Attorney Docket No.: 116983-5127-WO TREATMENT OF CANCER PATIENTS WITH TUMOR INFILTRATING LYMPHOCYTE THERAPIES IN COMBINATION WITH TROP-2 TARGETING ADC CROSS-REFERENCE TO RELATED APPLICATIONS [0001] This application claims priority to U.S. Provisional Patent Application No. 63/514,545, filed on July 19, 2023, which is hereby incorporated by reference in its entirety. BACKGROUND OF THE INVENTION [0002] Treatment of bulky, refractory cancers using adoptive autologous transfer of tumor infiltrating lymphocytes (TILs) represents a powerful approach to therapy for patients with poor prognoses. Gattinoni, et al., Nat. Rev. Immunol.2006, 6, 383-393. TILs are dominated by T cells, and IL-2-based TIL expansion followed by a “rapid expansion process” (REP) has become a preferred method for TIL expansion because of its speed and efficiency. Dudley, et al., Science 2002, 298, 850-54; Dudley, et al., J. Clin. Oncol.2005, 23, 2346-57; Dudley, et al., J. Clin. Oncol.2008, 26, 5233-39; Riddell, et al., Science 1992, 257, 238-41; Dudley, et al., J. Immunother.2003, 26, 332-42. A number of approaches to improve responses to TIL therapy in melanoma and to expand TIL therapy to other tumor types have been explored with limited success, and the field remains challenging. Goff, et al., J. Clin. Oncol.2016, 34, 2389-97; Dudley, et al., J. Clin. Oncol.2008, 26, 5233-39; Rosenberg, et al., Clin. Cancer Res.2011, 17, 4550-57. Combination studies with single immune checkpoint inhibitors have also been described, but further studies are ongoing and additional methods of treatment are needed (Kverneland, et al., Oncotarget, 2020, 11(22), 2092-2105). [0003] Furthermore, current TIL manufacturing and treatment processes are limited by length, cost, sterility concerns, and other factors described herein, such that the potential to treat cancer patients has been severely limited. There is an urgent need to provide TIL manufacturing processes and therapies based on such processes that are appropriate for use in treating cancer patients for whom very few or no viable treatment options remain. The present invention meets this need by providing manufacturing process for use in generating TILs which can then be utilized for the treatment of cancer patients in combination with Trop-2 targeting ADCs. 1
DB1/ 149202201.1 Attorney Docket No.: 116983-5127-WO [0004] TROP-2 is a transmembrane glycoprotein with both extracellular and intracellular components that is involved in calcium signal transduction (Shastry et al., Breast.2022 Dec; 66: 169–177). It was first discovered by Lipinski et al. who raised antibodies against the human choriocarcinoma cell line (Proc Natl Acad Sci U S A.1981;78(8):5147–5150). TROP- 2 has been implicated in several cell signaling pathways including intracellular calcium transduction, MAPK signaling pathway, RAF, NF-κB and Cyclin D/E among others (Goldenberg et al., Oncotarget. June 22, 2018;9(48) doi: 10.18632/oncotarget.25615.28989– 6). [0005] While initial investigation focused on TROP-2 expression in normal tissue, subsequent analysis showed that TROP-2 is upregulated in cancer cells when compared to normal cell counterparts (Trerotola et al., Oncogene.2013;32(2):222–233). This increased expression has been seen in many different tumor types including breast cancer, colon cancer, non-small cell lung cancer (NSCLC), esophageal squamous cell cancer, thyroid cancer and hepatobiliary cancers, raising the possibility of TROP-2 as a tumor agnostic biomarker (Zaman et al., OncoTargets Ther. March 1, 2019;12:1781–1790; Goldenberg et al., supra; Stepan et al., J Histochem Cytochem.2011 Jul;59(7):701–710). BRIEF SUMMARY OF THE INVENTION [0001] Some embodiments disclosed herein provide a method of treating a cancer in a patient in need thereof comprising administering a population of tumor infiltrating lymphocytes (TILs) and a Trop-2 targeting antibody drug conjugate (ADC). In some embodiments, the Trop-2 targeting ADC is selected from the group consisting of Trodelvy (sacituzumab govitecan), datopotamab deruxtecan, MK-2870/ SKB-264, ESG-401, BIO 106, DB-1305, JS108, BL-M02D1, and FZ-AD004. In some embodiments, the Trop-2 targeting ADC is administered at a dosage of about 1 mg/kg to about 100 mg/kg. In some embodiments, the Trop-2 targeting ADC is administered weekly, once every two weeks, or once every three weeks. In some embodiments, the Trop-2 targeting ADC is Trodelvy (sacituzumab govitecan). In some embodiments, Trodelvy (sacituzumab govitecan) is administered at a dosage of about 10 mg/kg. In some embodiments, Trodelvy (sacituzumab govitecan) is administered weekly. In some embodiments, the Trop-2 targeting ADC is datopotamab deruxtecan. In some embodiments, datopotamab deruxtecan is administered at a DB1/ 149202201.1 2 Attorney Docket No.: 116983-5127-WO dosage of about 6 mg/kg. In some embodiments, datopotamab deruxtecan is administered once every three weeks. In some embodiments, the Trop-2 targeting ADC is MK-2870/ SKB- 264. In some embodiments, MK-2870/ SKB-264is administered at a dosage of about 5 mg/m
2. In some embodiments, MK-2870/ SKB-264is administered once every two weeks. In some embodiments, the Trop-2 targeting ADC is ESG-401. In some embodiments, the Trop- 2 targeting ADC is BIO 106. In some embodiments, the Trop-2 targeting ADC is JS108. In some embodiments, the Trop-2 targeting ADC is BL-M02D1. In some embodiments, the Trop-2 targeting ADC is FZ-AD004. [0002] In some embodiments, the method comprises the steps of: (a) obtaining and/or receiving a first population of TILs from a tumor resected from the patient by processing a tumor sample obtained from the subject into multiple tumor fragments; (b) expanding the first population of TILs into a therapeutic population of TILs; (c) administering the therapeutic population of TILs to the subject; and (d) administering the Trop-2 targeting ADC to the subject. In some embodiments, the Trop-2 targeting ADC is provided to the patient at the same time as the TIL infusion. In some embodiments, the Trop-2 targeting ADC is provided to the patient after the TIL infusion. In some embodiments, the Trop-2 targeting ADC is provided to the patient contemporaneously with the TIL infusion and also provided after TIL infusion. In some embodiments, the Trop-2 targeting ADC is administered prior to the resection of the tumor from the patient. In some embodiments, the patient is refractory to pre-treatment with a Trop-2 targeting ADC. In some embodiments, the cancer has been previously treated with a PD-1 inhibitor and/or PD-L1 inhibitor or a biosimilar thereof. In some embodiments, the cancer has been previously treated with a PD-1 inhibitor or a biosimilar thereof. In some embodiments, the PD-1 inhibitor is selected from the group consisting of nivolumab, pembrolizumab, and biosimilars thereof. In some embodiments, the patient has been further previously treated with a PD-L1 inhibitor or a biosimilar thereof. In some embodiments, the PD-L1 inhibitor is selected from the group consisting of avelumab, atezolizumab, durvalumab, and biosimilars thereof. In some embodiments, the cancer has been previously treated with a chemotherapeutic regimen. In some embodiments, the chemotherapeutic regimen comprises carboplatin, paclitaxel, pemetrexed, and/or cisplatin. In some embodiments, the cancer has been previously treated with an angiogenesis inhibitor. In some embodiments, the angiogenesis inhibitor is bevacizumab. DB1/ 149202201.1 3 Attorney Docket No.: 116983-5127-WO [0003] In some embodiments, the method further comprises the step of treating the patient with a non-myeloablative lymphodepletion regimen prior to administering the TILs to the patient. In some embodiments, the non-myeloablative lymphodepletion regimen comprises the steps of administration of cyclophosphamide at a dose of 60 mg/m
2/day for two days followed by administration of fludarabine at a dose of 25 mg/m
2/day for five days. In some embodiments, the non-myeloablative lymphodepletion regimen comprises the steps of administration of cyclophosphamide at a dose of 60 mg/m
2/day and fludarabine at a dose of 25 mg/m
2/day for two days followed by administration of fludarabine at a dose of 25 mg/m
2/day for three days. In some embodiments, the cyclophosphamide is administered with mesna. In some embodiments, the method further comprises the step of treating the patient with an IL-2 regimen starting on the day after the administration of the TILs to the patient. In some embodiments, the method further comprises the step of treating the patient with an IL-2 regimen starting on the same day as administration of the TILs to the patient. In some embodiments, the IL-2 regimen is a high-dose IL-2 regimen comprising 600,000 or 720,000 IU/kg of aldesleukin, or a biosimilar or variant thereof, administered as a 15-minute bolus intravenous infusion every eight hours until tolerance. In some embodiments, a therapeutically effective population of TILs is administered and comprises from about 2.3×10
10 to about 13.7×10
10 TILs, optionally about 7.5×10
9 to about 72×10
9 TILs, optionally about 1 x 10
9 to about 100 x 10
9 TILs, or optionally about 7.5 x 10
9 to about 100 x 10
9 TILs. In some embodiments, the cancer is non-small-cell lung cancer (NSCLC). In some embodiments, the patient has no EGFR, ALK, or ROS1 genomic alterations. In some embodiments, the patient has one or more actionable genetic mutations in MET, HER2, RET, BRAF, and/or KRAS. In some embodiments, the cancer is selected from the group consisting of breast cancer (including triple-negative breast cancer (TNBC)), urothelial cancer, ovarian cancer, endometrial cancer, pancreatic cancer, bladder cancer, and head and neck cancer. BRIEF DESCRIPTION OF THE DRAWINGS [0004] Figure 1: Exemplary Gen 2 (process 2A) chart providing an overview of Steps A through F. [0005] Figure 2A-2C: Process flow chart of an embodiment of Gen 2 (process 2A) for TIL manufacturing. DB1/ 149202201.1 4 Attorney Docket No.: 116983-5127-WO DETAILED DESCRIPTION OF THE INVENTION I. Definitions [0001] Unless defined otherwise, all technical and scientific terms used herein have the same meaning as is commonly understood by one of skill in the art to which this invention belongs. All patents and publications referred to herein are incorporated by reference in their entireties. [0002] The terms “co-administration,” “co-administering,” “administered in combination with,” “administering in combination with,” “simultaneous,” and “concurrent,” as used herein, encompass administration of two or more active pharmaceutical ingredients (in a preferred embodiment of the present invention, for example, a plurality of TILs) to a subject so that both active pharmaceutical ingredients and/or their metabolites are present in the subject at the same time. Co-administration includes simultaneous administration in separate compositions, administration at different times in separate compositions, or administration in a composition in which two or more active pharmaceutical ingredients are present. Simultaneous administration in separate compositions and administration in a composition in which both agents are present are preferred. [0003] The term “in vivo” refers to an event that takes place in a subject's body. [0004] The term “in vitro” refers to an event that takes places outside of a subject's body. In vitro assays encompass cell-based assays in which cells alive or dead are employed and may also encompass a cell-free assay in which no intact cells are employed. [0005] The term “ex vivo” refers to an event which involves treating or performing a procedure on a cell, tissue and/or organ which has been removed from a subject’s body. Aptly, the cell, tissue and/or organ may be returned to the subject’s body in a method of surgery or treatment. [0006] By “tumor infiltrating lymphocytes” or “TILs” herein is meant a population of cells originally obtained as white blood cells that have left the bloodstream of a subject and migrated into a tumor. TILs include, but are not limited to, CD8
+ cytotoxic T cells (lymphocytes), Th1 and Th17 CD4
+ T cells, natural killer cells, dendritic cells and M1 macrophages. TILs include both primary and secondary TILs. “Primary TILs” are those that are obtained from patient tissue samples as outlined herein (sometimes referred to as “freshly DB1/ 149202201.1 5 Attorney Docket No.: 116983-5127-WO harvested”), and “secondary TILs” are any TIL cell populations that have been expanded or proliferated as discussed herein, including, but not limited to bulk TILs and expanded TILs (“REP TILs” or “post-REP TILs”). TIL cell populations can include genetically modified TILs. [0007] By “population of cells” (including TILs) herein is meant a number of cells that share common traits. In general, populations generally range from 1 X 10
6 to 1 X 10
10 in number, with different TIL populations comprising different numbers. For example, initial growth of primary TILs in the presence of IL-2 results in a population of bulk TILs of roughly 1 × 10
8 cells. REP expansion is generally done to provide populations of 1.5 × 10
9 to 1.5 × 10
10 cells for infusion. [0008] By “cryopreserved TILs” herein is meant that TILs, either primary, bulk, or expanded (REP TILs), are treated and stored in the range of about -150°C to -60°C. General methods for cryopreservation are also described elsewhere herein, including in the Examples. For clarity, “cryopreserved TILs” are distinguishable from frozen tissue samples which may be used as a source of primary TILs. [0009] By “thawed cryopreserved TILs” herein is meant a population of TILs that was previously cryopreserved and then treated to return to room temperature or higher, including but not limited to cell culture temperatures or temperatures wherein TILs may be administered to a patient. [0010] TILs can generally be defined either biochemically, using cell surface markers, or functionally, by their ability to infiltrate tumors and effect treatment. TILs can be generally categorized by expressing one or more of the following biomarkers: CD4, CD8, TCR αβ, CD27, CD28, CD56, CCR7, CD45Ra, CD95, PD-1, and CD25. Additionally and alternatively, TILs can be functionally defined by their ability to infiltrate solid tumors upon reintroduction into a patient. [0011] The term “cryopreservation media” or “cryopreservation medium” refers to any medium that can be used for cryopreservation of cells. Such media can include media comprising 7% to 10% DMSO. Exemplary media include CryoStor CS10, Hyperthermasol, as well as combinations thereof. The term “CS10” refers to a cryopreservation medium which is obtained from Stemcell Technologies or from Biolife Solutions. The CS10 medium may be referred to by the trade name “CryoStor® CS10”. The CS10 medium is a serum-free, animal DB1/ 149202201.1 6 Attorney Docket No.: 116983-5127-WO component-free medium which comprises DMSO. In some embodiments, the CS10 medium comprises 10% DMSO. [0012] The term “closed system” refers to a system that is closed to the outside environment. Any closed system appropriate for cell culture methods can be employed with the methods of the present invention. Closed systems include, for example, but are not limited to, closed G-containers. Once a tumor segment is added to the closed system, the system is no opened to the outside environment until the TILs are ready to be administered to the patient. [0013] The terms “fragmenting,” “fragment,” and “fragmented,” as used herein to describe processes for disrupting a tumor, includes mechanical fragmentation methods such as crushing, slicing, dividing, and morcellating tumor tissue as well as any other method for disrupting the physical structure of tumor tissue. [0014] The term “anti-CD3 antibody” refers to an antibody or variant thereof, e.g., a monoclonal antibody and including human, humanized, chimeric or murine antibodies which are directed against the CD3 receptor in the T cell antigen receptor of mature T cells. Anti- CD3 antibodies include OKT-3, also known as muromonab. Anti-CD3 antibodies also include the UHCT1 clone, also known as T3 and CD3ε. Other anti-CD3 antibodies include, for example, otelixizumab, teplizumab, and visilizumab. [0015] The term “OKT-3” (also referred to herein as “OKT3”) refers to a monoclonal antibody or biosimilar or variant thereof, including human, humanized, chimeric, or murine antibodies, directed against the CD3 receptor in the T cell antigen receptor of mature T cells, and includes commercially-available forms such as OKT-3 (30 ng/mL, MACS GMP CD3 pure, Miltenyi Biotech, Inc., San Diego, CA, USA) and muromonab or variants, conservative amino acid substitutions, glycoforms, or biosimilars thereof. The amino acid sequences of the heavy and light chains of muromonab are given in Table 1 (SEQ ID NO:1 and SEQ ID NO:2). A hybridoma capable of producing OKT-3 is deposited with the American Type Culture Collection and assigned the ATCC accession number CRL 8001. A hybridoma capable of producing OKT-3 is also deposited with European Collection of Authenticated Cell Cultures (ECACC) and assigned Catalogue No.86022706. DB1/ 149202201.1 7 Attorney Docket No.: 116983-5127-WO TABLE 1. Amino acid sequences of muromonab (exemplary OKT-3 antibody).

[0016] The term “IL-2” (also referred to herein as “IL2”) refers to the T cell growth factor known as interleukin-2, and includes all forms of IL-2 including human and mammalian forms, conservative amino acid substitutions, glycoforms, biosimilars, and variants thereof. IL-2 is described, e.g., in Nelson, J. Immunol.2004, 172, 3983-88 and Malek, Annu. Rev. Immunol.2008, 26, 453-79, the disclosures of which are incorporated by reference herein. The amino acid sequence of recombinant human IL-2 suitable for use in the invention is given in Table 2 (SEQ ID NO:3). For example, the term IL-2 encompasses human, recombinant forms of IL-2 such as aldesleukin (PROLEUKIN, available commercially from multiple suppliers in 22 million IU per single use vials), as well as the form of recombinant IL-2 commercially supplied by CellGenix, Inc., Portsmouth, NH, USA (CELLGRO GMP) or ProSpec-Tany TechnoGene Ltd., East Brunswick, NJ, USA (Cat. No. CYT-209-b) and other commercial equivalents from other vendors. Aldesleukin (des-alanyl-1, serine-125 human IL- 2) is a nonglycosylated human recombinant form of IL-2 with a molecular weight of approximately 15 kDa. The amino acid sequence of aldesleukin suitable for use in the invention is given in Table 2 (SEQ ID NO:4). The term IL-2 also encompasses pegylated forms of IL-2, as described herein, including the pegylated IL2 prodrug bempegaldesleukin (NKTR-214, pegylated human recombinant IL-2 as in SEQ ID NO:4 in which an average of 6 lysine residues are N
6 substituted with [(2,7-bis{[methylpoly(oxyethylene)]carbamoyl}-9H- fluoren-9-yl)methoxy]carbonyl), which is available from Nektar Therapeutics, South San Francisco, CA, USA, or which may be prepared by methods known in the art, such as the methods described in Example 19 of International Patent Application Publication No. WO 2018/132496 A1 or the method described in Example 1 of U.S. Patent Application Publication No. US 2019/0275133 A1, the disclosures of which are incorporated by reference herein. Bempegaldesleukin (NKTR-214) and other pegylated IL-2 molecules suitable for use DB1/ 149202201.1 8 Attorney Docket No.: 116983-5127-WO in the invention are described in U.S. Patent Application Publication No. US 2014/0328791 A1 and International Patent Application Publication No. WO 2012/065086 A1, the disclosures of which are incorporated by reference herein. Alternative forms of conjugated IL-2 suitable for use in the invention are described in U.S. Patent Nos.4,766,106, 5,206,344, 5,089,261 and 4,902,502, the disclosures of which are incorporated by reference herein. Formulations of IL-2 suitable for use in the invention are described in U.S. Patent No. 6,706,289, the disclosure of which is incorporated by reference herein. [0001] In some embodiments, an IL-2 form suitable for use in the present invention is THOR-707, available from Synthorx, Inc. The preparation and properties of THOR-707 and additional alternative forms of IL-2 suitable for use in the invention are described in U.S. Patent Application Publication Nos. US 2020/0181220 A1 and US 2020/0330601 A1, the disclosures of which are incorporated by reference herein. In some embodiments, and IL-2 form suitable for use in the invention is an interleukin 2 (IL-2) conjugate comprising: an isolated and purified IL-2 polypeptide; and a conjugating moiety that binds to the isolated and purified IL-2 polypeptide at an amino acid position selected from K35, T37, R38, T41, F42, K43, F44, Y45, E61, E62, E68, K64, P65, V69, L72, and Y107, wherein the numbering of the amino acid residues corresponds to SEQ ID NO:5. In some embodiments, the amino acid position is selected from T37, R38, T41, F42, F44, Y45, E61, E62, E68, K64, P65, V69, L72, and Y107. In some embodiments, the amino acid position is selected from T37, R38, T41, F42, F44, Y45, E61, E62, E68, P65, V69, L72, and Y107. In some embodiments, the amino acid position is selected from T37, T41, F42, F44, Y45, P65, V69, L72, and Y107. In some embodiments, the amino acid position is selected from R38 and K64. In some embodiments, the amino acid position is selected from E61, E62, and E68. In some embodiments, the amino acid position is at E62. In some embodiments, the amino acid residue selected from K35, T37, R38, T41, F42, K43, F44, Y45, E61, E62, E68, K64, P65, V69, L72, and Y107 is further mutated to lysine, cysteine, or histidine. In some embodiments, the amino acid residue is mutated to cysteine. In some embodiments, the amino acid residue is mutated to lysine. In some embodiments, the amino acid residue selected from K35, T37, R38, T41, F42, K43, F44, Y45, E61, E62, E68, K64, P65, V69, L72, and Y107 is further mutated to an unnatural amino acid. In some embodiments, the unnatural amino acid comprises N6-azidoethoxy-L- lysine (AzK), N6-propargylethoxy-L-lysine (PraK), BCN-L-lysine, norbornene lysine, TCO- lysine, methyltetrazine lysine, allyloxycarbonyllysine, 2-amino-8-oxononanoic acid, 2- DB1/ 149202201.1 9 Attorney Docket No.: 116983-5127-WO amino-8-oxooctanoic acid, p-acetyl-L-phenylalanine, p-azidomethyl-L-phenylalanine (pAMF), p-iodo-L-phenylalanine, m-acetylphenylalanine, 2-amino-8-oxononanoic acid, p- propargyloxyphenylalanine, p-propargyl-phenylalanine, 3-methyl-phenylalanine, L-Dopa, fluorinated phenylalanine, isopropyl-L-phenylalanine, p-azido-L-phenylalanine, p-acyl-L- phenylalanine, p-benzoyl-L-phenylalanine, p-bromophenylalanine, p-amino-L-phenylalanine, isopropyl-L-phenylalanine, O-allyltyrosine, O-methyl-L-tyrosine, O-4-allyl-L-tyrosine, 4- propyl-L-tyrosine, phosphonotyrosine, tri-O-acetyl-GlcNAcp-serine, L-phosphoserine, phosphonoserine, L-3-(2-naphthyl)alanine, 2-amino-3-((2-((3-(benzyloxy)-3- oxopropyl)amino)ethyl)selanyl)propanoic acid, 2-amino-3-(phenylselanyl)propanoic, or selenocysteine. In some embodiments, the IL-2 conjugate has a decreased affinity to IL-2 receptor α (IL-2Rα) subunit relative to a wild-type IL-2 polypeptide. In some embodiments, the decreased affinity is about 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80%, 90%, 95%, 99%, or greater than 99% decrease in binding affinity to IL-2Rα relative to a wild-type IL-2 polypeptide. In some embodiments, the decreased affinity is about 1-fold, 2-fold, 3-fold, 4- fold, 5-fold, 6-fold, 7-fold, 8-fold, 9-fold, 10-fold, 30-fold, 50-fold, 100-fold, 200-fold, 300- fold, 500-fold, 1000-fold, or more relative to a wild-type IL-2 polypeptide. In some embodiments, the conjugating moiety impairs or blocks the binding of IL-2 with IL-2Rα. In some embodiments, the conjugating moiety comprises a water-soluble polymer. In some embodiments, the additional conjugating moiety comprises a water-soluble polymer. In some embodiments, each of the water-soluble polymers independently comprises polyethylene glycol (PEG), poly(propylene glycol) (PPG), copolymers of ethylene glycol and propylene glycol, poly(oxyethylated polyol), poly(olefinic alcohol), poly(vinylpyrrolidone), poly(hydroxyalkylmethacrylamide), poly(hydroxyalkylmethacrylate), poly(saccharides), poly(α-hydroxy acid), poly(vinyl alcohol), polyphosphazene, polyoxazolines (POZ), poly(N- acryloylmorpholine), or a combination thereof. In some embodiments, each of the water- soluble polymers independently comprises PEG. In some embodiments, the PEG is a linear PEG or a branched PEG. In some embodiments, each of the water-soluble polymers independently comprises a polysaccharide. In some embodiments, the polysaccharide comprises dextran, polysialic acid (PSA), hyaluronic acid (HA), amylose, heparin, heparan sulfate (HS), dextrin, or hydroxyethyl-starch (HES). In some embodiments, each of the water-soluble polymers independently comprises a glycan. In some embodiments, each of the water-soluble polymers independently comprises polyamine. In some embodiments, the conjugating moiety comprises a protein. In some embodiments, the additional conjugating DB1/ 149202201.1 10 Attorney Docket No.: 116983-5127-WO moiety comprises a protein. In some embodiments, each of the proteins independently comprises an albumin, a transferrin, or a transthyretin. In some embodiments, each of the proteins independently comprises an Fc portion. In some embodiments, each of the proteins independently comprises an Fc portion of IgG. In some embodiments, the conjugating moiety comprises a polypeptide. In some embodiments, the additional conjugating moiety comprises a polypeptide. In some embodiments, each of the polypeptides independently comprises a XTEN peptide, a glycine-rich homoamino acid polymer (HAP), a PAS polypeptide, an elastin-like polypeptide (ELP), a CTP peptide, or a gelatin-like protein (GLK) polymer. In some embodiments, the isolated and purified IL-2 polypeptide is modified by glutamylation. In some embodiments, the conjugating moiety is directly bound to the isolated and purified IL-2 polypeptide. In some embodiments, the conjugating moiety is indirectly bound to the isolated and purified IL-2 polypeptide through a linker. In some embodiments, the linker comprises a homobifunctional linker. In some embodiments, the homobifunctional linker comprises Lomant's reagent dithiobis (succinimidylpropionate) DSP, 3′3′- dithiobis(sulfosuccinimidyl proprionate) (DTSSP), disuccinimidyl suberate (DSS), bis(sulfosuccinimidyl)suberate (BS), disuccinimidyl tartrate (DST), disulfosuccinimidyl tartrate (sulfo DST), ethylene glycobis(succinimidylsuccinate) (EGS), disuccinimidyl glutarate (DSG), N,N′-disuccinimidyl carbonate (DSC), dimethyl adipimidate (DMA), dimethyl pimelimidate (DMP), dimethyl suberimidate (DMS), dimethyl-3,3′- dithiobispropionimidate (DTBP), 1,4-di-(3′-(2′-pyridyldithio)propionamido)butane (DPDPB), bismaleimidohexane (BMH), aryl halide-containing compound (DFDNB), such as e.g.1,5- difluoro-2,4-dinitrobenzene or 1,3-difluoro-4,6-dinitrobenzene, 4,4′-difluoro-3,3′- dinitrophenylsulfone (DFDNPS), bis-[β-(4-azidosalicylamido)ethyl]disulfide (BASED), formaldehyde, glutaraldehyde, 1,4-butanediol diglycidyl ether, adipic acid dihydrazide, carbohydrazide, o-toluidine, 3,3′-dimethylbenzidine, benzidine, α,α′-p-diaminodiphenyl, diiodo-p-xylene sulfonic acid, N,N′-ethylene-bis(iodoacetamide), or N,N′-hexamethylene- bis(iodoacetamide). In some embodiments, the linker comprises a heterobifunctional linker. In some embodiments, the heterobifunctional linker comprises N-succinimidyl 3-(2- pyridyldithio)propionate (sPDP), long-chain N-succinimidyl 3-(2-pyridyldithio)propionate (LC-sPDP), water-soluble-long-chain N-succinimidyl 3-(2-pyridyldithio) propionate (sulfo- LC-sPDP), succinimidyloxycarbonyl-α-methyl-α-(2-pyridyldithio)toluene (sMPT), sulfosuccinimidyl-6-[α-methyl-α-(2-pyridyldithio)toluamido]hexanoate (sulfo-LC-sMPT), succinimidyl-4-(N-maleimidomethyl)cyclohexane-1-carboxylate (sMCC), sulfosuccinimidyl- DB1/ 149202201.1 11 Attorney Docket No.: 116983-5127-WO 4-(N-maleimidomethyl)cyclohexane-1-carboxylate (sulfo-sMCC), m-maleimidobenzoyl-N- hydroxysuccinimide ester (MBs), m-maleimidobenzoyl-N-hydroxysulfosuccinimide ester (sulfo-MBs), N-succinimidyl(4-iodoacteyl)aminobenzoate (sIAB), sulfosuccinimidyl(4- iodoacteyl)aminobenzoate (sulfo-sIAB), succinimidyl-4-(p-maleimidophenyl)butyrate (sMPB), sulfosuccinimidyl-4-(p-maleimidophenyl)butyrate (sulfo-sMPB), N-(γ- maleimidobutyryloxy)succinimide ester (GMBs), N-(γ-maleimidobutyryloxy) sulfosuccinimide ester (sulfo-GMBs), succinimidyl 6-((iodoacetyl)amino)hexanoate (sIAX), succinimidyl 6-[6-(((iodoacetyl)amino)hexanoyl)amino]hexanoate (slAXX), succinimidyl 4- (((iodoacetyl)amino)methyl)cyclohexane-1-carboxylate (sIAC), succinimidyl 6-(((((4- iodoacetyl)amino)methyl)cyclohexane-1-carbonyl)amino) hexanoate (sIACX), p-nitrophenyl iodoacetate (NPIA), carbonyl-reactive and sulfhydryl-reactive cross-linkers such as 4-(4-N- maleimidophenyl)butyric acid hydrazide (MPBH), 4-(N-maleimidomethyl)cyclohexane-1- carboxyl-hydrazide-8 (M2C2H), 3-(2-pyridyldithio)propionyl hydrazide (PDPH), N- hydroxysuccinimidyl-4-azidosalicylic acid (NHs-AsA), N-hydroxysulfosuccinimidyl-4- azidosalicylic acid (sulfo-NHs-AsA), sulfosuccinimidyl-(4-azidosalicylamido)hexanoate (sulfo-NHs-LC-AsA), sulfosuccinimidyl-2-(p-azidosalicylamido)ethyl-1,3′-dithiopropionate (sAsD), N-hydroxysuccinimidyl-4-azidobenzoate (HsAB), N-hydroxysulfosuccinimidyl-4- azidobenzoate (sulfo-HsAB), N-succinimidyl-6-(4′-azido-2′-nitrophenyl amino)hexanoate (sANPAH), sulfosuccinimidyl-6-(4′-azido-2′-nitrophenylamino)hexanoate (sulfo-sANPAH), N-5-azido-2-nitrobenzoyloxysuccinimide (ANB-NOs), sulfosuccinimidyl-2-(m-azido-o- nitrobenzamido)-ethyl-1,3′-dithiopropionate (sAND), N-succinimidyl-4(4-azidophenyl)1,3′- dithiopropionate (sADP), N-sulfosuccinimidyl(4-azidophenyl)-1,3′-dithiopropionate (sulfo- sADP), sulfosuccinimidyl 4-(ρ-azidophenyl)butyrate (sulfo-sAPB), sulfosuccinimidyl 2-(7- azido-4-methylcoumarin-3-acetamide)ethyl-1,3′-dithiopropionate (sAED), sulfosuccinimidyl 7-azido-4-methylcoumain-3-acetate (sulfo-sAMCA), p-nitrophenyl diazopyruvate (pNPDP), p-nitrophenyl-2-diazo-3,3,3-trifluoropropionate (PNP-DTP), 1-(ρ-azidosalicylamido)-4- (iodoacetamido)butane (AsIB), N-[4-(ρ-azidosalicylamido)butyl]-3′-(2′-pyridyldithio) propionamide (APDP), benzophenone-4-iodoacetamide, p-azidobenzoyl hydrazide (ABH), 4- (ρ-azidosalicylamido)butylamine (AsBA), or p-azidophenyl glyoxal (APG). In some embodiments, the linker comprises a cleavable linker, optionally comprising a dipeptide linker. In some embodiments, the dipeptide linker comprises Val-Cit, Phe-Lys, Val-Ala, or Val-Lys. In some embodiments, the linker comprises a non-cleavable linker. In some embodiments, the linker comprises a maleimide group, optionally comprising DB1/ 149202201.1 12 Attorney Docket No.: 116983-5127-WO maleimidocaproyl (mc), succinimidyl-4-(N-maleimidomethyl)cyclohexane-1-carboxylate (sMCC), or sulfosuccinimidyl-4-(N-maleimidomethyl)cyclohexane-1-carboxylate (sulfo- sMCC). In some embodiments, the linker further comprises a spacer. In some embodiments, the spacer comprises p-aminobenzyl alcohol (PAB), p-aminobenzyoxycarbonyl (PABC), a derivative, or an analog thereof. In some embodiments, the conjugating moiety is capable of extending the serum half-life of the IL-2 conjugate. In some embodiments, the additional conjugating moiety is capable of extending the serum half-life of the IL-2 conjugate. In some embodiments, the IL-2 form suitable for use in the invention is a fragment of any of the IL-2 forms described herein. In some embodiments, the IL-2 form suitable for use in the invention is pegylated as disclosed in U.S. Patent Application Publication No. US 2020/0181220 A1 and U.S. Patent Application Publication No. US 2020/0330601 A1. In some embodiments, the IL-2 form suitable for use in the invention is an IL-2 conjugate comprising: an IL-2 polypeptide comprising an N6-azidoethoxy-L-lysine (AzK) covalently attached to a conjugating moiety comprising a polyethylene glycol (PEG), wherein: the IL-2 polypeptide comprises an amino acid sequence having at least 80% sequence identity to SEQ ID NO:5; and the AzK substitutes for an amino acid at position K35, F42, F44, K43, E62, P65, R38, T41, E68, Y45, V69, or L72 in reference to the amino acid positions within SEQ ID NO:5. In some embodiments, the IL-2 polypeptide comprises an N-terminal deletion of one residue relative to SEQ ID NO:5. In some embodiments, the IL-2 form suitable for use in the invention lacks IL-2R alpha chain engagement but retains normal binding to the intermediate affinity IL-2R beta-gamma signaling complex. In some embodiments, the IL-2 form suitable for use in the invention is an IL-2 conjugate comprising: an IL-2 polypeptide comprising an N6-azidoethoxy-L-lysine (AzK) covalently attached to a conjugating moiety comprising a polyethylene glycol (PEG), wherein: the IL-2 polypeptide comprises an amino acid sequence having at least 90% sequence identity to SEQ ID NO:5; and the AzK substitutes for an amino acid at position K35, F42, F44, K43, E62, P65, R38, T41, E68, Y45, V69, or L72 in reference to the amino acid positions within SEQ ID NO:5. In some embodiments, the IL-2 form suitable for use in the invention is an IL-2 conjugate comprising: an IL-2 polypeptide comprising an N6-azidoethoxy-L-lysine (AzK) covalently attached to a conjugating moiety comprising a polyethylene glycol (PEG), wherein: the IL-2 polypeptide comprises an amino acid sequence having at least 95% sequence identity to SEQ ID NO:5; and the AzK substitutes for an amino acid at position K35, F42, F44, K43, E62, P65, R38, T41, E68, Y45, V69, or L72 in reference to the amino acid positions within SEQ ID NO:5. In some DB1/ 149202201.1 13 Attorney Docket No.: 116983-5127-WO embodiments, the IL-2 form suitable for use in the invention is an IL-2 conjugate comprising: an IL-2 polypeptide comprising an N6-azidoethoxy-L-lysine (AzK) covalently attached to a conjugating moiety comprising a polyethylene glycol (PEG), wherein: the IL-2 polypeptide comprises an amino acid sequence having at least 98% sequence identity to SEQ ID NO:5; and the AzK substitutes for an amino acid at position K35, F42, F44, K43, E62, P65, R38, T41, E68, Y45, V69, or L72 in reference to the amino acid positions within SEQ ID NO:5. [0002] In some embodiments, an IL-2 form suitable for use in the invention is nemvaleukin alfa, also known as ALKS-4230 (SEQ ID NO:6), which is available from Alkermes, Inc. Nemvaleukin alfa is also known as human interleukin 2 fragment (1-59), variant (Cys
125>Ser
51), fused via peptidyl linker (
60GG
61) to human interleukin 2 fragment (62-132), fused via peptidyl linker (
133GSGGGS
138) to human interleukin 2 receptor α-chain fragment (139-303), produced in Chinese hamster ovary (CHO) cells, glycosylated; human interleukin 2 (IL-2) (75-133)-peptide [Cys
125(51)>Ser]-mutant (1-59), fused via a G2 peptide linker (60- 61) to human interleukin 2 (IL-2) (4-74)-peptide (62-132) and via a GSG
3S peptide linker (133-138) to human interleukin 2 receptor α-chain (IL2R subunit alpha, IL2Rα, IL2RA) (1- 165)-peptide (139-303), produced in Chinese hamster ovary (CHO) cells, glycoform alfa. The amino acid sequence of nemvaleukin alfa is given in SEQ ID NO:6. In some embodiments, nemvaleukin alfa exhibits the following post-translational modifications: disulfide bridges at positions: 31-116, 141-285, 184-242, 269-301, 166-197 or 166-199, 168- 199 or 168-197 (using the numbering in SEQ ID NO:6), and glycosylation sites at positions: N187, N206, T212 using the numbering in SEQ ID NO:6. The preparation and properties of nemvaleukin alfa, as well as additional alternative forms of IL-2 suitable for use in the invention, is described in U.S. Patent Application Publication No. US 2021/0038684 A1 and U.S. Patent No.10,183,979, the disclosures of which are incorporated by reference herein. In some embodiments, an IL-2 form suitable for use in the invention is a protein having at least 80%, at least 90%, at least 95%, or at least 90% sequence identity to SEQ ID NO:6. In some embodiments, an IL-2 form suitable for use in the invention has the amino acid sequence given in SEQ ID NO:6 or conservative amino acid substitutions thereof. In some embodiments, an IL-2 form suitable for use in the invention is a fusion protein comprising amino acids 24-452 of SEQ ID NO:7, or variants, fragments, or derivatives thereof. In some embodiments, an IL-2 form suitable for use in the invention is a fusion protein comprising an amino acid sequence having at least 80%, at least 90%, at least 95%, or at least 90% sequence DB1/ 149202201.1 14 Attorney Docket No.: 116983-5127-WO identity to amino acids 24-452 of SEQ ID NO:7, or variants, fragments, or derivatives thereof. Other IL-2 forms suitable for use in the present invention are described in U.S. Patent No.10,183,979, the disclosures of which are incorporated by reference herein. Optionally, in some embodiments, an IL-2 form suitable for use in the invention is a fusion protein comprising a first fusion partner that is linked to a second fusion partner by a mucin domain polypeptide linker, wherein the first fusion partner is IL-1Rα or a protein having at least 98% amino acid sequence identity to IL-1Rα and having the receptor antagonist activity of IL-Rα, and wherein the second fusion partner comprises all or a portion of an immunoglobulin comprising an Fc region, wherein the mucin domain polypeptide linker comprises SEQ ID NO:8 or an amino acid sequence having at least 90% sequence identity to SEQ ID NO:8 and wherein the half-life of the fusion protein is improved as compared to a fusion of the first fusion partner to the second fusion partner in the absence of the mucin domain polypeptide linker. TABLE 2. Amino acid sequences of interleukins.

DB1/ 149202201.1 15 Attorney Docket No.: 116983-5127-WO
[0017] In some embodiments, an IL-2 form suitable for use in the invention includes a antibody cytokine engrafted protein comprises a heavy chain variable region (VH), comprising complementarity determining regions HCDR1, HCDR2, HCDR3; a light chain variable region (VL), comprising LCDR1, LCDR2, LCDR3; and an IL-2 molecule or a fragment thereof engrafted into a CDR of the VH or the VL, wherein the antibody cytokine engrafted protein preferentially expands T effector cells over regulatory T cells. In some embodiments, the antibody cytokine engrafted protein comprises a heavy chain variable region (VH), comprising complementarity determining regions HCDR1, HCDR2, HCDR3; a light chain variable region (V
L), comprising LCDR1, LCDR2, LCDR3; and an IL-2 molecule or a fragment thereof engrafted into a CDR of the V
H or the V
L, wherein the IL-2 molecule is a mutein, and wherein the antibody cytokine engrafted protein preferentially expands T effector cells over regulatory T cells. In some embodiments, the IL-2 regimen comprises administration of an antibody described in U.S. Patent Application Publication No. US 2020/0270334 A1, the disclosures of which are incorporated by reference herein. In some embodiments, the antibody cytokine engrafted protein comprises a heavy chain variable region (VH), comprising complementarity determining regions HCDR1, HCDR2, HCDR3; a light chain variable region (VL), comprising LCDR1, LCDR2, LCDR3; and an IL-2 molecule or a fragment thereof engrafted into a CDR of the VH or the VL, wherein the IL-2 molecule is a mutein, wherein the antibody cytokine engrafted protein preferentially expands T effector cells over regulatory T cells, and wherein the antibody further comprises an IgG class heavy chain and an IgG class light chain selected from the group consisting of: a IgG class light chain comprising SEQ ID NO:39 and a IgG class heavy chain comprising SEQ ID NO:38; a IgG class light chain comprising SEQ ID NO:37 and a IgG class heavy chain comprising SEQ ID NO:29; a IgG class light chain comprising SEQ ID NO:39 and a IgG class heavy chain comprising SEQ ID NO:29; and a IgG class light chain comprising SEQ ID NO:37 and a IgG class heavy chain comprising SEQ ID NO:38. DB1/ 149202201.1 16 Attorney Docket No.: 116983-5127-WO [0018] In some embodiments, an IL-2 molecule or a fragment thereof is engrafted into HCDR1 of the VH, wherein the IL-2 molecule is a mutein. In some embodiments, an IL-2 molecule or a fragment thereof is engrafted into HCDR2 of the VH, wherein the IL-2 molecule is a mutein. In some embodiments, an IL-2 molecule or a fragment thereof is engrafted into HCDR3 of the V
H, wherein the IL-2 molecule is a mutein. In some embodiments, an IL-2 molecule or a fragment thereof is engrafted into LCDR1 of the VL, wherein the IL-2 molecule is a mutein. In some embodiments, an IL-2 molecule or a fragment thereof is engrafted into LCDR2 of the V
L, wherein the IL-2 molecule is a mutein. In some embodiments, an IL-2 molecule or a fragment thereof is engrafted into LCDR3 of the VL, wherein the IL-2 molecule is a mutein. [0019] The insertion of the IL-2 molecule can be at or near the N-terminal region of the CDR, in the middle region of the CDR or at or near the C-terminal region of the CDR. In some embodiments, the antibody cytokine engrafted protein comprises an IL-2 molecule incorporated into a CDR, wherein the IL2 sequence does not frameshift the CDR sequence. In some embodiments, the antibody cytokine engrafted protein comprises an IL-2 molecule incorporated into a CDR, wherein the IL-2 sequence replaces all or part of a CDR sequence. The replacement by the IL-2 molecule can be the N-terminal region of the CDR, in the middle region of the CDR or at or near the C-terminal region the CDR. A replacement by the IL-2 molecule can be as few as one or two amino acids of a CDR sequence, or the entire CDR sequences. [0020] In some embodiments, an IL-2 molecule is engrafted directly into a CDR without a peptide linker, with no additional amino acids between the CDR sequence and the IL-2 sequence. In some embodiments, an IL-2 molecule is engrafted indirectly into a CDR with a peptide linker, with one or more additional amino acids between the CDR sequence and the IL-2 sequence. [0021] In some embodiments, the IL-2 molecule described herein is an IL-2 mutein. In some instances, the IL-2 mutein comprising an R67A substitution. In some embodiments, the IL-2 mutein comprises the amino acid sequence SEQ ID NO:14 or SEQ ID NO:15. In some embodiments, the IL-2 mutein comprises an amino acid sequence in Table 1 in U.S. Patent Application Publication No. US 2020/0270334 A1, the disclosure of which is incorporated by reference herein. DB1/ 149202201.1 17 Attorney Docket No.: 116983-5127-WO [0022] In some embodiments, the antibody cytokine engrafted protein comprises an HCDR1 selected from the group consisting of SEQ ID NO:16, SEQ ID NO:19, SEQ ID NO:22 and SEQ ID NO:25. In some embodiments, the antibody cytokine engrafted protein comprises an HCDR1 selected from the group consisting of SEQ ID NO:7, SEQ ID NO:10, SEQ ID NO:13 and SEQ ID NO:16. In some embodiments, the antibody cytokine engrafted protein comprises an HCDR1 selected from the group consisting of HCDR2 selected from the group consisting of SEQ ID NO:17, SEQ ID NO:20, SEQ ID NO:23, and SEQ ID NO:26. In some embodiments, the antibody cytokine engrafted protein comprises an HCDR3 selected from the group consisting of SEQ ID NO:18, SEQ ID NO:21, SEQ ID NO:24, and SEQ ID NO:27. In some embodiments, the antibody cytokine engrafted protein comprises a VH region comprising the amino acid sequence of SEQ ID NO:28. In some embodiments, the antibody cytokine engrafted protein comprises a heavy chain comprising the amino acid sequence of SEQ ID NO:29. In some embodiments, the antibody cytokine engrafted protein comprises a V
L region comprising the amino acid sequence of SEQ ID NO:36. In some embodiments, the antibody cytokine engrafted protein comprises a light chain comprising the amino acid sequence of SEQ ID NO:37. In some embodiments, the antibody cytokine engrafted protein comprises a V
H region comprising the amino acid sequence of SEQ ID NO:28 and a V
L region comprising the amino acid sequence of SEQ ID NO:36. In some embodiments, the antibody cytokine engrafted protein comprises a heavy chain region comprising the amino acid sequence of SEQ ID NO:29 and a light chain region comprising the amino acid sequence of SEQ ID NO:37. In some embodiments, the antibody cytokine engrafted protein comprises a heavy chain region comprising the amino acid sequence of SEQ ID NO:29 and a light chain region comprising the amino acid sequence of SEQ ID NO:39. In some embodiments, the antibody cytokine engrafted protein comprises a heavy chain region comprising the amino acid sequence of SEQ ID NO:38 and a light chain region comprising the amino acid sequence of SEQ ID NO:37. In some embodiments, the antibody cytokine engrafted protein comprises a heavy chain region comprising the amino acid sequence of SEQ ID NO:38 and a light chain region comprising the amino acid sequence of SEQ ID NO:39. In some embodiments, the antibody cytokine engrafted protein comprises IgG.IL2F71A.H1 or IgG.IL2R67A.H1 of U.S. Patent Application Publication No. 2020/0270334 A1, or variants, derivatives, or fragments thereof, or conservative amino acid substitutions thereof, or proteins with at least 80%, at least 90%, at least 95%, or at least 98% sequence identity thereto. In some embodiments, the antibody components of the antibody DB1/ 149202201.1 18 Attorney Docket No.: 116983-5127-WO cytokine engrafted protein described herein comprise immunoglobulin sequences, framework sequences, or CDR sequences of palivizumab. In some embodiments, the antibody cytokine engrafted protein described herein has a longer serum half-life that a wild-type IL-2 molecule such as, but not limited to, aldesleukin or a comparable molecule. In some embodiments, the antibody cytokine engrafted protein described herein has a sequence as set forth in Table 3. TABLE 3: Sequences of exemplary palivizumab antibody-IL-2 engrafted proteins Identifier Sequence (One-Letter Amino Acid Symbols) SEQ ID NO:13 MYRMQLLSCI ALSLALVTNS APTSSSTKKT QLQLEHLLLD LQMILNGINN YKNPKLTRML IL-2 60 TFKFYMPKKA TELKHLQCLE EELKPLEEVL NLAQSKNFHL RPRDLISNIN VIVLELKGSE 120 TTFMCEYADE TATIVEFLNR WITFCQSIIS TLT 153 SEQ ID NO:14 APTSSSTKKT QLQLEHLLLD LQMILNGINN YKNPKLTAML TFKFYMPKKA TELKHLQCLE IL-2 mutein 60 EELKPLEEVL NLAQSKNFHL RPRDLISNIN VIVLELKGSE TTFMCEYADE TATIVEFLNR 120 WITFCQSIIS TLT 133 SEQ ID NO:15 APTSSSTKKT QLQLEHLLLD LQMILNGINN YKNPKLTRML TAKFYMPKKA TELKHLQCLE IL-2 mutein 60 EELKPLEEVL NLAQSKNFHL RPRDLISNIN VIVLELKGSE TTFMCEYADE TATIVEFLNR 120 WITFCQSIIS TLT 133 SEQ ID NO:16 GFSLAPTSSS TKKTQLQLEH LLLDLQMILN GINNYKNPKL TAMLTFKFYM PKKATELKHL HCDR1_IL-2 60 QCLEEELKPL EEVLNLAQSK NFHLRPRDLI SNINVIVLEL KGSETTFMCE YADETATIVE 120 FLNRWITFCQ SIISTLTSTS GMSVG 145 SEQ ID NO:17 DIWWDDKKDY NPSLKS 16 HCDR2 SEQ ID NO:18 SMITNWYFDV 10 HCDR3 SEQ ID NO:19 APTSSSTKKT QLQLEHLLLD LQMILNGINN YKNPKLTAML TFKFYMPKKA TELKHLQCLE HCDR1_IL-2 60 kabat EELKPLEEVL NLAQSKNFHL RPRDLISNIN VIVLELKGSE TTFMCEYADE TATIVEFLNR 120 WITFCQSIIS TLTSTSGMSV G 141 SEQ ID NO:20 DIWWDDKKDY NPSLKS 16 HCDR2 kabat SEQ ID NO:21 SMITNWYFDV 10 HCDR3 kabat SEQ ID NO:22 GFSLAPTSSS TKKTQLQLEH LLLDLQMILN GINNYKNPKL TAMLTFKFYM PKKATELKHL HCDR1_IL-2 60 clothia QCLEEELKPL EEVLNLAQSK NFHLRPRDLI SNINVIVLEL KGSETTFMCE YADETATIVE 120 FLNRWITFCQ SIISTLTSTS GM 142 SEQ ID NO:23 WWDDK HCDR2 clothia 5 SEQ ID NO:24 SMITNWYFDV 10 HCDR3 clothia SEQ ID NO:25 GFSLAPTSSS TKKTQLQLEH LLLDLQMILN GINNYKNPKL TAMLTFKFYM PKKATELKHL HCDR1_IL-2 60 IMGT QCLEEELKPL EEVLNLAQSK NFHLRPRDLI SNINVIVLEL KGSETTFMCE YADETATIVE 120 FLNRWITFCQ SIISTLTSTS GMS 143 SEQ ID NO:26 IWWDDKK HCDR2 IMGT 7 SEQ ID NO:27 ARSMITNWYF DV 12 HCDR3 IMGT SEQ ID NO:28 QVTLRESGPA LVKPTQTLTL TCTFSGFSLA PTSSSTKKTQ LQLEHLLLDL QMILNGINNY V 60 KNPKLTAMLT FKFYMPKKAT ELKHLQCLEE ELKPLEEVLN LAQSKNFHLR PRDLISNINV 120 IVLELKGSET TFMCEYADET ATIVEFLNRW ITFCQSIIST LTSTSGMSVG WIRQPPGKAL 180 DB1/ 149202201.1 19 Attorney Docket No.: 116983-5127-WO EWLADIWWDD KKDYNPSLKS RLTISKDTSK NQVVLKVTNM DPADTATYYC ARSMITNWYF 240 DVWGAGTTVT VSS 253 SEQ ID NO:29 QMILNGINNY KNPKLTAMLT FKFYMPKKAT ELKHLQCLEE ELKPLEEVLN LAQSKNFHLR Heavy chain 60 PRDLISNINV IVLELKGSET TFMCEYADET ATIVEFLNRW ITFCQSIIST LTSTSGMSVG 120 WIRQPPGKAL EWLADIWWDD KKDYNPSLKS RLTISKDTSK NQVVLKVTNM DPADTATYYC 180 ARSMITNWYF DVWGAGTTVT VSSASTKGPS VFPLAPSSKS TSGGTAALGC LVKDYFPEPV 240 TVSWNSGALT SGVHTFPAVL QSSGLYSLSS VVTVPSSSLG TQTYICNVNH KPSNTKVDKR 300 VEPKSCDKTH TCPPCPAPEL LGGPSVFLFP PKPKDTLMIS RTPEVTCVVV AVSHEDPEVK 360 FNWYVDGVEV HNAKTKPREE QYNSTYRVVS VLTVLHQDWL NGKEYKCKVS NKALAAPIEK 420 TISKAKGQPR EPQVYTLPPS REEMTKNQVS LTCLVKGFYP SDIAVEWESN GQPENNYKTT 480 PPVLDSDGSF FLYSKLTVDK SRWQQGNVFS CSVMHEALHN HYTQKSLSLS PGK 533 SEQ ID NO:30 KAQLSVGYMH 10 LCDR1 kabat SEQ ID NO:31 DTSKLAS 7 LCDR2 kabat SEQ ID NO:32 FQGSGYPFT 9 LCDR3 kabat SEQ ID NO:33 QLSVGY 6 LCDR1 chothia SEQ ID NO:34 DTS 3 LCDR2 chothia SEQ ID NO:35 GSGYPF 6 LCDR3 chothia SEQ ID NO:36 DIQMTQSPST LSASVGDRVT ITCKAQLSVG YMHWYQQKPG KAPKLLIYDT SKLASGVPSR 60 V FSGSGSGTEF TLTISSLQPD DFATYYCFQG SGYPFTFGGG TKLEIK 106 SEQ ID NO:37 DIQMTQSPST LSASVGDRVT ITCKAQLSVG YMHWYQQKPG KAPKLLIYDT SKLASGVPSR 60 Light chain FSGSGSGTEF TLTISSLQPD DFATYYCFQG SGYPFTFGGG TKLEIKRTVA APSVFIFPPS 120 DEQLKSGTAS VVCLLNNFYP REAKVQWKVD NALQSGNSQE SVTEQDSKDS TYSLSSTLTL 180 SKADYEKHKV YACEVTHQGL SSPVTKSFNR GEC 213 SEQ ID NO:38 QVTLRESGPA LVKPTQTLTL TCTFSGFSLA PTSSSTKKTQ LQLEHLLLDL QMILNGINNY 60 Light chain KNPKLTRMLT AKFYMPKKAT ELKHLQCLEE ELKPLEEVLN LAQSKNFHLR PRDLISNINV 120 IVLELKGSET TFMCEYADET ATIVEFLNRW ITFCQSIIST LTSTSGMSVG WIRQPPGKAL 180 EWLADIWWDD KKDYNPSLKS RLTISKDTSK NQVVLKVTNM DPADTATYYC ARSMITNWYF 240 DVWGAGTTVT VSSASTKGPS VFPLAPSSKS TSGGTAALGC LVKDYFPEPV TVSWNSGALT 300 SGVHTFPAVL QSSGLYSLSS VVTVPSSSLG TQTYICNVNH KPSNTKVDKR VEPKSCDKTH 360 TCPPCPAPEL LGGPSVFLFP PKPKDTLMIS RTPEVTCVVV AVSHEDPEVK FNWYVDGVEV 420 HNAKTKPREE QYNSTYRVVS VLTVLHQDWL NGKEYKCKVS NKALAAPIEK TISKAKGQPR 480 EPQVYTLPPS REEMTKNQVS LTCLVKGFYP SDIAVEWESN GQPENNYKTT PPVLDSDGSF 540 FLYSKLTVDK SRWQQGNVFS CSVMHEALHN HYTQKSLSLS PGK 583 SEQ ID NO:39 DIQMTQSPST LSASVGDRVT ITCKAQLSVG YMHWYQQKPG KAPKLLIYDT SKLASGVPSR 60 Light chain FSGSGSGTEF TLTISSLQPD DFATYYCFQG SGYPFTFGGG TKLEIKRTVA APSVFIFPPS 120 DEQLKSGTAS VVCLLNNFYP REAKVQWKVD NALQSGNSQE SVTEQDSKDS TYSLSSTLTL 180 SKADYEKHKV YACEVTHQGL SSPVTKSFNR GEC 213 [0023] The term “IL-4” (also referred to herein as “IL4”) refers to the cytokine known as interleukin 4, which is produced by Th2 T cells and by eosinophils, basophils, and mast cells. IL-4 regulates the differentiation of naïve helper T cells (Th0 cells) to Th2 T cells. Steinke and Borish, Respir. Res.2001, 2, 66-70. Upon activation by IL-4, Th2 T cells subsequently produce additional IL-4 in a positive feedback loop. IL-4 also stimulates B cell proliferation and class II MHC expression, and induces class switching to IgE and IgG
1 expression from B cells. Recombinant human IL-4 suitable for use in the invention is commercially available from multiple suppliers, including ProSpec-Tany TechnoGene Ltd., East Brunswick, NJ, USA (Cat. No. CYT-211) and ThermoFisher Scientific, Inc., Waltham, MA, USA (human DB1/ 149202201.1 20 Attorney Docket No.: 116983-5127-WO IL-15 recombinant protein, Cat. No. Gibco CTP0043). The amino acid sequence of recombinant human IL-4 suitable for use in the invention is given in Table 2 (SEQ ID NO:9). [0024] The term “IL-7” (also referred to herein as “IL7”) refers to a glycosylated tissue- derived cytokine known as interleukin 7, which may be obtained from stromal and epithelial cells, as well as from dendritic cells. Fry and Mackall, Blood 2002, 99, 3892-904. IL-7 can stimulate the development of T cells. IL-7 binds to the IL-7 receptor, a heterodimer consisting of IL-7 receptor alpha and common gamma chain receptor, which in a series of signals important for T cell development within the thymus and survival within the periphery. Recombinant human IL-7 suitable for use in the invention is commercially available from multiple suppliers, including ProSpec-Tany TechnoGene Ltd., East Brunswick, NJ, USA (Cat. No. CYT-254) and ThermoFisher Scientific, Inc., Waltham, MA, USA (human IL-15 recombinant protein, Cat. No. Gibco PHC0071). The amino acid sequence of recombinant human IL-7 suitable for use in the invention is given in Table 2 (SEQ ID NO:10). [0025] The term “IL-15” (also referred to herein as “IL15”) refers to the T cell growth factor known as interleukin-15, and includes all forms of IL-2 including human and mammalian forms, conservative amino acid substitutions, glycoforms, biosimilars, and variants thereof. IL-15 is described, e.g., in Fehniger and Caligiuri, Blood 2001, 97, 14-32, the disclosure of which is incorporated by reference herein. IL-15 shares β and γ signaling receptor subunits with IL-2. Recombinant human IL-15 is a single, non-glycosylated polypeptide chain containing 114 amino acids (and an N-terminal methionine) with a molecular mass of 12.8 kDa. Recombinant human IL-15 is commercially available from multiple suppliers, including ProSpec-Tany TechnoGene Ltd., East Brunswick, NJ, USA (Cat. No. CYT-230-b) and ThermoFisher Scientific, Inc., Waltham, MA, USA (human IL-15 recombinant protein, Cat. No.34-8159-82). The amino acid sequence of recombinant human IL-15 suitable for use in the invention is given in Table 2 (SEQ ID NO:11). [0026] The term “IL-21” (also referred to herein as “IL21”) refers to the pleiotropic cytokine protein known as interleukin-21, and includes all forms of IL-21 including human and mammalian forms, conservative amino acid substitutions, glycoforms, biosimilars, and variants thereof. IL-21 is described, e.g., in Spolski and Leonard, Nat. Rev. Drug. Disc.2014, 13, 379-95, the disclosure of which is incorporated by reference herein. IL-21 is primarily produced by natural killer T cells and activated human CD4
+ T cells. Recombinant human IL- 21 is a single, non-glycosylated polypeptide chain containing 132 amino acids with a DB1/ 149202201.1 21 Attorney Docket No.: 116983-5127-WO molecular mass of 15.4 kDa. Recombinant human IL-21 is commercially available from multiple suppliers, including ProSpec-Tany TechnoGene Ltd., East Brunswick, NJ, USA (Cat. No. CYT-408-b) and ThermoFisher Scientific, Inc., Waltham, MA, USA (human IL-21 recombinant protein, Cat. No.14-8219-80). The amino acid sequence of recombinant human IL-21 suitable for use in the invention is given in Table 2 (SEQ ID NO:21). [0027] When “an anti-tumor effective amount”, “a tumor-inhibiting effective amount”, or “therapeutic amount” is indicated, the precise amount of the compositions of the present invention to be administered can be determined by a physician with consideration of individual differences in age, weight, tumor size, extent of infection or metastasis, and condition of the patient (subject). It can generally be stated that a pharmaceutical composition comprising the tumor infiltrating lymphocytes (e.g. secondary TILs or genetically modified cytotoxic lymphocytes) described herein may be administered at a dosage of 10
4 to 10
11 cells/kg body weight (e.g., 10
5 to 10
6, 10
5 to 10
10, 10
5 to 10
11, 10
6 to 10
10, 10
6 to 10
11,10
7 to 10
11, 10
7 to 10
10, 10
8 to 10
11, 10
8 to 10
10, 10
9 to 10
11, or 10
9 to 10
10 cells/kg body weight), including all integer values within those ranges. TILs (including in some cases, genetically modified cytotoxic lymphocytes) compositions may also be administered multiple times at these dosages. The TILs (including, in some cases, genetically engineered TILs) can be administered by using infusion techniques that are commonly known in immunotherapy (see, e.g., Rosenberg, et al., New Eng. J. of Med.1988, 319, 1676). The optimal dosage and treatment regime for a particular patient can readily be determined by one skilled in the art of medicine by monitoring the patient for signs of disease and adjusting the treatment accordingly. [0028] The term “microenvironment,” as used herein, may refer to the solid or hematological tumor microenvironment as a whole or to an individual subset of cells within the microenvironment. The tumor microenvironment, as used herein, refers to a complex mixture of “cells, soluble factors, signaling molecules, extracellular matrices, and mechanical cues that promote neoplastic transformation, support tumor growth and invasion, protect the tumor from host immunity, foster therapeutic resistance, and provide niches for dominant metastases to thrive,” as described in Swartz, et al., Cancer Res., 2012, 72, 2473. Although tumors express antigens that should be recognized by T cells, tumor clearance by the immune system is rare because of immune suppression by the microenvironment. DB1/ 149202201.1 22 Attorney Docket No.: 116983-5127-WO [0029] In some embodiments, the invention includes a method of treating a cancer with a population of TILs, wherein a patient is pre-treated with non-myeloablative chemotherapy prior to an infusion of TILs according to the invention. In some embodiments, the population of TILs may be provided wherein a patient is pre-treated with nonmyeloablative chemotherapy prior to an infusion of TILs according to the present invention. In some embodiments, the non-myeloablative chemotherapy is cyclophosphamide 60 mg/kg/d for 2 days (days 27 and 26 prior to TIL infusion) and fludarabine 25 mg/m2/d for 5 days (days 27 to 23 prior to TIL infusion). In some embodiments, after non-myeloablative chemotherapy and TIL infusion (at day 0) according to the invention, the patient receives an intravenous infusion of IL-2 intravenously at 720,000 IU/kg every 8 hours to physiologic tolerance. [0030] Experimental findings indicate that lymphodepletion prior to adoptive transfer of tumor-specific T lymphocytes plays a key role in enhancing treatment efficacy by eliminating regulatory T cells and competing elements of the immune system (“cytokine sinks”). Accordingly, some embodiments of the invention utilize a lymphodepletion step (sometimes also referred to as “immunosuppressive conditioning”) on the patient prior to the introduction of the TILs of the invention. [0031] The term “effective amount” or “therapeutically effective amount” refers to that amount of a compound or combination of compounds as described herein that is sufficient to effect the intended application including, but not limited to, disease treatment. A therapeutically effective amount may vary depending upon the intended application (in vitro or in vivo), or the subject and disease condition being treated (e.g., the weight, age and gender of the subject), the severity of the disease condition, or the manner of administration. The term also applies to a dose that will induce a particular response in target cells (e.g., the reduction of platelet adhesion and/or cell migration). The specific dose will vary depending on the particular compounds chosen, the dosing regimen to be followed, whether the compound is administered in combination with other compounds, timing of administration, the tissue to which it is administered, and the physical delivery system in which the compound is carried. [0032] The terms “treatment”, “treating”, “treat”, and the like, refer to obtaining a desired pharmacologic and/or physiologic effect. The effect may be prophylactic in terms of completely or partially preventing a disease or symptom thereof and/or may be therapeutic in terms of a partial or complete cure for a disease and/or adverse effect attributable to the DB1/ 149202201.1 23 Attorney Docket No.: 116983-5127-WO disease. “Treatment”, as used herein, covers any treatment of a disease in a mammal, particularly in a human, and includes: (a) preventing the disease from occurring in a subject which may be predisposed to the disease but has not yet been diagnosed as having it; (b) inhibiting the disease, i.e., arresting its development or progression; and (c) relieving the disease, i.e., causing regression of the disease and/or relieving one or more disease symptoms. “Treatment” is also meant to encompass delivery of an agent in order to provide for a pharmacologic effect, even in the absence of a disease or condition. For example, “treatment” encompasses delivery of a composition that can elicit an immune response or confer immunity in the absence of a disease condition, e.g., in the case of a vaccine. [0033] The terms “non-myeloablative chemotherapy,” “non-myeloablative lymphodepletion,” “NMALD,” “NMA LD,” “NMA-LD,” and any variants of the foregoing, are used interchangeably to indicate a chemotherapeutic regimen designed to deplete the patient’s lymphoid immune cells while avoiding depletion of the patient’s myeloid immune cells. Typically, the patient receives a course of non-myeloablative chemotherapy prior to the administration of tumor infiltrating lymphocytes to the patient as described herein. [0034] The term “heterologous” when used with reference to portions of a nucleic acid or protein indicates that the nucleic acid or protein comprises two or more subsequences that are not found in the same relationship to each other in nature. For instance, the nucleic acid is typically recombinantly produced, having two or more sequences from unrelated genes arranged to make a new functional nucleic acid, e.g., a promoter from one source and a coding region from another source, or coding regions from different sources. Similarly, a heterologous protein indicates that the protein comprises two or more subsequences that are not found in the same relationship to each other in nature (e.g., a fusion protein). [0035] The terms “sequence identity,” “percent identity,” and “sequence percent identity” (or synonyms thereof, e.g., “99% identical”) in the context of two or more nucleic acids or polypeptides, refer to two or more sequences or subsequences that are the same or have a specified percentage of nucleotides or amino acid residues that are the same, when compared and aligned (introducing gaps, if necessary) for maximum correspondence, not considering any conservative amino acid substitutions as part of the sequence identity. The percent identity can be measured using sequence comparison software or algorithms or by visual inspection. Various algorithms and software are known in the art that can be used to obtain alignments of amino acid or nucleotide sequences. Suitable programs to determine percent DB1/ 149202201.1 24 Attorney Docket No.: 116983-5127-WO sequence identity include for example the BLAST suite of programs available from the U.S. Government’s National Center for Biotechnology Information BLAST web site. Comparisons between two sequences can be carried using either the BLASTN or BLASTP algorithm. BLASTN is used to compare nucleic acid sequences, while BLASTP is used to compare amino acid sequences. ALIGN, ALIGN-2 (Genentech, South San Francisco, California) or MegAlign, available from DNASTAR, are additional publicly available software programs that can be used to align sequences. One skilled in the art can determine appropriate parameters for maximal alignment by particular alignment software. In certain embodiments, the default parameters of the alignment software are used. [0036] As used herein, the term “variant” encompasses but is not limited to antibodies or fusion proteins which comprise an amino acid sequence which differs from the amino acid sequence of a reference antibody by way of one or more substitutions, deletions and/or additions at certain positions within or adjacent to the amino acid sequence of the reference antibody. The variant may comprise one or more conservative substitutions in its amino acid sequence as compared to the amino acid sequence of a reference antibody. Conservative substitutions may involve, e.g., the substitution of similarly charged or uncharged amino acids. The variant retains the ability to specifically bind to the antigen of the reference antibody. The term variant also includes pegylated antibodies or proteins. [0037] By “tumor infiltrating lymphocytes” or “TILs” herein is meant a population of cells originally obtained as white blood cells that have left the bloodstream of a subject and migrated into a tumor. TILs include, but are not limited to, CD8
+ cytotoxic T cells (lymphocytes), Th1 and Th17 CD4
+ T cells, natural killer cells, dendritic cells and M1 macrophages. TILs include both primary and secondary TILs. “Primary TILs” are those that are obtained from patient tissue samples as outlined herein (sometimes referred to as “freshly harvested”), and “secondary TILs” are any TIL cell populations that have been expanded or proliferated as discussed herein, including, but not limited to bulk TILs, expanded TILs (“REP TILs”) as well as “reREP TILs” as discussed herein. reREP TILs can include for example second expansion TILs or second additional expansion TILs. [0038] TILs can generally be defined either biochemically, using cell surface markers, or functionally, by their ability to infiltrate tumors and effect treatment. TILs can be generally categorized by expressing one or more of the following biomarkers: CD4, CD8, TCR αβ, CD27, CD28, CD56, CCR7, CD45Ra, CD95, PD-1, and CD25. Additionally, and DB1/ 149202201.1 25 Attorney Docket No.: 116983-5127-WO alternatively, TILs can be functionally defined by their ability to infiltrate solid tumors upon reintroduction into a patient. TILs may further be characterized by potency – for example, TILs may be considered potent if, for example, interferon (IFN) release is greater than about 50 pg/mL, greater than about 100 pg/mL, greater than about 150 pg/mL, or greater than about 200 pg/mL. TILs may be considered potent if, for example, interferon (IFNγ) release is greater than about 50 pg/mL, greater than about 100 pg/mL, greater than about 150 pg/mL, or greater than about 200 pg/mL, greater than about 300 pg/mL, greater than about 400 pg/mL, greater than about 500 pg/mL, greater than about 600 pg/mL, greater than about 700 pg/mL, greater than about 800 pg/mL, greater than about 900 pg/mL, greater than about 1000 pg/mL. [0039] The term “deoxyribonucleotide” encompasses natural and synthetic, unmodified and modified deoxyribonucleotides. Modifications include changes to the sugar moiety, to the base moiety and/or to the linkages between deoxyribonucleotide in the oligonucleotide. [0040] The term “RNA” defines a molecule comprising at least one ribonucleotide residue. The term “ribonucleotide” defines a nucleotide with a hydroxyl group at the 2' position of a b-D-ribofuranose moiety. The term RNA includes double-stranded RNA, single-stranded RNA, isolated RNA such as partially purified RNA, essentially pure RNA, synthetic RNA, recombinantly produced RNA, as well as altered RNA that differs from naturally occurring RNA by the addition, deletion, substitution and/or alteration of one or more nucleotides. Nucleotides of the RNA molecules described herein may also comprise non-standard nucleotides, such as non-naturally occurring nucleotides or chemically synthesized nucleotides or deoxynucleotides. These altered RNAs can be referred to as analogs or analogs of naturally-occurring RNA. [0041] The terms “pharmaceutically acceptable carrier” or “pharmaceutically acceptable excipient” are intended to include any and all solvents, dispersion media, coatings, antibacterial and antifungal agents, isotonic and absorption delaying agents, and inert ingredients. The use of such pharmaceutically acceptable carriers or pharmaceutically acceptable excipients for active pharmaceutical ingredients is well known in the art. Except insofar as any conventional pharmaceutically acceptable carrier or pharmaceutically acceptable excipient is incompatible with the active pharmaceutical ingredient, its use in therapeutic compositions of the invention is contemplated. Additional active pharmaceutical ingredients, such as other drugs, can also be incorporated into the described compositions and methods. DB1/ 149202201.1 26 Attorney Docket No.: 116983-5127-WO [0042] The terms “about” and “approximately” mean within a statistically meaningful range of a value. Such a range can be within an order of magnitude, preferably within 50%, more preferably within 20%, more preferably still within 10%, and even more preferably within 5% of a given value or range. The allowable variation encompassed by the terms “about” or “approximately” depends on the particular system under study, and can be readily appreciated by one of ordinary skill in the art. Moreover, as used herein, the terms “about” and “approximately” mean that dimensions, sizes, formulations, parameters, shapes and other quantities and characteristics are not and need not be exact, but may be approximate and/or larger or smaller, as desired, reflecting tolerances, conversion factors, rounding off, measurement error and the like, and other factors known to those of skill in the art. In general, a dimension, size, formulation, parameter, shape or other quantity or characteristic is “about” or “approximate” whether or not expressly stated to be such. It is noted that embodiments of very different sizes, shapes and dimensions may employ the described arrangements. [0043] The transitional terms “comprising,” “consisting essentially of,” and “consisting of,” when used in the appended claims, in original and amended form, define the claim scope with respect to what unrecited additional claim elements or steps, if any, are excluded from the scope of the claim(s). The term “comprising” is intended to be inclusive or open-ended and does not exclude any additional, unrecited element, method, step or material. The term “consisting of” excludes any element, step or material other than those specified in the claim and, in the latter instance, impurities ordinary associated with the specified material(s). The term “consisting essentially of” limits the scope of a claim to the specified elements, steps or material(s) and those that do not materially affect the basic and novel characteristic(s) of the claimed invention. All compositions, methods, and kits described herein that embody the present invention can, in alternate embodiments, be more specifically defined by any of the transitional terms “comprising,” “consisting essentially of,” and “consisting of.” [0044] The terms “antibody” and its plural form “antibodies” refer to whole immunoglobulins and any antigen-binding fragment (“antigen-binding portion”) or single chains thereof. An “antibody” further refers to a glycoprotein comprising at least two heavy (H) chains and two light (L) chains inter-connected by disulfide bonds, or an antigen-binding portion thereof. Each heavy chain is comprised of a heavy chain variable region (abbreviated herein as VH) and a heavy chain constant region. The heavy chain constant region is DB1/ 149202201.1 27 Attorney Docket No.: 116983-5127-WO comprised of three domains, CH1, CH2 and CH3. Each light chain is comprised of a light chain variable region (abbreviated herein as VL) and a light chain constant region. The light chain constant region is comprised of one domain, CL. The VH and VL regions of an antibody may be further subdivided into regions of hypervariability, which are referred to as complementarity determining regions (CDR) or hypervariable regions (HVR), and which can be interspersed with regions that are more conserved, termed framework regions (FR). Each V
H and V
L is composed of three CDRs and four FRs, arranged from amino-terminus to carboxy-terminus in the following order: FR1, CDR1, FR2, CDR2, FR3, CDR3, FR4. The variable regions of the heavy and light chains contain a binding domain that interacts with an antigen epitope or epitopes. The constant regions of the antibodies may mediate the binding of the immunoglobulin to host tissues or factors, including various cells of the immune system (e.g., effector cells) and the first component (Clq) of the classical complement system. [0045] The term “antigen” refers to a substance that induces an immune response. In some embodiments, an antigen is a molecule capable of being bound by an antibody or a TCR if presented by major histocompatibility complex (MHC) molecules. The term “antigen”, as used herein, also encompasses T cell epitopes. An antigen is additionally capable of being recognized by the immune system. In some embodiments, an antigen is capable of inducing a humoral immune response or a cellular immune response leading to the activation of B lymphocytes and/or T lymphocytes. In some cases, this may require that the antigen contains or is linked to a Th cell epitope. An antigen can also have one or more epitopes (e.g., B- and T-epitopes). In some embodiments, an antigen will preferably react, typically in a highly specific and selective manner, with its corresponding antibody or TCR and not with the multitude of other antibodies or TCRs which may be induced by other antigens. [0046] The terms “monoclonal antibody,” “mAb,” “monoclonal antibody composition,” or their plural forms refer to a preparation of antibody molecules of single molecular composition. A monoclonal antibody composition displays a single binding specificity and affinity for a particular epitope. Monoclonal antibodies specific to certain receptors can be made using knowledge and skill in the art of injecting test subjects with suitable antigen and then isolating hybridomas expressing antibodies having the desired sequence or functional characteristics. DNA encoding the monoclonal antibodies is readily isolated and sequenced using conventional procedures (e.g., by using oligonucleotide probes that are capable of binding specifically to genes encoding the heavy and light chains of the monoclonal DB1/ 149202201.1 28 Attorney Docket No.: 116983-5127-WO antibodies). The hybridoma cells serve as a preferred source of such DNA. Once isolated, the DNA may be placed into expression vectors, which are then transfected into host cells such as E. coli cells, simian COS cells, Chinese hamster ovary (CHO) cells, or myeloma cells that do not otherwise produce immunoglobulin protein, to obtain the synthesis of monoclonal antibodies in the recombinant host cells. Recombinant production of antibodies will be described in more detail below. [0047] The terms “antigen-binding portion” or “antigen-binding fragment” of an antibody (or simply “antibody portion” or “fragment”), as used herein, refers to one or more fragments of an antibody that retain the ability to specifically bind to an antigen. It has been shown that the antigen-binding function of an antibody can be performed by fragments of a full-length antibody. Examples of binding fragments encompassed within the term “antigen-binding portion” of an antibody include (i) a Fab fragment, a monovalent fragment consisting of the VL, VH, CL and CH1 domains; (ii) a F(ab′)2 fragment, a bivalent fragment comprising two Fab fragments linked by a disulfide bridge at the hinge region; (iii) a Fd fragment consisting of the V
H and CH1 domains; (iv) a Fv fragment consisting of the V
L and V
H domains of a single arm of an antibody, (v) a domain antibody (dAb) fragment (Ward, et al., Nature, 1989, 341, 544-546), which may consist of a V
H or a V
L domain; and (vi) an isolated complementarity determining region (CDR). Furthermore, although the two domains of the Fv fragment, VL and VH, are coded for by separate genes, they can be joined, using recombinant methods, by a synthetic linker that enables them to be made as a single protein chain in which the V
L and V
H regions pair to form monovalent molecules known as single chain Fv (scFv); see, e.g., Bird, et al., Science 1988, 242, 423-426; and Huston, et al., Proc. Natl. Acad. Sci. USA 1988, 85, 5879-5883). Such scFv antibodies are also intended to be encompassed within the terms “antigen-binding portion” or “antigen-binding fragment” of an antibody. These antibody fragments are obtained using conventional techniques known to those with skill in the art, and the fragments are screened for utility in the same manner as are intact antibodies. In some embodiments, a scFv protein domain comprises a V
H portion and a V
L portion. A scFv molecule is denoted as either V
L-L-V
H if the V
L domain is the N-terminal part of the scFv molecule, or as VH-L-VL if the VH domain is the N-terminal part of the scFv molecule. Methods for making scFv molecules and designing suitable peptide linkers are described in U.S. Pat. No.4,704,692, U.S. Pat. No.4,946,778, R. Raag and M. Whitlow, “Single Chain Fvs.” FASEB Vol 9:73-80 (1995) and R. E. Bird and B. W. Walker, Single DB1/ 149202201.1 29 Attorney Docket No.: 116983-5127-WO Chain Antibody Variable Regions, TIBTECH, Vol 9: 132-137 (1991), the disclosures of which are incorporated by reference herein. [0048] The term “human antibody,” as used herein, is intended to include antibodies having variable regions in which both the framework and CDR regions are derived from human germline immunoglobulin sequences. Furthermore, if the antibody contains a constant region, the constant region also is derived from human germline immunoglobulin sequences. The human antibodies of the invention may include amino acid residues not encoded by human germline immunoglobulin sequences (e.g., mutations introduced by random or site-specific mutagenesis in vitro or by somatic mutation in vivo). The term “human antibody”, as used herein, is not intended to include antibodies in which CDR sequences derived from the germline of another mammalian species, such as a mouse, have been grafted onto human framework sequences. [0049] The term “human monoclonal antibody” refers to antibodies displaying a single binding specificity which have variable regions in which both the framework and CDR regions are derived from human germline immunoglobulin sequences. In some embodiments, the human monoclonal antibodies are produced by a hybridoma which includes a B cell obtained from a transgenic nonhuman animal, e.g., a transgenic mouse, having a genome comprising a human heavy chain transgene and a light chain transgene fused to an immortalized cell. [0050] The term “recombinant human antibody”, as used herein, includes all human antibodies that are prepared, expressed, created or isolated by recombinant means, such as (a) antibodies isolated from an animal (such as a mouse) that is transgenic or transchromosomal for human immunoglobulin genes or a hybridoma prepared therefrom (described further below), (b) antibodies isolated from a host cell transformed to express the human antibody, e.g., from a transfectoma, (c) antibodies isolated from a recombinant, combinatorial human antibody library, and (d) antibodies prepared, expressed, created or isolated by any other means that involve splicing of human immunoglobulin gene sequences to other DNA sequences. Such recombinant human antibodies have variable regions in which the framework and CDR regions are derived from human germline immunoglobulin sequences. In certain embodiments, however, such recombinant human antibodies can be subjected to in vitro mutagenesis (or, when an animal transgenic for human Ig sequences is used, in vivo somatic mutagenesis) and thus the amino acid sequences of the VH and VL regions of the DB1/ 149202201.1 30 Attorney Docket No.: 116983-5127-WO recombinant antibodies are sequences that, while derived from and related to human germline VH and VL sequences, may not naturally exist within the human antibody germline repertoire in vivo. [0051] As used herein, “isotype” refers to the antibody class (e.g., IgM or IgG1) that is encoded by the heavy chain constant region genes. [0052] The phrases “an antibody recognizing an antigen” and “an antibody specific for an antigen” are used interchangeably herein with the term “an antibody which binds specifically to an antigen.” [0053] The term “human antibody derivatives” refers to any modified form of the human antibody, including a conjugate of the antibody and another active pharmaceutical ingredient or antibody. The terms “conjugate,” “antibody-drug conjugate”, “ADC,” or “immunoconjugate” refers to an antibody, or a fragment thereof, conjugated to another therapeutic moiety, which can be conjugated to antibodies described herein using methods available in the art. [0054] The terms “humanized antibody,” “humanized antibodies,” and “humanized” are intended to refer to antibodies in which CDR sequences derived from the germline of another mammalian species, such as a mouse, have been grafted onto human framework sequences. Additional framework region modifications may be made within the human framework sequences. Humanized forms of non-human (for example, murine) antibodies are chimeric antibodies that contain minimal sequence derived from non-human immunoglobulin. For the most part, humanized antibodies are human immunoglobulins (recipient antibody) in which residues from a hypervariable region of the recipient are replaced by residues from a 15 hypervariable region of a non-human species (donor antibody) such as mouse, rat, rabbit or nonhuman primate having the desired specificity, affinity, and capacity. In some instances, Fv framework region (FR) residues of the human immunoglobulin are replaced by corresponding non-human residues. Furthermore, humanized antibodies may comprise residues that are not found in the recipient antibody or in the donor antibody. These modifications are made to further refine antibody performance. In general, the humanized antibody will comprise substantially all of at least one, and typically two, variable domains, in which all or substantially all of the hypervariable loops correspond to those of a non- human immunoglobulin and all or substantially all of the FR regions are those of a human immunoglobulin sequence. The humanized antibody optionally also will comprise at least a DB1/ 149202201.1 31 Attorney Docket No.: 116983-5127-WO portion of an immunoglobulin constant region (Fc), typically that of a human immunoglobulin. For further details, see Jones, et al., Nature 1986, 321, 522-525; Riechmann, et al., Nature 1988, 332, 323-329; and Presta, Curr. Op. Struct. Biol.1992, 2, 593-596. The antibodies described herein may also be modified to employ any Fc variant which is known to impart an improvement (e.g., reduction) in effector function and/or FcR binding. The Fc variants may include, for example, any one of the amino acid substitutions disclosed in International Patent Application Publication Nos. WO 1988/07089 A1, WO 1996/14339 A1, WO 1998/05787 A1, WO 1998/23289 A1, WO 1999/51642 A1, WO 99/58572 A1, WO 2000/09560 A2, WO 2000/32767 A1, WO 2000/42072 A2, WO 2002/44215 A2, WO 2002/060919 A2, WO 2003/074569 A2, WO 2004/016750 A2, WO 2004/029207 A2, WO 2004/035752 A2, WO 2004/063351 A2, WO 2004/074455 A2, WO 2004/099249 A2, WO 2005/040217 A2, WO 2005/070963 A1, WO 2005/077981 A2, WO 2005/092925 A2, WO 2005/123780 A2, WO 2006/019447 A1, WO 2006/047350 A2, and WO 2006/085967 A2; and U.S. Patent Nos.5,648,260; 5,739,277; 5,834,250; 5,869,046; 6,096,871; 6,121,022; 6,194,551; 6,242,195; 6,277,375; 6,528,624; 6,538,124; 6,737,056; 6,821,505; 6,998,253; and 7,083,784; the disclosures of which are incorporated by reference herein. [0055] The term “chimeric antibody” is intended to refer to antibodies in which the variable region sequences are derived from one species and the constant region sequences are derived from another species, such as an antibody in which the variable region sequences are derived from a mouse antibody and the constant region sequences are derived from a human antibody. [0056] A “diabody” is a small antibody fragment with two antigen-binding sites. The fragments comprises a heavy chain variable domain (V
H) connected to a light chain variable domain (VL) in the same polypeptide chain (VH-VL or VL-VH). By using a linker that is too short to allow pairing between the two domains on the same chain, the domains are forced to pair with the complementary domains of another chain and create two antigen-binding sites. Diabodies are described more fully in, e.g., European Patent No. EP 404,097, International Patent Publication No. WO 93/11161; and Bolliger, et al., Proc. Natl. Acad. Sci. USA 1993, 90, 6444-6448. [0057] The term “glycosylation” refers to a modified derivative of an antibody. An aglycoslated antibody lacks glycosylation. Glycosylation can be altered to, for example, DB1/ 149202201.1 32 Attorney Docket No.: 116983-5127-WO increase the affinity of the antibody for antigen. Such carbohydrate modifications can be accomplished by, for example, altering one or more sites of glycosylation within the antibody sequence. For example, one or more amino acid substitutions can be made that result in elimination of one or more variable region framework glycosylation sites to thereby eliminate glycosylation at that site. Aglycosylation may increase the affinity of the antibody for antigen, as described in U.S. Patent Nos.5,714,350 and 6,350,861. Additionally or alternatively, an antibody can be made that has an altered type of glycosylation, such as a hypofucosylated antibody having reduced amounts of fucosyl residues or an antibody having increased bisecting GlcNac structures. Such altered glycosylation patterns have been demonstrated to increase the ability of antibodies. Such carbohydrate modifications can be accomplished by, for example, expressing the antibody in a host cell with altered glycosylation machinery. Cells with altered glycosylation machinery have been described in the art and can be used as host cells in which to express recombinant antibodies of the invention to thereby produce an antibody with altered glycosylation. For example, the cell lines Ms704, Ms705, and Ms709 lack the fucosyltransferase gene, FUT8 (alpha (1,6) fucosyltransferase), such that antibodies expressed in the Ms704, Ms705, and Ms709 cell lines lack fucose on their carbohydrates. The Ms704, Ms705, and Ms709 FUT8−/− cell lines were created by the targeted disruption of the FUT8 gene in CHO/DG44 cells using two replacement vectors (see e.g. U.S. Patent Publication No.2004/0110704 or Yamane-Ohnuki, et al., Biotechnol. Bioeng., 2004, 87, 614-622). As another example, European Patent No. EP 1,176,195 describes a cell line with a functionally disrupted FUT8 gene, which encodes a fucosyl transferase, such that antibodies expressed in such a cell line exhibit hypofucosylation by reducing or eliminating the alpha 1,6 bond-related enzyme, and also describes cell lines which have a low enzyme activity for adding fucose to the N- acetylglucosamine that binds to the Fc region of the antibody or does not have the enzyme activity, for example the rat myeloma cell line YB2/0 (ATCC CRL 1662). International Patent Publication WO 03/035835 describes a variant CHO cell line, Lec 13 cells, with reduced ability to attach fucose to Asn(297)-linked carbohydrates, also resulting in hypofucosylation of antibodies expressed in that host cell (see also Shields, et al., J. Biol. Chem.2002, 277, 26733-26740. International Patent Publication WO 99/54342 describes cell lines engineered to express glycoprotein-modifying glycosyl transferases (e.g., beta(1,4)-N- acetylglucosaminyltransferase III (GnTIII)) such that antibodies expressed in the engineered cell lines exhibit increased bisecting GlcNac structures which results in increased ADCC DB1/ 149202201.1 33 Attorney Docket No.: 116983-5127-WO activity of the antibodies (see also Umana, et al., Nat. Biotech.1999, 17, 176-180). Alternatively, the fucose residues of the antibody may be cleaved off using a fucosidase enzyme. For example, the fucosidase alpha-L-fucosidase removes fucosyl residues from antibodies as described in Tarentino, et al., Biochem.1975, 14, 5516-5523. [0058] “Pegylation” refers to a modified antibody, or a fragment thereof, that typically is reacted with polyethylene glycol (PEG), such as a reactive ester or aldehyde derivative of PEG, under conditions in which one or more PEG groups become attached to the antibody or antibody fragment. Pegylation may, for example, increase the biological (e.g., serum) half life of the antibody. Preferably, the pegylation is carried out via an acylation reaction or an alkylation reaction with a reactive PEG molecule (or an analogous reactive water-soluble polymer). As used herein, the term “polyethylene glycol” is intended to encompass any of the forms of PEG that have been used to derivatize other proteins, such as mono (C1-C10)alkoxy- or aryloxy-polyethylene glycol or polyethylene glycol-maleimide. The antibody to be pegylated may be an aglycosylated antibody. Methods for pegylation are known in the art and can be applied to the antibodies of the invention, as described for example in European Patent Nos. EP 0154316 and EP 0401384 and U.S. Patent No.5,824,778, the disclosures of each of which are incorporated by reference herein. [0059] The term “biosimilar” means a biological product, including a monoclonal antibody or protein, that is highly similar to a U.S. licensed reference biological product notwithstanding minor differences in clinically inactive components, and for which there are no clinically meaningful differences between the biological product and the reference product in terms of the safety, purity, and potency of the product. Furthermore, a similar biological or “biosimilar” medicine is a biological medicine that is similar to another biological medicine that has already been authorized for use by the European Medicines Agency. The term “biosimilar” is also used synonymously by other national and regional regulatory agencies. Biological products or biological medicines are medicines that are made by or derived from a biological source, such as a bacterium or yeast. They can consist of relatively small molecules such as human insulin or erythropoietin, or complex molecules such as monoclonal antibodies. For example, if the reference IL-2 protein is aldesleukin (PROLEUKIN), a protein approved by drug regulatory authorities with reference to aldesleukin is a “biosimilar to” aldesleukin or is a “biosimilar thereof” of aldesleukin. In Europe, a similar biological or “biosimilar” medicine is a biological medicine that is similar DB1/ 149202201.1 34 Attorney Docket No.: 116983-5127-WO to another biological medicine that has already been authorized for use by the European Medicines Agency (EMA). The relevant legal basis for similar biological applications in Europe is Article 6 of Regulation (EC) No 726/2004 and Article 10(4) of Directive 2001/83/EC, as amended and therefore in Europe, the biosimilar may be authorized, approved for authorization or subject of an application for authorization under Article 6 of Regulation (EC) No 726/2004 and Article 10(4) of Directive 2001/83/EC. The already authorized original biological medicinal product may be referred to as a “reference medicinal product” in Europe. Some of the requirements for a product to be considered a biosimilar are outlined in the CHMP Guideline on Similar Biological Medicinal Products. In addition, product specific guidelines, including guidelines relating to monoclonal antibody biosimilars, are provided on a product-by-product basis by the EMA and published on its website. A biosimilar as described herein may be similar to the reference medicinal product by way of quality characteristics, biological activity, mechanism of action, safety profiles and/or efficacy. In addition, the biosimilar may be used or be intended for use to treat the same conditions as the reference medicinal product. Thus, a biosimilar as described herein may be deemed to have similar or highly similar quality characteristics to a reference medicinal product. Alternatively, or in addition, a biosimilar as described herein may be deemed to have similar or highly similar biological activity to a reference medicinal product. Alternatively, or in addition, a biosimilar as described herein may be deemed to have a similar or highly similar safety profile to a reference medicinal product. Alternatively, or in addition, a biosimilar as described herein may be deemed to have similar or highly similar efficacy to a reference medicinal product. As described herein, a biosimilar in Europe is compared to a reference medicinal product which has been authorized by the EMA. However, in some instances, the biosimilar may be compared to a biological medicinal product which has been authorized outside the European Economic Area (a non-EEA authorized “comparator”) in certain studies. Such studies include for example certain clinical and in vivo non-clinical studies. As used herein, the term “biosimilar” also relates to a biological medicinal product which has been or may be compared to a non-EEA authorized comparator. Certain biosimilars are proteins such as antibodies, antibody fragments (for example, antigen binding portions) and fusion proteins. A protein biosimilar may have an amino acid sequence that has minor modifications in the amino acid structure (including for example deletions, additions, and/or substitutions of amino acids) which do not significantly affect the function of the polypeptide. The biosimilar may comprise an amino acid sequence having a sequence DB1/ 149202201.1 35 Attorney Docket No.: 116983-5127-WO identity of 97% or greater to the amino acid sequence of its reference medicinal product, e.g., 97%, 98%, 99% or 100%. The biosimilar may comprise one or more post-translational modifications, for example, although not limited to, glycosylation, oxidation, deamidation, and/or truncation which is/are different to the post-translational modifications of the reference medicinal product, provided that the differences do not result in a change in safety and/or efficacy of the medicinal product. The biosimilar may have an identical or different glycosylation pattern to the reference medicinal product. Particularly, although not exclusively, the biosimilar may have a different glycosylation pattern if the differences address or are intended to address safety concerns associated with the reference medicinal product. Additionally, the biosimilar may deviate from the reference medicinal product in for example its strength, pharmaceutical form, formulation, excipients and/or presentation, providing safety and efficacy of the medicinal product is not compromised. The biosimilar may comprise differences in for example pharmacokinetic (PK) and/or pharmacodynamic (PD) profiles as compared to the reference medicinal product but is still deemed sufficiently similar to the reference medicinal product as to be authorized or considered suitable for authorization. In certain circumstances, the biosimilar exhibits different binding characteristics as compared to the reference medicinal product, wherein the different binding characteristics are considered by a Regulatory Authority such as the EMA not to be a barrier for authorization as a similar biological product. The term “biosimilar” is also used synonymously by other national and regional regulatory agencies. II. Trop-2 Targeting ADC [0060] An antibody drug conjugate (ADC) has three core components: a monoclonal antibody (MAb) targeting a specific tumor antigen, a cytotoxic payload, and a chemical linker connecting them. The basic mechanism involves binding of the antibody to the tumor antigen on the cell surface, followed by internalization of the ADC and lysosomal degradation. This results in release of the active cytotoxic agent into the cytoplasm which results in tumor cell death. Some ADCs also induce antibody dependent cellular cytotoxicity (ADCC) and complement dependent cytotoxicity (CDC) contributing to the antitumor efficacy of these agents (Junttila et al., Breast Cancer Res Treat.2011 Jul;128(2):347–356). DB1/ 149202201.1 36 Attorney Docket No.: 116983-5127-WO [0061] As used herein, a “Trop-2 targeting ADC” or “anti-Trop-2 ADC” refers to a conjugated molecule comprising an anti-Trop-2 antibody linked to a drug moiety that combines the specificity of the anti-Trop-2 antibody with potency of the drug moiety. [0062] In one aspect, the invention provides a Trop-2 targeting ADC of Formula (I): Ab-(L-(D)
m)
n Wherein Ab represents an anti-Trop-2 antibody described herein; L is a linker; D is a drug moiety; m is an integer from 1 to 8; and n is an integer from 1-20. [0063] In one embodiment, n is an integer from 1 to 10, 2 to 8, or 2 to 5. In a specific embodiment, n is 2, 3, or 4. In some embodiments, m is 1; in other embodiments, m is 2, 3 or 4. [0064] While the drug to antibody ratio (DAR) has an exact value for a specific conjugate molecule (e.g., n multiplied by m in Formula (I)), it is understood that the value will often be an average value when used to describe a sample containing many molecules, due to some degree of heterogeneity, typically associated with the conjugation step. In some embodiments, the DAR is between about 2 and about 10, and typically is about 3, 3.5, 4, 4.5, 5, 5.5, 6, 6.5, 7.0, 7.5, 8.0. In some embodiments, at least 50% of a sample by weight is compound having the average DAR plus or minus 2, and preferably at least 50% of the sample is a conjugate that contains the average DAR plus or minus 1. Embodiments include Trop-2 targeting ADCs wherein the DAR is about 3.5, 3.6, 3.7, 3.8 or 3.9. In some embodiments, a DAR of ‘about n’ means the measured value for DAR is within 20% of n. A. Anti-Trop-2 Antibody [0065] The Trop-2 targeting ADCs include at least one antibody or fragment thereof that binds to Trop-2. Anti-Trop-2 antibodies are known and/or publicly available and in alternative embodiments may be utilized in the Trop-2 targeting ADCs. While humanized or human antibodies are preferred for reduced immunogenicity, in alternative embodiments a chimeric antibody may be of use. As discussed below, methods of antibody humanization are DB1/ 149202201.1 37 Attorney Docket No.: 116983-5127-WO well known in the art and may be utilized to convert an available murine or chimeric antibody into a humanized form. [0066] Anti-Trop-2 antibodies are commercially available from a number of sources and include LS-C126418, LS-C178765, LS-C126416, LS-C126417 (LifeSpan Biosciences, Inc., Seattle, WA); 10428-MM01, 10428-MM02, 10428-R001, 10428-R030 (Sino Biological Inc., Beijing, China); MR54 (eBioscience, San Diego, CA); sc-376181, sc-376746, Santa Cruz Biotechnology (Santa Cruz, CA); MM0588-49D6, (Novus Biologicals, Littleton, CO); ab79976, and ab89928 (ABCAM®, Cambridge, MA). [0067] Other anti-Trop-2 antibodies have been disclosed in the patent literature. For example, U.S. Publ. No.2013/0089872 discloses anti-Trop-2 antibodies K5-70 (Accession No. FERM BP-11251), K5-107 (Accession No. FERM BP-11252), K5-116-2-1 (Accession No. FERM BP-11253), T6-16 (Accession No. FERM BP-11346), and T5-86 (Accession No. FERM BP- 11254), deposited with the International Patent Organism Depositary, Tsukuba, Japan. U.S. Patent No.5,840,854 disclosed the anti-Trop-2 monoclonal antibody BR110 (ATCC No. HB11698). U.S. Patent No.7,420,040 disclosed an anti-Trop-2 antibody produced by hybridoma cell line AR47A6.4.2, deposited with the IDAC (International Depository Authority of Canada, Winnipeg, Canada) as accession number 141205-05. U.S. Patent No. 7,420,041 disclosed an anti-Trop-2 antibody produced by hybridoma cell line AR52A301.5, deposited with the IDAC as accession number 141205-03. U.S. Publ. No.2013/0122020 disclosed anti-Trop-2 antibodies 3E9, 6G11, 7E6, 15E2, 18B1. Hybridomas encoding a representative antibody were deposited with the American Type Culture Collection (ATCC), Accession Nos. PTA-12871 and PTA-12872. U.S. Patent No.8,715,662 discloses anti-Trop-2 antibodies produced by hybridomas deposited at the AID-ICLC (Genoa, Italy) with deposit numbers PD 08019, PD 08020 and PD 08021. U.S. Patent Application Publ. No. 20120237518 discloses anti-Trop-2 antibodies 77220, KM4097 and KM4590. U.S. Patent No.8,309,094 (Wyeth) discloses antibodies A1 and A3, identified by sequence listing. The Examples section of each patent or patent application cited above in this paragraph is incorporated herein by reference. Non-patent publication Lipinski et al. (1981, Proc Natl. Acad Sci USA, 78:5147-50) disclosed anti-Trop-2 antibodies 162-25.3 and 162-46.2. [0068] Numerous anti-Trop-2 antibodies are known in the art and/or publicly available. As discussed below, methods for preparing antibodies against known antigens were routine in the art. The sequence of the human Trop-2 protein was also known in the art (see, e.g., DB1/ 149202201.1 38 Attorney Docket No.: 116983-5127-WO GenBank Accession No. CAA54801.1). Methods for producing humanized, human or chimeric antibodies were also known. The person of ordinary skill, reading the instant disclosure in light of general knowledge in the art, would have been able to make and use the genus of anti-Trop-2 antibodies in the subject ADCs. In an embodiment, the anti-Trop-2 antibody may be a humanized RS7 antibody (see, e.g., U.S. Patent No.7,238,785, incorporated herein by reference in its entirety). B. Drug Moiety [0069] The drug to be conjugated to the anti-Trop-2 antibody or antibody fragment may be selected from the group consisting of an anthracycline, a camptothecin, a tubulin inhibitor, a maytansinoid, a calicheamycin, an auristatin, a nitrogen mustard, an ethylenimine derivative, an alkyl sulfonate, a nitrosourea, a triazene, a folic acid analog, a taxane, a COX-2 inhibitor, a pyrimidine analog, a purine analog, an antibiotic, an enzyme inhibitor, an epipodophyllotoxin, a platinum coordination complex, a vinca alkaloid, a substituted urea, a methyl hydrazine derivative, an adrenocortical suppressant, a hormone antagonist, an antimetabolite, an alkylating agent, an antimitotic, an anti- angiogenic agent, a tyrosine kinase inhibitor, an mTOR inhibitor, a heat shock protein (HSP90) inhibitor, a proteosome inhibitor, an HDAC inhibitor, a pro-apoptotic agent, and a combination thereof. [0070] Specific drugs of use may be selected from the group consisting of 5-fluorouracil, afatinib, aplidin, azaribine, anastrozole, anthracyclines, axitinib, AVL-101, AVL-291, bendamustine, bleomycin, bortezomib, bosutinib, bryostatin-1 , busulfan, calicheamycin, camptothecin, carboplatin, 10-hydroxycamptothecin, carmustine, celecoxib, chlorambucil, cisplatinum, COX-2 inhibitors, irinotecan (CPT-11), SN-38, carboplatin, cladribine, camptothecans, crizotinib, cyclophosphamide, cytarabine, dacarbazine, dasatinib, dinaciclib, docetaxel, dactinomycin, daunorubicin, DM1, DM3, DM4, doxorubicin, 2- pyrrolinodoxorubicine (2-PDox), a pro-drug form of 2-PDox (pro-2-PDox), cyano- morpholino doxorubicin, doxorubicin glucuronide, endostatin, epirubicin glucuronide, erlotinib, estramustine, epidophyllotoxin, erlotinib, entinostat, estrogen receptor binding agents, etoposide (VP16), etoposide glucuronide, etoposide phosphate, exemestane, fingolimod, floxuridine (FUdR), 3',5'-0-dioleoyl-FudR (FUdR-dO), fludarabine, flutamide, farnesyl-protein transferase inhibitors, flavopiridol, fostamatinib, ganetespib, GDC-0834, GS- 1101, gefitinib, gemcitabine, hydroxyurea, ibrutinib, idarubicin, idelalisib, ifosfamide, imatinib, lapatinib, lenolidamide, leucovorin, LFM-A13, lomustine, mechlorethamine, DB1/ 149202201.1 39 Attorney Docket No.: 116983-5127-WO melphalan, mercaptopurine, 6-mercaptopurine, methotrexate, mitoxantrone, mithramycin, mitomycin, mitotane, monomethylauristatin F (MMAF), monomethylauristatin D (MMAD), monomethylauristatin E (MMAE), navelbine, neratinib, nilotinib, nitrosurea, olaparib, plicomycin, procarbazine, paclitaxel, PCI-32765, pentostatin, PSI-341, raloxifene, semustine, SN-38, sorafenib, streptozocin, SUl 1248, sunitinib, tamoxifen, temazolomide, transplatinum, thalidomide, thioguanine, thiotepa, teniposide, topotecan, uracil mustard, vatalanib, vinorelbine, vinblastine, vincristine, vinca alkaloids and ZD1839. In particularly preferred embodiments, the drug to be conjugated to the anti-Trop-2 antibody may be SN-38, pro-2- PDox or paclitaxel. [0071] The drug moiety D can be linked to the antibody Ab through linker L. L is any chemical moiety capable of linking the drug moiety to the antibody through covalent bonds. A cross-linking reagent is a bifunctional or multifunctional reagent that can be used to link a drug moiety and an antibody to form antibody drug conjugates. Antibody drug conjugates can be prepared using a cross-linking reagent having a reactive functionality capable of binding to both the drug moiety and the antibody. For example, a cysteine, thiol or an amine, e.g. N- terminus or an amino acid side chain, such as lysine of the antibody, can form a bond with a functional group of a cross-linking reagent. Alternatively, the Antibody drug conjugates can be prepared by pre-forming a linker-drug moiety (or drug-linker moiety, both terms being used interchangeably), and reacting the linker-drug moiety with the antibody. In some instant, the linker moiety is built onto the drug stepwise using several linking moieties until obtaining the desired linker-drug moiety. [0072] In one embodiment, L is a cleavable linker. In another embodiment, L is a non- cleavable linker. In some embodiments, L is an acid-labile linker, photo-labile linker, peptidase cleavable linker, esterase cleavable linker, a disulfide bond cleavable linker, a hydrophilic linker, a procharged linker, a glycosidase cleavable linker, a phosphodiesterase cleavable linker, a phosphatase cleavable linker, or a dicarboxylic acid based linker. [0073] Suitable cross-linking reagents that form a non-cleavable linker between the drug moiety, for example maytansinoid, and the antibody are well known in the art, and can form non-cleavable linkers that comprise a sulfur atom (such as SMCC) or those that are without a sulfur atom. Preferred cross-linking reagents that form non-cleavable linkers between the drug moiety, for example maytansinoid, and the antibody comprise a maleimido- or DB1/ 149202201.1 40 Attorney Docket No.: 116983-5127-WO haloacetyl-based moiety. According to the present invention, such non-cleavable linkers are said to be derived from maleimido- or haloacetyl-based moieties. [0074] Cross-linking reagents comprising a maleimido-based moiety include but not limited to, N-succinimidyl-4-(maleimidomethyl)cyclohexanecarboxylate (SMCC), sulfosuccinimidyl 4-(N-maleimidomethyl) cyclohexane-1-carboxylate (sulfo-SMCC), N-succinimidyl-4- (maleimidomethyl)cyclohexane-1-carboxy-(6-amidocaproate), which is a “long chain” analog of SMCC (LC-SMCC), κ-maleimidoundeconoic acid N-succinimidyl ester (KMUA), γ-maleimidobutyric acid N-succinimidyl ester (GMBS), ε-maleimidocaproic acid N- succinimidyl ester (EMCS), m-maleimidobenzoyl-N-hydroxysuccinimide ester (MBS), N-(α- maleimidoacetoxy)-succinimide ester (AMSA), succinimidyl-6-(β- maleimidopropionamido)hexanoate (SMPH), N-succinimidyl-4-(p-maleimidophenyl)- butyrate (SMPB), N-(-p-maleomidophenyl)isocyanate (PMIP) and maleimido-based cross- linking reagents containing a polyethythene glycol spacer, such as MAL-PEG-NHS. These cross-linking reagents form non-cleavable linkers derived from maleimido-based moieties. Representative structures of maleimido-based cross-linking reagents are shown below. DB1/ 149202201.1 41 Attorney Docket No.: 116983-5127-WO

[0075] In another embodiment, the linker L is derived from N-succinimidyl-4- (maleimidomethyl)cyclohexanecarboxylate (SMCC), sulfosuccinimidyl 4-(N- maleimidomethyl) cyclohexane-1-carboxylate (sulfo-SMCC) or MAL-PEG-NHS. [0076] Cross-linking reagents comprising a haloacetyle-based moiety include N-succinimidyl iodoacetate (SIA), N-succinimidyl(4-iodoacetyl)aminobenzoate (SIAB), N-succinimidyl bromoacetate (SBA) and N-succinimidyl 3-(bromoacetamido)propionate (SBAP). These cross-linking reagents form a non-cleavable linker derived from haloacetyl-based moieties. Representative structures of haloacetyl-based cross-linking reagents are shown below. DB1/ 149202201.1 42 Attorney Docket No.: 116983-5127-WO
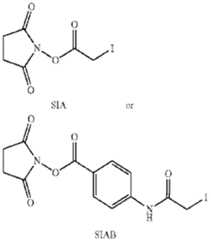
[0077] In one embodiment, the linker L is derived from N-succinimidyl iodoacetate (SIA) or N-succinimidyl(4-iodoacetyl)aminobenzoate (SIAB). [0078] Suitable cross-linking reagents that form a cleavable linker between the drug moiety, for example maytansinoid, and the antibody are well known in the art. Disulfide containing linkers are linkers cleavable through disulfide exchange, which can occur under physiological conditions. According to the present invention, such cleavable linkers are said to be derived from disulfide-based moieties. Suitable disulfide cross-linking reagents include N- succinimidyl-3-(2-pyridyldithio)propionate (SPDP), N-succinimidyl-4-(2- pyridyldithio)pentanoate (SPP), N-succinimidyl-4-(2-pyridyldithio)butanoate (SPDB) and N- succinimidyl-4-(2-pyridyldithio)-2-sulfo-butanoate (sulfo-SPDB), the structures of which are shown below. These disulfide cross-linking reagents form a cleavable linker derived from disulfide-based moieties. DB1/ 149202201.1 43 Attorney Docket No.: 116983-5127-WO
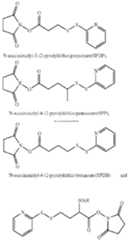
[0079] In one embodiment, the linker L is derived from N-succinimidyl-4-(2-pyridyldithio) butanoate (SPDB). [0080] [0081] Suitable cross-linking reagents that form a charged linker between the drug moiety, for example maytansinoid, and the antibody are known as procharged cross-linking reagents. DB1/ 149202201.1 44 Attorney Docket No.: 116983-5127-WO In one embodiment, the linker L is derived from the procharged cross-linking reagent CX1-1. The structure of CX1-1 is below.

