WO2025019352A2 - Mers-cov mrna vaccines - Google Patents
Mers-cov mrna vaccinesDownload PDFInfo
- Publication number
- WO2025019352A2 WO2025019352A2PCT/US2024/037879US2024037879WWO2025019352A2WO 2025019352 A2WO2025019352 A2WO 2025019352A2US 2024037879 WUS2024037879 WUS 2024037879WWO 2025019352 A2WO2025019352 A2WO 2025019352A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- mers
- cov
- protein
- composition
- domain
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Classifications
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/12—Viral antigens
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/51—Medicinal preparations containing antigens or antibodies comprising whole cells, viruses or DNA/RNA
- A61K2039/53—DNA (RNA) vaccination
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/57—Medicinal preparations containing antigens or antibodies characterised by the type of response, e.g. Th1, Th2
- A61K2039/575—Medicinal preparations containing antigens or antibodies characterised by the type of response, e.g. Th1, Th2 humoral response
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/60—Medicinal preparations containing antigens or antibodies characteristics by the carrier linked to the antigen
- A61K2039/6018—Lipids, e.g. in lipopeptides
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2770/00—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA ssRNA viruses positive-sense
- C12N2770/00011—Details
- C12N2770/20011—Coronaviridae
- C12N2770/20034—Use of virus or viral component as vaccine, e.g. live-attenuated or inactivated virus, VLP, viral protein
Definitions
- MERS-CoVMiddle East Respiratory Syndrome Coronavirus
- Symptoms of MERS-CoV infectioncan range from mild, such as those similar to a common cold (nasal discharge, sore throat, low fever), to severe, including acute respiratory distress syndrome, which may be fatal.
- MERS-CoVis endemic in dromedary camels of East Africa and the Arabian Peninsula and was first reported to infect humans in 2012.
- Some aspectsrelate to fusion proteins comprising one or more domains (e.g., N-terminal domain (NTD) and/or receptor-binding domain (RBD)) of a full-length MERS-CoV Spike protein.
- Some aspectsrelate to RNAs (e.g., mRNAs) encoding mutant MERS-CoV Spike proteins and/or fusion proteins as described herein.
- the proteins and RNAs described hereinare based, at least in part, on the findings that administration of lipid nanoparticle compositions containing mRNA encoding modified betacoronavirus Spike proteins elicited robust virus-neutralizing antibody titers (FIG.2).
- mRNA encoding a fusion NTD-RBD-TM proteincomprising an N-terminal domain (NTD) and receptor-binding domain (RBD) of a betacoronavirus, and a transmembrane (TM) domain
- NTDN-terminal domain
- RBDreceptor-binding domain
- TMtransmembrane domain
- FIG. 2Multiple T cell epitopes have been identified in the NTD of MERS-CoV Spike protein, and NTD-specific antibodies contribute to protective immunity against MERS- CoV infection.
- RBD of MERS-CoVbinds the receptor DPP4, contributing to viral attachment to host cells, and RBD-specific antibodies inhibit viral attachment, and consequently inhibit cellular infection and viral replication.
- compositionscomprising a lipid nanoparticle (LNP) and a messenger ribonucleic acid (mRNA) comprising an open reading frame encoding a Middle East Respiratory Syndrome (MERS)-CoV fusion protein comprising: (a) an N-terminal domain (NTD) of a full-length MERS-CoV Spike (S) protein; and (b) a receptor-binding domain (RBD) of the full-length MERS-CoV-S protein, wherein the MERS-CoV fusion protein does not comprise an NTD or an RBD of an S protein of a second coronavirus that is not MERS-CoV.
- NTDN-terminal domain
- RBDreceptor-binding domain
- the NTD and RBDare connected by a linker.
- the linkeris a glycine linker or a glycine-serine linker.
- the linkercomprises a pan-HLA-DR-binding epitope (PADRE).
- PADREpan-HLA-DR-binding epitope
- the MERS-CoV fusion proteinfurther comprises a transmembrane (TM) domain.
- the TM domainis a MERS-CoV S protein TM domain or derivative thereof.
- the TM domainis heterologous to MERS-CoV.
- the TM domainis an influenza virus hemagglutinin (HA) TM domain or derivative thereof.
- the MERS-CoV fusion proteincomprises, in N-to-C-terminal order, the NTD, the RBD, and the TM domain. In some embodiments, the MERS-CoV fusion protein does not comprise a transmembrane (TM) domain. In some embodiments, the MERS-CoV fusion protein is soluble. In some embodiments, the MERS-CoV fusion protein further comprises a signal peptide. In some embodiments, the signal peptide is a MERS-CoV S protein signal peptide. In some embodiments, the signal peptide is heterologous to MERS-CoV.
- the disclosurerelates to compositions comprising a lipid nanoparticle (LNP) and a messenger ribonucleic acid (mRNA) comprising an open reading frame encoding a mutant Middle East Respiratory Syndrome (MERS)-CoV S protein, wherein the mutant MERS-CoV S protein comprises (a) one or more proline substitutions or insertions, and (b) one or more substitutions or deletions in a furin cleavage site, relative to a full-length MERS-CoV S protein.
- the mutant MERS-CoV S proteincomprises prolines at positions corresponding to residues 1061 and 1061 of the full-length MERS-CoV S protein.
- the disclosurerelates to compositions comprising a lipid nanoparticle (LNP) and a messenger ribonucleic acid (mRNA) comprising an open reading frame encoding a mutant Middle East Respiratory Syndrome (MERS)-CoV S protein, wherein the mutant MERS-CoV S protein comprises (a) one or more substitutions or deletions in a furin cleavage site, and (b) a truncated endoplasmic reticulum (ER) retention signal, relative to a full-length MERS-CoV S protein.
- the mutant MERS-CoV S proteindoes not comprise an ER retention signal.
- the disclosurerelates to compositions comprising a lipid nanoparticle (LNP) and a messenger ribonucleic acid (mRNA) comprising an open reading frame encoding a mutant Middle East Respiratory Syndrome (MERS)-CoV S protein, wherein the mutant MERS-CoV S protein comprises (a) one or more substitutions or deletions in a furin cleavage site, and (b) a truncated cytoplasmic tail, relative to a full-length MERS-CoV S protein.
- the mutant MERS-CoV S proteindoes not comprise a cytoplasmic tail, or comprises a cytoplasmic tail comprising 21 or fewer amino acids.
- compositionscomprising a lipid nanoparticle (LNP) and a messenger ribonucleic acid (mRNA) comprising an open reading frame encoding a Middle East Respiratory Syndrome (MERS)-CoV fusion protein, wherein the MERS-CoV fusion protein consists essentially of (a) an N-terminal domain (NTD) of a full-length MERS-CoV S protein, (b) a receptor-binding domain of the full-length MERS-CoV protein, and (c) a transmembrane (TM) domain.
- NTDN-terminal domain
- TMtransmembrane
- the disclosurerelates to compositions comprising a lipid nanoparticle (LNP) and a messenger ribonucleic acid (mRNA) comprising an open reading frame encoding a Middle East Respiratory Syndrome (MERS)-CoV fusion protein, wherein the MERS-CoV fusion protein consists essentially of (a) an N-terminal domain (NTD) of a full-length MERS-CoV S protein, and (b) a transmembrane (TM) domain.
- NTDN-terminal domain
- TMtransmembrane
- the NTDcomprises amino acids 18–338 of a full-length MERS- CoV S protein.
- the disclosurerelates to compositions comprising a lipid nanoparticle (LNP) and a messenger ribonucleic acid (mRNA) comprising an open reading frame encoding a Middle East Respiratory Syndrome (MERS)-CoV fusion protein, wherein the MERS-CoV fusion protein consists essentially of (a) a receptor-binding domain (RBD) of a full-length MERS-CoV S protein, and (b) a transmembrane (TM) domain.
- the RBDcomprises amino acids 376–589 of a full-length MERS- CoV S protein.
- the TM domainis heterologous to MERS-CoV.
- the TM domainis an influenza virus hemagglutinin (HA) TM domain.
- the full-length MERS-CoV- S proteincomprises the amino acid sequence of SEQ ID NO: 84.
- the disclosurerelates to compositions comprising a lipid nanoparticle (LNP) and a messenger ribonucleic acid (mRNA) comprising an open reading frame encoding a protein comprising an amino acid sequence with at least 90%, at least 95%, at least 97%, at least 98%, at least 99%, or 100% sequence identity to the amino acid sequence of any one of SEQ ID NOs: 89–98.
- the mRNAcomprises a 5′ untranslated region (UTR), wherein the 5′ UTR comprises a nucleotide sequence with at least 90% sequence identity to a nucleotide sequence selected from SEQ ID NOs: 1, 2, 5–35, 66, 67, 70–72, 75, 76, and 81.
- the 5′ UTRcomprises a nucleotide sequence selected from SEQ ID NOs: 1, 2, 5–35, 66, 67, 70–72, 75, 76, and 81.
- the mRNAcomprises a 3′ untranslated region (UTR), wherein the 5′ UTR comprises a nucleotide sequence with at least 90% sequence identity to a nucleotide sequence selected from SEQ ID NOs: 3–4, 36–44, 68, 69, 73, 74, 77–79, and 82.
- the 3′ UTRcomprises a nucleotide sequence selected from SEQ ID NOs: 3–4, 36–44, 68, 69, 73, 74, 77–79, and 82.
- the ORFcomprises one or more stop codons immediately following the last amino acid-encoding codon.
- the one or more stop codonscomprise the nucleotide sequence UGAUGA. In some embodiments, the one or more stop codons comprise the nucleotide sequence UGAUAAUAG. In some embodiments, the mRNA comprises a polyadenosine (polyA) sequence comprising 20 or more consecutive adenosine nucleotides. In some embodiments, the polyA sequence comprises 100 consecutive adenosine nucleotides.
- polyApolyadenosine
- the polyA sequencecomprises, in 5′-to-3′ order, a first nucleotide sequence comprising 30 consecutive adenosine nucleotides, an intervening sequence comprising no more than three adenosine nucleotides, and a second nucleotide sequence comprising 70 consecutive adenosine nucleotides.
- the polyA sequencecomprises the nucleotide sequence of SEQ ID NO: 80.
- the mRNAfurther comprises a polycytidine (polyC) sequence comprising 20 or more consecutive cytidine nucleotides.
- the polyC sequencecomprises 30 consecutive cytidine nucleotides.
- the polyC sequenceis downstream from the polyA sequence, wherein the polyA sequence comprises 64 consecutive adenosine nucleotides. In some embodiments, the polyA sequence comprises 109 consecutive adenosine nucleotides.
- the mRNAcomprises a 5′ cap analog. In some embodiments, the 5′ cap analog comprises a 7mG(5′)ppp(5′)NlmpNp cap. In some embodiments, the 5′ cap analog comprises an m 2 3′O,7 G + (5′)ppp(5′)Am cap having the structure: .
- the lipid nanoparticlecomprises 40-55 mol% ionizable amino lipid, 30-45 mol% sterol, 5-15 mol% neutral lipid, and 1-5 mol% PEG-modified lipid.
- the ionizable amino lipidcomprises a compound of Formula (I): salt or isomer thereof, wherein: R 1 is R”M’R’ or C 5-20 alkenyl; R2 and R3 are each independently selected from C1-14 alkyl and C2-14 alkenyl; R4 is -(CH2)nQ, wherein Q is OH and n is selected from 3, 4, and 5; M and M’ are each independently -OC(O)- or -C(O)O-; R 5, R 6, and R 7 are each H; R’ is a linear C1-12 alkyl, or C1-12 alkyl substituted with C6-9 alkyl; R” is C3-14 alkyl; m is selected from 5, 6, 7, 8, 9, 10, 11, 12, and 13.
- the ionizable amino lipidcomprises Compound 1: In some embodiments, the ionizable amino lipid comprises a compound of the structure In some embodiments, the neutral lipid is 1,2 distearoyl-sn-glycero-3-phosphocholine (DSPC). In some embodiments, the sterol is cholesterol. In some embodiments, the PEG-modified lipid is PEG2000-DMG. In some embodiments, the open reading frame comprises one or more chemically modified nucleotides. In some embodiments, the open reading frame comprises N1-methylpseudouridine. In some embodiments, at least 80% of uracil nucleotides in the open reading frame comprise N1-methylpseudouridine.
- 100% of uracil nucleotides in the open reading framecomprise N1-methylpseudouridine.
- the open reading framecomprises 5-methylcytidine.
- at least 80% of cytosine nucleotides in the open reading framecomprise 5-methylcytidine.
- 100% of cytosine nucleotides in the open reading framecomprise 5-methylcytidine.
- the open reading framecomprises 5-methyluridine.
- at least 80% of uracil nucleotides in the open reading framecomprise 5-methyluridine.
- 100% of uracil nucleotides in the open reading framecomprise 5- methyluridine.
- the disclosurerelates to pharmaceutical compositions comprising a composition as described herein, and a pharmaceutically acceptable excipient.
- the disclosurerelates to methods comprising administering to a subject a composition as described herein.
- the compositionis administered intramuscularly.
- the compositionis effective to elicit, in the subject, CD4+ and/or CD8+ T cells specific to one or more epitopes of the protein.
- the compositionis effective to elicit, in the subject, neutralizing antibodies to MERS-CoV.
- the compositionis effective to elicit, in the subject, antibodies that mediate antibody-dependent cell-mediated cytotoxicity (ADCC) against MERS-CoV-infected cells.
- ADCCantibody-dependent cell-mediated cytotoxicity
- the methodcomprising administering a first dose and a second dose of the composition.
- the disclosurerelates to a MERS-CoV fusion protein comprising: (a) an N-terminal domain (NTD) of a MERS-CoV Spike (S) protein; and (b) a receptor-binding domain (RBD) of the MERS-CoV-S protein, wherein the MERS-CoV fusion protein does not comprise an NTD or an RBD of an S protein of a coronavirus that is not MERS-CoV.
- the NTD and RBDare connected by a linker.
- the linkeris a glycine linker or a glycine-serine linker.
- the linkercomprises a pan-HLA-DR-binding epitope (PADRE).
- the MERS-CoV fusion proteinfurther comprises a transmembrane (TM) domain.
- the TM domainis a MERS-CoV S protein TM domain or derivative thereof.
- the TM domainis heterologous to MERS-CoV.
- the TM domainis an influenza virus hemagglutinin (HA) TM domain or derivative thereof.
- the MERS-CoV fusion proteincomprises, in N-to-C-terminal order, the NTD, the RBD, and the TM domain.
- the MERS-CoV fusion proteindoes not comprise a transmembrane (TM) domain. In some embodiments, the MERS-CoV fusion protein is soluble. In some embodiments, the MERS-CoV fusion protein further comprises a signal peptide. In some embodiments, the signal peptide is a MERS-CoV S protein signal peptide. In some embodiments, the signal peptide is heterologous to MERS-CoV.
- the disclosurerelates to mutant Middle East Respiratory Syndrome (MERS)-CoV S proteins, comprising (a) one or more proline substitutions or insertions, and (b) one or more substitutions or deletions in a furin cleavage site, relative to a full-length MERS- CoV S protein.
- the mutant MERS-CoV S proteincomprises prolines at positions corresponding to residues 1060 and 1061 of the full-length MERS-CoV S protein.
- the disclosurerelates to mutant Middle East Respiratory Syndrome (MERS)-CoV S proteins comprising (a) one or more substitutions or deletions in a furin cleavage site, and (b) a truncated endoplasmic reticulum (ER) retention signal, relative to a full-length MERS-CoV S protein.
- the mutant MERS-CoV S proteindoes not comprise an ER retention signal.
- the disclosurerelates to mutant MERS-CoV S proteins comprising (a) one or more substitutions or deletions in a furin cleavage site, and (b) a truncated cytoplasmic tail, relative to a full-length MERS-CoV S protein.
- the mutant MERS-CoV S proteindoes not comprise a cytoplasmic tail, or comprises a cytoplasmic tail comprising 21 or fewer amino acids.
- the disclosurerelates to MERS-CoV fusion proteins consisting essentially of (a) an N-terminal domain (NTD) of a full-length MERS-CoV S protein, (b) a receptor-binding domain of the full-length MERS-CoV protein, and (c) a transmembrane (TM) domain.
- the disclosurerelates to Middle East Respiratory Syndrome (MERS)- CoV fusion proteins consisting essentially of (a) an N-terminal domain (NTD) of a full-length MERS-CoV S protein, and (b) a transmembrane (TM) domain.
- NTDN-terminal domain
- TMtransmembrane
- the NTDcomprises amino acids 18–338 of a full-length MERS- CoV S protein.
- the disclosurerelates to MERS-CoV fusion proteins consisting essentially of (a) a receptor-binding domain (RBD) of a full-length MERS-CoV S protein, and (b) a transmembrane (TM) domain.
- the RBDcomprises amino acids 376–589 of a full-length MERS- CoV S protein.
- the TM domainis heterologous to MERS-CoV.
- the TM domainis an influenza virus hemagglutinin (HA) TM domain.
- the full-length MERS-CoV- S proteincomprises the amino acid sequence of SEQ ID NO: 84.
- the disclosurerelates to proteins comprising an amino acid sequence with at least 95%, at least 97%, at least 98%, at least 99%, or 100% sequence identity to the amino acid sequence of any one of SEQ ID NOs: 87–98.
- the disclosurerelates to ribonucleic acids (RNA) comprising an open reading frame encoding a protein as described herein.
- the disclosurerelates to messenger ribonucleic acids (mRNA) comprising an open reading frame encoding a protein as described herein.
- the mRNAcomprises one or more chemically modified nucleotides.
- 100% of the uracil nucleotides of the mRNAcomprise are chemically modified nucleotides.
- 100% of uracil nucleotides of the mRNAcomprise N1- methylpseudouridine.
- the disclosurerelates to self-amplifying ribonucleic acids (saRNA) comprising an open reading frame encoding a protein comprising: (a) an N-terminal domain (NTD) of a MERS-CoV Spike (S) protein; and (b) a receptor-binding domain (RBD) of the MERS-CoV-S protein, wherein the MERS-CoV fusion protein does not comprise an NTD or an RBD of an S protein of a coronavirus that is not MERS-CoV.
- the NTD and RBDare connected by a linker.
- the linkeris a glycine linker or a glycine-serine linker.
- the linkercomprises a pan-HLA-DR-binding epitope (PADRE).
- the MERS-CoV fusion proteinfurther comprises a transmembrane (TM) domain.
- the TM domainis a MERS-CoV S protein TM domain or derivative thereof.
- the TM domainis heterologous to MERS-CoV.
- the TM domainis an influenza virus hemagglutinin (HA) TM domain or derivative thereof.
- the MERS-CoV fusion proteincomprises, in N-to-C-terminal order, the NTD, the RBD, and the TM domain.
- the MERS-CoV fusion proteindoes not comprise a transmembrane (TM) domain. In some embodiments, the MERS-CoV fusion protein is soluble. In some embodiments, the MERS-CoV fusion protein further comprises a signal peptide. In some embodiments, the signal peptide is a MERS-CoV S protein signal peptide. In some embodiments, the signal peptide is heterologous to MERS-CoV.
- the disclosurerelates to self-amplifying ribonucleic acids (saRNA) comprising an open reading frame encoding a mutant Middle East Respiratory Syndrome (MERS)-CoV S protein, wherein the mutant MERS-CoV S protein comprises (a) one or more proline substitutions or insertions, and (b) one or more substitutions or deletions in a furin cleavage site, relative to a full-length MERS-CoV S protein.
- the mutant MERS-CoV S proteincomprises prolines at positions corresponding to residues 1060 and 1061 of the full-length MERS-CoV S protein.
- the disclosurerelates to self-amplifying ribonucleic acids (saRNA) comprising an open reading frame encoding a mutant Middle East Respiratory Syndrome (MERS)-CoV S protein, wherein the mutant MERS-CoV S protein comprises (a) one or more substitutions or deletions in a furin cleavage site, and (b) a truncated endoplasmic reticulum (ER) retention signal, relative to a full-length MERS-CoV S protein.
- the mutant MERS-CoV S proteindoes not comprise an ER retention signal.
- the disclosurerelates to self-amplifying ribonucleic acids (saRNA) comprising an open reading frame encoding a mutant Middle East Respiratory Syndrome (MERS)-CoV S protein, wherein the mutant MERS-CoV S protein comprises (a) one or more substitutions or deletions in a furin cleavage site, and (b) a truncated cytoplasmic tail, relative to a full-length MERS-CoV S protein.
- the mutant MERS-CoV S proteindoes not comprise a cytoplasmic tail, or comprises a cytoplasmic tail comprising 21 or fewer amino acids.
- the disclosurerelates to self-amplifying ribonucleic acids (saRNA) comprising an open reading frame encoding a Middle East Respiratory Syndrome (MERS)-CoV fusion protein, wherein the MERS-CoV fusion protein consists essentially of (a) an N-terminal domain (NTD) of a full-length MERS-CoV S protein, (b) a receptor-binding domain of the full-length MERS-CoV protein, and (c) a transmembrane (TM) domain.
- NTDN-terminal domain
- TMtransmembrane
- the disclosurerelates to self-amplifying ribonucleic acids (saRNA) comprising an open reading frame encoding a Middle East Respiratory Syndrome (MERS)-CoV fusion protein, wherein the MERS-CoV fusion protein consists essentially of (a) an N-terminal domain (NTD) of a full-length MERS-CoV S protein, and (b) a transmembrane (TM) domain.
- the NTDcomprises amino acids 18–338 of a full-length MERS- CoV S protein.
- the disclosurerelates to self-amplifying ribonucleic acids (saRNA) comprising an open reading frame encoding a Middle East Respiratory Syndrome (MERS)-CoV fusion protein, wherein the MERS-CoV fusion protein consists essentially of (a) a receptor-binding domain (RBD) of a full-length MERS-CoV S protein, and (b) a transmembrane (TM) domain.
- the RBDcomprises amino acids 376–589 of a full-length MERS- CoV S protein.
- the TM domainis heterologous to MERS-CoV.
- the TM domainis an influenza virus hemagglutinin (HA) TM domain.
- the full-length MERS-CoV- S proteincomprises the amino acid sequence of SEQ ID NO: 84.
- FIG.1is a structural schematic of a betacoronavirus S protein, including the N-terminal domain (NTD), the receptor-binding domain (RBD), the Spike subdomain 1 (SD1), the Spike subdomain 2 (SD2), and the S2 subunit of the spike protein (S2) (left).
- the schematic on the rightshows elements of the NTD-RBD-TM construct design, including the linked RBD and NTD.
- FIG.2shows the neutralizing antibody (NAb) titers (y-axis) elicited in mice in response to immunization with either one or two doses of varying amounts of S-2P (2P-stabilized full- length S protein), NTD-RBD-TM (S protein NTD and RBD linked to a transmembrane (TM) domain), RBD-TM (S protein RBD linked to a TM domain), and NTD-TM (S protein NTD linked to a TM domain) (x-axis).
- S-2P2P-stabilized full- length S protein
- NTD-RBD-TMS protein NTD and RBD linked to a transmembrane (TM) domain
- RBD-TMS protein RBD linked to a TM domain
- NTD-TMS protein NTD linked to a TM domain
- Coronavirus genomesinclude a variable number of open reading frames (ORFs) that encode accessory proteins, nonstructural proteins, and structural proteins (Song et al. 2019 Viruses;11(1):p.59). Many antigenic peptides are located in the structural proteins (Cui et al.2019 Nat. Rev. Microbiol., 17(3):181–192). Spike glycoprotein (S), a small envelope protein (E), matrix protein (M), and nucleocapsid protein (N) are four such structural proteins. Since S protein contributes to cell tropism and virus particle entry, and also elicits neutralizing antibodies (NAb) and protective immunity, it is considered an important target in coronavirus vaccine development.
- ORFsopen reading frames
- Ssmall envelope protein
- Mmatrix protein
- Nnucleocapsid protein
- nucleic acidsin particular mRNA(s)
- appropriate carriers or delivery vehiclese.g., lipid nanoparticles
- nucleic acidupon administration to cells, tissues or subjects, nucleic acid is taken up by cells which, in turn, express protein(s) encoded by the nucleic acids, e.g., mRNAs.
- Antigensas used herein, are proteins capable of inducing an immune response (e.g., causing an immune system to produce antibodies against the antigens).
- the vaccines as described hereinprovide a unique advantage over traditional protein-based vaccination approaches, in which protein antigens are purified or produced in vitro, e.g., recombinant protein production technologies.
- the vaccines of the present disclosurefeature mRNA encoding the desired antigens, which when introduced into the body, i.e., administered to a mammalian subject (for example a human) in vivo, cause the cells of the body to express the desired antigens.
- a mammalian subjectfor example a human
- the mRNAsare encapsulated in lipid nanoparticles (LNPs).
- LNPslipid nanoparticles
- the mRNAsUpon delivery and uptake by cells of the body, the mRNAs are translated in the cytosol and protein antigens are generated by the host cell machinery.
- the protein antigensare presented and elicit an adaptive humoral and cellular immune response.
- Neutralizing antibodiesare directed against the expressed protein antigens and hence the protein antigens are considered relevant target antigens for vaccine development.
- antigenencompasses immunogenic proteins and immunogenic fragments (an immunogenic fragment that induces (or is capable of inducing) an immune response to a (at least one) MERS-CoV variant), unless otherwise stated.
- proteinencompasses peptides and the term “antigen” encompasses antigenic fragments.
- MERS-CoVMERS-CoV
- proteinsmay be antigenic such as bacterial polysaccharides or combinations of protein and polysaccharide structures, but for the viral vaccines included herein, viral proteins, fragments of viral proteins and designed and or mutated proteins derived from MERS-CoV are the antigens described herein. Many proteins have a quaternary or three-dimensional structure, which consists of more than one polypeptide or several polypeptide chains that associate into an oligomeric molecule.
- subunitrefers to a single protein molecule, for example, a polypeptide or polypeptide chain resulting from processing of a nascent protein molecule, which subunit assembles (or “coassembles”) with other protein molecules (e.g., subunits or chains) to form a protein complex.
- Proteinscan have a relatively small number of subunits and therefore be described as “oligomeric” or can consist of a large number of subunits and therefore be described as “multimeric”.
- the subunits of an oligomeric or multimeric proteinmay be identical, homologous or totally dissimilar and dedicated to disparate tasks. Proteins or protein subunits can further comprise domains.
- domainrefers to a distinct functional and/or structural unit within a protein.
- a “domain”is responsible for a particular function or interaction, contributing to the overall role of a protein.
- Domainscan exist in a variety of biological contexts. Similar domains (i.e., domains sharing structural, functional and/or sequence homology) can exist within a single protein or can exist within distinct proteins having similar or different functions.
- a protein domainis often a conserved part of a given protein tertiary structure or sequence that can function and exist independently of the rest of the protein or subunit thereof. In structural and molecular biology, identical, homologous or similar subunits or domains can help to classify newly identified or novel proteins.
- antigenis distinct from the term “epitope” which is a substructure of an antigen, e.g., a polypeptide, such as 7-10 amino acids, or carbohydrate structure, which may be recognized by an antigen binding site.
- an antigene.g., a polypeptide, such as 7-10 amino acids, or carbohydrate structure, which may be recognized by an antigen binding site.
- the artdescribes protein antigens that are delivered to subjects or immune cells in isolated form, e.g., isolated protein, polypeptide or peptide antigens, however, the design, testing, validation, and production of protein antigens can be costly and time-consuming, especially when producing proteins at large scale.
- mRNA technologyis amenable to rapid design and testing of mRNA constructs encoding a variety of antigens.
- mRNA coupled with inclusion in appropriate delivery vehiclescan proceed quickly and can rapidly produce mRNA vaccines at large scale.
- appropriate delivery vehiclese.g., lipid nanoparticles
- Potential benefitalso arises from the fact that antigens encoded by the mRNAs are expressed by the cells of the subject, e.g., are expressed by the human body, and thus the subject, e.g., the human body, serves as the “factory” to produce the antigens which, in turn, elicit the desired immune response.
- Other immune cellsfor example, B cells and T cells, are then able to recognize and mount and immune response develop an immune response against the encoded protein and ultimately create a long -lasting protective response against the coronavirus.
- compositionsmay include an RNA or multiple RNAs encoding two or more antigens of the same or different MERS-CoV strains or genotypes.
- combination vaccinesthat include RNA encoding one or more MERS-CoV antigens and one or more antigen(s) of a different organism.
- the vaccines of the present disclosuremay be combination vaccines that target one or more antigens of the same MERS-CoV genotype or strain, or one or more antigens of different MERS-CoV genotypes or different species, e.g., antigens which induce immunity to organisms which are found in the same geographic areas where the risk of coronavirus infection is high or organisms to which an individual is likely to be exposed to when exposed to a coronavirus (e.g., MERS-CoV).
- the second or subsequent circulating MERS-CoV antigenis an immunodominant antigen from an emerging strain.
- An immunodominant antigen of an emerging strainis assessed with respect to the strain from which the antigen is derived, relative to a different strain of the virus, such as the original strain or other variant thereof.
- An immunodominant antigen of the emerging straininduces a stronger immune response against the emerging strain than against the different strain.
- an immunodominant antigen of the emerging strainis more infective than a different strain of the virus, such as the original strain or other variant thereof.
- an RNAcomprises an open reading frame encoding a protein having at least 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity to an amino acid sequence of SEQ ID NO: 84.
- an RNAcomprises an open reading frame encoding a protein having at least 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity to an amino acid sequence of SEQ ID NO: 85.
- an RNAcomprises an open reading frame encoding a protein having at least 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity to an amino acid sequence of SEQ ID NO: 86.
- an RNAcomprises an open reading frame encoding a protein having at least 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity to an amino acid sequence of SEQ ID NO: 87.
- an RNAcomprises an open reading frame encoding a protein having at least 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity to an amino acid sequence of SEQ ID NO: 88.
- an RNAcomprises an open reading frame encoding a protein having at least 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity to an amino acid sequence of SEQ ID NO: 89.
- an RNAcomprises an open reading frame encoding a protein having at least 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity to an amino acid sequence of SEQ ID NO: 90.
- an RNAcomprises an open reading frame encoding a protein having at least 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity to an amino acid sequence of SEQ ID NO: 91.
- an RNAcomprises an open reading frame encoding a protein having at least 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity to an amino acid sequence of SEQ ID NO: 92.
- an RNAcomprises an open reading frame encoding a protein having at least 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity to an amino acid sequence of SEQ ID NO: 93.
- an RNAcomprises an open reading frame encoding a protein having at least 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity to an amino acid sequence of SEQ ID NO: 94.
- an RNAcomprises an open reading frame encoding a protein having at least 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity to an amino acid sequence of SEQ ID NO: 95.
- an RNAcomprises an open reading frame encoding a protein having at least 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity to an amino acid sequence of SEQ ID NO: 96.
- an RNAcomprises an open reading frame encoding a protein having at least 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity to an amino acid sequence of SEQ ID NO: 97.
- an RNA(e.g., mRNA) comprises an open reading frame encoding a protein having at least 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity to an amino acid sequence of SEQ ID NO: 98.
- an mRNA vaccinecomprises 1, 2, 3, 4, 5, or 6 mRNAs encoding different proteins, wherein each protein comprises at least one mutation and/or at least one deletion.
- the mRNA vaccinefurther comprises an mRNA encoding a wild-type MERS-CoV S protein or the antigenic fragment thereof.
- the mRNA vaccinein some embodiments, is in a lipid nanoparticle (that is, the lipid nanoparticle comprises 1, 2, 3, 4, 5, or 6 mRNAs encoding different protein).
- a compositioncomprises a first mRNA encoding a protein or variant thereof of a first MERS-CoV virus and a second mRNA encoding a second protein or variant thereof of a second MERS-CoV virus.
- the first MERS-CoV virusis a first circulating MERS-CoV virus.
- the second MERS-CoV virusis a second circulating MERS-CoV virus.
- “Circulating viruses” as used hereinrefer to viruses that have been in circulation for 1 month, 2 months, 3 months, 4 months, 5 months, 6 months, 7 months, 8 months, 9 months, 10 months, 11 months, a portion of a year, 1 year, 1.5 years, 2 years, 3 years, or longer.
- the first and second mRNAsare present in the composition in a 1:1, 1:2, 1:3, or 1:4 ratio.
- the first and second mRNAsare present in the composition in a 2:1, 3:1, or 4:1 ratio.
- the first and second mRNAsare present in the composition in a 1:1 ratio.
- a compositionfurther comprises a third mRNA encoding a protein or variant thereof of a third MERS-CoV virus, wherein the protein comprises at least one addition, substitution, or deletion relative to the amino acid sequence of SEQ ID NO: 84.
- the first and third mRNAsare present in the composition in a 1:1, 1:2, 1:3, or 1:4 ratio.
- the first and third mRNAsare present in the composition in a 2:1, 3:1, or 4:1 ratio.
- the first and third mRNAsare present in the composition in a 1:1 ratio.
- a compositionfurther comprises a fourth mRNA encoding a protein or variant thereof of a fourth MERS-CoV virus, wherein the protein comprises at least one addition, substitution, or deletion relative to the amino acid sequence of SEQ ID NO: 84.
- the first and fourth mRNAsare present in the composition in a 1:1, 1:2, 1:3, or 1:4 ratio.
- the first and fourth mRNAsare present in the composition in a 2:1, 3:1, or 4:1 ratio.
- the first and fourth mRNAsare present in the composition in a 1:1 ratio.
- a compositionfurther comprises a fifth mRNA encoding a protein or variant thereof of a fifth MERS-CoV virus, wherein the protein comprises at least one addition, substitution, or deletion relative to the amino acid sequence of SEQ ID NO: 84.
- the first and fifth mRNAsare present in the composition in a 1:1, 1:2, 1:3, or 1:4 ratio.
- the first and fifth mRNAsare present in the composition in a 2:1, 3:1, or 4:1 ratio.
- the first and fifth mRNAsare present in the composition in a 1:1 ratio.
- a compositionfurther comprises a sixth mRNA encoding a protein or variant thereof of a sixth MERS-CoV virus, wherein the protein comprises at least one addition, substitution, or deletion relative to the amino acid sequence of SEQ ID NO: 84.
- the first and sixth mRNAsare present in the composition in a 1:1, 1:2, 1:3, or 1:4 ratio.
- the first and sixth mRNAsare present in the composition in a 2:1, 3:1, or 4:1 ratio.
- the first and sixth mRNAsare present in the composition in a 1:1 ratio.
- the mRNAsare present in the composition in an equal amount (e.g., a 1:1 weight/weight ratio or a 1:1 molar ratio), for example, a ratio of 1:1 (:1:1:1:1) of mRNA encoding distinct coronavirus antigens.
- a “weight/weight ratio” or wt/wt ratio or wt:wt ratiorefers to the ratio between the weights (masses) of the different components.
- a “molar ratio”refers to the ratio between different components (e.g., the number of mRNA encoding each antigen). In some embodiments, the ratio is 1:1, 1:2, 1:3, 1:4, 2:1, 3:1, or 4:1.
- the featured vaccinesinclude the mRNAs encapsulated within LNPs. While it is possible to encapsulate each unique mRNA in its own LNP, the mRNA vaccine technology enjoys the significant technological advantage of being able to encapsulate several mRNAs in a single LNP product. In other embodiments the vaccines are separate vaccines that are not co-formulated, but may be admixed separately before administration or simply administered separately.
- Spike (S) ProteinsThe envelope spike (S) proteins of known betacoronaviruses determine the virus host tropism and entry into host cells. Coronavirus spike (S) protein is a choice antigen for the vaccine design as it can induce neutralizing antibodies and protective immunity.
- S proteinis critical for MERS-CoV infection.
- the organization of the S proteinis similar among betacoronaviruses, such as MERS-CoV, SARS-CoV-2, SARS-CoV, HKU1-CoV, MHV-CoV and NL63-CoV.
- the organization of the Spike (S) proteinis similar among betacoronaviruses, such as SARS-CoV-2, SARS-CoV, MERS-CoV, HKUl-CoV, MHV-CoV and NL63-CoV, including two subunits, S1 and S2, which mediate attachment and membrane fusion, respectively.
- the S1 subunitincludes an N terminal domain (NTD), a receptor binding domain (RBD), and two subdomains (SD1 and SD2); the S2 subunit participates in fusion with the host cell membrane and viral entry.
- NTDN terminal domain
- RBDreceptor binding domain
- SD1 and SD2subdomains
- the term “Spike protein”refers to a glycoprotein that forms homotrimers protruding from the envelope (viral surface) of viruses including betacoronaviruses. Trimerized Spike protein facilitates entry of the virion into a host cell by binding to a receptor on the surface of a host cell followed by fusion of the viral and host cell membranes.
- the S proteinis a highly glycosylated and large type I transmembrane fusion protein that is made up of 1,160 to 1,400 amino acids, depending upon the type of virus.
- Betacoronavirus Spike proteinscomprise between about 1100 to 1500 amino acids.
- MERS-CoV spike (S) proteinis a primary antigen choice for vaccine design, as it can induce neutralizing antibodies and protective immunity.
- mRNAs as described hereinare designed to produce MERS-CoV Spike proteins (i.e., encode Spike proteins such that Spike protein is expressed when the mRNA is delivered to a cell or tissue, for example a cell or tissue in a subject), as well as variants thereof.
- Spike proteinmay be necessary for a virus, e.g., a betacoronavirus, to perform its intended function of facilitating virus entry into a host cell
- a certain amount of variation in Spike protein structure and/or sequenceis tolerated when seeking primarily to elicit an immune response against Spike protein.
- minor truncatione.g., of one to a few, possibly up to 5 or up to 10 amino acids from the N- or C-terminus of the encoded Spike protein, e.g., encoded Spike protein antigen, may be tolerated without changing the antigenic properties of the protein.
- the Spike proteinis a stabilized Spike protein, for example, the Spike protein is stabilized by two proline substitutions (a 2P mutation).
- the Spike proteinis not a stabilized Spike protein, for example, the Spike protein is not stabilized by two proline substitutions (a 2P mutation).
- the Spike proteinis from a different virus strain.
- a strainis a genetic variant of a microorganism (e.g., a virus).
- New viral strainscan be created due to mutation, which may be selected due to enhanced replication, transmissibility, and/or evasion of pre-existing immune responses (e.g., antigenic drift), or recombination of genetic components when two or more viruses infect the same cell, with such recombinant viruses being selected due to enhanced replication, transmissibility, and/or evasion of pre-existing immune responses.
- Antigenic driftis a process that generates genetic and antigenic variation in viruses, by the accumulation of mutations in the virus genes that code for virus-surface proteins recognized by host immune responses (antibodies and T cells).
- Antigenic shiftis the process by which two or more different strains of a virus, or strains of two or more different viruses, combine to form a new subtype having a mixture of the surface antigens of the two or more original strains, which may create virus with a novel combination of surface antigens that did not previously exist in nature.
- the termis often applied specifically to influenza viruses, where segmentation of the viral genome into distinct RNA segments, and reassortment of genome segments during virion production, allows the production of reassortant progeny with novel combinations of genome segments from co-infected cells.
- genetic recombinationmay occur between non-segmented viruses (e.g., MERS-CoV) where multiple viral genotypes replicate in the same cell, e.g., by switching between two template genomes during replication, resulting in progeny genomes with combinations of sequences from two or more viral genotypes.
- Antigenic shiftis contrasted with antigenic drift (in which individual mutations accumulate over time, and may lead to a loss of immunity, or in vaccine mismatch).
- a virus strain as used hereinis a genetic variant or of a virus that is characterized by a differing isoform of one or more surface proteins of the virus.
- MERS-CoVfor example, a different amino acid sequence in the MERS-CoV spike protein where the immune response in an individual to the new strain is less effective than to the strain used to immunize or first infect the individual.
- a new virus strainmay arise from natural mutation or a combination of natural mutation and immune selection due to an ongoing immune response in an immunized or previously infected individual.
- a new virus straincan differ by one, two, three or more amino acid mutations in regions of the spike protein responsible for a viral function such as receptor binding or viral fusion with a target cell.
- a spike protein from a new strainmay differ from the parental strain by as much as 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity at the amino acid level.
- a natural virus strainis a variant of a given virus that is recognizable because it possesses some “unique phenotypic characteristics” that remain stable (e.g., stable and heritable biological, serological, and/or molecular characters) under natural conditions.
- Such “unique phenotypic characteristics”are biological properties different from the compared reference virus, such as unique antigenic properties, host range (e.g., infecting a different kind of host), symptoms of disease caused by the strain, different type of disease caused by the strain (e.g., transmitted by different means), etc.
- a “unique phenotypic characteristic”can be detected clinically (e.g., clinical manifestations detected in a host infected with the strain) or within a comparative animal experiment in which a researcher skilled in the art of virology can distinguish between the reference control virus-infected animal and the animal infected with the alleged new strain, without knowing which animal received which virus and without having any information about the differences between the two viruses.
- a virus variant with a simple difference in genome sequenceis not a separate strain if there is no recognizable distinct viral phenotype. The extent of genomic sequence variation is irrelevant for the classification of a variant as a strain since a distinct phenotype sometimes arises from few mutations.
- S proteins of coronavirusescan be divided into two important functional subunits, of which include the N-terminal S1 subunit, which forms of the globular head of the S protein, and the C-terminal S2 region that forms the stalk of the protein and is directly embedded into the viral envelope.
- the S1 subunitUpon interaction with a potential host cell, the S1 subunit will recognize and bind to receptors on the host cell, specifically dipeptidyl peptidase 4 (DPP4), whereas the S2 subunit, which is the most conserved component of the S protein, will be responsible for fusing the envelope of the virus with the host cell membrane.
- DPP4dipeptidyl peptidase 4
- Each monomer of trimeric S protein trimercontains the two subunits, S1 and S2, mediating attachment and membrane fusion, respectively.
- the two subunitsare separated from each other by an enzymatic cleavage process.
- S proteinis first cleaved by furin-mediated cleavage at the S1/S2 site in infected cells.
- a subsequent serine protease-mediated cleavage eventoccurs at the S2′ site within S1.
- the S1/S2 cleavage siteis at amino acids 748–RSVR–751 (SEQ ID NO: 45).
- the S2′ cleavage siteis at amino acids 884–RSAR–887 (SEQ ID NO: 46).
- S1 subunite.g., S1 subunit antigen
- S2 subunite.g., S2 subunit antigen
- Spike protein S1 or S2 subunitmay be necessary for receptor binding or membrane fusion, respectively, a certain amount of variation in S1 or S2 structure and/or sequence is tolerated when seeking primarily to elicit an immune response against Spike protein subunits.
- minor truncatione.g., of one to a few, possibly up to 4, 5, 6, 7, 8, 9 or 10 amino acids from the N- or C-terminus of the encoded subunit, e.g., encoded S1 or S2 protein antigens, may be tolerated without changing the antigenic properties of the protein.
- the compositioncomprises an mRNA encoding a Spike protein associated with the Kingdom of Saudi Arabia (KSA) 2019 lineage.
- KSAKingdom of Saudi Arabia
- An exemplary Spike protein of the MERS-CoV KSA 2019 lineageis provided as amino acid sequence of SEQ ID NO: 84.
- the compositioncomprises an mRNA encoding a Spike protein associated with the Riyadh 2013 lineage.
- an exemplary Spike protein of the MERS-CoV Riyadh 2013 lineageis provided as amino acid sequence of SEQ ID NO: 85.
- the compositioncomprises an mRNA encoding a Spike protein associated with the England 2012 lineage.
- An exemplary Spike protein of the MERS-CoV England 2012 lineageis provided as amino acid sequence of SEQ ID NO: 84.
- a Spike protein, e.g., an encoded Spike protein antigenhas the amino acid sequence of SEQ ID NO: 84.
- a Spike protein, e.g., an encoded Spike protein antigenhas the amino acid sequence of SEQ ID NO: 85.
- a Spike proteine.g., an encoded Spike protein antigen
- a Spike proteine.g., an encoded Spike protein antigen
- the variantpreferably has the same activity as the comparator Spike protein sequence and/or has the same immune specificity as the comparator Spike protein, as determined for example, in immunoassays (e.g., enzyme-linked immunosorbent assays (ELISA assays).
- a Spike proteine.g., an encoded Spike protein antigen
- ELISA assaysenzyme-linked immunosorbent assays
- a Spike proteine.g., an encoded Spike protein antigen
- a Spike protein, e.g., an encoded Spike protein antigenhas the amino acid sequence of SEQ ID NO: 90.
- a Spike proteine.g., an encoded Spike protein antigen
- a Spike proteine.g., an encoded Spike protein antigen
- a Spike proteine.g., an encoded Spike protein antigen
- a Spike proteine.g., an encoded Spike protein antigen, comprises one or more (e.g., two consecutive) proline substitutions or insertions at or near the boundary between a heptad repeat 1 (HR1) domain and a central helix (CH) domain. Introduction (e.g., by insertion or substitution) of proline residues in this region stabilize the Spike protein in a prefusion conformation.
- the Spike proteincomprises 1, 2, 3, 4, 5, 6, 7, 8, 9, or 10 proline residues that are inserted and/or substituted, relative to a wild-type (naturally occurring) MERS-CoV S protein.
- an introduced proline residueis up to 15 amino acids N-terminal to the last (C-terminal) residue of the HR1 domain, and up to 5 amino acids C-terminal to the first (N-terminal) residue of the CH domain of the wild-type MERS-CoV S protein.
- the Spike proteincomprises a proline residue at one or more positions corresponding to 1058, 1059, 1060, and 1061 of SEQ ID NO: 84.
- the Spike proteincomprises an L1058P, D1059P, V1060P, and/or L1061P substitution relative to the amino acid sequence of SEQ ID NO: 84.
- the Spike proteincomprises proline residues at positions corresponding to 1060 and 1061 of SEQ ID NO: 84. In some embodiments, the Spike protein comprises V1060P and L1061P substitutions, relative to the amino acid sequence of SEQ ID NO: 84. In some embodiments, the Spike protein has an amino acid sequence with at least 90%, at least 95%, at least 96%, at least 97%, at least 98%, or at least 99% identity to the amino acid sequence of SEQ ID NO: 89, and the Spike protein comprises proline residues at positions corresponding to amino acids 1060 and 1061 of SEQ ID NO: 84.
- a Spike proteine.g., an encoded Spike protein antigen, comprises one or more mutations in the S1/S2 cleavage site.
- the Spike proteindoes not comprise the amino acid sequence RSVR (SEQ ID NO: 45) at positions corresponding to amino acids 748–751 of SEQ ID NO: 84.
- the Spike proteindoes not comprise the amino acid sequence RXXR at positions corresponding to amino acids 748–751 of SEQ ID NO: 84.
- the Spike proteincomprises one or more mutations in the S2′ cleavage site.
- the Spike proteindoes not comprise the amino acid sequence RSAR (SEQ ID NO: 46) at positions corresponding to amino acids 884–887 of SEQ ID NO: 84. In some embodiments, the Spike protein does not comprise the amino acid sequence RXXR at positions corresponding to amino acids 884–887 of SEQ ID NO: 84. In some embodiments, one or more arginine (R) residues at positions corresponding to amino acids 748, 751, 884, and 887 of SEQ ID NO: 84 are mutated in the Spike protein. In some embodiments, the Spike protein does not comprise a basic amino acid at 1, 2, 3, or 4 positions corresponding to amino acids 748, 751, 884, and 887 of SEQ ID NO: 84.
- the Spike proteincomprises a neutral amino acid (e.g., glycine, alanine, or serine) at 1, 2, 3, or 4 positions corresponding to amino acids 748, 751, 884, and 887 of SEQ ID NO: 84. In some embodiments, the Spike protein comprises glycine residues at positions corresponding to 748 and 751 of SEQ ID NO: 84. In some embodiments, the Spike protein comprises glycine residues at positions corresponding to 884 and 887 of SEQ ID NO: 84. In some embodiments, the Spike protein does not comprise an active S1/S2 cleavage site. In some embodiments, the Spike protein does not comprise an active S2′ cleavage site.
- a neutral amino acide.g., glycine, alanine, or serine
- the Spike proteinhas an amino acid sequence with at least 90%, at least 95%, at least 96%, at least 97%, at least 98%, or at least 99% identity to the amino acid sequence of SEQ ID NO: 90, and does not comprise the amino acid sequence RSVR (SEQ ID NO: 45) at positions corresponding to amino acids 748–751 of SEQ ID NO: 84.
- the Spike proteinhas an amino acid sequence with at least 90%, at least 95%, at least 96%, at least 97%, at least 98%, or at least 99% identity to the amino acid sequence of SEQ ID NO: 90, and does not comprise the amino acid sequence RSAR (SEQ ID NO: 46) at positions corresponding to amino acids 748–751 of SEQ ID NO: 84.
- a Spike proteine.g., an encoded Spike protein antigen, comprises a mutated, truncated, or deleted endoplasmic reticulum (ER) retention signal.
- ER retention signalslocated at the C-terminus of betacoronavirus S proteins, allow retrieval of S proteins from the Golgi apparatus to the ER in retrograde, resulting in intracellular retention of the S protein. Without wishing to be bound by theory, it is expected that mutation of the ER retention signal increases expression of encoded Spike proteins on the cell surface, allowing increased exposure of the Spike protein and enhanced immunogenicity.
- ER retention signalsinclude dibasic (KXHXX) amino acid sequences at the C-terminus of a protein (e.g., KXHXX-COOH).
- a lysine (K) residue at a position corresponding to amino acid 1349, or a histidine (H) residue at a position corresponding to amino acid 1351, of SEQ ID NO: 84is substituted or deleted in the Spike protein.
- the Spike proteindoes not comprise a lysine residue at a position corresponding to amino acid 1349 of SEQ ID NO: 84.
- the Spike proteindoes not comprise a histidine residue at a position corresponding to amino acid 1351 of SEQ ID NO: 84.
- the Spike proteindoes not comprise the amino acid sequence KXH within the 20, 15, 10, or 5 C-terminal amino acids of the Spike protein.
- the Spike proteindoes not comprise the amino acid sequence KXH. In some embodiments, the Spike protein does not comprise the amino acid sequence KXHXX. In some embodiments, the Spike protein has an amino acid sequence with at least 90%, at least 95%, at least 96%, at least 97%, at least 98%, or at least 99% identity to the amino acid sequence of SEQ ID NO: 91, and does not comprise the amino acid sequence KXHXX at its C-terminus. In some embodiments, a Spike protein, e.g., an encoded Spike protein antigen, has a truncated C-terminus.
- a Spike protein with a truncated C-terminusrefers to a Spike protein that lacks one or more amino acids that are present at the C-terminus of a wild-type (naturally occurring) Spike protein.
- the Spike proteincomprises a truncated cytoplasmic tail.
- the Spike proteinhas a truncated cytoplasmic tail relative to a wild-type MERS-CoV Spike protein.
- a wild-type MERS-CoV Spike protein having the amino acid sequence of SEQ ID NO: 84comprises a cytoplasmic tail that is 36 amino acids (amino acids 1318–1353 of SEQ ID NO: 84), and so a modified Spike protein with a truncated C-terminus relative to SEQ ID NO: 84 comprises (i) a cytoplasmic tail with fewer than 36 amino acids; or (ii) no cytoplasmic tail.
- a Spike proteincomprises a cytoplasmic tail that comprises no more than 35, no more than 30, no more than 25, no more than 20, no more than 15, no more than 10, no more than 9, no more than 8, no more than 7, no more than 6, no more than 5, no more than 4, no more than 3, no more than 2, or no more than 1 amino acid.
- the cytoplasmic tailcomprises no more than 21 amino acids.
- a Spike proteincomprises a cytoplasmic tail that is 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, or 35 amino acids in length.
- a Spike proteincomprises a cytoplasmic tail that is 1–5, 5–10, 10–15, 15–20, 20–25, 25–30, or 30–35 amino acids in length. In some embodiments, the Spike protein comprises a cytoplasmic tail comprising no more than 35 amino acids. In some embodiments, the Spike protein comprises a cytoplasmic tail comprising no more than 30 amino acids. In some embodiments, the Spike protein comprises a cytoplasmic tail comprising no more than 25 amino acids. In some embodiments, the Spike protein comprises a cytoplasmic tail comprising no more than 21 amino acids. In some embodiments, the Spike protein comprises a cytoplasmic tail comprising no more than 20 amino acids.
- the Spike proteincomprises a cytoplasmic tail comprising no more than 10 amino acids. In some embodiments, the Spike protein comprises a cytoplasmic tail comprising no more than 8 amino acids. In some embodiments, the Spike protein comprises a cytoplasmic tail comprising no more than 6 amino acids. In some embodiments, the Spike protein comprises a cytoplasmic tail comprising no more than 5 amino acids. In some embodiments, the Spike protein comprises a cytoplasmic tail comprising no more than 4 amino acids. In some embodiments, the Spike protein comprises a cytoplasmic tail comprising no more than 3 amino acids. In some embodiments, the Spike protein comprises a cytoplasmic tail comprising no more than 2 amino acids.
- the Spike proteincomprises a cytoplasmic tail comprising no more than 1 amino acid. Truncations may be introduced by deleting one or more amino acids from the C-terminus of the cytoplasmic tail (e.g., amino acids 1318–1353 or 1339–1353 of SEQ ID NO: 84). In some embodiments, the Spike protein as described herein, or encoded by an RNA as described herein, lacks one or more amino acids present at the C-terminus of a wild-type sequence.
- the Spike proteinlacks 1–35, 1–30, 1–25, 1–20, 1–15, 1–10, 1–5, 5–35, 5–30, 5– 25, 5–20, 5–15, 5–10, 10–35, 10–30, 10–25, 10–20, 10–15, 15–35, 15–30, 15–25, 15–20, 20–35, 20–30, 20–25, 25–35, 25–30, or 30–35 amino acids that are present at the C-terminus of a wild- type MERS-CoV S protein amino acid sequence.
- a Spike protein as described herein, or encoded by an RNA as described hereinhas a truncated cytoplasmic tail relative to SEQ ID NO: 84.
- the Spike proteincomprises an extracellular domain and a transmembrane domain, and has a truncated cytoplasmic tail relative to SEQ ID NO: 84.
- the Spike proteinhas an amino acid sequence with at least 90%, at least 95%, at least 96%, at least 97%, at least 98%, or at least 99% identity to the amino acid sequence of SEQ ID NO: 92, and has a truncated cytoplasmic tail relative to SEQ ID NO: 84.
- a Spike proteine.g., an encoded Spike protein antigen, has the amino acid sequence of SEQ ID NO: 93.
- the Spike proteinhas an amino acid sequence with at least 90%, at least 95%, at least 96%, at least 97%, at least 98%, or at least 99% identity to the amino acid sequence of SEQ ID NO: 93, comprises proline residues at positions corresponding to amino acids 1060 and 1061 of SEQ ID NO: 84, and does not comprise the amino acid sequence RSVR (SEQ ID NO: 45) at positions corresponding to 748– 751 of SEQ ID NO: 84.
- the Spike proteinis stabilized in a prefusion conformation.
- the Spike proteindoes not comprise an active S1/S2 cleavage site.
- a Spike proteine.g., an encoded Spike protein antigen
- the Spike proteinhas an amino acid sequence with at least 90%, at least 95%, at least 96%, at least 97%, at least 98%, or at least 99% identity to the amino acid sequence of SEQ ID NO: 94, does not comprise the amino acid sequence RSVR (SEQ ID NO: 45) at positions corresponding to amino acids 748–751 of SEQ ID NO: 84, and does not comprise the amino acid sequence KXHXX at its C-terminus.
- the Spike proteindoes not comprise an active S1/S2 cleavage site. In some embodiments, the Spike protein does not comprise an ER retention signal. In some embodiments, a Spike protein, e.g., an encoded Spike protein antigen, has the amino acid sequence of SEQ ID NO: 95.
- the Spike proteinhas an amino acid sequence with at least 90%, at least 95%, at least 96%, at least 97%, at least 98%, or at least 99% identity to the amino acid sequence of SEQ ID NO: 95, does not comprise the amino acid sequence RSVR (SEQ ID NO: 45) at positions corresponding to amino acids 748–751 of SEQ ID NO: 84, and lacks one or more amino acids corresponding to amino acids 1339–1353 of SEQ ID NO: 84.
- the Spike proteindoes not comprise an active S1/S2 cleavage site.
- the Spike proteinhas a truncated cytoplasmic tail relative to SEQ ID NO: 84.
- a Spike proteine.g., an encoded Spike protein antigen
- MERS-CoV fusion proteins and RNAse.g., mRNAs or self-amplifying RNAs
- MERS-CoV fusion proteinswhich comprise (i) an N-terminal domain (NTD) of a full-length MERS-CoV Spike protein, and/or (ii) a receptor- binding domain (RBD) of a full-length MERS-CoV Spike protein, and do not comprise an NTD or RBD of a different human coronavirus that is not MERS-CoV (i.e., the protein does not comprise an NTD, and does not comprise an RBD, of a Spike protein of any of the following coronaviruses: SARS-CoV, SARS-CoV-2, 229E-CoV, NL63-CoV, OC43-CoV, and HKU1-CoV).
- the NTD and RBDare from the same full-length MERS-CoV Spike protein. In some embodiments, the NTD and RBD are from different full-length MERS-CoV Spike proteins.
- the S1 and S2 subunits of the MERS-CoV Spike proteinfurther include domains readily discernable by structure and function, which in turn can be featured in designing antigens to be encoded by the nucleic acid vaccines, in particular, mRNA vaccines.
- domainsinclude the N-terminal domain (NTD) and the receptor-binding domain (RBD), said RBD domain further including a receptor-binding motif (RBM)
- domainsinclude fusion peptide (FP), heptad repeat 1 (HR1), heptad repeat 2 (HR2), transmembrane domain (TM), and cytoplasm domain, also known as cytoplasmic tail (CT).
- the HR1 and HR2 domainscan be referred to as the “fusion core region” of MERS-CoV Spike protein.
- the S1 subunitincludes an N terminal domain (NTD), a linker region, a receptor binding domain (RBD), a first subdomain (SD1), and a second subdomain (SD2).
- the S2 subunitincludes, inter alia, a first heptad repeat (HR1), a second heptad repeat (HR2), a transmembrane domain (TM), and a cytoplasmic tail.
- HR1first heptad repeat
- HR2second heptad repeat
- TMtransmembrane domain
- cytoplasmic taila cytoplasmic tail.
- the NTD and RBD of S1have been shown to be the targets of neutralizing antibodies in betacoronavirus-infected individuals.
- N-terminal domainrefers to a domain within the MERS-CoV S1 subunit comprising approximately 321 amino acids in length, having identity to amino acids 18–338 of the S1 subunit of the Spike protein having the amino acid sequence set forth as SEQ ID NO: 84.
- receptor binding domainrefers to a domain within the S1 subunit of MERS-CoV comprising approximately 214 amino acids in length, having identity to amino acids 376–589 of the Spike protein having the amino acid sequence set forth as SEQ ID NO: 84.
- receptor binding motifrefers to the portion of the RBD that directly contacts the DPP4 receptor.
- compositions provided hereininclude RNAs (e.g., mRNAs) that may encode any one or more full-length or partial (truncated or other deletion of sequence) S protein subunit (e.g., S1 or S2 subunit), one or more domain or combination of domains of an S protein subunit (e.g., NTD, RBD, or NTD-RBD fusions, with or without an SD1 and/or SD2), or chimeras of full- length or partial and S2 protein subunits.
- S protein subunite.g., S1 or S2 subunit
- S1 or S2 subunite.g., S1 or S2 subunit
- domain or combination of domains of an S protein subunite.g., NTD, RBD, or NTD-RBD fusions, with or without an SD1 and/or SD2
- chimeras of full- length or partial and S2 protein subunitse.g., NTD, RBD, or NTD-RBD
- an RNAcomprises an open reading frame encoding a protein having at least 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity to an amino acid sequence of SEQ ID NO: 96.
- an RNAcomprises an open reading frame encoding a protein having at least 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity to an amino acid sequence of SEQ ID NO: 97. In some embodiments, an RNA comprises an open reading frame encoding a protein having at least 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity to an amino acid sequence of SEQ ID NO: 98.
- Any pair of protein portionsmay be contiguous in a protein as described herein (e.g., without an intervening amino acid sequence), or the two portions may be separated by a linker.
- the linkermay be a 2A or GS linker described herein in the section entitled “Linkers and Cleavable Peptides.”
- the linkermay also be another linker known in the art.
- a linker of a fusion proteinmay be present in place of an internal truncation relative to a wild-type sequence of a MERS-CoV Spike protein (e.g., the amino acid sequence of the protein comprises deletion of one or more amino acids, relative to the wild-type sequence, and insertion of the linker at the position previously occupied by the deleted amino acids).
- the linkercomprises 2–10 glycine residues.
- the linkercomprises 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, or 20 glycine residues.
- the linkercomprises 2–20, 2–15, 2–10, 2–5, 2–3, 3– 5, 5–7, 7–10, 10–15, 15–20, 3–10, 4–8, or 5–6 glycine residues. In some embodiments, the linker comprises 5–6 glycine residues. Where multiple linkers separate multiple pairs of portions, the multiple linkers may each comprise the same amino acid sequence (e.g., AAY). Multiple linkers may comprise different amino acid sequences (e.g., a first linker comprises the amino acid sequence GGS, and a second linker comprises the amino acid sequence GGS). Linkers connecting different pairs of portions may be the same length, or different lengths.
- two or more portions of a protein portionare connected by a glycine linker or a glycine-serine linker. In some embodiments, each pair of protein portions of a protein are connected by a glycine linker or a glycine-serine linker. In some embodiments, two or more portions of a protein are connected by a linker comprising the amino acid sequence AAY. In some embodiments, each pair of protein portions are connected by the amino acid sequence AAY. In some embodiments, no linkers are present between protein portions in the protein. In some embodiments, a protein comprising one or more domains of a MERS-CoV Spike protein comprises a transmembrane (TM) domain.
- TMtransmembrane
- the TM domainis a TM domain of a coronavirus. In some embodiments, the TM domain is a TM domain of a full- length MERS-CoV Spike protein.
- the full-length MERS-CoV Spike proteinmay the same Spike protein from which the NTD and/or RBD are derived, or a different full-length MERS-CoV Spike protein.
- An exemplary MERS-CoV Spike protein TM domain sequenceis provided as SEQ ID NO: 101.
- the TM domainis a TM domain is from a protein that is heterologous to MERS-CoV (i.e., a TM domain of a protein that is not encoded by a MERS-CoV genome).
- the transmembrane domainmay be from an influenza hemagglutinin transmembrane domain, which has been demonstrated to effectively anchor proteins at the cell surface.
- the TM domainis an influenza virus hemagglutinin (HA) TM domain.
- the influenza virus HA proteinmay have a sequence from any influenza virus species (e.g., influenza A virus, influenza B virus, or influenza C virus) or HA subtype (e.g., H1, H2, H3, H4, H5, H6, H7, H8, H9, H10, H11, H12, H13, H14, H15, H16, H17, or H18).
- An exemplary influenza A virus HA TM domain sequenceis provided as SEQ ID NO: 102.
- a protein as described herein or encoded by an RNA as described hereinconsists essentially of an NTD of a full-length MERS-CoV Spike protein, an RBD of a full- length MERS-CoV, and a TM domain.
- a protein consisting essentially of NTD-RBD-TM domainsmay include a linker between the NTD and RBD.
- a protein consisting essentially of NTD-RBD-TM domainsmay include a linker between the RBD and TM domain. Each pair of the NTD-RBD and RBD-TM domains may be connected by a linker.
- the NTDmay include a signal peptide of the full-length MERS-CoV Spike protein.
- the proteinincludes a signal peptide from a protein other than the MERS-CoV Spike protein.
- the proteincomprises the amino acid sequence of SEQ ID NO: 96.
- a protein as described herein or encoded by an RNA as described hereincomprises an NTD of a full-length MERS-CoV Spike protein, an RBD of a full-length MERS-CoV, and a TM domain.
- the NTD and RBDmay be connected by a linker.
- the RBD and TM domainmay be connected by a linker.
- the proteincomprises the amino acid sequence of SEQ ID NO: 96.
- the proteinhas no greater than 100, no greater than 90, no greater than 80, no greater than 70, no greater than 60, no greater than 50, no greater than 40, no greater than 30, no greater than 20, no greater than 10, or no greater than 5 amino acid substitutions and/or deletions as compared to (when aligned with) a protein having the amino acid sequence of SEQ ID NO: 96.
- the fusion proteinhas an amino acid sequence with at least 90%, at least 95%, at least 96%, at least 97%, at least 98%, or at least 99% identity to the amino acid sequence of SEQ ID NO: 96, and comprises an NTD and RBD of a full-length MERS-CoV Spike protein and a TM domain.
- the full-length MERS-CoV Spike proteincomprises the amino acid sequence of SEQ ID NO: 84. In some embodiments, the full-length MERS-CoV Spike protein comprises the amino acid sequence of SEQ ID NO: 85. In some embodiments, the full-length MERS-CoV Spike protein comprises the amino acid sequence of SEQ ID NO: 86.
- a protein as described herein or encoded by an RNA as described hereinconsists essentially of an NTD of a full-length MERS-CoV Spike protein, a TM domain. A protein consisting essentially of NTD and TM domains may include a between the NTD and TM domain. In some embodiments, the protein comprises the amino acid sequence of SEQ ID NO: 97.
- the NTDmay include a signal peptide of the full-length MERS-CoV Spike protein.
- the proteinincludes a signal peptide from a protein other than the MERS-CoV Spike protein.
- a protein as described herein or encoded by an RNA as described hereincomprises an NTD of a full-length MERS-CoV Spike protein and a TM domain. The NTD and TM domain may be connected by a linker.
- the proteincomprises the amino acid sequence of SEQ ID NO: 97.
- the proteinhas no greater than 100, no greater than 90, no greater than 80, no greater than 70, no greater than 60, no greater than 50, no greater than 40, no greater than 30, no greater than 20, no greater than 10, or no greater than 5 amino acid substitutions and/or deletions as compared to (when aligned with) a protein having the amino acid sequence of SEQ ID NO: 97.
- the fusion proteinhas an amino acid sequence with at least 90%, at least 95%, at least 96%, at least 97%, at least 98%, or at least 99% identity to the amino acid sequence of SEQ ID NO: 97, and comprises an NTD of a full-length MERS-CoV Spike protein and a TM domain.
- the full-length MERS-CoV Spike proteincomprises the amino acid sequence of SEQ ID NO: 84. In some embodiments, the full-length MERS-CoV Spike protein comprises the amino acid sequence of SEQ ID NO: 85. In some embodiments, the full-length MERS-CoV Spike protein comprises the amino acid sequence of SEQ ID NO: 86.
- a protein as described herein or encoded by an RNA as described hereinconsists essentially of an RBD of a full-length MERS-CoV Spike protein and a TM domain. A protein consisting essentially of RBD and TM domains may include a linker between the RBD and TM domain. In some embodiments, the protein includes a signal peptide.
- the signal peptidemay be from a MERS-CoV Spike protein.
- the proteinincludes a signal peptide from a protein other than the MERS-CoV Spike protein.
- the proteincomprises the amino acid sequence of SEQ ID NO: 98.
- a protein as described herein or encoded by an RNA as described hereincomprises an RBD of a full-length MERS-CoV Spike protein and a TM domain. The RBD and TM domain may be connected by a linker.
- the proteincomprises the amino acid sequence of SEQ ID NO: 98.
- the proteinhas no greater than 100, no greater than 90, no greater than 80, no greater than 70, no greater than 60, no greater than 50, no greater than 40, no greater than 30, no greater than 20, no greater than 10, or no greater than 5 amino acid substitutions and/or deletions as compared to (when aligned with) a protein having the amino acid sequence of SEQ ID NO: 98.
- the fusion proteinhas an amino acid sequence with at least 90%, at least 95%, at least 96%, at least 97%, at least 98%, or at least 99% identity to the amino acid sequence of SEQ ID NO: 98 and comprises an RBD of a full-length MERS-CoV Spike protein and a TM domain.
- the full-length MERS-CoV Spike proteincomprises the amino acid sequence of SEQ ID NO: 84. In some embodiments, the full-length MERS-CoV Spike protein comprises the amino acid sequence of SEQ ID NO: 85. In some embodiments, the full-length MERS-CoV Spike protein comprises the amino acid sequence of SEQ ID NO: 86.
- the compositions of the present disclosureinclude RNA that encodes a MERS-CoV antigen variant. Antigen variants or other polypeptide variants refers to molecules that differ in their amino acid sequence from a wild-type, native, or naturally occurring sequence.
- the antigen/polypeptide variantsmay possess substitutions, deletions, and/or insertions at certain positions within the amino acid sequence, as compared to a wild-type, native or naturally occurring sequence.
- MERS-CoV protein variantsare provided in the Sequence Listing. Ordinarily, variants possess at least 50% identity to a wild-type, native or naturally occurring sequence. In some embodiments, variants share at least 80%, or at least 90% identity with a wild-type, native, or naturally occurring sequence.
- the nucleic acid vaccines described hereinencode MERS-CoV variants comprising 1, 2, 3, 4, or more mutations relative to a naturally occurring sequence.
- the nucleic acid vaccines described hereinencode MERS-CoV variants comprising less than 20, 18, 15, 12, or 10 mutations relative to a naturally occurring sequence. In some embodiments, the nucleic acid vaccines described herein encode MERS-CoV variants having 1-501-40, 1-30, 1-25, 1-20, 1-15, 1-10, 5-50, 5-40, 5-30, 5-25, 5-20, 5-15, 5-10, 10-50, 10-40, 10-30, 10-25, 10-20, 10-15, 20-50, 20-40, 20-30, 20-25, 25-50, 25-40, 25-30, 30-50, 30-40, 40-50 mutations (e.g., substitutions).
- mutationrefers to an amino acid substitution, insertion, or deletion, relative to a comparator sequence.
- Variant antigens/polypeptides encoded by nucleic acids of the disclosuremay contain amino acid changes that confer any of a number of desirable properties, e.g., that enhance their immunogenicity, enhance their expression, and/or improve their stability or PK/PD properties in a subject.
- Variant antigens/polypeptidescan be made using routine mutagenesis techniques and assayed as appropriate to determine whether they possess the desired property. Assays to determine expression levels and immunogenicity are well known in the art and exemplary such assays are set forth in the Examples section.
- PK/PD properties of a protein variantcan be measured using art recognized techniques, e.g., by determining expression of antigens in a vaccinated subject over time and/or by looking at the durability of the induced immune response.
- the stability of protein(s) encoded by a variant nucleic acidmay be measured by assaying thermal stability or stability upon urea denaturation or may be measured using in silico prediction. Methods for such experiments and in silico determinations are known in the art.
- a compositioncomprises an RNA or an RNA ORF that comprises a nucleotide sequence of any one of the sequences provided herein, or comprises a nucleotide sequence at least 90%, at least 95%, at least 96%, at least 97%, at least 98%, or at least 99% identical to a nucleotide sequence of any one of the sequences provided herein.
- identityrefers to a relationship between the sequences of two or more polypeptides (e.g. antigens) or polynucleotides (nucleic acids), as determined by comparing the sequences.
- Identityalso refers to the degree of sequence relatedness between or among sequences as determined by the number of matches between strings of two or more amino acid residues or nucleic acid residues. Identity measures the percent of identical matches between the smaller of two or more sequences with gap alignments (if any) addressed by a particular mathematical model or computer program (e.g., “algorithms”). Identity of related antigens or nucleic acids can be readily calculated by known methods.
- Percent (%) identityas it applies to polypeptide or polynucleotide sequences is defined as the percentage of residues (amino acid residues or nucleic acid residues) in the candidate amino acid or nucleic acid sequence that are identical with the residues in the amino acid sequence or nucleic acid sequence of a second sequence after aligning the sequences and introducing gaps, if necessary, to achieve the maximum percent identity. Methods and computer programs for the alignment are well known in the art. It is understood that identity depends on a calculation of percent identity but may differ in value due to gaps and penalties introduced in the calculation.
- variants of a particular polynucleotide or polypeptidehave at least 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, 99% but less than 100% sequence identity to that particular comparator polynucleotide or polypeptide as determined by sequence alignment programs and parameters described herein and known to those skilled in the art.
- sequence alignment programs and parameters described herein and known to those skilled in the artinclude those of the BLAST suite (Stephen F. Altschul, et al (1997), "Gapped BLAST and PSI-BLAST: a new generation of protein database search programs", Nucleic Acids Res.25:3389-3402).
- Another popular local alignment techniqueis based on the Smith-Waterman algorithm (Smith, T.F.
- sequence tags or amino acidssuch as one or more lysines
- Sequence tagscan be used for peptide detection, purification or localization.
- Lysinescan be used to increase peptide solubility or to allow for biotinylation.
- amino acid residues located at the carboxy and amino terminal regions of the amino acid sequence of a peptide or proteinmay optionally be deleted providing for truncated sequences.
- Certain amino acidse.g., C-terminal or N-terminal residues
- sequences for (or encoding) signal sequences, termination sequences, transmembrane domains, linkers, multimerization domains (such as, e.g., foldon regions) and the likemay be substituted with alternative sequences that achieve the same or a similar function.
- cavities in the core of proteinscan be filled to improve stability, e.g., by introducing larger amino acids.
- buried hydrogen bond networksmay be replaced with hydrophobic resides to improve stability.
- glycosylation sitesmay be removed and replaced with appropriate residues.
- sequencesare readily identifiable to one of skill in the art. It should also be understood that some of the sequences provided herein contain sequence tags or terminal peptide sequences (e.g., at the N-terminal or C-terminal ends) that may be deleted, for example, prior to use in the preparation of an mRNA vaccine.
- protein fragments, functional protein domains, and homologous proteinsare also considered to be within the scope of coronavirus antigens of interest.
- any protein fragmentmeaning a polypeptide sequence at least one amino acid residue shorter than a full-length antigen sequence but otherwise identical
- an antigenincludes 2, 3, 4, 5, 6, 7, 8, 9, 10, or more mutations, as shown in any of the sequences provided or referenced herein.
- Antigens/antigenic polypeptidescan range in length from about 4, 6, or 8 amino acids to full length proteins.
- Linkers and Cleavable Peptidese.g., an RNA (e.g., mRNA) that encodes a protein further encodes a linker located between at least two, or between each domain of the protein.
- the linkermay be, for example, a cleavable linker or protease-sensitive linker.
- the linkeris selected from the group consisting of F2A linker, P2A linker, T2A linker, E2A linker, and combinations thereof (see, e.g., WO 2017/127750).
- the linkeris an F2A linker.
- the linkeris a GS linker.
- GS linkersare polypeptide linkers that include glycine and serine amino acids repeats. They comprise flexible and hydrophilic residues and can be used to perform fusion of protein subunits without interfering in the folding and function of the protein domains, and without formation of secondary structures.
- an RNAencodes a protein that comprises a GS linker that is 3 to 20 amino acids long.
- the GS linkermay have a length of (or have a length of at least) 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, or 20 amino acids.
- a GS linkeris (or is at least) 15 amino acids long (e.g., GGSGGSGGSGGSGGG (SEQ ID NO: 47)).
- a GS linkeris (or is at least) 8 amino acids long (e.g., GGGSGGGS (SEQ ID NO: 48)).
- a GS linkeris (or is at least) 7 amino acids long (e.g., GGGSGGG (SEQ ID NO: 49)). In some embodiments, a GS linker comprises the amino acid sequence GGGSGG (SEQ ID NO: 50). In some embodiments, a GS linker is (or is at least) 4 amino acid long (e.g., GGGS (SEQ ID NO: 51)). In some embodiments, the GS linker comprises (GGGS)n (SEQ ID NO: 51), where n is any integer from 1-5. In some embodiments, a GS linker is (or is at least) 4 amino acid long (e.g., GSGG (SEQ ID NO: 52)).
- the GS linkercomprises (GSGG)n (SEQ ID NO: 52), where n is any integer from 1-5.
- a GS linkercomprises the amino acid sequence SGGGS (SEQ ID NO: 53).
- a linkeris a glycine linker, for example having a length of (or a length of at least) 3 amino acids (e.g., GGG).
- a protein encoded by an RNAe.g., mRNA
- a linkercomprises two or more linkers, which may be the same or different from each other.
- a linkercomprises a pan-HLA-DR-binding epitope (PADRE).
- RNAe.g., mRNA
- an RNAhas an ORF that encodes a signal peptide fused to a protein.
- Signal peptidescomprising the N-terminal 15-60 amino acids of proteins, are typically involved for the translocation across the membrane on the secretory pathway and, thus, control the entry of proteins both in eukaryotes and prokaryotes to the secretory pathway.
- the signal peptide of a nascent precursor proteindirects the ribosome to the rough endoplasmic reticulum (ER) membrane and initiates the transport of the growing peptide chain across it for processing.
- ER processingproduces mature proteins, wherein the signal peptide is cleaved from precursor proteins, typically by an ER-resident signal peptidase of the host cell, or they remain uncleaved and function as a membrane anchor.
- a signal peptidemay also facilitate the targeting of the protein to the cell membrane.
- a signal peptidemay have a length of 15-60 amino acids.
- a signal peptidemay have a length of 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49, 50, 51, 52, 53, 54, 55, 56, 57, 58, 59, or 60 amino acids.
- a signal peptidehas a length of 20-60, 25-60, 30-60, 35- 60, 40-60, 45- 60, 50-60, 55-60, 15-55, 20-55, 25-55, 30-55, 35-55, 40-55, 45-55, 50-55, 15-50, 20-50, 25-50, 30-50, 35-50, 40-50, 45-50, 15-45, 20-45, 25-45, 30-45, 35-45, 40-45, 15-40, 20- 40, 25-40, 30-40, 35-40, 15-35, 20-35, 25-35, 30-35, 15-30, 20-30, 25-30, 15-25, 20-25, or 15-20 amino acids.
- an RNAe.g., mRNA
- an RNAcomprises an open reading frame that encodes a protein fused to a signal peptide comprising an amino acid sequence that has at least 50%, at least 60%, at least 70%, at least 75%, at least 80%, at least 85%, at least 90%, or at least 95% identity to the amino acid sequence of any one of the sequences provided herein, such as those reproduced below in Table 3.
- an RNAcomprises an open reading frame that encodes MERS-CoV protein including an endogenous signal peptide of the wild-type protein (e.g., an mRNA encoding a (wild-type or modified) MERS-CoV protein or variant thereof encodes a MERS-CoV S protein signal peptide).
- Nucleic Acids Encoding MERS-CoV ProteinsNucleic acids comprise a polymer of nucleotides (nucleotide monomers). Thus, nucleic acids are also referred to as polynucleotides.
- Nucleic acidsmay be or may include, for example, deoxyribonucleic acid (DNA), ribonucleic acid (RNA), threose nucleic acid (TNA), glycol nucleic acid (GNA), peptide nucleic acid (PNA), locked nucleic acid (LNA, including LNA having a ⁇ -D-ribo configuration, ⁇ -LNA having an ⁇ -L-ribo configuration (a diastereomer of LNA), 2′-amino-LNA having a 2′-amino functionalization, and 2′-amino- ⁇ -LNA having a 2′- amino functionalization), ethylene nucleic acid (ENA), cyclohexenyl nucleic acid (CeNA) and/or chimeras and/or combinations thereof.
- DNAdeoxyribonucleic acid
- RNAribonucleic acid
- TAAglycol nucleic acid
- PNApeptide nucleic acid
- LNAlocked nucleic
- the RNAe.g., mRNA
- the RNAfurther comprises a 5 ⁇ untranslated region (UTR), 3 ⁇ UTR, a poly(A) tail and/or a 5 ⁇ cap analog.
- Messenger RNA (mRNA)Messenger RNA (mRNA) is RNA that encodes a (at least one) protein (a naturally- occurring, non-naturally-occurring, or modified polymer of amino acids) and can be translated to produce the encoded protein in vitro, in vivo, in situ, or ex vivo.
- mRNAis not self-amplifying RNA (saRNA) (see, e.g., Bloom K et al. Gene Therapy 2021; 28: 117–129 for a comparison of mRNA and saRNA).
- saRNAsinclude alphavirus replicase sequences that encode an RNA-dependent RNA polymerase. mRNA does not include alphavirus replicase sequences.
- nucleic acid sequences set forth in the instant applicationmay recite “T”s in a representative DNA sequence but where the sequence represents mRNA, the “T”s would be substituted for “U”s.
- any of the DNAs disclosed and identified by a particular sequence identification number hereinalso disclose the corresponding mRNA sequence complementary to the DNA, where each “T” of the DNA sequence is substituted with “U.”
- Naturally-occurring eukaryotic mRNA moleculescan contain stabilizing elements, including, but not limited to, UTRs at their 5′-end (5′ UTR) and/or at their 3′-end (3′ UTR), in addition to other structural features, such as a 5′-cap structure or a 3′-poly(A) tail. Both the 5′ UTR and the 3′ UTR are typically transcribed from the genomic DNA and are elements of the premature mRNA.
- UTRsUntranslated Regions
- the mRNAs of the present disclosuremay comprise one or more regions or parts which act or function as an untranslated region.
- a “5′ untranslated region”refers to a region of an mRNA that is directly upstream (i.e., 5′) from the start codon (i.e., the first codon of an mRNA transcript translated by a ribosome) that does not encode a polypeptide.
- a “3′ untranslated region”refers to a region of an mRNA that is directly downstream (i.e., 3′) from the open reading frame (e.g., downstream from the last amino acid-encoding codon of an open reading frame, where the stop codon is considered part of the 3′ UTR, or downstream from the first stop codon signaling translation termination, where that stop codon is considered part of the open reading frame), and which does not encode a polypeptide.
- the 5’ UTRmay comprise a promoter sequence. Such promoter sequences are known in the art. It should be understood that such promoter sequences will not be present in a vaccine of the disclosure.
- the mRNAmay comprise a 5’ UTR and/or 3’ UTR.
- UTRs of an mRNAare transcribed but not translated.
- the 5′ UTRstarts at the transcription start site and continues to the start codon but does not include the start codon; the 3′ UTR starts immediately following the open reading frame and continues until the transcriptional termination signal.
- the 3′ UTRbegins with a stop codon, such that no amino acids are added to a polypeptide beyond the last amino acid encoded by the open reading frame.
- a 3′ UTRmay further comprise one or more stop codons.
- UTRnucleic acid molecule
- the regulatory features of a UTRcan be incorporated into the polynucleotides of the present disclosure to, among other things, enhance the stability of the molecule.
- the specific featurescan also be incorporated to ensure controlled down-regulation of the transcript in case they are misdirected to undesired organs sites.
- a variety of 5’ UTR and 3’ UTR sequencesare known.
- the 5′ UTRcomprises a sequence provided in Table 1 or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to a 5′ UTR sequence provided in Table 1, or a variant or a fragment thereof.
- the 3′ UTRcomprises a sequence provided in Table 2 or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to a 3′ UTR sequence provided in Table 2, or a variant or a fragment thereof. It should also be understood that the mRNA of the present disclosure may include any 5’ UTR and/or any 3’ UTR.
- a 5' UTRcomprises a sequence selected from: GGGAAAUAAGAGAGAAAAGAAGAGUAAGAAGAAAUAUAAGAGCCACC (SEQ ID NO: 1), GGGAAAUAAGAGAGAAAAGAAGAGUAAGAAGAAAUAUAAGACCCCGGCGCCGCCACC (SEQ ID NO: 2), GAGGAAAUCGCAAAAUUUGCUCUUCGCGUUAGAUUUCUUUUAGUUUUCUCGCAACUAGCAAGCUUUUGUUCUCGCC (SEQ ID NO: 66), and GGAAAUCGCAAAAUUUGCUCUUCGCGUUAGAUUUCUUUAGUUUUCUCGCAACUAGCAAGCUUUUGUUCUCGCC (SEQ ID NO: 66), and GGAAAUCGCAAAAUUUGCUCUUCGCGUUAGAUUUCUUUAGUUUUCUCGCAACUAGCAAGCUUUUGUUCUCGCC (SEQ ID NO: 67).
- a 3′ UTRcomprises, in 5′-to-3′ order: (a) the nucleic acid sequence UAAAGCUCCCCGGGGGCCUCGGUGGCCUAGCUUCUUGCCCCUUGGGCCUCCCCCCAGCCCCUCCUCCCCUUCCUGCAG (SEQ ID NO: 68), (b) an identification and ratio determination (IDR) sequence, and (c) the nucleic acid sequence UGGUCUUUGAAUAAAGUCUGAGUGGGCGGC (SEQ ID NO: 69).
- each mRNA encoding a distinct proteincomprises a 3′ UTR comprising, in 5′-to-3′ order: (a) the nucleotide sequence of SEQ ID NO: 68; (b) a distinct IDR sequence; and (c) the nucleotide sequence of SEQ ID NO: 69.
- a 5′ UTRcomprises a sequence derived from a 5′ UTR of a gene selected from HSD17B4, RPL32, ASAH1, ATP5A1, MP68, NDUFA4, NOSIP, RPL31, SLC7A3, TUBB4B and UBQLN2.
- the 5′ UTRcomprises a sequence derived from the 5′ UTR of human hydroxysteroid 17-beta dehydrogenase 4 (HSD17B4).
- a 5′ UTRcomprises the sequence GGGAGAGUCCCGCAGUCGGCGUCCAGCGGCUCUGCUUGUUCGUGUGUGUCGUUGCAGGCCUUAUUCAAGCUUACC (SEQ ID NO: 70). In some embodiments, a 5′ UTR comprises the sequence GUCCCGCAGUCGGCGUCCAGCGGCUCUGCUUGUUCGUGUGUGUCGUUGCAGGCCUUAUUC (SEQ ID NO: 71). In some embodiments, a 5′ UTR comprises the sequence GGGAGAAAGCUUACC (SEQ ID NO: 72).
- a 3′ UTRcomprises a sequence derived from a 3′ UTR of a gene selected from PSMB3, ALB7, alpha-globin, CASP1, COX6B1, GNAS, NDUFA1 and RPS9.
- a 3′ UTRcomprises a sequence derived from a 3′ UTR of PSMB3 (proteasome 20S subunit beta 3).
- a 3′ UTRcomprises a sequence derived from a 3′ UTR of alpha-globin (MUAG).
- a 3′ UTRcomprises the sequence AGGACUAGUCCCUGUUCCCAGAGCCCACUUUUUUUCUUUUUUGAAAUAAAAUAGCCUGUCUUUCAGAUCU (SEQ ID NO: 73). In some embodiments, a 3′ UTR comprises the sequence GGACUAGUUAUAAGACUGACUAGCCCGAUGGGCCUCCCAACGGGCCCUCCUCCCCUCCUUGCACCGAGAUUAAU (SEQ ID NO: 74).
- the mRNAcomprises a 5′ UTR comprising the nucleotide sequence of any one of SEQ ID NOs: 70–72, an open reading frame, one or more stop codons, and a 3′ UTR comprising the nucleotide sequence of SEQ ID NO: 73 or SEQ ID NO: 74.
- the mRNAfurther comprises a polyA sequence comprising at least 64 consecutive adenosine nucleotides.
- the mRNAfurther comprises a polyC sequence comprising at least 30 consecutive cytidine nucleotides.
- a 5′ UTRcomprises the sequence GAAUAAACUAGUAUUCUUCUGGUCCCCACAGACUCAGAGAACCCGCCACC (SEQ ID NO: 75). In some embodiments, a 5′ UTR comprises the sequence GAGAAUAAACUAGUAUUCUUCUGGUCCCCACAGACUCAGAGAACCCGCCACC (SEQ ID NO: 76).
- a 3′ UTRcomprises the sequence CUGGUACUGCAUGCACGCAAUGCUAGCUGCCCCUUUCCCGUCCUGGGUACCCCGAGUCUCCCCCGACCUCGGGUCCCA GGUAUGCUCCCACCUCCACCUGCCCCACUCACCACCUCUGCUAGUUCCAGACACCUCCCAAGCACGCAGCAAUGCAGC UCAAAACGCUUAGCCUAGCCACACCCCCACGGGAAACAGCAGUGAUUAACCUUUAGCAAUAAACGAAAGUUUAACUAA GCUAUACUAACCCCAGGGUUGGUCAAUUUCGUGCCAGCCACACC (SEQ ID NO: 77).
- a 3′ UTRcomprises the sequence CUCGAGCUGGUACUGCAUGCACGCAAUGCUAGCUGCCCCUUUCCCGUCCUGGGUACCCCGAGUCUCCCCCGACCUCGG GUCCCAGGUAUGCUCCCACCUCCACCUGCCCCACUCACCACCUCUGCUAGUUCCAGACACCUCCCAAGCACGCAGCAA UGCAGCUCAAAACGCUUAGCCUAGCCACACCCCCACGGGAAACAGCAGUGAUUAACCUUUAGCAAUAAACGAAAGUUU AACUAAGCUAUACUAACCCCAGGGUUGGUCAAUUUCGUGCCAGCCACACCCUGGAGCUAGC (SEQ ID NO: 78).
- a 3′ UTRcomprises the sequence CUGGUACUGCAUGCACGCAAUGCUAGCUGCCCCUUUCCCGUCCUGGGUACCCCGAGUCUCCCCCGACCUCGGGUCCCA GGUAUGCUCCCACCUCCACCUGCCCCACUCACCACCUCUGCUAGUUCCAGACACCUCCCAAGCACGCAGCAAUGCAGC UCAAAACGCUUAGCCUAGCCACACCCCCACGGGAAACAGCAGUGAUUAACCUUUAGCAAUAAACGAAAGUUUAACUAA GCUAUACUAACCCCAGGGUUGGUCAAUUUCGUGCCAGCCACACCCUGGAGCUAGC (SEQ ID NO: 79).
- an mRNAcomprises a 5′ UTR comprising the nucleotide sequence of SEQ ID NO: 75 or SEQ ID NO: 76, an open reading frame, one or more stop codons, and a 3′ UTR comprising the nucleotide sequence of any one of SEQ ID NOs: 77–79.
- an mRNAcomprises a 5′ UTR comprising the nucleotide sequence of SEQ ID NO: 76, an open reading frame, one or more stop codons, and a 3′ UTR comprising the nucleotide sequence of SEQ ID NO: 78.
- an mRNAcomprises a 5′ UTR comprising the nucleotide sequence of SEQ ID NO: 76, an open reading frame, the nucleotide sequence UGAUGA, and a 3′ UTR comprising the nucleotide sequence of SEQ ID NO: 78.
- the mRNAfurther comprises two poly(A) sequences separated by an intervening nucleotide sequence.
- the mRNAfurther comprises the nucleotide sequence of SEQ ID NO: 80.
- a 5′ UTRcomprises the sequence GAGGAGACCCAAGCUACAUUUGCUUCUGACACAACUGUGUUCACUAGCAACCUCAAACAGACACCGCCACC (SEQ ID NO: 81).
- a 3′ UTRcomprises the sequence GCUCGCUUUCUUGCUGUCCAAUUUCUAUUAAAGGUUCCUUUGUUCCCUAAGUCCAACUACUAAACUGGGGGAUAUUAU GAAGGGCCUUGAGCAUCUGGAUUCUGCCUAAUAAAAAACAUUUAUUUUCAUUGC (SEQ ID NO: 82).
- an mRNAcomprises a 5′ UTR comprising the nucleotide sequence of SEQ ID NO: 81, an open reading frame, one or more stop codons, and a 3′ UTR comprising the nucleotide sequence of SEQ ID NO: 82.
- the mRNAfurther comprises a polyA tail comprising 109 consecutive adenosine nucleotides.
- UTRsmay also be omitted from the mRNA described herein.
- a 5 ⁇ UTRdoes not encode a protein (is non-coding).
- Natural 5′ UTRshave features that play roles in translation initiation. They harbor signatures like Kozak sequences which are commonly known to be involved in the process by which the ribosome initiates translation of many genes.
- a 5’ UTRis a heterologous UTR, i.e., is a UTR found in nature associated with a different ORF.
- a 5’ UTRis a synthetic UTR, i.e., does not occur in nature. Synthetic UTRs include UTRs that have been mutated to improve their properties, e.g., which increase gene expression as well as those which are completely synthetic.
- Exemplary 5’ UTRsinclude Xenopus or human derived a-globin or b- globin (8278063; 9012219), human cytochrome b-245 a polypeptide, and hydroxysteroid (17b) dehydrogenase, and Tobacco etch virus (US8278063, US9012219).
- CMV immediate-early 1 (IE1) gene(US2014/0206753, WO2013/185069), the sequence GGGAUCCUACC (SEQ ID NO: 54) (WO 2014/144196) may also be used.
- a 5' UTRis a 5' UTR of a TOP gene lacking the 5' TOP motif (the oligopyrimidine tract) (e.g., WO2015/101414, WO2015/101415, WO2015/062738, WO2015/024667, WO2015/024667); 5' UTR element derived from ribosomal protein Large 32 (L32) gene (WO/2015101414, WO2015101415, WO/2015/062738), 5' UTR element derived from the 5' UTR of an hydroxysteroid (17- ⁇ ) dehydrogenase 4 gene (HSD17B4) (WO201/5024667), or a 5' UTR element derived from the 5' UTR of ATP5A1 (WO2015/024667) can be used.
- L32ribosomal protein Large 32
- an internal ribosome entry siteis used instead of a 5' UTR.
- a 3 ⁇ UTRdoes not encode a protein (is non-coding).
- Natural or wild type 3′ UTRsare known to have stretches of adenosines and uridines embedded in them. These AU rich signatures are particularly prevalent in genes with high rates of turnover. Based on their sequence features and functional properties, the AU rich elements (AREs) can be separated into three classes (Chen et al, 1995): Class I AREs contain several dispersed copies of an AUUUA motif within U-rich regions. C-Myc and MyoD contain class I AREs.
- AREspossess two or more overlapping UUAUUUA(U/A)(U/A) nonamers. Molecules containing this type of AREs include GM-CSF and TNF-a. Class III ARES are less well defined. These U rich regions do not contain an AUUUA motif. c-Jun and Myogenin are two well-studied examples of this class. Most proteins binding to the AREs are known to destabilize the messenger, whereas members of the ELAV family, most notably HuR, have been documented to increase the stability of mRNA. HuR binds to AREs of all the three classes.
- AREs3′ UTR AU rich elements
- AREs3′ UTR AU rich elements
- one or more copies of an AREcan be introduced to make nucleic acids of the disclosure less stable and thereby curtail translation and decrease production of the resultant protein.
- AREscan be identified and removed or mutated to increase the intracellular stability and thus increase translation and production of the resultant protein. Transfection experiments can be conducted in relevant cell lines, using nucleic acids of the disclosure and protein production can be assayed at various time points post-transfection.
- cellscan be transfected with different ARE- engineering molecules and by using an ELISA kit to the relevant protein and assaying protein produced at 6 hours, 12 hours, 1 day, 2 days, and 7 days post-transfection.
- 5’ UTRs that are heterologous or syntheticmay be used with any desired 3’ UTR sequence.
- a heterologous or synthetic 5’ UTRmay be used with a synthetic 3’ UTR or with a heterologous 3’ UTR.
- Non-UTR sequencesmay also be used as regions or subregions within a nucleic acid.
- introns or portions of introns sequencesmay be incorporated into regions of nucleic acid of the disclosure.
- the ORFmay be flanked by a 5′ UTR which may contain a strong Kozak translational initiation signal and/or a 3' UTR which may include an oligo(dT) sequence for templated addition of a poly-A tail.
- a 5′ UTRmay comprise a first polynucleotide fragment and a second polynucleotide fragment from the same and/or different genes such as the 5′ UTRs described in US2010/0293625 and WO2015/085318A2, each of which is herein incorporated by reference.
- any UTR from any genemay be incorporated into the regions of a nucleic acid.
- multiple wild-type UTRs of any known genemay be utilized. It is also within the scope of the present disclosure to provide artificial UTRs which are not variants of wild type regions. These UTRs or portions thereof may be placed in the same orientation as in the transcript from which they were selected or may be altered in orientation or location. Hence a 5′ or 3′ UTR may be inverted, shortened, lengthened, made with one or more other 5′ UTRs or 3′ UTRs.
- the term “altered” as it relates to a UTR sequencemeans that the UTR has been changed in some way in relation to a naturally occurring or comparator sequence.
- a 3′ UTR or 5′ UTRmay be altered relative to a wild- type/native UTR by the change in orientation or location as taught above or may be altered by the inclusion of additional nucleotides, deletion of nucleotides, swapping or transposition of nucleotides. Any of these changes producing an “altered” UTR (whether 3′ or 5′) comprise a variant UTR.
- a double, triple or quadruple UTRsuch as a 5′ UTR or 3′ UTR may be used.
- a “double” UTRis one in which two copies of the same UTR are encoded either in series or substantially in series.
- a double beta-globin 3′ UTRmay be used as described in US2010/0129877, which is incorporated herein by reference. It is also within the scope of the present disclosure to have patterned UTRs. As used herein “patterned UTRs” are those UTRs which reflect a repeating or alternating pattern, such as ABABAB or AABBAABBAABB or ABCABCABC or variants thereof repeated once, twice, or more than 3 times. In these patterns, each letter, A, B, or C represent a different UTR at the nucleotide level. In some embodiments, flanking regions are selected from a family of transcripts whose proteins share a common function, structure, feature, or property.
- polypeptides of interestmay belong to a family of proteins which are expressed in a particular cell, tissue or at some time during development.
- the UTRs from any of these genesmay be swapped for any other UTR of the same or different family of proteins to create a new polynucleotide.
- a “family of proteins”is used in the broadest sense to refer to a group of two or more polypeptides of interest which share at least one function, structure, feature, localization, origin, or expression pattern.
- the untranslated regionmay also include translation enhancer elements (TEE).
- TEEtranslation enhancer elements
- the TEEmay include those described in US 2009/0226470, herein incorporated by reference, and those known in the art.
- Open Reading FramesAn open reading frame (ORF) is a continuous stretch of DNA or RNA that (1) begins with a start codon (e.g., ATG or AUG, encoding methionine), and (2) ends with a stop codon (e.g., TAA, TAG or TGA, or UAA, UAG or UGA) or is immediately followed by a stop codon.
- a stop codondoes not encode an amino acid, such that translation of an ORF terminates when a ribosome reaches the stop codon immediately following the last amino acid-encoding codon in the ORF.
- a stop codon that results in translation terminationmay be considered part of the ORF, in which case the ORF ends with the stop codon.
- the first stop codon immediately following the last amino acid-encoding codon of an ORFmay considered part of the 3′ untranslated region (3′ UTR) of a DNA or RNA, rather than part of the ORF.
- 3′ UTR3′ untranslated region
- an ORF sequence that ends in a codon encoding amino acidwill be followed by one or more stop codons in a DNA or RNA.
- An ORFmay be followed by multiple stop codons.
- stop codonsreduces the extent of continued translation that may occur if a stop codon is mutated to a codon encoding an amino acid (readthrough), as a second stop codon may terminate translation even if a first stop codon is mutated and encodes an amino acid, such that only one amino acid is added to the C-terminus of the translated protein.
- the multiple stop codonsmay comprise the same stop codon (e.g., UGAUGA).
