Attorney Docket No. 2013237‐0757 RNA THERAPEUTICS WITH REDUCED TOXICITY Technical Field The disclosure provides RNA therapeutics, e.g. RNA molecules for treating or preventing an infection, and methods for reducing inherent toxicity of antigens or cytotoxicity exhibited by certain microbial antigens or immunogenic variants thereof when expressed intracellularly from RNA, as well as compositions comprising RNA therapeutics produced by such methods. The RNA encoding the antigens, immunogenic variants or fragments thereof is formulated and administered in a way that the antigens, variants or fragments are produced by cells of a subject, in particular after intramuscular or intravenous administration of the RNA and that, at the same time, toxicity or cytotoxicity is avoided. Background The use of RNA to deliver foreign genetic information into target cells offers an attractive alternative to DNA. The advantages of RNA include transient expression and non-transforming character. RNA does not require nucleus infiltration for expression and moreover cannot integrate into the host genome, thereby eliminating the risk of oncogenesis. The COVID-19 pandemic has showcased the utility and advantages of RNA technology for vaccination, as out of all COVID-19 vaccines under development, the first two to have received emergency use authorization by the FDA were RNA-based. The biotechnology response to the COVID-19 pandemic has highlighted the speed and flexibility of RNA vaccines, and reveals RNA therapeutics to be a powerful tool to address epidemic outbreaks caused by newly emerging viruses. The relative simplicity of the development process and flexibility of the manufacturing platform can markedly accelerate clinical development. A current limitation of known RNA therapeutics is the usability of known toxic proteins for RNA therapeutics as well as the usability of antigens that are cytotoxic as a result of the intracellular translation from RNA in mammalian cells. Summary The present disclosure provides solutions to overcome toxicity of toxic antigens to allow their usage in RNA therapeutics as well as solutions to overcome cytotoxicity resulting from the intracellular translation of antigens from RNA in mammalian cells. There are three groups of antigens not accessible to RNA therapeutics. The first group involves antigens known to exhibit cytotoxicity when administered in antigen form to patients. In view of the strong adverse effects of those antigens, either locally at an injection site or systemically, administration in the form of either native antigen or RNA encoding such antigen is strongly counterindicated. Since cytotoxic antigens often lead to particularly robust immune responses, the inability of utilizing them for vaccination or treatment severely hampers efforts to generate efficacious vaccines or treatments against their host-organisms. A second group of antigens not accessible to RNA therapeutics are antigens with previously unknown cytotoxic qualities. It has been observed that certain peptides or antigens previously unknown to be cytotoxic start exhibiting cytotoxicity when expressed from RNA intracellularly, in particular in mammalian cells. This observation can be explained by the fact that many antigens produced for pharmaceutical purposes are expressed in heterologous organisms, which may disrupt their natural structure, e.g. the tertinary structure. For instance, many antigen therapeutics are produced in 1 11529421v1 Attorney Docket No. 2013237‐0757 bacteria as inclusion bodies of unsoluble antigen and, later in the production process, are resolubilized using, e.g., denaturing agents and certain physical conditions (pH, temperature, pressure and the like). The resulting structures of antigens produced and treated this way, more often than not, does not fully conform to the structure exhibited by the same antigens upon expression in their native hosts. Such malconformation may mask a cytotoxic quality of those antigens present only in correctly folded antigens. Such antigens are particularly challenging for the development of RNA therapeutics, because the antigens may adopt a correct structure once expressed inside mammalian cells, exhibiting a previously unknown cytotoxicity. Such cytotoxicity, as with the antigens of the first group, raises significant safety concerns and, therefore, excludes these antigens from use in the context of RNA therapeutics. A third group of antigens exhibits malconformation as the consequence of being expressed in the non-native environment of a mammalian cell (e.g. in the context of RNA vaccination). In rare cases, such malconformation may induce cytotoxicity in antigens which are non-cytotoxic in their native, properly folded state. Also such antigens are excluded from use in the context of RNA therapeutics due to safety concerns. Solving the problem of inherent toxicity or cytotoxicity resulting from the translation of a protein, such as e.g. an antigen, in a mammalian cell from RNA broadens the scope of proteins available for RNA therapeutic, e.g. for vaccination and treatment against microbial infections, while avoiding cytotoxicity and providing improved safety. The disclosure provides RNA therapeutics and methods for reducing inherent toxicity or cytotoxicity exhibited by certain proteins, in particular antigens, or immunogenic variants thereof, e.g. microbial antigens of immunogenic variants thereof, when expressed intracellularly from RNA in mammalian cells as well as compositions comprising RNA therapeutics produced by such methods. In one aspect of the invention, reducing inherent toxicity or cytotoxicity exhibited by antigens or immunogenic variants thereof when expressed intracellularly from RNA in mammalian cells in the otherwise toxic or cytotoxic antigens is accomplished by separating the toxic or cytotoxic antigens into antigen fragments (also termed “chunks”). In some embodiments, chunks may be designed such that their borders intersect with certain secondary structures, such as beta-sheets and alpha-helices, disrupting their formation. Furthermore, in some embodiments chunks may be selected such that their borders interfere with the formation of tertiary structures, either by disrupting secondary structures or by otherwise interfering with the formation of bonds between neighboring parts of the amino acid chain. In some embodiments, these chunks can be further designed and optimized, such as avoiding sequences which might give rise to off-target immunity, in particular towards human proteins, and sequences that might affect solubility of the resulting encoded polypeptide, such as transmembrane regions. In some embodiments, the chunks may be arranged in an RNA molecule suitable to allow translation of the polypeptide in a mammalian cell (also termed “string”) such that their sequences are not in the same order as within the full-length sequence of the antigen. This re-ordering of sequences within the polypeptide leads to an at least partial disruption of secondary structures of the antigen, in particular where fragment borders intersect with those secondary structures, effectively removing a large portion of the secondary structure to an entirely different part of the resulting polypeptide. Furthermore, the reordering of sequences leads to an at least partial disruption of tertiary structures, because sequences that were closely associated with each other in the native antigen sequence are set further apart, or because other parts of the reordered polypeptide sterically interfere with the formation of cysteine bridges, hydrogen bonds and van der Waals interactions between certain parts of the amino acid chain three dimensionally associated with each 2 11529421v1 Attorney Docket No. 2013237‐0757 other in the native antigen. The disruption of tertiary structures, in turn, disrupts the formation of quaternary structures, because the changed surface conformation of the polypeptide no longer allows for attachment between protein complex subunits in the correct orientation. Consequently, the native functions of the antigen are disrupted as well, e.g., because the antigen is no longer able to form an enzymatic pocked allowing for entry and/or conversion of its substrate, or because binding specificity to other proteins or intracellular structures is lost due to the structural changes. Alternatively or in addition to the re-arrangement, in some embodiments chunks are combined by stringing them together interspersed by polypeptide linkers. The insertion of polypeptide linkers into the structure of the antigen allows for disruption of secondary, tertiary and quaternary structures much in the same way as the reordering of antigen fragments inside the RNA molecule encoding a polypeptide. The additional sequences may allow additional flexibility in antigen structures which are rigid in the native antigen. Conversely rigid polypeptide linkers may sterically disrupt interactions between sequences that, in the native antigen sequence, are closely spatially arranged. An added benefit of using polypeptide linkers is, that they also function to minimize the risk of creating neoepitopes. It was surprisingly found, that the disruption of the native antigen structures by creating chunks and by the disruptions did not abolish the immunogenic potential or immune response in a mammalian host. In some embodiments, the toxic or cytotoxic antigens to be made available for RNA therapeutics may be microbial antigens or immunogenic variants thereof. In some embodiments, the RNA therapeutic may be directed to induce a T cell response. Protective immunity to many infectious diseases is primarily shaped by T cell responses in most individuals. Also treatment of chronic infections may be T cell response driven. T cell responses are more desirable than B cell responses in many important infectious disease including e.g. infections caused by Mycobacterium tuberculosis (Mtb), human immunodeficiency virus (HIV), hepatitis C virus (HCV), Staphylococcus aureus or for liver- stage malaria infections. Presented here is a platform of RNA design specifically aimed at optimizing T cell responses in RNA therapeutics while, at the same time, reducing toxicity or cytotoxicity exhibited by certain microbial antigens or immunogenic variants thereof. This platform is utilized here to specifically design potent T cell inducing RNA therapeutics to prevent or treat infectious diseases like, e.g. tuberculosis or bacterial infections. The above approaches, individually or in combination, enable the generation of RNA therapeutics that represent a large or even the full range of epitopes from a microbial antigen, while at the same time reducing or even completely removing cytotoxic effects this microbial antigen or certain domains thereof may exhibit in its native form. Thereby, the methods and compositions of the invention enable increasing efficacy and, at the same time, improving safety for the RNA therapeutics technology, broadening the repertoire of antigens available for use in RNA therapeutics and reducing or removing the risk of previously undiscovered cytotoxicity exhibited by microbial antigens or immunogenic variants thereof when expressed intracellularly. In one aspect, the disclosure provides an RNA molecule encoding a polypeptide comprising epitopes of a microbial antigen or an immunogenic variant thereof, which microbial antigen or immunogenic variant thereof is cytotoxic when expressed in mammalian cells from RNA as a full-length sequence, wherein: a) the microbial antigen or immunogenic variant thereof is represented by two or more antigen fragments, 3 11529421v1 Attorney Docket No. 2013237‐0757 b) each antigen fragment comprises one or more epitopes, and c) cytotoxicity in mammalian cells is reduced by arranging the antigen fragments in the polypeptide such that their sequences are not in the same order as within the full-length sequence of the microbial antigen or immunogenic variant thereof, and/or by separating each antigen fragment from other antigen fragments in the polypeptide by a polypeptide linker and/or a protein sequence not part of the microbial antigen or immunogenic variant thereof. In some embodiments of the RNA molecule, antigen fragments comprising epitopes of a cytotoxic domain of the microbial antigen or immunogenic variant consist of an incomplete antigen domain of the antigen. In some embodiments, the incomplete antigen domain has a different secondary, tertiary and/or quaternary structure compared to the same sequence in the context of the full domain. In some embodiments, the incomplete antigen domain is interrupted within a secondary structure selected from an alpha helix or beta sheet. In some embodiments, the incomplete antigen domain is interrupted within a sequence or structure forming an active center. In some embodiments, the incomplete antigen domain is interrupted within a sequence or structure forming a multimerization domain. In some embodiments, the polypeptide has a reduced enzymatic function compared to the microbial antigen or an immunogenic variant thereof. In some embodiments, polypeptide has reduced or changed protein binding characteristics compared to the microbial antigen or an immunogenic variant thereof. In some embodiments, the polypeptide is less cytotoxic than the microbial antigen or immunogenic variant thereof when expressed in mammalian cells by at least 20%, at least 40%, at least 60% or at least 80%. In some embodiments, the incomplete antigen domain is not cytotoxic when expressed in mammalian cells. In some embodiments, the sequences of antigen fragments are partially overlapping. The advantage of a partial overlap between antigen fragments is, that a full representation of epitopes of the antigen can be achieved while at the same time reducing its toxicity or cytotoxicity when expressed in mammalian cells. 4 11529421v1 Attorney Docket No. 2013237‐0757 In some embodiments, the sequences of antigen fragments are overlapping by 12 or less, 8 or less or 4 or less amino acids. In some embodiments, the epitopes comprise B-cell epitopes. In some embodiments, the epitopes comprise T-cell epitopes. In some embodiments, the at least one cytotoxic microbial antigen is selected from the group consisting of the Mycobacterium tuberculosis antigens EspA, PlcA, PlcB, PlcC, PlcD, TlyA, Esat-6 and CFP10, the Pseudomonas aeruginosa antigens ExoS, ExoT, ExoU, ExoY, ExoA, Azurin and ExlA, the Staphylococcus aureus antigens Hla, Hlb, Hld, HlgA, HlgB, LukD and LukE, the Clostridioides difficile antigens TcdA and TcdB, the Bacillus anthracis antigens Lef and Cya, the Corynebacterium diphteriae antigen diphteria toxin and the Vibrio cholerae antigens CtxA and CtxB. In some embodiments: a) the EspA antigen comprises the amino acid sequence of SEQ ID NO: 1 and an immunogenic variant thereof has an amino acid sequence having at least 99%, 98%, 97%, 96%, 95%, 90%, 85%, or 80% identity to the amino acid sequence of SEQ ID NO: 1; b) the PlcA antigen comprises the amino acid sequence of SEQ ID NO: 2 and an immunogenic variant thereof has an amino acid sequence having at least 99%, 98%, 97%, 96%, 95%, 90%, 85%, or 80% identity to the amino acid sequence of SEQ ID NO: 2; c) the PlcB antigen comprises the amino acid sequence of SEQ ID NO: 3 and an immunogenic variant thereof has an amino acid sequence having at least 99%, 98%, 97%, 96%, 95%, 90%, 85%, or 80% identity to the amino acid sequence of SEQ ID NO: 3; d) the PlcC antigen comprises the amino acid sequence of SEQ ID NO: 46 and an immunogenic variant thereof has an amino acid sequence having at least 99%, 98%, 97%, 96%, 95%, 90%, 85%, or 80% identity to the amino acid sequence of SEQ ID NO: 46; e) the PlcD antigen comprises the amino acid sequence of SEQ ID NO: 47 and an immunogenic variant thereof has an amino acid sequence having at least 99%, 98%, 97%, 96%, 95%, 90%, 85%, or 80% identity to the amino acid sequence of SEQ ID NO: 47; f) the TlyA antigen comprises the amino acid sequence of SEQ ID NO: 48 and an immunogenic variant thereof has an amino acid sequence having at least 99%, 98%, 97%, 96%, 95%, 90%, 85%, or 80% identity to the amino acid sequence of SEQ ID NO: 48; g) the ExoS antigen comprises the amino acid sequence of SEQ ID NO: 49 and an immunogenic variant thereof has an amino acid sequence having at least 99%, 98%, 97%, 96%, 95%, 90%, 85%, or 80% identity to the amino acid sequence of SEQ ID NO: 49; h) the ExoT antigen comprises the amino acid sequence of SEQ ID NO: 50 and an immunogenic variant thereof has an amino acid sequence having at least 99%, 98%, 97%, 96%, 95%, 90%, 85%, or 80% identity to the amino acid sequence of SEQ ID NO: 50; 5 11529421v1 Attorney Docket No. 2013237‐0757 i) the ExoU antigen comprises the amino acid sequence of SEQ ID NO: 51 and an immunogenic variant thereof has an amino acid sequence having at least 99%, 98%, 97%, 96%, 95%, 90%, 85%, or 80% identity to the amino acid sequence of SEQ ID NO: 51; j) the ExoY antigen comprises the amino acid sequence of SEQ ID NO: 52 and an immunogenic variant thereof has an amino acid sequence having at least 99%, 98%, 97%, 96%, 95%, 90%, 85%, or 80% identity to the amino acid sequence of SEQ ID NO: 52; k) the ExoA antigen comprises the amino acid sequence of SEQ ID NO: 53 and an immunogenic variant thereof has an amino acid sequence having at least 99%, 98%, 97%, 96%, 95%, 90%, 85%, or 80% identity to the amino acid sequence of SEQ ID NO: 53; l) the Azurin antigen comprises the amino acid sequence of SEQ ID NO: 54 and an immunogenic variant thereof has an amino acid sequence having at least 99%, 98%, 97%, 96%, 95%, 90%, 85%, or 80% identity to the amino acid sequence of SEQ ID NO: 54; m) the ExlA antigen comprises the amino acid sequence of SEQ ID NO: 55 and an immunogenic variant thereof has an amino acid sequence having at least 99%, 98%, 97%, 96%, 95%, 90%, 85%, or 80% identity to the amino acid sequence of SEQ ID NO: 55; n) the Hla antigen comprises the amino acid sequence of SEQ ID NO: 56 and an immunogenic variant thereof has an amino acid sequence having at least 99%, 98%, 97%, 96%, 95%, 90%, 85%, or 80% identity to the amino acid sequence of SEQ ID NO: 56; o) the Hlb antigen comprises the amino acid sequence of SEQ ID NO: 57 and an immunogenic variant thereof has an amino acid sequence having at least 99%, 98%, 97%, 96%, 95%, 90%, 85%, or 80% identity to the amino acid sequence of SEQ ID NO: 57; p) the Hld antigen comprises the amino acid sequence of SEQ ID NO: 58 and an immunogenic variant thereof has an amino acid sequence having at least 99%, 98%, 97%, 96%, 95%, 90%, 85%, or 80% identity to the amino acid sequence of SEQ ID NO: 58; q) the HlgA antigen comprises the amino acid sequence of SEQ ID NO: 59 and an immunogenic variant thereof has an amino acid sequence having at least 99%, 98%, 97%, 96%, 95%, 90%, 85%, or 80% identity to the amino acid sequence of SEQ ID NO: 59; r) the HlgB antigen comprises the amino acid sequence of SEQ ID NO: 60 and an immunogenic variant thereof has an amino acid sequence having at least 99%, 98%, 97%, 96%, 95%, 90%, 85%, or 80% identity to the amino acid sequence of SEQ ID NO: 60; s) the HlgC antigen comprises the amino acid sequence of SEQ ID NO: 61 and an immunogenic variant thereof has an amino acid sequence having at least 99%, 98%, 97%, 96%, 95%, 90%, 85%, or 80% identity to the amino acid sequence of SEQ ID NO: 61; t) the LukD antigen comprises the amino acid sequence of SEQ ID NO: 62 and an immunogenic variant thereof has an amino acid sequence having at least 99%, 98%, 97%, 96%, 95%, 90%, 85%, or 80% identity to the amino acid sequence of SEQ ID NO: 62; 6 11529421v1 Attorney Docket No. 2013237‐0757 u) the LukE antigen comprises the amino acid sequence of SEQ ID NO: 63 and an immunogenic variant thereof has an amino acid sequence having at least 99%, 98%, 97%, 96%, 95%, 90%, 85%, or 80% identity to the amino acid sequence of SEQ ID NO: 63; v) the TcdA antigen comprises the amino acid sequence of SEQ ID NO: 64 and an immunogenic variant thereof has an amino acid sequence having at least 99%, 98%, 97%, 96%, 95%, 90%, 85%, or 80% identity to the amino acid sequence of SEQ ID NO: 64; w) the TcdB antigen comprises the amino acid sequence of SEQ ID NO: 65 and an immunogenic variant thereof has an amino acid sequence having at least 99%, 98%, 97%, 96%, 95%, 90%, 85%, or 80% identity to the amino acid sequence of SEQ ID NO: 65; x) the Lef antigen comprises the amino acid sequence of SEQ ID NO: 66 and an immunogenic variant thereof has an amino acid sequence having at least 99%, 98%, 97%, 96%, 95%, 90%, 85%, or 80% identity to the amino acid sequence of SEQ ID NO: 66; y) the Cya antigen comprises the amino acid sequence of SEQ ID NO: 67 and an immunogenic variant thereof has an amino acid sequence having at least 99%, 98%, 97%, 96%, 95%, 90%, 85%, or 80% identity to the amino acid sequence of SEQ ID NO: 67; z) the diphteria toxin antigen comprises the amino acid sequence of SEQ ID NO: 68 and an immunogenic variant thereof has an amino acid sequence having at least 99%, 98%, 97%, 96%, 95%, 90%, 85%, or 80% identity to the amino acid sequence of SEQ ID NO: 68; aa) the CtxA antigen comprises the amino acid sequence of SEQ ID NO: 69 and an immunogenic variant thereof has an amino acid sequence having at least 99%, 98%, 97%, 96%, 95%, 90%, 85%, or 80% identity to the amino acid sequence of SEQ ID NO: 69; bb) the CtxB antigen comprises the amino acid sequence of SEQ ID NO: 70 and an immunogenic variant thereof has an amino acid sequence having at least 99%, 98%, 97%, 96%, 95%, 90%, 85%, or 80% identity to the amino acid sequence of SEQ ID NO: 70; cc) the Esat-6 antigen comprises the amino acid sequence of SEQ ID NO: 71 and an immunogenic variant thereof has an amino acid sequence having at least 99%, 98%, 97%, 96%, 95%, 90%, 85%, or 80% identity to the amino acid sequence of SEQ ID NO: 71; and dd) the CFP10 antigen comprises the amino acid sequence of SEQ ID NO: 72 and an immunogenic variant thereof has an amino acid sequence having at least 99%, 98%, 97%, 96%, 95%, 90%, 85%, or 80% identity to the amino acid sequence of SEQ ID NO: 72. In some embodiments, the microbial antigen or immunogenic variant thereof is represented by three or more, four or more, five or more, six or more, seven or more, eight or more, nine or more or ten or more antigen fragments. In some embodiments, the antigen fragments are arranged in the polypeptide such that their sequences are not in the same order as within the full-length sequence of the microbial antigen or immunogenic variant thereof. In some embodiments, one or more antigen fragment is separated from other antigen fragments in the polypeptide by a polypeptide linker and/or a protein sequence not part of the microbial antigen or immunogenic variant thereof. 7 11529421v1 Attorney Docket No. 2013237‐0757 In some embodiments, the two or more antigen fragments of the microbial antigen or immunogenic variant thereof that is cytotoxic when expressed in mammalian cells from RNA as a full-length sequence are combined in the polypeptide with a) one or more antigen fragments of representing one or more different microbial antigens or immunogenic variants thereof that are cytotoxic when expressed in mammalian cells from RNA as a full-length sequence and/or b) one or more antigen fragments and/or full-length antigens representing one or more microbial antigens or immunogenic variants thereof that are not cytotoxic when expressed in mammalian cells from RNA as a full-length sequence. In some embodiments: a) a first antigen fragment of EspA comprises the amino acid sequence of positions 1 to 166 of SEQ ID NO: 1 or an amino acid sequence having at least 96%, 92%, 88%, 84% or 80% identity to the amino acid sequence of positions 1 to 166 of SEQ ID NO: 1; b) a second antigen fragment of EspA comprises the amino acid sequence of positions 177 to 280 of SEQ ID NO: 1 or an amino acid sequence having at least 96%, 92%, 88%, 84% or 80% identity to the amino acid sequence of positions 177 to 280 of SEQ ID NO: 1; and c) a third antigen fragment of EspA comprises the amino acid sequence of positions 291 to 392 of SEQ ID NO: 1 or an amino acid sequence having at least 96%, 92%, 88%, 84% or 80% identity to the amino acid sequence of positions 135 to 166 of SEQ ID NO: 1. In some embodiments, antigen fragments in the polypeptide are arranged in the following order: first antigen fragment of EspA – linker – third antigen fragment of EspA – linker – second antigen fragment of EspA. In some embodiments: d) a first antigen fragment of EspA comprises the amino acid sequence of positions 1 to 60 of SEQ ID NO: 1 or an amino acid sequence having at least 96%, 92%, 88%, 84% or 80% identity to the amino acid sequence of positions 1 to 60 of SEQ ID NO: 1; e) a second antigen fragment of EspA comprises the amino acid sequence of positions 62 to 131 of SEQ ID NO: 1 or an amino acid sequence having at least 96%, 92%, 88%, 84% or 80% identity to the amino acid sequence of positions 62 to 131 of SEQ ID NO: 1; f) a third antigen fragment of EspA comprises the amino acid sequence of positions 135 to 166 of SEQ ID NO: 1 or an amino acid sequence having at least 96%, 92%, 88%, 84% or 80% identity to the amino acid sequence of positions 135 to 166 of SEQ ID NO: 1; g) a fourth antigen fragment of EspA comprises the amino acid sequence of positions 177 to 239 of SEQ ID NO: 1 or an amino acid sequence having at least 96%, 92%, 88%, 84% or 80% identity to the amino acid sequence of positions 177 to 239 of SEQ ID NO: 1; 8 11529421v1 Attorney Docket No. 2013237‐0757 h) a fifth antigen fragment of EspA comprises the amino acid sequence of positions 241 to 279 of SEQ ID NO: 1 or an amino acid sequence having at least 96%, 92%, 88%, 84% or 80% identity to the amino acid sequence of positions 241 to 279 of SEQ ID NO: 1; i) a sixth antigen fragment of EspA comprises the amino acid sequence of positions 291 to 341 of SEQ ID NO: 1 or an amino acid sequence having at least 96%, 92%, 88%, 84% or 80% identity to the amino acid sequence of positions 291 to 341 of SEQ ID NO: 1; and j) a seventh antigen fragment of EspA comprises the amino acid sequence of positions 343 to 392 of SEQ ID NO: 1 or an amino acid sequence having at least 96%, 92%, 88%, 84% or 80% identity to the amino acid sequence of positions 343 to 392 of SEQ ID NO: 1. In some embodiments, the antigen fragments in the polypeptide are arranged in the following order: seventh antigen fragment of EspA – linker – first antigen fragment of EspA – linker – third antigen fragment of EspA – linker - fifth antigen fragment of EspA – linker – second antigen fragment of EspA – linker – fourth antigen fragment of EspA – linker – sixth antigen fragment of EspA. In some embodiments, the polypeptide comprises one, two, three, four, five, six or seven of the antigen fragments. In some embodiments, the polypeptide additionally comprises one or more further full-length antigens or antigen fragments. In some embodiments, one or more of the polypeptide linkers comprises one or more glycine and/or one or more serine amino acid. In some embodiments, one or more of the polypeptide linkers is at least 1, at least 5 or at least 10 amino acids in length. In some embodiments, one or more of the polypeptide linkers has the amino acid sequence of SEQ ID NO: 43. In some embodiments, the polypeptide comprises a signal peptide at its N-terminus. In some embodiments, the polypeptide comprises a heterologous human, bacterial or viral signal peptide at its N- terminus. In some embodiments, the heterologous signal peptide comprises a secretory signal. In some embodiments, the heterologous signal peptide is functional in mammalian cells. In some embodiments, the heterologous signal peptide comprises an amino acid sequence selected from the group of SEQ ID NOs: 4 to 24, amino acid sequences having at least 96%, 92%, 88%, 84% or 80% identity to SEQ ID NOs: 4 9 11529421v1 Attorney Docket No. 2013237‐0757 to 24, amino acid sequences encoded by a nucleotide sequence selected from the group of SEQ ID NOs: 25 to 38 and amino acid sequences having at least 96%, 92%, 88%, 84% or 80% identity to amino acid sequences encoded by a nucleotide sequence selected from the group of SEQ ID NOs: 25 to 38. In some embodiments, the heterologous signal peptide is a viral signal peptide. In some embodiments, the heterologous signal peptide is a HSV-1 glycoprotein D signal peptide. In some embodiments, the polypeptide comprises a heterologous trafficking domain at its C-terminus. In some embodiments, the heterologous trafficking domain is an MHC class I trafficking domain. In some embodiments, the MHC class I trafficking domain comprises the amino acid sequence of SEQ ID NO: 39 or an amino acid sequence having at least 98%, 96%, 90%, or 80% identity to the amino acid sequence of SEQ ID NO: 39. In some embodiments, the RNA molecule comprises a 5’ cap. In some embodiments, the 5’ cap comprises a cap1 structure. In some embodiments, the 5’-cap comprises m
27,3’-OGppp(m
12’-O)ApG. In some embodiments, the RNA molecule comprises a 5’-UTR. In some embodiments, the 5’-UTR comprises a modified human alpha-globin 5’-UTR. In some embodiments, the 5’-UTR comprises the nucleotide sequence of SEQ ID NO: 40, or a nucleotide sequence having at least 99%, 98%, 97%, 96%, 95%, 90%, 85%, or 80% identity to the nucleotide sequence of SEQ ID NO: 40. In some embodiments, the RNA comprises a 3’-UTR. In some embodiments, the 3’-UTR comprises a first sequence from the amino terminal enhancer of split (AES) messenger RNA and a second sequence from the mitochondrial encoded 12S ribosomal RNA. In some embodiments, the 3’-UTR comprises the nucleotide sequence of SEQ ID NO: 41, or a nucleotide sequence having at least 99%, 98%, 97%, 96%, 95%, 90%, 85%, or 80% identity to the nucleotide sequence of SEQ ID NO: 41. In some embodiments, the RNA molecule comprises a polyA sequence. In some embodiments, the polyA sequence is an interrupted sequence of A nucleotides. 10 11529421v1 Attorney Docket No. 2013237‐0757 In some embodiments, the polyA sequence comprises 30 adenine nucleotides followed by 70 adenine nucleotides, wherein the 30 adenine nucleotides and 70 adenine nucleotides are separated by a nucleotide linker sequence of 10 nucleotides. In some embodiments, the polyA sequence comprises the nucleotide sequence of SEQ ID NO: 42, or a nucleotide sequence having at least 99%, 98%, 97%, 96%, 95%, 90%, 85%, or 80% identity to the nucleotide sequence of SEQ ID NO: 42. In some embodiments, the RNA molecule comprises a 5’-cap, a 5’-UTR, a 3’-UTR and a polyA sequence. In some embodiments, the RNA molecule comprises modified nucleotides, nucleosides or nucleobases. In some embodiments, the RNA molecule comprises modified uridines. In some embodiments, the RNA molecule comprises modified uridines in place of all uridines. In some embodiments, the modified uridines are N1-methyl-pseudouridine. In some embodiments, the coding sequence of the RNA molecule is codon-optimized and/or is characterized in that its G/C content is increased compared to the parental sequence. In one aspect, the disclosure provides a method for generating an RNA molecule disclosed herein, including the steps of a) identifying two or more antigen fragments comprising one or more epitopes of a microbial antigen or immunogenic variant thereof, said microbial antigen or immunogenic variant thereof being cytotoxic when expressed in mammalian cells from RNA as a full-length sequence; b) arranging RNA sequences encoding the two or more antigen fragments such that the antigen fragments in a polypeptide encoded by the RNA molecule are arranged such that their sequences are not in the same order as within the full-length sequence of the microbial antigen or immunogenic variant thereof, and/or each antigen fragment is separated from other antigen fragments in the polypeptide by a polypeptide linker and/or a protein sequence not part of the microbial antigen or immunogenic variant thereof, thereby reducing cytotoxicity in mammalian cells. In some embodiments of the method, the antigen fragments consist of an incomplete antigen domain of the antigen. In some embodiments, the incomplete antigen domain causes a different secondary, tertiary and/or quaternary structure compared to the same sequence in the context of the full domain. In some embodiments, the incomplete antigen domain causes an interruption within a secondary structure selected from an alpha helix or beta sheet. 11 11529421v1 Attorney Docket No. 2013237‐0757 In some embodiments, the incomplete antigen domain is interrupted within a sequence or structure forming an active center. In some embodiments, the incomplete antigen domain is interrupted within a sequence or structure forming a multimerization domain. In some embodiments, the polypeptide encoded by the RNA molecule has reduced enzymatic function compared to the full-length microbial antigen or an immunogenic variant thereof. In some embodiments, the polypeptide encoded by the RNA molecule has reduced or changed protein binding characteristics compared to the full-length microbial antigen or an immunogenic variant thereof. In one aspect, the disclosure provides a polypeptide encoded by the RNA molecule disclosed herein. In one aspect, the disclosure provides a DNA molecule encoding the RNA molecule disclosed herein. In one aspect, the disclosure provides a pharmaceutical composition comprising one or more RNA molecules disclosed herein. In some embodiments of the pharmaceutical composition, the one or more RNA molecules are formulated in a lipid formulation, such as in lipid nanoparticles or liposomes. In some embodiments, the lipid formulation comprises each of: a) a cationically ionizable lipid; b) a steroid; c) a neutral lipid; and d) a polymer-conjugated lipid. In some embodiments, the cationically ionizable lipid is present in a concentration ranging from about 40 to about 60 mol percent of the total lipids. In some embodiments, the steroid is present in a concentration ranging from about 30 to about 50 mol percent of the total lipids. In some embodiments, the neutral lipid is present in a concentration ranging from about 5 to about 15 mol percent of the total lipids. In some embodiments, the polymer-conjugated lipid is present in a concentration ranging from about 1 to about 10 mol percent of the total lipids. 12 11529421v1 Attorney Docket No. 2013237‐0757 In some embodiments, the cationically ionizable lipid is within a range of about 40 to about 60 mole percent, the steroid is within a range of about 30 to about 50 mole percent, the neutral lipid is within a range of about 5 to about 15 mole percent, and the polymer-conjugated lipid is within a range of about 1 to about 10 mole percent. In some embodiments, the cationically ionizable lipid comprises ((4-hydroxybutyl)azanediyl)bis(hexane-6,1-diyl)bis(2- hexyldecanoate). In some embodiments, the steroid comprises cholesterol. In some embodiments, the neutral lipid comprises a phospholipid. In some embodiments, the phospholipid comprises distearoylphosphatidylcholine (DSPC). In some embodiments, the polymer-conjugated lipid comprises a polyethylene glycol (PEG)-lipid. In some embodiments, the PEG-lipid comprises 2-[(polyethylene glycol)-2000]-N,N-ditetradecylacetamide. In some embodiments, the lipid formulation comprises: (a) ((4-hydroxybutyl)azanediyl)bis(hexane-6,1-diyl)bis(2-hexyldecanoate); (b) cholesterol; (c) distearoylphosphatidylcholine (DSPC); and (d) 2-[(polyethylene glycol)-2000]-N,N-ditetradecylacetamide. In some embodiments, ((4-hydroxybutyl)azanediyl)bis(hexane-6,1-diyl)bis(2-hexyldecanoate) is within a range of about 40 to about 60 mole percent, cholesterol is within a range of about 30 to about 50 mole percent, distearoylphosphatidylcholine (DSPC) is within a range of about 5 to about 15 mole percent, and 2-[(polyethylene glycol)-2000]-N,N-ditetradecylacetamide is within a range of about 1 to about 10 mole percent. In some embodiments, the pharmaceutical composition further comprises one or more pharmaceutically acceptable carriers, diluents and/or excipients. In some embodiments, the one or more RNA molecules are in a liquid formulation. In some embodiments, the one or more RNA molecules are in a frozen formulation. In some embodiments, the one or more RNA molecules are in a lyophilized formulation. In some embodiments, the one or more RNA molecules are formulated for injection. 13 11529421v1 Attorney Docket No. 2013237‐0757 In some embodiments, the one or more RNA molecules are formulated for intramuscular administration. In some embodiments, the pharmaceutical composition is formulated for administration in human. In one aspect, the disclosure provides a kit comprising one or more pharmaceutical compositions disclosed herein. In some embodiments of the kit, two or more pharmaceutical compositions comprising the same or different RNA molecules are in separate vials. In some embodiments, the kit further comprises instructions for use of the one or more pharmaceutical composition for treating or preventing an infection. In one aspect, the disclosure provides an RNA molecule, polypeptide, DNA molecule, pharmaceutical composition or kit disclosed herein for use as a medicament. In some embodiments of the RNA molecule, polypeptide, DNA molecule, pharmaceutical composition or kit for use, the use comprises a therapeutic or prophylactic treatment of a disease or disorder in a subject. In some embodiments, the use comprises the use as a vaccine against a disease or disorder in a subject. In some embodiments, the subject is a human infected with the disease or disorder or in danger of contracting the disease or disorder. In one aspect, the disclosure provides an RNA molecule, polypeptide, DNA molecule, pharmaceutical composition or kit disclosed herein for use in treating or preventing a disease in a subject, wherein a) the RNA encodes an antigen of Mycobacterium tuberculosis and the disease to be treated or prevented is tuberculosis, b) the RNA encodes an antigen of Pseudomonas aeruginosa and the disease to be treated or prevented is an infection with Pseudomonas aeruginosa, c) the RNA encodes an antigen of Staphylococcus aureus and the disease to be treated or prevented is an infection with Staphylococcus aureus, d) the RNA encodes an antigen of Clostridioides difficile and the disease to be treated or prevented is an infection with Clostridioides difficile, e) the RNA encodes an antigen of Bacillus anthracis and the disease to be treated or prevented is an infection with Bacillus anthracis, f) the RNA encodes an antigen of Corynebacterium diphteriae and the disease to be treated or prevented is an infection with Corynebacterium diphteriae, and/or g) the RNA encodes an antigen of Vibrio cholerae and the disease to be treated or prevented is an infection with Vibrio cholerae. 14 11529421v1 Attorney Docket No. 2013237‐0757 In some embodiments of the RNA molecule, polypeptide, pharmaceutical composition or kit, the subject is a human suffering from tuberculosis or in danger of contracting a disease selected from tuberculosis or an infection with Pseudomonas aeruginosa, Staphylococcus aureus, Clostridioides difficile, Bacillus anthracis, Corynebacterium diphteriae or Vibrio cholerae. In some embodiments, the use is as a vaccine for preventing tuberculosis or an infection with Pseudomonas aeruginosa, Staphylococcus aureus, Clostridioides difficile, Bacillus anthracis, Corynebacterium diphteriae or Vibrio cholerae. In one aspect, the disclosure provides a use of the RNA molecule, polypeptide, DNA molecule, pharmaceutical composition or kit disclosed herein for the manufacture of a medicament for preventing or treating a disease in a subject, wherein a) the RNA encodes an antigen of Mycobacterium tuberculosis and the disease to be treated or prevented is tuberculosis, b) the RNA encodes an antigen of Pseudomonas aeruginosa and the disease to be treated or prevented is an infection with Pseudomonas aeruginosa, c) the RNA encodes an antigen of Staphylococcus aureus and the disease to be treated or prevented is an infection with Staphylococcus aureus, d) the RNA encodes an antigen of Clostridioides difficile and the disease to be treated or prevented is an infection with Clostridioides difficile, e) the RNA encodes an antigen of Bacillus anthracis and the disease to be treated or prevented is an infection with Bacillus anthracis, f) the RNA encodes an antigen of Corynebacterium diphteriae and the disease to be treated or prevented is an infection with Corynebacterium diphteriae, and/or g) the RNA encodes an antigen of Vibrio cholerae and the disease to be treated or prevented is an infection with Vibrio cholerae. In one aspect, the disclosure provides a method for reducing the cytotoxicity of a microbial antigen or immunogenic variant thereof, which is cytotoxic when expressed in mammalian cells from RNA as a full-length sequence, the method comprising the steps of a) identifying two or more antigen fragments comprising one or more epitopes of the microbial antigen; b) arranging RNA sequences encoding the two or more antigen fragments such that the antigen fragments in a polypeptide encoded by an RNA molecule are arranged such that their sequences are not in the same order as within the full-length sequence of the microbial antigen or immunogenic variant thereof, and/or each antigen fragment is separated from other antigen fragments in the polypeptide by a polypeptide linker and/or a protein sequence not part of the microbial antigen or immunogenic variant thereof. In some embodiments of the method, the antigen fragments consist of an incomplete antigen domain of the antigen. 15 11529421v1 Attorney Docket No. 2013237‐0757 In some embodiments, the incomplete antigen domain causes a different secondary, tertiary and/or quaternary structure compared to the same sequence in the context of the full domain. In some embodiments, the incomplete antigen domain causes an interruption within a secondary structure selected from an alpha helix or beta sheet. In some embodiments, the incomplete antigen domain is interrupted within a sequence or structure forming an active center. In some embodiments, the incomplete antigen domain is interrupted within a sequence or structure forming a multimerization domain. In some embodiments, the polypeptide encoded by the RNA molecule has reduced enzymatic function compared to the full-length microbial antigen or an immunogenic variant thereof. In some embodiments, the polypeptide encoded by the RNA molecule has reduced or changed protein binding characteristics compared to the full-length microbial antigen or an immunogenic variant thereof. In one aspect, the disclosure provides a method of vaccinating a subject comprising administering the RNA molecule, polypeptide, DNA molecule, pharmaceutical composition or kit disclosed herein to the subject. In some embodiments of the method: a) the RNA encodes an antigen of Mycobacterium tuberculosis and the vaccination is against tuberculosis, b) the RNA encodes an antigen of Pseudomonas aeruginosa and the vaccination is against an infection with Pseudomonas aeruginosa, c) the RNA encodes an antigen of Staphylococcus aureus and the vaccination is against an infection with Staphylococcus aureus, d) the RNA encodes an antigen of Clostridioides difficile and the vaccination is against an infection with Clostridioides difficile, e) the RNA encodes an antigen of Bacillus anthracis and the vaccination is against an infection with Bacillus anthracis, f) the RNA encodes an antigen of Corynebacterium diphteriae and the vaccination is against an infection with Corynebacterium diphteriae, and/or g) the RNA encodes an antigen of Vibrio cholerae and the vaccination is against an infection with Vibrio cholerae. In some embodiments, wherein administration is by intramuscular administration. 16 11529421v1 Attorney Docket No. 2013237‐0757 In some embodiments, the method comprises administering to the subject at least one dose of the RNA molecule, polypeptide or pharmaceutical composition. In some embodiments, the method comprises administering to the subject at least two doses of the RNA molecule, polypeptide or pharmaceutical composition. In some embodiments, an amount of the RNA molecule of at least 10 µg per dose is administered. In some embodiments, the subject is a human. In some embodiments of the RNA molecule, polypeptide, pharmaceutical composition or kit for use, the use or the method for vaccination disclosed herein, the tuberculosis is caused by an infection with a Mycobacterium. In some embodiments, the Mycobacterium is selected from the group of Mycobacterium tuberculosis, Mycobacterium bovis, Mycobacterium caprae, Mycobacterium orygis, Mycobacterium africanum, Mycobacterium microti, Mycobacterium canetti and Mycobacterium pinnipedii. In some embodiments, the Mycobacterium is Mycobacterium tuberculosis. 17 11529421v1 Attorney Docket No. 2013237‐0757 Brief description of the Figures Figure 1: Initial finding of cytotoxicity. Reduced confluency of HEK293T cells after transfection with EspA mRNA constructs. HEK293T cells were transfected with Ribojuice using 1 µg mRNA encoding EspA or control protein 1. As a negative control, cells were left untreated. At 18 h post transfection, cells were inspected visually by bright field microscopy. Figure 2: Initial finding of cytotoxicity. EspA mRNA-construct is not expressed in HEK293T cells 18h after transfection. HEK293T cells were transfected with Ribojuice using 1 µg mRNA encoding EspA or control protein 1 (each fused to a N-terminal signal sequence followed by a FLAG-tag for detection). As a negative control, cells were left untreated. At 18 h post transfection, cells were lysed. Lysate was subject to SDS-PAGE (8-16% polyacrylamide gel) and western blot using a FLAG-tag-specific monoclonal antibody. Staining with an alpha-Tubulin-specific (Tub) monoclonal antibody served as loading control. Figure 3: EspA mRNA-construct expression is detected early after transfection. HEK293T cells were transfected with Ribojuice using 25 ng, 100 ng, 250 ng or 1 µg mRNA encoding EspA or control protein 2 (each fused to a N-terminal signal sequence followed by a FLAG-tag for detection). Non-transfected cells served as a negative control. At 6 or 24 h post transfection, cells were lysed. Lysate was subject to SDS-PAGE (8-16% polyacrylamide gel) and western blot using a FLAG-tag specific monoclonal antibody. Staining with an alpha-Tubulin-specific monoclonal antibody served as loading control. Figure 4: Cytotoxicity as imaged by the Incucyte®. HEK293T cells were transfected with Ribojuice using 25 ng, 100 ng, 250 ng or 1 µg mRNA encoding EspA or control protein 2 incubated in the Incucyte® in presence of Cytotox-dye to stain dead cells for 24h. Non-transfected cells served as negative control. Cell confluency and Cytotox-positivity was detected at 2-hour intervals. (A) Exemplary overlay of bright field and fluorescent microscopy (Cytotox Red Dye) at 4h post transfection with 250 ng RNA. White bar = 400 μm, n = 4 pictures per well. (B) Normalized fold-change in the percentage of confluency for all samples over 24h post transfection (C) Normalized fold-change in cytotoxic intensity for all samples over 24h post transfection. Figure 5: EspA antigen chunks were selected based on bioinformatic analysis. Bioinformatic analysis was performed to map regions with (A) human proteome 8AA shortmer overlap (B, C, D) MHC-I neonmhc predicted epitope density T cell epitopes or accumulative number of start/end sites of epitope:HLA-allele pairs and (E, F, G) MHC-II neonmhc predicted epitope density T cell epitopes (neonmhc1 database) or accumulative number of start/end sites of epitope:HLA-allele pairs. X-axis: amino acid indices of target protein. (B, C, D, E, F, G) y-axis: HLA-allele pairs overlapping with amino acid position indicated by x-axis. Boxed regions indicate selected chunks of target protein for vaccine design (optimized to cover high predicted epitope number; include start/end site of epitopes; avoid human proteome overlaps.). This analysis, paired with structure-guided design, allowed EspA regions to be selected for inclusion in T cell strings. Figure 6: EspA construct design. Constructs were generated that contain different fragments or shuffled versions of the wild type, full length EspA (EspA_FL), EspA_A, EspA_B, EspA_C, EspA_AB, and EspA_BC represent smaller fragments. Negative controls are control protein 2, which is of the same length as EspA, and the slightly smaller control protein 3. All constructs are N-terminally fused to a SP followed by a FLAG-tag. 18 11529421v1 Attorney Docket No. 2013237‐0757 Figure 7: Assessment of cell viability and expression of the FLAG-tagged EspA constructs using flow cytometry. HEK293T cells were transfected with 250 ng RNA encoding the respective EspA constructs (EspA_FL, EspA_A, EspA_B, EspA_C, EspA_AB, EspA_BC) or control protein 2 and incubated for 4h. As negative control served non-transfected cells. Cells were stained for viability with a fixable viability dye and for presence of the antigen with a FLAG-tag specific fluorophore-conjugated monoclonal antibody. (A) Gating strategy. (B) Quadrant plots showing dead cells (eFluor 780+) and expression of the transfected FLAG-tagged antigen (BV421+). (C) Left Bar graphs of the percentage of dead cells (viability dye positive). Right Percentage of FLAG-positive cells. Statistical analysis: 1 = Mann-Whitney test, 2 = Kruskal- Wallis test with the bold bar indicating the control sample used for multiple comparison. * = p<0.05, ** = p<0.01, ns = p≥0.05. Bars represent mean ± SD, n = 6 measurements per sample, with open circles indicating one experiment and closed circles indicating the other. Abbreviations: FSC: forward-scatter, FL: Full-length; ns: non-significant; SSC: side-scatter. Figure 8: Assessment of confluency and viability of HEK293T cells via Incucyte® after transfection of EspA constructs. HEK293T cells were transfected with 250 ng of RNA encoding the respective EspA constructs (EspA_FL, EspA_A, EspA_B, EspA_C, EspA_AB, EspA_BC) or control proteins 2 and 3. As negative control served non-transfected cells. Cells were incubated in the Incucyte® in presence of Cytotox-dye to stain dead cells for 24h. (A) Normalized fold- change in Cytotox-dye intensity (RCU) at 4h post transfection. (B) Confluency at 8h post transfection. Baseline (dashed line) calibrated at y = 1. Statistical analysis 1 = Mann-Whitney test, 2 = Kruskal-Wallis test with the bold bar indicating the control sample used for multiple comparison. * = p<0.05, ** = p<0.01, *** = p<0.001, **** = p<0.0001, ns = p≥0.05. Bars represent mean ± SD, dots represent nine images taken per well at every timepoint. Abbreviations: ns: non-significant; RCU: Red calibrated units. Figure 9: Structure-guided T-cell string design to disrupt toxicity of EspA. A) To visualize the domain architecture of EspA, AlphaFold2 was used to generate a structural model. The N- and C-termini are shown in “sphere” representation. The EspA protein sequence was divided into subdomains containing three – EspA_3-1 (white), EspA_3-2 (light grey), EspA_3-3 (dark grey) – or seven regions in a manner that disrupted tertiary and secondary structure elements and function, respectively. Areas of human overlap (black) were avoided when selecting EspA chunks for inclusion in the T cell string. B) Adjacent EspA regions were reshuffled in the T cell string designs (EspA string 1, EspA string 2) to ensure that EspA chunks could not properly assemble into a functional protein during co-translation folding. It was hypothesized that this strategy would reduce toxicity and paired with the MITD and signal peptide (SP), increase protein turnover and generation of immunogenic T-cell epitopes. C) Exemplary EspA string. D) Exemplary EspA string 2. Figure 10: Mass-spectrometry analysis to determine expression. Expression of the 7-part string (but not the 3-part string) is clearly detected by MS. RNA drug substance were transfected into HEK293T cells in the presence or absence of proteasome inhibitor MG132. Following lysis, total protein content was normalized, cysteines were reduced and alkylated, and proteins were subjected to tryptic digestion. The samples were analyzed by targeted and discovery MS to look for tryptic peptides derived uniquely from the transfected RNA strings. Evidence of EspA string 2 expression was observed only in the presence of MG132, indicating that the encoded proteins is likely rapidly degraded following translation. Abbreviations: UT: Untreated. Figure 11: Processing and presentation of predicted HLA-epitopes. RNA drug substance were transfected into A375 cells stably expressing an affinity-tagged version of either HLA-B*07:02 or HLA-A*02:01. Following lysis, tagged HLA molecules were labeled with biotin and enriched on NeutrAvidin beads. Following washing, peptides were eluted from HLA complexes using acid and filtered through molecular weight cutoff filters. Cysteines were reduced and alkylated, 19 11529421v1 Attorney Docket No. 2013237‐0757 and peptides were cleaned up by C18 desalting. Heavy isotope-labeled synthetic peptides were spiked into each sample, and targeted and discovery MS was performed to look for epitope peptides derived uniquely from transfected mRNA constructs. Six Unique Epitopes Span EspA String 2 (7 parts). Six different epitopes were observed by targeted mass spectrometry from the EspA String 2. All epitopes are confirmed to bind B*07:02 based on the mono-allelic pulldown. Figure 12: Assessment of viability and confluency of HEK293T cells via Incucyte® after transfection of EspA constructs and strings. HEK293T cells were transfected with 250 ng of RNA encoding the respective EspA constructs (EspA_FL, EspA_A, EspA_B, EspA_C), EspA strings 1 and 2 or control protein 2. As negative control served non-transfected cells. Cells were incubated in the Incucyte® in presence of Cytotox-dye to stain dead cells for 24h. (A) Normalized fold- change in cytotoxic intensity (RCU) at 4h post transfection. (B) Fold change in confluency at 8h post transfection. Baseline (dashed line) calibrated at y = 1. Statistical analysis 1 = Mann-Whitney test, 2 = Kruskal-Wallis test with the bold bar indicating the control sample used for multiple comparison. * = p<0.05, ** = p<0.01, *** = p<0.001, **** = p<0.0001, ns = p≥0.05. Data has been normalized to t = 0. No datapoints available at 13 and 14h due to imaging error. Abbreviations: RCU: Red calibrated units. Figure 13: Assessment of cellular responses by splenocytes from buffer-injected animals. Five mice were injected with a buffer control (NaCl). Splenocytes were isolated and pooled. Cells were stimulated with a positive control (Concanavalin A, ConA), negative control (unspecific peptide, TRP1), or one of the seven peptide pools representative of the fragments used in the strings EspA_7.1 - EspA_7-7. Cytokine secretion (IFN-γ, IL-2, and TNF-α) was detected by a FluoroSpot assay as spot forming units (SFU). Bars represent mean values ± standard deviation; dots represent technical triplicates. Figure 14: Assessment of cellular responses by splenocytes from animals immunized with LNP-RNA encoding EspA string 1 or EspA string 2. Five mice per group were immunized with LNP-RNA encoding EspA string 1 or EspA string 2. Splenocytes were isolated and stimulated with a positive control (Concanavalin A, ConA), negative control (unspecific peptide, TRP1), or one of the seven peptide pools representative of the fragments used in the strings EspA_7.1 to EspA_7-7. Cytokine secretion (IFN-γ, IL-2, and TNF-α) was detected by a FluoroSpot assay as spot forming units (SFU). Bars represent mean values ± standard deviation; dots represent values from individual animals. Figure 15: Assessment of CD4+ and CD8+ T-cell responses from animals immunized with LNP-RNA encoding EspA string 1 or EspA string 2. Five mice per group were immunized with LNP-RNA encoding EspA string 1 or EspA string 2. Splenocytes were isolated, pooled and CD4+ and CD8+ T cells were separated by magnetic-activated cell sorting. T cells were stimulated with a positive control (Concanavalin A, ConA), negative control (unspecific peptide, TRP1), or one of the seven peptide pools representative of the fragments used in the strings EspA_7.1 to EspA_7-7 in presence of autologous bone marrow-derived dendritic cells. Cytokine secretion (IFN-γ, IL-2, and TNF-α) was detected by a FluoroSpot assay as spot forming units (SFU). Bars represent mean values ± standard deviation; dots represent technical triplicates. Figure 16: Assessment of CD4+ and CD8+ T-cell responses from animals immunized with LNP-RNA encoding EspA string 1 or EspA string 2. Five mice per group were immunized with LNP-RNA encoding EspA string 1 or EspA string 2. Splenocytes were isolated, pooled and CD4+ and CD8+ T cells were separated by magnetic-activated cell sorting. T cells were stimulated with a positive control (Concanavalin A, ConA), negative control (unspecific peptide, TRP1), or one of the seven peptide pools representative of the fragments used in the strings EspA_7.1 to EspA_7-7 in presence of autologous bone marrow-derived dendritic cells. Cytokine secretion (IFN-γ, IL-2, and TNF-α) was 20 11529421v1 Attorney Docket No. 2013237‐0757 detected by a FluoroSpot assay as spot forming units (SFU). Bars represent mean values ± standard deviation; dots represent technical triplicates. 21 11529421v1 Attorney Docket No. 2013237‐0757 Detailed Description Although the present disclosure is further described in more detail below, it is to be understood that this disclosure is not limited to the particular methodologies, protocols and reagents described herein as these may vary. It is also to be understood that the terminology used herein is for the purpose of describing particular embodiments only, and is not intended to limit the scope of the present disclosure which will be limited only by the appended claims. Unless defined otherwise, all technical and scientific terms used herein have the same meanings as commonly understood by one of ordinary skill in the art. In the following, the elements of the present disclosure will be described in more detail. These elements are listed with specific embodiments, however, it should be understood that they may be combined in any manner and in any number to create additional embodiments. The variously described examples and preferred embodiments should not be construed to limit the present disclosure to only the explicitly described embodiments. This description should be understood to support and encompass embodiments which combine the explicitly described embodiments with any number of the disclosed and/or preferred elements. Furthermore, any permutations and combinations of all described elements in this application should be considered disclosed by the description of the present application unless the context indicates otherwise. For example, the present disclosure describes combinations of sequence molecules which may have different levels of sequence identity to a specified sequence, e.g., (i) sequence molecule A comprising the sequence of SEQ ID NO: a, or a sequence having at least 99%, 98%, 97%, 96%, 95%, 90%, 85%, or 80% identity to the sequence of SEQ ID NO: a, (ii) sequence molecule B comprising the sequence of SEQ ID NO: b, or a sequence having at least 99%, 98%, 97%, 96%, 95%, 90%, 85%, or 80% identity to the sequence of SEQ ID NO: b etc. It should be understood that the sequence molecules may be combined in any of the identity levels specified. In some embodiments, the sequence molecules are combined such that the identity levels are identical; e.g., (i) sequence molecule A comprising the sequence of SEQ ID NO: a, (ii) sequence molecule B comprising the sequence of SEQ ID NO: b etc., or (i) sequence molecule A comprising a sequence having at least 90% identity to the sequence of SEQ ID NO: a, (ii) sequence molecule B comprising a sequence having at least 90% identity to the sequence of SEQ ID NO: b etc. In some embodiments, the identity levels are independently selected and are partially or entirely different from each other, i.e., the sequence molecules are combined such that the identity levels are not identical; e.g., (i) sequence molecule A comprising the sequence of SEQ ID NO: a, (ii) sequence molecule B comprising a sequence having at least 90% identity to the sequence of SEQ ID NO: b etc., or (i) sequence molecule A comprising a sequence having at least 90% identity to the sequence of SEQ ID NO: a, (ii) sequence molecule B comprising a sequence having at least 85% identity to the sequence of SEQ ID NO: b etc. The practice of the present disclosure will employ, unless otherwise indicated, conventional chemistry, biochemistry, cell biology, immunology, and recombinant DNA techniques which are explained in the literature in the field. Throughout this specification and the claims which follow, unless the context requires otherwise, the word "comprise", and variations such as "comprises" and "comprising", will be understood to imply the inclusion of a stated feature, element, member, integer or step or group of features, elements, members, integers or steps but not the exclusion of any other feature, element, member, integer or step or group of features, elements, members, integers or steps. The term "consisting essentially of" limits the scope of a claim or disclosure to the specified features, elements, members, integers, or steps and those that do not materially affect the basic and novel characteristic(s) of the claim or disclosure. The term “consisting of” limits the scope of a claim or disclosure to the specified features, elements, members, integers, 22 11529421v1 Attorney Docket No. 2013237‐0757 or steps. The term "comprising" encompasses the term "consisting essentially of" which, in turn, encompasses the term "consisting of". Thus, at each occurrence in the present application, the term "comprising" may be replaced with the term "consisting essentially of" or "consisting of". Likewise, at each occurrence in the present application, the term "consisting essentially of" may be replaced with the term "consisting of". The terms "a", "an" and "the" and similar references used in the context of describing the present disclosure (especially in the context of the claims) are to be construed to cover both the singular and the plural, unless otherwise indicated herein or clearly contradicted by the context. All methods described herein can be performed in any suitable order unless otherwise indicated herein or otherwise clearly contradicted by the context. The use of any and all examples, or exemplary language (e.g., "such as"), provided herein is intended merely to better illustrate the present disclosure and does not pose a limitation on the scope of the present disclosure otherwise claimed. No language in the specification should be construed as indicating any non-claimed element essential to the practice of the present disclosure. The term "optional" or "optionally" as used herein means that the subsequently described event, circumstance or condition may or may not occur, and that the description includes instances where said event, circumstance, or condition occurs and instances in which it does not occur. Where used herein, "and/or" is to be taken as specific disclosure of each of the two specified features or components with or without the other. For example, "X and/or Y" is to be taken as specific disclosure of each of (i) X, (ii) Y, and (iii) X and Y, just as if each is set out individually herein. In the context of the present disclosure, the term "about" denotes an interval of accuracy that the person of ordinary skill will understand to still ensure the technical effect of the feature in question. The term typically indicates deviation from the indicated numerical value by ±10%, ±5%, ±4%, ±3%, ±2%, ±1%, ±0.9%, ±0.8%, ±0.7%, ±0.6%, ±0.5%, ±0.4%, ±0.3%, ±0.2%, ±0.1%, ±0.05%, and for example ±0.01%. In some embodiments, “about” indicates deviation from the indicated numerical value by ±10%. In some embodiments, “about” indicates deviation from the indicated numerical value by ±5%. In some embodiments, “about” indicates deviation from the indicated numerical value by ±4%. In some embodiments, “about” indicates deviation from the indicated numerical value by ±3%. In some embodiments, “about” indicates deviation from the indicated numerical value by ±2%. In some embodiments, “about” indicates deviation from the indicated numerical value by ±1%. In some embodiments, “about” indicates deviation from the indicated numerical value by ±0.9%. In some embodiments, “about” indicates deviation from the indicated numerical value by ±0.8%. In some embodiments, “about” indicates deviation from the indicated numerical value by ±0.7%. In some embodiments, “about” indicates deviation from the indicated numerical value by ±0.6%. In some embodiments, “about” indicates deviation from the indicated numerical value by ±0.5%. In some embodiments, “about” indicates deviation from the indicated numerical value by ±0.4%. In some embodiments, “about” indicates deviation from the indicated numerical value by ±0.3%. In some embodiments, “about” indicates deviation from the indicated numerical value by ±0.2%. In some embodiments, “about” indicates deviation from the indicated numerical value by ±0.1%. In some embodiments, “about” indicates deviation from the indicated numerical value by ±0.05%. In some embodiments, “about” indicates deviation from the indicated numerical value by ±0.01%. As will be appreciated by the person of ordinary skill, the specific such deviation for a numerical value for a given technical effect 23 11529421v1 Attorney Docket No. 2013237‐0757 will depend on the nature of the technical effect. For example, a natural or biological technical effect may generally have a larger such deviation than one for a man-made or engineering technical effect. Recitation of ranges of values herein is merely intended to serve as a shorthand method of referring individually to each separate value falling within the range. Unless otherwise indicated herein, each individual value is incorporated into the specification as if it were individually recited herein. Several documents are cited throughout the text of this specification. Each of the documents cited herein (including all patents, patent applications, scientific publications, manufacturer's specifications, instructions, etc.), whether supra or infra, are hereby incorporated by reference in their entirety. Nothing herein is to be construed as an admission that the invention is not entitled to antedate such disclosure by virtue of prior invention. It should be noted for unambiguousness that whenever a sequence is referred to as being the sequence between the nucleotide at position x and the nucleotide at position y, the resulting sequence includes both the nucleotide at position x and the nucleotide at position y. Similarly, whenever a sequence is referred to as being the sequence between the amino acid at position x and the amino acid at position y, the resulting sequence includes both the amino acid at position x and the amino acid at position y. Moreover, while the sequences described herein, in particular in the sequence listing, refer to DNA molecules, it is clear that when it is stated in the description or the claims that an RNA comprises a nucleotide sequence as described herein, in particular in the sequence listing, the nucleotide sequence referred to is actually identical to the base-sequence of the DNA molecule described herein, in particular in the sequence listing, e.g., represented in a SEQ ID NO referred to, except that thymine is replaced by uracil. In the following, definitions and embodiments will be provided which apply to all aspects of the present disclosure. Terms which are defined in the following have the meanings as defined, unless otherwise indicated. Any undefined terms have their art recognized meanings. A “microbial infection” refers to an infection with a microbial organism, such as an eukaryotic parasite, a bacterium, fungus or virus. Within the scope of the present invention, the term “antigen domain” refers to a region of the antigen’s polypeptide chain that is self-stabilizing and that folds independently of the remainder of the polypeptide chain, forming a particular secondary and/or tertiary structure. An “incomplete antigen domain”, within the scope of the present invention, refers to a domain being truncated or otherwise shortened on its N-terminal or C-terminal end. In some embodiments, the “incomplete antigen domain” is shortened by at least 5, at least 10, at least 15, at least 20, at least 25, at least 30, at least 40, at least 50, at least 60, at least 100, at least 200, at least 300, at least 400, at least 500 or more amino acids. In some embodiments, the truncation or shortening results in partial or full loss of self-stabilization. Within the scope of the present invention, the term “the incomplete antigen domain is interrupted within” refers to the fact that the ends of a chunk or fragment of an antigen domain are selected such that that they intersect a structural feature of the domain, whereupon the remainder of the structural feature (outside the chunk or fragment in question) is missing from the antigen domain, rendering it incomplete and/or inoperable in a structurally or functionally relevant fashion. 24 11529421v1 Attorney Docket No. 2013237‐0757 The terms “toxicity”, “toxic” and the like refer to the quality of a polypeptide or antigen of interfering with an organisms normal metabolism. In some embodiments, toxic polypeptides or antigens exhibit toxicity when the organism is exposed to the polypeptides or antigens expressed outside of the organism’s own cells, e.g. when the toxic polypeptides or antigens are expressed and excreted by a microbial organism during a microbial infection of the organism. In some embodiments, toxic polypeptides or antigens induce cell death. More specifically, the terms "cytotoxicity", "cytotoxic" and the like refer to the quality of a polypeptide or antigen of being toxic to cells. Exposure of a cell to a cytotoxic polypeptide or antigen, e.g. when the polypeptide or antigen is expressed within the cell, can result in a variety of cell fates such as necrosis or apoptosis. The occurrence of necrosis or apoptosis in cells, and thus the presence of cytotoxicity, can be quantified and/or statistically analyzed by various means known to the skilled person, including observations of changes in cell morphology and/or detection of apoptotic markers. For instance, bright field microscopy images can be overlaid with images detecting fluorescent dyes which are membrane-impermeant and/or selectively bind to physiological structures or markers specific to necrotic and/or apoptotic cell death. Also indirect methods, such as the degradation of fluorescent probes due to caspase activity, are contemplated. Furthermore, factors specific for necrosis or apoptosis can be quantified in cell lysates using conventional biochemical assays for quantification, including antibody-based techniques such as western-blotting or ELISA or methods based on physical separation and detection such as mass-spectrometry. In the same manner, the strength of cytotoxicity of a given polypeptide or antigen can be determined by quantifying and/or statistically analyzing the prevalence of necrosis and/or apoptosis upon exposure of a cell population to the polypeptide or antigen and comparing the observed prevalence of necrosis and/or apoptosis to a control population of cells exposed to a different non-cytotoxic protein. In addition, partial or full removal of cytotoxicity can be determined by quantifying – in the manner described above – the prevalence of necrosis and/or apoptosis in a cell population expressing a fragment of a cytotoxic microbial antigen. The prevalence of necrosis and/or apoptosis in said cell population can then be compared to the prevalence of necrosis and/or apoptosis in a cell population expressing the full-length microbial antigen. This way, a reduction in cytotoxicity can be quantified to be reduced, e.g., by at least 20%, at least 40%, at least 60% or at least 80%. If the prevalence of necrosis and/or apoptosis is the same as in a control population of cells exposed to a different non-cytotoxic protein, cytotoxicity has been removed. The term “secondary structure” of a polypeptide or protein refers to structure such as alpha-helices or beta-sheets. The term “tertiary structure” of a polypeptide or protein refers to higher order folding of peptides which comprise e.g. one or more secondary structures and form the overall three dimensional shape of the peptide or protein. The term “quaternary structure” describes the number and arrangement of multiple folded polypeptide or protein subunits in a multi-subunit complex. The presence of secondary, tertiary and quaternary structures in a polypeptide, protein or protein complex can be determined either experimentally – using methods known to the skilled person such as X-ray protein crystallography or nuclear magnetic resonance (NMR) – or can be predicted theoretically. A large number of bioinformatics tools exists for the prediction of secondary, tertiary and quaternary structures, such as AlphaFold2 (https://alphafold.ebi.ac.uk/), Rosetta (https://www.rosettacommons.org/software/servers), RFdiffusion (https://colab.research.google.com/github/sokrypton/ColabDesign/blob/v1.1.1/rf/examples/diffusion.ipynb), PSSpred (https://zhanggroup.org/PSSpred/), Jpred4 (http://www.compbio.dundee.ac.uk /jpred4/index.html), Phyre