[0082] Each of the cross-linking reagents depicted above contains, at one end of the cross- linking reagent, a NHS-ester which reacts with a primary amine of the antibody to form an amide bond and, at the other end, a maleimide group or pyridinyldisulfide group which reacts with the sulfhydryl of the maytansionoid drug moiety to form a thioether or disulfide bond. [0083] In another embodiment, suitable cross-linking moieties that form a cleavable linker between the drug moiety (for example maytansinoid) and the antibody are represented by the following formula (II):
wherein:L
1 is a C
1-6 alkylene wherein one of the methylene groups may be replaced with oxygen; L2 is a C1-6 alkylene or is —(CH2 CH2 O)y —CH2 —CH2 — wherein y is 1 to 11; and X is —C(O)—NH—, —NHC(O)— or a triazole; wherein the alkylene is linear or branched. [0084] In one aspect of this embodiment y is 5, 7, 9 or 11. In another aspect of this embodiment y is less than 5. [0085] In yet another embodiment, suitable cross-linking moieties according to formula I are selected from the group consisting of: DB1/ 149202201.1 45 Attorney Docket No.: 116983-5127-WO
DB1/ 149202201.1 46 Attorney Docket No.: 116983-5127-WO
wherein y is 1 to 11. [0086] For the cross-linking moieties depicted above (i.e. MBT, MPET, MEPET; MMTBT, MPPT, MPBT), the maleimide group allows for reaction with the sulfhydryl (or thiol) of a Cysteine in an antibody thereby forming a thioether bond; and the thiol functionality of the cross-linking moiety is connected to the thiol of the maytansinoid drug moiety to form a cleavable disulfide bond. In view of the cross-reactional nature of the linking moiety of Formula (I) (thiol and maleimide could cross react), one of ordinary skill in the art would appreciate that the linking moiety has to be built stepwise onto the drug moiety as depicted in Scheme 1. [0087] According to the above embodiment, the linkers resulting from the cross linking moieties (i.e. MBT, MPET, MEPET) can be depicted as follow:

wherein * is linked to the thiol functionality on the antibody, and ** is linked to the thiol functionality of a drug moiety (e.g. maytansinoid drug moiety DM1, DM3 or DM4). [0088] According to the above embodiment, the linkers resulting from the cross linking moieties (i.e. MBT, MPET, MEPET) can be depicted as follows: DB1/ 149202201.1 47 Attorney Docket No.: 116983-5127-WO
DB1/ 149202201.1 48 Attorney Docket No.: 116983-5127-WO
DB1/ 149202201.1 49 Attorney Docket No.: 116983-5127-WO wherein y is 1 to 11; * is linked to the thiol functionality on the antibody, and ** is linked to the thiol functionality of the drug moiety (e.g. maytansinoid drug DM1, DM3 or DM4). [0089] In a preferred embodiment, the linker has the following formula:
wherein * is linked to the thiol functionality on the antibody, and ** is linked to the thiol functionality of the maytansinoid drug (DM1, DM3 or DM4). [0090] In one embodiment, the invention relates to the linker-drug moiety of Formula:
Wherein L
1 is a C
1-6 alkylene wherein one of the methylene groups may be replaced with oxygen; L2 is a C1-6 alkylene or is —(CH2 CH2O)y —CH2 —CH2— wherein y is 1 to 11; and X is —C(O)—NH—, —NHC(O)— or a triazole; wherein the alkylene is linear or branched. C. Linker [0091] As used herein, a “linker” is any chemical moiety that is capable of linking an antibody, antibody fragment (e.g., antigen binding fragments) or functional equivalent to another moiety, such as a drug moiety. Linkers can be susceptible to cleavage (cleavable linker), such as, acid-induced cleavage, photo-induced cleavage, peptidase-induced cleavage, esterase-induced cleavage, glycosidase induced cleavage, phosphodiesterase induced cleavage, phosphatase induced cleavage and disulfide bond cleavage, at conditions under which the compound or the antibody remains active. Alternatively, linkers can be DB1/ 149202201.1 50 Attorney Docket No.: 116983-5127-WO substantially resistant to cleavage (e.g., stable linker or noncleavable linker). In some aspects, the linker is a procharged linker, a hydrophilic linker, or a dicarboxylic acid based linker. [0092] In one aspect, the linker used in the present invention is derived from a crosslinking reagent such as N-succinimidyl-3-(2-pyridyldithio)propionate (SPDP), N-succinimidyl 4-(2- pyridyldithio)pentanoate (SPP), N-succinimidyl 4-(2-pyridyldithio)butanoate (SPDB), N- succinimidyl-4-(2-pyridyldithio)-2-sulfo-butanoate (sulfo-SPDB), N-succinimidyl iodoacetate (SIA), N-succinimidyl(4-iodoacetyl)aminobenzoate (SIAB), maleimide PEG NHS, N-succinimidyl 4-(maleimidomethyl) cyclohexanecarboxylate (SMCC), N- sulfosuccinimidyl 4-(maleimidomethyl) cyclohexanecarboxylate (sulfo-SMCC) or 2,5- dioxopyrrolidin-1-yl 17-(2,5-dioxo-2,5-dihydro-1H-pyrrol-1-yl)-5,8,11,14-tetraoxo- 4,7,10,13-tetraazaheptadecan-1-oate (CX1-1). [0093] Non-cleavable linkers are any chemical moiety capable of linking a drug, such as a maytansinoid, to an antibody in a stable, covalent manner and does not fall under the categories listed above for cleavable linkers. Thus, non-cleavable linkers are substantially resistant to acid-induced cleavage, photo-induced cleavage, peptidase-induced cleavage, esterase-induced cleavage and disulfide bond cleavage. Furthermore, non-cleavable refers to the ability of the chemical bond in the linker or adjoining to the linker to withstand cleavage induced by an acid, photolabile-cleaving agent, a peptidase, an esterase, or a chemical or physiological compound that cleaves a disulfide bond, at conditions under which the drug, such as maytansionoid or the antibody does not lose its activity. [0094] Acid-labile linkers are linkers cleavable at acidic pH. For example, certain intracellular compartments, such as endosomes and lysosomes, have an acidic pH (pH 4-5), and provide conditions suitable to cleave acid-labile linkers. [0095] Photo-labile linkers are linkers that are useful at the body surface and in many body cavities that are accessible to light. Furthermore, infrared light can penetrate tissue. [0096] Some linkers can be cleaved by peptidases, i.e., peptidase cleavable linkers. Only certain peptides are readily cleaved inside or outside cells, see e.g., Trout et al., Proc. Natl. Acad. Sci. USA, 626-629 (1982) and Umemoto et al. Int. J. Cancer, 677-684 (1989). Furthermore, peptides are composed of α-amino acids and peptidic bonds, which chemically are amide bonds between the carboxylate of one amino acid and the amino group of a second DB1/ 149202201.1 51 Attorney Docket No.: 116983-5127-WO amino acid. Other amide bonds, such as the bond between a carboxylate and the ε-amino group of lysine, are understood not to be peptidic bonds and are considered non-cleavable. [0097] Some linkers can be cleaved by esterases, i.e., esterase cleavable linkers. Again, only certain esters can be cleaved by esterases present inside or outside of cells. Esters are formed by the condensation of a carboxylic acid and an alcohol. Simple esters are esters produced with simple alcohols, such as aliphatic alcohols, and small cyclic and small aromatic alcohols. [0098] Procharged linkers are derived from charged cross-linking reagents that retain their charge after incorporation into an antibody drug conjugate. Examples of procharged linkers can be found in US 2009/0274713. D. Conjugation and Preparation of ADCs [0099] Numerous methods of conjugating linker-payloads to antigen binding moiety are known in the art (reviewed in for example: Antibody-Drug Conjugate, Methods in Molecular Biology, Vol.1045, Editor L. Ducry, Humana Press (2013)). Traditionally, drugs are conjugated to native lysine or native cysteine residues of the antibody. The resulting preparations are complex mixtures. More recently, site-specific conjugation methods are being employed to improve the therapeutic index and homogeneity of ADC preparations (For review: Panowski, et al., mAbs 2014, 6, 34). Besides glycoengineering, (Zhou, Q. et al. Bioconjugate chemistry 2014, 25, 510; Zhu, Z. et al. mAbs 2014, 6, 1190); some of the more common methods of preparing site-specific ADCs are based on the incorporation of engineered cysteines, (Junutula, J. R. et al., Nature biotechnology 2008, 26, 925; Shinmi, D. et al., Bioconjugate chemistry 2016, 27, 1324), non-canonical amino acids (Tian, F. et al., Proceedings National Academy of Sciences USA 2014, 111, 1766; Axup, J. Y. et al., Proceedings National Academy of Sciences USA 2012, 109, 16101) or short peptide sequences into the antibody backbone (Drake, P. M. et al., Bioconjugate chemistry 2014, 25, 1331; Strop, P. et al., Chemistry & biology 2013, 20, 161; Beerli, R. R. et al., PloS one 2015, 10, e0131177; Grunewald, J. et al., Bioconjugate chemistry 2015, 26, 2554). These methods provide control over stoichiometry and attachment site of the cytotoxin resulting in better pharmacokinetic (PK), safety, and efficacy profiles of the conjugates relative to traditionally prepared ADCs. DB1/ 149202201.1 52 Attorney Docket No.: 116983-5127-WO [00100] The conjugates of the present invention can be prepared by any methods known in the art, such as those described in U.S. Pat. Nos.7,811,572, 6,411,163, 7,368,565, and 8,163,888, US application publications 2011/0003969, 2011/0166319, 2012/0253021 and 2012/0259100, and PCT publications WO2014/124316 and WO2015/138615. The entire teachings of these patents and patent application publications are herein incorporated by reference. E. Process For Conjugation To Native Cysteine Antibody Residues [00101] Linker-drug moieties as described herein can be conjugated to native cysteine residues of non-engineered antibodies using a procedure that involves partial reduction of the antibodies (Doronina, S. O., et al., (2003) Nat. Biotechnol. 21, 778-784). The following protocol is a non-limiting example how such conjugates can be prepared: Inter- and intra- chain disulfides bonds of the antibody (at a concentration of typically 5 to 10 mg/ml) are first partially reduced in PBS containing 2 mM EDTA by adding TCEP to a final concentration of 10 mM and incubating the mixture at 37° C. for 1 hour. After desalting and addition of 1% w/v PS-20 detergent, the partially reduced antibodies (1-2 mg/ml) is reacted overnight at 4° C. with 0.5 to 1 mg of a maleimide containing linker payload compound per 10 mg antibody. Resulting conjugates are purified by Protein A chromatography by standard methods and buffer exchanged to PBS, and are profiled typically by mass-spectrometry (MS), analytical size-exclusion chromatography (AnSEC), and analytical hydrophobic interaction chromatography (AnHIC) for their drug-to-antibody-ratio, aggregation propensity, and hydrophobicity as well as by activity assays. F. One-Step Process for Cross-Linking to Lysine Antibody Residues [00102] In one embodiment, the conjugates of the present invention can be prepared by a one-step process for cross-linking the drug to lysine residues on the antibody. The process comprises combining the antibody, drug and cross-linking agent in a substantially aqueous medium, optionally containing one or more co-solvents, at a suitable pH. In one embodiment, the process comprises the step of contacting the antibody of the present invention with a drug (e.g., DM1 or DM4) to form a first mixture comprising the antibody and the drug, and then contacting the first mixture comprising the antibody and the drug with a cross-linking agent (e.g., SMCC, Sulfo-SMCC, SPDB, Sulfo-SPDB or CX1-1) in a solution having a pH of about 4 to about 9 to provide a mixture comprising (i) the conjugate (e.g., Ab-MCC-DM1, Ab- DB1/ 149202201.1 53 Attorney Docket No.: 116983-5127-WO SPDB-DM4, or Ab-CX1-1-DM1), (ii) free drug (e.g., DM1 or DM4), and (iii) reaction by- products. [00103] In one embodiment, the one-step process comprises contacting the antibody with the drug (e.g., DM1 or DM4) and then the cross-linking agent (e.g., SMCC, Sulfo- SMCC, SPDB, Sulfo-SPDB or CX1-1) in a solution having a pH of about 6 or greater (e.g., about 6 to about 9, about 6 to about 7, about 7 to about 9, about 7 to about 8.5, about 7.5 to about 8.5, about 7.5 to about 8.0, about 8.0 to about 9.0, or about 8.5 to about 9.0). For example, the inventive process comprises contacting a cell-binding agent with the drug (DM1 or DM4) and then the cross-linking agent (e.g., SMCC, Sulfo-SMCC, SPDB, Sulfo-SPDB or CX1-1) in a solution having a pH of about 6.0, about 6.1, about 6.2, about 6.3, about 6.4, about 6.5, about 6.6, about 6.7, about 6.8, about 6.9, about 7.0, about 7.1, about 7.2, about 7.3, about 7.4, about 7.5, about 7.6, about 7.7, about 7.8, about 7.9, about 8.0, about 8.1, about 8.2, about 8.3, about 8.4, about 8.5, about 8.6, about 8.7, about 8.8, about 8.9, or about 9.0. In a specific embodiment, the inventive process comprises contacting a cell-binding agent with the drug (e.g., DM1 or DM4) and then the cross-linking agent (e.g., SMCC, Sulfo- SMCC, SPDB, Sulfo-SPDB or CX1-1) in a solution having a pH of about 7.8 (e.g., a pH of 7.6 to 8.0 or a pH of 7.7 to 7.9). [00104] The one-step process (i.e., contacting the antibody with the drug (e.g., DM1 or DM4) and then the cross-linking agent (e.g., SMCC, Sulfo-SMCC, SPDB, Sulfo-SPDB or CX1-1) can be carried out at any suitable temperature known in the art. For example, the one- step process can occur at about 20° C. or less (e.g., about −10° C. (provided that the solution is prevented from freezing, e.g., by the presence of organic solvent used to dissolve the cytotoxic agent and the bifunctional crosslinking reagent) to about 20° C., about 0° C. to about 18° C., about 4° C. to about 16° C.), at room temperature (e.g., about 20° C. to about 30° C. or about 20° C. to about 25° C.), or at an elevated temperature (e.g., about 30° C. to about 37° C.). In one embodiment, the one-step process occurs at a temperature of about 16° C. to about 24° C. (e.g., about 16° C., about 17° C., about 18° C., about 19° C., about 20° C., about 21° C., about 22° C., about 23° C., about 24° C., or about 25° C.). In another embodiment, the one-step process is carried out at a temperature of about 15° C. or less (e.g., about −10° C. to about 15° C., or about 0° C. to about 15° C.). For example, the process comprises contacting the antibody with the drug (e.g., DM1 or DM4) and then the cross- linking agent (e.g., SMCC, Sulfo-SMCC, SPDB, Sulfo-SPDB or CX1-1) at a temperature of DB1/ 149202201.1 54 Attorney Docket No.: 116983-5127-WO about 15° C., about 14° C., about 13° C., about 12° C., about 11° C., about 10° C., about 9° C., about 8° C., about 7° C., about 6° C., about 5° C., about 4° C., about 3° C., about 2° C., about 1° C., about 0° C., about −1° C., about −2° C., about −3° C., about −4° C., about −5° C., about −6° C., about −7° C., about −8° C., about −9° C., or about −10° C., provided that the solution is prevented from freezing, e.g., by the presence of organic solvent(s) used to dissolve the cross-linking agent (e.g., SMCC, Sulfo-SMCC, Sulfo-SPDB SPDB, or CX1-1). In one embodiment, the process comprises contacting the antibody with the drug (e.g., DM1 or DM4) and then the cross-linking agent (e.g., SMCC, Sulfo-SMCC, SPDB, Sulfo-SPDB or CX1-1) at a temperature of about −10° C. to about 15° C., about 0° C. to about 15° C., about 0° C. to about 10° C., about 0° C. to about 5° C., about 5° C. to about 15° C., about 10° C. to about 15° C., or about 5° C. to about 10° C. In another embodiment, the process comprises contacting the antibody with the drug (e.g., DM1 or DM4) and then the cross-linking agent (e.g., SMCC, Sulfo-SMCC, SPDB, Sulfo-SPDB or CX1-1) at a temperature of about 10° C. (e.g., a temperature of 8° C. to 12° C. or a temperature of 9° C. to 11° C.). [00105] In one embodiment, the contacting described above is effected by providing the antibody, then contacting the antibody with the drug (e.g., DM1 or DM4) to form a first mixture comprising the antibody and the drug (e.g., DM1 or DM4), and then contacting the first mixture comprising the antibody and the drug (e.g., DM1 or DM4) with the cross-linking agent (e.g., SMCC, Sulfo-SMCC, SPDB, Sulfo-SPDB or CX1-1). For example, in one embodiment, the antibody is provided in a reaction vessel, the drug (e.g., DM1 or DM4) is added to the reaction vessel (thereby contacting the antibody), and then the cross-linking agent (e.g., SMCC, Sulfo-SMCC, SPDB, Sulfo-SPDB or CX1-1) is added to the mixture comprising the antibody and the drug (e.g., DM1 or DM4) (thereby contacting the mixture comprising the antibody and the drug). In one embodiment, the antibody is provided in a reaction vessel, and the drug (e.g., DM1 or DM4) is added to the reaction vessel immediately following providing the antibody to the vessel. In another embodiment, the antibody is provided in a reaction vessel, and the drug (e.g., DM1 or DM4) is added to the reaction vessel after a time interval following providing the antibody to the vessel (e.g., about 5 minutes, about 10 minutes, about 20 minutes, about 30 minutes, about 40 minutes, about 50 minutes, about 1 hour, about 1 day or longer after providing the cell-binding agent to the space). The drug (e.g., DM1 or DM4) can be added quickly (i.e., within a short time interval, such as about 5 minutes, about 10 minutes) or slowly (such as by using a pump). DB1/ 149202201.1 55 Attorney Docket No.: 116983-5127-WO [00106] The mixture comprising the antibody and the drug (e.g., DM1 or DM4) can then be contacted with the cross-linking agent (e.g., SMCC, Sulfo-SMCC, SPDB, Sulfo- SPDB or CX1-1) either immediately after contacting the antibody with the drug (e.g., DM1 or DM4) or at some later point (e.g., about 5 minutes to about 8 hours or longer) after contacting the antibody with the drug (e.g., DM1 or DM4). For example, in one embodiment, the cross-linking agent (e.g., SMCC, Sulfo-SMCC, SPDB, Sulfo-SPDB or CX1-1) is added to the mixture comprising the antibody and the drug (e.g., DM1 or DM4) immediately after the addition of the drug (e.g., DM1 or DM4) to the reaction vessel comprising the antibody. Alternatively, the mixture comprising the antibody and the drug (e.g., DM1 or DM4) can be contacted with the cross-linking agent (e.g., SMCC, Sulfo-SMCC, SPDB, Sulfo-SPDB or CX1-1) at about 5 minutes, about 10 minutes, about 20 minutes, about 30 minutes, about 1 hour, about 2 hours, about 3 hours, about 4 hours, about 5 hours, about 6 hours, about 7 hours, about 8 hours, or longer after contacting the antibody with the drug (e.g., DM1 or DM4). [00107] After the mixture comprising the antibody and the drug (e.g., DM1 or DM4) is contacted with the cross-linking agent (e.g., SMCC, Sulfo-SMCC, SPDB, Sulfo-SPDB or CX1-1) the reaction is allowed to proceed for about 1 hour, about 2 hours, about 3 hours, about 4 hours, about 5 hours, about 6 hours, about 7 hours, about 8 hours, about 9 hours, about 10 hours, about 11 hours, about 12 hours, about 13 hours, about 14 hours, about 15 hours, about 16 hours, about 17 hours, about 18 hours, about 19 hours, about 20 hours, about 21 hours, about 22 hours, about 23 hours, about 24 hours, or longer (e.g., about 30 hours, about 35 hours, about 40 hours, about 45 hours, or about 48 hrs). [00108] In one embodiment, the one-step process further comprises a quenching step to quench any unreacted drug (e.g., DM1 or DM4) and/or unreacted cross-linking agent (e.g., SMCC, Sulfo-SMCC, SPDB, Sulfo-SPDB or CX1-1). The quenching step is typically performed prior to purification of the conjugate. In one embodiment, the mixture is quenched by contacting the mixture with a quenching reagent. As used herein, the “quenching reagent” refers to a reagent that reacts with the free drug (e.g., DM1 or DM4) and/or cross-linking agent (e.g., SMCC, Sulfo-SMCC, SPDB, Sulfo-SPDB or CX1-1). In one embodiment, maleimide or haloacetamide quenching reagents, such as 4-maleimidobutyric acid, 3- maleimidopropionic acid, N-ethylmaleimide, iodoacetamide, or iodoacetamidopropionic acid, can be used to ensure that any unreacted group (such as thiol) in the drug (e.g., DM1 or DB1/ 149202201.1 56 Attorney Docket No.: 116983-5127-WO DM4) is quenched. The quenching step can help prevent the dimerization of the drug (e.g., DM1). The dimerized DM1 can be difficult to remove. Upon quenching with polar, charged thiol-quenching reagents (such as 4-maleimidobutyric acid or 3-maleimidopropionic acid), the excess, unreacted DM1 is converted into a polar, charged, water-soluble adduct that can be easily separated from the covalently-linked conjugate during the purification step. Quenching with non-polar and neutral thiol-quenching reagents can also be used. In one embodiment, the mixture is quenched by contacting the mixture with a quenching reagent that reacts with the unreacted cross-linking agent (e.g., SMCC, Sulfo-SMCC, SPDB, Sulfo- SPDB or CX1-1). For example, nucleophiles can be added to the mixture in order to quench any unreacted SMCC. The nucleophile preferably is an amino group containing nucleophile, such as lysine, taurine and hydroxylamine. [00109] In a preferred embodiment, the reaction (i.e., contacting the antibody with the drug (e.g., DM1 or DM4) and then cross-linking agent (e.g., SMCC, Sulfo-SMCC, SPDB, Sulfo-SPDB or CX1-1)) is allowed to proceed to completion prior to contacting the mixture with a quenching reagent. In this regard, the quenching reagent is added to the mixture about 1 hour to about 48 hours (e.g., about 1 hour, about 2 hours, about 3 hours, about 4 hours, about 5 hours, about 6 hours, about 7 hours, about 8 hours, about 9 hours, about 10 hours, about 11 hours, about 12 hours, about 13 hours, about 14 hours, about 15 hours, about 16 hours, about 17 hours, about 18 hours, about 19 hours, about 20 hours, about 21 hours, about 22 hours, about 23 hours, about 24 hours, or about 25 hours to about 48 hours) after the mixture comprising the antibody and the drug (e.g., DM1 or DM4) is contacted with the cross-linking agent (e.g., SMCC, Sulfo-SMCC, SPDB, Sulfo-SPDB or CX1-1). [00110] Alternatively, the mixture is quenched by lowering the pH of the mixture to about 5.0 (e.g., 4.8, 4.9, 5.0, 5.1 or 5.2). In another embodiment, the mixture is quenched by lowering the pH to less than 6.0, less than 5.5, less than 5.0, less than 4.8, less than 4.6, less than 4.4, less than 4.2, less than 4.0. Alternatively, the pH is lowered to about 4.0 (e.g., 3.8, 3.9, 4.0, 4.1 or 4.2) to about 6.0 (e.g., 5.8, 5.9, 6.0, 6.1 or 6.2), about 4.0 to about 5.0, about 4.5 (e.g., 4.3, 4.4, 4.5, 4.6 or 4.7) to about 5.0. In one embodiment, the mixture is quenched by lowering the pH of the mixture to 4.8. In another embodiment, the mixture is quenched by lowering the pH of the mixture to 5.5. [00111] In one embodiment, the one-step process further comprises a holding step to release the unstably bound linkers from the antibody. The holding step comprises holding the DB1/ 149202201.1 57 Attorney Docket No.: 116983-5127-WO mixture prior to purification of the conjugate (e.g., after the reaction step, between the reaction step and the quenching step, or after the quenching step). For example, the process comprises (a) contacting the antibody with the drug (e.g., DM1, DM3 or DM4) to form a mixture comprising the antibody and the drug (e.g., DM1, DM3 or DM4); and then contacting the mixture comprising the antibody and drug (e.g., DM1, DM3 or DM4) with the cross-linking agent (e.g., SMCC, Sulfo-SMCC, SPDB, Sulfo-SPDB or CX1-1), in a solution having a pH of about 4 to about 9 to provide a mixture comprising (i) the conjugate (e.g., Ab- MCC-DM1, Ab-SPDB-DM4 or Ab-CX1-1-DM1), (ii) free drug (e.g., DM1, DM3 or DM4), and (iii) reaction by-products, (b) holding the mixture prepared in step (a) to release the unstably bound linkers from the cell-binding agent, and (c) purifying the mixture to provide a purified conjugate. [00112] In another embodiment, the process comprises (a) contacting the antibody with the drug (e.g., DM1, DM3 or DM4) to form a mixture comprising the antibody and the drug (e.g., DM1, DM3 or DM4); and then contacting the mixture comprising the antibody and the drug (e.g., DM1, DM3 or DM4) with the cross-linking agent (e.g., SMCC, Sulfo- SMCC, SPDB, Sulfo-SPDB or CX1-1), in a solution having a pH of about 4 to about 9 to provide a mixture comprising (i) the conjugate, (ii) free drug (e.g., DM1, DM3 or DM4), and (iii) reaction by-products, (b) quenching the mixture prepared in step (a) to quench any unreacted drug (e.g., DM1, DM3 or DM4) and/or unreacted cross-linking agent (e.g., SMCC, Sulfo-SMCC, SPDB, Sulfo-SPDB or CX1-1), (c) holding the mixture prepared in step (b) to release the unstably bound linkers from the cell-binding agent, and (d) purifying the mixture to provide a purified conjugate (e.g., Ab-MCC-DM1, Ab-SPDB-DM4 or Ab-CX1-1-DM1). [00113] Alternatively, the holding step can be performed after purification of the conjugate, followed by an additional purification step. [00114] In a preferred embodiment, the reaction is allowed to proceed to completion prior to the holding step. In this regard, the holding step can be performed about 1 hour to about 48 hours (e.g., about 1 hour, about 2 hours, about 3 hours, about 4 hours, about 5 hours, about 6 hours, about 7 hours, about 8 hours, about 9 hours, about 10 hours, about 11 hours, about 12 hours, about 13 hours, about 14 hours, about 15 hours, about 16 hours, about 17 hours, about 18 hours, about 19 hours, about 20 hours, about 21 hours, about 22 hours, about 23 hours, about 24 hours, or about 24 hours to about 48 hours) after the mixture comprising DB1/ 149202201.1 58 Attorney Docket No.: 116983-5127-WO the antibody and the drug (e.g., DM1, DM3 or DM4) is contacted with the cross-linking agent (e.g., SMCC, Sulfo-SMCC, SPDB, Sulfo-SPDB or CX1-1). [00115] The holding step comprises maintaining the solution at a suitable temperature (e.g., about 0° C. to about 37° C.) for a suitable period of time (e.g., about 1 hour to about 1 week, about 1 hour to about 24 hours, about 1 hour to about 8 hours, or about 1 hour to about 4 hours) to release the unstably bound linkers from the antibody while not substantially releasing the stably bound linkers from the antibody. In one embodiment, the holding step comprises maintaining the solution at about 20° C. or less (e.g., about 0° C. to about 18° C., about 4° C. to about 16° C.), at room temperature (e.g., about 20° C. to about 30° C. or about 20° C. to about 25° C.), or at an elevated temperature (e.g., about 30° C. to about 37° C.). In one embodiment, the holding step comprises maintaining the solution at a temperature of about 16° C. to about 24° C. (e.g., about 15° C., about 16° C., about 17° C., about 18° C., about 19° C., about 20° C., about 21° C., about 22° C., about 23° C., about 24° C., or about 25° C.). In another embodiment, the holding step comprises maintaining the solution at a temperature of about 2° C. to about 8° C. (e.g., about 0° C., about 1° C., about 2° C., about 3° C., about 4° C., about 5° C., about 6° C., about 7° C., about 8° C., about 9° C., or about 10° C.). In another embodiment, the holding step comprises maintaining the solution at a temperature of about 37° C. (e.g., about 34° C., about 35° C., about 36° C., about 37° C., about 38° C., about 39° C., or about 40° C.). [00116] The duration of the holding step depends on the temperature and the pH at which the holding step is performed. For example, the duration of the holding step can be substantially reduced by performing the holding step at elevated temperature, with the maximum temperature limited by the stability of the cell-binding agent-cytotoxic agent conjugate. The holding step can comprise maintaining the solution for about 1 hour to about 1 day (e.g., about 1 hour, about 2 hours, about 3 hours, about 4 hours, about 5 hours, about 6 hours, about 7 hours, about 8 hours, about 9 hours, about 10 hours, about 12 hours, about 14 hours, about 16 hours, about 18 hours, about 20 hours, about 22 hours, or about 24 hours), about 10 hours to about 24 hours, about 12 hours to about 24 hours, about 14 hours to about 24 hours, about 16 hours to about 24 hours, about 18 hours to about 24 hours, about 20 hours to about 24 hours, about 5 hours to about 1 week, about 20 hours to about 1 week, about 12 hours to about 1 week (e.g., about 12 hours, about 16 hours, about 20 hours, about 24 hours, DB1/ 149202201.1 59 Attorney Docket No.: 116983-5127-WO about 2 days, about 3 days, about 4 days, about 5 days, about 6 days, or about 7 days), or about 1 day to about 1 week. [00117] In one embodiment, the holding step comprises maintaining the solution at a temperature of about 2° C. to about 8° C. for a period of at least about 12 hours for up to a week. In another embodiment, the holding step comprises maintaining the solution at a temperature of about 2° C. to about 8° C. overnight (e.g., about 12 to about 24 hours, preferably about 20 hours). [00118] The pH value for the holding step preferably is about 4 to about 10. In one embodiment, the pH value for the holding step is about 4 or more, but less than about 6 (e.g., 4 to 5.9) or about 5 or more, but less than about 6 (e.g., 5 to 5.9). In another embodiment, the pH values for the holding step range from about 6 to about 10 (e.g., about 6.5 to about 9, about 6 to about 8). For example, pH values for the holding step can be about 6, about 6.5, about 7, about 7.5, about 8, about 8.5, about 9, about 9.5, or about 10. [00119] In specific embodiments, the holding step can comprise incubating the mixture at 25° C. at a pH of about 6-7.5 for about 12 hours to about 1 week, incubating the mixture at 4° C. at a pH of about 4.5-5.9 for about 5 hours to about 5 days, or incubating the mixture at 25° C. at a pH of about 4.5-5.9 for about 5 hours to about 1 day. [00120] The one-step process may optionally include the addition of sucrose to the reaction step to increase solubility and recovery of the conjugates. Desirably, sucrose is added at a concentration of about 0.1% (w/v) to about 20% (w/v) (e.g., about 0.1% (w/v), 1% (w/v), 5% (w/v), 10% (w/v), 15% (w/v), or 20% (w/v)). Preferably, sucrose is added at a concentration of about 1% (w/v) to about 10% (w/v) (e.g., about 0.5% (w/v), about 1% (w/v), about 1.5% (w/v), about 2% (w/v), about 3% (w/v), about 4% (w/v), about 5% (w/v), about 6% (w/v), about 7% (w/v), about 8% (w/v), about 9% (w/v), about 10% (w/v), or about 11% (w/v)). In addition, the reaction step also can comprise the addition of a buffering agent. Any suitable buffering agent known in the art can be used. Suitable buffering agents include, for example, a citrate buffer, an acetate buffer, a succinate buffer, and a phosphate buffer. In one embodiment, the buffering agent is selected from the group consisting of HEPPSO (N-(2- hydroxyethyl)piperazine-N′-(2-hydroxypropanesulfonic acid)), POPSO (piperazine-1,4-bis- (2-hydroxy-propane-sulfonic acid) dehydrate), HEPES (4-(2-hydroxyethyl)piperazine-1- ethanesulfonic acid), HEPPS (EPPS) (4-(2-hydroxyethyl)piperazine-1-propanesulfonic acid), DB1/ 149202201.1 60 Attorney Docket No.: 116983-5127-WO TES (N-[tris(hydroxymethyl)methyl]-2-aminoethanesulfonic acid), and a combination thereof. [00121] In one embodiment, the one-step process can further comprise the step of purifying the mixture to provide purified conjugate (e.g., Ab-MCC-DM1, Ab-SPDB-DM4 or Ab-CX1-1-DM1). Any purification methods known in the art can be used to purify the conjugates of the present invention. In one embodiment, the conjugates of the present invention using tangential flow filtration (TFF), non-adsorptive chromatography, adsorptive chromatography, adsorptive filtration, selective precipitation, or any other suitable purification process, as well as combinations thereof. In another embodiment, prior to subjecting the conjugates to purification process described above, the conjugates are first filtered through one or more PVDF membranes. Alternatively, the conjugates are filtered through one or more PVDF membranes after subjecting the conjugates to the purification process described above. For example, in one embodiment, the conjugates are filtered through one or more PVDF membranes and then purified using tangential flow filtration. Alternatively, the conjugates are purified using tangential flow filtration and then filtered through one or more PVDF membranes. [00122] Any suitable TFF systems may be utilized for purification, including a Pellicon type system (Millipore, Billerica, Mass.), a Sartocon Cassette system (Sartorius A G, Edgewood, N.Y.), and a Centrasette type system (Pall Corp., East Hills, N.Y.). [00123] Any suitable adsorptive chromatography resin may be utilized for purification. Preferred adsorptive chromatography resins include hydroxyapatite chromatography, hydrophobic charge induction chromatography (HCIC), hydrophobic interaction chromatography (HIC), ion exchange chromatography, mixed mode ion exchange chromatography, immobilized metal affinity chromatography (IMAC), dye ligand chromatography, affinity chromatography, reversed phase chromatography, and combinations thereof. Examples of suitable hydroxyapatite resins include ceramic hydroxyapatite (CHT Type I and Type II, Bio-Rad Laboratories, Hercules, Calif.), HA Ultrogel hydroxyapatite (Pall Corp., East Hills, N.Y.), and ceramic fluoroapatite (CFT Type I and Type II, Bio-Rad Laboratories, Hercules, Calif.). An example of a suitable HCIC resin is MEP Hypercel resin (Pall Corp., East Hills, N.Y.). Examples of suitable HIC resins include Butyl-Sepharose, Hexyl-Sepaharose, Phenyl-Sepharose, and Octyl Sepharose resins (all from GE Healthcare, Piscataway, N.J.), as well as Macro-prep Methyl and Macro-Prep t-Butyl resins (Biorad DB1/ 149202201.1 61 Attorney Docket No.: 116983-5127-WO Laboratories, Hercules, Calif.). Examples of suitable ion exchange resins include SP- Sepharose, CM-Sepharose, and Q-Sepharose resins (all from GE Healthcare, Piscataway, N.J.), and Unosphere S resin (Bio-Rad Laboratories, Hercules, Calif.). Examples of suitable mixed mode ion exchangers include Bakerbond ABx resin (JT Baker, Phillipsburg N.J.). Examples of suitable IMAC resins include Chelating Sepharose resin (GE Healthcare, Piscataway, N.J.) and Profinity IMAC resin (Bio-Rad Laboratories, Hercules, Calif.). Examples of suitable dye ligand resins include Blue Sepharose resin (GE Healthcare, Piscataway, N.J.) and Affi-gel Blue resin (Bio-Rad Laboratories, Hercules, Calif.). Examples of suitable affinity resins include Protein A Sepharose resin (e.g., MabSelect, GE Healthcare, Piscataway, N.J.) and lectin affinity resins, e.g. Lentil Lectin Sepharose resin (GE Healthcare, Piscataway, N.J.), where the antibody bears appropriate lectin binding sites. Examples of suitable reversed phase resins include C4, C8, and C18 resins (Grace Vydac, Hesperia, Calif.). [00124] Any suitable non-adsorptive chromatography resin may be utilized for purification. Examples of suitable non-adsorptive chromatography resins include, but are not limited to, SEPHADEX™ G-25, G-50, G-100, SEPHACRYL™ resins (e.g., S-200 and S- 300), SUPERDEX™ resins (e.g., SUPERDEX™ 75 and SUPERDEX™ 200), BIO-GEL® resins (e.g., P-6, P-10, P-30, P-60, and P-100), and others known to those of ordinary skill in the art. G. Two-Step Process and One-Pot Process for Cross-Linking to Lysine Antibody Residues [00125] In one embodiment, the conjugates of the present invention can be prepared as described in the U.S. Pat. No.7,811,572 and U.S. Patent Application Publication No. 2006/0182750. The process comprises the steps of (a) contacting the antibody of the present invention with the cross-linking agent (e.g., SMCC, Sulfo-SMCC, SPDB, Sulfo-SPDB or CX1-1) to covalently attach the linker (i.e., Ab-SMCC, Ab-SPDB or Ab-CX1-1) to the antibody and thereby prepare a first mixture comprising the antibody having the linker bound thereto; (b) optionally subjecting the first mixture to a purification process to prepare a purified first mixture of the antibody having the linker bound thereto; (c) conjugating the drug (e.g., DM1, DM3, or DM4) to the antibody having the linker bound thereto in the first mixture by reacting the antibody having the linker bound thereto with the drug (e.g., DM1, DM3, or DM4) in a solution having a pH of about 4 to about 9 to prepare a second mixture DB1/ 149202201.1 62 Attorney Docket No.: 116983-5127-WO comprising (i) conjugate (e.g., Ab-MCC-DM1, Ab-SPDB-DM4 or Ab-CX1-1-DM1), (ii) free drug (e.g., DM1, DM3 or DM4); and (iii) reaction by-products; and (d) subjecting the second mixture to a purification process to purify the conjugate from the other components of the second mixture. Alternatively, the purification step (b) can be omitted. Any purification methods described herein can be used for steps (b) and (d). In one embodiment, TFF is used for both steps (b) and (d). In another embodiment, TFF is used for step (b) and absorptive chromatography (e.g., CHT) is used for step (d). H. Antibody Preparation [00126] Techniques for preparing monoclonal antibodies against virtually any target antigen, such as Trop-2, are well known in the art. See, for example, Kohler and Milstein, Nature 256: 495 (1975), and Coligan et al. (eds.), CURRENT PROTOCOLS IN IMMUNOLOGY, VOL.1, pages 2.5.1-2.6.7 (John Wiley & Sons 1991). Briefly, monoclonal antibodies can be obtained by injecting mice with a composition comprising an antigen, removing the spleen to obtain B-lymphocytes, fusing the B-lymphocytes with myeloma cells to produce hybridomas, cloning the hybridomas, selecting positive clones which produce antibodies to the antigen, culturing the clones that produce antibodies to the antigen, and isolating the antibodies from the hybridoma cultures. [00127] MAbs can be isolated and purified from hybridoma cultures by a variety of well- established techniques. Such isolation techniques include affinity chromatography with Protein-A or Protein-G Sepharose, size-exclusion chromatography, and ion-exchange chromatography. See, for example, Coligan at pages 2.7.1-2.7.12 and pages 2.9.1-2.9.3. Also, see Baines et al., "Purification of Immunoglobulin G (IgG)," in METHODS IN MOLECULAR BIOLOGY, VOL.10, pages 79-104 (The Humana Press, Inc.1992). [00128] After the initial raising of antibodies to the immunogen, the heavy and light chain genes of the antibodies can be sequenced and subsequently cloned by recombinant techniques. See, e.g., Kopitar-Jerala et al., Pflugers Arch – Eur J Physiol, 2000, 439 [Suppl]: R79-R80. Humanization and chimerization of murine antibodies and antibody fragments are well known to those skilled in the art, as discussed below. [00129] Chimeric Antibodies [00130] A chimeric antibody is a recombinant protein in which the variable regions of a human antibody have been replaced by the variable regions of, for example, a mouse DB1/ 149202201.1 63 Attorney Docket No.: 116983-5127-WO antibody, including the complementarity- determining regions (CDRs) of the mouse antibody. Chimeric antibodies exhibit decreased immunogenicity and increased stability when administered to a subject. General techniques for cloning murine immunoglobulin variable domains are disclosed, for example, in Orlandi et al, Proc. Nat'l Acad. Sci. USA 6: 3833 (1989). Techniques for constructing chimeric antibodies are well known to those of skill in the art. As an example, Leung et al, Hybridoma 13:469 (1994), produced an LL2 chimera by combining DNA sequences encoding the VK and VH domains of murine LL2, an anti- CD22 monoclonal antibody, with respective human κ and IgG
I constant region domains. [00131] Humanized Antibodies [00132] Techniques for producing humanized MAbs are well known in the art (see, e.g., Jones et al, Nature 321: 522 (1986), Riechmann et al, Nature 332: 323 (1988), Verhoeyen et al, Science 239: 1534 (1988), Carter et al, Proc. Nat'l Acad. Sci. USA 89: 4285 (1992), Sandhu, Crit. Rev. Biotech.12: 437 (1992), and Singer et al., J. Immun.150: 2844 (1993)). A chimeric or murine monoclonal antibody may be humanized by transferring the mouse CDRs from the heavy and light variable chains of the mouse immunoglobulin into the corresponding variable domains of a human antibody. The mouse framework regions (FR) in the chimeric monoclonal antibody are also replaced with human FR sequences. As simply transferring mouse CDRs into human FRs often results in a reduction or even loss of antibody affinity, additional modification might be required in order to restore the original affinity of the murine antibody. This can be accomplished by the replacement of one or more human residues in the FR regions with their murine counterparts to obtain an antibody that possesses good binding affinity to its epitope. See, for example, Tempest et al., Biotechnology 9:266 (1991) and Verhoeyen et al., Science 239: 1534 (1988). Preferred residues for substitution include FR residues that are located within 1, 2, or 3 Angstroms of a CDR residue side chain, that are located adjacent to a CDR sequence, or that are predicted to interact with a CDR residue. [00133] Human Antibodies [00134] Methods for producing fully human antibodies using either combinatorial approaches or transgenic animals transformed with human immunoglobulin loci are known in the art (e.g., Mancini et al., 2004, New Microbiol.27:315-28; Conrad and Scheller, 2005, Comb. Chem. High Throughput Screen.8: 117-26; Brekke and Loset, 2003, Curr. Opin. Pharmacol.3:544-50). A fully human antibody also can be constructed by genetic or DB1/ 149202201.1 64 Attorney Docket No.: 116983-5127-WO chromosomal transfection methods, as well as phage display technology, all of which are known in the art. See for example, McCafferty et al, Nature 348:552-553 (1990). Such fully human antibodies are expected to exhibit even fewer side effects than chimeric or humanized antibodies and to function in vivo as essentially endogenous human antibodies. [00135] In one alternative, the phage display technique may be used to generate human antibodies (e.g., Dantas-Barbosa et al., 2005, Genet. Mol. Res.4: 126-40). Human antibodies may be generated from normal humans or from humans that exhibit a particular disease state, such as cancer (Dantas-Barbosa et al., supra). The advantage to constructing human antibodies from a diseased individual is that the circulating antibody repertoire may be biased towards antibodies against disease-associated antigens. [00136] In one non-limiting example of this methodology, Dantas-Barbosa et al. constructed a phage display library of human Fab antibody fragments from osteosarcoma patients. Generally, total RNA was obtained from circulating blood lymphocytes (Id.). [00137] Recombinant Fab were cloned from the μ, γ and κ chain antibody repertoires and inserted into a phage display library (Id.). RNAs were converted to cDNAs and used to make Fab cDNA libraries using specific primers against the heavy and light chain immunoglobulin sequences (Marks et al., 1991, J. Mol. Biol.222:581-97). Library construction was performed according to Andris-Widhopf et al. (2000, In: Phage Display Laboratory Manual, Barbas et al. (eds), 1st edition, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY pp.9.1 to 9.22). The final Fab fragments were digested with restriction endonucleases and inserted into the bacteriophage genome to make the phage display library. Such libraries may be screened by standard phage display methods, as known in the art. Phage display can be performed in a variety of formats, for their review, see e.g. Johnson and Chiswell, Current Opinion in Structural Biology 3:5564-571 (1993). [00138] Human antibodies may also be generated by in vitro activated B-cells. See U.S. Patent Nos.5,567,610 and 5,229,275, incorporated herein by reference in their entirety. The skilled artisan will realize that these techniques are exemplary and any known method for making and screening human antibodies or antibody fragments may be utilized. [00139] In another alternative, transgenic animals that have been genetically engineered to produce human antibodies may be used to generate antibodies against essentially any immunogenic target, using standard immunization protocols. Methods for DB1/ 149202201.1 65 Attorney Docket No.: 116983-5127-WO obtaining human antibodies from transgenic mice are disclosed by Green et al, Nature Genet. 7:13 (1994), Lonberg et al, Nature 355:856 (1994), and Taylor et al, Int. Immun.6:579 (1994). A non- limiting example of such a system is the XenoMouse® (e.g., Green et al., 1999, J. Immunol. Methods 231 : 11-23, incorporated herein by reference) from Abgenix (Fremont, CA). In the XenoMouse® and similar animals, the mouse antibody genes have been inactivated and replaced by functional human antibody genes, while the remainder of the mouse immune system remains intact. [00140] The XenoMouse® was transformed with germline- configured YACs (yeast artificial chromosomes) that contained portions of the human IgH and Igkappa loci, including the majority of the variable region sequences, along with accessory genes and regulatory sequences. The human variable region repertoire may be used to generate antibody producing B-cells, which may be processed into hybridomas by known techniques. A XenoMouse® immunized with a target antigen will produce human antibodies by the normal immune response, which may be harvested and/or produced by standard techniques discussed above. A variety of strains of XenoMouse® are available, each of which is capable of producing a different class of antibody. Transgenically produced human antibodies have been shown to have therapeutic potential, while retaining the pharmacokinetic properties of normal human antibodies (Green et al., 1999, supra). The skilled artisan will realize that the claimed compositions and methods are not limited to use of the XenoMouse® system but may utilize any transgenic animal that has been genetically engineered to produce human antibodies. Cloning and production of human antibodies can be carried out using the usual hybridoma technology. See Little et al., Immunol. Today, 2000, 21:364-370. [00141] The host cells for harboring and expressing the anti-Trop-2 antibody chains can be either prokaryotic or eukaryotic. E. coli is one prokaryotic host useful for cloning and expressing the polynucleotides of the present invention. Other microbial hosts suitable for use include bacilli, such as Bacillus subtilis, and other enterobacteriaceae, such as Salmonella, Serratia , and various Pseudomonas species. In these prokaryotic hosts, one can also make expression vectors, which typically contain expression control sequences compatible with the host cell (e.g., an origin of replication). In addition, any number of a variety of well-known promoters will be present, such as the lactose promoter system, a tryptophan (trp) promoter system, a beta-lactamase promoter system, or a promoter system from phage lambda. The promoters typically control expression, optionally with an operator sequence, and have DB1/ 149202201.1 66 Attorney Docket No.: 116983-5127-WO ribosome binding site sequences and the like, for initiating and completing transcription and translation. Other microbes, such as yeast, can also be employed to express anti-Trop-2 polypeptides of the invention. Insect cells in combination with baculovirus vectors can also be used. [00142] In some preferred embodiments, mammalian host cells are used to express and produce the anti-Trop-2 polypeptides of the present invention. For example, they can be either a hybridoma cell line expressing endogenous immunoglobulin genes (e.g., the myeloma hybridoma clones as described in the Examples) or a mammalian cell line harboring an exogenous expression vector (e.g., the SP2/0 myeloma cells exemplified below). These include any normal mortal or normal or abnormal immortal animal or human cell. For example, a number of suitable host cell lines capable of secreting intact immunoglobulins have been developed, including the CHO cell lines, various Cos cell lines, HeLa cells, myeloma cell lines, transformed B-cells and hybridomas. The use of mammalian tissue cell culture to express polypeptides is discussed generally in, e.g., Winnacker, From Genes to Clones, VCH Publishers, N.Y., N.Y., 1987. Expression vectors for mammalian host cells can include expression control sequences, such as an origin of replication, a promoter, and an enhancer (see, e.g., Queen et al., Immunol. Rev.89:49-68, 1986), and necessary processing information sites, such as ribosome binding sites, RNA splice sites, polyadenylation sites, and transcriptional terminator sequences. These expression vectors usually contain promoters derived from mammalian genes or from mammalian viruses. Suitable promoters may be constitutive, cell type-specific, stage-specific, and/or modulatable or regulatable. Useful promoters include, but are not limited to, the metallothionein promoter, the constitutive adenovirus major late promoter, the dexamethasone-inducible MMTV promoter, the SV40 promoter, the MRP polIII promoter, the constitutive MPSV promoter, the tetracycline-inducible CMV promoter (such as the human immediate-early CMV promoter), the constitutive CMV promoter, and promoter-enhancer combinations known in the art. [00143] Methods for introducing expression vectors containing the polynucleotide sequences of interest vary depending on the type of cellular host. For example, calcium chloride transfection is commonly utilized for prokaryotic cells, whereas calcium phosphate treatment or electroporation may be used for other cellular hosts (see generally Sambrook et al., Molecular Cloning: A Laboratory Manual, 4th ed.). Other methods include, e.g., electroporation, calcium phosphate treatment, liposome-mediated transformation, injection DB1/ 149202201.1 67 Attorney Docket No.: 116983-5127-WO and microinjection, ballistic methods, virosomes, immunoliposomes, polycation:nucleic acid conjugates, naked DNA, artificial virions, fusion to the herpes virus structural protein VP22 (Elliot and O'Hare, Cell 88:223, 1997), agent-enhanced uptake of DNA, and ex vivo transduction. For long-term, high-yield production of recombinant proteins, stable expression will often be desired. For example, cell lines which stably express anti-Trop-2 antibody chains or binding fragments can be prepared using expression vectors of the invention which contain viral origins of replication or endogenous expression elements and a selectable marker gene. Following introduction of the vector, cells may be allowed to grow for 1-2 days in an enriched media before they are switched to selective media. The purpose of the selectable marker is to confer resistance to selection, and its presence allows growth of cells which successfully express the introduced sequences in selective media. Resistant, stably transfected cells can be proliferated using tissue culture techniques appropriate to the cell type. III. Gen 2 TIL Manufacturing Processes [0006] An exemplary family of TIL processes known as Gen 2 (also known as process 2A) containing some of these features is depicted in Figures 1 and 2. An embodiment of Gen 2 is shown in Figure 2. [0007] As discussed herein, the present invention can include a step relating to the restimulation of cryopreserved TILs to increase their metabolic activity and thus relative health prior to transplant into a patient, and methods of testing said metabolic health. As generally outlined herein, TILs are generally taken from a patient sample and manipulated to expand their number prior to transplant into a patient. In some embodiments, the TILs may be optionally genetically manipulated as discussed below. [0008] In some embodiments, the TILs may be cryopreserved. Once thawed, they may also be restimulated to increase their metabolism prior to infusion into a patient. [0009] In some embodiments, the first expansion (including processes referred to as the pre-REP as well as processes shown in Figure 1 as Step A) is shortened to 3 to 14 days and the second expansion (including processes referred to as the REP as well as processes shown in Figure 1 as Step B) is shorted to 7 to 14 days, as discussed in detail below as well as in the examples and figures. In some embodiments, the first expansion (for example, an expansion described as Step B in Figure 1) is shortened to 11 days and the second expansion (for DB1/ 149202201.1 68 Attorney Docket No.: 116983-5127-WO example, an expansion as described in Step D in Figure 1) is shortened to 11 days. In some embodiments, the combination of the first expansion and second expansion (for example, expansions described as Step B and Step D in Figure 1) is shortened to 22 days, as discussed in detail below and in the examples and figures. [0010] The “Step” Designations A, B, C, etc., below are in reference to Figure 1 and in reference to certain embodiments described herein. The ordering of the Steps below and in Figure 1 is exemplary and any combination or order of steps, as well as additional steps, repetition of steps, and/or omission of steps is contemplated by the present application and the methods disclosed herein. A. STEP A: Obtain Patient Tumor Sample [0011] In general, TILs are initially obtained from a patient tumor sample and then expanded into a larger population for further manipulation as described herein, optionally cryopreserved, restimulated as outlined herein and optionally evaluated for phenotype and metabolic parameters as an indication of TIL health. [0012] A patient tumor sample may be obtained using methods known in the art, generally via surgical resection, needle biopsy, core biopsy, small biopsy, or other means for obtaining a sample that contains a mixture of tumor and TIL cells. In some embodiments, multilesional sampling is used. In some embodiments, surgical resection, needle biopsy, core biopsy, small biopsy, or other means for obtaining a sample that contains a mixture of tumor and TIL cells includes multilesional sampling (i.e., obtaining samples from one or more tumor sites and/or locations in the patient, as well as one or more tumors in the same location or in close proximity). In general, the tumor sample may be from any solid tumor, including primary tumors, invasive tumors or metastatic tumors. The tumor sample may also be a liquid tumor, such as a tumor obtained from a hematological malignancy. The solid tumor may be of lung tissue. In some embodiments, useful TILs are obtained from non-small cell lung carcinoma (NSCLC). The solid tumor may be of skin tissue. In some embodiments, useful TILs are obtained from a melanoma. [0013] Once obtained, the tumor sample is generally fragmented using sharp dissection into small pieces of between 1 to about 8 mm
3, with from about 2-3 mm
3 being particularly useful. In some embodiments, the TILs are cultured from these fragments using enzymatic tumor digests. Such tumor digests may be produced by incubation in enzymatic media (e.g., DB1/ 149202201.1 69 Attorney Docket No.: 116983-5127-WO Roswell Park Memorial Institute (RPMI) 1640 buffer, 2 mM glutamate, 10 mcg/mL gentamicine, 30 units/mL of DNase and 1.0 mg/mL of collagenase) followed by mechanical dissociation (e.g., using a tissue dissociator). Tumor digests may be produced by placing the tumor in enzymatic media and mechanically dissociating the tumor for approximately 1 minute, followed by incubation for 30 minutes at 37 °C in 5% CO
2, followed by repeated cycles of mechanical dissociation and incubation under the foregoing conditions until only small tissue pieces are present. At the end of this process, if the cell suspension contains a large number of red blood cells or dead cells, a density gradient separation using FICOLL branched hydrophilic polysaccharide may be performed to remove these cells. Alternative methods known in the art may be used, such as those described in U.S. Patent Application Publication No.2012/0244133 A1, the disclosure of which is incorporated by reference herein. Any of the foregoing methods may be used in any of the embodiments described herein for methods of expanding TILs or methods treating a cancer. [0014] Tumor dissociating enzyme mixtures can include one or more dissociating (digesting) enzymes such as, but not limited to, collagenase (including any blend or type of collagenase), Accutase™, Accumax™, hyaluronidase, neutral protease (dispase), chymotrypsin, chymopapain, trypsin, caseinase, elastase, papain, protease type XIV (pronase), deoxyribonuclease I (DNase), trypsin inhibitor, any other dissociating or proteolytic enzyme, and any combination thereof. [0015] In some embodiments, the dissociating enzymes are reconstituted from lyophilized enzymes. In some embodiments, lyophilized enzymes are reconstituted in an amount of sterile buffer such as HBSS. [0016] In some instances, collagenase (such as animal free- type 1 collagenase) is reconstituted in 10 mL of sterile HBSS or another buffer. The lyophilized stock enzyme may be at a concentration of 2892 PZ U/vial. In some embodiments, collagenase is reconstituted in 5 mL to 15 mL buffer. In some embodiment, after reconstitution the collagenase stock ranges from about 100 PZ U/mL-about 400 PZ U/mL, e.g., about 100 PZ U/mL-about 400 PZ U/mL, about 100 PZ U/mL-about 350 PZ U/mL, about 100 PZ U/mL-about 300 PZ U/mL, about 150 PZ U/mL-about 400 PZ U/mL, about 100 PZ U/mL, about 150 PZ U/mL, about 200 PZ U/mL, about 210 PZ U/mL, about 220 PZ U/mL, about 230 PZ U/mL, about 240 PZ U/mL, about 250 PZ U/mL, about 260 PZ U/mL, about 270 PZ U/mL, about DB1/ 149202201.1 70 Attorney Docket No.: 116983-5127-WO 280 PZ U/mL, about 289.2 PZ U/mL, about 300 PZ U/mL, about 350 PZ U/mL, or about 400 PZ U/mL. [0017] In some embodiments, neutral protease is reconstituted in 1 mL of sterile HBSS or another buffer. The lyophilized stock enzyme may be at a concentration of 175 DMC U/vial. In some embodiments, after reconstitution the neutral protease stock ranges from about 100 DMC/mL-about 400 DMC/mL, e.g., about 100 DMC/mL-about 400 DMC/mL, about 100 DMC/mL-about 350 DMC/mL, about 100 DMC/mL-about 300 DMC/mL, about 150 DMC/mL-about 400 DMC/mL, about 100 DMC/mL, about 110 DMC/mL, about 120 DMC/mL, about 130 DMC/mL, about 140 DMC/mL, about 150 DMC/mL, about 160 DMC/mL, about 170 DMC/mL, about 175 DMC/mL, about 180 DMC/mL, about 190 DMC/mL, about 200 DMC/mL, about 250 DMC/mL, about 300 DMC/mL, about 350 DMC/mL, or about 400 DMC/mL. [0018] In some embodiments, DNAse I is reconstituted in 1 mL of sterile HBSS or another buffer. The lyophilized stock enzyme was at a concentration of 4 KU/vial. In some embodiments, after reconstitution the DNase I stock ranges from about 1 KU/mL-10 KU/mL, e.g., about 1 KU/mL, about 2 KU/mL, about 3 KU/mL, about 4 KU/mL, about 5 KU/mL, about 6 KU/mL, about 7 KU/mL, about 8 KU/mL, about 9 KU/mL, or about 10 KU/mL. [0019] In some embodiments, the stock of enzymes is variable and the concentrations may need to be determined. In some embodiments, the concentration of the lyophilized stock can be verified. In some embodiments, the final amount of enzyme added to the digest cocktail is adjusted based on the determined stock concentration. [0020] In some embodiment, the enzyme mixture includes about 10.2-ul of neutral protease (0.36 DMC U/mL), 21.3 µL of collagenase (1.2 PZ/mL) and 250-ul of DNAse I (200 U/mL) in about 4.7 mL of sterile HBSS. [0021] As indicated above, in some embodiments, the TILs are derived from solid tumors. In some embodiments, the solid tumors are not fragmented. In some embodiments, the solid tumors are not fragmented and are subjected to enzymatic digestion as whole tumors. In some embodiments, the tumors are digested in in an enzyme mixture comprising collagenase, DNase, and hyaluronidase. In some embodiments, the tumors are digested in in an enzyme mixture comprising collagenase, DNase, and hyaluronidase for 1-2 hours. In some embodiments, the tumors are digested in in an enzyme mixture comprising collagenase, DB1/ 149202201.1 71 Attorney Docket No.: 116983-5127-WO DNase, and hyaluronidase for 1-2 hours at 37°C, 5% CO
2. In some embodiments, the tumors are digested in in an enzyme mixture comprising collagenase, DNase, and hyaluronidase for 1-2 hours at 37°C, 5% CO2 with rotation. In some embodiments, the tumors are digested overnight with constant rotation. In some embodiments, the tumors are digested overnight at 37°C, 5% CO
2 with constant rotation. In some embodiments, the whole tumor is combined with the enzymes to form a tumor digest reaction mixture. [0022] In some embodiments, the tumor is reconstituted with the lyophilized enzymes in a sterile buffer. In some embodiments, the buffer is sterile HBSS. [0023] In some embodiments, the enzyme mixture comprises collagenase. In some embodiments, the collagenase is collagenase IV. In some embodiments, the working stock for the collagenase is a 100 mg/mL 10X working stock. [0024] In some embodiments, the enzyme mixture comprises DNAse. In some embodiments, the working stock for the DNAse is a 10,000 IU/mL 10X working stock. [0025] In some embodiments, the enzyme mixture comprises hyaluronidase. In some embodiments, the working stock for the hyaluronidase is a 10 mg/mL 10X working stock. [0026] In some embodiments, the enzyme mixture comprises 10 mg/mL collagenase, 1000 IU/mL DNAse, and 1 mg/mL hyaluronidase. [0027] In some embodiments, the enzyme mixture comprises 10 mg/mL collagenase, 500 IU/mL DNAse, and 1 mg/mL hyaluronidase. [0028] In general, the harvested cell suspension is called a “primary cell population” or a “freshly harvested” cell population. [0029] In some embodiments, fragmentation includes physical fragmentation, including for example, dissection as well as digestion. In some embodiments, the fragmentation is physical fragmentation. In some embodiments, the fragmentation is dissection. In some embodiments, the fragmentation is by digestion. In some embodiments, TILs can be initially cultured from enzymatic tumor digests and tumor fragments obtained from digesting or fragmenting a tumor sample obtained from a patient. [0030] In some embodiments, where the tumor is a solid tumor, the tumor undergoes physical fragmentation after the tumor sample is obtained in, for example, Step A (as provided in Figure 1). In some embodiments, the fragmentation occurs before DB1/ 149202201.1 72 Attorney Docket No.: 116983-5127-WO cryopreservation. In some embodiments, the fragmentation occurs after cryopreservation. In some embodiments, the fragmentation occurs after obtaining the tumor and in the absence of any cryopreservation. In some embodiments, the tumor is fragmented and 10, 20, 30, 40 or more fragments or pieces are placed in each container for the first expansion. In some embodiments, the tumor is fragmented and 30 or 40 fragments or pieces are placed in each container for the first expansion. In some embodiments, the tumor is fragmented and 40 fragments or pieces are placed in each container for the first expansion. In some embodiments, the multiple fragments comprise about 4 to about 50 fragments, wherein each fragment has a volume of about 27 mm
3. In some embodiments, the multiple fragments comprise about 30 to about 60 fragments with a total volume of about 1300 mm
3 to about 1500 mm
3. In some embodiments, the multiple fragments comprise about 50 fragments with a total volume of about 1350 mm
3. In some embodiments, the multiple fragments comprise about 50 fragments with a total mass of about 1 gram to about 1.5 grams. In some embodiments, the multiple fragments comprise about 4 fragments. [0031] In some embodiments, the TILs are obtained from tumor fragments. In some embodiments, the tumor fragment is obtained by sharp dissection. In some embodiments, the tumor fragment is between about 1 mm
3 and 10 mm
3. In some embodiments, the tumor fragment is between about 1 mm
3 and 8 mm
3. In some embodiments, the tumor fragment is about 1 mm
3. In some embodiments, the tumor fragment is about 2 mm
3. In some embodiments, the tumor fragment is about 3 mm
3. In some embodiments, the tumor fragment is about 4 mm
3. In some embodiments, the tumor fragment is about 5 mm
3. In some embodiments, the tumor fragment is about 6 mm
3. In some embodiments, the tumor fragment is about 7 mm
3. In some embodiments, the tumor fragment is about 8 mm
3. In some embodiments, the tumor fragment is about 9 mm
3. In some embodiments, the tumor fragment is about 10 mm
3. In some embodiments, the tumors are 1-4 mm × 1-4 mm × 1-4 mm. In some embodiments, the tumors are 1 mm × 1 mm × 1 mm. In some embodiments, the tumors are 2 mm × 2 mm × 2 mm. In some embodiments, the tumors are 3 mm × 3 mm × 3 mm. In some embodiments, the tumors are 4 mm × 4 mm × 4 mm. [0032] In some embodiments, the tumors are resected in order to minimize the amount of hemorrhagic, necrotic, and/or fatty tissues on each piece. In some embodiments, the tumors are resected in order to minimize the amount of hemorrhagic tissue on each piece. In some embodiments, the tumors are resected in order to minimize the amount of necrotic tissue on DB1/ 149202201.1 73 Attorney Docket No.: 116983-5127-WO each piece. In some embodiments, the tumors are resected in order to minimize the amount of fatty tissue on each piece. [0033] In some embodiments, the tumor fragmentation is performed in order to maintain the tumor internal structure. In some embodiments, the tumor fragmentation is performed without performing a sawing motion with a scalpel. In some embodiments, the TILs are obtained from tumor digests. In some embodiments, tumor digests were generated by incubation in enzyme media, for example but not limited to RPMI 1640, 2 mM GlutaMAX, 10 mg/mL gentamicin, 30 U/mL DNase, and 1.0 mg/mL collagenase, followed by mechanical dissociation (GentleMACS, Miltenyi Biotec, Auburn, CA). After placing the tumor in enzyme media, the tumor can be mechanically dissociated for approximately 1 minute. The solution can then be incubated for 30 minutes at 37 °C in 5% CO
2 and it then mechanically disrupted again for approximately 1 minute. After being incubated again for 30 minutes at 37 °C in 5% CO2, the tumor can be mechanically disrupted a third time for approximately 1 minute. In some embodiments, after the third mechanical disruption if large pieces of tissue were present, 1 or 2 additional mechanical dissociations were applied to the sample, with or without 30 additional minutes of incubation at 37 °C in 5% CO2. In some embodiments, at the end of the final incubation if the cell suspension contains a large number of red blood cells or dead cells, a density gradient separation using Ficoll can be performed to remove these cells. [0034] In some embodiments, the harvested cell suspension prior to the first expansion step is called a “primary cell population” or a “freshly harvested” cell population. [0035] In some embodiments, cells can be optionally frozen after sample harvest and stored frozen prior to entry into the expansion described in Step B, which is described in further detail below, as well as exemplified in Figure 1, as well as Figure 8. 1. Pleural effusion T-cells and TILs [0036] In some embodiments, the sample is a pleural fluid sample. In some embodiments, the source of the T-cells or TILs for expansion according to the processes described herein is a pleural fluid sample. In some embodiments, the sample is a pleural effusion derived sample. In some embodiments, the source of the T-cells or TILs for expansion according to the processes described herein is a pleural effusion derived sample. See, for example, methods DB1/ 149202201.1 74 Attorney Docket No.: 116983-5127-WO described in U.S. Patent Publication US 2014/0295426, incorporated herein by reference in its entirety for all purposes. [0037] In some embodiments, any pleural fluid or pleural effusion suspected of and/or containing TILs can be employed. Such a sample may be derived from a primary or metastatic lung cancer, such as NSCLC or SCLC. In some embodiments, the sample may be derived from secondary metastatic cancer cells which originated from another organ, e.g., breast, ovary, colon or prostate. In some embodiments, the sample for use in the expansion methods described herein is a pleural exudate. In some embodiments, the sample for use in the expansion methods described herein is a pleural transudate. Other biological samples may include other serous fluids containing TILs, including, e.g., ascites fluid from the abdomen or pancreatic cyst fluid. Ascites fluid and pleural fluids involve very similar chemical systems; both the abdomen and lung have mesothelial lines and fluid forms in the pleural space and abdominal spaces in the same matter in malignancies and such fluids in some embodiments contain TILs. In some embodiments, wherein the disclosed methods utilize pleural fluid, the same methods may be performed with similar results using ascites or other cyst fluids containing TILs. [0038] In some embodiments, the pleural fluid is in unprocessed form, directly as removed from the patient. In some embodiments, the unprocessed pleural fluid is placed in a standard blood collection tube, such as an EDTA or Heparin tube, prior to further processing steps. In some embodiments, the unprocessed pleural fluid is placed in a standard CellSave® tube (Veridex) prior to further processing steps. In some embodiments, the sample is placed in the CellSave tube immediately after collection from the patient to avoid a decrease in the number of viable TILs. The number of viable TILs can decrease to a significant extent within 24 hours, if left in the untreated pleural fluid, even at 4°C. In some embodiments, the sample is placed in the appropriate collection tube within 1 hour, 5 hours, 10 hours, 15 hours, or up to 24 hours after removal from the patient. In some embodiments, the sample is placed in the appropriate collection tube within 1 hour, 5 hours, 10 hours, 15 hours, or up to 24 hours after removal from the patient at 4°C. [0039] In some embodiments, the pleural fluid sample from the chosen subject may be diluted. In some embodiments, the dilution is 1:10 pleural fluid to diluent. In other embodiments, the dilution is 1:9 pleural fluid to diluent. In other embodiments, the dilution is 1:8 pleural fluid to diluent. In other embodiments, the dilution is 1:5 pleural fluid to diluent. DB1/ 149202201.1 75 Attorney Docket No.: 116983-5127-WO In other embodiments, the dilution is 1:2 pleural fluid to diluent. In other embodiments, the dilution is 1:1 pleural fluid to diluent. In some embodiments, diluents include saline, phosphate buffered saline, another buffer or a physiologically acceptable diluent. In some embodiments, the sample is placed in the CellSave tube immediately after collection from the patient and dilution to avoid a decrease in the viable TILs, which may occur to a significant extent within 24-48 hours, if left in the untreated pleural fluid, even at 4°C. In some embodiments, the pleural fluid sample is placed in the appropriate collection tube within 1 hour, 5 hours, 10 hours, 15 hours, 24 hours, 36 hours, up to 48 hours after removal from the patient, and dilution. In some embodiments, the pleural fluid sample is placed in the appropriate collection tube within 1 hour, 5 hours, 10 hours, 15 hours, 24 hours, 36 hours, up to 48 hours after removal from the patient, and dilution at 4°C. [0040] In still other embodiments, pleural fluid samples are concentrated by conventional means prior to further processing steps. In some embodiments, this pre-treatment of the pleural fluid is preferable in circumstances in which the pleural fluid must be cryopreserved for shipment to a laboratory performing the method or for later analysis (e.g., later than 24-48 hours post-collection). In some embodiments, the pleural fluid sample is prepared by centrifuging the pleural fluid sample after its withdrawal from the subject and resuspending the centrifugate or pellet in buffer. In some embodiments, the pleural fluid sample is subjected to multiple centrifugations and resuspensions, before it is cryopreserved for transport or later analysis and/or processing. [0041] In some embodiments, pleural fluid samples are concentrated prior to further processing steps by using a filtration method. In some embodiments, the pleural fluid sample used in further processing is prepared by filtering the fluid through a filter containing a known and essentially uniform pore size that allows for passage of the pleural fluid through the membrane but retains the tumor cells. In some embodiments, the diameter of the pores in the membrane may be at least 4 μM. In other embodiments the pore diameter may be 5 μM or more, and in other embodiment, any of 6, 7, 8, 9, or 10 μM. After filtration, the cells, including TILs, retained by the membrane may be rinsed off the membrane into a suitable physiologically acceptable buffer. Cells, including TILs, concentrated in this way may then be used in the further processing steps of the method. [0042] In some embodiments, pleural fluid sample (including, for example, the untreated pleural fluid), diluted pleural fluid, or the resuspended cell pellet, is contacted with a lytic DB1/ 149202201.1 76 Attorney Docket No.: 116983-5127-WO reagent that differentially lyses non-nucleated red blood cells present in the sample. In some embodiments, this step is performed prior to further processing steps in circumstances in which the pleural fluid contains substantial numbers of RBCs. Suitable lysing reagents include a single lytic reagent or a lytic reagent and a quench reagent, or a lytic agent, a quench reagent and a fixation reagent. Suitable lytic systems are marketed commercially and include the BD Pharm Lyse™ system (Becton Dickenson). Other lytic systems include the Versalyse™ system, the FACSlyse™ system (Becton Dickenson), the Immunoprep™ system or Erythrolyse II system (Beckman Coulter, Inc.), or an ammonium chloride system. In some embodiments, the lytic reagent can vary with the primary requirements being efficient lysis of the red blood cells, and the conservation of the TILs and phenotypic properties of the TILs in the pleural fluid. In addition to employing a single reagent for lysis, the lytic systems useful in methods described herein can include a second reagent, e.g., one that quenches or retards the effect of the lytic reagent during the remaining steps of the method, e.g., Stabilyse™ reagent (Beckman Coulter, Inc.). A conventional fixation reagent may also be employed depending upon the choice of lytic reagents or the preferred implementation of the method. [0043] In some embodiments, the pleural fluid sample, unprocessed, diluted or multiply centrifuged or processed as described herein above is cryopreserved at a temperature of about −140°C prior to being further processed and/or expanded as provided herein. B. STEP B: First Expansion [0044] In some embodiments, the present methods provide for obtaining young TILs, which are capable of increased replication cycles upon administration to a subject/patient and as such may provide additional therapeutic benefits over older TILs (i.e., TILs which have further undergone more rounds of replication prior to administration to a subject/patient). Features of young TILs have been described in the literature, for example in Donia, et al., Scand. J. Immunol.2012, 75, 157–167; Dudley, et al., Clin. Cancer Res.2010, 16, 6122- 6131; Huang, et al., J. Immunother.2005, 28, 258–267; Besser, et al., Clin. Cancer Res. 2013, 19, OF1-OF9; Besser, et al., J. Immunother.2009, 32:415–423; Robbins, et al., J. Immunol.2004, 173, 7125-7130; Shen, et al., J. Immunother., 2007, 30, 123–129; Zhou, et al., J. Immunother.2005, 28, 53–62; and Tran, et al., J. Immunother., 2008, 31, 742–751, each of which is incorporated herein by reference. DB1/ 149202201.1 77 Attorney Docket No.: 116983-5127-WO [0045] The diverse antigen receptors of T and B lymphocytes are produced by somatic recombination of a limited, but large number of gene segments. These gene segments: V (variable), D (diversity), J (joining), and C (constant), determine the binding specificity and downstream applications of immunoglobulins and T-cell receptors (TCRs). The present invention provides a method for generating TILs which exhibit and increase the T-cell repertoire diversity. In some embodiments, the TILs obtained by the present method exhibit an increase in the T-cell repertoire diversity. In some embodiments, the TILs obtained by the present method exhibit an increase in the T-cell repertoire diversity as compared to freshly harvested TILs and/or TILs prepared using other methods than those provide herein including for example, methods other than those embodied in Figure 1. In some embodiments, the TILs obtained by the present method exhibit an increase in the T-cell repertoire diversity as compared to freshly harvested TILs and/or TILs prepared using methods referred to as process 1C, as exemplified in Figure 5 and/or Figure 6. In some embodiments, the TILs obtained in the first expansion exhibit an increase in the T-cell repertoire diversity. In some embodiments, the increase in diversity is an increase in the immunoglobulin diversity and/or the T-cell receptor diversity. In some embodiments, the diversity is in the immunoglobulin is in the immunoglobulin heavy chain. In some embodiments, the diversity is in the immunoglobulin is in the immunoglobulin light chain. In some embodiments, the diversity is in the T-cell receptor. In some embodiments, the diversity is in one of the T-cell receptors selected from the group consisting of alpha, beta, gamma, and delta receptors. In some embodiments, there is an increase in the expression of T-cell receptor (TCR) alpha and/or beta. In some embodiments, there is an increase in the expression of T-cell receptor (TCR) alpha. In some embodiments, there is an increase in the expression of T-cell receptor (TCR) beta. In some embodiments, there is an increase in the expression of TCRab (i.e., TCRα/β). [0046] After dissection or digestion of tumor fragments, for example such as described in Step A of Figure 1, the resulting cells are cultured in serum containing IL-2 under conditions that favor the growth of TILs over tumor and other cells. In some embodiments, the tumor digests are incubated in 2 mL wells in media comprising inactivated human AB serum with 6000 IU/mL of IL-2. This primary cell population is cultured for a period of days, generally from 3 to 14 days, resulting in a bulk TIL population, generally about 1 × 108 bulk TIL cells. In some embodiments, this primary cell population is cultured for a period of 7 to 14 days, resulting in a bulk TIL population, generally about 1 × 108 bulk TIL cells. In some DB1/ 149202201.1 78 Attorney Docket No.: 116983-5127-WO embodiments, this primary cell population is cultured for a period of 10 to 14 days, resulting in a bulk TIL population, generally about 1 × 108 bulk TIL cells. In some embodiments, this primary cell population is cultured for a period of about 11 days, resulting in a bulk TIL population, generally about 1 × 108 bulk TIL cells. [0047] In some embodiments, expansion of TILs may be performed using an initial bulk TIL expansion step (for example such as those described in Step B of Figure 1, which can include processes referred to as pre-REP) as described below and herein, followed by a second expansion (Step D, including processes referred to as rapid expansion protocol (REP) steps) as described below under Step D and herein, followed by optional cryopreservation, and followed by a second Step D (including processes referred to as restimulation REP steps) as described below and herein. The TILs obtained from this process may be optionally characterized for phenotypic characteristics and metabolic parameters as described herein. [0048] In embodiments where TIL cultures are initiated in 24-well plates, for example, using Costar 24-well cell culture cluster, flat bottom (Corning Incorporated, Corning, NY, each well can be seeded with 1 × 10
6 tumor digest cells or one tumor fragment in 2 mL of complete medium (CM) with IL-2 (6000 IU/mL; Chiron Corp., Emeryville, CA). In some embodiments, the tumor fragment is between about 1 mm
3 and 10 mm
3. [0049] In some embodiments, the first expansion culture medium is referred to as “CM”, an abbreviation for culture media. In some embodiments, CM for Step B consists of RPMI 1640 with GlutaMAX, supplemented with 10% human AB serum, 25 mM Hepes, and 10 mg/mL gentamicin. In embodiments where cultures are initiated in gas-permeable flasks with a 40 mL capacity and a 10 cm
2 gas-permeable silicon bottom (for example, G-REX10; Wilson Wolf Manufacturing, New Brighton, MN), each flask was loaded with 10–40 × 10
6 viable tumor digest cells or 5–30 tumor fragments in 10–40 mL of CM with IL-2. Both the G- REX10 and 24-well plates were incubated in a humidified incubator at 37°C in 5% CO2 and 5 days after culture initiation, half the media was removed and replaced with fresh CM and IL- 2 and after day 5, half the media was changed every 2–3 days. [0050] In some embodiments, the culture medium used in the expansion processes disclosed herein is a serum-free medium or a defined medium. In some embodiments, the serum-free or defined medium comprises a basal cell medium and a serum supplement and/or a serum replacement. In some embodiments, the serum-free or defined medium is used to DB1/ 149202201.1 79 Attorney Docket No.: 116983-5127-WO prevent and/or decrease experimental variation due in part to the lot-to-lot variation of serum- containing media. [0051] In some embodiments, the serum-free or defined medium comprises a basal cell medium and a serum supplement and/or serum replacement. In some embodiments, the basal cell medium includes, but is not limited to CTS™ OpTmizer™ T-cell Expansion Basal Medium , CTS™ OpTmizer™ T-Cell Expansion SFM, CTS™ AIM-V Medium, CTS™ AIM-V SFM, LymphoONE™ T-Cell Expansion Xeno-Free Medium, Dulbecco's Modified Eagle's Medium (DMEM), Minimal Essential Medium (MEM), Basal Medium Eagle (BME), RPMI 1640, F-10, F-12, Minimal Essential Medium (αMEM), Glasgow's Minimal Essential Medium (G-MEM), RPMI growth medium, and Iscove's Modified Dulbecco's Medium. [0052] In some embodiments, the serum supplement or serum replacement includes, but is not limited to one or more of CTS™ OpTmizer T-Cell Expansion Serum Supplement, CTS™ Immune Cell Serum Replacement, one or more albumins or albumin substitutes, one or more amino acids, one or more vitamins, one or more transferrins or transferrin substitutes, one or more antioxidants, one or more insulins or insulin substitutes, one or more collagen precursors, one or more antibiotics, and one or more trace elements. In some embodiments, the defined medium comprises albumin and one or more ingredients selected from the group consisting of glycine, L- histidine, L-isoleucine, L-methionine, L-phenylalanine, L-proline, L- hydroxyproline, L-serine, L-threonine, L-tryptophan, L-tyrosine, L-valine, thiamine, reduced glutathione, L-ascorbic acid-2-phosphate, iron saturated transferrin, insulin, and compounds containing the trace element moieties Ag+, Al3+, Ba2+, Cd2+, Co2+, Cr3+, Ge4+, Se4+, Br, T, Mn2+, P, Si4+, V5+, Mo6+, Ni2+, Rb+, Sn2+ and Zr4+. In some embodiments, the defined medium further comprises L-glutamine, sodium bicarbonate and/or 2-mercaptoethanol. [0053] In some embodiments, the CTS™OpTmizer™ T-cell Immune Cell Serum Replacement is used with conventional growth media, including but not limited to CTS™ OpTmizer™ T-cell Expansion Basal Medium, CTS™ OpTmizer™ T-cell Expansion SFM, CTS™ AIM-V Medium, CST™ AIM-V SFM, LymphoONE™ T-Cell Expansion Xeno-Free Medium, Dulbecco's Modified Eagle's Medium (DMEM), Minimal Essential Medium (MEM), Basal Medium Eagle (BME), RPMI 1640, F-10, F-12, Minimal Essential Medium (αMEM), Glasgow's Minimal Essential Medium (G-MEM), RPMI growth medium, and Iscove's Modified Dulbecco's Medium. DB1/ 149202201.1 80 Attorney Docket No.: 116983-5127-WO [0054] In some embodiments, the total serum replacement concentration (vol%) in the serum-free or defined medium is from about 1%, 2%, 3%, 4%, 5%, 6%, 7%, 8%, 9%, 10%, 11%, 12%, 13%, 14%, 15%, 16%, 17%, 18%, 19%, or 20% by volume of the total serum-free or defined medium. In some embodiments, the total serum replacement concentration is about 3% of the total volume of the serum-free or defined medium. In some embodiments, the total serum replacement concentration is about 5% of the total volume of the serum-free or defined medium. In some embodiments, the total serum replacement concentration is about 10% of the total volume of the serum-free or defined medium. [0055] In some embodiments, the serum-free or defined medium is CTS™ OpTmizer™ T- cell Expansion SFM (ThermoFisher Scientific). Any formulation of CTS™ OpTmizer™ is useful in the present invention. CTS™ OpTmizer™ T-cell Expansion SFM is a combination of 1L CTS™ OpTmizer™ T-cell Expansion Basal Medium and 26 mL CTS™ OpTmizer™ T-Cell Expansion Supplement, which are mixed together prior to use. In some embodiments, the CTS™ OpTmizer™ T-cell Expansion SFM is supplemented with about 3% of the CTS™ Immune Cell Serum Replacement (SR) (ThermoFisher Scientific). In some embodiments, the CTS™ OpTmizer™ T-cell Expansion SFM is supplemented with about 3% of the CTS™ Immune Cell Serum Replacement (SR) (ThermoFisher Scientific), along with 2- mercaptoethanol at 55mM. In some embodiments, the CTS™ OpTmizer™ T-cell Expansion SFM is supplemented with about 3% of the CTS™ Immune Cell Serum Replacement (SR) (ThermoFisher Scientific) and the final concentration of 2-mercaptoethanol in the media is 55µM. [0056] In some embodiments, the defined medium is CTS™ OpTmizer™ T-cell Expansion SFM (ThermoFisher Scientific). Any formulation of CTS™ OpTmizer™ is useful in the present invention. CTS™ OpTmizer™ T-cell Expansion SFM is a combination of 1L CTS™ OpTmizer™ T-cell Expansion Basal Medium and 26 mL CTS™ OpTmizer™ T-Cell Expansion Supplement, which are mixed together prior to use. In some embodiments, the CTS™ OpTmizer™ T-cell Expansion SFM is supplemented with about 3% of the CTS™ Immune Cell Serum Replacement (SR) (ThermoFisher Scientific), along with 2- mercaptoethanol at 55mM. In some embodiments, the CTS™OpTmizer™ T-cell Expansion SFM is supplemented with about 3% of the CTS™ Immune Cell Serum Replacement (SR) (ThermoFisher Scientific), 55mM of 2-mercaptoethanol, and 2mM of L-glutamine. In some embodiments, the CTS™OpTmizer™ T-cell Expansion SFM is supplemented with about 3% DB1/ 149202201.1 81 Attorney Docket No.: 116983-5127-WO of the CTS™ Immune Cell Serum Replacement (SR) (ThermoFisher Scientific), 55mM of 2- mercaptoethanol, and 2mM of L-glutamine, and further comprises about 1000 IU/mL to about 8000 IU/mL of IL-2. In some embodiments, the CTS™OpTmizer™ T-cell Expansion SFM is supplemented with about 3% of the CTS™ Immune Cell Serum Replacement (SR) (ThermoFisher Scientific), 55mM of 2-mercaptoethanol, and 2mM of L-glutamine, and further comprises about 3000 IU/mL of IL-2. In some embodiments, the CTS™OpTmizer™ T-cell Expansion SFM is supplemented with about 3% of the CTS™ Immune Cell Serum Replacement (SR) (ThermoFisher Scientific), 55mM of 2-mercaptoethanol, and 2mM of L- glutamine, and further comprises about 6000 IU/mL of IL-2. In some embodiments, the CTS™OpTmizer™ T-cell Expansion SFM is supplemented with about 3% of the CTS™ Immune Cell Serum Replacement (SR) (ThermoFisher Scientific) and 55mM of 2- mercaptoethanol, and further comprises about 1000 IU/mL to about 8000 IU/mL of IL-2. In some embodiments, the CTS™OpTmizer™ T-cell Expansion SFM is supplemented with about 3% of the CTS™ Immune Cell Serum Replacement (SR) (ThermoFisher Scientific) and 55mM of 2-mercaptoethanol, and further comprises about 3000 IU/mL of IL-2. In some embodiments, the CTS™OpTmizer™ T-cell Expansion SFM is supplemented with about 3% of the CTS™ Immune Cell Serum Replacement (SR) (ThermoFisher Scientific) and 55mM of 2-mercaptoethanol, and further comprises about 1000 IU/mL to about 6000 IU/mL of IL-2. In some embodiments, the CTS™OpTmizer™ T-cell Expansion SFM is supplemented with about 3% of the CTS™ Immune Cell Serum Replacement (SR) (ThermoFisher Scientific) and about 2mM glutamine, and further comprises about 1000 IU/mL to about 8000 IU/mL of IL-2. In some embodiments, the CTS™OpTmizer™ T-cell Expansion SFM is supplemented with about 3% of the CTS™ Immune Cell Serum Replacement (SR) (ThermoFisher Scientific) and about 2mM glutamine, and further comprises about 3000 IU/mL of IL-2. In some embodiments, the CTS™OpTmizer™ T-cell Expansion SFM is supplemented with about 3% of the CTS™ Immune Cell Serum Replacement (SR) (ThermoFisher Scientific) and about 2mM glutamine, and further comprises about 6000 IU/mL of IL-2. In some embodiments, the CTS™ OpTmizer™ T-cell Expansion SFM is supplemented with about 3% of the CTS™ Immune Cell Serum Replacement (SR) (ThermoFisher Scientific) and the final concentration of 2-mercaptoethanol in the media is 55µM. [0057] In some embodiments, the serum-free medium or defined medium is supplemented with glutamine (i.e., GlutaMAX®) at a concentration of from about 0.1mM to about 10mM, DB1/ 149202201.1 82 Attorney Docket No.: 116983-5127-WO 0.5mM to about 9mM, 1mM to about 8mM, 2mM to about 7mM, 3mM to about 6mM, or 4mM to about 5 mM. In some embodiments, the serum-free medium or defined medium is supplemented with glutamine (i.e., GlutaMAX®) at a concentration of about 2mM. [0058] In some embodiments, the serum-free medium or defined medium is supplemented with 2-mercaptoethanol at a concentration of from about 5mM to about 150mM, 10mM to about 140mM, 15mM to about 130mM, 20mM to about 120mM, 25mM to about 110mM, 30mM to about 100mM, 35mM to about 95mM, 40mM to about 90mM, 45mM to about 85mM, 50mM to about 80mM, 55mM to about 75mM, 60mM to about 70mM, or about 65mM. In some embodiments, the serum-free medium or defined medium is supplemented with 2-mercaptoethanol at a concentration of about 55mM. In some embodiments, the final concentration of 2-mercaptoethanol in the media is 55µM. [0059] In some embodiments, the defined media described in International PCT Publication No. WO/1998/030679, which is herein incorporated by reference, are useful in the present invention. In that publication, serum-free eukaryotic cell culture media are described. The serum-free, eukaryotic cell culture medium includes a basal cell culture medium supplemented with a serum-free supplement capable of supporting the growth of cells in serum- free culture. The serum-free eukaryotic cell culture medium supplement comprises or is obtained by combining one or more ingredients selected from the group consisting of one or more albumins or albumin substitutes, one or more amino acids, one or more vitamins, one or more transferrins or transferrin substitutes, one or more antioxidants, one or more insulins or insulin substitutes, one or more collagen precursors, one or more trace elements, and one or more antibiotics. In some embodiments, the defined medium further comprises L- glutamine, sodium bicarbonate and/or beta-mercaptoethanol. In some embodiments, the defined medium comprises an albumin or an albumin substitute and one or more ingredients selected from group consisting of one or more amino acids, one or more vitamins, one or more transferrins or transferrin substitutes, one or more antioxidants, one or more insulins or insulin substitutes, one or more collagen precursors, and one or more trace elements. In some embodiments, the defined medium comprises albumin and one or more ingredients selected from the group consisting of glycine, L- histidine, L-isoleucine, L-methionine, L- phenylalanine, L-proline, L- hydroxyproline, L-serine, L-threonine, L-tryptophan, L-tyrosine, L-valine, thiamine, reduced glutathione, L-ascorbic acid-2-phosphate, iron saturated transferrin, insulin, and compounds containing the trace element moieties Ag+, Al3+, Ba2+, DB1/ 149202201.1 83 Attorney Docket No.: 116983-5127-WO Cd2+, Co2+, Cr3+, Ge4+, Se4+, Br, T, Mn2+, P, Si4+, V5+, Mo6+, Ni2+, Rb+, Sn2+ and Zr4+. In some embodiments, the basal cell media is selected from the group consisting of Dulbecco's Modified Eagle's Medium (DMEM), Minimal Essential Medium (MEM), Basal Medium Eagle (BME), RPMI 1640, F-10, F-12, Minimal Essential Medium (αMEM), Glasgow's Minimal Essential Medium (G-MEM), RPMI growth medium, and Iscove's Modified Dulbecco's Medium. [0060] In some embodiments, the concentration of glycine in the defined medium is in the range of from about 5-200 mg/L, the concentration of L- histidine is about 5-250 mg/L, the concentration of L-isoleucine is about 5-300 mg/L, the concentration of L-methionine is about 5-200 mg/L, the concentration of L-phenylalanine is about 5-400 mg/L, the concentration of L-proline is about 1-1000 mg/L, the concentration of L- hydroxyproline is about 1-45 mg/L, the concentration of L-serine is about 1-250 mg/L, the concentration of L- threonine is about 10-500 mg/L, the concentration of L-tryptophan is about 2-110 mg/L, the concentration of L-tyrosine is about 3-175 mg/L, the concentration of L-valine is about 5-500 mg/L, the concentration of thiamine is about 1-20 mg/L, the concentration of reduced glutathione is about 1-20 mg/L, the concentration of L-ascorbic acid-2-phosphate is about 1- 200 mg/L, the concentration of iron saturated transferrin is about 1-50 mg/L, the concentration of insulin is about 1-100 mg/L, the concentration of sodium selenite is about 0.000001-0.0001 mg/L, and the concentration of albumin (e.g., AlbuMAX® I) is about 5000- 50,000 mg/L. [0061] In some embodiments, the non-trace element moiety ingredients in the defined medium are present in the concentration ranges listed in the column under the heading “Concentration Range in 1X Medium” in Table 4 below. In other embodiments, the non-trace element moiety ingredients in the defined medium are present in the final concentrations listed in the column under the heading “A Preferred Embodiment of the 1X Medium” in Table 4. In other embodiments, the defined medium is a basal cell medium comprising a serum free supplement. In some of these embodiments, the serum free supplement comprises non-trace moiety ingredients of the type and in the concentrations listed in the column under the heading “A Preferred Embodiment in Supplement” in Table 4 below. TABLE 4: Concentrations of Non-Trace Element Moiety Ingredients DB1/ 149202201.1 84 Attorney Docket No.: 116983-5127-WO