- Multiple stop codonsmay comprise different stop codons in series (e.g., UGAUAAUAG).
- an ORFtypically encodes a protein. It will be understood that the sequences disclosed herein may further comprise additional elements, e.g., 5’ and/or 3’ UTRs, but that those elements, unlike the ORF, need not necessarily be present in an RNA (e.g., mRNA) as described herein.
- an RNAcomprises a 5′ terminal cap.
- 5′-capping of polynucleotidesmay be completed concomitantly during an in vitro transcription reaction using, for example, the following chemical RNA cap analogs to generate the 5′-guanosine cap structure according to manufacturer protocols: 3 ⁇ -O-Me-m7G(5')ppp(5') G [the ARCA cap];G(5')ppp(5')A; G(5')ppp(5')G; m7G(5')ppp(5')A; m7G(5')ppp(5')G (New England BioLabs, Ipswich, MA).
- 5′- capping of modified RNAmay be completed post-transcriptionally using, for example, a Vaccinia Virus Capping Enzyme to generate the “Cap 0” structure: m7G(5')ppp
- Cap 1 structuremay be generated using both Vaccinia Virus Capping Enzyme and a 2′-O methyl-transferase to generate: m7G(5')ppp(5')G-2′-O-methyl.
- Cap 2 structuremay be generated from the Cap 1 structure followed by the 2′-O-methylation of the 5′-antepenultimate nucleotide using a 2′-O methyl-transferase.
- Cap 3 structuremay be generated from the Cap 2 structure followed by the 2′-O-methylation of the 5′- preantepenultimate nucleotide using a 2′-O methyl-transferase.
- Enzymesmay be derived from a recombinant source. Other cap analogs may be used.
- a cap analogmay be, for example, a dinucleotide cap, a trinucleotide cap, or a tetranucleotide cap.
- a cap analogis a dinucleotide cap.
- a cap analogis a trinucleotide cap.
- a cap analogis a tetranucleotide cap.
- a nucleotide cap(e.g., a trinucleotide cap or tetranucleotide cap), in some embodiments, comprises a compound of formula (I) tautomer or salt thereof, wherein ring B1 is a modified or unmodified Guanine; ring B2 and ring B3 each independently is a nucleobase or a modified nucleobase; X 2 is O, S(O) p , NR 24 or CR 25 R 26 in which p is 0, 1, or 2; Y0 is O or CR6R7; Y1 is O, S(O)n, CR6R7, or NR8, in which n is 0, 1 , or 2; each --- is a single bond or absent, wherein when each --- is a single bond, Yi is O, S(O) n , CR 6 R 7 , or NR 8 ; and when each --- is absent, Y 1 is void; Y 2 is (OP(O)R
- a cap analogmay include any of the cap analogs described in international publication WO 2017/066797, published on 20 April 2017, incorporated by reference herein in its entirety.
- the B2 middle positioncan be a non-ribose molecule, such as arabinose.
- R2is ethyl-based.
- a tetranucleotide capcomprises the following structure: In other embodiments, a tetranucleotide cap comprises the following structure: In yet other embodiments, a tetranucleotide cap comprises the following structure: In yet other embodiments, a tetranucleotide cap comprises the following structure:
- Ris an alkyl (e.g., C 1 -C 6 alkyl). In some embodiments, R is a methyl group (e.g., C1 alkyl). In some embodiments, R is an ethyl group (e.g., C2 alkyl). In some embodiments, R is a hydrogen. In some embodiments, a tetranucleotide cap comprises GGAG. In some embodiments, a tetranucleotide cap comprises any one of the following structures: In some embodiments, an mRNA comprises an m2 3′O,7 G + (5′)ppp(5′)Am cap having the structure: .
- poly(A) tailis a region of mRNA that is downstream, e.g., directly downstream (i.e., 3′), from the 3′ UTR that contains multiple, consecutive adenosine monophosphates.
- a poly(A) tailmay contain 10 to 300 adenosine monophosphates. It can, in some instances, comprise up to about 400 adenine nucleotides.
- a poly(A) tailmay contain 10, 20, 30, 40, 50, 60, 70, 80, 90, 100, 110, 120, 130, 140, 150, 160, 170, 180, 190, 200, 210, 220, 230, 240, 250, 260, 270, 280, 290 or 300 adenosine monophosphates.
- a poly(A) tailcontains 50 to 250 adenosine monophosphates.
- the poly(A) tailfunctions to protect mRNA from enzymatic degradation, e.g., in the cytoplasm, and aids in transcription termination, and/or export of the mRNA from the nucleus and translation.
- the length of the 3′-poly(A) tailmay be an essential element with respect to the stability of the individual mRNA.
- a poly(A) tailhas a length of about 50, about 100, about 150, about 200, about 250, about 300, about 350, or about 400 nucleotides.
- a poly(A) tailhas a length of 100 nucleotides.
- an mRNAcomprises a poly(A) sequence that has a length of 50– 75 nucleotides.
- an mRNAcomprises a poly(A) sequence that comprises 60, 61, 62, 63, 64, 65, 66, 67, 68, 69, or 70 consecutive adenosine nucleotides.
- an mRNAcomprises a poly(A) sequence comprising 64 consecutive adenosine nucleotides.
- the consecutive adenosine nucleotides of a poly(A) sequenceare flanked at the 5′ and 3′ end by nucleotides that are not adenosine nucleotides.
- an mRNAcomprises a poly(C) sequence, which may comprise 10 to 300 cytidine nucleotides.
- the poly(C) sequencecomprises 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, or 40 consecutive cytidine nucleotides.
- the poly(C) sequencecomprises 30 cytidine nucleotides.
- the consecutive cytidine nucleotides of a poly(C) sequenceare flanked at the 5′ and 3′ end by nucleotides that are not cytidine nucleotides.
- an mRNAcomprises two poly(A) sequences separated by an intervening nucleotide sequence.
- the intervening nucleotide sequencecomprises no more than 3, no more than two, no more than 1, or no adenosine nucleotides. In some embodiments, the intervening sequence comprises 3 adenosine nucleotides. In some embodiments, the intervening sequence does not comprise an adenosine nucleotide. In some embodiments, the intervening sequence is no more than 30, no more than 25, no more than 20, no more than 15, or no more than 10 nucleotides long. In some embodiments, the intervening sequence consists of 10 nucleotides. In some embodiments, the intervening sequence comprises the sequence of GCAUAUGACU (SEQ ID NO: 55).
- the intervening sequencedoes not begin with an adenosine nucleotide, and does not end with an adenosine nucleotide.
- the first poly(A) sequencescomprises at least 15, at least 20, at least 25, or at least 30 consecutive adenosine nucleotides.
- the second poly(A) sequencescomprises at least 55, at least 60, at least 65, or at least 70 consecutive adenosine nucleotides.
- the first poly(A) sequencecomprises 30 consecutive adenosine nucleotides.
- the second poly(A) sequencecomprises 70 adenosine nucleotides.
- an mRNAcomprises the nucleotide sequence AAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA (SEQ ID NO: 80). In some embodiments, an mRNA comprises a poly(A) sequence that has a length of 90– 120 nucleotides. In some embodiments, an mRNA comprises a poly(A) sequence that comprises 100, 101, 102, 103, 104, 105, 106, 107, 108, 109, 110, 111, 112, 113, 114, 115, 116, 117, 118, 190, or 120 consecutive adenosine nucleotides.
- an mRNAcomprises a poly(A) sequence that comprises at least 109 consecutive adenosine nucleotides. In some embodiments, an mRNA comprises a poly(A) sequence that comprises 109 consecutive adenosine nucleotides. In some embodiments, an mRNA comprises a poly(A) sequence that consists of 109 consecutive adenosine nucleotides.
- Self-amplifying RNASome aspects of the disclosure relate to self-amplifying RNA (e.g., an RNA replicon) encoding a MERS-CoV S protein or variant thereof (e.g., fusion protein comprising one or more domains of a MERS-CoV S protein).
- An self-amplifying RNArefers to an RNA encoding one or more molecules (e.g., proteins) that, individually or in conjunction, are capable of replicating the self-amplifying RNA.
- the proteins encoded by the self-amplifying RNAare non-structural proteins nsP1, nsP2, nsP3, and nsP4, which form an RNA-dependent RNA polymerase (RdRp), or replicase, that is capable of replicating the self-amplifying RNA.
- a self-amplifying RNAis capable of self-amplification in a cell, provided that the cell can translate the RNA and produce the encoded protein(s).
- a self-amplifying RNAmay be referred to as an RNA replicon.
- an RNA replicon or self-amplifying RNAis translated, the one or more encoded viral non-structural proteins are translated.
- a “viral non-structural protein”is a protein encoded by a virus but that is not part of the virus particle.
- the viral non-structural proteinsin the context of self-amplifying RNA, replicate the nucleotide sequences encoding the vaccine antigen or therapeutic protein (e.g., MERS-CoV protein) from the self-amplifying RNA via the sub- genomic viral promoters. Such replication driven by the viral sub-genomic promoter using the viral non- structural proteins enhances the expression level of the encoded protein.
- the viral non-structural proteinsare from a single-strand positive-sense RNA viruses. In some embodiments, the viral non-structural proteins are from an Alphavirus, belonging to the Togaviridae family. In some embodiments, the alphavirus is Sindbis or Venezuelan equine encephalitis virus. In some embodiments, the viral non-structural protein is an RNA-dependent RNA polymerase (RdRp) polyprotein P1234 (also termed NSP1-4). Upon translation, P1234 is rapidly cleaved into P123 and nsP4 by autoproteolytic activity originating from the nsP2 (proteinase) portion of the polyprotein.
- RdRpRNA-dependent RNA polymerase
- Alphaviral RNA synthesisoccurs at the plasma membrane of a cell, where the nsPs, together with alphaviral RNA, form membrane invaginations (or “spherules”). These spherules contain dsRNA created by replication of “+” strand viral genomic RNA into “–“ strand anti-genomic RNA. The “–“ strand serves as a template from which additional “+” strand genomic RNA (synthesized from the 5’UTR) or a shorter subsequence of the genomic RNA (termed subgenomic RNA) is synthesized from the subgenomic viral promoter region located near the end of the nonstructural protein ORF.
- the “+” strand genomic RNA and the subgenomic RNAare exported out of the spherules into the cytoplasm where they are translated by endogenous ribosomes.
- the exported “+” strand genomic RNAcan associate with nsPs and form additional spherules, thus resulting in exponential increase of replicon RNA.
- the viral non-structural proteinsfacilitate the replication of the nucleotide sequences encoding the MERS-CoV protein via the subgenomic viral promoters (also referred to as “subgenomic promoters” herein).
- a “subgenomic viral promoter”refers to a promoter the drives the transcription of subgenomic mRNAs.
- an mRNAis transcribed from genomic DNAs and episomal DNAs (e.g., plasmids). Some viruses may transcribe subgenomic mRNAs from a RNA replicon that is produced from its genomic RNA. Many positive-sense RNA viruses produce subgenomic mRNAs as one of the common infection techniques used by these viruses and generally transcribe late viral genes. Subgenomic viral promoters range from 20 nucleotide (Sindbis virus) to over 100 nucleotides (Beet necrotic yellow vein virus) and are usually found upstream of the transcription start.
- the subgenomic viral promoteris 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49, 50, 51, 52, 53, 54, 55, 56, 57, 58, 59, 60, 61, 62, 63, 64, 65, 66, 67, 68, 69, 70, 71, 72, 73, 74, 75, 76, 77, 78, 79, 80, 81, 82, 83, 84, 85, 86, 87, 88, 89, 90, 91, 92, 93, 94, 95, 96, 97, 98, 99, 100 nucleotides long, or longer.
- RNAe.g., mRNA
- Stabilizing elementsmay include, for example, a histone stem-loop.
- a stem- loop binding protein (SLBP)a 32 kDa protein has been identified. It is associated with the histone stem-loop at the 3'-end of the histone messages in both the nucleus and the cytoplasm.
- SLBPRNA binding domain of SLBP is conserved through metazoa and protozoa; its binding to the histone stem-loop depends on the structure of the loop.
- the minimum binding siteincludes at least three nucleotides 5’ and two nucleotides 3 ⁇ relative to the stem-loop.
- an RNA(e.g., mRNA) includes an open reading frame (coding region), a histone stem-loop, and optionally, a poly(A) sequence or polyadenylation signal.
- the poly(A) sequence or polyadenylation signalgenerally should enhance the expression level of the encoded protein.
- the encoded proteinin some embodiments, is not a histone protein, a reporter protein (e.g., Luciferase, GFP, EGFP, ⁇ -Galactosidase, EGFP), or a marker or selection protein (e.g., alpha-Globin, Galactokinase and Xanthine:guanine phosphoribosyl transferase (GPT)).
- a reporter proteine.g., Luciferase, GFP, EGFP, ⁇ -Galactosidase, EGFP
- a marker or selection proteine.g., alpha-Globin, Galactokina
- an RNAe.g., mRNA
- an RNAincludes the combination of a poly(A) sequence or polyadenylation signal and at least one histone stem-loop, even though both represent alternative mechanisms in nature, they act synergistically to increase the protein expression beyond the level observed with either of the individual elements.
- the synergistic effect of the combination of poly(A) and a histone stem-loopdoes not depend on the order of the elements or the length of the poly(A) sequence.
- an RNAe.g., mRNA
- HDEhistone downstream element
- Histone downstream elementincludes a purine-rich polynucleotide stretch of approximately 15 to 20 nucleotides 3′ of naturally-occurring stem-loops, representing the binding site for the U7 snRNA, which is involved in processing of histone pre-mRNA into mature histone mRNA.
- the nucleic aciddoes not include an intron.
- An RNAe.g., mRNA
- the histone stem-loopis generally derived from histone genes and includes an intramolecular base pairing of two neighbored partially or entirely reverse complementary sequences separated by a spacer, consisting of a short sequence, which forms the loop of the structure.
- the unpaired loop regionis typically unable to base pair with either of the stem loop elements. It occurs more often in RNA, as is a key component of many RNA secondary structures but may be present in single-stranded DNA as well. Stability of the stem-loop structure generally depends on the length, number of mismatches or bulges, and base composition of the paired region. In some embodiments, wobble base pairing (non-Watson-Crick base pairing) may result.
- the at least one histone stem-loop sequencecomprises a length of 15 to 45 nucleotides.
- an RNAe.g., mRNA
- AURESAU-rich sequences removed. These sequences, sometimes referred to as AURES are destabilizing sequences found in the 3 ’UTR. The AURES may be removed from the mRNA. Alternatively, the AURES may remain in the mRNA. Sequence Modification
- an open reading frame encoding a protein of the disclosureis codon optimized. Codon optimization methods are known in the art. An open reading frame of any one or more of the sequences provided herein may be codon optimized.
- Codon optimizationmay be used to match codon frequencies in target and host organisms to ensure proper folding; bias GC content to increase RNA (e.g., mRNA) stability or reduce secondary structures; minimize tandem repeat codons or base runs that may impair gene construction or expression; customize transcriptional and translational control regions; insert or remove protein trafficking sequences; remove/add post translation modification sites in encoded protein (e.g., glycosylation sites); add, remove or shuffle protein domains; insert or delete restriction sites; modify ribosome binding sites and RNA (e.g., mRNA) degradation sites; adjust translational rates to allow the various domains of the protein to fold properly; or reduce or eliminate problem secondary structures within the polynucleotide.
- RNAe.g., mRNA
- Codon optimization tools, algorithms and servicesare known in the art – non-limiting examples include services from GeneArt (Life Technologies), DNA2.0 (Menlo Park CA) and/or proprietary methods.
- the open reading frame sequenceis optimized using optimization algorithms.
- a codon optimized sequenceshares less than 95% sequence identity to a naturally-occurring or wild-type sequence open reading frame (e.g., a naturally- occurring or wild-type RNA (e.g., mRNA) sequence encoding a MERS-CoV protein antigen).
- a codon optimized sequenceshares less than 90% sequence identity to a naturally-occurring or wild-type sequence (e.g., a naturally-occurring or wild-type RNA (e.g., mRNA) sequence encoding a MERS-CoV protein). In some embodiments, a codon optimized sequence shares less than 85% sequence identity to a naturally-occurring or wild-type sequence (e.g., a naturally-occurring or wild-type RNA (e.g., mRNA) sequence encoding a MERS-CoV protein).
- a naturally-occurring or wild-type sequencee.g., a naturally-occurring or wild-type RNA (e.g., mRNA) sequence encoding a MERS-CoV protein.
- a codon optimized sequenceshares less than 80% sequence identity to a naturally-occurring or wild-type sequence (e.g., a naturally-occurring or wild-type RNA (e.g., mRNA) sequence encoding a MERS-CoV protein). In some embodiments, a codon optimized sequence shares less than 75% sequence identity to a naturally-occurring or wild-type sequence (e.g., a naturally-occurring or wild-type RNA (e.g., mRNA) sequence encoding a MERS-CoV protein).
- a codon optimized sequenceshares between 65% and 85% (e.g., between about 67% and about 85% or between about 67% and about 80%) sequence identity to a naturally-occurring or wild-type sequence (e.g., a naturally-occurring or wild-type RNA (e.g., mRNA) sequence encoding a MERS-CoV protein).
- a codon optimized sequenceshares between 65% and 75% or about 80% sequence identity to a naturally-occurring or wild-type sequence (e.g., a naturally-occurring or wild-type RNA (e.g., mRNA) sequence encoding a MERS-CoV protein).
- a codon-optimized sequenceencodes an antigen that is as immunogenic as, or more immunogenic than (e.g., at least 10%, at least 20%, at least 30%, at least 40%, at least 50%, at least 100%, or at least 200% more), than a MERS-CoV protein encoded by a non-codon-optimized sequence.
- the modified mRNAsWhen transfected into mammalian host cells, the modified mRNAs have a stability of between 12-18 hours, or greater than 18 hours, e.g., 24, 36, 48, 60, 72, or greater than 72 hours and are capable of being expressed by the mammalian host cells.
- a codon optimized RNAmay be one in which the levels of G/C are enhanced.
- the G/C-content of nucleic acid moleculesmay influence the stability of the RNA.
- RNAe.g., mRNA
- having an increased amount of guanine (G) and/or cytosine (C) residuesmay be functionally more stable than RNA containing a large amount of adenine (A) and thymine (T) or uracil (U) nucleotides.
- WO02/098443discloses a pharmaceutical composition containing an RNA (e.g., mRNA) stabilized by sequence modifications in the translated region.
- RNAs described hereincomprise a sequence with a %G/C content of 30% – 80%, 40% – 70%, 50% – 60%, 35% – 50%, 50% – 65%, 65% – 70%, 40% – 45%, 45% – 50%, 50% – 55%, 55% – 70%, 70% – 75%, or 75% – 80%.
- the nucleic acid sequence of the full-length mRNAcomprises a %G/C content of 30% to 80%, 40% – 70%, 50% – 60%, 35% – 50%, 50% – 65%, 65% – 70%, 40% – 45%, 45% – 50%, 50% – 55%, 55% – 70%, 70% – 75%, or 75% – 80%.
- the mRNAcomprises an ORF with a %G/C content from about 30% to about 80%, about 35% to about 70%, about 40% to about 60%, about 45% to about 55%, about 40% to about 70%, about 50% to about 60%, about 35% to about 50%, about 50% to about 50% to about 65%, about 65% to about 70%, about 40% to about 45%, about 45% to about 50%, about 50% to about 55%, about 55% to about 70%, about 70% to about 75%, or about 75% to about 80%.
- the mRNAcomprises 5′ UTR with a %G/C content from about 30% to about 80%, about 35% to about 70%, about 40% to about 60%, about 45% to about 55%, about 40% to about 70%, about 50% to about 60%, about 35% to about 50%, about 50% to about 50% to about 65%, about 65% to about 70%, about 40% to about 45%, about 45% to about 50%, about 50% to about 55%, about 55% to about 70%, about 70% to about 75%, or about 75% to about 80%.
- the mRNAcomprises 3′ UTR with a %G/C content from about 30% to about 80%, about 35% to about 70%, about 40% to about 60%, about 45% to about 55%, about 40% to about 70%, about 50% to about 60%, about 35% to about 50%, about 50% to about 50% to about 65%, about 65% to about 70%, about 40% to about 45%, about 45% to about 50%, about 50% to about 55%, about 55% to about 70%, about 70% to about 75%, or about 75% to about 80%.
- a modified mRNA made by a method described hereincomprises a higher %G/C content than a wild-type mRNA sequence.
- the %G/C content of the modified mRNA sequenceis 2% or more, 3% or more, 4% or more, 5% or more, 6% or more, 7% or more, 8% or more, 9% or more, 10% or more, 12% or more, 15% or more, or 20% or more than the %G/C content of the wild-type RNA sequence.
- the %G/C content of the modified ORF sequenceis 2% or more, 3% or more, 4% or more, 5% or more, 6% or more, 7% or more, 8% or more, 9% or more, 10% or more, 12% or more, 15% or more, or 20% or more than the %G/C content of the wild-type ORF sequence.
- the %G/C content of the modified 5′ UTR sequenceis 2% or more, 3% or more, 4% or more, 5% or more, 6% or more, 7% or more, 8% or more, 9% or more, 10% or more, 12% or more, 15% or more, or 20% or more than the %G/C content of the wild-type 3′ UTR sequence.
- Chemically Modified Nucleotidescomprise, in some embodiments, an RNA having an open reading frame encoding a protein, wherein the nucleic acid comprises nucleotides and/or nucleosides that can be standard (unmodified) or modified as is known in the art.
- nucleotides and nucleosides of the present disclosurecomprise modified nucleotides or nucleosides.
- modified nucleotides and nucleosidescan be naturally- occurring modified nucleotides and nucleosides or non-naturally-occurring modified nucleotides and nucleosides.
- modificationscan include those at the sugar, backbone, or nucleobase portion of the nucleotide and/or nucleoside as are recognized in the art.
- a naturally-occurring modified nucleotide or nucleotide of the disclosureis one as is generally known or recognized in the art.
- Non-limiting examples of such naturally-occurring modified nucleotides and nucleotidescan be found, inter alia, in the widely recognized MODOMICS database.
- a non-naturally-occurring modified nucleotide or nucleoside of the disclosureis one as is generally known or recognized in the art.
- Non-limiting examples of such non-naturally-occurring modified nucleotides and nucleosidescan be found, inter alia, in international publication numbers WO2013/052523A1; WO2014/093924A1; WO2015/051173A2; WO2015/051169A2; WO2015/089511A2; or WO2017/153936A1, each of which is herein incorporated by reference.
- nucleic acids of the disclosurecan comprise standard nucleotides and nucleosides, naturally-occurring nucleotides and nucleosides, non-naturally-occurring nucleotides and nucleosides, or any combination thereof.
- Nucleic acids of the disclosuree.g., DNA and RNA, such as mRNA
- nucleic acids of the disclosurein some embodiments, comprise various (more than one) different types of standard and/or modified nucleotides and nucleosides.
- a particular region of a nucleic acidcontains one, two or more (optionally different) types of standard and/or modified nucleotides and nucleosides.
- a modified RNAintroduced to a cell or organism, exhibits reduced degradation in the cell or organism, respectively, relative to an unmodified nucleic acid comprising standard nucleotides and nucleosides.
- a modified RNAintroduced into a cell or organism, may exhibit reduced immunogenicity in the cell or organism, respectively (e.g., a reduced innate response) relative to an unmodified nucleic acid comprising standard nucleotides and nucleosides.
- Nucleic acidsin some embodiments, comprise non-natural modified nucleotides that are introduced during synthesis or post-synthesis of the nucleic acids to achieve desired functions or properties.
- the modificationsmay be present on internucleotide linkages, purine or pyrimidine bases, or sugars.
- the modificationmay be introduced with chemical synthesis or with a polymerase enzyme at the terminal of a chain or anywhere else in the chain. Any of the regions of a nucleic acid may be chemically modified.
- the present disclosureprovides for modified nucleosides and nucleotides of a nucleic acid (e.g., RNA nucleic acids, such as mRNA nucleic acids).
- nucleosiderefers to a compound containing a sugar molecule (e.g., a pentose or ribose) or a derivative thereof in combination with an organic base (e.g., a purine or pyrimidine) or a derivative thereof (also referred to herein as “nucleobase”).
- organic basee.g., a purine or pyrimidine
- nucleobasealso referred to herein as “nucleobase”.
- nucleotiderefers to a nucleoside, including a phosphate group. Modified nucleotides may by synthesized by any useful method, such as, for example, chemically, enzymatically, or recombinantly, to include one or more modified or non-natural nucleosides.
- Nucleic acidscan comprise a region or regions of linked nucleosides.
- Such regionsmay have variable backbone linkages.
- the linkagescan be standard phosphodiester linkages, in which case the nucleic acids would comprise regions of nucleotides.
- Modified nucleotide base pairingencompasses not only the standard adenosine-thymine, adenosine-uracil, or guanosine-cytosine base pairs, but also base pairs formed between nucleotides and/or modified nucleotides comprising non-standard or modified bases, wherein the arrangement of hydrogen bond donors and hydrogen bond acceptors permits hydrogen bonding between a non-standard base and a standard base or between two complementary non-standard base structures, such as, for example, in those nucleic acids having at least one chemical modification.
- modified nucleobases in nucleic acidscomprise 1-methyl-pseudouridine (m1 ⁇ ), 1-ethyl-pseudouridine (e1 ⁇ ), 5-methoxy-uridine (mo5U), 5-methyl-cytidine (m5C), and/or pseudouridine ( ⁇ ).
- modified nucleobases in nucleic acidscomprise 5-methoxymethyl uridine, 5-methylthio uridine, 1-methoxymethyl pseudouridine, 5-methyl cytidine, and/or 5-methoxy cytidine.
- the polyribonucleotideincludes a combination of at least two (e.g., 2, 3, 4 or more) of any of the aforementioned modified nucleobases, including but not limited to chemical modifications.
- a mRNA of the disclosurecomprises 1-methyl-pseudouridine (m1 ⁇ ) substitutions at one or more or all uridine positions of the nucleic acid.
- a mRNA of the disclosurecomprises 1-methyl-pseudouridine (m1 ⁇ ) substitutions at one or more or all uridine positions of the nucleic acid and 5-methyl cytidine substitutions at one or more or all cytidine positions of the nucleic acid.
- a mRNA of the disclosurecomprises pseudouridine ( ⁇ ) substitutions at one or more or all uridine positions of the nucleic acid.
- a mRNA of the disclosurecomprises pseudouridine ( ⁇ ) substitutions at one or more or all uridine positions of the nucleic acid and 5-methyl cytidine substitutions at one or more or all cytidine positions of the nucleic acid.
- a mRNA of the disclosurecomprises uridine at one or more or all uridine positions of the nucleic acid.
- mRNAsare uniformly modified (e.g., fully modified, modified throughout the entire sequence) for a particular modification.
- a nucleic acidcan be uniformly modified with 1-methyl-pseudouridine, meaning that all uridine residues in the mRNA sequence are replaced with 1-methyl-pseudouridine.
- a nucleic acidcan be uniformly modified for any type of nucleoside residue present in the sequence by replacement with a modified residue such as those set forth above.
- the nucleic acids of the present disclosuremay be partially or fully modified along the entire length of the molecule.
- one or more or all or a given type of nucleotidemay be uniformly modified in a nucleic acid of the disclosure, or in a predetermined sequence region thereof (e.g., in the mRNA including or excluding the poly(A) tail).
- nucleotides X in a nucleic acid of the present disclosureare modified nucleotides, wherein X may be any one of nucleotides A, G, U, C, or any one of the combinations A+G, A+U, A+C, G+U, G+C, U+C, A+G+U, A+G+C, G+U+C or A+G+C.
- the nucleic acidmay contain from about 1% to about 100% modified nucleotides (either in relation to overall nucleotide content, or in relation to one or more types of nucleotide, i.e., any one or more of A, G, U or C) or any intervening percentage (e.g., from 1% to 20%, from 1% to 25%, from 1% to 50%, from 1% to 60%, from 1% to 70%, from 1% to 80%, from 1% to 90%, from 1% to 95%, from 10% to 20%, from 10% to 25%, from 10% to 50%, from 10% to 60%, from 10% to 70%, from 10% to 80%, from 10% to 90%, from 10% to 95%, from 10% to 100%, from 20% to 25%, from 20% to 50%, from 20% to 60%, from 20% to 70%, from 20% to 80%, from 20% to 90%, from 20% to 95%, from 20% to 100%, from 50% to 60%, from 50% to 70%, from 50% to 80%, from 50% to 90%, from 50% to 95%, from 50% to 100%, from 70% to
- the mRNAsmay contain at a minimum 1% and at maximum 100% modified nucleotides, or any intervening percentage, such as at least 5% modified nucleotides, at least 10% modified nucleotides, at least 25% modified nucleotides, at least 50% modified nucleotides, at least 80% modified nucleotides, or at least 90% modified nucleotides.
- the nucleic acidsmay contain a modified pyrimidine such as a modified uracil or cytosine.
- At least 5%, at least 10%, at least 25%, at least 50%, at least 80%, at least 90% or 100% of the uracil in the nucleic acidis replaced with a modified uracil (e.g., a 5-substituted uracil).
- the modified uracilcan be replaced by a compound having a single unique structure or can be replaced by a plurality of compounds having different structures (e.g., 2, 3, 4 or more unique structures).
- cytosine in the nucleic acidis replaced with a modified cytosine (e.g., a 5-substituted cytosine).
- the modified cytosinecan be replaced by a compound having a single unique structure or can be replaced by a plurality of compounds having different structures (e.g., 2, 3, 4 or more unique structures).
- Modified nucleotidesmay include modified nucleobases.
- an RNA transcript(e.g., mRNA transcript) described herein may include a modified uracil nucleobase selected from pseudouracil ( ⁇ ), N1-methylpseudouracil (m1 ⁇ ), 1-ethylpseudouracil, 2-thiouracil, 4′-thiouracil, 2-thio-1-methyl-1-deaza-pseudouracil, 2-thio-1-methyl-pseudouracil, 2-thio-5-aza-uracil, 2-thio- dihydropseudouracil, 2-thio-dihydrouracil, 2-thio-pseudouracil, 4-methoxy-2-thio-pseudouracil, 4-methoxy-pseudouracil, 4-thio-1-methyl-pseudouracil, 4-thio-pseudouracil, 5-aza-uracil, dihydropseudouracil, 5-methyluracil,
- an RNA transcript(e.g., mRNA transcript) includes a modified guanine nucleobase selected from digoxigeninated guanine, 6-thioguanine, 7-deazaguanine, 7-deaza-7- propargylaminoguanine, 8-oxoguanine, araguanine, biotin-16-7-deaza-7-propargylaminoguanine, isoguanine, N2-methylguanine, O6-methylguanine, thienoguanine, and 2,6-daminoguanine.
- a modified guanine nucleobaseselected from digoxigeninated guanine, 6-thioguanine, 7-deazaguanine, 7-deaza-7- propargylaminoguanine, 8-oxoguanine, araguanine, biotin-16-7-deaza-7-propargylaminoguanine, isoguanine, N2-methylguanine, O6-methylgu
- an RNA transcriptmay include a modified cytosine nucleobase selected from digoxigeninated cytosine, 2-thiocytosine, 5-aminoallylcytosine, 5-bromocytosine, 5- carboxycytosine, 5-formylcytosine, 5-hydroxycytosine, 5-hydroxymethylcytosine, 5- methoxycytosine, 5-methylcytosine, 5-propargylaminocytosine, 5-propynylcytosine, 6- azacytosine, aracytosine, cyanine 3-5-propargylaminocytosine, cyanine 3-aminoallylcytosine, cyanine 5-6-propargylaminocytosine, cyanine 5-aminoallylcytosine, desthiobiotin-6- aminoallylcytosine, N4-biotin-OBEA-cytosine, N4-methylcytosine, pseudoisocytosine, and thienocytosine.
- an RNA transcript(e.g., mRNA transcript) includes a modified adenine nucleobase selected from digoxigeninated adenine, N6-methyladenine, 7- deazaadenine, 7-deaza-7-propargylaminoadenine, 8-azaadenine, 8-azidoadenine, 8- chloroadenine, 8-oxoadenine, araadenine, N1-methyladenine, N6-methyladenine, 3- deazaadenine, 2,6-diaminoadenine, 2-methyl-thio-N6-isopentenyladenine (ms2i6A), 2- methylthio-N6-methyladenine (ms2m6A), N6-(cis-hydroxyisopentenyl)adenine (io6A), 2- methylthio-N6-(cis-hydroxyisopentenyl)adenine (ms2io6A), N6-gly
- an RNA transcript(e.g., mRNA transcript) includes a combination of at least two (e.g., 2, 3, 4 or more) of the foregoing modified nucleobases.
- Modified nucleotidesmay include modified sugars.
- an RNA transcript(e.g., mRNA transcript) described herein may include a modified sugar selected from 2′-thioribose, 2′,3′-dideoxyribose, 2′-amino-2′-deoxyribose, 2′ deoxyribose, 2′-azido-2′-deoxyribose, 2′-fluoro- 2′-deoxyribose, 2′-O-methylribose, 2′-O-methyldeoxyribose, 3′-amino-2′,3′-dideoxyribose, 3′- azido-2′,3′-dideoxyribose, 3′-deoxyribose, 3′-O-(2-nitrobenzyl)-2′-deoxyribose, 3′-O- methylribose, 5′-aminoribose, 5′-thioribose, 5-nitro-1-indolyl-2′-deoxy
- an RNA transcript(e.g., mRNA transcript) includes a combination of at least two (e.g., 2, 3, 4 or more) of the foregoing modified sugars.
- Modified nucleotidesmay include modified phosphates.
- a modified phosphate groupis a phosphate group that differs from the canonical structure of phosphate.
- An example of a canonical structure of a phosphateis shown below: , where R5 and R3 are atoms or molecules to which the canonical phosphate is bonded.
- R5may refer to the upstream nucleotide of the nucleic acid
- R 3may refer to the downstream nucleotide of the nucleic acid.
- the canonical structure of phosphatealso refers to structures in which one or more hydroxyl groups of the phosphate are deprotonated, or in which an oxygen atom of the phosphate is bonded to an adjacent nucleotide in a nucleic acid sequence.
- an RNA transcript(e.g., mRNA transcript) described herein may include a modified phosphate selected from phosphorothioate (PS), thiophosphate, 5′-O-methylphosphonate, 3′-O-methylphosphonate, 5′- hydroxyphosphonate, hydroxyphosphanate, phosphoroselenoate, selenophosphate, phosphoramidate, carbophosphonate, methylphosphonate, phenylphosphonate, ethylphosphonate, H-phosphonate, guanidinium ring, triazole ring, boranophosphate (BP), methylphosphonate, and guanidinopropyl phosphoramidate.
- PSphosphorothioate
- thiophosphate5′-O-methylphosphonate
- 3′-O-methylphosphonate5′- hydroxyphosphonate
- hydroxyphosphanatephosphoroselenoate
- selenophosphateselenophosphate
- an RNA transcript(e.g., mRNA transcript) includes a combination of at least two (e.g., 2, 3, 4 or more) of the foregoing modified phosphates.
- an mRNAincludes N1-methylpseudouridine.
- at least 70%, at least 80%, at least 85%, at least 90%, at least 95%, at least 97%, at least 98%, or at least 99% of uracil nucleotides in an mRNAcomprise N1-methylpseudouridine.
- each uracil nucleotide of an mRNA transcriptcomprises N1- methylpseudouridine.
- an mRNAincludes 5-methylcytidine. In some embodiments, at least 70%, at least 80%, at least 85%, at least 90%, at least 95%, at least 97%, at least 98%, or at least 99% of cytosine nucleotides in an mRNA comprise 5-methylcytidine. In some embodiments, each cytosine nucleotide of an mRNA transcript comprises 5- methylcytidine. In some embodiments, an mRNA includes 5-methyluridine. In some embodiments, at least 70%, at least 80%, at least 85%, at least 90%, at least 95%, at least 97%, at least 98%, or at least 99% of uracil nucleotides in an mRNA comprise 5-methyluridine.
- each uracil nucleotide of an mRNA transcriptcomprises 5-methyluridine.
- an mRNAincludes 5-methylcytidine and 5-methyluridine.
- at least 70%, at least 80%, at least 85%, at least 90%, at least 95%, at least 97%, at least 98%, or at least 99% of uracil nucleotides in an mRNAcomprise 5-methyluridine and at least 70%, at least 80%, at least 85%, at least 90%, at least 95%, at least 97%, at least 98%, or at least 99% of cytosine nucleotides in an mRNA comprise 5-methylcytidine.
- each cytosine nucleotide of an mRNA transcriptcomprises 5-methylcytidine and each uracil nucleotide of an mRNA transcript comprises 5-methyluridine.
- an RNAe.g., mRNA
- nucleotides and nucleosides of the present disclosurecomprise standard nucleoside residues such as those present in transcribed RNA (e.g., A, G, C, or U).
- nucleotides and nucleosides of the present disclosurecomprise standard deoxyribonucleosides such as those present in DNA (e.g., dA, dG, dC, or dT).
- IVTIn Vitro Transcription
- cDNA encoding the polynucleotides described hereinmay be transcribed using an in vitro transcription (IVT) system.
- IVTin vitro transcription
- the RNA of the present disclosureis prepared in accordance with any one or more of the methods described in WO 2018/053209 and WO 2019/036682, each of which is incorporated by reference herein.
- the RNA transcriptis generated using a non-amplified, linearized DNA template in an in vitro transcription reaction to generate the RNA transcript.
- the template DNAis isolated DNA.
- the template DNAis cDNA.
- the cDNAis formed by reverse transcription of a RNA polynucleotide, for example, but not limited to influenza virus mRNA.
- cellse.g., bacterial cells, e.g., E.
- colie.g., DH-1 cells are transfected with the plasmid DNA template.
- the transfected cellsare cultured to replicate the plasmid DNA which is then isolated and purified.
- the DNA templateincludes a RNA polymerase promoter, e.g., a T7 promoter located 5 ' to and operably linked to the gene of interest.
- an in vitro transcription templateencodes a 5′ untranslated (UTR) region, contains an open reading frame, and encodes a 3′ UTR and a poly(A) tail. The particular nucleic acid sequence composition and length of an in vitro transcription template will depend on the mRNA encoded by the template.
- An in vitro transcription systemtypically comprises a transcription buffer, nucleotide triphosphates (NTPs), an RNase inhibitor and a polymerase.
- NTPsmay be manufactured in house, may be selected from a supplier, or may be synthesized as described herein.
- the NTPsmay be selected from, but are not limited to, those described herein including natural and unnatural (modified) NTPs. Any number of RNA polymerases or variants may be used in the method of the present disclosure.
- the polymerasemay be selected from, but is not limited to, a phage RNA polymerase, e.g., a T7 RNA polymerase, a T3 RNA polymerase, a SP6 RNA polymerase, and/or mutant polymerases such as, but not limited to, polymerases able to incorporate modified nucleic acids and/or modified nucleotides, including chemically modified nucleic acids and/or nucleotides. Some embodiments exclude the use of DNase.
- the RNA transcriptis capped via enzymatic capping.
- the RNAcomprises 5' terminal cap, for example, 7mG(5’)ppp(5’)NlmpNp.
- RNA polymeraseis an RNA polymerase variant, such as those described in WO 2020/172239, incorporated herein by reference in its entirety.
- RNA polymerase variants of the present disclosureinclude at least one amino acid substitution, relative to the wild type (WT) RNA polymerase.
- a WT T7 RNA polymeraseis represented by SEQ ID NO: 83: MNTINIAKNDFSDIELAAIPFNTLADHYGERLAREQLALEHESYEMGEARFRKMFERQLKAGEVADNAAAKPLITTLL PKMIARINDWFEEVKAKRGKRPTAFQFLQEIKPEAVAYITIKTTLACLTSADNTTVQAVASAIGRAIEDEARFGRIRD LEAKHFKKNVEEQLNKRVGHVYKKAFMQVVEADMLSKGLLGGEAWSSWHKEDSIHVGVRCIEMLIESTGMVSLHRQNA GVVGQDSETIELAPEYAEAIATRAGALAGISPMFQPCVVPPKPWTGITGGGYWANGRRPLALVRTHSKKALMRYEDVY MPEVYKAINIAQNTAWKINKKVLAVANVITKWKHCPVEDIPAIEREELPMKPEDIDMNPEALTAWKRAAAAVYRKDKA RKSRRISLEFMLEQANKFANH
- the RNA polymerase variantis a T7 RNA polymerase variant comprising at least one (one or more) amino acid substitution relative to WT RNA polymerase (e.g., WT T7 RNA polymerase having an amino acid sequence of SEQ ID NO: 83).
- a RNA polymerase variantcomprises a RNA polymerase that includes an (at least one) amino acid modification causes a loop structure of the RNA polymerase variant to undergo a conformational change to a helix structure as the RNA polymerase variant transitions from an initiation complex to an elongation complex.
- the amino acid modificationis an amino acid substitution at one or more of positions 42, 43, 44, 45, 46, and 47, relative to the wild-type RNA polymerase, wherein the wild-type RNA polymerase comprises the amino acid sequence of SEQ ID NO: 83.
- the amino acid substitutionin some embodiments, is a high propensity amino acid substitution.
- RNA polymerase variantcomprise a RNA polymerase that includes an additional C-terminal amino acid, relative to the wild-type RNA polymerase.
- the additional C-terminal amino acidin some embodiments, is selected from glycine, alanine, threonine, proline, glutamine, and serine.
- the additional C-terminal amino acid(e.g., at position 884 relative to wild-type RNA polymerase comprising the amino acid sequence of SEQ ID NO: 83) is glycine.
- co-transcriptional capping methods for ribonucleic acid (RNA) synthesisusing any of the RNA polymerase variants described herein. That is, RNA is produced in a “one-pot” reaction, without the need for a separate capping reaction.
- the methodsin some embodiments, comprise reacting a polynucleotide template with a RNA polymerase variant, nucleoside triphosphates, and a cap analog under in vitro transcription reaction conditions to produce RNA transcript.
- one or more nucleic acidscomprises an Identification and Ratio Determination sequence.
- An Identification and Ratio Determination (IDR) sequenceis a sequence of a biological molecule (e.g., nucleic acid or protein) that, when combined with the sequence of a target biological molecule, serves to identify the target biological molecule.
- an IDR sequenceis a heterologous sequence that is incorporated within or appended to a sequence of a target biological molecule and can be used as a reference to identify the target molecule.
- a nucleic acidcomprises (i) a target sequence of interest (e.g., a coding sequence encoding a therapeutic and/or antigenic peptide or protein); and (ii) a unique IDR sequence.
- a target sequence of intereste.g., a coding sequence encoding a therapeutic and/or antigenic peptide or protein
- a unique IDR sequencee.g., an RNA species (e.g., RNA having a given coding sequence) may comprise an IDR sequence that differs from the IDR sequence of other RNA species (e.g., RNA(s) having different coding sequence(s)).
- Each IDR sequencethus identifies a particular RNA species, and so the abundance of IDR sequences may be measured to determine the abundance of each RNA species in a composition.
- RNA speciesUse of distinct IDR sequences to identify RNA species allows for analysis of multivalent RNA compositions (e.g., containing multiple RNA species) containing RNA species with similar coding sequences and/or lengths, which could otherwise be difficult to distinguish using PCR- or chromatography-based analysis of full-length RNAs.
- Each RNA species in a multivalent RNA compositionmay comprise an IDR sequence that is not a sequence isomer of an IDR sequence of another RNA species in a multivalent RNA composition (e.g., the IDR sequence does not have the same number of adenosine nucleotides, the same number of cytosine nucleotides, the same number of guanine nucleotides, and the same number of uracil nucleotides, as another IDR sequence in the composition, even if those sequences have different sequences).
- Having identical nucleotide compositionscauses sequence isomers to have the same mass, presenting a challenge to distinguishing sequence isomers using mass-based identification methods (e.g., mass spectrometry).
- Each RNA species in a multivalent RNA compositionmay comprise an IDR sequence having a mass that differs from the mass of IDR sequences of each other RNA species in a multivalent RNA composition.
- the mass of each IDR sequencemay differ from the mass of other IDR sequences by at least 9 Da, at least 25 Da, at least 25 Da, or at least 50 Da.
- Use of IDR sequences with distinct massesallows RNA fragments comprising different IDR sequences to be distinguished using mass-based analysis methods (e.g., mass spectrometry), which do not require reverse transcription, amplification, or sequencing of RNAs.
- Each RNA species in an RNA compositionmay comprises an IDR sequence with a different length.
- each IDR sequencemay have a length independently selected from 0 to 25 nucleotides.
- the length of a nucleic acidinfluences the rate at which the nucleic acid traverses a chromatography column, and so the use of IDR sequences of different lengths on different RNA species allows RNA fragments having different IDR sequences to be distinguished using chromatography-based methods (e.g., LC-UV).
- IDR sequencesmay be chosen such that no IDR sequence comprises a start codon, ‘AUG’. Lack of a start codon in an IDR sequence prevents undesired translation of nucleotide sequences within and/or downstream from the IDR sequence.
- IDR sequencesmay be chosen such that no IDR sequence comprises a recognition site for a restriction enzyme.
- no IDR sequencecomprises a recognition site for XbaI, ‘UCUAG’.
- Lack of a recognition site for a restriction enzymee.g., XbaI recognition site ‘UCUAG’) allows the restriction enzyme to be used in generating and modifying a DNA template for in vitro transcription, without affecting the IDR sequence or sequence of the transcribed RNA.
- each mRNA encoding a distinct proteincomprises a 3′ UTR comprising a distinct IDR sequence selected from SEQ ID NOs: 56–60.
- Nucleic Acid Production Chemical SynthesisNucleic acids as described herein may be manufactured in whole or in part using solid phase techniques.
- Solid-phase chemical synthesis of nucleic acidsis an automated method wherein molecules are immobilized on a solid support and synthesized step by step in a reactant solution. Solid-phase synthesis is useful in site-specific introduction of chemical modifications in the nucleic acid sequences.
- the synthesis of nucleic acids of the present disclosure by the sequential addition of monomer building blocksmay be carried out in a liquid phase.
- the synthetic methods discussed aboveeach has its own advantages and limitations. Attempts have been conducted to combine these methods to overcome the limitations. Such combinations of methods are within the scope of the present disclosure.
- the use of solid-phase or liquid-phase chemical synthesis in combination with enzymatic ligationprovides an efficient way to generate long chain nucleic acids that cannot be obtained by chemical synthesis alone.
- Ligation Assembling nucleic acids by a ligasemay also be used.
- DNA or RNA ligasespromote intermolecular ligation of the 5’ and 3’ ends of polynucleotide chains through the formation of a phosphodiester bond.
- Nucleic acidssuch as chimeric polynucleotides and/or circular nucleic acids may be prepared by ligation of one or more regions or subregions. DNA fragments can be joined by a ligase catalyzed reaction to create recombinant DNA with different functions. Two oligodeoxynucleotides, one with a 5’ phosphoryl group and another with a free 3’ hydroxyl group, serve as substrates for a DNA ligase.
- nucleic acid clean-upmay include, but is not limited to, nucleic acid clean-up, quality assurance and quality control. Clean-up may be performed by methods known in the arts such as, but not limited to, AGENCOURT® beads (Beckman Coulter Genomics, Danvers, MA), poly-T beads, LNATM oligo-T capture probes (EXIQON® Inc, Vedbaek, Denmark) or HPLC based purification methods such as, but not limited to, strong anion exchange HPLC, weak anion exchange HPLC, reverse phase HPLC (RP-HPLC), and hydrophobic interaction HPLC (HIC-HPLC).
- AGENCOURT® beadsBeckman Coulter Genomics, Danvers, MA
- poly-T beadspoly-T beads
- LNATM oligo-T capture probesEXIQON® Inc, Vedbaek, Denmark
- HPLC based purification methodssuch as, but not limited to, strong anion exchange HPLC, weak anion exchange HPLC, reverse phase HPLC (
- purifiedwhen used in relation to a nucleic acid such as a “purified nucleic acid” refers to one that is separated from at least one contaminant.
- a “contaminant”is any substance that makes another unfit, impure or inferior.
- a purified nucleic acide.g., DNA and RNA
- a quality assurance and/or quality control checkmay be conducted using methods such as, but not limited to, gel electrophoresis, UV absorbance, or analytical HPLC.
- the nucleic acidsmay be sequenced by methods including, but not limited to reverse-transcriptase-PCR. Quantification In some embodiments, the nucleic acids of the present disclosure may be quantified in exosomes or when derived from one or more bodily fluid.
- Bodily fluidsinclude peripheral blood, serum, plasma, ascites, urine, cerebrospinal fluid (CSF), sputum, saliva, bone marrow, synovial fluid, aqueous humor, amniotic fluid, cerumen, breast milk, broncheoalveolar lavage fluid, semen, prostatic fluid, cowper's fluid or pre-ejaculatory fluid, sweat, fecal matter, hair, tears, cyst fluid, pleural and peritoneal fluid, pericardial fluid, lymph, chyme, chyle, bile, interstitial fluid, menses, pus, sebum, vomit, vaginal secretions, mucosal secretion, stool water, pancreatic juice, lavage fluids from sinus cavities, bronchopulmonary aspirates, blastocyl cavity fluid, and umbilical cord blood.
- CSFcerebrospinal fluid
- salivaaqueous humor
- amniotic fluidcerumen
- breast milkbroncheoalveolar lavage fluid
- exosomesmay be retrieved from an organ selected from the group consisting of lung, heart, pancreas, stomach, intestine, bladder, kidney, ovary, testis, skin, colon, breast, prostate, brain, esophagus, liver, and placenta.
- Assaysmay be performed using construct specific probes, cytometry, qRT-PCR, real-time PCR, PCR, flow cytometry, electrophoresis, mass spectrometry, or combinations thereof while the exosomes may be isolated using immunohistochemical methods such as enzyme linked immunosorbent assay (ELISA) methods.
- ELISAenzyme linked immunosorbent assay
- Exosomesmay also be isolated by size exclusion chromatography, density gradient centrifugation, differential centrifugation, nanomembrane ultrafiltration, immunoabsorbent capture, affinity purification, microfluidic separation, or combinations thereof. These methods afford the investigator the ability to monitor, in real time, the level of nucleic acids remaining or delivered. This is possible because the nucleic acids of the present disclosure, in some embodiments, differ from the endogenous forms due to the structural or chemical modifications. In some embodiments, the nucleic acid may be quantified using methods such as, but not limited to, ultraviolet visible spectroscopy (UV/Vis).
- UV/Visultraviolet visible spectroscopy
- a non-limiting example of a UV/Vis spectrometeris a NANODROP® spectrometer (ThermoFisher, Waltham, MA).
- the quantified nucleic acidmay be analyzed in order to determine if the nucleic acid may be of proper size, check that no degradation of the nucleic acid has occurred.
- Degradation of the nucleic acidmay be checked by methods such as, but not limited to, agarose gel electrophoresis, HPLC based purification methods such as, but not limited to, strong anion exchange HPLC, weak anion exchange HPLC, reverse phase HPLC (RP-HPLC), and hydrophobic interaction HPLC (HIC- HPLC), liquid chromatography-mass spectrometry (LCMS), capillary electrophoresis (CE) and capillary gel electrophoresis (CGE).
- a lipid compositionsuch as a composition comprising a lipid nanoparticle, a liposome, and/or a lipoplex.
- nucleic acidsare formulated as lipid nanoparticle (LNP) compositions.
- LNPlipid nanoparticle
- Lipid nanoparticlestypically comprise amino lipid, non-cationic lipid, structural lipid, and PEG lipid components along with the nucleic acid cargo of interest.
- the lipid nanoparticlescan be generated using components, compositions, and methods as are generally known in the art, see for example PCT/US2016/052352; PCT/US2016/068300; PCT/US2017/037551; PCT/US2015/027400; PCT/US2016/047406; PCT/US2016/000129; PCT/US2016/014280; PCT/US2017/038426; PCT/US2014/027077; PCT/US2014/055394; PCT/US2016/052117; PCT/US2012/069610; PCT/US2017/027492; PCT/US2016/059575; PCT/US2016/069491; PCT/US2016/069493; and PCT/US2014/066242, all of which are incorporated by reference herein in their entirety.
- the lipid nanoparticlecomprises at least one ionizable amino lipid, at least one non-cationic lipid, at least one sterol, and/or at least one polyethylene glycol (PEG)- modified lipid.
- the lipid nanoparticlecomprises a molar ratio of 20-60% ionizable amino lipid, 5-25% non-cationic lipid, 25-55% structural lipid, and 0.5-15% PEG-modified lipid.
- the lipid nanoparticlecomprises a molar ratio of 20-60% ionizable amino lipid, 5-30% non-cationic lipid, 10-55% structural lipid, and 0.5-15% PEG-modified lipid.
- the lipid nanoparticlecomprises 40-50 mol% ionizable lipid, optionally 45-50 mol%, for example, 45-46 mol%, 46-47 mol%, 47-48 mol%, 48-49 mol%, or 49-50 mol% for example about 45 mol%, 45.5 mol%, 46 mol%, 46.5 mol%, 47 mol%, 47.5 mol%, 48 mol%, 48.5 mol%, 49 mol%, or 49.5 mol%.
- the lipid nanoparticlecomprises 20-60 mol% ionizable amino lipid.
- the lipid nanoparticlemay comprise 20-50 mol%, 20-40 mol%, 20-30 mol%, 30-60 mol%, 30-50 mol%, 30-40 mol%, 40-60 mol%, 40-50 mol%, or 50-60 mol% ionizable amino lipid.
- the lipid nanoparticlecomprises 20 mol%, 30 mol%, 40 mol%, 50 mol%, or 60 mol% ionizable amino lipid.
- the lipid nanoparticlecomprises 35 mol%, 36 mol%, 37 mol%, 38 mol%, 39 mol%, 40 mol%, 41 mol%, 42 mol%, 43 mol%, 44 mol%, 45 mol%, 46 mol%, 47 mol%, 48 mol%, 49 mol%, 50 mol%, 51 mol%, 52 mol%, 53 mol%, 54 mol%, or 55 mol% ionizable amino lipid. In some embodiments, the lipid nanoparticle comprises 45 – 55 mole percent (mol%) ionizable amino lipid.
- lipid nanoparticlemay comprise 45, 46, 47, 48, 49, 50, 51, 52, 53, 54, or 55 mol% ionizable amino lipid.
- Ionizable amino lipids Formula (AI)the ionizable amino lipid of a lipid nanoparticle is a compound of Formula (AI): its N-oxide, or a salt or isomer thereof, wherein R’ a is R’ branched ; wherein denotes a point of attachment; wherein R a ⁇ , R a ⁇ , R a ⁇ , and R a ⁇ are each independently selected from the group consisting of H, C 2-12 alkyl, and C 2-12 alkenyl; R 2 and R 3 are each independently selected from the group consisting of C1-14 alkyl and C2-14 alkenyl; R 4 is selected from the group consisting of -(CH 2 ) n OH, wherein n is selected from the group consisting wherein denotes a point of attachment
- R’ ais R’ branched ;
- R’ branchedis denotes a point of attachment;
- R a ⁇ , R a ⁇ , R a ⁇ , and R a ⁇are each H;
- R 2 and R 3are each C1-14 alkyl;
- R 4is -(CH2)nOH; n is 2;
- each R 5is H;
- each R 6is H;
- M and M’are each - C(O)O-;
- R’is a C 1-12 alkyl; l is 5; and
- mis 7.
- R’ ais R’ branched ;
- R’ branchedis denotes a point of attachment;
- R a ⁇ , R a ⁇ , R a ⁇ , and R a ⁇are each H;
- R 2 and R 3are each C 1-14 alkyl;
- R 4is -(CH 2 ) n OH; n is 2;
- each R 5is H;
- each R 6is H;
- M and M’are each - C(O)O-;
- R’is a C1-12 alkyl; l is 3; and
- mis 7.
- R’ ais R’ branched ;
- R’ branchedis denotes a point of attachment;
- R a ⁇is C 2-12 alkyl;
- R a ⁇ , R a ⁇ , and R a ⁇are each H;
- R 2 and R 3are each C1-14 alkyl; alkyl);
- n2is 2;
- R 5is H;
- each R 6is H;
- M and M’are each -C(O)O-;
- R’is a C 1-12 alkyl; l is 5; and
- mis 7.
- R’ ais R’ branched ;
- R’ branchedis denotes a point of attachment;
- R a ⁇ , R a ⁇ , and R a ⁇are each H;
- R a ⁇is C 2-12 alkyl;
- R 2 and R 3are each C 1-14 alkyl;
- R 4is -(CH 2 ) n OH; n is 2;
- each R 5is H; each R 6 is H;
- M and M’are each -C(O)O-;
- R’is a C1-12 alkyl; l is 5; and
- mis 7.
- the compound of Formula (AI)is selected from: , , .
- the ionizable amino lipid of Formula (AI)is a compound of Formula (AIa): its N-oxide, or a salt or isomer thereof, wherein R’ a is R’ branched ; wherein R’ branched denotes a point of attachment; wherein R a ⁇ , R a ⁇ , and R a ⁇ are each independently selected from the group consisting of H, C2-12 alkyl, and C2-12 alkenyl; R 2 and R 3 are each independently selected from the group consisting of C 1-14 alkyl and C 2-14 alkenyl; R 4 is selected from the group consisting of -(CH2)nOH wherein n is selected from the group consisting wherein denotes a point of attachment; wherein R 10 is N(R)2; each R is independently selected from the group consisting of C1-6 alkyl, C2-3 alkenyl, and H; and n2 is selected from the group consisting of 1, 2, 3, 4, 5, 6, 7, 8, 9, and 10;
- the ionizable amino lipid of Formula (AI)is a compound of Formula (AIb): its N-oxide, or a salt or isomer thereof, wherein R’ a is R’ branched ; wherein denotes a point of attachment; wherein R a ⁇ , R a ⁇ , R a ⁇ , and R a ⁇ are each independently selected from the group consisting of H, C 2-12 alkyl, and C 2-12 alkenyl; R 2 and R 3 are each independently selected from the group consisting of C1-14 alkyl and C 2-14 alkenyl; R 4 is -(CH 2 ) n OH, wherein n is selected from the group consisting of 1, 2, 3, 4, and 5; each R 5 is independently selected from the group consisting of C1-3 alkyl, C2-3 alkenyl, and H; each R 6 is independently selected from the group consisting of C 1-3 alkyl, C 2-3 alkenyl, and H; M and M’ are each independently selected from
- R’ ais R’ branched ;
- R’ branchedis denotes a point of attachment;
- R a ⁇ , R a ⁇ , and R a ⁇are each H;
- R 2 and R 3are each C 1-14 alkyl;
- R 4is -(CH 2 ) n OH; n is 2;
- each R 5is H;
- each R 6is H;
- M and M’are each - C(O)O-;
- R’is a C1-12 alkyl; l is 5; and
- mis 7.
- R’ ais R’ branched ;
- R’ branchedis denotes a point of attachment;
- R a ⁇ , R a ⁇ , and R a ⁇are each H;
- R 2 and R 3are each C1-14 alkyl;
- R 4is -(CH2)nOH;
- nis 2;
- each R 5is H;
- each R 6is H;
- M and M’are each - C(O)O-;
- R’is a C 1-12 alkyl; l is 3; and m is 7.
- R’ ais R’ branched ;
- R’ branchedis denotes a point of attachment;
- R a ⁇ and R a ⁇are each H;
- R a ⁇is C 2-12 alkyl;
- R 2 and R 3are each C1-14 alkyl;
- R 4is -(CH2)nOH;
- nis 2;
- each R 5is H;
- each R 6is H;
- M and M’are each -C(O)O-;
- R’is a C 1-12 alkyl; l is 5; and
- mis 7.
- the ionizable amino lipid of Formula (AI)is a compound of Formula (AIc): its N-oxide, or a salt or isomer thereof, wherein R’ a is R’ branched ; wherein denotes a point of attachment; wherein R a ⁇ , R a ⁇ , R a ⁇ , and R a ⁇ are each independently selected from the group consisting of H, C2-12 alkyl, and C2-12 alkenyl; R 2 and R 3 are each independently selected from the group consisting of C1-14 alkyl and C 2-14 alkenyl; denotes a point of attachment; wherein R 10 is N(R)2; each R is independently selected from the group consisting of C1-6 alkyl, C2-3 alkenyl, and H; n2 is selected from the group consisting of 1, 2, 3, 4, 5, 6, 7, 8, 9, and 10; each R 5 is independently selected from the group consisting of C1-3 alkyl, C2-3 alkenyl, and H; each
- R a ⁇ , R a ⁇ , and R a ⁇are each H; R a ⁇ is C 2-12 alkyl; R 2 and R 3 are each C 1-14 alkyl; denotes a point of attachment; R 10 is NH(C1-6 alkyl); n2 is 2; each R 5 is H; each R 6 is H; M and M’ are each -C(O)O-; R’ is a C 1-12 alkyl; l is 5; and m is 7.
- the compound of Formula (AIc)is:
- the ionizable amino lipidis a compound of Formula (AII): wherein R’ a is R’ branched or R’ cyclic ; wherein of attachment; R a ⁇ and R a ⁇ are each independently selected from the group consisting of H, C 1-12 alkyl, and C2-12 alkenyl, wherein at least one of R a ⁇ and R a ⁇ is selected from the group consisting of C1- 12 alkyl and C2-12 alkenyl; R b ⁇ and R b ⁇ are each independently selected from the group consisting of H, C 1-12 alkyl, and C 2-12 alkenyl, wherein at least one of R b ⁇ and R b ⁇ is selected from the group consisting of C 1- 12 alkyl and C2-12 alkenyl; R 2 and R 3 are each independently selected from the group consisting of C1-14 alkyl and C 2-14 alkenyl; R 4 is selected from the group consisting
- the ionizable amino lipid of Formula (AII)is a compound of Formula (AII-a): (AII-a), or its N-oxide, or a salt or isomer thereof, wherein R’ a is R’ branched or R’ cyclic ; wherein denotes a point of attachment; R a ⁇ and R a ⁇ are each independently selected from the group consisting of H, C 1-12 alkyl, and C2-12 alkenyl, wherein at least one of R a ⁇ and R a ⁇ is selected from the group consisting of C1- 12 alkyl and C2-12 alkenyl; R b ⁇ and R b ⁇ are each independently selected from the group consisting of H, C 1-12 alkyl, and C 2-12 alkenyl, wherein at least one of R b ⁇ and R b ⁇ is selected from the group consisting of C 1- 12 alkyl and C2-12 alkenyl; R 2 and R 3 are each independently selected from the group consisting of C1
- the ionizable amino lipid of Formula (AII)is a compound of wherein R’ a is R’ branched or R’ cyclic ; wherein denotes a point of attachment; R a ⁇ and R b ⁇ are each independently selected from the group consisting of C 1-12 alkyl and C 2-12 alkenyl; R 2 and R 3 are each independently selected from the group consisting of C1-14 alkyl and C 2-14 alkenyl; R 4 is selected from the group consisting of -(CH 2 ) n OH wherein n is selected from the group consisting wherein denotes a point of attachment; wherein R 10 is N(R)2; each R is independently selected from the group consisting of C 1-6 alkyl, C 2-3 alkenyl, and H; and n2 is selected from the group consisting of 1, 2, 3, 4, 5, 6, 7, 8, 9, and 10; each R’ independently is a C1-12 alkyl or C2-12 alkenyl; m is selected from 1, 2, 2, 3, 4, 5,
- the ionizable amino lipid of Formula (AII)is a compound of Formula (AII-c): its N-oxide, or a salt or isomer thereof, wherein R’ a is R’ branched or R’ cyclic ; wherein denotes a point of attachment; wherein R a ⁇ is selected from the group consisting of C 1-12 alkyl and C 2-12 alkenyl; R 2 and R 3 are each independently selected from the group consisting of C 1-14 alkyl and C2-14 alkenyl; R 4 is selected from the group consisting of -(CH 2 ) n OH wherein n is selected from the group consisting wherein denotes a point of attachment; wherein R 10 is N(R)2; each R is independently selected from the group consisting of C 1-6 alkyl, C 2-3 alkenyl, and H; and n2 is selected from the group consisting of 1, 2, 3, 4, 5, 6, 7, 8, 9, and 10; R’ is a C1-12 alky
- the ionizable amino lipid of Formula (AII)is a compound of Formula (AII-d): its N-oxide, or a salt or isomer thereof, wherein R’ a is R’ branched or R’ cyclic ; wherein denotes a point of attachment; wherein R a ⁇ and R b ⁇ are each independently selected from the group consisting of C1-12 alkyl and C 2-12 alkenyl; R 4 is selected from the group consisting of -(CH 2 ) n OH wherein n is selected from the group consisting wherein denotes a point of attachment; wherein R 10 is N(R) 2 ; each R is independently selected from the group consisting of C1-6 alkyl, C2-3 alkenyl, and H; and n2 is selected from the group consisting of 1, 2, 3, 4, 5, 6, 7, 8, 9, and 10; each R’ independently is a C 1-12 alkyl or C 2-12 alkenyl; m is selected from 1, 2, 3, 4, 5,
- the ionizable amino lipid of Formula (AII)is a compound of its N-oxide, or a salt or isomer thereof, wherein R’ a is R’ branched or R’ cyclic ; wherein denotes a point of attachment; wherein R a ⁇ is selected from the group consisting of C 1-12 alkyl and C 2-12 alkenyl; R 2 and R 3 are each independently selected from the group consisting of C 1-14 alkyl and C2-14 alkenyl; R 4 is -(CH2)nOH wherein n is selected from the group consisting of 1, 2, 3, 4, and 5; R’ is a C 1-12 alkyl or C 2-12 alkenyl; m is selected from 1, 2, 3, 4, 5, 6, 7, 8, and 9; l is selected from 1, 2, 3, 4, 5, 6, 7, 8, and 9.
- m and lare each independently selected from 4, 5, and 6. In some embodiments of the compound of Formula (AII), (AII-a), (AII-b), (AII-c), (AII-d), or (AII-e), m and l are each 5. In some embodiments of the compound of Formula (AII), (AII-a), (AII-b), (AII-c), (AII-d), or (AII-e), each R’ independently is a C 1-12 alkyl.
- each R’independently is a C2-5 alkyl.
- R’ bis: and R 2 and R 3 are each independently a C 1-14 alkyl.
- R’ bis: and R 2 and R 3 are each independently a C 6-10 alkyl.
- R 2 and R 3are each independently a C8 alkyl.
- R 3are each independently a C6-10 alkyl.
- R a ⁇is a C2-6 alkyl and R 2 and R 3 are each independently a C6-10 alkyl.
- R 2 and R 3are each independently a C6-10 alkyl.
- R 3are each independently a C6-10 alkyl.
- R a ⁇is a C2-6 alkyl and R 2 and R 3 are each independently a C6-10 alkyl.
- R 3are each independently a C6-10 alkyl.
- each a C2-6 alkylIn some embodiments of the compound of Formula (AII), (AII-a), (AII-b), (AII-c), each a C2-6 alkyl. In some embodiments of the compound of Formula (AII), (AII-a), (AII-b), (AII-c), (AII- d), or (AII-e), m and l are each independently selected from 4, 5, and 6 and each R’ independently is a C1-12 alkyl. In some embodiments of the compound of Formula (AII), (AII-a), (AII-b), (AII-c), (AII-d), or (AII-e), m and l are each 5 and each R’ independently is a C 2-5 alkyl.
- R’ branchedis: are each independently selected from 4, 5, and 6, each R’ independently is a C1-12 alkyl, and R a ⁇ and R b ⁇ are each a C1-12 alkyl.
- each 5, each R’independently is a C 2-5 alkyl, and R a ⁇ and R b ⁇ are each a C 2-6 alkyl.
- R’is a C1-12 alkyl
- R a ⁇is a C1-12 alkyl
- R 2 and R 3are each independently a C 6-10 alkyl.
- R’is a C2-5 alkyl
- R a ⁇is a C2-6 alkyl
- R 2 and R 3are each a C8 alkyl.
- each R’independently is a C 1-12 alkyl, R a ⁇ and R b ⁇ are each a C1-12 alkyl, wherein R 10 is NH(C1-6 alkyl), and n2 is 2.
- each R’independently is a C2-5 alkyl, R a ⁇ and R b ⁇ are each a C2-6 alkyl, wherein R 10 is NH(CH3) and n2 is 2.
- R’is a C1-12 alkyl
- R 2 and R 3are each independently a C6-10 alkyl
- R a ⁇is a C1-12 alkyl
- R 10is NH(C1-6 alkyl) and n2 is 2.
- R’is a C 2- 5 alkyl
- R a ⁇is a C2-6 alkyl
- R 2 and R 3are each a C8 alkyl
- R 10is NH(CH 3 ) and n2 is 2.
- R 4is -(CH2)nOH and n is 2, 3, or 4.
- R 4is -(CH2)nOH and n is 2.
- each R’independently is a C 1-12 alkyl
- R a ⁇ and R b ⁇are each a C1-12 alkyl
- R 4is -(CH2)nOH
- nis 2, 3, or 4.
- R’ bis: , m and l are each 5, each R’ independently is a C2-5 alkyl, R a ⁇ and R b ⁇ are each a C2-6 alkyl, R 4 is -(CH2)nOH, and n is 2.
- the ionizable amino lipid of Formula (AII)is a compound of Formula (AII-f): its N-oxide, or a salt or isomer thereof, wherein R’ a is R’ branched or R’ cyclic ; wherein ; wherein denotes a point of attachment; R a ⁇ is a C1-12 alkyl; R 2 and R 3 are each independently a C 1-14 alkyl; R 4 is -(CH2)nOH wherein n is selected from the group consisting of 1, 2, 3, 4, and 5; R’ is a C1-12 alkyl; m is selected from 4, 5, and 6; and l is selected from 4, 5, and 6.
- m and lare each 5, and n is 2, 3, or 4.
- R’is a C 2-5 alkyl, R a ⁇ is a C 2-6 alkyl, and R 2 and R 3 are each a C6-10 alkyl.
- m and lare each 5, n is 2, 3, or 4, R’ is a C2-5 alkyl, R a ⁇ is a C2-6 alkyl, and R 2 and R 3 are each a C6-10 alkyl.
- the ionizable amino lipid of Formula (AII)is a compound of Formula (AII-g): thereof; wherein R a ⁇ is a C2-6 alkyl; R’ is a C 2-5 alkyl; and R 4 is selected from the group consisting of -(CH 2 ) n OH wherein n is selected from the group consisting wherein denotes a point of attachment, R 10 is NH(C 1-6 alkyl), and n2 is selected from the group consisting of 1, 2, and 3.
- the ionizable amino lipid of Formula (AII)is a compound of Formula (AII-h): thereof; wherein R a ⁇ and R b ⁇ are each independently a C 2-6 alkyl; each R’ independently is a C2-5 alkyl; and R 4 is selected from the group consisting of -(CH2)nOH wherein n is selected from the group consisting wherein denotes a point of attachment, R 10 is NH(C1-6 alkyl), and n2 is selected from the group consisting of 1, 2, and 3.
- R 4is , wherein R 10 is NH(CH 3 ) and n2 is 2.
- R 4is -(CH 2 ) 2 OH.
- the ionizable amino lipids of a lipid nanoparticlemay be one or more of compounds of Formula (AIII): (AIII), or their N-oxides, or salts or isomers thereof, wherein: R1 is selected from the group consisting of C5-30 alkyl, C5-20 alkenyl, -R*YR”, -YR”, and -R”M’R’; R2 and R3 are independently selected from the group consisting of H, C1-14 alkyl, C2-14 alkenyl, -R*YR”, -YR”, and -R*OR”, or R2 and R3, together with the atom to which they are attached, form a heterocycle or carbocycle; R 4 is selected from the group consisting of hydrogen, a C 3-6 carbocycle, -(CH 2 )
- another subset of compounds of Formula (AIII)includes those in which: R1 is selected from the group consisting of C5-30 alkyl, C5-20 alkenyl, -R*YR”, -YR”, and -R”M’R’; R2 and R3 are independently selected from the group consisting of H, C1-14 alkyl, C2-14 alkenyl, -R*YR”, -YR”, and -R*OR”, or R2 and R3, together with the atom to which they are attached, form a heterocycle or carbocycle; R4 is selected from the group consisting of a C3-6 carbocycle, -(CH2)nQ, -(CH2)nCHQR, -CHQR, -CQ(R)2, and unsubstituted C1-6 alkyl, where Q is selected from a C3-6 carbocycle, a 5- to 14-membered heteroaryl having one or more heteroatoms selected from N, O, and S, -OR, -
- another subset of compounds of Formula (AIII)includes those in which: R 1 is selected from the group consisting of C 5-30 alkyl, C 5-20 alkenyl, -R*YR”, -YR”, and -R”M’R’; R2 and R3 are independently selected from the group consisting of H, C1-14 alkyl, C2-14 alkenyl, -R*YR”, -YR”, and -R*OR”, or R2 and R3, together with the atom to which they are attached, form a heterocycle or carbocycle; R4 is selected from the group consisting of a C3-6 carbocycle, -(CH2)nQ, -(CH2)nCHQR, -CHQR, -CQ(R)2, and unsubstituted C1-6 alkyl, where Q is selected from a C3-6 carbocycle, a 5- to 14-membered heterocycle having one or more heteroatoms selected from N, O, and S, -OR, -O
- another subset of compounds of Formula (AIII)includes those in which: R1 is selected from the group consisting of C5-30 alkyl, C5-20 alkenyl, -R*YR”, -YR”, and -R”M’R’; R 2 and R 3 are independently selected from the group consisting of H, C 1-14 alkyl, C 2-14 alkenyl, -R*YR”, -YR”, and -R*OR”, or R2 and R3, together with the atom to which they are attached, form a heterocycle or carbocycle; R 4 is selected from the group consisting of a C 3-6 carbocycle, -(CH 2 ) n Q, -(CH 2 ) n CHQR, -CHQR, -CQ(R) 2 , and unsubstituted C 1-6 alkyl, where Q is selected from a C 3-6 carbocycle, a 5- to 14-membered heteroaryl having one or more heteroatoms selected from N,
- another subset of compounds of Formula (AIII)includes those in which R1 is selected from the group consisting of C5-30 alkyl, C5-20 alkenyl, -R*YR”, -YR”, and -R”M’R’; R 2 and R 3 are independently selected from the group consisting of H, C 2-14 alkyl, C 2-14 alkenyl, -R*YR”, -YR”, and -R*OR”, or R2 and R3, together with the atom to which they are attached, form a heterocycle or carbocycle; R 4 is -(CH 2 ) n Q or -(CH 2 ) n CHQR, where Q is -N(R) 2 , and n is selected from 3, 4, and 5; each R5 is independently selected from the group consisting of C1-3 alkyl, C2-3 alkenyl, and H; each R 6 is independently selected from the group consisting of C 1-3 alkyl, C 2-3 alkenyl, and H; M and M
- another subset of compounds of Formula (AIII)includes those in which R 1 is selected from the group consisting of C 5-30 alkyl, C 5-20 alkenyl, -R*YR”, -YR”, and -R”M’R’; R2 and R3 are independently selected from the group consisting of C1-14 alkyl, C2-14 alkenyl, -R*YR”, -YR”, and -R*OR”, or R 2 and R 3 , together with the atom to which they are attached, form a heterocycle or carbocycle; R 4 is selected from the group consisting of -(CH 2 ) n Q, -(CH 2 ) n CHQR, -CHQR, and -CQ(R)2, where Q is -N(R)2, and n is selected from 1, 2, 3, 4, and 5; each R5 is independently selected from the group consisting of C1-3 alkyl, C2-3 alkenyl, and H; each R 6 is independently selected from the group consisting of C1-3
- mis 5, 7, or 9.
- Qis OH, -NHC(S)N(R)2, or -NHC(O)N(R)2.
- Qis -N(R)C(O)R, or -N(R)S(O)2R.
- a subset of compounds of Formula (AIII)includes those of Formula (AIII-B): its N-oxide, or a salt or isomer thereof in which all variables are as defined herein.
- mis selected from 5, 6, 7, 8, and 9;
- M and M’are independently selected from -C(O)O-, -OC(O)-, -OC(O)-M”-C(O)O-, -C(O)N(R’)-, -P(O)(OR’)O-, -S-S-
- mis 5, 7, or 9.
- Qis OH, -NHC(S)N(R)2, or -NHC(O)N(R)2.
- Qis -N(R)C(O)R, or -N(R)S(O)2R.
- the compounds of Formula (AIII)are of Formula (AIII-D), their N-oxides, or salts or isomers thereof, wherein R 4 is as described herein.
- the compounds of Formula (AIII)are of Formula (AIII-E), their N-oxides, or salts or isomers thereof, wherein R4 is as described herein.
- the compounds of Formula (AIII)are of Formula (AIII-F) or (AIII-G): their N-oxides, or salts or isomers thereof, wherein R4 is as described herein.
- the compounds of Formula (AIII)are of Formula (AIII-H): their N-oxides, or salts or isomers thereof, wherein M is -C(O)O- or –OC(O)-, M” is C 1-6 alkyl or C 2-6 alkenyl, R 2 and R 3 are independently selected from the group consisting of C 5-14 alkyl and C 5-14 alkenyl, and n is selected from 2, 3, and 4.
- the compounds of Formula (AIII)are of Formula (AIII-I): (AIII-I), or their N-oxides, or salts or isomers thereof, wherein n is 2, 3, or 4; and m, R’, R”, and R 2 through R 6 are as described herein.
- each of R 2 and R3may be independently selected from the group consisting of C5-14 alkyl and C5-14 alkenyl.
- an ionizable amino lipid of the disclosurecomprises a compound having structure: (Compound 1).
- an ionizable amino lipid of the disclosurecomprises a compound having structure: (Compound 2).
- the compounds of Formula (AIII)are of Formula (AIII-J), (AIII-J), or their N-oxides, or salts or isomers thereof, wherein l is selected from 1, 2, 3, 4, and 5; m is selected from 5, 6, 7, 8, and 9; M1 is a bond or M’; M and M’ are independently selected from -C(O)O-, -OC(O)-, -OC(O)-M”-C(O)O-, -C(O)N(R’)-, -P(O)(OR’)O-, -S-S-, an aryl group, and a heteroaryl group; and R 2 and R 3 are independently selected from the group consisting of H, C1-14 alkyl, and C2-14 alkenyl.
- Mis C1-6 alkyl (e.g., C 1-4 alkyl) or C 2-6 alkenyl (e.g. C 2-4 alkenyl).
- R 2 and R 3are independently selected from the group consisting of C 5-14 alkyl and C 5-14 alkenyl.
- the ionizable amino lipidsare one or more of the compounds described in U.S. Application Nos.
- the central amine moiety of a lipid according to Formula (AIII), (AIII-A), (AIII-B), (AIII-C), (AIII-D), (AIII-E), (AIII-F), (AIII-G), (AIII-H), (AIII-I), or (AIII-J)may be protonated at a physiological pH.
- a lipidmay have a positive or partial positive charge at physiological pH.
- Such amino lipidsmay be referred to as cationic lipids, ionizable lipids, cationic amino lipids, or ionizable amino lipids.
- Amino lipidsmay also be zwitterionic, i.e., neutral molecules having both a positive and a negative charge.
- the ionizable amino lipids of a lipid nanoparticlemay be one or more of compounds of formula (AIV), t is 1 or 2; A1 and A2 are each independently selected from CH or N; Z is CH 2 or absent wherein when Z is CH 2 , the dashed lines (1) and (2) each represent a single bond; and when Z is absent, the dashed lines (1) and (2) are both absent; R1, R2, R3, R4, and R5 are independently selected from the group consisting of C5-20 alkyl, C5-20 alkenyl, -R”MR’, -R*YR”, -YR”, and -R*OR”; R X1 and R X2 are each independently H or C 1-3 alkyl; each M is independently selected from the group consisting of -C(O)O-, -OC(O)-, -OC(O)O-, -C(O)N(R’)-, -N(R’)C(O)-
- the compoundis of any of formulae (AIVa)-(AIVh):
- the ionizable amino lipidis salt thereof.
- the central amine moiety of a lipid according to Formula (AIV), (AIVa), (AIVb), (AIVc), (AIVd), (AIVe), (AIVf), (AIVg), or (AIVh)may be protonated at a physiological pH.
- a lipidmay have a positive or partial positive charge at physiological pH.
- the lipid nanoparticlecomprises a lipid having the structure: pharmaceutically acceptable salt thereof, wherein: each R 1a is independently hydrogen, R 1c , or R 1d ; each R 1b is independently R 1c or R 1d ; each R 1c is independently –[CH2]2C(O)X 1 R 3 ; each R 1d Is independently -C(O)R 4 ; each R 2 is independently -[C(R 2a ) 2 ] c R 2b ; each R 2a is independently hydrogen or C1-C6 alkyl; R 2b is -N(L1-B)2; -(OCH2CH2)6OH; or -(OCH2CH2)bOCH3; each R 3 and R 4 is independently C 6 -C 30 aliphatic; each L1 is independently C1-C10 alkylene; each B is independently hydrogen or an ionizable nitrogen-containing group; each X 1 is independently a covalent bond or O; each
- the lipid nanoparticlecomprises a lipid having the structure: pharmaceutically acceptable salt thereof, wherein R 1 and R 2 are the same or different, each a linear or branched alkyl with 1-9 carbons, or as alkenyl or alkynyl with 2 to 11 carbon atoms, L1 and L2 are the same or different, each a linear alkyl having 5 to 18 carbon atoms, or form a heterocycle with N, X 1 is a bond, or is -CG-G- whereby L 2 -CO-O-R 2 is formed, X2 is S or O, L3 is a bond or a lower alkyl, or form a heterocycle with N, R 3 is a lower alkyl, and R4 and R5 are the same or different, each a lower alkyl.
- R 1 and R 2are the same or different, each a linear or branched alkyl with 1-9 carbons, or as alkenyl or alkynyl with 2 to 11 carbon atoms, L1 and
- the lipid nanoparticlecomprises an ionizable lipid having the structure: (A1), or a pharmaceutically acceptable salt thereof. In some embodiments, the lipid nanoparticle comprises a lipid having the structure: pharmaceutically acceptable salt thereof. In some embodiments, the lipid nanoparticle comprises a lipid having the structure: (A3), or a pharmaceutically acceptable salt thereof. In some embodiments, the lipid nanoparticle comprises a lipid having the structure: (A4), or a pharmaceutically acceptable salt thereof. In some embodiments, the lipid nanoparticle comprises a lipid having the structure: (A5), or a pharmaceutically acceptable salt thereof.
- the lipid nanoparticlecomprises a lipid having the structure: (A6), or a pharmaceutically acceptable salt thereof. In some embodiments, the lipid nanoparticle comprises a lipid having the structure: (A7), or a pharmaceutically acceptable salt thereof. In some embodiments, the lipid nanoparticle comprises a lipid having the structure: pharmaceutically acceptable salt thereof. In some embodiments, the lipid nanoparticle comprises a lipid having the structure: pharmaceutically acceptable salt thereof. In some embodiments, the lipid nanoparticle comprises a lipid having the structure: (A10), or a pharmaceutically acceptable salt thereof. In some embodiments, the lipid nanoparticle comprises a lipid having the structure: (A11), or a pharmaceutically acceptable salt thereof.
- Non-cationic lipidsin certain embodiments, the lipid nanoparticles described herein comprise one or more non-cationic lipids. Non-cationic lipids may be phospholipids. In some embodiments, the lipid nanoparticle comprises 5-25 mol% non-cationic lipid. For example, the lipid nanoparticle may comprise 5-20 mol%, 5-15 mol%, 5-10 mol%, 10-25 mol%, 10-20 mol%, 10-25 mol%, 15-25 mol%, 15-20 mol%, or 20-25 mol% non-cationic lipid. In some embodiments, the lipid nanoparticle comprises 5 mol%, 10 mol%, 15 mol%, 20 mol%, or 25 mol% non-cationic lipid.
- a non-cationic lipid of the disclosurecomprises 1,2-distearoyl-sn- glycero-3-phosphocholine (DSPC), 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE), 1,2-dilinoleoyl-sn-glycero-3-phosphocholine (DLPC), 1,2-dimyristoyl-sn-gly cero- phosphocholine (DMPC), 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC), l,2-dipalmitoyl- sn-glycero-3-phosphocholine (DPPC), 1,2-diundecanoyl-sn-glycero-phosphocholine (DUPC), 1- palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC), 1,2-di-O-octadecenyl-sn-glycero-3-phospho
- the lipid nanoparticlecomprises 5 – 15 mol%, 5 – 10 mol%, or 10 – 15 mol% DSPC.
- the lipid nanoparticlemay comprise 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, or 15 mol% DSPC.
- the lipid composition of the lipid nanoparticle composition disclosed hereincan comprise one or more phospholipids, for example, one or more saturated or (poly)unsaturated phospholipids or a combination thereof.
- phospholipidscomprise a phospholipid moiety and one or more fatty acid moieties.
- a phospholipid moietycan be selected, for example, from the non-limiting group consisting of phosphatidyl choline, phosphatidyl ethanolamine, phosphatidyl glycerol, phosphatidyl serine, phosphatidic acid, 2-lysophosphatidyl choline, and a sphingomyelin.
- a fatty acid moietycan be selected, for example, from the non-limiting group consisting of lauric acid, myristic acid, myristoleic acid, palmitic acid, palmitoleic acid, stearic acid, oleic acid, linoleic acid, alpha-linolenic acid, erucic acid, phytanoic acid, arachidic acid, arachidonic acid, eicosapentaenoic acid, behenic acid, docosapentaenoic acid, and docosahexaenoic acid.
- Particular phospholipidscan facilitate fusion to a membrane.
- a cationic phospholipidcan interact with one or more negatively charged phospholipids of a membrane (e.g., a cellular or intracellular membrane). Fusion of a phospholipid to a membrane can allow one or more elements (e.g., a therapeutic agent) of a lipid-containing composition (e.g., LNPs) to pass through the membrane permitting, e.g., delivery of the one or more elements to a target tissue.
- elementse.g., a therapeutic agent
- a lipid-containing compositione.g., LNPs
- Non-natural phospholipid speciesincluding natural species with modifications and substitutions including branching, oxidation, cyclization, and alkynes are also contemplated.
- a phospholipidcan be functionalized with or cross-linked to one or more alkynes (e.g., an alkenyl group in which one or more double bonds is replaced with a triple bond).
- alkynese.g., an alkenyl group in which one or more double bonds is replaced with a triple bond.
- an alkyne groupcan undergo a copper-catalyzed cycloaddition upon exposure to an azide.
- Such reactionscan be useful in functionalizing a lipid bilayer of a nanoparticle composition to facilitate membrane permeation or cellular recognition or in conjugating a nanoparticle composition to a useful component such as a targeting or imaging moiety (e.g., a dye).
- Phospholipidsinclude, but are not limited to, glycerophospholipids such as phosphatidylcholines, phosphatidylethanolamines, phosphatidylserines, phosphatidylinositols, phosphatidy glycerols, and phosphatidic acids. Phospholipids also include phosphosphingolipid, such as sphingomyelin.
- a phospholipidcomprises 1,2-distearoyl-sn-glycero-3- phosphocholine (DSPC), 1,2-Distearoyl-sn-glycero-3-phosphoethanolamine (DSPE), 1,2- dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE), 1,2-dilinoleoyl-sn-glycero-3- phosphocholine (DLPC), 1,2-dimyristoyl-sn-gly cero-phosphocholine (DMPC), 1,2-dioleoyl-sn- glycero-3-phosphocholine (DOPC), l,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC), 1,2- diundecanoyl-sn-glycero-phosphocholine (DUPC), 1-palmitoyl-2-oleoyl-sn-glycero-3- phosphocholine (POPC), 1,2-di-
- a phospholipidis an analog or variant of DSPC.
- a phospholipidis a compound of Formula (HI): (HI), or a salt thereof, wherein: each R 1 is independently optionally substituted alkyl; or optionally two R 1 are joined together with the intervening atoms to form optionally substituted monocyclic carbocyclyl or optionally substituted monocyclic heterocyclyl; or optionally three R 1 are joined together with the intervening atoms to form optionally substituted bicyclic carbocyclyl or optionally substitute bicyclic heterocyclyl; n is 1, 2, 3, 4, 5, 6, 7, 8, 9, or 10; m is 0, 1, 2, 3, 4, 5, 6, 7, 8, 9, or 10; A is of the formula: each instance of L 2 is independently a bond or optionally substituted C1-6 alkylene, wherein one methylene unit of the optionally substituted C 1-6 alkylene is optionally replaced with O, N(R N ), S, C(O), C(O)N
- the compoundis not of the formula: , wherein each instance of R 2 is independently unsubstituted alkyl, unsubstituted alkenyl, or unsubstituted alkynyl.
- the phospholipidsmay be one or more of the phospholipids described in PCT Application No. PCT/US2018/037922.
- the lipid nanoparticlecomprises a molar ratio of 5-25% non- cationic lipid relative to the other lipid components.
- the lipid nanoparticlemay comprise a molar ratio of 5-30%, 5-15%, 5-10%, 10-25%, 10-20%, 10-25%, 15-25%, 15-20%, 20-25%, or 25-30% non-cationic lipid.
- the lipid nanoparticlecomprises a molar ratio of 5%, 10%, 15%, 20%, 25%, or 30% non-cationic lipid.
- the lipid nanoparticlecomprises a molar ratio of 5-25% phospholipid relative to the other lipid components.
- the lipid nanoparticlemay comprise a molar ratio of 5-30%, 5-15%, 5-10%, 10-25%, 10-20%, 10-25%, 15-25%, 15-20%, 20-25%, or 25-30% phospholipid.
- the lipid nanoparticlecomprises a molar ratio of 5%, 10%, 15%, 20%, 25%, or 30% phospholipid lipid.
- Structural lipidsThe lipid composition of a pharmaceutical composition disclosed herein can comprise one or more structural lipids.
- structural lipidincludes sterols and also to lipids containing sterol moieties.
- Structural lipidscan be selected from the group including but not limited to, cholesterol, fecosterol, sitosterol, ergosterol, campesterol, stigmasterol, brassicasterol, tomatidine, tomatine, ursolic acid, alpha-tocopherol, hopanoids, phytosterols, steroids, and mixtures thereof.
- the structural lipidis a sterol.
- “sterols”are a subgroup of steroids consisting of steroid alcohols.
- the structural lipidis a steroid.
- the structural lipidis cholesterol.
- the structural lipidis an analog of cholesterol. In certain embodiments, the structural lipid is alpha-tocopherol. In some embodiments, the structural lipids may be one or more of the structural lipids described in U.S. Application No.16/493,814. In some embodiments, the lipid nanoparticle comprises a molar ratio of 25-55% structural lipid relative to the other lipid components.
- the lipid nanoparticlemay comprise a molar ratio of 10- 55%, 25-50%, 25-45%, 25-40%, 25-35%, 25-30%, 30-55%, 30-50%, 30-45%, 30-40%, 30-35%, 35-55%, 35-50%, 35-45%, 35-40%, 40-55%, 40-50%, 40-45%, 45-55%, 45- 50%, or 50-55% structural lipid.
- the lipid nanoparticlecomprises a molar ratio of 10%, 15%, 20%, 25%, 30%, 35%, 40%, 45%, 50%, or 55% structural lipid.
- the lipid nanoparticlecomprises 30-45 mol% sterol, optionally 35- 40 mol%, for example, 30-31 mol%, 31-32 mol%, 32-33 mol%, 33-34 mol%, 34-35 mol%, 35- 36 mol%, 36-37 mol%, 37-38 mol%, 38-39 mol%, or 39-40 mol%. In some embodiments, the lipid nanoparticle comprises 25-55 mol% sterol.
- the lipid nanoparticlemay comprise 25-50 mol%, 25-45 mol%, 25-40 mol%, 25-35 mol%, 25-30 mol%, 30-55 mol%, 30- 50 mol%, 30-45 mol%, 30-40 mol%, 30-35 mol%, 35-55 mol%, 35-50 mol%, 35-45 mol%, 35- 40 mol%, 40-55 mol%, 40-50 mol%, 40-45 mol%, 45-55 mol%, 45-50 mol%, or 50-55 mol% sterol.
- the lipid nanoparticlecomprises 25 mol%, 30 mol%, 35 mol%, 40 mol%, 45 mol%, 50 mol%, or 55 mol% sterol. In some embodiments, the lipid nanoparticle comprises 35 – 40 mol% cholesterol. For example, the lipid nanoparticle may comprise 35, 35.5, 36, 36.5, 37, 37.5, 38, 38.5, 39, 39.5, or 40 mol% cholesterol.
- Polyethylene glycol (PEG)-LipidsThe lipid composition of a pharmaceutical composition disclosed herein can comprise one or more polyethylene glycol (PEG) lipids.
- PEG-lipidor “PEG-modified lipid” refers to polyethylene glycol (PEG)-modified lipids.
- PEG-lipidsinclude PEG-modified phosphatidylethanolamine and phosphatidic acid, PEG-ceramide conjugates (e.g., PEG-CerC14 or PEG-CerC20), PEG-modified dialkylamines, and PEG-modified 1,2-diacyloxypropan-3- amines.
- PEG-lipidsinclude PEG-modified phosphatidylethanolamine and phosphatidic acid, PEG-ceramide conjugates (e.g., PEG-CerC14 or PEG-CerC20), PEG-modified dialkylamines, and PEG-modified 1,2-diacyloxypropan-3- amines.
- PEGylated lipidsPEGylated lipids.
- a PEG lipidcan be PEG-c-DOMG, PEG-DMG, PEG-DLPE, PEG-DMPE, PEG-DPPC, or a PEG-DSPE lipid.
- the PEG-lipidincludes, but not limited to 1,2-dimyristoyl-sn- glycerol methoxypolyethylene glycol (PEG-DMG), 1,2-distearoyl-sn-glycero-3- phosphoethanolamine-N-[amino(polyethylene glycol)] (PEG-DSPE), PEG-disteryl glycerol (PEG-DSG), PEG-dipalmetoleyl, PEG-dioleyl, PEG-distearyl, PEG-diacylglycamide (PEG- DAG), PEG-dipalmitoyl phosphatidylethanolamine (PEG-DPPE), or PEG-l,2- dimyristyloxlpropyl-3-amine
- the PEG-lipidis selected from the group consisting of a PEG- modified phosphatidylethanolamine, a PEG-modified phosphatidic acid, a PEG-modified ceramide, a PEG-modified dialkylamine, a PEG-modified diacylglycerol, a PEG-modified dialkylglycerol, and mixtures thereof.
- the PEG-modified lipidis PEG- DMG, PEG-c-DOMG (also referred to as PEG-DOMG), PEG-DSG, and/or PEG-DPG.
- the lipid moiety of the PEG-lipidsincludes those having lengths of from about C14 to about C22, preferably from about C14 to about C16.
- a PEG moietyfor example an mPEG-NH 2 , has a size of about 1000, 2000, 5000, 10,000, 15,000 or 20,000 daltons.
- the PEG-lipidis PEG2k-DMG.
- the lipid nanoparticles described hereincan comprise a PEG lipid which is a non-diffusible PEG.
- Non-limiting examples of non-diffusible PEGsinclude PEG-DSG and PEG-DSPE.
- PEG-lipidsare known in the art, such as those described in U.S. Patent No.8158601 and International Publ. No. WO 2015/130584 A2, which are incorporated herein by reference in their entirety.
- some of the other lipid components (e.g., PEG lipids) of various formulae described hereinmay be synthesized as described International Patent Application No. PCT/US2016/000129, filed December 10, 2016, entitled “Compositions and Methods for Delivery of Therapeutic Agents,” which is incorporated by reference in its entirety.
- the lipid component of a lipid nanoparticle compositionmay include one or more molecules comprising polyethylene glycol, such as PEG or PEG-modified lipids. Such species may be alternately referred to as PEGylated lipids.
- a PEG lipidis a lipid modified with polyethylene glycol.
- a PEG lipidmay be selected from the non-limiting group including PEG- modified phosphatidylethanolamines, PEG-modified phosphatidic acids, PEG-modified ceramides, PEG-modified dialkylamines, PEG-modified diacylglycerols, PEG-modified dialkylglycerols, and mixtures thereof.
- a PEG lipidmay be PEG-c-DOMG, PEG- DMG, PEG-DLPE, PEG-DMPE, PEG-DPPC, or a PEG-DSPE lipid.
- the PEG-modified lipidsare a modified form of PEG DMG.
- PEG- DMGhas the following structure:
- PEG lipidscan be PEGylated lipids described in International Publication No. WO2012/099755, the contents of which is herein incorporated by reference in its entirety. Any of these exemplary PEG lipids described herein may be modified to comprise a hydroxyl group on the PEG chain.
- the PEG lipidis a PEG-OH lipid.
- a “PEG-OH lipid”(also referred to herein as “hydroxy-PEGylated lipid”) is a PEGylated lipid having one or more hydroxyl (–OH) groups on the lipid.
- the PEG-OH lipidincludes one or more hydroxyl groups on the PEG chain.
- a PEG-OH or hydroxy-PEGylated lipidcomprises an –OH group at the terminus of the PEG chain.
- a PEG lipidis a compound of Formula (PI): (PI), or salts thereof, wherein: R 3 is –OR O ; R O is hydrogen, optionally substituted alkyl, or an oxygen protecting group; r is an integer between 1 and 100, inclusive; L 1 is optionally substituted C 1-10 alkylene, wherein at least one methylene of the optionally substituted C 1-10 alkylene is independently replaced with optionally substituted carbocyclylene, optionally substituted heterocyclylene, optionally substituted arylene, optionally substituted heteroarylene, O, N(R N ), S, C(O), C(O)N(R N ), NR N C(O), C(O)O, OC(O), OC(O)O, OC(O)N(R N ), NR N C(O)O, or NR N C(O)N(R N ); D is a moiety obtained by click chemistry or a moiety cleavable under physiological
- the compound of Fomula (PI)is a PEG-OH lipid (i.e., R 3 is – OR O , and R O is hydrogen).
- the compound of Formula (PI)is of Formula (PI-OH): (PI-OH), or a salt thereof.
- Formula (PII)In certain embodiments, a PEG lipid is a PEGylated fatty acid. In certain embodiments, a PEG lipid is a compound of Formula (PII).
- compounds of Formula (PII)have the following formula: (PII), or a salts thereof, wherein: R 3 is–OR O ; R O is hydrogen, optionally substituted alkyl or an oxygen protecting group; r is an integer between 1 and 100, inclusive; R 5 is optionally substituted C 10-40 alkyl, optionally substituted C 10-40 alkenyl, or optionally substituted C 10-40 alkynyl; and optionally one or more methylene groups of R 5 are replaced with optionally substituted carbocyclylene, optionally substituted heterocyclylene, optionally substituted arylene, optionally substituted heteroarylene, N(R N ), O, S, C(O), C(O)N(R N ), - NR N C(O), NR N C(O)N(R N ), C(O)O, OC(O), OC(O)O, OC(O)N(R N ), NR N C(O)O, C(O)S, SC(O), C
- the compound of Formula (PII)is of Formula (PII-OH): (PII-OH), or a salt thereof.