2 (http://www.sbg.bio.ic.ac.uk/phyre2/html/page.cgi?id =index), ORION (https://www.dsimb.inserm.fr/ORION/), QuatIdent (http://www.csbio.sjtu.edu.cn/bioinf/ Quaternary/) and QuaBingo (http://predictor.nchu.edu.tw/QuaBingo/). In addition, secondary, tertiary and quaternary structures of polypeptides 25 11529421v1 Attorney Docket No. 2013237‐0757 and proteins can be determined based on comparisons with structurally characterized, closely related polypeptides or proteins. Within the scope of the present invention, an RNA molecule, such as an mRNA molecule, polypeptide, DNA or pharmaceutical compositions may be used in various forms of prophylactic, acute or chronic therapy. Embodiments of prophylactic therapy comprise the use as vaccines for preventing a microbial infection. Alternatively or in addition, the RNA molecule, polypeptide, DNA or pharmaceutical compositions may be used in order to enhance the immune response of patients already suffering from a microbial infection. Mycobacterium tuberculosis (Mtb) is a non-motile, slowly growing and rod shaped (2-4 μm in length and 0.2-0.5 μm in width) bacterium. Mtb is gram-positive, obligate aerobe, requires a host for growth and reproduction, and does not form spores. The term "tuberculosis" or "TB" is used to describe the infection caused by infective agents from the genus "Mycobacterium ". Tuberculosis is a potentially fatal contagious disease that can affect almost any part of the body but is most frequently an infection of the lungs. While the majority of tuberculosis infections is caused by Mycobacterium tuberculosis, there are other Mycobacterium species that can cause tuberculosis as well. These species include Mycobacterium bovis, Mycobacterium caprae, Mycobacterium orygis, Mycobacterium africanum, Mycobacterium microti, Mycobacterium canetti and Mycobacterium pinnipedii. Mycobacterium tuberculosis and some other mycobacteria are transmitted by airborne droplet nuclei produced when an individual with active disease coughs, speaks, or sneezes. When inhaled, the droplet nuclei reach the alveoli of the lung. In susceptible individuals the organisms may then multiply and spread through lymphatics to the lymph nodes, and through the bloodstream to other sites such as the lung apices, bone marrow, kidneys, and meninges. Infections with other Mycobacterium species, such as Mycobacterium bovis or Mycobacterium caprae are also associated with the consumption of un-pasteurized milk from infected animals. The development of acquired immunity in 2 to 10 weeks results in a halt to bacterial multiplication. Lesions heal and the individual remains asymptomatic. Mycobacteria can remain dormant (latent TB) in the body after infection for years, concealed in the phagocytosed cells, and never develop into the disease. Such an individual is said to have tuberculous infection without disease, and will show a positive tuberculin test. The clinical status of latent TB is traditionally associated with the transition of Mtb to a dormant state in response to non-optimal growth conditions in vivo due to activation of the host immune response. Dormancy is a specific physiological state characterized by significant cessation of metabolic activity and growth, whereas resuscitation from dormancy is a process of restoring cell activity followed by bacterial multiplication, which in case of Mtb can lead to disease progression. The risk of developing active disease with clinical symptoms diminishes with time and may never occur, but is a lifelong risk. Approximately 5% of individuals with tuberculous infection progress to active disease. Terms such as "reduce" or "inhibit" as used herein means the ability to cause an overall decrease, for example, of about 5% or greater, about 10% or greater, about 15% or greater, about 20% or greater, about 25% or greater, about 30% or greater, about 40% or greater, about 50% or greater, or about 75% or greater, in the level. The term "inhibit" or similar phrases includes a complete or essentially complete inhibition, i.e. a reduction to zero or essentially to zero. Terms such as "enhance" as used herein means the ability to cause an overall increase, or enhancement, for example, by at least about 5% or greater, about 10% or greater, about 15% or greater, about 20% or greater, about 25% or greater, about 30% or greater, about 40% or greater, about 50% or greater, about 75% or greater, or about 100% or greater in the level. 26 11529421v1 Attorney Docket No. 2013237‐0757 "Physiological pH" as used herein refers to a pH of about 7.4. In some embodiments, physiological pH is from 7.3 to 7.5. In some embodiments, physiological pH is from 7.35 to 7.45. In some embodiments, physiological pH is 7.3, 7.35, 7.4, 7.45, or 7.5. As used in the present disclosure, "% w/v" refers to weight by volume percent, which is a unit of concentration measuring the amount of solute in grams (g) expressed as a percent of the total volume of solution in milliliters (mL). As used in the present disclosure, "% by weight" refers to weight percent, which is a unit of concentration measuring the amount of a substance in grams (g) expressed as a percent of the total weight of the total composition in grams (g). As used in the present disclosure, "mol %" is defined as the ratio of the number of moles of one component to the total number of moles of all components, multiplied by 100. As used in the present disclosure, "mol % of the total lipid" is defined as the ratio of the number of moles of one lipid component to the total number of moles of all lipids, multiplied by 100. In this context, in some embodiments, the term "total lipid" includes lipids and lipid-like material. The term "ionic strength" refers to the mathematical relationship between the number of different kinds of ionic species in a particular solution and their respective charges. Thus, ionic strength I is represented mathematically by the formula: ^
^ ൌ1 2
∙ ^ z^ଶ∙ ^^^ ^ in which c is the molar concentration of a

and z the absolute value of its charge. The sum Σ is taken over all the different kinds of ions (i) in solution. According to the disclosure, the term "ionic strength" in some embodiments relates to the presence of monovalent ions. Regarding the presence of divalent ions, in particular divalent cations, their concentration or effective concentration (presence of free ions) due to the presence of chelating agents is, in some embodiments, sufficiently low so as to prevent degradation of the nucleic acid. In some embodiments, the concentration or effective concentration of divalent ions is below the catalytic level for hydrolysis of the phosphodiester bonds between nucleotides such as RNA nucleotides. In some embodiments, the concentration of free divalent ions is 20 µM or less. In some embodiments, there are no or essentially no free divalent ions. "Osmolality" refers to the concentration of a particular solute expressed as the number of osmoles of solute per kilogram of solvent. The term "lyophilizing" or "lyophilization" refers to the freeze-drying of a substance by freezing it and then reducing the surrounding pressure (e.g., below 15 Pa, such as below 10 Pa, below 5 Pa, or 1 Pa or less) to allow the frozen medium in the substance to sublimate directly from the solid phase to the gas phase. Thus, the terms "lyophilizing" and "freeze-drying" are used herein interchangeably. The term "spray-drying" refers to spray-drying a substance by mixing (heated) gas with a fluid that is atomized (sprayed) within a vessel (spray dryer), where the solvent from the formed droplets evaporates, leading to a dry powder. 27 11529421v1 Attorney Docket No. 2013237‐0757 The term "reconstitute" relates to adding a solvent such as water to a dried product to return it to a liquid state such as its original liquid state. The term "recombinant" in the context of the present disclosure means "made through genetic engineering". In some embodiments, a "recombinant object" in the context of the present disclosure is not occurring naturally. The term "naturally occurring" as used herein refers to the fact that an object can be found in nature. For example, a peptide or nucleic acid that is present in an organism (including viruses) and can be isolated from a source in nature and which has not been intentionally modified by man in the laboratory is naturally occurring. The term "found in nature" means "present in nature" and includes known objects as well as objects that have not yet been discovered and/or isolated from nature, but that may be discovered and/or isolated in the future from a natural source. As used herein, the terms "room temperature" and "ambient temperature" are used interchangeably herein and refer to temperatures from at least about 15°C, e.g., from about 15°C to about 35°C, from about 15°C to about 30°C, from about 15°C to about 25°C, or from about 17°C to about 22°C. Such temperatures will include 15°C, 16°C, 17°C, 18°C, 19°C, 20°C, 21°C and 22°C. The term “EDTA” refers to ethylenediaminetetraacetic acid disodium salt. All concentrations are given with respect to the EDTA disodium salt. The term "cryoprotectant" relates to a substance that is added to a formulation in order to protect the active ingredients during the freezing stages. The term "lyoprotectant" relates to a substance that is added to a formulation in order to protect the active ingredients during the drying stages. According to the present disclosure, the term "peptide" refers to substances which comprise about two or more, about 3 or more, about 4 or more, about 6 or more, about 8 or more or about 10 consecutive amino acids linked to one another via peptide bonds. The term "polypeptide" refers to larger peptides, in particular peptides having at least about 11 amino acids. The term "biological activity" means the response of a biological system to a molecule. Such biological systems may be, for example, a cell or an organism. In some embodiments, such response is therapeutically or pharmaceutically useful. The term "portion" refers to a fraction. With respect to a particular structure such as an amino acid sequence or protein the term "portion" thereof may designate a continuous or a discontinuous fraction of said structure. The terms "chunk", "part" and "fragment" are used interchangeably herein and refer to a continuous element. For example, a chunk, part of a structure such as an amino acid sequence or antigen refers to a continuous element of said structure. "Chunk", "part" and "fragment", with reference to an amino acid sequence (antigen or polypeptide), relates to a part of an amino acid sequence, i.e. a sequence which represents the amino acid sequence shortened at the N-terminus and/or C-terminus. A fragment shortened at the C-terminus (N-terminal fragment) is obtainable, e.g., by translation of a truncated open reading frame that lacks the 3'-end of the open reading frame. A fragment shortened at the N-terminus (C-terminal fragment) is obtainable, e.g., by translation of a truncated open reading frame that lacks the 5'-end of the open reading frame, as long as the truncated open reading frame comprises a start codon that serves to initiate translation. A fragment of an amino acid sequence comprises, e.g., at least 50 %, at least 60 %, at least 70 28 11529421v1 Attorney Docket No. 2013237‐0757 %, at least 80%, at least 90% of the amino acid residues from an amino acid sequence. A fragment of an amino acid sequence comprises, e.g., at least 5, at least 6, at least 7, in particular at least 8, at least 10, at least 12, at least 15, at least 20, at least 30, at least 50, or at least 100 consecutive amino acids from an amino acid sequence. A fragment of an amino acid sequence comprises, e.g., a sequence of up to 8, in particular up to 10, up to 12, up to 15, up to 20, up to 30, up to 50, up to 80, up to 100, up to 150 or up to 200 consecutive amino acids of the amino acid sequence. When used in context of a composition, the term "part" means a portion of the composition. For example, a part of a composition may be any portion from 0.1% to 99.9% (such as 0.1%, 0.5%, 1%, 5%, 10%, 50%, 90%, or 99%) of said composition. An antigen or immunogenic variant thereof "represented" by one or more antigen fragments as used herein refers to the fact that the one or more antigen fragments, respectively, constitutes amino acid sequences forming a specific part of the full length antigen or an immunogenic variant of the full length antigen. Antigen fragments representing an antigen or immunogenic variant thereof may be may or may not be overlapping. The one or more fragments of an antigen or an immunogenic variant of an antigen are capable of inducing an immune response against the antigen when delivered to a subject, e.g. in the form of a polypeptide or an RNA transcribed by a cell of the subject. In some embodiments, a fragment of an antigen or an immunogenic variant of an antigen comprises at least one epitope, e.g., at least one T cell epitope, of an antigen or an immunologically equivalent variant of said at least one epitope. In some embodiments, a fragment of an antigen or an immunogenic variant of an antigen comprises a fragment of, e.g., at least 5, at least 6, at least 7, in particular at least 8, at least 10, at least 12, at least 15, at least 20, at least 30, at least 50, or at least 100 consecutive amino acids of said antigen or immunogenic variant thereof. If reference is made to RNA encoding a polypeptide or encoding at least two antigen fragments representing at least one antigen or immunogenic variant thereof, such disclosure encompasses monocistronic and polycistronic RNAs. If only one antigen or immunogenic variant thereof is represented, the RNA encodes two or more fragments of the antigen or immunogenic variant thereof. If the RNA encodes more than two fragments of the antigen or immunogenic variant thereof, additional fragments of the antigen or immunogenic variant thereof may be encoded by different open reading frames located on the same or on different RNA molecules. If more than one antigen or immunogenic variant thereof is represented, the RNA encodes one or more cytotoxic antigens or immunogenic variants thereof, respectively, as one or more fragments with reduced cytotoxicity (compared to the full-length antigen), whereas non-cytotoxic antigens or immunogenic variants thereof are encoded as the full length antigen and/or as one or more antigen fragments, respectively. In some embodiments, the RNA encodes one or more fragments of each of the more than one antigens or immunogenic variants thereof. In some embodiments, the RNA encodes the full-length antigen of some of the more than one non-cytotoxic antigen or immunogenic variant thereof and/or encodes one or more fragments of some of the more than one cytotoxic or non-cytotoxic antigens or immunogenic variants thereof, wherein the RNA may encode the full-length antigen as well as one or more fragments of the same antigen or immunogenic variant thereof. The full-length antigens and/or fragments discussed above may be encoded by the same or different open reading frames located on the same or on different RNA molecules. The term “polypeptide linker” describes either a single amino acid or a sequence of amino acids connecting the amino acid sequences of two adjacent subunits in a linear amino acid sequence of a polypeptide. A polypeptide linker may have a length of 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 12, 15 or more amino acids. Preferably, the polypeptide linker is 10 amino 29 11529421v1 Attorney Docket No. 2013237‐0757 acids in length. Exemplary linkers include glycine-serine-polypeptide linkers, glycine-proline-polypeptide linkers, and proline-alanine polypeptide linkers. In certain embodiments, the linker is a glycine-serine-polypeptide linker (GS linker), i.e., a peptide that consists of glycine and serine residues. In some embodiments, a GS linker comprises the amino acid sequence of SEQ ID NO: 43. By "wild type" or "WT" or "native" herein is meant an amino acid sequence that is found in nature, including allelic variations and/or naturally occurring mutations. A wild type amino acid sequence, peptide or polypeptide has an amino acid sequence that has not been intentionally modified by man. The term “heterologous” as used herein in conjunction with amino acid sequences is meant to refer to amino acid sequences not found in nature in the specific context they are encoded, i.e., that have been intentionally modified by man – either in sequence or in sequence context. In some embodiments, a heterologous signal peptide sequence fused or operatively linked to a protein denotes that said signal peptide in nature does not occur fused or operatively linked to said protein, either because said signal peptide can naturally be found fused or operatively linked only to other proteins or only in other organisms, such as mammals, e.g. human, bacteria or viruses. Embodiments for such heterologous signal peptides are provided herein. In another embodiment, a heterologous signal peptide has been mutated in a purposeful manner (e.g., by random mutagenesis and targeted selection or by guided mutagenesis techniques, including, e.g., sequence synthesis) in order to obtain certain functional properties or to eliminate certain functional properties, resulting in a signal peptide structurally and functionally distinct from a signal peptide found in nature fused or operatively linked to the protein in question. "Immunogenic variant” as used herein and with reference to an antigen comprises an amino acid sequence which is "immunologically equivalent" to said antigen and thus, is able to induce an immune reaction to said antigen, when delivered to a subject. In some embodiments, an immunogenic variant of the antigen comprises an amino acid sequence differing from its wild type sequence by one or more deletions, insertions or substitutions in the amino acid sequence while having at least 99%, 98%, 97%, 96%, 95%, 90%, 85%, or 80% identity to the wild type sequence (e.g. determined by sequence alignment using known sequence alignment algorithms such as BLASTP). A full-length antigen representing an immunogenic variant of the antigen is characterized in that it comprises the full-length amino acid sequence of the immunogenic variant, whereas an antigen fragment representing an immunogenic variant of the antigen is characterized in that it comprises an amino acid sequence which is only a part of the full-length amino acid sequence of the immunogenic variant but which still is able to induce an immune reaction to the antigen, when delivered to a subject. In some embodiments the degree of similarity, such as identity between a given amino acid sequence and an amino acid sequence which is a variant of said given amino acid sequence, will be at least about 60%, 70%, 80%, 81%, 82%, 83%, 84%, 85%, 86%, 87%, 88%, 89%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99%. In some embodiments, the degree of similarity or identity is given for an amino acid region which is at least about 10%, at least about 20%, at least about 30%, at least about 40%, at least about 50%, at least about 60%, at least about 70%, at least about 80%, at least about 90% or about 100% of the entire length of the reference amino acid sequence. For example, if the reference amino acid sequence consists of 200 amino acids, the degree of similarity or identity is given, e.g., for at least about 20, at least about 40, at least about 60, at least about 80, at least about 100, at least about 120, at least about 140, at least about 160, at least about 180, or about 200 amino acids, in some embodiments, continuous amino acids. In some embodiments, the degree of similarity or identity is given for the entire length of the reference amino acid sequence. The alignment for determining sequence similarity, such as sequence identity, can be 30 11529421v1 Attorney Docket No. 2013237‐0757 done with art known tools, such as using the best sequence alignment, for example, using Align, using standard settings, preferably EMBOSS::needle, Matrix: Blosum62, Gap Open 10.0, Gap Extend 0.5. "Sequence similarity" indicates the percentage of amino acids that either are identical or that represent conservative amino acid substitutions. "Sequence identity" between two amino acid sequences indicates the percentage of amino acids that are identical between the sequences. "Sequence identity" between two nucleic acid sequences indicates the percentage of nucleotides that are identical between the sequences. The terms "% identical" and "% identity" or similar terms are intended to refer, in particular, to the percentage of nucleotides or amino acids which are identical in an optimal alignment between the sequences to be compared. Said percentage is purely statistical, and the differences between the two sequences may be but are not necessarily randomly distributed over the entire length of the sequences to be compared. Comparisons of two sequences are usually carried out by comparing the sequences, after optimal alignment, with respect to a segment or "window of comparison", in order to identify local regions of corresponding sequences. The optimal alignment for a comparison may be carried out manually or with the aid of the local homology algorithm by Smith and Waterman, 1981, Ads App. Math. 2, 482, with the aid of the local homology algorithm by Neddleman and Wunsch, 1970, J. Mol. Biol. 48, 443, with the aid of the similarity search algorithm by Pearson and Lipman, 1988, Proc. Natl Acad. Sci. USA 88, 2444, or with the aid of computer programs using said algorithms (GAP, BESTFIT, FASTA, BLAST P, BLAST N and TFASTA in Wisconsin Genetics Software Package, Genetics Computer Group, 575 Science Drive, Madison, Wis.). In some embodiments, percent identity of two sequences is determined using the BLASTN or BLASTP algorithm, as available on the United States National Center for Biotechnology Information (NCBI) website (e.g., at blast.ncbi.nlm.nih.gov/Blast.cgi?PAGE_TYPE=BlastSearch&BLAST_SPEC=blast2seq&LINK_LOC=align2seq). In some embodiments, the algorithm parameters used for BLASTN algorithm on the NCBI website include: (i) Expect Threshold set to 10; (ii) Word Size set to 28; (iii) Max matches in a query range set to 0; (iv) Match/Mismatch Scores set to 1, - 2; (v) Gap Costs set to Linear; and (vi) the filter for low complexity regions being used. In some embodiments, the algorithm parameters used for BLASTP algorithm on the NCBI website include: (i) Expect Threshold set to 10; (ii) Word Size set to 3; (iii) Max matches in a query range set to 0; (iv) Matrix set to BLOSUM62; (v) Gap Costs set to Existence: 11 Extension: 1; and (vi) conditional compositional score matrix adjustment. Percentage identity is obtained by determining the number of identical positions at which the sequences to be compared correspond, dividing this number by the number of positions compared (e.g., the number of positions in the reference sequence) and multiplying this result by 100. In some embodiments, the degree of similarity or identity is given for a region which is at least about 50%, at least about 60%, at least about 70%, at least about 80%, at least about 90% or about 100% of the entire length of the reference sequence. For example, if the reference nucleic acid sequence consists of 200 nucleotides, the degree of identity is given for at least about 100, at least about 120, at least about 140, at least about 160, at least about 180, or about 200 nucleotides, in some embodiments continuous nucleotides. In some embodiments, the degree of similarity or identity is given for the entire length of the reference sequence. Homologous amino acid sequences exhibit according to the disclosure at least 40%, in particular at least 50%, at least 60%, at least 70%, at least 80%, at least 90% and, e.g., at least 95%, at least 98 or at least 99% identity of the amino acid residues. 31 11529421v1 Attorney Docket No. 2013237‐0757 The amino acid sequence variants described herein may readily be prepared by the skilled person, for example, by recombinant DNA manipulation. The manipulation of DNA sequences for preparing peptides or polypeptides having substitutions, additions, insertions or deletions, is described in detail in Molecular Cloning: A Laboratory Manual, 4
th Edition, M.R. Green and J. Sambrook eds., Cold Spring Harbor Laboratory Press, Cold Spring Harbor 2012, for example. Furthermore, the peptides, polypeptides and amino acid variants described herein may be readily prepared with the aid of known peptide synthesis techniques such as, for example, by solid phase synthesis and similar methods. In some embodiments, a fragment or variant of an amino acid sequence (peptide or polypeptide) is a "functional fragment" or "functional variant". The term "functional fragment" or "functional variant" of an amino acid sequence relates to any fragment or variant exhibiting one or more functional properties identical or similar to those of the amino acid sequence from which it is derived, i.e., it is functionally equivalent. With respect to antigens or antigenic sequences, one particular function is one or more immunogenic activities displayed by the amino acid sequence from which the fragment or variant is derived. The term "functional fragment" or "functional variant", as used herein, in particular refers to a variant molecule or sequence that comprises an amino acid sequence that is altered by one or more amino acids compared to the amino acid sequence of the parent molecule or sequence and that is still capable of fulfilling one or more of the functions of the parent molecule or sequence, e.g., inducing an immune response. In some embodiments, the modifications in the amino acid sequence of the parent molecule or sequence do not significantly affect or alter the characteristics of the molecule or sequence. In different embodiments, the function of the functional fragment or functional variant may be reduced but still significantly present, e.g., function of the functional fragment or functional variant may be at least 50%, at least 60%, at least 70%, at least 80%, or at least 90% of the parent molecule or sequence. However, in other embodiments, function of the functional fragment or functional variant may be enhanced compared to the parent molecule or sequence. An amino acid sequence (peptide or polypeptide) "derived from" a designated amino acid sequence (peptide or polypeptide) refers to the origin of the first amino acid sequence. In some embodiments, the amino acid sequence which is derived from a particular amino acid sequence has an amino acid sequence that is identical, essentially identical or homologous to that particular sequence or a fragment thereof. Amino acid sequences derived from a particular amino acid sequence may be variants of that particular sequence or a fragment thereof. For example, it will be understood by one of ordinary skill in the art that the polypeptides suitable for use herein may be altered such that they vary in sequence from the naturally occurring or native sequences from which they were derived, while retaining the desirable activity of the native sequences. In some embodiments, "isolated" means removed (e.g., purified) from the natural state or from an artificial composition, such as a composition from a production process. For example, a nucleic acid, peptide or polypeptide naturally present in a living animal is not "isolated", but the same nucleic acid, peptide or polypeptide partially or completely separated from the coexisting materials of its natural state is "isolated". An isolated nucleic acid, peptide or polypeptide can exist in substantially purified form, or can exist in a non-native environment such as, for example, a host cell. The term "transfection" relates to the introduction of nucleic acids, in particular RNA, into a cell. For purposes of the present disclosure, the term "transfection" also includes the introduction of a nucleic acid into a cell or the uptake of a nucleic acid by such cell, wherein the cell may be present in a subject, e.g., a patient, or the cell may be in vitro, e.g., outside of a patient. Thus, according to the present disclosure, a cell for transfection of a nucleic acid described herein can be present in vitro or in vivo, e.g. the cell can form part of an organ, a tissue and/or the body of a patient. 32 11529421v1 Attorney Docket No. 2013237‐0757 According to the disclosure, transfection can be transient or stable. For some applications of transfection, it is sufficient if the transfected genetic material is only transiently expressed. RNA can be transfected into cells to transiently express its coded polypeptide. Since the nucleic acid introduced in the transfection process is usually not integrated into the nuclear genome, the foreign nucleic acid will be diluted through mitosis or degraded. Cells allowing episomal amplification of nucleic acids greatly reduce the rate of dilution. If it is desired that the transfected nucleic acid actually remains in the genome of the cell and its daughter cells, a stable transfection must occur. Such stable transfection can be achieved by using virus-based systems or transposon-based systems for transfection, for example. Generally, nucleic acid encoding polypeptides is transiently transfected into cells. RNA can be transfected into cells to transiently express its coded polypeptide. The disclosure includes analogs of a peptide or polypeptide. According to the present disclosure, an analog of a peptide or polypeptide is a modified form of said peptide or polypeptide from which it has been derived and has at least one functional property of said peptide or polypeptide. E.g., a pharmacological active analog of a peptide or polypeptide has at least one of the pharmacological activities of the peptide or polypeptide from which the analog has been derived. Such modifications include any chemical modification and comprise single or multiple substitutions, deletions and/or additions of any molecules associated with the peptide or polypeptide, such as carbohydrates, lipids and/or peptides or polypeptides. In some embodiments, "analogs" of peptides or polypeptides include those modified forms resulting from glycosylation, acetylation, phosphorylation, amidation, palmitoylation, myristoylation, isoprenylation, lipidation, alkylation, derivatization, introduction of protective/blocking groups, proteolytic cleavage or binding to an antibody or to another cellular ligand. The term "analog" also extends to all functional chemical equivalents of said peptides and polypeptides. As used herein, the terms "linked", "fused", or "fusion" are used interchangeably. These terms refer to the joining together of two or more elements or components or domains. As used herein "endogenous" refers to any material from or produced inside an organism, cell, tissue or system. As used herein, the term "exogenous" refers to any material introduced from or produced outside an organism, cell, tissue or system. According to various embodiments of the present disclosure, a nucleic acid such as RNA encoding a peptide or polypeptide is taken up by or introduced, i.e. transfected or transduced, into a cell which cell may be present in vitro or in a subject, resulting in expression of said peptide or polypeptide. The cell may, e.g., express the encoded peptide or polypeptide intracellularly (e.g. in the cytoplasm and/or in the nucleus), may secrete the encoded peptide or polypeptide, and/or may express it on the surface. In some embodiments, the cell secretes the encoded peptide or polypeptide. In particular, the term "encoding" refers to the inherent property of specific sequences of nucleotides in a polynucleotide, such as a gene, a cDNA, or an RNA (in particular, mRNA), to serve as templates for synthesis of other polymers and macromolecules in biological processes having either a defined sequence of nucleotides (i.e., rRNA, tRNA and mRNA) or a defined sequence of amino acids and the biological properties resulting therefrom. Thus, a gene encodes a polypeptide if transcription and translation of mRNA corresponding to that gene produces the polypeptide in a cell or other biological system. Both the coding strand, the nucleotide sequence of which is identical to the mRNA sequence and is usually provided in sequence listings, and the non-coding strand, used as the template for transcription of a gene or cDNA, can be referred to as encoding the polypeptide or other product of that gene or cDNA. 33 11529421v1 Attorney Docket No. 2013237‐0757 In this respect, an "open reading frame" or "ORF" is a continuous stretch of codons beginning with a start codon and ending with a stop codon. The term "expression" as used herein includes the transcription and/or translation of a particular nucleotide sequence. In the context of the present disclosure, the term "transcription" relates to a process, wherein the genetic code in a DNA sequence is transcribed into RNA (especially mRNA). Subsequently, the RNA may be translated into peptide or polypeptide. With respect to RNA, the term "expression" or "translation" relates to the process in the ribosomes of a cell by which a strand of mRNA directs the assembly of a sequence of amino acids to make a peptide or polypeptide. A medical preparation, in particular kit, described herein may comprise instructional material or instructions. As used herein, "instructional material" or "instructions" includes a publication, a recording, a diagram, or any other medium of expression which can be used to communicate the usefulness of the compositions and methods of the present disclosure. The instructional material of the kit of the present disclosure may, for example, be affixed to a container which contains the compositions/formulations of the present disclosure or be shipped together with a container which contains the compositions/formulations. Alternatively, the instructional material may be shipped separately from the container with the intention that the instructional material and the compositions be used cooperatively by the recipient. In the present specification, a structural formula of a compound may represent a certain isomer of said compound. It is to be understood, however, that the present disclosure includes all isomers such as geometrical isomers, optical isomers based on an asymmetrical carbon, stereoisomers, tautomers and the like which occur structurally and isomer mixtures and is not limited to the description of the formula. Furthermore, in the present specification, a structural formula of a compound may represent a specific salt and/or solvate of said compound. It is to be understood, however, that the present disclosure includes all salts (e.g., pharmaceutically acceptable salts) and solvates (e.g., hydrates) and is not limited to the description of the specific salt and/or solvate. "Isomers" are compounds having the same molecular formula but differ in structure ("structural isomers") or in the geometrical (spatial) positioning of the functional groups and/or atoms ("stereoisomers"). "Enantiomers" are a pair of stereoisomers which are non-superimposable mirror-images of each other. A "racemic mixture" or "racemate" contains a pair of enantiomers in equal amounts and is denoted by the prefix (±). "Diastereomers" are stereoisomers which are non-superimposable and which are not mirror-images of each other. "Tautomers" are structural isomers of the same chemical substance that spontaneously and reversibly interconvert into each other, even when pure, due to the migration of individual atoms or groups of atoms; i.e., the tautomers are in a dynamic chemical equilibrium with each other. An example of tautomers are the isomers of the keto-enol-tautomerism. "Conformers" are stereoisomers that can be interconverted just by rotations about formally single bonds, and include - in particular - those leading to different 3-dimentional forms of (hetero)cyclic rings, such as chair, half-chair, boat, and twist-boat forms of cyclohexane. The term "solvate" as used herein refers to an addition complex of a dissolved material in a solvent (such as an organic solvent (e.g., an aliphatic alcohol (such as methanol, ethanol, n-propanol, isopropanol), acetone, acetonitrile, ether, and the like), water or a mixture of two or more of these liquids), wherein the addition complex exists in the form of a crystal or mixed crystal. The amount of solvent contained in the addition complex may be stoichiometric or non- stoichiometric. A "hydrate" is a solvate wherein the solvent is water. 34 11529421v1 Attorney Docket No. 2013237‐0757 In isotopically labeled compounds one or more atoms are replaced by a corresponding atom having the same number of protons but differing in the number of neutrons. For example, a hydrogen atom may be replaced by a deuterium or tritium atom. Exemplary isotopes which can be used in the present disclosure include deuterium, tritium,
11C,
13C,
14C,
15N,
18F,
32P,
32S,
35S,
36Cl, and
125I. The term "average diameter" refers to the mean hydrodynamic diameter of particles as measured by dynamic light scattering (DLS) with data analysis using the so-called cumulant algorithm, which provides as results the so-called Z
average with the dimension of a length, and the polydispersity index (PDI), which is dimensionless (Koppel, D., J. Chem. Phys. 57, 1972, pp 4814-4820, ISO 13321). Here "average diameter", "diameter" or "size" for particles is used synonymously with this value of the Z
average. In some embodiments, the "polydispersity index" is calculated based on dynamic light scattering measurements by the so-called cumulant analysis as mentioned in the definition of the "average diameter". Under certain prerequisites, it can be taken as a measure of the size distribution of an ensemble of nanoparticles. The "radius of gyration" (abbreviated herein as R
g) of a particle about an axis of rotation is the radial distance of a point from the axis of rotation at which, if the whole mass of the particle is assumed to be concentrated, its moment of inertia about the given axis would be the same as with its actual distribution of mass. Mathematically, R
g is the root mean square distance of the particle's components from either its center of mass or a given axis. For example, for a macromolecule composed of n mass elements, of masses m
i (i = 1, 2, 3, …, n), located at fixed distances s
i from the center of mass, R
g is the square-root of the mass average of s
i2 over all mass elements and can be calculated as follows: ^ ^ ^/ଶ ^^
^ ൌ ൭^ ^^
^ ∙ ^^
^ଶ ^ ^ ^^
^ ^ The radius of gyration can be determined
by using light scattering. In particular, for small scattering vectors ^^^ the structure function S is defined as follows: ^
∙ ^1 െ^^ଶ ∙ ଶ ^
^^ ^^^ ^ ^^^^ 3
^ wherein N is the number of components
The "hydrodynamic radius" (which is sometimes called "Stokes radius" or "Stokes-Einstein radius") of a particle is the radius of a hypothetical hard sphere that diffuses at the same rate as said particle. The hydrodynamic radius is related to the mobility of the particle, taking into account not only size but also solvent effects. For example, a smaller charged particle with stronger hydration may have a greater hydrodynamic radius than a larger charged particle with weaker hydration. This is because the smaller particle drags a greater number of water molecules with it as it moves through the solution. Since the actual dimensions of the particle in a solvent are not directly measurable, the hydrodynamic radius may be defined by the Stokes-Einstein equation: ^
^^ ∙ ^^ ^^^ ൌ 6
∙ ^^ ∙ ^^ ∙ ^^ wherein k
B is the Boltzmann constant; T is the temperature; η is the viscosity of the solvent; and D is the diffusion coefficient. The diffusion coefficient can be determined experimentally, e.g., by using dynamic light scattering (DLS). 35 11529421v1 Attorney Docket No. 2013237‐0757 Thus, one procedure to determine the hydrodynamic radius of a particle or a population of particles (such as the hydrodynamic radius of particles contained in a sample or control composition as disclosed herein or the hydrodynamic radius of a particle peak obtained from subjecting such a sample or control composition to field-flow fractionation) is to measure the DLS signal of said particle or population of particles (such as DLS signal of particles contained in a sample or control composition as disclosed herein or the DLS signal of a particle peak obtained from subjecting such a sample or control composition to field-flow fractionation). The expression "light scattering" as used herein refers to the physical process where light is forced to deviate from a straight trajectory by one or more paths due to localized non-uniformities in the medium through which the light passes. The term "UV" means ultraviolet and designates a band of the electromagnetic spectrum with a wavelength from 10 nm to 400 nm, i.e., shorter than that of visible light but longer than X-rays. The expression "multi-angle light scattering" or "MALS" as used herein relates to a technique for measuring the light scattered by a sample into a plurality of angles. "Multi-angle" means in this respect that scattered light can be detected at different discrete angles as measured, for example, by a single detector moved over a range including the specific angles selected or an array of detectors fixed at specific angular locations. In certain embodiments, the light source used in MALS is a laser source (MALLS: multi-angle laser light scattering). Based on the MALS signal of a composition comprising particles and by using an appropriate formalism (e.g., Zimm plot, Berry plot, or Debye plot), it is possible to determine the radius of gyration (R
g) and, thus, the size of said particles. Preferably, the Zimm plot is a graphical presentation using the following equation: ^^
ఏ ൌ ^^ ^^
^ ^^
^ െ 2 ^
ଶ ଶ ^
^∗ ^^ ௪ ^
ଶ ^^ ^^
௪ ^^ ^ ^^^ wherein c is the mass concentration of
2 A
2 is the second virial coefficient (mol∙mL/g ); P(θ) is a form factor relating to the dependence of scattered light intensity on angle; R
θ is the excess Rayleigh ratio (cm
-1); and K* is an optical constant that is equal to 4π
2η
o (dn/dc)
2λ
0-4N
A-1, where η
o is the refractive index of the solvent at the incident radiation (vacuum) wavelength, λ
0 is the incident radiation (vacuum) wavelength (nm), N
A is Avogadro’s number (mol
-1), and dn/dc is the differential refractive index increment (mL/g) (cf., e.g., Buchholz et al. (Electrophoresis 22 (2001), 4118-4128); B.H. Zimm (J. Chem. Phys.13 (1945), 141; P. Debye (J. Appl. Phys.15 (1944): 338; and W. Burchard (Anal. Chem.75 (2003), 4279-4291). Preferably, the Berry plot is calculated using the following term or the reciprocal thereof: ^
^^ఏ ^
^∗ ^^ wherein c, R
θ and K* are as defined above.
Debye plot is calculated using the following term or the reciprocal thereof: ^^
∗ ^^ ^
^ఏ wherein c, R
θ and K* are as defined above.
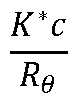
The expression "dynamic light scattering" or "DLS" as used herein refers to a technique to determine the size and size distribution profile of particles, in particular with respect to the hydrodynamic radius of the particles. A monochromatic 36 11529421v1 Attorney Docket No. 2013237‐0757 light source, usually a laser, is shot through a polarizer and into a sample. The scattered light then goes through a second polarizer where it is detected and the resulting image is projected onto a screen. The particles in the solution are being hit with the light and diffract the light in all directions. The diffracted light from the particles can either interfere constructively (light regions) or destructively (dark regions). This process is repeated at short time intervals and the resulting set of speckle patterns are analyzed by an autocorrelator that compares the intensity of light at each spot over time. The expression "static light scattering" or "SLS" as used herein refers to a technique to determine the size and size distribution profile of particles, in particular with respect to the radius of gyration of the particles, and/or the molar mass of particles. A high-intensity monochromatic light, usually a laser, is launched in a solution containing the particles. One or many detectors are used to measure the scattering intensity at one or many angles. The angular dependence is needed to obtain accurate measurements of both molar mass and size for all macromolecules of radius. Hence simultaneous measurements at several angles relative to the direction of incident light, known as multi-angle light scattering (MALS) or multi-angle laser light scattering (MALLS), is generally regarded as the standard implementation of static light scattering. Nucleic Acids The term "nucleic acid" comprises deoxyribonucleic acid (DNA), ribonucleic acid (RNA), combinations thereof, and modified forms thereof. The term comprises genomic DNA, cDNA, mRNA, recombinantly produced and chemically synthesized molecules. In some embodiments, a nucleic acid is DNA. In some embodiments, a nucleic acid is RNA. In some embodiments, a nucleic acid is a mixture of DNA and RNA. A nucleic acid may be present as a single-stranded or double-stranded and linear or covalently circularly closed molecule. A nucleic acid can be isolated. The term "isolated nucleic acid" means, according to the present disclosure, that the nucleic acid (i) was amplified in vitro, for example via polymerase chain reaction (PCR) for DNA or in vitro transcription (using, e.g., an RNA polymerase) for RNA, (ii) was produced recombinantly by cloning, (iii) was purified, for example, by cleavage and separation by gel electrophoresis, or (iv) was synthesized, for example, by chemical synthesis. The term "nucleoside" (abbreviated herein as "N") relates to compounds which can be thought of as nucleotides without a phosphate group. While a nucleoside is a nucleobase linked to a sugar (e.g., ribose or deoxyribose), a nucleotide is composed of a nucleoside and one or more phosphate groups. Examples of nucleosides include cytidine, uridine, pseudouridine, adenosine, and guanosine. The five standard nucleosides which usually make up naturally occurring nucleic acids are uridine, adenosine, thymidine, cytidine and guanosine. The five nucleosides are commonly abbreviated to their one letter codes U, A, T, C and G, respectively. However, thymidine is more commonly written as "dT" ("d" represents "deoxy") as it contains a 2'-deoxyribofuranose moiety rather than the ribofuranose ring found in uridine. This is because thymidine is found in deoxyribonucleic acid (DNA) and not ribonucleic acid (RNA). Conversely, uridine is found in RNA and not DNA. The remaining three nucleosides may be found in both RNA and DNA. In RNA, they would be represented as A, C and G, whereas in DNA they would be represented as dA, dC and dG. A modified purine (A or G) or pyrimidine (C, T, or U) base moiety is, in some embodiments, modified by one or more alkyl groups, e.g., one or more C
1-4 alkyl groups, e.g., one or more methyl groups. Particular examples of modified purine or pyrimidine base moieties include N
7-alkyl-guanine, N
6-alkyl-adenine, 5-alkyl-cytosine, 5-alkyl-uracil, and N(1)- alkyl-uracil, such as N
7-C
1-4 alkyl-guanine, N
6-C
1-4 alkyl-adenine, 5-C
1-4 alkyl-cytosine, 5-C
1-4 alkyl-uracil, and N(1)-C
1-4 37 11529421v1 Attorney Docket No. 2013237‐0757 alkyl-uracil, preferably N
7-methyl-guanine, N
6-methyl-adenine, 5-methyl-cytosine, 5-methyl-uracil, and N(1)-methyl- uracil. DNA Herein, the term "DNA" relates to a nucleic acid molecule which is entirely or at least substantially composed of deoxyribonucleotide residues. In preferred embodiments, the DNA contains all or a majority of deoxyribonucleotide residues. As used herein, "deoxyribonucleotide" refers to a nucleotide which lacks a hydroxyl group at the 2'-position of a β-D-ribofuranosyl group. DNA encompasses without limitation, double stranded DNA, single stranded DNA, isolated DNA such as partially purified DNA, essentially pure DNA, synthetic DNA, recombinantly produced DNA, as well as modified DNA that differs from naturally occurring DNA by the addition, deletion, substitution and/or alteration of one or more nucleotides. Such alterations may refer to addition of non-nucleotide material to internal DNA nucleotides or to the end(s) of DNA. It is also contemplated herein that nucleotides in DNA may be non-standard nucleotides, such as chemically synthesized nucleotides or ribonucleotides. For the present disclosure, these altered DNAs are considered analogs of naturally-occurring DNA. A molecule contains "a majority of deoxyribonucleotide residues" if the content of deoxyribonucleotide residues in the molecule is more than 50% (such as at least 55%, at least 60%, at least 65%, at least 70%, at least 75%, at least 80%, at least 85%, at least 90%, at least 95%, at least 96%, at least 97%, at least 98%, at least 99%), based on the total number of nucleotide residues in the molecule. The total number of nucleotide residues in a molecule is the sum of all nucleotide residues (irrespective of whether the nucleotide residues are standard (i.e., naturally occurring) nucleotide residues or analogs thereof). DNA may be recombinant DNA and may be obtained by cloning of a nucleic acid, in particular cDNA. The cDNA may be obtained by reverse transcription of RNA. RNA The term "RNA" relates to a nucleic acid molecule which includes ribonucleotide residues. In preferred embodiments, the RNA contains all or a majority of ribonucleotide residues. As used herein, "ribonucleotide" refers to a nucleotide with a hydroxyl group at the 2'-position of a β-D-ribofuranosyl group. RNA encompasses without limitation, double stranded RNA, single stranded RNA, isolated RNA such as partially purified RNA, essentially pure RNA, synthetic RNA, recombinantly produced RNA, as well as modified RNA that differs from naturally occurring RNA by the addition, deletion, substitution and/or alteration of one or more nucleotides. Such alterations may refer to addition of non- nucleotide material to internal RNA nucleotides or to the end(s) of RNA. It is also contemplated herein that nucleotides in RNA may be non-standard nucleotides, such as chemically synthesized nucleotides or deoxynucleotides. For the present disclosure, these altered/modified nucleotides can be referred to as analogs of naturally occurring nucleotides, and the corresponding RNAs containing such altered/modified nucleotides (i.e., altered/modified RNAs) can be referred to as analogs of naturally occurring RNAs. A molecule contains "a majority of ribonucleotide residues" if the content of ribonucleotide residues in the molecule is more than 50% (such as at least 55%, at least 60%, at least 65%, at least 70%, at least 75%, at least 80%, at least 85%, at least 90%, at least 95%, at least 96%, at least 97%, at least 98%, at least 99%), based on the total number of nucleotide residues in the molecule. The total number of nucleotide residues in a molecule is the sum of all nucleotide residues (irrespective of whether the nucleotide residues are standard (i.e., naturally occurring) nucleotide residues or analogs thereof). 38 11529421v1 Attorney Docket No. 2013237‐0757 "RNA" includes mRNA, tRNA, ribosomal RNA (rRNA), small nuclear RNA (snRNA), self-amplifying RNA (saRNA), trans- amplifying RNA (taRNA), single-stranded RNA (ssRNA), dsRNA, inhibitory RNA (such as antisense ssRNA, small interfering RNA (siRNA), or microRNA (miRNA)), activating RNA (such as small activating RNA) and immunostimulatory RNA (isRNA). In some embodiments, "RNA" refers to mRNA. The term "in vitro transcription" or "IVT" as used herein means that the transcription (i.e., the generation of RNA) is conducted in a cell-free manner. I.e., IVT does not use living/cultured cells but rather the transcription machinery extracted from cells (e.g., cell lysates or the isolated components thereof, including an RNA polymerase (preferably T7, T3 or SP6 polymerase)). According to the present disclosure, the term '"RNA" includes "mRNA". According to the present disclosure, the term "mRNA" means "messenger-RNA" and includes a "transcript" which may be generated by using a DNA template. Generally, mRNA encodes a peptide or polypeptide. mRNA is single-stranded but may contain self-complementary sequences that allow parts of the mRNA to fold and pair with itself to form double helices. According to the present disclosure, "dsRNA" means double-stranded RNA and is RNA with two partially or completely complementary strands. In preferred embodiments of the present disclosure, the mRNA relates to an RNA transcript which encodes a peptide or polypeptide. In some embodiments, the mRNA which preferably encodes a peptide or polypeptide has a length of at least 45 nucleotides (such as at least 60, at least 90, at least 100, at least 200, at least 300, at least 400, at least 500, at least 600, at least 700, at least 800, at least 900, at least 1,000, at least 1,500, at least 2,000, at least 2,500, at least 3,000, at least 3,500, at least 4,000, at least 4,500, at least 5,000, at least 6,000, at least 7,000, at least 8,000, at least 9,000 nucleotides), preferably up to 15,000, such as up to 14,000, up to 13,000, up to 12,000 nucleotides, up to 11,000 nucleotides or up to 10,000 nucleotides. As established in the art, mRNA generally contains a 5' untranslated region (5'-UTR), a peptide/polypeptide coding region and a 3' untranslated region (3'-UTR). In some embodiments, the mRNA is produced by in vitro transcription or chemical synthesis. In some embodiments, the mRNA is produced by in vitro transcription using a DNA template. The in vitro transcription methodology is known to the skilled person; cf., e.g., Molecular Cloning: A Laboratory Manual, 4
th Edition, M.R. Green and J. Sambrook eds., Cold Spring Harbor Laboratory Press, Cold Spring Harbor 2012. Furthermore, a variety of in vitro transcription kits is commercially available, e.g., from Thermo Fisher Scientific (such as TranscriptAid
TM T7 kit, MEGAscript® T7 kit, MAXIscript®), New England BioLabs Inc. (such as HiScribe™ T7 kit, HiScribe™ T7 ARCA mRNA kit), Promega (such as RiboMAX™, HeLaScribe®, Riboprobe® systems), Jena Bioscience (such as SP6 or T7 transcription kits), and Epicentre (such as AmpliScribe™). For providing modified mRNA, correspondingly modified nucleotides, such as modified naturally occurring nucleotides, non-naturally occurring nucleotides and/or modified non-naturally occurring nucleotides, can be incorporated during synthesis (preferably in vitro transcription), or modifications can be effected in and/or added to the mRNA after transcription. In some embodiments, RNA is in vitro transcribed RNA (IVT-RNA) and may be obtained by in vitro transcription of an appropriate DNA template. The promoter for controlling transcription can be any promoter for any RNA polymerase. Particular examples of RNA polymerases are the T7, T3, and SP6 RNA polymerases. Preferably, the in vitro transcription 39 11529421v1 Attorney Docket No. 2013237‐0757 is controlled by a T7 or SP6 promoter. A DNA template for in vitro transcription may be obtained by cloning of a nucleic acid, in particular cDNA, and introducing it into an appropriate vector for in vitro transcription. The cDNA may be obtained by reverse transcription of RNA. In some embodiments of the present disclosure, the RNA is "replicon RNA" or simply a "replicon", in particular "self- replicating RNA" or "self-amplifying RNA". In certain embodiments, the replicon or self-replicating RNA is derived from or comprises elements derived from an ssRNA virus, in particular a positive-stranded ssRNA virus such as an alphavirus. Alphaviruses are typical representatives of positive-stranded RNA viruses. Alphaviruses replicate in the cytoplasm of infected cells (for review of the alphaviral life cycle see José et al., Future Microbiol., 2009, vol. 4, pp. 837–856). The total genome length of many alphaviruses typically ranges between 11,000 and 12,000 nucleotides, and the genomic RNA typically has a 5’-cap, and a 3’ poly(A) tail. The genome of alphaviruses encodes non-structural proteins (involved in transcription, modification and replication of viral RNA and in protein modification) and structural proteins (forming the virus particle). There are typically two open reading frames (ORFs) in the genome. The four non-structural proteins (nsP1–nsP4) are typically encoded together by a first ORF beginning near the 5′ terminus of the genome, while alphavirus structural proteins are encoded together by a second ORF which is found downstream of the first ORF and extends near the 3’ terminus of the genome. Typically, the first ORF is larger than the second ORF, the ratio being roughly 2:1. In cells infected by an alphavirus, only the nucleic acid sequence encoding non-structural proteins is translated from the genomic RNA, while the genetic information encoding structural proteins is translatable from a subgenomic transcript, which is an RNA molecule that resembles eukaryotic messenger RNA (mRNA; Gould et al., 2010, Antiviral Res., vol.87 pp.111–124). Following infection, i.e. at early stages of the viral life cycle, the (+) stranded genomic RNA directly acts like a messenger RNA for the translation of the open reading frame encoding the non- structural poly-protein (nsP1234). Alphavirus-derived vectors have been proposed for delivery of foreign genetic information into target cells or target organisms. In simple approaches, the open reading frame encoding alphaviral structural proteins is replaced by an open reading frame encoding a polypeptide of interest. Alphavirus-based trans-replication (trans-amplification) systems rely on alphavirus nucleotide sequence elements on two separate nucleic acid molecules: one nucleic acid molecule encodes a viral replicase, and the other nucleic acid molecule is capable of being replicated by said replicase in trans (hence the designation trans-replication system). Trans-replication requires the presence of both these nucleic acid molecules in a given host cell. The nucleic acid molecule capable of being replicated by the replicase in trans must comprise certain alphaviral sequence elements to allow recognition and RNA synthesis by the alphaviral replicase. In some embodiments of the present disclosure, the RNA (in particular, mRNA) described herein (e.g., contained in the compositions/formulations of the present disclosure and/or used in the methods of the present disclosure) contains one or more modifications, e.g., in order to increase its stability and/or increase translation efficiency and/or decrease immunogenicity and/or decrease cytotoxicity. For example, in order to increase expression of the RNA (in particular, mRNA), it may be modified within the coding region, i.e., the sequence encoding the expressed peptide or polypeptide, preferably without altering the sequence of the expressed peptide or polypeptide. Such modifications are described, for example, in WO 2007/036366 and PCT/EP2019/056502, and include the following: a 5'-cap structure; an extension or truncation of the naturally occurring poly(A) tail; an alteration of the 5'- and/or 3'-untranslated regions (UTR) such as introduction of a UTR which is not related to the coding region of said RNA; the replacement of one or more naturally occurring nucleotides with synthetic nucleotides; and codon optimization (e.g., to alter, preferably increase, the GC content of the RNA). A combination of the above described modifications, i.e., incorporation of a 5'-cap structure, incorporation of a poly-A sequence, unmasking of a poly-A sequence, alteration of the 5'- and/or 3'-UTR (such as 40 11529421v1 Attorney Docket No. 2013237‐0757 incorporation of one or more 3'-UTRs), replacing one or more naturally occurring nucleotides with synthetic nucleotides (e.g., 5-methylcytidine for cytidine and/or pseudouridine (Ψ) or N(1)-methylpseudouridine (m1Ψ) or 5-methyluridine (m5U) for uridine), and codon optimization, has a synergistic influence on the stability of RNA (preferably mRNA) and increase in translation efficiency. Thus, in some embodiments, the RNA (in particular, mRNA) described in the present disclosure contains a combination of at least two, at least three, at least four or all five of the above-mentioned modifications, i.e., (i) incorporation of a 5'-cap structure, (ii) incorporation of a poly-A sequence, unmasking of a poly- A sequence; (iii) alteration of the 5'- and/or 3'-UTR (such as incorporation of one or more 3'-UTRs); (iv) replacing one or more naturally occurring nucleotides with synthetic nucleotides (e.g., 5-methylcytidine for cytidine and/or pseudouridine (Ψ) or N(1)-methylpseudouridine (m1Ψ) or 5-methyluridine (m5U) for uridine), and (v) codon optimization. 5'-Cap In some embodiments, the RNA (in particular, mRNA) described herein comprises a 5'-cap structure. In some embodiments, the RNA does not have uncapped 5'-triphosphates. In some embodiments, the RNA (in particular, mRNA) may comprise a conventional 5'-cap and/or a 5'-cap analog. The term "conventional 5'-cap" refers to a cap structure found on the 5'-end of an RNA molecule and generally comprises a guanosine 5'-triphosphate (Gppp) which is connected via its triphosphate moiety to the 5'-end of the next nucleotide of the RNA (i.e., the guanosine is connected via a 5' to 5' triphosphate linkage to the rest of the RNA). The guanosine may be methylated at position N
7 (resulting in the cap structure m
7Gppp). The term "5'-cap analog" includes a 5'-cap which is based on a conventional 5'-cap but which has been modified at either the 2'- or 3'-position of the m
7guanosine structure in order to avoid an integration of the 5'-cap analog in the reverse orientation (such 5'-cap analogs are also called anti-reverse cap analogs (ARCAs)). Particularly preferred 5'-cap analogs are those having one or more substitutions at the bridging and non-bridging oxygen in the phosphate bridge, such as phosphorothioate modified 5'-cap analogs at the β-phosphate (such as m
27,2'OG(5')ppSp(5')G (referred to as beta-S-ARCA or β-S-ARCA)), as described in PCT/EP2019/056502. Providing an RNA (in particular, mRNA) with a 5'-cap structure as described herein may be achieved by in vitro transcription of a DNA template in presence of a corresponding 5'-cap compound, wherein said 5'-cap structure is co-transcriptionally incorporated into the generated RNA (in particular, mRNA) strand, or the RNA (in particular, mRNA) may be generated, for example, by in vitro transcription, and the 5'-cap structure may be attached to the RNA post-transcriptionally using capping enzymes, for example, capping enzymes of vaccinia virus. In some embodiments, the RNA (in particular, mRNA) comprises a 5'-cap structure selected from the group consisting of m
27,2'OG(5’)ppSp(5')G (in particular its D1 diastereomer), m
27,3'OG(5')ppp(5')G, and m
27,3'-OGppp(m
12'-O)ApG. In some embodiments, RNA comprises m
27,2'OG(5’)ppSp(5')G (in particular its D1 diastereomer) as 5'-cap structure. In some embodiments, RNA comprises m2
7,3'-OGppp(m1
2'-O)ApG as 5'-cap structure. In some embodiments, the RNA (in particular, mRNA) comprises a cap0, cap1, or cap2, preferably cap1 or cap2. According to the present disclosure, the term "cap0" means the structure "m
7GpppN", wherein N is any nucleoside bearing an OH moiety at position 2'. According to the present disclosure, the term "cap1" means the structure "m
7GpppNm", wherein Nm is any nucleoside bearing an OCH
3 moiety at position 2'. According to the present disclosure, the term "cap2" means the structure "m
7GpppNmNm", wherein each Nm is independently any nucleoside bearing an OCH
3 moiety at position 2'. The 5'-cap analog beta-S-ARCA (β-S-ARCA) has the following structure: 41 11529421v1 Attorney Docket No. 2013237‐0757 H
3C O OH O .
The "D1 diastereomer of beta-S-ARCA" or "beta-S-ARCA(D1)" is the diastereomer of beta-S-ARCA which elutes first on an HPLC column compared to the D2 diastereomer of beta-S-ARCA (beta-S-ARCA(D2)) and thus exhibits a shorter retention time. The HPLC preferably is an analytical HPLC. In some embodiments, a Supelcosil LC-18-T RP column, preferably of the format: 5 μm, 4.6 x 250 mm is used for separation, whereby a flow rate of 1.3 ml/min can be applied. In some embodiments, a gradient of methanol in ammonium acetate, for example, a 0-25% linear gradient of methanol in 0.05 M ammonium acetate, pH = 5.9, within 15 min is used. UV-detection (VWD) can be performed at 260 nm and fluorescence detection (FLD) can be performed with excitation at 280 nm and detection at 337 nm. The 5'-cap analog m
27,3'-OGppp(m
12'-O)ApG (also referred to as m
27,3'OG(5')ppp(5')m
2'-OApG) which is a building block of a cap1 has the following structure: CH
3 H O O
NH2 2 . An
42 11529421v1 Attorney Docket No. 2013237‐0757 H
3C O OH O 2 .
An m
2 ppp structure: CH
3 H
OO O 2 .
An exemplary cap1 mRNA comprising m
27,3'-OGppp(m
12'-O)ApG and mRNA has the following structure: CH
3 H O O
NH2 2 .