[0062] In some embodiments, the osmolarity of the defined medium is between about 260 and 350 mOsmol. In some embodiments, the osmolarity is between about 280 and 310 mOsmol. In some embodiments, the defined medium is supplemented with up to about 3.7 g/L, or about 2.2 g/L sodium bicarbonate. The defined medium can be further supplemented with L-glutamine (final concentration of about 2 mM), one or more antibiotics, non-essential amino acids (NEAA; final concentration of about 100 μM), 2-mercaptoethanol (final concentration of about 100 μM). [0063] In some embodiments, the defined media described in Smith, et al., Clin Transl Immunology, 4(1) 2015 (doi: 10.1038/cti.2014.31) are useful in the present invention. Briefly, RPMI or CTS™ OpTmizer™ was used as the basal cell medium, and supplemented with either 0, 2%, 5%, or 10% CTS™ Immune Cell Serum Replacement. [0064] In some embodiments, the cell medium in the first and/or second gas permeable container is unfiltered. The use of unfiltered cell medium may simplify the procedures necessary to expand the number of cells. In some embodiments, the cell medium in the first DB1/ 149202201.1 85 Attorney Docket No.: 116983-5127-WO and/or second gas permeable container lacks beta-mercaptoethanol (BME or βME; also known as 2-mercaptoethanol, CAS 60-24-2). [0065] After preparation of the tumor fragments, the resulting cells (i.e., fragments) are cultured in serum containing IL-2 under conditions that favor the growth of TILs over tumor and other cells. In some embodiments, the tumor digests are incubated in 2 mL wells in media comprising inactivated human AB serum (or, in some cases, as outlined herein, in the presence of an APC cell population) with 6000 IU/mL of IL-2. This primary cell population is cultured for a period of days, generally from 10 to 14 days, resulting in a bulk TIL population, generally about 1×10
8 bulk TIL cells. In some embodiments, the growth media during the first expansion comprises IL-2 or a variant thereof. In some embodiments, the IL is recombinant human IL-2 (rhIL-2). In some embodiments the IL-2 stock solution has a specific activity of 20-30×10
6 IU/mg for a 1 mg vial. In some embodiments the IL-2 stock solution has a specific activity of 20×10
6 IU/mg for a 1 mg vial. In some embodiments the IL-2 stock solution has a specific activity of 25×10
6 IU/mg for a 1 mg vial. In some embodiments the IL-2 stock solution has a specific activity of 30×10
6 IU/mg for a 1 mg vial. In some embodiments, the IL- 2 stock solution has a final concentration of 4-8×10
6 IU/mg of IL-2. In some embodiments, the IL- 2 stock solution has a final concentration of 5-7×10
6 IU/mg of IL-2. In some embodiments, the IL- 2 stock solution has a final concentration of 6×10
6 IU/mg of IL-2. In some embodiments, the IL-2 stock solution is prepare as described in Example 5. In some embodiments, the first expansion culture media comprises about 10,000 IU/mL of IL-2, about 9,000 IU/mL of IL-2, about 8,000 IU/mL of IL-2, about 7,000 IU/mL of IL-2, about 6000 IU/mL of IL-2 or about 5,000 IU/mL of IL-2. In some embodiments, the first expansion culture media comprises about 9,000 IU/mL of IL-2 to about 5,000 IU/mL of IL-2. In some embodiments, the first expansion culture media comprises about 8,000 IU/mL of IL-2 to about 6,000 IU/mL of IL-2. In some embodiments, the first expansion culture media comprises about 7,000 IU/mL of IL-2 to about 6,000 IU/mL of IL-2. In some embodiments, the first expansion culture media comprises about 6,000 IU/mL of IL-2. In some embodiments, the cell culture medium further comprises IL-2. In some embodiments, the cell culture medium comprises about 3000 IU/mL of IL-2. In some embodiments, the cell culture medium further comprises IL-2. In some embodiments, the cell culture medium comprises about 3000 IU/mL of IL-2. In some embodiments, the cell culture medium comprises about 1000 IU/mL, about 1500 IU/mL, about 2000 IU/mL, about 2500 DB1/ 149202201.1 86 Attorney Docket No.: 116983-5127-WO IU/mL, about 3000 IU/mL, about 3500 IU/mL, about 4000 IU/mL, about 4500 IU/mL, about 5000 IU/mL, about 5500 IU/mL, about 6000 IU/mL, about 6500 IU/mL, about 7000 IU/mL, about 7500 IU/mL, or about 8000 IU/mL of IL-2. In some embodiments, the cell culture medium comprises between 1000 and 2000 IU/mL, between 2000 and 3000 IU/mL, between 3000 and 4000 IU/mL, between 4000 and 5000 IU/mL, between 5000 and 6000 IU/mL, between 6000 and 7000 IU/mL, between 7000 and 8000 IU/mL, or about 8000 IU/mL of IL- 2. [0066] In some embodiments, first expansion culture media comprises about 500 IU/mL of IL-15, about 400 IU/mL of IL-15, about 300 IU/mL of IL-15, about 200 IU/mL of IL-15, about 180 IU/mL of IL-15, about 160 IU/mL of IL-15, about 140 IU/mL of IL-15, about 120 IU/mL of IL-15, or about 100 IU/mL of IL-15. In some embodiments, the first expansion culture media comprises about 500 IU/mL of IL-15 to about 100 IU/mL of IL-15. In some embodiments, the first expansion culture media comprises about 400 IU/mL of IL-15 to about 100 IU/mL of IL-15. In some embodiments, the first expansion culture media comprises about 300 IU/mL of IL-15 to about 100 IU/mL of IL-15. In some embodiments, the first expansion culture media comprises about 200 IU/mL of IL-15. In some embodiments, the cell culture medium comprises about 180 IU/mL of IL-15. In some embodiments, the cell culture medium further comprises IL-15. In some embodiments, the cell culture medium comprises about 180 IU/mL of IL-15. [0067] In some embodiments, first expansion culture media comprises about 20 IU/mL of IL-21, about 15 IU/mL of IL-21, about 12 IU/mL of IL-21, about 10 IU/mL of IL-21, about 5 IU/mL of IL-21, about 4 IU/mL of IL-21, about 3 IU/mL of IL-21, about 2 IU/mL of IL-21, about 1 IU/mL of IL-21, or about 0.5 IU/mL of IL-21. In some embodiments, the first expansion culture media comprises about 20 IU/mL of IL-21 to about 0.5 IU/mL of IL-21. In some embodiments, the first expansion culture media comprises about 15 IU/mL of IL-21 to about 0.5 IU/mL of IL-21. In some embodiments, the first expansion culture media comprises about 12 IU/mL of IL-21 to about 0.5 IU/mL of IL-21. In some embodiments, the first expansion culture media comprises about 10 IU/mL of IL-21 to about 0.5 IU/mL of IL-21. In some embodiments, the first expansion culture media comprises about 5 IU/mL of IL-21 to about 1 IU/mL of IL-21. In some embodiments, the first expansion culture media comprises about 2 IU/mL of IL-21. In some embodiments, the cell culture medium comprises about 1 IU/mL of IL-21. In some embodiments, the cell culture medium comprises about 0.5 IU/mL DB1/ 149202201.1 87 Attorney Docket No.: 116983-5127-WO of IL-21. In some embodiments, the cell culture medium further comprises IL-21. In some embodiments, the cell culture medium comprises about 1 IU/mL of IL-21. [0068] In some embodiments, the cell culture medium comprises an anti-CD3 agonist antibody, e.g. OKT-3 antibody. In some embodiments, the cell culture medium comprises about 30 ng/mL of OKT-3 antibody. In some embodiments, the cell culture medium comprises about 0.1 ng/mL, about 0.5 ng/mL, about 1 ng/mL, about 2.5 ng/mL, about 5 ng/mL, about 7.5 ng/mL, about 10 ng/mL, about 15 ng/mL, about 20 ng/mL, about 25 ng/mL, about 30 ng/mL, about 35 ng/mL, about 40 ng/mL, about 50 ng/mL, about 60 ng/mL, about 70 ng/mL, about 80 ng/mL, about 90 ng/mL, about 100 ng/mL, about 200 ng/mL, about 500 ng/mL, and about 1 µg/mL of OKT-3 antibody. In some embodiments, the cell culture medium comprises between 0.1 ng/mL and 1 ng/mL, between 1 ng/mL and 5 ng/mL, between 5 ng/mL and 10 ng/mL, between 10 ng/mL and 20 ng/mL, between 20 ng/mL and 30 ng/mL, between 30 ng/mL and 40 ng/mL, between 40 ng/mL and 50 ng/mL, and between 50 ng/mL and 100 ng/mL of OKT-3 antibody. In some embodiments, the cell culture medium does not comprise OKT-3 antibody. In some embodiments, the OKT-3 antibody is muromonab. See, for example, Table 1. [0069] In some embodiments, the cell culture medium comprises one or more TNFRSF agonists in a cell culture medium. In some embodiments, the TNFRSF agonist comprises a 4- 1BB agonist. In some embodiments, the TNFRSF agonist is a 4-1BB agonist, and the 4-1BB agonist is selected from the group consisting of urelumab, utomilumab, EU-101, a fusion protein, and fragments, derivatives, variants, biosimilars, and combinations thereof. In some embodiments, the TNFRSF agonist is added at a concentration sufficient to achieve a concentration in the cell culture medium of between 0.1 µg/mL and 100 µg/mL. In some embodiments, the TNFRSF agonist is added at a concentration sufficient to achieve a concentration in the cell culture medium of between 20 µg/mL and 40 µg/mL. [0070] In some embodiments, in addition to one or more TNFRSF agonists, the cell culture medium further comprises IL-2 at an initial concentration of about 3000 IU/mL and OKT-3 antibody at an initial concentration of about 30 ng/mL, and wherein the one or more TNFRSF agonists comprises a 4-1BB agonist. [0071] In some embodiments, the first expansion culture medium is referred to as “CM”, an abbreviation for culture media. In some embodiments, it is referred to as CM1 (culture medium 1). In some embodiments, CM consists of RPMI 1640 with GlutaMAX, DB1/ 149202201.1 88 Attorney Docket No.: 116983-5127-WO supplemented with 10% human AB serum, 25 mM Hepes, and 10 mg/mL gentamicin. In embodiments where cultures are initiated in gas-permeable flasks with a 40 mL capacity and a 10cm
2 gas-permeable silicon bottom (for example, G-REX10; Wilson Wolf Manufacturing, New Brighton, MN), each flask was loaded with 10–40x10
6 viable tumor digest cells or 5–30 tumor fragments in 10–40mL of CM with IL-2. Both the G-REX10 and 24-well plates were incubated in a humidified incubator at 37°C in 5% CO2 and 5 days after culture initiation, half the media was removed and replaced with fresh CM and IL-2 and after day 5, half the media was changed every 2–3 days. In some embodiments, the CM is the CM1 described in the Examples, see, Example 1. In some embodiments, the first expansion occurs in an initial cell culture medium or a first cell culture medium. In some embodiments, the initial cell culture medium or the first cell culture medium comprises IL-2. [0072] In some embodiments, the first expansion (including processes such as for example those described in Step B of Figure 1, which can include those sometimes referred to as the pre-REP) process is shortened to 3-14 days, as discussed in the examples and figures. In some embodiments, the first expansion (including processes such as for example those described in Step B of Figure 1, which can include those sometimes referred to as the pre- REP) is shortened to 7 to 14 days, as discussed in the Examples and shown in Figures 4 and 5, as well as including for example, an expansion as described in Step B of Figure 1. In some embodiments, the first expansion of Step B is shortened to 10-14 days. In some embodiments, the first expansion is shortened to 11 days, as discussed in, for example, an expansion as described in Step B of Figure 1. [0073] In some embodiments, the first TIL expansion can proceed for 1 day, 2 days, 3 days, 4 days, 5 days, 6 days, 7 days, 8 days, 9 days, 10 days, 11 days, 12 days, 13 days, or 14 days. In some embodiments, the first TIL expansion can proceed for 1 day to 14 days. In some embodiments, the first TIL expansion can proceed for 2 days to 14 days. In some embodiments, the first TIL expansion can proceed for 3 days to 14 days. In some embodiments, the first TIL expansion can proceed for 4 days to 14 days. In some embodiments, the first TIL expansion can proceed for 5 days to 14 days. In some embodiments, the first TIL expansion can proceed for 6 days to 14 days. In some embodiments, the first TIL expansion can proceed for 7 days to 14 days. In some embodiments, the first TIL expansion can proceed for 8 days to 14 days. In some embodiments, the first TIL expansion can proceed for 9 days to 14 days. In some DB1/ 149202201.1 89 Attorney Docket No.: 116983-5127-WO embodiments, the first TIL expansion can proceed for 10 days to 14 days. In some embodiments, the first TIL expansion can proceed for 11 days to 14 days. In some embodiments, the first TIL expansion can proceed for 12 days to 14 days. In some embodiments, the first TIL expansion can proceed for 13 days to 14 days. In some embodiments, the first TIL expansion can proceed for 14 days. In some embodiments, the first TIL expansion can proceed for 1 day to 11 days. In some embodiments, the first TIL expansion can proceed for 2 days to 11 days. In some embodiments, the first TIL expansion can proceed for 3 days to 11 days. In some embodiments, the first TIL expansion can proceed for 4 days to 11 days. In some embodiments, the first TIL expansion can proceed for 5 days to 11 days. In some embodiments, the first TIL expansion can proceed for 6 days to 11 days. In some embodiments, the first TIL expansion can proceed for 7 days to 11 days. In some embodiments, the first TIL expansion can proceed for 8 days to 11 days. In some embodiments, the first TIL expansion can proceed for 9 days to 11 days. In some embodiments, the first TIL expansion can proceed for 10 days to 11 days. In some embodiments, the first TIL expansion can proceed for 11 days. [0074] In some embodiments, a combination of IL-2, IL-7, IL-15, and/or IL-21 are employed as a combination during the first expansion. In some embodiments, IL-2, IL-7, IL- 15, and/or IL-21 as well as any combinations thereof can be included during the first expansion, including for example during a Step B processes according to Figure 1, as well as described herein. In some embodiments, a combination of IL-2, IL-15, and IL-21 are employed as a combination during the first expansion. In some embodiments, IL-2, IL-15, and IL-21 as well as any combinations thereof can be included during Step B processes according to Figure 1 and as described herein. [0075] In some embodiments, the first expansion (including processes referred to as the pre-REP; for example, Step B according to Figure 1) process is shortened to 3 to 14 days, as discussed in the examples and figures. In some embodiments, the first expansion of Step B is shortened to 7 to 14 days. In some embodiments, the first expansion of Step B is shortened to 10 to 14 days. In some embodiments, the first expansion is shortened to 11 days. [0076] In some embodiments, the first expansion, for example, Step B according to Figure 1, is performed in a closed system bioreactor. In some embodiments, a closed system is employed for the TIL expansion, as described herein. In some embodiments, a single bioreactor is employed. In some embodiments, the single bioreactor employed is for example DB1/ 149202201.1 90 Attorney Docket No.: 116983-5127-WO a G-REX-10 or a G-REX-100. In some embodiments, the closed system bioreactor is a single bioreactor. 1. Cytokines and Other Additives [0077] The expansion methods described herein generally use culture media with high doses of a cytokine, in particular IL-2, as is known in the art. [0078] Alternatively, using combinations of cytokines for the rapid expansion and or second expansion of TILs is additionally possible, with combinations of two or more of IL-2, IL-15 and IL-21 as is described in U.S. Patent Application Publication No. US 2017/0107490 A1, the disclosure of which is incorporated by reference herein. Thus, possible combinations include IL-2 and IL-15, IL-2 and IL-21, IL-15 and IL-21 and IL-2, or IL-15 and IL-21, with the latter finding particular use in many embodiments. The use of combinations of cytokines specifically favors the generation of lymphocytes, and in particular T-cells as described therein. [0079] In some embodiments, Step B may also include the addition of OKT-3 antibody or muromonab to the culture media, as described elsewhere herein. In some embodiments, Step B may also include the addition of a 4-1BB agonist to the culture media, as described elsewhere herein. In some embodiments, Step B may also include the addition of an OX-40 agonist to the culture media, as described elsewhere herein. In other embodiments, additives such as peroxisome proliferator-activated receptor gamma coactivator I-alpha agonists, including proliferator-activated receptor (PPAR)-gamma agonists such as a thiazolidinedione compound, may be used in the culture media during Step B, as described in U.S. Patent Application Publication No. US 2019/0307796 A1, the disclosure of which is incorporated by reference herein. C. STEP C: First Expansion to Second Expansion Transition [0080] In some cases, the bulk TIL population obtained from the first expansion, including for example the TIL population obtained from for example, Step B as indicated in Figure 1, can be cryopreserved immediately, using the protocols discussed herein below. Alternatively, the TIL population obtained from the first expansion, referred to as the second TIL population, can be subjected to a second expansion (which can include expansions sometimes referred to as REP) and then cryopreserved as discussed below. Similarly, in the case where DB1/ 149202201.1 91 Attorney Docket No.: 116983-5127-WO genetically modified TILs will be used in therapy, the first TIL population (sometimes referred to as the bulk TIL population) or the second TIL population (which can in some embodiments include populations referred to as the REP TIL populations) can be subjected to genetic modifications for suitable treatments prior to expansion or after the first expansion and prior to the second expansion. [0081] In some embodiments, the TILs obtained from the first expansion (for example, from Step B as indicated in Figure 1) are stored until phenotyped for selection. In some embodiments, the TILs obtained from the first expansion (for example, from Step B as indicated in Figure 1) are not stored and proceed directly to the second expansion. In some embodiments, the TILs obtained from the first expansion are not cryopreserved after the first expansion and prior to the second expansion. In some embodiments, the transition from the first expansion to the second expansion occurs at about 3 days, 4, days, 5 days, 6 days, 7 days, 8 days, 9 days, 10 days, 11 days, 12 days, 13 days, or 14 days from when fragmentation occurs. In some embodiments, the transition from the first expansion to the second expansion occurs at about 3 days to 14 days from when fragmentation occurs. In some embodiments, the transition from the first expansion to the second expansion occurs at about 4 days to 14 days from when fragmentation occurs. In some embodiments, the transition from the first expansion to the second expansion occurs at about 4 days to 10 days from when fragmentation occurs. In some embodiments, the transition from the first expansion to the second expansion occurs at about 7 days to 14 days from when fragmentation occurs. In some embodiments, the transition from the first expansion to the second expansion occurs at about 14 days from when fragmentation occurs. [0082] In some embodiments, the transition from the first expansion to the second expansion occurs at 1 day, 2 days, 3 days, 4 days, 5 days, 6 days, 7 days, 8 days, 9 days, 10 days, 11 days, 12 days, 13 days, or 14 days from when fragmentation occurs. In some embodiments, the transition from the first expansion to the second expansion occurs 1 day to 14 days from when fragmentation occurs. In some embodiments, the first TIL expansion can proceed for 2 days to 14 days. In some embodiments, the transition from the first expansion to the second expansion occurs 3 days to 14 days from when fragmentation occurs. In some embodiments, the transition from the first expansion to the second expansion occurs 4 days to 14 days from when fragmentation occurs. In some embodiments, the transition from the first expansion to the second expansion occurs 5 days to 14 days from when fragmentation occurs. DB1/ 149202201.1 92 Attorney Docket No.: 116983-5127-WO In some embodiments, the transition from the first expansion to the second expansion occurs 6 days to 14 days from when fragmentation occurs. In some embodiments, the transition from the first expansion to the second expansion occurs 7 days to 14 days from when fragmentation occurs. In some embodiments, the transition from the first expansion to the second expansion occurs 8 days to 14 days from when fragmentation occurs. In some embodiments, the transition from the first expansion to the second expansion occurs 9 days to 14 days from when fragmentation occurs. In some embodiments, the transition from the first expansion to the second expansion occurs 10 days to 14 days from when fragmentation occurs. In some embodiments, the transition from the first expansion to the second expansion occurs 11 days to 14 days from when fragmentation occurs. In some embodiments, the transition from the first expansion to the second expansion occurs 12 days to 14 days from when fragmentation occurs. In some embodiments, the transition from the first expansion to the second expansion occurs 13 days to 14 days from when fragmentation occurs. In some embodiments, the transition from the first expansion to the second expansion occurs 14 days from when fragmentation occurs. In some embodiments, the transition from the first expansion to the second expansion occurs 1 day to 11 days from when fragmentation occurs. In some embodiments, the transition from the first expansion to the second expansion occurs 2 days to 11 days from when fragmentation occurs. In some embodiments, the transition from the first expansion to the second expansion occurs 3 days to 11 days from when fragmentation occurs. In some embodiments, the transition from the first expansion to the second expansion occurs 4 days to 11 days from when fragmentation occurs. In some embodiments, the transition from the first expansion to the second expansion occurs 5 days to 11 days from when fragmentation occurs. In some embodiments, the transition from the first expansion to the second expansion occurs 6 days to 11 days from when fragmentation occurs. In some embodiments, the transition from the first expansion to the second expansion occurs 7 days to 11 days from when fragmentation occurs. In some embodiments, the transition from the first expansion to the second expansion occurs 8 days to 11 days from when fragmentation occurs. In some embodiments, the transition from the first expansion to the second expansion occurs 9 days to 11 days from when fragmentation occurs. In some embodiments, the transition from the first expansion to the second expansion occurs 10 days to 11 days from when fragmentation occurs. In some embodiments, the transition from the first expansion to the second expansion occurs 11 days from when fragmentation occurs. DB1/ 149202201.1 93 Attorney Docket No.: 116983-5127-WO [0083] In some embodiments, the TILs are not stored after the first expansion and prior to the second expansion, and the TILs proceed directly to the second expansion (for example, in some embodiments, there is no storage during the transition from Step B to Step D as shown in Figure 1). In some embodiments, the transition occurs in closed system, as described herein. In some embodiments, the TILs from the first expansion, the second population of TILs, proceeds directly into the second expansion with no transition period. [0084] In some embodiments, the transition from the first expansion to the second expansion, for example, Step C according to Figure 1, is performed in a closed system bioreactor. In some embodiments, a closed system is employed for the TIL expansion, as described herein. In some embodiments, a single bioreactor is employed. In some embodiments, the single bioreactor employed is for example a G-REX-10 or a G-REX-100 bioreactor. In some embodiments, the closed system bioreactor is a single bioreactor. D. STEP D: Second Expansion [0085] In some embodiments, the TIL cell population is expanded in number after harvest and initial bulk processing for example, after Step A and Step B, and the transition referred to as Step C, as indicated in Figure 1). This further expansion is referred to herein as the second expansion, which can include expansion processes generally referred to in the art as a rapid expansion process (REP); as well as processes as indicated in Step D of Figure 1. The second expansion is generally accomplished using a culture media comprising a number of components, including feeder cells, a cytokine source, and an anti-CD3 antibody, in a gas- permeable container. [0086] In some embodiments, the second expansion or second TIL expansion (which can include expansions sometimes referred to as REP; as well as processes as indicated in Step D of Figure 1) of TIL can be performed using any TIL flasks or containers known by those of skill in the art. In some embodiments, the second TIL expansion can proceed for 7 days, 8 days, 9 days, 10 days, 11 days, 12 days, 13 days, or 14 days. In some embodiments, the second TIL expansion can proceed for about 7 days to about 14 days. In some embodiments, the second TIL expansion can proceed for about 8 days to about 14 days. In some embodiments, the second TIL expansion can proceed for about 9 days to about 14 days. In some embodiments, the second TIL expansion can proceed for about 10 days to about 14 days. In some embodiments, the second TIL expansion can proceed for about 11 days to DB1/ 149202201.1 94 Attorney Docket No.: 116983-5127-WO about 14 days. In some embodiments, the second TIL expansion can proceed for about 12 days to about 14 days. In some embodiments, the second TIL expansion can proceed for about 13 days to about 14 days. In some embodiments, the second TIL expansion can proceed for about 14 days. [0087] In some embodiments, the second expansion can be performed in a gas permeable container using the methods of the present disclosure (including for example, expansions referred to as REP; as well as processes as indicated in Step D of Figure 1). For example, TILs can be rapidly expanded using non-specific T-cell receptor stimulation in the presence of interleukin-2 (IL-2) or interleukin-15 (IL-15). The non-specific T-cell receptor stimulus can include, for example, an anti-CD3 antibody, such as about 30 ng/mL of OKT3, a mouse monoclonal anti-CD3 antibody (commercially available from Ortho-McNeil, Raritan, NJ or Miltenyi Biotech, Auburn, CA) or UHCT-1 (commercially available from BioLegend, San Diego, CA, USA). TILs can be expanded to induce further stimulation of the TILs in vitro by including one or more antigens during the second expansion, including antigenic portions thereof, such as epitope(s), of the cancer, which can be optionally expressed from a vector, such as a human leukocyte antigen A2 (HLA-A2) binding peptide, e.g., 0.3 μΜ MART-1 :26- 35 (27 L) or gpl 00:209-217 (210M), optionally in the presence of a T-cell growth factor, such as 300 IU/mL IL-2 or IL-15. Other suitable antigens may include, e.g., NY-ESO-1, TRP-1, TRP-2, tyrosinase cancer antigen, MAGE-A3, SSX-2, and VEGFR2, or antigenic portions thereof. TIL may also be rapidly expanded by re-stimulation with the same antigen(s) of the cancer pulsed onto HLA-A2-expressing antigen-presenting cells. Alternatively, the TILs can be further re-stimulated with, e.g., example, irradiated, autologous lymphocytes or with irradiated HLA-A2+ allogeneic lymphocytes and IL-2. In some embodiments, the re-stimulation occurs as part of the second expansion. In some embodiments, the second expansion occurs in the presence of irradiated, autologous lymphocytes or with irradiated HLA-A2+ allogeneic lymphocytes and IL-2. [0088] In some embodiments, the cell culture medium further comprises IL-2. In some embodiments, the cell culture medium comprises about 3000 IU/mL of IL-2. In some embodiments, the cell culture medium comprises about 1000 IU/mL, about 1500 IU/mL, about 2000 IU/mL, about 2500 IU/mL, about 3000 IU/mL, about 3500 IU/mL, about 4000 IU/mL, about 4500 IU/mL, about 5000 IU/mL, about 5500 IU/mL, about 6000 IU/mL, about 6500 IU/mL, about 7000 IU/mL, about 7500 IU/mL, or about 8000 IU/mL of IL-2. In some DB1/ 149202201.1 95 Attorney Docket No.: 116983-5127-WO embodiments, the cell culture medium comprises between 1000 and 2000 IU/mL, between 2000 and 3000 IU/mL, between 3000 and 4000 IU/mL, between 4000 and 5000 IU/mL, between 5000 and 6000 IU/mL, between 6000 and 7000 IU/mL, between 7000 and 8000 IU/mL, or between 8000 IU/mL of IL-2. [0089] In some embodiments, the cell culture medium comprises OKT-3 antibody. In some embodiments, the cell culture medium comprises about 30 ng/mL of OKT-3 antibody. In some embodiments, the cell culture medium comprises about 0.1 ng/mL, about 0.5 ng/mL, about 1 ng/mL, about 2.5 ng/mL, about 5 ng/mL, about 7.5 ng/mL, about 10 ng/mL, about 15 ng/mL, about 20 ng/mL, about 25 ng/mL, about 30 ng/mL, about 35 ng/mL, about 40 ng/mL, about 50 ng/mL, about 60 ng/mL, about 70 ng/mL, about 80 ng/mL, about 90 ng/mL, about 100 ng/mL, about 200 ng/mL, about 500 ng/mL, and about 1 µg/mL of OKT-3 antibody. In some embodiments, the cell culture medium comprises between 0.1 ng/mL and 1 ng/mL, between 1 ng/mL and 5 ng/mL, between 5 ng/mL and 10 ng/mL, between 10 ng/mL and 20 ng/mL, between 20 ng/mL and 30 ng/mL, between 30 ng/mL and 40 ng/mL, between 40 ng/mL and 50 ng/mL, and between 50 ng/mL and 100 ng/mL of OKT-3 antibody. In some embodiments, the cell culture medium does not comprise OKT-3 antibody. In some embodiments, the OKT-3 antibody is muromonab. [0090] In some embodiments, the cell culture medium comprises one or more TNFRSF agonists in a cell culture medium. In some embodiments, the TNFRSF agonist comprises a 4- 1BB agonist. In some embodiments, the TNFRSF agonist is a 4-1BB agonist, and the 4-1BB agonist is selected from the group consisting of urelumab, utomilumab, EU-101, a fusion protein, and fragments, derivatives, variants, biosimilars, and combinations thereof. In some embodiments, the TNFRSF agonist is added at a concentration sufficient to achieve a concentration in the cell culture medium of between 0.1 µg/mL and 100 µg/mL. In some embodiments, the TNFRSF agonist is added at a concentration sufficient to achieve a concentration in the cell culture medium of between 20 µg/mL and 40 µg/mL. [0091] In some embodiments, in addition to one or more TNFRSF agonists, the cell culture medium further comprises IL-2 at an initial concentration of about 3000 IU/mL and OKT-3 antibody at an initial concentration of about 30 ng/mL, and wherein the one or more TNFRSF agonists comprises a 4-1BB agonist. [0092] In some embodiments, a combination of IL-2, IL-7, IL-15, and/or IL-21 are employed as a combination during the second expansion. In some embodiments, IL-2, IL-7, DB1/ 149202201.1 96 Attorney Docket No.: 116983-5127-WO IL-15, and/or IL-21 as well as any combinations thereof can be included during the second expansion, including for example during a Step D processes according to Figure 1, as well as described herein. In some embodiments, a combination of IL-2, IL-15, and IL-21 are employed as a combination during the second expansion. In some embodiments, IL-2, IL-15, and IL-21 as well as any combinations thereof can be included during Step D processes according to Figure 1 and as described herein. [0093] In some embodiments, the second expansion can be conducted in a supplemented cell culture medium comprising IL-2, OKT-3, antigen-presenting feeder cells, and optionally a TNFRSF agonist. In some embodiments, the second expansion occurs in a supplemented cell culture medium. In some embodiments, the supplemented cell culture medium comprises IL-2, OKT-3, and antigen-presenting feeder cells. In some embodiments, the second cell culture medium comprises IL-2, OKT-3, and antigen-presenting cells (APCs; also referred to as antigen-presenting feeder cells). In some embodiments, the second expansion occurs in a cell culture medium comprising IL-2, OKT-3, and antigen-presenting feeder cells (i.e., antigen presenting cells). [0094] In some embodiments, the second expansion culture media comprises about 500 IU/mL of IL-15, about 400 IU/mL of IL-15, about 300 IU/mL of IL-15, about 200 IU/mL of IL-15, about 180 IU/mL of IL-15, about 160 IU/mL of IL-15, about 140 IU/mL of IL-15, about 120 IU/mL of IL-15, or about 100 IU/mL of IL-15. In some embodiments, the second expansion culture media comprises about 500 IU/mL of IL-15 to about 100 IU/mL of IL-15. In some embodiments, the second expansion culture media comprises about 400 IU/mL of IL-15 to about 100 IU/mL of IL-15. In some embodiments, the second expansion culture media comprises about 300 IU/mL of IL-15 to about 100 IU/mL of IL-15. In some embodiments, the second expansion culture media comprises about 200 IU/mL of IL-15. In some embodiments, the cell culture medium comprises about 180 IU/mL of IL-15. In some embodiments, the cell culture medium further comprises IL-15. In some embodiments, the cell culture medium comprises about 180 IU/mL of IL-15. [0095] In some embodiments, the second expansion culture media comprises about 20 IU/mL of IL-21, about 15 IU/mL of IL-21, about 12 IU/mL of IL-21, about 10 IU/mL of IL- 21, about 5 IU/mL of IL-21, about 4 IU/mL of IL-21, about 3 IU/mL of IL-21, about 2 IU/mL of IL-21, about 1 IU/mL of IL-21, or about 0.5 IU/mL of IL-21. In some embodiments, the second expansion culture media comprises about 20 IU/mL of IL-21 to about 0.5 IU/mL of DB1/ 149202201.1 97 Attorney Docket No.: 116983-5127-WO IL-21. In some embodiments, the second expansion culture media comprises about 15 IU/mL of IL-21 to about 0.5 IU/mL of IL-21. In some embodiments, the second expansion culture media comprises about 12 IU/mL of IL-21 to about 0.5 IU/mL of IL-21. In some embodiments, the second expansion culture media comprises about 10 IU/mL of IL-21 to about 0.5 IU/mL of IL-21. In some embodiments, the second expansion culture media comprises about 5 IU/mL of IL-21 to about 1 IU/mL of IL-21. In some embodiments, the second expansion culture media comprises about 2 IU/mL of IL-21. In some embodiments, the cell culture medium comprises about 1 IU/mL of IL-21. In some embodiments, the cell culture medium comprises about 0.5 IU/mL of IL-21. In some embodiments, the cell culture medium further comprises IL-21. In some embodiments, the cell culture medium comprises about 1 IU/mL of IL-21. [0096] In some embodiments the antigen-presenting feeder cells (APCs) are PBMCs. In some embodiments, the ratio of TILs to PBMCs and/or antigen-presenting cells in the rapid expansion and/or the second expansion is about 1 to 25, about 1 to 50, about 1 to 100, about 1 to 125, about 1 to 150, about 1 to 175, about 1 to 200, about 1 to 225, about 1 to 250, about 1 to 275, about 1 to 300, about 1 to 325, about 1 to 350, about 1 to 375, about 1 to 400, or about 1 to 500. In some embodiments, the ratio of TILs to PBMCs in the rapid expansion and/or the second expansion is between 1 to 50 and 1 to 300. In some embodiments, the ratio of TILs to PBMCs in the rapid expansion and/or the second expansion is between 1 to 100 and 1 to 200. [0097] In some embodiments, REP and/or the second expansion is performed in flasks with the bulk TILs being mixed with a 100- or 200-fold excess of inactivated feeder cells, 30 mg/mL OKT3 anti-CD3 antibody and 3000 IU/mL IL-2 in 150 mL media. Media replacement is done (generally 2/3 media replacement via respiration with fresh media) until the cells are transferred to an alternative growth chamber. Alternative growth chambers include G-REX flasks and gas permeable containers as more fully discussed below. [0098] In some embodiments, the second expansion (which can include processes referred to as the REP process) is shortened to 7-14 days, as discussed in the examples and figures. In some embodiments, the second expansion is shortened to 11 days. [0099] In some embodiments, REP and/or the second expansion may be performed using T- 175 flasks and gas permeable bags as previously described (Tran, et al., J. Immunother.2008, 31, 742-51; Dudley, et al., J. Immunother.2003, 26, 332-42) or gas permeable cultureware (G-REX flasks). In some embodiments, the second expansion (including expansions referred DB1/ 149202201.1 98 Attorney Docket No.: 116983-5127-WO to as rapid expansions) is performed in T-175 flasks, and about 1 x 10
6 TILs suspended in 150 mL of media may be added to each T-175 flask. The TILs may be cultured in a 1 to 1 mixture of CM and AIM-V medium, supplemented with 3000 IU per mL of IL-2 and 30 ng per mL of anti-CD3. The T-175 flasks may be incubated at 37° C in 5% CO
2. Half the media may be exchanged on day 5 using 50/50 medium with 3000 IU per mL of IL-2. In some embodiments, on day 7 cells from two T-175 flasks may be combined in a 3 L bag and 300 mL of AIM V with 5% human AB serum and 3000 IU per mL of IL-2 was added to the 300 mL of TIL suspension. The number of cells in each bag was counted every day or two and fresh media was added to keep the cell count between 0.5 and 2.0 x 10
6 cells/mL. [00100] In some embodiments, the second expansion (which can include expansions referred to as REP, as well as those referred to in Step D of Figure 1) may be performed in 500 mL capacity gas permeable flasks with 100 cm gas-permeable silicon bottoms (G-REX-100, commercially available from Wilson Wolf Manufacturing Corporation, New Brighton, MN, USA), 5 × 10
6 or 10 × 10
6 TIL may be cultured with PBMCs in 400 mL of 50/50 medium, supplemented with 5% human AB serum, 3000 IU per mL of IL-2 and 30 ng per mL of anti- CD3 (OKT3). The G-REX-100 flasks may be incubated at 37°C in 5% CO2. On day 5, 250 mL of supernatant may be removed and placed into centrifuge bottles and centrifuged at 1500 rpm (491 × g) for 10 minutes. The TIL pellets may be re-suspended with 150 mL of fresh medium with 5% human AB serum, 3000 IU per mL of IL-2, and added back to the original G-REX-100 flasks. When TIL are expanded serially in G-REX-100 flasks, on day 7 the TIL in each G-REX-100 may be suspended in the 300 mL of media present in each flask and the cell suspension may be divided into 3100 mL aliquots that may be used to seed 3 G-REX- 100 flasks. Then 150 mL of AIM-V with 5% human AB serum and 3000 IU per mL of IL-2 may be added to each flask. The G-REX-100 flasks may be incubated at 37° C in 5% CO
2 and after 4 days 150 mL of AIM-V with 3000 IU per mL of IL-2 may be added to each G- REX-100 flask. The cells may be harvested on day 14 of culture. [00101] In some embodiments, the second expansion (including expansions referred to as REP) is performed in flasks with the bulk TILs being mixed with a 100- or 200-fold excess of inactivated feeder cells, 30 mg/mL OKT3 anti-CD3 antibody and 3000 IU/mL IL-2 in 150 mL media. In some embodiments, media replacement is done until the cells are transferred to an alternative growth chamber. In some embodiments, 2/3 of the media is replaced by DB1/ 149202201.1 99 Attorney Docket No.: 116983-5127-WO respiration with fresh media. In some embodiments, alternative growth chambers include G- REX flasks and gas permeable containers as more fully discussed below. [00102] In some embodiments, the second expansion (including expansions referred to as REP) is performed and further comprises a step wherein TILs are selected for superior tumor reactivity. Any selection method known in the art may be used. For example, the methods described in U.S. Patent Application Publication No.2016/0010058 A1, the disclosures of which are incorporated herein by reference, may be used for selection of TILs for superior tumor reactivity. [00103] Optionally, a cell viability assay can be performed after the second expansion (including expansions referred to as the REP expansion), using standard assays known in the art. For example, a trypan blue exclusion assay can be done on a sample of the bulk TILs, which selectively labels dead cells and allows a viability assessment. In some embodiments, TIL samples can be counted and viability determined using a Cellometer K2 automated cell counter (Nexcelom Bioscience, Lawrence, MA). In some embodiments, viability is determined according to the standard Cellometer K2 Image Cytometer Automatic Cell Counter protocol. [00104] In some embodiments, the second expansion (including expansions referred to as REP) of TIL can be performed using T-175 flasks and gas-permeable bags as previously described (Tran, et al., 2008, J Immunother., 31, 742–751, and Dudley, et al.2003, J Immunother., 26, 332–342) or gas-permeable G-REX flasks. In some embodiments, the second expansion is performed using flasks. In some embodiments, the second expansion is performed using gas-permeable G-REX flasks. In some embodiments, the second expansion is performed in T-175 flasks, and about 1 × 10
6 TIL are suspended in about 150 mL of media and this is added to each T-175 flask. The TIL are cultured with irradiated (50 Gy) allogeneic PBMC as “feeder” cells at a ratio of 1 to 100 and the cells were cultured in a 1 to 1 mixture of CM and AIM-V medium (50/50 medium), supplemented with 3000 IU/mL of IL-2 and 30 ng/mL of anti-CD3. The T-175 flasks are incubated at 37°C in 5% CO
2. In some embodiments, half the media is changed on day 5 using 50/50 medium with 3000 IU/mL of IL-2. In some embodiments, on day 7, cells from 2 T-175 flasks are combined in a 3 L bag and 300 mL of AIM-V with 5% human AB serum and 3000 IU/mL of IL-2 is added to the 300 mL of TIL suspension. The number of cells in each bag can be counted every day or two DB1/ 149202201.1 100 Attorney Docket No.: 116983-5127-WO and fresh media can be added to keep the cell count between about 0.5 and about 2.0 × 10
6 cells/mL. [00105] In some embodiments, the second expansion (including expansions referred to as REP) are performed in 500 mL capacity flasks with 100 cm
2 gas-permeable silicon bottoms (G-REX-100, Wilson Wolf) about 5 × 10
6 or 10 × 10
6 TIL are cultured with irradiated allogeneic PBMC at a ratio of 1 to 100 in 400 mL of 50/50 medium, supplemented with 3000 IU/mL of IL-2 and 30 ng/ mL of anti-CD3. The G-REX-100 flasks are incubated at 37°C in 5% CO
2. In some embodiments, on day 5, 250mL of supernatant is removed and placed into centrifuge bottles and centrifuged at 1500 rpm (491 g) for 10 minutes. The TIL pellets can then be resuspended with 150 mL of fresh 50/50 medium with 3000 IU/ mL of IL-2 and added back to the original G-REX-100 flasks. In embodiments where TILs are expanded serially in G-REX-100 flasks, on day 7 the TIL in each G-REX-100 are suspended in the 300 mL of media present in each flask and the cell suspension was divided into three 100 mL aliquots that are used to seed 3 G-REX-100 flasks. Then 150 mL of AIM-V with 5% human AB serum and 3000 IU/mL of IL-2 is added to each flask. The G-REX-100 flasks are incubated at 37°C in 5% CO2 and after 4 days 150 mL of AIM-V with 3000 IU/mL of IL-2 is added to each G-REX-100 flask. The cells are harvested on day 14 of culture. [00106] The diverse antigen receptors of T and B lymphocytes are produced by somatic recombination of a limited, but large number of gene segments. These gene segments: V (variable), D (diversity), J (joining), and C (constant), determine the binding specificity and downstream applications of immunoglobulins and T-cell receptors (TCRs). The present invention provides a method for generating TILs which exhibit and increase the T-cell repertoire diversity. In some embodiments, the TILs obtained by the present method exhibit an increase in the T-cell repertoire diversity. In some embodiments, the TILs obtained in the second expansion exhibit an increase in the T-cell repertoire diversity. In some embodiments, the increase in diversity is an increase in the immunoglobulin diversity and/or the T-cell receptor diversity. In some embodiments, the diversity is in the immunoglobulin is in the immunoglobulin heavy chain. In some embodiments, the diversity is in the immunoglobulin is in the immunoglobulin light chain. In some embodiments, the diversity is in the T-cell receptor. In some embodiments, the diversity is in one of the T-cell receptors selected from the group consisting of alpha, beta, gamma, and delta receptors. In some embodiments, there is an increase in the expression of T-cell receptor (TCR) alpha and/or beta. In some DB1/ 149202201.1 101 Attorney Docket No.: 116983-5127-WO embodiments, there is an increase in the expression of T-cell receptor (TCR) alpha. In some embodiments, there is an increase in the expression of T-cell receptor (TCR) beta. In some embodiments, there is an increase in the expression of TCRab (i.e., TCRα/β). [00107] In some embodiments, the second expansion culture medium (e.g., sometimes referred to as CM2 or the second cell culture medium), comprises IL-2, OKT-3, as well as the antigen-presenting feeder cells (APCs), as discussed in more detail below. [00108] In some embodiments, the culture medium used in the expansion processes disclosed herein is a serum-free medium or a defined medium. In some embodiments, the serum-free or defined medium comprises a basal cell medium and a serum supplement and/or a serum replacement. In some embodiments, the serum-free or defined medium is used to prevent and/or decrease experimental variation due in part to the lot-to-lot variation of serum- containing media. [00109] In some embodiments, the serum-free or defined medium comprises a basal cell medium and a serum supplement and/or serum replacement. In some embodiments, the basal cell medium includes, but is not limited to CTS™ OpTmizer™ T-cell Expansion Basal Medium , CTS™ OpTmizer™ T-Cell Expansion SFM, CTS™ AIM-V Medium, CTS™ AIM-V SFM, LymphoONE™ T-Cell Expansion Xeno-Free Medium, Dulbecco's Modified Eagle's Medium (DMEM), Minimal Essential Medium (MEM), Basal Medium Eagle (BME), RPMI 1640, F-10, F-12, Minimal Essential Medium (αMEM), Glasgow's Minimal Essential Medium (G-MEM), RPMI growth medium, and Iscove's Modified Dulbecco's Medium. [00110] In some embodiments, the serum supplement or serum replacement includes, but is not limited to one or more of CTS™ OpTmizer T-Cell Expansion Serum Supplement, CTS™ Immune Cell Serum Replacement, one or more albumins or albumin substitutes, one or more amino acids, one or more vitamins, one or more transferrins or transferrin substitutes, one or more antioxidants, one or more insulins or insulin substitutes, one or more collagen precursors, one or more antibiotics, and one or more trace elements. In some embodiments, the defined medium comprises albumin and one or more ingredients selected from the group consisting of glycine, L- histidine, L-isoleucine, L-methionine, L-phenylalanine, L-proline, L- hydroxyproline, L-serine, L-threonine, L-tryptophan, L-tyrosine, L-valine, thiamine, reduced glutathione, L-ascorbic acid-2-phosphate, iron saturated transferrin, insulin, and compounds containing the trace element moieties Ag+, Al3+, Ba2+, Cd2+, Co2+, Cr3+, Ge4+, Se4+, Br, T, Mn2+, P, Si4+, V5+, Mo6+, Ni2+, Rb+, Sn2+ and Zr4+. In some DB1/ 149202201.1 102 Attorney Docket No.: 116983-5127-WO embodiments, the defined medium further comprises L-glutamine, sodium bicarbonate and/or 2-mercaptoethanol. [00111] In some embodiments, the CTS™OpTmizer™ T-cell Immune Cell Serum Replacement is used with conventional growth media, including but not limited to CTS™ OpTmizer™ T-cell Expansion Basal Medium, CTS™ OpTmizer™ T-cell Expansion SFM, CTS™ AIM-V Medium, CST™ AIM-V SFM, LymphoONE™ T-Cell Expansion Xeno-Free Medium, Dulbecco's Modified Eagle's Medium (DMEM), Minimal Essential Medium (MEM), Basal Medium Eagle (BME), RPMI 1640, F-10, F-12, Minimal Essential Medium (αMEM), Glasgow's Minimal Essential Medium (G-MEM), RPMI growth medium, and Iscove's Modified Dulbecco's Medium. [00112] In some embodiments, the total serum replacement concentration (vol%) in the serum-free or defined medium is from about 1%, 2%, 3%, 4%, 5%, 6%, 7%, 8%, 9%, 10%, 11%, 12%, 13%, 14%, 15%, 16%, 17%, 18%, 19%, or 20% by volume of the total serum-free or defined medium. In some embodiments, the total serum replacement concentration is about 3% of the total volume of the serum-free or defined medium. In some embodiments, the total serum replacement concentration is about 5% of the total volume of the serum-free or defined medium. In some embodiments, the total serum replacement concentration is about 10% of the total volume of the serum-free or defined medium. [00113] In some embodiments, the serum-free or defined medium is CTS™ OpTmizer™ T- cell Expansion SFM (ThermoFisher Scientific). Any formulation of CTS™ OpTmizer™ is useful in the present invention. CTS™ OpTmizer™ T-cell Expansion SFM is a combination of 1L CTS™ OpTmizer™ T-cell Expansion Basal Medium and 26 mL CTS™ OpTmizer™ T-Cell Expansion Supplement, which are mixed together prior to use. In some embodiments, the CTS™ OpTmizer™ T-cell Expansion SFM is supplemented with about 3% of the CTS™ Immune Cell Serum Replacement (SR) (ThermoFisher Scientific). In some embodiments, the CTS™ OpTmizer™ T-cell Expansion SFM is supplemented with about 3% of the CTS™ Immune Cell Serum Replacement (SR) (ThermoFisher Scientific), along with 2- mercaptoethanol at 55mM. In some embodiments, the CTS™ OpTmizer™ T-cell Expansion SFM is supplemented with about 3% of the CTS™ Immune Cell Serum Replacement (SR) (ThermoFisher Scientific) and the final concentration of 2-mercaptoethanol in the media is 55µM. DB1/ 149202201.1 103 Attorney Docket No.: 116983-5127-WO [00114] In some embodiments, the defined medium is CTS™ OpTmizer™ T-cell Expansion SFM (ThermoFisher Scientific). Any formulation of CTS™ OpTmizer™ is useful in the present invention. CTS™ OpTmizer™ T-cell Expansion SFM is a combination of 1L CTS™ OpTmizer™ T-cell Expansion Basal Medium and 26 mL CTS™ OpTmizer™ T-Cell Expansion Supplement, which are mixed together prior to use. In some embodiments, the CTS™ OpTmizer™ T-cell Expansion SFM is supplemented with about 3% of the CTS™ Immune Cell Serum Replacement (SR) (ThermoFisher Scientific), along with 2- mercaptoethanol at 55mM. In some embodiments, the CTS™OpTmizer™ T-cell Expansion SFM is supplemented with about 3% of the CTS™ Immune Cell Serum Replacement (SR) (ThermoFisher Scientific), 55mM of 2-mercaptoethanol, and 2mM of L-glutamine. In some embodiments, the CTS™OpTmizer™ T-cell Expansion SFM is supplemented with about 3% of the CTS™ Immune Cell Serum Replacement (SR) (ThermoFisher Scientific), 55mM of 2- mercaptoethanol, and 2mM of L-glutamine, and further comprises about 1000 IU/mL to about 8000 IU/mL of IL-2. In some embodiments, the CTS™OpTmizer™ T-cell Expansion SFM is supplemented with about 3% of the CTS™ Immune Cell Serum Replacement (SR) (ThermoFisher Scientific), 55mM of 2-mercaptoethanol, and 2mM of L-glutamine, and further comprises about 3000 IU/mL of IL-2. In some embodiments, the CTS™OpTmizer™ T-cell Expansion SFM is supplemented with about 3% of the CTS™ Immune Cell Serum Replacement (SR) (ThermoFisher Scientific), 55mM of 2-mercaptoethanol, and 2mM of L- glutamine, and further comprises about 6000 IU/mL of IL-2. In some embodiments, the CTS™OpTmizer™ T-cell Expansion SFM is supplemented with about 3% of the CTS™ Immune Cell Serum Replacement (SR) (ThermoFisher Scientific) and 55mM of 2- mercaptoethanol, and further comprises about 1000 IU/mL to about 8000 IU/mL of IL-2. In some embodiments, the CTS™OpTmizer™ T-cell Expansion SFM is supplemented with about 3% of the CTS™ Immune Cell Serum Replacement (SR) (ThermoFisher Scientific) and 55mM of 2-mercaptoethanol, and further comprises about 3000 IU/mL of IL-2. In some embodiments, the CTS™OpTmizer™ T-cell Expansion SFM is supplemented with about 3% of the CTS™ Immune Cell Serum Replacement (SR) (ThermoFisher Scientific) and 55mM of 2-mercaptoethanol, and further comprises about 1000 IU/mL to about 6000 IU/mL of IL-2. In some embodiments, the CTS™OpTmizer™ T-cell Expansion SFM is supplemented with about 3% of the CTS™ Immune Cell Serum Replacement (SR) (ThermoFisher Scientific) and about 2mM glutamine, and further comprises about 1000 IU/mL to about 8000 IU/mL of IL-2. In some embodiments, the CTS™OpTmizer™ T-cell Expansion SFM is supplemented DB1/ 149202201.1 104 Attorney Docket No.: 116983-5127-WO with about 3% of the CTS™ Immune Cell Serum Replacement (SR) (ThermoFisher Scientific) and about 2mM glutamine, and further comprises about 3000 IU/mL of IL-2. In some embodiments, the CTS™OpTmizer™ T-cell Expansion SFM is supplemented with about 3% of the CTS™ Immune Cell Serum Replacement (SR) (ThermoFisher Scientific) and about 2mM glutamine, and further comprises about 6000 IU/mL of IL-2. In some embodiments, the CTS™ OpTmizer™ T-cell Expansion SFM is supplemented with about 3% of the CTS™ Immune Cell Serum Replacement (SR) (ThermoFisher Scientific) and the final concentration of 2-mercaptoethanol in the media is 55µM. [00115] In some embodiments, the serum-free medium or defined medium is supplemented with glutamine (i.e., GlutaMAX®) at a concentration of from about 0.1mM to about 10mM, 0.5mM to about 9mM, 1mM to about 8mM, 2mM to about 7mM, 3mM to about 6mM, or 4mM to about 5 mM. In some embodiments, the serum-free medium or defined medium is supplemented with glutamine (i.e., GlutaMAX®) at a concentration of about 2mM. [00116] In some embodiments, the serum-free medium or defined medium is supplemented with 2-mercaptoethanol at a concentration of from about 5mM to about 150mM, 10mM to about 140mM, 15mM to about 130mM, 20mM to about 120mM, 25mM to about 110mM, 30mM to about 100mM, 35mM to about 95mM, 40mM to about 90mM, 45mM to about 85mM, 50mM to about 80mM, 55mM to about 75mM, 60mM to about 70mM, or about 65mM. In some embodiments, the serum-free medium or defined medium is supplemented with 2-mercaptoethanol at a concentration of about 55mM. In some embodiments, the final concentration of 2-mercaptoethanol in the media is 55µM. [00117] In some embodiments, the defined media described in International PCT Publication No. WO/1998/030679, which is herein incorporated by reference, are useful in the present invention. In that publication, serum-free eukaryotic cell culture media are described. The serum-free, eukaryotic cell culture medium includes a basal cell culture medium supplemented with a serum-free supplement capable of supporting the growth of cells in serum- free culture. The serum-free eukaryotic cell culture medium supplement comprises or is obtained by combining one or more ingredients selected from the group consisting of one or more albumins or albumin substitutes, one or more amino acids, one or more vitamins, one or more transferrins or transferrin substitutes, one or more antioxidants, one or more insulins or insulin substitutes, one or more collagen precursors, one or more trace elements, and one or more antibiotics. In some embodiments, the defined medium further comprises L- DB1/ 149202201.1 105 Attorney Docket No.: 116983-5127-WO glutamine, sodium bicarbonate and/or beta-mercaptoethanol. In some embodiments, the defined medium comprises an albumin or an albumin substitute and one or more ingredients selected from group consisting of one or more amino acids, one or more vitamins, one or more transferrins or transferrin substitutes, one or more antioxidants, one or more insulins or insulin substitutes, one or more collagen precursors, and one or more trace elements. In some embodiments, the defined medium comprises albumin and one or more ingredients selected from the group consisting of glycine, L- histidine, L-isoleucine, L-methionine, L- phenylalanine, L-proline, L- hydroxyproline, L-serine, L-threonine, L-tryptophan, L-tyrosine, L-valine, thiamine, reduced glutathione, L-ascorbic acid-2-phosphate, iron saturated transferrin, insulin, and compounds containing the trace element moieties Ag+, Al3+, Ba2+, Cd2+, Co2+, Cr3+, Ge4+, Se4+, Br, T, Mn2+, P, Si4+, V5+, Mo6+, Ni2+, Rb+, Sn2+ and Zr4+. In some embodiments, the basal cell media is selected from the group consisting of Dulbecco's Modified Eagle's Medium (DMEM), Minimal Essential Medium (MEM), Basal Medium Eagle (BME), RPMI 1640, F-10, F-12, Minimal Essential Medium (αMEM), Glasgow's Minimal Essential Medium (G-MEM), RPMI growth medium, and Iscove's Modified Dulbecco's Medium. [00118] In some embodiments, the concentration of glycine in the defined medium is in the range of from about 5-200 mg/L, the concentration of L- histidine is about 5-250 mg/L, the concentration of L-isoleucine is about 5-300 mg/L, the concentration of L-methionine is about 5-200 mg/L, the concentration of L-phenylalanine is about 5-400 mg/L, the concentration of L-proline is about 1-1000 mg/L, the concentration of L- hydroxyproline is about 1-45 mg/L, the concentration of L-serine is about 1-250 mg/L, the concentration of L- threonine is about 10-500 mg/L, the concentration of L-tryptophan is about 2-110 mg/L, the concentration of L-tyrosine is about 3-175 mg/L, the concentration of L-valine is about 5-500 mg/L, the concentration of thiamine is about 1-20 mg/L, the concentration of reduced glutathione is about 1-20 mg/L, the concentration of L-ascorbic acid-2-phosphate is about 1- 200 mg/L, the concentration of iron saturated transferrin is about 1-50 mg/L, the concentration of insulin is about 1-100 mg/L, the concentration of sodium selenite is about 0.000001-0.0001 mg/L, and the concentration of albumin (e.g., AlbuMAX® I) is about 5000- 50,000 mg/L. [00119] In some embodiments, the non-trace element moiety ingredients in the defined medium are present in the concentration ranges listed in the column under the heading DB1/ 149202201.1 106 Attorney Docket No.: 116983-5127-WO “Concentration Range in 1X Medium” in Table 4. In other embodiments, the non-trace element moiety ingredients in the defined medium are present in the final concentrations listed in the column under the heading “A Preferred Embodiment of the 1X Medium” in Table 4. In other embodiments, the defined medium is a basal cell medium comprising a serum free supplement. In some of these embodiments, the serum free supplement comprises non-trace moiety ingredients of the type and in the concentrations listed in the column under the heading “A Preferred Embodiment in Supplement” in Table 4. [00120] In some embodiments, the osmolarity of the defined medium is between about 260 and 350 mOsmol. In some embodiments, the osmolarity is between about 280 and 310 mOsmol. In some embodiments, the defined medium is supplemented with up to about 3.7 g/L, or about 2.2 g/L sodium bicarbonate. The defined medium can be further supplemented with L-glutamine (final concentration of about 2 mM), one or more antibiotics, non-essential amino acids (NEAA; final concentration of about 100 μM), 2-mercaptoethanol (final concentration of about 100 μM). [00121] In some embodiments, the defined media described in Smith, et al., Clin Transl Immunology, 4(1) 2015 (doi: 10.1038/cti.2014.31) are useful in the present invention. Briefly, RPMI or CTS™ OpTmizer™ was used as the basal cell medium, and supplemented with either 0, 2%, 5%, or 10% CTS™ Immune Cell Serum Replacement. [00122] In some embodiments, the cell medium in the first and/or second gas permeable container is unfiltered. The use of unfiltered cell medium may simplify the procedures necessary to expand the number of cells. In some embodiments, the cell medium in the first and/or second gas permeable container lacks beta-mercaptoethanol (BME or βME; also known as 2-mercaptoethanol, CAS 60-24-2). [00123] In some embodiments, the second expansion, for example, Step D according to Figure 1, is performed in a closed system bioreactor. In some embodiments, a closed system is employed for the TIL expansion, as described herein. In some embodiments, a single bioreactor is employed. In some embodiments, the single bioreactor employed is for example a G-REX -10 or a G-REX -100. In some embodiments, the closed system bioreactor is a single bioreactor. [00124] In some embodiments, the step of rapid or second expansion is split into a plurality of steps to achieve a scaling up of the culture by: (a) performing the rapid or second DB1/ 149202201.1 107 Attorney Docket No.: 116983-5127-WO expansion by culturing TILs in a small scale culture in a first container, e.g., a G-REX-100 MCS container, for a period of about 3 to 7 days, and then (b) effecting the transfer of the TILs in the small scale culture to a second container larger than the first container, e.g., a G- REX-500-MCS container, and culturing the TILs from the small scale culture in a larger scale culture in the second container for a period of about 4 to 7 days. [00125] In some embodiments, the step of rapid or second expansion is split into a plurality of steps to achieve a scaling out of the culture by: (a) performing the rapid or second expansion by culturing TILs in a first small scale culture in a first container, e.g., a G-REX- 100 MCS container, for a period of about 3 to 7 days, and then (b) effecting the transfer and apportioning of the TILs from the first small scale culture into and amongst at least 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, or 20 second containers that are equal in size to the first container, wherein in each second container the portion of the TILs from first small scale culture transferred to such second container is cultured in a second small scale culture for a period of about 4 to 7 days. [00126] In some embodiments, the first small scale TIL culture is apportioned into a plurality of about 2 to 5 subpopulations of TILs. [00127] In some embodiments, the step of rapid or second expansion is split into a plurality of steps to achieve a scaling out and scaling up of the culture by: (a) performing the rapid or second expansion by culturing TILs in a small scale culture in a first container, e.g., a G- REX-100 MCS container, for a period of about 3 to 7 days, and then (b) effecting the transfer and apportioning of the TILs from the small scale culture into and amongst at least 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, or 20 second containers that are larger in size than the first container, e.g., G-REX-500MCS containers, wherein in each second container the portion of the TILs from the small scale culture transferred to such second container is cultured in a larger scale culture for a period of about 4 to 7 days. [00128] In some embodiments, the step of rapid or second expansion is split into a plurality of steps to achieve a scaling out and scaling up of the culture by: (a) performing the rapid or second expansion by culturing TILs in a small scale culture in a first container, e.g., a G- REX-100 MCS container, for a period of about 5 days, and then (b) effecting the transfer and apportioning of the TILs from the small scale culture into and amongst 2, 3 or 4 second containers that are larger in size than the first container, e.g., G-REX-500 MCS containers, wherein in each second container the portion of the TILs from the small scale culture DB1/ 149202201.1 108 Attorney Docket No.: 116983-5127-WO transferred to such second container is cultured in a larger scale culture for a period of about 6 days. [00129] In some embodiments, upon the splitting of the rapid or second expansion, each second container comprises at least 10
8 TILs. In some embodiments, upon the splitting of the rapid or second expansion, each second container comprises at least 10
8 TILs, at least 10
9 TILs, or at least 10
10 TILs. In one exemplary embodiment, each second container comprises at least 10
10 TILs. [00130] In some embodiments, the first small scale TIL culture is apportioned into a plurality of subpopulations. In some embodiments, the first small scale TIL culture is apportioned into a plurality of about 2 to 5 subpopulations. In some embodiments, the first small scale TIL culture is apportioned into a plurality of about 2, 3, 4, or 5 subpopulations. [00131] In some embodiments, after the completion of the rapid or second expansion, the plurality of subpopulations comprises a therapeutically effective amount of TILs. In some embodiments, after the completion of the rapid or second expansion, one or more subpopulations of TILs are pooled together to produce a therapeutically effective amount of TILs. In some embodiments, after the completion of the rapid expansion, each subpopulation of TILs comprises a therapeutically effective amount of TILs. [00132] In some embodiments, the rapid or second expansion is performed for a period of about 3 to 7 days before being split into a plurality of steps. In some embodiments, the splitting of the rapid or second expansion occurs at about day 3, day 4, day 5, day 6, or day 7 after the initiation of the rapid or second expansion. [00133] In some embodiments, the splitting of the rapid or second expansion occurs at about day 7, day 8, day 9, day 10, day 11, day 12, day 13, day 14, day 15, or day 16 day 17, or day 18 after the initiation of the first expansion (i.e., pre-REP expansion). In one exemplary embodiment, the splitting of the rapid or second expansion occurs at about day 16 after the initiation of the first expansion. [00134] In some embodiments, the rapid or second expansion is further performed for a period of about 7 to 11 days after the splitting. In some embodiments, the rapid or second expansion is further performed for a period of about 5 days, 6 days, 7 days, 8 days, 9 days, 10 days, or 11 days after the splitting. DB1/ 149202201.1 109 Attorney Docket No.: 116983-5127-WO [00135] In some embodiments, the cell culture medium used for the rapid or second expansion before the splitting comprises the same components as the cell culture medium used for the rapid or second expansion after the splitting. In some embodiments, the cell culture medium used for the rapid or second expansion before the splitting comprises different components from the cell culture medium used for the rapid or second expansion after the splitting. [00136] In some embodiments, the cell culture medium used for the rapid or second expansion before the splitting comprises IL-2, optionally OKT-3 and further optionally APCs. In some embodiments, the cell culture medium used for the rapid or second expansion before the splitting comprises IL-2, OKT-3, and further optionally APCs. In some embodiments, the cell culture medium used for the rapid or second expansion before the splitting comprises IL-2, OKT-3 and APCs. [00137] In some embodiments, the cell culture medium used for the rapid or second expansion before the splitting is generated by supplementing the cell culture medium in the first expansion with fresh culture medium comprising IL-2, optionally OKT-3 and further optionally APCs. In some embodiments, the cell culture medium used for the rapid or second expansion before the splitting is generated by supplementing the cell culture medium in the first expansion with fresh culture medium comprising IL-2, OKT-3 and APCs. In some embodiments, the cell culture medium used for the rapid or second expansion before the splitting is generated by replacing the cell culture medium in the first expansion with fresh cell culture medium comprising IL-2, optionally OKT-3 and further optionally APCs. In some embodiments, the cell culture medium used for the rapid or second expansion before the splitting is generated by replacing the cell culture medium in the first expansion with fresh cell culture medium comprising IL-2, OKT-3 and APCs. [00138] In some embodiments, the cell culture medium used for the rapid or second expansion after the splitting comprises IL-2, and optionally OKT-3. In some embodiments, the cell culture medium used for the rapid or second expansion after the splitting comprises IL-2, and OKT-3. In some embodiments, the cell culture medium used for the rapid or second expansion after the splitting is generated by replacing the cell culture medium used for the rapid or second expansion before the splitting with fresh culture medium comprising IL-2 and optionally OKT-3. In some embodiments, the cell culture medium used for the rapid or second expansion after the splitting is generated by replacing the cell culture medium used DB1/ 149202201.1 110 Attorney Docket No.: 116983-5127-WO for the rapid or second expansion before the splitting with fresh culture medium comprising IL-2 and OKT-3. [00139] In some embodiments, the splitting of the rapid expansion occurs in a closed system. [00140] In some embodiments, the scaling up of the TIL culture during the rapid or second expansion comprises adding fresh cell culture medium to the TIL culture (also referred to as feeding the TILs). In some embodiments, the feeding comprises adding fresh cell culture medium to the TIL culture frequently. In some embodiments, the feeding comprises adding fresh cell culture medium to the TIL culture at a regular interval. In some embodiments, the fresh cell culture medium is supplied to the TILs via a constant flow. In some embodiments, an automated cell expansion system such as Xuri W25 is used for the rapid expansion and feeding. 1. Feeder Cells and Antigen Presenting Cells [00141] In some embodiments, the second expansion procedures described herein (for example including expansion such as those described in Step D from Figure 1, as well as those referred to as REP) require an excess of feeder cells during REP TIL expansion and/or during the second expansion. In many embodiments, the feeder cells are peripheral blood mononuclear cells (PBMCs) obtained from standard whole blood units from healthy blood donors. The PBMCs are obtained using standard methods such as Ficoll-Paque gradient separation. [00142] In general, the allogeneic PBMCs are inactivated, either via irradiation or heat treatment, and used in the REP procedures, as described in the examples, which provides an exemplary protocol for evaluating the replication incompetence of irradiate allogeneic PBMCs. [00143] In some embodiments, PBMCs are considered replication incompetent and accepted for use in the TIL expansion procedures described herein if the total number of viable cells on day 14 is less than the initial viable cell number put into culture on day 0 of the REP and/or day 0 of the second expansion (i.e., the start day of the second expansion). [00144] In some embodiments, PBMCs are considered replication incompetent and accepted for use in the TIL expansion procedures described herein if the total number of viable cells, cultured in the presence of OKT3 and IL-2, on day 7 and day 14 has not increased from the DB1/ 149202201.1 111 Attorney Docket No.: 116983-5127-WO initial viable cell number put into culture on day 0 of the REP and/or day 0 of the second expansion (i.e., the start day of the second expansion). In some embodiments, the PBMCs are cultured in the presence of 30 ng/mL OKT3 antibody and 3000 IU/mL IL-2. [00145] In some embodiments, PBMCs are considered replication incompetent and accepted for use in the TIL expansion procedures described herein if the total number of viable cells, cultured in the presence of OKT3 and IL-2, on day 7 and day 14 has not increased from the initial viable cell number put into culture on day 0 of the REP and/or day 0 of the second expansion (i.e., the start day of the second expansion). In some embodiments, the PBMCs are cultured in the presence of 5-60 ng/mL OKT3 antibody and 1000-6000 IU/mL IL-2. In some embodiments, the PBMCs are cultured in the presence of 10-50 ng/mL OKT3 antibody and 2000-5000 IU/mL IL-2. In some embodiments, the PBMCs are cultured in the presence of 20-40 ng/mL OKT3 antibody and 2000-4000 IU/mL IL-2. In some embodiments, the PBMCs are cultured in the presence of 25-35 ng/mL OKT3 antibody and 2500-3500 IU/mL IL-2. [00146] In some embodiments, the antigen-presenting feeder cells are PBMCs. In some embodiments, the antigen-presenting feeder cells are artificial antigen-presenting feeder cells. In some embodiments, the ratio of TILs to antigen-presenting feeder cells in the second expansion is about 1 to 25, about 1 to 50, about 1 to 100, about 1 to 125, about 1 to 150, about 1 to 175, about 1 to 200, about 1 to 225, about 1 to 250, about 1 to 275, about 1 to 300, about 1 to 325, about 1 to 350, about 1 to 375, about 1 to 400, or about 1 to 500. In some embodiments, the ratio of TILs to antigen-presenting feeder cells in the second expansion is between 1 to 50 and 1 to 300. In some embodiments, the ratio of TILs to antigen-presenting feeder cells in the second expansion is between 1 to 100 and 1 to 200. [00147] In some embodiments, the second expansion procedures described herein require a ratio of about 2.5x10
9 feeder cells to about 100x10
6 TIL. In other embodiments, the second expansion procedures described herein require a ratio of about 2.5x10
9 feeder cells to about 50x10
6 TIL. In yet other embodiments, the second expansion procedures described herein require about 2.5x10
9 feeder cells to about 25x10
6 TIL. [00148] In some embodiments, the second expansion procedures described herein require an excess of feeder cells during the second expansion. In many embodiments, the feeder cells are peripheral blood mononuclear cells (PBMCs) obtained from standard whole blood units from healthy blood donors. The PBMCs are obtained using standard methods such as Ficoll- DB1/ 149202201.1 112 Attorney Docket No.: 116983-5127-WO Paque gradient separation. In some embodiments, artificial antigen-presenting (aAPC) cells are used in place of PBMCs. [00149] In general, the allogeneic PBMCs are inactivated, either via irradiation or heat treatment, and used in the TIL expansion procedures described herein, including the exemplary procedures described in the figures and examples. [00150] In some embodiments, artificial antigen presenting cells are used in the second expansion as a replacement for, or in combination with, PBMCs. 2. Cytokines and Other Additives [00151] The expansion methods described herein generally use culture media with high doses of a cytokine, in particular IL-2, as is known in the art. [00152] Alternatively, using combinations of cytokines for the rapid expansion and or second expansion of TILs is additionally possible, with combinations of two or more of IL-2, IL-15 and IL-21 as is described in U.S. Patent Application Publication No. US 2017/0107490 A1, the disclosure of which is incorporated by reference herein. Thus, possible combinations include IL-2 and IL-15, IL-2 and IL-21, IL-15 and IL-21 and IL-2, IL-15 and IL-21, with the latter finding particular use in many embodiments. The use of combinations of cytokines specifically favors the generation of lymphocytes, and in particular T-cells as described therein. [00153] In some embodiments, Step D may also include the addition of OKT-3 antibody or muromonab to the culture media, as described elsewhere herein. In some embodiments, Step D may also include the addition of a 4-1BB agonist to the culture media, as described elsewhere herein. In some embodiments, Step D may also include the addition of an OX-40 agonist to the culture media, as described elsewhere herein. In addition, additives such as peroxisome proliferator-activated receptor gamma coactivator I-alpha agonists, including proliferator-activated receptor (PPAR)-gamma agonists such as a thiazolidinedione compound, may be used in the culture media during Step D, as described in U.S. Patent Application Publication No. US 2019/0307796 A1, the disclosure of which is incorporated by reference herein. E. STEP E: Harvest TILs DB1/ 149202201.1 113 Attorney Docket No.: 116983-5127-WO [00154] After the second expansion step, cells can be harvested. In some embodiments the TILs are harvested after one, two, three, four or more expansion steps, for example as provided in Figure 1. In some embodiments the TILs are harvested after two expansion steps, for example as provided in Figure 1. [00155] TILs can be harvested in any appropriate and sterile manner, including for example by centrifugation. Methods for TIL harvesting are well known in the art and any such know methods can be employed with the present process. In some embodiments, TILs are harvested using an automated system. [00156] Cell harvesters and/or cell processing systems are commercially available from a variety of sources, including, for example, Fresenius Kabi, Tomtec Life Science, Perkin Elmer, and Inotech Biosystems International, Inc. Any cell based harvester can be employed with the present methods. In some embodiments, the cell harvester and/or cell processing systems is a membrane-based cell harvester. In some embodiments, cell harvesting is via a cell processing system, such as the LOVO system (manufactured by Fresenius Kabi). The term “LOVO cell processing system” also refers to any instrument or device manufactured by any vendor that can pump a solution comprising cells through a membrane or filter such as a spinning membrane or spinning filter in a sterile and/or closed system environment, allowing for continuous flow and cell processing to remove supernatant or cell culture media without pelletization. In some embodiments, the cell harvester and/or cell processing system can perform cell separation, washing, fluid-exchange, concentration, and/or other cell processing steps in a closed, sterile system. [00157] In some embodiments, the harvest, for example, Step E according to Figure 1, is performed from a closed system bioreactor. In some embodiments, a closed system is employed for the TIL expansion, as described herein. In some embodiments, a single bioreactor is employed. In some embodiments, the single bioreactor employed is for example a G-REX-10 or a G-REX-100. In some embodiments, the closed system bioreactor is a single bioreactor. [00158] In some embodiments, Step E according to Figure 1, is performed according to the processes described herein. In some embodiments, the closed system is accessed via syringes under sterile conditions in order to maintain the sterility and closed nature of the system. In some embodiments, a closed system as described in the Examples is employed. DB1/ 149202201.1 114 Attorney Docket No.: 116983-5127-WO [00159] In some embodiments, TILs are harvested according to the methods described in the Examples. In some embodiments, TILs between days 1 and 11 are harvested using the methods as described in the steps referred herein, such as in the day 11 TIL harvest in the Examples. In some embodiments, TILs between days 12 and 24 are harvested using the methods as described in the steps referred herein, such as in the Day 22 TIL harvest in the Examples. In some embodiments, TILs between days 12 and 22 are harvested using the methods as described in the steps referred herein, such as in the Day 22 TIL harvest in the Examples. F. STEP F: Final Formulation and Transfer to Infusion Container [00160] After Steps A through E as provided in an exemplary order in Figure 1 and as outlined in detailed above and herein are complete, cells are transferred to a container for use in administration to a patient, such as an infusion bag or sterile vial. In some embodiments, once a therapeutically sufficient number of TILs are obtained using the expansion methods described above, they are transferred to a container for use in administration to a patient. [00161] In some embodiments, TILs expanded using APCs of the present disclosure are administered to a patient as a pharmaceutical composition. In some embodiments, the pharmaceutical composition is a suspension of TILs in a sterile buffer. TILs expanded using PBMCs of the present disclosure may be administered by any suitable route as known in the art. In some embodiments, the T-cells are administered as a single intra-arterial or intravenous infusion, which preferably lasts approximately 30 to 60 minutes. Other suitable routes of administration include intraperitoneal, intrathecal, and intralymphatic administration. IV. Pharmaceutical Compositions, Dosages, and Dosing Regimens [00162] Any suitable dose of TILs can be administered. In some embodiments, from about 2.3×10
10 to about 13.7×10
10 TILs are administered, with an average of around 7.8×10
10 TILs, particularly if the cancer is NSCLC or melanoma. In some embodiments, about 1.2×10
10 to about 4.3×10
10 of TILs are administered. In some embodiments, about 3×10
10 to about 12×10
10 TILs are administered. In some embodiments, about 4×10
10 to about 10×10
10 TILs are administered. In some embodiments, about 5×10
10 to about 8×10
10 TILs are administered. In some embodiments, about 6×10
10 to about 8×10
10 TILs are administered. In some embodiments, about 7×10
10 to about 8×10
10 TILs are administered. In some embodiments, the DB1/ 149202201.1 115 Attorney Docket No.: 116983-5127-WO therapeutically effective dosage is about 2.3×10
10 to about 13.7×10
10. In some embodiments, the therapeutically effective dosage is about 7.8×10
10 TILs, particularly of the cancer is melanoma. n some embodiments, the therapeutically effective dosage is about 7.8×10
10 TILs, particularly of the cancer is NSCLC. In some embodiments, the therapeutically effective dosage is about 1.2×10
