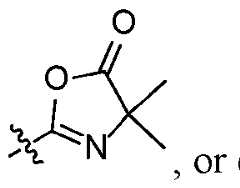
First Named Inventor: RAJA, Erum TP385332WO1 FUNCTIONALIZED MAGNETIC BEADS AND METHODS OF PREPARATION AND USE CROSS REFERENCE TO RELATED APPLICATION This application claims the benefit of the earlier filing date of U.S. provisional patent application No.63/517,010, filed August 1, 2023, and U.S. provisional patent application No.63/604,531, filed November 30, 2023, both of which are incorporated herein by reference in its entirety. FIELD This disclosure concerns magnetic beads that contain functional groups suitable for binding biological molecules including polypeptides, proteins, RNA, and/or DNA, and method for making the beads and using the beads for separating and/or isolating biological molecules for analysis, such as by mass spectrometry. BACKGROUND Mass spectrometry (MS)-based proteomics has become the most comprehensive approach for protein identification, quantitation, interactions, modifications, and structural characterization. It is also a challenging topic that requires expertise for sample preparation since a high-quality sample from a robust and reproducible sample preparation is critical for success. Proteomics sample preparation is highly complex and variable, with many different protocols available. For example, bottom-up proteomics sample preparation requires multiple steps including protein extraction, reduction and alkylation of cysteines, digestion of the proteins into peptides, cleanup, and concentration of the peptides for LC-MS analysis. The lack of standardization in proteomic sample preparation makes it difficult to accurately compare results from different laboratories and different protocols. SUMMARY First Named Inventor: RAJA, Erum TP385332WO1 Disclosed herein are aspects of a magnetic bead suitable for use in biological sample preparation protocols, for example, sample preparation for proteomic analysis, such as by MS. Also disclosed herein are methods for making and using the magnetic beads, and kits comprising the beads. In some aspects, the magnetic bead comprises a crosslinked polymer and a magnetic material, for example an iron source. The O O crosslinked polymer may comprise one or more azlactone , or one or more functional groups derived from an azlactone moiety, reaction

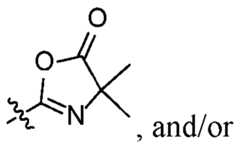
of the azlactone moiety with a nucleophilic reactive group. In certain aspects, the crosslinked polymer is a crosslinked azlactone polymer, and the magnetic bead is an azlactone bead. In some aspects, the magnetic material is or comprises iron oxide particles, such as Fe3O4 and/or Fe2O3 particles. Also disclosed are aspects of a method for using the disclosed magnetic beads. The method may comprise forming a first suspension comprising a plurality of magnetic beads and a first liquid that contains a biomolecule; removing the magnetic beads from the first suspension and optionally washing the magnetic beads using a second liquid; forming a second suspension comprising the magnetic beads and an elution solution; and removing the magnetic beads from the second suspension to leave a third liquid that comprises the biomolecule. In some aspects, the each magnetic bead comprises a crosslinked polymer and a magnetic material, where the crosslinked O polymer comprises one or more azlactone or one or more functional groups derived from an azlactone
from the reaction of the azlactone moiety with a nucleophilic reactive group. The foregoing and other objects, features, and advantages of the disclosure will become more apparent from the following detailed description, which proceeds with reference to the accompanying figures. First Named Inventor: RAJA, Erum TP385332WO1 BRIEF DESCRIPTION OF THE DRAWINGS FIG.1 is a flow chart illustrating one aspect of a standard workflow using the disclosed magnetic beads. FIG.2 is a flowchart illustrating an SP3 workflow that includes a PAC workflow and an SP2 workflow. FIG.3 is a digital image illustrating one aspect of the disclosed magnetic beads. FIG.4 is a graph of differential volume versus size, illustrating the average size of the disclosed magnetic beads, and a table providing the average size and standard deviation. FIG.5 is a digital image illustrating one aspect of the disclosed magnetic beads synthesized using iron oxide clusters as the magnetic material. FIG.6 is a digital image illustrating one aspect of the disclosed magnetic beads synthesized using iron oxide particles as the magnetic material. FIG.7 is a digital image illustrating one aspect of the disclosed magnetic beads synthesized using Dynabeads
TM as the magnetic material. FIGS.8A and 8B are a digital image of plates from various stages of an SP2 workflow (FIG.8A) and a table of results (FIG.8B), illustrating the surface absorption losses of different bead types. FIG.9 is a graph of peptide yield versus bead type, illustrating the peptide yields from different bead types, identified using a quantitative colorimetric peptide assay. FIG.10 is a graph of number of peptides versus bead types, illustrating the number of unique peptides isolated with different bead types and identified using nanoLC-MS analysis, 75 i.d. column, 150 min gradient, QE plus mass spectrometry, PD 2.5 data. FIG.11 is a graph of protein group versus bead types, illustrating the number of different protein groups isolated with different bead types and identified using nanoLC- MS analysis, 75 i.d. column, 150 min gradient, QE plus mass spectrometry, PD 2.5 data. First Named Inventor: RAJA, Erum TP385332WO1 FIG.12 is a graph of bound antibody versus magnetic bead, illustrating the capacity of the disclosed magnetic beads conjugated to alkali stable Protein A (asPA) to bind rabbit IgG. FIG.13 is a gel image comparing anion exchange performance magnetic beads with 3 different ligand chemistries. DETAILED DESCRIPTION I. Terms The following explanations of terms and methods are provided to better describe the present disclosure and to guide those of ordinary skill in the art in the practice of the present disclosure. The singular forms “a,” “an,” and “the” refer to one or more than one, unless the context clearly dictates otherwise. The term “or” refers to a single element of stated alternative elements or a combination of two or more elements, unless the context clearly indicates otherwise. As used herein, “comprises” means “includes.” Thus, “comprising A or B,” means “including A, B, or A and B,” without excluding additional elements. All references, including patents and patent applications cited herein, are incorporated by reference in their entirety, unless otherwise specified. Unless otherwise indicated, all numbers expressing quantities of components, molecular weights, percentages, temperatures, times, and so forth, as used in the specification or claims, are to be understood as being modified by the term “about.” Accordingly, unless otherwise indicated, implicitly or explicitly, the numerical parameters set forth are approximations that may depend on the desired properties sought and/or limits of detection under standard test conditions/methods. When directly and explicitly distinguishing embodiments from discussed prior art, the embodiment numbers are not approximates unless the word “about” is expressly recited. Unless explained otherwise, all technical and scientific terms used herein have the same meaning as commonly understood by one of ordinary skill in the art to which this disclosure pertains. Although methods and materials similar or equivalent to those described herein can be used in the practice or testing of the present disclosure, suitable First Named Inventor: RAJA, Erum TP385332WO1 methods and materials are described below. The materials, methods, and examples are illustrative only and not intended to be limiting. “Alkyl” refers to a saturated aliphatic hydrocarbyl group having from 1 to 25 (C1-25) or more carbon atoms, more typically 1 to 10 (C1-10) carbon atoms such as 1 to 6 (C
1-6) carbon atoms or 1 to 4 (C
1-4) carbon atoms. “Carboxy” refers to a -CO
2H functional group. “Ester” refers to a -CO2R functional group, where R is an alkyl group, such as a C1-6 alkyl group. “Nucleic acid” refers to a polynucleotide molecule. The polynucleotide may be a naturally occurring polynucleotide or a synthetic polynucleotide. A nucleic acid may be a DNA, RNA or mixture of DNA and RNA nucleotides. Typically, a nucleic acid contains from 20 to 10,000 nucleotides or more, such as from 100, 200, 300, 400, 500, 600, 700, 800, 900, 1000, 2000, 3000, 4000, or 5000 nucleotides to 10,000 nucleotides. “Peptide” refers to a compound comprising amino acid residues connected by peptide bonds. Typically, a peptide compound has from 2 to about 50 amino acid residues. “Polypeptide” refers to a compound comprising amino acid residues connected by peptide bonds. In some aspects, a polypeptide has from about 50 amino acid residues to 2000 or more amino acid residues. The amino acids may include alpha-amino acids, and may comprise the L-optical isomer, the D-optical isomer, or combinations thereof. “Protein” refers to a molecule or complex comprising one or more polypeptides having secondary, tertiary and/or quaternary structure. The secondary, tertiary and/or quaternary structure of a protein typically is stabilized using non-covalent bonds, such as hydrogen bonds, ionic bonds, hydrophobic interactions, and/or van der Walls interactions, and/or covalent bonds, for example, disulfide bonds, such as between the thiol groups of cysteine residues. II. Magnetic Beads Disclosed herein are aspects of a magnetic bead that comprises magnetic material dispersed in a polymer bead. In some aspects, the polymer bead comprises a First Named Inventor: RAJA, Erum TP385332WO1 O O crosslinked polymer that comprises one or more azlactone one or more functional groups derived from an azlactone moiety.
derived from an azlactone moiety may comprise moieties derived a between the azlactone moiety and a nucleophilic reactive group, such as, but not limited to, an amine-containing compound, thiol containing group, alcohol or water. Hydrolysis of azlactone or reaction of the azlactone group with an amine- containing compound can add functionality and/or alter the surface properties of the bead. For example, hydrolysis of azlactone rings may produce a bead having a plurality of carboxylic acid functional groups on its surface. Alternatively, the azlactone group are reacted with an amine-containing compound to increase or decrease the hydrophobicity or hydrophilicity of the bead. In certain aspects, azlactone beads are reacted with an amine-containing compound that includes primary, secondary, or tertiary amine group(s) and can, optionally, further include one or more additional functional groups. In other aspects, azlactone beads are reacted with a thiol or alcohol containing compound resulting in thioamide and ester functionalized beads. In other aspects, the azlactone bead are reacted with an amine-containing compound to provide a linker that provides one or more additional functional groups for further reaction with a ligand or biomolecule. For example, an amine-containing compound that includes an additional amine reactive group may be reacted with the azlactone bead, where the reactive amine group can be converted to an epoxide, maleimide, or iodoacetyl group using standard methods known in the art. In yet other aspects, azlactone beads are reacted with a compound to expand the number of functional groups attached to the bead. Such an approach can be an effective approach for increasing the binding capacity of the bead for a particular target molecule. For example, the beads may be reacted with an amine-containing compound (e.g., a polymer or dendrimer) that includes multiple reactive groups. First Named Inventor: RAJA, Erum TP385332WO1 In other aspects, the amine-containing compound are reacted with an azlactone group on the bead to provide a spacer to overcome steric hindrance upon ligand attachment or to provide a linker for subsequent reaction with a complementary reactive group using compounds and methods. For example, the amine containing compound can include a reactive functional group, such as an azide, alkyne (e.g., a cyclic alkyne such as dibenzocyclooctyne (DBCO)), maleimide, amine, hydroxyl, carboxylic acid, sulfonic acid, a chelator such as nitrilotriacetic acid (NTA), or iminodiacetic acid (IDA). Amine-containing compounds also include biomolecules (e.g., proteins, peptides, nucleic acids, nucleotides, and oligo- and polysaccharide), organic ligands, and large synthetic compounds, such as polymers (e.g., PEG or polyethylenimine (PEI)) and dendrimers. Representative amine-containing organic ligands include, without limitation, alkylamines (e.g., straight or branched C
4-C
12 alkyl amines such as 1,5- diaminopentane, diethylamine, butylamine, and octylamine), arylamines (e.g., benzylamine and 4-aminobenzoic acid), aminocaproic acid, ethanolamine, 5-amino-2- methyl benzene sulfonic acid, aminoethyl-trimethylammonium chloride (AETMA), taurine, tris(hydroxymethyl)aminomethane (Tris), and the like. Other examples of amine-containing compounds are positively or negatively charged ligands that can be used, e.g., in ion exchange (cationic and anionic) applications. Representative examples of ligands that can be used for anion exchange include, without limitation, alkylamines (e.g., straight or branched C4-C12 alkyl amines such as 1,5-diaminopentane (PDA) and butylamine), aryl amines (e.g. benzylamine and naphthylamine) and polymeric amines (e.g. PEG or polyethylenimine (PEI) such as PEI-25K, PEI-800K etc. In other embodiments, representative examples of ligands that can be used for cation exchange include, without limitation, any alkyl or aryl carboxylic acid, sulfonic acid and phosphonic acid. In some aspects, an amine containing moiety is a protein such as an antibody, an antibody binding protein (e.g., Protein A, Protein A/G, Protein G, and Protein L), Streptavidin, Neutravidin, glutathione, enzyme, or other biomolecule containing amines. In one aspect, azlactone beads are conjugated to an amine containing First Named Inventor: RAJA, Erum TP385332WO1 biomolecule such as an alkali stable Protein A (asPA) and the final conjugate can then be used to purify IgG. It is desired that when the biomolecule is an enzyme, it maintains its enzymatic selectivity once coupled to the bead. Examples of enzymes include proteolytic enzymes (e.g., trypsin) and other types of enzymes known in the art. FIG.12 shows similar performance with azlactone beads (asPA-magUL) and magnetic agarose beads (asPA-magAg). The assay was performed with duplicate samples of each magnetic bead. In some aspects, an amine-containing compound is or includes a metal charged chelate (e.g., nickel-charged nitrilotriacetic acid, Ni-NTA). In one aspect, azlactone beads are conjugated to Ni-NTA and the final conjugate is used to purify polyhistidine- tagged proteins from a soluble protein extract. In one aspect, azlactone beads are conjugated to metal charged chelate, such as Fe-NTA, Ti-NTA, Zr-NTA, or Ga-NTA, and the final conjugate is used to purify phosphorylated peptides or proteins from a biological sample (e.g., a cell lysate). In some aspects, an amine-containing compound is or includes a positively charged ligand. By way of example, the bead can be conjugated to a positively charged ligand, such as 1,5-diaminopentane. The positively charged bead conjugate can used to purify a negatively charged species (e.g., a nucleic acid, an extracellular vesicle, or a virus particle, or the like) from a biological sample (e.g., plasma, blood, cell culture supernatant, urine, and the like). In some aspects, the polymer bead is an azlactone bead formed from a vinyl azlactone and a crosslinker. The vinyl azlactone may be 4,4-dimethyl-2-vinyloxazol- 5(4H)-one, N 4,4-

5(4H)-one The cross linker maybe any crosslinker suitable to facilitate polymerization and form the bead. In some aspects, the crosslinker is a bisacrylamide, agarose, or a vinyl ether. In certain aspects, the crosslinker is methylene bisacrylamide. First Named Inventor: RAJA, Erum TP385332WO1 In particular aspects, the vinyl azlactone is 4,4-dimethyl-2-vinyloxazol-5(4H)- one and the crosslinker is a bisacrylamide, such as methylene bisacrylamide. In some aspects, the beads comprise a plurality of functional moieties on the O OH N surface of the bead. The functional moieties may be selected
, O H
N OH
. In certain aspects, the functional moieties
. First Named Inventor: RAJA, Erum TP385332WO1 In any aspects, the magnetic material in the magnetic beads may be in any suitable form, such as, but not limited to, a particle, powder, flake, cluster, bead, or combination thereof. In some embodiments, the magnetic material is or comprises magnetic particles. In any aspects, the magnetic material in the magnetic beads may comprise an iron source. The iron source may be any suitable iron source, such as an iron source that is added during the polymer bead formation reaction. The iron source may be in the form of iron-containing particles and/or may be an iron oxide, such as Fe3O4, Fe2O3, or a combination thereof. Additionally, or alternatively, the beads may include magnetic particles as the iron source, for example Dynabeads
TM (available from Thermo Fisher Scientific) and/or Sera-Mag
TM Speedbeads (available from Cytiva Life Sciences) and/or other magnetic beads or particles. In certain aspects, the magnetic material is, or comprise, Fe
3O
4 particles. In certain aspects, the magnetic material is an activated iron oxide source, such as Fe3O4-NHS, to provide an addition source of reactivity during particle synthesis. In some aspects, the magnetic material is an activated magnetic material and may contain one or more functional groups, such as, for example, a carboxy group or ester group. In certain aspects, the magnetic material is an activated iron source that comprises additional functional groups, such as, but not limited to, carboxy or ester groups. In some aspects, the magnetic material is contained within an agarose bead, and the agarose bead is contained within the crosslinked polymer to form the magnetic bead. The magnetic particles may have a size suitable for use in the disclosed beads, such as from 5 nm to 1,000 nm or more, such as from 5 nm to 200 nm or from 100 nm to 800 nm. In certain aspects, the magnetic particles have a size of from 15 nm to 100 nm, such as from 15 nm to 50 nm, or from 50 nm to 100 nm. In certain other aspects, the magnetic particles have a size of 100 nm to 800 nm, for example, iron clusters that may have an average particle size of from 100 nm to 800 nm, such as from 100 nm to 600 nm, or about 100 nm, 200 nm, 300 nm, 400 nm, 500 nm or 600 nm. First Named Inventor: RAJA, Erum TP385332WO1 The average particle size of the magnetic particles may be determined using a particle size analyzer. In some aspects, the magnetic particle is substantially spherical, but in other aspects, the particle is not substantially spherical. In certain aspects, the magnetic particle is a substantially spherical iron cluster. In certain other aspects, the magnetic particle is not an iron cluster and is not substantially spherical. However, such magnetic particles may be coated with another material that provides a substantially spherical shape. The magnetic beads have an average size suitable for use in separation technology. The average particle size is determined by a particle analyzer (e.g. Beckman Coulter
TM LS13320XR) or by scanning electron microscopy (SEM). In some aspects the magnetic beads are substantially spherical and the average particle size is the average diameter of the beads. In some aspects, the magnetic beads have an average size of from 20 μm to 100 μm or more, such as from 20 μm to 80 μm, from 30 μm to 60 μm, from 30 μm to 50 μm, or from 40 μm to 80 μm. The magnetic beads may be porous or non-porous. In some aspects, the magnetic beads are porous and may comprise pores having a size of from 100 Angstroms to 200 Angstroms. And in some aspects, the pores extend from the surface of the particle into the interior, but do not pass completely though the particle. In some aspects, at least a portion, and may be substantially all, of the magnetic material is located in the pores. The magnetic beads may have a hydrated state and a non-hydrated state. Typically, the disclosed bead size is the bead size in the hydrated state, and the bead size of the magnetic beads in the hydrated state (the hydrated bead size) is larger than the non-hydrated bead. These magnetic beads have the tendency to swell, from 3 times larger to 10 times larger or more, from 5 times larger to 8 times larger, or from 5 times to 6 times larger. By way of comparison, nonmagnetic beads typically swell to about 8x larger when hydrated, compared to their non-hydrated state. And to the inventors’ knowledge, other magnetic beads do not swell to a measurable extent. The swelling of the disclosed magnetic beads may allow the functional moieties on the bead surface, such as azlactone moieties, to be more accessible, and/or allow large biomolecules to First Named Inventor: RAJA, Erum TP385332WO1 bind to the functional moieties more easily because steric problems due to their size are reduced due to the swelling increasing the space between the functional moieties. In some aspects, the disclosed magnetic beads demonstrate a reduced bead loss during use in an automated process, for example, a Kingfisher apparatus, when compared with other magnetic beads. The bead loss may be due, at least in part, to surface adsorption onto the apparatus, for example, onto the tip plate and sample plate. In some aspects, a population of the disclosed magnetic beads exhibits a bead loss of less than about 5% by weight when used in an automated process. In a comparative experiment, the disclosed magnetic beads demonstrated a bead loss of 4.7% by weight, where as Cytiva Sera-Mag
TM beads had a loss of 6.0% by weight, Invitrogen
TM DynaGreen
TM silica beads lost 11.5% by weight and Dynabeads
TM MyOne
TM Silane beads lost 6.8% by weight. III. Synthesis of the Magnetic Beads


