e.g., contiguous with correction of a prior mutation). For example, in some embodiments a genetically engineered hematopoietic cell comprises a genetic modification corresponding to correction of a prior mutation which substitutes a single prior mutation, e.g., a single nucleotide point mutation, for the corresponding base(s) present in a wild-type cell, and one or more silent mutations contiguous with the prior mutation. Accordingly, the disclosure provides a genetically engineered hematopoietic cell comprising a genetic modification corresponding to integration of a donor sequence as described herein, e.g., a donor sequence described herein. In some embodiments, integration of the donor sequence into a genetically engineered T cell results in the expression of a chimeric antigen receptor (CAR). In some embodiments, the CAR binds to at least one antigen present on a cell surface protein. In some embodiments, the cell surface protein is CD33. It will be understood that, upon engrafting donor cells into a recipient host organism, the relative levels of the engrafted donor cells (and descendants thereof) and the host cells, e.g., in a given niche (e.g., bone marrow), are important for physiological and/or therapeutic outcomes for the host organism. The level of engrafted donor cells, or descendants thereof, relative to host cells in a given tissue or niche is referred to herein as chimerism. In some embodiments, a cell described herein (e.g., an HSC or HPC) is capable of engrafting in a human subject and does not exhibit any difference in chimerism as compared to a hematopoietic cell of the same cell type, but not comprising a genomic modification that results in expression of a variant form (e.g., a wild-type form or having wild-type functionality) of a gene or a loss of expression of a gene. In some embodiments, a cell described herein (e.g., an HSC or HPC) is capable of engrafting in a human subject exhibits no more than a 1, no more than a 2, no more than a 5, no more than a 10, no more than a 15, no more than a 20, no more than a 25, no more than a 30, no more than a 35, no more than a 40, no more than a 45, or no more than a 50% difference in chimerism as compared to a hematopoietic cell of the same cell type, but not comprising a genomic modification that results in expression of a variant form (e.g., a wild-type form or having wild-type functionality) of a gene or a loss of expression of a gene. In some embodiments, a genetic modification is produced using HDR. In some embodiments, producing a genetic modification using HDR comprises contacting cells with a template polynucleotide, a CRISPR/Cas system, and one or more other agents (e.g., one or more HDR-promoting agents or expansion agents), e.g., contacting cells with a genetic modification mixture described herein. The disclosure provides, in part, methods and compositions that achieve unexpectedly high editing efficiencies utilizing HDR. In some embodiments, efficiency of HDR-mediated editing and/or efficiency of total/overall editing (HDR- and non-HDR-mediated) is determined by a method described herein. In some embodiments, the efficiency of HDR is at least 20, 30, 40, 50, 60, 70, 80, 90, 95, or 99% (e.g., 50%, 60%, 70%, 80%, 90% or higher). In some embodiments, contacting cells to produce a genetic modification using HDR comprises contacting cells with one or more HDR-promoting agents as described herein. Without wishing to be bound by theory, the disclosure is directed, in part, to the discovery that the presence of one or more HDR- promoting agents may result in unexpectedly and advantageously high efficiency of HDR. Accordingly, methods describing contacting a cell herein also contemplate contacting a population of cells to produce a population of genetically modified cells, e.g., an editing efficiency, percent viability, and/or HDR efficiency described herein. In some embodiments, producing a genetic modification using HDR comprises contacting a cell with a genetic modification mixture. As used herein, a genetic modification mixture refers to a mixture comprising a plurality of components that may be used to genetically modify a target DNA, e.g., in a cell. In some embodiments, a genetic modification mixture comprises one, two, three, or all of a CRISPR/Cas system, a template polynucleotide, one or more HDR-promoting agents, and one or more expansion agents. In some embodiments, a genetic modification mixture promotes HDR and HDR-mediated genetic modification (e.g., relative to another DNA repair pathway or genetic modifications utilizing another DNA repair pathway). In some embodiments, contacting a cell with the genetic modification mixture comprises adding the genetic modification mixture directly to media comprising the cell. In some embodiments, contacting a cell with the genetic modification mixture comprises adding media comprising the genetic modification mixture to the cell or adding the cell to media comprising the genetic modification mixture. In some embodiments, the media is a growth media, e.g., a growth media suited to hematopoietic cells (e.g., hematopoietic stem cells (HSCs)). Examples of growth media include, but are not limited to, a Stromal cell Growth Media (SCGMTM, e.g., as available from Lonza Bioscience) or serum- and feeder-free media (SFFM). In some embodiments, contacting a cell with the genetic modification mixture comprises electroporating the genetic modification mixture or one or more components of the mixture into the cell. In some embodiments, contacting a cell with the genetic modification mixture comprises solvating the mixture in a lipid-permeable buffer, e.g., to serve as a carrier for movement of mixture components across the cell membrane. Examples of lipid- permeable buffers include, but are not limited to, DMSO and lipofectamine. In some embodiments, the genetic modification mixture comprises a template polynucleotide, e.g., a single-strand donor oligonucleotide (ssODN), a double-stranded donor oligonucleotide (dsODN), a minicircle plasmid, or a nanoplasmid, comprising a donor sequence, a first flanking sequence and a second flanking sequence. In some embodiments, the genetic modification mixture comprises a dsODN comprising “capped” or “closed-ends” in order to minimize the chances that the NHEJ pathway is used in the genetic modification process. In some embodiments, the dsODN comprising “capped” or “closed-ends” is a GenWand® double-stranded DNA. In some embodiments, the genetic modification mixture comprises a CRISPR/Cas system capable of producing a break, e.g., a double-stranded break, at a target site in the genome of the cell. In some embodiments, the genetic modification mixture comprises one or more other agents (e.g., an expansion agent and/or HDR-promoting agent) that promote genetic modification. In some embodiments, the template polynucleotide, e.g., ssODN, dsODN, minicircle plasmid, or nanoplasmid, and the CRISPR/Cas system of the genetic modification mixture is mixed with the one or more other agents that promote genetic modification. Base Editing In some embodiments, generating a genetically engineered hematopoietic cell comprises contacting a hematopoietic cell with a CRISPR/Cas system comprising a Cas nuclease, wherein the Cas nuclease comprises a base editor. In some embodiments, a base editor is used to a create a genomic modification resulting in an artificial PAM. In some embodiments, a base editor is used to a create a genomic modification resulting in a loss of expression of a protein antigen of interest (e.g., a cell surface protein antigen ), or in expression of a protein antigen variant not targeted by an immunotherapy. A base editor nuclease generally comprises a catalytically inactive Cas9 nuclease fused to a functional domain, e.g., a deaminase domain. See, e.g., Eid et al. Biochem. J. (2018) 475(11): 1955-1964; Rees et al. Nature Reviews Genetics (2018) 19:770-788. In some embodiments, the catalytically inactive Cas9 nuclease is referred to as “dead Cas” or “dCas9.” In some embodiments, the catalytically inactive Cas nuclease has reduced nuclease activity and is, e.g., a nickase (referred to as “nCas”). In some embodiments, the nuclease comprises a dCas9 fused to one or more uracil glycosylase inhibitor (UGI) domains. In some embodiments, the nuclease comprises a dCas9 fused to an adenine base editor (ABE), for example an ABE evolved from the RNA adenine deaminase TadA. In some embodiments, the nuclease comprises a dCas9 fused to cytidine deaminase enzyme (e.g., APOBEC deaminase, pmCDA1, activation-induced cytidine deaminase (AID)). In some embodiments, the catalytically inactive Cas9 nuclease has reduced activity and is nCas9. In some embodiments, the catalytically inactive Cas9 nuclease (dCas9) is fused to one or more uracil glycosylase inhibitor (UGI) domains. In some embodiments, the Cas9 nuclease comprises an inactive Cas9 nuclease (dCas9) fused to an adenine base editor (ABE), for example an ABE evolved from the RNA adenine deaminase TadA. In some embodiments, the Cas9 nuclease comprises a nCas9 fused to an adenine base editor (ABE), for example an ABE evolved from the RNA adenine deaminase TadA. In some embodiments, the Cas9 nuclease comprises a dCas9 fused to cytidine deaminase enzyme (e.g., APOBEC deaminase, pmCDA1, activation- induced cytidine deaminase (AID)). In some embodiments, the Cas9 nuclease comprises a nCas9 fused to cytidine deaminase enzyme (e.g., APOBEC deaminase, pmCDA1, activation- induced cytidine deaminase (AID)). Examples of base editors include, without limitation, BE1, BE2, BE3, HF-BE3, BE4, BE4max, BE4-Gam, YE1-BE3, EE-BE3, YE2-BE3, YEE-CE3, VQR-BE3, VRER-BE3, SaBE3, SaBE4, SaBE4-Gam, Sa(KKH)-BE3, Target-AID, Target-AID-NG, xBE3, eA3A- BE3, BE-PLUS, TAM, CRISPR-X, ABE7.9, ABE7.10, ABE7.10*, ABE8, ABE8e, ABE8.20-m, xABE, ABESa, VQR-ABE, VRER-ABE, Sa(KKH)-ABE, and CRISPR-SKIP. Additional examples of base editors can be found, for example, in US Publication No. 2018/0312825A1, US Publication No.2018/0312828A1, International Publication No. WO 2018/165629A1, International Publication No. WO 2021/030666A1, International Publication No. WO 2022/261509A1, International Publication No. WO 2021/158921A1, and Gaudelli et al., Nat Biotechnol.2020;38(7):892-900, which are incorporated by reference herein in their entireties. In some embodiments, the base editor is a cytosine base editor (CBE). In some embodiments, the CBE is CBE1, CBE2, CBE3, or CBE4. In some embodiments, the CBE is nCas9-2xUGI; BE4-rAPOBEC1; BE4-rAPOBEC1 K34A H122A; BE4-PpAPOBEC1; BE4- PpAPOBEC1 R33A; BE4-PpAPOBEC1 H122A; BE4-RrA3F; BE4-AmAPOBEC1; or BE4- SsAPOBEC3B. In some embodiments, the base editor is an adenine base editor (ABE). In some embodiments, the ABE is ABE1, ABE2, ABE3, ABE4, ABE5, ABE6, ABE7, or ABE8. In some embodiments, the ABE is ABE7.10-m; ABE7.10-d; ABE8e; ABE8.8-m; ABE8.8-d; ABE8.13-m; ABE8.13-d; ABE8.17-m; ABE8.17-d; ABE8.20-m; or ABE8.20-d. In some embodiments, the base editors includes, without limitation, BE1, BE2, BE3, HF-BE3, BE4, BE4max, BE4-Gam, YE1-BE3, EE-BE3, YE2-BE3, YEE-CE3, VQR-BE3, VRER-BE3, SaBE3, SaBE4, SaBE4-Gam, Sa(KKH)-BE3, Target-AID, Target-AID-NG, xBE3, eA3A-BE3, BE-PLUS, TAM, CRISPR-X, ABE7.9, ABE7.10, ABE7.10*, xABE, ABESa, VQR-ABE, VRER-ABE, Sa(KKH)-ABE, and CRISPR-SKIP. In preferred embodiments, the base editor is ABE8e or ABE8.20-m. Additional examples of base editors can be found, for example, in US Publication No. 2018/0312825A1, US Publication No.2018/0312828A1, International Publication No. WO 2018/165629A1, Yu et al. Nat Commun. (2020) 11(1):2052, and Gaudelli et al. Nat Biotechnol. (2020) 38(7):892-900. which are incorporated by reference herein in their entireties. An exemplary abe8_20m sequence is provided below: atgtccgaagtcgagttttcccatgagtactggatgagacacgcattgactctcgcaaagagggctcgagatgaa cgcgaggtgcccgtgggggcagtactcgtgctcaacaatcgcgtaatcggcgaaggttggaatagggcaatcgga ctccacgaccccactgcacatgcggaaatcatggcccttcgacagggagggcttgtgatgcagaattatcgactt tatgatgcgacgctgtactcgacgtttgaaccttgcgtaatgtgcgcgggagctatgattcactcccgcattgga cgagttgtattcggtgttcgcaacgccaagacgggtgccgcaggttcactgatggacgtgctgcatcatccaggc atgaaccaccgggtagaaatcacagaaggcatattggcggacgaatgtgcggcgctgttgtgtcgtttttttcgc atgcccaggcgggtctttaacgcccagaaaaaagcacaatcctctactgactctggtggttcttctggtggttct agcggcagcgagactcccgggacctcagagtccgccacacccgaaagttctggtggttcttctggtggttctgac aagaagtacagcatcggcctggccatcggcaccaactctgtgggctgggccgtgatcaccgacgagtacaaggtg cccagcaagaaattcaaggtgctgggcaacaccgaccggcacagcatcaagaagaacctgatcggagccctgctg ttcgacagcggcgaaacagccgaggccacccggctgaagagaaccgccagaagaagatacaccagacggaagaac cggatctgctatctgcaagagatcttcagcaacgagatggccaaggtggacgacagcttcttccacagactggaa gagtccttcctggtggaagaggataagaagcacgagcggcaccccatcttcggcaacatcgtggacgaggtggcc taccacgagaagtaccccaccatctaccacctgagaaagaaactggtggacagcaccgacaaggccgacctgcgg ctgatctatctggccctggcccacatgatcaagttccggggccacttcctgatcgagggcgacctgaaccccgac aacagcgacgtggacaagctgttcatccagctggtgcagacctacaaccagctgttcgaggaaaaccccatcaac gccagcggcgtggacgccaaggccatcctgtctgccagactgagcaagagcagacggctggaaaatctgatcgcc cagctgcccggcgagaagaagaatggcctgttcggaaacctgattgccctgagcctgggcctgacccccaacttc aagagcaacttcgacctggccgaggatgccaaactgcagctgagcaaggacacctacgacgacgacctggacaac ctgctggcccagatcggcgaccagtacgccgacctgtttctggccgccaagaacctgtccgacgccatcctgctg agcgacatcctgagagtgaacaccgagatcaccaaggcccccctgagcgcctctatgatcaagagatacgacgag caccaccaggacctgaccctgctgaaagctctcgtgcggcagcagctgcctgagaagtacaaagagattttcttc gaccagagcaagaacggctacgccggctacattgacggcggagccagccaggaagagttctacaagttcatcaag cccatcctggaaaagatggacggcaccgaggaactgctcgtgaagctgaacagagaggacctgctgcggaagcag cggaccttcgacaacggcagcatcccccaccagatccacctgggagagctgcacgccattctgcggcggcaggaa gatttttacccattcctgaaggacaaccgggaaaagatcgagaagatcctgaccttccgcatcccctactacgtg ggccctctggccaggggaaacagcagattcgcctggatgaccagaaagagcgaggaaaccatcaccccctggaac ttcgaggaagtggtggacaagggcgcttccgcccagagcttcatcgagcggatgaccaacttcgataagaacctg cccaacgagaaggtgctgcccaagcacagcctgctgtacgagtacttcaccgtgtataacgagctgaccaaagtg aaatacgtgaccgagggaatgagaaagcccgccttcctgagcggcgagcagaaaaaggccatcgtggacctgctg ttcaagaccaaccggaaagtgaccgtgaagcagctgaaagaggactacttcaagaaaatcgagtgcttcgactcc gtggaaatctccggcgtggaagatcggttcaacgcctccctgggcacataccacgatctgctgaaaattatcaag gacaaggacttcctggacaatgaggaaaacgaggacattctggaagatatcgtgctgaccctgacactgtttgag gacagagagatgatcgaggaacggctgaaaacctatgcccacctgttcgacgacaaagtgatgaagcagctgaag cggcggagatacaccggctggggcaggctgagccggaagctgatcaacggcatccgggacaagcagtccggcaag acaatcctggatttcctgaagtccgacggcttcgccaacagaaacttcatgcagctgatccacgacgacagcctg acctttaaagaggacatccagaaagcccaggtgtccggccagggcgatagcctgcacgagcacattgccaatctg gccggcagccccgccattaagaagggcatcctgcagacagtgaaggtggtggacgagctcgtgaaagtgatgggc cggcacaagcccgagaacatcgtgatcgaaatggccagagagaaccagaccacccagaagggacagaagaacagc cgcgagagaatgaagcggatcgaagagggcatcaaagagctgggcagccagatcctgaaagaacaccccgtggaa aacacccagctgcagaacgagaagctgtacctgtactacctgcagaatgggcgggatatgtacgtggaccaggaa ctggacatcaaccggctgtccgactacgatgtggaccatatcgtgcctcagagctttctgaaggacgactccatcgacaacaaggtgctgaccagaagcgacaagaaccggggcaagagcgacaacgtgccctccgaagaggtcgtgaag aagatgaagaactactggcggcagctgctgaacgccaagctgattacccagagaaagttcgacaatctgaccaag gccgagagaggcggcctgagcgaactggataaggccggcttcatcaagagacagctggtggaaacccggcagatc acaaagcacgtggcacagatcctggactcccggatgaacactaagtacgacgagaatgacaagctgatccgggaa gtgaaagtgatcaccctgaagtccaagctggtgtccgatttccggaaggatttccagttttacaaagtgcgcgag atcaacaactaccaccacgcccacgacgcctacctgaacgccgtcgtgggaaccgccctgatcaaaaagtaccct aagctggaaagcgagttcgtgtacggcgactacaaggtgtacgacgtgcggaagatgatcgccaagagcgagcag gaaatcggcaaggctaccgccaagtacttcttctacagcaacatcatgaactttttcaagaccgagattaccctg gccaacggcgagatccggaagcggcctctgatcgagacaaacggcgaaaccggggagatcgtgtgggataagggc cgggattttgccaccgtgcggaaagtgctgagcatgccccaagtgaatatcgtgaaaaagaccgaggtgcagaca ggcggcttcagcaaagagtctatcctgcccaagaggaacagcgataagctgatcgccagaaagaaggactgggac cctaagaagtacggcggcttcgacagccccaccgtggcctattctgtgctggtggtggccaaagtggaaaagggc aagtccaagaaactgaagagtgtgaaagagctgctggggatcaccatcatggaaagaagcagcttcgagaagaat cccatcgactttctggaagccaagggctacaaagaagtgaaaaaggacctgatcatcaagctgcctaagtactcc ctgttcgagctggaaaacggccggaagagaatgctggcctctgccggcgaactgcagaagggaaacgaactggccctgccctccaaatatgtgaacttcctgtacctggccagccactatgagaagctgaagggctcccccgaggataat gagcagaaacagctgtttgtggaacagcacaagcactacctggacgagatcatcgagcagatcagcgagttctcc aagagagtgatcctggccgacgctaatctggacaaagtgctgtccgcctacaacaagcaccgggataagcccatc agagagcaggccgagaatatcatccacctgtttaccctgaccaatctgggagcccctgccgccttcaagtacttt gacaccaccatcgaccggaagaggtacaccagcaccaaagaggtgctggacgccaccctgatccaccagagcatc accggcctgtacgagacacggatcgacctgtctcagctgggaggtgacgagggagctgataagcgcaccgccgatggttccgagttcgaaagccccaagaagaagaggaaagtc (SEQ ID NO: 31) An exemplary ppabobec1-orf-r33a sequence is provided below: atgacctctgagaagggccctagcacaggcgaccccaccctgcggcggagaatcgagagctgggagttcgacgtgttctacgaccctagagaactggccaaggaaacctgcctgctgtacgagatcaagtggggcatgagcagaaagatc tggcggagctctggcaagaacaccaccaaccacgtggaagtgaatttcatcaagaagttcaccagcgagagaagg ttccacagcagcatcagctgcagcatcacctggttcctgagctggtccccttgctgggaatgcagccaggccatc agagagttcctgagccaacaccccggagtgacactggtgatctacgtggccagactgttctggcacatggaccag agaaacagacagggcctgagagatctggtcaacagcggcgtgactatccagatcatgcgggccagcgagtactac cactgttggcggaacttcgtgaactacccccccggcgatgaggcccactggcctcagtaccctcctctgtggatg atgctgtacgccctggaactgcactgcatcatcctgtctctgcctccatgtctgaagatctctagaagatggcag aaccacctggccttcttcagactgcacctgcagaattgccactaccagaccatccccccccacatcctgctggct acaggcctgatccacccttctgtgacctggagacttaagagcggaggatctagcggcggctctagcggatctgag acacctggcacaagcgagtctgccacacctgagagtagcggcggatcttctggtggctctgacaagaagtacagc atcggcctggccatcggcaccaactctgtgggctgggccgtgatcaccgacgagtacaaggtgcccagcaagaaa ttcaaggtgctgggcaacaccgaccggcacagcatcaagaagaacctgatcggagccctgctgttcgacagcggc gaaacagccgaggccacccggctgaagagaaccgccagaagaagatacaccagacggaagaaccggatctgctat ctgcaagagatcttcagcaacgagatggccaaggtggacgacagcttcttccacagactggaagagtccttcctg gtggaagaggataagaagcacgagcggcaccccatcttcggcaacatcgtggacgaggtggcctaccacgagaag taccccaccatctaccacctgagaaagaaactggtggacagcaccgacaaggccgacctgcggctgatctatctg gccctggcccacatgatcaagttccggggccacttcctgatcgagggcgacctgaaccccgacaacagcgacgtg gacaagctgttcatccagctggtgcagacctacaaccagctgttcgaggaaaaccccatcaacgccagcggcgtg gacgccaaggccatcctgtctgccagactgagcaagagcagacggctggaaaatctgatcgcccagctgcccggc gagaagaagaatggcctgttcggaaacctgattgccctgagcctgggcctgacccccaacttcaagagcaacttc gacctggccgaggatgccaaactgcagctgagcaaggacacctacgacgacgacctggacaacctgctggcccag atcggcgaccagtacgccgacctgtttctggccgccaagaacctgtccgacgccatcctgctgagcgacatcctg agagtgaacaccgagatcaccaaggcccccctgagcgcctctatgatcaagagatacgacgagcaccaccaggac ctgaccctgctgaaagctctcgtgcggcagcagctgcctgagaagtacaaagagattttcttcgaccagagcaag aacggctacgccggctacattgacggcggagccagccaggaagagttctacaagttcatcaagcccatcctggaa aagatggacggcaccgaggaactgctcgtgaagctgaacagagaggacctgctgcggaagcagcggaccttcgac aacggcagcatcccccaccagatccacctgggagagctgcacgccattctgcggcggcaggaagatttttaccca ttcctgaaggacaaccgggaaaagatcgagaagatcctgaccttccgcatcccctactacgtgggccctctggcc aggggaaacagcagattcgcctggatgaccagaaagagcgaggaaaccatcaccccctggaacttcgaggaagtg gtggacaagggcgcttccgcccagagcttcatcgagcggatgaccaacttcgataagaacctgcccaacgagaag gtgctgcccaagcacagcctgctgtacgagtacttcaccgtgtataacgagctgaccaaagtgaaatacgtgacc gagggaatgagaaagcccgccttcctgagcggcgagcagaaaaaggccatcgtggacctgctgttcaagaccaac cggaaagtgaccgtgaagcagctgaaagaggactacttcaagaaaatcgagtgcttcgactccgtggaaatctcc ggcgtggaagatcggttcaacgcctccctgggcacataccacgatctgctgaaaattatcaaggacaaggacttc ctggacaatgaggaaaacgaggacattctggaagatatcgtgctgaccctgacactgtttgaggacagagagatg atcgaggaacggctgaaaacctatgcccacctgttcgacgacaaagtgatgaagcagctgaagcggcggagatacaccggctggggcaggctgagccggaagctgatcaacggcatccgggacaagcagtccggcaagacaatcctggat ttcctgaagtccgacggcttcgccaacagaaacttcatgcagctgatccacgacgacagcctgacctttaaagag gacatccagaaagcccaggtgtccggccagggcgatagcctgcacgagcacattgccaatctggccggcagcccc gccattaagaagggcatcctgcagacagtgaaggtggtggacgagctcgtgaaagtgatgggccggcacaagccc gagaacatcgtgatcgaaatggccagagagaaccagaccacccagaagggacagaagaacagccgcgagagaatg aagcggatcgaagagggcatcaaagagctgggcagccagatcctgaaagaacaccccgtggaaaacacccagctg cagaacgagaagctgtacctgtactacctgcagaatgggcgggatatgtacgtggaccaggaactggacatcaac cggctgtccgactacgatgtggaccatatcgtgcctcagagctttctgaaggacgactccatcgacaacaaggtg ctgaccagaagcgacaagaaccggggcaagagcgacaacgtgccctccgaagaggtcgtgaagaagatgaagaac tactggcggcagctgctgaacgccaagctgattacccagagaaagttcgacaatctgaccaaggccgagagaggc ggcctgagcgaactggataaggccggcttcatcaagagacagctggtggaaacccggcagatcacaaagcacgtg gcacagatcctggactcccggatgaacactaagtacgacgagaatgacaagctgatccgggaagtgaaagtgatc accctgaagtccaagctggtgtccgatttccggaaggatttccagttttacaaagtgcgcgagatcaacaactac caccacgcccacgacgcctacctgaacgccgtcgtgggaaccgccctgatcaaaaagtaccctaagctggaaagc gagttcgtgtacggcgactacaaggtgtacgacgtgcggaagatgatcgccaagagcgagcaggaaatcggcaaggctaccgccaagtacttcttctacagcaacatcatgaactttttcaagaccgagattaccctggccaacggcgag atccggaagcggcctctgatcgagacaaacggcgaaaccggggagatcgtgtgggataagggccgggattttgcc accgtgcggaaagtgctgagcatgccccaagtgaatatcgtgaaaaagaccgaggtgcagacaggcggcttcagc aaagagtctatcctgcccaagaggaacagcgataagctgatcgccagaaagaaggactgggaccctaagaagtac ggcggcttcgacagccccaccgtggcctattctgtgctggtggtggccaaagtggaaaagggcaagtccaagaaa ctgaagagtgtgaaagagctgctggggatcaccatcatggaaagaagcagcttcgagaagaatcccatcgacttt ctggaagccaagggctacaaagaagtgaaaaaggacctgatcatcaagctgcctaagtactccctgttcgagctg gaaaacggccggaagagaatgctggcctctgccggcgaactgcagaagggaaacgaactggccctgccctccaaa tatgtgaacttcctgtacctggccagccactatgagaagctgaagggctcccccgaggataatgagcagaaacag ctgtttgtggaacagcacaagcactacctggacgagatcatcgagcagatcagcgagttctccaagagagtgatc ctggccgacgctaatctggacaaagtgctgtccgcctacaacaagcaccgggataagcccatcagagagcaggcc gagaatatcatccacctgtttaccctgaccaatctgggagcccctgccgccttcaagtactttgacaccaccatc gaccggaagaggtacaccagcaccaaagaggtgctggacgccaccctgatccaccagagcatcaccggcctgtac gagacacggatcgacctgtctcagctgggaggtgactctggtggaagcggaggatctggcggcagcaccaatctg agcgacatcatcgagaaagagacaggcaagcagctggtcatccaagagtccatcctgatgctgcctgaagaggtg gaagaagtgatcggcaacaagcccgagtccgacatcctggtgcacaccgcctacgatgagagcaccgacgagaac gtgatgctgctgacctctgacgcccctgagtacaagccttgggctctcgtgatccaggacagcaacggcgagaac aagatcaagatgctgagcggcggctctggtggctctggcggatctacaaacctgtccgatattattgagaaagaa accgggaaacagctcgtgattcaagagtctattctcatgctcccggaagaagtcgaggaagtcattggaaacaag cctgagagcgatattctggtccatacagcctacgacgagtctaccgatgagaatgtcatgctcctcaccagcgac gctcccgagtataagccatgggcacttgtcattcaggactccaatggggaaaacaaaatcaaaatgctcccaaag aaaaaacgcaaggtg (SEQ ID NO: 32) An exemplary ppabobec1-orf-wt sequence is provided below: atgacctctgagaagggccctagcacaggcgaccccaccctgcggcggagaatcgagagctgggagttcgacgtg ttctacgaccctagagaactgagaaaggaaacctgcctgctgtacgagatcaagtggggcatgagcagaaagatc tggcggagctctggcaagaacaccaccaaccacgtggaagtgaatttcatcaagaagttcaccagcgagagaagg ttccacagcagcatcagctgcagcatcacctggttcctgagctggtccccttgctgggaatgcagccaggccatc agagagttcctgagccaacaccccggagtgacactggtgatctacgtggccagactgttctggcacatggaccag agaaacagacagggcctgagagatctggtcaacagcggcgtgactatccagatcatgcgggccagcgagtactac cactgttggcggaacttcgtgaactacccccccggcgatgaggcccactggcctcagtaccctcctctgtggatg atgctgtacgccctggaactgcactgcatcatcctgtctctgcctccatgtctgaagatctctagaagatggcag aaccacctggccttcttcagactgcacctgcagaattgccactaccagaccatccccccccacatcctgctggct acaggcctgatccacccttctgtgacctggagacttaagagcggaggatctagcggcggctctagcggatctgag acacctggcacaagcgagtctgccacacctgagagtagcggcggatcttctggtggctctgacaagaagtacagc atcggcctggccatcggcaccaactctgtgggctgggccgtgatcaccgacgagtacaaggtgcccagcaagaaa ttcaaggtgctgggcaacaccgaccggcacagcatcaagaagaacctgatcggagccctgctgttcgacagcggc gaaacagccgaggccacccggctgaagagaaccgccagaagaagatacaccagacggaagaaccggatctgctat ctgcaagagatcttcagcaacgagatggccaaggtggacgacagcttcttccacagactggaagagtccttcctg gtggaagaggataagaagcacgagcggcaccccatcttcggcaacatcgtggacgaggtggcctaccacgagaag taccccaccatctaccacctgagaaagaaactggtggacagcaccgacaaggccgacctgcggctgatctatctg gccctggcccacatgatcaagttccggggccacttcctgatcgagggcgacctgaaccccgacaacagcgacgtg gacaagctgttcatccagctggtgcagacctacaaccagctgttcgaggaaaaccccatcaacgccagcggcgtg gacgccaaggccatcctgtctgccagactgagcaagagcagacggctggaaaatctgatcgcccagctgcccggc gagaagaagaatggcctgttcggaaacctgattgccctgagcctgggcctgacccccaacttcaagagcaacttc gacctggccgaggatgccaaactgcagctgagcaaggacacctacgacgacgacctggacaacctgctggcccagatcggcgaccagtacgccgacctgtttctggccgccaagaacctgtccgacgccatcctgctgagcgacatcctg agagtgaacaccgagatcaccaaggcccccctgagcgcctctatgatcaagagatacgacgagcaccaccaggac ctgaccctgctgaaagctctcgtgcggcagcagctgcctgagaagtacaaagagattttcttcgaccagagcaag aacggctacgccggctacattgacggcggagccagccaggaagagttctacaagttcatcaagcccatcctggaa aagatggacggcaccgaggaactgctcgtgaagctgaacagagaggacctgctgcggaagcagcggaccttcgac aacggcagcatcccccaccagatccacctgggagagctgcacgccattctgcggcggcaggaagatttttaccca ttcctgaaggacaaccgggaaaagatcgagaagatcctgaccttccgcatcccctactacgtgggccctctggcc aggggaaacagcagattcgcctggatgaccagaaagagcgaggaaaccatcaccccctggaacttcgaggaagtg gtggacaagggcgcttccgcccagagcttcatcgagcggatgaccaacttcgataagaacctgcccaacgagaag gtgctgcccaagcacagcctgctgtacgagtacttcaccgtgtataacgagctgaccaaagtgaaatacgtgacc gagggaatgagaaagcccgccttcctgagcggcgagcagaaaaaggccatcgtggacctgctgttcaagaccaac cggaaagtgaccgtgaagcagctgaaagaggactacttcaagaaaatcgagtgcttcgactccgtggaaatctcc ggcgtggaagatcggttcaacgcctccctgggcacataccacgatctgctgaaaattatcaaggacaaggacttc ctggacaatgaggaaaacgaggacattctggaagatatcgtgctgaccctgacactgtttgaggacagagagatg atcgaggaacggctgaaaacctatgcccacctgttcgacgacaaagtgatgaagcagctgaagcggcggagatacaccggctggggcaggctgagccggaagctgatcaacggcatccgggacaagcagtccggcaagacaatcctggat ttcctgaagtccgacggcttcgccaacagaaacttcatgcagctgatccacgacgacagcctgacctttaaagag gacatccagaaagcccaggtgtccggccagggcgatagcctgcacgagcacattgccaatctggccggcagcccc gccattaagaagggcatcctgcagacagtgaaggtggtggacgagctcgtgaaagtgatgggccggcacaagccc gagaacatcgtgatcgaaatggccagagagaaccagaccacccagaagggacagaagaacagccgcgagagaatg aagcggatcgaagagggcatcaaagagctgggcagccagatcctgaaagaacaccccgtggaaaacacccagctg cagaacgagaagctgtacctgtactacctgcagaatgggcgggatatgtacgtggaccaggaactggacatcaac cggctgtccgactacgatgtggaccatatcgtgcctcagagctttctgaaggacgactccatcgacaacaaggtg ctgaccagaagcgacaagaaccggggcaagagcgacaacgtgccctccgaagaggtcgtgaagaagatgaagaac tactggcggcagctgctgaacgccaagctgattacccagagaaagttcgacaatctgaccaaggccgagagaggc ggcctgagcgaactggataaggccggcttcatcaagagacagctggtggaaacccggcagatcacaaagcacgtg gcacagatcctggactcccggatgaacactaagtacgacgagaatgacaagctgatccgggaagtgaaagtgatc accctgaagtccaagctggtgtccgatttccggaaggatttccagttttacaaagtgcgcgagatcaacaactac caccacgcccacgacgcctacctgaacgccgtcgtgggaaccgccctgatcaaaaagtaccctaagctggaaagc gagttcgtgtacggcgactacaaggtgtacgacgtgcggaagatgatcgccaagagcgagcaggaaatcggcaag gctaccgccaagtacttcttctacagcaacatcatgaactttttcaagaccgagattaccctggccaacggcgag atccggaagcggcctctgatcgagacaaacggcgaaaccggggagatcgtgtgggataagggccgggattttgcc accgtgcggaaagtgctgagcatgccccaagtgaatatcgtgaaaaagaccgaggtgcagacaggcggcttcagc aaagagtctatcctgcccaagaggaacagcgataagctgatcgccagaaagaaggactgggaccctaagaagtac ggcggcttcgacagccccaccgtggcctattctgtgctggtggtggccaaagtggaaaagggcaagtccaagaaa ctgaagagtgtgaaagagctgctggggatcaccatcatggaaagaagcagcttcgagaagaatcccatcgacttt ctggaagccaagggctacaaagaagtgaaaaaggacctgatcatcaagctgcctaagtactccctgttcgagctg gaaaacggccggaagagaatgctggcctctgccggcgaactgcagaagggaaacgaactggccctgccctccaaa tatgtgaacttcctgtacctggccagccactatgagaagctgaagggctcccccgaggataatgagcagaaacag ctgtttgtggaacagcacaagcactacctggacgagatcatcgagcagatcagcgagttctccaagagagtgatc ctggccgacgctaatctggacaaagtgctgtccgcctacaacaagcaccgggataagcccatcagagagcaggcc gagaatatcatccacctgtttaccctgaccaatctgggagcccctgccgccttcaagtactttgacaccaccatc gaccggaagaggtacaccagcaccaaagaggtgctggacgccaccctgatccaccagagcatcaccggcctgtac gagacacggatcgacctgtctcagctgggaggtgactctggtggaagcggaggatctggcggcagcaccaatctg agcgacatcatcgagaaagagacaggcaagcagctggtcatccaagagtccatcctgatgctgcctgaagaggtg gaagaagtgatcggcaacaagcccgagtccgacatcctggtgcacaccgcctacgatgagagcaccgacgagaac gtgatgctgctgacctctgacgcccctgagtacaagccttgggctctcgtgatccaggacagcaacggcgagaac aagatcaagatgctgagcggcggctctggtggctctggcggatctacaaacctgtccgatattattgagaaagaa accgggaaacagctcgtgattcaagagtctattctcatgctcccggaagaagtcgaggaagtcattggaaacaag cctgagagcgatattctggtccatacagcctacgacgagtctaccgatgagaatgtcatgctcctcaccagcgac gctcccgagtataagccatgggcacttgtcattcaggactccaatggggaaaacaaaatcaaaatgctcccaaag aaaaaacgcaaggtg (SEQ ID NO: 33) An exemplary prtn_SzW8eqL7-abe8_20m sequence is provided below: MSEVEFSHEYWMRHALTLAKRARDEREVPVGAVLVLNNRVIGEGWNRAIGLHDPTAHAEIMALRQGGLVMQNYRL YDATLYSTFEPCVMCAGAMIHSRIGRVVFGVRNAKTGAAGSLMDVLHHPGMNHRVEITEGILADECAALLCRFFR MPRRVFNAQKKAQSSTDSGGSSGGSSGSETPGTSESATPESSGGSSGGSDKKYSIGLAIGTNSVGWAVITDEYKV PSKKFKVLGNTDRHSIKKNLIGALLFDSGETAEATRLKRTARRRYTRRKNRICYLQEIFSNEMAKVDDSFFHRLE ESFLVEEDKKHERHPIFGNIVDEVAYHEKYPTIYHLRKKLVDSTDKADLRLIYLALAHMIKFRGHFLIEGDLNPD NSDVDKLFIQLVQTYNQLFEENPINASGVDAKAILSARLSKSRRLENLIAQLPGEKKNGLFGNLIALSLGLTPNF KSNFDLAEDAKLQLSKDTYDDDLDNLLAQIGDQYADLFLAAKNLSDAILLSDILRVNTEITKAPLSASMIKRYDE HHQDLTLLKALVRQQLPEKYKEIFFDQSKNGYAGYIDGGASQEEFYKFIKPILEKMDGTEELLVKLNREDLLRKQ RTFDNGSIPHQIHLGELHAILRRQEDFYPFLKDNREKIEKILTFRIPYYVGPLARGNSRFAWMTRKSEETITPWN FEEVVDKGASAQSFIERMTNFDKNLPNEKVLPKHSLLYEYFTVYNELTKVKYVTEGMRKPAFLSGEQKKAIVDLL FKTNRKVTVKQLKEDYFKKIECFDSVEISGVEDRFNASLGTYHDLLKIIKDKDFLDNEENEDILEDIVLTLTLFE DREMIEERLKTYAHLFDDKVMKQLKRRRYTGWGRLSRKLINGIRDKQSGKTILDFLKSDGFANRNFMQLIHDDSL TFKEDIQKAQVSGQGDSLHEHIANLAGSPAIKKGILQTVKVVDELVKVMGRHKPENIVIEMARENQTTQKGQKNS RERMKRIEEGIKELGSQILKEHPVENTQLQNEKLYLYYLQNGRDMYVDQELDINRLSDYDVDHIVPQSFLKDDSI DNKVLTRSDKNRGKSDNVPSEEVVKKMKNYWRQLLNAKLITQRKFDNLTKAERGGLSELDKAGFIKRQLVETRQI TKHVAQILDSRMNTKYDENDKLIREVKVITLKSKLVSDFRKDFQFYKVREINNYHHAHDAYLNAVVGTALIKKYP KLESEFVYGDYKVYDVRKMIAKSEQEIGKATAKYFFYSNIMNFFKTEITLANGEIRKRPLIETNGETGEIVWDKG RDFATVRKVLSMPQVNIVKKTEVQTGGFSKESILPKRNSDKLIARKKDWDPKKYGGFDSPTVAYSVLVVAKVEKGKSKKLKSVKELLGITIMERSSFEKNPIDFLEAKGYKEVKKDLIIKLPKYSLFELENGRKRMLASAGELQKGNELA LPSKYVNFLYLASHYEKLKGSPEDNEQKQLFVEQHKHYLDEIIEQISEFSKRVILADANLDKVLSAYNKHRDKPI REQAENIIHLFTLTNLGAPAAFKYFDTTIDRKRYTSTKEVLDATLIHQSITGLYETRIDLSQLGGDEGADKRTADGSEFESPKKKRKV (SEQ ID NO: 34) An exemplary prtn_ZJVPExXY-ppabobec1-r33a-protein sequence is provided below: MTSEKGPSTGDPTLRRRIESWEFDVFYDPRELAKETCLLYEIKWGMSRKIWRSSGKNTTNHVEVNFIKKFTSERR FHSSISCSITWFLSWSPCWECSQAIREFLSQHPGVTLVIYVARLFWHMDQRNRQGLRDLVNSGVTIQIMRASEYY HCWRNFVNYPPGDEAHWPQYPPLWMMLYALELHCIILSLPPCLKISRRWQNHLAFFRLHLQNCHYQTIPPHILLA TGLIHPSVTWRLKSGGSSGGSSGSETPGTSESATPESSGGSSGGSDKKYSIGLAIGTNSVGWAVITDEYKVPSKKFKVLGNTDRHSIKKNLIGALLFDSGETAEATRLKRTARRRYTRRKNRICYLQEIFSNEMAKVDDSFFHRLEESFL VEEDKKHERHPIFGNIVDEVAYHEKYPTIYHLRKKLVDSTDKADLRLIYLALAHMIKFRGHFLIEGDLNPDNSDV DKLFIQLVQTYNQLFEENPINASGVDAKAILSARLSKSRRLENLIAQLPGEKKNGLFGNLIALSLGLTPNFKSNF DLAEDAKLQLSKDTYDDDLDNLLAQIGDQYADLFLAAKNLSDAILLSDILRVNTEITKAPLSASMIKRYDEHHQD LTLLKALVRQQLPEKYKEIFFDQSKNGYAGYIDGGASQEEFYKFIKPILEKMDGTEELLVKLNREDLLRKQRTFD NGSIPHQIHLGELHAILRRQEDFYPFLKDNREKIEKILTFRIPYYVGPLARGNSRFAWMTRKSEETITPWNFEEV VDKGASAQSFIERMTNFDKNLPNEKVLPKHSLLYEYFTVYNELTKVKYVTEGMRKPAFLSGEQKKAIVDLLFKTN RKVTVKQLKEDYFKKIECFDSVEISGVEDRFNASLGTYHDLLKIIKDKDFLDNEENEDILEDIVLTLTLFEDREM IEERLKTYAHLFDDKVMKQLKRRRYTGWGRLSRKLINGIRDKQSGKTILDFLKSDGFANRNFMQLIHDDSLTFKE DIQKAQVSGQGDSLHEHIANLAGSPAIKKGILQTVKVVDELVKVMGRHKPENIVIEMARENQTTQKGQKNSRERM KRIEEGIKELGSQILKEHPVENTQLQNEKLYLYYLQNGRDMYVDQELDINRLSDYDVDHIVPQSFLKDDSIDNKV LTRSDKNRGKSDNVPSEEVVKKMKNYWRQLLNAKLITQRKFDNLTKAERGGLSELDKAGFIKRQLVETRQITKHV AQILDSRMNTKYDENDKLIREVKVITLKSKLVSDFRKDFQFYKVREINNYHHAHDAYLNAVVGTALIKKYPKLES EFVYGDYKVYDVRKMIAKSEQEIGKATAKYFFYSNIMNFFKTEITLANGEIRKRPLIETNGETGEIVWDKGRDFA TVRKVLSMPQVNIVKKTEVQTGGFSKESILPKRNSDKLIARKKDWDPKKYGGFDSPTVAYSVLVVAKVEKGKSKKLKSVKELLGITIMERSSFEKNPIDFLEAKGYKEVKKDLIIKLPKYSLFELENGRKRMLASAGELQKGNELALPSK YVNFLYLASHYEKLKGSPEDNEQKQLFVEQHKHYLDEIIEQISEFSKRVILADANLDKVLSAYNKHRDKPIREQA ENIIHLFTLTNLGAPAAFKYFDTTIDRKRYTSTKEVLDATLIHQSITGLYETRIDLSQLGGDSGGSGGSGGSTNL SDIIEKETGKQLVIQESILMLPEEVEEVIGNKPESDILVHTAYDESTDENVMLLTSDAPEYKPWALVIQDSNGEN KIKMLSGGSGGSGGSTNLSDIIEKETGKQLVIQESILMLPEEVEEVIGNKPESDILVHTAYDESTDENVMLLTSD APEYKPWALVIQDSNGENKIKMLPKKKRKV (SEQ ID NO: 35) An exemplary prtn_ZyqE8AYc-ppabobec1-wt-protein sequence is provided below: MTSEKGPSTGDPTLRRRIESWEFDVFYDPRELRKETCLLYEIKWGMSRKIWRSSGKNTTNHVEVNFIKKFTSERR FHSSISCSITWFLSWSPCWECSQAIREFLSQHPGVTLVIYVARLFWHMDQRNRQGLRDLVNSGVTIQIMRASEYY HCWRNFVNYPPGDEAHWPQYPPLWMMLYALELHCIILSLPPCLKISRRWQNHLAFFRLHLQNCHYQTIPPHILLA TGLIHPSVTWRLKSGGSSGGSSGSETPGTSESATPESSGGSSGGSDKKYSIGLAIGTNSVGWAVITDEYKVPSKK FKVLGNTDRHSIKKNLIGALLFDSGETAEATRLKRTARRRYTRRKNRICYLQEIFSNEMAKVDDSFFHRLEESFL VEEDKKHERHPIFGNIVDEVAYHEKYPTIYHLRKKLVDSTDKADLRLIYLALAHMIKFRGHFLIEGDLNPDNSDV DKLFIQLVQTYNQLFEENPINASGVDAKAILSARLSKSRRLENLIAQLPGEKKNGLFGNLIALSLGLTPNFKSNF DLAEDAKLQLSKDTYDDDLDNLLAQIGDQYADLFLAAKNLSDAILLSDILRVNTEITKAPLSASMIKRYDEHHQD LTLLKALVRQQLPEKYKEIFFDQSKNGYAGYIDGGASQEEFYKFIKPILEKMDGTEELLVKLNREDLLRKQRTFD NGSIPHQIHLGELHAILRRQEDFYPFLKDNREKIEKILTFRIPYYVGPLARGNSRFAWMTRKSEETITPWNFEEV VDKGASAQSFIERMTNFDKNLPNEKVLPKHSLLYEYFTVYNELTKVKYVTEGMRKPAFLSGEQKKAIVDLLFKTN RKVTVKQLKEDYFKKIECFDSVEISGVEDRFNASLGTYHDLLKIIKDKDFLDNEENEDILEDIVLTLTLFEDREM IEERLKTYAHLFDDKVMKQLKRRRYTGWGRLSRKLINGIRDKQSGKTILDFLKSDGFANRNFMQLIHDDSLTFKE DIQKAQVSGQGDSLHEHIANLAGSPAIKKGILQTVKVVDELVKVMGRHKPENIVIEMARENQTTQKGQKNSRERM KRIEEGIKELGSQILKEHPVENTQLQNEKLYLYYLQNGRDMYVDQELDINRLSDYDVDHIVPQSFLKDDSIDNKV LTRSDKNRGKSDNVPSEEVVKKMKNYWRQLLNAKLITQRKFDNLTKAERGGLSELDKAGFIKRQLVETRQITKHV AQILDSRMNTKYDENDKLIREVKVITLKSKLVSDFRKDFQFYKVREINNYHHAHDAYLNAVVGTALIKKYPKLES EFVYGDYKVYDVRKMIAKSEQEIGKATAKYFFYSNIMNFFKTEITLANGEIRKRPLIETNGETGEIVWDKGRDFATVRKVLSMPQVNIVKKTEVQTGGFSKESILPKRNSDKLIARKKDWDPKKYGGFDSPTVAYSVLVVAKVEKGKSKK LKSVKELLGITIMERSSFEKNPIDFLEAKGYKEVKKDLIIKLPKYSLFELENGRKRMLASAGELQKGNELALPSK YVNFLYLASHYEKLKGSPEDNEQKQLFVEQHKHYLDEIIEQISEFSKRVILADANLDKVLSAYNKHRDKPIREQA ENIIHLFTLTNLGAPAAFKYFDTTIDRKRYTSTKEVLDATLIHQSITGLYETRIDLSQLGGDSGGSGGSGGSTNL SDIIEKETGKQLVIQESILMLPEEVEEVIGNKPESDILVHTAYDESTDENVMLLTSDAPEYKPWALVIQDSNGEN KIKMLSGGSGGSGGSTNLSDIIEKETGKQLVIQESILMLPEEVEEVIGNKPESDILVHTAYDESTDENVMLLTSDAPEYKPWALVIQDSNGENKIKMLPKKKRKV (SEQ ID NO: 36) In some embodiments, the base editor has been further modified to inhibit base excision repair at the target site and induce cellular mismatch repair. Any of the Cas9 nucleases described herein may be fused to a Gam domain (bacteriophage Mu protein) to protect the Cas9 nuclease from degradation and exonuclease activity. See, e.g., Eid et al. Biochem. J. (2018) 475(11): 1955-1964. In some embodiments, the Cas nuclease belongs to class 2 type V of Cas endonuclease. Class 2 type V Cas endonucleases can be further categorized as type V-A, type V-B, type V-C, and type V-U. See, e.g., Stella et al. Nature Structural & Molecular Biology (2017) 24: 882-892. In some embodiments, the Cas nuclease is a type V-A Cas endonuclease, such as a Cpf1 (Cas12a) nuclease. In some embodiments, the Cas9 nuclease is a type V-B Cas endonuclease, such as a C2c1 endonuclease. See, e.g., Shmakov et al. Mol Cell (2015) 60: 385-397. In some embodiments, the Cas nuclease is MAD7TM. Alternatively, or in addition, the Cas9 nuclease is a Cpf1 nuclease or a variant thereof. As will be appreciated by one of skill in the art, the Cpf1 nuclease may also be referred to as Cas12a. See, e.g., Strohkendl et al. Mol. Cell (2018) 71: 1-9. In some embodiments, a composition or method described herein involves, or a host cell expresses a Cpf1 nuclease derived from Provetella spp. or Francisella spp., Acidaminococcus sp. (AsCpf1), Lachnospiraceae bacterium (LpCpf1), or Eubacterium rectale. In some embodiments, the nucleotide sequence encoding the Cpf1 nuclease may be codon optimized for expression in a host cell. In some embodiments, the nucleotide sequence encoding the Cpf1 endonuclease is further modified to alter the activity of the protein. Both naturally occurring and modified variants of CRISPR/Cas nucleases are suitable for use according to aspects of this disclosure. For example, dCas or nickase variants, Cas variants having altered PAM specificities, and Cas variants having improved nuclease activities are embraced by some embodiments of this disclosure. In some embodiments, catalytically inactive variants of Cas nucleases (e.g., of Cas9 or Cas12a) are used according to the methods described herein. A catalytically inactive variant of Cpf1 (Cas12a) may be referred to dCas12a. As described herein, catalytically inactive variants of Cpf1 maybe fused to a function domain to form a base editor. See, e.g., Rees et al. Nature Reviews Genetics (2018) 19:770-788. In some embodiments, the catalytically inactive Cas9 nuclease is dCas9. In some embodiments, the nuclease comprises a dCas12a fused to one or more uracil glycosylase inhibitor (UGI) domains. In some embodiments, the Cas9 nuclease comprises a dCas12a fused to an adenine base editor (ABE), for example an ABE evolved from the RNA adenine deaminase TadA. In some embodiments, the Cas nuclease comprises a dCas12a fused to cytidine deaminase enzyme (e.g., APOBEC deaminase, pmCDA1, activation- induced cytidine deaminase (AID)). Alternatively or in addition, the Cas9 nuclease may be a Cas14 nuclease or variant thereof. Cas14 nucleases are derived from archaea and tend to be smaller in size (e.g., 400– 700 amino acids). Additionally Cas14 nucleases do not require a PAM sequence. See, e.g., Harrington et al. Science (2018). Any of the Cas9 nucleases described herein may be modulated to regulate levels of expression and/or activity of the Cas9 nuclease at a desired time. For example, it may be advantageous to increase levels of expression and/or activity of the Cas9 nuclease during particular phase(s) of the cell cycle. It has been demonstrated that levels of homology- directed repair are reduced during the G1 phase of the cell cycle, therefore increasing levels of expression and/or activity of the Cas9 nuclease during the S phase, G2 phase, and/or M phase may increase homology-directed repair following the Cas endonuclease editing. In some embodiments, levels of expression and/or activity of the Cas9 nuclease are increased during the S phase, G2 phase, and/or M phase of the cell cycle. In one example, the Cas9 nuclease fused to the N-terminal region of human Geminin. See, e.g., Gutschner et al. Cell Rep. (2016) 14(6): 1555-1566. In some embodiments, levels of expression and/or activity of the Cas9 nuclease are reduced during the G1 phase. In one example, the Cas9 nuclease is modified such that it has reduced activity during the G1 phase. See, e.g., Lomova et al. Stem Cells (2018). Alternatively or in addition, any of the Cas9 nucleases described herein may be fused to an epigenetic modifier (e.g., a chromatin-modifying enzyme, e.g., DNA methylase, histone deacetylase). See, e.g., Kungulovski et al. Trends Genet. (2016) 32(2):101-113. Cas9 nuclease fused to an epigenetic modifier may be referred to as “epieffectors” and may allow for temporal and/or transient endonuclease activity. In some embodiments, the Cas9 nuclease is a dCas9 fused to a chromatin-modifying enzyme. III. Cytotoxic Agents Cytotoxic agents targeting cells (e.g., cancer cells) expressing a cell surface protein can be co-used with the genetically engineered hematopoietic cells as described herein. As used herein, the term “cytotoxic agent” refers to any agent that can directly or indirectly induce cytotoxicity of a target cell, which expresses the cell surface protein (e.g., a target cancer cell). Such a cytotoxic agent may comprise a protein-binding fragment that binds and targets an epitope of the lineage-specific cell surface antigen. In some instances, the cytotoxic agent may comprise an antibody, which may be conjugated to a drug (e.g., an anti- cancer drug) to form an antibody-drug conjugate (ADC). The cytotoxic agent for use in the methods described herein may directly cause cell death of a target cell. For example, the cytotoxic agent can be an immune cell (e.g., a cytotoxic T cell) expressing a chimeric receptor. Upon engagement of the protein binding domain of the chimeric receptor with the corresponding epitope in a lineage-specific cell surface antigen, a signal (e.g., activation signal) may be transduced to the immune cell resulting in release of cytotoxic molecules, such as perforins and granzymes, as well as activation of effector functions, leading to death of the target cell. In another example, the cytotoxic agent may be an ADC molecule. Upon binding to a target cell, the drug moiety in the ADC would exert cytotoxic activity, leading to target cell death. In other embodiments, the cytotoxic agent may indirectly induce cell death of the target cell. For example, the cytotoxic agent may be an antibody, which, upon binding to the target cell, would trigger effector activities (e.g., ADCC) and/or recruit other factors (e.g., complements), resulting in target cell death. Any of the cytotoxic agents described herein target a lineage-specific cell surface antigen, e.g., comprising a protein-binding fragment that specifically binds an epitope in the cell surface protein. Non-limiting examples of cytotoxic agents that can be utilized in the compositions and methods described herein include those described in PCT Publication Nos. WO2022/056497, WO2022/056490, and WO2019/178382, WO2023/201238, and International Patent Application No. PCT/US2024/029308. For leukemias that become resistance to CAR-T therapy, an emerging strategy is to simultaneously target alternative or multiple antigens (see e.g., Nature Reviews Immunology (2019), Volume 19, pages 73–74 and Cancer Discov. (2018) Oct;8(10):1219-1226). In some embodiments, more than one (e.g., 2, 3, 4, 5 or more) cytotoxic agent is used to target more than one (e.g., 1, 2, 3, 4, 5 or more) epitopes of a lineage-specific cell surface antigen. In some embodiments, more than one (e.g., 1, 2, 3, 4, 5 or more) cytotoxic agent is used to target an epitope(s) of one or more lineage-specific cell surface antigen(s) (e.g., additional/alternative antigens). In some embodiments, targeting of more than one cell surface protein reduces relapse of a hematopoietic malignancy. In one embodiment, two or more cytotoxic agents are used in the methods described herein. In some embodiments, the two or more cytotoxic agents are administered concurrently. In some embodiments, the two or more cytotoxic agents are administered sequentially. Examples of additional cell surface proteins that may be targeted are known in the art (see, e.g., Tasian, Ther. Adv. Hematol. (2018) 9(6): 135-148; Hoseini and Cheung, Blood Cancer Journal (2017) 7, e522; doi:10.1038/bcj.2017.2; Taraseviciute et al. Hematology and Oncology (2019) 31(1)). In some embodiments, the methods described herein involve targeting a cell surface protein and one or more additional cell surface proteins. In some embodiments, a genetically engineered cell of the present disclosure may be contacted with a cytotoxic agent comprising an antigen-binding fragment that recognizes a cell surface protein selected from a group consisting of: CD10; CD101; CD117; CD11A; CD11C; CD120B; CD123; CD13; CD133; CD152; CD200; CD205 (LY75); CD226; CD244 (SLAMF4); CD25; CD274; CD305 (LAIR1); CD32; CD33; CD34; CD366; CD38; CD41; CD42B; CD44; CD45RA; CD47; CD48; CD49D (ITGA4); CD49F; CD52; CD58; CD64; CD7; CD70; CD82; CD84; CD85; CD85K; CD86; CD9; CD96; CD99; CLL-1; EMR2 (ADGRE2); FR-B; GPR56; and IL1RAP. In some embodiments, a genetically engineered cell of the present disclosure may be contacted with a cytotoxic agent comprising an antigen-binding fragment that recognizes a cell surface protein selected from a group consisting of: CD10; CD101; CD11A; CD11C; CD123; CD133; CD152; CD200; CD205; CD244; CD25; CD305; CD33; CD34; CD366; CD38; CD42B; CD44; CD45RA; CD47; CD48; CD49D; CD49F; CD52; CD58; CD64; CD7; CD84; CD85K; CD9; CD99; CLL-1; EMR2; GPR56; IL1RAP; and CD274. In some embodiments, a genetically engineered cell of the present disclosure may be contacted with a cytotoxic agent comprising an antigen-binding fragment that recognizes a cell surface protein selected from a group consisting of: CD10; CD117; CD11A; CD11C; CD120B; CD123; CD13; CD133; CD152; CD205; CD22; CD226; CD244; CD25; CD305; CD32; CD33; CD34; CD38; CD41; CD44; CD45RA; CD47; CD48; CD49D; CD49F; CD52; CD56; CD58; CD64; CD7; CD70; CD71; CD82; CD84; CD85K; CD86; CD96; CD99; CLL- 1; EMR2; FR-B; GPR56; HLA-DR; and IL1RAP. In some embodiments, a genetically engineered cell of the present disclosure may be contacted with a first cytotoxic agent comprising an antigen-binding fragment that recognizes a first cell surface protein and a second cytotoxic agent comprising an antigen-binding fragment that recognizes a second cell surface protein selected from a group consisting of: CD274 and CD152; CD274 and CD226; CD152 and CD226; CD10 and CD49F; CD49F and GPR56; CD200 and CD34; CD34 and CD52; CD34 and CD133; GPR56 and CD133; CD42B and CD41; CD85K and CD64; CD85K and CD32; CD64 and CD32; CD64 and CD11C; CD64 and CD86; CD45RA and CD96; CD45RA and IL1RAP; CD45RA and CD123; CD84 and CD33; CD11C and IL1RAP; CD11C and CD33; CD11C and CD86; CD9 and IL1RAP; IL1RAP and CD33; IL1RAP and CD205; IL1RAP and CD305; IL1RAP and CD123; CD33 and CLL-1; CD33 and CD305; CD11A and CD86; CLL-1 and CD305; CD58 and CD305; CD49D and CD47; CD49D and CD99; CD49D and CD44; CD49D and CD305; CD47 and CD99; CD47 and EMR2; CD99 and CD44; CD99 and CD305; CD99 and EMR2; CD205 and CD305; CD205 and CD123; CD44 and CD305; CD44 and EMR2; CD244 and CD117; CD123 and EMR2; EMR2 and CD82; CD152 and CD120B; CD48 and CD11A; CD200 and CD25; CD34 and CD7; CD34 and CD13; CD52 and GPR56; CD52and CD133; CD52 and CD7; GPR56 and CD7; GPR56 and CD82; CD133 and CD7; CD133 and CD82; CD7 and CD13; CD101 and CD64; CD85 and CD70; CD85K and CD86; CD64 and FR-B; CD366 and CD96; CD25 and CD45RA; CD25 and CD96; CD9 and CD82; IL1RAP and CD58; CD11A and CD38; CD11A and CD44; CD11A and CD244; CD38 and CD305; CD38 and EMR2; CLL-1 and CD49D; CD58 and CD49D; CD47 and CD205; and CD47 and CD44. In some embodiments, a genetically engineered cell of the present disclosure may be contacted with a first cytotoxic agent comprising an antigen-binding fragment that recognizes a first cell surface protein and a second cytotoxic agent comprising an antigen-binding fragment that recognizes a second cell surface protein selected from a group consisting of: CD33 and CD205 (LY75); CD33 and CD244 (SLAMF4); CD33 and CD305 (LAIR1); CD33 and CD49D (ITGA4); CLL-1 and CD205 (LY75); CLL-1 and CD244 (SLAMF4); CLL-1 and CD305 (LAIR1); CLL-1 and CD49D (ITGA4); EMR2 and CD205 (LY75); EMR2 and CD244 (SLAMF4); EMR2 and CD305 (LAIR1); EMR2 and CD49D (ITGA4); CD38 and CD205 (LY75); CD38 and CD244 (SLAMF4); CD38 and CD305 (LAIR1); CD38 and CD49D (ITGA4); CD305 (LAIR1) and CD49D (ITGA4); CD305 (LAIR1) and CD205 (LY75); CD305 (LAIR1) and CD244 (SLAMF4); CD49D (ITGA4) and CD205 (LY75); CD49D (ITGA4) and CD244 (SLAMF4); and CD205 (LY75) and CD244 (SLAMF4). In some embodiments, a genetically engineered cell of the present disclosure may be contacted with a first cytotoxic agent comprising an antigen-binding fragment that recognizes a first cell surface protein and a second cytotoxic agent comprising an antigen-binding fragment that recognizes a second cell surface protein selected from a group consisting of: CD85K and CD64; CD45RA and IL1RAP; CD34 and CD133; IL1RAP and CD33; CD47 and CD99; CLL-1 and CD305; CD152 and CD226; CD45RA and CD96; CD45RA and CD123; CD49D and CD305; IL1RAP and CD305; CD58 and CD305; CD49D and CD47; CD33 and CD305; CD49D and CD44; CD64 and CD86; GPR56 and CD133; CD34 and CD52; EMR2 and CD82; CD64 and CD32; CD42B and CD41; CD64 and CD11C; CD99 and CD44; CD200 and CD34; CD11C and CD33; IL1RAP and CD123; CD11C and IL1RAP; CD44 and EMR2; CD205 and CD305; CD44 and CD305; CD49D and CD99; CD274 and CD226; CD49F and GPR56; CD9 and IL1RAP; CD99 and CD305; CD10 and CD49F; CD11A and CD86; CD33 and CLL-1; CD84 and CD33; CD244 and CD117; CD85K and CD32; CD47 and EMR2; CD274 and CD152; IL1RAP and CD205; CD11C and CD86; CD99 and EMR2; CD205 and CD123; and CD123 and EMR2. In some embodiments, a genetically engineered cell of the present disclosure may be contacted with a first cytotoxic agent comprising an antigen-binding fragment that recognizes a first cell surface protein and a second cytotoxic agent comprising an antigen-binding fragment that recognizes a second cell surface protein selected from a group consisting of: CD274 and CD152; CD274 and CD226; CD152 and CD226; CD10 and CD49F; CD49F and GPR56; CD200 and CD34; CD34 and CD52; CD34 and CD133; GPR56 and CD133; CD42B and CD41; CD85K and CD64; CD85K and CD32; CD64 and CD32; CD64 and CD11C; CD64 and CD86; CD45RA and CD96; CD45RA and IL1RAP; CD45RA and CD123; CD84 and CD33; CD11C and IL1RAP; CD11C and CD33; CD11C and CD86; CD9 and IL1RAP; IL1RAP and CD33; IL1RAP and CD205; IL1RAP and CD305; IL1RAP and CD123; CD33 and CLL-1; CD33 and CD305; CD11A and CD86; CLL-1 and CD305; CD58 and CD305; CD49D and CD47; CD49D and CD99; CD49D and CD44; CD49D and CD305; CD47 and CD99; CD47 and EMR2; CD99 and CD44; CD99 and CD305; CD99 and EMR2; CD205 and CD305; CD205 and CD123; CD44 and CD305; CD44 and EMR2; CD244 and CD117; CD123 and EMR2; and EMR2 and CD82. In some embodiments, a genetically engineered cell of the present disclosure may be contacted with a first cytotoxic agent comprising an antigen-binding fragment that recognizes a first cell surface protein and a second cytotoxic agent comprising an antigen-binding fragment that recognizes a second cell surface protein selected from a group consisting of: CD9 and CD82; CD45RA and CD123; CD133 and CD82; CD85K and CD70; CD47 and CD205; CD64 and FR-B; CD11A and CD44; CD85K and CD86; CD366 and CD96; CD101 and CD64; CLL-1 and CD49D; CD49D and CD47; EMR2 and CD82; CD38 and CD305; CD34 and CD7; CD200 and CD25; CD25 and CD45RA; CD49D and CD44; CD11A and CD244; CD133 and CD7; CD48 and CD11A; CD52 and CD133; GPR56 and CD82; CD11A and CD38; CD34 and CD13; CD58 and CD49D; CD47 and CD99; IL1RAP and CD58; CD47 and CD44; CD33 and CD305; CD64 and CD86; CD152 and CD120B; CD42B and CD41; CD34 and CD52; CD152 and CD226; CD7 and CD13; CD38 and EMR2; IL1RAP and CD33; CLL-1 and CD305; CD45RA and IL1RAP; CD34 and CD133; GPR56 and CD133; CD49D and CD305; CD25 and CD96; CD52 and CD7; CD64 and CD32; CD58 and CD305; CD45RA and CD96; IL1RAP and CD305; CD52 and GPR56; GPR56 and CD7; and CD85K and CD64. Non-limiting examples of agents (e.g., cytotoxic agents) comprising antigen-binding fragments that may be used to target malignant hematopoietic cells in a subject are further provided herein. Therapeutic Antibodies Any antibody or an antigen-binding fragment thereof can be used as a cytotoxic agent or for constructing a cytotoxic agent that targets an epitope of a lineage-specific cell surface antigen, as described herein. Such an antibody or antigen-binding fragment can be prepared by a conventional method, for example, the hybridoma technology or recombinant technology. As used herein, the term "antibody" refers to a glycoprotein comprising at least two heavy (H) chains and two light (L) chains inter-connected by disulfide bonds, i.e., covalent heterotetramers comprised of two identical Ig H chains and two identical L chains that are encoded by different genes. Each heavy chain is comprised of a heavy chain variable region (abbreviated herein as HCVR or VH) and a heavy chain constant region. The heavy chain constant region is comprised of three domains, CH1, CH2 and CH3. Each light chain is comprised of a light chain variable region (abbreviated herein as LCVR or VL) and a light chain constant region. The light chain constant region is comprised of one domain, CL. The VH and VL regions can be further subdivided into regions of hypervariability, termed complementarity determining regions (CDR), interspersed with regions that are more conserved, termed framework regions (FR). Each VH and VL is composed of three CDRs and four FRs, arranged from amino-terminus to carboxy-terminus in the following order: FR1, CDR1, FR2, CDR2, FR3, CDR3, FR4. The variable regions of the heavy and light chains contain a binding domain that interacts with an antigen. The constant regions of the antibodies may mediate the binding of the immunoglobulin to host tissues or factors, including various cells of the immune system (e.g., effector cells) and the first component (Clq) of the classical complement system. Formation of a mature functional antibody molecule can be accomplished when two proteins are expressed in stoichiometric quantities and self-assemble with the proper configuration. In some embodiments, the antigen-binding fragment is a single-chain antibody fragment (scFv) that specifically binds the epitope of the lineage-specific cell surface antigen. In other embodiments, the antigen-binding fragment is a full-length antibody that specifically binds the epitope of the lineage-specific cell surface antigen. As described herein and as will be evident to a skilled artisan, the CDRs of an antibody specifically bind to the epitope of a target protein/antigen (the cell surface protein/antigen). In some embodiments, the antibodies are full-length antibodies, meaning the antibodies comprise a fragment crystallizable (Fc) portion and a fragment antigen-binding (Fab) portion. In some embodiments, the antibodies are of the isotype IgG, IgA, IgM, IgA, or IgD. In some embodiments, a population of antibodies comprises one isotype of antibody. In some embodiments, the antibodies are IgG antibodies. In some embodiments, the antibodies are IgM antibodies. In some embodiments, a population of antibodies comprises more than one isotype of antibody. In some embodiments, a population of antibodies is comprised of a majority of one isotype of antibodies but also contains one or more other isotypes of antibodies. In some embodiments, the antibodies are selected from the group consisting of IgGl, IgG2, IgG3, IgG4, IgM, IgAl, IgA2, IgAsec, IgD, and IgE. The antibodies described herein may specifically bind to a target protein. As used herein, “specific binding” refers to antibody binding to a predetermined protein, such as a cancer antigen. “Specific binding” involves more frequent, more rapid, greater duration of interaction, and/or greater affinity to a target protein relative to alternative proteins. In some embodiments, a population of antibodies specifically binds to a particular epitope of a target protein, meaning the antibodies bind to the particular protein with more frequently, more rapidly, for greater duration of interaction, and/or with greater affinity to the epitope relative to alternative epitopes of the same target protein or to epitopes of another protein. In some embodiments, the antibodies that specifically bind to a particular epitope of a target protein may not bind to other epitopes of the same protein. Antibodies may be selected based on the binding affinity of the antibody to the target protein or epitope. Alternatively or in additional, the antibodies may be mutated to introduce one or more mutations to modify (e.g., enhance or reduce) the binding affinity of the antibody to the target protein or epitope. The present antibodies or antigen-binding portions can specifically bind with a dissociation constant (KD) of less than about 10-7 M, less than about 10-8 M, less than about 10-9 M, less than about 10-10 M, less than about 10-11 M, or less than about 10-12 M. Affinities of the antibodies according to the present disclosure can be readily determined using conventional techniques (see, e.g., Scatchard et al., Ann. N.Y. Acad. Sci. (1949) 51:660; and U.S. Patent Nos.5,283,173, 5,468,614, or the equivalent). The binding affinity or binding specificity for an epitope or protein can be determined by a variety of methods including equilibrium dialysis, equilibrium binding, gel filtration, ELISA, surface plasmon resonance, or spectroscopy. For example, antibodies (of antigen-binding fragments thereof) specific to an epitope of a cell surface protein of interest can be made by the conventional hybridoma technology. The cell surface protein, which may be coupled to a carrier protein such as KLH, can be used to immunize a host animal for generating antibodies binding to that complex. The route and schedule of immunization of the host animal are generally in keeping with established and conventional techniques for antibody stimulation and production, as further described herein. General techniques for production of mouse, humanized, and human antibodies are known in the art and are described herein. It is contemplated that any mammalian subject including humans or antibody producing cells therefrom can be manipulated to serve as the basis for production of mammalian, including human hybridoma cell lines. Typically, the host animal is inoculated intraperitoneally, intramuscularly, orally, subcutaneously, intraplantar, and/or intradermally with an amount of immunogen, including as described herein. Hybridomas can be prepared from the lymphocytes and immortalized myeloma cells using the general somatic cell hybridization technique of Kohler, B. and Milstein, C. (1975) Nature 256:495-497 or as modified by Buck, D. W., et al., In Vitro, 18:377-381 (1982). Available myeloma lines, including but not limited to X63-Ag8.653 and those from the Salk Institute, Cell Distribution Center, San Diego, Calif., USA, may be used in the hybridization. Generally, the technique involves fusing myeloma cells and lymphoid cells using a fusogen such as polyethylene glycol, or by electrical means well known to those skilled in the art. After the fusion, the cells are separated from the fusion medium and grown in a selective growth medium, such as hypoxanthine-aminopterin-thymidine (HAT) medium, to eliminate unhybridized parent cells. Any of the media described herein, supplemented with or without serum, can be used for culturing hybridomas that secrete monoclonal antibodies. As another alternative to the cell fusion technique, EBV immortalized B cells may be used to produce the TCR-like monoclonal antibodies described herein. The hybridomas are expanded and subcloned, if desired, and supernatants are assayed for anti-immunogen activity by conventional immunoassay procedures (e.g., radioimmunoassay, enzyme immunoassay, or fluorescence immunoassay). Hybridomas that may be used as source of antibodies encompass all derivatives, progeny cells of the parent hybridomas that produce monoclonal antibodies capable of binding to a cell surface protein. Hybridomas that produce such antibodies may be grown in vitro or in vivo using known procedures. The monoclonal antibodies may be isolated from the culture media or body fluids, by conventional immunoglobulin purification procedures such as ammonium sulfate precipitation, gel electrophoresis, dialysis, chromatography, and ultrafiltration, if desired. Undesired activity if present, can be removed, for example, by running the preparation over adsorbents made of the immunogen attached to a solid phase and eluting or releasing the desired antibodies off the immunogen. Immunization of a host animal with a target protein or a fragment containing the target amino acid sequence conjugated to a protein that is immunogenic in the species to be immunized, e.g., keyhole limpet hemocyanin, serum albumin, bovine thyroglobulin, or soybean trypsin inhibitor using a bifunctional or derivatizing agent, for example maleimidobenzoyl sulfosuccinimide ester (conjugation through cysteine residues), N-hydroxysuccinimide (through lysine residues), glutaraldehyde, succinic anhydride, SOCl, or R1N=C=NR, where R and R1 are different alkyl groups, can yield a population of antibodies (e.g., monoclonal antibodies). If desired, an antibody of interest (e.g., produced by a hybridoma) may be sequenced and the polynucleotide sequence may then be cloned into a vector for expression or propagation. The sequence encoding the antibody of interest may be maintained in vector in a host cell and the host cell can then be expanded and frozen for future use. In an alternative, the polynucleotide sequence may be used for genetic manipulation to "humanize" the antibody or to improve the affinity (affinity maturation), or other characteristics of the antibody. For example, the constant region may be engineered to more resemble human constant regions to avoid immune response if the antibody is used in clinical trials and treatments in humans. It may be desirable to genetically manipulate the antibody sequence to obtain greater affinity to the cell surface protein. In some examples, the antibody sequence is manipulated to increase binding affinity of the antibody to the cell surface protein such that lower levels of the cell surface protein are detected by the antibody. In some embodiments, antibodies that have increased binding to the cell surface protein may be used to reduce or prevent relapse of a hematopoietic malignancy. It will be apparent to one of skill in the art that one or more polynucleotide changes can be made to the antibody and still maintain its binding specificity to the target protein. In other embodiments, fully human antibodies can be obtained by using commercially available mice that have been engineered to express specific human immunoglobulin proteins. Transgenic animals that are designed to produce a more desirable (e.g., fully human antibodies) or more robust immune response may also be used for generation of humanized or human antibodies. Examples of such technology are XenomouseRTM from Amgen, Inc. (Fremont, Calif.) and HuMAb-MouseRTM and TC MouseTM from Medarex, Inc. (Princeton, N.J.). In another alternative, antibodies may be made recombinantly by phage display or yeast technology. See, for example, U.S. Pat. Nos.5,565,332; 5,580,717; 5,733,743; and 6,265,150; and Winter et al., (1994) Annu. Rev. Immunol.12:433-455. Alternatively, the phage display technology (McCafferty et al., (1990) Nature 348:552-553) can be used to produce human antibodies and antibody fragments in vitro, from immunoglobulin variable (V) domain gene repertoires from unimmunized donors. Antigen-binding fragments of an intact antibody (full-length antibody) can be prepared via routine methods. For example, F(ab')2 fragments can be produced by pepsin digestion of an antibody molecule, and Fab fragments that can be generated by reducing the disulfide bridges of F(ab')2 fragments. Genetically engineered antibodies, such as humanized antibodies, chimeric antibodies, single-chain antibodies, and bi-specific antibodies, can be produced via, e.g., conventional recombinant technology. In one example, DNA encoding a monoclonal antibodies specific to a target protein can be readily isolated and sequenced using conventional procedures (e.g., by using oligonucleotide probes that are capable of binding specifically to genes encoding the heavy and light chains of the monoclonal antibodies). The hybridoma cells serve as a preferred source of such DNA. Once isolated, the DNA may be placed into one or more expression vectors, which are then transfected into host cells such as E. coli cells, simian COS cells, Chinese hamster ovary (CHO) cells, or myeloma cells that do not otherwise produce immunoglobulin protein, to obtain the synthesis of monoclonal antibodies in the recombinant host cells. See, e.g., PCT Publication No. WO 87/04462. The DNA can then be modified, for example, by substituting the coding sequence for human heavy and light chain constant domains in place of the homologous murine sequences, Morrison et al., (1984) Proc. Nat. Acad. Sci.81:6851, or by covalently joining to the immunoglobulin coding sequence all or part of the coding sequence for a non- immunoglobulin polypeptide. In that manner, genetically engineered antibodies, such as “chimeric” or “hybrid” antibodies; can be prepared that have the binding specificity of a target protein. Techniques developed for the production of “chimeric antibodies” are well known in the art. See, e.g., Morrison et al. (1984) Proc. Natl. Acad. Sci. USA 81, 6851; Neuberger et al. (1984) Nature 312, 604; and Takeda et al. (1984) Nature 314:452. Methods for constructing humanized antibodies are also well known in the art. See, e.g., Queen et al., Proc. Natl. Acad. Sci. USA, 86:10029-10033 (1989). In one example, variable regions of VH and VL of a parent non-human antibody are subjected to three- dimensional molecular modeling analysis following methods known in the art. Next, framework amino acid residues predicted to be important for the formation of the correct CDR structures are identified using the same molecular modeling analysis. In parallel, human VH and VL chains having amino acid sequences that are homologous to those of the parent non-human antibody are identified from any antibody gene database using the parent VH and VL sequences as search queries. Human VH and VL acceptor genes are then selected. The CDR regions within the selected human acceptor genes can be replaced with the CDR regions from the parent non-human antibody or functional variants thereof. When necessary, residues within the framework regions of the parent chain that are predicted to be important in interacting with the CDR regions (see above description) can be used to substitute for the corresponding residues in the human acceptor genes. A single-chain antibody can be prepared via recombinant technology by linking a nucleotide sequence coding for a heavy chain variable region and a nucleotide sequence coding for a light chain variable region. Preferably, a flexible linker is incorporated between the two variable regions. Alternatively, techniques described for the production of single chain antibodies (U.S. Patent Nos.4,946,778 and 4,704,692) can be adapted to produce a phage or yeast scFv library and scFv clones specific to a cell surface protein can be identified from the library following routine procedures. Positive clones can be subjected to further screening to identify those that bind cell surface protein. In some instances, the cytotoxic agent for use in the methods described herein comprises an antigen-binding fragment that targets CD33. In other examples, the cytotoxic agent for use in the methods described herein comprises an antigen-binding fragment that targets IL1RAP. In other example, two or more cytotoxic agents are used in the methods described herein. In some embodiments, the two or more cytotoxic agents are administered concurrently. In some embodiments, the two or more cytotoxic agents are administered sequentially. In one non-limiting example, antibodies or antigen-binding fragments thereof targeting CD33 and IL1RAP in combination are used in the methods described herein. In one non-limiting example, antibodies or antigen-binding fragments thereof targeting CD44 and EMR2 in combination are used in the methods described herein. In one non-limiting example, antibodies or antigen-binding fragments thereof targeting CD33 are used in combination with a cytotoxic agent (e.g., antibodies, immune cells expressing chimeric antigen receptors, antibody-drug conjugates) that targets a second cell surface protein or an additional cell surface protein. In one non-limiting example, antibodies or antigen-binding fragments thereof targeting IL1RAP are used in combination with a cytotoxic agent (e.g., antibodies, immune cells expressing chimeric antigen receptors, antibody-drug conjugates) that targets a second cell surface protein or an additional cell surface protein. In some embodiments, bispecific or multi-specific antibodies may be used to target more than one epitope (e.g., more than one epitope of a lineage-specific cell surface antigen, epitopes of more than one lineage-specific cell surface antigen, an epitope of cell surface protein and an epitope of an additional cell surface antigen). See, e.g., Hoseini et al. Blood Cancer Journal (2017) 7, e552. Non-limiting examples of bispecific antibodies include tandem double scFv (e.g., single-chain bispecific tandem fragment variable (scBsTaFv), bispecific T-cell engager (BiTE), bispecific single-chain Fv (bsscFv), bispecific killer-cell engager (BiKE), dual-affinity re-targeting (DART), diabody, tandem diabodies (TandAb), single-chain Fv triplebody (sctb), bispecific scFv immunofusion (Blf), Fabsc, dual-variable- domain immunoglobulin (DVD-Ig), CrossMab (CH1-CL), modular bispecific antibody (IgG- scFv). See, e.g., Marin-Acevedo et al. J. Hematol. Oncol.