10 to about 4.3×10
10 of TILs. In some embodiments, the therapeutically effective dosage is about 3×10
10 to about 12×10
10 TILs. In some embodiments, the therapeutically effective dosage is about 4×10
10 to about 10×10
10 TILs. In some embodiments, the therapeutically effective dosage is about 5×10
10 to about 8×10
10 TILs. In some embodiments, the therapeutically effective dosage is about 6×10
10 to about 8×10
10 TILs. In some embodiments, the therapeutically effective dosage is about 7×10
10 to about 8×10
10 TILs. In some embodiments, the therapeutically effective dosage is about 7.5×10
9 to about 72×10
9 TILs. In some embodiments, the therapeutically effective dosage is about 1 x 10
9 to about 100 x 10
9 TILs. In some embodiments, the therapeutically effective dosage is about 7.5 x 10
9 to about 100 x 10
9 TILs. [00163] In some embodiments, the number of the TILs provided in the pharmaceutical compositions of the invention is about 1×10
6, 2×10
6, 3×10
6, 4×10
6, 5×10
6, 6×10
6, 7×10
6, 8×10
6, 9×10
6, 1×10
7, 2×10
7, 3×10
7, 4×10
7, 5×10
7, 6×10
7, 7×10
7, 8×10
7, 9×10
7, 1×10
8, 2×10
8, 3×10
8, 4×10
8, 5×10
8, 6×10
8, 7×10
8, 8×10
8, 9×10
8, 1×10
9, 2×10
9, 3×10
9, 4×10
9, 5×10
9, 6×10
9, 7×10
9, 8×10
9, 9×10
9, 1×10
10, 2×10
10, 3×10
10, 4×10
10, 5×10
10, 6×10
10, 7×10
10, 8×10
10, 9×10
10, 1×10
11, 2×10
11, 3×10
11, 4×10
11, 5×10
11, 6×10
11, 7×10
11, 8×10
11, 9×10
11, 1×10
12, 2×10
12, 3×10
12, 4×10
12, 5×10
12, 6×10
12, 7×10
12, 8×10
12, 9×10
12, 1×10
13, 2×10
13, 3×10
13, 4×10
13, 5×10
13, 6×10
13, 7×10
13, 8×10
13, and 9×10
13. In some embodiments, the number of the TILs provided in the pharmaceutical compositions of the invention is in the range of 1×10
6 to 5×10
6, 5×10
6 to 1×10
7, 1×10
7 to 5×10
7, 5×10
7 to 1×10
8, 1×10
8 to 5×10
8, 5×10
8 to 1×10
9, 1×10
9 to 5×10
9, 5×10
9 to 1×10
10, 1×10
10 to 5×10
10, 5×10
10 to 1×10
11, 5×10
11 to 1×10
12, 1×10
12 to 5×10
12, and 5×10
12 to 1×10
13. V. Methods of Treating Cancer Using TIL and Trop-2 Targeting ADC [00144] Some embodiments of the present disclosure provide a method of treating a cancer in a patient in need thereof comprising administering a population of tumor infiltrating lymphocytes (TILs) and a Trop-2 targeting ADC. In some embodiments, the population of TILs is Lifileucel. DB1/ 149202201.1 116 Attorney Docket No.: 116983-5127-WO [00145] In some embodiments, the Trop-2 targeting ADC is Trodelvy (sacituzumab govitecan). In some embodiments, the Trop-2 targeting ADC is datopotamab deruxtecan. In some embodiments, the Trop-2 targeting ADC is MK-2870/ SKB-264. In some embodiments, the Trop-2 targeting ADC is ESG-401. In some embodiments, the Trop-2 targeting ADC is BIO 106. In some embodiments, the Trop-2 targeting ADC is DB-1305. In some embodiments, the Trop-2 targeting ADC is JS108. In some embodiments, the Trop-2 targeting ADC is BL-M02D1. In some embodiments, the Trop-2 targeting ADC is FZ- AD004. [00146] In some embodiments, the Trop-2 targeting ADC is provided to the patient with the TIL infusion. In some embodiments, the Trop-2 targeting ADC is not provided to the patient at the same time as the TIL infusion. In some embodiments, the Trop-2 targeting ADC is provided to the patient after TIL infusion. In some embodiments, the Trop-2 targeting ADC is provided to the patient contemporaneously with the TIL infusion and also provided after TIL infusion. In particular embodiments, the Trop-2 targeting ADC is provided to the patient about 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 16, or 24 hours after TIL infusion. In certain embodiments, the Trop-2 targeting ADC is provided to the patient about 1, 2, 3, 4, 5, 6, or 7 days after TIL infusion. In certain embodiments, the Trop-2 targeting ADC is provided to the patient about 1, 2, 3, 4, 5, 6, or 7 days after TIL infusion. In exemplary embodiments, the Trop-2 targeting ADC is provided to the patient about 1, 2, 3, or 4, weeks after TIL infusion. In particular embodiments, the Trop-2 targeting ADC is provided to the patient about 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11 or 12 months after TIL infusion. In some embodiments, the patient administered the subject TIL treatment was previously administered a Trop-2 targeting ADC and continues to receive the Trop-2 targeting ADC post treatment. In exemplary embodiments, the patient continues to receive a Trop-2 targeting ADC treatment for at least 1, 2, 3, or 4 weeks after receiving the subject TIL treatment. In exemplary embodiments, the patient continues to receive a Trop-2 targeting ADC treatment for at least 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, or 12 months after receiving the subject TIL treatment. In exemplary embodiments, the patient continues to receive a Trop-2 targeting ADC treatment for at least 1, 2, 3, 4, 5, 6, 7, 8, 9, or 10, years after receiving the subject TIL treatment. In some embodiments, the patient continues to receive a Trop-2 targeting ADC treatment until progression of the patient’s cancer or unacceptable toxicity occurs after receiving the subject DB1/ 149202201.1 117 Attorney Docket No.: 116983-5127-WO TIL treatment. In some embodiments, the patient continues to receive a Trop-2 targeting ADC treatment for life after the subject TIL treatment. [00147] In some embodiments, a cancer patient in need of is treated with both the TILs provided herein and a Trop-2 targeting ADC. In some embodiments, the Trop-2 targeting ADC is administered to the patient contemporaneously with the TILs. In some embodiments, the Trop-2 targeting ADC and the TILs are administered sequentially. In some embodiments, the Trop-2 targeting ADC is administered before the administering of the TILs. In some embodiments, the Trop-2 targeting ADC is administered after the administering of the TILs. In some embodiments, the Trop-2 targeting ADC is administered both before and after the administering of the TILs. In some embodiments, the administering of the Trop-2 targeting ADC is maintained after the administering of the TILs. [00148] In some embodiments, prior to the administering of the TILs, the cancer patient has been treated with a Trop-2 targeting ADC. In some embodiments, the cancer patient has bene treated with a Trop-2 targeting ADC prior to tumor harvest. In some embodiments, a Trop-2 targeting ADC is administered prior to the resection of the tumor from the patient. In some embodiments, a Trop-2 targeting ADC is administered prior to surgical resection, needle biopsy, core biopsy, small biopsy, or other means for obtaining a tumor sample from the patient. [00149] In some embodiments, the patient is refractory to pre-treatment with aTrop-2 targeting ADC. In some embodiments, the patient is responsive to the pretreatment with a Trop-2 targeting ADC. [00150] In some embodiments, the Trop-2 targeting ADC provided herein is administered at a dosage of about 1 mg/kg, 2 mg/kg, 3 mg/kg, 4 mg/kg, 5 mg/kg, 6 mg/kg, 7 mg/kg, 8 mg/kg, 9 mg/kg, 10 mg/kg, 20 mg/kg, 30 mg/kg, 40 mg/kg, 50 mg/kg, 60 mg/kg, 70 mg/kg, 80 mg/kg, 90 mg/kg, or 100 mg/kg. In some embodiments, the Trop-2 targeting ADC is administered at a dosage of at least 1 mg/kg, 2 mg/kg, 3 mg/kg, 4 mg/kg, 5 mg/kg, 6 mg/kg, 7 mg/kg, 8 mg/kg, 9 mg/kg, 10 mg/kg, 20 mg/kg, 30 mg/kg, 40 mg/kg, 50 mg/kg, 60 mg/kg, 70 mg/kg, 80 mg/kg, 90 mg/kg, or 100 mg/kg. In some embodiments, the Trop-2 targeting ADC is administered 1, 2, 3, 4, 5, 6, or 7 times weekly. In exemplary embodiments, the Trop-2 targeting ADC is administered once weekly. In exemplary embodiments, the Trop-2 targeting ADC is administered once every two weeks. In exemplary embodiments, the DB1/ 149202201.1 118 Attorney Docket No.: 116983-5127-WO Trop-2 targeting ADC is administered once every three weeks. In exemplary embodiments, the Trop-2 targeting ADC is administered once every four weeks. In exemplary embodiments, the Trop-2 targeting ADC is administered once every five weeks. [00151] In some embodiments, the Trop-2 targeting ADC is Trodelvy (sacituzumab govitecan), and is administered at a dosage of about 1 mg/kg, 2 mg/kg, 3 mg/kg, 4 mg/kg, 5 mg/kg, 6 mg/kg, 7 mg/kg, 8 mg/kg, 9 mg/kg, 10 mg/kg, 20 mg/kg, 30 mg/kg, 40 mg/kg, 50 mg/kg, 60 mg/kg, 70 mg/kg, 80 mg/kg, 90 mg/kg, or 100 mg/kg. In some embodiments, Trodelvy (sacituzumab govitecan) is administered at a dosage of at least 1 mg/kg, 2 mg/kg, 3 mg/kg, 4 mg/kg, 5 mg/kg, 6 mg/kg, 7 mg/kg, 8 mg/kg, 9 mg/kg, 10 mg/kg, 20 mg/kg, 30 mg/kg, 40 mg/kg, 50 mg/kg, 60 mg/kg, 70 mg/kg, 80 mg/kg, 90 mg/kg, or 100 mg/kg. In exemplary embodiments, Trodelvy (sacituzumab govitecan) is administered at a dosage of about 10 mg/kg. In exemplary embodiments, Trodelvy (sacituzumab govitecan) is administered once weekly. [00152] In some embodiments, the Trop-2 targeting ADC is datopotamab deruxtecan, and is administered at a dosage of about 1 mg/kg, 2 mg/kg, 3 mg/kg, 4 mg/kg, 5 mg/kg, 6 mg/kg, 7 mg/kg, 8 mg/kg, 9 mg/kg, 10 mg/kg, 20 mg/kg, 30 mg/kg, 40 mg/kg, 50 mg/kg, 60 mg/kg, 70 mg/kg, 80 mg/kg, 90 mg/kg, or 100 mg/kg. In some embodiments, datopotamab deruxtecan is administered at a dosage of at least 1 mg/kg, 2 mg/kg, 3 mg/kg, 4 mg/kg, 5 mg/kg, 6 mg/kg, 7 mg/kg, 8 mg/kg, 9 mg/kg, 10 mg/kg, 20 mg/kg, 30 mg/kg, 40 mg/kg, 50 mg/kg, 60 mg/kg, 70 mg/kg, 80 mg/kg, 90 mg/kg, or 100 mg/kg. In exemplary embodiments, datopotamab deruxtecan is administered at a dosage of about 6 mg/kg. In exemplary embodiments, datopotamab deruxtecan is administered once every three weeks. [00153] In some embodiments, the Trop-2 targeting ADC is MK-2870/ SKB-264, and is administered at a dosage of about 1 mg/m
2, 2 mg/m
2, 3 mg/m
2, 4 mg/m
2, 5 mg/m
2, 6 mg/m
2, 7 mg/m
2, 8 mg/m
2, 9 mg/m
2, 10 mg/m
2, 20 mg/m
2, 30 mg/m
2, 40 mg/m
2, 50 mg/m
2, 60 mg/m
2, 70 mg/m
2, 80 mg/m
2, 90 mg/m
2, or 100 mg/m
2. In some embodiments, MK-2870/ SKB-264 is administered at a dosage of at least 1 mg/m
2, 2 mg/m
2, 3 mg/m
2, 4 mg/m
2, 5 mg/m
2, 6 mg/m
2, 7 mg/m
2, 8 mg/m
2, 9 mg/m
2, 10 mg/m
2, 20 mg/m
2, 30 mg/m
2, 40 mg/m
2, 50 mg/m
2, 60 mg/m
2, 70 mg/m
2, 80 mg/m
2, 90 mg/m
2, or 100 mg/m
2. In exemplary embodiments, MK-2870/ SKB-264 is administered at a dosage of about 5 mg/m
2. In exemplary embodiments, MK-2870/ SKB-264 is administered once every two weeks. DB1/ 149202201.1 119 Attorney Docket No.: 116983-5127-WO [00154] In some embodiments, the Trop-2 targeting ADC is ESG-401, and is administered at a dosage of about 1 mg/kg, 2 mg/kg, 3 mg/kg, 4 mg/kg, 5 mg/kg, 6 mg/kg, 7 mg/kg, 8 mg/kg, 9 mg/kg, 10 mg/kg, 20 mg/kg, 30 mg/kg, 40 mg/kg, 50 mg/kg, 60 mg/kg, 70 mg/kg, 80 mg/kg, 90 mg/kg, or 100 mg/kg. In some embodiments, ESG-401is administered at a dosage of at least 1 mg/kg, 2 mg/kg, 3 mg/kg, 4 mg/kg, 5 mg/kg, 6 mg/kg, 7 mg/kg, 8 mg/kg, 9 mg/kg, 10 mg/kg, 20 mg/kg, 30 mg/kg, 40 mg/kg, 50 mg/kg, 60 mg/kg, 70 mg/kg, 80 mg/kg, 90 mg/kg, or 100 mg/kg. In some embodiments, ESG-401 is administered 1, 2, 3, 4, 5, 6, or 7 times weekly. In exemplary embodiments, ESG-401 is administered once weekly. In exemplary embodiments, ESG-401 is administered once every two weeks. In exemplary embodiments, ESG-401 is administered once every three weeks. In exemplary embodiments, ESG-401 is administered once every four weeks. In exemplary embodiments, ESG-401 is administered once every five weeks. [00155] In some embodiments, the Trop-2 targeting ADC is BIO 106, and is administered at a dosage of about 1 mg/kg, 2 mg/kg, 3 mg/kg, 4 mg/kg, 5 mg/kg, 6 mg/kg, 7 mg/kg, 8 mg/kg, 9 mg/kg, 10 mg/kg, 20 mg/kg, 30 mg/kg, 40 mg/kg, 50 mg/kg, 60 mg/kg, 70 mg/kg, 80 mg/kg, 90 mg/kg, or 100 mg/kg. In some embodiments, BIO 106 is administered at a dosage of at least 1 mg/kg, 2 mg/kg, 3 mg/kg, 4 mg/kg, 5 mg/kg, 6 mg/kg, 7 mg/kg, 8 mg/kg, 9 mg/kg, 10 mg/kg, 20 mg/kg, 30 mg/kg, 40 mg/kg, 50 mg/kg, 60 mg/kg, 70 mg/kg, 80 mg/kg, 90 mg/kg, or 100 mg/kg. In some embodiments, BIO 106 is administered 1, 2, 3, 4, 5, 6, or 7 times weekly. In exemplary embodiments, BIO 106 is administered once weekly. In exemplary embodiments, BIO 106 is administered once every two weeks. In exemplary embodiments, BIO 106 is administered once every three weeks. In exemplary embodiments, BIO 106 is administered once every four weeks. In exemplary embodiments, BIO 106 is administered once every five weeks. [00156] In some embodiments, the Trop-2 targeting ADC is DB-1305, and is administered at a dosage of about 1 mg/kg, 2 mg/kg, 3 mg/kg, 4 mg/kg, 5 mg/kg, 6 mg/kg, 7 mg/kg, 8 mg/kg, 9 mg/kg, 10 mg/kg, 20 mg/kg, 30 mg/kg, 40 mg/kg, 50 mg/kg, 60 mg/kg, 70 mg/kg, 80 mg/kg, 90 mg/kg, or 100 mg/kg. In some embodiments, DB-1305 is administered at a dosage of at least 1 mg/kg, 2 mg/kg, 3 mg/kg, 4 mg/kg, 5 mg/kg, 6 mg/kg, 7 mg/kg, 8 mg/kg, 9 mg/kg, 10 mg/kg, 20 mg/kg, 30 mg/kg, 40 mg/kg, 50 mg/kg, 60 mg/kg, 70 mg/kg, 80 mg/kg, 90 mg/kg, or 100 mg/kg. In some embodiments, DB-1305 is administered 1, 2, 3, 4, 5, 6, or 7 times weekly. In exemplary embodiments, DB-1305 is administered once DB1/ 149202201.1 120 Attorney Docket No.: 116983-5127-WO weekly. In exemplary embodiments, DB-1305 is administered once every two weeks. In exemplary embodiments, DB-1305 is administered once every three weeks. In exemplary embodiments, DB-1305 is administered once every four weeks. In exemplary embodiments, DB-1305 is administered once every five weeks. [00157] In some embodiments, the Trop-2 targeting ADC is JS108, and is administered at a dosage of about 1 mg/kg, 2 mg/kg, 3 mg/kg, 4 mg/kg, 5 mg/kg, 6 mg/kg, 7 mg/kg, 8 mg/kg, 9 mg/kg, 10 mg/kg, 20 mg/kg, 30 mg/kg, 40 mg/kg, 50 mg/kg, 60 mg/kg, 70 mg/kg, 80 mg/kg, 90 mg/kg, or 100 mg/kg. In some embodiments, JS108 is administered at a dosage of at least 1 mg/kg, 2 mg/kg, 3 mg/kg, 4 mg/kg, 5 mg/kg, 6 mg/kg, 7 mg/kg, 8 mg/kg, 9 mg/kg, 10 mg/kg, 20 mg/kg, 30 mg/kg, 40 mg/kg, 50 mg/kg, 60 mg/kg, 70 mg/kg, 80 mg/kg, 90 mg/kg, or 100 mg/kg. In exemplary embodiments, JS108 is administered once every three weeks. [00158] In some embodiments, the Trop-2 targeting ADC is BL-M02D1, and is administered at a dosage of about 1 mg/kg, 2 mg/kg, 3 mg/kg, 4 mg/kg, 5 mg/kg, 6 mg/kg, 7 mg/kg, 8 mg/kg, 9 mg/kg, 10 mg/kg, 20 mg/kg, 30 mg/kg, 40 mg/kg, 50 mg/kg, 60 mg/kg, 70 mg/kg, 80 mg/kg, 90 mg/kg, or 100 mg/kg. In some embodiments, BL-M02D1 is administered at a dosage of at least 1 mg/kg, 2 mg/kg, 3 mg/kg, 4 mg/kg, 5 mg/kg, 6 mg/kg, 7 mg/kg, 8 mg/kg, 9 mg/kg, 10 mg/kg, 20 mg/kg, 30 mg/kg, 40 mg/kg, 50 mg/kg, 60 mg/kg, 70 mg/kg, 80 mg/kg, 90 mg/kg, or 100 mg/kg. In some embodiments, BL-M02D1 is administered 1, 2, 3, 4, 5, 6, or 7 times weekly. In exemplary embodiments, BL-M02D1 is administered once weekly. In exemplary embodiments, BL-M02D1 is administered once every two weeks. In exemplary embodiments, BL-M02D1 is administered once every three weeks. In exemplary embodiments, BL-M02D1 is administered once every four weeks. In exemplary embodiments, BL-M02D1 is administered once every five weeks. [00159] In some embodiments, the Trop-2 targeting ADC is FZ-AD004, and is administered at a dosage of about 1 mg/kg, 2 mg/kg, 3 mg/kg, 4 mg/kg, 5 mg/kg, 6 mg/kg, 7 mg/kg, 8 mg/kg, 9 mg/kg, 10 mg/kg, 20 mg/kg, 30 mg/kg, 40 mg/kg, 50 mg/kg, 60 mg/kg, 70 mg/kg, 80 mg/kg, 90 mg/kg, or 100 mg/kg. In some embodiments, FZ-AD004 is administered at a dosage of at least 1 mg/kg, 2 mg/kg, 3 mg/kg, 4 mg/kg, 5 mg/kg, 6 mg/kg, 7 mg/kg, 8 mg/kg, 9 mg/kg, 10 mg/kg, 20 mg/kg, 30 mg/kg, 40 mg/kg, 50 mg/kg, 60 mg/kg, 70 mg/kg, 80 mg/kg, 90 mg/kg, or 100 mg/kg. In some embodiments, FZ-AD004 is administered 1, 2, 3, 4, 5, 6, or 7 times weekly. In exemplary embodiments, FZ-AD004 is DB1/ 149202201.1 121 Attorney Docket No.: 116983-5127-WO administered once weekly. In exemplary embodiments, FZ-AD004 is administered once every two weeks. In exemplary embodiments, FZ-AD004 is administered once every three weeks. In exemplary embodiments, FZ-AD004 is administered once every four weeks. In exemplary embodiments, FZ-AD004 is administered once every five weeks. [00160] In some embodiments, the method comprises the steps of: (a) obtaining and/or receiving a first population of TILs from a tumor resected from the patient by processing a tumor sample obtained from the subject into multiple tumor fragments; (b) expanding the first population of TILs into a therapeutic population of TILs; (c) administering the therapeutic population of TILs to the subject; and (d) administering the Trop-2 targeting ADC to the subject. [00161] In exemplary embodiments, the Trop-2 targeting ADC is provided to the patient prior to resection of the source tumor from which the autologous TIL therapeutic is derived. Without being bound by any particular theory of operation, it is believed that the Trop-2 targeting ADC treatment prior to resection of the source tumor leads to antigen remodeling of TILs obtained from the tumor, thus providing more robust TILs for expansion and downstream use in the TIL therapeutics provided herein. In some embodiments, the patient is provided the Trop-2 targeting ADC for at least 1, 2, 3, 4, 5, 6, or 7 days prior to resection of the source tumor. In some embodiments, the patient has received the Trop-2 targeting ADC for at least 1, 2, 3, 4 weeks prior to resection of the source tumor. In some embodiments, the patient has received the Trop-2 targeting ADC for at least 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, or 12 months prior to resection of the source tumor. In some embodiments, the patient has received the Trop-2 targeting ADC for at least 1, 2, 3, 4, 5, 6, 7, 8, 9, or 10 years prior to resection of the source tumor. In some embodiments, the Trop-2 targeting ADC is provided post resection of the source tumor. [00162] In some embodiments, the cancer is refractory or resistant to prior treatment with an anti-PD-1 or anti-PD-L1 antibody. In some embodiments, the patient is a primary refractory patient to an anti-PD-1 or anti-PD-L1 antibody. In some embodiments, the patient shows no prior response to an anti-PD-1 or anti-PD-L1 antibody. In some embodiments, the patient shows a prior response to an anti-PD-1 or anti-PD-L1 antibody, follow by progression of the patient’s cancer. DB1/ 149202201.1 122 Attorney Docket No.: 116983-5127-WO [00163] In some embodiments, the PD-1 inhibitor is selected from the group consisting of nivolumab, pembrolizumab, and biosimilars thereof. In some embodiments, the PD-L1 inhibitor is selected from the group consisting of avelumab, atezolizumab, durvalumab, and biosimilars thereof. [00164] In some embodiments, the cancer is refractory to a chemotherapeutic agent. In some embodiments, the prior chemotherapeutic agent is carboplatin, paclitaxel, pemetrexed, and/or cisplatin. In some embodiments, the chemotherapeutic agent(s) is a platinum doublet chemotherapeutic agent. In some embodiments, the platinum doublet therapy comprises a first chemotherapeutic agent selected from the group consisting of cisplatin and carboplatin and a second chemotherapeutic agent selected from the group consisting of vinorelbine, gemcitabine and a taxane (including for example, paclitaxel, docetaxel or nab-paclitaxel). In some embodiments, the platinum doublet chemotherapeutic agent is in combination with pemetrexed. [00165] In some embodiments, the cancer has been previously treated with an angiogenesis inhibitor. In some embodiments, the angiogenesis inhibitor is bevacizumab. [00166] In some embodiments, the cancer is a solid tumor cancer. In some embodiments, the solid tumor cancer is selected from the group consisting of anal cancer, bladder cancer, breast cancer (including triple-negative breast cancer), bone cancer, cancer caused by human papilloma virus (HPV), central nervous system associated cancer (including ependymoma, medulloblastoma, neuroblastoma, pineoblastoma, and primitive neuroectodermal tumor), cervical cancer (including squamous cell cervical cancer, adenosquamous cervical cancer, and cervical adenocarcinoma), colon cancer, colorectal cancer, endometrial cancer, esophageal cancer, esophagogastric junction cancer, gastric cancer, gastrointestinal cancer, gastrointestinal stromal tumor, glioblastoma, glioma, head and neck cancer (including head and neck squamous cell carcinoma (HNSCC), hypopharynx cancer, larynx cancer, nasopharynx cancer, oropharynx cancer, and pharynx cancer), kidney cancer, liver cancer, lung cancer (including non-small-cell lung cancer (NSCLC) and small- cell lung cancer), melanoma (including uveal melanoma, choroidal melanoma, ciliary body melanoma, or iris melanoma), mesothelioma (including malignant pleural mesothelioma), ovarian cancer, pancreatic cancer (including pancreatic ductal adenocarcinoma), penile cancer, rectal cancer, renal cancer, renal cell carcinoma, sarcoma (including Ewing sarcoma, DB1/ 149202201.1 123 Attorney Docket No.: 116983-5127-WO osteosarcoma, rhabdomyosarcoma, and other bone and soft tissue sarcomas), thyroid cancer (including anaplastic thyroid cancer), urothelial cancer, uterine cancer, and vaginal cancer. [00167] In some embodiments, the cancer is a hematological malignancy. In some embodiments, the hematological malignancy is selected from the group consisting of chronic lymphocytic leukemia, acute lymphoblastic leukemia, diffuse large B cell lymphoma, non- Hodgkin’s lymphoma, Hodgkin’s lymphoma, follicular lymphoma, mantle cell lymphoma, and multiple myeloma. [00168] In some embodiments, the cancer is non-small-cell lung cancer (NSCLC). In some embodiments, the NSCLC patient has no EGFR, ALK, or ROS1 genomic alterations. In some embodiments, the NSCLC patient has one or more actionable genetic mutations in MET, HER2, RET, BRAF, and/or KRAS. [00169] In some embodiments, the cancer is breast cancer (including triple-negative breast cancer (TNBC)). [00170] In some embodiments, the TILs administered to the patient is Lifileucel. In some embodiments, TILs are administered to a patient as a pharmaceutical composition. In some embodiments, the pharmaceutical composition is a suspension of TILs in a sterile buffer. TILs may be administered by any suitable route as known in the art. In some embodiments, TILs are administered as a single intra-arterial or intravenous infusion, which preferably lasts approximately 30 to 60 minutes. Other suitable routes of administration include intraperitoneal, intrathecal, and intralymphatic administration. [00171] In some embodiments, the number of the TILs provided in the pharmaceutical compositions of the invention is about 1×10
6, 2×10
6, 3×10
6, 4×10
6, 5×10
6, 6×10
6, 7×10
6, 8×10
6, 9×10
6, 1×10
7, 2×10
7, 3×10
7, 4×10
7, 5×10
7, 6×10
7, 7×10
7, 8×10
7, 9×10
7, 1×10
8, 2×10
8, 3×10
8, 4×10
8, 5×10
8, 6×10
8, 7×10
8, 8×10
8, 9×10
8, 1×10
9, 2×10
9, 3×10
9, 4×10
9, 5×10
9, 6×10
9, 7×10
9, 8×10
9, 9×10
9, 1×10
10, 2×10
10, 3×10
10, 4×10
10, 5×10
10, 6×10
10, 7×10
10, 8×10
10, 9×10
10, 1×10
11, 2×10
11, 3×10
11, 4×10
11, 5×10
11, 6×10
11, 7×10
11, 8×10
11, 9×10
11, 1×10
12, 2×10
12, 3×10
12, 4×10
12, 5×10
12, 6×10
12, 7×10
12, 8×10
12, 9×10
12, 1×10
13, 2×10
13, 3×10
13, 4×10
13, 5×10
13, 6×10
13, 7×10
13, 8×10
13, and 9×10
13. In some embodiments, the number of the TILs provided in the pharmaceutical compositions of the invention is in the range of 1×10
6 to 5×10
6, 5×10
6 to 1×10
7, 1×10
7 to 5×10
7, 5×10
7 to 1×10
8, 1×10
8 to 5×10
8, 5×10
8 to 1×10
9, DB1/ 149202201.1 124 Attorney Docket No.: 116983-5127-WO 1×10
9 to 5×10
9, 5×10
9 to 1×10
10, 1×10
10 to 5×10
10, 5×10
10 to 1×10
11, 5×10
11 to 1×10
12, 1×10
12 to 5×10
12, and 5×10
12 to 1×10
13. [00172] In some embodiments, the concentration of the TILs provided in the pharmaceutical compositions of the invention is less than, for example, 100%, 90%, 80%, 70%, 60%, 50%, 40%, 30%, 20%, 19%, 18%, 17%, 16%, 15%, 14%, 13%, 12%, 11%, 10%, 9%, 8%, 7%, 6%, 5%, 4%, 3%, 2%, 1%, 0.5%, 0.4%, 0.3%, 0.2%, 0.1%, 0.09%, 0.08%, 0.07%, 0.06%, 0.05%, 0.04%, 0.03%, 0.02%, 0.01%, 0.009%, 0.008%, 0.007%, 0.006%, 0.005%, 0.004%, 0.003%, 0.002%, 0.001%, 0.0009%, 0.0008%, 0.0007%, 0.0006%, 0.0005%, 0.0004%, 0.0003%, 0.0002% or 0.0001% w/w, w/v or v/v of the pharmaceutical composition. [00173] In some embodiments, the concentration of the TILs provided in the pharmaceutical compositions of the invention is greater than 90%, 80%, 70%, 60%, 50%, 40%, 30%, 20%, 19.75%, 19.50%, 19.25% 19%, 18.75%, 18.50%, 18.25% 18%, 17.75%, 17.50%, 17.25% 17%, 16.75%, 16.50%, 16.25% 16%, 15.75%, 15.50%, 15.25% 15%, 14.75%, 14.50%, 14.25% 14%, 13.75%, 13.50%, 13.25% 13%, 12.75%, 12.50%, 12.25% 12%, 11.75%, 11.50%, 11.25% 11%, 10.75%, 10.50%, 10.25% 10%, 9.75%, 9.50%, 9.25% 9%, 8.75%, 8.50%, 8.25% 8%, 7.75%, 7.50%, 7.25% 7%, 6.75%, 6.50%, 6.25% 6%, 5.75%, 5.50%, 5.25% 5%, 4.75%, 4.50%, 4.25%, 4%, 3.75%, 3.50%, 3.25%, 3%, 2.75%, 2.50%, 2.25%, 2%, 1.75%, 1.50%, 125%, 1%, 0.5%, 0.4%, 0.3%, 0.2%, 0.1%, 0.09%, 0.08%, 0.07%, 0.06%, 0.05%, 0.04%, 0.03%, 0.02%, 0.01%, 0.009%, 0.008%, 0.007%, 0.006%, 0.005%, 0.004%, 0.003%, 0.002%, 0.001%, 0.0009%, 0.0008%, 0.0007%, 0.0006%, 0.0005%, 0.0004%, 0.0003%, 0.0002% or 0.0001% w/w, w/v, or v/v of the pharmaceutical composition. [00174] In some embodiments, the concentration of the TILs provided in the pharmaceutical compositions of the invention is in the range from about 0.0001% to about 50%, about 0.001% to about 40%, about 0.01% to about 30%, about 0.02% to about 29%, about 0.03% to about 28%, about 0.04% to about 27%, about 0.05% to about 26%, about 0.06% to about 25%, about 0.07% to about 24%, about 0.08% to about 23%, about 0.09% to about 22%, about 0.1% to about 21%, about 0.2% to about 20%, about 0.3% to about 19%, about 0.4% to about 18%, about 0.5% to about 17%, about 0.6% to about 16%, about 0.7% to about 15%, about 0.8% to about 14%, about 0.9% to about 12% or about 1% to about 10% w/w, w/v or v/v of the pharmaceutical composition. DB1/ 149202201.1 125 Attorney Docket No.: 116983-5127-WO [00175] In some embodiments, the concentration of the TILs provided in the pharmaceutical compositions of the invention is in the range from about 0.001% to about 10%, about 0.01% to about 5%, about 0.02% to about 4.5%, about 0.03% to about 4%, about 0.04% to about 3.5%, about 0.05% to about 3%, about 0.06% to about 2.5%, about 0.07% to about 2%, about 0.08% to about 1.5%, about 0.09% to about 1%, about 0.1% to about 0.9% w/w, w/v or v/v of the pharmaceutical composition. [00176] In some embodiments, the amount of the TILs provided in the pharmaceutical compositions of the invention is equal to or less than 10 g, 9.5 g, 9.0 g, 8.5 g, 8.0 g, 7.5 g, 7.0 g, 6.5 g, 6.0 g, 5.5 g, 5.0 g, 4.5 g, 4.0 g, 3.5 g, 3.0 g, 2.5 g, 2.0 g, 1.5 g, 1.0 g, 0.95 g, 0.9 g, 0.85 g, 0.8 g, 0.75 g, 0.7 g, 0.65 g, 0.6 g, 0.55 g, 0.5 g, 0.45 g, 0.4 g, 0.35 g, 0.3 g, 0.25 g, 0.2 g, 0.15 g, 0.1 g, 0.09 g, 0.08 g, 0.07 g, 0.06 g, 0.05 g, 0.04 g, 0.03 g, 0.02 g, 0.01 g, 0.009 g, 0.008 g, 0.007 g, 0.006 g, 0.005 g, 0.004 g, 0.003 g, 0.002 g, 0.001 g, 0.0009 g, 0.0008 g, 0.0007 g, 0.0006 g, 0.0005 g, 0.0004 g, 0.0003 g, 0.0002 g, or 0.0001 g. [00177] In some embodiments, the amount of the TILs provided in the pharmaceutical compositions of the invention is more than 0.0001 g, 0.0002 g, 0.0003 g, 0.0004 g, 0.0005 g, 0.0006 g, 0.0007 g, 0.0008 g, 0.0009 g, 0.001 g, 0.0015 g, 0.002 g, 0.0025 g, 0.003 g, 0.0035 g, 0.004 g, 0.0045 g, 0.005 g, 0.0055 g, 0.006 g, 0.0065 g, 0.007 g, 0.0075 g, 0.008 g, 0.0085 g, 0.009 g, 0.0095 g, 0.01 g, 0.015 g, 0.02 g, 0.025 g, 0.03 g, 0.035 g, 0.04 g, 0.045 g, 0.05 g, 0.055 g, 0.06 g, 0.065 g, 0.07 g, 0.075 g, 0.08 g, 0.085 g, 0.09 g, 0.095 g, 0.1 g, 0.15 g, 0.2 g, 0.25 g, 0.3 g, 0.35 g, 0.4 g, 0.45 g, 0.5 g, 0.55 g, 0.6 g, 0.65 g, 0.7 g, 0.75 g, 0.8 g, 0.85 g, 0.9 g, 0.95 g, 1 g, 1.5 g, 2 g, 2.5, 3 g, 3.5, 4 g, 4.5 g, 5 g, 5.5 g, 6 g, 6.5 g, 7 g, 7.5 g, 8 g, 8.5 g, 9 g, 9.5 g, or 10 g. [00178] The TILs provided in the pharmaceutical compositions of the invention are effective over a wide dosage range. The exact dosage will depend upon the route of administration, the form in which the compound is administered, the gender and age of the subject to be treated, the body weight of the subject to be treated, and the preference and experience of the attending physician. The clinically-established dosages of the TILs may also be used if appropriate. The amounts of the pharmaceutical compositions administered using the methods herein, such as the dosages of TILs, will be dependent on the human or mammal being treated, the severity of the disorder or condition, the rate of administration, the disposition of the active pharmaceutical ingredients and the discretion of the prescribing physician. DB1/ 149202201.1 126 Attorney Docket No.: 116983-5127-WO [00179] In some embodiments, TILs may be administered in a single dose. Such administration may be by injection, e.g., intravenous injection. In some embodiments, TILs may be administered in multiple doses. Dosing may be once, twice, three times, four times, five times, six times, or more than six times per year. Dosing may be once a month, once every two weeks, once a week, or once every other day. Administration of TILs may continue as long as necessary. [00180] In some embodiments, an effective dosage of TILs is in the range of about 0.01 mg/kg to about 4.3 mg/kg, about 0.15 mg/kg to about 3.6 mg/kg, about 0.3 mg/kg to about 3.2 mg/kg, about 0.35 mg/kg to about 2.85 mg/kg, about 0.15 mg/kg to about 2.85 mg/kg, about 0.3 mg to about 2.15 mg/kg, about 0.45 mg/kg to about 1.7 mg/kg, about 0.15 mg/kg to about 1.3 mg/kg, about 0.3 mg/kg to about 1.15 mg/kg, about 0.45 mg/kg to about 1 mg/kg, about 0.55 mg/kg to about 0.85 mg/kg, about 0.65 mg/kg to about 0.8 mg/kg, about 0.7 mg/kg to about 0.75 mg/kg, about 0.7 mg/kg to about 2.15 mg/kg, about 0.85 mg/kg to about 2 mg/kg, about 1 mg/kg to about 1.85 mg/kg, about 1.15 mg/kg to about 1.7 mg/kg, about 1.3 mg/kg mg to about 1.6 mg/kg, about 1.35 mg/kg to about 1.5 mg/kg, about 2.15 mg/kg to about 3.6 mg/kg, about 2.3 mg/kg to about 3.4 mg/kg, about 2.4 mg/kg to about 3.3 mg/kg, about 2.6 mg/kg to about 3.15 mg/kg, about 2.7 mg/kg to about 3 mg/kg, about 2.8 mg/kg to about 3 mg/kg, or about 2.85 mg/kg to about 2.95 mg/kg. [00181] In some embodiments, an effective dosage of TILs is in the range of about 1 mg to about 500 mg, about 10 mg to about 300 mg, about 20 mg to about 250 mg, about 25 mg to about 200 mg, about 1 mg to about 50 mg, about 5 mg to about 45 mg, about 10 mg to about 40 mg, about 15 mg to about 35 mg, about 20 mg to about 30 mg, about 23 mg to about 28 mg, about 50 mg to about 150 mg, about 60 mg to about 140 mg, about 70 mg to about 130 mg, about 80 mg to about 120 mg, about 90 mg to about 110 mg, or about 95 mg to about 105 mg, about 98 mg to about 102 mg, about 150 mg to about 250 mg, about 160 mg to about 240 mg, about 170 mg to about 230 mg, about 180 mg to about 220 mg, about 190 mg to about 210 mg, about 195 mg to about 205 mg, or about 198 to about 207 mg. [00182] An effective amount of the TILs may be administered in either single or multiple doses by any of the accepted modes of administration of agents having similar utilities, including intranasal and transdermal routes, by intra-arterial injection, intravenously, intraperitoneally, parenterally, intramuscularly, subcutaneously, topically, by transplantation, or by inhalation. DB1/ 149202201.1 127 Attorney Docket No.: 116983-5127-WO A. Lymphodepletion Preconditioning of Patients [00183] In some embodiments, the invention includes a method of treating a cancer with a population of TILs, wherein a patient is pre-treated with non-myeloablative chemotherapy prior to an infusion of TILs according to the present disclosure. In some embodiments, the invention includes a population of TILs for use in the treatment of cancer in a patient which has been pre-treated with non-myeloablative chemotherapy. In some embodiments, the population of TILs is for administration by infusion. In some embodiments, the non- myeloablative chemotherapy is cyclophosphamide 60 mg/kg/d for 2 days (days 27 and 26 prior to TIL infusion) and fludarabine 25 mg/m
2/d for 5 days (days 27 to 23 prior to TIL infusion). In some embodiments, after non-myeloablative chemotherapy and TIL infusion (at day 0) according to the present disclosure, the patient receives an intravenous infusion of IL- 2 (aldesleukin, commercially available as PROLEUKIN) intravenously at 720,000 IU/kg every 8 hours to physiologic tolerance. In certain embodiments, the population of TILs is for use in treating cancer in combination with IL-2, wherein the IL-2 is administered after the population of TILs. [00184] Experimental findings indicate that lymphodepletion prior to adoptive transfer of tumor-specific T lymphocytes plays a key role in enhancing treatment efficacy by eliminating regulatory T cells and competing elements of the immune system (‘cytokine sinks’). Accordingly, some embodiments of the invention utilize a lymphodepletion step (sometimes also referred to as “immunosuppressive conditioning”) on the patient prior to the introduction of the TILs of the invention. [00185] In general, lymphodepletion is achieved using administration of fludarabine or cyclophosphamide (the active form being referred to as mafosfamide) and combinations thereof. Such methods are described in Gassner, et al., Cancer Immunol. Immunother.2011, 60, 75–85, Muranski, et al., Nat. Clin. Pract. Oncol., 2006, 3, 668–681, Dudley, et al., J. Clin. Oncol.2008, 26, 5233-5239, and Dudley, et al., J. Clin. Oncol.2005, 23, 2346–2357, all of which are incorporated by reference herein in their entireties. [00186] In some embodiments, the fludarabine is administered at a concentration of 0.5 μg/mL to 10 μg/mL fludarabine. In some embodiments, the fludarabine is administered at a concentration of 1 μg/mL fludarabine. In some embodiments, the fludarabine treatment is administered for 1 day, 2 days, 3 days, 4 days, 5 days, 6 days, or 7 days or more. In some embodiments, the fludarabine is administered at a dosage of 10 mg/kg/day, 15 mg/kg/day, DB1/ 149202201.1 128 Attorney Docket No.: 116983-5127-WO 20 mg/kg/day¸ 25 mg/kg/day, 30 mg/kg/day, 35 mg/kg/day, 40 mg/kg/day, or 45 mg/kg/day. In some embodiments, the fludarabine treatment is administered for 2-7 days at 35 mg/kg/day. In some embodiments, the fludarabine treatment is administered for 4-5 days at 35 mg/kg/day. In some embodiments, the fludarabine treatment is administered for 4- 5 days at 25 mg/kg/day. [00187] In some embodiments, the mafosfamide, the active form of cyclophosphamide, is obtained at a concentration of 0.5 μg/mL to 10 μg/mL by administration of cyclophosphamide. In some embodiments, mafosfamide, the active form of cyclophosphamide, is obtained at a concentration of 1 μg/mL by administration of cyclophosphamide. In some embodiments, the cyclophosphamide treatment is administered for 1 day, 2 days, 3 days, 4 days, 5 days, 6 days, or 7 days or more. In some embodiments, the cyclophosphamide is administered at a dosage of 100 mg/m
2/day, 150 mg/m
2/day, 175 mg/m
2/day¸ 200 mg/m
2/day, 225 mg/m
2/day, 250 mg/m
2/day, 275 mg/m
2/day, or 300 mg/m
2/day. In some embodiments, the cyclophosphamide is administered intravenously (i.e., i.v.) In some embodiments, the cyclophosphamide treatment is administered for 2- 7 days at 35 mg/kg/day. In some embodiments, the cyclophosphamide treatment is administered for 4-5 days at 250 mg/m
2/day i.v. In some embodiments, the cyclophosphamide treatment is administered for 4 days at 250 mg/m
2/day i.v. [00188] In some embodiments, lymphodepletion is performed by administering the fludarabine and the cyclophosphamide together to a patient. In some embodiments, fludarabine is administered at 25 mg/m
2/day i.v. and cyclophosphamide is administered at 250 mg/m
2/day i.v. over 4 days. [00189] In some embodiments, the lymphodepletion is performed by administration of cyclophosphamide at a dose of 60 mg/m
2/day for two days followed by administration of fludarabine at a dose of 25 mg/m
2/day for five days. [00190] In some embodiments, the lymphodepletion is performed by administration of cyclophosphamide at a dose of 60 mg/m
2/day for two days and administration of fludarabine at a dose of 25 mg/m
2/day for five days, wherein cyclophosphamide and fludarabine are both administered on the first two days, and wherein the lymphodepletion is performed in five days in total. DB1/ 149202201.1 129 Attorney Docket No.: 116983-5127-WO [00191] In some embodiments, the lymphodepletion is performed by administration of cyclophosphamide at a dose of about 50 mg/m
2/day for two days and administration of fludarabine at a dose of about 25 mg/m
2/day for five days, wherein cyclophosphamide and fludarabine are both administered on the first two days, and wherein the lymphodepletion is performed in five days in total. [00192] In some embodiments, the lymphodepletion is performed by administration of cyclophosphamide at a dose of about 50 mg/m
2/day for two days and administration of fludarabine at a dose of about 20 mg/m
2/day for five days, wherein cyclophosphamide and fludarabine are both administered on the first two days, and wherein the lymphodepletion is performed in five days in total. [00193] In some embodiments, the lymphodepletion is performed by administration of cyclophosphamide at a dose of about 40 mg/m
2/day for two days and administration of fludarabine at a dose of about 20 mg/m
2/day for five days, wherein cyclophosphamide and fludarabine are both administered on the first two days, and wherein the lymphodepletion is performed in five days in total. [00194] In some embodiments, the lymphodepletion is performed by administration of cyclophosphamide at a dose of about 40 mg/m
2/day for two days and administration of fludarabine at a dose of about 15 mg/m
2/day for five days, wherein cyclophosphamide and fludarabine are both administered on the first two days, and wherein the lymphodepletion is performed in five days in total. [00195] In some embodiments, the lymphodepletion is performed by administration of cyclophosphamide at a dose of 60 mg/m
2/day and fludarabine at a dose of 25 mg/m
2/day for two days followed by administration of fludarabine at a dose of 25 mg/m
2/day for three days. [00196] In some embodiments, the cyclophosphamide is administered with mesna. In some embodiments, mesna is administered at 15 mg/kg. In some embodiments where mesna is infused, and if infused continuously, mesna can be infused over approximately 2 hours with cyclophosphamide (on Days -5 and/or -4), then at a rate of 3 mg/kg/hour for the remaining 22 hours over the 24 hours starting concomitantly with each cyclophosphamide dose. [00197] In some embodiments, the lymphodepletion comprises the step of treating the patient with an IL-2 regimen starting on the day after administration of the population of TILs to the patient. DB1/ 149202201.1 130 Attorney Docket No.: 116983-5127-WO [00198] In some embodiments, the lymphodepletion comprises the step of treating the patient with an IL-2 regimen starting on the same day as administration of population of TILs to the patient. [00199] In some embodiments, the lymphodeplete comprises 5 days of preconditioning treatment. In some embodiments, the days are indicated as days -5 through -1, or Day 0 through Day 4. In some embodiments, the regimen comprises cyclophosphamide on days -5 and -4 (i.e., days 0 and 1). In some embodiments, the regimen comprises intravenous cyclophosphamide on days -5 and -4 (i.e., days 0 and 1). In some embodiments, the regimen comprises 60 mg/kg intravenous cyclophosphamide on days -5 and -4 (i.e., days 0 and 1). In some embodiments, the cyclophosphamide is administered with mesna. In some embodiments, the regimen further comprises fludarabine. In some embodiments, the regimen further comprises intravenous fludarabine. In some embodiments, the regimen further comprises 25 mg/m
2 intravenous fludarabine. In some embodiments, the regimen further comprises 25 mg/m
2 intravenous fludarabine on days -5 and -1 (i.e., days 0 through 4). In some embodiments, the regimen further comprises 25 mg/m
2 intravenous fludarabine on days -5 and -1 (i.e., days 0 through 4). [00200] In some embodiments, the non-myeloablative lymphodepletion regimen comprises the steps of administration of cyclophosphamide at a dose of 60 mg/m
2/day and fludarabine at a dose of 25 mg/m
2/day for two days followed by administration of fludarabine at a dose of 25 mg/m
2/day for five days. [00201] In some embodiments, the non-myeloablative lymphodepletion regimen comprises the steps of administration of cyclophosphamide at a dose of 60 mg/m
2/day for two days followed by administration of fludarabine at a dose of 25 mg/m
2/day for five days. [00202] In some embodiments, the non-myeloablative lymphodepletion regimen comprises the steps of administration of cyclophosphamide at a dose of 60 mg/m
2/day for two days followed by administration of fludarabine at a dose of 25 mg/m
2/day for three days. [00203] In some embodiments, the non-myeloablative lymphodepletion regimen comprises the steps of administration of cyclophosphamide at a dose of 60 mg/m
2/day and fludarabine at a dose of 25 mg/m
2/day for two days followed by administration of fludarabine at a dose of 25 mg/m
2/day for three days. DB1/ 149202201.1 131 Attorney Docket No.: 116983-5127-WO [00204] In some embodiments, the non-myeloablative lymphodepletion regimen comprises the steps of administration of cyclophosphamide at a dose of 60 mg/m
2/day and fludarabine at a dose of 25 mg/m
2/day for two days followed by administration of fludarabine at a dose of 25 mg/m
2/day for one day. [00205] In some embodiments, the non-myeloablative lymphodepletion regimen comprises the steps of administration of cyclophosphamide at a dose of 60 mg/m
2/day for two days followed by administration of fludarabine at a dose of 25 mg/m
2/day for three days. [00206] In some embodiments, the non-myeloablative lymphodepletion regimen comprises the steps of administration of cyclophosphamide at a dose of 60 mg/m
2/day and fludarabine at a dose of 25 mg/m
2/day for two days followed by administration of fludarabine at a dose of 25 mg/m
2/day for three days. [00207] In some embodiments, the non-myeloablative lymphodepletion regimen is administered according to Table 5. TABLE 5. Exemplary lymphodepletion and treatment regimen.
[00208] In some embodiments, the non-myeloablative lymphodepletion regimen is administered according to Table 6. TABLE 6. Exemplary lymphodepletion and treatment regimen.
[00209] In some embodiments, the non-myeloablative lymphodepletion regimen is administered according to Table 7. DB1/ 149202201.1 132 Attorney Docket No.: 116983-5127-WO TABLE 7. Exemplary lymphodepletion and treatment regimen.
[00210] In some embodiments, the non-myeloablative lymphodepletion regimen is administered according to Table 8. TABLE 8. Exemplary lymphodepletion and treatment regimen.
[00211] In some embodiments, the non-myeloablative lymphodepletion regimen is administered according to Table 9. TABLE 9. Exemplary lymphodepletion and treatment regimen.
[00212] In some embodiments, the non-myeloablative lymphodepletion regimen is administered according to Table 10. TABLE 10. Exemplary lymphodepletion and treatment regimen.
DB1/ 149202201.1 133 Attorney Docket No.: 116983-5127-WO [00213] In some embodiments, the non-myeloablative lymphodepletion regimen is administered according to Table 11. TABLE 11. Exemplary lymphodepletion and treatment regimen.
[00214] In some embodiments, the non-myeloablative lymphodepletion regimen is administered according to Table 12. TABLE 12. Exemplary lymphodepletion and treatment regimen.

[0003] In some embodiments, the TIL infusion used with the foregoing embodiments of myeloablative lymphodepletion regimens may be any TIL composition described herein, as well as the addition of IL-2 regimens and administration of co-therapies (such as Trop-2 targeting ADCs) as described herein. B. IL-2 Regimens [00215] In some embodiments, the IL-2 regimen comprises a high-dose IL-2 regimen, wherein the high-dose IL-2 regimen comprises aldesleukin, or a biosimilar or variant thereof, administered intravenously starting on the day after administering a therapeutically effective portion of the therapeutic population of TILs, wherein the aldesleukin or a biosimilar or variant thereof is administered at a dose of 0.037 mg/kg or 0.044 mg/kg IU/kg (patient body mass) using 15-minute bolus intravenous infusions every eight hours until tolerance, for a maximum of 14 doses. Following 9 days of rest, this schedule may be repeated for another 14 doses, for a maximum of 28 doses in total. In some embodiments, IL-2 is administered in 1, DB1/ 149202201.1 134 Attorney Docket No.: 116983-5127-WO 2, 3, 4, 5, or 6 doses. In some embodiments, IL-2 is administered at a maximum dosage of up to 6 doses. [00216] In some embodiments, the IL-2 regimen comprises a decrescendo IL-2 regimen. Decrescendo IL-2 regimens have been described in O’Day, et al., J. Clin. Oncol.1999, 17, 2752-61 and Eton, et al., Cancer 2000, 88, 1703-9, the disclosures of which are incorporated herein by reference. In some embodiments, a decrescendo IL-2 regimen comprises 18 × 10
6 IU/m
2 aldesleukin, or a biosimilar or variant thereof, administered intravenously over 6 hours, followed by 18 × 10
6 IU/m
2 administered intravenously over 12 hours, followed by 18 × 10
6 IU/m
2 administered intravenously over 24 hours, followed by 4.5 × 10
6 IU/m
2 administered intravenously over 72 hours. This treatment cycle may be repeated every 28 days for a maximum of four cycles. In some embodiments, a decrescendo IL-2 regimen comprises 18,000,000 IU/m
2 on day 1, 9,000,000 IU/m
2 on day 2, and 4,500,000 IU/m
2 on days 3 and 4. [0004] In some embodiments, the IL-2 regimen comprises a low-dose IL-2 regimen. Any low-dose IL-2 regimen known in the art may be used, including the low-dose IL-2 regimens described in Dominguez-Villar and Hafler, Nat. Immunology 2000, 19, 665-673; Hartemann, et al., Lancet Diabetes Endocrinol.2013, 1, 295-305; and Rosenzwaig, et al., Ann. Rheum. Dis.2019, 78, 209–217, the disclosures of which are incorporated herein by reference. In some embodiments, a low-dose IL-2 regimen comprises 18 × 10
6 IU per m
2 of aldesleukin, or a biosimilar or variant thereof, per 24 hours, administered as a continuous infusion for 5 days, followed by 2-6 days without IL-2 therapy, optionally followed by an additional 5 days of intravenous aldesleukin or a biosimilar or variant thereof, as a continuous infusion of 18 x 10
6 IU per m
2 per 24 hours, optionally followed by 3 weeks without IL-2 therapy, after which additional cycles may be administered. [00217] In some embodiments, IL-2 is administered at a maximum dosage of up to 6 doses. In some embodiments, the high-dose IL-2 regimen is adapted for pediatric use. In some embodiments, a dose of 600,000 international units (IU)/kg of aldesleukin every 8–12 hours for up to a maximum of 6 doses is used. In some embodiments, a dose of 500,000 international units (IU)/kg of aldesleukin every 8–12 hours for up to a maximum of 6 doses is used. In some embodiments, a dose of 400,000 international units (IU)/kg of aldesleukin every 8–12 hours for up to a maximum of 6 doses is used. In some embodiments, a dose of 500,000 international units (IU)/kg of aldesleukin every 8–12 hours for up to a maximum of 6 DB1/ 149202201.1 135 Attorney Docket No.: 116983-5127-WO doses is used. In some embodiments, a dose of 300,000 international units (IU)/kg of aldesleukin every 8–12 hours for up to a maximum of 6 doses is used. In some embodiments, a dose of 200,000 international units (IU)/kg of aldesleukin every 8–12 hours for up to a maximum of 6 doses is used. In some embodiments, a dose of 100,000 international units (IU)/kg of aldesleukin every 8–12 hours for up to a maximum of 6 doses is used. [00218] In some embodiments, the IL-2 regimen comprises administration of pegylated IL-2 every 1, 2, 4, 6, 7, 14 or 21 days at a dose of 0.10 mg/day to 50 mg/day. In some embodiments, the IL-2 regimen comprises administration of bempegaldesleukin, or a fragment, variant, or biosimilar thereof, every 1, 2, 4, 6, 7, 14 or 21 days at a dose of 0.10 mg/day to 50 mg/day. [00219] In some embodiments, the IL-2 regimen comprises administration of THOR- 707, or a fragment, variant, or biosimilar thereof, every 1, 2, 4, 6, 7, 14 or 21 days at a dose of 0.10 mg/day to 50 mg/day. [00220] In some embodiments, the IL-2 regimen comprises administration of nemvaleukin alfa, or a fragment, variant, or biosimilar thereof, following administration of TIL. In certain embodiments, the patient the nemvaleukin is administered every 1, 2, 4, 6, 7, 14 or 21 days at a dose of 0.10 mg/day to 50 mg/day. [00221] In some embodiments, the IL-2 regimen comprises administration of an IL-2 fragment engrafted onto an antibody backbone. In some embodiments, the IL-2 regimen comprises administration of an antibody-cytokine engrafted protein that binds the IL-2 low affinity receptor. In some embodiments, the antibody cytokine engrafted protein comprises a heavy chain variable region (VH), comprising complementarity determining regions HCDR1, HCDR2, HCDR3; a light chain variable region (VL), comprising LCDR1, LCDR2, LCDR3; and an IL-2 molecule or a fragment thereof engrafted into a CDR of the V
H or the V
L, wherein the antibody cytokine engrafted protein preferentially expands T effector cells over regulatory T cells. In some embodiments, the antibody cytokine engrafted protein comprises a heavy chain variable region (V
H), comprising complementarity determining regions HCDR1, HCDR2, HCDR3; a light chain variable region (V
L), comprising LCDR1, LCDR2, LCDR3; and an IL-2 molecule or a fragment thereof engrafted into a CDR of the VH or the V
L, wherein the IL-2 molecule is a mutein, and wherein the antibody cytokine engrafted protein preferentially expands T effector cells over regulatory T cells. In some embodiments, the IL-2 regimen comprises administration of an antibody comprising a heavy chain selected DB1/ 149202201.1 136 Attorney Docket No.: 116983-5127-WO from the group consisting of SEQ ID NO:29 and SEQ ID NO:38 and a light chain selected from the group consisting of SEQ ID NO:37 and SEQ ID NO:39, or a fragment, variant, or biosimilar thereof, every 1, 2, 4, 6, 7, 14 or 21 days at a dose of 0.10 mg/day to 50 mg/day [00222] In some embodiments, the antibody cytokine engrafted protein described herein has a longer serum half-life that a wild-type IL-2 molecule such as, but not limited to, aldesleukin (Proleukin®) or a comparable molecule. [00223] In some embodiments, the TIL infusion used with the foregoing embodiments of myeloablative lymphodepletion regimens may be any TIL composition described herein, as well as the addition of IL-2 regimens and administration of co-therapies (such as Trop-2 targeting ADCs) as described herein. EXAMPLES [00224] The embodiments encompassed herein are now described with reference to the following examples. These examples are provided for the purpose of illustration only and the disclosure encompassed herein should in no way be construed as being limited to these examples, but rather should be construed to encompass any and all variations which become evident as a result of the teachings provided herein. EXAMPLE 1: PREPARATION OF MEDIA FOR PRE-REP AND REP PROCESSES [00164] This example describes the procedure for the preparation of tissue culture media for use in protocols involving the culture of tumor infiltrating lymphocytes (TIL) derived from various solid tumors. This media can be used for preparation of any of the TILs described in the present application and other examples. [00165] Preparation of CM1. Removed the following reagents from cold storage and warm them in a 37°C water bath: (RPMI1640, Human AB serum, 200 mM L-glutamine). Prepared CM1 medium according to Table 13 below by adding each of the ingredients into the top section of a 0.2 µm filter unit appropriate to the volume to be filtered. Store at 4°C. TABLE 13. Preparation of CM1

DB1/ 149202201.1 137 Attorney Docket No.: 116983-5127-WO
[00166] On the day of use, prewarmed required amount of CM1 in 37°C water bath and add 6000 IU/mL IL-2. [00167] Additional supplementation may be performed as needed according to Table 14. TABLE 14. Additional supplementation of CM1, as needed.
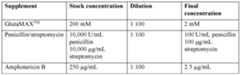
Preparation of CM2 [00168] Removed prepared CM1 from refrigerator or prepare fresh CM1. Removed AIM- V® from refrigerator and prepared the amount of CM2 needed by mixing prepared CM1 with an equal volume of AIM-V® in a sterile media bottle. Added 3000 IU/mL IL-2 to CM2 medium on the day of usage. Made sufficient amount of CM2 with 3000 IU/mL IL-2 on the day of usage. Labeled the CM2 media bottle with its name, the initials of the preparer, the date it was filtered/prepared, the two-week expiration date and store at 4°C until needed for tissue culture. Preparation of CM3 [00169] Prepared CM3 on the day it was required for use. CM3 was the same as AIM-V® medium, supplemented with 3000 IU/mL IL-2 on the day of use. Prepared an amount of CM3 sufficient to experimental needs by adding IL-2 stock solution directly to the bottle or bag of AIM-V. Mixed well by gentle shaking. Label bottle with “3000 IU/mL IL-2” immediately after adding to the AIM-V. If there was excess CM3, stored it in bottles at 4°C labeled with the media name, the initials of the preparer, the date the media was prepared, and its DB1/ 149202201.1 138 Attorney Docket No.: 116983-5127-WO expiration date (7 days after preparation). Discarded media supplemented with IL-2 after 7 days storage at 4°C. Preparation of CM4 [00170] CM4 was the same as CM3, with the additional supplement of 2mM GlutaMAX
TM (final concentration). For every 1L of CM3, add 10 mL of 200 mM GlutaMAX
TM. Prepare an amount of CM4 sufficient to experimental needs by adding IL-2 stock solution and GlutaMAX
TM stock solution directly to the bottle or bag of AIM-V. Mixed well by gentle shaking. Labeled bottle with “3000 IL/mL IL-2 and GlutaMAX” immediately after adding to the AIM-V. If there was excess CM4, stored it in bottles at 4°C labeled with the media name, “GlutaMAX”, and its expiration date (7 days after preparation). Discarded media supplemented with IL-2 after more than 7-days storage at 4°C. EXAMPLE 2: EXEMPLARY GEN 2 PRODUCTION OF A CRYOPRESERVED TIL CELL THERAPY [00171] This example describes the cGMP manufacture of Iovance Biotherapeutics, Inc. TIL Cell Therapy Process in G-REX Flasks according to current Good Tissue Practices and current Good Manufacturing Practices. This example describes an exemplary cGMP manufacture of TIL Cell Therapy Process in G-REX Flasks according to current Good Tissue Practices and current Good Manufacturing Practices. TABLE 15. Process Expansion Exemplary Plan.
DB1/ 149202201.1 139 Attorney Docket No.: 116983-5127-WO TABLE 16. Flask Volumes.
[00172] Day 0 CM1 Media Preparation. In the BSC added reagents to RPMI 1640 Media bottle. Added the following reagents t Added per bottle: Heat Inactivated Human AB Serum (100.0 mL); GlutaMax™ (10.0 mL); Gentamicin sulfate, 50 mg/mL (1.0 mL); 2- mercaptoethanol (1.0 mL) [00173] Removed unnecessary materials from BSC. Passed out media reagents from BSC, left Gentamicin Sulfate and HBSS in BSC for Formulated Wash Media preparation. [00174] Thawed IL-2 aliquot. Thawed one 1.1 mL IL-2 aliquot (6x10
6 IU/mL) (BR71424) until all ice had melted. Recorded IL-2: Lot # and Expiry [00175] Transferred IL-2 stock solution to media. In the BSC, transferred 1.0 mL of IL-2 stock solution to the CM1 Day 0 Media Bottle prepared. Added CM1 Day 0 Media 1 bottle and IL-2 (6x10
6 IU/mL) 1.0 mL. [00176] Passed G-REX100MCS into BSC. Aseptically passed G-REX100MCS (W3013130) into the BSC. [00177] Pumped all Complete CM1 Day 0 Media into G-REX100MCS flask. Tissue Fragments Conical or GRex100MCS . [00178] Day 0 Tumor Wash Media Preparation. In the BSC, added 5.0 mL Gentamicin (W3009832 or W3012735) to 1 x 500 mL HBSS Media (W3013128) bottle. Added per bottle: HBSS (500.0 mL); Gentamicin sulfate, 50 mg/mL (5.0 mL). Filtered HBSS containing gentamicin prepared through a 1L 0.22-micron filter unit (W1218810). [00179] Day 0 Tumor Processing. Obtained tumor specimen and transferred into suite at 2-8 ºC immediately for processing. Aliquoted tumor wash media. Tumor wash 1 is performed using 8” forceps (W3009771). The tumor is removed from the specimen bottle and transferred to the “Wash 1” dish prepared. This is followed by tumor wash 2 and tumor wash 3. Measured and assessed tumor. Assessed whether > 30% of entire tumor area observed to be necrotic and/or fatty tissue. Clean up dissection if applicable. If tumor was DB1/ 149202201.1 140 Attorney Docket No.: 116983-5127-WO large and >30% of tissue exterior was observed to be necrotic/fatty, performed “clean up dissection” by removing necrotic/fatty tissue while preserving tumor inner structure using a combination of scalpel and/or forceps. Dissect tumor. Using a combination of scalpel and/or forceps, cut the tumor specimen into even, appropriately sized fragments (up to 6 intermediate fragments). Transferred intermediate tumor fragments. Dissected tumor fragments into pieces approximately 3x3x3mm in size. Stored Intermediate Fragments to prevent drying. Repeated intermediate fragment dissection. Determined number of pieces collected. If desirable tissue remains, selected additional favorable tumor pieces from the “favorable intermediate fragments” 6-well plate to fill the drops for a maximum of 50 pieces. [00180] Prepared conical tube. Transferred tumor pieces to 50 mL conical tube. Prepared BSC for G-REX100MCS. Removed G-REX100MCS from incubator. Aseptically passed G-REX100MCS flask into the BSC. Added tumor fragments to G-REX100MCS flask. Evenly distributed pieces. [00181] Incubated G-REX100MCS at the following parameters: Incubated G-REX flask: Temperature LED Display: 37.0±2.0 ºC; CO
2 Percentage: 5.0±1.5 %CO
2. Calculations: Time of incubation; lower limit = time of incubation + 252 hours; upper limit = time of incubation + 276 hours. [00182] After process was complete, discarded any remaining warmed media and thawed aliquots of IL-2. [00183] Day 11 – Media Preparation. Monitored incubator. Incubator parameters: Temperature LED Display: 37.0±2.0 ºC; CO
2 Percentage: 5.0±1.5 %CO
2. [00184] Warmed 3× 1000 mL RPMI 1640 Media (W3013112) bottles and 3× 1000 mL AIM-V (W3009501) bottles in an incubator for ≥ 30 minutes. Removed RPMI 1640 Media from incubator. Prepared RPMI 1640 Media. Filter Media. Thawed 3 x 1.1 mL aliquots of IL-2 (6x10
6 IU/mL) (BR71424). Removed AIM-V Media from the incubator. Add IL-2 to AIM-V. Aseptically transferred a 10 L Labtainer Bag and a repeater pump transfer set into the BSC. [00185] Prepared 10 L Labtainer media bag. Prepared Baxa pump. Prepared 10L Labtainer media bag. Pumped media into 10 L Labtainer. Removed pumpmatic from Labtainer bag. DB1/ 149202201.1 141 Attorney Docket No.: 116983-5127-WO [00186] Mixed media. Gently massaged the bag to mix. Sample media per sample plan. Removed 20.0 mL of media and place in a 50 mL conical tube. Prepared cell count dilution tubes. In the BSC, added 4.5 mL of AIM-V Media that had been labelled with “For Cell Count Dilutions” and lot number to four 15 mL conical tubes. Transferred reagents from the BSC to 2-8°C. Prepared 1 L Transfer Pack. Outside of the BSC weld (per Process Note 5.11) a 1L Transfer Pack to the transfer set attached to the “Complete CM2 Day 11 Media” bag prepared. Prepared feeder cell transfer pack. Incubated Complete CM2 Day 11 Media. [00187] Day 11 - TIL Harvest. Preprocessing table. Incubator parameters: Temperature LED display: 37.0±2.0 ºC; CO2 Percentage: 5.0±1.5 % CO2. Removed G-REX100MCS from incubator. Prepared 300 mL Transfer Pack. Welded transfer packs to G-REX100MCS. [00188] Prepare flask for TIL Harvest and initiation of TIL Harvest. TIL Harvested. Using the GatheRex, transferred the cell suspension through the blood filter into the 300 mL transfer pack. Inspect membrane for adherent cells. [00189] Rinsed flask membrane. Closed clamps on G-REX100MCS. Ensured all clamps are closed. Heat sealed the TIL and the “Supernatant” transfer pack. Calculated volume of TIL suspension. Prepared Supernatant Transfer Pack for Sampling. [00190] Pulled Bac-T Sample. In the BSC, draw up approximately 20.0 mL of supernatant from the 1L “Supernatant” transfer pack and dispense into a sterile 50 mL conical tube. [00191] Inoculated BacT per Sample Plan. Removed a 1.0 mL sample from the 50 mL conical labeled BacT prepared using an appropriately sized syringe and inoculated the anaerobic bottle. [00192] Incubated TIL. Placed TIL transfer pack in incubator until needed. Performed cell counts and calculations. Determined the Average of Viable Cell Concentration and Viability of the cell counts performed. Viability ÷ 2. Viable Cell Concentration ÷ 2. Determined Upper and Lower Limit for counts. Lower Limit: Average of Viable Cell Concentration x 0.9. Upper Limit: Average of Viable Cell Concentration x 1.1. Confirmed both counts within acceptable limits. Determined an average Viable Cell Concentration from all four counts performed. DB1/ 149202201.1 142 Attorney Docket No.: 116983-5127-WO [00193] Adjusted Volume of TIL Suspension: Calculate the adjusted volume of TIL suspension after removal of cell count samples. Total TIL Cell Volume (A). Volume of Cell Count Sample Removed (4.0 mL) (B) Adjusted Total TIL Cell Volume C=A-B. [00194] Calculated Total Viable TIL Cells. Average Viable Cell Concentration*: Total Volume; Total Viable Cells: C = A x B. [00195] Calculation for flow cytometry: if the Total Viable TIL Cell count from was ≥ 4.0x10
7, calculated the volume to obtain 1.0×10
7cells for the flow cytometry sample. [00196] Total viable cells required for flow cytometry: 1.0×10
7cells. Volume of cells required for flow cytometry: Viable cell concentration divided by 1.0×10
7cells A. [00197] Calculated the volume of TIL suspension equal to 2.0×10
8viable cells. As needed, calculated the excess volume of TIL cells to remove and removed excess TIL and placed TIL in incubator as needed. Calculated total excess TIL removed, as needed. [00198] Calculated amount of CS-10 media to add to excess TIL cells with the target cell concentration for freezing is 1.0×10
8 cells/mL. Centrifuged excess TILs, as needed. Observed conical tube and added CS-10. [00199] Filled Vials. Aliquoted 1.0 mL cell suspension, into appropriately sized cryovials. Aliquoted residual volume into appropriately sized cryovial. If volume is ≤0.5 mL, add CS10 to vial until volume is 0.5 mL. [00200] Calculated the volume of cells required to obtain 1x10
7cells for cryopreservation. Removed sample for cryopreservation. Placed TIL in incubator. [00201] Cryopreservation of sample. Observed conical tube and added CS-10 slowly and record volume of 0.5 mL of CS10 added. [00202] Day 11 - Feeder Cells. Obtained feeder cells. Obtained 3 bags of feeder cells with at least two different lot numbers from LN2 freezer. Kept cells on dry ice until ready to thaw. Prepared water bath or cryotherm. Thawed feeder cells at 37.0 ± 2.0°C in the water bath or cytotherm for ~3-5 minutes or until ice has just disappeared. Removed media from incubator. Pooled thawed feeder cells. Added feeder cells to transfer pack. Dispensed the feeder cells from the syringe into the transfer pack. Mixed pooled feeder cells and labeled transfer pack. DB1/ 149202201.1 143 Attorney Docket No.: 116983-5127-WO [00203] Calculated total volume of feeder cell suspension in transfer pack. Removed cell count samples. Using a separate 3 mL syringe for each sample, pulled 4x1.0 mL cell count samples from Feeder Cell Suspension Transfer Pack using the needless injection port. Aliquoted each sample into the cryovials labeled. Performed cell counts and determine multiplication factors, elected protocols and entered multiplication factors. Determined the average of viable cell concentration and viability of the cell counts performed. Determined upper and lower limit for counts and confirm within limits. [00204] Adjusted volume of feeder cell suspension. Calculated the adjusted volume of feeder cell suspension after removal of cell count samples. Calculated total viable feeder cells. Obtained additional feeder cells as needed. Thawed additional feeder cells as needed. Placed the 4th feeder cell bag into a zip top bag and thaw in a 37.0 ± 2.0°C water bath or cytotherm for ~3-5 minutes and pooled additional feeder cells. Measured volume. Measured the volume of the feeder cells in the syringe and recorded below (B). Calculated the new total volume of feeder cells. Added feeder cells to transfer pack. [00205] Prepared dilutions as needed, adding 4.5 mL of AIM-V Media to four 15 mL conical tubes. Prepared cell counts. Using a separate 3 mL syringe for each sample, removed 4 x 1.0 mL cell count samples from Feeder Cell Suspension transfer pack, using the needless injection port. Performed cell counts and calculations. Determined an average viable cell concentration from all four counts performed. Adjusted volume of feeder cell suspension and calculated the adjusted volume of feeder cell suspension after removal of cell count samples. Total Feeder Cell Volume minues 4.0 mL removed. Calculated the volume of Feeder Cell Suspension that was required to obtain 5x10
9viable feeder cells. Calculated excess feeder cell volume. Calculated the volume of excess feeder cells to remove. Removed excess feeder cells. [00206] Using a 1.0 mL syringe and 16G needle, drew up 0.15 mL of OKT3 and added OKT3. Heat sealed the feeder cell suspension transfer pack. [00207] Day 11 G-REX Fill and Seed Set up G-REX500MCS. Removed “Complete CM2 Day 11 Media”, from incubator and pumped media into G-REX500MCS. Pumped 4.5L of media into the G-REX500MCS, filling to the line marked on the flask. Heat sealed and incubated flask as needed. Welded the Feeder Cell suspension transfer pack to the G- REX500MCS. Added Feeder Cells to G-REX500MCS. Heat sealed. Welded the TIL DB1/ 149202201.1 144 Attorney Docket No.: 116983-5127-WO Suspension transfer pack to the flask. Added TIL to G-REX500MCS. Heat sealed. Incubated G-REX500MCS at 37.0±2.0 ºC, CO2 Percentage: 5.0±1.5 %CO2. [00208] Calculated incubation window. Performed calculations to determine the proper time to remove G-REX500MCS from incubator on Day 16. Lower limit: Time of incubation + 108 hours. Upper limit: Time of incubation + 132 hours. [00209] Day 11 Excess TIL Cryopreservation. Applicable: Froze Excess TIL Vials. Verified the CRF has been set up prior to freeze. Perform Cryopreservation. Transferred vials from Controlled Rate Freezer to the appropriate storage. Upon completion of freeze, transfer vials from CRF to the appropriate storage container. Transferred vials to appropriate storage. Recorded storage location in LN2. [00210] Day 16 Media Preparation. Pre-warmed AIM-V Media. Calculated time Media was warmed for media bags 1, 2, and 3. Ensured all bags have been warmed for a duration between 12 and 24 hours. Setup 10L Labtainer for Supernatant. Attached the larger diameter end of a fluid pump transfer set to one of the female ports of a 10L Labtainer bag using the Luer connectors. Setup 10L Labtainer for Supernatant and label. Setup 10L Labtainer for Supernatant. Ensure all clamps were closed prior to removing from the BSC. NOTE: Supernatant bag was used during TIL Harvest, which may be performed concurrently with media preparation. [00211] Thawed IL-2. Thawed 5×1.1 mL aliquots of IL-2 (6×10
6 IU/mL) (BR71424) per bag of CTS AIM V media until all ice had melted. Aliquoted 100.0 mL GlutaMax™. Added IL-2 to GlutaMax™. Prepared CTS AIM V media bag for formulation. Prepared CTS AIM V media bag for formulation. Stage Baxa Pump. Prepared to formulate media. Pumped GlutaMax™ +IL-2 into bag. Monitored parameters: Temperature LED Display: 37.0±2.0 ºC, CO
2 Percentage: 5.0±1.5% CO
2. Warmed Complete CM4 Day 16 Media. Prepared Dilutions. [00212] Day 16 REP Spilt. Monitored Incubator parameters: Temperature LED display: 37.0±2.0 ºC, CO
2 Percentage: 5.0±1.5 %CO
2. Removed G-REX500MCS from the incubator. Prepared and labeled 1 L Transfer Pack as TIL Suspension and weighed 1L. [00213] Volume Reduction of G-REX500MCS. Transferred ~4.5L of culture supernatant from the G-REX500MCS to the 10L Labtainer. DB1/ 149202201.1 145 Attorney Docket No.: 116983-5127-WO [00214] Prepared flask for TIL harvest. After removal of the supernatant, closed all clamps to the red line. [00215] Initiation of TIL Harvest. Vigorously tap flask and swirl media to release cells and ensure all cells have detached. [00216] TIL Harvest. Released all clamps leading to the TIL suspension transfer pack. Using the GatheRex transferred the cell suspension into the TIL Suspension transfer pack. NOTE: Be sure to maintain the tilted edge until all cells and media are collected. Inspected membrane for adherent cells. Rinsed flask membrane. Closed clamps on G-REX500MCS. Heat sealed the Transfer Pack containing the TIL. Heat sealed the 10L Labtainer containing the supernatant. Recorded weight of Transfer Pack with cell suspension and calculate the volume suspension. Prepared transfer pack for sample removal. Removed testing samples from cell supernatant. [00217] Sterility & BacT testing sampling. Removed a 1.0 mL sample from the 15 mL conical labeled BacT prepared. Removed Cell Count Samples. In the BSC, using separate 3 mL syringes for each sample, removed 4x1.0 mL cell count samples from “TIL Suspension” transfer pack. [00218] Removed mycoplasma samples. Using a 3 mL syringe, removed 1.0 mL from TIL Suspension transfer pack and place into 15 mL conical labeled “Mycoplasma diluent” prepared. [00219] Prepared transfer pack for seeding. Placed TIL in incubator. Removed cell suspension from the BSC and place in incubator until needed. Performed cell counts and calculations. Diluted cell count samples initially by adding 0.5 mL of cell suspension into 4.5 mL of AIM-V media prepared which gave a 1:10 dilution. Determined the average of viable cell concentration and viability of the cell counts performed. Determined upper and lower limit for counts. Note: dilution may be adjusted according based off the expected concentration of cells. Determined an average viable cell concentration from all four counts performed. Adjusted volume of TIL suspension. Calculated the adjusted volume of TIL suspension after removal of cell count samples. Total TIL cell volume minus 5.0 mL removed for testing. [00220] Calculated total viable TIL cells. Calculated the total number of flasks to seed. NOTE: The maximum number of G-REX500MCS flasks to seed was five. If the calculated DB1/ 149202201.1 146 Attorney Docket No.: 116983-5127-WO number of flasks to seed exceeded five, only five were seeded using the entire volume of cell suspension available. [00221] Calculate number of flasks for subculture. Calculated the number of media bags required in addition to the bag prepared. Prepared one 10L bag of “CM4 Day 16 Media” for every two G-REX-500M flask needed as calculated. Proceeded to seed the first GREX- 500M flask(s) while additional media is prepared and warmed. Prepared and warmed the calculated number of additional media bags determined. Filled G-REX500MCS. Prepared to pump media and pumped 4.5L of media into G-REX500MCS. Heat Sealed. Repeated Fill. Incubated flask. Calculated the target volume of TIL suspension to add to the new G- REX500MCS flasks. If the calculated number of flasks exceeds five only five will be seeded, USING THE ENTIRE VOLUME OF CELL SUSPENSION. Prepared Flasks for Seeding. Removed G-REX500MCS from the incubator. Prepared G-REX500MCS for pumping. Closed all clamps on except large filter line. Removed TIL from incubator. Prepared cell suspension for seeding. Sterile welded (per Process Note 5.11) “TIL Suspension” transfer pack to pump inlet line. Placed TIL suspension bag on a scale. [00222] Seeded flask with TIL Suspension. Pump the volume of TIL suspension calculated into flask. Heat sealed. Filled remaining flasks. [00223] Monitored Incubator. Incubator parameters: Temperature LED Display: 37.0±2.0 ºC, CO2 Percentage: 5.0±1.5 % CO2. Incubated Flasks. [00224] Determined the time range to remove G-REX500MCS from incubator on Day 22. [00225] Day 22 Wash Buffer Preparation. Prepared 10 L Labtainer Bag. In BSC, attach a 4” plasma transfer set to a 10L Labtainer Bag via luer connection. Prepared 10 L Labtainer Bag. Closed all clamps before transferring out of the BSC. NOTE: Prepared one 10L Labtainer Bag for every two G-REX500MCS flasks to be harvested. Pumped Plasmalyte into 3000 mL bag and removed air from 3000 mL Origen bag by reversing the pump and manipulating the position of the bag. Added human albumin 25% to 3000 mL Bag. Obtain a final volumeof 120.0 mL of human albumin 25%. [00226] Prepared IL-2 diluent. Using a 10 mL syringe, removed 5.0 mL of LOVO Wash Buffer using the needleless injection port on the LOVO Wash Buffer bag. Dispensed LOVO wash buffer into a 50 mL conical tube. DB1/ 149202201.1 147 Attorney Docket No.: 116983-5127-WO [00227] CRF blank bag LOVO wash buffer aliquotted. Using a 100 mL syringe, drew up 70.0 mL of LOVO Wash Buffer from the needleless injection port. [00228] Thawed one 1.1 mL of IL-2 (6x10
6 IU/mL), until all ice has melted. Added 50 µL IL-2 stock (6×10
6 IU/mL) to the 50 mL conical tube labeled “IL-2 Diluent.” [00229] Cryopreservation preparation. Placed 5 cryo-cassettes at 2-8°C to precondition them for final product cryopreservation. [00230] Prepared cell count dilutions. In the BSC, added 4.5 mL of AIM-V Media that has been labelled with lot number and “For Cell Count Dilutions” to 4 separate 15 mL conical tubes. Prepared cell counts. Labeled 4 cryovials with vial number (1-4). Kept vials under BSC to be used. [00231] Day 22 TIL Harvest. Monitored Incubator. Incubator Parameters Temperature LED display: 37 ± 2.0°C, CO2 Percentage: 5%±1.5%. Removed G-REX500MCS Flasks from Incubator. Prepared TIL collection bag and labeled. Sealed off extra connections. Volume Reduction: Transferred ~4.5L of supernatant from the G-REX500MCS to the Supernatant bag. [00232] Prepared flask for TIL harvest. Initiated collection of TIL. Vigorously tap flask and swirl media to release cells. Ensure all cells have detached. Initiated collection of TIL. Released all clamps leading to the TIL suspension collection bag. TIL Harvest. Using the GatheRex, transferred the TIL suspension into the 3000 mL collection bag. Inspect membrane for adherent cells. Rinsed flask membrane. Closed clamps on G- Rex500MCS and ensured all clamps are closed. Transferred cell suspension into LOVO source bag. Closed all clamps. Heat Sealed. Removed 4x1.0 mL Cell Counts Samples [00233] Performed Cell Counts. Performed cell counts and calculations utilizing NC- 200 and Process Note 5.14. Diluted cell count samples initially by adding 0.5 mL of cell suspension into 4.5 mL of AIM-V media prepared. This gave a 1:10 dilution. Determined the average viability, viable cell concentration, and total nucleated cell concentration of the cell counts performed. Determined Upper and Lower Limit for counts. Determined the average viability, viable cell concentration, and total nucleated cell concentration of the cell counts performed. Weighed LOVO source bag. Calculated total viable TIL Cells. Calculated total nucleated cells. DB1/ 149202201.1 148 Attorney Docket No.: 116983-5127-WO [00234] Prepared Mycoplasma Diluent. Removed 10.0 mL from one supernatant bag via luer sample port and placed in a 15 mL conical. [00235] Performed “TIL G-REX Harvest” protocol and determined the final product target volume. Loaded disposable kit. Removed filtrate bag. Entered Filtrate capacity. Placed Filtrate container on benchtop. Attached PlasmaLyte. Verified that the PlasmaLyte was attached and observed that the PlasmaLyte is moving. Attached Source container to tubing and verified Source container was attached. Confirmed PlasmaLyte was moving. [00236] Final Formulation and Fill. Target volume/bag calculation. Calculated volume of CS-10 and LOVO wash buffer to formulate blank bag. Prepared CRF Blank. [00237] Calculated the volume of IL-2 to add to the Final Product. Final IL-2 Concentration desired (IU/mL) – 300IU/mL. IL-2 working stock: 6 × 10
4 IU/mL. Assembled connect apparatus. Sterile welded a 4S-4M60 to a CC2 cell connection. Sterile welded the CS750 cryobags to the harness prepared. Welded CS-10 bags to spikes of the 4S-4M60. Prepared TIL with IL-2. Using an appropriately sized syringe, removed amount of IL-2 determined from the “IL-26x10
4” aliquot. Labeled forumlated TIL Bag. Added the formulated TIL bag to the apparatus. Added CS10. Switched Syringes. Drew ~10 mL of air into a 100 mL syringe and replaced the 60 mL syringe on the apparatus. Added CS10. Prepared CS-750 bags. Dispensed cells. [00238] Removed air from final product bags and take retain. Once the last final product bag was filled, closed all clamps. Drew 10 mL of air into a new 100 mL syringe and replace the syringe on the apparatus. Dispensed retain into a 50 mL conical tube and label tube as “Retain” and lot number. Repeat air removal step for each bag. [00239] Prepared final product for cryopreservation, including visual inspection. Held the cryobags on cold pack or at 2-8°C until cryopreservation. [00240] Removed cell count sample. Using an appropriately sized pipette, remove 2.0 mL of retain and place in a 15 mL conical tube to be used for cell counts. Performed cell counts and calculations. NOTE: Diluted only one sample to appropriate dilution to verify dilution is sufficient. Diluted additional samples to appropriate dilution factor and proceed with counts. Determined the Average of Viable Cell Concentration and Viability of the cell counts performed. Determined Upper and Lower Limit for counts. NOTE: Dilution may be adjusted according based off the expected concentration of cells. Determined the Average of DB1/ 149202201.1 149 Attorney Docket No.: 116983-5127-WO Viable Cell Concentration and Viability. Determined Upper and Lower Limit for counts. Calculated IFN-γ. Heat Sealed Final Product bags. [00241] Labeled and collected samples per exemplary sample plan below. TABLE 17. Sample plan.

[00242] Sterility and BacT testing. Testing Sampling. In the BSC, remove a 1.0 mL sample from the retained cell suspension collected using an appropriately sized syringe and inoculate the anaerobic bottle. Repeat the above for the aerobic bottle. [00243] Final Product Cryopreservation. Prepared controlled rate freezer (CRF). Verified the CRF had been set up. Set up CRF probes. Placed final product and samples in CRF. Determined the time needed to reach 4 ºC ± 1.5 ºC and proceed with the CRF run. CRF completed and stored. Stopped the CRF after the completion of the run. Remove cassettes and vials from CRF. Transferred cassettes and vials to vapor phase LN2 for storage. Recorded storage location. [00244] Post-Processing and analysis of final drug product included the following tests: (Day 22) Determination of CD3+ cells on Day 22 REP by flow cytometry; (Day 22) Gram staining method (GMP); (Day 22) Bacterial endotoxin test by Gel Clot LAL Assay (GMP); (Day 16) BacT Sterility Assay (GMP); (Day 16) Mycoplasma DNA detection by TD-PCR (GMP); Acceptable appearance attributes; (Day 22) BacT sterility assay DB1/ 149202201.1 150 Attorney Docket No.: 116983-5127-WO (GMP)(Day 22); (Day 22) IFN-gamma assay. Other potency assay as described herein are also employed to analyze TIL products. EXAMPLE 3: STUDY OF AUTOLOGOUS TUMOR INFILTRATING LYMPHOCYTES PLUS SACITUZUMAB GOVITECAN IN PATIENTS WITH METASTATIC NON-SMALL-CELL LUNG CANCER [00225] This example describes a Phase 1/2 clinical trial for treating patients with metastatic non-small-cell lung cancer (NSCLC) with lifileucel, an autologous TIL product (commercially available as Amtagvi
TM from Iovance Biotherapeutics, Inc.) and the TROP-2 ADC sacituzumab govitecan (commercially available as Trodelvy from Gilead Sciences, Inc.). TIL products may also be expanded from patient tumor samples using the Gen 2 process as described in Example 2 above, or from other processes or sources known in the art, e.g., as described in International Patent Publication No. WO 2023/039488 A1, and U.S. Patent Nos.8,556,882 and 11,384,337, the contents of which are hereby incorporated by reference in their entireties. Other TROP-2 ADCs including datopotamab deruxtecan may be substituted for sacituzumab govitecan with suitable adjustments known to those of skill in the art. [00226] The indication for the trial is patients with metastatic non-small-cell lung cancer (NSCLC) without an actionable driver mutation who have disease progression on or following systemic therapy with immune checkpoint inhibitor (CPI) and chemotherapy. [00227] The primary objectives of the trial are to evaluate the safety of the combination of lifileucel and sacituzumab govitecan in patients with metastatic NSCLC without an actionable driver mutation who have disease progression on or following systemic therapy with immune checkpoint inhibitor (CPI) and chemotherapy and to evaluate the efficacy of combination of lifileucel and sacituzumab govitecan, as determined by objective response rate (ORR) using RECIST v1.1. Secondary objectives include the further evaluatation of the efficacy combination of lifileucel and sacituzumab govitecan using complete response (CR) rate, duration of response (DOR) or median DOR, disease control rate (DCR), progression-free survival (PFS), and overall survival (OS). [00228] This example describes a Phase 1b/2, open-label, dose-escalation study of lifileucel and sacituzumab govitecan in patients with pretreated, advanced or metastatic non- small cell lung cancer. This study may determine a recommended phase 2 dose (RP2D) of the DB1/ 149202201.1 151 Attorney Docket No.: 116983-5127-WO combination of lifileucel and sacituzumab govitecan and then proceed immediately to a Phase 2 portion. Up to three dose levels may be evaluated in the Phase 1b portion. Once the MTD/RP2D is determined or confirmed, additional patients will be enrolled in the dose expansion cohort. Up to 150 patients may be enrolled in total for both dose escalation and expansion cohorts. The study will be conducted in conformity with Good Clinical Practice (GCP). [00229] All patients will receive lifileucel and sacituzumab govitecan therapy, consisting of these steps: 1. Harvest (surgical resection) to provide the autologous tissue that serves as the source of TILs. A fraction of the tissue will be digested and frozen for later characterization of neo-antigen reactive TILs. 2. Production of lifileucel (TIL) investigational product (IP) at a central Good Manufacturing Practice (GMP) facility. 3. A 5-day nonmyeloablative lymphodepletion (NMA-LD) preconditioning regimen (Days -5 to -1). 4. Infusion of the TIL product (Day 0). 5. Administration of IV sacituzumab govitecan (see below). [00230] The following general sequential periods will occur in all patients, unless otherwise specified: 1. Screening Period: From informed consent form (ICF) signature to enrollment. 2. Pre-treatment Period: From enrollment to initiation of preparative NMA-LD regimen. 3. Treatment Period: From initiation of preparative NMA-LD to End-of- Treatment (EOT) Visit, including NMA-LD (Days -5 to -1), TIL infusion (Day 0), followed by sacituzumab govitecan administrations. The EOT occurs approximately 65 days after Day 0. 4. Posttreatment Follow-up Period, which is composed of: a. Post-treatment Efficacy Follow-up Period (TEFU): From the EOT visit to the last efficacy assessment visit (End-of-Efficacy Assessment [EOEA] DB1/ 149202201.1 152 Attorney Docket No.: 116983-5127-WO visit), which will be defined as a visit occurring either at the time of disease progression or start of a new anticancer therapy, whichever comes first. b. Long-Term Follow-up Period (LTFU): From EOEA, as described above, to study completion. [00231] Study participants (enrolled patients) may transition early to LTFU (e.g., at partial withdrawal of consent, or if is determined that they will not receive the combination of sacituzumab govitecan therapy for any reason). Early study discontinuation is prompted by either consent withdrawal, death, lost to follow-up, or study termination. Doses and Treatment Schedule: Combination of Lifileucel and Sacituzumab Govitecan [00232] Tumor resection for TIL production may be performed by excisional biopsy or by core biopsies, as described above and in US 2020/0277573 A1, the disclosure of which is incorporated by reference herein in its entirety. [00233] Sacituzumab govitecan administration may begin at reduced doses, such as 1 mg/kg, 2 mg/kg, 3 mg/kg, or 5 mg/kg, up to 10 mg/kg, during the Phase 1b portion of the trial. Sacituzumab govitecan administration occurs by intravenous infusion on day 1 and day 8 of continuous 21-day cycles. During the Phase 2 portion of the trial, if the expected RP2D is reached, 10 mg/kg of sacituzumab govitecan is administered on days 1 and 8 of continuous 21-day cycles. The days of the 21-day cycle may be adjusted relative to the days for the lifileucel administration in three alternative schedules: 1. Resection before the first 21-day cycle, before the first day 1 of sacituzumab govitecan administration, and administration of lifileucel during the first 21-day cycle: resection of tumor for lifileucel (TIL) production can be performed prior to commencement of sacituzumab govitecan. For example, lifileucel production, release testing, and shipping can be performed in approximately 30 to 34 days. The five-day lymphodepletion cycle can be started once lifileucel arrives at the clinical site, followed by a rest day if warranted, followed by infusion of lifileucel and up to six courses of high-dose aldesleukin (e.g., 600,000 IU/kg every 8 to 12 hours) as described elsewhere herein, for a total of 37 to 43 days. As a result, tumor resection can occur on approximately day -27 to day -33 (relative to day 1 of sacituzumab govitecan), allowing lifileucel infusion to occur on day 10 to 14 (relative to day 1 of DB1/ 149202201.1 153 Attorney Docket No.: 116983-5127-WO sacituzumab govitecan). Adjustments can be made based on the health status of the patient. 2. Resection during the first 21-day cycle, between the period between day 8 and day 21, and administration of lifileucel during the second 21-day cycle: resection of tumor for lifileucel (TIL) production can be performed during the first 21-day cycle, for example between days 1 and 8. Administration of lifileucel can then be performed following lymphodepletion on approximately day 18 of the second 21-day cycle of sacituzumab govitecan (i.e., 38 days from the first dose of sacituzumab govitecan). The 21-day cycle of sacituzumab govitecan can also be extended slightly to accommodate delays in receipt of the TIL product, lymphodepletion, or patient health factors. 3. Resection during the second or later 21-day cycle, between the period between day 8 and day 21, and administration of lifileucel during the third or later 21-day cycle: resection of tumor for lifileucel (TIL) production can be performed during the second or a later sacituzumab govitecan, following the approach described above, with adjustments as necessary. [00234] Patients will undergo a 5-day preconditioning NMA-LD regimen that will be initiated prior to the planned combination of lifileucel and sacituzumab govitecan infusion. The NMA-LD regimen consists of 2 days of intravenous (IV) cyclophosphamide (60 mg/kg) with mesna (per site standard of care or USPI/SmPC) on days -5 and -4, and 5 days of fludarabine IV (25 mg/m
2, days -5 through -1), as shown in Table 18. TABLE 18. Lymphodepletion and treatment regimen.