- ris 40-50.
- the compound of Formula (PII)is: or a salt thereof.
- the compound of Formula (PII)is .
- the lipid composition of the pharmaceutical compositions disclosed hereindoes not comprise a PEG-lipid.
- the PEG-lipidsmay be one or more of the PEG lipids described in U.S. Application No. US15/674,872.
- the lipid nanoparticlecomprises a molar ratio of 0.5-15% PEG lipid relative to the other lipid components.
- the lipid nanoparticlemay comprise a molar ratio of 0.5-10%, 0.5-5%, 1-15%, 1-10%, 1-5%, 2-15%, 2-10%, 2-5%, 5-15%, 5-10%, or 10-15% PEG lipid.
- the lipid nanoparticlecomprises a molar ratio of 0.5%, 1%, 2%, 3%, 4%, 5%, 6%, 7%, 8%, 9%, 10%, 11%, 12%, 13%, 14%, or 15% PEG- lipid.
- the lipid nanoparticlecomprises 1-5% PEG-modified lipid, optionally 1-3 mol%, for example 1.5 to 2.5 mol%, 1-2 mol%, 2-3 mol%, 3-4 mol%, or 4-5 mol%.
- the lipid nanoparticlecomprises 0.5-15 mol% PEG-modified lipid.
- the lipid nanoparticlemay comprise 0.5-10 mol%, 0.5-5 mol%, 1-15 mol%, 1-10 mol%, 1-5 mol%, 2-15 mol%, 2-10 mol%, 2-5 mol%, 5-15 mol%, 5-10 mol%, or 10-15 mol%.
- the lipid nanoparticlecomprises 0.5 mol%, 1 mol%, 2 mol%, 3 mol%, 4 mol%, 5 mol%, 6 mol%, 7 mol%, 8 mol%, 9 mol%, 10 mol%, 11 mol%, 12 mol%, 13 mol%, 14 mol%, or 15 mol% PEG-modified lipid.
- Some embodimentscomprise adding PEG to a composition comprising an LNP encapsulating a nucleic acid (e.g., which already includes PEG in the amounts listed above).
- the lipid nanoparticlecomprises 20-60 mol% ionizable amino lipid, 5-25 mol% non-cationic lipid, 25-55 mol% sterol, and 0.5-15 mol% PEG-modified lipid.
- a LNP of the disclosurecomprises an ionizable amino lipid of Compound 1, wherein the non-cationic lipid is DSPC, the structural lipid that is cholesterol, and the PEG lipid is DMG-PEG.
- a LNP of the disclosurecomprises an ionizable amino lipid of Compound 2, wherein the non-cationic lipid is DSPC, the structural lipid that is cholesterol, and the PEG lipid is DMG-PEG.
- a LNPcomprises an ionizable amino lipid of any of Formula (AIII), (AIV), or (AV), a phospholipid comprising DSPC, a structural lipid, and a PEG lipid comprising PEG-DMG.
- a LNPcomprises an ionizable amino lipid of any of Formula (AIII), (AIV), or (AV), a phospholipid comprising DSPC, a structural lipid, and a PEG lipid comprising a compound having Formula (PII).
- a LNPcomprises an ionizable amino lipid of Formula (AIII), (AIV), or (AV), a phospholipid comprising a compound having Formula (HI), a structural lipid, and the PEG lipid comprising a compound having Formula (PI) or (PII).
- a LNPcomprises an ionizable amino lipid of Formula (AIII), (AIV), or (AV), a phospholipid comprising a compound having Formula (HI), a structural lipid, and the PEG lipid comprising a compound having Formula (PI) or (PII).
- a LNPcomprises an ionizable amino lipid of Formula (AIII), (AIV), or (AV), a phospholipid having Formula (HI), a structural lipid, and a PEG lipid comprising a compound having Formula (PII).
- the lipid nanoparticlecomprises 49 mol% ionizable amino lipid, 10 mol% DSPC, 38.5 mol% cholesterol, and 2.5 mol% DMG-PEG. In some embodiments, the lipid nanoparticle comprises 49 mol% ionizable amino lipid, 11 mol% DSPC, 38.5 mol% cholesterol, and 1.5 mol% DMG-PEG. In some embodiments, the lipid nanoparticle comprises 48 mol% ionizable amino lipid, 11 mol% DSPC, 38.5 mol% cholesterol, and 2.5 mol% DMG-PEG. In some embodiments, a LNP comprises an N:P ratio of from about 2:1 to about 30:1.
- a LNPcomprises an N:P ratio of about 6:1. In some embodiments, a LNP comprises an N:P ratio of about 3:1, 4:1, or 5:1. In some embodiments, a LNP comprises a wt/wt ratio of the ionizable amino lipid component to the RNA of from about 10:1 to about 100:1. In some embodiments, a LNP comprises a wt/wt ratio of the ionizable amino lipid component to the RNA of about 20:1. In some embodiments, a LNP comprises a wt/wt ratio of the ionizable amino lipid component to the RNA of about 10:1.
- Some embodimentscomprise a composition having one or more LNPs having a diameter of about 150 nm or less, such as about 140 nm, 130 nm, 120 nm, 110 nm, 100 nm, 90 nm, 80 nm, 70 nm, 60 nm, 50 nm, 40 nm, 30 nm, or 20 nm or less.
- Some embodimentscomprise a composition having a mean LNP diameter of about 150 nm or less, such as about 140 nm, 130 nm, 120 nm, 110 nm, 100 nm, 90 nm, 80 nm, 70 nm, 60 nm, 50 nm, 40 nm, 30 nm, or 20 nm or less.
- the compositionhas a mean LNP diameter from about 30nm to about 150nm, or a mean diameter from about 60nm to about 120nm.
- a LNPmay comprise or one or more types of lipids, including but not limited to amino lipids (e.g., ionizable amino lipids), neutral lipids, non-cationic lipids, charged lipids, PEG- modified lipids, phospholipids, structural lipids and sterols.
- a LNPmay further comprise one or more cargo molecules, including but not limited to nucleic acids (e.g., mRNA, plasmid DNA, DNA or RNA oligonucleotides, siRNA, shRNA, snRNA, snoRNA, lncRNA, etc.), small molecules, proteins and peptides.
- the compositioncomprises a liposome.
- a liposomeis a lipid particle comprising lipids arranged into one or more concentric lipid bilayers around a central region. The central region of a liposome may comprise an aqueous solution, suspension, or other aqueous composition.
- the compositioncomprises a lipoplex.
- a lipoplexis a lipid particle comprising a cationic liposome and a nucleic acid (e.g., mRNA). Lipoplexes may be formed by contacting a liposome comprising a cationic lipid with a nucleic acid.
- a lipoplexmay comprise multiple concentric lipid bilayers, each concentric bilayer separated by one or more nucleic acids.
- the central region of the lipoplexmay comprise an aqueous solution, suspension, or other aqueous composition.
- the compositioncomprises a lipopolyplex.
- a lipopolyplexis a lipid particle comprising a lipid bilayer surrounding a complex of a cationic polymer and a nucleic acid (e.g., mRNA).
- a lipopolyplexmay be formed by contacting a cationic liposome (e.g., liposome comprising a cationic lipid) with the complex of nucleic acid and cationic polymer.
- the central region of the lipopolyplexmay comprise an aqueous solution, suspension, or other aqueous composition.
- the compositioncomprises a cationic nanoemulsion.
- a cationic nanoemulsioncomprises a cationic lipid, hydrophilic surfactant, and hydrophobic surfactant.
- a liposome, lipoplex, lipopolyplex, or cationic nanoemulsionmay comprise a sterol.
- a liposome, lipoplex, lipopolyplex, or cationic nanoemulsionmay comprise a neutral lipid.
- a liposome, lipoplex, lipopolyplex, or cationic nanoemulsionmay comprise a PEG-modified lipid.
- a lipid nanoparticlemay comprise two or more components (e.g., amino lipid and nucleic acid, PEG-lipid, phospholipid, structural lipid).
- a lipid nanoparticlemay comprise an amino lipid and a nucleic acid.
- Compositions comprising the lipid nanoparticles, such as those described herein,may be used for a wide variety of applications, including the stealth delivery of therapeutic payloads with minimal adverse innate immune response.
- nucleic acidsi.e., originating from outside of a cell or organism
- a particulate carriere.g., lipid nanoparticles
- the particulate carriershould be formulated to have minimal particle aggregation, be relatively stable prior to intracellular delivery, effectively deliver nucleic acids intracellularly, and illicit no or minimal immune response.
- many conventional particulate carriershave relied on the presence and/or concentration of certain components (e.g., PEG-lipid).
- the lipid nanoparticlescomprise one or more of ionizable molecules, polynucleotides, and optional components, such as structural lipids, sterols, neutral lipids, phospholipids and a molecule capable of reducing particle aggregation (e.g., polyethylene glycol (PEG), PEG-modified lipid), such as those described above.
- PEGpolyethylene glycol
- a LNP described hereinmay include one or more ionizable molecules (e.g., amino lipids or ionizable lipids).
- the ionizable moleculemay comprise a charged group and may have a certain pKa.
- the pKa of the ionizable moleculemay be greater than or equal to about 6, greater than or equal to about 6.2, greater than or equal to about 6.5, greater than or equal to about 6.8, greater than or equal to about 7, greater than or equal to about 7.2, greater than or equal to about 7.5, greater than or equal to about 7.8, greater than or equal to about 8.
- the pKa of the ionizable moleculemay be less than or equal to about 10, less than or equal to about 9.8, less than or equal to about 9.5, less than or equal to about 9.2, less than or equal to about 9.0, less than or equal to about 8.8, or less than or equal to about 8.5. Combinations of the above referenced ranges are also possible (e.g., greater than or equal to 6 and less than or equal to about 8.5). Other ranges are also possible. In embodiments in which more than one type of ionizable molecule are present in a particle, each type of ionizable molecule may independently have a pKa in one or more of the ranges described above.
- an ionizable moleculecomprises one or more charged groups.
- an ionizable moleculemay be positively charged or negatively charged.
- an ionizable moleculemay be positively charged.
- an ionizable moleculemay comprise an amine group.
- the term “ionizable molecule”has its ordinary meaning in the art and may refer to a molecule or matrix comprising one or more charged moiety.
- a “charged moiety”is a chemical moiety that carries a formal electronic charge, e.g., monovalent (+1, or -1), divalent (+2, or -2), trivalent (+3, or -3), etc.
- the charged moietymay be anionic (i.e., negatively charged) or cationic (i.e., positively charged).
- positively-charged moietiesinclude amine groups (e.g., primary, secondary, and/or tertiary amines), ammonium groups, pyridinium group, guanidine groups, and imidizolium groups.
- the charged moietiescomprise amine groups.
- negatively- charged groups or precursors thereofinclude carboxylate groups, sulfonate groups, sulfate groups, phosphonate groups, phosphate groups, hydroxyl groups, and the like.
- the charge of the charged moietymay vary, in some cases, with the environmental conditions, for example, changes in pH may alter the charge of the moiety, and/or cause the moiety to become charged or uncharged.
- the charge density of the molecule and/or matrixmay be selected as desired.
- an ionizable moleculee.g., an amino lipid or ionizable lipid
- the ionizable moleculemay include a neutral moiety that can be hydrolyzed to form a charged moiety, such as those described above.
- the molecule or matrixmay include an amide, which can be hydrolyzed to form an amine, respectively.
- an amidewhich can be hydrolyzed to form an amine, respectively.
- Those of ordinary skill in the artwill be able to determine whether a given chemical moiety carries a formal electronic charge (for example, by inspection, pH titration, ionic conductivity measurements, etc.), and/or whether a given chemical moiety can be reacted (e.g., hydrolyzed) to form a chemical moiety that carries a formal electronic charge.
- the ionizable moleculee.g., amino lipid or ionizable lipid
- the molecular weight of an ionizable moleculeis less than or equal to about 2,500 g/mol, less than or equal to about 2,000 g/mol, less than or equal to about 1,500 g/mol, less than or equal to about 1,250 g/mol, less than or equal to about 1,000 g/mol, less than or equal to about 900 g/mol, less than or equal to about 800 g/mol, less than or equal to about 700 g/mol, less than or equal to about 600 g/mol, less than or equal to about 500 g/mol, less than or equal to about 400 g/mol, less than or equal to about 300 g/mol, less than or equal to about 200 g/mol, or less than or equal to about 100 g/mol.
- the molecular weight of an ionizable moleculeis greater than or equal to about 100 g/mol, greater than or equal to about 200 g/mol, greater than or equal to about 300 g/mol, greater than or equal to about 400 g/mol, greater than or equal to about 500 g/mol, greater than or equal to about 600 g/mol, greater than or equal to about 700 g/mol, greater than or equal to about 1000 g/mol, greater than or equal to about 1,250 g/mol, greater than or equal to about 1,500 g/mol, greater than or equal to about 1,750 g/mol, greater than or equal to about 2,000 g/mol, or greater than or equal to about 2,250 g/mol.
- each type of ionizable moleculemay independently have a molecular weight in one or more of the ranges described above.
- the percentage (e.g., by weight, or by mole) of a single type of ionizable molecule (e.g., amino lipid or ionizable lipid) and/or of all the ionizable molecules within a particlemay be greater than or equal to about 15%, greater than or equal to about 16%, greater than or equal to about 17%, greater than or equal to about 18%, greater than or equal to about 19%, greater than or equal to about 20%, greater than or equal to about 21%, greater than or equal to about 22%, greater than or equal to about 23%, greater than or equal to about 24%, greater than or equal to about 25%, greater than or equal to about 30%, greater than or equal to about 35%, greater than or equal to about 40%, greater than or equal to about 42%, greater than or equal to about 45%, greater than or equal to about 48%, greater than or equal to about 50%, greater than or equal to about 52%, greater than or equal to about 55%, greater than or equal to about 58%, greater than
- the percentage(e.g., by weight, or by mole) may be less than or equal to about 70%, less than or equal to about 68%, less than or equal to about 65%, less than or equal to about 62%, less than or equal to about 60%, less than or equal to about 58%, less than or equal to about 55%, less than or equal to about 52%, less than or equal to about 50%, or less than or equal to about 48%. Combinations of the above referenced ranges are also possible (e.g., greater than or equal to 20% and less than or equal to about 60%, greater than or equal to 40% and less than or equal to about 55%, etc.).
- each type of ionizable moleculemay independently have a percentage (e.g., by weight, or by mole) in one or more of the ranges described above.
- the percentagee.g., by weight, or by mole
- the percentagemay be determined by extracting the ionizable molecule(s) from the dried particles using, e.g., organic solvents, and measuring the quantity of the agent using high pressure liquid chromatography (i.e., HPLC), liquid chromatography-mass spectrometry (LC-MS), nuclear magnetic resonance (NMR), or mass spectrometry (MS).
- HPLCmay be used to quantify the amount of a component, by, e.g., comparing the area under the curve of a HPLC chromatogram to a standard curve.
- chargeor “charged moiety” does not refer to a “partial negative charge” or “partial positive charge” on a molecule.
- partial negative chargeand “partial positive charge” are given their ordinary meaning in the art.
- a “partial negative charge”may result when a functional group comprises a bond that becomes polarized such that electron density is pulled toward one atom of the bond, creating a partial negative charge on the atom.
- a lipid compositionmay comprise one or more lipids as described herein.
- Such lipidsmay include those useful in the preparation of lipid nanoparticle formulations as described above or as known in the art.
- Stabilizing CompoundsSome embodiments of the compositions described herein are stabilized pharmaceutical compositions.
- Various non-viral delivery systems, including nanoparticle formulationspresent attractive opportunities to overcome many challenges associated with mRNA delivery.
- Lipid nanoparticlesLNPs
- LNPsLipid nanoparticles
- lipidshave been shown to degrade nucleic acids, including mRNA, and lipid nanoparticle formulations undergo rapid loss of purity when stored as refrigerated liquids. Moreover, the storage stability of mRNA encapsulated within LNPs is lower than that of unencapsulated mRNA.
- a class of compoundshas been found to stabilize nucleic acids within a lipid carrier such as an LNP, an unexpected and unprecedented discovery which enables applications including extended refrigerated liquid shelf-life, extended in-use periods at room temperature, and extended in-use stability at physiological temperatures up to higher temperatures such as 40°C. Such stabilizing compounds solve a critical problem, as current manufacturing processes and formulations experience a 5-10% purity loss during LNP formation and processing that is typical with current large-scale LNP production.
- the stabilized pharmaceutical compositioncomprises a nucleic acid formulation comprising a nucleic acid and a stabilizing compound (e.g., a compound of Formula (I), of Formula (II), or a tautomer or solvate thereof).
- a stabilizing compounde.g., a compound of Formula (I), of Formula (II), or a tautomer or solvate thereof.
- the stabilized pharmaceutical compositioncomprises a nucleic acid formulation comprising a nucleic acid and a lipid, and a compound of Formula (I): tautomer or solvate thereof, wherein: is a single bond or a double bond; R 1 is H; R 2 is OCH3, or together with R 3 is OCH2O; R 3 is OCH3, or together with R 2 is OCH 2 O; R 4 is H; R 5 is H or OCH 3 ; R 6 is OCH 3 ; R 7 is H or OCH 3 ; R 8 is H; R 9 is H or CH 3 ; and X is a pharmaceutically acceptable anion, e.g., a halide such as chloride.
- R 1is H
- R 2is OCH3, or together with R 3 is OCH2O
- R 3is OCH3, or together with R 2 is OCH 2 O
- R 4is H
- R 5is H or OCH 3
- R 6is OCH 3
- R 7is H or OCH 3
- the compound of Formula (I)has the structure of: Formula (Ia) Formula (Ib) Formula (Ic) or a tautomer or solvate thereof.
- the stabilized pharmaceutical compositioncomprises a nucleic acid formulation comprising a nucleic acid and a lipid, and a compound of Formula (II): tautomer or solvate thereof, wherein: R 10 is H; R 11 is H; R 12 together with R 13 is OCH2O; R 14 is H; R 15 together with R 16 is OCH 2 O; R 17 is H; and X is a pharmaceutically acceptable anion, e.g., a halide such as chloride.
- the compound of Formula (II)has the structure of: Formula (IIa), or a tautomer or solvate thereof. Stabilizing compounds of Formulas (I), (Ia), (Ib), (Ic), (II), and (Iia) are described in International Application No. PCT/US2022/025967, which is incorporated by reference herein in its entirety.
- the nucleic acid formulationcomprises lipid nanoparticles.
- the nucleic acidis mRNA.
- the stabilizing compound (“the compound”)has a purity of at least 70%, 80%, 90%, 95%, or 99%. In some embodiments, the compound contains fewer than 100ppm of elemental metals.
- the stabilized pharmaceutical composition(“the composition”) comprises a pharmaceutically acceptable metal chelator, e.g., EDTA (ethylenediaminetetraacetic acid) or DTPA (diethylenetriaminepentaacetic acid).
- the compositionis an aqueous solution.
- the compoundis present at a concentration between about 0.1mM and about 10mM in the aqueous solution.
- the aqueous solutionhas a pH of or about 5 to 8, including pH of about 5, 5.5, 6, 6.5, 7, 7.5, or 8.
- the aqueous solutiondoes not comprise NaCl.
- the aqueous solutioncomprises NaCl in a concentration of or about 150mM. In some embodiments, the aqueous solution comprises a phosphate buffer, a tris buffer, an acetate buffer, a histidine buffer, or a citrate buffer. In some embodiments, microbial growth in the composition is inhibited by the compound. In some embodiments, the composition is characterized as having a mRNA purity level of greater than 60%, greater than 70%, greater than 80%, or greater than 90% main peak mRNA purity after at least thirty days of storage. In some embodiments, the composition comprises a mRNA purity level of greater than 50% main peak mRNA purity after at least six months of storage. In some embodiments, the storage is at room temperature.
- the compositioncomprises a lipid nanoparticle encapsulating a mRNA, and the composition comprises less than 50%, less than 60%, less than 70%, less than 80%, less than 90%, or less than 95% RNA fragments after at least thirty days of storage.
- the storage temperatureis greater than room temperature. In some embodiments, the storage temperature is about 4°C.
- the compoundinteracts with the nucleic acid comprised within a lipid nanostructure (e.g., a lipid nanoparticle, liposome, or lipoplex), e.g., via pi-pi stacking and/or by changing backbone helicity of the nucleic acid.
- the compoundintercalates with a nucleic acid.
- the compoundbinds with a nucleic acid, e.g., reversible binding, and/or binding to the stranded regions of the nucleic acid.
- the compoundself-associates, binds to nucleic acid ribose contacts, and/or binds to nucleic acid base contacts.
- the compounddoes not substantially bind to nucleic acid phosphate contacts.
- the positive charge of the compoundcontributes to nucleic acid binding.
- the compoundinteracts with a nucleic acid and provides shielding from solvent, e.g., water.
- the compoundshields ribose from solvent more than the compound shields the phosphate groups of the nucleic acid.
- the solvent exposureis measured by the solvent accessible surface area (SASA).
- a stabilizing compounddecreases the solvent accessible area of ribose to about 5- 10 nm2.
- a stabilizing compounddecreases the solvent accessible area of ribose to about 6-8 nm2. In some embodiments, a stabilizing compound decreases the solvent accessible area of phosphate to about 9-12 nm2. In some embodiments, a stabilizing compound decreases the solvent accessible area of phosphate to about 10-11 nm2.
- a nucleic acid that is conformationally stabilized by the compoundexhibits thermal unfolding temperatures (measured by circular dichroism or DSC, for example) that are higher than in the absence of the compound. In some embodiments, the compound confers increased stability, e.g., thermal stability, to the nucleic acid in a folded structure, e.g., relative to its unfolded or less folded or more linear form.
- the compoundcauses compaction of the nucleic acid upon interaction with the nucleic acid. In some embodiments, the compound causes a decrease in the hydrodynamic radius of the nucleic acid molecule upon interaction with the nucleic acid. In some embodiments, a stabilizing compound causes compaction or a decrease in the hydrodynamic radius of a nucleic acid molecule by 5%, 10%, 15%, 20%, 25%, 30%, 35%, 40%, 45%, 50%, 55%, 60%, or more.
- a stabilizing compoundcauses compaction or a decrease in the hydrodynamic radius of a nucleic acid molecule when the compound is in a concentration of 1 ⁇ M, 2 ⁇ M, 3 ⁇ M, 4 ⁇ M, 5 ⁇ M, 6 ⁇ M, 7 ⁇ M, 8 ⁇ M, 9 ⁇ M, 10 ⁇ M, 15 ⁇ M, 20 ⁇ M, 25 ⁇ M, 30 ⁇ M, 35 ⁇ M, 40 ⁇ M, 45 ⁇ M, 50 ⁇ M, 60 ⁇ M, 70 ⁇ M, 80 ⁇ M, 90 ⁇ M, or 100 ⁇ M.
- compositionse.g., pharmaceutical compositions
- methods, kits and reagents for prevention or treatment of coronavirus infections in humans and other mammalsfor example.
- the compositions provided hereincan be used as therapeutic or prophylactic agents. They may be used in medicine to prevent and/or treat a coronavirus infection.
- the MERS-CoV vaccine containing RNA as described hereincan be administered to a subject (e.g., a mammalian subject, such as a human subject), and the RNA polynucleotides are translated in vivo to produce an antigenic polypeptide (antigen).
- an “effective amount” of a compositionis based, at least in part, on the target tissue, target cell type, means of administration, physical characteristics of the RNA (e.g., length, nucleotide composition, and/or extent of modified nucleosides), other components of the vaccine, and other determinants, such as age, body weight, height, sex and general health of the subject.
- an effective amount of a compositionprovides an induced or boosted immune response as a function of antigen production in the cells of the subject.
- an effective amount of the composition containing RNA polynucleotides having at least one chemical modificationsare more efficient than a composition containing a corresponding unmodified polynucleotide encoding the same antigen or a peptide antigen.
- Increased antigen productionmay be demonstrated by increased cell transfection (the percentage of cells transfected with the RNA vaccine), increased protein translation and/or expression from the polynucleotide, decreased nucleic acid degradation (as demonstrated, for example, by increased duration of protein translation from a modified polynucleotide), or altered antigen specific immune response of the host cell.
- compositionrefers to the combination of an active agent with a carrier, inert or active, making the composition especially suitable for diagnostic or therapeutic use in vivo or ex vivo.
- a “pharmaceutically acceptable carrier,” after administered to or upon a subject,does not cause undesirable physiological effects.
- the carrier in the pharmaceutical compositionmust be “acceptable” also in the sense that it is compatible with the active ingredient and can be capable of stabilizing it.
- One or more solubilizing agentscan be utilized as pharmaceutical carriers for delivery of an active agent.
- a pharmaceutically acceptable carrierinclude, but are not limited to, biocompatible vehicles, adjuvants, additives, and diluents to achieve a composition usable as a dosage form.
- compositionscomprising polynucleotides and their encoded polypeptides in accordance with the present disclosure may be used for treatment or prevention of a coronavirus infection.
- a compositionmay be administered prophylactically or therapeutically as part of an active immunization scheme to healthy individuals or early in infection during the incubation phase or during active infection after onset of symptoms.
- the amount of RNA provided to a cell, a tissue or a subjectmay be an amount effective for immune prophylaxis.
- a compositionmay be administered with other prophylactic or therapeutic compounds.
- a prophylactic or therapeutic compoundmay be an adjuvant or a booster.
- boostrefers to an extra administration of the vaccine composition and may include a traditional boost, seasonal boost or a pandemic shift boost.
- a booster(or booster vaccine) may be given after an earlier administration of the prophylactic composition.
- the time of administration between the initial administration of the prophylactic composition and the boostermay be, but is not limited to, 1 minute, 2 minutes, 3 minutes, 4 minutes, 5 minutes, 6 minutes, 7 minutes, 8 minutes, 9 minutes, 10 minutes, 15 minutes, 20 minutes 35 minutes, 40 minutes, 45 minutes, 50 minutes, 55 minutes, 1 hour, 2 hours, 3 hours, 4 hours, 5 hours, 6 hours, 7 hours, 8 hours, 9 hours, 10 hours, 11 hours, 12 hours, 13 hours, 14 hours, 15 hours, 16 hours, 17 hours, 18 hours, 19 hours, 20 hours, 21 hours, 22 hours, 23 hours, 1 day, 36 hours, 2 days, 3 days, 4 days, 5 days, 6 days, 1 week, 10 days, 2 weeks, 3 weeks, 1 month, 2 months, 3 months, 4 months, 5 months, or 6 months, 7 months, 8 months, 9 months, 10 months, 11 months, one year, or more.
- the time of administration between the initial administration of the prophylactic composition and the boosteris at least 6 months.
- the time of administration between the initial administration of the prophylactic composition and the boostermay be, but is not limited to, 1 week, 2 weeks, 3 weeks, 1 month, 2 months, 3 months, or 6 months.
- the boostermay comprise the same or different mRNAs as compared to the earlier administration of the prophylactic composition.
- the boostermay comprise a combination of the same mRNA from the earlier administration of the prophylactic composition and at least one different mRNA.
- the ratio of the mRNA from the earlier administration of the prophylactic composition and the at least one different mRNAis 1:1, 1:2, 1:4, 4:1, or 2:1. In one embodiment, the ratio is 1:1.
- the boostermay comprise different mRNAs as compared to the earlier administration of the prophylactic compositions. In some embodiments, such a booster may comprise 1, 2, 3, 4 or more mRNAs that were not present in the prophylactic composition.
- the ratio of two mRNA polynucleotides (none of which were in the prophylactic composition) in the boosteris 1:1, 1:2, 1:4, 4:1, or 2:1. In one embodiment, the ratio is 1:1.
- a boost or booster dosemay be administered more than once, for example 2, 3, 4, 5, 6 or more times after the initial prophylactic (prime) dose.
- a subsequent boostis administered within weeks, e.g., within 3-4 weeks of the first (or previous) boost.
- a second boostis administered 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, or more weeks after the first (or previous) boost.
- the boosterin some embodiments is monovalent (e.g., the mRNA encodes a single antigen). In some embodiments, the booster is multivalent (e.g., the mRNA encodes more than one antigen).
- the booster doseis 5 ⁇ g-30 ⁇ g, 5 ⁇ g-25 ⁇ g, 5 ⁇ g-20 ⁇ g, 5 ⁇ g-15 ⁇ g, 5 ⁇ g-10 ⁇ g, 10 ⁇ g-30 ⁇ g, 10 ⁇ g-25 ⁇ g, 10 ⁇ g-20 ⁇ g, 10 ⁇ g-15 ⁇ g, 15 ⁇ g-30 ⁇ g, 15 ⁇ g-25 ⁇ g, 15 ⁇ g-20 ⁇ g, 20 ⁇ g-30 ⁇ g, 25 ⁇ g-30 ⁇ g, or 25 ⁇ g-300 ⁇ g.
- the booster doseis 10 ⁇ g-60 ⁇ g, 10 ⁇ g-55 ⁇ g, 10 ⁇ g-50 ⁇ g, 10 ⁇ g-45 ⁇ g, 10 ⁇ g-40 ⁇ g, 10 ⁇ g-35 ⁇ g, 10 ⁇ g- 30 ⁇ g, 10 ⁇ g-25 ⁇ g, 10 ⁇ g-20 ⁇ g, 15 ⁇ g-60 ⁇ g, 15 ⁇ g-55 ⁇ g, 15 ⁇ g-50 ⁇ g, 15 ⁇ g-45 ⁇ g, 15 ⁇ g- 40 ⁇ g, 15 ⁇ g-35 ⁇ g, 15 ⁇ g-30 ⁇ g, 15 ⁇ g-25 ⁇ g, 15 ⁇ g-20 ⁇ g, 20 ⁇ g-60 ⁇ g, 20 ⁇ g-55 ⁇ g, 20 ⁇ g- 50 ⁇ g, 20 ⁇ g-45 ⁇ g, 20 ⁇ g-40 ⁇ g, 20 ⁇ g-35 ⁇ g, 20 ⁇ g-30 ⁇ g, 20 ⁇ g-25 ⁇ g, 20 ⁇ g
- the booster doseis at least 10 ⁇ g and less than 25 ⁇ g of the composition. In some embodiments, the booster dose is at least 5 ⁇ g and less than 25 ⁇ g of the composition. For example, the booster dose is 5 ⁇ g, 10 ⁇ g, 15 ⁇ g, 20 ⁇ g, 25 ⁇ g, 30 ⁇ g, 35 ⁇ g, 40 ⁇ g, 45 ⁇ g, 50 ⁇ g, 55 ⁇ g, 60 ⁇ g, 65 ⁇ g, 70 ⁇ g, 75 ⁇ g, 80 ⁇ g, 85 ⁇ g, 90 ⁇ g, 95 ⁇ g, 100 ⁇ g, 110 ⁇ g, 120 ⁇ g, 130 ⁇ g, 140 ⁇ g, 150 ⁇ g, 160 ⁇ g, 170 ⁇ g, 180 ⁇ g, 190 ⁇ g, 200 ⁇ g, 250 ⁇ g, or 300 ⁇ g.
- a compositionmay be administered intramuscularly, intranasally or intradermally, similarly to the administration of inactivated vaccines known in the art.
- a compositionmay be utilized in various settings depending on the prevalence of the infection or the degree or level of unmet medical need.
- the RNA vaccinesmay be utilized to treat and/or prevent a variety of infectious disease. RNA vaccines have superior properties in that they produce much larger antibody titers, better neutralizing immunity, produce more durable immune responses, and/or produce responses earlier than commercially available vaccines.
- pharmaceutical compositionsincluding RNA and/or complexes optionally in combination with one or more pharmaceutically acceptable excipients.
- RNAmay be formulated or administered alone or in conjunction with one or more other components.
- an immunizing compositionmay comprise other components including, but not limited to, adjuvants.
- an immunizing compositiondoes not include an adjuvant (it is adjuvant free).
- An RNAmay be formulated or administered in combination with one or more pharmaceutically-acceptable excipients.
- vaccine compositionscomprise at least one additional active substances, such as, for example, a therapeutically-active substance, a prophylactically-active substance, or a combination of both.
- Vaccine compositionsmay be sterile, pyrogen-free or both sterile and pyrogen-free.
- an immunizing compositionis administered to humans, human patients or subjects.
- the phrase “active ingredient”generally refers to the RNA vaccines or the polynucleotides contained therein, for example, RNA polynucleotides (e.g., mRNA polynucleotides) encoding antigens.
- Formulations of the vaccine compositions described hereinmay be prepared by any method known or hereafter developed in the art of pharmacology.
- such preparatory methodsinclude the step of bringing the active ingredient (e.g., mRNA polynucleotide) into association with an excipient and/or one or more other accessory ingredients, and then, if necessary and/or desirable, dividing, shaping and/or packaging the product into a desired single- or multi-dose unit.
- the active ingrediente.g., mRNA polynucleotide
- Relative amounts of the active ingredient, the pharmaceutically acceptable excipient, and/or any additional ingredients in a pharmaceutical composition in accordance with the disclosurewill vary, depending upon the identity, size, and/or condition of the subject treated and further depending upon the route by which the composition is to be administered.
- the compositionmay comprise between 0.1% and 100%, e.g., between 0.5 and 50%, between 1-30%, between 5-80%, at least 80% (w/w) active ingredient.
- an RNAis formulated using one or more excipients to: (1) increase stability; (2) increase cell transfection; (3) permit the sustained or delayed release (e.g., from a depot formulation); (4) alter the biodistribution (e.g., target to specific tissues or cell types); (5) increase the translation of encoded protein in vivo; and/or (6) alter the release profile of encoded protein (antigen) in vivo.
- excipientscan include, without limitation, lipidoids, liposomes, lipid nanoparticles, polymers, lipoplexes, core-shell nanoparticles, peptides, proteins, cells transfected with the RNA (e.g., for transplantation into a subject), hyaluronidase, nanoparticle mimics and combinations thereof.
- immunizing compositionse.g., RNA vaccines
- methods, kits and reagents for prevention and/or treatment of coronavirus infection in humans and other mammalscan be used as therapeutic or prophylactic agents.
- immunizing compositionsare used to provide prophylactic protection from coronavirus infection.
- immunizing compositionsare used to treat a coronavirus infection.
- immunizing compositionsare used in the priming of immune effector cells, for example, to activate peripheral blood mononuclear cells (PBMCs) ex vivo, which are then infused (re-infused) into a subject.
- PBMCsperipheral blood mononuclear cells
- a subjectmay be any mammal, including non-human primate and human subjects.
- a subjectis a human subject.
- an immunizing compositione.g., RNA vaccine
- a subjecte.g., a mammalian subject, such as a human subject
- an immunizing compositionis administered to a subject (e.g., a mammalian subject, such as a human subject) in an effective amount to induce an antigen-specific immune response.
- the RNA encoding the coronavirus spike protein antigenis expressed and translated in vivo to produce the antigen, which then stimulates an immune response in the subject.
- Prophylactic protection from a coronaviruscan be achieved following administration of an immunizing composition (e.g., an RNA vaccine) of the present disclosure.
- Immunizing compositionscan be administered once, twice, three times, four times or more but it is likely sufficient to administer the vaccine once (optionally followed by a single booster). It is possible, although less desirable, to administer an immunizing composition to an infected individual to achieve a therapeutic response. Dosing may need to be adjusted accordingly.
- a method of eliciting an immune response in a subject against a coronavirus antigen (or multiple antigens)is provided in aspects of the present disclosure.
- a methodinvolves administering to the subject an immunizing composition comprising a mRNA having an open reading frame encoding a coronavirus antigen, thereby inducing in the subject an immune response specific to the coronavirus antigen, wherein anti-antigen antibody titer in the subject is increased following vaccination relative to anti-antigen antibody titer in a subject vaccinated with a prophylactically effective dose of a traditional vaccine against the antigen.
- An “anti-antigen antibody”is a serum antibody the binds specifically to the antigen.
- a prophylactically effective doseis an effective dose that prevents infection with the virus at a clinically acceptable level.
- the effective doseis a dose listed in a package insert for the vaccine.
- a traditional vaccinerefers to a vaccine other than the mRNA vaccines of the present disclosure.
- a traditional vaccineincludes, but is not limited, to live microorganism vaccines, killed microorganism vaccines, subunit vaccines, protein antigen vaccines, DNA vaccines, virus like particle (VLP) vaccines, etc.
- a traditional vaccineis a vaccine that has achieved regulatory approval and/or is registered by a national drug regulatory body, for example the Food and Drug Administration (FDA) in the United States or the European Medicines Agency (EMA).
- FDAFood and Drug Administration
- EMAEuropean Medicines Agency
- the anti-antigen antibody titer in the subjectis increased 1 log to 10 log following vaccination relative to anti-antigen antibody titer in a subject vaccinated with a prophylactically effective dose of a traditional vaccine against the coronavirus or an unvaccinated subject. In some embodiments, the anti-antigen antibody titer in the subject is increased 1 log, 2 log, 3 log, 4 log, 5 log, or 10 log following vaccination relative to anti-antigen antibody titer in a subject vaccinated with a prophylactically effective dose of a traditional vaccine against the coronavirus or an unvaccinated subject.
- a method of eliciting an immune response in a subject against a coronavirusis provided in other aspects of the disclosure.
- the methodinvolves administering to the subject a composition comprising an mRNA comprising an open reading frame encoding a coronavirus antigen, thereby inducing in the subject an immune response specific to the coronavirus, wherein the immune response in the subject is equivalent to an immune response in a subject vaccinated with a traditional vaccine against the coronavirus at 2 times to 100 times the dosage level relative to the composition.
- the immune response in the subjectis equivalent to an immune response in a subject vaccinated with a traditional vaccine at twice the dosage level relative to a composition of the present disclosure.
- the immune response in the subjectis equivalent to an immune response in a subject vaccinated with a traditional vaccine at three times the dosage level relative to a composition of the present disclosure.
- the immune response in the subjectis equivalent to an immune response in a subject vaccinated with a traditional vaccine at 4 times, 5 times, 10 times, 50 times, or 100 times the dosage level relative to a composition of the present disclosure. In some embodiments, the immune response in the subject is equivalent to an immune response in a subject vaccinated with a traditional vaccine at 10 times to 1000 times the dosage level relative to a composition of the present disclosure. In some embodiments, the immune response in the subject is equivalent to an immune response in a subject vaccinated with a traditional vaccine at 100 times to 1000 times the dosage level relative to a composition of the present disclosure. In other embodiments, the immune response is assessed by determining [protein] antibody titer in the subject.
- the ability of serum or antibody from an immunized subjectis tested for its ability to neutralize viral uptake or reduce coronavirus transformation of human B lymphocytes.
- the ability to promote a robust T cell response(s)is measured using art recognized techniques.
- the disclosureprovide methods of eliciting an immune response in a subject against a coronavirus by administering to the subject composition comprising an mRNA having an open reading frame encoding a coronavirus antigen, thereby inducing in the subject an immune response specific to the coronavirus antigen, wherein the immune response in the subject is induced 2 days to 10 weeks earlier relative to an immune response induced in a subject vaccinated with a prophylactically effective dose of a traditional vaccine against the coronavirus.
- the immune response in the subjectis induced in a subject vaccinated with a prophylactically effective dose of a traditional vaccine at 2 times to 100 times the dosage level relative to a composition of the present disclosure.
- the immune response in the subjectis induced 2 days, 3 days, 1 week, 2 weeks, 3 weeks, 5 weeks, or 10 weeks earlier relative to an immune response induced in a subject vaccinated with a prophylactically effective dose of a traditional vaccine.
- methods of eliciting an immune response in a subject against a coronavirusby administering to the subject an mRNA having an open reading frame encoding a first antigen, wherein the RNA does not include a stabilization element, and wherein an adjuvant is not co-formulated or co-administered with the vaccine.
- a compositionmay be administered by any route that results in a therapeutically effective outcome.
- RNA vaccinesare administered to a subject in need thereof.
- the exact amount requiredwill vary from subject to subject, depending on the species, age, and general condition of the subject, the severity of the disease, the particular composition, its mode of administration, its mode of activity, and the like.
- the RNAis typically formulated in dosage unit form for ease of administration and uniformity of dosage. It will be understood, however, that the total daily usage of the RNA may be decided by the attending physician within the scope of sound medical judgment.
- the specific therapeutically effective, prophylactically effective, or appropriate imaging dose level for any particular patientwill depend upon a variety of factors including the disorder being treated and the severity of the disorder; the activity of the specific compound employed; the specific composition employed; the age, body weight, general health, sex and diet of the patient; the time of administration, route of administration, and rate of excretion of the specific compound employed; the duration of the treatment; drugs used in combination or coincidental with the specific compound employed; and like factors well known in the medical arts.
- the effective amount (e.g., effective dose) of the RNA, as provided herein,may be as low as 20 ⁇ g, administered for example as a single dose or as two 10 ⁇ g doses.
- the effective amountis a total dose of 20 ⁇ g-300 ⁇ g5 ⁇ g-30 ⁇ g, 5 ⁇ g -25 ⁇ g, 5 ⁇ g -20 ⁇ g, 5 ⁇ g -15 ⁇ g, 5 ⁇ g -10 ⁇ g, 10 ⁇ g -30 ⁇ g, 10 ⁇ g -25 ⁇ g, 10 ⁇ g-20 ⁇ g, 10 ⁇ g -15 ⁇ g, 15 ⁇ g -30 ⁇ g, 15 ⁇ g -25 ⁇ g, 15 ⁇ g -20 ⁇ g, 20 ⁇ g -30 ⁇ g, 25 ⁇ g -30 ⁇ g, or 25 ⁇ g-300 ⁇ g.
- the effective dose(e.g., effective amount) is at least 10 ⁇ g and less than 25 ⁇ g of the composition. In some embodiments, the effective dose (e.g., effective amount) is at least 5 ⁇ g and less than 25 ⁇ g of the composition.
- the effective amountmay be a total dose of 5 ⁇ g, 10 ⁇ g, 15 ⁇ g, 20 ⁇ g, 25 ⁇ g, 30 ⁇ g, 35 ⁇ g, 40 ⁇ g, 45 ⁇ g, 50 ⁇ g, 55 ⁇ g, 60 ⁇ g, 65 ⁇ g, 70 ⁇ g, 75 ⁇ g, 80 ⁇ g, 85 ⁇ g, 90 ⁇ g, 95 ⁇ g, 100 ⁇ g, 110 ⁇ g, 120 ⁇ g, 130 ⁇ g, 140 ⁇ g, 150 ⁇ g, 160 ⁇ g, 170 ⁇ g, 180 ⁇ g, 190 ⁇ g, 200 ⁇ g, 250 ⁇ g, or 300 ⁇ g.
- the effective amount(e.g., effective dose) is a total dose of 10 ⁇ g. In some embodiments, the effective amount is a total dose of 20 ⁇ g (e.g., two 10 ⁇ g doses). In some embodiments, the effective amount is a total dose of 25 ⁇ g. In some embodiments, the effective amount is a total dose of 30 ⁇ g. In some embodiments, the effective amount is a total dose of 50 ⁇ g. In some embodiments, the effective amount is a total dose of 60 ⁇ g (e.g., two 30 ⁇ g doses). In some embodiments, the effective amount is a total dose of 75 ⁇ g. In some embodiments, the effective amount is a total dose of 100 ⁇ g.
- the effective amountis a total dose of 150 ⁇ g. In some embodiments, the effective amount is a total dose of 200 ⁇ g. In some embodiments, the effective amount is a total dose of 250 ⁇ g. In some embodiments, the effective amount is a total dose of 300 ⁇ g. Any of the doses provided above may be an effective amount for a booster dose; for example, in some embodiments, the booster dose is a total dose of 50 ⁇ g. In some embodiments, the composition comprises two or more mRNA polynucleotides and effective amount is a total dose of 20 ⁇ g (e.g., 10 ⁇ g of a first mRNA and 10 ⁇ g of a second mRNA).
- the compositioncomprises two or more mRNA polynucleotides and effective amount is a total dose of 50 ⁇ g (e.g., 25 ⁇ g of a first mRNA and 25 ⁇ g of a second mRNA). In some embodiments, the composition comprises two or more mRNA polynucleotides and effective amount is a total dose of 100 ⁇ g (e.g., 50 ⁇ g of a first mRNA and 50 ⁇ g of a second mRNA).
- RNA described hereincan be formulated into a dosage form described herein, such as an intranasal, intratracheal, or injectable (e.g., intravenous, intraocular, intravitreal, intramuscular, intradermal, intracardiac, intraperitoneal, and subcutaneous).
- Vaccine EfficacySome aspects of the present disclosure provide formulations of the compositions (e.g., RNA vaccines), wherein the RNA is formulated in an effective amount to produce an antigen specific immune response in a subject (e.g., production of antibodies specific to a coronavirus antigen). “An effective amount” is a dose of the RNA effective to produce an antigen-specific immune response.
- an immune response to a vaccine or LNP of the present disclosureis the development in a subject of a humoral and/or a cellular immune response to a (one or more) coronavirus protein(s) present in the vaccine.
- a “humoral” immune responserefers to an immune response mediated by antibody molecules, including, e.g., secretory (IgA) or IgG molecules, while a “cellular” immune response is one mediated by T- lymphocytes (e.g., CD4+ helper and/or CD8+ T cells (e.g., CTLs) and/or other white blood cells.
- CTLscytolytic T- cells
- MHCmajor histocompatibility complex
- helper T-cellsact to help stimulate the function and focus the activity nonspecific effector cells against cells displaying peptide antigens in association with MHC molecules on their surface.
- a cellular immune responsealso leads to the production of cytokines, chemokines, and other such molecules produced by activated T-cells and/or other white blood cells including those derived from CD4+ and CD8+ T-cells.
- the antigen-specific immune responseis characterized by measuring an anti-coronavirus antigen antibody titer produced in a subject administered a composition as provided herein.
- An antibody titeris a measurement of the amount of antibodies within a subject, for example, antibodies that are specific to a particular antigen or epitope of an antigen.
- Antibody titeris typically expressed as the inverse of the greatest dilution that provides a positive result.
- Enzyme-linked immunosorbent assayis a common assay for determining antibody titers, for example.
- a variety of serological testscan be used to measure antibody against encoded antigen of interest, for example, SAR-CoV-2 virus or SAR-CoV-2 viral antigen, e.g., SAR-CoV-2 spike or S protein, of domain thereof. These tests include the hemagglutination-inhibition test, complement fixation test, fluorescent antibody test, enzyme-linked immunosorbent assay (ELISA), and plaque reduction neutralization test (PRNT). Each of these tests measures different antibody activities.
- a plaque reduction neutralization test, or PRNTis used as a serological correlate of protection.
- PRNTmeasures the biological parameter of in vitro virus neutralization and is the most serologically virus-specific test among certain classes of viruses, correlating well to serum levels of protection from virus infection.
- the basic design of the PRNTallows for virus-antibody interaction to occur in a test tube or microtiter plate, and then measuring antibody effects on viral infectivity by plating the mixture on virus-susceptible cells, preferably cells of mammalian origin. The cells are overlaid with a semi-solid media that restricts spread of progeny virus.
- virus that initiates a productive infectionproduces a localized area of infection (a plaque), that can be detected in a variety of ways. Plaques are counted and compared back to the starting concentration of virus to determine the percent reduction in total virus infectivity.
- the serum sample being testedis usually subjected to serial dilutions prior to mixing with a standardized amount of virus.
- concentration of virusis held constant such that, when added to susceptible cells and overlaid with semi-solid media, individual plaques can be discerned and counted.
- PRNT end- point titerscan be calculated for each serum sample at any selected percent reduction of virus activity.
- the serum sample dilution series for antibody titrationshould ideally start below the “seroprotective” threshold titer.
- a seropositivity threshold of 1:10can be considered a seroprotection threshold in certain embodiments.
- a neutralizing immune responseis an immune response that produces a level of antibodies that meet or exceed a seroprotection threshold.
- PRNT end-point titersare expressed as the reciprocal of the last serum dilution showing the desired percent reduction in plaque counts. The PRNT titer can be calculated based on a 50% or greater reduction in plaque counts (PRNT50).
- a PRNT50 titeris preferred over titers using higher cut-offs (e.g., PRNT90) for vaccine sera, providing more accurate results from the linear portion of the titration curve.
- PRNT90cut-offs
- PRNT titersThere are several ways to calculate PRNT titers. The simplest and most widely used way to calculate titers is to count plaques and report the titer as the reciprocal of the last serum dilution to show >50% reduction of the input plaque count as based on the back-titration of input plaques. Use of curve fitting methods from several serum dilutions may permit calculation of a more precise result. There are a variety of computer analysis programs available for this (e.g., SPSS or GraphPad Prism).
- an antibody titeris used to assess whether a subject has had an infection or to determine whether immunizations are required. In some embodiments, an antibody titer is used to determine the strength of an autoimmune response, to determine whether a booster immunization is needed, to determine whether a previous vaccine was effective, and to identify any recent or prior infections. In accordance with the present disclosure, an antibody titer may be used to determine the strength of an immune response induced in a subject by a composition (e.g., RNA vaccine). In some embodiments, an anti-coronavirus antigen antibody titer produced in a subject is increased by at least 1 log relative to a control.
- anti-coronavirus antigen antibody titer produced in a subjectmay be increased by at least 1.5, at least 2, at least 2.5, or at least 3 log relative to a control.
- the anti-coronavirus antigen antibody titer produced in the subjectis increased by 1, 1.5, 2, 2.5 or 3 log relative to a control.
- the anti-coronavirus antigen antibody titer produced in the subjectis increased by 1-3 log relative to a control.
- the anti-coronavirus antigen antibody titer produced in a subjectmay be increased by 1-1.5, 1-2, 1-2.5, 1-3, 1.5-2, 1.5-2.5, 1.5-3, 2-2.5, 2-3, or 2.5-3 log relative to a control.
- the anti-coronavirus antigen antibody titer produced in a subjectis increased at least 2 times relative to a control.
- the anti-coronavirus antigen n antibody titer produced in a subjectmay be increased at least 3 times, at least 4 times, at least 5 times, at least 6 times, at least 7 times, at least 8 times, at least 9 times, or at least 10 times relative to a control.
- the anti-coronavirus antigen antibody titer produced in the subjectis increased 2, 3, 4, 5, 6, 7, 8, 9, or 10 times relative to a control.
- the anti-coronavirus antigen antibody titer produced in a subjectis increased 2-10 times relative to a control.
- the anti-coronavirus antigen antibody titer produced in a subjectmay be increased 2-10, 2-9, 2-8, 2-7, 2-6, 2-5, 2-4, 2-3, 3-10, 3-9, 3-8, 3-7, 3-6, 3-5, 3-4, 4-10, 4-9, 4-8, 4-7, 4-6, 4-5, 5-10, 5-9, 5-8, 5-7, 5-6, 6-10, 6-9, 6-8, 6-7, 7-10, 7-9, 7-8, 8-10, 8-9, or 9-10 times relative to a control.
- an antigen-specific immune responseis measured as a ratio of geometric mean titer (GMT), referred to as a geometric mean ratio (GMR), of serum neutralizing antibody titers to coronavirus.
- GTTgeometric mean titer
- a geometric mean titer (GMT)is the average antibody titer for a group of subjects calculated by multiplying all values and taking the nth root of the number, where n is the number of subjects with available data.
- a controlin some embodiments, is an anti-coronavirus antigen antibody titer produced in a subject who has not been administered a composition (e.g., RNA vaccine).
- a controlis an anti-coronavirus antigen antibody titer produced in a subject administered a recombinant or purified protein vaccine.
- Recombinant protein vaccinestypically include protein antigens that either have been produced in a heterologous expression system (e.g., bacteria or yeast) or purified from large amounts of the pathogenic organism.
- the ability of a compositione.g., RNA vaccine
- a compositionmay be administered to a murine model and the murine model assayed for induction of neutralizing antibody titers. Viral challenge studies may also be used to assess the efficacy of a vaccine of the present disclosure.
- a compositionmay be administered to a murine model, the murine model challenged with virus, and the murine model assayed for survival and/or immune response (e.g., neutralizing antibody response, T cell response (e.g., cytokine response)).
- an effective amount of a compositione.g., RNA vaccine
- a “standard of care,” as provided herein,refers to a medical or psychological treatment guideline and can be general or specific. “Standard of care” specifies appropriate treatment based on scientific evidence and collaboration between medical professionals involved in the treatment of a given condition.
- a “standard of care dose,” as provided herein,refers to the dose of a recombinant or purified protein vaccine, or a live attenuated or inactivated vaccine, or a VLP vaccine, that a physician/clinician or other medical professional would administer to a subject to treat or prevent coronavirus infection or a related condition, while following the standard of care guideline for treating or preventing coronavirus infection or a related condition.
- the anti-coronavirus antigen antibody titer produced in a subject administered an effective amount of an compositionis equivalent to an anti-coronavirus antigen antibody titer produced in a control subject administered a standard of care dose of a recombinant or purified protein vaccine, or a live attenuated or inactivated vaccine, or a VLP vaccine.
- Vaccine efficacymay be assessed using standard analyses (see, e.g., Weinberg et al., J Infect Dis.2010 Jun 1;201(11):1607-10). For example, vaccine efficacy may be measured by double-blind, randomized, clinical controlled trials.
- ARdisease attack rate
- RRrelative risk
- vaccine effectivenessmay be assessed using standard analyses (see, e.g., Weinberg et al., J Infect Dis.2010 Jun 1;201(11):1607-10).
- Vaccine effectivenessis an assessment of how a vaccine (which may have already proven to have high vaccine efficacy) reduces disease in a population.
- Vaccine effectivenessis proportional to vaccine efficacy (potency) but is also affected by how well target groups in the population are immunized, as well as by other non-vaccine-related factors that influence the ‘real-world’ outcomes of hospitalizations, ambulatory visits, or costs.
- a retrospective case control analysismay be used, in which the rates of vaccination among a set of infected cases and appropriate controls are compared.
- efficacy of the compositionis at least 60% relative to unvaccinated control subjects.
- efficacy of the compositionmay be at least 65%, at least 70%, at least 75%, at least 80%, at least 85%, at least 95%, at least 98%, or 100% relative to unvaccinated control subjects.
- Sterilizing Immunityrefers to a unique immune status that prevents effective pathogen infection into the host.
- the effective amount of a composition of the present disclosureis sufficient to provide sterilizing immunity in the subject for at least 1 year.
- the effective amount of a composition of the present disclosureis sufficient to provide sterilizing immunity in the subject for at least 2 years, at least 3 years, at least 4 years, or at least 5 years.
- the effective amount of a composition of the present disclosureis sufficient to provide sterilizing immunity in the subject at an at least 5-fold lower dose relative to control.
- the effective amountmay be sufficient to provide sterilizing immunity in the subject at an at least 10-fold lower, 15-fold, or 20-fold lower dose relative to a control.
- Detectable Antigenis sufficient to produce detectable levels of coronavirus antigen as measured in serum of the subject at 1-72 hours post administration. Titer.
- An antibody titeris a measurement of the number of antibodies within a subject, for example, antibodies that are specific to a particular antigen (e.g., an anti-coronavirus antigen). Antibody titer is typically expressed as the inverse of the greatest dilution that provides a positive result. Enzyme-linked immunosorbent assay (ELISA) is a common assay for determining antibody titers, for example.
- ELISAEnzyme-linked immunosorbent assay
- a neutralizing immune responseis an immune response that is a neutralizing antibody response and/or an effective neutralizing T cell response. In some embodiments a neutralizing antibody response produces a level of antibodies that meet or exceed a seroprotection threshold.
- An effective T cell responseis a response which produces a baseline level of viral activated or viral specific T cells including CD8+ and CD4+ T helper type 1 cells.
- CD8+ cytotoxic T lymphocytestypically clear the intracellular virus compartment and CD4+ T cells exert various functions in the body such as helping B and other T cells, promoting memory generation and indirect or direct cytotoxic activity.
- the effective T cellscomprises a high proportion of CD8+ T cells and/or CD4+ T cells, relative to a baseline level (in a na ⁇ ve subject). In some embodiments these T cells are differentiated towards an early- differentiated memory phenotype with co-expression of CD27 and CD28.
- the effective amount of a composition of the present disclosureis sufficient to produce a 1,000-10,000 neutralizing antibody titer produced by neutralizing antibody against the coronavirus antigen as measured in serum of the subject at 1-72 hours post administration. In some embodiments, the effective amount is sufficient to produce a 1,000-5,000 neutralizing antibody titer produced by neutralizing antibody against the coronavirus antigen as measured in serum of the subject at 1-72 hours post administration. In some embodiments, the effective amount is sufficient to produce a 5,000-10,000 neutralizing antibody titer produced by neutralizing antibody against the coronavirus antigen as measured in serum of the subject at 1-72 hours post administration. In some embodiments, the neutralizing antibody titer is at least 100 NT 50 .
- the neutralizing antibody titermay be at least 200, 300, 400, 500, 600, 700, 800, 900 or 1000 NT50. In some embodiments, the neutralizing antibody titer is at least 10,000 NT50. In some embodiments, the neutralizing antibody titer is at least 100 neutralizing units per milliliter (NU/mL). For example, the neutralizing antibody titer may be at least 200, 300, 400, 500, 600, 700, 800, 900 or 1000 NU/mL. In some embodiments, the neutralizing antibody titer is at least 10,000 NU/mL. In some embodiments, an anti-coronavirus antigen antibody titer produced in the subject is increased by at least 1 log relative to a control.
- an anti-coronavirus antigen antibody titer produced in the subjectmay be increased by at least 2, 3, 4, 5, 6, 7, 8, 9 or 10 log relative to a control.
- an anti-coronavirus antigen antibody titer produced in the subjectis increased at least 2 times relative to a control.
- an anti-coronavirus antigen antibody titer produced in the subjectis increased by at least 3, 4, 5, 6, 7, 8, 9 or 10 times relative to a control.
- a geometric meanwhich is the nth root of the product of n numbers, is generally used to describe proportional growth. Geometric mean, in some embodiments, is used to characterize antibody titer produced in a subject.
- a controlmay be, for example, an unvaccinated subject, or a subject administered a live attenuated viral vaccine, an inactivated viral vaccine, or a protein subunit vaccine.
- EXAMPLES Methods Manufacture of polynucleotides described herein, and/or parts or regions thereof,may be accomplished using the methods taught in PCT Publication No. WO 2014/152027, entitled “Manufacturing Methods for Production of RNA Transcripts,” the contents of which are incorporated herein by reference in their entirety. Purification methods may include those described in PCT Publication No. WO 2014/152030 and PCT Publication No. WO 2014/152031, the contents of each of which are incorporated herein by reference in their entirety.
- Detection and characterization methods of the polynucleotidesmay be performed as described in PCT Publication No. WO 2014/144039, the contents of which are incorporated herein by reference in their entirety. Characterization of the polynucleotides of the disclosure may be accomplished using polynucleotide mapping, reverse transcriptase sequencing, charge distribution analysis, detection of RNA impurities, or any combination of two or more of the foregoing. “Characterizing” comprises determining the RNA transcript sequence, determining the purity of the RNA transcript, or determining the charge heterogeneity of the RNA transcript, for example. Such methods are taught in, for example, PCT Publication No. WO 2014/144711 and PCT Publication No.
- lipid nanoparticle (LNP) formulationincluded or includes 48 mol% lipid of Compound 1, 11 mol% 1,2 distearoyl-sn- glycero-3-phosphocholine (DSPC), 38.5 mol% cholesterol, and 2.5 mol% PEG-modified 1,2 dimyristoyl-sn-glycerol, methoxypolyethyleneglycol (PEG2000 DMG).
- DSPCdistearoyl-sn- glycero-3-phosphocholine
- PEG2000 DMGmethoxypolyethyleneglycol
- miceare immunized with two doses of a given composition, receiving the first dose on day 0, and the second dose on day 22.
- Seraare collected on day 21, three weeks after the first (prime) dose but before administration of the second (boost) dose, and day 36, two weeks after administration of the second (boost) dose.
- miceare euthanized on day 36, and spleens are collected and processed to harvest splenocytes.
- Splenocytesare stimulated with one of a panel of peptide pools, each pool containing peptides from a single MERS-CoV antigen, in the presence of a Golgi blocker, so that cells producing cytokines in response to stimulation retain cytokines instead of secreting them.
- Cell surfacesare stained for lymphocyte markers, including CD3, CD4, CD8, and/or CD107a, and cells are permeabilized and stained for multiple cytokines, including TNF- ⁇ , IFN- ⁇ , IL-2, IL-4, IL-5, IL-10, and/or IL-13. Stained cells are incubated with a viability dye and analyzed by flow cytometry.
- Neutralization AssaysAntibodies in serum, when bound to a viral surface protein that is essential for infection, can prevent a virus from infecting a target cell, an activity referred to as “neutralization.” To determine the ability of mRNA compositions to generate neutralizing antibodies against MERS- CoV, the neutralization activity of sera is quantified using a neutralization assay. For each assay, ARPE-19 cells are plated in 96-well plates, at a density of 2*10 4 cells/well and incubated for 20– 24 hours. Then, serial 3-fold dilutions of each serum sample are prepared in phenol red-free cDMEM.
- MERS-CoV reporter viruscontaining a gene encoding GFP
- a consistent amount of MERS-CoV reporter virus, containing a gene encoding GFPis incubated with each serum dilution sample, to allow for binding of any MERS-CoV-specific antibodies to the virus.
- cellsare washed, and incubated with MERS-CoV /serum mixtures.24 hours after incubation, GFP fluorescence in each well is measured to determine the extent of infection.
- the 50% neutralization titer (NT50)is calculated as the reciprocal of the serum dilution at which 50% of GFP+ cells are observed.
- NT5050% neutralization titer
- Codon-optimized nucleotide sequences encoding MERS-CoV Spike proteinsare cloned into pCAGGS vectors.
- MERS-CoV Spike proteinpseudotyped recombinant VSV- ⁇ G-firefly luciferase virusBHK- 21/WI-2 cells (Kerafast, EH1011) are transfected with the Spike protein expression plasmid, and subsequently infected with VSV ⁇ G-firefly-luciferase as previously described (Whitt, 2010, Journal of Virological Methods 169, 365–374).
- serially diluted serum samplesare mixed with pseudovirus and incubated at 37°C for 45 minutes.
- the virus- serum mixis subsequently used to infect Vero-81 or Huh7.5s cells for 18 hours at 37°C before adding ONE-Glo reagent (Promega E6120) for measurement of luciferase signal (relative luminescence unit; RLU).
- RLUrelative luminescence unit
- the percentage of neutralizationis calculated based on RLU of the virus only control, and subsequently analyzed using four-parameter logistic curve (Prism 8).
- Antibody-dependent cell-mediated cytotoxicity assaysAntibodies in serum, when bound to viral protein expressed on the surface of an infected cell, can be recognized by effector cells, such as natural killer (NK) cells.
- NKnatural killer
- ADCCantibody-dependent cell-mediated cytotoxicity
- Vero cellsare plated in 96-well plates, at a density of 2.5*10 4 cells/well, incubated for 20–24 hours, then inoculated with MERS-CoV at a multiplicity of infection (MOI) of 5 plaque-forming units (PFU) per cell.16 hours after inoculation, serial 3-fold dilutions of serum samples are prepared in RPMI + 4% fetal bovine serum (FBS) containing only minimal amounts of IgG, to reduce background. Serum samples are added to each well, to allow antibodies to bind to infected Vero cells expressing viral surface proteins.
- MOImultiplicity of infection
- FBSfetal bovine serum
- reporter effector cellsare serially diluted in RPMI + 4% low-IgG FBS, added to wells, and incubated for 6 hours to allow for recognition of surface- bound antibodies and expression of luciferase.
- a luciferase substrateis added to wells, so that any luciferase present can react with the substrate to produce light.
- Light emitted from wellsis measured to quantify the amount of luciferase activity as a measurement of ADCC activity.
- Serum-depletion AssaysEpitope- or domain-level specificity of antibodies in serum is evaluated by depleting antibodies with a given specificity from serum, and comparing such antigen-depleted (Ag- depleted) serum with mock-depleted serum.
- Antibodies specific to a given epitope or antigenmay be depleted by incubating serum with a solid support (e.g., surface of a 96-well plate) on which the epitope or antigen is immobilized. Incubation allows antibodies with the desired specificity to bind to the immobilized epitope or antigen, such that the serum collected after incubation does not contain the surface-bound antibodies.
- Mock-depleted serumis prepared by incubating serum under similar conditions, where the solid support does not contain an immobilized epitope or antigen.
- Antigen-depleted seramay be compared to mock-depleted sera in multiple in vitro assays, such as ELISA, neutralization assays, ADCC assays, and/or in vivo, such as by passive transfer of sera before, during, or after inoculation with a virus.
- Example 1Design of mRNA encoding MERS-CoV S proteins, fusion proteins, and variants thereof. MERS-CoV S protein sequences were modified with one or more changes as described in Table E1-1.
- Modifications included(i) introduction of two proline substitutions to stabilize the S protein in a prefusion conformation (S2P); (ii) inactivation of a furin cleavage site; (iii) deletion of an endoplasmic reticulum (ER) retention signal; and/or truncation of a cytoplasmic tail Fusion proteins comprising one or more domains of MERS-CoV S protein, and optionally one or more linkers and/or a transmembrane domain, were designed as described in Table E1-2.
- NTDN-terminal domain
- RBDreceptor-binding domain
- LNPslipid nanoparticles containing mRNAs encoding a 2P-stabilized betacoronavirus S protein having two proline substitutions (S-2P) or a fusion protein comprising an NTD and RBD of the same betacoronavirus S protein, linked to a transmembrane (TM) domain (NTD-RBD-TM), were administered to mice in one dose containing 1 ug mRNA, or in two doses, each dose containing the same amount of mRNA, at either 0.001, 0.01 ug, 0.1 ug, or 1 ug. The first dose was administered on day 0, and second dose was administered on day 22.
- S-2P2P-stabilized betacoronavirus S protein having two proline substitutions
- NTD-RBD-TMtransmembrane domain
- compositions containing mRNA encoding the NTD- RBD fusion protein(NTD-RBD-TM) elicited significantly higher neutralizing antibody titers, relative to those containing mRNA encoding the 2P-stabilized prefusion S protein (S-2P) (FIG. 2).
- NTD-RBD-TMNTD- RBD fusion protein
- S-2P2P-stabilized prefusion S protein
- Example 2Immunization of mice with compositions containing mRNAs encoding MERS- CoV proteins. Mice are administered lipid nanoparticles (LNPs) containing the mRNAs encoding MERS-CoV Spike proteins and/or fusion proteins.
- LNPslipid nanoparticles
- miceare also euthanized to collect spleens for analysis of T cells by cell surface marker and intracellular cytokine staining.
- T cell phenotypes and epitope-specificityis evaluated as described above in “Immunization Methods.” T cells are incubated with peptides from the MERS-CoV S protein NTD, RBD, S1 subunit, S2 subunit, and/or full-length S protein to evaluate specificity to different regions of the S protein. Sera are evaluated for antiviral activities such as neutralization, ADCC activity, and/or prevention of cell-cell spread by MERS-CoV.
- Seraare incubated with immobilized MERS-CoV S protein NTD, RBD, S1 subunit, S2 subunit, and/or full-length S protein to generate sera depleted of NTD-specific, RBD-specific, S1 subunit-specific, S2 subunit-specific, and/or full- length S protein-specific antibodies, and depleted sera are compared to mock-depleted sera to evaluate the contribution of antibody specificity to neutralization, ADCC activity, and/or prevention of cell-cell spread by MERS-CoV.
- Table E2-1MERS-CoV Proteins Example 3.
- LNPslipid nanoparticles
- MERS-CoV S protein-specific IgG titersare measured by ELISA at Day 21 post-vaccination.
- Seraare evaluated for antiviral activities such as neutralization, ADCC activity, and/or prevention of cell-cell spread by MERS- CoV.
- Sera depleted of specific antibodiesare prepared and compared to mock-depleted sera to evaluate the contribution of specific antibodies to neutralization, ADCC activity, and/or prevention of cell-cell spread by MERS-CoV.
- Example 4
- any of the mRNA sequences described hereinmay include a 5’ UTR and/or a 3’ UTR.
- the UTR sequencesmay be selected from the following sequences, or other known UTR sequences may be used.
- any of the mRNA constructs described hereinmay further comprise a poly(A) tail and/or cap (e.g., 7mG(5’)ppp(5’)NlmpNp).
- RNAs and encoded antigen sequences described hereininclude a signal peptide and/or a peptide tag (e.g., C-terminal His tag), it should be understood that the indicated signal peptide and/or peptide tag may be substituted for a different signal peptide and/or peptide tag, or the signal peptide and/or peptide tag may be omitted.
- a signal peptide and/or a peptide tage.g., C-terminal His tag
- any combination of two or more such features, systems, articles, materials, kits, and/or methods, if such features, systems, articles, materials, kits, and/or methods are not mutually inconsistent,is included within the inventive scope of the present disclosure.
- All definitions, as defined and used herein,should be understood to control over dictionary definitions, definitions in documents incorporated by reference, and/or ordinary meanings of the defined terms. All references, patents and patent applications disclosed herein are incorporated by reference with respect to the subject matter for which each is cited, which in some cases may encompass the entirety of the document.
- a reference to “A and/or B”, when used in conjunction with open-ended language such as “comprising”can refer, in some embodiments, to A only (optionally including elements other than B); in another embodiment, to B only (optionally including elements other than A); in yet another embodiment, to both A and B (optionally including other elements); etc.
- “or”should be understood to have the same meaning as “and/or” as defined above.
- At least one of A and Bcan refer, in some embodiments, to at least one, optionally including more than one, A, with no B present (and optionally including elements other than B); in another embodiment, to at least one, optionally including more than one, B, with no A present (and optionally including elements other than A); in yet another embodiment, to at least one, optionally including more than one, A, and at least one, optionally including more than one, B (and optionally including other elements); etc.