(2018)11: 8; Slaney et al. Cancer Discovery (2018) 8(8): 924-934, and Elgundi et al. Advanced Drug Discovery Reviews (2017) 122: 2-19. In some embodiments, the antibody is a bispecific T-cell engager (BiTE) comprising two linked scFv molecules. In some embodiments, at least of the linked scFv of the BiTE binds an epitope of a lineage-specific cell surface protein (e.g., CD33 or IL1RAP). In one example, the BiTE is blinatumomab. See, e.g. Slaney et al. Cancer Discovery (2018) 8(8): 924-934. For example, an antibody that targets both CD33 and IL1RAP may be used in the methods described herein. Antibodies and antigen-binding fragments targeting CD33 or IL1RAP or a combination thereof can be prepared by routine practice. For example, an antibody that targets both CD44 and EMR2 may be used in the methods described herein. Antibodies and antigen-binding fragments targeting CD44 or EMR2 or a combination thereof can be prepared by routine practice. In some embodiments, a bispecific antibody may be used in which one molecule targets an epitope of a lineage-specific cell surface protein on a target cell and the other molecule targets a surface antigen on an effector cell (e.g., T cell, NK cell) such that the target cell is brought into proximity with the effector cell. See, e.g., Hoseini et al. Blood Cancer Journal (2017) 7, e552. In some embodiments, two or more (e.g., 2, 3, 4, 5 or more) epitopes of a cell surface protein have been modified, enabling two or more (e.g., 2, 3, 4, 5 or more) different cytotoxic agents (e.g., two antibodies) to be targeted to the two or more epitopes. In some embodiments, the antibodies could work synergistically to enhance efficacy. In some embodiments, epitopes of two or more (e.g., 2, 3, 4, 5 or more) cell surface protein have been modified, enabling two or more (e.g., 2, 3, 4, 5 or more) different cytotoxic agents (e.g., two antibodies) to be targeted to epitopes of the two or more cell surface proteins. In some embodiments, one or more (e.g., 1, 2, 3, 4, 5 or more) epitopes of a cell surface protein have been modified and one or more (e.g., 1, 2, 3, 4, 5 or more) epitopes of an additional cell surface protein have been modified, enabling two or more (e.g., 2, 3, 4, 5 or more) different cytotoxic agents (e.g., two antibodies) to be targeted to epitopes of the cell surface protein and epitopes of the additional cell surface protein. In some embodiments, targeting of two or more cell surface protein may reduce relapse of a hematopoietic malignancy. In some embodiments, the methods described herein involve administering a cytotoxic agent that targets an epitope of a cell surface protein that is mutated in the population of genetically engineered hematopoietic cells. In some embodiments, the methods described herein involve administering a cytotoxic agent that targets an epitope of a cell surface protein that is mutated in the population of genetically engineered hematopoietic cells and one or more additional cytotoxic agents that target one or more additional cell surface proteins. In some embodiments, the antibodies work synergistically to enhance efficacy by targeting more than one cell surface protein. In some embodiments, the methods described herein involve administering to the subject a population of genetically engineered cells lacking a non-essential epitope in a cell surface protein and one or more immunotherapeutic agents (e.g., antibodies) that target cells expressing the lineage-specific cell surface antigen. In some embodiments, the methods described herein involve administering to the subject a population of genetically engineered cells lacking a non-essential epitope in a type 1 cell surface protein and one or more immunotherapeutic agents (e.g., antibodies) that target cells expressing the lineage-specific cell surface antigen. In some embodiments, the methods described herein involve administering to the subject a population of genetically engineered cells lacking a non- essential epitope in a type 2 cell surface protein and one or more immunotherapeutic agents (e.g., antibodies) that target cells expressing the lineage-specific cell surface antigen. In any of the embodiments described herein, one or more additional immunotherapeutic agents may be further administered to the subject (e.g., targeting one or more additional epitopes and/or antigens), for example if the hematopoietic malignancy relapses. Antibody-Drug Conjugates In some embodiments, the cytotoxic agent targeting an epitope of a cell surface protein is an antibody-drug conjugate (ADC). As will be evident to one of ordinary skill in the art, the term “antibody-drug conjugate” can be used interchangeably with “immunotoxin” and refers to a fusion molecule comprising an antibody (or antigen-binding fragment thereof) conjugated to a toxin or drug molecule. Binding of the antibody to the corresponding epitope of the target protein allows for delivery of the toxin or drug molecule to a cell that presents the protein (and epitope thereof) on the cell surface (e.g., target cell), thereby resulting in death of the target cell. In some embodiments, the antibody-drug conjugate (or antigen- binding fragment thereof) binds to its corresponding epitope of a cell surface protein but does not bind to a cell surface protein that lacks the epitope or in which the epitope has been mutated. In some embodiments, the agent is an antibody-drug conjugate. In some embodiments, the antibody-drug conjugate comprises an antigen-binding fragment and a toxin or drug that induces cytotoxicity in a target cell. In some embodiments, the antibody- drug conjugate binds to an epitope in an exon of IL1RAP. In some embodiments, the antibody-drug conjugate targets a type 2 protein. In some embodiments, the antibody-drug conjugate targets CD33. In some embodiments, the antibody-drug conjugate binds to an epitope in exon 2 or exon 3 of CD33. In some embodiments, the antibody-drug conjugate targets a type 1 protein. In some embodiments, the antibody-drug conjugate targets IL1RAP. In some embodiments, the antibody-drug conjugate targets a type 0 protein. Toxins or drugs compatible for use in antibody-drug conjugates are well known in the art and will be evident to one of ordinary skill in the art. See, e.g., Peters et al. Biosci. Rep.(2015) 35(4): e00225, Beck et al. Nature Reviews Drug Discovery (2017) 16:315-337; Marin-Acevedo et al. J. Hematol. Oncol.(2018)11: 8; Elgundi et al. Advanced Drug Delivery Reviews (2017) 122: 2-19. In some embodiments, the antibody-drug conjugate may further comprise a linker (e.g., a peptide linker, such as a cleavable linker or a non-cleavable linker) attaching the antibody and drug molecule. Examples of antibody-drug conjugates include, without limitation, brentuximab vedotin, glembatumumab vedotin/CDX-011, depatuxizumab mafodotin/ABT-414, PSMA ADC, polatuzumab vedotin/RG7596/DCDS4501A, denintuzumab mafodotin/SGN-CD19A, AGS-16C3F, CDX-014, RG7841/DLYE5953A, RG7882/DMUC406A, RG7986/DCDS0780A, SGN-LIV1A, enfortumab vedotin/ASG- 22ME, AG-15ME, AGS67E, telisotuzumab vedotin/ABBV-399, ABBV-221, ABBV-085, GSK-2857916, tisotumab vedotin/HuMax-TF-ADC, HuMax-Axl-ADC, pinatuzumab veodtin/RG7593/DCDT2980S, lifastuzumab vedotin/RG7599/DNIB0600A, indusatumab vedotin/MLN-0264/TAK-264, vandortuzumab vedotin/RG7450/DSTP3086S, sofituzumab vedotin/RG7458/DMUC5754A, RG7600/DMOT4039A, RG7336/DEDN6526A, ME1547, PF-06263507/ADC 5T4, trastuzumab emtansine/T-DM1, mirvetuximab soravtansine/ IMGN853, coltuximab ravtansine/SAR3419, naratuximab emtansine/IMGN529, indatuximab ravtansine/BT-062, anetumab ravtansine/BAY 94–9343, SAR408701, SAR428926, AMG 224, PCA062, HKT288, LY3076226, SAR566658, lorvotuzumab mertansine/IMGN901, cantuzumab mertansine/SB-408075, cantuzumab ravtansine/IMGN242, laprituximab emtansine/IMGN289, IMGN388, bivatuzumab mertansine, AVE9633, BIIB015, MLN2704, AMG 172, AMG 595, LOP 628, vadastuximab talirine/SGN-CD33A, SGN-CD70A, SGN- CD19B, SGN-CD123A, SGN-CD352A, rovalpituzumab tesirine/SC16LD6.5, SC-002, SC- 003, ADCT-301/HuMax-TAC-PBD, ADCT-402, MEDI3726/ADC-401, IMGN779, IMGN632, gemtuzumab ozogamicin, inotuzumab ozogamicin/ CMC-544, PF-06647263, CMD-193, CMB-401, trastuzumab duocarmazine/SYD985, BMS-936561/MDX-1203, sacituzumab govitecan/IMMU-132, labetuzumab govitecan/IMMU-130, DS-8201a, U3- 1402, milatuzumab doxorubicin/IMMU-110/hLL1-DOX, BMS-986148, RC48- ADC/hertuzumab-vc–MMAE, PF-06647020, PF-06650808, PF-06664178/RN927C, lupartumab amadotin/ BAY1129980, aprutumab ixadotin/BAY1187982, ARX788, AGS62P1, XMT-1522, AbGn-107, MEDI4276, DSTA4637S/RG7861. In one example, the antibody- drug conjugate is gemtuzumab ozogamicin. In some embodiments, binding of the antibody-drug conjugate to the epitope of the cell surface protein induces internalization of the antibody-drug conjugate, and the drug (or toxin) may be released intracellularly. In some embodiments, binding of the antibody-drug conjugate to the epitope of a cell surface protein induces internalization of the toxin or drug, which allows the toxin or drug to kill the cells expressing the cell surface protein (target cells). In some embodiments, binding of the antibody-drug conjugate to the epitope of a cell surface protein induces internalization of the toxin or drug, which may regulate the activity of the cell expressing the cell surface protein (target cells). The type of toxin or drug used in the antibody-drug conjugates described herein is not limited to any specific type. In some embodiments, two or more (e.g., 2, 3, 4, 5 or more) epitopes of a cell surface protein have been modified, enabling two or more (e.g., 2, 3, 4, 5 or more) different cytotoxic agents (e.g., two ADCs) to be targeted to the two or more epitopes. In some embodiments, the toxins carried by the ADCs could work synergistically to enhance efficacy (e.g., death of the target cells). In some embodiments, epitopes of two or more (e.g., 2, 3, 4, 5 or more) lineage-specific cell surface protein have been modified, enabling two or more (e.g., 2, 3, 4, 5 or more) different cytotoxic agents (e.g., two ADCs) to be targeted to epitopes of the two or more cell surface proteins. In some embodiments, one or more (e.g., 1, 2, 3, 4, 5 or more) epitopes of a cell surface protein have been modified and one or more (e.g., 1, 2, 3, 4, 5 or more) epitopes of an additional cell surface protein have been modified, enabling two or more (e.g., 2, 3, 4, 5 or more) different cytotoxic agents (e.g., two ADCs) to be targeted to epitopes of the cell surface protein and epitopes of additional cell surface antigen. In some embodiments, targeting of more than one cell surface protein or a cell surface protein and one or more additional cell surface protein/antigen may reduce relapse of a hematopoietic malignancy. In some embodiments, the methods described herein involve administering ADCs that target an epitope of a cell surface protein that is mutated in the population of genetically engineered hematopoietic cells. In some embodiments, the methods described herein involve administering ADCs that target an epitope of a cell surface protein that is mutated in the population of genetically engineered hematopoietic cells and one or more additional cytotoxic agents that may target one or more additional cell surface proteins. In some embodiments, the agents could work synergistically to enhance efficacy by targeting more than one cell surface protein. An ADC described herein may be used as a follow-on treatment to subjects who have been undergone the combined therapy as described herein. In some embodiments, the methods described herein involve administering to the subject a population of genetically engineered cells lacking a non-essential epitope in a cell surface protein and one or more immunotherapeutic agents (e.g., ADCs) that target cells expressing the lineage-specific cell surface antigen. In some embodiments, the methods described herein involve administering to the subject a population of genetically engineered cells lacking a non-essential epitope in a type 1 cell surface protein and one or more immunotherapeutic agents (e.g., ADCs) that target cells expressing the lineage-specific cell surface antigen. In some embodiments, the methods described herein involve administering to the subject a population of genetically engineered cells lacking a non-essential epitope in a type 2 cell surface protein and one or more immunotherapeutic agents (e.g., ADCs) that target cells expressing the lineage-specific cell surface antigen. In any of the embodiments described herein, one or more additional immunotherapeutic agents may be further administered to the subject (e.g., targeting one or more additional epitopes and/or antigens), for example if the hematopoietic malignancy relapses. In some examples, the methods described herein involve administering to the subject a population of genetically engineered cells lacking a non-essential epitope of CD33 and one or more antibody-drug conjugates that target cells expressing CD33. In some examples, the methods described herein involve administering to the subject a population of genetically engineered cells lacking an epitope in exon 2 or exon 3 of CD33 and one or more antibodies antibody-drug conjugates that target cells expressing CD33. In some examples, the methods described herein involve administering to the subject a population of genetically engineered cells expressing a mutated CD33 comprising the amino acid sequence of SEQ ID NO: 56 or SEQ ID NO: 58 and one or more antibody-drug conjugates that target cells expressing CD33. In some examples, the methods described herein involve administering to the subject a population of genetically engineered cells lacking a non-essential epitope of IL1RAP and one or more antibody-drug conjugates that target cells expressing IL1RAP. In some examples, the methods described herein involve administering to the subject a population of genetically engineered cells lacking an epitope in and of IL1RAP and one or more antibody-drug conjugates that target cells expressing IL1RAP. Immune Cells Expressing Chimeric Antigen Receptors In some embodiments, the cytotoxic agent that targets an epitope of a cell surface protein as described herein is an immune cell that expresses a chimeric receptor, which comprises an antigen-binding fragment (e.g., a single-chain antibody) capable of binding to the epitope of the cell surface protein (e.g., CD33 or IL1RAP). Recognition of a target cell (e.g., a cancer cell) having the epitope of the cell surface protein on its cell surface by the antigen-binding fragment of the chimeric receptor transduces an activation signal to the signaling domain(s) (e.g., co-stimulatory signaling domain and/or the cytoplasmic signaling domain) of the chimeric receptor, which may activate an effector function in the immune cell expressing the chimeric receptor. In some embodiments, the immune cell expresses more than one chimeric receptor (e.g., 2, 3, 4, 5 or more), referred to as a bispecific or multi- specific immune cell. In some embodiments, the immune cell expresses more than one chimeric receptor, at least one of which targets an epitope of a lineage-specific cell surface antigen. In some embodiments, the immune cell expresses more than one chimeric receptor, each of which targets an epitope of a lineage-specific cell surface antigen. In some embodiments, the immune cell expresses more than one chimeric receptor, at least one of which targets an epitope of a cell surface protein and at least one of which targets an epitope of an additional cell surface antigen. In some embodiments, targeting of more than one lineage-specific cell surface protein or a lineage-specific cell surface protein and one or more additional cell surface protein may reduce relapse of a hematopoietic malignancy. In some embodiments, the immune cell expresses a chimeric receptor that targets more than one epitopes (e.g., more than one epitopes of one cell surface protein or epitopes of more than one cell surface protein), referred to as a bispecific chimeric receptor. In some embodiments, epitopes of two or more lineage-specific cell surface proteins are targeted by cytotoxic agents. In some embodiments, two or more chimeric receptors are expressed in the same immune cell, e.g., bispecific chimeric receptors. Such cells can be used in any of the methods described herein. In some embodiments, cells expressing a chimeric receptor are “pooled,, i.e., two or more groups of cells express two or more different chimeric receptors. In some embodiments, two or more cells expressing different chimeric antigen receptors are administered concurrently. In some embodiments, two or more cells expressing different chimeric antigen receptors are administered sequentially. In some embodiments, epitopes of CD33 and IL1RAP are targeted by cytotoxic agents. In some embodiments, the chimeric receptors targeting CD33 and IL1RAP are expressed in the same immune cell (i.e., a bispecific immune cell). Such cells can be used in any of the methods described herein. In some embodiments, cells expressing chimeric receptors targeting CD33 and IL1RAP “pooled,” i.e., two or more groups of cells express two or more different chimeric receptors. In some embodiments, two or more groups of cells expressing chimeric receptors targeting CD33 and IL1RAP are administered concurrently. In some embodiments, two or more groups of cells expressing chimeric receptors targeting CD33 and IL1RAP are administered sequentially. In some embodiments, epitopes of CD44 and EMR2 are targeted by cytotoxic agents. In some embodiments, the chimeric receptors targeting CD44 and EMR2 are expressed in the same immune cell (i.e., a bispecific immune cell). Such cells can be used in any of the methods described herein. In some embodiments, cells expressing chimeric receptors targeting CD44 and EMR2 “pooled,” i.e., two or more groups of cells express two or more different chimeric receptors. In some embodiments, two or more groups of cells expressing chimeric receptors targeting CD44 and EMR2 are administered concurrently. In some embodiments, two or more groups of cells expressing chimeric receptors targeting CD44 and EMR2 are administered sequentially. As used herein, a chimeric receptor refers to a non-naturally occurring molecule that can be expressed on the surface of a host cell and comprises binding domain that provides specificity of the chimeric receptor (e.g., an antigen-binding fragment that binds to an epitope of a cell surface protein). In general, chimeric receptors comprise at least two domains that are derived from different molecules. In addition to the epitope-binding fragment described herein, the chimeric receptor may further comprise one or more of the following: a hinge domain, a transmembrane domain, a co-stimulatory domain, a cytoplasmic signaling domain, and combinations thereof. In some embodiments, the chimeric receptor comprises from N terminus to C terminus, an antigen-binding fragment that binds to a cell surface protein, a hinge domain, a transmembrane domain, and a cytoplasmic signaling domain. In some embodiments, the chimeric receptor further comprises at least one co-stimulatory domain. See, e.g., Marin-Acevedo et al. J. Hematol. Oncol.(2018)11: 8. Alternatively or in addition, the chimeric receptor may be a switchable chimeric receptor. See, e.g., Rodger et al. PNAS (2016) 113: 459-468; Cao et al. Angew. Chem. Int. Ed. (2016) 55: 7520-7524. In general, a switchable chimeric receptor comprises a binding domain that binds a soluble antigen-binding fragment, which has antigen binding specificity and may be administered concomitantly with the immune cells. In some embodiments, the chimeric receptor may be a masked chimeric receptor, which is maintained in an “off” state until the immune cell expressing the chimeric receptor is localized to a desired location in the subject. For example, the binding domain of the chimeric receptor (e.g., antigen-binding fragment) may be blocked by an inhibitory peptide that is cleaved by a protease present at a desired location in the subject. In some embodiments, it may be advantageous to modulate the binding affinity of the binding domain (e.g., antigen-binding fragment). For example, in some instances, relapse of hematopoietic malignancies results due to the reduced expression of the targeted cell surface protein on the surface of target cells (e.g., antigen escape) and the lower levels of cell surface protein any be inefficient or less efficient in stimulating cytotoxicity of the target cells. See, e.g., Majzner et al. Cancer Discovery (2018) 8(10). In some embodiments, the binding affinity of the binding domain (e.g., antigen-binding fragment) may be enhanced, for example by mutating one or more amino acid residues of the binding domain. Binding domains having enhanced binding affinity to a cell surface protein may result in immune cells that response to lower levels of cell surface protein (lower cell surface protein density) and reduce or prevent relapse. Hinge Domains of Chimeric Antigen Receptors In some embodiments, the chimeric receptors described herein comprise one or more hinge domain(s). In some embodiments, the hinge domain may be located between the antigen-binding fragment and a transmembrane domain. A hinge domain is an amino acid segment that is generally found between two domains of a protein and may allow for flexibility of the protein and movement of one or both of the domains relative to one another. Any amino acid sequence that provides such flexibility and movement of the antigen-binding fragment relative to another domain of the chimeric receptor can be used. The hinge domain may contain about 10-200 amino acids, e.g., 15-150 amino acids, 20-100 amino acids, or 30-60 amino acids. In some embodiments, the hinge domain may be of about 10, 11, 12, 13, 14,15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 35, 40, 45, 50, 55, 60, 65, 70, 75, 80, 85, 90, 95, 100, 110, 120, 130, 140, 150, 160, 170, 180, 190, or 200 amino acids in length. In some embodiments, the hinge domain is a hinge domain of a naturally occurring protein. Hinge domains of any protein known in the art to comprise a hinge domain are compatible for use in the chimeric receptors described herein. In some embodiments, the hinge domain is at least a portion of a hinge domain of a naturally occurring protein and confers flexibility to the chimeric receptor. In some embodiments, the hinge domain is of CD8α or CD28. In some embodiments, the hinge domain is a portion of the hinge domain of CD8α, e.g., a fragment containing at least 15 (e.g., 20, 25, 30, 35, or 40) consecutive amino acids of the hinge domain of CD8α or CD28. Hinge domains of antibodies, such as an IgG, IgA, IgM, IgE, or IgD antibody, are also compatible for use in the chimeric receptors described herein. In some embodiments, the hinge domain is the hinge domain that joins the constant domains CH1 and CH2 of an antibody. In some embodiments, the hinge domain is of an antibody and comprises the hinge domain of the antibody and one or more constant regions of the antibody. In some embodiments, the hinge domain comprises the hinge domain of an antibody and the CH3 constant region of the antibody. In some embodiments, the hinge domain comprises the hinge domain of an antibody and the CH2 and CH3 constant regions of the antibody. In some embodiments, the antibody is an IgG, IgA, IgM, IgE, or IgD antibody. In some embodiments, the antibody is an IgG antibody. In some embodiments, the antibody is an IgG1, IgG2, IgG3, or IgG4 antibody. In some embodiments, the hinge region comprises the hinge region and the CH2 and CH3 constant regions of an IgG1 antibody. In some embodiments, the hinge region comprises the hinge region and the CH3 constant region of an IgG1 antibody. Also within the scope of the present disclosure are chimeric receptors comprising a hinge domain that is a non-naturally occurring peptide. In some embodiments, the hinge domain between the C-terminus of the extracellular ligand-binding domain of an Fc receptor and the N-terminus of the transmembrane domain is a peptide linker, such as a (GlyxSer)n linker, wherein x and n, independently can be an integer between 3 and 12, including 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, or more. Additional peptide linkers that may be used in a hinge domain of the chimeric receptors described herein are known in the art. See, e.g., Wriggers et al. Current Trends in Peptide Science (2005) 80(6): 736-746 and PCT Publication WO 2012/088461. Transmembrane Domains of Chimeric Antigen Receptors In some embodiments, the chimeric receptors described herein may comprise one or more transmembrane domain(s). The transmembrane domain for use in the chimeric receptors can be in any form known in the art. As used herein, a “transmembrane domain” refers to any protein structure that is thermodynamically stable in a cell membrane, preferably a eukaryotic cell membrane. Transmembrane domains compatible for use in the chimeric receptors used herein may be obtained from a naturally occurring protein. Alternatively, the transmembrane domain may be a synthetic, non-naturally occurring protein segment, e.g., a hydrophobic protein segment that is thermodynamically stable in a cell membrane. Transmembrane domains are classified based on the transmembrane domain topology, including the number of passes that the transmembrane domain makes across the membrane and the orientation of the protein. For example, single-pass membrane proteins cross the cell membrane once, and multi-pass membrane proteins cross the cell membrane at least twice (e.g., 2, 3, 4, 5, 6, 7 or more times). In some embodiments, the transmembrane domain is a single-pass transmembrane domain. In some embodiments, the transmembrane domain is a single-pass transmembrane domain that orients the N terminus of the chimeric receptor to the extracellular side of the cell and the C terminus of the chimeric receptor to the intracellular side of the cell. In some embodiments, the transmembrane domain is obtained from a single pass transmembrane protein. In some embodiments, the transmembrane domain is of CD8α. In some embodiments, the transmembrane domain is of CD28. In some embodiments, the transmembrane domain is of ICOS. Costimulatory and Signaling Domains of Chimeric Antigen Receptors In some embodiments, the chimeric receptors described herein comprise one or more costimulatory signaling domains. The term “co-stimulatory signaling domain,” as used herein, refers to at least a portion of a protein that mediates signal transduction within a cell to induce an immune response, such as an effector function. The co-stimulatory signaling domain of the chimeric receptor described herein can be a cytoplasmic signaling domain from a co-stimulatory protein, which transduces a signal and modulates responses mediated by immune cells, such as T cells, NK cells, macrophages, neutrophils, or eosinophils. In some embodiments, the chimeric receptor comprises more than one (at least 2, 3, 4, or more) co-stimulatory signaling domains. In some embodiments, the chimeric receptor comprises more than one co-stimulatory signaling domains obtained from different costimulatory proteins. In some embodiments, the chimeric receptor does not comprise a co- stimulatory signaling domain. In general, many immune cells require co-stimulation, in addition to stimulation of an antigen-specific signal, to promote cell proliferation, differentiation and survival, and to activate effector functions of the cell. Activation of a co-stimulatory signaling domain in a host cell (e.g., an immune cell) may induce the cell to increase or decrease the production and secretion of cytokines, phagocytic properties, proliferation, differentiation, survival, and/or cytotoxicity. The co-stimulatory signaling domain of any co-stimulatory protein may be compatible for use in the chimeric receptors described herein. The type(s) of co-stimulatory signaling domain is selected based on factors such as the type of the immune cells in which the chimeric receptors would be expressed (e.g., primary T cells, T cell lines, NK cell lines) and the desired immune effector function (e.g., cytotoxicity). Examples of co-stimulatory signaling domains for use in the chimeric receptors can be the cytoplasmic signaling domain of co-stimulatory proteins, including, without limitation, CD27, CD28, 4-1BB, OX40, CD30, ICOS, CD2, CD7, LIGHT, NKG2C, B7-H3. In some embodiments, the co-stimulatory domain is derived from 4-1BB, CD28, or ICOS. In some embodiments, the costimulatory domain is derived from CD28 and chimeric receptor comprises a second co-stimulatory domain from 4-1BB or ICOS. In some embodiments, the costimulatory domain is a fusion domain comprising more than one costimulatory domain or portions of more than one costimulatory domains. In some embodiments, the costimulatory domain is a fusion of costimulatory domains from CD28 and ICOS. In some embodiments, the chimeric receptors described herein comprise one or more cytoplasmic signaling domain(s). Any cytoplasmic signaling domain can be used in the chimeric receptors described herein. In general, a cytoplasmic signaling domain relays a signal, such as interaction of an extracellular ligand-binding domain with its ligand, to stimulate a cellular response, such as inducing an effector function of the cell (e.g., cytotoxicity). As will be evident to one of ordinary skill in the art, a factor involved in T cell activation is the phosphorylation of immunoreceptor tyrosine-based activation motif (ITAM) of a cytoplasmic signaling domain. Any ITAM-containing domain known in the art may be used to construct the chimeric receptors described herein. In general, an ITAM motif may comprise two repeats of the amino acid sequence YxxL/I separated by 6-8 amino acids, wherein each x is independently any amino acid, producing the conserved motif YxxL/Ix(6- 8)YxxL/I. In some embodiments, the cytoplasmic signaling domain is from CD3ζ. Exemplary Uses and Methods Related to Chimeric Antigen Receptors In some embodiments, the chimeric receptor described herein targets a type 2 protein. In some embodiments, the chimeric receptor targets CD33. In some embodiments, the chimeric receptor described herein targets a type 1 protein. In some embodiments, the chimeric receptor targets IL1RAP. In some embodiments, the chimeric receptor targets a type 0 protein. Such a chimeric receptor may comprise an antigen-binding fragment (e.g., an scFv) comprising a heavy chain variable region and a light chain variable region that bind to IL1RAP. Alternatively, the chimeric receptor may comprise an antigen-binding fragment (e.g., scFv) comprising a heavy chain variable region and a light chain variable region that bind to CD33. In some embodiments, the immune cells described herein express a chimeric receptor (e.g., bispecific chimeric receptor) that targets a type 2 protein and a chimeric receptor that targets a type 1 protein. In some embodiments, the immune cells described herein express a chimeric receptor (e.g., bispecific chimeric receptor) that targets a type 2 protein and at least one additional cell surface protein, such as any of those described herein. In some embodiments, the immune cells described herein express a chimeric receptor that targets CD33 and at least one additional cell surface protein, such as any of those described herein. In some embodiments, the immune cells described herein express a chimeric receptor that targets a type 1 protein and at least one additional cell surface protein, such as any of those described herein. In some embodiments, the immune cells described herein express a chimeric receptor that targets IL1RAP and at least one additional cell surface protein, such as any of those described herein. In some embodiments, the chimeric receptor described herein targets CD33 and IL1RAP. In some embodiments, the immune cells described herein express a chimeric receptor that targets EMR2 and at least one additional cell surface protein, such as any of those described herein. In some embodiments, the chimeric receptor described herein targets CD44 and EMR2. In some embodiments, the immune cells described herein express a chimeric receptor that targets a type 2 protein and at least one additional chimeric receptor that targets an additional cell surface protein, such as any of those described herein. In some embodiments, the immune cells described herein express a chimeric receptor that targets CD33 and at least one additional chimeric receptor that targets another cell surface protein, such as any of those described herein. In some embodiments, the immune cells described herein express a chimeric receptor that targets a type 1 protein and at least one additional chimeric receptor that targets another cell surface protein, such as any of those described herein. In some embodiments, the immune cells described herein express a chimeric receptor that targets IL1RAP and at least one additional chimeric receptor that targets another cell surface protein, such as any of those described herein. In some embodiments, the immune cells express a chimeric receptor that targets CD33 and a chimeric receptor that targets IL1RAP. A chimeric receptor construct targeting CD33 and/or IL1RAP may further comprise at least a hinge domain (e.g., from CD28, CD8α, or an antibody), a transmembrane domain (e.g., from CD8α, CD28 or ICOS), one or more co-stimulatory domains (from one or more of CD28, ICOS, or 4-1BB) and a cytoplasmic signaling domain (e.g., from CD3ζ), or a combination thereof. In some examples, the methods described herein involve administering to a subject a population of genetically engineered hematopoietic cells and an immune cell expressing a chimeric receptor that targets CD33 and/or IL1RAP, which may further comprise at least a hinge domain (e.g., from CD28, CD8α, or an antibody), a transmembrane domain (e.g., from CD8α, CD28 or ICOS), one or more co-stimulatory domains (from one or more of CD28, ICOS, or 4-1BB) and a cytoplasmic signaling domain (e.g., from CD3ζ), or a combination thereof Any of the chimeric receptors described herein can be prepared by routine methods, such as recombinant technology. Methods for preparing the chimeric receptors herein involve generation of a nucleic acid that encodes a polypeptide comprising each of the domains of the chimeric receptors, including the antigen-binding fragment and optionally, the hinge domain, the transmembrane domain, at least one co-stimulatory signaling domain, and the cytoplasmic signaling domain. In some embodiments, nucleic acids encoding the components of a chimeric receptor are joined together using recombinant technology. Sequences of each of the components of the chimeric receptors may be obtained via routine technology, e.g., PCR amplification from any one of a variety of sources known in the art. In some embodiments, sequences of one or more of the components of the chimeric receptors are obtained from a human cell. Alternatively, the sequences of one or more components of the chimeric receptors can be synthesized. Sequences of each of the components (e.g., domains) can be joined directly or indirectly (e.g., using a nucleic acid sequence encoding a peptide linker) to form a nucleic acid sequence encoding the chimeric receptor, using methods such as PCR amplification or ligation. Alternatively, the nucleic acid encoding the chimeric receptor may be synthesized. In some embodiments, the nucleic acid is DNA. In other embodiments, the nucleic acid is RNA. Mutation of one or more residues within one or more of the components of the chimeric receptor (e.g., the antigen-binding fragment, etc) may be performed prior to or after joining the sequences of each of the components. In some embodiments, one or more mutations in a component of the chimeric receptor may be made to modulate (increase or decrease) the affinity of the component for an epitope (e.g., the antigen-binding fragment for the target protein) and/or modulate the activity of the component. Any of the chimeric receptors described herein can be introduced into a suitable immune cell for expression via conventional technology. In some embodiments, the immune cells are T cells, such as primary T cells or T cell lines. Alternatively, the immune cells can be NK cells, such as established NK cell lines (e.g., NK-92 cells). In some embodiments, the immune cells are T cells that express CD8 (CD8+) or CD8 and CD4 (CD8+/CD4+). In some embodiments, the T cells are T cells of an established T cell line, for example, 293T cells or Jurkat cells. Primary T cells may be obtained from any source, such as peripheral blood mononuclear cells (PBMCs), bone marrow, tissues such as spleen, lymph node, thymus, or tumor tissue. A source suitable for obtaining the type of immune cells desired would be evident to one of skill in the art. In some embodiments, the population of immune cells is derived from a human patient having a hematopoietic malignancy, such as from the bone marrow or from PBMCs obtained from the patient. In some embodiments, the population of immune cells is derived from a healthy donor. In some embodiments, the immune cells are obtained from the subject to whom the immune cells expressing the chimeric receptors will be subsequently administered. Immune cells that are administered to the same subject from which the cells were obtained are referred to as autologous cells, whereas immune cells that are obtained from a subject who is not the subject to whom the cells will be administered are referred to as allogeneic cells. The type of host cells desired may be expanded within the population of cells obtained by co-incubating the cells with stimulatory molecules, for example, anti-CD3 and anti-CD28 antibodies may be used for expansion of T cells. To construct the immune cells that express any of the chimeric receptor constructs described herein, expression vectors for stable or transient expression of the chimeric receptor construct may be constructed via conventional methods as described herein and introduced into immune host cells. For example, nucleic acids encoding the chimeric receptors may be cloned into a suitable expression vector, such as a viral vector in operable linkage to a suitable promoter. The nucleic acids and the vector may be contacted, under suitable conditions, with a restriction enzyme to create complementary ends on each molecule that can pair with each other and be joined with a ligase. Alternatively, synthetic nucleic acid linkers can be ligated to the termini of the nucleic acid encoding the chimeric receptors. The synthetic linkers may contain nucleic acid sequences that correspond to a particular restriction site in the vector. The selection of expression vectors/plasmids/viral vectors would depend on the type of host cells for expression of the chimeric receptors, but should be suitable for integration and replication in eukaryotic cells. In some embodiments, the chimeric receptors are expressed using a non-integrating transient expression system. In some embodiments, the chimeric receptors are integrated into the genome of the immune cell. In some embodiments, the chimeric receptors are integrated into a specific loci of the genome of immune cell using gene editing (e.g., zinc-finger nucleases, meganucleases, TALENs, CRISPR systems). A variety of promoters can be used for expression of the chimeric receptors described herein, including, without limitation, cytomegalovirus (CMV) intermediate early promoter, a viral LTR such as the Rous sarcoma virus LTR, HIV-LTR, HTLV-1 LTR, Maloney murine leukemia virus (MMLV) LTR, myeloproliferative sarcoma virus (MPSV) LTR, spleen focus- forming virus (SFFV) LTR, the simian virus 40 (SV40) early promoter, herpes simplex tk virus promoter, elongation factor 1-alpha (EF1-α) promoter with or without the EF1-α intron. Additional promoters for expression of the chimeric receptors include any constitutively active promoter in an immune cell. Alternatively, any regulatable promoter may be used, such that its expression can be modulated within an immune cell. In some embodiments, the promoter regulating expression of a chimeric receptor is an inducible promoter. In general, the activity of “inducible promoters” may be regulated based on the presence (or absence) of a signal, such as an endogenous signal or an exogenous signal, for example a small molecule or drug that is administered to the subject. Additionally, the vector may contain, for example, some or all of the following: a selectable marker gene, such as the neomycin gene for selection of stable or transient transfectants in host cells; enhancer/promoter sequences from the immediate early gene of human CMV for high levels of transcription; transcription termination and RNA processing signals from SV40 for mRNA stability; 5’-and 3’-untranslated regions for mRNA stability and translation efficiency from highly-expressed genes like α-globin or β-globin; SV40 polyoma origins of replication and ColE1 for proper episomal replication; internal ribosome binding sites (IRESes), versatile multiple cloning sites; T7 and SP6 RNA promoters for in vitro transcription of sense and antisense RNA; a “suicide switch” or “suicide gene” which when triggered causes cells carrying the vector to die (e.g., HSV thymidine kinase, an inducible caspase such as iCasp9, drug-induced suicide switch, monoclonal antibody mediated suicide switch), and reporter gene for assessing expression of the chimeric receptor. Suitable vectors and methods for producing vectors containing transgenes are well known and available in the art. Examples of the preparation of vectors for expression of chimeric receptors can be found, for example, in US2014/0106449, herein incorporated by reference in its entirety. As will be appreciated by one of ordinary skill in the art, in some embodiments, it may be advantageous to quickly and efficiently reduce or eliminate the immune cells expressing chimeric receptors, for example at a time point following administration to a subject. Mechanisms of killing immune cells expressing chimeric receptors, or inducing cytotoxicity of such cells, are known in the art, see, e.g., Labanieh et al. Nature Biomedical Engineering (2018)2: 337-391; Slaney et al. Cancer Discovery (2018) 8(8): 924-934. In some embodiments, the immune cell expressing the chimeric receptor may also express a suicide switch” or “suicide gene,” which may or may not be encoded by the same vector as the chimeric receptor, as described above. In some embodiments, the immune cell expressing chimeric receptors further express an epitope tag such that upon engagement of the epitope tag, the immune cell is killed through antibody-dependent cell-mediated cytotoxicity and/or complement-mediated cytotoxicity. See, e.g., Paszkiewicz et al. J. Clin. Invest. (2016) 126: 4262-4272; Wang et al. Blood (2011) 118: 1255-1263; Tasian et al. Blood (2017) 129: 2395- 2407; Philip et al. Blood (2014) 124: 1277-1287. In some embodiments, the chimeric receptor construct or the nucleic acid encoding said chimeric receptor is a DNA molecule. In some embodiments, chimeric receptor construct or the nucleic acid encoding said chimeric receptor is a DNA vector and may be electroporated to immune cells (see, e.g., Till, et al. Blood (2012) 119(17): 3940-3950). In some embodiments, the nucleic acid encoding the chimeric receptor is an RNA molecule, which may be electroporated to immune cells. Any of the vectors comprising a nucleic acid sequence that encodes a chimeric receptor construct described herein is also within the scope of the present disclosure. Such a vector may be delivered into host cells such as host immune cells by a suitable method. Methods of delivering vectors to immune cells are well known in the art and may include DNA, RNA, or transposon electroporation, transfection reagents such as liposomes or nanoparticles to delivery DNA, RNA, or transposons; delivery of DNA, RNA, or transposons or protein by mechanical deformation (see, e.g., Sharei et al. Proc. Natl. Acad. Sci. USA (2013) 110(6): 2082-2087); or viral transduction. In some embodiments, the vectors for expression of the chimeric receptors are delivered to host cells by viral transduction. Exemplary viral methods for delivery include, but are not limited to, recombinant retroviruses (see, e.g., PCT Publication Nos. WO 90/07936; WO 94/03622; WO 93/25698; WO 93/25234; WO 93/11230; WO 93/10218; WO 91/02805; U.S. Pat. Nos.5,219,740 and 4,777,127; GB Patent No.2,200,651; and EP Patent No.0345242), alphavirus-based vectors, and adeno- associated virus (AAV) vectors (see, e.g., PCT Publication Nos. WO 94/12649, WO 93/03769; WO 93/19191; WO 94/28938; WO 95/11984 and WO 95/00655). In some embodiments, the vectors for expression of the chimeric receptors are retroviruses. In some embodiments, the vectors for expression of the chimeric receptors are lentiviruses. In some embodiments, the vectors for expression of the chimeric receptors are adeno-associated viruses. In examples in which the vectors encoding chimeric receptors are introduced to the host cells using a viral vector, viral particles that are capable of infecting the immune cells and carry the vector may be produced by any method known in the art and can be found, for example in PCT Application No. WO 1991/002805A2, WO 1998/009271 A1, and U.S. Patent 6,194,191. The viral particles are harvested from the cell culture supernatant and may be isolated and/or purified prior to contacting the viral particles with the immune cells. In some embodiments, the methods described herein involve use of immune cells that express more than one chimeric receptor (e.g., chimeric receptors that target first epitope and chimeric receptors that target a second epitope). In some embodiments, more than one chimeric receptor (e.g., chimeric receptors that target first epitope and chimeric receptors that target a second epitope) are expressed from a single vector. In some embodiments, more than one chimeric receptor (e.g., chimeric receptors that target first epitope and chimeric receptors that target a second epitope) are expressed from a more than one vector. Such immune cells may be prepared using methods known in the art, for example by delivering a vector that encodes a first chimeric receptor simultaneously or sequentially with a second vector that encodes a second chimeric receptor. In some embodiments, the resulting immune cell population is a mixed population comprising a subset of immune cells that express one chimeric receptor and a subset of immune cells that express both chimeric receptors. In some embodiments, the domains of the chimeric receptor are encoded by and expressed from a single vector. Alternatively, the domains of the chimeric receptor may be encoded by and expressed from more than one vector. In some embodiments, activity of immune cells expressing the chimeric receptors may be regulated by controlling assembly of the chimeric receptor. The domains of a chimeric receptor may be expressed as two or more non-functional segments of the chimeric receptor and induced to assemble at a desired time and/or location, for example through use of a dimerization agent (e.g., dimerizing drug). The methods of preparing host cells expressing any of the chimeric receptors described herein may comprise activating and/or expanding the immune cells ex vivo. Activating a host cell means stimulating a host cell into an activate state in which the cell may be able to perform effector functions (e.g., cytotoxicity). Methods of activating a host cell will depend on the type of host cell used for expression of the chimeric receptors. Expanding host cells may involve any method that results in an increase in the number of cells expressing chimeric receptors, for example, allowing the host cells to proliferate or stimulating the host cells to proliferate. Methods for stimulating expansion of host cells will depend on the type of host cell used for expression of the chimeric receptors and will be evident to one of skill in the art. In some embodiments, the host cells expressing any of the chimeric receptors described herein are activated and/or expanded ex vivo prior to administration to a subject. It has been appreciated that administration of immunostimulatory cytokines may enhance proliferation of immune cells expressing chimeric receptors following administration to a subject and/or promote engraftment of the immune cells. See, e.g., Pegram et al. Leukemia (2015) 29:415-422; Chinnasamy et al. Clin. Cancer Res. (2012) 18: 1672-1683; Krenciute et al. Cancer Immunol. Res. (2017) 5: 571-581; Hu et al. Cell Rep. (2017) 20: 3025-3033; Markley et al. Blood (2010) 115: 3508-3519). Any of the methods described herein may further involve administering one or more immunostimulatory cytokines concurrently with any of the immune cells expressing chimeric receptors. Non-limiting example of immunostimulatory cytokines include IL-12, IL-15, IL-1, IL-21, and combinations thereof. Any anti- IL1RAP CAR and anti-CD33 CAR molecules known in the art can be used together with the genetically engineered HSCs described herein. Exemplary anti- IL1RAP CARs include axicabtagene and tisagenlecleucel. Exemplary anti-CD33 CARs include those disclosed in PCT Publication Nos. WO2017/066760 and WO2017/079400. In one specific example, primary human CD8+ T cells are isolated from patients’ peripheral blood by immunomagnetic separation (Miltenyi Biotec). T cells are cultured and stimulated with anti-CD3 and anti-CD28 mAbs–coated beads (Invitrogen) as previously described (Levine et al., J. Immunol. (1997) 159(12):5921). Chimeric receptors that target a lineage-specific cell surface proteins (e.g., IL1RAP or CD33) are generated using conventional recombinant DNA technologies and inserted into a lentiviral vector. The vectors containing the chimeric receptors are used to generate lentiviral particles, which are used to transduce primary CD8+ T cells. Human recombinant IL-2 may be added every other day (50 IU/mL). T cells are cultured for ~14 days after stimulation. Expression of the chimeric receptors can be confirmed using methods, such as Western blotting and flow cytometry. T cells expressing the chimeric receptors are selected and assessed for their ability to bind to the lineage-specific cell surface protein such as IL1RAP or CD33 and to induce cytotoxicity of cells expressing the cell surface protein. Immune cells expressing the chimeric receptor are also evaluated for their ability to induce cytotoxicity of cells expressing the cell surface protein in mutated form. Preferably, immune cells expressing chimeric receptors that binds to the wild-type cell surface protein but not the mutated form. (FIGURE 3, using CD33 as an example). The cells (e.g., hematopoietic stem cells) that express the mutated cell surface protein are also assessed for various characteristics, including proliferation, erythropoietic differentiation, and colony formation to confirm that the mutated cell surface protein retained the bioactivity as the wild-type counterpart. The immune cells expressing one or more CAR-T receptors may be further modified genetically, for example, by knock-out of the native T-cell receptor (TCR) or an MHC chain and/or by introducing a new TCR. Alternatively, the immune cells may retain the native TCR loci. In some embodiments, the methods described herein involve administering to the subject a population of genetically engineered cells lacking a non-essential epitope in a cell surface protein and one or more immunotherapeutic agents (e.g., immune cells expressing chimeric receptor(s)) that target cells expressing the lineage-specific cell surface antigen. In some embodiments, the methods described herein involve administering to the subject a population of genetically engineered cells lacking a non-essential epitope in a type 1 cell surface protein and one or more immunotherapeutic agents (e.g., immune cells expressing chimeric receptor(s)) that target cells expressing the lineage-specific cell surface antigen. In some embodiments, the methods described herein involve administering to the subject a population of genetically engineered cells lacking a non-essential epitope in a type 2 cell surface protein and one or more immunotherapeutic agents (e.g., immune cells expressing chimeric receptor(s)) that target cells expressing the lineage-specific cell surface antigen. In any of the embodiments described herein, one or more additional immunotherapeutic agents may be further administered to the subject (e.g., targeting one or more additional epitopes and/or antigens), for example if the hematopoietic malignancy relapses. In some embodiments, the methods described herein involve administering immune cells expressing chimeric receptors that target an epitope of a lineage-specific cell surface protein that is mutated in the population of genetically engineered hematopoietic cells. In some embodiments, the methods described herein involve administering immune cells expressing chimeric receptors that target an epitope of a lineage-specific cell surface protein that is mutated in the population of genetically engineered hematopoietic cells and one or more additional cytotoxic agents that may target one or more additional cell surface proteins. In some embodiments, the agents could work synergistically to enhance efficacy by targeting more than one cell surface protein. In some examples, the methods described herein involve administering to the subject a population of genetically engineered cells lacking a non-essential epitope of CD33 and one or more immune cells expressing chimeric receptor(s) that target cells expressing CD33. In some examples, the methods described herein involve administering to the subject a population of genetically engineered cells lacking an epitope in exon 2 or exon 3 of CD33 and one or more immune cells expressing chimeric receptor(s) that target cells expressing CD33. In some examples, the methods described herein involve administering to the subject a population of genetically engineered cells expressing a mutated CD33 comprising the amino acid sequence of SEQ ID NO: 56 or SEQ ID NO: 58 and one or more immune cells expressing chimeric receptor(s) that target cells expressing CD33. In some examples, the methods described herein involve administering to the subject a population of genetically engineered cells lacking a non-essential epitope of IL1RAP and one or more immune cells expressing chimeric receptor(s) that target cells expressing IL1RAP. In some examples, the methods described herein involve administering to the subject a population of genetically engineered cells lacking an epitope in an exon of IL1RAP and one or more immune cells expressing chimeric receptor(s) that target cells expressing IL1RAP. IV. Methods of Treatment and Combination Therapies The genetically engineered hematopoietic cells such as HSCs may be administered to a subject in need of the treatment, either taken alone or in combination of one or more cytotoxic agents that target one or more antigens as described herein. Since the hematopoietic cells are genetically edited in the genes of the one or more antigens, the hematopoietic cells and/or descendant cells thereof would express the one or more antigens in mutated form (e.g., but functional) such that they can escape being targeted by the cytotoxic agents, for example, CAR-T cells. Thus, the present disclosure provides methods for treating a hematopoietic malignancy, the method comprising administering to a subject in need thereof (i) a population of the genetically engineered hematopoietic cells described herein, and optionally (ii) a cytotoxic agent such as CAR-T cells that target an antigen, the gene of which is genetically edited in the hematopoietic cells such that the cytotoxic agent does not target hematopoietic cells or descendant cells thereof. The administration of (i) and (ii) may be concurrently or in any order. In some embodiments, the cytotoxic agents and/or the hematopoietic cells may be mixed with a pharmaceutically acceptable carrier to form a pharmaceutical composition, which is also within the scope of the present disclosure. In some embodiments, the genetically engineered hematopoietic stem cells have genetic editing in genes of at least two antigens A and B. Such hematopoietic stem cells can be administered to a subject, who has been or will be treated with a first cytotoxic agent specific to A (e.g., anti-protein A CAR-T cells), or who is at risk of a hematopoietic malignancy that would need treatment of the cytotoxic agent. When the subject developed resistance to the cytotoxic agent after the treatment or has relapse of the hematopoietic malignancy, a second cytotoxic agent specific to B (e.g., anti-protein B CAR-T cells) may be administered to the subject. Because the genetically engineered hematopoietic cells have genetic editing in both A and B genes, those cells and descendant cells thereof would also be resistant to cytotoxicity induced by the second cytotoxic agent. As such, administering once the genetically engineered hematopoietic cells would be sufficient to compensate loss of normal cells expressing at least lineage-specific cell surface proteins/antigens A and B due to multiple treatment by cytotoxic agents specific to at least proteins/antigens A and B. As used herein, “subject,” “individual,” and “patient” are used interchangeably, and refer to a vertebrate, preferably a mammal such as a human. Mammals include, but are not limited to, human primates, non-human primates or murine, bovine, equine, canine or feline species. In some embodiments, the subject is a human patient having a hematopoietic malignancy. To perform the methods described herein, an effective amount of the genetically engineered hematopoietic cells can be administered to a subject in need of the treatment. Optionally, the hematopoietic cells can be co-used with a cytotoxic agent as described herein. As used herein the term “effective amount” may be used interchangeably with the term “therapeutically effective amount” and refers to that quantity of a cytotoxic agent, hematopoietic cell population, or pharmaceutical composition (e.g., a composition comprising cytotoxic agents and/or hematopoietic cells) that is sufficient to result in a desired activity upon administration to a subject in need thereof. Within the context of the present disclosure, the term “effective amount” refers to that quantity of a compound, cell population, or pharmaceutical composition that is sufficient to delay the manifestation, arrest the progression, relieve or alleviate at least one symptom of a disorder treated by the methods of the present disclosure. Note that when a combination of active ingredients is administered the effective amount of the combination may or may not include amounts of each ingredient that would have been effective if administered individually. Effective amounts vary, as recognized by those skilled in the art, depending on the particular condition being treated, the severity of the condition, the individual patient parameters including age, physical condition, size, gender and weight, the duration of the treatment, the nature of concurrent therapy (if any), the specific route of administration and like factors within the knowledge and expertise of the health practitioner. In some embodiments, the effective amount alleviates, relieves, ameliorates, improves, reduces the symptoms, or delays the progression of any disease or disorder in the subject. In some embodiments, the subject is a human. In some embodiments, the subject is a human patient having a hematopoietic malignancy. As described herein, the hematopoietic cells and/or immune cells expressing chimeric receptors may be autologous to the subject, i.e., the cells are obtained from the subject in need of the treatment, manipulated such that the cells do not bind the cytotoxic agents, and then administered to the same subject. Administration of autologous cells to a subject may result in reduced rejection of the host cells as compared to administration of non-autologous cells. Alternatively, the host cells are allogeneic cells, i.e., the cells are obtained from a first subject, manipulated such that the cells do not bind the cytotoxic agents, and then administered to a second subject that is different from the first subject but of the same species. For example, allogeneic immune cells may be derived from a human donor and administered to a human recipient who is different from the donor. In some embodiments, engineered hematopoietic cells comprising one or more genetically engineered gene(s) encoding lineage-specific cell surface protein(s) (e.g., CD33 and IL1RAP or CD44 and EMR2) and the immune cells engineered to target an epitope of the antigen(s) are both allogeneic to the subject. In some embodiments, engineered hematopoietic cells comprising one or more genetically engineered gene(s) encoding antigen(s) (e.g., CD33 and IL1RAP or CD44 and EMR2 or any other combination of antigens described herein) and the immune cells engineered to target an epitope of the antigen(s) are from the same allogeneic donor. In some embodiments, engineered hematopoietic cells comprising one or more genetically engineered gene(s) encoding antigen(s) (e.g., CD33 and IL1RAP or CD44 and EMR2 or any other combination of antigens described herein) and the immune cells engineered to target an epitope of the antigen(s) are from two different allogeneic donors. In some embodiments, engineered hematopoietic cells comprising one or more genetically engineered gene(s) encoding antigen(s) (e.g., CD33 and IL1RAP or CD44 and EMR2 or any other combination of antigens described herein) and the immune cells engineered to target an epitope of the antigen(s) are both autologous to the subject. In some embodiments, engineered hematopoietic cells comprising one or more genetically engineered gene(s) encoding antigen(s) (e.g., CD33 and IL1RAP or CD44 and EMR2 or any other combination of antigens described herein) are autologous to the subject and the immune cells engineered to target an epitope of the antigen(s) are from an allogeneic donor. In some embodiments, engineered hematopoietic cells comprising one or more genetically engineered gene(s) encoding antigen(s) (e.g., CD33 and IL1RAP or CD44 and EMR2 or any other combination of antigens described herein) are from an allogeneic donor and the immune cells engineered to target an epitope of the lineage-specific cell surface protein(s) are autologous to the subject. In some embodiments, the immune cells and/or