[00235] Sacituzumab govitecan may continue indefinitely, or for up to 10 to 12 cycles. Sacituzumab govitecan should be withheld for absolute neutrophil counts below DB1/ 149202201.1 154 Attorney Docket No.: 116983-5127-WO 1500/mm
3 on Day 1 of any cycle or neutrophil count below 1000/mm
3 on Day 8 of any cycle and for neutropenic fever. Administer granulocyte-colony stimulating factor as clinically needed. Discontinue for grade 4 infusion reactions. [00236] Overall, the study participation time will be up to 5 years from treatment to completion. Inclusion Criteria [00237] To be eligible for study participation, patients must meet all of the following inclusion criteria prior to enrollment: 1. Provide written informed consent and written authorization for use and disclosure of protected health information. 2. Be ≥ 18 years of age at the time of consent. 3. Have histologically or pathologically confirmed diagnosis of NSCLC (squamous, nonsquamous, adenocarcinoma, large cell, or mixed histologies), and must have documented PD-L1 expression status, as determined by the tumor proportion score (TPS) prior to the CPI treatment that they received. 4. Have received a systemic therapy that included checkpoint inhibitor(s) and chemotherapy with documented radiographic disease progression thereon. (Prior systemic therapy in the adjuvant or neoadjuvant setting, or as part of definitive chemoradiotherapy, will not be counted as a line of therapy if the disease has not progressed during or within 12 months of the completion of such therapy.) 5. Have documented no signs or symptoms of ischemia or clinically significant arrhythmias. 6. Have Eastern Cooperative Oncology Group (ECOG) performance status of 0 or 1 and an estimated life expectancy of > 6 months. 7. Have at least one resectable lesion (or aggregate lesions) of a minimum 1.5 cm in diameter for TIL production. If the lesion considered for harvest is within a previously irradiated field, the lesion must have demonstrated radiographic progression prior to harvest, and the irradiation must have been completed at least 3 months prior to enrollment. DB1/ 149202201.1 155 Attorney Docket No.: 116983-5127-WO Patients must have an adequate histopathology specimen for protocol-required testing. 8. Following tumor harvest for TIL manufacturing, all patients must have at least one remaining measurable lesion, as defined by RECIST v1.1, with the following considerations: a. Lesions in previously irradiated areas should not be selected as target lesions unless progression has been demonstrated in those lesions and the irradiation has been completed at least 3 months prior to enrollment. b. Lesions that are surgically partially resected for TIL generation that are still measurable per RECIST v1.1 may be selected as nontarget lesions but cannot serve as a target lesion for response assessment. 9. Have the following hematologic parameters independent of transfusions and/or blood product support for at least 5 days prior to laboratory testing: a. Absolute neutrophil count (ANC) ≥ 1000/mm
3 b. Hemoglobin ≥ 9.0 g/dL c. Platelet count ≥ 100,000/mm
3 10. Have adequate organ function with the following laboratory values: a. Serum alanine aminotransferase (ALT) and aspartate aminotransferase (AST) ≤ 3 times the upper limit of normal (≤ 3 × ULN); patients with liver metastasis ≤ 5 × ULN b. Estimated creatinine clearance (eCrCl) ≥ 40 mL/min using the Cockcroft- Gault formula at Screening c. Total bilirubin ≤ 2 mg/dL; patients with Gilbert’s Syndrome must have a total bilirubin ≤ 3 mg/dL 11. Have a left ventricular ejection fraction (LVEF) > 45% and be New York Heart Association (NYHA) Class 1; a cardiac stress test is required for all patients and must demonstrate no irreversible wall movement abnormality. Patients with an abnormal cardiac stress test may be enrolled if they have adequate ejection fraction and cardiology clearance. DB1/ 149202201.1 156 Attorney Docket No.: 116983-5127-WO 12. Screening pulmonary function testing (PFT) is required for patients having any of the following: a. History of cigarette smoking of ≥ 20 pack-years b. Ceased smoking within the past 2 years or still smoking c. History of chronic obstructive pulmonary disease (COPD) d. Any signs or symptoms of respiratory dysfunction Post-bronchodilator values: forced expiratory volume (FEV1)/forced vital capacity (FVC) > 70% or FEV1 > 50% of predicted normal are required for study entry. If a patient is unable to perform reliable spirometry due to abnormal upper airway anatomy (ie, tracheostomy), a 6-minute walk test may be used to assess pulmonary function. Patients who are unable to walk a distance of at least 80% predicted for age and sex or who demonstrate evidence of hypoxia at any point during the test (SpO
2 < 90%) are excluded. 13. Must have a washout period from prior systemic anticancer therapy of a minimum duration of 21 days prior to enrollment. Palliative radiation therapy: prior external beam radiation is allowed, provided all radiation-related toxicities are resolved to Grade 1 or baseline, excluding alopecia, skin pigmentation change, or other clinically insignificant events, eg, small area radiation dermatitis or rectal or urinary urgency. Surgery/pre-planned procedure(s): previous surgical procedure(s) is/are permitted, provided that wound healing has occurred, all complications have resolved, and at least 14 days have elapsed (for major operative procedures) prior to the tumor resection. 14. Must have recovered from all prior anticancer treatment-related adverse events (TRAEs) to Grade ≤ 1 (per Common Terminology Criteria for Adverse Events [CTCAE], Version 5.0), except for alopecia or vitiligo. Patients with stable Grade ≥ 2 toxicity from prior anticancer therapy may be considered on a case-by-case basis. DB1/ 149202201.1 157 Attorney Docket No.: 116983-5127-WO 15. Patients of childbearing potential or those with partners of childbearing potential must be willing to practice an approved method of highly effective birth control during treatment and for 12 months after receiving all protocol- related therapy. Approved methods of birth control are as follows: a. Combined (estrogen and progesterone containing) hormonal birth control associated with inhibition of ovulation: oral, intravaginal, transdermal b. Progesterone-only hormonal birth control associated with inhibition of ovulation: oral, injectable, implantable c. Intrauterine device (IUD) d. Intrauterine hormone-releasing system (IUS) e. Bilateral tubal occlusion f. Vasectomized partner g. True absolute sexual abstinence when this is in line with the preferred and usual lifestyle of the patient. Periodic abstinence (e.g., calendar ovulation, symptothermal, post-ovulation methods) is unacceptable. Exclusion Criteria [00238] To be eligible for study participation, patients must meet none of the following criteria prior to enrollment: 1. Have received an organ allograft or prior cell transfer therapy that included a nonmyeloablative or myeloablative chemotherapy regimen within the past 20 years. 2. Have known oncogene driver mutations (eg, EGFR, ALK, ROS) which are sensitive to targeted therapies. 3. Have symptomatic and/or untreated brain metastases, with the following considerations: a. Patients with historical (prior to consenting for study participation) definitively treated brain metastases will be considered for enrollment if, prior to enrollment the patient is clinically stable for ≥ 2 weeks, there are no DB1/ 149202201.1 158 Attorney Docket No.: 116983-5127-WO new brain lesions via screening magnetic resonance imaging (MRI), and the patient does not require ongoing corticosteroid treatment. b. Patients with recently (within 28 days prior to enrollment), definitively treated brain metastases will be considered for enrollment if, prior to enrollment the metastases are asymptomatic, and the patient is clinically stable for ≥ 2 weeks. Prior to initiation of NMA-LD, the following are required: a repeat brain MRI at least 4 weeks posttreatment demonstrating that there are no new or increasing brain lesions, and confirmation that patient is clinically stable for ≥ 2 weeks and does not require ongoing steroid treatment. 4. Require systemic steroid therapy > 10 mg/day of prednisone or other steroid equivalent dose. Patients receiving steroids as replacement therapy for adrenocortical insufficiency at ≤ 10 mg/day of prednisone or another steroid equivalent dose are not excluded. 5. Have weight loss > 10% since metastatic NSCLC diagnosis. 6. Have serologic evidence of any of the following: a. Human immunodeficiency virus (HIV)-1 or HIV-2 b. Serologic evidence of active or chronic hepatitis B virus (HBV) or hepatitis C virus (HCV) infection. Patients with acute or chronic hepatitis infections may be enrolled if the viral load by polymerase chain reaction (PCR) is undetectable with/without active treatment c. Syphilis (rapid plasma reagin [RPR] test or venereal disease research laboratory [VDRL]) test) d. Cytomegalovirus (CMV) immunoglobulin M (IgM) antibody and viral load PCR; and Epstein-Barr virus (EBV) IgM and viral load PCR, indicating active infection e. Herpes simplex virus (HSV)-1 and HSV-2 IgM serology and viral load PCR. 7. Be pregnant or breastfeeding. 8. Have active medical illness(es) that would pose increased risks for study participation, such as systemic infections (eg, syphilis or any other infection DB1/ 149202201.1 159 Attorney Docket No.: 116983-5127-WO requiring antibiotics); coagulation disorders; other active major medical illnesses of the cardiovascular, respiratory, or immune systems; or any history of COVID- 19 infection with residual pulmonary compromise. 9. Have received a live or attenuated vaccination within 28 days prior to the start of NMA-LD. 10. Have any form of primary immunodeficiency (eg, severe combined immunodeficiency disease [SCID] or acquired immune deficiency syndrome [AIDS]). 11. Have a history of hypersensitivity to any component of the study drugs. Combination of TIL and sacituzumab govitecan should not be administered to patients with a known hypersensitivity to any component of the autologous TIL product formulation, including, but not limited to, any of the following: a. NMA-LD (cyclophosphamide, mesna, and fludarabine) b. Sacituzumab govitecan c. Antibiotics of the aminoglycoside group (ie, streptomycin, gentamicin [excluding those who are skin-test negative for gentamicin hypersensitivity]) d. Any component of the TIL product formulation, including dimethyl sulfoxide (DMSO), human serum albumin (HSA), or dextran-40 12. Have had another primary malignancy within the previous 3 years (except for those that do not require treatment or have been curatively treated > 1 year ago, and does not pose a significant risk of recurrence including, but not limited to, non-melanoma skin cancer, ductal carcinoma in situ (DCIS), lobular carcinoma in situ (LCIS), prostate cancer Gleason score ≤ 6, or superficial bladder cancer). 13. Participated in another clinical study with an investigational product within 21 days of the initiation of treatment. [00239] Efficacy assessment: The following efficacy parameters will be determined: ORR, CR rate, DOR, DCR, PFS, and OS. [00240] Safety assessment: For all study participants, AEs/SAEs are collected and graded as per CTCAE v5.0, during the following study periods: Screening, Pre-treatment, DB1/ 149202201.1 160 Attorney Docket No.: 116983-5127-WO Treatment, and Posttreatment Follow-up Period (as applicable). Analyses includes all study periods and will be performed separately for each cohort. [00241] A trial design as shown above may be used for a registrational trial for the combination of sacituzumab govitecan, or another TROP-2 ADC, and lifileucel, or another TIL therapy. The registrational trial may be performed in NSCLC patients, as described above, or in other indications described herein, such as breast cancer, including triple- negative breast cancer (TNBC), urothelial cancer, ovarian cancer, endometrial cancer, pancreatic cancer, bladder cancer, or head and neck cancer. Patient subpopulations may be enrolled as the trial population as well. For example, TNBC patients with unresectable locally advanced or metastatic triple-negative breast cancer who have received two or more prior systemic therapies, at least one of them for metastatic disease. Similarly, breast cancer patients with unresectable locally advanced or metastatic hormone receptor (HR)- positive, human epidermal growth factor receptor 2 (HER2)-negative (IHC 0, IHC 1+ or IHC 2+/ISH–) breast cancer who have received endocrine-ebased therapy and at least two additional systemic therapies in the metastatic setting may be enrolled as the trial population. * * * [00242] The examples set forth above are provided to give those of ordinary skill in the art a complete disclosure and description of how to make and use the embodiments of the compositions, systems and methods of the invention, and are not intended to limit the scope of what the inventors regard as their invention. Modifications of the above-described modes for carrying out the invention that are obvious to persons of skill in the art are intended to be within the scope of the following claims. All patents and publications mentioned in the specification are indicative of the levels of skill of those skilled in the art to which the invention pertains. [00243] All headings and section designations are used for clarity and reference purposes only and are not to be considered limiting in any way. For example, those of skill in the art will appreciate the usefulness of combining various aspects from different headings and sections as appropriate according to the spirit and scope of the invention described herein. DB1/ 149202201.1 161 Attorney Docket No.: 116983-5127-WO [00244] All references cited herein are hereby incorporated by reference herein in their entireties and for all purposes to the same extent as if each individual publication or patent or patent application was specifically and individually indicated to be incorporated by reference in its entirety for all purposes. [00245] Many modifications and variations of this application can be made without departing from its spirit and scope, as will be apparent to those skilled in the art. The specific embodiments and examples described herein are offered by way of example only, and the application is to be limited only by the terms of the appended claims, along with the full scope of equivalents to which the claims are entitled. DB1/ 149202201.1 162
 [0017] In some embodiments, an IL-2 form suitable for use in the invention includes a antibody cytokine engrafted protein comprises a heavy chain variable region (VH), comprising complementarity determining regions HCDR1, HCDR2, HCDR3; a light chain variable region (VL), comprising LCDR1, LCDR2, LCDR3; and an IL-2 molecule or a fragment thereof engrafted into a CDR of the VH or the VL, wherein the antibody cytokine engrafted protein preferentially expands T effector cells over regulatory T cells. In some embodiments, the antibody cytokine engrafted protein comprises a heavy chain variable region (VH), comprising complementarity determining regions HCDR1, HCDR2, HCDR3; a light chain variable region (VL), comprising LCDR1, LCDR2, LCDR3; and an IL-2 molecule or a fragment thereof engrafted into a CDR of the VH or the VL, wherein the IL-2 molecule is a mutein, and wherein the antibody cytokine engrafted protein preferentially expands T effector cells over regulatory T cells. In some embodiments, the IL-2 regimen comprises administration of an antibody described in U.S. Patent Application Publication No. US 2020/0270334 A1, the disclosures of which are incorporated by reference herein. In some embodiments, the antibody cytokine engrafted protein comprises a heavy chain variable region (VH), comprising complementarity determining regions HCDR1, HCDR2, HCDR3; a light chain variable region (VL), comprising LCDR1, LCDR2, LCDR3; and an IL-2 molecule or a fragment thereof engrafted into a CDR of the VH or the VL, wherein the IL-2 molecule is a mutein, wherein the antibody cytokine engrafted protein preferentially expands T effector cells over regulatory T cells, and wherein the antibody further comprises an IgG class heavy chain and an IgG class light chain selected from the group consisting of: a IgG class light chain comprising SEQ ID NO:39 and a IgG class heavy chain comprising SEQ ID NO:38; a IgG class light chain comprising SEQ ID NO:37 and a IgG class heavy chain comprising SEQ ID NO:29; a IgG class light chain comprising SEQ ID NO:39 and a IgG class heavy chain comprising SEQ ID NO:29; and a IgG class light chain comprising SEQ ID NO:37 and a IgG class heavy chain comprising SEQ ID NO:38. DB1/ 149202201.1 16 Attorney Docket No.: 116983-5127-WO [0018] In some embodiments, an IL-2 molecule or a fragment thereof is engrafted into HCDR1 of the VH, wherein the IL-2 molecule is a mutein. In some embodiments, an IL-2 molecule or a fragment thereof is engrafted into HCDR2 of the VH, wherein the IL-2 molecule is a mutein. In some embodiments, an IL-2 molecule or a fragment thereof is engrafted into HCDR3 of the VH, wherein the IL-2 molecule is a mutein. In some embodiments, an IL-2 molecule or a fragment thereof is engrafted into LCDR1 of the VL, wherein the IL-2 molecule is a mutein. In some embodiments, an IL-2 molecule or a fragment thereof is engrafted into LCDR2 of the VL, wherein the IL-2 molecule is a mutein. In some embodiments, an IL-2 molecule or a fragment thereof is engrafted into LCDR3 of the VL, wherein the IL-2 molecule is a mutein. [0019] The insertion of the IL-2 molecule can be at or near the N-terminal region of the CDR, in the middle region of the CDR or at or near the C-terminal region of the CDR. In some embodiments, the antibody cytokine engrafted protein comprises an IL-2 molecule incorporated into a CDR, wherein the IL2 sequence does not frameshift the CDR sequence. In some embodiments, the antibody cytokine engrafted protein comprises an IL-2 molecule incorporated into a CDR, wherein the IL-2 sequence replaces all or part of a CDR sequence. The replacement by the IL-2 molecule can be the N-terminal region of the CDR, in the middle region of the CDR or at or near the C-terminal region the CDR. A replacement by the IL-2 molecule can be as few as one or two amino acids of a CDR sequence, or the entire CDR sequences. [0020] In some embodiments, an IL-2 molecule is engrafted directly into a CDR without a peptide linker, with no additional amino acids between the CDR sequence and the IL-2 sequence. In some embodiments, an IL-2 molecule is engrafted indirectly into a CDR with a peptide linker, with one or more additional amino acids between the CDR sequence and the IL-2 sequence. [0021] In some embodiments, the IL-2 molecule described herein is an IL-2 mutein. In some instances, the IL-2 mutein comprising an R67A substitution. In some embodiments, the IL-2 mutein comprises the amino acid sequence SEQ ID NO:14 or SEQ ID NO:15. In some embodiments, the IL-2 mutein comprises an amino acid sequence in Table 1 in U.S. Patent Application Publication No. US 2020/0270334 A1, the disclosure of which is incorporated by reference herein. DB1/ 149202201.1 17 Attorney Docket No.: 116983-5127-WO [0022] In some embodiments, the antibody cytokine engrafted protein comprises an HCDR1 selected from the group consisting of SEQ ID NO:16, SEQ ID NO:19, SEQ ID NO:22 and SEQ ID NO:25. In some embodiments, the antibody cytokine engrafted protein comprises an HCDR1 selected from the group consisting of SEQ ID NO:7, SEQ ID NO:10, SEQ ID NO:13 and SEQ ID NO:16. In some embodiments, the antibody cytokine engrafted protein comprises an HCDR1 selected from the group consisting of HCDR2 selected from the group consisting of SEQ ID NO:17, SEQ ID NO:20, SEQ ID NO:23, and SEQ ID NO:26. In some embodiments, the antibody cytokine engrafted protein comprises an HCDR3 selected from the group consisting of SEQ ID NO:18, SEQ ID NO:21, SEQ ID NO:24, and SEQ ID NO:27. In some embodiments, the antibody cytokine engrafted protein comprises a VH region comprising the amino acid sequence of SEQ ID NO:28. In some embodiments, the antibody cytokine engrafted protein comprises a heavy chain comprising the amino acid sequence of SEQ ID NO:29. In some embodiments, the antibody cytokine engrafted protein comprises a VL region comprising the amino acid sequence of SEQ ID NO:36. In some embodiments, the antibody cytokine engrafted protein comprises a light chain comprising the amino acid sequence of SEQ ID NO:37. In some embodiments, the antibody cytokine engrafted protein comprises a VH region comprising the amino acid sequence of SEQ ID NO:28 and a VL region comprising the amino acid sequence of SEQ ID NO:36. In some embodiments, the antibody cytokine engrafted protein comprises a heavy chain region comprising the amino acid sequence of SEQ ID NO:29 and a light chain region comprising the amino acid sequence of SEQ ID NO:37. In some embodiments, the antibody cytokine engrafted protein comprises a heavy chain region comprising the amino acid sequence of SEQ ID NO:29 and a light chain region comprising the amino acid sequence of SEQ ID NO:39. In some embodiments, the antibody cytokine engrafted protein comprises a heavy chain region comprising the amino acid sequence of SEQ ID NO:38 and a light chain region comprising the amino acid sequence of SEQ ID NO:37. In some embodiments, the antibody cytokine engrafted protein comprises a heavy chain region comprising the amino acid sequence of SEQ ID NO:38 and a light chain region comprising the amino acid sequence of SEQ ID NO:39. In some embodiments, the antibody cytokine engrafted protein comprises IgG.IL2F71A.H1 or IgG.IL2R67A.H1 of U.S. Patent Application Publication No. 2020/0270334 A1, or variants, derivatives, or fragments thereof, or conservative amino acid substitutions thereof, or proteins with at least 80%, at least 90%, at least 95%, or at least 98% sequence identity thereto. In some embodiments, the antibody components of the antibody DB1/ 149202201.1 18 Attorney Docket No.: 116983-5127-WO cytokine engrafted protein described herein comprise immunoglobulin sequences, framework sequences, or CDR sequences of palivizumab. In some embodiments, the antibody cytokine engrafted protein described herein has a longer serum half-life that a wild-type IL-2 molecule such as, but not limited to, aldesleukin or a comparable molecule. In some embodiments, the antibody cytokine engrafted protein described herein has a sequence as set forth in Table 3. TABLE 3: Sequences of exemplary palivizumab antibody-IL-2 engrafted proteins Identifier Sequence (One-Letter Amino Acid Symbols) SEQ ID NO:13 MYRMQLLSCI ALSLALVTNS APTSSSTKKT QLQLEHLLLD LQMILNGINN YKNPKLTRML IL-2 60 TFKFYMPKKA TELKHLQCLE EELKPLEEVL NLAQSKNFHL RPRDLISNIN VIVLELKGSE 120 TTFMCEYADE TATIVEFLNR WITFCQSIIS TLT 153 SEQ ID NO:14 APTSSSTKKT QLQLEHLLLD LQMILNGINN YKNPKLTAML TFKFYMPKKA TELKHLQCLE IL-2 mutein 60 EELKPLEEVL NLAQSKNFHL RPRDLISNIN VIVLELKGSE TTFMCEYADE TATIVEFLNR 120 WITFCQSIIS TLT 133 SEQ ID NO:15 APTSSSTKKT QLQLEHLLLD LQMILNGINN YKNPKLTRML TAKFYMPKKA TELKHLQCLE IL-2 mutein 60 EELKPLEEVL NLAQSKNFHL RPRDLISNIN VIVLELKGSE TTFMCEYADE TATIVEFLNR 120 WITFCQSIIS TLT 133 SEQ ID NO:16 GFSLAPTSSS TKKTQLQLEH LLLDLQMILN GINNYKNPKL TAMLTFKFYM PKKATELKHL HCDR1_IL-2 60 QCLEEELKPL EEVLNLAQSK NFHLRPRDLI SNINVIVLEL KGSETTFMCE YADETATIVE 120 FLNRWITFCQ SIISTLTSTS GMSVG 145 SEQ ID NO:17 DIWWDDKKDY NPSLKS 16 HCDR2 SEQ ID NO:18 SMITNWYFDV 10 HCDR3 SEQ ID NO:19 APTSSSTKKT QLQLEHLLLD LQMILNGINN YKNPKLTAML TFKFYMPKKA TELKHLQCLE HCDR1_IL-2 60 kabat EELKPLEEVL NLAQSKNFHL RPRDLISNIN VIVLELKGSE TTFMCEYADE TATIVEFLNR 120 WITFCQSIIS TLTSTSGMSV G 141 SEQ ID NO:20 DIWWDDKKDY NPSLKS 16 HCDR2 kabat SEQ ID NO:21 SMITNWYFDV 10 HCDR3 kabat SEQ ID NO:22 GFSLAPTSSS TKKTQLQLEH LLLDLQMILN GINNYKNPKL TAMLTFKFYM PKKATELKHL HCDR1_IL-2 60 clothia QCLEEELKPL EEVLNLAQSK NFHLRPRDLI SNINVIVLEL KGSETTFMCE YADETATIVE 120 FLNRWITFCQ SIISTLTSTS GM 142 SEQ ID NO:23 WWDDK HCDR2 clothia 5 SEQ ID NO:24 SMITNWYFDV 10 HCDR3 clothia SEQ ID NO:25 GFSLAPTSSS TKKTQLQLEH LLLDLQMILN GINNYKNPKL TAMLTFKFYM PKKATELKHL HCDR1_IL-2 60 IMGT QCLEEELKPL EEVLNLAQSK NFHLRPRDLI SNINVIVLEL KGSETTFMCE YADETATIVE 120 FLNRWITFCQ SIISTLTSTS GMS 143 SEQ ID NO:26 IWWDDKK HCDR2 IMGT 7 SEQ ID NO:27 ARSMITNWYF DV 12 HCDR3 IMGT SEQ ID NO:28 QVTLRESGPA LVKPTQTLTL TCTFSGFSLA PTSSSTKKTQ LQLEHLLLDL QMILNGINNY V 60 KNPKLTAMLT FKFYMPKKAT ELKHLQCLEE ELKPLEEVLN LAQSKNFHLR PRDLISNINV 120 IVLELKGSET TFMCEYADET ATIVEFLNRW ITFCQSIIST LTSTSGMSVG WIRQPPGKAL 180 DB1/ 149202201.1 19 Attorney Docket No.: 116983-5127-WO EWLADIWWDD KKDYNPSLKS RLTISKDTSK NQVVLKVTNM DPADTATYYC ARSMITNWYF 240 DVWGAGTTVT VSS 253 SEQ ID NO:29 QMILNGINNY KNPKLTAMLT FKFYMPKKAT ELKHLQCLEE ELKPLEEVLN LAQSKNFHLR Heavy chain 60 PRDLISNINV IVLELKGSET TFMCEYADET ATIVEFLNRW ITFCQSIIST LTSTSGMSVG 120 WIRQPPGKAL EWLADIWWDD KKDYNPSLKS RLTISKDTSK NQVVLKVTNM DPADTATYYC 180 ARSMITNWYF DVWGAGTTVT VSSASTKGPS VFPLAPSSKS TSGGTAALGC LVKDYFPEPV 240 TVSWNSGALT SGVHTFPAVL QSSGLYSLSS VVTVPSSSLG TQTYICNVNH KPSNTKVDKR 300 VEPKSCDKTH TCPPCPAPEL LGGPSVFLFP PKPKDTLMIS RTPEVTCVVV AVSHEDPEVK 360 FNWYVDGVEV HNAKTKPREE QYNSTYRVVS VLTVLHQDWL NGKEYKCKVS NKALAAPIEK 420 TISKAKGQPR EPQVYTLPPS REEMTKNQVS LTCLVKGFYP SDIAVEWESN GQPENNYKTT 480 PPVLDSDGSF FLYSKLTVDK SRWQQGNVFS CSVMHEALHN HYTQKSLSLS PGK 533 SEQ ID NO:30 KAQLSVGYMH 10 LCDR1 kabat SEQ ID NO:31 DTSKLAS 7 LCDR2 kabat SEQ ID NO:32 FQGSGYPFT 9 LCDR3 kabat SEQ ID NO:33 QLSVGY 6 LCDR1 chothia SEQ ID NO:34 DTS 3 LCDR2 chothia SEQ ID NO:35 GSGYPF 6 LCDR3 chothia SEQ ID NO:36 DIQMTQSPST LSASVGDRVT ITCKAQLSVG YMHWYQQKPG KAPKLLIYDT SKLASGVPSR 60 V FSGSGSGTEF TLTISSLQPD DFATYYCFQG SGYPFTFGGG TKLEIK 106 SEQ ID NO:37 DIQMTQSPST LSASVGDRVT ITCKAQLSVG YMHWYQQKPG KAPKLLIYDT SKLASGVPSR 60 Light chain FSGSGSGTEF TLTISSLQPD DFATYYCFQG SGYPFTFGGG TKLEIKRTVA APSVFIFPPS 120 DEQLKSGTAS VVCLLNNFYP REAKVQWKVD NALQSGNSQE SVTEQDSKDS TYSLSSTLTL 180 SKADYEKHKV YACEVTHQGL SSPVTKSFNR GEC 213 SEQ ID NO:38 QVTLRESGPA LVKPTQTLTL TCTFSGFSLA PTSSSTKKTQ LQLEHLLLDL QMILNGINNY 60 Light chain KNPKLTRMLT AKFYMPKKAT ELKHLQCLEE ELKPLEEVLN LAQSKNFHLR PRDLISNINV 120 IVLELKGSET TFMCEYADET ATIVEFLNRW ITFCQSIIST LTSTSGMSVG WIRQPPGKAL 180 EWLADIWWDD KKDYNPSLKS RLTISKDTSK NQVVLKVTNM DPADTATYYC ARSMITNWYF 240 DVWGAGTTVT VSSASTKGPS VFPLAPSSKS TSGGTAALGC LVKDYFPEPV TVSWNSGALT 300 SGVHTFPAVL QSSGLYSLSS VVTVPSSSLG TQTYICNVNH KPSNTKVDKR VEPKSCDKTH 360 TCPPCPAPEL LGGPSVFLFP PKPKDTLMIS RTPEVTCVVV AVSHEDPEVK FNWYVDGVEV 420 HNAKTKPREE QYNSTYRVVS VLTVLHQDWL NGKEYKCKVS NKALAAPIEK TISKAKGQPR 480 EPQVYTLPPS REEMTKNQVS LTCLVKGFYP SDIAVEWESN GQPENNYKTT PPVLDSDGSF 540 FLYSKLTVDK SRWQQGNVFS CSVMHEALHN HYTQKSLSLS PGK 583 SEQ ID NO:39 DIQMTQSPST LSASVGDRVT ITCKAQLSVG YMHWYQQKPG KAPKLLIYDT SKLASGVPSR 60 Light chain FSGSGSGTEF TLTISSLQPD DFATYYCFQG SGYPFTFGGG TKLEIKRTVA APSVFIFPPS 120 DEQLKSGTAS VVCLLNNFYP REAKVQWKVD NALQSGNSQE SVTEQDSKDS TYSLSSTLTL 180 SKADYEKHKV YACEVTHQGL SSPVTKSFNR GEC 213 [0023] The term “IL-4” (also referred to herein as “IL4”) refers to the cytokine known as interleukin 4, which is produced by Th2 T cells and by eosinophils, basophils, and mast cells. IL-4 regulates the differentiation of naïve helper T cells (Th0 cells) to Th2 T cells. Steinke and Borish, Respir. Res.2001, 2, 66-70. Upon activation by IL-4, Th2 T cells subsequently produce additional IL-4 in a positive feedback loop. IL-4 also stimulates B cell proliferation and class II MHC expression, and induces class switching to IgE and IgG1 expression from B cells. Recombinant human IL-4 suitable for use in the invention is commercially available from multiple suppliers, including ProSpec-Tany TechnoGene Ltd., East Brunswick, NJ, USA (Cat. No. CYT-211) and ThermoFisher Scientific, Inc., Waltham, MA, USA (human DB1/ 149202201.1 20 Attorney Docket No.: 116983-5127-WO IL-15 recombinant protein, Cat. No. Gibco CTP0043). The amino acid sequence of recombinant human IL-4 suitable for use in the invention is given in Table 2 (SEQ ID NO:9). [0024] The term “IL-7” (also referred to herein as “IL7”) refers to a glycosylated tissue- derived cytokine known as interleukin 7, which may be obtained from stromal and epithelial cells, as well as from dendritic cells. Fry and Mackall, Blood 2002, 99, 3892-904. IL-7 can stimulate the development of T cells. IL-7 binds to the IL-7 receptor, a heterodimer consisting of IL-7 receptor alpha and common gamma chain receptor, which in a series of signals important for T cell development within the thymus and survival within the periphery. Recombinant human IL-7 suitable for use in the invention is commercially available from multiple suppliers, including ProSpec-Tany TechnoGene Ltd., East Brunswick, NJ, USA (Cat. No. CYT-254) and ThermoFisher Scientific, Inc., Waltham, MA, USA (human IL-15 recombinant protein, Cat. No. Gibco PHC0071). The amino acid sequence of recombinant human IL-7 suitable for use in the invention is given in Table 2 (SEQ ID NO:10). [0025] The term “IL-15” (also referred to herein as “IL15”) refers to the T cell growth factor known as interleukin-15, and includes all forms of IL-2 including human and mammalian forms, conservative amino acid substitutions, glycoforms, biosimilars, and variants thereof. IL-15 is described, e.g., in Fehniger and Caligiuri, Blood 2001, 97, 14-32, the disclosure of which is incorporated by reference herein. IL-15 shares β and γ signaling receptor subunits with IL-2. Recombinant human IL-15 is a single, non-glycosylated polypeptide chain containing 114 amino acids (and an N-terminal methionine) with a molecular mass of 12.8 kDa. Recombinant human IL-15 is commercially available from multiple suppliers, including ProSpec-Tany TechnoGene Ltd., East Brunswick, NJ, USA (Cat. No. CYT-230-b) and ThermoFisher Scientific, Inc., Waltham, MA, USA (human IL-15 recombinant protein, Cat. No.34-8159-82). The amino acid sequence of recombinant human IL-15 suitable for use in the invention is given in Table 2 (SEQ ID NO:11). [0026] The term “IL-21” (also referred to herein as “IL21”) refers to the pleiotropic cytokine protein known as interleukin-21, and includes all forms of IL-21 including human and mammalian forms, conservative amino acid substitutions, glycoforms, biosimilars, and variants thereof. IL-21 is described, e.g., in Spolski and Leonard, Nat. Rev. Drug. Disc.2014, 13, 379-95, the disclosure of which is incorporated by reference herein. IL-21 is primarily produced by natural killer T cells and activated human CD4+ T cells. Recombinant human IL- 21 is a single, non-glycosylated polypeptide chain containing 132 amino acids with a DB1/ 149202201.1 21 Attorney Docket No.: 116983-5127-WO molecular mass of 15.4 kDa. Recombinant human IL-21 is commercially available from multiple suppliers, including ProSpec-Tany TechnoGene Ltd., East Brunswick, NJ, USA (Cat. No. CYT-408-b) and ThermoFisher Scientific, Inc., Waltham, MA, USA (human IL-21 recombinant protein, Cat. No.14-8219-80). The amino acid sequence of recombinant human IL-21 suitable for use in the invention is given in Table 2 (SEQ ID NO:21). [0027] When “an anti-tumor effective amount”, “a tumor-inhibiting effective amount”, or “therapeutic amount” is indicated, the precise amount of the compositions of the present invention to be administered can be determined by a physician with consideration of individual differences in age, weight, tumor size, extent of infection or metastasis, and condition of the patient (subject). It can generally be stated that a pharmaceutical composition comprising the tumor infiltrating lymphocytes (e.g. secondary TILs or genetically modified cytotoxic lymphocytes) described herein may be administered at a dosage of 104 to 1011 cells/kg body weight (e.g., 105 to 106, 105 to 1010, 105 to 1011, 106 to 1010, 106 to 1011,107 to 1011, 107 to 1010, 108 to 1011, 108 to 1010, 109 to 1011, or 109 to 1010 cells/kg body weight), including all integer values within those ranges. TILs (including in some cases, genetically modified cytotoxic lymphocytes) compositions may also be administered multiple times at these dosages. The TILs (including, in some cases, genetically engineered TILs) can be administered by using infusion techniques that are commonly known in immunotherapy (see, e.g., Rosenberg, et al., New Eng. J. of Med.1988, 319, 1676). The optimal dosage and treatment regime for a particular patient can readily be determined by one skilled in the art of medicine by monitoring the patient for signs of disease and adjusting the treatment accordingly. [0028] The term “microenvironment,” as used herein, may refer to the solid or hematological tumor microenvironment as a whole or to an individual subset of cells within the microenvironment. The tumor microenvironment, as used herein, refers to a complex mixture of “cells, soluble factors, signaling molecules, extracellular matrices, and mechanical cues that promote neoplastic transformation, support tumor growth and invasion, protect the tumor from host immunity, foster therapeutic resistance, and provide niches for dominant metastases to thrive,” as described in Swartz, et al., Cancer Res., 2012, 72, 2473. Although tumors express antigens that should be recognized by T cells, tumor clearance by the immune system is rare because of immune suppression by the microenvironment. DB1/ 149202201.1 22 Attorney Docket No.: 116983-5127-WO [0029] In some embodiments, the invention includes a method of treating a cancer with a population of TILs, wherein a patient is pre-treated with non-myeloablative chemotherapy prior to an infusion of TILs according to the invention. In some embodiments, the population of TILs may be provided wherein a patient is pre-treated with nonmyeloablative chemotherapy prior to an infusion of TILs according to the present invention. In some embodiments, the non-myeloablative chemotherapy is cyclophosphamide 60 mg/kg/d for 2 days (days 27 and 26 prior to TIL infusion) and fludarabine 25 mg/m2/d for 5 days (days 27 to 23 prior to TIL infusion). In some embodiments, after non-myeloablative chemotherapy and TIL infusion (at day 0) according to the invention, the patient receives an intravenous infusion of IL-2 intravenously at 720,000 IU/kg every 8 hours to physiologic tolerance. [0030] Experimental findings indicate that lymphodepletion prior to adoptive transfer of tumor-specific T lymphocytes plays a key role in enhancing treatment efficacy by eliminating regulatory T cells and competing elements of the immune system (“cytokine sinks”). Accordingly, some embodiments of the invention utilize a lymphodepletion step (sometimes also referred to as “immunosuppressive conditioning”) on the patient prior to the introduction of the TILs of the invention. [0031] The term “effective amount” or “therapeutically effective amount” refers to that amount of a compound or combination of compounds as described herein that is sufficient to effect the intended application including, but not limited to, disease treatment. A therapeutically effective amount may vary depending upon the intended application (in vitro or in vivo), or the subject and disease condition being treated (e.g., the weight, age and gender of the subject), the severity of the disease condition, or the manner of administration. The term also applies to a dose that will induce a particular response in target cells (e.g., the reduction of platelet adhesion and/or cell migration). The specific dose will vary depending on the particular compounds chosen, the dosing regimen to be followed, whether the compound is administered in combination with other compounds, timing of administration, the tissue to which it is administered, and the physical delivery system in which the compound is carried. [0032] The terms “treatment”, “treating”, “treat”, and the like, refer to obtaining a desired pharmacologic and/or physiologic effect. The effect may be prophylactic in terms of completely or partially preventing a disease or symptom thereof and/or may be therapeutic in terms of a partial or complete cure for a disease and/or adverse effect attributable to the DB1/ 149202201.1 23 Attorney Docket No.: 116983-5127-WO disease. “Treatment”, as used herein, covers any treatment of a disease in a mammal, particularly in a human, and includes: (a) preventing the disease from occurring in a subject which may be predisposed to the disease but has not yet been diagnosed as having it; (b) inhibiting the disease, i.e., arresting its development or progression; and (c) relieving the disease, i.e., causing regression of the disease and/or relieving one or more disease symptoms. “Treatment” is also meant to encompass delivery of an agent in order to provide for a pharmacologic effect, even in the absence of a disease or condition. For example, “treatment” encompasses delivery of a composition that can elicit an immune response or confer immunity in the absence of a disease condition, e.g., in the case of a vaccine. [0033] The terms “non-myeloablative chemotherapy,” “non-myeloablative lymphodepletion,” “NMALD,” “NMA LD,” “NMA-LD,” and any variants of the foregoing, are used interchangeably to indicate a chemotherapeutic regimen designed to deplete the patient’s lymphoid immune cells while avoiding depletion of the patient’s myeloid immune cells. Typically, the patient receives a course of non-myeloablative chemotherapy prior to the administration of tumor infiltrating lymphocytes to the patient as described herein. [0034] The term “heterologous” when used with reference to portions of a nucleic acid or protein indicates that the nucleic acid or protein comprises two or more subsequences that are not found in the same relationship to each other in nature. For instance, the nucleic acid is typically recombinantly produced, having two or more sequences from unrelated genes arranged to make a new functional nucleic acid, e.g., a promoter from one source and a coding region from another source, or coding regions from different sources. Similarly, a heterologous protein indicates that the protein comprises two or more subsequences that are not found in the same relationship to each other in nature (e.g., a fusion protein). [0035] The terms “sequence identity,” “percent identity,” and “sequence percent identity” (or synonyms thereof, e.g., “99% identical”) in the context of two or more nucleic acids or polypeptides, refer to two or more sequences or subsequences that are the same or have a specified percentage of nucleotides or amino acid residues that are the same, when compared and aligned (introducing gaps, if necessary) for maximum correspondence, not considering any conservative amino acid substitutions as part of the sequence identity. The percent identity can be measured using sequence comparison software or algorithms or by visual inspection. Various algorithms and software are known in the art that can be used to obtain alignments of amino acid or nucleotide sequences. Suitable programs to determine percent DB1/ 149202201.1 24 Attorney Docket No.: 116983-5127-WO sequence identity include for example the BLAST suite of programs available from the U.S. Government’s National Center for Biotechnology Information BLAST web site. Comparisons between two sequences can be carried using either the BLASTN or BLASTP algorithm. BLASTN is used to compare nucleic acid sequences, while BLASTP is used to compare amino acid sequences. ALIGN, ALIGN-2 (Genentech, South San Francisco, California) or MegAlign, available from DNASTAR, are additional publicly available software programs that can be used to align sequences. One skilled in the art can determine appropriate parameters for maximal alignment by particular alignment software. In certain embodiments, the default parameters of the alignment software are used. [0036] As used herein, the term “variant” encompasses but is not limited to antibodies or fusion proteins which comprise an amino acid sequence which differs from the amino acid sequence of a reference antibody by way of one or more substitutions, deletions and/or additions at certain positions within or adjacent to the amino acid sequence of the reference antibody. The variant may comprise one or more conservative substitutions in its amino acid sequence as compared to the amino acid sequence of a reference antibody. Conservative substitutions may involve, e.g., the substitution of similarly charged or uncharged amino acids. The variant retains the ability to specifically bind to the antigen of the reference antibody. The term variant also includes pegylated antibodies or proteins. [0037] By “tumor infiltrating lymphocytes” or “TILs” herein is meant a population of cells originally obtained as white blood cells that have left the bloodstream of a subject and migrated into a tumor. TILs include, but are not limited to, CD8+ cytotoxic T cells (lymphocytes), Th1 and Th17 CD4+ T cells, natural killer cells, dendritic cells and M1 macrophages. TILs include both primary and secondary TILs. “Primary TILs” are those that are obtained from patient tissue samples as outlined herein (sometimes referred to as “freshly harvested”), and “secondary TILs” are any TIL cell populations that have been expanded or proliferated as discussed herein, including, but not limited to bulk TILs, expanded TILs (“REP TILs”) as well as “reREP TILs” as discussed herein. reREP TILs can include for example second expansion TILs or second additional expansion TILs. [0038] TILs can generally be defined either biochemically, using cell surface markers, or functionally, by their ability to infiltrate tumors and effect treatment. TILs can be generally categorized by expressing one or more of the following biomarkers: CD4, CD8, TCR αβ, CD27, CD28, CD56, CCR7, CD45Ra, CD95, PD-1, and CD25. Additionally, and DB1/ 149202201.1 25 Attorney Docket No.: 116983-5127-WO alternatively, TILs can be functionally defined by their ability to infiltrate solid tumors upon reintroduction into a patient. TILs may further be characterized by potency – for example, TILs may be considered potent if, for example, interferon (IFN) release is greater than about 50 pg/mL, greater than about 100 pg/mL, greater than about 150 pg/mL, or greater than about 200 pg/mL. TILs may be considered potent if, for example, interferon (IFNγ) release is greater than about 50 pg/mL, greater than about 100 pg/mL, greater than about 150 pg/mL, or greater than about 200 pg/mL, greater than about 300 pg/mL, greater than about 400 pg/mL, greater than about 500 pg/mL, greater than about 600 pg/mL, greater than about 700 pg/mL, greater than about 800 pg/mL, greater than about 900 pg/mL, greater than about 1000 pg/mL. [0039] The term “deoxyribonucleotide” encompasses natural and synthetic, unmodified and modified deoxyribonucleotides. Modifications include changes to the sugar moiety, to the base moiety and/or to the linkages between deoxyribonucleotide in the oligonucleotide. [0040] The term “RNA” defines a molecule comprising at least one ribonucleotide residue. The term “ribonucleotide” defines a nucleotide with a hydroxyl group at the 2' position of a b-D-ribofuranose moiety. The term RNA includes double-stranded RNA, single-stranded RNA, isolated RNA such as partially purified RNA, essentially pure RNA, synthetic RNA, recombinantly produced RNA, as well as altered RNA that differs from naturally occurring RNA by the addition, deletion, substitution and/or alteration of one or more nucleotides. Nucleotides of the RNA molecules described herein may also comprise non-standard nucleotides, such as non-naturally occurring nucleotides or chemically synthesized nucleotides or deoxynucleotides. These altered RNAs can be referred to as analogs or analogs of naturally-occurring RNA. [0041] The terms “pharmaceutically acceptable carrier” or “pharmaceutically acceptable excipient” are intended to include any and all solvents, dispersion media, coatings, antibacterial and antifungal agents, isotonic and absorption delaying agents, and inert ingredients. The use of such pharmaceutically acceptable carriers or pharmaceutically acceptable excipients for active pharmaceutical ingredients is well known in the art. Except insofar as any conventional pharmaceutically acceptable carrier or pharmaceutically acceptable excipient is incompatible with the active pharmaceutical ingredient, its use in therapeutic compositions of the invention is contemplated. Additional active pharmaceutical ingredients, such as other drugs, can also be incorporated into the described compositions and methods. DB1/ 149202201.1 26 Attorney Docket No.: 116983-5127-WO [0042] The terms “about” and “approximately” mean within a statistically meaningful range of a value. Such a range can be within an order of magnitude, preferably within 50%, more preferably within 20%, more preferably still within 10%, and even more preferably within 5% of a given value or range. The allowable variation encompassed by the terms “about” or “approximately” depends on the particular system under study, and can be readily appreciated by one of ordinary skill in the art. Moreover, as used herein, the terms “about” and “approximately” mean that dimensions, sizes, formulations, parameters, shapes and other quantities and characteristics are not and need not be exact, but may be approximate and/or larger or smaller, as desired, reflecting tolerances, conversion factors, rounding off, measurement error and the like, and other factors known to those of skill in the art. In general, a dimension, size, formulation, parameter, shape or other quantity or characteristic is “about” or “approximate” whether or not expressly stated to be such. It is noted that embodiments of very different sizes, shapes and dimensions may employ the described arrangements. [0043] The transitional terms “comprising,” “consisting essentially of,” and “consisting of,” when used in the appended claims, in original and amended form, define the claim scope with respect to what unrecited additional claim elements or steps, if any, are excluded from the scope of the claim(s). The term “comprising” is intended to be inclusive or open-ended and does not exclude any additional, unrecited element, method, step or material. The term “consisting of” excludes any element, step or material other than those specified in the claim and, in the latter instance, impurities ordinary associated with the specified material(s). The term “consisting essentially of” limits the scope of a claim to the specified elements, steps or material(s) and those that do not materially affect the basic and novel characteristic(s) of the claimed invention. All compositions, methods, and kits described herein that embody the present invention can, in alternate embodiments, be more specifically defined by any of the transitional terms “comprising,” “consisting essentially of,” and “consisting of.” [0044] The terms “antibody” and its plural form “antibodies” refer to whole immunoglobulins and any antigen-binding fragment (“antigen-binding portion”) or single chains thereof. An “antibody” further refers to a glycoprotein comprising at least two heavy (H) chains and two light (L) chains inter-connected by disulfide bonds, or an antigen-binding portion thereof. Each heavy chain is comprised of a heavy chain variable region (abbreviated herein as VH) and a heavy chain constant region. The heavy chain constant region is DB1/ 149202201.1 27 Attorney Docket No.: 116983-5127-WO comprised of three domains, CH1, CH2 and CH3. Each light chain is comprised of a light chain variable region (abbreviated herein as VL) and a light chain constant region. The light chain constant region is comprised of one domain, CL. The VH and VL regions of an antibody may be further subdivided into regions of hypervariability, which are referred to as complementarity determining regions (CDR) or hypervariable regions (HVR), and which can be interspersed with regions that are more conserved, termed framework regions (FR). Each VH and VL is composed of three CDRs and four FRs, arranged from amino-terminus to carboxy-terminus in the following order: FR1, CDR1, FR2, CDR2, FR3, CDR3, FR4. The variable regions of the heavy and light chains contain a binding domain that interacts with an antigen epitope or epitopes. The constant regions of the antibodies may mediate the binding of the immunoglobulin to host tissues or factors, including various cells of the immune system (e.g., effector cells) and the first component (Clq) of the classical complement system. [0045] The term “antigen” refers to a substance that induces an immune response. In some embodiments, an antigen is a molecule capable of being bound by an antibody or a TCR if presented by major histocompatibility complex (MHC) molecules. The term “antigen”, as used herein, also encompasses T cell epitopes. An antigen is additionally capable of being recognized by the immune system. In some embodiments, an antigen is capable of inducing a humoral immune response or a cellular immune response leading to the activation of B lymphocytes and/or T lymphocytes. In some cases, this may require that the antigen contains or is linked to a Th cell epitope. An antigen can also have one or more epitopes (e.g., B- and T-epitopes). In some embodiments, an antigen will preferably react, typically in a highly specific and selective manner, with its corresponding antibody or TCR and not with the multitude of other antibodies or TCRs which may be induced by other antigens. [0046] The terms “monoclonal antibody,” “mAb,” “monoclonal antibody composition,” or their plural forms refer to a preparation of antibody molecules of single molecular composition. A monoclonal antibody composition displays a single binding specificity and affinity for a particular epitope. Monoclonal antibodies specific to certain receptors can be made using knowledge and skill in the art of injecting test subjects with suitable antigen and then isolating hybridomas expressing antibodies having the desired sequence or functional characteristics. DNA encoding the monoclonal antibodies is readily isolated and sequenced using conventional procedures (e.g., by using oligonucleotide probes that are capable of binding specifically to genes encoding the heavy and light chains of the monoclonal DB1/ 149202201.1 28 Attorney Docket No.: 116983-5127-WO antibodies). The hybridoma cells serve as a preferred source of such DNA. Once isolated, the DNA may be placed into expression vectors, which are then transfected into host cells such as E. coli cells, simian COS cells, Chinese hamster ovary (CHO) cells, or myeloma cells that do not otherwise produce immunoglobulin protein, to obtain the synthesis of monoclonal antibodies in the recombinant host cells. Recombinant production of antibodies will be described in more detail below. [0047] The terms “antigen-binding portion” or “antigen-binding fragment” of an antibody (or simply “antibody portion” or “fragment”), as used herein, refers to one or more fragments of an antibody that retain the ability to specifically bind to an antigen. It has been shown that the antigen-binding function of an antibody can be performed by fragments of a full-length antibody. Examples of binding fragments encompassed within the term “antigen-binding portion” of an antibody include (i) a Fab fragment, a monovalent fragment consisting of the VL, VH, CL and CH1 domains; (ii) a F(ab′)2 fragment, a bivalent fragment comprising two Fab fragments linked by a disulfide bridge at the hinge region; (iii) a Fd fragment consisting of the VH and CH1 domains; (iv) a Fv fragment consisting of the VL and VH domains of a single arm of an antibody, (v) a domain antibody (dAb) fragment (Ward, et al., Nature, 1989, 341, 544-546), which may consist of a VH or a VL domain; and (vi) an isolated complementarity determining region (CDR). Furthermore, although the two domains of the Fv fragment, VL and VH, are coded for by separate genes, they can be joined, using recombinant methods, by a synthetic linker that enables them to be made as a single protein chain in which the VL and VH regions pair to form monovalent molecules known as single chain Fv (scFv); see, e.g., Bird, et al., Science 1988, 242, 423-426; and Huston, et al., Proc. Natl. Acad. Sci. USA 1988, 85, 5879-5883). Such scFv antibodies are also intended to be encompassed within the terms “antigen-binding portion” or “antigen-binding fragment” of an antibody. These antibody fragments are obtained using conventional techniques known to those with skill in the art, and the fragments are screened for utility in the same manner as are intact antibodies. In some embodiments, a scFv protein domain comprises a VH portion and a VL portion. A scFv molecule is denoted as either VL-L-VH if the VL domain is the N-terminal part of the scFv molecule, or as VH-L-VL if the VH domain is the N-terminal part of the scFv molecule. Methods for making scFv molecules and designing suitable peptide linkers are described in U.S. Pat. No.4,704,692, U.S. Pat. No.4,946,778, R. Raag and M. Whitlow, “Single Chain Fvs.” FASEB Vol 9:73-80 (1995) and R. E. Bird and B. W. Walker, Single DB1/ 149202201.1 29 Attorney Docket No.: 116983-5127-WO Chain Antibody Variable Regions, TIBTECH, Vol 9: 132-137 (1991), the disclosures of which are incorporated by reference herein. [0048] The term “human antibody,” as used herein, is intended to include antibodies having variable regions in which both the framework and CDR regions are derived from human germline immunoglobulin sequences. Furthermore, if the antibody contains a constant region, the constant region also is derived from human germline immunoglobulin sequences. The human antibodies of the invention may include amino acid residues not encoded by human germline immunoglobulin sequences (e.g., mutations introduced by random or site-specific mutagenesis in vitro or by somatic mutation in vivo). The term “human antibody”, as used herein, is not intended to include antibodies in which CDR sequences derived from the germline of another mammalian species, such as a mouse, have been grafted onto human framework sequences. [0049] The term “human monoclonal antibody” refers to antibodies displaying a single binding specificity which have variable regions in which both the framework and CDR regions are derived from human germline immunoglobulin sequences. In some embodiments, the human monoclonal antibodies are produced by a hybridoma which includes a B cell obtained from a transgenic nonhuman animal, e.g., a transgenic mouse, having a genome comprising a human heavy chain transgene and a light chain transgene fused to an immortalized cell. [0050] The term “recombinant human antibody”, as used herein, includes all human antibodies that are prepared, expressed, created or isolated by recombinant means, such as (a) antibodies isolated from an animal (such as a mouse) that is transgenic or transchromosomal for human immunoglobulin genes or a hybridoma prepared therefrom (described further below), (b) antibodies isolated from a host cell transformed to express the human antibody, e.g., from a transfectoma, (c) antibodies isolated from a recombinant, combinatorial human antibody library, and (d) antibodies prepared, expressed, created or isolated by any other means that involve splicing of human immunoglobulin gene sequences to other DNA sequences. Such recombinant human antibodies have variable regions in which the framework and CDR regions are derived from human germline immunoglobulin sequences. In certain embodiments, however, such recombinant human antibodies can be subjected to in vitro mutagenesis (or, when an animal transgenic for human Ig sequences is used, in vivo somatic mutagenesis) and thus the amino acid sequences of the VH and VL regions of the DB1/ 149202201.1 30 Attorney Docket No.: 116983-5127-WO recombinant antibodies are sequences that, while derived from and related to human germline VH and VL sequences, may not naturally exist within the human antibody germline repertoire in vivo. [0051] As used herein, “isotype” refers to the antibody class (e.g., IgM or IgG1) that is encoded by the heavy chain constant region genes. [0052] The phrases “an antibody recognizing an antigen” and “an antibody specific for an antigen” are used interchangeably herein with the term “an antibody which binds specifically to an antigen.” [0053] The term “human antibody derivatives” refers to any modified form of the human antibody, including a conjugate of the antibody and another active pharmaceutical ingredient or antibody. The terms “conjugate,” “antibody-drug conjugate”, “ADC,” or “immunoconjugate” refers to an antibody, or a fragment thereof, conjugated to another therapeutic moiety, which can be conjugated to antibodies described herein using methods available in the art. [0054] The terms “humanized antibody,” “humanized antibodies,” and “humanized” are intended to refer to antibodies in which CDR sequences derived from the germline of another mammalian species, such as a mouse, have been grafted onto human framework sequences. Additional framework region modifications may be made within the human framework sequences. Humanized forms of non-human (for example, murine) antibodies are chimeric antibodies that contain minimal sequence derived from non-human immunoglobulin. For the most part, humanized antibodies are human immunoglobulins (recipient antibody) in which residues from a hypervariable region of the recipient are replaced by residues from a 15 hypervariable region of a non-human species (donor antibody) such as mouse, rat, rabbit or nonhuman primate having the desired specificity, affinity, and capacity. In some instances, Fv framework region (FR) residues of the human immunoglobulin are replaced by corresponding non-human residues. Furthermore, humanized antibodies may comprise residues that are not found in the recipient antibody or in the donor antibody. These modifications are made to further refine antibody performance. In general, the humanized antibody will comprise substantially all of at least one, and typically two, variable domains, in which all or substantially all of the hypervariable loops correspond to those of a non- human immunoglobulin and all or substantially all of the FR regions are those of a human immunoglobulin sequence. The humanized antibody optionally also will comprise at least a DB1/ 149202201.1 31 Attorney Docket No.: 116983-5127-WO portion of an immunoglobulin constant region (Fc), typically that of a human immunoglobulin. For further details, see Jones, et al., Nature 1986, 321, 522-525; Riechmann, et al., Nature 1988, 332, 323-329; and Presta, Curr. Op. Struct. Biol.1992, 2, 593-596. The antibodies described herein may also be modified to employ any Fc variant which is known to impart an improvement (e.g., reduction) in effector function and/or FcR binding. The Fc variants may include, for example, any one of the amino acid substitutions disclosed in International Patent Application Publication Nos. WO 1988/07089 A1, WO 1996/14339 A1, WO 1998/05787 A1, WO 1998/23289 A1, WO 1999/51642 A1, WO 99/58572 A1, WO 2000/09560 A2, WO 2000/32767 A1, WO 2000/42072 A2, WO 2002/44215 A2, WO 2002/060919 A2, WO 2003/074569 A2, WO 2004/016750 A2, WO 2004/029207 A2, WO 2004/035752 A2, WO 2004/063351 A2, WO 2004/074455 A2, WO 2004/099249 A2, WO 2005/040217 A2, WO 2005/070963 A1, WO 2005/077981 A2, WO 2005/092925 A2, WO 2005/123780 A2, WO 2006/019447 A1, WO 2006/047350 A2, and WO 2006/085967 A2; and U.S. Patent Nos.5,648,260; 5,739,277; 5,834,250; 5,869,046; 6,096,871; 6,121,022; 6,194,551; 6,242,195; 6,277,375; 6,528,624; 6,538,124; 6,737,056; 6,821,505; 6,998,253; and 7,083,784; the disclosures of which are incorporated by reference herein. [0055] The term “chimeric antibody” is intended to refer to antibodies in which the variable region sequences are derived from one species and the constant region sequences are derived from another species, such as an antibody in which the variable region sequences are derived from a mouse antibody and the constant region sequences are derived from a human antibody. [0056] A “diabody” is a small antibody fragment with two antigen-binding sites. The fragments comprises a heavy chain variable domain (VH) connected to a light chain variable domain (VL) in the same polypeptide chain (VH-VL or VL-VH). By using a linker that is too short to allow pairing between the two domains on the same chain, the domains are forced to pair with the complementary domains of another chain and create two antigen-binding sites. Diabodies are described more fully in, e.g., European Patent No. EP 404,097, International Patent Publication No. WO 93/11161; and Bolliger, et al., Proc. Natl. Acad. Sci. USA 1993, 90, 6444-6448. [0057] The term “glycosylation” refers to a modified derivative of an antibody. An aglycoslated antibody lacks glycosylation. Glycosylation can be altered to, for example, DB1/ 149202201.1 32 Attorney Docket No.: 116983-5127-WO increase the affinity of the antibody for antigen. Such carbohydrate modifications can be accomplished by, for example, altering one or more sites of glycosylation within the antibody sequence. For example, one or more amino acid substitutions can be made that result in elimination of one or more variable region framework glycosylation sites to thereby eliminate glycosylation at that site. Aglycosylation may increase the affinity of the antibody for antigen, as described in U.S. Patent Nos.5,714,350 and 6,350,861. Additionally or alternatively, an antibody can be made that has an altered type of glycosylation, such as a hypofucosylated antibody having reduced amounts of fucosyl residues or an antibody having increased bisecting GlcNac structures. Such altered glycosylation patterns have been demonstrated to increase the ability of antibodies. Such carbohydrate modifications can be accomplished by, for example, expressing the antibody in a host cell with altered glycosylation machinery. Cells with altered glycosylation machinery have been described in the art and can be used as host cells in which to express recombinant antibodies of the invention to thereby produce an antibody with altered glycosylation. For example, the cell lines Ms704, Ms705, and Ms709 lack the fucosyltransferase gene, FUT8 (alpha (1,6) fucosyltransferase), such that antibodies expressed in the Ms704, Ms705, and Ms709 cell lines lack fucose on their carbohydrates. The Ms704, Ms705, and Ms709 FUT8−/− cell lines were created by the targeted disruption of the FUT8 gene in CHO/DG44 cells using two replacement vectors (see e.g. U.S. Patent Publication No.2004/0110704 or Yamane-Ohnuki, et al., Biotechnol. Bioeng., 2004, 87, 614-622). As another example, European Patent No. EP 1,176,195 describes a cell line with a functionally disrupted FUT8 gene, which encodes a fucosyl transferase, such that antibodies expressed in such a cell line exhibit hypofucosylation by reducing or eliminating the alpha 1,6 bond-related enzyme, and also describes cell lines which have a low enzyme activity for adding fucose to the N- acetylglucosamine that binds to the Fc region of the antibody or does not have the enzyme activity, for example the rat myeloma cell line YB2/0 (ATCC CRL 1662). International Patent Publication WO 03/035835 describes a variant CHO cell line, Lec 13 cells, with reduced ability to attach fucose to Asn(297)-linked carbohydrates, also resulting in hypofucosylation of antibodies expressed in that host cell (see also Shields, et al., J. Biol. Chem.2002, 277, 26733-26740. International Patent Publication WO 99/54342 describes cell lines engineered to express glycoprotein-modifying glycosyl transferases (e.g., beta(1,4)-N- acetylglucosaminyltransferase III (GnTIII)) such that antibodies expressed in the engineered cell lines exhibit increased bisecting GlcNac structures which results in increased ADCC DB1/ 149202201.1 33 Attorney Docket No.: 116983-5127-WO activity of the antibodies (see also Umana, et al., Nat. Biotech.1999, 17, 176-180). Alternatively, the fucose residues of the antibody may be cleaved off using a fucosidase enzyme. For example, the fucosidase alpha-L-fucosidase removes fucosyl residues from antibodies as described in Tarentino, et al., Biochem.1975, 14, 5516-5523. [0058] “Pegylation” refers to a modified antibody, or a fragment thereof, that typically is reacted with polyethylene glycol (PEG), such as a reactive ester or aldehyde derivative of PEG, under conditions in which one or more PEG groups become attached to the antibody or antibody fragment. Pegylation may, for example, increase the biological (e.g., serum) half life of the antibody. Preferably, the pegylation is carried out via an acylation reaction or an alkylation reaction with a reactive PEG molecule (or an analogous reactive water-soluble polymer). As used herein, the term “polyethylene glycol” is intended to encompass any of the forms of PEG that have been used to derivatize other proteins, such as mono (C1-C10)alkoxy- or aryloxy-polyethylene glycol or polyethylene glycol-maleimide. The antibody to be pegylated may be an aglycosylated antibody. Methods for pegylation are known in the art and can be applied to the antibodies of the invention, as described for example in European Patent Nos. EP 0154316 and EP 0401384 and U.S. Patent No.5,824,778, the disclosures of each of which are incorporated by reference herein. [0059] The term “biosimilar” means a biological product, including a monoclonal antibody or protein, that is highly similar to a U.S. licensed reference biological product notwithstanding minor differences in clinically inactive components, and for which there are no clinically meaningful differences between the biological product and the reference product in terms of the safety, purity, and potency of the product. Furthermore, a similar biological or “biosimilar” medicine is a biological medicine that is similar to another biological medicine that has already been authorized for use by the European Medicines Agency. The term “biosimilar” is also used synonymously by other national and regional regulatory agencies. Biological products or biological medicines are medicines that are made by or derived from a biological source, such as a bacterium or yeast. They can consist of relatively small molecules such as human insulin or erythropoietin, or complex molecules such as monoclonal antibodies. For example, if the reference IL-2 protein is aldesleukin (PROLEUKIN), a protein approved by drug regulatory authorities with reference to aldesleukin is a “biosimilar to” aldesleukin or is a “biosimilar thereof” of aldesleukin. In Europe, a similar biological or “biosimilar” medicine is a biological medicine that is similar DB1/ 149202201.1 34 Attorney Docket No.: 116983-5127-WO to another biological medicine that has already been authorized for use by the European Medicines Agency (EMA). The relevant legal basis for similar biological applications in Europe is Article 6 of Regulation (EC) No 726/2004 and Article 10(4) of Directive 2001/83/EC, as amended and therefore in Europe, the biosimilar may be authorized, approved for authorization or subject of an application for authorization under Article 6 of Regulation (EC) No 726/2004 and Article 10(4) of Directive 2001/83/EC. The already authorized original biological medicinal product may be referred to as a “reference medicinal product” in Europe. Some of the requirements for a product to be considered a biosimilar are outlined in the CHMP Guideline on Similar Biological Medicinal Products. In addition, product specific guidelines, including guidelines relating to monoclonal antibody biosimilars, are provided on a product-by-product basis by the EMA and published on its website. A biosimilar as described herein may be similar to the reference medicinal product by way of quality characteristics, biological activity, mechanism of action, safety profiles and/or efficacy. In addition, the biosimilar may be used or be intended for use to treat the same conditions as the reference medicinal product. Thus, a biosimilar as described herein may be deemed to have similar or highly similar quality characteristics to a reference medicinal product. Alternatively, or in addition, a biosimilar as described herein may be deemed to have similar or highly similar biological activity to a reference medicinal product. Alternatively, or in addition, a biosimilar as described herein may be deemed to have a similar or highly similar safety profile to a reference medicinal product. Alternatively, or in addition, a biosimilar as described herein may be deemed to have similar or highly similar efficacy to a reference medicinal product. As described herein, a biosimilar in Europe is compared to a reference medicinal product which has been authorized by the EMA. However, in some instances, the biosimilar may be compared to a biological medicinal product which has been authorized outside the European Economic Area (a non-EEA authorized “comparator”) in certain studies. Such studies include for example certain clinical and in vivo non-clinical studies. As used herein, the term “biosimilar” also relates to a biological medicinal product which has been or may be compared to a non-EEA authorized comparator. Certain biosimilars are proteins such as antibodies, antibody fragments (for example, antigen binding portions) and fusion proteins. A protein biosimilar may have an amino acid sequence that has minor modifications in the amino acid structure (including for example deletions, additions, and/or substitutions of amino acids) which do not significantly affect the function of the polypeptide. The biosimilar may comprise an amino acid sequence having a sequence DB1/ 149202201.1 35 Attorney Docket No.: 116983-5127-WO identity of 97% or greater to the amino acid sequence of its reference medicinal product, e.g., 97%, 98%, 99% or 100%. The biosimilar may comprise one or more post-translational modifications, for example, although not limited to, glycosylation, oxidation, deamidation, and/or truncation which is/are different to the post-translational modifications of the reference medicinal product, provided that the differences do not result in a change in safety and/or efficacy of the medicinal product. The biosimilar may have an identical or different glycosylation pattern to the reference medicinal product. Particularly, although not exclusively, the biosimilar may have a different glycosylation pattern if the differences address or are intended to address safety concerns associated with the reference medicinal product. Additionally, the biosimilar may deviate from the reference medicinal product in for example its strength, pharmaceutical form, formulation, excipients and/or presentation, providing safety and efficacy of the medicinal product is not compromised. The biosimilar may comprise differences in for example pharmacokinetic (PK) and/or pharmacodynamic (PD) profiles as compared to the reference medicinal product but is still deemed sufficiently similar to the reference medicinal product as to be authorized or considered suitable for authorization. In certain circumstances, the biosimilar exhibits different binding characteristics as compared to the reference medicinal product, wherein the different binding characteristics are considered by a Regulatory Authority such as the EMA not to be a barrier for authorization as a similar biological product. The term “biosimilar” is also used synonymously by other national and regional regulatory agencies. II. Trop-2 Targeting ADC [0060] An antibody drug conjugate (ADC) has three core components: a monoclonal antibody (MAb) targeting a specific tumor antigen, a cytotoxic payload, and a chemical linker connecting them. The basic mechanism involves binding of the antibody to the tumor antigen on the cell surface, followed by internalization of the ADC and lysosomal degradation. This results in release of the active cytotoxic agent into the cytoplasm which results in tumor cell death. Some ADCs also induce antibody dependent cellular cytotoxicity (ADCC) and complement dependent cytotoxicity (CDC) contributing to the antitumor efficacy of these agents (Junttila et al., Breast Cancer Res Treat.2011 Jul;128(2):347–356). DB1/ 149202201.1 36 Attorney Docket No.: 116983-5127-WO [0061] As used herein, a “Trop-2 targeting ADC” or “anti-Trop-2 ADC” refers to a conjugated molecule comprising an anti-Trop-2 antibody linked to a drug moiety that combines the specificity of the anti-Trop-2 antibody with potency of the drug moiety. [0062] In one aspect, the invention provides a Trop-2 targeting ADC of Formula (I): Ab-(L-(D)m)n Wherein Ab represents an anti-Trop-2 antibody described herein; L is a linker; D is a drug moiety; m is an integer from 1 to 8; and n is an integer from 1-20. [0063] In one embodiment, n is an integer from 1 to 10, 2 to 8, or 2 to 5. In a specific embodiment, n is 2, 3, or 4. In some embodiments, m is 1; in other embodiments, m is 2, 3 or 4. [0064] While the drug to antibody ratio (DAR) has an exact value for a specific conjugate molecule (e.g., n multiplied by m in Formula (I)), it is understood that the value will often be an average value when used to describe a sample containing many molecules, due to some degree of heterogeneity, typically associated with the conjugation step. In some embodiments, the DAR is between about 2 and about 10, and typically is about 3, 3.5, 4, 4.5, 5, 5.5, 6, 6.5, 7.0, 7.5, 8.0. In some embodiments, at least 50% of a sample by weight is compound having the average DAR plus or minus 2, and preferably at least 50% of the sample is a conjugate that contains the average DAR plus or minus 1. Embodiments include Trop-2 targeting ADCs wherein the DAR is about 3.5, 3.6, 3.7, 3.8 or 3.9. In some embodiments, a DAR of ‘about n’ means the measured value for DAR is within 20% of n. A. Anti-Trop-2 Antibody [0065] The Trop-2 targeting ADCs include at least one antibody or fragment thereof that binds to Trop-2. Anti-Trop-2 antibodies are known and/or publicly available and in alternative embodiments may be utilized in the Trop-2 targeting ADCs. While humanized or human antibodies are preferred for reduced immunogenicity, in alternative embodiments a chimeric antibody may be of use. As discussed below, methods of antibody humanization are DB1/ 149202201.1 37 Attorney Docket No.: 116983-5127-WO well known in the art and may be utilized to convert an available murine or chimeric antibody into a humanized form. [0066] Anti-Trop-2 antibodies are commercially available from a number of sources and include LS-C126418, LS-C178765, LS-C126416, LS-C126417 (LifeSpan Biosciences, Inc., Seattle, WA); 10428-MM01, 10428-MM02, 10428-R001, 10428-R030 (Sino Biological Inc., Beijing, China); MR54 (eBioscience, San Diego, CA); sc-376181, sc-376746, Santa Cruz Biotechnology (Santa Cruz, CA); MM0588-49D6, (Novus Biologicals, Littleton, CO); ab79976, and ab89928 (ABCAM®, Cambridge, MA). [0067] Other anti-Trop-2 antibodies have been disclosed in the patent literature. For example, U.S. Publ. No.2013/0089872 discloses anti-Trop-2 antibodies K5-70 (Accession No. FERM BP-11251), K5-107 (Accession No. FERM BP-11252), K5-116-2-1 (Accession No. FERM BP-11253), T6-16 (Accession No. FERM BP-11346), and T5-86 (Accession No. FERM BP- 11254), deposited with the International Patent Organism Depositary, Tsukuba, Japan. U.S. Patent No.5,840,854 disclosed the anti-Trop-2 monoclonal antibody BR110 (ATCC No. HB11698). U.S. Patent No.7,420,040 disclosed an anti-Trop-2 antibody produced by hybridoma cell line AR47A6.4.2, deposited with the IDAC (International Depository Authority of Canada, Winnipeg, Canada) as accession number 141205-05. U.S. Patent No. 7,420,041 disclosed an anti-Trop-2 antibody produced by hybridoma cell line AR52A301.5, deposited with the IDAC as accession number 141205-03. U.S. Publ. No.2013/0122020 disclosed anti-Trop-2 antibodies 3E9, 6G11, 7E6, 15E2, 18B1. Hybridomas encoding a representative antibody were deposited with the American Type Culture Collection (ATCC), Accession Nos. PTA-12871 and PTA-12872. U.S. Patent No.8,715,662 discloses anti-Trop-2 antibodies produced by hybridomas deposited at the AID-ICLC (Genoa, Italy) with deposit numbers PD 08019, PD 08020 and PD 08021. U.S. Patent Application Publ. No. 20120237518 discloses anti-Trop-2 antibodies 77220, KM4097 and KM4590. U.S. Patent No.8,309,094 (Wyeth) discloses antibodies A1 and A3, identified by sequence listing. The Examples section of each patent or patent application cited above in this paragraph is incorporated herein by reference. Non-patent publication Lipinski et al. (1981, Proc Natl. Acad Sci USA, 78:5147-50) disclosed anti-Trop-2 antibodies 162-25.3 and 162-46.2. [0068] Numerous anti-Trop-2 antibodies are known in the art and/or publicly available. As discussed below, methods for preparing antibodies against known antigens were routine in the art. The sequence of the human Trop-2 protein was also known in the art (see, e.g., DB1/ 149202201.1 38 Attorney Docket No.: 116983-5127-WO GenBank Accession No. CAA54801.1). Methods for producing humanized, human or chimeric antibodies were also known. The person of ordinary skill, reading the instant disclosure in light of general knowledge in the art, would have been able to make and use the genus of anti-Trop-2 antibodies in the subject ADCs. In an embodiment, the anti-Trop-2 antibody may be a humanized RS7 antibody (see, e.g., U.S. Patent No.7,238,785, incorporated herein by reference in its entirety). B. Drug Moiety [0069] The drug to be conjugated to the anti-Trop-2 antibody or antibody fragment may be selected from the group consisting of an anthracycline, a camptothecin, a tubulin inhibitor, a maytansinoid, a calicheamycin, an auristatin, a nitrogen mustard, an ethylenimine derivative, an alkyl sulfonate, a nitrosourea, a triazene, a folic acid analog, a taxane, a COX-2 inhibitor, a pyrimidine analog, a purine analog, an antibiotic, an enzyme inhibitor, an epipodophyllotoxin, a platinum coordination complex, a vinca alkaloid, a substituted urea, a methyl hydrazine derivative, an adrenocortical suppressant, a hormone antagonist, an antimetabolite, an alkylating agent, an antimitotic, an anti- angiogenic agent, a tyrosine kinase inhibitor, an mTOR inhibitor, a heat shock protein (HSP90) inhibitor, a proteosome inhibitor, an HDAC inhibitor, a pro-apoptotic agent, and a combination thereof. [0070] Specific drugs of use may be selected from the group consisting of 5-fluorouracil, afatinib, aplidin, azaribine, anastrozole, anthracyclines, axitinib, AVL-101, AVL-291, bendamustine, bleomycin, bortezomib, bosutinib, bryostatin-1 , busulfan, calicheamycin, camptothecin, carboplatin, 10-hydroxycamptothecin, carmustine, celecoxib, chlorambucil, cisplatinum, COX-2 inhibitors, irinotecan (CPT-11), SN-38, carboplatin, cladribine, camptothecans, crizotinib, cyclophosphamide, cytarabine, dacarbazine, dasatinib, dinaciclib, docetaxel, dactinomycin, daunorubicin, DM1, DM3, DM4, doxorubicin, 2- pyrrolinodoxorubicine (2-PDox), a pro-drug form of 2-PDox (pro-2-PDox), cyano- morpholino doxorubicin, doxorubicin glucuronide, endostatin, epirubicin glucuronide, erlotinib, estramustine, epidophyllotoxin, erlotinib, entinostat, estrogen receptor binding agents, etoposide (VP16), etoposide glucuronide, etoposide phosphate, exemestane, fingolimod, floxuridine (FUdR), 3',5'-0-dioleoyl-FudR (FUdR-dO), fludarabine, flutamide, farnesyl-protein transferase inhibitors, flavopiridol, fostamatinib, ganetespib, GDC-0834, GS- 1101, gefitinib, gemcitabine, hydroxyurea, ibrutinib, idarubicin, idelalisib, ifosfamide, imatinib, lapatinib, lenolidamide, leucovorin, LFM-A13, lomustine, mechlorethamine, DB1/ 149202201.1 39 Attorney Docket No.: 116983-5127-WO melphalan, mercaptopurine, 6-mercaptopurine, methotrexate, mitoxantrone, mithramycin, mitomycin, mitotane, monomethylauristatin F (MMAF), monomethylauristatin D (MMAD), monomethylauristatin E (MMAE), navelbine, neratinib, nilotinib, nitrosurea, olaparib, plicomycin, procarbazine, paclitaxel, PCI-32765, pentostatin, PSI-341, raloxifene, semustine, SN-38, sorafenib, streptozocin, SUl 1248, sunitinib, tamoxifen, temazolomide, transplatinum, thalidomide, thioguanine, thiotepa, teniposide, topotecan, uracil mustard, vatalanib, vinorelbine, vinblastine, vincristine, vinca alkaloids and ZD1839. In particularly preferred embodiments, the drug to be conjugated to the anti-Trop-2 antibody may be SN-38, pro-2- PDox or paclitaxel. [0071] The drug moiety D can be linked to the antibody Ab through linker L. L is any chemical moiety capable of linking the drug moiety to the antibody through covalent bonds. A cross-linking reagent is a bifunctional or multifunctional reagent that can be used to link a drug moiety and an antibody to form antibody drug conjugates. Antibody drug conjugates can be prepared using a cross-linking reagent having a reactive functionality capable of binding to both the drug moiety and the antibody. For example, a cysteine, thiol or an amine, e.g. N- terminus or an amino acid side chain, such as lysine of the antibody, can form a bond with a functional group of a cross-linking reagent. Alternatively, the Antibody drug conjugates can be prepared by pre-forming a linker-drug moiety (or drug-linker moiety, both terms being used interchangeably), and reacting the linker-drug moiety with the antibody. In some instant, the linker moiety is built onto the drug stepwise using several linking moieties until obtaining the desired linker-drug moiety. [0072] In one embodiment, L is a cleavable linker. In another embodiment, L is a non- cleavable linker. In some embodiments, L is an acid-labile linker, photo-labile linker, peptidase cleavable linker, esterase cleavable linker, a disulfide bond cleavable linker, a hydrophilic linker, a procharged linker, a glycosidase cleavable linker, a phosphodiesterase cleavable linker, a phosphatase cleavable linker, or a dicarboxylic acid based linker. [0073] Suitable cross-linking reagents that form a non-cleavable linker between the drug moiety, for example maytansinoid, and the antibody are well known in the art, and can form non-cleavable linkers that comprise a sulfur atom (such as SMCC) or those that are without a sulfur atom. Preferred cross-linking reagents that form non-cleavable linkers between the drug moiety, for example maytansinoid, and the antibody comprise a maleimido- or DB1/ 149202201.1 40 Attorney Docket No.: 116983-5127-WO haloacetyl-based moiety. According to the present invention, such non-cleavable linkers are said to be derived from maleimido- or haloacetyl-based moieties. [0074] Cross-linking reagents comprising a maleimido-based moiety include but not limited to, N-succinimidyl-4-(maleimidomethyl)cyclohexanecarboxylate (SMCC), sulfosuccinimidyl 4-(N-maleimidomethyl) cyclohexane-1-carboxylate (sulfo-SMCC), N-succinimidyl-4- (maleimidomethyl)cyclohexane-1-carboxy-(6-amidocaproate), which is a “long chain” analog of SMCC (LC-SMCC), κ-maleimidoundeconoic acid N-succinimidyl ester (KMUA), γ-maleimidobutyric acid N-succinimidyl ester (GMBS), ε-maleimidocaproic acid N- succinimidyl ester (EMCS), m-maleimidobenzoyl-N-hydroxysuccinimide ester (MBS), N-(α- maleimidoacetoxy)-succinimide ester (AMSA), succinimidyl-6-(β- maleimidopropionamido)hexanoate (SMPH), N-succinimidyl-4-(p-maleimidophenyl)- butyrate (SMPB), N-(-p-maleomidophenyl)isocyanate (PMIP) and maleimido-based cross- linking reagents containing a polyethythene glycol spacer, such as MAL-PEG-NHS. These cross-linking reagents form non-cleavable linkers derived from maleimido-based moieties. Representative structures of maleimido-based cross-linking reagents are shown below. DB1/ 149202201.1 41 Attorney Docket No.: 116983-5127-WO
[0017] In some embodiments, an IL-2 form suitable for use in the invention includes a antibody cytokine engrafted protein comprises a heavy chain variable region (VH), comprising complementarity determining regions HCDR1, HCDR2, HCDR3; a light chain variable region (VL), comprising LCDR1, LCDR2, LCDR3; and an IL-2 molecule or a fragment thereof engrafted into a CDR of the VH or the VL, wherein the antibody cytokine engrafted protein preferentially expands T effector cells over regulatory T cells. In some embodiments, the antibody cytokine engrafted protein comprises a heavy chain variable region (VH), comprising complementarity determining regions HCDR1, HCDR2, HCDR3; a light chain variable region (VL), comprising LCDR1, LCDR2, LCDR3; and an IL-2 molecule or a fragment thereof engrafted into a CDR of the VH or the VL, wherein the IL-2 molecule is a mutein, and wherein the antibody cytokine engrafted protein preferentially expands T effector cells over regulatory T cells. In some embodiments, the IL-2 regimen comprises administration of an antibody described in U.S. Patent Application Publication No. US 2020/0270334 A1, the disclosures of which are incorporated by reference herein. In some embodiments, the antibody cytokine engrafted protein comprises a heavy chain variable region (VH), comprising complementarity determining regions HCDR1, HCDR2, HCDR3; a light chain variable region (VL), comprising LCDR1, LCDR2, LCDR3; and an IL-2 molecule or a fragment thereof engrafted into a CDR of the VH or the VL, wherein the IL-2 molecule is a mutein, wherein the antibody cytokine engrafted protein preferentially expands T effector cells over regulatory T cells, and wherein the antibody further comprises an IgG class heavy chain and an IgG class light chain selected from the group consisting of: a IgG class light chain comprising SEQ ID NO:39 and a IgG class heavy chain comprising SEQ ID NO:38; a IgG class light chain comprising SEQ ID NO:37 and a IgG class heavy chain comprising SEQ ID NO:29; a IgG class light chain comprising SEQ ID NO:39 and a IgG class heavy chain comprising SEQ ID NO:29; and a IgG class light chain comprising SEQ ID NO:37 and a IgG class heavy chain comprising SEQ ID NO:38. DB1/ 149202201.1 16 Attorney Docket No.: 116983-5127-WO [0018] In some embodiments, an IL-2 molecule or a fragment thereof is engrafted into HCDR1 of the VH, wherein the IL-2 molecule is a mutein. In some embodiments, an IL-2 molecule or a fragment thereof is engrafted into HCDR2 of the VH, wherein the IL-2 molecule is a mutein. In some embodiments, an IL-2 molecule or a fragment thereof is engrafted into HCDR3 of the VH, wherein the IL-2 molecule is a mutein. In some embodiments, an IL-2 molecule or a fragment thereof is engrafted into LCDR1 of the VL, wherein the IL-2 molecule is a mutein. In some embodiments, an IL-2 molecule or a fragment thereof is engrafted into LCDR2 of the VL, wherein the IL-2 molecule is a mutein. In some embodiments, an IL-2 molecule or a fragment thereof is engrafted into LCDR3 of the VL, wherein the IL-2 molecule is a mutein. [0019] The insertion of the IL-2 molecule can be at or near the N-terminal region of the CDR, in the middle region of the CDR or at or near the C-terminal region of the CDR. In some embodiments, the antibody cytokine engrafted protein comprises an IL-2 molecule incorporated into a CDR, wherein the IL2 sequence does not frameshift the CDR sequence. In some embodiments, the antibody cytokine engrafted protein comprises an IL-2 molecule incorporated into a CDR, wherein the IL-2 sequence replaces all or part of a CDR sequence. The replacement by the IL-2 molecule can be the N-terminal region of the CDR, in the middle region of the CDR or at or near the C-terminal region the CDR. A replacement by the IL-2 molecule can be as few as one or two amino acids of a CDR sequence, or the entire CDR sequences. [0020] In some embodiments, an IL-2 molecule is engrafted directly into a CDR without a peptide linker, with no additional amino acids between the CDR sequence and the IL-2 sequence. In some embodiments, an IL-2 molecule is engrafted indirectly into a CDR with a peptide linker, with one or more additional amino acids between the CDR sequence and the IL-2 sequence. [0021] In some embodiments, the IL-2 molecule described herein is an IL-2 mutein. In some instances, the IL-2 mutein comprising an R67A substitution. In some embodiments, the IL-2 mutein comprises the amino acid sequence SEQ ID NO:14 or SEQ ID NO:15. In some embodiments, the IL-2 mutein comprises an amino acid sequence in Table 1 in U.S. Patent Application Publication No. US 2020/0270334 A1, the disclosure of which is incorporated by reference herein. DB1/ 149202201.1 17 Attorney Docket No.: 116983-5127-WO [0022] In some embodiments, the antibody cytokine engrafted protein comprises an HCDR1 selected from the group consisting of SEQ ID NO:16, SEQ ID NO:19, SEQ ID NO:22 and SEQ ID NO:25. In some embodiments, the antibody cytokine engrafted protein comprises an HCDR1 selected from the group consisting of SEQ ID NO:7, SEQ ID NO:10, SEQ ID NO:13 and SEQ ID NO:16. In some embodiments, the antibody cytokine engrafted protein comprises an HCDR1 selected from the group consisting of HCDR2 selected from the group consisting of SEQ ID NO:17, SEQ ID NO:20, SEQ ID NO:23, and SEQ ID NO:26. In some embodiments, the antibody cytokine engrafted protein comprises an HCDR3 selected from the group consisting of SEQ ID NO:18, SEQ ID NO:21, SEQ ID NO:24, and SEQ ID NO:27. In some embodiments, the antibody cytokine engrafted protein comprises a VH region comprising the amino acid sequence of SEQ ID NO:28. In some embodiments, the antibody cytokine engrafted protein comprises a heavy chain comprising the amino acid sequence of SEQ ID NO:29. In some embodiments, the antibody cytokine engrafted protein comprises a VL region comprising the amino acid sequence of SEQ ID NO:36. In some embodiments, the antibody cytokine engrafted protein comprises a light chain comprising the amino acid sequence of SEQ ID NO:37. In some embodiments, the antibody cytokine engrafted protein comprises a VH region comprising the amino acid sequence of SEQ ID NO:28 and a VL region comprising the amino acid sequence of SEQ ID NO:36. In some embodiments, the antibody cytokine engrafted protein comprises a heavy chain region comprising the amino acid sequence of SEQ ID NO:29 and a light chain region comprising the amino acid sequence of SEQ ID NO:37. In some embodiments, the antibody cytokine engrafted protein comprises a heavy chain region comprising the amino acid sequence of SEQ ID NO:29 and a light chain region comprising the amino acid sequence of SEQ ID NO:39. In some embodiments, the antibody cytokine engrafted protein comprises a heavy chain region comprising the amino acid sequence of SEQ ID NO:38 and a light chain region comprising the amino acid sequence of SEQ ID NO:37. In some embodiments, the antibody cytokine engrafted protein comprises a heavy chain region comprising the amino acid sequence of SEQ ID NO:38 and a light chain region comprising the amino acid sequence of SEQ ID NO:39. In some embodiments, the antibody cytokine engrafted protein comprises IgG.IL2F71A.H1 or IgG.IL2R67A.H1 of U.S. Patent Application Publication No. 2020/0270334 A1, or variants, derivatives, or fragments thereof, or conservative amino acid substitutions thereof, or proteins with at least 80%, at least 90%, at least 95%, or at least 98% sequence identity thereto. In some embodiments, the antibody components of the antibody DB1/ 149202201.1 18 Attorney Docket No.: 116983-5127-WO cytokine engrafted protein described herein comprise immunoglobulin sequences, framework sequences, or CDR sequences of palivizumab. In some embodiments, the antibody cytokine engrafted protein described herein has a longer serum half-life that a wild-type IL-2 molecule such as, but not limited to, aldesleukin or a comparable molecule. In some embodiments, the antibody cytokine engrafted protein described herein has a sequence as set forth in Table 3. TABLE 3: Sequences of exemplary palivizumab antibody-IL-2 engrafted proteins Identifier Sequence (One-Letter Amino Acid Symbols) SEQ ID NO:13 MYRMQLLSCI ALSLALVTNS APTSSSTKKT QLQLEHLLLD LQMILNGINN YKNPKLTRML IL-2 60 TFKFYMPKKA TELKHLQCLE EELKPLEEVL NLAQSKNFHL RPRDLISNIN VIVLELKGSE 120 TTFMCEYADE TATIVEFLNR WITFCQSIIS TLT 153 SEQ ID NO:14 APTSSSTKKT QLQLEHLLLD LQMILNGINN YKNPKLTAML TFKFYMPKKA TELKHLQCLE IL-2 mutein 60 EELKPLEEVL NLAQSKNFHL RPRDLISNIN VIVLELKGSE TTFMCEYADE TATIVEFLNR 120 WITFCQSIIS TLT 133 SEQ ID NO:15 APTSSSTKKT QLQLEHLLLD LQMILNGINN YKNPKLTRML TAKFYMPKKA TELKHLQCLE IL-2 mutein 60 EELKPLEEVL NLAQSKNFHL RPRDLISNIN VIVLELKGSE TTFMCEYADE TATIVEFLNR 120 WITFCQSIIS TLT 133 SEQ ID NO:16 GFSLAPTSSS TKKTQLQLEH LLLDLQMILN GINNYKNPKL TAMLTFKFYM PKKATELKHL HCDR1_IL-2 60 QCLEEELKPL EEVLNLAQSK NFHLRPRDLI SNINVIVLEL KGSETTFMCE YADETATIVE 120 FLNRWITFCQ SIISTLTSTS GMSVG 145 SEQ ID NO:17 DIWWDDKKDY NPSLKS 16 HCDR2 SEQ ID NO:18 SMITNWYFDV 10 HCDR3 SEQ ID NO:19 APTSSSTKKT QLQLEHLLLD LQMILNGINN YKNPKLTAML TFKFYMPKKA TELKHLQCLE HCDR1_IL-2 60 kabat EELKPLEEVL NLAQSKNFHL RPRDLISNIN VIVLELKGSE TTFMCEYADE TATIVEFLNR 120 WITFCQSIIS TLTSTSGMSV G 141 SEQ ID NO:20 DIWWDDKKDY NPSLKS 16 HCDR2 kabat SEQ ID NO:21 SMITNWYFDV 10 HCDR3 kabat SEQ ID NO:22 GFSLAPTSSS TKKTQLQLEH LLLDLQMILN GINNYKNPKL TAMLTFKFYM PKKATELKHL HCDR1_IL-2 60 clothia QCLEEELKPL EEVLNLAQSK NFHLRPRDLI SNINVIVLEL KGSETTFMCE YADETATIVE 120 FLNRWITFCQ SIISTLTSTS GM 142 SEQ ID NO:23 WWDDK HCDR2 clothia 5 SEQ ID NO:24 SMITNWYFDV 10 HCDR3 clothia SEQ ID NO:25 GFSLAPTSSS TKKTQLQLEH LLLDLQMILN GINNYKNPKL TAMLTFKFYM PKKATELKHL HCDR1_IL-2 60 IMGT QCLEEELKPL EEVLNLAQSK NFHLRPRDLI SNINVIVLEL KGSETTFMCE YADETATIVE 120 FLNRWITFCQ SIISTLTSTS GMS 143 SEQ ID NO:26 IWWDDKK HCDR2 IMGT 7 SEQ ID NO:27 ARSMITNWYF DV 12 HCDR3 IMGT SEQ ID NO:28 QVTLRESGPA LVKPTQTLTL TCTFSGFSLA PTSSSTKKTQ LQLEHLLLDL QMILNGINNY V 60 KNPKLTAMLT FKFYMPKKAT ELKHLQCLEE ELKPLEEVLN LAQSKNFHLR PRDLISNINV 120 IVLELKGSET TFMCEYADET ATIVEFLNRW ITFCQSIIST LTSTSGMSVG WIRQPPGKAL 180 DB1/ 149202201.1 19 Attorney Docket No.: 116983-5127-WO EWLADIWWDD KKDYNPSLKS RLTISKDTSK NQVVLKVTNM DPADTATYYC ARSMITNWYF 240 DVWGAGTTVT VSS 253 SEQ ID NO:29 QMILNGINNY KNPKLTAMLT FKFYMPKKAT ELKHLQCLEE ELKPLEEVLN LAQSKNFHLR Heavy chain 60 PRDLISNINV IVLELKGSET TFMCEYADET ATIVEFLNRW ITFCQSIIST LTSTSGMSVG 120 WIRQPPGKAL EWLADIWWDD KKDYNPSLKS RLTISKDTSK NQVVLKVTNM DPADTATYYC 180 ARSMITNWYF DVWGAGTTVT VSSASTKGPS VFPLAPSSKS TSGGTAALGC LVKDYFPEPV 240 TVSWNSGALT SGVHTFPAVL QSSGLYSLSS VVTVPSSSLG TQTYICNVNH KPSNTKVDKR 300 VEPKSCDKTH TCPPCPAPEL LGGPSVFLFP PKPKDTLMIS RTPEVTCVVV AVSHEDPEVK 360 FNWYVDGVEV HNAKTKPREE QYNSTYRVVS VLTVLHQDWL NGKEYKCKVS NKALAAPIEK 420 TISKAKGQPR EPQVYTLPPS REEMTKNQVS LTCLVKGFYP SDIAVEWESN GQPENNYKTT 480 PPVLDSDGSF FLYSKLTVDK SRWQQGNVFS CSVMHEALHN HYTQKSLSLS PGK 533 SEQ ID NO:30 KAQLSVGYMH 10 LCDR1 kabat SEQ ID NO:31 DTSKLAS 7 LCDR2 kabat SEQ ID NO:32 FQGSGYPFT 9 LCDR3 kabat SEQ ID NO:33 QLSVGY 6 LCDR1 chothia SEQ ID NO:34 DTS 3 LCDR2 chothia SEQ ID NO:35 GSGYPF 6 LCDR3 chothia SEQ ID NO:36 DIQMTQSPST LSASVGDRVT ITCKAQLSVG YMHWYQQKPG KAPKLLIYDT SKLASGVPSR 60 V FSGSGSGTEF TLTISSLQPD DFATYYCFQG SGYPFTFGGG TKLEIK 106 SEQ ID NO:37 DIQMTQSPST LSASVGDRVT ITCKAQLSVG YMHWYQQKPG KAPKLLIYDT SKLASGVPSR 60 Light chain FSGSGSGTEF TLTISSLQPD DFATYYCFQG SGYPFTFGGG TKLEIKRTVA APSVFIFPPS 120 DEQLKSGTAS VVCLLNNFYP REAKVQWKVD NALQSGNSQE SVTEQDSKDS TYSLSSTLTL 180 SKADYEKHKV YACEVTHQGL SSPVTKSFNR GEC 213 SEQ ID NO:38 QVTLRESGPA LVKPTQTLTL TCTFSGFSLA PTSSSTKKTQ LQLEHLLLDL QMILNGINNY 60 Light chain KNPKLTRMLT AKFYMPKKAT ELKHLQCLEE ELKPLEEVLN LAQSKNFHLR PRDLISNINV 120 IVLELKGSET TFMCEYADET ATIVEFLNRW ITFCQSIIST LTSTSGMSVG WIRQPPGKAL 180 EWLADIWWDD KKDYNPSLKS RLTISKDTSK NQVVLKVTNM DPADTATYYC ARSMITNWYF 240 DVWGAGTTVT VSSASTKGPS VFPLAPSSKS TSGGTAALGC LVKDYFPEPV TVSWNSGALT 300 SGVHTFPAVL QSSGLYSLSS VVTVPSSSLG TQTYICNVNH KPSNTKVDKR VEPKSCDKTH 360 TCPPCPAPEL LGGPSVFLFP PKPKDTLMIS RTPEVTCVVV AVSHEDPEVK FNWYVDGVEV 420 HNAKTKPREE QYNSTYRVVS VLTVLHQDWL NGKEYKCKVS NKALAAPIEK TISKAKGQPR 480 EPQVYTLPPS REEMTKNQVS LTCLVKGFYP SDIAVEWESN GQPENNYKTT PPVLDSDGSF 540 FLYSKLTVDK SRWQQGNVFS CSVMHEALHN HYTQKSLSLS PGK 583 SEQ ID NO:39 DIQMTQSPST LSASVGDRVT ITCKAQLSVG YMHWYQQKPG KAPKLLIYDT SKLASGVPSR 60 Light chain FSGSGSGTEF TLTISSLQPD DFATYYCFQG SGYPFTFGGG TKLEIKRTVA APSVFIFPPS 120 DEQLKSGTAS VVCLLNNFYP REAKVQWKVD NALQSGNSQE SVTEQDSKDS TYSLSSTLTL 180 SKADYEKHKV YACEVTHQGL SSPVTKSFNR GEC 213 [0023] The term “IL-4” (also referred to herein as “IL4”) refers to the cytokine known as interleukin 4, which is produced by Th2 T cells and by eosinophils, basophils, and mast cells. IL-4 regulates the differentiation of naïve helper T cells (Th0 cells) to Th2 T cells. Steinke and Borish, Respir. Res.2001, 2, 66-70. Upon activation by IL-4, Th2 T cells subsequently produce additional IL-4 in a positive feedback loop. IL-4 also stimulates B cell proliferation and class II MHC expression, and induces class switching to IgE and IgG1 expression from B cells. Recombinant human IL-4 suitable for use in the invention is commercially available from multiple suppliers, including ProSpec-Tany TechnoGene Ltd., East Brunswick, NJ, USA (Cat. No. CYT-211) and ThermoFisher Scientific, Inc., Waltham, MA, USA (human DB1/ 149202201.1 20 Attorney Docket No.: 116983-5127-WO IL-15 recombinant protein, Cat. No. Gibco CTP0043). The amino acid sequence of recombinant human IL-4 suitable for use in the invention is given in Table 2 (SEQ ID NO:9). [0024] The term “IL-7” (also referred to herein as “IL7”) refers to a glycosylated tissue- derived cytokine known as interleukin 7, which may be obtained from stromal and epithelial cells, as well as from dendritic cells. Fry and Mackall, Blood 2002, 99, 3892-904. IL-7 can stimulate the development of T cells. IL-7 binds to the IL-7 receptor, a heterodimer consisting of IL-7 receptor alpha and common gamma chain receptor, which in a series of signals important for T cell development within the thymus and survival within the periphery. Recombinant human IL-7 suitable for use in the invention is commercially available from multiple suppliers, including ProSpec-Tany TechnoGene Ltd., East Brunswick, NJ, USA (Cat. No. CYT-254) and ThermoFisher Scientific, Inc., Waltham, MA, USA (human IL-15 recombinant protein, Cat. No. Gibco PHC0071). The amino acid sequence of recombinant human IL-7 suitable for use in the invention is given in Table 2 (SEQ ID NO:10). [0025] The term “IL-15” (also referred to herein as “IL15”) refers to the T cell growth factor known as interleukin-15, and includes all forms of IL-2 including human and mammalian forms, conservative amino acid substitutions, glycoforms, biosimilars, and variants thereof. IL-15 is described, e.g., in Fehniger and Caligiuri, Blood 2001, 97, 14-32, the disclosure of which is incorporated by reference herein. IL-15 shares β and γ signaling receptor subunits with IL-2. Recombinant human IL-15 is a single, non-glycosylated polypeptide chain containing 114 amino acids (and an N-terminal methionine) with a molecular mass of 12.8 kDa. Recombinant human IL-15 is commercially available from multiple suppliers, including ProSpec-Tany TechnoGene Ltd., East Brunswick, NJ, USA (Cat. No. CYT-230-b) and ThermoFisher Scientific, Inc., Waltham, MA, USA (human IL-15 recombinant protein, Cat. No.34-8159-82). The amino acid sequence of recombinant human IL-15 suitable for use in the invention is given in Table 2 (SEQ ID NO:11). [0026] The term “IL-21” (also referred to herein as “IL21”) refers to the pleiotropic cytokine protein known as interleukin-21, and includes all forms of IL-21 including human and mammalian forms, conservative amino acid substitutions, glycoforms, biosimilars, and variants thereof. IL-21 is described, e.g., in Spolski and Leonard, Nat. Rev. Drug. Disc.2014, 13, 379-95, the disclosure of which is incorporated by reference herein. IL-21 is primarily produced by natural killer T cells and activated human CD4+ T cells. Recombinant human IL- 21 is a single, non-glycosylated polypeptide chain containing 132 amino acids with a DB1/ 149202201.1 21 Attorney Docket No.: 116983-5127-WO molecular mass of 15.4 kDa. Recombinant human IL-21 is commercially available from multiple suppliers, including ProSpec-Tany TechnoGene Ltd., East Brunswick, NJ, USA (Cat. No. CYT-408-b) and ThermoFisher Scientific, Inc., Waltham, MA, USA (human IL-21 recombinant protein, Cat. No.14-8219-80). The amino acid sequence of recombinant human IL-21 suitable for use in the invention is given in Table 2 (SEQ ID NO:21). [0027] When “an anti-tumor effective amount”, “a tumor-inhibiting effective amount”, or “therapeutic amount” is indicated, the precise amount of the compositions of the present invention to be administered can be determined by a physician with consideration of individual differences in age, weight, tumor size, extent of infection or metastasis, and condition of the patient (subject). It can generally be stated that a pharmaceutical composition comprising the tumor infiltrating lymphocytes (e.g. secondary TILs or genetically modified cytotoxic lymphocytes) described herein may be administered at a dosage of 104 to 1011 cells/kg body weight (e.g., 105 to 106, 105 to 1010, 105 to 1011, 106 to 1010, 106 to 1011,107 to 1011, 107 to 1010, 108 to 1011, 108 to 1010, 109 to 1011, or 109 to 1010 cells/kg body weight), including all integer values within those ranges. TILs (including in some cases, genetically modified cytotoxic lymphocytes) compositions may also be administered multiple times at these dosages. The TILs (including, in some cases, genetically engineered TILs) can be administered by using infusion techniques that are commonly known in immunotherapy (see, e.g., Rosenberg, et al., New Eng. J. of Med.1988, 319, 1676). The optimal dosage and treatment regime for a particular patient can readily be determined by one skilled in the art of medicine by monitoring the patient for signs of disease and adjusting the treatment accordingly. [0028] The term “microenvironment,” as used herein, may refer to the solid or hematological tumor microenvironment as a whole or to an individual subset of cells within the microenvironment. The tumor microenvironment, as used herein, refers to a complex mixture of “cells, soluble factors, signaling molecules, extracellular matrices, and mechanical cues that promote neoplastic transformation, support tumor growth and invasion, protect the tumor from host immunity, foster therapeutic resistance, and provide niches for dominant metastases to thrive,” as described in Swartz, et al., Cancer Res., 2012, 72, 2473. Although tumors express antigens that should be recognized by T cells, tumor clearance by the immune system is rare because of immune suppression by the microenvironment. DB1/ 149202201.1 22 Attorney Docket No.: 116983-5127-WO [0029] In some embodiments, the invention includes a method of treating a cancer with a population of TILs, wherein a patient is pre-treated with non-myeloablative chemotherapy prior to an infusion of TILs according to the invention. In some embodiments, the population of TILs may be provided wherein a patient is pre-treated with nonmyeloablative chemotherapy prior to an infusion of TILs according to the present invention. In some embodiments, the non-myeloablative chemotherapy is cyclophosphamide 60 mg/kg/d for 2 days (days 27 and 26 prior to TIL infusion) and fludarabine 25 mg/m2/d for 5 days (days 27 to 23 prior to TIL infusion). In some embodiments, after non-myeloablative chemotherapy and TIL infusion (at day 0) according to the invention, the patient receives an intravenous infusion of IL-2 intravenously at 720,000 IU/kg every 8 hours to physiologic tolerance. [0030] Experimental findings indicate that lymphodepletion prior to adoptive transfer of tumor-specific T lymphocytes plays a key role in enhancing treatment efficacy by eliminating regulatory T cells and competing elements of the immune system (“cytokine sinks”). Accordingly, some embodiments of the invention utilize a lymphodepletion step (sometimes also referred to as “immunosuppressive conditioning”) on the patient prior to the introduction of the TILs of the invention. [0031] The term “effective amount” or “therapeutically effective amount” refers to that amount of a compound or combination of compounds as described herein that is sufficient to effect the intended application including, but not limited to, disease treatment. A therapeutically effective amount may vary depending upon the intended application (in vitro or in vivo), or the subject and disease condition being treated (e.g., the weight, age and gender of the subject), the severity of the disease condition, or the manner of administration. The term also applies to a dose that will induce a particular response in target cells (e.g., the reduction of platelet adhesion and/or cell migration). The specific dose will vary depending on the particular compounds chosen, the dosing regimen to be followed, whether the compound is administered in combination with other compounds, timing of administration, the tissue to which it is administered, and the physical delivery system in which the compound is carried. [0032] The terms “treatment”, “treating”, “treat”, and the like, refer to obtaining a desired pharmacologic and/or physiologic effect. The effect may be prophylactic in terms of completely or partially preventing a disease or symptom thereof and/or may be therapeutic in terms of a partial or complete cure for a disease and/or adverse effect attributable to the DB1/ 149202201.1 23 Attorney Docket No.: 116983-5127-WO disease. “Treatment”, as used herein, covers any treatment of a disease in a mammal, particularly in a human, and includes: (a) preventing the disease from occurring in a subject which may be predisposed to the disease but has not yet been diagnosed as having it; (b) inhibiting the disease, i.e., arresting its development or progression; and (c) relieving the disease, i.e., causing regression of the disease and/or relieving one or more disease symptoms. “Treatment” is also meant to encompass delivery of an agent in order to provide for a pharmacologic effect, even in the absence of a disease or condition. For example, “treatment” encompasses delivery of a composition that can elicit an immune response or confer immunity in the absence of a disease condition, e.g., in the case of a vaccine. [0033] The terms “non-myeloablative chemotherapy,” “non-myeloablative lymphodepletion,” “NMALD,” “NMA LD,” “NMA-LD,” and any variants of the foregoing, are used interchangeably to indicate a chemotherapeutic regimen designed to deplete the patient’s lymphoid immune cells while avoiding depletion of the patient’s myeloid immune cells. Typically, the patient receives a course of non-myeloablative chemotherapy prior to the administration of tumor infiltrating lymphocytes to the patient as described herein. [0034] The term “heterologous” when used with reference to portions of a nucleic acid or protein indicates that the nucleic acid or protein comprises two or more subsequences that are not found in the same relationship to each other in nature. For instance, the nucleic acid is typically recombinantly produced, having two or more sequences from unrelated genes arranged to make a new functional nucleic acid, e.g., a promoter from one source and a coding region from another source, or coding regions from different sources. Similarly, a heterologous protein indicates that the protein comprises two or more subsequences that are not found in the same relationship to each other in nature (e.g., a fusion protein). [0035] The terms “sequence identity,” “percent identity,” and “sequence percent identity” (or synonyms thereof, e.g., “99% identical”) in the context of two or more nucleic acids or polypeptides, refer to two or more sequences or subsequences that are the same or have a specified percentage of nucleotides or amino acid residues that are the same, when compared and aligned (introducing gaps, if necessary) for maximum correspondence, not considering any conservative amino acid substitutions as part of the sequence identity. The percent identity can be measured using sequence comparison software or algorithms or by visual inspection. Various algorithms and software are known in the art that can be used to obtain alignments of amino acid or nucleotide sequences. Suitable programs to determine percent DB1/ 149202201.1 24 Attorney Docket No.: 116983-5127-WO sequence identity include for example the BLAST suite of programs available from the U.S. Government’s National Center for Biotechnology Information BLAST web site. Comparisons between two sequences can be carried using either the BLASTN or BLASTP algorithm. BLASTN is used to compare nucleic acid sequences, while BLASTP is used to compare amino acid sequences. ALIGN, ALIGN-2 (Genentech, South San Francisco, California) or MegAlign, available from DNASTAR, are additional publicly available software programs that can be used to align sequences. One skilled in the art can determine appropriate parameters for maximal alignment by particular alignment software. In certain embodiments, the default parameters of the alignment software are used. [0036] As used herein, the term “variant” encompasses but is not limited to antibodies or fusion proteins which comprise an amino acid sequence which differs from the amino acid sequence of a reference antibody by way of one or more substitutions, deletions and/or additions at certain positions within or adjacent to the amino acid sequence of the reference antibody. The variant may comprise one or more conservative substitutions in its amino acid sequence as compared to the amino acid sequence of a reference antibody. Conservative substitutions may involve, e.g., the substitution of similarly charged or uncharged amino acids. The variant retains the ability to specifically bind to the antigen of the reference antibody. The term variant also includes pegylated antibodies or proteins. [0037] By “tumor infiltrating lymphocytes” or “TILs” herein is meant a population of cells originally obtained as white blood cells that have left the bloodstream of a subject and migrated into a tumor. TILs include, but are not limited to, CD8+ cytotoxic T cells (lymphocytes), Th1 and Th17 CD4+ T cells, natural killer cells, dendritic cells and M1 macrophages. TILs include both primary and secondary TILs. “Primary TILs” are those that are obtained from patient tissue samples as outlined herein (sometimes referred to as “freshly harvested”), and “secondary TILs” are any TIL cell populations that have been expanded or proliferated as discussed herein, including, but not limited to bulk TILs, expanded TILs (“REP TILs”) as well as “reREP TILs” as discussed herein. reREP TILs can include for example second expansion TILs or second additional expansion TILs. [0038] TILs can generally be defined either biochemically, using cell surface markers, or functionally, by their ability to infiltrate tumors and effect treatment. TILs can be generally categorized by expressing one or more of the following biomarkers: CD4, CD8, TCR αβ, CD27, CD28, CD56, CCR7, CD45Ra, CD95, PD-1, and CD25. Additionally, and DB1/ 149202201.1 25 Attorney Docket No.: 116983-5127-WO alternatively, TILs can be functionally defined by their ability to infiltrate solid tumors upon reintroduction into a patient. TILs may further be characterized by potency – for example, TILs may be considered potent if, for example, interferon (IFN) release is greater than about 50 pg/mL, greater than about 100 pg/mL, greater than about 150 pg/mL, or greater than about 200 pg/mL. TILs may be considered potent if, for example, interferon (IFNγ) release is greater than about 50 pg/mL, greater than about 100 pg/mL, greater than about 150 pg/mL, or greater than about 200 pg/mL, greater than about 300 pg/mL, greater than about 400 pg/mL, greater than about 500 pg/mL, greater than about 600 pg/mL, greater than about 700 pg/mL, greater than about 800 pg/mL, greater than about 900 pg/mL, greater than about 1000 pg/mL. [0039] The term “deoxyribonucleotide” encompasses natural and synthetic, unmodified and modified deoxyribonucleotides. Modifications include changes to the sugar moiety, to the base moiety and/or to the linkages between deoxyribonucleotide in the oligonucleotide. [0040] The term “RNA” defines a molecule comprising at least one ribonucleotide residue. The term “ribonucleotide” defines a nucleotide with a hydroxyl group at the 2' position of a b-D-ribofuranose moiety. The term RNA includes double-stranded RNA, single-stranded RNA, isolated RNA such as partially purified RNA, essentially pure RNA, synthetic RNA, recombinantly produced RNA, as well as altered RNA that differs from naturally occurring RNA by the addition, deletion, substitution and/or alteration of one or more nucleotides. Nucleotides of the RNA molecules described herein may also comprise non-standard nucleotides, such as non-naturally occurring nucleotides or chemically synthesized nucleotides or deoxynucleotides. These altered RNAs can be referred to as analogs or analogs of naturally-occurring RNA. [0041] The terms “pharmaceutically acceptable carrier” or “pharmaceutically acceptable excipient” are intended to include any and all solvents, dispersion media, coatings, antibacterial and antifungal agents, isotonic and absorption delaying agents, and inert ingredients. The use of such pharmaceutically acceptable carriers or pharmaceutically acceptable excipients