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Virology (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Chemical & Material Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- General Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Molecular Biology (AREA)
- Pulmonology (AREA)
- Communicable Diseases (AREA)
- Oncology (AREA)
- Immunology (AREA)
- Microbiology (AREA)
- Mycology (AREA)
- Epidemiology (AREA)
- Medicines Containing Antibodies Or Antigens For Use As Internal Diagnostic Agents (AREA)
- Peptides Or Proteins (AREA)
Abstract
Description
MERS-COV MRNA VACCINES RELATED APPLICATION This application claims the benefit under 35 U.S.C. § 119(e) of U.S. Provisional Application No.63/513,614, filed July 14, 2023, the contents of which are incorporated by reference herein in their entirety. REFERENCE TO AN ELECTRONIC SEQUENCE LISTING The contents of the electronic sequence listing (M137870284WO00-SEQ-NTJ.xml; Size: 113,075; and Date of Creation: July 10, 2024) are incorporated by reference herein in their entirety. BACKGROUND Human coronaviruses are highly contagious enveloped, positive-sense single-stranded RNA viruses of the Coronaviridae family. Two sub-families of Coronaviridae are known to cause human disease. The most important being the β-coronaviruses (betacoronaviruses). The β- coronaviruses are common etiological agents of mild to moderate upper respiratory tract infections. Middle East Respiratory Syndrome Coronavirus (MERS-CoV) is a betacoronavirus that infects the human respiratory tract. Symptoms of MERS-CoV infection can range from mild, such as those similar to a common cold (nasal discharge, sore throat, low fever), to severe, including acute respiratory distress syndrome, which may be fatal. MERS-CoV is endemic in dromedary camels of East Africa and the Arabian Peninsula and was first reported to infect humans in 2012. The case fatality rate of MERS-CoV infection varies depending on factors that impact an individual’s overall health but was found to be an average of 34.4% according to the World Health Organization (WHO). Though zoonotic in origin, human-to-human transmission of MERS-CoV is possible. See, e.g., Ramadan et al., Germs.2019.9(1):35–42. SUMMARY Some aspects of the disclosure relate to mutant MERS-CoV Spike (S) proteins having one or more insertions, deletions, and/or substitutions relative to a naturally occurring full-length MERS-CoV Spike protein. Some aspects relate to fusion proteins comprising one or more domains (e.g., N-terminal domain (NTD) and/or receptor-binding domain (RBD)) of a full-length MERS-CoV Spike protein. Some aspects relate to RNAs (e.g., mRNAs) encoding mutant MERS-CoV Spike proteins and/or fusion proteins as described herein. The proteins and RNAs described herein are based, at least in part, on the findings that administration of lipid nanoparticle compositions containing mRNA encoding modified betacoronavirus Spike proteins elicited robust virus-neutralizing antibody titers (FIG.2). Additionally, mRNA encoding a fusion NTD-RBD-TM protein comprising an N-terminal domain (NTD) and receptor-binding domain (RBD) of a betacoronavirus, and a transmembrane (TM) domain, exhibited improved expression and elicited significantly higher titers of virus-neutralizing antibodies, compared to a full-length Spike protein (FIG.2). Multiple T cell epitopes have been identified in the NTD of MERS-CoV Spike protein, and NTD-specific antibodies contribute to protective immunity against MERS- CoV infection. RBD of MERS-CoV binds the receptor DPP4, contributing to viral attachment to host cells, and RBD-specific antibodies inhibit viral attachment, and consequently inhibit cellular infection and viral replication. Accordingly, the disclosure relates, in some aspects, to compositions comprising a lipid nanoparticle (LNP) and a messenger ribonucleic acid (mRNA) comprising an open reading frame encoding a Middle East Respiratory Syndrome (MERS)-CoV fusion protein comprising: (a) an N-terminal domain (NTD) of a full-length MERS-CoV Spike (S) protein; and (b) a receptor-binding domain (RBD) of the full-length MERS-CoV-S protein, wherein the MERS-CoV fusion protein does not comprise an NTD or an RBD of an S protein of a second coronavirus that is not MERS-CoV. In some embodiments, the NTD and RBD are connected by a linker. In some embodiments, the linker is a glycine linker or a glycine-serine linker. In some embodiments, the linker comprises a pan-HLA-DR-binding epitope (PADRE). In some embodiments, the MERS-CoV fusion protein further comprises a transmembrane (TM) domain. In some embodiments, the TM domain is a MERS-CoV S protein TM domain or derivative thereof. In some embodiments, the TM domain is heterologous to MERS-CoV. In some embodiments, the TM domain is an influenza virus hemagglutinin (HA) TM domain or derivative thereof. In some embodiments, the MERS-CoV fusion protein comprises, in N-to-C-terminal order, the NTD, the RBD, and the TM domain. In some embodiments, the MERS-CoV fusion protein does not comprise a transmembrane (TM) domain. In some embodiments, the MERS-CoV fusion protein is soluble. In some embodiments, the MERS-CoV fusion protein further comprises a signal peptide. In some embodiments, the signal peptide is a MERS-CoV S protein signal peptide. In some embodiments, the signal peptide is heterologous to MERS-CoV. In some aspects, the disclosure relates to compositions comprising a lipid nanoparticle (LNP) and a messenger ribonucleic acid (mRNA) comprising an open reading frame encoding a mutant Middle East Respiratory Syndrome (MERS)-CoV S protein, wherein the mutant MERS-CoV S protein comprises (a) one or more proline substitutions or insertions, and (b) one or more substitutions or deletions in a furin cleavage site, relative to a full-length MERS-CoV S protein. In some embodiments, the mutant MERS-CoV S protein comprises prolines at positions corresponding to residues 1061 and 1061 of the full-length MERS-CoV S protein. In some aspects, the disclosure relates to compositions comprising a lipid nanoparticle (LNP) and a messenger ribonucleic acid (mRNA) comprising an open reading frame encoding a mutant Middle East Respiratory Syndrome (MERS)-CoV S protein, wherein the mutant MERS-CoV S protein comprises (a) one or more substitutions or deletions in a furin cleavage site, and (b) a truncated endoplasmic reticulum (ER) retention signal, relative to a full-length MERS-CoV S protein. In some embodiments, the mutant MERS-CoV S protein does not comprise an ER retention signal. In some aspects, the disclosure relates to compositions comprising a lipid nanoparticle (LNP) and a messenger ribonucleic acid (mRNA) comprising an open reading frame encoding a mutant Middle East Respiratory Syndrome (MERS)-CoV S protein, wherein the mutant MERS-CoV S protein comprises (a) one or more substitutions or deletions in a furin cleavage site, and (b) a truncated cytoplasmic tail, relative to a full-length MERS-CoV S protein. In some embodiments, the mutant MERS-CoV S protein does not comprise a cytoplasmic tail, or comprises a cytoplasmic tail comprising 21 or fewer amino acids. In some aspects, the disclosure relates to compositions comprising a lipid nanoparticle (LNP) and a messenger ribonucleic acid (mRNA) comprising an open reading frame encoding a Middle East Respiratory Syndrome (MERS)-CoV fusion protein, wherein the MERS-CoV fusion protein consists essentially of (a) an N-terminal domain (NTD) of a full-length MERS-CoV S protein, (b) a receptor-binding domain of the full-length MERS-CoV protein, and (c) a transmembrane (TM) domain. In some aspects, the disclosure relates to compositions comprising a lipid nanoparticle (LNP) and a messenger ribonucleic acid (mRNA) comprising an open reading frame encoding a Middle East Respiratory Syndrome (MERS)-CoV fusion protein, wherein the MERS-CoV fusion protein consists essentially of (a) an N-terminal domain (NTD) of a full-length MERS-CoV S protein, and (b) a transmembrane (TM) domain. In some embodiments, the NTD comprises amino acids 18–338 of a full-length MERS- CoV S protein. In some aspects, the disclosure relates to compositions comprising a lipid nanoparticle (LNP) and a messenger ribonucleic acid (mRNA) comprising an open reading frame encoding a Middle East Respiratory Syndrome (MERS)-CoV fusion protein, wherein the MERS-CoV fusion protein consists essentially of (a) a receptor-binding domain (RBD) of a full-length MERS-CoV S protein, and (b) a transmembrane (TM) domain. In some embodiments, the RBD comprises amino acids 376–589 of a full-length MERS- CoV S protein. In some embodiments, the TM domain is heterologous to MERS-CoV. In some embodiments, the TM domain is an influenza virus hemagglutinin (HA) TM domain. In some embodiments, the full-length MERS-CoV- S protein comprises the amino acid sequence of SEQ ID NO: 84. In some aspects, the disclosure relates to compositions comprising a lipid nanoparticle (LNP) and a messenger ribonucleic acid (mRNA) comprising an open reading frame encoding a protein comprising an amino acid sequence with at least 90%, at least 95%, at least 97%, at least 98%, at least 99%, or 100% sequence identity to the amino acid sequence of any one of SEQ ID NOs: 89–98. In some embodiments, the mRNA comprises a 5′ untranslated region (UTR), wherein the 5′ UTR comprises a nucleotide sequence with at least 90% sequence identity to a nucleotide sequence selected from SEQ ID NOs: 1, 2, 5–35, 66, 67, 70–72, 75, 76, and 81. In some embodiments, the 5′ UTR comprises a nucleotide sequence selected from SEQ ID NOs: 1, 2, 5–35, 66, 67, 70–72, 75, 76, and 81. In some embodiments, the mRNA comprises a 3′ untranslated region (UTR), wherein the 5′ UTR comprises a nucleotide sequence with at least 90% sequence identity to a nucleotide sequence selected from SEQ ID NOs: 3–4, 36–44, 68, 69, 73, 74, 77–79, and 82. In some embodiments, the 3′ UTR comprises a nucleotide sequence selected from SEQ ID NOs: 3–4, 36–44, 68, 69, 73, 74, 77–79, and 82. In some embodiments, the ORF comprises one or more stop codons immediately following the last amino acid-encoding codon. In some embodiments, the one or more stop codons comprise the nucleotide sequence UGAUGA. In some embodiments, the one or more stop codons comprise the nucleotide sequence UGAUAAUAG. In some embodiments, the mRNA comprises a polyadenosine (polyA) sequence comprising 20 or more consecutive adenosine nucleotides. In some embodiments, the polyA sequence comprises 100 consecutive adenosine nucleotides. In some embodiments, the polyA sequence comprises, in 5′-to-3′ order, a first nucleotide sequence comprising 30 consecutive adenosine nucleotides, an intervening sequence comprising no more than three adenosine nucleotides, and a second nucleotide sequence comprising 70 consecutive adenosine nucleotides. In some embodiments, the polyA sequence comprises the nucleotide sequence of SEQ ID NO: 80. In some embodiments, the mRNA further comprises a polycytidine (polyC) sequence comprising 20 or more consecutive cytidine nucleotides. In some embodiments, the polyC sequence comprises 30 consecutive cytidine nucleotides. In some embodiments, the polyC sequence is downstream from the polyA sequence, wherein the polyA sequence comprises 64 consecutive adenosine nucleotides. In some embodiments, the polyA sequence comprises 109 consecutive adenosine nucleotides. In some embodiments, the mRNA comprises a 5′ cap analog. In some embodiments, the 5′ cap analog comprises a 7mG(5′)ppp(5′)NlmpNp cap. In some embodiments, the 5′ cap analog comprises an m23′O,7 G+(5′)ppp(5′)Am cap having the structure: . In some embodiments, the lipid nanoparticle comprises 40-55 mol% ionizable amino lipid, 30-45 mol% sterol, 5-15 mol% neutral lipid, and 1-5 mol% PEG-modified lipid. In some embodiments, the ionizable amino lipid comprises a compound of Formula (I):
having the structure: . In some embodiments, the lipid nanoparticle comprises 40-55 mol% ionizable amino lipid, 30-45 mol% sterol, 5-15 mol% neutral lipid, and 1-5 mol% PEG-modified lipid. In some embodiments, the ionizable amino lipid comprises a compound of Formula (I): salt or isomer thereof, wherein: R1 is R”M’R’ or C5-20 alkenyl; R2 and R3 are each independently selected from C1-14 alkyl and C2-14 alkenyl; R4 is -(CH2)nQ, wherein Q is OH and n is selected from 3, 4, and 5; M and M’ are each independently -OC(O)- or -C(O)O-; R5, R6, and R7 are each H; R’ is a linear C1-12 alkyl, or C1-12 alkyl substituted with C6-9 alkyl; R” is C3-14 alkyl; m is selected from 5, 6, 7, 8, 9, 10, 11, 12, and 13. In some embodiments, the ionizable amino lipid comprises Compound 1:
salt or isomer thereof, wherein: R1 is R”M’R’ or C5-20 alkenyl; R2 and R3 are each independently selected from C1-14 alkyl and C2-14 alkenyl; R4 is -(CH2)nQ, wherein Q is OH and n is selected from 3, 4, and 5; M and M’ are each independently -OC(O)- or -C(O)O-; R5, R6, and R7 are each H; R’ is a linear C1-12 alkyl, or C1-12 alkyl substituted with C6-9 alkyl; R” is C3-14 alkyl; m is selected from 5, 6, 7, 8, 9, 10, 11, 12, and 13. In some embodiments, the ionizable amino lipid comprises Compound 1: In some embodiments, the ionizable amino lipid comprises a compound of the structure
In some embodiments, the ionizable amino lipid comprises a compound of the structure In some embodiments, the neutral lipid is 1,2 distearoyl-sn-glycero-3-phosphocholine (DSPC). In some embodiments, the sterol is cholesterol. In some embodiments, the PEG-modified lipid is PEG2000-DMG. In some embodiments, the open reading frame comprises one or more chemically modified nucleotides. In some embodiments, the open reading frame comprises N1-methylpseudouridine. In some embodiments, at least 80% of uracil nucleotides in the open reading frame comprise N1-methylpseudouridine. In some embodiments, 100% of uracil nucleotides in the open reading frame comprise N1-methylpseudouridine. In some embodiments, the open reading frame comprises 5-methylcytidine. In some embodiments, at least 80% of cytosine nucleotides in the open reading frame comprise 5-methylcytidine. In some embodiments, 100% of cytosine nucleotides in the open reading frame comprise 5-methylcytidine. In some embodiments, the open reading frame comprises 5-methyluridine. In some embodiments, at least 80% of uracil nucleotides in the open reading frame comprise 5-methyluridine. In some embodiments, 100% of uracil nucleotides in the open reading frame comprise 5- methyluridine. In some aspects, the disclosure relates to pharmaceutical compositions comprising a composition as described herein, and a pharmaceutically acceptable excipient. In some aspects, the disclosure relates to methods comprising administering to a subject a composition as described herein. In some embodiments, the composition is administered intramuscularly. In some embodiments, the composition is effective to elicit, in the subject, CD4+ and/or CD8+ T cells specific to one or more epitopes of the protein. In some embodiments, the composition is effective to elicit, in the subject, neutralizing antibodies to MERS-CoV. In some embodiments, the composition is effective to elicit, in the subject, antibodies that mediate antibody-dependent cell-mediated cytotoxicity (ADCC) against MERS-CoV-infected cells. In some embodiments, the method comprising administering a first dose and a second dose of the composition. In some aspects, the disclosure relates to a MERS-CoV fusion protein comprising: (a) an N-terminal domain (NTD) of a MERS-CoV Spike (S) protein; and (b) a receptor-binding domain (RBD) of the MERS-CoV-S protein, wherein the MERS-CoV fusion protein does not comprise an NTD or an RBD of an S protein of a coronavirus that is not MERS-CoV. In some embodiments, the NTD and RBD are connected by a linker. In some embodiments, the linker is a glycine linker or a glycine-serine linker. In some embodiments, the linker comprises a pan-HLA-DR-binding epitope (PADRE). In some embodiments, the MERS-CoV fusion protein further comprises a transmembrane (TM) domain. In some embodiments, the TM domain is a MERS-CoV S protein TM domain or derivative thereof. In some embodiments, the TM domain is heterologous to MERS-CoV. In some embodiments, the TM domain is an influenza virus hemagglutinin (HA) TM domain or derivative thereof. In some embodiments, the MERS-CoV fusion protein comprises, in N-to-C-terminal order, the NTD, the RBD, and the TM domain. In some embodiments, the MERS-CoV fusion protein does not comprise a transmembrane (TM) domain. In some embodiments, the MERS-CoV fusion protein is soluble. In some embodiments, the MERS-CoV fusion protein further comprises a signal peptide. In some embodiments, the signal peptide is a MERS-CoV S protein signal peptide. In some embodiments, the signal peptide is heterologous to MERS-CoV. In some aspects, the disclosure relates to mutant Middle East Respiratory Syndrome (MERS)-CoV S proteins, comprising (a) one or more proline substitutions or insertions, and (b) one or more substitutions or deletions in a furin cleavage site, relative to a full-length MERS- CoV S protein. In some embodiments, the mutant MERS-CoV S protein comprises prolines at positions corresponding to residues 1060 and 1061 of the full-length MERS-CoV S protein. In some aspects, the disclosure relates to mutant Middle East Respiratory Syndrome (MERS)-CoV S proteins comprising (a) one or more substitutions or deletions in a furin cleavage site, and (b) a truncated endoplasmic reticulum (ER) retention signal, relative to a full-length MERS-CoV S protein. In some embodiments, the mutant MERS-CoV S protein does not comprise an ER retention signal. In some aspects, the disclosure relates to mutant MERS-CoV S proteins comprising (a) one or more substitutions or deletions in a furin cleavage site, and (b) a truncated cytoplasmic tail, relative to a full-length MERS-CoV S protein. In some embodiments, the mutant MERS-CoV S protein does not comprise a cytoplasmic tail, or comprises a cytoplasmic tail comprising 21 or fewer amino acids. In some aspects, the disclosure relates to MERS-CoV fusion proteins consisting essentially of (a) an N-terminal domain (NTD) of a full-length MERS-CoV S protein, (b) a receptor-binding domain of the full-length MERS-CoV protein, and (c) a transmembrane (TM) domain. In some aspects, the disclosure relates to Middle East Respiratory Syndrome (MERS)- CoV fusion proteins consisting essentially of (a) an N-terminal domain (NTD) of a full-length MERS-CoV S protein, and (b) a transmembrane (TM) domain. In some embodiments, the NTD comprises amino acids 18–338 of a full-length MERS- CoV S protein. In some aspects, the disclosure relates to MERS-CoV fusion proteins consisting essentially of (a) a receptor-binding domain (RBD) of a full-length MERS-CoV S protein, and (b) a transmembrane (TM) domain. In some embodiments, the RBD comprises amino acids 376–589 of a full-length MERS- CoV S protein. In some embodiments, the TM domain is heterologous to MERS-CoV. In some embodiments, the TM domain is an influenza virus hemagglutinin (HA) TM domain. In some embodiments, the full-length MERS-CoV- S protein comprises the amino acid sequence of SEQ ID NO: 84. In some aspects the disclosure relates to proteins comprising an amino acid sequence with at least 95%, at least 97%, at least 98%, at least 99%, or 100% sequence identity to the amino acid sequence of any one of SEQ ID NOs: 87–98. In some aspects, the disclosure relates to ribonucleic acids (RNA) comprising an open reading frame encoding a protein as described herein. In some aspects, the disclosure relates to messenger ribonucleic acids (mRNA) comprising an open reading frame encoding a protein as described herein. In some embodiments, the mRNA comprises one or more chemically modified nucleotides. In some embodiments, 100% of the uracil nucleotides of the mRNA comprise are chemically modified nucleotides. In some embodiments, 100% of uracil nucleotides of the mRNA comprise N1- methylpseudouridine. In some aspects, the disclosure relates to self-amplifying ribonucleic acids (saRNA) comprising an open reading frame encoding a protein comprising: (a) an N-terminal domain (NTD) of a MERS-CoV Spike (S) protein; and (b) a receptor-binding domain (RBD) of the MERS-CoV-S protein, wherein the MERS-CoV fusion protein does not comprise an NTD or an RBD of an S protein of a coronavirus that is not MERS-CoV. In some embodiments, the NTD and RBD are connected by a linker. In some embodiments, the linker is a glycine linker or a glycine-serine linker. In some embodiments, the linker comprises a pan-HLA-DR-binding epitope (PADRE). In some embodiments, the MERS-CoV fusion protein further comprises a transmembrane (TM) domain. In some embodiments, the TM domain is a MERS-CoV S protein TM domain or derivative thereof. In some embodiments, the TM domain is heterologous to MERS-CoV. In some embodiments, the TM domain is an influenza virus hemagglutinin (HA) TM domain or derivative thereof. In some embodiments, the MERS-CoV fusion protein comprises, in N-to-C-terminal order, the NTD, the RBD, and the TM domain. In some embodiments, the MERS-CoV fusion protein does not comprise a transmembrane (TM) domain. In some embodiments, the MERS-CoV fusion protein is soluble. In some embodiments, the MERS-CoV fusion protein further comprises a signal peptide. In some embodiments, the signal peptide is a MERS-CoV S protein signal peptide. In some embodiments, the signal peptide is heterologous to MERS-CoV. In some aspects, the disclosure relates to self-amplifying ribonucleic acids (saRNA) comprising an open reading frame encoding a mutant Middle East Respiratory Syndrome (MERS)-CoV S protein, wherein the mutant MERS-CoV S protein comprises (a) one or more proline substitutions or insertions, and (b) one or more substitutions or deletions in a furin cleavage site, relative to a full-length MERS-CoV S protein. In some embodiments, the mutant MERS-CoV S protein comprises prolines at positions corresponding to residues 1060 and 1061 of the full-length MERS-CoV S protein. In some aspects, the disclosure relates to self-amplifying ribonucleic acids (saRNA) comprising an open reading frame encoding a mutant Middle East Respiratory Syndrome (MERS)-CoV S protein, wherein the mutant MERS-CoV S protein comprises (a) one or more substitutions or deletions in a furin cleavage site, and (b) a truncated endoplasmic reticulum (ER) retention signal, relative to a full-length MERS-CoV S protein. In some embodiments, the mutant MERS-CoV S protein does not comprise an ER retention signal. In some aspects, the disclosure relates to self-amplifying ribonucleic acids (saRNA) comprising an open reading frame encoding a mutant Middle East Respiratory Syndrome (MERS)-CoV S protein, wherein the mutant MERS-CoV S protein comprises (a) one or more substitutions or deletions in a furin cleavage site, and (b) a truncated cytoplasmic tail, relative to a full-length MERS-CoV S protein. In some embodiments, the mutant MERS-CoV S protein does not comprise a cytoplasmic tail, or comprises a cytoplasmic tail comprising 21 or fewer amino acids. In some aspects, the disclosure relates to self-amplifying ribonucleic acids (saRNA) comprising an open reading frame encoding a Middle East Respiratory Syndrome (MERS)-CoV fusion protein, wherein the MERS-CoV fusion protein consists essentially of (a) an N-terminal domain (NTD) of a full-length MERS-CoV S protein, (b) a receptor-binding domain of the full-length MERS-CoV protein, and (c) a transmembrane (TM) domain. In some aspects, the disclosure relates to self-amplifying ribonucleic acids (saRNA) comprising an open reading frame encoding a Middle East Respiratory Syndrome (MERS)-CoV fusion protein, wherein the MERS-CoV fusion protein consists essentially of (a) an N-terminal domain (NTD) of a full-length MERS-CoV S protein, and (b) a transmembrane (TM) domain. In some embodiments, the NTD comprises amino acids 18–338 of a full-length MERS- CoV S protein. In some aspects, the disclosure relates to self-amplifying ribonucleic acids (saRNA) comprising an open reading frame encoding a Middle East Respiratory Syndrome (MERS)-CoV fusion protein, wherein the MERS-CoV fusion protein consists essentially of (a) a receptor-binding domain (RBD) of a full-length MERS-CoV S protein, and (b) a transmembrane (TM) domain. In some embodiments, the RBD comprises amino acids 376–589 of a full-length MERS- CoV S protein. In some embodiments, the TM domain is heterologous to MERS-CoV. In some embodiments, the TM domain is an influenza virus hemagglutinin (HA) TM domain. In some embodiments, the full-length MERS-CoV- S protein comprises the amino acid sequence of SEQ ID NO: 84. BRIEF DESCRIPTION OF THE DRAWINGS FIG.1 is a structural schematic of a betacoronavirus S protein, including the N-terminal domain (NTD), the receptor-binding domain (RBD), the Spike subdomain 1 (SD1), the Spike subdomain 2 (SD2), and the S2 subunit of the spike protein (S2) (left). The schematic on the right shows elements of the NTD-RBD-TM construct design, including the linked RBD and NTD. FIG.2 shows the neutralizing antibody (NAb) titers (y-axis) elicited in mice in response to immunization with either one or two doses of varying amounts of S-2P (2P-stabilized full- length S protein), NTD-RBD-TM (S protein NTD and RBD linked to a transmembrane (TM) domain), RBD-TM (S protein RBD linked to a TM domain), and NTD-TM (S protein NTD linked to a TM domain) (x-axis). DETAILED DESCRIPTION MERS-CoV The genome of MERS-CoV is a single-stranded positive-sense RNA (+ssRNA) with the size of about 30.1 kb. Coronavirus genomes include a variable number of open reading frames (ORFs) that encode accessory proteins, nonstructural proteins, and structural proteins (Song et al. 2019 Viruses;11(1):p.59). Many antigenic peptides are located in the structural proteins (Cui et al.2019 Nat. Rev. Microbiol., 17(3):181–192). Spike glycoprotein (S), a small envelope protein (E), matrix protein (M), and nucleocapsid protein (N) are four such structural proteins. Since S protein contributes to cell tropism and virus particle entry, and also elicits neutralizing antibodies (NAb) and protective immunity, it is considered an important target in coronavirus vaccine development. Moreover, amino acid sequence analysis has shown that the S protein contains regions conserved among coronaviruses, which may be the basis for universal vaccine development. MERS-CoV Proteins Some aspects of the disclosure relate to compositions comprising nucleic acids (e.g., mRNAs) encoding proteins of interest, e.g., a protein derived from a betacoronavirus structural protein such as a MERS-CoV Spike (S) protein. Such compositions do not comprise antigens per se, but rather comprise nucleic acids, in particular, mRNA(s) that encode antigens or antigenic sequences once delivered to a cell, tissue or subject. Delivery of nucleic acids, in particular mRNA(s), is achieved by inclusion of nucleic acids in appropriate carriers or delivery vehicles (e.g., lipid nanoparticles) such that upon administration to cells, tissues or subjects, nucleic acid is taken up by cells which, in turn, express protein(s) encoded by the nucleic acids, e.g., mRNAs. Antigens, as used herein, are proteins capable of inducing an immune response (e.g., causing an immune system to produce antibodies against the antigens). The vaccines as described herein provide a unique advantage over traditional protein-based vaccination approaches, in which protein antigens are purified or produced in vitro, e.g., recombinant protein production technologies. The vaccines of the present disclosure feature mRNA encoding the desired antigens, which when introduced into the body, i.e., administered to a mammalian subject (for example a human) in vivo, cause the cells of the body to express the desired antigens. To facilitate delivery of the mRNAs of the present disclosure to the cells of the body, the mRNAs are encapsulated in lipid nanoparticles (LNPs). Upon delivery and uptake by cells of the body, the mRNAs are translated in the cytosol and protein antigens are generated by the host cell machinery. The protein antigens are presented and elicit an adaptive humoral and cellular immune response. Neutralizing antibodies are directed against the expressed protein antigens and hence the protein antigens are considered relevant target antigens for vaccine development. Herein, use of the term “antigen” encompasses immunogenic proteins and immunogenic fragments (an immunogenic fragment that induces (or is capable of inducing) an immune response to a (at least one) MERS-CoV variant), unless otherwise stated. It should be understood that the term “protein” encompasses peptides and the term “antigen” encompasses antigenic fragments. Other molecules may be antigenic such as bacterial polysaccharides or combinations of protein and polysaccharide structures, but for the viral vaccines included herein, viral proteins, fragments of viral proteins and designed and or mutated proteins derived from MERS-CoV are the antigens described herein. Many proteins have a quaternary or three-dimensional structure, which consists of more than one polypeptide or several polypeptide chains that associate into an oligomeric molecule. As used herein the term “subunit” refers to a single protein molecule, for example, a polypeptide or polypeptide chain resulting from processing of a nascent protein molecule, which subunit assembles (or “coassembles”) with other protein molecules (e.g., subunits or chains) to form a protein complex. Proteins can have a relatively small number of subunits and therefore be described as “oligomeric” or can consist of a large number of subunits and therefore be described as “multimeric”. The subunits of an oligomeric or multimeric protein may be identical, homologous or totally dissimilar and dedicated to disparate tasks. Proteins or protein subunits can further comprise domains. As used herein, the term “domain” refers to a distinct functional and/or structural unit within a protein. Typically, a “domain” is responsible for a particular function or interaction, contributing to the overall role of a protein. Domains can exist in a variety of biological contexts. Similar domains (i.e., domains sharing structural, functional and/or sequence homology) can exist within a single protein or can exist within distinct proteins having similar or different functions. A protein domain is often a conserved part of a given protein tertiary structure or sequence that can function and exist independently of the rest of the protein or subunit thereof. In structural and molecular biology, identical, homologous or similar subunits or domains can help to classify newly identified or novel proteins. As used herein, the term antigen is distinct from the term “epitope” which is a substructure of an antigen, e.g., a polypeptide, such as 7-10 amino acids, or carbohydrate structure, which may be recognized by an antigen binding site. The art describes protein antigens that are delivered to subjects or immune cells in isolated form, e.g., isolated protein, polypeptide or peptide antigens, however, the design, testing, validation, and production of protein antigens can be costly and time-consuming, especially when producing proteins at large scale. By contrast, mRNA technology is amenable to rapid design and testing of mRNA constructs encoding a variety of antigens. Moreover, rapid production of mRNA coupled with inclusion in appropriate delivery vehicles (e.g., lipid nanoparticles), can proceed quickly and can rapidly produce mRNA vaccines at large scale. Potential benefit also arises from the fact that antigens encoded by the mRNAs are expressed by the cells of the subject, e.g., are expressed by the human body, and thus the subject, e.g., the human body, serves as the “factory” to produce the antigens which, in turn, elicit the desired immune response. Other immune cells, for example, B cells and T cells, are then able to recognize and mount and immune response develop an immune response against the encoded protein and ultimately create a long -lasting protective response against the coronavirus. Low immunogenicity, a drawback in protein vaccine development due to poor presentation to the immune system or incorrect folding of the antigens, is avoided through the use of the highly effective mRNA vaccines encoding spike protein, subunits and domains thereof described herein. The compositions, as described herein, may include an RNA or multiple RNAs encoding two or more antigens of the same or different MERS-CoV strains or genotypes. Also provided herein are combination vaccines that include RNA encoding one or more MERS-CoV antigens and one or more antigen(s) of a different organism. Thus, the vaccines of the present disclosure may be combination vaccines that target one or more antigens of the same MERS-CoV genotype or strain, or one or more antigens of different MERS-CoV genotypes or different species, e.g., antigens which induce immunity to organisms which are found in the same geographic areas where the risk of coronavirus infection is high or organisms to which an individual is likely to be exposed to when exposed to a coronavirus (e.g., MERS-CoV). In some embodiments, the second or subsequent circulating MERS-CoV antigen is an immunodominant antigen from an emerging strain. An immunodominant antigen of an emerging strain is assessed with respect to the strain from which the antigen is derived, relative to a different strain of the virus, such as the original strain or other variant thereof. An immunodominant antigen of the emerging strain induces a stronger immune response against the emerging strain than against the different strain. In some embodiments, an immunodominant antigen of the emerging strain is more infective than a different strain of the virus, such as the original strain or other variant thereof. Exemplary sequences of coronavirus proteins and variants thereof (e.g., comprising one or more domains of a coronavirus protein) encoded by mRNAs as described herein are provided in EXEMPLARY SEQUENCES as SEQ ID NOs: 84–98. In some embodiments, an RNA (e.g., mRNA) comprises an open reading frame encoding a protein having at least 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity to an amino acid sequence of SEQ ID NO: 84. In some embodiments, an RNA (e.g., mRNA) comprises an open reading frame encoding a protein having at least 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity to an amino acid sequence of SEQ ID NO: 85. In some embodiments, an RNA (e.g., mRNA) comprises an open reading frame encoding a protein having at least 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity to an amino acid sequence of SEQ ID NO: 86. In some embodiments, an RNA (e.g., mRNA) comprises an open reading frame encoding a protein having at least 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity to an amino acid sequence of SEQ ID NO: 87. In some embodiments, an RNA (e.g., mRNA) comprises an open reading frame encoding a protein having at least 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity to an amino acid sequence of SEQ ID NO: 88. In some embodiments, an RNA (e.g., mRNA) comprises an open reading frame encoding a protein having at least 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity to an amino acid sequence of SEQ ID NO: 89. In some embodiments, an RNA (e.g., mRNA) comprises an open reading frame encoding a protein having at least 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity to an amino acid sequence of SEQ ID NO: 90. In some embodiments, an RNA (e.g., mRNA) comprises an open reading frame encoding a protein having at least 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity to an amino acid sequence of SEQ ID NO: 91. In some embodiments, an RNA (e.g., mRNA) comprises an open reading frame encoding a protein having at least 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity to an amino acid sequence of SEQ ID NO: 92. In some embodiments, an RNA (e.g., mRNA) comprises an open reading frame encoding a protein having at least 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity to an amino acid sequence of SEQ ID NO: 93. In some embodiments, an RNA (e.g., mRNA) comprises an open reading frame encoding a protein having at least 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity to an amino acid sequence of SEQ ID NO: 94. In some embodiments, an RNA (e.g., mRNA) comprises an open reading frame encoding a protein having at least 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity to an amino acid sequence of SEQ ID NO: 95. In some embodiments, an RNA (e.g., mRNA) comprises an open reading frame encoding a protein having at least 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity to an amino acid sequence of SEQ ID NO: 96. In some embodiments, an RNA (e.g., mRNA) comprises an open reading frame encoding a protein having at least 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity to an amino acid sequence of SEQ ID NO: 97. In some embodiments, an RNA (e.g., mRNA) comprises an open reading frame encoding a protein having at least 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity to an amino acid sequence of SEQ ID NO: 98. In some embodiments, an mRNA vaccine comprises 1, 2, 3, 4, 5, or 6 mRNAs encoding different proteins, wherein each protein comprises at least one mutation and/or at least one deletion. In some embodiments, the mRNA vaccine further comprises an mRNA encoding a wild-type MERS-CoV S protein or the antigenic fragment thereof. The mRNA vaccine, in some embodiments, is in a lipid nanoparticle (that is, the lipid nanoparticle comprises 1, 2, 3, 4, 5, or 6 mRNAs encoding different protein). In some embodiments, a composition comprises a first mRNA encoding a protein or variant thereof of a first MERS-CoV virus and a second mRNA encoding a second protein or variant thereof of a second MERS-CoV virus. In some embodiments, the first MERS-CoV virus is a first circulating MERS-CoV virus. In some embodiments, the second MERS-CoV virus is a second circulating MERS-CoV virus. “Circulating viruses” as used herein refer to viruses that have been in circulation for 1 month, 2 months, 3 months, 4 months, 5 months, 6 months, 7 months, 8 months, 9 months, 10 months, 11 months, a portion of a year, 1 year, 1.5 years, 2 years, 3 years, or longer. In some embodiments, the first and second mRNAs are present in the composition in a 1:1, 1:2, 1:3, or 1:4 ratio. In some embodiments, the first and second mRNAs are present in the composition in a 2:1, 3:1, or 4:1 ratio. In some embodiments, the first and second mRNAs are present in the composition in a 1:1 ratio. In some embodiments, a composition further comprises a third mRNA encoding a protein or variant thereof of a third MERS-CoV virus, wherein the protein comprises at least one addition, substitution, or deletion relative to the amino acid sequence of SEQ ID NO: 84. In some embodiments, the first and third mRNAs are present in the composition in a 1:1, 1:2, 1:3, or 1:4 ratio. In some embodiments, the first and third mRNAs are present in the composition in a 2:1, 3:1, or 4:1 ratio. In some embodiments, the first and third mRNAs are present in the composition in a 1:1 ratio. In some embodiments, a composition further comprises a fourth mRNA encoding a protein or variant thereof of a fourth MERS-CoV virus, wherein the protein comprises at least one addition, substitution, or deletion relative to the amino acid sequence of SEQ ID NO: 84. In some embodiments, the first and fourth mRNAs are present in the composition in a 1:1, 1:2, 1:3, or 1:4 ratio. In some embodiments, the first and fourth mRNAs are present in the composition in a 2:1, 3:1, or 4:1 ratio. In some embodiments, the first and fourth mRNAs are present in the composition in a 1:1 ratio. In some embodiments, a composition further comprises a fifth mRNA encoding a protein or variant thereof of a fifth MERS-CoV virus, wherein the protein comprises at least one addition, substitution, or deletion relative to the amino acid sequence of SEQ ID NO: 84. In some embodiments, the first and fifth mRNAs are present in the composition in a 1:1, 1:2, 1:3, or 1:4 ratio. In some embodiments, the first and fifth mRNAs are present in the composition in a 2:1, 3:1, or 4:1 ratio. In some embodiments, the first and fifth mRNAs are present in the composition in a 1:1 ratio. In some embodiments, a composition further comprises a sixth mRNA encoding a protein or variant thereof of a sixth MERS-CoV virus, wherein the protein comprises at least one addition, substitution, or deletion relative to the amino acid sequence of SEQ ID NO: 84. In some embodiments, the first and sixth mRNAs are present in the composition in a 1:1, 1:2, 1:3, or 1:4 ratio. In some embodiments, the first and sixth mRNAs are present in the composition in a 2:1, 3:1, or 4:1 ratio. In some embodiments, the first and sixth mRNAs are present in the composition in a 1:1 ratio. In some embodiments, the mRNAs are present in the composition in an equal amount (e.g., a 1:1 weight/weight ratio or a 1:1 molar ratio), for example, a ratio of 1:1 (:1:1:1:1) of mRNA encoding distinct coronavirus antigens. As used herein, a “weight/weight ratio” or wt/wt ratio or wt:wt ratio refers to the ratio between the weights (masses) of the different components. A “molar ratio” refers to the ratio between different components (e.g., the number of mRNA encoding each antigen). In some embodiments, the ratio is 1:1, 1:2, 1:3, 1:4, 2:1, 3:1, or 4:1. In each embodiment or aspect described herein, it is understood that the featured vaccines include the mRNAs encapsulated within LNPs. While it is possible to encapsulate each unique mRNA in its own LNP, the mRNA vaccine technology enjoys the significant technological advantage of being able to encapsulate several mRNAs in a single LNP product. In other embodiments the vaccines are separate vaccines that are not co-formulated, but may be admixed separately before administration or simply administered separately. Spike (S) Proteins The envelope spike (S) proteins of known betacoronaviruses determine the virus host tropism and entry into host cells. Coronavirus spike (S) protein is a choice antigen for the vaccine design as it can induce neutralizing antibodies and protective immunity. S protein is critical for MERS-CoV infection. The organization of the S protein is similar among betacoronaviruses, such as MERS-CoV, SARS-CoV-2, SARS-CoV, HKU1-CoV, MHV-CoV and NL63-CoV. The organization of the Spike (S) protein is similar among betacoronaviruses, such as SARS-CoV-2, SARS-CoV, MERS-CoV, HKUl-CoV, MHV-CoV and NL63-CoV, including two subunits, S1 and S2, which mediate attachment and membrane fusion, respectively. The S1 subunit includes an N terminal domain (NTD), a receptor binding domain (RBD), and two subdomains (SD1 and SD2); the S2 subunit participates in fusion with the host cell membrane and viral entry. As used herein, the term “Spike protein” refers to a glycoprotein that forms homotrimers protruding from the envelope (viral surface) of viruses including betacoronaviruses. Trimerized Spike protein facilitates entry of the virion into a host cell by binding to a receptor on the surface of a host cell followed by fusion of the viral and host cell membranes. The S protein is a highly glycosylated and large type I transmembrane fusion protein that is made up of 1,160 to 1,400 amino acids, depending upon the type of virus. Betacoronavirus Spike proteins comprise between about 1100 to 1500 amino acids. MERS-CoV spike (S) protein is a primary antigen choice for vaccine design, as it can induce neutralizing antibodies and protective immunity. mRNAs as described herein are designed to produce MERS-CoV Spike proteins (i.e., encode Spike proteins such that Spike protein is expressed when the mRNA is delivered to a cell or tissue, for example a cell or tissue in a subject), as well as variants thereof. The skilled artisan will understand that, while an essentially full length or complete Spike protein may be necessary for a virus, e.g., a betacoronavirus, to perform its intended function of facilitating virus entry into a host cell, a certain amount of variation in Spike protein structure and/or sequence is tolerated when seeking primarily to elicit an immune response against Spike protein. For example, minor truncation, e.g., of one to a few, possibly up to 5 or up to 10 amino acids from the N- or C-terminus of the encoded Spike protein, e.g., encoded Spike protein antigen, may be tolerated without changing the antigenic properties of the protein. Likewise, variation (e.g., conservative substitution) of one to a few, possibly up to 5 or up to 10 amino acids (or more) of the encoded Spike protein, e.g., encoded Spike protein antigen, may be tolerated without changing the antigenic properties of the protein. In some embodiments, the Spike protein is a stabilized Spike protein, for example, the Spike protein is stabilized by two proline substitutions (a 2P mutation). In some embodiments, the Spike protein is not a stabilized Spike protein, for example, the Spike protein is not stabilized by two proline substitutions (a 2P mutation). In some embodiments, the Spike protein is from a different virus strain. A strain is a genetic variant of a microorganism (e.g., a virus). New viral strains can be created due to mutation, which may be selected due to enhanced replication, transmissibility, and/or evasion of pre-existing immune responses (e.g., antigenic drift), or recombination of genetic components when two or more viruses infect the same cell, with such recombinant viruses being selected due to enhanced replication, transmissibility, and/or evasion of pre-existing immune responses. Antigenic drift is a process that generates genetic and antigenic variation in viruses, by the accumulation of mutations in the virus genes that code for virus-surface proteins recognized by host immune responses (antibodies and T cells). This results in selection for new virus strains that are not effectively inhibited by the antibodies and/or T cell responses that prevented or mitigated infection by previous (wild-type or ancestral) strains. This makes it easier for the changed virus to spread throughout a partially immune population. Antigenic shift is the process by which two or more different strains of a virus, or strains of two or more different viruses, combine to form a new subtype having a mixture of the surface antigens of the two or more original strains, which may create virus with a novel combination of surface antigens that did not previously exist in nature. The term is often applied specifically to influenza viruses, where segmentation of the viral genome into distinct RNA segments, and reassortment of genome segments during virion production, allows the production of reassortant progeny with novel combinations of genome segments from co-infected cells. However, genetic recombination may occur between non-segmented viruses (e.g., MERS-CoV) where multiple viral genotypes replicate in the same cell, e.g., by switching between two template genomes during replication, resulting in progeny genomes with combinations of sequences from two or more viral genotypes. Antigenic shift is contrasted with antigenic drift (in which individual mutations accumulate over time, and may lead to a loss of immunity, or in vaccine mismatch). In contrast to accumulation of mutations on an ancestral genome (antigenic drift), genetic recombination (through reassortment or molecular recombination of genomes) is often associated with a major reorganization of viral genomes, resulting in novel combination of genes, which may cause a more immediate and drastic change in viral phenotype. A virus strain as used herein is a genetic variant or of a virus that is characterized by a differing isoform of one or more surface proteins of the virus. In the case of MERS-CoV, for example, a different amino acid sequence in the MERS-CoV spike protein where the immune response in an individual to the new strain is less effective than to the strain used to immunize or first infect the individual. A new virus strain may arise from natural mutation or a combination of natural mutation and immune selection due to an ongoing immune response in an immunized or previously infected individual. A new virus strain can differ by one, two, three or more amino acid mutations in regions of the spike protein responsible for a viral function such as receptor binding or viral fusion with a target cell. A spike protein from a new strain may differ from the parental strain by as much as 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity at the amino acid level. A natural virus strain is a variant of a given virus that is recognizable because it possesses some “unique phenotypic characteristics” that remain stable (e.g., stable and heritable biological, serological, and/or molecular characters) under natural conditions. Such “unique phenotypic characteristics” are biological properties different from the compared reference virus, such as unique antigenic properties, host range (e.g., infecting a different kind of host), symptoms of disease caused by the strain, different type of disease caused by the strain (e.g., transmitted by different means), etc. A “unique phenotypic characteristic” can be detected clinically (e.g., clinical manifestations detected in a host infected with the strain) or within a comparative animal experiment in which a researcher skilled in the art of virology can distinguish between the reference control virus-infected animal and the animal infected with the alleged new strain, without knowing which animal received which virus and without having any information about the differences between the two viruses. Importantly, a virus variant with a simple difference in genome sequence is not a separate strain if there is no recognizable distinct viral phenotype. The extent of genomic sequence variation is irrelevant for the classification of a variant as a strain since a distinct phenotype sometimes arises from few mutations. S proteins of coronaviruses can be divided into two important functional subunits, of which include the N-terminal S1 subunit, which forms of the globular head of the S protein, and the C-terminal S2 region that forms the stalk of the protein and is directly embedded into the viral envelope. Upon interaction with a potential host cell, the S1 subunit will recognize and bind to receptors on the host cell, specifically dipeptidyl peptidase 4 (DPP4), whereas the S2 subunit, which is the most conserved component of the S protein, will be responsible for fusing the envelope of the virus with the host cell membrane. (See e.g., Raj et al., Nature.2013. 495(7440):251–254.). Each monomer of trimeric S protein trimer contains the two subunits, S1 and S2, mediating attachment and membrane fusion, respectively. As part of the infection process in vivo, the two subunits are separated from each other by an enzymatic cleavage process. S protein is first cleaved by furin-mediated cleavage at the S1/S2 site in infected cells. In vivo, a subsequent serine protease-mediated cleavage event occurs at the S2′ site within S1. In MERS- CoV, the S1/S2 cleavage site is at amino acids 748–RSVR–751 (SEQ ID NO: 45). The S2′ cleavage site is at amino acids 884–RSAR–887 (SEQ ID NO: 46). As used herein, for example in the context of designing MERS-CoV S proteins, subunits, and/or fusion proteins encoded by the nucleic acids, e.g., mRNAs, as described herein, the term “S1 subunit” (e.g., S1 subunit antigen) refers to the N-terminal subunit of the Spike protein beginning at the S protein N-terminus and ending at the S1/S2 cleavage site whereas the term “S2 subunit” (e.g., S2 subunit antigen) refers to the C-terminal subunit of the Spike protein beginning at the S1/S2 cleavage site and ending at the C-terminus of the Spike protein. As described supra, the skilled artisan will understand that, while an essentially full length or complete Spike protein S1 or S2 subunit may be necessary for receptor binding or membrane fusion, respectively, a certain amount of variation in S1 or S2 structure and/or sequence is tolerated when seeking primarily to elicit an immune response against Spike protein subunits. For example, minor truncation, e.g., of one to a few, possibly up to 4, 5, 6, 7, 8, 9 or 10 amino acids from the N- or C-terminus of the encoded subunit, e.g., encoded S1 or S2 protein antigens, may be tolerated without changing the antigenic properties of the protein. Likewise, variation (e.g., conservative substitution) of one to a few, possibly up to 4, 5, 6, 7, 8, 9 or 10 amino acids (or more) of the encoded Spike protein subunits, e.g., encoded S1 or S2 protein antigen, may be tolerated without changing the antigenic properties of the protein(s). In some embodiments, the composition comprises an mRNA encoding a Spike protein associated with the Kingdom of Saudi Arabia (KSA) 2019 lineage. An exemplary Spike protein of the MERS-CoV KSA 2019 lineage is provided as amino acid sequence of SEQ ID NO: 84. In some embodiments, the composition comprises an mRNA encoding a Spike protein associated with the Riyadh 2013 lineage. An exemplary Spike protein of the MERS-CoV Riyadh 2013 lineage is provided as amino acid sequence of SEQ ID NO: 85. In some embodiments, the composition comprises an mRNA encoding a Spike protein associated with the England 2012 lineage. An exemplary Spike protein of the MERS-CoV England 2012 lineage is provided as amino acid sequence of SEQ ID NO: 84. In some embodiments, a Spike protein, e.g., an encoded Spike protein antigen, has the amino acid sequence of SEQ ID NO: 84. In some embodiments, a Spike protein, e.g., an encoded Spike protein antigen, has the amino acid sequence of SEQ ID NO: 85. In some embodiments, a Spike protein, e.g., an encoded Spike protein antigen, has the amino acid sequence of SEQ ID NO: 86. In some embodiments, a Spike protein, e.g., an encoded Spike protein antigen, has no greater than 100, no greater than 90, no greater than 80, no greater than 70, no greater than 60, no greater than 50, no greater than 40, no greater than 30, no greater than 20, no greater than 10, or no greater than 5 amino acid substitutions and/or deletions as compared to (when aligned with) a Spike protein having the amino acid sequence as provided in any one of SEQ ID NOs: 84–86. Where minor variations are made in encoded Spike protein sequences, the variant preferably has the same activity as the comparator Spike protein sequence and/or has the same immune specificity as the comparator Spike protein, as determined for example, in immunoassays (e.g., enzyme-linked immunosorbent assays (ELISA assays). In some embodiments, a Spike protein, e.g., an encoded Spike protein antigen, has the amino acid sequence of SEQ ID NO: 89. In some embodiments, a Spike protein, e.g., an encoded Spike protein antigen, has the amino acid sequence of SEQ ID NO: 90. In some embodiments, a Spike protein, e.g., an encoded Spike protein antigen, has the amino acid sequence of SEQ ID NO: 91. In some embodiments, a Spike protein, e.g., an encoded Spike protein antigen, has the amino acid sequence of SEQ ID NO: 92. In some embodiments, a Spike protein, e.g., an encoded Spike protein antigen, has no greater than 100, no greater than 90, no greater than 80, no greater than 70, no greater than 60, no greater than 50, no greater than 40, no greater than 30, no greater than 20, no greater than 10, or no greater than 5 amino acid substitutions and/or deletions as compared to (when aligned with) a Spike protein having the amino acid sequence as provided in any one of SEQ ID NOs: 89–92. In some embodiments, a Spike protein, e.g., an encoded Spike protein antigen, comprises one or more (e.g., two consecutive) proline substitutions or insertions at or near the boundary between a heptad repeat 1 (HR1) domain and a central helix (CH) domain. Introduction (e.g., by insertion or substitution) of proline residues in this region stabilize the Spike protein in a prefusion conformation. In some embodiments, the Spike protein comprises 1, 2, 3, 4, 5, 6, 7, 8, 9, or 10 proline residues that are inserted and/or substituted, relative to a wild-type (naturally occurring) MERS-CoV S protein. In some embodiments, an introduced proline residue is up to 15 amino acids N-terminal to the last (C-terminal) residue of the HR1 domain, and up to 5 amino acids C-terminal to the first (N-terminal) residue of the CH domain of the wild-type MERS-CoV S protein. In some embodiments, the Spike protein comprises a proline residue at one or more positions corresponding to 1058, 1059, 1060, and 1061 of SEQ ID NO: 84. In some embodiments, the Spike protein comprises an L1058P, D1059P, V1060P, and/or L1061P substitution relative to the amino acid sequence of SEQ ID NO: 84. In some embodiments, the Spike protein comprises proline residues at positions corresponding to 1060 and 1061 of SEQ ID NO: 84. In some embodiments, the Spike protein comprises V1060P and L1061P substitutions, relative to the amino acid sequence of SEQ ID NO: 84. In some embodiments, the Spike protein has an amino acid sequence with at least 90%, at least 95%, at least 96%, at least 97%, at least 98%, or at least 99% identity to the amino acid sequence of SEQ ID NO: 89, and the Spike protein comprises proline residues at positions corresponding to amino acids 1060 and 1061 of SEQ ID NO: 84. In some embodiments, a Spike protein, e.g., an encoded Spike protein antigen, comprises one or more mutations in the S1/S2 cleavage site. In some embodiments, the Spike protein does not comprise the amino acid sequence RSVR (SEQ ID NO: 45) at positions corresponding to amino acids 748–751 of SEQ ID NO: 84. In some embodiments, the Spike protein does not comprise the amino acid sequence RXXR at positions corresponding to amino acids 748–751 of SEQ ID NO: 84. In some embodiments, the Spike protein comprises one or more mutations in the S2′ cleavage site. In some embodiments, the Spike protein does not comprise the amino acid sequence RSAR (SEQ ID NO: 46) at positions corresponding to amino acids 884–887 of SEQ ID NO: 84. In some embodiments, the Spike protein does not comprise the amino acid sequence RXXR at positions corresponding to amino acids 884–887 of SEQ ID NO: 84. In some embodiments, one or more arginine (R) residues at positions corresponding to amino acids 748, 751, 884, and 887 of SEQ ID NO: 84 are mutated in the Spike protein. In some embodiments, the Spike protein does not comprise a basic amino acid at 1, 2, 3, or 4 positions corresponding to amino acids 748, 751, 884, and 887 of SEQ ID NO: 84. In some embodiments, the Spike protein comprises a neutral amino acid (e.g., glycine, alanine, or serine) at 1, 2, 3, or 4 positions corresponding to amino acids 748, 751, 884, and 887 of SEQ ID NO: 84. In some embodiments, the Spike protein comprises glycine residues at positions corresponding to 748 and 751 of SEQ ID NO: 84. In some embodiments, the Spike protein comprises glycine residues at positions corresponding to 884 and 887 of SEQ ID NO: 84. In some embodiments, the Spike protein does not comprise an active S1/S2 cleavage site. In some embodiments, the Spike protein does not comprise an active S2′ cleavage site. Any suitable method may be used to determine whether an S1/S2 or S2′ cleavage site is active. See, e.g., Millet and Whittaker, Proc Natl Acad Sci U S A. 2014.111(42):15214–15219. In some embodiments, the Spike protein has an amino acid sequence with at least 90%, at least 95%, at least 96%, at least 97%, at least 98%, or at least 99% identity to the amino acid sequence of SEQ ID NO: 90, and does not comprise the amino acid sequence RSVR (SEQ ID NO: 45) at positions corresponding to amino acids 748–751 of SEQ ID NO: 84. In some embodiments, the Spike protein has an amino acid sequence with at least 90%, at least 95%, at least 96%, at least 97%, at least 98%, or at least 99% identity to the amino acid sequence of SEQ ID NO: 90, and does not comprise the amino acid sequence RSAR (SEQ ID NO: 46) at positions corresponding to amino acids 748–751 of SEQ ID NO: 84. In some embodiments, a Spike protein, e.g., an encoded Spike protein antigen, comprises a mutated, truncated, or deleted endoplasmic reticulum (ER) retention signal. ER retention signals, located at the C-terminus of betacoronavirus S proteins, allow retrieval of S proteins from the Golgi apparatus to the ER in retrograde, resulting in intracellular retention of the S protein. Without wishing to be bound by theory, it is expected that mutation of the ER retention signal increases expression of encoded Spike proteins on the cell surface, allowing increased exposure of the Spike protein and enhanced immunogenicity. ER retention signals include dibasic (KXHXX) amino acid sequences at the C-terminus of a protein (e.g., KXHXX-COOH). Thus, in some embodiments, a lysine (K) residue at a position corresponding to amino acid 1349, or a histidine (H) residue at a position corresponding to amino acid 1351, of SEQ ID NO: 84, is substituted or deleted in the Spike protein. In some embodiments, the Spike protein does not comprise a lysine residue at a position corresponding to amino acid 1349 of SEQ ID NO: 84. In some embodiments, the Spike protein does not comprise a histidine residue at a position corresponding to amino acid 1351 of SEQ ID NO: 84. In some embodiments, the Spike protein does not comprise the amino acid sequence KXH within the 20, 15, 10, or 5 C-terminal amino acids of the Spike protein. In some embodiments, the Spike protein does not comprise the amino acid sequence KXH. In some embodiments, the Spike protein does not comprise the amino acid sequence KXHXX. In some embodiments, the Spike protein has an amino acid sequence with at least 90%, at least 95%, at least 96%, at least 97%, at least 98%, or at least 99% identity to the amino acid sequence of SEQ ID NO: 91, and does not comprise the amino acid sequence KXHXX at its C-terminus. In some embodiments, a Spike protein, e.g., an encoded Spike protein antigen, has a truncated C-terminus. A Spike protein with a truncated C-terminus refers to a Spike protein that lacks one or more amino acids that are present at the C-terminus of a wild-type (naturally occurring) Spike protein. In some embodiments, the Spike protein comprises a truncated cytoplasmic tail. In some embodiments, the Spike protein has a truncated cytoplasmic tail relative to a wild-type MERS-CoV Spike protein. For example, a wild-type MERS-CoV Spike protein having the amino acid sequence of SEQ ID NO: 84 comprises a cytoplasmic tail that is 36 amino acids (amino acids 1318–1353 of SEQ ID NO: 84), and so a modified Spike protein with a truncated C-terminus relative to SEQ ID NO: 84 comprises (i) a cytoplasmic tail with fewer than 36 amino acids; or (ii) no cytoplasmic tail. In some embodiments, a Spike protein comprises a cytoplasmic tail that comprises no more than 35, no more than 30, no more than 25, no more than 20, no more than 15, no more than 10, no more than 9, no more than 8, no more than 7, no more than 6, no more than 5, no more than 4, no more than 3, no more than 2, or no more than 1 amino acid. In some embodiments, the cytoplasmic tail comprises no more than 21 amino acids. In some embodiments, a Spike protein comprises a cytoplasmic tail that is 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, or 35 amino acids in length. In some embodiments, a Spike protein comprises a cytoplasmic tail that is 1–5, 5–10, 10–15, 15–20, 20–25, 25–30, or 30–35 amino acids in length. In some embodiments, the Spike protein comprises a cytoplasmic tail comprising no more than 35 amino acids. In some embodiments, the Spike protein comprises a cytoplasmic tail comprising no more than 30 amino acids. In some embodiments, the Spike protein comprises a cytoplasmic tail comprising no more than 25 amino acids. In some embodiments, the Spike protein comprises a cytoplasmic tail comprising no more than 21 amino acids. In some embodiments, the Spike protein comprises a cytoplasmic tail comprising no more than 20 amino acids. In some embodiments, the Spike protein comprises a cytoplasmic tail comprising no more than 10 amino acids. In some embodiments, the Spike protein comprises a cytoplasmic tail comprising no more than 8 amino acids. In some embodiments, the Spike protein comprises a cytoplasmic tail comprising no more than 6 amino acids. In some embodiments, the Spike protein comprises a cytoplasmic tail comprising no more than 5 amino acids. In some embodiments, the Spike protein comprises a cytoplasmic tail comprising no more than 4 amino acids. In some embodiments, the Spike protein comprises a cytoplasmic tail comprising no more than 3 amino acids. In some embodiments, the Spike protein comprises a cytoplasmic tail comprising no more than 2 amino acids. In some embodiments, the Spike protein comprises a cytoplasmic tail comprising no more than 1 amino acid. Truncations may be introduced by deleting one or more amino acids from the C-terminus of the cytoplasmic tail (e.g., amino acids 1318–1353 or 1339–1353 of SEQ ID NO: 84). In some embodiments, the Spike protein as described herein, or encoded by an RNA as described herein, lacks one or more amino acids present at the C-terminus of a wild-type sequence. In some embodiments, the Spike protein lacks 1–35, 1–30, 1–25, 1–20, 1–15, 1–10, 1–5, 5–35, 5–30, 5– 25, 5–20, 5–15, 5–10, 10–35, 10–30, 10–25, 10–20, 10–15, 15–35, 15–30, 15–25, 15–20, 20–35, 20–30, 20–25, 25–35, 25–30, or 30–35 amino acids that are present at the C-terminus of a wild- type MERS-CoV S protein amino acid sequence. In some embodiments, a Spike protein as described herein, or encoded by an RNA as described herein, has a truncated cytoplasmic tail relative to SEQ ID NO: 84. In some embodiments, the Spike protein comprises an extracellular domain and a transmembrane domain, and has a truncated cytoplasmic tail relative to SEQ ID NO: 84. In some embodiments, the Spike protein has an amino acid sequence with at least 90%, at least 95%, at least 96%, at least 97%, at least 98%, or at least 99% identity to the amino acid sequence of SEQ ID NO: 92, and has a truncated cytoplasmic tail relative to SEQ ID NO: 84. In some embodiments, a Spike protein, e.g., an encoded Spike protein antigen, has the amino acid sequence of SEQ ID NO: 93. In some embodiments, the Spike protein has an amino acid sequence with at least 90%, at least 95%, at least 96%, at least 97%, at least 98%, or at least 99% identity to the amino acid sequence of SEQ ID NO: 93, comprises proline residues at positions corresponding to amino acids 1060 and 1061 of SEQ ID NO: 84, and does not comprise the amino acid sequence RSVR (SEQ ID NO: 45) at positions corresponding to 748– 751 of SEQ ID NO: 84. In some embodiments, the Spike protein is stabilized in a prefusion conformation. In some embodiments, the Spike protein does not comprise an active S1/S2 cleavage site. In some embodiments, a Spike protein, e.g., an encoded Spike protein antigen, has the amino acid sequence of SEQ ID NO: 94. In some embodiments, the Spike protein has an amino acid sequence with at least 90%, at least 95%, at least 96%, at least 97%, at least 98%, or at least 99% identity to the amino acid sequence of SEQ ID NO: 94, does not comprise the amino acid sequence RSVR (SEQ ID NO: 45) at positions corresponding to amino acids 748–751 of SEQ ID NO: 84, and does not comprise the amino acid sequence KXHXX at its C-terminus. In some embodiments, the Spike protein does not comprise an active S1/S2 cleavage site. In some embodiments, the Spike protein does not comprise an ER retention signal. In some embodiments, a Spike protein, e.g., an encoded Spike protein antigen, has the amino acid sequence of SEQ ID NO: 95. In some embodiments, the Spike protein has an amino acid sequence with at least 90%, at least 95%, at least 96%, at least 97%, at least 98%, or at least 99% identity to the amino acid sequence of SEQ ID NO: 95, does not comprise the amino acid sequence RSVR (SEQ ID NO: 45) at positions corresponding to amino acids 748–751 of SEQ ID NO: 84, and lacks one or more amino acids corresponding to amino acids 1339–1353 of SEQ ID NO: 84. In some embodiments, the Spike protein does not comprise an active S1/S2 cleavage site. In some embodiments, the Spike protein has a truncated cytoplasmic tail relative to SEQ ID NO: 84. In some embodiments, a Spike protein, e.g., an encoded Spike protein antigen, has no greater than 100, no greater than 90, no greater than 80, no greater than 70, no greater than 60, no greater than 50, no greater than 40, no greater than 30, no greater than 20, no greater than 10, or no greater than 5 amino acid substitutions and/or deletions as compared to (when aligned with) a Spike protein having the amino acid sequence as provided in any one of SEQ ID NOs: 93–95. Domains and Fusion Proteins Some aspects of the disclosure relate to MERS-CoV fusion proteins and RNAs (e.g., mRNAs or self-amplifying RNAs) encoding MERS-CoV fusion proteins, which comprise (i) an N-terminal domain (NTD) of a full-length MERS-CoV Spike protein, and/or (ii) a receptor- binding domain (RBD) of a full-length MERS-CoV Spike protein, and do not comprise an NTD or RBD of a different human coronavirus that is not MERS-CoV (i.e., the protein does not comprise an NTD, and does not comprise an RBD, of a Spike protein of any of the following coronaviruses: SARS-CoV, SARS-CoV-2, 229E-CoV, NL63-CoV, OC43-CoV, and HKU1-CoV). In some embodiments, the NTD and RBD are from the same full-length MERS-CoV Spike protein. In some embodiments, the NTD and RBD are from different full-length MERS-CoV Spike proteins. The S1 and S2 subunits of the MERS-CoV Spike protein further include domains readily discernable by structure and function, which in turn can be featured in designing antigens to be encoded by the nucleic acid vaccines, in particular, mRNA vaccines. Within the S1 subunit, domains include the N-terminal domain (NTD) and the receptor-binding domain (RBD), said RBD domain further including a receptor-binding motif (RBM) Within the S2 subunit, domains include fusion peptide (FP), heptad repeat 1 (HR1), heptad repeat 2 (HR2), transmembrane domain (TM), and cytoplasm domain, also known as cytoplasmic tail (CT). The HR1 and HR2 domains can be referred to as the “fusion core region” of MERS-CoV Spike protein. The S1 subunit includes an N terminal domain (NTD), a linker region, a receptor binding domain (RBD), a first subdomain (SD1), and a second subdomain (SD2). The S2 subunit includes, inter alia, a first heptad repeat (HR1), a second heptad repeat (HR2), a transmembrane domain (TM), and a cytoplasmic tail. The NTD and RBD of S1 have been shown to be the targets of neutralizing antibodies in betacoronavirus-infected individuals. As used herein, for example, in the context of an antigen design (said antigen encoded by an RNA and to be expressed, for example, from an RNA vaccine), the term “N-terminal domain” or “NTD” refers to a domain within the MERS-CoV S1 subunit comprising approximately 321 amino acids in length, having identity to amino acids 18–338 of the S1 subunit of the Spike protein having the amino acid sequence set forth as SEQ ID NO: 84. As used herein, for example, in the context of an antigen design (said antigen encoded by an mRNA and to be expressed, for example, from an mRNA vaccine), the term “receptor binding domain” or “RBD” refers to a domain within the S1 subunit of MERS-CoV comprising approximately 214 amino acids in length, having identity to amino acids 376–589 of the Spike protein having the amino acid sequence set forth as SEQ ID NO: 84. As used herein, the term “receptor binding motif” (RBM) refers to the portion of the RBD that directly contacts the DPP4 receptor. Expressed RBDs are predicted to specifically bind to dipeptidyl peptidase 4 (DPP4) as its receptor and/or specifically react with RBD-binding and/or neutralizing antibodies, e.g., KNIH90-F1. The compositions provided herein include RNAs (e.g., mRNAs) that may encode any one or more full-length or partial (truncated or other deletion of sequence) S protein subunit (e.g., S1 or S2 subunit), one or more domain or combination of domains of an S protein subunit (e.g., NTD, RBD, or NTD-RBD fusions, with or without an SD1 and/or SD2), or chimeras of full- length or partial and S2 protein subunits. Other S protein subunit and/or domain configurations are contemplated herein. Exemplary sequences of proteins comprising one or more domains of a MERS-CoV Spike protein (e.g., NTD-RBD fusion proteins) as described herein are provided in EXEMPLARY SEQUENCES as SEQ ID NOS: 96–98. In some embodiments, an RNA comprises an open reading frame encoding a protein having at least 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity to an amino acid sequence of SEQ ID NO: 96. In some embodiments, an RNA comprises an open reading frame encoding a protein having at least 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity to an amino acid sequence of SEQ ID NO: 97. In some embodiments, an RNA comprises an open reading frame encoding a protein having at least 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity to an amino acid sequence of SEQ ID NO: 98. Any pair of protein portions (e.g., NTD and RBD, NTD and TM domain, RBD and TM domain) may be contiguous in a protein as described herein (e.g., without an intervening amino acid sequence), or the two portions may be separated by a linker. The linker may be a 2A or GS linker described herein in the section entitled “Linkers and Cleavable Peptides.” The linker may also be another linker known in the art. A linker of a fusion protein may be present in place of an internal truncation relative to a wild-type sequence of a MERS-CoV Spike protein (e.g., the amino acid sequence of the protein comprises deletion of one or more amino acids, relative to the wild-type sequence, and insertion of the linker at the position previously occupied by the deleted amino acids). In some embodiments, the linker comprises 2–10 glycine residues. In some embodiments, the linker comprises 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, or 20 glycine residues. In some embodiments, the linker comprises 2–20, 2–15, 2–10, 2–5, 2–3, 3– 5, 5–7, 7–10, 10–15, 15–20, 3–10, 4–8, or 5–6 glycine residues. In some embodiments, the linker comprises 5–6 glycine residues. Where multiple linkers separate multiple pairs of portions, the multiple linkers may each comprise the same amino acid sequence (e.g., AAY). Multiple linkers may comprise different amino acid sequences (e.g., a first linker comprises the amino acid sequence GGS, and a second linker comprises the amino acid sequence GGS). Linkers connecting different pairs of portions may be the same length, or different lengths. In some embodiments, two or more portions of a protein portion are connected by a glycine linker or a glycine-serine linker. In some embodiments, each pair of protein portions of a protein are connected by a glycine linker or a glycine-serine linker. In some embodiments, two or more portions of a protein are connected by a linker comprising the amino acid sequence AAY. In some embodiments, each pair of protein portions are connected by the amino acid sequence AAY. In some embodiments, no linkers are present between protein portions in the protein. In some embodiments, a protein comprising one or more domains of a MERS-CoV Spike protein comprises a transmembrane (TM) domain. In some embodiments, the TM domain is a TM domain of a coronavirus. In some embodiments, the TM domain is a TM domain of a full- length MERS-CoV Spike protein. The full-length MERS-CoV Spike protein may the same Spike protein from which the NTD and/or RBD are derived, or a different full-length MERS-CoV Spike protein. An exemplary MERS-CoV Spike protein TM domain sequence is provided as SEQ ID NO: 101. In some embodiments, the TM domain is a TM domain is from a protein that is heterologous to MERS-CoV (i.e., a TM domain of a protein that is not encoded by a MERS-CoV genome). For example, the transmembrane domain may be from an influenza hemagglutinin transmembrane domain, which has been demonstrated to effectively anchor proteins at the cell surface. In some embodiments, the TM domain is an influenza virus hemagglutinin (HA) TM domain. The influenza virus HA protein may have a sequence from any influenza virus species (e.g., influenza A virus, influenza B virus, or influenza C virus) or HA subtype (e.g., H1, H2, H3, H4, H5, H6, H7, H8, H9, H10, H11, H12, H13, H14, H15, H16, H17, or H18). An exemplary influenza A virus HA TM domain sequence is provided as SEQ ID NO: 102. In some aspects, a protein as described herein or encoded by an RNA as described herein consists essentially of an NTD of a full-length MERS-CoV Spike protein, an RBD of a full- length MERS-CoV, and a TM domain. A protein consisting essentially of NTD-RBD-TM domains may include a linker between the NTD and RBD. A protein consisting essentially of NTD-RBD-TM domains may include a linker between the RBD and TM domain. Each pair of the NTD-RBD and RBD-TM domains may be connected by a linker. The NTD may include a signal peptide of the full-length MERS-CoV Spike protein. In some embodiments, the protein includes a signal peptide from a protein other than the MERS-CoV Spike protein. In some embodiments, the protein comprises the amino acid sequence of SEQ ID NO: 96. In some embodiments, a protein as described herein or encoded by an RNA as described herein comprises an NTD of a full-length MERS-CoV Spike protein, an RBD of a full-length MERS-CoV, and a TM domain. The NTD and RBD may be connected by a linker. The RBD and TM domain may be connected by a linker. In some embodiments, the protein comprises the amino acid sequence of SEQ ID NO: 96. In some embodiments, the protein has no greater than 100, no greater than 90, no greater than 80, no greater than 70, no greater than 60, no greater than 50, no greater than 40, no greater than 30, no greater than 20, no greater than 10, or no greater than 5 amino acid substitutions and/or deletions as compared to (when aligned with) a protein having the amino acid sequence of SEQ ID NO: 96. In some embodiments, the fusion protein has an amino acid sequence with at least 90%, at least 95%, at least 96%, at least 97%, at least 98%, or at least 99% identity to the amino acid sequence of SEQ ID NO: 96, and comprises an NTD and RBD of a full-length MERS-CoV Spike protein and a TM domain. In some embodiments, the full-length MERS-CoV Spike protein comprises the amino acid sequence of SEQ ID NO: 84. In some embodiments, the full-length MERS-CoV Spike protein comprises the amino acid sequence of SEQ ID NO: 85. In some embodiments, the full-length MERS-CoV Spike protein comprises the amino acid sequence of SEQ ID NO: 86. In some aspects, a protein as described herein or encoded by an RNA as described herein consists essentially of an NTD of a full-length MERS-CoV Spike protein, a TM domain. A protein consisting essentially of NTD and TM domains may include a between the NTD and TM domain. In some embodiments, the protein comprises the amino acid sequence of SEQ ID NO: 97. The NTD may include a signal peptide of the full-length MERS-CoV Spike protein. In some embodiments, the protein includes a signal peptide from a protein other than the MERS-CoV Spike protein. In some embodiments, a protein as described herein or encoded by an RNA as described herein comprises an NTD of a full-length MERS-CoV Spike protein and a TM domain. The NTD and TM domain may be connected by a linker. In some embodiments, the protein comprises the amino acid sequence of SEQ ID NO: 97. In some embodiments, the protein has no greater than 100, no greater than 90, no greater than 80, no greater than 70, no greater than 60, no greater than 50, no greater than 40, no greater than 30, no greater than 20, no greater than 10, or no greater than 5 amino acid substitutions and/or deletions as compared to (when aligned with) a protein having the amino acid sequence of SEQ ID NO: 97. In some embodiments, the fusion protein has an amino acid sequence with at least 90%, at least 95%, at least 96%, at least 97%, at least 98%, or at least 99% identity to the amino acid sequence of SEQ ID NO: 97, and comprises an NTD of a full-length MERS-CoV Spike protein and a TM domain. In some embodiments, the full-length MERS-CoV Spike protein comprises the amino acid sequence of SEQ ID NO: 84. In some embodiments, the full-length MERS-CoV Spike protein comprises the amino acid sequence of SEQ ID NO: 85. In some embodiments, the full-length MERS-CoV Spike protein comprises the amino acid sequence of SEQ ID NO: 86. In some aspects, a protein as described herein or encoded by an RNA as described herein consists essentially of an RBD of a full-length MERS-CoV Spike protein and a TM domain. A protein consisting essentially of RBD and TM domains may include a linker between the RBD and TM domain. In some embodiments, the protein includes a signal peptide. The signal peptide may be from a MERS-CoV Spike protein. In some embodiments, the protein includes a signal peptide from a protein other than the MERS-CoV Spike protein. In some embodiments, the protein comprises the amino acid sequence of SEQ ID NO: 98. In some embodiments, a protein as described herein or encoded by an RNA as described herein comprises an RBD of a full-length MERS-CoV Spike protein and a TM domain. The RBD and TM domain may be connected by a linker. In some embodiments, the protein comprises the amino acid sequence of SEQ ID NO: 98. In some embodiments, the protein has no greater than 100, no greater than 90, no greater than 80, no greater than 70, no greater than 60, no greater than 50, no greater than 40, no greater than 30, no greater than 20, no greater than 10, or no greater than 5 amino acid substitutions and/or deletions as compared to (when aligned with) a protein having the amino acid sequence of SEQ ID NO: 98. In some embodiments, the fusion protein has an amino acid sequence with at least 90%, at least 95%, at least 96%, at least 97%, at least 98%, or at least 99% identity to the amino acid sequence of SEQ ID NO: 98 and comprises an RBD of a full-length MERS-CoV Spike protein and a TM domain. In some embodiments, the full-length MERS-CoV Spike protein comprises the amino acid sequence of SEQ ID NO: 84. In some embodiments, the full-length MERS-CoV Spike protein comprises the amino acid sequence of SEQ ID NO: 85. In some embodiments, the full-length MERS-CoV Spike protein comprises the amino acid sequence of SEQ ID NO: 86. Variants In some embodiments, the compositions of the present disclosure include RNA that encodes a MERS-CoV antigen variant. Antigen variants or other polypeptide variants refers to molecules that differ in their amino acid sequence from a wild-type, native, or naturally occurring sequence. The antigen/polypeptide variants may possess substitutions, deletions, and/or insertions at certain positions within the amino acid sequence, as compared to a wild-type, native or naturally occurring sequence. Examples of MERS-CoV protein variants are provided in the Sequence Listing. Ordinarily, variants possess at least 50% identity to a wild-type, native or naturally occurring sequence. In some embodiments, variants share at least 80%, or at least 90% identity with a wild-type, native, or naturally occurring sequence. In some embodiments, the nucleic acid vaccines described herein encode MERS-CoV variants comprising 1, 2, 3, 4, or more mutations relative to a naturally occurring sequence. In some embodiments, the nucleic acid vaccines described herein encode MERS-CoV variants comprising less than 20, 18, 15, 12, or 10 mutations relative to a naturally occurring sequence. In some embodiments, the nucleic acid vaccines described herein encode MERS-CoV variants having 1-501-40, 1-30, 1-25, 1-20, 1-15, 1-10, 5-50, 5-40, 5-30, 5-25, 5-20, 5-15, 5-10, 10-50, 10-40, 10-30, 10-25, 10-20, 10-15, 20-50, 20-40, 20-30, 20-25, 25-50, 25-40, 25-30, 30-50, 30-40, 40-50 mutations (e.g., substitutions). As used herein, “mutation” refers to an amino acid substitution, insertion, or deletion, relative to a comparator sequence. Variant antigens/polypeptides encoded by nucleic acids of the disclosure may contain amino acid changes that confer any of a number of desirable properties, e.g., that enhance their immunogenicity, enhance their expression, and/or improve their stability or PK/PD properties in a subject. Variant antigens/polypeptides can be made using routine mutagenesis techniques and assayed as appropriate to determine whether they possess the desired property. Assays to determine expression levels and immunogenicity are well known in the art and exemplary such assays are set forth in the Examples section. Similarly, PK/PD properties of a protein variant can be measured using art recognized techniques, e.g., by determining expression of antigens in a vaccinated subject over time and/or by looking at the durability of the induced immune response. The stability of protein(s) encoded by a variant nucleic acid may be measured by assaying thermal stability or stability upon urea denaturation or may be measured using in silico prediction. Methods for such experiments and in silico determinations are known in the art. In some embodiments, a composition comprises an RNA or an RNA ORF that comprises a nucleotide sequence of any one of the sequences provided herein, or comprises a nucleotide sequence at least 90%, at least 95%, at least 96%, at least 97%, at least 98%, or at least 99% identical to a nucleotide sequence of any one of the sequences provided herein. The term “identity” refers to a relationship between the sequences of two or more polypeptides (e.g. antigens) or polynucleotides (nucleic acids), as determined by comparing the sequences. Identity also refers to the degree of sequence relatedness between or among sequences as determined by the number of matches between strings of two or more amino acid residues or nucleic acid residues. Identity measures the percent of identical matches between the smaller of two or more sequences with gap alignments (if any) addressed by a particular mathematical model or computer program (e.g., “algorithms”). Identity of related antigens or nucleic acids can be readily calculated by known methods. “Percent (%) identity” as it applies to polypeptide or polynucleotide sequences is defined as the percentage of residues (amino acid residues or nucleic acid residues) in the candidate amino acid or nucleic acid sequence that are identical with the residues in the amino acid sequence or nucleic acid sequence of a second sequence after aligning the sequences and introducing gaps, if necessary, to achieve the maximum percent identity. Methods and computer programs for the alignment are well known in the art. It is understood that identity depends on a calculation of percent identity but may differ in value due to gaps and penalties introduced in the calculation. Generally, variants of a particular polynucleotide or polypeptide (e.g., antigen) have at least 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, 99% but less than 100% sequence identity to that particular comparator polynucleotide or polypeptide as determined by sequence alignment programs and parameters described herein and known to those skilled in the art. Such tools for alignment include those of the BLAST suite (Stephen F. Altschul, et al (1997), "Gapped BLAST and PSI-BLAST: a new generation of protein database search programs", Nucleic Acids Res.25:3389-3402). Another popular local alignment technique is based on the Smith-Waterman algorithm (Smith, T.F. & Waterman, M.S. (1981) “Identification of common molecular subsequences.” J. Mol. Biol. 147:195-197). A general global alignment technique based on dynamic programming is the Needleman–Wunsch algorithm (Needleman, S.B. & Wunsch, C.D. (1970) “A general method applicable to the search for similarities in the amino acid sequences of two proteins.” J. Mol. Biol.48:443-453). More recently a Fast Optimal Global Sequence Alignment Algorithm (FOGSAA) has been developed that purportedly produces global alignment of nucleotide and protein sequences faster than other optimal global alignment methods, including the Needleman– Wunsch algorithm. As such, polynucleotides encoding peptides or polypeptides containing substitutions, insertions and/or additions, deletions and covalent modifications with respect to reference sequences, in particular the polypeptide (e.g., antigen) sequences disclosed herein, are included within the scope of this disclosure. For example, sequence tags or amino acids, such as one or more lysines, can be added to peptide sequences (e.g., at the N-terminal or C-terminal ends). Sequence tags can be used for peptide detection, purification or localization. Lysines can be used to increase peptide solubility or to allow for biotinylation. Alternatively, amino acid residues located at the carboxy and amino terminal regions of the amino acid sequence of a peptide or protein may optionally be deleted providing for truncated sequences. Certain amino acids (e.g., C-terminal or N-terminal residues) may alternatively be deleted depending on the use of the sequence, as for example, expression of the sequence as part of a larger sequence which is soluble or linked to a solid support. In some embodiments, sequences for (or encoding) signal sequences, termination sequences, transmembrane domains, linkers, multimerization domains (such as, e.g., foldon regions) and the like may be substituted with alternative sequences that achieve the same or a similar function. In some embodiments, cavities in the core of proteins can be filled to improve stability, e.g., by introducing larger amino acids. In other embodiments, buried hydrogen bond networks may be replaced with hydrophobic resides to improve stability. In yet other embodiments, glycosylation sites may be removed and replaced with appropriate residues. Such sequences are readily identifiable to one of skill in the art. It should also be understood that some of the sequences provided herein contain sequence tags or terminal peptide sequences (e.g., at the N-terminal or C-terminal ends) that may be deleted, for example, prior to use in the preparation of an mRNA vaccine. As recognized by those skilled in the art, protein fragments, functional protein domains, and homologous proteins are also considered to be within the scope of coronavirus antigens of interest. For example, provided herein is any protein fragment (meaning a polypeptide sequence at least one amino acid residue shorter than a full-length antigen sequence but otherwise identical) of a full-length protein, provided that the fragment is immunogenic and confers a protective immune response to the coronavirus. In addition to variants that are identical to the full-length protein but are truncated, in some embodiments, an antigen includes 2, 3, 4, 5, 6, 7, 8, 9, 10, or more mutations, as shown in any of the sequences provided or referenced herein. Antigens/antigenic polypeptides can range in length from about 4, 6, or 8 amino acids to full length proteins. Linkers and Cleavable Peptides In some embodiments, an RNA (e.g., mRNA) that encodes a protein further encodes a linker located between at least two, or between each domain of the protein. The linker may be, for example, a cleavable linker or protease-sensitive linker. In some embodiments, the linker is selected from the group consisting of F2A linker, P2A linker, T2A linker, E2A linker, and combinations thereof (see, e.g., WO 2017/127750). This family of self-cleaving peptide linkers, referred to as 2A peptides, has been described in the art (see, e.g., Kim, J.H. et al. PLoS ONE 2011;6:e18556). In some embodiments, the linker is an F2A linker. In some embodiments, the linker is a GS linker. GS linkers are polypeptide linkers that include glycine and serine amino acids repeats. They comprise flexible and hydrophilic residues and can be used to perform fusion of protein subunits without interfering in the folding and function of the protein domains, and without formation of secondary structures. In some embodiments, an RNA (e.g., mRNA) encodes a protein that comprises a GS linker that is 3 to 20 amino acids long. For example, the GS linker may have a length of (or have a length of at least) 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, or 20 amino acids. In some embodiments, a GS linker is (or is at least) 15 amino acids long (e.g., GGSGGSGGSGGSGGG (SEQ ID NO: 47)). In some embodiments, a GS linker is (or is at least) 8 amino acids long (e.g., GGGSGGGS (SEQ ID NO: 48)). In some embodiments, a GS linker is (or is at least) 7 amino acids long (e.g., GGGSGGG (SEQ ID NO: 49)). In some embodiments, a GS linker comprises the amino acid sequence GGGSGG (SEQ ID NO: 50). In some embodiments, a GS linker is (or is at least) 4 amino acid long (e.g., GGGS (SEQ ID NO: 51)). In some embodiments, the GS linker comprises (GGGS)n (SEQ ID NO: 51), where n is any integer from 1-5. In some embodiments, a GS linker is (or is at least) 4 amino acid long (e.g., GSGG (SEQ ID NO: 52)). In some embodiments, the GS linker comprises (GSGG)n (SEQ ID NO: 52), where n is any integer from 1-5. In some embodiments, a GS linker comprises the amino acid sequence SGGGS (SEQ ID NO: 53). In some embodiments, a linker is a glycine linker, for example having a length of (or a length of at least) 3 amino acids (e.g., GGG). In some embodiments, a protein encoded by an RNA (e.g., mRNA) includes two or more linkers, which may be the same or different from each other. In some embodiments, a linker comprises a pan-HLA-DR-binding epitope (PADRE). See, e.g., Alexander et al., J Immunol.2000.164(3):1625–1633. The skilled artisan will appreciate that other art-recognized linkers may be suitable for use in the constructs of the disclosure (e.g., encoded by the nucleic acids of the disclosure). The skilled artisan will likewise appreciate that other polycistronic constructs (RNA (e.g., mRNA) encoding more than one protein separately within the same molecule) may be suitable for use as provided herein. Signal Peptides In some embodiments, an RNA (e.g., mRNA) has an ORF that encodes a signal peptide fused to a protein. Signal peptides, comprising the N-terminal 15-60 amino acids of proteins, are typically involved for the translocation across the membrane on the secretory pathway and, thus, control the entry of proteins both in eukaryotes and prokaryotes to the secretory pathway. In eukaryotes, the signal peptide of a nascent precursor protein (pre-protein) directs the ribosome to the rough endoplasmic reticulum (ER) membrane and initiates the transport of the growing peptide chain across it for processing. ER processing produces mature proteins, wherein the signal peptide is cleaved from precursor proteins, typically by an ER-resident signal peptidase of the host cell, or they remain uncleaved and function as a membrane anchor. A signal peptide may also facilitate the targeting of the protein to the cell membrane. A signal peptide may have a length of 15-60 amino acids. For example, a signal peptide may have a length of 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49, 50, 51, 52, 53, 54, 55, 56, 57, 58, 59, or 60 amino acids. In some embodiments, a signal peptide has a length of 20-60, 25-60, 30-60, 35- 60, 40-60, 45- 60, 50-60, 55-60, 15-55, 20-55, 25-55, 30-55, 35-55, 40-55, 45-55, 50-55, 15-50, 20-50, 25-50, 30-50, 35-50, 40-50, 45-50, 15-45, 20-45, 25-45, 30-45, 35-45, 40-45, 15-40, 20- 40, 25-40, 30-40, 35-40, 15-35, 20-35, 25-35, 30-35, 15-30, 20-30, 25-30, 15-25, 20-25, or 15-20 amino acids. Signal peptides from heterologous genes (which regulate expression of genes other than MERS-CoV proteins in nature) are known in the art and can be tested for desired properties and then incorporated into a nucleic acid of the disclosure. In some embodiments, an RNA (e.g., mRNA) comprises an open reading frame that encodes a protein fused to a signal peptide comprising an amino acid sequence that has at least 50%, at least 60%, at least 70%, at least 75%, at least 80%, at least 85%, at least 90%, or at least 95% identity to the amino acid sequence of any one of the sequences provided herein, such as those reproduced below in Table 3. In some embodiments, an RNA comprises an open reading frame that encodes MERS-CoV protein including an endogenous signal peptide of the wild-type protein (e.g., an mRNA encoding a (wild-type or modified) MERS-CoV protein or variant thereof encodes a MERS-CoV S protein signal peptide). Nucleic Acids Encoding MERS-CoV Proteins Nucleic acids comprise a polymer of nucleotides (nucleotide monomers). Thus, nucleic acids are also referred to as polynucleotides. Nucleic acids may be or may include, for example, deoxyribonucleic acid (DNA), ribonucleic acid (RNA), threose nucleic acid (TNA), glycol nucleic acid (GNA), peptide nucleic acid (PNA), locked nucleic acid (LNA, including LNA having a β-D-ribo configuration, α-LNA having an α-L-ribo configuration (a diastereomer of LNA), 2′-amino-LNA having a 2′-amino functionalization, and 2′-amino- α-LNA having a 2′- amino functionalization), ethylene nucleic acid (ENA), cyclohexenyl nucleic acid (CeNA) and/or chimeras and/or combinations thereof. RNA (e.g., mRNA) of the present disclosure comprises an open reading frame (ORF) encoding a MERS-CoV protein or variant thereof. In some embodiments, the RNA (e.g., mRNA) further comprises a 5 ^ untranslated region (UTR), 3 ^ UTR, a poly(A) tail and/or a 5 ^ cap analog. Messenger RNA (mRNA) Messenger RNA (mRNA) is RNA that encodes a (at least one) protein (a naturally- occurring, non-naturally-occurring, or modified polymer of amino acids) and can be translated to produce the encoded protein in vitro, in vivo, in situ, or ex vivo. It is understood that mRNA is not self-amplifying RNA (saRNA) (see, e.g., Bloom K et al. Gene Therapy 2021; 28: 117–129 for a comparison of mRNA and saRNA). saRNAs include alphavirus replicase sequences that encode an RNA-dependent RNA polymerase. mRNA does not include alphavirus replicase sequences. The skilled artisan will appreciate that, except where otherwise noted, nucleic acid sequences set forth in the instant application may recite “T”s in a representative DNA sequence but where the sequence represents mRNA, the “T”s would be substituted for “U”s. Thus, any of the DNAs disclosed and identified by a particular sequence identification number herein also disclose the corresponding mRNA sequence complementary to the DNA, where each “T” of the DNA sequence is substituted with “U.” Naturally-occurring eukaryotic mRNA molecules can contain stabilizing elements, including, but not limited to, UTRs at their 5′-end (5′ UTR) and/or at their 3′-end (3′ UTR), in addition to other structural features, such as a 5′-cap structure or a 3′-poly(A) tail. Both the 5′ UTR and the 3′ UTR are typically transcribed from the genomic DNA and are elements of the premature mRNA. Characteristic structural features of mature mRNA, such as the 5′-cap and the 3′-poly(A) tail are usually added to the transcribed (premature) mRNA during mRNA processing. Untranslated Regions (UTRs) The mRNAs of the present disclosure may comprise one or more regions or parts which act or function as an untranslated region. A “5′ untranslated region” (UTR) refers to a region of an mRNA that is directly upstream (i.e., 5′) from the start codon (i.e., the first codon of an mRNA transcript translated by a ribosome) that does not encode a polypeptide. A “3′ untranslated region” (UTR) refers to a region of an mRNA that is directly downstream (i.e., 3′) from the open reading frame (e.g., downstream from the last amino acid-encoding codon of an open reading frame, where the stop codon is considered part of the 3′ UTR, or downstream from the first stop codon signaling translation termination, where that stop codon is considered part of the open reading frame), and which does not encode a polypeptide. When RNA transcripts are being generated, the 5’ UTR may comprise a promoter sequence. Such promoter sequences are known in the art. It should be understood that such promoter sequences will not be present in a vaccine of the disclosure. Where mRNAs encode a (at least one) protein, the mRNA may comprise a 5’ UTR and/or 3’ UTR. UTRs of an mRNA are transcribed but not translated. In mRNA, the 5′ UTR starts at the transcription start site and continues to the start codon but does not include the start codon; the 3′ UTR starts immediately following the open reading frame and continues until the transcriptional termination signal. Where an open reading frame ends with a codon encoding an amino acid, the 3′ UTR begins with a stop codon, such that no amino acids are added to a polypeptide beyond the last amino acid encoded by the open reading frame. A 3′ UTR may further comprise one or more stop codons. There is a growing body of evidence about the regulatory roles played by the UTRs in terms of stability of the nucleic acid molecule and translation. The regulatory features of a UTR can be incorporated into the polynucleotides of the present disclosure to, among other things, enhance the stability of the molecule. The specific features can also be incorporated to ensure controlled down-regulation of the transcript in case they are misdirected to undesired organs sites. A variety of 5’ UTR and 3’ UTR sequences are known. In some embodiments, the 5′ UTR comprises a sequence provided in Table 1 or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to a 5′ UTR sequence provided in Table 1, or a variant or a fragment thereof. In some embodiments, the 3′ UTR comprises a sequence provided in Table 2 or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to a 3′ UTR sequence provided in Table 2, or a variant or a fragment thereof. It should also be understood that the mRNA of the present disclosure may include any 5’ UTR and/or any 3’ UTR. Exemplary UTR sequences include SEQ ID NOs: 1-44, 66-79 and 81- 82; however, other UTR sequences may be