hematopoietic cells are allogeneic cells and have been further genetically engineered to reduce graft-versus-host disease. For example, as described herein, the hematopoietic stem cells may be genetically engineered (e.g., using genome editing) to have reduced expression of CD45RA. Methods for reducing graft-versus-host disease are known in the art, see, e.g., Yang et al. Curr. Opin. Hematol. (2015) 22(6): 509-515. In some embodiments, the immune cells (e.g., T cells) may be genetically engineered to reduce or eliminate expression of the T cell receptor (TCR) or reduce or eliminate cell surface localization of the TCR. In some examples, the gene encoding the TCR is knocked out or silenced (e.g., using gene editing methods, or shRNAs). In some embodiments, the TCR is silenced using peptide inhibitors of the TCR. In some embodiments, the immune cells (e.g., T cells) are subjected to a selection process to select for immune cells or a population of immune cells that do not contain an alloreactive TCR. Alternatively, in some embodiments, immune cells that naturally do not express TCRs (e.g., NK cells) may be used in any of the methods described herein. In some embodiments, the immune cells and/or hematopoietic cells have been further genetically engineered to reduce host-versus-graft effects. For example, in some embodiments, immune cells and/or hematopoietic cells may be subjected to gene editing or silencing methods to reduce or eliminate expression of one or more proteins involved in inducing host immune responses, e.g., CD52, MHC molecules, and/or MHC beta-2 microglobulin. In some embodiments, the immune cells expressing any of the chimeric receptors described herein are administered to a subject in an amount effective in to reduce the number of target cells (e.g., cancer cells) by least 20%, e.g., 50%, 80%, 100%, 2-fold, 5-fold, 10-fold, 20-fold, 50-fold, 100-fold or more. A typical amount of cells, i.e., immune cells or hematopoietic cells, administered to a mammal (e.g., a human) can be, for example, in the range of about 106 to 1011 cells. In some embodiments it may be desirable to administer fewer than 106 cells to the subject. In some embodiments, it may be desirable to administer more than 1011 cells to the subject. In some embodiments, one or more doses of cells includes about 106 cells to about 1011 cells, about 107 cells to about 1010 cells, about 108 cells to about 109 cells, about 106 cells to about 108 cells, about 107 cells to about 109 cells, about 107 cells to about 1010 cells, about 107 cells to about 1011 cells, about 108 cells to about 1010 cells, about 108 cells to about 1011 cells, about 109 cells to about 1010 cells, about 109 cells to about 1011 cells, or about 1010 cells to about 1011 cells. In some embodiments, the subject is preconditioned prior to administration of the cytotoxic agent and/or hematopoietic cells. In some embodiments, the method further comprises pre-conditioning the subject. In general, preconditioning a subject involves subjecting the patient to one or more therapy, such as a chemotherapy or other type of therapy, such as irradiation. In some embodiments, the preconditioning may induce or enhance the patient’s tolerance of one or more subsequent therapy (e.g., a targeted therapy), as described herein. In some embodiments, the pre-conditioning involves administering one or more chemotherapeutic agents to the subject. Non-limiting examples of chemotherapeutic agents include actinomycin, azacitidine, azathioprine, bleomycin, bortezomib, carboplatin, capecitabine, cisplatin, chlorambucil, cyclophosphamide, cytarabine, daunorubicin, docetaxel, doxifluridine, doxorubicin, epirubicin, epothilone, etoposide, fludarabine, fluorouracil, gemcitabine, hydroxyurea, idarubicin, imatinib, irinotecan, mechlorethamine, mercaptopurine, methotrexate, mitoxantrone, oxaliplatin, paclitaxel, pemetrexed, teniposide, tioguanine, topotecan, valrubicin, vinblastine, vincristine, vindesine, and vinorelbine. In some embodiments, the subject is preconditioned at least one day, two days, three days, four days, 5 days, 6 days, 7 days, 8 days, 9 days, 10 days, 11 days, 12 days, 13 days, two weeks, three weeks, four weeks, five weeks, six weeks, seven weeks, eight weeks, nine weeks, ten weeks, two months, three months, four months, five months, or at least six months prior to administering the cytotoxic agent and/or hematopoietic cells. In other embodiments, the chemotherapy(ies) or other therapy(ies) are administered concurrently with the cytotoxic agent and manipulated hematopoietic cells. In other embodiments, the chemotherapy(ies) or other therapy(ies) are administered after the cytotoxic agent and manipulated hematopoietic cells. In one embodiment, the chimeric receptor (e.g., a nucleic acid encoding the chimeric receptor) is introduced into an immune cell, and the subject (e.g., human patient) receives an initial administration or dose of the immune cells expressing the chimeric receptor. One or more subsequent administrations of the cytotoxic agent (e.g., immune cells expressing the chimeric receptor) may be provided to the patient at intervals of 15 days, 14, 13, 12, 11, 10, 9, 8, 7, 6, 5, 4, 3, or 2 days after the previous administration. More than one dose of the cytotoxic agent can be administered to the subject per week, e.g., 2, 3, 4, or more administrations of the agent. The subject may receive more than one doses of the cytotoxic agent (e.g., an immune cell expressing a chimeric receptor) per week, followed by a week of no administration of the agent, and finally followed by one or more additional doses of the cytotoxic agent (e.g., more than one administration of immune cells expressing a chimeric receptor per week). The immune cells expressing a chimeric receptor may be administered every other day for 3 administrations per week for two, three, four, five, six, seven, eight or more weeks. Any of the methods described herein may be for the treatment of a hematological malignancy in a subject. As used herein, the terms “treat,” “treating,” and “treatment” mean to relieve or alleviate at least one symptom associated with the disease or disorder, or to slow or reverse the progression of the disease or disorder. Within the meaning of the present disclosure, the term “treat” also denotes to arrest, delay the onset (i.e., the period prior to clinical manifestation of a disease) and/or reduce the risk of developing or worsening a disease. For example, in connection with cancer, the term “treat” may mean eliminate or reduce the number or replication of cancer cells, and/or prevent, delay or inhibit metastasis, etc. In some embodiments, a population of genetically engineered hematopoietic cells (e.g., carrying genetic edits in genes encoding antigens or expressing those proteins in mutated form) and a cytotoxic agent(s) specific to the cell surface protein are co-administered to a subject via a suitable route (e.g., infusion). In such a combined therapeutic method, the cytotoxic agent recognizes (binds) a target cell expressing the cell surface protein for targeted killing. The hematopoietic cells that express the protein in mutated form, which has reduced binding activity or do not bind the cytotoxic acid (e.g., because of lacking binding epitope) allow for repopulation of a cell type that is targeted by the cytotoxic agent. In some embodiments, the methods described herein involve administering a population of genetically engineered hematopoietic cells to a subject and administering one or more immunotherapeutic agents (e.g., cytotoxic agents). As will be appreciated by one of ordinary skill in the art, the immunotherapeutic agents may be of the same or different type (e.g., therapeutic antibodies, populations of immune cells expressing chimeric antigen receptor(s), and/or antibody-drug conjugates). In some embodiments, the treatment of the patient can involve the following steps: (1) administering a therapeutically effective amount of the cytotoxic agent to the patient and (2) infusing or reinfusing the patient with hematopoietic stem cells, either autologous or allogenic. In some embodiments, the treatment of the patient can involve the following steps: (1) administering a therapeutically effective amount of an immune cell expressing a chimeric receptor to the patient, wherein the immune cell comprises a nucleic acid sequence encoding a chimeric receptor that binds an epitope of a cell surface lineage-specific, disease-associated protein; and (2) infusing or reinfusing the patient with hematopoietic cells (e.g., hematopoietic stem cells), either autologous or allogenic. In each of the methods described herein, the cytotoxic agent (e.g., CAR-T cells) and the genetically engineered hematopoietic cells can be administered to the subject in any order. In some instances, the hematopoietic cells are given to the subject prior to the cytotoxic agent. In some instances, a second cytotoxic agent can be administered to the subject after treatment with the first cytotoxic agent, e.g., when the patient develops resistance or disease relapse. The hematopoietic cells given the same subject may have multiple edited genes expressing lineage-specific cell surface proteins in mutated form such that the cytotoxic agents can target wild-type proteins but not the mutated form. The efficacy of the therapeutic methods using a population of genetically engineered hematopoietic cells described herein, optionally in combination with a cytotoxic agent (e.g., CART) may be assessed by any method known in the art and would be evident to a skilled medical professional. For example, the efficacy of the therapy may be assessed by survival of the subject or cancer burden in the subject or tissue or sample thereof. In some embodiments, the efficacy of the therapy is assessed by quantifying the absolute number of a particular cell surface protein on a cell or population of cells. In some embodiments, the efficacy of the therapy is assessed by quantifying the number of cells presenting the antigen. In some embodiments, the cytotoxic agent comprising an antigen-binding fragment that binds to the epitope of the cell surface protein and the population of hematopoietic cells is administered concomitantly. In some embodiments, the cytotoxic agent comprising an antigen-binding fragment that binds an epitope of the cell surface protein (e.g., immune cells expressing a chimeric receptor as described herein) is administered prior to administration of the hematopoietic cells. In some embodiments, the agent comprising an antigen-binding fragment that binds an epitope of the cell surface protein (e.g., immune cells expressing a chimeric receptor as described herein) is administered at least about 1 day, 2 days, 3 days, 4 days, 5 days, 6 days, 1 week, 2 weeks, 3 weeks, 4 weeks, 5 weeks, 6 weeks, 7 weeks, 8 weeks, 9 weeks, 10 weeks, 11 weeks, 12 weeks, 3 months, 4 months, 5 months, 6 months or more prior to administration of the hematopoietic cells. In some embodiments, the hematopoietic cells are administered prior to the cytotoxic agent comprising an antigen-binding fragment that binds an epitope of the cell surface protein (e.g., immune cells expressing a chimeric receptor as described herein). In some embodiments, the population of hematopoietic cells is administered at least about 1 day, 2 days, 3 days, 4 days, 5 days, 6 days, 1 week, 2 weeks, 3 weeks, 4 weeks, 5 weeks, 6 weeks, 7 weeks, 8 weeks, 9 weeks, 10 weeks, 11 weeks, 12 weeks, 3 months, 4 months, 5 months, 6 months or more prior to administration of the cytotoxic agent comprising an antigen-binding fragment that binds to an epitope of the antigen. In some embodiments, the cytotoxic agent targeting the cell surface protein and the population of hematopoietic cells are administered at substantially the same time. In some embodiments, the cytotoxic agent targeting the cell surface protein is administered and the patient is assessed for a period of time, after which the population of hematopoietic cells is administered. In some embodiments, the population of hematopoietic cells is administered and the patient is assessed for a period of time, after which the cytotoxic agent targeting the cell surface protein is administered. Also within the scope of the present disclosure are multiple administrations (e.g., doses) of the cytotoxic agents and/or populations of hematopoietic cells. In some embodiments, the cytotoxic agents and/or populations of hematopoietic cells are administered to the subject once. In some embodiments, cytotoxic agents and/or populations of hematopoietic cells are administered to the subject more than once (e.g., at least 2, 3, 4, 5, or more times). In some embodiments, the cytotoxic agents and/or populations of hematopoietic cells are administered to the subject at a regular interval, e.g., every six months. In some embodiments, the subject is a human subject having a hematopoietic malignancy. As used herein a hematopoietic malignancy refers to a malignant abnormality involving hematopoietic cells (e.g., blood cells, including progenitor and stem cells). Examples of hematopoietic malignancies include, without limitation, Hodgkin’s lymphoma, non-Hodgkin’s lymphoma, leukemia, or multiple myeloma. Exemplary leukemias include, without limitation, acute myeloid leukemia, acute lymphoid leukemia, chronic myelogenous leukemia, acute lymphoblastic leukemia or chronic lymphoblastic leukemia, and chronic lymphoid leukemia. In some embodiments, cells involved in the hematopoietic malignancy are resistant to convention or standard therapeutics used to treat the malignancy. For example, the cells (e.g., cancer cells) may be resistant to a chemotherapeutic agent and/or CAR T cells used to treat the malignancy. Examples of hematopoietic malignancies include, but are not limited to, B cell malignancies such as non-Hodgkin’s lymphoma, Hodgkin’s lymphoma, leukemia, multiple myeloma, acute lymphoblastic leukemia, acute lymphoid leukemia, acute lymphocytic leukemia, chronic lymphoblastic leukemia, chronic lymphoid leukemia, and chronic lymphocytic leukemia. In some embodiments, the hematopoietic malignancy is a relapsing hematopoietic malignancy. In some embodiments, the leukemia is acute myeloid leukemia (AML). AML is characterized as a heterogeneous, clonal, neoplastic disease that originates from transformed cells that have progressively acquired critical genetic changes that disrupt key differentiation and growth-regulatory pathways. (Dohner et al., NEJM, (2015) 373:1136). CD33 glycoprotein is expressed on the majority of myeloid leukemia cells as well as on normal myeloid and monocytic precursors and has been considered to be an attractive target for AML therapy (Laszlo et al., Blood Rev. (2014) 28(4):143-53). While clinical trials using anti-CD33 monoclonal antibody based therapy have shown improved survival in a subset of AML patients when combined with standard chemotherapy, these effects were also accompanied by safety and efficacy concerns. In some cases, a subject may initially respond to a therapy (e.g., for a hematopoietic malignancy) and subsequently experience relapse. Any of the methods or populations of genetically engineered hematopoietic cells described herein may be used to reduce or prevent relapse of a hematopoietic malignancy. Alternatively or in addition, any of the methods described herein may involve administering any of the populations of genetically engineered hematopoietic cells described herein and an immunotherapeutic agent (e.g., cytotoxic agent) that targets cells associated with the hematopoietic malignancy and further administering one or more additional immunotherapeutic agents when the hematopoietic malignancy relapses. As used herein, the term “relapse” refers to the reemergence or reappearance of cells associated with a hematopoietic malignancy following a period of responsiveness to a therapy. Methods of determining whether a hematopoietic malignancy has relapsed in a subject will be appreciated by one of ordinary skill in the art. In some embodiments, the period of responsiveness to a therapy involves the level or quantity of cells associated with the hematopoietic malignancy the falling below a threshold, e.g., below 20%, 15%, 10%, 5%, 4%, 3%, 2%, or 1% of the level or quantity of cells prior to administration of the therapy. In some embodiments, a relapse is characterized by the level or quantity of cells associated with the hematopoietic malignancy above a threshold, e.g., above 20%, 15%, 10%, 5%, 4%, 3%, 2%, or 1% higher than the level or quantity of cells during the period of responsiveness. Methods of determining the minimal residual disease in a subject are known in the art and may be used, for example to assess whether a hematopoietic malignancy has relapsed or is likely to relapse. See, e.g., Taraseviciute et al. Hematology and Oncology (2019) 31(1)). In some embodiments, the subject has or is susceptible to relapse of a hematopoietic malignancy (e.g., AML) following administration of one or more previous therapies. In some embodiments, the methods described herein reduce the subject’s risk of relapse or the severity of relapse. Without wishing to be bound by any particular theory, some cancers, including hematopoietic malignancies, are thought to relapse after an initial period of responsiveness to a therapy due to mechanisms such as antigen loss/antigen escape or lineage switch. In general, antigen loss/antigen escape results in relapse with a phenotypically similar hematopoietic malignancy characterized by cells that lack surface expression of the antigen targeted by the previous therapy (e.g., immunotherapeutic agent) such that the cells are no longer targeted by the previous therapy. In contrast, lineage switch presents as a genetically related but phenotypically different malignancy in which the cells lack surface expression of the antigen targeted by the previous therapy (e.g., immunotherapeutic agent) such that the cells are no longer targeted by the previous therapy. See, e.g., Brown et al. Nature Reviews Immunology (2019) 19:73-74; Majzner et al. Cancer Discovery (2018) 8(10). Antigen loss/antigen escape in which the target antigen is no longer present on the target cells (e.g., cells of the hematopoietic malignancy) frequently occurs as a result of genetic mutation and/or enrichment of cells that express a variant of the antigen (e.g., lineage-specific cell surface antigen) that is not targeted by the immunotherapeutic agent (e.g., cytotoxic agent). In some embodiments, the hematopoietic malignancy has relapsed due to antigen loss/antigen escape. In some embodiments, the target cells have lost the targeted epitope (e.g., of a lineage-specific cell surface antigen) or have reduced expression of the antigen (e.g., lineage-specific cell surface antigen) such that the targeted epitope is not recognized by the immunotherapeutic agent or is not sufficient to induce cytotoxicity. In some embodiments, the hematopoietic malignancy has relapsed due to lineage switch. In some embodiments, the methods described herein reduce or avoid relapse of a hematopoietic malignancy by targeting more than one antigen. In some embodiments, the populations of genetically engineered hematopoietic cells express mutants of more than one antigens such that the mutated antigens are not targeted by an immunotherapeutic agent(s). In some embodiments, a cancer treated with the methods herein comprises a first sub- population of cancer cells and a second sub-population of cancer cells. One of the sub- populations may be cancer stem cells. One of the sub-populations may be cancer bulk cells. One of the sub-populations may have one or more (e.g., at least 2, 3, 4, 5, or all) markers of differentiated hematopoietic cells. One of the sub-populations may have one or more (e.g., at least 2, 3, 4, 5, or all) markers of earlier lineage cells, e.g., HSCs or HPCs. Any of the immune cells expressing chimeric receptors described herein may be administered in a pharmaceutically acceptable carrier or excipient as a pharmaceutical composition. The phrase “pharmaceutically acceptable,” as used in connection with compositions and/or cells of the present disclosure, refers to molecular entities and other ingredients of such compositions that are physiologically tolerable and do not typically produce untoward reactions when administered to a mammal (e.g., a human). Preferably, as used herein, the term “pharmaceutically acceptable” means approved by a regulatory agency of the Federal or a state government or listed in the U.S. Pharmacopeia or other generally recognized pharmacopeia for use in mammals, and more particularly in humans. “Acceptable” means that the carrier is compatible with the active ingredient of the composition (e.g., the nucleic acids, vectors, cells, or therapeutic antibodies) and does not negatively affect the subject to which the composition(s) are administered. Any of the pharmaceutical compositions and/or cells to be used in the present methods can comprise pharmaceutically acceptable carriers, excipients, or stabilizers in the form of lyophilized formations or aqueous solutions. Pharmaceutically acceptable carriers, including buffers, are well known in the art, and may comprise phosphate, citrate, and other organic acids; antioxidants including ascorbic acid and methionine; preservatives; low molecular weight polypeptides; proteins, such as serum albumin, gelatin, or immunoglobulins; amino acids; hydrophobic polymers; monosaccharides; disaccharides; and other carbohydrates; metal complexes; and/or non-ionic surfactants. See, e.g. Remington: The Science and Practice of Pharmacy 20th Ed. (2000) Lippincott Williams and Wilkins, Ed. K. E. Hoover. V. Diagnostic and/or Prognostic Methods In some embodiments, aspects of the present disclosure relate to methods for determining if a subject is at risk of relapse of a disease. In some embodiments, such methods comprise providing a first biological sample obtained from a subject at a first time point and a second biological sample obtained from the subject at a second time point, measuring a level of a first cell surface protein in the first biological sample and in the second biological sample, identifying the subject as at risk for relapse of the disease if the level of the first cell surface protein in the first biological sample deviates from the level of the first cell surface protein in the second biological sample. In some embodiments, the cell surface protein is one selected from Table 3 or Table 4. In some embodiments, aspects of the present disclosure relate to methods for identifying a subject as a candidate for treatment with an agent, such as a cytotoxic agent described herein. In some embodiments, such methods comprise providing a first biological sample obtained from a subject at a first time point and a second biological sample obtained from the subject at a second time point, and measuring a level of a first cell surface protein in the first biological sample and in the second biological sample. In some embodiments, the cell surface protein is one selected from Table 3 or Table 4. In some embodiments, methods of identifying subjects as being at risk of relapse of a disease and/or as a candidate for treatment with an agent further comprises administering to the subject a therapeutically effective amount of an agent comprising an antigen-binding fragment that binds to the first cell surface protein, such as a cytotoxic agent described herein. In some embodiments, methods of identifying subjects as being at risk of relapse of a disease and/or as a candidate for treatment with an agent further comprises administering to the subject a population of genetically engineered hematopoietic cells having reduced or eliminated expression of the first cell surface protein, such as a genetically engineered hematopoietic cell described herein, or a cell population thereof. In some embodiments, the first biological sample and/or the second biological sample comprises AML blast cells. In some embodiments, first time point is at or near the time the subject is diagnosed with the disease. Subjects In some embodiments, the subject is a human subject having a hematopoietic malignancy. In some embodiments, a subject has, is suspected of having, or at risk of developing Hodgkin’s lymphoma, non-Hodgkin’s lymphoma, leukemia, or multiple myeloma. Exemplary leukemias include, without limitation, acute myeloid leukemia, acute lymphoid leukemia, chronic myelogenous leukemia, acute lymphoblastic leukemia or chronic lymphoblastic leukemia, or chronic lymphoid leukemia. In some cases, a subject may initially respond to a therapy (e.g., for a hematopoietic malignancy) and subsequently experience relapse. In some embodiments, the subject has or is susceptible to relapse of a hematopoietic malignancy (e.g., AML) following administration of one or more previous therapies. In some embodiments, a subject exhibits an initial period of responsiveness to a therapy due to mechanisms such as antigen loss/antigen escape or lineage switch. In some embodiments, a subject may have, be suspect of having, or at risk of developing antigen loss/antigen escape which results in relapse with a phenotypically similar hematopoietic malignancy characterized by cells that lack surface expression of the cell surface protein targeted by the previous therapy (e.g., immunotherapeutic agent) such that the cells are no longer targeted by the previous therapy. In some embodiments, a subject may have, be suspect of having, or at risk of developing lineage switch wherein a genetically related but phenotypically different malignancy in which the cells lack surface expression of the cell surface protein targeted by the previous therapy (e.g., immunotherapeutic agent) such that the cells are no longer targeted by the previous therapy. See, e.g., Brown et al. Nature Reviews Immunology (2019) 19:73-74; Majzner et al. Cancer Discovery (2018) 8(10). In some embodiments, antigen loss/antigen escape comprises the target antigen no longer being present on the target cells (e.g., cells of the hematopoietic malignancy) and may occur as a result of genetic mutation and/or enrichment of cells that express a variant of the cell surface protein (e.g., lineage-specific cell surface antigen) that is not targeted by the immunotherapeutic agent (e.g., cytotoxic agent). In some embodiments, the hematopoietic malignancy has relapsed due to antigen loss/antigen escape. In some embodiments, the target cells have lost the targeted epitope (e.g., of a lineage-specific cell surface antigen) or have reduced expression of the cell surface protein (e.g., lineage-specific cell surface antigen) such that the targeted epitope is not recognized by the immunotherapeutic agent or is not sufficient to induce cytotoxicity. In some embodiments, the hematopoietic malignancy has relapsed due to lineage switch. In some embodiments, the methods described herein reduce or avoid relapse of a hematopoietic malignancy by targeting more than one antigen. In some embodiments, the populations of genetically engineered hematopoietic cells express mutants of more than one antigens such that the mutated antigens are not targeted by an immunotherapeutic agent(s). In some embodiments, the methods described herein reduce the subject’s risk of relapse or the severity of relapse. In some embodiments, the period of responsiveness to a therapy involves the level or quantity of cells associated with the hematopoietic malignancy the falling below a threshold, e.g., below 20%, 15%, 10%, 5%, 4%, 3%, 2%, or 1% of the level or quantity of cells prior to administration of the therapy. In some embodiments, a relapse is characterized by the level or quantity of cells associated with the hematopoietic malignancy above a threshold, e.g., above 20%, 15%, 10%, 5%, 4%, 3%, 2%, or 1% higher than the level or quantity of cells during the period of responsiveness. Methods of determining the minimal residual disease in a subject are known in the art and may be used, for example to assess whether a hematopoietic malignancy has relapsed or is likely to relapse. See, e.g., Taraseviciute et al. Hematology and Oncology (2019) 31(1)). Mutated Genes Encoding Antigens In some embodiments, a subject has, is suspected of having, or is at risk of developing a mutation in a gene encoding a lineage-specific cell surface antigen. In some embodiments, the mutation is associated with altered function a lineage-specific cell surface antigen. In some embodiments, the mutation is associated with upregulating a lineage-specific cell surface antigen. In some embodiments, the mutation is associated with downregulating a lineage-specific cell surface antigen. In some embodiments, the subject has, is suspected of having, or is at risk of developing more than one (e.g., two, three, four, or more) mutated gene encoding cell surface proteins. In some embodiments, the subject may comprise a first mutated gene that results in downregulation of a first cell surface protein and second mutated gene that results in downregulation of a second lineage-specific cell surface antigen. In some embodiments, the subject may comprise a first mutated gene that results in upregulation of a first cell surface protein and second mutated gene that results in downregulation of a second lineage-specific cell surface antigen. In some embodiments, the subject may comprise a first mutated gene that results in upregulation of a first cell surface protein and second mutated gene that results in upregulation of a second lineage-specific cell surface antigen. In some embodiments, a subject has, is suspected of having, or is at risk of developing a mutation in a gene encoding a lineage-specific cell surface antigen, such as CD1a, CD1b, CD1c, CD1d, CD1e, CD2, CD3, CD3d, CD3e, CD3g, CD4, CD5, CD6, CD7, CD8a, CD8b, CD9, CD10, CD11a, CD11b, CD11c, CD11d, CDw12, CD13, CD14, CD15, CD16, CD16b, CD17, CD18, CD19, CD20, CD21, CD22, CD23, CD24, CD25, CD26, CD27, CD28, CD29, CD30, CD31, CD32a, CD32b, CD32c, CD34, CD35, CD36, CD37, CD38, CD39, CD40, CD41, CD42a, CD42b, CD42c, CD42d, CD43, CD44, CD45, CD45RA, CD45RB, CD45RC, CD45RO, CD46, CD47, CD48, CD49a, CD49b, CD49c, CD49d, CD49e, CD49f, CD50, CD51, CD52, CD53, CD54, CD55, CD56, CD57, CD58, CD59, CD60a, CD61, CD62E, CD62L, CD62P, CD63, CD64a, CD65, CD65s, CD66a, CD66b, CD66c, CD66F, CD68, CD69, CD70, CD71, CD72, CD73, CD74, CD75, CD75S, CD77, CD79a, CD79b, CD80, CD81, CD82, CD83, CD84, CD85A, CD85C, CD85D, CD85E, CD85F, CD85G, CD85H, CD85I, CD85J, CD85K, CD86, CD87, CD88, CD89, CD90, CD91, CD92, CD93, CD94, CD95, CD96, CD97, CD98, CD99, CD99R, CD100, CD101, CD102, CD103, CD104, CD105, CD106, CD107a, CD107b, CD108, CD109, CD110, CD111, CD112, CD113, CD114, CD115, CD116, CD117, CD118, CD119, CD120a, CD120b, CD121a, CD121b, CD121a, CD121b, CD122, CD123, CD124, CD125, CD126, CD127, CD129, CD130, CD131, CD132, CD133, CD134, CD135, CD136, CD137, CD138, CD139, CD140a, CD140b, CD141, CD142, CD143, CD14, CDw145, CD146, CD147, CD148, CD150, CD152, CD152, CD153, CD154, CD155, CD156a, CD156b, CD156c, CD157, CD158b1, CD158b2, CD158d, CD158e1/e2, CD158f, CD158g, CD158h, CD158i, CD158j, CD158k, CD159a, CD159c, CD160, CD161, CD163, CD164, CD165, CD166, CD167a, CD168, CD169, CD170, CD171, CD172a, CD172b, CD172g, CD173, CD174, CD175, CD175s, CD176, CD177, CD178, CD179a, CD179b, CD180, CD181, CD182, CD183, CD184, CD185, CD186, CD191, CD192, CD193, CD194, CD195, CD196, CD197, CDw198, CDw199, CD200, CD201, CD202b, CD203c, CD204, CD205, CD206, CD207, CD208, CD209, CD210a, CDw210b, CD212, CD213a1, CD213a2, CD215, CD217, CD218a, CD218b, CD220, CD221, CD222, CD223, CD224, CD225, CD226, CD227, CD228, CD229, CD230, CD231, CD232, CD233, CD234, CD235a, CD235b, CD236, CD236R, CD238, CD239, CD240, CD241, CD242, CD243, CD244, CD245, CD246, CD247, CD248, CD249, CD252, CD253, CD254, CD256, CD257, CD258, CD261, CD262, CD263, CD264, CD265, CD266, CD267, CD268, CD269, CD270, CD272, CD272, CD273, CD274, CD275, CD276, CD277, CD278, CD279, CD280, CD281, CD282, CD283, CD284, CD286, CD288, CD289, CD290, CD292, CDw293, CD294, CD295, CD296, CD297, CD298, CD299, CD300a, CD300c, CD300e, CD301, CD302, CD303, CD304, CD305, CD306, CD307a, CD307b, CD307c, CD307d, CD307e, CD309, CD312, CD314, CD315, CD316, CD317, CD318, CD319, CD320, CD321, CD322, CD324, CD325, CD326, CD327, CD328, CD329, CD331, CD332, CD333, CD334, CD335, CD336, CD337, CD338, CD339, CD340, CD344, CD349, CD350, CD351, CD352, CD353, CD354, CD355, CD357, CD358, CD359, CD360, CD361, CD362, CD363, CD364, CD365, CD366, CD367, CD368, CD369, CD370, and CD371. In some embodiments, a subject has, is suspected of having, or is at risk of developing a mutation in a gene encoding a cell surface protein selected from the group consisting of: CD19; CD123; CD22; CD30; CD171; CS-1 (also referred to as CD2 subset 1, CRACC, SLAMF7, CD319, and 19A24); C-type lectin-like molecule-1 (CLECL1); epidermal growth factor receptor variant III (EGFRvIII); ganglioside G2 (CD2); ganglioside GD3 (aNeu5Ac(2- 8)aNeu5Ac(2-3)bDGalp(1-4)bDGlep(1-1)Cer); TNF receptor family member B cell maturation (BCMA), Tn antigen ((Tn Ag) or (GalNAcα-Ser/Thr)); prostate-specific membrane antigen (PSMA); Receptor tyrosine kinase-like orphan receptor 1 (ROR1); Fms- Like tyrosine Kinase 3 (FLT3); Tumor-associated glycoprotein 72 (TAG72); CD38; CD44v6; Carcinoembryonic antigen (CEA); Epithelial cell adhesion molecule (EPCAM); B7H3 (CD276); KIT (CD117); Interleukin-13 receptor subunit alpha-2 (IL-13Ra2 or CD213A2); Mesothelin; Interleukin 11 receptor alpha (IL-11Ra); prostate stem cell antigen (PSCA); Protease Serine 21 (Testisin or PRSS21); vascular endothelial growth factor receptor 2 (VEGFR2); Lewis(Y) antigen; CD24; Platelet-derived growth factor receptor beta (PDGFR- beta); Stage-specific embryonic antigen-4 (SSEA-4); CD20; Folate receptor alpha; Receptor tyrosine-protein kinase ERBB2 (Her2/neu); Mucin 1, cell surface associated (MUC1); epidermal growth factor receptor (EGFR); neural cell adhesion molecule (NCAM); Prostase; prostatic acid phosphatase (PAP); elongation factor 2 mutated (ELF2M); Ephrin B2; fibroblast activation protein alpha (FAP); insulin-like growth factor I receptor (IGF-I receptor), carbonic anhydrase IX (CAIX), Proteasome (Prosome, Macropain) Subunit, Beta Type 9 (LMP2); glycoprotein 100 (gp100); oncogene fusion protein consisting of breakpoint cluster region (BCR) and Abelson murine leukemia viral oncogene homolog 1 (Abl) (bcr- abl); tyrosinase; ephrin type-A receptor 2 (EphA2); Fucosyl GM1; sialyl Lewis adhesion molecule (sLe); ganglioside GM3 (aNeu5Ac(2-3)bDGalp(1-4)bDGlcp(1-1)Cer); transglutaminase 5 (TGS5); high molecular weight-melanoma-associated antigen (HMWMAA); o-acetyl-GD2 ganglioside (OAcGD2); Folate receptor beta; tumor endothelial marker 1 (TEM1/CD248); tumor endothelial marker 7-related (TEM7R); claudin 6 (CLDN6); thyroid stimulating hormone receptor (TSHR); G protein-coupled receptor class C group 5, member D (GPRC5D); chromosome X open reading frame 61 (CXORF61); CD97; CD179a; anaplastic lymphoma kinase (ALK); Polysialic acid; placenta-specific 1 (PLAC1); hexasaccharide portion of globoH glycoceramide (GloboH); mammary gland differentiation antigen (NY-BR-1); uroplakin 2 (UPK2); Hepatitis A virus cellular receptor 1 (HAVCR1); adrenoceptor beta 3 (ADRB3); pannexin 3 (PANX3); G protein-coupled receptor 20 (GPR20); lymphocyte antigen 6 complex; locus K 9 (LY6K); Olfactory receptor 51E2 (OR51E2); TCR Gamma Alternate Reading Frame Protein (TARP); Wilms tumor protein (WT1); Cancer/testis antigen 1 (NY-ESO-1); Cancer/testis antigen 2 (LAGE-1a); Melanoma- associated antigen 1 (MAGE-A1), ETS translocation-variant gene 6, located on chromosome 12p (ETV6-AML); sperm protein 17 (SPA17); X Antigen Family, member 1A (XAGE1); angiopoietin-binding cell surface receptor 2 (Tie 2); melanoma cancer testis antigen-1 (MAD-CT-1); melanoma cancer testis antigen-2 (MAD-CT-2); Fos-related antigen 1; tumor protein p53 (p53); p53 mutant; prostein; surviving; telomerase; prostate carcinoma tumor antigen-1 (PCTA-1 or Galectin 8), melanoma antigen recognized by T cells 1 (MelanA or MART1); Rat sarcoma (Ras) mutant; human Telomerase reverse transcriptase (hTERT); sarcoma translocation breakpoints; melanoma inhibitor of apoptosis (ML-1AP); ERG (transmembrane protease, serine 2 (TMPRSS2) ETS fusion gene); N-Acetyl glucosaminyl- transferase V (NA17); paired box protein Pax-3 (PAX3); Androgen receptor; Cyclin B1; v- myc avian myelocytomatosis viral oncogene neuroblastoma derived homolog (MYCN); Ras Homolog Family Member C (RhoC); Tyrosinase-related protein 2 (TRP-2); Cytochrome P4501B1 (CYP1B1); CCCTC-Binding Factor (Zinc Finger Protein)-Like (BORIS or Brother of the Regulator of Imprinted Sites), Squamous Cell Carcinoma Antigen Recognized By T Cells 3 (SART3); Paired box protein Pax-5 (PAX5); proacrosin binding protein sp32 (OY- TES1); lymphocyte-specific protein tyrosine kinase (LCK); A kinase anchor protein 4 (AKAP-4); synovial sarcoma, X breakpoint 2 (SSX2); Receptor for Advanced Glycation Endproducts (RAGE-1); renal ubiquitous 1 (RU1); renal ubiquitous 2 (RU2); legumain; human papilloma virus E6 (HPV E6); human papilloma virus E7 (HPV E7); intestinal carboxy esterase; heat shock protein 70-2 mutated (mut hsp70-2); CD79a; CD79b; CD72; Leukocyte-associated immunoglobulin-like receptor 1 (LAIR1); Fc fragment of IgA receptor (FCAR or CD89); Leukocyte immunoglobulin-like receptor subfamily A member 2 (LILRA2); CD300 molecule-like family member f (CD300LF); C-type lectin domain family 12 member A (CLEC12A); bone marrow stromal cell antigen 2 (BST2); EGF-like module- containing mucin-like hormone receptor-like 2 (EMR2), lymphocyte antigen 75 (LY75); Glypican-3 (GPC3); Fc receptor-like 5 (FCRL5); and immunoglobulin lambda-like polypeptide 1 (IGLL1). In some embodiments, a subject has, is suspected of having, or is at risk of developing a mutation in a gene encoding a cell surface protein known to be associated with a cancer, such as CD20, CD22 (Non-Hodgkin's lymphoma, B-cell lymphoma, chronic lymphocytic leukemia (CLL)), CD52 (B-cell CLL), CD33 (Acute myelogenous leukemia (AML)), CD10 (gp100) (Common (pre-B) acute lymphocytic leukemia and malignant melanoma), CD3/T- cell receptor (TCR) (T-cell lymphoma and leukemia), CD79/B-cell receptor (BCR) (B-cell lymphoma and leukemia), CD26 (epithelial and lymphoid malignancies), human leukocyte antigen (HLA)-DR, HLA-DP, and HLA-DQ (lymphoid malignancies), RCAS1 (gynecological carcinomas, biliary adenocarcinomas and ductal adenocarcinomas of the pancreas) as well as prostate specific membrane antigen. In some embodiments, a subject has, is suspected of having, or is at risk of developing a mutation in a gene encoding a cell surface protein selected from a group consisting of: CD10; CD101; CD117; CD11A; CD11C; CD120B; CD123; CD13; CD133; CD152; CD200; CD205 (LY75); CD226; CD244 (SLAMF4); CD25; CD274; CD305 (LAIR1); CD32; CD33; CD34; CD366; CD38; CD41; CD42B; CD44; CD45RA; CD47; CD48; CD49D (ITGA4); CD49F; CD52; CD58; CD64; CD7; CD70; CD82; CD84; CD85; CD85K; CD86; CD9; CD96; CD99; CLL-1; EMR2 (ADGRE2); FR-B; GPR56; and IL1RAP. In some embodiments, a subject has, is suspected of having, or is at risk of developing a mutation in a gene encoding a cell surface protein selected from a group consisting of: CD10; CD101; CD11A; CD11C; CD123; CD133; CD152; CD200; CD205; CD244; CD25; CD305; CD33; CD34; CD366; CD38; CD42B; CD44; CD45RA; CD47; CD48; CD49D; CD49F; CD52; CD58; CD64; CD7; CD84; CD85K; CD9; CD99; CLL-1; EMR2; GPR56; IL1RAP; and CD274. In some embodiments, a subject has, is suspected of having, or is at risk of developing a mutation in a gene encoding a cell surface protein selected from a group consisting of: CD10; CD117; CD11A; CD11C; CD120B; CD123; CD13; CD133; CD152; CD205; CD22; CD226; CD244; CD25; CD305; CD32; CD33; CD34; CD38; CD41; CD44; CD45RA; CD47; CD48; CD49D; CD49F; CD52; CD56; CD58; CD64; CD7; CD70; CD71; CD82; CD84; CD85K; CD86; CD96; CD99; CLL-1; EMR2; FR-B; GPR56; HLA-DR; and IL1RAP. In some embodiments, a subject has, is suspected of having, or is at risk of developing a mutation in a first gene encoding a first cell surface protein and a second gene encoding a second cell surface protein selected from a group consisting of: CD274 and CD152; CD274 and CD226; CD152 and CD226; CD10 and CD49F; CD49F and GPR56; CD200 and CD34; CD34 and CD52; CD34 and CD133; GPR56 and CD133; CD42B and CD41; CD85K and CD64; CD85K and CD32; CD64 and CD32; CD64 and CD11C; CD64 and CD86; CD45RA and CD96; CD45RA and IL1RAP; CD45RA and CD123; CD84 and CD33; CD11C and IL1RAP; CD11C and CD33; CD11C and CD86; CD9 and IL1RAP; IL1RAP and CD33; IL1RAP and CD205; IL1RAP and CD305; IL1RAP and CD123; CD33 and CLL-1; CD33 and CD305; CD11A and CD86; CLL-1 and CD305; CD58 and CD305; CD49D and CD47; CD49D and CD99; CD49D and CD44; CD49D and CD305; CD47 and CD99; CD47 and EMR2; CD99 and CD44; CD99 and CD305; CD99 and EMR2; CD205 and CD305; CD205 and CD123; CD44 and CD305; CD44 and EMR2; CD244 and CD117; CD123 and EMR2; EMR2 and CD82; CD152 and CD120B; CD48 and CD11A; CD200 and CD25; CD34 and CD7; CD34 and CD13; CD52 and GPR56; CD52and CD133; CD52 and CD7; GPR56 and CD7; GPR56 and CD82; CD133 and CD7; CD133 and CD82; CD7 and CD13; CD101 and CD64; CD85 and CD70; CD85K and CD86; CD64 and FR-B; CD366 and CD96; CD25 and CD45RA; CD25 and CD96; CD9 and CD82; IL1RAP and CD58; CD11A and CD38; CD11A and CD44; CD11A and CD244; CD38 and CD305; CD38 and EMR2; CLL-1 and CD49D; CD58 and CD49D; CD47 and CD205; and CD47 and CD44. In some embodiments, a subject has, is suspected of having, or is at risk of developing a mutation in a first gene encoding a first cell surface protein and a second gene encoding a second cell surface protein selected from a group consisting of: CD85K and CD64; CD45RA and IL1RAP; CD34 and CD133; IL1RAP and CD33; CD47 and CD99; CLL-1 and CD305; CD152 and CD226; CD45RA and CD96; CD45RA and CD123; CD49D and CD305; IL1RAP and CD305; CD58 and CD305; CD49D and CD47; CD33 and CD305; CD49D and CD44; CD64 and CD86; GPR56 and CD133; CD34 and CD52; EMR2 and CD82; CD64 and CD32; CD42B and CD41; CD64 and CD11C; CD99 and CD44; CD200 and CD34; CD11C and CD33; IL1RAP and CD123; CD11C and IL1RAP; CD44 and EMR2; CD205 and CD305; CD44 and CD305; CD49D and CD99; CD274 and CD226; CD49F and GPR56; CD9 and IL1RAP; CD99 and CD305; CD10 and CD49F; CD11A and CD86; CD33 and CLL-1; CD84 and CD33; CD244 and CD117; CD85K and CD32; CD47 and EMR2; CD274 and CD152; IL1RAP and CD205; CD11C and CD86; CD99 and EMR2; CD205 and CD123; and CD123 and EMR2. In some embodiments, a subject has, is suspected of having, or is at risk of developing a mutation in a first gene encoding a first cell surface protein and a second gene encoding a second cell surface protein selected from a group consisting of: CD33 and CD205 (LY75); CD33 and CD244 (SLAMF4); CD33 and CD305 (LAIR1); CD33 and CD49D (ITGA4); CLL-1 and CD205 (LY75); CLL-1 and CD244 (SLAMF4); CLL-1 and CD305 (LAIR1); CLL-1 and CD49D (ITGA4); EMR2 and CD205 (LY75); EMR2 and CD244 (SLAMF4); EMR2 and CD305 (LAIR1); EMR2 and CD49D (ITGA4); CD38 and CD205 (LY75); CD38 and CD244 (SLAMF4); CD38 and CD305 (LAIR1); CD38 and CD49D (ITGA4); CD305 (LAIR1) and CD49D (ITGA4); CD305 (LAIR1) and CD205 (LY75); CD305 (LAIR1) and CD244 (SLAMF4); CD49D (ITGA4) and CD205 (LY75); CD49D (ITGA4) and CD244 (SLAMF4); and CD205 (LY75) and CD244 (SLAMF4). In some embodiments, a subject has, is suspected of having, or is at risk of developing a mutation in a first gene encoding a first cell surface protein and a second gene encoding a second lineage-specific cell surface antigen, wherein mutation of the first gene and second gene results in dysregulated expression of the first cell surface protein and the second cell surface protein which is indicative (e.g., diagnostic) of the subject having a hematopoietic malignancy. In some embodiments, the first gene and second gene encode combinations of antigens selected from a group consisting of: CD274 and CD152; CD274 and CD226; CD152 and CD226; CD10 and CD49F; CD49F and GPR56; CD200 and CD34; CD34 and CD52; CD34 and CD133; GPR56 and CD133; CD42B and CD41; CD85K and CD64; CD85K and CD32; CD64 and CD32; CD64 and CD11C; CD64 and CD86; CD45RA and CD96; CD45RA and IL1RAP; CD45RA and CD123; CD84 and CD33; CD11C and IL1RAP; CD11C and CD33; CD11C and CD86; CD9 and IL1RAP; IL1RAP and CD33; IL1RAP and CD205; IL1RAP and CD305; IL1RAP and CD123; CD33 and CLL-1; CD33 and CD305; CD11A and CD86; CLL-1 and CD305; CD58 and CD305; CD49D and CD47; CD49D and CD99; CD49D and CD44; CD49D and CD305; CD47 and CD99; CD47 and EMR2; CD99 and CD44; CD99 and CD305; CD99 and EMR2; CD205 and CD305; CD205 and CD123; CD44 and CD305; CD44 and EMR2; CD244 and CD117; CD123 and EMR2; and EMR2 and CD82. In some embodiments, the first gene and second gene encode combinations of antigens selected from a group consisting of: CD33 and CD205 (LY75); CD33 and CD244 (SLAMF4); CD33 and CD305 (LAIR1); CD33 and CD49D (ITGA4); CLL-1 and CD205 (LY75); CLL-1 and CD244 (SLAMF4); CLL-1 and CD305 (LAIR1); CLL-1 and CD49D (ITGA4); EMR2 and CD205 (LY75); EMR2 and CD244 (SLAMF4); EMR2 and CD305 (LAIR1); EMR2 and CD49D (ITGA4); CD38 and CD205 (LY75); CD38 and CD244 (SLAMF4); CD38 and CD305 (LAIR1); CD38 and CD49D (ITGA4); CD305 (LAIR1) and CD49D (ITGA4); CD305 (LAIR1) and CD205 (LY75); CD305 (LAIR1) and CD244 (SLAMF4); CD49D (ITGA4) and CD205 (LY75); CD49D (ITGA4) and CD244 (SLAMF4); and CD205 (LY75) and CD244 (SLAMF4). In some embodiments, a subject has, is suspected of having, or is at risk of developing a mutation in a first gene encoding a first cell surface protein and a second gene encoding a second lineage-specific cell surface antigen, wherein mutation of the first gene and second gene results in dysregulated expression of the first cell surface protein and the second cell surface protein which is indicative (e.g., diagnostic) of the subject having experienced a relapse of a hematopoietic malignancy. In some embodiments, the first gene and second gene encode combinations of antigens selected from a group consisting of: CD9 and CD82; CD45RA and CD123; CD133 and CD82; CD85K and CD70; CD47 and CD205; CD64 and FR-B; CD11A and CD44; CD85K and CD86; CD366 and CD96; CD101 and CD64; CLL-1 and CD49D; CD49D and CD47; EMR2 and CD82; CD38 and CD305; CD34 and CD7; CD200 and CD25; CD25 and CD45RA; CD49D and CD44; CD11A and CD244; CD133 and CD7; CD48 and CD11A; CD52 and CD133; GPR56 and CD82; CD11A and CD38; CD34 and CD13; CD58 and CD49D; CD47 and CD99; IL1RAP and CD58; CD47 and CD44; CD33 and CD305; CD64 and CD86; CD152 and CD120B; CD42B and CD41; CD34 and CD52; CD152 and CD226; CD7 and CD13; CD38 and EMR2; IL1RAP and CD33; CLL-1 and CD305; CD45RA and IL1RAP; CD34 and CD133; GPR56 and CD133; CD49D and CD305; CD25 and CD96; CD52 and CD7; CD64 and CD32; CD58 and CD305; CD45RA and CD96; IL1RAP and CD305; CD52 and GPR56; GPR56 and CD7; and CD85K and CD64. In some embodiments, the first gene and second gene encode combinations of antigens selected from a group consisting of: CD33 and CD205 (LY75); CD33 and CD244 (SLAMF4); CD33 and CD305 (LAIR1); CD33 and CD49D (ITGA4); CLL-1 and CD205 (LY75); CLL-1 and CD244 (SLAMF4); CLL-1 and CD305 (LAIR1); CLL-1 and CD49D (ITGA4); EMR2 and CD205 (LY75); EMR2 and CD244 (SLAMF4); EMR2 and CD305 (LAIR1); EMR2 and CD49D (ITGA4); CD38 and CD205 (LY75); CD38 and CD244 (SLAMF4); CD38 and CD305 (LAIR1); CD38 and CD49D (ITGA4); CD305 (LAIR1) and CD49D (ITGA4); CD305 (LAIR1) and CD205 (LY75); CD305 (LAIR1) and CD244 (SLAMF4); CD49D (ITGA4) and CD205 (LY75); CD49D (ITGA4) and CD244 (SLAMF4); and CD205 (LY75) and CD244 (SLAMF4). Biological Samples The term “biological sample” refers to any sample including tissue samples (such as tissue sections and needle biopsies of a tissue); cell samples (e.g., cytological smears (such as Pap or blood smears) or samples of cells obtained by microdissection); samples of whole organisms (such as samples of yeasts or bacteria); or cell fractions, fragments or organelles (such as obtained by lysing cells and separating the components thereof by centrifugation or otherwise). Other examples of biological samples include blood, serum, urine, semen, fecal matter, cerebrospinal fluid, interstitial fluid, mucous, tears, sweat, pus, biopsied tissue (e.g., obtained by a surgical biopsy or needle biopsy), nipple aspirates, milk, vaginal fluid, saliva, swabs (such as buccal swabs), or any material containing biomolecules that is derived from a first biological sample. In some embodiments, a biological sample is derived from a subject who has, is suspected of having, or is at risk of developing a hematopoietic malignancy. In some embodiments, a biological sample is obtained at a time during or near the time the subject is diagnosed with a hematopoietic malignancy. In some embodiments, a biological sample is obtained at a time during or near when the subject has or is suspected to have experienced relapse of the hematopoietic malignancy. In some embodiments, a biological sample is obtained after the subject has undergone has received at least one treatment for a hematopoietic malignancy. In some embodiments, a biological sample comprises malignant hematopoietic cells. In some embodiments, a biological sample comprises AML blast cells. In some embodiments, methods of the present disclosure further comprise determining the expression level of a first cell surface protein and the expression level of a second cell surface protein in a first biological sample by obtaining or having obtained the first biological sample from a subject and performing or having performed an assay comprising detecting the presence or absence of expression of the first cell surface protein and the second cell surface protein in the first biological sample. In some embodiments, methods of the present disclosure further comprise determining the expression level of the first cell surface protein and the expression level of the second cell surface protein in a second biological sample by obtaining or having obtained the second biological sample from the subject and performing or having performed an assay comprising detecting the presence or absence of expression of the first cell surface protein and the second cell surface protein in the second biological sample. In some embodiments, methods of the present disclosure further comprise identifying or having identified the second biological sample as comprising altered expression of the first cell surface protein and the second cell surface protein when the expression levels of the first cell surface protein and the second cell surface protein are different in the second biological sample relative to the first biological sample. In some embodiments, identifying or having identified the second biological sample as comprising altered expression of the first cell surface protein and the second cell surface protein also identifies the subject as having experienced relapse of the hematopoietic malignancy. In some embodiments, such methods further comprise administering to the subject a at least one of a genetically engineered hematopoietic, or cell population thereof, a cytotoxic agent described herein, or both. In some embodiments, identifying or having identified the second biological sample as comprising altered expression of the first cell surface protein and the second cell surface protein also identifies subject as being in need of administration to the subject a at least one of a genetically engineered hematopoietic, or cell population thereof, a cytotoxic agent described herein, or both, In some embodiments, a biological sample is derived from a subject who has, is suspected of having, or is at risk of developing a mutation in a gene encoding a cell surface protein selected from a group consisting of: CD10; CD101; CD117; CD11A; CD11C; CD120B; CD123; CD13; CD133; CD152; CD200; CD205 (LY75); CD226; CD244 (SLAMF4); CD25; CD274; CD305 (LAIR1); CD32; CD33; CD34; CD366; CD38; CD41; CD42B; CD44; CD45RA; CD47; CD48; CD49D (ITGA4); CD49F; CD52; CD58; CD64; CD7; CD70; CD82; CD84; CD85; CD85K; CD86; CD9; CD96; CD99; CLL-1; EMR2 (ADGRE2); FR-B; GPR56; and IL1RAP. In some embodiments, a biological sample is derived from a subject who has, is suspected of having, or is at risk of developing a mutation in a gene encoding a cell surface protein selected from a group consisting of: CD10; CD101; CD11A; CD11C; CD123; CD133; CD152; CD200; CD205; CD244; CD25; CD305; CD33; CD34; CD366; CD38; CD42B; CD44; CD45RA; CD47; CD48; CD49D; CD49F; CD52; CD58; CD64; CD7; CD84; CD85K; CD9; CD99; CLL-1; EMR2; GPR56; IL1RAP; and CD274. In some embodiments, a biological sample is derived from a subject who has, is suspected of having, or is at risk of developing a mutation in a gene encoding a cell surface protein selected from a group consisting of: CD10; CD117; CD11A; CD11C; CD120B; CD123; CD13; CD133; CD152; CD205; CD22; CD226; CD244; CD25; CD305; CD32; CD33; CD34; CD38; CD41; CD44; CD45RA; CD47; CD48; CD49D; CD49F; CD52; CD56; CD58; CD64; CD7; CD70; CD71; CD82; CD84; CD85K; CD86; CD96; CD99; CLL-1; EMR2; FR-B; GPR56; HLA-DR; and IL1RAP. In some embodiments, a biological sample is derived from a subject who has, is suspected of having, or is at risk of developing a mutation in a first gene encoding a first cell surface protein and a second gene encoding a second cell surface protein selected from a group consisting of: CD274 and CD152; CD274 and CD226; CD152 and CD226; CD10 and CD49F; CD49F and GPR56; CD200 and CD34; CD34 and CD52; CD34 and CD133; GPR56 and CD133; CD42B and CD41; CD85K and CD64; CD85K and CD32; CD64 and CD32; CD64 and CD11C; CD64 and CD86; CD45RA and CD96; CD45RA and IL1RAP; CD45RA and CD123; CD84 and CD33; CD11C and IL1RAP; CD11C and CD33; CD11C and CD86; CD9 and IL1RAP; IL1RAP and CD33; IL1RAP and CD205; IL1RAP and CD305; IL1RAP and CD123; CD33 and CLL-1; CD33 and CD305; CD11A and CD86; CLL-1 and CD305; CD58 and CD305; CD49D and CD47; CD49D and CD99; CD49D and CD44; CD49D and CD305; CD47 and CD99; CD47 and EMR2; CD99 and CD44; CD99 and CD305; CD99 and EMR2; CD205 and CD305; CD205 and CD123; CD44 and CD305; CD44 and EMR2; CD244 and CD117; CD123 and EMR2; EMR2 and CD82; CD152 and CD120B; CD48 and CD11A; CD200 and CD25; CD34 and CD7; CD34 and CD13; CD52 and GPR56; CD52and CD133; CD52 and CD7; GPR56 and CD7; GPR56 and CD82; CD133 and CD7; CD133 and CD82; CD7 and CD13; CD101 and CD64; CD85 and CD70; CD85K and CD86; CD64 and FR-B; CD366 and CD96; CD25 and CD45RA; CD25 and CD96; CD9 and CD82; IL1RAP and CD58; CD11A and CD38; CD11A and CD44; CD11A and CD244; CD38 and CD305; CD38 and EMR2; CLL-1 and CD49D; CD58 and CD49D; CD47 and CD205; and CD47 and CD44. In some embodiments, a biological sample is derived from a subject who has, is suspected of having, or is at risk of developing a mutation in a first gene encoding a first cell surface protein and a second gene encoding a second cell surface protein selected from a group consisting of: CD33 and CD205 (LY75); CD33 and CD244 (SLAMF4); CD33 and CD305 (LAIR1); CD33 and CD49D (ITGA4); CLL-1 and CD205 (LY75); CLL-1 and CD244 (SLAMF4); CLL-1 and CD305 (LAIR1); CLL-1 and CD49D (ITGA4); EMR2 and CD205 (LY75); EMR2 and CD244 (SLAMF4); EMR2 and CD305 (LAIR1); EMR2 and CD49D (ITGA4); CD38 and CD205 (LY75); CD38 and CD244 (SLAMF4); CD38 and CD305 (LAIR1); CD38 and CD49D (ITGA4); CD305 (LAIR1) and CD49D (ITGA4); CD305 (LAIR1) and CD205 (LY75); CD305 (LAIR1) and CD244 (SLAMF4); CD49D (ITGA4) and CD205 (LY75); CD49D (ITGA4) and CD244 (SLAMF4); and CD205 (LY75) and CD244 (SLAMF4). VI. Kits for Therapeutic Uses Also within the scope of the present disclosure are kits for use in treating hematopoietic malignancy. Such a kit may comprise the genetically engineered hematopoietic cells such as HSCs, and optionally one or more cytotoxic agents targeting cell surface proteins, the genes of which are edited in the hematopoietic cells. Such kits may include a container comprising a first pharmaceutical composition that comprises any of the genetically engineered hematopoietic cells as described herein, and optionally one or more additional containers comprising one or more cytotoxic agents (e.g., immune cells expressing chimeric receptors described herein) targeting the cell surface proteins as also described herein. In some embodiments, the kit can comprise instructions for use in any of the methods described herein. The included instructions can comprise a description of administration of the genetically engineered hematopoietic cells and optionally descriptions of administration of the one or more cytotoxic agents to a subject to achieve the intended activity in a subject. The kit may further comprise a description of selecting a subject suitable for treatment based on identifying whether the subject is in need of the treatment. In some embodiments, the instructions comprise a description of administering the genetically engineered hematopoietic cells and optionally the one or more cytotoxic agents to a subject who is in need of the treatment. The instructions relating to the use of the genetically engineered hematopoietic cells and optionally the cytotoxic agents described herein generally include information as to dosage, dosing schedule, and route of administration for the intended treatment. The containers may be unit doses, bulk packages (e.g., multi-dose packages) or sub-unit doses. Instructions supplied in the kits of the disclosure are typically written instructions on a label or package insert. The label or package insert indicates that the pharmaceutical compositions are used for treating, delaying the onset, and/or alleviating a disease or disorder in a subject. The kits provided herein are in suitable packaging. Suitable packaging includes, but is not limited to, vials, bottles, jars, flexible packaging, and the like. Also contemplated are packages for use in combination with a specific device, such as an inhaler, nasal administration device, or an infusion device. A kit may have a sterile access port (for example, the container may be an intravenous solution bag or a vial having a stopper pierceable by a hypodermic injection needle). The container may also have a sterile access port. . Kits optionally may provide additional components such as buffers and interpretive information. Normally, the kit comprises a container and a label or package insert(s) on or associated with the container. In some embodiment, the disclosure provides articles of manufacture comprising contents of the kits described above. General techniques The practice of the present disclosure will employ, unless otherwise indicated, conventional techniques of molecular biology (including recombinant techniques), microbiology, cell biology, biochemistry, and immunology, which are within the skill of the art. Such techniques are explained fully in the literature, such as Molecular Cloning: A Laboratory Manual, second edition (Sambrook, et al., 1989) Cold Spring Harbor Press; Oligonucleotide Synthesis (M. J. Gait, ed.1984); Methods in Molecular Biology, Humana Press; Cell Biology: A Laboratory Notebook (J. E. Cellis, ed., 1989) Academic Press; Animal Cell Culture (R. I. Freshney, ed.1987); Introduction to Cell and Tissue Culture (J. P. Mather and P. E. Roberts, 1998) Plenum Press; Cell and Tissue Culture: Laboratory Procedures (A. Doyle, J. B. Griffiths, and D. G. Newell, eds.1993-8) J. Wiley and Sons; Methods in Enzymology (Academic Press, Inc.); Handbook of Experimental Immunology (D. M. Weir and C. C. Blackwell, eds.): Gene Transfer Vectors for Mammalian Cells (J. M. Miller and M. P. Calos, eds., 1987); Current Protocols in Molecular Biology (F. M. Ausubel, et al. eds. 1987); PCR: The Polymerase Chain Reaction, (Mullis, et al., eds.1994); Current Protocols in Immunology (J. E. Coligan et al., eds., 1991); Short Protocols in Molecular Biology (Wiley and Sons, 1999); Immunobiology (C. A. Janeway and P. Travers, 1997); Antibodies (P. Finch, 1997); Antibodies: a practice approach (D. Catty., ed., IRL Press, 1988-1989); Monoclonal antibodies: a practical approach (P. Shepherd and C. Dean, eds., Oxford University Press, 2000); Using antibodies: a laboratory manual (E. Harlow and D. Lane (Cold Spring Harbor Laboratory Press, 1999); The Antibodies (M. Zanetti and J. D. Capra, eds. Harwood Academic Publishers, 1995); DNA Cloning: A practical Approach, Volumes I and II (D.N. Glover ed.1985); Nucleic Acid Hybridization (B.D. Hames & S.J. Higgins eds.(1985»; Transcription and Translation (B.D. Hames & S.J. Higgins, eds. (1984»; Animal Cell Culture (R.I. Freshney, ed. (1986»; Immobilized Cells and Enzymes (lRL Press, (1986»; and B. Perbal, A practical Guide To Molecular Cloning (1984); F.M. Ausubel et al. (eds.). Without further elaboration, it is believed that one skilled in the art can, based on the above description, utilize the present invention to its fullest extent. The following specific embodiments are, therefore, to be construed as merely illustrative, and not limitative of the remainder of the disclosure in any way whatsoever. All publications cited herein are incorporated by reference for the purposes or subject matter referenced herein. EXAMPLES Example 1: Longitudinal multimodal atlas of AML to enable novel targeting strategies Acute myeloid leukemia (AML) is an aggressive clonal malignancy characterized by combinations of chromosomal abnormalities, gene mutations, and cell surface antigen (Ag) expression profiles. This heterogeneity contributes to refractory or relapsed disease that presents a major challenge when treating AML. Previous single cell sequencing studies of primary AML samples have provided remarkable insight into the clonal architecture of AML cell populations and how their mutational, transcriptomic, and surface Ag profiles vary between patients. However, information addressing clonal shifts during progression from diagnosis to relapse in large cohorts is lacking. Here, single cell RNA sequencing was adapted with surface Ag feature barcoding to analyze more than 450,000 cells from 28 paired AML patient bone marrow mononuclear cell samples collected at diagnosis and relapse (56 samples) to generate the largest and most comprehensive single cell AML atlas to date. This atlas contains rich clinical metadata including cytogenetics, mutation status of canonical AML genes, treatment history, and survival information. These data were leveraged to identify potential correlations with clonal heterogeneity during progression to relapse and to propose novel strategies for targeting the surface of AML cells. The feature barcoding panel consisted of a comprehensive list of 81 surface Ags reported in clinicaltrials.gov or mined from literature, including large proteomics mass spectrometry datasets of AML patient bone marrow samples (DeBoer et. al., Cancer Cell 2018; Jayavelu et al, Cancer Cell 2022). Feature barcoding produced antibody derived tag (ADT) counts which are interpreted as relative values but alone does not indicate absolute number of Ags per cell. This limitation was addressed by measuring absolute Ag density of AML Ags, CD33, CLL-1, CD123, and EMR2 in patient samples using the flow cytometric QuantiBRITE assay. Focusing on these four Ags, the relationship between ADT expression and antibodies per cell (ABC) Ag density from QuantiBRITE was modeled. The model was applied to impute absolute Ag density for the remaining 77 Ags in all samples. UMAP visualization after sample transcriptome integration revealed distinct clustering of CD45-dim myeloblasts, T cells, B cells, and erythroid cells. Longitudinal analysis identified 28 surface Ags that were differentially expressed (absolute log2FC > 1, p < 0.01) between relapse and diagnosis myeloblasts across multiple patients (see, FIG.4A). An additional 19 Ags were differentially expressed in no more than one patient while the other 19 had no significant change in any patient, highlighting the inter-and intra- heterogeneity of AML at diagnosis and relapse in these patients. Targeting multiple antigens simultaneously may help address the issue of antigen heterogeneity of tumor cells and help avoid potential antigen escape. Because multi-targeting therapies are a potential solution to this, the single-cell resolution of the atlas was leveraged to systematically identify Ags that in combination provide maximum coverage and are significantly co-expressed in myeloblasts. From this analysis, 48 and 52 co-expressed Ag pairs were found in diagnosis and relapse blasts, respectively (r > 0.4, p < 0.01; see, FIG.4B). Of note, CD44 and EMR2 were co- expressed at both diagnosis (r = 0.49 p < 0.01 ) and relapse (r = 0.3, p < 0.01) with >90% of their blasts expressing CD44, EMR2, or both at targetable antigen levels of >1000 Ags/cell (see, Table 3). Furthermore, 10 samples had >40% of their blasts co-expressing both Ags simultaneously at >1000 Ags/cell at diagnosis or relapse.6 of these patients had a >10% increase in co-expression at relapse, 3 had >10% decrease at relapse, and one patient had no co-expression change. Table 3. Correlation of Cell surface Proteins That Exhibited Dysregulated Expression at Diagnosis and/or Relapse.