for active pharmaceutical ingredients is well known in the art. Except insofar as any conventional pharmaceutically acceptable carrier or pharmaceutically acceptable excipient is incompatible with the active pharmaceutical ingredient, its use in therapeutic compositions of the invention is contemplated. Additional active pharmaceutical ingredients, such as other drugs, can also be incorporated into the described compositions and methods. DB1/ 149202201.1 26 Attorney Docket No.: 116983-5127-WO [0042] The terms “about” and “approximately” mean within a statistically meaningful range of a value. Such a range can be within an order of magnitude, preferably within 50%, more preferably within 20%, more preferably still within 10%, and even more preferably within 5% of a given value or range. The allowable variation encompassed by the terms “about” or “approximately” depends on the particular system under study, and can be readily appreciated by one of ordinary skill in the art. Moreover, as used herein, the terms “about” and “approximately” mean that dimensions, sizes, formulations, parameters, shapes and other quantities and characteristics are not and need not be exact, but may be approximate and/or larger or smaller, as desired, reflecting tolerances, conversion factors, rounding off, measurement error and the like, and other factors known to those of skill in the art. In general, a dimension, size, formulation, parameter, shape or other quantity or characteristic is “about” or “approximate” whether or not expressly stated to be such. It is noted that embodiments of very different sizes, shapes and dimensions may employ the described arrangements. [0043] The transitional terms “comprising,” “consisting essentially of,” and “consisting of,” when used in the appended claims, in original and amended form, define the claim scope with respect to what unrecited additional claim elements or steps, if any, are excluded from the scope of the claim(s). The term “comprising” is intended to be inclusive or open-ended and does not exclude any additional, unrecited element, method, step or material. The term “consisting of” excludes any element, step or material other than those specified in the claim and, in the latter instance, impurities ordinary associated with the specified material(s). The term “consisting essentially of” limits the scope of a claim to the specified elements, steps or material(s) and those that do not materially affect the basic and novel characteristic(s) of the claimed invention. All compositions, methods, and kits described herein that embody the present invention can, in alternate embodiments, be more specifically defined by any of the transitional terms “comprising,” “consisting essentially of,” and “consisting of.” [0044] The terms “antibody” and its plural form “antibodies” refer to whole immunoglobulins and any antigen-binding fragment (“antigen-binding portion”) or single chains thereof. An “antibody” further refers to a glycoprotein comprising at least two heavy (H) chains and two light (L) chains inter-connected by disulfide bonds, or an antigen-binding portion thereof. Each heavy chain is comprised of a heavy chain variable region (abbreviated herein as VH) and a heavy chain constant region. The heavy chain constant region is DB1/ 149202201.1 27 Attorney Docket No.: 116983-5127-WO comprised of three domains, CH1, CH2 and CH3. Each light chain is comprised of a light chain variable region (abbreviated herein as VL) and a light chain constant region. The light chain constant region is comprised of one domain, CL. The VH and VL regions of an antibody may be further subdivided into regions of hypervariability, which are referred to as complementarity determining regions (CDR) or hypervariable regions (HVR), and which can be interspersed with regions that are more conserved, termed framework regions (FR). Each VH and VL is composed of three CDRs and four FRs, arranged from amino-terminus to carboxy-terminus in the following order: FR1, CDR1, FR2, CDR2, FR3, CDR3, FR4. The variable regions of the heavy and light chains contain a binding domain that interacts with an antigen epitope or epitopes. The constant regions of the antibodies may mediate the binding of the immunoglobulin to host tissues or factors, including various cells of the immune system (e.g., effector cells) and the first component (Clq) of the classical complement system. [0045] The term “antigen” refers to a substance that induces an immune response. In some embodiments, an antigen is a molecule capable of being bound by an antibody or a TCR if presented by major histocompatibility complex (MHC) molecules. The term “antigen”, as used herein, also encompasses T cell epitopes. An antigen is additionally capable of being recognized by the immune system. In some embodiments, an antigen is capable of inducing a humoral immune response or a cellular immune response leading to the activation of B lymphocytes and/or T lymphocytes. In some cases, this may require that the antigen contains or is linked to a Th cell epitope. An antigen can also have one or more epitopes (e.g., B- and T-epitopes). In some embodiments, an antigen will preferably react, typically in a highly specific and selective manner, with its corresponding antibody or TCR and not with the multitude of other antibodies or TCRs which may be induced by other antigens. [0046] The terms “monoclonal antibody,” “mAb,” “monoclonal antibody composition,” or their plural forms refer to a preparation of antibody molecules of single molecular composition. A monoclonal antibody composition displays a single binding specificity and affinity for a particular epitope. Monoclonal antibodies specific to certain receptors can be made using knowledge and skill in the art of injecting test subjects with suitable antigen and then isolating hybridomas expressing antibodies having the desired sequence or functional characteristics. DNA encoding the monoclonal antibodies is readily isolated and sequenced using conventional procedures (e.g., by using oligonucleotide probes that are capable of binding specifically to genes encoding the heavy and light chains of the monoclonal DB1/ 149202201.1 28 Attorney Docket No.: 116983-5127-WO antibodies). The hybridoma cells serve as a preferred source of such DNA. Once isolated, the DNA may be placed into expression vectors, which are then transfected into host cells such as E. coli cells, simian COS cells, Chinese hamster ovary (CHO) cells, or myeloma cells that do not otherwise produce immunoglobulin protein, to obtain the synthesis of monoclonal antibodies in the recombinant host cells. Recombinant production of antibodies will be described in more detail below. [0047] The terms “antigen-binding portion” or “antigen-binding fragment” of an antibody (or simply “antibody portion” or “fragment”), as used herein, refers to one or more fragments of an antibody that retain the ability to specifically bind to an antigen. It has been shown that the antigen-binding function of an antibody can be performed by fragments of a full-length antibody. Examples of binding fragments encompassed within the term “antigen-binding portion” of an antibody include (i) a Fab fragment, a monovalent fragment consisting of the VL, VH, CL and CH1 domains; (ii) a F(ab′)2 fragment, a bivalent fragment comprising two Fab fragments linked by a disulfide bridge at the hinge region; (iii) a Fd fragment consisting of the VH and CH1 domains; (iv) a Fv fragment consisting of the VL and VH domains of a single arm of an antibody, (v) a domain antibody (dAb) fragment (Ward, et al., Nature, 1989, 341, 544-546), which may consist of a VH or a VL domain; and (vi) an isolated complementarity determining region (CDR). Furthermore, although the two domains of the Fv fragment, VL and VH, are coded for by separate genes, they can be joined, using recombinant methods, by a synthetic linker that enables them to be made as a single protein chain in which the VL and VH regions pair to form monovalent molecules known as single chain Fv (scFv); see, e.g., Bird, et al., Science 1988, 242, 423-426; and Huston, et al., Proc. Natl. Acad. Sci. USA 1988, 85, 5879-5883). Such scFv antibodies are also intended to be encompassed within the terms “antigen-binding portion” or “antigen-binding fragment” of an antibody. These antibody fragments are obtained using conventional techniques known to those with skill in the art, and the fragments are screened for utility in the same manner as are intact antibodies. In some embodiments, a scFv protein domain comprises a VH portion and a VL portion. A scFv molecule is denoted as either VL-L-VH if the VL domain is the N-terminal part of the scFv molecule, or as VH-L-VL if the VH domain is the N-terminal part of the scFv molecule. Methods for making scFv molecules and designing suitable peptide linkers are described in U.S. Pat. No.4,704,692, U.S. Pat. No.4,946,778, R. Raag and M. Whitlow, “Single Chain Fvs.” FASEB Vol 9:73-80 (1995) and R. E. Bird and B. W. Walker, Single DB1/ 149202201.1 29 Attorney Docket No.: 116983-5127-WO Chain Antibody Variable Regions, TIBTECH, Vol 9: 132-137 (1991), the disclosures of which are incorporated by reference herein. [0048] The term “human antibody,” as used herein, is intended to include antibodies having variable regions in which both the framework and CDR regions are derived from human germline immunoglobulin sequences. Furthermore, if the antibody contains a constant region, the constant region also is derived from human germline immunoglobulin sequences. The human antibodies of the invention may include amino acid residues not encoded by human germline immunoglobulin sequences (e.g., mutations introduced by random or site-specific mutagenesis in vitro or by somatic mutation in vivo). The term “human antibody”, as used herein, is not intended to include antibodies in which CDR sequences derived from the germline of another mammalian species, such as a mouse, have been grafted onto human framework sequences. [0049] The term “human monoclonal antibody” refers to antibodies displaying a single binding specificity which have variable regions in which both the framework and CDR regions are derived from human germline immunoglobulin sequences. In some embodiments, the human monoclonal antibodies are produced by a hybridoma which includes a B cell obtained from a transgenic nonhuman animal, e.g., a transgenic mouse, having a genome comprising a human heavy chain transgene and a light chain transgene fused to an immortalized cell. [0050] The term “recombinant human antibody”, as used herein, includes all human antibodies that are prepared, expressed, created or isolated by recombinant means, such as (a) antibodies isolated from an animal (such as a mouse) that is transgenic or transchromosomal for human immunoglobulin genes or a hybridoma prepared therefrom (described further below), (b) antibodies isolated from a host cell transformed to express the human antibody, e.g., from a transfectoma, (c) antibodies isolated from a recombinant, combinatorial human antibody library, and (d) antibodies prepared, expressed, created or isolated by any other means that involve splicing of human immunoglobulin gene sequences to other DNA sequences. Such recombinant human antibodies have variable regions in which the framework and CDR regions are derived from human germline immunoglobulin sequences. In certain embodiments, however, such recombinant human antibodies can be subjected to in vitro mutagenesis (or, when an animal transgenic for human Ig sequences is used, in vivo somatic mutagenesis) and thus the amino acid sequences of the VH and VL regions of the DB1/ 149202201.1 30 Attorney Docket No.: 116983-5127-WO recombinant antibodies are sequences that, while derived from and related to human germline VH and VL sequences, may not naturally exist within the human antibody germline repertoire in vivo. [0051] As used herein, “isotype” refers to the antibody class (e.g., IgM or IgG1) that is encoded by the heavy chain constant region genes. [0052] The phrases “an antibody recognizing an antigen” and “an antibody specific for an antigen” are used interchangeably herein with the term “an antibody which binds specifically to an antigen.” [0053] The term “human antibody derivatives” refers to any modified form of the human antibody, including a conjugate of the antibody and another active pharmaceutical ingredient or antibody. The terms “conjugate,” “antibody-drug conjugate”, “ADC,” or “immunoconjugate” refers to an antibody, or a fragment thereof, conjugated to another therapeutic moiety, which can be conjugated to antibodies described herein using methods available in the art. [0054] The terms “humanized antibody,” “humanized antibodies,” and “humanized” are intended to refer to antibodies in which CDR sequences derived from the germline of another mammalian species, such as a mouse, have been grafted onto human framework sequences. Additional framework region modifications may be made within the human framework sequences. Humanized forms of non-human (for example, murine) antibodies are chimeric antibodies that contain minimal sequence derived from non-human immunoglobulin. For the most part, humanized antibodies are human immunoglobulins (recipient antibody) in which residues from a hypervariable region of the recipient are replaced by residues from a 15 hypervariable region of a non-human species (donor antibody) such as mouse, rat, rabbit or nonhuman primate having the desired specificity, affinity, and capacity. In some instances, Fv framework region (FR) residues of the human immunoglobulin are replaced by corresponding non-human residues. Furthermore, humanized antibodies may comprise residues that are not found in the recipient antibody or in the donor antibody. These modifications are made to further refine antibody performance. In general, the humanized antibody will comprise substantially all of at least one, and typically two, variable domains, in which all or substantially all of the hypervariable loops correspond to those of a non- human immunoglobulin and all or substantially all of the FR regions are those of a human immunoglobulin sequence. The humanized antibody optionally also will comprise at least a DB1/ 149202201.1 31 Attorney Docket No.: 116983-5127-WO portion of an immunoglobulin constant region (Fc), typically that of a human immunoglobulin. For further details, see Jones, et al., Nature 1986, 321, 522-525; Riechmann, et al., Nature 1988, 332, 323-329; and Presta, Curr. Op. Struct. Biol.1992, 2, 593-596. The antibodies described herein may also be modified to employ any Fc variant which is known to impart an improvement (e.g., reduction) in effector function and/or FcR binding. The Fc variants may include, for example, any one of the amino acid substitutions disclosed in International Patent Application Publication Nos. WO 1988/07089 A1, WO 1996/14339 A1, WO 1998/05787 A1, WO 1998/23289 A1, WO 1999/51642 A1, WO 99/58572 A1, WO 2000/09560 A2, WO 2000/32767 A1, WO 2000/42072 A2, WO 2002/44215 A2, WO 2002/060919 A2, WO 2003/074569 A2, WO 2004/016750 A2, WO 2004/029207 A2, WO 2004/035752 A2, WO 2004/063351 A2, WO 2004/074455 A2, WO 2004/099249 A2, WO 2005/040217 A2, WO 2005/070963 A1, WO 2005/077981 A2, WO 2005/092925 A2, WO 2005/123780 A2, WO 2006/019447 A1, WO 2006/047350 A2, and WO 2006/085967 A2; and U.S. Patent Nos.5,648,260; 5,739,277; 5,834,250; 5,869,046; 6,096,871; 6,121,022; 6,194,551; 6,242,195; 6,277,375; 6,528,624; 6,538,124; 6,737,056; 6,821,505; 6,998,253; and 7,083,784; the disclosures of which are incorporated by reference herein. [0055] The term “chimeric antibody” is intended to refer to antibodies in which the variable region sequences are derived from one species and the constant region sequences are derived from another species, such as an antibody in which the variable region sequences are derived from a mouse antibody and the constant region sequences are derived from a human antibody. [0056] A “diabody” is a small antibody fragment with two antigen-binding sites. The fragments comprises a heavy chain variable domain (VH) connected to a light chain variable domain (VL) in the same polypeptide chain (VH-VL or VL-VH). By using a linker that is too short to allow pairing between the two domains on the same chain, the domains are forced to pair with the complementary domains of another chain and create two antigen-binding sites. Diabodies are described more fully in, e.g., European Patent No. EP 404,097, International Patent Publication No. WO 93/11161; and Bolliger, et al., Proc. Natl. Acad. Sci. USA 1993, 90, 6444-6448. [0057] The term “glycosylation” refers to a modified derivative of an antibody. An aglycoslated antibody lacks glycosylation. Glycosylation can be altered to, for example, DB1/ 149202201.1 32 Attorney Docket No.: 116983-5127-WO increase the affinity of the antibody for antigen. Such carbohydrate modifications can be accomplished by, for example, altering one or more sites of glycosylation within the antibody sequence. For example, one or more amino acid substitutions can be made that result in elimination of one or more variable region framework glycosylation sites to thereby eliminate glycosylation at that site. Aglycosylation may increase the affinity of the antibody for antigen, as described in U.S. Patent Nos.5,714,350 and 6,350,861. Additionally or alternatively, an antibody can be made that has an altered type of glycosylation, such as a hypofucosylated antibody having reduced amounts of fucosyl residues or an antibody having increased bisecting GlcNac structures. Such altered glycosylation patterns have been demonstrated to increase the ability of antibodies. Such carbohydrate modifications can be accomplished by, for example, expressing the antibody in a host cell with altered glycosylation machinery. Cells with altered glycosylation machinery have been described in the art and can be used as host cells in which to express recombinant antibodies of the invention to thereby produce an antibody with altered glycosylation. For example, the cell lines Ms704, Ms705, and Ms709 lack the fucosyltransferase gene, FUT8 (alpha (1,6) fucosyltransferase), such that antibodies expressed in the Ms704, Ms705, and Ms709 cell lines lack fucose on their carbohydrates. The Ms704, Ms705, and Ms709 FUT8−/− cell lines were created by the targeted disruption of the FUT8 gene in CHO/DG44 cells using two replacement vectors (see e.g. U.S. Patent Publication No.2004/0110704 or Yamane-Ohnuki, et al., Biotechnol. Bioeng., 2004, 87, 614-622). As another example, European Patent No. EP 1,176,195 describes a cell line with a functionally disrupted FUT8 gene, which encodes a fucosyl transferase, such that antibodies expressed in such a cell line exhibit hypofucosylation by reducing or eliminating the alpha 1,6 bond-related enzyme, and also describes cell lines which have a low enzyme activity for adding fucose to the N- acetylglucosamine that binds to the Fc region of the antibody or does not have the enzyme activity, for example the rat myeloma cell line YB2/0 (ATCC CRL 1662). International Patent Publication WO 03/035835 describes a variant CHO cell line, Lec 13 cells, with reduced ability to attach fucose to Asn(297)-linked carbohydrates, also resulting in hypofucosylation of antibodies expressed in that host cell (see also Shields, et al., J. Biol. Chem.2002, 277, 26733-26740. International Patent Publication WO 99/54342 describes cell lines engineered to express glycoprotein-modifying glycosyl transferases (e.g., beta(1,4)-N- acetylglucosaminyltransferase III (GnTIII)) such that antibodies expressed in the engineered cell lines exhibit increased bisecting GlcNac structures which results in increased ADCC DB1/ 149202201.1 33 Attorney Docket No.: 116983-5127-WO activity of the antibodies (see also Umana, et al., Nat. Biotech.1999, 17, 176-180). Alternatively, the fucose residues of the antibody may be cleaved off using a fucosidase enzyme. For example, the fucosidase alpha-L-fucosidase removes fucosyl residues from antibodies as described in Tarentino, et al., Biochem.1975, 14, 5516-5523. [0058] “Pegylation” refers to a modified antibody, or a fragment thereof, that typically is reacted with polyethylene glycol (PEG), such as a reactive ester or aldehyde derivative of PEG, under conditions in which one or more PEG groups become attached to the antibody or antibody fragment. Pegylation may, for example, increase the biological (e.g., serum) half life of the antibody. Preferably, the pegylation is carried out via an acylation reaction or an alkylation reaction with a reactive PEG molecule (or an analogous reactive water-soluble polymer). As used herein, the term “polyethylene glycol” is intended to encompass any of the forms of PEG that have been used to derivatize other proteins, such as mono (C1-C10)alkoxy- or aryloxy-polyethylene glycol or polyethylene glycol-maleimide. The antibody to be pegylated may be an aglycosylated antibody. Methods for pegylation are known in the art and can be applied to the antibodies of the invention, as described for example in European Patent Nos. EP 0154316 and EP 0401384 and U.S. Patent No.5,824,778, the disclosures of each of which are incorporated by reference herein. [0059] The term “biosimilar” means a biological product, including a monoclonal antibody or protein, that is highly similar to a U.S. licensed reference biological product notwithstanding minor differences in clinically inactive components, and for which there are no clinically meaningful differences between the biological product and the reference product in terms of the safety, purity, and potency of the product. Furthermore, a similar biological or “biosimilar” medicine is a biological medicine that is similar to another biological medicine that has already been authorized for use by the European Medicines Agency. The term “biosimilar” is also used synonymously by other national and regional regulatory agencies. Biological products or biological medicines are medicines that are made by or derived from a biological source, such as a bacterium or yeast. They can consist of relatively small molecules such as human insulin or erythropoietin, or complex molecules such as monoclonal antibodies. For example, if the reference IL-2 protein is aldesleukin (PROLEUKIN), a protein approved by drug regulatory authorities with reference to aldesleukin is a “biosimilar to” aldesleukin or is a “biosimilar thereof” of aldesleukin. In Europe, a similar biological or “biosimilar” medicine is a biological medicine that is similar DB1/ 149202201.1 34 Attorney Docket No.: 116983-5127-WO to another biological medicine that has already been authorized for use by the European Medicines Agency (EMA). The relevant legal basis for similar biological applications in Europe is Article 6 of Regulation (EC) No 726/2004 and Article 10(4) of Directive 2001/83/EC, as amended and therefore in Europe, the biosimilar may be authorized, approved for authorization or subject of an application for authorization under Article 6 of Regulation (EC) No 726/2004 and Article 10(4) of Directive 2001/83/EC. The already authorized original biological medicinal product may be referred to as a “reference medicinal product” in Europe. Some of the requirements for a product to be considered a biosimilar are outlined in the CHMP Guideline on Similar Biological Medicinal Products. In addition, product specific guidelines, including guidelines relating to monoclonal antibody biosimilars, are provided on a product-by-product basis by the EMA and published on its website. A biosimilar as described herein may be similar to the reference medicinal product by way of quality characteristics, biological activity, mechanism of action, safety profiles and/or efficacy. In addition, the biosimilar may be used or be intended for use to treat the same conditions as the reference medicinal product. Thus, a biosimilar as described herein may be deemed to have similar or highly similar quality characteristics to a reference medicinal product. Alternatively, or in addition, a biosimilar as described herein may be deemed to have similar or highly similar biological activity to a reference medicinal product. Alternatively, or in addition, a biosimilar as described herein may be deemed to have a similar or highly similar safety profile to a reference medicinal product. Alternatively, or in addition, a biosimilar as described herein may be deemed to have similar or highly similar efficacy to a reference medicinal product. As described herein, a biosimilar in Europe is compared to a reference medicinal product which has been authorized by the EMA. However, in some instances, the biosimilar may be compared to a biological medicinal product which has been authorized outside the European Economic Area (a non-EEA authorized “comparator”) in certain studies. Such studies include for example certain clinical and in vivo non-clinical studies. As used herein, the term “biosimilar” also relates to a biological medicinal product which has been or may be compared to a non-EEA authorized comparator. Certain biosimilars are proteins such as antibodies, antibody fragments (for example, antigen binding portions) and fusion proteins. A protein biosimilar may have an amino acid sequence that has minor modifications in the amino acid structure (including for example deletions, additions, and/or substitutions of amino acids) which do not significantly affect the function of the polypeptide. The biosimilar may comprise an amino acid sequence having a sequence DB1/ 149202201.1 35 Attorney Docket No.: 116983-5127-WO identity of 97% or greater to the amino acid sequence of its reference medicinal product, e.g., 97%, 98%, 99% or 100%. The biosimilar may comprise one or more post-translational modifications, for example, although not limited to, glycosylation, oxidation, deamidation, and/or truncation which is/are different to the post-translational modifications of the reference medicinal product, provided that the differences do not result in a change in safety and/or efficacy of the medicinal product. The biosimilar may have an identical or different glycosylation pattern to the reference medicinal product. Particularly, although not exclusively, the biosimilar may have a different glycosylation pattern if the differences address or are intended to address safety concerns associated with the reference medicinal product. Additionally, the biosimilar may deviate from the reference medicinal product in for example its strength, pharmaceutical form, formulation, excipients and/or presentation, providing safety and efficacy of the medicinal product is not compromised. The biosimilar may comprise differences in for example pharmacokinetic (PK) and/or pharmacodynamic (PD) profiles as compared to the reference medicinal product but is still deemed sufficiently similar to the reference medicinal product as to be authorized or considered suitable for authorization. In certain circumstances, the biosimilar exhibits different binding characteristics as compared to the reference medicinal product, wherein the different binding characteristics are considered by a Regulatory Authority such as the EMA not to be a barrier for authorization as a similar biological product. The term “biosimilar” is also used synonymously by other national and regional regulatory agencies. II. Trop-2 Targeting ADC [0060] An antibody drug conjugate (ADC) has three core components: a monoclonal antibody (MAb) targeting a specific tumor antigen, a cytotoxic payload, and a chemical linker connecting them. The basic mechanism involves binding of the antibody to the tumor antigen on the cell surface, followed by internalization of the ADC and lysosomal degradation. This results in release of the active cytotoxic agent into the cytoplasm which results in tumor cell death. Some ADCs also induce antibody dependent cellular cytotoxicity (ADCC) and complement dependent cytotoxicity (CDC) contributing to the antitumor efficacy of these agents (Junttila et al., Breast Cancer Res Treat.2011 Jul;128(2):347–356). DB1/ 149202201.1 36 Attorney Docket No.: 116983-5127-WO [0061] As used herein, a “Trop-2 targeting ADC” or “anti-Trop-2 ADC” refers to a conjugated molecule comprising an anti-Trop-2 antibody linked to a drug moiety that combines the specificity of the anti-Trop-2 antibody with potency of the drug moiety. [0062] In one aspect, the invention provides a Trop-2 targeting ADC of Formula (I): Ab-(L-(D)m)n Wherein Ab represents an anti-Trop-2 antibody described herein; L is a linker; D is a drug moiety; m is an integer from 1 to 8; and n is an integer from 1-20. [0063] In one embodiment, n is an integer from 1 to 10, 2 to 8, or 2 to 5. In a specific embodiment, n is 2, 3, or 4. In some embodiments, m is 1; in other embodiments, m is 2, 3 or 4. [0064] While the drug to antibody ratio (DAR) has an exact value for a specific conjugate molecule (e.g., n multiplied by m in Formula (I)), it is understood that the value will often be an average value when used to describe a sample containing many molecules, due to some degree of heterogeneity, typically associated with the conjugation step. In some embodiments, the DAR is between about 2 and about 10, and typically is about 3, 3.5, 4, 4.5, 5, 5.5, 6, 6.5, 7.0, 7.5, 8.0. In some embodiments, at least 50% of a sample by weight is compound having the average DAR plus or minus 2, and preferably at least 50% of the sample is a conjugate that contains the average DAR plus or minus 1. Embodiments include Trop-2 targeting ADCs wherein the DAR is about 3.5, 3.6, 3.7, 3.8 or 3.9. In some embodiments, a DAR of ‘about n’ means the measured value for DAR is within 20% of n. A. Anti-Trop-2 Antibody [0065] The Trop-2 targeting ADCs include at least one antibody or fragment thereof that binds to Trop-2. Anti-Trop-2 antibodies are known and/or publicly available and in alternative embodiments may be utilized in the Trop-2 targeting ADCs. While humanized or human antibodies are preferred for reduced immunogenicity, in alternative embodiments a chimeric antibody may be of use. As discussed below, methods of antibody humanization are DB1/ 149202201.1 37 Attorney Docket No.: 116983-5127-WO well known in the art and may be utilized to convert an available murine or chimeric antibody into a humanized form. [0066] Anti-Trop-2 antibodies are commercially available from a number of sources and include LS-C126418, LS-C178765, LS-C126416, LS-C126417 (LifeSpan Biosciences, Inc., Seattle, WA); 10428-MM01, 10428-MM02, 10428-R001, 10428-R030 (Sino Biological Inc., Beijing, China); MR54 (eBioscience, San Diego, CA); sc-376181, sc-376746, Santa Cruz Biotechnology (Santa Cruz, CA); MM0588-49D6, (Novus Biologicals, Littleton, CO); ab79976, and ab89928 (ABCAM®, Cambridge, MA). [0067] Other anti-Trop-2 antibodies have been disclosed in the patent literature. For example, U.S. Publ. No.2013/0089872 discloses anti-Trop-2 antibodies K5-70 (Accession No. FERM BP-11251), K5-107 (Accession No. FERM BP-11252), K5-116-2-1 (Accession No. FERM BP-11253), T6-16 (Accession No. FERM BP-11346), and T5-86 (Accession No. FERM BP- 11254), deposited with the International Patent Organism Depositary, Tsukuba, Japan. U.S. Patent No.5,840,854 disclosed the anti-Trop-2 monoclonal antibody BR110 (ATCC No. HB11698). U.S. Patent No.7,420,040 disclosed an anti-Trop-2 antibody produced by hybridoma cell line AR47A6.4.2, deposited with the IDAC (International Depository Authority of Canada, Winnipeg, Canada) as accession number 141205-05. U.S. Patent No. 7,420,041 disclosed an anti-Trop-2 antibody produced by hybridoma cell line AR52A301.5, deposited with the IDAC as accession number 141205-03. U.S. Publ. No.2013/0122020 disclosed anti-Trop-2 antibodies 3E9, 6G11, 7E6, 15E2, 18B1. Hybridomas encoding a representative antibody were deposited with the American Type Culture Collection (ATCC), Accession Nos. PTA-12871 and PTA-12872. U.S. Patent No.8,715,662 discloses anti-Trop-2 antibodies produced by hybridomas deposited at the AID-ICLC (Genoa, Italy) with deposit numbers PD 08019, PD 08020 and PD 08021. U.S. Patent Application Publ. No. 20120237518 discloses anti-Trop-2 antibodies 77220, KM4097 and KM4590. U.S. Patent No.8,309,094 (Wyeth) discloses antibodies A1 and A3, identified by sequence listing. The Examples section of each patent or patent application cited above in this paragraph is incorporated herein by reference. Non-patent publication Lipinski et al. (1981, Proc Natl. Acad Sci USA, 78:5147-50) disclosed anti-Trop-2 antibodies 162-25.3 and 162-46.2. [0068] Numerous anti-Trop-2 antibodies are known in the art and/or publicly available. As discussed below, methods for preparing antibodies against known antigens were routine in the art. The sequence of the human Trop-2 protein was also known in the art (see, e.g., DB1/ 149202201.1 38 Attorney Docket No.: 116983-5127-WO GenBank Accession No. CAA54801.1). Methods for producing humanized, human or chimeric antibodies were also known. The person of ordinary skill, reading the instant disclosure in light of general knowledge in the art, would have been able to make and use the genus of anti-Trop-2 antibodies in the subject ADCs. In an embodiment, the anti-Trop-2 antibody may be a humanized RS7 antibody (see, e.g., U.S. Patent No.7,238,785, incorporated herein by reference in its entirety). B. Drug Moiety [0069] The drug to be conjugated to the anti-Trop-2 antibody or antibody fragment may be selected from the group consisting of an anthracycline, a camptothecin, a tubulin inhibitor, a maytansinoid, a calicheamycin, an auristatin, a nitrogen mustard, an ethylenimine derivative, an alkyl sulfonate, a nitrosourea, a triazene, a folic acid analog, a taxane, a COX-2 inhibitor, a pyrimidine analog, a purine analog, an antibiotic, an enzyme inhibitor, an epipodophyllotoxin, a platinum coordination complex, a vinca alkaloid, a substituted urea, a methyl hydrazine derivative, an adrenocortical suppressant, a hormone antagonist, an antimetabolite, an alkylating agent, an antimitotic, an anti- angiogenic agent, a tyrosine kinase inhibitor, an mTOR inhibitor, a heat shock protein (HSP90) inhibitor, a proteosome inhibitor, an HDAC inhibitor, a pro-apoptotic agent, and a combination thereof. [0070] Specific drugs of use may be selected from the group consisting of 5-fluorouracil, afatinib, aplidin, azaribine, anastrozole, anthracyclines, axitinib, AVL-101, AVL-291, bendamustine, bleomycin, bortezomib, bosutinib, bryostatin-1 , busulfan, calicheamycin, camptothecin, carboplatin, 10-hydroxycamptothecin, carmustine, celecoxib, chlorambucil, cisplatinum, COX-2 inhibitors, irinotecan (CPT-11), SN-38, carboplatin, cladribine, camptothecans, crizotinib, cyclophosphamide, cytarabine, dacarbazine, dasatinib, dinaciclib, docetaxel, dactinomycin, daunorubicin, DM1, DM3, DM4, doxorubicin, 2- pyrrolinodoxorubicine (2-PDox), a pro-drug form of 2-PDox (pro-2-PDox), cyano- morpholino doxorubicin, doxorubicin glucuronide, endostatin, epirubicin glucuronide, erlotinib, estramustine, epidophyllotoxin, erlotinib, entinostat, estrogen receptor binding agents, etoposide (VP16), etoposide glucuronide, etoposide phosphate, exemestane, fingolimod, floxuridine (FUdR), 3',5'-0-dioleoyl-FudR (FUdR-dO), fludarabine, flutamide, farnesyl-protein transferase inhibitors, flavopiridol, fostamatinib, ganetespib, GDC-0834, GS- 1101, gefitinib, gemcitabine, hydroxyurea, ibrutinib, idarubicin, idelalisib, ifosfamide, imatinib, lapatinib, lenolidamide, leucovorin, LFM-A13, lomustine, mechlorethamine, DB1/ 149202201.1 39 Attorney Docket No.: 116983-5127-WO melphalan, mercaptopurine, 6-mercaptopurine, methotrexate, mitoxantrone, mithramycin, mitomycin, mitotane, monomethylauristatin F (MMAF), monomethylauristatin D (MMAD), monomethylauristatin E (MMAE), navelbine, neratinib, nilotinib, nitrosurea, olaparib, plicomycin, procarbazine, paclitaxel, PCI-32765, pentostatin, PSI-341, raloxifene, semustine, SN-38, sorafenib, streptozocin, SUl 1248, sunitinib, tamoxifen, temazolomide, transplatinum, thalidomide, thioguanine, thiotepa, teniposide, topotecan, uracil mustard, vatalanib, vinorelbine, vinblastine, vincristine, vinca alkaloids and ZD1839. In particularly preferred embodiments, the drug to be conjugated to the anti-Trop-2 antibody may be SN-38, pro-2- PDox or paclitaxel. [0071] The drug moiety D can be linked to the antibody Ab through linker L. L is any chemical moiety capable of linking the drug moiety to the antibody through covalent bonds. A cross-linking reagent is a bifunctional or multifunctional reagent that can be used to link a drug moiety and an antibody to form antibody drug conjugates. Antibody drug conjugates can be prepared using a cross-linking reagent having a reactive functionality capable of binding to both the drug moiety and the antibody. For example, a cysteine, thiol or an amine, e.g. N- terminus or an amino acid side chain, such as lysine of the antibody, can form a bond with a functional group of a cross-linking reagent. Alternatively, the Antibody drug conjugates can be prepared by pre-forming a linker-drug moiety (or drug-linker moiety, both terms being used interchangeably), and reacting the linker-drug moiety with the antibody. In some instant, the linker moiety is built onto the drug stepwise using several linking moieties until obtaining the desired linker-drug moiety. [0072] In one embodiment, L is a cleavable linker. In another embodiment, L is a non- cleavable linker. In some embodiments, L is an acid-labile linker, photo-labile linker, peptidase cleavable linker, esterase cleavable linker, a disulfide bond cleavable linker, a hydrophilic linker, a procharged linker, a glycosidase cleavable linker, a phosphodiesterase cleavable linker, a phosphatase cleavable linker, or a dicarboxylic acid based linker. [0073] Suitable cross-linking reagents that form a non-cleavable linker between the drug moiety, for example maytansinoid, and the antibody are well known in the art, and can form non-cleavable linkers that comprise a sulfur atom (such as SMCC) or those that are without a sulfur atom. Preferred cross-linking reagents that form non-cleavable linkers between the drug moiety, for example maytansinoid, and the antibody comprise a maleimido- or DB1/ 149202201.1 40 Attorney Docket No.: 116983-5127-WO haloacetyl-based moiety. According to the present invention, such non-cleavable linkers are said to be derived from maleimido- or haloacetyl-based moieties. [0074] Cross-linking reagents comprising a maleimido-based moiety include but not limited to, N-succinimidyl-4-(maleimidomethyl)cyclohexanecarboxylate (SMCC), sulfosuccinimidyl 4-(N-maleimidomethyl) cyclohexane-1-carboxylate (sulfo-SMCC), N-succinimidyl-4- (maleimidomethyl)cyclohexane-1-carboxy-(6-amidocaproate), which is a “long chain” analog of SMCC (LC-SMCC), κ-maleimidoundeconoic acid N-succinimidyl ester (KMUA), γ-maleimidobutyric acid N-succinimidyl ester (GMBS), ε-maleimidocaproic acid N- succinimidyl ester (EMCS), m-maleimidobenzoyl-N-hydroxysuccinimide ester (MBS), N-(α- maleimidoacetoxy)-succinimide ester (AMSA), succinimidyl-6-(β- maleimidopropionamido)hexanoate (SMPH), N-succinimidyl-4-(p-maleimidophenyl)- butyrate (SMPB), N-(-p-maleomidophenyl)isocyanate (PMIP) and maleimido-based cross- linking reagents containing a polyethythene glycol spacer, such as MAL-PEG-NHS. These cross-linking reagents form non-cleavable linkers derived from maleimido-based moieties. Representative structures of maleimido-based cross-linking reagents are shown below. DB1/ 149202201.1 41 Attorney Docket No.: 116983-5127-WO wherein:L1 is a C1-6 alkylene wherein one of the methylene groups may be replaced with oxygen; L2 is a C1-6 alkylene or is —(CH2 CH2 O)y —CH2 —CH2 — wherein y is 1 to 11; and X is —C(O)—NH—, —NHC(O)— or a triazole; wherein the alkylene is linear or branched. [0084] In one aspect of this embodiment y is 5, 7, 9 or 11. In another aspect of this embodiment y is less than 5. [0085] In yet another embodiment, suitable cross-linking moieties according to formula I are selected from the group consisting of: DB1/ 149202201.1 45 Attorney Docket No.: 116983-5127-WO
wherein:L1 is a C1-6 alkylene wherein one of the methylene groups may be replaced with oxygen; L2 is a C1-6 alkylene or is —(CH2 CH2 O)y —CH2 —CH2 — wherein y is 1 to 11; and X is —C(O)—NH—, —NHC(O)— or a triazole; wherein the alkylene is linear or branched. [0084] In one aspect of this embodiment y is 5, 7, 9 or 11. In another aspect of this embodiment y is less than 5. [0085] In yet another embodiment, suitable cross-linking moieties according to formula I are selected from the group consisting of: DB1/ 149202201.1 45 Attorney Docket No.: 116983-5127-WO DB1/ 149202201.1 46 Attorney Docket No.: 116983-5127-WO
DB1/ 149202201.1 46 Attorney Docket No.: 116983-5127-WO wherein y is 1 to 11. [0086] For the cross-linking moieties depicted above (i.e. MBT, MPET, MEPET; MMTBT, MPPT, MPBT), the maleimide group allows for reaction with the sulfhydryl (or thiol) of a Cysteine in an antibody thereby forming a thioether bond; and the thiol functionality of the cross-linking moiety is connected to the thiol of the maytansinoid drug moiety to form a cleavable disulfide bond. In view of the cross-reactional nature of the linking moiety of Formula (I) (thiol and maleimide could cross react), one of ordinary skill in the art would appreciate that the linking moiety has to be built stepwise onto the drug moiety as depicted in Scheme 1. [0087] According to the above embodiment, the linkers resulting from the cross linking moieties (i.e. MBT, MPET, MEPET) can be depicted as follow:
wherein y is 1 to 11. [0086] For the cross-linking moieties depicted above (i.e. MBT, MPET, MEPET; MMTBT, MPPT, MPBT), the maleimide group allows for reaction with the sulfhydryl (or thiol) of a Cysteine in an antibody thereby forming a thioether bond; and the thiol functionality of the cross-linking moiety is connected to the thiol of the maytansinoid drug moiety to form a cleavable disulfide bond. In view of the cross-reactional nature of the linking moiety of Formula (I) (thiol and maleimide could cross react), one of ordinary skill in the art would appreciate that the linking moiety has to be built stepwise onto the drug moiety as depicted in Scheme 1. [0087] According to the above embodiment, the linkers resulting from the cross linking moieties (i.e. MBT, MPET, MEPET) can be depicted as follow: DB1/ 149202201.1 48 Attorney Docket No.: 116983-5127-WO
DB1/ 149202201.1 48 Attorney Docket No.: 116983-5127-WO DB1/ 149202201.1 49 Attorney Docket No.: 116983-5127-WO wherein y is 1 to 11; * is linked to the thiol functionality on the antibody, and ** is linked to the thiol functionality of the drug moiety (e.g. maytansinoid drug DM1, DM3 or DM4). [0089] In a preferred embodiment, the linker has the following formula:
DB1/ 149202201.1 49 Attorney Docket No.: 116983-5127-WO wherein y is 1 to 11; * is linked to the thiol functionality on the antibody, and ** is linked to the thiol functionality of the drug moiety (e.g. maytansinoid drug DM1, DM3 or DM4). [0089] In a preferred embodiment, the linker has the following formula: wherein * is linked to the thiol functionality on the antibody, and ** is linked to the thiol functionality of the maytansinoid drug (DM1, DM3 or DM4). [0090] In one embodiment, the invention relates to the linker-drug moiety of Formula:
wherein * is linked to the thiol functionality on the antibody, and ** is linked to the thiol functionality of the maytansinoid drug (DM1, DM3 or DM4). [0090] In one embodiment, the invention relates to the linker-drug moiety of Formula: Wherein L1 is a C1-6 alkylene wherein one of the methylene groups may be replaced with oxygen; L2 is a C1-6 alkylene or is —(CH2 CH2O)y —CH2 —CH2— wherein y is 1 to 11; and X is —C(O)—NH—, —NHC(O)— or a triazole; wherein the alkylene is linear or branched. C. Linker [0091] As used herein, a “linker” is any chemical moiety that is capable of linking an antibody, antibody fragment (e.g., antigen binding fragments) or functional equivalent to another moiety, such as a drug moiety. Linkers can be susceptible to cleavage (cleavable linker), such as, acid-induced cleavage, photo-induced cleavage, peptidase-induced cleavage, esterase-induced cleavage, glycosidase induced cleavage, phosphodiesterase induced cleavage, phosphatase induced cleavage and disulfide bond cleavage, at conditions under which the compound or the antibody remains active. Alternatively, linkers can be DB1/ 149202201.1 50 Attorney Docket No.: 116983-5127-WO substantially resistant to cleavage (e.g., stable linker or noncleavable linker). In some aspects, the linker is a procharged linker, a hydrophilic linker, or a dicarboxylic acid based linker. [0092] In one aspect, the linker used in the present invention is derived from a crosslinking reagent such as N-succinimidyl-3-(2-pyridyldithio)propionate (SPDP), N-succinimidyl 4-(2- pyridyldithio)pentanoate (SPP), N-succinimidyl 4-(2-pyridyldithio)butanoate (SPDB), N- succinimidyl-4-(2-pyridyldithio)-2-sulfo-butanoate (sulfo-SPDB), N-succinimidyl iodoacetate (SIA), N-succinimidyl(4-iodoacetyl)aminobenzoate (SIAB), maleimide PEG NHS, N-succinimidyl 4-(maleimidomethyl) cyclohexanecarboxylate (SMCC), N- sulfosuccinimidyl 4-(maleimidomethyl) cyclohexanecarboxylate (sulfo-SMCC) or 2,5- dioxopyrrolidin-1-yl 17-(2,5-dioxo-2,5-dihydro-1H-pyrrol-1-yl)-5,8,11,14-tetraoxo- 4,7,10,13-tetraazaheptadecan-1-oate (CX1-1). [0093] Non-cleavable linkers are any chemical moiety capable of linking a drug, such as a maytansinoid, to an antibody in a stable, covalent manner and does not fall under the categories listed above for cleavable linkers. Thus, non-cleavable linkers are substantially resistant to acid-induced cleavage, photo-induced cleavage, peptidase-induced cleavage, esterase-induced cleavage and disulfide bond cleavage. Furthermore, non-cleavable refers to the ability of the chemical bond in the linker or adjoining to the linker to withstand cleavage induced by an acid, photolabile-cleaving agent, a peptidase, an esterase, or a chemical or physiological compound that cleaves a disulfide bond, at conditions under which the drug, such as maytansionoid or the antibody does not lose its activity. [0094] Acid-labile linkers are linkers cleavable at acidic pH. For example, certain intracellular compartments, such as endosomes and lysosomes, have an acidic pH (pH 4-5), and provide conditions suitable to cleave acid-labile linkers. [0095] Photo-labile linkers are linkers that are useful at the body surface and in many body cavities that are accessible to light. Furthermore, infrared light can penetrate tissue. [0096] Some linkers can be cleaved by peptidases, i.e., peptidase cleavable linkers. Only certain peptides are readily cleaved inside or outside cells, see e.g., Trout et al., Proc. Natl. Acad. Sci. USA, 626-629 (1982) and Umemoto et al. Int. J. Cancer, 677-684 (1989). Furthermore, peptides are composed of α-amino acids and peptidic bonds, which chemically are amide bonds between the carboxylate of one amino acid and the amino group of a second DB1/ 149202201.1 51 Attorney Docket No.: 116983-5127-WO amino acid. Other amide bonds, such as the bond between a carboxylate and the ε-amino group of lysine, are understood not to be peptidic bonds and are considered non-cleavable. [0097] Some linkers can be cleaved by esterases, i.e., esterase cleavable linkers. Again, only certain esters can be cleaved by esterases present inside or outside of cells. Esters are formed by the condensation of a carboxylic acid and an alcohol. Simple esters are esters produced with simple alcohols, such as aliphatic alcohols, and small cyclic and small aromatic alcohols. [0098] Procharged linkers are derived from charged cross-linking reagents that retain their charge after incorporation into an antibody drug conjugate. Examples of procharged linkers can be found in US 2009/0274713. D. Conjugation and Preparation of ADCs [0099] Numerous methods of conjugating linker-payloads to antigen binding moiety are known in the art (reviewed in for example: Antibody-Drug Conjugate, Methods in Molecular Biology, Vol.1045, Editor L. Ducry, Humana Press (2013)). Traditionally, drugs are conjugated to native lysine or native cysteine residues of the antibody. The resulting preparations are complex mixtures. More recently, site-specific conjugation methods are being employed to improve the therapeutic index and homogeneity of ADC preparations (For review: Panowski, et al., mAbs 2014, 6, 34). Besides glycoengineering, (Zhou, Q. et al. Bioconjugate chemistry 2014, 25, 510; Zhu, Z. et al. mAbs 2014, 6, 1190); some of the more common methods of preparing site-specific ADCs are based on the incorporation of engineered cysteines, (Junutula, J. R. et al., Nature biotechnology 2008, 26, 925; Shinmi, D. et al., Bioconjugate chemistry 2016, 27, 1324), non-canonical amino acids (Tian, F. et al., Proceedings National Academy of Sciences USA 2014, 111, 1766; Axup, J. Y. et al., Proceedings National Academy of Sciences USA 2012, 109, 16101) or short peptide sequences into the antibody backbone (Drake, P. M. et al., Bioconjugate chemistry 2014, 25, 1331; Strop, P. et al., Chemistry & biology 2013, 20, 161; Beerli, R. R. et al., PloS one 2015, 10, e0131177; Grunewald, J. et al., Bioconjugate chemistry 2015, 26, 2554). These methods provide control over stoichiometry and attachment site of the cytotoxin resulting in better pharmacokinetic (PK), safety, and efficacy profiles of the conjugates relative to traditionally prepared ADCs. DB1/ 149202201.1 52 Attorney Docket No.: 116983-5127-WO [00100] The conjugates of the present invention can be prepared by any methods known in the art, such as those described in U.S. Pat. Nos.7,811,572, 6,411,163, 7,368,565, and 8,163,888, US application publications 2011/0003969, 2011/0166319, 2012/0253021 and 2012/0259100, and PCT publications WO2014/124316 and WO2015/138615. The entire teachings of these patents and patent application publications are herein incorporated by reference. E. Process For Conjugation To Native Cysteine Antibody Residues [00101] Linker-drug moieties as described herein can be conjugated to native cysteine residues of non-engineered antibodies using a procedure that involves partial reduction of the antibodies (Doronina, S. O., et al., (2003) Nat. Biotechnol. 21, 778-784). The following protocol is a non-limiting example how such conjugates can be prepared: Inter- and intra- chain disulfides bonds of the antibody (at a concentration of typically 5 to 10 mg/ml) are first partially reduced in PBS containing 2 mM EDTA by adding TCEP to a final concentration of 10 mM and incubating the mixture at 37° C. for 1 hour. After desalting and addition of 1% w/v PS-20 detergent, the partially reduced antibodies (1-2 mg/ml) is reacted overnight at 4° C. with 0.5 to 1 mg of a maleimide containing linker payload compound per 10 mg antibody. Resulting conjugates are purified by Protein A chromatography by standard methods and buffer exchanged to PBS, and are profiled typically by mass-spectrometry (MS), analytical size-exclusion chromatography (AnSEC), and analytical hydrophobic interaction chromatography (AnHIC) for their drug-to-antibody-ratio, aggregation propensity, and hydrophobicity as well as by activity assays. F. One-Step Process for Cross-Linking to Lysine Antibody Residues [00102] In one embodiment, the conjugates of the present invention can be prepared by a one-step process for cross-linking the drug to lysine residues on the antibody. The process comprises combining the antibody, drug and cross-linking agent in a substantially aqueous medium, optionally containing one or more co-solvents, at a suitable pH. In one embodiment, the process comprises the step of contacting the antibody of the present invention with a drug (e.g., DM1 or DM4) to form a first mixture comprising the antibody and the drug, and then contacting the first mixture comprising the antibody and the drug with a cross-linking agent (e.g., SMCC, Sulfo-SMCC, SPDB, Sulfo-SPDB or CX1-1) in a solution having a pH of about 4 to about 9 to provide a mixture comprising (i) the conjugate (e.g., Ab-MCC-DM1, Ab- DB1/ 149202201.1 53 Attorney Docket No.: 116983-5127-WO SPDB-DM4, or Ab-CX1-1-DM1), (ii) free drug (e.g., DM1 or DM4), and (iii) reaction by- products. [00103] In one embodiment, the one-step process comprises contacting the antibody with the drug (e.g., DM1 or DM4) and then the cross-linking agent (e.g., SMCC, Sulfo- SMCC, SPDB, Sulfo-SPDB or CX1-1) in a solution having a pH of about 6 or greater (e.g., about 6 to about 9, about 6 to about 7, about 7 to about 9, about 7 to about 8.5, about 7.5 to about 8.5, about 7.5 to about 8.0, about 8.0 to about 9.0, or about 8.5 to about 9.0). For example, the inventive process comprises contacting a cell-binding agent with the drug (DM1 or DM4) and then the cross-linking agent (e.g., SMCC, Sulfo-SMCC, SPDB, Sulfo-SPDB or CX1-1) in a solution having a pH of about 6.0, about 6.1, about 6.2, about 6.3, about 6.4, about 6.5, about 6.6, about 6.7, about 6.8, about 6.9, about 7.0, about 7.1, about 7.2, about 7.3, about 7.4, about 7.5, about 7.6, about 7.7, about 7.8, about 7.9, about 8.0, about 8.1, about 8.2, about 8.3, about 8.4, about 8.5, about 8.6, about 8.7, about 8.8, about 8.9, or about 9.0. In a specific embodiment, the inventive process comprises contacting a cell-binding agent with the drug (e.g., DM1 or DM4) and then the cross-linking agent (e.g., SMCC, Sulfo- SMCC, SPDB, Sulfo-SPDB or CX1-1) in a solution having a pH of about 7.8 (e.g., a pH of 7.6 to 8.0 or a pH of 7.7 to 7.9). [00104] The one-step process (i.e., contacting the antibody with the drug (e.g., DM1 or DM4) and then the cross-linking agent (e.g., SMCC, Sulfo-SMCC, SPDB, Sulfo-SPDB or CX1-1) can be carried out at any suitable temperature known in the art. For example, the one- step process can occur at about 20° C. or less (e.g., about −10° C. (provided that the solution is prevented from freezing, e.g., by the presence of organic solvent used to dissolve the cytotoxic agent and the bifunctional crosslinking reagent) to about 20° C., about 0° C. to about 18° C., about 4° C. to about 16° C.), at room temperature (e.g., about 20° C. to about 30° C. or about 20° C. to about 25° C.), or at an elevated temperature (e.g., about 30° C. to about 37° C.). In one embodiment, the one-step process occurs at a temperature of about 16° C. to about 24° C. (e.g., about 16° C., about 17° C., about 18° C., about 19° C., about 20° C., about 21° C., about 22° C., about 23° C., about 24° C., or about 25° C.). In another embodiment, the one-step process is carried out at a temperature of about 15° C. or less (e.g., about −10° C. to about 15° C., or about 0° C. to about 15° C.). For example, the process comprises contacting the antibody with the drug (e.g., DM1 or DM4) and then the cross- linking agent (e.g., SMCC, Sulfo-SMCC, SPDB, Sulfo-SPDB or CX1-1) at a temperature of DB1/ 149202201.1 54 Attorney Docket No.: 116983-5127-WO about 15° C., about 14° C., about 13° C., about 12° C., about 11° C., about 10° C., about 9° C., about 8° C., about 7° C., about 6° C., about 5° C., about 4° C., about 3° C., about 2° C., about 1° C., about 0° C., about −1° C., about −2° C., about −3° C., about −4° C., about −5° C., about −6° C., about −7° C., about −8° C., about −9° C., or about −10° C., provided that the solution is prevented from freezing, e.g., by the presence of organic solvent(s) used to dissolve the cross-linking agent (e.g., SMCC, Sulfo-SMCC, Sulfo-SPDB SPDB, or CX1-1). In one embodiment, the process comprises contacting the antibody with the drug (e.g., DM1 or DM4) and then the cross-linking agent (e.g., SMCC, Sulfo-SMCC, SPDB, Sulfo-SPDB or CX1-1) at a temperature of about −10° C. to about 15° C., about 0° C. to about 15° C., about 0° C. to about 10° C., about 0° C. to about 5° C., about 5° C. to about 15° C., about 10° C. to about 15° C., or about 5° C. to about 10° C. In another embodiment, the process comprises contacting the antibody with the drug (e.g., DM1 or DM4) and then the cross-linking agent (e.g., SMCC, Sulfo-SMCC, SPDB, Sulfo-SPDB or CX1-1) at a temperature of about 10° C. (e.g., a temperature of 8° C. to 12° C. or a temperature of 9° C. to 11° C.). [00105] In one embodiment, the contacting described above is effected by providing the antibody, then contacting the antibody with the drug (e.g., DM1 or DM4) to form a first mixture comprising the antibody and the drug (e.g., DM1 or DM4), and then contacting the first mixture comprising the antibody and the drug (e.g., DM1 or DM4) with the cross-linking agent (e.g., SMCC, Sulfo-SMCC, SPDB, Sulfo-SPDB or CX1-1). For example, in one embodiment, the antibody is provided in a reaction vessel, the drug (e.g., DM1 or DM4) is added to the reaction vessel (thereby contacting the antibody), and then the cross-linking agent (e.g., SMCC, Sulfo-SMCC, SPDB, Sulfo-SPDB or CX1-1) is added to the mixture comprising the antibody and the drug (e.g., DM1 or DM4) (thereby contacting the mixture comprising the antibody and the drug). In one embodiment, the antibody is provided in a reaction vessel, and the drug (e.g., DM1 or DM4) is added to the reaction vessel immediately following providing the antibody to the vessel. In another embodiment, the antibody is provided in a reaction vessel, and the drug (e.g., DM1 or DM4) is added to the reaction vessel after a time interval following providing the antibody to the vessel (e.g., about 5 minutes, about 10 minutes, about 20 minutes, about 30 minutes, about 40 minutes, about 50 minutes, about 1 hour, about 1 day or longer after providing the cell-binding agent to the space). The drug (e.g., DM1 or DM4) can be added quickly (i.e., within a short time interval, such as about 5 minutes, about 10 minutes) or slowly (such as by using a pump). DB1/ 149202201.1 55 Attorney Docket No.: 116983-5127-WO [00106] The mixture comprising the antibody and the drug (e.g., DM1 or DM4) can then be contacted with the cross-linking agent (e.g., SMCC, Sulfo-SMCC, SPDB, Sulfo- SPDB or CX1-1) either immediately after contacting the antibody with the drug (e.g., DM1 or DM4) or at some later point (e.g., about 5 minutes to about 8 hours or longer) after contacting the antibody with the drug (e.g., DM1 or DM4). For example, in one embodiment, the cross-linking agent (e.g., SMCC, Sulfo-SMCC, SPDB, Sulfo-SPDB or CX1-1) is added to the mixture comprising the antibody and the drug (e.g., DM1 or DM4) immediately after the addition of the drug (e.g., DM1 or DM4) to the reaction vessel comprising the antibody. Alternatively, the mixture comprising the antibody and the drug (e.g., DM1 or DM4) can be contacted with the cross-linking agent (e.g., SMCC, Sulfo-SMCC, SPDB, Sulfo-SPDB or CX1-1) at about 5 minutes, about 10 minutes, about 20 minutes, about 30 minutes, about 1 hour, about 2 hours, about 3 hours, about 4 hours, about 5 hours, about 6 hours, about 7 hours, about 8 hours, or longer after contacting the antibody with the drug (e.g., DM1 or DM4). [00107] After the mixture comprising the antibody and the drug (e.g., DM1 or DM4) is contacted with the cross-linking agent (e.g., SMCC, Sulfo-SMCC, SPDB, Sulfo-SPDB or CX1-1) the reaction is allowed to proceed for about 1 hour, about 2 hours, about 3 hours, about 4 hours, about 5 hours, about 6 hours, about 7 hours, about 8 hours, about 9 hours, about 10 hours, about 11 hours, about 12 hours, about 13 hours, about 14 hours, about 15 hours, about 16 hours, about 17 hours, about 18 hours, about 19 hours, about 20 hours, about 21 hours, about 22 hours, about 23 hours, about 24 hours, or longer (e.g., about 30 hours, about 35 hours, about 40 hours, about 45 hours, or about 48 hrs). [00108] In one embodiment, the one-step process further comprises a quenching step to quench any unreacted drug (e.g., DM1 or DM4) and/or unreacted cross-linking agent (e.g., SMCC, Sulfo-SMCC, SPDB, Sulfo-SPDB or CX1-1). The quenching step is typically performed prior to purification of the conjugate. In one embodiment, the mixture is quenched by contacting the mixture with a quenching reagent. As used herein, the “quenching reagent” refers to a reagent that reacts with the free drug (e.g., DM1 or DM4) and/or cross-linking agent (e.g., SMCC, Sulfo-SMCC, SPDB, Sulfo-SPDB or CX1-1). In one embodiment, maleimide or haloacetamide quenching reagents, such as 4-maleimidobutyric acid, 3- maleimidopropionic acid, N-ethylmaleimide, iodoacetamide, or iodoacetamidopropionic acid, can be used to ensure that any unreacted group (such as thiol) in the drug (e.g., DM1 or DB1/ 149202201.1 56 Attorney Docket No.: 116983-5127-WO DM4) is quenched. The quenching step can help prevent the dimerization of the drug (e.g., DM1). The dimerized DM1 can be difficult to remove. Upon quenching with polar, charged thiol-quenching reagents (such as 4-maleimidobutyric acid or 3-maleimidopropionic acid), the excess, unreacted DM1 is converted into a polar, charged, water-soluble adduct that can be easily separated from the covalently-linked conjugate during the purification step. Quenching with non-polar and neutral thiol-quenching reagents can also be used. In one embodiment, the mixture is quenched by contacting the mixture with a quenching reagent that reacts with the unreacted cross-linking agent (e.g., SMCC, Sulfo-SMCC, SPDB, Sulfo- SPDB or CX1-1). For example, nucleophiles can be added to the mixture in order to quench any unreacted SMCC. The nucleophile preferably is an amino group containing nucleophile, such as lysine, taurine and hydroxylamine. [00109] In a preferred embodiment, the reaction (i.e., contacting the antibody with the drug (e.g., DM1 or DM4) and then cross-linking agent (e.g., SMCC, Sulfo-SMCC, SPDB, Sulfo-SPDB or CX1-1)) is allowed to proceed to completion prior to contacting the mixture with a quenching reagent. In this regard, the quenching reagent is added to the mixture about 1 hour to about 48 hours (e.g., about 1 hour, about 2 hours, about 3 hours, about 4 hours, about 5 hours, about 6 hours, about 7 hours, about 8 hours, about 9 hours, about 10 hours, about 11 hours, about 12 hours, about 13 hours, about 14 hours, about 15 hours, about 16 hours, about 17 hours, about 18 hours, about 19 hours, about 20 hours, about 21 hours, about 22 hours, about 23 hours, about 24 hours, or about 25 hours to about 48 hours) after the mixture comprising the antibody and the drug (e.g., DM1 or DM4) is contacted with the cross-linking agent (e.g., SMCC, Sulfo-SMCC, SPDB, Sulfo-SPDB or CX1-1). [00110] Alternatively, the mixture is quenched by lowering the pH of the mixture to about 5.0 (e.g., 4.8, 4.9, 5.0, 5.1 or 5.2). In another embodiment, the mixture is quenched by lowering the pH to less than 6.0, less than 5.5, less than 5.0, less than 4.8, less than 4.6, less than 4.4, less than 4.2, less than 4.0. Alternatively, the pH is lowered to about 4.0 (e.g., 3.8, 3.9, 4.0, 4.1 or 4.2) to about 6.0 (e.g., 5.8, 5.9, 6.0, 6.1 or 6.2), about 4.0 to about 5.0, about 4.5 (e.g., 4.3, 4.4, 4.5, 4.6 or 4.7) to about 5.0. In one embodiment, the mixture is quenched by lowering the pH of the mixture to 4.8. In another embodiment, the mixture is quenched by lowering the pH of the mixture to 5.5. [00111] In one embodiment, the one-step process further comprises a holding step to release the unstably bound linkers from the antibody. The holding step comprises holding the DB1/ 149202201.1 57 Attorney Docket No.: 116983-5127-WO mixture prior to purification of the conjugate (e.g., after the reaction step, between the reaction step and the quenching step, or after the quenching step). For example, the process comprises (a) contacting the antibody with the drug (e.g., DM1, DM3 or DM4) to form a mixture comprising the antibody and the drug (e.g., DM1, DM3 or DM4); and then contacting the mixture comprising the antibody and drug (e.g., DM1, DM3 or DM4) with the cross-linking agent (e.g., SMCC, Sulfo-SMCC, SPDB, Sulfo-SPDB or CX1-1), in a solution having a pH of about 4 to about 9 to provide a mixture comprising (i) the conjugate (e.g., Ab- MCC-DM1, Ab-SPDB-DM4 or Ab-CX1-1-DM1), (ii) free drug (e.g., DM1, DM3 or DM4), and (iii) reaction by-products, (b) holding the mixture prepared in step (a) to release the unstably bound linkers from the cell-binding agent, and (c) purifying the mixture to provide a purified conjugate. [00112] In another embodiment, the process comprises (a) contacting the antibody with the drug (e.g., DM1, DM3 or DM4) to form a mixture comprising the antibody and the drug (e.g., DM1, DM3 or DM4); and then contacting the mixture comprising the antibody and the drug (e.g., DM1, DM3 or DM4) with the cross-linking agent (e.g., SMCC, Sulfo- SMCC, SPDB, Sulfo-SPDB or CX1-1), in a solution having a pH of about 4 to about 9 to provide a mixture comprising (i) the conjugate, (ii) free drug (e.g., DM1, DM3 or DM4), and (iii) reaction by-products, (b) quenching the mixture prepared in step (a) to quench any unreacted drug (e.g., DM1, DM3 or DM4) and/or unreacted cross-linking agent (e.g., SMCC, Sulfo-SMCC, SPDB, Sulfo-SPDB or CX1-1), (c) holding the mixture prepared in step (b) to release the unstably bound linkers from the cell-binding agent, and (d) purifying the mixture to provide a purified conjugate (e.g., Ab-MCC-DM1, Ab-SPDB-DM4 or Ab-CX1-1-DM1). [00113] Alternatively, the holding step can be performed after purification of the conjugate, followed by an additional purification step. [00114] In a preferred embodiment, the reaction is allowed to proceed to completion prior to the holding step. In this regard, the holding step can be performed about 1 hour to about 48 hours (e.g., about 1 hour, about 2 hours, about 3 hours, about 4 hours, about 5 hours, about 6 hours, about 7 hours, about 8 hours, about 9 hours, about 10 hours, about 11 hours, about 12 hours, about 13 hours, about 14 hours, about 15 hours, about 16 hours, about 17 hours, about 18 hours, about 19 hours, about 20 hours, about 21 hours, about 22 hours, about 23 hours, about 24 hours, or about 24 hours to about 48 hours) after the mixture comprising DB1/ 149202201.1 58 Attorney Docket No.: 116983-5127-WO the antibody and the drug (e.g., DM1, DM3 or DM4) is contacted with the cross-linking agent (e.g., SMCC, Sulfo-SMCC, SPDB, Sulfo-SPDB or CX1-1). [00115] The holding step comprises maintaining the solution at a suitable temperature (e.g., about 0° C. to about 37° C.) for a suitable period of time (e.g., about 1 hour to about 1 week, about 1 hour to about 24 hours, about 1 hour to about 8 hours, or about 1 hour to about 4 hours) to release the unstably bound linkers from the antibody while not substantially releasing the stably bound linkers from the antibody. In one embodiment, the holding step comprises maintaining the solution at about 20° C. or less (e.g., about 0° C. to about 18° C., about 4° C. to about 16° C.), at room temperature (e.g., about 20° C. to about 30° C. or about 20° C. to about 25° C.), or at an elevated temperature (e.g., about 30° C. to about 37° C.). In one embodiment, the holding step comprises maintaining the solution at a temperature of about 16° C. to about 24° C. (e.g., about 15° C., about 16° C., about 17° C., about 18° C., about 19° C., about 20° C., about 21° C., about 22° C., about 23° C., about 24° C., or about 25° C.). In another embodiment, the holding step comprises maintaining the solution at a temperature of about 2° C. to about 8° C. (e.g., about 0° C., about 1° C., about 2° C., about 3° C., about 4° C., about 5° C., about 6° C., about 7° C., about 8° C., about 9° C., or about 10° C.). In another embodiment, the holding step comprises maintaining the solution at a temperature of about 37° C. (e.g., about 34° C., about 35° C., about 36° C., about 37° C., about 38° C., about 39° C., or about 40° C.). [00116] The duration of the holding step depends on the temperature and the pH at which the holding step is performed. For example, the duration of the holding step can be substantially reduced by performing the holding step at elevated temperature, with the maximum temperature limited by the stability of the cell-binding agent-cytotoxic agent conjugate. The holding step can comprise maintaining the solution for about 1 hour to about 1 day (e.g., about 1 hour, about 2 hours, about 3 hours, about 4 hours, about 5 hours, about 6 hours, about 7 hours, about 8 hours, about 9 hours, about 10 hours, about 12 hours, about 14 hours, about 16 hours, about 18 hours, about 20 hours, about 22 hours, or about 24 hours), about 10 hours to about 24 hours, about 12 hours to about 24 hours, about 14 hours to about 24 hours, about 16 hours to about 24 hours, about 18 hours to about 24 hours, about 20 hours to about 24 hours, about 5 hours to about 1 week, about 20 hours to about 1 week, about 12 hours to about 1 week (e.g., about 12 hours, about 16 hours, about 20 hours, about 24 hours, DB1/ 149202201.1 59 Attorney Docket No.: 116983-5127-WO about 2 days, about 3 days, about 4 days, about 5 days, about 6 days, or about 7 days), or about 1 day to about 1 week. [00117] In one embodiment, the holding step comprises maintaining the solution at a temperature of about 2° C. to about 8° C. for a period of at least about 12 hours for up to a week. In another embodiment, the holding step comprises maintaining the solution at a temperature of about 2° C. to about 8° C. overnight (e.g., about 12 to about 24 hours, preferably about 20 hours). [00118] The pH value for the holding step preferably is about 4 to about 10. In one embodiment, the pH value for the holding step is about 4 or more, but less than about 6 (e.g., 4 to 5.9) or about 5 or more, but less than about 6 (e.g., 5 to 5.9). In another embodiment, the pH values for the holding step range from about 6 to about 10 (e.g., about 6.5 to about 9, about 6 to about 8). For example, pH values for the holding step can be about 6, about 6.5, about 7, about 7.5, about 8, about 8.5, about 9, about 9.5, or about 10. [00119] In specific embodiments, the holding step can comprise incubating the mixture at 25° C. at a pH of about 6-7.5 for about 12 hours to about 1 week, incubating the mixture at 4° C. at a pH of about 4.5-5.9 for about 5 hours to about 5 days, or incubating the mixture at 25° C. at a pH of about 4.5-5.9 for about 5 hours to about 1 day. [00120] The one-step process may optionally include the addition of sucrose to the reaction step to increase solubility and recovery of the conjugates. Desirably, sucrose is added at a concentration of about 0.1% (w/v) to about 20% (w/v) (e.g., about 0.1% (w/v), 1% (w/v), 5% (w/v), 10% (w/v), 15% (w/v), or 20% (w/v)). Preferably, sucrose is added at a concentration of about 1% (w/v) to about 10% (w/v) (e.g., about 0.5% (w/v), about 1% (w/v), about 1.5% (w/v), about 2% (w/v), about 3% (w/v), about 4% (w/v), about 5% (w/v), about 6% (w/v), about 7% (w/v), about 8% (w/v), about 9% (w/v), about 10% (w/v), or about 11% (w/v)). In addition, the reaction step also can comprise the addition of a buffering agent. Any suitable buffering agent known in the art can be used. Suitable buffering agents include, for example, a citrate buffer, an acetate buffer, a succinate buffer, and a phosphate buffer. In one embodiment, the buffering agent is selected from the group consisting of HEPPSO (N-(2- hydroxyethyl)piperazine-N′-(2-hydroxypropanesulfonic acid)), POPSO (piperazine-1,4-bis- (2-hydroxy-propane-sulfonic acid) dehydrate), HEPES (4-(2-hydroxyethyl)piperazine-1- ethanesulfonic acid), HEPPS (EPPS) (4-(2-hydroxyethyl)piperazine-1-propanesulfonic acid), DB1/ 149202201.1 60 Attorney Docket No.: 116983-5127-WO TES (N-[tris(hydroxymethyl)methyl]-2-aminoethanesulfonic acid), and a combination thereof. [00121] In one embodiment, the one-step process can further comprise the step of purifying the mixture to provide purified conjugate (e.g., Ab-MCC-DM1, Ab-SPDB-DM4 or Ab-CX1-1-DM1). Any purification methods known in the art can be used to purify the conjugates of the present invention. In one embodiment, the conjugates of the present invention using tangential flow filtration (TFF), non-adsorptive chromatography, adsorptive chromatography, adsorptive filtration, selective precipitation, or any other suitable purification process, as well as combinations thereof. In another embodiment, prior to subjecting the conjugates to purification process described above, the conjugates are first filtered through one or more PVDF membranes. Alternatively, the conjugates are filtered through one or more PVDF membranes after subjecting the conjugates to the purification process described above. For example, in one embodiment, the conjugates are filtered through one or more PVDF membranes and then purified using tangential flow filtration. Alternatively, the conjugates are purified using tangential flow filtration and then filtered through one or more PVDF membranes. [00122] Any suitable TFF systems may be utilized for purification, including a Pellicon type system (Millipore, Billerica, Mass.), a Sartocon Cassette system (Sartorius A G, Edgewood, N.Y.), and a Centrasette type system (Pall Corp., East Hills, N.Y.). [00123] Any suitable adsorptive chromatography resin may be utilized for purification. Preferred adsorptive chromatography resins include hydroxyapatite chromatography, hydrophobic charge induction chromatography (HCIC), hydrophobic interaction chromatography (HIC), ion exchange chromatography, mixed mode ion exchange chromatography, immobilized metal affinity chromatography (IMAC), dye ligand chromatography, affinity chromatography, reversed phase chromatography, and combinations thereof. Examples of suitable hydroxyapatite resins include ceramic hydroxyapatite (CHT Type I and Type II, Bio-Rad Laboratories, Hercules, Calif.), HA Ultrogel hydroxyapatite (Pall Corp., East Hills, N.Y.), and ceramic fluoroapatite (CFT Type I and Type II, Bio-Rad Laboratories, Hercules, Calif.). An example of a suitable HCIC resin is MEP Hypercel resin (Pall Corp., East Hills, N.Y.). Examples of suitable HIC resins include Butyl-Sepharose, Hexyl-Sepaharose, Phenyl-Sepharose, and Octyl Sepharose resins (all from GE Healthcare, Piscataway, N.J.), as well as Macro-prep Methyl and Macro-Prep t-Butyl resins (Biorad DB1/ 149202201.1 61 Attorney Docket No.: 116983-5127-WO Laboratories, Hercules, Calif.). Examples of suitable ion exchange resins include SP- Sepharose, CM-Sepharose, and Q-Sepharose resins (all from GE Healthcare, Piscataway, N.J.), and Unosphere S resin (Bio-Rad Laboratories, Hercules, Calif.). Examples of suitable mixed mode ion exchangers include Bakerbond ABx resin (JT Baker, Phillipsburg N.J.). Examples of suitable IMAC resins include Chelating Sepharose resin (GE Healthcare, Piscataway, N.J.) and Profinity IMAC resin (Bio-Rad Laboratories, Hercules, Calif.). Examples of suitable dye ligand resins include Blue Sepharose resin (GE Healthcare, Piscataway, N.J.) and Affi-gel Blue resin (Bio-Rad Laboratories, Hercules, Calif.). Examples of suitable affinity resins include Protein A Sepharose resin (e.g., MabSelect, GE Healthcare, Piscataway, N.J.) and lectin affinity resins, e.g. Lentil Lectin Sepharose resin (GE Healthcare, Piscataway, N.J.), where the antibody bears appropriate lectin binding sites. Examples of suitable reversed phase resins include C4, C8, and C18 resins (Grace Vydac, Hesperia, Calif.). [00124] Any suitable non-adsorptive chromatography resin may be utilized for purification. Examples of suitable non-adsorptive chromatography resins include, but are not limited to, SEPHADEX™ G-25, G-50, G-100, SEPHACRYL™ resins (e.g., S-200 and S- 300), SUPERDEX™ resins (e.g., SUPERDEX™ 75 and SUPERDEX™ 200), BIO-GEL® resins (e.g., P-6, P-10, P-30, P-60, and P-100), and others known to those of ordinary skill in the art. G. Two-Step Process and One-Pot Process for Cross-Linking to Lysine Antibody Residues [00125] In one embodiment, the conjugates of the present invention can be prepared as described in the U.S. Pat. No.7,811,572 and U.S. Patent Application Publication No. 2006/0182750. The process comprises the steps of (a) contacting the antibody of the present invention with the cross-linking agent (e.g., SMCC, Sulfo-SMCC, SPDB, Sulfo-SPDB or CX1-1) to covalently attach the linker (i.e., Ab-SMCC, Ab-SPDB or Ab-CX1-1) to the antibody and thereby prepare a first mixture comprising the antibody having the linker bound thereto; (b) optionally subjecting the first mixture to a purification process to prepare a purified first mixture of the antibody having the linker bound thereto; (c) conjugating the drug (e.g., DM1, DM3, or DM4) to the antibody having the linker bound thereto in the first mixture by reacting the antibody having the linker bound thereto with the drug (e.g., DM1, DM3, or DM4) in a solution having a pH of about 4 to about 9 to prepare a second mixture DB1/ 149202201.1 62 Attorney Docket No.: 116983-5127-WO comprising (i) conjugate (e.g., Ab-MCC-DM1, Ab-SPDB-DM4 or Ab-CX1-1-DM1), (ii) free drug (e.g., DM1, DM3 or DM4); and (iii) reaction by-products; and (d) subjecting the second mixture to a purification process to purify the conjugate from the other components of the second mixture. Alternatively, the purification step (b) can be omitted. Any purification methods described herein can be used for steps (b) and (d). In one embodiment, TFF is used for both steps (b) and (d). In another embodiment, TFF is used for step (b) and absorptive chromatography (e.g., CHT) is used for step (d). H. Antibody Preparation [00126] Techniques for preparing monoclonal antibodies against virtually any target antigen, such as Trop-2, are well known in the art. See, for example, Kohler and Milstein, Nature 256: 495 (1975), and Coligan et al. (eds.), CURRENT PROTOCOLS IN IMMUNOLOGY, VOL.1, pages 2.5.1-2.6.7 (John Wiley & Sons 1991). Briefly, monoclonal antibodies can be obtained by injecting mice with a composition comprising an antigen, removing the spleen to obtain B-lymphocytes, fusing the B-lymphocytes with myeloma cells to produce hybridomas, cloning the hybridomas, selecting positive clones which produce antibodies to the antigen, culturing the clones that produce antibodies to the antigen, and isolating the antibodies from the hybridoma cultures. [00127] MAbs can be isolated and purified from hybridoma cultures by a variety of well- established techniques. Such isolation techniques include affinity chromatography with Protein-A or Protein-G Sepharose, size-exclusion chromatography, and ion-exchange chromatography. See, for example, Coligan at pages 2.7.1-2.7.12 and pages 2.9.1-2.9.3. Also, see Baines et al., "Purification of Immunoglobulin G (IgG)," in METHODS IN MOLECULAR BIOLOGY, VOL.10, pages 79-104 (The Humana Press, Inc.1992). [00128] After the initial raising of antibodies to the immunogen, the heavy and light chain genes of the antibodies can be sequenced and subsequently cloned by recombinant techniques. See, e.g., Kopitar-Jerala et al., Pflugers Arch – Eur J Physiol, 2000, 439 [Suppl]: R79-R80. Humanization and chimerization of murine antibodies and antibody fragments are well known to those skilled in the art, as discussed below. [00129] Chimeric Antibodies [00130] A chimeric antibody is a recombinant protein in which the variable regions of a human antibody have been replaced by the variable regions of, for example, a mouse DB1/ 149202201.1 63 Attorney Docket No.: 116983-5127-WO antibody, including the complementarity- determining regions (CDRs) of the mouse antibody. Chimeric antibodies exhibit decreased immunogenicity and increased stability when administered to a subject. General techniques for cloning murine immunoglobulin variable domains are disclosed, for example, in Orlandi et al, Proc. Nat'l Acad. Sci. USA 6: 3833 (1989). Techniques for constructing chimeric antibodies are well known to those of skill in the art. As an example, Leung et al, Hybridoma 13:469 (1994), produced an LL2 chimera by combining DNA sequences encoding the VK and VH domains of murine LL2, an anti- CD22 monoclonal antibody, with respective human κ and IgGI constant region domains. [00131] Humanized Antibodies [00132] Techniques for producing humanized MAbs are well known in the art (see, e.g., Jones et al, Nature 321: 522 (1986), Riechmann et al, Nature 332: 323 (1988), Verhoeyen et al, Science 239: 1534 (1988), Carter et al, Proc. Nat'l Acad. Sci. USA 89: 4285 (1992), Sandhu, Crit. Rev. Biotech.12: 437 (1992), and Singer et al., J. Immun.150: 2844 (1993)). A chimeric or murine monoclonal antibody may be humanized by transferring the mouse CDRs from the heavy and light variable chains of the mouse immunoglobulin into the corresponding variable domains of a human antibody. The mouse framework regions (FR) in the chimeric monoclonal antibody are also replaced with human FR sequences. As simply transferring mouse CDRs into human FRs often results in a reduction or even loss of antibody affinity, additional modification might be required in order to restore the original affinity of the murine antibody. This can be accomplished by the replacement of one or more human residues in the FR regions with their murine counterparts to obtain an antibody that possesses good binding affinity to its epitope. See, for example, Tempest et al., Biotechnology 9:266 (1991) and Verhoeyen et al., Science 239: 1534 (1988). Preferred residues for substitution include FR residues that are located within 1, 2, or 3 Angstroms of a CDR residue side chain, that are located adjacent to a CDR sequence, or that are predicted to interact with a CDR residue. [00133] Human Antibodies [00134] Methods for producing fully human antibodies using either combinatorial approaches or transgenic animals transformed with human immunoglobulin loci are known in the art (e.g., Mancini et al., 2004, New Microbiol.27:315-28; Conrad and Scheller, 2005, Comb. Chem. High Throughput Screen.8: 117-26; Brekke and Loset, 2003, Curr. Opin. Pharmacol.3:544-50). A fully human antibody also can be constructed by genetic or DB1/ 149202201.1 64 Attorney Docket No.: 116983-5127-WO chromosomal transfection methods, as well as phage display technology, all of which are known in the art. See for example, McCafferty et al, Nature 348:552-553 (1990). Such fully human antibodies are expected to exhibit even fewer side effects than chimeric or humanized antibodies and to function in vivo as essentially endogenous human antibodies. [00135] In one alternative, the phage display technique may be used to generate human antibodies (e.g., Dantas-Barbosa et al., 2005, Genet. Mol. Res.4: 126-40). Human antibodies may be generated from normal humans or from humans that exhibit a particular disease state, such as cancer (Dantas-Barbosa et al., supra). The advantage to constructing human antibodies from a diseased individual is that the circulating antibody repertoire may be biased towards antibodies against disease-associated antigens. [00136] In one non-limiting example of this methodology, Dantas-Barbosa et al. constructed a phage display library of human Fab antibody fragments from osteosarcoma patients. Generally, total RNA was obtained from circulating blood lymphocytes (Id.). [00137] Recombinant Fab were cloned from the μ, γ and κ chain antibody repertoires and inserted into a phage display library (Id.). RNAs were converted to cDNAs and used to make Fab cDNA libraries using specific primers against the heavy and light chain immunoglobulin sequences (Marks et al., 1991, J. Mol. Biol.222:581-97). Library construction was performed according to Andris-Widhopf et al. (2000, In: Phage Display Laboratory Manual, Barbas et al. (eds), 1st edition, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY pp.9.1 to 9.22). The final Fab fragments were digested with restriction endonucleases and inserted into the bacteriophage genome to make the phage display library. Such libraries may be screened by standard phage display methods, as known in the art. Phage display can be performed in a variety of formats, for their review, see e.g. Johnson and Chiswell, Current Opinion in Structural Biology 3:5564-571 (1993). [00138] Human antibodies may also be generated by in vitro activated B-cells. See U.S. Patent Nos.5,567,610 and 5,229,275, incorporated herein by reference in their entirety. The skilled artisan will realize that these techniques are exemplary and any known method for making and screening human antibodies or antibody fragments may be utilized. [00139] In another alternative, transgenic animals that have been genetically engineered to produce human antibodies may be used to generate antibodies against essentially any immunogenic target, using standard immunization protocols. Methods for DB1/ 149202201.1 65 Attorney Docket No.: 116983-5127-WO obtaining human antibodies from transgenic mice are disclosed by Green et al, Nature Genet. 7:13 (1994), Lonberg et al, Nature 355:856 (1994), and Taylor et al, Int. Immun.6:579 (1994). A non- limiting example of such a system is the XenoMouse® (e.g., Green et al., 1999, J. Immunol. Methods 231 : 11-23, incorporated herein by reference) from Abgenix (Fremont, CA). In the XenoMouse® and similar animals, the mouse antibody genes have been inactivated and replaced by functional human antibody genes, while the remainder of the mouse immune system remains intact. [00140] The XenoMouse® was transformed with germline- configured YACs (yeast artificial chromosomes) that contained portions of the human IgH and Igkappa loci, including the majority of the variable region sequences, along with accessory genes and regulatory sequences. The human variable region repertoire may be used to generate antibody producing B-cells, which may be processed into hybridomas by known techniques. A XenoMouse® immunized with a target antigen will produce human antibodies by the normal immune response, which may be harvested and/or produced by standard techniques discussed above. A variety of strains of XenoMouse® are available, each of which is capable of producing a different class of antibody. Transgenically produced human antibodies have been shown to have therapeutic potential, while retaining the pharmacokinetic properties of normal human antibodies (Green et al., 1999, supra). The skilled artisan will realize that the claimed compositions and methods are not limited to use of the XenoMouse® system but may utilize any transgenic animal that has been genetically engineered to produce human antibodies. Cloning and production of human antibodies can be carried out using the usual hybridoma technology. See Little et al., Immunol. Today, 2000, 21:364-370. [00141] The host cells for harboring and expressing the anti-Trop-2 antibody chains can be either prokaryotic or eukaryotic. E. coli is one prokaryotic host useful for cloning and expressing the polynucleotides of the present invention. Other microbial hosts suitable for use include bacilli, such as Bacillus subtilis, and other enterobacteriaceae, such as Salmonella, Serratia , and various Pseudomonas species. In these prokaryotic hosts, one can also make expression vectors, which typically contain expression control sequences compatible with the host cell (e.g., an origin of replication). In addition, any number of a variety of well-known promoters will be present, such as the lactose promoter system, a tryptophan (trp) promoter system, a beta-lactamase promoter system, or a promoter system from phage lambda. The promoters typically control expression, optionally with an operator sequence, and have DB1/ 149202201.1 66 Attorney Docket No.: 116983-5127-WO ribosome binding site sequences and the like, for initiating and completing transcription and translation. Other microbes, such as yeast, can also be employed to express anti-Trop-2 polypeptides of the invention. Insect cells in combination with baculovirus vectors can also be used. [00142] In some preferred embodiments, mammalian host cells are used to express and produce the anti-Trop-2 polypeptides of the present invention. For example, they can be either a hybridoma cell line expressing endogenous immunoglobulin genes (e.g., the myeloma hybridoma clones as described in the Examples) or a mammalian cell line harboring an exogenous expression vector (e.g., the SP2/0 myeloma cells exemplified below). These include any normal mortal or normal or abnormal immortal animal or human cell. For example, a number of suitable host cell lines capable of secreting intact immunoglobulins have been developed, including the CHO cell lines, various Cos cell lines, HeLa cells, myeloma cell lines, transformed B-cells and hybridomas. The use of mammalian tissue cell culture to express polypeptides is discussed generally in, e.g., Winnacker, From Genes to Clones, VCH Publishers, N.Y., N.Y., 1987. Expression vectors for mammalian host cells can include expression control sequences, such as an origin of replication, a promoter, and an enhancer (see, e.g., Queen et al., Immunol. Rev.89:49-68, 1986), and necessary processing information sites, such as ribosome binding sites, RNA splice sites, polyadenylation sites, and transcriptional terminator sequences. These expression vectors usually contain promoters derived from mammalian genes or from mammalian viruses. Suitable promoters may be constitutive, cell type-specific, stage-specific, and/or modulatable or regulatable. Useful promoters include, but are not limited to, the metallothionein promoter, the constitutive adenovirus major late promoter, the dexamethasone-inducible MMTV promoter, the SV40 promoter, the MRP polIII promoter, the constitutive MPSV promoter, the tetracycline-inducible CMV promoter (such as the human immediate-early CMV promoter), the constitutive CMV promoter, and promoter-enhancer combinations known in the art. [00143] Methods for introducing expression vectors containing the polynucleotide sequences of interest vary depending on the type of cellular host. For example, calcium chloride transfection is commonly utilized for prokaryotic cells, whereas calcium phosphate treatment or electroporation may be used for other cellular hosts (see generally Sambrook et al., Molecular Cloning: A Laboratory Manual, 4th ed.). Other methods include, e.g., electroporation, calcium phosphate treatment, liposome-mediated transformation, injection DB1/ 149202201.1 67 Attorney Docket No.: 116983-5127-WO and microinjection, ballistic methods, virosomes, immunoliposomes, polycation:nucleic acid conjugates, naked DNA, artificial virions, fusion to the herpes virus structural protein VP22 (Elliot and O'Hare, Cell 88:223, 1997), agent-enhanced uptake of DNA, and ex vivo transduction. For long-term, high-yield production of recombinant proteins, stable expression will often be desired. For example, cell lines which stably express anti-Trop-2 antibody chains or binding fragments can be prepared using expression vectors of the invention which contain viral origins of replication or endogenous expression elements and a selectable marker gene. Following introduction of the vector, cells may be allowed to grow for 1-2 days in an enriched media before they are switched to selective media. The purpose of the selectable marker is to confer resistance to selection, and its presence allows growth of cells which successfully express the introduced sequences in selective media. Resistant, stably transfected cells can be proliferated using tissue culture techniques appropriate to the cell type. III. Gen 2 TIL Manufacturing Processes [0006] An exemplary family of TIL processes known as Gen 2 (also known as process 2A) containing some of these features is depicted in Figures 1 and 2. An embodiment of Gen 2 is shown in Figure 2. [0007] As discussed herein, the present invention can include a step relating to the restimulation of cryopreserved TILs to increase their metabolic activity and thus relative health prior to transplant into a patient, and methods of testing said metabolic health. As generally outlined herein, TILs are generally taken from a patient sample and manipulated to expand their number prior to transplant into a patient. In some embodiments, the TILs may be optionally genetically manipulated as discussed below. [0008] In some embodiments, the TILs may be cryopreserved. Once thawed, they may also be restimulated to increase their metabolism prior to infusion into a patient. [0009] In some embodiments, the first expansion (including processes referred to as the pre-REP as well as processes shown in Figure 1 as Step A) is shortened to 3 to 14 days and the second expansion (including processes referred to as the REP as well as processes shown in Figure 1 as Step B) is shorted to 7 to 14 days, as discussed in detail below as well as in the examples and figures. In some embodiments, the first expansion (for example, an expansion described as Step B in Figure 1) is shortened to 11 days and the second expansion (for DB1/ 149202201.1 68 Attorney Docket No.: 116983-5127-WO example, an expansion as described in Step D in Figure 1) is shortened to 11 days. In some embodiments, the combination of the first expansion and second expansion (for example, expansions described as Step B and Step D in Figure 1) is shortened to 22 days, as discussed in detail below and in the examples and figures. [0010] The “Step” Designations A, B, C, etc., below are in reference to Figure 1 and in reference to certain embodiments described herein. The ordering of the Steps below and in Figure 1 is exemplary and any combination or order of steps, as well as additional steps, repetition of steps, and/or omission of steps is contemplated by the present application and the methods disclosed herein. A. STEP A: Obtain Patient Tumor Sample [0011] In general, TILs are initially obtained from a patient tumor sample and then expanded into a larger population for further manipulation as described herein, optionally cryopreserved, restimulated as outlined herein and optionally evaluated for phenotype and metabolic parameters as an indication of TIL health. [0012] A patient tumor sample may be obtained using methods known in the art, generally via surgical resection, needle biopsy, core biopsy, small biopsy, or other means for obtaining a sample that contains a mixture of tumor and TIL cells. In some embodiments, multilesional sampling is used. In some embodiments, surgical resection, needle biopsy, core biopsy, small biopsy, or other means for obtaining a sample that contains a mixture of tumor and TIL cells includes multilesional sampling (i.e., obtaining samples from one or more tumor sites and/or locations in the patient, as well as one or more tumors in the same location or in close proximity). In general, the tumor sample may be from any solid tumor, including primary tumors, invasive tumors or metastatic tumors. The tumor sample may also be a liquid tumor, such as a tumor obtained from a hematological malignancy. The solid tumor may be of lung tissue. In some embodiments, useful TILs are obtained from non-small cell lung carcinoma (NSCLC). The solid tumor may be of skin tissue. In some embodiments, useful TILs are obtained from a melanoma. [0013] Once obtained, the tumor sample is generally fragmented using sharp dissection into small pieces of between 1 to about 8 mm3, with from about 2-3 mm3 being particularly useful. In some embodiments, the TILs are cultured from these fragments using enzymatic tumor digests. Such tumor digests may be produced by incubation in enzymatic media (e.g., DB1/ 149202201.1 69 Attorney Docket No.: 116983-5127-WO Roswell Park Memorial Institute (RPMI) 1640 buffer, 2 mM glutamate, 10 mcg/mL gentamicine, 30 units/mL of DNase and 1.0 mg/mL of collagenase) followed by mechanical dissociation (e.g., using a tissue dissociator). Tumor digests may be produced by placing the tumor in enzymatic media and mechanically dissociating the tumor for approximately 1 minute, followed by incubation for 30 minutes at 37 °C in 5% CO2, followed by repeated cycles of mechanical dissociation and incubation under the foregoing conditions until only small tissue pieces are present. At the end of this process, if the cell suspension contains a large number of red blood cells or dead cells, a density gradient separation using FICOLL branched hydrophilic polysaccharide may be performed to remove these cells. Alternative methods known in the art may be used, such as those described in U.S. Patent Application Publication No.2012/0244133 A1, the disclosure of which is incorporated by reference herein. Any of the foregoing methods may be used in any of the embodiments described herein for methods of expanding TILs or methods treating a cancer. [0014] Tumor dissociating enzyme mixtures can include one or more dissociating (digesting) enzymes such as, but not limited to, collagenase (including any blend or type of collagenase), Accutase™, Accumax™, hyaluronidase, neutral protease (dispase), chymotrypsin, chymopapain, trypsin, caseinase, elastase, papain, protease type XIV (pronase), deoxyribonuclease I (DNase), trypsin inhibitor, any other dissociating or proteolytic enzyme, and any combination thereof. [0015] In some embodiments, the dissociating enzymes are reconstituted from lyophilized enzymes. In some embodiments, lyophilized enzymes are reconstituted in an amount of sterile buffer such as HBSS. [0016] In some instances, collagenase (such as animal free- type 1 collagenase) is reconstituted in 10 mL of sterile HBSS or another buffer. The lyophilized stock enzyme may be at a concentration of 2892 PZ U/vial. In some embodiments, collagenase is reconstituted in 5 mL to 15 mL buffer. In some embodiment, after reconstitution the collagenase stock ranges from about 100 PZ U/mL-about 400 PZ U/mL, e.g., about 100 PZ U/mL-about 400 PZ U/mL, about 100 PZ U/mL-about 350 PZ U/mL, about 100 PZ U/mL-about 300 PZ U/mL, about 150 PZ U/mL-about 400 PZ U/mL, about 100 PZ U/mL, about 150 PZ U/mL, about 200 PZ U/mL, about 210 PZ U/mL, about 220 PZ U/mL, about 230 PZ U/mL, about 240 PZ U/mL, about 250 PZ U/mL, about 260 PZ U/mL, about 270 PZ U/mL, about DB1/ 149202201.1 70 Attorney Docket No.: 116983-5127-WO 280 PZ U/mL, about 289.2 PZ U/mL, about 300 PZ U/mL, about 350 PZ U/mL, or about 400 PZ U/mL. [0017] In some embodiments, neutral protease is reconstituted in 1 mL of sterile HBSS or another buffer. The lyophilized stock enzyme may be at a concentration of 175 DMC U/vial. In some embodiments, after reconstitution the neutral protease stock ranges from about 100 DMC/mL-about 400 DMC/mL, e.g., about 100 DMC/mL-about 400 DMC/mL, about 100 DMC/mL-about 350 DMC/mL, about 100 DMC/mL-about 300 DMC/mL, about 150 DMC/mL-about 400 DMC/mL, about 100 DMC/mL, about 110 DMC/mL, about 120 DMC/mL, about 130 DMC/mL, about 140 DMC/mL, about 150 DMC/mL, about 160 DMC/mL, about 170 DMC/mL, about 175 DMC/mL, about 180 DMC/mL, about 190 DMC/mL, about 200 DMC/mL, about 250 DMC/mL, about 300 DMC/mL, about 350 DMC/mL, or about 400 DMC/mL. [0018] In some embodiments, DNAse I is reconstituted in 1 mL of sterile HBSS or another buffer. The lyophilized stock enzyme was at a concentration of 4 KU/vial. In some embodiments, after reconstitution the DNase I stock ranges from about 1 KU/mL-10 KU/mL, e.g., about 1 KU/mL, about 2 KU/mL, about 3 KU/mL, about 4 KU/mL, about 5 KU/mL, about 6 KU/mL, about 7 KU/mL, about 8 KU/mL, about 9 KU/mL, or about 10 KU/mL. [0019] In some embodiments, the stock of enzymes is variable and the concentrations may need to be determined. In some embodiments, the concentration of the lyophilized stock can be verified. In some embodiments, the final amount of enzyme added to the digest cocktail is adjusted based on the determined stock concentration. [0020] In some embodiment, the enzyme mixture includes about 10.2-ul of neutral protease (0.36 DMC U/mL), 21.3 µL of collagenase (1.2 PZ/mL) and 250-ul of DNAse I (200 U/mL) in about 4.7 mL of sterile HBSS. [0021] As indicated above, in some embodiments, the TILs are derived from solid tumors. In some embodiments, the solid tumors are not fragmented. In some embodiments, the solid tumors are not fragmented and are subjected to enzymatic digestion as whole tumors. In some embodiments, the tumors are digested in in an enzyme mixture comprising collagenase, DNase, and hyaluronidase. In some embodiments, the tumors are digested in in an enzyme mixture comprising collagenase, DNase, and hyaluronidase for 1-2 hours. In some embodiments, the tumors are digested in in an enzyme mixture comprising collagenase, DB1/ 149202201.1 71 Attorney Docket No.: 116983-5127-WO DNase, and hyaluronidase for 1-2 hours at 37°C, 5% CO2. In some embodiments, the tumors are digested in in an enzyme mixture comprising collagenase, DNase, and hyaluronidase for 1-2 hours at 37°C, 5% CO2 with rotation. In some embodiments, the tumors are digested overnight with constant rotation. In some embodiments, the tumors are digested overnight at 37°C, 5% CO2 with constant rotation. In some embodiments, the whole tumor is combined with the enzymes to form a tumor digest reaction mixture. [0022] In some embodiments, the tumor is reconstituted with the lyophilized enzymes in a sterile buffer. In some embodiments, the buffer is sterile HBSS. [0023] In some embodiments, the enzyme mixture comprises collagenase. In some embodiments, the collagenase is collagenase IV. In some embodiments, the working stock for the collagenase is a 100 mg/mL 10X working stock. [0024] In some embodiments, the enzyme mixture comprises DNAse. In some embodiments, the working stock for the DNAse is a 10,000 IU/mL 10X working stock. [0025] In some embodiments, the enzyme mixture comprises hyaluronidase. In some embodiments, the working stock for the hyaluronidase is a 10 mg/mL 10X working stock. [0026] In some embodiments, the enzyme mixture comprises 10 mg/mL collagenase, 1000 IU/mL DNAse, and 1 mg/mL hyaluronidase. [0027] In some embodiments, the enzyme mixture comprises 10 mg/mL collagenase, 500 IU/mL DNAse, and 1 mg/mL hyaluronidase. [0028] In general, the harvested cell suspension is called a “primary cell population” or a “freshly harvested” cell population. [0029] In some embodiments, fragmentation includes physical fragmentation, including for example, dissection as well as digestion. In some embodiments, the fragmentation is physical fragmentation. In some embodiments, the fragmentation is dissection. In some embodiments, the fragmentation is by digestion. In some embodiments, TILs can be initially cultured from enzymatic tumor digests and tumor fragments obtained from digesting or fragmenting a tumor sample obtained from a patient. [0030] In some embodiments, where the tumor is a solid tumor, the tumor undergoes physical fragmentation after the tumor sample is obtained in, for example, Step A (as provided in Figure 1). In some embodiments, the fragmentation occurs before DB1/ 149202201.1 72 Attorney Docket No.: 116983-5127-WO cryopreservation. In some embodiments, the fragmentation occurs after cryopreservation. In some embodiments, the fragmentation occurs after obtaining the tumor and in the absence of any cryopreservation. In some embodiments, the tumor is fragmented and 10, 20, 30, 40 or more fragments or pieces are placed in each container for the first expansion. In some embodiments, the tumor is fragmented and 30 or 40 fragments or pieces are placed in each container for the first expansion. In some embodiments, the tumor is fragmented and 40 fragments or pieces are placed in each container for the first expansion. In some embodiments, the multiple fragments comprise about 4 to about 50 fragments, wherein each fragment has a volume of about 27 mm3. In some embodiments, the multiple fragments comprise about 30 to about 60 fragments with a total volume of about 1300 mm3 to about 1500 mm3. In some embodiments, the multiple fragments comprise about 50 fragments with a total volume of about 1350 mm3. In some embodiments, the multiple fragments comprise about 50 fragments with a total mass of about 1 gram to about 1.5 grams. In some embodiments, the multiple fragments comprise about 4 fragments. [0031] In some embodiments, the TILs are obtained from tumor fragments. In some embodiments, the tumor fragment is obtained by sharp dissection. In some embodiments, the tumor fragment is between about 1 mm3 and 10 mm3. In some embodiments, the tumor fragment is between about 1 mm3 and 8 mm3. In some embodiments, the tumor fragment is about 1 mm3. In some embodiments, the tumor fragment is about 2 mm3. In some embodiments, the tumor fragment is about 3 mm3. In some embodiments, the tumor fragment is about 4 mm3. In some embodiments, the tumor fragment is about 5 mm3. In some embodiments, the tumor fragment is about 6 mm3. In some embodiments, the tumor fragment is about 7 mm3. In some embodiments, the tumor fragment is about 8 mm3. In some embodiments, the tumor fragment is about 9 mm3. In some embodiments, the tumor fragment is about 10 mm3. In some embodiments, the tumors are 1-4 mm × 1-4 mm × 1-4 mm. In some embodiments, the tumors are 1 mm × 1 mm × 1 mm. In some embodiments, the tumors are 2 mm × 2 mm × 2 mm. In some embodiments, the tumors are 3 mm × 3 mm × 3 mm. In some embodiments, the tumors are 4 mm × 4 mm × 4 mm. [0032] In some embodiments, the tumors are resected in order to minimize the amount of hemorrhagic, necrotic, and/or fatty tissues on each piece. In some embodiments, the tumors are resected in order to minimize the amount of hemorrhagic tissue on each piece. In some embodiments, the tumors are resected in order to minimize the amount of necrotic tissue on DB1/ 149202201.1 73 Attorney Docket No.: 116983-5127-WO each piece. In some embodiments, the tumors are resected in order to minimize the amount of fatty tissue on each piece. [0033] In some embodiments, the tumor fragmentation is performed in order to maintain the tumor internal structure. In some embodiments, the tumor fragmentation is performed without performing a sawing motion with a scalpel. In some embodiments, the TILs are obtained from tumor digests. In some embodiments, tumor digests were generated by incubation in enzyme media, for example but not limited to RPMI 1640, 2 mM GlutaMAX, 10 mg/mL gentamicin, 30 U/mL DNase, and 1.0 mg/mL collagenase, followed by mechanical dissociation (GentleMACS, Miltenyi Biotec, Auburn, CA). After placing the tumor in enzyme media, the tumor can be mechanically dissociated for approximately 1 minute. The solution can then be incubated for 30 minutes at 37 °C in 5% CO2 and it then mechanically disrupted again for approximately 1 minute. After being incubated again for 30 minutes at 37 °C in 5% CO2, the tumor can be mechanically disrupted a third time for approximately 1 minute. In some embodiments, after the third mechanical disruption if large pieces of tissue were present, 1 or 2 additional mechanical dissociations were applied to the sample, with or without 30 additional minutes of incubation at 37 °C in 5% CO2. In some embodiments, at the end of the final incubation if the cell suspension contains a large number of red blood cells or dead cells, a density gradient separation using Ficoll can be performed to remove these cells. [0034] In some embodiments, the harvested cell suspension prior to the first expansion step is called a “primary cell population” or a “freshly harvested” cell population. [0035] In some embodiments, cells can be optionally frozen after sample harvest and stored frozen prior to entry into the expansion described in Step B, which is described in further detail below, as well as exemplified in Figure 1, as well as Figure 8. 