A first solution of the monomer and optionally a polymeric stabilizer is prepared in a suitable solvent, such as an alkane solvent, for example, heptane or hexanes, or a chlorinated solvent, such as carbon tetrachloride, chloroform, or dichloromethane. The monomer may be an azlactone monomer, such as a vinyl azlactone, for example, 4,4- dimethyl-2-vinyloxazol-5(4H)-one. The polymeric stabilizer may be any polymeric stabilizer suitable for use in the polymerization reaction. Example, polymeric stabilizers include, but are not limited to, copoly(isooctylacrylate/acrylic acid), copoly(isobutylmethacrylate/acrylic acid) or copoly(hexylacrylate/sodium acrylate). After agitation, the magnetic material is added in a suitable form, such as, but not limited to, a particle, powder, flake, cluster, bead, or combination thereof. In some First Named Inventor: RAJA, Erum TP385332WO1 aspects, the magnetic material comprises an iron source, such as iron oxide. In certain aspects, iron oxide particles are used. And in some aspects, the first solution is prepared and maintained at a temperature of from 35 °C to 50 °C, such as from 40 °C to 45 °C. A second solution is prepared comprising a crosslinker in a suitable solvent. The crosslinker is any crosslinker suitable to facilitate bead formation. Exemplary crosslinkers include, but are not limited to, an acrylamide, bisacrylamide, agarose, or a vinyl ether. In certain aspects, the crosslinker is methylene bisacrylamide. The solvent may be any solvent suitable to facilitate the polymerization reaction, such as an alcohol (for example, methanol, ethanol, 2-propanol, 1-propanol, or a combination thereof), water, or a combination thereof. In some aspects, a radical initiator, for example, sodium persulfate, is added to the second solution. And in some aspects, the second solution is prepared and maintained at a temperature of from 25 °C to 40 °C, such as from 30 °C to 35 °C. The first solution and the second solution are combined and the mixture is agitated such as by stirring, shaking, swirling, etc, at a temperature suitable to facilitate polymerization and bead formation, followed by addition of a polymerization initiator, such as tetramethylethylenediamine (TEMED). In some aspects, the temperature is from 30 °C to 50 °C. After cooling to room temperature, the beads are filtered and washed with a suitable solvent, such as acetone. The beads then are resuspended in a suitable solvent, such as acetone, and agitated, such as by shaking, stirring, swirling, or sonication, with cooling, such as in an ice bath. The beads are then screened to select beads of a desired size range. Screening the beads may comprise using one or more sieves, such as two sieves, to select beads having the desired size or size range. In some aspects, a 63 μm sieve and a 25 μm sieve are used to select a population of magnetic beads having a size range of from 63-25 μm. The magnetic beads then are placed in a magnetic separate system to remove any non-magnetic residue. After filtering, the beads are dried, for example, under a vacuum and/or with warming at from 30 °C to 60 °C First Named Inventor: RAJA, Erum TP385332WO1 Before use, the beads are typically swelled in an aqueous liquid, such as water, a buffer solution, or an aqueous/organic solvent mixture. Additional information concerning synthesizing crosslinked beads can be found in United States Patent Nos.5,403,902 and 5,292,840, which are incorporated herein by reference in their entirety. IV. Kit Comprising the Magnetic Beads Also disclosed herein are aspects of a kit comprising the disclosed magnetic beads. The components of the kit may depend on the intended use. In some aspects, the kit comprises, in addition to the disclosed magnetic beads, a lysis solution, universal nuclease, reduction solution, alkylation solution, enzyme reconstitution solution, Trypsin/Lys-C protease mix, bead wash/binding solution, wash solution, elution solution, or any combination thereof. In certain aspects, a kit comprises the disclosed magnetic beads, a lysis solution, a universal nuclease, a reduction solution, an alkylation solution, an enzyme reconstitution solution, and a Trypsin/Lys-C protease mix. In other aspects, a kit comprises the disclosed magnetic beads, a bead wash/binding solution, a wash solution, and an elution solution. V. Biomolecules and Reagents Successful mass spectrometry analysis of peptides requires high-quality input samples. Sample preparation prior to mass spectrometry (MS) often requires the introduction of chemicals which are incompatible with downstream MS analysis and must be removed from the sample prior to LC-MS analysis. However, complete cleanup of salts, detergents, and other chemical and non-chemical contaminants which simultaneously retains all peptide species and demonstrates reasonable recovery is very difficult, often requiring special equipment, highly complex processes, and/or large time commitments. The disclosed magnetic beads may be used to simplify and accelerate sample cleanup while improving sample quality in terms of cleanliness and introducing First Named Inventor: RAJA, Erum TP385332WO1 amenability to automation. The beads also are compatible with plastics and solvents that are frequently used in biological assay. These features make the beads amenable to downstream applications prior to MS, and also other applications, such as chemical labeling, enrichment, and fractionation. The disclosed magnetic beads are suitable for use with a wide range of biomolecules, including proteins, polypeptides, peptides, DNA, RNA, and combinations thereof. Additionally, the disclosed magnetic beads are suitable for use with isotopically labeled biomolecules. The isotopic labelling may include labeling with deuterium, carbon-13, nitrogen-15, oxygen-18, or a combination thereof. Additionally, or alternatively, the biomolecules may be labelled with an isotopically-enriched mass tag, such as, but not limited to, Tandem Mass Tag (TMT and TMTpro) Reagents (available from Thermo Fisher Scientific
TM); stable isotopically labeled peptides and/or proteins prepared using SILAC Metabolic Labeling Systems (available Thermo Fisher Scientific
TM); rare-earth metals (e.g., lanthanide); fluorescent labels (e.g., dyes); tissues and/or cells grown with heavy amino acids; or a combination thereof. In some aspects, the disclosed magnetic beads are useful in multi- analyte analysis applications because of their distinct binding profiles to different types of biomolecules. For example, the disclosed magnetic beads typically bind DNA at low levels of organic solvent, such as about 30% organic, and are not affected by the presence of guanidine. In contrast, the disclosed beads bind to RNA at higher levels of organic solvent, such as about 90% organic. And the disclosed magnetic beads can bind to proteins in a solvent up to 45% organic in the presence of guanidine hydrochloride, or at about 80% without guanidine. These combinations of guanidine and organic presence can be utilized uniquely in the disclosed magnetic beads for sequential extraction of DNA, protein, and RNA from a single sample, with potential for much less cross-contamination of other analytes. Furthermore, the disclosed magnetic beads are compatible with a variety of organic solvents, including, but not limited to, alcohols such as methanol, ethanol, and isopropanol; acetonitrile; esters, such as ethyl acetate; and acids, such as trifluoroacetic acid and formic acid. First Named Inventor: RAJA, Erum TP385332WO1 VI. Applications In some aspects, a general method of using the magnetic beads disclosed herein comprises forming a first suspension comprising a plurality of the disclosed magnetic beads and a first liquid that contains a biomolecule of interest. After the biomolecule has bound to the magnetic beads, the beads are removed from the first suspension and optionally washed to remove any unwanted or unbound material. A second suspension then is formed comprising the magnetic beads with the bound biomolecule and an elution solution. And the magnetic beads are then removed from the second suspension to leave a solution comprising the biomolecule of interest. The biomolecule may be any biomolecule suitable for binding to the disclosed magnetic beads. In some aspects, the biomolecule is a protein, peptide, polypeptide, DNA or RNA molecule. And in some aspects, the biomolecule is a composite molecule that comprises a combination of a protein, peptide, polypeptide, DNA, and/or RNA moiety. By appropriate choice of ligand, magnetic beads disclosed herein can be adapted for use in anion and cation exchange purification methods. A general ion exchange method that uses the magnetic beads disclosed herein includes forming a first suspension that includes a plurality of the disclosed magnetic beads and a first liquid that contains a species of interest that carries a negative charge or a positive charge. The species of interest can bind to the ligand on the bead and then can be separated from the other components of the suspension. In certain embodiments, the species of interest can be released from the bead. In any aspects, the biomolecule may be labeled. In some aspects, the label may be any label suitable for use in an analytical technique, such as mass spectrometry. Example labels include, but are not limited to, stable isotopes, such as deuterium, carbon-13, nitrogen-15, oxygen-18; isotopically-enriched mass tags, such as, but not limited to, Tandem Mass Tag (TMT and TMTpro) Reagents (available from Thermo Fisher Scientific
TM); stable isotopically labeled peptides and/or proteins prepared using SILAC Metabolic Labeling Systems (available Thermo Fisher Scientific
TM); rare-earth First Named Inventor: RAJA, Erum TP385332WO1 metals (e.g., lanthanide); fluorescent labels (e.g., dyes); tissues and/or cells grown with heavy amino acids; or any combination thereof. FIG.2 provide a flowchart of an SP3 workflow using the disclosed magnetic beads, that includes a PAC process and an SP2 process which are discussed below in more detail. A. Cleaning-up Peptides and/or Polypeptides In some aspects, the disclosed magnetic beads are used to clean up peptides and/or polypeptides. In some aspects, a mixture is formed comprising the disclosed beads and a sample containing the peptides and/or polypeptides to be cleaned-up. The sample may be a protein digest sample. Optionally, the magnetic beads may be washed before being combined with the sample, for example, to remove a storage buffer solution. The mixture may further comprise a bead wash/binding solution. In some aspects, the bead wash/binding solution is 100% acetonitrile with 0.1% formic acid. The bead wash/binding solution may be added to the magnetic beads to form a bead slurry before the beads are added to the sample. The amount of the bead slurry that is added to the sample may vary depending on the amount of proteins in the sample. In some aspects, the total volume of the bead slurry for input proteins of 15-30 µg, 30-60 µg, or 60-150 µg should be 250 µL, 500 µL, or 1000 µL, respectively. In some aspects, the bead slurry has a bead concentration of 25%. In certain aspects, about 20 μL of a 25% bead slurry is used that provides 5 mg of beads per sample. In some aspects, from 3 mg to 10 mg of beads are used for 10-25 μg of proteins, such as about 5 mg of beads per 10-25 μg proteins. After combining the disclosed magnetic beads with the sample, the resulting mixture may be incubated at a temperature suitable to facilitate protein binding, such as from 20 °C to 30 °C or at about room temperature. The incubation is allowed to proceed for a time period suitable to facilitate protein binding, such as from 15 minutes to 180 minutes or more, or from 30 minutes to 60 minutes. The mixture may be agitated during incubation, such as by shaking, swirling, stirring, sonication, etc. First Named Inventor: RAJA, Erum TP385332WO1 After incubation, the magnetic beads are removed from the mixture and any unbound peptides or polypeptides by a suitable technique, such as using a magnetic stand. The magnetic beads may be washed to remove any remaining unbound material, solvents, etc. In some aspect s, the wash solution is from 90% to 99% organic in water with from 0.1% to 1% organic acid, and may be about 85% acetonitrile, 10% ethanol, 0.5% formic acid and 4.5% water. The magnetic beads then are treated with an elution solution to remove the bound peptides and/or polypeptides from the magnetic beads. In aspects, the elution solution is from 1% to 5% organic with from 0.05% to 0.25% acid in water, such as 2% acetonitrile/0.1% formic acid in water. The magnetic beads are incubated with the elution solution at a temperature and for a time period suitable to facilitate the peptides and/or polypeptides being eluted from the beads. Incubation may proceed at a temperature of from 20 °C to 30 °C or at about room temperature, and/or for from 1 minute to 10 minutes or more, such as from 2 minute to 5 minutes, or about 3 minutes. After incubation, the magnetic beads are removed from the eluted peptide and/or polypeptide solution using a suitable technique, such as a magnetic stand. The eluted peptide and/or polypeptide solution then may be transferred to a suitable container for analysis, such as by mass spectrometry. In one aspect, a method for obtaining a peptide or polypeptide for LC-MS analysis comprises: forming a first suspension comprising a plurality of the disclosed magnetic beads and a first liquid that contains a protein digest sample comprising the peptide or polypeptide of interest; agitating the first suspension at room temperature for from 30 to 60 minutes; removing the magnetic beads from the first suspension and washing the magnetic beads using a second liquid; forming a second suspension comprising the magnetic beads and an elution solution and agitating the second suspension at room temperature for from 1 to 5 minutes; and First Named Inventor: RAJA, Erum TP385332WO1 separating the magnetic beads from the second suspension to leave a third liquid comprising the peptide or the polypeptide of interest. In some aspects, each magnetic bead in the plurality of magnetic beads comprises iron oxide particles distributed throughout an azlactone bead and a plurality O OH N m
oieties located on the surface of the magnetic bead.
B. Protein Aggregation Capture (PAC) Aspects of the disclosed magnetic beads are useful for protein aggregation capture (PAC) applications. In some aspects, a mixture is formed comprising the disclosed beads and a sample containing the proteins. Optionally, the magnetic beads may be washed before being combined with the sample, for example, to remove a storage buffer solution. In some aspects, the washing solution may comprise from 70% to 95% by volume organic solvent, such as from 75% to 85% by volume organic solvent (for example, an alcohol such as 2-propanol, or acetonitrile), and also may comprise an organic acid, such as trifluoroacetic acid (TFA), in an amount of from greater than zero to 1% by volume, such as 0.25% to 1% by volume organic acid, such as TFA. In some aspects, the washed magnetic beads are reconstituted in a solution comprising from 30% to 95% organic solvent, such as from 75% to 85% organic solvent (for example, an alcohol such as 2-propanol, or acetonitrile), and an organic acid, such as TFA, in an amount of from greater than zero to 1%, such as 0.25% to 0.75% organic acid. In some aspects, about 80% 2-propanol with 0.5% TFA is used for the washing solution, reconstitution solution, or both. Organic solvent and optionally organic acid may be added to the protein sample to produce a final organic concentration of from 70% to 95% or more organic solvent in the protein sample, such as from 75% to 85% organic solvent, and optionally from First Named Inventor: RAJA, Erum TP385332WO1 greater than zero to 1% organic acid concentration, such as from 0.25% to 0.57% or about 0.5% organic acid concentration. In any aspects of the PAC method, unless otherwise specified, the organic solvent may be any suitable organic solvent, such as an alcohol (for example, 2- propanol, ethanol, methanol, 1-propanol), an ester solvent (for example, ethyl acetate), or acetonitrile, of a combination thereof. In any aspects of the PAC method, unless otherwise specified, the organic acid may be any suitable organic acid, such as trifluoroacetic acid (TFA) or formic acid (FA). In other aspects, the organic acid is replaced by an organic base, such as a trialkylamine, for example triethylamine. The disclosed magnetic beads are then added to the protein sample. In some aspects, the magnetic beads are added in an amount of from 5:1 w/w beads:protein to 15:1 w/w beads:protein, such as from 8:1 w/w beads:protein to 12:1 w/w beads:protein, or about 10:1 w/w beads:protein. The magnetic beads may be added in a buffer at from 70% to 95% organic concentration, such as from 75% to 85% organic concentration. And/or the protein concentration may be from greater than zero to 5 mg/mL, such as from 0.5 mg/mL to 3 mg/mL, or from 1 mg/mL to 2 mg/mL. After combining the disclosed magnetic beads with the sample, the resulting mixture may be incubated at a temperature suitable to facilitate protein binding, such as from 20 °C to 30 °C or at about room temperature. The incubation is allowed to proceed for a time period suitable to facilitate protein binding, such as from 15 minutes to 180 minutes or more, or from 30 minutes to 60 minutes. The mixture may be agitated during incubation, such as by shaking, swirling, stirring, sonication, etc. After incubation, the magnetic beads are separated from the mixture. The separated beads are washed in a solution comprising from 65% to 100% organic solvent. The beads may be washed once, or more than once. In some aspects, the beads are washed with 100% organic solvent and followed by 65% to 95% organic solvent, such as 70% to 80% organic solvent. The organic solvent may be any organic solvent suitable for use in the PAC method. In certain aspects, the organic solvent may be First Named Inventor: RAJA, Erum TP385332WO1 acetonitrile or an alcohol. In particular aspects, the beads are washed with 100% acetonitrile followed by 70% alcohol, such as ethanol. After washing, the magnetic beads are exposed to a digestion mixture. In some aspects, from 10 μL to 25 μL digestion mixture is added for a 1-2 mg/mL protein sample. The digestion mixture may be any suitable digestion mixture. In some aspects, the digestion mixture comprises a buffer, such as a trialkyl ammonium buffer (for example, triethylammonium bicarbonate (TEAB)), an inorganic salt, such as calcium chloride, and one or more enzymes (for example, Trypsin and/or LysC). The enzyme or enzyme mixture may be present in an amount of from 1:7 to 1:30 w/w enzyme:protein, such as 1:10 to 1:25 w/w enzyme:protein. The magnetic beads are incubated in the digestion mixture for a time period suitable to facilitate digestion, such as from 1 hour to 5 hours or more, or from 2.4 hours to 3.5 hours. The mixture may be agitated during incubation, such as by shaking, swirling, stirring, sonication, etc. The incubation may proceed at a temperature suitable to facilitate digestion, such as from 25 °C to 50 °C, from 30 °C to 40 °C, or from 35 °C to 40°C. After incubation, organic solvent is added to the mixture to achieve an organic concentration in the mixture of greater than 90%, such as greater than 92%, or greater than 92.5%. The magnetic beads may be separated and washed with a solution having a high organic concentration, such as an organic concentration of greater than 90%, greater than 92% or at least 95% organic. In some aspects, the washing solution comprises acetonitrile, an alcohol such as ethanol, and water, and may further comprise an organic acid such as formic acid. In one example, the washing solution comprises about 85% acetonitrile, about 10% ethanol, and about 5% water, and may further comprise 0.5% formic acid. After washing, the magnetic beads are eluted with an aqueous solution comprising from 1% to 5% organic solvent, such as from 2% to 4% organic solvent, First Named Inventor: RAJA, Erum TP385332WO1 and from 0.1% to 0.5% organic acid, such as from 0.2% to 0.3% organic acid. In some aspects, the organic solvent is acetonitrile and/or the organic acid is formic acid. After elution, the magnetic beads are removed from the mixture. In one aspect, a method for obtaining a protein comprises: forming a first suspension comprising the plurality of magnetic beads and a first liquid that contains the protein, wherein the first liquid is an aqueous solution comprising 0.5% trifluoroacetic acid, and 80% of an organic solvent selected from 2- propanol and acetonitrile; agitating the first suspension at room temperature for from 30 to 60 minutes; removing the magnetic beads from the first suspension and washing the magnetic beads with a second liquid comprising an organic solvent; incubating the magnetic beads with a digestion mixture comprising Trypsin and LysC for from 1 to 4 hours; washing the beads with a washing solution comprising water, an organic solvent and formic acid, to remove the digestion mixture; forming a second suspension comprising the magnetic beads and an elution solution and agitating the second suspension at room temperature for from 1 to 5 minutes; and separating the magnetic beads from the second suspension to leave a third liquid comprising the protein. And in some aspects of the method, each magnetic bead comprises iron oxide O OH N H particles distributed throughout an azlactone bead and a plurality of O moieties located on the surface of the magnetic bead. C. Multi-Omic Sequential Aggregation Capture Aspects of the disclosed magnetic beads are useful for sequential aggregation capture applications for samples comprising combinations of nucleic acids and proteins. In some aspects, a mixture is formed comprising the disclosed beads and a sample First Named Inventor: RAJA, Erum TP385332WO1 containing the molecules of interest. The sample may be a cell lysate. In some aspects, from 10 mg to 20 mg of the disclosed magnetic beads are used per 1 x 10
6 cells of lysate, such as about 15 mg of the disclosed magnetic beads per 1 x 10