43 11529421v1 Attorney Docket No. 2013237‐0757 Poly-A tail As used herein, the term "poly-A tail" or "poly-A sequence" refers to an uninterrupted or interrupted sequence of adenylate residues which is typically located at the 3'-end of an RNA (in particular, mRNA) molecule. Poly-A tails or poly-A sequences are known to those of skill in the art and may follow the 3’-UTR in the RNAs (in particular, mRNAs) described herein. An uninterrupted poly-A tail is characterized by consecutive adenylate residues. In nature, an uninterrupted poly-A tail is typical. RNAs (in particular, mRNAs) disclosed herein can have a poly-A tail attached to the free 3'-end of the RNA by a template-independent RNA polymerase after transcription or a poly-A tail encoded by DNA and transcribed by a template-dependent RNA polymerase. It has been demonstrated that a poly-A tail of about 120 A nucleotides has a beneficial influence on the levels of RNA in transfected eukaryotic cells, as well as on the levels of polypeptide that is translated from an open reading frame that is present upstream (5’) of the poly-A tail (Holtkamp et al., 2006, Blood, vol. 108, pp. 4009-4017). The poly-A tail may be of any length. In some embodiments, a poly-A tail comprises, essentially consists of, or consists of at least 20, at least 30, at least 40, at least 80, or at least 100 and up to 500, up to 400, up to 300, up to 200, or up to 150 A nucleotides, and, in particular, about 120 A nucleotides. In this context, "essentially consists of" means that most nucleotides in the poly-A tail, typically at least 75%, at least 80%, at least 85%, at least 90%, at least 95%, at least 96%, at least 97%, at least 98%, or at least 99% by number of nucleotides in the poly-A tail are A nucleotides, but permits that remaining nucleotides are nucleotides other than A nucleotides, such as U nucleotides (uridylate), G nucleotides (guanylate), or C nucleotides (cytidylate). In this context, "consists of" means that all nucleotides in the poly-A tail, i.e., 100% by number of nucleotides in the poly-A tail, are A nucleotides. The term "A nucleotide" or "A" refers to adenylate. In some embodiments, a poly-A tail is attached during RNA transcription, e.g., during preparation of in vitro transcribed RNA, based on a DNA template comprising repeated dT nucleotides (deoxythymidylate) in the strand complementary to the coding strand. The DNA sequence encoding a poly-A tail (coding strand) is referred to as poly(A) cassette. In some embodiments, the poly(A) cassette present in the coding strand of DNA essentially consists of dA nucleotides, but is interrupted by a random sequence of the four nucleotides (dA, dC, dG, and dT). Such random sequence may be 5 to 50, 10 to 30, or 10 to 20 nucleotides in length. Such a cassette is disclosed in WO 2016/005324 A1, hereby incorporated by reference. Any poly(A) cassette disclosed in WO 2016/005324 A1 may be used in the present disclosure. A poly(A) cassette that essentially consists of dA nucleotides, but is interrupted by a random sequence having an equal distribution of the four nucleotides (dA, dC, dG, dT) and having a length of e.g., 5 to 50 nucleotides shows, on DNA level, constant propagation of plasmid DNA in E. coli and is still associated, on RNA level, with the beneficial properties with respect to supporting RNA stability and translational efficiency is encompassed. Consequently, in some embodiments, the poly-A tail contained in an RNA (in particular, mRNA) molecule described herein essentially consists of A nucleotides, but is interrupted by a random sequence of the four nucleotides (A, C, G, U). Such random sequence may be 5 to 50, 10 to 30, or 10 to 20 nucleotides in length. In some embodiments, the poly(A) tail comprises 30 adenine nucleotides followed by 70 adenine nucleotides, wherein the 30 adenine nucleotides and 70 adenine nucleotides are separated by a linker sequence of 10 nucleotides. In some embodiments, no nucleotides other than A nucleotides flank a poly-A tail at its 3'-end, i.e., the poly-A tail is not masked or followed at its 3'-end by a nucleotide other than A. 44 11529421v1 Attorney Docket No. 2013237‐0757 In some embodiments, a poly-A tail may comprise at least 20, at least 30, at least 40, at least 80, or at least 100 and up to 500, up to 400, up to 300, up to 200, or up to 150 nucleotides. In some embodiments, the poly-A tail may essentially consist of at least 20, at least 30, at least 40, at least 80, or at least 100 and up to 500, up to 400, up to 300, up to 200, or up to 150 nucleotides. In some embodiments, the poly-A tail may consist of at least 20, at least 30, at least 40, at least 80, or at least 100 and up to 500, up to 400, up to 300, up to 200, or up to 150 nucleotides. In some embodiments, the poly-A tail comprises the poly-A tail shown in SEQ ID NO: 42. In some embodiments, the poly- A tail comprises at least 100 nucleotides. In some embodiments, the poly-A tail comprises about 150 nucleotides. In some embodiments, the poly-A tail comprises about 120 nucleotides. Untranslated regions (UTR) In some embodiments, RNA (in particular, mRNA) described in present disclosure comprises a 5'-UTR and/or a 3'-UTR. The term "untranslated region" or "UTR" relates to a region in a DNA molecule which is transcribed but is not translated into an amino acid sequence, or to the corresponding region in an RNA molecule, such as an mRNA molecule. An untranslated region (UTR) can be present 5' (upstream) of an open reading frame (5'-UTR) and/or 3' (downstream) of an open reading frame (3'-UTR). A 5'-UTR, if present, is located at the 5'-end, upstream of the start codon of a protein- encoding region. A 5'-UTR is downstream of the 5'-cap (if present), e.g., directly adjacent to the 5'-cap. A 3'-UTR, if present, is located at the 3'-end, downstream of the termination codon of a protein-encoding region, but the term "3'- UTR" does generally not include the poly-A sequence. Thus, the 3'-UTR is upstream of the poly-A sequence (if present), e.g., directly adjacent to the poly-A sequence. Incorporation of a 3'-UTR into the 3'-non translated region of an RNA (preferably mRNA) molecule can result in an enhancement in translation efficiency. A synergistic effect may be achieved by incorporating two or more of such 3'-UTRs (which are preferably arranged in a head-to-tail orientation; cf., e.g., Holtkamp et al., Blood 108, 4009-4017 (2006)). The 3'-UTRs may be autologous or heterologous to the RNA (e.g., mRNA) into which they are introduced. In certain embodiments, the 3'-UTR is derived from a globin gene or mRNA, such as a gene or mRNA of alpha2-globin, alpha1-globin, or beta-globin, e.g., beta-globin, e.g., human beta-globin. For example, the RNA (e.g., mRNA) may be modified by the replacement of the existing 3'-UTR with or the insertion of one or more, e.g., two copies of a 3'-UTR derived from a globin gene, such as alpha2-globin, alpha1-globin, beta- globin, e.g., beta-globin, e.g., human beta-globin. In some embodiments, a 5’-UTR is or comprises a modified human alpha-globin 5’-UTR. A particularly preferred 5’-UTR comprises the nucleotide sequence of SEQ ID NO: 40. In some embodiments, a 3’-UTR comprises a first sequence from the amino terminal enhancer of split (AES) messenger RNA and a second sequence from the mitochondrial encoded 12S ribosomal RNA. A particularly preferred 3’-UTR comprises the nucleotide sequence of SEQ ID NO: 41. In some embodiments, RNA comprises a 5’-UTR comprising the nucleotide sequence of SEQ ID NO: 40, or a nucleotide sequence having at least 99%, 98%, 97%, 96%, 95%, 90%, 85%, or 80% identity to the nucleotide sequence of SEQ ID NO: 40. In some embodiments, RNA comprises a 3’-UTR comprising the nucleotide sequence of SEQ ID NO: 41, or a nucleotide sequence having at least 99%, 98%, 97%, 96%, 95%, 90%, 85%, or 80% identity to the nucleotide sequence of SEQ ID NO: 41. 45 11529421v1 Attorney Docket No. 2013237‐0757 Table 1: Exemplary untranslated RNA sequences Type Sequence 5’-UTR aactagtatt cttctggtcc ccacagactc agagagaacc cgccacc [SEQ ID NO: 40] 3’-UTR ctggtactgc atgcacgcaa tgctagctgc ccctttcccg tcctgggtac cccgagtctc ccccgacctc gggtcccagg tatgctccca cctccacctg ccccactcac cacctctgct agttccagac acctcccaag cacgcagcaa tgcagctcaa aacgcttagc ctagccacac ccccacggga aacagcagtg attaaccttt agcaataaac gaaagtttaa ctaagctata ctaaccccag ggttggtcaa tttcgtgcca gccacacc [SEQ ID NO: 41] polyA aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa gcatatgact aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa [SEQ ID NO: 42] Chemical modification The RNA (in particular, mRNA) described herein may have modified ribonucleotides in order to increase its stability and/or decrease immunogenicity and/or decrease cytotoxicity. For example, in some embodiments, uridine in the RNA (in particular, mRNA) described herein is replaced (partially or completely, preferably completely) by a modified nucleoside. In some embodiments, the modified nucleoside is a modified uridine. In some embodiments, the modified uridine replacing uridine is selected from the group consisting of pseudouridine (ψ), N1-methyl-pseudouridine (m1ψ), 5-methyl-uridine (m5U), and combinations thereof. In some embodiments, the modified nucleoside replacing (partially or completely, preferably completely) uridine in the RNA may be any one or more of 3-methyl-uridine (m3U), 5-methoxy-uridine (mo5U), 5-aza-uridine, 6-aza-uridine, 2- thio-5-aza-uridine, 2-thio-uridine (s2U), 4-thio-uridine (s4U), 4-thio-pseudouridine, 2-thio-pseudouridine, 5-hydroxy- uridine (ho5U), 5-aminoallyl-uridine, 5-halo-uridine (e.g., 5-iodo-uridineor 5-bromo-uridine), uridine 5-oxyacetic acid (cmo5U), uridine 5-oxyacetic acid methyl ester (mcmo5U), 5-carboxymethyl-uridine (cm5U), 1-carboxymethyl- pseudouridine, 5-carboxyhydroxymethyl-uridine (chm5U), 5-carboxyhydroxymethyl-uridine methyl ester (mchm5U), 5- methoxycarbonylmethyl-uridine (mcm5U), 5-methoxycarbonylmethyl-2-thio-uridine (mcm5s2U), 5-aminomethyl-2- thio-uridine (nm5s2U), 5-methylaminomethyl-uridine (mnm5U), 1-ethyl-pseudouridine, 5-methylaminomethyl-2-thio- uridine (mnm5s2U), 5-methylaminomethyl-2-seleno-uridine (mnm5se2U), 5-carbamoylmethyl-uridine (ncm5U), 5- carboxymethylaminomethyl-uridine (cmnm5U), 5-carboxymethylaminomethyl-2-thio-uridine (cmnm5s2U), 5-propynyl- uridine, 1-propynyl-pseudouridine, 5-taurinomethyl-uridine (τm5U), 1-taurinomethyl-pseudouridine, 5-taurinomethyl- 2-thio-uridine(τm5s2U), 1-taurinomethyl-4-thio-pseudouridine), 5-methyl-2-thio-uridine (m5s2U), 1-methyl-4-thio- pseudouridine (m1s4ψ), 4-thio-1-methyl-pseudouridine, 3-methyl-pseudouridine (m3ψ), 2-thio-1-methyl- pseudouridine, 1-methyl-1-deaza-pseudouridine, 2-thio-1-methyl-1-deaza-pseudouridine, dihydrouridine (D), dihydropseudouridine, 5,6-dihydrouridine, 5-methyl-dihydrouridine (m5D), 2-thio-dihydrouridine, 2-thio- dihydropseudouridine, 2-methoxy-uridine, 2-methoxy-4-thio-uridine, 4-methoxy-pseudouridine, 4-methoxy-2-thio- pseudouridine, N1-methyl-pseudouridine, 3-(3-amino-3-carboxypropyl)uridine (acp3U), 1-methyl-3-(3-amino-3- carboxypropyl)pseudouridine (acp3 ψ), 5-(isopentenylaminomethyl)uridine (inm5U), 5-(isopentenylaminomethyl)-2- thio-uridine (inm5s2U), α-thio-uridine, 2′-O-methyl-uridine (Um), 5,2′-O-dimethyl-uridine (m5Um), 2′-O-methyl- pseudouridine (ψm), 2-thio-2′-O-methyl-uridine (s2Um), 5-methoxycarbonylmethyl-2′-O-methyl-uridine (mcm5Um), 5- carbamoylmethyl-2′-O-methyl-uridine (ncm5Um), 5-carboxymethylaminomethyl-2′-O-methyl-uridine (cmnm5Um), 46 11529421v1 Attorney Docket No. 2013237‐0757 3,2′-O-dimethyl-uridine (m3Um), 5-(isopentenylaminomethyl)-2′-O-methyl-uridine (inm5Um), 1-thio-uridine, deoxythymidine, 2′-F-ara-uridine, 2′-F-uridine, 2′-OH-ara-uridine, 5-(2-carbomethoxyvinyl) uridine, 5-[3-(1-E- propenylamino)uridine, or any other modified uridine known in the art. An RNA (preferably mRNA) which is modified by pseudouridine (replacing partially or completely, preferably completely, uridine) is referred to herein as "Ψ-modified", whereas the term "m1Ψ-modified" means that the RNA (preferably mRNA) contains N(1)-methylpseudouridine (replacing partially or completely, preferably completely, uridine). Furthermore, the term "m5U-modified" means that the RNA (preferably mRNA) contains 5-methyluridine (replacing partially or completely, preferably completely, uridine). Such Ψ- or m1Ψ- or m5U-modified RNAs usually exhibit decreased immunogenicity compared to their unmodified forms and, thus, are preferred in applications where the induction of an immune response is to be avoided or minimized. In some embodiments, the RNA (preferably mRNA) contains N(1)-methylpseudouridine replacing completely uridine. Codon optimization and GC enrichment The codons of the RNA (in particular, mRNA) described in the present disclosure may further be optimized, e.g., to increase the GC content of the RNA and/or to replace codons which are rare in the cell (or subject) in which the peptide or polypeptide of interest is to be expressed by codons which are synonymous frequent codons in said cell (or subject). In some embodiments, the amino acid sequence encoded by the RNA (in particular, mRNA) described in the present disclosure is encoded by a coding sequence which is codon-optimized and/or the G/C content of which is increased compared to wild type coding sequence. This also includes embodiments, wherein one or more sequence regions of the coding sequence are codon-optimized and/or increased in the G/C content compared to the corresponding sequence regions of the wild type coding sequence. In some embodiments, the codon-optimization and/or the increase in the G/C content preferably does not change the sequence of the encoded amino acid sequence. The term "codon-optimized" refers to the alteration of codons in the coding region of a nucleic acid molecule to reflect the typical codon usage of a host organism without preferably altering the amino acid sequence encoded by the nucleic acid molecule. Within the context of the present disclosure, coding regions may be codon-optimized for optimal expression in a subject to be treated using the RNA (in particular, mRNA) described herein. Codon-optimization is based on the finding that the translation efficiency is also determined by a different frequency in the occurrence of tRNAs in cells. Thus, the sequence of RNA (in particular, mRNA) may be modified such that codons for which frequently occurring tRNAs are available are inserted in place of "rare codons". In some embodiments, the guanosine/cytosine (G/C) content of the coding region of the RNA (in particular, mRNA) described herein is increased compared to the G/C content of the corresponding coding sequence of the wild type RNA, wherein the amino acid sequence encoded by the RNA is preferably not modified compared to the amino acid sequence encoded by the wild type RNA. This modification of the RNA sequence is based on the fact that the sequence of any RNA region to be translated is important for efficient translation of that RNA. Sequences having an increased G (guanosine)/C (cytosine) content are more stable than sequences having an increased A (adenosine)/U (uracil) content. In respect to the fact that several codons code for one and the same amino acid (so-called degeneration of the genetic code), the most favorable codons for the stability can be determined (so-called alternative codon usage). Depending on the amino acid to be encoded by the RNA, there are various possibilities for modification of the RNA sequence, compared to its wild type sequence. In particular, codons which contain A and/or U nucleotides can be modified by 47 11529421v1 Attorney Docket No. 2013237‐0757 substituting these codons by other codons, which code for the same amino acids but contain no A and/or U or contain a lower content of A and/or U nucleotides. In various embodiments, the G/C content of the coding region of the RNA (in particular, mRNA) described herein is increased by at least 10%, at least 20%, at least 30%, at least 40%, at least 50%, at least 55%, or even more compared to the G/C content of the coding region of the wild type RNA. Non-immunogenic RNA The term "non-immunogenic RNA" (such as "non-immunogenic mRNA") as used herein refers to RNA that does not induce a response by the immune system upon administration, e.g., to a mammal, or induces a weaker response than would have been induced by the same RNA that differs only in that it has not been subjected to the modifications and treatments that render the non-immunogenic RNA non-immunogenic, i.e., than would have been induced by standard RNA (stdRNA). In certain embodiments, non-immunogenic RNA is rendered non-immunogenic by incorporating modified nucleosides suppressing RNA-mediated activation of innate immune receptors into the RNA and/or limiting the amount of double-stranded RNA (dsRNA), e.g., by limiting the formation of double-stranded RNA (dsRNA), e.g., during in vitro transcription, and/or by removing double-stranded RNA (dsRNA), e.g., following in vitro transcription. In certain embodiments, non-immunogenic RNA is rendered non-immunogenic by incorporating modified nucleosides suppressing RNA-mediated activation of innate immune receptors into the RNA and/or by removing double-stranded RNA (dsRNA), e.g., following in vitro transcription. For rendering the non-immunogenic RNA (especially mRNA) non-immunogenic by the incorporation of modified nucleosides, any modified nucleoside may be used as long as it lowers or suppresses immunogenicity of the RNA. Particularly preferred are modified nucleosides that suppress RNA-mediated activation of innate immune receptors. In some embodiments, the modified nucleosides comprise a replacement of one or more uridines with a nucleoside comprising a modified nucleobase. In some embodiments, the modified nucleobase is a modified uracil. In some embodiments, the nucleoside comprising a modified nucleobase is selected from the group consisting of 3-methyl- uridine (m
3U), 5-methoxy-uridine (mo
5U), 5-aza-uridine, 6-aza-uridine, 2-thio-5-aza-uridine, 2-thio-uridine (s
2U), 4- thio-uridine (s
4U), 4-thio-pseudouridine, 2-thio-pseudouridine, 5-hydroxy-uridine (ho
5U), 5-aminoallyl-uridine, 5-halo- uridine (e.g., 5-iodo-uridine or 5-bromo-uridine), uridine 5-oxyacetic acid (cmo
5U), uridine 5-oxyacetic acid methyl ester (mcmo
5U), 5-carboxymethyl-uridine (cm
5U), 1-carboxymethyl-pseudouridine, 5-carboxyhydroxymethyl-uridine (chm
5U), 5-carboxyhydroxymethyl-uridine methyl ester (mchm
5U), 5-methoxycarbonylmethyl-uridine (mcm
5U), 5- methoxycarbonylmethyl-2-thio-uridine (mcm
5s
2U), 5-aminomethyl-2-thio-uridine (nm
5s
2U), 5-methylaminomethyl- uridine (mnm
5U), 1-ethyl-pseudouridine, 5-methylaminomethyl-2-thio-uridine (mnm
5s
2U), 5-methylaminomethyl-2- seleno-uridine (mnm
5se
2U), 5-carbamoylmethyl-uridine (ncm
5U), 5-carboxymethylaminomethyl-uridine (cmnm
5U), 5- carboxymethylaminomethyl-2-thio-uridine (cmnm
5s
2U), 5-propynyl-uridine, 1-propynyl-pseudouridine, 5- taurinomethyl-uridine (τm
5U), 1-taurinomethyl-pseudouridine, 5-taurinomethyl-2-thio-uridine(τm5s2U), 1- taurinomethyl-4-thio-pseudouridine), 5-methyl-2-thio-uridine (m
5s
2U), 1-methyl-4-thio-pseudouridine (m
1s
4ψ), 4-thio- 1-methyl-pseudouridine, 3-methyl-pseudouridine (m
3ψ), 2-thio-1-methyl-pseudouridine, 1-methyl-1-deaza- pseudouridine, 2-thio-1-methyl-1-deaza-pseudouridine, dihydrouridine (D), dihydropseudouridine, 5,6-dihydrouridine, 5-methyl-dihydrouridine (m
5D), 2-thio-dihydrouridine, 2-thio-dihydropseudouridine, 2-methoxy-uridine, 2-methoxy-4- thio-uridine, 4-methoxy-pseudouridine, 4-methoxy-2-thio-pseudouridine, N1-methyl-pseudouridine, 3-(3-amino-3- carboxypropyl)uridine (acp
3U), 1-methyl-3-(3-amino-3-carboxypropyl)pseudouridine (acp
3 ψ), 5- (isopentenylaminomethyl)uridine (inm
5U), 5-(isopentenylaminomethyl)-2-thio-uridine (inm
5s
2U), α-thio-uridine, 2′-O- 48 11529421v1 Attorney Docket No. 2013237‐0757 methyl-uridine (Um), 5,2′-O-dimethyl-uridine (m
5Um), 2′-O-methyl-pseudouridine (ψm), 2-thio-2′-O-methyl-uridine (s
2Um), 5-methoxycarbonylmethyl-2′-O-methyl-uridine (mcm
5Um), 5-carbamoylmethyl-2′-O-methyl-uridine (ncm
5Um), 5-carboxymethylaminomethyl-2′-O-methyl-uridine (cmnm
5Um), 3,2′-O-dimethyl-uridine (m
3Um), 5- (isopentenylaminomethyl)-2′-O-methyl-uridine (inm
5Um), 1-thio-uridine, deoxythymidine, 2′-F-ara-uridine, 2′-F- uridine, 2′-OH-ara-uridine, 5-(2-carbomethoxyvinyl) uridine, and 5-[3-(1-E-propenylamino)uridine. In certain embodiments, the nucleoside comprising a modified nucleobase is pseudouridine (ψ), N1-methyl-pseudouridine (m1ψ) or 5-methyl-uridine (m5U), in particular N1-methyl-pseudouridine. In some embodiments, the replacement of one or more uridines with a nucleoside comprising a modified nucleobase comprises a replacement of at least 1%, at least 2%, at least 3%, at least 4%, at least 5%, at least 10%, at least 25%, at least 50%, at least 75%, at least 90%, at least 95%, at least 96%, at least 97%, at least 98%, at least 99% or 100% of the uridines. During synthesis of mRNA by in vitro transcription (IVT) using T7 RNA polymerase significant amounts of aberrant products, including double-stranded RNA (dsRNA) are produced due to unconventional activity of the enzyme. dsRNA induces inflammatory cytokines and activates effector enzymes leading to protein synthesis inhibition. Formation of dsRNA can be limited during synthesis of mRNA by in vitro transcription (IVT), for example, by limiting the amount of uridine triphosphate (UTP) during synthesis. Optionally, UTP may be added once or several times during synthesis of mRNA. Also, dsRNA can be removed from RNA such as IVT RNA, for example, by ion-pair reversed phase HPLC using a non-porous or porous C-18 polystyrene-divinylbenzene (PS-DVB) matrix. Alternatively, an enzymatic based method using E. coli RNaseIII that specifically hydrolyzes dsRNA but not ssRNA, thereby eliminating dsRNA contaminants from IVT RNA preparations can be used. Furthermore, dsRNA can be separated from ssRNA by using a cellulose material. In some embodiments, an RNA preparation is contacted with a cellulose material and the ssRNA is separated from the cellulose material under conditions which allow binding of dsRNA to the cellulose material and do not allow binding of ssRNA to the cellulose material. Suitable methods for providing ssRNA are disclosed, for example, in WO 2017/182524. As the term is used herein, "remove" or "removal" refers to the characteristic of a population of first substances, such as non-immunogenic RNA, being separated from the proximity of a population of second substances, such as dsRNA, wherein the population of first substances is not necessarily devoid of the second substance, and the population of second substances is not necessarily devoid of the first substance. However, a population of first substances characterized by the removal of a population of second substances has a measurably lower content of second substances as compared to the non-separated mixture of first and second substances. In some embodiments, the amount of double-stranded RNA (dsRNA) is limited, e.g., dsRNA (especially dsmRNA) is removed from non-immunogenic RNA , such that less than 10%, less than 5%, less than 4%, less than 3%, less than 2%, less than 1%, less than 0.5%, less than 0.3%, less than 0.1%, less than 0.05%, less than 0.03%, less than 0.01%, less than 0.005%, less than 0.004%, less than 0.003%, less than 0.002%, less than 0.001%, or less than 0.0005% of the RNA in the non-immunogenic RNA composition is dsRNA. In some embodiments, the non-immunogenic RNA (especially mRNA) is free or essentially free of dsRNA. In some embodiments, the non-immunogenic RNA (especially mRNA) composition comprises a purified preparation of single-stranded nucleoside modified RNA. In some embodiments, the non-immunogenic RNA (especially mRNA) composition comprises single-stranded nucleoside modified RNA (especially mRNA) and is substantially free of double stranded RNA (dsRNA). In some embodiments, the non-immunogenic RNA (especially mRNA) composition comprises at least 90%, at least 91%, at least 92%, at least 93 %, at least 94%, at least 95%, at least 96%, at least 97%, at least 98%, at least 99%, at least 99.5%, at least 99.9%, 49 11529421v1 Attorney Docket No. 2013237‐0757 at least 99.99%, at least 99.991%, at least 99.992%, , at least 99.993%,, at least 99.994%, , at least 99.995%, at least 99.996%, at least 99.997%, or at least 99.998% single stranded nucleoside modified RNA, relative to all other nucleic acid molecules (DNA, dsRNA, etc.). Various methods can be used to determine the amount of dsRNA. For example, a sample may be contacted with dsRNA-specific antibody and the amount of antibody binding to RNA may be taken as a measure for the amount of dsRNA in the sample. A sample containing a known amount of dsRNA may be used as a reference. For example, RNA may be spotted onto a membrane, e.g., nylon blotting membrane. The membrane may be blocked, e.g., in TBS-T buffer (20 mM TRIS pH 7.4, 137 mM NaCl, 0.1% (v/v) TWEEN-20) containing 5% (w/v) skim milk powder. For detection of dsRNA, the membrane may be incubated with dsRNA-specific antibody, e.g., dsRNA-specific mouse mAb (English & Scientific Consulting, Szirák, Hungary). After washing, e.g., with TBS-T, the membrane may be incubated with a secondary antibody, e.g., HRP-conjugated donkey anti-mouse IgG (Jackson ImmunoResearch, Cat #715-035-150), and the signal provided by the secondary antibody may be detected. In some embodiments, the non-immunogenic RNA (especially mRNA) is translated in a cell more efficiently than standard RNA with the same sequence. In some embodiments, translation is enhanced by a factor of 2-fold relative to its unmodified counterpart. In some embodiments, translation is enhanced by a 3-fold factor. In some embodiments, translation is enhanced by a 4-fold factor. In some embodiments, translation is enhanced by a 5-fold factor. In some embodiments, translation is enhanced by a 6-fold factor. In some embodiments, translation is enhanced by a 7-fold factor. In some embodiments, translation is enhanced by an 8-fold factor. In some embodiments, translation is enhanced by a 9-fold factor. In some embodiments, translation is enhanced by a 10-fold factor. In some embodiments, translation is enhanced by a 15-fold factor. In some embodiments, translation is enhanced by a 20-fold factor. In some embodiments, translation is enhanced by a 50-fold factor. In some embodiments, translation is enhanced by a 100- fold factor. In some embodiments, translation is enhanced by a 200-fold factor. In some embodiments, translation is enhanced by a 500-fold factor. In some embodiments, translation is enhanced by a 1000-fold factor. In some embodiments, translation is enhanced by a 2000-fold factor. In some embodiments, the factor is 10-1000-fold. In some embodiments, the factor is 10-100-fold. In some embodiments, the factor is 10-200-fold. In some embodiments, the factor is 10-300-fold. In some embodiments, the factor is 10-500-fold. In some embodiments, the factor is 20-1000- fold. In some embodiments, the factor is 30-1000-fold. In some embodiments, the factor is 50-1000-fold. In some embodiments, the factor is 100-1000-fold. In some embodiments, the factor is 200-1000-fold. In some embodiments, translation is enhanced by any other significant amount or range of amounts. In some embodiments, the non-immunogenic RNA (especially mRNA) exhibits significantly less innate immunogenicity than standard RNA with the same sequence. In some embodiments, the non-immunogenic RNA (especially mRNA) exhibits an innate immune response that is 2-fold less than its unmodified counterpart. In some embodiments, innate immunogenicity is reduced by a 3-fold factor. In some embodiments, innate immunogenicity is reduced by a 4-fold factor. In some embodiments, innate immunogenicity is reduced by a 5-fold factor. In some embodiments, innate immunogenicity is reduced by a 6-fold factor. In some embodiments, innate immunogenicity is reduced by a 7-fold factor. In some embodiments, innate immunogenicity is reduced by an 8-fold factor. In some embodiments, innate immunogenicity is reduced by a 9-fold factor. In some embodiments, innate immunogenicity is reduced by a 10-fold factor. In some embodiments, innate immunogenicity is reduced by a 15-fold factor. In some embodiments, innate immunogenicity is reduced by a 20-fold factor. In some embodiments, innate immunogenicity is reduced by a 50-fold factor. In some embodiments, innate immunogenicity is reduced by a 100-fold factor. In some embodiments, innate 50 11529421v1 Attorney Docket No. 2013237‐0757 immunogenicity is reduced by a 200-fold factor. In some embodiments, innate immunogenicity is reduced by a 500- fold factor. In some embodiments, innate immunogenicity is reduced by a 1000-fold factor. In some embodiments, innate immunogenicity is reduced by a 2000-fold factor. The term "exhibits significantly less innate immunogenicity" refers to a detectable decrease in innate immunogenicity. In some embodiments, the term refers to a decrease such that an effective amount of the non-immunogenic RNA (especially mRNA) can be administered without triggering a detectable innate immune response. In some embodiments, the term refers to a decrease such that the non-immunogenic RNA (especially mRNA) can be repeatedly administered without eliciting an innate immune response sufficient to detectably reduce production of the polypeptide encoded by the non-immunogenic RNA. In some embodiments, the decrease is such that the non-immunogenic RNA (especially mRNA) can be repeatedly administered without eliciting an innate immune response sufficient to eliminate detectable production of the polypeptide encoded by the non-immunogenic RNA. "Immunogenicity" is the ability of a foreign substance, such as RNA, to provoke an immune response in the body of a human or other animal. The innate immune system is the component of the immune system that is relatively unspecific and immediate. It is one of two main components of the vertebrate immune system, along with the adaptive immune system. Antigen-coding RNA and use thereof for inducing an immune response Generally, RNA (in particular, mRNA) described in the present disclosure comprises a nucleic acid sequence encoding a polypeptide comprising two or more fragments of antigens or immunogenic variants, for inducing an immune response in a subject. In some embodiments, the RNA (in particular, mRNA) is translated into the respective polypeptide upon entering cells of a subject being administered the RNA, e.g., muscle cells or antigen-presenting cells (APCs). In some embodiments, the RNA encoding the polypeptide is expressed in cells of the subject to provide the polypeptide. In some embodiments, the RNA encoding the polypeptide is transiently expressed in cells of the subject. In some embodiments, the polypeptide is presented in the context of MHC. In some embodiments, the polypeptide is secreted by cells of the subject. In some embodiments, the RNA encoding the polypeptide is administered intramuscularly. In some embodiments, the RNA encoding the polypeptide is administered systemically, e.g., intravenously. In some embodiments, after systemic administration of the RNA encoding the polypeptide, expression of the RNA encoding the polypeptide in spleen occurs. In some embodiments, after systemic administration of the RNA encoding the polypeptide, expression of the RNA encoding the polypeptide in antigen presenting cells, preferably professional antigen presenting cells occurs. In some embodiments, the antigen presenting cells are selected from the group consisting of dendritic cells, macrophages and B cells. In some embodiments, after systemic administration of the RNA encoding the polypeptide, no or essentially no expression of the RNA encoding the polypeptide in lung and/or liver occurs. In some embodiments, after systemic administration of the RNA encoding the polypeptide, expression of the RNA encoding the polypeptide in spleen is at least 5-fold the amount of expression in lung. A polypeptide of the invention comprises an epitope for inducing an immune response against a disease-associated antigen, e.g., a protein of an infectious agent (e.g., antigens from Mycobacterium tuberculosis, Pseudomonas aeruginosa, Staphylococcus aureus, Clostridioides difficile, Bacillus anthracis, Corynebacterium diphteriae and Vibrio Cholerae), in a subject. Accordingly, the polypeptide comprises an antigenic sequence for inducing an immune response 51 11529421v1 Attorney Docket No. 2013237‐0757 against a disease-associated antigen in a subject. Such antigenic sequence may correspond to a target antigen or disease-associated antigen, an immunogenic variant thereof or the immunogenic variant thereof. Thus, the antigenic sequence may comprise at least an epitope of a target antigen or disease-associated antigen or an immunogenic variant thereof. The antigenic sequences, e.g., epitopes, suitable for use according to the disclosure typically may be derived from a target antigen, i.e. the antigen against which an immune response is to be elicited. For example, the antigenic sequences contained within the polypeptide may be a fragment of the target antigen a of or variant of a target antigen. The antigenic sequence or a procession product thereof, e.g., a fragment thereof, may bind to an antigen receptor such as TCR carried by immune effector cells. A polypeptide which may be provided to a subject according to the present disclosure by administering RNA encoding the polypeptide, preferably results in the induction of an immune response, e.g., in the stimulation, priming and/or expansion of immune effector cells, in the subject being provided the polypeptide. Said immune response, e.g., stimulated, primed and/or expanded immune effector cells, is preferably directed against a target antigen, in particular a target antigen expressed in diseased cells, tissues and/or organs, i.e., a disease-associated antigen. Thus, a polypeptide may comprise two or more fragments of the disease-associated antigen or of a variant thereof. In some embodiments, such fragment is immunologically equivalent to the disease-associated antigen. The term "immunologically equivalent" means that the immunologically equivalent molecule such as the immunologically equivalent amino acid sequence exhibits the same or essentially the same immunological properties and/or exerts the same or essentially the same immunological effects, e.g., with respect to the type of the immunological effect. In the context of the present disclosure, the term "immunologically equivalent" is preferably used with respect to the immunological effects or properties of antigens or antigen variants used for immunization. For example, an amino acid sequence is immunologically equivalent to a reference amino acid sequence if said amino acid sequence when exposed to the immune system of a subject induces an immune reaction having a specificity of reacting with the reference amino acid sequence. Thus, in some embodiments, a molecule which is immunologically equivalent to an antigen exhibits the same or essentially the same properties and/or exerts the same or essentially the same effects regarding the stimulation, priming and/or expansion of T cells as the antigen to which the T cells are targeted. In the context of the present disclosure, the term "fragment of an antigen" or "fragment of an immunogenic variant of an antigen" means an agent which results in the induction of an immune response, e.g., in the stimulation, priming and/or expansion of immune effector cells, which immune response, e.g., stimulated, primed and/or expanded immune effector cells, targets the antigen, i.e. a disease-associated antigen, in particular when presented by diseased cells, tissues and/or organs. An "immunogenic fragment of an antigen" according to the disclosure preferably relates to a fragment of an antigen which is capable of inducing an immune response against, e.g., stimulating, priming and/or expanding immune effector cells carrying an antigen receptor binding to, the antigen or cells expressing the antigen. It is preferred that the polypeptide (similar to the disease-associated antigen) provides the relevant epitope for binding by the antigen receptor present on the immune effector cells. In some embodiments, the polypeptide or a fragment thereof (similar to the disease-associated antigen) is expressed on the surface of a cell such as an antigen-presenting cell (optionally in the context of MHC) so as to provide the relevant epitope for binding by immune effector cells. 52 11529421v1 Attorney Docket No. 2013237‐0757 In some embodiments of all aspects described herein, the RNA encoding the polypeptide is expressed in cells of a subject to provide the antigen or a procession product thereof for binding by the antigen receptor expressed by immune effector cells, said binding resulting in stimulation, priming and/or expansion of the immune effector cells. An "antigen" according to the present disclosure covers any full-length protein (i.e. a protein in the form it is present in after translation and post-translational modification) that will elicit an immune response and/or against which an immune response or an immune mechanism such as a cellular response and/or humoral response is directed. This also includes situations wherein the antigen is processed into antigen peptides and an immune response or an immune mechanism is directed against one or more antigen peptides, in particular if presented in the context of MHC molecules. In particular, an "antigen" relates to a full-length protein that reacts specifically with antibodies or T-lymphocytes (T- cells). "Antigens" may comprise at least one epitope, such as a T cell epitope. In some embodiments, an antigen is a molecule which induces an immune reaction specific for the antigen (including cells expressing the antigen). In some embodiments, an antigen is a disease-associated antigen, such as an antigen from Mycobacterium tuberculosis, Pseudomonas aeruginosa, Staphylococcus aureus, Clostridioides difficile, Bacillus anthracis, Corynebacterium diphteriae and Vibrio Cholerae. In some embodiments, peptides of an antigen are presented or present on the surface of cells of the immune system such as antigen presenting cells like dendritic cells or macrophages. An antigen or a procession product thereof such as a T cell epitope is in some embodiments bound by an antigen receptor. Accordingly, an antigen or a procession product thereof may react specifically with immune effector cells such as T-lymphocytes (T cells). According to the present disclosure, an antigen or a combination of antigens described herein may induce an immune response, wherein the immune response may comprise a humoral or cellular immune response, or both. In the context of some embodiments of the present disclosure, the antigen is presented by a cell, such as by an antigen presenting cell, in the context of MHC molecules, which results in an immune response against the antigen. An antigen may be a product which corresponds to or is derived from a naturally occurring antigen. According to the present disclosure, an antigen may correspond to a naturally occurring product. The term "disease-associated antigen" is used in its broadest sense to refer to any antigen associated with a disease. In some embodiments, a disease-associated antigen is a molecule which contains epitopes that will stimulate a host's immune system to make a cellular antigen-specific immune response and/or a humoral antibody response against the disease. Disease-associated antigens include pathogen-associated antigens, i.e., antigens which are associated with infection by microbes, typically microbial antigens (such as antigens from eukaryotic parasites, bacterial, fungal or viral antigens), or antigens associated with cancer, typically tumors, such as tumor antigens. The term "bacterial antigen" refers to any bacterial component having antigenic properties, i.e. being able to provoke an immune response in an individual. The bacterial antigen may be derived from the cell wall or cytoplasm membrane of the bacterium. The term "bacterial antigen" includes, e.g., antigens from Mycobacterium tuberculosis, Pseudomonas aeruginosa, Staphylococcus aureus, Clostridioides difficile, Bacillus anthracis, Corynebacterium diphteriae and Vibrio Cholerae. The term "epitope" refers to an antigenic determinant in a molecule such as an antigen, i.e., to a part in or fragment of the molecule that is recognized by the immune system, for example, that is recognized by antibodies, T cells or B cells, in particular when presented in the context of MHC molecules. An epitope of an antigen may comprise a continuous or discontinuous portion of said antigen and, e.g., may be between about 5 and about 100, between about 53 11529421v1 Attorney Docket No. 2013237‐0757 5 and about 50, between about 8 and about 30, or about 10 and about 25 amino acids in length, for example, the epitope may be preferably 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, or 25 amino acids in length. In some embodiments, the epitope in the context of the present disclosure is a T cell epitope. Terms such as "epitope", "fragment of an antigen", "immunogenic peptide" and "antigen peptide" are used interchangeably herein and, e.g., may relate to an incomplete representation of an antigen which is, e.g., capable of eliciting an immune response against the antigen or a cell expressing or comprising and presenting the antigen. In some embodiments, the terms relate to an immunogenic portion of an antigen. In some embodiments, it is a portion of an antigen that is recognized (i.e., specifically bound) by a T cell receptor, in particular if presented in the context of MHC molecules. Certain preferred immunogenic portions bind to an MHC class I or class II molecule. The term "epitope" refers to a part or fragment of a molecule such as an antigen that is recognized by the immune system. For example, the epitope may be recognized by T cells, B cells or antibodies. An epitope of an antigen may include a continuous or discontinuous portion of the antigen and may be between about 5 and about 100, such as between about 5 and about 50, between about 8 and about 30, or between about 8 and about 25 amino acids in length, for example, the epitope may be 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, or 25 amino acids in length. In some embodiments, an epitope is between about 10 and about 25 amino acids in length. The term "epitope" includes T cell epitopes. The term "T cell epitope" refers to a part or fragment of a antigen that is recognized by a T cell when presented in the context of MHC molecules, including epitopes predicted by bioinformatic means. The term "major histocompatibility complex" and the abbreviation "MHC" includes MHC class I and MHC class II molecules and relates to a complex of genes which is present in all vertebrates. MHC proteins or molecules are important for signaling between lymphocytes and antigen presenting cells or diseased cells in immune reactions, wherein the MHC proteins or molecules bind peptide epitopes and present them for recognition by T cell receptors on T cells. The proteins encoded by the MHC are expressed on the surface of cells, and display both self-antigens (peptide fragments from the cell itself) and non-self- antigens (e.g., fragments of invading microorganisms) to a T cell. In the case of class I MHC/peptide complexes, the binding peptides are typically about 8 to about 10 amino acids long although longer or shorter peptides may be effective. In the case of class II MHC/peptide complexes, the binding peptides are typically about 10 to about 25 amino acids long and are in particular about 13 to about 18 amino acids long, whereas longer and shorter peptides may be effective. In some embodiments, a polypeptide whose inoculation into a subject induces an immune response, is recognized by an immune effector cell. In some embodiments, the polypeptide if recognized by an immune effector cell is able to induce in the presence of appropriate co-stimulatory signals, stimulation, priming and/or expansion of the immune effector cell carrying an antigen receptor recognizing the polypeptide. In the context of the embodiments of the present disclosure, the polypeptide may be, e.g., presented or present on the surface of a cell, such as an antigen presenting cell. In some embodiments, an antigen is expressed in a diseased cell (such as an infected cell). In some embodiments, an antigen is presented by a diseased cell (such as an infected cell). In some embodiments, an antigen receptor is a TCR which binds to an epitope of an antigen presented in the context of MHC. In some embodiments, binding of a TCR when expressed by T cells and/or present on T cells to an antigen presented by cells such as antigen presenting cells results in stimulation, priming and/or expansion of said T cells. In some embodiments, binding of a TCR when expressed by T cells and/or present on T cells to an antigen presented on diseased cells results 54 11529421v1 Attorney Docket No. 2013237‐0757 in cytolysis and/or apoptosis of the diseased cells, wherein said T cells release cytotoxic factors, e.g., perforins and granzymes. In some embodiments, an antigen receptor is an antibody or B cell receptor which binds to an epitope in an antigen. In some embodiments, an antibody or B cell receptor binds to native epitopes of an antigen. The terms "T cell" and "T lymphocyte" are used interchangeably herein and include T helper cells (CD4+ T cells) and cytotoxic T cells (CTLs, CD8+ T cells) which comprise cytolytic T cells. The term "antigen-specific T cell" or similar terms relate to a T cell which recognizes the antigen to which the T cell is targeted, in particular when presented on the surface of antigen presenting cells or diseased cells in the context of MHC molecules and preferably exerts effector functions of T cells. T cells are considered to be specific for antigen if the cells kill target cells expressing an antigen. T cell specificity may be evaluated using any of a variety of standard techniques, for example, within a chromium release assay or proliferation assay. Alternatively, synthesis of lymphokines (such as interferon-γ) can be measured. In some embodiments, the term "target" shall mean an agent such as a cell or tissue which is a target for an immune response such as a cellular immune response. Targets include cells that present an antigen or an antigen epitope, i.e., a peptide fragment derived from an antigen. In some embodiments, the target cell is a cell expressing an antigen and presenting said antigen with class I MHC. "Antigen processing" refers to the degradation of an antigen into processing products which are fragments of said antigen (e.g., the degradation of a polypeptide into peptides) and the association of one or more of these fragments (e.g., via binding) with MHC molecules for presentation by cells, such as antigen-presenting cells to specific T-cells. Antigen-presenting cells can be distinguished in professional antigen presenting cells and non-professional antigen presenting cells. The term "professional antigen presenting cells" relates to antigen presenting cells which constitutively express the Major Histocompatibility Complex class II (MHC class II) molecules required for interaction with naive T cells. If a T cell interacts with the MHC class II molecule complex on the membrane of the antigen presenting cell, the antigen presenting cell produces a co-stimulatory molecule inducing activation of the T cell. Professional antigen presenting cells comprise dendritic cells and macrophages. The term "non-professional antigen presenting cells" relates to antigen presenting cells which do not constitutively express MHC class II molecules, but upon stimulation by certain cytokines such as interferon-gamma. Exemplary, non- professional antigen presenting cells include fibroblasts, thymic epithelial cells, thyroid epithelial cells, glial cells, pancreatic beta cells or vascular endothelial cells. The term "dendritic cell" (DC) refers to a subtype of phagocytic cells belonging to the class of antigen presenting cells. In some embodiments, dendritic cells are derived from hematopoietic bone marrow progenitor cells. These progenitor cells initially transform into immature dendritic cells. These immature cells are characterized by high phagocytic activity and low T cell activation potential. Immature dendritic cells constantly sample the surrounding environment for pathogens such as viruses and bacteria. Once they have come into contact with a presentable antigen, they become activated into mature dendritic cells and begin to migrate to the spleen or to the lymph node. Immature dendritic cells phagocytose pathogens and degrade their antigens into small pieces and upon maturation present those fragments at their cell surface using MHC molecules. Simultaneously, they upregulate cell-surface receptors that act as co-receptors in T cell activation such as CD80, CD86, and CD40 greatly enhancing their ability to activate T cells. They also upregulate CCR7, a chemotactic receptor that induces the dendritic cell to travel through the blood stream to the spleen or through 55 11529421v1 Attorney Docket No. 2013237‐0757 the lymphatic system to a lymph node. Here they act as antigen-presenting cells and activate helper T cells and killer T cells as well as B cells by presenting them antigens, alongside non-antigen specific co-stimulatory signals. Thus, dendritic cells can actively induce a T cell- or B cell-related immune response. In some embodiments, the dendritic cells are splenic dendritic cells. The term "macrophage" refers to a subgroup of phagocytic cells produced by the differentiation of monocytes. Macrophages which are activated by inflammation, immune cytokines or microbial products nonspecifically engulf and kill foreign pathogens within the macrophage by hydrolytic and oxidative attack resulting in degradation of the pathogen. Peptides from degraded antigens are displayed on the macrophage cell surface where they can be recognized by T cells, and they can directly interact with antibodies on the B cell surface, resulting in T and B cell activation and further stimulation of the immune response. Macrophages belong to the class of antigen presenting cells. In some embodiments, the macrophages are splenic macrophages. By "antigen-responsive CTL" is meant a CD8
+ T-cell that is responsive to an antigen or a peptide derived from said antigen, which is presented with class I MHC on the surface of antigen presenting cells. According to the disclosure, CTL responsiveness may include sustained calcium flux, cell division, production of cytokines such as IFN- ^ and TNF-α, up-regulation of activation markers such as CD44 and CD69, and specific cytolytic killing of tumor antigen expressing target cells. CTL responsiveness may also be determined using an artificial reporter that accurately indicates CTL responsiveness. "Activation" or "stimulation", as used herein, refers to the state of a cell that has been sufficiently stimulated to induce detectable cellular proliferation, such as an immune effector cell such as T cell. Activation can also be associated with initiation of signaling pathways, induced cytokine production, and detectable effector functions. The term "activated immune effector cells" refers to, among other things, immune effector cells that are undergoing cell division. The term "priming" refers to a process wherein an immune effector cell such as a T cell has its first contact with its specific antigen and causes differentiation into effector cells such as effector T cells. The term "expansion" refers to a process wherein a specific entity is multiplied. In some embodiments, the term is used in the context of an immunological response in which immune effector cells are stimulated by an antigen, proliferate, and the specific immune effector cell recognizing said antigen is amplified. In some embodiments, expansion leads to differentiation of the immune effector cells. The terms "immune response" and "immune reaction" are used herein interchangeably in their conventional meaning and refer to an integrated bodily response to an antigen and may refer to a cellular immune response, a humoral immune response, or both. According to the disclosure, the term "immune response to" or "immune response against" with respect to an agent such as an antigen, cell or tissue, relates to an immune response such as a cellular response directed against the agent. An immune response may comprise one or more reactions selected from the group consisting of developing antibodies against one or more antigens and expansion of antigen-specific T-lymphocytes, such as CD4
+ and CD8
+ T-lymphocytes, e.g. CD8
+ T-lymphocytes, which may be detected in various proliferation or cytokine production tests in vitro. The terms "inducing an immune response" and "eliciting an immune response" and similar terms in the context of the present disclosure refer to the induction of an immune response, such as the induction of a cellular immune response, a humoral immune response, or both. The immune response may be protective/preventive/prophylactic and/or 56 11529421v1 Attorney Docket No. 2013237‐0757 therapeutic. The immune response may be directed against any immunogen or antigen or antigen peptide, such as against a pathogen-associated antigen (e.g., an antigen of Mtb). "Inducing" in this context may mean that there was no immune response against a particular antigen or pathogen before induction, but it may also mean that there was a certain level of immune response against a particular antigen or pathogen before induction and after induction said immune response is enhanced. Thus, "inducing the immune response" in this context also includes "enhancing the immune response". In some embodiments, after inducing an immune response in an individual, said individual is protected from developing a disease such as an infectious disease or the disease condition is ameliorated by inducing an immune response. The terms "cellular immune response", "cellular response", "cell-mediated immunity" or similar terms are meant to include a cellular response directed to cells characterized by expression of an antigen and/or presentation of an antigen with class I or class II MHC. The cellular response relates to cells called T cells or T lymphocytes which act as either "helpers" or "killers". The helper T cells (also termed CD4
+ T cells) play a central role by regulating the immune response and the killer cells (also termed cytotoxic T cells, cytolytic T cells, CD8
+ T cells or CTLs) kill cells such as diseased cells. The term "humoral immune response" refers to a process in living organisms wherein antibodies are produced in response to agents and organisms, which they ultimately neutralize and/or eliminate. The specificity of the antibody response is mediated by T and/or B cells through membrane-associated receptors that bind antigen of a single specificity. Following binding of an appropriate antigen and receipt of various other activating signals, B lymphocytes divide, which produces memory B cells as well as antibody secreting plasma cell clones, each producing antibodies that recognize the identical antigenic epitope as was recognized by its antigen receptor. Memory B lymphocytes remain dormant until they are subsequently activated by their specific antigen. These lymphocytes provide the cellular basis of memory and the resulting escalation in antibody response when re-exposed to a specific antigen. The term "antibody" as used herein, refers to an immunoglobulin molecule, which is able to specifically bind to an epitope on an antigen. In particular, the term "antibody" refers to a glycoprotein comprising at least two heavy (H) chains and two light (L) chains inter-connected by disulfide bonds. The term "antibody" includes monoclonal antibodies, recombinant antibodies, human antibodies, humanized antibodies, chimeric antibodies and combinations of any of the foregoing. Each heavy chain is comprised of a heavy chain variable region (VH) and a heavy chain constant region (CH). Each light chain is comprised of a light chain variable region (VL) and a light chain constant region (CL). The variable regions and constant regions are also referred to herein as variable domains and constant domains, respectively. The VH and VL regions can be further subdivided into regions of hypervariability, termed complementarity determining regions (CDRs), interspersed with regions that are more conserved, termed framework regions (FRs). Each VH and VL is composed of three CDRs and four FRs, arranged from amino-terminus to carboxy-terminus in the following order: FR1, CDR1, FR2, CDR2, FR3, CDR3, FR4. The CDRs of a VH are termed HCDR1, HCDR2 and HCDR3, the CDRs of a VL are termed LCDR1, LCDR2 and LCDR3. The variable regions of the heavy and light chains contain a binding domain that interacts with an antigen. The constant regions of an antibody comprise the heavy chain constant region (CH) and the light chain constant region (CL), wherein CH can be further subdivided into constant domain CH1, a hinge region, and constant domains CH2 and CH3 (arranged from amino-terminus to carboxy-terminus in the following order: CH1, CH2, CH3). The constant regions of the antibodies may mediate the binding of the immunoglobulin to host tissues or factors, including various cells of the immune system (e.g., effector cells) and the first component (C1q) of the classical complement system. Antibodies can be intact immunoglobulins derived from natural sources or from recombinant sources and can be immunoactive portions of intact immunoglobulins. Antibodies are typically tetramers 57 11529421v1 Attorney Docket No. 2013237‐0757 of immunoglobulin molecules. Antibodies may exist in a variety of forms including, for example, polyclonal antibodies, monoclonal antibodies, Fv, Fab and F(ab)
2, as well as single chain antibodies and humanized antibodies. The term "immunoglobulin" relates to proteins of the immunoglobulin superfamily, such as to antigen receptors such as antibodies or the B cell receptor (BCR). The immunoglobulins are characterized by a structural domain, i.e., the immunoglobulin domain, having a characteristic immunoglobulin (Ig) fold. The term encompasses membrane bound immunoglobulins as well as soluble immunoglobulins. Membrane bound immunoglobulins are also termed surface immunoglobulins or membrane immunoglobulins, which are generally part of the BCR. Soluble immunoglobulins are generally termed antibodies. Immunoglobulins generally comprise several chains, typically two identical heavy chains and two identical light chains which are linked via disulfide bonds. These chains are primarily composed of immunoglobulin domains, such as the V
L (variable light chain) domain, C
L (constant light chain) domain, V
H (variable heavy chain) domain, and the C
H (constant heavy chain) domains C
H1, C
H2, C
H3, and C
H4. There are five types of mammalian immunoglobulin heavy chains, i.e., α, ^, ε, ^, and µ which account for the different classes of antibodies, i.e., IgA, IgD, IgE, IgG, and IgM. As opposed to the heavy chains of soluble immunoglobulins, the heavy chains of membrane or surface immunoglobulins comprise a transmembrane domain and a short cytoplasmic domain at their carboxy-terminus. In mammals there are two types of light chains, i.e., lambda and kappa. The immunoglobulin chains comprise a variable region and a constant region. The constant region is essentially conserved within the different isotypes of the immunoglobulins, wherein the variable part is highly divers and accounts for antigen recognition. The terms "vaccination" and "immunization" describe the process of treating an individual for therapeutic or prophylactic reasons and relate to the procedure of administering one or more immunogen(s) or antigen(s) or derivatives thereof, in particular in the form of RNA (especially mRNA) coding therefor, as described herein to an individual and stimulating an immune response against said one or more immunogen(s) or antigen(s) or cells characterized by presentation of said one or more immunogen(s) or antigen(s). By "cell characterized by presentation of an antigen" or "cell presenting an antigen" or "MHC molecules which present an antigen on the surface of an antigen presenting cell" or similar expressions is meant a cell such as a diseased cell, in particular an infected cell, or an antigen presenting cell presenting the antigen or an antigen peptide, either directly or following processing, in the context of MHC molecules, such as MHC class I and/or MHC class II molecules. In some embodiments, the MHC molecules are MHC class I molecules. Embodiments of antigen-coding RNA Generally, at least four formats useful for RNA pharmaceutical compositions may be used herein, namely non-modified uridine containing mRNA (uRNA), nucleoside modified mRNA (modRNA), self-amplifying RNA (saRNA), and trans- amplifying RNAs. Features of modified uridine (e.g., pseudouridine) platform may include reduced adjuvant effect, blunted immune innate immune sensor activating capacity and thus augmented polypeptide (e.g., protein) expression. Features of self-amplifying platform may include, for example, long duration of polypeptide (e.g., protein) expression, good tolerability and safety, higher likelihood for efficacy with very low RNA dose. In some embodiments, a self-amplifying platform (e.g., RNA) comprises two nucleic acid molecules, wherein one nucleic acid molecule encodes a replicase (e.g., a viral replicase) and the other nucleic acid molecule is capable of being replicated (e.g., a replicon) by said replicase in trans (trans-replication system). In some embodiments, a self- 58 11529421v1 Attorney Docket No. 2013237‐0757 amplifying platform (e.g., RNA) comprises a plurality of nucleic acid molecules, wherein said nucleic acids encode a plurality of replicases and/or replicons. In some embodiments, a trans-replication system comprises the presence of both nucleic acid molecules in a single host cell. In some such embodiments, a nucleic acid encoding a replicase (e.g., a viral replicase) is not capable of self-replication in a target cell and/or target organism. In some such embodiments, a nucleic acid encoding a replicase (e.g., a viral replicase) lacks at least one conserved sequence element important for (-) strand synthesis based on a (+) strand template and/or for (+) strand synthesis based on a (-) strand template. In some embodiments, a self-amplifying RNA comprises a 3’ untranslated region (UTR), a 5’ UTR, a cap structure, a poly adenine (polyA) tail, and any combinations thereof. In some embodiments, a self-amplifying platform does not require propagation of virus particles (e.g., is not associated with undesired virus-particle formation). In some embodiments, a self-amplifying platform is not capable of forming virus particles. In some embodiments, RNA (e.g., a single stranded RNA) described herein has a length of at least 500 ribonucleotides (such as, e.g., at least 600 ribonucleotides, at least 700 ribonucleotides, at least 800 ribonucleotides, at least 900 ribonucleotides, at least 1000 ribonucleotides, at least 1250 ribonucleotides, at least 1500 ribonucleotides, at least 1750 ribonucleotides, at least 2000 ribonucleotides, at least 2500 ribonucleotides, at least 3000 ribonucleotides, at least 3500 ribonucleotides, at least 4000 ribonucleotides, at least 4500 ribonucleotides, at least 5000 ribonucleotides, or longer). In some embodiments, RNA described herein is single-stranded RNA having a length of about 800 ribonucleotides to 5000 ribonucleotides. In some embodiments, a relevant RNA includes a polypeptide-encoding portion or a plurality of polypeptide-encoding portions. In some particular embodiments, such a portion or portions encode one or more polypeptides which are not endogenous (i.e., it is foreign) to the subject treated. In some embodiments, the RNA described herein (e.g., contained in the compositions/formulations of the present disclosure and/or used in the methods of the present disclosure) is single-stranded RNA (in particular, mRNA) that may be translated into the respective polypeptide upon entering cells, e.g., cells of a recipient, e.g., muscle cells or antigen- presenting cells (APCs). In addition to wild-type or codon-optimized sequences encoding an antigen sequence, the RNA may contain one or more structural elements optimized for maximal efficacy of the RNA with respect to stability and translational efficiency (5' cap, 5' UTR, 3' UTR, poly(A)‐tail). In some embodiments, the RNA contains all of these elements. In some embodiments, beta-S-ARCA(D1) (m
27,2'-OGppSpG) or m
27,3’-OGppp(m