1. Pleural effusion T-cells and TILs [0036] In some embodiments, the sample is a pleural fluid sample. In some embodiments, the source of the T-cells or TILs for expansion according to the processes described herein is a pleural fluid sample. In some embodiments, the sample is a pleural effusion derived sample. In some embodiments, the source of the T-cells or TILs for expansion according to the processes described herein is a pleural effusion derived sample. See, for example, methods DB1/ 149202201.1 74 Attorney Docket No.: 116983-5127-WO described in U.S. Patent Publication US 2014/0295426, incorporated herein by reference in its entirety for all purposes. [0037] In some embodiments, any pleural fluid or pleural effusion suspected of and/or containing TILs can be employed. Such a sample may be derived from a primary or metastatic lung cancer, such as NSCLC or SCLC. In some embodiments, the sample may be derived from secondary metastatic cancer cells which originated from another organ, e.g., breast, ovary, colon or prostate. In some embodiments, the sample for use in the expansion methods described herein is a pleural exudate. In some embodiments, the sample for use in the expansion methods described herein is a pleural transudate. Other biological samples may include other serous fluids containing TILs, including, e.g., ascites fluid from the abdomen or pancreatic cyst fluid. Ascites fluid and pleural fluids involve very similar chemical systems; both the abdomen and lung have mesothelial lines and fluid forms in the pleural space and abdominal spaces in the same matter in malignancies and such fluids in some embodiments contain TILs. In some embodiments, wherein the disclosed methods utilize pleural fluid, the same methods may be performed with similar results using ascites or other cyst fluids containing TILs. [0038] In some embodiments, the pleural fluid is in unprocessed form, directly as removed from the patient. In some embodiments, the unprocessed pleural fluid is placed in a standard blood collection tube, such as an EDTA or Heparin tube, prior to further processing steps. In some embodiments, the unprocessed pleural fluid is placed in a standard CellSave® tube (Veridex) prior to further processing steps. In some embodiments, the sample is placed in the CellSave tube immediately after collection from the patient to avoid a decrease in the number of viable TILs. The number of viable TILs can decrease to a significant extent within 24 hours, if left in the untreated pleural fluid, even at 4°C. In some embodiments, the sample is placed in the appropriate collection tube within 1 hour, 5 hours, 10 hours, 15 hours, or up to 24 hours after removal from the patient. In some embodiments, the sample is placed in the appropriate collection tube within 1 hour, 5 hours, 10 hours, 15 hours, or up to 24 hours after removal from the patient at 4°C. [0039] In some embodiments, the pleural fluid sample from the chosen subject may be diluted. In some embodiments, the dilution is 1:10 pleural fluid to diluent. In other embodiments, the dilution is 1:9 pleural fluid to diluent. In other embodiments, the dilution is 1:8 pleural fluid to diluent. In other embodiments, the dilution is 1:5 pleural fluid to diluent. DB1/ 149202201.1 75 Attorney Docket No.: 116983-5127-WO In other embodiments, the dilution is 1:2 pleural fluid to diluent. In other embodiments, the dilution is 1:1 pleural fluid to diluent. In some embodiments, diluents include saline, phosphate buffered saline, another buffer or a physiologically acceptable diluent. In some embodiments, the sample is placed in the CellSave tube immediately after collection from the patient and dilution to avoid a decrease in the viable TILs, which may occur to a significant extent within 24-48 hours, if left in the untreated pleural fluid, even at 4°C. In some embodiments, the pleural fluid sample is placed in the appropriate collection tube within 1 hour, 5 hours, 10 hours, 15 hours, 24 hours, 36 hours, up to 48 hours after removal from the patient, and dilution. In some embodiments, the pleural fluid sample is placed in the appropriate collection tube within 1 hour, 5 hours, 10 hours, 15 hours, 24 hours, 36 hours, up to 48 hours after removal from the patient, and dilution at 4°C. [0040] In still other embodiments, pleural fluid samples are concentrated by conventional means prior to further processing steps. In some embodiments, this pre-treatment of the pleural fluid is preferable in circumstances in which the pleural fluid must be cryopreserved for shipment to a laboratory performing the method or for later analysis (e.g., later than 24-48 hours post-collection). In some embodiments, the pleural fluid sample is prepared by centrifuging the pleural fluid sample after its withdrawal from the subject and resuspending the centrifugate or pellet in buffer. In some embodiments, the pleural fluid sample is subjected to multiple centrifugations and resuspensions, before it is cryopreserved for transport or later analysis and/or processing. [0041] In some embodiments, pleural fluid samples are concentrated prior to further processing steps by using a filtration method. In some embodiments, the pleural fluid sample used in further processing is prepared by filtering the fluid through a filter containing a known and essentially uniform pore size that allows for passage of the pleural fluid through the membrane but retains the tumor cells. In some embodiments, the diameter of the pores in the membrane may be at least 4 μM. In other embodiments the pore diameter may be 5 μM or more, and in other embodiment, any of 6, 7, 8, 9, or 10 μM. After filtration, the cells, including TILs, retained by the membrane may be rinsed off the membrane into a suitable physiologically acceptable buffer. Cells, including TILs, concentrated in this way may then be used in the further processing steps of the method. [0042] In some embodiments, pleural fluid sample (including, for example, the untreated pleural fluid), diluted pleural fluid, or the resuspended cell pellet, is contacted with a lytic DB1/ 149202201.1 76 Attorney Docket No.: 116983-5127-WO reagent that differentially lyses non-nucleated red blood cells present in the sample. In some embodiments, this step is performed prior to further processing steps in circumstances in which the pleural fluid contains substantial numbers of RBCs. Suitable lysing reagents include a single lytic reagent or a lytic reagent and a quench reagent, or a lytic agent, a quench reagent and a fixation reagent. Suitable lytic systems are marketed commercially and include the BD Pharm Lyse™ system (Becton Dickenson). Other lytic systems include the Versalyse™ system, the FACSlyse™ system (Becton Dickenson), the Immunoprep™ system or Erythrolyse II system (Beckman Coulter, Inc.), or an ammonium chloride system. In some embodiments, the lytic reagent can vary with the primary requirements being efficient lysis of the red blood cells, and the conservation of the TILs and phenotypic properties of the TILs in the pleural fluid. In addition to employing a single reagent for lysis, the lytic systems useful in methods described herein can include a second reagent, e.g., one that quenches or retards the effect of the lytic reagent during the remaining steps of the method, e.g., Stabilyse™ reagent (Beckman Coulter, Inc.). A conventional fixation reagent may also be employed depending upon the choice of lytic reagents or the preferred implementation of the method. [0043] In some embodiments, the pleural fluid sample, unprocessed, diluted or multiply centrifuged or processed as described herein above is cryopreserved at a temperature of about −140°C prior to being further processed and/or expanded as provided herein. B. STEP B: First Expansion [0044] In some embodiments, the present methods provide for obtaining young TILs, which are capable of increased replication cycles upon administration to a subject/patient and as such may provide additional therapeutic benefits over older TILs (i.e., TILs which have further undergone more rounds of replication prior to administration to a subject/patient). Features of young TILs have been described in the literature, for example in Donia, et al., Scand. J. Immunol.2012, 75, 157–167; Dudley, et al., Clin. Cancer Res.2010, 16, 6122- 6131; Huang, et al., J. Immunother.2005, 28, 258–267; Besser, et al., Clin. Cancer Res. 2013, 19, OF1-OF9; Besser, et al., J. Immunother.2009, 32:415–423; Robbins, et al., J. Immunol.2004, 173, 7125-7130; Shen, et al., J. Immunother., 2007, 30, 123–129; Zhou, et al., J. Immunother.2005, 28, 53–62; and Tran, et al., J. Immunother., 2008, 31, 742–751, each of which is incorporated herein by reference. DB1/ 149202201.1 77 Attorney Docket No.: 116983-5127-WO [0045] The diverse antigen receptors of T and B lymphocytes are produced by somatic recombination of a limited, but large number of gene segments. These gene segments: V (variable), D (diversity), J (joining), and C (constant), determine the binding specificity and downstream applications of immunoglobulins and T-cell receptors (TCRs). The present invention provides a method for generating TILs which exhibit and increase the T-cell repertoire diversity. In some embodiments, the TILs obtained by the present method exhibit an increase in the T-cell repertoire diversity. In some embodiments, the TILs obtained by the present method exhibit an increase in the T-cell repertoire diversity as compared to freshly harvested TILs and/or TILs prepared using other methods than those provide herein including for example, methods other than those embodied in Figure 1. In some embodiments, the TILs obtained by the present method exhibit an increase in the T-cell repertoire diversity as compared to freshly harvested TILs and/or TILs prepared using methods referred to as process 1C, as exemplified in Figure 5 and/or Figure 6. In some embodiments, the TILs obtained in the first expansion exhibit an increase in the T-cell repertoire diversity. In some embodiments, the increase in diversity is an increase in the immunoglobulin diversity and/or the T-cell receptor diversity. In some embodiments, the diversity is in the immunoglobulin is in the immunoglobulin heavy chain. In some embodiments, the diversity is in the immunoglobulin is in the immunoglobulin light chain. In some embodiments, the diversity is in the T-cell receptor. In some embodiments, the diversity is in one of the T-cell receptors selected from the group consisting of alpha, beta, gamma, and delta receptors. In some embodiments, there is an increase in the expression of T-cell receptor (TCR) alpha and/or beta. In some embodiments, there is an increase in the expression of T-cell receptor (TCR) alpha. In some embodiments, there is an increase in the expression of T-cell receptor (TCR) beta. In some embodiments, there is an increase in the expression of TCRab (i.e., TCRα/β). [0046] After dissection or digestion of tumor fragments, for example such as described in Step A of Figure 1, the resulting cells are cultured in serum containing IL-2 under conditions that favor the growth of TILs over tumor and other cells. In some embodiments, the tumor digests are incubated in 2 mL wells in media comprising inactivated human AB serum with 6000 IU/mL of IL-2. This primary cell population is cultured for a period of days, generally from 3 to 14 days, resulting in a bulk TIL population, generally about 1 × 108 bulk TIL cells. In some embodiments, this primary cell population is cultured for a period of 7 to 14 days, resulting in a bulk TIL population, generally about 1 × 108 bulk TIL cells. In some DB1/ 149202201.1 78 Attorney Docket No.: 116983-5127-WO embodiments, this primary cell population is cultured for a period of 10 to 14 days, resulting in a bulk TIL population, generally about 1 × 108 bulk TIL cells. In some embodiments, this primary cell population is cultured for a period of about 11 days, resulting in a bulk TIL population, generally about 1 × 108 bulk TIL cells. [0047] In some embodiments, expansion of TILs may be performed using an initial bulk TIL expansion step (for example such as those described in Step B of Figure 1, which can include processes referred to as pre-REP) as described below and herein, followed by a second expansion (Step D, including processes referred to as rapid expansion protocol (REP) steps) as described below under Step D and herein, followed by optional cryopreservation, and followed by a second Step D (including processes referred to as restimulation REP steps) as described below and herein. The TILs obtained from this process may be optionally characterized for phenotypic characteristics and metabolic parameters as described herein. [0048] In embodiments where TIL cultures are initiated in 24-well plates, for example, using Costar 24-well cell culture cluster, flat bottom (Corning Incorporated, Corning, NY, each well can be seeded with 1 × 106 tumor digest cells or one tumor fragment in 2 mL of complete medium (CM) with IL-2 (6000 IU/mL; Chiron Corp., Emeryville, CA). In some embodiments, the tumor fragment is between about 1 mm3 and 10 mm3. [0049] In some embodiments, the first expansion culture medium is referred to as “CM”, an abbreviation for culture media. In some embodiments, CM for Step B consists of RPMI 1640 with GlutaMAX, supplemented with 10% human AB serum, 25 mM Hepes, and 10 mg/mL gentamicin. In embodiments where cultures are initiated in gas-permeable flasks with a 40 mL capacity and a 10 cm2 gas-permeable silicon bottom (for example, G-REX10; Wilson Wolf Manufacturing, New Brighton, MN), each flask was loaded with 10–40 × 106 viable tumor digest cells or 5–30 tumor fragments in 10–40 mL of CM with IL-2. Both the G- REX10 and 24-well plates were incubated in a humidified incubator at 37°C in 5% CO2 and 5 days after culture initiation, half the media was removed and replaced with fresh CM and IL- 2 and after day 5, half the media was changed every 2–3 days. [0050] In some embodiments, the culture medium used in the expansion processes disclosed herein is a serum-free medium or a defined medium. In some embodiments, the serum-free or defined medium comprises a basal cell medium and a serum supplement and/or a serum replacement. In some embodiments, the serum-free or defined medium is used to DB1/ 149202201.1 79 Attorney Docket No.: 116983-5127-WO prevent and/or decrease experimental variation due in part to the lot-to-lot variation of serum- containing media. [0051] In some embodiments, the serum-free or defined medium comprises a basal cell medium and a serum supplement and/or serum replacement. In some embodiments, the basal cell medium includes, but is not limited to CTS™ OpTmizer™ T-cell Expansion Basal Medium , CTS™ OpTmizer™ T-Cell Expansion SFM, CTS™ AIM-V Medium, CTS™ AIM-V SFM, LymphoONE™ T-Cell Expansion Xeno-Free Medium, Dulbecco's Modified Eagle's Medium (DMEM), Minimal Essential Medium (MEM), Basal Medium Eagle (BME), RPMI 1640, F-10, F-12, Minimal Essential Medium (αMEM), Glasgow's Minimal Essential Medium (G-MEM), RPMI growth medium, and Iscove's Modified Dulbecco's Medium. [0052] In some embodiments, the serum supplement or serum replacement includes, but is not limited to one or more of CTS™ OpTmizer T-Cell Expansion Serum Supplement, CTS™ Immune Cell Serum Replacement, one or more albumins or albumin substitutes, one or more amino acids, one or more vitamins, one or more transferrins or transferrin substitutes, one or more antioxidants, one or more insulins or insulin substitutes, one or more collagen precursors, one or more antibiotics, and one or more trace elements. In some embodiments, the defined medium comprises albumin and one or more ingredients selected from the group consisting of glycine, L- histidine, L-isoleucine, L-methionine, L-phenylalanine, L-proline, L- hydroxyproline, L-serine, L-threonine, L-tryptophan, L-tyrosine, L-valine, thiamine, reduced glutathione, L-ascorbic acid-2-phosphate, iron saturated transferrin, insulin, and compounds containing the trace element moieties Ag+, Al3+, Ba2+, Cd2+, Co2+, Cr3+, Ge4+, Se4+, Br, T, Mn2+, P, Si4+, V5+, Mo6+, Ni2+, Rb+, Sn2+ and Zr4+. In some embodiments, the defined medium further comprises L-glutamine, sodium bicarbonate and/or 2-mercaptoethanol. [0053] In some embodiments, the CTS™OpTmizer™ T-cell Immune Cell Serum Replacement is used with conventional growth media, including but not limited to CTS™ OpTmizer™ T-cell Expansion Basal Medium, CTS™ OpTmizer™ T-cell Expansion SFM, CTS™ AIM-V Medium, CST™ AIM-V SFM, LymphoONE™ T-Cell Expansion Xeno-Free Medium, Dulbecco's Modified Eagle's Medium (DMEM), Minimal Essential Medium (MEM), Basal Medium Eagle (BME), RPMI 1640, F-10, F-12, Minimal Essential Medium (αMEM), Glasgow's Minimal Essential Medium (G-MEM), RPMI growth medium, and Iscove's Modified Dulbecco's Medium. DB1/ 149202201.1 80 Attorney Docket No.: 116983-5127-WO [0054] In some embodiments, the total serum replacement concentration (vol%) in the serum-free or defined medium is from about 1%, 2%, 3%, 4%, 5%, 6%, 7%, 8%, 9%, 10%, 11%, 12%, 13%, 14%, 15%, 16%, 17%, 18%, 19%, or 20% by volume of the total serum-free or defined medium. In some embodiments, the total serum replacement concentration is about 3% of the total volume of the serum-free or defined medium. In some embodiments, the total serum replacement concentration is about 5% of the total volume of the serum-free or defined medium. In some embodiments, the total serum replacement concentration is about 10% of the total volume of the serum-free or defined medium. [0055] In some embodiments, the serum-free or defined medium is CTS™ OpTmizer™ T- cell Expansion SFM (ThermoFisher Scientific). Any formulation of CTS™ OpTmizer™ is useful in the present invention. CTS™ OpTmizer™ T-cell Expansion SFM is a combination of 1L CTS™ OpTmizer™ T-cell Expansion Basal Medium and 26 mL CTS™ OpTmizer™ T-Cell Expansion Supplement, which are mixed together prior to use. In some embodiments, the CTS™ OpTmizer™ T-cell Expansion SFM is supplemented with about 3% of the CTS™ Immune Cell Serum Replacement (SR) (ThermoFisher Scientific). In some embodiments, the CTS™ OpTmizer™ T-cell Expansion SFM is supplemented with about 3% of the CTS™ Immune Cell Serum Replacement (SR) (ThermoFisher Scientific), along with 2- mercaptoethanol at 55mM. In some embodiments, the CTS™ OpTmizer™ T-cell Expansion SFM is supplemented with about 3% of the CTS™ Immune Cell Serum Replacement (SR) (ThermoFisher Scientific) and the final concentration of 2-mercaptoethanol in the media is 55µM. [0056] In some embodiments, the defined medium is CTS™ OpTmizer™ T-cell Expansion SFM (ThermoFisher Scientific). Any formulation of CTS™ OpTmizer™ is useful in the present invention. CTS™ OpTmizer™ T-cell Expansion SFM is a combination of 1L CTS™ OpTmizer™ T-cell Expansion Basal Medium and 26 mL CTS™ OpTmizer™ T-Cell Expansion Supplement, which are mixed together prior to use. In some embodiments, the CTS™ OpTmizer™ T-cell Expansion SFM is supplemented with about 3% of the CTS™ Immune Cell Serum Replacement (SR) (ThermoFisher Scientific), along with 2- mercaptoethanol at 55mM. In some embodiments, the CTS™OpTmizer™ T-cell Expansion SFM is supplemented with about 3% of the CTS™ Immune Cell Serum Replacement (SR) (ThermoFisher Scientific), 55mM of 2-mercaptoethanol, and 2mM of L-glutamine. In some embodiments, the CTS™OpTmizer™ T-cell Expansion SFM is supplemented with about 3% DB1/ 149202201.1 81 Attorney Docket No.: 116983-5127-WO of the CTS™ Immune Cell Serum Replacement (SR) (ThermoFisher Scientific), 55mM of 2- mercaptoethanol, and 2mM of L-glutamine, and further comprises about 1000 IU/mL to about 8000 IU/mL of IL-2. In some embodiments, the CTS™OpTmizer™ T-cell Expansion SFM is supplemented with about 3% of the CTS™ Immune Cell Serum Replacement (SR) (ThermoFisher Scientific), 55mM of 2-mercaptoethanol, and 2mM of L-glutamine, and further comprises about 3000 IU/mL of IL-2. In some embodiments, the CTS™OpTmizer™ T-cell Expansion SFM is supplemented with about 3% of the CTS™ Immune Cell Serum Replacement (SR) (ThermoFisher Scientific), 55mM of 2-mercaptoethanol, and 2mM of L- glutamine, and further comprises about 6000 IU/mL of IL-2. In some embodiments, the CTS™OpTmizer™ T-cell Expansion SFM is supplemented with about 3% of the CTS™ Immune Cell Serum Replacement (SR) (ThermoFisher Scientific) and 55mM of 2- mercaptoethanol, and further comprises about 1000 IU/mL to about 8000 IU/mL of IL-2. In some embodiments, the CTS™OpTmizer™ T-cell Expansion SFM is supplemented with about 3% of the CTS™ Immune Cell Serum Replacement (SR) (ThermoFisher Scientific) and 55mM of 2-mercaptoethanol, and further comprises about 3000 IU/mL of IL-2. In some embodiments, the CTS™OpTmizer™ T-cell Expansion SFM is supplemented with about 3% of the CTS™ Immune Cell Serum Replacement (SR) (ThermoFisher Scientific) and 55mM of 2-mercaptoethanol, and further comprises about 1000 IU/mL to about 6000 IU/mL of IL-2. In some embodiments, the CTS™OpTmizer™ T-cell Expansion SFM is supplemented with about 3% of the CTS™ Immune Cell Serum Replacement (SR) (ThermoFisher Scientific) and about 2mM glutamine, and further comprises about 1000 IU/mL to about 8000 IU/mL of IL-2. In some embodiments, the CTS™OpTmizer™ T-cell Expansion SFM is supplemented with about 3% of the CTS™ Immune Cell Serum Replacement (SR) (ThermoFisher Scientific) and about 2mM glutamine, and further comprises about 3000 IU/mL of IL-2. In some embodiments, the CTS™OpTmizer™ T-cell Expansion SFM is supplemented with about 3% of the CTS™ Immune Cell Serum Replacement (SR) (ThermoFisher Scientific) and about 2mM glutamine, and further comprises about 6000 IU/mL of IL-2. In some embodiments, the CTS™ OpTmizer™ T-cell Expansion SFM is supplemented with about 3% of the CTS™ Immune Cell Serum Replacement (SR) (ThermoFisher Scientific) and the final concentration of 2-mercaptoethanol in the media is 55µM. [0057] In some embodiments, the serum-free medium or defined medium is supplemented with glutamine (i.e., GlutaMAX®) at a concentration of from about 0.1mM to about 10mM, DB1/ 149202201.1 82 Attorney Docket No.: 116983-5127-WO 0.5mM to about 9mM, 1mM to about 8mM, 2mM to about 7mM, 3mM to about 6mM, or 4mM to about 5 mM. In some embodiments, the serum-free medium or defined medium is supplemented with glutamine (i.e., GlutaMAX®) at a concentration of about 2mM. [0058] In some embodiments, the serum-free medium or defined medium is supplemented with 2-mercaptoethanol at a concentration of from about 5mM to about 150mM, 10mM to about 140mM, 15mM to about 130mM, 20mM to about 120mM, 25mM to about 110mM, 30mM to about 100mM, 35mM to about 95mM, 40mM to about 90mM, 45mM to about 85mM, 50mM to about 80mM, 55mM to about 75mM, 60mM to about 70mM, or about 65mM. In some embodiments, the serum-free medium or defined medium is supplemented with 2-mercaptoethanol at a concentration of about 55mM. In some embodiments, the final concentration of 2-mercaptoethanol in the media is 55µM. [0059] In some embodiments, the defined media described in International PCT Publication No. WO/1998/030679, which is herein incorporated by reference, are useful in the present invention. In that publication, serum-free eukaryotic cell culture media are described. The serum-free, eukaryotic cell culture medium includes a basal cell culture medium supplemented with a serum-free supplement capable of supporting the growth of cells in serum- free culture. The serum-free eukaryotic cell culture medium supplement comprises or is obtained by combining one or more ingredients selected from the group consisting of one or more albumins or albumin substitutes, one or more amino acids, one or more vitamins, one or more transferrins or transferrin substitutes, one or more antioxidants, one or more insulins or insulin substitutes, one or more collagen precursors, one or more trace elements, and one or more antibiotics. In some embodiments, the defined medium further comprises L- glutamine, sodium bicarbonate and/or beta-mercaptoethanol. In some embodiments, the defined medium comprises an albumin or an albumin substitute and one or more ingredients selected from group consisting of one or more amino acids, one or more vitamins, one or more transferrins or transferrin substitutes, one or more antioxidants, one or more insulins or insulin substitutes, one or more collagen precursors, and one or more trace elements. In some embodiments, the defined medium comprises albumin and one or more ingredients selected from the group consisting of glycine, L- histidine, L-isoleucine, L-methionine, L- phenylalanine, L-proline, L- hydroxyproline, L-serine, L-threonine, L-tryptophan, L-tyrosine, L-valine, thiamine, reduced glutathione, L-ascorbic acid-2-phosphate, iron saturated transferrin, insulin, and compounds containing the trace element moieties Ag+, Al3+, Ba2+, DB1/ 149202201.1 83 Attorney Docket No.: 116983-5127-WO Cd2+, Co2+, Cr3+, Ge4+, Se4+, Br, T, Mn2+, P, Si4+, V5+, Mo6+, Ni2+, Rb+, Sn2+ and Zr4+. In some embodiments, the basal cell media is selected from the group consisting of Dulbecco's Modified Eagle's Medium (DMEM), Minimal Essential Medium (MEM), Basal Medium Eagle (BME), RPMI 1640, F-10, F-12, Minimal Essential Medium (αMEM), Glasgow's Minimal Essential Medium (G-MEM), RPMI growth medium, and Iscove's Modified Dulbecco's Medium. [0060] In some embodiments, the concentration of glycine in the defined medium is in the range of from about 5-200 mg/L, the concentration of L- histidine is about 5-250 mg/L, the concentration of L-isoleucine is about 5-300 mg/L, the concentration of L-methionine is about 5-200 mg/L, the concentration of L-phenylalanine is about 5-400 mg/L, the concentration of L-proline is about 1-1000 mg/L, the concentration of L- hydroxyproline is about 1-45 mg/L, the concentration of L-serine is about 1-250 mg/L, the concentration of L- threonine is about 10-500 mg/L, the concentration of L-tryptophan is about 2-110 mg/L, the concentration of L-tyrosine is about 3-175 mg/L, the concentration of L-valine is about 5-500 mg/L, the concentration of thiamine is about 1-20 mg/L, the concentration of reduced glutathione is about 1-20 mg/L, the concentration of L-ascorbic acid-2-phosphate is about 1- 200 mg/L, the concentration of iron saturated transferrin is about 1-50 mg/L, the concentration of insulin is about 1-100 mg/L, the concentration of sodium selenite is about 0.000001-0.0001 mg/L, and the concentration of albumin (e.g., AlbuMAX® I) is about 5000- 50,000 mg/L. [0061] In some embodiments, the non-trace element moiety ingredients in the defined medium are present in the concentration ranges listed in the column under the heading “Concentration Range in 1X Medium” in Table 4 below. In other embodiments, the non-trace element moiety ingredients in the defined medium are present in the final concentrations listed in the column under the heading “A Preferred Embodiment of the 1X Medium” in Table 4. In other embodiments, the defined medium is a basal cell medium comprising a serum free supplement. In some of these embodiments, the serum free supplement comprises non-trace moiety ingredients of the type and in the concentrations listed in the column under the heading “A Preferred Embodiment in Supplement” in Table 4 below. TABLE 4: Concentrations of Non-Trace Element Moiety Ingredients DB1/ 149202201.1 84 Attorney Docket No.: 116983-5127-WO
Wherein L1 is a C1-6 alkylene wherein one of the methylene groups may be replaced with oxygen; L2 is a C1-6 alkylene or is —(CH2 CH2O)y —CH2 —CH2— wherein y is 1 to 11; and X is —C(O)—NH—, —NHC(O)— or a triazole; wherein the alkylene is linear or branched. C. Linker [0091] As used herein, a “linker” is any chemical moiety that is capable of linking an antibody, antibody fragment (e.g., antigen binding fragments) or functional equivalent to another moiety, such as a drug moiety. Linkers can be susceptible to cleavage (cleavable linker), such as, acid-induced cleavage, photo-induced cleavage, peptidase-induced cleavage, esterase-induced cleavage, glycosidase induced cleavage, phosphodiesterase induced cleavage, phosphatase induced cleavage and disulfide bond cleavage, at conditions under which the compound or the antibody remains active. Alternatively, linkers can be DB1/ 149202201.1 50 Attorney Docket No.: 116983-5127-WO substantially resistant to cleavage (e.g., stable linker or noncleavable linker). In some aspects, the linker is a procharged linker, a hydrophilic linker, or a dicarboxylic acid based linker. [0092] In one aspect, the linker used in the present invention is derived from a crosslinking reagent such as N-succinimidyl-3-(2-pyridyldithio)propionate (SPDP), N-succinimidyl 4-(2- pyridyldithio)pentanoate (SPP), N-succinimidyl 4-(2-pyridyldithio)butanoate (SPDB), N- succinimidyl-4-(2-pyridyldithio)-2-sulfo-butanoate (sulfo-SPDB), N-succinimidyl iodoacetate (SIA), N-succinimidyl(4-iodoacetyl)aminobenzoate (SIAB), maleimide PEG NHS, N-succinimidyl 4-(maleimidomethyl) cyclohexanecarboxylate (SMCC), N- sulfosuccinimidyl 4-(maleimidomethyl) cyclohexanecarboxylate (sulfo-SMCC) or 2,5- dioxopyrrolidin-1-yl 17-(2,5-dioxo-2,5-dihydro-1H-pyrrol-1-yl)-5,8,11,14-tetraoxo- 4,7,10,13-tetraazaheptadecan-1-oate (CX1-1). [0093] Non-cleavable linkers are any chemical moiety capable of linking a drug, such as a maytansinoid, to an antibody in a stable, covalent manner and does not fall under the categories listed above for cleavable linkers. Thus, non-cleavable linkers are substantially resistant to acid-induced cleavage, photo-induced cleavage, peptidase-induced cleavage, esterase-induced cleavage and disulfide bond cleavage. Furthermore, non-cleavable refers to the ability of the chemical bond in the linker or adjoining to the linker to withstand cleavage induced by an acid, photolabile-cleaving agent, a peptidase, an esterase, or a chemical or physiological compound that cleaves a disulfide bond, at conditions under which the drug, such as maytansionoid or the antibody does not lose its activity. [0094] Acid-labile linkers are linkers cleavable at acidic pH. For example, certain intracellular compartments, such as endosomes and lysosomes, have an acidic pH (pH 4-5), and provide conditions suitable to cleave acid-labile linkers. [0095] Photo-labile linkers are linkers that are useful at the body surface and in many body cavities that are accessible to light. Furthermore, infrared light can penetrate tissue. [0096] Some linkers can be cleaved by peptidases, i.e., peptidase cleavable linkers. Only certain peptides are readily cleaved inside or outside cells, see e.g., Trout et al., Proc. Natl. Acad. Sci. USA, 626-629 (1982) and Umemoto et al. Int. J. Cancer, 677-684 (1989). Furthermore, peptides are composed of α-amino acids and peptidic bonds, which chemically are amide bonds between the carboxylate of one amino acid and the amino group of a second DB1/ 149202201.1 51 Attorney Docket No.: 116983-5127-WO amino acid. Other amide bonds, such as the bond between a carboxylate and the ε-amino group of lysine, are understood not to be peptidic bonds and are considered non-cleavable. [0097] Some linkers can be cleaved by esterases, i.e., esterase cleavable linkers. Again, only certain esters can be cleaved by esterases present inside or outside of cells. Esters are formed by the condensation of a carboxylic acid and an alcohol. Simple esters are esters produced with simple alcohols, such as aliphatic alcohols, and small cyclic and small aromatic alcohols. [0098] Procharged linkers are derived from charged cross-linking reagents that retain their charge after incorporation into an antibody drug conjugate. Examples of procharged linkers can be found in US 2009/0274713. D. Conjugation and Preparation of ADCs [0099] Numerous methods of conjugating linker-payloads to antigen binding moiety are known in the art (reviewed in for example: Antibody-Drug Conjugate, Methods in Molecular Biology, Vol.1045, Editor L. Ducry, Humana Press (2013)). Traditionally, drugs are conjugated to native lysine or native cysteine residues of the antibody. The resulting preparations are complex mixtures. More recently, site-specific conjugation methods are being employed to improve the therapeutic index and homogeneity of ADC preparations (For review: Panowski, et al., mAbs 2014, 6, 34). Besides glycoengineering, (Zhou, Q. et al. Bioconjugate chemistry 2014, 25, 510; Zhu, Z. et al. mAbs 2014, 6, 1190); some of the more common methods of preparing site-specific ADCs are based on the incorporation of engineered cysteines, (Junutula, J. R. et al., Nature biotechnology 2008, 26, 925; Shinmi, D. et al., Bioconjugate chemistry 2016, 27, 1324), non-canonical amino acids (Tian, F. et al., Proceedings National Academy of Sciences USA 2014, 111, 1766; Axup, J. Y. et al., Proceedings National Academy of Sciences USA 2012, 109, 16101) or short peptide sequences into the antibody backbone (Drake, P. M. et al., Bioconjugate chemistry 2014, 25, 1331; Strop, P. et al., Chemistry & biology 2013, 20, 161; Beerli, R. R. et al., PloS one 2015, 10, e0131177; Grunewald, J. et al., Bioconjugate chemistry 2015, 26, 2554). These methods provide control over stoichiometry and attachment site of the cytotoxin resulting in better pharmacokinetic (PK), safety, and efficacy profiles of the conjugates relative to traditionally prepared ADCs. DB1/ 149202201.1 52 Attorney Docket No.: 116983-5127-WO [00100] The conjugates of the present invention can be prepared by any methods known in the art, such as those described in U.S. Pat. Nos.7,811,572, 6,411,163, 7,368,565, and 8,163,888, US application publications 2011/0003969, 2011/0166319, 2012/0253021 and 2012/0259100, and PCT publications WO2014/124316 and WO2015/138615. The entire teachings of these patents and patent application publications are herein incorporated by reference. E. Process For Conjugation To Native Cysteine Antibody Residues [00101] Linker-drug moieties as described herein can be conjugated to native cysteine residues of non-engineered antibodies using a procedure that involves partial reduction of the antibodies (Doronina, S. O., et al., (2003) Nat. Biotechnol. 21, 778-784). The following protocol is a non-limiting example how such conjugates can be prepared: Inter- and intra- chain disulfides bonds of the antibody (at a concentration of typically 5 to 10 mg/ml) are first partially reduced in PBS containing 2 mM EDTA by adding TCEP to a final concentration of 10 mM and incubating the mixture at 37° C. for 1 hour. After desalting and addition of 1% w/v PS-20 detergent, the partially reduced antibodies (1-2 mg/ml) is reacted overnight at 4° C. with 0.5 to 1 mg of a maleimide containing linker payload compound per 10 mg antibody. Resulting conjugates are purified by Protein A chromatography by standard methods and buffer exchanged to PBS, and are profiled typically by mass-spectrometry (MS), analytical size-exclusion chromatography (AnSEC), and analytical hydrophobic interaction chromatography (AnHIC) for their drug-to-antibody-ratio, aggregation propensity, and hydrophobicity as well as by activity assays. F. One-Step Process for Cross-Linking to Lysine Antibody Residues [00102] In one embodiment, the conjugates of the present invention can be prepared by a one-step process for cross-linking the drug to lysine residues on the antibody. The process comprises combining the antibody, drug and cross-linking agent in a substantially aqueous medium, optionally containing one or more co-solvents, at a suitable pH. In one embodiment, the process comprises the step of contacting the antibody of the present invention with a drug (e.g., DM1 or DM4) to form a first mixture comprising the antibody and the drug, and then contacting the first mixture comprising the antibody and the drug with a cross-linking agent (e.g., SMCC, Sulfo-SMCC, SPDB, Sulfo-SPDB or CX1-1) in a solution having a pH of about 4 to about 9 to provide a mixture comprising (i) the conjugate (e.g., Ab-MCC-DM1, Ab- DB1/ 149202201.1 53 Attorney Docket No.: 116983-5127-WO SPDB-DM4, or Ab-CX1-1-DM1), (ii) free drug (e.g., DM1 or DM4), and (iii) reaction by- products. [00103] In one embodiment, the one-step process comprises contacting the antibody with the drug (e.g., DM1 or DM4) and then the cross-linking agent (e.g., SMCC, Sulfo- SMCC, SPDB, Sulfo-SPDB or CX1-1) in a solution having a pH of about 6 or greater (e.g., about 6 to about 9, about 6 to about 7, about 7 to about 9, about 7 to about 8.5, about 7.5 to about 8.5, about 7.5 to about 8.0, about 8.0 to about 9.0, or about 8.5 to about 9.0). For example, the inventive process comprises contacting a cell-binding agent with the drug (DM1 or DM4) and then the cross-linking agent (e.g., SMCC, Sulfo-SMCC, SPDB, Sulfo-SPDB or CX1-1) in a solution having a pH of about 6.0, about 6.1, about 6.2, about 6.3, about 6.4, about 6.5, about 6.6, about 6.7, about 6.8, about 6.9, about 7.0, about 7.1, about 7.2, about 7.3, about 7.4, about 7.5, about 7.6, about 7.7, about 7.8, about 7.9, about 8.0, about 8.1, about 8.2, about 8.3, about 8.4, about 8.5, about 8.6, about 8.7, about 8.8, about 8.9, or about 9.0. In a specific embodiment, the inventive process comprises contacting a cell-binding agent with the drug (e.g., DM1 or DM4) and then the cross-linking agent (e.g., SMCC, Sulfo- SMCC, SPDB, Sulfo-SPDB or CX1-1) in a solution having a pH of about 7.8 (e.g., a pH of 7.6 to 8.0 or a pH of 7.7 to 7.9). [00104] The one-step process (i.e., contacting the antibody with the drug (e.g., DM1 or DM4) and then the cross-linking agent (e.g., SMCC, Sulfo-SMCC, SPDB, Sulfo-SPDB or CX1-1) can be carried out at any suitable temperature known in the art. For example, the one- step process can occur at about 20° C. or less (e.g., about −10° C. (provided that the solution is prevented from freezing, e.g., by the presence of organic solvent used to dissolve the cytotoxic agent and the bifunctional crosslinking reagent) to about 20° C., about 0° C. to about 18° C., about 4° C. to about 16° C.), at room temperature (e.g., about 20° C. to about 30° C. or about 20° C. to about 25° C.), or at an elevated temperature (e.g., about 30° C. to about 37° C.). In one embodiment, the one-step process occurs at a temperature of about 16° C. to about 24° C. (e.g., about 16° C., about 17° C., about 18° C., about 19° C., about 20° C., about 21° C., about 22° C., about 23° C., about 24° C., or about 25° C.). In another embodiment, the one-step process is carried out at a temperature of about 15° C. or less (e.g., about −10° C. to about 15° C., or about 0° C. to about 15° C.). For example, the process comprises contacting the antibody with the drug (e.g., DM1 or DM4) and then the cross- linking agent (e.g., SMCC, Sulfo-SMCC, SPDB, Sulfo-SPDB or CX1-1) at a temperature of DB1/ 149202201.1 54 Attorney Docket No.: 116983-5127-WO about 15° C., about 14° C., about 13° C., about 12° C., about 11° C., about 10° C., about 9° C., about 8° C., about 7° C., about 6° C., about 5° C., about 4° C., about 3° C., about 2° C., about 1° C., about 0° C., about −1° C., about −2° C., about −3° C., about −4° C., about −5° C., about −6° C., about −7° C., about −8° C., about −9° C., or about −10° C., provided that the solution is prevented from freezing, e.g., by the presence of organic solvent(s) used to dissolve the cross-linking agent (e.g., SMCC, Sulfo-SMCC, Sulfo-SPDB SPDB, or CX1-1). In one embodiment, the process comprises contacting the antibody with the drug (e.g., DM1 or DM4) and then the cross-linking agent (e.g., SMCC, Sulfo-SMCC, SPDB, Sulfo-SPDB or CX1-1) at a temperature of about −10° C. to about 15° C., about 0° C. to about 15° C., about 0° C. to about 10° C., about 0° C. to about 5° C., about 5° C. to about 15° C., about 10° C. to about 15° C., or about 5° C. to about 10° C. In another embodiment, the process comprises contacting the antibody with the drug (e.g., DM1 or DM4) and then the cross-linking agent (e.g., SMCC, Sulfo-SMCC, SPDB, Sulfo-SPDB or CX1-1) at a temperature of about 10° C. (e.g., a temperature of 8° C. to 12° C. or a temperature of 9° C. to 11° C.). [00105] In one embodiment, the contacting described above is effected by providing the antibody, then contacting the antibody with the drug (e.g., DM1 or DM4) to form a first mixture comprising the antibody and the drug (e.g., DM1 or DM4), and then contacting the first mixture comprising the antibody and the drug (e.g., DM1 or DM4) with the cross-linking agent (e.g., SMCC, Sulfo-SMCC, SPDB, Sulfo-SPDB or CX1-1). For example, in one embodiment, the antibody is provided in a reaction vessel, the drug (e.g., DM1 or DM4) is added to the reaction vessel (thereby contacting the antibody), and then the cross-linking agent (e.g., SMCC, Sulfo-SMCC, SPDB, Sulfo-SPDB or CX1-1) is added to the mixture comprising the antibody and the drug (e.g., DM1 or DM4) (thereby contacting the mixture comprising the antibody and the drug). In one embodiment, the antibody is provided in a reaction vessel, and the drug (e.g., DM1 or DM4) is added to the reaction vessel immediately following providing the antibody to the vessel. In another embodiment, the antibody is provided in a reaction vessel, and the drug (e.g., DM1 or DM4) is added to the reaction vessel after a time interval following providing the antibody to the vessel (e.g., about 5 minutes, about 10 minutes, about 20 minutes, about 30 minutes, about 40 minutes, about 50 minutes, about 1 hour, about 1 day or longer after providing the cell-binding agent to the space). The drug (e.g., DM1 or DM4) can be added quickly (i.e., within a short time interval, such as about 5 minutes, about 10 minutes) or slowly (such as by using a pump). DB1/ 149202201.1 55 Attorney Docket No.: 116983-5127-WO [00106] The mixture comprising the antibody and the drug (e.g., DM1 or DM4) can then be contacted with the cross-linking agent (e.g., SMCC, Sulfo-SMCC, SPDB, Sulfo- SPDB or CX1-1) either immediately after contacting the antibody with the drug (e.g., DM1 or DM4) or at some later point (e.g., about 5 minutes to about 8 hours or longer) after contacting the antibody with the drug (e.g., DM1 or DM4). For example, in one embodiment, the cross-linking agent (e.g., SMCC, Sulfo-SMCC, SPDB, Sulfo-SPDB or CX1-1) is added to the mixture comprising the antibody and the drug (e.g., DM1 or DM4) immediately after the addition of the drug (e.g., DM1 or DM4) to the reaction vessel comprising the antibody. Alternatively, the mixture comprising the antibody and the drug (e.g., DM1 or DM4) can be contacted with the cross-linking agent (e.g., SMCC, Sulfo-SMCC, SPDB, Sulfo-SPDB or CX1-1) at about 5 minutes, about 10 minutes, about 20 minutes, about 30 minutes, about 1 hour, about 2 hours, about 3 hours, about 4 hours, about 5 hours, about 6 hours, about 7 hours, about 8 hours, or longer after contacting the antibody with the drug (e.g., DM1 or DM4). [00107] After the mixture comprising the antibody and the drug (e.g., DM1 or DM4) is contacted with the cross-linking agent (e.g., SMCC, Sulfo-SMCC, SPDB, Sulfo-SPDB or CX1-1) the reaction is allowed to proceed for about 1 hour, about 2 hours, about 3 hours, about 4 hours, about 5 hours, about 6 hours, about 7 hours, about 8 hours, about 9 hours, about 10 hours, about 11 hours, about 12 hours, about 13 hours, about 14 hours, about 15 hours, about 16 hours, about 17 hours, about 18 hours, about 19 hours, about 20 hours, about 21 hours, about 22 hours, about 23 hours, about 24 hours, or longer (e.g., about 30 hours, about 35 hours, about 40 hours, about 45 hours, or about 48 hrs). [00108] In one embodiment, the one-step process further comprises a quenching step to quench any unreacted drug (e.g., DM1 or DM4) and/or unreacted cross-linking agent (e.g., SMCC, Sulfo-SMCC, SPDB, Sulfo-SPDB or CX1-1). The quenching step is typically performed prior to purification of the conjugate. In one embodiment, the mixture is quenched by contacting the mixture with a quenching reagent. As used herein, the “quenching reagent” refers to a reagent that reacts with the free drug (e.g., DM1 or DM4) and/or cross-linking agent (e.g., SMCC, Sulfo-SMCC, SPDB, Sulfo-SPDB or CX1-1). In one embodiment, maleimide or haloacetamide quenching reagents, such as 4-maleimidobutyric acid, 3- maleimidopropionic acid, N-ethylmaleimide, iodoacetamide, or iodoacetamidopropionic acid, can be used to ensure that any unreacted group (such as thiol) in the drug (e.g., DM1 or DB1/ 149202201.1 56 Attorney Docket No.: 116983-5127-WO DM4) is quenched. The quenching step can help prevent the dimerization of the drug (e.g., DM1). The dimerized DM1 can be difficult to remove. Upon quenching with polar, charged thiol-quenching reagents (such as 4-maleimidobutyric acid or 3-maleimidopropionic acid), the excess, unreacted DM1 is converted into a polar, charged, water-soluble adduct that can be easily separated from the covalently-linked conjugate during the purification step. Quenching with non-polar and neutral thiol-quenching reagents can also be used. In one embodiment, the mixture is quenched by contacting the mixture with a quenching reagent that reacts with the unreacted cross-linking agent (e.g., SMCC, Sulfo-SMCC, SPDB, Sulfo- SPDB or CX1-1). For example, nucleophiles can be added to the mixture in order to quench any unreacted SMCC. The nucleophile preferably is an amino group containing nucleophile, such as lysine, taurine and hydroxylamine. [00109] In a preferred embodiment, the reaction (i.e., contacting the antibody with the drug (e.g., DM1 or DM4) and then cross-linking agent (e.g., SMCC, Sulfo-SMCC, SPDB, Sulfo-SPDB or CX1-1)) is allowed to proceed to completion prior to contacting the mixture with a quenching reagent. In this regard, the quenching reagent is added to the mixture about 1 hour to about 48 hours (e.g., about 1 hour, about 2 hours, about 3 hours, about 4 hours, about 5 hours, about 6 hours, about 7 hours, about 8 hours, about 9 hours, about 10 hours, about 11 hours, about 12 hours, about 13 hours, about 14 hours, about 15 hours, about 16 hours, about 17 hours, about 18 hours, about 19 hours, about 20 hours, about 21 hours, about 22 hours, about 23 hours, about 24 hours, or about 25 hours to about 48 hours) after the mixture comprising the antibody and the drug (e.g., DM1 or DM4) is contacted with the cross-linking agent (e.g., SMCC, Sulfo-SMCC, SPDB, Sulfo-SPDB or CX1-1). [00110] Alternatively, the mixture is quenched by lowering the pH of the mixture to about 5.0 (e.g., 4.8, 4.9, 5.0, 5.1 or 5.2). In another embodiment, the mixture is quenched by lowering the pH to less than 6.0, less than 5.5, less than 5.0, less than 4.8, less than 4.6, less than 4.4, less than 4.2, less than 4.0. Alternatively, the pH is lowered to about 4.0 (e.g., 3.8, 3.9, 4.0, 4.1 or 4.2) to about 6.0 (e.g., 5.8, 5.9, 6.0, 6.1 or 6.2), about 4.0 to about 5.0, about 4.5 (e.g., 4.3, 4.4, 4.5, 4.6 or 4.7) to about 5.0. In one embodiment, the mixture is quenched by lowering the pH of the mixture to 4.8. In another embodiment, the mixture is quenched by lowering the pH of the mixture to 5.5. [00111] In one embodiment, the one-step process further comprises a holding step to release the unstably bound linkers from the antibody. The holding step comprises holding the DB1/ 149202201.1 57 Attorney Docket No.: 116983-5127-WO mixture prior to purification of the conjugate (e.g., after the reaction step, between the reaction step and the quenching step, or after the quenching step). For example, the process comprises (a) contacting the antibody with the drug (e.g., DM1, DM3 or DM4) to form a mixture comprising the antibody and the drug (e.g., DM1, DM3 or DM4); and then contacting the mixture comprising the antibody and drug (e.g., DM1, DM3 or DM4) with the cross-linking agent (e.g., SMCC, Sulfo-SMCC, SPDB, Sulfo-SPDB or CX1-1), in a solution having a pH of about 4 to about 9 to provide a mixture comprising (i) the conjugate (e.g., Ab- MCC-DM1, Ab-SPDB-DM4 or Ab-CX1-1-DM1), (ii) free drug (e.g., DM1, DM3 or DM4), and (iii) reaction by-products, (b) holding the mixture prepared in step (a) to release the unstably bound linkers from the cell-binding agent, and (c) purifying the mixture to provide a purified conjugate. [00112] In another embodiment, the process comprises (a) contacting the antibody with the drug (e.g., DM1, DM3 or DM4) to form a mixture comprising the antibody and the drug (e.g., DM1, DM3 or DM4); and then contacting the mixture comprising the antibody and the drug (e.g., DM1, DM3 or DM4) with the cross-linking agent (e.g., SMCC, Sulfo- SMCC, SPDB, Sulfo-SPDB or CX1-1), in a solution having a pH of about 4 to about 9 to provide a mixture comprising (i) the conjugate, (ii) free drug (e.g., DM1, DM3 or DM4), and (iii) reaction by-products, (b) quenching the mixture prepared in step (a) to quench any unreacted drug (e.g., DM1, DM3 or DM4) and/or unreacted cross-linking agent (e.g., SMCC, Sulfo-SMCC, SPDB, Sulfo-SPDB or CX1-1), (c) holding the mixture prepared in step (b) to release the unstably bound linkers from the cell-binding agent, and (d) purifying the mixture to provide a purified conjugate (e.g., Ab-MCC-DM1, Ab-SPDB-DM4 or Ab-CX1-1-DM1). [00113] Alternatively, the holding step can be performed after purification of the conjugate, followed by an additional purification step. [00114] In a preferred embodiment, the reaction is allowed to proceed to completion prior to the holding step. In this regard, the holding step can be performed about 1 hour to about 48 hours (e.g., about 1 hour, about 2 hours, about 3 hours, about 4 hours, about 5 hours, about 6 hours, about 7 hours, about 8 hours, about 9 hours, about 10 hours, about 11 hours, about 12 hours, about 13 hours, about 14 hours, about 15 hours, about 16 hours, about 17 hours, about 18 hours, about 19 hours, about 20 hours, about 21 hours, about 22 hours, about 23 hours, about 24 hours, or about 24 hours to about 48 hours) after the mixture comprising DB1/ 149202201.1 58 Attorney Docket No.: 116983-5127-WO the antibody and the drug (e.g., DM1, DM3 or DM4) is contacted with the cross-linking agent (e.g., SMCC, Sulfo-SMCC, SPDB, Sulfo-SPDB or CX1-1). [00115] The holding step comprises maintaining the solution at a suitable temperature (e.g., about 0° C. to about 37° C.) for a suitable period of time (e.g., about 1 hour to about 1 week, about 1 hour to about 24 hours, about 1 hour to about 8 hours, or about 1 hour to about 4 hours) to release the unstably bound linkers from the antibody while not substantially releasing the stably bound linkers from the antibody. In one embodiment, the holding step comprises maintaining the solution at about 20° C. or less (e.g., about 0° C. to about 18° C., about 4° C. to about 16° C.), at room temperature (e.g., about 20° C. to about 30° C. or about 20° C. to about 25° C.), or at an elevated temperature (e.g., about 30° C. to about 37° C.). In one embodiment, the holding step comprises maintaining the solution at a temperature of about 16° C. to about 24° C. (e.g., about 15° C., about 16° C., about 17° C., about 18° C., about 19° C., about 20° C., about 21° C., about 22° C., about 23° C., about 24° C., or about 25° C.). In another embodiment, the holding step comprises maintaining the solution at a temperature of about 2° C. to about 8° C. (e.g., about 0° C., about 1° C., about 2° C., about 3° C., about 4° C., about 5° C., about 6° C., about 7° C., about 8° C., about 9° C., or about 10° C.). In another embodiment, the holding step comprises maintaining the solution at a temperature of about 37° C. (e.g., about 34° C., about 35° C., about 36° C., about 37° C., about 38° C., about 39° C., or about 40° C.). [00116] The duration of the holding step depends on the temperature and the pH at which the holding step is performed. For example, the duration of the holding step can be substantially reduced by performing the holding step at elevated temperature, with the maximum temperature limited by the stability of the cell-binding agent-cytotoxic agent conjugate. The holding step can comprise maintaining the solution for about 1 hour to about 1 day (e.g., about 1 hour, about 2 hours, about 3 hours, about 4 hours, about 5 hours, about 6 hours, about 7 hours, about 8 hours, about 9 hours, about 10 hours, about 12 hours, about 14 hours, about 16 hours, about 18 hours, about 20 hours, about 22 hours, or about 24 hours), about 10 hours to about 24 hours, about 12 hours to about 24 hours, about 14 hours to about 24 hours, about 16 hours to about 24 hours, about 18 hours to about 24 hours, about 20 hours to about 24 hours, about 5 hours to about 1 week, about 20 hours to about 1 week, about 12 hours to about 1 week (e.g., about 12 hours, about 16 hours, about 20 hours, about 24 hours, DB1/ 149202201.1 59 Attorney Docket No.: 116983-5127-WO about 2 days, about 3 days, about 4 days, about 5 days, about 6 days, or about 7 days), or about 1 day to about 1 week. [00117] In one embodiment, the holding step comprises maintaining the solution at a temperature of about 2° C. to about 8° C. for a period of at least about 12 hours for up to a week. In another embodiment, the holding step comprises maintaining the solution at a temperature of about 2° C. to about 8° C. overnight (e.g., about 12 to about 24 hours, preferably about 20 hours). [00118] The pH value for the holding step preferably is about 4 to about 10. In one embodiment, the pH value for the holding step is about 4 or more, but less than about 6 (e.g., 4 to 5.9) or about 5 or more, but less than about 6 (e.g., 5 to 5.9). In another embodiment, the pH values for the holding step range from about 6 to about 10 (e.g., about 6.5 to about 9, about 6 to about 8). For example, pH values for the holding step can be about 6, about 6.5, about 7, about 7.5, about 8, about 8.5, about 9, about 9.5, or about 10. [00119] In specific embodiments, the holding step can comprise incubating the mixture at 25° C. at a pH of about 6-7.5 for about 12 hours to about 1 week, incubating the mixture at 4° C. at a pH of about 4.5-5.9 for about 5 hours to about 5 days, or incubating the mixture at 25° C. at a pH of about 4.5-5.9 for about 5 hours to about 1 day. [00120] The one-step process may optionally include the addition of sucrose to the reaction step to increase solubility and recovery of the conjugates. Desirably, sucrose is added at a concentration of about 0.1% (w/v) to about 20% (w/v) (e.g., about 0.1% (w/v), 1% (w/v), 5% (w/v), 10% (w/v), 15% (w/v), or 20% (w/v)). Preferably, sucrose is added at a concentration of about 1% (w/v) to about 10% (w/v) (e.g., about 0.5% (w/v), about 1% (w/v), about 1.5% (w/v), about 2% (w/v), about 3% (w/v), about 4% (w/v), about 5% (w/v), about 6% (w/v), about 7% (w/v), about 8% (w/v), about 9% (w/v), about 10% (w/v), or about 11% (w/v)). In addition, the reaction step also can comprise the addition of a buffering agent. Any suitable buffering agent known in the art can be used. Suitable buffering agents include, for example, a citrate buffer, an acetate buffer, a succinate buffer, and a phosphate buffer. In one embodiment, the buffering agent is selected from the group consisting of HEPPSO (N-(2- hydroxyethyl)piperazine-N′-(2-hydroxypropanesulfonic acid)), POPSO (piperazine-1,4-bis- (2-hydroxy-propane-sulfonic acid) dehydrate), HEPES (4-(2-hydroxyethyl)piperazine-1- ethanesulfonic acid), HEPPS (EPPS) (4-(2-hydroxyethyl)piperazine-1-propanesulfonic acid), DB1/ 149202201.1 60 Attorney Docket No.: 116983-5127-WO TES (N-[tris(hydroxymethyl)methyl]-2-aminoethanesulfonic acid), and a combination thereof. [00121] In one embodiment, the one-step process can further comprise the step of purifying the mixture to provide purified conjugate (e.g., Ab-MCC-DM1, Ab-SPDB-DM4 or Ab-CX1-1-DM1). Any purification methods known in the art can be used to purify the conjugates of the present invention. In one embodiment, the conjugates of the present invention using tangential flow filtration (TFF), non-adsorptive chromatography, adsorptive chromatography, adsorptive filtration, selective precipitation, or any other suitable purification process, as well as combinations thereof. In another embodiment, prior to subjecting the conjugates to purification process described above, the conjugates are first filtered through one or more PVDF membranes. Alternatively, the conjugates are filtered through one or more PVDF membranes after subjecting the conjugates to the purification process described above. For example, in one embodiment, the conjugates are filtered through one or more PVDF membranes and then purified using tangential flow filtration. Alternatively, the conjugates are purified using tangential flow filtration and then filtered through one or more PVDF membranes. [00122] Any suitable TFF systems may be utilized for purification, including a Pellicon type system (Millipore, Billerica, Mass.), a Sartocon Cassette system (Sartorius A G, Edgewood, N.Y.), and a Centrasette type system (Pall Corp., East Hills, N.Y.). [00123] Any suitable adsorptive chromatography resin may be utilized for purification. Preferred adsorptive chromatography resins include hydroxyapatite chromatography, hydrophobic charge induction chromatography (HCIC), hydrophobic interaction chromatography (HIC), ion exchange chromatography, mixed mode ion exchange chromatography, immobilized metal affinity chromatography (IMAC), dye ligand chromatography, affinity chromatography, reversed phase chromatography, and combinations thereof. Examples of suitable hydroxyapatite resins include ceramic hydroxyapatite (CHT Type I and Type II, Bio-Rad Laboratories, Hercules, Calif.), HA Ultrogel hydroxyapatite (Pall Corp., East Hills, N.Y.), and ceramic fluoroapatite (CFT Type I and Type II, Bio-Rad Laboratories, Hercules, Calif.). An example of a suitable HCIC resin is MEP Hypercel resin (Pall Corp., East Hills, N.Y.). Examples of suitable HIC resins include Butyl-Sepharose, Hexyl-Sepaharose, Phenyl-Sepharose, and Octyl Sepharose resins (all from GE Healthcare, Piscataway, N.J.), as well as Macro-prep Methyl and Macro-Prep t-Butyl resins (Biorad DB1/ 149202201.1 61 Attorney Docket No.: 116983-5127-WO Laboratories, Hercules, Calif.). Examples of suitable ion exchange resins include SP- Sepharose, CM-Sepharose, and Q-Sepharose resins (all from GE Healthcare, Piscataway, N.J.), and Unosphere S resin (Bio-Rad Laboratories, Hercules, Calif.). Examples of suitable mixed mode ion exchangers include Bakerbond ABx resin (JT Baker, Phillipsburg N.J.). Examples of suitable IMAC resins include Chelating Sepharose resin (GE Healthcare, Piscataway, N.J.) and Profinity IMAC resin (Bio-Rad Laboratories, Hercules, Calif.). Examples of suitable dye ligand resins include Blue Sepharose resin (GE Healthcare, Piscataway, N.J.) and Affi-gel Blue resin (Bio-Rad Laboratories, Hercules, Calif.). Examples of suitable affinity resins include Protein A Sepharose resin (e.g., MabSelect, GE Healthcare, Piscataway, N.J.) and lectin affinity resins, e.g. Lentil Lectin Sepharose resin (GE Healthcare, Piscataway, N.J.), where the antibody bears appropriate lectin binding sites. Examples of suitable reversed phase resins include C4, C8, and C18 resins (Grace Vydac, Hesperia, Calif.). [00124] Any suitable non-adsorptive chromatography resin may be utilized for purification. Examples of suitable non-adsorptive chromatography resins include, but are not limited to, SEPHADEX™ G-25, G-50, G-100, SEPHACRYL™ resins (e.g., S-200 and S- 300), SUPERDEX™ resins (e.g., SUPERDEX™ 75 and SUPERDEX™ 200), BIO-GEL® resins (e.g., P-6, P-10, P-30, P-60, and P-100), and others known to those of ordinary skill in the art. G. Two-Step Process and One-Pot Process for Cross-Linking to Lysine Antibody Residues [00125] In one embodiment, the conjugates of the present invention can be prepared as described in the U.S. Pat. No.7,811,572 and U.S. Patent Application Publication No. 2006/0182750. The process comprises the steps of (a) contacting the antibody of the present invention with the cross-linking agent (e.g., SMCC, Sulfo-SMCC, SPDB, Sulfo-SPDB or CX1-1) to covalently attach the linker (i.e., Ab-SMCC, Ab-SPDB or Ab-CX1-1) to the antibody and thereby prepare a first mixture comprising the antibody having the linker bound thereto; (b) optionally subjecting the first mixture to a purification process to prepare a purified first mixture of the antibody having the linker bound thereto; (c) conjugating the drug (e.g., DM1, DM3, or DM4) to the antibody having the linker bound thereto in the first mixture by reacting the antibody having the linker bound thereto with the drug (e.g., DM1, DM3, or DM4) in a solution having a pH of about 4 to about 9 to prepare a second mixture DB1/ 149202201.1 62 Attorney Docket No.: 116983-5127-WO comprising (i) conjugate (e.g., Ab-MCC-DM1, Ab-SPDB-DM4 or Ab-CX1-1-DM1), (ii) free drug (e.g., DM1, DM3 or DM4); and (iii) reaction by-products; and (d) subjecting the second mixture to a purification process to purify the conjugate from the other components of the second mixture. Alternatively, the purification step (b) can be omitted. Any purification methods described herein can be used for steps (b) and (d). In one embodiment, TFF is used for both steps (b) and (d). In another embodiment, TFF is used for step (b) and absorptive chromatography (e.g., CHT) is used for step (d). H. Antibody Preparation [00126] Techniques for preparing monoclonal antibodies against virtually any target antigen, such as Trop-2, are well known in the art. See, for example, Kohler and Milstein, Nature 256: 495 (1975), and Coligan et al. (eds.), CURRENT PROTOCOLS IN IMMUNOLOGY, VOL.1, pages 2.5.1-2.6.7 (John Wiley & Sons 1991). Briefly, monoclonal antibodies can be obtained by injecting mice with a composition comprising an antigen, removing the spleen to obtain B-lymphocytes, fusing the B-lymphocytes with myeloma cells to produce hybridomas, cloning the hybridomas, selecting positive clones which produce antibodies to the antigen, culturing the clones that produce antibodies to the antigen, and isolating the antibodies from the hybridoma cultures. [00127] MAbs can be isolated and purified from hybridoma cultures by a variety of well- established techniques. Such isolation techniques include affinity chromatography with Protein-A or Protein-G Sepharose, size-exclusion chromatography, and ion-exchange chromatography. See, for example, Coligan at pages 2.7.1-2.7.12 and pages 2.9.1-2.9.3. Also, see Baines et al., "Purification of Immunoglobulin G (IgG)," in METHODS IN MOLECULAR BIOLOGY, VOL.10, pages 79-104 (The Humana Press, Inc.1992). [00128] After the initial raising of antibodies to the immunogen, the heavy and light chain genes of the antibodies can be sequenced and subsequently cloned by recombinant techniques. See, e.g., Kopitar-Jerala et al., Pflugers Arch – Eur J Physiol, 2000, 439 [Suppl]: R79-R80. Humanization and chimerization of murine antibodies and antibody fragments are well known to those skilled in the art, as discussed below. [00129] Chimeric Antibodies [00130] A chimeric antibody is a recombinant protein in which the variable regions of a human antibody have been replaced by the variable regions of, for example, a mouse DB1/ 149202201.1 63 Attorney Docket No.: 116983-5127-WO antibody, including the complementarity- determining regions (CDRs) of the mouse antibody. Chimeric antibodies exhibit decreased immunogenicity and increased stability when administered to a subject. General techniques for cloning murine immunoglobulin variable domains are disclosed, for example, in Orlandi et al, Proc. Nat'l Acad. Sci. USA 6: 3833 (1989). Techniques for constructing chimeric antibodies are well known to those of skill in the art. As an example, Leung et al, Hybridoma 13:469 (1994), produced an LL2 chimera by combining DNA sequences encoding the VK and VH domains of murine LL2, an anti- CD22 monoclonal antibody, with respective human κ and IgGI constant region domains. [00131] Humanized Antibodies [00132] Techniques for producing humanized MAbs are well known in the art (see, e.g., Jones et al, Nature 321: 522 (1986), Riechmann et al, Nature 332: 323 (1988), Verhoeyen et al, Science 239: 1534 (1988), Carter et al, Proc. Nat'l Acad. Sci. USA 89: 4285 (1992), Sandhu, Crit. Rev. Biotech.12: 437 (1992), and Singer et al., J. Immun.150: 2844 (1993)). A chimeric or murine monoclonal antibody may be humanized by transferring the mouse CDRs from the heavy and light variable chains of the mouse immunoglobulin into the corresponding variable domains of a human antibody. The mouse framework regions (FR) in the chimeric monoclonal antibody are also replaced with human FR sequences. As simply transferring mouse CDRs into human FRs often results in a reduction or even loss of antibody affinity, additional modification might be required in order to restore the original affinity of the murine antibody. This can be accomplished by the replacement of one or more human residues in the FR regions with their murine counterparts to obtain an antibody that possesses good binding affinity to its epitope. See, for example, Tempest et al., Biotechnology 9:266 (1991) and Verhoeyen et al., Science 239: 1534 (1988). Preferred residues for substitution include FR residues that are located within 1, 2, or 3 Angstroms of a CDR residue side chain, that are located adjacent to a CDR sequence, or that are predicted to interact with a CDR residue. [00133] Human Antibodies [00134] Methods for producing fully human antibodies using either combinatorial approaches or transgenic animals transformed with human immunoglobulin loci are known in the art (e.g., Mancini et al., 2004, New Microbiol.27:315-28; Conrad and Scheller, 2005, Comb. Chem. High Throughput Screen.8: 117-26; Brekke and Loset, 2003, Curr. Opin. Pharmacol.3:544-50). A fully human antibody also can be constructed by genetic or DB1/ 149202201.1 64 Attorney Docket No.: 116983-5127-WO chromosomal transfection methods, as well as phage display technology, all of which are known in the art. See for example, McCafferty et al, Nature 348:552-553 (1990). Such fully human antibodies are expected to exhibit even fewer side effects than chimeric or humanized antibodies and to function in vivo as essentially endogenous human antibodies. [00135] In one alternative, the phage display technique may be used to generate human antibodies (e.g., Dantas-Barbosa et al., 2005, Genet. Mol. Res.4: 126-40). Human antibodies may be generated from normal humans or from humans that exhibit a particular disease state, such as cancer (Dantas-Barbosa et al., supra). The advantage to constructing human antibodies from a diseased individual is that the circulating antibody repertoire may be biased towards antibodies against disease-associated antigens. [00136] In one non-limiting example of this methodology, Dantas-Barbosa et al. constructed a phage display library of human Fab antibody fragments from osteosarcoma patients. Generally, total RNA was obtained from circulating blood lymphocytes (Id.). [00137] Recombinant Fab were cloned from the μ, γ and κ chain antibody repertoires and inserted into a phage display library (Id.). RNAs were converted to cDNAs and used to make Fab cDNA libraries using specific primers against the heavy and light chain immunoglobulin sequences (Marks et al., 1991, J. Mol. Biol.222:581-97). Library construction was performed according to Andris-Widhopf et al. (2000, In: Phage Display Laboratory Manual, Barbas et al. (eds), 1st edition, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY pp.9.1 to 9.22). The final Fab fragments were digested with restriction endonucleases and inserted into the bacteriophage genome to make the phage display library. Such libraries may be screened by standard phage display methods, as known in the art. Phage display can be performed in a variety of formats, for their review, see e.g. Johnson and Chiswell, Current Opinion in Structural Biology 3:5564-571 (1993). [00138] Human antibodies may also be generated by in vitro activated B-cells. See U.S. Patent Nos.5,567,610 and 5,229,275, incorporated herein by reference in their entirety. The skilled artisan will realize that these techniques are exemplary and any known method for making and screening human antibodies or antibody fragments may be utilized. [00139] In another alternative, transgenic animals that have been genetically engineered to produce human antibodies may be used to generate antibodies against essentially any immunogenic target, using standard immunization protocols. Methods for DB1/ 149202201.1 65 Attorney Docket No.: 116983-5127-WO obtaining human antibodies from transgenic mice are disclosed by Green et al, Nature Genet. 7:13 (1994), Lonberg et al, Nature 355:856 (1994), and Taylor et al, Int. Immun.6:579 (1994). A non- limiting example of such a system is the XenoMouse® (e.g., Green et al., 1999, J. Immunol. Methods 231 : 11-23, incorporated herein by reference) from Abgenix (Fremont, CA). In the XenoMouse® and similar animals, the mouse antibody genes have been inactivated and replaced by functional human antibody genes, while the remainder of the mouse immune system remains intact. [00140] The XenoMouse® was transformed with germline- configured YACs (yeast artificial chromosomes) that contained portions of the human IgH and Igkappa loci, including the majority of the variable region sequences, along with accessory genes and regulatory sequences. The human variable region repertoire may be used to generate antibody producing B-cells, which may be processed into hybridomas by known techniques. A XenoMouse® immunized with a target antigen will produce human antibodies by the normal immune response, which may be harvested and/or produced by standard techniques discussed above. A variety of strains of XenoMouse® are available, each of which is capable of producing a different class of antibody. Transgenically produced human antibodies have been shown to have therapeutic potential, while retaining the pharmacokinetic properties of normal human antibodies (Green et al., 1999, supra). The skilled artisan will realize that the claimed compositions and methods are not limited to use of the XenoMouse® system but may utilize any transgenic animal that has been genetically engineered to produce human antibodies. Cloning and production of human antibodies can be carried out using the usual hybridoma technology. See Little et al., Immunol. Today, 2000, 21:364-370. [00141] The host cells for harboring and expressing the anti-Trop-2 antibody chains can be either prokaryotic or eukaryotic. E. coli is one prokaryotic host useful for cloning and expressing the polynucleotides of the present invention. Other microbial hosts suitable for use include bacilli, such as Bacillus subtilis, and other enterobacteriaceae, such as Salmonella, Serratia , and various Pseudomonas species. In these prokaryotic hosts, one can also make expression vectors, which typically contain expression control sequences compatible with the host cell (e.g., an origin of replication). In addition, any number of a variety of well-known promoters will be present, such as the lactose promoter system, a tryptophan (trp) promoter system, a beta-lactamase promoter system, or a promoter system from phage lambda. The promoters typically control expression, optionally with an operator sequence, and have DB1/ 149202201.1 66 Attorney Docket No.: 116983-5127-WO ribosome binding site sequences and the like, for initiating and completing transcription and translation. Other microbes, such as yeast, can also be employed to express anti-Trop-2 polypeptides of the invention. Insect cells in combination with baculovirus vectors can also be used. [00142] In some preferred embodiments, mammalian host cells are used to express and produce the anti-Trop-2 polypeptides of the present invention. For example, they can be either a hybridoma cell line expressing endogenous immunoglobulin genes (e.g., the myeloma hybridoma clones as described in the Examples) or a mammalian cell line harboring an exogenous expression vector (e.g., the SP2/0 myeloma cells exemplified below). These include any normal mortal or normal or abnormal immortal animal or human cell. For example, a number of suitable host cell lines capable of secreting intact immunoglobulins have been developed, including the CHO cell lines, various Cos cell lines, HeLa cells, myeloma cell lines, transformed B-cells and hybridomas. The use of mammalian tissue cell culture to express polypeptides is discussed generally in, e.g., Winnacker, From Genes to Clones, VCH Publishers, N.Y., N.Y., 1987. Expression vectors for mammalian host cells can include expression control sequences, such as an origin of replication, a promoter, and an enhancer (see, e.g., Queen et al., Immunol. Rev.89:49-68, 1986), and necessary processing information sites, such as ribosome binding sites, RNA splice sites, polyadenylation sites, and transcriptional terminator sequences. These expression vectors usually contain promoters derived from mammalian genes or from mammalian viruses. Suitable promoters may be constitutive, cell type-specific, stage-specific, and/or modulatable or regulatable. Useful promoters include, but are not limited to, the metallothionein promoter, the constitutive adenovirus major late promoter, the dexamethasone-inducible MMTV promoter, the SV40 promoter, the MRP polIII promoter, the constitutive MPSV promoter, the tetracycline-inducible CMV promoter (such as the human immediate-early CMV promoter), the constitutive CMV promoter, and promoter-enhancer combinations known in the art. [00143] Methods for introducing expression vectors containing the polynucleotide sequences of interest vary depending on the type of cellular host. For example, calcium chloride transfection is commonly utilized for prokaryotic cells, whereas calcium phosphate treatment or electroporation may be used for other cellular hosts (see generally Sambrook et al., Molecular Cloning: A Laboratory Manual, 4th ed.). Other methods include, e.g., electroporation, calcium phosphate treatment, liposome-mediated transformation, injection DB1/ 149202201.1 67 Attorney Docket No.: 116983-5127-WO and microinjection, ballistic methods, virosomes, immunoliposomes, polycation:nucleic acid conjugates, naked DNA, artificial virions, fusion to the herpes virus structural protein VP22 (Elliot and O'Hare, Cell 88:223, 1997), agent-enhanced uptake of DNA, and ex vivo transduction. For long-term, high-yield production of recombinant proteins, stable expression will often be desired. For example, cell lines which stably express anti-Trop-2 antibody chains or binding fragments can be prepared using expression vectors of the invention which contain viral origins of replication or endogenous expression elements and a selectable marker gene. Following introduction of the vector, cells may be allowed to grow for 1-2 days in an enriched media before they are switched to selective media. The purpose of the selectable marker is to confer resistance to selection, and its presence allows growth of cells which successfully express the introduced sequences in selective media. Resistant, stably transfected cells can be proliferated using tissue culture techniques appropriate to the cell type. III. Gen 2 TIL Manufacturing Processes [0006] An exemplary family of TIL processes known as Gen 2 (also known as process 2A) containing some of these features is depicted in Figures 1 and 2. An embodiment of Gen 2 is shown in Figure 2. [0007] As discussed herein, the present invention can include a step relating to the restimulation of cryopreserved TILs to increase their metabolic activity and thus relative health prior to transplant into a patient, and methods of testing said metabolic health. As generally outlined herein, TILs are generally taken from a patient sample and manipulated to expand their number prior to transplant into a patient. In some embodiments, the TILs may be optionally genetically manipulated as discussed below. [0008] In some embodiments, the TILs may be cryopreserved. Once thawed, they may also be restimulated to increase their metabolism prior to infusion into a patient. [0009] In some embodiments, the first expansion (including processes referred to as the pre-REP as well as processes shown in Figure 1 as Step A) is shortened to 3 to 14 days and the second expansion (including processes referred to as the REP as well as processes shown in Figure 1 as Step B) is shorted to 7 to 14 days, as discussed in detail below as well as in the examples and figures. In some embodiments, the first expansion (for example, an expansion described as Step B in Figure 1) is shortened to 11 days and the second expansion (for DB1/ 149202201.1 68 Attorney Docket No.: 116983-5127-WO example, an expansion as described in Step D in Figure 1) is shortened to 11 days. In some embodiments, the combination of the first expansion and second expansion (for example, expansions described as Step B and Step D in Figure 1) is shortened to 22 days, as discussed in detail below and in the examples and figures. [0010] The “Step” Designations A, B, C, etc., below are in reference to Figure 1 and in reference to certain embodiments described herein. The ordering of the Steps below and in Figure 1 is exemplary and any combination or order of steps, as well as additional steps, repetition of steps, and/or omission of steps is contemplated by the present application and the methods disclosed herein. A. STEP A: Obtain Patient Tumor Sample [0011] In general, TILs are initially obtained from a patient tumor sample and then expanded into a larger population for further manipulation as described herein, optionally cryopreserved, restimulated as outlined herein and optionally evaluated for phenotype and metabolic parameters as an indication of TIL health. [0012] A patient tumor sample may be obtained using methods known in the art, generally via surgical resection, needle biopsy, core biopsy, small biopsy, or other means for obtaining a sample that contains a mixture of tumor and TIL cells. In some embodiments, multilesional sampling is used. In some embodiments, surgical resection, needle biopsy, core biopsy, small biopsy, or other means for obtaining a sample that contains a mixture of tumor and TIL cells includes multilesional sampling (i.e., obtaining samples from one or more tumor sites and/or locations in the patient, as well as one or more tumors in the same location or in close proximity). In general, the tumor sample may be from any solid tumor, including primary tumors, invasive tumors or metastatic tumors. The tumor sample may also be a liquid tumor, such as a tumor obtained from a hematological malignancy. The solid tumor may be of lung tissue. In some embodiments, useful TILs are obtained from non-small cell lung carcinoma (NSCLC). The solid tumor may be of skin tissue. In some embodiments, useful TILs are obtained from a melanoma. [0013] Once obtained, the tumor sample is generally fragmented using sharp dissection into small pieces of between 1 to about 8 mm3, with from about 2-3 mm3 being particularly useful. In some embodiments, the TILs are cultured from these fragments using enzymatic tumor digests. Such tumor digests may be produced by incubation in enzymatic media (e.g., DB1/ 149202201.1 69 Attorney Docket No.: 116983-5127-WO Roswell Park Memorial Institute (RPMI) 1640 buffer, 2 mM glutamate, 10 mcg/mL gentamicine, 30 units/mL of DNase and 1.0 mg/mL of collagenase) followed by mechanical dissociation (e.g., using a tissue dissociator). Tumor digests may be produced by placing the tumor in enzymatic media and mechanically dissociating the tumor for approximately 1 minute, followed by incubation for 30 minutes at 37 °C in 5% CO2, followed by repeated cycles of mechanical dissociation and incubation under the foregoing conditions until only small tissue pieces are present. At the end of this process, if the cell suspension contains a large number of red blood cells or dead cells, a density gradient separation using FICOLL branched hydrophilic polysaccharide may be performed to remove these cells. Alternative methods known in the art may be used, such as those described in U.S. Patent Application Publication No.2012/0244133 A1, the disclosure of which is incorporated by reference herein. Any of the foregoing methods may be used in any of the embodiments described herein for methods of expanding TILs or methods treating a cancer. [0014] Tumor dissociating enzyme mixtures can include one or more dissociating (digesting) enzymes such as, but not limited to, collagenase (including any blend or type of collagenase), Accutase™, Accumax™, hyaluronidase, neutral protease (dispase), chymotrypsin, chymopapain, trypsin, caseinase, elastase, papain, protease type XIV (pronase), deoxyribonuclease I (DNase), trypsin inhibitor, any other dissociating or proteolytic enzyme, and any combination thereof. [0015] In some embodiments, the dissociating enzymes are reconstituted from lyophilized enzymes. In some embodiments, lyophilized enzymes are reconstituted in an amount of sterile buffer such as HBSS. [0016] In some instances, collagenase (such as animal free- type 1 collagenase) is reconstituted in 10 mL of sterile HBSS or another buffer. The lyophilized stock enzyme may be at a concentration of 2892 PZ U/vial. In some embodiments, collagenase is reconstituted in 5 mL to 15 mL buffer. In some embodiment, after reconstitution the collagenase stock ranges from about 100 PZ U/mL-about 400 PZ U/mL, e.g., about 100 PZ U/mL-about 400 PZ U/mL, about 100 PZ U/mL-about 350 PZ U/mL, about 100 PZ U/mL-about 300 PZ U/mL, about 150 PZ U/mL-about 400 PZ U/mL, about 100 PZ U/mL, about 150 PZ U/mL, about 200 PZ U/mL, about 210 PZ U/mL, about 220 PZ U/mL, about 230 PZ U/mL, about 240 PZ U/mL, about 250 PZ U/mL, about 260 PZ U/mL, about 270 PZ U/mL, about DB1/ 149202201.1 70 Attorney Docket No.: 116983-5127-WO 280 PZ U/mL, about 289.2 PZ U/mL, about 300 PZ U/mL, about 350 PZ U/mL, or about 400 PZ U/mL. [0017] In some embodiments, neutral protease is reconstituted in 1 mL of sterile HBSS or another buffer. The lyophilized stock enzyme may be at a concentration of 175 DMC U/vial. In some embodiments, after reconstitution the neutral protease stock ranges from about 100 DMC/mL-about 400 DMC/mL, e.g., about 100 DMC/mL-about 400 DMC/mL, about 100 DMC/mL-about 350 DMC/mL, about 100 DMC/mL-about 300 DMC/mL, about 150 DMC/mL-about 400 DMC/mL, about 100 DMC/mL, about 110 DMC/mL, about 120 DMC/mL, about 130 DMC/mL, about 140 DMC/mL, about 150 DMC/mL, about 160 DMC/mL, about 170 DMC/mL, about 175 DMC/mL, about 180 DMC/mL, about 190 DMC/mL, about 200 DMC/mL, about 250 DMC/mL, about 300 DMC/mL, about 350 DMC/mL, or about 400 DMC/mL. [0018] In some embodiments, DNAse I is reconstituted in 1 mL of sterile HBSS or another buffer. The lyophilized stock enzyme was at a concentration of 4 KU/vial. In some embodiments, after reconstitution the DNase I stock ranges from about 1 KU/mL-10 KU/mL, e.g., about 1 KU/mL, about 2 KU/mL, about 3 KU/mL, about 4 KU/mL, about 5 KU/mL, about 6 KU/mL, about 7 KU/mL, about 8 KU/mL, about 9 KU/mL, or about 10 KU/mL. [0019] In some embodiments, the stock of enzymes is variable and the concentrations may need to be determined. In some embodiments, the concentration of the lyophilized stock can be verified. In some embodiments, the final amount of enzyme added to the digest cocktail is adjusted based on the determined stock concentration. [0020] In some embodiment, the enzyme mixture includes about 10.2-ul of neutral protease (0.36 DMC U/mL), 21.3 µL of collagenase (1.2 PZ/mL) and 250-ul of DNAse I (200 U/mL) in about 4.7 mL of sterile HBSS. [0021] As indicated above, in some embodiments, the TILs are derived from solid tumors. In some embodiments, the solid tumors are not fragmented. In some embodiments, the solid tumors are not fragmented and are subjected to enzymatic digestion as whole tumors. In some embodiments, the tumors are digested in in an enzyme mixture comprising collagenase, DNase, and hyaluronidase. In some embodiments, the tumors are digested in in an enzyme mixture comprising collagenase, DNase, and hyaluronidase for 1-2 hours. In some embodiments, the tumors are digested in in an enzyme mixture comprising collagenase, DB1/ 149202201.1 71 Attorney Docket No.: 116983-5127-WO DNase, and hyaluronidase for 1-2 hours at 37°C, 5% CO2. In some embodiments, the tumors are digested in in an enzyme mixture comprising collagenase, DNase, and hyaluronidase for 1-2 hours at 37°C, 5% CO2 with rotation. In some embodiments, the tumors are digested overnight with constant rotation. In some embodiments, the tumors are digested overnight at 37°C, 5% CO2 with constant rotation. In some embodiments, the whole tumor is combined with the enzymes to form a tumor digest reaction mixture. [0022] In some embodiments, the tumor is reconstituted with the lyophilized enzymes in a sterile buffer. In some embodiments, the buffer is sterile HBSS. [0023] In some embodiments, the enzyme mixture comprises collagenase. In some embodiments, the collagenase is collagenase IV. In some embodiments, the working stock for the collagenase is a 100 mg/mL 10X working stock. [0024] In some embodiments, the enzyme mixture comprises DNAse. In some embodiments, the working stock for the DNAse is a 10,000 IU/mL 10X working stock. [0025] In some embodiments, the enzyme mixture comprises hyaluronidase. In some embodiments, the working stock for the hyaluronidase is a 10 mg/mL 10X working stock. [0026] In some embodiments, the enzyme mixture comprises 10 mg/mL collagenase, 1000 IU/mL DNAse, and 1 mg/mL hyaluronidase. [0027] In some embodiments, the enzyme mixture comprises 10 mg/mL collagenase, 500 IU/mL DNAse, and 1 mg/mL hyaluronidase. [0028] In general, the harvested cell suspension is called a “primary cell population” or a “freshly harvested” cell population. [0029] In some embodiments, fragmentation includes physical fragmentation, including for example, dissection as well as digestion. In some embodiments, the fragmentation is physical fragmentation. In some embodiments, the fragmentation is dissection. In some embodiments, the fragmentation is by digestion. In some embodiments, TILs can be initially cultured from enzymatic tumor digests and tumor fragments obtained from digesting or fragmenting a tumor sample obtained from a patient. [0030] In some embodiments, where the tumor is a solid tumor, the tumor undergoes physical fragmentation after the tumor sample is obtained in, for example, Step A (as provided in Figure 1). In some embodiments, the fragmentation occurs before DB1/ 149202201.1 72 Attorney Docket No.: 116983-5127-WO cryopreservation. In some embodiments, the fragmentation occurs after cryopreservation. In some embodiments, the fragmentation occurs after obtaining the tumor and in the absence of any cryopreservation. In some embodiments, the tumor is fragmented and 10, 20, 30, 40 or more fragments or pieces are placed in each container for the first expansion. In some embodiments, the tumor is fragmented and 30 or 40 fragments or pieces are placed in each container for the first expansion. In some embodiments, the tumor is fragmented and 40 fragments or pieces are placed in each container for the first expansion. In some embodiments, the multiple fragments comprise about 4 to about 50 fragments, wherein each fragment has a volume of about 27 mm3. In some embodiments, the multiple fragments comprise about 30 to about 60 fragments with a total volume of about 1300 mm3 to about 1500 mm3. In some embodiments, the multiple fragments comprise about 50 fragments with a total volume of about 1350 mm3. In some embodiments, the multiple fragments comprise about 50 fragments with a total mass of about 1 gram to about 1.5 grams. In some embodiments, the multiple fragments comprise about 4 fragments. [0031] In some embodiments, the TILs are obtained from tumor fragments. In some embodiments, the tumor fragment is obtained by sharp dissection. In some embodiments, the tumor fragment is between about 1 mm3 and 10 mm3. In some embodiments, the tumor fragment is between about 1 mm3 and 8 mm3. In some embodiments, the tumor fragment is about 1 mm3. In some embodiments, the tumor fragment is about 2 mm3. In some embodiments, the tumor fragment is about 3 mm3. In some embodiments, the tumor fragment is about 4 mm3. In some embodiments, the tumor fragment is about 5 mm3. In some embodiments, the tumor fragment is about 6 mm3. In some embodiments, the tumor fragment is about 7 mm3. In some embodiments, the tumor fragment is about 8 mm3. In some embodiments, the tumor fragment is about 9 mm3. In some embodiments, the tumor fragment is about 10 mm3. In some embodiments, the tumors are 1-4 mm × 1-4 mm × 1-4 mm. In some embodiments, the tumors are 1 mm × 1 mm × 1 mm. In some embodiments, the tumors are 2 mm × 2 mm × 2 mm. In some embodiments, the tumors are 3 mm × 3 mm × 3 mm. In some embodiments, the tumors are 4 mm × 4 mm × 4 mm. [0032] In some embodiments, the tumors are resected in order to minimize the amount of hemorrhagic, necrotic, and/or fatty tissues on each piece. In some embodiments, the tumors are resected in order to minimize the amount of hemorrhagic tissue on each piece. In some embodiments, the tumors are resected in order to minimize the amount of necrotic tissue on DB1/ 149202201.1 73 Attorney Docket No.: 116983-5127-WO each piece. In some embodiments, the tumors are resected in order to minimize the amount of fatty tissue on each piece. [0033] In some embodiments, the tumor fragmentation is performed in order to maintain the tumor internal structure. In some embodiments, the tumor fragmentation is performed without performing a sawing motion with a scalpel. In some embodiments, the TILs are obtained from tumor digests. In some embodiments, tumor digests were generated by incubation in enzyme media, for example but not limited to RPMI 1640, 2 mM GlutaMAX, 10 mg/mL gentamicin, 30 U/mL DNase, and 1.0 mg/mL collagenase, followed by mechanical dissociation (GentleMACS, Miltenyi Biotec, Auburn, CA). After placing the tumor in enzyme media, the tumor can be mechanically dissociated for approximately 1 minute. The solution can then be incubated for 30 minutes at 37 °C in 5% CO2 and it then mechanically disrupted again for approximately 1 minute. After being incubated again for 30 minutes at 37 °C in 5% CO2, the tumor can be mechanically disrupted a third time for approximately 1 minute. In some embodiments, after the third mechanical disruption if large pieces of tissue were present, 1 or 2 additional mechanical dissociations were applied to the sample, with or without 30 additional minutes of incubation at 37 °C in 5% CO2. In some embodiments, at the end of the final incubation if the cell suspension contains a large number of red blood cells or dead cells, a density gradient separation using Ficoll can be performed to remove these cells. [0034] In some embodiments, the harvested cell suspension prior to the first expansion step is called a “primary cell population” or a “freshly harvested” cell population. [0035] In some embodiments, cells can be optionally frozen after sample harvest and stored frozen prior to entry into the expansion described in Step B, which is described in further detail below, as well as exemplified in Figure 1, as well as Figure 8. 1. Pleural effusion T-cells and TILs [0036] In some embodiments, the sample is a pleural fluid sample. In some embodiments, the source of the T-cells or TILs for expansion according to the processes described herein is a pleural fluid sample. In some embodiments, the sample is a pleural effusion derived sample. In some embodiments, the source of the T-cells or TILs for expansion according to the processes described herein is a pleural effusion derived sample. See, for example, methods DB1/ 149202201.1 74 Attorney Docket No.: 116983-5127-WO described in U.S. Patent Publication US 2014/0295426, incorporated herein by reference in its entirety for all purposes. [0037] In some embodiments, any pleural fluid or pleural effusion suspected of and/or containing TILs can be employed. Such a sample may be derived from a primary or metastatic lung cancer, such as NSCLC or SCLC. In some embodiments, the sample may be derived from secondary metastatic cancer cells which originated from another organ, e.g., breast, ovary, colon or prostate. In some embodiments, the sample for use in the expansion methods described herein is a pleural exudate. In some embodiments, the sample for use in the expansion methods described herein is a pleural transudate. Other biological samples may include other serous fluids containing TILs, including, e.g., ascites fluid from the abdomen or pancreatic cyst fluid. Ascites fluid and pleural fluids involve very similar chemical systems; both the abdomen and lung have mesothelial lines and fluid forms in the pleural space and abdominal spaces in the same matter in malignancies and such fluids in some embodiments contain TILs. In some embodiments, wherein the disclosed methods utilize pleural fluid, the same methods may be performed with similar results using ascites or other cyst fluids containing TILs. [0038] In some embodiments, the pleural fluid is in unprocessed form, directly as removed from the patient. In some embodiments, the unprocessed pleural fluid is placed in a standard blood collection tube, such as an EDTA or Heparin tube, prior to further processing steps. In some embodiments, the unprocessed pleural fluid is placed in a standard CellSave® tube (Veridex) prior to further processing steps. In some embodiments, the sample is placed in the CellSave tube immediately after collection from the patient to avoid a decrease in the number of viable TILs. The number of viable TILs can decrease to a significant extent within 24 hours, if left in the untreated pleural fluid, even at 4°C. In some embodiments, the sample is placed in the appropriate collection tube within 1 hour, 5 hours, 10 hours, 15 hours, or up to 24 hours after removal from the patient. In some embodiments, the sample is placed in the appropriate collection tube within 1 hour, 5 hours, 10 hours, 15 hours, or up to 24 hours after removal from the patient at 4°C. [0039] In some embodiments, the pleural fluid sample from the chosen subject may be diluted. In some embodiments, the dilution is 1:10 pleural fluid to diluent. In other embodiments, the dilution is 1:9 pleural fluid to diluent. In other embodiments, the dilution is 1:8 pleural fluid to diluent. In other embodiments, the dilution is 1:5 pleural fluid to diluent. DB1/ 149202201.1 75 Attorney Docket No.: 116983-5127-WO In other embodiments, the dilution is 1:2 pleural fluid to diluent. In other embodiments, the dilution is 1:1 pleural fluid to diluent. In some embodiments, diluents include saline, phosphate buffered saline, another buffer or a physiologically acceptable diluent. In some embodiments, the sample is placed in the CellSave tube immediately after collection from the patient and dilution to avoid a decrease in the viable TILs, which may occur to a significant extent within 24-48 hours, if left in the untreated pleural fluid, even at 4°C. In some embodiments, the pleural fluid sample is placed in the appropriate collection tube within 1 hour, 5 hours, 10 hours, 15 hours, 24 hours, 36 hours, up to 48 hours after removal from the patient, and dilution. In some embodiments, the pleural fluid sample is placed in the appropriate collection tube within 1 hour, 5 hours, 10 hours, 15 hours, 24 hours, 36 hours, up to 48 hours after removal from the patient, and dilution at 4°C. [0040] In still other embodiments, pleural fluid samples are concentrated by conventional means prior to further processing steps. In some embodiments, this pre-treatment of the pleural fluid is preferable in circumstances in which the pleural fluid must be cryopreserved for shipment to a laboratory performing the method or for later analysis (e.g., later than 24-48 hours post-collection). In some embodiments, the pleural fluid sample is prepared by centrifuging the pleural fluid sample after its withdrawal from the subject and resuspending the centrifugate or pellet in buffer. In some embodiments, the pleural fluid sample is subjected to multiple centrifugations and resuspensions, before it is cryopreserved for transport or later analysis and/or processing. [0041] In some embodiments, pleural fluid samples are concentrated prior to further processing steps by using a filtration method. In some embodiments, the pleural fluid sample used in further processing is prepared by filtering the fluid through a filter containing a known and essentially uniform pore size that allows for passage of the pleural fluid through the membrane but retains the tumor cells. In some embodiments, the diameter of the pores in the membrane may be at least 4 μM. In other embodiments the pore diameter may be 5 μM or more, and in other embodiment, any of 6, 7, 8, 9, or 10 μM. After filtration, the cells, including TILs, retained by the membrane may be rinsed off the membrane into a suitable physiologically acceptable buffer. Cells, including TILs, concentrated in this way may then be used in the further processing steps of the method. [0042] In some embodiments, pleural fluid sample (including, for example, the untreated pleural fluid), diluted pleural fluid, or the resuspended cell pellet, is contacted with a lytic DB1/ 149202201.1 76 Attorney Docket No.: 116983-5127-WO reagent that differentially lyses non-nucleated red blood cells present in the sample. In some embodiments, this step is performed prior to further processing steps in circumstances in which the pleural fluid contains substantial numbers of RBCs. Suitable lysing reagents include a single lytic reagent or a lytic reagent and a quench reagent, or a lytic agent, a quench reagent and a fixation reagent. Suitable lytic systems are marketed commercially and include the BD Pharm Lyse™ system (Becton Dickenson). Other lytic systems include the Versalyse™ system, the FACSlyse™ system (Becton Dickenson), the Immunoprep™ system or Erythrolyse II system (Beckman Coulter, Inc.), or an ammonium chloride system. In some embodiments, the lytic reagent can vary with the primary requirements being efficient lysis of the red blood cells, and the conservation of the TILs and phenotypic properties of the TILs in the pleural fluid. In addition to employing a single reagent for lysis, the lytic systems useful in methods described herein can include a second reagent, e.g., one that quenches or retards the effect of the lytic reagent during the remaining steps of the method, e.g., Stabilyse™ reagent (Beckman Coulter, Inc.). A conventional fixation reagent may also be employed depending upon the choice of lytic reagents or the preferred implementation of the method. [0043] In some embodiments, the pleural fluid sample, unprocessed, diluted or multiply centrifuged or processed as described herein above is cryopreserved at a temperature of about −140°C prior to being further processed and/or expanded as provided herein. B. STEP B: First Expansion [0044] In some embodiments, the present methods provide for obtaining young TILs, which are capable of increased replication cycles upon administration to a subject/patient and as such may provide additional therapeutic benefits over older TILs (i.e., TILs which have further undergone more rounds of replication prior to administration to a subject/patient). Features of young TILs have been described in the literature, for example in Donia, et al., Scand. J. Immunol.2012, 75, 157–167; Dudley, et al., Clin. Cancer Res.2010, 16, 6122- 6131; Huang, et al., J. Immunother.2005, 28, 258–267; Besser, et al., Clin. Cancer Res. 2013, 19, OF1-OF9; Besser, et al., J. Immunother.2009, 32:415–423; Robbins, et al., J. Immunol.2004, 173, 7125-7130; Shen, et al., J. Immunother., 2007, 30, 123–129; Zhou, et al., J. Immunother.2005, 28, 53–62; and Tran, et al., J. Immunother., 2008, 31, 742–751, each of which is incorporated herein by reference. DB1/ 149202201.1 77 Attorney Docket No.: 116983-5127-WO [0045] The diverse antigen receptors of T and B lymphocytes are produced by somatic recombination of a limited, but large number of gene segments. These gene segments: V (variable), D (diversity), J (joining), and C (constant), determine the binding specificity and downstream applications of immunoglobulins and T-cell receptors (TCRs). The present invention provides a method for generating TILs which exhibit and increase the T-cell repertoire diversity. In some embodiments, the TILs obtained by the present method exhibit an increase in the T-cell repertoire diversity. In some embodiments, the TILs obtained by the present method exhibit an increase in the T-cell repertoire diversity as compared to freshly harvested TILs and/or TILs prepared using other methods than those provide herein including for example, methods other than those embodied in Figure 1. In some embodiments, the TILs obtained by the present method exhibit an increase in the T-cell repertoire diversity as compared to freshly harvested TILs and/or TILs prepared using methods referred to as process 1C, as exemplified in Figure 5 and/or Figure 6. In some embodiments, the TILs obtained in the first expansion exhibit an increase in the T-cell repertoire diversity. In some embodiments, the increase in diversity is an increase in the immunoglobulin diversity and/or the T-cell receptor diversity. In some embodiments, the diversity is in the immunoglobulin is in the immunoglobulin heavy chain. In some embodiments, the diversity is in the immunoglobulin is in the immunoglobulin light chain. In some embodiments, the diversity is in the T-cell receptor. In some embodiments, the diversity is in one of the T-cell receptors selected from the group consisting of alpha, beta, gamma, and delta receptors. In some embodiments, there is an increase in the expression of T-cell receptor (TCR) alpha and/or beta. In some embodiments, there is an increase in the expression of T-cell receptor (TCR) alpha. In some embodiments, there is an increase in the expression of T-cell receptor (TCR) beta. In some embodiments, there is an increase in the expression of TCRab (i.e., TCRα/β). [0046] After dissection or digestion of tumor fragments, for example such as described in Step A of Figure 1, the resulting cells are cultured in serum containing IL-2 under conditions that favor the growth of TILs over tumor and other cells. In some embodiments, the tumor digests are incubated in 2 mL wells in media comprising inactivated human AB serum with 6000 IU/mL of IL-2. This primary cell population is cultured for a period of days, generally from 3 to 14 days, resulting in a bulk TIL population, generally about 1 × 108 bulk TIL cells. In some embodiments, this primary cell population is cultured for a period of 7 to 14 days, resulting in a bulk TIL population, generally about 1 × 108 bulk TIL cells. In some DB1/ 149202201.1 78 Attorney Docket No.: 116983-5127-WO embodiments, this primary cell population is cultured for a period of 10 to 14 days, resulting in a bulk TIL population, generally about 1 × 108 bulk TIL cells. In some embodiments, this primary cell population is cultured for a period of about 11 days, resulting in a bulk TIL population, generally about 1 × 108 bulk TIL cells. [0047] In some embodiments, expansion of TILs may be performed using an initial bulk TIL expansion step (for example such as those described in Step B of Figure 1, which can include processes referred to as pre-REP) as described below and herein, followed by a second expansion (Step D, including processes referred to as rapid expansion protocol (REP) steps) as described below under Step D and herein, followed by optional cryopreservation, and followed by a second Step D (including processes referred to as restimulation REP steps) as described below and herein. The TILs obtained from this process may be optionally characterized for phenotypic characteristics and metabolic parameters as described herein. [0048] In embodiments where TIL cultures are initiated in 24-well plates, for example, using Costar 24-well cell culture cluster, flat bottom (Corning Incorporated, Corning, NY, each well can be seeded with 1 × 106 tumor digest cells or one tumor fragment in 2 mL of complete medium (CM) with IL-2 (6000 IU/mL; Chiron Corp., Emeryville, CA). In some embodiments, the tumor fragment is between about 1 mm3 and 10 mm3. [0049] In some embodiments, the first expansion culture medium is referred to as “CM”, an abbreviation for culture media. In some embodiments, CM for Step B consists of RPMI 1640 with GlutaMAX, supplemented with 10% human AB serum, 25 mM Hepes, and 10 mg/mL gentamicin. In embodiments where cultures are initiated in gas-permeable flasks with a 40 mL capacity and a 10 cm2 gas-permeable silicon bottom (for example, G-REX10; Wilson Wolf Manufacturing, New Brighton, MN), each flask was loaded with 10–40 × 106 viable tumor digest cells or 5–30 tumor fragments in 10–40 mL of CM with IL-2. Both the G- REX10 and 24-well plates were incubated in a humidified incubator at 37°C in 5% CO2 and 5 days after culture initiation, half the media was removed and replaced with fresh CM and IL- 2 and after day 5, half the media was changed every 2–3 days. [0050] In some embodiments, the culture medium used in the expansion processes disclosed herein is a serum-free medium or a defined medium. In some embodiments, the serum-free or defined medium comprises a basal cell medium and a serum supplement and/or a serum replacement. In some embodiments, the serum-free or defined medium is used to DB1/ 149202201.1 79 Attorney Docket No.: 116983-5127-WO prevent and/or decrease experimental variation due in part to the lot-to-lot variation of serum- containing media. [0051] In some embodiments, the serum-free or defined medium comprises a basal cell medium and a serum supplement and/or serum replacement. In some embodiments, the basal cell medium includes, but is not limited to CTS™ OpTmizer™ T-cell Expansion Basal Medium , CTS™ OpTmizer™ T-Cell Expansion SFM, CTS™ AIM-V Medium, CTS™ AIM-V SFM, LymphoONE™ T-Cell Expansion Xeno-Free Medium, Dulbecco's Modified Eagle's Medium (DMEM), Minimal Essential Medium (MEM), Basal Medium Eagle (BME), RPMI 1640, F-10, F-12, Minimal Essential Medium (αMEM), Glasgow's Minimal Essential Medium (G-MEM), RPMI growth medium, and Iscove's Modified Dulbecco's Medium. [0052] In some embodiments, the serum supplement or serum replacement includes, but is not limited to one or more of CTS™ OpTmizer T-Cell Expansion Serum Supplement, CTS™ Immune Cell Serum Replacement, one or more albumins or albumin substitutes, one or more amino acids, one or more vitamins, one or more transferrins or transferrin substitutes, one or more antioxidants, one or more insulins or insulin substitutes, one or more collagen precursors, one or more antibiotics, and one or more trace elements. In some embodiments, the defined medium comprises albumin and one or more ingredients selected from the group consisting of glycine, L- histidine, L-isoleucine, L-methionine, L-phenylalanine, L-proline, L- hydroxyproline, L-serine, L-threonine, L-tryptophan, L-tyrosine, L-valine, thiamine, reduced glutathione, L-ascorbic acid-2-phosphate, iron saturated transferrin, insulin, and compounds containing the trace element moieties Ag+, Al3+, Ba2+, Cd2+, Co2+, Cr3+, Ge4+, Se4+, Br, T, Mn2+, P, Si4+, V5+, Mo6+, Ni2+, Rb+, Sn2+ and Zr4+. In some embodiments, the defined medium further comprises L-glutamine, sodium bicarbonate and/or 2-mercaptoethanol. [0053] In some embodiments, the CTS™OpTmizer™ T-cell Immune Cell Serum Replacement is used with conventional growth media, including but not limited to CTS™ OpTmizer™ T-cell Expansion Basal Medium, CTS™ OpTmizer™ T-cell Expansion SFM, CTS™ AIM-V Medium, CST™ AIM-V SFM, LymphoONE™ T-Cell Expansion Xeno-Free Medium, Dulbecco's Modified Eagle's Medium (DMEM), Minimal Essential Medium (MEM), Basal Medium Eagle (BME), RPMI 1640, F-10, F-12, Minimal Essential Medium (αMEM), Glasgow's Minimal Essential Medium (G-MEM), RPMI growth medium, and Iscove's Modified Dulbecco's Medium. DB1/ 149202201.1 80 Attorney Docket No.: 116983-5127-WO [0054] In some embodiments, the total serum replacement concentration (vol%) in the serum-free or defined medium is from about 1%, 2%, 3%, 4%, 5%, 6%, 7%, 8%, 9%, 10%, 11%, 12%, 13%, 14%, 15%, 16%, 17%, 18%, 19%, or 20% by volume of the total serum-free or defined medium. In some embodiments, the total serum replacement concentration is about 3% of the total volume of the serum-free or defined medium. In some embodiments, the total serum replacement concentration is about 5% of the total volume of the serum-free or defined medium. In some embodiments, the total serum replacement concentration is about 10% of the total volume of the serum-free or defined medium. [0055] In some embodiments, the serum-free or defined medium is CTS™ OpTmizer™ T- cell Expansion SFM (ThermoFisher Scientific). Any formulation of CTS™ OpTmizer™ is useful in the present invention. CTS™ OpTmizer™ T-cell Expansion SFM is a combination of 1L CTS™ OpTmizer™ T-cell Expansion Basal Medium and 26 mL CTS™ OpTmizer™ T-Cell Expansion Supplement, which are mixed together prior to use. In some embodiments, the CTS™ OpTmizer™ T-cell Expansion SFM is supplemented with about 3% of the CTS™ Immune Cell Serum Replacement (SR) (ThermoFisher Scientific). In some embodiments, the CTS™ OpTmizer™ T-cell Expansion SFM is supplemented with about 3% of the CTS™ Immune Cell Serum Replacement (SR) (ThermoFisher Scientific), along with 2- mercaptoethanol at 55mM. In some embodiments, the CTS™ OpTmizer™ T-cell Expansion SFM is supplemented with about 3% of the CTS™ Immune Cell Serum Replacement (SR) (ThermoFisher Scientific) and the final concentration of 2-mercaptoethanol in the media is 55µM. [0056] In some embodiments, the defined medium is CTS™ OpTmizer™ T-cell Expansion SFM (ThermoFisher Scientific). Any formulation of CTS™ OpTmizer™ is useful in the present invention. CTS™ OpTmizer™ T-cell Expansion SFM is a combination of 1L CTS™ OpTmizer™ T-cell Expansion Basal Medium and 26 mL CTS™ OpTmizer™ T-Cell Expansion Supplement, which are mixed together prior to use. In some embodiments, the CTS™ OpTmizer™ T-cell Expansion SFM is supplemented with about 3% of the CTS™ Immune Cell Serum Replacement (SR) (ThermoFisher Scientific), along with 2- mercaptoethanol at 55mM. In some embodiments, the CTS™OpTmizer™ T-cell Expansion SFM is supplemented with about 3% of the CTS™ Immune Cell Serum Replacement (SR) (ThermoFisher Scientific), 55mM of 2-mercaptoethanol, and 2mM of L-glutamine. In some embodiments, the CTS™OpTmizer™ T-cell Expansion SFM is supplemented with about 3% DB1/ 149202201.1 81 Attorney Docket No.: 116983-5127-WO of the CTS™ Immune Cell Serum Replacement (SR) (ThermoFisher Scientific), 55mM of 2- mercaptoethanol, and 2mM of L-glutamine, and further comprises about 1000 IU/mL to about 8000 IU/mL of IL-2. In some embodiments, the CTS™OpTmizer™ T-cell Expansion SFM is supplemented with about 3% of the CTS™ Immune Cell Serum Replacement (SR) (ThermoFisher Scientific), 55mM of 2-mercaptoethanol, and 2mM of L-glutamine, and further comprises about 3000 IU/mL of IL-2. In some embodiments, the CTS™OpTmizer™ T-cell Expansion SFM is supplemented with about 3% of the CTS™ Immune Cell Serum Replacement (SR) (ThermoFisher Scientific), 55mM of 2-mercaptoethanol, and 2mM of L- glutamine, and further comprises about 6000 IU/mL of IL-2. In some embodiments, the CTS™OpTmizer™ T-cell Expansion SFM is supplemented with about 3% of the CTS™ Immune Cell Serum Replacement (SR) (ThermoFisher Scientific) and 55mM of 2- mercaptoethanol, and further comprises about 1000 IU/mL to about 8000 IU/mL of IL-2. In some embodiments, the CTS™OpTmizer™ T-cell Expansion SFM is supplemented with about 3% of the CTS™ Immune Cell Serum Replacement (SR) (ThermoFisher Scientific) and 55mM of 2-mercaptoethanol, and further comprises about 3000 IU/mL of IL-2. In some embodiments, the CTS™OpTmizer™ T-cell Expansion SFM is supplemented with about 3% of the CTS™ Immune Cell Serum Replacement (SR) (ThermoFisher Scientific) and 55mM of 2-mercaptoethanol, and further comprises about 1000 IU/mL to about 6000 IU/mL of IL-2. In some embodiments, the CTS™OpTmizer™ T-cell Expansion SFM is supplemented with about 3% of the CTS™ Immune Cell Serum Replacement (SR) (ThermoFisher Scientific) and about 2mM glutamine, and further comprises about 1000 IU/mL to about 8000 IU/mL of IL-2. In some embodiments, the CTS™OpTmizer™ T-cell Expansion SFM is supplemented with about 3% of the CTS™ Immune Cell Serum Replacement (SR) (ThermoFisher Scientific) and about 2mM glutamine, and further comprises about 3000 IU/mL of IL-2. In some embodiments, the CTS™OpTmizer™ T-cell Expansion SFM is supplemented with about 3% of the CTS™ Immune Cell Serum Replacement (SR) (ThermoFisher Scientific) and about 2mM glutamine, and further comprises about 6000 IU/mL of IL-2. In some embodiments, the CTS™ OpTmizer™ T-cell Expansion SFM is supplemented with about 3% of the CTS™ Immune Cell Serum Replacement (SR) (ThermoFisher Scientific) and the final concentration of 2-mercaptoethanol in the media is 55µM. [0057] In some embodiments, the serum-free medium or defined medium is supplemented with glutamine (i.e., GlutaMAX®) at a concentration of from about 0.1mM to about 10mM, DB1/ 149202201.1 82 Attorney Docket No.: 116983-5127-WO 0.5mM to about 9mM, 1mM to about 8mM, 2mM to about 7mM, 3mM to about 6mM, or 4mM to about 5 mM. In some embodiments, the serum-free medium or defined medium is supplemented with glutamine (i.e., GlutaMAX®) at a concentration of about 2mM. [0058] In some embodiments, the serum-free medium or defined medium is supplemented with 2-mercaptoethanol at a concentration of from about 5mM to about 150mM, 10mM to about 140mM, 15mM to about 130mM, 20mM to about 120mM, 25mM to about 110mM, 30mM to about 100mM, 35mM to about 95mM, 40mM to about 90mM, 45mM to about 85mM, 50mM to about 80mM, 55mM to about 75mM, 60mM to about 70mM, or about 65mM. In some embodiments, the serum-free medium or defined medium is supplemented with 2-mercaptoethanol at a concentration of about 55mM. In some embodiments, the final concentration of 2-mercaptoethanol in the media is 55µM. [0059] In some embodiments, the defined media described in International PCT Publication No. WO/1998/030679, which is herein incorporated by reference, are useful in the present invention. In that publication, serum-free eukaryotic cell culture media are described. The serum-free, eukaryotic cell culture medium includes a basal cell culture medium supplemented with a serum-free supplement capable of supporting the growth of cells in serum- free culture. The serum-free eukaryotic cell culture medium supplement comprises or is obtained by combining one or more ingredients selected from the group consisting of one or more albumins or albumin substitutes, one or more amino acids, one or more vitamins, one or more transferrins or transferrin substitutes, one or more antioxidants, one or more insulins or insulin substitutes, one or more collagen precursors, one or more trace elements, and one or more antibiotics. In some embodiments, the defined medium further comprises L- glutamine, sodium bicarbonate and/or beta-mercaptoethanol. In some embodiments, the defined medium comprises an albumin or an albumin substitute and one or more ingredients selected from group consisting of one or more amino acids, one or more vitamins, one or more transferrins or transferrin substitutes, one or more antioxidants, one or more insulins or insulin substitutes, one or more collagen precursors, and one or more trace elements. In some embodiments, the defined medium comprises albumin and one or more ingredients selected from the group consisting of glycine, L- histidine, L-isoleucine, L-methionine, L- phenylalanine, L-proline, L- hydroxyproline, L-serine, L-threonine, L-tryptophan, L-tyrosine, L-valine, thiamine, reduced glutathione, L-ascorbic acid-2-phosphate, iron saturated transferrin, insulin, and compounds containing the trace element moieties Ag+, Al3+, Ba2+, DB1/ 149202201.1 83 Attorney Docket No.: 116983-5127-WO Cd2+, Co2+, Cr3+, Ge4+, Se4+, Br, T, Mn2+, P, Si4+, V5+, Mo6+, Ni2+, Rb+, Sn2+ and Zr4+. In some embodiments, the basal cell media is selected from the group consisting of Dulbecco's Modified Eagle's Medium (DMEM), Minimal Essential Medium (MEM), Basal Medium Eagle (BME), RPMI 1640, F-10, F-12, Minimal Essential Medium (αMEM), Glasgow's Minimal Essential Medium (G-MEM), RPMI growth medium, and Iscove's Modified Dulbecco's Medium. [0060] In some embodiments, the concentration of glycine in the defined medium is in the range of from about 5-200 mg/L, the concentration of L- histidine is about 5-250 mg/L, the concentration of L-isoleucine is about 5-300 mg/L, the concentration of L-methionine is about 5-200 mg/L, the concentration of L-phenylalanine is about 5-400 mg/L, the concentration of L-proline is about 1-1000 mg/L, the concentration of L- hydroxyproline is about 1-45 mg/L, the concentration of L-serine is about 1-250 mg/L, the concentration of L- threonine is about 10-500 mg/L, the concentration of L-tryptophan is about 2-110 mg/L, the concentration of L-tyrosine is about 3-175 mg/L, the concentration of L-valine is about 5-500 mg/L, the concentration of thiamine is about 1-20 mg/L, the concentration of reduced glutathione is about 1-20 mg/L, the concentration of L-ascorbic acid-2-phosphate is about 1- 200 mg/L, the concentration of iron saturated transferrin is about 1-50 mg/L, the concentration of insulin is about 1-100 mg/L, the concentration of sodium selenite is about 0.000001-0.0001 mg/L, and the concentration of albumin (e.g., AlbuMAX® I) is about 5000- 50,000 mg/L. [0061] In some embodiments, the non-trace element moiety ingredients in the defined medium are present in the concentration ranges listed in the column under the heading “Concentration Range in 1X Medium” in Table 4 below. In other embodiments, the non-trace element moiety ingredients in the defined medium are present in the final concentrations listed in the column under the heading “A Preferred Embodiment of the 1X Medium” in Table 4. In other embodiments, the defined medium is a basal cell medium comprising a serum free supplement. In some of these embodiments, the serum free supplement comprises non-trace moiety ingredients of the type and in the concentrations listed in the column under the heading “A Preferred Embodiment in Supplement” in Table 4 below. TABLE 4: Concentrations of Non-Trace Element Moiety Ingredients DB1/ 149202201.1 84 Attorney Docket No.: 116983-5127-WO [00208] In some embodiments, the non-myeloablative lymphodepletion regimen is administered according to Table 6. TABLE 6. Exemplary lymphodepletion and treatment regimen.