6 cells. Optionally, the magnetic beads may be washed before being combined with the sample, for example, to remove a storage buffer solution. Described below are aspects of a method for separating DNA, protein and RNA from a mixed sample. A person of ordinary skill in the art understands that the method steps can be used in any order, or omitted, depending on the sample provided and the nature of the desired molecules. a. DNA binding After the magnetic beads are added to the sample, organic solvent is added to produce an organic concentration in the sample of from 10% to 40%, such as from 20% to 35% or from 25% to 35%, or about 30% organic concentration. The organic solvent can be any organic solvent suitable for facilitate DNA binding. Unless otherwise specified, the organic solvent may be any suitable organic solvent, such as an alcohol (for example, 2-propanol, ethanol, methanol, 1-propanol), and ester solvent (for example, an acetate ester, such as ethyl acetate), or acetonitrile, of a combination thereof. After combining the disclosed magnetic beads with the sample, the resulting mixture may be incubated at a temperature suitable to facilitate DNA binding, such as from 20 °C to 30 °C or at about room temperature. The incubation is allowed to proceed for a time period suitable to facilitate DNA binding, such as from 15 minutes to 180 minutes or more, or from 30 minutes to 60 minutes. The mixture may be agitated during incubation, such as by shaking, swirling, stirring, sonication, etc. After incubation, the magnetic beads with the DNA bound thereto are separated from the mixture by a suitable technique, such as a magnetic rack. The remaining lysate may be treated as necessary with additional magnetic beads to remove RNA and/or proteins as disclosed herein. The magnetic beads with the DNA bound thereto is treated with guanidine hydrochloride, a buffer, and a chelating agent, in an aqueous solvent comprising about First Named Inventor: RAJA, Erum TP385332WO1 from 20% to 40% organic solvent, such as from 25% to 35% organic solvent. The organic solvent may be an alcohol, such as 2-propanol, ethanol, methanol or 1-propanol. The resulting mixture is incubated for from 5 minutes to 30 minutes with agitation. After incubation, the beads are removed from the liquid, by a suitable technique, such as a magnetic rack, and are then treated with a second guanidine hydrochloride solution comprising a buffer, surfactant, and chelating agent. The buffer(s) may be Tris HCL, PBS, HEPES, triethylammonium bicarbonate (TEAB), or a combination thereof. The surfactant may be Tween, Triton X-100, SDS, or a combination thereof. And/or the chelating agent may be ethylenediaminetetraacetic acid (EDTA). After incubating, the supernatant is removed and proteinase K and/or RNase may be added to the supernatant if desired. The mixture is incubated at from 40 °C to 65°C, such as from 50 °C to 60 °C, for from 5 minutes to 30 minutes, with water added to the beads to prevent drying out. After incubation, the water is removed and the supernatant is returned to the beads. The mixture is treated with a further portion of guanidine hydrochloride in a buffer solution containing EDTA and about 60% organic solvent, and the resulting mixture is incubated for from 30 minutes to 60 minutes at room temperature with agitation. After washing the beads with a solution comprising a buffer, such as a Tris hydrochloride buffer, and about 60% to 80% organic solvent, the DNA is eluted with an aqueous solution comprising a buffer such as Tris HCl and a chelating agent, such as EDTA. The magnetic beads are then removed by a suitable technique, and the DNA can be used or stored for future applications. b. Protein separation A lysate sample, such as the lysate sample from the DNA binding described above, is treated with fresh magnetic beads as disclosed herein, along with sufficient organic solvent to produce a total organic concentration of about 45%. Organic acid, such as TFA or formic acid, also may be added. The resultant mixture is incubated for from 30 minutes to 60 minutes or more with agitation to facilitate protein binding. First Named Inventor: RAJA, Erum TP385332WO1 After incubation the magnetic beads are removed by a suitable technique, such as magnetic rack. The RNA-containing supernatant may be used in an RNA-binding protocol described herein. The magnetic beads with the proteins bound thereto are washed one or more times, such as 1, 2, 3 or more times with from 70% to 100% organic solvent, for example, 70% ethanol and/or 100% acetonitrile. After washing the magnetic beads optionally may be reduced and/or alkylated, such as by treating with tris (2- carboxyethyl)phosphine (TCEP) and/or 2-chloroacetamide. The beads then are treated with universal nuclease followed by Trypsin/LysC. After digestion, the proteins are eluted as described herein with respect to the PAC method. c. RNA binding Guanidine hydrochloride is added to a liquid sample containing RNA, such as the RNA-containing supernatant described herein, and the mixture is incubated at from 30 °C to 40 °C for from 1 minute to 20 minutes. A clean sample of the disclosed magnetic beads is added along with sufficient organic solvent such that the sample is at least 85% organic solvent, such as 90% organic solvent. Suitable organic solvents include, but are not limited to, acetonitrile or an alcohol, such as 2-propanol, 1- propanol, ethanol, methanol, or a combination thereof. The mixture is incubated at room temperature for a sufficient time to facilitate RNA binding to the magnetic beads, such as for 30 minutes to 60 minutes. After incubation, the magnetic beads are separated from the supernatant by a suitable technique, such as magnetic rack. The magnetic beads are treated with a mixture of buffer, guanidine hydrochloride, surfactant and a chelating agent. The buffer maybe PBS, Tris HCl, or a combination thereof. And/or the surfactant is 10% Tween, Triton X-100, or a combination thereof. And/or the chelating agent may be EDTA. After incubating, the supernatant is removed and proteinase K and/or RNase may be added to the supernatant if desired. The mixture is incubated at from 40 °C to First Named Inventor: RAJA, Erum TP385332WO1 65°C, such as from 50 °C to 60 °C, for from 5 minutes to 30 minutes, with water added to the beads to prevent drying out. After incubation, the water is removed and the supernatant is returned to the beads. Organic solvent is added to the bead/sample mixture such that sample is 90% final organic concentration. Suitable organic solvents include, but are not limited to, acetonitrile or an alcohol, such as 2-propanol, 1-propanol, ethanol, methanol, or a combination thereof. The mixture is incubated with agitation at room temperature for a time sufficient to facilitate binding, such as from 30 minutes to 60 minutes. The magnetic beads are then washed with a mixture of from 60% to 80% organic solvent, such as from 65% to 75% organic solvent, a chelating agent, and a buffer. The chelating agent may be EDTA, and/or the buffer may be Tris HCl, PBS, or a combination thereof. Suitable organic solvents include, but are not limited to, acetonitrile or an alcohol, such as 2-propanol, 1-propanol, ethanol, methanol, or a combination thereof. The RNA is then eluted with water and a chelating agent, such as EDTA, and the magnetic beads are separated from the RNA-containing mixture. D. Ion Exchange Applications The disclosed magnetic beads can be useful in applications involving capture of charged species from a biological sample. In an exemplary ion exchange application, a mixture is formed that includes beads bearing an appropriate ligand, as disclosed herein, and a sample containing the charged species of interest. By way of example, in one method about 10 mg to about 20 mg of the disclosed magnetic beads are used per 1 x 10
6 cells of cell lysate, such that there are about 15 mg of the disclosed magnetic beads per 1 x 10
6 cells. Optionally, the magnetic beads can be washed before combining with the sample to remove a storage buffer solution or other contaminants. In certain embodiments, the charged species of interest can be released from the beads for potential use in further downstream processes. Described herein are aspects of a method for separating DNA, protein and RNA from a mixed sample, and the method steps can First Named Inventor: RAJA, Erum TP385332WO1 be used in any order, or omitted, depending on the sample provided and the nature of the desired molecules. VII. Examples Example 1 Preparation of Magnetic Beads A one-liter, Morton type round bottom flask (Flask 1) equipped with an overhead stirrer, nitrogen inlet and a thermocouple was charged with toluene (100-150 mL) and heated to 40 °C with a stirring rate of 200 rpm. A polymeric stabilizer was added (1 g/ml, 282 μl) after which the temperature increased to 45 °C. Heptane (400 mL) was slowly added to the reaction ensuring that the temperature remained above 40 °C at all times. In parallel, a 250-ml round bottom flask (Flask 2), equipped with a magnetic stirrer, nitrogen inlet and a thermocouple, was charged with methylene bisacrylamide (10.5 g, 68 mmol), isopropanol (72.5 mL) and product water (40 mL) and was heated to 30 °C. Vinyl dimethylazlactone (VDM or VDMA) (1.0 g, 7.2 mmol) was added to Flask 1 after the addition of heptane. The remaining steps were performed within 15 minutes. Stir rate was increased followed by the addition of iron oxide (3 g, 13 mmol) (Fe
3O
4). Sodium persulfate (0.435 g, 1.8 mmol), dissolved in 8 mL of DI water (which was purged with nitrogen for 1 hour prior to use) was added to Flask 2 and stirred for 1- 2 minutes. The magnetic stir bar was removed, and contents of Flask 2 were transferred to Flask 1. The final mixture was stirred for 5-10 minutes at 35 °C, after which time neat TEMED (0.435 mL, 3 mmol) was added to initiate polymerization. Within a minute, an exotherm of about 5-8 °C was observed followed by formation of gray/black beads. The reaction was stirred at 40 °C for 2 hours. Once the reaction had cooled down to room temperature, the beads were filtered and washed with acetone. The beads were resuspended in acetone and sonicated for 2-3 hours in an ice bath. The beads were then sieved using a 63 μm screen on top and 25 μm screen at the bottom with majority of the beads being collected on top of the 25 μm screen. The population of beads between 63-25 μm was isolated, transferred to a 1L First Named Inventor: RAJA, Erum TP385332WO1 Pyrex Bottle and placed in a magnet, such as a magnetic separation system from Sepmag
TM or equivalent, to remove any non-magnetic residue from the magnetic beads. The beads then were filtered using a Buchner funnel and transferred to a glass vial to be dried in a vacuum oven overnight at 35-40 °C. The magnetic beads were swelled in water and analyzed under a microscope. FIG.3 shows that the process produced round, spherical beads with iron oxide being distributed throughout the body of the beads. Particle size analysis was performed using a LS13-320 XRSW Particle Size Analyzer from Beckman Coulter (Indianapolis, IN). FIG.4 shows that the mean particle size was about 48 μm. FIGS.5-7 provide digital images showing beads made using different sources of magnetic material, including iron oxide clusters (FIG.5), iron oxide particles (FIG.6) and Dynabeads
TM (FIG.7). FIGS.5-7 demonstrate that despite the different magnetic materials used, the magnetic beads were all substantially round and spherical, and had an average size of from 30 μm to 60 μm. Example 2 Preparation of Magnetic Beads A one-liter, Morton type round bottom flask (Flask 1) equipped with an overhead stirrer, nitrogen inlet and a thermocouple was charged with toluene (160 mL) and heated to 40 °C with a stirring rate of 200 rpm. A polymeric stabilizer was added (1 g/ml, 282 μl) after which the temperature increased to 45 °C. Heptane (400 mL) was slowly added to the reaction ensuring that the temperature remained above 40 °C at all times. In parallel, a 250-ml round bottom flask (Flask 2), equipped with a magnetic stirrer, nitrogen inlet and a thermocouple, was charged with methylene bisacrylamide (20 g), isopropanol (72.5 mL) and product water (40 mL) and was heated to 30 °C. Vinyl dimethylazlactone (VDM or VDMA) (1.0 g, 7.2 mmol) was added to Flask 1 after the addition of heptane. Stir rate was increased to 500 rpm. Iron source and sodium sulfate were added to flask 2 containing MBA and added to flask 1. The final mixture was stirred for 5-10 minutes after which time neat TEMED was added to initiate polymerization. Within a minute, an exotherm of about 5-8 °C was observed First Named Inventor: RAJA, Erum TP385332WO1 followed by formation of gray/black beads. The reaction was stirred at 600 rpm and 40 °C for 2 hours. Once the reaction had cooled down to room temperature, magnetic beads were filtered and washed with acetone. The beads were sonicated, sieved and tested for microscopic analysis as well as particle size analysis using the procedure described in Example 1. Example 3 Preparation of Magnetic Beads A one-liter, Morton type round bottom flask (Flask 1) equipped with an overhead stirrer, nitrogen inlet and a thermocouple was charged with toluene (160 mL) and heated to 40 °C with a stirring rate of 400 rpm. A polymeric stabilizer was added (1 g/ml, 282 μl) after which the temperature increased to 45 °C. Heptane (200 mL) was slowly added to the reaction ensuring that the temperature remained above 40 °C at all times. In parallel, a 250-ml round bottom flask (Flask 2), equipped with a magnetic stirrer, nitrogen inlet and a thermocouple, was charged with methylene bisacrylamide (20 g), isopropanol (72.5 mL) and product water (40 mL) and was heated to 30 °C. Vinyl dimethylazlactone (VDM or VDMA) (1.0 g, 7.2 mmol) was added to Flask 1 after the addition of heptane. Stir rate was increased to 500 rpm after which Dynabeads
TM (1g) and sodium sulfate were added to flask 2 containing MBA. The mixture was stirred for 2 minutes and added to flask 1. The final mixture was stirred for 5-10 minutes after which time neat TEMED was added to initiate polymerization. Within a minute, an exotherm of about 5-8 °C was observed followed by formation of gray/black beads. The reaction was stirred at 850 rpm and 40 °C for 2 hours. Once the reaction had cooled down to room temperature, magnetic beads were filtered and washed with acetone. The beads were sonicated, sieved and tested for microscopic analysis as well as particle size analysis using the procedure described in Example 1. Example 4 First Named Inventor: RAJA, Erum TP385332WO1 Hydrolysis of Magnetic Beads O N R
Beesaind HydrolysisR B
ee
sa
ind N
OH O O H O
Magnetic beads were hydrolyzed according to the following procedure. Magnetic beads (0.50 g) were added to a 15 ml conical tube containing water (10 mL) and rotated overnight at room temperature. The beads were washed the next day with water using an appropriate magnetic stand. The hydrolyzed beads were then adjusted to a 25% slurry in water. Solutions used in Examples 5-8 are provided in Table 1 unless otherwise specified. Table 1. List of solutions
First Named Inventor: RAJA, Erum TP385332WO1 Example 5 Example Procedure (SP2) using the Disclosed Magnetic Beads Use 15-150 µg of protein per sample preparation. Rinse cultured cells or tissues 2-3 times with 1 × PBS to remove cell culture media or excess blood, respectively. Resuspend proteins, cells, or tissues in Lysis Solution without additional buffers. A. Extract protein, reduce, and alkylate 1. For cultured cells, add 50 µL of Lysis Buffer and 1 µL of Universal Nuclease to a minimum of 1 × 10
6 cells. Pipet up and down (with P200 tip) for 10-15 cycles until sample viscosity is reduced. Note: Lyse the cells in an appropriate volume of lysis buffer to have the final concentration of 2-5 µg/µl with a final volume of 7.5 µL, 15 µL, or 30 µL for input proteins of 15-30 µg, 30-60 µg, or 60-150 µg, respectively. Centrifugation of cultured cell lysates is typically not required after aspiration using a pipet. 2. For tissue samples, add 50 µL of Lysis Solution (containing 1 µL Universal Nuclease) per 5 mg of tissue and disrupt with tissue homogenizer until sample is homogenized. Centrifuge tissue lysates at 16,000 × g for 10 minutes. 3. For purified proteins, depleted and undepleted plasma, and serum samples, dilute samples directly in Lysis Solution to 2-5 µg/µL with a final volume of 7.5 µL, 15 µL, or 30 µL for input proteins of 10-25 µg, 25-50 µg, or 50-100 µg, respectively. Note: The concentration of plasma is around 60-70 µg/µL. For purified proteins and plasma samples, addition of Universal Nuclease is not required. 4. Determine the protein concentration of the supernatant using established methods such as the Pierce™ BCA Protein Assay Kit (Product No.23227) or Pierce™ Rapid Gold BCA Protein Assay Kit (Product No. A53226). Note: Dilution with Lysis Solution may be required for concentrated samples for BCA assay. 5. Transfer 15-150 µg of protein sample into a 1.5-mL low protein binding tube or a 96-well plate and adjust the final volume to 7.5 µL, 15 µL, or 30 µL for input proteins of 15-30 µg, 30-60 µg, or 60-150 µg, respectively with Lysis Solution. First Named Inventor: RAJA, Erum TP385332WO1 6. Add 3.75 µL, 7.5 µL, or 15 µL of Reduction Solution for input proteins of 15-30 µg, 30-60 µg, or 60-150 µg, respectively to the sample and gently mix. 7. Add 3.75 µL, 7.5 µL, or 15 µL of Alkylation Solution for input proteins of 15- 30 µg, 30-60 µg, or 60-150 µg, respectively to the sample and gently mix. 8. Incubate the sample at 95 °C using a heat block for 10 minutes or 50 °C for 20- 30 minutes to reduce and alkylate the protein sample. 9. After incubation, allow the sample to cool to room temperature. 10. Briefly centrifuge the tube or plate before digestion. B. Digest protein 1. Add 150 µL of Enzyme Reconstitution Solution to 1 vial of Trypsin/Lys-C Protease Mix to prepare 0.67 µg/µL enzyme mix. 2. Add 3.75 µL, 7.5 µL, or 15 µL of the reconstituted enzyme solution for input proteins of 15-30 µg, 30-60 µg, or 60-150 µg, respectively to the reduced and alkylated protein sample solution. 3. Incubate with shaking at 37°C for 1-2 hours to digest the protein sample. Note: Optional labeling with TMT reagents can be performed at this point before peptide clean-up. C. Clean-up peptides 1. Transfer 20 µL of Magnetic Bead slurry to a microcentrifuge tube or a 96-well plate and remove the storage buffer using a magnetic stand. 2. Wash the resin by adding 200 µL of Bead Wash/Binding Solution, then vortex briefly and remove the buffer using a magnetic stand. 3. Add Bead Wash/Binding Solution to the resin and transfer the bead slurry to the protein digest sample. The total volume of the diluted sample for input proteins of 15- 30 µg, 30-60 µg, or 60-150 µg should be 250 µL, 500 µL, or 1000 µL, respectively. 4. Incubate the sample at room temperature for 30-60 minutes with mixing. 5. Collect the bead using a magnetic stand and remove the unbound peptide solution. 6. Wash the resin by adding 500 µL of Wash Solution, then vortex briefly and remove the buffer using a magnetic stand. First Named Inventor: RAJA, Erum TP385332WO1 7. Add 30-100 µL of Elution Solution, then vortex briefly and incubate for 3 minutes with mixing at room temperature. 8. Transfer the eluted peptide solution using a magnetic stand to a new container for LC-MS analysis. Example 6 Example Procedure (PAC or Mutli-analyte Aggregation Capture, with SP3 for protein) Note: Any amount of protein can be used as long as bead: protein ratios are scaled accordingly. Rinse cultured cells or tissues 2-3 times with 1 × PBS to remove cell culture media or excess blood, respectively. Resuspend proteins, cells, or tissues in a lysis solution, such as the Lysis Solution described herein. For protein extraction, many buffer components are allowable, including various detergents, salts, and chaotropic agents. For DNA extraction, chaotropic agents should be avoided. For RNA extraction, guanidine hydrochloride should be added to lysis buffer. Lysis and Reduction/Alkylation – General Notes and Getting Started: 1. For cultured cells, add lysis buffer to a minimum of 1 × 10
6 cells. Use enough lysis buffer for an anticipated concentration between 2-12 mg/mL protein. Let sit on ice for 15 minutes. Note: for 1 x 10