used or exchanged for any of the UTR sequences described herein. In some embodiments, a 5' UTR comprises a sequence selected from: GGGAAAUAAGAGAGAAAAGAAGAGUAAGAAGAAAUAUAAGAGCCACC (SEQ ID NO: 1), GGGAAAUAAGAGAGAAAAGAAGAGUAAGAAGAAAUAUAAGACCCCGGCGCCGCCACC (SEQ ID NO: 2), GAGGAAAUCGCAAAAUUUGCUCUUCGCGUUAGAUUUCUUUUAGUUUUCUCGCAACUAGCAAGCUUUUUGUUCUCGCC (SEQ ID NO: 66), and GGAAAUCGCAAAAUUUGCUCUUCGCGUUAGAUUUCUUUUAGUUUUCUCGCAACUAGCAAGCUUUUUGUUCUCGCC (SEQ ID NO: 67). In some embodiments, a 3′ UTR comprises, in 5′-to-3′ order: (a) the nucleic acid sequence UAAAGCUCCCCGGGGGCCUCGGUGGCCUAGCUUCUUGCCCCUUGGGCCUCCCCCCAGCCCCUCCUCCCCUUCCUGCAG (SEQ ID NO: 68), (b) an identification and ratio determination (IDR) sequence, and (c) the nucleic acid sequence UGGUCUUUGAAUAAAGUCUGAGUGGGCGGC (SEQ ID NO: 69). In some embodiments, each mRNA encoding a distinct protein (i.e., having a different amino acid sequence from proteins encoded by other mRNAs in a composition) comprises a 3′ UTR comprising, in 5′-to-3′ order: (a) the nucleotide sequence of SEQ ID NO: 68; (b) a distinct IDR sequence; and (c) the nucleotide sequence of SEQ ID NO: 69. IDR sequences are described herein in the section entitled “Identification and Ratio Determination (IDR) Sequences.” In some embodiments, a 5′ UTR comprises a sequence derived from a 5′ UTR of a gene selected from HSD17B4, RPL32, ASAH1, ATP5A1, MP68, NDUFA4, NOSIP, RPL31, SLC7A3, TUBB4B and UBQLN2. In some embodiments, the 5′ UTR comprises a sequence derived from the 5′ UTR of human hydroxysteroid 17-beta dehydrogenase 4 (HSD17B4). In some embodiments, a 5′ UTR comprises the sequence GGGAGAGUCCCGCAGUCGGCGUCCAGCGGCUCUGCUUGUUCGUGUGUGUGUCGUUGCAGGCCUUAUUCAAGCUUACC (SEQ ID NO: 70). In some embodiments, a 5′ UTR comprises the sequenceGUCCCGCAGUCGGCGUCCAGCGGCUCUGCUUGUUCGUGUGUGUGUCGUUGCAGGCCUUAUUC (SEQ ID NO: 71). In some embodiments, a 5′ UTR comprises the sequence GGGAGAAAGCUUACC (SEQ ID NO: 72). In some embodiments, a 3′ UTR comprises a sequence derived from a 3′ UTR of a gene selected from PSMB3, ALB7, alpha-globin, CASP1, COX6B1, GNAS, NDUFA1 and RPS9. In some embodiments, a 3′ UTR comprises a sequence derived from a 3′ UTR of PSMB3 (proteasome 20S subunit beta 3). In some embodiments, a 3′ UTR comprises a sequence derived from a 3′ UTR of alpha-globin (MUAG). In some embodiments, a 3′ UTR comprises the sequence AGGACUAGUCCCUGUUCCCAGAGCCCACUUUUUUUUCUUUUUUUGAAAUAAAAUAGCCUGUCUUUCAGAUCU (SEQ ID NO: 73). In some embodiments, a 3′ UTR comprises the sequenceGGACUAGUUAUAAGACUGACUAGCCCGAUGGGCCUCCCAACGGGCCCUCCUCCCCUCCUUGCACCGAGAUUAAU (SEQ ID NO: 74). In some embodiments, the mRNA comprises a 5′ UTR comprising the nucleotide sequence of any one of SEQ ID NOs: 70–72, an open reading frame, one or more stop codons, and a 3′ UTR comprising the nucleotide sequence of SEQ ID NO: 73 or SEQ ID NO: 74. In some embodiments, the mRNA further comprises a polyA sequence comprising at least 64 consecutive adenosine nucleotides. In some embodiments, the mRNA further comprises a polyC sequence comprising at least 30 consecutive cytidine nucleotides. In some embodiments, a 5′ UTR comprises the sequence GAAUAAACUAGUAUUCUUCUGGUCCCCACAGACUCAGAGAGAACCCGCCACC (SEQ ID NO: 75). In some embodiments, a 5′ UTR comprises the sequence GAGAAUAAACUAGUAUUCUUCUGGUCCCCACAGACUCAGAGAGAACCCGCCACC (SEQ ID NO: 76). In some embodiments, a 3′ UTR comprises the sequence CUGGUACUGCAUGCACGCAAUGCUAGCUGCCCCUUUCCCGUCCUGGGUACCCCGAGUCUCCCCCGACCUCGGGUCCCA GGUAUGCUCCCACCUCCACCUGCCCCACUCACCACCUCUGCUAGUUCCAGACACCUCCCAAGCACGCAGCAAUGCAGC UCAAAACGCUUAGCCUAGCCACACCCCCACGGGAAACAGCAGUGAUUAACCUUUAGCAAUAAACGAAAGUUUAACUAA GCUAUACUAACCCCAGGGUUGGUCAAUUUCGUGCCAGCCACACC (SEQ ID NO: 77). In some embodiments, a 3′ UTR comprises the sequence CUCGAGCUGGUACUGCAUGCACGCAAUGCUAGCUGCCCCUUUCCCGUCCUGGGUACCCCGAGUCUCCCCCGACCUCGG GUCCCAGGUAUGCUCCCACCUCCACCUGCCCCACUCACCACCUCUGCUAGUUCCAGACACCUCCCAAGCACGCAGCAA UGCAGCUCAAAACGCUUAGCCUAGCCACACCCCCACGGGAAACAGCAGUGAUUAACCUUUAGCAAUAAACGAAAGUUUAACUAAGCUAUACUAACCCCAGGGUUGGUCAAUUUCGUGCCAGCCACACCCUGGAGCUAGC (SEQ ID NO: 78). In some embodiments, a 3′ UTR comprises the sequence CUGGUACUGCAUGCACGCAAUGCUAGCUGCCCCUUUCCCGUCCUGGGUACCCCGAGUCUCCCCCGACCUCGGGUCCCA GGUAUGCUCCCACCUCCACCUGCCCCACUCACCACCUCUGCUAGUUCCAGACACCUCCCAAGCACGCAGCAAUGCAGC UCAAAACGCUUAGCCUAGCCACACCCCCACGGGAAACAGCAGUGAUUAACCUUUAGCAAUAAACGAAAGUUUAACUAA GCUAUACUAACCCCAGGGUUGGUCAAUUUCGUGCCAGCCACACCCUGGAGCUAGC (SEQ ID NO: 79). In some embodiments, an mRNA comprises a 5′ UTR comprising the nucleotide sequence of SEQ ID NO: 75 or SEQ ID NO: 76, an open reading frame, one or more stop codons, and a 3′ UTR comprising the nucleotide sequence of any one of SEQ ID NOs: 77–79. In some embodiments, an mRNA comprises a 5′ UTR comprising the nucleotide sequence of SEQ ID NO: 76, an open reading frame, one or more stop codons, and a 3′ UTR comprising the nucleotide sequence of SEQ ID NO: 78. In some embodiments, an mRNA comprises a 5′ UTR comprising the nucleotide sequence of SEQ ID NO: 76, an open reading frame, the nucleotide sequence UGAUGA, and a 3′ UTR comprising the nucleotide sequence of SEQ ID NO: 78. In some embodiments, the mRNA further comprises two poly(A) sequences separated by an intervening nucleotide sequence. In some embodiments, the mRNA further comprises the nucleotide sequence of SEQ ID NO: 80. In some embodiments, a 5′ UTR comprises the sequence GAGGAGACCCAAGCUACAUUUGCUUCUGACACAACUGUGUUCACUAGCAACCUCAAACAGACACCGCCACC (SEQ ID NO: 81). In some embodiments, a 3′ UTR comprises the sequence GCUCGCUUUCUUGCUGUCCAAUUUCUAUUAAAGGUUCCUUUGUUCCCUAAGUCCAACUACUAAACUGGGGGAUAUUAU GAAGGGCCUUGAGCAUCUGGAUUCUGCCUAAUAAAAAACAUUUAUUUUCAUUGC (SEQ ID NO: 82). In some embodiments, an mRNA comprises a 5′ UTR comprising the nucleotide sequence of SEQ ID NO: 81, an open reading frame, one or more stop codons, and a 3′ UTR comprising the nucleotide sequence of SEQ ID NO: 82. In some embodiments, the mRNA further comprises a polyA tail comprising 109 consecutive adenosine nucleotides. UTRs may also be omitted from the mRNA described herein. A 5 ^ UTR does not encode a protein (is non-coding). Natural 5′ UTRs have features that play roles in translation initiation. They harbor signatures like Kozak sequences which are commonly known to be involved in the process by which the ribosome initiates translation of many genes. Kozak sequences have the consensus CCRCCAUGG, where R is a purine (adenine or guanine) three bases upstream of the start codon (AUG), which is followed by another 'G'.5′UTR also have been known to form secondary structures which are involved in elongation factor binding. In some embodiments of the disclosure, a 5’ UTR is a heterologous UTR, i.e., is a UTR found in nature associated with a different ORF. In other embodiments, a 5’ UTR is a synthetic UTR, i.e., does not occur in nature. Synthetic UTRs include UTRs that have been mutated to improve their properties, e.g., which increase gene expression as well as those which are completely synthetic. Exemplary 5’ UTRs include Xenopus or human derived a-globin or b- globin (8278063; 9012219), human cytochrome b-245 a polypeptide, and hydroxysteroid (17b) dehydrogenase, and Tobacco etch virus (US8278063, US9012219). CMV immediate-early 1 (IE1) gene (US2014/0206753, WO2013/185069), the sequence GGGAUCCUACC (SEQ ID NO: 54) (WO 2014/144196) may also be used. In other embodiments, a 5' UTR is a 5' UTR of a TOP gene lacking the 5' TOP motif (the oligopyrimidine tract) (e.g., WO2015/101414, WO2015/101415, WO2015/062738, WO2015/024667, WO2015/024667); 5' UTR element derived from ribosomal protein Large 32 (L32) gene (WO/2015101414, WO2015101415, WO/2015/062738), 5' UTR element derived from the 5' UTR of an hydroxysteroid (17-β) dehydrogenase 4 gene (HSD17B4) (WO201/5024667), or a 5' UTR element derived from the 5' UTR of ATP5A1 (WO2015/024667) can be used. In some embodiments, an internal ribosome entry site (IRES) is used instead of a 5' UTR. A 3 ^ UTR does not encode a protein (is non-coding). Natural or wild type 3′ UTRs are known to have stretches of adenosines and uridines embedded in them. These AU rich signatures are particularly prevalent in genes with high rates of turnover. Based on their sequence features and functional properties, the AU rich elements (AREs) can be separated into three classes (Chen et al, 1995): Class I AREs contain several dispersed copies of an AUUUA motif within U-rich regions. C-Myc and MyoD contain class I AREs. Class II AREs possess two or more overlapping UUAUUUA(U/A)(U/A) nonamers. Molecules containing this type of AREs include GM-CSF and TNF-a. Class III ARES are less well defined. These U rich regions do not contain an AUUUA motif. c-Jun and Myogenin are two well-studied examples of this class. Most proteins binding to the AREs are known to destabilize the messenger, whereas members of the ELAV family, most notably HuR, have been documented to increase the stability of mRNA. HuR binds to AREs of all the three classes. Engineering the HuR specific binding sites into the 3′ UTR of nucleic acid molecules will lead to HuR binding and thus, stabilization of the message in vivo. Introduction, removal or modification of 3′ UTR AU rich elements (AREs) can be used to modulate the stability of mRNA of the disclosure. When engineering specific nucleic acids, one or more copies of an ARE can be introduced to make nucleic acids of the disclosure less stable and thereby curtail translation and decrease production of the resultant protein. Likewise, AREs can be identified and removed or mutated to increase the intracellular stability and thus increase translation and production of the resultant protein. Transfection experiments can be conducted in relevant cell lines, using nucleic acids of the disclosure and protein production can be assayed at various time points post-transfection. For example, cells can be transfected with different ARE- engineering molecules and by using an ELISA kit to the relevant protein and assaying protein produced at 6 hours, 12 hours, 1 day, 2 days, and 7 days post-transfection. Those of ordinary skill in the art will understand that 5’ UTRs that are heterologous or synthetic may be used with any desired 3’ UTR sequence. For example, a heterologous or synthetic 5’ UTR may be used with a synthetic 3’ UTR or with a heterologous 3’ UTR. Non-UTR sequences may also be used as regions or subregions within a nucleic acid. For example, introns or portions of introns sequences may be incorporated into regions of nucleic acid of the disclosure. Incorporation of intronic sequences may increase protein production as well as nucleic acid levels. Combinations of features may be included in flanking regions and may be contained within other features. For example, the ORF may be flanked by a 5′ UTR which may contain a strong Kozak translational initiation signal and/or a 3' UTR which may include an oligo(dT) sequence for templated addition of a poly-A tail. A 5′ UTR may comprise a first polynucleotide fragment and a second polynucleotide fragment from the same and/or different genes such as the 5′ UTRs described in US2010/0293625 and WO2015/085318A2, each of which is herein incorporated by reference. It should be understood that any UTR from any gene may be incorporated into the regions of a nucleic acid. Furthermore, multiple wild-type UTRs of any known gene may be utilized. It is also within the scope of the present disclosure to provide artificial UTRs which are not variants of wild type regions. These UTRs or portions thereof may be placed in the same orientation as in the transcript from which they were selected or may be altered in orientation or location. Hence a 5′ or 3′ UTR may be inverted, shortened, lengthened, made with one or more other 5′ UTRs or 3′ UTRs. As used herein, the term “altered” as it relates to a UTR sequence, means that the UTR has been changed in some way in relation to a naturally occurring or comparator sequence. For example, a 3′ UTR or 5′ UTR may be altered relative to a wild- type/native UTR by the change in orientation or location as taught above or may be altered by the inclusion of additional nucleotides, deletion of nucleotides, swapping or transposition of nucleotides. Any of these changes producing an “altered” UTR (whether 3′ or 5′) comprise a variant UTR. In some embodiments, a double, triple or quadruple UTR such as a 5′ UTR or 3′ UTR may be used. As used herein, a “double” UTR is one in which two copies of the same UTR are encoded either in series or substantially in series. For example, a double beta-globin 3′ UTR may be used as described in US2010/0129877, which is incorporated herein by reference. It is also within the scope of the present disclosure to have patterned UTRs. As used herein “patterned UTRs” are those UTRs which reflect a repeating or alternating pattern, such as ABABAB or AABBAABBAABB or ABCABCABC or variants thereof repeated once, twice, or more than 3 times. In these patterns, each letter, A, B, or C represent a different UTR at the nucleotide level. In some embodiments, flanking regions are selected from a family of transcripts whose proteins share a common function, structure, feature, or property. For example, polypeptides of interest may belong to a family of proteins which are expressed in a particular cell, tissue or at some time during development. The UTRs from any of these genes may be swapped for any other UTR of the same or different family of proteins to create a new polynucleotide. As used herein, a “family of proteins” is used in the broadest sense to refer to a group of two or more polypeptides of interest which share at least one function, structure, feature, localization, origin, or expression pattern. The untranslated region may also include translation enhancer elements (TEE). As a non- limiting example, the TEE may include those described in US 2009/0226470, herein incorporated by reference, and those known in the art. Open Reading Frames An open reading frame (ORF) is a continuous stretch of DNA or RNA that (1) begins with a start codon (e.g., ATG or AUG, encoding methionine), and (2) ends with a stop codon (e.g., TAA, TAG or TGA, or UAA, UAG or UGA) or is immediately followed by a stop codon. A stop codon does not encode an amino acid, such that translation of an ORF terminates when a ribosome reaches the stop codon immediately following the last amino acid-encoding codon in the ORF. A stop codon that results in translation termination may be considered part of the ORF, in which case the ORF ends with the stop codon. Alternatively, the first stop codon immediately following the last amino acid-encoding codon of an ORF may considered part of the 3′ untranslated region (3′ UTR) of a DNA or RNA, rather than part of the ORF. Those skilled in the art will understand that an ORF sequence that ends in a codon encoding amino acid will be followed by one or more stop codons in a DNA or RNA. An ORF may be followed by multiple stop codons. Inclusion of multiple consecutive stop codons reduces the extent of continued translation that may occur if a stop codon is mutated to a codon encoding an amino acid (readthrough), as a second stop codon may terminate translation even if a first stop codon is mutated and encodes an amino acid, such that only one amino acid is added to the C-terminus of the translated protein. Where multiple stop codons are present at the end, or immediately following, an ORF, the multiple stop codons may comprise the same stop codon (e.g., UGAUGA). Multiple stop codons may comprise different stop codons in series (e.g., UGAUAAUAG). In addition to reducing the extent of readthrough if a first stop codon is mutated, the presence of multiple different stop codons reduces the extent of readthrough if the first stop codon fails to allow translation termination (e.g., if a suppressor tRNA with an anticodon complementary to the first stop codon is present in the cell). An ORF typically encodes a protein. It will be understood that the sequences disclosed herein may further comprise additional elements, e.g., 5’ and/or 3’ UTRs, but that those elements, unlike the ORF, need not necessarily be present in an RNA (e.g., mRNA) as described herein. 5′ Caps In some embodiments, an RNA (e.g., mRNA) comprises a 5′ terminal cap.5′-capping of polynucleotides may be completed concomitantly during an in vitro transcription reaction using, for example, the following chemical RNA cap analogs to generate the 5′-guanosine cap structure according to manufacturer protocols: 3´-O-Me-m7G(5')ppp(5') G [the ARCA cap];G(5')ppp(5')A; G(5')ppp(5')G; m7G(5')ppp(5')A; m7G(5')ppp(5')G (New England BioLabs, Ipswich, MA).5′- capping of modified RNA (e.g., mRNA) may be completed post-transcriptionally using, for example, a Vaccinia Virus Capping Enzyme to generate the “Cap 0” structure: m7G(5')ppp(5')G (New England BioLabs, Ipswich, MA). Cap 1 structure may be generated using both Vaccinia Virus Capping Enzyme and a 2′-O methyl-transferase to generate: m7G(5')ppp(5')G-2′-O-methyl. Cap 2 structure may be generated from the Cap 1 structure followed by the 2′-O-methylation of the 5′-antepenultimate nucleotide using a 2′-O methyl-transferase. Cap 3 structure may be generated from the Cap 2 structure followed by the 2′-O-methylation of the 5′- preantepenultimate nucleotide using a 2′-O methyl-transferase. Enzymes may be derived from a recombinant source. Other cap analogs may be used. A cap analog may be, for example, a dinucleotide cap, a trinucleotide cap, or a tetranucleotide cap. In some embodiments, a cap analog is a dinucleotide cap. In some embodiments, a cap analog is a trinucleotide cap. In some embodiments, a cap analog is a tetranucleotide cap. A nucleotide cap (e.g., a trinucleotide cap or tetranucleotide cap), in some embodiments, comprises a compound of formula (I)
In some embodiments, the neutral lipid is 1,2 distearoyl-sn-glycero-3-phosphocholine (DSPC). In some embodiments, the sterol is cholesterol. In some embodiments, the PEG-modified lipid is PEG2000-DMG. In some embodiments, the open reading frame comprises one or more chemically modified nucleotides. In some embodiments, the open reading frame comprises N1-methylpseudouridine. In some embodiments, at least 80% of uracil nucleotides in the open reading frame comprise N1-methylpseudouridine. In some embodiments, 100% of uracil nucleotides in the open reading frame comprise N1-methylpseudouridine. In some embodiments, the open reading frame comprises 5-methylcytidine. In some embodiments, at least 80% of cytosine nucleotides in the open reading frame comprise 5-methylcytidine. In some embodiments, 100% of cytosine nucleotides in the open reading frame comprise 5-methylcytidine. In some embodiments, the open reading frame comprises 5-methyluridine. In some embodiments, at least 80% of uracil nucleotides in the open reading frame comprise 5-methyluridine. In some embodiments, 100% of uracil nucleotides in the open reading frame comprise 5- methyluridine. In some aspects, the disclosure relates to pharmaceutical compositions comprising a composition as described herein, and a pharmaceutically acceptable excipient. In some aspects, the disclosure relates to methods comprising administering to a subject a composition as described herein. In some embodiments, the composition is administered intramuscularly. In some embodiments, the composition is effective to elicit, in the subject, CD4+ and/or CD8+ T cells specific to one or more epitopes of the protein. In some embodiments, the composition is effective to elicit, in the subject, neutralizing antibodies to MERS-CoV. In some embodiments, the composition is effective to elicit, in the subject, antibodies that mediate antibody-dependent cell-mediated cytotoxicity (ADCC) against MERS-CoV-infected cells. In some embodiments, the method comprising administering a first dose and a second dose of the composition. In some aspects, the disclosure relates to a MERS-CoV fusion protein comprising: (a) an N-terminal domain (NTD) of a MERS-CoV Spike (S) protein; and (b) a receptor-binding domain (RBD) of the MERS-CoV-S protein, wherein the MERS-CoV fusion protein does not comprise an NTD or an RBD of an S protein of a coronavirus that is not MERS-CoV. In some embodiments, the NTD and RBD are connected by a linker. In some embodiments, the linker is a glycine linker or a glycine-serine linker. In some embodiments, the linker comprises a pan-HLA-DR-binding epitope (PADRE). In some embodiments, the MERS-CoV fusion protein further comprises a transmembrane (TM) domain. In some embodiments, the TM domain is a MERS-CoV S protein TM domain or derivative thereof. In some embodiments, the TM domain is heterologous to MERS-CoV. In some embodiments, the TM domain is an influenza virus hemagglutinin (HA) TM domain or derivative thereof. In some embodiments, the MERS-CoV fusion protein comprises, in N-to-C-terminal order, the NTD, the RBD, and the TM domain. In some embodiments, the MERS-CoV fusion protein does not comprise a transmembrane (TM) domain. In some embodiments, the MERS-CoV fusion protein is soluble. In some embodiments, the MERS-CoV fusion protein further comprises a signal peptide. In some embodiments, the signal peptide is a MERS-CoV S protein signal peptide. In some embodiments, the signal peptide is heterologous to MERS-CoV. In some aspects, the disclosure relates to mutant Middle East Respiratory Syndrome (MERS)-CoV S proteins, comprising (a) one or more proline substitutions or insertions, and (b) one or more substitutions or deletions in a furin cleavage site, relative to a full-length MERS- CoV S protein. In some embodiments, the mutant MERS-CoV S protein comprises prolines at positions corresponding to residues 1060 and 1061 of the full-length MERS-CoV S protein. In some aspects, the disclosure relates to mutant Middle East Respiratory Syndrome (MERS)-CoV S proteins comprising (a) one or more substitutions or deletions in a furin cleavage site, and (b) a truncated endoplasmic reticulum (ER) retention signal, relative to a full-length MERS-CoV S protein. In some embodiments, the mutant MERS-CoV S protein does not comprise an ER retention signal. In some aspects, the disclosure relates to mutant MERS-CoV S proteins comprising (a) one or more substitutions or deletions in a furin cleavage site, and (b) a truncated cytoplasmic tail, relative to a full-length MERS-CoV S protein. In some embodiments, the mutant MERS-CoV S protein does not comprise a cytoplasmic tail, or comprises a cytoplasmic tail comprising 21 or fewer amino acids. In some aspects, the disclosure relates to MERS-CoV fusion proteins consisting essentially of (a) an N-terminal domain (NTD) of a full-length MERS-CoV S protein, (b) a receptor-binding domain of the full-length MERS-CoV protein, and (c) a transmembrane (TM) domain. In some aspects, the disclosure relates to Middle East Respiratory Syndrome (MERS)- CoV fusion proteins consisting essentially of (a) an N-terminal domain (NTD) of a full-length MERS-CoV S protein, and (b) a transmembrane (TM) domain. In some embodiments, the NTD comprises amino acids 18–338 of a full-length MERS- CoV S protein. In some aspects, the disclosure relates to MERS-CoV fusion proteins consisting essentially of (a) a receptor-binding domain (RBD) of a full-length MERS-CoV S protein, and (b) a transmembrane (TM) domain. In some embodiments, the RBD comprises amino acids 376–589 of a full-length MERS- CoV S protein. In some embodiments, the TM domain is heterologous to MERS-CoV. In some embodiments, the TM domain is an influenza virus hemagglutinin (HA) TM domain. In some embodiments, the full-length MERS-CoV- S protein comprises the amino acid sequence of SEQ ID NO: 84. In some aspects the disclosure relates to proteins comprising an amino acid sequence with at least 95%, at least 97%, at least 98%, at least 99%, or 100% sequence identity to the amino acid sequence of any one of SEQ ID NOs: 87–98. In some aspects, the disclosure relates to ribonucleic acids (RNA) comprising an open reading frame encoding a protein as described herein. In some aspects, the disclosure relates to messenger ribonucleic acids (mRNA) comprising an open reading frame encoding a protein as described herein. In some embodiments, the mRNA comprises one or more chemically modified nucleotides. In some embodiments, 100% of the uracil nucleotides of the mRNA comprise are chemically modified nucleotides. In some embodiments, 100% of uracil nucleotides of the mRNA comprise N1- methylpseudouridine. In some aspects, the disclosure relates to self-amplifying ribonucleic acids (saRNA) comprising an open reading frame encoding a protein comprising: (a) an N-terminal domain (NTD) of a MERS-CoV Spike (S) protein; and (b) a receptor-binding domain (RBD) of the MERS-CoV-S protein, wherein the MERS-CoV fusion protein does not comprise an NTD or an RBD of an S protein of a coronavirus that is not MERS-CoV. In some embodiments, the NTD and RBD are connected by a linker. In some embodiments, the linker is a glycine linker or a glycine-serine linker. In some embodiments, the linker comprises a pan-HLA-DR-binding epitope (PADRE). In some embodiments, the MERS-CoV fusion protein further comprises a transmembrane (TM) domain. In some embodiments, the TM domain is a MERS-CoV S protein TM domain or derivative thereof. In some embodiments, the TM domain is heterologous to MERS-CoV. In some embodiments, the TM domain is an influenza virus hemagglutinin (HA) TM domain or derivative thereof. In some embodiments, the MERS-CoV fusion protein comprises, in N-to-C-terminal order, the NTD, the RBD, and the TM domain. In some embodiments, the MERS-CoV fusion protein does not comprise a transmembrane (TM) domain. In some embodiments, the MERS-CoV fusion protein is soluble. In some embodiments, the MERS-CoV fusion protein further comprises a signal peptide. In some embodiments, the signal peptide is a MERS-CoV S protein signal peptide. In some embodiments, the signal peptide is heterologous to MERS-CoV. In some aspects, the disclosure relates to self-amplifying ribonucleic acids (saRNA) comprising an open reading frame encoding a mutant Middle East Respiratory Syndrome (MERS)-CoV S protein, wherein the mutant MERS-CoV S protein comprises (a) one or more proline substitutions or insertions, and (b) one or more substitutions or deletions in a furin cleavage site, relative to a full-length MERS-CoV S protein. In some embodiments, the mutant MERS-CoV S protein comprises prolines at positions corresponding to residues 1060 and 1061 of the full-length MERS-CoV S protein. In some aspects, the disclosure relates to self-amplifying ribonucleic acids (saRNA) comprising an open reading frame encoding a mutant Middle East Respiratory Syndrome (MERS)-CoV S protein, wherein the mutant MERS-CoV S protein comprises (a) one or more substitutions or deletions in a furin cleavage site, and (b) a truncated endoplasmic reticulum (ER) retention signal, relative to a full-length MERS-CoV S protein. In some embodiments, the mutant MERS-CoV S protein does not comprise an ER retention signal. In some aspects, the disclosure relates to self-amplifying ribonucleic acids (saRNA) comprising an open reading frame encoding a mutant Middle East Respiratory Syndrome (MERS)-CoV S protein, wherein the mutant MERS-CoV S protein comprises (a) one or more substitutions or deletions in a furin cleavage site, and (b) a truncated cytoplasmic tail, relative to a full-length MERS-CoV S protein. In some embodiments, the mutant MERS-CoV S protein does not comprise a cytoplasmic tail, or comprises a cytoplasmic tail comprising 21 or fewer amino acids. In some aspects, the disclosure relates to self-amplifying ribonucleic acids (saRNA) comprising an open reading frame encoding a Middle East Respiratory Syndrome (MERS)-CoV fusion protein, wherein the MERS-CoV fusion protein consists essentially of (a) an N-terminal domain (NTD) of a full-length MERS-CoV S protein, (b) a receptor-binding domain of the full-length MERS-CoV protein, and (c) a transmembrane (TM) domain. In some aspects, the disclosure relates to self-amplifying ribonucleic acids (saRNA) comprising an open reading frame encoding a Middle East Respiratory Syndrome (MERS)-CoV fusion protein, wherein the MERS-CoV fusion protein consists essentially of (a) an N-terminal domain (NTD) of a full-length MERS-CoV S protein, and (b) a transmembrane (TM) domain. In some embodiments, the NTD comprises amino acids 18–338 of a full-length MERS- CoV S protein. In some aspects, the disclosure relates to self-amplifying ribonucleic acids (saRNA) comprising an open reading frame encoding a Middle East Respiratory Syndrome (MERS)-CoV fusion protein, wherein the MERS-CoV fusion protein consists essentially of (a) a receptor-binding domain (RBD) of a full-length MERS-CoV S protein, and (b) a transmembrane (TM) domain. In some embodiments, the RBD comprises amino acids 376–589 of a full-length MERS- CoV S protein. In some embodiments, the TM domain is heterologous to MERS-CoV. In some embodiments, the TM domain is an influenza virus hemagglutinin (HA) TM domain. In some embodiments, the full-length MERS-CoV- S protein comprises the amino acid sequence of SEQ ID NO: 84. BRIEF DESCRIPTION OF THE DRAWINGS FIG.1 is a structural schematic of a betacoronavirus S protein, including the N-terminal domain (NTD), the receptor-binding domain (RBD), the Spike subdomain 1 (SD1), the Spike subdomain 2 (SD2), and the S2 subunit of the spike protein (S2) (left). The schematic on the right shows elements of the NTD-RBD-TM construct design, including the linked RBD and NTD. FIG.2 shows the neutralizing antibody (NAb) titers (y-axis) elicited in mice in response to immunization with either one or two doses of varying amounts of S-2P (2P-stabilized full- length S protein), NTD-RBD-TM (S protein NTD and RBD linked to a transmembrane (TM) domain), RBD-TM (S protein RBD linked to a TM domain), and NTD-TM (S protein NTD linked to a TM domain) (x-axis). DETAILED DESCRIPTION MERS-CoV The genome of MERS-CoV is a single-stranded positive-sense RNA (+ssRNA) with the size of about 30.1 kb. Coronavirus genomes include a variable number of open reading frames (ORFs) that encode accessory proteins, nonstructural proteins, and structural proteins (Song et al. 2019 Viruses;11(1):p.59). Many antigenic peptides are located in the structural proteins (Cui et al.2019 Nat. Rev. Microbiol., 17(3):181–192). Spike glycoprotein (S), a small envelope protein (E), matrix protein (M), and nucleocapsid protein (N) are four such structural proteins. Since S protein contributes to cell tropism and virus particle entry, and also elicits neutralizing antibodies (NAb) and protective immunity, it is considered an important target in coronavirus vaccine development. Moreover, amino acid sequence analysis has shown that the S protein contains regions conserved among coronaviruses, which may be the basis for universal vaccine development. MERS-CoV Proteins Some aspects of the disclosure relate to compositions comprising nucleic acids (e.g., mRNAs) encoding proteins of interest, e.g., a protein derived from a betacoronavirus structural protein such as a MERS-CoV Spike (S) protein. Such compositions do not comprise antigens per se, but rather comprise nucleic acids, in particular, mRNA(s) that encode antigens or antigenic sequences once delivered to a cell, tissue or subject. Delivery of nucleic acids, in particular mRNA(s), is achieved by inclusion of nucleic acids in appropriate carriers or delivery vehicles (e.g., lipid nanoparticles) such that upon administration to cells, tissues or subjects, nucleic acid is taken up by cells which, in turn, express protein(s) encoded by the nucleic acids, e.g., mRNAs. Antigens, as used herein, are proteins capable of inducing an immune response (e.g., causing an immune system to produce antibodies against the antigens). The vaccines as described herein provide a unique advantage over traditional protein-based vaccination approaches, in which protein antigens are purified or produced in vitro, e.g., recombinant protein production technologies. The vaccines of the present disclosure feature mRNA encoding the desired antigens, which when introduced into the body, i.e., administered to a mammalian subject (for example a human) in vivo, cause the cells of the body to express the desired antigens. To facilitate delivery of the mRNAs of the present disclosure to the cells of the body, the mRNAs are encapsulated in lipid nanoparticles (LNPs). Upon delivery and uptake by cells of the body, the mRNAs are translated in the cytosol and protein antigens are generated by the host cell machinery. The protein antigens are presented and elicit an adaptive humoral and cellular immune response. Neutralizing antibodies are directed against the expressed protein antigens and hence the protein antigens are considered relevant target antigens for vaccine development. Herein, use of the term “antigen” encompasses immunogenic proteins and immunogenic fragments (an immunogenic fragment that induces (or is capable of inducing) an immune response to a (at least one) MERS-CoV variant), unless otherwise stated. It should be understood that the term “protein” encompasses peptides and the term “antigen” encompasses antigenic fragments. Other molecules may be antigenic such as bacterial polysaccharides or combinations of protein and polysaccharide structures, but for the viral vaccines included herein, viral proteins, fragments of viral proteins and designed and or mutated proteins derived from MERS-CoV are the antigens described herein. Many proteins have a quaternary or three-dimensional structure, which consists of more than one polypeptide or several polypeptide chains that associate into an oligomeric molecule. As used herein the term “subunit” refers to a single protein molecule, for example, a polypeptide or polypeptide chain resulting from processing of a nascent protein molecule, which subunit assembles (or “coassembles”) with other protein molecules (e.g., subunits or chains) to form a protein complex. Proteins can have a relatively small number of subunits and therefore be described as “oligomeric” or can consist of a large number of subunits and therefore be described as “multimeric”. The subunits of an oligomeric or multimeric protein may be identical, homologous or totally dissimilar and dedicated to disparate tasks. Proteins or protein subunits can further comprise domains. As used herein, the term “domain” refers to a distinct functional and/or structural unit within a protein. Typically, a “domain” is responsible for a particular function or interaction, contributing to the overall role of a protein. Domains can exist in a variety of biological contexts. Similar domains (i.e., domains sharing structural, functional and/or sequence homology) can exist within a single protein or can exist within distinct proteins having similar or different functions. A protein domain is often a conserved part of a given protein tertiary structure or sequence that can function and exist independently of the rest of the protein or subunit thereof. In structural and molecular biology, identical, homologous or similar subunits or domains can help to classify newly identified or novel proteins. As used herein, the term antigen is distinct from the term “epitope” which is a substructure of an antigen, e.g., a polypeptide, such as 7-10 amino acids, or carbohydrate structure, which may be recognized by an antigen binding site. The art describes protein antigens that are delivered to subjects or immune cells in isolated form, e.g., isolated protein, polypeptide or peptide antigens, however, the design, testing, validation, and production of protein antigens can be costly and time-consuming, especially when producing proteins at large scale. By contrast, mRNA technology is amenable to rapid design and testing of mRNA constructs encoding a variety of antigens. Moreover, rapid production of mRNA coupled with inclusion in appropriate delivery vehicles (e.g., lipid nanoparticles), can proceed quickly and can rapidly produce mRNA vaccines at large scale. Potential benefit also arises from the fact that antigens encoded by the mRNAs are expressed by the cells of the subject, e.g., are expressed by the human body, and thus the subject, e.g., the human body, serves as the “factory” to produce the antigens which, in turn, elicit the desired immune response. Other immune cells, for example, B cells and T cells, are then able to recognize and mount and immune response develop an immune response against the encoded protein and ultimately create a long -lasting protective response against the coronavirus. Low immunogenicity, a drawback in protein vaccine development due to poor presentation to the immune system or incorrect folding of the antigens, is avoided through the use of the highly effective mRNA vaccines encoding spike protein, subunits and domains thereof described herein. The compositions, as described herein, may include an RNA or multiple RNAs encoding two or more antigens of the same or different MERS-CoV strains or genotypes. Also provided herein are combination vaccines that include RNA encoding one or more MERS-CoV antigens and one or more antigen(s) of a different organism. Thus, the vaccines of the present disclosure may be combination vaccines that target one or more antigens of the same MERS-CoV genotype or strain, or one or more antigens of different MERS-CoV genotypes or different species, e.g., antigens which induce immunity to organisms which are found in the same geographic areas where the risk of coronavirus infection is high or organisms to which an individual is likely to be exposed to when exposed to a coronavirus (e.g., MERS-CoV). In some embodiments, the second or subsequent circulating MERS-CoV antigen is an immunodominant antigen from an emerging strain. An immunodominant antigen of an emerging strain is assessed with respect to the strain from which the antigen is derived, relative to a different strain of the virus, such as the original strain or other variant thereof. An immunodominant antigen of the emerging strain induces a stronger immune response against the emerging strain than against the different strain. In some embodiments, an immunodominant antigen of the emerging strain is more infective than a different strain of the virus, such as the original strain or other variant thereof. Exemplary sequences of coronavirus proteins and variants thereof (e.g., comprising one or more domains of a coronavirus protein) encoded by mRNAs as described herein are provided in EXEMPLARY SEQUENCES as SEQ ID NOs: 84–98. In some embodiments, an RNA (e.g., mRNA) comprises an open reading frame encoding a protein having at least 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity to an amino acid sequence of SEQ ID NO: 84. In some embodiments, an RNA (e.g., mRNA) comprises an open reading frame encoding a protein having at least 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity to an amino acid sequence of SEQ ID NO: 85. In some embodiments, an RNA (e.g., mRNA) comprises an open reading frame encoding a protein having at least 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity to an amino acid sequence of SEQ ID NO: 86. In some embodiments, an RNA (e.g., mRNA) comprises an open reading frame encoding a protein having at least 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity to an amino acid sequence of SEQ ID NO: 87. In some embodiments, an RNA (e.g., mRNA) comprises an open reading frame encoding a protein having at least 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity to an amino acid sequence of SEQ ID NO: 88. In some embodiments, an RNA (e.g., mRNA) comprises an open reading frame encoding a protein having at least 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity to an amino acid sequence of SEQ ID NO: 89. In some embodiments, an RNA (e.g., mRNA) comprises an open reading frame encoding a protein having at least 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity to an amino acid sequence of SEQ ID NO: 90. In some embodiments, an RNA (e.g., mRNA) comprises an open reading frame encoding a protein having at least 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity to an amino acid sequence of SEQ ID NO: 91. In some embodiments, an RNA (e.g., mRNA) comprises an open reading frame encoding a protein having at least 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity to an amino acid sequence of SEQ ID NO: 92. In some embodiments, an RNA (e.g., mRNA) comprises an open reading frame encoding a protein having at least 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity to an amino acid sequence of SEQ ID NO: 93. In some embodiments, an RNA (e.g., mRNA) comprises an open reading frame encoding a protein having at least 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity to an amino acid sequence of SEQ ID NO: 94. In some embodiments, an RNA (e.g., mRNA) comprises an open reading frame encoding a protein having at least 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity to an amino acid sequence of SEQ ID NO: 95. In some embodiments, an RNA (e.g., mRNA) comprises an open reading frame encoding a protein having at least 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity to an amino acid sequence of SEQ ID NO: 96. In some embodiments, an RNA (e.g., mRNA) comprises an open reading frame encoding a protein having at least 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity to an amino acid sequence of SEQ ID NO: 97. In some embodiments, an RNA (e.g., mRNA) comprises an open reading frame encoding a protein having at least 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity to an amino acid sequence of SEQ ID NO: 98. In some embodiments, an mRNA vaccine comprises 1, 2, 3, 4, 5, or 6 mRNAs encoding different proteins, wherein each protein comprises at least one mutation and/or at least one deletion. In some embodiments, the mRNA vaccine further comprises an mRNA encoding a wild-type MERS-CoV S protein or the antigenic fragment thereof. The mRNA vaccine, in some embodiments, is in a lipid nanoparticle (that is, the lipid nanoparticle comprises 1, 2, 3, 4, 5, or 6 mRNAs encoding different protein). In some embodiments, a composition comprises a first mRNA encoding a protein or variant thereof of a first MERS-CoV virus and a second mRNA encoding a second protein or variant thereof of a second MERS-CoV virus. In some embodiments, the first MERS-CoV virus is a first circulating MERS-CoV virus. In some embodiments, the second MERS-CoV virus is a second circulating MERS-CoV virus. “Circulating viruses” as used herein refer to viruses that have been in circulation for 1 month, 2 months, 3 months, 4 months, 5 months, 6 months, 7 months, 8 months, 9 months, 10 months, 11 months, a portion of a year, 1 year, 1.5 years, 2 years, 3 years, or longer. In some embodiments, the first and second mRNAs are present in the composition in a 1:1, 1:2, 1:3, or 1:4 ratio. In some embodiments, the first and second mRNAs are present in the composition in a 2:1, 3:1, or 4:1 ratio. In some embodiments, the first and second mRNAs are present in the composition in a 1:1 ratio. In some embodiments, a composition further comprises a third mRNA encoding a protein or variant thereof of a third MERS-CoV virus, wherein the protein comprises at least one addition, substitution, or deletion relative to the amino acid sequence of SEQ ID NO: 84. In some embodiments, the first and third mRNAs are present in the composition in a 1:1, 1:2, 1:3, or 1:4 ratio. In some embodiments, the first and third mRNAs are present in the composition in a 2:1, 3:1, or 4:1 ratio. In some embodiments, the first and third mRNAs are present in the composition in a 1:1 ratio. In some embodiments, a composition further comprises a fourth mRNA encoding a protein or variant thereof of a fourth MERS-CoV virus, wherein the protein comprises at least one addition, substitution, or deletion relative to the amino acid sequence of SEQ ID NO: 84. In some embodiments, the first and fourth mRNAs are present in the composition in a 1:1, 1:2, 1:3, or 1:4 ratio. In some embodiments, the first and fourth mRNAs are present in the composition in a 2:1, 3:1, or 4:1 ratio. In some embodiments, the first and fourth mRNAs are present in the composition in a 1:1 ratio. In some embodiments, a composition further comprises a fifth mRNA encoding a protein or variant thereof of a fifth MERS-CoV virus, wherein the protein comprises at least one addition, substitution, or deletion relative to the amino acid sequence of SEQ ID NO: 84. In some embodiments, the first and fifth mRNAs are present in the composition in a 1:1, 1:2, 1:3, or 1:4 ratio. In some embodiments, the first and fifth mRNAs are present in the composition in a 2:1, 3:1, or 4:1 ratio. In some embodiments, the first and fifth mRNAs are present in the composition in a 1:1 ratio. In some embodiments, a composition further comprises a sixth mRNA encoding a protein or variant thereof of a sixth MERS-CoV virus, wherein the protein comprises at least one addition, substitution, or deletion relative to the amino acid sequence of SEQ ID NO: 84. In some embodiments, the first and sixth mRNAs are present in the composition in a 1:1, 1:2, 1:3, or 1:4 ratio. In some embodiments, the first and sixth mRNAs are present in the composition in a 2:1, 3:1, or 4:1 ratio. In some embodiments, the first and sixth mRNAs are present in the composition in a 1:1 ratio. In some embodiments, the mRNAs are present in the composition in an equal amount (e.g., a 1:1 weight/weight ratio or a 1:1 molar ratio), for example, a ratio of 1:1 (:1:1:1:1) of mRNA encoding distinct coronavirus antigens. As used herein, a “weight/weight ratio” or wt/wt ratio or wt:wt ratio refers to the ratio between the weights (masses) of the different components. A “molar ratio” refers to the ratio between different components (e.g., the number of mRNA encoding each antigen). In some embodiments, the ratio is 1:1, 1:2, 1:3, 1:4, 2:1, 3:1, or 4:1. In each embodiment or aspect described herein, it is understood that the featured vaccines include the mRNAs encapsulated within LNPs. While it is possible to encapsulate each unique mRNA in its own LNP, the mRNA vaccine technology enjoys the significant technological advantage of being able to encapsulate several mRNAs in a single LNP product. In other embodiments the vaccines are separate vaccines that are not co-formulated, but may be admixed separately before administration or simply administered separately. Spike (S) Proteins The envelope spike (S) proteins of known betacoronaviruses determine the virus host tropism and entry into host cells. Coronavirus spike (S) protein is a choice antigen for the vaccine design as it can induce neutralizing antibodies and protective immunity. S protein is critical for MERS-CoV infection. The organization of the S protein is similar among betacoronaviruses, such as MERS-CoV, SARS-CoV-2, SARS-CoV, HKU1-CoV, MHV-CoV and NL63-CoV. The organization of the Spike (S) protein is similar among betacoronaviruses, such as SARS-CoV-2, SARS-CoV, MERS-CoV, HKUl-CoV, MHV-CoV and NL63-CoV, including two subunits, S1 and S2, which mediate attachment and membrane fusion, respectively. The S1 subunit includes an N terminal domain (NTD), a receptor binding domain (RBD), and two subdomains (SD1 and SD2); the S2 subunit participates in fusion with the host cell membrane and viral entry. As used herein, the term “Spike protein” refers to a glycoprotein that forms homotrimers protruding from the envelope (viral surface) of viruses including betacoronaviruses. Trimerized Spike protein facilitates entry of the virion into a host cell by binding to a receptor on the surface of a host cell followed by fusion of the viral and host cell membranes. The S protein is a highly glycosylated and large type I transmembrane fusion protein that is made up of 1,160 to 1,400 amino acids, depending upon the type of virus. Betacoronavirus Spike proteins comprise between about 1100 to 1500 amino acids. MERS-CoV spike (S) protein is a primary antigen choice for vaccine design, as it can induce neutralizing antibodies and protective immunity. mRNAs as described herein are designed to produce MERS-CoV Spike proteins (i.e., encode Spike proteins such that Spike protein is expressed when the mRNA is delivered to a cell or tissue, for example a cell or tissue in a subject), as well as variants thereof. The skilled artisan will understand that, while an essentially full length or complete Spike protein may be necessary for a virus, e.g., a betacoronavirus, to perform its intended function of facilitating virus entry into a host cell, a certain amount of variation in Spike protein structure and/or sequence is tolerated when seeking primarily to elicit an immune response against Spike protein. For example, minor truncation, e.g., of one to a few, possibly up to 5 or up to 10 amino acids from the N- or C-terminus of the encoded Spike protein, e.g., encoded Spike protein antigen, may be tolerated without changing the antigenic properties of the protein. Likewise, variation (e.g., conservative substitution) of one to a few, possibly up to 5 or up to 10 amino acids (or more) of the encoded Spike protein, e.g., encoded Spike protein antigen, may be tolerated without changing the antigenic properties of the protein. In some embodiments, the Spike protein is a stabilized Spike protein, for example, the Spike protein is stabilized by two proline substitutions (a 2P mutation). In some embodiments, the Spike protein is not a stabilized Spike protein, for example, the Spike protein is not stabilized by two proline substitutions (a 2P mutation). In some embodiments, the Spike protein is from a different virus strain. A strain is a genetic variant of a microorganism (e.g., a virus). New viral strains can be created due to mutation, which may be selected due to enhanced replication, transmissibility, and/or evasion of pre-existing immune responses (e.g., antigenic drift), or recombination of genetic components when two or more viruses infect the same cell, with such recombinant viruses being selected due to enhanced replication, transmissibility, and/or evasion of pre-existing immune responses. Antigenic drift is a process that generates genetic and antigenic variation in viruses, by the accumulation of mutations in the virus genes that code for virus-surface proteins recognized by host immune responses (antibodies and T cells). This results in selection for new virus strains that are not effectively inhibited by the antibodies and/or T cell responses that prevented or mitigated infection by previous (wild-type or ancestral) strains. This makes it easier for the changed virus to spread throughout a partially immune population. Antigenic shift is the process by which two or more different strains of a virus, or strains of two or more different viruses, combine to form a new subtype having a mixture of the surface antigens of the two or more original strains, which may create virus with a novel combination of surface antigens that did not previously exist in nature. The term is often applied specifically to influenza viruses, where segmentation of the viral genome into distinct RNA segments, and reassortment of genome segments during virion production, allows the production of reassortant progeny with novel combinations of genome segments from co-infected cells. However, genetic recombination may occur between non-segmented viruses (e.g., MERS-CoV) where multiple viral genotypes replicate in the same cell, e.g., by switching between two template genomes during replication, resulting in progeny genomes with combinations of sequences from two or more viral genotypes. Antigenic shift is contrasted with antigenic drift (in which individual mutations accumulate over time, and may lead to a loss of immunity, or in vaccine mismatch). In contrast to accumulation of mutations on an ancestral genome (antigenic drift), genetic recombination (through reassortment or molecular recombination of genomes) is often associated with a major reorganization of viral genomes, resulting in novel combination of genes, which may cause a more immediate and drastic change in viral phenotype. A virus strain as used herein is a genetic variant or of a virus that is characterized by a differing isoform of one or more surface proteins of the virus. In the case of MERS-CoV, for example, a different amino acid sequence in the MERS-CoV spike protein where the immune response in an individual to the new strain is less effective than to the strain used to immunize or first infect the individual. A new virus strain may arise from natural mutation or a combination of natural mutation and immune selection due to an ongoing immune response in an immunized or previously infected individual. A new virus strain can differ by one, two, three or more amino acid mutations in regions of the spike protein responsible for a viral function such as receptor binding or viral fusion with a target cell. A spike protein from a new strain may differ from the parental strain by as much as 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity at the amino acid level. A natural virus strain is a variant of a given virus that is recognizable because it possesses some “unique phenotypic characteristics” that remain stable (e.g., stable and heritable biological, serological, and/or molecular characters) under natural conditions. Such “unique phenotypic characteristics” are biological properties different from the compared reference virus, such as unique antigenic properties, host range (e.g., infecting a different kind of host), symptoms of disease caused by the strain, different type of disease caused by the strain (e.g., transmitted by different means), etc. A “unique phenotypic characteristic” can be detected clinically (e.g., clinical manifestations detected in a host infected with the strain) or within a comparative animal experiment in which a researcher skilled in the art of virology can distinguish between the reference control virus-infected animal and the animal infected with the alleged new strain, without knowing which animal received which virus and without having any information about the differences between the two viruses. Importantly, a virus variant with a simple difference in genome sequence is not a separate strain if there is no recognizable distinct viral phenotype. The extent of genomic sequence variation is irrelevant for the classification of a variant as a strain since a distinct phenotype sometimes arises from few mutations. S proteins of coronaviruses can be divided into two important functional subunits, of which include the N-terminal S1 subunit, which forms of the globular head of the S protein, and the C-terminal S2 region that forms the stalk of the protein and is directly embedded into the viral envelope. Upon interaction with a potential host cell, the S1 subunit will recognize and bind to receptors on the host cell, specifically dipeptidyl peptidase 4 (DPP4), whereas the S2 subunit, which is the most conserved component of the S protein, will be responsible for fusing the envelope of the virus with the host cell membrane. (See e.g., Raj et al., Nature.2013. 495(7440):251–254.). Each monomer of trimeric S protein trimer contains the two subunits, S1 and S2, mediating attachment and membrane fusion, respectively. As part of the infection process in vivo, the two subunits are separated from each other by an enzymatic cleavage process. S protein is first cleaved by furin-mediated cleavage at the S1/S2 site in infected cells. In vivo, a subsequent serine protease-mediated cleavage event occurs at the S2′ site within S1. In MERS- CoV, the S1/S2 cleavage site is at amino acids 748–RSVR–751 (SEQ ID NO: 45). The S2′ cleavage site is at amino acids 884–RSAR–887 (SEQ ID NO: 46). As used herein, for example in the context of designing MERS-CoV S proteins, subunits, and/or fusion proteins encoded by the nucleic acids, e.g., mRNAs, as described herein, the term “S1 subunit” (e.g., S1 subunit antigen) refers to the N-terminal subunit of the Spike protein beginning at the S protein N-terminus and ending at the S1/S2 cleavage site whereas the term “S2 subunit” (e.g., S2 subunit antigen) refers to the C-terminal subunit of the Spike protein beginning at the S1/S2 cleavage site and ending at the C-terminus of the Spike protein. As described supra, the skilled artisan will understand that, while an essentially full length or complete Spike protein S1 or S2 subunit may be necessary for receptor binding or membrane fusion, respectively, a certain amount of variation in S1 or S2 structure and/or sequence is tolerated when seeking primarily to elicit an immune response against Spike protein subunits. For example, minor truncation, e.g., of one to a few, possibly up to 4, 5, 6, 7, 8, 9 or 10 amino acids from the N- or C-terminus of the encoded subunit, e.g., encoded S1 or S2 protein antigens, may be tolerated without changing the antigenic properties of the protein. Likewise, variation (e.g., conservative substitution) of one to a few, possibly up to 4, 5, 6, 7, 8, 9 or 10 amino acids (or more) of the encoded Spike protein subunits, e.g., encoded S1 or S2 protein antigen, may be tolerated without changing the antigenic properties of the protein(s). In some embodiments, the composition comprises an mRNA encoding a Spike protein associated with the Kingdom of Saudi Arabia (KSA) 2019 lineage. An exemplary Spike protein of the MERS-CoV KSA 2019 lineage is provided as amino acid sequence of SEQ ID NO: 84. In some embodiments, the composition comprises an mRNA encoding a Spike protein associated with the Riyadh 2013 lineage. An exemplary Spike protein of the MERS-CoV Riyadh 2013 lineage is provided as amino acid sequence of SEQ ID NO: 85. In some embodiments, the composition comprises an mRNA encoding a Spike protein associated with the England 2012 lineage. An exemplary Spike protein of the MERS-CoV England 2012 lineage is provided as amino acid sequence of SEQ ID NO: 84. In some embodiments, a Spike protein, e.g., an encoded Spike protein antigen, has the amino acid sequence of SEQ ID NO: 84. In some embodiments, a Spike protein, e.g., an encoded Spike protein antigen, has the amino acid sequence of SEQ ID NO: 85. In some embodiments, a Spike protein, e.g., an encoded Spike protein antigen, has the amino acid sequence of SEQ ID NO: 86. In some embodiments, a Spike protein, e.g., an encoded Spike protein antigen, has no greater than 100, no greater than 90, no greater than 80, no greater than 70, no greater than 60, no greater than 50, no greater than 40, no greater than 30, no greater than 20, no greater than 10, or no greater than 5 amino acid substitutions and/or deletions as compared to (when aligned with) a Spike protein having the amino acid sequence as provided in any one of SEQ ID NOs: 84–86. Where minor variations are made in encoded Spike protein sequences, the variant preferably has the same activity as the comparator Spike protein sequence and/or has the same immune specificity as the comparator Spike protein, as determined for example, in immunoassays (e.g., enzyme-linked immunosorbent assays (ELISA assays). In some embodiments, a Spike protein, e.g., an encoded Spike protein antigen, has the amino acid sequence of SEQ ID NO: 89. In some embodiments, a Spike protein, e.g., an encoded Spike protein antigen, has the amino acid sequence of SEQ ID NO: 90. In some embodiments, a Spike protein, e.g., an encoded Spike protein antigen, has the amino acid sequence of SEQ ID NO: 91. In some embodiments, a Spike protein, e.g., an encoded Spike protein antigen, has the amino acid sequence of SEQ ID NO: 92. In some embodiments, a Spike protein, e.g., an encoded Spike protein antigen, has no greater than 100, no greater than 90, no greater than 80, no greater than 70, no greater than 60, no greater than 50, no greater than 40, no greater than 30, no greater than 20, no greater than 10, or no greater than 5 amino acid substitutions and/or deletions as compared to (when aligned with) a Spike protein having the amino acid sequence as provided in any one of SEQ ID NOs: 89–92. In some embodiments, a Spike protein, e.g., an encoded Spike protein antigen, comprises one or more (e.g., two consecutive) proline substitutions or insertions at or near the boundary between a heptad repeat 1 (HR1) domain and a central helix (CH) domain. Introduction (e.g., by insertion or substitution) of proline residues in this region stabilize the Spike protein in a prefusion conformation. In some embodiments, the Spike protein comprises 1, 2, 3, 4, 5, 6, 7, 8, 9, or 10 proline residues that are inserted and/or substituted, relative to a wild-type (naturally occurring) MERS-CoV S protein. In some embodiments, an introduced proline residue is up to 15 amino acids N-terminal to the last (C-terminal) residue of the HR1 domain, and up to 5 amino acids C-terminal to the first (N-terminal) residue of the CH domain of the wild-type MERS-CoV S protein. In some embodiments, the Spike protein comprises a proline residue at one or more positions corresponding to 1058, 1059, 1060, and 1061 of SEQ ID NO: 84. In some embodiments, the Spike protein comprises an L1058P, D1059P, V1060P, and/or L1061P substitution relative to the amino acid sequence of SEQ ID NO: 84. In some embodiments, the Spike protein comprises proline residues at positions corresponding to 1060 and 1061 of SEQ ID NO: 84. In some embodiments, the Spike protein comprises V1060P and L1061P substitutions, relative to the amino acid sequence of SEQ ID NO: 84. In some embodiments, the Spike protein has an amino acid sequence with at least 90%, at least 95%, at least 96%, at least 97%, at least 98%, or at least 99% identity to the amino acid sequence of SEQ ID NO: 89, and the Spike protein comprises proline residues at positions corresponding to amino acids 1060 and 1061 of SEQ ID NO: 84. In some embodiments, a Spike protein, e.g., an encoded Spike protein antigen, comprises one or more mutations in the S1/S2 cleavage site. In some embodiments, the Spike protein does not comprise the amino acid sequence RSVR (SEQ ID NO: 45) at positions corresponding to amino acids 748–751 of SEQ ID NO: 84. In some embodiments, the Spike protein does not comprise the amino acid sequence RXXR at positions corresponding to amino acids 748–751 of SEQ ID NO: 84. In some embodiments, the Spike protein comprises one or more mutations in the S2′ cleavage site. In some embodiments, the Spike protein does not comprise the amino acid sequence RSAR (SEQ ID NO: 46) at positions corresponding to amino acids 884–887 of SEQ ID NO: 84. In some embodiments, the Spike protein does not comprise the amino acid sequence RXXR at positions corresponding to amino acids 884–887 of SEQ ID NO: 84. In some embodiments, one or more arginine (R) residues at positions corresponding to amino acids 748, 751, 884, and 887 of SEQ ID NO: 84 are mutated in the Spike protein. In some embodiments, the Spike protein does not comprise a basic amino acid at 1, 2, 3, or 4 positions corresponding to amino acids 748, 751, 884, and 887 of SEQ ID NO: 84. In some embodiments, the Spike protein comprises a neutral amino acid (e.g., glycine, alanine, or serine) at 1, 2, 3, or 4 positions corresponding to amino acids 748, 751, 884, and 887 of SEQ ID NO: 84. In some embodiments, the Spike protein comprises glycine residues at positions corresponding to 748 and 751 of SEQ ID NO: 84. In some embodiments, the Spike protein comprises glycine residues at positions corresponding to 884 and 887 of SEQ ID NO: 84. In some embodiments, the Spike protein does not comprise an active S1/S2 cleavage site. In some embodiments, the Spike protein does not comprise an active S2′ cleavage site. Any suitable method may be used to determine whether an S1/S2 or S2′ cleavage site is active. See, e.g., Millet and Whittaker, Proc Natl Acad Sci U S A. 2014.111(42):15214–15219. In some embodiments, the Spike protein has an amino acid sequence with at least 90%, at least 95%, at least 96%, at least 97%, at least 98%, or at least 99% identity to the amino acid sequence of SEQ ID NO: 90, and does not comprise the amino acid sequence RSVR (SEQ ID NO: 45) at positions corresponding to amino acids 748–751 of SEQ ID NO: 84. In some embodiments, the Spike protein has an amino acid sequence with at least 90%, at least 95%, at least 96%, at least 97%, at least 98%, or at least 99% identity to the amino acid sequence of SEQ ID NO: 90, and does not comprise the amino acid sequence RSAR (SEQ ID NO: 46) at positions corresponding to amino acids 748–751 of SEQ ID NO: 84. In some embodiments, a Spike protein, e.g., an encoded Spike protein antigen, comprises a mutated, truncated, or deleted endoplasmic reticulum (ER) retention signal. ER retention signals, located at the C-terminus of betacoronavirus S proteins, allow retrieval of S proteins from the Golgi apparatus to the ER in retrograde, resulting in intracellular retention of the S protein. Without wishing to be bound by theory, it is expected that mutation of the ER retention signal increases expression of encoded Spike proteins on the cell surface, allowing increased exposure of the Spike protein and enhanced immunogenicity. ER retention signals include dibasic (KXHXX) amino acid sequences at the C-terminus of a protein (e.g., KXHXX-COOH). Thus, in some embodiments, a lysine (K) residue at a position corresponding to amino acid 1349, or a histidine (H) residue at a position corresponding to amino acid 1351, of SEQ ID NO: 84, is substituted or deleted in the Spike protein. In some embodiments, the Spike protein does not comprise a lysine residue at a position corresponding to amino acid 1349 of SEQ ID NO: 84. In some embodiments, the Spike protein does not comprise a histidine residue at a position corresponding to amino acid 1351 of SEQ ID NO: 84. In some embodiments, the Spike protein does not comprise the amino acid sequence KXH within the 20, 15, 10, or 5 C-terminal amino acids of the Spike protein. In some embodiments, the Spike protein does not comprise the amino acid sequence KXH. In some embodiments, the Spike protein does not comprise the amino acid sequence KXHXX. In some embodiments, the Spike protein has an amino acid sequence with at least 90%, at least 95%, at least 96%, at least 97%, at least 98%, or at least 99% identity to the amino acid sequence of SEQ ID NO: 91, and does not comprise the amino acid sequence KXHXX at its C-terminus. In some embodiments, a Spike protein, e.g., an encoded Spike protein antigen, has a truncated C-terminus. A Spike protein with a truncated C-terminus refers to a Spike protein that lacks one or more amino acids that are present at the C-terminus of a wild-type (naturally occurring) Spike protein. In some embodiments, the Spike protein comprises a truncated cytoplasmic tail. In some embodiments, the Spike protein has a truncated cytoplasmic tail relative to a wild-type MERS-CoV Spike protein. For example, a wild-type MERS-CoV Spike protein having the amino acid sequence of SEQ ID NO: 84 comprises a cytoplasmic tail that is 36 amino acids (amino acids 1318–1353 of SEQ ID NO: 84), and so a modified Spike protein with a truncated C-terminus relative to SEQ ID NO: 84 comprises (i) a cytoplasmic tail with fewer than 36 amino acids; or (ii) no cytoplasmic tail. In some embodiments, a Spike protein comprises a cytoplasmic tail that comprises no more than 35, no more than 30, no more than 25, no more than 20, no more than 15, no more than 10, no more than 9, no more than 8, no more than 7, no more than 6, no more than 5, no more than 4, no more than 3, no more than 2, or no more than 1 amino acid. In some embodiments, the cytoplasmic tail comprises no more than 21 amino acids. In some embodiments, a Spike protein comprises a cytoplasmic tail that is 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, or 35 amino acids in length. In some embodiments, a Spike protein comprises a cytoplasmic tail that is 1–5, 5–10, 10–15, 15–20, 20–25, 25–30, or 30–35 amino acids in length. In some embodiments, the Spike protein comprises a cytoplasmic tail comprising no more than 35 amino acids. In some embodiments, the Spike protein comprises a cytoplasmic tail comprising no more than 30 amino acids. In some embodiments, the Spike protein comprises a cytoplasmic tail comprising no more than 25 amino acids. In some embodiments, the Spike protein comprises a cytoplasmic tail comprising no more than 21 amino acids. In some embodiments, the Spike protein comprises a cytoplasmic tail comprising no more than 20 amino acids. In some embodiments, the Spike protein comprises a cytoplasmic tail comprising no more than 10 amino acids. In some embodiments, the Spike protein comprises a cytoplasmic tail comprising no more than 8 amino acids. In some embodiments, the Spike protein comprises a cytoplasmic tail comprising no more than 6 amino acids. In some embodiments, the Spike protein comprises a cytoplasmic tail comprising no more than 5 amino acids. In some embodiments, the Spike protein comprises a cytoplasmic tail comprising no more than 4 amino acids. In some embodiments, the Spike protein comprises a cytoplasmic tail comprising no more than 3 amino acids. In some embodiments, the Spike protein comprises a cytoplasmic tail comprising no more than 2 amino acids. In some embodiments, the Spike protein comprises a cytoplasmic tail comprising no more than 1 amino acid. Truncations may be introduced by deleting one or more amino acids from the C-terminus of the cytoplasmic tail (e.g., amino acids 1318–1353 or 1339–1353 of SEQ ID NO: 84). In some embodiments, the Spike protein as described herein, or encoded by an RNA as described herein, lacks one or more amino acids present at the C-terminus of a wild-type sequence. In some embodiments, the Spike protein lacks 1–35, 1–30, 1–25, 1–20, 1–15, 1–10, 1–5, 5–35, 5–30, 5– 25, 5–20, 5–15, 5–10, 10–35, 10–30, 10–25, 10–20, 10–15, 15–35, 15–30, 15–25, 15–20, 20–35, 20–30, 20–25, 25–35, 25–30, or 30–35 amino acids that are present at the C-terminus of a wild- type MERS-CoV S protein amino acid sequence. In some embodiments, a Spike protein as described herein, or encoded by an RNA as described herein, has a truncated cytoplasmic tail relative to SEQ ID NO: 84. In some embodiments, the Spike protein comprises an extracellular domain and a transmembrane domain, and has a truncated cytoplasmic tail relative to SEQ ID NO: 84. In some embodiments, the Spike protein has an amino acid sequence with at least 90%, at least 95%, at least 96%, at least 97%, at least 98%, or at least 99% identity to the amino acid sequence of SEQ ID NO: 92, and has a truncated cytoplasmic tail relative to SEQ ID NO: 84. In some embodiments, a Spike protein, e.g., an encoded Spike protein antigen, has the amino acid sequence of SEQ ID NO: 93. In some embodiments, the Spike protein has an amino acid sequence with at least 90%, at least 95%, at least 96%, at least 97%, at least 98%, or at least 99% identity to the amino acid sequence of SEQ ID NO: 93, comprises proline residues at positions corresponding to amino acids 1060 and 1061 of SEQ ID NO: 84, and does not comprise the amino acid sequence RSVR (SEQ ID NO: 45) at positions corresponding to 748– 751 of SEQ ID NO: 84. In some embodiments, the Spike protein is stabilized in a prefusion conformation. In some embodiments, the Spike protein does not comprise an active S1/S2 cleavage site. In some embodiments, a Spike protein, e.g., an encoded Spike protein antigen, has the amino acid sequence of SEQ ID NO: 94. In some embodiments, the Spike protein has an amino acid sequence with at least 90%, at least 95%, at least 96%, at least 97%, at least 98%, or at least 99% identity to the amino acid sequence of SEQ ID NO: 94, does not comprise the amino acid sequence RSVR (SEQ ID NO: 45) at positions corresponding to amino acids 748–751 of SEQ ID NO: 84, and does not comprise the amino acid sequence KXHXX at its C-terminus. In some embodiments, the Spike protein does not comprise an active S1/S2 cleavage site. In some embodiments, the Spike protein does not comprise an ER retention signal. In some embodiments, a Spike protein, e.g., an encoded Spike protein antigen, has the amino acid sequence of SEQ ID NO: 95. In some embodiments, the Spike protein has an amino acid sequence with at least 90%, at least 95%, at least 96%, at least 97%, at least 98%, or at least 99% identity to the amino acid sequence of SEQ ID NO: 95, does not comprise the amino acid sequence RSVR (SEQ ID NO: 45) at positions corresponding to amino acids 748–751 of SEQ ID NO: 84, and lacks one or more amino acids corresponding to amino acids 1339–1353 of SEQ ID NO: 84. In some embodiments, the Spike protein does not comprise an active S1/S2 cleavage site. In some embodiments, the Spike protein has a truncated cytoplasmic tail relative to SEQ ID NO: 84. In some embodiments, a Spike protein, e.g., an encoded Spike protein antigen, has no greater than 100, no greater than 90, no greater than 80, no greater than 70, no greater than 60, no greater than 50, no greater than 40, no greater than 30, no greater than 20, no greater than 10, or no greater than 5 amino acid substitutions and/or deletions as compared to (when aligned with) a Spike protein having the amino acid sequence as provided in any one of SEQ ID NOs: 93–95. Domains and Fusion Proteins Some aspects of the disclosure relate to MERS-CoV fusion proteins and RNAs (e.g., mRNAs or self-amplifying RNAs) encoding MERS-CoV fusion proteins, which comprise (i) an N-terminal domain (NTD) of a full-length MERS-CoV Spike protein, and/or (ii) a receptor- binding domain (RBD) of a full-length MERS-CoV Spike protein, and do not comprise an NTD or RBD of a different human coronavirus that is not MERS-CoV (i.e., the protein does not comprise an NTD, and does not comprise an RBD, of a Spike protein of any of the following coronaviruses: SARS-CoV, SARS-CoV-2, 229E-CoV, NL63-CoV, OC43-CoV, and HKU1-CoV). In some embodiments, the NTD and RBD are from the same full-length MERS-CoV Spike protein. In some embodiments, the NTD and RBD are from different full-length MERS-CoV Spike proteins. The S1 and S2 subunits of the MERS-CoV Spike protein further include domains readily discernable by structure and function, which in turn can be featured in designing antigens to be encoded by the nucleic acid vaccines, in particular, mRNA vaccines. Within the S1 subunit, domains include the N-terminal domain (NTD) and the receptor-binding domain (RBD), said RBD domain further including a receptor-binding motif (RBM) Within the S2 subunit, domains include fusion peptide (FP), heptad repeat 1 (HR1), heptad repeat 2 (HR2), transmembrane domain (TM), and cytoplasm domain, also known as cytoplasmic tail (CT). The HR1 and HR2 domains can be referred to as the “fusion core region” of MERS-CoV Spike protein. The S1 subunit includes an N terminal domain (NTD), a linker region, a receptor binding domain (RBD), a first subdomain (SD1), and a second subdomain (SD2). The S2 subunit includes, inter alia, a first heptad repeat (HR1), a second heptad repeat (HR2), a transmembrane domain (TM), and a cytoplasmic tail. The NTD and RBD of S1 have been shown to be the targets of neutralizing antibodies in betacoronavirus-infected individuals. As used herein, for example, in the context of an antigen design (said antigen encoded by an RNA and to be expressed, for example, from an RNA vaccine), the term “N-terminal domain” or “NTD” refers to a domain within the MERS-CoV S1 subunit comprising approximately 321 amino acids in length, having identity to amino acids 18–338 of the S1 subunit of the Spike protein having the amino acid sequence set forth as SEQ ID NO: 84. As used herein, for example, in the context of an antigen design (said antigen encoded by an mRNA and to be expressed, for example, from an mRNA vaccine), the term “receptor binding domain” or “RBD” refers to a domain within the S1 subunit of MERS-CoV comprising approximately 214 amino acids in length, having identity to amino acids 376–589 of the Spike protein having the amino acid sequence set forth as SEQ ID NO: 84. As used herein, the term “receptor binding motif” (RBM) refers to the portion of the RBD that directly contacts the DPP4 receptor. Expressed RBDs are predicted to specifically bind to dipeptidyl peptidase 4 (DPP4) as its receptor and/or specifically react with RBD-binding and/or neutralizing antibodies, e.g., KNIH90-F1. The compositions provided herein include RNAs (e.g., mRNAs) that may encode any one or more full-length or partial (truncated or other deletion of sequence) S protein subunit (e.g., S1 or S2 subunit), one or more domain or combination of domains of an S protein subunit (e.g., NTD, RBD, or NTD-RBD fusions, with or without an SD1 and/or SD2), or chimeras of full- length or partial and S2 protein subunits. Other S protein subunit and/or domain configurations are contemplated herein. Exemplary sequences of proteins comprising one or more domains of a MERS-CoV Spike protein (e.g., NTD-RBD fusion proteins) as described herein are provided in EXEMPLARY SEQUENCES as SEQ ID NOS: 96–98. In some embodiments, an RNA comprises an open reading frame encoding a protein having at least 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity to an amino acid sequence of SEQ ID NO: 96. In some embodiments, an RNA comprises an open reading frame encoding a protein having at least 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity to an amino acid sequence of SEQ ID NO: 97. In some embodiments, an RNA comprises an open reading frame encoding a protein having at least 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity to an amino acid sequence of SEQ ID NO: 98. Any pair of protein portions (e.g., NTD and RBD, NTD and TM domain, RBD and TM domain) may be contiguous in a protein as described herein (e.g., without an intervening amino acid sequence), or the two portions may be separated by a linker. The linker may be a 2A or GS linker described herein in the section entitled “Linkers and Cleavable Peptides.” The linker may also be another linker known in the art. A linker of a fusion protein may be present in place of an internal truncation relative to a wild-type sequence of a MERS-CoV Spike protein (e.g., the amino acid sequence of the protein comprises deletion of one or more amino acids, relative to the wild-type sequence, and insertion of the linker at the position previously occupied by the deleted amino acids). In some embodiments, the linker comprises 2–10 glycine residues. In some embodiments, the linker comprises 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, or 20 glycine residues. In some embodiments, the linker comprises 2–20, 2–15, 2–10, 2–5, 2–3, 3– 5, 5–7, 7–10, 10–15, 15–20, 3–10, 4–8, or 5–6 glycine residues. In some embodiments, the linker comprises 5–6 glycine residues. Where multiple linkers separate multiple pairs of portions, the multiple linkers may each comprise the same amino acid sequence (e.g., AAY). Multiple linkers may comprise different amino acid sequences (e.g., a first linker comprises the amino acid sequence GGS, and a second linker comprises the amino acid sequence GGS). Linkers connecting different pairs of portions may be the same length, or different lengths. In some embodiments, two or more portions of a protein portion are connected by a glycine linker or a glycine-serine linker. In some embodiments, each pair of protein portions of a protein are connected by a glycine linker or a glycine-serine linker. In some embodiments, two or more portions of a protein are connected by a linker comprising the amino acid sequence AAY. In some embodiments, each pair of protein portions are connected by the amino acid sequence AAY. In some embodiments, no linkers are present between protein portions in the protein. In some embodiments, a protein comprising one or more domains of a MERS-CoV Spike protein comprises a transmembrane (TM) domain. In some embodiments, the TM domain is a TM domain of a coronavirus. In some embodiments, the TM domain is a TM domain of a full- length MERS-CoV Spike protein. The full-length MERS-CoV Spike protein may the same Spike protein from which the NTD and/or RBD are derived, or a different full-length MERS-CoV Spike protein. An exemplary MERS-CoV Spike protein TM domain sequence is provided as SEQ ID NO: 101. In some embodiments, the TM domain is a TM domain is from a protein that is heterologous to MERS-CoV (i.e., a TM domain of a protein that is not encoded by a MERS-CoV genome). For example, the transmembrane domain may be from an influenza hemagglutinin transmembrane domain, which has been demonstrated to effectively anchor proteins at the cell surface. In some embodiments, the TM domain is an influenza virus hemagglutinin (HA) TM domain. The influenza virus HA protein may have a sequence from any influenza virus species (e.g., influenza A virus, influenza B virus, or influenza C virus) or HA subtype (e.g., H1, H2, H3, H4, H5, H6, H7, H8, H9, H10, H11, H12, H13, H14, H15, H16, H17, or H18). An exemplary influenza A virus HA TM domain sequence is provided as SEQ ID NO: 102. In some aspects, a protein as described herein or encoded by an RNA as described herein consists essentially of an NTD of a full-length MERS-CoV Spike protein, an RBD of a full- length MERS-CoV, and a TM domain. A protein consisting essentially of NTD-RBD-TM domains may include a linker between the NTD and RBD. A protein consisting essentially of NTD-RBD-TM domains may include a linker between the RBD and TM domain. Each pair of the NTD-RBD and RBD-TM domains may be connected by a linker. The NTD may include a signal peptide of the full-length MERS-CoV Spike protein. In some embodiments, the protein includes a signal peptide from a protein other than the MERS-CoV Spike protein. In some embodiments, the protein comprises the amino acid sequence of SEQ ID NO: 96. In some embodiments, a protein as described herein or encoded by an RNA as described herein comprises an NTD of a full-length MERS-CoV Spike protein, an RBD of a full-length MERS-CoV, and a TM domain. The NTD and RBD may be connected by a linker. The RBD and TM domain may be connected by a linker. In some embodiments, the protein comprises the amino acid sequence of SEQ ID NO: 96. In some embodiments, the protein has no greater than 100, no greater than 90, no greater than 80, no greater than 70, no greater than 60, no greater than 50, no greater than 40, no greater than 30, no greater than 20, no greater than 10, or no greater than 5 amino acid substitutions and/or deletions as compared to (when aligned with) a protein having the amino acid sequence of SEQ ID NO: 96. In some embodiments, the fusion protein has an amino acid sequence with at least 90%, at least 95%, at least 96%, at least 97%, at least 98%, or at least 99% identity to the amino acid sequence of SEQ ID NO: 96, and comprises an NTD and RBD of a full-length MERS-CoV Spike protein and a TM domain. In some embodiments, the full-length MERS-CoV Spike protein comprises the amino acid sequence of SEQ ID NO: 84. In some embodiments, the full-length MERS-CoV Spike protein comprises the amino acid sequence of SEQ ID NO: 85. In some embodiments, the full-length MERS-CoV Spike protein comprises the amino acid sequence of SEQ ID NO: 86. In some aspects, a protein as described herein or encoded by an RNA as described herein consists essentially of an NTD of a full-length MERS-CoV Spike protein, a TM domain. A protein consisting essentially of NTD and TM domains may include a between the NTD and TM domain. In some embodiments, the protein comprises the amino acid sequence of SEQ ID NO: 97. The NTD may include a signal peptide of the full-length MERS-CoV Spike protein. In some embodiments, the protein includes a signal peptide from a protein other than the MERS-CoV Spike protein. In some embodiments, a protein as described herein or encoded by an RNA as described herein comprises an NTD of a full-length MERS-CoV Spike protein and a TM domain. The NTD and TM domain may be connected by a linker. In some embodiments, the protein comprises the amino acid sequence of SEQ ID NO: 97. In some embodiments, the protein has no greater than 100, no greater than 90, no greater than 80, no greater than 70, no greater than 60, no greater than 50, no greater than 40, no greater than 30, no greater than 20, no greater than 10, or no greater than 5 amino acid substitutions and/or deletions as compared to (when aligned with) a protein having the amino acid sequence of SEQ ID NO: 97. In some embodiments, the fusion protein has an amino acid sequence with at least 90%, at least 95%, at least 96%, at least 97%, at least 98%, or at least 99% identity to the amino acid sequence of SEQ ID NO: 97, and comprises an NTD of a full-length MERS-CoV Spike protein and a TM domain. In some embodiments, the full-length MERS-CoV Spike protein comprises the amino acid sequence of SEQ ID NO: 84. In some embodiments, the full-length MERS-CoV Spike protein comprises the amino acid sequence of SEQ ID NO: 85. In some embodiments, the full-length MERS-CoV Spike protein comprises the amino acid sequence of SEQ ID NO: 86. In some aspects, a protein as described herein or encoded by an RNA as described herein consists essentially of an RBD of a full-length MERS-CoV Spike protein and a TM domain. A protein consisting essentially of RBD and TM domains may include a linker between the RBD and TM domain. In some embodiments, the protein includes a signal peptide. The signal peptide may be from a MERS-CoV Spike protein. In some embodiments, the protein includes a signal peptide from a protein other than the MERS-CoV Spike protein. In some embodiments, the protein comprises the amino acid sequence of SEQ ID NO: 98. In some embodiments, a protein as described herein or encoded by an RNA as described herein comprises an RBD of a full-length MERS-CoV Spike protein and a TM domain. The RBD and TM domain may be connected by a linker. In some embodiments, the protein comprises the amino acid sequence of SEQ ID NO: 98. In some embodiments, the protein has no greater than 100, no greater than 90, no greater than 80, no greater than 70, no greater than 60, no greater than 50, no greater than 40, no greater than 30, no greater than 20, no greater than 10, or no greater than 5 amino acid substitutions and/or deletions as compared to (when aligned with) a protein having the amino acid sequence of SEQ ID NO: 98. In some embodiments, the fusion protein has an amino acid sequence with at least 90%, at least 95%, at least 96%, at least 97%, at least 98%, or at least 99% identity to the amino acid sequence of SEQ ID NO: 98 and comprises an RBD of a full-length MERS-CoV Spike protein and a TM domain. In some embodiments, the full-length MERS-CoV Spike protein comprises the amino acid sequence of SEQ ID NO: 84. In some embodiments, the full-length MERS-CoV Spike protein comprises the amino acid sequence of SEQ ID NO: 85. In some embodiments, the full-length MERS-CoV Spike protein comprises the amino acid sequence of SEQ ID NO: 86. Variants In some embodiments, the compositions of the present disclosure include RNA that encodes a MERS-CoV antigen variant. Antigen variants or other polypeptide variants refers to molecules that differ in their amino acid sequence from a wild-type, native, or naturally occurring sequence. The antigen/polypeptide variants may possess substitutions, deletions, and/or insertions at certain positions within the amino acid sequence, as compared to a wild-type, native or naturally occurring sequence. Examples of MERS-CoV protein variants are provided in the Sequence Listing. Ordinarily, variants possess at least 50% identity to a wild-type, native or naturally occurring sequence. In some embodiments, variants share at least 80%, or at least 90% identity with a wild-type, native, or naturally occurring sequence. In some embodiments, the nucleic acid vaccines described herein encode MERS-CoV variants comprising 1, 2, 3, 4, or more mutations relative to a naturally occurring sequence. In some embodiments, the nucleic acid vaccines described herein encode MERS-CoV variants comprising less than 20, 18, 15, 12, or 10 mutations relative to a naturally occurring sequence. In some embodiments, the nucleic acid vaccines described herein encode MERS-CoV variants having 1-501-40, 1-30, 1-25, 1-20, 1-15, 1-10, 5-50, 5-40, 5-30, 5-25, 5-20, 5-15, 5-10, 10-50, 10-40, 10-30, 10-25, 10-20, 10-15, 20-50, 20-40, 20-30, 20-25, 25-50, 25-40, 25-30, 30-50, 30-40, 40-50 mutations (e.g., substitutions). As used herein, “mutation” refers to an amino acid substitution, insertion, or deletion, relative to a comparator sequence. Variant antigens/polypeptides encoded by nucleic acids of the disclosure may contain amino acid changes that confer any of a number of desirable properties, e.g., that enhance their immunogenicity, enhance their expression, and/or improve their stability or PK/PD properties in a subject. Variant antigens/polypeptides can be made using routine mutagenesis techniques and assayed as appropriate to determine whether they possess the desired property. Assays to determine expression levels and immunogenicity are well known in the art and exemplary such assays are set forth in the Examples section. Similarly, PK/PD properties of a protein variant can be measured using art recognized techniques, e.g., by determining expression of antigens in a vaccinated subject over time and/or by looking at the durability of the induced immune response. The stability of protein(s) encoded by a variant nucleic acid may be measured by assaying thermal stability or stability upon urea denaturation or may be measured using in silico prediction. Methods for such experiments and in silico determinations are known in the art. In some embodiments, a composition comprises an RNA or an RNA ORF that comprises a nucleotide sequence of any one of the sequences provided herein, or comprises a nucleotide sequence at least 90%, at least 95%, at least 96%, at least 97%, at least 98%, or at least 99% identical to a nucleotide sequence of any one of the sequences provided herein. The term “identity” refers to a relationship between the sequences of two or more polypeptides (e.g. antigens) or polynucleotides (nucleic acids), as determined by comparing the sequences. Identity also refers to the degree of sequence relatedness between or among sequences as determined by the number of matches between strings of two or more amino acid residues or nucleic acid residues. Identity measures the percent of identical matches between the smaller of two or more sequences with gap alignments (if any) addressed by a particular mathematical model or computer program (e.g., “algorithms”). Identity of related antigens or nucleic acids can be readily calculated by known methods. “Percent (%) identity” as it applies to polypeptide or polynucleotide sequences is defined as the percentage of residues (amino acid residues or nucleic acid residues) in the candidate amino acid or nucleic acid sequence that are identical with the residues in the amino acid sequence or nucleic acid sequence of a second sequence after aligning the sequences and introducing gaps, if necessary, to achieve the maximum percent identity. Methods and computer programs for the alignment are well known in the art. It is understood that identity depends on a calculation of percent identity but may differ in value due to gaps and penalties introduced in the calculation. Generally, variants of a particular polynucleotide or polypeptide (e.g., antigen) have at least 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, 99% but less than 100% sequence identity to that particular comparator polynucleotide or polypeptide as determined by sequence alignment programs and parameters described herein and known to those skilled in the art. Such tools for alignment include those of the BLAST suite (Stephen F. Altschul, et al (1997), "Gapped BLAST and PSI-BLAST: a new generation of protein database search programs", Nucleic Acids Res.25:3389-3402). Another popular local alignment technique is based on the Smith-Waterman algorithm (Smith, T.F. & Waterman, M.S. (1981) “Identification of common molecular subsequences.” J. Mol. Biol. 147:195-197). A general global alignment technique based on dynamic programming is the Needleman–Wunsch algorithm (Needleman, S.B. & Wunsch, C.D. (1970) “A general method applicable to the search for similarities in the amino acid sequences of two proteins.” J. Mol. Biol.48:443-453). More recently a Fast Optimal Global Sequence Alignment Algorithm (FOGSAA) has been developed that purportedly produces global alignment of nucleotide and protein sequences faster than other optimal global alignment methods, including the Needleman– Wunsch algorithm. As such, polynucleotides encoding peptides or polypeptides containing substitutions, insertions and/or additions, deletions and covalent modifications with respect to reference sequences, in particular the polypeptide (e.g., antigen) sequences disclosed herein, are included within the scope of this disclosure. For example, sequence tags or amino acids, such as one or more lysines, can be added to peptide sequences (e.g., at the N-terminal or C-terminal ends). Sequence tags can be used for peptide detection, purification or localization. Lysines can be used to increase peptide solubility or to allow for biotinylation. Alternatively, amino acid residues located at the carboxy and amino terminal regions of the amino acid sequence of a peptide or protein may optionally be deleted providing for truncated sequences. Certain amino acids (e.g., C-terminal or N-terminal residues) may alternatively be deleted depending on the use of the sequence, as for example, expression of the sequence as part of a larger sequence which is soluble or linked to a solid support. In some embodiments, sequences for (or encoding) signal sequences, termination sequences, transmembrane domains, linkers, multimerization domains (such as, e.g., foldon regions) and the like may be substituted with alternative sequences that achieve the same or a similar function. In some embodiments, cavities in the core of proteins can be filled to improve stability, e.g., by introducing larger amino acids. In other embodiments, buried hydrogen bond networks may be replaced with hydrophobic resides to improve stability. In yet other embodiments, glycosylation sites may be removed and replaced with appropriate residues. Such sequences are readily identifiable to one of skill in the art. It should also be understood that some of the sequences provided herein contain sequence tags or terminal peptide sequences (e.g., at the N-terminal or C-terminal ends) that may be deleted, for example, prior to use in the preparation of an mRNA vaccine. As recognized by those skilled in the art, protein fragments, functional protein domains, and homologous proteins are also considered to be within the scope of coronavirus antigens of interest. For example, provided herein is any protein fragment (meaning a polypeptide sequence at least one amino acid residue shorter than a full-length antigen sequence but otherwise identical) of a full-length protein, provided that the fragment is immunogenic and confers a protective immune response to the coronavirus. In addition to variants that are identical to the full-length protein but are truncated, in some embodiments, an antigen includes 2, 3, 4, 5, 6, 7, 8, 9, 10, or more mutations, as shown in any of the sequences provided or referenced herein. Antigens/antigenic polypeptides can range in length from about 4, 6, or 8 amino acids to full length proteins. Linkers and Cleavable Peptides In some embodiments, an RNA (e.g., mRNA) that encodes a protein further encodes a linker located between at least two, or between each domain of the protein. The linker may be, for example, a cleavable linker or protease-sensitive linker. In some embodiments, the linker is selected from the group consisting of F2A linker, P2A linker, T2A linker, E2A linker, and combinations thereof (see, e.g., WO 2017/127750). This family of self-cleaving peptide linkers, referred to as 2A peptides, has been described in the art (see, e.g., Kim, J.H. et al. PLoS ONE 2011;6:e18556). In some embodiments, the linker is an F2A linker. In some embodiments, the linker is a GS linker. GS linkers are polypeptide linkers that include glycine and serine amino acids repeats. They comprise flexible and hydrophilic residues and can be used to perform fusion of protein subunits without interfering in the folding and function of the protein domains, and without formation of secondary structures. In some embodiments, an RNA (e.g., mRNA) encodes a protein that comprises a GS linker that is 3 to 20 amino acids long. For example, the GS linker may have a length of (or have a length of at least) 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, or 20 amino acids. In some embodiments, a GS linker is (or is at least) 15 amino acids long (e.g., GGSGGSGGSGGSGGG (SEQ ID NO: 47)). In some embodiments, a GS linker is (or is at least) 8 amino acids long (e.g., GGGSGGGS (SEQ ID NO: 48)). In some embodiments, a GS linker is (or is at least) 7 amino acids long (e.g., GGGSGGG (SEQ ID NO: 49)). In some embodiments, a GS linker comprises the amino acid sequence GGGSGG (SEQ ID NO: 50). In some embodiments, a GS linker is (or is at least) 4 amino acid long (e.g., GGGS (SEQ ID NO: 51)). In some embodiments, the GS linker comprises (GGGS)n (SEQ ID NO: 51), where n is any integer from 1-5. In some embodiments, a GS linker is (or is at least) 4 amino acid long (e.g., GSGG (SEQ ID NO: 52)). In some embodiments, the GS linker comprises (GSGG)n (SEQ ID NO: 52), where n is any integer from 1-5. In some embodiments, a GS linker comprises the amino acid sequence SGGGS (SEQ ID NO: 53). In some embodiments, a linker is a glycine linker, for example having a length of (or a length of at least) 3 amino acids (e.g., GGG). In some embodiments, a protein encoded by an RNA (e.g., mRNA) includes two or more linkers, which may be the same or different from each other. In some embodiments, a linker comprises a pan-HLA-DR-binding epitope (PADRE). See, e.g., Alexander et al., J Immunol.2000.164(3):1625–1633. The skilled artisan will appreciate that other art-recognized linkers may be suitable for use in the constructs of the disclosure (e.g., encoded by the nucleic acids of the disclosure). The skilled artisan will likewise appreciate that other polycistronic constructs (RNA (e.g., mRNA) encoding more than one protein separately within the same molecule) may be suitable for use as provided herein. Signal Peptides In some embodiments, an RNA (e.g., mRNA) has an ORF that encodes a signal peptide fused to a protein. Signal peptides, comprising the N-terminal 15-60 amino acids of proteins, are typically involved for the translocation across the membrane on the secretory pathway and, thus, control the entry of proteins both in eukaryotes and prokaryotes to the secretory pathway. In eukaryotes, the signal peptide of a nascent precursor protein (pre-protein) directs the ribosome to the rough endoplasmic reticulum (ER) membrane and initiates the transport of the growing peptide chain across it for processing. ER processing produces mature proteins, wherein the signal peptide is cleaved from precursor proteins, typically by an ER-resident signal peptidase of the host cell, or they remain uncleaved and function as a membrane anchor. A signal peptide may also facilitate the targeting of the protein to the cell membrane. A signal peptide may have a length of 15-60 amino acids. For example, a signal peptide may have a length of 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49, 50, 51, 52, 53, 54, 55, 56, 57, 58, 59, or 60 amino acids. In some embodiments, a signal peptide has a length of 20-60, 25-60, 30-60, 35- 60, 40-60, 45- 60, 50-60, 55-60, 15-55, 20-55, 25-55, 30-55, 35-55, 40-55, 45-55, 50-55, 15-50, 20-50, 25-50, 30-50, 35-50, 40-50, 45-50, 15-45, 20-45, 25-45, 30-45, 35-45, 40-45, 15-40, 20- 40, 25-40, 30-40, 35-40, 15-35, 20-35, 25-35, 30-35, 15-30, 20-30, 25-30, 15-25, 20-25, or 15-20 amino acids. Signal peptides from heterologous genes (which regulate expression of genes other than MERS-CoV proteins in nature) are known in the art and can be tested for desired properties and then incorporated into a nucleic acid of the disclosure. In some embodiments, an RNA (e.g., mRNA) comprises an open reading frame that encodes a protein fused to a signal peptide comprising an amino acid sequence that has at least 50%, at least 60%, at least 70%, at least 75%, at least 80%, at least 85%, at least 90%, or at least 95% identity to the amino acid sequence of any one of the sequences provided herein, such as those reproduced below in Table 3. In some embodiments, an RNA comprises an open reading frame that encodes MERS-CoV protein including an endogenous signal peptide of the wild-type protein (e.g., an mRNA encoding a (wild-type or modified) MERS-CoV protein or variant thereof encodes a MERS-CoV S protein signal peptide). Nucleic Acids Encoding MERS-CoV Proteins Nucleic acids comprise a polymer of nucleotides (nucleotide monomers). Thus, nucleic acids are also referred to as polynucleotides. Nucleic acids may be or may include, for example, deoxyribonucleic acid (DNA), ribonucleic acid (RNA), threose nucleic acid (TNA), glycol nucleic acid (GNA), peptide nucleic acid (PNA), locked nucleic acid (LNA, including LNA having a β-D-ribo configuration, α-LNA having an α-L-ribo configuration (a diastereomer of LNA), 2′-amino-LNA having a 2′-amino functionalization, and 2′-amino- α-LNA having a 2′- amino functionalization), ethylene nucleic acid (ENA), cyclohexenyl nucleic acid (CeNA) and/or chimeras and/or combinations thereof. RNA (e.g., mRNA) of the present disclosure comprises an open reading frame (ORF) encoding a MERS-CoV protein or variant thereof. In some embodiments, the RNA (e.g., mRNA) further comprises a 5 ^ untranslated region (UTR), 3 ^ UTR, a poly(A) tail and/or a 5 ^ cap analog. Messenger RNA (mRNA) Messenger RNA (mRNA) is RNA