12’-O)ApG may be utilized as specific capping structure at the 5'-end of the RNA. As 5'-UTR sequence, the 5'-UTR sequence of the human alpha- globin mRNA, optionally with an optimized ʻKozak sequenceʼ to increase translational efficiency may be used. As 3'- UTR sequence, a combination of two sequence elements (FI element) derived from the "amino terminal enhancer of split" (AES) mRNA (called F) and the mitochondrial encoded 12S ribosomal RNA (called I) placed between the coding sequence and the poly(A)-tail to assure higher maximum polypeptide levels and prolonged persistence of the mRNA may be used (see WO 2017/060314, herein incorporated by reference). Furthermore, a poly(A)-tail measuring 110 nucleotides in length, consisting of a stretch of 30 adenosine residues, followed by a 10 nucleotide linker sequence (of random nucleotides) and another 70 adenosine residues may be used. In some embodiments, the 5’-UTR comprises the nucleotide sequence of SEQ ID NO: 40, or a nucleotide sequence having at least 99%, 98%, 97%, 96%, 95%, 59 11529421v1 Attorney Docket No. 2013237‐0757 90%, 85%, or 80% identity to the nucleotide sequence of SEQ ID NO: 40. In some embodiments, the 3’-UTR comprises the nucleotide sequence of SEQ ID NO: 41, or a nucleotide sequence having at least 99%, 98%, 97%, 96%, 95%, 90%, 85%, or 80% identity to the nucleotide sequence of SEQ ID NO: 41. In some embodiments, the poly(A) sequence comprises the nucleotide sequence of SEQ ID NO: 42, or a nucleotide sequence having at least 99%, 98%, 97%, 96%, 95%, 90%, 85%, or 80% identity to the nucleotide sequence of SEQ ID NO: 42. In some embodiments, the RNA described herein is not chemically modified, i.e. it solely contains naturally occurring nucleosides, and preferably has the composition of naturally occurring RNA. In some embodiments, the RNA described herein is modified for optimized efficacy of the RNA (e.g., increased translation efficacy, decreased immunogenicity, and/or decreased cytotoxicity) (e.g., by replacing (partially or completely, preferably completely) naturally occurring nucleosides (in particular uridine) with synthetic nucleosides (e.g., modified nucleosides, e.g., selected from the group consisting of pseudouridine (ψ), N1-methyl-pseudouridine (m1ψ), and 5-methyl-uridine); and/or codon-optimization). In some embodiments, the RNA comprises a modified nucleoside in place of uridine. In some embodiments, the modified nucleoside replacing (partially or completely, preferably completely) uridine is selected from the group consisting of pseudouridine (ψ), N1-methyl-pseudouridine (m1ψ), and 5-methyl-uridine. In some embodiments, the RNA encoding the polypeptide has a coding sequence (a) which is codon-optimized, (b) the G/C content of which is increased compared to the wild type coding sequence, or (c) both (a) and (b). In some embodiments, the RNA described herein comprises a 5' cap, a 5' UTR, a 3' UTR, and a poly(A) sequence (e.g., as described above); is modified by replacing (partially or completely, preferably completely) uridine with modified nucleosides, e.g., selected from the group consisting of pseudouridine (ψ), N1-methyl-pseudouridine (m1ψ), and 5- methyl-uridine; and has a coding sequence which is codon-optimized, and the G/C content of which is increased compared to the wild type coding sequence. In some embodiments, if the present disclosure provides for a mixture of different RNA molecules, a composition comprising different RNA molecules or an administration of different RNA molecules, these different RNA molecules are present in approximately the same amount. Such different RNA molecules may be formulated in individual particulate formulations, mixed particulate formulations, or combined particulate formulations as described herein. The present disclosure provides RNA (in particular, mRNA) comprising a nucleic acid sequence encoding a polypeptide comprising fragments of an antigen or an immunogenic variant thereof. In some embodiments, RNA (in particular, mRNA) described in the present disclosure comprises a nucleic acid sequence encoding two or more fragments of aantigen or an immunogenic variant thereof, and is capable of expressing said fragments of an antigen or immunogenic variant thereof, in particular if transferred into a cell or subject, preferably a human cell or subject. Thus, in some embodiments, the RNA (in particular, mRNA) described in the present disclosure contains a coding region (open reading frame (ORF)) encoding two or more fragments of an antigen or an immunogenic variant thereof. In some embodiments, RNA comprises a nucleic acid sequence encoding one or more fragments of more than one antigen or immunogenic variant thereof, e.g., two, three, four or more antigens or immunogenic variants thereof. In some embodiments, one or more fragments of two or more of such antigens or immunogenic variants thereof, are present as a fusion protein. 60 11529421v1 Attorney Docket No. 2013237‐0757 In some embodiments, the one or more antigens, immunogenic variants thereof, or immunogenic fragments of the antigens or the immunogenic variants thereof may comprise or consist of naturally occurring sequences, may comprise or consist of variants of naturally occurring sequences, or may comprise or consist of sequences which are not naturally occurring, e.g., recombinant sequences. In some embodiments, the polypeptide encoded by the RNA described herein may consist of two or more fragments of at least one of the one or more antigens or immunogenic variants thereof, or may comprise two or more fragments of at least one of the one or more antigens or immunogenic variants thereof and may comprise additional sequences such as secretion signals, extended-PK groups, tags and any other sequences. In some embodiments, the additional sequences are fused to the two or more fragments of the one or more antigens or immunogenic variants thereof, in some embodiments, separated by a linker. In these embodiments, the two or more fragments of at least one of the one or more antigens or immunogenic variants thereof may be considered the pharmaceutically active peptide or polypeptide even if additional sequences support the function or effect of the one or more fragments of the one or more antigens or immunogenic variants thereof. According to the present disclosure, the term "pharmaceutically active peptide or polypeptide" means a peptide or polypeptide that can be used in the treatment of an individual where the expression of the peptide or polypeptide would be of benefit, e.g., in ameliorating the symptoms of a disease. Preferably, a pharmaceutically active peptide or polypeptide has curative or palliative properties and may be administered to ameliorate, relieve, alleviate, reverse, delay onset of or lessen the severity of one or more symptoms of a disease. In some embodiments, a pharmaceutically active peptide or polypeptide has a positive or advantageous effect on the condition or disease state of an individual when administered to the individual in a therapeutically effective amount. A pharmaceutically active peptide or polypeptide may have prophylactic properties and may be used to delay the onset of a disease or to lessen the severity of such disease. The term "pharmaceutically active peptide or polypeptide" includes entire peptides or polypeptides, and can also refer to pharmaceutically active fragments thereof. It can also include pharmaceutically active variants and/or analogs of a peptide or polypeptide. Specific examples of pharmaceutically active peptides and polypeptides include, but are not limited to, antigens for vaccination such as Mtb antigens, immunogenic variants thereof, or immunogenic fragments of the Mtb antigens or the immunogenic variants thereof. Fragments of antigens or immunogenic variants thereof described herein can be prepared as fusion or chimeric polypeptides that include a portion which corresponds to one or more, two or more, three or more, four or more, five or more, six or more, seven or more or eight or more fragments of one or more, two or more, three or more, four or more, five or more, six or more, seven or more or eight or more antigens or immunogenic variants originating from a certain microbial species and one or more heterologous polypeptides (e.g., polypeptides that are an antigens from different microbial species or an immunogenic variants of such antigens). According to certain embodiments, a “signal peptide” (or signal sequence) is fused, either directly or through a linker, to the N-terminus of a polypeptide described herein. In some embodiments, an open reading frame of the RNA described herein encodes a polypeptide that includes a signal sequence, e.g., that is functional in mammalian cells. In some embodiments, a utilized signal sequence is “intrinsic” in that it is, in nature, associated with (e.g., linked to) the full-length antigen or antigen fragment at the N-terminus of the polypeptide. 61 11529421v1 Attorney Docket No. 2013237‐0757 In some embodiments, a utilized signal sequence is heterologous to the encoded polypeptide – e.g., is not naturally part of a full-length antigen or antigen fragment whose sequences are included in the encoded polypeptide. In some embodiments, signal peptides are sequences, which are typically characterized by a length of about 15 to 30 amino acids. In many embodiments, signal peptides are positioned at the N-terminus of an encoded polypeptide as described herein, without being limited thereto. In some embodiments, signal peptides preferably allow the transport of the polypeptide encoded by RNAs of the present disclosure with which they are associated into a defined cellular compartment, preferably the cell surface, the endoplasmic reticulum (ER) or the endosomal-lysosomal compartment. In some embodiments, an RNA sequence encodes a peptidoglycan hydrolase, e.g., an endolysin, that may comprise or otherwise be linked to a signal sequence (e.g., secretory sequence), such as those listed in Table 2 and 3, or a sequence having 1, 2, 3, 4, or 5 amino acid differences relative thereto. In some embodiments, a signal sequence such as MRVMAPRTLILLLSGALALTETWAGS [SEQ ID NO: 4], or a sequence having 1, 2, 3, 4, or at the most 5 amino acid differences relative thereto is utilized. In some embodiments, a sequence such as MRVMAPRTLILLLSGALALTETWAGS, or a sequence having 1, 2, 3, 4, or at the most 5 amino acid differences relative thereto, is utilized. In some embodiments, a signal peptide is selected from those included in the Table 2 below and/or those encoded by the sequences in Table 3 below or a sequence having 1, 2, 3, 4, or 5 amino acid differences relative thereto: Table 2: Exemplary signal sequences Signal Sequence (Amino Acid) HSV-1 gD SP MGGAAARLGAVILFVVIVGLHGVRSKY [SEQ ID NO: 5] HSV-1 gD SP MGGAAARLGAVILFVVIVGLHGVRGKY [SEQ ID NO: 6] HSV-2 gD SP MGRLTSGVGTAALLVVAVGLRVVCA [SEQ ID NO: 7] HSV-2 MGRLTSGVGTAALLVVAVGLRVVCAKYA [SEQ ID NO: 8] SARS-CoV-2-S MFVFLVLLPLVSSQCVNLT [SEQ ID NO: 9] human Ig heavy MDWIWRILFLVGAATGAHSQM [SEQ ID NO: 10] chain signal peptide (huSec) HuIgGk signal METPAQLLFLLLLWLPDTTG [SEQ ID NO: 11] peptide IgE heavy chain MDWTWILFLVAAATRVHS [SEQ ID NO: 12] epsilon-1signal peptide Japanese MLGSNSGQRVVFTILLLLVAPAYS [SEQ ID NO: 13] encephalitis PRM signal sequence VSVg protein MKCLLYLAFLFIGVNCA [SEQ ID NO: 14] signal sequence human Ig heavy MELGLSWIFLLAILKGVQC [SEQ ID NO: 15] chain signal peptide 62 11529421v1 Attorney Docket No. 2013237‐0757 Signal Sequence (Amino Acid) human Ig heavy MELGLRWVFLVAILEGVQC [SEQ ID NO: 16] chain signal peptide human Ig heavy MKHLWFFLLLVAAPRWVLS [SEQ ID NO: 17] chain signal peptide human Ig heavy MDWTWRILFLVAAATGAHS [SEQ ID NO: 18] chain signal peptide human Ig heavy MDWTWRFLFVVAAATGVQS [SEQ ID NO: 19] chain signal peptide human Ig heavy MEFGLSWLFLVAILKGVQC [SEQ ID NO: 20] chain signal peptide human Ig heavy MEFGLSWVFLVALFRGVQC [SEQ ID NO: 21] chain signal peptide human Ig heavy MDLLHKNMKHLWFFLLLVAAPRWVLS [SEQ ID NO: 22] chain signal peptide human Ig kappa MDMRVPAQLLGLLLLWLSGARC [SEQ ID NO: 23] chain signal peptide human Ig kappa MKYLLPTAAAGLLLLAAQPAMA [SEQ ID NO: 24] chain signal peptide Table 3: Exemplary nucleotide sequences encoding signal sequences Signal Sequence (Nucleotide) HSV-1 gD SP ATGGGGGGGGCTGCCGCCAGGTTGGGGGCCGTGATTTTGTTTGTCGTCATAGTGGG wild-type CCTCCATGGGGTCCGCAGCAAATAT [SEQ ID NO: 25] HSV-1 gD SP ATGggaggagccGCCGCCagactgggaGCCGTGatcctgttcgtggtgatcGTGggactgCATgga gtgagaAGCaagtac [SEQ ID NO: 26] optimized nucleotide sequence SARS-CoV-2-S ATGTTTGTGTTTCTTGTGCTGCTGCCTCTTGTGTCTTCTCAGTGTGTGAATTTGACA [SEQ ID NO: 27] human Ig heavy ATGGATTGGATTTGGAGAATCCTGTTCCTCGTGGGAGCCGCTACAGGAGCCCACTCC chain signal CAGATG [SEQ ID NO: 28] peptide (huSec) 63 11529421v1 Attorney Docket No. 2013237‐0757 Signal Sequence (Nucleotide) human Ig heavy ATGGAGTTGGGACTGAGCTGGATTTTCCTTTTGGCTATTTTAAAAGGTGTCCAGTGT chain signal [SEQ ID NO: 29] peptide human Ig heavy ATGGAACTGGGGCTCCGCTGGGTTTTCCTTGTTGCTATTTTAGAAGGTGTCCAGTGT chain signal [SEQ ID NO: 30] peptide human Ig heavy ATGAAACACCTGTGGTTCTTCCTCCTGCTGGTGGCAGCTCCCAGATGGGTCCTGTCC chain signal [SEQ ID NO: 31] peptide human Ig heavy ATGGACTGGACCTGGAGGATCCTCTTCTTGGTGGCAGCAGCAACAGGTGCCCACTC chain signal G [SEQ ID NO: 32] peptide human Ig heavy ATGGACTGGACCTGGAGGTTCCTCTTTGTGGTGGCAGCAGCTACAGGTGTCCAGTC chain signal C [SEQ ID NO: 33] peptide human Ig heavy ATGGAGTTTGGGCTGAGCTGGCTTTTTCTTGTGGCGATTCTAAAAGGTGTCCAGTG chain signal T [SEQ ID NO: 34] peptide human Ig heavy ATGGAGTTTGGGCTGAGCTGGGTTTTCCTCGTTGCTCTTTTTAGAGGTGTCCAGTGT chain signal [SEQ ID NO: 35] peptide human Ig heavy ATGGACCTCCTGCACAAGAACATGAAACACCTGTGGTTCTTCCTCCTCCTGGTGGCA chain signal GCTCCCAGATGGGTGCTGTCC [SEQ ID NO: 36] peptide human Ig kappa ATGGACATGAGGGTCCCTGCTCAGCTCCTGGGGCTCCTGCTGCTCTGGCTCTCAGG chain signal TGCCAGATGT [SEQ ID NO: 37] peptide human Ig kappa ATGAAATACCTATTGCCTACGGCAGCCGCTGGATTGTTATTACTCGCGGCCCAGCCG chain signal GCCATGGCC [SEQ ID NO: 38] peptide According to some embodiments, an amino acid sequence enhancing antigen processing and/or presentation is fused, either directly or through a linker, to an antigenic peptide or polypeptide (antigenic sequence), e.g., one or more antigens, immunogenic variants thereof, or immunogenic fragments of the antigens or the immunogenic variants thereof. Accordingly, in some embodiments, the RNA described herein comprises at least one coding region encoding an antigenic peptide or polypeptide and an amino acid sequence enhancing antigen processing and/or presentation. Such amino acid sequences enhancing antigen processing and/or presentation are preferably located at the C-terminus of the or without limited thereto. Amino acid

11529421v1 Attorney Docket No. 2013237‐0757 In some embodiments, an amino acid sequence enhancing antigen processing and/or presentation comprises the amino acid sequence of SEQ ID NO: 39, an amino acid sequence having at least 99%, 98%, 97%, 96%, 95%, 90%, 85%, or 80% identity to the amino acid sequence of SEQ ID NO: 39, or a functional fragment of the amino acid sequence of SEQ ID NO: 39, or the amino acid sequence having at least 99%, 98%, 97%, 96%, 95%, 90%, 85%, or 80% identity to the amino acid sequence of SEQ ID NO: 39. In some embodiments, an amino acid sequence enhancing antigen processing and/or presentation comprises the amino acid sequence of SEQ ID NO: 39. Accordingly, in some embodiments, the RNA described herein comprises at least one coding region encoding an antigenic peptide or polypeptide and an amino acid sequence enhancing antigen processing and/or presentation, said amino acid sequence enhancing antigen processing and/or presentation preferably being fused to the antigenic peptide or polypeptide, more preferably to the C-terminus of the antigenic peptide or polypeptide as described herein. Furthermore, a secretory sequence, e.g., a sequence comprising the amino acid sequence selected from SEQ ID NOs: 4 to 24 or encoded by the nucleotide sequence selected from SEQ ID NOs: 25 to 38, may be fused to the N-terminus of the antigenic peptide or polypeptide. Antigens, immunogenic variants thereof, or immunogenic fragments of the antigens or the immunogenic variants thereof may be fused to an extended-PK group, which increases circulation half-life. Non-limiting examples of extended- PK groups are described herein. It should be understood that other PK groups that increase the circulation half-life of peptides or polypeptides such as antigens, immunogenic variants thereof, or immunogenic fragments of the antigens or the immunogenic variants thereof are also applicable to the present disclosure. In certain embodiments, the extended-PK group is a serum albumin domain (e.g., mouse serum albumin, human serum albumin, or recombinant serum albumin). As used herein, the term "PK" is an acronym for "pharmacokinetic" and encompasses properties of a compound including, by way of example, absorption, distribution, metabolism, and elimination by a subject. As used herein, an "extended-PK group" refers to a protein, peptide, or moiety that increases the circulation half-life of a biologically active molecule when fused to or administered together with the biologically active molecule. Examples of an extended-PK group include serum albumin (e.g., HSA), Immunoglobulin Fc or Fc fragments and variants thereof, transferrin and variants thereof, and human serum albumin (HSA) binders (as disclosed in U.S. Publication Nos. 2005/0287153 and 2007/0003549). Other exemplary extended-PK groups are disclosed in Kontermann, Expert Opin Biol Ther, 2016 Jul;16(7):903-15 which is herein incorporated by reference in its entirety. As used herein, an "extended-PK" polypeptide refers to a polypeptide moiety such as an antigen or immunogenic variant thereof in combination with an extended-PK group. In some embodiments, the extended-PK polypeptide is a fusion protein in which a polypeptide moiety is linked or fused to an extended-PK group. In certain embodiments, the serum half-life of an extended-PK polypeptide is increased relative to the polypeptide alone (i.e., the polypeptide not fused to an extended-PK group). In certain embodiments, the serum half-life of the extended-PK polypeptide is at least 20%, at least 40%, at least 60%, at least 80%, at least 100%, at least 120%, at least 150%, at least 180%, at least 200%, at least 400%, at least 600%, at least 800%, or at least 1000% longer relative to the serum half-life of the polypeptide alone. In certain embodiments, the serum half-life of the extended- PK polypeptide is at least 1.5-fold, 2-fold, 2.5-fold, 3-fold, 3.5-fold, 4-fold, 4.5-fold, 5-fold, 6-fold, 7-fold, 8-fold, 10- fold, 12-fold, 13-fold, 15-fold, 17-fold, 20-fold, 22-fold, 25-fold, 27-fold, 30-fold, 35-fold, 40-fold, or 50-fold greater than the serum half-life of the polypeptide alone. In certain embodiments, the serum half-life of the extended-PK polypeptide is at least 10 hours, 15 hours, 20 hours, 25 hours, 30 hours, 35 hours, 40 hours, 50 hours, 60 hours, 70 65 11529421v1 Attorney Docket No. 2013237‐0757 hours, 80 hours, 90 hours, 100 hours, 110 hours, 120 hours, 130 hours, 135 hours, 140 hours, 150 hours, 160 hours, or 200 hours. As used herein, "half-life" refers to the time taken for the serum or plasma concentration of a compound such as a peptide or polypeptide to reduce by 50%, in vivo, for example due to degradation and/or clearance or sequestration by natural mechanisms. An extended-PK polypeptide suitable for use herein is stabilized in vivo and its half-life increased by, e.g., fusion to serum albumin (e.g., human serum albumin (HSA) or mouse serum albumin (MSA)), which resist degradation and/or clearance or sequestration. The half-life can be determined in any manner known per se, such as by pharmacokinetic analysis. Suitable techniques will be clear to the person skilled in the art, and may for example generally involve the steps of suitably administering a suitable dose of the amino acid sequence or compound to a subject; collecting blood samples or other samples from said subject at regular intervals; determining the level or concentration of the amino acid sequence or compound in said blood sample; and calculating, from (a plot of) the data thus obtained, the time until the level or concentration of the amino acid sequence or compound has been reduced by 50% compared to the initial level upon dosing. Further details are provided in, e.g., standard handbooks, such as Kenneth, A. et al., Chemical Stability of Pharmaceuticals: A Handbook for Pharmacists and in Peters et al., Pharmacokinetic Analysis: A Practical Approach (1996). Reference is also made to Gibaldi, M. et al., Pharmacokinetics, 2nd Rev. Edition, Marcel Dekker (1982). In certain embodiments, the extended-PK group includes serum albumin, or fragments thereof or variants of the serum albumin or fragments thereof (all of which for the purpose of the present disclosure are comprised by the term "albumin"). Polypeptides described herein may be fused to albumin (or a fragment or variant thereof) to form albumin fusion proteins. Such albumin fusion proteins are described in U.S. Publication No. 20070048282. As used herein, "albumin fusion protein" refers to a protein formed by the fusion of at least one molecule of albumin (or a fragment or variant thereof) to at least one molecule of a protein such as a therapeutic protein, in particular an antigen, immunogenic variant thereof, or immunogenic fragment of the antigen or the immunogenic variant thereof. The albumin fusion protein may be generated by translation of a nucleic acid in which a polynucleotide encoding a therapeutic protein is joined in-frame with a polynucleotide encoding an albumin. The therapeutic protein and albumin, once part of the albumin fusion protein, may each be referred to as a “portion”, “region” or “moiety” of the albumin fusion protein (e.g., a “therapeutic protein portion” or an “albumin protein portion”). In a highly preferred embodiment, an albumin fusion protein comprises at least one molecule of a therapeutic protein (including, but not limited to a mature form of the therapeutic protein) and at least one molecule of albumin (including but not limited to a mature form of albumin). In some embodiments, an albumin fusion protein is processed by a host cell such as a cell of the target organ for administered RNA, e.g. a liver cell, and secreted into the circulation. Processing of the nascent albumin fusion protein that occurs in the secretory pathways of the host cell used for expression of the RNA may include, but is not limited to signal peptide cleavage; formation of disulfide bonds; proper folding; addition and processing of carbohydrates (such as for example, N- and O-linked glycosylation); specific proteolytic cleavages; and/or assembly into multimeric proteins. An albumin fusion protein is preferably encoded by RNA in a non-processed form which in particular has a signal peptide at its N-terminus and following secretion by a cell is preferably present in the processed form wherein in particular the signal peptide has been cleaved off. In a most preferred embodiment, the “processed form of an albumin fusion protein” refers to an albumin fusion protein product which has undergone N-terminal signal peptide cleavage, herein also referred to as a “mature albumin fusion protein”. 66 11529421v1 Attorney Docket No. 2013237‐0757 In preferred embodiments, albumin fusion proteins comprising a therapeutic protein have a higher plasma stability compared to the plasma stability of the same therapeutic protein when not fused to albumin. Plasma stability typically refers to the time period between when the therapeutic protein is administered in vivo and carried into the bloodstream and when the therapeutic protein is degraded and cleared from the bloodstream, into an organ, such as the kidney or liver, that ultimately clears the therapeutic protein from the body. Plasma stability is calculated in terms of the half-life of the therapeutic protein in the bloodstream. The half-life of the therapeutic protein in the bloodstream can be readily determined by common assays known in the art. As used herein, “albumin” refers collectively to albumin protein or amino acid sequence, or an albumin fragment or variant, having one or more functional activities (e.g., biological activities) of albumin. In particular, “albumin” refers to human albumin or fragments or variants thereof especially the mature form of human albumin, or albumin from other vertebrates or fragments thereof, or variants of these molecules. The albumin may be derived from any vertebrate, especially any mammal, for example human, cow, sheep, or pig. Non-mammalian albumins include, but are not limited to, hen and salmon. The albumin portion of the albumin fusion protein may be from a different animal than the therapeutic protein portion. In certain embodiments, the albumin is human serum albumin (HSA), or fragments or variants thereof, such as those disclosed in US 5,876,969, WO 2011/124718, WO 2013/075066, and WO 2011/0514789. The terms, human serum albumin (HSA) and human albumin (HA) are used interchangeably herein. The terms, “albumin and “serum albumin” are broader, and encompass human serum albumin (and fragments and variants thereof) as well as albumin from other species (and fragments and variants thereof). As used herein, a fragment of albumin sufficient to prolong the therapeutic activity or plasma stability of the therapeutic protein refers to a fragment of albumin sufficient in length or structure to stabilize or prolong the therapeutic activity or plasma stability of the protein so that the plasma stability of the therapeutic protein portion of the albumin fusion protein is prolonged or extended compared to the plasma stability in the non-fusion state. The albumin portion of the albumin fusion proteins may comprise the full length of the albumin sequence, or may include one or more fragments thereof that are capable of stabilizing or prolonging the therapeutic activity or plasma stability. Such fragments may be of 10 or more amino acids in length or may include about 15, 20, 25, 30, 50, or more contiguous amino acids from the albumin sequence or may include part or all of specific domains of albumin. For instance, one or more fragments of HSA spanning the first two immunoglobulin-like domains may be used. In a preferred embodiment, the HSA fragment is the mature form of HSA. Generally speaking, an albumin fragment or variant will be at least 100 amino acids long, preferably at least 150 amino acids long. According to the disclosure, albumin may be naturally occurring albumin or a fragment or variant thereof. Albumin may be human albumin and may be derived from any vertebrate, especially any mammal. Preferably, the albumin fusion protein comprises albumin as the N-terminal portion, and a therapeutic protein as the C-terminal portion. Alternatively, an albumin fusion protein comprising albumin as the C-terminal portion, and a therapeutic protein as the N-terminal portion may also be used. In other embodiments, the albumin fusion protein has a therapeutic protein fused to both the N-terminus and the C-terminus of albumin. In a preferred embodiment, the therapeutic proteins fused at the N- and C-termini are the same therapeutic proteins. In another preferred embodiment, 67 11529421v1 Attorney Docket No. 2013237‐0757 the therapeutic proteins fused at the N- and C-termini are different therapeutic proteins. In some embodiments, the different therapeutic proteins both comprise one or more fragments of antigens or immunogenic variants thereof. In some embodiments, the therapeutic protein(s) is (are) joined to the albumin through (a) peptide linker(s). A peptide linker between the fused portions may provide greater physical separation between the moieties and thus maximize the accessibility of the therapeutic protein portion, for instance, for binding to its cognate receptor. The peptide linker may consist of amino acids such that it is flexible or more rigid. The linker sequence may be cleavable by a protease or chemically. As used herein, the term "Fc region" refers to the portion of a native immunoglobulin formed by the respective Fc domains (or Fc moieties) of its two heavy chains. As used herein, the term "Fc domain" refers to a portion or fragment of a single immunoglobulin (Ig) heavy chain wherein the Fc domain does not comprise an Fv domain. In certain embodiments, an Fc domain begins in the hinge region just upstream of the papain cleavage site and ends at the C- terminus of the antibody. Accordingly, a complete Fc domain comprises at least a hinge domain, a CH2 domain, and a CH3 domain. In certain embodiments, an Fc domain comprises at least one of: a hinge (e.g., upper, middle, and/or lower hinge region) domain, a CH2 domain, a CH3 domain, a CH4 domain, or a variant, portion, or fragment thereof. In certain embodiments, an Fc domain comprises a complete Fc domain (i.e., a hinge domain, a CH2 domain, and a CH3 domain). In certain embodiments, an Fc domain comprises a hinge domain (or portion thereof) fused to a CH3 domain (or portion thereof). In certain embodiments, an Fc domain comprises a CH2 domain (or portion thereof) fused to a CH3 domain (or portion thereof). In certain embodiments, an Fc domain consists of a CH3 domain or portion thereof. In certain embodiments, an Fc domain consists of a hinge domain (or portion thereof) and a CH3 domain (or portion thereof). In certain embodiments, an Fc domain consists of a CH2 domain (or portion thereof) and a CH3 domain. In certain embodiments, an Fc domain consists of a hinge domain (or portion thereof) and a CH2 domain (or portion thereof). In certain embodiments, an Fc domain lacks at least a portion of a CH2 domain (e.g., all or part of a CH2 domain). An Fc domain herein generally refers to a polypeptide comprising all or part of the Fc domain of an immunoglobulin heavy-chain. This includes, but is not limited to, polypeptides comprising the entire CH1, hinge, CH2, and/or CH3 domains as well as fragments of such peptides comprising only, e.g., the hinge, CH2, and CH3 domain. The Fc domain may be derived from an immunoglobulin of any species and/or any subtype, including, but not limited to, a human IgG1, IgG2, IgG3, IgG4, IgD, IgA, IgE, or IgM antibody. The Fc domain encompasses native Fc and Fc variant molecules. As set forth herein, it will be understood by one of ordinary skill in the art that any Fc domain may be modified such that it varies in amino acid sequence from the native Fc domain of a naturally occurring immunoglobulin molecule. In certain embodiments, the Fc domain has reduced effector function (e.g., FcγR binding). The Fc domains of a polypeptide described herein may be derived from different immunoglobulin molecules. For example, an Fc domain of a polypeptide may comprise a CH2 and/or CH3 domain derived from an IgG1 molecule and a hinge region derived from an IgG3 molecule. In another example, an Fc domain can comprise a chimeric hinge region derived, in part, from an IgG1 molecule and, in part, from an IgG3 molecule. In another example, an Fc domain can comprise a chimeric hinge derived, in part, from an IgG1 molecule and, in part, from an IgG4 molecule. In certain embodiments, an extended-PK group includes an Fc domain or fragments thereof or variants of the Fc domain or fragments thereof (all of which for the purpose of the present disclosure are comprised by the term "Fc domain"). The Fc domain does not contain a variable region that binds to antigen. Fc domains suitable for use in the present disclosure may be obtained from a number of different sources. In certain embodiments, an Fc domain is derived from a human immunoglobulin. In certain embodiments, the Fc domain is from a human IgG1 constant region. 68 11529421v1 Attorney Docket No. 2013237‐0757 It is understood, however, that the Fc domain may be derived from an immunoglobulin of another mammalian species, including for example, a rodent (e.g. a mouse, rat, rabbit, guinea pig) or non-human primate (e.g. chimpanzee, macaque) species. Moreover, the Fc domain (or a fragment or variant thereof) may be derived from any immunoglobulin class, including IgM, IgG, IgD, IgA, and IgE, and any immunoglobulin isotype, including IgG1, IgG2, IgG3, and IgG4. A variety of Fc domain gene sequences (e.g., mouse and human constant region gene sequences) are available in the form of publicly accessible deposits. Constant region domains comprising an Fc domain sequence can be selected lacking a particular effector function and/or with a particular modification to reduce immunogenicity. Many sequences of antibodies and antibody-encoding genes have been published and suitable Fc domain sequences (e.g. hinge, CH2, and/or CH3 sequences, or fragments or variants thereof) can be derived from these sequences using art recognized techniques. In certain embodiments, the extended-PK group is a serum albumin binding protein such as those described in US2005/0287153, US2007/0003549, US2007/0178082, US2007/0269422, US2010/0113339, WO2009/083804, and WO2009/133208, which are herein incorporated by reference in their entirety. In certain embodiments, the extended- PK group is transferrin, as disclosed in US 7,176,278 and US 8,158,579, which are herein incorporated by reference in their entirety. In certain embodiments, the extended-PK group is a serum immunoglobulin binding protein such as those disclosed in US2007/0178082, US2014/0220017, and US2017/0145062, which are herein incorporated by reference in their entirety. In certain embodiments, the extended-PK group is a fibronectin (Fn)-based scaffold domain protein that binds to serum albumin, such as those disclosed in US2012/0094909, which is herein incorporated by reference in its entirety. Methods of making fibronectin-based scaffold domain proteins are also disclosed in US2012/0094909. A non-limiting example of a Fn3-based extended-PK group is Fn3(HSA), i.e., a Fn3 protein that binds to human serum albumin. In certain aspects, the extended-PK polypeptide, suitable for use according to the disclosure, can employ one or more peptide linkers. As used herein, the term "peptide linker" refers to a peptide or polypeptide sequence which connects two or more domains (e.g., the extended-PK moiety and a polypeptide moiety, e.g., a fragment of an antigen or immunogenic variant thereof) in a linear amino acid sequence of a polypeptide chain. For example, peptide linkers may be used to connect a fragment of an antigen or immunogenic variant thereof to a HSA domain. Linkers suitable for fusing the extended-PK group to, e.g., a fragment of an antigen or immunogenic variant thereof are well known in the art and described herein above. In the following, embodiments of vaccine RNAs are described, wherein certain terms used when describing elements thereof have the following meanings: cap: 5'-cap structure, e.g., selected from the group consisting of m
27,2'OG(5’)ppSp(5')G (in particular its D1 diastereomer), m
27,3'OG(5')ppp(5')G, and m
27,3'-OGppp(m
12'-O)ApG. hAg-Kozak: 5'-UTR sequence of the human alpha-globin mRNA with an optimized ʻKozak sequenceʼ to increase translational efficiency. sec/MITD: Fusion-protein tags derived from the sequence encoding the human MHC class I complex (HLA-B51, haplotype A2, B27/B51, Cw2/Cw3), which have been shown to improve antigen processing and presentation. Sec corresponds to the 78 bp fragment coding for the secretory signal peptide, which guides translocation of the nascent 69 11529421v1 Attorney Docket No. 2013237‐0757 polypeptide chain into the endoplasmatic reticulum. MITD corresponds to the transmembrane and cytoplasmic domain of the MHC class I molecule, also called MHC class I trafficking domain. Antigen: Sequences encoding the respective two or more fragments of the antigen(s)/epitope(s), i.e., of one or more antigens or immunogenic variants thereof. Polypeptide linker: Sequences coding for short peptide linkers predominantly. FI element: The 3'-UTR is a combination of two sequence elements derived from the “amino terminal enhancer of split” (AES) mRNA (called F) and the mitochondrial encoded 12S ribosomal RNA (called I). These were identified by an ex vivo selection process for sequences that confer RNA stability and augment total protein expression. A30L70: A poly(A)-tail measuring 110 nucleotides in length, consisting of a stretch of 30 adenosine residues, followed by a 10 nucleotide linker sequence and another 70 adenosine residues designed to enhance RNA stability and translational efficiency in dendritic cells. In some embodiments, vaccine RNA described herein has one of the following structures: cap-hAg-Kozak-Antigen(s)-FI-A30L70 cap-hAg-Kozak-sec-Antigen(s)-FI-A30L70 cap-hAg-Kozak-sec-Antigen(s)-MITD-FI-A30L70 In some embodiments, polypeptide described herein has the structure: sec-Antigen sec-Antigen-MITD In some embodiments, hAg-Kozak comprises the nucleotide sequence of SEQ ID NO: 40. In some embodiments, sec of the encoded polypeptide/epitope comprises an amino acid sequence selected from SEQ ID NOs: 4 to 24 or encoded by the nucleotide sequence selected from SEQ ID NOs: 25 to 38. In some embodiments, MITD of the encoded polypeptide/epitope comprises the amino acid sequence of SEQ ID NO: 39. In some embodiments, FI comprises the nucleotide sequence of SEQ ID NO: 41. In some embodiments, A30L70 comprises the nucleotide sequence of SEQ ID NO: 42. In some embodiments, the different elements (sec, Antigen, MITD) may be linked by one or more GS linkers. In some embodiments, a GS linker of the encoded polypeptide/epitope comprises the amino acid sequence of SEQ ID NO: 43. In some embodiments, the sequence encoding the polypeptide/epitope comprises a modified nucleoside replacing (partially or completely, preferably completely) uridine, wherein the modified nucleoside is selected from the group consisting of pseudouridine (ψ), N1-methyl-pseudouridine (m1ψ), and 5-methyl-uridine. In some embodiments, the sequence encoding the polypeptide/epitope is codon-optimized. In some embodiments, the G/C content of the sequence encoding the polypeptide/epitope is increased compared to the wild type coding sequence. In some embodiments, the RNA (in particular, mRNA) described herein comprises: ‐ a 5’ UTR comprising the nucleotide sequence of SEQ ID NO: 40, or a nucleotide sequence having at least 99%, 98%, 97%, 96%, 95%, 90%, 85%, or 80% identity to the nucleotide sequence of SEQ ID NO: 40; 70 11529421v1 Attorney Docket No. 2013237‐0757 ‐ a 3’ UTR comprising the nucleotide sequence of SEQ ID NO: 41, or a nucleotide sequence having at least 99%, 98%, 97%, 96%, 95%, 90%, 85%, or 80% identity to the nucleotide sequence of SEQ ID NO: 41; and a poly-A sequence comprising the nucleotide sequence of SEQ ID NO: 42. In some embodiments, the RNA (in particular, mRNA) described herein comprises: ‐ m
27,3’-OGppp(m
12’-O) ApG as capping structure at the 5'-end of the mRNA; ‐ a 5’ UTR comprising the nucleotide sequence of SEQ ID NO: 40, or a nucleotide sequence having at least 99%, 98%, 97%, 96%, 95%, 90%, 85%, or 80% identity to the nucleotide sequence of SEQ ID NO: 40; ‐ a 3’ UTR comprising the nucleotide sequence of SEQ ID NO: 41, or a nucleotide sequence having at least 99%, 98%, 97%, 96%, 95%, 90%, 85%, or 80% identity to the nucleotide sequence of SEQ ID NO: 41; and ‐ a poly-A sequence comprising the nucleotide sequence of SEQ ID NO: 42. In some embodiments, the RNA is unmodified. In some embodiments, the RNA is modified. In some embodiments, the RNA comprises N1-methyl-pseudouridine (m1ψ) in place of at least one uridine (e.g., in place of each uridine). In some embodiments, the RNA (in particular, mRNA) described herein comprises: ‐ m2
7,3’-OGppp(m1
2’-O) ApG as capping structure at the 5'-end of the mRNA; ‐ a 5’ UTR comprising the nucleotide sequence of SEQ ID NO: 40, or a nucleotide sequence having at least 99%, 98%, 97%, 96%, 95%, 90%, 85%, or 80% identity to the nucleotide sequence of SEQ ID NO: 40; ‐ a 3’ UTR comprising the nucleotide sequence of SEQ ID NO: 41, or a nucleotide sequence having at least 99%, 98%, 97%, 96%, 95%, 90%, 85%, or 80% identity to the nucleotide sequence of SEQ ID NO: 41; and ‐ a poly-A sequence comprising the nucleotide sequence of SEQ ID NO: 42; and ‐ N1-methyl-pseudouridine (m1ψ) in place of at least one uridine (e.g., in place of each uridine). In some embodiments, a polypeptide or epitope described herein is derived from Mycobacterium tuberculosis, Pseudomonas aeruginosa, Staphylococcus aureus, Clostridioides difficile, Bacillus anthracis, Corynebacterium diphteriae or Vibrio Cholerae. In some embodiments, a polypeptide or epitope described herein is derived from a Mycobacterium tuberculosis protein, an immunogenic variant thereof, or an immunogenic fragment of the Mycobacterium tuberculosis protein or the immunogenic variant thereof. Thus, in some embodiments, the RNA, e.g., mRNA, used in the present disclosure encodes an amino acid sequence comprising an Mtb protein, an immunogenic variant thereof, or an immunogenic fragment of the Mtb protein or the immunogenic variant thereof. In some embodiments, a polypeptide or epitope described herein is derived from an Mtb protein from the acute phase of the Mtb life cycle, an immunogenic variant thereof, or an immunogenic fragment of the Mtb protein from the acute phase of the Mtb life cycle or the immunogenic variant thereof. In some embodiments, a polypeptide or epitope described herein is derived from an Mtb protein from the latent phase of the Mtb life cycle, an immunogenic variant thereof, or an immunogenic fragment of the Mtb protein from the latent phase of the Mtb life cycle or the immunogenic variant thereof. 71 11529421v1 Attorney Docket No. 2013237‐0757 In some embodiments, a polypeptide or epitope described herein is derived from an Mtb protein from the resuscitation phase of the Mtb life cycle, an immunogenic variant thereof, or an immunogenic fragment of the Mtb protein from the resuscitation phase of the Mtb life cycle or the immunogenic variant thereof. In some embodiments, a polypeptide or epitope described herein is derived from an EspA. In some embodiments, RNA (in particular, mRNA) described herein (e.g., contained in the compositions/formulations of the present disclosure and/or used in the methods of the present disclosure) may be presented as a product containing the vaccine RNA as active substance and other ingredients comprising: ALC-0315 ((4- hydroxybutyl)azanediyl)bis(hexane-6,1-diyl)bis(2-hexyldecanoate), ALC-0159 (2-[(polyethylene glycol)-2000]-N,N- ditetradecylacetamide), 1,2-Distearoyl-sn-glycero-3-phosphocholine (DSPC), and cholesterol. In some embodiments, the RNA (in particular, mRNA) described herein is formulated or is to be formulated as a liquid, a solid, or a combination thereof. In some embodiments, the RNA (in particular, mRNA) described herein is formulated or is to be formulated for injection. In some embodiments, the RNA (in particular, mRNA) described herein is formulated or is to be formulated for intramuscular administration. In some embodiments, the RNA (in particular, mRNA) described herein is formulated or is to be formulated as a composition, e.g., a pharmaceutical composition. In some embodiments, the composition comprises a cationically ionizable lipid. In some embodiments, the composition comprises a cationically ionizable lipid and one or more additional lipids. In some embodiments, the one or more additional lipids are selected from polymer-conjugated lipids, neutral lipids, and combinations thereof. In some embodiments, the neutral lipids include phospholipids, steroid lipids, and combinations thereof. In some embodiments, the one or more additional lipids are a combination of a polymer-conjugated lipid, a phospholipid, and a steroid lipid. In some embodiments, the composition comprises a cationically ionizable lipid; a polymer-conjugated lipid which is a PEG-conjugated lipid; cholesterol; and a phospholipid. In some embodiments, the phospholipid is DSPC. In some embodiments, the phospholipid is DOPE. In some embodiments, the composition comprises a cationically ionizable lipid; a polymer-conjugated lipid which is 2- [(polyethylene glycol)-2000]-N,N-ditetradecylacetamide; cholesterol; and a phospholipid. In some embodiments, the phospholipid is DSPC. In some embodiments, the phospholipid is DOPE. In some embodiments, the composition comprises a cationically ionizable lipid which is ((4- hydroxybutyl)azanediyl)bis(hexane-6,1-diyl)bis(2-hexyldecanoate); a polymer-conjugated lipid which is 2- [(polyethylene glycol)-2000]-N,N-ditetradecylacetamide; cholesterol; and a phospholipid. In some embodiments, the phospholipid is DSPC. In some embodiments, the phospholipid is DOPE. In some embodiments, at least a portion of (i) the RNA, (ii) the cationically ionizable lipid, and if present, (iii) the one or more additional lipids is present in particles. In some embodiments, the particles are nanoparticles, such as lipid nanoparticles (LNPs). In some embodiments, the composition, in particular the pharmaceutical composition, is a vaccine. 72 11529421v1 Attorney Docket No. 2013237‐0757 In some embodiments, the composition, in particular the pharmaceutical composition, further comprises one or more pharmaceutically acceptable carriers, diluents and/or excipients. In some embodiments, the RNA and/or the composition, in particular the pharmaceutical composition, is/are a component of a kit. In some embodiments, the kit further comprises instructions for use of the RNA for inducing an immune response against Mycobacterium tuberculosis in a subject. In some embodiments, the kit further comprises instructions for use of the RNA for therapeutically or prophylactically treating a Mycobacterium tuberculosis infection in a subject. In some embodiments, the subject is a human. In some embodiments, the RNA (in particular, mRNA), e.g., RNA encoding polypeptide, described in the present disclosure is non-immunogenic. RNA encoding an immunostimulant may be administered according to the present disclosure to provide an adjuvant effect. The RNA encoding an immunostimulant may be standard RNA or non- immunogenic RNA. Embodiments of toxic or cytotoxic microbioal antigens The present disclosure describes antigens and immunogenic variants thereof, (referred to as "antigens" herein) and RNA encoding these antigens. EspA In some embodiments, the Mtb antigen EspA comprises the amino acid sequence according to SEQ ID NO: 1. A full- length antigen representing the antigen EspA is characterized in that it comprises the full-length amino acid sequence according to SEQ ID NO: 1, whereas an antigen fragment representing the antigen EspA is characterized in that it comprises an amino acid sequence which is only a part of SEQ ID NO: 1 but which still is able to induce an immune reaction to EspA, when delivered to a subject. An immunogenic variant of the Mtb antigen EspA comprises an amino acid sequence which is "immunologically equivalent" to EspA and thus, is able to induce an immune reaction to EspA, when delivered to a subject. In some embodiments, an immunogenic variant of the Mtb antigen EspA comprises an amino acid sequence differing from SEQ ID NO: 1 by one or more deletions, insertions or substitutions in the amino acid sequence while having at least 99%, 98%, 97%, 96%, 95%, 90%, 85%, or 80% identity to SEQ ID NO: 1 (e.g. determined by sequence alignment using known sequence alignment algorithms such as BLASTP). A full-length antigen representing an immunogenic variant of the antigen EspA is characterized in that it comprises the full-length amino acid sequence of the immunogenic variant, whereas an antigen fragment representing an immunogenic variant of the antigen EspA is characterized in that it comprises an amino acid sequence which is only a part of the full-length amino acid sequence of the immunogenic variant but which still is able to induce an immune reaction to EspA, when delivered to a subject. PlcA In some embodiments, the Mtb antigen PlcA comprises the amino acid sequence according to SEQ ID NO: 2. A full- length antigen representing the antigen PlcA is characterized in that it comprises the full-length amino acid sequence according to SEQ ID NO: 2, whereas an antigen fragment representing the antigen PlcA is characterized in that it 73 11529421v1 Attorney Docket No. 2013237‐0757 comprises an amino acid sequence which is only a part of SEQ ID NO: 2 but which still is able to induce an immune reaction to PlcA, when delivered to a subject. An immunogenic variant of the Mtb antigen PlcA comprises an amino acid sequence which is "immunologically equivalent" to PlcA and thus, is able to induce an immune reaction to PlcA, when delivered to a subject. In some embodiments, an immunogenic variant of the Mtb antigen PlcA comprises an amino acid sequence differing from SEQ ID NO: 2 by one or more deletions, insertions or substitutions in the amino acid sequence while having at least 99%, 98%, 97%, 96%, 95%, 90%, 85%, or 80% identity to SEQ ID NO: 2 (e.g. determined by sequence alignment using known sequence alignment algorithms such as BLASTP). A full-length antigen representing an immunogenic variant of the antigen PlcA is characterized in that it comprises the full-length amino acid sequence of the immunogenic variant, whereas an antigen fragment representing an immunogenic variant of the antigen PlcA is characterized in that it comprises an amino acid sequence which is only a part of the full-length amino acid sequence of the immunogenic variant but which still is able to induce an immune reaction to PlcA, when delivered to a subject. PlcB In some embodiments, the Mtb antigen PlcB comprises the amino acid sequence according to SEQ ID NO: 3. A full- length antigen representing the antigen PlcB is characterized in that it comprises the full-length amino acid sequence according to SEQ ID NO: 3, whereas an antigen fragment representing the antigen PlcB is characterized in that it comprises an amino acid sequence which is only a part of SEQ ID NO: 3 but which still is able to induce an immune reaction to PlcB, when delivered to a subject. An immunogenic variant of the Mtb antigen PlcB comprises an amino acid sequence which is "immunologically equivalent" to PlcB and thus, is able to induce an immune reaction to PlcB, when delivered to a subject. In some embodiments, an immunogenic variant of the Mtb antigen PlcB comprises an amino acid sequence differing from SEQ ID NO: 3 by one or more deletions, insertions or substitutions in the amino acid sequence while having at least 99%, 98%, 97%, 96%, 95%, 90%, 85%, or 80% identity to SEQ ID NO: 3 (e.g. determined by sequence alignment using known sequence alignment algorithms such as BLASTP). A full-length antigen representing an immunogenic variant of the antigen PlcB is characterized in that it comprises the full-length amino acid sequence of the immunogenic variant, whereas an antigen fragment representing an immunogenic variant of the antigen PlcB is characterized in that it comprises an amino acid sequence which is only a part of the full-length amino acid sequence of the immunogenic variant but which still is able to induce an immune reaction to PlcB, when delivered to a subject. PlcC In some embodiments, the Mtb antigen PlcC comprises the amino acid sequence according to SEQ ID NO: 46. A full- length antigen representing the antigen PlcC is characterized in that it comprises the full-length amino acid sequence according to SEQ ID NO: 46, whereas an antigen fragment representing the antigen PlcC is characterized in that it comprises an amino acid sequence which is only a part of SEQ ID NO: 46 but which still is able to induce an immune reaction to PlcC, when delivered to a subject. An immunogenic variant of the Mtb antigen PlcC comprises an amino acid sequence which is "immunologically equivalent" to PlcC and thus, is able to induce an immune reaction to PlcC, when delivered to a subject. In some embodiments, an immunogenic variant of the Mtb antigen PlcC comprises an amino acid sequence differing from SEQ ID NO: 46 by one or more deletions, insertions or substitutions in the amino acid sequence while having at least 99%, 98%, 97%, 96%, 95%, 90%, 85%, or 80% identity to SEQ ID NO: 46 (e.g. determined by sequence alignment using known sequence alignment algorithms such as BLASTP). A full-length antigen representing an immunogenic variant of 74 11529421v1 Attorney Docket No. 2013237‐0757 the antigen PlcC is characterized in that it comprises the full-length amino acid sequence of the immunogenic variant, whereas an antigen fragment representing an immunogenic variant of the antigen PlcC is characterized in that it comprises an amino acid sequence which is only a part of the full-length amino acid sequence of the immunogenic variant but which still is able to induce an immune reaction to PlcC, when delivered to a subject. PlcD In some embodiments, the Mtb antigen PlcD comprises the amino acid sequence according to SEQ ID NO: 47. A full- length antigen representing the antigen PlcD is characterized in that it comprises the full-length amino acid sequence according to SEQ ID NO: 47, whereas an antigen fragment representing the antigen PlcD is characterized in that it comprises an amino acid sequence which is only a part of SEQ ID NO: 47 but which still is able to induce an immune reaction to PlcD, when delivered to a subject. An immunogenic variant of the Mtb antigen PlcD comprises an amino acid sequence which is "immunologically equivalent" to PlcD and thus, is able to induce an immune reaction to PlcD, when delivered to a subject. In some embodiments, an immunogenic variant of the Mtb antigen PlcD comprises an amino acid sequence differing from SEQ ID NO: 47 by one or more deletions, insertions or substitutions in the amino acid sequence while having at least 99%, 98%, 97%, 96%, 95%, 90%, 85%, or 80% identity to SEQ ID NO: 47 (e.g. determined by sequence alignment using known sequence alignment algorithms such as BLASTP). A full-length antigen representing an immunogenic variant of the antigen PlcD is characterized in that it comprises the full-length amino acid sequence of the immunogenic variant, whereas an antigen fragment representing an immunogenic variant of the antigen PlcD is characterized in that it comprises an amino acid sequence which is only a part of the full-length amino acid sequence of the immunogenic variant but which still is able to induce an immune reaction to PlcD, when delivered to a subject. TlyA In some embodiments, the Mtb antigen TlyA comprises the amino acid sequence according to SEQ ID NO: 48. A full- length antigen representing the antigen TlyA is characterized in that it comprises the full-length amino acid sequence according to SEQ ID NO: 48, whereas an antigen fragment representing the antigen TlyA is characterized in that it comprises an amino acid sequence which is only a part of SEQ ID NO: 48 but which still is able to induce an immune reaction to TlyA, when delivered to a subject. An immunogenic variant of the Mtb antigen TlyA comprises an amino acid sequence which is "immunologically equivalent" to TlyA and thus, is able to induce an immune reaction to TlyA, when delivered to a subject. In some embodiments, an immunogenic variant of the Mtb antigen TlyA comprises an amino acid sequence differing from SEQ ID NO: 48 by one or more deletions, insertions or substitutions in the amino acid sequence while having at least 99%, 98%, 97%, 96%, 95%, 90%, 85%, or 80% identity to SEQ ID NO: 48 (e.g. determined by sequence alignment using known sequence alignment algorithms such as BLASTP). A full-length antigen representing an immunogenic variant of the antigen TlyA is characterized in that it comprises the full-length amino acid sequence of the immunogenic variant, whereas an antigen fragment representing an immunogenic variant of the antigen TlyA is characterized in that it comprises an amino acid sequence which is only a part of the full-length amino acid sequence of the immunogenic variant but which still is able to induce an immune reaction to TlyA, when delivered to a subject. ExoS In some embodiments, the Pseudomonas aeruginosa antigen ExoS comprises the amino acid sequence according to SEQ ID NO: 49. A full-length antigen representing the antigen ExoS is characterized in that it comprises the full-length amino acid sequence according to SEQ ID NO: 49, whereas an antigen fragment representing the antigen ExoS is 75 11529421v1 Attorney Docket No. 2013237‐0757 characterized in that it comprises an amino acid sequence which is only a part of SEQ ID NO: 49 but which still is able to induce an immune reaction to ExoS, when delivered to a subject. An immunogenic variant of the Pseudomonas aeruginosa antigen ExoS comprises an amino acid sequence which is "immunologically equivalent" to ExoS and thus, is able to induce an immune reaction to ExoS, when delivered to a subject. In some embodiments, an immunogenic variant of the Pseudomonas aeruginosa antigen ExoS comprises an amino acid sequence differing from SEQ ID NO: 49 by one or more deletions, insertions or substitutions in the amino acid sequence while having at least 99%, 98%, 97%, 96%, 95%, 90%, 85%, or 80% identity to SEQ ID NO: 49 (e.g. determined by sequence alignment using known sequence alignment algorithms such as BLASTP). A full-length antigen representing an immunogenic variant of the antigen ExoS is characterized in that it comprises the full-length amino acid sequence of the immunogenic variant, whereas an antigen fragment representing an immunogenic variant of the antigen ExoS is characterized in that it comprises an amino acid sequence which is only a part of the full-length amino acid sequence of the immunogenic variant but which still is able to induce an immune reaction to ExoS, when delivered to a subject. ExoT In some embodiments, the Pseudomonas aeruginosa antigen ExoT comprises the amino acid sequence according to SEQ ID NO: 50. A full-length antigen representing the antigen ExoT is characterized in that it comprises the full-length amino acid sequence according to SEQ ID NO: 50, whereas an antigen fragment representing the antigen ExoT is characterized in that it comprises an amino acid sequence which is only a part of SEQ ID NO: 50 but which still is able to induce an immune reaction to ExoT, when delivered to a subject. An immunogenic variant of the Pseudomonas aeruginosa antigen ExoT comprises an amino acid sequence which is "immunologically equivalent" to ExoT and thus, is able to induce an immune reaction to ExoT, when delivered to a subject. In some embodiments, an immunogenic variant of the Pseudomonas aeruginosa antigen ExoT comprises an amino acid sequence differing from SEQ ID NO: 50 by one or more deletions, insertions or substitutions in the amino acid sequence while having at least 99%, 98%, 97%, 96%, 95%, 90%, 85%, or 80% identity to SEQ ID NO: 50 (e.g. determined by sequence alignment using known sequence alignment algorithms such as BLASTP). A full-length antigen representing an immunogenic variant of the antigen ExoT is characterized in that it comprises the full-length amino acid sequence of the immunogenic variant, whereas an antigen fragment representing an immunogenic variant of the antigen ExoT is characterized in that it comprises an amino acid sequence which is only a part of the full-length amino acid sequence of the immunogenic variant but which still is able to induce an immune reaction to ExoT, when delivered to a subject. ExoU In some embodiments, the Pseudomonas aeruginosa antigen ExoU comprises the amino acid sequence according to SEQ ID NO: 51. A full-length antigen representing the antigen ExoU is characterized in that it comprises the full-length amino acid sequence according to SEQ ID NO: 51, whereas an antigen fragment representing the antigen ExoU is characterized in that it comprises an amino acid sequence which is only a part of SEQ ID NO: 51 but which still is able to induce an immune reaction to ExoU, when delivered to a subject. An immunogenic variant of the Pseudomonas aeruginosa antigen ExoU comprises an amino acid sequence which is "immunologically equivalent" to ExoU and thus, is able to induce an immune reaction to ExoU, when delivered to a subject. In some embodiments, an immunogenic variant of the Pseudomonas aeruginosa antigen ExoU comprises an amino acid sequence differing from SEQ ID NO: 51 by one or more deletions, insertions or substitutions in the amino 76 11529421v1 Attorney Docket No. 2013237‐0757 acid sequence while having at least 99%, 98%, 97%, 96%, 95%, 90%, 85%, or 80% identity to SEQ ID NO: 51 (e.g. determined by sequence alignment using known sequence alignment algorithms such as BLASTP). A full-length antigen representing an immunogenic variant of the antigen ExoU is characterized in that it comprises the full-length amino acid sequence of the immunogenic variant, whereas an antigen fragment representing an immunogenic variant of the antigen ExoU is characterized in that it comprises an amino acid sequence which is only a part of the full-length amino acid sequence of the immunogenic variant but which still is able to induce an immune reaction to ExoU, when delivered to a subject. ExoY In some embodiments, the Pseudomonas aeruginosa antigen ExoY comprises the amino acid sequence according to SEQ ID NO: 52. A full-length antigen representing the antigen ExoY is characterized in that it comprises the full-length amino acid sequence according to SEQ ID NO: 52, whereas an antigen fragment representing the antigen ExoY is characterized in that it comprises an amino acid sequence which is only a part of SEQ ID NO: 52 but which still is able to induce an immune reaction to ExoY, when delivered to a subject. An immunogenic variant of the Pseudomonas aeruginosa antigen ExoY comprises an amino acid sequence which is "immunologically equivalent" to ExoY and thus, is able to induce an immune reaction to ExoY, when delivered to a subject. In some embodiments, an immunogenic variant of the Pseudomonas aeruginosa antigen ExoY comprises an amino acid sequence differing from SEQ ID NO: 52 by one or more deletions, insertions or substitutions in the amino acid sequence while having at least 99%, 98%, 97%, 96%, 95%, 90%, 85%, or 80% identity to SEQ ID NO: 52 (e.g. determined by sequence alignment using known sequence alignment algorithms such as BLASTP). A full-length antigen representing an immunogenic variant of the antigen ExoY is characterized in that it comprises the full-length amino acid sequence of the immunogenic variant, whereas an antigen fragment representing an immunogenic variant of the antigen ExoY is characterized in that it comprises an amino acid sequence which is only a part of the full-length amino acid sequence of the immunogenic variant but which still is able to induce an immune reaction to ExoY, when delivered to a subject. ExoA In some embodiments, the Pseudomonas aeruginosa antigen ExoA comprises the amino acid sequence according to SEQ ID NO: 53. A full-length antigen representing the antigen ExoA is characterized in that it comprises the full-length amino acid sequence according to SEQ ID NO: 53, whereas an antigen fragment representing the antigen ExoA is characterized in that it comprises an amino acid sequence which is only a part of SEQ ID NO: 53 but which still is able to induce an immune reaction to ExoA, when delivered to a subject. An immunogenic variant of the Pseudomonas aeruginosa antigen ExoA comprises an amino acid sequence which is "immunologically equivalent" to ExoA and thus, is able to induce an immune reaction to ExoA, when delivered to a subject. In some embodiments, an immunogenic variant of the Pseudomonas aeruginosa antigen ExoA comprises an amino acid sequence differing from SEQ ID NO: 53 by one or more deletions, insertions or substitutions in the amino acid sequence while having at least 99%, 98%, 97%, 96%, 95%, 90%, 85%, or 80% identity to SEQ ID NO: 53 (e.g. determined by sequence alignment using known sequence alignment algorithms such as BLASTP). A full-length antigen representing an immunogenic variant of the antigen ExoA is characterized in that it comprises the full-length amino acid sequence of the immunogenic variant, whereas an antigen fragment representing an immunogenic variant of the antigen ExoA is characterized in that it comprises an amino acid sequence which is only a part of the full-length amino acid sequence of the immunogenic variant but which still is able to induce an immune reaction to ExoA, when delivered to a subject. 77 11529421v1 Attorney Docket No. 2013237‐0757 Azurin In some embodiments, the Pseudomonas aeruginosa antigen Azurin comprises the amino acid sequence according to SEQ ID NO: 54. A full-length antigen representing the antigen Azurin is characterized in that it comprises the full-length amino acid sequence according to SEQ ID NO: 54, whereas an antigen fragment representing the antigen Azurin is characterized in that it comprises an amino acid sequence which is only a part of SEQ ID NO: 54 but which still is able to induce an immune reaction to Azurin, when delivered to a subject. An immunogenic variant of the Pseudomonas aeruginosa antigen Azurin comprises an amino acid sequence which is "immunologically equivalent" to Azurin and thus, is able to induce an immune reaction to Azurin, when delivered to a subject. In some embodiments, an immunogenic variant of the Pseudomonas aeruginosa antigen Azurin comprises an amino acid sequence differing from SEQ ID NO: 54 by one or more deletions, insertions or substitutions in the amino acid sequence while having at least 99%, 98%, 97%, 96%, 95%, 90%, 85%, or 80% identity to SEQ ID NO: 54 (e.g. determined by sequence alignment using known sequence alignment algorithms such as BLASTP). A full-length antigen representing an immunogenic variant of the antigen Azurin is characterized in that it comprises the full-length amino acid sequence of the immunogenic variant, whereas an antigen fragment representing an immunogenic variant of the antigen Azurin is characterized in that it comprises an amino acid sequence which is only a part of the full-length amino acid sequence of the immunogenic variant but which still is able to induce an immune reaction to Azurin, when delivered to a subject. ExlA In some embodiments, the Pseudomonas aeruginosa antigen ExlA comprises the amino acid sequence according to SEQ ID NO: 55. A full-length antigen representing the antigen ExlA is characterized in that it comprises the full-length amino acid sequence according to SEQ ID NO: 55, whereas an antigen fragment representing the antigen ExlA is characterized in that it comprises an amino acid sequence which is only a part of SEQ ID NO: 55 but which still is able to induce an immune reaction to ExlA, when delivered to a subject. An immunogenic variant of the Pseudomonas aeruginosa antigen ExlA comprises an amino acid sequence which is "immunologically equivalent" to ExlA and thus, is able to induce an immune reaction to ExlA, when delivered to a subject. In some embodiments, an immunogenic variant of the Pseudomonas aeruginosa antigen ExlA comprises an amino acid sequence differing from SEQ ID NO: 55 by one or more deletions, insertions or substitutions in the amino acid sequence while having at least 99%, 98%, 97%, 96%, 95%, 90%, 85%, or 80% identity to SEQ ID NO: 55 (e.g. determined by sequence alignment using known sequence alignment algorithms such as BLASTP). A full-length antigen representing an immunogenic variant of the antigen ExlA is characterized in that it comprises the full-length amino acid sequence of the immunogenic variant, whereas an antigen fragment representing an immunogenic variant of the antigen ExlA is characterized in that it comprises an amino acid sequence which is only a part of the full-length amino acid sequence of the immunogenic variant but which still is able to induce an immune reaction to ExlA, when delivered to a subject. Hla In some embodiments, the Staphylococcus aureus antigen Hla comprises the amino acid sequence according to SEQ ID NO: 56. A full-length antigen representing the antigen Hla is characterized in that it comprises the full-length amino acid sequence according to SEQ ID NO: 56, whereas an antigen fragment representing the antigen Hla is characterized in that it comprises an amino acid sequence which is only a part of SEQ ID NO: 56 but which still is able to induce an immune reaction to Hla, when delivered to a subject. 