6 cells, assume 450 μg protein, 5 μg DNA, and 10 μg RNA and use 15 mg of the disclosed magnetic beads per each sample type. Note: For tissue samples, add lysis buffer to tissue and disrupt with tissue homogenizer until sample is homogenized. Note: For purposes of protein extraction only, add 1% (by volume) Universal nuclease to lysis buffer. Do not add nuclease or sonicate sample if the intention is to perform multi-omic sequential extractions of any combination of RNA, DNA, or protein. For this purpose, immediately move to “Bind DNA to Beads” section described in Example 5. 2. Centrifuge lysates at 16,000 × g for 15 minutes at 4 °C. First Named Inventor: RAJA, Erum TP385332WO1 3. Determine the protein concentration of the supernatant using established methods such as the Pierce™ BCA Protein Assay Kit (Product No.23227) or Pierce™ Rapid Gold BCA Protein Assay Kit (Product No. A53226). 4. Proceed with reduction and alkylation, if preferred, to a final concentration of 10 mM TCEP and 20 mM 2-chloroacetamide (or equivalent protocol using dithiothreitol and iodoacetamide). TCEP and 2-chloroacetamide can be used together, but DTT and iodoacetamide must be used sequentially with DTT quench after incubation periods are complete. Note: Reduction and alkylation can be done before or after protein binding to beads. If reduction and alkylation are done after protein binding to beads, heat at 50 °C for 45 minutes. If reduction and alkylation are done prior to protein binding to beads, heat at 95 °C for 10 minutes. After incubation, allow the sample to cool to room temperature before adding beads or enzymes. Bind Protein to Beads (SP3 for protein ONLY) Note: This protocol is written with Reduction and Alkylation occurring prior to protein binding on bead at 95 °C for 10 minutes, with 5-minute cooling period prior to adding beads. Note: for protein only applications, 2-propanol and acetonitrile and inter- changeable. Note: Upstream of PAC workflow, lysis, reduction and alkylation chemistry is compatible. 1. Vortex the disclosed magnetic beads to ensure total suspension. 2. Remove supernatant from beads and wash in 500uL of 80% 2-propanol, 0.5% TFA. Remove wash. 3. Recon beads in 80% 2-propanol 0.5% TFA for [50 μg/μL]. 4. Add 99.5% 2-propanol 0.5% TFA to protein solution for a final organic concentration of 80% 2-propanol, 0.5% TFA. 5. Beads (10:1 bead: protein w/w) are added to protein in buffer for 80% organic concentration and 1-2mg/mL protein concentration. First Named Inventor: RAJA, Erum TP385332WO1 6. Incubate 45 minutes room temp with sufficient agitation to keep beads in suspension, then remove the flow through. 7. Add 4x bead volume of 100% MeCN and vortex thoroughly. Remove wash solution. 8. Add 4x bead volume of 70% EtOH and vortex thoroughly. Remove wash solution. 9. The tubes are removed from the magnetic rack and 10-25uL digestion mixture is added to beads, comprising 50mM TEAB, 10mM CaCl2, and 1:10- 1:25 (enzyme: protein (w/w)) Trypsin and LysC. 10. Vortex briefly and incubate 3 hours in a Thermomixer at 37 °C, 1200 rpm. 11. After incubation, centrifuge briefly to collect any evaporated liquid. 12. Add 100% acetonitrile to sample and beads eluate for final organic concentration at least 92.5%. Note: from this point, SP2 is also compatible. 13. Sample vortexed 30 seconds. Total peptide concentration is at least 0.2 mg/mL. 14. Sample was incubated 10 minutes at room temperature. 15. Beads are collected and the supernatant is aspirated to waste. 16. Sample is washed twice with 4x bead volume of 85% acetonitrile, 10% ethanol, 5% water, 0.5% formic acid. 17. Beads are collected and the supernatant was aspirated to waste. 18. Sample is eluted with 2x bead volume of 2-4% MeCN, 0.2%FA in water for 2 minutes with vortexing (per 15 mg beads). 19. The tube is placed on the magnetic rack for 2 minutes, and the supernatant is transferred to a clean tube. Samples are centrifuged at 10,000xg for 30 seconds, and then the sample is moved to a new tube without disturbing pellet. 20. Quantitate with fluorometric or colorimetric peptide assay, or UV-Vis and/or move to downstream application. Example 7 Multi-OMIC Sequential Aggregation Capture First Named Inventor: RAJA, Erum TP385332WO1 Bind DNA to Beads Note: this protocol is for sequential elution of DNA-Protein-RNA. Other sequences are possible with these beads. For extracting Protein-DNA only, start with 2% SDS, 100 mM TEAB, 1 mM EDTA, then reduce and alkylate with TCEP (10 mM) and 2-chloroacetamide (20mM), and bind protein at 80% 2-propanol, 0.5% TFA, take flow through and dilute with water to 30-40% 2-propanol for DNA binding. Presence of CH6ClN3 (guanidine hydrochloride) substantially alters the binding properties of each analyte on this magnetic bead and therefore organic concentrations are variable depending on when CH
6ClN
3 is introduced and the concentration thereof. 1. Dilute sample 1:1 with 0.2% SDS, 100 mM TEAB. Let sit on ice 15 minutes. 2. Wash 15 mg of the disclosed magnetic beads with 100% 2-propanol (per each 1 x 10
6 cells of lysate). 3. Add the required amount of beads to the lysates. 4. Add enough 2-propanol to the sample to achieve a total concentration of 30% organic. 5. Bind for 45 minutes at room temperature with sufficient agitation to keep beads in suspension. 6. Place on a magnetic rack and remove the lysate, which now contains RNA and protein, to “Bind Protein” section of this protocol. Move forward with DNA processing steps below, using only beads. 7. For each 15 mg of beads, add about 250 μL of 30% 2-propanol, 4.2 M CH6ClN3, 150 mM Tris HCl, 20 mM EDTA, remove from magnet and incubate 10 minutes at room temperature with sufficient agitation to keep beads in suspension. 8. Place on magnetic rack and discard supernatant to waste. 9. Add 100 μL of the following formulation per each 15 mg beads: 0.5x PBS, 28% CH
6ClN
3, 10% Tween, 0.5% Tris HCl, 0.5% EDTA, 0.5% Triton X-100. Invert several times to mix with beads and put on magnetic rack. 10. Remove supernatant to a new tube. Add proteinase K and RNase if desired, invert several times to mix, and incubate for 10 minutes at 55 °C, 400 rpm. Note: add water to beads during incubation so they do not dry out. First Named Inventor: RAJA, Erum TP385332WO1 11. After incubation, remove water from beads and add sample back to beads. 12. Add a 1x volume of 60% 2-propanol, 2.4M CH
6ClN
3, 150 mM Tris HCl, 20 mM EDTA to bead/sample mixture and bind for 45 minutes at room temperature with sufficient agitation to keep beads in suspension. 13. Wash DNA-bound beads 2x with 200 μL 70% EtOH, 1 mM EDTA, and 25 mM Tris HCl (per 15 mg beads). 14. Elute in 50 μL 25 mM Tris-HCl, 0.1 mM EDTA (per 15 mg beads). 15. Place on magnetic rack, and transfer supernatant to new 1.5 mL tubes 16. Centrifuge 12,000 xg at 4 °C for 2 minutes. 17. Move to new tubes without beads. 18. Add EDTA to 1 mM and store at 4 °C until downstream application. Bind Protein to Beads (Sequential Method ONLY) 1. Retrieve flowthrough from “Bind DNA to Beads” or from “Lysis and Reduction/Alkylation”. 2. Wash a new set of 30 μL (15 mg) of the disclosed magnetic beads with 200uL 99.5% 2-propanol, 0.5% trifluoroacetic acid, then remove wash solution from beads. 3. Transfer samples to 15mg of the disclosed magnetic beads (per 1 x 10
6 cells). 4. Add enough 99% 2-Propanol, 1% TFA to a total concentration of 45% organic. 5. Bind for 45 minutes at room temperature with sufficient agitation to keep beads in suspension. 6. Place on magnetic rack and transfer RNA-containing supernatant to new 1.5 mL tubes. a. Add 0.1mM EDTA to RNA-containing supernatant and store at 4 °C for short term storage. 7. Wash with 200 μL 70% EtOH, vortex briefly (per 15 mg beads). 8. Wash with 200 μL 100% MeCN, vortex briefly (per 15 mg beads). 9. Resuspend bead-bound proteins in 40 μL 100mM TEAB, 10 mM CaCl
2. First Named Inventor: RAJA, Erum TP385332WO1 a. Reduce and alkylate with 10 μL total of TCEP and 2-chloroacetamide, if not done prior to bead binding. Final concentrations are 20 mM 2-chloroacetamide, 10 mM TCEP. Vortex sample and incubate at 50 °C for 45 minutes. b. If the samples have been previously reduced and alkylated, add 50uL 100 mM TEAB, 10mM CaCl
2 to beads instead of 40 μL. 10. Add 0.6 μL universal nuclease. Pipette up and down repeatedly. 11. Take 1 vial of Trypsin/LysC from -20 °C and let thaw 5 minutes. a. Reconstitute vial in 50 μL 0.1% CH3COOH for 2.0 mg/mL. Mix gently and let sit 5 minutes. b. Add 1:10-1:25 of enzyme:protein (w/w). c. Incubate 2 hours in a Thermomixer at 37 °C, 1200rpm. Continue with the steps below for complete SP3 workflow of protein preparation. Alternatively, SP2 preparation is also compatible and can be used in place of this protocol, and still be SP3 (scaled for 15 mg beads) 12. Begin peptide cleanup by adding enough 2-propanol for >92.75% organic concentration. a. Sample is incubated 10 minutes at room temperature 1200 rpm. b. Beads are collected and the supernatant was aspirated to waste. c. Sample is washed twice with 200 μL of 100% HPLC-grade MeCN (or 85% MeCN, 10% EtOH, 5.0% water, 0.5% formic acid for improved salt removal, such as in TMT- or TMTpro-labeled samples). d. Beads are collected and the supernatant is aspirated to waste. e. Sample is eluted with 50 μL of 2-4% MeCN, 0.2%FA in water for 2 minutes with occasional vortex. f. Place on the magnetic rack for 2 minutes, and transfer supernatant to a clean tube. g. Samples are centrifuged at 10,000xg for 30 seconds. h. Sample is moved to a new tube without disturbing pellet. First Named Inventor: RAJA, Erum TP385332WO1 13. Quantitate with fluorometric or colorimetric peptide assay, or UV-Vis and/or move to downstream application. Bind RNA to Beads 1. Retrieve RNA flowthru from 4 °C storage (45% 2-propanol with RNA). 2. Add 50 mg dry CH
6ClN
3 to 200 μL sample to reach 2.5M CH
6ClN
3 and incubate 10 minutes, 37 °C, 1200 rpm. 3. Wash 30 μL fresh magnetic beads disclosed herein with 200 μL 2-propanol. 4. Transfer 200 μL sample to 15 mg dry magnetic beads. 5. Add about 1200 μL 2-propanol such that sample is 90% final organic concentration. 6. Bind for 45 minutes at room temperature with sufficient agitation to keep beads in suspension. 7. Place on magnetic rack and discard supernatant. 8. Add 100 μL of the following formulation per each 15 mg beads: 0.5x PBS, 28% CH
6ClN
3, 10% Tween, 0.5% Tris HCl, 0.5% EDTA, 0.5% Triton X-100. Invert several times to mix with beads and put on magnetic rack. 9. Remove RNA-containing supernatant from beads and transfer to new 1.5 mL tube. a. Resuspend beads in 200 μL water. 10. Remove RNA-containing supernatant to a new tube. Add proteinase K and RNase if desired, invert several times to mix, and incubate for 10 minutes at 55 °C, 400 rpm. Note: add water to beads during incubation so they do not dry out. 11. After incubation, remove water from beads and add sample back to beads. 12. Add 2-propanol to bead/sample mixture such that sample is 90% final organic concentration and bind for 45 minutes at room temperature with sufficient agitation to keep beads in suspension. 13. Wash RNA-bound beads 2x with 400 μL 70% EtOH, 1 mM EDTA, and 25 mM Tris HCl (per 15mg bead, don’t vortex!). First Named Inventor: RAJA, Erum TP385332WO1 14. Elute in 50 μL water, 0.1 mM EDTA (per 15mg bead). 15. Extract purified RNA from beads and place in new 1.5 mL tube and move to downstream application. Example 8 Comparison Experiment To compare the disclosed magnetic beads with other, commercially available beads, a comparative workflow was developed using a KingFisher
TM Sample Purification System from Thermo Fisher Scientific
TM. Table 2 provides the beads that were used for the comparison experiment. Table 2. Beads used in the comparison experiment Bead Bead Description Diameter Mass Used T
M TM A 25 μg HeLa lysate sample was used for each bead type. The comparative workflow was as follows: A. Protein Digestion (Off KingFisher) • Reduction/alkylation: 95 °C for 10 minutes, cooled to room temperature, and centrifuged for seconds. • Digestion: 37 °C for 1 hour with shaking at 1,000 rpm. • Trypsin/Lys-C: 0.67 μg/μL (100 μg of Trypsin/Lys-C reconstituted in 150 μL of 0.1% acetic acid/10 mM CaCl2 for use, sample to trypsin ratio: 10:1) First Named Inventor: RAJA, Erum TP385332WO1 Table 3. Sample/reagent volume for protein digestion Input Sample Sample/reagent vol. (Sample + Digest volume reduction + alkylation + enzyme) B.

• Bead conditioning: Load 20 μL of beads (5.0 mg) with 80 μL of Bead Wash/Binding Solution. Wash the beads with 200 μL of Bead Wash/Binding Solution. • Dilution: Transfer beads into the diluted sample. The total volume is 500 μL (>9% organic phase after dilution). • Binding: Mix the sample/beads at room temperature for 45 minutes at a medium rate. • Wash: Transfer the sample/beads into 500 μL of Wash Solution for peptide wash. Mix the plate at room temperature for 2 minutes at a medium rate. • Elution: Transfer the sample/beads into 100 μL of Elution Solution for peptide elution. Mix the plate at room temperature for 5 minutes at a medium rate. Collect the output peptide samples for LC-MS analysis. Table 4. Peptide cleanup volumes Input sample Dilution Peptide Wash volume Elution volume A)
Results from the experiment are provided in FIGS.8A, 8B, and 9-11. FIGS.8A and 8B demonstrate that the disclosed magnetic beads exhibit less surface absorption loss than the Sera-Mag
TM carboxylate modified particles, MyOne
TM, or silica beads in the SP2 workflow. The SeraMag
TM, MyOne
TM, and silica beads all demonstrated significant bead loss on the surfaces of the comb tip plate and sample plate (bead First Named Inventor: RAJA, Erum TP385332WO1 transfer/mix and binding steps). In contrast, the disclosed magnetic beads did not exhibit significant bead loss. The data provided in FIGS.8A and 8B with respect to the disclosed magnetic beads refers to hydrolyzed beads, that is beads where the azlactone moieties on the surface of the beads have been hydrolyzed. However, similar results were achieved using disclosed magnetic beads where the azlactone moieties were treated with aminobenzoic acid or aminoethyl-trimethylammonium chloride (AETMA) (data not shown). FIG.9 provides data demonstrating that the peptide yield from the KingFisher SP2 workflow for the disclosed magnetic beads was substantially higher than the peptide yield for the Sera-Mag
TM, MyOne
TM Silane or silica beads, using a quantitative colorimetric peptide assay. No peptides were recovered using silica beads. FIG.10 demonstrates that using the disclosed magnetic beads, substantially more unique peptides were identified by nano-LCMS analysis, than were identified using the other bead types. And FIG.11 demonstrates that substantially more protein groups were identified using the disclosed magnetic beads. Example 9 An experiment was designed to demonstrate the superior binding of the disclosed magnetic beads coupled to alkali stable Protein A (asPA), compared to a commercial magnetic bead that comprises asPA conjugates. Conjugate asPA to the disclosed magnetic beads Alkali stable Protein A was conjugated to the disclosed azlactone beads according to the following procedure: 1. Weigh out 77mg of the disclosed azlactone beads. 2. Dilute asPA (20mg/mL in 250mM Sodium Acetate, pH 5.5, 0.5M NaCl, 0.05% Tween-20) into Coupling Buffer (0.1M Sodium Carbonate, 1.2M Sodium Citrate, pH 9) 1:3 (v/v); final concentration of components is 5mg/mL asPA, 0.075M Sodium Carbonate, 0.9M Sodium Carbonate, pH 9 3. Add 3.6mL of asPA in Coupling Buffer to azlactone beads. First Named Inventor: RAJA, Erum TP385332WO1 4. Couple asPA to beads overnight at room temperature. 5. Wash asPA-beads with 2-3 mL water. 6. Block asPA-beads with 2-3 mL of 3M ethanolamine for 2 hours. 7. Wash asPA-beads with 2-3 mL water. 8. Wash asPA-beads twice with 2-3 mL 0.1M Glycine, pH 2 (Pierce IgG Elution Buffer, #21028) 9. Wash asPA-beads three times with 2-3 mL water. 10. Slurry asPA-beads to 25% using 20% ethanol. Measure the binding capacity of asPA-azlactone beads compared to asPA- magnetic agarose The procedure for measuring the binding capacity of asPA-azlactone beads compared to asPA-magnetic agarose (Pierce High Capacity Protein A MagBeads, alkali stable, #A53036, 25% slurry) was as follows: 1. Prepare 3mg/mL Rabbit IgG (RIgG) in PBS. 2. Add 2 x 50uL of 25% magnetic bead slurry (asPA-azlactone beads and asPA- magnetic agarose) to 1.5mL microcentrifuge tubes. 3. Wash beads twice with 500uL of PBS. 4. Add 500uL of 3mg/mL (RIgG) to beads and incubate for 1hr at room temperature with shaking/mixing. 5. Collect the flowthrough and save for analysis. 6. Create a RIgG standard curve from 0 to 3mg/mL by measuring absorbance at 280nm. 7. Measure absorbance of starting solution of RIgG and Flowthrough from each bead sample. 8. Calculate concentrations of start and flowthrough using standard curve. 9. Calculate RIgG bound to the magnetic beads by subtracting the RIgG in each Flowthrough from the starting RIgG. Extrapolate to amount RIgG bound per mL of settled beads. First Named Inventor: RAJA, Erum TP385332WO1 Results The results clearly demonstrated that the asPA conjugated disclosed magnetic beads had about a 15% higher RIgG binding capacity that asPA-magentic agarose beads (FIG.12). Without being bound to a particular theory, this may be due, at least in part, to the disclosed azlactone magnetic beads being able to conjugate a larger amount of asPA than an amount asPA that the same amount of magnetic agarose beads can conjugate. And in some aspects, the amount of asPA bound to the disclosed azlactone magnetic beads is about 2-fold higher than the amount of asPA bound to the same amount of magnetic agarose beads. 2 a 50

ml conical tube containing a citrate/carbonate buffer (pH = 8-12, 30 mL) and rotated overnight at room temperature. The following day, beads were washed three times with water using an appropriate magnetic stand. Once washed, the beads were adjusted to a 25% slurry in water. Other types of anion exchange beads can be prepared using different types of diamines. Cation exchange beads can be prepared similarly using a different ligand such as sulfonic acid. Example 11 Preparation of Magnetic Beads for Phosphopeptide Enrichment First Named Inventor: RAJA, Erum TP385332WO1 HO O

Magnetic-NTA beads were prepared according to the following procedure. Magnetic beads (0.50g) and amino-linker-NTA or amino-linker-IDA (1.0g) such as 6- amino-carboxylhexyl iminodiacetic acid were added to a 50 ml conical tube containing an appropriate buffer (pH 8-12) and rotated overnight at room temperature. The following day, beads were washed three times with water using an appropriate magnetic stand. Magnetic-NTA beads can then be loaded with an appropriate metal such as Fe, Ti, Zr, or Ga for enrichment of phosphorylated peptides from simple or complex mixtures via immobilized metal affinity chromatography (IMAC) or Metal Oxide Affinity Chromatography (MOAC). Magnetic-NTA beads were loaded with Fe using FeCl2 and/or FeCl3 to yield magnetic Fe-NTA beads. Magnetic Fe-NTA beads can be bound to a phosphorylated peptides in a protein digest (such as from cells, tissues, cerebrospinal fluid, peptide mixtures or other sources) using a binding buffer, and phosphopeptides can be eluted using an elution buffer using buffers known to those skilled in the art. Samples then can be analyzed with a mass spectrometer and phospho-modifications searched using Proteome Discover software to measure phosphopeptides and phosphospecificity in a given sample. Magnetic-NTA beads can also be immobilized with an appropriate metal such as nickel (Ni) or cobalt (Co) for purifying His-tagged proteins and analyzed using the procedure described above. Example 12 Extracellular Vesicle Capture First Named Inventor: RAJA, Erum TP385332WO1 Isolation of extracellular vesicles (EV) from cell culture supernatant 1. Add 2mL Equilibration Buffer (EquilB, 50mM HEPES, 100mM NaCl, pH 8) to a 5mL tube. 2. Add 100-200uL of 25% anion exchange magnetic beads to the EquilB. Vortex for 5 seconds, collect beads on a magnetic stand, and remove EquilB. 3. Add 2mL EquilB to beads and incubate for 2 min at room temperature. Collect beads and remove EquilB. 4. Add 3mL clarified cell culture supernatant (Expi293F) to the beads, and incubate for 1 hr while mixing end-over-end on a tube rotator at 25 rpm. 5. Collect beads and remove flowthrough. 6. Wash beads once with 3mL Wash Buffer (WB, 50mM HEPES, 250mM NaCl, pH 7. Gently mix end-over-end 3-5x until beads are fully suspended. 8. Collect beads and remove WB. Transfer beads to a 1.5mL microcentrifuge tube using 2 x 0.3mL of WB. 9. Collect beads and remove WB. 10. Add 0.3mL Elution Buffer to beads (EB, 50mM HEPES, 1M NaCl, pH 8). Mix at 1200rpm in a Thermomixer for 10min at room temperature. 11. Collect the beads and save the eluate for analysis. Identify EV marker (CD81) and contaminant (Calnexin) in isolated EV by Western Blotting 1. Prepare gel sample by mixing eluate with 5X SDS sample loading buffer. 2. Resolve eluates, normalized by volume, on a 4-20% SDS-PAGE gel. 3. Transfer the gel to a nitrocellulose membrane using the Invitrogen Power Blotter for 10min at constant 5A and 25V. 4. Cut blot in half, one for each target. 5. Block the blots with 10% BSA Blocker mixed 1:1 with TBS containing 0.05% Tween-20 (TBST) for 1hr at room temperature. 6. Incubate blots with primary antibody (anti-calnexin or CD81) overnight at 4C. 7. Wash blots 5 x 5min with TBST First Named Inventor: RAJA, Erum TP385332WO1 8. Dilute Goat anti-rabbit or Goat anti-mouse (H+L) secondary antibody conjugated to horseradish peroxidase (HRP) (Thermo Fisher Scientific, #32460 and #32430, respectively) 1:50,000 into Block:TBST (1:4). Incubate blots in secondary antibody for 30 min at room temperature. 9. Wash blots 5 x 5min with TBST 10. Incubate blots with SuperSignal West Dura Extended Duration Substrate (Thermo Fisher Scientific, #34076) for 5min at room temperature 11. Scan blots on the Invitrogen iBright FL1500 Imaging System (Thermo Fisher Scientific, #A44115) with chemiluminescent detection. Identify total protein in isolated EV by gel analysis 1. Prepare gel sample by mixing eluate with 5X sample loading buffer. 2. Resolve eluate, normalized by volume, on a 4-20% SDS-PAGE gel. 3. Stain gel for 1 hr with GelCode Blue Safe Protein Stain (Thermo Fisher Scientific, #24594) 4. Destain gel in water and scan. Results The above protocol was used to compare magnetic beads that were conjugated to three types of ligands (1,5-pentanediamine (PDA), PEI-800 and PEI-25K ) at two different pH levels (pH 7 and pH 9) as described in Example 10. The results demonstrate that a variety of ligands, including but not limited to 1,5-pentanediamine (PDA), PEI-800 and PEI-25K, can be used successfully for EV isolation. As seen in the gel image (FIG.13), anion exchange magnetic beads with the three different ligand chemistries successfully isolated EV from Expi293 cell culture supernatant. It was found that the buffer type and pH that was used to prepare the bead-ligand conjugates affected the yield of isolated EV. Higher yield of the EV marker, CD81, was obtained when sodium citrate/sodium carbonate, pH 9 buffer was used for conjugation of PDA to the magnetic beads in comparison to PBS, pH 7. In contrast, higher yield of CD81 was obtained when PBS at neutral pH (pH ~7) was used for conjugation of PEI ligands to the magnetic beads. Without wishing to be bound by theory, the higher yield of EV capture by the bead can be attributed to having a higher ligand density on the bead. First Named Inventor: RAJA, Erum TP385332WO1 Although the ligands tested in this experiment were effective for EV isolation, the smaller organic ligand (PDA) was found to have certain advantages over the polymeric ligands, PEI-800 or PEI-25K. Overall, the PDA magnetic beads resulted in the highest yield of EV with the lowest background. Calnexin, a contaminant, was found to be present with the PEI ligands, but negligible with the PDA ligand. In addition, many protein bands were seen in the PEI samples in a Coomassie-stained gel, whereas only a negligible number of bands were seen with the PDA sample. In view of the many possible embodiments to which the principles of the disclosure may be applied, it should be recognized that the illustrated embodiments are only preferred examples of the disclosure and should not be taken as limiting the scope of the disclosure. Rather, the scope of the disclosure is defined by the following claims. We therefore claim as our disclosure all that comes within the scope and spirit of these claims.
 N OH
N OH . In certain aspects, the functional moieties.