[00208] In some embodiments, the non-myeloablative lymphodepletion regimen is administered according to Table 6. TABLE 6. Exemplary lymphodepletion and treatment regimen. [00209] In some embodiments, the non-myeloablative lymphodepletion regimen is administered according to Table 7. DB1/ 149202201.1 132 Attorney Docket No.: 116983-5127-WO TABLE 7. Exemplary lymphodepletion and treatment regimen.
[00209] In some embodiments, the non-myeloablative lymphodepletion regimen is administered according to Table 7. DB1/ 149202201.1 132 Attorney Docket No.: 116983-5127-WO TABLE 7. Exemplary lymphodepletion and treatment regimen. [00210] In some embodiments, the non-myeloablative lymphodepletion regimen is administered according to Table 8. TABLE 8. Exemplary lymphodepletion and treatment regimen.
[00210] In some embodiments, the non-myeloablative lymphodepletion regimen is administered according to Table 8. TABLE 8. Exemplary lymphodepletion and treatment regimen. [00211] In some embodiments, the non-myeloablative lymphodepletion regimen is administered according to Table 9. TABLE 9. Exemplary lymphodepletion and treatment regimen.
[00211] In some embodiments, the non-myeloablative lymphodepletion regimen is administered according to Table 9. TABLE 9. Exemplary lymphodepletion and treatment regimen. [00212] In some embodiments, the non-myeloablative lymphodepletion regimen is administered according to Table 10. TABLE 10. Exemplary lymphodepletion and treatment regimen.
[00212] In some embodiments, the non-myeloablative lymphodepletion regimen is administered according to Table 10. TABLE 10. Exemplary lymphodepletion and treatment regimen. DB1/ 149202201.1 133 Attorney Docket No.: 116983-5127-WO [00213] In some embodiments, the non-myeloablative lymphodepletion regimen is administered according to Table 11. TABLE 11. Exemplary lymphodepletion and treatment regimen.
DB1/ 149202201.1 133 Attorney Docket No.: 116983-5127-WO [00213] In some embodiments, the non-myeloablative lymphodepletion regimen is administered according to Table 11. TABLE 11. Exemplary lymphodepletion and treatment regimen. [00214] In some embodiments, the non-myeloablative lymphodepletion regimen is administered according to Table 12. TABLE 12. Exemplary lymphodepletion and treatment regimen.
[00214] In some embodiments, the non-myeloablative lymphodepletion regimen is administered according to Table 12. TABLE 12. Exemplary lymphodepletion and treatment regimen. [00166] On the day of use, prewarmed required amount of CM1 in 37°C water bath and add 6000 IU/mL IL-2. [00167] Additional supplementation may be performed as needed according to Table 14. TABLE 14. Additional supplementation of CM1, as needed.
[00166] On the day of use, prewarmed required amount of CM1 in 37°C water bath and add 6000 IU/mL IL-2. [00167] Additional supplementation may be performed as needed according to Table 14. TABLE 14. Additional supplementation of CM1, as needed. DB1/ 149202201.1 139 Attorney Docket No.: 116983-5127-WO TABLE 16. Flask Volumes.
DB1/ 149202201.1 139 Attorney Docket No.: 116983-5127-WO TABLE 16. Flask Volumes. [00172] Day 0 CM1 Media Preparation. In the BSC added reagents to RPMI 1640 Media bottle. Added the following reagents t Added per bottle: Heat Inactivated Human AB Serum (100.0 mL); GlutaMax™ (10.0 mL); Gentamicin sulfate, 50 mg/mL (1.0 mL); 2- mercaptoethanol (1.0 mL) [00173] Removed unnecessary materials from BSC. Passed out media reagents from BSC, left Gentamicin Sulfate and HBSS in BSC for Formulated Wash Media preparation. [00174] Thawed IL-2 aliquot. Thawed one 1.1 mL IL-2 aliquot (6x106 IU/mL) (BR71424) until all ice had melted. Recorded IL-2: Lot # and Expiry [00175] Transferred IL-2 stock solution to media. In the BSC, transferred 1.0 mL of IL-2 stock solution to the CM1 Day 0 Media Bottle prepared. Added CM1 Day 0 Media 1 bottle and IL-2 (6x106 IU/mL) 1.0 mL. [00176] Passed G-REX100MCS into BSC. Aseptically passed G-REX100MCS (W3013130) into the BSC. [00177] Pumped all Complete CM1 Day 0 Media into G-REX100MCS flask. Tissue Fragments Conical or GRex100MCS . [00178] Day 0 Tumor Wash Media Preparation. In the BSC, added 5.0 mL Gentamicin (W3009832 or W3012735) to 1 x 500 mL HBSS Media (W3013128) bottle. Added per bottle: HBSS (500.0 mL); Gentamicin sulfate, 50 mg/mL (5.0 mL). Filtered HBSS containing gentamicin prepared through a 1L 0.22-micron filter unit (W1218810). [00179] Day 0 Tumor Processing. Obtained tumor specimen and transferred into suite at 2-8 ºC immediately for processing. Aliquoted tumor wash media. Tumor wash 1 is performed using 8” forceps (W3009771). The tumor is removed from the specimen bottle and transferred to the “Wash 1” dish prepared. This is followed by tumor wash 2 and tumor wash 3. Measured and assessed tumor. Assessed whether > 30% of entire tumor area observed to be necrotic and/or fatty tissue. Clean up dissection if applicable. If tumor was DB1/ 149202201.1 140 Attorney Docket No.: 116983-5127-WO large and >30% of tissue exterior was observed to be necrotic/fatty, performed “clean up dissection” by removing necrotic/fatty tissue while preserving tumor inner structure using a combination of scalpel and/or forceps. Dissect tumor. Using a combination of scalpel and/or forceps, cut the tumor specimen into even, appropriately sized fragments (up to 6 intermediate fragments). Transferred intermediate tumor fragments. Dissected tumor fragments into pieces approximately 3x3x3mm in size. Stored Intermediate Fragments to prevent drying. Repeated intermediate fragment dissection. Determined number of pieces collected. If desirable tissue remains, selected additional favorable tumor pieces from the “favorable intermediate fragments” 6-well plate to fill the drops for a maximum of 50 pieces. [00180] Prepared conical tube. Transferred tumor pieces to 50 mL conical tube. Prepared BSC for G-REX100MCS. Removed G-REX100MCS from incubator. Aseptically passed G-REX100MCS flask into the BSC. Added tumor fragments to G-REX100MCS flask. Evenly distributed pieces. [00181] Incubated G-REX100MCS at the following parameters: Incubated G-REX flask: Temperature LED Display: 37.0±2.0 ºC; CO2 Percentage: 5.0±1.5 %CO2. Calculations: Time of incubation; lower limit = time of incubation + 252 hours; upper limit = time of incubation + 276 hours. [00182] After process was complete, discarded any remaining warmed media and thawed aliquots of IL-2. [00183] Day 11 – Media Preparation. Monitored incubator. Incubator parameters: Temperature LED Display: 37.0±2.0 ºC; CO2 Percentage: 5.0±1.5 %CO2. [00184] Warmed 3× 1000 mL RPMI 1640 Media (W3013112) bottles and 3× 1000 mL AIM-V (W3009501) bottles in an incubator for ≥ 30 minutes. Removed RPMI 1640 Media from incubator. Prepared RPMI 1640 Media. Filter Media. Thawed 3 x 1.1 mL aliquots of IL-2 (6x106 IU/mL) (BR71424). Removed AIM-V Media from the incubator. Add IL-2 to AIM-V. Aseptically transferred a 10 L Labtainer Bag and a repeater pump transfer set into the BSC. [00185] Prepared 10 L Labtainer media bag. Prepared Baxa pump. Prepared 10L Labtainer media bag. Pumped media into 10 L Labtainer. Removed pumpmatic from Labtainer bag. DB1/ 149202201.1 141 Attorney Docket No.: 116983-5127-WO [00186] Mixed media. Gently massaged the bag to mix. Sample media per sample plan. Removed 20.0 mL of media and place in a 50 mL conical tube. Prepared cell count dilution tubes. In the BSC, added 4.5 mL of AIM-V Media that had been labelled with “For Cell Count Dilutions” and lot number to four 15 mL conical tubes. Transferred reagents from the BSC to 2-8°C. Prepared 1 L Transfer Pack. Outside of the BSC weld (per Process Note 5.11) a 1L Transfer Pack to the transfer set attached to the “Complete CM2 Day 11 Media” bag prepared. Prepared feeder cell transfer pack. Incubated Complete CM2 Day 11 Media. [00187] Day 11 - TIL Harvest. Preprocessing table. Incubator parameters: Temperature LED display: 37.0±2.0 ºC; CO2 Percentage: 5.0±1.5 % CO2. Removed G-REX100MCS from incubator. Prepared 300 mL Transfer Pack. Welded transfer packs to G-REX100MCS. [00188] Prepare flask for TIL Harvest and initiation of TIL Harvest. TIL Harvested. Using the GatheRex, transferred the cell suspension through the blood filter into the 300 mL transfer pack. Inspect membrane for adherent cells. [00189] Rinsed flask membrane. Closed clamps on G-REX100MCS. Ensured all clamps are closed. Heat sealed the TIL and the “Supernatant” transfer pack. Calculated volume of TIL suspension. Prepared Supernatant Transfer Pack for Sampling. [00190] Pulled Bac-T Sample. In the BSC, draw up approximately 20.0 mL of supernatant from the 1L “Supernatant” transfer pack and dispense into a sterile 50 mL conical tube. [00191] Inoculated BacT per Sample Plan. Removed a 1.0 mL sample from the 50 mL conical labeled BacT prepared using an appropriately sized syringe and inoculated the anaerobic bottle. [00192] Incubated TIL. Placed TIL transfer pack in incubator until needed. Performed cell counts and calculations. Determined the Average of Viable Cell Concentration and Viability of the cell counts performed. Viability ÷ 2. Viable Cell Concentration ÷ 2. Determined Upper and Lower Limit for counts. Lower Limit: Average of Viable Cell Concentration x 0.9. Upper Limit: Average of Viable Cell Concentration x 1.1. Confirmed both counts within acceptable limits. Determined an average Viable Cell Concentration from all four counts performed. DB1/ 149202201.1 142 Attorney Docket No.: 116983-5127-WO [00193] Adjusted Volume of TIL Suspension: Calculate the adjusted volume of TIL suspension after removal of cell count samples. Total TIL Cell Volume (A). Volume of Cell Count Sample Removed (4.0 mL) (B) Adjusted Total TIL Cell Volume C=A-B. [00194] Calculated Total Viable TIL Cells. Average Viable Cell Concentration*: Total Volume; Total Viable Cells: C = A x B. [00195] Calculation for flow cytometry: if the Total Viable TIL Cell count from was ≥ 4.0x107, calculated the volume to obtain 1.0×107cells for the flow cytometry sample. [00196] Total viable cells required for flow cytometry: 1.0×107cells. Volume of cells required for flow cytometry: Viable cell concentration divided by 1.0×107cells A. [00197] Calculated the volume of TIL suspension equal to 2.0×108viable cells. As needed, calculated the excess volume of TIL cells to remove and removed excess TIL and placed TIL in incubator as needed. Calculated total excess TIL removed, as needed. [00198] Calculated amount of CS-10 media to add to excess TIL cells with the target cell concentration for freezing is 1.0×108 cells/mL. Centrifuged excess TILs, as needed. Observed conical tube and added CS-10. [00199] Filled Vials. Aliquoted 1.0 mL cell suspension, into appropriately sized cryovials. Aliquoted residual volume into appropriately sized cryovial. If volume is ≤0.5 mL, add CS10 to vial until volume is 0.5 mL. [00200] Calculated the volume of cells required to obtain 1x107cells for cryopreservation. Removed sample for cryopreservation. Placed TIL in incubator. [00201] Cryopreservation of sample. Observed conical tube and added CS-10 slowly and record volume of 0.5 mL of CS10 added. [00202] Day 11 - Feeder Cells. Obtained feeder cells. Obtained 3 bags of feeder cells with at least two different lot numbers from LN2 freezer. Kept cells on dry ice until ready to thaw. Prepared water bath or cryotherm. Thawed feeder cells at 37.0 ± 2.0°C in the water bath or cytotherm for ~3-5 minutes or until ice has just disappeared. Removed media from incubator. Pooled thawed feeder cells. Added feeder cells to transfer pack. Dispensed the feeder cells from the syringe into the transfer pack. Mixed pooled feeder cells and labeled transfer pack. DB1/ 149202201.1 143 Attorney Docket No.: 116983-5127-WO [00203] Calculated total volume of feeder cell suspension in transfer pack. Removed cell count samples. Using a separate 3 mL syringe for each sample, pulled 4x1.0 mL cell count samples from Feeder Cell Suspension Transfer Pack using the needless injection port. Aliquoted each sample into the cryovials labeled. Performed cell counts and determine multiplication factors, elected protocols and entered multiplication factors. Determined the average of viable cell concentration and viability of the cell counts performed. Determined upper and lower limit for counts and confirm within limits. [00204] Adjusted volume of feeder cell suspension. Calculated the adjusted volume of feeder cell suspension after removal of cell count samples. Calculated total viable feeder cells. Obtained additional feeder cells as needed. Thawed additional feeder cells as needed. Placed the 4th feeder cell bag into a zip top bag and thaw in a 37.0 ± 2.0°C water bath or cytotherm for ~3-5 minutes and pooled additional feeder cells. Measured volume. Measured the volume of the feeder cells in the syringe and recorded below (B). Calculated the new total volume of feeder cells. Added feeder cells to transfer pack. [00205] Prepared dilutions as needed, adding 4.5 mL of AIM-V Media to four 15 mL conical tubes. Prepared cell counts. Using a separate 3 mL syringe for each sample, removed 4 x 1.0 mL cell count samples from Feeder Cell Suspension transfer pack, using the needless injection port. Performed cell counts and calculations. Determined an average viable cell concentration from all four counts performed. Adjusted volume of feeder cell suspension and calculated the adjusted volume of feeder cell suspension after removal of cell count samples. Total Feeder Cell Volume minues 4.0 mL removed. Calculated the volume of Feeder Cell Suspension that was required to obtain 5x109viable feeder cells. Calculated excess feeder cell volume. Calculated the volume of excess feeder cells to remove. Removed excess feeder cells. [00206] Using a 1.0 mL syringe and 16G needle, drew up 0.15 mL of OKT3 and added OKT3. Heat sealed the feeder cell suspension transfer pack. [00207] Day 11 G-REX Fill and Seed Set up G-REX500MCS. Removed “Complete CM2 Day 11 Media”, from incubator and pumped media into G-REX500MCS. Pumped 4.5L of media into the G-REX500MCS, filling to the line marked on the flask. Heat sealed and incubated flask as needed. Welded the Feeder Cell suspension transfer pack to the G- REX500MCS. Added Feeder Cells to G-REX500MCS. Heat sealed. Welded the TIL DB1/ 149202201.1 144 Attorney Docket No.: 116983-5127-WO Suspension transfer pack to the flask. Added TIL to G-REX500MCS. Heat sealed. Incubated G-REX500MCS at 37.0±2.0 ºC, CO2 Percentage: 5.0±1.5 %CO2. [00208] Calculated incubation window. Performed calculations to determine the proper time to remove G-REX500MCS from incubator on Day 16. Lower limit: Time of incubation + 108 hours. Upper limit: Time of incubation + 132 hours. [00209] Day 11 Excess TIL Cryopreservation. Applicable: Froze Excess TIL Vials. Verified the CRF has been set up prior to freeze. Perform Cryopreservation. Transferred vials from Controlled Rate Freezer to the appropriate storage. Upon completion of freeze, transfer vials from CRF to the appropriate storage container. Transferred vials to appropriate storage. Recorded storage location in LN2. [00210] Day 16 Media Preparation. Pre-warmed AIM-V Media. Calculated time Media was warmed for media bags 1, 2, and 3. Ensured all bags have been warmed for a duration between 12 and 24 hours. Setup 10L Labtainer for Supernatant. Attached the larger diameter end of a fluid pump transfer set to one of the female ports of a 10L Labtainer bag using the Luer connectors. Setup 10L Labtainer for Supernatant and label. Setup 10L Labtainer for Supernatant. Ensure all clamps were closed prior to removing from the BSC. NOTE: Supernatant bag was used during TIL Harvest, which may be performed concurrently with media preparation. [00211] Thawed IL-2. Thawed 5×1.1 mL aliquots of IL-2 (6×106 IU/mL) (BR71424) per bag of CTS AIM V media until all ice had melted. Aliquoted 100.0 mL GlutaMax™. Added IL-2 to GlutaMax™. Prepared CTS AIM V media bag for formulation. Prepared CTS AIM V media bag for formulation. Stage Baxa Pump. Prepared to formulate media. Pumped GlutaMax™ +IL-2 into bag. Monitored parameters: Temperature LED Display: 37.0±2.0 ºC, CO2 Percentage: 5.0±1.5% CO2. Warmed Complete CM4 Day 16 Media. Prepared Dilutions. [00212] Day 16 REP Spilt. Monitored Incubator parameters: Temperature LED display: 37.0±2.0 ºC, CO2 Percentage: 5.0±1.5 %CO2. Removed G-REX500MCS from the incubator. Prepared and labeled 1 L Transfer Pack as TIL Suspension and weighed 1L. [00213] Volume Reduction of G-REX500MCS. Transferred ~4.5L of culture supernatant from the G-REX500MCS to the 10L Labtainer. DB1/ 149202201.1 145 Attorney Docket No.: 116983-5127-WO [00214] Prepared flask for TIL harvest. After removal of the supernatant, closed all clamps to the red line. [00215] Initiation of TIL Harvest. Vigorously tap flask and swirl media to release cells and ensure all cells have detached. [00216] TIL Harvest. Released all clamps leading to the TIL suspension transfer pack. Using the GatheRex transferred the cell suspension into the TIL Suspension transfer pack. NOTE: Be sure to maintain the tilted edge until all cells and media are collected. Inspected membrane for adherent cells. Rinsed flask membrane. Closed clamps on G-REX500MCS. Heat sealed the Transfer Pack containing the TIL. Heat sealed the 10L Labtainer containing the supernatant. Recorded weight of Transfer Pack with cell suspension and calculate the volume suspension. Prepared transfer pack for sample removal. Removed testing samples from cell supernatant. [00217] Sterility & BacT testing sampling. Removed a 1.0 mL sample from the 15 mL conical labeled BacT prepared. Removed Cell Count Samples. In the BSC, using separate 3 mL syringes for each sample, removed 4x1.0 mL cell count samples from “TIL Suspension” transfer pack. [00218] Removed mycoplasma samples. Using a 3 mL syringe, removed 1.0 mL from TIL Suspension transfer pack and place into 15 mL conical labeled “Mycoplasma diluent” prepared. [00219] Prepared transfer pack for seeding. Placed TIL in incubator. Removed cell suspension from the BSC and place in incubator until needed. Performed cell counts and calculations. Diluted cell count samples initially by adding 0.5 mL of cell suspension into 4.5 mL of AIM-V media prepared which gave a 1:10 dilution. Determined the average of viable cell concentration and viability of the cell counts performed. Determined upper and lower limit for counts. Note: dilution may be adjusted according based off the expected concentration of cells. Determined an average viable cell concentration from all four counts performed. Adjusted volume of TIL suspension. Calculated the adjusted volume of TIL suspension after removal of cell count samples. Total TIL cell volume minus 5.0 mL removed for testing. [00220] Calculated total viable TIL cells. Calculated the total number of flasks to seed. NOTE: The maximum number of G-REX500MCS flasks to seed was five. If the calculated DB1/ 149202201.1 146 Attorney Docket No.: 116983-5127-WO number of flasks to seed exceeded five, only five were seeded using the entire volume of cell suspension available. [00221] Calculate number of flasks for subculture. Calculated the number of media bags required in addition to the bag prepared. Prepared one 10L bag of “CM4 Day 16 Media” for every two G-REX-500M flask needed as calculated. Proceeded to seed the first GREX- 500M flask(s) while additional media is prepared and warmed. Prepared and warmed the calculated number of additional media bags determined. Filled G-REX500MCS. Prepared to pump media and pumped 4.5L of media into G-REX500MCS. Heat Sealed. Repeated Fill. Incubated flask. Calculated the target volume of TIL suspension to add to the new G- REX500MCS flasks. If the calculated number of flasks exceeds five only five will be seeded, USING THE ENTIRE VOLUME OF CELL SUSPENSION. Prepared Flasks for Seeding. Removed G-REX500MCS from the incubator. Prepared G-REX500MCS for pumping. Closed all clamps on except large filter line. Removed TIL from incubator. Prepared cell suspension for seeding. Sterile welded (per Process Note 5.11) “TIL Suspension” transfer pack to pump inlet line. Placed TIL suspension bag on a scale. [00222] Seeded flask with TIL Suspension. Pump the volume of TIL suspension calculated into flask. Heat sealed. Filled remaining flasks. [00223] Monitored Incubator. Incubator parameters: Temperature LED Display: 37.0±2.0 ºC, CO2 Percentage: 5.0±1.5 % CO2. Incubated Flasks. [00224] Determined the time range to remove G-REX500MCS from incubator on Day 22. [00225] Day 22 Wash Buffer Preparation. Prepared 10 L Labtainer Bag. In BSC, attach a 4” plasma transfer set to a 10L Labtainer Bag via luer connection. Prepared 10 L Labtainer Bag. Closed all clamps before transferring out of the BSC. NOTE: Prepared one 10L Labtainer Bag for every two G-REX500MCS flasks to be harvested. Pumped Plasmalyte into 3000 mL bag and removed air from 3000 mL Origen bag by reversing the pump and manipulating the position of the bag. Added human albumin 25% to 3000 mL Bag. Obtain a final volumeof 120.0 mL of human albumin 25%. [00226] Prepared IL-2 diluent. Using a 10 mL syringe, removed 5.0 mL of LOVO Wash Buffer using the needleless injection port on the LOVO Wash Buffer bag. Dispensed LOVO wash buffer into a 50 mL conical tube. DB1/ 149202201.1 147 Attorney Docket No.: 116983-5127-WO [00227] CRF blank bag LOVO wash buffer aliquotted. Using a 100 mL syringe, drew up 70.0 mL of LOVO Wash Buffer from the needleless injection port. [00228] Thawed one 1.1 mL of IL-2 (6x106 IU/mL), until all ice has melted. Added 50 µL IL-2 stock (6×106 IU/mL) to the 50 mL conical tube labeled “IL-2 Diluent.” [00229] Cryopreservation preparation. Placed 5 cryo-cassettes at 2-8°C to precondition them for final product cryopreservation. [00230] Prepared cell count dilutions. In the BSC, added 4.5 mL of AIM-V Media that has been labelled with lot number and “For Cell Count Dilutions” to 4 separate 15 mL conical tubes. Prepared cell counts. Labeled 4 cryovials with vial number (1-4). Kept vials under BSC to be used. [00231] Day 22 TIL Harvest. Monitored Incubator. Incubator Parameters Temperature LED display: 37 ± 2.0°C, CO2 Percentage: 5%±1.5%. Removed G-REX500MCS Flasks from Incubator. Prepared TIL collection bag and labeled. Sealed off extra connections. Volume Reduction: Transferred ~4.5L of supernatant from the G-REX500MCS to the Supernatant bag. [00232] Prepared flask for TIL harvest. Initiated collection of TIL. Vigorously tap flask and swirl media to release cells. Ensure all cells have detached. Initiated collection of TIL. Released all clamps leading to the TIL suspension collection bag. TIL Harvest. Using the GatheRex, transferred the TIL suspension into the 3000 mL collection bag. Inspect membrane for adherent cells. Rinsed flask membrane. Closed clamps on G- Rex500MCS and ensured all clamps are closed. Transferred cell suspension into LOVO source bag. Closed all clamps. Heat Sealed. Removed 4x1.0 mL Cell Counts Samples [00233] Performed Cell Counts. Performed cell counts and calculations utilizing NC- 200 and Process Note 5.14. Diluted cell count samples initially by adding 0.5 mL of cell suspension into 4.5 mL of AIM-V media prepared. This gave a 1:10 dilution. Determined the average viability, viable cell concentration, and total nucleated cell concentration of the cell counts performed. Determined Upper and Lower Limit for counts. Determined the average viability, viable cell concentration, and total nucleated cell concentration of the cell counts performed. Weighed LOVO source bag. Calculated total viable TIL Cells. Calculated total nucleated cells. DB1/ 149202201.1 148 Attorney Docket No.: 116983-5127-WO [00234] Prepared Mycoplasma Diluent. Removed 10.0 mL from one supernatant bag via luer sample port and placed in a 15 mL conical. [00235] Performed “TIL G-REX Harvest” protocol and determined the final product target volume. Loaded disposable kit. Removed filtrate bag. Entered Filtrate capacity. Placed Filtrate container on benchtop. Attached PlasmaLyte. Verified that the PlasmaLyte was attached and observed that the PlasmaLyte is moving. Attached Source container to tubing and verified Source container was attached. Confirmed PlasmaLyte was moving. [00236] Final Formulation and Fill. Target volume/bag calculation. Calculated volume of CS-10 and LOVO wash buffer to formulate blank bag. Prepared CRF Blank. [00237] Calculated the volume of IL-2 to add to the Final Product. Final IL-2 Concentration desired (IU/mL) – 300IU/mL. IL-2 working stock: 6 × 104 IU/mL. Assembled connect apparatus. Sterile welded a 4S-4M60 to a CC2 cell connection. Sterile welded the CS750 cryobags to the harness prepared. Welded CS-10 bags to spikes of the 4S-4M60. Prepared TIL with IL-2. Using an appropriately sized syringe, removed amount of IL-2 determined from the “IL-26x104” aliquot. Labeled forumlated TIL Bag. Added the formulated TIL bag to the apparatus. Added CS10. Switched Syringes. Drew ~10 mL of air into a 100 mL syringe and replaced the 60 mL syringe on the apparatus. Added CS10. Prepared CS-750 bags. Dispensed cells. [00238] Removed air from final product bags and take retain. Once the last final product bag was filled, closed all clamps. Drew 10 mL of air into a new 100 mL syringe and replace the syringe on the apparatus. Dispensed retain into a 50 mL conical tube and label tube as “Retain” and lot number. Repeat air removal step for each bag. [00239] Prepared final product for cryopreservation, including visual inspection. Held the cryobags on cold pack or at 2-8°C until cryopreservation. [00240] Removed cell count sample. Using an appropriately sized pipette, remove 2.0 mL of retain and place in a 15 mL conical tube to be used for cell counts. Performed cell counts and calculations. NOTE: Diluted only one sample to appropriate dilution to verify dilution is sufficient. Diluted additional samples to appropriate dilution factor and proceed with counts. Determined the Average of Viable Cell Concentration and Viability of the cell counts performed. Determined Upper and Lower Limit for counts. NOTE: Dilution may be adjusted according based off the expected concentration of cells. Determined the Average of DB1/ 149202201.1 149 Attorney Docket No.: 116983-5127-WO Viable Cell Concentration and Viability. Determined Upper and Lower Limit for counts. Calculated IFN-γ. Heat Sealed Final Product bags. [00241] Labeled and collected samples per exemplary sample plan below. TABLE 17. Sample plan.
[00172] Day 0 CM1 Media Preparation. In the BSC added reagents to RPMI 1640 Media bottle. Added the following reagents t Added per bottle: Heat Inactivated Human AB Serum (100.0 mL); GlutaMax™ (10.0 mL); Gentamicin sulfate, 50 mg/mL (1.0 mL); 2- mercaptoethanol (1.0 mL) [00173] Removed unnecessary materials from BSC. Passed out media reagents from BSC, left Gentamicin Sulfate and HBSS in BSC for Formulated Wash Media preparation. [00174] Thawed IL-2 aliquot. Thawed one 1.1 mL IL-2 aliquot (6x106 IU/mL) (BR71424) until all ice had melted. Recorded IL-2: Lot # and Expiry [00175] Transferred IL-2 stock solution to media. In the BSC, transferred 1.0 mL of IL-2 stock solution to the CM1 Day 0 Media Bottle prepared. Added CM1 Day 0 Media 1 bottle and IL-2 (6x106 IU/mL) 1.0 mL. [00176] Passed G-REX100MCS into BSC. Aseptically passed G-REX100MCS (W3013130) into the BSC. [00177] Pumped all Complete CM1 Day 0 Media into G-REX100MCS flask. Tissue Fragments Conical or GRex100MCS . [00178] Day 0 Tumor Wash Media Preparation. In the BSC, added 5.0 mL Gentamicin (W3009832 or W3012735) to 1 x 500 mL HBSS Media (W3013128) bottle. Added per bottle: HBSS (500.0 mL); Gentamicin sulfate, 50 mg/mL (5.0 mL). Filtered HBSS containing gentamicin prepared through a 1L 0.22-micron filter unit (W1218810). [00179] Day 0 Tumor Processing. Obtained tumor specimen and transferred into suite at 2-8 ºC immediately for processing. Aliquoted tumor wash media. Tumor wash 1 is performed using 8” forceps (W3009771). The tumor is removed from the specimen bottle and transferred to the “Wash 1” dish prepared. This is followed by tumor wash 2 and tumor wash 3. Measured and assessed tumor. Assessed whether > 30% of entire tumor area observed to be necrotic and/or fatty tissue. Clean up dissection if applicable. If tumor was DB1/ 149202201.1 140 Attorney Docket No.: 116983-5127-WO large and >30% of tissue exterior was observed to be necrotic/fatty, performed “clean up dissection” by removing necrotic/fatty tissue while preserving tumor inner structure using a combination of scalpel and/or forceps. Dissect tumor. Using a combination of scalpel and/or forceps, cut the tumor specimen into even, appropriately sized fragments (up to 6 intermediate fragments). Transferred intermediate tumor fragments. Dissected tumor fragments into pieces approximately 3x3x3mm in size. Stored Intermediate Fragments to prevent drying. Repeated intermediate fragment dissection. Determined number of pieces collected. If desirable tissue remains, selected additional favorable tumor pieces from the “favorable intermediate fragments” 6-well plate to fill the drops for a maximum of 50 pieces. [00180] Prepared conical tube. Transferred tumor pieces to 50 mL conical tube. Prepared BSC for G-REX100MCS. Removed G-REX100MCS from incubator. Aseptically passed G-REX100MCS flask into the BSC. Added tumor fragments to G-REX100MCS flask. Evenly distributed pieces. [00181] Incubated G-REX100MCS at the following parameters: Incubated G-REX flask: Temperature LED Display: 37.0±2.0 ºC; CO2 Percentage: 5.0±1.5 %CO2. Calculations: Time of incubation; lower limit = time of incubation + 252 hours; upper limit = time of incubation + 276 hours. [00182] After process was complete, discarded any remaining warmed media and thawed aliquots of IL-2. [00183] Day 11 – Media Preparation. Monitored incubator. Incubator parameters: Temperature LED Display: 37.0±2.0 ºC; CO2 Percentage: 5.0±1.5 %CO2. [00184] Warmed 3× 1000 mL RPMI 1640 Media (W3013112) bottles and 3× 1000 mL AIM-V (W3009501) bottles in an incubator for ≥ 30 minutes. Removed RPMI 1640 Media from incubator. Prepared RPMI 1640 Media. Filter Media. Thawed 3 x 1.1 mL aliquots of IL-2 (6x106 IU/mL) (BR71424). Removed AIM-V Media from the incubator. Add IL-2 to AIM-V. Aseptically transferred a 10 L Labtainer Bag and a repeater pump transfer set into the BSC. [00185] Prepared 10 L Labtainer media bag. Prepared Baxa pump. Prepared 10L Labtainer media bag. Pumped media into 10 L Labtainer. Removed pumpmatic from Labtainer bag. DB1/ 149202201.1 141 Attorney Docket No.: 116983-5127-WO [00186] Mixed media. Gently massaged the bag to mix. Sample media per sample plan. Removed 20.0 mL of media and place in a 50 mL conical tube. Prepared cell count dilution tubes. In the BSC, added 4.5 mL of AIM-V Media that had been labelled with “For Cell Count Dilutions” and lot number to four 15 mL conical tubes. Transferred reagents from the BSC to 2-8°C. Prepared 1 L Transfer Pack. Outside of the BSC weld (per Process Note 5.11) a 1L Transfer Pack to the transfer set attached to the “Complete CM2 Day 11 Media” bag prepared. Prepared feeder cell transfer pack. Incubated Complete CM2 Day 11 Media. [00187] Day 11 - TIL Harvest. Preprocessing table. Incubator parameters: Temperature LED display: 37.0±2.0 ºC; CO2 Percentage: 5.0±1.5 % CO2. Removed G-REX100MCS from incubator. Prepared 300 mL Transfer Pack. Welded transfer packs to G-REX100MCS. [00188] Prepare flask for TIL Harvest and initiation of TIL Harvest. TIL Harvested. Using the GatheRex, transferred the cell suspension through the blood filter into the 300 mL transfer pack. Inspect membrane for adherent cells. [00189] Rinsed flask membrane. Closed clamps on G-REX100MCS. Ensured all clamps are closed. Heat sealed the TIL and the “Supernatant” transfer pack. Calculated volume of TIL suspension. Prepared Supernatant Transfer Pack for Sampling. [00190] Pulled Bac-T Sample. In the BSC, draw up approximately 20.0 mL of supernatant from the 1L “Supernatant” transfer pack and dispense into a sterile 50 mL conical tube. [00191] Inoculated BacT per Sample Plan. Removed a 1.0 mL sample from the 50 mL conical labeled BacT prepared using an appropriately sized syringe and inoculated the anaerobic bottle. [00192] Incubated TIL. Placed TIL transfer pack in incubator until needed. Performed cell counts and calculations. Determined the Average of Viable Cell Concentration and Viability of the cell counts performed. Viability ÷ 2. Viable Cell Concentration ÷ 2. Determined Upper and Lower Limit for counts. Lower Limit: Average of Viable Cell Concentration x 0.9. Upper Limit: Average of Viable Cell Concentration x 1.1. Confirmed both counts within acceptable limits. Determined an average Viable Cell Concentration from all four counts performed. DB1/ 149202201.1 142 Attorney Docket No.: 116983-5127-WO [00193] Adjusted Volume of TIL Suspension: Calculate the adjusted volume of TIL suspension after removal of cell count samples. Total TIL Cell Volume (A). Volume of Cell Count Sample Removed (4.0 mL) (B) Adjusted Total TIL Cell Volume C=A-B. [00194] Calculated Total Viable TIL Cells. Average Viable Cell Concentration*: Total Volume; Total Viable Cells: C = A x B. [00195] Calculation for flow cytometry: if the Total Viable TIL Cell count from was ≥ 4.0x107, calculated the volume to obtain 1.0×107cells for the flow cytometry sample. [00196] Total viable cells required for flow cytometry: 1.0×107cells. Volume of cells required for flow cytometry: Viable cell concentration divided by 1.0×107cells A. [00197] Calculated the volume of TIL suspension equal to 2.0×108viable cells. As needed, calculated the excess volume of TIL cells to remove and removed excess TIL and placed TIL in incubator as needed. Calculated total excess TIL removed, as needed. [00198] Calculated amount of CS-10 media to add to excess TIL cells with the target cell concentration for freezing is 1.0×108 cells/mL. Centrifuged excess TILs, as needed. Observed conical tube and added CS-10. [00199] Filled Vials. Aliquoted 1.0 mL cell suspension, into appropriately sized cryovials. Aliquoted residual volume into appropriately sized cryovial. If volume is ≤0.5 mL, add CS10 to vial until volume is 0.5 mL. [00200] Calculated the volume of cells required to obtain 1x107cells for cryopreservation. Removed sample for cryopreservation. Placed TIL in incubator. [00201] Cryopreservation of sample. Observed conical tube and added CS-10 slowly and record volume of 0.5 mL of CS10 added. [00202] Day 11 - Feeder Cells. Obtained feeder cells. Obtained 3 bags of feeder cells with at least two different lot numbers from LN2 freezer. Kept cells on dry ice until ready to thaw. Prepared water bath or cryotherm. Thawed feeder cells at 37.0 ± 2.0°C in the water bath or cytotherm for ~3-5 minutes or until ice has just disappeared. Removed media from incubator. Pooled thawed feeder cells. Added feeder cells to transfer pack. Dispensed the feeder cells from the syringe into the transfer pack. Mixed pooled feeder cells and labeled transfer pack. DB1/ 149202201.1 143 Attorney Docket No.: 116983-5127-WO [00203] Calculated total volume of feeder cell suspension in transfer pack. Removed cell count samples. Using a separate 3 mL syringe for each sample, pulled 4x1.0 mL cell count samples from Feeder Cell Suspension Transfer Pack using the needless injection port. Aliquoted each sample into the cryovials labeled. Performed cell counts and determine multiplication factors, elected protocols and entered multiplication factors. Determined the average of viable cell concentration and viability of the cell counts performed. Determined upper and lower limit for counts and confirm within limits. [00204] Adjusted volume of feeder cell suspension. Calculated the adjusted volume of feeder cell suspension after removal of cell count samples. Calculated total viable feeder cells. Obtained additional feeder cells as needed. Thawed additional feeder cells as needed. Placed the 4th feeder cell bag into a zip top bag and thaw in a 37.0 ± 2.0°C water bath or cytotherm for ~3-5 minutes and pooled additional feeder cells. Measured volume. Measured the volume of the feeder cells in the syringe and recorded below (B). Calculated the new total volume of feeder cells. Added feeder cells to transfer pack. [00205] Prepared dilutions as needed, adding 4.5 mL of AIM-V Media to four 15 mL conical tubes. Prepared cell counts. Using a separate 3 mL syringe for each sample, removed 4 x 1.0 mL cell count samples from Feeder Cell Suspension transfer pack, using the needless injection port. Performed cell counts and calculations. Determined an average viable cell concentration from all four counts performed. Adjusted volume of feeder cell suspension and calculated the adjusted volume of feeder cell suspension after removal of cell count samples. Total Feeder Cell Volume minues 4.0 mL removed. Calculated the volume of Feeder Cell Suspension that was required to obtain 5x109viable feeder cells. Calculated excess feeder cell volume. Calculated the volume of excess feeder cells to remove. Removed excess feeder cells. [00206] Using a 1.0 mL syringe and 16G needle, drew up 0.15 mL of OKT3 and added OKT3. Heat sealed the feeder cell suspension transfer pack. [00207] Day 11 G-REX Fill and Seed Set up G-REX500MCS. Removed “Complete CM2 Day 11 Media”, from incubator and pumped media into G-REX500MCS. Pumped 4.5L of media into the G-REX500MCS, filling to the line marked on the flask. Heat sealed and incubated flask as needed. Welded the Feeder Cell suspension transfer pack to the G- REX500MCS. Added Feeder Cells to G-REX500MCS. Heat sealed. Welded the TIL DB1/ 149202201.1 144 Attorney Docket No.: 116983-5127-WO Suspension transfer pack to the flask. Added TIL to G-REX500MCS. Heat sealed. Incubated G-REX500MCS at 37.0±2.0 ºC, CO2 Percentage: 5.0±1.5 %CO2. [00208] Calculated incubation window. Performed calculations to determine the proper time to remove G-REX500MCS from incubator on Day 16. Lower limit: Time of incubation + 108 hours. Upper limit: Time of incubation + 132 hours. [00209] Day 11 Excess TIL Cryopreservation. Applicable: Froze Excess TIL Vials. Verified the CRF has been set up prior to freeze. Perform Cryopreservation. Transferred vials from Controlled Rate Freezer to the appropriate storage. Upon completion of freeze, transfer vials from CRF to the appropriate storage container. Transferred vials to appropriate storage. Recorded storage location in LN2. [00210] Day 16 Media Preparation. Pre-warmed AIM-V Media. Calculated time Media was warmed for media bags 1, 2, and 3. Ensured all bags have been warmed for a duration between 12 and 24 hours. Setup 10L Labtainer for Supernatant. Attached the larger diameter end of a fluid pump transfer set to one of the female ports of a 10L Labtainer bag using the Luer connectors. Setup 10L Labtainer for Supernatant and label. Setup 10L Labtainer for Supernatant. Ensure all clamps were closed prior to removing from the BSC. NOTE: Supernatant bag was used during TIL Harvest, which may be performed concurrently with media preparation. [00211] Thawed IL-2. Thawed 5×1.1 mL aliquots of IL-2 (6×106 IU/mL) (BR71424) per bag of CTS AIM V media until all ice had melted. Aliquoted 100.0 mL GlutaMax™. Added IL-2 to GlutaMax™. Prepared CTS AIM V media bag for formulation. Prepared CTS AIM V media bag for formulation. Stage Baxa Pump. Prepared to formulate media. Pumped GlutaMax™ +IL-2 into bag. Monitored parameters: Temperature LED Display: 37.0±2.0 ºC, CO2 Percentage: 5.0±1.5% CO2. Warmed Complete CM4 Day 16 Media. Prepared Dilutions. [00212] Day 16 REP Spilt. Monitored Incubator parameters: Temperature LED display: 37.0±2.0 ºC, CO2 Percentage: 5.0±1.5 %CO2. Removed G-REX500MCS from the incubator. Prepared and labeled 1 L Transfer Pack as TIL Suspension and weighed 1L. [00213] Volume Reduction of G-REX500MCS. Transferred ~4.5L of culture supernatant from the G-REX500MCS to the 10L Labtainer. DB1/ 149202201.1 145 Attorney Docket No.: 116983-5127-WO [00214] Prepared flask for TIL harvest. After removal of the supernatant, closed all clamps to the red line. [00215] Initiation of TIL Harvest. Vigorously tap flask and swirl media to release cells and ensure all cells have detached. [00216] TIL Harvest. Released all clamps leading to the TIL suspension transfer pack. Using the GatheRex transferred the cell suspension into the TIL Suspension transfer pack. NOTE: Be sure to maintain the tilted edge until all cells and media are collected. Inspected membrane for adherent cells. Rinsed flask membrane. Closed clamps on G-REX500MCS. Heat sealed the Transfer Pack containing the TIL. Heat sealed the 10L Labtainer containing the supernatant. Recorded weight of Transfer Pack with cell suspension and calculate the volume suspension. Prepared transfer pack for sample removal. Removed testing samples from cell supernatant. [00217] Sterility & BacT testing sampling. Removed a 1.0 mL sample from the 15 mL conical labeled BacT prepared. Removed Cell Count Samples. In the BSC, using separate 3 mL syringes for each sample, removed 4x1.0 mL cell count samples from “TIL Suspension” transfer pack. [00218] Removed mycoplasma samples. Using a 3 mL syringe, removed 1.0 mL from TIL Suspension transfer pack and place into 15 mL conical labeled “Mycoplasma diluent” prepared. [00219] Prepared transfer pack for seeding. Placed TIL in incubator. Removed cell suspension from the BSC and place in incubator until needed. Performed cell counts and calculations. Diluted cell count samples initially by adding 0.5 mL of cell suspension into 4.5 mL of AIM-V media prepared which gave a 1:10 dilution. Determined the average of viable cell concentration and viability of the cell counts performed. Determined upper and lower limit for counts. Note: dilution may be adjusted according based off the expected concentration of cells. Determined an average viable cell concentration from all four counts performed. Adjusted volume of TIL suspension. Calculated the adjusted volume of TIL suspension after removal of cell count samples. Total TIL cell volume minus 5.0 mL removed for testing. [00220] Calculated total viable TIL cells. Calculated the total number of flasks to seed. NOTE: The maximum number of G-REX500MCS flasks to seed was five. If the calculated DB1/ 149202201.1 146 Attorney Docket No.: 116983-5127-WO number of flasks to seed exceeded five, only five were seeded using the entire volume of cell suspension available. [00221] Calculate number of flasks for subculture. Calculated the number of media bags required in addition to the bag prepared. Prepared one 10L bag of “CM4 Day 16 Media” for every two G-REX-500M flask needed as calculated. Proceeded to seed the first GREX- 500M flask(s) while additional media is prepared and warmed. Prepared and warmed the calculated number of additional media bags determined. Filled G-REX500MCS. Prepared to pump media and pumped 4.5L of media into G-REX500MCS. Heat Sealed. Repeated Fill. Incubated flask. Calculated the target volume of TIL suspension to add to the new G- REX500MCS flasks. If the calculated number of flasks exceeds five only five will be seeded, USING THE ENTIRE VOLUME OF CELL SUSPENSION. Prepared Flasks for Seeding. Removed G-REX500MCS from the incubator. Prepared G-REX500MCS for pumping. Closed all clamps on except large filter line. Removed TIL from incubator. Prepared cell suspension for seeding. Sterile welded (per Process Note 5.11) “TIL Suspension” transfer pack to pump inlet line. Placed TIL suspension bag on a scale. [00222] Seeded flask with TIL Suspension. Pump the volume of TIL suspension calculated into flask. Heat sealed. Filled remaining flasks. [00223] Monitored Incubator. Incubator parameters: Temperature LED Display: 37.0±2.0 ºC, CO2 Percentage: 5.0±1.5 % CO2. Incubated Flasks. [00224] Determined the time range to remove G-REX500MCS from incubator on Day 22. [00225] Day 22 Wash Buffer Preparation. Prepared 10 L Labtainer Bag. In BSC, attach a 4” plasma transfer set to a 10L Labtainer Bag via luer connection. Prepared 10 L Labtainer Bag. Closed all clamps before transferring out of the BSC. NOTE: Prepared one 10L Labtainer Bag for every two G-REX500MCS flasks to be harvested. Pumped Plasmalyte into 3000 mL bag and removed air from 3000 mL Origen bag by reversing the pump and manipulating the position of the bag. Added human albumin 25% to 3000 mL Bag. Obtain a final volumeof 120.0 mL of human albumin 25%. [00226] Prepared IL-2 diluent. Using a 10 mL syringe, removed 5.0 mL of LOVO Wash Buffer using the needleless injection port on the LOVO Wash Buffer bag. Dispensed LOVO wash buffer into a 50 mL conical tube. DB1/ 149202201.1 147 Attorney Docket No.: 116983-5127-WO [00227] CRF blank bag LOVO wash buffer aliquotted. Using a 100 mL syringe, drew up 70.0 mL of LOVO Wash Buffer from the needleless injection port. [00228] Thawed one 1.1 mL of IL-2 (6x106 IU/mL), until all ice has melted. Added 50 µL IL-2 stock (6×106 IU/mL) to the 50 mL conical tube labeled “IL-2 Diluent.” [00229] Cryopreservation preparation. Placed 5 cryo-cassettes at 2-8°C to precondition them for final product cryopreservation. [00230] Prepared cell count dilutions. In the BSC, added 4.5 mL of AIM-V Media that has been labelled with lot number and “For Cell Count Dilutions” to 4 separate 15 mL conical tubes. Prepared cell counts. Labeled 4 cryovials with vial number (1-4). Kept vials under BSC to be used. [00231] Day 22 TIL Harvest. Monitored Incubator. Incubator Parameters Temperature LED display: 37 ± 2.0°C, CO2 Percentage: 5%±1.5%. Removed G-REX500MCS Flasks from Incubator. Prepared TIL collection bag and labeled. Sealed off extra connections. Volume Reduction: Transferred ~4.5L of supernatant from the G-REX500MCS to the Supernatant bag. [00232] Prepared flask for TIL harvest. Initiated collection of TIL. Vigorously tap flask and swirl media to release cells. Ensure all cells have detached. Initiated collection of TIL. Released all clamps leading to the TIL suspension collection bag. TIL Harvest. Using the GatheRex, transferred the TIL suspension into the 3000 mL collection bag. Inspect membrane for adherent cells. Rinsed flask membrane. Closed clamps on G- Rex500MCS and ensured all clamps are closed. Transferred cell suspension into LOVO source bag. Closed all clamps. Heat Sealed. Removed 4x1.0 mL Cell Counts Samples [00233] Performed Cell Counts. Performed cell counts and calculations utilizing NC- 200 and Process Note 5.14. Diluted cell count samples initially by adding 0.5 mL of cell suspension into 4.5 mL of AIM-V media prepared. This gave a 1:10 dilution. Determined the average viability, viable cell concentration, and total nucleated cell concentration of the cell counts performed. Determined Upper and Lower Limit for counts. Determined the average viability, viable cell concentration, and total nucleated cell concentration of the cell counts performed. Weighed LOVO source bag. Calculated total viable TIL Cells. Calculated total nucleated cells. DB1/ 149202201.1 148 Attorney Docket No.: 116983-5127-WO [00234] Prepared Mycoplasma Diluent. Removed 10.0 mL from one supernatant bag via luer sample port and placed in a 15 mL conical. [00235] Performed “TIL G-REX Harvest” protocol and determined the final product target volume. Loaded disposable kit. Removed filtrate bag. Entered Filtrate capacity. Placed Filtrate container on benchtop. Attached PlasmaLyte. Verified that the PlasmaLyte was attached and observed that the PlasmaLyte is moving. Attached Source container to tubing and verified Source container was attached. Confirmed PlasmaLyte was moving. [00236] Final Formulation and Fill. Target volume/bag calculation. Calculated volume of CS-10 and LOVO wash buffer to formulate blank bag. Prepared CRF Blank. [00237] Calculated the volume of IL-2 to add to the Final Product. Final IL-2 Concentration desired (IU/mL) – 300IU/mL. IL-2 working stock: 6 × 104 IU/mL. Assembled connect apparatus. Sterile welded a 4S-4M60 to a CC2 cell connection. Sterile welded the CS750 cryobags to the harness prepared. Welded CS-10 bags to spikes of the 4S-4M60. Prepared TIL with IL-2. Using an appropriately sized syringe, removed amount of IL-2 determined from the “IL-26x104” aliquot. Labeled forumlated TIL Bag. Added the formulated TIL bag to the apparatus. Added CS10. Switched Syringes. Drew ~10 mL of air into a 100 mL syringe and replaced the 60 mL syringe on the apparatus. Added CS10. Prepared CS-750 bags. Dispensed cells. [00238] Removed air from final product bags and take retain. Once the last final product bag was filled, closed all clamps. Drew 10 mL of air into a new 100 mL syringe and replace the syringe on the apparatus. Dispensed retain into a 50 mL conical tube and label tube as “Retain” and lot number. Repeat air removal step for each bag. [00239] Prepared final product for cryopreservation, including visual inspection. Held the cryobags on cold pack or at 2-8°C until cryopreservation. [00240] Removed cell count sample. Using an appropriately sized pipette, remove 2.0 mL of retain and place in a 15 mL conical tube to be used for cell counts. Performed cell counts and calculations. NOTE: Diluted only one sample to appropriate dilution to verify dilution is sufficient. Diluted additional samples to appropriate dilution factor and proceed with counts. Determined the Average of Viable Cell Concentration and Viability of the cell counts performed. Determined Upper and Lower Limit for counts. NOTE: Dilution may be adjusted according based off the expected concentration of cells. Determined the Average of DB1/ 149202201.1 149 Attorney Docket No.: 116983-5127-WO Viable Cell Concentration and Viability. Determined Upper and Lower Limit for counts. Calculated IFN-γ. Heat Sealed Final Product bags. [00241] Labeled and collected samples per exemplary sample plan below. TABLE 17. Sample plan. [00235] Sacituzumab govitecan may continue indefinitely, or for up to 10 to 12 cycles. Sacituzumab govitecan should be withheld for absolute neutrophil counts below DB1/ 149202201.1 154 Attorney Docket No.: 116983-5127-WO 1500/mm3 on Day 1 of any cycle or neutrophil count below 1000/mm3 on Day 8 of any cycle and for neutropenic fever. Administer granulocyte-colony stimulating factor as clinically needed. Discontinue for grade 4 infusion reactions. [00236] Overall, the study participation time will be up to 5 years from treatment to completion. Inclusion Criteria [00237] To be eligible for study participation, patients must meet all of the following inclusion criteria prior to enrollment: 1. Provide written informed consent and written authorization for use and disclosure of protected health information. 2. Be ≥ 18 years of age at the time of consent. 3. Have histologically or pathologically confirmed diagnosis of NSCLC (squamous, nonsquamous, adenocarcinoma, large cell, or mixed histologies), and must have documented PD-L1 expression status, as determined by the tumor proportion score (TPS) prior to the CPI treatment that they received. 4. Have received a systemic therapy that included checkpoint inhibitor(s) and chemotherapy with documented radiographic disease progression thereon. (Prior systemic therapy in the adjuvant or neoadjuvant setting, or as part of definitive chemoradiotherapy, will not be counted as a line of therapy if the disease has not progressed during or within 12 months of the completion of such therapy.) 5. Have documented no signs or symptoms of ischemia or clinically significant arrhythmias. 6. Have Eastern Cooperative Oncology Group (ECOG) performance status of 0 or 1 and an estimated life expectancy of > 6 months. 7. Have at least one resectable lesion (or aggregate lesions) of a minimum 1.5 cm in diameter for TIL production. If the lesion considered for harvest is within a previously irradiated field, the lesion must have demonstrated radiographic progression prior to harvest, and the irradiation must have been completed at least 3 months prior to enrollment. DB1/ 149202201.1 155 Attorney Docket No.: 116983-5127-WO Patients must have an adequate histopathology specimen for protocol-required testing. 8. Following tumor harvest for TIL manufacturing, all patients must have at least one remaining measurable lesion, as defined by RECIST v1.1, with the following considerations: a. Lesions in previously irradiated areas should not be selected as target lesions unless progression has been demonstrated in those lesions and the irradiation has been completed at least 3 months prior to enrollment. b. Lesions that are surgically partially resected for TIL generation that are still measurable per RECIST v1.1 may be selected as nontarget lesions but cannot serve as a target lesion for response assessment. 9. Have the following hematologic parameters independent of transfusions and/or blood product support for at least 5 days prior to laboratory testing: a. Absolute neutrophil count (ANC) ≥ 1000/mm3 b. Hemoglobin ≥ 9.0 g/dL c. Platelet count ≥ 100,000/mm3 10. Have adequate organ function with the following laboratory values: a. Serum alanine aminotransferase (ALT) and aspartate aminotransferase (AST) ≤ 3 times the upper limit of normal (≤ 3 × ULN); patients with liver metastasis ≤ 5 × ULN b. Estimated creatinine clearance (eCrCl) ≥ 40 mL/min using the Cockcroft- Gault formula at Screening c. Total bilirubin ≤ 2 mg/dL; patients with Gilbert’s Syndrome must have a total bilirubin ≤ 3 mg/dL 11. Have a left ventricular ejection fraction (LVEF) > 45% and be New York Heart Association (NYHA) Class 1; a cardiac stress test is required for all patients and must demonstrate no irreversible wall movement abnormality. Patients with an abnormal cardiac stress test may be enrolled if they have adequate ejection fraction and cardiology clearance. DB1/ 149202201.1 156 Attorney Docket No.: 116983-5127-WO 12. Screening pulmonary function testing (PFT) is required for patients having any of the following: a. History of cigarette smoking of ≥ 20 pack-years b. Ceased smoking within the past 2 years or still smoking c. History of chronic obstructive pulmonary disease (COPD) d. Any signs or symptoms of respiratory dysfunction Post-bronchodilator values: forced expiratory volume (FEV1)/forced vital capacity (FVC) > 70% or FEV1 > 50% of predicted normal are required for study entry. If a patient is unable to perform reliable spirometry due to abnormal upper airway anatomy (ie, tracheostomy), a 6-minute walk test may be used to assess pulmonary function. Patients who are unable to walk a distance of at least 80% predicted for age and sex or who demonstrate evidence of hypoxia at any point during the test (SpO2 < 90%) are excluded. 13. Must have a washout period from prior systemic anticancer therapy of a minimum duration of 21 days prior to enrollment. Palliative radiation therapy: prior external beam radiation is allowed, provided all radiation-related toxicities are resolved to Grade 1 or baseline, excluding alopecia, skin pigmentation change, or other clinically insignificant events, eg, small area radiation dermatitis or rectal or urinary urgency. Surgery/pre-planned procedure(s): previous surgical procedure(s) is/are permitted, provided that wound healing has occurred, all complications have resolved, and at least 14 days have elapsed (for major operative procedures) prior to the tumor resection. 14. Must have recovered from all prior anticancer treatment-related adverse events (TRAEs) to Grade ≤ 1 (per Common Terminology Criteria for Adverse Events [CTCAE], Version 5.0), except for alopecia or vitiligo. Patients with stable Grade ≥ 2 toxicity from prior anticancer therapy may be considered on a case-by-case basis. DB1/ 149202201.1 157 Attorney Docket No.: 116983-5127-WO 15. Patients of childbearing potential or those with partners of childbearing potential must be willing to practice an approved method of highly effective birth control during treatment and for 12 months after receiving all protocol- related therapy. Approved methods of birth control are as follows: a. Combined (estrogen and progesterone containing) hormonal birth control associated with inhibition of ovulation: oral, intravaginal, transdermal b. Progesterone-only hormonal birth control associated with inhibition of ovulation: oral, injectable, implantable c. Intrauterine device (IUD) d. Intrauterine hormone-releasing system (IUS) e. Bilateral tubal occlusion f. Vasectomized partner g. True absolute sexual abstinence when this is in line with the preferred and usual lifestyle of the patient. Periodic abstinence (e.g., calendar ovulation, symptothermal, post-ovulation methods) is unacceptable. Exclusion Criteria [00238] To be eligible for study participation, patients must meet none of the following criteria prior to enrollment: 1. Have received an organ allograft or prior cell transfer therapy that included a nonmyeloablative or myeloablative chemotherapy regimen within the past 20 years. 2. Have known oncogene driver mutations (eg, EGFR, ALK, ROS) which are sensitive to targeted therapies. 3. Have symptomatic and/or untreated brain metastases, with the following considerations: a. Patients with historical (prior to consenting for study participation) definitively treated brain metastases will be considered for enrollment if, prior to enrollment the patient is clinically stable for ≥ 2 weeks, there are no DB1/ 149202201.1 158 Attorney Docket No.: 116983-5127-WO new brain lesions via screening magnetic resonance imaging (MRI), and the patient does not require ongoing corticosteroid treatment. b. Patients with recently (within 28 days prior to enrollment), definitively treated brain metastases will be considered for enrollment if, prior to enrollment the metastases are asymptomatic, and the patient is clinically stable for ≥ 2 weeks. Prior to initiation of NMA-LD, the following are required: a repeat brain MRI at least 4 weeks posttreatment demonstrating that there are no new or increasing brain lesions, and confirmation that patient is clinically stable for ≥ 2 weeks and does not require ongoing steroid treatment. 4. Require systemic steroid therapy > 10 mg/day of prednisone or other steroid equivalent dose. Patients receiving steroids as replacement therapy for adrenocortical insufficiency at ≤ 10 mg/day of prednisone or another steroid equivalent dose are not excluded. 5. Have weight loss > 10% since metastatic NSCLC diagnosis. 6. Have serologic evidence of any of the following: a. Human immunodeficiency virus (HIV)-1 or HIV-2 b. Serologic evidence of active or chronic hepatitis B virus (HBV) or hepatitis C virus (HCV) infection. Patients with acute or chronic hepatitis infections may be enrolled if the viral load by polymerase chain reaction (PCR) is undetectable with/without active treatment c. Syphilis (rapid plasma reagin [RPR] test or venereal disease research laboratory [VDRL]) test) d. Cytomegalovirus (CMV) immunoglobulin M (IgM) antibody and viral load PCR; and Epstein-Barr virus (EBV) IgM and viral load PCR, indicating active infection e. Herpes simplex virus (HSV)-1 and HSV-2 IgM serology and viral load PCR. 7. Be pregnant or breastfeeding. 8. Have active medical illness(es) that would pose increased risks for study participation, such as systemic infections (eg, syphilis or any other infection DB1/ 149202201.1 159 Attorney Docket No.: 116983-5127-WO requiring antibiotics); coagulation disorders; other active major medical illnesses of the cardiovascular, respiratory, or immune systems; or any history of COVID- 19 infection with residual pulmonary compromise. 9. Have received a live or attenuated vaccination within 28 days prior to the start of NMA-LD. 10. Have any form of primary immunodeficiency (eg, severe combined immunodeficiency disease [SCID] or acquired immune deficiency syndrome [AIDS]). 11. Have a history of hypersensitivity to any component of the study drugs. Combination of TIL and sacituzumab govitecan should not be administered to patients with a known hypersensitivity to any component of the autologous TIL product formulation, including, but not limited to, any of the following: a. NMA-LD (cyclophosphamide, mesna, and fludarabine) b. Sacituzumab govitecan c. Antibiotics of the aminoglycoside group (ie, streptomycin, gentamicin [excluding those who are skin-test negative for gentamicin hypersensitivity]) d. Any component of the TIL product formulation, including dimethyl sulfoxide (DMSO), human serum albumin (HSA), or dextran-40 12. Have had another primary malignancy within the previous 3 years (except for those that do not require treatment or have been curatively treated > 1 year ago, and does not pose a significant risk of recurrence including, but not limited to, non-melanoma skin cancer, ductal carcinoma in situ (DCIS), lobular carcinoma in situ (LCIS), prostate cancer Gleason score ≤ 6, or superficial bladder cancer). 13. Participated in another clinical study with an investigational product within 21 days of the initiation of treatment. [00239] Efficacy assessment: The following efficacy parameters will be determined: ORR, CR rate, DOR, DCR, PFS, and OS. [00240] Safety assessment: For all study participants, AEs/SAEs are collected and graded as per CTCAE v5.0, during the following study periods: Screening, Pre-treatment, DB1/ 149202201.1 160 Attorney Docket No.: 116983-5127-WO Treatment, and Posttreatment Follow-up Period (as applicable). Analyses includes all study periods and will be performed separately for each cohort. [00241] A trial design as shown above may be used for a registrational trial for the combination of sacituzumab govitecan, or another TROP-2 ADC, and lifileucel, or another TIL therapy. The registrational trial may be performed in NSCLC patients, as described above, or in other indications described herein, such as breast cancer, including triple- negative breast cancer (TNBC), urothelial cancer, ovarian cancer, endometrial cancer, pancreatic cancer, bladder cancer, or head and neck cancer. Patient subpopulations may be enrolled as the trial population as well. For example, TNBC patients with unresectable locally advanced or metastatic triple-negative breast cancer who have received two or more prior systemic therapies, at least one of them for metastatic disease. Similarly, breast cancer patients with unresectable locally advanced or metastatic hormone receptor (HR)- positive, human epidermal growth factor receptor 2 (HER2)-negative (IHC 0, IHC 1+ or IHC 2+/ISH–) breast cancer who have received endocrine-ebased therapy and at least two additional systemic therapies in the metastatic setting may be enrolled as the trial population. * * * [00242] The examples set forth above are provided to give those of ordinary skill in the art a complete disclosure and description of how to make and use the embodiments of the compositions, systems and methods of the invention, and are not intended to limit the scope of what the inventors regard as their invention. Modifications of the above-described modes for carrying out the invention that are obvious to persons of skill in the art are intended to be within the scope of the following claims. All patents and publications mentioned in the specification are indicative of the levels of skill of those skilled in the art to which the invention pertains. [00243] All headings and section designations are used for clarity and reference purposes only and are not to be considered limiting in any way. For example, those of skill in the art will appreciate the usefulness of combining various aspects from different headings and sections as appropriate according to the spirit and scope of the invention described herein. DB1/ 149202201.1 161 Attorney Docket No.: 116983-5127-WO [00244] All references cited herein are hereby incorporated by reference herein in their entireties and for all purposes to the same extent as if each individual publication or patent or patent application was specifically and individually indicated to be incorporated by reference in its entirety for all purposes. [00245] Many modifications and variations of this application can be made without departing from its spirit and scope, as will be apparent to those skilled in the art. The specific embodiments and examples described herein are offered by way of example only, and the application is to be limited only by the terms of the appended claims, along with the full scope of equivalents to which the claims are entitled. DB1/ 149202201.1 162
[00235] Sacituzumab govitecan may continue indefinitely, or for up to 10 to 12 cycles. Sacituzumab govitecan should be withheld for absolute neutrophil counts below DB1/ 149202201.1 154 Attorney Docket No.: 116983-5127-WO 1500/mm3 on Day 1 of any cycle or neutrophil count below 1000/mm3 on Day 8 of any cycle and for neutropenic fever. Administer granulocyte-colony stimulating factor as clinically needed. Discontinue for grade 4 infusion reactions. [00236] Overall, the study participation time will be up to 5 years from treatment to completion. Inclusion Criteria [00237] To be eligible for study participation, patients must meet all of the following inclusion criteria prior to enrollment: 1. Provide written informed consent and written authorization for use and disclosure of protected health information. 2. Be ≥ 18 years of age at the time of consent. 3. Have histologically or pathologically confirmed diagnosis of NSCLC (squamous, nonsquamous, adenocarcinoma, large cell, or mixed histologies), and must have documented PD-L1 expression status, as determined by the tumor proportion score (TPS) prior to the CPI treatment that they received. 4. Have received a systemic therapy that included checkpoint inhibitor(s) and chemotherapy with documented radiographic disease progression thereon. (Prior systemic therapy in the adjuvant or neoadjuvant setting, or as part of definitive chemoradiotherapy, will not be counted as a line of therapy if the disease has not progressed during or within 12 months of the completion of such therapy.) 5. Have documented no signs or symptoms of ischemia or clinically significant arrhythmias. 6. Have Eastern Cooperative Oncology Group (ECOG) performance status of 0 or 1 and an estimated life expectancy of > 6 months. 7. Have at least one resectable lesion (or aggregate lesions) of a minimum 1.5 cm in diameter for TIL production. If the lesion considered for harvest is within a previously irradiated field, the lesion must have demonstrated radiographic progression prior to harvest, and the irradiation must have been completed at least 3 months prior to enrollment. DB1/ 149202201.1 155 Attorney Docket No.: 116983-5127-WO Patients must have an adequate histopathology specimen for protocol-required testing. 8. Following tumor harvest for TIL manufacturing, all patients must have at least one remaining measurable lesion, as defined by RECIST v1.1, with the following considerations: a. Lesions in previously irradiated areas should not be selected as target lesions unless progression has been demonstrated in those lesions and the irradiation has been completed at least 3 months prior to enrollment. b. Lesions that are surgically partially resected for TIL generation that are still measurable per RECIST v1.1 may be selected as nontarget lesions but cannot serve as a target lesion for response assessment. 9. Have the following hematologic parameters independent of transfusions and/or blood product support for at least 5 days prior to laboratory testing: a. Absolute neutrophil count (ANC) ≥ 1000/mm3 b. Hemoglobin ≥ 9.0 g/dL c. Platelet count ≥ 100,000/mm3 10. Have adequate organ function with the following laboratory values: a. Serum alanine aminotransferase (ALT) and aspartate aminotransferase (AST) ≤ 3 times the upper limit of normal (≤ 3 × ULN); patients with liver metastasis ≤ 5 × ULN b. Estimated creatinine clearance (eCrCl) ≥ 40 mL/min using the Cockcroft- Gault formula at Screening c. Total bilirubin ≤ 2 mg/dL; patients with Gilbert’s Syndrome must have a total bilirubin ≤ 3 mg/dL 11. Have a left ventricular ejection fraction (LVEF) > 45% and be New York Heart Association (NYHA) Class 1; a cardiac stress test is required for all patients and must demonstrate no irreversible wall movement abnormality. Patients with an abnormal cardiac stress test may be enrolled if they have adequate ejection fraction and cardiology clearance. DB1/ 149202201.1 156 Attorney Docket No.: 116983-5127-WO 12. Screening pulmonary function testing (PFT) is required for patients having any of the following: a. History of cigarette smoking of ≥ 20 pack-years b. Ceased smoking within the past 2 years or still smoking c. History of chronic obstructive pulmonary disease (COPD) d. Any signs or symptoms of respiratory dysfunction Post-bronchodilator values: forced expiratory volume (FEV1)/forced vital capacity (FVC) > 70% or FEV1 > 50% of predicted normal are required for study entry. If a patient is unable to perform reliable spirometry due to abnormal upper airway anatomy (ie, tracheostomy), a 6-minute walk test may be used to assess pulmonary function. Patients who are unable to walk a distance of at least 80% predicted for age and sex or who demonstrate evidence of hypoxia at any point during the test (SpO2 < 90%) are excluded. 13. Must have a washout period from prior systemic anticancer therapy of a minimum duration of 21 days prior to enrollment. Palliative radiation therapy: prior external beam radiation is allowed, provided all radiation-related toxicities are resolved to Grade 1 or baseline, excluding alopecia, skin pigmentation change, or other clinically insignificant events, eg, small area radiation dermatitis or rectal or urinary urgency. Surgery/pre-planned procedure(s): previous surgical procedure(s) is/are permitted, provided that wound healing has occurred, all complications have resolved, and at least 14 days have elapsed (for major operative procedures) prior to the tumor resection. 14. Must have recovered from all prior anticancer treatment-related adverse events (TRAEs) to Grade ≤ 1 (per Common Terminology Criteria for Adverse Events [CTCAE], Version 5.0), except for alopecia or vitiligo. Patients with stable Grade ≥ 2 toxicity from prior anticancer therapy may be considered on a case-by-case basis. DB1/ 149202201.1 157 Attorney Docket No.: 116983-5127-WO 15. Patients of childbearing potential or those with partners of childbearing potential must be willing to practice an approved method of highly effective birth control during treatment and for 12 months after receiving all protocol- related therapy. Approved methods of birth control are as follows: a. Combined (estrogen and progesterone containing) hormonal birth control associated with inhibition of ovulation: oral, intravaginal, transdermal b. Progesterone-only hormonal birth control associated with inhibition of ovulation: oral, injectable, implantable c. Intrauterine device (IUD) d. Intrauterine hormone-releasing system (IUS) e. Bilateral tubal occlusion f. Vasectomized partner g. True absolute sexual abstinence when this is in line with the preferred and usual lifestyle of the patient. Periodic abstinence (e.g., calendar ovulation, symptothermal, post-ovulation methods) is unacceptable. Exclusion Criteria [00238] To be eligible for study participation, patients must meet none of the following criteria prior to enrollment: 1. Have received an organ allograft or prior cell transfer therapy that included a nonmyeloablative or myeloablative chemotherapy regimen within the past 20 years. 2. Have known oncogene driver mutations (eg, EGFR, ALK, ROS) which are sensitive to targeted therapies. 3. Have symptomatic and/or untreated brain metastases, with the following considerations: a. Patients with historical (prior to consenting for study participation) definitively treated brain metastases will be considered for enrollment if, prior to enrollment the patient is clinically stable for ≥ 2 weeks, there are no DB1/ 149202201.1 158 Attorney Docket No.: 116983-5127-WO new brain lesions via screening magnetic resonance imaging (MRI), and the patient does not require ongoing corticosteroid treatment. b. Patients with recently (within 28 days prior to enrollment), definitively treated brain metastases will be considered for enrollment if, prior to enrollment the metastases are asymptomatic, and the patient is clinically stable for ≥ 2 weeks. Prior to initiation of NMA-LD, the following are required: a repeat brain MRI at least 4 weeks posttreatment demonstrating that there are no new or increasing brain lesions, and confirmation that patient is clinically stable for ≥ 2 weeks and does not require ongoing steroid treatment. 4. Require systemic steroid therapy > 10 mg/day of prednisone or other steroid equivalent dose. Patients receiving steroids as replacement therapy for adrenocortical insufficiency at ≤ 10 mg/day of prednisone or another steroid equivalent dose are not excluded. 5. Have weight loss > 10% since metastatic NSCLC diagnosis. 6. Have serologic evidence of any of the following: a. Human immunodeficiency virus (HIV)-1 or HIV-2 b. Serologic evidence of active or chronic hepatitis B virus (HBV) or hepatitis C virus (HCV) infection. Patients with acute or chronic hepatitis infections may be enrolled if the viral load by polymerase chain reaction (PCR) is undetectable with/without active treatment c. Syphilis (rapid plasma reagin [RPR] test or venereal disease research laboratory [VDRL]) test) d. Cytomegalovirus (CMV) immunoglobulin M (IgM) antibody and viral load PCR; and Epstein-Barr virus (EBV) IgM and viral load PCR, indicating active infection e. Herpes simplex virus (HSV)-1 and HSV-2 IgM serology and viral load PCR. 7. Be pregnant or breastfeeding. 8. Have active medical illness(es) that would pose increased risks for study participation, such as systemic infections (eg, syphilis or any other infection DB1/ 149202201.1 159 Attorney Docket No.: 116983-5127-WO requiring antibiotics); coagulation disorders; other active major medical illnesses of the cardiovascular, respiratory, or immune systems; or any history of COVID- 19 infection with residual pulmonary compromise. 9. Have received a live or attenuated vaccination within 28 days prior to the start of NMA-LD. 10. Have any form of primary immunodeficiency (eg, severe combined immunodeficiency disease [SCID] or acquired immune deficiency syndrome [AIDS]). 11. Have a history of hypersensitivity to any component of the study drugs. Combination of TIL and sacituzumab govitecan should not be administered to patients with a known hypersensitivity to any component of the autologous TIL product formulation, including, but not limited to, any of the following: a. NMA-LD (cyclophosphamide, mesna, and fludarabine) b. Sacituzumab govitecan c. Antibiotics of the aminoglycoside group (ie, streptomycin, gentamicin [excluding those who are skin-test negative for gentamicin hypersensitivity]) d. Any component of the TIL product formulation, including dimethyl sulfoxide (DMSO), human serum albumin (HSA), or dextran-40 12. Have had another primary malignancy within the previous 3 years (except for those that do not require treatment or have been curatively treated > 1 year ago, and does not pose a significant risk of recurrence including, but not limited to, non-melanoma skin cancer, ductal carcinoma in situ (DCIS), lobular carcinoma in situ (LCIS), prostate cancer Gleason score ≤ 6, or superficial bladder cancer). 13. Participated in another clinical study with an investigational product within 21 days of the initiation of treatment. [00239] Efficacy assessment: The following efficacy parameters will be determined: ORR, CR rate, DOR, DCR, PFS, and OS. [00240] Safety assessment: For all study participants, AEs/SAEs are collected and graded as per CTCAE v5.0, during the following study periods: Screening, Pre-treatment, DB1/ 149202201.1 160 Attorney Docket No.: 116983-5127-WO Treatment, and Posttreatment Follow-up Period (as applicable). Analyses includes all study periods and will be performed separately for each cohort. [00241] A trial design as shown above may be used for a registrational trial for the combination of sacituzumab govitecan, or another TROP-2 ADC, and lifileucel, or another TIL therapy. The registrational trial may be performed in NSCLC patients, as described above, or in other indications described herein, such as breast cancer, including triple- negative breast cancer (TNBC), urothelial cancer, ovarian cancer, endometrial cancer, pancreatic cancer, bladder cancer, or head and neck cancer. Patient subpopulations may be enrolled as the trial population as well. For example, TNBC patients with unresectable locally advanced or metastatic triple-negative breast cancer who have received two or more prior systemic therapies, at least one of them for metastatic disease. Similarly, breast cancer patients with unresectable locally advanced or metastatic hormone receptor (HR)- positive, human epidermal growth factor receptor 2 (HER2)-negative (IHC 0, IHC 1+ or IHC 2+/ISH–) breast cancer who have received endocrine-ebased therapy and at least two additional systemic therapies in the metastatic setting may be enrolled as the trial population. * * * [00242] The examples set forth above are provided to give those of ordinary skill in the art a complete disclosure and description of how to make and use the embodiments of the compositions, systems and methods of the invention, and are not intended to limit the scope of what the inventors regard as their invention. Modifications of the above-described modes for carrying out the invention that are obvious to persons of skill in the art are intended to be within the scope of the following claims. All patents and publications mentioned in the specification are indicative of the levels of skill of those skilled in the art to which the invention pertains. [00243] All headings and section designations are used for clarity and reference purposes only and are not to be considered limiting in any way. For example, those of skill in the art will appreciate the usefulness of combining various aspects from different headings and sections as appropriate according to the spirit and scope of the invention described herein. DB1/ 149202201.1 161 Attorney Docket No.: 116983-5127-WO [00244] All references cited herein are hereby incorporated by reference herein in their entireties and for all purposes to the same extent as if each individual publication or patent or patent application was specifically and individually indicated to be incorporated by reference in its entirety for all purposes. [00245] Many modifications and variations of this application can be made without departing from its spirit and scope, as will be apparent to those skilled in the art. The specific embodiments and examples described herein are offered by way of example only, and the application is to be limited only by the terms of the appended claims, along with the full scope of equivalents to which the claims are entitled. DB1/ 149202201.1 162





