78 11529421v1 Attorney Docket No. 2013237‐0757 An immunogenic variant of the Staphylococcus aureus antigen Hla comprises an amino acid sequence which is "immunologically equivalent" to Hla and thus, is able to induce an immune reaction to Hla, when delivered to a subject. In some embodiments, an immunogenic variant of the Staphylococcus aureus antigen Hla comprises an amino acid sequence differing from SEQ ID NO: 56 by one or more deletions, insertions or substitutions in the amino acid sequence while having at least 99%, 98%, 97%, 96%, 95%, 90%, 85%, or 80% identity to SEQ ID NO: 56 (e.g. determined by sequence alignment using known sequence alignment algorithms such as BLASTP). A full-length antigen representing an immunogenic variant of the antigen Hla is characterized in that it comprises the full-length amino acid sequence of the immunogenic variant, whereas an antigen fragment representing an immunogenic variant of the antigen Hla is characterized in that it comprises an amino acid sequence which is only a part of the full-length amino acid sequence of the immunogenic variant but which still is able to induce an immune reaction to Hla, when delivered to a subject. Hlb In some embodiments, the Staphylococcus aureus antigen Hlb comprises the amino acid sequence according to SEQ ID NO: 57. A full-length antigen representing the antigen Hlb is characterized in that it comprises the full-length amino acid sequence according to SEQ ID NO: 57, whereas an antigen fragment representing the antigen Hlb is characterized in that it comprises an amino acid sequence which is only a part of SEQ ID NO: 57 but which still is able to induce an immune reaction to Hlb, when delivered to a subject. An immunogenic variant of the Staphylococcus aureus antigen Hlb comprises an amino acid sequence which is "immunologically equivalent" to Hlb and thus, is able to induce an immune reaction to Hlb, when delivered to a subject. In some embodiments, an immunogenic variant of the Staphylococcus aureus antigen Hlb comprises an amino acid sequence differing from SEQ ID NO: 57 by one or more deletions, insertions or substitutions in the amino acid sequence while having at least 99%, 98%, 97%, 96%, 95%, 90%, 85%, or 80% identity to SEQ ID NO: 57 (e.g. determined by sequence alignment using known sequence alignment algorithms such as BLASTP). A full-length antigen representing an immunogenic variant of the antigen Hlb is characterized in that it comprises the full-length amino acid sequence of the immunogenic variant, whereas an antigen fragment representing an immunogenic variant of the antigen Hlb is characterized in that it comprises an amino acid sequence which is only a part of the full-length amino acid sequence of the immunogenic variant but which still is able to induce an immune reaction to Hlb, when delivered to a subject. Hld In some embodiments, the Staphylococcus aureus antigen Hld comprises the amino acid sequence according to SEQ ID NO: 58. A full-length antigen representing the antigen Hld is characterized in that it comprises the full-length amino acid sequence according to SEQ ID NO: 58, whereas an antigen fragment representing the antigen Hld is characterized in that it comprises an amino acid sequence which is only a part of SEQ ID NO: 58 but which still is able to induce an immune reaction to Hld, when delivered to a subject. An immunogenic variant of the Staphylococcus aureus antigen Hld comprises an amino acid sequence which is "immunologically equivalent" to Hld and thus, is able to induce an immune reaction to Hld, when delivered to a subject. In some embodiments, an immunogenic variant of the Staphylococcus aureus antigen Hld comprises an amino acid sequence differing from SEQ ID NO: 58 by one or more deletions, insertions or substitutions in the amino acid sequence while having at least 99%, 98%, 97%, 96%, 95%, 90%, 85%, or 80% identity to SEQ ID NO: 58 (e.g. determined by sequence alignment using known sequence alignment algorithms such as BLASTP). A full-length antigen representing an immunogenic variant of the antigen Hld is characterized in that it comprises the full-length amino acid sequence of the immunogenic variant, whereas an antigen fragment representing an immunogenic variant of the antigen Hld is 79 11529421v1 Attorney Docket No. 2013237‐0757 characterized in that it comprises an amino acid sequence which is only a part of the full-length amino acid sequence of the immunogenic variant but which still is able to induce an immune reaction to Hld, when delivered to a subject. HlgA In some embodiments, the Staphylococcus aureus antigen HlgA comprises the amino acid sequence according to SEQ ID NO: 59. A full-length antigen representing the antigen HlgA is characterized in that it comprises the full-length amino acid sequence according to SEQ ID NO: 59, whereas an antigen fragment representing the antigen HlgA is characterized in that it comprises an amino acid sequence which is only a part of SEQ ID NO: 59 but which still is able to induce an immune reaction to HlgA, when delivered to a subject. An immunogenic variant of the Staphylococcus aureus antigen HlgA comprises an amino acid sequence which is "immunologically equivalent" to HlgA and thus, is able to induce an immune reaction to HlgA, when delivered to a subject. In some embodiments, an immunogenic variant of the Staphylococcus aureus antigen HlgA comprises an amino acid sequence differing from SEQ ID NO: 59 by one or more deletions, insertions or substitutions in the amino acid sequence while having at least 99%, 98%, 97%, 96%, 95%, 90%, 85%, or 80% identity to SEQ ID NO: 59 (e.g. determined by sequence alignment using known sequence alignment algorithms such as BLASTP). A full-length antigen representing an immunogenic variant of the antigen HlgA is characterized in that it comprises the full-length amino acid sequence of the immunogenic variant, whereas an antigen fragment representing an immunogenic variant of the antigen HlgA is characterized in that it comprises an amino acid sequence which is only a part of the full-length amino acid sequence of the immunogenic variant but which still is able to induce an immune reaction to HlgA, when delivered to a subject. HlgB In some embodiments, the Staphylococcus aureus antigen HlgB comprises the amino acid sequence according to SEQ ID NO: 60. A full-length antigen representing the antigen HlgB is characterized in that it comprises the full-length amino acid sequence according to SEQ ID NO: 60, whereas an antigen fragment representing the antigen HlgB is characterized in that it comprises an amino acid sequence which is only a part of SEQ ID NO: 60 but which still is able to induce an immune reaction to HlgB, when delivered to a subject. An immunogenic variant of the Staphylococcus aureus antigen HlgB comprises an amino acid sequence which is "immunologically equivalent" to HlgB and thus, is able to induce an immune reaction to HlgB, when delivered to a subject. In some embodiments, an immunogenic variant of the Staphylococcus aureus antigen HlgB comprises an amino acid sequence differing from SEQ ID NO: 60 by one or more deletions, insertions or substitutions in the amino acid sequence while having at least 99%, 98%, 97%, 96%, 95%, 90%, 85%, or 80% identity to SEQ ID NO: 60 (e.g. determined by sequence alignment using known sequence alignment algorithms such as BLASTP). A full-length antigen representing an immunogenic variant of the antigen HlgB is characterized in that it comprises the full-length amino acid sequence of the immunogenic variant, whereas an antigen fragment representing an immunogenic variant of the antigen HlgB is characterized in that it comprises an amino acid sequence which is only a part of the full-length amino acid sequence of the immunogenic variant but which still is able to induce an immune reaction to HlgB, when delivered to a subject. HlgC In some embodiments, the Staphylococcus aureus antigen HlgC comprises the amino acid sequence according to SEQ ID NO: 61. A full-length antigen representing the antigen HlgC is characterized in that it comprises the full-length amino acid sequence according to SEQ ID NO: 61, whereas an antigen fragment representing the antigen HlgC is 80 11529421v1 Attorney Docket No. 2013237‐0757 characterized in that it comprises an amino acid sequence which is only a part of SEQ ID NO: 61 but which still is able to induce an immune reaction to HlgC, when delivered to a subject. An immunogenic variant of the Staphylococcus aureus antigen HlgC comprises an amino acid sequence which is "immunologically equivalent" to HlgC and thus, is able to induce an immune reaction to HlgC, when delivered to a subject. In some embodiments, an immunogenic variant of the Staphylococcus aureus antigen HlgC comprises an amino acid sequence differing from SEQ ID NO: 61 by one or more deletions, insertions or substitutions in the amino acid sequence while having at least 99%, 98%, 97%, 96%, 95%, 90%, 85%, or 80% identity to SEQ ID NO: 61 (e.g. determined by sequence alignment using known sequence alignment algorithms such as BLASTP). A full-length antigen representing an immunogenic variant of the antigen HlgC is characterized in that it comprises the full-length amino acid sequence of the immunogenic variant, whereas an antigen fragment representing an immunogenic variant of the antigen HlgC is characterized in that it comprises an amino acid sequence which is only a part of the full-length amino acid sequence of the immunogenic variant but which still is able to induce an immune reaction to HlgC, when delivered to a subject. LukD In some embodiments, the Staphylococcus aureus antigen LukD comprises the amino acid sequence according to SEQ ID NO: 62. A full-length antigen representing the antigen LukD is characterized in that it comprises the full-length amino acid sequence according to SEQ ID NO: 62, whereas an antigen fragment representing the antigen LukD is characterized in that it comprises an amino acid sequence which is only a part of SEQ ID NO: 62 but which still is able to induce an immune reaction to LukD, when delivered to a subject. An immunogenic variant of the Staphylococcus aureus antigen LukD comprises an amino acid sequence which is "immunologically equivalent" to LukD and thus, is able to induce an immune reaction to LukD, when delivered to a subject. In some embodiments, an immunogenic variant of the Staphylococcus aureus antigen LukD comprises an amino acid sequence differing from SEQ ID NO: 62 by one or more deletions, insertions or substitutions in the amino acid sequence while having at least 99%, 98%, 97%, 96%, 95%, 90%, 85%, or 80% identity to SEQ ID NO: 62 (e.g. determined by sequence alignment using known sequence alignment algorithms such as BLASTP). A full-length antigen representing an immunogenic variant of the antigen LukD is characterized in that it comprises the full-length amino acid sequence of the immunogenic variant, whereas an antigen fragment representing an immunogenic variant of the antigen LukD is characterized in that it comprises an amino acid sequence which is only a part of the full-length amino acid sequence of the immunogenic variant but which still is able to induce an immune reaction to LukD, when delivered to a subject. LukE In some embodiments, the Staphylococcus aureus antigen LukE comprises the amino acid sequence according to SEQ ID NO: 63. A full-length antigen representing the antigen LukE is characterized in that it comprises the full-length amino acid sequence according to SEQ ID NO: 63, whereas an antigen fragment representing the antigen LukE is characterized in that it comprises an amino acid sequence which is only a part of SEQ ID NO: 63 but which still is able to induce an immune reaction to LukE, when delivered to a subject. An immunogenic variant of the Staphylococcus aureus antigen LukE comprises an amino acid sequence which is "immunologically equivalent" to LukE and thus, is able to induce an immune reaction to LukE, when delivered to a subject. In some embodiments, an immunogenic variant of the Staphylococcus aureus antigen LukE comprises an amino acid sequence differing from SEQ ID NO: 63 by one or more deletions, insertions or substitutions in the amino 81 11529421v1 Attorney Docket No. 2013237‐0757 acid sequence while having at least 99%, 98%, 97%, 96%, 95%, 90%, 85%, or 80% identity to SEQ ID NO: 63 (e.g. determined by sequence alignment using known sequence alignment algorithms such as BLASTP). A full-length antigen representing an immunogenic variant of the antigen LukE is characterized in that it comprises the full-length amino acid sequence of the immunogenic variant, whereas an antigen fragment representing an immunogenic variant of the antigen LukE is characterized in that it comprises an amino acid sequence which is only a part of the full-length amino acid sequence of the immunogenic variant but which still is able to induce an immune reaction to LukE, when delivered to a subject. TcdA In some embodiments, the Clostridioides difficile antigen TcdA comprises the amino acid sequence according to SEQ ID NO: 64. A full-length antigen representing the antigen TcdA is characterized in that it comprises the full-length amino acid sequence according to SEQ ID NO: 64, whereas an antigen fragment representing the antigen TcdA is characterized in that it comprises an amino acid sequence which is only a part of SEQ ID NO: 64 but which still is able to induce an immune reaction to TcdA, when delivered to a subject. An immunogenic variant of the Clostridioides difficile antigen TcdA comprises an amino acid sequence which is "immunologically equivalent" to TcdA and thus, is able to induce an immune reaction to TcdA, when delivered to a subject. In some embodiments, an immunogenic variant of the Clostridioides difficile antigen TcdA comprises an amino acid sequence differing from SEQ ID NO: 64 by one or more deletions, insertions or substitutions in the amino acid sequence while having at least 99%, 98%, 97%, 96%, 95%, 90%, 85%, or 80% identity to SEQ ID NO: 64 (e.g. determined by sequence alignment using known sequence alignment algorithms such as BLASTP). A full-length antigen representing an immunogenic variant of the antigen TcdA is characterized in that it comprises the full-length amino acid sequence of the immunogenic variant, whereas an antigen fragment representing an immunogenic variant of the antigen TcdA is characterized in that it comprises an amino acid sequence which is only a part of the full-length amino acid sequence of the immunogenic variant but which still is able to induce an immune reaction to TcdA, when delivered to a subject. TcdB In some embodiments, the Clostridioides difficile antigen TcdB comprises the amino acid sequence according to SEQ ID NO: 65. A full-length antigen representing the antigen TcdB is characterized in that it comprises the full-length amino acid sequence according to SEQ ID NO: 65, whereas an antigen fragment representing the antigen TcdB is characterized in that it comprises an amino acid sequence which is only a part of SEQ ID NO: 65 but which still is able to induce an immune reaction to TcdB, when delivered to a subject. An immunogenic variant of the Clostridioides difficile antigen TcdB comprises an amino acid sequence which is "immunologically equivalent" to TcdB and thus, is able to induce an immune reaction to TcdB, when delivered to a subject. In some embodiments, an immunogenic variant of the Clostridioides difficile antigen TcdB comprises an amino acid sequence differing from SEQ ID NO: 65 by one or more deletions, insertions or substitutions in the amino acid sequence while having at least 99%, 98%, 97%, 96%, 95%, 90%, 85%, or 80% identity to SEQ ID NO: 65 (e.g. determined by sequence alignment using known sequence alignment algorithms such as BLASTP). A full-length antigen representing an immunogenic variant of the antigen TcdB is characterized in that it comprises the full-length amino acid sequence of the immunogenic variant, whereas an antigen fragment representing an immunogenic variant of the antigen TcdB is characterized in that it comprises an amino acid sequence which is only a part of the full-length amino acid sequence of the immunogenic variant but which still is able to induce an immune reaction to TcdB, when delivered to a subject. 82 11529421v1 Attorney Docket No. 2013237‐0757 Lef In some embodiments, the Bacillus anthracis antigen Lef comprises the amino acid sequence according to SEQ ID NO: 66. A full-length antigen representing the antigen Lef is characterized in that it comprises the full-length amino acid sequence according to SEQ ID NO: 66, whereas an antigen fragment representing the antigen Lef is characterized in that it comprises an amino acid sequence which is only a part of SEQ ID NO: 66 but which still is able to induce an immune reaction to Lef, when delivered to a subject. An immunogenic variant of the Bacillus anthracis antigen Lef comprises an amino acid sequence which is "immunologically equivalent" to Lef and thus, is able to induce an immune reaction to Lef, when delivered to a subject. In some embodiments, an immunogenic variant of the Bacillus anthracis antigen Lef comprises an amino acid sequence differing from SEQ ID NO: 66 by one or more deletions, insertions or substitutions in the amino acid sequence while having at least 99%, 98%, 97%, 96%, 95%, 90%, 85%, or 80% identity to SEQ ID NO: 66 (e.g. determined by sequence alignment using known sequence alignment algorithms such as BLASTP). A full-length antigen representing an immunogenic variant of the antigen Lef is characterized in that it comprises the full-length amino acid sequence of the immunogenic variant, whereas an antigen fragment representing an immunogenic variant of the antigen Lef is characterized in that it comprises an amino acid sequence which is only a part of the full-length amino acid sequence of the immunogenic variant but which still is able to induce an immune reaction to Lef, when delivered to a subject. Cya In some embodiments, the Bacillus anthracis antigen Cya comprises the amino acid sequence according to SEQ ID NO: 67. A full-length antigen representing the antigen Cya is characterized in that it comprises the full-length amino acid sequence according to SEQ ID NO: 67, whereas an antigen fragment representing the antigen Cya is characterized in that it comprises an amino acid sequence which is only a part of SEQ ID NO: 67 but which still is able to induce an immune reaction to Cya, when delivered to a subject. An immunogenic variant of the Bacillus anthracis antigen Cya comprises an amino acid sequence which is "immunologically equivalent" to Cya and thus, is able to induce an immune reaction to Cya, when delivered to a subject. In some embodiments, an immunogenic variant of the Bacillus anthracis antigen Cya comprises an amino acid sequence differing from SEQ ID NO: 67 by one or more deletions, insertions or substitutions in the amino acid sequence while having at least 99%, 98%, 97%, 96%, 95%, 90%, 85%, or 80% identity to SEQ ID NO: 67 (e.g. determined by sequence alignment using known sequence alignment algorithms such as BLASTP). A full-length antigen representing an immunogenic variant of the antigen Cya is characterized in that it comprises the full-length amino acid sequence of the immunogenic variant, whereas an antigen fragment representing an immunogenic variant of the antigen Cya is characterized in that it comprises an amino acid sequence which is only a part of the full-length amino acid sequence of the immunogenic variant but which still is able to induce an immune reaction to Cya, when delivered to a subject. Diphteria toxin In some embodiments, the Corynebacterium diphteriae antigen Diphteria toxin comprises the amino acid sequence according to SEQ ID NO: 68. A full-length antigen representing the antigen Diphteria toxin is characterized in that it comprises the full-length amino acid sequence according to SEQ ID NO: 68, whereas an antigen fragment representing the antigen Diphteria toxin is characterized in that it comprises an amino acid sequence which is only a part of SEQ ID NO: 68 but which still is able to induce an immune reaction to Diphteria toxin, when delivered to a subject. An immunogenic variant of the Corynebacterium diphteriae antigen Diphteria toxin comprises an amino acid sequence which is "immunologically equivalent" to Diphteria toxin and thus, is able to induce an immune reaction to Diphteria toxin, when delivered to a subject. In some embodiments, an immunogenic variant of the Corynebacterium diphteriae 83 11529421v1 Attorney Docket No. 2013237‐0757 antigen Diphteria toxin comprises an amino acid sequence differing from SEQ ID NO: 68 by one or more deletions, insertions or substitutions in the amino acid sequence while having at least 99%, 98%, 97%, 96%, 95%, 90%, 85%, or 80% identity to SEQ ID NO: 68 (e.g. determined by sequence alignment using known sequence alignment algorithms such as BLASTP). A full-length antigen representing an immunogenic variant of the antigen Diphteria toxin is characterized in that it comprises the full-length amino acid sequence of the immunogenic variant, whereas an antigen fragment representing an immunogenic variant of the antigen Diphteria toxin is characterized in that it comprises an amino acid sequence which is only a part of the full-length amino acid sequence of the immunogenic variant but which still is able to induce an immune reaction to Diphteria toxin, when delivered to a subject. CtxA In some embodiments, the Vibrio Cholerae antigen CtxA comprises the amino acid sequence according to SEQ ID NO: 69. A full-length antigen representing the antigen CtxA is characterized in that it comprises the full-length amino acid sequence according to SEQ ID NO: 69, whereas an antigen fragment representing the antigen CtxA is characterized in that it comprises an amino acid sequence which is only a part of SEQ ID NO: 69 but which still is able to induce an immune reaction to CtxA, when delivered to a subject. An immunogenic variant of the Vibrio Cholerae antigen CtxA comprises an amino acid sequence which is "immunologically equivalent" to CtxA and thus, is able to induce an immune reaction to CtxA, when delivered to a subject. In some embodiments, an immunogenic variant of the Vibrio Cholerae antigen CtxA comprises an amino acid sequence differing from SEQ ID NO: 69 by one or more deletions, insertions or substitutions in the amino acid sequence while having at least 99%, 98%, 97%, 96%, 95%, 90%, 85%, or 80% identity to SEQ ID NO: 69 (e.g. determined by sequence alignment using known sequence alignment algorithms such as BLASTP). A full-length antigen representing an immunogenic variant of the antigen CtxA is characterized in that it comprises the full-length amino acid sequence of the immunogenic variant, whereas an antigen fragment representing an immunogenic variant of the antigen CtxA is characterized in that it comprises an amino acid sequence which is only a part of the full-length amino acid sequence of the immunogenic variant but which still is able to induce an immune reaction to CtxA, when delivered to a subject. CtxB In some embodiments, the Vibrio Cholerae antigen CtxB comprises the amino acid sequence according to SEQ ID NO: 70. A full-length antigen representing the antigen CtxB is characterized in that it comprises the full-length amino acid sequence according to SEQ ID NO: 70, whereas an antigen fragment representing the antigen CtxB is characterized in that it comprises an amino acid sequence which is only a part of SEQ ID NO: 70 but which still is able to induce an immune reaction to CtxB, when delivered to a subject. An immunogenic variant of the Vibrio Cholerae antigen CtxB comprises an amino acid sequence which is "immunologically equivalent" to CtxB and thus, is able to induce an immune reaction to CtxB, when delivered to a subject. In some embodiments, an immunogenic variant of the Vibrio Cholerae antigen CtxB comprises an amino acid sequence differing from SEQ ID NO: 70 by one or more deletions, insertions or substitutions in the amino acid sequence while having at least 99%, 98%, 97%, 96%, 95%, 90%, 85%, or 80% identity to SEQ ID NO: 70 (e.g. determined by sequence alignment using known sequence alignment algorithms such as BLASTP). A full-length antigen representing an immunogenic variant of the antigen CtxB is characterized in that it comprises the full-length amino acid sequence of the immunogenic variant, whereas an antigen fragment representing an immunogenic variant of the antigen CtxB is characterized in that it comprises an amino acid sequence which is only a part of the full-length amino acid sequence of the immunogenic variant but which still is able to induce an immune reaction to CtxB, when delivered to a subject. 84 11529421v1 Attorney Docket No. 2013237‐0757 Esat-6 In some embodiments, the Mtb antigen Esat-6 comprises the amino acid sequence according to SEQ ID NO: 71. A full- length antigen representing the antigen Esat-6 is characterized in that it comprises the full-length amino acid sequence according to SEQ ID NO: 71, whereas an antigen fragment representing the antigen Esat-6 is characterized in that it comprises an amino acid sequence which is only a part of SEQ ID NO: 71 but which still is able to induce an immune reaction to Esat-6, when delivered to a subject. An immunogenic variant of the Mtb antigen Esat-6 comprises an amino acid sequence which is "immunologically equivalent" to Esat-6 and thus, is able to induce an immune reaction to Esat-6, when delivered to a subject. In some embodiments, an immunogenic variant of the Mtb antigen Esat-6 comprises an amino acid sequence differing from SEQ ID NO: 71 by one or more deletions, insertions or substitutions in the amino acid sequence while having at least 99%, 98%, 97%, 96%, 95%, 90%, 85%, or 80% identity to SEQ ID NO: 71 (e.g. determined by sequence alignment using known sequence alignment algorithms such as BLASTP). A full-length antigen representing an immunogenic variant of the antigen Esat-6 is characterized in that it comprises the full-length amino acid sequence of the immunogenic variant, whereas an antigen fragment representing an immunogenic variant of the antigen Esat-6 is characterized in that it comprises an amino acid sequence which is only a part of the full-length amino acid sequence of the immunogenic variant but which still is able to induce an immune reaction to Esat-6, when delivered to a subject. CFP10 In some embodiments, the Mtb antigen CFP10 comprises the amino acid sequence according to SEQ ID NO: 72. A full- length antigen representing the antigen CFP10 is characterized in that it comprises the full-length amino acid sequence according to SEQ ID NO: 72, whereas an antigen fragment representing the antigen CFP10 is characterized in that it comprises an amino acid sequence which is only a part of SEQ ID NO: 72 but which still is able to induce an immune reaction to CFP10, when delivered to a subject. An immunogenic variant of the Mtb antigen CFP10 comprises an amino acid sequence which is "immunologically equivalent" to CFP10 and thus, is able to induce an immune reaction to CFP10, when delivered to a subject. In some embodiments, an immunogenic variant of the Mtb antigen CFP10 comprises an amino acid sequence differing from SEQ ID NO: 72 by one or more deletions, insertions or substitutions in the amino acid sequence while having at least 99%, 98%, 97%, 96%, 95%, 90%, 85%, or 80% identity to SEQ ID NO: 72 (e.g. determined by sequence alignment using known sequence alignment algorithms such as BLASTP). A full-length antigen representing an immunogenic variant of the antigen CFP10 is characterized in that it comprises the full-length amino acid sequence of the immunogenic variant, whereas an antigen fragment representing an immunogenic variant of the antigen CFP10 is characterized in that it comprises an amino acid sequence which is only a part of the full-length amino acid sequence of the immunogenic variant but which still is able to induce an immune reaction to CFP10, when delivered to a subject. 85 11529421v1
7570‐7 T E AV AGP
32T W
L L T YS N M
CFF I D
L Q
GYAD
PV3QVS VQ VEP KT LLT GPPI P GL Q M
SPAV NYR A
PKP TE LL PAV CK A
C G W
DL V T
L A
PSS IK D
AIS C V
NtD II SeHGD M
DCAL V L A
AFTD DAP T
HQDCDNPQ R
kI DcLoSAVGA GE QI IV TNSTV G V V
AA Q
GDL QI FT S R
I VPDI PQF F R FELL PL PLKPPNP LP FT PWE HyLDS GeQRIIAA A LREPT SDP D
GP H
TYF HD PTKTD DAGPDL A
ALAPT SLT D
IP NPSP PT AL A Q
T Nnro D DttL AAPAALA YQGP N D L VL P V
NP DL FYKGEPPAD I A
ALS DE NF HRP F D F PR GR GV PGHIFSP A
SP L AAS TI A G GPAT TSR R NHA T YD A D V
PIG V
T D E A Q
V S F
L AA NI TD Q V
PP NT D
YFGV FFF N
ASDL P PYP P F
E L AA SS QR L NAGT V LS D
GVPPSL GVT G G
FT L RG N
TVAA E LT TYNHTAT G
MT NNS T A T
GYRSH D
F N
HK A
A N
LHSY GYNS DF F N
FYV NL E D
M FYY G
LSF R
LA FFA L
K F NK K YGIF M
PGFP AHV A G
Y N
DH GDM V
T GST KLGH S TDTLT N
GSD N TF GLL VIVFD G S VNHM GL T AQST FSP I NS V F EM RQL K N
LT AAR PT L G
E D G
YTA K
AS G D
NL K R N
YPR G
DYD NEPS QT K
DN RE NAGD LYT AIL GS A
E GL G
SVGV QF Y P
VEWQL VS LA KIV IS A
T AAG Q S KGS MA R
KILV G
ML V KWV A T
CL RT S LA AR F S LLT N T
QATDRLVQAW
S AP L N
VI R NYT K
W GV L PV AYK I PV P HAVE P L V GHI VEW
HLGNV E QKT WLY GATM V
GHQ EI AK P W
WA V
TEI D
WAS ND L E
L P I
VI D
A TP W
WL LSI VN AP D
LQGDLS AM
TD LR GLPV G
A PGS FGL M G
TL WV PN L N
GNP HN A
NLAFR HW G
GANP V
AV G AV S V T
P HDASL VPG A G GP NDP GEPD ARGSD N
DS RLL GF L
AA AE LI V G
CPG NM
IVIAILCPAL N
VR K
E MGGC M
P GL N
RL FR RGW V KLGAG G
QR PIL G E AT GGE I LAGLGANP A AG T AA G
S W N
SA G
TSCLGYNM
NL K
P QS GWM
IV RIS Y A
HY H
VT H
GSAW
I SR VI QL EC FFVG Y A
ALRMS L K
CL KM
N GQVD PI KEL W
T V
QS YP EAG A
Q L
QF A K S
EHW I
VL M
SF AQG EI ILPPF FT GI V
HATF AT GS SP IP W
S QIAD R T
PAITLGV HCI SFFS V SA YAE SLL VPG Q
AVQLT H
TGP AHNHHV PAAW
KADF GS SAWQL A P
VP D G F
RLSDP6 P
EE IS M
A LTE W
QP VP RPI8 I
HQI F D L LNQWQLLT V D LLD S HP GAS W
GDHE FAD PT M
FNVLF P G
QTTL DPAP P VRA G
H G
GG M
P GP P H
SV TEA G
VCP G
GP R P VD EVHE M
L P Q
V RV L SQ F D N
EV ES M
Q AE GL SYM
A SAN V
I SE GGS s
NPVP FE AAVLQLPT TT G A
D T
VPS QAC QP V GR E n
eI IYLA A
ACE QLIGP M
AV GWSIVPF G A
E G
GTS QV g
G iI TGTA GGP G
NR t
G nLL VGVA G
DGPS VAL APD VYQ AGPV L PGV GL DKPCPV R
AAGQP YP P D VV S TT aLD W LD LP P KR I L V YN I le VP T a
c DYAGTG L F K
P T GW
SIYPAF TT W
R KP G NRFPI AL GD inLVPLDA] T G R
ASV VIV R
FDPWV Q SFCPY FASNGAF GNV P P b
eu GRK D
R 1 : LF T PKC YQRRM
R L TK ARTT V QPV V o
rq DIV ci es AF P SI E W L
L ROE T DI PPVPI E L A
NR R
DRY SL NL R
ASRR S] TP W
V G YD IPT V
]RSNITALR NF PS TFR GYP ]6 L
2 M
L T
LAAGT3 M
RYI NL L4 m dTGKQD I
FL AGL A: GVR F RIGL: GP WNIGIGE: ci ic PK x
a D oI KLR A
G N
TQ M E
GP L
ITWYRVGLGO D DN V GN P APD TL GVO AL P GAN FATY IL V P R
D GNO LN to I n
FAGEL S A
ASLV L D P T D A[ V
PS GR A A
GT I
HP E
DT YS N D VGT GGG PP V I
QTNR P
IR FIF D GGL I o
tiR LINHVAAPNFRIGLQ SAGPV CQ S P M
D GVQ y
mSDTI V NSDPL D GVESGQLLSI S QE V DL T WS VE c
A M
R F Q R M
L T G G V [ M
S S f
T S G D P [ M
A G P G N [ o
sec n e
u) qsi)s esi)si)si s
ol s u
os s lo o ulul dic cr ae crcu rcr be e e o
u n t
bubub i t tut m m m a
uiru m
u m
u e iririr e v
tetetet icacacac taba N
oc bob b 1 : cococv 4ny eMy y y1 (
M M M2 eg 4 l i b
tA ( ( ( p
A9 a
n scl Bcl Ccl 25 T A E P P P117570‐7M3V2APAGLI GQE31A02 P CF V KGRD R. NYV G
QQP AG Q V
AT LT T ANQGNG LEIo W
R F D R
I VTGAE DR F QGNKR YS W
HSINt GDC V DS De KV L PV L A
L C
Q SPWL QD NL SP Q Q
QYESk QT DTH R P K
QPQAIA GQLAE G
VcoQFASR VYL G
RV LYTI G
L GAHI S AGNDIDFyLPEPP T I V AGTGeP DnS ArV LDGYAV R GGDI V YS L
AS P V T
D AI A ID HGI TYG HAS L G
R G
ot TYQGL N
AAtANF HIKF A GP P P F A
DS S RGR L G
S R
RS QE LF L AL PL EL LAGF GLL M
T T D
IS T
A TD TWS EL SAGLKL RM
VLRV K
AE IT QVL APS N
A G
C G
ESGL GVETSI GAA QD G GSR LTS G
V PP VI A L VAWQA D
T L W
TSYQ G
AL T
YYV GY HVKL A
R H
DIRS G Q
AL EI RSP AFDL F
GTL D AF HAPL V MISH YM
VEI GDI A
AE G D
S G S
FF NF GVTAA DVEG G
FSL G
VIR P R A
R H D
V S GT R EFV RF HVM F
T SPI GSALPQPAR PSER RTP NST VLS GRSQ HGLE R VM R
RAG D
GLGP L R
D M W
IFKV AL GAGGP LLARI YDR L NEAT DAS SARLNRQS D Q
ASKY NSVGR L TELAIYVAGEQ SDAAGL GKELS DGL VL AETS E MR F
TRYILVSD VR D
R ALATSQERA F KV AT L L Y
QP T
RT RVVLTAL AT DALS YQ LAGF GK V AK F
QW
KWVTDQTL G
SGGVEL G
LS HEA D
GS HEA IP DLSE VWA LVRR LF G
VTLATGLE G
SR D D
GSE G
RL V HR D
G TW W KRL K V
HR KAL G
GV T G L
NAAPNPT D
ILFPA G
L G V
EPP A
G D
D QT H
D DSNAVPSAEYA D
D G D
KKSEGA S
LGE AL GNLV V
NL V APT EVQGS ALGE AQRAAKKV D S LIG T LNRAE L QLL CGQ F AG QA V D AR D AA PGP R G M
LI R P R GSAL A
NLR K D
ALALT VPEF DL W
IRNTEKVA N
P G
QS H
ASNF DQH D
E G
AL GS VL ATPQALQK DG Y W
IL PLAEVLAAKRE AE K
HGAF RLLREPER A
RSD G
GE QRL VEGG H
EI M
F R
AQCG D
L V
SA SPLV V AEGSPRE SQ ETG P V RG VAS T
P I R
VKTGESRT HT A
AVGSR7 G A G VVAPE G VTAR WP HS V RGV T GSL FE8 ASQA F
P DFPIKPVQL VARF QQRTAHRTGL S
HF DTVT GG A
DS MLPPA NH R A
T A
QT E G
AL LK LVT S
P D R T
GIRW
EILERLK V
LE V
ES AVAE A
D GNQHH AVLE V P L ES MTTE L A
SGQS L A
V A A
QAVKQEE WLE GL LAKVQE EE GLK G
DAV G
CAPELQP E A Q
AE LQR DSALR L G
MP VGR R
GGG G
V GQ T
QQAR GSL G
MQV GR G A
GASKT A eADN cVLNPR F
QIS P R K
PVYMIS GRAK R YLV Q
SR GA V
AT V LTHLEV G
QT HKEV A Q
LF G
GPQAQ DW
PS QPAL V AL HML FGSLVAAS G n e
A uLFRT PSI V
D GDAV L PLQ D
A A
QE LE AKI M
DK T
DI V R
F P V
RAS E R
V ATISDAQE R
GA q E e
R DTPCP R] RV VA ] AQEM
S] VF L L R SL ] s RSFA RS L L
RVRY7 PL VR 8 FSQL R GA I 4 :E H A
L 4 :P V R QS 9 L
4 S M
QD :P M
EPILL V
05 diV Y cGP W
N a
G YAF LGLG R
OA ND V
AQ G
POS M
LEDI L ONQS AR: O
DNQQP L VRNQKI AQM N o
I M n
HTL V VR DGIN R R i S PITD I
AD H Q V
D I
L KS S
FVAK Q G
M Q D
D I
S E S S
ALG VAD DT G
D I Q
GN TPA LAFGGLQR L R
LVV G
QQI VDLVATEQQIAS HLEK Q mS R E A E HA D E HL Q D E A M
A D
TPSAI G
AAS[ M
ETGPS[ M
SSL Q
LEK N
S[ M
SASAR G
YLS[ )
si) ssi os ) ) lo a ul sas cu o rc erno ini be g ub ugu t
u t
rer aea m
u m
s s iruia a e
rn t
e no o c
t ac m ba omo ob d d 1 n co u uev e yc g Mye MsPsP 1 (
2 i ( ( (
4 t n
DcAySoTo 92 A
lPlTxExE 5117570‐7LK
3EKLQPL LYF L GS
I Q HDG
L P
2T S RTTDAANGR GFG G
1G0 GER EERL ID PTT L IE2NI.EoFEFD S M
PDLDL V EAFI NS P Q
AAGS QQEE K D
NQS AASAL Q
TTAT T M
DL A
MLS RAAP G
L G
tR IeLkc THT I
QGV TE FTQSSDR AG NEV NGFEL TI SA I
G H A
HD LPNL G
M H
DI SEGE REFT A D
MoQFL NDR LE REV N
KE GR CAS G
AL HPF SEGL GyPKL TGE SeADFKS PSP KLAR HL AAH Y
RLDTn GKKKIFE QA H
RLLr AHAN S
NQLG A D
VF RAGPLVV S PLILRNF LGQVA D
NFTHS L T
VFP H
GottDATLS I TD YL N
KEERD L AV Q V
D G G
AIV L A
VYGQ I SD LAA M H
ASCI AS R
LEH ELD R
TP L R
GLR D S
S LN RR ELY E
IL DR GW
NF G
SHF V AL KI ARA QE EV A G
D LLPV ALDVAR G
M M
SK A
DL E E P
L E TS QMD GRE R IQLE YL E DPRR HL DQG HQR A LV G
KGS QP D FQ TD L PTNL GSI HEL V
E D
KEHA ARAE W S
SL VF N
RE D T WR ST G Q NYG R
V G Q
GP IAQLP A
N H
KSKQLAL NISSL G
GKR GME N G
E G
L R
ALG F GDI AKD L Q NIALT M R
F VAS MH A
D EGP C F T TLD EL V DAQE KL A SP R P KE D N
F D
D YI VV R GT G T
N K
VSLRPVIE QSTVSQ R
L LAI PS Y SEM
SAP D IGS W
R K PL P
LAA F G
AEA D
AEAP MKG PG P N Q
YFL D
N L
SIV ANM
S HR KF VLQFDVHS GE ST GERGP KALAI M
TR F M
L Q
TPEYQS S LA QWL G R
PL PPH GSLVT V NA LA IAP G G
LTEE F G
RKRTPRM DE A
E GR LR NT PS GAD A D
NT SS KSLK R
SF RPGPN V
LT G
TI QR D GL K
R EIS P S LP T V R NLIV SI V Q TL P
TGA S
GVQFGL LRLTGI N
N V LEN C
QD G
YVSAFT QG A
E E MR G
LA TV QAGW D
G D
HIRDVGRLY KK K
LSP A
SHT GS ALVI R
TLRAPKL FQ Q
QDLDPCS ML VGTWE F N G
SAIV D GSELM
HAD VKAL NGL A LM
GNDVP RGL A
KA V DS ETF L V
I N
YWSFE NSD IVH GS QG GAR ASP Q
RI PR HCSN QRYL I LQES VIGRARI ATI GP P
GWV GYYAKPKVAG T
R G
QEAF G
S GV A
YP KL AF P
DVR V GFG N
AE E L KQPHG D
ALPR KN T
TC V
TYLT N
DGC NG EGLYY LNADPNF FF Q
MRT DSV LES TAVEKINIP P R GELDAR D L S VE P ISQM T
TP 8 FFQLSLCE WLVL LL VLL ESMY 8 SKE D R
VQAI AEV R S L LAGFP VL D WCRL A
Q D
P D
QQE L
K KAQE R
DREK V ND RKL F A
NLV Q V
D QL D N
G H
L RFLAR AMEL V AAPEGE P
E KIA Q
VL A G
SF ELSKPGSAYD G
GEK L G
QRNEMEL SKEAWGS Y G
D G ARL AL P T
R AYG QILK S
S G
ACAP AV N
D S Q
G S
PAFSE P YLFAI V
ARY R
D A
ARW
EQEGLV V ARS D T ARS GSQVAHE QV V FS A Q
S LAC P P D
CEFT E
ADI I H
L A G
VKVPQSI ATVC GRW
QEE D G
TAV e
cV GKRAME DR VRNV GATR YKWG A
AIPI ALS D n
PE APL AR TI R Q
G V
L D
R E D
LL SEGAYAS LPK e
uQGV Q qN AA Q
EP A GT RAFGYR R R
RPVP FPI AE G
eLG G
NL VE LNEAQQDLRV LS VPQC G T
RV SLS sSSSLLLEL AQ D PL ] V QA 1 AT 5 DF YS ] AG G
E 25A V
PKQRL N
AHSWV IT ] R
3LG 5
SL I ] L
45 dAV iT ca ALSSFPGAA : LGS E G
:LP P QART V A GTLGTV VS A GHS VT LO D VT N QG D
G H
VO I E RN WV MF V VTEDDE :LK : A
LA DLOS P
NV T O A
HAN o
LP I AWLPS FE DL A
MAE HEDHG G
V L S QWDAI D n S Q
P KNGM
S QK I
R GI I
PT E S V
P AS G I
LKV R I i
I RSN G
F KL D VN Q AKSPM Q L LGL H
ARAL Q RSQ mHL LV V
A TR E QAV AA R
E HRI ACG A
RI T
E LDE A M
Q G L GDL Q W
S[ M
A V PS[ M
T H A E V ES[ M
DS[ )
) a
) ) sa a as o
soso o n
ininini g
ug g g ru u ur e
rere e a
a a a sas s sa n
a a on n no m
o o om m mo d
o od u
dudu ue 1 n esev ePse Ps s P
P (
1 g ( ( (
2 i
tU ni4 noY A r92 A
xo Exo u ExEzA 5117570‐7331 GQGRGLRD SDS R V
TKTAAT DR VV LAS K02 A. QID R
TREAHRT H R
RAGAGGKAP T
T RKKQY GDEDTo LQLGDVN AR GR RE K NA S QQAE A A
QL EL LV A Q
Y Q
D D N D
S FI G
DI YTAVQD SS Y R
GSRR LS T T NAD GPDR F Q T
YtekNVGcLoVAV EG P W
E Q L
S AS QQT GGI V A
D DE G
HEAISEV AE HDL S
GAIEASGEDFNS TGGAS Q T LI RE NT G
HEDLDyGNGTIS ReQTVRGI A V
VE ELLLDRD R NAAR SGA D
QS KKS GKTYIFI IP L
GQL DAGL D G
FE T R
KFV V V
V H
K G
nr RNLV TS S V A
GV N
A Q
AQV AL AI RFLDotDK ItVAG A
NR Q
DKAKND YLL D
SVREVV I VD LP DI D VN Q
NTN N
NKPYANQ YLGSE ATE DKE L
GAWAGSDG VV GGSLD NNEGF LKVYQ KIGN R SS V ALSTKLD G
GS SAA Q
R D
AAL KKQYNKYK F
DTSEGTLL QA AAG LLI RS K D
AAE GRKI V
IA GND N H
LT KS SSEI AI G
GDV L L GAQR E T A
S NGVP NHAK QK T
S N H
E N SP LRI E GR RLW N
DGAAVAGIEVASD G
EL V K
SRK D
GIR GI YNS G
NAAKE VVSAL KS G A
V F
GGH K
L N
QTK G
A G
SVDIS FV DL NMDV AF Q G
P GV AR KRLD S QAGQRANS T D A A
L NLQSEW
LRSS G
EG A
S K A
QI GS H K
T Q
LYA G
IV RKK N
W PS M A
VHGRIA G
EV LQAD NL RL DKGL F V
IL TA Y Q
S TS A
A H
GS G
KRQ Q
AIKY VRAEAV AV D
FADDA KKGFGD N
N V
IP NTFL LT HL V
NT D TA G
QS GVR HRV NNVRHGI DP W
DFH G I
ISAL A G
KAAL D
QAVA Q
EE D V
E A
VYY D
MK G
SF NPGA P
F D VS INR QGS S GT T
Q Q
ATSDKK T
S G
GR GGK G N
T D
GYL C G
YL S
DR D RLG A
LS ETDR V R
AR EG L TA ES KDSVS V D
GGRE KE G D
TLK V
GG R
QSS D
GI AKK N
LA D GL TL TS KF N QN NV GL D
T R
GNSAQASAQ SGNV DKPRTYT VSS LV H
V N
LS LIP Q
GR V T V
R EHSDLSADRNA K
F N
AYLPSLE VLN G
A M
YQL D
GT GS FRA G
DI HS ESHGDGR Q
V R
D NK N
V N
IKN K
PNP GGSAAFVR EGL IE SDL S V
SR T K
KS G T
F G
P HE K
L T G
NSP V LLS G
SLSNL G G
QL S GALGYS LG AGG KYDLSV IM PD P
ID D GYREAR GA ATRSGT D V
I G
GNDSL SL L VTTLF N
LYKF D
VLNGSAG V
SL AS KS P A
K N
R D V
T TK SD L KSEP G
DAVKAL GDSSNN RVNRASGVLD G
K NS M
A A
DT VITK A A
AGSLGSGSD TQPL N
NL TE TTGL G D
GI YE K DAV G E QKSY G
SP D SVYS KSEE ST QELGAKP AE SA DKM
S DKGNA L G
F G
IYG ED R KNR RRALD NQKD GR GQ GNGSITT T D
GKGVKI A
F QI AL T
DQRRVHDGNVS NKKER GIS NS RE DE NLP Y A
PR H GN R
L ARSS SS G
GASE D
T R
E LL T AGT A T
SP GAE GGV A AGL A T Y
GTKI N R
T P
KNAA T
KAV D H9 T G G
DS 8 Q
P N L
IGV AV KHSKQDP TN G
VQYL A M
GL GLR G R
L NIYMFM
E SSS IYF TRAIDNEHR GSFLLAAQT DS Y D
LE N D
YVD HS PAL SASAG A
NDLDS ASF Q F
YDVG RIR KGGGVD V
RAD DS Q N
K RVSL GGNDL AE TS G
T GA AI E NY A
QGL WAEDV LR E R GKV KAGW
VKK AYTKY LL SR IA L I S
IAES SD D GQQ A
HAA D
ALNKS VG V DGK Y D NDVAQQE AVE P IK VLD PIY GV V KRGV L N IR F LI V N L NP AEAAD S L
NEIR S VGSG G NPNY W
KF e C LL GKYY IKAR QEI E QR GR c
RAG Q
NLAA AQS LNR GVAEL S S SKHSGE E AS AA R
GV G R
GA ND WRL ENGKA QQ M
LPLS D
SASKY S
ARSE DPVV E
R L Q
I QRSL A
QSFD n L e
KSAS S LSDLHGPGAEIGSSHSAQP GQS L DR D
LRDW u
GAD NADAT RPS ADSERSNTI DGLQ V
YPTT q RDVAG e P RDP D GTE QDRAT S R QEVE GR]LLKGW
] V L SI P VEKKP] s S G L
RL PALAARR E E AS A
V GL I N AG G
GEQ ARAL DAIV GRYS N LS A 5 TF ER5 TAS W
K6 KP ND5 KL T M
K7 Q
ET 5 diH cF QL V PLQ AGSDIVG DDIY A
TR YYA E
RNQL GDGS TG G
:T P QKT: S QYE V: A
AD NGR V VOV NSWNNO N
NP AVO a P N A
ISI RSAR DFYFLDQL GGIV YRSALV TSKYRIEN o
N nAGSL GGLS GGNAHELKARL NL D I
VI MG D I i
DT EKN D I R
L Q
DRD HEL GKRTGGIRAAG LGELSGK DERSA VG QR G T
SNK KN QKK K
KRV DL Q mH GQN G AVG L E K FW
E V V Q E A M
GANSASKSLRGSHRTTD G
A D
N G
AL QV Q
LRHSNFS[ M
NAIVNTS[ M
NSH G
KPS[ )
asoni ) g
s) us eu urrer ea uaua s
s s an ucuc oc c mo oco d
oc lol nu yhyh 1v ees papa 1 g P i( t tStS2 A
( (49 n
l a b2 A
xEl H
l H
5117570‐7 D
AEA
32Y D
V GW
10P TN Y V V2 YF T P N
WK GGYV V R
. KS VPo K KVNNYGH G
N L N A
N R
V N KW R
YTWLDt DekS KNF KGY N
I Q
TRVS LYcYTV G
FS N
T YL SSKS So T A I NDI NND RWKD NR F GyeSSV HKGS T
LMEGQY Q
QHnrIoF G K
R D
ATTR Q M
Ett GS KV TSA QNV L RYYTKL ES RK M
QA KS LVL SEV I T
L VIA V
E R TQI AYH V
V H DKK D NT L
E R KYS KQV A
T T DNK NQD D
V V YI K YT K
YY ST A KYM N
NNA KE G KS KY DS D D ISR Y T KI F Q KKG KQAF ATR VSY KN DYD KS M NF NH FYT S
DNIE VI FTN R
TLG N
L F Q
FSFE DK FSGI LVI YQGSL NGS T
QY INTI SIGF QN KS E T GDS NF QSEFFL G
P N
V G
GLSS G
TGEK I
S SDNSF N W K
P A
KE AGKS I L R
GIDD SI SR NS Q
HS K
SGA KT DS S SP GAGL TKF N
VT T
ARTTGL FP D GAI IS E TM
E GI F DQHKYQTNS QF TNSY LL R
GTGLHR QKIYP N K
I GT ITI V F
KT AIIE GF I LT G EDNF S INS QN DS QQ A
Y G
P D V N
V QQS GKGV VL GLF KF NKNP Q G
KKE AGTPL I QGS V D SSE N
YANEI D
SEE 0 E
IV Q D
ILVPQSS TEEIKS D
9 AP TI N K
QKT TDNKP K N
ESPL KP RN AGD AP V L
F Y
ADIKQ GEYAL LL K
AKYD K K
KS NN K
D A P
NDV F V
LE V K
AN NILRP N T I F
E PV AE S D Q K
IFLF T G
N V
N G
H N
L V KSG N
PY V
NNY I
D SSYELPYKHKV eTA c
DLL R LL D D ALLNT S
P W ND P H
DIALN T
KK YIE n
I eI P A
V A INPAQF G
S E P uWKLS D
TKMS PS SD YTAKGH LL VFT T IRS LG K SI L qeV ]GKP T]ASGK] VNDW
] sL D
8V 5AL TKD Q
IS 95 V VYF0 S NPT6SYS N LQD V 16 di G c
I : O
TL AK AS F IK : O
S YGR K
K T: O
TF F E TP AY: O
a TS o
INTI L I D LNYVE N VLA G
W G
K NTWS K N I
IYQHD I
KWNF N
L K
D I
I R K Q
VTYD nDKQFDTNYNY K NM A
GN I i
Q QNPYKQ ML MI NQKSRQ mA EKI AS WE KKKAEKLVE E A M
S[ M
WI V NS[ M
SF H GS[ M
HTANVS[ )) ss) u
us)su eeu r
rerer uu u u aa a a s
s uus s c
cu uc ccc o
ococo cc oo l lc y yolcol h hy y phphp1 npav eatatat1 gtS2 i
tS( (
S (
S (
4 A B C9 n
dlglg g2 A
H Hl H
l H
5117570‐732310 K2PWS R
G K
V A N
.NG SIN V D
G W
HoS W
S KKNNKQ F RtKek YHN SVS E A
c GNo STTYTRKYID NKRR SS NNGy Ie NDI DIF QT KRnrGoATM
E GDPtt AT F R
R Q
QVELTA KLY M V
SYKSVA LETV V TQKIILYY STI D
KK AKD K
Y DN YIS D QL K
KT N ST KEK N D
ST YYSR IG K
FD K K
TY KS ISF F
KL DSF SK KYKL N
H V FGS F NN TNL D F V LL V F S R IGS GI I
QNL Q
GFVGK F
I SENGS NL L
GP N
QI G TS K KN D
S F I
N VP E GAH NN DIG S S R W A
KQFLT S
D K TGL NTI A
GLS T
YPS GFS T
S I KYQ V G D
I N
P N Y
GHT ET Y G
F L
LPRL G T
I TLKT S KQF D QI V NV QQ V Q
VE GL D
V QGGAVPP K
FAPG D
DTL K
E E
E NSNTGTQ N R
NS EI N
EIKD1 KQS K
KSEI T PA 9 PTN IKD D N
P A
G G
TNK GT N
P F L
Y T
E Q A
YLFILA A
DVLR ISR A E
KLSNIS D
V LE LG V
NNIQT T N
VGNIS PVP G
N S
AY DT Q SNPNH e
LLNP SDW D
ALP DP G KS K cLYV QV nLE e
A uI SSEA SDYI T V K TL TLF IE qS eS VR SG L GS s
A V
VYT ]2 V G S
IY R
H T
] dNPF i S GT 6 :L N S
YD K 3 H
W 6 : c S Y K
K V AQ G
WT N
OAFP AFN V
O aGGGN NQL W
KKN o
L n
ND I M i
KE W
KI KY D I I
YN FMIKYQKL GR K
R DPV Q mKKNE F KTV VFE A M
QS H NS[ M
TL NS[ )su)s e
ru uer a
u sa ucs cu occ c
oo lc yol hy ph 1 n at pav eSt1 g2 i
(S(4 t
D E9 n
k Auk2 L
uL 5117570‐7 VV
K3 F TF I
ILANI V
NS
2 PL VLDEYEKYTGA T
SIS P F
I KI S IVNE
ED
AEE NIFWG
F G
LS M
IY R
S N
FTNE F G
F ANAP TG T
DI YY3HL KDS NS A
SIVS NKS N
FAPI F D F NI NISY NDYTQYAFNVTV10NNIH2 K ITLKLD KAKNL Q
IP HN R
KEL NNIF P G
RNTLI YT N
AV VK G
. E QGYSTD Po VSLKS ALLSTN D
TSEFLS SKKS G
NAEFFYAW G
NN PSKPL GKG EQIK DNI YLFI SI D K
ID GTF F V
NFYQ G
GYT T I T
t S YekT YMQIT LLDDE QTSP DD G
NF YN YL LS GVTY K
EINPKN KTAAQcNND KS NQIIKI ISFSVKVESSLKSF F KDL P MQQKNGGNFKW
oS IF V D
ILE YTDFILV AF IKD ELWF AI YM
WS D
NKFDIYSDN NGT KRLAQLHVDLFS GF IYT I HII T
I N
SIIL GVGIA TD V TY K
NVyeInLKrIQY V
Q N
NIYFSYKESLGD NTQKNVKLS GK DSQT GTQKGA K
oVRGIVFNSI EV KD D II SNDNS GDDL NLKNII LNAVW G
GF Y
StEDGtKYFSDYKSESL N
NALI L AGEYII KF IYKINFATGTP M
T DIYI N I
F NGLYGNS SLNV VT TV V L KYL N
RD YD VAF KSAT ARNN G
DE K KM
LAA KAFS AELS DLKDKAT P K
K N
HI M
DI KFF H
P N
A K
YE ELD H
FY L
RKLL AI A K
YE A
VD S NV MK S ILNAI H G
LTKI F N
GYDIKF SEF N N
AT GTLY A
ESS SL STI NI S K
S LSG N
YYN N
G N
ANVYL IK N
SK LYRVIE GP NSLEYLYS G G
ADI QT N
PEM
IIK N
D P DFLA NKF R
GL AF RKI D
L D
GS PR IAE F
KE D
DILM N
IEY M
DK FSS LT K
HY H
S K F
NK FYNF DYG YKY F
FFYS D
NL QH NM
S SLKE NIS SA F S SS I K NYS YL R NA DAVS A
NL I N T
NSTW
V N
QA IFLV LI AV YD IG KAD D S
LS QT TLV KV A
QGGN I
RK K
GFRF Y
IR S
F LS L NKEGNF I GT L NQEIGAN S
T QNNER LYF KPE GS LLFAVCT R L K
FYNKSINSA G
GLM
TI DI Y K
QQY YNGRLKA S
E N P
KDHN K
AFA ENKAE GLWYG K
KV NFD LITV D
EGNI AT AL I F
GYTKG GQGEI RI K
QN D GLLLKFKNGKI KDNW
NNA N
IELLFI MF L A
HLE DIT N
EISK G
ITSL EYTIR DFAS T N
QGLTNQ M
F E V
EE E LQS GTGIKL D
V K K
NPGDSS DPIKYNQ YNYF LIT A
LF A D
GE DLS KEV Q
NKFR G
DI QS LL N N
YL NET Y
YNQKP VIYF GAKTI N I
S QY E LI S K
L Y
NT FVE KSS N
TP HD F D
D R
IV I SI NYS Y E
IL PI K K
GNFYA Q
HKDYE SANN T QAN PA AN N
L S PD VNDHI EAE KNYGGF YD L E L EIAI V KIR HII LEESS D
RIN NLV I KT K
TEILKG N
EKV NQI DLI V
LL VEKYL VN VS TKE E QE INI YNL LI AFT NNITYKNAYE NA L SQ SEKT E FLK Q
L ETFNFD NYV NL DEL D
RNS GNREL W
NIN N
YDFYQ G
FP M
ESKLQPE LKIDT G
DI DLNVGYGTI YGTN N
YKEI G D
AF YVIL K
AS D KNKLS N
LSRN EAL L N
AIPDNF GLKNR V
VR E
GP LW
T N
S ITKNPYE I GIKE GA G
VL NEITV Q
GS LI NGAAIG AC N DAG K
F N
EKA N
LR Q N
TI QAT GT RTIYYNVL IKLS W
LNINFI F FETPATL H
IDFG VN D
DAYYKS IN YD I DR D KE KSTSDS IP VAFYNIT AQT N
GS NN TVI L T
LG H
KS D
NDNN F
RV LKDTIG E
NGDDE L G
D IISLITPKI S
T VLKGE DKSE T N
AI W
AI Q
G K L
FL NSI SS T FYNF G
FYAGP T
AM
IF V
KANF N
ES NLS YKLI V D
S N Y
SKTKL V A
NSLSS T
SFL NNYN NEDESE P SE ISVPGI NV FYGGG DI ST HF FNNF W Q
GL T
I N
TSIP GYTE T SI DI KKS DFST K K
PAE D I Q
YI 2 S WYN N
YYSGNKS F G
TPG 9 E D
YKR YIKYDS LWLSN N
T KADS TILKI LEI KGKW
S E KI LYKEE S
WPYNDTGF IKF FS V DIFYDNNF L G
PA SNI KGI T YKN D
P G
YF NK NL TK LINIYT D
SLF K D
NTITI ES F V
S K D
NYIS L G
G N
KQKFKHRF V G I
NT R
KT A Q
YMNL HETP LR SL GNDI DL KL TYYK TVVW G
NY NYF V
K G
YD Y
F N
DSKIS IF S L H N
SP LLSL DVNNVL L CT II KGISIF N
V K
E YASE KNF E
NSALL KTYKV N
FAS LL YKI N
K N
IL SF K
DS AK ARGQI KGF VM
IDS W
SELD SFITY N
NY PT S YN KNVD S DE W
NM
TYN D
FY E
D TAE R
AEE I LNK G
PS IED VLGYS GDK K
E D
QYFYA T
GTLTI G
GTIT I PWDL AS NM N
PYVKN V
VS M
G A
KATI CIHF YVIFNV LFD S KV G R
I QRKIP S KNNIERT SAETATMI GV TF NET YYAHKT N
A K
KT N
AIYDI YI YMLQQI EE DENSAKKKDF L N
IV Y
NP T Q G
GFS N Q
F SG TNE AYEE HETII V LL VYILD S SIED D
GNK N
EDI Y Y
DS YYF NAFL ] L
E K
LS W R
Q N
L S NF L T
V NHLF D
YSIIKLLI V DNTKGVIHTG M
G 46 I
AVL DLQ L
I E
DHSLFSN K
DKSEV ILL L L M
N T
IIII NK Y
K G
FYAK G
NF AGA: E SGQL P YI L SD GQENEK N
SIFV KE KFNGDYQ G
NYYTAO KVKIYNN SIEIISS RNKFE NSISNT RIK D
NIGVPYGQTNF IT KKEIS KNP AN N
E S MSI KDN V
EI E D D L YL H
NAI QGI N
T KE M F
A D I L
GD PKP SGIDIRS VFF KLN NCS GAN LIY FILSGINLEE TFE N
DAP AWNNY G
G N
YT R
T PT A IAT C IFY K
S R AT GLT NT I T
Y R
DPQE M W
TPITQEFECTLLGP VNL GSYITTL D DYYDLFYDTTALFHTSA Q N M
S[ )eliciffid sedioidirts1 ol v C
1 (
2 A
4 d
9 c
2 T
5117570‐7 L
EEP Y
S S3 NL A
2 KKHE L P S L F
GAYS Y RG
V L F S Y
T V G
KD S S KP QR
DK
T A
VYNK3 EQIDIN A
GP KS YIL AT SI EI NGT S KE SFI P G
E R
T N
NKI KI I
GSIT D N
FYYEFY EL NQV Q
NE L Q S
LTIL F PS I K
QL I YV FLKIKD K
I R
DT QIGKV1 V KLSDKSSDL AY V II NKAM
E SL KSK0 PII KKTQLT DGSV NVV S QKLENS TL2. TYo LYMN NNDLS D G
P IVAS VI KS AP QM
EQIIV T KI YSLS SFV D
KI V IS GF V LGGGD A A
IELL D DHVREt SL P I R
NISL NKI D
N Q
KDEYI EI E D R
LSE V K
LIKQA N
FDLD KL D D FQANE QI AS IE E MSNVQSF QL D
NNLKKSL L N
E DASKEYHekcKN Q
YII TVIKE K
FSVS KF FFNT H N
S V
E CK SD YM
IKD N
Y VAI K
IEE GPID EToLDEL QVy I KGS G
LLYALPIFMGS HDE D FLLF DS Y N
GWVG Y
EGE Q
SLLTV V
DS H
DGIKADKH V
DL NFHAAe EIDI YLIIGS IF M
GNKDS G V
T N
Q EI GE E S AKQMnrT YoV II NKF YYI T
SI SE VS GD RPKEKLYL FSGNF V AMKYYNFDtL LA V
AGRTLA D
H T
L R ER I YEL L N
LSDT KNTS D
YID KIIYATPAE G
EVEV K D
I NI R
YFtYQIAE M K
FRAKGAEE E VV KES S V LKDEF C
SKD TF K
F FYDSIIKN EYI SL MDINI I S SS SS NLSTIYQEAE I TTI ISNTQ NSSIE G
D S D
KE N
V VAYQNE AIA K
KRLL KR LF IPTP KLS I Q Q
EAH G
VLI KFTF N
T VD G
D Y L I G
F Q HKENFLKM N
KE NFF D
S GTSD DKKKS TT SS GDSGS NGINS VGAI FYD Y Y
A KT AKLES PLE A
AF KATSV AES IL S NT S VIIL D
S N
AGAI LIEKTKPQ N
YISHT D M
INEEP FE YL QKTIE I
IKDLSEYLYAT E FNE K G
K N
SS F V
G G
LKT D
N D
PE K
V AKYI II DNT RH GNG S
FAG E
LGTE GAEKAGI L N
MRAFIL EMI D
NNGKN N
F V
EDFLY D
YL EEKQTFR N
LPTL NL FINDSLS VW
F R
D DFT KGFDPFF GF T
YH YEE QL TNYV D
VF G Y
KPW
L K
DRFI AYE DI Y
E EPKTWI ESQ EIAIEWG K
D M
LISRINA R
NLI Q
ILEESS V SLLT KI RS I
F M
I CYKIF TNL E V
SYSED D
KF VYGI G
VL GE G
QTQID R
F D
SELGT NL D
IN F DTNFGS LFAGIKF GQKYAVL QYT D
S GTRDI KVLHSG SIGGNS Y
E LEK D
GV EAHKFAK A
LGDSPIGNYSK VDTY SSNE MIIVNR VQPDS Q
I N
D G T
R DF QSVT TS ERNKD YKSLQGL EN H
L NLETI EYE KINKNENPKD FL A
SGS EEPEEI I Q
HRVLEE I
TI S NE NHT QDLENE GIY Y
EIYNEGT D Y G
YI R
LL QLY N
K L
E SL L A T
NFL Y V
S S G
F GE IRYVQ I DRNDR V D
LEYVIET T
VGEFYD A K R
EY A
KAILTFI L LRNI E H
LN LF IR R SV GM
D T
FE D
E NFYAI KK A
FL AL DIKVKDI N
IDV D
N KS ES GEK FNTL GEST PDI D VVNLNN Q
SGTDN G
D D
YYENE YKEKNG QNKIFK S
AI DKI SD LANS A D
DSE LL NGI L K
R MLIL LTL D
I KI D D VYAFEIELL KS KS TKSFSDEKP ITL L TSI KTDV YI T
NIFNQEN Q
D D
TFK N
HVE Q
DRYDS N LEI F QVI N
GLR RVS TVESVL GDIDINGVDKIPWED MI Y KE H
E Q
SKYG A
QIT YI K
ETE KEFE EEADS EK YAKKLED DV K Q
K D
YL S G
P IH H
GV N
YREKLLVLSQR NVL V
NE D
NGEI R
QEE V KD N IQE WQQTD KFA A
ASEI G
KEEAE N
HI KT D
NKDS VKL R L T
T TAIF D NI R
L K
SP LST D
VS I T
Y K
NS Y NNV ID DFAY YRF YYVIKDK V
DKS I D
RVP WQ GNP KIA RS V
EEGA N
GM L M
R I
LNE NF SST LELWL NK V
S DFGY SAL KDH L M
GN YQEDKDS LLE N QNK ELNHL YI V V GNN TDGD K GI L KNIIL D SFG M
AS KSE KYKISV T SV K D
P NNS T
GE TL EDI DQ T
DF GDWEIASIYG M
LKI NFRNIFD A
SFAW G
V D KISYYKF PE SRILLL Y
T YF EPT D G
IE KV IPPS W K
YAY NSEYF F E QE I DI LND I LV VN DYSI D G H TE N S I TI L GI TD F DLF V FHKFILS A YEDYVP E
LFKINQIQI ISNDT TFRE NKL K NYMLYPNGN T
KAES PSIKL AI A M
SEV E N
YNI GK WNEYQI N
VDG DD QL LRE E V
Y II NYSEGAETP KKG G
NID ND39 A
V N
LEKT R DM T
L V N
KS VPGEGYGS YI N
I NM
IL QI T
K D
GEGSLHLLTDLSV D
LVSEYN IN FEE PES IEIE L SE FIQSILGT NI KM
KME VF SEFQ V
AE EKGD AAYA H
AYEI K VD MD NNG YVC EE AR D
LSVKIII DKEGL G T
GGI NNEL Y
SYAIPLLRQLPRRG A
NF Q
YK D
E A
TI SLLSP L YVD SINF NEQESD ISI Q N
GER Q
EN NHFE ENNE G QW
YAKL HT DV LE LPI H
A QSNA Y
S K
D I LD CI YN NI DI AL F S
VVF M RM V
NAL GP T SL KF Y I D T VHKTESNIVHI E
T F AE YGYIN P G
TIFLLI D SD V NFF E
D QKD V R
WI M
I DE P NESLLK E GE YE L D
F Y
LVG G
L TIF DLKE GFYSWQL H S DDDYITI GE LKEDEL ESF I T
DM
S DV VKQ G
VINEL R D N
G T
A L T
GQSP Q
MYLKS D F
QKSWT QIK G
LEIEVVL D
GYEI VIF VYL NEG D
FYEI FY I A
DV GWI D
NSTN R
N V
IYYE DD ETNKP I D
A K
DLNKS FS V DKT EPSI D IE YTKF TIYDS H Q
L SI Y K
RLS NANI NVN Y S AL AQT SE T
I K
QT AK A
LYA N
LT G
YTL LT T K G
YSG VLIKDS YKL SPENI SFLEES N
E TSE DL SL I G
D N
SNGL D Y G
DEDCSGAH Q
KKYKL G
V IF M
A K
T L E
D YK T
VYI I L
NSR SV SVF V I K V
DNN QV K
D Y
NNI L A E
RLEKE SII ND AS NI GF PT]5MGINKL FL GKK IEE EI ] R LIYF6 NAEY N
PAKISN 6S P VIAKI I K
F SN R
SE V L KI ENA Y
SY KKSP Q6 F
GT HL FYMS VI YV F V
:VKYTEKKNGD : Q K
QI R GDP DKI P NQF NYIEGV K
E N
V ETFPDLV L G
KGGI DG O N
KIMNL W
E K
FE WHNIO N
IKEFKSF QTE ANDLTT LSP IP LT E F S S A
EI NQFE ELVKE E NELL F N
G V
III DIL NAFM
LLKA D
V V IVKKI N
VYG D
DE VM
D I
KVDLYSLFIQ N
ISQFD I L
SWIL KL R L NTDI TL F
DPFLDSLEN C
C SLF ISDYVP FS T
PII VIGNAID N
D NSEYE F A DSPT YFTR I NVLGQKI D F DE NSPKR Q
AP L
NGI R T Q SR NLSEKPE M
H N D C S AGL ERI N GSL F G M
K T TSF T E G ES[ M
V Y M
SNI Q P AS[ )eliciff
id)s s i e
ca di r o
iht d
n ir a t
s s o
lul1 lv C
i 1 (
ca2 B
B4 d
( cf 92 T
eL 5117570‐7L D
KI
E L I
E S3E2S SEFV N
ERT V
IIARGR
T L
E G
GA3YE FL NKENTE T K
S CS W
L RL A Y1I S0E2 LKIK LLHD DKK A
F VE N DVF N
L E I T YE VM
TR AI. VD EP FEDSYIE S F N
RI QHE RLoND NtKLD FIIS AFE DT L L KEGE E YKRGVPP T ER D
KKTLEDNE A A
VSP H
AAG L H
F G
DKePIP KNPKN D
GSTF DYLAELF G
N V
ASAL M
kcKoKDLE V
EL LV QE D NL KKATI D
SI R G REDLL SI KGEN G
GE T KYQ NA DQQVL H LSY G
AyeDnQD KrT KKKG E VHV G
KY TS E HF E TI GKSDKKoSttQILSV N K
NV V LL YW N
ESTA E VKSAQSAQI IDEAL AV NS KFF G
NI L L Y
P NV VYPADI K
LS D
SIQM Q
V D G
SR EIKYI E QADGI D
H LIK T
GEE F GI GRI NS W D
V KAQ A
L K
D D
L N
QS EKGH NG TIGKNRTYSS KDS DS KE V GH M
PI RSY GP F
EKKS W
GTL N
Q H
YS G
ESLSTPVFYR VE EYEL LN T
Y KVKDFYKKITL N G
E A
VTSLLLGTDV NQKEVAFELL Q VE TFP K N
AIV QRY QFI LKMDFGLE N
ASQV GLS P SG T GL QA GR EA NS NQYE PIVTL TPQ SKL V
SE AIPR IEFANDG KN S
SY D
NM
GIKR D
IT N T
PKV S VILPHR H
IKF K
ID AVGLNI IR QIRSMKEET N
DYL R Q
NTI FAP A
PEKM Q
KE K
Y D
YLGT AK GA G
NKT N
E A
L N
E HTIA W
KKL K QKY QD I G
P HTI IDL DI M E
KE Y TIQ IGHK NVE KEI N
LVDSQW N
PF S D
FR HV A
I D
M Q
GDR F SK TKIIT SV SDYV KIEGQG N
LR KE K
KP HGNLT TN W
K Y KH T H D
HNEGE GF L DG GV Y G
AL IYE P EESEAI SEG R D
RYSS ETP VKP KT I G LF FI E TEAND T YA TGK GV W
T K H
R K
F LS N
KYGIS A K
I GV T M K
VQFGIYE GYQR YS R
E YKRF DP NQ K
HYQE T K
I F SKS G
GS S Q
I KV FI V FT A
E Y
KRGP S K
SS MD IT D GR S GQS KK D
F AV VE AI L F LP RI PIISIRP Y SR L ES SGSQI G
KV N
NE EPSD QN T
V LIS D
E N
MI G
YLT TAF YF AKI N N
YKNYEIML DSLE SLE GGDYI HP HLSEILS R
EVF SLW D
AV ATHIGLALFQ EV Q NKFNTL E ELL NNYS K
GLL N
TNKE S Q
ST VK N TALYNTFYA SEKIT CKW
SSKV IE GH4 M A
GL KYKV N
R V
S NGYLQIS D
K E
NVKISES LV SEQELS YE 9 GKYGRVL VT SNAYR H
DPD QAC E NK IIP R
NQ K
DKFIDE A
S G
DAGFPP H
TKL A
MSF N
FL DIELGSI D NVTD AR E
HVSP ED T
QSNI I SK SEDIYE STIK T KKNRA G
DVSRSLFH PNKK AK L DS AYD C
IN IE LRV DIP NRATN Q
A M A
ES F
SKNL GT EI KEH A
ARVQTAYL GLNSP S L
LNE KLI N V
FQ SPL V
NQAKH V
KVT I DM GA LDNLRAIV R VQE GSF P KGAVH LIKGGGD D
NSSH N
K D
TI H
A G
TLCN V
PN G
NVAAI NR YA S
L FE KKE S
WMEN I GP A Q
ASYVAYFPAASHP I KE I
KF A
KL E W
ASGL YAWAY PVS SH FM
N SLTK F
E F KVLKDE Q
AEK IQNN]L P
KY76 AT GV MA KYAYRP]SN N
SP G K 86 S AC ] G
9L N ] 6V N 07 NPDISH FER I
PLIT H
QL Y: I V EYNYP :LFTP : TF KI W: F
DS O LIVMAS R O IF AI P OFV O FD KTL N
FLV Y
DLD KE L NS SGL I GYK SK D AGAG F
G A
DAN FHC VFN TFV A D VYIHNVC N H D GF L K
D R ILETE GAE T
ES APD I
LKQV V V I
FIYI I
KE I G
DSKILPI I KSNI KIQK K
ERSGSGRP QD QI K
N T
FLG EKY V
TWPQL EKV I
KQE M
G DLA GPAIFKV QS[ M
LP G G NDIS[ M
S H
EES[ M
AES[ )
eair )
e s
t ic h a
pi) r
de) ae h
ra t
e n m a
ul rel i
oho s
ni ulx rCh C l
io e t
tc o o 1 cairi a
ai Brbebr ibv i1 (
etnyV (
V (
24 a
h r yp oA9 xB 2 C
i D
C(tx CtC 5117570‐ F L
7F GA
Q3 MS A
2 GA Q
NG T T
P G G
GVV V
SEVLL DGELG A
VA31T QHGPGS SVII S I G
DFF GI0V N
QE NGA RL RT A
DSFAGR SV2G E KER L T EV.o EN TD NL A
KAGGS KT LVt S GL DTL Q A
MLLQVNS ALekAR Q S
ATETGP S G G
QTAW
V A
c M A
Y YKA N
GAV DS SAN A
GR LoD Q Q V
DyeGG AILT AG A
SAG G
NIQS GA A
AKA G
FF A GL T
DF HL VInArEQ S G
VE SEQSGSKIL GT GoStI RtT I L VL SYL YV SGVSI V VIAR N WY A T GAKKNS GGVLYL WG G
L SIDL A
TW
GKV AQG LR TS NEG Q
L DPGTT G A LFGGRGN GR AD LG G
L KG EAVKSFSV D D SL GA G G
NQAV AAS A
LV V GK KVLW ELS NK L GTR QAQEG LQ E M
SVR GM
KG EK EAS CFFLG TLGP WMGANGV A NAAHGVGGNP T AKCT TAAV LG A AE EF M F
PGLKADC AF H N
A N
G DQYAGV EPR T M W
FEQ D V VANIG K Q
RV LS FAM G
WLA G
PAG G
KFE QS Q VA SYASVPEKIVRE S G G
AWG VAL QL GL VYVQ G I G
AGGVA D
G G
KE G
AS G
AP S AI PI GI GAAGII Q4 GGI YTQFEAV A G
AS GDAG: GR GLAIES VGO GLDS W
GS GGV N W W
L VV A Q D VAL Q
EV GI YL G
KW
GD LSQ II EAD I A A
G Q
YL VPS Q
LTG K T
G D
AP DAQ L
L GR STGL AAE K
TS DI VG LG GG K
G G
T G
P S A
I A
GVS [ S
A A QT SFEVIIGKFASSGP * K
S IA TL G
QED A
GS A
AAAGS G D
T GEP KA EV Q
DIK A
AV S G
GL L A
DS GSS E SG E DGALS 5 A
GRP V 9 D
LDI L
IFGG S
QAE RL VIIALPL A
HGRLDS I
P DAAP H M
KAL S G
AG HT IKS D G D
IALSGEF GGQQ AR A SL M T
D GR R
T AGA T RGS GYS GE SST RL ALSS D
VG PS SA S SAAS NS GIV G
Q V
RL GL V G
SYL G
HA H
SA QR IEH FL A N
AG Q
GS G
D H A
LLIAG GAV V
V Q
A Q
AGARSGGGPAS SN AGecV nIVQ D
TS GH G
A V L
IVQFL IYL Y T
S S AAV HAVNGL P G AE seeu F IAVV GS ]FPIT D L G
G EQ A
] d q LILS HNS 44 LIGV VGK I G
] A1 F7 L :
T2 A7ite :
ps V e dAILV Q
RA VSVIQ i
GQRAL LL : G
O AG N GL SA L LV SG G
P GG GSS N OA D
O N
pcL R
D S VKAL DSR V
GK W
N T yl a AL PQADIRAYL T F GR Q
DKF Q I M
GD o M I
p oAD L nAALG G
GG A GFL QE AGA D
GE S R GL G
GC E
TQEEQQy iGE S GGS GIGKGLMV M
ASA [ M
SE SS[ral mGQGAT[ GAS S KGT p
A M
F G D V * M
I G A P A m e
x )
e s
i) ssi f o o
s los u
lu e c
rc c er n bebe u uu t t
qe m m
s uirui d er teic ctca aba obo n
co yci e m
di1 My v (
M(A t :p1 62 -t0 a15e e
p 1 2 y
g g4 sP lni n92 E
F l C
b a
orir 5 T PtStS117570‐7323102.oNtekcoDyenrottA 6
9 1
v12492511 Attorney Docket No. 2013237‐0757 RNA delivery RNA described herein may be delivered for therapeutic applications described herein using any appropriate methods known in the art, including, e.g., delivery as naked RNA, or delivery mediated by delivery vehicles. Some aspects of the disclosure involve the targeted delivery of the RNA disclosed herein to certain cells or tissues. In some embodiments, after administration of the RNA (in particular, mRNA) compositions/formulations described herein, at least a portion of the RNA is delivered to a target cell or target organ. In some embodiments, at least a portion of the RNA is delivered to the cytosol of the target cell. In some embodiments, the RNA (in particular, mRNA) is translated by the target cell to produce the encoded peptide or polypeptide. In some embodiments, the target cell is a muscle cell. In some embodiments, the target cell is a cell in the liver. In some embodiments, the target cell is a cell in the lung. In some embodiments, the disclosure involves targeting the lymphatic system, in particular secondary lymphoid organs, more specifically spleen. In some embodiments, the target cell is a cell in the lymph nodes. In some embodiments, the target cell is a spleen cell. In some embodiments, the target cell is an antigen presenting cell such as a professional antigen presenting cell in the spleen. In some embodiments, the target cell is a dendritic cell in the spleen. Thus, RNA (in particular, mRNA) compositions/formulations described herein may be used for delivering RNA to such target cell. The "lymphatic system" is part of the circulatory system and an important part of the immune system, comprising a network of lymphatic vessels that carry lymph. The lymphatic system consists of lymphatic organs, a conducting network of lymphatic vessels, and the circulating lymph. The primary or central lymphoid organs generate lymphocytes from immature progenitor cells. The thymus and the bone marrow constitute the primary lymphoid organs. Secondary or peripheral lymphoid organs, which include lymph nodes and the spleen, maintain mature naive lymphocytes and initiate an adaptive immune response. Lipid-based RNA delivery systems have an inherent preference to the liver, where, depending on the composition of the RNA delivery systems used, RNA expression in the liver can be obtained. Liver accumulation is caused by the discontinuous nature of the hepatic vasculature or the lipid metabolism (liposomes and lipid or cholesterol conjugates). In some embodiments, the target organ for RNA expression is liver and the target tissue is liver tissue. The delivery to such target tissue is preferred, in particular, if presence of RNA or of the encoded peptide or polypeptide in this organ or tissue is desired and/or if it is desired to express large amounts of the encoded peptide or polypeptide and/or if systemic presence of the encoded peptide or polypeptide, in particular in significant amounts, is desired or required. Delivery vehicles To overcome the barriers to safe and effective RNA delivery, RNA may be administered with one or more delivery vehicles that protect the RNA from degradation, maximize delivery to on-target cells and minimize exposure to off- target cells. Such RNA delivery vehicles may complex or encapsulate RNA and include a range of materials, including polymers and lipids. In some embodiments, such RNA delivery vehicles may form particles with RNA. Suitable formulations for such delivery vehicles are described, e.g., in WO2023/126404, WO2023/126053 and WO 2023/094713, the contents of which are incorporated herein by reference. 