. In certain aspects, the functional moieties. First Named Inventor: RAJA, Erum TP385332WO1 In any aspects, the magnetic material in the magnetic beads may be in any suitable form, such as, but not limited to, a particle, powder, flake, cluster, bead, or combination thereof. In some embodiments, the magnetic material is or comprises magnetic particles. In any aspects, the magnetic material in the magnetic beads may comprise an iron source. The iron source may be any suitable iron source, such as an iron source that is added during the polymer bead formation reaction. The iron source may be in the form of iron-containing particles and/or may be an iron oxide, such as Fe3O4, Fe2O3, or a combination thereof. Additionally, or alternatively, the beads may include magnetic particles as the iron source, for example DynabeadsTM (available from Thermo Fisher Scientific) and/or Sera-MagTM Speedbeads (available from Cytiva Life Sciences) and/or other magnetic beads or particles. In certain aspects, the magnetic material is, or comprise, Fe3O4 particles. In certain aspects, the magnetic material is an activated iron oxide source, such as Fe3O4-NHS, to provide an addition source of reactivity during particle synthesis. In some aspects, the magnetic material is an activated magnetic material and may contain one or more functional groups, such as, for example, a carboxy group or ester group. In certain aspects, the magnetic material is an activated iron source that comprises additional functional groups, such as, but not limited to, carboxy or ester groups. In some aspects, the magnetic material is contained within an agarose bead, and the agarose bead is contained within the crosslinked polymer to form the magnetic bead. The magnetic particles may have a size suitable for use in the disclosed beads, such as from 5 nm to 1,000 nm or more, such as from 5 nm to 200 nm or from 100 nm to 800 nm. In certain aspects, the magnetic particles have a size of from 15 nm to 100 nm, such as from 15 nm to 50 nm, or from 50 nm to 100 nm. In certain other aspects, the magnetic particles have a size of 100 nm to 800 nm, for example, iron clusters that may have an average particle size of from 100 nm to 800 nm, such as from 100 nm to 600 nm, or about 100 nm, 200 nm, 300 nm, 400 nm, 500 nm or 600 nm. First Named Inventor: RAJA, Erum TP385332WO1 The average particle size of the magnetic particles may be determined using a particle size analyzer. In some aspects, the magnetic particle is substantially spherical, but in other aspects, the particle is not substantially spherical. In certain aspects, the magnetic particle is a substantially spherical iron cluster. In certain other aspects, the magnetic particle is not an iron cluster and is not substantially spherical. However, such magnetic particles may be coated with another material that provides a substantially spherical shape. The magnetic beads have an average size suitable for use in separation technology. The average particle size is determined by a particle analyzer (e.g. Beckman CoulterTM LS13320XR) or by scanning electron microscopy (SEM). In some aspects the magnetic beads are substantially spherical and the average particle size is the average diameter of the beads. In some aspects, the magnetic beads have an average size of from 20 μm to 100 μm or more, such as from 20 μm to 80 μm, from 30 μm to 60 μm, from 30 μm to 50 μm, or from 40 μm to 80 μm. The magnetic beads may be porous or non-porous. In some aspects, the magnetic beads are porous and may comprise pores having a size of from 100 Angstroms to 200 Angstroms. And in some aspects, the pores extend from the surface of the particle into the interior, but do not pass completely though the particle. In some aspects, at least a portion, and may be substantially all, of the magnetic material is located in the pores. The magnetic beads may have a hydrated state and a non-hydrated state. Typically, the disclosed bead size is the bead size in the hydrated state, and the bead size of the magnetic beads in the hydrated state (the hydrated bead size) is larger than the non-hydrated bead. These magnetic beads have the tendency to swell, from 3 times larger to 10 times larger or more, from 5 times larger to 8 times larger, or from 5 times to 6 times larger. By way of comparison, nonmagnetic beads typically swell to about 8x larger when hydrated, compared to their non-hydrated state. And to the inventors’ knowledge, other magnetic beads do not swell to a measurable extent. The swelling of the disclosed magnetic beads may allow the functional moieties on the bead surface, such as azlactone moieties, to be more accessible, and/or allow large biomolecules to First Named Inventor: RAJA, Erum TP385332WO1 bind to the functional moieties more easily because steric problems due to their size are reduced due to the swelling increasing the space between the functional moieties. In some aspects, the disclosed magnetic beads demonstrate a reduced bead loss during use in an automated process, for example, a Kingfisher apparatus, when compared with other magnetic beads. The bead loss may be due, at least in part, to surface adsorption onto the apparatus, for example, onto the tip plate and sample plate. In some aspects, a population of the disclosed magnetic beads exhibits a bead loss of less than about 5% by weight when used in an automated process. In a comparative experiment, the disclosed magnetic beads demonstrated a bead loss of 4.7% by weight, where as Cytiva Sera-MagTM beads had a loss of 6.0% by weight, InvitrogenTM DynaGreenTM silica beads lost 11.5% by weight and DynabeadsTM MyOneTM Silane beads lost 6.8% by weight. III. Synthesis of the Magnetic Beads
First Named Inventor: RAJA, Erum TP385332WO1 In any aspects, the magnetic material in the magnetic beads may be in any suitable form, such as, but not limited to, a particle, powder, flake, cluster, bead, or combination thereof. In some embodiments, the magnetic material is or comprises magnetic particles. In any aspects, the magnetic material in the magnetic beads may comprise an iron source. The iron source may be any suitable iron source, such as an iron source that is added during the polymer bead formation reaction. The iron source may be in the form of iron-containing particles and/or may be an iron oxide, such as Fe3O4, Fe2O3, or a combination thereof. Additionally, or alternatively, the beads may include magnetic particles as the iron source, for example DynabeadsTM (available from Thermo Fisher Scientific) and/or Sera-MagTM Speedbeads (available from Cytiva Life Sciences) and/or other magnetic beads or particles. In certain aspects, the magnetic material is, or comprise, Fe3O4 particles. In certain aspects, the magnetic material is an activated iron oxide source, such as Fe3O4-NHS, to provide an addition source of reactivity during particle synthesis. In some aspects, the magnetic material is an activated magnetic material and may contain one or more functional groups, such as, for example, a carboxy group or ester group. In certain aspects, the magnetic material is an activated iron source that comprises additional functional groups, such as, but not limited to, carboxy or ester groups. In some aspects, the magnetic material is contained within an agarose bead, and the agarose bead is contained within the crosslinked polymer to form the magnetic bead. The magnetic particles may have a size suitable for use in the disclosed beads, such as from 5 nm to 1,000 nm or more, such as from 5 nm to 200 nm or from 100 nm to 800 nm. In certain aspects, the magnetic particles have a size of from 15 nm to 100 nm, such as from 15 nm to 50 nm, or from 50 nm to 100 nm. In certain other aspects, the magnetic particles have a size of 100 nm to 800 nm, for example, iron clusters that may have an average particle size of from 100 nm to 800 nm, such as from 100 nm to 600 nm, or about 100 nm, 200 nm, 300 nm, 400 nm, 500 nm or 600 nm. First Named Inventor: RAJA, Erum TP385332WO1 The average particle size of the magnetic particles may be determined using a particle size analyzer. In some aspects, the magnetic particle is substantially spherical, but in other aspects, the particle is not substantially spherical. In certain aspects, the magnetic particle is a substantially spherical iron cluster. In certain other aspects, the magnetic particle is not an iron cluster and is not substantially spherical. However, such magnetic particles may be coated with another material that provides a substantially spherical shape. The magnetic beads have an average size suitable for use in separation technology. The average particle size is determined by a particle analyzer (e.g. Beckman CoulterTM LS13320XR) or by scanning electron microscopy (SEM). In some aspects the magnetic beads are substantially spherical and the average particle size is the average diameter of the beads. In some aspects, the magnetic beads have an average size of from 20 μm to 100 μm or more, such as from 20 μm to 80 μm, from 30 μm to 60 μm, from 30 μm to 50 μm, or from 40 μm to 80 μm. The magnetic beads may be porous or non-porous. In some aspects, the magnetic beads are porous and may comprise pores having a size of from 100 Angstroms to 200 Angstroms. And in some aspects, the pores extend from the surface of the particle into the interior, but do not pass completely though the particle. In some aspects, at least a portion, and may be substantially all, of the magnetic material is located in the pores. The magnetic beads may have a hydrated state and a non-hydrated state. Typically, the disclosed bead size is the bead size in the hydrated state, and the bead size of the magnetic beads in the hydrated state (the hydrated bead size) is larger than the non-hydrated bead. These magnetic beads have the tendency to swell, from 3 times larger to 10 times larger or more, from 5 times larger to 8 times larger, or from 5 times to 6 times larger. By way of comparison, nonmagnetic beads typically swell to about 8x larger when hydrated, compared to their non-hydrated state. And to the inventors’ knowledge, other magnetic beads do not swell to a measurable extent. The swelling of the disclosed magnetic beads may allow the functional moieties on the bead surface, such as azlactone moieties, to be more accessible, and/or allow large biomolecules to First Named Inventor: RAJA, Erum TP385332WO1 bind to the functional moieties more easily because steric problems due to their size are reduced due to the swelling increasing the space between the functional moieties. In some aspects, the disclosed magnetic beads demonstrate a reduced bead loss during use in an automated process, for example, a Kingfisher apparatus, when compared with other magnetic beads. The bead loss may be due, at least in part, to surface adsorption onto the apparatus, for example, onto the tip plate and sample plate. In some aspects, a population of the disclosed magnetic beads exhibits a bead loss of less than about 5% by weight when used in an automated process. In a comparative experiment, the disclosed magnetic beads demonstrated a bead loss of 4.7% by weight, where as Cytiva Sera-MagTM beads had a loss of 6.0% by weight, InvitrogenTM DynaGreenTM silica beads lost 11.5% by weight and DynabeadsTM MyOneTM Silane beads lost 6.8% by weight. III. Synthesis of the Magnetic Beads R Beesaind NOH O O H O
R Beesaind NOH O O H O Magnetic beads were hydrolyzed according to the following procedure. Magnetic beads (0.50 g) were added to a 15 ml conical tube containing water (10 mL) and rotated overnight at room temperature. The beads were washed the next day with water using an appropriate magnetic stand. The hydrolyzed beads were then adjusted to a 25% slurry in water. Solutions used in Examples 5-8 are provided in Table 1 unless otherwise specified. Table 1. List of solutions
Magnetic beads were hydrolyzed according to the following procedure. Magnetic beads (0.50 g) were added to a 15 ml conical tube containing water (10 mL) and rotated overnight at room temperature. The beads were washed the next day with water using an appropriate magnetic stand. The hydrolyzed beads were then adjusted to a 25% slurry in water. Solutions used in Examples 5-8 are provided in Table 1 unless otherwise specified. Table 1. List of solutions First Named Inventor: RAJA, Erum TP385332WO1 Example 5 Example Procedure (SP2) using the Disclosed Magnetic Beads Use 15-150 µg of protein per sample preparation. Rinse cultured cells or tissues 2-3 times with 1 × PBS to remove cell culture media or excess blood, respectively. Resuspend proteins, cells, or tissues in Lysis Solution without additional buffers. A. Extract protein, reduce, and alkylate 1. For cultured cells, add 50 µL of Lysis Buffer and 1 µL of Universal Nuclease to a minimum of 1 × 106 cells. Pipet up and down (with P200 tip) for 10-15 cycles until sample viscosity is reduced. Note: Lyse the cells in an appropriate volume of lysis buffer to have the final concentration of 2-5 µg/µl with a final volume of 7.5 µL, 15 µL, or 30 µL for input proteins of 15-30 µg, 30-60 µg, or 60-150 µg, respectively. Centrifugation of cultured cell lysates is typically not required after aspiration using a pipet. 2. For tissue samples, add 50 µL of Lysis Solution (containing 1 µL Universal Nuclease) per 5 mg of tissue and disrupt with tissue homogenizer until sample is homogenized. Centrifuge tissue lysates at 16,000 × g for 10 minutes. 3. For purified proteins, depleted and undepleted plasma, and serum samples, dilute samples directly in Lysis Solution to 2-5 µg/µL with a final volume of 7.5 µL, 15 µL, or 30 µL for input proteins of 10-25 µg, 25-50 µg, or 50-100 µg, respectively. Note: The concentration of plasma is around 60-70 µg/µL. For purified proteins and plasma samples, addition of Universal Nuclease is not required. 4. Determine the protein concentration of the supernatant using established methods such as the Pierce™ BCA Protein Assay Kit (Product No.23227) or Pierce™ Rapid Gold BCA Protein Assay Kit (Product No. A53226). Note: Dilution with Lysis Solution may be required for concentrated samples for BCA assay. 5. Transfer 15-150 µg of protein sample into a 1.5-mL low protein binding tube or a 96-well plate and adjust the final volume to 7.5 µL, 15 µL, or 30 µL for input proteins of 15-30 µg, 30-60 µg, or 60-150 µg, respectively with Lysis Solution. First Named Inventor: RAJA, Erum TP385332WO1 6. Add 3.75 µL, 7.5 µL, or 15 µL of Reduction Solution for input proteins of 15-30 µg, 30-60 µg, or 60-150 µg, respectively to the sample and gently mix. 7. Add 3.75 µL, 7.5 µL, or 15 µL of Alkylation Solution for input proteins of 15- 30 µg, 30-60 µg, or 60-150 µg, respectively to the sample and gently mix. 8. Incubate the sample at 95 °C using a heat block for 10 minutes or 50 °C for 20- 30 minutes to reduce and alkylate the protein sample. 9. After incubation, allow the sample to cool to room temperature. 10. Briefly centrifuge the tube or plate before digestion. B. Digest protein 1. Add 150 µL of Enzyme Reconstitution Solution to 1 vial of Trypsin/Lys-C Protease Mix to prepare 0.67 µg/µL enzyme mix. 2. Add 3.75 µL, 7.5 µL, or 15 µL of the reconstituted enzyme solution for input proteins of 15-30 µg, 30-60 µg, or 60-150 µg, respectively to the reduced and alkylated protein sample solution. 3. Incubate with shaking at 37°C for 1-2 hours to digest the protein sample. Note: Optional labeling with TMT reagents can be performed at this point before peptide clean-up. C. Clean-up peptides 1. Transfer 20 µL of Magnetic Bead slurry to a microcentrifuge tube or a 96-well plate and remove the storage buffer using a magnetic stand. 2. Wash the resin by adding 200 µL of Bead Wash/Binding Solution, then vortex briefly and remove the buffer using a magnetic stand. 3. Add Bead Wash/Binding Solution to the resin and transfer the bead slurry to the protein digest sample. The total volume of the diluted sample for input proteins of 15- 30 µg, 30-60 µg, or 60-150 µg should be 250 µL, 500 µL, or 1000 µL, respectively. 4. Incubate the sample at room temperature for 30-60 minutes with mixing. 5. Collect the bead using a magnetic stand and remove the unbound peptide solution. 6. Wash the resin by adding 500 µL of Wash Solution, then vortex briefly and remove the buffer using a magnetic stand. First Named Inventor: RAJA, Erum TP385332WO1 7. Add 30-100 µL of Elution Solution, then vortex briefly and incubate for 3 minutes with mixing at room temperature. 8. Transfer the eluted peptide solution using a magnetic stand to a new container for LC-MS analysis. Example 6 Example Procedure (PAC or Mutli-analyte Aggregation Capture, with SP3 for protein) Note: Any amount of protein can be used as long as bead: protein ratios are scaled accordingly. Rinse cultured cells or tissues 2-3 times with 1 × PBS to remove cell culture media or excess blood, respectively. Resuspend proteins, cells, or tissues in a lysis solution, such as the Lysis Solution described herein. For protein extraction, many buffer components are allowable, including various detergents, salts, and chaotropic agents. For DNA extraction, chaotropic agents should be avoided. For RNA extraction, guanidine hydrochloride should be added to lysis buffer. Lysis and Reduction/Alkylation – General Notes and Getting Started: 1. For cultured cells, add lysis buffer to a minimum of 1 × 106 cells. Use enough lysis buffer for an anticipated concentration between 2-12 mg/mL protein. Let sit on ice for 15 minutes. Note: for 1 x 106 cells, assume 450 μg protein, 5 μg DNA, and 10 μg RNA and use 15 mg of the disclosed magnetic beads per each sample type. Note: For tissue samples, add lysis buffer to tissue and disrupt with tissue homogenizer until sample is homogenized. Note: For purposes of protein extraction only, add 1% (by volume) Universal nuclease to lysis buffer. Do not add nuclease or sonicate sample if the intention is to perform multi-omic sequential extractions of any combination of RNA, DNA, or protein. For this purpose, immediately move to “Bind DNA to Beads” section described in Example 5. 2. Centrifuge lysates at 16,000 × g for 15 minutes at 4 °C. First Named Inventor: RAJA, Erum TP385332WO1 3. Determine the protein concentration of the supernatant using established methods such as the Pierce™ BCA Protein Assay Kit (Product No.23227) or Pierce™ Rapid Gold BCA Protein Assay Kit (Product No. A53226). 4. Proceed with reduction and alkylation, if preferred, to a final concentration of 10 mM TCEP and 20 mM 2-chloroacetamide (or equivalent protocol using dithiothreitol and iodoacetamide). TCEP and 2-chloroacetamide can be used together, but DTT and iodoacetamide must be used sequentially with DTT quench after incubation periods are complete. Note: Reduction and alkylation can be done before or after protein binding to beads. If reduction and alkylation are done after protein binding to beads, heat at 50 °C for 45 minutes. If reduction and alkylation are done prior to protein binding to beads, heat at 95 °C for 10 minutes. After incubation, allow the sample to cool to room temperature before adding beads or enzymes. Bind Protein to Beads (SP3 for protein ONLY) Note: This protocol is written with Reduction and Alkylation occurring prior to protein binding on bead at 95 °C for 10 minutes, with 5-minute cooling period prior to adding beads. Note: for protein only applications, 2-propanol and acetonitrile and inter- changeable. Note: Upstream of PAC workflow, lysis, reduction and alkylation chemistry is compatible. 1. Vortex the disclosed magnetic beads to ensure total suspension. 2. Remove supernatant from beads and wash in 500uL of 80% 2-propanol, 0.5% TFA. Remove wash. 3. Recon beads in 80% 2-propanol 0.5% TFA for [50 μg/μL]. 4. Add 99.5% 2-propanol 0.5% TFA to protein solution for a final organic concentration of 80% 2-propanol, 0.5% TFA. 5. Beads (10:1 bead: protein w/w) are added to protein in buffer for 80% organic concentration and 1-2mg/mL protein concentration. First Named Inventor: RAJA, Erum TP385332WO1 6. Incubate 45 minutes room temp with sufficient agitation to keep beads in suspension, then remove the flow through. 7. Add 4x bead volume of 100% MeCN and vortex thoroughly. Remove wash solution. 8. Add 4x bead volume of 70% EtOH and vortex thoroughly. Remove wash solution. 9. The tubes are removed from the magnetic rack and 10-25uL digestion mixture is added to beads, comprising 50mM TEAB, 10mM CaCl2, and 1:10- 1:25 (enzyme: protein (w/w)) Trypsin and LysC. 10. Vortex briefly and incubate 3 hours in a Thermomixer at 37 °C, 1200 rpm. 11. After incubation, centrifuge briefly to collect any evaporated liquid. 12. Add 100% acetonitrile to sample and beads eluate for final organic concentration at least 92.5%. Note: from this point, SP2 is also compatible. 13. Sample vortexed 30 seconds. Total peptide concentration is at least 0.2 mg/mL. 14. Sample was incubated 10 minutes at room temperature. 15. Beads are collected and the supernatant is aspirated to waste. 16. Sample is washed twice with 4x bead volume of 85% acetonitrile, 10% ethanol, 5% water, 0.5% formic acid. 17. Beads are collected and the supernatant was aspirated to waste. 18. Sample is eluted with 2x bead volume of 2-4% MeCN, 0.2%FA in water for 2 minutes with vortexing (per 15 mg beads). 19. The tube is placed on the magnetic rack for 2 minutes, and the supernatant is transferred to a clean tube. Samples are centrifuged at 10,000xg for 30 seconds, and then the sample is moved to a new tube without disturbing pellet. 20. Quantitate with fluorometric or colorimetric peptide assay, or UV-Vis and/or move to downstream application. Example 7 Multi-OMIC Sequential Aggregation Capture First Named Inventor: RAJA, Erum TP385332WO1 Bind DNA to Beads Note: this protocol is for sequential elution of DNA-Protein-RNA. Other sequences are possible with these beads. For extracting Protein-DNA only, start with 2% SDS, 100 mM TEAB, 1 mM EDTA, then reduce and alkylate with TCEP (10 mM) and 2-chloroacetamide (20mM), and bind protein at 80% 2-propanol, 0.5% TFA, take flow through and dilute with water to 30-40% 2-propanol for DNA binding. Presence of CH6ClN3 (guanidine hydrochloride) substantially alters the binding properties of each analyte on this magnetic bead and therefore organic concentrations are variable depending on when CH6ClN3 is introduced and the concentration thereof. 1. Dilute sample 1:1 with 0.2% SDS, 100 mM TEAB. Let sit on ice 15 minutes. 2. Wash 15 mg of the disclosed magnetic beads with 100% 2-propanol (per each 1 x 106 cells of lysate). 3. Add the required amount of beads to the lysates. 4. Add enough 2-propanol to the sample to achieve a total concentration of 30% organic. 5. Bind for 45 minutes at room temperature with sufficient agitation to keep beads in suspension. 6. Place on a magnetic rack and remove the lysate, which now contains RNA and protein, to “Bind Protein” section of this protocol. Move forward with DNA processing steps below, using only beads. 7. For each 15 mg of beads, add about 250 μL of 30% 2-propanol, 4.2 M CH6ClN3, 150 mM Tris HCl, 20 mM EDTA, remove from magnet and incubate 10 minutes at room temperature with sufficient agitation to keep beads in suspension. 8. Place on magnetic rack and discard supernatant to waste. 