GIFPGDGSGKQETRAGVVHGAIGGAGVTALLALCLCLIFFIVKTHRRKAARTAVGRNDTH PTTGSASPKHQKKSKLHGPTETSSCSGAAPTVEMDEELHYASLNFHGMNPSKDTSTEYSE VRTQ (SEQ ID NO: 1) (Uniprot No. P20138) Non-limiting examples of CD33 gRNAs may be found in International Patent Application Publication Nos. WO 2017/066760, WO 2020/150478, WO 2020/237217, WO 2018/160768, WO 2019/046285, WO2023/015182, WO2024/015925, WO2023/196816, WO2023/043858, and WO2022/217086, and WO2020/047164 each of which are incorporated by reference herein in their entirety. In some embodiments, a targeting domain which is complementary to a target nucleic acid in a cell surface protein is complementary to a target nucleic acid encoding human CLL- 1 or a portion thereof, e.g., having a sequence at least 70%, 75%, 80%, 85%, 90%, 95%, 96%, 97%, 98%, or 99% identical thereto to the CLL-1 amino acid sequence set forth below: MSEEVTYADLQFQNSSEMEKIPEIGKFGEKAPPAPSHVWRPAALFLTLLCLLLLIGLGVL ASMFHVTLKIEMKKMNKLQNISEELQRNISLQLMSNMNISNKIRNLSTTLQTIATKLCRE LYSKEQEHKCKPCPRRWIWHKDSCYFLSDDVQTWQESKMACAAQNASLLKINNKNALEFI KSQSRSYDYWLGLSPEEDSTRGMRVDNIINSSAWVIRNAPDLNNMYCGYINRLYVQYYHC TYKKRMICEKMANPVQLGSTYFREA(SEQ ID NO: 26)(Uniprot No. Q5QGZ9) Non-limiting examples of CLL-1 gRNAs may be found in PCT Publication Nos. WO 2021/041971 WO2020/047164, WO2023/015182, WO2024/015925, WO2023/196816, WO2023/043858, and WO2022/047168, each of which are incorporated by reference herein in their entirety. In some embodiments, a targeting domain which is complementary to a target nucleic acid in a cell surface protein is complementary to a target nucleic acid encoding human CD123 or a portion thereof, e.g., having a sequence at least 70%, 75%, 80%, 85%, 90%, 95%, 96%, 97%, 98%, or 99% identical thereto to the CD123 amino acid sequence set forth below: MVLLWLTLLLIALPCLLQTKEDPNPPITNLRMKAKAQQLTWDLNRNVTDIECVKDADYSM PAVNNSYCQFGAISLCEVTNYTVRVANPPFSTWILFPENSGKPWAGAENLTCWIHDVDFL SCSWAVGPGAPADVQYDLYLNVANRRQQYECLHYKTDAQGTRIGCRFDDISRLSSGSQSS HILVRGRSAAFGIPCTDKFVVFSQIEILTPPNMTAKCNKTHSFMHWKMRSHFNRKFRYEL QIQKRMQPVITEQVRDRTSFQLLNPGTYTVQIRARERVYEFLSAWSTPQRFECDQEEGAN TRAWRTSLLIALGTLLALVCVFVICRRYLVMQRLFPRIPHMKDPIGDSFQNDKLVVWEAG KAGLEECLVTEVQVVQKT (SEQ ID NO: 27)(Uniport No. P26951) Non-limiting examples of CD123 gRNAs may be found in PCT Publication Nos. WO 2021/041977, WO2023/015182, WO2024015925, WO2023196816, WO2023043858, and WO2022/047165, each of which are incorporated by reference herein in their entirety. In some embodiments, a targeting domain which is complementary to a target nucleic acid in a cell surface protein is complementary to a target nucleic acid encoding human CD38 or a portion thereof, e.g., having a sequence at least 70%, 75%, 80%, 85%, 90%, 95%, 96%, 97%, 98%, or 99% identical thereto to the CD38 amino acid sequence set forth below: MANCEFSPVSGDKPCCRLSRRAQLCLGVSILVLILVVVLAVVVPRWRQQWSGPGTTKRFP ETVLARCVKYTEIHPEMRHVDCQSVWDAFKGAFISKHPCNITEEDYQPLMKLGTQTVPCN KILLWSRIKDLAHQFTQVQRDMFTLEDTLLGYLADDLTWCGEFNTSKINYQSCPDWRKDC SNNPVSVFWKTVSRRFAEAACDVVHVMLNGSRSKIFDKNSTFGSVEVHNLQPEKVQTLEA WVIHGGREDSRDLCQDPTIKELESIISKRNIQFSCKNIYRPDKFLQCVKNPEDSSCTSEI (SEQ ID NO: 28) (Uniprot No. P28907) Non-limiting examples of CD38 gRNAs may be found in PCT Publication Nos. WO 2022/056489, WO2023/015182, and WO2023/196816, which are incorporated by reference herein in their entirety. In some embodiments, a targeting domain which is complementary to a target nucleic acid in a cell surface protein is complementary to a target nucleic acid encoding human EMR2 or a portion thereof, e.g., having a sequence at least 70%, 75%, 80%, 85%, 90%, 95%, 96%, 97%, 98%, or 99% identical thereto to the EMR2 amino acid sequence set forth below: MGGRVFLVFLAFCVWLTLPGAETQDSRGCARWCPQDSSCVNATACRCNPGFSSFSEIITT PMETCDDINECATLSKVSCGKFSDCWNTEGSYDCVCSPGYEPVSGAKTFKNESENTCQDV DECQQNPRLCKSYGTCVNTLGSYTCQCLPGFKLKPEDPKLCTDVNECTSGQNPCHSSTHC LNNVGSYQCRCRPGWQPIPGSPNGPNNTVCEDVDECSSGQHQCDSSTVCFNTVGSYSCRC RPGWKPRHGIPNNQKDTVCEDMTFSTWTPPPGVHSQTLSRFFDKVQDLGRDYKPGLANNT IQSILQALDELLEAPGDLETLPRLQQHCVASHLLDGLEDVLRGLSKNLSNGLLNFSYPAG TELSLEVQKQVDRSVTLRQNQAVMQLDWNQAQKSGDPGPSVVGLVSIPGMGKLLAEAPLV LEPEKQMLLHETHQGLLQDGSPILLSDVISAFLSNNDTQNLSSPVTFTFSHRSVIPRQKV LCVFWEHGQNGCGHWATTGCSTIGTRDTSTICRCTHLSSFAVLMAHYDVQEEDPVLTVIT YMGLSVSLLCLLLAALTFLLCKAIQNTSTSLHLQLSLCLFLAHLLFLVAIDQTGHKVLCS IIAGTLHYLYLATLTWMLLEALYLFLTARNLTVVNYSSINRFMKKLMFPVGYGVPAVTVA ISAASRPHLYGTPSRCWLQPEKGFIWGFLGPVCAIFSVNLVLFLVTLWILKNRLSSLNSE VSTLRNTRMLAFKATAQLFILGCTWCLGILQVGPAARVMAYLFTIINSLQGVFIFLVYCL LSQQVREQYGKWSKGIRKLKTESEMHTLSSSAKADTSKPSTVN (SEQ ID NO: 29)(Uniprot No. Q9UHX3) Non-limiting examples of EMR2 gRNAs may be found in PCT Publication Nos. WO 2023/086422, WO2024/015925, WO2023/196816, and WO2023/043858, which are incorporated by reference herein in their entirety. In some embodiments, a targeting domain which is complementary to a target nucleic acid in a cell surface protein is complementary to a target nucleic acid encoding human CD117 or a portion thereof, e.g., having a sequence at least 70%, 75%, 80%, 85%, 90%, 95%, 96%, 97%, 98%, or 99% identical thereto to the CD117 amino acid sequence set forth below: MRGARGAWDFLCVLLLLLRVQTGSSQPSVSPGEPSPPSIHPGKSDLIVRVGDEIRLLCTD PGFVKWTFEILDETNENKQNEWITEKAEATNTGKYTCTNKHGLSNSIYVFVRDPAKLFLV DRSLYGKEDNDTLVRCPLTDPEVTNYSLKGCQGKPLPKDLRFIPDPKAGIMIKSVKRAYH RLCLHCSVDQEGKSVLSEKFILKVRPAFKAVPVVSVSKASYLLREGEEFTVTCTIKDVSS SVYSTWKRENSQTKLQEKYNSWHHGDFNYERQATLTISSARVNDSGVFMCYANNTFGSAN VTTTLEVVDKGFINIFPMINTTVFVNDGENVDLIVEYEAFPKPEHQQWIYMNRTFTDKWE DYPKSENESNIRYVSELHLTRLKGTEGGTYTFLVSNSDVNAAIAFNVYVNTKPEILTYDR LVNGMLQCVAAGFPEPTIDWYFCPGTEQRCSASVLPVDVQTLNSSGPPFGKLVVQSSIDS SAFKHNGTVECKAYNDVGKTSAYFNFAFKGNNKEQIHPHTLFTPLLIGFVIVAGMMCIIV MILTYKYLQKPMYEVQWKVVEEINGNNYVYIDPTQLPYDHKWEFPRNRLSFGKTLGAGAF GKVVEATAYGLIKSDAAMTVAVKMLKPSAHLTEREALMSELKVLSYLGNHMNIVNLLGAC TIGGPTLVITEYCCYGDLLNFLRRKRDSFICSKQEDHAEAALYKNLLHSKESSCSDSTNE YMDMKPGVSYVVPTKADKRRSVRIGSYIERDVTPAIMEDDELALDLEDLLSFSYQVAKGM AFLASKNCIHRDLAARNILLTHGRITKICDFGLARDIKNDSNYVVKGNARLPVKWMAPES IFNCVYTFESDVWSYGIFLWELFSLGSSPYPGMPVDSKFYKMIKEGFRMLSPEHAPAEMY DIMKTCWDADPLKRPTFKQIVQLIEKQISESTNHIYSNLANCSPNRQKPVVDHSVRINSV GSTASSSQPLLVHDDV (SEQ ID NO: 30) (Uniprot No. P10721) In some embodiments, a targeting domain which is complementary to a target nucleic acid in a cell surface protein is complementary to a target nucleic acid encoding human IL1RAP or a portion thereof, e.g., having a sequence at least 70%, 75%, 80%, 85%, 90%, 95%, 96%, 97%, 98%, or 99% identical thereto to the IL1RAP amino acid sequence set forth below: MTLLWCVVSLYFYGILQSDASERCDDWGLDTMRQIQVFEDEPARIKCPLFEHFLKFNYSTAHSAGLTLIWYWTRQ DRDLEEPINFRLPENRISKEKDVLWFRPTLLNDTGNYTCMLRNTTYCSKVAFPLEVVQKDSCFNSPMKLPVHKLYIEYGIQRITCPNVDGYFPSSVKPTITWYMGCYKIQNFNNVIPEGMNLSFLIALISNNGNYTCVVTYPENGRTFHL TRTLTVKVVGSPKNAVPPVIHSPNDHVVYEKEPGEELLIPCTVYFSFLMDSRNEVWWTIDGKKPDDITIDVTINE SISHSRTEDETRTQILSIKKVTSEDLKRSYVCHARSAKGEVAKAAKVKQKVPAPRYTVELACGFGATVLLVVILI VVYHVYWLEMVLFYRAHFGTDETILDGKEYDIYVSYARNAEEEEFVLLTLRGVLENEFGYKLCIFDRDSLPGGIV TDETLSFIQKSRRLLVVLSPNYVLQGTQALLELKAGLENMASRGNINVILVQYKAVKETKVKELKRAKTVLTVIK WKGEKSKYPQGRFWKQLQVAMPVKKSPRRSSSDEQGLSYSSLKNV (SEQ ID NO: 8)(UniPort ID: Q9NPH3-1) MTLLWCVVSLYFYGILQSDASERCDDWGLDTMRQIQVFEDEPARIKCPLFEHFLKFNYSTAHSAGLTLIWYWTRQ DRDLEEPINFRLPENRISKEKDVLWFRPTLLNDTGNYTCMLRNTTYCSKVAFPLEVVQKDSCFNSPMKLPVHKLY IEYGIQRITCPNVDGYFPSSVKPTITWYMGCYKIQNFNNVIPEGMNLSFLIALISNNGNYTCVVTYPENGRTFHL TRTLTVKVVGSPKNAVPPVIHSPNDHVVYEKEPGEELLIPCTVYFSFLMDSRNEVWWTIDGKKPDDITIDVTINE SISHSRTEDETRTQILSIKKVTSEDLKRSYVCHARSAKGEVAKAAKVKQKGNRCGQ (SEQ ID NO: 9) (UniPort ID: Q9NPH3-2) MTLLWCVVSLYFYGILQSDASERCDDWGLDTMRQIQVFEDEPARIKCPLFEHFLKFNYSTAHSAGLTLIWYWTRQ DRDLEEPINFRLPENRISKEKDVLWFRPTLLNDTGNYTCMLRNTTYCSKVAFPLEVVQKDSCFNSPMKLPVHKLY IEYGIQRITCPNVDGYFPSSVKPTITWYMGCYKIQNFNNVIPEGMNLSFLIALISNNGNYTCVVTYPENGRTFHL TRTLTVKVVGSPKNAVPPVIHSPNDHVVYEKEPGEELLIPCTVYFSFLMDSRNEVWWTIDGKKPDDITIDVTINE SASSKIHSGTGLWFWSHSPASGDSHCCLPCLLARDGPILPGSFWNR(SEQ ID NO: 10) ((UniPort ID: Q9NPH3-3) Additional non-limiting examples of gRNAs which, in some embodiments, may be useful for editing genes encoding cell surface proteins are found in PCT Publication Nos. WO2022/061115, WO2022/067240, WO2022/072643, WO2022/094245, WO2022/147347, WO2018/160768, WO2019/046285, WO2020/047164, WO2020/150478, WO2021/041971, WO2021/041977, WO2022/056489, WO2023/015182, WO2024/015925, WO2023/196816, WO2023/086422, WO2023/043858, and WO2022/217086, which are incorporated by reference herein in their entirety. For example, in some embodiments, a genetically engineered hematopoietic cell comprises genetic engineering in a first gene encoding a first cell surface protein as described herein and genetic engineering in a second gene encoding a second cell surface protein as described herein, wherein the first gene and the second gene each comprise a mutation (e.g., a substitution, a deletion, or an insertion at one or more nucleotide positions) which is located in a sequence (e.g., an exon and/or an intron) that hybridizes with a gRNA, or hybridizes with a portion of a gRNA (e.g., a sequence of at least 10 nucleotides in the gRNA), described in any of the foregoing references (e.g., see: disclosures related to CD47 editing in WO2023/196816; disclosures related to CD7 editing in WO2022/061115 and WO2023/015182; disclosures related to CD34 editing in WO2022/147347; disclosures related to CD33 editing in WO 2020/237217, WO2018/160768, WO2020/047164, WO2019/046285, WO2020/150478, WO2023/015182, WO2024/015925, WO2023/196816, WO2023/043858, and WO2022/217086; disclosures related to CD123 editing in WO2020/047164, WO2021/041977, WO2023/015182, WO2024/015925, WO2023/196816, WO2023/043858, and WO2022/047165; disclosures related to CLL-1 editing in WO2020/047164, WO2021/041971, WO2023/015182, WO2024/015925, WO2023/196816, WO2023/043858, and WO2022/047168; disclosures related to CD38 editing in WO2022/056489, WO2023/196816, WO2023/015182; disclosures related to CD30 editing in WO2023/015182; and disclosures related to EMR2 editing in WO2024/015925, WO2023/086422, WO2023/043858, and WO2023/196816). Homology-Directed Repair (HDR) Using Template Polynucleotides In some embodiments, the present disclosure provides genetically engineered cells and cell populations, and methods of producing genetically engineered cells and cell populations using HDR-mediated gene editing, e.g., CRISPR/Cas-based HDR-mediated gene editing. Without being bound by any particular theory, HDR is a process wherein damage to DNA (e.g., a break in the DNA) is repaired using a donor sequence with flanking sequences comprising homology to the site of DNA damage. In some embodiments, a CRISPR/Cas system is used to introduce a break in the DNA (e.g., a double-stranded break (DSB)). Without wishing to be bound by theory, by providing a donor sequence (e.g., via a template polynucleotide) in the presence of a DSB, it is thought that HDR can be promoted (e.g., relative to other DNA repair pathways, e.g., NHEJ). HDR can result in substitution or insertion mutations that replace endogenous or naturally occurring sequences with those of the donor sequence. For example, methods described herein can be used to introduce a mutation into a target DNA, e.g., to correct a disease-associated genetic mutation. As a further example, methods described herein can be used to introduce a mutation, e.g., to insert a nucleotide sequence (e.g., a nucleotide sequence correcting a mutation in a genomic sequence). HDR may be induced by a DNA damage event that is capable of being mutagenic if left unrepaired or unprocessed, e.g., a double-stranded break. In some embodiments, the DNA damage event is induced by a CRISPR/Cas system, e.g., comprising a Cas nuclease, e.g., Cas9. Examples of DNA damage capable of producing a mutation include, but are not limited to, DNA alkylation, base deamination, base depurination, incidence of abasic sites, single-stranded breaks, and double-stranded breaks. Once DNA is damaged, the damage is repaired in multiple steps wherein cellular nucleases degrade nucleotide sequences at and proximal to the sites of the damage on one strand of the DNA. As used in this context, sequence “proximal” to the sites of damage is defined as a sequence that is found anywhere 10, 20, 30, 40, 50, 60, 70, 80, 90, 100, 150, 200, 250, 300, 350, 400, 450, or 500 nucleotides in the 5’ or 3’ direction of site of damage. Processing by nucleases, in turn, generates single- stranded overhangs comprised of a stretch of nucleotides that are not participating in base pairing interactions with nucleotides on the cognate strand to which the strand bearing the overhang is hybridized. Strand invasion follows, wherein the overhangs transiently base pair with a donor sequence that is located in close physical proximity to the damaged DNA molecule. In this way, template polynucleotide homology to a target site provided by the flanking sequences directs template polynucleotide participation in HDR. Strand invasion is followed by cellular polymerase-dependent recombination wherein the donor sequence serves as the template to direct the repair of the damaged DNA. Recombination between the donor sequence and the damaged DNA can incorporate the sequence of the donor sequence into the damaged DNA molecule. Following recombination, the repair is completed by a cellular ligase enzyme. In some embodiments, the donor sequence is provided by, for example, a template polynucleotide. When the donor sequence differs at one or more positions relative to a target DNA, integration of the donor sequence by HDR results in a mutation. In some embodiments, the target DNA comprises a mutation relative to a reference sequence (e.g., a wild-type sequence, or a sequence dominant in a population of subjects, or a sequence not characteristic of, or causally associated with, a disease or disorder); such a mutation may be referred to herein as a prior mutation (as distinguished from a mutation introduced by a method described herein). In some embodiments, the prior mutation is characteristic of, or causally associated with, a disease, e.g., a mutation that is known to cause a genetic disease, or is a mutation that is known to convey increased risk of a genetic disease. In some embodiments, a method described herein alters a genomic sequence comprising a mutation characteristic for and/or causally associated with a disease or disorder, changing the genomic sequence to a sequence that is not characteristic for and/or causally associated with that disease or disorder. In some embodiments, such alteration comprises correcting a prior mutation. In some embodiments, such alteration comprises introducing a silent mutation, a restriction site, or a tag sequence. In some embodiments, a donor sequence differs from a sequence in the target DNA in one or more nucleotides, and integration of the donor sequence into the target DNA produces a genetic modification in the target DNA. In some embodiments, the donor sequence differs from a target DNA in a manner that integration of the donor sequence corrects a prior mutation in the target DNA, e.g., integration of the donor sequence into a target DNA comprising a prior mutation results in a modification of the mutation within the target DNA, e.g., in a modification of the target DNA sequence to the wild-type sequence, to the dominant sequence in a population of subjects, or, where the prior mutation is characteristic of, or causally associated with, a disease or disorder, in a modification to a sequence that is not characteristic of, or causally associated with a disease or disorder. In some embodiments, the prior mutation is characteristic for, or causally associated with, a disease or disorder. In some embodiments, the prior mutation is not characteristic for, or not causally associated with, a disease or disorder. In some such embodiments, a template polynucleotide comprising the donor sequence is referred to as a template for HDR of the mutation. In some embodiments, the donor sequence comprises a gene or a portion thereof, e.g., a gene, or portion thereof, that (e.g., prior to any genetic modification described herein) is mutated or non-functional in the target DNA or the genome of a cell. In some such embodiments, a template polynucleotide comprising the donor sequence is referred to as a template for HDR insertion of a gene, or portion thereof, in the target DNA. In some embodiments, a template polynucleotide is single-stranded, e.g., a single- strand donor oligonucleotide (ssODN). In some embodiments, a template polynucleotide is double-stranded, e.g., a plasmid or a double-stranded donor oligonucleotide (dsODN). In some embodiments, a template polynucleotide is a minicircle plasmid. In some embodiments, a template polynucleotide is a nanoplasmid. Those of ordinary skill in the art will recognize that a minicircle plasmid is a plasmid that contains double stranded donor template without conventional plasmid backbones. Minicircle plasmids may be processed via a single DSB leading to linearization, whereas larger plasmids might require two DSBs which flank the template polynucleotide to excise the donor. Those of ordinary skill in the art will also recognize that a nanoplasmid comprises a circular DNA molecule of 500 base pairs or less and can be generated using services provided by Aldevron®. As such, minicircle plasmids and nanoplasmids are, in some embodiments, cut in a host cell prior to HDR via, for example, an exogenous nuclease (e.g., Cas9) targeting gRNA cut sites engineered into the plasmid sequence. However, in some embodiments, minicircle plasmids and nanoplasmids comprising a template polynucleotide need not be cut by an exogenous prior to HDR. As used herein, a template polynucleotide refers to a nucleic acid that is a template for HDR, e.g., HDR of a mutation in the target DNA. In some embodiments, a template polynucleotide is approximately 10, 20, 30, 40, 50, 60, 70, 80, 90, 100, 110, 120, 130, 140, 150, 160, 170, 180, 190, or 200 nucleotides long +/- 1, 2, 3, 4, 5, 6, 7, 8, 9, or 10 nucleotides long. In some embodiments, a template polynucleotide is approximately 300, 400, 500, 600, 700, 800, 900, 1000, 1100, 1200, 1300, 1400, 1500, 1600, 1700, 1800, 1900, 2000, 2100, 2200, 2300, 2400, 2500, 2600, 2700, 2800, 2900, 3000, 3100, 3200, 3300, 3400, or 3500 nucleotides +/- 10, 25, 50, or 75 nucleotides long. In some embodiments, the donor sequence comprises a modification as compared to the target DNA, for example, a mutation, e.g., an insertion, deletion, or substitution as compared to the target DNA nucleotide sequence. In some embodiments, the donor sequence comprises a substitution of a single nucleotide as compared to the target DNA. Such donor sequences are useful, for example, to effect genetic modifications that correct a single nucleotide mutation in a target DNA sequence that is characteristic for, or causally associated with, a disease or disorder. In some embodiments, the donor sequence comprises a substitution of two or more nucleotides as compared to the target DNA. Such donor sequences are useful, for example, to effect genetic modifications that correct more complex mutations, e.g., affecting two or more nucleotides, in a target DNA sequence that are characteristic for, or causally associated with, a disease or disorder. In some embodiments, the donor sequence comprises one or more insertions (e.g., of one or more nucleotides) as compared to the target DNA. Such donor sequences are useful, for example, to effect genetic modifications that create insertion mutations in a target DNA sequence. In some embodiments, the donor sequence comprises one or more deletions (e.g., of one or more nucleotides) as compared to the target DNA. Such donor sequences are useful, for example, to effect genetic modifications that create deletion mutations in a target DNA sequence. In some embodiments, the donor sequence comprises two or more substitutions as compared to the target DNA, wherein, if integrated into the target DNA, at least one such substitution results in the correction of a mutation that is characteristic of, or causally associated with, a disease or mutation, and wherein at least one such substitution results in a silent mutation in the target DNA, e.g., a substitution of a wobble base within an amino acid-encoding codon of a target DNA. Such donor sequences are useful, for example, to effect genetic modifications that correct disease-associated mutations in a target DNA sequence, while at the same time creating a sequence tag, e.g., a non-naturally occurring sequence or a sequence that did was not previously present in the target DNA, which is useful for identification and/or tracking of the modified cells. In some embodiments, the donor sequence comprises a restriction site or a unique sequence tag, for example, a unique primer binding site. In some embodiments, the sequence comprising the restriction site or a unique sequence tag is an insertion relative to the target DNA, e.g., the target DNA does not comprise a restriction site or a unique sequence tag where the donor sequence comprises one. In some embodiments, the sequence comprising the restriction site or a unique sequence tag is not an insertion relative to the target DNA. For example, in some embodiments, the sequence comprising the restriction site or a unique sequence tag comprises a mutation (e.g., a substitution) as compared to the target DNA that, upon integration into the target DNA, produces a restriction site or a unique sequence tag. In some embodiments, the sequence comprising the restriction site or a unique sequence tag does not alter an amino acid sequence encoded by the target DNA. A restriction site or a unique sequence tag thus introduced may be used, e.g., to confirm the success of integration of the donor sequence (e.g., in an experiment where the modified target DNA is cleaved and fragments or sequences thereof are analyzed). In some embodiments, the restriction endonuclease site comprises a Pvu1 site, e.g., 5’-CGATCG-3’. In some embodiments, the target DNA comprises a prior mutation and the donor sequence differs from the target DNA in a manner such that integration of the donor sequence corrects the prior mutation and produces one or more additional mutations (e.g., a second, third, fourth, or fifth mutation relative to the correction of the prior mutation (the first mutation)). In some embodiments, the one or more additional mutations comprise one or more silent mutations that do not alter the amino acid encoded by the nucleic acid sequence of the target DNA. In some embodiments, the one or more silent mutations are contiguous (i.e., directly adjacent) to the prior mutation or the codon containing the prior mutation. In some embodiments, silent mutations are used, e.g., as identifiers (e.g., “tags” or “bar codes”) of a given correction of a prior mutation or to facilitate confirmation of integration of the donor sequence (e.g., in an experiment where the modified target DNA sequences are analyzed). In some embodiments, methods and compositions provided by the present disclosure are applied to a target DNA, e.g., in order to modify the target DNA. For example, in some embodiments, the target DNA comprises a nucleotide sequence that is characteristic for, or causally associated with, a disease or disorder. Where such a target DNA sequence is different from the wild-type DNA sequence, or from a dominant DNA sequence at this locus within a population of subjects not affected by the disease or disorder, the divergence between the target DNA and the wild-type or dominant DNA sequence is also sometimes referred to herein as a prior mutation. In some embodiments, a template polynucleotide comprises a first flanking sequence and a second flanking sequence, also referred to herein as a first homology sequence and a second homology sequence. In some embodiments, the first flanking sequence and second flanking sequence direct the binding of the template polynucleotide to a target DNA sequence in the cell. In some embodiments, a first flanking sequence is at least 50, at least 60, at least 70, at least 80, at least 90, at least 100, at least 110, at least 120, at least 130, at least 140, at least 150, at least 160, at least 170, at least 180, at least 190, at least 200, at least 210, at least 220, at least 230, at least 240, or at least 250 nucleotides long (and optionally no more than 1000, no more than 750, no more than 500, no more than 400, no more than 300, or no more than 250 nucleotides long). In some embodiments, a first flanking sequence comprises 25- 300, 50-300, 75-300, 100-300, 125-300, 150-300, 175-300, 200-300, 225-300, 250-300, 275- 300, 100-400, 125-400, 150-400, 175-400, 200-400, 225-400, 250-400, 275-400, 300-400, 325, 400, 350-400, 375-400, 200-500, 225-500, 250-500, 275-500, 300-500, 325-500, 350- 500, 375-500, 400-500, 425-500, 450-500, or 475-500 nucleotides in length. In some embodiments, a first flanking sequence comprises 500-2000, 600-2000, 700-2000, 800-2000, 900-2000, 1000-2000, 1100-2000, 1200-2000, 1300-2000, 1400-2000, 1500-2000, 1600- 2000, 1700-2000, 1800-2000, or 1900-2000 nucleotides in length. In some embodiments, the first flanking sequence has at least 50%, at least 60%, at least 70%, at least at least 80%, at least 90%, at least 91%, at least 92%, at least 93%, at least 94%, at least 95%, at least 96%, at least 97%, at least 98%, at least 99%, or at least 100% identity to a sequence upstream of a DSB in the target DNA (e.g., upstream of a site where a DSB is produced by a CRISPR/Cas system described herein), or a sequence complementary thereto. In some embodiments, the first flanking sequence has 100% identity to a sequence upstream of a DSB in the target DNA (e.g., upstream of a site where a DSB is produced by a CRISPR/Cas system described herein), or a sequence complementary thereto. As used in this context, sequence “upstream” and “downstream” refer to a region within 10, within 20, within 30, within 40, within 50, within 60, within 70, within 80, within 90, or within 100 nucleotides of a feature in the DNA (e.g., a DSB), with each term referring to a different direction from the target site, and, in the case where the target DNA is a gene or portion thereof upstream is toward the transcription start site for the gene and downstream is away from the transcription start site for the gene. In some embodiments, the first flanking sequence is a 5’ homology arm of a template polynucleotide and is 5’ of a donor sequence, e.g., in an ssODN, dsODN, minicircle plasmid, or nanoplasmid. In some embodiments, a second flanking sequence is at least 50, at least 60, at least 70, at least 80, at least 90, at least 100, at least 110, at least 120, at least 130, at least 140, at least 150, at least 160, at least 170, at least 180, at least 190, at least 200, at least 210, at least 220, at least 230, at least 240, or at least 250 nucleotides long (and optionally no more than 1000, no more than 750, no more than 500, no more than 400, no more than 300, or no more than 250 nucleotides long). In some embodiments, a second flanking sequence comprises 25-300, 50-300, 75-300, 100-300, 125-300, 150-300, 175-300, 200-300, 225-300, 250-300, 275-300, 100-400, 125-400, 150-400, 175-400, 200-400, 225-400, 250-400, 275- 400, 300-400, 325, 400, 350-400, 375-400, 200-500, 225-500, 250-500, 275-500, 300-500, 325-500, 350-500, 375-500, 400-500, 425-500, 450-500, or 475-500 nucleotides in length. In some embodiments, a second flanking sequence comprises 500-2000, 600-2000, 700-2000, 800-2000, 900-2000, 1000-2000, 1100-2000, 1200-2000, 1300-2000, 1400-2000, 1500-2000, 1600-2000, 1700-2000, 1800-2000, or 1900-2000 nucleotides in length. In some embodiments, the second flanking sequence has at least 50%, at least 60%, at least 70%, at least 80%, at least 90%, at least 91%, at least 92%, at least 93%, at least 94%, at least 95%, at least 96%, at least 97%, at least 98%, at least 99%, or at least 100% identity to a sequence downstream of a target site (e.g., downstream of a DSB produced by a CRISPR/Cas system in the target site), or a sequence complementary thereto. In some embodiments, the second flanking sequence has 100% identity to a sequence downstream of a DSB in the target DNA (e.g., downstream of a site where a DSB is produced by a CRISPR/Cas system described herein), or a sequence complementary thereto. In some embodiments, the second flanking sequence is a 3’ homology arm of a template polynucleotide and is 3’ of a donor sequence, e.g., in an ssODN, dsODN, minicircle plasmid, or nanoplasmid. In some embodiments, the first flanking sequence and the second flanking sequence have identity or complementarity to different sequences within or proximal to the target DNA. For example, in some embodiments the first flanking sequence has identity or complementarity to a first target sequence within or proximal to a target DNA and the second flanking sequence has identity or complementarity to a second target sequence within or proximal to the target DNA. In some embodiments, the first target sequence and second target sequence are no more than 5, no more than 10, no more than 20, no more than 30, no more than 40, no more than 50, no more than 100, no more than 150, no more than 200, no more than 250, no more than 300, no more than 500, or no more than 1000 bases apart in the nucleic acid molecule comprising the target DNA. In some embodiments, the first flanking sequence has 100% identity to a sequence upstream of a DSB in the target DNA, or a sequence complementary thereto, and the second flanking sequence has 100% identity to a sequence downstream of a DSB in the target DNA, or a sequence complementary thereto. In some embodiments, a flanking sequence comprises 500-2000, 600-2000, 700-2000, 800-2000, 900-2000, 1000-2000, 1100-2000, 1200-2000, 1300-2000, 1400-2000, 1500-2000, 1600-2000, 1700-2000, 1800-2000, or 1900-2000 consecutive nucleotides that are 100% identical to a target sequence within a target DNA. In some embodiments, a second flanking sequence comprises 500-2000, 600-2000, 700-2000, 800-2000, 900-2000, 1000-2000, 1100- 2000, 1200-2000, 1300-2000, 1400-2000, 1500-2000, 1600-2000, 1700-2000, 1800-2000, or 1900-2000 nucleotides in length. In some embodiments, a flanking sequence (e.g., a 3’ homology arm or 5’ homology arm) comprises 25-300, 50-300, 75-300, 100-300, 125-300, 150-300, 175-300, 200-300, 225-300, 250-300, 275-300, 100-400, 125-400, 150-400, 175- 400, 200-400, 225-400, 250-400, 275-400, 300-400, 325, 400, 350-400, 375-400, 200-500, 225-500, 250-500, 275-500, 300-500, 325-500, 350-500, 375-500, 400-500, 425-500, 450- 500, or 475-500 consecutive nucleotides that are 100% identical to a target sequence within a target DNA. In some embodiments, a flanking sequence (e.g., a 3’ homology arm or 5’ homology arm) comprises 2-100, 10-100, 20-100, 30-100, 40-100, 50-100, 60-100, 70-100, 80-100, 90-100, 2-150, 2-200, 2-250, 10-150, 10-200, 10-250, 50-150, 50-200, 50-250, 100- 150, 100-200, 100-250, 150-200, 150-200, or 200-250 consecutive nucleotides that are 100% identical to a target sequence within a target DNA. In some embodiments, a flanking sequence (e.g., a 3’ homology arm or 5’ homology arm) comprises at least 2, at least 10, at least 20, at least 30, at least 40, at least 50, at least 60, at least 70, at least 80, at least 90, or at least 100 consecutive nucleotides that are 100% identical to a target sequence within a target DNA (and optionally no more than 200, no more than 180, no more than 160, no more than 140, no more than 120, or no more than 100 consecutive nucleotides that are 100% identical to a target sequence within a target DNA). In some embodiments, a flanking sequence (e.g., a 3’ homology arm or a 5’ homology arm) comprises a nucleotide sequence that is 100% identical to a PAM sequence in the target DNA. In some embodiments, the nucleotide sequence identical to the PAM sequence is 2-3, 2-4, 2-5, 2-6, 3-4, 3-5, 3-6, 4-5, 4-6, or 5-6 nucleotides in length (e.g., 2, 3, 4, 5, or 6 nucleotides in length). In some embodiments, a template polynucleotide comprises a donor sequence. In some embodiments, the donor sequence is integrated into a target DNA at the site of a DSB. In some embodiments, the donor sequence is homologous to the target DNA or a portion thereof, e.g., the sequence of the target DNA surrounding or adjacent to the DSB. In some embodiments, the donor sequence is contiguous with the first and second flanking sequences in a template polynucleotide. For example, in some embodiments a target DNA comprises a gene or a portion thereof, and the donor sequence is homologous to the target DNA or a portion thereof (e.g., in proximity to a DSB or a site targeted for a DSB by a CRISPR/Cas system as described herein). In some embodiments, the first and second flanking sequences guide binding of the template polynucleotide to a target DNA, facilitating interaction of the donor sequence with its homologous sequence in the target DNA and/or with cellular DNA repair (e.g., HDR) pathway components. In some embodiments, the donor sequence differs from a homologous sequence of the target DNA at 1, 2, 3, 4, 5, 6, 7, 8, 9, or 10 bases (e.g., 1- 10, 1-9, 1-8, 1-7, 1-6, 1-5, 1-4, 1-3, 1-2, 2-10, 2-9, 2-8, 2-7, 2-6, 2-5, 2-4, 2-3, 3-10, 3-9, 3-8, 3-7, 3-6, 3-5, 3-4, 4-10, 4-9, 4-8, 4-7, 4-6, 4-5, 5-10, 5-9, 5-8, 5-7, 5-6, 6-10, 6-9, 6-8, 6-7, 7- 10, 7-9, 7-8, 8-10, 8-9, or 9-10 bases), or at a number of positions corresponding to up to 1, 5, 10, 15, or 20% of the length of the donor sequence. In some embodiments, a donor sequence is 1-100, 1-80, 1-60, 1-40, 1-20, 1-15, 1-10, 1-9, 1-8, 1-7, 1-6, 1-5, 1-4, 1-3, 1-2, 5-100, 5-80, 5-60, 5-40, 5-20, 5-15, 5-10, 5-9, 5-8, 5-7, 5-6, 10-100, 10-80, 10-60, 10-40, 10-20, 10-15, 20-100, 20-80, 20-60, 20-40, 60-100, or 60-80 nucleotides in length. In some embodiments, a donor sequence is no more than 100, no more than 90, no more than 80, no more than 70, no more than 60, no more than 50, no more than 45, no more than 40, no more than 35, no more than 30, no more than 25, no more than 20, no more than 15, no more than 14, no more than 13, no more than 12, no more than 11, no more than 10, no more than 9, no more than 8, no more than 7, no more than 6, no more than 5, no more than 4, no more than 3, no more than 2, or no more than 1 bases long. In some embodiments, a donor sequence is 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49, 50, 55, 60, 65, 70, 75, 80, 85, 90, 95, or 100 bases long. In some embodiments, a donor sequence differs from a homologous sequence of the target DNA at a position or positions corresponding to a prior mutation in the target DNA (e.g., characteristic of, or causally associated with, a disease or disorder, or risk of developing a disease or disorder), e.g., a prior point mutation. In some embodiments, the donor sequence comprises sequence corresponding to the wild-type, functional, and/or naturally-occurring sequence at a position or positions corresponding to a prior mutation in the target DNA. In some embodiments, the donor sequence comprises an artificial or heterologous sequence. In some embodiments, a donor sequence is 200-2000, 200-1900, 200-1800, 200-1700, 200-1600, 200-1500, 200-1400, 200-1300, 200-1200, 200- 1100, 100-1000, 100-900, 100-800, 100-700, 100-600, 100-500, 100-400, 100-300, or 100- 200 nucleotides in length. In some embodiments, a donor sequence is no more than 2000, no more than 1900, no more than 1800, no more than 1700, no more than 1600, no more than 1500, no more than 1400, no more than 1300, no more than 1200, no more than 1100, no more than 1000, no more than 900, no more than 800, no more than 700, no more than 600, no more than 500, no more than 400, no more than 300, or no more than 200 nucleotides in length. A schematic of an exemplary template polynucleotide, such as an ssODN, a double- stranded dsODN, minicircle plasmid, or nanoplasmid is provided below: [5’-homology arm] – [donor sequence] – [3’ homology arm] Each homology arm (e.g., a flanking sequence described herein) has homology to a sequence in the target DNA proximal to the sequence homologous to the donor sequence. In some embodiments, a homology arm comprises a sequence homologous to a PAM sequence in the target DNA. In some embodiments, a CRISPR/Cas system for use in a method of the disclosure comprises a Cas nuclease that recognizes a PAM sequence in the target DNA and cuts the target DNA at a position near to the PAM sequence (e.g., 5’ or 3’ of the PAM sequence). Accordingly, in some embodiments a PAM homologous sequence is present in a 3’ homology arm or a 5’ homology arm of a template polynucleotide. In some embodiments, the PAM homologous sequence is positioned such that HDR of a DSB produced by a Cas nuclease promotes integration of a donor sequence. In some embodiments, the DSB is positioned in a target DNA sequence homologous to the donor sequence. A schematic of an exemplary 3’ homology arm (e.g., where a CRISPR/Cas system (e.g., comprising Cas9) cuts a target DNA 5’ of a PAM sequence) is provided below: [N]x – [PAM] – [N]y For example, an exemplary Cas nuclease, Cas9, cuts a target DNA 3-4 nucleotides 5’ of a PAM sequence. In some embodiments, x is 3-4, and y is the number of nucleotides in the remaining length of the homology arm (e.g., wherein the length of the homology arm is described herein). For example, for x = 3, and a homology arm length of 100 nucleotides, y would be 100 minus 3 and minus the length of the PAM homologous sequence (e.g., where the PAM sequence is 3 nucleotides long, y would be 94 (100-3-3). In some embodiments, x is 2 and the homology arm is 50-60 nucleotides long. In some embodiments, x is 2 and the homology arm is 60-70 nucleotides long. In some embodiments, x is 2 and the homology arm is 70-80 nucleotides long. In some embodiments, x is 2 and the homology arm is 80-90 nucleotides long. In some embodiments, x is 2 and the homology arm is 90-100 nucleotides long. In some embodiments, x is 2 and the homology arm is 100-110 nucleotides long. In some embodiments, x is 2 and the homology arm is 110-120 nucleotides long. In some embodiments, x is 2 and the homology arm is 120-130 nucleotides long. In some embodiments, x is 2 and the homology arm is 130-140 nucleotides long. In some embodiments, x is 2 and the homology arm is 140-150 nucleotides long. In some embodiments, x is 2 and the homology arm is 150-160 nucleotides long. In some embodiments, x is 2 and the homology arm is 160-170 nucleotides long. In some embodiments, x is 2 and the homology arm is 170-180 nucleotides long. In some embodiments, x is 2 and the homology arm is 180-190 nucleotides long. In some embodiments, x is 2 and the homology arm is 190-200 nucleotides long. In some embodiments, x is 2 and the homology arm is 210-220 nucleotides long. In some embodiments, x is 2 and the homology arm is 220-230 nucleotides long. In some embodiments, x is 2 and the homology arm is 230-240 nucleotides long. In some embodiments, x is 2 and the homology arm is 240-250 nucleotides long. In some embodiments, x is 3 and the homology arm is 50-60 nucleotides long. In some embodiments, x is 3 and the homology arm is 60-70 nucleotides long. In some embodiments, x is 3 and the homology arm is 70-80 nucleotides long. In some embodiments, x is 3 and the homology arm is 80-90 nucleotides long. In some embodiments, x is 3 and the homology arm is 90-100 nucleotides long. In some embodiments, x is 3 and the homology arm is 100-110 nucleotides long. In some embodiments, x is 3 and the homology arm is 110-120 nucleotides long. In some embodiments, x is 3 and the homology arm is 120-130 nucleotides long. In some embodiments, x is 3 and the homology arm is 130-140 nucleotides long. In some embodiments, x is 3 and the homology arm is 140-150 nucleotides long. In some embodiments, x is 3 and the homology arm is 150-160 nucleotides long. In some embodiments, x is 3 and the homology arm is 160-170 nucleotides long. In some embodiments, x is 3 and the homology arm is 170-180 nucleotides long. In some embodiments, x is 3 and the homology arm is 180-190 nucleotides long. In some embodiments, x is 3 and the homology arm is 190-200 nucleotides long. In some embodiments, x is 3 and the homology arm is 210-220 nucleotides long. In some embodiments, x is 3 and the homology arm is 220-230 nucleotides long. In some embodiments, x is 3 and the homology arm is 230-240 nucleotides long. In some embodiments, x is 3 and the homology arm is 240-250 nucleotides long. In some embodiments, x is 4 and the homology arm is 50-60 nucleotides long. In some embodiments, x is 4 and the homology arm is 60-70 nucleotides long. In some embodiments, x is 4 and the homology arm is 70-80 nucleotides long. In some embodiments, x is 4 and the homology arm is 80-90 nucleotides long. In some embodiments, x is 4 and the homology arm is 90-100 nucleotides long. In some embodiments, x is 4 and the homology arm is 100-110 nucleotides long. In some embodiments, x is 4 and the homology arm is 110-120 nucleotides long. In some embodiments, x is 4 and the homology arm is 120-130 nucleotides long. In some embodiments, x is 4 and the homology arm is 130-140 nucleotides long. In some embodiments, x is 4 and the homology arm is 140-150 nucleotides long. In some embodiments, x is 4 and the homology arm is 150-160 nucleotides long. In some embodiments, x is 4 and the homology arm is 160-170 nucleotides long. In some embodiments, x is 4 and the homology arm is 170-180 nucleotides long. In some embodiments, x is 4 and the homology arm is 180-190 nucleotides long. In some embodiments, x is 4 and the homology arm is 190-200 nucleotides long. In some embodiments, x is 4 and the homology arm is 210-220 nucleotides long. In some embodiments, x is 4 and the homology arm is 220-230 nucleotides long. In some embodiments, x is 4 and the homology arm is 230-240 nucleotides long. In some embodiments, x is 4 and the homology arm is 240-250 nucleotides long. A schematic of an exemplary 5’ homology arm (e.g., where a CRISPR/Cas system (e.g., comprising Cas12a) cuts a target DNA 3’ of a PAM sequence) is provided below: [N]a – [PAM] – [N]b As a further example, another exemplary Cas nuclease, Cas12a, cuts a target DNA 18-19 nucleotides 3’ of a PAM sequence. In some embodiments, b is 18-19, and a is the number of nucleotides in the remaining length of the homology arm (e.g., wherein the length of the homology arm is described herein). For example, for b = 18, and a homology arm length of 100 nucleotides, a would be 100 minus 18 and minus the length of the PAM homologous sequence (e.g., where the PAM sequence is 3 nucleotides long, a would be 79 (100-18-3). In some embodiments, b is 17 and the homology arm is 50-60 nucleotides long. In some embodiments, b is 17 and the homology arm is 60-70 nucleotides long. In some embodiments, b is 17 and the homology arm is 70-80 nucleotides long. In some embodiments, b is 17 and the homology arm is 80-90 nucleotides long. In some embodiments, b is 17 and the homology arm is 90-100 nucleotides long. In some embodiments, b is 17 and the homology arm is 100-110 nucleotides long. In some embodiments, b is 17 and the homology arm is 110-120 nucleotides long. In some embodiments, b is 17 and the homology arm is 120-130 nucleotides long. In some embodiments, b is 17 and the homology arm is 130-140 nucleotides long. In some embodiments, b is 17 and the homology arm is 140-150 nucleotides long. In some embodiments, b is 17 and the homology arm is 150-160 nucleotides long. In some embodiments, b is 17 and the homology arm is 160-170 nucleotides long. In some embodiments, b is 17 and the homology arm is 170-180 nucleotides long. In some embodiments, b is 17 and the homology arm is 180-190 nucleotides long. In some embodiments, b is 17 and the homology arm is 190-200 nucleotides long. In some embodiments, b is 17 and the homology arm is 210-220 nucleotides long. In some embodiments, b is 17 and the homology arm is 220-230 nucleotides long. In some embodiments, b is 17 and the homology arm is 230-240 nucleotides long. In some embodiments, b is 17 and the homology arm is 240-250 nucleotides long. In some embodiments, b is 18 and the homology arm is 50-60 nucleotides long. In some embodiments, b is 18 and the homology arm is 60-70 nucleotides long. In some embodiments, b is 18 and the homology arm is 70-80 nucleotides long. In some embodiments, b is 18 and the homology arm is 80-90 nucleotides long. In some embodiments, b is 18 and the homology arm is 90-100 nucleotides long. In some embodiments, b is 18 and the homology arm is 100-110 nucleotides long. In some embodiments, b is 18 and the homology arm is 110-120 nucleotides long. In some embodiments, b is 18 and the homology arm is 120-130 nucleotides long. In some embodiments, b is 18 and the homology arm is 130-140 nucleotides long. In some embodiments, b is 18 and the homology arm is 140-150 nucleotides long. In some embodiments, x is 3 and the homology arm is 150-160 nucleotides long. In some embodiments, b is 18 and the homology arm is 160-170 nucleotides long. In some embodiments, b is 18 and the homology arm is 170-180 nucleotides long. In some embodiments, b is 18 and the homology arm is 180-190 nucleotides long. In some embodiments, b is 18 and the homology arm is 190-200 nucleotides long. In some embodiments, b is 18 and the homology arm is 210-220 nucleotides long. In some embodiments, b is 18 and the homology arm is 220-230 nucleotides long. In some embodiments, b is 18 and the homology arm is 230-240 nucleotides long. In some embodiments, b is 18 and the homology arm is 240-250 nucleotides long. In some embodiments, b is 19 and the homology arm is 50-60 nucleotides long. In some embodiments, b is 19 and the homology arm is 60-70 nucleotides long. In some embodiments, b is 19 and the homology arm is 70-80 nucleotides long. In some embodiments, b is 19 and the homology arm is 80-90 nucleotides long. In some embodiments, b is 19 and the homology arm is 90-100 nucleotides long. In some embodiments, b is 19 and the homology arm is 100-110 nucleotides long. In some embodiments, b is 19 and the homology arm is 110-120 nucleotides long. In some embodiments, b is 19 and the homology arm is 120-130 nucleotides long. In some embodiments, b is 19 and the homology arm is 130-140 nucleotides long. In some embodiments, b is 19 and the homology arm is 140-150 nucleotides long. In some embodiments, b is 19 and the homology arm is 150-160 nucleotides long. In some embodiments, b is 19 and the homology arm is 160-170 nucleotides long. In some embodiments, b is 19 and the homology arm is 170-180 nucleotides long. In some embodiments, b is 19 and the homology arm is 180-190 nucleotides long. In some embodiments, b is 19 and the homology arm is 190-200 nucleotides long. In some embodiments, b is 19 and the homology arm is 210-220 nucleotides long. In some embodiments, b is 19 and the homology arm is 220-230 nucleotides long. In some embodiments, b is 19 and the homology arm is 230-240 nucleotides long. In some embodiments, b is 19 and the homology arm is 240-250 nucleotides long. In some embodiments, a genetically engineered hematopoietic cell comprises a genetic modification corresponding to integration of a donor sequence (e.g., from a template polynucleotide described herein) into a target DNA in the hematopoietic cell. In some embodiments, the genetic modification corresponds to a position or positions where the donor sequence differs from the sequence of the target DNA. In some embodiments, integration of the donor sequence results in modification at 1, 2, 3, 4, 5, 6, 7, 8, 9, or 10 bases (e.g., 1-10, 1- 9, 1-8, 1-7, 1-6, 1-5, 1-4, 1-3, 1-2, 2-10, 2-9, 2-8, 2-7, 2-6, 2-5, 2-4, 2-3, 3-10, 3-9, 3-8, 3-7, 3-6, 3-5, 3-4, 4-10, 4-9, 4-8, 4-7, 4-6, 4-5, 5-10, 5-9, 5-8, 5-7, 5-6, 6-10, 6-9, 6-8, 6-7, 7-10, 7-9, 7-8, 8-10, 8-9, or 9-10 bases) in the target DNA. In some embodiments, integration of the donor sequence results in an insertion of 1, 2, 3, 4, 5, 6, 7, 8, 9, or 10 bases (e.g., 1-10, 1- 9, 1-8, 1-7, 1-6, 1-5, 1-4, 1-3, 1-2, 2-10, 2-9, 2-8, 2-7, 2-6, 2-5, 2-4, 2-3, 3-10, 3-9, 3-8, 3-7, 3-6, 3-5, 3-4, 4-10, 4-9, 4-8, 4-7, 4-6, 4-5, 5-10, 5-9, 5-8, 5-7, 5-6, 6-10, 6-9, 6-8, 6-7, 7-10, 7-9, 7-8, 8-10, 8-9, or 9-10 bases) in the target DNA. In some embodiments, integration of the donor sequence results in substitution of 1, 2, 3, 4, 5, 6, 7, 8, 9, or 10 bases (e.g., 1-10, 1- 9, 1-8, 1-7, 1-6, 1-5, 1-4, 1-3, 1-2, 2-10, 2-9, 2-8, 2-7, 2-6, 2-5, 2-4, 2-3, 3-10, 3-9, 3-8, 3-7, 3-6, 3-5, 3-4, 4-10, 4-9, 4-8, 4-7, 4-6, 4-5, 5-10, 5-9, 5-8, 5-7, 5-6, 6-10, 6-9, 6-8, 6-7, 7-10, 7-9, 7-8, 8-10, 8-9, or 9-10 bases) in the target DNA. In some embodiments, integration of the donor sequence results in modification at a number of positions in the target DNA corresponding to up to 1, 5, 10, 15, 20, 25, 30, 35, 40, 45, 50, 55, 60, 65, 70, 75, 80, 85, 90, 95, or 100% of the length of the donor sequence. In some embodiments, integration of the donor sequence results in insertion of a number of bases in the target DNA corresponding to up to 1, 5, 10, 15, 20, 25, 30, 35, 40, 45, 50, 55, 60, 65, 70, 75, 80, 85, 90, 95, or 100% of the length of the donor sequence. In some embodiments, a donor sequence is 200-2000, 200- 1900, 200-1800, 200-1700, 200-1600, 200-1500, 200-1400, 200-1300, 200-1200, 200-1100, 100-1000, 100-900, 100-800, 100-700, 100-600, 100-500, 100-400, 100-300, or 100-200 nucleotides in length. In some embodiments, a donor sequence is no more than 2000, no more than 1900, no more than 1800, no more than 1700, no more than 1600, no more than 1500, no more than 1400, no more than 1300, no more than 1200, no more than 1100, no more than 1000, no more than 900, no more than 800, no more than 700, no more than 600, no more than 500, no more than 400, no more than 300, or no more than 200 nucleotides in length. In some embodiments, the donor sequence is 1-100, 1-80, 1-60, 1-40, 1-20, 1-15, 1-10, 1-9, 1-8, 1-7, 1-6, 1-5, 1-4, 1-3, 1-2, 5-100, 5-80, 5-60, 5-40, 5-20, 5-15, 5-10, 5-9, 5-8, 5-7, 5-6, 10- 100, 10-80, 10-60, 10-40, 10-20, 10-15, 20-100, 20-80, 20-60, 20-40, 60-100, or 60-80 nucleotides in length. In some embodiments, a donor sequence is no more than 100, no more than 90, no more than 80, no more than 70, no more than 60, no more than 50, no more than 45, no more than 40, no more than 35, no more than 30, no more than 25, no more than 20, no more than 15, no more than 14, no more than 13, no more than 12, no more than 11, no more than 10, no more than 9, no more than 8, no more than 7, no more than 6, no more than 5, no more than 4, no more than 3, no more than 2, or no more than 1 bases long. In some embodiments, a donor sequence is 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49, 50, 55, 60, 65, 70, 75, 80, 85, 90, 95, or 100 bases long. In some embodiments, integration of the donor sequence into the genetically engineered hematopoietic cell corrected a prior mutation in the target DNA (e.g., a disease-associated mutation (a mutation characteristic of, or causally associated with, a disease or disorder, or risk of developing a disease or disorder) or a non-disease-associated mutation), e.g., as described herein. In some embodiments, the donor sequence integrated comprises a sequence corresponding to the wild-type, functional, and/or naturally-occurring sequence at a position or positions corresponding to a prior mutation in the target DNA. In some embodiments, the donor sequence comprises an artificial or heterologous sequence. In some embodiments, integration of the donor sequence produces a restriction nuclease site or a unique sequence tag in the target DNA of the genetically engineered hematopoietic cell. In some embodiments, integration of the donor sequence into the target DNA of the genetically engineered hematopoietic cell produces one or more silent mutations along with a non-silent mutation (e.g., correction of a prior mutation, e.g., in a coding sequence). In some embodiments, the one or more silent mutations are contiguous with another mutation described herein (e.g., contiguous with correction of a prior mutation). For example, in some embodiments a genetically engineered hematopoietic cell comprises a genetic modification corresponding to correction of a prior mutation which substitutes a single prior mutation, e.g., a single nucleotide point mutation, for the corresponding base(s) present in a wild-type cell, and one or more silent mutations contiguous with the prior mutation. Accordingly, the disclosure provides a genetically engineered hematopoietic cell comprising a genetic modification corresponding to integration of a donor sequence as described herein, e.g., a donor sequence described herein. In some embodiments, integration of the donor sequence into a genetically engineered T cell results in the expression of a chimeric antigen receptor (CAR). In some embodiments, the CAR binds to at least one antigen present on a cell surface protein. In some embodiments, the cell surface protein is CD33. It will be understood that, upon engrafting donor cells into a recipient host organism, the relative levels of the engrafted donor cells (and descendants thereof) and the host cells, e.g., in a given niche (e.g., bone marrow), are important for physiological and/or therapeutic outcomes for the host organism. The level of engrafted donor cells, or descendants thereof, relative to host cells in a given tissue or niche is referred to herein as chimerism. In some embodiments, a cell described herein (e.g., an HSC or HPC) is capable of engrafting in a human subject and does not exhibit any difference in chimerism as compared to a hematopoietic cell of the same cell type, but not comprising a genomic modification that results in expression of a variant form (e.g., a wild-type form or having wild-type functionality) of a gene or a loss of expression of a gene. In some embodiments, a cell described herein (e.g., an HSC or HPC) is capable of engrafting in a human subject exhibits no more than a 1, no more than a 2, no more than a 5, no more than a 10, no more than a 15, no more than a 20, no more than a 25, no more than a 30, no more than a 35, no more than a 40, no more than a 45, or no more than a 50% difference in chimerism as compared to a hematopoietic cell of the same cell type, but not comprising a genomic modification that results in expression of a variant form (e.g., a wild-type form or having wild-type functionality) of a gene or a loss of expression of a gene. In some embodiments, a genetic modification is produced using HDR. In some embodiments, producing a genetic modification using HDR comprises contacting cells with a template polynucleotide, a CRISPR/Cas system, and one or more other agents (e.g., one or more HDR-promoting agents or expansion agents), e.g., contacting cells with a genetic modification mixture described herein. The disclosure provides, in part, methods and compositions that achieve unexpectedly high editing efficiencies utilizing HDR. In some embodiments, efficiency of HDR-mediated editing and/or efficiency of total/overall editing (HDR- and non-HDR-mediated) is determined by a method described herein. In some embodiments, the efficiency of HDR is at least 20, 30, 40, 50, 60, 70, 80, 90, 95, or 99% (e.g., 50%, 60%, 70%, 80%, 90% or higher). In some embodiments, contacting cells to produce a genetic modification using HDR comprises contacting cells with one or more HDR-promoting agents as described herein. Without wishing to be bound by theory, the disclosure is directed, in part, to the discovery that the presence of one or more HDR- promoting agents may result in unexpectedly and advantageously high efficiency of HDR. Accordingly, methods describing contacting a cell herein also contemplate contacting a population of cells to produce a population of genetically modified cells, e.g., an editing efficiency, percent viability, and/or HDR efficiency described herein. In some embodiments, producing a genetic modification using HDR comprises contacting a cell with a genetic modification mixture. As used herein, a genetic modification mixture refers to a mixture comprising a plurality of components that may be used to genetically modify a target DNA, e.g., in a cell. In some embodiments, a genetic modification mixture comprises one, two, three, or all of a CRISPR/Cas system, a template polynucleotide, one or more HDR-promoting agents, and one or more expansion agents. In some embodiments, a genetic modification mixture promotes HDR and HDR-mediated genetic modification (e.g., relative to another DNA repair pathway or genetic modifications utilizing another DNA repair pathway). In some embodiments, contacting a cell with the genetic modification mixture comprises adding the genetic modification mixture directly to media comprising the cell. In some embodiments, contacting a cell with the genetic modification mixture comprises adding media comprising the genetic modification mixture to the cell or adding the cell to media comprising the genetic modification mixture. In some embodiments, the media is a growth media, e.g., a growth media suited to hematopoietic cells (e.g., hematopoietic stem cells (HSCs)). Examples of growth media include, but are not limited to, a Stromal cell Growth Media (SCGMTM, e.g., as available from Lonza Bioscience) or serum- and feeder-free media (SFFM). In some embodiments, contacting a cell with the genetic modification mixture comprises electroporating the genetic modification mixture or one or more components of the mixture into the cell. In some embodiments, contacting a cell with the genetic modification mixture comprises solvating the mixture in a lipid-permeable buffer, e.g., to serve as a carrier for movement of mixture components across the cell membrane. Examples of lipid- permeable buffers include, but are not limited to, DMSO and lipofectamine. In some embodiments, the genetic modification mixture comprises a template polynucleotide, e.g., a single-strand donor oligonucleotide (ssODN), a double-stranded donor oligonucleotide (dsODN), a minicircle plasmid, or a nanoplasmid, comprising a donor sequence, a first flanking sequence and a second flanking sequence. In some embodiments, the genetic modification mixture comprises a dsODN comprising “capped” or “closed-ends” in order to minimize the chances that the NHEJ pathway is used in the genetic modification process. In some embodiments, the dsODN comprising “capped” or “closed-ends” is a GenWand® double-stranded DNA. In some embodiments, the genetic modification mixture comprises a CRISPR/Cas system capable of producing a break, e.g., a double-stranded break, at a target site in the genome of the cell. In some embodiments, the genetic modification mixture comprises one or more other agents (e.g., an expansion agent and/or HDR-promoting agent) that promote genetic modification. In some embodiments, the template polynucleotide, e.g., ssODN, dsODN, minicircle plasmid, or nanoplasmid, and the CRISPR/Cas system of the genetic modification mixture is mixed with the one or more other agents that promote genetic modification. Base Editing In some embodiments, generating a genetically engineered hematopoietic cell comprises contacting a hematopoietic cell with a CRISPR/Cas system comprising a Cas nuclease, wherein the Cas nuclease comprises a base editor. In some embodiments, a base editor is used to a create a genomic modification resulting in an artificial PAM. In some embodiments, a base editor is used to a create a genomic modification resulting in a loss of expression of a protein antigen of interest (e.g., a cell surface protein antigen ), or in expression of a protein antigen variant not targeted by an immunotherapy. A base editor nuclease generally comprises a catalytically inactive Cas9 nuclease fused to a functional domain, e.g., a deaminase domain. See, e.g., Eid et al. Biochem. J. (2018) 475(11): 1955-1964; Rees et al. Nature Reviews Genetics (2018) 19:770-788. In some embodiments, the catalytically inactive Cas9 nuclease is referred to as “dead Cas” or “dCas9.” In some embodiments, the catalytically inactive Cas nuclease has reduced nuclease activity and is, e.g., a nickase (referred to as “nCas”). In some embodiments, the nuclease comprises a dCas9 fused to one or more uracil glycosylase inhibitor (UGI) domains. In some embodiments, the nuclease comprises a dCas9 fused to an adenine base editor (ABE), for example an ABE evolved from the RNA adenine deaminase TadA. In some embodiments, the nuclease comprises a dCas9 fused to cytidine deaminase enzyme (e.g., APOBEC deaminase, pmCDA1, activation-induced cytidine deaminase (AID)). In some embodiments, the catalytically inactive Cas9 nuclease has reduced activity and is nCas9. In some embodiments, the catalytically inactive Cas9 nuclease (dCas9) is fused to one or more uracil glycosylase inhibitor (UGI) domains. In some embodiments, the Cas9 nuclease comprises an inactive Cas9 nuclease (dCas9) fused to an adenine base editor (ABE), for example an ABE evolved from the RNA adenine deaminase TadA. In some embodiments, the Cas9 nuclease comprises a nCas9 fused to an adenine base editor (ABE), for example an ABE evolved from the RNA adenine deaminase TadA. In some embodiments, the Cas9 nuclease comprises a dCas9 fused to cytidine deaminase enzyme (e.g., APOBEC deaminase, pmCDA1, activation- induced cytidine deaminase (AID)). In some embodiments, the Cas9 nuclease comprises a nCas9 fused to cytidine deaminase enzyme (e.g., APOBEC deaminase, pmCDA1, activation- induced cytidine deaminase (AID)). Examples of base editors include, without limitation, BE1, BE2, BE3, HF-BE3, BE4, BE4max, BE4-Gam, YE1-BE3, EE-BE3, YE2-BE3, YEE-CE3, VQR-BE3, VRER-BE3, SaBE3, SaBE4, SaBE4-Gam, Sa(KKH)-BE3, Target-AID, Target-AID-NG, xBE3, eA3A- BE3, BE-PLUS, TAM, CRISPR-X, ABE7.9, ABE7.10, ABE7.10*, ABE8, ABE8e, ABE8.20-m, xABE, ABESa, VQR-ABE, VRER-ABE, Sa(KKH)-ABE, and CRISPR-SKIP. Additional examples of base editors can be found, for example, in US Publication No. 2018/0312825A1, US Publication No.2018/0312828A1, International Publication No. WO 2018/165629A1, International Publication No. WO 2021/030666A1, International Publication No. WO 2022/261509A1, International Publication No. WO 2021/158921A1, and Gaudelli et al., Nat Biotechnol.2020;38(7):892-900, which are incorporated by reference herein in their entireties. In some embodiments, the base editor is a cytosine base editor (CBE). In some embodiments, the CBE is CBE1, CBE2, CBE3, or CBE4. In some embodiments, the CBE is nCas9-2xUGI; BE4-rAPOBEC1; BE4-rAPOBEC1 K34A H122A; BE4-PpAPOBEC1; BE4- PpAPOBEC1 R33A; BE4-PpAPOBEC1 H122A; BE4-RrA3F; BE4-AmAPOBEC1; or BE4- SsAPOBEC3B. In some embodiments, the base editor is an adenine base editor (ABE). In some embodiments, the ABE is ABE1, ABE2, ABE3, ABE4, ABE5, ABE6, ABE7, or ABE8. In some embodiments, the ABE is ABE7.10-m; ABE7.10-d; ABE8e; ABE8.8-m; ABE8.8-d; ABE8.13-m; ABE8.13-d; ABE8.17-m; ABE8.17-d; ABE8.20-m; or ABE8.20-d. In some embodiments, the base editors includes, without limitation, BE1, BE2, BE3, HF-BE3, BE4, BE4max, BE4-Gam, YE1-BE3, EE-BE3, YE2-BE3, YEE-CE3, VQR-BE3, VRER-BE3, SaBE3, SaBE4, SaBE4-Gam, Sa(KKH)-BE3, Target-AID, Target-AID-NG, xBE3, eA3A-BE3, BE-PLUS, TAM, CRISPR-X, ABE7.9, ABE7.10, ABE7.10*, xABE, ABESa, VQR-ABE, VRER-ABE, Sa(KKH)-ABE, and CRISPR-SKIP. In preferred embodiments, the base editor is ABE8e or ABE8.20-m. Additional examples of base editors can be found, for example, in US Publication No. 2018/0312825A1, US Publication No.2018/0312828A1, International Publication No. WO 2018/165629A1, Yu et al. Nat Commun. (2020) 11(1):2052, and Gaudelli et al. Nat Biotechnol. (2020) 38(7):892-900. which are incorporated by reference herein in their entireties. An exemplary abe8_20m sequence is provided below: atgtccgaagtcgagttttcccatgagtactggatgagacacgcattgactctcgcaaagagggctcgagatgaa cgcgaggtgcccgtgggggcagtactcgtgctcaacaatcgcgtaatcggcgaaggttggaatagggcaatcgga ctccacgaccccactgcacatgcggaaatcatggcccttcgacagggagggcttgtgatgcagaattatcgactt tatgatgcgacgctgtactcgacgtttgaaccttgcgtaatgtgcgcgggagctatgattcactcccgcattgga cgagttgtattcggtgttcgcaacgccaagacgggtgccgcaggttcactgatggacgtgctgcatcatccaggc atgaaccaccgggtagaaatcacagaaggcatattggcggacgaatgtgcggcgctgttgtgtcgtttttttcgc atgcccaggcgggtctttaacgcccagaaaaaagcacaatcctctactgactctggtggttcttctggtggttct agcggcagcgagactcccgggacctcagagtccgccacacccgaaagttctggtggttcttctggtggttctgac aagaagtacagcatcggcctggccatcggcaccaactctgtgggctgggccgtgatcaccgacgagtacaaggtg cccagcaagaaattcaaggtgctgggcaacaccgaccggcacagcatcaagaagaacctgatcggagccctgctg ttcgacagcggcgaaacagccgaggccacccggctgaagagaaccgccagaagaagatacaccagacggaagaac cggatctgctatctgcaagagatcttcagcaacgagatggccaaggtggacgacagcttcttccacagactggaa gagtccttcctggtggaagaggataagaagcacgagcggcaccccatcttcggcaacatcgtggacgaggtggcc taccacgagaagtaccccaccatctaccacctgagaaagaaactggtggacagcaccgacaaggccgacctgcgg ctgatctatctggccctggcccacatgatcaagttccggggccacttcctgatcgagggcgacctgaaccccgac aacagcgacgtggacaagctgttcatccagctggtgcagacctacaaccagctgttcgaggaaaaccccatcaac gccagcggcgtggacgccaaggccatcctgtctgccagactgagcaagagcagacggctggaaaatctgatcgcc cagctgcccggcgagaagaagaatggcctgttcggaaacctgattgccctgagcctgggcctgacccccaacttc aagagcaacttcgacctggccgaggatgccaaactgcagctgagcaaggacacctacgacgacgacctggacaac ctgctggcccagatcggcgaccagtacgccgacctgtttctggccgccaagaacctgtccgacgccatcctgctg agcgacatcctgagagtgaacaccgagatcaccaaggcccccctgagcgcctctatgatcaagagatacgacgag caccaccaggacctgaccctgctgaaagctctcgtgcggcagcagctgcctgagaagtacaaagagattttcttc gaccagagcaagaacggctacgccggctacattgacggcggagccagccaggaagagttctacaagttcatcaag cccatcctggaaaagatggacggcaccgaggaactgctcgtgaagctgaacagagaggacctgctgcggaagcag cggaccttcgacaacggcagcatcccccaccagatccacctgggagagctgcacgccattctgcggcggcaggaa gatttttacccattcctgaaggacaaccgggaaaagatcgagaagatcctgaccttccgcatcccctactacgtg ggccctctggccaggggaaacagcagattcgcctggatgaccagaaagagcgaggaaaccatcaccccctggaac ttcgaggaagtggtggacaagggcgcttccgcccagagcttcatcgagcggatgaccaacttcgataagaacctg cccaacgagaaggtgctgcccaagcacagcctgctgtacgagtacttcaccgtgtataacgagctgaccaaagtg aaatacgtgaccgagggaatgagaaagcccgccttcctgagcggcgagcagaaaaaggccatcgtggacctgctg ttcaagaccaaccggaaagtgaccgtgaagcagctgaaagaggactacttcaagaaaatcgagtgcttcgactcc gtggaaatctccggcgtggaagatcggttcaacgcctccctgggcacataccacgatctgctgaaaattatcaag gacaaggacttcctggacaatgaggaaaacgaggacattctggaagatatcgtgctgaccctgacactgtttgag gacagagagatgatcgaggaacggctgaaaacctatgcccacctgttcgacgacaaagtgatgaagcagctgaag cggcggagatacaccggctggggcaggctgagccggaagctgatcaacggcatccgggacaagcagtccggcaag acaatcctggatttcctgaagtccgacggcttcgccaacagaaacttcatgcagctgatccacgacgacagcctg acctttaaagaggacatccagaaagcccaggtgtccggccagggcgatagcctgcacgagcacattgccaatctg gccggcagccccgccattaagaagggcatcctgcagacagtgaaggtggtggacgagctcgtgaaagtgatgggc cggcacaagcccgagaacatcgtgatcgaaatggccagagagaaccagaccacccagaagggacagaagaacagc cgcgagagaatgaagcggatcgaagagggcatcaaagagctgggcagccagatcctgaaagaacaccccgtggaa aacacccagctgcagaacgagaagctgtacctgtactacctgcagaatgggcgggatatgtacgtggaccaggaa ctggacatcaaccggctgtccgactacgatgtggaccatatcgtgcctcagagctttctgaaggacgactccatcgacaacaaggtgctgaccagaagcgacaagaaccggggcaagagcgacaacgtgccctccgaagaggtcgtgaag aagatgaagaactactggcggcagctgctgaacgccaagctgattacccagagaaagttcgacaatctgaccaag gccgagagaggcggcctgagcgaactggataaggccggcttcatcaagagacagctggtggaaacccggcagatc acaaagcacgtggcacagatcctggactcccggatgaacactaagtacgacgagaatgacaagctgatccgggaa gtgaaagtgatcaccctgaagtccaagctggtgtccgatttccggaaggatttccagttttacaaagtgcgcgag atcaacaactaccaccacgcccacgacgcctacctgaacgccgtcgtgggaaccgccctgatcaaaaagtaccct aagctggaaagcgagttcgtgtacggcgactacaaggtgtacgacgtgcggaagatgatcgccaagagcgagcag gaaatcggcaaggctaccgccaagtacttcttctacagcaacatcatgaactttttcaagaccgagattaccctg gccaacggcgagatccggaagcggcctctgatcgagacaaacggcgaaaccggggagatcgtgtgggataagggc cgggattttgccaccgtgcggaaagtgctgagcatgccccaagtgaatatcgtgaaaaagaccgaggtgcagaca ggcggcttcagcaaagagtctatcctgcccaagaggaacagcgataagctgatcgccagaaagaaggactgggac cctaagaagtacggcggcttcgacagccccaccgtggcctattctgtgctggtggtggccaaagtggaaaagggc aagtccaagaaactgaagagtgtgaaagagctgctggggatcaccatcatggaaagaagcagcttcgagaagaat cccatcgactttctggaagccaagggctacaaagaagtgaaaaaggacctgatcatcaagctgcctaagtactcc ctgttcgagctggaaaacggccggaagagaatgctggcctctgccggcgaactgcagaagggaaacgaactggccctgccctccaaatatgtgaacttcctgtacctggccagccactatgagaagctgaagggctcccccgaggataat gagcagaaacagctgtttgtggaacagcacaagcactacctggacgagatcatcgagcagatcagcgagttctcc aagagagtgatcctggccgacgctaatctggacaaagtgctgtccgcctacaacaagcaccgggataagcccatc agagagcaggccgagaatatcatccacctgtttaccctgaccaatctgggagcccctgccgccttcaagtacttt gacaccaccatcgaccggaagaggtacaccagcaccaaagaggtgctggacgccaccctgatccaccagagcatc accggcctgtacgagacacggatcgacctgtctcagctgggaggtgacgagggagctgataagcgcaccgccgatggttccgagttcgaaagccccaagaagaagaggaaagtc (SEQ ID NO: 31) An exemplary ppabobec1-orf-r33a sequence is provided below: atgacctctgagaagggccctagcacaggcgaccccaccctgcggcggagaatcgagagctgggagttcgacgtgttctacgaccctagagaactggccaaggaaacctgcctgctgtacgagatcaagtggggcatgagcagaaagatc tggcggagctctggcaagaacaccaccaaccacgtggaagtgaatttcatcaagaagttcaccagcgagagaagg ttccacagcagcatcagctgcagcatcacctggttcctgagctggtccccttgctgggaatgcagccaggccatc agagagttcctgagccaacaccccggagtgacactggtgatctacgtggccagactgttctggcacatggaccag agaaacagacagggcctgagagatctggtcaacagcggcgtgactatccagatcatgcgggccagcgagtactac cactgttggcggaacttcgtgaactacccccccggcgatgaggcccactggcctcagtaccctcctctgtggatg atgctgtacgccctggaactgcactgcatcatcctgtctctgcctccatgtctgaagatctctagaagatggcag