97 11529421v1 Attorney Docket No. 2013237‐0757 Compositions comprising nucleic acid A composition comprising one or more RNAs described herein, e.g., in the form of RNA particles, may comprise salts, buffers, or other components as further described below. In some embodiments, a salt for use in the compositions described herein comprises sodium chloride. Without wishing to be bound by theory, sodium chloride functions as an ionic osmolality agent for preconditioning RNA prior to mixing with lipids. In some embodiments, the compositions described herein may comprise alternative organic or inorganic salts. Alternative salts include, without limitation, potassium chloride, dipotassium phosphate, monopotassium phosphate, potassium acetate, potassium bicarbonate, potassium sulfate, disodium phosphate, monosodium phosphate, sodium acetate, sodium bicarbonate, sodium sulfate, lithium chloride, magnesium chloride, magnesium phosphate, calcium chloride, and sodium salts of ethylenediaminetetraacetic acid (EDTA). Generally, compositions for storing RNA particles such as for freezing RNA particles comprise low sodium chloride concentrations, or comprises a low ionic strength. In some embodiments, the sodium chloride is at a concentration from 0 mM to about 50 mM, from 0 mM to about 40 mM, or from about 10 mM to about 50 mM. According to the present disclosure, the RNA particle compositions described herein have a pH suitable for the stability of the RNA particles and, in particular, for the stability of the RNA. Without wishing to be bound by theory, the use of a buffer system maintains the pH of the particle compositions described herein during manufacturing, storage and use of the compositions. In some embodiments of the present disclosure, the buffer system may comprise a solvent (in particular, water, such as deionized water, in particular water for injection) and a buffering substance. The buffering substance may be selected from 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid (HEPES), 2-amino-2-(hydroxymethyl)propane-1,3-diol (Tris), acetate, and histidine. In some embodiments, the buffering substance is HEPES. In some embodiments, the buffering substance is Tris. Compositions (in particular, RNA compositions/formulations) described herein may also comprise a cryoprotectant and/or a surfactant as stabilizer to avoid substantial loss of the product quality and, in particular, substantial loss of RNA activity during storage, freezing, and/or lyophilization, for example to reduce or prevent aggregation, particle collapse, RNA degradation and/or other types of damage. In some embodiments, the cryoprotectant is a carbohydrate. The term "carbohydrate", as used herein, refers to and encompasses monosaccharides, disaccharides, trisaccharides, oligosaccharides and polysaccharides. In some embodiments, the cryoprotectant is a monosaccharide. The term "monosaccharide", as used herein refers to a single carbohydrate unit (e.g., a simple sugar) that cannot be hydrolyzed to simpler carbohydrate units. Exemplary monosaccharide cryoprotectants include glucose, fructose, galactose, xylose, ribose and the like. In some embodiments, the cryoprotectant is a disaccharide. The term "disaccharide", as used herein refers to a compound or a chemical moiety formed by 2 monosaccharide units that are bonded together through a glycosidic linkage, for example through 1-4 linkages or 1-6 linkages. A disaccharide may be hydrolyzed into two 98 11529421v1 Attorney Docket No. 2013237‐0757 monosaccharides. Exemplary disaccharide cryoprotectants include sucrose, trehalose, lactose, maltose and the like. In some embodiments, the cryoprotectant is sucrose. The term "trisaccharide" means three sugars linked together to form one molecule. Examples of a trisaccharides include raffinose and melezitose. In some embodiments, the cryoprotectant is an oligosaccharide. The term "oligosaccharide", as used herein refers to a compound or a chemical moiety formed by 3 to about 15, such as 3 to about 10 monosaccharide units that are bonded together through glycosidic linkages, for example through 1-4 linkages or 1-6 linkages, to form a linear, branched or cyclic structure. Exemplary oligosaccharide cryoprotectants include cyclodextrins, raffinose, melezitose, maltotriose, stachyose, acarbose, and the like. An oligosaccharide can be oxidized or reduced. In an embodiment, the cryoprotectant is a cyclic oligosaccharide. The term "cyclic oligosaccharide", as used herein refers to a compound or a chemical moiety formed by 3 to about 15, such as 6, 7, 8, 9, or 10 monosaccharide units that are bonded together through glycosidic linkages, for example through 1-4 linkages or 1-6 linkages, to form a cyclic structure. Exemplary cyclic oligosaccharide cryoprotectants include cyclic oligosaccharides that are discrete compounds, such as α cyclodextrin, β cyclodextrin, or γ cyclodextrin. Other exemplary cyclic oligosaccharide cryoprotectants include compounds which include a cyclodextrin moiety in a larger molecular structure, such as a polymer that contains a cyclic oligosaccharide moiety. A cyclic oligosaccharide can be oxidized or reduced, for example, oxidized to dicarbonyl forms. The term "cyclodextrin moiety", as used herein refers to cyclodextrin (e.g., an α, β, or γ cyclodextrin) radical that is incorporated into, or a part of, a larger molecular structure, such as a polymer. A cyclodextrin moiety can be bonded to one or more other moieties directly, or through an optional linker. A cyclodextrin moiety can be oxidized or reduced, for example, oxidized to dicarbonyl forms. Carbohydrate cryoprotectants, e.g., cyclic oligosaccharide cryoprotectants, can be derivatized carbohydrates. For example, in an embodiment, the cryoprotectant is a derivatized cyclic oligosaccharide, e.g., a derivatized cyclodextrin, e.g., 2-hydroxypropyl-β-cyclodextrin, e.g., partially etherified cyclodextrins (e.g., partially etherified β cyclodextrins). An exemplary cryoprotectant is a polysaccharide. The term "polysaccharide", as used herein refers to a compound or a chemical moiety formed by at least 16 monosaccharide units that are bonded together through glycosidic linkages, for example through 1-4 linkages or 1-6 linkages, to form a linear, branched or cyclic structure, and includes polymers that comprise polysaccharides as part of their backbone structure. In backbones, the polysaccharide can be linear or cyclic. Exemplary polysaccharide cryoprotectants include glycogen, amylase, cellulose, dextran, maltodextrin and the like. In some embodiments, RNA particle compositions may include sucrose. Without wishing to be bound by theory, sucrose functions to promote cryoprotection of the compositions, thereby preventing RNA (especially mRNA) particle aggregation and maintaining chemical and physical stability of the composition. In some embodiments, RNA particle compositions may include alternative cryoprotectants to sucrose. Alternative stabilizers include, without 99 11529421v1 Attorney Docket No. 2013237‐0757 limitation, trehalose and glucose. In a specific embodiment, an alternative stabilizer to sucrose is trehalose or a mixture of sucrose and trehalose. A preferred cryoprotectant is selected from the group consisting of sucrose, trehalose, glucose, and a combination thereof, such as a combination of sucrose and trehalose. In a preferred embodiment, the cryoprotectant is sucrose. Some embodiments of the present disclosure contemplate the use of a chelating agent in an RNA composition described herein. Chelating agents refer to chemical compounds that are capable of forming at least two coordinate covalent bonds with a metal ion, thereby generating a stable, water-soluble complex. Without wishing to be bound by theory, chelating agents reduce the concentration of free divalent ions, which may otherwise induce accelerated RNA degradation in the present disclosure. Examples of suitable chelating agents include, without limitation, ethylenediaminetetraacetic acid (EDTA), a salt of EDTA, desferrioxamine B, deferoxamine, dithiocarb sodium, penicillamine, pentetate calcium, a sodium salt of pentetic acid, succimer, trientine, nitrilotriacetic acid, trans- diaminocyclohexanetetraacetic acid (DCTA), diethylenetriaminepentaacetic acid (DTPA), and bis(aminoethyl)glycolether-N,N,N',N'-tetraacetic acid. In some embodiments, the chelating agent is EDTA or a salt of EDTA. In some embodiments, the chelating agent is EDTA disodium dihydrate. In some embodiments, the EDTA is at a concentration from about 0.05 mM to about 5 mM, from about 0.1 mM to about 2.5 mM or from about 0.25 mM to about 1 mM. In an alternative embodiment, the RNA particle compositions described herein do not comprise a chelating agent. Pharmaceutical compositions The agents described herein may be administered in pharmaceutical compositions or medicaments and may be administered in the form of any suitable pharmaceutical composition. In some embodiments, the pharmaceutical composition is for therapeutic or prophylactic treatments, e.g., for use in treating or preventing a disease involving an antigen, in particular infections with Mycobacterium tuberculosis, Pseudomonas aeruginosa, Staphylococcus aureus, Clostridioides difficile, Bacillus anthracis, Corynebacterium diphteriae or Vibrio Cholerae. The term "pharmaceutical composition" relates to a composition comprising a therapeutically effective agent, preferably together with pharmaceutically acceptable carriers, diluents and/or excipients. Said pharmaceutical composition is useful for treating, preventing, or reducing the severity of a disease by administration of said pharmaceutical composition to a subject. The pharmaceutical compositions of the present disclosure may comprise one or more adjuvants or may be administered with one or more adjuvants. The term "adjuvant" relates to a compound which prolongs, enhances or accelerates an immune response. Adjuvants comprise a heterogeneous group of compounds such as oil emulsions (e.g., Freund’s adjuvants), mineral compounds (such as alum), bacterial products (such as Bordetella pertussis toxin), or immune-stimulating complexes. Examples of adjuvants include, without limitation, LPS, GP96, CpG oligodeoxynucleotides, growth factors, and cytokines, such as monokines, lymphokines, interleukins, chemokines. The chemokines may be IL-1, IL-2, IL-3, IL-4, IL-5, IL-6, IL-7, IL-8, IL-9, IL-10, IL-12, INFa, INF-γ, 100 11529421v1 Attorney Docket No. 2013237‐0757 GM-CSF, LT-a. Further known adjuvants are aluminum hydroxide, Freund's adjuvant or oil such as Montanide® ISA51. Other suitable adjuvants for use in the present disclosure include lipopeptides, such as Pam3Cys, as well as lipophilic components, such as saponins, trehalose-6,6-dibehenate (TDB), monophosphoryl lipid-A (MPL), monomycoloyl glycerol (MMG), or glucopyranosyl lipid adjuvant (GLA). The pharmaceutical compositions of the present disclosure may be in a storable form (e.g., in a frozen or lyophilized/freeze-dried form) or in a "ready-to-use form" (i.e., in a form which can be immediately administered to a subject, e.g., without any processing such as diluting). Thus, prior to administration of a storable form of a pharmaceutical composition, this storable form has to be processed or transferred into a ready-to-use or administrable form. E.g., a frozen pharmaceutical composition has to be thawed, or a freeze-dried pharmaceutical composition has to be reconstituted, e.g. by using a suitable solvent (e.g., deionized water, such as water for injection) or liquid (e.g., an aqueous solution). The pharmaceutical compositions according to the present disclosure are generally applied in a "pharmaceutically effective amount" and in "a pharmaceutically acceptable preparation". The term "pharmaceutically acceptable" refers to the non-toxicity of a material which does not interact with the action of the active component of the pharmaceutical composition. The term "pharmaceutically effective amount" refers to the amount which achieves a desired reaction or a desired effect alone or together with further doses. In some embodiments relating to the treatment of a particular disease, the desired reaction may relate to inhibition of the course of the disease. This comprises slowing down the progress of the disease and, in some embodiments, interrupting or reversing the progress of the disease. The desired reaction in a treatment of a disease may also be delay of the onset or a prevention of the onset of said disease or said condition, or symptoms thereof. An effective amount of the pharmaceutical compositions described herein will depend on the condition to be treated, the severeness of the disease, the individual parameters of the patient, including age, physiological condition, size and weight, the duration of treatment, the type of an accompanying therapy (if present), the specific route of administration and similar factors. Accordingly, the doses administered of the pharmaceutical compositions described herein may depend on various of such parameters. In the case that a reaction in a patient is insufficient with an initial dose, higher doses (or effectively higher doses achieved by a different, more localized route of administration) may be used. The pharmaceutical compositions of the present disclosure may contain buffers, preservatives, and optionally other therapeutic agents. In some embodiments, the pharmaceutical compositions of the present disclosure comprise one or more pharmaceutically acceptable carriers, diluents and/or excipients. Suitable preservatives for use in the pharmaceutical compositions of the present disclosure include, without limitation, benzalkonium chloride, chlorobutanol, paraben and thimerosal. The term "excipient" as used herein refers to a substance which may be present in a pharmaceutical composition of the present disclosure but is not an active ingredient. Examples of excipients, include without limitation, carriers, 101 11529421v1 Attorney Docket No. 2013237‐0757 binders, diluents, lubricants, thickeners, surface active agents, preservatives, stabilizers, emulsifiers, buffers, flavoring agents, or colorants. The term "diluent" relates a diluting and/or thinning agent. Moreover, the term "diluent" includes any one or more of fluid, liquid or solid suspension and/or mixing media. Examples of suitable diluents include ethanol, glycerol and water. The term "carrier" refers to a component which may be natural, synthetic, organic, inorganic in which the active component is combined in order to facilitate, enhance or enable administration of the pharmaceutical composition. A carrier as used herein may be one or more compatible solid or liquid fillers, diluents or encapsulating substances, which are suitable for administration to subject. Suitable carriers include, without limitation, sterile water, Ringer, Ringer lactate, sterile sodium chloride solution, isotonic saline, polyalkylene glycols, hydrogenated naphthalenes and, in particular, biocompatible lactide polymers, lactide/glycolide copolymers or polyoxyethylene/polyoxy- propylene copolymers. In some embodiments, the pharmaceutical composition of the present disclosure includes isotonic saline. Pharmaceutically acceptable carriers, excipients or diluents for therapeutic use are well known in the pharmaceutical art, and are described, for example, in Remington's Pharmaceutical Sciences, Mack Publishing Co. (A. R Gennaro edit. 1985). Pharmaceutical carriers, excipients or diluents can be selected with regard to the intended route of administration and standard pharmaceutical practice. Routes of administration of pharmaceutical compositions In some embodiments, the pharmaceutical compositions described herein may be administered intravenously, intraarterially, subcutaneously, intradermally, dermally, intranodally, or intramuscularly. In some embodiments, the pharmaceutical compositions described herein may be administered intramuscularly. In some embodiments, the pharmaceutical composition is formulated for local administration or systemic administration. Systemic administration may include enteral administration, which involves absorption through the gastrointestinal tract, or parenteral administration. As used herein, "parenteral administration" refers to the administration in any manner other than through the gastrointestinal tract, such as by intravenous injection. In some embodiments, the pharmaceutical compositions are formulated for systemic administration. In some embodiments, the systemic administration is by intravenous administration. In some embodiments, the pharmaceutical compositions are formulated for intramuscular administration. In some embodiments, intramuscular administration comprises administration into the upper arm, in particular into the musculus deltoideus. If more than one dose, e.g., three doses, of a pharmaceutical composition described herein is administered, the different administrations may be into the same arm. 102 11529421v1 Attorney Docket No. 2013237‐0757 Use of compositions Compositions described herein may be used in the therapeutic or prophylactic treatment of diseases wherein provision of one or more peptides or polypeptides, i.e., polypeptides, described herein to a subject results in a therapeutic or prophylactic effect. In some embodiments, the disease is infection with Mycobacterium tuberculosis. In some embodiments, the disease is tuberculosis. The term "disease" (also referred to as "disorder" herein) refers to an abnormal condition that affects the body of an individual. A disease is often construed as a medical condition associated with specific symptoms and signs. A disease may be caused by factors originally from an external source, such as infectious disease, or it may be caused by internal dysfunctions, such as autoimmune diseases. In humans, "disease" is often used more broadly to refer to any condition that causes pain, dysfunction, distress, social problems, or death to the individual afflicted, or similar problems for those in contact with the individual. In this broader sense, it sometimes includes injuries, disabilities, disorders, syndromes, infections, isolated symptoms, deviant behaviors, and atypical variations of structure and function, while in other contexts and for other purposes these may be considered distinguishable categories. Diseases usually affect individuals not only physically, but also emotionally, as contracting and living with many diseases can alter one's perspective on life, and one's personality. The term "disease involving an antigen" refers to any disease which implicates an antigen, e.g. a disease which is characterized by the presence of an antigen. The disease involving an antigen can be an infectious disease. The antigen may be a disease-associated antigen, such as a bacterial antigen. In some embodiments, a disease involving an antigen is a disease involving cells comprising and/or expressing an antigen, and preferably presenting the antigen on the cell surface, e.g., in the context of MHC. The term "infectious disease" refers to any disease which can be transmitted from individual to individual or from organism to organism, and is caused by a microbial agent (e.g. common cold). Infectious diseases are known in the art and include, for example, a viral disease, a bacterial disease, or a parasitic disease, which diseases are caused by a virus, a bacterium, and a parasite, respectively. In this regard, the infectious disease can be, for example, hepatitis, sexually transmitted diseases (e.g. chlamydia or gonorrhea), tuberculosis, malaria, HIV/acquired immune deficiency syndrome (AIDS), diphtheria, hepatitis B, hepatitis C, cholera, severe acute respiratory syndrome (SARS), the bird flu, and influenza. In the present context, the term "treatment", "treating" or "therapeutic intervention" relates to the management and care of a subject for the purpose of combating a condition such as a disease. The term is intended to include the full spectrum of treatments for a given condition from which the subject is suffering, such as administration of the therapeutically effective compound to alleviate the symptoms or complications, to delay the progression of the disease, disorder or condition, to alleviate or relief the symptoms and complications, and/or to cure or eliminate the disease, disorder or condition as well as to prevent the condition, wherein prevention is to be understood as the management and care of an individual for the purpose of combating the disease, condition or disorder and includes the administration of the active compounds to prevent the onset of the symptoms or complications. 103 11529421v1 Attorney Docket No. 2013237‐0757 The term "therapeutic treatment" relates to any treatment which improves the health status and/or prolongs (increases) the lifespan of an individual. Said treatment may eliminate the disease in an individual, arrest or slow the development of a disease in an individual, inhibit or slow the development of a disease in an individual, decrease the frequency or severity of symptoms in an individual, and/or decrease the recurrence in an individual who currently has or who previously has had a disease. The terms "prophylactic treatment" or "preventive treatment" relate to any treatment that is intended to prevent a disease from occurring in an individual. The terms "prophylactic treatment" or "preventive treatment" are used herein interchangeably. The terms "individual" and "subject" are used herein interchangeably. They refer to a human or another mammal (e.g., mouse, rat, rabbit, dog, cat, cattle, swine, sheep, horse or primate), or any other non-mammal-animal, including birds (chicken), fish or any other animal species that can be afflicted with or is susceptible to a disease (e.g., cancer, infectious diseases) but may or may not have the disease, or may have a need for prophylactic intervention such as vaccination, or may have a need for interventions such as by protein replacement. In many embodiments, the individual is a human being. Unless otherwise stated, the terms "individual" and "subject" do not denote a particular age, and thus encompass adults, elderlies, children, and newborns. In some embodiments of the present disclosure, the "individual" or "subject" is a "patient". In some embodiments, the terms "individual" and "subject" relate to pregnant women and immunocompromised persons. The term "patient" means an individual or subject for treatment, in particular a diseased individual or subject. RNA described herein may be administered to a subject for delivering the RNA to cells of the subject. RNA described herein may be administered to a subject for delivering a therapeutic or prophylactic peptide or polypeptide (e.g., a pharmaceutically active peptide or polypeptide) to the subject, wherein the RNA encodes a therapeutic or prophylactic peptide or polypeptide. RNA described herein may be administered to a subject for treating or preventing a disease in a subject, wherein delivering the RNA to cells of the subject is beneficial in treating or preventing the disease. RNA described herein may be administered to a subject for treating or preventing a disease in a subject, wherein the RNA encodes a therapeutic or prophylactic peptide or polypeptide and wherein delivering the therapeutic or prophylactic peptide or polypeptide to the subject is beneficial in treating or preventing the disease. In some embodiments of the disclosure, the aim is to induce an immune response by providing RNA described herein. A person skilled in the art will know that one of the principles of immunotherapy and vaccination is based on the fact that an immunoprotective reaction to a disease is produced by immunizing a subject with a polypeptide or an epitope, which is immunologically relevant with respect to the disease to be treated. Accordingly, RNA described herein is applicable for inducing or enhancing an immune response. RNA described herein is thus useful in a prophylactic and/or therapeutic treatment of a disease involving a polypeptide or epitope. 104 11529421v1 Attorney Docket No. 2013237‐0757 In some embodiments of the disclosure, the aim is to provide an immune response against cells comprising an antigen, e.g., Mtb antigen. In some embodiments of the disclosure, the aim is to prophylactically or therapeutically treat tuberculosis by vaccination. Due to the high degree of sequence conservation of the disclosed antigens between different Mycobacterium species, including Mycobacterium tuberculosis, Mycobacterium bovis, Mycobacterium caprae, Mycobacterium orygis, Mycobacterium africanum, Mycobacterium microti, Mycobacterium canetti and Mycobacterium pinnipedii, exposure of a subject to Mycobacterium tuberculosis antigens, in the context of a polypeptide of the invention, will result in a high degree of cross-reactivity with antigens of other Mycobacterium species. Therefore, a vaccine based on or directed at Mycobacterium tuberculosis antigens will elicit a robust immune response against other Mycobacterium species, in particular Mycobacterium bovis, Mycobacterium caprae, Mycobacterium orygis, Mycobacterium africanum, Mycobacterium microti, Mycobacterium canetti and Mycobacterium pinnipedii as well. In some embodiments of the disclosure, the aim is to provide an immune response against a Mycobacterium selected from Mycobacterium tuberculosis, Mycobacterium bovis, Mycobacterium caprae, Mycobacterium orygis, Mycobacterium africanum, Mycobacterium microti, Mycobacterium canetti and Mycobacterium pinnipedii and to prevent or treat tuberculosis. In preferred embodiments of the disclosure, the aim is to provide an immune response against Mycobacterium tuberculosis. In some embodiments of the disclosure, the aim is to treat an infection with Mycobacterium tuberculosis. In some embodiments of the disclosure, the aim is to prevent or treat disease symptoms caused by an infection with Mycobacterium tuberculosis. In some embodiments of the disclosure, the aim is to provide protection against an infection with Mycobacterium tuberculosis by vaccination. In some embodiments of the disclosure, the aim is to provide protection against an outbreak of disease in a subject infected with Mycobacterium tuberculosis. In some embodiments of the disclosure, the aim is to provide protection against symptoms of tuberculosis in a subjected infected with Mycobacterium tuberculosis. In some embodiments, the RNA is present in a composition as described herein. In some embodiments, the RNA is administered in a pharmaceutically effective amount. In some embodiments, the subject is a mammal. In some embodiments, the mammal is a human. In some embodiments, the subject treated had been exposed to Mycobacterium tuberculosis. In some embodiments, the subject treated had not been exposed to Mycobacterium tuberculosis. 105 11529421v1 Attorney Docket No. 2013237‐0757 In some embodiments, the treatments described herein involve pre- or post-exposure vaccination against Mycobacterium tuberculosis, or a combination thereof. Citation of documents and studies referenced herein is not intended as an admission that any of the foregoing is pertinent prior art. All statements as to the contents of these documents are based on the information available to the applicants and do not constitute any admission as to the correctness of the contents of these documents. The description (including the following examples) is presented to enable a person of ordinary skill in the art to make and use the various embodiments. Descriptions of specific devices, techniques, and applications are provided only as examples. Various modifications to the examples described herein will be readily apparent to those of ordinary skill in the art, and the general principles defined herein may be applied to other examples and applications without departing from the spirit and scope of the various embodiments. Thus, the various embodiments are not intended to be limited to the examples described herein and shown, but are to be accorded the scope consistent with the claims. Examples Example 1: Observation that EspA is toxic when expressed as mRNA in HEK293T cells. EspA (Rv3616c) was encoded on mRNA using its wild-type protein sequence modified by an N-terminal signal peptide (human HLA-A1 derived, hereafter SP) and a FLAG-epitope tag. This construct was investigated for expression in HEK293T cells by RiboJuiceTM (Sigma-Aldrich) transfection of 1 µg of RNA. After overnight incubation (approximately 18h) cells were inspected visually under a bright field microscope. Strikingly, a large reduction in cell confluency could be observed in cells transfected with EspA compared to the non-transfected control or compared to control protein 1 which was also FLAG-tagged and known to be well expressed (Figure 1). Lysates were generated and analyzed by western blot for expression of the FLAG-tag, which could be well detected for control protein 1. However, no expression was found for EspA (Figure 2). These results were our first indication that expression of EspA could be highly toxic for cells when expressed on mRNA. Example 2: Confirmation of EspA expression and toxicity and introducing live imaging as a method to quantify toxicity. Wild type EspA or a control protein with a similar predicted molecular weight and a similar predicted tertiary structure (control protein 2), were expressed in HEK293T cells by RiboJuiceTM transfection of different amounts of RNA (25 ng, 100 ng, 250 ng or 1000 ng). Both proteins were FLAG-tagged and fused with a N-terminal signal peptide (SP). Lysates of the cells were generated 6 hours post transfection or 24h post transfection. Lysates from transfected cells were separated by SDS-PAGE on a 6-18% polyacrylamide gel and western blot using an FLAG- specific monoclonal antibody conjugated to horseradish peroxidase (HRP; clone M2 from Sigma-Aldrich) and a α- Tubulin-specific HRP-conjugated rabbit monoclonal antibody (clone 11H10 from Cell Signaling Technology). The 106 11529421v1 Attorney Docket No. 2013237‐0757 FLAG-tagged control protein 2 was detected at both timepoints (250ng and 1000ng of RNA; Figure 3). In contrast, and similar to example 1, EspA expression could not be detected 24 hours post transfection. However, at 6 hours post transfection a faint band could be seen at the indicated size confirming that EspA is expressed early after mRNA transfection. In parallel HEK293T cells were transfected with the same constructs and incubated with Incucyte® Cytotox Red Dye for Counting Dead Cells in the Incucyte® S3 Live-Cell Analysis System (Sartorius). Pictures were taken in 2-hour intervals for 24 hours to measure cell confluence (bright field images) and Cytotox- staining of cells with impaired membrane potential as measure of cell death (Red channel, excitation 567-607 nm, emission 622-704 nm). Analysis was performed using the Incucyte® Live-Cell Analysis software provided by Sartorius. The Basic Analyzer analysis type was used. Confluence of cells was determined based on shape and size, where small blebs were excluded (Area (μm2) min = 200.00, Cleanup Adjust size (pixels) = -6). Cytotox positivity was defined based on size and signal intensity (Segmentation: Surface fit, Threshold (RCU) = 4.0000, Edge Split Off), where disproportionately small and large spots were removed as well as very weak signals (Area (μm2) min = 12.000, Mean Intensity min = 8.0000, Integrated Intensity min = 600.00). Confluency was defined in percentage (%), while Cytotox+ spots were defined in Red calibrated units (RCU) times square μm per image (RCU*μm2/image). Both parameters were normalized to a fold change compared to t = 0. Confluency decreased while the signal for the Cytotox dye increased when cells were transfected with EspA (Figure 4A-C). The opposite was true for control protein 2. Especially the concentration of 250 ng RNA clearly indicated the differences between confluency and Cytotox-stained cells upon transfection of both constructs. Example 3: EspA toxicity is primarily driven by the N-terminal A domain. Since it was found that EspA was toxic when expressed as a wild type, full-length protein, a bioinformatic analysis was performed to separate the protein into domains to determine whether this would impair cytotoxicity and if the part of the protein could be determined, which mediated its cytotoxicity. Based on sequence comparison with the human proteome, two regions of potential overlap with the human proteome were identified and excluded moving forward. Therefore, three major domains termed EspA_A (aa 2-166), EspA_B (aa 177-280), and EspA_C (aa 291- 392) were selected (Figure 5). Furthermore, to make sure that specifically these excluded overlapping-regions were not essential for determining toxicity of EspA, two constructs were designed, that combined the N-terminal two parts and the region of human overlap bridging them, dubbed EspA_AB, and a similar construct for the C- terminal parts called EspA_BC (Figure 6). To assess expression of the generated constructs, cells were seeded and incubated overnight before transfection with 250 ng RNA/well as described above. The negative control consists of non-transfected cells. Cells were incubated for 4h at 37°C by 5% CO¬2 and transferred to a 96-well plate and washed. Samples were stained with Fixable Viability Dye eFluor™ 780 (Thermo Fisher Scientific) for 15 minutes at room temperature. Cells were washed and fixed for 12 minutes at room temperature, followed by permeabilization for 5 minutes at 4°C. Permeabilized cells were stained with a FLAG-tag specific antibody (BioLegend) for 30 min at 4°C. Cells were washed and acquired with a FACS Celesta (BD Biosciences). Data was analyzed using FACS Diva and Flow Jo software (both BD Biosciences). Gating excluded doublet cells and debris (Figure 7A). Cell viability was measured on the B-A-780-A spectrum, while FLAG-positive cells were measured on the BV421-A spectrum. Antigen expression as reflected by 107 11529421v1 Attorney Docket No. 2013237‐0757 FLAG-tag binding was obtained from the viable cell population. Transfected constructs containing the A fragment of EspA showed a higher percentage of dead cells compared to control protein 2 (p<0.01) (Figure 7B and C). Upon transfection with EspA_FL, approximately 5% more cells were dead as compared to transfection of the control protein 2 encoding construct. Transfection with EspA_A and EspA_AB resulted in approximately 30% (p<0.01) and over 10% (p<0.05) more cell death, respectively. All other constructs had a similar viability as the negative construct control (p>0.05). As expected, in the non-transfected control, no cells of the viable cell population were staining positive for the FLAG-tag (Figure 7B and C). On the contrary, constructs encoding control protein 2, EspA_B, EspA_C, and EspA_BC did stain positive. The constructs with high cell death barely showed any positivity for the tag in either of the cell populations. This is most likely due to the viable cells not being transfected with the construct, and the cells that were transfected were either currently dying, accounting for the slight positivity, or were already dead and the FLAG-tagged protein was already degraded. To investigate the cytotoxicity of the generated EspA constructs (Figure 6) HEK293T cells were seeded and incubated as stated above (Example 2). Cells were transfected with 250 ng of RNA constructs encoding EspA_FL, EspA_A, EspA_B, EspA_C, EspA_AB, EspA_BC, or control proteins 2 and 3. Control protein 3 is non-toxic, of the same size as EspA-FL and control protein 2 and was added to further validate the assay results. Non-transfected cells served as a negative control. Immediately after transfection, cells were imaged in bright field and for fluorescence signals every hour for 24h. Cells transfected with constructs containing the EspA-A-fragment all showed increased Cytotox+ integrated intensity and decreased confluency indicating cytotoxicity (Figure 8A and B). EspA_A induced slightly more cytotoxicity when compared to EspA_FL. Both control proteins 2 and 3 behaved similarly to the non-transfected control and did not differ between each other (p<0.05). EspA_FL, EspA_A, and EspA_AB differed from control proteins 2 and 3 (p<0.001). Additionally, slightly higher values for EspA_B were observed as well (p<0.05). EspA_C and EspA_BC did not differ from the negative controls (p>0.05). Clearly, the highest toxicity values are indicated by constructs containing the A fragment, hence confirming the flow cytometer data. Example 4: A structure-guided T-cell string design to disrupt toxicity of EspA To visualize the domain architecture of EspA, AlphaFold2 was used to generate a model of EspA (Figure 9, panel A). There is high confidence in the EspA model due to high homology with EspB, which has high resolution crystal structures (https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4520771/) and a cryo-electron microscopy higher order pore structure (https://www.sciencedirect.com/science/article/pii/S2590152420300118?via%3Dihub) deposited in the Protein Data Bank (PDB). The model was juxtaposed against the bioinformatics data described above showing regions of human overlap. The EspA protein sequence was divided into subdomains in a manner that disrupted secondary structure elements and tertiary protein structure and function (Figure 9, panel B). Fragments were selected such that areas of human overlap were avoided when selecting EspA chunks for inclusion in the T cell string. In one approach, the EspA protein sequence was divided into subdomains containing three regions of EspA: EspA_3-1 (amino acids 1-166 of FL EspA), EspA_3-2 (amino acids 177-280 of FL EspA), and EspA_3-3 (amino acids 291-392 of FL EspA). The fragments were rearranged in a manner that disrupted tertiary and secondary structure 108 11529421v1 Attorney Docket No. 2013237‐0757 elements and function (Fig.9B and 9C). In another approach, the three fragments were further split up to generate seven fragments in total: EspA_3-1 was further fragmented into EspA_7-1 (amino acids 1-60 of FL EspA), EspA_7- 2 (amino acids 62-131 of FL EspA), and EspA_7-3 (amino acids 135-166 of FL EspA), EspA_3-2 was further fragmented into EspA_7-4 (amino acids 177-239 of FL EspA) and EspA_7-5 (amino acids 241-279 of FL EspA), and EspA_3-3 was further fragmented into EspA_7-6 (amino acids 291-341 of FL EspA), and EspA_7-7 (amino acids 343-392 of FL EspA). Again, the fragments were rearranged in a manner that disrupted tertiary and secondary structure elements and function (Fig. 9B and 9D). In both approaches, adjacent EspA regions were reshuffled to ensure that EspA chunks could not properly assemble into a functional protein during co-translation folding (Figure 9, C and D). It was hypothesized that this strategy would reduce toxicity and combined with the MITD, increase protein turnover and generation of immunogenic T cell epitopes. Example 5: Analysis of expression of the 3-part and 7-part EspA strings in vitro by mass-spectrometry RNA drug substance encoding string 2 (where EspA was separated and shuffled in 7 parts with linkers – Figure 10, panel A) was transfected into HEK293T cells in the presence (Light grey bars) or absence (Dark grey bars) of proteasome inhibitor MG132. Following lysis, total protein content was normalized, cysteines were reduced and alkylated, and proteins were subjected to tryptic digestion. The samples were analyzed by targeted and discovery MS to look for tryptic peptides derived uniquely from the transfected RNA strings (Figure 10, panel B). The indicated peptide (NHVNFFQELADLDR) was synthesized as a heavy peptide and its counterparts could be detected in the total trypsinated lysate from transfected with EspA string 2 in the presence of MG132 proteasome inhibitor. No expression was detected from non-transfected cells independently of the presence of MG132. These data confirm expression of EspA string 2 after overnight expression and the proteasome inhibitor data suggest that this string is efficiently turned over and degraded in normal conditions. Example 6: Structure-guided T-cell string approach allows processing and presentation of predicted HLA-epitopes RNA drug substance (EspA_String2) was transfected into A375 cells stably expressing an affinity-tagged version of HLA-B*07:02. Following lysis, tagged HLA molecules were labeled with biotin and enriched on NeutrAvidin beads. Following washing, peptides were eluted from HLA complexes using acid and filtered through molecular weight cutoff filters. Cysteines were reduced and alkylated, and peptides were cleaned up by C18 desalting. Heavy isotope- labeled synthetic peptides were spiked into each sample, and targeted and discovery MS was performed to look for epitope peptides derived uniquely from transfected mRNA constructs. The different detected peptides are depicted in the visual representation of the T cell string as well as in the table (Figure 11) depicting their exact sequence. Note that the detected epitopes were derived from different parts of the T-cell string indicating that the whole construct has the potential to be processed and presented on human HLA molecules. Example 7: T-cell string approach detoxifies EspA Cells were seeded and incubated as described above and transfected with RNA constructs encoding EspA_FL, EspA_A, EspA_B, EspA_C, EspA string1, EspA string2 or control protein 2 at a concentration of 250 ng/well. Wells 109 11529421v1 Attorney Docket No. 2013237‐0757 were stained with 1:4000 diluted Incucyte® Cytotox Red Dye for Counting Dead Cells. Negative controls consisted of non-transfected cells. The plate was placed in the Incucyte® S3 Live-Cell Analysis System directly post transfection. Wells were imaged in bright field and fluorescence every hour for 24h. All analyses were performed identically to those described in example 3. As before, the EspA_FL and EspA_A proteins were cytotoxic to the cells (Figure 12, panel A). However, the Cytotox+ of the two strings was significantly lower than EspA itself (p<0.05) and EspA_String2 is even lower than control protein 2 (p<0.001). The confluency of EspA string1 matched that of control protein 2 (p>0.05), while that of EspA string2 was more comparable with the non-transfected control (p<0.01 when compared to control protein 2) (Figure 12, panel B). Both show better confluence than EspA (p<0.05). These data indicate that both string designs, but especially the structure-guided approach used for EspA_String2, are highly effective in detoxifying EspA. Example 8: T-cell strings of EspA are immunogenic in C57BL/6 mice and leads to polyfunctional T- cell responses Adult female C57BL/6 mice were immunized with 4 µg of EspA String1 or String2 (structure-guided detoxification) RNA constructs formulated in a lipid nanoparticle formulation (in 20 µl of volume) or with 0.9% NaCl solution as a control. 21 days after the primary immunization the mice were boosted with the same dose. 14 days after the boosting dose (35 days post primary immunization) mice were sacrificed and splenocytes were isolated. In addition, a portion of the splenocytes was pooled and separated into CD4+ and CD8+ fractions by magnetic associated cell- sorting (MACS). Both the total splenocytes and the CD4+ and CD8+ fractions were assessed for T-cell reactogenicity by restimulation with peptide pools specific for individual chunks (or parts of chunks) of the T-cell string. The peptide libraries consist of 15 amino acid long peptides with a 5 amino acid interval. In other words, the first peptide will cover amino acid 1-15 of the antigen and the second peptide will cover amino acids 16-20, and so forth. Fluorospot was used as a read-out to determine IFN-γ, IL-2 and TNF-α production. The peptides libraries were not immunogenic above baseline (Figure 13). In both EspA String1 and String2, immunogenic regions were identified especially with peptide libraries of EspA_7.2 and 7.3 which correspond to the sequences of Chunk EspA_A which was found to be cytotoxic when expressed in itself (Figure 14). Some responses above baseline could be detected in response to peptide library EspA_7.5, 7.6 and 7.7, but these were all markedly lower than the responses detected toward 7.2 and 7.3. Separation of splenocytes into CD4+ and CD8+ T cells confirmed the results above and clearly indicated that EspA_7.2 includes both CD4+ and CD8+ epitopes while the responses detected to EspA_7.3 are determined by CD8+ responses (Figure 15). These data also confirmed the immunogenicity of EspA_7.5-7.7 and indicated that these relatively low levels of immune responses were primarily mediated by CD4+ T cells. CD4+ and CD8+ T cells can also be subdivided into different cytokine-secreting classes, called polyfunctional cells. Combinations of two or three cytokines were identified as well (Figure 16). For both strings, CD4+ T cells mostly secreted IL-2 in combination with one of the other two cytokines, whereas some also secreted all three. In contrast, CD8+ T cells mostly secreted IFN-γ and TNF-α (which is known for CD8+ T cells). Additionally, the cells from both string-immunized study groups induced a strong immune response against EspA_7.2 and EspA_7.3 as seen before (Figure 15). EspA_7.2 induced strong IFN-γ+ IL-2+ TNF-α+ CD4+ T cell responses, but no such responses were 110 11529421v1 Attorney Docket No. 2013237‐0757 observed for EspA_7.3. In contrast, the IFN-γ+ TNF-α+ CD8+ T cell responses were higher for EspA_7.3 compared to EspA_7.2. Example 9: Polyfunctional T cell responses Together, these data show that EspA_A (The N-terminal 166 amino acids) contains the most immunogenic regions of EspA. Therefore, classic truncation of the protein to only detect non-toxic section of the antigen would also result in a predicted loss of immunogenicity of EspA. In contrast, the T-cell string approach has successfully reduced toxicity of EspA_A in vitro and is able to induce strong T-cell responses. Finally, structure-guided detoxification led to further reduction of in vitro toxicity and increased in vitro expression levels as detected in examples 3 and 4 respectively, but also increased immunogenicity. Especially as measured by polyfunctional T-cell responses, which in murine studies are considered to be advantageous in the protection against TB infection. Confirming that the T-cell string approach was successful and specific in inducing immunogenicity of multiple different antigens simultaneously. Analysis of MACS´ed cells reveals that T Cell strings can induce potent CD4+ and CD8+ responses. 111 11529421v1
 by using light scattering. In particular, for small scattering vectors ^^^ the structure function S is defined as follows: ^ ∙ ^1 െ^^ଶ ∙ ଶ ^^^ ^^^ ^ ^^^^ 3 ^ wherein N is the number of components
by using light scattering. In particular, for small scattering vectors ^^^ the structure function S is defined as follows: ^ ∙ ^1 െ^^ଶ ∙ ଶ ^^^ ^^^ ^ ^^^^ 3 ^ wherein N is the number of components The "hydrodynamic radius" (which is sometimes called "Stokes radius" or "Stokes-Einstein radius") of a particle is the radius of a hypothetical hard sphere that diffuses at the same rate as said particle. The hydrodynamic radius is related to the mobility of the particle, taking into account not only size but also solvent effects. For example, a smaller charged particle with stronger hydration may have a greater hydrodynamic radius than a larger charged particle with weaker hydration. This is because the smaller particle drags a greater number of water molecules with it as it moves through the solution. Since the actual dimensions of the particle in a solvent are not directly measurable, the hydrodynamic radius may be defined by the Stokes-Einstein equation: ^^^ ∙ ^^ ^^^ ൌ 6 ∙ ^^ ∙ ^^ ∙ ^^ wherein kB is the Boltzmann constant; T is the temperature; η is the viscosity of the solvent; and D is the diffusion coefficient. The diffusion coefficient can be determined experimentally, e.g., by using dynamic light scattering (DLS). 35 11529421v1 Attorney Docket No. 2013237‐0757 Thus, one procedure to determine the hydrodynamic radius of a particle or a population of particles (such as the hydrodynamic radius of particles contained in a sample or control composition as disclosed herein or the hydrodynamic radius of a particle peak obtained from subjecting such a sample or control composition to field-flow fractionation) is to measure the DLS signal of said particle or population of particles (such as DLS signal of particles contained in a sample or control composition as disclosed herein or the DLS signal of a particle peak obtained from subjecting such a sample or control composition to field-flow fractionation). The expression "light scattering" as used herein refers to the physical process where light is forced to deviate from a straight trajectory by one or more paths due to localized non-uniformities in the medium through which the light passes. The term "UV" means ultraviolet and designates a band of the electromagnetic spectrum with a wavelength from 10 nm to 400 nm, i.e., shorter than that of visible light but longer than X-rays. The expression "multi-angle light scattering" or "MALS" as used herein relates to a technique for measuring the light scattered by a sample into a plurality of angles. "Multi-angle" means in this respect that scattered light can be detected at different discrete angles as measured, for example, by a single detector moved over a range including the specific angles selected or an array of detectors fixed at specific angular locations. In certain embodiments, the light source used in MALS is a laser source (MALLS: multi-angle laser light scattering). Based on the MALS signal of a composition comprising particles and by using an appropriate formalism (e.g., Zimm plot, Berry plot, or Debye plot), it is possible to determine the radius of gyration (Rg) and, thus, the size of said particles. Preferably, the Zimm plot is a graphical presentation using the following equation: ^^ఏ ൌ ^^ ^^^ ^^^ െ 2 ^ଶ ଶ ^^∗ ^^ ௪ ^ଶ ^^ ^^௪ ^^ ^ ^^^ wherein c is the mass concentration of2
The "hydrodynamic radius" (which is sometimes called "Stokes radius" or "Stokes-Einstein radius") of a particle is the radius of a hypothetical hard sphere that diffuses at the same rate as said particle. The hydrodynamic radius is related to the mobility of the particle, taking into account not only size but also solvent effects. For example, a smaller charged particle with stronger hydration may have a greater hydrodynamic radius than a larger charged particle with weaker hydration. This is because the smaller particle drags a greater number of water molecules with it as it moves through the solution. Since the actual dimensions of the particle in a solvent are not directly measurable, the hydrodynamic radius may be defined by the Stokes-Einstein equation: ^^^ ∙ ^^ ^^^ ൌ 6 ∙ ^^ ∙ ^^ ∙ ^^ wherein kB is the Boltzmann constant; T is the temperature; η is the viscosity of the solvent; and D is the diffusion coefficient. The diffusion coefficient can be determined experimentally, e.g., by using dynamic light scattering (DLS). 35 11529421v1 Attorney Docket No. 2013237‐0757 Thus, one procedure to determine the hydrodynamic radius of a particle or a population of particles (such as the hydrodynamic radius of particles contained in a sample or control composition as disclosed herein or the hydrodynamic radius of a particle peak obtained from subjecting such a sample or control composition to field-flow fractionation) is to measure the DLS signal of said particle or population of particles (such as DLS signal of particles contained in a sample or control composition as disclosed herein or the DLS signal of a particle peak obtained from subjecting such a sample or control composition to field-flow fractionation). The expression "light scattering" as used herein refers to the physical process where light is forced to deviate from a straight trajectory by one or more paths due to localized non-uniformities in the medium through which the light passes. The term "UV" means ultraviolet and designates a band of the electromagnetic spectrum with a wavelength from 10 nm to 400 nm, i.e., shorter than that of visible light but longer than X-rays. The expression "multi-angle light scattering" or "MALS" as used herein relates to a technique for measuring the light scattered by a sample into a plurality of angles. "Multi-angle" means in this respect that scattered light can be detected at different discrete angles as measured, for example, by a single detector moved over a range including the specific angles selected or an array of detectors fixed at specific angular locations. In certain embodiments, the light source used in MALS is a laser source (MALLS: multi-angle laser light scattering). Based on the MALS signal of a composition comprising particles and by using an appropriate formalism (e.g., Zimm plot, Berry plot, or Debye plot), it is possible to determine the radius of gyration (Rg) and, thus, the size of said particles. Preferably, the Zimm plot is a graphical presentation using the following equation: ^^ఏ ൌ ^^ ^^^ ^^^ െ 2 ^ଶ ଶ ^^∗ ^^ ௪ ^ଶ ^^ ^^௪ ^^ ^ ^^^ wherein c is the mass concentration of2 A2 is the second virial coefficient (mol∙mL/g ); P(θ) is a form factor relating to the dependence of scattered light intensity on angle; Rθ is the excess Rayleigh ratio (cm-1); and K* is an optical constant that is equal to 4π2ηo (dn/dc)2λ0-4NA-1, where ηo is the refractive index of the solvent at the incident radiation (vacuum) wavelength, λ0 is the incident radiation (vacuum) wavelength (nm), NA is Avogadro’s number (mol-1), and dn/dc is the differential refractive index increment (mL/g) (cf., e.g., Buchholz et al. (Electrophoresis 22 (2001), 4118-4128); B.H. Zimm (J. Chem. Phys.13 (1945), 141; P. Debye (J. Appl. Phys.15 (1944): 338; and W. Burchard (Anal. Chem.75 (2003), 4279-4291). Preferably, the Berry plot is calculated using the following term or the reciprocal thereof: ^ ^^ఏ ^^∗ ^^ wherein c, Rθ and K* are as defined above.
A2 is the second virial coefficient (mol∙mL/g ); P(θ) is a form factor relating to the dependence of scattered light intensity on angle; Rθ is the excess Rayleigh ratio (cm-1); and K* is an optical constant that is equal to 4π2ηo (dn/dc)2λ0-4NA-1, where ηo is the refractive index of the solvent at the incident radiation (vacuum) wavelength, λ0 is the incident radiation (vacuum) wavelength (nm), NA is Avogadro’s number (mol-1), and dn/dc is the differential refractive index increment (mL/g) (cf., e.g., Buchholz et al. (Electrophoresis 22 (2001), 4118-4128); B.H. Zimm (J. Chem. Phys.13 (1945), 141; P. Debye (J. Appl. Phys.15 (1944): 338; and W. Burchard (Anal. Chem.75 (2003), 4279-4291). Preferably, the Berry plot is calculated using the following term or the reciprocal thereof: ^ ^^ఏ ^^∗ ^^ wherein c, Rθ and K* are as defined above. Debye plot is calculated using the following term or the reciprocal thereof: ^^∗ ^^ ^^ఏ wherein c, Rθ and K* are as defined above.
Debye plot is calculated using the following term or the reciprocal thereof: ^^∗ ^^ ^^ఏ wherein c, Rθ and K* are as defined above. The "D1 diastereomer of beta-S-ARCA" or "beta-S-ARCA(D1)" is the diastereomer of beta-S-ARCA which elutes first on an HPLC column compared to the D2 diastereomer of beta-S-ARCA (beta-S-ARCA(D2)) and thus exhibits a shorter retention time. The HPLC preferably is an analytical HPLC. In some embodiments, a Supelcosil LC-18-T RP column, preferably of the format: 5 μm, 4.6 x 250 mm is used for separation, whereby a flow rate of 1.3 ml/min can be applied. In some embodiments, a gradient of methanol in ammonium acetate, for example, a 0-25% linear gradient of methanol in 0.05 M ammonium acetate, pH = 5.9, within 15 min is used. UV-detection (VWD) can be performed at 260 nm and fluorescence detection (FLD) can be performed with excitation at 280 nm and detection at 337 nm. The 5'-cap analog m27,3'-OGppp(m12'-O)ApG (also referred to as m27,3'OG(5')ppp(5')m2'-OApG) which is a building block of a cap1 has the following structure: CH3 H O ONH2 2 . An
The "D1 diastereomer of beta-S-ARCA" or "beta-S-ARCA(D1)" is the diastereomer of beta-S-ARCA which elutes first on an HPLC column compared to the D2 diastereomer of beta-S-ARCA (beta-S-ARCA(D2)) and thus exhibits a shorter retention time. The HPLC preferably is an analytical HPLC. In some embodiments, a Supelcosil LC-18-T RP column, preferably of the format: 5 μm, 4.6 x 250 mm is used for separation, whereby a flow rate of 1.3 ml/min can be applied. In some embodiments, a gradient of methanol in ammonium acetate, for example, a 0-25% linear gradient of methanol in 0.05 M ammonium acetate, pH = 5.9, within 15 min is used. UV-detection (VWD) can be performed at 260 nm and fluorescence detection (FLD) can be performed with excitation at 280 nm and detection at 337 nm. The 5'-cap analog m27,3'-OGppp(m12'-O)ApG (also referred to as m27,3'OG(5')ppp(5')m2'-OApG) which is a building block of a cap1 has the following structure: CH3 H O ONH2 2 . An 42 11529421v1 Attorney Docket No. 2013237‐0757 H3C O OH O 2 .
42 11529421v1 Attorney Docket No. 2013237‐0757 H3C O OH O 2 . An m2 ppp structure: CH3 H OO O 2 .
An m2 ppp structure: CH3 H OO O 2 . An exemplary cap1 mRNA comprising m27,3'-OGppp(m12'-O)ApG and mRNA has the following structure: CH3 H O ONH2 2 .
An exemplary cap1 mRNA comprising m27,3'-OGppp(m12'-O)ApG and mRNA has the following structure: CH3 H O ONH2 2 . IS C V
IS C V NtD II SeHGD MDCAL V L AAFTD DAP THQDCDNPQ RkI DcLoSAVGA GE QI IV TNSTV G V VAA QGDL QI FT S RI VPDI PQF F R FELL PL PLKPPNP LP FT PWE HyLDS GeQRIIAA A LREPT SDP D GP H TYF HD PTKTD DAGPDL AALAPT SLT DIP NPSP PT AL A QT Nnro D DttL AAPAALA YQGP N D L VL P VNP DL FYKGEPPAD I AALS DE NF HRP F D F PR GR GV PGHIFSP ASP L AAS TI A G GPAT TSR R NHA T YD A D VPIG VT D E A QV S F L AA NI TD Q V PP NT D YFGV FFF N ASDL P PYP P F E L AA SS QR L NAGT V LS D GVPPSL GVT G GFT L RG NTVAA E LT TYNHTAT GMT NNS T A T GYRSH DF N HK AA NLHSY GYNS DF F NFYV NL E D M FYY GLSF RLA FFA L K F NK K YGIF MPGFP AHV A GY NDH GDM V T GST KLGH S TDTLT NGSD N TF GLL VIVFD G S VNHM GL T AQST FSP I NS V F EM RQL K NLT AAR PT L GE D G YTA KAS G DNL K R NYPR GDYD NEPS QT KDN RE NAGD LYT AIL GS A E GL GSVGV QF Y P VEWQL VS LA KIV IS AT AAG Q S KGS MA R KILV GML V KWV A TCL RT S LA AR F S LLT N T QATDRLVQAWS AP L NVI R NYT KW GV L PV AYK I PV P HAVE P L V GHI VEWHLGNV E QKT WLY GATM VGHQ EI AK P WWA V TEI D WAS ND L E L P I VI DA TP WWL LSI VN AP DLQGDLS AMTD LR GLPV GA PGS FGL M G TL WV PN L NGNP HN ANLAFR HW GGANP V AV G AV S V T P HDASL VPG A G GP NDP GEPD ARGSD NDS RLL GF L AA AE LI V GCPG NMIVIAILCPAL NVR K E MGGC MP GL NRL FR RGW V KLGAG GQR PIL G E AT GGE I LAGLGANP A AG T AA GS W NSA GTSCLGYNMNL KP QS GWMIV RIS Y AHY HVT HGSAWI SR VI QL EC FFVG Y AALRMS L KCL KMN GQVD PI KEL WT V QS YP EAG AQ LQF A K S EHW IVL MSF AQG EI ILPPF FT GI V HATF AT GS SP IP WS QIAD R TPAITLGV HCI SFFS V SA YAE SLL VPG QAVQLT HTGP AHNHHV PAAWKADF GS SAWQL A P VP D G F RLSDP6 PEE IS MA LTE WQP VP RPI8 IHQI F D L LNQWQLLT V D LLD S HP GAS WGDHE FAD PT MFNVLF P GQTTL DPAP P VRA GH GGG MP GP P HSV TEA GVCP GGP R P VD EVHE ML P QV RV L SQ F D NEV ES MQ AE GL SYMA SAN V I SE GGS sNPVP FE AAVLQLPT TT G AD TVPS QAC QP V GR E neI IYLA A ACE QLIGP MAV GWSIVPF G AE GGTS QV gG iI TGTA GGP GNR tG nLL VGVA GDGPS VAL APD VYQ AGPV L PGV GL DKPCPV R AAGQP YP P D VV S TT aLD W LD LP P KR I L V YN I le VP T ac DYAGTG L F KP T GWSIYPAF TT WR KP G NRFPI AL GD inLVPLDA] T G R ASV VIV RFDPWV Q SFCPY FASNGAF GNV P P beu GRK DR 1 : LF T PKC YQRRMR L TK ARTT V QPV V orq DIV ci es AF P SI E W LL ROE T DI PPVPI E L A NR RDRY SL NL R ASRR S] TP WV G YD IPT V]RSNITALR NF PS TFR GYP ]6 L 2 ML TLAAGT3 MRYI NL L4 m dTGKQD IFL AGL A: GVR F RIGL: GP WNIGIGE: ci ic PK xa D oI KLR AG NTQ M E GP L ITWYRVGLGO D DN V GN P APD TL GVO AL P GAN FATY IL V P R D GNO LN to I n FAGEL S AASLV L D P T D A[ V PS GR A AGT IHP E DT YS N D VGT GGG PP V IQTNR P IR FIF D GGL I otiR LINHVAAPNFRIGLQ SAGPV CQ S P MD GVQ ymSDTI V NSDPL D GVESGQLLSI S QE V DL T WS VE cA MR F Q R ML T G G V [ MS S f T S G D P [ MA G P G N [ o sec n eu) qsi)s esi)si)si sol s u os s lo o ulul dic cr ae crcu rcr be e e ou n tbubub i t tut m m m auiru mu mu e iririr e vtetetet icacacac taba Noc bob b 1 : cococv 4ny eMy y y1 (M M M2 eg 4 l i b tA ( ( ( pA9 a n scl Bcl Ccl 25 T A E P P P117570‐7M3V2
NtD II SeHGD MDCAL V L AAFTD DAP THQDCDNPQ RkI DcLoSAVGA GE QI IV TNSTV G V VAA QGDL QI FT S RI VPDI PQF F R FELL PL PLKPPNP LP FT PWE HyLDS GeQRIIAA A LREPT SDP D GP H TYF HD PTKTD DAGPDL AALAPT SLT DIP NPSP PT AL A QT Nnro D DttL AAPAALA YQGP N D L VL P VNP DL FYKGEPPAD I AALS DE NF HRP F D F PR GR GV PGHIFSP ASP L AAS TI A G GPAT TSR R NHA T YD A D VPIG VT D E A QV S F L AA NI TD Q V PP NT D YFGV FFF N ASDL P PYP P F E L AA SS QR L NAGT V LS D GVPPSL GVT G GFT L RG NTVAA E LT TYNHTAT GMT NNS T A T GYRSH DF N HK AA NLHSY GYNS DF F NFYV NL E D M FYY GLSF RLA FFA L K F NK K YGIF MPGFP AHV A GY NDH GDM V T GST KLGH S TDTLT NGSD N TF GLL VIVFD G S VNHM GL T AQST FSP I NS V F EM RQL K NLT AAR PT L GE D G YTA KAS G DNL K R NYPR GDYD NEPS QT KDN RE NAGD LYT AIL GS A E GL GSVGV QF Y P VEWQL VS LA KIV IS AT AAG Q S KGS MA R KILV GML V KWV A TCL RT S LA AR F S LLT N T QATDRLVQAWS AP L NVI R NYT KW GV L PV AYK I PV P HAVE P L V GHI VEWHLGNV E QKT WLY GATM VGHQ EI AK P WWA V TEI D WAS ND L E L P I VI DA TP WWL LSI VN AP DLQGDLS AMTD LR GLPV GA PGS FGL M G TL WV PN L NGNP HN ANLAFR HW GGANP V AV G AV S V T P HDASL VPG A G GP NDP GEPD ARGSD NDS RLL GF L AA AE LI V GCPG NMIVIAILCPAL NVR K E MGGC MP GL NRL FR RGW V KLGAG GQR PIL G E AT GGE I LAGLGANP A AG T AA GS W NSA GTSCLGYNMNL KP QS GWMIV RIS Y AHY HVT HGSAWI SR VI QL EC FFVG Y AALRMS L KCL KMN GQVD PI KEL WT V QS YP EAG AQ LQF A K S EHW IVL MSF AQG EI ILPPF FT GI V HATF AT GS SP IP WS QIAD R TPAITLGV HCI SFFS V SA YAE SLL VPG QAVQLT HTGP AHNHHV PAAWKADF GS SAWQL A P VP D G F RLSDP6 PEE IS MA LTE WQP VP RPI8 IHQI F D L LNQWQLLT V D LLD S HP GAS WGDHE FAD PT MFNVLF P GQTTL DPAP P VRA GH GGG MP GP P HSV TEA GVCP GGP R P VD EVHE ML P QV RV L SQ F D NEV ES MQ AE GL SYMA SAN V I SE GGS sNPVP FE AAVLQLPT TT G AD TVPS QAC QP V GR E neI IYLA A ACE QLIGP MAV GWSIVPF G AE GGTS QV gG iI TGTA GGP GNR tG nLL VGVA GDGPS VAL APD VYQ AGPV L PGV GL DKPCPV R AAGQP YP P D VV S TT aLD W LD LP P KR I L V YN I le VP T ac DYAGTG L F KP T GWSIYPAF TT WR KP G NRFPI AL GD inLVPLDA] T G R ASV VIV RFDPWV Q SFCPY FASNGAF GNV P P beu GRK DR 1 : LF T PKC YQRRMR L TK ARTT V QPV V orq DIV ci es AF P SI E W LL ROE T DI PPVPI E L A NR RDRY SL NL R ASRR S] TP WV G YD IPT V]RSNITALR NF PS TFR GYP ]6 L 2 ML TLAAGT3 MRYI NL L4 m dTGKQD IFL AGL A: GVR F RIGL: GP WNIGIGE: ci ic PK xa D oI KLR AG NTQ M E GP L ITWYRVGLGO D DN V GN P APD TL GVO AL P GAN FATY IL V P R D GNO LN to I n FAGEL S AASLV L D P T D A[ V PS GR A AGT IHP E DT YS N D VGT GGG PP V IQTNR P IR FIF D GGL I otiR LINHVAAPNFRIGLQ SAGPV CQ S P MD GVQ ymSDTI V NSDPL D GVESGQLLSI S QE V DL T WS VE cA MR F Q R ML T G G V [ MS S f T S G D P [ MA G P G N [ o sec n eu) qsi)s esi)si)si sol s u os s lo o ulul dic cr ae crcu rcr be e e ou n tbubub i t tut m m m auiru mu mu e iririr e vtetetet icacacac taba Noc bob b 1 : cococv 4ny eMy y y1 (M M M2 eg 4 l i b tA ( ( ( pA9 a n scl Bcl Ccl 25 T A E P P P117570‐7M3V2 APAGLI GQE
APAGLI GQE 31A02 P CF V KGRD R. NYV GQQP AG Q V AT LT T ANQGNG LEIo WR F D R I VTGAE DR F QGNKR YS WHSINt GDC V DS De KV L PV L AL C Q SPWL QD NL SP Q QQYESk QT DTH R P K QPQAIA GQLAE GVcoQFASR VYL GRV LYTI GL GAHI S AGNDIDFyLPEPP T I V AGTGeP DnS ArV LDGYAV R GGDI V YS LAS P V T D AI A ID HGI TYG HAS L GR Got TYQGL N AAtANF HIKF A GP P P F ADS S RGR L GS R RS QE LF L AL PL EL LAGF GLL MT T DIS T A TD TWS EL SAGLKL RMVLRV KAE IT QVL APS N A GC GESGL GVETSI GAA QD G GSR LTS GV PP VI A L VAWQA D T L WTSYQ GAL TYYV GY HVKL AR H DIRS G QAL EI RSP AFDL FGTL D AF HAPL V MISH YMVEI GDI AAE G D S G SFF NF GVTAA DVEG GFSL GVIR P R AR H DV S GT R EFV RF HVM FT SPI GSALPQPAR PSER RTP NST VLS GRSQ HGLE R VM R RAG D GLGP L R D M WIFKV AL GAGGP LLARI YDR L NEAT DAS SARLNRQS D QASKY NSVGR L TELAIYVAGEQ SDAAGL GKELS DGL VL AETS E MR F TRYILVSD VR DR ALATSQERA F KV AT L L YQP TRT RVVLTAL AT DALS YQ LAGF GK V AK F QWKWVTDQTL GSGGVEL GLS HEA DGS HEA IP DLSE VWA LVRR LF GVTLATGLE GSR D DGSE GRL V HR D G TW W KRL K V HR KAL GGV T G LNAAPNPT DILFPA GL G VEPP AG D D QT HD DSNAVPSAEYA DD G D KKSEGA S LGE AL GNLV VNL V APT EVQGS ALGE AQRAAKKV D S LIG T LNRAE L QLL CGQ F AG QA V D AR D AA PGP R G MLI R P R GSAL ANLR K D ALALT VPEF DL WIRNTEKVA NP GQS HASNF DQH D E GAL GS VL ATPQALQK DG Y WIL PLAEVLAAKRE AE KHGAF RLLREPER ARSD GGE QRL VEGG H EI MF RAQCG DL V SA SPLV V AEGSPRE SQ ETG P V RG VAS TP I RVKTGESRT HT AAVGSR7 G A G VVAPE G VTAR WP HS V RGV T GSL FE8 ASQA F P DFPIKPVQL VARF QQRTAHRTGL SHF DTVT GG ADS MLPPA NH R AT AQT E GAL LK LVT SP D R TGIRWEILERLK V LE VES AVAE AD GNQHH AVLE V P L ES MTTE L ASGQS L AV A AQAVKQEE WLE GL LAKVQE EE GLK GDAV GCAPELQP E A QAE LQR DSALR L GMP VGR R GGG GV GQ T QQAR GSL GMQV GR G AGASKT A eADN cVLNPR FQIS P R KPVYMIS GRAK R YLV QSR GA V AT V LTHLEV G QT HKEV A QLF GGPQAQ DWPS QPAL V AL HML FGSLVAAS G n eA uLFRT PSI V D GDAV L PLQ DA AQE LE AKI MDK T DI V RF P VRAS E R V ATISDAQE R GA q E e R DTPCP R] RV VA ] AQEMS] VF L L R SL ] s RSFA RS L L RVRY7 PL VR 8 FSQL R GA I 4 :E H AL 4 :P V R QS 9 L 4 S MQD :P MEPILL V05 diV Y cGP WN aG YAF LGLG ROA ND VAQ GPOS MLEDI L ONQS AR: O DNQQP L VRNQKI AQM N o I M n HTL V VR DGIN R R i S PITD IAD H Q VD IL KS S FVAK Q GM Q DD IS E S S ALG VAD DT GD I QGN TPA LAFGGLQR L R LVV GQQI VDLVATEQQIAS HLEK Q mS R E A E HA D E HL Q D E A MA DTPSAI GAAS[ METGPS[ MSSL QLEK NS[ MSASAR GYLS[ )si) ssi os ) ) lo a ul sas cu o rc erno ini be g ub ugu tu trer aea mu ms s iruia a ern te no o ct ac m ba omo ob d d 1 n co u uev e yc g Mye MsPsP 1 (2 i ( ( (4 t n DcAySoTo 92 AlPlTxExE 5117570‐7LK3EKLQPL LYF L GSI Q HDGL P2T S RT
31A02 P CF V KGRD R. NYV GQQP AG Q V AT LT T ANQGNG LEIo WR F D R I VTGAE DR F QGNKR YS WHSINt GDC V DS De KV L PV L AL C Q SPWL QD NL SP Q QQYESk QT DTH R P K QPQAIA GQLAE GVcoQFASR VYL GRV LYTI GL GAHI S AGNDIDFyLPEPP T I V AGTGeP DnS ArV LDGYAV R GGDI V YS LAS P V T D AI A ID HGI TYG HAS L GR Got TYQGL N AAtANF HIKF A GP P P F ADS S RGR L GS R RS QE LF L AL PL EL LAGF GLL MT T DIS T A TD TWS EL SAGLKL RMVLRV KAE IT QVL APS N A GC GESGL GVETSI GAA QD G GSR LTS GV PP VI A L VAWQA D T L WTSYQ GAL TYYV GY HVKL AR H DIRS G QAL EI RSP AFDL FGTL D AF HAPL V MISH YMVEI GDI AAE G D S G SFF NF GVTAA DVEG GFSL GVIR P R AR H DV S GT R EFV RF HVM FT SPI GSALPQPAR PSER RTP NST VLS GRSQ HGLE R VM R RAG D GLGP L R D M WIFKV AL GAGGP LLARI YDR L NEAT DAS SARLNRQS D QASKY NSVGR L TELAIYVAGEQ SDAAGL GKELS DGL VL AETS E MR F TRYILVSD VR DR ALATSQERA F KV AT L L YQP TRT RVVLTAL AT DALS YQ LAGF GK V AK F QWKWVTDQTL GSGGVEL GLS HEA DGS HEA IP DLSE VWA LVRR LF GVTLATGLE GSR D DGSE GRL V HR D G TW W KRL K V HR KAL GGV T G LNAAPNPT DILFPA GL G VEPP AG D D QT HD DSNAVPSAEYA DD G D KKSEGA S LGE AL GNLV VNL V APT EVQGS ALGE AQRAAKKV D S LIG T LNRAE L QLL CGQ F AG QA V D AR D AA PGP R G MLI R P R GSAL ANLR K D ALALT VPEF DL WIRNTEKVA NP GQS HASNF DQH D E GAL GS VL ATPQALQK DG Y WIL PLAEVLAAKRE AE KHGAF RLLREPER ARSD GGE QRL VEGG H EI MF RAQCG DL V SA SPLV V AEGSPRE SQ ETG P V RG VAS TP I RVKTGESRT HT AAVGSR7 G A G VVAPE G VTAR WP HS V RGV T GSL FE8 ASQA F P DFPIKPVQL VARF QQRTAHRTGL SHF DTVT GG ADS MLPPA NH R AT AQT E GAL LK LVT SP D R TGIRWEILERLK V LE VES AVAE AD GNQHH AVLE V P L ES MTTE L ASGQS L AV A AQAVKQEE WLE GL LAKVQE EE GLK GDAV GCAPELQP E A QAE LQR DSALR L GMP VGR R GGG GV GQ T QQAR GSL GMQV GR G AGASKT A eADN cVLNPR FQIS P R KPVYMIS GRAK R YLV QSR GA V AT V LTHLEV G QT HKEV A QLF GGPQAQ DWPS QPAL V AL HML FGSLVAAS G n eA uLFRT PSI V D GDAV L PLQ DA AQE LE AKI MDK T DI V RF P VRAS E R V ATISDAQE R GA q E e R DTPCP R] RV VA ] AQEMS] VF L L R SL ] s RSFA RS L L RVRY7 PL VR 8 FSQL R GA I 4 :E H AL 4 :P V R QS 9 L 4 S MQD :P MEPILL V05 diV Y cGP WN aG YAF LGLG ROA ND VAQ GPOS MLEDI L ONQS AR: O DNQQP L VRNQKI AQM N o I M n HTL V VR DGIN R R i S PITD IAD H Q VD IL KS S FVAK Q GM Q DD IS E S S ALG VAD DT GD I QGN TPA LAFGGLQR L R LVV GQQI VDLVATEQQIAS HLEK Q mS R E A E HA D E HL Q D E A MA DTPSAI GAAS[ METGPS[ MSSL QLEK NS[ MSASAR GYLS[ )si) ssi os ) ) lo a ul sas cu o rc erno ini be g ub ugu tu trer aea mu ms s iruia a ern te no o ct ac m ba omo ob d d 1 n co u uev e yc g Mye MsPsP 1 (2 i ( ( (4 t n DcAySoTo 92 AlPlTxExE 5117570‐7LK3EKLQPL LYF L GSI Q HDGL P2T S RT TDAANGR GFG G
TDAANGR GFG G 1G0 GER EERL ID PTT L IE2NI.EoFEFD S MPDLDL V EAFI NS P QAAGS QQEE K DNQS AASAL QTTAT T MDL AMLS RAAP G L GtR IeLkc THT IQGV TE FTQSSDR AG NEV NGFEL TI SA IG H AHD LPNL GM HDI SEGE REFT A D MoQFL NDR LE REV NKE GR CAS GAL HPF SEGL GyPKL TGE SeADFKS PSP KLAR HL AAH YRLDTn GKKKIFE QA HRLLr AHAN S NQLG A DVF RAGPLVV S PLILRNF LGQVA D NFTHS L T VFP HGottDATLS I TD YL NKEERD L AV Q VD G GAIV L AVYGQ I SD LAA M HASCI AS R LEH ELD R TP L R GLR D S S LN RR ELY E IL DR GWNF GSHF V AL KI ARA QE EV A GD LLPV ALDVAR GM MSK ADL E E P L E TS QMD GRE R IQLE YL E DPRR HL DQG HQR A LV GKGS QP D FQ TD L PTNL GSI HEL VE D KEHA ARAE W S SL VF NRE D T WR ST G Q NYG RV G QGP IAQLP AN H KSKQLAL NISSL GGKR GME N GE GL RALG F GDI AKD L Q NIALT M RF VAS MH AD EGP C F T TLD EL V DAQE KL A SP R P KE D NF DD YI VV R GT G TN K VSLRPVIE QSTVSQ R L LAI PS Y SEMSAP D IGS WR K PL PLAA F GAEA DAEAP MKG PG P N QYFL D N LSIV ANMS HR KF VLQFDVHS GE ST GERGP KALAI MTR F ML QTPEYQS S LA QWL G RPL PPH GSLVT V NA LA IAP G GLTEE F GRKRTPRM DE A E GR LR NT PS GAD A D NT SS KSLK R SF RPGPN V LT GTI QR D GL KR EIS P S LP T V R NLIV SI V Q TL PTGA SGVQFGL LRLTGI NN V LEN CQD GYVSAFT QG AE E MR GLA TV QAGW D G D HIRDVGRLY KK KLSP ASHT GS ALVI R TLRAPKL FQ QQDLDPCS ML VGTWE F N GSAIV D GSELMHAD VKAL NGL A LMGNDVP RGL AKA V DS ETF L V I NYWSFE NSD IVH GS QG GAR ASP QRI PR HCSN QRYL I LQES VIGRARI ATI GP PGWV GYYAKPKVAG T R GQEAF GS GV AYP KL AF P DVR V GFG NAE E L KQPHG D ALPR KN T TC V TYLT NDGC NG EGLYY LNADPNF FF QMRT DSV LES TAVEKINIP P R GELDAR D L S VE P ISQM T TP 8 FFQLSLCE WLVL LL VLL ESMY 8 SKE D RVQAI AEV R S L LAGFP VL D WCRL AQ D P D QQE L K KAQE R DREK V ND RKL F ANLV Q VD QL D NG HL RFLAR AMEL V AAPEGE P E KIA QVL A GSF ELSKPGSAYD G GEK L GQRNEMEL SKEAWGS Y GD G ARL AL P T R AYG QILK S S GACAP AV N D S QG S PAFSE P YLFAI VARY RD A ARWEQEGLV V ARS D T ARS GSQVAHE QV V FS A QS LAC P P D CEFT E ADI I HL A GVKVPQSI ATVC GRWQEE D GTAV ecV GKRAME DR VRNV GATR YKWG AAIPI ALS D nPE APL AR TI R QG V L D R E D LL SEGAYAS LPK euQGV Q qN AA QEP A GT RAFGYR R R RPVP FPI AE G eLG GNL VE LNEAQQDLRV LS VPQC G T RV SLS sSSSLLLEL AQ D PL ] V QA 1 AT 5 DF YS ] AG GE 25A VPKQRL NAHSWV IT ] R 3LG 5SL I ] L 45 dAV iT ca ALSSFPGAA : LGS E G:LP P QART V A GTLGTV VS A GHS VT LO D VT N QG D G HVO I E RN WV MF V VTEDDE :LK : ALA DLOS PNV T O AHAN o LP I AWLPS FE DL AMAE HEDHG GV L S QWDAI D n S QP KNGMS QK IR GI IPT E S VP AS G ILKV R I iI RSN GF KL D VN Q AKSPM Q L LGL HARAL Q RSQ mHL LV V A TR E QAV AA RE HRI ACG ARI TE LDE A MQ G L GDL Q WS[ MA V PS[ MT H A E V ES[ MDS[ ) ) a) ) sa a as ososo o nininini gug g g ru u ur erere e a a a a sas s sa na a on n no mo o om m mo do od ududu ue 1 n esev ePse Ps s P P (1 g ( ( (2 itU ni4 noY A r92 Axo Exo u ExEzA 5117570‐73
1G0 GER EERL ID PTT L IE2NI.EoFEFD S MPDLDL V EAFI NS P QAAGS QQEE K DNQS AASAL QTTAT T MDL AMLS RAAP G L GtR IeLkc THT IQGV TE FTQSSDR AG NEV NGFEL TI SA IG H AHD LPNL GM HDI SEGE REFT A D MoQFL NDR LE REV NKE GR CAS GAL HPF SEGL GyPKL TGE SeADFKS PSP KLAR HL AAH YRLDTn GKKKIFE QA HRLLr AHAN S NQLG A DVF RAGPLVV S PLILRNF LGQVA D NFTHS L T VFP HGottDATLS I TD YL NKEERD L AV Q VD G GAIV L AVYGQ I SD LAA M HASCI AS R LEH ELD R TP L R GLR D S S LN RR ELY E IL DR GWNF GSHF V AL KI ARA QE EV A GD LLPV ALDVAR GM MSK ADL E E P L E TS QMD GRE R IQLE YL E DPRR HL DQG HQR A LV GKGS QP D FQ TD L PTNL GSI HEL VE D KEHA ARAE W S SL VF NRE D T WR ST G Q NYG RV G QGP IAQLP AN H KSKQLAL NISSL GGKR GME N GE GL RALG F GDI AKD L Q NIALT M RF VAS MH AD EGP C F T TLD EL V DAQE KL A SP R P KE D NF DD YI VV R GT G TN K VSLRPVIE QSTVSQ R L LAI PS Y SEMSAP D IGS WR K PL PLAA F GAEA DAEAP MKG PG P N QYFL D N LSIV ANMS HR KF VLQFDVHS GE ST GERGP KALAI MTR F ML QTPEYQS S LA QWL G RPL PPH GSLVT V NA LA IAP G GLTEE F GRKRTPRM DE A E GR LR NT PS GAD A D NT SS KSLK R SF RPGPN V LT GTI QR D GL KR EIS P S LP T V R NLIV SI V Q TL PTGA SGVQFGL LRLTGI NN V LEN CQD GYVSAFT QG AE E MR GLA TV QAGW D G D HIRDVGRLY KK KLSP ASHT GS ALVI R TLRAPKL FQ QQDLDPCS ML VGTWE F N GSAIV D GSELMHAD VKAL NGL A LMGNDVP RGL AKA V DS ETF L V I NYWSFE NSD IVH GS QG GAR ASP QRI PR HCSN QRYL I LQES VIGRARI ATI GP PGWV GYYAKPKVAG T R GQEAF GS GV AYP KL AF P DVR V GFG NAE E L KQPHG D ALPR KN T TC V TYLT NDGC NG EGLYY LNADPNF FF QMRT DSV LES TAVEKINIP P R GELDAR D L S VE P ISQM T TP 8 FFQLSLCE WLVL LL VLL ESMY 8 SKE D RVQAI AEV R S L LAGFP VL D WCRL AQ D P D QQE L K KAQE R DREK V ND RKL F ANLV Q VD QL D NG HL RFLAR AMEL V AAPEGE P E KIA QVL A GSF ELSKPGSAYD G GEK L GQRNEMEL SKEAWGS Y GD G ARL AL P T R AYG QILK S S GACAP AV N D S QG S PAFSE P YLFAI VARY RD A ARWEQEGLV V ARS D T ARS GSQVAHE QV V FS A QS LAC P P D CEFT E ADI I HL A GVKVPQSI ATVC GRWQEE D GTAV ecV GKRAME DR VRNV GATR YKWG AAIPI ALS D nPE APL AR TI R QG V L D R E D LL SEGAYAS LPK euQGV Q qN AA QEP A GT RAFGYR R R RPVP FPI AE G eLG GNL VE LNEAQQDLRV LS VPQC G T RV SLS sSSSLLLEL AQ D PL ] V QA 1 AT 5 DF YS ] AG GE 25A VPKQRL NAHSWV IT ] R 3LG 5SL I ] L 45 dAV iT ca ALSSFPGAA : LGS E G:LP P QART V A GTLGTV VS A GHS VT LO D VT N QG D G HVO I E RN WV MF V VTEDDE :LK : ALA DLOS PNV T O AHAN o LP I AWLPS FE DL AMAE HEDHG GV L S QWDAI D n S QP KNGMS QK IR GI IPT E S VP AS G ILKV R I iI RSN GF KL D VN Q AKSPM Q L LGL HARAL Q RSQ mHL LV V A TR E QAV AA RE HRI ACG ARI TE LDE A MQ G L GDL Q WS[ MA V PS[ MT H A E V ES[ MDS[ ) ) a) ) sa a as ososo o nininini gug g g ru u ur erere e a a a a sas s sa na a on n no mo o om m mo do od ududu ue 1 n esev ePse Ps s P P (1 g ( ( (2 itU ni4 noY A r92 Axo Exo u ExEzA 5117570‐73 31 GQGRGLRD SDS R V TKTAAT DR VV LAS K02 A. QID R TREAHRT H R RAGAGGKAP T T RKKQY GDEDTo LQLGDVN AR GR RE K NA S QQAE A AQL EL LV A QY QD D N D S FI GDI YTAVQD SS Y R GSRR LS T T NAD GPDR F Q T YtekNVGcLoVAV EG P WE Q L S AS QQT GGI V AD DE GHEAISEV AE HDL S GAIEASGEDFNS TGGAS Q T LI RE NT GHEDLDyGNGTIS ReQTVRGI A VVE ELLLDRD R NAAR SGA D QS KKS GKTYIFI IP L GQL DAGL D GFE T RKFV V VV HK Gnr RNLV TS S V AGV NA QAQV AL AI RFLDotDK ItVAG ANR QDKAKND YLL D SVREVV I VD LP DI D VN QNTN NNKPYANQ YLGSE ATE DKE LGAWAGSDG VV GGSLD NNEGF LKVYQ KIGN R SS V ALSTKLD GGS SAA QR DAAL KKQYNKYK FDTSEGTLL QA AAG LLI RS K D AAE GRKI VIA GND N HLT KS SSEI AI GGDV L L GAQR E T AS NGVP NHAK QK T S N HE N SP LRI E GR RLW NDGAAVAGIEVASD GEL V KSRK DGIR GI YNS GNAAKE VVSAL KS G AV F GGH KL NQTK GA GSVDIS FV DL NMDV AF Q GP GV AR KRLD S QAGQRANS T D A AL NLQSEWLRSS GEG AS K AQI GS H KT QLYA GIV RKK NW PS M AVHGRIA GEV LQAD NL RL DKGL F VIL TA Y QS TS AA HGS GKRQ QAIKY VRAEAV AV DFADDA KKGFGD NN VIP NTFL LT HL VNT D TA GQS GVR HRV NNVRHGI DP WDFH G IISAL A GKAAL DQAVA QEE D VE AVYY D MK GSF NPGA PF D VS INR QGS S GT T Q QATSDKK T S GGR GGK G NT D GYL C GYL SDR D RLG ALS ETDR V R AR EG L TA ES KDSVS V D GGRE KE G D TLK VGG R QSS D GI AKK NLA D GL TL TS KF N QN NV GL DT R GNSAQASAQ SGNV DKPRTYT VSS LV HV N LS LIP QGR V T V R EHSDLSADRNA KF NAYLPSLE VLN GA MYQL D GT GS FRA GDI HS ESHGDGR QV R D NK NV NIKN K PNP GGSAAFVR EGL IE SDL S V SR T KKS G TF GP HE KL T GNSP V LLS GSLSNL G GQL S GALGYS LG AGG KYDLSV IM PD P ID D GYREAR GA ATRSGT D V I GGNDSL SL L VTTLF NLYKF DVLNGSAG V SL AS KS P AK NR D V T TK SD L KSEP GDAVKAL GDSSNN RVNRASGVLD GK NS MA ADT VITK A AAGSLGSGSD TQPL NNL TE TTGL G D GI YE K DAV G E QKSY GSP D SVYS KSEE ST QELGAKP AE SA DKMS DKGNA L GF GIYG ED R KNR RRALD NQKD GR GQ GNGSITT T DGKGVKI AF QI AL TDQRRVHDGNVS NKKER GIS NS RE DE NLP Y APR H GN RL ARSS SS GGASE DT R E LL T AGT A T SP GAE GGV A AGL A T YGTKI N R T P KNAA TKAV D H9 T G GDS 8 QP N L IGV AV KHSKQDP TN GVQYL A MGL GLR G R L NIYMFME SSS IYF TRAIDNEHR GSFLLAAQT DS Y D LE N DYVD HS PAL SASAG ANDLDS ASF Q F YDVG RIR KGGGVD V RAD DS Q NK RVSL GGNDL AE TS GT GA AI E NY AQGL WAEDV LR E R GKV KAGWVKK AYTKY LL SR IA L I SIAES SD D GQQ AHAA DALNKS VG V DGK Y D NDVAQQE AVE P IK VLD PIY GV V KRGV L N IR F LI V N L NP AEAAD S L NEIR S VGSG G NPNY WKF e C LL GKYY IKAR QEI E QR GR c RAG QNLAA AQS LNR GVAEL S S SKHSGE E AS AA R GV G R GA ND WRL ENGKA QQ MLPLS DSASKY SARSE DPVV E R L QI QRSL AQSFD n L e KSAS S LSDLHGPGAEIGSSHSAQP GQS L DR DLRDW u GAD NADAT RPS ADSERSNTI DGLQ VYPTT q RDVAG e P RDP D GTE QDRAT S R QEVE GR]LLKGW] V L SI P VEKKP] s S G L RL PALAARR E E AS AV GL I N AG GGEQ ARAL DAIV GRYS N LS A 5 TF ER5 TAS WK6 KP ND5 KL T MK7 QET 5 diH cF QL V PLQ AGSDIVG DDIY ATR YYA E RNQL GDGS TG G:T P QKT: S QYE V: AAD NGR V VOV NSWNNO NNP AVO a P N AISI RSAR DFYFLDQL GGIV YRSALV TSKYRIEN oN nAGSL GGLS GGNAHELKARL NL D IVI MG D I i DT EKN D I R L QDRD HEL GKRTGGIRAAG LGELSGK DERSA VG QR G T SNK KN QKK KKRV DL Q mH GQN G AVG L E K FWE V V Q E A MGANSASKSLRGSHRTTD GA DN GAL QV QLRHSNFS[ MNAIVNTS[ MNSH GKPS[ )asoni ) g s) us eu urrer ea uaua ss s an ucuc oc c mo oco doc lol nu yhyh 1v ees papa 1 g P i( t tStS2 A( (49 nl a b2 AxEl Hl H 5117570‐7 DAEA32