9. Add 100 μL of the following formulation per each 15 mg beads: 0.5x PBS, 28% CH6ClN3, 10% Tween, 0.5% Tris HCl, 0.5% EDTA, 0.5% Triton X-100. Invert several times to mix with beads and put on magnetic rack. 10. Remove supernatant to a new tube. Add proteinase K and RNase if desired, invert several times to mix, and incubate for 10 minutes at 55 °C, 400 rpm. Note: add water to beads during incubation so they do not dry out. First Named Inventor: RAJA, Erum TP385332WO1 11. After incubation, remove water from beads and add sample back to beads. 12. Add a 1x volume of 60% 2-propanol, 2.4M CH6ClN3, 150 mM Tris HCl, 20 mM EDTA to bead/sample mixture and bind for 45 minutes at room temperature with sufficient agitation to keep beads in suspension. 13. Wash DNA-bound beads 2x with 200 μL 70% EtOH, 1 mM EDTA, and 25 mM Tris HCl (per 15 mg beads). 14. Elute in 50 μL 25 mM Tris-HCl, 0.1 mM EDTA (per 15 mg beads). 15. Place on magnetic rack, and transfer supernatant to new 1.5 mL tubes 16. Centrifuge 12,000 xg at 4 °C for 2 minutes. 17. Move to new tubes without beads. 18. Add EDTA to 1 mM and store at 4 °C until downstream application. Bind Protein to Beads (Sequential Method ONLY) 1. Retrieve flowthrough from “Bind DNA to Beads” or from “Lysis and Reduction/Alkylation”. 2. Wash a new set of 30 μL (15 mg) of the disclosed magnetic beads with 200uL 99.5% 2-propanol, 0.5% trifluoroacetic acid, then remove wash solution from beads. 3. Transfer samples to 15mg of the disclosed magnetic beads (per 1 x 106 cells). 4. Add enough 99% 2-Propanol, 1% TFA to a total concentration of 45% organic. 5. Bind for 45 minutes at room temperature with sufficient agitation to keep beads in suspension. 6. Place on magnetic rack and transfer RNA-containing supernatant to new 1.5 mL tubes. a. Add 0.1mM EDTA to RNA-containing supernatant and store at 4 °C for short term storage. 7. Wash with 200 μL 70% EtOH, vortex briefly (per 15 mg beads). 8. Wash with 200 μL 100% MeCN, vortex briefly (per 15 mg beads). 9. Resuspend bead-bound proteins in 40 μL 100mM TEAB, 10 mM CaCl2. First Named Inventor: RAJA, Erum TP385332WO1 a. Reduce and alkylate with 10 μL total of TCEP and 2-chloroacetamide, if not done prior to bead binding. Final concentrations are 20 mM 2-chloroacetamide, 10 mM TCEP. Vortex sample and incubate at 50 °C for 45 minutes. b. If the samples have been previously reduced and alkylated, add 50uL 100 mM TEAB, 10mM CaCl2 to beads instead of 40 μL. 10. Add 0.6 μL universal nuclease. Pipette up and down repeatedly. 11. Take 1 vial of Trypsin/LysC from -20 °C and let thaw 5 minutes. a. Reconstitute vial in 50 μL 0.1% CH3COOH for 2.0 mg/mL. Mix gently and let sit 5 minutes. b. Add 1:10-1:25 of enzyme:protein (w/w). c. Incubate 2 hours in a Thermomixer at 37 °C, 1200rpm. Continue with the steps below for complete SP3 workflow of protein preparation. Alternatively, SP2 preparation is also compatible and can be used in place of this protocol, and still be SP3 (scaled for 15 mg beads) 12. Begin peptide cleanup by adding enough 2-propanol for >92.75% organic concentration. a. Sample is incubated 10 minutes at room temperature 1200 rpm. b. Beads are collected and the supernatant was aspirated to waste. c. Sample is washed twice with 200 μL of 100% HPLC-grade MeCN (or 85% MeCN, 10% EtOH, 5.0% water, 0.5% formic acid for improved salt removal, such as in TMT- or TMTpro-labeled samples). d. Beads are collected and the supernatant is aspirated to waste. e. Sample is eluted with 50 μL of 2-4% MeCN, 0.2%FA in water for 2 minutes with occasional vortex. f. Place on the magnetic rack for 2 minutes, and transfer supernatant to a clean tube. g. Samples are centrifuged at 10,000xg for 30 seconds. h. Sample is moved to a new tube without disturbing pellet. First Named Inventor: RAJA, Erum TP385332WO1 13. Quantitate with fluorometric or colorimetric peptide assay, or UV-Vis and/or move to downstream application. Bind RNA to Beads 1. Retrieve RNA flowthru from 4 °C storage (45% 2-propanol with RNA). 2. Add 50 mg dry CH6ClN3 to 200 μL sample to reach 2.5M CH6ClN3 and incubate 10 minutes, 37 °C, 1200 rpm. 3. Wash 30 μL fresh magnetic beads disclosed herein with 200 μL 2-propanol. 4. Transfer 200 μL sample to 15 mg dry magnetic beads. 5. Add about 1200 μL 2-propanol such that sample is 90% final organic concentration. 6. Bind for 45 minutes at room temperature with sufficient agitation to keep beads in suspension. 7. Place on magnetic rack and discard supernatant. 8. Add 100 μL of the following formulation per each 15 mg beads: 0.5x PBS, 28% CH6ClN3, 10% Tween, 0.5% Tris HCl, 0.5% EDTA, 0.5% Triton X-100. Invert several times to mix with beads and put on magnetic rack. 9. Remove RNA-containing supernatant from beads and transfer to new 1.5 mL tube. a. Resuspend beads in 200 μL water. 10. Remove RNA-containing supernatant to a new tube. Add proteinase K and RNase if desired, invert several times to mix, and incubate for 10 minutes at 55 °C, 400 rpm. Note: add water to beads during incubation so they do not dry out. 11. After incubation, remove water from beads and add sample back to beads. 12. Add 2-propanol to bead/sample mixture such that sample is 90% final organic concentration and bind for 45 minutes at room temperature with sufficient agitation to keep beads in suspension. 13. Wash RNA-bound beads 2x with 400 μL 70% EtOH, 1 mM EDTA, and 25 mM Tris HCl (per 15mg bead, don’t vortex!). First Named Inventor: RAJA, Erum TP385332WO1 14. Elute in 50 μL water, 0.1 mM EDTA (per 15mg bead). 15. Extract purified RNA from beads and place in new 1.5 mL tube and move to downstream application. Example 8 Comparison Experiment To compare the disclosed magnetic beads with other, commercially available beads, a comparative workflow was developed using a KingFisherTM Sample Purification System from Thermo Fisher ScientificTM. Table 2 provides the beads that were used for the comparison experiment. Table 2. Beads used in the comparison experiment Bead Bead Description Diameter Mass Used TM TM
First Named Inventor: RAJA, Erum TP385332WO1 Example 5 Example Procedure (SP2) using the Disclosed Magnetic Beads Use 15-150 µg of protein per sample preparation. Rinse cultured cells or tissues 2-3 times with 1 × PBS to remove cell culture media or excess blood, respectively. Resuspend proteins, cells, or tissues in Lysis Solution without additional buffers. A. Extract protein, reduce, and alkylate 1. For cultured cells, add 50 µL of Lysis Buffer and 1 µL of Universal Nuclease to a minimum of 1 × 106 cells. Pipet up and down (with P200 tip) for 10-15 cycles until sample viscosity is reduced. Note: Lyse the cells in an appropriate volume of lysis buffer to have the final concentration of 2-5 µg/µl with a final volume of 7.5 µL, 15 µL, or 30 µL for input proteins of 15-30 µg, 30-60 µg, or 60-150 µg, respectively. Centrifugation of cultured cell lysates is typically not required after aspiration using a pipet. 2. For tissue samples, add 50 µL of Lysis Solution (containing 1 µL Universal Nuclease) per 5 mg of tissue and disrupt with tissue homogenizer until sample is homogenized. Centrifuge tissue lysates at 16,000 × g for 10 minutes. 3. For purified proteins, depleted and undepleted plasma, and serum samples, dilute samples directly in Lysis Solution to 2-5 µg/µL with a final volume of 7.5 µL, 15 µL, or 30 µL for input proteins of 10-25 µg, 25-50 µg, or 50-100 µg, respectively. Note: The concentration of plasma is around 60-70 µg/µL. For purified proteins and plasma samples, addition of Universal Nuclease is not required. 4. Determine the protein concentration of the supernatant using established methods such as the Pierce™ BCA Protein Assay Kit (Product No.23227) or Pierce™ Rapid Gold BCA Protein Assay Kit (Product No. A53226). Note: Dilution with Lysis Solution may be required for concentrated samples for BCA assay. 5. Transfer 15-150 µg of protein sample into a 1.5-mL low protein binding tube or a 96-well plate and adjust the final volume to 7.5 µL, 15 µL, or 30 µL for input proteins of 15-30 µg, 30-60 µg, or 60-150 µg, respectively with Lysis Solution. First Named Inventor: RAJA, Erum TP385332WO1 6. Add 3.75 µL, 7.5 µL, or 15 µL of Reduction Solution for input proteins of 15-30 µg, 30-60 µg, or 60-150 µg, respectively to the sample and gently mix. 7. Add 3.75 µL, 7.5 µL, or 15 µL of Alkylation Solution for input proteins of 15- 30 µg, 30-60 µg, or 60-150 µg, respectively to the sample and gently mix. 8. Incubate the sample at 95 °C using a heat block for 10 minutes or 50 °C for 20- 30 minutes to reduce and alkylate the protein sample. 9. After incubation, allow the sample to cool to room temperature. 10. Briefly centrifuge the tube or plate before digestion. B. Digest protein 1. Add 150 µL of Enzyme Reconstitution Solution to 1 vial of Trypsin/Lys-C Protease Mix to prepare 0.67 µg/µL enzyme mix. 2. Add 3.75 µL, 7.5 µL, or 15 µL of the reconstituted enzyme solution for input proteins of 15-30 µg, 30-60 µg, or 60-150 µg, respectively to the reduced and alkylated protein sample solution. 3. Incubate with shaking at 37°C for 1-2 hours to digest the protein sample. Note: Optional labeling with TMT reagents can be performed at this point before peptide clean-up. C. Clean-up peptides 1. Transfer 20 µL of Magnetic Bead slurry to a microcentrifuge tube or a 96-well plate and remove the storage buffer using a magnetic stand. 2. Wash the resin by adding 200 µL of Bead Wash/Binding Solution, then vortex briefly and remove the buffer using a magnetic stand. 3. Add Bead Wash/Binding Solution to the resin and transfer the bead slurry to the protein digest sample. The total volume of the diluted sample for input proteins of 15- 30 µg, 30-60 µg, or 60-150 µg should be 250 µL, 500 µL, or 1000 µL, respectively. 4. Incubate the sample at room temperature for 30-60 minutes with mixing. 5. Collect the bead using a magnetic stand and remove the unbound peptide solution. 6. Wash the resin by adding 500 µL of Wash Solution, then vortex briefly and remove the buffer using a magnetic stand. First Named Inventor: RAJA, Erum TP385332WO1 7. Add 30-100 µL of Elution Solution, then vortex briefly and incubate for 3 minutes with mixing at room temperature. 8. Transfer the eluted peptide solution using a magnetic stand to a new container for LC-MS analysis. Example 6 Example Procedure (PAC or Mutli-analyte Aggregation Capture, with SP3 for protein) Note: Any amount of protein can be used as long as bead: protein ratios are scaled accordingly. Rinse cultured cells or tissues 2-3 times with 1 × PBS to remove cell culture media or excess blood, respectively. Resuspend proteins, cells, or tissues in a lysis solution, such as the Lysis Solution described herein. For protein extraction, many buffer components are allowable, including various detergents, salts, and chaotropic agents. For DNA extraction, chaotropic agents should be avoided. For RNA extraction, guanidine hydrochloride should be added to lysis buffer. Lysis and Reduction/Alkylation – General Notes and Getting Started: 1. For cultured cells, add lysis buffer to a minimum of 1 × 106 cells. Use enough lysis buffer for an anticipated concentration between 2-12 mg/mL protein. Let sit on ice for 15 minutes. Note: for 1 x 106 cells, assume 450 μg protein, 5 μg DNA, and 10 μg RNA and use 15 mg of the disclosed magnetic beads per each sample type. Note: For tissue samples, add lysis buffer to tissue and disrupt with tissue homogenizer until sample is homogenized. Note: For purposes of protein extraction only, add 1% (by volume) Universal nuclease to lysis buffer. Do not add nuclease or sonicate sample if the intention is to perform multi-omic sequential extractions of any combination of RNA, DNA, or protein. For this purpose, immediately move to “Bind DNA to Beads” section described in Example 5. 2. Centrifuge lysates at 16,000 × g for 15 minutes at 4 °C. First Named Inventor: RAJA, Erum TP385332WO1 3. Determine the protein concentration of the supernatant using established methods such as the Pierce™ BCA Protein Assay Kit (Product No.23227) or Pierce™ Rapid Gold BCA Protein Assay Kit (Product No. A53226). 4. Proceed with reduction and alkylation, if preferred, to a final concentration of 10 mM TCEP and 20 mM 2-chloroacetamide (or equivalent protocol using dithiothreitol and iodoacetamide). TCEP and 2-chloroacetamide can be used together, but DTT and iodoacetamide must be used sequentially with DTT quench after incubation periods are complete. Note: Reduction and alkylation can be done before or after protein binding to beads. If reduction and alkylation are done after protein binding to beads, heat at 50 °C for 45 minutes. If reduction and alkylation are done prior to protein binding to beads, heat at 95 °C for 10 minutes. After incubation, allow the sample to cool to room temperature before adding beads or enzymes. Bind Protein to Beads (SP3 for protein ONLY) Note: This protocol is written with Reduction and Alkylation occurring prior to protein binding on bead at 95 °C for 10 minutes, with 5-minute cooling period prior to adding beads. Note: for protein only applications, 2-propanol and acetonitrile and inter- changeable. Note: Upstream of PAC workflow, lysis, reduction and alkylation chemistry is compatible. 1. Vortex the disclosed magnetic beads to ensure total suspension. 2. Remove supernatant from beads and wash in 500uL of 80% 2-propanol, 0.5% TFA. Remove wash. 3. Recon beads in 80% 2-propanol 0.5% TFA for [50 μg/μL]. 4. Add 99.5% 2-propanol 0.5% TFA to protein solution for a final organic concentration of 80% 2-propanol, 0.5% TFA. 5. Beads (10:1 bead: protein w/w) are added to protein in buffer for 80% organic concentration and 1-2mg/mL protein concentration. First Named Inventor: RAJA, Erum TP385332WO1 6. Incubate 45 minutes room temp with sufficient agitation to keep beads in suspension, then remove the flow through. 7. Add 4x bead volume of 100% MeCN and vortex thoroughly. Remove wash solution. 8. Add 4x bead volume of 70% EtOH and vortex thoroughly. Remove wash solution. 9. The tubes are removed from the magnetic rack and 10-25uL digestion mixture is added to beads, comprising 50mM TEAB, 10mM CaCl2, and 1:10- 1:25 (enzyme: protein (w/w)) Trypsin and LysC. 10. Vortex briefly and incubate 3 hours in a Thermomixer at 37 °C, 1200 rpm. 11. After incubation, centrifuge briefly to collect any evaporated liquid. 12. Add 100% acetonitrile to sample and beads eluate for final organic concentration at least 92.5%. Note: from this point, SP2 is also compatible. 13. Sample vortexed 30 seconds. Total peptide concentration is at least 0.2 mg/mL. 14. Sample was incubated 10 minutes at room temperature. 15. Beads are collected and the supernatant is aspirated to waste. 16. Sample is washed twice with 4x bead volume of 85% acetonitrile, 10% ethanol, 5% water, 0.5% formic acid. 17. Beads are collected and the supernatant was aspirated to waste. 18. Sample is eluted with 2x bead volume of 2-4% MeCN, 0.2%FA in water for 2 minutes with vortexing (per 15 mg beads). 19. The tube is placed on the magnetic rack for 2 minutes, and the supernatant is transferred to a clean tube. Samples are centrifuged at 10,000xg for 30 seconds, and then the sample is moved to a new tube without disturbing pellet. 20. Quantitate with fluorometric or colorimetric peptide assay, or UV-Vis and/or move to downstream application. Example 7 Multi-OMIC Sequential Aggregation Capture First Named Inventor: RAJA, Erum TP385332WO1 Bind DNA to Beads Note: this protocol is for sequential elution of DNA-Protein-RNA. Other sequences are possible with these beads. For extracting Protein-DNA only, start with 2% SDS, 100 mM TEAB, 1 mM EDTA, then reduce and alkylate with TCEP (10 mM) and 2-chloroacetamide (20mM), and bind protein at 80% 2-propanol, 0.5% TFA, take flow through and dilute with water to 30-40% 2-propanol for DNA binding. Presence of CH6ClN3 (guanidine hydrochloride) substantially alters the binding properties of each analyte on this magnetic bead and therefore organic concentrations are variable depending on when CH6ClN3 is introduced and the concentration thereof. 1. Dilute sample 1:1 with 0.2% SDS, 100 mM TEAB. Let sit on ice 15 minutes. 2. Wash 15 mg of the disclosed magnetic beads with 100% 2-propanol (per each 1 x 106 cells of lysate). 3. Add the required amount of beads to the lysates. 4. Add enough 2-propanol to the sample to achieve a total concentration of 30% organic. 5. Bind for 45 minutes at room temperature with sufficient agitation to keep beads in suspension. 6. Place on a magnetic rack and remove the lysate, which now contains RNA and protein, to “Bind Protein” section of this protocol. Move forward with DNA processing steps below, using only beads. 7. For each 15 mg of beads, add about 250 μL of 30% 2-propanol, 4.2 M CH6ClN3, 150 mM Tris HCl, 20 mM EDTA, remove from magnet and incubate 10 minutes at room temperature with sufficient agitation to keep beads in suspension. 8. Place on magnetic rack and discard supernatant to waste. 9. Add 100 μL of the following formulation per each 15 mg beads: 0.5x PBS, 28% CH6ClN3, 10% Tween, 0.5% Tris HCl, 0.5% EDTA, 0.5% Triton X-100. Invert several times to mix with beads and put on magnetic rack. 10. Remove supernatant to a new tube. Add proteinase K and RNase if desired, invert several times to mix, and incubate for 10 minutes at 55 °C, 400 rpm. Note: add water to beads during incubation so they do not dry out. First Named Inventor: RAJA, Erum TP385332WO1 11. After incubation, remove water from beads and add sample back to beads. 12. Add a 1x volume of 60% 2-propanol, 2.4M CH6ClN3, 150 mM Tris HCl, 20 mM EDTA to bead/sample mixture and bind for 45 minutes at room temperature with sufficient agitation to keep beads in suspension. 13. Wash DNA-bound beads 2x with 200 μL 70% EtOH, 1 mM EDTA, and 25 mM Tris HCl (per 15 mg beads). 14. Elute in 50 μL 25 mM Tris-HCl, 0.1 mM EDTA (per 15 mg beads). 15. Place on magnetic rack, and transfer supernatant to new 1.5 mL tubes 16. Centrifuge 12,000 xg at 4 °C for 2 minutes. 17. Move to new tubes without beads. 18. Add EDTA to 1 mM and store at 4 °C until downstream application. Bind Protein to Beads (Sequential Method ONLY) 1. Retrieve flowthrough from “Bind DNA to Beads” or from “Lysis and Reduction/Alkylation”. 2. Wash a new set of 30 μL (15 mg) of the disclosed magnetic beads with 200uL 99.5% 2-propanol, 0.5% trifluoroacetic acid, then remove wash solution from beads. 3. Transfer samples to 15mg of the disclosed magnetic beads (per 1 x 106 cells). 4. Add enough 99% 2-Propanol, 1% TFA to a total concentration of 45% organic. 5. Bind for 45 minutes at room temperature with sufficient agitation to keep beads in suspension. 6. Place on magnetic rack and transfer RNA-containing supernatant to new 1.5 mL tubes. a. Add 0.1mM EDTA to RNA-containing supernatant and store at 4 °C for short term storage. 7. Wash with 200 μL 70% EtOH, vortex briefly (per 15 mg beads). 8. Wash with 200 μL 100% MeCN, vortex briefly (per 15 mg beads). 9. Resuspend bead-bound proteins in 40 μL 100mM TEAB, 10 mM CaCl2. First Named Inventor: RAJA, Erum TP385332WO1 a. Reduce and alkylate with 10 μL total of TCEP and 2-chloroacetamide, if not done prior to bead binding. Final concentrations are 20 mM 2-chloroacetamide, 10 mM TCEP. Vortex sample and incubate at 50 °C for 45 minutes. b. If the samples have been previously reduced and alkylated, add 50uL 100 mM TEAB, 10mM CaCl2 to beads instead of 40 μL. 10. Add 0.6 μL universal nuclease. Pipette up and down repeatedly. 11. Take 1 vial of Trypsin/LysC from -20 °C and let thaw 5 minutes. a. Reconstitute vial in 50 μL 0.1% CH3COOH for 2.0 mg/mL. Mix gently and let sit 5 minutes. b. Add 1:10-1:25 of enzyme:protein (w/w). c. Incubate 2 hours in a Thermomixer at 37 °C, 1200rpm. Continue with the steps below for complete SP3 workflow of protein preparation. Alternatively, SP2 preparation is also compatible and can be used in place of this protocol, and still be SP3 (scaled for 15 mg beads) 12. Begin peptide cleanup by adding enough 2-propanol for >92.75% organic concentration. a. Sample is incubated 10 minutes at room temperature 1200 rpm. b. Beads are collected and the supernatant was aspirated to waste. c. Sample is washed twice with 200 μL of 100% HPLC-grade MeCN (or 85% MeCN, 10% EtOH, 5.0% water, 0.5% formic acid for improved salt removal, such as in TMT- or TMTpro-labeled samples). d. Beads are collected and the supernatant is aspirated to waste. e. Sample is eluted with 50 μL of 2-4% MeCN, 0.2%FA in water for 2 minutes with occasional vortex. f. Place on the magnetic rack for 2 minutes, and transfer supernatant to a clean tube. g. Samples are centrifuged at 10,000xg for 30 seconds. h. Sample is moved to a new tube without disturbing pellet. First Named Inventor: RAJA, Erum TP385332WO1 13. Quantitate with fluorometric or colorimetric peptide assay, or UV-Vis and/or move to downstream application. Bind RNA to Beads 1. Retrieve RNA flowthru from 4 °C storage (45% 2-propanol with RNA). 2. Add 50 mg dry CH6ClN3 to 200 μL sample to reach 2.5M CH6ClN3 and incubate 10 minutes, 37 °C, 1200 rpm. 3. Wash 30 μL fresh magnetic beads disclosed herein with 200 μL 2-propanol. 4. Transfer 200 μL sample to 15 mg dry magnetic beads. 5. Add about 1200 μL 2-propanol such that sample is 90% final organic concentration. 6. Bind for 45 minutes at room temperature with sufficient agitation to keep beads in suspension. 7. Place on magnetic rack and discard supernatant. 8. Add 100 μL of the following formulation per each 15 mg beads: 0.5x PBS, 28% CH6ClN3, 10% Tween, 0.5% Tris HCl, 0.5% EDTA, 0.5% Triton X-100. Invert several times to mix with beads and put on magnetic rack. 9. Remove RNA-containing supernatant from beads and transfer to new 1.5 mL tube. a. Resuspend beads in 200 μL water. 10. Remove RNA-containing supernatant to a new tube. Add proteinase K and RNase if desired, invert several times to mix, and incubate for 10 minutes at 55 °C, 400 rpm. Note: add water to beads during incubation so they do not dry out. 11. After incubation, remove water from beads and add sample back to beads. 12. Add 2-propanol to bead/sample mixture such that sample is 90% final organic concentration and bind for 45 minutes at room temperature with sufficient agitation to keep beads in suspension. 13. Wash RNA-bound beads 2x with 400 μL 70% EtOH, 1 mM EDTA, and 25 mM Tris HCl (per 15mg bead, don’t vortex!). First Named Inventor: RAJA, Erum TP385332WO1 14. Elute in 50 μL water, 0.1 mM EDTA (per 15mg bead). 15. Extract purified RNA from beads and place in new 1.5 mL tube and move to downstream application. Example 8 Comparison Experiment To compare the disclosed magnetic beads with other, commercially available beads, a comparative workflow was developed using a KingFisherTM Sample Purification System from Thermo Fisher ScientificTM. Table 2 provides the beads that were used for the comparison experiment. Table 2. Beads used in the comparison experiment Bead Bead Description Diameter Mass Used TM TM A 25 μg HeLa lysate sample was used for each bead type. The comparative workflow was as follows: A. Protein Digestion (Off KingFisher) • Reduction/alkylation: 95 °C for 10 minutes, cooled to room temperature, and centrifuged for seconds. • Digestion: 37 °C for 1 hour with shaking at 1,000 rpm. • Trypsin/Lys-C: 0.67 μg/μL (100 μg of Trypsin/Lys-C reconstituted in 150 μL of 0.1% acetic acid/10 mM CaCl2 for use, sample to trypsin ratio: 10:1) First Named Inventor: RAJA, Erum TP385332WO1 Table 3. Sample/reagent volume for protein digestion Input Sample Sample/reagent vol. (Sample + Digest volume reduction + alkylation + enzyme) B.