aaccacctggccttcttcagactgcacctgcagaattgccactaccagaccatccccccccacatcctgctggct acaggcctgatccacccttctgtgacctggagacttaagagcggaggatctagcggcggctctagcggatctgag acacctggcacaagcgagtctgccacacctgagagtagcggcggatcttctggtggctctgacaagaagtacagc atcggcctggccatcggcaccaactctgtgggctgggccgtgatcaccgacgagtacaaggtgcccagcaagaaa ttcaaggtgctgggcaacaccgaccggcacagcatcaagaagaacctgatcggagccctgctgttcgacagcggc gaaacagccgaggccacccggctgaagagaaccgccagaagaagatacaccagacggaagaaccggatctgctat ctgcaagagatcttcagcaacgagatggccaaggtggacgacagcttcttccacagactggaagagtccttcctg gtggaagaggataagaagcacgagcggcaccccatcttcggcaacatcgtggacgaggtggcctaccacgagaag taccccaccatctaccacctgagaaagaaactggtggacagcaccgacaaggccgacctgcggctgatctatctg gccctggcccacatgatcaagttccggggccacttcctgatcgagggcgacctgaaccccgacaacagcgacgtg gacaagctgttcatccagctggtgcagacctacaaccagctgttcgaggaaaaccccatcaacgccagcggcgtg gacgccaaggccatcctgtctgccagactgagcaagagcagacggctggaaaatctgatcgcccagctgcccggc gagaagaagaatggcctgttcggaaacctgattgccctgagcctgggcctgacccccaacttcaagagcaacttc gacctggccgaggatgccaaactgcagctgagcaaggacacctacgacgacgacctggacaacctgctggcccag atcggcgaccagtacgccgacctgtttctggccgccaagaacctgtccgacgccatcctgctgagcgacatcctg agagtgaacaccgagatcaccaaggcccccctgagcgcctctatgatcaagagatacgacgagcaccaccaggac ctgaccctgctgaaagctctcgtgcggcagcagctgcctgagaagtacaaagagattttcttcgaccagagcaag aacggctacgccggctacattgacggcggagccagccaggaagagttctacaagttcatcaagcccatcctggaa aagatggacggcaccgaggaactgctcgtgaagctgaacagagaggacctgctgcggaagcagcggaccttcgac aacggcagcatcccccaccagatccacctgggagagctgcacgccattctgcggcggcaggaagatttttaccca ttcctgaaggacaaccgggaaaagatcgagaagatcctgaccttccgcatcccctactacgtgggccctctggcc aggggaaacagcagattcgcctggatgaccagaaagagcgaggaaaccatcaccccctggaacttcgaggaagtg gtggacaagggcgcttccgcccagagcttcatcgagcggatgaccaacttcgataagaacctgcccaacgagaag gtgctgcccaagcacagcctgctgtacgagtacttcaccgtgtataacgagctgaccaaagtgaaatacgtgacc gagggaatgagaaagcccgccttcctgagcggcgagcagaaaaaggccatcgtggacctgctgttcaagaccaac cggaaagtgaccgtgaagcagctgaaagaggactacttcaagaaaatcgagtgcttcgactccgtggaaatctcc ggcgtggaagatcggttcaacgcctccctgggcacataccacgatctgctgaaaattatcaaggacaaggacttc ctggacaatgaggaaaacgaggacattctggaagatatcgtgctgaccctgacactgtttgaggacagagagatg atcgaggaacggctgaaaacctatgcccacctgttcgacgacaaagtgatgaagcagctgaagcggcggagatacaccggctggggcaggctgagccggaagctgatcaacggcatccgggacaagcagtccggcaagacaatcctggat ttcctgaagtccgacggcttcgccaacagaaacttcatgcagctgatccacgacgacagcctgacctttaaagag gacatccagaaagcccaggtgtccggccagggcgatagcctgcacgagcacattgccaatctggccggcagcccc gccattaagaagggcatcctgcagacagtgaaggtggtggacgagctcgtgaaagtgatgggccggcacaagccc gagaacatcgtgatcgaaatggccagagagaaccagaccacccagaagggacagaagaacagccgcgagagaatg aagcggatcgaagagggcatcaaagagctgggcagccagatcctgaaagaacaccccgtggaaaacacccagctg cagaacgagaagctgtacctgtactacctgcagaatgggcgggatatgtacgtggaccaggaactggacatcaac cggctgtccgactacgatgtggaccatatcgtgcctcagagctttctgaaggacgactccatcgacaacaaggtg ctgaccagaagcgacaagaaccggggcaagagcgacaacgtgccctccgaagaggtcgtgaagaagatgaagaac tactggcggcagctgctgaacgccaagctgattacccagagaaagttcgacaatctgaccaaggccgagagaggc ggcctgagcgaactggataaggccggcttcatcaagagacagctggtggaaacccggcagatcacaaagcacgtg gcacagatcctggactcccggatgaacactaagtacgacgagaatgacaagctgatccgggaagtgaaagtgatc accctgaagtccaagctggtgtccgatttccggaaggatttccagttttacaaagtgcgcgagatcaacaactac caccacgcccacgacgcctacctgaacgccgtcgtgggaaccgccctgatcaaaaagtaccctaagctggaaagc gagttcgtgtacggcgactacaaggtgtacgacgtgcggaagatgatcgccaagagcgagcaggaaatcggcaaggctaccgccaagtacttcttctacagcaacatcatgaactttttcaagaccgagattaccctggccaacggcgag atccggaagcggcctctgatcgagacaaacggcgaaaccggggagatcgtgtgggataagggccgggattttgcc accgtgcggaaagtgctgagcatgccccaagtgaatatcgtgaaaaagaccgaggtgcagacaggcggcttcagc aaagagtctatcctgcccaagaggaacagcgataagctgatcgccagaaagaaggactgggaccctaagaagtac ggcggcttcgacagccccaccgtggcctattctgtgctggtggtggccaaagtggaaaagggcaagtccaagaaa ctgaagagtgtgaaagagctgctggggatcaccatcatggaaagaagcagcttcgagaagaatcccatcgacttt ctggaagccaagggctacaaagaagtgaaaaaggacctgatcatcaagctgcctaagtactccctgttcgagctg gaaaacggccggaagagaatgctggcctctgccggcgaactgcagaagggaaacgaactggccctgccctccaaa tatgtgaacttcctgtacctggccagccactatgagaagctgaagggctcccccgaggataatgagcagaaacag ctgtttgtggaacagcacaagcactacctggacgagatcatcgagcagatcagcgagttctccaagagagtgatc ctggccgacgctaatctggacaaagtgctgtccgcctacaacaagcaccgggataagcccatcagagagcaggcc gagaatatcatccacctgtttaccctgaccaatctgggagcccctgccgccttcaagtactttgacaccaccatc gaccggaagaggtacaccagcaccaaagaggtgctggacgccaccctgatccaccagagcatcaccggcctgtac gagacacggatcgacctgtctcagctgggaggtgactctggtggaagcggaggatctggcggcagcaccaatctg agcgacatcatcgagaaagagacaggcaagcagctggtcatccaagagtccatcctgatgctgcctgaagaggtg gaagaagtgatcggcaacaagcccgagtccgacatcctggtgcacaccgcctacgatgagagcaccgacgagaac gtgatgctgctgacctctgacgcccctgagtacaagccttgggctctcgtgatccaggacagcaacggcgagaac aagatcaagatgctgagcggcggctctggtggctctggcggatctacaaacctgtccgatattattgagaaagaa accgggaaacagctcgtgattcaagagtctattctcatgctcccggaagaagtcgaggaagtcattggaaacaag cctgagagcgatattctggtccatacagcctacgacgagtctaccgatgagaatgtcatgctcctcaccagcgac gctcccgagtataagccatgggcacttgtcattcaggactccaatggggaaaacaaaatcaaaatgctcccaaag aaaaaacgcaaggtg (SEQ ID NO: 32) An exemplary ppabobec1-orf-wt sequence is provided below: atgacctctgagaagggccctagcacaggcgaccccaccctgcggcggagaatcgagagctgggagttcgacgtg ttctacgaccctagagaactgagaaaggaaacctgcctgctgtacgagatcaagtggggcatgagcagaaagatc tggcggagctctggcaagaacaccaccaaccacgtggaagtgaatttcatcaagaagttcaccagcgagagaagg ttccacagcagcatcagctgcagcatcacctggttcctgagctggtccccttgctgggaatgcagccaggccatc agagagttcctgagccaacaccccggagtgacactggtgatctacgtggccagactgttctggcacatggaccag agaaacagacagggcctgagagatctggtcaacagcggcgtgactatccagatcatgcgggccagcgagtactac cactgttggcggaacttcgtgaactacccccccggcgatgaggcccactggcctcagtaccctcctctgtggatg atgctgtacgccctggaactgcactgcatcatcctgtctctgcctccatgtctgaagatctctagaagatggcag aaccacctggccttcttcagactgcacctgcagaattgccactaccagaccatccccccccacatcctgctggct acaggcctgatccacccttctgtgacctggagacttaagagcggaggatctagcggcggctctagcggatctgag acacctggcacaagcgagtctgccacacctgagagtagcggcggatcttctggtggctctgacaagaagtacagc atcggcctggccatcggcaccaactctgtgggctgggccgtgatcaccgacgagtacaaggtgcccagcaagaaa ttcaaggtgctgggcaacaccgaccggcacagcatcaagaagaacctgatcggagccctgctgttcgacagcggc gaaacagccgaggccacccggctgaagagaaccgccagaagaagatacaccagacggaagaaccggatctgctat ctgcaagagatcttcagcaacgagatggccaaggtggacgacagcttcttccacagactggaagagtccttcctg gtggaagaggataagaagcacgagcggcaccccatcttcggcaacatcgtggacgaggtggcctaccacgagaag taccccaccatctaccacctgagaaagaaactggtggacagcaccgacaaggccgacctgcggctgatctatctg gccctggcccacatgatcaagttccggggccacttcctgatcgagggcgacctgaaccccgacaacagcgacgtg gacaagctgttcatccagctggtgcagacctacaaccagctgttcgaggaaaaccccatcaacgccagcggcgtg gacgccaaggccatcctgtctgccagactgagcaagagcagacggctggaaaatctgatcgcccagctgcccggc gagaagaagaatggcctgttcggaaacctgattgccctgagcctgggcctgacccccaacttcaagagcaacttc gacctggccgaggatgccaaactgcagctgagcaaggacacctacgacgacgacctggacaacctgctggcccagatcggcgaccagtacgccgacctgtttctggccgccaagaacctgtccgacgccatcctgctgagcgacatcctg agagtgaacaccgagatcaccaaggcccccctgagcgcctctatgatcaagagatacgacgagcaccaccaggac ctgaccctgctgaaagctctcgtgcggcagcagctgcctgagaagtacaaagagattttcttcgaccagagcaag aacggctacgccggctacattgacggcggagccagccaggaagagttctacaagttcatcaagcccatcctggaa aagatggacggcaccgaggaactgctcgtgaagctgaacagagaggacctgctgcggaagcagcggaccttcgac aacggcagcatcccccaccagatccacctgggagagctgcacgccattctgcggcggcaggaagatttttaccca ttcctgaaggacaaccgggaaaagatcgagaagatcctgaccttccgcatcccctactacgtgggccctctggcc aggggaaacagcagattcgcctggatgaccagaaagagcgaggaaaccatcaccccctggaacttcgaggaagtg gtggacaagggcgcttccgcccagagcttcatcgagcggatgaccaacttcgataagaacctgcccaacgagaag gtgctgcccaagcacagcctgctgtacgagtacttcaccgtgtataacgagctgaccaaagtgaaatacgtgacc gagggaatgagaaagcccgccttcctgagcggcgagcagaaaaaggccatcgtggacctgctgttcaagaccaac cggaaagtgaccgtgaagcagctgaaagaggactacttcaagaaaatcgagtgcttcgactccgtggaaatctcc ggcgtggaagatcggttcaacgcctccctgggcacataccacgatctgctgaaaattatcaaggacaaggacttc ctggacaatgaggaaaacgaggacattctggaagatatcgtgctgaccctgacactgtttgaggacagagagatg atcgaggaacggctgaaaacctatgcccacctgttcgacgacaaagtgatgaagcagctgaagcggcggagatacaccggctggggcaggctgagccggaagctgatcaacggcatccgggacaagcagtccggcaagacaatcctggat ttcctgaagtccgacggcttcgccaacagaaacttcatgcagctgatccacgacgacagcctgacctttaaagag gacatccagaaagcccaggtgtccggccagggcgatagcctgcacgagcacattgccaatctggccggcagcccc gccattaagaagggcatcctgcagacagtgaaggtggtggacgagctcgtgaaagtgatgggccggcacaagccc gagaacatcgtgatcgaaatggccagagagaaccagaccacccagaagggacagaagaacagccgcgagagaatg aagcggatcgaagagggcatcaaagagctgggcagccagatcctgaaagaacaccccgtggaaaacacccagctg cagaacgagaagctgtacctgtactacctgcagaatgggcgggatatgtacgtggaccaggaactggacatcaac cggctgtccgactacgatgtggaccatatcgtgcctcagagctttctgaaggacgactccatcgacaacaaggtg ctgaccagaagcgacaagaaccggggcaagagcgacaacgtgccctccgaagaggtcgtgaagaagatgaagaac tactggcggcagctgctgaacgccaagctgattacccagagaaagttcgacaatctgaccaaggccgagagaggc ggcctgagcgaactggataaggccggcttcatcaagagacagctggtggaaacccggcagatcacaaagcacgtg gcacagatcctggactcccggatgaacactaagtacgacgagaatgacaagctgatccgggaagtgaaagtgatc accctgaagtccaagctggtgtccgatttccggaaggatttccagttttacaaagtgcgcgagatcaacaactac caccacgcccacgacgcctacctgaacgccgtcgtgggaaccgccctgatcaaaaagtaccctaagctggaaagc gagttcgtgtacggcgactacaaggtgtacgacgtgcggaagatgatcgccaagagcgagcaggaaatcggcaag gctaccgccaagtacttcttctacagcaacatcatgaactttttcaagaccgagattaccctggccaacggcgag atccggaagcggcctctgatcgagacaaacggcgaaaccggggagatcgtgtgggataagggccgggattttgcc accgtgcggaaagtgctgagcatgccccaagtgaatatcgtgaaaaagaccgaggtgcagacaggcggcttcagc aaagagtctatcctgcccaagaggaacagcgataagctgatcgccagaaagaaggactgggaccctaagaagtac ggcggcttcgacagccccaccgtggcctattctgtgctggtggtggccaaagtggaaaagggcaagtccaagaaa ctgaagagtgtgaaagagctgctggggatcaccatcatggaaagaagcagcttcgagaagaatcccatcgacttt ctggaagccaagggctacaaagaagtgaaaaaggacctgatcatcaagctgcctaagtactccctgttcgagctg gaaaacggccggaagagaatgctggcctctgccggcgaactgcagaagggaaacgaactggccctgccctccaaa tatgtgaacttcctgtacctggccagccactatgagaagctgaagggctcccccgaggataatgagcagaaacag ctgtttgtggaacagcacaagcactacctggacgagatcatcgagcagatcagcgagttctccaagagagtgatc ctggccgacgctaatctggacaaagtgctgtccgcctacaacaagcaccgggataagcccatcagagagcaggcc gagaatatcatccacctgtttaccctgaccaatctgggagcccctgccgccttcaagtactttgacaccaccatc gaccggaagaggtacaccagcaccaaagaggtgctggacgccaccctgatccaccagagcatcaccggcctgtac gagacacggatcgacctgtctcagctgggaggtgactctggtggaagcggaggatctggcggcagcaccaatctg agcgacatcatcgagaaagagacaggcaagcagctggtcatccaagagtccatcctgatgctgcctgaagaggtg gaagaagtgatcggcaacaagcccgagtccgacatcctggtgcacaccgcctacgatgagagcaccgacgagaac gtgatgctgctgacctctgacgcccctgagtacaagccttgggctctcgtgatccaggacagcaacggcgagaac aagatcaagatgctgagcggcggctctggtggctctggcggatctacaaacctgtccgatattattgagaaagaa accgggaaacagctcgtgattcaagagtctattctcatgctcccggaagaagtcgaggaagtcattggaaacaag cctgagagcgatattctggtccatacagcctacgacgagtctaccgatgagaatgtcatgctcctcaccagcgac gctcccgagtataagccatgggcacttgtcattcaggactccaatggggaaaacaaaatcaaaatgctcccaaag aaaaaacgcaaggtg (SEQ ID NO: 33) An exemplary prtn_SzW8eqL7-abe8_20m sequence is provided below: MSEVEFSHEYWMRHALTLAKRARDEREVPVGAVLVLNNRVIGEGWNRAIGLHDPTAHAEIMALRQGGLVMQNYRL YDATLYSTFEPCVMCAGAMIHSRIGRVVFGVRNAKTGAAGSLMDVLHHPGMNHRVEITEGILADECAALLCRFFR MPRRVFNAQKKAQSSTDSGGSSGGSSGSETPGTSESATPESSGGSSGGSDKKYSIGLAIGTNSVGWAVITDEYKV PSKKFKVLGNTDRHSIKKNLIGALLFDSGETAEATRLKRTARRRYTRRKNRICYLQEIFSNEMAKVDDSFFHRLE ESFLVEEDKKHERHPIFGNIVDEVAYHEKYPTIYHLRKKLVDSTDKADLRLIYLALAHMIKFRGHFLIEGDLNPD NSDVDKLFIQLVQTYNQLFEENPINASGVDAKAILSARLSKSRRLENLIAQLPGEKKNGLFGNLIALSLGLTPNF KSNFDLAEDAKLQLSKDTYDDDLDNLLAQIGDQYADLFLAAKNLSDAILLSDILRVNTEITKAPLSASMIKRYDE HHQDLTLLKALVRQQLPEKYKEIFFDQSKNGYAGYIDGGASQEEFYKFIKPILEKMDGTEELLVKLNREDLLRKQ RTFDNGSIPHQIHLGELHAILRRQEDFYPFLKDNREKIEKILTFRIPYYVGPLARGNSRFAWMTRKSEETITPWN FEEVVDKGASAQSFIERMTNFDKNLPNEKVLPKHSLLYEYFTVYNELTKVKYVTEGMRKPAFLSGEQKKAIVDLL FKTNRKVTVKQLKEDYFKKIECFDSVEISGVEDRFNASLGTYHDLLKIIKDKDFLDNEENEDILEDIVLTLTLFE DREMIEERLKTYAHLFDDKVMKQLKRRRYTGWGRLSRKLINGIRDKQSGKTILDFLKSDGFANRNFMQLIHDDSL TFKEDIQKAQVSGQGDSLHEHIANLAGSPAIKKGILQTVKVVDELVKVMGRHKPENIVIEMARENQTTQKGQKNS RERMKRIEEGIKELGSQILKEHPVENTQLQNEKLYLYYLQNGRDMYVDQELDINRLSDYDVDHIVPQSFLKDDSI DNKVLTRSDKNRGKSDNVPSEEVVKKMKNYWRQLLNAKLITQRKFDNLTKAERGGLSELDKAGFIKRQLVETRQI TKHVAQILDSRMNTKYDENDKLIREVKVITLKSKLVSDFRKDFQFYKVREINNYHHAHDAYLNAVVGTALIKKYP KLESEFVYGDYKVYDVRKMIAKSEQEIGKATAKYFFYSNIMNFFKTEITLANGEIRKRPLIETNGETGEIVWDKG RDFATVRKVLSMPQVNIVKKTEVQTGGFSKESILPKRNSDKLIARKKDWDPKKYGGFDSPTVAYSVLVVAKVEKGKSKKLKSVKELLGITIMERSSFEKNPIDFLEAKGYKEVKKDLIIKLPKYSLFELENGRKRMLASAGELQKGNELA LPSKYVNFLYLASHYEKLKGSPEDNEQKQLFVEQHKHYLDEIIEQISEFSKRVILADANLDKVLSAYNKHRDKPI REQAENIIHLFTLTNLGAPAAFKYFDTTIDRKRYTSTKEVLDATLIHQSITGLYETRIDLSQLGGDEGADKRTADGSEFESPKKKRKV (SEQ ID NO: 34) An exemplary prtn_ZJVPExXY-ppabobec1-r33a-protein sequence is provided below: MTSEKGPSTGDPTLRRRIESWEFDVFYDPRELAKETCLLYEIKWGMSRKIWRSSGKNTTNHVEVNFIKKFTSERR FHSSISCSITWFLSWSPCWECSQAIREFLSQHPGVTLVIYVARLFWHMDQRNRQGLRDLVNSGVTIQIMRASEYY HCWRNFVNYPPGDEAHWPQYPPLWMMLYALELHCIILSLPPCLKISRRWQNHLAFFRLHLQNCHYQTIPPHILLA TGLIHPSVTWRLKSGGSSGGSSGSETPGTSESATPESSGGSSGGSDKKYSIGLAIGTNSVGWAVITDEYKVPSKKFKVLGNTDRHSIKKNLIGALLFDSGETAEATRLKRTARRRYTRRKNRICYLQEIFSNEMAKVDDSFFHRLEESFL VEEDKKHERHPIFGNIVDEVAYHEKYPTIYHLRKKLVDSTDKADLRLIYLALAHMIKFRGHFLIEGDLNPDNSDV DKLFIQLVQTYNQLFEENPINASGVDAKAILSARLSKSRRLENLIAQLPGEKKNGLFGNLIALSLGLTPNFKSNF DLAEDAKLQLSKDTYDDDLDNLLAQIGDQYADLFLAAKNLSDAILLSDILRVNTEITKAPLSASMIKRYDEHHQD LTLLKALVRQQLPEKYKEIFFDQSKNGYAGYIDGGASQEEFYKFIKPILEKMDGTEELLVKLNREDLLRKQRTFD NGSIPHQIHLGELHAILRRQEDFYPFLKDNREKIEKILTFRIPYYVGPLARGNSRFAWMTRKSEETITPWNFEEV VDKGASAQSFIERMTNFDKNLPNEKVLPKHSLLYEYFTVYNELTKVKYVTEGMRKPAFLSGEQKKAIVDLLFKTN RKVTVKQLKEDYFKKIECFDSVEISGVEDRFNASLGTYHDLLKIIKDKDFLDNEENEDILEDIVLTLTLFEDREM IEERLKTYAHLFDDKVMKQLKRRRYTGWGRLSRKLINGIRDKQSGKTILDFLKSDGFANRNFMQLIHDDSLTFKE DIQKAQVSGQGDSLHEHIANLAGSPAIKKGILQTVKVVDELVKVMGRHKPENIVIEMARENQTTQKGQKNSRERM KRIEEGIKELGSQILKEHPVENTQLQNEKLYLYYLQNGRDMYVDQELDINRLSDYDVDHIVPQSFLKDDSIDNKV LTRSDKNRGKSDNVPSEEVVKKMKNYWRQLLNAKLITQRKFDNLTKAERGGLSELDKAGFIKRQLVETRQITKHV AQILDSRMNTKYDENDKLIREVKVITLKSKLVSDFRKDFQFYKVREINNYHHAHDAYLNAVVGTALIKKYPKLES EFVYGDYKVYDVRKMIAKSEQEIGKATAKYFFYSNIMNFFKTEITLANGEIRKRPLIETNGETGEIVWDKGRDFA TVRKVLSMPQVNIVKKTEVQTGGFSKESILPKRNSDKLIARKKDWDPKKYGGFDSPTVAYSVLVVAKVEKGKSKKLKSVKELLGITIMERSSFEKNPIDFLEAKGYKEVKKDLIIKLPKYSLFELENGRKRMLASAGELQKGNELALPSK YVNFLYLASHYEKLKGSPEDNEQKQLFVEQHKHYLDEIIEQISEFSKRVILADANLDKVLSAYNKHRDKPIREQA ENIIHLFTLTNLGAPAAFKYFDTTIDRKRYTSTKEVLDATLIHQSITGLYETRIDLSQLGGDSGGSGGSGGSTNL SDIIEKETGKQLVIQESILMLPEEVEEVIGNKPESDILVHTAYDESTDENVMLLTSDAPEYKPWALVIQDSNGEN KIKMLSGGSGGSGGSTNLSDIIEKETGKQLVIQESILMLPEEVEEVIGNKPESDILVHTAYDESTDENVMLLTSD APEYKPWALVIQDSNGENKIKMLPKKKRKV (SEQ ID NO: 35) An exemplary prtn_ZyqE8AYc-ppabobec1-wt-protein sequence is provided below: MTSEKGPSTGDPTLRRRIESWEFDVFYDPRELRKETCLLYEIKWGMSRKIWRSSGKNTTNHVEVNFIKKFTSERR FHSSISCSITWFLSWSPCWECSQAIREFLSQHPGVTLVIYVARLFWHMDQRNRQGLRDLVNSGVTIQIMRASEYY HCWRNFVNYPPGDEAHWPQYPPLWMMLYALELHCIILSLPPCLKISRRWQNHLAFFRLHLQNCHYQTIPPHILLA TGLIHPSVTWRLKSGGSSGGSSGSETPGTSESATPESSGGSSGGSDKKYSIGLAIGTNSVGWAVITDEYKVPSKK FKVLGNTDRHSIKKNLIGALLFDSGETAEATRLKRTARRRYTRRKNRICYLQEIFSNEMAKVDDSFFHRLEESFL VEEDKKHERHPIFGNIVDEVAYHEKYPTIYHLRKKLVDSTDKADLRLIYLALAHMIKFRGHFLIEGDLNPDNSDV DKLFIQLVQTYNQLFEENPINASGVDAKAILSARLSKSRRLENLIAQLPGEKKNGLFGNLIALSLGLTPNFKSNF DLAEDAKLQLSKDTYDDDLDNLLAQIGDQYADLFLAAKNLSDAILLSDILRVNTEITKAPLSASMIKRYDEHHQD LTLLKALVRQQLPEKYKEIFFDQSKNGYAGYIDGGASQEEFYKFIKPILEKMDGTEELLVKLNREDLLRKQRTFD NGSIPHQIHLGELHAILRRQEDFYPFLKDNREKIEKILTFRIPYYVGPLARGNSRFAWMTRKSEETITPWNFEEV VDKGASAQSFIERMTNFDKNLPNEKVLPKHSLLYEYFTVYNELTKVKYVTEGMRKPAFLSGEQKKAIVDLLFKTN RKVTVKQLKEDYFKKIECFDSVEISGVEDRFNASLGTYHDLLKIIKDKDFLDNEENEDILEDIVLTLTLFEDREM IEERLKTYAHLFDDKVMKQLKRRRYTGWGRLSRKLINGIRDKQSGKTILDFLKSDGFANRNFMQLIHDDSLTFKE DIQKAQVSGQGDSLHEHIANLAGSPAIKKGILQTVKVVDELVKVMGRHKPENIVIEMARENQTTQKGQKNSRERM KRIEEGIKELGSQILKEHPVENTQLQNEKLYLYYLQNGRDMYVDQELDINRLSDYDVDHIVPQSFLKDDSIDNKV LTRSDKNRGKSDNVPSEEVVKKMKNYWRQLLNAKLITQRKFDNLTKAERGGLSELDKAGFIKRQLVETRQITKHV AQILDSRMNTKYDENDKLIREVKVITLKSKLVSDFRKDFQFYKVREINNYHHAHDAYLNAVVGTALIKKYPKLES EFVYGDYKVYDVRKMIAKSEQEIGKATAKYFFYSNIMNFFKTEITLANGEIRKRPLIETNGETGEIVWDKGRDFATVRKVLSMPQVNIVKKTEVQTGGFSKESILPKRNSDKLIARKKDWDPKKYGGFDSPTVAYSVLVVAKVEKGKSKK LKSVKELLGITIMERSSFEKNPIDFLEAKGYKEVKKDLIIKLPKYSLFELENGRKRMLASAGELQKGNELALPSK YVNFLYLASHYEKLKGSPEDNEQKQLFVEQHKHYLDEIIEQISEFSKRVILADANLDKVLSAYNKHRDKPIREQA ENIIHLFTLTNLGAPAAFKYFDTTIDRKRYTSTKEVLDATLIHQSITGLYETRIDLSQLGGDSGGSGGSGGSTNL SDIIEKETGKQLVIQESILMLPEEVEEVIGNKPESDILVHTAYDESTDENVMLLTSDAPEYKPWALVIQDSNGEN KIKMLSGGSGGSGGSTNLSDIIEKETGKQLVIQESILMLPEEVEEVIGNKPESDILVHTAYDESTDENVMLLTSDAPEYKPWALVIQDSNGENKIKMLPKKKRKV (SEQ ID NO: 36) In some embodiments, the base editor has been further modified to inhibit base excision repair at the target site and induce cellular mismatch repair. Any of the Cas9 nucleases described herein may be fused to a Gam domain (bacteriophage Mu protein) to protect the Cas9 nuclease from degradation and exonuclease activity. See, e.g., Eid et al. Biochem. J. (2018) 475(11): 1955-1964. In some embodiments, the Cas nuclease belongs to class 2 type V of Cas endonuclease. Class 2 type V Cas endonucleases can be further categorized as type V-A, type V-B, type V-C, and type V-U. See, e.g., Stella et al. Nature Structural & Molecular Biology (2017) 24: 882-892. In some embodiments, the Cas nuclease is a type V-A Cas endonuclease, such as a Cpf1 (Cas12a) nuclease. In some embodiments, the Cas9 nuclease is a type V-B Cas endonuclease, such as a C2c1 endonuclease. See, e.g., Shmakov et al. Mol Cell (2015) 60: 385-397. In some embodiments, the Cas nuclease is MAD7TM. Alternatively, or in addition, the Cas9 nuclease is a Cpf1 nuclease or a variant thereof. As will be appreciated by one of skill in the art, the Cpf1 nuclease may also be referred to as Cas12a. See, e.g., Strohkendl et al. Mol. Cell (2018) 71: 1-9. In some embodiments, a composition or method described herein involves, or a host cell expresses a Cpf1 nuclease derived from Provetella spp. or Francisella spp., Acidaminococcus sp. (AsCpf1), Lachnospiraceae bacterium (LpCpf1), or Eubacterium rectale. In some embodiments, the nucleotide sequence encoding the Cpf1 nuclease may be codon optimized for expression in a host cell. In some embodiments, the nucleotide sequence encoding the Cpf1 endonuclease is further modified to alter the activity of the protein. Both naturally occurring and modified variants of CRISPR/Cas nucleases are suitable for use according to aspects of this disclosure. For example, dCas or nickase variants, Cas variants having altered PAM specificities, and Cas variants having improved nuclease activities are embraced by some embodiments of this disclosure. In some embodiments, catalytically inactive variants of Cas nucleases (e.g., of Cas9 or Cas12a) are used according to the methods described herein. A catalytically inactive variant of Cpf1 (Cas12a) may be referred to dCas12a. As described herein, catalytically inactive variants of Cpf1 maybe fused to a function domain to form a base editor. See, e.g., Rees et al. Nature Reviews Genetics (2018) 19:770-788. In some embodiments, the catalytically inactive Cas9 nuclease is dCas9. In some embodiments, the nuclease comprises a dCas12a fused to one or more uracil glycosylase inhibitor (UGI) domains. In some embodiments, the Cas9 nuclease comprises a dCas12a fused to an adenine base editor (ABE), for example an ABE evolved from the RNA adenine deaminase TadA. In some embodiments, the Cas nuclease comprises a dCas12a fused to cytidine deaminase enzyme (e.g., APOBEC deaminase, pmCDA1, activation- induced cytidine deaminase (AID)). Alternatively or in addition, the Cas9 nuclease may be a Cas14 nuclease or variant thereof. Cas14 nucleases are derived from archaea and tend to be smaller in size (e.g., 400– 700 amino acids). Additionally Cas14 nucleases do not require a PAM sequence. See, e.g., Harrington et al. Science (2018). Any of the Cas9 nucleases described herein may be modulated to regulate levels of expression and/or activity of the Cas9 nuclease at a desired time. For example, it may be advantageous to increase levels of expression and/or activity of the Cas9 nuclease during particular phase(s) of the cell cycle. It has been demonstrated that levels of homology- directed repair are reduced during the G1 phase of the cell cycle, therefore increasing levels of expression and/or activity of the Cas9 nuclease during the S phase, G2 phase, and/or M phase may increase homology-directed repair following the Cas endonuclease editing. In some embodiments, levels of expression and/or activity of the Cas9 nuclease are increased during the S phase, G2 phase, and/or M phase of the cell cycle. In one example, the Cas9 nuclease fused to the N-terminal region of human Geminin. See, e.g., Gutschner et al. Cell Rep. (2016) 14(6): 1555-1566. In some embodiments, levels of expression and/or activity of the Cas9 nuclease are reduced during the G1 phase. In one example, the Cas9 nuclease is modified such that it has reduced activity during the G1 phase. See, e.g., Lomova et al. Stem Cells (2018). Alternatively or in addition, any of the Cas9 nucleases described herein may be fused to an epigenetic modifier (e.g., a chromatin-modifying enzyme, e.g., DNA methylase, histone deacetylase). See, e.g., Kungulovski et al. Trends Genet. (2016) 32(2):101-113. Cas9 nuclease fused to an epigenetic modifier may be referred to as “epieffectors” and may allow for temporal and/or transient endonuclease activity. In some embodiments, the Cas9 nuclease is a dCas9 fused to a chromatin-modifying enzyme. III. Cytotoxic Agents Cytotoxic agents targeting cells (e.g., cancer cells) expressing a cell surface protein can be co-used with the genetically engineered hematopoietic cells as described herein. As used herein, the term “cytotoxic agent” refers to any agent that can directly or indirectly induce cytotoxicity of a target cell, which expresses the cell surface protein (e.g., a target cancer cell). Such a cytotoxic agent may comprise a protein-binding fragment that binds and targets an epitope of the lineage-specific cell surface antigen. In some instances, the cytotoxic agent may comprise an antibody, which may be conjugated to a drug (e.g., an anti- cancer drug) to form an antibody-drug conjugate (ADC). The cytotoxic agent for use in the methods described herein may directly cause cell death of a target cell. For example, the cytotoxic agent can be an immune cell (e.g., a cytotoxic T cell) expressing a chimeric receptor. Upon engagement of the protein binding domain of the chimeric receptor with the corresponding epitope in a lineage-specific cell surface antigen, a signal (e.g., activation signal) may be transduced to the immune cell resulting in release of cytotoxic molecules, such as perforins and granzymes, as well as activation of effector functions, leading to death of the target cell. In another example, the cytotoxic agent may be an ADC molecule. Upon binding to a target cell, the drug moiety in the ADC would exert cytotoxic activity, leading to target cell death. In other embodiments, the cytotoxic agent may indirectly induce cell death of the target cell. For example, the cytotoxic agent may be an antibody, which, upon binding to the target cell, would trigger effector activities (e.g., ADCC) and/or recruit other factors (e.g., complements), resulting in target cell death. Any of the cytotoxic agents described herein target a lineage-specific cell surface antigen, e.g., comprising a protein-binding fragment that specifically binds an epitope in the cell surface protein. Non-limiting examples of cytotoxic agents that can be utilized in the compositions and methods described herein include those described in PCT Publication Nos. WO2022/056497, WO2022/056490, and WO2019/178382, WO2023/201238, and International Patent Application No. PCT/US2024/029308. For leukemias that become resistance to CAR-T therapy, an emerging strategy is to simultaneously target alternative or multiple antigens (see e.g., Nature Reviews Immunology (2019), Volume 19, pages 73–74 and Cancer Discov. (2018) Oct;8(10):1219-1226). In some embodiments, more than one (e.g., 2, 3, 4, 5 or more) cytotoxic agent is used to target more than one (e.g., 1, 2, 3, 4, 5 or more) epitopes of a lineage-specific cell surface antigen. In some embodiments, more than one (e.g., 1, 2, 3, 4, 5 or more) cytotoxic agent is used to target an epitope(s) of one or more lineage-specific cell surface antigen(s) (e.g., additional/alternative antigens). In some embodiments, targeting of more than one cell surface protein reduces relapse of a hematopoietic malignancy. In one embodiment, two or more cytotoxic agents are used in the methods described herein. In some embodiments, the two or more cytotoxic agents are administered concurrently. In some embodiments, the two or more cytotoxic agents are administered sequentially. Examples of additional cell surface proteins that may be targeted are known in the art (see, e.g., Tasian, Ther. Adv. Hematol. (2018) 9(6): 135-148; Hoseini and Cheung, Blood Cancer Journal (2017) 7, e522; doi:10.1038/bcj.2017.2; Taraseviciute et al. Hematology and Oncology (2019) 31(1)). In some embodiments, the methods described herein involve targeting a cell surface protein and one or more additional cell surface proteins. In some embodiments, a genetically engineered cell of the present disclosure may be contacted with a cytotoxic agent comprising an antigen-binding fragment that recognizes a cell surface protein selected from a group consisting of: CD10; CD101; CD117; CD11A; CD11C; CD120B; CD123; CD13; CD133; CD152; CD200; CD205 (LY75); CD226; CD244 (SLAMF4); CD25; CD274; CD305 (LAIR1); CD32; CD33; CD34; CD366; CD38; CD41; CD42B; CD44; CD45RA; CD47; CD48; CD49D (ITGA4); CD49F; CD52; CD58; CD64; CD7; CD70; CD82; CD84; CD85; CD85K; CD86; CD9; CD96; CD99; CLL-1; EMR2 (ADGRE2); FR-B; GPR56; and IL1RAP. In some embodiments, a genetically engineered cell of the present disclosure may be contacted with a cytotoxic agent comprising an antigen-binding fragment that recognizes a cell surface protein selected from a group consisting of: CD10; CD101; CD11A; CD11C; CD123; CD133; CD152; CD200; CD205; CD244; CD25; CD305; CD33; CD34; CD366; CD38; CD42B; CD44; CD45RA; CD47; CD48; CD49D; CD49F; CD52; CD58; CD64; CD7; CD84; CD85K; CD9; CD99; CLL-1; EMR2; GPR56; IL1RAP; and CD274. In some embodiments, a genetically engineered cell of the present disclosure may be contacted with a cytotoxic agent comprising an antigen-binding fragment that recognizes a cell surface protein selected from a group consisting of: CD10; CD117; CD11A; CD11C; CD120B; CD123; CD13; CD133; CD152; CD205; CD22; CD226; CD244; CD25; CD305; CD32; CD33; CD34; CD38; CD41; CD44; CD45RA; CD47; CD48; CD49D; CD49F; CD52; CD56; CD58; CD64; CD7; CD70; CD71; CD82; CD84; CD85K; CD86; CD96; CD99; CLL- 1; EMR2; FR-B; GPR56; HLA-DR; and IL1RAP. In some embodiments, a genetically engineered cell of the present disclosure may be contacted with a first cytotoxic agent comprising an antigen-binding fragment that recognizes a first cell surface protein and a second cytotoxic agent comprising an antigen-binding fragment that recognizes a second cell surface protein selected from a group consisting of: CD274 and CD152; CD274 and CD226; CD152 and CD226; CD10 and CD49F; CD49F and GPR56; CD200 and CD34; CD34 and CD52; CD34 and CD133; GPR56 and CD133; CD42B and CD41; CD85K and CD64; CD85K and CD32; CD64 and CD32; CD64 and CD11C; CD64 and CD86; CD45RA and CD96; CD45RA and IL1RAP; CD45RA and CD123; CD84 and CD33; CD11C and IL1RAP; CD11C and CD33; CD11C and CD86; CD9 and IL1RAP; IL1RAP and CD33; IL1RAP and CD205; IL1RAP and CD305; IL1RAP and CD123; CD33 and CLL-1; CD33 and CD305; CD11A and CD86; CLL-1 and CD305; CD58 and CD305; CD49D and CD47; CD49D and CD99; CD49D and CD44; CD49D and CD305; CD47 and CD99; CD47 and EMR2; CD99 and CD44; CD99 and CD305; CD99 and EMR2; CD205 and CD305; CD205 and CD123; CD44 and CD305; CD44 and EMR2; CD244 and CD117; CD123 and EMR2; EMR2 and CD82; CD152 and CD120B; CD48 and CD11A; CD200 and CD25; CD34 and CD7; CD34 and CD13; CD52 and GPR56; CD52and CD133; CD52 and CD7; GPR56 and CD7; GPR56 and CD82; CD133 and CD7; CD133 and CD82; CD7 and CD13; CD101 and CD64; CD85 and CD70; CD85K and CD86; CD64 and FR-B; CD366 and CD96; CD25 and CD45RA; CD25 and CD96; CD9 and CD82; IL1RAP and CD58; CD11A and CD38; CD11A and CD44; CD11A and CD244; CD38 and CD305; CD38 and EMR2; CLL-1 and CD49D; CD58 and CD49D; CD47 and CD205; and CD47 and CD44. In some embodiments, a genetically engineered cell of the present disclosure may be contacted with a first cytotoxic agent comprising an antigen-binding fragment that recognizes a first cell surface protein and a second cytotoxic agent comprising an antigen-binding fragment that recognizes a second cell surface protein selected from a group consisting of: CD33 and CD205 (LY75); CD33 and CD244 (SLAMF4); CD33 and CD305 (LAIR1); CD33 and CD49D (ITGA4); CLL-1 and CD205 (LY75); CLL-1 and CD244 (SLAMF4); CLL-1 and CD305 (LAIR1); CLL-1 and CD49D (ITGA4); EMR2 and CD205 (LY75); EMR2 and CD244 (SLAMF4); EMR2 and CD305 (LAIR1); EMR2 and CD49D (ITGA4); CD38 and CD205 (LY75); CD38 and CD244 (SLAMF4); CD38 and CD305 (LAIR1); CD38 and CD49D (ITGA4); CD305 (LAIR1) and CD49D (ITGA4); CD305 (LAIR1) and CD205 (LY75); CD305 (LAIR1) and CD244 (SLAMF4); CD49D (ITGA4) and CD205 (LY75); CD49D (ITGA4) and CD244 (SLAMF4); and CD205 (LY75) and CD244 (SLAMF4). In some embodiments, a genetically engineered cell of the present disclosure may be contacted with a first cytotoxic agent comprising an antigen-binding fragment that recognizes a first cell surface protein and a second cytotoxic agent comprising an antigen-binding fragment that recognizes a second cell surface protein selected from a group consisting of: CD85K and CD64; CD45RA and IL1RAP; CD34 and CD133; IL1RAP and CD33; CD47 and CD99; CLL-1 and CD305; CD152 and CD226; CD45RA and CD96; CD45RA and CD123; CD49D and CD305; IL1RAP and CD305; CD58 and CD305; CD49D and CD47; CD33 and CD305; CD49D and CD44; CD64 and CD86; GPR56 and CD133; CD34 and CD52; EMR2 and CD82; CD64 and CD32; CD42B and CD41; CD64 and CD11C; CD99 and CD44; CD200 and CD34; CD11C and CD33; IL1RAP and CD123; CD11C and IL1RAP; CD44 and EMR2; CD205 and CD305; CD44 and CD305; CD49D and CD99; CD274 and CD226; CD49F and GPR56; CD9 and IL1RAP; CD99 and CD305; CD10 and CD49F; CD11A and CD86; CD33 and CLL-1; CD84 and CD33; CD244 and CD117; CD85K and CD32; CD47 and EMR2; CD274 and CD152; IL1RAP and CD205; CD11C and CD86; CD99 and EMR2; CD205 and CD123; and CD123 and EMR2. In some embodiments, a genetically engineered cell of the present disclosure may be contacted with a first cytotoxic agent comprising an antigen-binding fragment that recognizes a first cell surface protein and a second cytotoxic agent comprising an antigen-binding fragment that recognizes a second cell surface protein selected from a group consisting of: CD274 and CD152; CD274 and CD226; CD152 and CD226; CD10 and CD49F; CD49F and GPR56; CD200 and CD34; CD34 and CD52; CD34 and CD133; GPR56 and CD133; CD42B and CD41; CD85K and CD64; CD85K and CD32; CD64 and CD32; CD64 and CD11C; CD64 and CD86; CD45RA and CD96; CD45RA and IL1RAP; CD45RA and CD123; CD84 and CD33; CD11C and IL1RAP; CD11C and CD33; CD11C and CD86; CD9 and IL1RAP; IL1RAP and CD33; IL1RAP and CD205; IL1RAP and CD305; IL1RAP and CD123; CD33 and CLL-1; CD33 and CD305; CD11A and CD86; CLL-1 and CD305; CD58 and CD305; CD49D and CD47; CD49D and CD99; CD49D and CD44; CD49D and CD305; CD47 and CD99; CD47 and EMR2; CD99 and CD44; CD99 and CD305; CD99 and EMR2; CD205 and CD305; CD205 and CD123; CD44 and CD305; CD44 and EMR2; CD244 and CD117; CD123 and EMR2; and EMR2 and CD82. In some embodiments, a genetically engineered cell of the present disclosure may be contacted with a first cytotoxic agent comprising an antigen-binding fragment that recognizes a first cell surface protein and a second cytotoxic agent comprising an antigen-binding fragment that recognizes a second cell surface protein selected from a group consisting of: CD9 and CD82; CD45RA and CD123; CD133 and CD82; CD85K and CD70; CD47 and CD205; CD64 and FR-B; CD11A and CD44; CD85K and CD86; CD366 and CD96; CD101 and CD64; CLL-1 and CD49D; CD49D and CD47; EMR2 and CD82; CD38 and CD305; CD34 and CD7; CD200 and CD25; CD25 and CD45RA; CD49D and CD44; CD11A and CD244; CD133 and CD7; CD48 and CD11A; CD52 and CD133; GPR56 and CD82; CD11A and CD38; CD34 and CD13; CD58 and CD49D; CD47 and CD99; IL1RAP and CD58; CD47 and CD44; CD33 and CD305; CD64 and CD86; CD152 and CD120B; CD42B and CD41; CD34 and CD52; CD152 and CD226; CD7 and CD13; CD38 and EMR2; IL1RAP and CD33; CLL-1 and CD305; CD45RA and IL1RAP; CD34 and CD133; GPR56 and CD133; CD49D and CD305; CD25 and CD96; CD52 and CD7; CD64 and CD32; CD58 and CD305; CD45RA and CD96; IL1RAP and CD305; CD52 and GPR56; GPR56 and CD7; and CD85K and CD64. Non-limiting examples of agents (e.g., cytotoxic agents) comprising antigen-binding fragments that may be used to target malignant hematopoietic cells in a subject are further provided herein. Therapeutic Antibodies Any antibody or an antigen-binding fragment thereof can be used as a cytotoxic agent or for constructing a cytotoxic agent that targets an epitope of a lineage-specific cell surface antigen, as described herein. Such an antibody or antigen-binding fragment can be prepared by a conventional method, for example, the hybridoma technology or recombinant technology. As used herein, the term "antibody" refers to a glycoprotein comprising at least two heavy (H) chains and two light (L) chains inter-connected by disulfide bonds, i.e., covalent heterotetramers comprised of two identical Ig H chains and two identical L chains that are encoded by different genes. Each heavy chain is comprised of a heavy chain variable region (abbreviated herein as HCVR or VH) and a heavy chain constant region. The heavy chain constant region is comprised of three domains, CH1, CH2 and CH3. Each light chain is comprised of a light chain variable region (abbreviated herein as LCVR or VL) and a light chain constant region. The light chain constant region is comprised of one domain, CL. The VH and VL regions can be further subdivided into regions of hypervariability, termed complementarity determining regions (CDR), interspersed with regions that are more conserved, termed framework regions (FR). Each VH and VL is composed of three CDRs and four FRs, arranged from amino-terminus to carboxy-terminus in the following order: FR1, CDR1, FR2, CDR2, FR3, CDR3, FR4. The variable regions of the heavy and light chains contain a binding domain that interacts with an antigen. The constant regions of the antibodies may mediate the binding of the immunoglobulin to host tissues or factors, including various cells of the immune system (e.g., effector cells) and the first component (Clq) of the classical complement system. Formation of a mature functional antibody molecule can be accomplished when two proteins are expressed in stoichiometric quantities and self-assemble with the proper configuration. In some embodiments, the antigen-binding fragment is a single-chain antibody fragment (scFv) that specifically binds the epitope of the lineage-specific cell surface antigen. In other embodiments, the antigen-binding fragment is a full-length antibody that specifically binds the epitope of the lineage-specific cell surface antigen. As described herein and as will be evident to a skilled artisan, the CDRs of an antibody specifically bind to the epitope of a target protein/antigen (the cell surface protein/antigen). In some embodiments, the antibodies are full-length antibodies, meaning the antibodies comprise a fragment crystallizable (Fc) portion and a fragment antigen-binding (Fab) portion. In some embodiments, the antibodies are of the isotype IgG, IgA, IgM, IgA, or IgD. In some embodiments, a population of antibodies comprises one isotype of antibody. In some embodiments, the antibodies are IgG antibodies. In some embodiments, the antibodies are IgM antibodies. In some embodiments, a population of antibodies comprises more than one isotype of antibody. In some embodiments, a population of antibodies is comprised of a majority of one isotype of antibodies but also contains one or more other isotypes of antibodies. In some embodiments, the antibodies are selected from the group consisting of IgGl, IgG2, IgG3, IgG4, IgM, IgAl, IgA2, IgAsec, IgD, and IgE. The antibodies described herein may specifically bind to a target protein. As used herein, “specific binding” refers to antibody binding to a predetermined protein, such as a cancer antigen. “Specific binding” involves more frequent, more rapid, greater duration of interaction, and/or greater affinity to a target protein relative to alternative proteins. In some embodiments, a population of antibodies specifically binds to a particular epitope of a target protein, meaning the antibodies bind to the particular protein with more frequently, more rapidly, for greater duration of interaction, and/or with greater affinity to the epitope relative to alternative epitopes of the same target protein or to epitopes of another protein. In some embodiments, the antibodies that specifically bind to a particular epitope of a target protein may not bind to other epitopes of the same protein. Antibodies may be selected based on the binding affinity of the antibody to the target protein or epitope. Alternatively or in additional, the antibodies may be mutated to introduce one or more mutations to modify (e.g., enhance or reduce) the binding affinity of the antibody to the target protein or epitope. The present antibodies or antigen-binding portions can specifically bind with a dissociation constant (KD) of less than about 10-7 M, less than about 10-8 M, less than about 10-9 M, less than about 10-10 M, less than about 10-11 M, or less than about 10-12 M. Affinities of the antibodies according to the present disclosure can be readily determined using conventional techniques (see, e.g., Scatchard et al., Ann. N.Y. Acad. Sci. (1949) 51:660; and U.S. Patent Nos.5,283,173, 5,468,614, or the equivalent). The binding affinity or binding specificity for an epitope or protein can be determined by a variety of methods including equilibrium dialysis, equilibrium binding, gel filtration, ELISA, surface plasmon resonance, or spectroscopy. For example, antibodies (of antigen-binding fragments thereof) specific to an epitope of a cell surface protein of interest can be made by the conventional hybridoma technology. The cell surface protein, which may be coupled to a carrier protein such as KLH, can be used to immunize a host animal for generating antibodies binding to that complex. The route and schedule of immunization of the host animal are generally in keeping with established and conventional techniques for antibody stimulation and production, as further described herein. General techniques for production of mouse, humanized, and human antibodies are known in the art and are described herein. It is contemplated that any mammalian subject including humans or antibody producing cells therefrom can be manipulated to serve as the basis for production of mammalian, including human hybridoma cell lines. Typically, the host animal is inoculated intraperitoneally, intramuscularly, orally, subcutaneously, intraplantar, and/or intradermally with an amount of immunogen, including as described herein. Hybridomas can be prepared from the lymphocytes and immortalized myeloma cells using the general somatic cell hybridization technique of Kohler, B. and Milstein, C. (1975) Nature 256:495-497 or as modified by Buck, D. W., et al., In Vitro, 18:377-381 (1982). Available myeloma lines, including but not limited to X63-Ag8.653 and those from the Salk Institute, Cell Distribution Center, San Diego, Calif., USA, may be used in the hybridization. Generally, the technique involves fusing myeloma cells and lymphoid cells using a fusogen such as polyethylene glycol, or by electrical means well known to those skilled in the art. After the fusion, the cells are separated from the fusion medium and grown in a selective growth medium, such as hypoxanthine-aminopterin-thymidine (HAT) medium, to eliminate unhybridized parent cells. Any of the media described herein, supplemented with or without serum, can be used for culturing hybridomas that secrete monoclonal antibodies. As another alternative to the cell fusion technique, EBV immortalized B cells may be used to produce the TCR-like monoclonal antibodies described herein. The hybridomas are expanded and subcloned, if desired, and supernatants are assayed for anti-immunogen activity by conventional immunoassay procedures (e.g., radioimmunoassay, enzyme immunoassay, or fluorescence immunoassay). Hybridomas that may be used as source of antibodies encompass all derivatives, progeny cells of the parent hybridomas that produce monoclonal antibodies capable of binding to a cell surface protein. Hybridomas that produce such antibodies may be grown in vitro or in vivo using known procedures. The monoclonal antibodies may be isolated from the culture media or body fluids, by conventional immunoglobulin purification procedures such as ammonium sulfate precipitation, gel electrophoresis, dialysis, chromatography, and ultrafiltration, if desired. Undesired activity if present, can be removed, for example, by running the preparation over adsorbents made of the immunogen attached to a solid phase and eluting or releasing the desired antibodies off the immunogen. Immunization of a host animal with a target protein or a fragment containing the target amino acid sequence conjugated to a protein that is immunogenic in the species to be immunized, e.g., keyhole limpet hemocyanin, serum albumin, bovine thyroglobulin, or soybean trypsin inhibitor using a bifunctional or derivatizing agent, for example maleimidobenzoyl sulfosuccinimide ester (conjugation through cysteine residues), N-hydroxysuccinimide (through lysine residues), glutaraldehyde, succinic anhydride, SOCl, or R1N=C=NR, where R and R1 are different alkyl groups, can yield a population of antibodies (e.g., monoclonal antibodies). If desired, an antibody of interest (e.g., produced by a hybridoma) may be sequenced and the polynucleotide sequence may then be cloned into a vector for expression or propagation. The sequence encoding the antibody of interest may be maintained in vector in a host cell and the host cell can then be expanded and frozen for future use. In an alternative, the polynucleotide sequence may be used for genetic manipulation to "humanize" the antibody or to improve the affinity (affinity maturation), or other characteristics of the antibody. For example, the constant region may be engineered to more resemble human constant regions to avoid immune response if the antibody is used in clinical trials and treatments in humans. It may be desirable to genetically manipulate the antibody sequence to obtain greater affinity to the cell surface protein. In some examples, the antibody sequence is manipulated to increase binding affinity of the antibody to the cell surface protein such that lower levels of the cell surface protein are detected by the antibody. In some embodiments, antibodies that have increased binding to the cell surface protein may be used to reduce or prevent relapse of a hematopoietic malignancy. It will be apparent to one of skill in the art that one or more polynucleotide changes can be made to the antibody and still maintain its binding specificity to the target protein. In other embodiments, fully human antibodies can be obtained by using commercially available mice that have been engineered to express specific human immunoglobulin proteins. Transgenic animals that are designed to produce a more desirable (e.g., fully human antibodies) or more robust immune response may also be used for generation of humanized or human antibodies. Examples of such technology are XenomouseRTM from Amgen, Inc. (Fremont, Calif.) and HuMAb-MouseRTM and TC MouseTM from Medarex, Inc. (Princeton, N.J.). In another alternative, antibodies may be made recombinantly by phage display or yeast technology. See, for example, U.S. Pat. Nos.5,565,332; 5,580,717; 5,733,743; and 6,265,150; and Winter et al., (1994) Annu. Rev. Immunol.12:433-455. Alternatively, the phage display technology (McCafferty et al., (1990) Nature 348:552-553) can be used to produce human antibodies and antibody fragments in vitro, from immunoglobulin variable (V) domain gene repertoires from unimmunized donors. Antigen-binding fragments of an intact antibody (full-length antibody) can be prepared via routine methods. For example, F(ab')2 fragments can be produced by pepsin digestion of an antibody molecule, and Fab fragments that can be generated by reducing the disulfide bridges of F(ab')2 fragments. Genetically engineered antibodies, such as humanized antibodies, chimeric antibodies, single-chain antibodies, and bi-specific antibodies, can be produced via, e.g., conventional recombinant technology. In one example, DNA encoding a monoclonal antibodies specific to a target protein can be readily isolated and sequenced using conventional procedures (e.g., by using oligonucleotide probes that are capable of binding specifically to genes encoding the heavy and light chains of the monoclonal antibodies). The hybridoma cells serve as a preferred source of such DNA. Once isolated, the DNA may be placed into one or more expression vectors, which are then transfected into host cells such as E. coli cells, simian COS cells, Chinese hamster ovary (CHO) cells, or myeloma cells that do not otherwise produce immunoglobulin protein, to obtain the synthesis of monoclonal antibodies in the recombinant host cells. See, e.g., PCT Publication No. WO 87/04462. The DNA can then be modified, for example, by substituting the coding sequence for human heavy and light chain constant domains in place of the homologous murine sequences, Morrison et al., (1984) Proc. Nat. Acad. Sci.81:6851, or by covalently joining to the immunoglobulin coding sequence all or part of the coding sequence for a non- immunoglobulin polypeptide. In that manner, genetically engineered antibodies, such as “chimeric” or “hybrid” antibodies; can be prepared that have the binding specificity of a target protein. Techniques developed for the production of “chimeric antibodies” are well known in the art. See, e.g., Morrison et al. (1984) Proc. Natl. Acad. Sci. USA 81, 6851; Neuberger et al. (1984) Nature 312, 604; and Takeda et al. (1984) Nature 314:452. Methods for constructing humanized antibodies are also well known in the art. See, e.g., Queen et al., Proc. Natl. Acad. Sci. USA, 86:10029-10033 (1989). In one example, variable regions of VH and VL of a parent non-human antibody are subjected to three- dimensional molecular modeling analysis following methods known in the art. Next, framework amino acid residues predicted to be important for the formation of the correct CDR structures are identified using the same molecular modeling analysis. In parallel, human VH and VL chains having amino acid sequences that are homologous to those of the parent non-human antibody are identified from any antibody gene database using the parent VH and VL sequences as search queries. Human VH and VL acceptor genes are then selected. The CDR regions within the selected human acceptor genes can be replaced with the CDR regions from the parent non-human antibody or functional variants thereof. When necessary, residues within the framework regions of the parent chain that are predicted to be important in interacting with the CDR regions (see above description) can be used to substitute for the corresponding residues in the human acceptor genes. A single-chain antibody can be prepared via recombinant technology by linking a nucleotide sequence coding for a heavy chain variable region and a nucleotide sequence coding for a light chain variable region. Preferably, a flexible linker is incorporated between the two variable regions. Alternatively, techniques described for the production of single chain antibodies (U.S. Patent Nos.4,946,778 and 4,704,692) can be adapted to produce a phage or yeast scFv library and scFv clones specific to a cell surface protein can be identified from the library following routine procedures. Positive clones can be subjected to further screening to identify those that bind cell surface protein. In some instances, the cytotoxic agent for use in the methods described herein comprises an antigen-binding fragment that targets CD33. In other examples, the cytotoxic agent for use in the methods described herein comprises an antigen-binding fragment that targets IL1RAP. In other example, two or more cytotoxic agents are used in the methods described herein. In some embodiments, the two or more cytotoxic agents are administered concurrently. In some embodiments, the two or more cytotoxic agents are administered sequentially. In one non-limiting example, antibodies or antigen-binding fragments thereof targeting CD33 and IL1RAP in combination are used in the methods described herein. In one non-limiting example, antibodies or antigen-binding fragments thereof targeting CD44 and EMR2 in combination are used in the methods described herein. In one non-limiting example, antibodies or antigen-binding fragments thereof targeting CD33 are used in combination with a cytotoxic agent (e.g., antibodies, immune cells expressing chimeric antigen receptors, antibody-drug conjugates) that targets a second cell surface protein or an additional cell surface protein. In one non-limiting example, antibodies or antigen-binding fragments thereof targeting IL1RAP are used in combination with a cytotoxic agent (e.g., antibodies, immune cells expressing chimeric antigen receptors, antibody-drug conjugates) that targets a second cell surface protein or an additional cell surface protein. In some embodiments, bispecific or multi-specific antibodies may be used to target more than one epitope (e.g., more than one epitope of a lineage-specific cell surface antigen, epitopes of more than one lineage-specific cell surface antigen, an epitope of cell surface protein and an epitope of an additional cell surface antigen). See, e.g., Hoseini et al. Blood Cancer Journal (2017) 7, e552. Non-limiting examples of bispecific antibodies include tandem double scFv (e.g., single-chain bispecific tandem fragment variable (scBsTaFv), bispecific T-cell engager (BiTE), bispecific single-chain Fv (bsscFv), bispecific killer-cell engager (BiKE), dual-affinity re-targeting (DART), diabody, tandem diabodies (TandAb), single-chain Fv triplebody (sctb), bispecific scFv immunofusion (Blf), Fabsc, dual-variable- domain immunoglobulin (DVD-Ig), CrossMab (CH1-CL), modular bispecific antibody (IgG- scFv). See, e.g., Marin-Acevedo et al. J. Hematol. Oncol.(2018)11: 8; Slaney et al. Cancer Discovery (2018) 8(8): 924-934, and Elgundi et al. Advanced Drug Discovery Reviews (2017) 122: 2-19. In some embodiments, the antibody is a bispecific T-cell engager (BiTE) comprising two linked scFv molecules. In some embodiments, at least of the linked scFv of the BiTE binds an epitope of a lineage-specific cell surface protein (e.g., CD33 or IL1RAP). In one example, the BiTE is blinatumomab. See, e.g. Slaney et al. Cancer Discovery (2018) 8(8): 924-934. For example, an antibody that targets both CD33 and IL1RAP may be used in the methods described herein. Antibodies and antigen-binding fragments targeting CD33 or IL1RAP or a combination thereof can be prepared by routine practice. For example, an antibody that targets both CD44 and EMR2 may be used in the methods described herein. Antibodies and antigen-binding fragments targeting CD44 or EMR2 or a combination thereof can be prepared by routine practice. In some embodiments, a bispecific antibody may be used in which one molecule targets an epitope of a lineage-specific cell surface protein on a target cell and the other molecule targets a surface antigen on an effector cell (e.g., T cell, NK cell) such that the target cell is brought into proximity with the effector cell. See, e.g., Hoseini et al. Blood Cancer Journal (2017) 7, e552. In some embodiments, two or more (e.g., 2, 3, 4, 5 or more) epitopes of a cell surface protein have been modified, enabling two or more (e.g., 2, 3, 4, 5 or more) different cytotoxic agents (e.g., two antibodies) to be targeted to the two or more epitopes. In some embodiments, the antibodies could work synergistically to enhance efficacy. In some embodiments, epitopes of two or more (e.g., 2, 3, 4, 5 or more) cell surface protein have been modified, enabling two or more (e.g., 2, 3, 4, 5 or more) different cytotoxic agents (e.g., two antibodies) to be targeted to epitopes of the two or more cell surface proteins. In some embodiments, one or more (e.g., 1, 2, 3, 4, 5 or more) epitopes of a cell surface protein have been modified and one or more (e.g., 1, 2, 3, 4, 5 or more) epitopes of an additional cell surface protein have been modified, enabling two or more (e.g., 2, 3, 4, 5 or more) different cytotoxic agents (e.g., two antibodies) to be targeted to epitopes of the cell surface protein and epitopes of the additional cell surface protein. In some embodiments, targeting of two or more cell surface protein may reduce relapse of a hematopoietic malignancy. In some embodiments, the methods described herein involve administering a cytotoxic agent that targets an epitope of a cell surface protein that is mutated in the population of genetically engineered hematopoietic cells. In some embodiments, the methods described herein involve administering a cytotoxic agent that targets an epitope of a cell surface protein that is mutated in the population of genetically engineered hematopoietic cells and one or more additional cytotoxic agents that target one or more additional cell surface proteins. In some embodiments, the antibodies work synergistically to enhance efficacy by targeting more than one cell surface protein. In some embodiments, the methods described herein involve administering to the subject a population of genetically engineered cells lacking a non-essential epitope in a cell surface protein and one or more immunotherapeutic agents (e.g., antibodies) that target cells expressing the lineage-specific cell surface antigen. In some embodiments, the methods described herein involve administering to the subject a population of genetically engineered cells lacking a non-essential epitope in a type 1 cell surface protein and one or more immunotherapeutic agents (e.g., antibodies) that target cells expressing the lineage-specific cell surface antigen. In some embodiments, the methods described herein involve administering to the subject a population of genetically engineered cells lacking a non- essential epitope in a type 2 cell surface protein and one or more immunotherapeutic agents (e.g., antibodies) that target cells expressing the lineage-specific cell surface antigen. In any of the embodiments described herein, one or more additional immunotherapeutic agents may be further administered to the subject (e.g., targeting one or more additional epitopes and/or antigens), for example if the hematopoietic malignancy relapses. Antibody-Drug Conjugates In some embodiments, the cytotoxic agent targeting an epitope of a cell surface protein is an antibody-drug conjugate (ADC). As will be evident to one of ordinary skill in the art, the term “antibody-drug conjugate” can be used interchangeably with “immunotoxin” and refers to a fusion molecule comprising an antibody (or antigen-binding fragment thereof) conjugated to a toxin or drug molecule. Binding of the antibody to the corresponding epitope of the target protein allows for delivery of the toxin or drug molecule to a cell that presents the protein (and epitope thereof) on the cell surface (e.g., target cell), thereby resulting in death of the target cell. In some embodiments, the antibody-drug conjugate (or antigen- binding fragment thereof) binds to its corresponding epitope of a cell surface protein but does not bind to a cell surface protein that lacks the epitope or in which the epitope has been mutated. In some embodiments, the agent is an antibody-drug conjugate. In some embodiments, the antibody-drug conjugate comprises an antigen-binding fragment and a toxin or drug that induces cytotoxicity in a target cell. In some embodiments, the antibody- drug conjugate binds to an epitope in an exon of IL1RAP. In some embodiments, the antibody-drug conjugate targets a type 2 protein. In some embodiments, the antibody-drug conjugate targets CD33. In some embodiments, the antibody-drug conjugate binds to an epitope in exon 2 or exon 3 of CD33. In some embodiments, the antibody-drug conjugate targets a type 1 protein. In some embodiments, the antibody-drug conjugate targets IL1RAP. In some embodiments, the antibody-drug conjugate targets a type 0 protein. Toxins or drugs compatible for use in antibody-drug conjugates are well known in the art and will be evident to one of ordinary skill in the art. See, e.g., Peters et al. Biosci. Rep.(2015) 35(4): e00225, Beck et al. Nature Reviews Drug Discovery (2017) 16:315-337; Marin-Acevedo et al. J. Hematol. Oncol.(2018)11: 8; Elgundi et al. Advanced Drug Delivery Reviews (2017) 122: 2-19. In some embodiments, the antibody-drug conjugate may further comprise a linker (e.g., a peptide linker, such as a cleavable linker or a non-cleavable linker) attaching the antibody and drug molecule. Examples of antibody-drug conjugates include, without limitation, brentuximab vedotin, glembatumumab vedotin/CDX-011, depatuxizumab mafodotin/ABT-414, PSMA ADC, polatuzumab vedotin/RG7596/DCDS4501A, denintuzumab mafodotin/SGN-CD19A, AGS-16C3F, CDX-014, RG7841/DLYE5953A, RG7882/DMUC406A, RG7986/DCDS0780A, SGN-LIV1A, enfortumab vedotin/ASG- 22ME, AG-15ME, AGS67E, telisotuzumab vedotin/ABBV-399, ABBV-221, ABBV-085, GSK-2857916, tisotumab vedotin/HuMax-TF-ADC, HuMax-Axl-ADC, pinatuzumab veodtin/RG7593/DCDT2980S, lifastuzumab vedotin/RG7599/DNIB0600A, indusatumab vedotin/MLN-0264/TAK-264, vandortuzumab vedotin/RG7450/DSTP3086S, sofituzumab vedotin/RG7458/DMUC5754A, RG7600/DMOT4039A, RG7336/DEDN6526A, ME1547, PF-06263507/ADC 5T4, trastuzumab emtansine/T-DM1, mirvetuximab soravtansine/ IMGN853, coltuximab ravtansine/SAR3419, naratuximab emtansine/IMGN529, indatuximab ravtansine/BT-062, anetumab ravtansine/BAY 94–9343, SAR408701, SAR428926, AMG 224, PCA062, HKT288, LY3076226, SAR566658, lorvotuzumab mertansine/IMGN901, cantuzumab mertansine/SB-408075, cantuzumab ravtansine/IMGN242, laprituximab emtansine/IMGN289, IMGN388, bivatuzumab mertansine, AVE9633, BIIB015, MLN2704, AMG 172, AMG 595, LOP 628, vadastuximab talirine/SGN-CD33A, SGN-CD70A, SGN- CD19B, SGN-CD123A, SGN-CD352A, rovalpituzumab tesirine/SC16LD6.5, SC-002, SC- 003, ADCT-301/HuMax-TAC-PBD, ADCT-402, MEDI3726/ADC-401, IMGN779, IMGN632, gemtuzumab ozogamicin, inotuzumab ozogamicin/ CMC-544, PF-06647263, CMD-193, CMB-401, trastuzumab duocarmazine/SYD985, BMS-936561/MDX-1203, sacituzumab govitecan/IMMU-132, labetuzumab govitecan/IMMU-130, DS-8201a, U3- 1402, milatuzumab doxorubicin/IMMU-110/hLL1-DOX, BMS-986148, RC48- ADC/hertuzumab-vc–MMAE, PF-06647020, PF-06650808, PF-06664178/RN927C, lupartumab amadotin/ BAY1129980, aprutumab ixadotin/BAY1187982, ARX788, AGS62P1, XMT-1522, AbGn-107, MEDI4276, DSTA4637S/RG7861. In one example, the antibody- drug conjugate is gemtuzumab ozogamicin. In some embodiments, binding of the antibody-drug conjugate to the epitope of the cell surface protein induces internalization of the antibody-drug conjugate, and the drug (or toxin) may be released intracellularly. In some embodiments, binding of the antibody-drug conjugate to the epitope of a cell surface protein induces internalization of the toxin or drug, which allows the toxin or drug to kill the cells expressing the cell surface protein (target cells). In some embodiments, binding of the antibody-drug conjugate to the epitope of a cell surface protein induces internalization of the toxin or drug, which may regulate the activity of the cell expressing the cell surface protein (target cells). The type of toxin or drug used in the antibody-drug conjugates described herein is not limited to any specific type. In some embodiments, two or more (e.g., 2, 3, 4, 5 or more) epitopes of a cell surface protein have been modified, enabling two or more (e.g., 2, 3, 4, 5 or more) different cytotoxic agents (e.g., two ADCs) to be targeted to the two or more epitopes. In some embodiments, the toxins carried by the ADCs could work synergistically to enhance efficacy (e.g., death of the target cells). In some embodiments, epitopes of two or more (e.g., 2, 3, 4, 5 or more) lineage-specific cell surface protein have been modified, enabling two or more (e.g., 2, 3, 4, 5 or more) different cytotoxic agents (e.g., two ADCs) to be targeted to epitopes of the two or more cell surface proteins. In some embodiments, one or more (e.g., 1, 2, 3, 4, 5 or more) epitopes of a cell surface protein have been modified and one or more (e.g., 1, 2, 3, 4, 5 or more) epitopes of an additional cell surface protein have been modified, enabling two or more (e.g., 2, 3, 4, 5 or more) different cytotoxic agents (e.g., two ADCs) to be targeted to epitopes of the cell surface protein and epitopes of additional cell surface antigen. In some embodiments, targeting of more than one cell surface protein or a cell surface protein and one or more additional cell surface protein/antigen may reduce relapse of a hematopoietic malignancy. In some embodiments, the methods described herein involve administering ADCs that target an epitope of a cell surface protein that is mutated in the population of genetically engineered hematopoietic cells. In some embodiments, the methods described herein involve administering ADCs that target an epitope of a cell surface protein that is mutated in the population of genetically engineered hematopoietic cells and one or more additional cytotoxic agents that may target one or more additional cell surface proteins. In some embodiments, the agents could work synergistically to enhance efficacy by targeting more than one cell surface protein. An ADC described herein may be used as a follow-on treatment to subjects who have been undergone the combined therapy as described herein. In some embodiments, the methods described herein involve administering to the subject a population of genetically engineered cells lacking a non-essential epitope in a cell surface protein and one or more immunotherapeutic agents (e.g., ADCs) that target cells expressing the lineage-specific cell surface antigen. In some embodiments, the methods described herein involve administering to the subject a population of genetically engineered cells lacking a non-essential epitope in a type 1 cell surface protein and one or more immunotherapeutic agents (e.g., ADCs) that target cells expressing the lineage-specific cell surface antigen. In some embodiments, the methods described herein involve administering to the subject a population of genetically engineered cells lacking a non-essential epitope in a type 2 cell surface protein and one or more immunotherapeutic agents (e.g., ADCs) that target cells expressing the lineage-specific cell surface antigen. In any of the embodiments described herein, one or more additional immunotherapeutic agents may be further administered to the subject (e.g., targeting one or more additional epitopes and/or antigens), for example if the hematopoietic malignancy relapses. In some examples, the methods described herein involve administering to the subject a population of genetically engineered cells lacking a non-essential epitope of CD33 and one or more antibody-drug conjugates that target cells expressing CD33. In some examples, the methods described herein involve administering to the subject a population of genetically engineered cells lacking an epitope in exon 2 or exon 3 of CD33 and one or more antibodies antibody-drug conjugates that target cells expressing CD33. In some examples, the methods described herein involve administering to the subject a population of genetically engineered cells expressing a mutated CD33 comprising the amino acid sequence of SEQ ID NO: 56 or SEQ ID NO: 58 and one or more antibody-drug conjugates that target cells expressing CD33. In some examples, the methods described herein involve administering to the subject a population of genetically engineered cells lacking a non-essential epitope of IL1RAP and one or more antibody-drug conjugates that target cells expressing IL1RAP. In some examples, the methods described herein involve administering to the subject a population of genetically engineered cells lacking an epitope in and of IL1RAP and one or more antibody-drug conjugates that target cells expressing IL1RAP. Immune Cells Expressing Chimeric Antigen Receptors In some embodiments, the cytotoxic agent that targets an epitope of a cell surface protein as described herein is an immune cell that expresses a chimeric receptor, which comprises an antigen-binding fragment (e.g., a single-chain antibody) capable of binding to the epitope of the cell surface protein (e.g., CD33 or IL1RAP). Recognition of a target cell (e.g., a cancer cell) having the epitope of the cell surface protein on its cell surface by the antigen-binding fragment of the chimeric receptor transduces an activation signal to the signaling domain(s) (e.g., co-stimulatory signaling domain and/or the cytoplasmic signaling domain) of the chimeric receptor, which may activate an effector function in the immune cell expressing the chimeric receptor. In some embodiments, the immune cell expresses more than one chimeric receptor (e.g., 2, 3, 4, 5 or more), referred to as a bispecific or multi- specific immune cell. In some embodiments, the immune cell expresses more than one chimeric receptor, at least one of which targets an epitope of a lineage-specific cell surface antigen. In some embodiments, the immune cell expresses more than one chimeric receptor, each of which targets an epitope of a lineage-specific cell surface antigen. In some embodiments, the immune cell expresses more than one chimeric receptor, at least one of which targets an epitope of a cell surface protein and at least one of which targets an epitope of an additional cell surface antigen. In some embodiments, targeting of more than one lineage-specific cell surface protein or a lineage-specific cell surface protein and one or more additional cell surface protein may reduce relapse of a hematopoietic malignancy. In some embodiments, the immune cell expresses a chimeric receptor that targets more than one epitopes (e.g., more than one epitopes of one cell surface protein or epitopes of more than one cell surface protein), referred to as a bispecific chimeric receptor. In some embodiments, epitopes of two or more lineage-specific cell surface proteins are targeted by cytotoxic agents. In some embodiments, two or more chimeric receptors are expressed in the same immune cell, e.g., bispecific chimeric receptors. Such cells can be used in any of the methods described herein. In some embodiments, cells expressing a chimeric receptor are “pooled,, i.e., two or more groups of cells express two or more different chimeric receptors. In some embodiments, two or more cells expressing different chimeric antigen receptors are administered concurrently. In some embodiments, two or more cells expressing different chimeric antigen receptors are administered sequentially. In some embodiments, epitopes of CD33 and IL1RAP are targeted by cytotoxic agents. In some embodiments, the chimeric receptors targeting CD33 and IL1RAP are expressed in the same immune cell (i.e., a bispecific immune cell). Such cells can be used in any of the methods described herein. In some embodiments, cells expressing chimeric receptors targeting CD33 and IL1RAP “pooled,” i.e., two or more groups of cells express two or more different chimeric receptors. In some embodiments, two or more groups of cells expressing chimeric receptors targeting CD33 and IL1RAP are administered concurrently. In some embodiments, two or more groups of cells expressing chimeric receptors targeting CD33 and IL1RAP are administered sequentially. In some embodiments, epitopes of CD44 and EMR2 are targeted by cytotoxic agents. In some embodiments, the chimeric receptors targeting CD44 and EMR2 are expressed in the same immune cell (i.e., a bispecific immune cell). Such cells can be used in any of the methods described herein. In some embodiments, cells expressing chimeric receptors targeting CD44 and EMR2 “pooled,” i.e., two or more groups of cells express two or more different chimeric receptors. In some embodiments, two or more groups of cells expressing chimeric receptors targeting CD44 and EMR2 are administered concurrently. In some embodiments, two or more groups of cells expressing chimeric receptors targeting CD44 and EMR2 are administered sequentially. As used herein, a chimeric receptor refers to a non-naturally occurring molecule that can be expressed on the surface of a host cell and comprises binding domain that provides specificity of the chimeric receptor (e.g., an antigen-binding fragment that binds to an epitope of a cell surface protein). In general, chimeric receptors comprise at least two domains that are derived from different molecules. In addition to the epitope-binding fragment described herein, the chimeric receptor may further comprise one or more of the following: a hinge domain, a transmembrane domain, a co-stimulatory domain, a cytoplasmic signaling domain, and combinations thereof. In some embodiments, the chimeric receptor comprises from N terminus to C terminus, an antigen-binding fragment that binds to a cell surface protein, a hinge domain, a transmembrane domain, and a cytoplasmic signaling domain. In some embodiments, the chimeric receptor further comprises at least one co-stimulatory domain. See, e.g., Marin-Acevedo et al. J. Hematol. Oncol.