that encodes a (at least one) protein (a naturally- occurring, non-naturally-occurring, or modified polymer of amino acids) and can be translated to produce the encoded protein in vitro, in vivo, in situ, or ex vivo. It is understood that mRNA is not self-amplifying RNA (saRNA) (see, e.g., Bloom K et al. Gene Therapy 2021; 28: 117–129 for a comparison of mRNA and saRNA). saRNAs include alphavirus replicase sequences that encode an RNA-dependent RNA polymerase. mRNA does not include alphavirus replicase sequences. The skilled artisan will appreciate that, except where otherwise noted, nucleic acid sequences set forth in the instant application may recite “T”s in a representative DNA sequence but where the sequence represents mRNA, the “T”s would be substituted for “U”s. Thus, any of the DNAs disclosed and identified by a particular sequence identification number herein also disclose the corresponding mRNA sequence complementary to the DNA, where each “T” of the DNA sequence is substituted with “U.” Naturally-occurring eukaryotic mRNA molecules can contain stabilizing elements, including, but not limited to, UTRs at their 5′-end (5′ UTR) and/or at their 3′-end (3′ UTR), in addition to other structural features, such as a 5′-cap structure or a 3′-poly(A) tail. Both the 5′ UTR and the 3′ UTR are typically transcribed from the genomic DNA and are elements of the premature mRNA. Characteristic structural features of mature mRNA, such as the 5′-cap and the 3′-poly(A) tail are usually added to the transcribed (premature) mRNA during mRNA processing. Untranslated Regions (UTRs) The mRNAs of the present disclosure may comprise one or more regions or parts which act or function as an untranslated region. A “5′ untranslated region” (UTR) refers to a region of an mRNA that is directly upstream (i.e., 5′) from the start codon (i.e., the first codon of an mRNA transcript translated by a ribosome) that does not encode a polypeptide. A “3′ untranslated region” (UTR) refers to a region of an mRNA that is directly downstream (i.e., 3′) from the open reading frame (e.g., downstream from the last amino acid-encoding codon of an open reading frame, where the stop codon is considered part of the 3′ UTR, or downstream from the first stop codon signaling translation termination, where that stop codon is considered part of the open reading frame), and which does not encode a polypeptide. When RNA transcripts are being generated, the 5’ UTR may comprise a promoter sequence. Such promoter sequences are known in the art. It should be understood that such promoter sequences will not be present in a vaccine of the disclosure. Where mRNAs encode a (at least one) protein, the mRNA may comprise a 5’ UTR and/or 3’ UTR. UTRs of an mRNA are transcribed but not translated. In mRNA, the 5′ UTR starts at the transcription start site and continues to the start codon but does not include the start codon; the 3′ UTR starts immediately following the open reading frame and continues until the transcriptional termination signal. Where an open reading frame ends with a codon encoding an amino acid, the 3′ UTR begins with a stop codon, such that no amino acids are added to a polypeptide beyond the last amino acid encoded by the open reading frame. A 3′ UTR may further comprise one or more stop codons. There is a growing body of evidence about the regulatory roles played by the UTRs in terms of stability of the nucleic acid molecule and translation. The regulatory features of a UTR can be incorporated into the polynucleotides of the present disclosure to, among other things, enhance the stability of the molecule. The specific features can also be incorporated to ensure controlled down-regulation of the transcript in case they are misdirected to undesired organs sites. A variety of 5’ UTR and 3’ UTR sequences are known. In some embodiments, the 5′ UTR comprises a sequence provided in Table 1 or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to a 5′ UTR sequence provided in Table 1, or a variant or a fragment thereof. In some embodiments, the 3′ UTR comprises a sequence provided in Table 2 or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to a 3′ UTR sequence provided in Table 2, or a variant or a fragment thereof. It should also be understood that the mRNA of the present disclosure may include any 5’ UTR and/or any 3’ UTR. Exemplary UTR sequences include SEQ ID NOs: 1-44, 66-79 and 81- 82; however, other UTR sequences may be used or exchanged for any of the UTR sequences described herein. In some embodiments, a 5' UTR comprises a sequence selected from: GGGAAAUAAGAGAGAAAAGAAGAGUAAGAAGAAAUAUAAGAGCCACC (SEQ ID NO: 1), GGGAAAUAAGAGAGAAAAGAAGAGUAAGAAGAAAUAUAAGACCCCGGCGCCGCCACC (SEQ ID NO: 2), GAGGAAAUCGCAAAAUUUGCUCUUCGCGUUAGAUUUCUUUUAGUUUUCUCGCAACUAGCAAGCUUUUUGUUCUCGCC (SEQ ID NO: 66), and GGAAAUCGCAAAAUUUGCUCUUCGCGUUAGAUUUCUUUUAGUUUUCUCGCAACUAGCAAGCUUUUUGUUCUCGCC (SEQ ID NO: 67). In some embodiments, a 3′ UTR comprises, in 5′-to-3′ order: (a) the nucleic acid sequence UAAAGCUCCCCGGGGGCCUCGGUGGCCUAGCUUCUUGCCCCUUGGGCCUCCCCCCAGCCCCUCCUCCCCUUCCUGCAG (SEQ ID NO: 68), (b) an identification and ratio determination (IDR) sequence, and (c) the nucleic acid sequence UGGUCUUUGAAUAAAGUCUGAGUGGGCGGC (SEQ ID NO: 69). In some embodiments, each mRNA encoding a distinct protein (i.e., having a different amino acid sequence from proteins encoded by other mRNAs in a composition) comprises a 3′ UTR comprising, in 5′-to-3′ order: (a) the nucleotide sequence of SEQ ID NO: 68; (b) a distinct IDR sequence; and (c) the nucleotide sequence of SEQ ID NO: 69. IDR sequences are described herein in the section entitled “Identification and Ratio Determination (IDR) Sequences.” In some embodiments, a 5′ UTR comprises a sequence derived from a 5′ UTR of a gene selected from HSD17B4, RPL32, ASAH1, ATP5A1, MP68, NDUFA4, NOSIP, RPL31, SLC7A3, TUBB4B and UBQLN2. In some embodiments, the 5′ UTR comprises a sequence derived from the 5′ UTR of human hydroxysteroid 17-beta dehydrogenase 4 (HSD17B4). In some embodiments, a 5′ UTR comprises the sequence GGGAGAGUCCCGCAGUCGGCGUCCAGCGGCUCUGCUUGUUCGUGUGUGUGUCGUUGCAGGCCUUAUUCAAGCUUACC (SEQ ID NO: 70). In some embodiments, a 5′ UTR comprises the sequenceGUCCCGCAGUCGGCGUCCAGCGGCUCUGCUUGUUCGUGUGUGUGUCGUUGCAGGCCUUAUUC (SEQ ID NO: 71). In some embodiments, a 5′ UTR comprises the sequence GGGAGAAAGCUUACC (SEQ ID NO: 72). In some embodiments, a 3′ UTR comprises a sequence derived from a 3′ UTR of a gene selected from PSMB3, ALB7, alpha-globin, CASP1, COX6B1, GNAS, NDUFA1 and RPS9. In some embodiments, a 3′ UTR comprises a sequence derived from a 3′ UTR of PSMB3 (proteasome 20S subunit beta 3). In some embodiments, a 3′ UTR comprises a sequence derived from a 3′ UTR of alpha-globin (MUAG). In some embodiments, a 3′ UTR comprises the sequence AGGACUAGUCCCUGUUCCCAGAGCCCACUUUUUUUUCUUUUUUUGAAAUAAAAUAGCCUGUCUUUCAGAUCU (SEQ ID NO: 73). In some embodiments, a 3′ UTR comprises the sequenceGGACUAGUUAUAAGACUGACUAGCCCGAUGGGCCUCCCAACGGGCCCUCCUCCCCUCCUUGCACCGAGAUUAAU (SEQ ID NO: 74). In some embodiments, the mRNA comprises a 5′ UTR comprising the nucleotide sequence of any one of SEQ ID NOs: 70–72, an open reading frame, one or more stop codons, and a 3′ UTR comprising the nucleotide sequence of SEQ ID NO: 73 or SEQ ID NO: 74. In some embodiments, the mRNA further comprises a polyA sequence comprising at least 64 consecutive adenosine nucleotides. In some embodiments, the mRNA further comprises a polyC sequence comprising at least 30 consecutive cytidine nucleotides. In some embodiments, a 5′ UTR comprises the sequence GAAUAAACUAGUAUUCUUCUGGUCCCCACAGACUCAGAGAGAACCCGCCACC (SEQ ID NO: 75). In some embodiments, a 5′ UTR comprises the sequence GAGAAUAAACUAGUAUUCUUCUGGUCCCCACAGACUCAGAGAGAACCCGCCACC (SEQ ID NO: 76). In some embodiments, a 3′ UTR comprises the sequence CUGGUACUGCAUGCACGCAAUGCUAGCUGCCCCUUUCCCGUCCUGGGUACCCCGAGUCUCCCCCGACCUCGGGUCCCA GGUAUGCUCCCACCUCCACCUGCCCCACUCACCACCUCUGCUAGUUCCAGACACCUCCCAAGCACGCAGCAAUGCAGC UCAAAACGCUUAGCCUAGCCACACCCCCACGGGAAACAGCAGUGAUUAACCUUUAGCAAUAAACGAAAGUUUAACUAA GCUAUACUAACCCCAGGGUUGGUCAAUUUCGUGCCAGCCACACC (SEQ ID NO: 77). In some embodiments, a 3′ UTR comprises the sequence CUCGAGCUGGUACUGCAUGCACGCAAUGCUAGCUGCCCCUUUCCCGUCCUGGGUACCCCGAGUCUCCCCCGACCUCGG GUCCCAGGUAUGCUCCCACCUCCACCUGCCCCACUCACCACCUCUGCUAGUUCCAGACACCUCCCAAGCACGCAGCAA UGCAGCUCAAAACGCUUAGCCUAGCCACACCCCCACGGGAAACAGCAGUGAUUAACCUUUAGCAAUAAACGAAAGUUUAACUAAGCUAUACUAACCCCAGGGUUGGUCAAUUUCGUGCCAGCCACACCCUGGAGCUAGC (SEQ ID NO: 78). In some embodiments, a 3′ UTR comprises the sequence CUGGUACUGCAUGCACGCAAUGCUAGCUGCCCCUUUCCCGUCCUGGGUACCCCGAGUCUCCCCCGACCUCGGGUCCCA GGUAUGCUCCCACCUCCACCUGCCCCACUCACCACCUCUGCUAGUUCCAGACACCUCCCAAGCACGCAGCAAUGCAGC UCAAAACGCUUAGCCUAGCCACACCCCCACGGGAAACAGCAGUGAUUAACCUUUAGCAAUAAACGAAAGUUUAACUAA GCUAUACUAACCCCAGGGUUGGUCAAUUUCGUGCCAGCCACACCCUGGAGCUAGC (SEQ ID NO: 79). In some embodiments, an mRNA comprises a 5′ UTR comprising the nucleotide sequence of SEQ ID NO: 75 or SEQ ID NO: 76, an open reading frame, one or more stop codons, and a 3′ UTR comprising the nucleotide sequence of any one of SEQ ID NOs: 77–79. In some embodiments, an mRNA comprises a 5′ UTR comprising the nucleotide sequence of SEQ ID NO: 76, an open reading frame, one or more stop codons, and a 3′ UTR comprising the nucleotide sequence of SEQ ID NO: 78. In some embodiments, an mRNA comprises a 5′ UTR comprising the nucleotide sequence of SEQ ID NO: 76, an open reading frame, the nucleotide sequence UGAUGA, and a 3′ UTR comprising the nucleotide sequence of SEQ ID NO: 78. In some embodiments, the mRNA further comprises two poly(A) sequences separated by an intervening nucleotide sequence. In some embodiments, the mRNA further comprises the nucleotide sequence of SEQ ID NO: 80. In some embodiments, a 5′ UTR comprises the sequence GAGGAGACCCAAGCUACAUUUGCUUCUGACACAACUGUGUUCACUAGCAACCUCAAACAGACACCGCCACC (SEQ ID NO: 81). In some embodiments, a 3′ UTR comprises the sequence GCUCGCUUUCUUGCUGUCCAAUUUCUAUUAAAGGUUCCUUUGUUCCCUAAGUCCAACUACUAAACUGGGGGAUAUUAU GAAGGGCCUUGAGCAUCUGGAUUCUGCCUAAUAAAAAACAUUUAUUUUCAUUGC (SEQ ID NO: 82). In some embodiments, an mRNA comprises a 5′ UTR comprising the nucleotide sequence of SEQ ID NO: 81, an open reading frame, one or more stop codons, and a 3′ UTR comprising the nucleotide sequence of SEQ ID NO: 82. In some embodiments, the mRNA further comprises a polyA tail comprising 109 consecutive adenosine nucleotides. UTRs may also be omitted from the mRNA described herein. A 5 ^ UTR does not encode a protein (is non-coding). Natural 5′ UTRs have features that play roles in translation initiation. They harbor signatures like Kozak sequences which are commonly known to be involved in the process by which the ribosome initiates translation of many genes. Kozak sequences have the consensus CCRCCAUGG, where R is a purine (adenine or guanine) three bases upstream of the start codon (AUG), which is followed by another 'G'.5′UTR also have been known to form secondary structures which are involved in elongation factor binding. In some embodiments of the disclosure, a 5’ UTR is a heterologous UTR, i.e., is a UTR found in nature associated with a different ORF. In other embodiments, a 5’ UTR is a synthetic UTR, i.e., does not occur in nature. Synthetic UTRs include UTRs that have been mutated to improve their properties, e.g., which increase gene expression as well as those which are completely synthetic. Exemplary 5’ UTRs include Xenopus or human derived a-globin or b- globin (8278063; 9012219), human cytochrome b-245 a polypeptide, and hydroxysteroid (17b) dehydrogenase, and Tobacco etch virus (US8278063, US9012219). CMV immediate-early 1 (IE1) gene (US2014/0206753, WO2013/185069), the sequence GGGAUCCUACC (SEQ ID NO: 54) (WO 2014/144196) may also be used. In other embodiments, a 5' UTR is a 5' UTR of a TOP gene lacking the 5' TOP motif (the oligopyrimidine tract) (e.g., WO2015/101414, WO2015/101415, WO2015/062738, WO2015/024667, WO2015/024667); 5' UTR element derived from ribosomal protein Large 32 (L32) gene (WO/2015101414, WO2015101415, WO/2015/062738), 5' UTR element derived from the 5' UTR of an hydroxysteroid (17-β) dehydrogenase 4 gene (HSD17B4) (WO201/5024667), or a 5' UTR element derived from the 5' UTR of ATP5A1 (WO2015/024667) can be used. In some embodiments, an internal ribosome entry site (IRES) is used instead of a 5' UTR. A 3 ^ UTR does not encode a protein (is non-coding). Natural or wild type 3′ UTRs are known to have stretches of adenosines and uridines embedded in them. These AU rich signatures are particularly prevalent in genes with high rates of turnover. Based on their sequence features and functional properties, the AU rich elements (AREs) can be separated into three classes (Chen et al, 1995): Class I AREs contain several dispersed copies of an AUUUA motif within U-rich regions. C-Myc and MyoD contain class I AREs. Class II AREs possess two or more overlapping UUAUUUA(U/A)(U/A) nonamers. Molecules containing this type of AREs include GM-CSF and TNF-a. Class III ARES are less well defined. These U rich regions do not contain an AUUUA motif. c-Jun and Myogenin are two well-studied examples of this class. Most proteins binding to the AREs are known to destabilize the messenger, whereas members of the ELAV family, most notably HuR, have been documented to increase the stability of mRNA. HuR binds to AREs of all the three classes. Engineering the HuR specific binding sites into the 3′ UTR of nucleic acid molecules will lead to HuR binding and thus, stabilization of the message in vivo. Introduction, removal or modification of 3′ UTR AU rich elements (AREs) can be used to modulate the stability of mRNA of the disclosure. When engineering specific nucleic acids, one or more copies of an ARE can be introduced to make nucleic acids of the disclosure less stable and thereby curtail translation and decrease production of the resultant protein. Likewise, AREs can be identified and removed or mutated to increase the intracellular stability and thus increase translation and production of the resultant protein. Transfection experiments can be conducted in relevant cell lines, using nucleic acids of the disclosure and protein production can be assayed at various time points post-transfection. For example, cells can be transfected with different ARE- engineering molecules and by using an ELISA kit to the relevant protein and assaying protein produced at 6 hours, 12 hours, 1 day, 2 days, and 7 days post-transfection. Those of ordinary skill in the art will understand that 5’ UTRs that are heterologous or synthetic may be used with any desired 3’ UTR sequence. For example, a heterologous or synthetic 5’ UTR may be used with a synthetic 3’ UTR or with a heterologous 3’ UTR. Non-UTR sequences may also be used as regions or subregions within a nucleic acid. For example, introns or portions of introns sequences may be incorporated into regions of nucleic acid of the disclosure. Incorporation of intronic sequences may increase protein production as well as nucleic acid levels. Combinations of features may be included in flanking regions and may be contained within other features. For example, the ORF may be flanked by a 5′ UTR which may contain a strong Kozak translational initiation signal and/or a 3' UTR which may include an oligo(dT) sequence for templated addition of a poly-A tail. A 5′ UTR may comprise a first polynucleotide fragment and a second polynucleotide fragment from the same and/or different genes such as the 5′ UTRs described in US2010/0293625 and WO2015/085318A2, each of which is herein incorporated by reference. It should be understood that any UTR from any gene may be incorporated into the regions of a nucleic acid. Furthermore, multiple wild-type UTRs of any known gene may be utilized. It is also within the scope of the present disclosure to provide artificial UTRs which are not variants of wild type regions. These UTRs or portions thereof may be placed in the same orientation as in the transcript from which they were selected or may be altered in orientation or location. Hence a 5′ or 3′ UTR may be inverted, shortened, lengthened, made with one or more other 5′ UTRs or 3′ UTRs. As used herein, the term “altered” as it relates to a UTR sequence, means that the UTR has been changed in some way in relation to a naturally occurring or comparator sequence. For example, a 3′ UTR or 5′ UTR may be altered relative to a wild- type/native UTR by the change in orientation or location as taught above or may be altered by the inclusion of additional nucleotides, deletion of nucleotides, swapping or transposition of nucleotides. Any of these changes producing an “altered” UTR (whether 3′ or 5′) comprise a variant UTR. In some embodiments, a double, triple or quadruple UTR such as a 5′ UTR or 3′ UTR may be used. As used herein, a “double” UTR is one in which two copies of the same UTR are encoded either in series or substantially in series. For example, a double beta-globin 3′ UTR may be used as described in US2010/0129877, which is incorporated herein by reference. It is also within the scope of the present disclosure to have patterned UTRs. As used herein “patterned UTRs” are those UTRs which reflect a repeating or alternating pattern, such as ABABAB or AABBAABBAABB or ABCABCABC or variants thereof repeated once, twice, or more than 3 times. In these patterns, each letter, A, B, or C represent a different UTR at the nucleotide level. In some embodiments, flanking regions are selected from a family of transcripts whose proteins share a common function, structure, feature, or property. For example, polypeptides of interest may belong to a family of proteins which are expressed in a particular cell, tissue or at some time during development. The UTRs from any of these genes may be swapped for any other UTR of the same or different family of proteins to create a new polynucleotide. As used herein, a “family of proteins” is used in the broadest sense to refer to a group of two or more polypeptides of interest which share at least one function, structure, feature, localization, origin, or expression pattern. The untranslated region may also include translation enhancer elements (TEE). As a non- limiting example, the TEE may include those described in US 2009/0226470, herein incorporated by reference, and those known in the art. Open Reading Frames An open reading frame (ORF) is a continuous stretch of DNA or RNA that (1) begins with a start codon (e.g., ATG or AUG, encoding methionine), and (2) ends with a stop codon (e.g., TAA, TAG or TGA, or UAA, UAG or UGA) or is immediately followed by a stop codon. A stop codon does not encode an amino acid, such that translation of an ORF terminates when a ribosome reaches the stop codon immediately following the last amino acid-encoding codon in the ORF. A stop codon that results in translation termination may be considered part of the ORF, in which case the ORF ends with the stop codon. Alternatively, the first stop codon immediately following the last amino acid-encoding codon of an ORF may considered part of the 3′ untranslated region (3′ UTR) of a DNA or RNA, rather than part of the ORF. Those skilled in the art will understand that an ORF sequence that ends in a codon encoding amino acid will be followed by one or more stop codons in a DNA or RNA. An ORF may be followed by multiple stop codons. Inclusion of multiple consecutive stop codons reduces the extent of continued translation that may occur if a stop codon is mutated to a codon encoding an amino acid (readthrough), as a second stop codon may terminate translation even if a first stop codon is mutated and encodes an amino acid, such that only one amino acid is added to the C-terminus of the translated protein. Where multiple stop codons are present at the end, or immediately following, an ORF, the multiple stop codons may comprise the same stop codon (e.g., UGAUGA). Multiple stop codons may comprise different stop codons in series (e.g., UGAUAAUAG). In addition to reducing the extent of readthrough if a first stop codon is mutated, the presence of multiple different stop codons reduces the extent of readthrough if the first stop codon fails to allow translation termination (e.g., if a suppressor tRNA with an anticodon complementary to the first stop codon is present in the cell). An ORF typically encodes a protein. It will be understood that the sequences disclosed herein may further comprise additional elements, e.g., 5’ and/or 3’ UTRs, but that those elements, unlike the ORF, need not necessarily be present in an RNA (e.g., mRNA) as described herein. 5′ Caps In some embodiments, an RNA (e.g., mRNA) comprises a 5′ terminal cap.5′-capping of polynucleotides may be completed concomitantly during an in vitro transcription reaction using, for example, the following chemical RNA cap analogs to generate the 5′-guanosine cap structure according to manufacturer protocols: 3´-O-Me-m7G(5')ppp(5') G [the ARCA cap];G(5')ppp(5')A; G(5')ppp(5')G; m7G(5')ppp(5')A; m7G(5')ppp(5')G (New England BioLabs, Ipswich, MA).5′- capping of modified RNA (e.g., mRNA) may be completed post-transcriptionally using, for example, a Vaccinia Virus Capping Enzyme to generate the “Cap 0” structure: m7G(5')ppp(5')G (New England BioLabs, Ipswich, MA). Cap 1 structure may be generated using both Vaccinia Virus Capping Enzyme and a 2′-O methyl-transferase to generate: m7G(5')ppp(5')G-2′-O-methyl. Cap 2 structure may be generated from the Cap 1 structure followed by the 2′-O-methylation of the 5′-antepenultimate nucleotide using a 2′-O methyl-transferase. Cap 3 structure may be generated from the Cap 2 structure followed by the 2′-O-methylation of the 5′- preantepenultimate nucleotide using a 2′-O methyl-transferase. Enzymes may be derived from a recombinant source. Other cap analogs may be used. A cap analog may be, for example, a dinucleotide cap, a trinucleotide cap, or a tetranucleotide cap. In some embodiments, a cap analog is a dinucleotide cap. In some embodiments, a cap analog is a trinucleotide cap. In some embodiments, a cap analog is a tetranucleotide cap. A nucleotide cap (e.g., a trinucleotide cap or tetranucleotide cap), in some embodiments, comprises a compound of formula (I) tautomer or salt thereof, wherein
tautomer or salt thereof, wherein ring B1 is a modified or unmodified Guanine; ring B2 and ring B3 each independently is a nucleobase or a modified nucleobase; X2 is O, S(O)p, NR24 or CR25R26 in which p is 0, 1, or 2; Y0 is O or CR6R7; Y1 is O, S(O)n, CR6R7, or NR8, in which n is 0, 1 , or 2; each --- is a single bond or absent, wherein when each --- is a single bond, Yi is O, S(O)n, CR6R7, or NR8; and when each --- is absent, Y1 is void; Y2 is (OP(O)R4)m in which m is 0, 1, or 2, or -O-(CR40R41)u-Q0-(CR42R43)v-, in which Q0 is a bond, O, S(O)r, NR44, or CR45R46, r is 0, 1 , or 2, and each of u and v independently is 1, 2, 3 or 4; each R2 and R2' independently is halo, LNA, or OR3; each R3 independently is H, C1-C6 alkyl, C2-C6 alkenyl, or C2-C6 alkynyl and R3, when being C1-C6 alkyl, C2-C6 alkenyl, or C2-C6 alkynyl, is optionally substituted with one or more of halo, OH and C1-C6 alkoxyl that is optionally substituted with one or more OH or OC(O)-C1-C6 alkyl; each R4 and R4' independently is H, halo, C1-C6 alkyl, OH, SH, SeH, or BH3-; each of R6, R7, and R8, independently, is -Q1-T1, in which Q1 is a bond or C1-C3 alkyl linker optionally substituted with one or more of halo, cyano, OH and C1-C6 alkoxy, and T1 is H, halo, OH, COOH, cyano, or Rs1, in which Rs1 is C1-C3 alkyl, C2-C6 alkenyl, C2-C6 alkynyl, C1- C6 alkoxyl, C(O)O-C1-C6 alkyl, C3-C8 cycloalkyl, C6-C10 aryl, NR31R32, (NR31R32R33)+, 4 to 12- membered heterocycloalkyl, or 5- or 6-membered heteroaryl, and Rs1 is optionally substituted with one or more substituents selected from the group consisting of halo, OH, oxo, C1-C6 alkyl, COOH, C(O)O-C1-C6 alkyl, cyano, C1-C6 alkoxyl, NR31R32, (NR31R32R33)+, C3-C8 cycloalkyl, C6-C10 aryl, 4 to 12-membered heterocycloalkyl, and 5- or 6-membered heteroaryl; each of R10, R11, R12, R13 R14, and R15, independently, is -Q2-T2, in which Q2 is a bond or C1-C3 alkyl linker optionally substituted with one or more of halo, cyano, OH and C1-C6 alkoxy, and T2 is H, halo, OH, NH2, cyano, NO2, N3, Rs2, or ORs2, in which Rs2 is C1-C6 alkyl, C2-C6 alkenyl, C2-C6 alkynyl, C3-C8 cycloalkyl, C6-C10 aryl, NHC(O)-C1-C6 alkyl, NR31R32, (NR31R32R33)+, 4 to 12-membered heterocycloalkyl, or 5- or 6-membered heteroaryl, and Rs2 is optionally substituted with one or more substituents selected from the group consisting of halo, OH, oxo, C1-C6 alkyl, COOH, C(O)O-C1-C6 alkyl, cyano, C1 - C6 alkoxyl, NR31R32, (NR31R32R33)+, C3-C8 cycloalkyl, C6-C10 aryl, 4 to 12-membered heterocycloalkyl, and 5- or 6- membered heteroaryl; or alternatively R12 together with R14 is oxo, or R13 together with R15 is oxo, each of R20, R21, R22, and R23 independently is -Q3-T3, in which Q3 is a bond or C1-C3 alkyl linker optionally substituted with one or more of halo, cyano, OH and C1-C6 alkoxy, and T3 is H, halo, OH, NH2, cyano, NO2, N3, RS3, or ORS3, in which RS3 is C1-C6 alkyl, C2- C6 alkenyl, C2-C6 alkynyl, C3-C8 cycloalkyl, C6-C10 aryl, NHC(O)-C1-C6 alkyl, mono-C1- C6 alkylamino, di-C1-C6 alkylamino, 4 to 12-membered heterocycloalkyl, or 5- or 6-membered heteroaryl, and Rs3 is optionally substituted with one or more substituents selected from the group consisting of halo, OH, oxo, C1-C6 alkyl, COOH, C(O)O-C1-C6 alkyl, cyano, C1-C6 alkoxyl, amino, mono-C1-C6 alkylamino, di-C1-C6 alkylamino, C3-C8 cycloalkyl, C6-C10 aryl, 4 to 12-membered heterocycloalkyl, and 5- or 6-membered heteroaryl; each of R24, R25, and R26 independently is H or C1-C6 alkyl; each of R27 and R28 independently is H or OR29; or R27 and R28 together form O-R30-O; each R29 independently is H, C1-C6 alkyl, C2-C6 alkenyl, or C2-C6 alkynyl and R29, when being C1-C6 alkyl, C2-C6 alkenyl, or C2-C6 alkynyl, is optionally substituted with one or more of halo, OH and C1-C6 alkoxyl that is optionally substituted with one or more OH or OC(O)-C1-C6 alkyl; R30 is C1-C6 alkylene optionally substituted with one or more of halo, OH and C1-C6 alkoxyl; each of R31, R32, and R33, independently is H, C1-C6 alkyl, C3-C8 cycloalkyl, C6-C10 aryl, 4 to 12-membered heterocycloalkyl, or 5- or 6-membered heteroaryl; each of R40, R41, R42, and R43 independently is H, halo, OH, cyano, N3, OP(O)R47R48, or C1-C6 alkyl optionally substituted with one or more OP(O)R47R48, or one R41 and one R43, together with the carbon atoms to which they are attached and Q0, form C4-C10 cycloalkyl, 4- to 14-membered heterocycloalkyl, C6-C10 aryl, or 5- to 14-membered heteroaryl, and each of the cycloalkyl, heterocycloalkyl, phenyl, or 5- to 6-membered heteroaryl is optionally substituted with one or more of OH, halo, cyano, N3, oxo, OP(O)R47R48, C1-C6 alkyl, C1-C6 haloalkyl, COOH, C(O)O-C1-C6 alkyl, C1-C6 alkoxyl, C1-C6 haloalkoxyl, amino, mono-C1-C6 alkylamino, and di-C1-C6 alkylamino; R44 is H, C1-C6 alkyl, or an amine protecting group; each of R45 and R46 independently is H, OP(O)R47R48, or C1-C6 alkyl optionally substituted with one or more OP(O)R47R48, and each of R47 and R48, independently is H, halo, C1-C6 alkyl, OH, SH, SeH, or BH3. It should be understood that a cap analog, as provided herein, may include any of the cap analogs described in international publication WO 2017/066797, published on 20 April 2017, incorporated by reference herein in its entirety. In some embodiments, the B2 middle position can be a non-ribose molecule, such as arabinose. In some embodiments R2 is ethyl-based. Thus, in some embodiments, a tetranucleotide cap comprises the following structure:
ring B1 is a modified or unmodified Guanine; ring B2 and ring B3 each independently is a nucleobase or a modified nucleobase; X2 is O, S(O)p, NR24 or CR25R26 in which p is 0, 1, or 2; Y0 is O or CR6R7; Y1 is O, S(O)n, CR6R7, or NR8, in which n is 0, 1 , or 2; each --- is a single bond or absent, wherein when each --- is a single bond, Yi is O, S(O)n, CR6R7, or NR8; and when each --- is absent, Y1 is void; Y2 is (OP(O)R4)m in which m is 0, 1, or 2, or -O-(CR40R41)u-Q0-(CR42R43)v-, in which Q0 is a bond, O, S(O)r, NR44, or CR45R46, r is 0, 1 , or 2, and each of u and v independently is 1, 2, 3 or 4; each R2 and R2' independently is halo, LNA, or OR3; each R3 independently is H, C1-C6 alkyl, C2-C6 alkenyl, or C2-C6 alkynyl and R3, when being C1-C6 alkyl, C2-C6 alkenyl, or C2-C6 alkynyl, is optionally substituted with one or more of halo, OH and C1-C6 alkoxyl that is optionally substituted with one or more OH or OC(O)-C1-C6 alkyl; each R4 and R4' independently is H, halo, C1-C6 alkyl, OH, SH, SeH, or BH3-; each of R6, R7, and R8, independently, is -Q1-T1, in which Q1 is a bond or C1-C3 alkyl linker optionally substituted with one or more of halo, cyano, OH and C1-C6 alkoxy, and T1 is H, halo, OH, COOH, cyano, or Rs1, in which Rs1 is C1-C3 alkyl, C2-C6 alkenyl, C2-C6 alkynyl, C1- C6 alkoxyl, C(O)O-C1-C6 alkyl, C3-C8 cycloalkyl, C6-C10 aryl, NR31R32, (NR31R32R33)+, 4 to 12- membered heterocycloalkyl, or 5- or 6-membered heteroaryl, and Rs1 is optionally substituted with one or more substituents selected from the group consisting of halo, OH, oxo, C1-C6 alkyl, COOH, C(O)O-C1-C6 alkyl, cyano, C1-C6 alkoxyl, NR31R32, (NR31R32R33)+, C3-C8 cycloalkyl, C6-C10 aryl, 4 to 12-membered heterocycloalkyl, and 5- or 6-membered heteroaryl; each of R10, R11, R12, R13 R14, and R15, independently, is -Q2-T2, in which Q2 is a bond or C1-C3 alkyl linker optionally substituted with one or more of halo, cyano, OH and C1-C6 alkoxy, and T2 is H, halo, OH, NH2, cyano, NO2, N3, Rs2, or ORs2, in which Rs2 is C1-C6 alkyl, C2-C6 alkenyl, C2-C6 alkynyl, C3-C8 cycloalkyl, C6-C10 aryl, NHC(O)-C1-C6 alkyl, NR31R32, (NR31R32R33)+, 4 to 12-membered heterocycloalkyl, or 5- or 6-membered heteroaryl, and Rs2 is optionally substituted with one or more substituents selected from the group consisting of halo, OH, oxo, C1-C6 alkyl, COOH, C(O)O-C1-C6 alkyl, cyano, C1 - C6 alkoxyl, NR31R32, (NR31R32R33)+, C3-C8 cycloalkyl, C6-C10 aryl, 4 to 12-membered heterocycloalkyl, and 5- or 6- membered heteroaryl; or alternatively R12 together with R14 is oxo, or R13 together with R15 is oxo, each of R20, R21, R22, and R23 independently is -Q3-T3, in which Q3 is a bond or C1-C3 alkyl linker optionally substituted with one or more of halo, cyano, OH and C1-C6 alkoxy, and T3 is H, halo, OH, NH2, cyano, NO2, N3, RS3, or ORS3, in which RS3 is C1-C6 alkyl, C2- C6 alkenyl, C2-C6 alkynyl, C3-C8 cycloalkyl, C6-C10 aryl, NHC(O)-C1-C6 alkyl, mono-C1- C6 alkylamino, di-C1-C6 alkylamino, 4 to 12-membered heterocycloalkyl, or 5- or 6-membered heteroaryl, and Rs3 is optionally substituted with one or more substituents selected from the group consisting of halo, OH, oxo, C1-C6 alkyl, COOH, C(O)O-C1-C6 alkyl, cyano, C1-C6 alkoxyl, amino, mono-C1-C6 alkylamino, di-C1-C6 alkylamino, C3-C8 cycloalkyl, C6-C10 aryl, 4 to 12-membered heterocycloalkyl, and 5- or 6-membered heteroaryl; each of R24, R25, and R26 independently is H or C1-C6 alkyl; each of R27 and R28 independently is H or OR29; or R27 and R28 together form O-R30-O; each R29 independently is H, C1-C6 alkyl, C2-C6 alkenyl, or C2-C6 alkynyl and R29, when being C1-C6 alkyl, C2-C6 alkenyl, or C2-C6 alkynyl, is optionally substituted with one or more of halo, OH and C1-C6 alkoxyl that is optionally substituted with one or more OH or OC(O)-C1-C6 alkyl; R30 is C1-C6 alkylene optionally substituted with one or more of halo, OH and C1-C6 alkoxyl; each of R31, R32, and R33, independently is H, C1-C6 alkyl, C3-C8 cycloalkyl, C6-C10 aryl, 4 to 12-membered heterocycloalkyl, or 5- or 6-membered heteroaryl; each of R40, R41, R42, and R43 independently is H, halo, OH, cyano, N3, OP(O)R47R48, or C1-C6 alkyl optionally substituted with one or more OP(O)R47R48, or one R41 and one R43, together with the carbon atoms to which they are attached and Q0, form C4-C10 cycloalkyl, 4- to 14-membered heterocycloalkyl, C6-C10 aryl, or 5- to 14-membered heteroaryl, and each of the cycloalkyl, heterocycloalkyl, phenyl, or 5- to 6-membered heteroaryl is optionally substituted with one or more of OH, halo, cyano, N3, oxo, OP(O)R47R48, C1-C6 alkyl, C1-C6 haloalkyl, COOH, C(O)O-C1-C6 alkyl, C1-C6 alkoxyl, C1-C6 haloalkoxyl, amino, mono-C1-C6 alkylamino, and di-C1-C6 alkylamino; R44 is H, C1-C6 alkyl, or an amine protecting group; each of R45 and R46 independently is H, OP(O)R47R48, or C1-C6 alkyl optionally substituted with one or more OP(O)R47R48, and each of R47 and R48, independently is H, halo, C1-C6 alkyl, OH, SH, SeH, or BH3. It should be understood that a cap analog, as provided herein, may include any of the cap analogs described in international publication WO 2017/066797, published on 20 April 2017, incorporated by reference herein in its entirety. In some embodiments, the B2 middle position can be a non-ribose molecule, such as arabinose. In some embodiments R2 is ethyl-based. Thus, in some embodiments, a tetranucleotide cap comprises the following structure: In other embodiments, a tetranucleotide cap comprises the following structure:
In other embodiments, a tetranucleotide cap comprises the following structure: In yet other embodiments, a tetranucleotide cap comprises the following structure:
In yet other embodiments, a tetranucleotide cap comprises the following structure: In yet other embodiments, a tetranucleotide cap comprises the following structure:
In yet other embodiments, a tetranucleotide cap comprises the following structure:









In some embodiments, R is an alkyl (e.g., C1-C6 alkyl). In some embodiments, R is a methyl group (e.g., C1 alkyl). In some embodiments, R is an ethyl group (e.g., C2 alkyl). In some embodiments, R is a hydrogen. In some embodiments, a tetranucleotide cap comprises GGAG. In some embodiments, a tetranucleotide cap comprises any one of the following structures: In some embodiments, an mRNA comprises an m23′O,7 G+(5′)ppp(5′)Am cap having the
In some embodiments, an mRNA comprises an m23′O,7 G+(5′)ppp(5′)Am cap having the structure: . Polyadenylation A “poly(A) tail” is a region of mRNA that is downstream, e.g., directly downstream (i.e., 3′), from the 3′ UTR that contains multiple, consecutive adenosine monophosphates. A poly(A) tail may contain 10 to 300 adenosine monophosphates. It can, in some instances, comprise up to about 400 adenine nucleotides. For example, a poly(A) tail may contain 10, 20, 30, 40, 50, 60, 70, 80, 90, 100, 110, 120, 130, 140, 150, 160, 170, 180, 190, 200, 210, 220, 230, 240, 250, 260, 270, 280, 290 or 300 adenosine monophosphates. In some embodiments, a poly(A) tail contains 50 to 250 adenosine monophosphates. In a relevant biological setting (e.g., in cells, in vivo) the poly(A) tail functions to protect mRNA from enzymatic degradation, e.g., in the cytoplasm, and aids in transcription termination, and/or export of the mRNA from the nucleus and translation. In some embodiments, the length of the 3′-poly(A) tail may be an essential element with respect to the stability of the individual mRNA. In some embodiments, a poly(A) tail has a length of about 50, about 100, about 150, about 200, about 250, about 300, about 350, or about 400 nucleotides. In some embodiments, a poly(A) tail has a length of 100 nucleotides. In some embodiments, an mRNA comprises a poly(A) sequence that has a length of 50– 75 nucleotides. In some embodiments, an mRNA comprises a poly(A) sequence that comprises 60, 61, 62, 63, 64, 65, 66, 67, 68, 69, or 70 consecutive adenosine nucleotides. In some embodiments, an mRNA comprises a poly(A) sequence comprising 64 consecutive adenosine nucleotides. In some embodiments, the consecutive adenosine nucleotides of a poly(A) sequence are flanked at the 5′ and 3′ end by nucleotides that are not adenosine nucleotides. In some embodiments, an mRNA comprises a poly(C) sequence, which may comprise 10 to 300 cytidine nucleotides. In some embodiments, the poly(C) sequence comprises 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, or 40 consecutive cytidine nucleotides. In some embodiments, the poly(C) sequence comprises 30 cytidine nucleotides. In some embodiments, the consecutive cytidine nucleotides of a poly(C) sequence are flanked at the 5′ and 3′ end by nucleotides that are not cytidine nucleotides. In some embodiments, an mRNA comprises two poly(A) sequences separated by an intervening nucleotide sequence. In some embodiments, the intervening nucleotide sequence comprises no more than 3, no more than two, no more than 1, or no adenosine nucleotides. In some embodiments, the intervening sequence comprises 3 adenosine nucleotides. In some embodiments, the intervening sequence does not comprise an adenosine nucleotide. In some embodiments, the intervening sequence is no more than 30, no more than 25, no more than 20, no more than 15, or no more than 10 nucleotides long. In some embodiments, the intervening sequence consists of 10 nucleotides. In some embodiments, the intervening sequence comprises the sequence of GCAUAUGACU (SEQ ID NO: 55). In some embodiments, the intervening sequence does not begin with an adenosine nucleotide, and does not end with an adenosine nucleotide. In some embodiments, the first poly(A) sequences comprises at least 15, at least 20, at least 25, or at least 30 consecutive adenosine nucleotides. In some embodiments, the second poly(A) sequences comprises at least 55, at least 60, at least 65, or at least 70 consecutive adenosine nucleotides. In some embodiments, the first poly(A) sequence comprises 30 consecutive adenosine nucleotides. In some embodiments, the second poly(A) sequence comprises 70 adenosine nucleotides. In some embodiments, an mRNA comprises the nucleotide sequence
structure: . Polyadenylation A “poly(A) tail” is a region of mRNA that is downstream, e.g., directly downstream (i.e., 3′), from the 3′ UTR that contains multiple, consecutive adenosine monophosphates. A poly(A) tail may contain 10 to 300 adenosine monophosphates. It can, in some instances, comprise up to about 400 adenine nucleotides. For example, a poly(A) tail may contain 10, 20, 30, 40, 50, 60, 70, 80, 90, 100, 110, 120, 130, 140, 150, 160, 170, 180, 190, 200, 210, 220, 230, 240, 250, 260, 270, 280, 290 or 300 adenosine monophosphates. In some embodiments, a poly(A) tail contains 50 to 250 adenosine monophosphates. In a relevant biological setting (e.g., in cells, in vivo) the poly(A) tail functions to protect mRNA from enzymatic degradation, e.g., in the cytoplasm, and aids in transcription termination, and/or export of the mRNA from the nucleus and translation. In some embodiments, the length of the 3′-poly(A) tail may be an essential element with respect to the stability of the individual mRNA. In some embodiments, a poly(A) tail has a length of about 50, about 100, about 150, about 200, about 250, about 300, about 350, or about 400 nucleotides. In some embodiments, a poly(A) tail has a length of 100 nucleotides. In some embodiments, an mRNA comprises a poly(A) sequence that has a length of 50– 75 nucleotides. In some embodiments, an mRNA comprises a poly(A) sequence that comprises 60, 61, 62, 63, 64, 65, 66, 67, 68, 69, or 70 consecutive adenosine nucleotides. In some embodiments, an mRNA comprises a poly(A) sequence comprising 64 consecutive adenosine nucleotides. In some embodiments, the consecutive adenosine nucleotides of a poly(A) sequence are flanked at the 5′ and 3′ end by nucleotides that are not adenosine nucleotides. In some embodiments, an mRNA comprises a poly(C) sequence, which may comprise 10 to 300 cytidine nucleotides. In some embodiments, the poly(C) sequence comprises 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, or 40 consecutive cytidine nucleotides. In some embodiments, the poly(C) sequence comprises 30 cytidine nucleotides. In some embodiments, the consecutive cytidine nucleotides of a poly(C) sequence are flanked at the 5′ and 3′ end by nucleotides that are not cytidine nucleotides. In some embodiments, an mRNA comprises two poly(A) sequences separated by an intervening nucleotide sequence. In some embodiments, the intervening nucleotide sequence comprises no more than 3, no more than two, no more than 1, or no adenosine nucleotides. In some embodiments, the intervening sequence comprises 3 adenosine nucleotides. In some embodiments, the intervening sequence does not comprise an adenosine nucleotide. In some embodiments, the intervening sequence is no more than 30, no more than 25, no more than 20, no more than 15, or no more than 10 nucleotides long. In some embodiments, the intervening sequence consists of 10 nucleotides. In some embodiments, the intervening sequence comprises the sequence of GCAUAUGACU (SEQ ID NO: 55). In some embodiments, the intervening sequence does not begin with an adenosine nucleotide, and does not end with an adenosine nucleotide. In some embodiments, the first poly(A) sequences comprises at least 15, at least 20, at least 25, or at least 30 consecutive adenosine nucleotides. In some embodiments, the second poly(A) sequences comprises at least 55, at least 60, at least 65, or at least 70 consecutive adenosine nucleotides. In some embodiments, the first poly(A) sequence comprises 30 consecutive adenosine nucleotides. In some embodiments, the second poly(A) sequence comprises 70 adenosine nucleotides. In some embodiments, an mRNA comprises the nucleotide sequence AAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA (SEQ ID NO: 80). In some embodiments, an mRNA comprises a poly(A) sequence that has a length of 90– 120 nucleotides. In some embodiments, an mRNA comprises a poly(A) sequence that comprises 100, 101, 102, 103, 104, 105, 106, 107, 108, 109, 110, 111, 112, 113, 114, 115, 116, 117, 118, 190, or 120 consecutive adenosine nucleotides. In some embodiments, an mRNA comprises a poly(A) sequence that comprises at least 109 consecutive adenosine nucleotides. In some embodiments, an mRNA comprises a poly(A) sequence that comprises 109 consecutive adenosine nucleotides. In some embodiments, an mRNA comprises a poly(A) sequence that consists of 109 consecutive adenosine nucleotides. Self-amplifying RNA Some aspects of the disclosure relate to self-amplifying RNA (e.g., an RNA replicon) encoding a MERS-CoV S protein or variant thereof (e.g., fusion protein comprising one or more domains of a MERS-CoV S protein). An self-amplifying RNA refers to an RNA encoding one or more molecules (e.g., proteins) that, individually or in conjunction, are capable of replicating the self-amplifying RNA. In some embodiments, the proteins encoded by the self-amplifying RNA are non-structural proteins nsP1, nsP2, nsP3, and nsP4, which form an RNA-dependent RNA polymerase (RdRp), or replicase, that is capable of replicating the self-amplifying RNA. By encoding proteins that are capable of replicating the RNA, a self-amplifying RNA is capable of self-amplification in a cell, provided that the cell can translate the RNA and produce the encoded protein(s). A self-amplifying RNA may be referred to as an RNA replicon. When an RNA replicon or self-amplifying RNA is translated, the one or more encoded viral non-structural proteins are translated. A “viral non-structural protein” is a protein encoded by a virus but that is not part of the virus particle. The viral non-structural proteins, in the context of self-amplifying RNA, replicate the nucleotide sequences encoding the vaccine antigen or therapeutic protein (e.g., MERS-CoV protein) from the self-amplifying RNA via the sub- genomic viral promoters. Such replication driven by the viral sub-genomic promoter using the viral non- structural proteins enhances the expression level of the encoded protein. In some embodiments, the viral non-structural proteins are from a single-strand positive-sense RNA viruses. In some embodiments, the viral non-structural proteins are from an Alphavirus, belonging to the Togaviridae family. In some embodiments, the alphavirus is Sindbis or Venezuelan equine encephalitis virus. In some embodiments, the viral non-structural protein is an RNA-dependent RNA polymerase (RdRp) polyprotein P1234 (also termed NSP1-4). Upon translation, P1234 is rapidly cleaved into P123 and nsP4 by autoproteolytic activity originating from the nsP2 (proteinase) portion of the polyprotein. Alphaviral RNA synthesis occurs at the plasma membrane of a cell, where the nsPs, together with alphaviral RNA, form membrane invaginations (or “spherules”). These spherules contain dsRNA created by replication of “+” strand viral genomic RNA into “–“ strand anti-genomic RNA. The “–“ strand serves as a template from which additional “+” strand genomic RNA (synthesized from the 5’UTR) or a shorter subsequence of the genomic RNA (termed subgenomic RNA) is synthesized from the subgenomic viral promoter region located near the end of the nonstructural protein ORF. The “+” strand genomic RNA and the subgenomic RNA are exported out of the spherules into the cytoplasm where they are translated by endogenous ribosomes. The exported “+” strand genomic RNA can associate with nsPs and form additional spherules, thus resulting in exponential increase of replicon RNA. The viral non-structural proteins facilitate the replication of the nucleotide sequences encoding the MERS-CoV protein via the subgenomic viral promoters (also referred to as “subgenomic promoters” herein). A “subgenomic viral promoter” refers to a promoter the drives the transcription of subgenomic mRNAs. Typically, an mRNA is transcribed from genomic DNAs and episomal DNAs (e.g., plasmids). Some viruses may transcribe subgenomic mRNAs from a RNA replicon that is produced from its genomic RNA. Many positive-sense RNA viruses produce subgenomic mRNAs as one of the common infection techniques used by these viruses and generally transcribe late viral genes. Subgenomic viral promoters range from 20 nucleotide (Sindbis virus) to over 100 nucleotides (Beet necrotic yellow vein virus) and are usually found upstream of the transcription start. In some embodiments, the subgenomic viral promoter is 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49, 50, 51, 52, 53, 54, 55, 56, 57, 58, 59, 60, 61, 62, 63, 64, 65, 66, 67, 68, 69, 70, 71, 72, 73, 74, 75, 76, 77, 78, 79, 80, 81, 82, 83, 84, 85, 86, 87, 88, 89, 90, 91, 92, 93, 94, 95, 96, 97, 98, 99, 100 nucleotides long, or longer. Subgenomic viral promoters have been described in the art, e.g., in PCT Application Publication No. WO 2016/040359, and Wagner et al., Nature Chemical Biology, DOI: 10.1038/s41589-018-0146-9 (2018). Additional Stabilizing Elements RNA (e.g., mRNA) provided herein, in some embodiments, includes an additional stabilizing element. Stabilizing elements may include, for example, a histone stem-loop. A stem- loop binding protein (SLBP), a 32 kDa protein has been identified. It is associated with the histone stem-loop at the 3'-end of the histone messages in both the nucleus and the cytoplasm. Its expression level is regulated by the cell cycle; it peaks during the S-phase, when histone mRNA levels are also elevated. The protein has been shown to be essential for efficient 3'-end processing of histone pre-mRNA by the U7 snRNP. SLBP continues to be associated with the stem-loop after processing, and then stimulates the translation of mature histone mRNAs into histone proteins in the cytoplasm. The RNA binding domain of SLBP is conserved through metazoa and protozoa; its binding to the histone stem-loop depends on the structure of the loop. The minimum binding site includes at least three nucleotides 5’ and two nucleotides 3 ^ relative to the stem-loop. In some embodiments, an RNA (e.g., mRNA) includes an open reading frame (coding region), a histone stem-loop, and optionally, a poly(A) sequence or polyadenylation signal. The poly(A) sequence or polyadenylation signal generally should enhance the expression level of the encoded protein. The encoded protein, in some embodiments, is not a histone protein, a reporter protein (e.g., Luciferase, GFP, EGFP, β-Galactosidase, EGFP), or a marker or selection protein (e.g., alpha-Globin, Galactokinase and Xanthine:guanine phosphoribosyl transferase (GPT)). In some embodiments, an RNA (e.g., mRNA) includes the combination of a poly(A) sequence or polyadenylation signal and at least one histone stem-loop, even though both represent alternative mechanisms in nature, they act synergistically to increase the protein expression beyond the level observed with either of the individual elements. The synergistic effect of the combination of poly(A) and a histone stem-loop does not depend on the order of the elements or the length of the poly(A) sequence. In some embodiments, an RNA (e.g., mRNA) does not include a histone downstream element (HDE). “Histone downstream element” (HDE) includes a purine-rich polynucleotide stretch of approximately 15 to 20 nucleotides 3′ of naturally-occurring stem-loops, representing the binding site for the U7 snRNA, which is involved in processing of histone pre-mRNA into mature histone mRNA. In some embodiments, the nucleic acid does not include an intron. An RNA (e.g., mRNA) may or may not contain an enhancer and/or promoter sequence, which may be modified or unmodified or which may be activated or inactivated. In some embodiments, the histone stem-loop is generally derived from histone genes and includes an intramolecular base pairing of two neighbored partially or entirely reverse complementary sequences separated by a spacer, consisting of a short sequence, which forms the loop of the structure. The unpaired loop region is typically unable to base pair with either of the stem loop elements. It occurs more often in RNA, as is a key component of many RNA secondary structures but may be present in single-stranded DNA as well. Stability of the stem-loop structure generally depends on the length, number of mismatches or bulges, and base composition of the paired region. In some embodiments, wobble base pairing (non-Watson-Crick base pairing) may result. In some embodiments, the at least one histone stem-loop sequence comprises a length of 15 to 45 nucleotides. In some embodiments, an RNA (e.g., mRNA) has one or more AU-rich sequences removed. These sequences, sometimes referred to as AURES are destabilizing sequences found in the 3 ’UTR. The AURES may be removed from the mRNA. Alternatively, the AURES may remain in the mRNA. Sequence Modification In some embodiments, an open reading frame encoding a protein of the disclosure is codon optimized. Codon optimization methods are known in the art. An open reading frame of any one or more of the sequences provided herein may be codon optimized. Codon optimization, in some embodiments, may be used to match codon frequencies in target and host organisms to ensure proper folding; bias GC content to increase RNA (e.g., mRNA) stability or reduce secondary structures; minimize tandem repeat codons or base runs that may impair gene construction or expression; customize transcriptional and translational control regions; insert or remove protein trafficking sequences; remove/add post translation modification sites in encoded protein (e.g., glycosylation sites); add, remove or shuffle protein domains; insert or delete restriction sites; modify ribosome binding sites and RNA (e.g., mRNA) degradation sites; adjust translational rates to allow the various domains of the protein to fold properly; or reduce or eliminate problem secondary structures within the polynucleotide. Codon optimization tools, algorithms and services are known in the art – non-limiting examples include services from GeneArt (Life Technologies), DNA2.0 (Menlo Park CA) and/or proprietary methods. In some embodiments, the open reading frame sequence is optimized using optimization algorithms. In some embodiments, a codon optimized sequence shares less than 95% sequence identity to a naturally-occurring or wild-type sequence open reading frame (e.g., a naturally- occurring or wild-type RNA (e.g., mRNA) sequence encoding a MERS-CoV protein antigen). In some embodiments, a codon optimized sequence shares less than 90% sequence identity to a naturally-occurring or wild-type sequence (e.g., a naturally-occurring or wild-type RNA (e.g., mRNA) sequence encoding a MERS-CoV protein). In some embodiments, a codon optimized sequence shares less than 85% sequence identity to a naturally-occurring or wild-type sequence (e.g., a naturally-occurring or wild-type RNA (e.g., mRNA) sequence encoding a MERS-CoV protein). In some embodiments, a codon optimized sequence shares less than 80% sequence identity to a naturally-occurring or wild-type sequence (e.g., a naturally-occurring or wild-type RNA (e.g., mRNA) sequence encoding a MERS-CoV protein). In some embodiments, a codon optimized sequence shares less than 75% sequence identity to a naturally-occurring or wild-type sequence (e.g., a naturally-occurring or wild-type RNA (e.g., mRNA) sequence encoding a MERS-CoV protein). In some embodiments, a codon optimized sequence shares between 65% and 85% (e.g., between about 67% and about 85% or between about 67% and about 80%) sequence identity to a naturally-occurring or wild-type sequence (e.g., a naturally-occurring or wild-type RNA (e.g., mRNA) sequence encoding a MERS-CoV protein). In some embodiments, a codon optimized sequence shares between 65% and 75% or about 80% sequence identity to a naturally-occurring or wild-type sequence (e.g., a naturally-occurring or wild-type RNA (e.g., mRNA) sequence encoding a MERS-CoV protein). In some embodiments, a codon-optimized sequence encodes an antigen that is as immunogenic as, or more immunogenic than (e.g., at least 10%, at least 20%, at least 30%, at least 40%, at least 50%, at least 100%, or at least 200% more), than a MERS-CoV protein encoded by a non-codon-optimized sequence. When transfected into mammalian host cells, the modified mRNAs have a stability of between 12-18 hours, or greater than 18 hours, e.g., 24, 36, 48, 60, 72, or greater than 72 hours and are capable of being expressed by the mammalian host cells. In some embodiments, a codon optimized RNA (e.g., mRNA) may be one in which the levels of G/C are enhanced. The G/C-content of nucleic acid molecules (e.g., mRNA) may influence the stability of the RNA. RNA (e.g., mRNA) having an increased amount of guanine (G) and/or cytosine (C) residues may be functionally more stable than RNA containing a large amount of adenine (A) and thymine (T) or uracil (U) nucleotides. As an example, WO02/098443 discloses a pharmaceutical composition containing an RNA (e.g., mRNA) stabilized by sequence modifications in the translated region. Due to the degeneracy of the genetic code, the modifications work by substituting existing codons for those that promote greater RNA stability without changing the resulting amino acid. The approach is limited to coding regions of the RNA (e.g., mRNA). Some embodiments of mRNAs described herein, comprise a sequence with a %G/C content of 30% – 80%, 40% – 70%, 50% – 60%, 35% – 50%, 50% – 65%, 65% – 70%, 40% – 45%, 45% – 50%, 50% – 55%, 55% – 70%, 70% – 75%, or 75% – 80%. In some embodiments, the nucleic acid sequence of the full-length mRNA comprises a %G/C content of 30% to 80%, 40% – 70%, 50% – 60%, 35% – 50%, 50% – 65%, 65% – 70%, 40% – 45%, 45% – 50%, 50% – 55%, 55% – 70%, 70% – 75%, or 75% – 80%. In some embodiments, the mRNA comprises an ORF with a %G/C content from about 30% to about 80%, about 35% to about 70%, about 40% to about 60%, about 45% to about 55%, about 40% to about 70%, about 50% to about 60%, about 35% to about 50%, about 50% to about 50% to about 65%, about 65% to about 70%, about 40% to about 45%, about 45% to about 50%, about 50% to about 55%, about 55% to about 70%, about 70% to about 75%, or about 75% to about 80%. In some embodiments, the mRNA comprises 5′ UTR with a %G/C content from about 30% to about 80%, about 35% to about 70%, about 40% to about 60%, about 45% to about 55%, about 40% to about 70%, about 50% to about 60%, about 35% to about 50%, about 50% to about 50% to about 65%, about 65% to about 70%, about 40% to about 45%, about 45% to about 50%, about 50% to about 55%, about 55% to about 70%, about 70% to about 75%, or about 75% to about 80%. In some embodiments, the mRNA comprises 3′ UTR with a %G/C content from about 30% to about 80%, about 35% to about 70%, about 40% to about 60%, about 45% to about 55%, about 40% to about 70%, about 50% to about 60%, about 35% to about 50%, about 50% to about 50% to about 65%, about 65% to about 70%, about 40% to about 45%, about 45% to about 50%, about 50% to about 55%, about 55% to about 70%, about 70% to about 75%, or about 75% to about 80%. In some embodiments, a modified mRNA made by a method described herein comprises a higher %G/C content than a wild-type mRNA sequence. In some embodiments, the %G/C content of the modified mRNA sequence is 2% or more, 3% or more, 4% or more, 5% or more, 6% or more, 7% or more, 8% or more, 9% or more, 10% or more, 12% or more, 15% or more, or 20% or more than the %G/C content of the wild-type RNA sequence. In some embodiments, the %G/C content of the modified ORF sequence is 2% or more, 3% or more, 4% or more, 5% or more, 6% or more, 7% or more, 8% or more, 9% or more, 10% or more, 12% or more, 15% or more, or 20% or more than the %G/C content of the wild-type ORF sequence. In some embodiments, the %G/C content of the modified 5′ UTR sequence is 2% or more, 3% or more, 4% or more, 5% or more, 6% or more, 7% or more, 8% or more, 9% or more, 10% or more, 12% or more, 15% or more, or 20% or more than the %G/C content of the wild-type 3′ UTR sequence. Chemically Modified Nucleotides The compositions of the present disclosure comprise, in some embodiments, an RNA having an open reading frame encoding a protein, wherein the nucleic acid comprises nucleotides and/or nucleosides that can be standard (unmodified) or modified as is known in the art. In some embodiments, nucleotides and nucleosides of the present disclosure comprise modified nucleotides or nucleosides. Such modified nucleotides and nucleosides can be naturally- occurring modified nucleotides and nucleosides or non-naturally-occurring modified nucleotides and nucleosides. Such modifications can include those at the sugar, backbone, or nucleobase portion of the nucleotide and/or nucleoside as are recognized in the art. In some embodiments, a naturally-occurring modified nucleotide or nucleotide of the disclosure is one as is generally known or recognized in the art. Non-limiting examples of such naturally-occurring modified nucleotides and nucleotides can be found, inter alia, in the widely recognized MODOMICS database. In some embodiments, a non-naturally-occurring modified nucleotide or nucleoside of the disclosure is one as is generally known or recognized in the art. Non-limiting examples of such non-naturally-occurring modified nucleotides and nucleosides can be found, inter alia, in international publication numbers WO2013/052523A1; WO2014/093924A1; WO2015/051173A2; WO2015/051169A2; WO2015/089511A2; or WO2017/153936A1, each of which is herein incorporated by reference. Hence, nucleic acids of the disclosure (e.g., DNA and RNA, such as mRNA) can comprise standard nucleotides and nucleosides, naturally-occurring nucleotides and nucleosides, non-naturally-occurring nucleotides and nucleosides, or any combination thereof. Nucleic acids of the disclosure (e.g., DNA and RNA, such as mRNA), in some embodiments, comprise various (more than one) different types of standard and/or modified nucleotides and nucleosides. In some embodiments, a particular region of a nucleic acid contains one, two or more (optionally different) types of standard and/or modified nucleotides and nucleosides. In some embodiments, a modified RNA (e.g., mRNA) introduced to a cell or organism, exhibits reduced degradation in the cell or organism, respectively, relative to an unmodified nucleic acid comprising standard nucleotides and nucleosides. In some embodiments, a modified RNA (e.g., mRNA) introduced into a cell or organism, may exhibit reduced immunogenicity in the cell or organism, respectively (e.g., a reduced innate response) relative to an unmodified nucleic acid comprising standard nucleotides and nucleosides. Nucleic acids (e.g., RNA, such as mRNA), in some embodiments, comprise non-natural modified nucleotides that are introduced during synthesis or post-synthesis of the nucleic acids to achieve desired functions or properties. The modifications may be present on internucleotide linkages, purine or pyrimidine bases, or sugars. The modification may be introduced with chemical synthesis or with a polymerase enzyme at the terminal of a chain or anywhere else in the chain. Any of the regions of a nucleic acid may be chemically modified. The present disclosure provides for modified nucleosides and nucleotides of a nucleic acid (e.g., RNA nucleic acids, such as mRNA nucleic acids). A “nucleoside” refers to a compound containing a sugar molecule (e.g., a pentose or ribose) or a derivative thereof in combination with an organic base (e.g., a purine or pyrimidine) or a derivative thereof (also referred to herein as “nucleobase”). A “nucleotide” refers to a nucleoside, including a phosphate group. Modified nucleotides may by synthesized by any useful method, such as, for example, chemically, enzymatically, or recombinantly, to include one or more modified or non-natural nucleosides. Nucleic acids can comprise a region or regions of linked nucleosides. Such regions may have variable backbone linkages. The linkages can be standard phosphodiester linkages, in which case the nucleic acids would comprise regions of nucleotides. Modified nucleotide base pairing encompasses not only the standard adenosine-thymine, adenosine-uracil, or guanosine-cytosine base pairs, but also base pairs formed between nucleotides and/or modified nucleotides comprising non-standard or modified bases, wherein the arrangement of hydrogen bond donors and hydrogen bond acceptors permits hydrogen bonding between a non-standard base and a standard base or between two complementary non-standard base structures, such as, for example, in those nucleic acids having at least one chemical modification. One example of such non-standard base pairing is the base pairing between the modified nucleotide inosine and adenine, cytosine or uracil. Any combination of base/sugar or linker may be incorporated into nucleic acids of the present disclosure. In some embodiments, modified nucleobases in nucleic acids (e.g., RNA nucleic acids, such as mRNA nucleic acids) comprise 1-methyl-pseudouridine (m1ψ), 1-ethyl-pseudouridine (e1ψ), 5-methoxy-uridine (mo5U), 5-methyl-cytidine (m5C), and/or pseudouridine (ψ). In some embodiments, modified nucleobases in nucleic acids (e.g., RNA nucleic acids, such as mRNA nucleic acids) comprise 5-methoxymethyl uridine, 5-methylthio uridine, 1-methoxymethyl pseudouridine, 5-methyl cytidine, and/or 5-methoxy cytidine. In some embodiments, the polyribonucleotide includes a combination of at least two (e.g., 2, 3, 4 or more) of any of the aforementioned modified nucleobases, including but not limited to chemical modifications. In some embodiments, a mRNA of the disclosure comprises 1-methyl-pseudouridine (m1ψ) substitutions at one or more or all uridine positions of the nucleic acid. In some embodiments, a mRNA of the disclosure comprises 1-methyl-pseudouridine (m1ψ) substitutions at one or more or all uridine positions of the nucleic acid and 5-methyl cytidine substitutions at one or more or all cytidine positions of the nucleic acid. In some embodiments, a mRNA of the disclosure comprises pseudouridine (ψ) substitutions at one or more or all uridine positions of the nucleic acid. In some embodiments, a mRNA of the disclosure comprises pseudouridine (ψ) substitutions at one or more or all uridine positions of the nucleic acid and 5-methyl cytidine substitutions at one or more or all cytidine positions of the nucleic acid. In some embodiments, a mRNA of the disclosure comprises uridine at one or more or all uridine positions of the nucleic acid. In some embodiments, mRNAs are uniformly modified (e.g., fully modified, modified throughout the entire sequence) for a particular modification. For example, a nucleic acid can be uniformly modified with 1-methyl-pseudouridine, meaning that all uridine residues in the mRNA sequence are replaced with 1-methyl-pseudouridine. Similarly, a nucleic acid can be uniformly modified for any type of nucleoside residue present in the sequence by replacement with a modified residue such as those set forth above. The nucleic acids of the present disclosure may be partially or fully modified along the entire length of the molecule. For example, one or more or all or a given type of nucleotide (e.g., purine or pyrimidine, or any one or more or all of A, G, U, C) may be uniformly modified in a nucleic acid of the disclosure, or in a predetermined sequence region thereof (e.g., in the mRNA including or excluding the poly(A) tail). In some embodiments, all nucleotides X in a nucleic acid of the present disclosure (or in a sequence region thereof) are modified nucleotides, wherein X may be any one of nucleotides A, G, U, C, or any one of the combinations A+G, A+U, A+C, G+U, G+C, U+C, A+G+U, A+G+C, G+U+C or A+G+C. The nucleic acid may contain from about 1% to about 100% modified nucleotides (either in relation to overall nucleotide content, or in relation to one or more types of nucleotide, i.e., any one or more of A, G, U or C) or any intervening percentage (e.g., from 1% to 20%, from 1% to 25%, from 1% to 50%, from 1% to 60%, from 1% to 70%, from 1% to 80%, from 1% to 90%, from 1% to 95%, from 10% to 20%, from 10% to 25%, from 10% to 50%, from 10% to 60%, from 10% to 70%, from 10% to 80%, from 10% to 90%, from 10% to 95%, from 10% to 100%, from 20% to 25%, from 20% to 50%, from 20% to 60%, from 20% to 70%, from 20% to 80%, from 20% to 90%, from 20% to 95%, from 20% to 100%, from 50% to 60%, from 50% to 70%, from 50% to 80%, from 50% to 90%, from 50% to 95%, from 50% to 100%, from 70% to 80%, from 70% to 90%, from 70% to 95%, from 70% to 100%, from 80% to 90%, from 80% to 95%, from 80% to 100%, from 90% to 95%, from 90% to 100%, and from 95% to 100%). It will be understood that any remaining percentage is accounted for by the presence of unmodified A, G, U, or C. The mRNAs may contain at a minimum 1% and at maximum 100% modified nucleotides, or any intervening percentage, such as at least 5% modified nucleotides, at least 10% modified nucleotides, at least 25% modified nucleotides, at least 50% modified nucleotides, at least 80% modified nucleotides, or at least 90% modified nucleotides. For example, the nucleic acids may contain a modified pyrimidine such as a modified uracil or cytosine. In some embodiments, at least 5%, at least 10%, at least 25%, at least 50%, at least 80%, at least 90% or 100% of the uracil in the nucleic acid is replaced with a modified uracil (e.g., a 5-substituted uracil). The modified uracil can be replaced by a compound having a single unique structure or can be replaced by a plurality of compounds having different structures (e.g., 2, 3, 4 or more unique structures). In some embodiments, at least 5%, at least 10%, at least 25%, at least 50%, at least 80%, at least 90% or 100% of the cytosine in the nucleic acid is replaced with a modified cytosine (e.g., a 5-substituted cytosine). The modified cytosine can be replaced by a compound having a single unique structure or can be replaced by a plurality of compounds having different structures (e.g., 2, 3, 4 or more unique structures). Modified nucleotides may include modified nucleobases. For example, an RNA transcript (e.g., mRNA transcript) described herein may include a modified uracil nucleobase selected from pseudouracil (ψ), N1-methylpseudouracil (m1ψ), 1-ethylpseudouracil, 2-thiouracil, 4′-thiouracil, 2-thio-1-methyl-1-deaza-pseudouracil, 2-thio-1-methyl-pseudouracil, 2-thio-5-aza-uracil, 2-thio- dihydropseudouracil, 2-thio-dihydrouracil, 2-thio-pseudouracil, 4-methoxy-2-thio-pseudouracil, 4-methoxy-pseudouracil, 4-thio-1-methyl-pseudouracil, 4-thio-pseudouracil, 5-aza-uracil, dihydropseudouracil, 5-methyluracil, 5-methoxyuracil (mo5U) and 2′-O-methyluracil. In some embodiments, an RNA transcript (e.g., mRNA transcript) includes a modified guanine nucleobase selected from digoxigeninated guanine, 6-thioguanine, 7-deazaguanine, 7-deaza-7- propargylaminoguanine, 8-oxoguanine, araguanine, biotin-16-7-deaza-7-propargylaminoguanine, isoguanine, N2-methylguanine, O6-methylguanine, thienoguanine, and 2,6-daminoguanine. In some embodiments, an RNA transcript may include a modified cytosine nucleobase selected from digoxigeninated cytosine, 2-thiocytosine, 5-aminoallylcytosine, 5-bromocytosine, 5- carboxycytosine, 5-formylcytosine, 5-hydroxycytosine, 5-hydroxymethylcytosine, 5- methoxycytosine, 5-methylcytosine, 5-propargylaminocytosine, 5-propynylcytosine, 6- azacytosine, aracytosine, cyanine 3-5-propargylaminocytosine, cyanine 3-aminoallylcytosine, cyanine 5-6-propargylaminocytosine, cyanine 5-aminoallylcytosine, desthiobiotin-6- aminoallylcytosine, N4-biotin-OBEA-cytosine, N4-methylcytosine, pseudoisocytosine, and thienocytosine. In some embodiments, an RNA transcript (e.g., mRNA transcript) includes a modified adenine nucleobase selected from digoxigeninated adenine, N6-methyladenine, 7- deazaadenine, 7-deaza-7-propargylaminoadenine, 8-azaadenine, 8-azidoadenine, 8- chloroadenine, 8-oxoadenine, araadenine, N1-methyladenine, N6-methyladenine, 3- deazaadenine, 2,6-diaminoadenine, 2-methyl-thio-N6-isopentenyladenine (ms2i6A), 2- methylthio-N6-methyladenine (ms2m6A), N6-(cis-hydroxyisopentenyl)adenine (io6A), 2- methylthio-N6-(cis-hydroxyisopentenyl)adenine (ms2io6A), N6-glycinylcarbamoyladenine (g6A), N6-threonylcarbamoyladenine (t6A), 2-methylthio-N6-threonyl carbamoyladenine (ms2t6A), N6-methyl-N6-threonylcarbamoyladenine (m6t6A), N6- hydroxynorvalylcarbamoyladenine (hn6A), 2-methylthio-N6-hydroxynorvalyl carbamoyladenine (ms2hn6A), N6,N6-dimethyladenine (m62A), and N6-acetyladenine (ac6A). In some embodiments, an RNA transcript (e.g., mRNA transcript) includes a combination of at least two (e.g., 2, 3, 4 or more) of the foregoing modified nucleobases. Modified nucleotides may include modified sugars. For example, an RNA transcript (e.g., mRNA transcript) described herein may include a modified sugar selected from 2′-thioribose, 2′,3′-dideoxyribose, 2′-amino-2′-deoxyribose, 2′ deoxyribose, 2′-azido-2′-deoxyribose, 2′-fluoro- 2′-deoxyribose, 2′-O-methylribose, 2′-O-methyldeoxyribose, 3′-amino-2′,3′-dideoxyribose, 3′- azido-2′,3′-dideoxyribose, 3′-deoxyribose, 3′-O-(2-nitrobenzyl)-2′-deoxyribose, 3′-O- methylribose, 5′-aminoribose, 5′-thioribose, 5-nitro-1-indolyl-2′-deoxyribose, 5′-biotin-ribose, 2′- O,4′-C-methylene-linked, 2′-O,4′-C-amino-linked ribose, and 2′-O,4′-C-thio-linked ribose. In some embodiments, an RNA transcript (e.g., mRNA transcript) includes a combination of at least two (e.g., 2, 3, 4 or more) of the foregoing modified sugars. Modified nucleotides may include modified phosphates. A modified phosphate group is a phosphate group that differs from the canonical structure of phosphate. An example of a canonical structure of a phosphate is shown below: , where R5 and R3 are atoms or molecules to which the canonical phosphate is bonded. For example, for a phosphate in a nucleic acid sequence, R5 may refer to the upstream nucleotide of the nucleic acid, and R3 may refer to the downstream nucleotide of the nucleic acid. The canonical structure of phosphate also refers to structures in which one or more hydroxyl groups of the phosphate are deprotonated, or in which an oxygen atom of the phosphate is bonded to an adjacent nucleotide in a nucleic acid sequence. In some embodiments, an RNA transcript (e.g., mRNA transcript) described herein may include a modified phosphate selected from phosphorothioate (PS), thiophosphate, 5′-O-methylphosphonate, 3′-O-methylphosphonate, 5′- hydroxyphosphonate, hydroxyphosphanate, phosphoroselenoate, selenophosphate, phosphoramidate, carbophosphonate, methylphosphonate, phenylphosphonate, ethylphosphonate, H-phosphonate, guanidinium ring, triazole ring, boranophosphate (BP), methylphosphonate, and guanidinopropyl phosphoramidate. In some embodiments, an RNA transcript (e.g., mRNA transcript) includes a combination of at least two (e.g., 2, 3, 4 or more) of the foregoing modified phosphates. In some embodiments, an mRNA includes N1-methylpseudouridine. In some embodiments, at least 70%, at least 80%, at least 85%, at least 90%, at least 95%, at least 97%, at least 98%, or at least 99% of uracil nucleotides in an mRNA comprise N1-methylpseudouridine. In some embodiments, each uracil nucleotide of an mRNA transcript comprises N1- methylpseudouridine. In some embodiments, an mRNA includes 5-methylcytidine. In some embodiments, at least 70%, at least 80%, at least 85%, at least 90%, at least 95%, at least 97%, at least 98%, or at least 99% of cytosine nucleotides in an mRNA comprise 5-methylcytidine. In some embodiments, each cytosine nucleotide of an mRNA transcript comprises 5- methylcytidine. In some embodiments, an mRNA includes 5-methyluridine. In some embodiments, at least 70%, at least 80%, at least 85%, at least 90%, at least 95%, at least 97%, at least 98%, or at least 99% of uracil nucleotides in an mRNA comprise 5-methyluridine. In some embodiments, each uracil nucleotide of an mRNA transcript comprises 5-methyluridine. In some embodiments, an mRNA includes 5-methylcytidine and 5-methyluridine. In some embodiments, at least 70%, at least 80%, at least 85%, at least 90%, at least 95%, at least 97%, at least 98%, or at least 99% of uracil nucleotides in an mRNA comprise 5-methyluridine and at least 70%, at least 80%, at least 85%, at least 90%, at least 95%, at least 97%, at least 98%, or at least 99% of cytosine nucleotides in an mRNA comprise 5-methylcytidine. In some embodiments, each cytosine nucleotide of an mRNA transcript comprises 5-methylcytidine and each uracil nucleotide of an mRNA transcript comprises 5-methyluridine. Unmodified Nucleotides In some embodiments, an RNA (e.g., mRNA) is not chemically modified and comprises the standard ribonucleotides consisting of adenosine, guanosine, cytosine and uridine. In some embodiments, nucleotides and nucleosides of the present disclosure comprise standard nucleoside residues such as those present in transcribed RNA (e.g., A, G, C, or U). In some embodiments, nucleotides and nucleosides of the present disclosure comprise standard deoxyribonucleosides such as those present in DNA (e.g., dA, dG, dC, or dT). In Vitro Transcription (IVT) cDNA encoding the polynucleotides described herein may be transcribed using an in vitro transcription (IVT) system. In vitro transcription of RNA is known in the art and is described in International Publication WO 2014/152027, which is incorporated by reference herein in its entirety. In some embodiments, the RNA of the present disclosure is prepared in accordance with any one or more of the methods described in WO 2018/053209 and WO 2019/036682, each of which is incorporated by reference herein. In some embodiments, the RNA transcript is generated using a non-amplified, linearized DNA template in an in vitro transcription reaction to generate the RNA transcript. In some embodiments, the template DNA is isolated DNA. In some embodiments, the template DNA is cDNA. In some embodiments, the cDNA is formed by reverse transcription of a RNA polynucleotide, for example, but not limited to influenza virus mRNA. In some embodiments, cells, e.g., bacterial cells, e.g., E. coli, e.g., DH-1 cells are transfected with the plasmid DNA template. In some embodiments, the transfected cells are cultured to replicate the plasmid DNA which is then isolated and purified. In some embodiments, the DNA template includes a RNA polymerase promoter, e.g., a T7 promoter located 5 ' to and operably linked to the gene of interest. In some embodiments, an in vitro transcription template encodes a 5′ untranslated (UTR) region, contains an open reading frame, and encodes a 3′ UTR and a poly(A) tail. The particular nucleic acid sequence composition and length of an in vitro transcription template will depend on the mRNA encoded by the template. An in vitro transcription system typically comprises a transcription buffer, nucleotide triphosphates (NTPs), an RNase inhibitor and a polymerase. The NTPs may be manufactured in house, may be selected from a supplier, or may be synthesized as described herein. The NTPs may be selected from, but are not limited to, those described herein including natural and unnatural (modified) NTPs. Any number of RNA polymerases or variants may be used in the method of the present disclosure. The polymerase may be selected from, but is not limited to, a phage RNA polymerase, e.g., a T7 RNA polymerase, a T3 RNA polymerase, a SP6 RNA polymerase, and/or mutant polymerases such as, but not limited to, polymerases able to incorporate modified nucleic acids and/or modified nucleotides, including chemically modified nucleic acids and/or nucleotides. Some embodiments exclude the use of DNase. In some embodiments, the RNA transcript is capped via enzymatic capping. In some embodiments, the RNA comprises 5' terminal cap, for example, 7mG(5’)ppp(5’)NlmpNp. In some embodiments, the RNA polymerase is an RNA polymerase variant, such as those described in WO 2020/172239, incorporated herein by reference in its entirety. RNA polymerase variants of the present disclosure include at least one amino acid substitution, relative to the wild type (WT) RNA polymerase. A WT T7 RNA polymerase is represented by SEQ ID NO: 83: MNTINIAKNDFSDIELAAIPFNTLADHYGERLAREQLALEHESYEMGEARFRKMFERQLKAGEVADNAAAKPLITTLL PKMIARINDWFEEVKAKRGKRPTAFQFLQEIKPEAVAYITIKTTLACLTSADNTTVQAVASAIGRAIEDEARFGRIRD LEAKHFKKNVEEQLNKRVGHVYKKAFMQVVEADMLSKGLLGGEAWSSWHKEDSIHVGVRCIEMLIESTGMVSLHRQNA GVVGQDSETIELAPEYAEAIATRAGALAGISPMFQPCVVPPKPWTGITGGGYWANGRRPLALVRTHSKKALMRYEDVY MPEVYKAINIAQNTAWKINKKVLAVANVITKWKHCPVEDIPAIEREELPMKPEDIDMNPEALTAWKRAAAAVYRKDKA RKSRRISLEFMLEQANKFANHKAIWFPYNMDWRGRVYAVSMFNPQGNDMTKGLLTLAKGKPIGKEGYYWLKIHGANCA GVDKVPFPERIKFIEENHENIMACAKSPLENTWWAEQDSPFCFLAFCFEYAGVQHHGLSYNCSLPLAFDGSCSGIQHF SAMLRDEVGGRAVNLLPSETVQDIYGIVAKKVNEILQADAINGTDNEVVTVTDENTGEISEKVKLGTKALAGQWLAYG VTRSVTKRSVMTLAYGSKEFGFRQQVLEDTIQPAIDSGKGLMFTQPNQAAGYMAKLIWESVSVTVVAAVEAMNWLKSA AKLLAAEVKDKKTGEILRKRCAVHWVTPDGFPVWQEYKKPIQTRLNLMFLGQFRLQPTINTNKDSEIDAHKQESGIAP NFVHSQDGSHLRKTVVWAHEKYGIESFALIHDSFGTIPADAANLFKAVRETMVDTYESCDVLADFYDQFADQLHESQL DKMPALPAKGNLNLRDILESDFAFA (SEQ ID NO: 83). For example, with reference to WT T7 RNA polymerase having an amino acid sequence of SEQ ID NO: 83, the glycine at position 47 is considered a “wild-type amino acid,” whereas a substitution of the glycine for alanine at position 47 is considered an “amino acid substitution” that has a high-helix propensity. In some embodiments, the RNA polymerase variant is a T7 RNA polymerase variant comprising at least one (one or more) amino acid substitution relative to WT RNA polymerase (e.g., WT T7 RNA polymerase having an amino acid sequence of SEQ ID NO: 83). In some embodiments, a RNA polymerase variant comprises a RNA polymerase that includes an (at least one) amino acid modification causes a loop structure of the RNA polymerase variant to undergo a conformational change to a helix structure as the RNA polymerase variant transitions from an initiation complex to an elongation complex. In some embodiments, the amino acid modification is an amino acid substitution at one or more of positions 42, 43, 44, 45, 46, and 47, relative to the wild-type RNA polymerase, wherein the wild-type RNA polymerase comprises the amino acid sequence of SEQ ID NO: 83. The amino acid substitution, in some embodiments, is a high propensity amino acid substitution. Examples of high-helix propensity amino acids include alanine, isoleucine, leucine, arginine, methionine, lysine, glutamine, and/or glutamate. In some embodiments, the amino acid substitution at position 47 is G47A. In some embodiments, a RNA polymerase variant comprise a RNA polymerase that includes an additional C-terminal amino acid, relative to the wild-type RNA polymerase. The additional C-terminal amino acid, in some embodiments, is selected from glycine, alanine, threonine, proline, glutamine, and serine. In some embodiments, the additional C-terminal amino acid (e.g., at position 884 relative to wild-type RNA polymerase comprising the amino acid sequence of SEQ ID NO: 83) is glycine. Also provided herein are co-transcriptional capping methods for ribonucleic acid (RNA) synthesis, using any of the RNA polymerase variants described herein. That is, RNA is produced in a “one-pot” reaction, without the need for a separate capping reaction. Thus, the methods, in some embodiments, comprise reacting a polynucleotide template with a RNA polymerase variant, nucleoside triphosphates, and a cap analog under in vitro transcription reaction conditions to produce RNA transcript. Identification and Ratio Determination (IDR) Sequences In some embodiments, one or more nucleic acids comprises an Identification and Ratio Determination sequence. An Identification and Ratio Determination (IDR) sequence is a sequence of a biological molecule (e.g., nucleic acid or protein) that, when combined with the sequence of a target biological molecule, serves to identify the target biological molecule. Typically, an IDR sequence is a heterologous sequence that is incorporated within or appended to a sequence of a target biological molecule and can be used as a reference to identify the target molecule. Thus, in some embodiments, a nucleic acid (e.g., mRNA) comprises (i) a target sequence of interest (e.g., a coding sequence encoding a therapeutic and/or antigenic peptide or protein); and (ii) a unique IDR sequence. An RNA species (e.g., RNA having a given coding sequence) may comprise an IDR sequence that differs from the IDR sequence of other RNA species (e.g., RNA(s) having different coding sequence(s)). Each IDR sequence thus identifies a particular RNA species, and so the abundance of IDR sequences may be measured to determine the abundance of each RNA species in a composition. Use of distinct IDR sequences to identify RNA species allows for analysis of multivalent RNA compositions (e.g., containing multiple RNA species) containing RNA species with similar coding sequences and/or lengths, which could otherwise be difficult to distinguish using PCR- or chromatography-based analysis of full-length RNAs. Each RNA species in a multivalent RNA composition may comprise an IDR sequence that is not a sequence isomer of an IDR sequence of another RNA species in a multivalent RNA composition (e.g., the IDR sequence does not have the same number of adenosine nucleotides, the same number of cytosine nucleotides, the same number of guanine nucleotides, and the same number of uracil nucleotides, as another IDR sequence in the composition, even if those sequences have different sequences). Having identical nucleotide compositions causes sequence isomers to have the same mass, presenting a challenge to distinguishing sequence isomers using mass-based identification methods (e.g., mass spectrometry). Each RNA species in a multivalent RNA composition may comprise an IDR sequence having a mass that differs from the mass of IDR sequences of each other RNA species in a multivalent RNA composition. For example, the mass of each IDR sequence may differ from the mass of other IDR sequences by at least 9 Da, at least 25 Da, at least 25 Da, or at least 50 Da. Use of IDR sequences with distinct masses allows RNA fragments comprising different IDR sequences to be distinguished using mass-based analysis methods (e.g., mass spectrometry), which do not require reverse transcription, amplification, or sequencing of RNAs. Each RNA species in an RNA composition may comprises an IDR sequence with a different length. For example, each IDR sequence may have a length independently selected from 0 to 25 nucleotides. The length of a nucleic acid influences the rate at which the nucleic acid traverses a chromatography column, and so the use of IDR sequences of different lengths on different RNA species allows RNA fragments having different IDR sequences to be distinguished using chromatography-based methods (e.g., LC-UV). IDR sequences may be chosen such that no IDR sequence comprises a start codon, ‘AUG’. Lack of a start codon in an IDR sequence prevents undesired translation of nucleotide sequences within and/or downstream from the IDR sequence. IDR sequences may be chosen such that no IDR sequence comprises a recognition site for a restriction enzyme. In one example, no IDR sequence comprises a recognition site for XbaI, ‘UCUAG’. Lack of a recognition site for a restriction enzyme (e.g., XbaI recognition site ‘UCUAG’) allows the restriction enzyme to be used in generating and modifying a DNA template for in vitro transcription, without affecting the IDR sequence or sequence of the transcribed RNA. Non-limiting examples of distinct IDR sequences include: GAGAUUGAGUGUAGUGACUAG (SEQ ID NO: 56), GAGAUUGAGUGUAGUGAC (SEQ ID NO: 57), GAGAUUGAGUGUAGUG (SEQ ID NO: 58), GAUUGAGACUACGGG (SEQ ID NO: 59), and CAUAGACACUACG (SEQ ID NO: 60). In some embodiments of the compositions described herein, each mRNA encoding a distinct protein comprises a 3′ UTR comprising a distinct IDR sequence selected from SEQ ID NOs: 56–60. Nucleic Acid Production Chemical Synthesis Nucleic acids as described herein may be manufactured in whole or in part using solid phase techniques. Solid-phase chemical synthesis of nucleic acids is an automated method wherein molecules are immobilized on a solid support and synthesized step by step in a reactant solution. Solid-phase synthesis is useful in site-specific introduction of chemical modifications in the nucleic acid sequences. The synthesis of nucleic acids of the present disclosure by the sequential addition of monomer building blocks may be carried out in a liquid phase. The synthetic methods discussed above each has its own advantages and limitations. Attempts have been conducted to combine these methods to overcome the limitations. Such combinations of methods are within the scope of the present disclosure. The use of solid-phase or liquid-phase chemical synthesis in combination with enzymatic ligation provides an efficient way to generate long chain nucleic acids that cannot be obtained by chemical synthesis alone. Ligation Assembling nucleic acids by a ligase may also be used. DNA or RNA ligases promote intermolecular ligation of the 5’ and 3’ ends of polynucleotide chains through the formation of a phosphodiester bond. Nucleic acids such as chimeric polynucleotides and/or circular nucleic acids may be prepared by ligation of one or more regions or subregions. DNA fragments can be joined by a ligase catalyzed reaction to create recombinant DNA with different functions. Two oligodeoxynucleotides, one with a 5’ phosphoryl group and another with a free 3’ hydroxyl group, serve as substrates for a DNA ligase. Purification Purification of the nucleic acids described herein may include, but is not limited to, nucleic acid clean-up, quality assurance and quality control. Clean-up may be performed by methods known in the arts such as, but not limited to, AGENCOURT® beads (Beckman Coulter Genomics, Danvers, MA), poly-T beads, LNATM oligo-T capture probes (EXIQON® Inc, Vedbaek, Denmark) or HPLC based purification methods such as, but not limited to, strong anion exchange HPLC, weak anion exchange HPLC, reverse phase HPLC (RP-HPLC), and hydrophobic interaction HPLC (HIC-HPLC). The term “purified” when used in relation to a nucleic acid such as a “purified nucleic acid” refers to one that is separated from at least one contaminant. A “contaminant” is any substance that makes another unfit, impure or inferior. Thus, a purified nucleic acid (e.g., DNA and RNA) is present in a form or setting different from that in which it is found in nature, or a form or setting different from that which existed prior to subjecting it to a treatment or purification method. A quality assurance and/or quality control check may be conducted using methods such as, but not limited to, gel electrophoresis, UV absorbance, or analytical HPLC. In some embodiments, the nucleic acids may be sequenced by methods including, but not limited to reverse-transcriptase-PCR. Quantification In some embodiments, the nucleic acids of the present disclosure may be quantified in exosomes or when derived from one or more bodily fluid. Bodily fluids include peripheral blood, serum, plasma, ascites, urine, cerebrospinal fluid (CSF), sputum, saliva, bone marrow, synovial fluid, aqueous humor, amniotic fluid, cerumen, breast milk, broncheoalveolar lavage fluid, semen, prostatic fluid, cowper's fluid or pre-ejaculatory fluid, sweat, fecal matter, hair, tears, cyst fluid, pleural and peritoneal fluid, pericardial fluid, lymph, chyme, chyle, bile, interstitial fluid, menses, pus, sebum, vomit, vaginal secretions, mucosal secretion, stool water, pancreatic juice, lavage fluids from sinus cavities, bronchopulmonary aspirates, blastocyl cavity fluid, and umbilical cord blood. Alternatively, exosomes may be retrieved from an organ selected from the group consisting of lung, heart, pancreas, stomach, intestine, bladder, kidney, ovary, testis, skin, colon, breast, prostate, brain, esophagus, liver, and placenta. Assays may be performed using construct specific probes, cytometry, qRT-PCR, real-time PCR, PCR, flow cytometry, electrophoresis, mass spectrometry, or combinations thereof while the exosomes may be isolated using immunohistochemical methods such as enzyme linked immunosorbent assay (ELISA) methods. Exosomes may also be isolated by size exclusion chromatography, density gradient centrifugation, differential centrifugation, nanomembrane ultrafiltration, immunoabsorbent capture, affinity purification, microfluidic separation, or combinations thereof. These methods afford the investigator the ability to monitor, in real time, the level of nucleic acids remaining or delivered. This is possible because the nucleic acids of the present disclosure, in some embodiments, differ from the endogenous forms due to the structural or chemical modifications. In some embodiments, the nucleic acid may be quantified using methods such as, but not limited to, ultraviolet visible spectroscopy (UV/Vis). A non-limiting example of a UV/Vis spectrometer is a NANODROP® spectrometer (ThermoFisher, Waltham, MA). The quantified nucleic acid may be analyzed in order to determine if the nucleic acid may be of proper size, check that no degradation of the nucleic acid has occurred. Degradation of the nucleic acid may be checked by methods such as, but not limited to, agarose gel electrophoresis, HPLC based purification methods such as, but not limited to, strong anion exchange HPLC, weak anion exchange HPLC, reverse phase HPLC (RP-HPLC), and hydrophobic interaction HPLC (HIC- HPLC), liquid chromatography-mass spectrometry (LCMS), capillary electrophoresis (CE) and capillary gel electrophoresis (CGE). Lipid Compositions In some embodiments, the nucleic acids are formulated as a lipid composition, such as a composition comprising a lipid nanoparticle, a liposome, and/or a lipoplex. In some embodiments, nucleic acids are formulated as lipid nanoparticle (LNP) compositions. Lipid nanoparticles typically comprise amino lipid, non-cationic lipid, structural lipid, and PEG lipid components along with the nucleic acid cargo of interest. The lipid nanoparticles can be generated using components, compositions, and methods as are generally known in the art, see for example PCT/US2016/052352; PCT/US2016/068300; PCT/US2017/037551; PCT/US2015/027400; PCT/US2016/047406; PCT/US2016/000129; PCT/US2016/014280; PCT/US2017/038426; PCT/US2014/027077; PCT/US2014/055394; PCT/US2016/052117; PCT/US2012/069610; PCT/US2017/027492; PCT/US2016/059575; PCT/US2016/069491; PCT/US2016/069493; and PCT/US2014/066242, all of which are incorporated by reference herein in their entirety. In some embodiments, the lipid nanoparticle comprises at least one ionizable amino lipid, at least one non-cationic lipid, at least one sterol, and/or at least one polyethylene glycol (PEG)- modified lipid. In some embodiments, the lipid nanoparticle comprises a molar ratio of 20-60% ionizable amino lipid, 5-25% non-cationic lipid, 25-55% structural lipid, and 0.5-15% PEG-modified lipid. In some embodiments, the lipid nanoparticle comprises a molar ratio of 20-60% ionizable amino lipid, 5-30% non-cationic lipid, 10-55% structural lipid, and 0.5-15% PEG-modified lipid. In some embodiments, the lipid nanoparticle comprises 40-50 mol% ionizable lipid, optionally 45-50 mol%, for example, 45-46 mol%, 46-47 mol%, 47-48 mol%, 48-49 mol%, or 49-50 mol% for example about 45 mol%, 45.5 mol%, 46 mol%, 46.5 mol%, 47 mol%, 47.5 mol%, 48 mol%, 48.5 mol%, 49 mol%, or 49.5 mol%. In some embodiments, the lipid nanoparticle comprises 20-60 mol% ionizable amino lipid. For example, the lipid nanoparticle may comprise 20-50 mol%, 20-40 mol%, 20-30 mol%, 30-60 mol%, 30-50 mol%, 30-40 mol%, 40-60 mol%, 40-50 mol%, or 50-60 mol% ionizable amino lipid. In some embodiments, the lipid nanoparticle comprises 20 mol%, 30 mol%, 40 mol%, 50 mol%, or 60 mol% ionizable amino lipid. In some embodiments, the lipid nanoparticle comprises 35 mol%, 36 mol%, 37 mol%, 38 mol%, 39 mol%, 40 mol%, 41 mol%, 42 mol%, 43 mol%, 44 mol%, 45 mol%, 46 mol%, 47 mol%, 48 mol%, 49 mol%, 50 mol%, 51 mol%, 52 mol%, 53 mol%, 54 mol%, or 55 mol% ionizable amino lipid. In some embodiments, the lipid nanoparticle comprises 45 – 55 mole percent (mol%) ionizable amino lipid. For example, lipid nanoparticle may comprise 45, 46, 47, 48, 49, 50, 51, 52, 53, 54, or 55 mol% ionizable amino lipid. Ionizable amino lipids Formula (AI) In some embodiments, the ionizable amino lipid of a lipid nanoparticle is a compound of Formula (AI):
AAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA (SEQ ID NO: 80). In some embodiments, an mRNA comprises a poly(A) sequence that has a length of 90– 120 nucleotides. In some embodiments, an mRNA comprises a poly(A) sequence that comprises 100, 101, 102, 103, 104, 105, 106, 107, 108, 109, 110, 111, 112, 113, 114, 115, 116, 117, 118, 190, or 120 consecutive adenosine nucleotides. In some embodiments, an mRNA comprises a poly(A) sequence that comprises at least 109 consecutive adenosine nucleotides. In some embodiments, an mRNA comprises a poly(A) sequence that comprises 109 consecutive adenosine nucleotides. In some embodiments, an mRNA comprises a poly(A) sequence that consists of 109 consecutive adenosine nucleotides. Self-amplifying RNA Some aspects of the disclosure relate to self-amplifying RNA (e.g., an RNA replicon) encoding a MERS-CoV S protein or variant thereof (e.g., fusion protein comprising one or more domains of a MERS-CoV S protein). An self-amplifying RNA refers to an RNA encoding one or more molecules (e.g., proteins) that, individually or in conjunction, are capable of replicating the self-amplifying RNA. In some embodiments, the proteins encoded by the self-amplifying RNA are non-structural proteins nsP1, nsP2, nsP3, and nsP4, which form an RNA-dependent RNA polymerase (RdRp), or replicase, that is capable of replicating the self-amplifying RNA. By encoding proteins that are capable of replicating the RNA, a self-amplifying RNA is capable of self-amplification in a cell, provided that the cell can translate the RNA and produce the encoded protein(s). A self-amplifying RNA may be referred to as an RNA replicon. When an RNA replicon or self-amplifying RNA is translated, the one or more encoded viral non-structural proteins are translated. A “viral non-structural protein” is a protein encoded by a virus but that is not part of the virus particle. The viral non-structural proteins, in the context of self-amplifying RNA, replicate the nucleotide sequences encoding the vaccine antigen or therapeutic protein (e.g., MERS-CoV protein) from the self-amplifying RNA via the sub- genomic viral promoters. Such replication driven by the viral sub-genomic promoter using the viral non- structural proteins enhances the expression level of the encoded protein. In some embodiments, the viral non-structural proteins are from a single-strand positive-sense RNA viruses. In some embodiments, the viral non-structural proteins are from an Alphavirus, belonging to the Togaviridae family. In some embodiments, the alphavirus is Sindbis or Venezuelan equine encephalitis virus. In some embodiments, the viral non-structural protein is an RNA-dependent RNA polymerase (RdRp) polyprotein P1234 (also termed NSP1-4). Upon translation, P1234 is rapidly cleaved into P123 and nsP4 by autoproteolytic activity originating from the nsP2 (proteinase) portion of the polyprotein. Alphaviral RNA synthesis occurs at the plasma membrane of a cell, where the nsPs, together with alphaviral RNA, form membrane invaginations (or “spherules”). These spherules contain dsRNA created by replication of “+” strand viral genomic RNA into “–“ strand anti-genomic RNA. The “–“ strand serves as a template from which additional “+” strand genomic RNA (synthesized from the 5’UTR) or a shorter subsequence of the genomic RNA (termed subgenomic RNA) is synthesized from the subgenomic viral promoter region located near the end of the nonstructural protein ORF. The “+” strand genomic RNA and the subgenomic RNA are exported out of the spherules into the cytoplasm where they are translated by endogenous ribosomes. The exported “+” strand genomic RNA can associate with nsPs and form additional spherules, thus resulting in exponential increase of replicon RNA. The viral non-structural proteins facilitate the replication of the nucleotide sequences encoding the MERS-CoV protein via the subgenomic viral promoters (also referred to as “subgenomic promoters” herein). A “subgenomic viral promoter” refers to a promoter the drives the transcription of subgenomic mRNAs. Typically, an mRNA is transcribed from genomic DNAs and episomal DNAs (e.g., plasmids). Some viruses may transcribe subgenomic mRNAs from a RNA replicon that is produced from its genomic RNA. Many positive-sense RNA viruses produce subgenomic mRNAs as one of the common infection techniques used by these viruses and generally transcribe late viral genes. Subgenomic viral promoters range from 20 nucleotide (Sindbis virus) to over 100 nucleotides (Beet necrotic yellow vein virus) and are usually found upstream of the transcription start. In some embodiments, the subgenomic viral promoter is 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49, 50, 51, 52, 53, 54, 55, 56, 57, 58, 59, 60, 61, 62, 63, 64, 65, 66, 67, 68, 69, 70, 71, 72, 73, 74, 75, 76, 77, 78, 79, 80, 81, 82, 83, 84, 85, 86, 87, 88, 89, 90, 91, 92, 93, 94, 95, 96, 97, 98, 99, 100 nucleotides long, or longer. Subgenomic viral promoters have been described in the art, e.g., in PCT Application Publication No. WO 2016/040359, and Wagner et al., Nature Chemical Biology, DOI: 10.1038/s41589-018-0146-9 (2018). Additional Stabilizing Elements RNA (e.g., mRNA) provided herein, in some embodiments, includes an additional stabilizing element. Stabilizing elements may include, for example, a histone stem-loop. A stem- loop binding protein (SLBP), a 32 kDa protein has been identified. It is associated with the histone stem-loop at the 3'-end of the histone messages in both the nucleus and the cytoplasm. Its expression level is regulated by the cell cycle; it peaks during the S-phase, when histone mRNA levels are also elevated. The protein has been shown to be essential for efficient 3'-end processing of histone pre-mRNA by the U7 snRNP. SLBP continues to be associated with the stem-loop after processing, and then stimulates the translation of mature histone mRNAs into histone proteins in the cytoplasm. The RNA binding domain of SLBP is conserved through metazoa and protozoa; its binding to the histone stem-loop depends on the structure of the loop. The minimum binding site includes at least three nucleotides 5’ and two nucleotides 3 ^ relative to the stem-loop. In some embodiments, an RNA (e.g., mRNA) includes an open reading frame (coding region), a histone stem-loop, and optionally, a poly(A) sequence or polyadenylation signal. The poly(A) sequence or polyadenylation signal generally should enhance the expression level of the encoded protein. The encoded protein, in some embodiments, is not a histone protein, a reporter protein (e.g., Luciferase, GFP, EGFP, β-Galactosidase, EGFP), or a marker or selection protein (e.g., alpha-Globin, Galactokinase and Xanthine:guanine phosphoribosyl transferase (GPT)). In some embodiments, an RNA (e.g., mRNA) includes the combination of a poly(A) sequence or polyadenylation signal and at least one histone stem-loop, even though both represent alternative mechanisms in nature, they act synergistically to increase the protein expression beyond the level observed with either of the individual elements. The synergistic effect of the combination of poly(A) and a histone stem-loop does not depend on the order of the elements or the length of the poly(A) sequence. In some embodiments, an RNA (e.g., mRNA) does not include a histone downstream element (HDE). “Histone downstream element” (HDE) includes a purine-rich polynucleotide stretch of approximately 15 to 20 nucleotides 3′ of naturally-occurring stem-loops, representing the binding site for the U7 snRNA, which is involved in processing of histone pre-mRNA into mature histone mRNA. In some embodiments, the nucleic acid does not include an intron. An RNA (e.g., mRNA) may or may not contain an enhancer and/or promoter sequence, which may be modified or unmodified or which may be activated or inactivated. In some embodiments, the histone stem-loop is generally derived from histone genes and includes an intramolecular base pairing of two neighbored partially or entirely reverse complementary sequences separated by a spacer, consisting of a short sequence, which forms the loop of the structure. The unpaired loop region is typically unable to base pair with either of the stem loop elements. It occurs more often in RNA, as is a key component of many RNA secondary structures but may be present in single-stranded DNA as well. Stability of the stem-loop structure generally depends on the length, number of mismatches or bulges, and base composition of the paired region. In some embodiments, wobble base pairing (non-Watson-Crick base pairing) may result. In some embodiments, the at least one histone stem-loop sequence comprises a length of 15 to 45 nucleotides. In some embodiments, an RNA (e.g., mRNA) has one or more AU-rich sequences removed. These sequences, sometimes referred to as AURES are destabilizing sequences found in the 3 ’UTR. The AURES may be removed from the mRNA. Alternatively, the AURES may remain in the mRNA. Sequence Modification In some embodiments, an open reading frame encoding a protein of the disclosure is codon optimized. Codon optimization methods are known in the art. An open reading frame of any one or more of the sequences provided herein may be codon optimized. Codon optimization, in some embodiments, may be used to match codon frequencies in target and host organisms to ensure proper folding; bias GC content to increase RNA (e.g., mRNA) stability or reduce secondary structures; minimize tandem repeat codons or base runs that may impair gene construction or expression; customize transcriptional and translational control regions; insert or remove protein trafficking sequences; remove/add post translation modification sites in encoded protein (e.g., glycosylation sites); add, remove or shuffle protein domains; insert or delete restriction sites; modify ribosome binding sites and RNA (e.g., mRNA) degradation sites; adjust translational rates to allow the various domains of the protein to fold properly; or reduce or eliminate problem secondary structures within the polynucleotide. Codon optimization tools, algorithms and services are known in the art – non-limiting examples include services from GeneArt (Life Technologies), DNA2.0 (Menlo Park CA) and/or proprietary methods. In some embodiments, the open reading frame sequence is optimized using optimization algorithms. In some embodiments, a codon optimized sequence shares less than 95% sequence identity to a naturally-occurring or wild-type sequence open reading frame (e.g., a naturally- occurring or wild-type RNA (e.g., mRNA) sequence encoding a MERS-CoV protein antigen). In some embodiments, a codon optimized sequence shares less than 90% sequence identity to a naturally-occurring or wild-type sequence (e.g., a naturally-occurring or wild-type RNA (e.g., mRNA) sequence encoding a MERS-CoV protein). In some embodiments, a codon optimized sequence shares less than 85% sequence identity to a naturally-occurring or wild-type sequence (e.g., a naturally-occurring or wild-type RNA (e.g., mRNA) sequence encoding a MERS-CoV protein). In some embodiments, a codon optimized sequence shares less than 80% sequence identity to a naturally-occurring or wild-type sequence (e.g., a naturally-occurring or wild-type RNA (e.g., mRNA) sequence encoding a MERS-CoV protein). In some embodiments, a codon optimized sequence shares less than 75% sequence identity to a naturally-occurring or wild-type sequence (e.g., a naturally-occurring or wild-type RNA (e.g., mRNA) sequence encoding a MERS-CoV protein). In some embodiments, a codon optimized sequence shares between 65% and 85% (e.g., between about 67% and about 85% or between about 67% and about 80%) sequence identity to a naturally-occurring or wild-type sequence (e.g., a naturally-occurring or wild-type RNA (e.g., mRNA) sequence encoding a MERS-CoV protein). In some embodiments, a codon optimized sequence shares between 65% and 75% or about 80% sequence identity to a naturally-occurring or wild-type sequence (e.g., a naturally-occurring or wild-type RNA (e.g., mRNA) sequence encoding a MERS-CoV protein). In some embodiments, a codon-optimized sequence encodes an antigen that is as immunogenic as, or more immunogenic than (e.g., at least 10%, at least 20%, at least 30%, at least 40%, at least 50%, at least 100%, or at least 200% more), than a MERS-CoV protein encoded by a non-codon-optimized sequence. When transfected into mammalian host cells, the modified mRNAs have a stability of between 12-18 hours, or greater than 18 hours, e.g., 24, 36, 48, 60, 72, or greater than 72 hours and are capable of being expressed by the mammalian host cells. In some embodiments, a codon optimized RNA (e.g., mRNA) may be one in which the levels of G/C are enhanced. The G/C-content of nucleic acid molecules (e.g., mRNA) may influence the stability of the RNA. RNA (e.g., mRNA) having an increased amount of guanine (G) and/or cytosine (C) residues may be functionally more stable than RNA containing a large amount of adenine (A) and thymine (T) or uracil (U) nucleotides. As an example, WO02/098443 discloses a pharmaceutical composition containing an RNA (e.g., mRNA) stabilized by sequence modifications in the translated region. Due to the degeneracy of the genetic code, the modifications work by substituting existing codons for those that promote greater RNA stability without changing the resulting amino acid. The approach is limited to coding regions of the RNA (e.g., mRNA). Some embodiments of mRNAs described herein, comprise a sequence with a %G/C content of 30% – 80%, 40% – 70%, 50% – 60%, 35% – 50%, 50% – 65%, 65% – 70%, 40% – 45%, 45% – 50%, 50% – 55%, 55% – 70%, 70% – 75%, or 75% – 80%. In some embodiments, the nucleic acid sequence of the full-length mRNA comprises a %G/C content of 30% to 80%, 40% – 70%, 50% – 60%, 35% – 50%, 50% – 65%, 65% – 70%, 40% – 45%, 45% – 50%, 50% – 55%, 55% – 70%, 70% – 75%, or 75% – 80%. In some embodiments, the mRNA comprises an ORF with a %G/C content from about 30% to about 80%, about 35% to about 70%, about 40% to about 60%, about 45% to about 55%, about 40% to about 70%, about 50% to about 60%, about 35% to about 50%, about 50% to about 50% to about 65%, about 65% to about 70%, about 40% to about 45%, about 45% to about 50%, about 50% to about 55%, about 55% to about 70%, about 70% to about 75%, or about 75% to about 80%. In some embodiments, the mRNA comprises 5′ UTR with a %G/C content from about 30% to about 80%, about 35% to about 70%, about 40% to about 60%, about 45% to about 55%, about 40% to about 70%, about 50% to about 60%, about 35% to about 50%, about 50% to about 50% to about 65%, about 65% to about 70%, about 40% to about 45%, about 45% to about 50%, about 50% to about 55%, about 55% to about 70%, about 70% to about 75%, or about 75% to about 80%. In some embodiments, the mRNA comprises 3′ UTR with a %G/C content from about 30% to about 80%, about 35% to about 70%, about 40% to about 60%, about 45% to about 55%, about 40% to about 70%, about 50% to about 60%, about 35% to about 50%, about 50% to about 50% to about 65%, about 65% to about 70%, about 40% to about 45%, about 45% to about 50%, about 50% to about 55%, about 55% to about 70%, about 70% to about 75%, or about 75% to about 80%. In some embodiments, a modified mRNA made by a method described herein comprises a higher %G/C content than a wild-type mRNA sequence. In some embodiments, the %G/C content of the modified mRNA sequence is 2% or more, 3% or more, 4% or more, 5% or more, 6% or more, 7% or more, 8% or more, 9% or more, 10% or more, 12% or more, 15% or more, or 20% or more than the %G/C content of the wild-type RNA sequence. In some embodiments, the %G/C content of the modified ORF sequence is 2% or more, 3% or more, 4% or more, 5% or more, 6% or more, 7% or more, 8% or more, 9% or more, 10% or more, 12% or more, 15% or more, or 20% or more than the %G/C content of the wild-type ORF sequence. In some embodiments, the %G/C content of the modified 5′ UTR sequence is 2% or more, 3% or more, 4% or more, 5% or more, 6% or more, 7% or more, 8% or more, 9% or more, 10% or more, 12% or more, 15% or more, or 20% or more than the %G/C content of the wild-type 3′ UTR sequence. Chemically Modified Nucleotides The compositions of the present disclosure comprise, in some embodiments, an RNA having an open reading frame encoding a protein, wherein the nucleic acid comprises nucleotides and/or nucleosides that can be standard (unmodified) or modified as is known in the art. In some embodiments, nucleotides and nucleosides of the present disclosure comprise modified nucleotides or nucleosides. Such modified nucleotides and nucleosides can be naturally- occurring modified nucleotides and nucleosides or non-naturally-occurring modified nucleotides and nucleosides. Such modifications can include those at the sugar, backbone, or nucleobase portion of the nucleotide and/or nucleoside as are recognized in the art. In some embodiments, a naturally-occurring modified nucleotide or nucleotide of the disclosure is one as is generally known or recognized in the art. Non-limiting examples of such naturally-occurring modified nucleotides and nucleotides can be found, inter alia, in the widely recognized MODOMICS database. In some embodiments, a non-naturally-occurring modified nucleotide or nucleoside of the disclosure is one as is generally known or recognized in the art. Non-limiting examples of such non-naturally-occurring modified nucleotides and nucleosides can be found, inter alia, in international publication numbers WO2013/052523A1; WO2014/093924A1; WO2015/051173A2; WO2015/051169A2; WO2015/089511A2; or WO2017/153936A1, each of which is herein incorporated by reference. Hence, nucleic acids of the disclosure (e.g., DNA and RNA, such as mRNA) can comprise standard nucleotides and nucleosides, naturally-occurring nucleotides and nucleosides, non-naturally-occurring nucleotides and nucleosides, or any combination thereof. Nucleic acids of the disclosure (e.g., DNA and RNA, such as mRNA), in some embodiments, comprise various (more than one) different types of standard and/or modified nucleotides and nucleosides. In some embodiments, a particular region of a nucleic acid contains one, two or more (optionally different) types of standard and/or modified nucleotides and nucleosides. In some embodiments, a modified RNA (e.g., mRNA) introduced to a cell or organism, exhibits reduced degradation in the cell or organism, respectively, relative to an unmodified nucleic acid comprising standard nucleotides and nucleosides. In some embodiments, a modified RNA (e.g., mRNA) introduced into a cell or organism, may exhibit reduced immunogenicity in the cell or organism, respectively (e.g., a reduced innate response) relative to an unmodified nucleic acid comprising standard nucleotides and nucleosides. Nucleic acids (e.g., RNA, such as mRNA), in some embodiments, comprise non-natural modified nucleotides that are introduced during synthesis or post-synthesis of the nucleic acids to achieve desired functions or properties. The modifications may be present on internucleotide linkages, purine or pyrimidine bases, or sugars. The modification may be introduced with chemical synthesis or with a polymerase enzyme at the terminal of a chain or anywhere else in the chain. Any of the regions of a nucleic acid may be chemically modified. The present disclosure provides for modified nucleosides and nucleotides of a nucleic acid (e.g., RNA nucleic acids, such as mRNA nucleic acids). A “nucleoside” refers to a compound containing a sugar molecule (e.g., a pentose or ribose) or a derivative thereof in combination with an organic base (e.g., a purine or pyrimidine) or a derivative thereof (also referred to herein as “nucleobase”). A “nucleotide” refers to a nucleoside, including a phosphate group. Modified nucleotides may by synthesized by any useful method, such as, for example, chemically, enzymatically, or recombinantly, to include one or more modified or non-natural nucleosides. Nucleic acids can comprise a region or regions of linked nucleosides. Such regions may have variable backbone linkages. The linkages can be standard phosphodiester linkages, in which case the nucleic acids would comprise regions of nucleotides. Modified nucleotide base pairing encompasses not only the standard adenosine-thymine, adenosine-uracil, or guanosine-cytosine base pairs, but also base pairs formed between nucleotides and/or modified nucleotides comprising non-standard or modified bases, wherein the arrangement of hydrogen bond donors and hydrogen bond acceptors permits hydrogen bonding between a non-standard base and a standard base or between two complementary non-standard base structures, such as, for example, in those nucleic acids having at least one chemical modification. One example of such non-standard base pairing is the base pairing between the modified nucleotide inosine and adenine, cytosine or uracil. Any combination of base/sugar or linker may be incorporated into nucleic acids of the present disclosure. In some embodiments, modified nucleobases in nucleic acids (e.g., RNA nucleic acids, such as mRNA nucleic acids) comprise 1-methyl-pseudouridine (m1ψ), 1-ethyl-pseudouridine (e1ψ), 5-methoxy-uridine (mo5U), 5-methyl-cytidine (m5C), and/or pseudouridine (ψ). In some embodiments, modified nucleobases in nucleic acids (e.g., RNA nucleic acids, such as mRNA nucleic acids) comprise 5-methoxymethyl uridine, 5-methylthio uridine, 1-methoxymethyl pseudouridine, 5-methyl cytidine, and/or 5-methoxy cytidine. In some embodiments, the polyribonucleotide includes a combination of at least two (e.g., 2, 3, 4 or more) of any of the aforementioned modified nucleobases, including but not limited to chemical modifications. In some embodiments, a mRNA of the disclosure comprises 1-methyl-pseudouridine (m1ψ) substitutions at one or more or all uridine positions of the nucleic acid. In some embodiments, a mRNA of the disclosure comprises 1-methyl-pseudouridine (m1ψ) substitutions at one or more or all uridine positions of the nucleic acid and 5-methyl cytidine substitutions at one or more or all cytidine positions of the nucleic acid. In some embodiments, a mRNA of the disclosure comprises pseudouridine (ψ) substitutions at one or more or all uridine positions of the nucleic acid. In some embodiments, a mRNA of the disclosure comprises pseudouridine (ψ) substitutions at one or more or all uridine positions of the nucleic acid and 5-methyl cytidine substitutions at one or more or all cytidine positions of the nucleic acid. In some embodiments, a mRNA of the disclosure comprises uridine at one or more or all uridine positions of the nucleic acid. In some embodiments, mRNAs are uniformly modified (e.g., fully modified, modified throughout the entire sequence) for a particular modification. For example, a nucleic acid can be uniformly modified with 1-methyl-pseudouridine, meaning that all uridine residues in the mRNA sequence are replaced with 1-methyl-pseudouridine. Similarly, a nucleic acid can be uniformly modified for any type of nucleoside residue present in the sequence by replacement with a modified residue such as those set forth above. The nucleic acids of the present disclosure may be partially or fully modified along the entire length of the molecule. For example, one or more or all or a given type of nucleotide (e.g., purine or pyrimidine, or any one or more or all of A, G, U, C) may be uniformly modified in a nucleic acid of the disclosure, or in a predetermined sequence region thereof (e.g., in the mRNA including or excluding the poly(A) tail). In some embodiments, all nucleotides X in a nucleic acid of the present disclosure (or in a sequence region thereof) are modified nucleotides, wherein X may be any one of nucleotides A, G, U, C, or any one of the combinations A+G, A+U, A+C, G+U, G+C, U+C, A+G+U, A+G+C, G+U+C or A+G+C. The nucleic acid may contain from about 1% to about 100% modified nucleotides (either in relation to overall nucleotide content, or in relation to one or more types of nucleotide, i.e., any one or more of A, G, U or C) or any intervening percentage (e.g., from 1% to 20%, from 1% to 25%, from 1% to 50%, from 1% to 60%, from 1% to 70%, from 1% to 80%, from 1% to 90%, from 1% to 95%, from 10% to 20%, from 10% to 25%, from 10% to 50%, from 10% to 60%, from 10% to 70%, from 10% to 80%, from 10% to 90%, from 10% to 95%, from 10% to 100%, from 20% to 25%, from 20% to 50%, from 20% to 60%, from 20% to 70%, from 20% to 80%, from 20% to 90%, from 20% to 95%, from 20% to 100%, from 50% to 60%, from 50% to 70%, from 50% to 80%, from 50% to 90%, from 50% to 95%, from 50% to 100%, from 70% to 80%, from 70% to 90%, from 70% to 95%, from 70% to 100%, from 80% to 90%, from 80% to 95%, from 80% to 100%, from 90% to 95%, from 90% to 100%, and from 95% to 100%). It will be understood that any remaining percentage is accounted for by the presence of unmodified A, G, U, or C. The mRNAs may contain at a minimum 1% and at maximum 100% modified nucleotides, or any intervening percentage, such as at least 5% modified nucleotides, at least 10% modified nucleotides, at least 25% modified nucleotides, at least 50% modified nucleotides, at least 80% modified nucleotides, or at least 90% modified nucleotides. For example, the nucleic acids may contain a modified pyrimidine such as a modified uracil or cytosine. In some embodiments, at least 5%, at least 10%, at least 25%, at least 50%, at least 80%, at least 90% or 100% of the uracil in the nucleic acid is replaced with a modified uracil (e.g., a 5-substituted uracil). The modified uracil can be replaced by a compound having a single unique structure or can be replaced by a plurality of compounds having different structures (e.g., 2, 3, 4 or more unique structures). In some embodiments, at least 5%, at least 10%, at least 25%, at least 50%, at least 80%, at least 90% or 100% of the cytosine in the nucleic acid is replaced with a modified cytosine (e.g., a 5-substituted cytosine). The modified cytosine can be replaced by a compound having a single unique structure or can be replaced by a plurality of compounds having different structures (e.g., 2, 3, 4 or more unique structures). Modified nucleotides may include modified nucleobases. For example, an RNA transcript (e.g., mRNA transcript) described herein may include a modified uracil nucleobase selected from pseudouracil (ψ), N1-methylpseudouracil (m1ψ), 1-ethylpseudouracil, 2-thiouracil, 4′-thiouracil, 2-thio-1-methyl-1-deaza-pseudouracil, 2-thio-1-methyl-pseudouracil, 2-thio-5-aza-uracil, 2-thio- dihydropseudouracil, 2-thio-dihydrouracil, 2-thio-pseudouracil, 4-methoxy-2-thio-pseudouracil, 4-methoxy-pseudouracil, 4-thio-1-methyl-pseudouracil, 4-thio-pseudouracil, 5-aza-uracil, dihydropseudouracil, 5-methyluracil, 5-methoxyuracil (mo5U) and 2′-O-methyluracil. In some embodiments, an RNA transcript (e.g., mRNA transcript) includes a modified guanine nucleobase selected from digoxigeninated guanine, 6-thioguanine, 7-deazaguanine, 7-deaza-7- propargylaminoguanine, 8-oxoguanine, araguanine, biotin-16-7-deaza-7-propargylaminoguanine, isoguanine, N2-methylguanine, O6-methylguanine, thienoguanine, and 2,6-daminoguanine. In some embodiments, an RNA transcript may include a modified cytosine nucleobase selected from digoxigeninated cytosine, 2-thiocytosine, 5-aminoallylcytosine, 5-bromocytosine, 5- carboxycytosine, 5-formylcytosine, 5-hydroxycytosine, 5-hydroxymethylcytosine, 5- methoxycytosine, 5-methylcytosine, 5-propargylaminocytosine, 5-propynylcytosine, 6- azacytosine, aracytosine, cyanine 3-5-propargylaminocytosine, cyanine 3-aminoallylcytosine, cyanine 5-6-propargylaminocytosine, cyanine 5-aminoallylcytosine, desthiobiotin-6- aminoallylcytosine, N4-biotin-OBEA-cytosine, N4-methylcytosine, pseudoisocytosine, and thienocytosine. In some embodiments, an RNA transcript (e.g., mRNA transcript) includes a modified adenine nucleobase selected from digoxigeninated adenine, N6-methyladenine, 7- deazaadenine, 7-deaza-7-propargylaminoadenine, 8-azaadenine, 8-azidoadenine, 8- chloroadenine, 8-oxoadenine, araadenine, N1-methyladenine, N6-methyladenine, 3- deazaadenine, 2,6-diaminoadenine, 2-methyl-thio-N6-isopentenyladenine (ms2i6A), 2- methylthio-N6-methyladenine (ms2m6A), N6-(cis-hydroxyisopentenyl)adenine (io6A), 2- methylthio-N6-(cis-hydroxyisopentenyl)adenine (ms2io6A), N6-glycinylcarbamoyladenine (g6A), N6-threonylcarbamoyladenine (t6A), 2-methylthio-N6-threonyl carbamoyladenine (ms2t6A), N6-methyl-N6-threonylcarbamoyladenine (m6t6A), N6- hydroxynorvalylcarbamoyladenine (hn6A), 2-methylthio-N6-hydroxynorvalyl carbamoyladenine (ms2hn6A), N6,N6-dimethyladenine (m62A), and N6-acetyladenine (ac6A). In some embodiments, an RNA transcript (e.g., mRNA transcript) includes a combination of at least two (e.g., 2, 3, 4 or more) of the foregoing modified nucleobases. Modified nucleotides may include modified sugars. For example, an RNA transcript (e.g., mRNA transcript) described herein may include a modified sugar selected from 2′-thioribose, 2′,3′-dideoxyribose, 2′-amino-2′-deoxyribose, 2′ deoxyribose, 2′-azido-2′-deoxyribose, 2′-fluoro- 2′-deoxyribose, 2′-O-methylribose, 2′-O-methyldeoxyribose, 3′-amino-2′,3′-dideoxyribose, 3′- azido-2′,3′-dideoxyribose, 3′-deoxyribose, 3′-O-(2-nitrobenzyl)-2′-deoxyribose, 3′-O- methylribose, 5′-aminoribose, 5′-thioribose, 5-nitro-1-indolyl-2′-deoxyribose, 5′-biotin-ribose, 2′- O,4′-C-methylene-linked, 2′-O,4′-C-amino-linked ribose, and 2′-O,4′-C-thio-linked ribose. In some embodiments, an RNA transcript (e.g., mRNA transcript) includes a combination of at least two (e.g., 2, 3, 4 or more) of the foregoing modified sugars. Modified nucleotides may include modified phosphates. A modified phosphate group is a phosphate group that differs from the canonical structure of phosphate. An example of a canonical structure of a phosphate is shown below: , where R5 and R3 are atoms or molecules to which the canonical phosphate is bonded. For example, for a phosphate in a nucleic acid sequence, R5 may refer to the upstream nucleotide of the nucleic acid, and R3 may refer to the downstream nucleotide of the nucleic acid. The canonical structure of phosphate also refers to structures in which one or more hydroxyl groups of the phosphate are deprotonated, or in which an oxygen atom of the phosphate is bonded to an adjacent nucleotide in a nucleic acid sequence. In some embodiments, an RNA transcript (e.g., mRNA transcript) described herein may include a modified phosphate selected from phosphorothioate (PS), thiophosphate, 5′-O-methylphosphonate, 3′-O-methylphosphonate, 5′- hydroxyphosphonate, hydroxyphosphanate, phosphoroselenoate, selenophosphate, phosphoramidate, carbophosphonate, methylphosphonate, phenylphosphonate, ethylphosphonate, H-phosphonate, guanidinium ring, triazole ring, boranophosphate (BP), methylphosphonate, and guanidinopropyl phosphoramidate. In some embodiments, an RNA transcript (e.g., mRNA transcript) includes a combination of at least two (e.g., 2, 3, 4 or more) of the foregoing modified phosphates. In some embodiments, an mRNA includes N1-methylpseudouridine. In some embodiments, at least 70%, at least 80%, at least 85%, at least 90%, at least 95%, at least 97%, at least 98%, or at least 99% of uracil nucleotides in an mRNA comprise N1-methylpseudouridine. In some embodiments, each uracil nucleotide of an mRNA transcript comprises N1- methylpseudouridine. In some embodiments, an mRNA includes 5-methylcytidine. In some embodiments, at least 70%, at least 80%, at least 85%, at least 90%, at least 95%, at least 97%, at least 98%, or at least 99% of cytosine nucleotides in an mRNA comprise 5-methylcytidine. In some embodiments, each cytosine nucleotide of an mRNA transcript comprises 5- methylcytidine. In some embodiments, an mRNA includes 5-methyluridine. In some embodiments, at least 70%, at least 80%, at least 85%, at least 90%, at least 95%, at least 97%, at least 98%, or at least 99% of uracil nucleotides in an mRNA comprise 5-methyluridine. In some embodiments, each uracil nucleotide of an mRNA transcript comprises 5-methyluridine. In some embodiments, an mRNA includes 5-methylcytidine and 5-methyluridine. In some embodiments, at least 70%, at least 80%, at least 85%, at least 90%, at least 95%, at least 97%, at least 98%, or at least 99% of uracil nucleotides in an mRNA comprise 5-methyluridine and at least 70%, at least 80%, at least 85%, at least 90%, at least 95%, at least 97%, at least 98%, or at least 99% of cytosine nucleotides in an mRNA comprise 5-methylcytidine. In some embodiments, each cytosine nucleotide of an mRNA transcript comprises 5-methylcytidine and each uracil nucleotide of an mRNA transcript comprises 5-methyluridine. Unmodified Nucleotides In some embodiments, an RNA (e.g., mRNA) is not chemically modified and comprises the standard ribonucleotides consisting of adenosine, guanosine, cytosine and uridine. In some embodiments, nucleotides and nucleosides of the present disclosure comprise standard nucleoside residues such as those present in transcribed RNA (e.g., A, G, C, or U). In some embodiments, nucleotides and nucleosides of the present disclosure comprise standard deoxyribonucleosides such as those present in DNA (e.g., dA, dG, dC, or dT). In Vitro Transcription (IVT) cDNA encoding the polynucleotides described herein may be transcribed using an in vitro transcription (IVT) system. In vitro transcription of RNA is known in the art and is described in International Publication WO 2014/152027, which is incorporated by reference herein in its entirety. In some embodiments, the RNA of the present disclosure is prepared in accordance with any one or more of the methods described in WO 2018/053209 and WO 2019/036682, each of which is incorporated by reference herein. In some embodiments, the RNA transcript is generated using a non-amplified, linearized DNA template in an in vitro transcription reaction to generate the RNA transcript. In some embodiments, the template DNA is isolated DNA. In some embodiments, the template DNA is cDNA. In some embodiments, the cDNA is formed by reverse transcription of a RNA polynucleotide, for example, but not limited to influenza virus mRNA. In some embodiments, cells, e.g., bacterial cells, e.g., E. coli, e.g., DH-1 cells are transfected with the plasmid DNA template. In some embodiments, the transfected cells are cultured to replicate the plasmid DNA which is then isolated and purified. In some embodiments, the DNA template includes a RNA polymerase promoter, e.g., a T7 promoter located 5 ' to and operably linked to the gene of interest. In some embodiments, an in vitro transcription template encodes a 5′ untranslated (UTR) region, contains an open reading frame, and encodes a 3′ UTR and a poly(A) tail. The particular nucleic acid sequence composition and length of an in vitro transcription template will depend on the mRNA encoded by the template. An in vitro transcription system typically comprises a transcription buffer, nucleotide triphosphates (NTPs), an RNase inhibitor and a polymerase. The NTPs may be manufactured in house, may be selected from a supplier, or may be synthesized as described herein. The NTPs may be selected from, but are not limited to, those described herein including natural and unnatural (modified) NTPs. Any number of RNA polymerases or variants may be used in the method of the present disclosure. The polymerase may be selected from, but is not limited to, a phage RNA polymerase, e.g., a T7 RNA polymerase, a T3 RNA polymerase, a SP6 RNA polymerase, and/or mutant polymerases such as, but not limited to, polymerases able to incorporate modified nucleic acids and/or modified nucleotides, including chemically modified nucleic acids and/or nucleotides. Some embodiments exclude the use of DNase. In some embodiments, the RNA transcript is capped via enzymatic capping. In some embodiments, the RNA comprises 5' terminal cap, for example, 7mG(5’)ppp(5’)NlmpNp. In some embodiments, the RNA polymerase is an RNA polymerase variant, such as those described in WO 2020/172239, incorporated herein by reference in its entirety. RNA polymerase variants of the present disclosure include at least one amino acid substitution, relative to the wild type (WT) RNA polymerase. A WT T7 RNA polymerase is represented by SEQ ID NO: 83: MNTINIAKNDFSDIELAAIPFNTLADHYGERLAREQLALEHESYEMGEARFRKMFERQLKAGEVADNAAAKPLITTLL PKMIARINDWFEEVKAKRGKRPTAFQFLQEIKPEAVAYITIKTTLACLTSADNTTVQAVASAIGRAIEDEARFGRIRD LEAKHFKKNVEEQLNKRVGHVYKKAFMQVVEADMLSKGLLGGEAWSSWHKEDSIHVGVRCIEMLIESTGMVSLHRQNA GVVGQDSETIELAPEYAEAIATRAGALAGISPMFQPCVVPPKPWTGITGGGYWANGRRPLALVRTHSKKALMRYEDVY MPEVYKAINIAQNTAWKINKKVLAVANVITKWKHCPVEDIPAIEREELPMKPEDIDMNPEALTAWKRAAAAVYRKDKA RKSRRISLEFMLEQANKFANHKAIWFPYNMDWRGRVYAVSMFNPQGNDMTKGLLTLAKGKPIGKEGYYWLKIHGANCA GVDKVPFPERIKFIEENHENIMACAKSPLENTWWAEQDSPFCFLAFCFEYAGVQHHGLSYNCSLPLAFDGSCSGIQHF SAMLRDEVGGRAVNLLPSETVQDIYGIVAKKVNEILQADAINGTDNEVVTVTDENTGEISEKVKLGTKALAGQWLAYG VTRSVTKRSVMTLAYGSKEFGFRQQVLEDTIQPAIDSGKGLMFTQPNQAAGYMAKLIWESVSVTVVAAVEAMNWLKSA AKLLAAEVKDKKTGEILRKRCAVHWVTPDGFPVWQEYKKPIQTRLNLMFLGQFRLQPTINTNKDSEIDAHKQESGIAP NFVHSQDGSHLRKTVVWAHEKYGIESFALIHDSFGTIPADAANLFKAVRETMVDTYESCDVLADFYDQFADQLHESQL DKMPALPAKGNLNLRDILESDFAFA (SEQ ID NO: 83). For example, with reference to WT T7 RNA polymerase having an amino acid sequence of SEQ ID NO: 83, the glycine at position 47 is considered a “wild-type amino acid,” whereas a substitution of the glycine for alanine at position 47 is considered an “amino acid substitution” that has a high-helix propensity. In some embodiments, the RNA polymerase variant is a T7 RNA polymerase variant comprising at least one (one or more) amino acid substitution relative to WT RNA polymerase (e.g., WT T7 RNA polymerase having an amino acid sequence of SEQ ID NO: 83). In some embodiments, a RNA polymerase variant comprises a RNA polymerase that includes an (at least one) amino acid modification causes a loop structure of the RNA polymerase variant to undergo a conformational change to a helix structure as the RNA polymerase variant transitions from an initiation complex to an elongation complex. In some embodiments, the amino acid modification is an amino acid substitution at one or more of positions 42, 43, 44, 45, 46, and 47, relative to the wild-type RNA polymerase, wherein the wild-type RNA polymerase comprises the amino acid sequence of SEQ ID NO: 83. The amino acid substitution, in some embodiments, is a high propensity amino acid substitution. Examples of high-helix propensity amino acids include alanine, isoleucine, leucine, arginine, methionine, lysine, glutamine, and/or glutamate. In some embodiments, the amino acid substitution at position 47 is G47A. In some embodiments, a RNA polymerase variant comprise a RNA polymerase that includes an additional C-terminal amino acid, relative to the wild-type RNA polymerase. The additional C-terminal amino acid, in some embodiments, is selected from glycine, alanine, threonine, proline, glutamine, and serine. In some embodiments, the additional C-terminal amino acid (e.g., at position 884 relative to wild-type RNA polymerase comprising the amino acid sequence of SEQ ID NO: 83) is glycine. Also provided herein are co-transcriptional capping methods for ribonucleic acid (RNA) synthesis, using any of the RNA polymerase variants described herein. That is, RNA is produced in a “one-pot” reaction, without the need for a separate capping reaction. Thus, the methods, in some embodiments, comprise reacting a polynucleotide template with a RNA polymerase variant, nucleoside triphosphates, and a cap analog under in vitro transcription reaction conditions to produce RNA transcript. Identification and Ratio Determination (IDR) Sequences In some embodiments, one or more nucleic acids comprises an Identification and Ratio Determination sequence. An Identification and Ratio Determination (IDR) sequence is a sequence of a biological molecule (e.g., nucleic acid or protein) that, when combined with the sequence of a target biological molecule, serves to identify the target biological molecule. Typically, an IDR sequence is a heterologous sequence that is incorporated within or appended to a sequence of a target biological molecule and can be used as a reference to identify the target molecule. Thus, in some embodiments, a nucleic acid (e.g., mRNA) comprises (i) a target sequence of interest (e.g., a coding sequence encoding a therapeutic and/or antigenic peptide or protein); and (ii) a unique IDR sequence. An RNA species (e.g., RNA having a given coding sequence) may comprise an IDR sequence that differs from the IDR sequence of other RNA species (e.g., RNA(s) having different coding sequence(s)). Each IDR sequence thus identifies a particular RNA species, and so the abundance of IDR sequences may be measured to determine the abundance of each RNA species in a composition. Use of distinct IDR sequences to identify RNA species allows for analysis of multivalent RNA compositions (e.g., containing multiple RNA species) containing RNA species with similar coding sequences and/or lengths, which could otherwise be difficult to distinguish using PCR- or chromatography-based analysis of full-length RNAs. Each RNA species in a multivalent RNA composition may comprise an IDR sequence that is not a sequence isomer of an IDR sequence of another RNA species in a multivalent RNA composition (e.g., the IDR sequence does not have the same number of adenosine nucleotides, the same number of cytosine nucleotides, the same number of guanine nucleotides, and the same number of uracil nucleotides, as another IDR sequence in the composition, even if those sequences have different sequences). Having identical nucleotide compositions causes sequence isomers to have the same mass, presenting a challenge to distinguishing sequence isomers using mass-based identification methods (e.g., mass spectrometry). Each RNA species in a multivalent RNA composition may comprise an IDR sequence having a mass that differs from the mass of IDR sequences of each other RNA species in a multivalent RNA composition. For example, the mass of each IDR sequence may differ from the mass of other IDR sequences by at least 9 Da, at least 25 Da, at least 25 Da, or at least 50 Da. Use of IDR sequences with distinct masses allows RNA fragments comprising different IDR sequences to be distinguished using mass-based analysis methods (e.g., mass spectrometry), which do not require reverse transcription, amplification, or sequencing of RNAs. Each RNA species in an RNA composition may comprises an IDR sequence with a different length. For example, each IDR sequence may have a length independently selected from 0 to 25 nucleotides. The length of a nucleic acid influences the rate at which the nucleic acid traverses a chromatography column, and so the use of IDR sequences of different lengths on different RNA species allows RNA fragments having different IDR sequences to be distinguished using chromatography-based methods (e.g., LC-UV). IDR sequences may be chosen such that no IDR sequence comprises a start codon, ‘AUG’. Lack of a start codon in an IDR sequence prevents undesired translation of nucleotide sequences within and/or downstream from the IDR sequence. IDR sequences may be chosen such that no IDR sequence comprises a recognition site for a restriction enzyme. In one example, no IDR sequence comprises a recognition site for XbaI, ‘UCUAG’. Lack of a recognition site for a restriction enzyme (e.g., XbaI recognition site ‘UCUAG’) allows the restriction enzyme to be used in generating and modifying a DNA template for in vitro transcription, without affecting the IDR sequence or sequence of the transcribed RNA. Non-limiting examples of distinct IDR sequences include: GAGAUUGAGUGUAGUGACUAG (SEQ ID NO: 56), GAGAUUGAGUGUAGUGAC (SEQ ID NO: 57), GAGAUUGAGUGUAGUG (SEQ ID NO: 58), GAUUGAGACUACGGG (SEQ ID NO: 59), and CAUAGACACUACG (SEQ ID NO: 60). In some embodiments of the compositions described herein, each mRNA encoding a distinct protein comprises a 3′ UTR comprising a distinct IDR sequence selected from SEQ ID NOs: 56–60. Nucleic Acid Production Chemical Synthesis Nucleic acids as described herein may be manufactured in whole or in part using solid phase techniques. Solid-phase chemical synthesis of nucleic acids is an automated method wherein molecules are immobilized on a solid support and synthesized step by step in a reactant solution. Solid-phase synthesis is useful in site-specific introduction of chemical modifications in the nucleic acid sequences. The synthesis of nucleic acids of the present disclosure by the sequential addition of monomer building blocks may be carried out in a liquid phase. The synthetic methods discussed above each has its own advantages and limitations. Attempts have been conducted to combine these methods to overcome the limitations. Such combinations of methods are within the scope of the present disclosure. The use of solid-phase or liquid-phase chemical synthesis in combination with enzymatic ligation provides an efficient way to generate long chain nucleic acids that cannot be obtained by chemical synthesis alone. Ligation Assembling nucleic acids by a ligase may also be used. DNA or RNA ligases promote intermolecular ligation of the 5’ and 3’ ends of polynucleotide chains through the formation of a phosphodiester bond. Nucleic acids such as chimeric polynucleotides and/or circular nucleic acids may be prepared by ligation of one or more regions or subregions. DNA fragments can be joined by a ligase catalyzed reaction to create recombinant DNA with different functions. Two oligodeoxynucleotides, one with a 5’ phosphoryl group and another with a free 3’ hydroxyl group, serve as substrates for a DNA ligase. Purification Purification of the nucleic acids described herein may include, but is not limited to, nucleic acid clean-up, quality assurance and quality control. Clean-up may be performed by methods known in the arts such as, but not limited to, AGENCOURT® beads (Beckman Coulter Genomics, Danvers, MA), poly-T beads, LNATM oligo-T capture probes (EXIQON® Inc, Vedbaek, Denmark) or HPLC based purification methods such as, but not limited to, strong anion exchange HPLC, weak anion exchange HPLC, reverse phase HPLC (RP-HPLC), and hydrophobic interaction HPLC (HIC-HPLC). The term “purified” when used in relation to a nucleic acid such as a “purified nucleic acid” refers to one that is separated from at least one contaminant. A “contaminant” is any substance that makes another unfit, impure or inferior. Thus, a purified nucleic acid (e.g., DNA and RNA) is present in a form or setting different from that in which it is found in nature, or a form or setting different from that which existed prior to subjecting it to a treatment or purification method. A quality assurance and/or quality control check may be conducted using methods such as, but not limited to, gel electrophoresis, UV absorbance, or analytical HPLC. In some embodiments, the nucleic acids may be sequenced by methods including, but not limited to reverse-transcriptase-PCR. Quantification In some embodiments, the nucleic acids of the present disclosure may be quantified in exosomes or when derived from one or more bodily fluid. Bodily fluids include peripheral blood, serum, plasma, ascites, urine, cerebrospinal fluid (CSF), sputum, saliva, bone marrow, synovial fluid, aqueous humor, amniotic fluid, cerumen, breast milk, broncheoalveolar lavage fluid, semen, prostatic fluid, cowper's fluid or pre-ejaculatory fluid, sweat, fecal matter, hair, tears, cyst fluid, pleural and peritoneal fluid, pericardial fluid, lymph, chyme, chyle, bile, interstitial fluid, menses, pus, sebum, vomit, vaginal secretions, mucosal secretion, stool water, pancreatic juice, lavage fluids from sinus cavities, bronchopulmonary aspirates, blastocyl cavity fluid, and umbilical cord blood. Alternatively, exosomes may be retrieved from an organ selected from the group consisting of lung, heart, pancreas, stomach, intestine, bladder, kidney, ovary, testis, skin, colon, breast, prostate, brain, esophagus, liver, and placenta. Assays may be performed using construct specific probes, cytometry, qRT-PCR, real-time PCR, PCR, flow cytometry, electrophoresis, mass spectrometry, or combinations thereof while the exosomes may be isolated using immunohistochemical methods such as enzyme linked immunosorbent assay (ELISA) methods. Exosomes may also be isolated by size exclusion chromatography, density gradient centrifugation, differential centrifugation, nanomembrane ultrafiltration, immunoabsorbent capture, affinity purification, microfluidic separation, or combinations thereof. These methods afford the investigator the ability to monitor, in real time, the level of nucleic acids remaining or delivered. This is possible because the nucleic acids of the present disclosure, in some embodiments, differ from the endogenous forms due to the structural or chemical modifications. In some embodiments, the nucleic acid may be quantified using methods such as, but not limited to, ultraviolet visible spectroscopy (UV/Vis). A non-limiting example of a UV/Vis spectrometer is a NANODROP® spectrometer (ThermoFisher, Waltham, MA). The quantified nucleic acid may be analyzed in order to determine if the nucleic acid may be of proper size, check that no degradation of the nucleic acid has occurred. Degradation of the nucleic acid may be checked by methods such as, but not limited to, agarose gel electrophoresis, HPLC based purification methods such as, but not limited to, strong anion exchange HPLC, weak anion exchange HPLC, reverse phase HPLC (RP-HPLC), and hydrophobic interaction HPLC (HIC- HPLC), liquid chromatography-mass spectrometry (LCMS), capillary electrophoresis (CE) and capillary gel electrophoresis (CGE). Lipid Compositions In some embodiments, the nucleic acids are formulated as a lipid composition, such as a composition comprising a lipid nanoparticle, a liposome, and/or a lipoplex. In some embodiments, nucleic acids are formulated as lipid nanoparticle (LNP) compositions. Lipid nanoparticles typically comprise amino lipid, non-cationic lipid, structural lipid, and PEG lipid components along with the nucleic acid cargo of interest. The lipid nanoparticles can be generated using components, compositions, and methods as are generally known in the art, see for example PCT/US2016/052352; PCT/US2016/068300; PCT/US2017/037551; PCT/US2015/027400; PCT/US2016/047406; PCT/US2016/000129; PCT/US2016/014280; PCT/US2017/038426; PCT/US2014/027077; PCT/US2014/055394; PCT/US2016/052117; PCT/US2012/069610; PCT/US2017/027492; PCT/US2016/059575; PCT/US2016/069491; PCT/US2016/069493; and PCT/US2014/066242, all of which are incorporated by reference herein in their entirety. In some embodiments, the lipid nanoparticle comprises at least one ionizable amino lipid, at least one non-cationic lipid, at least one sterol, and/or at least one polyethylene glycol (PEG)- modified lipid. In some embodiments, the lipid nanoparticle comprises a molar ratio of 20-60% ionizable amino lipid, 5-25% non-cationic lipid, 25-55% structural lipid, and 0.5-15% PEG-modified lipid. In some embodiments, the lipid nanoparticle comprises a molar ratio of 20-60% ionizable amino lipid, 5-30% non-cationic lipid, 10-55% structural lipid, and 0.5-15% PEG-modified lipid. In some embodiments, the lipid nanoparticle comprises 40-50 mol% ionizable lipid, optionally 45-50 mol%, for example, 45-46 mol%, 46-47 mol%, 47-48 mol%, 48-49 mol%, or 49-50 mol% for example about 45 mol%, 45.5 mol%, 46 mol%, 46.5 mol%, 47 mol%, 47.5 mol%, 48 mol%, 48.5 mol%, 49 mol%, or 49.5 mol%. In some embodiments, the lipid nanoparticle comprises 20-60 mol% ionizable amino lipid. For example, the lipid nanoparticle may comprise 20-50 mol%, 20-40 mol%, 20-30 mol%, 30-60 mol%, 30-50 mol%, 30-40 mol%, 40-60 mol%, 40-50 mol%, or 50-60 mol% ionizable amino lipid. In some embodiments, the lipid nanoparticle comprises 20 mol%, 30 mol%, 40 mol%, 50 mol%, or 60 mol% ionizable amino lipid. In some embodiments, the lipid nanoparticle comprises 35 mol%, 36 mol%, 37 mol%, 38 mol%, 39 mol%, 40 mol%, 41 mol%, 42 mol%, 43 mol%, 44 mol%, 45 mol%, 46 mol%, 47 mol%, 48 mol%, 49 mol%, 50 mol%, 51 mol%, 52 mol%, 53 mol%, 54 mol%, or 55 mol% ionizable amino lipid. In some embodiments, the lipid nanoparticle comprises 45 – 55 mole percent (mol%) ionizable amino lipid. For example, lipid nanoparticle may comprise 45, 46, 47, 48, 49, 50, 51, 52, 53, 54, or 55 mol% ionizable amino lipid. Ionizable amino lipids Formula (AI) In some embodiments, the ionizable amino lipid of a lipid nanoparticle is a compound of Formula (AI): its N-oxide, or a salt or isomer thereof, wherein R’a is R’branched; wherein
its N-oxide, or a salt or isomer thereof, wherein R’a is R’branched; wherein denotes a point of attachment; wherein Raα, Raβ, Raγ, and Raδ are each independently selected from the group consisting of H, C2-12 alkyl, and C2-12 alkenyl; R2 and R3 are each independently selected from the group consisting of C1-14 alkyl and C2-14 alkenyl; R4 is selected from the group consisting of -(CH2)nOH, wherein n is selected from the group consisting
denotes a point of attachment; wherein Raα, Raβ, Raγ, and Raδ are each independently selected from the group consisting of H, C2-12 alkyl, and C2-12 alkenyl; R2 and R3 are each independently selected from the group consisting of C1-14 alkyl and C2-14 alkenyl; R4 is selected from the group consisting of -(CH2)nOH, wherein n is selected from the group consisting wherein denotes a point of attachment; wherein R10 is N(R)2; each R is independently selected from the group consisting of C1-6 alkyl, C2-3 alkenyl, and H; and n2 is selected from the group consisting of 1, 2, 3, 4, 5, 6, 7, 8, 9, and 10; each R5 is independently selected from the group consisting of C1-3 alkyl, C2-3 alkenyl, and H; each R6 is independently selected from the group consisting of C1-3 alkyl, C2-3 alkenyl, and H; M and M’ are each independently selected from the group consisting of -C(O)O- and -OC(O)-; R’ is a C1-12 alkyl or C2-12 alkenyl; l is selected from the group consisting of 1, 2, 3, 4, and 5; and m is selected from the group consisting of 5, 6, 7, 8, 9, 10, 11, 12, and 13. In some embodiments of the compounds of Formula (AI), R’a is R’branched; R’branched is
wherein denotes a point of attachment; wherein R10 is N(R)2; each R is independently selected from the group consisting of C1-6 alkyl, C2-3 alkenyl, and H; and n2 is selected from the group consisting of 1, 2, 3, 4, 5, 6, 7, 8, 9, and 10; each R5 is independently selected from the group consisting of C1-3 alkyl, C2-3 alkenyl, and H; each R6 is independently selected from the group consisting of C1-3 alkyl, C2-3 alkenyl, and H; M and M’ are each independently selected from the group consisting of -C(O)O- and -OC(O)-; R’ is a C1-12 alkyl or C2-12 alkenyl; l is selected from the group consisting of 1, 2, 3, 4, and 5; and m is selected from the group consisting of 5, 6, 7, 8, 9, 10, 11, 12, and 13. In some embodiments of the compounds of Formula (AI), R’a is R’branched; R’branched is denotes a point of attachment; Raα, Raβ, Raγ, and Raδ are each H; R2 and R3 are each C1-14 alkyl; R4 is -(CH2)nOH; n is 2; each R5 is H; each R6 is H; M and M’ are each - C(O)O-; R’ is a C1-12 alkyl; l is 5; and m is 7. In some embodiments of the compounds of Formula (AI), R’a is R’branched; R’branched is
denotes a point of attachment; Raα, Raβ, Raγ, and Raδ are each H; R2 and R3 are each C1-14 alkyl; R4 is -(CH2)nOH; n is 2; each R5 is H; each R6 is H; M and M’ are each - C(O)O-; R’ is a C1-12 alkyl; l is 5; and m is 7. In some embodiments of the compounds of Formula (AI), R’a is R’branched; R’branched is denotes a point of attachment; Raα, Raβ, Raγ, and Raδ are each H; R2 and R3 are each C1-14 alkyl; R4 is -(CH2)nOH; n is 2; each R5 is H; each R6 is H; M and M’ are each - C(O)O-; R’ is a C1-12 alkyl; l is 3; and m is 7. In some embodiments of the compounds of Formula (AI), R’a is R’branched; R’branched is
denotes a point of attachment; Raα, Raβ, Raγ, and Raδ are each H; R2 and R3 are each C1-14 alkyl; R4 is -(CH2)nOH; n is 2; each R5 is H; each R6 is H; M and M’ are each - C(O)O-; R’ is a C1-12 alkyl; l is 3; and m is 7. In some embodiments of the compounds of Formula (AI), R’a is R’branched; R’branched is denotes a point of attachment; Raα is C2-12 alkyl; Raβ, Raγ, and Raδ are each H; R2 and R3 are each C1-14 alkyl;
denotes a point of attachment; Raα is C2-12 alkyl; Raβ, Raγ, and Raδ are each H; R2 and R3 are each C1-14 alkyl; alkyl); n2 is 2; R5 is H; each R6 is H; M and M’ are each -C(O)O-; R’ is a C1-12 alkyl; l is 5; and m is 7. In some embodiments of the compounds of Formula (AI), R’a is R’branched; R’branched is
alkyl); n2 is 2; R5 is H; each R6 is H; M and M’ are each -C(O)O-; R’ is a C1-12 alkyl; l is 5; and m is 7. In some embodiments of the compounds of Formula (AI), R’a is R’branched; R’branched is denotes a point of attachment; Raα, Raβ, and Raδ are each H; Raγ is C2-12 alkyl; R2 and R3 are each C1-14 alkyl; R4 is -(CH2)nOH; n is 2; each R5 is H; each R6 is H; M and M’ are each -C(O)O-; R’ is a C1-12 alkyl; l is 5; and m is 7. In some embodiments, the compound of Formula (AI) is selected from: ,
denotes a point of attachment; Raα, Raβ, and Raδ are each H; Raγ is C2-12 alkyl; R2 and R3 are each C1-14 alkyl; R4 is -(CH2)nOH; n is 2; each R5 is H; each R6 is H; M and M’ are each -C(O)O-; R’ is a C1-12 alkyl; l is 5; and m is 7. In some embodiments, the compound of Formula (AI) is selected from: , , . In some embodiments, the ionizable amino lipid of Formula (AI) is a compound of Formula (AIa):
, . In some embodiments, the ionizable amino lipid of Formula (AI) is a compound of Formula (AIa): its N-oxide, or a salt or isomer thereof, wherein R’a is R’branched; wherein R’branched
its N-oxide, or a salt or isomer thereof, wherein R’a is R’branched; wherein R’branched denotes a point of attachment; wherein Raβ, Raγ, and Raδ are each independently selected from the group consisting of H, C2-12 alkyl, and C2-12 alkenyl; R2 and R3 are each independently selected from the group consisting of C1-14 alkyl and C2-14 alkenyl; R4 is selected from the group consisting of -(CH2)nOH wherein n is selected from the group consisting
denotes a point of attachment; wherein Raβ, Raγ, and Raδ are each independently selected from the group consisting of H, C2-12 alkyl, and C2-12 alkenyl; R2 and R3 are each independently selected from the group consisting of C1-14 alkyl and C2-14 alkenyl; R4 is selected from the group consisting of -(CH2)nOH wherein n is selected from the group consisting wherein denotes a point of attachment; wherein R10 is N(R)2; each R is independently selected from the group consisting of C1-6 alkyl, C2-3 alkenyl, and H; and n2 is selected from the group consisting of 1, 2, 3, 4, 5, 6, 7, 8, 9, and 10; each R5 is independently selected from the group consisting of C1-3 alkyl, C2-3 alkenyl, and H; each R6 is independently selected from the group consisting of C1-3 alkyl, C2-3 alkenyl, and H; M and M’ are each independently selected from the group consisting of -C(O)O- and -OC(O)-; R’ is a C1-12 alkyl or C2-12 alkenyl; l is selected from the group consisting of 1, 2, 3, 4, and 5; and m is selected from the group consisting of 5, 6, 7, 8, 9, 10, 11, 12, and 13. In some embodiments, the ionizable amino lipid of Formula (AI) is a compound of Formula (AIb):
wherein denotes a point of attachment; wherein R10 is N(R)2; each R is independently selected from the group consisting of C1-6 alkyl, C2-3 alkenyl, and H; and n2 is selected from the group consisting of 1, 2, 3, 4, 5, 6, 7, 8, 9, and 10; each R5 is independently selected from the group consisting of C1-3 alkyl, C2-3 alkenyl, and H; each R6 is independently selected from the group consisting of C1-3 alkyl, C2-3 alkenyl, and H; M and M’ are each independently selected from the group consisting of -C(O)O- and -OC(O)-; R’ is a C1-12 alkyl or C2-12 alkenyl; l is selected from the group consisting of 1, 2, 3, 4, and 5; and m is selected from the group consisting of 5, 6, 7, 8, 9, 10, 11, 12, and 13. In some embodiments, the ionizable amino lipid of Formula (AI) is a compound of Formula (AIb): its N-oxide, or a salt or isomer thereof, wherein R’a is R’branched; wherein
its N-oxide, or a salt or isomer thereof, wherein R’a is R’branched; wherein denotes a point of attachment; wherein Raα, Raβ, Raγ, and Raδ are each independently selected from the group consisting of H, C2-12 alkyl, and C2-12 alkenyl; R2 and R3 are each independently selected from the group consisting of C1-14 alkyl and C2-14 alkenyl; R4 is -(CH2)nOH, wherein n is selected from the group consisting of 1, 2, 3, 4, and 5; each R5 is independently selected from the group consisting of C1-3 alkyl, C2-3 alkenyl, and H; each R6 is independently selected from the group consisting of C1-3 alkyl, C2-3 alkenyl, and H; M and M’ are each independently selected from the group consisting of -C(O)O- and -OC(O)-; R’ is a C1-12 alkyl or C2-12 alkenyl; l is selected from the group consisting of 1, 2, 3, 4, and 5; and m is selected from the group consisting of 5, 6, 7, 8, 9, 10, 11, 12, and 13. In some embodiments of Formula (AI) or (AIb), R’a is R’branched; R’branched is
denotes a point of attachment; wherein Raα, Raβ, Raγ, and Raδ are each independently selected from the group consisting of H, C2-12 alkyl, and C2-12 alkenyl; R2 and R3 are each independently selected from the group consisting of C1-14 alkyl and C2-14 alkenyl; R4 is -(CH2)nOH, wherein n is selected from the group consisting of 1, 2, 3, 4, and 5; each R5 is independently selected from the group consisting of C1-3 alkyl, C2-3 alkenyl, and H; each R6 is independently selected from the group consisting of C1-3 alkyl, C2-3 alkenyl, and H; M and M’ are each independently selected from the group consisting of -C(O)O- and -OC(O)-; R’ is a C1-12 alkyl or C2-12 alkenyl; l is selected from the group consisting of 1, 2, 3, 4, and 5; and m is selected from the group consisting of 5, 6, 7, 8, 9, 10, 11, 12, and 13. In some embodiments of Formula (AI) or (AIb), R’a is R’branched; R’branched is denotes a point of attachment; Raβ, Raγ, and Raδ are each H; R2 and R3 are each C1-14 alkyl; R4 is -(CH2)nOH; n is 2; each R5 is H; each R6 is H; M and M’ are each - C(O)O-; R’ is a C1-12 alkyl; l is 5; and m is 7. In some embodiments of Formula (AI) or (AIb), R’a is R’branched; R’branched is
denotes a point of attachment; Raβ, Raγ, and Raδ are each H; R2 and R3 are each C1-14 alkyl; R4 is -(CH2)nOH; n is 2; each R5 is H; each R6 is H; M and M’ are each - C(O)O-; R’ is a C1-12 alkyl; l is 5; and m is 7. In some embodiments of Formula (AI) or (AIb), R’a is R’branched; R’branched is denotes a point of attachment; Raβ, Raγ, and Raδ are each H; R2 and R3 are each C1-14 alkyl; R4 is -(CH2)nOH; n is 2; each R5 is H; each R6 is H; M and M’ are each - C(O)O-; R’ is a C1-12 alkyl; l is 3; and m is 7. In some embodiments of Formula (AI) or (AIb), R’a is R’branched; R’branched is
denotes a point of attachment; Raβ, Raγ, and Raδ are each H; R2 and R3 are each C1-14 alkyl; R4 is -(CH2)nOH; n is 2; each R5 is H; each R6 is H; M and M’ are each - C(O)O-; R’ is a C1-12 alkyl; l is 3; and m is 7. In some embodiments of Formula (AI) or (AIb), R’a is R’branched; R’branched is denotes a point of attachment; Raβ and Raδ are each H; Raγ is C2-12 alkyl; R2 and R3 are each C1-14 alkyl; R4 is -(CH2)nOH; n is 2; each R5 is H; each R6 is H; M and M’ are each -C(O)O-; R’ is a C1-12 alkyl; l is 5; and m is 7. In some embodiments, the ionizable amino lipid of Formula (AI) is a compound of Formula (AIc):
denotes a point of attachment; Raβ and Raδ are each H; Raγ is C2-12 alkyl; R2 and R3 are each C1-14 alkyl; R4 is -(CH2)nOH; n is 2; each R5 is H; each R6 is H; M and M’ are each -C(O)O-; R’ is a C1-12 alkyl; l is 5; and m is 7. In some embodiments, the ionizable amino lipid of Formula (AI) is a compound of Formula (AIc): its N-oxide, or a salt or isomer thereof, wherein R’a is R’branched; wherein
its N-oxide, or a salt or isomer thereof, wherein R’a is R’branched; wherein denotes a point of attachment; wherein Raα, Raβ, Raγ, and Raδ are each independently selected from the group consisting of H, C2-12 alkyl, and C2-12 alkenyl; R2 and R3 are each independently selected from the group consisting of C1-14 alkyl and C2-14 alkenyl;
denotes a point of attachment; wherein Raα, Raβ, Raγ, and Raδ are each independently selected from the group consisting of H, C2-12 alkyl, and C2-12 alkenyl; R2 and R3 are each independently selected from the group consisting of C1-14 alkyl and C2-14 alkenyl; denotes a point of attachment; wherein R10 is N(R)2; each R is independently selected from the group consisting of C1-6 alkyl, C2-3 alkenyl, and H; n2 is selected from the group consisting of 1, 2, 3, 4, 5, 6, 7, 8, 9, and 10; each R5 is independently selected from the group consisting of C1-3 alkyl, C2-3 alkenyl, and H; each R6 is independently selected from the group consisting of C1-3 alkyl, C2-3 alkenyl, and H; M and M’ are each independently selected from the group consisting of -C(O)O- and -OC(O)-; R’ is a C1-12 alkyl or C2-12 alkenyl; l is selected from the group consisting of 1, 2, 3, 4, and 5; and m is selected from the group consisting of 5, 6, 7, 8, 9, 10, 11, 12, and 13. In some embodiments,
denotes a point of attachment; wherein R10 is N(R)2; each R is independently selected from the group consisting of C1-6 alkyl, C2-3 alkenyl, and H; n2 is selected from the group consisting of 1, 2, 3, 4, 5, 6, 7, 8, 9, and 10; each R5 is independently selected from the group consisting of C1-3 alkyl, C2-3 alkenyl, and H; each R6 is independently selected from the group consisting of C1-3 alkyl, C2-3 alkenyl, and H; M and M’ are each independently selected from the group consisting of -C(O)O- and -OC(O)-; R’ is a C1-12 alkyl or C2-12 alkenyl; l is selected from the group consisting of 1, 2, 3, 4, and 5; and m is selected from the group consisting of 5, 6, 7, 8, 9, 10, 11, 12, and 13. In some embodiments, denotes a point of attachment; Raβ, Raγ, and Raδ are each H; Raα is C2-12 alkyl; R2 and R3 are each C1-14 alkyl;
denotes a point of attachment; Raβ, Raγ, and Raδ are each H; Raα is C2-12 alkyl; R2 and R3 are each C1-14 alkyl; denotes a point of attachment; R10 is NH(C1-6 alkyl); n2 is 2; each R5 is H; each R6 is H; M and M’ are each -C(O)O-; R’ is a C1-12 alkyl; l is 5; and m is 7. In some embodiments, the compound of Formula (AIc) is:
denotes a point of attachment; R10 is NH(C1-6 alkyl); n2 is 2; each R5 is H; each R6 is H; M and M’ are each -C(O)O-; R’ is a C1-12 alkyl; l is 5; and m is 7. In some embodiments, the compound of Formula (AIc) is: In some embodiments, the ionizable amino lipid is a compound of Formula (AII):
In some embodiments, the ionizable amino lipid is a compound of Formula (AII): wherein R’a is R’branched or R’cyclic; wherein
wherein R’a is R’branched or R’cyclic; wherein of attachment; Raγ and Raδ are each independently selected from the group consisting of H, C1-12 alkyl, and C2-12 alkenyl, wherein at least one of Raγ and Raδ is selected from the group consisting of C1- 12 alkyl and C2-12 alkenyl; Rbγ and Rbδ are each independently selected from the group consisting of H, C1-12 alkyl, and C2-12 alkenyl, wherein at least one of Rbγ and Rbδ is selected from the group consisting of C1- 12 alkyl and C2-12 alkenyl; R2 and R3 are each independently selected from the group consisting of C1-14 alkyl and C2-14 alkenyl; R4 is selected from the group consisting of -(CH2)nOH wherein n is selected from the group consisting
of attachment; Raγ and Raδ are each independently selected from the group consisting of H, C1-12 alkyl, and C2-12 alkenyl, wherein at least one of Raγ and Raδ is selected from the group consisting of C1- 12 alkyl and C2-12 alkenyl; Rbγ and Rbδ are each independently selected from the group consisting of H, C1-12 alkyl, and C2-12 alkenyl, wherein at least one of Rbγ and Rbδ is selected from the group consisting of C1- 12 alkyl and C2-12 alkenyl; R2 and R3 are each independently selected from the group consisting of C1-14 alkyl and C2-14 alkenyl; R4 is selected from the group consisting of -(CH2)nOH wherein n is selected from the group consisting wherein denotes a point of attachment; wherein R10 is N(R)2; each R is independently selected from the group consisting of C1-6 alkyl, C2-3 alkenyl, and H; and n2 is selected from the group consisting of 1, 2, 3, 4, 5, 6, 7, 8, 9, and 10; each R’ independently is a C1-12 alkyl or C2-12 alkenyl; Ya is a C3-6 carbocycle; R*”a is selected from the group consisting of C1-15 alkyl and C2-15 alkenyl; and s is 2 or 3; m is selected from 1, 2, 3, 4, 5, 6, 7, 8, and 9; l is selected from 1, 2, 3, 4, 5, 6, 7, 8, and 9. In some embodiments, the ionizable amino lipid of Formula (AII) is a compound of Formula (AII-a): (AII-a), or its N-oxide, or a salt or isomer thereof, wherein R’a is R’branched or R’cyclic; wherein
wherein denotes a point of attachment; wherein R10 is N(R)2; each R is independently selected from the group consisting of C1-6 alkyl, C2-3 alkenyl, and H; and n2 is selected from the group consisting of 1, 2, 3, 4, 5, 6, 7, 8, 9, and 10; each R’ independently is a C1-12 alkyl or C2-12 alkenyl; Ya is a C3-6 carbocycle; R*”a is selected from the group consisting of C1-15 alkyl and C2-15 alkenyl; and s is 2 or 3; m is selected from 1, 2, 3, 4, 5, 6, 7, 8, and 9; l is selected from 1, 2, 3, 4, 5, 6, 7, 8, and 9. In some embodiments, the ionizable amino lipid of Formula (AII) is a compound of Formula (AII-a): (AII-a), or its N-oxide, or a salt or isomer thereof, wherein R’a is R’branched or R’cyclic; wherein denotes a point of attachment; Raγ and Raδ are each independently selected from the group consisting of H, C1-12 alkyl, and C2-12 alkenyl, wherein at least one of Raγ and Raδ is selected from the group consisting of C1- 12 alkyl and C2-12 alkenyl; Rbγ and Rbδ are each independently selected from the group consisting of H, C1-12 alkyl, and C2-12 alkenyl, wherein at least one of Rbγ and Rbδ is selected from the group consisting of C1- 12 alkyl and C2-12 alkenyl; R2 and R3 are each independently selected from the group consisting of C1-14 alkyl and C2-14 alkenyl; R4 is selected from the group consisting of -(CH2)nOH wherein n is selected from the group consisting
denotes a point of attachment; Raγ and Raδ are each independently selected from the group consisting of H, C1-12 alkyl, and C2-12 alkenyl, wherein at least one of Raγ and Raδ is selected from the group consisting of C1- 12 alkyl and C2-12 alkenyl; Rbγ and Rbδ are each independently selected from the group consisting of H, C1-12 alkyl, and C2-12 alkenyl, wherein at least one of Rbγ and Rbδ is selected from the group consisting of C1- 12 alkyl and C2-12 alkenyl; R2 and R3 are each independently selected from the group consisting of C1-14 alkyl and C2-14 alkenyl; R4 is selected from the group consisting of -(CH2)nOH wherein n is selected from the group consisting wherein denotes a point of attachment; wherein R10 is N(R)2; each R is independently selected from the group consisting of C1-6 alkyl, C2-3 alkenyl, and H; and n2 is selected from the group consisting of 1, 2, 3, 4, 5, 6, 7, 8, 9, and 10; each R’ independently is a C1-12 alkyl or C2-12 alkenyl; m is selected from 1, 2, 3, 4, 5, 6, 7, 8, and 9; l is selected from 1, 2, 3, 4, 5, 6, 7, 8, and 9. In some embodiments, the ionizable amino lipid of Formula (AII) is a compound of
wherein denotes a point of attachment; wherein R10 is N(R)2; each R is independently selected from the group consisting of C1-6 alkyl, C2-3 alkenyl, and H; and n2 is selected from the group consisting of 1, 2, 3, 4, 5, 6, 7, 8, 9, and 10; each R’ independently is a C1-12 alkyl or C2-12 alkenyl; m is selected from 1, 2, 3, 4, 5, 6, 7, 8, and 9; l is selected from 1, 2, 3, 4, 5, 6, 7, 8, and 9. In some embodiments, the ionizable amino lipid of Formula (AII) is a compound of wherein R’a is R’branched or R’cyclic; wherein
wherein R’a is R’branched or R’cyclic; wherein denotes a point of attachment; Raγ and Rbγ are each independently selected from the group consisting of C1-12 alkyl and C2-12 alkenyl; R2 and R3 are each independently selected from the group consisting of C1-14 alkyl and C2-14 alkenyl; R4 is selected from the group consisting of -(CH2)nOH wherein n is selected from the group consisting
denotes a point of attachment; Raγ and Rbγ are each independently selected from the group consisting of C1-12 alkyl and C2-12 alkenyl; R2 and R3 are each independently selected from the group consisting of C1-14 alkyl and C2-14 alkenyl; R4 is selected from the group consisting of -(CH2)nOH wherein n is selected from the group consisting wherein denotes a point of attachment; wherein R10 is N(R)2; each R is independently selected from the group consisting of C1-6 alkyl, C2-3 alkenyl, and H; and n2 is selected from the group consisting of 1, 2, 3, 4, 5, 6, 7, 8, 9, and 10; each R’ independently is a C1-12 alkyl or C2-12 alkenyl; m is selected from 1, 2, 3, 4, 5, 6, 7, 8, and 9; l is selected from 1, 2, 3, 4, 5, 6, 7, 8, and 9. In some embodiments, the ionizable amino lipid of Formula (AII) is a compound of Formula (AII-c):
wherein denotes a point of attachment; wherein R10 is N(R)2; each R is independently selected from the group consisting of C1-6 alkyl, C2-3 alkenyl, and H; and n2 is selected from the group consisting of 1, 2, 3, 4, 5, 6, 7, 8, 9, and 10; each R’ independently is a C1-12 alkyl or C2-12 alkenyl; m is selected from 1, 2, 3, 4, 5, 6, 7, 8, and 9; l is selected from 1, 2, 3, 4, 5, 6, 7, 8, and 9. In some embodiments, the ionizable amino lipid of Formula (AII) is a compound of Formula (AII-c): its N-oxide, or a salt or isomer thereof, wherein R’a is R’branched or R’cyclic; wherein
its N-oxide, or a salt or isomer thereof, wherein R’a is R’branched or R’cyclic; wherein denotes a point of attachment; wherein Raγ is selected from the group consisting of C1-12 alkyl and C2-12 alkenyl; R2 and R3 are each independently selected from the group consisting of C1-14 alkyl and C2-14 alkenyl; R4 is selected from the group consisting of -(CH2)nOH wherein n is selected from the group consisting
denotes a point of attachment; wherein Raγ is selected from the group consisting of C1-12 alkyl and C2-12 alkenyl; R2 and R3 are each independently selected from the group consisting of C1-14 alkyl and C2-14 alkenyl; R4 is selected from the group consisting of -(CH2)nOH wherein n is selected from the group consisting wherein denotes a point of attachment; wherein R10 is N(R)2; each R is independently selected from the group consisting of C1-6 alkyl, C2-3 alkenyl, and H; and n2 is selected from the group consisting of 1, 2, 3, 4, 5, 6, 7, 8, 9, and 10; R’ is a C1-12 alkyl or C2-12 alkenyl; m is selected from 1, 2, 3, 4, 5, 6, 7, 8, and 9; l is selected from 1, 2, 3, 4, 5, 6, 7, 8, and 9. In some embodiments, the ionizable amino lipid of Formula (AII) is a compound of Formula (AII-d):
wherein denotes a point of attachment; wherein R10 is N(R)2; each R is independently selected from the group consisting of C1-6 alkyl, C2-3 alkenyl, and H; and n2 is selected from the group consisting of 1, 2, 3, 4, 5, 6, 7, 8, 9, and 10; R’ is a C1-12 alkyl or C2-12 alkenyl; m is selected from 1, 2, 3, 4, 5, 6, 7, 8, and 9; l is selected from 1, 2, 3, 4, 5, 6, 7, 8, and 9. In some embodiments, the ionizable amino lipid of Formula (AII) is a compound of Formula (AII-d): its N-oxide, or a salt or isomer thereof, wherein R’a is R’branched or R’cyclic; wherein
its N-oxide, or a salt or isomer thereof, wherein R’a is R’branched or R’cyclic; wherein denotes a point of attachment; wherein Raγ and Rbγ are each independently selected from the group consisting of C1-12 alkyl and C2-12 alkenyl; R4 is selected from the group consisting of -(CH2)nOH wherein n is selected from the group consisting
denotes a point of attachment; wherein Raγ and Rbγ are each independently selected from the group consisting of C1-12 alkyl and C2-12 alkenyl; R4 is selected from the group consisting of -(CH2)nOH wherein n is selected from the group consisting wherein denotes a point of attachment; wherein R10 is N(R)2; each R is independently selected from the group consisting of C1-6 alkyl, C2-3 alkenyl, and H; and n2 is selected from the group consisting of 1, 2, 3, 4, 5, 6, 7, 8, 9, and 10; each R’ independently is a C1-12 alkyl or C2-12 alkenyl; m is selected from 1, 2, 3, 4, 5, 6, 7, 8, and 9; l is selected from 1, 2, 3, 4, 5, 6, 7, 8, and 9. In some embodiments, the ionizable amino lipid of Formula (AII) is a compound of
wherein denotes a point of attachment; wherein R10 is N(R)2; each R is independently selected from the group consisting of C1-6 alkyl, C2-3 alkenyl, and H; and n2 is selected from the group consisting of 1, 2, 3, 4, 5, 6, 7, 8, 9, and 10; each R’ independently is a C1-12 alkyl or C2-12 alkenyl; m is selected from 1, 2, 3, 4, 5, 6, 7, 8, and 9; l is selected from 1, 2, 3, 4, 5, 6, 7, 8, and 9. In some embodiments, the ionizable amino lipid of Formula (AII) is a compound of its N-oxide, or a salt or isomer thereof, wherein R’a is R’branched or R’cyclic; wherein
its N-oxide, or a salt or isomer thereof, wherein R’a is R’branched or R’cyclic; wherein denotes a point of attachment; wherein Raγ is selected from the group consisting of C1-12 alkyl and C2-12 alkenyl; R2 and R3 are each independently selected from the group consisting of C1-14 alkyl and C2-14 alkenyl; R4 is -(CH2)nOH wherein n is selected from the group consisting of 1, 2, 3, 4, and 5; R’ is a C1-12 alkyl or C2-12 alkenyl; m is selected from 1, 2, 3, 4, 5, 6, 7, 8, and 9; l is selected from 1, 2, 3, 4, 5, 6, 7, 8, and 9. In some embodiments of the compound of Formula (AII), (AII-a), (AII-b), (AII-c), (AII- d), or (AII-e), m and l are each independently selected from 4, 5, and 6. In some embodiments of the compound of Formula (AII), (AII-a), (AII-b), (AII-c), (AII-d), or (AII-e), m and l are each 5. In some embodiments of the compound of Formula (AII), (AII-a), (AII-b), (AII-c), (AII- d), or (AII-e), each R’ independently is a C1-12 alkyl. In some embodiments of the compound of Formula (AII), (AII-a), (AII-b), (AII-c), (AII-d), or (AII-e), each R’ independently is a C2-5 alkyl. In some embodiments of the compound of Formula (AII), (AII-a), (AII-b), (AII-c), (AII- d), or (AII-e), R’b is: and R2 and R3 are each independently a C1-14 alkyl. In some embodiments of the compound of Formula (AII), (AII-a), (AII-b), (AII-c), (AII-d), or (AII-e), R’b is: and R2 and R3 are each independently a C6-10 alkyl. In some embodiments of the compound of Formula (AII), (AII-a), (AII-b), (AII-c), (AII-d), or (AII-e), R’b is: and R2 and R3 are each a C8 alkyl. In some embodiments of the compound of Formula (AII), (AII-a), (AII-b), (AII-c), (AII-
denotes a point of attachment; wherein Raγ is selected from the group consisting of C1-12 alkyl and C2-12 alkenyl; R2 and R3 are each independently selected from the group consisting of C1-14 alkyl and C2-14 alkenyl; R4 is -(CH2)nOH wherein n is selected from the group consisting of 1, 2, 3, 4, and 5; R’ is a C1-12 alkyl or C2-12 alkenyl; m is selected from 1, 2, 3, 4, 5, 6, 7, 8, and 9; l is selected from 1, 2, 3, 4, 5, 6, 7, 8, and 9. In some embodiments of the compound of Formula (AII), (AII-a), (AII-b), (AII-c), (AII- d), or (AII-e), m and l are each independently selected from 4, 5, and 6. In some embodiments of the compound of Formula (AII), (AII-a), (AII-b), (AII-c), (AII-d), or (AII-e), m and l are each 5. In some embodiments of the compound of Formula (AII), (AII-a), (AII-b), (AII-c), (AII- d), or (AII-e), each R’ independently is a C1-12 alkyl. In some embodiments of the compound of Formula (AII), (AII-a), (AII-b), (AII-c), (AII-d), or (AII-e), each R’ independently is a C2-5 alkyl. In some embodiments of the compound of Formula (AII), (AII-a), (AII-b), (AII-c), (AII- d), or (AII-e), R’b is: and R2 and R3 are each independently a C1-14 alkyl. In some embodiments of the compound of Formula (AII), (AII-a), (AII-b), (AII-c), (AII-d), or (AII-e), R’b is: and R2 and R3 are each independently a C6-10 alkyl. In some embodiments of the compound of Formula (AII), (AII-a), (AII-b), (AII-c), (AII-d), or (AII-e), R’b is: and R2 and R3 are each a C8 alkyl. In some embodiments of the compound of Formula (AII), (AII-a), (AII-b), (AII-c), (AII- and R3 are each independently a C6-10 alkyl. In some embodiments of the compound of Formula
and R3 are each independently a C6-10 alkyl. In some embodiments of the compound of Formula , Raγ is a C2-6 alkyl and R2 and R3 are each independently a C6-10 alkyl. In some embodiments of the compound of Formula (AII), (AII-a), (AII-b), (AII-c), (AII-d), or (AII-e),
, Raγ is a C2-6 alkyl and R2 and R3 are each independently a C6-10 alkyl. In some embodiments of the compound of Formula (AII), (AII-a), (AII-b), (AII-c), (AII-d), or (AII-e), C8 alkyl. In some embodiments of the compound of Formula (AII), (AII-a), (AII-b), (AII-c), (AII- d), or (AII-e), R’branched is:
C8 alkyl. In some embodiments of the compound of Formula (AII), (AII-a), (AII-b), (AII-c), (AII- d), or (AII-e), R’branched is: Rbγ
Rbγ a C1-12 alkyl. In some embodiments of the compound of Formula (AII), (AII-a), (AII-b), (AII-c),
a C1-12 alkyl. In some embodiments of the compound of Formula (AII), (AII-a), (AII-b), (AII-c), each a C2-6 alkyl. In some embodiments of the compound of Formula (AII), (AII-a), (AII-b), (AII-c), (AII- d), or (AII-e), m and l are each independently selected from 4, 5, and 6 and each R’ independently is a C1-12 alkyl. In some embodiments of the compound of Formula (AII), (AII-a), (AII-b), (AII-c), (AII-d), or (AII-e), m and l are each 5 and each R’ independently is a C2-5 alkyl. In some embodiments of the compound of (AII), (AII-a), (AII-b), (AII-c), (AII-d), or (AII-e), R’branched is:
each a C2-6 alkyl. In some embodiments of the compound of Formula (AII), (AII-a), (AII-b), (AII-c), (AII- d), or (AII-e), m and l are each independently selected from 4, 5, and 6 and each R’ independently is a C1-12 alkyl. In some embodiments of the compound of Formula (AII), (AII-a), (AII-b), (AII-c), (AII-d), or (AII-e), m and l are each 5 and each R’ independently is a C2-5 alkyl. In some embodiments of the compound of (AII), (AII-a), (AII-b), (AII-c), (AII-d), or (AII-e), R’branched is: is:
is: are each independently selected from 4, 5, and 6, each R’ independently is a C1-12 alkyl, and Raγ and Rbγ are each a C1-12 alkyl. In some embodiments of the compound of Formula (AII), (AII-a), (AII-b),
are each independently selected from 4, 5, and 6, each R’ independently is a C1-12 alkyl, and Raγ and Rbγ are each a C1-12 alkyl. In some embodiments of the compound of Formula (AII), (AII-a), (AII-b), each 5, each R’ independently is a C2-5 alkyl, and Raγ and Rbγ are each a C2-6 alkyl. In some embodiments of the compound of Formula (AII), (AII-a), (AII-b), (AII-c), (AII-
each 5, each R’ independently is a C2-5 alkyl, and Raγ and Rbγ are each a C2-6 alkyl. In some embodiments of the compound of Formula (AII), (AII-a), (AII-b), (AII-c), (AII- each independently selected from 4, 5, and 6, R’ is a C1-12 alkyl, Raγ is a C1-12 alkyl and R2 and R3 are each independently a C6-10 alkyl. In some embodiments of the compound of Formula (AII), (AII-a), (AII-b), (AII-c), (AII-
each independently selected from 4, 5, and 6, R’ is a C1-12 alkyl, Raγ is a C1-12 alkyl and R2 and R3 are each independently a C6-10 alkyl. In some embodiments of the compound of Formula (AII), (AII-a), (AII-b), (AII-c), (AII- are each 5, R’ is a C2-5 alkyl, Raγ is a C2-6 alkyl, and R2 and R3 are each a C8 alkyl. In some embodiments of the compound of (AII), (AII-a), (AII-b), (AII-c), (AII-d), or
are each 5, R’ is a C2-5 alkyl, Raγ is a C2-6 alkyl, and R2 and R3 are each a C8 alkyl. In some embodiments of the compound of (AII), (AII-a), (AII-b), (AII-c), (AII-d), or wherein R10 is NH(C1-6 alkyl) and n2 is 2. In some embodiments of the compound of Formula (AII), (AII-a), (AII-b), (AII-c), (AII-d), or (AII-e), R4
wherein R10 is NH(C1-6 alkyl) and n2 is 2. In some embodiments of the compound of Formula (AII), (AII-a), (AII-b), (AII-c), (AII-d), or (AII-e), R4 wherein R10 is NH(CH3) and n2 is 2. In some embodiments of the compound of Formula (AII), (AII-a), (AII-b), (AII-c), (AII-
wherein R10 is NH(CH3) and n2 is 2. In some embodiments of the compound of Formula (AII), (AII-a), (AII-b), (AII-c), (AII- independently selected from 4, 5, and 6, each R’ independently is a C1-12 alkyl, Raγ and Rbγ are each a C1-12 alkyl,
independently selected from 4, 5, and 6, each R’ independently is a C1-12 alkyl, Raγ and Rbγ are each a C1-12 alkyl, wherein R10 is NH(C1-6 alkyl), and n2 is 2. In some embodiments of the compound of Formula (AII), (AII-a), (AII-b), (AII-c), (AII-d), or (AII-
wherein R10 is NH(C1-6 alkyl), and n2 is 2. In some embodiments of the compound of Formula (AII), (AII-a), (AII-b), (AII-c), (AII-d), or (AII- are each 5, each R’ independently is a C2-5 alkyl, Raγ and Rbγ are each a C2-6 alkyl,
are each 5, each R’ independently is a C2-5 alkyl, Raγ and Rbγ are each a C2-6 alkyl, wherein R10 is NH(CH3) and n2 is 2. In some embodiments of the compound of Formula (AII), (AII-a), (AII-b), (AII-c), (AII-
wherein R10 is NH(CH3) and n2 is 2. In some embodiments of the compound of Formula (AII), (AII-a), (AII-b), (AII-c), (AII- are each independently selected from 4, 5, and 6, R’ is a C1-12 alkyl, R2 and R3 are each independently a C6-10 alkyl, Raγ is a C1-12 alkyl,
are each independently selected from 4, 5, and 6, R’ is a C1-12 alkyl, R2 and R3 are each independently a C6-10 alkyl, Raγ is a C1-12 alkyl, wherein R10 is NH(C1-6 alkyl) and n2 is 2. In some embodiments of the compound of Formula (AII), (AII-a), (AII-b), (AII-c), (AII-
wherein R10 is NH(C1-6 alkyl) and n2 is 2. In some embodiments of the compound of Formula (AII), (AII-a), (AII-b), (AII-c), (AII- are each 5, R’ is a C2- 5 alkyl, Raγ is a C2-6 alkyl, R2 and R3 are each a C8 alkyl,
are each 5, R’ is a C2- 5 alkyl, Raγ is a C2-6 alkyl, R2 and R3 are each a C8 alkyl, wherein R10 is NH(CH3) and n2 is 2. In some embodiments of the compound of Formula (AII), (AII-a), (AII-b), (AII-c), (AII- d), or (AII-e), R4 is -(CH2)nOH and n is 2, 3, or 4. In some embodiments of the compound of Formula (AII), (AII-a), (AII-b), (AII-c), (AII-d), or (AII-e), R4 is -(CH2)nOH and n is 2. In some embodiments of the compound of Formula (AII), (AII-a), (AII-b), (AII-c), (AII-
wherein R10 is NH(CH3) and n2 is 2. In some embodiments of the compound of Formula (AII), (AII-a), (AII-b), (AII-c), (AII- d), or (AII-e), R4 is -(CH2)nOH and n is 2, 3, or 4. In some embodiments of the compound of Formula (AII), (AII-a), (AII-b), (AII-c), (AII-d), or (AII-e), R4 is -(CH2)nOH and n is 2. In some embodiments of the compound of Formula (AII), (AII-a), (AII-b), (AII-c), (AII- independently selected from 4, 5, and 6, each R’ independently is a C1-12 alkyl, Raγ and Rbγ are each a C1-12 alkyl, R4 is -(CH2)nOH, and n is 2, 3, or 4. In some embodiments of the compound of Formula
independently selected from 4, 5, and 6, each R’ independently is a C1-12 alkyl, Raγ and Rbγ are each a C1-12 alkyl, R4 is -(CH2)nOH, and n is 2, 3, or 4. In some embodiments of the compound of Formula R’b is:
R’b is: , m and l are each 5, each R’ independently is a C2-5 alkyl, Raγ and Rbγ are each a C2-6 alkyl, R4 is -(CH2)nOH, and n is 2. In some embodiments, the ionizable amino lipid of Formula (AII) is a compound of Formula (AII-f):
, m and l are each 5, each R’ independently is a C2-5 alkyl, Raγ and Rbγ are each a C2-6 alkyl, R4 is -(CH2)nOH, and n is 2. In some embodiments, the ionizable amino lipid of Formula (AII) is a compound of Formula (AII-f): its N-oxide, or a salt or isomer thereof, wherein R’a is R’branched or R’cyclic; wherein
its N-oxide, or a salt or isomer thereof, wherein R’a is R’branched or R’cyclic; wherein ; wherein denotes a point of attachment; Raγ is a C1-12 alkyl; R2 and R3 are each independently a C1-14 alkyl; R4 is -(CH2)nOH wherein n is selected from the group consisting of 1, 2, 3, 4, and 5; R’ is a C1-12 alkyl; m is selected from 4, 5, and 6; and l is selected from 4, 5, and 6. In some embodiments of the compound of Formula (AII-f), m and l are each 5, and n is 2, 3, or 4. In some embodiments of the compound of Formula (AII-f) R’ is a C2-5 alkyl, Raγ is a C2-6 alkyl, and R2 and R3 are each a C6-10 alkyl. In some embodiments of the compound of Formula (AII-f), m and l are each 5, n is 2, 3, or 4, R’ is a C2-5 alkyl, Raγ is a C2-6 alkyl, and R2 and R3 are each a C6-10 alkyl. In some embodiments, the ionizable amino lipid of Formula (AII) is a compound of Formula (AII-g):
; wherein denotes a point of attachment; Raγ is a C1-12 alkyl; R2 and R3 are each independently a C1-14 alkyl; R4 is -(CH2)nOH wherein n is selected from the group consisting of 1, 2, 3, 4, and 5; R’ is a C1-12 alkyl; m is selected from 4, 5, and 6; and l is selected from 4, 5, and 6. In some embodiments of the compound of Formula (AII-f), m and l are each 5, and n is 2, 3, or 4. In some embodiments of the compound of Formula (AII-f) R’ is a C2-5 alkyl, Raγ is a C2-6 alkyl, and R2 and R3 are each a C6-10 alkyl. In some embodiments of the compound of Formula (AII-f), m and l are each 5, n is 2, 3, or 4, R’ is a C2-5 alkyl, Raγ is a C2-6 alkyl, and R2 and R3 are each a C6-10 alkyl. In some embodiments, the ionizable amino lipid of Formula (AII) is a compound of Formula (AII-g): thereof; wherein Raγ is a C2-6 alkyl; R’ is a C2-5 alkyl; and R4 is selected from the group consisting of -(CH2)nOH wherein n is selected from the group consisting
thereof; wherein Raγ is a C2-6 alkyl; R’ is a C2-5 alkyl; and R4 is selected from the group consisting of -(CH2)nOH wherein n is selected from the group consisting wherein
wherein denotes a point of attachment, R10 is NH(C1-6 alkyl), and n2 is selected from the group consisting of 1, 2, and 3. In some embodiments, the ionizable amino lipid of Formula (AII) is a compound of Formula (AII-h):
denotes a point of attachment, R10 is NH(C1-6 alkyl), and n2 is selected from the group consisting of 1, 2, and 3. In some embodiments, the ionizable amino lipid of Formula (AII) is a compound of Formula (AII-h): thereof; wherein Raγ and Rbγ are each independently a C2-6 alkyl; each R’ independently is a C2-5 alkyl; and R4 is selected from the group consisting of -(CH2)nOH wherein n is selected from the group consisting
thereof; wherein Raγ and Rbγ are each independently a C2-6 alkyl; each R’ independently is a C2-5 alkyl; and R4 is selected from the group consisting of -(CH2)nOH wherein n is selected from the group consisting wherein denotes a point of attachment, R10 is NH(C1-6 alkyl), and n2 is selected from the group consisting of 1, 2, and 3. In some embodiments of the compound of Formula (AII-g) or (AII-h), R4 is
wherein denotes a point of attachment, R10 is NH(C1-6 alkyl), and n2 is selected from the group consisting of 1, 2, and 3. In some embodiments of the compound of Formula (AII-g) or (AII-h), R4 is , wherein R10 is NH(CH3) and n2 is 2. In some embodiments of the compound of Formula (AII-g) or (AII-h), R4 is -(CH2)2OH. Formula (AIII) In some embodiments, the ionizable amino lipids of a lipid nanoparticle may be one or more of compounds of Formula (AIII):
, wherein R10 is NH(CH3) and n2 is 2. In some embodiments of the compound of Formula (AII-g) or (AII-h), R4 is -(CH2)2OH. Formula (AIII) In some embodiments, the ionizable amino lipids of a lipid nanoparticle may be one or more of compounds of Formula (AIII): (AIII), or their N-oxides, or salts or isomers thereof, wherein: R1 is selected from the group consisting of C5-30 alkyl, C5-20 alkenyl, -R*YR”, -YR”, and -R”M’R’; R2 and R3 are independently selected from the group consisting of H, C1-14 alkyl, C2-14 alkenyl, -R*YR”, -YR”, and -R*OR”, or R2 and R3, together with the atom to which they are attached, form a heterocycle or carbocycle; R4 is selected from the group consisting of hydrogen, a C3-6 carbocycle, -(CH2)nQ, -(CH2)nCHQR, -CHQR, -CQ(R)2, and unsubstituted C1-6 alkyl, where Q is selected from a carbocycle, heterocycle, -OR, -O(CH2)nN(R)2, -C(O)OR, -OC(O)R, -CX3, -CX2H, -CXH2, -CN, -N(R)2, -C(O)N(R)2, -N(R)C(O)R, -N(R)S(O)2R, -N(R)C(O)N(R)2, -N(R)C(S)N(R)2, -N(R)R8, -N(R)S(O)2R8, -O(CH2)nOR, -N(R)C(=NR9)N(R)2, -N(R)C(=CHR9)N(R)2, -OC(O)N(R)2, -N(R)C(O)OR, -N(OR)C(O)R, -N(OR)S(O)2R, -N(OR)C(O)OR, -N(OR)C(O)N(R)2, -N(OR)C(S)N(R)2, -N(OR)C(=NR9)N(R)2, -N(OR)C(=CHR9)N(R)2, -C(=NR9)N(R)2, -C(=NR9)R, -C(O)N(R)OR, and –C(R)N(R)2C(O)OR, and each n is independently selected from 1, 2, 3, 4, and 5; each R5 is independently selected from the group consisting of C1-3 alkyl, C2-3 alkenyl, and H; each R6 is independently selected from the group consisting of C1-3 alkyl, C2-3 alkenyl, and H; M and M’ are independently selected from -C(O)O-, -OC(O)-, -OC(O)-M”-C(O)O-, -C(O)N(R’)-, -N(R’)C(O)-, -C(O)-, -C(S)-, -C(S)S-, -SC(S)-, -CH(OH)-, -P(O)(OR’)O-, -S(O)2-, -S-S-, an aryl group, and a heteroaryl group, in which M” is a bond, C1-13 alkyl or C2-13 alkenyl; R7 is selected from the group consisting of C1-3 alkyl, C2-3 alkenyl, and H; R8 is selected from the group consisting of C3-6 carbocycle and heterocycle; R9 is selected from the group consisting of H, CN, NO2, C1-6 alkyl, -OR, -S(O)2R, -S(O)2N(R)2, C2-6 alkenyl, C3-6 carbocycle and heterocycle; each R is independently selected from the group consisting of C1-3 alkyl, C2-3 alkenyl, and H; each R’ is independently selected from the group consisting of C1-18 alkyl, C2-18 alkenyl, -R*YR”, -YR”, and H; each R” is independently selected from the group consisting of C3-15 alkyl and C3-15 alkenyl; each R* is independently selected from the group consisting of C1-12 alkyl and C2-12 alkenyl; each Y is independently a C3-6 carbocycle; each X is independently selected from the group consisting of F, Cl, Br, and I; and m is selected from 5, 6, 7, 8, 9, 10, 11, 12, and 13; and wherein when R4 is -(CH2)nQ, -(CH2)nCHQR, –CHQR, or -CQ(R)2, then (i) Q is not -N(R)2 when n is 1, 2, 3, 4 or 5, or (ii) Q is not 5, 6, or 7-membered heterocycloalkyl when n is 1 or 2. In some embodiments, another subset of compounds of Formula (AIII) includes those in which: R1 is selected from the group consisting of C5-30 alkyl, C5-20 alkenyl, -R*YR”, -YR”, and -R”M’R’; R2 and R3 are independently selected from the group consisting of H, C1-14 alkyl, C2-14 alkenyl, -R*YR”, -YR”, and -R*OR”, or R2 and R3, together with the atom to which they are attached, form a heterocycle or carbocycle; R4 is selected from the group consisting of a C3-6 carbocycle, -(CH2)nQ, -(CH2)nCHQR, -CHQR, -CQ(R)2, and unsubstituted C1-6 alkyl, where Q is selected from a C3-6 carbocycle, a 5- to 14-membered heteroaryl having one or more heteroatoms selected from N, O, and S, -OR, -O(CH2)nN(R)2, -C(O)OR, -OC(O)R, -CX3, -CX2H, -CXH2, -CN, -C(O)N(R)2, -N(R)C(O)R, -N(R)S(O)2R, -N(R)C(O)N(R)2, -N(R)C(S)N(R)2, -CRN(R)2C(O)OR, -N(R)R8, -O(CH2)nOR, -N(R)C(=NR9)N(R)2, -N(R)C(=CHR9)N(R)2, -OC(O)N(R)2, -N(R)C(O)OR, -N(OR)C(O)R, -N(OR)S(O)2R, -N(OR)C(O)OR, -N(OR)C(O)N(R)2, -N(OR)C(S)N(R)2, -N(OR)C(=NR9)N(R)2, -N(OR)C(=CHR9)N(R)2, -C(=NR9)N(R)2, -C(=NR9)R, -C(O)N(R)OR, and a 5- to 14-membered heterocycloalkyl having one or more heteroatoms selected from N, O, and S which is substituted with one or more substituents selected from oxo (=O), OH, amino, mono- or di-alkylamino, and C1-3 alkyl, and each n is independently selected from 1, 2, 3, 4, and 5; each R5 is independently selected from the group consisting of C1-3 alkyl, C2-3 alkenyl, and H; each R6 is independently selected from the group consisting of C1-3 alkyl, C2-3 alkenyl, and H; M and M’ are independently selected from -C(O)O-, -OC(O)-, -C(O)N(R’)-, -N(R’)C(O)-, -C(O)-, -C(S)-, -C(S)S-, -SC(S)-, -CH(OH)-, -P(O)(OR’)O-, -S(O)2-, -S-S-, an aryl group, and a heteroaryl group; R7 is selected from the group consisting of C1-3 alkyl, C2-3 alkenyl, and H; R8 is selected from the group consisting of C3-6 carbocycle and heterocycle; R9 is selected from the group consisting of H, CN, NO2, C1-6 alkyl, -OR, -S(O)2R, -S(O)2N(R)2, C2-6 alkenyl, C3-6 carbocycle and heterocycle; each R is independently selected from the group consisting of C1-3 alkyl, C2-3 alkenyl, and H; each R’ is independently selected from the group consisting of C1-18 alkyl, C2-18 alkenyl, -R*YR”, -YR”, and H; each R” is independently selected from the group consisting of C3-14 alkyl and C3-14 alkenyl; each R* is independently selected from the group consisting of C1-12 alkyl and C2-12 alkenyl; each Y is independently a C3-6 carbocycle; each X is independently selected from the group consisting of F, Cl, Br, and I; and m is selected from 5, 6, 7, 8, 9, 10, 11, 12, and 13, or salts or isomers thereof. In some embodiments, another subset of compounds of Formula (AIII) includes those in which: R1 is selected from the group consisting of C5-30 alkyl, C5-20 alkenyl, -R*YR”, -YR”, and -R”M’R’; R2 and R3 are independently selected from the group consisting of H, C1-14 alkyl, C2-14 alkenyl, -R*YR”, -YR”, and -R*OR”, or R2 and R3, together with the atom to which they are attached, form a heterocycle or carbocycle; R4 is selected from the group consisting of a C3-6 carbocycle, -(CH2)nQ, -(CH2)nCHQR, -CHQR, -CQ(R)2, and unsubstituted C1-6 alkyl, where Q is selected from a C3-6 carbocycle, a 5- to 14-membered heterocycle having one or more heteroatoms selected from N, O, and S, -OR, -O(CH2)nN(R)2, -C(O)OR, -OC(O)R, -CX3, -CX2H, -CXH2, -CN, -C(O)N(R)2, -N(R)C(O)R, -N(R)S(O)2R, -N(R)C(O)N(R)2, -N(R)C(S)N(R)2, -CRN(R)2C(O)OR, -N(R)R8, -O(CH2)nOR, -N(R)C(=NR9)N(R)2, -N(R)C(=CHR9)N(R)2, -OC(O)N(R)2, -N(R)C(O)OR, -N(OR)C(O)R, -N(OR)S(O)2R, -N(OR)C(O)OR, -N(OR)C(O)N(R)2, -N(OR)C(S)N(R)2, -N(OR)C(=NR9)N(R)2, -N(OR)C(=CHR9)N(R)2, -C(=NR9)R, -C(O)N(R)OR, and -C(=NR9)N(R)2, and each n is independently selected from 1, 2, 3, 4, and 5; and when Q is a 5- to 14-membered heterocycle and (i) R4 is -(CH2)nQ in which n is 1 or 2, or (ii) R4 is -(CH2)nCHQR in which n is 1, or (iii) R4 is -CHQR, and -CQ(R)2, then Q is either a 5- to 14-membered heteroaryl or 8- to 14-membered heterocycloalkyl; each R5 is independently selected from the group consisting of C1-3 alkyl, C2-3 alkenyl, and H; each R6 is independently selected from the group consisting of C1-3 alkyl, C2-3 alkenyl, and H; M and M’ are independently selected from -C(O)O-, -OC(O)-, -C(O)N(R’)-, -N(R’)C(O)-, -C(O)-, -C(S)-, -C(S)S-, -SC(S)-, -CH(OH)-, -P(O)(OR’)O-, -S(O)2-, -S-S-, an aryl group, and a heteroaryl group; R7 is selected from the group consisting of C1-3 alkyl, C2-3 alkenyl, and H; R8 is selected from the group consisting of C3-6 carbocycle and heterocycle; R9 is selected from the group consisting of H, CN, NO2, C1-6 alkyl, -OR, -S(O)2R, - S(O)2N(R)2, C2-6 alkenyl, C3-6 carbocycle and heterocycle; each R is independently selected from the group consisting of C1-3 alkyl, C2-3 alkenyl, and H; each R’ is independently selected from the group consisting of C1-18 alkyl, C2-18 alkenyl, -R*YR”, -YR”, and H; each R” is independently selected from the group consisting of C3-14 alkyl and C3-14 alkenyl; each R* is independently selected from the group consisting of C1-12 alkyl and C2-12 alkenyl; each Y is independently a C3-6 carbocycle; each X is independently selected from the group consisting of F, Cl, Br, and I; and m is selected from 5, 6, 7, 8, 9, 10, 11, 12, and 13, or salts or isomers thereof. In some embodiments, another subset of compounds of Formula (AIII) includes those in which: R1 is selected from the group consisting of C5-30 alkyl, C5-20 alkenyl, -R*YR”, -YR”, and -R”M’R’; R2 and R3 are independently selected from the group consisting of H, C1-14 alkyl, C2-14 alkenyl, -R*YR”, -YR”, and -R*OR”, or R2 and R3, together with the atom to which they are attached, form a heterocycle or carbocycle; R4 is selected from the group consisting of a C3-6 carbocycle, -(CH2)nQ, -(CH2)nCHQR, -CHQR, -CQ(R)2, and unsubstituted C1-6 alkyl, where Q is selected from a C3-6 carbocycle, a 5- to 14-membered heteroaryl having one or more heteroatoms selected from N, O, and S, -OR, -O(CH2)nN(R)2, -C(O)OR, -OC(O)R, -CX3, -CX2H, -CXH2, -CN, -C(O)N(R)2, -N(R)C(O)R, -N(R)S(O)2R, -N(R)C(O)N(R)2, -N(R)C(S)N(R)2, -CRN(R)2C(O)OR, -N(R)R8, -O(CH2)nOR, -N(R)C(=NR9)N(R)2, -N(R)C(=CHR9)N(R)2, -OC(O)N(R)2, -N(R)C(O)OR, -N(OR)C(O)R, -N(OR)S(O)2R, -N(OR)C(O)OR, -N(OR)C(O)N(R)2, -N(OR)C(S)N(R)2, -N(OR)C(=NR9)N(R)2, -N(OR)C(=CHR9)N(R)2, -C(=NR9)R, -C(O)N(R)OR, and -C(=NR9)N(R)2, and each n is independently selected from 1, 2, 3, 4, and 5; each R5 is independently selected from the group consisting of C1-3 alkyl, C2-3 alkenyl, and H; each R6 is independently selected from the group consisting of C1-3 alkyl, C2-3 alkenyl, and H; M and M’ are independently selected from -C(O)O-, -OC(O)-, -C(O)N(R’)-, -N(R’)C(O)-, -C(O)-, -C(S)-, -C(S)S-, -SC(S)-, -CH(OH)-, -P(O)(OR’)O-, -S(O)2-, -S-S-, an aryl group, and a heteroaryl group; R7 is selected from the group consisting of C1-3 alkyl, C2-3 alkenyl, and H; R8 is selected from the group consisting of C3-6 carbocycle and heterocycle; R9 is selected from the group consisting of H, CN, NO2, C1-6 alkyl, -OR, -S(O)2R, - S(O)2N(R)2, C2-6 alkenyl, C3-6 carbocycle and heterocycle; each R is independently selected from the group consisting of C1-3 alkyl, C2-3 alkenyl, and H; each R’ is independently selected from the group consisting of C1-18 alkyl, C2-18 alkenyl, -R*YR”, -YR”, and H; each R” is independently selected from the group consisting of C3-14 alkyl and C3-14 alkenyl; each R* is independently selected from the group consisting of C1-12 alkyl and C2-12 alkenyl; each Y is independently a C3-6 carbocycle; each X is independently selected from the group consisting of F, Cl, Br, and I; and m is selected from 5, 6, 7, 8, 9, 10, 11, 12, and 13, or salts or isomers thereof. In some embodiments, another subset of compounds of Formula (AIII) includes those in which R1 is selected from the group consisting of C5-30 alkyl, C5-20 alkenyl, -R*YR”, -YR”, and -R”M’R’; R2 and R3 are independently selected from the group consisting of H, C2-14 alkyl, C2-14 alkenyl, -R*YR”, -YR”, and -R*OR”, or R2 and R3, together with the atom to which they are attached, form a heterocycle or carbocycle; R4 is -(CH2)nQ or -(CH2)nCHQR, where Q is -N(R)2, and n is selected from 3, 4, and 5; each R5 is independently selected from the group consisting of C1-3 alkyl, C2-3 alkenyl, and H; each R6 is independently selected from the group consisting of C1-3 alkyl, C2-3 alkenyl, and H; M and M’ are independently selected from -C(O)O-, -OC(O)-, -C(O)N(R’)-, -N(R’)C(O)-, -C(O)-, -C(S)-, -C(S)S-, -SC(S)-, -CH(OH)-, -P(O)(OR’)O-, -S(O)2-, -S-S-, an aryl group, and a heteroaryl group; R7 is selected from the group consisting of C1-3 alkyl, C2-3 alkenyl, and H; each R is independently selected from the group consisting of C1-3 alkyl, C2-3 alkenyl, and H; each R’ is independently selected from the group consisting of C1-18 alkyl, C2-18 alkenyl, -R*YR”, -YR”, and H; each R” is independently selected from the group consisting of C3-14 alkyl and C3-14 alkenyl; each R* is independently selected from the group consisting of C1-12 alkyl and C1-12 alkenyl; each Y is independently a C3-6 carbocycle; each X is independently selected from the group consisting of F, Cl, Br, and I; and m is selected from 5, 6, 7, 8, 9, 10, 11, 12, and 13, or salts or isomers thereof. In some embodiments, another subset of compounds of Formula (AIII) includes those in which R1 is selected from the group consisting of C5-30 alkyl, C5-20 alkenyl, -R*YR”, -YR”, and -R”M’R’; R2 and R3 are independently selected from the group consisting of C1-14 alkyl, C2-14 alkenyl, -R*YR”, -YR”, and -R*OR”, or R2 and R3, together with the atom to which they are attached, form a heterocycle or carbocycle; R4 is selected from the group consisting of -(CH2)nQ, -(CH2)nCHQR, -CHQR, and -CQ(R)2, where Q is -N(R)2, and n is selected from 1, 2, 3, 4, and 5; each R5 is independently selected from the group consisting of C1-3 alkyl, C2-3 alkenyl, and H; each R6 is independently selected from the group consisting of C1-3 alkyl, C2-3 alkenyl, and H; M and M’ are independently selected from -C(O)O-, -OC(O)-, -C(O)N(R’)-, -N(R’)C(O)-, -C(O)-, -C(S)-, -C(S)S-, -SC(S)-, -CH(OH)-, -P(O)(OR’)O-, -S(O)2-, -S-S-, an aryl group, and a heteroaryl group; R7 is selected from the group consisting of C1-3 alkyl, C2-3 alkenyl, and H; each R is independently selected from the group consisting of C1-3 alkyl, C2-3 alkenyl, and H; each R’ is independently selected from the group consisting of C1-18 alkyl, C2-18 alkenyl, -R*YR”, -YR”, and H; each R” is independently selected from the group consisting of C3-14 alkyl and C3-14 alkenyl; each R* is independently selected from the group consisting of C1-12 alkyl and C1-12 alkenyl; each Y is independently a C3-6 carbocycle; each X is independently selected from the group consisting of F, Cl, Br, and I; and m is selected from 5, 6, 7, 8, 9, 10, 11, 12, and 13, or salts or isomers thereof. In certain embodiments, a subset of compounds of Formula (AIII) includes those of Formula (AIII-A):