A 25 μg HeLa lysate sample was used for each bead type. The comparative workflow was as follows: A. Protein Digestion (Off KingFisher) • Reduction/alkylation: 95 °C for 10 minutes, cooled to room temperature, and centrifuged for seconds. • Digestion: 37 °C for 1 hour with shaking at 1,000 rpm. • Trypsin/Lys-C: 0.67 μg/μL (100 μg of Trypsin/Lys-C reconstituted in 150 μL of 0.1% acetic acid/10 mM CaCl2 for use, sample to trypsin ratio: 10:1) First Named Inventor: RAJA, Erum TP385332WO1 Table 3. Sample/reagent volume for protein digestion Input Sample Sample/reagent vol. (Sample + Digest volume reduction + alkylation + enzyme) B. Results from the experiment are provided in FIGS.8A, 8B, and 9-11. FIGS.8A and 8B demonstrate that the disclosed magnetic beads exhibit less surface absorption loss than the Sera-MagTM carboxylate modified particles, MyOneTM, or silica beads in the SP2 workflow. The SeraMagTM, MyOneTM, and silica beads all demonstrated significant bead loss on the surfaces of the comb tip plate and sample plate (bead First Named Inventor: RAJA, Erum TP385332WO1 transfer/mix and binding steps). In contrast, the disclosed magnetic beads did not exhibit significant bead loss. The data provided in FIGS.8A and 8B with respect to the disclosed magnetic beads refers to hydrolyzed beads, that is beads where the azlactone moieties on the surface of the beads have been hydrolyzed. However, similar results were achieved using disclosed magnetic beads where the azlactone moieties were treated with aminobenzoic acid or aminoethyl-trimethylammonium chloride (AETMA) (data not shown). FIG.9 provides data demonstrating that the peptide yield from the KingFisher SP2 workflow for the disclosed magnetic beads was substantially higher than the peptide yield for the Sera-MagTM, MyOneTM Silane or silica beads, using a quantitative colorimetric peptide assay. No peptides were recovered using silica beads. FIG.10 demonstrates that using the disclosed magnetic beads, substantially more unique peptides were identified by nano-LCMS analysis, than were identified using the other bead types. And FIG.11 demonstrates that substantially more protein groups were identified using the disclosed magnetic beads. Example 9 An experiment was designed to demonstrate the superior binding of the disclosed magnetic beads coupled to alkali stable Protein A (asPA), compared to a commercial magnetic bead that comprises asPA conjugates. Conjugate asPA to the disclosed magnetic beads Alkali stable Protein A was conjugated to the disclosed azlactone beads according to the following procedure: 1. Weigh out 77mg of the disclosed azlactone beads. 2. Dilute asPA (20mg/mL in 250mM Sodium Acetate, pH 5.5, 0.5M NaCl, 0.05% Tween-20) into Coupling Buffer (0.1M Sodium Carbonate, 1.2M Sodium Citrate, pH 9) 1:3 (v/v); final concentration of components is 5mg/mL asPA, 0.075M Sodium Carbonate, 0.9M Sodium Carbonate, pH 9 3. Add 3.6mL of asPA in Coupling Buffer to azlactone beads. First Named Inventor: RAJA, Erum TP385332WO1 4. Couple asPA to beads overnight at room temperature. 5. Wash asPA-beads with 2-3 mL water. 6. Block asPA-beads with 2-3 mL of 3M ethanolamine for 2 hours. 7. Wash asPA-beads with 2-3 mL water. 8. Wash asPA-beads twice with 2-3 mL 0.1M Glycine, pH 2 (Pierce IgG Elution Buffer, #21028) 9. Wash asPA-beads three times with 2-3 mL water. 10. Slurry asPA-beads to 25% using 20% ethanol. Measure the binding capacity of asPA-azlactone beads compared to asPA- magnetic agarose The procedure for measuring the binding capacity of asPA-azlactone beads compared to asPA-magnetic agarose (Pierce High Capacity Protein A MagBeads, alkali stable, #A53036, 25% slurry) was as follows: 1. Prepare 3mg/mL Rabbit IgG (RIgG) in PBS. 2. Add 2 x 50uL of 25% magnetic bead slurry (asPA-azlactone beads and asPA- magnetic agarose) to 1.5mL microcentrifuge tubes. 3. Wash beads twice with 500uL of PBS. 4. Add 500uL of 3mg/mL (RIgG) to beads and incubate for 1hr at room temperature with shaking/mixing. 5. Collect the flowthrough and save for analysis. 6. Create a RIgG standard curve from 0 to 3mg/mL by measuring absorbance at 280nm. 7. Measure absorbance of starting solution of RIgG and Flowthrough from each bead sample. 8. Calculate concentrations of start and flowthrough using standard curve. 9. Calculate RIgG bound to the magnetic beads by subtracting the RIgG in each Flowthrough from the starting RIgG. Extrapolate to amount RIgG bound per mL of settled beads. First Named Inventor: RAJA, Erum TP385332WO1 Results The results clearly demonstrated that the asPA conjugated disclosed magnetic beads had about a 15% higher RIgG binding capacity that asPA-magentic agarose beads (FIG.12). Without being bound to a particular theory, this may be due, at least in part, to the disclosed azlactone magnetic beads being able to conjugate a larger amount of asPA than an amount asPA that the same amount of magnetic agarose beads can conjugate. And in some aspects, the amount of asPA bound to the disclosed azlactone magnetic beads is about 2-fold higher than the amount of asPA bound to the same amount of magnetic agarose beads. 2 a 50
Results from the experiment are provided in FIGS.8A, 8B, and 9-11. FIGS.8A and 8B demonstrate that the disclosed magnetic beads exhibit less surface absorption loss than the Sera-MagTM carboxylate modified particles, MyOneTM, or silica beads in the SP2 workflow. The SeraMagTM, MyOneTM, and silica beads all demonstrated significant bead loss on the surfaces of the comb tip plate and sample plate (bead First Named Inventor: RAJA, Erum TP385332WO1 transfer/mix and binding steps). In contrast, the disclosed magnetic beads did not exhibit significant bead loss. The data provided in FIGS.8A and 8B with respect to the disclosed magnetic beads refers to hydrolyzed beads, that is beads where the azlactone moieties on the surface of the beads have been hydrolyzed. However, similar results were achieved using disclosed magnetic beads where the azlactone moieties were treated with aminobenzoic acid or aminoethyl-trimethylammonium chloride (AETMA) (data not shown). FIG.9 provides data demonstrating that the peptide yield from the KingFisher SP2 workflow for the disclosed magnetic beads was substantially higher than the peptide yield for the Sera-MagTM, MyOneTM Silane or silica beads, using a quantitative colorimetric peptide assay. No peptides were recovered using silica beads. FIG.10 demonstrates that using the disclosed magnetic beads, substantially more unique peptides were identified by nano-LCMS analysis, than were identified using the other bead types. And FIG.11 demonstrates that substantially more protein groups were identified using the disclosed magnetic beads. Example 9 An experiment was designed to demonstrate the superior binding of the disclosed magnetic beads coupled to alkali stable Protein A (asPA), compared to a commercial magnetic bead that comprises asPA conjugates. Conjugate asPA to the disclosed magnetic beads Alkali stable Protein A was conjugated to the disclosed azlactone beads according to the following procedure: 1. Weigh out 77mg of the disclosed azlactone beads. 2. Dilute asPA (20mg/mL in 250mM Sodium Acetate, pH 5.5, 0.5M NaCl, 0.05% Tween-20) into Coupling Buffer (0.1M Sodium Carbonate, 1.2M Sodium Citrate, pH 9) 1:3 (v/v); final concentration of components is 5mg/mL asPA, 0.075M Sodium Carbonate, 0.9M Sodium Carbonate, pH 9 3. Add 3.6mL of asPA in Coupling Buffer to azlactone beads. First Named Inventor: RAJA, Erum TP385332WO1 4. Couple asPA to beads overnight at room temperature. 5. Wash asPA-beads with 2-3 mL water. 6. Block asPA-beads with 2-3 mL of 3M ethanolamine for 2 hours. 7. Wash asPA-beads with 2-3 mL water. 8. Wash asPA-beads twice with 2-3 mL 0.1M Glycine, pH 2 (Pierce IgG Elution Buffer, #21028) 9. Wash asPA-beads three times with 2-3 mL water. 10. Slurry asPA-beads to 25% using 20% ethanol. Measure the binding capacity of asPA-azlactone beads compared to asPA- magnetic agarose The procedure for measuring the binding capacity of asPA-azlactone beads compared to asPA-magnetic agarose (Pierce High Capacity Protein A MagBeads, alkali stable, #A53036, 25% slurry) was as follows: 1. Prepare 3mg/mL Rabbit IgG (RIgG) in PBS. 2. Add 2 x 50uL of 25% magnetic bead slurry (asPA-azlactone beads and asPA- magnetic agarose) to 1.5mL microcentrifuge tubes. 3. Wash beads twice with 500uL of PBS. 4. Add 500uL of 3mg/mL (RIgG) to beads and incubate for 1hr at room temperature with shaking/mixing. 5. Collect the flowthrough and save for analysis. 6. Create a RIgG standard curve from 0 to 3mg/mL by measuring absorbance at 280nm. 7. Measure absorbance of starting solution of RIgG and Flowthrough from each bead sample. 8. Calculate concentrations of start and flowthrough using standard curve. 9. Calculate RIgG bound to the magnetic beads by subtracting the RIgG in each Flowthrough from the starting RIgG. Extrapolate to amount RIgG bound per mL of settled beads. First Named Inventor: RAJA, Erum TP385332WO1 Results The results clearly demonstrated that the asPA conjugated disclosed magnetic beads had about a 15% higher RIgG binding capacity that asPA-magentic agarose beads (FIG.12). Without being bound to a particular theory, this may be due, at least in part, to the disclosed azlactone magnetic beads being able to conjugate a larger amount of asPA than an amount asPA that the same amount of magnetic agarose beads can conjugate. And in some aspects, the amount of asPA bound to the disclosed azlactone magnetic beads is about 2-fold higher than the amount of asPA bound to the same amount of magnetic agarose beads. 2 a 50

















 a reaction of the azlactone moiety with a nucleophilic reactive group. 2. The method of claim 1, wherein the magnetic material comprises an iron source. 3. The method of claim 2, wherein the iron source comprises iron oxide. 4. The method of any one of claims 1-3, wherein the magnetic material is contained within an agarose bead, and the agarose bead is contained within the crosslinked polymer. 5. The method of any one of claims 1-4, wherein the magnetic material is in the form of a particle, powder, flake, or cluster. First Named Inventor: RAJA, Erum TP385332WO1 6. The method of any one of claims 1-5, wherein the crosslinked polymer is a crosslinked azlactone polymer, and the magnetic bead is an azlactone bead. 7. The method of any one of claims 1-6, wherein the magnetic bead comprises one or more functional groups derived from a reaction of the azlactone moiety with an amine-containing compound selected from a biomolecule, organic ligand, polymer, dendrimer, or a combination thereof. 8. The method of claim 6, comprising: iron oxide particles distributed throughout an azlactone bead; and a plurality of functional moieties located on the surface of the magnetic bead; O OH wherein the functional moieties are selected, O H
a reaction of the azlactone moiety with a nucleophilic reactive group. 2. The method of claim 1, wherein the magnetic material comprises an iron source. 3. The method of claim 2, wherein the iron source comprises iron oxide. 4. The method of any one of claims 1-3, wherein the magnetic material is contained within an agarose bead, and the agarose bead is contained within the crosslinked polymer. 5. The method of any one of claims 1-4, wherein the magnetic material is in the form of a particle, powder, flake, or cluster. First Named Inventor: RAJA, Erum TP385332WO1 6. The method of any one of claims 1-5, wherein the crosslinked polymer is a crosslinked azlactone polymer, and the magnetic bead is an azlactone bead. 7. The method of any one of claims 1-6, wherein the magnetic bead comprises one or more functional groups derived from a reaction of the azlactone moiety with an amine-containing compound selected from a biomolecule, organic ligand, polymer, dendrimer, or a combination thereof. 8. The method of claim 6, comprising: iron oxide particles distributed throughout an azlactone bead; and a plurality of functional moieties located on the surface of the magnetic bead; O OH wherein the functional moieties are selected, O H N OH
N OH
 First Named Inventor: RAJA, Erum TP385332WO1 10. The method of any one of claims 1-9, wherein the magnetic material comprises Fe3O4, Fe2O3, or a combination thereof. 11. The method of any one of claims 1-10, wherein the azlactone bead is formed from a vinyl azlactone and a crosslinker selected from a bisacrylamide, agarose, and a vinyl ether. 12. The method of claim 11, wherein the vinyl azlactone is 4,4-dimethyl-2- vinyloxazol-5(4H)-one. 13. The method of claim 11 or claim 12, wherein the crosslinker is methylene bisacrylamide. 14. The method of any one of claims 1-13, wherein the biomolecule is a protein, peptide, polypeptide, DNA or RNA molecule. 15. The method of claim 14, wherein the biomolecule is labeled with a stable isotope, isotopically-enriched mass tag, stable isotopically labeled peptide and/or protein, rare-earth metal, fluorescent label, tissue and/or cell grown with heavy amino acids, or a combination thereof. 16. The method of claim 15, wherein the biomolecule is labeled with a stable isotope selected from deuterium, carbon-13, nitrogen-15, oxygen-18, or a combination thereof. 17. The method of any one of claims 1-16, wherein the biomolecule is a protein, polypeptide or a peptide and the first liquid has an organic concentration of 80% or more. First Named Inventor: RAJA, Erum TP385332WO1 18. The method of claim 17, wherein the first liquid comprises 80% or more of 2-propanol, acetonitrile, or a combination thereof. 19. The method of claim 17 or claim 18, wherein the elution solution comprises from greater than zero to 0.5% formic acid in water. 20. The method of any one of claims 17-19, wherein the first liquid is a protein sample. 21. The method of claim 20, wherein after washing the magnetic beads using the second liquid and before adding the elution solution, the method further comprises: incubating the magnetic beads with a digestion mixture; and after incubation, washing the beads to remove the digestion mixture. 22. The method of any one of claims 17-19, wherein the first liquid is a protein digest sample. 23. The method of any one of claims 1-16, wherein the biomolecule is DNA. 24. The method of claim 23, wherein the second liquid comprises from 25% to 35% organic solvent and a chelating agent, guanidinium chloride, a buffer, or a combination thereof. 25. The method of claim 24, wherein prior to forming the second suspension, the method further comprises: separate the magnetic beads from the second liquid; add to the magnetic beads a surfactant mixture comprising a buffer, guanidinium chloride, a chelating agent, and a surfactant and agitate; First Named Inventor: RAJA, Erum TP385332WO1 separate the magnetic beads from the surfactant mixture and to the surfactant mixture add proteinase K and/or RNase, agitate and recombine with the magnetic beads; add a further mixture comprising from 50% to 70% organic solvent in water, a chelating agent, guanidinium chloride, and a buffer; and wash the magnetic beads with a mixture comprising 65% to 80% organic solvent, a chelating agent and a buffer. 26. The method of any one of claims 23-25, wherein the elution solvent comprises a buffer and a chelating agent. 27. The method of any one of claims 24-26, wherein the chelating agent is EDTA. 28. The method of any one of claims 24-27, wherein the buffer is tris HCl buffer, PBS, or a combination thereof. 29. The method of any one of claims 24-28, wherein the organic solvent is 2- propanol, ethanol, or a combination thereof. 30. The method of any one of claims 24-29, wherein the surfactant is Tween, Triton X-100, or a combination thereof. 31. The method of any one of claims 1-16, wherein the biomolecule is RNA. 32. The method of claim 31, wherein the first liquid has an organic concentration of from 85% or more. 33. The method of claim 31 or claim 32, wherein the elution solution comprises a buffer, guanidinium chloride, a chelating agent, and a surfactant. First Named Inventor: RAJA, Erum TP385332WO1 34. The method of claim 33, wherein: the buffer is PBS, Tris HCl, or a combination thereof; the chelating agent is ETDA; the surfactant is Tween, Triton X-100, or a combination thereof; or a combination thereof. 35. The method of claim 33 or claim 34, further comprising adding proteinase K and/or RNase to the third liquid. 36. The method of claim 35, further comprising recombining the third liquid with the magnetic beads to form a third suspension and adjusting an organic concentration of the third suspension to be at least 85% organic. 37. The method of claim 36, further comprising washing the magnetic beads with a solution comprising an organic solvent, a chelating agent and a buffer. 38. The method of claim 37, wherein the organic solvent is ethanol, the chelating agent is EDTA, the buffer is Tris HCl, or a combination thereof. 39. The method of claim 37 or claim 38, further comprising eluting the RNA from the magnetic beads with a solution of EDTA in water. 40. The method of any one of claims 1-39, therein the method further comprises analyzing the biomolecule by mass spectrometry. 41. A method for obtaining a peptide or polypeptide for LC-MS analysis, the method comprising: form a first suspension comprising a plurality of magnetic beads and a first liquid that contains a protein digest sample comprising the peptide or polypeptide; First Named Inventor: RAJA, Erum TP385332WO1 agitate the first suspension at room temperature for from 30 to 60 minutes; remove the magnetic beads from the first suspension and wash the magnetic beads using a second liquid; form a second suspension comprising the magnetic beads and an elution solution and agitate the second suspension at room temperature for from 1 to 5 minutes; and separate the magnetic beads from the second suspension to leave a third liquid comprising the peptide or the polypeptide; wherein each magnetic bead comprises iron oxide particles distributed O OH N H throughout an azlactone bead and a plurality ofOmoieties located on the surface of the magnetic bead. 42. A method for obtaining a protein, the method comprising: form a first suspension comprising the plurality of magnetic beads and a first liquid that contains the protein, wherein the first liquid is an aqueous solution comprising 0.5% trifluoroacetic acid, and 80% of an organic solvent selected from 2- propanol and acetonitrile; agitate the first suspension at room temperature for from 30 to 60 minutes; remove the magnetic beads from the first suspension and wash the magnetic beads with a second liquid comprising an organic solvent; incubate the magnetic beads with a digestion mixture comprising Trypsin and LysC for from 1 to 4 hours; wash the beads with a washing solution comprising water, an organic solvent and formic acid, to remove the digestion mixture; form a second suspension comprising the magnetic beads and an elution solution and agitate the second suspension at room temperature for from 1 to 5 minutes; and separate the magnetic beads from the second suspension to leave a third liquid comprising the protein; First Named Inventor: RAJA, Erum TP385332WO1 wherein each magnetic bead comprises iron oxide particles distributed O OH N throughout an azlactone bead and a pluralitymoieties located on the surface of the magnetic bead.