(2018)11: 8. Alternatively or in addition, the chimeric receptor may be a switchable chimeric receptor. See, e.g., Rodger et al. PNAS (2016) 113: 459-468; Cao et al. Angew. Chem. Int. Ed. (2016) 55: 7520-7524. In general, a switchable chimeric receptor comprises a binding domain that binds a soluble antigen-binding fragment, which has antigen binding specificity and may be administered concomitantly with the immune cells. In some embodiments, the chimeric receptor may be a masked chimeric receptor, which is maintained in an “off” state until the immune cell expressing the chimeric receptor is localized to a desired location in the subject. For example, the binding domain of the chimeric receptor (e.g., antigen-binding fragment) may be blocked by an inhibitory peptide that is cleaved by a protease present at a desired location in the subject. In some embodiments, it may be advantageous to modulate the binding affinity of the binding domain (e.g., antigen-binding fragment). For example, in some instances, relapse of hematopoietic malignancies results due to the reduced expression of the targeted cell surface protein on the surface of target cells (e.g., antigen escape) and the lower levels of cell surface protein any be inefficient or less efficient in stimulating cytotoxicity of the target cells. See, e.g., Majzner et al. Cancer Discovery (2018) 8(10). In some embodiments, the binding affinity of the binding domain (e.g., antigen-binding fragment) may be enhanced, for example by mutating one or more amino acid residues of the binding domain. Binding domains having enhanced binding affinity to a cell surface protein may result in immune cells that response to lower levels of cell surface protein (lower cell surface protein density) and reduce or prevent relapse. Hinge Domains of Chimeric Antigen Receptors In some embodiments, the chimeric receptors described herein comprise one or more hinge domain(s). In some embodiments, the hinge domain may be located between the antigen-binding fragment and a transmembrane domain. A hinge domain is an amino acid segment that is generally found between two domains of a protein and may allow for flexibility of the protein and movement of one or both of the domains relative to one another. Any amino acid sequence that provides such flexibility and movement of the antigen-binding fragment relative to another domain of the chimeric receptor can be used. The hinge domain may contain about 10-200 amino acids, e.g., 15-150 amino acids, 20-100 amino acids, or 30-60 amino acids. In some embodiments, the hinge domain may be of about 10, 11, 12, 13, 14,15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 35, 40, 45, 50, 55, 60, 65, 70, 75, 80, 85, 90, 95, 100, 110, 120, 130, 140, 150, 160, 170, 180, 190, or 200 amino acids in length. In some embodiments, the hinge domain is a hinge domain of a naturally occurring protein. Hinge domains of any protein known in the art to comprise a hinge domain are compatible for use in the chimeric receptors described herein. In some embodiments, the hinge domain is at least a portion of a hinge domain of a naturally occurring protein and confers flexibility to the chimeric receptor. In some embodiments, the hinge domain is of CD8α or CD28. In some embodiments, the hinge domain is a portion of the hinge domain of CD8α, e.g., a fragment containing at least 15 (e.g., 20, 25, 30, 35, or 40) consecutive amino acids of the hinge domain of CD8α or CD28. Hinge domains of antibodies, such as an IgG, IgA, IgM, IgE, or IgD antibody, are also compatible for use in the chimeric receptors described herein. In some embodiments, the hinge domain is the hinge domain that joins the constant domains CH1 and CH2 of an antibody. In some embodiments, the hinge domain is of an antibody and comprises the hinge domain of the antibody and one or more constant regions of the antibody. In some embodiments, the hinge domain comprises the hinge domain of an antibody and the CH3 constant region of the antibody. In some embodiments, the hinge domain comprises the hinge domain of an antibody and the CH2 and CH3 constant regions of the antibody. In some embodiments, the antibody is an IgG, IgA, IgM, IgE, or IgD antibody. In some embodiments, the antibody is an IgG antibody. In some embodiments, the antibody is an IgG1, IgG2, IgG3, or IgG4 antibody. In some embodiments, the hinge region comprises the hinge region and the CH2 and CH3 constant regions of an IgG1 antibody. In some embodiments, the hinge region comprises the hinge region and the CH3 constant region of an IgG1 antibody. Also within the scope of the present disclosure are chimeric receptors comprising a hinge domain that is a non-naturally occurring peptide. In some embodiments, the hinge domain between the C-terminus of the extracellular ligand-binding domain of an Fc receptor and the N-terminus of the transmembrane domain is a peptide linker, such as a (GlyxSer)n linker, wherein x and n, independently can be an integer between 3 and 12, including 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, or more. Additional peptide linkers that may be used in a hinge domain of the chimeric receptors described herein are known in the art. See, e.g., Wriggers et al. Current Trends in Peptide Science (2005) 80(6): 736-746 and PCT Publication WO 2012/088461. Transmembrane Domains of Chimeric Antigen Receptors In some embodiments, the chimeric receptors described herein may comprise one or more transmembrane domain(s). The transmembrane domain for use in the chimeric receptors can be in any form known in the art. As used herein, a “transmembrane domain” refers to any protein structure that is thermodynamically stable in a cell membrane, preferably a eukaryotic cell membrane. Transmembrane domains compatible for use in the chimeric receptors used herein may be obtained from a naturally occurring protein. Alternatively, the transmembrane domain may be a synthetic, non-naturally occurring protein segment, e.g., a hydrophobic protein segment that is thermodynamically stable in a cell membrane. Transmembrane domains are classified based on the transmembrane domain topology, including the number of passes that the transmembrane domain makes across the membrane and the orientation of the protein. For example, single-pass membrane proteins cross the cell membrane once, and multi-pass membrane proteins cross the cell membrane at least twice (e.g., 2, 3, 4, 5, 6, 7 or more times). In some embodiments, the transmembrane domain is a single-pass transmembrane domain. In some embodiments, the transmembrane domain is a single-pass transmembrane domain that orients the N terminus of the chimeric receptor to the extracellular side of the cell and the C terminus of the chimeric receptor to the intracellular side of the cell. In some embodiments, the transmembrane domain is obtained from a single pass transmembrane protein. In some embodiments, the transmembrane domain is of CD8α. In some embodiments, the transmembrane domain is of CD28. In some embodiments, the transmembrane domain is of ICOS. Costimulatory and Signaling Domains of Chimeric Antigen Receptors In some embodiments, the chimeric receptors described herein comprise one or more costimulatory signaling domains. The term “co-stimulatory signaling domain,” as used herein, refers to at least a portion of a protein that mediates signal transduction within a cell to induce an immune response, such as an effector function. The co-stimulatory signaling domain of the chimeric receptor described herein can be a cytoplasmic signaling domain from a co-stimulatory protein, which transduces a signal and modulates responses mediated by immune cells, such as T cells, NK cells, macrophages, neutrophils, or eosinophils. In some embodiments, the chimeric receptor comprises more than one (at least 2, 3, 4, or more) co-stimulatory signaling domains. In some embodiments, the chimeric receptor comprises more than one co-stimulatory signaling domains obtained from different costimulatory proteins. In some embodiments, the chimeric receptor does not comprise a co- stimulatory signaling domain. In general, many immune cells require co-stimulation, in addition to stimulation of an antigen-specific signal, to promote cell proliferation, differentiation and survival, and to activate effector functions of the cell. Activation of a co-stimulatory signaling domain in a host cell (e.g., an immune cell) may induce the cell to increase or decrease the production and secretion of cytokines, phagocytic properties, proliferation, differentiation, survival, and/or cytotoxicity. The co-stimulatory signaling domain of any co-stimulatory protein may be compatible for use in the chimeric receptors described herein. The type(s) of co-stimulatory signaling domain is selected based on factors such as the type of the immune cells in which the chimeric receptors would be expressed (e.g., primary T cells, T cell lines, NK cell lines) and the desired immune effector function (e.g., cytotoxicity). Examples of co-stimulatory signaling domains for use in the chimeric receptors can be the cytoplasmic signaling domain of co-stimulatory proteins, including, without limitation, CD27, CD28, 4-1BB, OX40, CD30, ICOS, CD2, CD7, LIGHT, NKG2C, B7-H3. In some embodiments, the co-stimulatory domain is derived from 4-1BB, CD28, or ICOS. In some embodiments, the costimulatory domain is derived from CD28 and chimeric receptor comprises a second co-stimulatory domain from 4-1BB or ICOS. In some embodiments, the costimulatory domain is a fusion domain comprising more than one costimulatory domain or portions of more than one costimulatory domains. In some embodiments, the costimulatory domain is a fusion of costimulatory domains from CD28 and ICOS. In some embodiments, the chimeric receptors described herein comprise one or more cytoplasmic signaling domain(s). Any cytoplasmic signaling domain can be used in the chimeric receptors described herein. In general, a cytoplasmic signaling domain relays a signal, such as interaction of an extracellular ligand-binding domain with its ligand, to stimulate a cellular response, such as inducing an effector function of the cell (e.g., cytotoxicity). As will be evident to one of ordinary skill in the art, a factor involved in T cell activation is the phosphorylation of immunoreceptor tyrosine-based activation motif (ITAM) of a cytoplasmic signaling domain. Any ITAM-containing domain known in the art may be used to construct the chimeric receptors described herein. In general, an ITAM motif may comprise two repeats of the amino acid sequence YxxL/I separated by 6-8 amino acids, wherein each x is independently any amino acid, producing the conserved motif YxxL/Ix(6- 8)YxxL/I. In some embodiments, the cytoplasmic signaling domain is from CD3ζ. Exemplary Uses and Methods Related to Chimeric Antigen Receptors In some embodiments, the chimeric receptor described herein targets a type 2 protein. In some embodiments, the chimeric receptor targets CD33. In some embodiments, the chimeric receptor described herein targets a type 1 protein. In some embodiments, the chimeric receptor targets IL1RAP. In some embodiments, the chimeric receptor targets a type 0 protein. Such a chimeric receptor may comprise an antigen-binding fragment (e.g., an scFv) comprising a heavy chain variable region and a light chain variable region that bind to IL1RAP. Alternatively, the chimeric receptor may comprise an antigen-binding fragment (e.g., scFv) comprising a heavy chain variable region and a light chain variable region that bind to CD33. In some embodiments, the immune cells described herein express a chimeric receptor (e.g., bispecific chimeric receptor) that targets a type 2 protein and a chimeric receptor that targets a type 1 protein. In some embodiments, the immune cells described herein express a chimeric receptor (e.g., bispecific chimeric receptor) that targets a type 2 protein and at least one additional cell surface protein, such as any of those described herein. In some embodiments, the immune cells described herein express a chimeric receptor that targets CD33 and at least one additional cell surface protein, such as any of those described herein. In some embodiments, the immune cells described herein express a chimeric receptor that targets a type 1 protein and at least one additional cell surface protein, such as any of those described herein. In some embodiments, the immune cells described herein express a chimeric receptor that targets IL1RAP and at least one additional cell surface protein, such as any of those described herein. In some embodiments, the chimeric receptor described herein targets CD33 and IL1RAP. In some embodiments, the immune cells described herein express a chimeric receptor that targets EMR2 and at least one additional cell surface protein, such as any of those described herein. In some embodiments, the chimeric receptor described herein targets CD44 and EMR2. In some embodiments, the immune cells described herein express a chimeric receptor that targets a type 2 protein and at least one additional chimeric receptor that targets an additional cell surface protein, such as any of those described herein. In some embodiments, the immune cells described herein express a chimeric receptor that targets CD33 and at least one additional chimeric receptor that targets another cell surface protein, such as any of those described herein. In some embodiments, the immune cells described herein express a chimeric receptor that targets a type 1 protein and at least one additional chimeric receptor that targets another cell surface protein, such as any of those described herein. In some embodiments, the immune cells described herein express a chimeric receptor that targets IL1RAP and at least one additional chimeric receptor that targets another cell surface protein, such as any of those described herein. In some embodiments, the immune cells express a chimeric receptor that targets CD33 and a chimeric receptor that targets IL1RAP. A chimeric receptor construct targeting CD33 and/or IL1RAP may further comprise at least a hinge domain (e.g., from CD28, CD8α, or an antibody), a transmembrane domain (e.g., from CD8α, CD28 or ICOS), one or more co-stimulatory domains (from one or more of CD28, ICOS, or 4-1BB) and a cytoplasmic signaling domain (e.g., from CD3ζ), or a combination thereof. In some examples, the methods described herein involve administering to a subject a population of genetically engineered hematopoietic cells and an immune cell expressing a chimeric receptor that targets CD33 and/or IL1RAP, which may further comprise at least a hinge domain (e.g., from CD28, CD8α, or an antibody), a transmembrane domain (e.g., from CD8α, CD28 or ICOS), one or more co-stimulatory domains (from one or more of CD28, ICOS, or 4-1BB) and a cytoplasmic signaling domain (e.g., from CD3ζ), or a combination thereof Any of the chimeric receptors described herein can be prepared by routine methods, such as recombinant technology. Methods for preparing the chimeric receptors herein involve generation of a nucleic acid that encodes a polypeptide comprising each of the domains of the chimeric receptors, including the antigen-binding fragment and optionally, the hinge domain, the transmembrane domain, at least one co-stimulatory signaling domain, and the cytoplasmic signaling domain. In some embodiments, nucleic acids encoding the components of a chimeric receptor are joined together using recombinant technology. Sequences of each of the components of the chimeric receptors may be obtained via routine technology, e.g., PCR amplification from any one of a variety of sources known in the art. In some embodiments, sequences of one or more of the components of the chimeric receptors are obtained from a human cell. Alternatively, the sequences of one or more components of the chimeric receptors can be synthesized. Sequences of each of the components (e.g., domains) can be joined directly or indirectly (e.g., using a nucleic acid sequence encoding a peptide linker) to form a nucleic acid sequence encoding the chimeric receptor, using methods such as PCR amplification or ligation. Alternatively, the nucleic acid encoding the chimeric receptor may be synthesized. In some embodiments, the nucleic acid is DNA. In other embodiments, the nucleic acid is RNA. Mutation of one or more residues within one or more of the components of the chimeric receptor (e.g., the antigen-binding fragment, etc) may be performed prior to or after joining the sequences of each of the components. In some embodiments, one or more mutations in a component of the chimeric receptor may be made to modulate (increase or decrease) the affinity of the component for an epitope (e.g., the antigen-binding fragment for the target protein) and/or modulate the activity of the component. Any of the chimeric receptors described herein can be introduced into a suitable immune cell for expression via conventional technology. In some embodiments, the immune cells are T cells, such as primary T cells or T cell lines. Alternatively, the immune cells can be NK cells, such as established NK cell lines (e.g., NK-92 cells). In some embodiments, the immune cells are T cells that express CD8 (CD8+) or CD8 and CD4 (CD8+/CD4+). In some embodiments, the T cells are T cells of an established T cell line, for example, 293T cells or Jurkat cells. Primary T cells may be obtained from any source, such as peripheral blood mononuclear cells (PBMCs), bone marrow, tissues such as spleen, lymph node, thymus, or tumor tissue. A source suitable for obtaining the type of immune cells desired would be evident to one of skill in the art. In some embodiments, the population of immune cells is derived from a human patient having a hematopoietic malignancy, such as from the bone marrow or from PBMCs obtained from the patient. In some embodiments, the population of immune cells is derived from a healthy donor. In some embodiments, the immune cells are obtained from the subject to whom the immune cells expressing the chimeric receptors will be subsequently administered. Immune cells that are administered to the same subject from which the cells were obtained are referred to as autologous cells, whereas immune cells that are obtained from a subject who is not the subject to whom the cells will be administered are referred to as allogeneic cells. The type of host cells desired may be expanded within the population of cells obtained by co-incubating the cells with stimulatory molecules, for example, anti-CD3 and anti-CD28 antibodies may be used for expansion of T cells. To construct the immune cells that express any of the chimeric receptor constructs described herein, expression vectors for stable or transient expression of the chimeric receptor construct may be constructed via conventional methods as described herein and introduced into immune host cells. For example, nucleic acids encoding the chimeric receptors may be cloned into a suitable expression vector, such as a viral vector in operable linkage to a suitable promoter. The nucleic acids and the vector may be contacted, under suitable conditions, with a restriction enzyme to create complementary ends on each molecule that can pair with each other and be joined with a ligase. Alternatively, synthetic nucleic acid linkers can be ligated to the termini of the nucleic acid encoding the chimeric receptors. The synthetic linkers may contain nucleic acid sequences that correspond to a particular restriction site in the vector. The selection of expression vectors/plasmids/viral vectors would depend on the type of host cells for expression of the chimeric receptors, but should be suitable for integration and replication in eukaryotic cells. In some embodiments, the chimeric receptors are expressed using a non-integrating transient expression system. In some embodiments, the chimeric receptors are integrated into the genome of the immune cell. In some embodiments, the chimeric receptors are integrated into a specific loci of the genome of immune cell using gene editing (e.g., zinc-finger nucleases, meganucleases, TALENs, CRISPR systems). A variety of promoters can be used for expression of the chimeric receptors described herein, including, without limitation, cytomegalovirus (CMV) intermediate early promoter, a viral LTR such as the Rous sarcoma virus LTR, HIV-LTR, HTLV-1 LTR, Maloney murine leukemia virus (MMLV) LTR, myeloproliferative sarcoma virus (MPSV) LTR, spleen focus- forming virus (SFFV) LTR, the simian virus 40 (SV40) early promoter, herpes simplex tk virus promoter, elongation factor 1-alpha (EF1-α) promoter with or without the EF1-α intron. Additional promoters for expression of the chimeric receptors include any constitutively active promoter in an immune cell. Alternatively, any regulatable promoter may be used, such that its expression can be modulated within an immune cell. In some embodiments, the promoter regulating expression of a chimeric receptor is an inducible promoter. In general, the activity of “inducible promoters” may be regulated based on the presence (or absence) of a signal, such as an endogenous signal or an exogenous signal, for example a small molecule or drug that is administered to the subject. Additionally, the vector may contain, for example, some or all of the following: a selectable marker gene, such as the neomycin gene for selection of stable or transient transfectants in host cells; enhancer/promoter sequences from the immediate early gene of human CMV for high levels of transcription; transcription termination and RNA processing signals from SV40 for mRNA stability; 5’-and 3’-untranslated regions for mRNA stability and translation efficiency from highly-expressed genes like α-globin or β-globin; SV40 polyoma origins of replication and ColE1 for proper episomal replication; internal ribosome binding sites (IRESes), versatile multiple cloning sites; T7 and SP6 RNA promoters for in vitro transcription of sense and antisense RNA; a “suicide switch” or “suicide gene” which when triggered causes cells carrying the vector to die (e.g., HSV thymidine kinase, an inducible caspase such as iCasp9, drug-induced suicide switch, monoclonal antibody mediated suicide switch), and reporter gene for assessing expression of the chimeric receptor. Suitable vectors and methods for producing vectors containing transgenes are well known and available in the art. Examples of the preparation of vectors for expression of chimeric receptors can be found, for example, in US2014/0106449, herein incorporated by reference in its entirety. As will be appreciated by one of ordinary skill in the art, in some embodiments, it may be advantageous to quickly and efficiently reduce or eliminate the immune cells expressing chimeric receptors, for example at a time point following administration to a subject. Mechanisms of killing immune cells expressing chimeric receptors, or inducing cytotoxicity of such cells, are known in the art, see, e.g., Labanieh et al. Nature Biomedical Engineering (2018)2: 337-391; Slaney et al. Cancer Discovery (2018) 8(8): 924-934. In some embodiments, the immune cell expressing the chimeric receptor may also express a suicide switch” or “suicide gene,” which may or may not be encoded by the same vector as the chimeric receptor, as described above. In some embodiments, the immune cell expressing chimeric receptors further express an epitope tag such that upon engagement of the epitope tag, the immune cell is killed through antibody-dependent cell-mediated cytotoxicity and/or complement-mediated cytotoxicity. See, e.g., Paszkiewicz et al. J. Clin. Invest. (2016) 126: 4262-4272; Wang et al. Blood (2011) 118: 1255-1263; Tasian et al. Blood (2017) 129: 2395- 2407; Philip et al. Blood (2014) 124: 1277-1287. In some embodiments, the chimeric receptor construct or the nucleic acid encoding said chimeric receptor is a DNA molecule. In some embodiments, chimeric receptor construct or the nucleic acid encoding said chimeric receptor is a DNA vector and may be electroporated to immune cells (see, e.g., Till, et al. Blood (2012) 119(17): 3940-3950). In some embodiments, the nucleic acid encoding the chimeric receptor is an RNA molecule, which may be electroporated to immune cells. Any of the vectors comprising a nucleic acid sequence that encodes a chimeric receptor construct described herein is also within the scope of the present disclosure. Such a vector may be delivered into host cells such as host immune cells by a suitable method. Methods of delivering vectors to immune cells are well known in the art and may include DNA, RNA, or transposon electroporation, transfection reagents such as liposomes or nanoparticles to delivery DNA, RNA, or transposons; delivery of DNA, RNA, or transposons or protein by mechanical deformation (see, e.g., Sharei et al. Proc. Natl. Acad. Sci. USA (2013) 110(6): 2082-2087); or viral transduction. In some embodiments, the vectors for expression of the chimeric receptors are delivered to host cells by viral transduction. Exemplary viral methods for delivery include, but are not limited to, recombinant retroviruses (see, e.g., PCT Publication Nos. WO 90/07936; WO 94/03622; WO 93/25698; WO 93/25234; WO 93/11230; WO 93/10218; WO 91/02805; U.S. Pat. Nos.5,219,740 and 4,777,127; GB Patent No.2,200,651; and EP Patent No.0345242), alphavirus-based vectors, and adeno- associated virus (AAV) vectors (see, e.g., PCT Publication Nos. WO 94/12649, WO 93/03769; WO 93/19191; WO 94/28938; WO 95/11984 and WO 95/00655). In some embodiments, the vectors for expression of the chimeric receptors are retroviruses. In some embodiments, the vectors for expression of the chimeric receptors are lentiviruses. In some embodiments, the vectors for expression of the chimeric receptors are adeno-associated viruses. In examples in which the vectors encoding chimeric receptors are introduced to the host cells using a viral vector, viral particles that are capable of infecting the immune cells and carry the vector may be produced by any method known in the art and can be found, for example in PCT Application No. WO 1991/002805A2, WO 1998/009271 A1, and U.S. Patent 6,194,191. The viral particles are harvested from the cell culture supernatant and may be isolated and/or purified prior to contacting the viral particles with the immune cells. In some embodiments, the methods described herein involve use of immune cells that express more than one chimeric receptor (e.g., chimeric receptors that target first epitope and chimeric receptors that target a second epitope). In some embodiments, more than one chimeric receptor (e.g., chimeric receptors that target first epitope and chimeric receptors that target a second epitope) are expressed from a single vector. In some embodiments, more than one chimeric receptor (e.g., chimeric receptors that target first epitope and chimeric receptors that target a second epitope) are expressed from a more than one vector. Such immune cells may be prepared using methods known in the art, for example by delivering a vector that encodes a first chimeric receptor simultaneously or sequentially with a second vector that encodes a second chimeric receptor. In some embodiments, the resulting immune cell population is a mixed population comprising a subset of immune cells that express one chimeric receptor and a subset of immune cells that express both chimeric receptors. In some embodiments, the domains of the chimeric receptor are encoded by and expressed from a single vector. Alternatively, the domains of the chimeric receptor may be encoded by and expressed from more than one vector. In some embodiments, activity of immune cells expressing the chimeric receptors may be regulated by controlling assembly of the chimeric receptor. The domains of a chimeric receptor may be expressed as two or more non-functional segments of the chimeric receptor and induced to assemble at a desired time and/or location, for example through use of a dimerization agent (e.g., dimerizing drug). The methods of preparing host cells expressing any of the chimeric receptors described herein may comprise activating and/or expanding the immune cells ex vivo. Activating a host cell means stimulating a host cell into an activate state in which the cell may be able to perform effector functions (e.g., cytotoxicity). Methods of activating a host cell will depend on the type of host cell used for expression of the chimeric receptors. Expanding host cells may involve any method that results in an increase in the number of cells expressing chimeric receptors, for example, allowing the host cells to proliferate or stimulating the host cells to proliferate. Methods for stimulating expansion of host cells will depend on the type of host cell used for expression of the chimeric receptors and will be evident to one of skill in the art. In some embodiments, the host cells expressing any of the chimeric receptors described herein are activated and/or expanded ex vivo prior to administration to a subject. It has been appreciated that administration of immunostimulatory cytokines may enhance proliferation of immune cells expressing chimeric receptors following administration to a subject and/or promote engraftment of the immune cells. See, e.g., Pegram et al. Leukemia (2015) 29:415-422; Chinnasamy et al. Clin. Cancer Res. (2012) 18: 1672-1683; Krenciute et al. Cancer Immunol. Res. (2017) 5: 571-581; Hu et al. Cell Rep. (2017) 20: 3025-3033; Markley et al. Blood (2010) 115: 3508-3519). Any of the methods described herein may further involve administering one or more immunostimulatory cytokines concurrently with any of the immune cells expressing chimeric receptors. Non-limiting example of immunostimulatory cytokines include IL-12, IL-15, IL-1, IL-21, and combinations thereof. Any anti- IL1RAP CAR and anti-CD33 CAR molecules known in the art can be used together with the genetically engineered HSCs described herein. Exemplary anti- IL1RAP CARs include axicabtagene and tisagenlecleucel. Exemplary anti-CD33 CARs include those disclosed in PCT Publication Nos. WO2017/066760 and WO2017/079400. In one specific example, primary human CD8+ T cells are isolated from patients’ peripheral blood by immunomagnetic separation (Miltenyi Biotec). T cells are cultured and stimulated with anti-CD3 and anti-CD28 mAbs–coated beads (Invitrogen) as previously described (Levine et al., J. Immunol. (1997) 159(12):5921). Chimeric receptors that target a lineage-specific cell surface proteins (e.g., IL1RAP or CD33) are generated using conventional recombinant DNA technologies and inserted into a lentiviral vector. The vectors containing the chimeric receptors are used to generate lentiviral particles, which are used to transduce primary CD8+ T cells. Human recombinant IL-2 may be added every other day (50 IU/mL). T cells are cultured for ~14 days after stimulation. Expression of the chimeric receptors can be confirmed using methods, such as Western blotting and flow cytometry. T cells expressing the chimeric receptors are selected and assessed for their ability to bind to the lineage-specific cell surface protein such as IL1RAP or CD33 and to induce cytotoxicity of cells expressing the cell surface protein. Immune cells expressing the chimeric receptor are also evaluated for their ability to induce cytotoxicity of cells expressing the cell surface protein in mutated form. Preferably, immune cells expressing chimeric receptors that binds to the wild-type cell surface protein but not the mutated form. (FIGURE 3, using CD33 as an example). The cells (e.g., hematopoietic stem cells) that express the mutated cell surface protein are also assessed for various characteristics, including proliferation, erythropoietic differentiation, and colony formation to confirm that the mutated cell surface protein retained the bioactivity as the wild-type counterpart. The immune cells expressing one or more CAR-T receptors may be further modified genetically, for example, by knock-out of the native T-cell receptor (TCR) or an MHC chain and/or by introducing a new TCR. Alternatively, the immune cells may retain the native TCR loci. In some embodiments, the methods described herein involve administering to the subject a population of genetically engineered cells lacking a non-essential epitope in a cell surface protein and one or more immunotherapeutic agents (e.g., immune cells expressing chimeric receptor(s)) that target cells expressing the lineage-specific cell surface antigen. In some embodiments, the methods described herein involve administering to the subject a population of genetically engineered cells lacking a non-essential epitope in a type 1 cell surface protein and one or more immunotherapeutic agents (e.g., immune cells expressing chimeric receptor(s)) that target cells expressing the lineage-specific cell surface antigen. In some embodiments, the methods described herein involve administering to the subject a population of genetically engineered cells lacking a non-essential epitope in a type 2 cell surface protein and one or more immunotherapeutic agents (e.g., immune cells expressing chimeric receptor(s)) that target cells expressing the lineage-specific cell surface antigen. In any of the embodiments described herein, one or more additional immunotherapeutic agents may be further administered to the subject (e.g., targeting one or more additional epitopes and/or antigens), for example if the hematopoietic malignancy relapses. In some embodiments, the methods described herein involve administering immune cells expressing chimeric receptors that target an epitope of a lineage-specific cell surface protein that is mutated in the population of genetically engineered hematopoietic cells. In some embodiments, the methods described herein involve administering immune cells expressing chimeric receptors that target an epitope of a lineage-specific cell surface protein that is mutated in the population of genetically engineered hematopoietic cells and one or more additional cytotoxic agents that may target one or more additional cell surface proteins. In some embodiments, the agents could work synergistically to enhance efficacy by targeting more than one cell surface protein. In some examples, the methods described herein involve administering to the subject a population of genetically engineered cells lacking a non-essential epitope of CD33 and one or more immune cells expressing chimeric receptor(s) that target cells expressing CD33. In some examples, the methods described herein involve administering to the subject a population of genetically engineered cells lacking an epitope in exon 2 or exon 3 of CD33 and one or more immune cells expressing chimeric receptor(s) that target cells expressing CD33. In some examples, the methods described herein involve administering to the subject a population of genetically engineered cells expressing a mutated CD33 comprising the amino acid sequence of SEQ ID NO: 56 or SEQ ID NO: 58 and one or more immune cells expressing chimeric receptor(s) that target cells expressing CD33. In some examples, the methods described herein involve administering to the subject a population of genetically engineered cells lacking a non-essential epitope of IL1RAP and one or more immune cells expressing chimeric receptor(s) that target cells expressing IL1RAP. In some examples, the methods described herein involve administering to the subject a population of genetically engineered cells lacking an epitope in an exon of IL1RAP and one or more immune cells expressing chimeric receptor(s) that target cells expressing IL1RAP. IV. Methods of Treatment and Combination Therapies The genetically engineered hematopoietic cells such as HSCs may be administered to a subject in need of the treatment, either taken alone or in combination of one or more cytotoxic agents that target one or more antigens as described herein. Since the hematopoietic cells are genetically edited in the genes of the one or more antigens, the hematopoietic cells and/or descendant cells thereof would express the one or more antigens in mutated form (e.g., but functional) such that they can escape being targeted by the cytotoxic agents, for example, CAR-T cells. Thus, the present disclosure provides methods for treating a hematopoietic malignancy, the method comprising administering to a subject in need thereof (i) a population of the genetically engineered hematopoietic cells described herein, and optionally (ii) a cytotoxic agent such as CAR-T cells that target an antigen, the gene of which is genetically edited in the hematopoietic cells such that the cytotoxic agent does not target hematopoietic cells or descendant cells thereof. The administration of (i) and (ii) may be concurrently or in any order. In some embodiments, the cytotoxic agents and/or the hematopoietic cells may be mixed with a pharmaceutically acceptable carrier to form a pharmaceutical composition, which is also within the scope of the present disclosure. In some embodiments, the genetically engineered hematopoietic stem cells have genetic editing in genes of at least two antigens A and B. Such hematopoietic stem cells can be administered to a subject, who has been or will be treated with a first cytotoxic agent specific to A (e.g., anti-protein A CAR-T cells), or who is at risk of a hematopoietic malignancy that would need treatment of the cytotoxic agent. When the subject developed resistance to the cytotoxic agent after the treatment or has relapse of the hematopoietic malignancy, a second cytotoxic agent specific to B (e.g., anti-protein B CAR-T cells) may be administered to the subject. Because the genetically engineered hematopoietic cells have genetic editing in both A and B genes, those cells and descendant cells thereof would also be resistant to cytotoxicity induced by the second cytotoxic agent. As such, administering once the genetically engineered hematopoietic cells would be sufficient to compensate loss of normal cells expressing at least lineage-specific cell surface proteins/antigens A and B due to multiple treatment by cytotoxic agents specific to at least proteins/antigens A and B. As used herein, “subject,” “individual,” and “patient” are used interchangeably, and refer to a vertebrate, preferably a mammal such as a human. Mammals include, but are not limited to, human primates, non-human primates or murine, bovine, equine, canine or feline species. In some embodiments, the subject is a human patient having a hematopoietic malignancy. To perform the methods described herein, an effective amount of the genetically engineered hematopoietic cells can be administered to a subject in need of the treatment. Optionally, the hematopoietic cells can be co-used with a cytotoxic agent as described herein. As used herein the term “effective amount” may be used interchangeably with the term “therapeutically effective amount” and refers to that quantity of a cytotoxic agent, hematopoietic cell population, or pharmaceutical composition (e.g., a composition comprising cytotoxic agents and/or hematopoietic cells) that is sufficient to result in a desired activity upon administration to a subject in need thereof. Within the context of the present disclosure, the term “effective amount” refers to that quantity of a compound, cell population, or pharmaceutical composition that is sufficient to delay the manifestation, arrest the progression, relieve or alleviate at least one symptom of a disorder treated by the methods of the present disclosure. Note that when a combination of active ingredients is administered the effective amount of the combination may or may not include amounts of each ingredient that would have been effective if administered individually. Effective amounts vary, as recognized by those skilled in the art, depending on the particular condition being treated, the severity of the condition, the individual patient parameters including age, physical condition, size, gender and weight, the duration of the treatment, the nature of concurrent therapy (if any), the specific route of administration and like factors within the knowledge and expertise of the health practitioner. In some embodiments, the effective amount alleviates, relieves, ameliorates, improves, reduces the symptoms, or delays the progression of any disease or disorder in the subject. In some embodiments, the subject is a human. In some embodiments, the subject is a human patient having a hematopoietic malignancy. As described herein, the hematopoietic cells and/or immune cells expressing chimeric receptors may be autologous to the subject, i.e., the cells are obtained from the subject in need of the treatment, manipulated such that the cells do not bind the cytotoxic agents, and then administered to the same subject. Administration of autologous cells to a subject may result in reduced rejection of the host cells as compared to administration of non-autologous cells. Alternatively, the host cells are allogeneic cells, i.e., the cells are obtained from a first subject, manipulated such that the cells do not bind the cytotoxic agents, and then administered to a second subject that is different from the first subject but of the same species. For example, allogeneic immune cells may be derived from a human donor and administered to a human recipient who is different from the donor. In some embodiments, engineered hematopoietic cells comprising one or more genetically engineered gene(s) encoding lineage-specific cell surface protein(s) (e.g., CD33 and IL1RAP or CD44 and EMR2) and the immune cells engineered to target an epitope of the antigen(s) are both allogeneic to the subject. In some embodiments, engineered hematopoietic cells comprising one or more genetically engineered gene(s) encoding antigen(s) (e.g., CD33 and IL1RAP or CD44 and EMR2 or any other combination of antigens described herein) and the immune cells engineered to target an epitope of the antigen(s) are from the same allogeneic donor. In some embodiments, engineered hematopoietic cells comprising one or more genetically engineered gene(s) encoding antigen(s) (e.g., CD33 and IL1RAP or CD44 and EMR2 or any other combination of antigens described herein) and the immune cells engineered to target an epitope of the antigen(s) are from two different allogeneic donors. In some embodiments, engineered hematopoietic cells comprising one or more genetically engineered gene(s) encoding antigen(s) (e.g., CD33 and IL1RAP or CD44 and EMR2 or any other combination of antigens described herein) and the immune cells engineered to target an epitope of the antigen(s) are both autologous to the subject. In some embodiments, engineered hematopoietic cells comprising one or more genetically engineered gene(s) encoding antigen(s) (e.g., CD33 and IL1RAP or CD44 and EMR2 or any other combination of antigens described herein) are autologous to the subject and the immune cells engineered to target an epitope of the antigen(s) are from an allogeneic donor. In some embodiments, engineered hematopoietic cells comprising one or more genetically engineered gene(s) encoding antigen(s) (e.g., CD33 and IL1RAP or CD44 and EMR2 or any other combination of antigens described herein) are from an allogeneic donor and the immune cells engineered to target an epitope of the lineage-specific cell surface protein(s) are autologous to the subject. In some embodiments, the immune cells and/or hematopoietic cells are allogeneic cells and have been further genetically engineered to reduce graft-versus-host disease. For example, as described herein, the hematopoietic stem cells may be genetically engineered (e.g., using genome editing) to have reduced expression of CD45RA. Methods for reducing graft-versus-host disease are known in the art, see, e.g., Yang et al. Curr. Opin. Hematol. (2015) 22(6): 509-515. In some embodiments, the immune cells (e.g., T cells) may be genetically engineered to reduce or eliminate expression of the T cell receptor (TCR) or reduce or eliminate cell surface localization of the TCR. In some examples, the gene encoding the TCR is knocked out or silenced (e.g., using gene editing methods, or shRNAs). In some embodiments, the TCR is silenced using peptide inhibitors of the TCR. In some embodiments, the immune cells (e.g., T cells) are subjected to a selection process to select for immune cells or a population of immune cells that do not contain an alloreactive TCR. Alternatively, in some embodiments, immune cells that naturally do not express TCRs (e.g., NK cells) may be used in any of the methods described herein. In some embodiments, the immune cells and/or hematopoietic cells have been further genetically engineered to reduce host-versus-graft effects. For example, in some embodiments, immune cells and/or hematopoietic cells may be subjected to gene editing or silencing methods to reduce or eliminate expression of one or more proteins involved in inducing host immune responses, e.g., CD52, MHC molecules, and/or MHC beta-2 microglobulin. In some embodiments, the immune cells expressing any of the chimeric receptors described herein are administered to a subject in an amount effective in to reduce the number of target cells (e.g., cancer cells) by least 20%, e.g., 50%, 80%, 100%, 2-fold, 5-fold, 10-fold, 20-fold, 50-fold, 100-fold or more. A typical amount of cells, i.e., immune cells or hematopoietic cells, administered to a mammal (e.g., a human) can be, for example, in the range of about 106 to 1011 cells. In some embodiments it may be desirable to administer fewer than 106 cells to the subject. In some embodiments, it may be desirable to administer more than 1011 cells to the subject. In some embodiments, one or more doses of cells includes about 106 cells to about 1011 cells, about 107 cells to about 1010 cells, about 108 cells to about 109 cells, about 106 cells to about 108 cells, about 107 cells to about 109 cells, about 107 cells to about 1010 cells, about 107 cells to about 1011 cells, about 108 cells to about 1010 cells, about 108 cells to about 1011 cells, about 109 cells to about 1010 cells, about 109 cells to about 1011 cells, or about 1010 cells to about 1011 cells. In some embodiments, the subject is preconditioned prior to administration of the cytotoxic agent and/or hematopoietic cells. In some embodiments, the method further comprises pre-conditioning the subject. In general, preconditioning a subject involves subjecting the patient to one or more therapy, such as a chemotherapy or other type of therapy, such as irradiation. In some embodiments, the preconditioning may induce or enhance the patient’s tolerance of one or more subsequent therapy (e.g., a targeted therapy), as described herein. In some embodiments, the pre-conditioning involves administering one or more chemotherapeutic agents to the subject. Non-limiting examples of chemotherapeutic agents include actinomycin, azacitidine, azathioprine, bleomycin, bortezomib, carboplatin, capecitabine, cisplatin, chlorambucil, cyclophosphamide, cytarabine, daunorubicin, docetaxel, doxifluridine, doxorubicin, epirubicin, epothilone, etoposide, fludarabine, fluorouracil, gemcitabine, hydroxyurea, idarubicin, imatinib, irinotecan, mechlorethamine, mercaptopurine, methotrexate, mitoxantrone, oxaliplatin, paclitaxel, pemetrexed, teniposide, tioguanine, topotecan, valrubicin, vinblastine, vincristine, vindesine, and vinorelbine. In some embodiments, the subject is preconditioned at least one day, two days, three days, four days, 5 days, 6 days, 7 days, 8 days, 9 days, 10 days, 11 days, 12 days, 13 days, two weeks, three weeks, four weeks, five weeks, six weeks, seven weeks, eight weeks, nine weeks, ten weeks, two months, three months, four months, five months, or at least six months prior to administering the cytotoxic agent and/or hematopoietic cells. In other embodiments, the chemotherapy(ies) or other therapy(ies) are administered concurrently with the cytotoxic agent and manipulated hematopoietic cells. In other embodiments, the chemotherapy(ies) or other therapy(ies) are administered after the cytotoxic agent and manipulated hematopoietic cells. In one embodiment, the chimeric receptor (e.g., a nucleic acid encoding the chimeric receptor) is introduced into an immune cell, and the subject (e.g., human patient) receives an initial administration or dose of the immune cells expressing the chimeric receptor. One or more subsequent administrations of the cytotoxic agent (e.g., immune cells expressing the chimeric receptor) may be provided to the patient at intervals of 15 days, 14, 13, 12, 11, 10, 9, 8, 7, 6, 5, 4, 3, or 2 days after the previous administration. More than one dose of the cytotoxic agent can be administered to the subject per week, e.g., 2, 3, 4, or more administrations of the agent. The subject may receive more than one doses of the cytotoxic agent (e.g., an immune cell expressing a chimeric receptor) per week, followed by a week of no administration of the agent, and finally followed by one or more additional doses of the cytotoxic agent (e.g., more than one administration of immune cells expressing a chimeric receptor per week). The immune cells expressing a chimeric receptor may be administered every other day for 3 administrations per week for two, three, four, five, six, seven, eight or more weeks. Any of the methods described herein may be for the treatment of a hematological malignancy in a subject. As used herein, the terms “treat,” “treating,” and “treatment” mean to relieve or alleviate at least one symptom associated with the disease or disorder, or to slow or reverse the progression of the disease or disorder. Within the meaning of the present disclosure, the term “treat” also denotes to arrest, delay the onset (i.e., the period prior to clinical manifestation of a disease) and/or reduce the risk of developing or worsening a disease. For example, in connection with cancer, the term “treat” may mean eliminate or reduce the number or replication of cancer cells, and/or prevent, delay or inhibit metastasis, etc. In some embodiments, a population of genetically engineered hematopoietic cells (e.g., carrying genetic edits in genes encoding antigens or expressing those proteins in mutated form) and a cytotoxic agent(s) specific to the cell surface protein are co-administered to a subject via a suitable route (e.g., infusion). In such a combined therapeutic method, the cytotoxic agent recognizes (binds) a target cell expressing the cell surface protein for targeted killing. The hematopoietic cells that express the protein in mutated form, which has reduced binding activity or do not bind the cytotoxic acid (e.g., because of lacking binding epitope) allow for repopulation of a cell type that is targeted by the cytotoxic agent. In some embodiments, the methods described herein involve administering a population of genetically engineered hematopoietic cells to a subject and administering one or more immunotherapeutic agents (e.g., cytotoxic agents). As will be appreciated by one of ordinary skill in the art, the immunotherapeutic agents may be of the same or different type (e.g., therapeutic antibodies, populations of immune cells expressing chimeric antigen receptor(s), and/or antibody-drug conjugates). In some embodiments, the treatment of the patient can involve the following steps: (1) administering a therapeutically effective amount of the cytotoxic agent to the patient and (2) infusing or reinfusing the patient with hematopoietic stem cells, either autologous or allogenic. In some embodiments, the treatment of the patient can involve the following steps: (1) administering a therapeutically effective amount of an immune cell expressing a chimeric receptor to the patient, wherein the immune cell comprises a nucleic acid sequence encoding a chimeric receptor that binds an epitope of a cell surface lineage-specific, disease-associated protein; and (2) infusing or reinfusing the patient with hematopoietic cells (e.g., hematopoietic stem cells), either autologous or allogenic. In each of the methods described herein, the cytotoxic agent (e.g., CAR-T cells) and the genetically engineered hematopoietic cells can be administered to the subject in any order. In some instances, the hematopoietic cells are given to the subject prior to the cytotoxic agent. In some instances, a second cytotoxic agent can be administered to the subject after treatment with the first cytotoxic agent, e.g., when the patient develops resistance or disease relapse. The hematopoietic cells given the same subject may have multiple edited genes expressing lineage-specific cell surface proteins in mutated form such that the cytotoxic agents can target wild-type proteins but not the mutated form. The efficacy of the therapeutic methods using a population of genetically engineered hematopoietic cells described herein, optionally in combination with a cytotoxic agent (e.g., CART) may be assessed by any method known in the art and would be evident to a skilled medical professional. For example, the efficacy of the therapy may be assessed by survival of the subject or cancer burden in the subject or tissue or sample thereof. In some embodiments, the efficacy of the therapy is assessed by quantifying the absolute number of a particular cell surface protein on a cell or population of cells. In some embodiments, the efficacy of the therapy is assessed by quantifying the number of cells presenting the antigen. In some embodiments, the cytotoxic agent comprising an antigen-binding fragment that binds to the epitope of the cell surface protein and the population of hematopoietic cells is administered concomitantly. In some embodiments, the cytotoxic agent comprising an antigen-binding fragment that binds an epitope of the cell surface protein (e.g., immune cells expressing a chimeric receptor as described herein) is administered prior to administration of the hematopoietic cells. In some embodiments, the agent comprising an antigen-binding fragment that binds an epitope of the cell surface protein (e.g., immune cells expressing a chimeric receptor as described herein) is administered at least about 1 day, 2 days, 3 days, 4 days, 5 days, 6 days, 1 week, 2 weeks, 3 weeks, 4 weeks, 5 weeks, 6 weeks, 7 weeks, 8 weeks, 9 weeks, 10 weeks, 11 weeks, 12 weeks, 3 months, 4 months, 5 months, 6 months or more prior to administration of the hematopoietic cells. In some embodiments, the hematopoietic cells are administered prior to the cytotoxic agent comprising an antigen-binding fragment that binds an epitope of the cell surface protein (e.g., immune cells expressing a chimeric receptor as described herein). In some embodiments, the population of hematopoietic cells is administered at least about 1 day, 2 days, 3 days, 4 days, 5 days, 6 days, 1 week, 2 weeks, 3 weeks, 4 weeks, 5 weeks, 6 weeks, 7 weeks, 8 weeks, 9 weeks, 10 weeks, 11 weeks, 12 weeks, 3 months, 4 months, 5 months, 6 months or more prior to administration of the cytotoxic agent comprising an antigen-binding fragment that binds to an epitope of the antigen. In some embodiments, the cytotoxic agent targeting the cell surface protein and the population of hematopoietic cells are administered at substantially the same time. In some embodiments, the cytotoxic agent targeting the cell surface protein is administered and the patient is assessed for a period of time, after which the population of hematopoietic cells is administered. In some embodiments, the population of hematopoietic cells is administered and the patient is assessed for a period of time, after which the cytotoxic agent targeting the cell surface protein is administered. Also within the scope of the present disclosure are multiple administrations (e.g., doses) of the cytotoxic agents and/or populations of hematopoietic cells. In some embodiments, the cytotoxic agents and/or populations of hematopoietic cells are administered to the subject once. In some embodiments, cytotoxic agents and/or populations of hematopoietic cells are administered to the subject more than once (e.g., at least 2, 3, 4, 5, or more times). In some embodiments, the cytotoxic agents and/or populations of hematopoietic cells are administered to the subject at a regular interval, e.g., every six months. In some embodiments, the subject is a human subject having a hematopoietic malignancy. As used herein a hematopoietic malignancy refers to a malignant abnormality involving hematopoietic cells (e.g., blood cells, including progenitor and stem cells). Examples of hematopoietic malignancies include, without limitation, Hodgkin’s lymphoma, non-Hodgkin’s lymphoma, leukemia, or multiple myeloma. Exemplary leukemias include, without limitation, acute myeloid leukemia, acute lymphoid leukemia, chronic myelogenous leukemia, acute lymphoblastic leukemia or chronic lymphoblastic leukemia, and chronic lymphoid leukemia. In some embodiments, cells involved in the hematopoietic malignancy are resistant to convention or standard therapeutics used to treat the malignancy. For example, the cells (e.g., cancer cells) may be resistant to a chemotherapeutic agent and/or CAR T cells used to treat the malignancy. Examples of hematopoietic malignancies include, but are not limited to, B cell malignancies such as non-Hodgkin’s lymphoma, Hodgkin’s lymphoma, leukemia, multiple myeloma, acute lymphoblastic leukemia, acute lymphoid leukemia, acute lymphocytic leukemia, chronic lymphoblastic leukemia, chronic lymphoid leukemia, and chronic lymphocytic leukemia. In some embodiments, the hematopoietic malignancy is a relapsing hematopoietic malignancy. In some embodiments, the leukemia is acute myeloid leukemia (AML). AML is characterized as a heterogeneous, clonal, neoplastic disease that originates from transformed cells that have progressively acquired critical genetic changes that disrupt key differentiation and growth-regulatory pathways. (Dohner et al., NEJM, (2015) 373:1136). CD33 glycoprotein is expressed on the majority of myeloid leukemia cells as well as on normal myeloid and monocytic precursors and has been considered to be an attractive target for AML therapy (Laszlo et al., Blood Rev. (2014) 28(4):143-53). While clinical trials using anti-CD33 monoclonal antibody based therapy have shown improved survival in a subset of AML patients when combined with standard chemotherapy, these effects were also accompanied by safety and efficacy concerns. In some cases, a subject may initially respond to a therapy (e.g., for a hematopoietic malignancy) and subsequently experience relapse. Any of the methods or populations of genetically engineered hematopoietic cells described herein may be used to reduce or prevent relapse of a hematopoietic malignancy. Alternatively or in addition, any of the methods described herein may involve administering any of the populations of genetically engineered hematopoietic cells described herein and an immunotherapeutic agent (e.g., cytotoxic agent) that targets cells associated with the hematopoietic malignancy and further administering one or more additional immunotherapeutic agents when the hematopoietic malignancy relapses. As used herein, the term “relapse” refers to the reemergence or reappearance of cells associated with a hematopoietic malignancy following a period of responsiveness to a therapy. Methods of determining whether a hematopoietic malignancy has relapsed in a subject will be appreciated by one of ordinary skill in the art. In some embodiments, the period of responsiveness to a therapy involves the level or quantity of cells associated with the hematopoietic malignancy the falling below a threshold, e.g., below 20%, 15%, 10%, 5%, 4%, 3%, 2%, or 1% of the level or quantity of cells prior to administration of the therapy. In some embodiments, a relapse is characterized by the level or quantity of cells associated with the hematopoietic malignancy above a threshold, e.g., above 20%, 15%, 10%, 5%, 4%, 3%, 2%, or 1% higher than the level or quantity of cells during the period of responsiveness. Methods of determining the minimal residual disease in a subject are known in the art and may be used, for example to assess whether a hematopoietic malignancy has relapsed or is likely to relapse. See, e.g., Taraseviciute et al. Hematology and Oncology (2019) 31(1)). In some embodiments, the subject has or is susceptible to relapse of a hematopoietic malignancy (e.g., AML) following administration of one or more previous therapies. In some embodiments, the methods described herein reduce the subject’s risk of relapse or the severity of relapse. Without wishing to be bound by any particular theory, some cancers, including hematopoietic malignancies, are thought to relapse after an initial period of responsiveness to a therapy due to mechanisms such as antigen loss/antigen escape or lineage switch. In general, antigen loss/antigen escape results in relapse with a phenotypically similar hematopoietic malignancy characterized by cells that lack surface expression of the antigen targeted by the previous therapy (e.g., immunotherapeutic agent) such that the cells are no longer targeted by the previous therapy. In contrast, lineage switch presents as a genetically related but phenotypically different malignancy in which the cells lack surface expression of the antigen targeted by the previous therapy (e.g., immunotherapeutic agent) such that the cells are no longer targeted by the previous therapy. See, e.g., Brown et al. Nature Reviews Immunology (2019) 19:73-74; Majzner et al. Cancer Discovery (2018) 8(10). Antigen loss/antigen escape in which the target antigen is no longer present on the target cells (e.g., cells of the hematopoietic malignancy) frequently occurs as a result of genetic mutation and/or enrichment of cells that express a variant of the antigen (e.g., lineage-specific cell surface antigen) that is not targeted by the immunotherapeutic agent (e.g., cytotoxic agent). In some embodiments, the hematopoietic malignancy has relapsed due to antigen loss/antigen escape. In some embodiments, the target cells have lost the targeted epitope (e.g., of a lineage-specific cell surface antigen) or have reduced expression of the antigen (e.g., lineage-specific cell surface antigen) such that the targeted epitope is not recognized by the immunotherapeutic agent or is not sufficient to induce cytotoxicity. In some embodiments, the hematopoietic malignancy has relapsed due to lineage switch. In some embodiments, the methods described herein reduce or avoid relapse of a hematopoietic malignancy by targeting more than one antigen. In some embodiments, the populations of genetically engineered hematopoietic cells express mutants of more than one antigens such that the mutated antigens are not targeted by an immunotherapeutic agent(s). In some embodiments, a cancer treated with the methods herein comprises a first sub- population of cancer cells and a second sub-population of cancer cells. One of the sub- populations may be cancer stem cells. One of the sub-populations may be cancer bulk cells. One of the sub-populations may have one or more (e.g., at least 2, 3, 4, 5, or all) markers of differentiated hematopoietic cells. One of the sub-populations may have one or more (e.g., at least 2, 3, 4, 5, or all) markers of earlier lineage cells, e.g., HSCs or HPCs. Any of the immune cells expressing chimeric receptors described herein may be administered in a pharmaceutically acceptable carrier or excipient as a pharmaceutical composition. The phrase “pharmaceutically acceptable,” as used in connection with compositions and/or cells of the present disclosure, refers to molecular entities and other ingredients of such compositions that are physiologically tolerable and do not typically produce untoward reactions when administered to a mammal (e.g., a human). Preferably, as used herein, the term “pharmaceutically acceptable” means approved by a regulatory agency of the Federal or a state government or listed in the U.S. Pharmacopeia or other generally recognized pharmacopeia for use in mammals, and more particularly in humans. “Acceptable” means that the carrier is compatible with the active ingredient of the composition (e.g., the nucleic acids, vectors, cells, or therapeutic antibodies) and does not negatively affect the subject to which the composition(s) are administered. Any of the pharmaceutical compositions and/or cells to be used in the present methods can comprise pharmaceutically acceptable carriers, excipients, or stabilizers in the form of lyophilized formations or aqueous solutions. Pharmaceutically acceptable carriers, including buffers, are well known in the art, and may comprise phosphate, citrate, and other organic acids; antioxidants including ascorbic acid and methionine; preservatives; low molecular weight polypeptides; proteins, such as serum albumin, gelatin, or immunoglobulins; amino acids; hydrophobic polymers; monosaccharides; disaccharides; and other carbohydrates; metal complexes; and/or non-ionic surfactants. See, e.g. Remington: The Science and Practice of Pharmacy 20th Ed. (2000) Lippincott Williams and Wilkins, Ed. K. E. Hoover. V. Diagnostic and/or Prognostic Methods In some embodiments, aspects of the present disclosure relate to methods for determining if a subject is at risk of relapse of a disease. In some embodiments, such methods comprise providing a first biological sample obtained from a subject at a first time point and a second biological sample obtained from the subject at a second time point, measuring a level of a first cell surface protein in the first biological sample and in the second biological sample, identifying the subject as at risk for relapse of the disease if the level of the first cell surface protein in the first biological sample deviates from the level of the first cell surface protein in the second biological sample. In some embodiments, the cell surface protein is one selected from Table 3 or Table 4. In some embodiments, aspects of the present disclosure relate to methods for identifying a subject as a candidate for treatment with an agent, such as a cytotoxic agent described herein. In some embodiments, such methods comprise providing a first biological sample obtained from a subject at a first time point and a second biological sample obtained from the subject at a second time point, and measuring a level of a first cell surface protein in the first biological sample and in the second biological sample. In some embodiments, the cell surface protein is one selected from Table 3 or Table 4. In some embodiments, methods of identifying subjects as being at risk of relapse of a disease and/or as a candidate for treatment with an agent further comprises administering to the subject a therapeutically effective amount of an agent comprising an antigen-binding fragment that binds to the first cell surface protein, such as a cytotoxic agent described herein. In some embodiments, methods of identifying subjects as being at risk of relapse of a disease and/or as a candidate for treatment with an agent further comprises administering to the subject a population of genetically engineered hematopoietic cells having reduced or eliminated expression of the first cell surface protein, such as a genetically engineered hematopoietic cell described herein, or a cell population thereof. In some embodiments, the first biological sample and/or the second biological sample comprises AML blast cells. In some embodiments, first time point is at or near the time the subject is diagnosed with the disease. Subjects In some embodiments, the subject is a human subject having a hematopoietic malignancy. In some embodiments, a subject has, is suspected of having, or at risk of developing Hodgkin’s lymphoma, non-Hodgkin’s lymphoma, leukemia, or multiple myeloma. Exemplary leukemias include, without limitation, acute myeloid leukemia, acute lymphoid leukemia, chronic myelogenous leukemia, acute lymphoblastic leukemia or chronic lymphoblastic leukemia, or chronic lymphoid leukemia. In some cases, a subject may initially respond to a therapy (e.g., for a hematopoietic malignancy) and subsequently experience relapse. In some embodiments, the subject has or is susceptible to relapse of a hematopoietic malignancy (e.g., AML) following administration of one or more previous therapies. In some embodiments, a subject exhibits an initial period of responsiveness to a therapy due to mechanisms such as antigen loss/antigen escape or lineage switch. In some embodiments, a subject may have, be suspect of having, or at risk of developing antigen loss/antigen escape which results in relapse with a phenotypically similar hematopoietic malignancy characterized by cells that