31 GQGRGLRD SDS R V TKTAAT DR VV LAS K02 A. QID R TREAHRT H R RAGAGGKAP T T RKKQY GDEDTo LQLGDVN AR GR RE K NA S QQAE A AQL EL LV A QY QD D N D S FI GDI YTAVQD SS Y R GSRR LS T T NAD GPDR F Q T YtekNVGcLoVAV EG P WE Q L S AS QQT GGI V AD DE GHEAISEV AE HDL S GAIEASGEDFNS TGGAS Q T LI RE NT GHEDLDyGNGTIS ReQTVRGI A VVE ELLLDRD R NAAR SGA D QS KKS GKTYIFI IP L GQL DAGL D GFE T RKFV V VV HK Gnr RNLV TS S V AGV NA QAQV AL AI RFLDotDK ItVAG ANR QDKAKND YLL D SVREVV I VD LP DI D VN QNTN NNKPYANQ YLGSE ATE DKE LGAWAGSDG VV GGSLD NNEGF LKVYQ KIGN R SS V ALSTKLD GGS SAA QR DAAL KKQYNKYK FDTSEGTLL QA AAG LLI RS K D AAE GRKI VIA GND N HLT KS SSEI AI GGDV L L GAQR E T AS NGVP NHAK QK T S N HE N SP LRI E GR RLW NDGAAVAGIEVASD GEL V KSRK DGIR GI YNS GNAAKE VVSAL KS G AV F GGH KL NQTK GA GSVDIS FV DL NMDV AF Q GP GV AR KRLD S QAGQRANS T D A AL NLQSEWLRSS GEG AS K AQI GS H KT QLYA GIV RKK NW PS M AVHGRIA GEV LQAD NL RL DKGL F VIL TA Y QS TS AA HGS GKRQ QAIKY VRAEAV AV DFADDA KKGFGD NN VIP NTFL LT HL VNT D TA GQS GVR HRV NNVRHGI DP WDFH G IISAL A GKAAL DQAVA QEE D VE AVYY D MK GSF NPGA PF D VS INR QGS S GT T Q QATSDKK T S GGR GGK G NT D GYL C GYL SDR D RLG ALS ETDR V R AR EG L TA ES KDSVS V D GGRE KE G D TLK VGG R QSS D GI AKK NLA D GL TL TS KF N QN NV GL DT R GNSAQASAQ SGNV DKPRTYT VSS LV HV N LS LIP QGR V T V R EHSDLSADRNA KF NAYLPSLE VLN GA MYQL D GT GS FRA GDI HS ESHGDGR QV R D NK NV NIKN K PNP GGSAAFVR EGL IE SDL S V SR T KKS G TF GP HE KL T GNSP V LLS GSLSNL G GQL S GALGYS LG AGG KYDLSV IM PD P ID D GYREAR GA ATRSGT D V I GGNDSL SL L VTTLF NLYKF DVLNGSAG V SL AS KS P AK NR D V T TK SD L KSEP GDAVKAL GDSSNN RVNRASGVLD GK NS MA ADT VITK A AAGSLGSGSD TQPL NNL TE TTGL G D GI YE K DAV G E QKSY GSP D SVYS KSEE ST QELGAKP AE SA DKMS DKGNA L GF GIYG ED R KNR RRALD NQKD GR GQ GNGSITT T DGKGVKI AF QI AL TDQRRVHDGNVS NKKER GIS NS RE DE NLP Y APR H GN RL ARSS SS GGASE DT R E LL T AGT A T SP GAE GGV A AGL A T YGTKI N R T P KNAA TKAV D H9 T G GDS 8 QP N L IGV AV KHSKQDP TN GVQYL A MGL GLR G R L NIYMFME SSS IYF TRAIDNEHR GSFLLAAQT DS Y D LE N DYVD HS PAL SASAG ANDLDS ASF Q F YDVG RIR KGGGVD V RAD DS Q NK RVSL GGNDL AE TS GT GA AI E NY AQGL WAEDV LR E R GKV KAGWVKK AYTKY LL SR IA L I SIAES SD D GQQ AHAA DALNKS VG V DGK Y D NDVAQQE AVE P IK VLD PIY GV V KRGV L N IR F LI V N L NP AEAAD S L NEIR S VGSG G NPNY WKF e C LL GKYY IKAR QEI E QR GR c RAG QNLAA AQS LNR GVAEL S S SKHSGE E AS AA R GV G R GA ND WRL ENGKA QQ MLPLS DSASKY SARSE DPVV E R L QI QRSL AQSFD n L e KSAS S LSDLHGPGAEIGSSHSAQP GQS L DR DLRDW u GAD NADAT RPS ADSERSNTI DGLQ VYPTT q RDVAG e P RDP D GTE QDRAT S R QEVE GR]LLKGW] V L SI P VEKKP] s S G L RL PALAARR E E AS AV GL I N AG GGEQ ARAL DAIV GRYS N LS A 5 TF ER5 TAS WK6 KP ND5 KL T MK7 QET 5 diH cF QL V PLQ AGSDIVG DDIY ATR YYA E RNQL GDGS TG G:T P QKT: S QYE V: AAD NGR V VOV NSWNNO NNP AVO a P N AISI RSAR DFYFLDQL GGIV YRSALV TSKYRIEN oN nAGSL GGLS GGNAHELKARL NL D IVI MG D I i DT EKN D I R L QDRD HEL GKRTGGIRAAG LGELSGK DERSA VG QR G T SNK KN QKK KKRV DL Q mH GQN G AVG L E K FWE V V Q E A MGANSASKSLRGSHRTTD GA DN GAL QV QLRHSNFS[ MNAIVNTS[ MNSH GKPS[ )asoni ) g s) us eu urrer ea uaua ss s an ucuc oc c mo oco doc lol nu yhyh 1v ees papa 1 g P i( t tStS2 A( (49 nl a b2 AxEl Hl H 5117570‐7 DAEA32 Y DV GW
Y DV GW 10P TN Y V V2 YF T P NWK GGYV V R. KS VPo K KVNNYGH GN L N AN RV N KW R YTWLDt DekS KNF KGY NI Q TRVS LYcYTV GFS NT YL SSKS So T A I NDI NND RWKD NR F GyeSSV HKGS T LMEGQY QQHnrIoF G KR DATTR Q MEtt GS KV TSA QNV L RYYTKL ES RK MQA KS LVL SEV I T L VIA VE R TQI AYH VV H DKK D NT LE R KYS KQV AT T DNK NQD DV V YI K YT KYY ST A KYM N NNA KE G KS KY DS D D ISR Y T KI F Q KKG KQAF ATR VSY KN DYD KS M NF NH FYT S DNIE VI FTN R TLG NL F QFSFE DK FSGI LVI YQGSL NGS T QY INTI SIGF QN KS E T GDS NF QSEFFL GP NV GGLSS G TGEK IS SDNSF N W KP AKE AGKS I L R GIDD SI SR NS QHS KSGA KT DS S SP GAGL TKF NVT TARTTGL FP D GAI IS E TME GI F DQHKYQTNS QF TNSY LL RGTGLHR QKIYP N KI GT ITI V F KT AIIE GF I LT G EDNF S INS QN DS QQ AY GP D V N V QQS GKGV VL GLF KF NKNP Q GKKE AGTPL I QGS V D SSE NYANEI D SEE 0 EIV Q D ILVPQSS TEEIKS D9 AP TI N KQKT TDNKP K NESPL KP RN AGD AP V L F Y ADIKQ GEYAL LL K AKYD K KKS NN KD A P NDV F V LE V K AN NILRP N T I F E PV AE S D Q K IFLF T GN V N GH NL V KSG NPY VNNY I D SSYELPYKHKV eTA cDLL R LL D D ALLNT S P W ND P HDIALN TKK YIE nI eI P AV A INPAQF G S E P uWKLS D TKMS PS SD YTAKGH LL VFT T IRS LG K SI L qeV ]GKP T]ASGK] VNDW] sL D8V 5AL TKD QIS 95 V VYF0 S NPT6SYS N LQD V 16 di G c I : O TL AK AS F IK : OS YGR KK T: O TF F E TP AY: O a TS o INTI L I D LNYVE N VLA GW GK NTWS K N IIYQHD IKWNF N L KD II R K QVTYD nDKQFDTNYNY K NM AGN I iQ QNPYKQ ML MI NQKSRQ mA EKI AS WE KKKAEKLVE E A MS[ MWI V NS[ MSF H GS[ MHTANVS[ )) ss) uus)su eeu rrerer uu u u aa a a ss uus s ccu uc ccc oococo cc oo l lc y yolcol h hy y phphp1 npav eatatat1 gtS2 itS( (S (S (4 A B C9 ndlglg g2 A H Hl Hl H 5117570‐732
10P TN Y V V2 YF T P NWK GGYV V R. KS VPo K KVNNYGH GN L N AN RV N KW R YTWLDt DekS KNF KGY NI Q TRVS LYcYTV GFS NT YL SSKS So T A I NDI NND RWKD NR F GyeSSV HKGS T LMEGQY QQHnrIoF G KR DATTR Q MEtt GS KV TSA QNV L RYYTKL ES RK MQA KS LVL SEV I T L VIA VE R TQI AYH VV H DKK D NT LE R KYS KQV AT T DNK NQD DV V YI K YT KYY ST A KYM N NNA KE G KS KY DS D D ISR Y T KI F Q KKG KQAF ATR VSY KN DYD KS M NF NH FYT S DNIE VI FTN R TLG NL F QFSFE DK FSGI LVI YQGSL NGS T QY INTI SIGF QN KS E T GDS NF QSEFFL GP NV GGLSS G TGEK IS SDNSF N W KP AKE AGKS I L R GIDD SI SR NS QHS KSGA KT DS S SP GAGL TKF NVT TARTTGL FP D GAI IS E TME GI F DQHKYQTNS QF TNSY LL RGTGLHR QKIYP N KI GT ITI V F KT AIIE GF I LT G EDNF S INS QN DS QQ AY GP D V N V QQS GKGV VL GLF KF NKNP Q GKKE AGTPL I QGS V D SSE NYANEI D SEE 0 EIV Q D ILVPQSS TEEIKS D9 AP TI N KQKT TDNKP K NESPL KP RN AGD AP V L F Y ADIKQ GEYAL LL K AKYD K KKS NN KD A P NDV F V LE V K AN NILRP N T I F E PV AE S D Q K IFLF T GN V N GH NL V KSG NPY VNNY I D SSYELPYKHKV eTA cDLL R LL D D ALLNT S P W ND P HDIALN TKK YIE nI eI P AV A INPAQF G S E P uWKLS D TKMS PS SD YTAKGH LL VFT T IRS LG K SI L qeV ]GKP T]ASGK] VNDW] sL D8V 5AL TKD QIS 95 V VYF0 S NPT6SYS N LQD V 16 di G c I : O TL AK AS F IK : OS YGR KK T: O TF F E TP AY: O a TS o INTI L I D LNYVE N VLA GW GK NTWS K N IIYQHD IKWNF N L KD II R K QVTYD nDKQFDTNYNY K NM AGN I iQ QNPYKQ ML MI NQKSRQ mA EKI AS WE KKKAEKLVE E A MS[ MWI V NS[ MSF H GS[ MHTANVS[ )) ss) uus)su eeu rrerer uu u u aa a a ss uus s ccu uc ccc oococo cc oo l lc y yolcol h hy y phphp1 npav eatatat1 gtS2 itS( (S (S (4 A B C9 ndlglg g2 A H Hl Hl H 5117570‐732
 310 K2PWS RG KV A N.NG SIN V DG WHoS WS KKNNKQ F RtKek YHN SVS E Ac GNo STTYTRKYID NKRR SS NNGy Ie NDI DIF QT KRnrGoATME GDPtt AT F RR QQVELTA KLY M VSYKSVA LETV V TQKIILYY STI DKK AKD KY DN YIS D QL K KT N ST KEK N DST YYSR IG KFD K KTY KS ISF FKL DSF SK KYKL NH V FGS F NN TNL D F V LL V F S R IGS GI IQNL QGFVGK FI SENGS NL L GP N QI G TS K KN D S F I N VP E GAH NN DIG S S R W AKQFLT SD K TGL NTI AGLS TYPS GFS TS I KYQ V G D I NP N YGHT ET Y GF LLPRL G TI TLKT S KQF D QI V NV QQ V QVE GL DV QGGAVPP KFAPG DDTL KE E E NSNTGTQ N R NS EI NEIKD1 KQS KKSEI T PA 9 PTN IKD D NP AG GTNK GT NP F L Y T E Q AYLFILA ADVLR ISR A EKLSNIS D V LE LG V NNIQT T NVGNIS PVP GN SAY DT Q SNPNH e LLNP SDW D ALP DP G KS K cLYV QV nLE eA uI SSEA SDYI T V K TL TLF IE qS eS VR SG L GS sA V VYT ]2 V G S IY RH T ] dNPF i S GT 6 :L N S YD K 3 HW 6 : c S Y KK V AQ GWT N OAFP AFN VO aGGGN NQL WKKN o L nND I M iKE WKI KY D I I YN FMIKYQKL GR KR DPV Q mKKNE F KTV VFE A MQS H NS[ MTL NS[ )su)s eru uer a u sa ucs cu occ coo lc yol hy ph 1 n at pav eSt1 g2 i(S(4 tD E9 nk Auk2 LuL 5117570‐7 VVK3 F TF IILANI VNS2 PL VLDEYEKYTGA T SIS P FI KI S IVNEEDAEE NIFWGF GLS MIY RS NFTNE F GF ANAP TG T DI YY3HL KDS NS ASIVS NKS NFAPI F D F NI NISY NDYTQYAFNVTV10NNIH2 K ITLKLD KAKNL QIP HN RKEL NNIF P GRNTLI YT NAV VK G. E QGYSTD Po VSLKS ALLSTN D TSEFLS SKKS GNAEFFYAW GNN PSKPL GKG EQIK DNI YLFI SI D KID GTF F V NFYQ GGYT T I Tt S YekT YMQIT LLDDE QTSP DD GNF YN YL LS GVTY KEINPKN KTAAQcNND KS NQIIKI ISFSVKVESSLKSF F KDL P MQQKNGGNFKWoS IF V D ILE YTDFILV AF IKD ELWF AI YMWS DNKFDIYSDN NGT KRLAQLHVDLFS GF IYT I HII T I NSIIL GVGIA TD V TY KNVyeInLKrIQY V Q NNIYFSYKESLGD NTQKNVKLS GK DSQT GTQKGA KoVRGIVFNSI EV KD D II SNDNS GDDL NLKNII LNAVW GGF YStEDGtKYFSDYKSESL NNALI L AGEYII KF IYKINFATGTP MT DIYI N IF NGLYGNS SLNV VT TV V L KYL NRD YD VAF KSAT ARNN G DE K KMLAA KAFS AELS DLKDKAT P KK NHI MDI KFF HP NA KYE ELD HFY LRKLL AI A KYE AVD S NV MK S ILNAI H GLTKI F NGYDIKF SEF N NAT GTLY AESS SL STI NI S KS LSG NYYN NG NANVYL IK NSK LYRVIE GP NSLEYLYS G GADI QT NPEMIIK ND P DFLA NKF R GL AF RKI DL D GS PR IAE F KE D DILM NIEY MDK FSS LT KHY HS K F NK FYNF DYG YKY F FFYS DNL QH NMS SLKE NIS SA F S SS I K NYS YL R NA DAVS ANL I N T NSTWV NQA IFLV LI AV YD IG KAD D S LS QT TLV KV AQGGN I RK KGFRF YIR SF LS L NKEGNF I GT L NQEIGAN ST QNNER LYF KPE GS LLFAVCT R L KFYNKSINSA GGLMTI DI Y KQQY YNGRLKA S E N P KDHN KAFA ENKAE GLWYG KKV NFD LITV DEGNI AT AL I F GYTKG GQGEI RI KQN D GLLLKFKNGKI KDNWNNA NIELLFI MF L AHLE DIT NEISK GITSL EYTIR DFAS T NQGLTNQ MF E V EE E LQS GTGIKL DV K KNPGDSS DPIKYNQ YNYF LIT ALF A D GE DLS KEV QNKFR GDI QS LL N NYL NET YYNQKP VIYF GAKTI N IS QY E LI S KL YNT FVE KSS NTP HD F DD R IV I SI NYS Y E IL PI K KGNFYA QHKDYE SANN T QAN PA AN N L S PD VNDHI EAE KNYGGF YD L E L EIAI V KIR HII LEESS DRIN NLV I KT KTEILKG NEKV NQI DLI VLL VEKYL VN VS TKE E QE INI YNL LI AFT NNITYKNAYE NA L SQ SEKT E FLK QL ETFNFD NYV NL DEL DRNS GNREL WNIN NYDFYQ GFP MESKLQPE LKIDT GDI DLNVGYGTI YGTN NYKEI G D AF YVIL KAS D KNKLS NLSRN EAL L NAIPDNF GLKNR V VR EGP LWT NS ITKNPYE I GIKE GA GVL NEITV QGS LI NGAAIG AC N DAG KF NEKA NLR Q NTI QAT GT RTIYYNVL IKLS WLNINFI F FETPATL HIDFG VN D DAYYKS IN YD I DR D KE KSTSDS IP VAFYNIT AQT NGS NN TVI L TLG HKS D NDNN F RV LKDTIG ENGDDE L GD IISLITPKI S T VLKGE DKSE T NAI WAI QG K LFL NSI SS T FYNF GFYAGP T AMIF V KANF NES NLS YKLI V D S N YSKTKL V ANSLSS TSFL NNYN NEDESE P SE ISVPGI NV FYGGG DI ST HF FNNF W QGL T I NTSIP GYTE T SI DI KKS DFST K KPAE D I QYI 2 S WYN NYYSGNKS F GTPG 9 E DYKR YIKYDS LWLSN NT KADS TILKI LEI KGKWS E KI LYKEE SWPYNDTGF IKF FS V DIFYDNNF L GPA SNI KGI T YKN D P GYF NK NL TK LINIYT D SLF K D NTITI ES F V S K DNYIS L GG NKQKFKHRF V G INT R KT A QYMNL HETP LR SL GNDI DL KL TYYK TVVW GNY NYF VK GYD YF NDSKIS IF S L H NSP LLSL DVNNVL L CT II KGISIF NV KE YASE KNF ENSALL KTYKV NFAS LL YKI NK NIL SF KDS AK ARGQI KGF VMIDS WSELD SFITY NNY PT S YN KNVD S DE WNMTYN D FY ED TAE RAEE I LNK GPS IED VLGYS GDK KE DQYFYA T GTLTI GGTIT I PWDL AS NM NPYVKN VVS MG AKATI CIHF YVIFNV LFD S KV G RI QRKIP S KNNIERT SAETATMI GV TF NET YYAHKT NA KKT NAIYDI YI YMLQQI EE DENSAKKKDF L NIV YNP T Q GGFS N QF SG TNE AYEE HETII V LL VYILD S SIED DGNK NEDI Y YDS YYF NAFL ] L E KLS W R Q NL S NF L T V NHLF DYSIIKLLI V DNTKGVIHTG MG 46 I AVL DLQ L I E DHSLFSN KDKSEV ILL L L MN TIIII NK YK GFYAK GNF AGA: E SGQL P YI L SD GQENEK NSIFV KE KFNGDYQ GNYYTAO KVKIYNN SIEIISS RNKFE NSISNT RIK DNIGVPYGQTNF IT KKEIS KNP AN NE S MSI KDN VEI E D D L YL HNAI QGI NT KE M FA D I LGD PKP SGIDIRS VFF KLN NCS GAN LIY FILSGINLEE TFE NDAP AWNNY GG NYT RT PT A IAT C IFY KS R AT GLT NT I TY R DPQE M WTPITQEFECTLLGP VNL GSYITTL D DYYDLFYDTTALFHTSA Q N MS[ )eliciffid sedioidirts1 ol v C 1 (2 A 4 d 9 c 2 T 5117570‐7 LEEP YS S3 NL A2 KKHE L P S L F GAYS Y RGV L F S YT V GKD S S KP QRDKT AVYNK3 EQIDIN AGP KS YIL AT SI EI NGT S KE SFI P GE RT NNKI KI I GSIT D NFYYEFY EL NQV QNE L Q S LTIL F PS I KQL I YV FLKIKD KI R DT QIGKV1 V KLSDKSSDL AY V II NKAME SL KSK0 PII KKTQLT DGSV NVV S QKLENS TL2. TYo LYMN NNDLS D GP IVAS VI KS AP QMEQIIV T KI YSLS SFV D KI V IS GF V LGGGD A AIELL D DHVREt SL P I R NISL NKI DN QKDEYI EI E D R LSE V KLIKQA NFDLD KL D D FQANE QI AS IE E MSNVQSF QL DNNLKKSL L NE DASKEYHekcKN QYII TVIKE KFSVS KF FFNT H NS V E CK SD YMIKD NY VAI KIEE GPID EToLDEL QVy I KGS GLLYALPIFMGS HDE D FLLF DS Y NGWVG Y EGE QSLLTV VDS H DGIKADKH VDL NFHAAe EIDI YLIIGS IF MGNKDS G VT NQ EI GE E S AKQMnrT YoV II NKF YYI T SI SE VS GD RPKEKLYL FSGNF V AMKYYNFDtL LA VAGRTLA D H TL R ER I YEL L NLSDT KNTS DYID KIIYATPAE GEVEV K DI NI R YFtYQIAE M KFRAKGAEE E VV KES S V LKDEF CSKD TF KF FYDSIIKN EYI SL MDINI I S SS SS NLSTIYQEAE I TTI ISNTQ NSSIE GD S D KE NV VAYQNE AIA KKRLL KR LF IPTP KLS I Q QEAH GVLI KFTF NT VD GD Y L I GF Q HKENFLKM NKE NFF DS GTSD DKKKS TT SS GDSGS NGINS VGAI FYD Y YA KT AKLES PLE A AF KATSV AES IL S NT S VIIL DS NAGAI LIEKTKPQ NYISHT D MINEEP FE YL QKTIE I IKDLSEYLYAT E FNE K GK NSS F VG GLKT D N DPE KV AKYI II DNT RH GNG SFAG ELGTE GAEKAGI L NMRAFIL EMI D NNGKN NF V EDFLY DYL EEKQTFR NLPTL NL FINDSLS VWF RD DFT KGFDPFF GF TYH YEE QL TNYV D VF G Y KPWL KDRFI AYE DI YE EPKTWI ESQ EIAIEWG KD MLISRINA R NLI QILEESS V SLLT KI RS IF MI CYKIF TNL E V SYSED DKF VYGI GVL GE G QTQID R F DSELGT NL DIN F DTNFGS LFAGIKF GQKYAVL QYT DS GTRDI KVLHSG SIGGNS YE LEK DGV EAHKFAK ALGDSPIGNYSK VDTY SSNE MIIVNR VQPDS QI ND G TR DF QSVT TS ERNKD YKSLQGL EN HL NLETI EYE KINKNENPKD FL ASGS EEPEEI I QHRVLEE I TI S NE NHT QDLENE GIY YEIYNEGT D Y GYI R LL QLY NK LE SL L A T NFL Y V S S GF GE IRYVQ I DRNDR V DLEYVIET TVGEFYD A K R EY AKAILTFI L LRNI E HLN LF IR R SV GMD TFE DE NFYAI KK AFL AL DIKVKDI NIDV DN KS ES GEK FNTL GEST PDI D VVNLNN QSGTDN GD D YYENE YKEKNG QNKIFK S AI DKI SD LANS A DDSE LL NGI L KR MLIL LTL DI KI D D VYAFEIELL KS KS TKSFSDEKP ITL L TSI KTDV YI TNIFNQEN QD DTFK NHVE QDRYDS N LEI F QVI NGLR RVS TVESVL GDIDINGVDKIPWED MI Y KE HE QSKYG AQIT YI KETE KEFE EEADS EK YAKKLED DV K QK DYL S GP IH HGV NYREKLLVLSQR NVL V NE D NGEI R QEE V KD N IQE WQQTD KFA AASEI GKEEAE NHI KT DNKDS VKL R L T T TAIF D NI RL KSP LST D VS I TY KNS Y NNV ID DFAY YRF YYVIKDK V DKS I DRVP WQ GNP KIA RS VEEGA NGM L MR I LNE NF SST LELWL NK VS DFGY SAL KDH L MGN YQEDKDS LLE N QNK ELNHL YI V V GNN TDGD K GI L KNIIL D SFG MAS KSE KYKISV T SV K D P NNS T GE TL EDI DQ TDF GDWEIASIYG MLKI NFRNIFD ASFAW G V D KISYYKF PE SRILLL YT YF EPT D GIE KV IPPS W KYAY NSEYF F E QE I DI LND I LV VN DYSI D G H TE N S I TI L GI TD F DLF V FHKFILS A YEDYVP ELFKINQIQI ISNDT TFRE NKL K NYMLYPNGN TKAES PSIKL AI A MSEV E NYNI GK WNEYQI NVDG DD QL LRE E VY II NYSEGAETP KKG GNID ND39 AV NLEKT R DM TL V NKS VPGEGYGS YI NI NMIL QI T K DGEGSLHLLTDLSV DLVSEYN IN FEE PES IEIE L SE FIQSILGT NI KMKME VF SEFQ VAE EKGD AAYA H AYEI K VD MD NNG YVC EE AR DLSVKIII DKEGL G T GGI NNEL YSYAIPLLRQLPRRG ANF QYK D E ATI SLLSP L YVD SINF NEQESD ISI Q NGER QEN NHFE ENNE G QWYAKL HT DV LE LPI HA QSNA YS KD I LD CI YN NI DI AL F SVVF M RM V NAL GP T SL KF Y I D T VHKTESNIVHI E T F AE YGYIN P GTIFLLI D SD V NFF ED QKD V R WI MI DE P NESLLK E GE YE L DF YLVG GL TIF DLKE GFYSWQL H S DDDYITI GE LKEDEL ESF I TDMS DV VKQ GVINEL R D NG TA L TGQSP QMYLKS D F QKSWT QIK GLEIEVVL D GYEI VIF VYL NEG D FYEI FY I ADV GWI DNSTN R N V IYYE DD ETNKP I D A KDLNKS FS V DKT EPSI D IE YTKF TIYDS H QL SI Y KRLS NANI NVN Y S AL AQT SE TI KQT AK ALYA NLT GYTL LT T K GYSG VLIKDS YKL SPENI SFLEES NE TSE DL SL I GD NSNGL D Y GDEDCSGAH QKKYKL GV IF MA KT L E D YK T VYI I L NSR SV SVF V I K V DNN QV KD YNNI L A E RLEKE SII ND AS NI GF PT]5MGINKL FL GKK IEE EI ] R LIYF6 NAEY NPAKISN 6S P VIAKI I KF SN RSE V L KI ENA YSY KKSP Q6 FGT HL FYMS VI YV F V :VKYTEKKNGD : Q KQI R GDP DKI P NQF NYIEGV KE NV ETFPDLV L GKGGI DG O N KIMNL WE KFE WHNIO N IKEFKSF QTE ANDLTT LSP IP LT E F S S AEI NQFE ELVKE E NELL F NG V III DIL NAFMLLKA DV V IVKKI NVYG DDE VMD IKVDLYSLFIQ NISQFD I LSWIL KL R L NTDI TL FDPFLDSLEN CC SLF ISDYVP FS TPII VIGNAID ND NSEYE F A DSPT YFTR I NVLGQKI D F DE NSPKR QAP L NGI R T Q SR NLSEKPE MH N D C S AGL ERI N GSL F G MK T TSF T E G ES[ MV Y MSNI Q P AS[ )eliciffid)s s i e ca di r oiht d n ir a ts s olul1 lv C i 1 (ca2 BB4 d( cf 92 TeL 5117570‐7L DKIE L IE S3E2S SEFV NERT V IIARGRT L E GGA3YE FL NKENTE T KS CS WL RL A Y1I S0E2 LKIK LLHD DKK AF VE N DVF NL E I T YE VMTR AI. VD EP FEDSYIE S F NRI QHE RLoND NtKLD FIIS AFE DT L L KEGE E YKRGVPP T ER DKKTLEDNE A AVSP HAAG L HF GDKePIP KNPKN D GSTF DYLAELF GN V ASAL MkcKoKDLE VEL LV QE D NL KKATI D SI R G REDLL SI KGEN GGE T KYQ NA DQQVL H LSY GAyeDnQD KrT KKKG E VHV GKY TS E HF E TI GKSDKKoSttQILSV N KNV V LL YW NESTA E VKSAQSAQI IDEAL AV NS KFF GNI L L YP NV VYPADI KLS DSIQM QV D GSR EIKYI E QADGI DH LIK TGEE F GI GRI NS W DV KAQ AL K D DL N QS EKGH NG TIGKNRTYSS KDS DS KE V GH MPI RSY GP FEKKS WGTL NQ HYS GESLSTPVFYR VE EYEL LN TY KVKDFYKKITL N GE AVTSLLLGTDV NQKEVAFELL Q VE TFP K NAIV QRY QFI LKMDFGLE NASQV GLS P SG T GL QA GR EA NS NQYE PIVTL TPQ SKL VSE AIPR IEFANDG KN SSY DNMGIKR D IT N T PKV S VILPHR HIKF KID AVGLNI IR QIRSMKEET N DYL R QNTI FAP APEKM QKE KY DYLGT AK GA GNKT NE A L NE HTIA WKKL K QKY QD I GP HTI IDL DI M E KE Y TIQ IGHK NVE KEI NLVDSQW NPF S DFR HV AI DM QGDR F SK TKIIT SV SDYV KIEGQG NLR KE KKP HGNLT TN WK Y KH T H D HNEGE GF L DG GV Y G AL IYE P EESEAI SEG R DRYSS ETP VKP KT I G LF FI E TEAND T YA TGK GV WT K HR KF LS NKYGIS A KI GV T M KVQFGIYE GYQR YS RE YKRF DP NQ KHYQE T KI F SKS GGS S QI KV FI V FT AE YKRGP S KSS MD IT D GR S GQS KK DF AV VE AI L F LP RI PIISIRP Y SR L ES SGSQI GKV NNE EPSD QN T V LIS DE NMI GYLT TAF YF AKI N NYKNYEIML DSLE SLE GGDYI HP HLSEILS R EVF SLW DAV ATHIGLALFQ EV Q NKFNTL E ELL NNYS KGLL NTNKE S QST VK N TALYNTFYA SEKIT CKWSSKV IE GH4 M AGL KYKV NR V S NGYLQIS DK ENVKISES LV SEQELS YE 9 GKYGRVL VT SNAYR H DPD QAC E NK IIP RNQ KDKFIDE AS GDAGFPP HTKL AMSF NFL DIELGSI D NVTD AR EHVSP ED T QSNI I SK SEDIYE STIK T KKNRA GDVSRSLFH PNKK AK L DS AYD CIN IE LRV DIP NRATN QA M AES FSKNL GT EI KEH AARVQTAYL GLNSP S LLNE KLI N V FQ SPL VNQAKH VKVT I DM GA LDNLRAIV R VQE GSF P KGAVH LIKGGGD DNSSH NK D TI HA GTLCN VPN GNVAAI NR YA SL FE KKE S WMEN I GP A QASYVAYFPAASHP I KE I KF AKL E WASGL YAWAY PVS SH FMN SLTK FE F KVLKDE QAEK IQNN]L PKY76 AT GV MA KYAYRP]SN NSP G K 86 S AC ] G 9L N ] 6V N 07 NPDISH FER IPLIT HQL Y: I V EYNYP :LFTP : TF KI W: FDS O LIVMAS R O IF AI P OFV O FD KTL NFLV YDLD KE L NS SGL I GYK SK D AGAG F G ADAN FHC VFN TFV A D VYIHNVC N H D GF L K D R ILETE GAE T ES APD ILKQV V V IFIYI IKE I GDSKILPI I KSNI KIQK KERSGSGRP QD QI KN TFLG EKY V TWPQL EKV I KQE MG DLA GPAIFKV QS[ MLP G G NDIS[ MS HEES[ MAES[ )eair ) e s t ic h a pi) rde) ae hra te n m aul rel i oho sni ulx rCh C lio e t tc o o 1 cairi aai Brbebr ibv i1 (etnyV (V (24 ah r yp oA9 xB 2 Ci DC(tx CtC 5117570‐ F L7F GAQ3 MS A2 GA Q NG T TP G GGVV VSEVLL DGELG AVA31T QHGPGS SVII S I GDFF GI0V NQE NGA RL RT ADSFAGR SV2G E KER L T EV.o EN TD NL A KAGGS KT LVt S GL DTL Q A MLLQVNS ALekAR Q S ATETGP S G GQTAWV Ac M A Y YKA NGAV DS SAN AGR LoD Q Q V DyeGG AILT AG ASAG GNIQS GA A AKA GFF A GL T DF HL VInArEQ S GVE SEQSGSKIL GT GoStI RtT I L VL SYL YV SGVSI V VIAR N WY A T GAKKNS GGVLYL WG G L SIDL ATWGKV AQG LR TS NEG QL DPGTT G A LFGGRGN GR AD LG GL KG EAVKSFSV D D SL GA G G NQAV AAS ALV V GK KVLW ELS NK L GTR QAQEG LQ E MSVR GMKG EK EAS CFFLG TLGP WMGANGV A NAAHGVGGNP T AKCT TAAV LG A AE EF M FPGLKADC AF H NA NG DQYAGV EPR T M WFEQ D V VANIG K QRV LS FAM GWLA GPAG GKFE QS Q VA SYASVPEKIVRE S G GAWG VAL QL GL VYVQ G I GAGGVA D G GKE GAS
310 K2PWS RG KV A N.NG SIN V DG WHoS WS KKNNKQ F RtKek YHN SVS E Ac GNo STTYTRKYID NKRR SS NNGy Ie NDI DIF QT KRnrGoATME GDPtt AT F RR QQVELTA KLY M VSYKSVA LETV V TQKIILYY STI DKK AKD KY DN YIS D QL K KT N ST KEK N DST YYSR IG KFD K KTY KS ISF FKL DSF SK KYKL NH V FGS F NN TNL D F V LL V F S R IGS GI IQNL QGFVGK FI SENGS NL L GP N QI G TS K KN D S F I N VP E GAH NN DIG S S R W AKQFLT SD K TGL NTI AGLS TYPS GFS TS I KYQ V G D I NP N YGHT ET Y GF LLPRL G TI TLKT S KQF D QI V NV QQ V QVE GL DV QGGAVPP KFAPG DDTL KE E E NSNTGTQ N R NS EI NEIKD1 KQS KKSEI T PA 9 PTN IKD D NP AG GTNK GT NP F L Y T E Q AYLFILA ADVLR ISR A EKLSNIS D V LE LG V NNIQT T NVGNIS PVP GN SAY DT Q SNPNH e LLNP SDW D ALP DP G KS K cLYV QV nLE eA uI SSEA SDYI T V K TL TLF IE qS eS VR SG L GS sA V VYT ]2 V G S IY RH T ] dNPF i S GT 6 :L N S YD K 3 HW 6 : c S Y KK V AQ GWT N OAFP AFN VO aGGGN NQL WKKN o L nND I M iKE WKI KY D I I YN FMIKYQKL GR KR DPV Q mKKNE F KTV VFE A MQS H NS[ MTL NS[ )su)s eru uer a u sa ucs cu occ coo lc yol hy ph 1 n at pav eSt1 g2 i(S(4 tD E9 nk Auk2 LuL 5117570‐7 VVK3 F TF IILANI VNS2 PL VLDEYEKYTGA T SIS P FI KI S IVNEEDAEE NIFWGF GLS MIY RS NFTNE F GF ANAP TG T DI YY3HL KDS NS ASIVS NKS NFAPI F D F NI NISY NDYTQYAFNVTV10NNIH2 K ITLKLD KAKNL QIP HN RKEL NNIF P GRNTLI YT NAV VK G. E QGYSTD Po VSLKS ALLSTN D TSEFLS SKKS GNAEFFYAW GNN PSKPL GKG EQIK DNI YLFI SI D KID GTF F V NFYQ GGYT T I Tt S YekT YMQIT LLDDE QTSP DD GNF YN YL LS GVTY KEINPKN KTAAQcNND KS NQIIKI ISFSVKVESSLKSF F KDL P MQQKNGGNFKWoS IF V D ILE YTDFILV AF IKD ELWF AI YMWS DNKFDIYSDN NGT KRLAQLHVDLFS GF IYT I HII T I NSIIL GVGIA TD V TY KNVyeInLKrIQY V Q NNIYFSYKESLGD NTQKNVKLS GK DSQT GTQKGA KoVRGIVFNSI EV KD D II SNDNS GDDL NLKNII LNAVW GGF YStEDGtKYFSDYKSESL NNALI L AGEYII KF IYKINFATGTP MT DIYI N IF NGLYGNS SLNV VT TV V L KYL NRD YD VAF KSAT ARNN G DE K KMLAA KAFS AELS DLKDKAT P KK NHI MDI KFF HP NA KYE ELD HFY LRKLL AI A KYE AVD S NV MK S ILNAI H GLTKI F NGYDIKF SEF N NAT GTLY AESS SL STI NI S KS LSG NYYN NG NANVYL IK NSK LYRVIE GP NSLEYLYS G GADI QT NPEMIIK ND P DFLA NKF R GL AF RKI DL D GS PR IAE F KE D DILM NIEY MDK FSS LT KHY HS K F NK FYNF DYG YKY F FFYS DNL QH NMS SLKE NIS SA F S SS I K NYS YL R NA DAVS ANL I N T NSTWV NQA IFLV LI AV YD IG KAD D S LS QT TLV KV AQGGN I RK KGFRF YIR SF LS L NKEGNF I GT L NQEIGAN ST QNNER LYF KPE GS LLFAVCT R L KFYNKSINSA GGLMTI DI Y KQQY YNGRLKA S E N P KDHN KAFA ENKAE GLWYG KKV NFD LITV DEGNI AT AL I F GYTKG GQGEI RI KQN D GLLLKFKNGKI KDNWNNA NIELLFI MF L AHLE DIT NEISK GITSL EYTIR DFAS T NQGLTNQ MF E V EE E LQS GTGIKL DV K KNPGDSS DPIKYNQ YNYF LIT ALF A D GE DLS KEV QNKFR GDI QS LL N NYL NET YYNQKP VIYF GAKTI N IS QY E LI S KL YNT FVE KSS NTP HD F DD R IV I SI NYS Y E IL PI K KGNFYA QHKDYE SANN T QAN PA AN N L S PD VNDHI EAE KNYGGF YD L E L EIAI V KIR HII LEESS DRIN NLV I KT KTEILKG NEKV NQI DLI VLL VEKYL VN VS TKE E QE INI YNL LI AFT NNITYKNAYE NA L SQ SEKT E FLK QL ETFNFD NYV NL DEL DRNS GNREL WNIN NYDFYQ GFP MESKLQPE LKIDT GDI DLNVGYGTI YGTN NYKEI G D AF YVIL KAS D KNKLS NLSRN EAL L NAIPDNF GLKNR V VR EGP LWT NS ITKNPYE I GIKE GA GVL NEITV QGS LI NGAAIG AC N DAG KF NEKA NLR Q NTI QAT GT RTIYYNVL IKLS WLNINFI F FETPATL HIDFG VN D DAYYKS IN YD I DR D KE KSTSDS IP VAFYNIT AQT NGS NN TVI L TLG HKS D NDNN F RV LKDTIG ENGDDE L GD IISLITPKI S T VLKGE DKSE T NAI WAI QG K LFL NSI SS T FYNF GFYAGP T AMIF V KANF NES NLS YKLI V D S N YSKTKL V ANSLSS TSFL NNYN NEDESE P SE ISVPGI NV FYGGG DI ST HF FNNF W QGL T I NTSIP GYTE T SI DI KKS DFST K KPAE D I QYI 2 S WYN NYYSGNKS F GTPG 9 E DYKR YIKYDS LWLSN NT KADS TILKI LEI KGKWS E KI LYKEE SWPYNDTGF IKF FS V DIFYDNNF L GPA SNI KGI T YKN D P GYF NK NL TK LINIYT D SLF K D NTITI ES F V S K DNYIS L GG NKQKFKHRF V G INT R KT A QYMNL HETP LR SL GNDI DL KL TYYK TVVW GNY NYF VK GYD YF NDSKIS IF S L H NSP LLSL DVNNVL L CT II KGISIF NV KE YASE KNF ENSALL KTYKV NFAS LL YKI NK NIL SF KDS AK ARGQI KGF VMIDS WSELD SFITY NNY PT S YN KNVD S DE WNMTYN D FY ED TAE RAEE I LNK GPS IED VLGYS GDK KE DQYFYA T GTLTI GGTIT I PWDL AS NM NPYVKN VVS MG AKATI CIHF YVIFNV LFD S KV G RI QRKIP S KNNIERT SAETATMI GV TF NET YYAHKT NA KKT NAIYDI YI YMLQQI EE DENSAKKKDF L NIV YNP T Q GGFS N QF SG TNE AYEE HETII V LL VYILD S SIED DGNK NEDI Y YDS YYF NAFL ] L E KLS W R Q NL S NF L T V NHLF DYSIIKLLI V DNTKGVIHTG MG 46 I AVL DLQ L I E DHSLFSN KDKSEV ILL L L MN TIIII NK YK GFYAK GNF AGA: E SGQL P YI L SD GQENEK NSIFV KE KFNGDYQ GNYYTAO KVKIYNN SIEIISS RNKFE NSISNT RIK DNIGVPYGQTNF IT KKEIS KNP AN NE S MSI KDN VEI E D D L YL HNAI QGI NT KE M FA D I LGD PKP SGIDIRS VFF KLN NCS GAN LIY FILSGINLEE TFE NDAP AWNNY GG NYT RT PT A IAT C IFY KS R AT GLT NT I TY R DPQE M WTPITQEFECTLLGP VNL GSYITTL D DYYDLFYDTTALFHTSA Q N MS[ )eliciffid sedioidirts1 ol v C 1 (2 A 4 d 9 c 2 T 5117570‐7 LEEP YS S3 NL A2 KKHE L P S L F GAYS Y RGV L F S YT V GKD S S KP QRDKT AVYNK3 EQIDIN AGP KS YIL AT SI EI NGT S KE SFI P GE RT NNKI KI I GSIT D NFYYEFY EL NQV QNE L Q S LTIL F PS I KQL I YV FLKIKD KI R DT QIGKV1 V KLSDKSSDL AY V II NKAME SL KSK0 PII KKTQLT DGSV NVV S QKLENS TL2. TYo LYMN NNDLS D GP IVAS VI KS AP QMEQIIV T KI YSLS SFV D KI V IS GF V LGGGD A AIELL D DHVREt SL P I R NISL NKI DN QKDEYI EI E D R LSE V KLIKQA NFDLD KL D D FQANE QI AS IE E MSNVQSF QL DNNLKKSL L NE DASKEYHekcKN QYII TVIKE KFSVS KF FFNT H NS V E CK SD YMIKD NY VAI KIEE GPID EToLDEL QVy I KGS GLLYALPIFMGS HDE D FLLF DS Y NGWVG Y EGE QSLLTV VDS H DGIKADKH VDL NFHAAe EIDI YLIIGS IF MGNKDS G VT NQ EI GE E S AKQMnrT YoV II NKF YYI T SI SE VS GD RPKEKLYL FSGNF V AMKYYNFDtL LA VAGRTLA D H TL R ER I YEL L NLSDT KNTS DYID KIIYATPAE GEVEV K DI NI R YFtYQIAE M KFRAKGAEE E VV KES S V LKDEF CSKD TF KF FYDSIIKN EYI SL MDINI I S SS SS NLSTIYQEAE I TTI ISNTQ NSSIE GD S D KE NV VAYQNE AIA KKRLL KR LF IPTP KLS I Q QEAH GVLI KFTF NT VD GD Y L I GF Q HKENFLKM NKE NFF DS GTSD DKKKS TT SS GDSGS NGINS VGAI FYD Y YA KT AKLES PLE A AF KATSV AES IL S NT S VIIL DS NAGAI LIEKTKPQ NYISHT D MINEEP FE YL QKTIE I IKDLSEYLYAT E FNE K GK NSS F VG GLKT D N DPE KV AKYI II DNT RH GNG SFAG ELGTE GAEKAGI L NMRAFIL EMI D NNGKN NF V EDFLY DYL EEKQTFR NLPTL NL FINDSLS VWF RD DFT KGFDPFF GF TYH YEE QL TNYV D VF G Y KPWL KDRFI AYE DI YE EPKTWI ESQ EIAIEWG KD MLISRINA R NLI QILEESS V SLLT KI RS IF MI CYKIF TNL E V SYSED DKF VYGI GVL GE G QTQID R F DSELGT NL DIN F DTNFGS LFAGIKF GQKYAVL QYT DS GTRDI KVLHSG SIGGNS YE LEK DGV EAHKFAK ALGDSPIGNYSK VDTY SSNE MIIVNR VQPDS QI ND G TR DF QSVT TS ERNKD YKSLQGL EN HL NLETI EYE KINKNENPKD FL ASGS EEPEEI I QHRVLEE I TI S NE NHT QDLENE GIY YEIYNEGT D Y GYI R LL QLY NK LE SL L A T NFL Y V S S GF GE IRYVQ I DRNDR V DLEYVIET TVGEFYD A K R EY AKAILTFI L LRNI E HLN LF IR R SV GMD TFE DE NFYAI KK AFL AL DIKVKDI NIDV DN KS ES GEK FNTL GEST PDI D VVNLNN QSGTDN GD D YYENE YKEKNG QNKIFK S AI DKI SD LANS A DDSE LL NGI L KR MLIL LTL DI KI D D VYAFEIELL KS KS TKSFSDEKP ITL L TSI KTDV YI TNIFNQEN QD DTFK NHVE QDRYDS N LEI F QVI NGLR RVS TVESVL GDIDINGVDKIPWED MI Y KE HE QSKYG AQIT YI KETE KEFE EEADS EK YAKKLED DV K QK DYL S GP IH HGV NYREKLLVLSQR NVL V NE D NGEI R QEE V KD N IQE WQQTD KFA AASEI GKEEAE NHI KT DNKDS VKL R L T T TAIF D NI RL KSP LST D VS I TY KNS Y NNV ID DFAY YRF YYVIKDK V DKS I DRVP WQ GNP KIA RS VEEGA NGM L MR I LNE NF SST LELWL NK VS DFGY SAL KDH L MGN YQEDKDS LLE N QNK ELNHL YI V V GNN TDGD K GI L KNIIL D SFG MAS KSE KYKISV T SV K D P NNS T GE TL EDI DQ TDF GDWEIASIYG MLKI NFRNIFD ASFAW G V D KISYYKF PE SRILLL YT YF EPT D GIE KV IPPS W KYAY NSEYF F E QE I DI LND I LV VN DYSI D G H TE N S I TI L GI TD F DLF V FHKFILS A YEDYVP ELFKINQIQI ISNDT TFRE NKL K NYMLYPNGN TKAES PSIKL AI A MSEV E NYNI GK WNEYQI NVDG DD QL LRE E VY II NYSEGAETP KKG GNID ND39 AV NLEKT R DM TL V NKS VPGEGYGS YI NI NMIL QI T K DGEGSLHLLTDLSV DLVSEYN IN FEE PES IEIE L SE FIQSILGT NI KMKME VF SEFQ VAE EKGD AAYA H AYEI K VD MD NNG YVC EE AR DLSVKIII DKEGL G T GGI NNEL YSYAIPLLRQLPRRG ANF QYK D E ATI SLLSP L YVD SINF NEQESD ISI Q NGER QEN NHFE ENNE G QWYAKL HT DV LE LPI HA QSNA YS KD I LD CI YN NI DI AL F SVVF M RM V NAL GP T SL KF Y I D T VHKTESNIVHI E T F AE YGYIN P GTIFLLI D SD V NFF ED QKD V R WI MI DE P NESLLK E GE YE L DF YLVG GL TIF DLKE GFYSWQL H S DDDYITI GE LKEDEL ESF I TDMS DV VKQ GVINEL R D NG TA L TGQSP QMYLKS D F QKSWT QIK GLEIEVVL D GYEI VIF VYL NEG D FYEI FY I ADV GWI DNSTN R N V IYYE DD ETNKP I D A KDLNKS FS V DKT EPSI D IE YTKF TIYDS H QL SI Y KRLS NANI NVN Y S AL AQT SE TI KQT AK ALYA NLT GYTL LT T K GYSG VLIKDS YKL SPENI SFLEES NE TSE DL SL I GD NSNGL D Y GDEDCSGAH QKKYKL GV IF MA KT L E D YK T VYI I L NSR SV SVF V I K V DNN QV KD YNNI L A E RLEKE SII ND AS NI GF PT]5MGINKL FL GKK IEE EI ] R LIYF6 NAEY NPAKISN 6S P VIAKI I KF SN RSE V L KI ENA YSY KKSP Q6 FGT HL FYMS VI YV F V :VKYTEKKNGD : Q KQI R GDP DKI P NQF NYIEGV KE NV ETFPDLV L GKGGI DG O N KIMNL WE KFE WHNIO N IKEFKSF QTE ANDLTT LSP IP LT E F S S AEI NQFE ELVKE E NELL F NG V III DIL NAFMLLKA DV V IVKKI NVYG DDE VMD IKVDLYSLFIQ NISQFD I LSWIL KL R L NTDI TL FDPFLDSLEN CC SLF ISDYVP FS TPII VIGNAID ND NSEYE F A DSPT YFTR I NVLGQKI D F DE NSPKR QAP L NGI R T Q SR NLSEKPE MH N D C S AGL ERI N GSL F G MK T TSF T E G ES[ MV Y MSNI Q P AS[ )eliciffid)s s i e ca di r oiht d n ir a ts s olul1 lv C i 1 (ca2 BB4 d( cf 92 TeL 5117570‐7L DKIE L IE S3E2S SEFV NERT V IIARGRT L E GGA3YE FL NKENTE T KS CS WL RL A Y1I S0E2 LKIK LLHD DKK AF VE N DVF NL E I T YE VMTR AI. VD EP FEDSYIE S F NRI QHE RLoND NtKLD FIIS AFE DT L L KEGE E YKRGVPP T ER DKKTLEDNE A AVSP HAAG L HF GDKePIP KNPKN D GSTF DYLAELF GN V ASAL MkcKoKDLE VEL LV QE D NL KKATI D SI R G REDLL SI KGEN GGE T KYQ NA DQQVL H LSY GAyeDnQD KrT KKKG E VHV GKY TS E HF E TI GKSDKKoSttQILSV N KNV V LL YW NESTA E VKSAQSAQI IDEAL AV NS KFF GNI L L YP NV VYPADI KLS DSIQM QV D GSR EIKYI E QADGI DH LIK TGEE F GI GRI NS W DV KAQ AL K D DL N QS EKGH NG TIGKNRTYSS KDS DS KE V GH MPI RSY GP FEKKS WGTL NQ HYS GESLSTPVFYR VE EYEL LN TY KVKDFYKKITL N GE AVTSLLLGTDV NQKEVAFELL Q VE TFP K NAIV QRY QFI LKMDFGLE NASQV GLS P SG T GL QA GR EA NS NQYE PIVTL TPQ SKL VSE AIPR IEFANDG KN SSY DNMGIKR D IT N T PKV S VILPHR HIKF KID AVGLNI IR QIRSMKEET N DYL R QNTI FAP APEKM QKE KY DYLGT AK GA GNKT NE A L NE HTIA WKKL K QKY QD I GP HTI IDL DI M E KE Y TIQ IGHK NVE KEI NLVDSQW NPF S DFR HV AI DM QGDR F SK TKIIT SV SDYV KIEGQG NLR KE KKP HGNLT TN WK Y KH T H D HNEGE GF L DG GV Y G AL IYE P EESEAI SEG R DRYSS ETP VKP KT I G LF FI E TEAND T YA TGK GV WT K HR KF LS NKYGIS A KI GV T M KVQFGIYE GYQR YS RE YKRF DP NQ KHYQE T KI F SKS GGS S QI KV FI V FT AE YKRGP S KSS MD IT D GR S GQS KK DF AV VE AI L F LP RI PIISIRP Y SR L ES SGSQI GKV NNE EPSD QN T V LIS DE NMI GYLT TAF YF AKI N NYKNYEIML DSLE SLE GGDYI HP HLSEILS R EVF SLW DAV ATHIGLALFQ EV Q NKFNTL E ELL NNYS KGLL NTNKE S QST VK N TALYNTFYA SEKIT CKWSSKV IE GH4 M AGL KYKV NR V S NGYLQIS DK ENVKISES LV SEQELS YE 9 GKYGRVL VT SNAYR H DPD QAC E NK IIP RNQ KDKFIDE AS GDAGFPP HTKL AMSF NFL DIELGSI D NVTD AR EHVSP ED T QSNI I SK SEDIYE STIK T KKNRA GDVSRSLFH PNKK AK L DS AYD CIN IE LRV DIP NRATN QA M AES FSKNL GT EI KEH AARVQTAYL GLNSP S LLNE KLI N V FQ SPL VNQAKH VKVT I DM GA LDNLRAIV R VQE GSF P KGAVH LIKGGGD DNSSH NK D TI HA GTLCN VPN GNVAAI NR YA SL FE KKE S WMEN I GP A QASYVAYFPAASHP I KE I KF AKL E WASGL YAWAY PVS SH FMN SLTK FE F KVLKDE QAEK IQNN]L PKY76 AT GV MA KYAYRP]SN NSP G K 86 S AC ] G 9L N ] 6V N 07 NPDISH FER IPLIT HQL Y: I V EYNYP :LFTP : TF KI W: FDS O LIVMAS R O IF AI P OFV O FD KTL NFLV YDLD KE L NS SGL I GYK SK D AGAG F G ADAN FHC VFN TFV A D VYIHNVC N H D GF L K D R ILETE GAE T ES APD ILKQV V V IFIYI IKE I GDSKILPI I KSNI KIQK KERSGSGRP QD QI KN TFLG EKY V TWPQL EKV I KQE MG DLA GPAIFKV QS[ MLP G G NDIS[ MS HEES[ MAES[ )eair ) e s t ic h a pi) rde) ae hra te n m aul rel i oho sni ulx rCh C lio e t tc o o 1 cairi aai Brbebr ibv i1 (etnyV (V (24 ah r yp oA9 xB 2 Ci DC(tx CtC 5117570‐ F L7F GAQ3 MS A2 GA Q NG T TP G GGVV VSEVLL DGELG AVA31T QHGPGS SVII S I GDFF GI0V NQE NGA RL RT ADSFAGR SV2G E KER L T EV.o EN TD NL A KAGGS KT LVt S GL DTL Q A MLLQVNS ALekAR Q S ATETGP S G GQTAWV Ac M A Y YKA NGAV DS SAN AGR LoD Q Q V DyeGG AILT AG ASAG GNIQS GA A AKA GFF A GL T DF HL VInArEQ S GVE SEQSGSKIL GT GoStI RtT I L VL SYL YV SGVSI V VIAR N WY A T GAKKNS GGVLYL WG G L SIDL ATWGKV AQG LR TS NEG QL DPGTT G A LFGGRGN GR AD LG GL KG EAVKSFSV D D SL GA G G NQAV AAS ALV V GK KVLW ELS NK L GTR QAQEG LQ E MSVR GMKG EK EAS CFFLG TLGP WMGANGV A NAAHGVGGNP T AKCT TAAV LG A AE EF M FPGLKADC AF H NA NG DQYAGV EPR T M WFEQ D V VANIG K QRV LS FAM GWLA GPAG GKFE QS Q VA SYASVPEKIVRE S G GAWG VAL QL GL VYVQ G I GAGGVA D G GKE GAS G AP S AI PI GI GAAGII Q4 GGI YTQFEAV A GAS GDAG: GR GLAIES VGO GLDS WGS GGV N W WL VV A Q D VAL QEV GI YL G KWGD LSQ II EAD I A AG QYL VPS QLTG K T G DAP DAQ L L GR STGL AAE KTS DI VG LG GG KG G T GP S AI AGVS [ SA A QT SFEVIIGKFASSGP * KS IA TL GQED AGS AAAAGS G D T GEP KA EV Q DIK AAV S GGL L ADS GSS E SG E DGALS 5 AGRP V 9 DLDI LIFGG SQAE RL VIIALPL AHGRLDS I P DAAP H MKAL S GAG HT IKS D G D IALSGEF GGQQ AR A SL M TD GR R T AGA T RGS GYS GE SST RL ALSS D VG PS SA S SAAS NS GIV GQ V RL GL V GSYL GHA HSA QR IEH FL A NAG QGS GD H A LLIAG GAV V V QA Q AGARSGGGPAS SN AGecV nIVQ D TS GH GA V L IVQFL IYL Y T S S AAV HAVNGL P G AE seeu F IAVV GS ]FPIT D L GG EQ A ] d q LILS HNS 44 LIGV VGK I G] A1 F7 L : T2 A7ite :ps V e dAILV QRA VSVIQ iGQRAL LL : GO AG N GL SA L LV SG GP GG GSS N OA D O NpcL R D S VKAL DSR V GK WN T yl a AL PQADIRAYL T F GR Q DKF Q I MGD o M Ip oAD L nAALG GGG A GFL QE AGA D GE S R GL GGC ETQEEQQy iGE S GGS GIGKGLMV MASA [ MSE SS[ral mGQGAT[ GAS S KGT p A MF G D V * MI G A P A m ex ) e si) ssi f o os los ulu e crc c er n bebe u uu t tqe m ms uirui d er teic ctca aba obo n co yci e mdi1 My v (M(A t :p1 62 -t0 a15e e p 1 2 yg g4 sP lni n92 EF l C b a orir 5 T PtStS117570‐7323102.oNtekcoDyenrottA 69 1v12492511 Attorney Docket No. 2013237‐0757 RNA delivery RNA described herein may be delivered for therapeutic applications described herein using any appropriate methods known in the art, including, e.g., delivery as naked RNA, or delivery mediated by delivery vehicles. Some aspects of the disclosure involve the targeted delivery of the RNA disclosed herein to certain cells or tissues. In some embodiments, after administration of the RNA (in particular, mRNA) compositions/formulations described herein, at least a portion of the RNA is delivered to a target cell or target organ. In some embodiments, at least a portion of the RNA is delivered to the cytosol of the target cell. In some embodiments, the RNA (in particular, mRNA) is translated by the target cell to produce the encoded peptide or polypeptide. In some embodiments, the target cell is a muscle cell. In some embodiments, the target cell is a cell in the liver. In some embodiments, the target cell is a cell in the lung. In some embodiments, the disclosure involves targeting the lymphatic system, in particular secondary lymphoid organs, more specifically spleen. In some embodiments, the target cell is a cell in the lymph nodes. In some embodiments, the target cell is a spleen cell. In some embodiments, the target cell is an antigen presenting cell such as a professional antigen presenting cell in the spleen. In some embodiments, the target cell is a dendritic cell in the spleen. Thus, RNA (in particular, mRNA) compositions/formulations described herein may be used for delivering RNA to such target cell. The "lymphatic system" is part of the circulatory system and an important part of the immune system, comprising a network of lymphatic vessels that carry lymph. The lymphatic system consists of lymphatic organs, a conducting network of lymphatic vessels, and the circulating lymph. The primary or central lymphoid organs generate lymphocytes from immature progenitor cells. The thymus and the bone marrow constitute the primary lymphoid organs. Secondary or peripheral lymphoid organs, which include lymph nodes and the spleen, maintain mature naive lymphocytes and initiate an adaptive immune response. Lipid-based RNA delivery systems have an inherent preference to the liver, where, depending on the composition of the RNA delivery systems used, RNA expression in the liver can be obtained. Liver accumulation is caused by the discontinuous nature of the hepatic vasculature or the lipid metabolism (liposomes and lipid or cholesterol conjugates). In some embodiments, the target organ for RNA expression is liver and the target tissue is liver tissue. The delivery to such target tissue is preferred, in particular, if presence of RNA or of the encoded peptide or polypeptide in this organ or tissue is desired and/or if it is desired to express large amounts of the encoded peptide or polypeptide and/or if systemic presence of the encoded peptide or polypeptide, in particular in significant amounts, is desired or required. Delivery vehicles To overcome the barriers to safe and effective RNA delivery, RNA may be administered with one or more delivery vehicles that protect the RNA from degradation, maximize delivery to on-target cells and minimize exposure to off- target cells. Such RNA delivery vehicles may complex or encapsulate RNA and include a range of materials, including polymers and lipids. In some embodiments, such RNA delivery vehicles may form particles with RNA. Suitable formulations for such delivery vehicles are described, e.g., in WO2023/126404, WO2023/126053 and WO 2023/094713, the contents of which are incorporated herein by reference. 97 11529421v1 Attorney Docket No. 2013237‐0757 Compositions comprising nucleic acid A composition comprising one or more RNAs described herein, e.g., in the form of RNA particles, may comprise salts, buffers, or other components as further described below. In some embodiments, a salt for use in the compositions described herein comprises sodium chloride. Without wishing to be bound by theory, sodium chloride functions as an ionic osmolality agent for preconditioning RNA prior to mixing with lipids. In some embodiments, the compositions described herein may comprise alternative organic or inorganic salts. Alternative salts include, without limitation, potassium chloride, dipotassium phosphate, monopotassium phosphate, potassium acetate, potassium bicarbonate, potassium sulfate, disodium phosphate, monosodium phosphate, sodium acetate, sodium bicarbonate, sodium sulfate, lithium chloride, magnesium chloride, magnesium phosphate, calcium chloride, and sodium salts of ethylenediaminetetraacetic acid (EDTA). Generally, compositions for storing RNA particles such as for freezing RNA particles comprise low sodium chloride concentrations, or comprises a low ionic strength. In some embodiments, the sodium chloride is at a concentration from 0 mM to about 50 mM, from 0 mM to about 40 mM, or from about 10 mM to about 50 mM. According to the present disclosure, the RNA particle compositions described herein have a pH suitable for the stability of the RNA particles and, in particular, for the stability of the RNA. Without wishing to be bound by theory, the use of a buffer system maintains the pH of the particle compositions described herein during manufacturing, storage and use of the compositions. In some embodiments of the present disclosure, the buffer system may comprise a solvent (in particular, water, such as deionized water, in particular water for injection) and a buffering substance. The buffering substance may be selected from 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid (HEPES), 2-amino-2-(hydroxymethyl)propane-1,3-diol (Tris), acetate, and histidine. In some embodiments, the buffering substance is HEPES. In some embodiments, the buffering substance is Tris. Compositions (in particular, RNA compositions/formulations) described herein may also comprise a cryoprotectant and/or a surfactant as stabilizer to avoid substantial loss of the product quality and, in particular, substantial loss of RNA activity during storage, freezing, and/or lyophilization, for example to reduce or prevent aggregation, particle collapse, RNA degradation and/or other types of damage. In some embodiments, the cryoprotectant is a carbohydrate. The term "carbohydrate", as used herein, refers to and encompasses monosaccharides, disaccharides, trisaccharides, oligosaccharides and polysaccharides. In some embodiments, the cryoprotectant is a monosaccharide. The term "monosaccharide", as used herein refers to a single carbohydrate unit (e.g., a simple sugar) that cannot be hydrolyzed to simpler carbohydrate units. Exemplary monosaccharide cryoprotectants include glucose, fructose, galactose, xylose, ribose and the like. In some embodiments, the cryoprotectant is a disaccharide. The term "disaccharide", as used herein refers to a compound or a chemical moiety formed by 2 monosaccharide units that are bonded together through a glycosidic linkage, for example through 1-4 linkages or 1-6 linkages. A disaccharide may be hydrolyzed into two 98 11529421v1 Attorney Docket No. 2013237‐0757 monosaccharides. Exemplary disaccharide cryoprotectants include sucrose, trehalose, lactose, maltose and the like. In some embodiments, the cryoprotectant is sucrose. The term "trisaccharide" means three sugars linked together to form one molecule. Examples of a trisaccharides include raffinose and melezitose. In some embodiments, the cryoprotectant is an oligosaccharide. The term "oligosaccharide", as used herein refers to a compound or a chemical moiety formed by 3 to about 15, such as 3 to about 10 monosaccharide units that are bonded together through glycosidic linkages, for example through 1-4 linkages or 1-6 linkages, to form a linear, branched or cyclic structure. Exemplary oligosaccharide cryoprotectants include cyclodextrins, raffinose, melezitose, maltotriose, stachyose, acarbose, and the like. An oligosaccharide can be oxidized or reduced. In an embodiment, the cryoprotectant is a cyclic oligosaccharide. The term "cyclic oligosaccharide", as used herein refers to a compound or a chemical moiety formed by 3 to about 15, such as 6, 7, 8, 9, or 10 monosaccharide units that are bonded together through glycosidic linkages, for example through 1-4 linkages or 1-6 linkages, to form a cyclic structure. Exemplary cyclic oligosaccharide cryoprotectants include cyclic oligosaccharides that are discrete compounds, such as α cyclodextrin, β cyclodextrin, or γ cyclodextrin. Other exemplary cyclic oligosaccharide cryoprotectants include compounds which include a cyclodextrin moiety in a larger molecular structure, such as a polymer that contains a cyclic oligosaccharide moiety. A cyclic oligosaccharide can be oxidized or reduced, for example, oxidized to dicarbonyl forms. The term "cyclodextrin moiety", as used herein refers to cyclodextrin (e.g., an α, β, or γ cyclodextrin) radical that is incorporated into, or a part of, a larger molecular structure, such as a polymer. A cyclodextrin moiety can be bonded to one or more other moieties directly, or through an optional linker. A cyclodextrin moiety can be oxidized or reduced, for example, oxidized to dicarbonyl forms. Carbohydrate cryoprotectants, e.g., cyclic oligosaccharide cryoprotectants, can be derivatized carbohydrates. For example, in an embodiment, the cryoprotectant is a derivatized cyclic oligosaccharide, e.g., a derivatized cyclodextrin, e.g., 2-hydroxypropyl-β-cyclodextrin, e.g., partially etherified cyclodextrins (e.g., partially etherified β cyclodextrins). An exemplary cryoprotectant is a polysaccharide. The term "polysaccharide", as used herein refers to a compound or a chemical moiety formed by at least 16 monosaccharide units that are bonded together through glycosidic linkages, for example through 1-4 linkages or 1-6 linkages, to form a linear, branched or cyclic structure, and includes polymers that comprise polysaccharides as part of their backbone structure. In backbones, the polysaccharide can be linear or cyclic. Exemplary polysaccharide cryoprotectants include glycogen, amylase, cellulose, dextran, maltodextrin and the like. In some embodiments, RNA particle compositions may include sucrose. Without wishing to be bound by theory, sucrose functions to promote cryoprotection of the compositions, thereby preventing RNA (especially mRNA) particle aggregation and maintaining chemical and physical stability of the composition. In some embodiments, RNA particle compositions may include alternative cryoprotectants to sucrose. Alternative stabilizers include, without 99 11529421v1 Attorney Docket No. 2013237‐0757 limitation, trehalose and glucose. In a specific embodiment, an alternative stabilizer to sucrose is trehalose or a mixture of sucrose and trehalose. A preferred cryoprotectant is selected from the group consisting of sucrose, trehalose, glucose, and a combination thereof, such as a combination of sucrose and trehalose. In a preferred embodiment, the cryoprotectant is sucrose. Some embodiments of the present disclosure contemplate the use of a chelating agent in an RNA composition described herein. Chelating agents refer to chemical compounds that are capable of forming at least two coordinate covalent bonds with a metal ion, thereby generating a stable, water-soluble complex. Without wishing to be bound by theory, chelating agents reduce the concentration of free divalent ions, which may otherwise induce accelerated RNA degradation in the present disclosure. Examples of suitable chelating agents include, without limitation, ethylenediaminetetraacetic acid (EDTA), a salt of EDTA, desferrioxamine B, deferoxamine, dithiocarb sodium, penicillamine, pentetate calcium, a sodium salt of pentetic acid, succimer, trientine, nitrilotriacetic acid, trans- diaminocyclohexanetetraacetic acid (DCTA), diethylenetriaminepentaacetic acid (DTPA), and bis(aminoethyl)glycolether-N,N,N',N'-tetraacetic acid. In some embodiments, the chelating agent is EDTA or a salt of EDTA. In some embodiments, the chelating agent is EDTA disodium dihydrate. In some embodiments, the EDTA is at a concentration from about 0.05 mM to about 5 mM, from about 0.1 mM to about 2.5 mM or from about 0.25 mM to about 1 mM. In an alternative embodiment, the RNA particle compositions described herein do not comprise a chelating agent. Pharmaceutical compositions The agents described herein may be administered in pharmaceutical compositions or medicaments and may be administered in the form of any suitable pharmaceutical composition. In some embodiments, the pharmaceutical composition is for therapeutic or prophylactic treatments, e.g., for use in treating or preventing a disease involving an antigen, in particular infections with Mycobacterium tuberculosis, Pseudomonas aeruginosa, Staphylococcus aureus, Clostridioides difficile, Bacillus anthracis, Corynebacterium diphteriae or Vibrio Cholerae. The term "pharmaceutical composition" relates to a composition comprising a therapeutically effective agent, preferably together with pharmaceutically acceptable carriers, diluents and/or excipients. Said pharmaceutical composition is useful for treating, preventing, or reducing the severity of a disease by administration of said pharmaceutical composition to a subject. The pharmaceutical compositions of the present disclosure may comprise one or more adjuvants or may be administered with one or more adjuvants. The term "adjuvant" relates to a compound which prolongs, enhances or accelerates an immune response. Adjuvants comprise a heterogeneous group of compounds such as oil emulsions (e.g., Freund’s adjuvants), mineral compounds (such as alum), bacterial products (such as Bordetella pertussis toxin), or immune-stimulating complexes. Examples of adjuvants include, without limitation, LPS, GP96, CpG oligodeoxynucleotides, growth factors, and cytokines, such as monokines, lymphokines, interleukins, chemokines. The chemokines may be IL-1, IL-2, IL-3, IL-4, IL-5, IL-6, IL-7, IL-8, IL-9, IL-10, IL-12, INFa, INF-γ, 100 11529421v1 Attorney Docket No. 2013237‐0757 GM-CSF, LT-a. Further known adjuvants are aluminum hydroxide, Freund's adjuvant or oil such as Montanide® ISA51. Other suitable adjuvants for use in the present disclosure include lipopeptides, such as Pam3Cys, as well as lipophilic components, such as saponins, trehalose-6,6-dibehenate (TDB), monophosphoryl lipid-A (MPL), monomycoloyl glycerol (MMG), or glucopyranosyl lipid adjuvant (GLA). The pharmaceutical compositions of the present disclosure may be in a storable form (e.g., in a frozen or lyophilized/freeze-dried form) or in a "ready-to-use form" (i.e., in a form which can be immediately administered to a subject, e.g., without any processing such as diluting). Thus, prior to administration of a storable form of a pharmaceutical composition, this storable form has to be processed or transferred into a ready-to-use or administrable form. E.g., a frozen pharmaceutical composition has to be thawed, or a freeze-dried pharmaceutical composition has to be reconstituted, e.g. by using a suitable solvent (e.g., deionized water, such as water for injection) or liquid (e.g., an aqueous solution). The pharmaceutical compositions according to the present disclosure are generally applied in a "pharmaceutically effective amount" and in "a pharmaceutically acceptable preparation". The term "pharmaceutically acceptable" refers to the non-toxicity of a material which does not interact with the action of the active component of the pharmaceutical composition. The term "pharmaceutically effective amount" refers to the amount which achieves a desired reaction or a desired effect alone or together with further doses. In some embodiments relating to the treatment of a particular disease, the desired reaction may relate to inhibition of the course of the disease. This comprises slowing down the progress of the disease and, in some embodiments, interrupting or reversing the progress of the disease. The desired reaction in a treatment of a disease may also be delay of the onset or a prevention of the onset of said disease or said condition, or symptoms thereof. An effective amount of the pharmaceutical compositions described herein will depend on the condition to be treated, the severeness of the disease, the individual parameters of the patient, including age, physiological condition, size and weight, the duration of treatment, the type of an accompanying therapy (if present), the specific route of administration and similar factors. Accordingly, the doses administered of the pharmaceutical compositions described herein may depend on various of such parameters. In the case that a reaction in a patient is insufficient with an initial dose, higher doses (or effectively higher doses achieved by a different, more localized route of administration) may be used. The pharmaceutical compositions of the present disclosure may contain buffers, preservatives, and optionally other therapeutic agents. In some embodiments, the pharmaceutical compositions of the present disclosure comprise one or more pharmaceutically acceptable carriers, diluents and/or excipients. Suitable preservatives for use in the pharmaceutical compositions of the present disclosure include, without limitation, benzalkonium chloride, chlorobutanol, paraben and thimerosal. The term "excipient" as used herein refers to a substance which may be present in a pharmaceutical composition of the present disclosure but is not an active ingredient. Examples of excipients, include without limitation, carriers, 101 11529421v1 Attorney Docket No. 2013237‐0757 binders, diluents, lubricants, thickeners, surface active agents, preservatives, stabilizers, emulsifiers, buffers, flavoring agents, or colorants. The term "diluent" relates a diluting and/or thinning agent. Moreover, the term "diluent" includes any one or more of fluid, liquid or solid suspension and/or mixing media. Examples of suitable diluents include ethanol, glycerol and water. The term "carrier" refers to a component which may be natural, synthetic, organic, inorganic in which the active component is combined in order to facilitate, enhance or enable administration of the pharmaceutical composition. A carrier as used herein may be one or more compatible solid or liquid fillers, diluents or encapsulating substances, which are suitable for administration to subject. Suitable carriers include, without limitation, sterile water, Ringer, Ringer lactate, sterile sodium chloride solution, isotonic saline, polyalkylene glycols, hydrogenated naphthalenes and, in particular, biocompatible lactide polymers, lactide/glycolide copolymers or polyoxyethylene/polyoxy- propylene copolymers. In some embodiments, the pharmaceutical composition of the present disclosure includes isotonic saline. Pharmaceutically acceptable carriers, excipients or diluents for therapeutic use are well known in the pharmaceutical art, and are described, for example, in Remington's Pharmaceutical Sciences, Mack Publishing Co. (A. R Gennaro edit. 1985). Pharmaceutical carriers, excipients or diluents can be selected with regard to the intended route of administration and standard pharmaceutical practice. Routes of administration of pharmaceutical compositions In some embodiments, the pharmaceutical compositions described herein may be administered intravenously, intraarterially, subcutaneously, intradermally, dermally, intranodally, or intramuscularly. In some embodiments, the pharmaceutical compositions described herein may be administered intramuscularly. In some embodiments, the pharmaceutical composition is formulated for local administration or systemic administration. Systemic administration may include enteral administration, which involves absorption through the gastrointestinal tract, or parenteral administration. As used herein, "parenteral administration" refers to the administration in any manner other than through the gastrointestinal tract, such as by intravenous injection. In some embodiments, the pharmaceutical compositions are formulated for systemic administration. In some embodiments, the systemic administration is by intravenous administration. In some embodiments, the pharmaceutical compositions are formulated for intramuscular administration. In some embodiments, intramuscular administration comprises administration into the upper arm, in particular into the musculus deltoideus. If more than one dose, e.g., three doses, of a pharmaceutical composition described herein is administered, the different administrations may be into the same arm. 102 11529421v1 Attorney Docket No. 2013237‐0757 Use of compositions Compositions described herein may be used in the therapeutic or prophylactic treatment of diseases wherein provision of one or more peptides or polypeptides, i.e., polypeptides, described herein to a subject results in a therapeutic or prophylactic effect. In some embodiments, the disease is infection with Mycobacterium tuberculosis. In some embodiments, the disease is tuberculosis. The term "disease" (also referred to as "disorder" herein) refers to an abnormal condition that affects the body of an individual. A disease is often construed as a medical condition associated with specific symptoms and signs. A disease may be caused by factors originally from an external source, such as infectious disease, or it may be caused by internal dysfunctions, such as autoimmune diseases. In humans, "disease" is often used more broadly to refer to any condition that causes pain, dysfunction, distress, social problems, or death to the individual afflicted, or similar problems for those in contact with the individual. In this broader sense, it sometimes includes injuries, disabilities, disorders, syndromes, infections, isolated symptoms, deviant behaviors, and atypical variations of structure and function, while in other contexts and for other purposes these may be considered distinguishable categories. Diseases usually affect individuals not only physically, but also emotionally, as contracting and living with many diseases can alter one's perspective on life, and one's personality. The term "disease involving an antigen" refers to any disease which implicates an antigen, e.g. a disease which is characterized by the presence of an antigen. The disease involving an antigen can be an infectious disease. The antigen may be a disease-associated antigen, such as a bacterial antigen. In some embodiments, a disease involving an antigen is a disease involving cells comprising and/or expressing an antigen, and preferably presenting the antigen on the cell surface, e.g., in the context of MHC. The term "infectious disease" refers to any disease which can be transmitted from individual to individual or from organism to organism, and is caused by a microbial agent (e.g. common cold). Infectious diseases are known in the art and include, for example, a viral disease, a bacterial disease, or a parasitic disease, which diseases are caused by a virus, a bacterium, and a parasite, respectively. In this regard, the infectious disease can be, for example, hepatitis, sexually transmitted diseases (e.g. chlamydia or gonorrhea), tuberculosis, malaria, HIV/acquired immune deficiency syndrome (AIDS), diphtheria, hepatitis B, hepatitis C, cholera, severe acute respiratory syndrome (SARS), the bird flu, and influenza. In the present context, the term "treatment", "treating" or "therapeutic intervention" relates to the management and care of a subject for the purpose of combating a condition such as a disease. The term is intended to include the full spectrum of treatments for a given condition from which the subject is suffering, such as administration of the therapeutically effective compound to alleviate the symptoms or complications, to delay the progression of the disease, disorder or condition, to alleviate or relief the symptoms and complications, and/or to cure or eliminate the disease, disorder or condition as well as to prevent the condition, wherein prevention is to be understood as the management and care of an individual for the purpose of combating the disease, condition or disorder and includes the administration of the active compounds to prevent the onset of the symptoms or complications. 103 11529421v1 Attorney Docket No. 2013237‐0757 The term "therapeutic treatment" relates to any treatment which improves the health status and/or prolongs (increases) the lifespan of an individual. Said treatment may eliminate the disease in an individual, arrest or slow the development of a disease in an individual, inhibit or slow the development of a disease in an individual, decrease the frequency or severity of symptoms in an individual, and/or decrease the recurrence in an individual who currently has or who previously has had a disease. The terms "prophylactic treatment" or "preventive treatment" relate to any treatment that is intended to prevent a disease from occurring in an individual. The terms "prophylactic treatment" or "preventive treatment" are used herein interchangeably. The terms "individual" and "subject" are used herein interchangeably. They refer to a human or another mammal (e.g., mouse, rat, rabbit, dog, cat, cattle, swine, sheep, horse or primate), or any other non-mammal-animal, including birds (chicken), fish or any other animal species that can be afflicted with or is susceptible to a disease (e.g., cancer, infectious diseases) but may or may not have the disease, or may have a need for prophylactic intervention such as vaccination, or may have a need for interventions such as by protein replacement. In many embodiments, the individual is a human being. Unless otherwise stated, the terms "individual" and "subject" do not denote a particular age, and thus encompass adults, elderlies, children, and newborns. In some embodiments of the present disclosure, the "individual" or "subject" is a "patient". In some embodiments, the terms "individual" and "subject" relate to pregnant women and immunocompromised persons. The term "patient" means an individual or subject for treatment, in particular a diseased individual or subject. RNA described herein may be administered to a subject for delivering the RNA to cells of the subject. RNA described herein may be administered to a subject for delivering a therapeutic or prophylactic peptide or polypeptide (e.g., a pharmaceutically active peptide or polypeptide) to the subject, wherein the RNA encodes a therapeutic or prophylactic peptide or polypeptide. RNA described herein may be administered to a subject for treating or preventing a disease in a subject, wherein delivering the RNA to cells of the subject is beneficial in treating or preventing the disease. RNA described herein may be administered to a subject for treating or preventing a disease in a subject, wherein the RNA encodes a therapeutic or prophylactic peptide or polypeptide and wherein delivering the therapeutic or prophylactic peptide or polypeptide to the subject is beneficial in treating or preventing the disease. In some embodiments of the disclosure, the aim is to induce an immune response by providing RNA described herein. A person skilled in the art will know that one of the principles of immunotherapy and vaccination is based on the fact that an immunoprotective reaction to a disease is produced by immunizing a subject with a polypeptide or an epitope, which is immunologically relevant with respect to the disease to be treated. Accordingly, RNA described herein is applicable for inducing or enhancing an immune response. RNA described herein is thus useful in a prophylactic and/or therapeutic treatment of a disease involving a polypeptide or epitope. 104 11529421v1 Attorney Docket No. 2013237‐0757 In some embodiments of the disclosure, the aim is to provide an immune response against cells comprising an antigen, e.g., Mtb antigen. In some embodiments of the disclosure, the aim is to prophylactically or therapeutically treat tuberculosis by vaccination. Due to the high degree of sequence conservation of the disclosed antigens between different Mycobacterium species, including Mycobacterium tuberculosis, Mycobacterium bovis, Mycobacterium caprae, Mycobacterium orygis, Mycobacterium africanum, Mycobacterium microti, Mycobacterium canetti and Mycobacterium pinnipedii, exposure of a subject to Mycobacterium tuberculosis antigens, in the context of a polypeptide of the invention, will result in a high degree of cross-reactivity with antigens of other Mycobacterium species. Therefore, a vaccine based on or directed at Mycobacterium tuberculosis antigens will elicit a robust immune response against other Mycobacterium species, in particular Mycobacterium bovis, Mycobacterium caprae, Mycobacterium orygis, Mycobacterium africanum, Mycobacterium microti, Mycobacterium canetti and Mycobacterium pinnipedii as well. In some embodiments of the disclosure, the aim is to provide an immune response against a Mycobacterium selected from Mycobacterium tuberculosis, Mycobacterium bovis, Mycobacterium caprae, Mycobacterium orygis, Mycobacterium africanum, Mycobacterium microti, Mycobacterium canetti and Mycobacterium pinnipedii and to prevent or treat tuberculosis. In preferred embodiments of the disclosure, the aim is to provide an immune response against Mycobacterium tuberculosis. In some embodiments of the disclosure, the aim is to treat an infection with Mycobacterium tuberculosis. In some embodiments of the disclosure, the aim is to prevent or treat disease symptoms caused by an infection with Mycobacterium tuberculosis. In some embodiments of the disclosure, the aim is to provide protection against an infection with Mycobacterium tuberculosis by vaccination. In some embodiments of the disclosure, the aim is to provide protection against an outbreak of disease in a subject infected with Mycobacterium tuberculosis. In some embodiments of the disclosure, the aim is to provide protection against symptoms of tuberculosis in a subjected infected with Mycobacterium tuberculosis. In some embodiments, the RNA is present in a composition as described herein. In some embodiments, the RNA is administered in a pharmaceutically effective amount. In some embodiments, the subject is a mammal. In some embodiments, the mammal is a human. In some embodiments, the subject treated had been exposed to Mycobacterium tuberculosis. In some embodiments, the subject treated had not been exposed to Mycobacterium tuberculosis. 