First Named Inventor: RAJA, Erum TP385332WO1 10. The method of any one of claims 1-9, wherein the magnetic material comprises Fe3O4, Fe2O3, or a combination thereof. 11. The method of any one of claims 1-10, wherein the azlactone bead is formed from a vinyl azlactone and a crosslinker selected from a bisacrylamide, agarose, and a vinyl ether. 12. The method of claim 11, wherein the vinyl azlactone is 4,4-dimethyl-2- vinyloxazol-5(4H)-one. 13. The method of claim 11 or claim 12, wherein the crosslinker is methylene bisacrylamide. 14. The method of any one of claims 1-13, wherein the biomolecule is a protein, peptide, polypeptide, DNA or RNA molecule. 15. The method of claim 14, wherein the biomolecule is labeled with a stable isotope, isotopically-enriched mass tag, stable isotopically labeled peptide and/or protein, rare-earth metal, fluorescent label, tissue and/or cell grown with heavy amino acids, or a combination thereof. 16. The method of claim 15, wherein the biomolecule is labeled with a stable isotope selected from deuterium, carbon-13, nitrogen-15, oxygen-18, or a combination thereof. 17. The method of any one of claims 1-16, wherein the biomolecule is a protein, polypeptide or a peptide and the first liquid has an organic concentration of 80% or more. First Named Inventor: RAJA, Erum TP385332WO1 18. The method of claim 17, wherein the first liquid comprises 80% or more of 2-propanol, acetonitrile, or a combination thereof. 19. The method of claim 17 or claim 18, wherein the elution solution comprises from greater than zero to 0.5% formic acid in water. 20. The method of any one of claims 17-19, wherein the first liquid is a protein sample. 21. The method of claim 20, wherein after washing the magnetic beads using the second liquid and before adding the elution solution, the method further comprises: incubating the magnetic beads with a digestion mixture; and after incubation, washing the beads to remove the digestion mixture. 22. The method of any one of claims 17-19, wherein the first liquid is a protein digest sample. 23. The method of any one of claims 1-16, wherein the biomolecule is DNA. 24. The method of claim 23, wherein the second liquid comprises from 25% to 35% organic solvent and a chelating agent, guanidinium chloride, a buffer, or a combination thereof. 25. The method of claim 24, wherein prior to forming the second suspension, the method further comprises: separate the magnetic beads from the second liquid; add to the magnetic beads a surfactant mixture comprising a buffer, guanidinium chloride, a chelating agent, and a surfactant and agitate; First Named Inventor: RAJA, Erum TP385332WO1 separate the magnetic beads from the surfactant mixture and to the surfactant mixture add proteinase K and/or RNase, agitate and recombine with the magnetic beads; add a further mixture comprising from 50% to 70% organic solvent in water, a chelating agent, guanidinium chloride, and a buffer; and wash the magnetic beads with a mixture comprising 65% to 80% organic solvent, a chelating agent and a buffer. 26. The method of any one of claims 23-25, wherein the elution solvent comprises a buffer and a chelating agent. 27. The method of any one of claims 24-26, wherein the chelating agent is EDTA. 28. The method of any one of claims 24-27, wherein the buffer is tris HCl buffer, PBS, or a combination thereof. 29. The method of any one of claims 24-28, wherein the organic solvent is 2- propanol, ethanol, or a combination thereof. 30. The method of any one of claims 24-29, wherein the surfactant is Tween, Triton X-100, or a combination thereof. 31. The method of any one of claims 1-16, wherein the biomolecule is RNA. 32. The method of claim 31, wherein the first liquid has an organic concentration of from 85% or more. 33. The method of claim 31 or claim 32, wherein the elution solution comprises a buffer, guanidinium chloride, a chelating agent, and a surfactant. First Named Inventor: RAJA, Erum TP385332WO1 34. The method of claim 33, wherein: the buffer is PBS, Tris HCl, or a combination thereof; the chelating agent is ETDA; the surfactant is Tween, Triton X-100, or a combination thereof; or a combination thereof. 35. The method of claim 33 or claim 34, further comprising adding proteinase K and/or RNase to the third liquid. 36. The method of claim 35, further comprising recombining the third liquid with the magnetic beads to form a third suspension and adjusting an organic concentration of the third suspension to be at least 85% organic. 37. The method of claim 36, further comprising washing the magnetic beads with a solution comprising an organic solvent, a chelating agent and a buffer. 38. The method of claim 37, wherein the organic solvent is ethanol, the chelating agent is EDTA, the buffer is Tris HCl, or a combination thereof. 39. The method of claim 37 or claim 38, further comprising eluting the RNA from the magnetic beads with a solution of EDTA in water. 40. The method of any one of claims 1-39, therein the method further comprises analyzing the biomolecule by mass spectrometry. 41. A method for obtaining a peptide or polypeptide for LC-MS analysis, the method comprising: form a first suspension comprising a plurality of magnetic beads and a first liquid that contains a protein digest sample comprising the peptide or polypeptide; First Named Inventor: RAJA, Erum TP385332WO1 agitate the first suspension at room temperature for from 30 to 60 minutes; remove the magnetic beads from the first suspension and wash the magnetic beads using a second liquid; form a second suspension comprising the magnetic beads and an elution solution and agitate the second suspension at room temperature for from 1 to 5 minutes; and separate the magnetic beads from the second suspension to leave a third liquid comprising the peptide or the polypeptide; wherein each magnetic bead comprises iron oxide particles distributed O OH N H throughout an azlactone bead and a plurality ofOmoieties located on the surface of the magnetic bead. 42. A method for obtaining a protein, the method comprising: form a first suspension comprising the plurality of magnetic beads and a first liquid that contains the protein, wherein the first liquid is an aqueous solution comprising 0.5% trifluoroacetic acid, and 80% of an organic solvent selected from 2- propanol and acetonitrile; agitate the first suspension at room temperature for from 30 to 60 minutes; remove the magnetic beads from the first suspension and wash the magnetic beads with a second liquid comprising an organic solvent; incubate the magnetic beads with a digestion mixture comprising Trypsin and LysC for from 1 to 4 hours; wash the beads with a washing solution comprising water, an organic solvent and formic acid, to remove the digestion mixture; form a second suspension comprising the magnetic beads and an elution solution and agitate the second suspension at room temperature for from 1 to 5 minutes; and separate the magnetic beads from the second suspension to leave a third liquid comprising the protein; First Named Inventor: RAJA, Erum TP385332WO1 wherein each magnetic bead comprises iron oxide particles distributed O OH N throughout an azlactone bead and a pluralitymoieties located on the surface of the magnetic bead. 43. A magnetic bead, comprising a crosslinked polymer and a magnetic material, wherein the crosslinked polymer comprises one or more azlactone moieties O O one or more functional groups derived from an azlactone moiety, the reaction of the azlactone moiety with a nucleophilic reactive group.
43. A magnetic bead, comprising a crosslinked polymer and a magnetic material, wherein the crosslinked polymer comprises one or more azlactone moieties O O one or more functional groups derived from an azlactone moiety, the reaction of the azlactone moiety with a nucleophilic reactive group. 44. The magnetic bead of claim 43, wherein the magnetic material comprises an iron source. 45. The magnetic bead of claim 44, wherein the iron source comprises iron oxide. 46. The magnetic bead of any one of claims 43-45, wherein the magnetic material is contained within an agarose bead, and the agarose bead is contained within the crosslinked polymer. 47. The magnetic bead of any one of claims 43-46, wherein the magnetic material is in the form of a particle, powder, flake, or cluster. 48. The magnetic bead of any one of claims 43-47, wherein the crosslinked polymer is a crosslinked azlactone polymer, and the magnetic bead is an azlactone bead. First Named Inventor: RAJA, Erum TP385332WO1 49. The magnetic bead of any one of claims 43-48, wherein the magnetic bead comprises one or more functional groups derived from a reaction of the azlactone moiety with an amine-containing compound selected from a biomolecule, organic ligand, polymer, dendrimer, or a combination thereof. 50. The magnetic bead of claim 48, comprising: iron oxide particles distributed throughout an azlactone bead; and a plurality of functional moieties located on the surface of the magnetic bead; O O OH O wherein the functional moieties are selected , O H N
44. The magnetic bead of claim 43, wherein the magnetic material comprises an iron source. 45. The magnetic bead of claim 44, wherein the iron source comprises iron oxide. 46. The magnetic bead of any one of claims 43-45, wherein the magnetic material is contained within an agarose bead, and the agarose bead is contained within the crosslinked polymer. 47. The magnetic bead of any one of claims 43-46, wherein the magnetic material is in the form of a particle, powder, flake, or cluster. 48. The magnetic bead of any one of claims 43-47, wherein the crosslinked polymer is a crosslinked azlactone polymer, and the magnetic bead is an azlactone bead. First Named Inventor: RAJA, Erum TP385332WO1 49. The magnetic bead of any one of claims 43-48, wherein the magnetic bead comprises one or more functional groups derived from a reaction of the azlactone moiety with an amine-containing compound selected from a biomolecule, organic ligand, polymer, dendrimer, or a combination thereof. 50. The magnetic bead of claim 48, comprising: iron oxide particles distributed throughout an azlactone bead; and a plurality of functional moieties located on the surface of the magnetic bead; O O OH O wherein the functional moieties are selected , O H N OH
OH 51. The magnetic bead of claim 43, wherein the functional moieties .
51. The magnetic bead of claim 43, wherein the functional moieties . 52. The magnetic bead of claim 43 or claim 51, wherein the azlactone bead is formed from a vinyl azlactone and a crosslinker selected from a bisacrylamide, agarose, and a vinyl ether. First Named Inventor: RAJA, Erum TP385332WO1 53. The magnetic bead of any one of claims 43-52, wherein: the vinyl azlactone is 4,4-dimethyl-2-vinyloxazol-5(4H)-one; the crosslinker is a bisacrylamide; or the vinyl azlactone is 4,4-dimethyl-2-vinyloxazol-5(4H)-one and the crosslinker is a bisacrylamide. 54. The magnetic bead of claim 52 or claim 53, wherein the crosslinker is methylene bisacrylamide. 55. The magnetic bead of any one of claims 43-54, wherein the magnetic material is Fe3O4 or Fe2O3 or a combination thereof. 56. The magnetic bead of any one of claims 43-55, wherein the bead has a bead size of from 20 to 80 microns. 57. The magnetic bead of claim 56, wherein the bead has a bead size of from 30 to 60 microns. 58. The magnetic bead of any one of claims 56-57, wherein the bead size is a bead size in a non-hydrated state, and the bead has a hydrated bead size that is from 6 to 8 times larger than the bead size in the non-hydrated state. 59. The magnetic bead of any one of claims 43-58, where the magnetic bead is a porous magnetic bead. 60. A population of the magnetic beads according to any one of claims 43- 59, wherein the population exhibits a bead loss of less than about 5% by weight when used in an automated process. First Named Inventor: RAJA, Erum TP385332WO1 61. A kit, comprising a plurality of magnetic beads according to any one of claims 43-59 or claims 64-70. 62. A method of removing contaminants from a sample, comprising: provide a sample comprising a liquid matrix and protein fragments or polypeptides, wherein the liquid matrix comprises contaminants; combine the sample with a population of magnetic beads according to any one of claims 36-45 in the presence of a binding solution, wherein the protein fragments or polypeptides bind to the magnetic beads; magnetically separate the population of magnetic beads from the liquid matrix; optionally wash the population of separated magnetic beads with a wash solution to remove any remaining contaminants from the population of magnetic beads; elute the protein fragments or polypeptides from the separated magnetic beads with an elution solution, to form a solution comprising the protein fragments or polypeptides; and magnetically separate the population of magnetic beads from the solution comprising the protein fragments or polypeptides. 63. A method for protein aggregation capture from a sample, the method comprising: provide a sample comprising a liquid matrix and a plurality of proteins; combine the sample with a population of magnetic beads according to any one of claims 36-45 in the presence of a binding solution, wherein at least a portion of the proteins bind to the magnetic beads to form bound proteins on the magnetic beads; magnetically separate the population of magnetic beads from the liquid matrix; optionally expose the population of magnetic beads with the bound proteins to a reduction reagent and/or an alkylation reagent and then separate the population of magnetic beads from the reduction reagent and/or the alkylation reagent; optionally wash the population of separated magnetic beads; First Named Inventor: RAJA, Erum TP385332WO1 elute the bound proteins from the population of magnetic beads with an elution solution to form a solution comprising unbound proteins; and optionally digest the unbound proteins with a digestion reagent. 64. The magnetic bead of claim 49, wherein the amine-containing compound further comprises a positively or negatively charged group. 65. The magnetic bead of claim 49, wherein the amine-containing compound is an alkyl or aryl amine group. 66. The magnetic bead of claim 65, wherein the amine-containing compound is pentanediamine, PEI-25K, or PEI-800. 67. The magnetic bead of claim 49, wherein the amine-containing compound is selected from the group consisting of carboxylates, sulfonates and phosphonates. 68. The magnetic bead of claim 49, wherein the amine-containing compound comprises an azide or alkyne group. 69. The magnetic bead of claim 49, wherein the amine-containing compound comprises a metal charged chelator selected from the group consisting of nitrilotriacetic acid (NTA), iminodiacetic acid (IDA) and ethylenediaminetetraacetic acid (EDTA). 70. The magnetic bead of claim 69, wherein the metal is selected from the group consisting of metals such as nickel, iron, cobalt, aluminum, titanium, zirconium, gallium, and any combination thereof. 71. A method for isolating a negatively-charged species from a biological sample, the method comprising: First Named Inventor: RAJA, Erum TP385332WO1 providing a magnetic bead, wherein each magnetic bead comprises iron oxide particles distributed throughout an azlactone bead and a positively-charged group located on the surface of the magnetic bead; combining the magnetic bead with a biological sample comprising a negatively- charged species to form a bead conjugate, wherein the conjugate comprises the negatively-charged species bound to the positively charged group on the bead; and separating the bead conjugate from the biological sample. 72. The method of claim 71, wherein the negatively-charged species is an extracellular vesicle, a nucleic acid, or a virus particle. 73. A method for isolating a positively-charged species from a biological sample, the method comprising: providing a magnetic bead, wherein each magnetic bead comprises iron oxide particles distributed throughout an azlactone bead and a negatively-charged group located on the surface of the magnetic bead; combining the magnetic bead with a biological sample comprising a positively- charged species to form a bead conjugate, wherein the conjugate comprises the positively-charged species bound to the negatively-charged group on the bead; and separating the bead conjugate from the biological sample. 74. A method of removing contaminants from a sample, comprising: provide a sample comprising a liquid matrix and proteins, protein fragments or polypeptides, wherein the liquid matrix comprises contaminants; combine the sample with a population of magnetic beads according to claim 69 or claim 70 in the presence of a binding solution, wherein the proteins, protein fragments or polypeptides bind to the magnetic beads; magnetically separate the population of magnetic beads from the liquid matrix; First Named Inventor: RAJA, Erum TP385332WO1 optionally wash the population of separated magnetic beads with a wash solution to remove any remaining contaminants or nonspecifically bound analytes from the population of magnetic beads; elute the proteins, protein fragments or polypeptides from the separated magnetic beads with an elution solution, to form a solution comprising the proteins, protein fragments or polypeptides; and magnetically separate the population of magnetic beads from the solution comprising the proteins, protein fragments or polypeptides.
52. The magnetic bead of claim 43 or claim 51, wherein the azlactone bead is formed from a vinyl azlactone and a crosslinker selected from a bisacrylamide, agarose, and a vinyl ether. First Named Inventor: RAJA, Erum TP385332WO1 53. The magnetic bead of any one of claims 43-52, wherein: the vinyl azlactone is 4,4-dimethyl-2-vinyloxazol-5(4H)-one; the crosslinker is a bisacrylamide; or the vinyl azlactone is 4,4-dimethyl-2-vinyloxazol-5(4H)-one and the crosslinker is a bisacrylamide. 54. The magnetic bead of claim 52 or claim 53, wherein the crosslinker is methylene bisacrylamide. 55. The magnetic bead of any one of claims 43-54, wherein the magnetic material is Fe3O4 or Fe2O3 or a combination thereof. 56. The magnetic bead of any one of claims 43-55, wherein the bead has a bead size of from 20 to 80 microns. 57. The magnetic bead of claim 56, wherein the bead has a bead size of from 30 to 60 microns. 58. The magnetic bead of any one of claims 56-57, wherein the bead size is a bead size in a non-hydrated state, and the bead has a hydrated bead size that is from 6 to 8 times larger than the bead size in the non-hydrated state. 59. The magnetic bead of any one of claims 43-58, where the magnetic bead is a porous magnetic bead. 60. A population of the magnetic beads according to any one of claims 43- 59, wherein the population exhibits a bead loss of less than about 5% by weight when used in an automated process. First Named Inventor: RAJA, Erum TP385332WO1 61. A kit, comprising a plurality of magnetic beads according to any one of claims 43-59 or claims 64-70. 62. A method of removing contaminants from a sample, comprising: provide a sample comprising a liquid matrix and protein fragments or polypeptides, wherein the liquid matrix comprises contaminants; combine the sample with a population of magnetic beads according to any one of claims 36-45 in the presence of a binding solution, wherein the protein fragments or polypeptides bind to the magnetic beads; magnetically separate the population of magnetic beads from the liquid matrix; optionally wash the population of separated magnetic beads with a wash solution to remove any remaining contaminants from the population of magnetic beads; elute the protein fragments or polypeptides from the separated magnetic beads with an elution solution, to form a solution comprising the protein fragments or polypeptides; and magnetically separate the population of magnetic beads from the solution comprising the protein fragments or polypeptides. 63. A method for protein aggregation capture from a sample, the method comprising: provide a sample comprising a liquid matrix and a plurality of proteins; combine the sample with a population of magnetic beads according to any one of claims 36-45 in the presence of a binding solution, wherein at least a portion of the proteins bind to the magnetic beads to form bound proteins on the magnetic beads; magnetically separate the population of magnetic beads from the liquid matrix; optionally expose the population of magnetic beads with the bound proteins to a reduction reagent and/or an alkylation reagent and then separate the population of magnetic beads from the reduction reagent and/or the alkylation reagent; optionally wash the population of separated magnetic beads; First Named Inventor: RAJA, Erum TP385332WO1 elute the bound proteins from the population of magnetic beads with an elution solution to form a solution comprising unbound proteins; and optionally digest the unbound proteins with a digestion reagent. 64. The magnetic bead of claim 49, wherein the amine-containing compound further comprises a positively or negatively charged group. 65. The magnetic bead of claim 49, wherein the amine-containing compound is an alkyl or aryl amine group. 66. The magnetic bead of claim 65, wherein the amine-containing compound is pentanediamine, PEI-25K, or PEI-800. 67. The magnetic bead of claim 49, wherein the amine-containing compound is selected from the group consisting of carboxylates, sulfonates and phosphonates. 68. The magnetic bead of claim 49, wherein the amine-containing compound comprises an azide or alkyne group. 69. The magnetic bead of claim 49, wherein the amine-containing compound comprises a metal charged chelator selected from the group consisting of nitrilotriacetic acid (NTA), iminodiacetic acid (IDA) and ethylenediaminetetraacetic acid (EDTA). 70. The magnetic bead of claim 69, wherein the metal is selected from the group consisting of metals such as nickel, iron, cobalt, aluminum, titanium, zirconium, gallium, and any combination thereof. 71. A method for isolating a negatively-charged species from a biological sample, the method comprising: First Named Inventor: RAJA, Erum TP385332WO1 providing a magnetic bead, wherein each magnetic bead comprises iron oxide particles distributed throughout an azlactone bead and a positively-charged group located on the surface of the magnetic bead; combining the magnetic bead with a biological sample comprising a negatively- charged species to form a bead conjugate, wherein the conjugate comprises the negatively-charged species bound to the positively charged group on the bead; and separating the bead conjugate from the biological sample. 72. The method of claim 71, wherein the negatively-charged species is an extracellular vesicle, a nucleic acid, or a virus particle. 73. A method for isolating a positively-charged species from a biological sample, the method comprising: providing a magnetic bead, wherein each magnetic bead comprises iron oxide particles distributed throughout an azlactone bead and a negatively-charged group located on the surface of the magnetic bead; combining the magnetic bead with a biological sample comprising a positively- charged species to form a bead conjugate, wherein the conjugate comprises the positively-charged species bound to the negatively-charged group on the bead; and separating the bead conjugate from the biological sample. 74. A method of removing contaminants from a sample, comprising: provide a sample comprising a liquid matrix and proteins, protein fragments or polypeptides, wherein the liquid matrix comprises contaminants; combine the sample with a population of magnetic beads according to claim 69 or claim 70 in the presence of a binding solution, wherein the proteins, protein fragments or polypeptides bind to the magnetic beads; magnetically separate the population of magnetic beads from the liquid matrix; First Named Inventor: RAJA, Erum TP385332WO1 optionally wash the population of separated magnetic beads with a wash solution to remove any remaining contaminants or nonspecifically bound analytes from the population of magnetic beads; elute the proteins, protein fragments or polypeptides from the separated magnetic beads with an elution solution, to form a solution comprising the proteins, protein fragments or polypeptides; and magnetically separate the population of magnetic beads from the solution comprising the proteins, protein fragments or polypeptides.