lack surface expression of the cell surface protein targeted by the previous therapy (e.g., immunotherapeutic agent) such that the cells are no longer targeted by the previous therapy. In some embodiments, a subject may have, be suspect of having, or at risk of developing lineage switch wherein a genetically related but phenotypically different malignancy in which the cells lack surface expression of the cell surface protein targeted by the previous therapy (e.g., immunotherapeutic agent) such that the cells are no longer targeted by the previous therapy. See, e.g., Brown et al. Nature Reviews Immunology (2019) 19:73-74; Majzner et al. Cancer Discovery (2018) 8(10). In some embodiments, antigen loss/antigen escape comprises the target antigen no longer being present on the target cells (e.g., cells of the hematopoietic malignancy) and may occur as a result of genetic mutation and/or enrichment of cells that express a variant of the cell surface protein (e.g., lineage-specific cell surface antigen) that is not targeted by the immunotherapeutic agent (e.g., cytotoxic agent). In some embodiments, the hematopoietic malignancy has relapsed due to antigen loss/antigen escape. In some embodiments, the target cells have lost the targeted epitope (e.g., of a lineage-specific cell surface antigen) or have reduced expression of the cell surface protein (e.g., lineage-specific cell surface antigen) such that the targeted epitope is not recognized by the immunotherapeutic agent or is not sufficient to induce cytotoxicity. In some embodiments, the hematopoietic malignancy has relapsed due to lineage switch. In some embodiments, the methods described herein reduce or avoid relapse of a hematopoietic malignancy by targeting more than one antigen. In some embodiments, the populations of genetically engineered hematopoietic cells express mutants of more than one antigens such that the mutated antigens are not targeted by an immunotherapeutic agent(s). In some embodiments, the methods described herein reduce the subject’s risk of relapse or the severity of relapse. In some embodiments, the period of responsiveness to a therapy involves the level or quantity of cells associated with the hematopoietic malignancy the falling below a threshold, e.g., below 20%, 15%, 10%, 5%, 4%, 3%, 2%, or 1% of the level or quantity of cells prior to administration of the therapy. In some embodiments, a relapse is characterized by the level or quantity of cells associated with the hematopoietic malignancy above a threshold, e.g., above 20%, 15%, 10%, 5%, 4%, 3%, 2%, or 1% higher than the level or quantity of cells during the period of responsiveness. Methods of determining the minimal residual disease in a subject are known in the art and may be used, for example to assess whether a hematopoietic malignancy has relapsed or is likely to relapse. See, e.g., Taraseviciute et al. Hematology and Oncology (2019) 31(1)). Mutated Genes Encoding Antigens In some embodiments, a subject has, is suspected of having, or is at risk of developing a mutation in a gene encoding a lineage-specific cell surface antigen. In some embodiments, the mutation is associated with altered function a lineage-specific cell surface antigen. In some embodiments, the mutation is associated with upregulating a lineage-specific cell surface antigen. In some embodiments, the mutation is associated with downregulating a lineage-specific cell surface antigen. In some embodiments, the subject has, is suspected of having, or is at risk of developing more than one (e.g., two, three, four, or more) mutated gene encoding cell surface proteins. In some embodiments, the subject may comprise a first mutated gene that results in downregulation of a first cell surface protein and second mutated gene that results in downregulation of a second lineage-specific cell surface antigen. In some embodiments, the subject may comprise a first mutated gene that results in upregulation of a first cell surface protein and second mutated gene that results in downregulation of a second lineage-specific cell surface antigen. In some embodiments, the subject may comprise a first mutated gene that results in upregulation of a first cell surface protein and second mutated gene that results in upregulation of a second lineage-specific cell surface antigen. In some embodiments, a subject has, is suspected of having, or is at risk of developing a mutation in a gene encoding a lineage-specific cell surface antigen, such as CD1a, CD1b, CD1c, CD1d, CD1e, CD2, CD3, CD3d, CD3e, CD3g, CD4, CD5, CD6, CD7, CD8a, CD8b, CD9, CD10, CD11a, CD11b, CD11c, CD11d, CDw12, CD13, CD14, CD15, CD16, CD16b, CD17, CD18, CD19, CD20, CD21, CD22, CD23, CD24, CD25, CD26, CD27, CD28, CD29, CD30, CD31, CD32a, CD32b, CD32c, CD34, CD35, CD36, CD37, CD38, CD39, CD40, CD41, CD42a, CD42b, CD42c, CD42d, CD43, CD44, CD45, CD45RA, CD45RB, CD45RC, CD45RO, CD46, CD47, CD48, CD49a, CD49b, CD49c, CD49d, CD49e, CD49f, CD50, CD51, CD52, CD53, CD54, CD55, CD56, CD57, CD58, CD59, CD60a, CD61, CD62E, CD62L, CD62P, CD63, CD64a, CD65, CD65s, CD66a, CD66b, CD66c, CD66F, CD68, CD69, CD70, CD71, CD72, CD73, CD74, CD75, CD75S, CD77, CD79a, CD79b, CD80, CD81, CD82, CD83, CD84, CD85A, CD85C, CD85D, CD85E, CD85F, CD85G, CD85H, CD85I, CD85J, CD85K, CD86, CD87, CD88, CD89, CD90, CD91, CD92, CD93, CD94, CD95, CD96, CD97, CD98, CD99, CD99R, CD100, CD101, CD102, CD103, CD104, CD105, CD106, CD107a, CD107b, CD108, CD109, CD110, CD111, CD112, CD113, CD114, CD115, CD116, CD117, CD118, CD119, CD120a, CD120b, CD121a, CD121b, CD121a, CD121b, CD122, CD123, CD124, CD125, CD126, CD127, CD129, CD130, CD131, CD132, CD133, CD134, CD135, CD136, CD137, CD138, CD139, CD140a, CD140b, CD141, CD142, CD143, CD14, CDw145, CD146, CD147, CD148, CD150, CD152, CD152, CD153, CD154, CD155, CD156a, CD156b, CD156c, CD157, CD158b1, CD158b2, CD158d, CD158e1/e2, CD158f, CD158g, CD158h, CD158i, CD158j, CD158k, CD159a, CD159c, CD160, CD161, CD163, CD164, CD165, CD166, CD167a, CD168, CD169, CD170, CD171, CD172a, CD172b, CD172g, CD173, CD174, CD175, CD175s, CD176, CD177, CD178, CD179a, CD179b, CD180, CD181, CD182, CD183, CD184, CD185, CD186, CD191, CD192, CD193, CD194, CD195, CD196, CD197, CDw198, CDw199, CD200, CD201, CD202b, CD203c, CD204, CD205, CD206, CD207, CD208, CD209, CD210a, CDw210b, CD212, CD213a1, CD213a2, CD215, CD217, CD218a, CD218b, CD220, CD221, CD222, CD223, CD224, CD225, CD226, CD227, CD228, CD229, CD230, CD231, CD232, CD233, CD234, CD235a, CD235b, CD236, CD236R, CD238, CD239, CD240, CD241, CD242, CD243, CD244, CD245, CD246, CD247, CD248, CD249, CD252, CD253, CD254, CD256, CD257, CD258, CD261, CD262, CD263, CD264, CD265, CD266, CD267, CD268, CD269, CD270, CD272, CD272, CD273, CD274, CD275, CD276, CD277, CD278, CD279, CD280, CD281, CD282, CD283, CD284, CD286, CD288, CD289, CD290, CD292, CDw293, CD294, CD295, CD296, CD297, CD298, CD299, CD300a, CD300c, CD300e, CD301, CD302, CD303, CD304, CD305, CD306, CD307a, CD307b, CD307c, CD307d, CD307e, CD309, CD312, CD314, CD315, CD316, CD317, CD318, CD319, CD320, CD321, CD322, CD324, CD325, CD326, CD327, CD328, CD329, CD331, CD332, CD333, CD334, CD335, CD336, CD337, CD338, CD339, CD340, CD344, CD349, CD350, CD351, CD352, CD353, CD354, CD355, CD357, CD358, CD359, CD360, CD361, CD362, CD363, CD364, CD365, CD366, CD367, CD368, CD369, CD370, and CD371. In some embodiments, a subject has, is suspected of having, or is at risk of developing a mutation in a gene encoding a cell surface protein selected from the group consisting of: CD19; CD123; CD22; CD30; CD171; CS-1 (also referred to as CD2 subset 1, CRACC, SLAMF7, CD319, and 19A24); C-type lectin-like molecule-1 (CLECL1); epidermal growth factor receptor variant III (EGFRvIII); ganglioside G2 (CD2); ganglioside GD3 (aNeu5Ac(2- 8)aNeu5Ac(2-3)bDGalp(1-4)bDGlep(1-1)Cer); TNF receptor family member B cell maturation (BCMA), Tn antigen ((Tn Ag) or (GalNAcα-Ser/Thr)); prostate-specific membrane antigen (PSMA); Receptor tyrosine kinase-like orphan receptor 1 (ROR1); Fms- Like tyrosine Kinase 3 (FLT3); Tumor-associated glycoprotein 72 (TAG72); CD38; CD44v6; Carcinoembryonic antigen (CEA); Epithelial cell adhesion molecule (EPCAM); B7H3 (CD276); KIT (CD117); Interleukin-13 receptor subunit alpha-2 (IL-13Ra2 or CD213A2); Mesothelin; Interleukin 11 receptor alpha (IL-11Ra); prostate stem cell antigen (PSCA); Protease Serine 21 (Testisin or PRSS21); vascular endothelial growth factor receptor 2 (VEGFR2); Lewis(Y) antigen; CD24; Platelet-derived growth factor receptor beta (PDGFR- beta); Stage-specific embryonic antigen-4 (SSEA-4); CD20; Folate receptor alpha; Receptor tyrosine-protein kinase ERBB2 (Her2/neu); Mucin 1, cell surface associated (MUC1); epidermal growth factor receptor (EGFR); neural cell adhesion molecule (NCAM); Prostase; prostatic acid phosphatase (PAP); elongation factor 2 mutated (ELF2M); Ephrin B2; fibroblast activation protein alpha (FAP); insulin-like growth factor I receptor (IGF-I receptor), carbonic anhydrase IX (CAIX), Proteasome (Prosome, Macropain) Subunit, Beta Type 9 (LMP2); glycoprotein 100 (gp100); oncogene fusion protein consisting of breakpoint cluster region (BCR) and Abelson murine leukemia viral oncogene homolog 1 (Abl) (bcr- abl); tyrosinase; ephrin type-A receptor 2 (EphA2); Fucosyl GM1; sialyl Lewis adhesion molecule (sLe); ganglioside GM3 (aNeu5Ac(2-3)bDGalp(1-4)bDGlcp(1-1)Cer); transglutaminase 5 (TGS5); high molecular weight-melanoma-associated antigen (HMWMAA); o-acetyl-GD2 ganglioside (OAcGD2); Folate receptor beta; tumor endothelial marker 1 (TEM1/CD248); tumor endothelial marker 7-related (TEM7R); claudin 6 (CLDN6); thyroid stimulating hormone receptor (TSHR); G protein-coupled receptor class C group 5, member D (GPRC5D); chromosome X open reading frame 61 (CXORF61); CD97; CD179a; anaplastic lymphoma kinase (ALK); Polysialic acid; placenta-specific 1 (PLAC1); hexasaccharide portion of globoH glycoceramide (GloboH); mammary gland differentiation antigen (NY-BR-1); uroplakin 2 (UPK2); Hepatitis A virus cellular receptor 1 (HAVCR1); adrenoceptor beta 3 (ADRB3); pannexin 3 (PANX3); G protein-coupled receptor 20 (GPR20); lymphocyte antigen 6 complex; locus K 9 (LY6K); Olfactory receptor 51E2 (OR51E2); TCR Gamma Alternate Reading Frame Protein (TARP); Wilms tumor protein (WT1); Cancer/testis antigen 1 (NY-ESO-1); Cancer/testis antigen 2 (LAGE-1a); Melanoma- associated antigen 1 (MAGE-A1), ETS translocation-variant gene 6, located on chromosome 12p (ETV6-AML); sperm protein 17 (SPA17); X Antigen Family, member 1A (XAGE1); angiopoietin-binding cell surface receptor 2 (Tie 2); melanoma cancer testis antigen-1 (MAD-CT-1); melanoma cancer testis antigen-2 (MAD-CT-2); Fos-related antigen 1; tumor protein p53 (p53); p53 mutant; prostein; surviving; telomerase; prostate carcinoma tumor antigen-1 (PCTA-1 or Galectin 8), melanoma antigen recognized by T cells 1 (MelanA or MART1); Rat sarcoma (Ras) mutant; human Telomerase reverse transcriptase (hTERT); sarcoma translocation breakpoints; melanoma inhibitor of apoptosis (ML-1AP); ERG (transmembrane protease, serine 2 (TMPRSS2) ETS fusion gene); N-Acetyl glucosaminyl- transferase V (NA17); paired box protein Pax-3 (PAX3); Androgen receptor; Cyclin B1; v- myc avian myelocytomatosis viral oncogene neuroblastoma derived homolog (MYCN); Ras Homolog Family Member C (RhoC); Tyrosinase-related protein 2 (TRP-2); Cytochrome P4501B1 (CYP1B1); CCCTC-Binding Factor (Zinc Finger Protein)-Like (BORIS or Brother of the Regulator of Imprinted Sites), Squamous Cell Carcinoma Antigen Recognized By T Cells 3 (SART3); Paired box protein Pax-5 (PAX5); proacrosin binding protein sp32 (OY- TES1); lymphocyte-specific protein tyrosine kinase (LCK); A kinase anchor protein 4 (AKAP-4); synovial sarcoma, X breakpoint 2 (SSX2); Receptor for Advanced Glycation Endproducts (RAGE-1); renal ubiquitous 1 (RU1); renal ubiquitous 2 (RU2); legumain; human papilloma virus E6 (HPV E6); human papilloma virus E7 (HPV E7); intestinal carboxy esterase; heat shock protein 70-2 mutated (mut hsp70-2); CD79a; CD79b; CD72; Leukocyte-associated immunoglobulin-like receptor 1 (LAIR1); Fc fragment of IgA receptor (FCAR or CD89); Leukocyte immunoglobulin-like receptor subfamily A member 2 (LILRA2); CD300 molecule-like family member f (CD300LF); C-type lectin domain family 12 member A (CLEC12A); bone marrow stromal cell antigen 2 (BST2); EGF-like module- containing mucin-like hormone receptor-like 2 (EMR2), lymphocyte antigen 75 (LY75); Glypican-3 (GPC3); Fc receptor-like 5 (FCRL5); and immunoglobulin lambda-like polypeptide 1 (IGLL1). In some embodiments, a subject has, is suspected of having, or is at risk of developing a mutation in a gene encoding a cell surface protein known to be associated with a cancer, such as CD20, CD22 (Non-Hodgkin's lymphoma, B-cell lymphoma, chronic lymphocytic leukemia (CLL)), CD52 (B-cell CLL), CD33 (Acute myelogenous leukemia (AML)), CD10 (gp100) (Common (pre-B) acute lymphocytic leukemia and malignant melanoma), CD3/T- cell receptor (TCR) (T-cell lymphoma and leukemia), CD79/B-cell receptor (BCR) (B-cell lymphoma and leukemia), CD26 (epithelial and lymphoid malignancies), human leukocyte antigen (HLA)-DR, HLA-DP, and HLA-DQ (lymphoid malignancies), RCAS1 (gynecological carcinomas, biliary adenocarcinomas and ductal adenocarcinomas of the pancreas) as well as prostate specific membrane antigen. In some embodiments, a subject has, is suspected of having, or is at risk of developing a mutation in a gene encoding a cell surface protein selected from a group consisting of: CD10; CD101; CD117; CD11A; CD11C; CD120B; CD123; CD13; CD133; CD152; CD200; CD205 (LY75); CD226; CD244 (SLAMF4); CD25; CD274; CD305 (LAIR1); CD32; CD33; CD34; CD366; CD38; CD41; CD42B; CD44; CD45RA; CD47; CD48; CD49D (ITGA4); CD49F; CD52; CD58; CD64; CD7; CD70; CD82; CD84; CD85; CD85K; CD86; CD9; CD96; CD99; CLL-1; EMR2 (ADGRE2); FR-B; GPR56; and IL1RAP. In some embodiments, a subject has, is suspected of having, or is at risk of developing a mutation in a gene encoding a cell surface protein selected from a group consisting of: CD10; CD101; CD11A; CD11C; CD123; CD133; CD152; CD200; CD205; CD244; CD25; CD305; CD33; CD34; CD366; CD38; CD42B; CD44; CD45RA; CD47; CD48; CD49D; CD49F; CD52; CD58; CD64; CD7; CD84; CD85K; CD9; CD99; CLL-1; EMR2; GPR56; IL1RAP; and CD274. In some embodiments, a subject has, is suspected of having, or is at risk of developing a mutation in a gene encoding a cell surface protein selected from a group consisting of: CD10; CD117; CD11A; CD11C; CD120B; CD123; CD13; CD133; CD152; CD205; CD22; CD226; CD244; CD25; CD305; CD32; CD33; CD34; CD38; CD41; CD44; CD45RA; CD47; CD48; CD49D; CD49F; CD52; CD56; CD58; CD64; CD7; CD70; CD71; CD82; CD84; CD85K; CD86; CD96; CD99; CLL-1; EMR2; FR-B; GPR56; HLA-DR; and IL1RAP. In some embodiments, a subject has, is suspected of having, or is at risk of developing a mutation in a first gene encoding a first cell surface protein and a second gene encoding a second cell surface protein selected from a group consisting of: CD274 and CD152; CD274 and CD226; CD152 and CD226; CD10 and CD49F; CD49F and GPR56; CD200 and CD34; CD34 and CD52; CD34 and CD133; GPR56 and CD133; CD42B and CD41; CD85K and CD64; CD85K and CD32; CD64 and CD32; CD64 and CD11C; CD64 and CD86; CD45RA and CD96; CD45RA and IL1RAP; CD45RA and CD123; CD84 and CD33; CD11C and IL1RAP; CD11C and CD33; CD11C and CD86; CD9 and IL1RAP; IL1RAP and CD33; IL1RAP and CD205; IL1RAP and CD305; IL1RAP and CD123; CD33 and CLL-1; CD33 and CD305; CD11A and CD86; CLL-1 and CD305; CD58 and CD305; CD49D and CD47; CD49D and CD99; CD49D and CD44; CD49D and CD305; CD47 and CD99; CD47 and EMR2; CD99 and CD44; CD99 and CD305; CD99 and EMR2; CD205 and CD305; CD205 and CD123; CD44 and CD305; CD44 and EMR2; CD244 and CD117; CD123 and EMR2; EMR2 and CD82; CD152 and CD120B; CD48 and CD11A; CD200 and CD25; CD34 and CD7; CD34 and CD13; CD52 and GPR56; CD52and CD133; CD52 and CD7; GPR56 and CD7; GPR56 and CD82; CD133 and CD7; CD133 and CD82; CD7 and CD13; CD101 and CD64; CD85 and CD70; CD85K and CD86; CD64 and FR-B; CD366 and CD96; CD25 and CD45RA; CD25 and CD96; CD9 and CD82; IL1RAP and CD58; CD11A and CD38; CD11A and CD44; CD11A and CD244; CD38 and CD305; CD38 and EMR2; CLL-1 and CD49D; CD58 and CD49D; CD47 and CD205; and CD47 and CD44. In some embodiments, a subject has, is suspected of having, or is at risk of developing a mutation in a first gene encoding a first cell surface protein and a second gene encoding a second cell surface protein selected from a group consisting of: CD85K and CD64; CD45RA and IL1RAP; CD34 and CD133; IL1RAP and CD33; CD47 and CD99; CLL-1 and CD305; CD152 and CD226; CD45RA and CD96; CD45RA and CD123; CD49D and CD305; IL1RAP and CD305; CD58 and CD305; CD49D and CD47; CD33 and CD305; CD49D and CD44; CD64 and CD86; GPR56 and CD133; CD34 and CD52; EMR2 and CD82; CD64 and CD32; CD42B and CD41; CD64 and CD11C; CD99 and CD44; CD200 and CD34; CD11C and CD33; IL1RAP and CD123; CD11C and IL1RAP; CD44 and EMR2; CD205 and CD305; CD44 and CD305; CD49D and CD99; CD274 and CD226; CD49F and GPR56; CD9 and IL1RAP; CD99 and CD305; CD10 and CD49F; CD11A and CD86; CD33 and CLL-1; CD84 and CD33; CD244 and CD117; CD85K and CD32; CD47 and EMR2; CD274 and CD152; IL1RAP and CD205; CD11C and CD86; CD99 and EMR2; CD205 and CD123; and CD123 and EMR2. In some embodiments, a subject has, is suspected of having, or is at risk of developing a mutation in a first gene encoding a first cell surface protein and a second gene encoding a second cell surface protein selected from a group consisting of: CD33 and CD205 (LY75); CD33 and CD244 (SLAMF4); CD33 and CD305 (LAIR1); CD33 and CD49D (ITGA4); CLL-1 and CD205 (LY75); CLL-1 and CD244 (SLAMF4); CLL-1 and CD305 (LAIR1); CLL-1 and CD49D (ITGA4); EMR2 and CD205 (LY75); EMR2 and CD244 (SLAMF4); EMR2 and CD305 (LAIR1); EMR2 and CD49D (ITGA4); CD38 and CD205 (LY75); CD38 and CD244 (SLAMF4); CD38 and CD305 (LAIR1); CD38 and CD49D (ITGA4); CD305 (LAIR1) and CD49D (ITGA4); CD305 (LAIR1) and CD205 (LY75); CD305 (LAIR1) and CD244 (SLAMF4); CD49D (ITGA4) and CD205 (LY75); CD49D (ITGA4) and CD244 (SLAMF4); and CD205 (LY75) and CD244 (SLAMF4). In some embodiments, a subject has, is suspected of having, or is at risk of developing a mutation in a first gene encoding a first cell surface protein and a second gene encoding a second lineage-specific cell surface antigen, wherein mutation of the first gene and second gene results in dysregulated expression of the first cell surface protein and the second cell surface protein which is indicative (e.g., diagnostic) of the subject having a hematopoietic malignancy. In some embodiments, the first gene and second gene encode combinations of antigens selected from a group consisting of: CD274 and CD152; CD274 and CD226; CD152 and CD226; CD10 and CD49F; CD49F and GPR56; CD200 and CD34; CD34 and CD52; CD34 and CD133; GPR56 and CD133; CD42B and CD41; CD85K and CD64; CD85K and CD32; CD64 and CD32; CD64 and CD11C; CD64 and CD86; CD45RA and CD96; CD45RA and IL1RAP; CD45RA and CD123; CD84 and CD33; CD11C and IL1RAP; CD11C and CD33; CD11C and CD86; CD9 and IL1RAP; IL1RAP and CD33; IL1RAP and CD205; IL1RAP and CD305; IL1RAP and CD123; CD33 and CLL-1; CD33 and CD305; CD11A and CD86; CLL-1 and CD305; CD58 and CD305; CD49D and CD47; CD49D and CD99; CD49D and CD44; CD49D and CD305; CD47 and CD99; CD47 and EMR2; CD99 and CD44; CD99 and CD305; CD99 and EMR2; CD205 and CD305; CD205 and CD123; CD44 and CD305; CD44 and EMR2; CD244 and CD117; CD123 and EMR2; and EMR2 and CD82. In some embodiments, the first gene and second gene encode combinations of antigens selected from a group consisting of: CD33 and CD205 (LY75); CD33 and CD244 (SLAMF4); CD33 and CD305 (LAIR1); CD33 and CD49D (ITGA4); CLL-1 and CD205 (LY75); CLL-1 and CD244 (SLAMF4); CLL-1 and CD305 (LAIR1); CLL-1 and CD49D (ITGA4); EMR2 and CD205 (LY75); EMR2 and CD244 (SLAMF4); EMR2 and CD305 (LAIR1); EMR2 and CD49D (ITGA4); CD38 and CD205 (LY75); CD38 and CD244 (SLAMF4); CD38 and CD305 (LAIR1); CD38 and CD49D (ITGA4); CD305 (LAIR1) and CD49D (ITGA4); CD305 (LAIR1) and CD205 (LY75); CD305 (LAIR1) and CD244 (SLAMF4); CD49D (ITGA4) and CD205 (LY75); CD49D (ITGA4) and CD244 (SLAMF4); and CD205 (LY75) and CD244 (SLAMF4). In some embodiments, a subject has, is suspected of having, or is at risk of developing a mutation in a first gene encoding a first cell surface protein and a second gene encoding a second lineage-specific cell surface antigen, wherein mutation of the first gene and second gene results in dysregulated expression of the first cell surface protein and the second cell surface protein which is indicative (e.g., diagnostic) of the subject having experienced a relapse of a hematopoietic malignancy. In some embodiments, the first gene and second gene encode combinations of antigens selected from a group consisting of: CD9 and CD82; CD45RA and CD123; CD133 and CD82; CD85K and CD70; CD47 and CD205; CD64 and FR-B; CD11A and CD44; CD85K and CD86; CD366 and CD96; CD101 and CD64; CLL-1 and CD49D; CD49D and CD47; EMR2 and CD82; CD38 and CD305; CD34 and CD7; CD200 and CD25; CD25 and CD45RA; CD49D and CD44; CD11A and CD244; CD133 and CD7; CD48 and CD11A; CD52 and CD133; GPR56 and CD82; CD11A and CD38; CD34 and CD13; CD58 and CD49D; CD47 and CD99; IL1RAP and CD58; CD47 and CD44; CD33 and CD305; CD64 and CD86; CD152 and CD120B; CD42B and CD41; CD34 and CD52; CD152 and CD226; CD7 and CD13; CD38 and EMR2; IL1RAP and CD33; CLL-1 and CD305; CD45RA and IL1RAP; CD34 and CD133; GPR56 and CD133; CD49D and CD305; CD25 and CD96; CD52 and CD7; CD64 and CD32; CD58 and CD305; CD45RA and CD96; IL1RAP and CD305; CD52 and GPR56; GPR56 and CD7; and CD85K and CD64. In some embodiments, the first gene and second gene encode combinations of antigens selected from a group consisting of: CD33 and CD205 (LY75); CD33 and CD244 (SLAMF4); CD33 and CD305 (LAIR1); CD33 and CD49D (ITGA4); CLL-1 and CD205 (LY75); CLL-1 and CD244 (SLAMF4); CLL-1 and CD305 (LAIR1); CLL-1 and CD49D (ITGA4); EMR2 and CD205 (LY75); EMR2 and CD244 (SLAMF4); EMR2 and CD305 (LAIR1); EMR2 and CD49D (ITGA4); CD38 and CD205 (LY75); CD38 and CD244 (SLAMF4); CD38 and CD305 (LAIR1); CD38 and CD49D (ITGA4); CD305 (LAIR1) and CD49D (ITGA4); CD305 (LAIR1) and CD205 (LY75); CD305 (LAIR1) and CD244 (SLAMF4); CD49D (ITGA4) and CD205 (LY75); CD49D (ITGA4) and CD244 (SLAMF4); and CD205 (LY75) and CD244 (SLAMF4). Biological Samples The term “biological sample” refers to any sample including tissue samples (such as tissue sections and needle biopsies of a tissue); cell samples (e.g., cytological smears (such as Pap or blood smears) or samples of cells obtained by microdissection); samples of whole organisms (such as samples of yeasts or bacteria); or cell fractions, fragments or organelles (such as obtained by lysing cells and separating the components thereof by centrifugation or otherwise). Other examples of biological samples include blood, serum, urine, semen, fecal matter, cerebrospinal fluid, interstitial fluid, mucous, tears, sweat, pus, biopsied tissue (e.g., obtained by a surgical biopsy or needle biopsy), nipple aspirates, milk, vaginal fluid, saliva, swabs (such as buccal swabs), or any material containing biomolecules that is derived from a first biological sample. In some embodiments, a biological sample is derived from a subject who has, is suspected of having, or is at risk of developing a hematopoietic malignancy. In some embodiments, a biological sample is obtained at a time during or near the time the subject is diagnosed with a hematopoietic malignancy. In some embodiments, a biological sample is obtained at a time during or near when the subject has or is suspected to have experienced relapse of the hematopoietic malignancy. In some embodiments, a biological sample is obtained after the subject has undergone has received at least one treatment for a hematopoietic malignancy. In some embodiments, a biological sample comprises malignant hematopoietic cells. In some embodiments, a biological sample comprises AML blast cells. In some embodiments, methods of the present disclosure further comprise determining the expression level of a first cell surface protein and the expression level of a second cell surface protein in a first biological sample by obtaining or having obtained the first biological sample from a subject and performing or having performed an assay comprising detecting the presence or absence of expression of the first cell surface protein and the second cell surface protein in the first biological sample. In some embodiments, methods of the present disclosure further comprise determining the expression level of the first cell surface protein and the expression level of the second cell surface protein in a second biological sample by obtaining or having obtained the second biological sample from the subject and performing or having performed an assay comprising detecting the presence or absence of expression of the first cell surface protein and the second cell surface protein in the second biological sample. In some embodiments, methods of the present disclosure further comprise identifying or having identified the second biological sample as comprising altered expression of the first cell surface protein and the second cell surface protein when the expression levels of the first cell surface protein and the second cell surface protein are different in the second biological sample relative to the first biological sample. In some embodiments, identifying or having identified the second biological sample as comprising altered expression of the first cell surface protein and the second cell surface protein also identifies the subject as having experienced relapse of the hematopoietic malignancy. In some embodiments, such methods further comprise administering to the subject a at least one of a genetically engineered hematopoietic, or cell population thereof, a cytotoxic agent described herein, or both. In some embodiments, identifying or having identified the second biological sample as comprising altered expression of the first cell surface protein and the second cell surface protein also identifies subject as being in need of administration to the subject a at least one of a genetically engineered hematopoietic, or cell population thereof, a cytotoxic agent described herein, or both, In some embodiments, a biological sample is derived from a subject who has, is suspected of having, or is at risk of developing a mutation in a gene encoding a cell surface protein selected from a group consisting of: CD10; CD101; CD117; CD11A; CD11C; CD120B; CD123; CD13; CD133; CD152; CD200; CD205 (LY75); CD226; CD244 (SLAMF4); CD25; CD274; CD305 (LAIR1); CD32; CD33; CD34; CD366; CD38; CD41; CD42B; CD44; CD45RA; CD47; CD48; CD49D (ITGA4); CD49F; CD52; CD58; CD64; CD7; CD70; CD82; CD84; CD85; CD85K; CD86; CD9; CD96; CD99; CLL-1; EMR2 (ADGRE2); FR-B; GPR56; and IL1RAP. In some embodiments, a biological sample is derived from a subject who has, is suspected of having, or is at risk of developing a mutation in a gene encoding a cell surface protein selected from a group consisting of: CD10; CD101; CD11A; CD11C; CD123; CD133; CD152; CD200; CD205; CD244; CD25; CD305; CD33; CD34; CD366; CD38; CD42B; CD44; CD45RA; CD47; CD48; CD49D; CD49F; CD52; CD58; CD64; CD7; CD84; CD85K; CD9; CD99; CLL-1; EMR2; GPR56; IL1RAP; and CD274. In some embodiments, a biological sample is derived from a subject who has, is suspected of having, or is at risk of developing a mutation in a gene encoding a cell surface protein selected from a group consisting of: CD10; CD117; CD11A; CD11C; CD120B; CD123; CD13; CD133; CD152; CD205; CD22; CD226; CD244; CD25; CD305; CD32; CD33; CD34; CD38; CD41; CD44; CD45RA; CD47; CD48; CD49D; CD49F; CD52; CD56; CD58; CD64; CD7; CD70; CD71; CD82; CD84; CD85K; CD86; CD96; CD99; CLL-1; EMR2; FR-B; GPR56; HLA-DR; and IL1RAP. In some embodiments, a biological sample is derived from a subject who has, is suspected of having, or is at risk of developing a mutation in a first gene encoding a first cell surface protein and a second gene encoding a second cell surface protein selected from a group consisting of: CD274 and CD152; CD274 and CD226; CD152 and CD226; CD10 and CD49F; CD49F and GPR56; CD200 and CD34; CD34 and CD52; CD34 and CD133; GPR56 and CD133; CD42B and CD41; CD85K and CD64; CD85K and CD32; CD64 and CD32; CD64 and CD11C; CD64 and CD86; CD45RA and CD96; CD45RA and IL1RAP; CD45RA and CD123; CD84 and CD33; CD11C and IL1RAP; CD11C and CD33; CD11C and CD86; CD9 and IL1RAP; IL1RAP and CD33; IL1RAP and CD205; IL1RAP and CD305; IL1RAP and CD123; CD33 and CLL-1; CD33 and CD305; CD11A and CD86; CLL-1 and CD305; CD58 and CD305; CD49D and CD47; CD49D and CD99; CD49D and CD44; CD49D and CD305; CD47 and CD99; CD47 and EMR2; CD99 and CD44; CD99 and CD305; CD99 and EMR2; CD205 and CD305; CD205 and CD123; CD44 and CD305; CD44 and EMR2; CD244 and CD117; CD123 and EMR2; EMR2 and CD82; CD152 and CD120B; CD48 and CD11A; CD200 and CD25; CD34 and CD7; CD34 and CD13; CD52 and GPR56; CD52and CD133; CD52 and CD7; GPR56 and CD7; GPR56 and CD82; CD133 and CD7; CD133 and CD82; CD7 and CD13; CD101 and CD64; CD85 and CD70; CD85K and CD86; CD64 and FR-B; CD366 and CD96; CD25 and CD45RA; CD25 and CD96; CD9 and CD82; IL1RAP and CD58; CD11A and CD38; CD11A and CD44; CD11A and CD244; CD38 and CD305; CD38 and EMR2; CLL-1 and CD49D; CD58 and CD49D; CD47 and CD205; and CD47 and CD44. In some embodiments, a biological sample is derived from a subject who has, is suspected of having, or is at risk of developing a mutation in a first gene encoding a first cell surface protein and a second gene encoding a second cell surface protein selected from a group consisting of: CD33 and CD205 (LY75); CD33 and CD244 (SLAMF4); CD33 and CD305 (LAIR1); CD33 and CD49D (ITGA4); CLL-1 and CD205 (LY75); CLL-1 and CD244 (SLAMF4); CLL-1 and CD305 (LAIR1); CLL-1 and CD49D (ITGA4); EMR2 and CD205 (LY75); EMR2 and CD244 (SLAMF4); EMR2 and CD305 (LAIR1); EMR2 and CD49D (ITGA4); CD38 and CD205 (LY75); CD38 and CD244 (SLAMF4); CD38 and CD305 (LAIR1); CD38 and CD49D (ITGA4); CD305 (LAIR1) and CD49D (ITGA4); CD305 (LAIR1) and CD205 (LY75); CD305 (LAIR1) and CD244 (SLAMF4); CD49D (ITGA4) and CD205 (LY75); CD49D (ITGA4) and CD244 (SLAMF4); and CD205 (LY75) and CD244 (SLAMF4). VI. Kits for Therapeutic Uses Also within the scope of the present disclosure are kits for use in treating hematopoietic malignancy. Such a kit may comprise the genetically engineered hematopoietic cells such as HSCs, and optionally one or more cytotoxic agents targeting cell surface proteins, the genes of which are edited in the hematopoietic cells. Such kits may include a container comprising a first pharmaceutical composition that comprises any of the genetically engineered hematopoietic cells as described herein, and optionally one or more additional containers comprising one or more cytotoxic agents (e.g., immune cells expressing chimeric receptors described herein) targeting the cell surface proteins as also described herein. In some embodiments, the kit can comprise instructions for use in any of the methods described herein. The included instructions can comprise a description of administration of the genetically engineered hematopoietic cells and optionally descriptions of administration of the one or more cytotoxic agents to a subject to achieve the intended activity in a subject. The kit may further comprise a description of selecting a subject suitable for treatment based on identifying whether the subject is in need of the treatment. In some embodiments, the instructions comprise a description of administering the genetically engineered hematopoietic cells and optionally the one or more cytotoxic agents to a subject who is in need of the treatment. The instructions relating to the use of the genetically engineered hematopoietic cells and optionally the cytotoxic agents described herein generally include information as to dosage, dosing schedule, and route of administration for the intended treatment. The containers may be unit doses, bulk packages (e.g., multi-dose packages) or sub-unit doses. Instructions supplied in the kits of the disclosure are typically written instructions on a label or package insert. The label or package insert indicates that the pharmaceutical compositions are used for treating, delaying the onset, and/or alleviating a disease or disorder in a subject. The kits provided herein are in suitable packaging. Suitable packaging includes, but is not limited to, vials, bottles, jars, flexible packaging, and the like. Also contemplated are packages for use in combination with a specific device, such as an inhaler, nasal administration device, or an infusion device. A kit may have a sterile access port (for example, the container may be an intravenous solution bag or a vial having a stopper pierceable by a hypodermic injection needle). The container may also have a sterile access port. . Kits optionally may provide additional components such as buffers and interpretive information. Normally, the kit comprises a container and a label or package insert(s) on or associated with the container. In some embodiment, the disclosure provides articles of manufacture comprising contents of the kits described above. General techniques The practice of the present disclosure will employ, unless otherwise indicated, conventional techniques of molecular biology (including recombinant techniques), microbiology, cell biology, biochemistry, and immunology, which are within the skill of the art. Such techniques are explained fully in the literature, such as Molecular Cloning: A Laboratory Manual, second edition (Sambrook, et al., 1989) Cold Spring Harbor Press; Oligonucleotide Synthesis (M. J. Gait, ed.1984); Methods in Molecular Biology, Humana Press; Cell Biology: A Laboratory Notebook (J. E. Cellis, ed., 1989) Academic Press; Animal Cell Culture (R. I. Freshney, ed.1987); Introduction to Cell and Tissue Culture (J. P. Mather and P. E. Roberts, 1998) Plenum Press; Cell and Tissue Culture: Laboratory Procedures (A. Doyle, J. B. Griffiths, and D. G. Newell, eds.1993-8) J. Wiley and Sons; Methods in Enzymology (Academic Press, Inc.); Handbook of Experimental Immunology (D. M. Weir and C. C. Blackwell, eds.): Gene Transfer Vectors for Mammalian Cells (J. M. Miller and M. P. Calos, eds., 1987); Current Protocols in Molecular Biology (F. M. Ausubel, et al. eds. 1987); PCR: The Polymerase Chain Reaction, (Mullis, et al., eds.1994); Current Protocols in Immunology (J. E. Coligan et al., eds., 1991); Short Protocols in Molecular Biology (Wiley and Sons, 1999); Immunobiology (C. A. Janeway and P. Travers, 1997); Antibodies (P. Finch, 1997); Antibodies: a practice approach (D. Catty., ed., IRL Press, 1988-1989); Monoclonal antibodies: a practical approach (P. Shepherd and C. Dean, eds., Oxford University Press, 2000); Using antibodies: a laboratory manual (E. Harlow and D. Lane (Cold Spring Harbor Laboratory Press, 1999); The Antibodies (M. Zanetti and J. D. Capra, eds. Harwood Academic Publishers, 1995); DNA Cloning: A practical Approach, Volumes I and II (D.N. Glover ed.1985); Nucleic Acid Hybridization (B.D. Hames & S.J. Higgins eds.(1985»; Transcription and Translation (B.D. Hames & S.J. Higgins, eds. (1984»; Animal Cell Culture (R.I. Freshney, ed. (1986»; Immobilized Cells and Enzymes (lRL Press, (1986»; and B. Perbal, A practical Guide To Molecular Cloning (1984); F.M. Ausubel et al. (eds.). Without further elaboration, it is believed that one skilled in the art can, based on the above description, utilize the present invention to its fullest extent. The following specific embodiments are, therefore, to be construed as merely illustrative, and not limitative of the remainder of the disclosure in any way whatsoever. All publications cited herein are incorporated by reference for the purposes or subject matter referenced herein. EXAMPLES Example 1: Longitudinal multimodal atlas of AML to enable novel targeting strategies Acute myeloid leukemia (AML) is an aggressive clonal malignancy characterized by combinations of chromosomal abnormalities, gene mutations, and cell surface antigen (Ag) expression profiles. This heterogeneity contributes to refractory or relapsed disease that presents a major challenge when treating AML. Previous single cell sequencing studies of primary AML samples have provided remarkable insight into the clonal architecture of AML cell populations and how their mutational, transcriptomic, and surface Ag profiles vary between patients. However, information addressing clonal shifts during progression from diagnosis to relapse in large cohorts is lacking. Here, single cell RNA sequencing was adapted with surface Ag feature barcoding to analyze more than 450,000 cells from 28 paired AML patient bone marrow mononuclear cell samples collected at diagnosis and relapse (56 samples) to generate the largest and most comprehensive single cell AML atlas to date. This atlas contains rich clinical metadata including cytogenetics, mutation status of canonical AML genes, treatment history, and survival information. These data were leveraged to identify potential correlations with clonal heterogeneity during progression to relapse and to propose novel strategies for targeting the surface of AML cells. The feature barcoding panel consisted of a comprehensive list of 81 surface Ags reported in clinicaltrials.gov or mined from literature, including large proteomics mass spectrometry datasets of AML patient bone marrow samples (DeBoer et. al., Cancer Cell 2018; Jayavelu et al, Cancer Cell 2022). Feature barcoding produced antibody derived tag (ADT) counts which are interpreted as relative values but alone does not indicate absolute number of Ags per cell. This limitation was addressed by measuring absolute Ag density of AML Ags, CD33, CLL-1, CD123, and EMR2 in patient samples using the flow cytometric QuantiBRITE assay. Focusing on these four Ags, the relationship between ADT expression and antibodies per cell (ABC) Ag density from QuantiBRITE was modeled. The model was applied to impute absolute Ag density for the remaining 77 Ags in all samples. UMAP visualization after sample transcriptome integration revealed distinct clustering of CD45-dim myeloblasts, T cells, B cells, and erythroid cells. Longitudinal analysis identified 28 surface Ags that were differentially expressed (absolute log2FC > 1, p < 0.01) between relapse and diagnosis myeloblasts across multiple patients (see, FIG.4A). An additional 19 Ags were differentially expressed in no more than one patient while the other 19 had no significant change in any patient, highlighting the inter-and intra- heterogeneity of AML at diagnosis and relapse in these patients. Targeting multiple antigens simultaneously may help address the issue of antigen heterogeneity of tumor cells and help avoid potential antigen escape. Because multi-targeting therapies are a potential solution to this, the single-cell resolution of the atlas was leveraged to systematically identify Ags that in combination provide maximum coverage and are significantly co-expressed in myeloblasts. From this analysis, 48 and 52 co-expressed Ag pairs were found in diagnosis and relapse blasts, respectively (r > 0.4, p < 0.01; see, FIG.4B). Of note, CD44 and EMR2 were co- expressed at both diagnosis (r = 0.49 p < 0.01 ) and relapse (r = 0.3, p < 0.01) with >90% of their blasts expressing CD44, EMR2, or both at targetable antigen levels of >1000 Ags/cell (see, Table 3). Furthermore, 10 samples had >40% of their blasts co-expressing both Ags simultaneously at >1000 Ags/cell at diagnosis or relapse.6 of these patients had a >10% increase in co-expression at relapse, 3 had >10% decrease at relapse, and one patient had no co-expression change. Table 3. Correlation of Cell surface Proteins That Exhibited Dysregulated Expression at Diagnosis and/or Relapse.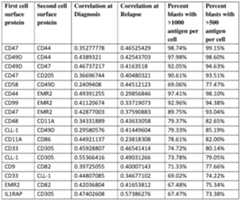
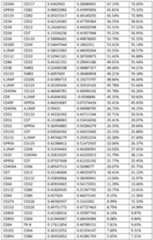
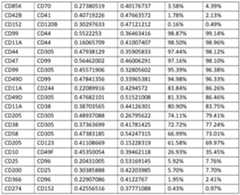 Flow cytometry was further employed to characterize surface expression of four key myeloid antigens including CD33, CLL-1, CD123, and EMR2. More than 80% of AML blasts expressed CD33 at both diagnosis and relapse. A similar trend was observed for CLL- 1, CD123, and EMR2 (FIG.5A). Antigen density, which refers to the number of molecules of an antigen present on a cell surface, can inform the development of effective targeted therapies. AML blasts were contacted with QuantiBRITE beads for quantification of the average number of antibodies bound per blast cell (ABCs). The results suggested that CD33- positive blasts had on average more than 1000 antigens per cell, with the same trend observed for the other 3 antigens (FIG.5B). The data also suggested that these 4 antigens were all expressed at a level that can be targeted by CAR T-cells which are capable of targeting cells expressing approximately 1000 antigens on the cell surface. As shown in FIG.6, mass spectrometry data generated from >200 patient samples in addition to genomic data from the Cancer Surfaceome Atlas was used to derive an initial set of 538 candidates cell surface antigens for further analyses. GTEx and Tabula Sapiens was used to exclude non-heme tissue antigens, reducing the list to 137 novel candidate antigens. The list was combined with antigens being explored in the pre-clinical or clinical context along with 24 cell-type markers to arrive at 196 antigens. Finally, 81 antigens were selected based on whether oligo-tagged antibodies for these antigens were commercially available. With this antigen panel, scRNAseq was performed on matched samples from diagnosis and relapse with feature barcoding on all 56 samples to construct the atlas which was used to characterize heterogeneity across patients and within patients across clinical timepoints (FIGs.6-7). Single-cell RNA sequencing analyses of samples derived from AML subjects were used to correlate cell surface antigen expression profiles to hematopoietic cell lineage (FIGs. 8A-9). A UMAP plot was generated to represent the entire atlas consisting of about 400,00 cells across all patients and timepoints (FIGs.8 and 10). The data was harmonized, clustered, and annotated into broad cell type categories using a combination of cell surface protein and RNA information. Blast cell populations were identified that were characterized by high CD33, CLL-1, CD34 surface expression and diminished CD45 expression (FIG.8B). Monocyte, dendritic cell, T cell, B cell, and erythroid cell populations were also identified in each sample (FIGs.8A and 9). To quantify antigen density, the raw antigen-derived counts (ADT) generated by feature barcoding were generated to provide relative values, and the QuantiBRITE™ data set was used to analyze absolute antigen counts. Then, the relationship between ADTs and QuantiBRITE data for CD33, CLL-1, CD123, and EMR2 was analyzed. A strong linear correlation between the two modalities was observed (FIG.10). This relationship was used to estimate antigen density on each blast based on its ADT expression. An unbiased analysis of the data described above was performed to rank the 81 antigens based on the average surface expression across all blasts. Heatmaps were generated to illustrate estimated antigen density across blasts in each patient at diagnosis and relapse. CD33 and CLL-1 were among the top antigens expressed in blasts at both time points. High expression of both CD99 and CD44 was also observed (FIG.11A). Antigen expression changes between timepoints within individual patients were also assessed which identified antigens with increased expression at relapse (FIG.11A). To determine whether simultaneous targeting of CD33 and CLL-1 could destroy a larger percentage of the blast population through synergy, the correlation between all possible pairs of antigens across blasts belonging to an individual sample was systematically computed to construct a correlation matrix (see data for “Patient 70” in FIGs.12A-12B). These analyses revealed that CD33 and CLL-1 expression was highly correlated (FIG.12A). At the 300-400 range for antigen density, both CD33 and CLL-1 were co-expressed in all blasts. When the antigen threshold criteria were increased, lower co-expression of both CD33 and CLL-1 was observed but the addition of CLL-1 encompasses additional blasts that express CD33 at a lower level (FIG.12B). These results suggest that a multi-targeting strategy, e.g., with multi-specific CAR T cells, can enhance blast killing and guard against antigen escape. Blasts from patient 26 were extracted and used to re-clustered the data. Clear separation between blasts collected from the two timepoints (diagnosis and relapse) was observed (FIG.13A). CD33 was upregulated 4-fold in the relapse blasts (FIG.13B and 13C). Using the RNA component of the atlas, cell cycle module analysis was performed, and it was found that 50% of relapse blasts were in the S and G2/M phases, compared to 13% in diagnosis blasts (FIG.13D), suggesting that relapse blasts were in a more proliferative cell state. The proliferating blasts were further analyzed using differential gene expression analysis between the two timepoints (AML diagnosis and relapse). This identified 256 upregulated genes and 772 down-regulated genes (FIG.13E). Pathway enrichment analysis using the Hallmark gene sets showed a significant downregulation of inflammatory response pathways including interferon gamma response, TNF alpha signaling, and interferon alpha response (FIG.14A). There was also significant upregulation of oxidative phosphorylation and an increased expression of MYC and E2F target genes (FIG.14B). Further, cellular indexing of transcriptomes and epitopes (CITE-seq) profiling was performed to assess expression of 81 candidate antigens across 400,000 cells from 28 paired diagnosis and relapse patient bone marrow samples (FIG.15A). One AML target, EMR2, is among the top 30 highest surface expressors in both diagnosis and relapsed samples (FIG. 15A and Table 4). This multimodal profiling was paired with flow cytometry surface expression analysis. Single cell fluorescent intensities of CD33, CLL-1, CD123, and EMR2 from the flow cytometry data were converted to antigen density based on a standard curve produced from the QuantiBRITE assay.5% quantiles of this antigen density distribution were calculated for each antigen for each sample.5% quantiles were then calculated for the Antigen Derived Tag (ADT) readout for same antigens and samples and matched with the former quantiles produced from QuantiBRITE. A random forest machine learning model (Ho, T. K. (1995). Random decision forests. Proceedings of 3rd international conference on document analysis and recognition (Vol.1, pp.278–282)) was trained to predict the quantile values from QuantiBRITE using the quantile values from ADT and antibody quality control metrics from Cellranger (10X Genomics). The trained model was then used to predict antigen density for the remaining 81 antigens across all cells in the atlas (FIG.15B). Table 4. Antigens Showing High Surface Expression on Patient Blasts at AML Diagnosis and Relapse from FIG.15A
Flow cytometry was further employed to characterize surface expression of four key myeloid antigens including CD33, CLL-1, CD123, and EMR2. More than 80% of AML blasts expressed CD33 at both diagnosis and relapse. A similar trend was observed for CLL- 1, CD123, and EMR2 (FIG.5A). Antigen density, which refers to the number of molecules of an antigen present on a cell surface, can inform the development of effective targeted therapies. AML blasts were contacted with QuantiBRITE beads for quantification of the average number of antibodies bound per blast cell (ABCs). The results suggested that CD33- positive blasts had on average more than 1000 antigens per cell, with the same trend observed for the other 3 antigens (FIG.5B). The data also suggested that these 4 antigens were all expressed at a level that can be targeted by CAR T-cells which are capable of targeting cells expressing approximately 1000 antigens on the cell surface. As shown in FIG.6, mass spectrometry data generated from >200 patient samples in addition to genomic data from the Cancer Surfaceome Atlas was used to derive an initial set of 538 candidates cell surface antigens for further analyses. GTEx and Tabula Sapiens was used to exclude non-heme tissue antigens, reducing the list to 137 novel candidate antigens. The list was combined with antigens being explored in the pre-clinical or clinical context along with 24 cell-type markers to arrive at 196 antigens. Finally, 81 antigens were selected based on whether oligo-tagged antibodies for these antigens were commercially available. With this antigen panel, scRNAseq was performed on matched samples from diagnosis and relapse with feature barcoding on all 56 samples to construct the atlas which was used to characterize heterogeneity across patients and within patients across clinical timepoints (FIGs.6-7). Single-cell RNA sequencing analyses of samples derived from AML subjects were used to correlate cell surface antigen expression profiles to hematopoietic cell lineage (FIGs. 8A-9). A UMAP plot was generated to represent the entire atlas consisting of about 400,00 cells across all patients and timepoints (FIGs.8 and 10). The data was harmonized, clustered, and annotated into broad cell type categories using a combination of cell surface protein and RNA information. Blast cell populations were identified that were characterized by high CD33, CLL-1, CD34 surface expression and diminished CD45 expression (FIG.8B). Monocyte, dendritic cell, T cell, B cell, and erythroid cell populations were also identified in each sample (FIGs.8A and 9). To quantify antigen density, the raw antigen-derived counts (ADT) generated by feature barcoding were generated to provide relative values, and the QuantiBRITE™ data set was used to analyze absolute antigen counts. Then, the relationship between ADTs and QuantiBRITE data for CD33, CLL-1, CD123, and EMR2 was analyzed. A strong linear correlation between the two modalities was observed (FIG.10). This relationship was used to estimate antigen density on each blast based on its ADT expression. An unbiased analysis of the data described above was performed to rank the 81 antigens based on the average surface expression across all blasts. Heatmaps were generated to illustrate estimated antigen density across blasts in each patient at diagnosis and relapse. CD33 and CLL-1 were among the top antigens expressed in blasts at both time points. High expression of both CD99 and CD44 was also observed (FIG.11A). Antigen expression changes between timepoints within individual patients were also assessed which identified antigens with increased expression at relapse (FIG.11A). To determine whether simultaneous targeting of CD33 and CLL-1 could destroy a larger percentage of the blast population through synergy, the correlation between all possible pairs of antigens across blasts belonging to an individual sample was systematically computed to construct a correlation matrix (see data for “Patient 70” in FIGs.12A-12B). These analyses revealed that CD33 and CLL-1 expression was highly correlated (FIG.12A). At the 300-400 range for antigen density, both CD33 and CLL-1 were co-expressed in all blasts. When the antigen threshold criteria were increased, lower co-expression of both CD33 and CLL-1 was observed but the addition of CLL-1 encompasses additional blasts that express CD33 at a lower level (FIG.12B). These results suggest that a multi-targeting strategy, e.g., with multi-specific CAR T cells, can enhance blast killing and guard against antigen escape. Blasts from patient 26 were extracted and used to re-clustered the data. Clear separation between blasts collected from the two timepoints (diagnosis and relapse) was observed (FIG.13A). CD33 was upregulated 4-fold in the relapse blasts (FIG.13B and 13C). Using the RNA component of the atlas, cell cycle module analysis was performed, and it was found that 50% of relapse blasts were in the S and G2/M phases, compared to 13% in diagnosis blasts (FIG.13D), suggesting that relapse blasts were in a more proliferative cell state. The proliferating blasts were further analyzed using differential gene expression analysis between the two timepoints (AML diagnosis and relapse). This identified 256 upregulated genes and 772 down-regulated genes (FIG.13E). Pathway enrichment analysis using the Hallmark gene sets showed a significant downregulation of inflammatory response pathways including interferon gamma response, TNF alpha signaling, and interferon alpha response (FIG.14A). There was also significant upregulation of oxidative phosphorylation and an increased expression of MYC and E2F target genes (FIG.14B). Further, cellular indexing of transcriptomes and epitopes (CITE-seq) profiling was performed to assess expression of 81 candidate antigens across 400,000 cells from 28 paired diagnosis and relapse patient bone marrow samples (FIG.15A). One AML target, EMR2, is among the top 30 highest surface expressors in both diagnosis and relapsed samples (FIG. 15A and Table 4). This multimodal profiling was paired with flow cytometry surface expression analysis. Single cell fluorescent intensities of CD33, CLL-1, CD123, and EMR2 from the flow cytometry data were converted to antigen density based on a standard curve produced from the QuantiBRITE assay.5% quantiles of this antigen density distribution were calculated for each antigen for each sample.5% quantiles were then calculated for the Antigen Derived Tag (ADT) readout for same antigens and samples and matched with the former quantiles produced from QuantiBRITE. A random forest machine learning model (Ho, T. K. (1995). Random decision forests. Proceedings of 3rd international conference on document analysis and recognition (Vol.1, pp.278–282)) was trained to predict the quantile values from QuantiBRITE using the quantile values from ADT and antibody quality control metrics from Cellranger (10X Genomics). The trained model was then used to predict antigen density for the remaining 81 antigens across all cells in the atlas (FIG.15B). Table 4. Antigens Showing High Surface Expression on Patient Blasts at AML Diagnosis and Relapse from FIG.15A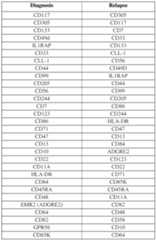 The comprehensive profiling of AML described herein has provided high resolution insight on cell surface proteins at diagnosis and during disease relapse and produced novel conclusions about targetable combinations of surface antigens based on expression changes between timepoints, co-expression, and surface densities. Furthermore, the reference atlas can be further dissected to characterize the transcriptome and mutational profiles of myeloblasts at diagnosis and relapse thereby providing a resource to identify correlations between molecular and immunophenotypic features during disease progression. Example 2: Multiplex Editing of Cell Surface Antigens Showing High Expression at AML Diagnosis and Relapse This Example relates to a strategy for multiplexed genetic editing of cell surface antigens (e.g., two or more of EMR2, CD33, CD123, and CLL-1) in healthy HSCs to enable combinatorial targeting of AML cells. Using multiplexed base editing, a “protected” HSC transplant can be generated which enables selective eradication of AML blast and leukemic stem cells by administration of combinatorial immunotherapy. Briefly, CD34+ cells were thawed and two days later electroporated with adenosine base editor mRNA and either a gRNA targeting splicing regulatory sequences in exon 19 of EMR2, a gRNA targeting splicing regulatory sequences in exon 5 of CD33, or both gRNAs simultaneously (FIG.16A and Table 5). For multiplex base editing conditions, equal amounts of EMR2 and CD33 gRNAs were electroporated. No electroporation, mock electroporation and electroporation with a control guide were included as electroporation controls. Sequence information for the gRNAs is shown in Table 5. Two days and five days post-EP, cells were taken for gDNA editing analysis via rhAmpSeq (NGS) and for surface protein analysis via flow cytometry (FIG.16B). Table 5. Examples of gRNAs for Multiplex Adenosine Base Editing of EMR2 and CD33
The comprehensive profiling of AML described herein has provided high resolution insight on cell surface proteins at diagnosis and during disease relapse and produced novel conclusions about targetable combinations of surface antigens based on expression changes between timepoints, co-expression, and surface densities. Furthermore, the reference atlas can be further dissected to characterize the transcriptome and mutational profiles of myeloblasts at diagnosis and relapse thereby providing a resource to identify correlations between molecular and immunophenotypic features during disease progression. Example 2: Multiplex Editing of Cell Surface Antigens Showing High Expression at AML Diagnosis and Relapse This Example relates to a strategy for multiplexed genetic editing of cell surface antigens (e.g., two or more of EMR2, CD33, CD123, and CLL-1) in healthy HSCs to enable combinatorial targeting of AML cells. Using multiplexed base editing, a “protected” HSC transplant can be generated which enables selective eradication of AML blast and leukemic stem cells by administration of combinatorial immunotherapy. Briefly, CD34+ cells were thawed and two days later electroporated with adenosine base editor mRNA and either a gRNA targeting splicing regulatory sequences in exon 19 of EMR2, a gRNA targeting splicing regulatory sequences in exon 5 of CD33, or both gRNAs simultaneously (FIG.16A and Table 5). For multiplex base editing conditions, equal amounts of EMR2 and CD33 gRNAs were electroporated. No electroporation, mock electroporation and electroporation with a control guide were included as electroporation controls. Sequence information for the gRNAs is shown in Table 5. Two days and five days post-EP, cells were taken for gDNA editing analysis via rhAmpSeq (NGS) and for surface protein analysis via flow cytometry (FIG.16B). Table 5. Examples of gRNAs for Multiplex Adenosine Base Editing of EMR2 and CD33 Editing frequencies analyzed after subjecting cells to single gene or multiplex adenosine base editing conditions demonstrated efficient splice site disruption in EMR2 and CD33 target genes (FIG.16C) and were correlated with reduced cell surface expression of the antigens (FIG.16D). In addition to genetically engineered cells comprising edited EMR2 and CD33 genes, multiplex base editing was utilized to genetically engineer the EMR2, CD33, CD123, and CLL-1 genes. Cells that were subjected to adenosine base editing conditions that utilized a non-targeting control gRNA or gRNAs targeting the EMR2, CD33, CD123, and CLL-1 genes. At 16 days (D16) post-electroporation under base editing conditions, flow cytometry was used to analyze the number of cells in the population having cell surface expression of one, two, three, or four of the antigens selected from CD33, CD123, EMR2, and CLL-1 (FIG. 17). Example 3: Multimodal Atlas of Paired Diagnosis and Relapse AML Reveals Surface Antigens for Multi-specific Immunotherapy This Example relates to an expanded cell surface antigen atlas that included additional samples from healthy donors and categorizations of blasts into hematopoietic cell lineages. The dataset of blast heterogeneity across 68 samples was used to identify novel cell surface antigens (Ags) that can be targeted with multi-specific immunotherapies. As described in the present disclosure, all samples in the atlas included both feature barcoding with antigen derived tags (ADT) on 81 Ags and QuantiBRITE flow cytometric data, measured as antibodies bound per cell (ABC), on four well-characterized AML Ags (CD33, CLL-1, CD123, and EMR2). Using patient-matched data from these two assays, random forests were trained to model the relationship between ABC and antigen derived tag (ADT) readouts for these Ags and was applied to estimate Ag count for the remaining 77 Ags on individual blasts for all samples. This represents a novel method for integrating sequencing and fluorescence data with machine learning to infer Ag count. Effective target Ag combinations are collectively expressed on blasts to enable recognition by multi-specific therapies. Moreover, co-expression of these Ags on individual blasts can guard against potential Ag escape driven by blast heterogeneity. As observed in the AML atlas, CD33 and CLL-1 cover a considerable fraction of blasts. However, to increase the likelihood of targeting all blasts, the Ags in this expanded cell surface antigen atlas were ranked which, when combined together, or with CD33 and CLL-1, label the maximum number of blasts at a sufficient therapeutic threshold for immunotherapy (Ag count >1000/cell) (Sharma and Kranz, 2016). Four additional Ag candidates were identified from the atlas: LAIR1, ITGA4, LY75, and CD244 that were expressed in 23, 26, 18, and 15 of 28 diagnosis samples, and 24, 25, 18, and 17 of 28 relapse samples, respectively, on >80% of blasts in the sample at >500 predicted Ags/blast (FIGs.18A-18B and FIG.19A). Additionally, six co-expressed antigen pairs (CD305 & CD49d; CD305 & CD205; CD305 & CD244; CD49d & CD205; CD49d & CD244; CD205 & CD244) were identified as effective for multi-targeted therapeutics because when combined, they are highly expressed in a large number of patient samples (FIG.19B). As validation, QuantiBRITE was used to measure ABCs of the four Ags on four samples selected from the atlas repository (FIGs.21A-21B). High consistency between the measured ABC readout and the predicted ABC was observed for all four Ags and samples (Pearson r = 0.87, p = 9.9e-6) (and FIG.22). Therapeutic efficacy of targeting these Ags individually was evaluated using in vitro cytotoxicity secondary ADC assays in MOLM-13 cells, providing a measure of sensitivity to the ADC as a function of Ag expression. Greater than 50% ADC-mediated cell killing at <0.1nM primary antibody for all four Ags was also observed (FIGs.20A-20B). In summary, rich multimodal data from the atlas, machine learning, and experimental validation were combined to identify promising combinations of Ag targets, including CD33, CLL-1, LAIR1, ITGA4, LY75, and CD244. These targets were widely present across blasts and exhibited co-expression on individual blast cells in a large cohort of matched AML patient samples. These novel combinations can be leveraged to develop future novel multi- targeting strategies with multi-specific agents such as CAR-T cells or ADCs that can combat Ag heterogeneity and guard against potential Ag escape. OTHER EMBODIMENTS All of the features disclosed in this specification may be combined in any combination. Each feature disclosed in this specification may be replaced by an alternative feature serving the same, equivalent, or similar purpose. Thus, unless expressly stated otherwise, each feature disclosed is only an example of a generic series of equivalent or similar features. From the above description, one skilled in the art can easily ascertain the essential characteristics of the present invention, and without departing from the spirit and scope thereof, can make various changes and modifications of the invention to adapt it to various usages and conditions. Thus, other embodiments are also within the claims. EQUIVALENTS While several inventive embodiments have been described and illustrated herein, those of ordinary skill in the art will readily envision a variety of other means and/or structures for performing the function and/or obtaining the results and/or one or more of the advantages described herein, and each of such variations and/or modifications is deemed to be within the scope of the inventive embodiments described herein. More generally, those skilled in the art will readily appreciate that all parameters, dimensions, materials, and configurations described herein are meant to be exemplary and that the actual parameters, dimensions, materials, and/or configurations will depend upon the specific application or applications for which the inventive teachings is/are used. Those skilled in the art will recognize, or be able to ascertain using no more than routine experimentation, many equivalents to the specific inventive embodiments described herein. It is, therefore, to be understood that the foregoing embodiments are presented by way of example only and that, within the scope of the appended claims and equivalents thereto, inventive embodiments may be practiced otherwise than as specifically described and claimed. Inventive embodiments of the present disclosure are directed to each individual feature, system, article, material, kit, and/or method described herein. In addition, any combination of two or more such features, systems, articles, materials, kits, and/or methods, if such features, systems, articles, materials, kits, and/or methods are not mutually inconsistent, is included within the inventive scope of the present disclosure. All definitions, as defined and used herein, should be understood to control over dictionary definitions, definitions in documents incorporated by reference, and/or ordinary meanings of the defined terms. All references, patents and patent applications disclosed herein are incorporated by reference with respect to the subject matter for which each is cited, which in some cases may encompass the entirety of the document. The indefinite articles “a” and “an,” as used herein in the specification and in the claims, unless clearly indicated to the contrary, should be understood to mean “at least one.” The phrase “and/or,” as used herein in the specification and in the claims, should be understood to mean “either or both” of the elements so conjoined, i.e., elements that are conjunctively present in some cases and disjunctively present in other cases. Multiple elements listed with “and/or” should be construed in the same fashion, i.e., “one or more” of the elements so conjoined. Other elements may optionally be present other than the elements specifically identified by the “and/or” clause, whether related or unrelated to those elements specifically identified. Thus, as a non-limiting example, a reference to “A and/or B,” when used in conjunction with open-ended language such as “comprising” can refer, in one embodiment, to A only (optionally including elements other than B); in another embodiment, to B only (optionally including elements other than A); in yet another embodiment, to both A and B (optionally including other elements); etc. As used herein in the specification and in the claims, “or” should be understood to have the same meaning as “and/or” as defined above. For example, when separating items in a list, “or” or “and/or” shall be interpreted as being inclusive, i.e., the inclusion of at least one, but also including more than one, of a number or list of elements, and, optionally, additional unlisted items. Only terms clearly indicated to the contrary, such as “only one of” or “exactly one of,” or, when used in the claims, “consisting of,” will refer to the inclusion of exactly one element of a number or list of elements. In general, the term “or” as used herein shall only be interpreted as indicating exclusive alternatives (i.e. “one or the other but not both”) when preceded by terms of exclusivity, such as “either,” “one of,” “only one of,” or “exactly one of.” “Consisting essentially of,” when used in the claims, shall have its ordinary meaning as used in the field of patent law. As used herein in the specification and in the claims, the phrase “at least one,” in reference to a list of one or more elements, should be understood to mean at least one element selected from any one or more of the elements in the list of elements, but not necessarily including at least one of each and every element specifically listed within the list of elements and not excluding any combinations of elements in the list of elements. This definition also allows that elements may optionally be present other than the elements specifically identified within the list of elements to which the phrase “at least one” refers, whether related or unrelated to those elements specifically identified. Thus, as a non-limiting example, “at least one of A and B” (or, equivalently, “at least one of A or B,” or, equivalently “at least one of A and/or B”) can refer, in one embodiment, to at least one, optionally including more than one, A, with no B present (and optionally including elements other than B); in another embodiment, to at least one, optionally including more than one, B, with no A present (and optionally including elements other than A); in yet another embodiment, to at least one, optionally including more than one, A, and at least one, optionally including more than one, B (and optionally including other elements); etc. It should also be understood that, unless clearly indicated to the contrary, in any methods claimed herein that include more than one step or act, the order of the steps or acts of the method is not necessarily limited to the order in which the steps or acts of the method are recited.