105 11529421v1 Attorney Docket No. 2013237‐0757 In some embodiments, the treatments described herein involve pre- or post-exposure vaccination against Mycobacterium tuberculosis, or a combination thereof. Citation of documents and studies referenced herein is not intended as an admission that any of the foregoing is pertinent prior art. All statements as to the contents of these documents are based on the information available to the applicants and do not constitute any admission as to the correctness of the contents of these documents. The description (including the following examples) is presented to enable a person of ordinary skill in the art to make and use the various embodiments. Descriptions of specific devices, techniques, and applications are provided only as examples. Various modifications to the examples described herein will be readily apparent to those of ordinary skill in the art, and the general principles defined herein may be applied to other examples and applications without departing from the spirit and scope of the various embodiments. Thus, the various embodiments are not intended to be limited to the examples described herein and shown, but are to be accorded the scope consistent with the claims. Examples Example 1: Observation that EspA is toxic when expressed as mRNA in HEK293T cells. EspA (Rv3616c) was encoded on mRNA using its wild-type protein sequence modified by an N-terminal signal peptide (human HLA-A1 derived, hereafter SP) and a FLAG-epitope tag. This construct was investigated for expression in HEK293T cells by RiboJuiceTM (Sigma-Aldrich) transfection of 1 µg of RNA. After overnight incubation (approximately 18h) cells were inspected visually under a bright field microscope. Strikingly, a large reduction in cell confluency could be observed in cells transfected with EspA compared to the non-transfected control or compared to control protein 1 which was also FLAG-tagged and known to be well expressed (Figure 1). Lysates were generated and analyzed by western blot for expression of the FLAG-tag, which could be well detected for control protein 1. However, no expression was found for EspA (Figure 2). These results were our first indication that expression of EspA could be highly toxic for cells when expressed on mRNA. Example 2: Confirmation of EspA expression and toxicity and introducing live imaging as a method to quantify toxicity. Wild type EspA or a control protein with a similar predicted molecular weight and a similar predicted tertiary structure (control protein 2), were expressed in HEK293T cells by RiboJuiceTM transfection of different amounts of RNA (25 ng, 100 ng, 250 ng or 1000 ng). Both proteins were FLAG-tagged and fused with a N-terminal signal peptide (SP). Lysates of the cells were generated 6 hours post transfection or 24h post transfection. Lysates from transfected cells were separated by SDS-PAGE on a 6-18% polyacrylamide gel and western blot using an FLAG- specific monoclonal antibody conjugated to horseradish peroxidase (HRP; clone M2 from Sigma-Aldrich) and a α- Tubulin-specific HRP-conjugated rabbit monoclonal antibody (clone 11H10 from Cell Signaling Technology). The 106 11529421v1 Attorney Docket No. 2013237‐0757 FLAG-tagged control protein 2 was detected at both timepoints (250ng and 1000ng of RNA; Figure 3). In contrast, and similar to example 1, EspA expression could not be detected 24 hours post transfection. However, at 6 hours post transfection a faint band could be seen at the indicated size confirming that EspA is expressed early after mRNA transfection. In parallel HEK293T cells were transfected with the same constructs and incubated with Incucyte® Cytotox Red Dye for Counting Dead Cells in the Incucyte® S3 Live-Cell Analysis System (Sartorius). Pictures were taken in 2-hour intervals for 24 hours to measure cell confluence (bright field images) and Cytotox- staining of cells with impaired membrane potential as measure of cell death (Red channel, excitation 567-607 nm, emission 622-704 nm). Analysis was performed using the Incucyte® Live-Cell Analysis software provided by Sartorius. The Basic Analyzer analysis type was used. Confluence of cells was determined based on shape and size, where small blebs were excluded (Area (μm2) min = 200.00, Cleanup Adjust size (pixels) = -6). Cytotox positivity was defined based on size and signal intensity (Segmentation: Surface fit, Threshold (RCU) = 4.0000, Edge Split Off), where disproportionately small and large spots were removed as well as very weak signals (Area (μm2) min = 12.000, Mean Intensity min = 8.0000, Integrated Intensity min = 600.00). Confluency was defined in percentage (%), while Cytotox+ spots were defined in Red calibrated units (RCU) times square μm per image (RCU*μm2/image). Both parameters were normalized to a fold change compared to t = 0. Confluency decreased while the signal for the Cytotox dye increased when cells were transfected with EspA (Figure 4A-C). The opposite was true for control protein 2. Especially the concentration of 250 ng RNA clearly indicated the differences between confluency and Cytotox-stained cells upon transfection of both constructs. Example 3: EspA toxicity is primarily driven by the N-terminal A domain. Since it was found that EspA was toxic when expressed as a wild type, full-length protein, a bioinformatic analysis was performed to separate the protein into domains to determine whether this would impair cytotoxicity and if the part of the protein could be determined, which mediated its cytotoxicity. Based on sequence comparison with the human proteome, two regions of potential overlap with the human proteome were identified and excluded moving forward. Therefore, three major domains termed EspA_A (aa 2-166), EspA_B (aa 177-280), and EspA_C (aa 291- 392) were selected (Figure 5). Furthermore, to make sure that specifically these excluded overlapping-regions were not essential for determining toxicity of EspA, two constructs were designed, that combined the N-terminal two parts and the region of human overlap bridging them, dubbed EspA_AB, and a similar construct for the C- terminal parts called EspA_BC (Figure 6). To assess expression of the generated constructs, cells were seeded and incubated overnight before transfection with 250 ng RNA/well as described above. The negative control consists of non-transfected cells. Cells were incubated for 4h at 37°C by 5% CO¬2 and transferred to a 96-well plate and washed. Samples were stained with Fixable Viability Dye eFluor™ 780 (Thermo Fisher Scientific) for 15 minutes at room temperature. Cells were washed and fixed for 12 minutes at room temperature, followed by permeabilization for 5 minutes at 4°C. Permeabilized cells were stained with a FLAG-tag specific antibody (BioLegend) for 30 min at 4°C. Cells were washed and acquired with a FACS Celesta (BD Biosciences). Data was analyzed using FACS Diva and Flow Jo software (both BD Biosciences). Gating excluded doublet cells and debris (Figure 7A). Cell viability was measured on the B-A-780-A spectrum, while FLAG-positive cells were measured on the BV421-A spectrum. Antigen expression as reflected by 107 11529421v1 Attorney Docket No. 2013237‐0757 FLAG-tag binding was obtained from the viable cell population. Transfected constructs containing the A fragment of EspA showed a higher percentage of dead cells compared to control protein 2 (p<0.01) (Figure 7B and C). Upon transfection with EspA_FL, approximately 5% more cells were dead as compared to transfection of the control protein 2 encoding construct. Transfection with EspA_A and EspA_AB resulted in approximately 30% (p<0.01) and over 10% (p<0.05) more cell death, respectively. All other constructs had a similar viability as the negative construct control (p>0.05). As expected, in the non-transfected control, no cells of the viable cell population were staining positive for the FLAG-tag (Figure 7B and C). On the contrary, constructs encoding control protein 2, EspA_B, EspA_C, and EspA_BC did stain positive. The constructs with high cell death barely showed any positivity for the tag in either of the cell populations. This is most likely due to the viable cells not being transfected with the construct, and the cells that were transfected were either currently dying, accounting for the slight positivity, or were already dead and the FLAG-tagged protein was already degraded. To investigate the cytotoxicity of the generated EspA constructs (Figure 6) HEK293T cells were seeded and incubated as stated above (Example 2). Cells were transfected with 250 ng of RNA constructs encoding EspA_FL, EspA_A, EspA_B, EspA_C, EspA_AB, EspA_BC, or control proteins 2 and 3. Control protein 3 is non-toxic, of the same size as EspA-FL and control protein 2 and was added to further validate the assay results. Non-transfected cells served as a negative control. Immediately after transfection, cells were imaged in bright field and for fluorescence signals every hour for 24h. Cells transfected with constructs containing the EspA-A-fragment all showed increased Cytotox+ integrated intensity and decreased confluency indicating cytotoxicity (Figure 8A and B). EspA_A induced slightly more cytotoxicity when compared to EspA_FL. Both control proteins 2 and 3 behaved similarly to the non-transfected control and did not differ between each other (p<0.05). EspA_FL, EspA_A, and EspA_AB differed from control proteins 2 and 3 (p<0.001). Additionally, slightly higher values for EspA_B were observed as well (p<0.05). EspA_C and EspA_BC did not differ from the negative controls (p>0.05). Clearly, the highest toxicity values are indicated by constructs containing the A fragment, hence confirming the flow cytometer data. Example 4: A structure-guided T-cell string design to disrupt toxicity of EspA To visualize the domain architecture of EspA, AlphaFold2 was used to generate a model of EspA (Figure 9, panel A). There is high confidence in the EspA model due to high homology with EspB, which has high resolution crystal structures (https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4520771/) and a cryo-electron microscopy higher order pore structure (https://www.sciencedirect.com/science/article/pii/S2590152420300118?via%3Dihub) deposited in the Protein Data Bank (PDB). The model was juxtaposed against the bioinformatics data described above showing regions of human overlap. The EspA protein sequence was divided into subdomains in a manner that disrupted secondary structure elements and tertiary protein structure and function (Figure 9, panel B). Fragments were selected such that areas of human overlap were avoided when selecting EspA chunks for inclusion in the T cell string. In one approach, the EspA protein sequence was divided into subdomains containing three regions of EspA: EspA_3-1 (amino acids 1-166 of FL EspA), EspA_3-2 (amino acids 177-280 of FL EspA), and EspA_3-3 (amino acids 291-392 of FL EspA). The fragments were rearranged in a manner that disrupted tertiary and secondary structure 108 11529421v1 Attorney Docket No. 2013237‐0757 elements and function (Fig.9B and 9C). In another approach, the three fragments were further split up to generate seven fragments in total: EspA_3-1 was further fragmented into EspA_7-1 (amino acids 1-60 of FL EspA), EspA_7- 2 (amino acids 62-131 of FL EspA), and EspA_7-3 (amino acids 135-166 of FL EspA), EspA_3-2 was further fragmented into EspA_7-4 (amino acids 177-239 of FL EspA) and EspA_7-5 (amino acids 241-279 of FL EspA), and EspA_3-3 was further fragmented into EspA_7-6 (amino acids 291-341 of FL EspA), and EspA_7-7 (amino acids 343-392 of FL EspA). Again, the fragments were rearranged in a manner that disrupted tertiary and secondary structure elements and function (Fig. 9B and 9D). In both approaches, adjacent EspA regions were reshuffled to ensure that EspA chunks could not properly assemble into a functional protein during co-translation folding (Figure 9, C and D). It was hypothesized that this strategy would reduce toxicity and combined with the MITD, increase protein turnover and generation of immunogenic T cell epitopes. Example 5: Analysis of expression of the 3-part and 7-part EspA strings in vitro by mass-spectrometry RNA drug substance encoding string 2 (where EspA was separated and shuffled in 7 parts with linkers – Figure 10, panel A) was transfected into HEK293T cells in the presence (Light grey bars) or absence (Dark grey bars) of proteasome inhibitor MG132. Following lysis, total protein content was normalized, cysteines were reduced and alkylated, and proteins were subjected to tryptic digestion. The samples were analyzed by targeted and discovery MS to look for tryptic peptides derived uniquely from the transfected RNA strings (Figure 10, panel B). The indicated peptide (NHVNFFQELADLDR) was synthesized as a heavy peptide and its counterparts could be detected in the total trypsinated lysate from transfected with EspA string 2 in the presence of MG132 proteasome inhibitor. No expression was detected from non-transfected cells independently of the presence of MG132. These data confirm expression of EspA string 2 after overnight expression and the proteasome inhibitor data suggest that this string is efficiently turned over and degraded in normal conditions. Example 6: Structure-guided T-cell string approach allows processing and presentation of predicted HLA-epitopes RNA drug substance (EspA_String2) was transfected into A375 cells stably expressing an affinity-tagged version of HLA-B*07:02. Following lysis, tagged HLA molecules were labeled with biotin and enriched on NeutrAvidin beads. Following washing, peptides were eluted from HLA complexes using acid and filtered through molecular weight cutoff filters. Cysteines were reduced and alkylated, and peptides were cleaned up by C18 desalting. Heavy isotope- labeled synthetic peptides were spiked into each sample, and targeted and discovery MS was performed to look for epitope peptides derived uniquely from transfected mRNA constructs. The different detected peptides are depicted in the visual representation of the T cell string as well as in the table (Figure 11) depicting their exact sequence. Note that the detected epitopes were derived from different parts of the T-cell string indicating that the whole construct has the potential to be processed and presented on human HLA molecules. Example 7: T-cell string approach detoxifies EspA Cells were seeded and incubated as described above and transfected with RNA constructs encoding EspA_FL, EspA_A, EspA_B, EspA_C, EspA string1, EspA string2 or control protein 2 at a concentration of 250 ng/well. Wells 109 11529421v1 Attorney Docket No. 2013237‐0757 were stained with 1:4000 diluted Incucyte® Cytotox Red Dye for Counting Dead Cells. Negative controls consisted of non-transfected cells. The plate was placed in the Incucyte® S3 Live-Cell Analysis System directly post transfection. Wells were imaged in bright field and fluorescence every hour for 24h. All analyses were performed identically to those described in example 3. As before, the EspA_FL and EspA_A proteins were cytotoxic to the cells (Figure 12, panel A). However, the Cytotox+ of the two strings was significantly lower than EspA itself (p<0.05) and EspA_String2 is even lower than control protein 2 (p<0.001). The confluency of EspA string1 matched that of control protein 2 (p>0.05), while that of EspA string2 was more comparable with the non-transfected control (p<0.01 when compared to control protein 2) (Figure 12, panel B). Both show better confluence than EspA (p<0.05). These data indicate that both string designs, but especially the structure-guided approach used for EspA_String2, are highly effective in detoxifying EspA. Example 8: T-cell strings of EspA are immunogenic in C57BL/6 mice and leads to polyfunctional T- cell responses Adult female C57BL/6 mice were immunized with 4 µg of EspA String1 or String2 (structure-guided detoxification) RNA constructs formulated in a lipid nanoparticle formulation (in 20 µl of volume) or with 0.9% NaCl solution as a control. 21 days after the primary immunization the mice were boosted with the same dose. 14 days after the boosting dose (35 days post primary immunization) mice were sacrificed and splenocytes were isolated. In addition, a portion of the splenocytes was pooled and separated into CD4+ and CD8+ fractions by magnetic associated cell- sorting (MACS). Both the total splenocytes and the CD4+ and CD8+ fractions were assessed for T-cell reactogenicity by restimulation with peptide pools specific for individual chunks (or parts of chunks) of the T-cell string. The peptide libraries consist of 15 amino acid long peptides with a 5 amino acid interval. In other words, the first peptide will cover amino acid 1-15 of the antigen and the second peptide will cover amino acids 16-20, and so forth. Fluorospot was used as a read-out to determine IFN-γ, IL-2 and TNF-α production. The peptides libraries were not immunogenic above baseline (Figure 13). In both EspA String1 and String2, immunogenic regions were identified especially with peptide libraries of EspA_7.2 and 7.3 which correspond to the sequences of Chunk EspA_A which was found to be cytotoxic when expressed in itself (Figure 14). Some responses above baseline could be detected in response to peptide library EspA_7.5, 7.6 and 7.7, but these were all markedly lower than the responses detected toward 7.2 and 7.3. Separation of splenocytes into CD4+ and CD8+ T cells confirmed the results above and clearly indicated that EspA_7.2 includes both CD4+ and CD8+ epitopes while the responses detected to EspA_7.3 are determined by CD8+ responses (Figure 15). These data also confirmed the immunogenicity of EspA_7.5-7.7 and indicated that these relatively low levels of immune responses were primarily mediated by CD4+ T cells. CD4+ and CD8+ T cells can also be subdivided into different cytokine-secreting classes, called polyfunctional cells. Combinations of two or three cytokines were identified as well (Figure 16). For both strings, CD4+ T cells mostly secreted IL-2 in combination with one of the other two cytokines, whereas some also secreted all three. In contrast, CD8+ T cells mostly secreted IFN-γ and TNF-α (which is known for CD8+ T cells). Additionally, the cells from both string-immunized study groups induced a strong immune response against EspA_7.2 and EspA_7.3 as seen before (Figure 15). EspA_7.2 induced strong IFN-γ+ IL-2+ TNF-α+ CD4+ T cell responses, but no such responses were 110 11529421v1 Attorney Docket No. 2013237‐0757 observed for EspA_7.3. In contrast, the IFN-γ+ TNF-α+ CD8+ T cell responses were higher for EspA_7.3 compared to EspA_7.2. Example 9: Polyfunctional T cell responses Together, these data show that EspA_A (The N-terminal 166 amino acids) contains the most immunogenic regions of EspA. Therefore, classic truncation of the protein to only detect non-toxic section of the antigen would also result in a predicted loss of immunogenicity of EspA. In contrast, the T-cell string approach has successfully reduced toxicity of EspA_A in vitro and is able to induce strong T-cell responses. Finally, structure-guided detoxification led to further reduction of in vitro toxicity and increased in vitro expression levels as detected in examples 3 and 4 respectively, but also increased immunogenicity. Especially as measured by polyfunctional T-cell responses, which in murine studies are considered to be advantageous in the protection against TB infection. Confirming that the T-cell string approach was successful and specific in inducing immunogenicity of multiple different antigens simultaneously. Analysis of MACS´ed cells reveals that T Cell strings can induce potent CD4+ and CD8+ responses. 111 11529421v1
G AP S AI PI GI GAAGII Q4 GGI YTQFEAV A GAS GDAG: GR GLAIES VGO GLDS WGS GGV N W WL VV A Q D VAL QEV GI YL G KWGD LSQ II EAD I A AG QYL VPS QLTG K T G DAP DAQ L L GR STGL AAE KTS DI VG LG GG KG G T GP S AI AGVS [ SA A QT SFEVIIGKFASSGP * KS IA TL GQED AGS AAAAGS G D T GEP KA EV Q DIK AAV S GGL L ADS GSS E SG E DGALS 5 AGRP V 9 DLDI LIFGG SQAE RL VIIALPL AHGRLDS I P DAAP H MKAL S GAG HT IKS D G D IALSGEF GGQQ AR A SL M TD GR R T AGA T RGS GYS GE SST RL ALSS D VG PS SA S SAAS NS GIV GQ V RL GL V GSYL GHA HSA QR IEH FL A NAG QGS GD H A LLIAG GAV V V QA Q AGARSGGGPAS SN AGecV nIVQ D TS GH GA V L IVQFL IYL Y T S S AAV HAVNGL P G AE seeu F IAVV GS ]FPIT D L GG EQ A ] d q LILS HNS 44 LIGV VGK I G] A1 F7 L : T2 A7ite :ps V e dAILV QRA VSVIQ iGQRAL LL : GO AG N GL SA L LV SG GP GG GSS N OA D O NpcL R D S VKAL DSR V GK WN T yl a AL PQADIRAYL T F GR Q DKF Q I MGD o M Ip oAD L nAALG GGG A GFL QE AGA D GE S R GL GGC ETQEEQQy iGE S GGS GIGKGLMV MASA [ MSE SS[ral mGQGAT[ GAS S KGT p A MF G D V * MI G A P A m ex ) e si) ssi f o os los ulu e crc c er n bebe u uu t tqe m ms uirui d er teic ctca aba obo n co yci e mdi1 My v (M(A t :p1 62 -t0 a15e e p 1 2 yg g4 sP lni n92 EF l C b a orir 5 T PtStS117570‐7323102.oNtekcoDyenrottA 69 1v12492511 Attorney Docket No. 2013237‐0757 RNA delivery RNA described herein may be delivered for therapeutic applications described herein using any appropriate methods known in the art, including, e.g., delivery as naked RNA, or delivery mediated by delivery vehicles. Some aspects of the disclosure involve the targeted delivery of the RNA disclosed herein to certain cells or tissues. In some embodiments, after administration of the RNA (in particular, mRNA) compositions/formulations described herein, at least a portion of the RNA is delivered to a target cell or target organ. In some embodiments, at least a portion of the RNA is delivered to the cytosol of the target cell. In some embodiments, the RNA (in particular, mRNA) is translated by the target cell to produce the encoded peptide or polypeptide. In some embodiments, the target cell is a muscle cell. In some embodiments, the target cell is a cell in the liver. In some embodiments, the target cell is a cell in the lung. In some embodiments, the disclosure involves targeting the lymphatic system, in particular secondary lymphoid organs, more specifically spleen. In some embodiments, the target cell is a cell in the lymph nodes. In some embodiments, the target cell is a spleen cell. In some embodiments, the target cell is an antigen presenting cell such as a professional antigen presenting cell in the spleen. In some embodiments, the target cell is a dendritic cell in the spleen. Thus, RNA (in particular, mRNA) compositions/formulations described herein may be used for delivering RNA to such target cell. The "lymphatic system" is part of the circulatory system and an important part of the immune system, comprising a network of lymphatic vessels that carry lymph. The lymphatic system consists of lymphatic organs, a conducting network of lymphatic vessels, and the circulating lymph. The primary or central lymphoid organs generate lymphocytes from immature progenitor cells. The thymus and the bone marrow constitute the primary lymphoid organs. Secondary or peripheral lymphoid organs, which include lymph nodes and the spleen, maintain mature naive lymphocytes and initiate an adaptive immune response. Lipid-based RNA delivery systems have an inherent preference to the liver, where, depending on the composition of the RNA delivery systems used, RNA expression in the liver can be obtained. Liver accumulation is caused by the discontinuous nature of the hepatic vasculature or the lipid metabolism (liposomes and lipid or cholesterol conjugates). In some embodiments, the target organ for RNA expression is liver and the target tissue is liver tissue. The delivery to such target tissue is preferred, in particular, if presence of RNA or of the encoded peptide or polypeptide in this organ or tissue is desired and/or if it is desired to express large amounts of the encoded peptide or polypeptide and/or if systemic presence of the encoded peptide or polypeptide, in particular in significant amounts, is desired or required. Delivery vehicles To overcome the barriers to safe and effective RNA delivery, RNA may be administered with one or more delivery vehicles that protect the RNA from degradation, maximize delivery to on-target cells and minimize exposure to off- target cells. Such RNA delivery vehicles may complex or encapsulate RNA and include a range of materials, including polymers and lipids. In some embodiments, such RNA delivery vehicles may form particles with RNA. Suitable formulations for such delivery vehicles are described, e.g., in WO2023/126404, WO2023/126053 and WO 2023/094713, the contents of which are incorporated herein by reference. 97 11529421v1 Attorney Docket No. 2013237‐0757 Compositions comprising nucleic acid A composition comprising one or more RNAs described herein, e.g., in the form of RNA particles, may comprise salts, buffers, or other components as further described below. In some embodiments, a salt for use in the compositions described herein comprises sodium chloride. Without wishing to be bound by theory, sodium chloride functions as an ionic osmolality agent for preconditioning RNA prior to mixing with lipids. In some embodiments, the compositions described herein may comprise alternative organic or inorganic salts. Alternative salts include, without limitation, potassium chloride, dipotassium phosphate, monopotassium phosphate, potassium acetate, potassium bicarbonate, potassium sulfate, disodium phosphate, monosodium phosphate, sodium acetate, sodium bicarbonate, sodium sulfate, lithium chloride, magnesium chloride, magnesium phosphate, calcium chloride, and sodium salts of ethylenediaminetetraacetic acid (EDTA). Generally, compositions for storing RNA particles such as for freezing RNA particles comprise low sodium chloride concentrations, or comprises a low ionic strength. In some embodiments, the sodium chloride is at a concentration from 0 mM to about 50 mM, from 0 mM to about 40 mM, or from about 10 mM to about 50 mM. According to the present disclosure, the RNA particle compositions described herein have a pH suitable for the stability of the RNA particles and, in particular, for the stability of the RNA. Without wishing to be bound by theory, the use of a buffer system maintains the pH of the particle compositions described herein during manufacturing, storage and use of the compositions. In some embodiments of the present disclosure, the buffer system may comprise a solvent (in particular, water, such as deionized water, in particular water for injection) and a buffering substance. The buffering substance may be selected from 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid (HEPES), 2-amino-2-(hydroxymethyl)propane-1,3-diol (Tris), acetate, and histidine. In some embodiments, the buffering substance is HEPES. In some embodiments, the buffering substance is Tris. Compositions (in particular, RNA compositions/formulations) described herein may also comprise a cryoprotectant and/or a surfactant as stabilizer to avoid substantial loss of the product quality and, in particular, substantial loss of RNA activity during storage, freezing, and/or lyophilization, for example to reduce or prevent aggregation, particle collapse, RNA degradation and/or other types of damage. In some embodiments, the cryoprotectant is a carbohydrate. The term "carbohydrate", as used herein, refers to and encompasses monosaccharides, disaccharides, trisaccharides, oligosaccharides and polysaccharides. In some embodiments, the cryoprotectant is a monosaccharide. The term "monosaccharide", as used herein refers to a single carbohydrate unit (e.g., a simple sugar) that cannot be hydrolyzed to simpler carbohydrate units. Exemplary monosaccharide cryoprotectants include glucose, fructose, galactose, xylose, ribose and the like. In some embodiments, the cryoprotectant is a disaccharide. The term "disaccharide", as used herein refers to a compound or a chemical moiety formed by 2 monosaccharide units that are bonded together through a glycosidic linkage, for example through 1-4 linkages or 1-6 linkages. A disaccharide may be hydrolyzed into two 98 11529421v1 Attorney Docket No. 2013237‐0757 monosaccharides. Exemplary disaccharide cryoprotectants include sucrose, trehalose, lactose, maltose and the like. In some embodiments, the cryoprotectant is sucrose. The term "trisaccharide" means three sugars linked together to form one molecule. Examples of a trisaccharides include raffinose and melezitose. In some embodiments, the cryoprotectant is an oligosaccharide. The term "oligosaccharide", as used herein refers to a compound or a chemical moiety formed by 3 to about 15, such as 3 to about 10 monosaccharide units that are bonded together through glycosidic linkages, for example through 1-4 linkages or 1-6 linkages, to form a linear, branched or cyclic structure. Exemplary oligosaccharide cryoprotectants include cyclodextrins, raffinose, melezitose, maltotriose, stachyose, acarbose, and the like. An oligosaccharide can be oxidized or reduced. In an embodiment, the cryoprotectant is a cyclic oligosaccharide. The term "cyclic oligosaccharide", as used herein refers to a compound or a chemical moiety formed by 3 to about 15, such as 6, 7, 8, 9, or 10 monosaccharide units that are bonded together through glycosidic linkages, for example through 1-4 linkages or 1-6 linkages, to form a cyclic structure. Exemplary cyclic oligosaccharide cryoprotectants include cyclic oligosaccharides that are discrete compounds, such as α cyclodextrin, β cyclodextrin, or γ cyclodextrin. Other exemplary cyclic oligosaccharide cryoprotectants include compounds which include a cyclodextrin moiety in a larger molecular structure, such as a polymer that contains a cyclic oligosaccharide moiety. A cyclic oligosaccharide can be oxidized or reduced, for example, oxidized to dicarbonyl forms. The term "cyclodextrin moiety", as used herein refers to cyclodextrin (e.g., an α, β, or γ cyclodextrin) radical that is incorporated into, or a part of, a larger molecular structure, such as a polymer. A cyclodextrin moiety can be bonded to one or more other moieties directly, or through an optional linker. A cyclodextrin moiety can be oxidized or reduced, for example, oxidized to dicarbonyl forms. Carbohydrate cryoprotectants, e.g., cyclic oligosaccharide cryoprotectants, can be derivatized carbohydrates. For example, in an embodiment, the cryoprotectant is a derivatized cyclic oligosaccharide, e.g., a derivatized cyclodextrin, e.g., 2-hydroxypropyl-β-cyclodextrin, e.g., partially etherified cyclodextrins (e.g., partially etherified β cyclodextrins). An exemplary cryoprotectant is a polysaccharide. The term "polysaccharide", as used herein refers to a compound or a chemical moiety formed by at least 16 monosaccharide units that are bonded together through glycosidic linkages, for example through 1-4 linkages or 1-6 linkages, to form a linear, branched or cyclic structure, and includes polymers that comprise polysaccharides as part of their backbone structure. In backbones, the polysaccharide can be linear or cyclic. Exemplary polysaccharide cryoprotectants include glycogen, amylase, cellulose, dextran, maltodextrin and the like. In some embodiments, RNA particle compositions may include sucrose. Without wishing to be bound by theory, sucrose functions to promote cryoprotection of the compositions, thereby preventing RNA (especially mRNA) particle aggregation and maintaining chemical and physical stability of the composition. In some embodiments, RNA particle compositions may include alternative cryoprotectants to sucrose. Alternative stabilizers include, without 99 11529421v1 Attorney Docket No. 2013237‐0757 limitation, trehalose and glucose. In a specific embodiment, an alternative stabilizer to sucrose is trehalose or a mixture of sucrose and trehalose. A preferred cryoprotectant is selected from the group consisting of sucrose, trehalose, glucose, and a combination thereof, such as a combination of sucrose and trehalose. In a preferred embodiment, the cryoprotectant is sucrose. Some embodiments of the present disclosure contemplate the use of a chelating agent in an RNA composition described herein. Chelating agents refer to chemical compounds that are capable of forming at least two coordinate covalent bonds with a metal ion, thereby generating a stable, water-soluble complex. Without wishing to be bound by theory, chelating agents reduce the concentration of free divalent ions, which may otherwise induce accelerated RNA degradation in the present disclosure. Examples of suitable chelating agents include, without limitation, ethylenediaminetetraacetic acid (EDTA), a salt of EDTA, desferrioxamine B, deferoxamine, dithiocarb sodium, penicillamine, pentetate calcium, a sodium salt of pentetic acid, succimer, trientine, nitrilotriacetic acid, trans- diaminocyclohexanetetraacetic acid (DCTA), diethylenetriaminepentaacetic acid (DTPA), and bis(aminoethyl)glycolether-N,N,N',N'-tetraacetic acid. In some embodiments, the chelating agent is EDTA or a salt of EDTA. In some embodiments, the chelating agent is EDTA disodium dihydrate. In some embodiments, the EDTA is at a concentration from about 0.05 mM to about 5 mM, from about 0.1 mM to about 2.5 mM or from about 0.25 mM to about 1 mM. In an alternative embodiment, the RNA particle compositions described herein do not comprise a chelating agent. Pharmaceutical compositions The agents described herein may be administered in pharmaceutical compositions or medicaments and may be administered in the form of any suitable pharmaceutical composition. In some embodiments, the pharmaceutical composition is for therapeutic or prophylactic treatments, e.g., for use in treating or preventing a disease involving an antigen, in particular infections with Mycobacterium tuberculosis, Pseudomonas aeruginosa, Staphylococcus aureus, Clostridioides difficile, Bacillus anthracis, Corynebacterium diphteriae or Vibrio Cholerae. The term "pharmaceutical composition" relates to a composition comprising a therapeutically effective agent, preferably together with pharmaceutically acceptable carriers, diluents and/or excipients. Said pharmaceutical composition is useful for treating, preventing, or reducing the severity of a disease by administration of said pharmaceutical composition to a subject. The pharmaceutical compositions of the present disclosure may comprise one or more adjuvants or may be administered with one or more adjuvants. The term "adjuvant" relates to a compound which prolongs, enhances or accelerates an immune response. Adjuvants comprise a heterogeneous group of compounds such as oil emulsions (e.g., Freund’s adjuvants), mineral compounds (such as alum), bacterial products (such as Bordetella pertussis toxin), or immune-stimulating complexes. Examples of adjuvants include, without limitation, LPS, GP96, CpG oligodeoxynucleotides, growth factors, and cytokines, such as monokines, lymphokines, interleukins, chemokines. The chemokines may be IL-1, IL-2, IL-3, IL-4, IL-5, IL-6, IL-7, IL-8, IL-9, IL-10, IL-12, INFa, INF-γ, 100 11529421v1 Attorney Docket No. 2013237‐0757 GM-CSF, LT-a. Further known adjuvants are aluminum hydroxide, Freund's adjuvant or oil such as Montanide® ISA51. Other suitable adjuvants for use in the present disclosure include lipopeptides, such as Pam3Cys, as well as lipophilic components, such as saponins, trehalose-6,6-dibehenate (TDB), monophosphoryl lipid-A (MPL), monomycoloyl glycerol (MMG), or glucopyranosyl lipid adjuvant (GLA). The pharmaceutical compositions of the present disclosure may be in a storable form (e.g., in a frozen or lyophilized/freeze-dried form) or in a "ready-to-use form" (i.e., in a form which can be immediately administered to a subject, e.g., without any processing such as diluting). Thus, prior to administration of a storable form of a pharmaceutical composition, this storable form has to be processed or transferred into a ready-to-use or administrable form. E.g., a frozen pharmaceutical composition has to be thawed, or a freeze-dried pharmaceutical composition has to be reconstituted, e.g. by using a suitable solvent (e.g., deionized water, such as water for injection) or liquid (e.g., an aqueous solution). The pharmaceutical compositions according to the present disclosure are generally applied in a "pharmaceutically effective amount" and in "a pharmaceutically acceptable preparation". The term "pharmaceutically acceptable" refers to the non-toxicity of a material which does not interact with the action of the active component of the pharmaceutical composition. The term "pharmaceutically effective amount" refers to the amount which achieves a desired reaction or a desired effect alone or together with further doses. In some embodiments relating to the treatment of a particular disease, the desired reaction may relate to inhibition of the course of the disease. This comprises slowing down the progress of the disease and, in some embodiments, interrupting or reversing the progress of the disease. The desired reaction in a treatment of a disease may also be delay of the onset or a prevention of the onset of said disease or said condition, or symptoms thereof. An effective amount of the pharmaceutical compositions described herein will depend on the condition to be treated, the severeness of the disease, the individual parameters of the patient, including age, physiological condition, size and weight, the duration of treatment, the type of an accompanying therapy (if present), the specific route of administration and similar factors. Accordingly, the doses administered of the pharmaceutical compositions described herein may depend on various of such parameters. In the case that a reaction in a patient is insufficient with an initial dose, higher doses (or effectively higher doses achieved by a different, more localized route of administration) may be used. The pharmaceutical compositions of the present disclosure may contain buffers, preservatives, and optionally other therapeutic agents. In some embodiments, the pharmaceutical compositions of the present disclosure comprise one or more pharmaceutically acceptable carriers, diluents and/or excipients. Suitable preservatives for use in the pharmaceutical compositions of the present disclosure include, without limitation, benzalkonium chloride, chlorobutanol, paraben and thimerosal. The term "excipient" as used herein refers to a substance which may be present in a pharmaceutical composition of the present disclosure but is not an active ingredient. Examples of excipients, include without limitation, carriers, 101 11529421v1 Attorney Docket No. 2013237‐0757 binders, diluents, lubricants, thickeners, surface active agents, preservatives, stabilizers, emulsifiers, buffers, flavoring agents, or colorants. The term "diluent" relates a diluting and/or thinning agent. Moreover, the term "diluent" includes any one or more of fluid, liquid or solid suspension and/or mixing media. Examples of suitable diluents include ethanol, glycerol and water. The term "carrier" refers to a component which may be natural, synthetic, organic, inorganic in which the active component is combined in order to facilitate, enhance or enable administration of the pharmaceutical composition. A carrier as used herein may be one or more compatible solid or liquid fillers, diluents or encapsulating substances, which are suitable for administration to subject. Suitable carriers include, without limitation, sterile water, Ringer, Ringer lactate, sterile sodium chloride solution, isotonic saline, polyalkylene glycols, hydrogenated naphthalenes and, in particular, biocompatible lactide polymers, lactide/glycolide copolymers or polyoxyethylene/polyoxy- propylene copolymers. In some embodiments, the pharmaceutical composition of the present disclosure includes isotonic saline. Pharmaceutically acceptable carriers, excipients or diluents for therapeutic use are well known in the pharmaceutical art, and are described, for example, in Remington's Pharmaceutical Sciences, Mack Publishing Co. (A. R Gennaro edit. 1985). Pharmaceutical carriers, excipients or diluents can be selected with regard to the intended route of administration and standard pharmaceutical practice. Routes of administration of pharmaceutical compositions In some embodiments, the pharmaceutical compositions described herein may be administered intravenously, intraarterially, subcutaneously, intradermally, dermally, intranodally, or intramuscularly. In some embodiments, the pharmaceutical compositions described herein may be administered intramuscularly. In some embodiments, the pharmaceutical composition is formulated for local administration or systemic administration. Systemic administration may include enteral administration, which involves absorption through the gastrointestinal tract, or parenteral administration. As used herein, "parenteral administration" refers to the administration in any manner other than through the gastrointestinal tract, such as by intravenous injection. In some embodiments, the pharmaceutical compositions are formulated for systemic administration. In some embodiments, the systemic administration is by intravenous administration. In some embodiments, the pharmaceutical compositions are formulated for intramuscular administration. In some embodiments, intramuscular administration comprises administration into the upper arm, in particular into the musculus deltoideus. If more than one dose, e.g., three doses, of a pharmaceutical composition described herein is administered, the different administrations may be into the same arm. 102 11529421v1 Attorney Docket No. 2013237‐0757 Use of compositions Compositions described herein may be used in the therapeutic or prophylactic treatment of diseases wherein provision of one or more peptides or polypeptides, i.e., polypeptides, described herein to a subject results in a therapeutic or prophylactic effect. In some embodiments, the disease is infection with Mycobacterium tuberculosis. In some embodiments, the disease is tuberculosis. The term "disease" (also referred to as "disorder" herein) refers to an abnormal condition that affects the body of an individual. A disease is often construed as a medical condition associated with specific symptoms and signs. A disease may be caused by factors originally from an external source, such as infectious disease, or it may be caused by internal dysfunctions, such as autoimmune diseases. In humans, "disease" is often used more broadly to refer to any condition that causes pain, dysfunction, distress, social problems, or death to the individual afflicted, or similar problems for those in contact with the individual. In this broader sense, it sometimes includes injuries, disabilities, disorders, syndromes, infections, isolated symptoms, deviant behaviors, and atypical variations of structure and function, while in other contexts and for other purposes these may be considered distinguishable categories. Diseases usually affect individuals not only physically, but also emotionally, as contracting and living with many diseases can alter one's perspective on life, and one's personality. The term "disease involving an antigen" refers to any disease which implicates an antigen, e.g. a disease which is characterized by the presence of an antigen. The disease involving an antigen can be an infectious disease. The antigen may be a disease-associated antigen, such as a bacterial antigen. In some embodiments, a disease involving an antigen is a disease involving cells comprising and/or expressing an antigen, and preferably presenting the antigen on the cell surface, e.g., in the context of MHC. The term "infectious disease" refers to any disease which can be transmitted from individual to individual or from organism to organism, and is caused by a microbial agent (e.g. common cold). Infectious diseases are known in the art and include, for example, a viral disease, a bacterial disease, or a parasitic disease, which diseases are caused by a virus, a bacterium, and a parasite, respectively. In this regard, the infectious disease can be, for example, hepatitis, sexually transmitted diseases (e.g. chlamydia or gonorrhea), tuberculosis, malaria, HIV/acquired immune deficiency syndrome (AIDS), diphtheria, hepatitis B, hepatitis C, cholera, severe acute respiratory syndrome (SARS), the bird flu, and influenza. In the present context, the term "treatment", "treating" or "therapeutic intervention" relates to the management and care of a subject for the purpose of combating a condition such as a disease. The term is intended to include the full spectrum of treatments for a given condition from which the subject is suffering, such as administration of the therapeutically effective compound to alleviate the symptoms or complications, to delay the progression of the disease, disorder or condition, to alleviate or relief the symptoms and complications, and/or to cure or eliminate the disease, disorder or condition as well as to prevent the condition, wherein prevention is to be understood as the management and care of an individual for the purpose of combating the disease, condition or disorder and includes the administration of the active compounds to prevent the onset of the symptoms or complications. 103 11529421v1 Attorney Docket No. 2013237‐0757 The term "therapeutic treatment" relates to any treatment which improves the health status and/or prolongs (increases) the lifespan of an individual. Said treatment may eliminate the disease in an individual, arrest or slow the development of a disease in an individual, inhibit or slow the development of a disease in an individual, decrease the frequency or severity of symptoms in an individual, and/or decrease the recurrence in an individual who currently has or who previously has had a disease. The terms "prophylactic treatment" or "preventive treatment" relate to any treatment that is intended to prevent a disease from occurring in an individual. The terms "prophylactic treatment" or "preventive treatment" are used herein interchangeably. The terms "individual" and "subject" are used herein interchangeably. They refer to a human or another mammal (e.g., mouse, rat, rabbit, dog, cat, cattle, swine, sheep, horse or primate), or any other non-mammal-animal, including birds (chicken), fish or any other animal species that can be afflicted with or is susceptible to a disease (e.g., cancer, infectious diseases) but may or may not have the disease, or may have a need for prophylactic intervention such as vaccination, or may have a need for interventions such as by protein replacement. In many embodiments, the individual is a human being. Unless otherwise stated, the terms "individual" and "subject" do not denote a particular age, and thus encompass adults, elderlies, children, and newborns. In some embodiments of the present disclosure, the "individual" or "subject" is a "patient". In some embodiments, the terms "individual" and "subject" relate to pregnant women and immunocompromised persons. The term "patient" means an individual or subject for treatment, in particular a diseased individual or subject. RNA described herein may be administered to a subject for delivering the RNA to cells of the subject. RNA described herein may be administered to a subject for delivering a therapeutic or prophylactic peptide or polypeptide (e.g., a pharmaceutically active peptide or polypeptide) to the subject, wherein the RNA encodes a therapeutic or prophylactic peptide or polypeptide. RNA described herein may be administered to a subject for treating or preventing a disease in a subject, wherein delivering the RNA to cells of the subject is beneficial in treating or preventing the disease. RNA described herein may be administered to a subject for treating or preventing a disease in a subject, wherein the RNA encodes a therapeutic or prophylactic peptide or polypeptide and wherein delivering the therapeutic or prophylactic peptide or polypeptide to the subject is beneficial in treating or preventing the disease. In some embodiments of the disclosure, the aim is to induce an immune response by providing RNA described herein. A person skilled in the art will know that one of the principles of immunotherapy and vaccination is based on the fact that an immunoprotective reaction to a disease is produced by immunizing a subject with a polypeptide or an epitope, which is immunologically relevant with respect to the disease to be treated. Accordingly, RNA described herein is applicable for inducing or enhancing an immune response. RNA described herein is thus useful in a prophylactic and/or therapeutic treatment of a disease involving a polypeptide or epitope. 104 11529421v1 Attorney Docket No. 2013237‐0757 In some embodiments of the disclosure, the aim is to provide an immune response against cells comprising an antigen, e.g., Mtb antigen. In some embodiments of the disclosure, the aim is to prophylactically or therapeutically treat tuberculosis by vaccination. Due to the high degree of sequence conservation of the disclosed antigens between different Mycobacterium species, including Mycobacterium tuberculosis, Mycobacterium bovis, Mycobacterium caprae, Mycobacterium orygis, Mycobacterium africanum, Mycobacterium microti, Mycobacterium canetti and Mycobacterium pinnipedii, exposure of a subject to Mycobacterium tuberculosis antigens, in the context of a polypeptide of the invention, will result in a high degree of cross-reactivity with antigens of other Mycobacterium species. Therefore, a vaccine based on or directed at Mycobacterium tuberculosis antigens will elicit a robust immune response against other Mycobacterium species, in particular Mycobacterium bovis, Mycobacterium caprae, Mycobacterium orygis, Mycobacterium africanum, Mycobacterium microti, Mycobacterium canetti and Mycobacterium pinnipedii as well. In some embodiments of the disclosure, the aim is to provide an immune response against a Mycobacterium selected from Mycobacterium tuberculosis, Mycobacterium bovis, Mycobacterium caprae, Mycobacterium orygis, Mycobacterium africanum, Mycobacterium microti, Mycobacterium canetti and Mycobacterium pinnipedii and to prevent or treat tuberculosis. In preferred embodiments of the disclosure, the aim is to provide an immune response against Mycobacterium tuberculosis. In some embodiments of the disclosure, the aim is to treat an infection with Mycobacterium tuberculosis. In some embodiments of the disclosure, the aim is to prevent or treat disease symptoms caused by an infection with Mycobacterium tuberculosis. In some embodiments of the disclosure, the aim is to provide protection against an infection with Mycobacterium tuberculosis by vaccination. In some embodiments of the disclosure, the aim is to provide protection against an outbreak of disease in a subject infected with Mycobacterium tuberculosis. In some embodiments of the disclosure, the aim is to provide protection against symptoms of tuberculosis in a subjected infected with Mycobacterium tuberculosis. In some embodiments, the RNA is present in a composition as described herein. In some embodiments, the RNA is administered in a pharmaceutically effective amount. In some embodiments, the subject is a mammal. In some embodiments, the mammal is a human. In some embodiments, the subject treated had been exposed to Mycobacterium tuberculosis. In some embodiments, the subject treated had not been exposed to Mycobacterium tuberculosis. 105 11529421v1 Attorney Docket No. 2013237‐0757 In some embodiments, the treatments described herein involve pre- or post-exposure vaccination against Mycobacterium tuberculosis, or a combination thereof. Citation of documents and studies referenced herein is not intended as an admission that any of the foregoing is pertinent prior art. All statements as to the contents of these documents are based on the information available to the applicants and do not constitute any admission as to the correctness of the contents of these documents. The description (including the following examples) is presented to enable a person of ordinary skill in the art to make and use the various embodiments. Descriptions of specific devices, techniques, and applications are provided only as examples. Various modifications to the examples described herein will be readily apparent to those of ordinary skill in the art, and the general principles defined herein may be applied to other examples and applications without departing from the spirit and scope of the various embodiments. Thus, the various embodiments are not intended to be limited to the examples described herein and shown, but are to be accorded the scope consistent with the claims. Examples Example 1: Observation that EspA is toxic when expressed as mRNA in HEK293T cells. EspA (Rv3616c) was encoded on mRNA using its wild-type protein sequence modified by an N-terminal signal peptide (human HLA-A1 derived, hereafter SP) and a FLAG-epitope tag. This construct was investigated for expression in HEK293T cells by RiboJuiceTM (Sigma-Aldrich) transfection of 1 µg of RNA. After overnight incubation (approximately 18h) cells were inspected visually under a bright field microscope. Strikingly, a large reduction in cell confluency could be observed in cells transfected with EspA compared to the non-transfected control or compared to control protein 1 which was also FLAG-tagged and known to be well expressed (Figure 1). Lysates were generated and analyzed by western blot for expression of the FLAG-tag, which could be well detected for control protein 1. However, no expression was found for EspA (Figure 2). These results were our first indication that expression of EspA could be highly toxic for cells when expressed on mRNA. Example 2: Confirmation of EspA expression and toxicity and introducing live imaging as a method to quantify toxicity. Wild type EspA or a control protein with a similar predicted molecular weight and a similar predicted tertiary structure (control protein 2), were expressed in HEK293T cells by RiboJuiceTM transfection of different amounts of RNA (25 ng, 100 ng, 250 ng or 1000 ng). Both proteins were FLAG-tagged and fused with a N-terminal signal peptide (SP). Lysates of the cells were generated 6 hours post transfection or 24h post transfection. Lysates from transfected cells were separated by SDS-PAGE on a 6-18% polyacrylamide gel and western blot using an FLAG- specific monoclonal antibody conjugated to horseradish peroxidase (HRP; clone M2 from Sigma-Aldrich) and a α- Tubulin-specific HRP-conjugated rabbit monoclonal antibody (clone 11H10 from Cell Signaling Technology). The 106 11529421v1 Attorney Docket No. 2013237‐0757 FLAG-tagged control protein 2 was detected at both timepoints (250ng and 1000ng of RNA; Figure 3). In contrast, and similar to example 1, EspA expression could not be detected 24 hours post transfection. However, at 6 hours post transfection a faint band could be seen at the indicated size confirming that EspA is expressed early after mRNA transfection. In parallel HEK293T cells were transfected with the same constructs and incubated with Incucyte® Cytotox Red Dye for Counting Dead Cells in the Incucyte® S3 Live-Cell Analysis System (Sartorius). Pictures were taken in 2-hour intervals for 24 hours to measure cell confluence (bright field images) and Cytotox- staining of cells with impaired membrane potential as measure of cell death (Red channel, excitation 567-607 nm, emission 622-704 nm). Analysis was performed using the Incucyte® Live-Cell Analysis software provided by Sartorius. The Basic Analyzer analysis type was used. Confluence of cells was determined based on shape and size, where small blebs were excluded (Area (μm2) min = 200.00, Cleanup Adjust size (pixels) = -6). Cytotox positivity was defined based on size and signal intensity (Segmentation: Surface fit, Threshold (RCU) = 4.0000, Edge Split Off), where disproportionately small and large spots were removed as well as very weak signals (Area (μm2) min = 12.000, Mean Intensity min = 8.0000, Integrated Intensity min = 600.00). Confluency was defined in percentage (%), while Cytotox+ spots were defined in Red calibrated units (RCU) times square μm per image (RCU*μm2/image). Both parameters were normalized to a fold change compared to t = 0. Confluency decreased while the signal for the Cytotox dye increased when cells were transfected with EspA (Figure 4A-C). The opposite was true for control protein 2. Especially the concentration of 250 ng RNA clearly indicated the differences between confluency and Cytotox-stained cells upon transfection of both constructs. Example 3: EspA toxicity is primarily driven by the N-terminal A domain. Since it was found that EspA was toxic when expressed as a wild type, full-length protein, a bioinformatic analysis was performed to separate the protein into domains to determine whether this would impair cytotoxicity and if the part of the protein could be determined, which mediated its cytotoxicity. Based on sequence comparison with the human proteome, two regions of potential overlap with the human proteome were identified and excluded moving forward. Therefore, three major domains termed EspA_A (aa 2-166), EspA_B (aa 177-280), and EspA_C (aa 291- 392) were selected (Figure 5). Furthermore, to make sure that specifically these excluded overlapping-regions were not essential for determining toxicity of EspA, two constructs were designed, that combined the N-terminal two parts and the region of human overlap bridging them, dubbed EspA_AB, and a similar construct for the C- terminal parts called EspA_BC (Figure 6). To assess expression of the generated constructs, cells were seeded and incubated overnight before transfection with 250 ng RNA/well as described above. The negative control consists of non-transfected cells. Cells were incubated for 4h at 37°C by 5% CO¬2 and transferred to a 96-well plate and washed. Samples were stained with Fixable Viability Dye eFluor™ 780 (Thermo Fisher Scientific) for 15 minutes at room temperature. Cells were washed and fixed for 12 minutes at room temperature, followed by permeabilization for 5 minutes at 4°C. Permeabilized cells were stained with a FLAG-tag specific antibody (BioLegend) for 30 min at 4°C. Cells were washed and acquired with a FACS Celesta (BD Biosciences). Data was analyzed using FACS Diva and Flow Jo software (both BD Biosciences). Gating excluded doublet cells and debris (Figure 7A). Cell viability was measured on the B-A-780-A spectrum, while FLAG-positive cells were measured on the BV421-A spectrum. Antigen expression as reflected by 107 11529421v1 Attorney Docket No. 2013237‐0757 FLAG-tag binding was obtained from the viable cell population. Transfected constructs containing the A fragment of EspA showed a higher percentage of dead cells compared to control protein 2 (p<0.01) (Figure 7B and C). Upon transfection with EspA_FL, approximately 5% more cells were dead as compared to transfection of the control protein 2 encoding construct. Transfection with EspA_A and EspA_AB resulted in approximately 30% (p<0.01) and over 10% (p<0.05) more cell death, respectively. All other constructs had a similar viability as the negative construct control (p>0.05). As expected, in the non-transfected control, no cells of the viable cell population were staining positive for the FLAG-tag (Figure 7B and C). On the contrary, constructs encoding control protein 2, EspA_B, EspA_C, and EspA_BC did stain positive. The constructs with high cell death barely showed any positivity for the tag in either of the cell populations. This is most likely due to the viable cells not being transfected with the construct, and the cells that were transfected were either currently dying, accounting for the slight positivity, or were already dead and the FLAG-tagged protein was already degraded. To investigate the cytotoxicity of the generated EspA constructs (Figure 6) HEK293T cells were seeded and incubated as stated above (Example 2). Cells were transfected with 250 ng of RNA constructs encoding EspA_FL, EspA_A, EspA_B, EspA_C, EspA_AB, EspA_BC, or control proteins 2 and 3. Control protein 3 is non-toxic, of the same size as EspA-FL and control protein 2 and was added to further validate the assay results. Non-transfected cells served as a negative control. Immediately after transfection, cells were imaged in bright field and for fluorescence signals every hour for 24h. Cells transfected with constructs containing the EspA-A-fragment all showed increased Cytotox+ integrated intensity and decreased confluency indicating cytotoxicity (Figure 8A and B). EspA_A induced slightly more cytotoxicity when compared to EspA_FL. Both control proteins 2 and 3 behaved similarly to the non-transfected control and did not differ between each other (p<0.05). EspA_FL, EspA_A, and EspA_AB differed from control proteins 2 and 3 (p<0.001). Additionally, slightly higher values for EspA_B were observed as well (p<0.05). EspA_C and EspA_BC did not differ from the negative controls (p>0.05). Clearly, the highest toxicity values are indicated by constructs containing the A fragment, hence confirming the flow cytometer data. Example 4: A structure-guided T-cell string design to disrupt toxicity of EspA To visualize the domain architecture of EspA, AlphaFold2 was used to generate a model of EspA (Figure 9, panel A). There is high confidence in the EspA model due to high homology with EspB, which has high resolution crystal structures (https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4520771/) and a cryo-electron microscopy higher order pore structure (https://www.sciencedirect.com/science/article/pii/S2590152420300118?via%3Dihub) deposited in the Protein Data Bank (PDB). The model was juxtaposed against the bioinformatics data described above showing regions of human overlap. The EspA protein sequence was divided into subdomains in a manner that disrupted secondary structure elements and tertiary protein structure and function (Figure 9, panel B). Fragments were selected such that areas of human overlap were avoided when selecting EspA chunks for inclusion in the T cell string. In one approach, the EspA protein sequence was divided into subdomains containing three regions of EspA: EspA_3-1 (amino acids 1-166 of FL EspA), EspA_3-2 (amino acids 177-280 of FL EspA), and EspA_3-3 (amino acids 291-392 of FL EspA). The fragments were rearranged in a manner that disrupted tertiary and secondary structure 108 11529421v1 Attorney Docket No. 2013237‐0757 elements and function (Fig.9B and 9C). In another approach, the three fragments were further split up to generate seven fragments in total: EspA_3-1 was further fragmented into EspA_7-1 (amino acids 1-60 of FL EspA), EspA_7- 2 (amino acids 62-131 of FL EspA), and EspA_7-3 (amino acids 135-166 of FL EspA), EspA_3-2 was further fragmented into EspA_7-4 (amino acids 177-239 of FL EspA) and EspA_7-5 (amino acids 241-279 of FL EspA), and EspA_3-3 was further fragmented into EspA_7-6 (amino acids 291-341 of FL EspA), and EspA_7-7 (amino acids 343-392 of FL EspA). Again, the fragments were rearranged in a manner that disrupted tertiary and secondary structure elements and function (Fig. 9B and 9D). In both approaches, adjacent EspA regions were reshuffled to ensure that EspA chunks could not properly assemble into a functional protein during co-translation folding (Figure 9, C and D). It was hypothesized that this strategy would reduce toxicity and combined with the MITD, increase protein turnover and generation of immunogenic T cell epitopes. Example 5: Analysis of expression of the 3-part and 7-part EspA strings in vitro by mass-spectrometry RNA drug substance encoding string 2 (where EspA was separated and shuffled in 7 parts with linkers – Figure 10, panel A) was transfected into HEK293T cells in the presence (Light grey bars) or absence (Dark grey bars) of proteasome inhibitor MG132. Following lysis, total protein content was normalized, cysteines were reduced and alkylated, and proteins were subjected to tryptic digestion. The samples were analyzed by targeted and discovery MS to look for tryptic peptides derived uniquely from the transfected RNA strings (Figure 10, panel B). The indicated peptide (NHVNFFQELADLDR) was synthesized as a heavy peptide and its counterparts could be detected in the total trypsinated lysate from transfected with EspA string 2 in the presence of MG132 proteasome inhibitor. No expression was detected from non-transfected cells independently of the presence of MG132. These data confirm expression of EspA string 2 after overnight expression and the proteasome inhibitor data suggest that this string is efficiently turned over and degraded in normal conditions. Example 6: Structure-guided T-cell string approach allows processing and presentation of predicted HLA-epitopes RNA drug substance (EspA_String2) was transfected into A375 cells stably expressing an affinity-tagged version of HLA-B*07:02. Following lysis, tagged HLA molecules were labeled with biotin and enriched on NeutrAvidin beads. Following washing, peptides were eluted from HLA complexes using acid and filtered through molecular weight cutoff filters. Cysteines were reduced and alkylated, and peptides were cleaned up by C18 desalting. Heavy isotope- labeled synthetic peptides were spiked into each sample, and targeted and discovery MS was performed to look for epitope peptides derived uniquely from transfected mRNA constructs. The different detected peptides are depicted in the visual representation of the T cell string as well as in the table (Figure 11) depicting their exact sequence. Note that the detected epitopes were derived from different parts of the T-cell string indicating that the whole construct has the potential to be processed and presented on human HLA molecules. Example 7: T-cell string approach detoxifies EspA Cells were seeded and incubated as described above and transfected with RNA constructs encoding EspA_FL, EspA_A, EspA_B, EspA_C, EspA string1, EspA string2 or control protein 2 at a concentration of 250 ng/well. Wells 109 11529421v1 Attorney Docket No. 2013237‐0757 were stained with 1:4000 diluted Incucyte® Cytotox Red Dye for Counting Dead Cells. Negative controls consisted of non-transfected cells. The plate was placed in the Incucyte® S3 Live-Cell Analysis System directly post transfection. Wells were imaged in bright field and fluorescence every hour for 24h. All analyses were performed identically to those described in example 3. As before, the EspA_FL and EspA_A proteins were cytotoxic to the cells (Figure 12, panel A). However, the Cytotox+ of the two strings was significantly lower than EspA itself (p<0.05) and EspA_String2 is even lower than control protein 2 (p<0.001). The confluency of EspA string1 matched that of control protein 2 (p>0.05), while that of EspA string2 was more comparable with the non-transfected control (p<0.01 when compared to control protein 2) (Figure 12, panel B). Both show better confluence than EspA (p<0.05). These data indicate that both string designs, but especially the structure-guided approach used for EspA_String2, are highly effective in detoxifying EspA. Example 8: T-cell strings of EspA are immunogenic in C57BL/6 mice and leads to polyfunctional T- cell responses Adult female C57BL/6 mice were immunized with 4 µg of EspA String1 or String2 (structure-guided detoxification) RNA constructs formulated in a lipid nanoparticle formulation (in 20 µl of volume) or with 0.9% NaCl solution as a control. 21 days after the primary immunization the mice were boosted with the same dose. 14 days after the boosting dose (35 days post primary immunization) mice were sacrificed and splenocytes were isolated. In addition, a portion of the splenocytes was pooled and separated into CD4+ and CD8+ fractions by magnetic associated cell- sorting (MACS). Both the total splenocytes and the CD4+ and CD8+ fractions were assessed for T-cell reactogenicity by restimulation with peptide pools specific for individual chunks (or parts of chunks) of the T-cell string. The peptide libraries consist of 15 amino acid long peptides with a 5 amino acid interval. In other words, the first peptide will cover amino acid 1-15 of the antigen and the second peptide will cover amino acids 16-20, and so forth. Fluorospot was used as a read-out to determine IFN-γ, IL-2 and TNF-α production. The peptides libraries were not immunogenic above baseline (Figure 13). In both EspA String1 and String2, immunogenic regions were identified especially with peptide libraries of EspA_7.2 and 7.3 which correspond to the sequences of Chunk EspA_A which was found to be cytotoxic when expressed in itself (Figure 14). Some responses above baseline could be detected in response to peptide library EspA_7.5, 7.6 and 7.7, but these were all markedly lower than the responses detected toward 7.2 and 7.3. Separation of splenocytes into CD4+ and CD8+ T cells confirmed the results above and clearly indicated that EspA_7.2 includes both CD4+ and CD8+ epitopes while the responses detected to EspA_7.3 are determined by CD8+ responses (Figure 15). These data also confirmed the immunogenicity of EspA_7.5-7.7 and indicated that these relatively low levels of immune responses were primarily mediated by CD4+ T cells. CD4+ and CD8+ T cells can also be subdivided into different cytokine-secreting classes, called polyfunctional cells. Combinations of two or three cytokines were identified as well (Figure 16). For both strings, CD4+ T cells mostly secreted IL-2 in combination with one of the other two cytokines, whereas some also secreted all three. In contrast, CD8+ T cells mostly secreted IFN-γ and TNF-α (which is known for CD8+ T cells). Additionally, the cells from both string-immunized study groups induced a strong immune response against EspA_7.2 and EspA_7.3 as seen before (Figure 15). EspA_7.2 induced strong IFN-γ+ IL-2+ TNF-α+ CD4+ T cell responses, but no such responses were 110 11529421v1 Attorney Docket No. 2013237‐0757 observed for EspA_7.3. In contrast, the IFN-γ+ TNF-α+ CD8+ T cell responses were higher for EspA_7.3 compared to EspA_7.2. Example 9: Polyfunctional T cell responses Together, these data show that EspA_A (The N-terminal 166 amino acids) contains the most immunogenic regions of EspA. Therefore, classic truncation of the protein to only detect non-toxic section of the antigen would also result in a predicted loss of immunogenicity of EspA. In contrast, the T-cell string approach has successfully reduced toxicity of EspA_A in vitro and is able to induce strong T-cell responses. Finally, structure-guided detoxification led to further reduction of in vitro toxicity and increased in vitro expression levels as detected in examples 3 and 4 respectively, but also increased immunogenicity. Especially as measured by polyfunctional T-cell responses, which in murine studies are considered to be advantageous in the protection against TB infection. Confirming that the T-cell string approach was successful and specific in inducing immunogenicity of multiple different antigens simultaneously. Analysis of MACS´ed cells reveals that T Cell strings can induce potent CD4+ and CD8+ responses. 111 11529421v1 






