Editing frequencies analyzed after subjecting cells to single gene or multiplex adenosine base editing conditions demonstrated efficient splice site disruption in EMR2 and CD33 target genes (FIG.16C) and were correlated with reduced cell surface expression of the antigens (FIG.16D). In addition to genetically engineered cells comprising edited EMR2 and CD33 genes, multiplex base editing was utilized to genetically engineer the EMR2, CD33, CD123, and CLL-1 genes. Cells that were subjected to adenosine base editing conditions that utilized a non-targeting control gRNA or gRNAs targeting the EMR2, CD33, CD123, and CLL-1 genes. At 16 days (D16) post-electroporation under base editing conditions, flow cytometry was used to analyze the number of cells in the population having cell surface expression of one, two, three, or four of the antigens selected from CD33, CD123, EMR2, and CLL-1 (FIG. 17). Example 3: Multimodal Atlas of Paired Diagnosis and Relapse AML Reveals Surface Antigens for Multi-specific Immunotherapy This Example relates to an expanded cell surface antigen atlas that included additional samples from healthy donors and categorizations of blasts into hematopoietic cell lineages. The dataset of blast heterogeneity across 68 samples was used to identify novel cell surface antigens (Ags) that can be targeted with multi-specific immunotherapies. As described in the present disclosure, all samples in the atlas included both feature barcoding with antigen derived tags (ADT) on 81 Ags and QuantiBRITE flow cytometric data, measured as antibodies bound per cell (ABC), on four well-characterized AML Ags (CD33, CLL-1, CD123, and EMR2). Using patient-matched data from these two assays, random forests were trained to model the relationship between ABC and antigen derived tag (ADT) readouts for these Ags and was applied to estimate Ag count for the remaining 77 Ags on individual blasts for all samples. This represents a novel method for integrating sequencing and fluorescence data with machine learning to infer Ag count. Effective target Ag combinations are collectively expressed on blasts to enable recognition by multi-specific therapies. Moreover, co-expression of these Ags on individual blasts can guard against potential Ag escape driven by blast heterogeneity. As observed in the AML atlas, CD33 and CLL-1 cover a considerable fraction of blasts. However, to increase the likelihood of targeting all blasts, the Ags in this expanded cell surface antigen atlas were ranked which, when combined together, or with CD33 and CLL-1, label the maximum number of blasts at a sufficient therapeutic threshold for immunotherapy (Ag count >1000/cell) (Sharma and Kranz, 2016). Four additional Ag candidates were identified from the atlas: LAIR1, ITGA4, LY75, and CD244 that were expressed in 23, 26, 18, and 15 of 28 diagnosis samples, and 24, 25, 18, and 17 of 28 relapse samples, respectively, on >80% of blasts in the sample at >500 predicted Ags/blast (FIGs.18A-18B and FIG.19A). Additionally, six co-expressed antigen pairs (CD305 & CD49d; CD305 & CD205; CD305 & CD244; CD49d & CD205; CD49d & CD244; CD205 & CD244) were identified as effective for multi-targeted therapeutics because when combined, they are highly expressed in a large number of patient samples (FIG.19B). As validation, QuantiBRITE was used to measure ABCs of the four Ags on four samples selected from the atlas repository (FIGs.21A-21B). High consistency between the measured ABC readout and the predicted ABC was observed for all four Ags and samples (Pearson r = 0.87, p = 9.9e-6) (and FIG.22). Therapeutic efficacy of targeting these Ags individually was evaluated using in vitro cytotoxicity secondary ADC assays in MOLM-13 cells, providing a measure of sensitivity to the ADC as a function of Ag expression. Greater than 50% ADC-mediated cell killing at <0.1nM primary antibody for all four Ags was also observed (FIGs.20A-20B). In summary, rich multimodal data from the atlas, machine learning, and experimental validation were combined to identify promising combinations of Ag targets, including CD33, CLL-1, LAIR1, ITGA4, LY75, and CD244. These targets were widely present across blasts and exhibited co-expression on individual blast cells in a large cohort of matched AML patient samples. These novel combinations can be leveraged to develop future novel multi- targeting strategies with multi-specific agents such as CAR-T cells or ADCs that can combat Ag heterogeneity and guard against potential Ag escape. OTHER EMBODIMENTS All of the features disclosed in this specification may be combined in any combination. Each feature disclosed in this specification may be replaced by an alternative feature serving the same, equivalent, or similar purpose. Thus, unless expressly stated otherwise, each feature disclosed is only an example of a generic series of equivalent or similar features. From the above description, one skilled in the art can easily ascertain the essential characteristics of the present invention, and without departing from the spirit and scope thereof, can make various changes and modifications of the invention to adapt it to various usages and conditions. Thus, other embodiments are also within the claims. EQUIVALENTS While several inventive embodiments have been described and illustrated herein, those of ordinary skill in the art will readily envision a variety of other means and/or structures for performing the function and/or obtaining the results and/or one or more of the advantages described herein, and each of such variations and/or modifications is deemed to be within the scope of the inventive embodiments described herein. More generally, those skilled in the art will readily appreciate that all parameters, dimensions, materials, and configurations described herein are meant to be exemplary and that the actual parameters, dimensions, materials, and/or configurations will depend upon the specific application or applications for which the inventive teachings is/are used. Those skilled in the art will recognize, or be able to ascertain using no more than routine experimentation, many equivalents to the specific inventive embodiments described herein. It is, therefore, to be understood that the foregoing embodiments are presented by way of example only and that, within the scope of the appended claims and equivalents thereto, inventive embodiments may be practiced otherwise than as specifically described and claimed. Inventive embodiments of the present disclosure are directed to each individual feature, system, article, material, kit, and/or method described herein. In addition, any combination of two or more such features, systems, articles, materials, kits, and/or methods, if such features, systems, articles, materials, kits, and/or methods are not mutually inconsistent, is included within the inventive scope of the present disclosure. All definitions, as defined and used herein, should be understood to control over dictionary definitions, definitions in documents incorporated by reference, and/or ordinary meanings of the defined terms. All references, patents and patent applications disclosed herein are incorporated by reference with respect to the subject matter for which each is cited, which in some cases may encompass the entirety of the document. The indefinite articles “a” and “an,” as used herein in the specification and in the claims, unless clearly indicated to the contrary, should be understood to mean “at least one.” The phrase “and/or,” as used herein in the specification and in the claims, should be understood to mean “either or both” of the elements so conjoined, i.e., elements that are conjunctively present in some cases and disjunctively present in other cases. Multiple elements listed with “and/or” should be construed in the same fashion, i.e., “one or more” of the elements so conjoined. Other elements may optionally be present other than the elements specifically identified by the “and/or” clause, whether related or unrelated to those elements specifically identified. Thus, as a non-limiting example, a reference to “A and/or B,” when used in conjunction with open-ended language such as “comprising” can refer, in one embodiment, to A only (optionally including elements other than B); in another embodiment, to B only (optionally including elements other than A); in yet another embodiment, to both A and B (optionally including other elements); etc. As used herein in the specification and in the claims, “or” should be understood to have the same meaning as “and/or” as defined above. For example, when separating items in a list, “or” or “and/or” shall be interpreted as being inclusive, i.e., the inclusion of at least one, but also including more than one, of a number or list of elements, and, optionally, additional unlisted items. Only terms clearly indicated to the contrary, such as “only one of” or “exactly one of,” or, when used in the claims, “consisting of,” will refer to the inclusion of exactly one element of a number or list of elements. In general, the term “or” as used herein shall only be interpreted as indicating exclusive alternatives (i.e. “one or the other but not both”) when preceded by terms of exclusivity, such as “either,” “one of,” “only one of,” or “exactly one of.” “Consisting essentially of,” when used in the claims, shall have its ordinary meaning as used in the field of patent law. As used herein in the specification and in the claims, the phrase “at least one,” in reference to a list of one or more elements, should be understood to mean at least one element selected from any one or more of the elements in the list of elements, but not necessarily including at least one of each and every element specifically listed within the list of elements and not excluding any combinations of elements in the list of elements. This definition also allows that elements may optionally be present other than the elements specifically identified within the list of elements to which the phrase “at least one” refers, whether related or unrelated to those elements specifically identified. Thus, as a non-limiting example, “at least one of A and B” (or, equivalently, “at least one of A or B,” or, equivalently “at least one of A and/or B”) can refer, in one embodiment, to at least one, optionally including more than one, A, with no B present (and optionally including elements other than B); in another embodiment, to at least one, optionally including more than one, B, with no A present (and optionally including elements other than A); in yet another embodiment, to at least one, optionally including more than one, A, and at least one, optionally including more than one, B (and optionally including other elements); etc. It should also be understood that, unless clearly indicated to the contrary, in any methods claimed herein that include more than one step or act, the order of the steps or acts of the method is not necessarily limited to the order in which the steps or acts of the method are recited.









Claims
CLAIMS What Is Claimed Is: 1. A genetically engineered hematopoietic cell, or descendant thereof, comprising: a) a mutation in a first gene encoding a first cell surface protein such that the genetically engineered hematopoietic cell has i) reduced or eliminated expression of the first cell surface protein relative to a wildtype counterpart cell or ii) expresses a mutant of the first cell surface protein; and b) a mutation in a second gene encoding a second cell surface protein such that the genetically engineered hematopoietic cell has i) reduced or eliminated expression of the second cell surface protein relative to the wildtype counterpart cell or ii) expresses a mutant of the second cell surface protein, and wherein the first cell surface protein and the second cell surface protein are of a combination as set forth in Table 3 or Table 4.
2. The genetically engineered hematopoietic cell, or descendant thereof, of claim 1, wherein the whole or a portion of the first gene and/or the second gene is deleted.
3. The genetically engineered hematopoietic cell, or descendant thereof, of claim 1 or 2, wherein the whole or a portion of the first gene and/or the second gene is deleted using genome editing.
4. The genetically engineered hematopoietic cell, or descendant thereof, of claim 3, wherein the genome editing carried out involves a zinc finger nuclease (ZFN), a transcription activator-like effector-based nuclease (TALEN), or a CRISPR-Cas system.
5. The genetically engineered hematopoietic cell, or descendant thereof, of claim 4, wherein the CRISPR-Cas system comprises a nucleic acid encoding a gRNA and an RNA- guided nuclease.
6. The genetically engineered hematopoietic cell, or descendant thereof, of claim 5, wherein the RNA-guided nuclease is a base editor.
7. The genetically engineered hematopoietic cell, or descendant thereof, of any one of claims 1-6, wherein the first cell surface protein and the second cell surface protein are selected from the group consisting of: CD10; CD101; CD117; CD11A; CD11C; CD120B; CD123; CD13; CD133; CD152; CD200; CD205 (LY75); CD226; CD244 (SLAMF4); CD25; CD274; CD305 (LAIR1); CD32; CD33; CD34; CD366; CD38; CD41; CD42B; CD44; CD45RA; CD47; CD48; CD49D (ITGA4); CD49F; CD52; CD58; CD64; CD7; CD70; CD82; CD84; CD85; CD85K; CD86; CD9; CD96; CD99; CLL-1; EMR2 (ADGRE2); FR-B; GPR56; and IL1RAP.
8. The genetically engineered hematopoietic cell, or descendant thereof, of any one of claims 1-7, wherein the first cell surface protein and the second cell surface protein are selected from the group consisting of: CD85K and CD64; CD45RA and IL1RAP; CD34 and CD133; IL1RAP and CD33; CD47 and CD99; CLL-1 and CD305 (LAIR1); CD152 and CD226; CD45RA and CD96; CD45RA and CD123; CD49D (ITGA4) and CD305 (LAIR1); IL1RAP and CD305 (LAIR1); CD58 and CD305 (LAIR1); CD49D (ITGA4) and CD47; CD33 and CD305 (LAIR1); CD49D (ITGA4) and CD44; CD64 and CD86; GPR56 and CD133; CD34 and CD52; EMR2 and CD82; CD64 and CD32; CD42B and CD41; CD64 and CD11C; CD99 and CD44; CD200 and CD34; CD11C and CD33; IL1RAP and CD123; CD11C and IL1RAP; CD44 and EMR2; CD205 (LY75) and CD305 (LAIR1); CD44 and CD305 (LAIR1); CD49D (ITGA4) and CD99; CD274 and CD226; CD49F and GPR56; CD9 and IL1RAP; CD99 and CD305 (LAIR1); CD10 and CD49F; CD11A and CD86; CD33 and CLL-1; CD84 and CD33; CD244 (SLAMF4) and CD117; CD85K and CD32; CD47 and EMR2; CD274 and CD152; IL1RAP and CD205 (LY75); CD11C and CD86; CD99 and EMR2; CD205 (LY75) and CD123; and CD123 and EMR2.
9. The genetically engineered hematopoietic cell, or descendant thereof, of any one of claims 1-7, wherein the first cell surface protein and the second cell surface protein are selected from the group consisting of: CD33 and CD205 (LY75); CD33 and CD244 (SLAMF4); CD33 and CD305 (LAIR1); CD33 and CD49D (ITGA4); CLL-1 and CD205 (LY75); CLL-1 and CD244 (SLAMF4); CLL-1 and CD305 (LAIR1); CLL-1 and CD49D (ITGA4); EMR2 and CD205 (LY75); EMR2 and CD244 (SLAMF4); EMR2 and CD305 (LAIR1); EMR2 and CD49D (ITGA4); CD38 and CD205 (LY75); CD38 and CD244 (SLAMF4); CD38 and CD305 (LAIR1); CD38 and CD49D (ITGA4); CD305 (LAIR1) and CD49D (ITGA4); CD305 (LAIR1) and CD205 (LY75); CD305 (LAIR1) and CD244 (SLAMF4); CD49D (ITGA4) and CD205 (LY75); CD49D (ITGA4) and CD244 (SLAMF4); and CD205 (LY75) and CD244 (SLAMF4).
10. The genetically engineered hematopoietic cell, or descendant thereof, of any one of claims 1-8, wherein the first cell surface protein is CD33 and the second cell surface protein is IL1RAP.
11. The genetically engineered hematopoietic cell, or descendant thereof, of any one of claims 1-8, wherein the first cell surface protein is CD44, and the second cell surface protein is EMR2.
12. The genetically engineered hematopoietic cell, or descendant thereof, of any one of claims 1-11, wherein the mutation of the first gene and/or the second gene comprises a substitution, deletion, or addition of at least one nucleotide or a combination thereof.
13. The genetically engineered hematopoietic cell, or descendant thereof, of any one of claims 1-12, wherein the mutation of the first gene and/or the second gene comprises a substitution, deletion, or addition of at least one nucleotide or a combination thereof in a sequence encoding a non-essential epitope of the first and/or the second cell surface protein.
14. The genetically engineered hematopoietic cell, or descendant thereof, of any one of claims 1-13, wherein expression of the first cell surface protein and/or the second cell surface protein is reduced by at least 20%, at least 30%, at least 40%, at least 50%, at least 60%, at least 70%, at least 80%, at least 90%, or at least 95% relative to the wildtype counterpart cell.
15. The genetically engineered hematopoietic cell, or descendant thereof, of any one of claims 1-14, wherein expression of the first cell surface protein and/or the second cell surface protein is not substantially altered, wherein the mutant of the first cell surface protein and/or the mutant of the second cell surface protein comprise a modified non-essential epitope.
16. The genetically engineered hematopoietic cell, or descendant thereof, of any one of claims 1-15, wherein the mutation alters 1, 2, 3, 4, or 5 amino acid residues of the first and/or second cell surface protein.
17. The genetically engineered hematopoietic cell, or descendant thereof, of any one of claims 1-16, wherein the mutation alters no more than 1, no more than 2, no more than 3, no more than 4, or no more than 5 amino acid residues of the first and/or second cell surface protein.
18. The genetically engineered hematopoietic cell, or descendant thereof, of any one of claims 1-17, wherein the mutant of the first cell surface protein and/or the mutant of the second cell surface protein comprises a substitution, deletion, or addition of at least one amino acid or a combination thereof.
19. The genetically engineered hematopoietic cell, or descendant thereof, of any one of claims 1-18, wherein the mutation of the first gene and/or the second gene comprises a substitution, deletion, or addition of at least one amino acid or a combination thereof in a non-essential epitope of the first and/or the second cell surface protein.
20. The genetically engineered hematopoietic cell, or descendant thereof, of any one of claims 1-19, wherein the cell has reduced or eliminated binding to a first agent comprising an antigen-binding fragment that binds to the first cell surface protein and/or a second agent comprising an antigen-binding fragment that binds to the second cell surface protein.
21. The genetically engineered hematopoietic cell, or descendant thereof, of claim 20, wherein the first agent and/or the second agent is a cytotoxic agent.
22. The genetically engineered hematopoietic cell, or descendant thereof, of claim 20 or 21, wherein the antigen-binding fragment of the cytotoxic agent is a single-chain antibody fragment (scFv) that specifically binds the cell surface protein or an epitope thereof.
23. The genetically engineered hematopoietic cell, or descendant thereof, of claim 21 or 22, wherein the cytotoxic agent is an antibody or an antibody-drug conjugate (ADC).
24. The genetically engineered hematopoietic cell, or descendant thereof, of claim 21 or 22, wherein the cytotoxic agent is an immune cell expressing a chimeric receptor that comprises the antigen-binding fragment.
25. The genetically engineered hematopoietic cell, or descendant thereof, of claim 24, wherein the immune cells are T cells.
26. The genetically engineered hematopoietic cell, or descendant thereof, of claim 24 or 25, wherein the chimeric receptor further comprises: (a) a hinge domain, (b) a transmembrane domain, (c) at least one co-stimulatory domain, (d) a cytoplasmic signaling domain, or (e) a combination thereof.
27. The genetically engineered hematopoietic cell, or descendant thereof, of claim 26, wherein the chimeric receptor comprises at least one co-stimulatory signaling domain, which is derived from a co-stimulatory receptor selected from the group consisting of CD27, CD28, 4-1BB, OX40, CD30, ICOS, CD2, CD7, LIGHT, NKG2C, B7-H3, GITR, HVEM, and a combination thereof.
28. The genetically engineered hematopoietic cell, or descendant thereof, of claim 26 or 27, wherein the chimeric receptor comprises a cytoplasmic signaling domain, which is from CD3ζ.
29. The genetically engineered hematopoietic cell, or descendant thereof, of any one of claims 26-28, wherein the chimeric receptor comprises a hinge domain, which is from CD8α or CD28.
30. The genetically engineered hematopoietic cell, or descendant thereof, of any one of claims 1-29, wherein the mutation in the first gene does not affect the bioactivity of the first cell surface protein and/or the mutation in the second gene does not affect the bioactivity of the second cell surface protein.
31. The genetically engineered hematopoietic cell, or descendant thereof, of any one of claims 1-30, wherein the cell retains the ability to differentiate normally.
32. The genetically engineered hematopoietic cell, or descendant thereof, of any one of claims 1-31, wherein the cell is a hematopoietic stem or progenitor cell, or a descendant thereof.
33. The genetically engineered hematopoietic cell, or descendant thereof, of any one of claims 1-32, wherein the hematopoietic cell is a hematopoietic stem cell (HSC).
34. The genetically engineered hematopoietic cell, or descendant thereof, of any one of claims 1-33, wherein the hematopoietic cell is a CD34+ cell.
35. The genetically engineered hematopoietic cell, or descendant thereof, of any one of claims 1-34, wherein the hematopoietic cell is from bone marrow, blood, umbilical cord, or peripheral blood mononuclear cells (PBMCs).
36. The genetically engineered hematopoietic cell, or descendant thereof, of any one of claims 1-35, wherein the immune cells, the genetically engineered hematopoietic cells, or both, are allogeneic or autologous.
37. The genetically engineered hematopoietic cell, or descendant thereof, of any one of claims 1-36, wherein the hematopoietic cell is a human cell.
38. A cell population comprising one or more of the genetically engineered hematopoietic cells, or descendant thereof, of any one of claims 1-37.
39. A pharmaceutical composition comprising the genetically engineered hematopoietic cell, or descendant thereof, of any one of claims 1-37 or the cell population of claim 36.
40. A method comprising administering to a subject in need thereof the genetically engineered hematopoietic cell, or descendant thereof, of any one of claims 1-37, the cell population of claim 38, or the pharmaceutical composition of claim 39.
41. The method of claim 40, further comprising administering to the subject a therapeutically effective amount of at least one agent that targets the first and/or second cell surface protein, wherein the agent comprises an antigen binding fragment that binds the first or second cell surface protein.
42. The method of claim 41, wherein the agent is an antibody-drug conjugate or an immune effector cell expressing a chimeric antigen receptor (CAR).
43. The method of any one of claims 40-42, wherein the subject has blastic plasmacytoid dendritic cell neoplasm, multiple myeloma, myelodysplastic syndrome, Hodgkin’s lymphoma, non-Hodgkin’s lymphoma, leukemia, multiple myeloma (MM), myelodysplastic syndrome (MDS), B-cell acute lymphoblastic leukemia (B-ALL), chronic myelogenous leukemia, acute lymphoblastic leukemia, or chronic lymphoblastic leukemia or blastic plasmacytoid dendritic cell neoplasm (BPDCN).
44. The method of any one of claims 40-43, wherein the subject has a hematopoietic malignancy or a hematopoietic pre-malignant disease.
45. The method of any one of claims 40-44, wherein the hematopoietic malignancy is acute myeloid leukemia (AML).
46. The method of any one of claims 40-44, wherein the hematopoietic pre-malignancy is myelodysplastic syndrome (MDS).
47. The method of any one of claims 40-45, wherein the subject has or is at risk of leukemia relapse.
48. A method for determining if a subject is at risk of relapse of a disease, comprising providing a first biological sample obtained from a subject at a first time point and a second biological sample obtained from the subject at a second time point; measuring a level of a first cell surface protein in the first biological sample and in the second biological sample; identifying the subject as at risk for relapse of the disease if the level of the first cell surface protein in the first biological sample deviates from the level of the first cell surface protein in the second biological sample; wherein the cell surface protein is selected from Table 3 or Table 4.
49. A method for identifying a subject as a candidate for treatment with an agent, comprising providing a first biological sample obtained from a subject at a first time point and a second biological sample obtained from the subject at a second time point; and measuring a level of a first cell surface protein in the first biological sample and in the second biological sample; wherein the cell surface protein is selected from Table 3 or Table 4.
50. The method of claim 48 or 49, further comprising administering to the subject a therapeutically effective amount of an agent comprising an antigen-binding fragment that binds to the first cell surface protein.
51. The method of any one of claims 48-50, further comprising administering to the subject a population of genetically engineered hematopoietic cells having reduced or eliminated expression of the first cell surface protein.
52. The method of any one of claims 48-51, wherein the first biological sample and/or the second biological sample comprises blast cells.
53. The method of any one of claims 48-52, wherein the first time point is at or near the time the subject is diagnosed with the disease.
Applications Claiming Priority (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US202363517109P | 2023-08-01 | 2023-08-01 | |
| US63/517,109 | 2023-08-01 | ||
| US202363607780P | 2023-12-08 | 2023-12-08 | |
| US63/607,780 | 2023-12-08 | ||
| US202463645618P | 2024-05-10 | 2024-05-10 | |
| US63/645,618 | 2024-05-10 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2025030010A1true WO2025030010A1 (en) | 2025-02-06 |
Family
ID=92672299
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2024/040528WO2025030010A1 (en) | 2023-08-01 | 2024-08-01 | Compositions comprising genetically engineered hematopoietic stem cells and methods of use thereof |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2025030010A1 (en) |
Citations (71)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1987004462A1 (en) | 1986-01-23 | 1987-07-30 | Celltech Limited | Recombinant dna sequences, vectors containing them and method for the use thereof |
| US4704692A (en) | 1986-09-02 | 1987-11-03 | Ladner Robert C | Computer based system and method for determining and displaying possible chemical structures for converting double- or multiple-chain polypeptides to single-chain polypeptides |
| GB2200651A (en) | 1987-02-07 | 1988-08-10 | Al Sumidaie Ayad Mohamed Khala | A method of obtaining a retrovirus-containing fraction from retrovirus-containing cells |
| US4777127A (en) | 1985-09-30 | 1988-10-11 | Labsystems Oy | Human retrovirus-related products and methods of diagnosing and treating conditions associated with said retrovirus |
| EP0345242A2 (en) | 1988-06-03 | 1989-12-06 | Smithkline Biologicals S.A. | Expression of gag proteins from retroviruses in eucaryotic cells |
| WO1990007936A1 (en) | 1989-01-23 | 1990-07-26 | Chiron Corporation | Recombinant therapies for infection and hyperproliferative disorders |
| US4946778A (en) | 1987-09-21 | 1990-08-07 | Genex Corporation | Single polypeptide chain binding molecules |
| WO1991002805A2 (en) | 1989-08-18 | 1991-03-07 | Viagene, Inc. | Recombinant retroviruses delivering vector constructs to target cells |
| WO1993003769A1 (en) | 1991-08-20 | 1993-03-04 | THE UNITED STATES OF AMERICA, represented by THE SECRETARY, DEPARTEMENT OF HEALTH AND HUMAN SERVICES | Adenovirus mediated transfer of genes to the gastrointestinal tract |
| WO1993010218A1 (en) | 1991-11-14 | 1993-05-27 | The United States Government As Represented By The Secretary Of The Department Of Health And Human Services | Vectors including foreign genes and negative selective markers |
| WO1993011230A1 (en) | 1991-12-02 | 1993-06-10 | Dynal As | Modified mammalian stem cell blocking viral replication |
| US5219740A (en) | 1987-02-13 | 1993-06-15 | Fred Hutchinson Cancer Research Center | Retroviral gene transfer into diploid fibroblasts for gene therapy |
| WO1993019191A1 (en) | 1992-03-16 | 1993-09-30 | Centre National De La Recherche Scientifique | Defective recombinant adenoviruses expressing cytokines for use in antitumoral treatment |
| WO1993025698A1 (en) | 1992-06-10 | 1993-12-23 | The United States Government As Represented By The | Vector particles resistant to inactivation by human serum |
| WO1993025234A1 (en) | 1992-06-08 | 1993-12-23 | The Regents Of The University Of California | Methods and compositions for targeting specific tissue |
| US5283173A (en) | 1990-01-24 | 1994-02-01 | The Research Foundation Of State University Of New York | System to detect protein-protein interactions |
| WO1994003622A1 (en) | 1992-07-31 | 1994-02-17 | Imperial College Of Science, Technology & Medicine | D-type retroviral vectors, based on mpmv |
| WO1994012649A2 (en) | 1992-12-03 | 1994-06-09 | Genzyme Corporation | Gene therapy for cystic fibrosis |
| WO1994028938A1 (en) | 1993-06-07 | 1994-12-22 | The Regents Of The University Of Michigan | Adenovirus vectors for gene therapy sponsorship |
| WO1995000655A1 (en) | 1993-06-24 | 1995-01-05 | Mc Master University | Adenovirus vectors for gene therapy |
| WO1995011984A2 (en) | 1993-10-25 | 1995-05-04 | Canji, Inc. | Recombinant adenoviral vector and methods of use |
| US5565332A (en) | 1991-09-23 | 1996-10-15 | Medical Research Council | Production of chimeric antibodies - a combinatorial approach |
| US5580717A (en) | 1990-05-01 | 1996-12-03 | Affymax Technologies N.V. | Recombinant library screening methods |
| WO1998009271A1 (en) | 1996-08-28 | 1998-03-05 | Burgett, Inc. | Method and apparatus for actuating solenoids in a player piano |
| US5733743A (en) | 1992-03-24 | 1998-03-31 | Cambridge Antibody Technology Limited | Methods for producing members of specific binding pairs |
| US6194191B1 (en) | 1996-11-20 | 2001-02-27 | Introgen Therapeutics, Inc. | Method for the production and purification of adenoviral vectors |
| US6265150B1 (en) | 1995-06-07 | 2001-07-24 | Becton Dickinson & Company | Phage antibodies |
| WO2012088461A2 (en) | 2010-12-23 | 2012-06-28 | Biogen Idec Inc. | Linker peptides and polypeptides comprising same |
| WO2013176772A1 (en) | 2012-05-25 | 2013-11-28 | The Regents Of The University Of California | Methods and compositions for rna-directed target dna modification and for rna-directed modulation of transcription |
| US20140106449A1 (en) | 2010-12-09 | 2014-04-17 | The Trustees Of The University Of Pennsylvania | Use of Chimeric Antigen Receptor-Modified T Cells to Treat Cancer |
| WO2014093694A1 (en) | 2012-12-12 | 2014-06-19 | The Broad Institute, Inc. | Crispr-cas nickase systems, methods and compositions for sequence manipulation in eukaryotes |
| WO2015157070A2 (en) | 2014-04-09 | 2015-10-15 | Editas Medicine, Inc. | Crispr/cas-related methods and compositions for treating cystic fibrosis |
| WO2016089433A1 (en) | 2014-12-03 | 2016-06-09 | Agilent Technologies, Inc. | Guide rna with chemical modifications |
| WO2016164356A1 (en) | 2015-04-06 | 2016-10-13 | The Board Of Trustees Of The Leland Stanford Junior University | Chemically modified guide rnas for crispr/cas-mediated gene regulation |
| WO2017066760A1 (en) | 2015-10-16 | 2017-04-20 | The Trustees Of Columbia University In The City Of New York | Compositions and methods for inhibition of lineage specific antigens |
| WO2017079400A1 (en) | 2015-11-04 | 2017-05-11 | The Trustees Of The University Of Pennsylvania | Methods and compositions for gene editing in hematopoietic stem cells |
| WO2017087800A1 (en) | 2015-11-19 | 2017-05-26 | Abbvie Stemcentrx Llc | Novel anti-emr2 antibodies and methods of use |
| WO2017214460A1 (en) | 2016-06-08 | 2017-12-14 | Agilent Technologies, Inc. | High specificity genome editing using chemically modified guide rnas |
| WO2018126176A1 (en) | 2016-12-30 | 2018-07-05 | Editas Medicine, Inc. | Synthetic guide molecules, compositions and methods relating thereto |
| WO2018160768A1 (en) | 2017-02-28 | 2018-09-07 | Vor Biopharma, Inc. | Compositions and methods for inhibition lineage specific proteins |
| WO2018165629A1 (en) | 2017-03-10 | 2018-09-13 | President And Fellows Of Harvard College | Cytosine to guanine base editor |
| US20180312825A1 (en) | 2015-10-23 | 2018-11-01 | President And Fellows Of Harvard College | Nucleobase editors and uses thereof |
| US20180312828A1 (en) | 2017-03-23 | 2018-11-01 | President And Fellows Of Harvard College | Nucleobase editors comprising nucleic acid programmable dna binding proteins |
| WO2019046285A1 (en) | 2017-08-28 | 2019-03-07 | The Trustees Of Columbia University In The City Of New York | Cd33 exon 2 deficient donor stem cells for use with cd33 targeting agents |
| WO2019178382A1 (en) | 2018-03-14 | 2019-09-19 | The United States Of America, As Represented By The Secretary, Department Of Health And Human Services | Anti-cd33 chimeric antigen receptors and their uses |
| WO2020047164A1 (en) | 2018-08-28 | 2020-03-05 | Vor Biopharma, Inc | Genetically engineered hematopoietic stem cells and uses thereof |
| WO2020150478A1 (en) | 2019-01-16 | 2020-07-23 | The Trustees Of Columbia University In The City Of New York | Compositions and methods for inhibition of lineage specific antigens |
| WO2020237217A1 (en) | 2019-05-23 | 2020-11-26 | Vor Biopharma, Inc | Compositions and methods for cd33 modification |
| WO2021030666A1 (en) | 2019-08-15 | 2021-02-18 | The Broad Institute, Inc. | Base editing by transglycosylation |
| WO2021041971A1 (en) | 2019-08-28 | 2021-03-04 | Vor Biopharma, Inc. | Compositions and methods for cll1 modification |
| WO2021041977A1 (en) | 2019-08-28 | 2021-03-04 | Vor Biopharma, Inc. | Compositions and methods for cd123 modification |
| WO2021158921A2 (en) | 2020-02-05 | 2021-08-12 | The Broad Institute, Inc. | Adenine base editors and uses thereof |
| WO2021247856A2 (en)* | 2020-06-03 | 2021-12-09 | The Trustees Of Columbia University In The City Of New York | Compositions and methods for inhibition of lineage specific antigens using crispr-based base editor systems |
| WO2022047168A1 (en) | 2020-08-28 | 2022-03-03 | Vor Biopharma Inc. | Compositions and methods for cll1 modification |
| WO2022047165A1 (en) | 2020-08-28 | 2022-03-03 | Vor Biopharma Inc. | Compositions and methods for cd123 modification |
| WO2022056490A1 (en) | 2020-09-14 | 2022-03-17 | Vor Biopharma, Inc. | Chimeric antigen receptors for treatment of cancer |
| WO2022056497A1 (en) | 2020-09-14 | 2022-03-17 | Vor Biopharma, Inc. | Single domain antibodies against cd33 |
| WO2022056489A1 (en) | 2020-09-14 | 2022-03-17 | Vor Biopharma, Inc. | Compositions and methods for cd38 modification |
| WO2022061115A1 (en) | 2020-09-18 | 2022-03-24 | Vor Biopharma Inc. | Compositions and methods for cd7 modification |
| WO2022067240A1 (en) | 2020-09-28 | 2022-03-31 | Vor Biopharma, Inc. | Compositions and methods for cd6 modification |
| WO2022072643A1 (en) | 2020-09-30 | 2022-04-07 | Vor Biopharma Inc. | Compositions and methods for cd30 gene modification |
| WO2022094245A1 (en) | 2020-10-30 | 2022-05-05 | Vor Biopharma, Inc. | Compositions and methods for bcma modification |
| WO2022147347A1 (en) | 2020-12-31 | 2022-07-07 | Vor Biopharma Inc. | Compositions and methods for cd34 gene modification |
| WO2022217086A1 (en) | 2021-04-09 | 2022-10-13 | Vor Biopharma Inc. | Photocleavable guide rnas and methods of use thereof |
| WO2022261509A1 (en) | 2021-06-11 | 2022-12-15 | The Broad Institute, Inc. | Improved cytosine to guanine base editors |
| WO2023015182A1 (en) | 2021-08-02 | 2023-02-09 | Vor Biopharma Inc. | Compositions and methods for gene modification |
| WO2023043858A1 (en) | 2021-09-14 | 2023-03-23 | Vor Biopharma Inc. | Compositions and methods for multiplex base editing in hematopoietic cells |
| WO2023086422A1 (en) | 2021-11-09 | 2023-05-19 | Vor Biopharma Inc. | Compositions and methods for erm2 modification |
| WO2023196816A1 (en) | 2022-04-04 | 2023-10-12 | Vor Biopharma Inc. | Compositions and methods for mediating epitope engineering |
| WO2023201238A1 (en) | 2022-04-11 | 2023-10-19 | Vor Biopharma Inc. | Binding agents and methods of use thereof |
| WO2024015925A2 (en) | 2022-07-13 | 2024-01-18 | Vor Biopharma Inc. | Compositions and methods for artificial protospacer adjacent motif (pam) generation |
- 2024
- 2024-08-01WOPCT/US2024/040528patent/WO2025030010A1/enunknown
Patent Citations (73)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4777127A (en) | 1985-09-30 | 1988-10-11 | Labsystems Oy | Human retrovirus-related products and methods of diagnosing and treating conditions associated with said retrovirus |
| WO1987004462A1 (en) | 1986-01-23 | 1987-07-30 | Celltech Limited | Recombinant dna sequences, vectors containing them and method for the use thereof |
| US4704692A (en) | 1986-09-02 | 1987-11-03 | Ladner Robert C | Computer based system and method for determining and displaying possible chemical structures for converting double- or multiple-chain polypeptides to single-chain polypeptides |
| GB2200651A (en) | 1987-02-07 | 1988-08-10 | Al Sumidaie Ayad Mohamed Khala | A method of obtaining a retrovirus-containing fraction from retrovirus-containing cells |
| US5219740A (en) | 1987-02-13 | 1993-06-15 | Fred Hutchinson Cancer Research Center | Retroviral gene transfer into diploid fibroblasts for gene therapy |
| US4946778A (en) | 1987-09-21 | 1990-08-07 | Genex Corporation | Single polypeptide chain binding molecules |
| EP0345242A2 (en) | 1988-06-03 | 1989-12-06 | Smithkline Biologicals S.A. | Expression of gag proteins from retroviruses in eucaryotic cells |
| WO1990007936A1 (en) | 1989-01-23 | 1990-07-26 | Chiron Corporation | Recombinant therapies for infection and hyperproliferative disorders |
| WO1991002805A2 (en) | 1989-08-18 | 1991-03-07 | Viagene, Inc. | Recombinant retroviruses delivering vector constructs to target cells |
| US5283173A (en) | 1990-01-24 | 1994-02-01 | The Research Foundation Of State University Of New York | System to detect protein-protein interactions |
| US5468614A (en) | 1990-01-24 | 1995-11-21 | The Research Foundation Of State University Of New York | System to detect protein-protein interactions |
| US5580717A (en) | 1990-05-01 | 1996-12-03 | Affymax Technologies N.V. | Recombinant library screening methods |
| WO1993003769A1 (en) | 1991-08-20 | 1993-03-04 | THE UNITED STATES OF AMERICA, represented by THE SECRETARY, DEPARTEMENT OF HEALTH AND HUMAN SERVICES | Adenovirus mediated transfer of genes to the gastrointestinal tract |
| US5565332A (en) | 1991-09-23 | 1996-10-15 | Medical Research Council | Production of chimeric antibodies - a combinatorial approach |
| WO1993010218A1 (en) | 1991-11-14 | 1993-05-27 | The United States Government As Represented By The Secretary Of The Department Of Health And Human Services | Vectors including foreign genes and negative selective markers |
| WO1993011230A1 (en) | 1991-12-02 | 1993-06-10 | Dynal As | Modified mammalian stem cell blocking viral replication |
| WO1993019191A1 (en) | 1992-03-16 | 1993-09-30 | Centre National De La Recherche Scientifique | Defective recombinant adenoviruses expressing cytokines for use in antitumoral treatment |
| US5733743A (en) | 1992-03-24 | 1998-03-31 | Cambridge Antibody Technology Limited | Methods for producing members of specific binding pairs |
| WO1993025234A1 (en) | 1992-06-08 | 1993-12-23 | The Regents Of The University Of California | Methods and compositions for targeting specific tissue |
| WO1993025698A1 (en) | 1992-06-10 | 1993-12-23 | The United States Government As Represented By The | Vector particles resistant to inactivation by human serum |
| WO1994003622A1 (en) | 1992-07-31 | 1994-02-17 | Imperial College Of Science, Technology & Medicine | D-type retroviral vectors, based on mpmv |
| WO1994012649A2 (en) | 1992-12-03 | 1994-06-09 | Genzyme Corporation | Gene therapy for cystic fibrosis |
| WO1994028938A1 (en) | 1993-06-07 | 1994-12-22 | The Regents Of The University Of Michigan | Adenovirus vectors for gene therapy sponsorship |
| WO1995000655A1 (en) | 1993-06-24 | 1995-01-05 | Mc Master University | Adenovirus vectors for gene therapy |
| WO1995011984A2 (en) | 1993-10-25 | 1995-05-04 | Canji, Inc. | Recombinant adenoviral vector and methods of use |
| US6265150B1 (en) | 1995-06-07 | 2001-07-24 | Becton Dickinson & Company | Phage antibodies |
| WO1998009271A1 (en) | 1996-08-28 | 1998-03-05 | Burgett, Inc. | Method and apparatus for actuating solenoids in a player piano |
| US6194191B1 (en) | 1996-11-20 | 2001-02-27 | Introgen Therapeutics, Inc. | Method for the production and purification of adenoviral vectors |
| US20140106449A1 (en) | 2010-12-09 | 2014-04-17 | The Trustees Of The University Of Pennsylvania | Use of Chimeric Antigen Receptor-Modified T Cells to Treat Cancer |
| WO2012088461A2 (en) | 2010-12-23 | 2012-06-28 | Biogen Idec Inc. | Linker peptides and polypeptides comprising same |
| WO2013176772A1 (en) | 2012-05-25 | 2013-11-28 | The Regents Of The University Of California | Methods and compositions for rna-directed target dna modification and for rna-directed modulation of transcription |
| WO2014093694A1 (en) | 2012-12-12 | 2014-06-19 | The Broad Institute, Inc. | Crispr-cas nickase systems, methods and compositions for sequence manipulation in eukaryotes |
| WO2015157070A2 (en) | 2014-04-09 | 2015-10-15 | Editas Medicine, Inc. | Crispr/cas-related methods and compositions for treating cystic fibrosis |
| WO2016089433A1 (en) | 2014-12-03 | 2016-06-09 | Agilent Technologies, Inc. | Guide rna with chemical modifications |
| WO2016164356A1 (en) | 2015-04-06 | 2016-10-13 | The Board Of Trustees Of The Leland Stanford Junior University | Chemically modified guide rnas for crispr/cas-mediated gene regulation |
| WO2017066760A1 (en) | 2015-10-16 | 2017-04-20 | The Trustees Of Columbia University In The City Of New York | Compositions and methods for inhibition of lineage specific antigens |
| US20170326179A1 (en)* | 2015-10-16 | 2017-11-16 | The Trustees Of Columbia University In The | Compositions and methods for inhibition of lineage specific antigens |
| US20180312825A1 (en) | 2015-10-23 | 2018-11-01 | President And Fellows Of Harvard College | Nucleobase editors and uses thereof |
| WO2017079400A1 (en) | 2015-11-04 | 2017-05-11 | The Trustees Of The University Of Pennsylvania | Methods and compositions for gene editing in hematopoietic stem cells |
| WO2017087800A1 (en) | 2015-11-19 | 2017-05-26 | Abbvie Stemcentrx Llc | Novel anti-emr2 antibodies and methods of use |
| WO2017214460A1 (en) | 2016-06-08 | 2017-12-14 | Agilent Technologies, Inc. | High specificity genome editing using chemically modified guide rnas |
| WO2018126176A1 (en) | 2016-12-30 | 2018-07-05 | Editas Medicine, Inc. | Synthetic guide molecules, compositions and methods relating thereto |
| WO2018160768A1 (en) | 2017-02-28 | 2018-09-07 | Vor Biopharma, Inc. | Compositions and methods for inhibition lineage specific proteins |
| WO2018165629A1 (en) | 2017-03-10 | 2018-09-13 | President And Fellows Of Harvard College | Cytosine to guanine base editor |
| US20180312828A1 (en) | 2017-03-23 | 2018-11-01 | President And Fellows Of Harvard College | Nucleobase editors comprising nucleic acid programmable dna binding proteins |
| WO2019046285A1 (en) | 2017-08-28 | 2019-03-07 | The Trustees Of Columbia University In The City Of New York | Cd33 exon 2 deficient donor stem cells for use with cd33 targeting agents |
| WO2019178382A1 (en) | 2018-03-14 | 2019-09-19 | The United States Of America, As Represented By The Secretary, Department Of Health And Human Services | Anti-cd33 chimeric antigen receptors and their uses |
| WO2020047164A1 (en) | 2018-08-28 | 2020-03-05 | Vor Biopharma, Inc | Genetically engineered hematopoietic stem cells and uses thereof |
| WO2020150478A1 (en) | 2019-01-16 | 2020-07-23 | The Trustees Of Columbia University In The City Of New York | Compositions and methods for inhibition of lineage specific antigens |
| WO2020237217A1 (en) | 2019-05-23 | 2020-11-26 | Vor Biopharma, Inc | Compositions and methods for cd33 modification |
| WO2021030666A1 (en) | 2019-08-15 | 2021-02-18 | The Broad Institute, Inc. | Base editing by transglycosylation |
| WO2021041971A1 (en) | 2019-08-28 | 2021-03-04 | Vor Biopharma, Inc. | Compositions and methods for cll1 modification |
| WO2021041977A1 (en) | 2019-08-28 | 2021-03-04 | Vor Biopharma, Inc. | Compositions and methods for cd123 modification |
| WO2021158921A2 (en) | 2020-02-05 | 2021-08-12 | The Broad Institute, Inc. | Adenine base editors and uses thereof |
| WO2021247856A2 (en)* | 2020-06-03 | 2021-12-09 | The Trustees Of Columbia University In The City Of New York | Compositions and methods for inhibition of lineage specific antigens using crispr-based base editor systems |
| WO2022047168A1 (en) | 2020-08-28 | 2022-03-03 | Vor Biopharma Inc. | Compositions and methods for cll1 modification |
| WO2022047165A1 (en) | 2020-08-28 | 2022-03-03 | Vor Biopharma Inc. | Compositions and methods for cd123 modification |
| WO2022056490A1 (en) | 2020-09-14 | 2022-03-17 | Vor Biopharma, Inc. | Chimeric antigen receptors for treatment of cancer |
| WO2022056497A1 (en) | 2020-09-14 | 2022-03-17 | Vor Biopharma, Inc. | Single domain antibodies against cd33 |
| WO2022056489A1 (en) | 2020-09-14 | 2022-03-17 | Vor Biopharma, Inc. | Compositions and methods for cd38 modification |
| WO2022061115A1 (en) | 2020-09-18 | 2022-03-24 | Vor Biopharma Inc. | Compositions and methods for cd7 modification |
| WO2022067240A1 (en) | 2020-09-28 | 2022-03-31 | Vor Biopharma, Inc. | Compositions and methods for cd6 modification |
| WO2022072643A1 (en) | 2020-09-30 | 2022-04-07 | Vor Biopharma Inc. | Compositions and methods for cd30 gene modification |
| WO2022094245A1 (en) | 2020-10-30 | 2022-05-05 | Vor Biopharma, Inc. | Compositions and methods for bcma modification |
| WO2022147347A1 (en) | 2020-12-31 | 2022-07-07 | Vor Biopharma Inc. | Compositions and methods for cd34 gene modification |
| WO2022217086A1 (en) | 2021-04-09 | 2022-10-13 | Vor Biopharma Inc. | Photocleavable guide rnas and methods of use thereof |
| WO2022261509A1 (en) | 2021-06-11 | 2022-12-15 | The Broad Institute, Inc. | Improved cytosine to guanine base editors |
| WO2023015182A1 (en) | 2021-08-02 | 2023-02-09 | Vor Biopharma Inc. | Compositions and methods for gene modification |
| WO2023043858A1 (en) | 2021-09-14 | 2023-03-23 | Vor Biopharma Inc. | Compositions and methods for multiplex base editing in hematopoietic cells |
| WO2023086422A1 (en) | 2021-11-09 | 2023-05-19 | Vor Biopharma Inc. | Compositions and methods for erm2 modification |
| WO2023196816A1 (en) | 2022-04-04 | 2023-10-12 | Vor Biopharma Inc. | Compositions and methods for mediating epitope engineering |
| WO2023201238A1 (en) | 2022-04-11 | 2023-10-19 | Vor Biopharma Inc. | Binding agents and methods of use thereof |
| WO2024015925A2 (en) | 2022-07-13 | 2024-01-18 | Vor Biopharma Inc. | Compositions and methods for artificial protospacer adjacent motif (pam) generation |
Non-Patent Citations (115)
| Title |
|---|
| "Cell and Tissue Culture: Laboratory Procedures", August 1993, J. WILEY AND SONS |
| "Current Protocols in Immunology", 1991 |
| "DNA Cloning: A practical Approach", 1985 |
| "Gene Transfer Vectors for Mammalian Cells", 1987, HUMANA PRESS |
| "Handbook of Experimental Immunology", 1994, ACADEMIC PRESS |
| "Immobilized Cells and Enzymes", 1986, LRL PRESS |
| "Introduction to Cell and Tissue Culture", 1998, PLENUM PRESS |
| "Remington: The Science and Practice of Pharmacy", 2000, LIPPINCOTT WILLIAMS AND WILKINS |
| ALAYED ET AL., AMERICAN JOURNAL OF HEMATOLOGY, vol. 88, no. 12, 2013, pages 1055 - 1061 |
| ALTSCHUL ET AL., J. MOL. BIOL., vol. 215, 1990, pages 403 - 10 |
| ALTSCHUL ET AL., NUCLEIC ACIDS RES., vol. 25, no. 17, 1997, pages 3389 - 3402 |
| ANGELOT-DELETTRE ET AL., HEMATOLOGICA, vol. 100, no. 2, 2015, pages 223 - 230 |
| ANZALONE ET AL., NAT. BIOTECHNOL., vol. 38, 2020, pages 824 - 844 |
| ANZALONE ET AL., NATURE, vol. 576, no. 7785, 2019, pages 149 - 157 |
| B. PERBAL, A PRACTICAL GUIDE TO MOLECULAR CLONING, 1984 |
| BECK ET AL., NATURE REVIEWS DRUG DISCOVERY, vol. 16, 2017, pages 315 - 337 |
| BRINKMAN-VAN DER LINDEN ET AL., MOL CELL. BIOL., vol. 23, 2003, pages 4199 - 4206 |
| BROWN ET AL., NATURE REVIEWS IMMUNOLOGY, vol. 19, 2019, pages 73 - 74 |
| BROWNFRAZIER, TRENDS IN CELL BIOLOGY, vol. 11, no. 3, 2001, pages 130 - 135 |
| BUCK, D. W. ET AL., IN VITRO, vol. 18, 1982, pages 377 - 381 |
| C. A. JANEWAYP. TRAVERS: "Immunobiology", 1997 |
| CANCER DISCOV., vol. 8, no. 10, 2018, pages 1219 - 1226 |
| CAO ET AL., ANGEW. CHEM. INT. ED., vol. 55, 2016, pages 7520 - 7524 |
| CHANG ET AL., FEBS LETTERS, vol. 547, no. 1-3, 2003, pages 145 - 150 |
| CHINNASAMY ET AL., CLIN. CANCER RES., vol. 18, 2012, pages 1672 - 1683 |
| CHOI ET AL., PLOS ONE, vol. 7, no. 10, 2012, pages e46688 |
| DABROWSKA ET AL., FRONTIERS IN NEUROSCIENCE, vol. 12, no. 75, 2018 |
| DEBOER, CANCER CELL, 2018 |
| DEL GIUDICE ET AL., HEMATOLOGICA, vol. 89, no. 3, 2004, pages 303 - 308 |
| DOHNER ET AL., NEIM, vol. 373, 2015, pages 1136 |
| DOHNER ET AL., NEJM, vol. 373, 2015, pages 1136 |
| E. HARLOWD. LANE: "Using antibodies: a laboratory manual", 1999, COLD SPRING HARBOR LABORATORY PRESS |
| EID ET AL., BIOCHEM. J., vol. 475, no. 11, 2018, pages 1955 - 1964 |
| ELGUNDI ET AL., ADVANCED DRUG DELIVERY REVIEWS, vol. 122, 2017, pages 2 - 19 |
| ELGUNDI ET AL., ADVANCED DRUG DISCOVERY REVIEWS, vol. 122, 2017, pages 2 - 19 |
| ELKINS, MOL. CANCER THER., vol. 10, 2012, pages 2222 - 32 |
| FU Y ET AL., NAT BIOTECHNOL, 2014 |
| GAO ET AL., NAT. BIOTECHNOL., vol. 35, no. 8, 2017, pages 789 - 792 |
| GAUDELLI ET AL., NAT BIOTECHNOL., vol. 38, no. 7, 2020, pages 892 - 900 |
| GUDMUNDSSON ET AL., ARXIV:2107.11458V3 |
| GUTSCHNER ET AL., CELL REP., vol. 14, no. 6, 2016, pages 1555 - 1566 |
| HARRINGTON ET AL., SCIENCE, 2018 |
| HENDEL ET AL., NAT BIOTECHNOL., vol. 33, no. 9, September 2015 (2015-09-01), pages 985 - 989 |
| HENDEL, A. ET AL., NATURE BIOTECH., vol. 33, no. 9, 2015 |
| HO, T. K.: "Random decision forests", PROCEEDINGS OF 3RD INTERNATIONAL CONFERENCE ON DOCUMENT ANALYSIS AND RECOGNITION, vol. 1, 1995, pages 1988 - 1989 |
| HOSEINI ET AL., BLOOD CANCER JOURNAL, vol. 7, 2017, pages e552 |
| HU ET AL., CELL REP., vol. 20, 2017, pages 3025 - 3033 |
| JASIN ET AL., DNA REPAIR, vol. 44, 2016, pages 6 - 16 |
| JAYAVELU ET AL., CANCER CELL, 2022 |
| JIANG ET AL., JOURNAL OF HEMATOLOGY & ONCOLOGY, vol. 14, 2021, pages 180 |
| JINEK ET AL., SCIENCE, vol. 337, no. 6096, 2012, pages 816 - 821 |
| JINEK ET AL., SCIENCE, vol. 343, no. 6176, 2014, pages 1247997 |
| KARLINALTSCHUL, PROC. NATL. ACAD. SCI. USA, vol. 87, 1990, pages 2264 - 68 |
| KARLINALTSCHUL, PROC. NATL. ACAD. SCI. USA, vol. 90, 1993, pages 5873 - 77 |
| KLEINSTIVER ET AL., NATURE, vol. 529, 2016, pages 490 - 495 |
| KOHLER, B.MILSTEIN, C., NATURE, vol. 256, 1975, pages 495 - 497 |
| KOMOR ET AL., CELL, vol. 168, 2017, pages 20 - 36 |
| KRENCIUTE ET AL., CANCER IMMUNOL. RES., vol. 5, 2017, pages 571 - 581 |
| KUAN-YU ET AL., FRONT. IMMUNOL., vol. 8, 2017, pages 373 |
| KUNGULOVSKI ET AL., TRENDS GENET., vol. 32, no. 2, 2016, pages 101 - 113 |
| LABANIEH ET AL., NATURE BIOMEDICAL ENGINEERING, vol. 2, 2018, pages 337 - 391 |
| LAMBLE ET AL., JOURNAL OF CLINICAL ONCOLOGY, vol. 40, no. 3, 2022, pages 252 - 261 |
| LANZE ET AL., JOURNAL OF BIOLOGICAL REGULATORS AND HOMEOSTATIC AGENTS, vol. 15, no. 1, 2001, pages 1 - 13 |
| LASZLO ET AL., BLOOD REV., vol. 28, no. 4, 2014, pages 143 - 53 |
| LEE ET AL., BLOOD, vol. 103, no. 6, 2004, pages 2308 - 2315 |
| LEVINE ET AL., J. IMMUNOL., vol. 159, no. 12, 1997, pages 5921 |
| LOMOVA ET AL., STEM CELLS, 2018 |
| MAJZNER ET AL., CANCER DISCOVERY, vol. 8, no. 10, 2018, pages 924 - 934 |
| MARIN-ACEVEDO ET AL., J. HEMATOL. ONCOL., vol. 11, 2018, pages 8 |
| MARKLEY ET AL., BLOOD, vol. 115, 2010, pages 3508 - 3519 |
| MCCAFFERTY ET AL., NATURE, vol. 348, 1990, pages 552 - 553 |
| MORRISON ET AL., PROC. NAT. ACAD. SCI., vol. 81, 1984, pages 6851 |
| MORRISON ET AL., PROC. NATL. ACAD. SCI. USA, vol. 81, 1984, pages 6851 |
| MUNOZ ET AL., HEMATOLOGICA, vol. 86, no. 12, 2001, pages 1261 - 1269 |
| NEUBERGER ET AL., NATURE, vol. 314, 1984, pages 452 |
| NIELSENMCNAGNY, JOURNAL OF CELL SCIENCE, vol. 121, no. 22, 2008, pages 3683 - 3692 |
| NISHIMASU ET AL., CELL, vol. 156, 2014, pages 1262 - 1278 |
| PARADANANI ET AL., LEUKEMIA, vol. 30, no. 4, 2016, pages 914 - 918 |
| PASZKIEWICZ ET AL., J. CLIN. INVEST., vol. 126, 2016, pages 4262 - 4272 |
| PEGRAM ET AL., LEUKEMIA, vol. 29, 2015, pages 415 - 422 |
| PETERS ET AL., BIOSCI. REP., vol. 35, no. 4, 2015, pages e00225 |
| PHILIP ET AL., BLOOD, vol. 124, 2014, pages 1277 - 1287 |
| QUEEN ET AL., PROC. NATL. ACAD. SCI. USA, vol. 86, 1989, pages 10029 - 10033 |
| RAHDAR ET AL., PNAS, vol. 112, no. 51, 22 December 2015 (2015-12-22), pages E7110 - E7117 |
| RAN ET AL., NATURE PROTOCOLS, vol. 8, 2013, pages 2281 - 2308 |
| REES ET AL., NAT. REV. GENET., vol. 19, no. 12, 2018, pages 770 - 788 |
| REES ET AL., NATURE REVIEWS GENETICS, vol. 19, 2018, pages 770 - 788 |
| RODGER ET AL., PNAS, vol. 113, 2016, pages 459 - 468 |
| SAFAEE ET AL., ONC. REV., vol. 8, no. 242, 2014, pages 20 - 24 |
| SAMBROOK ET AL.: "Molecular Cloning: A Laboratory Manual", 1989, COLD SPRING HARBOR PRESS |
| SARAI ET AL., CURRENTLY PHARMA. BIOTECHNOL., vol. 18, no. 13, 2017 |
| SCATCHARD ET AL., ANN. N.Y. ACAD. SCI., vol. 51, 1949, pages 660 |
| SFEIR ET AL., TRENDS BIOCHEM. SCI., vol. 40, 2015, pages 701 - 714 |
| SHAREI ET AL., PROC. NATL. ACAD. SCI. USA, vol. 110, no. 6, 2013, pages 2082 - 2087 |
| SHMAKOV ET AL., MOL CELL, vol. 60, 2015, pages 385 - 397 |
| SLAYMAKER ET AL., SCIENCE, vol. 351, no. 6268, 2016, pages 84 - 88 |
| STELLA ET AL., NATURE STRUCTURAL & MOLECULAR BIOLOGY, vol. 24, 2017, pages 882 - 892 |
| STERNBERG SH ET AL., NATURE, 2014 |
| STROHKENDL ET AL., MOL. CELL, vol. 71, 2018, pages 1 - 9 |
| SUTHERLANDKEATING, JOURNAL OF HEMATOTHERAPY, vol. 1, no. 2, 2009, pages 115 - 129 |
| SYDNEY ET AL., STEM CELLS, vol. 32, no. 6, 2014, pages 1380 - 1389 |
| TARASEVICIUTE ET AL., HEMATOLOGY AND ONCOLOGY, vol. 31, no. 1, 2019 |
| TASIAN ET AL., BLOOD, vol. 129, 2017, pages 2395 - 2407 |
| TASIAN, THER. ADV. HEMATOL., vol. 9, no. 6, 2018, pages 135 - 148 |
| TESTA ET AL., BIOMARKER RESEARCH, vol. 2, no. 4, 2014, pages 4 |
| TILL ET AL., BLOOD, vol. 119, no. 17, 2012, pages 3940 - 3950 |
| VAN DE DONK ET AL., BLOOD, vol. 131, no. 1, 2018, pages 13 - 29 |
| VANEGAS ET AL., FUNGAL BIOL BIOTECHNOL., vol. 6, 2019, pages 6 |
| WANG ET AL., BLOOD, vol. 118, 2011, pages 1255 - 1263 |
| WINTER ET AL., ANNU. REV. IMMUNOL., vol. 12, 1994, pages 433 - 455 |
| WRIGGERS ET AL., CURRENT TRENDS IN PEPTIDE SCIENCE, vol. 80, no. 6, 2005, pages 736 - 746 |
| YANG ET AL., CURR. OPIN. HEMATOL., vol. 22, no. 6, 2015, pages 509 - 515 |
| YEH ET AL., NAT. CELL. BIOL., vol. 21, 2019, pages 1468 - 1478 |
| YONA ET AL., FASEB J., vol. 22, no. 3, 2008, pages 741 - 751 |
| YU ET AL., NAT COMMUN., vol. 11, no. 1, 2020, pages 2052 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US11571445B2 (en) | Genetically engineered hematopoietic stem cells and uses thereof | |
| US20240350550A1 (en) | Compositions and methods for inhibition of lineage specific proteins | |
| JP2020531535A (en) | CD33 exon2-deficient donor stem cells for use with CD33 targeting agents | |
| EP4022063A1 (en) | Compositions and methods for cll1 modification | |
| JP2022517618A (en) | Compositions and Methods for Inhibition of Strain-Specific Antigens | |
| JP2023528415A (en) | Compositions and methods for inhibition of lineage-specific antigens using CRISPR-based base editor systems | |
| US20240366675A1 (en) | Compositions and methods for cd34 gene modification | |
| WO2024015925A2 (en) | Compositions and methods for artificial protospacer adjacent motif (pam) generation | |
| US20230414755A1 (en) | Methods and compositions relating to genetically engineered cells expressing chimeric antigen receptors | |
| WO2025030010A1 (en) | Compositions comprising genetically engineered hematopoietic stem cells and methods of use thereof | |
| CN116744963A (en) | Methods and compositions related to genetically engineered cells expressing chimeric antigen receptors |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | Ref document number:24765786 Country of ref document:EP Kind code of ref document:A1 |