(AIII), or their N-oxides, or salts or isomers thereof, wherein: R1 is selected from the group consisting of C5-30 alkyl, C5-20 alkenyl, -R*YR”, -YR”, and -R”M’R’; R2 and R3 are independently selected from the group consisting of H, C1-14 alkyl, C2-14 alkenyl, -R*YR”, -YR”, and -R*OR”, or R2 and R3, together with the atom to which they are attached, form a heterocycle or carbocycle; R4 is selected from the group consisting of hydrogen, a C3-6 carbocycle, -(CH2)nQ, -(CH2)nCHQR, -CHQR, -CQ(R)2, and unsubstituted C1-6 alkyl, where Q is selected from a carbocycle, heterocycle, -OR, -O(CH2)nN(R)2, -C(O)OR, -OC(O)R, -CX3, -CX2H, -CXH2, -CN, -N(R)2, -C(O)N(R)2, -N(R)C(O)R, -N(R)S(O)2R, -N(R)C(O)N(R)2, -N(R)C(S)N(R)2, -N(R)R8, -N(R)S(O)2R8, -O(CH2)nOR, -N(R)C(=NR9)N(R)2, -N(R)C(=CHR9)N(R)2, -OC(O)N(R)2, -N(R)C(O)OR, -N(OR)C(O)R, -N(OR)S(O)2R, -N(OR)C(O)OR, -N(OR)C(O)N(R)2, -N(OR)C(S)N(R)2, -N(OR)C(=NR9)N(R)2, -N(OR)C(=CHR9)N(R)2, -C(=NR9)N(R)2, -C(=NR9)R, -C(O)N(R)OR, and –C(R)N(R)2C(O)OR, and each n is independently selected from 1, 2, 3, 4, and 5; each R5 is independently selected from the group consisting of C1-3 alkyl, C2-3 alkenyl, and H; each R6 is independently selected from the group consisting of C1-3 alkyl, C2-3 alkenyl, and H; M and M’ are independently selected from -C(O)O-, -OC(O)-, -OC(O)-M”-C(O)O-, -C(O)N(R’)-, -N(R’)C(O)-, -C(O)-, -C(S)-, -C(S)S-, -SC(S)-, -CH(OH)-, -P(O)(OR’)O-, -S(O)2-, -S-S-, an aryl group, and a heteroaryl group, in which M” is a bond, C1-13 alkyl or C2-13 alkenyl; R7 is selected from the group consisting of C1-3 alkyl, C2-3 alkenyl, and H; R8 is selected from the group consisting of C3-6 carbocycle and heterocycle; R9 is selected from the group consisting of H, CN, NO2, C1-6 alkyl, -OR, -S(O)2R, -S(O)2N(R)2, C2-6 alkenyl, C3-6 carbocycle and heterocycle; each R is independently selected from the group consisting of C1-3 alkyl, C2-3 alkenyl, and H; each R’ is independently selected from the group consisting of C1-18 alkyl, C2-18 alkenyl, -R*YR”, -YR”, and H; each R” is independently selected from the group consisting of C3-15 alkyl and C3-15 alkenyl; each R* is independently selected from the group consisting of C1-12 alkyl and C2-12 alkenyl; each Y is independently a C3-6 carbocycle; each X is independently selected from the group consisting of F, Cl, Br, and I; and m is selected from 5, 6, 7, 8, 9, 10, 11, 12, and 13; and wherein when R4 is -(CH2)nQ, -(CH2)nCHQR, –CHQR, or -CQ(R)2, then (i) Q is not -N(R)2 when n is 1, 2, 3, 4 or 5, or (ii) Q is not 5, 6, or 7-membered heterocycloalkyl when n is 1 or 2. In some embodiments, another subset of compounds of Formula (AIII) includes those in which: R1 is selected from the group consisting of C5-30 alkyl, C5-20 alkenyl, -R*YR”, -YR”, and -R”M’R’; R2 and R3 are independently selected from the group consisting of H, C1-14 alkyl, C2-14 alkenyl, -R*YR”, -YR”, and -R*OR”, or R2 and R3, together with the atom to which they are attached, form a heterocycle or carbocycle; R4 is selected from the group consisting of a C3-6 carbocycle, -(CH2)nQ, -(CH2)nCHQR, -CHQR, -CQ(R)2, and unsubstituted C1-6 alkyl, where Q is selected from a C3-6 carbocycle, a 5- to 14-membered heteroaryl having one or more heteroatoms selected from N, O, and S, -OR, -O(CH2)nN(R)2, -C(O)OR, -OC(O)R, -CX3, -CX2H, -CXH2, -CN, -C(O)N(R)2, -N(R)C(O)R, -N(R)S(O)2R, -N(R)C(O)N(R)2, -N(R)C(S)N(R)2, -CRN(R)2C(O)OR, -N(R)R8, -O(CH2)nOR, -N(R)C(=NR9)N(R)2, -N(R)C(=CHR9)N(R)2, -OC(O)N(R)2, -N(R)C(O)OR, -N(OR)C(O)R, -N(OR)S(O)2R, -N(OR)C(O)OR, -N(OR)C(O)N(R)2, -N(OR)C(S)N(R)2, -N(OR)C(=NR9)N(R)2, -N(OR)C(=CHR9)N(R)2, -C(=NR9)N(R)2, -C(=NR9)R, -C(O)N(R)OR, and a 5- to 14-membered heterocycloalkyl having one or more heteroatoms selected from N, O, and S which is substituted with one or more substituents selected from oxo (=O), OH, amino, mono- or di-alkylamino, and C1-3 alkyl, and each n is independently selected from 1, 2, 3, 4, and 5; each R5 is independently selected from the group consisting of C1-3 alkyl, C2-3 alkenyl, and H; each R6 is independently selected from the group consisting of C1-3 alkyl, C2-3 alkenyl, and H; M and M’ are independently selected from -C(O)O-, -OC(O)-, -C(O)N(R’)-, -N(R’)C(O)-, -C(O)-, -C(S)-, -C(S)S-, -SC(S)-, -CH(OH)-, -P(O)(OR’)O-, -S(O)2-, -S-S-, an aryl group, and a heteroaryl group; R7 is selected from the group consisting of C1-3 alkyl, C2-3 alkenyl, and H; R8 is selected from the group consisting of C3-6 carbocycle and heterocycle; R9 is selected from the group consisting of H, CN, NO2, C1-6 alkyl, -OR, -S(O)2R, -S(O)2N(R)2, C2-6 alkenyl, C3-6 carbocycle and heterocycle; each R is independently selected from the group consisting of C1-3 alkyl, C2-3 alkenyl, and H; each R’ is independently selected from the group consisting of C1-18 alkyl, C2-18 alkenyl, -R*YR”, -YR”, and H; each R” is independently selected from the group consisting of C3-14 alkyl and C3-14 alkenyl; each R* is independently selected from the group consisting of C1-12 alkyl and C2-12 alkenyl; each Y is independently a C3-6 carbocycle; each X is independently selected from the group consisting of F, Cl, Br, and I; and m is selected from 5, 6, 7, 8, 9, 10, 11, 12, and 13, or salts or isomers thereof. In some embodiments, another subset of compounds of Formula (AIII) includes those in which: R1 is selected from the group consisting of C5-30 alkyl, C5-20 alkenyl, -R*YR”, -YR”, and -R”M’R’; R2 and R3 are independently selected from the group consisting of H, C1-14 alkyl, C2-14 alkenyl, -R*YR”, -YR”, and -R*OR”, or R2 and R3, together with the atom to which they are attached, form a heterocycle or carbocycle; R4 is selected from the group consisting of a C3-6 carbocycle, -(CH2)nQ, -(CH2)nCHQR, -CHQR, -CQ(R)2, and unsubstituted C1-6 alkyl, where Q is selected from a C3-6 carbocycle, a 5- to 14-membered heterocycle having one or more heteroatoms selected from N, O, and S, -OR, -O(CH2)nN(R)2, -C(O)OR, -OC(O)R, -CX3, -CX2H, -CXH2, -CN, -C(O)N(R)2, -N(R)C(O)R, -N(R)S(O)2R, -N(R)C(O)N(R)2, -N(R)C(S)N(R)2, -CRN(R)2C(O)OR, -N(R)R8, -O(CH2)nOR, -N(R)C(=NR9)N(R)2, -N(R)C(=CHR9)N(R)2, -OC(O)N(R)2, -N(R)C(O)OR, -N(OR)C(O)R, -N(OR)S(O)2R, -N(OR)C(O)OR, -N(OR)C(O)N(R)2, -N(OR)C(S)N(R)2, -N(OR)C(=NR9)N(R)2, -N(OR)C(=CHR9)N(R)2, -C(=NR9)R, -C(O)N(R)OR, and -C(=NR9)N(R)2, and each n is independently selected from 1, 2, 3, 4, and 5; and when Q is a 5- to 14-membered heterocycle and (i) R4 is -(CH2)nQ in which n is 1 or 2, or (ii) R4 is -(CH2)nCHQR in which n is 1, or (iii) R4 is -CHQR, and -CQ(R)2, then Q is either a 5- to 14-membered heteroaryl or 8- to 14-membered heterocycloalkyl; each R5 is independently selected from the group consisting of C1-3 alkyl, C2-3 alkenyl, and H; each R6 is independently selected from the group consisting of C1-3 alkyl, C2-3 alkenyl, and H; M and M’ are independently selected from -C(O)O-, -OC(O)-, -C(O)N(R’)-, -N(R’)C(O)-, -C(O)-, -C(S)-, -C(S)S-, -SC(S)-, -CH(OH)-, -P(O)(OR’)O-, -S(O)2-, -S-S-, an aryl group, and a heteroaryl group; R7 is selected from the group consisting of C1-3 alkyl, C2-3 alkenyl, and H; R8 is selected from the group consisting of C3-6 carbocycle and heterocycle; R9 is selected from the group consisting of H, CN, NO2, C1-6 alkyl, -OR, -S(O)2R, - S(O)2N(R)2, C2-6 alkenyl, C3-6 carbocycle and heterocycle; each R is independently selected from the group consisting of C1-3 alkyl, C2-3 alkenyl, and H; each R’ is independently selected from the group consisting of C1-18 alkyl, C2-18 alkenyl, -R*YR”, -YR”, and H; each R” is independently selected from the group consisting of C3-14 alkyl and C3-14 alkenyl; each R* is independently selected from the group consisting of C1-12 alkyl and C2-12 alkenyl; each Y is independently a C3-6 carbocycle; each X is independently selected from the group consisting of F, Cl, Br, and I; and m is selected from 5, 6, 7, 8, 9, 10, 11, 12, and 13, or salts or isomers thereof. In some embodiments, another subset of compounds of Formula (AIII) includes those in which: R1 is selected from the group consisting of C5-30 alkyl, C5-20 alkenyl, -R*YR”, -YR”, and -R”M’R’; R2 and R3 are independently selected from the group consisting of H, C1-14 alkyl, C2-14 alkenyl, -R*YR”, -YR”, and -R*OR”, or R2 and R3, together with the atom to which they are attached, form a heterocycle or carbocycle; R4 is selected from the group consisting of a C3-6 carbocycle, -(CH2)nQ, -(CH2)nCHQR, -CHQR, -CQ(R)2, and unsubstituted C1-6 alkyl, where Q is selected from a C3-6 carbocycle, a 5- to 14-membered heteroaryl having one or more heteroatoms selected from N, O, and S, -OR, -O(CH2)nN(R)2, -C(O)OR, -OC(O)R, -CX3, -CX2H, -CXH2, -CN, -C(O)N(R)2, -N(R)C(O)R, -N(R)S(O)2R, -N(R)C(O)N(R)2, -N(R)C(S)N(R)2, -CRN(R)2C(O)OR, -N(R)R8, -O(CH2)nOR, -N(R)C(=NR9)N(R)2, -N(R)C(=CHR9)N(R)2, -OC(O)N(R)2, -N(R)C(O)OR, -N(OR)C(O)R, -N(OR)S(O)2R, -N(OR)C(O)OR, -N(OR)C(O)N(R)2, -N(OR)C(S)N(R)2, -N(OR)C(=NR9)N(R)2, -N(OR)C(=CHR9)N(R)2, -C(=NR9)R, -C(O)N(R)OR, and -C(=NR9)N(R)2, and each n is independently selected from 1, 2, 3, 4, and 5; each R5 is independently selected from the group consisting of C1-3 alkyl, C2-3 alkenyl, and H; each R6 is independently selected from the group consisting of C1-3 alkyl, C2-3 alkenyl, and H; M and M’ are independently selected from -C(O)O-, -OC(O)-, -C(O)N(R’)-, -N(R’)C(O)-, -C(O)-, -C(S)-, -C(S)S-, -SC(S)-, -CH(OH)-, -P(O)(OR’)O-, -S(O)2-, -S-S-, an aryl group, and a heteroaryl group; R7 is selected from the group consisting of C1-3 alkyl, C2-3 alkenyl, and H; R8 is selected from the group consisting of C3-6 carbocycle and heterocycle; R9 is selected from the group consisting of H, CN, NO2, C1-6 alkyl, -OR, -S(O)2R, - S(O)2N(R)2, C2-6 alkenyl, C3-6 carbocycle and heterocycle; each R is independently selected from the group consisting of C1-3 alkyl, C2-3 alkenyl, and H; each R’ is independently selected from the group consisting of C1-18 alkyl, C2-18 alkenyl, -R*YR”, -YR”, and H; each R” is independently selected from the group consisting of C3-14 alkyl and C3-14 alkenyl; each R* is independently selected from the group consisting of C1-12 alkyl and C2-12 alkenyl; each Y is independently a C3-6 carbocycle; each X is independently selected from the group consisting of F, Cl, Br, and I; and m is selected from 5, 6, 7, 8, 9, 10, 11, 12, and 13, or salts or isomers thereof. In some embodiments, another subset of compounds of Formula (AIII) includes those in which R1 is selected from the group consisting of C5-30 alkyl, C5-20 alkenyl, -R*YR”, -YR”, and -R”M’R’; R2 and R3 are independently selected from the group consisting of H, C2-14 alkyl, C2-14 alkenyl, -R*YR”, -YR”, and -R*OR”, or R2 and R3, together with the atom to which they are attached, form a heterocycle or carbocycle; R4 is -(CH2)nQ or -(CH2)nCHQR, where Q is -N(R)2, and n is selected from 3, 4, and 5; each R5 is independently selected from the group consisting of C1-3 alkyl, C2-3 alkenyl, and H; each R6 is independently selected from the group consisting of C1-3 alkyl, C2-3 alkenyl, and H; M and M’ are independently selected from -C(O)O-, -OC(O)-, -C(O)N(R’)-, -N(R’)C(O)-, -C(O)-, -C(S)-, -C(S)S-, -SC(S)-, -CH(OH)-, -P(O)(OR’)O-, -S(O)2-, -S-S-, an aryl group, and a heteroaryl group; R7 is selected from the group consisting of C1-3 alkyl, C2-3 alkenyl, and H; each R is independently selected from the group consisting of C1-3 alkyl, C2-3 alkenyl, and H; each R’ is independently selected from the group consisting of C1-18 alkyl, C2-18 alkenyl, -R*YR”, -YR”, and H; each R” is independently selected from the group consisting of C3-14 alkyl and C3-14 alkenyl; each R* is independently selected from the group consisting of C1-12 alkyl and C1-12 alkenyl; each Y is independently a C3-6 carbocycle; each X is independently selected from the group consisting of F, Cl, Br, and I; and m is selected from 5, 6, 7, 8, 9, 10, 11, 12, and 13, or salts or isomers thereof. In some embodiments, another subset of compounds of Formula (AIII) includes those in which R1 is selected from the group consisting of C5-30 alkyl, C5-20 alkenyl, -R*YR”, -YR”, and -R”M’R’; R2 and R3 are independently selected from the group consisting of C1-14 alkyl, C2-14 alkenyl, -R*YR”, -YR”, and -R*OR”, or R2 and R3, together with the atom to which they are attached, form a heterocycle or carbocycle; R4 is selected from the group consisting of -(CH2)nQ, -(CH2)nCHQR, -CHQR, and -CQ(R)2, where Q is -N(R)2, and n is selected from 1, 2, 3, 4, and 5; each R5 is independently selected from the group consisting of C1-3 alkyl, C2-3 alkenyl, and H; each R6 is independently selected from the group consisting of C1-3 alkyl, C2-3 alkenyl, and H; M and M’ are independently selected from -C(O)O-, -OC(O)-, -C(O)N(R’)-, -N(R’)C(O)-, -C(O)-, -C(S)-, -C(S)S-, -SC(S)-, -CH(OH)-, -P(O)(OR’)O-, -S(O)2-, -S-S-, an aryl group, and a heteroaryl group; R7 is selected from the group consisting of C1-3 alkyl, C2-3 alkenyl, and H; each R is independently selected from the group consisting of C1-3 alkyl, C2-3 alkenyl, and H; each R’ is independently selected from the group consisting of C1-18 alkyl, C2-18 alkenyl, -R*YR”, -YR”, and H; each R” is independently selected from the group consisting of C3-14 alkyl and C3-14 alkenyl; each R* is independently selected from the group consisting of C1-12 alkyl and C1-12 alkenyl; each Y is independently a C3-6 carbocycle; each X is independently selected from the group consisting of F, Cl, Br, and I; and m is selected from 5, 6, 7, 8, 9, 10, 11, 12, and 13, or salts or isomers thereof. In certain embodiments, a subset of compounds of Formula (AIII) includes those of Formula (AIII-A): (AIII-A), or its N-oxide, or a salt or isomer thereof, wherein l is selected from 1, 2, 3, 4, and 5; m is selected from 5, 6, 7, 8, and 9; M1 is a bond or M’; R4 is hydrogen, unsubstituted C1-3 alkyl, or -(CH2)nQ, in which Q is -OH, -NHC(S)N(R)2, -NHC(O)N(R)2, -N(R)C(O)R, -N(R)S(O)2R, -N(R)R8, -NHC(=NR9)N(R)2, -NHC(=CHR9)N(R)2, -OC(O)N(R)2, -N(R)C(O)OR, heteroaryl or heterocycloalkyl; M and M’ are independently selected from -C(O)O-, -OC(O)-, -OC(O)-M”-C(O)O-, -C(O)N(R’)-, -P(O)(OR’)O-, -S-S-, an aryl group, and a heteroaryl group,; and R2 and R3 are independently selected from the group consisting of H, C1-14 alkyl, and C2-14 alkenyl. For example, m is 5, 7, or 9. For example, Q is OH, -NHC(S)N(R)2, or -NHC(O)N(R)2. For example, Q is -N(R)C(O)R, or -N(R)S(O)2R. In certain embodiments, a subset of compounds of Formula (AIII) includes those of Formula (AIII-B):
(AIII-A), or its N-oxide, or a salt or isomer thereof, wherein l is selected from 1, 2, 3, 4, and 5; m is selected from 5, 6, 7, 8, and 9; M1 is a bond or M’; R4 is hydrogen, unsubstituted C1-3 alkyl, or -(CH2)nQ, in which Q is -OH, -NHC(S)N(R)2, -NHC(O)N(R)2, -N(R)C(O)R, -N(R)S(O)2R, -N(R)R8, -NHC(=NR9)N(R)2, -NHC(=CHR9)N(R)2, -OC(O)N(R)2, -N(R)C(O)OR, heteroaryl or heterocycloalkyl; M and M’ are independently selected from -C(O)O-, -OC(O)-, -OC(O)-M”-C(O)O-, -C(O)N(R’)-, -P(O)(OR’)O-, -S-S-, an aryl group, and a heteroaryl group,; and R2 and R3 are independently selected from the group consisting of H, C1-14 alkyl, and C2-14 alkenyl. For example, m is 5, 7, or 9. For example, Q is OH, -NHC(S)N(R)2, or -NHC(O)N(R)2. For example, Q is -N(R)C(O)R, or -N(R)S(O)2R. In certain embodiments, a subset of compounds of Formula (AIII) includes those of Formula (AIII-B): its N-oxide, or a salt or isomer thereof in which all variables are as defined herein. For example, m is selected from 5, 6, 7, 8, and 9; R4 is hydrogen, unsubstituted C1-3 alkyl, or -(CH2)nQ, in which Q is H, -NHC(S)N(R)2, -NHC(O)N(R)2, -N(R)C(O)R, -N(R)S(O)2R, -N(R)R8, -NHC(=NR9)N(R)2, -NHC(=CHR9)N(R)2, -OC(O)N(R)2, -N(R)C(O)OR, heteroaryl or heterocycloalkyl; M and M’ are independently selected from -C(O)O-, -OC(O)-, -OC(O)-M”-C(O)O-, -C(O)N(R’)-, -P(O)(OR’)O-, -S-S-, an aryl group, and a heteroaryl group; and R2 and R3 are independently selected from the group consisting of H, C1-14 alkyl, and C2-14 alkenyl. For example, m is 5, 7, or 9. For example, Q is OH, -NHC(S)N(R)2, or -NHC(O)N(R)2. For example, Q is -N(R)C(O)R, or -N(R)S(O)2R. In certain embodiments, a subset of compounds of Formula (AIII) includes those of Formula (AIII-C):
its N-oxide, or a salt or isomer thereof in which all variables are as defined herein. For example, m is selected from 5, 6, 7, 8, and 9; R4 is hydrogen, unsubstituted C1-3 alkyl, or -(CH2)nQ, in which Q is H, -NHC(S)N(R)2, -NHC(O)N(R)2, -N(R)C(O)R, -N(R)S(O)2R, -N(R)R8, -NHC(=NR9)N(R)2, -NHC(=CHR9)N(R)2, -OC(O)N(R)2, -N(R)C(O)OR, heteroaryl or heterocycloalkyl; M and M’ are independently selected from -C(O)O-, -OC(O)-, -OC(O)-M”-C(O)O-, -C(O)N(R’)-, -P(O)(OR’)O-, -S-S-, an aryl group, and a heteroaryl group; and R2 and R3 are independently selected from the group consisting of H, C1-14 alkyl, and C2-14 alkenyl. For example, m is 5, 7, or 9. For example, Q is OH, -NHC(S)N(R)2, or -NHC(O)N(R)2. For example, Q is -N(R)C(O)R, or -N(R)S(O)2R. In certain embodiments, a subset of compounds of Formula (AIII) includes those of Formula (AIII-C): (AIII-C), or its N-oxide, or a salt or isomer thereof, wherein l is selected from 1, 2, 3, 4, and 5; M1 is a bond or M’; R4 is hydrogen, unsubstituted C1-3 alkyl, or -(CH2)nQ, in which n is 2, 3, or 4, and Q is OH, -NHC(S)N(R)2, -NHC(O)N(R)2, -N(R)C(O)R, -N(R)S(O)2R, -N(R)R8, -NHC(=NR9)N(R)2, -NHC(=CHR9)N(R)2, -OC(O)N(R)2, -N(R)C(O)OR, heteroaryl or heterocycloalkyl; M and M’ are independently selected from -C(O)O-, -OC(O)-, -OC(O)-M”-C(O)O-, -C(O)N(R’)-, -P(O)(OR’)O-, -S-S-, an aryl group, and a heteroaryl group; and R2 and R3 are independently selected from the group consisting of H, C1-14 alkyl, and C2-14 alkenyl. In some embodiments, the compounds of Formula (AIII) are of Formula (AIII-D), their N-oxides, or salts or isomers thereof, wherein R4 is as described herein. In another embodiment, the compounds of Formula (AIII) are of Formula (AIII-E),
(AIII-C), or its N-oxide, or a salt or isomer thereof, wherein l is selected from 1, 2, 3, 4, and 5; M1 is a bond or M’; R4 is hydrogen, unsubstituted C1-3 alkyl, or -(CH2)nQ, in which n is 2, 3, or 4, and Q is OH, -NHC(S)N(R)2, -NHC(O)N(R)2, -N(R)C(O)R, -N(R)S(O)2R, -N(R)R8, -NHC(=NR9)N(R)2, -NHC(=CHR9)N(R)2, -OC(O)N(R)2, -N(R)C(O)OR, heteroaryl or heterocycloalkyl; M and M’ are independently selected from -C(O)O-, -OC(O)-, -OC(O)-M”-C(O)O-, -C(O)N(R’)-, -P(O)(OR’)O-, -S-S-, an aryl group, and a heteroaryl group; and R2 and R3 are independently selected from the group consisting of H, C1-14 alkyl, and C2-14 alkenyl. In some embodiments, the compounds of Formula (AIII) are of Formula (AIII-D), their N-oxides, or salts or isomers thereof, wherein R4 is as described herein. In another embodiment, the compounds of Formula (AIII) are of Formula (AIII-E), their N-oxides, or salts or isomers thereof, wherein R4 is as described herein. In another embodiment, the compounds of Formula (AIII) are of Formula (AIII-F) or (AIII-G):
their N-oxides, or salts or isomers thereof, wherein R4 is as described herein. In another embodiment, the compounds of Formula (AIII) are of Formula (AIII-F) or (AIII-G): their N-oxides, or salts or isomers thereof, wherein R4 is as described herein. In another embodiment, the compounds of Formula (AIII) are of Formula (AIII-H):
their N-oxides, or salts or isomers thereof, wherein R4 is as described herein. In another embodiment, the compounds of Formula (AIII) are of Formula (AIII-H): their N-oxides, or salts or isomers thereof, wherein M is -C(O)O- or –OC(O)-, M” is C1-6 alkyl or C2-6 alkenyl, R2 and R3 are independently selected from the group consisting of C5-14 alkyl and C5-14 alkenyl, and n is selected from 2, 3, and 4. In a further embodiment, the compounds of Formula (AIII) are of Formula (AIII-I): (AIII-I), or their N-oxides, or salts or isomers thereof, wherein n is 2, 3, or 4; and m, R’, R”, and R2 through R6 are as described herein. For example, each of R2 and R3 may be independently selected from the group consisting of C5-14 alkyl and C5-14 alkenyl. In some embodiments, an ionizable amino lipid of the disclosure comprises a compound having structure:
their N-oxides, or salts or isomers thereof, wherein M is -C(O)O- or –OC(O)-, M” is C1-6 alkyl or C2-6 alkenyl, R2 and R3 are independently selected from the group consisting of C5-14 alkyl and C5-14 alkenyl, and n is selected from 2, 3, and 4. In a further embodiment, the compounds of Formula (AIII) are of Formula (AIII-I): (AIII-I), or their N-oxides, or salts or isomers thereof, wherein n is 2, 3, or 4; and m, R’, R”, and R2 through R6 are as described herein. For example, each of R2 and R3 may be independently selected from the group consisting of C5-14 alkyl and C5-14 alkenyl. In some embodiments, an ionizable amino lipid of the disclosure comprises a compound having structure: (Compound 1). In some embodiments, an ionizable amino lipid of the disclosure comprises a compound having structure:
(Compound 1). In some embodiments, an ionizable amino lipid of the disclosure comprises a compound having structure: (Compound 2). In a further embodiment, the compounds of Formula (AIII) are of Formula (AIII-J),
(Compound 2). In a further embodiment, the compounds of Formula (AIII) are of Formula (AIII-J), (AIII-J), or their N-oxides, or salts or isomers thereof, wherein l is selected from 1, 2, 3, 4, and 5; m is selected from 5, 6, 7, 8, and 9; M1 is a bond or M’; M and M’ are independently selected from -C(O)O-, -OC(O)-, -OC(O)-M”-C(O)O-, -C(O)N(R’)-, -P(O)(OR’)O-, -S-S-, an aryl group, and a heteroaryl group; and R2 and R3 are independently selected from the group consisting of H, C1-14 alkyl, and C2-14 alkenyl. For example, M” is C1-6 alkyl (e.g., C1-4 alkyl) or C2-6 alkenyl (e.g. C2-4 alkenyl). For example, R2 and R3 are independently selected from the group consisting of C5-14 alkyl and C5-14 alkenyl. In some embodiments, the ionizable amino lipids are one or more of the compounds described in U.S. Application Nos. 62/220,091, 62/252,316, 62/253,433, 62/266,460, 62/333,557, 62/382,740, 62/393,940, 62/471,937, 62/471,949, 62/475,140, and 62/475,166, and PCT Application No. PCT/US2016/052352. The central amine moiety of a lipid according to Formula (AIII), (AIII-A), (AIII-B), (AIII-C), (AIII-D), (AIII-E), (AIII-F), (AIII-G), (AIII-H), (AIII-I), or (AIII-J) may be protonated at a physiological pH. Thus, a lipid may have a positive or partial positive charge at physiological pH. Such amino lipids may be referred to as cationic lipids, ionizable lipids, cationic amino lipids, or ionizable amino lipids. Amino lipids may also be zwitterionic, i.e., neutral molecules having both a positive and a negative charge. Formula (AIV) In some embodiments, the ionizable amino lipids of a lipid nanoparticle may be one or more of compounds of formula (AIV),
(AIII-J), or their N-oxides, or salts or isomers thereof, wherein l is selected from 1, 2, 3, 4, and 5; m is selected from 5, 6, 7, 8, and 9; M1 is a bond or M’; M and M’ are independently selected from -C(O)O-, -OC(O)-, -OC(O)-M”-C(O)O-, -C(O)N(R’)-, -P(O)(OR’)O-, -S-S-, an aryl group, and a heteroaryl group; and R2 and R3 are independently selected from the group consisting of H, C1-14 alkyl, and C2-14 alkenyl. For example, M” is C1-6 alkyl (e.g., C1-4 alkyl) or C2-6 alkenyl (e.g. C2-4 alkenyl). For example, R2 and R3 are independently selected from the group consisting of C5-14 alkyl and C5-14 alkenyl. In some embodiments, the ionizable amino lipids are one or more of the compounds described in U.S. Application Nos. 62/220,091, 62/252,316, 62/253,433, 62/266,460, 62/333,557, 62/382,740, 62/393,940, 62/471,937, 62/471,949, 62/475,140, and 62/475,166, and PCT Application No. PCT/US2016/052352. The central amine moiety of a lipid according to Formula (AIII), (AIII-A), (AIII-B), (AIII-C), (AIII-D), (AIII-E), (AIII-F), (AIII-G), (AIII-H), (AIII-I), or (AIII-J) may be protonated at a physiological pH. Thus, a lipid may have a positive or partial positive charge at physiological pH. Such amino lipids may be referred to as cationic lipids, ionizable lipids, cationic amino lipids, or ionizable amino lipids. Amino lipids may also be zwitterionic, i.e., neutral molecules having both a positive and a negative charge. Formula (AIV) In some embodiments, the ionizable amino lipids of a lipid nanoparticle may be one or more of compounds of formula (AIV), t is 1 or 2; A1 and A2 are each independently selected from CH or N; Z is CH2 or absent wherein when Z is CH2, the dashed lines (1) and (2) each represent a single bond; and when Z is absent, the dashed lines (1) and (2) are both absent; R1, R2, R3, R4, and R5 are independently selected from the group consisting of C5-20 alkyl, C5-20 alkenyl, -R”MR’, -R*YR”, -YR”, and -R*OR”; RX1 and RX2 are each independently H or C1-3 alkyl; each M is independently selected from the group consisting of -C(O)O-, -OC(O)-, -OC(O)O-, -C(O)N(R’)-, -N(R’)C(O)-, -C(O)-, -C(S)-, -C(S)S-, -SC(S)-, -CH(OH)-, -P(O)(OR’)O-, -S(O)2-, -C(O)S-, -SC(O)-, an aryl group, and a heteroaryl group; M* is C1-C6 alkyl, W1 and W2 are each independently selected from the group consisting of -O- and -N(R6)-; each R6 is independently selected from the group consisting of H and C1-5 alkyl; X1, X2, and X3 are independently selected from the group consisting of a bond, -CH2-, -(CH2)2-, -CHR-, -CHY-, -C(O)-, -C(O)O-, -OC(O)-, -(CH2)n-C(O)-, -C(O)-(CH2)n-, -(CH2)n-C(O)O-, -OC(O)-(CH2)n-, -(CH2)n-OC(O)-, -C(O)O-(CH2)n-, -CH(OH)-, -C(S)-, and -CH(SH)-; each Y is independently a C3-6 carbocycle; each R* is independently selected from the group consisting of C1-12 alkyl and C2-12 alkenyl; each R is independently selected from the group consisting of C1-3 alkyl and a C3-6 carbocycle; each R’ is independently selected from the group consisting of C1-12 alkyl, C2-12 alkenyl, and H; each R” is independently selected from the group consisting of C3-12 alkyl, C3-12 alkenyl and -R*MR’; and n is an integer from 1-6; wherein when ring
t is 1 or 2; A1 and A2 are each independently selected from CH or N; Z is CH2 or absent wherein when Z is CH2, the dashed lines (1) and (2) each represent a single bond; and when Z is absent, the dashed lines (1) and (2) are both absent; R1, R2, R3, R4, and R5 are independently selected from the group consisting of C5-20 alkyl, C5-20 alkenyl, -R”MR’, -R*YR”, -YR”, and -R*OR”; RX1 and RX2 are each independently H or C1-3 alkyl; each M is independently selected from the group consisting of -C(O)O-, -OC(O)-, -OC(O)O-, -C(O)N(R’)-, -N(R’)C(O)-, -C(O)-, -C(S)-, -C(S)S-, -SC(S)-, -CH(OH)-, -P(O)(OR’)O-, -S(O)2-, -C(O)S-, -SC(O)-, an aryl group, and a heteroaryl group; M* is C1-C6 alkyl, W1 and W2 are each independently selected from the group consisting of -O- and -N(R6)-; each R6 is independently selected from the group consisting of H and C1-5 alkyl; X1, X2, and X3 are independently selected from the group consisting of a bond, -CH2-, -(CH2)2-, -CHR-, -CHY-, -C(O)-, -C(O)O-, -OC(O)-, -(CH2)n-C(O)-, -C(O)-(CH2)n-, -(CH2)n-C(O)O-, -OC(O)-(CH2)n-, -(CH2)n-OC(O)-, -C(O)O-(CH2)n-, -CH(OH)-, -C(S)-, and -CH(SH)-; each Y is independently a C3-6 carbocycle; each R* is independently selected from the group consisting of C1-12 alkyl and C2-12 alkenyl; each R is independently selected from the group consisting of C1-3 alkyl and a C3-6 carbocycle; each R’ is independently selected from the group consisting of C1-12 alkyl, C2-12 alkenyl, and H; each R” is independently selected from the group consisting of C3-12 alkyl, C3-12 alkenyl and -R*MR’; and n is an integer from 1-6; wherein when ring then i) at least one of X1, X2, and X3 is not -CH2-; and/or ii) at least one of R1, R2, R3, R4, and R5 is -R”MR’. In some embodiments, the compound is of any of formulae (AIVa)-(AIVh):
then i) at least one of X1, X2, and X3 is not -CH2-; and/or ii) at least one of R1, R2, R3, R4, and R5 is -R”MR’. In some embodiments, the compound is of any of formulae (AIVa)-(AIVh): In some embodiments, the ionizable amino lipid is
In some embodiments, the ionizable amino lipid is salt thereof. The central amine moiety of a lipid according to Formula (AIV), (AIVa), (AIVb), (AIVc), (AIVd), (AIVe), (AIVf), (AIVg), or (AIVh) may be protonated at a physiological pH. Thus, a lipid may have a positive or partial positive charge at physiological pH. Formula (AV) In some embodiments, the lipid nanoparticle comprises a lipid having the structure: pharmaceutically acceptable salt, tautomer, or stereoisomer thereof, wherein: R1 is optionally substituted C1-C24 alkyl or optionally substituted C2-C24 alkenyl; R2 and R3 are each independently optionally substituted C1-C36 alkyl; R4 and R5 are each independently optionally substituted C 4 1-C6 alkyl, or R and R5 join, along with the N to which they are attached, to form a heterocyclyl or heteroaryl; L1, L2, and L3 are each independently optionally substituted C1-C18 alkylene; G1 is a direct bond, -(CH2)nO(C=O)-, -(CH2)n(C=O)O-, or -(C=O)-; G2 and G3 are each independently –(C=O)O- or -0(C=O)-; and n is an integer greater than 0. Formula (AVI) In some embodiments, the lipid nanoparticle comprises a lipid having the structure:
salt thereof. The central amine moiety of a lipid according to Formula (AIV), (AIVa), (AIVb), (AIVc), (AIVd), (AIVe), (AIVf), (AIVg), or (AIVh) may be protonated at a physiological pH. Thus, a lipid may have a positive or partial positive charge at physiological pH. Formula (AV) In some embodiments, the lipid nanoparticle comprises a lipid having the structure: pharmaceutically acceptable salt, tautomer, or stereoisomer thereof, wherein: R1 is optionally substituted C1-C24 alkyl or optionally substituted C2-C24 alkenyl; R2 and R3 are each independently optionally substituted C1-C36 alkyl; R4 and R5 are each independently optionally substituted C 4 1-C6 alkyl, or R and R5 join, along with the N to which they are attached, to form a heterocyclyl or heteroaryl; L1, L2, and L3 are each independently optionally substituted C1-C18 alkylene; G1 is a direct bond, -(CH2)nO(C=O)-, -(CH2)n(C=O)O-, or -(C=O)-; G2 and G3 are each independently –(C=O)O- or -0(C=O)-; and n is an integer greater than 0. Formula (AVI) In some embodiments, the lipid nanoparticle comprises a lipid having the structure: pharmaceutically acceptable salt, tautomer, or stereoisomer thereof, wherein: G1 is -N(R3)R4 or -OR5; R1 is optionally substituted branched, saturated or unsaturated C12-C36 alkyl; R2 is optionally substituted branched or unbranched, saturated or unsaturated C12- C36 alkyl when L is -C(=O)-; or R2 is optionally substituted branched or unbranched, saturated or unsaturated C4-C36 alkyl when L is C6-C12 alkylene, C6-C12 alkenylene, or C2-C6 alkynylene; R3 and R4 are each independently H, optionally substituted branched or unbranched, saturated or unsaturated C1-C6 alkyl; or R3 and R4 are each independently optionally substituted branched or unbranched, saturated or unsaturated C1-C6 alkyl when L is C6-C12 alkylene, C6-C12 alkenylene, or C2-C6 alkynylene; or R3 and R4, together with the nitrogen to which they are attached, join to form a heterocyclyl; R5 is H or optionally substituted C1-C6 alkyl; L is -C(=O)-, C6-C12 alkylene, C6-C12 alkenylene, or C2-C6 alkynylene; and n is an integer from 1 to 12. Formula (AVII) In some embodiments, the lipid nanoparticle comprises a lipid having the structure: pharmaceutically acceptable salt thereof, wherein: each R1a is independently hydrogen, R1c, or R1d; each R1b is independently R1c or R1d; each R1c is independently –[CH2]2C(O)X1R3; each R1d Is independently -C(O)R4; each R2 is independently -[C(R2a)2]cR2b; each R2a is independently hydrogen or C1-C6 alkyl; R2b is -N(L1-B)2; -(OCH2CH2)6OH; or -(OCH2CH2)bOCH3; each R3 and R4 is independently C6-C30 aliphatic; each L1 is independently C1-C10 alkylene; each B is independently hydrogen or an ionizable nitrogen-containing group; each X1 is independently a covalent bond or O; each a is independently an integer of 1-10; each b is independently an integer of 1-10; and each c is independently an integer of 1-10. Formula (AVIII) In some embodiments, the lipid nanoparticle comprises a lipid having the structure:
pharmaceutically acceptable salt, tautomer, or stereoisomer thereof, wherein: G1 is -N(R3)R4 or -OR5; R1 is optionally substituted branched, saturated or unsaturated C12-C36 alkyl; R2 is optionally substituted branched or unbranched, saturated or unsaturated C12- C36 alkyl when L is -C(=O)-; or R2 is optionally substituted branched or unbranched, saturated or unsaturated C4-C36 alkyl when L is C6-C12 alkylene, C6-C12 alkenylene, or C2-C6 alkynylene; R3 and R4 are each independently H, optionally substituted branched or unbranched, saturated or unsaturated C1-C6 alkyl; or R3 and R4 are each independently optionally substituted branched or unbranched, saturated or unsaturated C1-C6 alkyl when L is C6-C12 alkylene, C6-C12 alkenylene, or C2-C6 alkynylene; or R3 and R4, together with the nitrogen to which they are attached, join to form a heterocyclyl; R5 is H or optionally substituted C1-C6 alkyl; L is -C(=O)-, C6-C12 alkylene, C6-C12 alkenylene, or C2-C6 alkynylene; and n is an integer from 1 to 12. Formula (AVII) In some embodiments, the lipid nanoparticle comprises a lipid having the structure: pharmaceutically acceptable salt thereof, wherein: each R1a is independently hydrogen, R1c, or R1d; each R1b is independently R1c or R1d; each R1c is independently –[CH2]2C(O)X1R3; each R1d Is independently -C(O)R4; each R2 is independently -[C(R2a)2]cR2b; each R2a is independently hydrogen or C1-C6 alkyl; R2b is -N(L1-B)2; -(OCH2CH2)6OH; or -(OCH2CH2)bOCH3; each R3 and R4 is independently C6-C30 aliphatic; each L1 is independently C1-C10 alkylene; each B is independently hydrogen or an ionizable nitrogen-containing group; each X1 is independently a covalent bond or O; each a is independently an integer of 1-10; each b is independently an integer of 1-10; and each c is independently an integer of 1-10. Formula (AVIII) In some embodiments, the lipid nanoparticle comprises a lipid having the structure: pharmaceutically acceptable salt, prodrug or stereoisomer thereof, wherein: X is N, and Y is absent; or X is CR, and Y is NR; L1 is -O(C-O)R1, -(C=O)OR1, -C(=O)R1, -OR1, -S(O)xR1, -S-SR1, -C(=O)SR1, -SC(=O)R1, -NRaC(=O)R1, -C(=O)NRbRc, -NRaC(=O)NRbRc, -OC(=O)NRbRc, or -NRaC(=O)OR1; L2 is -O(C=O)R2, -(C=O)OR2, -C(=O)R2, -OR2, -S(O)xR2, -S-SR2, -C(=O)SR2, -SC(=O)R2, -NRdC(=O)R2, -C(=O)NReRf, -NRdC(=O)NReRf, -OC(=O)NReRf; -NRdC(=O)OR2 or a direct bond to R2; L3 is -O(C=O)R3 or -(C=O)OR3; G1 and G2 are each independently C2-C12 alkylene or C2-C12 alkenylene; G3 is C1-C24 alkylene, C2-C24 alkenylene, C1-C24 heteroalkylene or C2-C24 heteroalkenylene when X is CR, and Y is NR; and G3 is C1-C24 heteroalkylene or C2-C24 heteroalkenylene when X is N, and Y is absent; Ra, Rb, Rd and Re are each independently H or C1-C12 alkyl or C1-C12 alkenyl; Rc and Rf are each independently C1-C12 alkyl or C2-C12 alkenyl; each R is independently H or C1-C12 alkyl; R1, R2 and R3 are each independently C1-C24 alkyl or C2-C24 alkenyl; and x is 0, 1 or 2, and wherein each alkyl, alkenyl, alkylene, alkenylene, heteroalkylene and heteroalkenylene is independently substituted or unsubstituted unless otherwise specified. Formula (AIX) In some embodiments, the lipid nanoparticle comprises a lipid having the structure:
pharmaceutically acceptable salt, prodrug or stereoisomer thereof, wherein: X is N, and Y is absent; or X is CR, and Y is NR; L1 is -O(C-O)R1, -(C=O)OR1, -C(=O)R1, -OR1, -S(O)xR1, -S-SR1, -C(=O)SR1, -SC(=O)R1, -NRaC(=O)R1, -C(=O)NRbRc, -NRaC(=O)NRbRc, -OC(=O)NRbRc, or -NRaC(=O)OR1; L2 is -O(C=O)R2, -(C=O)OR2, -C(=O)R2, -OR2, -S(O)xR2, -S-SR2, -C(=O)SR2, -SC(=O)R2, -NRdC(=O)R2, -C(=O)NReRf, -NRdC(=O)NReRf, -OC(=O)NReRf; -NRdC(=O)OR2 or a direct bond to R2; L3 is -O(C=O)R3 or -(C=O)OR3; G1 and G2 are each independently C2-C12 alkylene or C2-C12 alkenylene; G3 is C1-C24 alkylene, C2-C24 alkenylene, C1-C24 heteroalkylene or C2-C24 heteroalkenylene when X is CR, and Y is NR; and G3 is C1-C24 heteroalkylene or C2-C24 heteroalkenylene when X is N, and Y is absent; Ra, Rb, Rd and Re are each independently H or C1-C12 alkyl or C1-C12 alkenyl; Rc and Rf are each independently C1-C12 alkyl or C2-C12 alkenyl; each R is independently H or C1-C12 alkyl; R1, R2 and R3 are each independently C1-C24 alkyl or C2-C24 alkenyl; and x is 0, 1 or 2, and wherein each alkyl, alkenyl, alkylene, alkenylene, heteroalkylene and heteroalkenylene is independently substituted or unsubstituted unless otherwise specified. Formula (AIX) In some embodiments, the lipid nanoparticle comprises a lipid having the structure: pharmaceutically acceptable salt, tautomer, prodrug or stereoisomer thereof, wherein: L1 and L2 are each independently -O(C=O)-, -(C=O)O-, -C(=O)-, -O-, -S(O)x-, -S-S-, -C(=O)S-, -SC(=O)-, -NRaC(=O)-, -C(=O)NRa-, -NRaC(=O)NRa-, -OC(=O)NRa-, -NRaC(=O)O- or a direct bond; G1 is C1-C2 alkylene, -(C=O)-, -O(C=O)-, -SC(=O)-, -NRaC(=O)- or a direct bond; G2 is -C(O)-, -(CO)O-, -C(=O)S-, -C(=O)NRa- or a direct bond; G3 is C1-C6 alkylene; Ra is H or C1-C12 alkyl; R1a and R1b are, at each occurrence, independently either: (a) H or C1-C12 alkyl; or (b) R1a is H or C1-C12 alkyl, and R1b together with the carbon atom to which it is bound is taken together with an adjacent R1b and the carbon atom to which it is bound to form a carbon-carbon double bond; R2a and R2b are, at each occurrence, independently either: (a) H or C1-C12 alkyl; or (b) R2a is H or C1-C12 alkyl, and R2b together with the carbon atom to which it is bound is taken together with an adjacent R2b and the carbon atom to which it is bound to form a carbon-carbon double bond; R3a and R3b are, at each occurrence, independently either (a): H or C1-C12 alkyl; or (b) R3a is H or C1-C12 alkyl, and R3b together with the carbon atom to which it is bound is taken together with an adjacent R and the carbon atom to which it is bound to form a carbon-carbon double bond; R4a and R4b are, at each occurrence, independently either: (a) H or C1-C12 alkyl; or (b) R4a is H or C1-C12 alkyl, and R4b together with the carbon atom to which it is bound is taken together with an adjacent R4b and the carbon atom to which it is bound to form a carbon-carbon double bond; R5 and R6 are each independently H or methyl; R7 is H or C1-C20 alkyl; R8 is OH, -N(R9)(C=O)R10, -(C=O)NR9R10, -NR9R10, -(C=O)OR" or -O(C=O)R", provided that G3 is C 8 4-C6 alkylene when R is -NR9R10, R9 and R10 are each independently H or C1-C12 alkyl; R" is aralkyl; a, b, c and d are each independently an integer from 1 to 24; and x is 0, 1 or 2, wherein each alkyl, alkylene and aralkyl is optionally substituted. Formula (AX) In some embodiments, the lipid nanoparticle comprises a lipid having the structure:
pharmaceutically acceptable salt, tautomer, prodrug or stereoisomer thereof, wherein: L1 and L2 are each independently -O(C=O)-, -(C=O)O-, -C(=O)-, -O-, -S(O)x-, -S-S-, -C(=O)S-, -SC(=O)-, -NRaC(=O)-, -C(=O)NRa-, -NRaC(=O)NRa-, -OC(=O)NRa-, -NRaC(=O)O- or a direct bond; G1 is C1-C2 alkylene, -(C=O)-, -O(C=O)-, -SC(=O)-, -NRaC(=O)- or a direct bond; G2 is -C(O)-, -(CO)O-, -C(=O)S-, -C(=O)NRa- or a direct bond; G3 is C1-C6 alkylene; Ra is H or C1-C12 alkyl; R1a and R1b are, at each occurrence, independently either: (a) H or C1-C12 alkyl; or (b) R1a is H or C1-C12 alkyl, and R1b together with the carbon atom to which it is bound is taken together with an adjacent R1b and the carbon atom to which it is bound to form a carbon-carbon double bond; R2a and R2b are, at each occurrence, independently either: (a) H or C1-C12 alkyl; or (b) R2a is H or C1-C12 alkyl, and R2b together with the carbon atom to which it is bound is taken together with an adjacent R2b and the carbon atom to which it is bound to form a carbon-carbon double bond; R3a and R3b are, at each occurrence, independently either (a): H or C1-C12 alkyl; or (b) R3a is H or C1-C12 alkyl, and R3b together with the carbon atom to which it is bound is taken together with an adjacent R and the carbon atom to which it is bound to form a carbon-carbon double bond; R4a and R4b are, at each occurrence, independently either: (a) H or C1-C12 alkyl; or (b) R4a is H or C1-C12 alkyl, and R4b together with the carbon atom to which it is bound is taken together with an adjacent R4b and the carbon atom to which it is bound to form a carbon-carbon double bond; R5 and R6 are each independently H or methyl; R7 is H or C1-C20 alkyl; R8 is OH, -N(R9)(C=O)R10, -(C=O)NR9R10, -NR9R10, -(C=O)OR" or -O(C=O)R", provided that G3 is C 8 4-C6 alkylene when R is -NR9R10, R9 and R10 are each independently H or C1-C12 alkyl; R" is aralkyl; a, b, c and d are each independently an integer from 1 to 24; and x is 0, 1 or 2, wherein each alkyl, alkylene and aralkyl is optionally substituted. Formula (AX) In some embodiments, the lipid nanoparticle comprises a lipid having the structure: pharmaceutically acceptable salt, prodrug or stereoisomer thereof, wherein: X and X' are each independently N or CR; Y and Y' are each independently absent, -O(C=O)-, -(C=O)O- or NR, provided that: a) Y is absent when X is N; b) Y' is absent when X' is N; c) Y is -O(C=O)-, -(C=O)O- or NR when X is CR; and d) Y' is -O(C=O)-, -(C=O)O- or NR when X' is CR, L1 and L1' are each independently -O(C=O)R', -(C=O)OR', -C(=O)R', -OR1, -S(O)zR', -S-SR1, -C(=O)SR', -SC(=O)R', -NRaC(=O)R', -C(=O)NRbRc, -NRaC(=O)NRbRc, -OC(=O)NRbRc or -NRaC(=O)OR'; L2 and L2’ are each independently -O(C=O)R2, -(C=O)OR2, -C(=O)R2, -OR2, -S(O)zR2, -S-SR2, -C(=O)SR2, -SC(=O)R2, -NRdC(=O)R2, -C(=O)NReRf, -NRdC(=O)NReRf, -OC(=O)NReRf, -NRdC(=O)OR2 or a direct bond to R2; G1. G1’, G2 and G2’ are each independently C2-C12 alkylene or C2-C12 alkenylene; G is C2-C24 heteroalkylene or C2-C24 heteroalkenylene; Ra, Rb, Rd and Re are, at each occurrence, independently H, C1-C12 alkyl or C2-C12 alkenyl; Rc and Rf are, at each occurrence, independently C1-C12 alkyl or C2-C12 alkenyl; R is, at each occurrence, independently H or C1-C12 alkyl; R1 and R2 are, at each occurrence, independently branched C6-C24 alkyl or branched C6-C24 alkenyl; z is 0, 1 or 2, and wherein each alkyl, alkenyl, alkylene, alkenylene, heteroalkylene and heteroalkenylene is independently substituted or unsubstituted unless otherwise specified. Formula (AXI) In some embodiments, the lipid nanoparticle comprises a lipid having the structure:
pharmaceutically acceptable salt, prodrug or stereoisomer thereof, wherein: X and X' are each independently N or CR; Y and Y' are each independently absent, -O(C=O)-, -(C=O)O- or NR, provided that: a) Y is absent when X is N; b) Y' is absent when X' is N; c) Y is -O(C=O)-, -(C=O)O- or NR when X is CR; and d) Y' is -O(C=O)-, -(C=O)O- or NR when X' is CR, L1 and L1' are each independently -O(C=O)R', -(C=O)OR', -C(=O)R', -OR1, -S(O)zR', -S-SR1, -C(=O)SR', -SC(=O)R', -NRaC(=O)R', -C(=O)NRbRc, -NRaC(=O)NRbRc, -OC(=O)NRbRc or -NRaC(=O)OR'; L2 and L2’ are each independently -O(C=O)R2, -(C=O)OR2, -C(=O)R2, -OR2, -S(O)zR2, -S-SR2, -C(=O)SR2, -SC(=O)R2, -NRdC(=O)R2, -C(=O)NReRf, -NRdC(=O)NReRf, -OC(=O)NReRf, -NRdC(=O)OR2 or a direct bond to R2; G1. G1’, G2 and G2’ are each independently C2-C12 alkylene or C2-C12 alkenylene; G is C2-C24 heteroalkylene or C2-C24 heteroalkenylene; Ra, Rb, Rd and Re are, at each occurrence, independently H, C1-C12 alkyl or C2-C12 alkenyl; Rc and Rf are, at each occurrence, independently C1-C12 alkyl or C2-C12 alkenyl; R is, at each occurrence, independently H or C1-C12 alkyl; R1 and R2 are, at each occurrence, independently branched C6-C24 alkyl or branched C6-C24 alkenyl; z is 0, 1 or 2, and wherein each alkyl, alkenyl, alkylene, alkenylene, heteroalkylene and heteroalkenylene is independently substituted or unsubstituted unless otherwise specified. Formula (AXI) In some embodiments, the lipid nanoparticle comprises a lipid having the structure: pharmaceutically acceptable salt, prodrug or stereoisomer thereof, wherein: L1 is -O(C=O)R1, -(C=O)OR1, -C(=O)R1, -OR1, -S(O)xR1, -S-SR1, - C(=O)SR1, -SC(=O)R1, -NRaC(=O)R1, -C(=O)NRbRc, -NRaC(=O)NRbRc, -OC(=O)NRbRc or -NRaC(=O)OR1; L2 is -O(C=O)R2, -(C=O)OR2, -C(=O)R2, -OR2, -S(O)xR2, -S-SR2, -C(=O)SR2, -SC(=O)R2, -NRdC(=O)R2, -C(=O)NReRf, -NRdC(=O)NReRf, -OC(=O)NReRf; -NRdC(=O)OR2 or a direct bond to R2; G1 and G2 are each independently C2-C12 alkylene or C2-C12 alkenylene; G3 is C1-C24 alkylene, C2-C24 alkenylene, C3-C8 cycloalkylene or C3-C8 cycloalkenylene; Ra, Rb, Rd and Re are each independently H or C1-C12 alkyl or C1-C12 alkenyl; Rc and Rf are each independently C1-C12 alkyl or C2-C12 alkenyl; R1 and R2 are each independently branched C6-C24 alkyl or branched C6- C24 alkenyl; R3 is -N(R4)R5; R4 is C1-C12 alkyl; R5 is substituted C1-C12 alkyl; and x is 0, 1 or 2, and wherein each alkyl, alkenyl, alkylene, alkenylene, cycloalkylene, cycloalkenylene, aryl and aralkyl is independently substituted or unsubstituted unless otherwise specified. In some embodiments, the lipid nanoparticle comprises a lipid having the structure: pharmaceutically acceptable salt, prodrug or stereoisomer thereof, wherein: L1 is -O(C=O)R1, -(C=O)OR1, -C(=O)R1, -OR1, -S(O)xR1, -S-SR1, -C(=O)SR1, -SC(=O)R1, -NRaC(=O)R1, -C(=O)NRbRc, -NRaC(=O)NRbRc, -OC(=O)NRbRc or -NRaC(=O)OR1; L2 is -O(C=O)R2, -(C=O)OR2, -C(=O)R2, -OR2, -S(O)xR2, -S-SR2, -C(=O)SR2, -SC(=O)R2, -NRdC(=O)R2, -C(=O)NReRf, -NRdC(=O)NReRf, -OC(=O)NReRf; -NRdC(=O)OR2 or a direct bond to R2; G1a and G2b are each independently C2-C12 alkylene or C2-C12 alkenylene; G1b and G2b are each independently C1-C12 alkylene or C2-C12 alkenylene; G3 is C1-C24 alkylene, C2-C24 alkenylene, C3-C8 cycloalkylene or C3-C8 cycloalkenylene; Ra, Rb, Rd and Re are each independently H or C1-C12 alkyl or C2-C12 alkenyl; Rc and Rf are each independently C1-C12 alkyl or C2-C12 alkenyl; R1 and R2 are each independently branched C6-C24 alkyl or branched C6- C24 alkenyl; R3a is -C(=O)N(R4a)R5a or -C(=O)OR6; R3b is -NR4bC(=O)R5b; R4a is C1-C12 alkyl; R4b is H, C1-C12 alkyl or C2-C12 alkenyl; R5a is H, C1-C8 alkyl or C2-C8 alkenyl; R5b is C2-C12 alkyl or C2-C12 alkenyl when R4b is H; or R5b is C1-C12 alkyl or C2-C12 alkenyl when R4b is C1-C12 alkyl or C2-C12 alkenyl; R6 is H, aryl or aralkyl; and x is 0, 1 or 2, and wherein each alkyl, alkenyl, alkylene, alkenylene, cycloalkylene, cycloalkenylene, aryl and aralkyl is independently substituted or unsubstituted. Formula (AXII) In some embodiments, the lipid nanoparticle comprises a lipid having the structure:
pharmaceutically acceptable salt, prodrug or stereoisomer thereof, wherein: L1 is -O(C=O)R1, -(C=O)OR1, -C(=O)R1, -OR1, -S(O)xR1, -S-SR1, - C(=O)SR1, -SC(=O)R1, -NRaC(=O)R1, -C(=O)NRbRc, -NRaC(=O)NRbRc, -OC(=O)NRbRc or -NRaC(=O)OR1; L2 is -O(C=O)R2, -(C=O)OR2, -C(=O)R2, -OR2, -S(O)xR2, -S-SR2, -C(=O)SR2, -SC(=O)R2, -NRdC(=O)R2, -C(=O)NReRf, -NRdC(=O)NReRf, -OC(=O)NReRf; -NRdC(=O)OR2 or a direct bond to R2; G1 and G2 are each independently C2-C12 alkylene or C2-C12 alkenylene; G3 is C1-C24 alkylene, C2-C24 alkenylene, C3-C8 cycloalkylene or C3-C8 cycloalkenylene; Ra, Rb, Rd and Re are each independently H or C1-C12 alkyl or C1-C12 alkenyl; Rc and Rf are each independently C1-C12 alkyl or C2-C12 alkenyl; R1 and R2 are each independently branched C6-C24 alkyl or branched C6- C24 alkenyl; R3 is -N(R4)R5; R4 is C1-C12 alkyl; R5 is substituted C1-C12 alkyl; and x is 0, 1 or 2, and wherein each alkyl, alkenyl, alkylene, alkenylene, cycloalkylene, cycloalkenylene, aryl and aralkyl is independently substituted or unsubstituted unless otherwise specified. In some embodiments, the lipid nanoparticle comprises a lipid having the structure: pharmaceutically acceptable salt, prodrug or stereoisomer thereof, wherein: L1 is -O(C=O)R1, -(C=O)OR1, -C(=O)R1, -OR1, -S(O)xR1, -S-SR1, -C(=O)SR1, -SC(=O)R1, -NRaC(=O)R1, -C(=O)NRbRc, -NRaC(=O)NRbRc, -OC(=O)NRbRc or -NRaC(=O)OR1; L2 is -O(C=O)R2, -(C=O)OR2, -C(=O)R2, -OR2, -S(O)xR2, -S-SR2, -C(=O)SR2, -SC(=O)R2, -NRdC(=O)R2, -C(=O)NReRf, -NRdC(=O)NReRf, -OC(=O)NReRf; -NRdC(=O)OR2 or a direct bond to R2; G1a and G2b are each independently C2-C12 alkylene or C2-C12 alkenylene; G1b and G2b are each independently C1-C12 alkylene or C2-C12 alkenylene; G3 is C1-C24 alkylene, C2-C24 alkenylene, C3-C8 cycloalkylene or C3-C8 cycloalkenylene; Ra, Rb, Rd and Re are each independently H or C1-C12 alkyl or C2-C12 alkenyl; Rc and Rf are each independently C1-C12 alkyl or C2-C12 alkenyl; R1 and R2 are each independently branched C6-C24 alkyl or branched C6- C24 alkenyl; R3a is -C(=O)N(R4a)R5a or -C(=O)OR6; R3b is -NR4bC(=O)R5b; R4a is C1-C12 alkyl; R4b is H, C1-C12 alkyl or C2-C12 alkenyl; R5a is H, C1-C8 alkyl or C2-C8 alkenyl; R5b is C2-C12 alkyl or C2-C12 alkenyl when R4b is H; or R5b is C1-C12 alkyl or C2-C12 alkenyl when R4b is C1-C12 alkyl or C2-C12 alkenyl; R6 is H, aryl or aralkyl; and x is 0, 1 or 2, and wherein each alkyl, alkenyl, alkylene, alkenylene, cycloalkylene, cycloalkenylene, aryl and aralkyl is independently substituted or unsubstituted. Formula (AXII) In some embodiments, the lipid nanoparticle comprises a lipid having the structure: pharmaceutically acceptable salt, prodrug or stereoisomer thereof, wherein: G1 is -OH, -R3R4, -(C=O)R5 or -R3(C=O)R5; R is, at each occurrence, independently H or OH; R1 and R2 are each independently optionally substituted branched, saturated or unsaturated C12-C36 alkyl; R3 and R4 are each independently H or optionally substituted straight or branched, saturated or unsaturated C1-C6 alkyl; R5 is optionally substituted straight or branched, saturated or unsaturated C1-C6 alkyl; and n is an integer from 2 to 6. Formula (AXIII) In some embodiments, the lipid nanoparticle comprises a lipid having the structure:
pharmaceutically acceptable salt, prodrug or stereoisomer thereof, wherein: G1 is -OH, -R3R4, -(C=O)R5 or -R3(C=O)R5; R is, at each occurrence, independently H or OH; R1 and R2 are each independently optionally substituted branched, saturated or unsaturated C12-C36 alkyl; R3 and R4 are each independently H or optionally substituted straight or branched, saturated or unsaturated C1-C6 alkyl; R5 is optionally substituted straight or branched, saturated or unsaturated C1-C6 alkyl; and n is an integer from 2 to 6. Formula (AXIII) In some embodiments, the lipid nanoparticle comprises a lipid having the structure: pharmaceutically acceptable salt, prodrug or stereoisomer thereof, wherein: one of G1 or G2 is, at each occurrence, -O(C=O)-, -(C=O)O-, -C(=O)-, -O-, -S(O) , -S-S-, -C(=O)S-, SC(=O)-, -N(Ra)C(=O)-, -C(=O)N(Ra)-, -N(Ra)C(=O)N(Ra)-, -OC(=O)N(Ra)- or -N(Ra)C(=O)O-, and the other of G1 or G2 is, at each occurrence, -O(C=O)-, -(C=O)O-, -C(=O)-, -O-, -S(O), -S-S-, -C(=O)S-, -SC(=O)-, -N(Ra)C(=O)-, -C(=O)N(Ra)-, -N(Ra)C(=O)N(Ra)-, -OC(=O)N(Ra)- or -N(Ra)C(=O)O- or a direct bond; L is, at each occurrence, ~O(C=O)-, wherein ~ represents a covalent bond to X; X is CRa; Z is alkyl, cycloalkyl or a monovalent moiety comprising at least one polar functional group when n is 1; or Z is alkylene, cycloalkylene or a polyvalent moiety comprising at least one polar functional group when n is greater than 1; Ra is, at each occurrence, independently H, C1-C12 alkyl, C1-C12 hydroxylalkyl, C1-C12 aminoalkyl, C1-C12 alkylaminylalkyl, C1-C12 alkoxyalkyl, C1-C12 alkoxycarbonyl, C1-C12 alkylcarbonyloxy, C1-C12 alkylcarbonyloxyalkyl or C1-C12 alkylcarbonyl; R is, at each occurrence, independently either: (a) H or C1-C12 alkyl; or (b) R together with the carbon atom to which it is bound is taken together with an adjacent R and the carbon atom to which it is bound to form a carbon-carbon double bond; R1 and R2 have, at each occurrence, the following structure, respectively: a1 and a2 are, at each occurrence, independently an integer from 3 to 12; b1 and b2 are, at each occurrence, independently 0 or 1; c1 and c2 are, at each occurrence, independently an integer from 5 to 10; d1 and d2 are, at each occurrence, independently an integer from 5 to 10; y is, at each occurrence, independently an integer from 0 to 2; and n is an integer from 1 to 6, wherein each alkyl, alkylene, hydroxylalkyl, aminoalkyl, alkylaminylalkyl, alkoxyalkyl, alkoxycarbonyl, alkylcarbonyloxy, alkylcarbonyloxyalkyl and alkylcarbonyl is optionally substituted with one or more substituent. Formula (AXIV) In some embodiments, the lipid nanoparticle comprises a lipid having the structure:
pharmaceutically acceptable salt, prodrug or stereoisomer thereof, wherein: one of G1 or G2 is, at each occurrence, -O(C=O)-, -(C=O)O-, -C(=O)-, -O-, -S(O) , -S-S-, -C(=O)S-, SC(=O)-, -N(Ra)C(=O)-, -C(=O)N(Ra)-, -N(Ra)C(=O)N(Ra)-, -OC(=O)N(Ra)- or -N(Ra)C(=O)O-, and the other of G1 or G2 is, at each occurrence, -O(C=O)-, -(C=O)O-, -C(=O)-, -O-, -S(O), -S-S-, -C(=O)S-, -SC(=O)-, -N(Ra)C(=O)-, -C(=O)N(Ra)-, -N(Ra)C(=O)N(Ra)-, -OC(=O)N(Ra)- or -N(Ra)C(=O)O- or a direct bond; L is, at each occurrence, ~O(C=O)-, wherein ~ represents a covalent bond to X; X is CRa; Z is alkyl, cycloalkyl or a monovalent moiety comprising at least one polar functional group when n is 1; or Z is alkylene, cycloalkylene or a polyvalent moiety comprising at least one polar functional group when n is greater than 1; Ra is, at each occurrence, independently H, C1-C12 alkyl, C1-C12 hydroxylalkyl, C1-C12 aminoalkyl, C1-C12 alkylaminylalkyl, C1-C12 alkoxyalkyl, C1-C12 alkoxycarbonyl, C1-C12 alkylcarbonyloxy, C1-C12 alkylcarbonyloxyalkyl or C1-C12 alkylcarbonyl; R is, at each occurrence, independently either: (a) H or C1-C12 alkyl; or (b) R together with the carbon atom to which it is bound is taken together with an adjacent R and the carbon atom to which it is bound to form a carbon-carbon double bond; R1 and R2 have, at each occurrence, the following structure, respectively: a1 and a2 are, at each occurrence, independently an integer from 3 to 12; b1 and b2 are, at each occurrence, independently 0 or 1; c1 and c2 are, at each occurrence, independently an integer from 5 to 10; d1 and d2 are, at each occurrence, independently an integer from 5 to 10; y is, at each occurrence, independently an integer from 0 to 2; and n is an integer from 1 to 6, wherein each alkyl, alkylene, hydroxylalkyl, aminoalkyl, alkylaminylalkyl, alkoxyalkyl, alkoxycarbonyl, alkylcarbonyloxy, alkylcarbonyloxyalkyl and alkylcarbonyl is optionally substituted with one or more substituent. Formula (AXIV) In some embodiments, the lipid nanoparticle comprises a lipid having the structure: pharmaceutically acceptable salt, prodrug or stereoisomer thereof, wherein:
pharmaceutically acceptable salt, prodrug or stereoisomer thereof, wherein: -C(=O)Ra-, RaC(=O)Ra-, -OC(=O)Ra- or -NRaC(=O)O- or a direct bond; G1 and G2 are each independently unsubstituted C1-C12 alkylene or C1-C12 alkenylene; G3 is C1-C24 alkylene, C1-C24 alkenylene, C3-C8 cycloalkylene, C3-C8 cycloalkenylene; Ra is H or C1-C12 alkyl; R1 and R2 are each independently C6-C24 alkyl or C6-C24 alkenyl; R3 is H, OR5, CN, -C(=O)OR4, -OC(=O)R4 or -R5C(=O)R4; R4 is C1-C12 alkyl; R5 is H or C1-C6 alkyl; and x is 0, 1 or 2. Formula (AXV) In some embodiments, the lipid nanoparticle comprises a lipid having the structure: pharmaceutically acceptable salt, tautomer, prodrug or stereoisomer thereof, wherein: L1 and L2 are each independently -O(C=O)-, -(C=O)O-, -C(=O)-, -O-, -S(O)x-, -S-S-, -C(=O)S-, -SC(=O)-, -RaC(=O)-, -C(=O)Ra-, -RaC(=O)Ra-, -OC(=O)Ra-, -RaC(=O)O- or a direct bond; G1 is C1-C2 alkylene, -(C=O)-, -O(C=O)-, -SC(=O)-, -RaC(=O)- or a direct bond: G2 is -C(=O)-, -(C=O)O-, -C(=O)S-, -C(=O)NRa- or a direct bond; G3 is C1-C6 alkylene; Ra is H or C1-C12 alkyl; R1a and R1b are, at each occurrence, independently either: (a) H or C1-C12 alkyl; or (b) R1a is H or C1-C12 alkyl, and R1b together with the carbon atom to which it is bound is taken together with an adjacent R1b and the carbon atom to which it is bound to form a carbon-carbon double bond; R2a and R2b are, at each occurrence, independently either: (a) H or C1-C12 alkyl; or (b) R2a is H or C1-C12 alkyl, and R2b together with the carbon atom to which it is bound is taken together with an adjacent R2b and the carbon atom to which it is bound to form a carbon-carbon double bond; R3a and R3b are, at each occurrence, independently either (a): H or C1-C12 alkyl; or (b) R3a is H or C1-C12 alkyl, and R3b together with the carbon atom to which it is bound is taken together with an adjacent R and the carbon atom to which it is bound to form a carbon-carbon double bond; R4a and R4b are, at each occurrence, independently either: (a) H or C1-C12 alkyl; or (b) R4a is H or C1-C12 alkyl, and R4b together with the carbon atom to which it is bound is taken together with an adjacent R4b and the carbon atom to which it is bound to form a carbon-carbon double bond; R5 and R6 are each independently H or methyl; R7 is C4-C20 alkyl; R8 and R9 are each independently C1-C12 alkyl; or R8 and R9, together with the nitrogen atom to which they are attached, form a 5, 6 or 7-membered heterocyclic ring; a, b, c and d are each independently an integer from 1 to 24; and x is 0, 1 or 2. Formula (AXVI) In some embodiments, the lipid nanoparticle comprises a lipid having the structure:
-C(=O)Ra-, RaC(=O)Ra-, -OC(=O)Ra- or -NRaC(=O)O- or a direct bond; G1 and G2 are each independently unsubstituted C1-C12 alkylene or C1-C12 alkenylene; G3 is C1-C24 alkylene, C1-C24 alkenylene, C3-C8 cycloalkylene, C3-C8 cycloalkenylene; Ra is H or C1-C12 alkyl; R1 and R2 are each independently C6-C24 alkyl or C6-C24 alkenyl; R3 is H, OR5, CN, -C(=O)OR4, -OC(=O)R4 or -R5C(=O)R4; R4 is C1-C12 alkyl; R5 is H or C1-C6 alkyl; and x is 0, 1 or 2. Formula (AXV) In some embodiments, the lipid nanoparticle comprises a lipid having the structure: pharmaceutically acceptable salt, tautomer, prodrug or stereoisomer thereof, wherein: L1 and L2 are each independently -O(C=O)-, -(C=O)O-, -C(=O)-, -O-, -S(O)x-, -S-S-, -C(=O)S-, -SC(=O)-, -RaC(=O)-, -C(=O)Ra-, -RaC(=O)Ra-, -OC(=O)Ra-, -RaC(=O)O- or a direct bond; G1 is C1-C2 alkylene, -(C=O)-, -O(C=O)-, -SC(=O)-, -RaC(=O)- or a direct bond: G2 is -C(=O)-, -(C=O)O-, -C(=O)S-, -C(=O)NRa- or a direct bond; G3 is C1-C6 alkylene; Ra is H or C1-C12 alkyl; R1a and R1b are, at each occurrence, independently either: (a) H or C1-C12 alkyl; or (b) R1a is H or C1-C12 alkyl, and R1b together with the carbon atom to which it is bound is taken together with an adjacent R1b and the carbon atom to which it is bound to form a carbon-carbon double bond; R2a and R2b are, at each occurrence, independently either: (a) H or C1-C12 alkyl; or (b) R2a is H or C1-C12 alkyl, and R2b together with the carbon atom to which it is bound is taken together with an adjacent R2b and the carbon atom to which it is bound to form a carbon-carbon double bond; R3a and R3b are, at each occurrence, independently either (a): H or C1-C12 alkyl; or (b) R3a is H or C1-C12 alkyl, and R3b together with the carbon atom to which it is bound is taken together with an adjacent R and the carbon atom to which it is bound to form a carbon-carbon double bond; R4a and R4b are, at each occurrence, independently either: (a) H or C1-C12 alkyl; or (b) R4a is H or C1-C12 alkyl, and R4b together with the carbon atom to which it is bound is taken together with an adjacent R4b and the carbon atom to which it is bound to form a carbon-carbon double bond; R5 and R6 are each independently H or methyl; R7 is C4-C20 alkyl; R8 and R9 are each independently C1-C12 alkyl; or R8 and R9, together with the nitrogen atom to which they are attached, form a 5, 6 or 7-membered heterocyclic ring; a, b, c and d are each independently an integer from 1 to 24; and x is 0, 1 or 2. Formula (AXVI) In some embodiments, the lipid nanoparticle comprises a lipid having the structure: pharmaceutically acceptable salt, tautomer, prodrug or stereoisomer thereof, wherein: L1 and L2 are each independently -O(C=O)-, -(C=O)O- or a carbon-carbon double bond; R1a and R1b are, at each occurrence, independently either (a) H or C1-C12 alkyl, or (b) R1a is H or C1-C12 alkyl, and R1b together with the carbon atom to which it is bound is taken together with an adjacent R1b and the carbon atom to which it is bound to form a carbon-carbon double bond; R2a and R2b are, at each occurrence, independently either (a) H or C1-C12 alkyl, or (b) R2a is H or C1-C12 alkyl, and R2b together with the carbon atom to which it is bound is taken together with an adjacent R2b and the carbon atom to which it is bound to form a carbon-carbon double bond; R3a and R3b are, at each occurrence, independently either (a) H or C1-C12 alkyl, or (b) R3a is H or C1-C12 alkyl, and R3b together with the carbon atom to which it is bound is taken together with an adjacent R3b and the carbon atom to which it is bound to form a carbon-carbon double bond; R4a and R4b are, at each occurrence, independently either (a) H or C1-C12 alkyl, or (b) R4a is H or C1-C12 alkyl, and R4b together with the carbon atom to which it is bound is taken together with an adjacent R4b and the carbon atom to which it is bound to form a carbon-carbon double bond; R5 and R6 are each independently methyl or cycloalkyl; R7 is, at each occurrence, independently H or C1-C12 alkyl; R8 and R9 are each independently unsubstituted C1-C12 alkyl; or R8 and R9, together with the nitrogen atom to which they are attached, form a 5, 6 or 7- membered heterocyclic ring comprising one nitrogen atom; a and d are each independently an integer from 0 to 24; b and c are each independently an integer from 1 to 24; and e is 1 or 2, provided that: at least one of R1a, R2a, R3a or R4a is C1-C12 alkyl, or at least one of L1 or L2 is -O(C=O)- or -(C=O)O-; and R1a and R1b are not isopropyl when a is 6 or n-butyl when a is 8. Formula (AXVII) In some embodiments, the lipid nanoparticle comprises a lipid having the structure:
pharmaceutically acceptable salt, tautomer, prodrug or stereoisomer thereof, wherein: L1 and L2 are each independently -O(C=O)-, -(C=O)O- or a carbon-carbon double bond; R1a and R1b are, at each occurrence, independently either (a) H or C1-C12 alkyl, or (b) R1a is H or C1-C12 alkyl, and R1b together with the carbon atom to which it is bound is taken together with an adjacent R1b and the carbon atom to which it is bound to form a carbon-carbon double bond; R2a and R2b are, at each occurrence, independently either (a) H or C1-C12 alkyl, or (b) R2a is H or C1-C12 alkyl, and R2b together with the carbon atom to which it is bound is taken together with an adjacent R2b and the carbon atom to which it is bound to form a carbon-carbon double bond; R3a and R3b are, at each occurrence, independently either (a) H or C1-C12 alkyl, or (b) R3a is H or C1-C12 alkyl, and R3b together with the carbon atom to which it is bound is taken together with an adjacent R3b and the carbon atom to which it is bound to form a carbon-carbon double bond; R4a and R4b are, at each occurrence, independently either (a) H or C1-C12 alkyl, or (b) R4a is H or C1-C12 alkyl, and R4b together with the carbon atom to which it is bound is taken together with an adjacent R4b and the carbon atom to which it is bound to form a carbon-carbon double bond; R5 and R6 are each independently methyl or cycloalkyl; R7 is, at each occurrence, independently H or C1-C12 alkyl; R8 and R9 are each independently unsubstituted C1-C12 alkyl; or R8 and R9, together with the nitrogen atom to which they are attached, form a 5, 6 or 7- membered heterocyclic ring comprising one nitrogen atom; a and d are each independently an integer from 0 to 24; b and c are each independently an integer from 1 to 24; and e is 1 or 2, provided that: at least one of R1a, R2a, R3a or R4a is C1-C12 alkyl, or at least one of L1 or L2 is -O(C=O)- or -(C=O)O-; and R1a and R1b are not isopropyl when a is 6 or n-butyl when a is 8. Formula (AXVII) In some embodiments, the lipid nanoparticle comprises a lipid having the structure: pharmaceutically acceptable salt thereof, wherein R1 and R2 are the same or different, each a linear or branched alkyl with 1-9 carbons, or as alkenyl or alkynyl with 2 to 11 carbon atoms, L1 and L2 are the same or different, each a linear alkyl having 5 to 18 carbon atoms, or form a heterocycle with N, X1 is a bond, or is -CG-G- whereby L2-CO-O-R2 is formed, X2 is S or O, L3 is a bond or a lower alkyl, or form a heterocycle with N, R3 is a lower alkyl, and R4 and R5 are the same or different, each a lower alkyl. Compounds (A1)-(A11) In some embodiments, the lipid nanoparticle comprises an ionizable lipid having the structure:
pharmaceutically acceptable salt thereof, wherein R1 and R2 are the same or different, each a linear or branched alkyl with 1-9 carbons, or as alkenyl or alkynyl with 2 to 11 carbon atoms, L1 and L2 are the same or different, each a linear alkyl having 5 to 18 carbon atoms, or form a heterocycle with N, X1 is a bond, or is -CG-G- whereby L2-CO-O-R2 is formed, X2 is S or O, L3 is a bond or a lower alkyl, or form a heterocycle with N, R3 is a lower alkyl, and R4 and R5 are the same or different, each a lower alkyl. Compounds (A1)-(A11) In some embodiments, the lipid nanoparticle comprises an ionizable lipid having the structure: (A1), or a pharmaceutically acceptable salt thereof. In some embodiments, the lipid nanoparticle comprises a lipid having the structure:
(A1), or a pharmaceutically acceptable salt thereof. In some embodiments, the lipid nanoparticle comprises a lipid having the structure: pharmaceutically acceptable salt thereof. In some embodiments, the lipid nanoparticle comprises a lipid having the structure: (A3), or a pharmaceutically acceptable salt thereof. In some embodiments, the lipid nanoparticle comprises a lipid having the structure:
pharmaceutically acceptable salt thereof. In some embodiments, the lipid nanoparticle comprises a lipid having the structure: (A3), or a pharmaceutically acceptable salt thereof. In some embodiments, the lipid nanoparticle comprises a lipid having the structure: (A4), or a pharmaceutically acceptable salt thereof. In some embodiments, the lipid nanoparticle comprises a lipid having the structure:
(A4), or a pharmaceutically acceptable salt thereof. In some embodiments, the lipid nanoparticle comprises a lipid having the structure: (A5), or a pharmaceutically acceptable salt thereof. In some embodiments, the lipid nanoparticle comprises a lipid having the structure:
(A5), or a pharmaceutically acceptable salt thereof. In some embodiments, the lipid nanoparticle comprises a lipid having the structure: (A6), or a pharmaceutically acceptable salt thereof. In some embodiments, the lipid nanoparticle comprises a lipid having the structure:
(A6), or a pharmaceutically acceptable salt thereof. In some embodiments, the lipid nanoparticle comprises a lipid having the structure: (A7), or a pharmaceutically acceptable salt thereof. In some embodiments, the lipid nanoparticle comprises a lipid having the structure:
(A7), or a pharmaceutically acceptable salt thereof. In some embodiments, the lipid nanoparticle comprises a lipid having the structure: pharmaceutically acceptable salt thereof. In some embodiments, the lipid nanoparticle comprises a lipid having the structure:
pharmaceutically acceptable salt thereof. In some embodiments, the lipid nanoparticle comprises a lipid having the structure: pharmaceutically acceptable salt thereof. In some embodiments, the lipid nanoparticle comprises a lipid having the structure:
pharmaceutically acceptable salt thereof. In some embodiments, the lipid nanoparticle comprises a lipid having the structure: (A10), or a pharmaceutically acceptable salt thereof. In some embodiments, the lipid nanoparticle comprises a lipid having the structure:
(A10), or a pharmaceutically acceptable salt thereof. In some embodiments, the lipid nanoparticle comprises a lipid having the structure: (A11), or a pharmaceutically acceptable salt thereof. Non-cationic lipids In certain embodiments, the lipid nanoparticles described herein comprise one or more non-cationic lipids. Non-cationic lipids may be phospholipids. In some embodiments, the lipid nanoparticle comprises 5-25 mol% non-cationic lipid. For example, the lipid nanoparticle may comprise 5-20 mol%, 5-15 mol%, 5-10 mol%, 10-25 mol%, 10-20 mol%, 10-25 mol%, 15-25 mol%, 15-20 mol%, or 20-25 mol% non-cationic lipid. In some embodiments, the lipid nanoparticle comprises 5 mol%, 10 mol%, 15 mol%, 20 mol%, or 25 mol% non-cationic lipid. In some embodiments, a non-cationic lipid of the disclosure comprises 1,2-distearoyl-sn- glycero-3-phosphocholine (DSPC), 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE), 1,2-dilinoleoyl-sn-glycero-3-phosphocholine (DLPC), 1,2-dimyristoyl-sn-gly cero- phosphocholine (DMPC), 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC), l,2-dipalmitoyl- sn-glycero-3-phosphocholine (DPPC), 1,2-diundecanoyl-sn-glycero-phosphocholine (DUPC), 1- palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC), 1,2-di-O-octadecenyl-sn-glycero-3- phosphocholine (18:0 Diether PC), 1-oleoyl-2 cholesterylhemisuccinoyl-sn-glycero-3- phosphocholine (OChemsPC), 1-hexadecyl-sn-glycero-3-phosphocholine (C16 Lyso PC), 1,2- dilinolenoyl-sn-glycero-3-phosphocholine,1,2-diarachidonoyl-sn-glycero-3-phosphocholine, 1,2- didocosahexaenoyl-sn-glycero-3-phosphocholine, 1,2-diphytanoyl-sn-glycero-3- phosphoethanolamine (ME 16.0 PE), 1,2-distearoyl-sn-glycero-3-phosphoethanolamine, 1,2- dilinoleoyl-sn-glycero-3-phosphoethanolamine, 1,2-dilinolenoyl-sn-glycero-3- phosphoethanolamine, 1,2-diarachidonoyl-sn-glycero-3-phosphoethanolamine, 1,2- didocosahexaenoyl-sn-glycero-3-phosphoethanolamine, 1,2-dioleoyl-sn-glycero-3-phospho-rac- (1-glycerol) sodium salt (DOPG), sphingomyelin, or mixtures thereof. In some embodiments, the lipid nanoparticle comprises 5 – 15 mol%, 5 – 10 mol%, or 10 – 15 mol% DSPC. For example, the lipid nanoparticle may comprise 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, or 15 mol% DSPC. In certain embodiments, the lipid composition of the lipid nanoparticle composition disclosed herein can comprise one or more phospholipids, for example, one or more saturated or (poly)unsaturated phospholipids or a combination thereof. In general, phospholipids comprise a phospholipid moiety and one or more fatty acid moieties. A phospholipid moiety can be selected, for example, from the non-limiting group consisting of phosphatidyl choline, phosphatidyl ethanolamine, phosphatidyl glycerol, phosphatidyl serine, phosphatidic acid, 2-lysophosphatidyl choline, and a sphingomyelin. A fatty acid moiety can be selected, for example, from the non-limiting group consisting of lauric acid, myristic acid, myristoleic acid, palmitic acid, palmitoleic acid, stearic acid, oleic acid, linoleic acid, alpha-linolenic acid, erucic acid, phytanoic acid, arachidic acid, arachidonic acid, eicosapentaenoic acid, behenic acid, docosapentaenoic acid, and docosahexaenoic acid. Particular phospholipids can facilitate fusion to a membrane. For example, a cationic phospholipid can interact with one or more negatively charged phospholipids of a membrane (e.g., a cellular or intracellular membrane). Fusion of a phospholipid to a membrane can allow one or more elements (e.g., a therapeutic agent) of a lipid-containing composition (e.g., LNPs) to pass through the membrane permitting, e.g., delivery of the one or more elements to a target tissue. Non-natural phospholipid species including natural species with modifications and substitutions including branching, oxidation, cyclization, and alkynes are also contemplated. For example, a phospholipid can be functionalized with or cross-linked to one or more alkynes (e.g., an alkenyl group in which one or more double bonds is replaced with a triple bond). Under appropriate reaction conditions, an alkyne group can undergo a copper-catalyzed cycloaddition upon exposure to an azide. Such reactions can be useful in functionalizing a lipid bilayer of a nanoparticle composition to facilitate membrane permeation or cellular recognition or in conjugating a nanoparticle composition to a useful component such as a targeting or imaging moiety (e.g., a dye). Phospholipids include, but are not limited to, glycerophospholipids such as phosphatidylcholines, phosphatidylethanolamines, phosphatidylserines, phosphatidylinositols, phosphatidy glycerols, and phosphatidic acids. Phospholipids also include phosphosphingolipid, such as sphingomyelin. In some embodiments, a phospholipid comprises 1,2-distearoyl-sn-glycero-3- phosphocholine (DSPC), 1,2-Distearoyl-sn-glycero-3-phosphoethanolamine (DSPE), 1,2- dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE), 1,2-dilinoleoyl-sn-glycero-3- phosphocholine (DLPC), 1,2-dimyristoyl-sn-gly cero-phosphocholine (DMPC), 1,2-dioleoyl-sn- glycero-3-phosphocholine (DOPC), l,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC), 1,2- diundecanoyl-sn-glycero-phosphocholine (DUPC), 1-palmitoyl-2-oleoyl-sn-glycero-3- phosphocholine (POPC), 1,2-di-O-octadecenyl-sn-glycero-3-phosphocholine (18:0 Diether PC), 1-oleoyl-2 cholesterylhemisuccinoyl-sn-glycero-3-phosphocholine (OChemsPC), 1-hexadecyl- sn-glycero-3-phosphocholine (C16 Lyso PC), 1,2-dilinolenoyl-sn-glycero-3-phosphocholine,1,2- diarachidonoyl-sn-glycero-3-phosphocholine, 1,2-didocosahexaenoyl-sn-glycero-3- phosphocholine, 1,2-diphytanoyl-sn-glycero-3-phosphoethanolamine (ME 16.0 PE), 1,2- distearoyl-sn-glycero-3-phosphoethanolamine, 1,2-dilinoleoyl-sn-glycero-3- phosphoethanolamine, 1,2-dilinolenoyl-sn-glycero-3-phosphoethanolamine, 1,2-diarachidonoyl- sn-glycero-3-phosphoethanolamine, 1,2-didocosahexaenoyl-sn-glycero-3-phosphoethanolamine, 1,2-dioleoyl-sn-glycero-3-phospho-rac-(1-glycerol) sodium salt (DOPG), sphingomyelin, or mixtures thereof. Formula (HI) In certain embodiments, a phospholipid is an analog or variant of DSPC. In certain embodiments, a phospholipid is a compound of Formula (HI):
(A11), or a pharmaceutically acceptable salt thereof. Non-cationic lipids In certain embodiments, the lipid nanoparticles described herein comprise one or more non-cationic lipids. Non-cationic lipids may be phospholipids. In some embodiments, the lipid nanoparticle comprises 5-25 mol% non-cationic lipid. For example, the lipid nanoparticle may comprise 5-20 mol%, 5-15 mol%, 5-10 mol%, 10-25 mol%, 10-20 mol%, 10-25 mol%, 15-25 mol%, 15-20 mol%, or 20-25 mol% non-cationic lipid. In some embodiments, the lipid nanoparticle comprises 5 mol%, 10 mol%, 15 mol%, 20 mol%, or 25 mol% non-cationic lipid. In some embodiments, a non-cationic lipid of the disclosure comprises 1,2-distearoyl-sn- glycero-3-phosphocholine (DSPC), 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE), 1,2-dilinoleoyl-sn-glycero-3-phosphocholine (DLPC), 1,2-dimyristoyl-sn-gly cero- phosphocholine (DMPC), 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC), l,2-dipalmitoyl- sn-glycero-3-phosphocholine (DPPC), 1,2-diundecanoyl-sn-glycero-phosphocholine (DUPC), 1- palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC), 1,2-di-O-octadecenyl-sn-glycero-3- phosphocholine (18:0 Diether PC), 1-oleoyl-2 cholesterylhemisuccinoyl-sn-glycero-3- phosphocholine (OChemsPC), 1-hexadecyl-sn-glycero-3-phosphocholine (C16 Lyso PC), 1,2- dilinolenoyl-sn-glycero-3-phosphocholine,1,2-diarachidonoyl-sn-glycero-3-phosphocholine, 1,2- didocosahexaenoyl-sn-glycero-3-phosphocholine, 1,2-diphytanoyl-sn-glycero-3- phosphoethanolamine (ME 16.0 PE), 1,2-distearoyl-sn-glycero-3-phosphoethanolamine, 1,2- dilinoleoyl-sn-glycero-3-phosphoethanolamine, 1,2-dilinolenoyl-sn-glycero-3- phosphoethanolamine, 1,2-diarachidonoyl-sn-glycero-3-phosphoethanolamine, 1,2- didocosahexaenoyl-sn-glycero-3-phosphoethanolamine, 1,2-dioleoyl-sn-glycero-3-phospho-rac- (1-glycerol) sodium salt (DOPG), sphingomyelin, or mixtures thereof. In some embodiments, the lipid nanoparticle comprises 5 – 15 mol%, 5 – 10 mol%, or 10 – 15 mol% DSPC. For example, the lipid nanoparticle may comprise 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, or 15 mol% DSPC. In certain embodiments, the lipid composition of the lipid nanoparticle composition disclosed herein can comprise one or more phospholipids, for example, one or more saturated or (poly)unsaturated phospholipids or a combination thereof. In general, phospholipids comprise a phospholipid moiety and one or more fatty acid moieties. A phospholipid moiety can be selected, for example, from the non-limiting group consisting of phosphatidyl choline, phosphatidyl ethanolamine, phosphatidyl glycerol, phosphatidyl serine, phosphatidic acid, 2-lysophosphatidyl choline, and a sphingomyelin. A fatty acid moiety can be selected, for example, from the non-limiting group consisting of lauric acid, myristic acid, myristoleic acid, palmitic acid, palmitoleic acid, stearic acid, oleic acid, linoleic acid, alpha-linolenic acid, erucic acid, phytanoic acid, arachidic acid, arachidonic acid, eicosapentaenoic acid, behenic acid, docosapentaenoic acid, and docosahexaenoic acid. Particular phospholipids can facilitate fusion to a membrane. For example, a cationic phospholipid can interact with one or more negatively charged phospholipids of a membrane (e.g., a cellular or intracellular membrane). Fusion of a phospholipid to a membrane can allow one or more elements (e.g., a therapeutic agent) of a lipid-containing composition (e.g., LNPs) to pass through the membrane permitting, e.g., delivery of the one or more elements to a target tissue. Non-natural phospholipid species including natural species with modifications and substitutions including branching, oxidation, cyclization, and alkynes are also contemplated. For example, a phospholipid can be functionalized with or cross-linked to one or more alkynes (e.g., an alkenyl group in which one or more double bonds is replaced with a triple bond). Under appropriate reaction conditions, an alkyne group can undergo a copper-catalyzed cycloaddition upon exposure to an azide. Such reactions can be useful in functionalizing a lipid bilayer of a nanoparticle composition to facilitate membrane permeation or cellular recognition or in conjugating a nanoparticle composition to a useful component such as a targeting or imaging moiety (e.g., a dye). Phospholipids include, but are not limited to, glycerophospholipids such as phosphatidylcholines, phosphatidylethanolamines, phosphatidylserines, phosphatidylinositols, phosphatidy glycerols, and phosphatidic acids. Phospholipids also include phosphosphingolipid, such as sphingomyelin. In some embodiments, a phospholipid comprises 1,2-distearoyl-sn-glycero-3- phosphocholine (DSPC), 1,2-Distearoyl-sn-glycero-3-phosphoethanolamine (DSPE), 1,2- dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE), 1,2-dilinoleoyl-sn-glycero-3- phosphocholine (DLPC), 1,2-dimyristoyl-sn-gly cero-phosphocholine (DMPC), 1,2-dioleoyl-sn- glycero-3-phosphocholine (DOPC), l,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC), 1,2- diundecanoyl-sn-glycero-phosphocholine (DUPC), 1-palmitoyl-2-oleoyl-sn-glycero-3- phosphocholine (POPC), 1,2-di-O-octadecenyl-sn-glycero-3-phosphocholine (18:0 Diether PC), 1-oleoyl-2 cholesterylhemisuccinoyl-sn-glycero-3-phosphocholine (OChemsPC), 1-hexadecyl- sn-glycero-3-phosphocholine (C16 Lyso PC), 1,2-dilinolenoyl-sn-glycero-3-phosphocholine,1,2- diarachidonoyl-sn-glycero-3-phosphocholine, 1,2-didocosahexaenoyl-sn-glycero-3- phosphocholine, 1,2-diphytanoyl-sn-glycero-3-phosphoethanolamine (ME 16.0 PE), 1,2- distearoyl-sn-glycero-3-phosphoethanolamine, 1,2-dilinoleoyl-sn-glycero-3- phosphoethanolamine, 1,2-dilinolenoyl-sn-glycero-3-phosphoethanolamine, 1,2-diarachidonoyl- sn-glycero-3-phosphoethanolamine, 1,2-didocosahexaenoyl-sn-glycero-3-phosphoethanolamine, 1,2-dioleoyl-sn-glycero-3-phospho-rac-(1-glycerol) sodium salt (DOPG), sphingomyelin, or mixtures thereof. Formula (HI) In certain embodiments, a phospholipid is an analog or variant of DSPC. In certain embodiments, a phospholipid is a compound of Formula (HI): (HI), or a salt thereof, wherein: each R1 is independently optionally substituted alkyl; or optionally two R1 are joined together with the intervening atoms to form optionally substituted monocyclic carbocyclyl or optionally substituted monocyclic heterocyclyl; or optionally three R1 are joined together with the intervening atoms to form optionally substituted bicyclic carbocyclyl or optionally substitute bicyclic heterocyclyl; n is 1, 2, 3, 4, 5, 6, 7, 8, 9, or 10; m is 0, 1, 2, 3, 4, 5, 6, 7, 8, 9, or 10; A is of the formula:
(HI), or a salt thereof, wherein: each R1 is independently optionally substituted alkyl; or optionally two R1 are joined together with the intervening atoms to form optionally substituted monocyclic carbocyclyl or optionally substituted monocyclic heterocyclyl; or optionally three R1 are joined together with the intervening atoms to form optionally substituted bicyclic carbocyclyl or optionally substitute bicyclic heterocyclyl; n is 1, 2, 3, 4, 5, 6, 7, 8, 9, or 10; m is 0, 1, 2, 3, 4, 5, 6, 7, 8, 9, or 10; A is of the formula: each instance of L2 is independently a bond or optionally substituted C1-6 alkylene, wherein one methylene unit of the optionally substituted C1-6 alkylene is optionally replaced with O, N(RN), S, C(O), C(O)N(RN), NRNC(O), C(O)O, OC(O), OC(O)O, OC(O)N(RN), NRNC(O)O, or NRNC(O)N(RN); each instance of R2 is independently optionally substituted C1-30 alkyl, optionally substituted C1-30 alkenyl, or optionally substituted C1-30 alkynyl; optionally wherein one or more methylene units of R2 are independently replaced with optionally substituted carbocyclylene, optionally substituted heterocyclylene, optionally substituted arylene, optionally substituted heteroarylene, N(RN), O, S, C(O), C(O)N(RN), NRNC(O), NRNC(O)N(RN), C(O)O, OC(O), - OC(O)O, OC(O)N(RN), NRNC(O)O, C(O)S, SC(O), C(=NRN), C(=NRN)N(RN), NRNC(=NRN), NRNC(=NRN)N(RN), C(S), C(S)N(RN), NRNC(S), NRNC(S)N(RN), S(O), OS(O), S(O)O, - OS(O)O, OS(O)2, S(O)2O, OS(O)2O, N(RN)S(O), S(O)N(RN), N(RN)S(O)N(RN), OS(O)N(RN), N(RN)S(O)O, S(O)2, N(RN)S(O)2, S(O)2N(RN), N(RN)S(O)2N(RN), OS(O)2N(RN), or - N(RN)S(O)2O; each instance of RN is independently hydrogen, optionally substituted alkyl, or a nitrogen protecting group; Ring B is optionally substituted carbocyclyl, optionally substituted heterocyclyl, optionally substituted aryl, or optionally substituted heteroaryl; and p is 1 or 2. In certain embodiments, the compound is not of the formula:
each instance of L2 is independently a bond or optionally substituted C1-6 alkylene, wherein one methylene unit of the optionally substituted C1-6 alkylene is optionally replaced with O, N(RN), S, C(O), C(O)N(RN), NRNC(O), C(O)O, OC(O), OC(O)O, OC(O)N(RN), NRNC(O)O, or NRNC(O)N(RN); each instance of R2 is independently optionally substituted C1-30 alkyl, optionally substituted C1-30 alkenyl, or optionally substituted C1-30 alkynyl; optionally wherein one or more methylene units of R2 are independently replaced with optionally substituted carbocyclylene, optionally substituted heterocyclylene, optionally substituted arylene, optionally substituted heteroarylene, N(RN), O, S, C(O), C(O)N(RN), NRNC(O), NRNC(O)N(RN), C(O)O, OC(O), - OC(O)O, OC(O)N(RN), NRNC(O)O, C(O)S, SC(O), C(=NRN), C(=NRN)N(RN), NRNC(=NRN), NRNC(=NRN)N(RN), C(S), C(S)N(RN), NRNC(S), NRNC(S)N(RN), S(O), OS(O), S(O)O, - OS(O)O, OS(O)2, S(O)2O, OS(O)2O, N(RN)S(O), S(O)N(RN), N(RN)S(O)N(RN), OS(O)N(RN), N(RN)S(O)O, S(O)2, N(RN)S(O)2, S(O)2N(RN), N(RN)S(O)2N(RN), OS(O)2N(RN), or - N(RN)S(O)2O; each instance of RN is independently hydrogen, optionally substituted alkyl, or a nitrogen protecting group; Ring B is optionally substituted carbocyclyl, optionally substituted heterocyclyl, optionally substituted aryl, or optionally substituted heteroaryl; and p is 1 or 2. In certain embodiments, the compound is not of the formula: , wherein each instance of R2 is independently unsubstituted alkyl, unsubstituted alkenyl, or unsubstituted alkynyl. In some embodiments, the phospholipids may be one or more of the phospholipids described in PCT Application No. PCT/US2018/037922. In some embodiments, the lipid nanoparticle comprises a molar ratio of 5-25% non- cationic lipid relative to the other lipid components. For example, the lipid nanoparticle may comprise a molar ratio of 5-30%, 5-15%, 5-10%, 10-25%, 10-20%, 10-25%, 15-25%, 15-20%, 20-25%, or 25-30% non-cationic lipid. In some embodiments, the lipid nanoparticle comprises a molar ratio of 5%, 10%, 15%, 20%, 25%, or 30% non-cationic lipid. In some embodiments, the lipid nanoparticle comprises a molar ratio of 5-25% phospholipid relative to the other lipid components. For example, the lipid nanoparticle may comprise a molar ratio of 5-30%, 5-15%, 5-10%, 10-25%, 10-20%, 10-25%, 15-25%, 15-20%, 20-25%, or 25-30% phospholipid. In some embodiments, the lipid nanoparticle comprises a molar ratio of 5%, 10%, 15%, 20%, 25%, or 30% phospholipid lipid. Structural lipids The lipid composition of a pharmaceutical composition disclosed herein can comprise one or more structural lipids. As used herein, the term “structural lipid” includes sterols and also to lipids containing sterol moieties. Incorporation of structural lipids in the lipid nanoparticle may help mitigate aggregation of other lipids in the particle. Structural lipids can be selected from the group including but not limited to, cholesterol, fecosterol, sitosterol, ergosterol, campesterol, stigmasterol, brassicasterol, tomatidine, tomatine, ursolic acid, alpha-tocopherol, hopanoids, phytosterols, steroids, and mixtures thereof. In some embodiments, the structural lipid is a sterol. As defined herein, “sterols” are a subgroup of steroids consisting of steroid alcohols. In certain embodiments, the structural lipid is a steroid. In certain embodiments, the structural lipid is cholesterol. In certain embodiments, the structural lipid is an analog of cholesterol. In certain embodiments, the structural lipid is alpha-tocopherol. In some embodiments, the structural lipids may be one or more of the structural lipids described in U.S. Application No.16/493,814. In some embodiments, the lipid nanoparticle comprises a molar ratio of 25-55% structural lipid relative to the other lipid components. For example, the lipid nanoparticle may comprise a molar ratio of 10- 55%, 25-50%, 25-45%, 25-40%, 25-35%, 25-30%, 30-55%, 30-50%, 30-45%, 30-40%, 30-35%, 35-55%, 35-50%, 35-45%, 35-40%, 40-55%, 40-50%, 40-45%, 45-55%, 45- 50%, or 50-55% structural lipid. In some embodiments, the lipid nanoparticle comprises a molar ratio of 10%, 15%, 20%, 25%, 30%, 35%, 40%, 45%, 50%, or 55% structural lipid. In some embodiments, the lipid nanoparticle comprises 30-45 mol% sterol, optionally 35- 40 mol%, for example, 30-31 mol%, 31-32 mol%, 32-33 mol%, 33-34 mol%, 34-35 mol%, 35- 36 mol%, 36-37 mol%, 37-38 mol%, 38-39 mol%, or 39-40 mol%. In some embodiments, the lipid nanoparticle comprises 25-55 mol% sterol. For example, the lipid nanoparticle may comprise 25-50 mol%, 25-45 mol%, 25-40 mol%, 25-35 mol%, 25-30 mol%, 30-55 mol%, 30- 50 mol%, 30-45 mol%, 30-40 mol%, 30-35 mol%, 35-55 mol%, 35-50 mol%, 35-45 mol%, 35- 40 mol%, 40-55 mol%, 40-50 mol%, 40-45 mol%, 45-55 mol%, 45-50 mol%, or 50-55 mol% sterol. In some embodiments, the lipid nanoparticle comprises 25 mol%, 30 mol%, 35 mol%, 40 mol%, 45 mol%, 50 mol%, or 55 mol% sterol. In some embodiments, the lipid nanoparticle comprises 35 – 40 mol% cholesterol. For example, the lipid nanoparticle may comprise 35, 35.5, 36, 36.5, 37, 37.5, 38, 38.5, 39, 39.5, or 40 mol% cholesterol. Polyethylene glycol (PEG)-Lipids The lipid composition of a pharmaceutical composition disclosed herein can comprise one or more polyethylene glycol (PEG) lipids. As used herein, the term “PEG-lipid” or “PEG-modified lipid” refers to polyethylene glycol (PEG)-modified lipids. Non-limiting examples of PEG-lipids include PEG-modified phosphatidylethanolamine and phosphatidic acid, PEG-ceramide conjugates (e.g., PEG-CerC14 or PEG-CerC20), PEG-modified dialkylamines, and PEG-modified 1,2-diacyloxypropan-3- amines. Such lipids are also referred to as PEGylated lipids. For example, a PEG lipid can be PEG-c-DOMG, PEG-DMG, PEG-DLPE, PEG-DMPE, PEG-DPPC, or a PEG-DSPE lipid. In some embodiments, the PEG-lipid includes, but not limited to 1,2-dimyristoyl-sn- glycerol methoxypolyethylene glycol (PEG-DMG), 1,2-distearoyl-sn-glycero-3- phosphoethanolamine-N-[amino(polyethylene glycol)] (PEG-DSPE), PEG-disteryl glycerol (PEG-DSG), PEG-dipalmetoleyl, PEG-dioleyl, PEG-distearyl, PEG-diacylglycamide (PEG- DAG), PEG-dipalmitoyl phosphatidylethanolamine (PEG-DPPE), or PEG-l,2- dimyristyloxlpropyl-3-amine (PEG-c-DMA). In some embodiments, the PEG-lipid is selected from the group consisting of a PEG- modified phosphatidylethanolamine, a PEG-modified phosphatidic acid, a PEG-modified ceramide, a PEG-modified dialkylamine, a PEG-modified diacylglycerol, a PEG-modified dialkylglycerol, and mixtures thereof. In some embodiments, the PEG-modified lipid is PEG- DMG, PEG-c-DOMG (also referred to as PEG-DOMG), PEG-DSG, and/or PEG-DPG. In some embodiments, the lipid moiety of the PEG-lipids includes those having lengths of from about C14 to about C22, preferably from about C14 to about C16. In some embodiments, a PEG moiety, for example an mPEG-NH2, has a size of about 1000, 2000, 5000, 10,000, 15,000 or 20,000 daltons. In some embodiments, the PEG-lipid is PEG2k-DMG. In some embodiments, the lipid nanoparticles described herein can comprise a PEG lipid which is a non-diffusible PEG. Non-limiting examples of non-diffusible PEGs include PEG-DSG and PEG-DSPE. PEG-lipids are known in the art, such as those described in U.S. Patent No.8158601 and International Publ. No. WO 2015/130584 A2, which are incorporated herein by reference in their entirety. In general, some of the other lipid components (e.g., PEG lipids) of various formulae described herein may be synthesized as described International Patent Application No. PCT/US2016/000129, filed December 10, 2016, entitled “Compositions and Methods for Delivery of Therapeutic Agents,” which is incorporated by reference in its entirety. The lipid component of a lipid nanoparticle composition may include one or more molecules comprising polyethylene glycol, such as PEG or PEG-modified lipids. Such species may be alternately referred to as PEGylated lipids. A PEG lipid is a lipid modified with polyethylene glycol. A PEG lipid may be selected from the non-limiting group including PEG- modified phosphatidylethanolamines, PEG-modified phosphatidic acids, PEG-modified ceramides, PEG-modified dialkylamines, PEG-modified diacylglycerols, PEG-modified dialkylglycerols, and mixtures thereof. For example, a PEG lipid may be PEG-c-DOMG, PEG- DMG, PEG-DLPE, PEG-DMPE, PEG-DPPC, or a PEG-DSPE lipid. In some embodiments the PEG-modified lipids are a modified form of PEG DMG. PEG- DMG has the following structure:
, wherein each instance of R2 is independently unsubstituted alkyl, unsubstituted alkenyl, or unsubstituted alkynyl. In some embodiments, the phospholipids may be one or more of the phospholipids described in PCT Application No. PCT/US2018/037922. In some embodiments, the lipid nanoparticle comprises a molar ratio of 5-25% non- cationic lipid relative to the other lipid components. For example, the lipid nanoparticle may comprise a molar ratio of 5-30%, 5-15%, 5-10%, 10-25%, 10-20%, 10-25%, 15-25%, 15-20%, 20-25%, or 25-30% non-cationic lipid. In some embodiments, the lipid nanoparticle comprises a molar ratio of 5%, 10%, 15%, 20%, 25%, or 30% non-cationic lipid. In some embodiments, the lipid nanoparticle comprises a molar ratio of 5-25% phospholipid relative to the other lipid components. For example, the lipid nanoparticle may comprise a molar ratio of 5-30%, 5-15%, 5-10%, 10-25%, 10-20%, 10-25%, 15-25%, 15-20%, 20-25%, or 25-30% phospholipid. In some embodiments, the lipid nanoparticle comprises a molar ratio of 5%, 10%, 15%, 20%, 25%, or 30% phospholipid lipid. Structural lipids The lipid composition of a pharmaceutical composition disclosed herein can comprise one or more structural lipids. As used herein, the term “structural lipid” includes sterols and also to lipids containing sterol moieties. Incorporation of structural lipids in the lipid nanoparticle may help mitigate aggregation of other lipids in the particle. Structural lipids can be selected from the group including but not limited to, cholesterol, fecosterol, sitosterol, ergosterol, campesterol, stigmasterol, brassicasterol, tomatidine, tomatine, ursolic acid, alpha-tocopherol, hopanoids, phytosterols, steroids, and mixtures thereof. In some embodiments, the structural lipid is a sterol. As defined herein, “sterols” are a subgroup of steroids consisting of steroid alcohols. In certain embodiments, the structural lipid is a steroid. In certain embodiments, the structural lipid is cholesterol. In certain embodiments, the structural lipid is an analog of cholesterol. In certain embodiments, the structural lipid is alpha-tocopherol. In some embodiments, the structural lipids may be one or more of the structural lipids described in U.S. Application No.16/493,814. In some embodiments, the lipid nanoparticle comprises a molar ratio of 25-55% structural lipid relative to the other lipid components. For example, the lipid nanoparticle may comprise a molar ratio of 10- 55%, 25-50%, 25-45%, 25-40%, 25-35%, 25-30%, 30-55%, 30-50%, 30-45%, 30-40%, 30-35%, 35-55%, 35-50%, 35-45%, 35-40%, 40-55%, 40-50%, 40-45%, 45-55%, 45- 50%, or 50-55% structural lipid. In some embodiments, the lipid nanoparticle comprises a molar ratio of 10%, 15%, 20%, 25%, 30%, 35%, 40%, 45%, 50%, or 55% structural lipid. In some embodiments, the lipid nanoparticle comprises 30-45 mol% sterol, optionally 35- 40 mol%, for example, 30-31 mol%, 31-32 mol%, 32-33 mol%, 33-34 mol%, 34-35 mol%, 35- 36 mol%, 36-37 mol%, 37-38 mol%, 38-39 mol%, or 39-40 mol%. In some embodiments, the lipid nanoparticle comprises 25-55 mol% sterol. For example, the lipid nanoparticle may comprise 25-50 mol%, 25-45 mol%, 25-40 mol%, 25-35 mol%, 25-30 mol%, 30-55 mol%, 30- 50 mol%, 30-45 mol%, 30-40 mol%, 30-35 mol%, 35-55 mol%, 35-50 mol%, 35-45 mol%, 35- 40 mol%, 40-55 mol%, 40-50 mol%, 40-45 mol%, 45-55 mol%, 45-50 mol%, or 50-55 mol% sterol. In some embodiments, the lipid nanoparticle comprises 25 mol%, 30 mol%, 35 mol%, 40 mol%, 45 mol%, 50 mol%, or 55 mol% sterol. In some embodiments, the lipid nanoparticle comprises 35 – 40 mol% cholesterol. For example, the lipid nanoparticle may comprise 35, 35.5, 36, 36.5, 37, 37.5, 38, 38.5, 39, 39.5, or 40 mol% cholesterol. Polyethylene glycol (PEG)-Lipids The lipid composition of a pharmaceutical composition disclosed herein can comprise one or more polyethylene glycol (PEG) lipids. As used herein, the term “PEG-lipid” or “PEG-modified lipid” refers to polyethylene glycol (PEG)-modified lipids. Non-limiting examples of PEG-lipids include PEG-modified phosphatidylethanolamine and phosphatidic acid, PEG-ceramide conjugates (e.g., PEG-CerC14 or PEG-CerC20), PEG-modified dialkylamines, and PEG-modified 1,2-diacyloxypropan-3- amines. Such lipids are also referred to as PEGylated lipids. For example, a PEG lipid can be PEG-c-DOMG, PEG-DMG, PEG-DLPE, PEG-DMPE, PEG-DPPC, or a PEG-DSPE lipid. In some embodiments, the PEG-lipid includes, but not limited to 1,2-dimyristoyl-sn- glycerol methoxypolyethylene glycol (PEG-DMG), 1,2-distearoyl-sn-glycero-3- phosphoethanolamine-N-[amino(polyethylene glycol)] (PEG-DSPE), PEG-disteryl glycerol (PEG-DSG), PEG-dipalmetoleyl, PEG-dioleyl, PEG-distearyl, PEG-diacylglycamide (PEG- DAG), PEG-dipalmitoyl phosphatidylethanolamine (PEG-DPPE), or PEG-l,2- dimyristyloxlpropyl-3-amine (PEG-c-DMA). In some embodiments, the PEG-lipid is selected from the group consisting of a PEG- modified phosphatidylethanolamine, a PEG-modified phosphatidic acid, a PEG-modified ceramide, a PEG-modified dialkylamine, a PEG-modified diacylglycerol, a PEG-modified dialkylglycerol, and mixtures thereof. In some embodiments, the PEG-modified lipid is PEG- DMG, PEG-c-DOMG (also referred to as PEG-DOMG), PEG-DSG, and/or PEG-DPG. In some embodiments, the lipid moiety of the PEG-lipids includes those having lengths of from about C14 to about C22, preferably from about C14 to about C16. In some embodiments, a PEG moiety, for example an mPEG-NH2, has a size of about 1000, 2000, 5000, 10,000, 15,000 or 20,000 daltons. In some embodiments, the PEG-lipid is PEG2k-DMG. In some embodiments, the lipid nanoparticles described herein can comprise a PEG lipid which is a non-diffusible PEG. Non-limiting examples of non-diffusible PEGs include PEG-DSG and PEG-DSPE. PEG-lipids are known in the art, such as those described in U.S. Patent No.8158601 and International Publ. No. WO 2015/130584 A2, which are incorporated herein by reference in their entirety. In general, some of the other lipid components (e.g., PEG lipids) of various formulae described herein may be synthesized as described International Patent Application No. PCT/US2016/000129, filed December 10, 2016, entitled “Compositions and Methods for Delivery of Therapeutic Agents,” which is incorporated by reference in its entirety. The lipid component of a lipid nanoparticle composition may include one or more molecules comprising polyethylene glycol, such as PEG or PEG-modified lipids. Such species may be alternately referred to as PEGylated lipids. A PEG lipid is a lipid modified with polyethylene glycol. A PEG lipid may be selected from the non-limiting group including PEG- modified phosphatidylethanolamines, PEG-modified phosphatidic acids, PEG-modified ceramides, PEG-modified dialkylamines, PEG-modified diacylglycerols, PEG-modified dialkylglycerols, and mixtures thereof. For example, a PEG lipid may be PEG-c-DOMG, PEG- DMG, PEG-DLPE, PEG-DMPE, PEG-DPPC, or a PEG-DSPE lipid. In some embodiments the PEG-modified lipids are a modified form of PEG DMG. PEG- DMG has the following structure: In some embodiments, PEG lipids can be PEGylated lipids described in International Publication No. WO2012/099755, the contents of which is herein incorporated by reference in its entirety. Any of these exemplary PEG lipids described herein may be modified to comprise a hydroxyl group on the PEG chain. In certain embodiments, the PEG lipid is a PEG-OH lipid. As generally defined herein, a “PEG-OH lipid” (also referred to herein as “hydroxy-PEGylated lipid”) is a PEGylated lipid having one or more hydroxyl (–OH) groups on the lipid. In certain embodiments, the PEG-OH lipid includes one or more hydroxyl groups on the PEG chain. In certain embodiments, a PEG-OH or hydroxy-PEGylated lipid comprises an –OH group at the terminus of the PEG chain. Each possibility represents a separate embodiment. Formula (PI) In certain embodiments, a PEG lipid is a compound of Formula (PI):
In some embodiments, PEG lipids can be PEGylated lipids described in International Publication No. WO2012/099755, the contents of which is herein incorporated by reference in its entirety. Any of these exemplary PEG lipids described herein may be modified to comprise a hydroxyl group on the PEG chain. In certain embodiments, the PEG lipid is a PEG-OH lipid. As generally defined herein, a “PEG-OH lipid” (also referred to herein as “hydroxy-PEGylated lipid”) is a PEGylated lipid having one or more hydroxyl (–OH) groups on the lipid. In certain embodiments, the PEG-OH lipid includes one or more hydroxyl groups on the PEG chain. In certain embodiments, a PEG-OH or hydroxy-PEGylated lipid comprises an –OH group at the terminus of the PEG chain. Each possibility represents a separate embodiment. Formula (PI) In certain embodiments, a PEG lipid is a compound of Formula (PI): (PI), or salts thereof, wherein: R3 is –ORO; RO is hydrogen, optionally substituted alkyl, or an oxygen protecting group; r is an integer between 1 and 100, inclusive; L1 is optionally substituted C1-10 alkylene, wherein at least one methylene of the optionally substituted C1-10 alkylene is independently replaced with optionally substituted carbocyclylene, optionally substituted heterocyclylene, optionally substituted arylene, optionally substituted heteroarylene, O, N(RN), S, C(O), C(O)N(RN), NRNC(O), C(O)O, OC(O), OC(O)O, OC(O)N(RN), NRNC(O)O, or NRNC(O)N(RN); D is a moiety obtained by click chemistry or a moiety cleavable under physiological conditions; m is 0, 1, 2, 3, 4, 5, 6, 7, 8, 9, or 10; A is of the formula:
(PI), or salts thereof, wherein: R3 is –ORO; RO is hydrogen, optionally substituted alkyl, or an oxygen protecting group; r is an integer between 1 and 100, inclusive; L1 is optionally substituted C1-10 alkylene, wherein at least one methylene of the optionally substituted C1-10 alkylene is independently replaced with optionally substituted carbocyclylene, optionally substituted heterocyclylene, optionally substituted arylene, optionally substituted heteroarylene, O, N(RN), S, C(O), C(O)N(RN), NRNC(O), C(O)O, OC(O), OC(O)O, OC(O)N(RN), NRNC(O)O, or NRNC(O)N(RN); D is a moiety obtained by click chemistry or a moiety cleavable under physiological conditions; m is 0, 1, 2, 3, 4, 5, 6, 7, 8, 9, or 10; A is of the formula: each instance of L2 is independently a bond or optionally substituted C1-6 alkylene, wherein one methylene unit of the optionally substituted C1-6 alkylene is optionally replaced with O, N(RN), S, C(O), C(O)N(RN), NRNC(O), C(O)O, OC(O), OC(O)O, OC(O)N(RN), NRNC(O)O, or NRNC(O)N(RN); each instance of R2 is independently optionally substituted C1-30 alkyl, optionally substituted C1-30 alkenyl, or optionally substituted C1-30 alkynyl; optionally wherein one or more methylene units of R2 are independently replaced with optionally substituted carbocyclylene, optionally substituted heterocyclylene, optionally substituted arylene, optionally substituted heteroarylene, N(RN), O, S, C(O), C(O)N(RN), NRNC(O), NRNC(O)N(RN), C(O)O, OC(O), - OC(O)O, OC(O)N(RN), NRNC(O)O, C(O)S, SC(O), C(=NRN), C(=NRN)N(RN), NRNC(=NRN), NRNC(=NRN)N(RN), C(S), C(S)N(RN), NRNC(S), NRNC(S)N(RN), S(O) , OS(O), S(O)O, - OS(O)O, OS(O)2, S(O)2O, OS(O)2O, N(RN)S(O), S(O)N(RN), N(RN)S(O)N(RN), OS(O)N(RN), N(RN)S(O)O, S(O)2, N(RN)S(O)2, S(O)2N(RN), N(RN)S(O)2N(RN), OS(O)2N(RN), or - N(RN)S(O)2O; each instance of RN is independently hydrogen, optionally substituted alkyl, or a nitrogen protecting group; Ring B is optionally substituted carbocyclyl, optionally substituted heterocyclyl, optionally substituted aryl, or optionally substituted heteroaryl; and p is 1 or 2. In certain embodiments, the compound of Fomula (PI) is a PEG-OH lipid (i.e., R3 is – ORO, and RO is hydrogen). In certain embodiments, the compound of Formula (PI) is of Formula (PI-OH):
each instance of L2 is independently a bond or optionally substituted C1-6 alkylene, wherein one methylene unit of the optionally substituted C1-6 alkylene is optionally replaced with O, N(RN), S, C(O), C(O)N(RN), NRNC(O), C(O)O, OC(O), OC(O)O, OC(O)N(RN), NRNC(O)O, or NRNC(O)N(RN); each instance of R2 is independently optionally substituted C1-30 alkyl, optionally substituted C1-30 alkenyl, or optionally substituted C1-30 alkynyl; optionally wherein one or more methylene units of R2 are independently replaced with optionally substituted carbocyclylene, optionally substituted heterocyclylene, optionally substituted arylene, optionally substituted heteroarylene, N(RN), O, S, C(O), C(O)N(RN), NRNC(O), NRNC(O)N(RN), C(O)O, OC(O), - OC(O)O, OC(O)N(RN), NRNC(O)O, C(O)S, SC(O), C(=NRN), C(=NRN)N(RN), NRNC(=NRN), NRNC(=NRN)N(RN), C(S), C(S)N(RN), NRNC(S), NRNC(S)N(RN), S(O) , OS(O), S(O)O, - OS(O)O, OS(O)2, S(O)2O, OS(O)2O, N(RN)S(O), S(O)N(RN), N(RN)S(O)N(RN), OS(O)N(RN), N(RN)S(O)O, S(O)2, N(RN)S(O)2, S(O)2N(RN), N(RN)S(O)2N(RN), OS(O)2N(RN), or - N(RN)S(O)2O; each instance of RN is independently hydrogen, optionally substituted alkyl, or a nitrogen protecting group; Ring B is optionally substituted carbocyclyl, optionally substituted heterocyclyl, optionally substituted aryl, or optionally substituted heteroaryl; and p is 1 or 2. In certain embodiments, the compound of Fomula (PI) is a PEG-OH lipid (i.e., R3 is – ORO, and RO is hydrogen). In certain embodiments, the compound of Formula (PI) is of Formula (PI-OH): (PI-OH), or a salt thereof. Formula (PII) In certain embodiments, a PEG lipid is a PEGylated fatty acid. In certain embodiments, a PEG lipid is a compound of Formula (PII). In some embodiments, compounds of Formula (PII) have the following formula:
(PI-OH), or a salt thereof. Formula (PII) In certain embodiments, a PEG lipid is a PEGylated fatty acid. In certain embodiments, a PEG lipid is a compound of Formula (PII). In some embodiments, compounds of Formula (PII) have the following formula: (PII), or a salts thereof, wherein: R3 is–ORO; RO is hydrogen, optionally substituted alkyl or an oxygen protecting group; r is an integer between 1 and 100, inclusive; R5 is optionally substituted C10-40 alkyl, optionally substituted C10-40 alkenyl, or optionally substituted C10-40 alkynyl; and optionally one or more methylene groups of R5 are replaced with optionally substituted carbocyclylene, optionally substituted heterocyclylene, optionally substituted arylene, optionally substituted heteroarylene, N(RN), O, S, C(O), C(O)N(RN), - NRNC(O), NRNC(O)N(RN), C(O)O, OC(O), OC(O)O, OC(O)N(RN), NRNC(O)O, C(O)S, SC(O), C(=NRN), C(=NRN)N(RN), NRNC(=NRN), NRNC(=NRN)N(RN), C(S), C(S)N(RN), NRNC(S), - NRNC(S)N(RN), S(O), OS(O), S(O)O, OS(O)O, OS(O)2, S(O)2O, OS(O)2O, N(RN)S(O), - S(O)N(RN), N(RN)S(O)N(RN), OS(O)N(RN), N(RN)S(O)O, S(O)2, N(RN)S(O)2, S(O)2N(RN), - N(RN)S(O)2N(RN), OS(O)2N(RN), or N(RN)S(O)2O; and each instance of RN is independently hydrogen, optionally substituted alkyl, or a nitrogen protecting group. In certain embodiments, the compound of Formula (PII) is of Formula (PII-OH):
(PII), or a salts thereof, wherein: R3 is–ORO; RO is hydrogen, optionally substituted alkyl or an oxygen protecting group; r is an integer between 1 and 100, inclusive; R5 is optionally substituted C10-40 alkyl, optionally substituted C10-40 alkenyl, or optionally substituted C10-40 alkynyl; and optionally one or more methylene groups of R5 are replaced with optionally substituted carbocyclylene, optionally substituted heterocyclylene, optionally substituted arylene, optionally substituted heteroarylene, N(RN), O, S, C(O), C(O)N(RN), - NRNC(O), NRNC(O)N(RN), C(O)O, OC(O), OC(O)O, OC(O)N(RN), NRNC(O)O, C(O)S, SC(O), C(=NRN), C(=NRN)N(RN), NRNC(=NRN), NRNC(=NRN)N(RN), C(S), C(S)N(RN), NRNC(S), - NRNC(S)N(RN), S(O), OS(O), S(O)O, OS(O)O, OS(O)2, S(O)2O, OS(O)2O, N(RN)S(O), - S(O)N(RN), N(RN)S(O)N(RN), OS(O)N(RN), N(RN)S(O)O, S(O)2, N(RN)S(O)2, S(O)2N(RN), - N(RN)S(O)2N(RN), OS(O)2N(RN), or N(RN)S(O)2O; and each instance of RN is independently hydrogen, optionally substituted alkyl, or a nitrogen protecting group. In certain embodiments, the compound of Formula (PII) is of Formula (PII-OH): (PII-OH), or a salt thereof. In some embodiments, r is 40-50. In yet other embodiments the compound of Formula (PII) is:
(PII-OH), or a salt thereof. In some embodiments, r is 40-50. In yet other embodiments the compound of Formula (PII) is: or a salt thereof. In some embodiments, the compound of Formula (PII) is
or a salt thereof. In some embodiments, the compound of Formula (PII) is . In some embodiments, the lipid composition of the pharmaceutical compositions disclosed herein does not comprise a PEG-lipid. In some embodiments, the PEG-lipids may be one or more of the PEG lipids described in U.S. Application No. US15/674,872. In some embodiments, the lipid nanoparticle comprises a molar ratio of 0.5-15% PEG lipid relative to the other lipid components. For example, the lipid nanoparticle may comprise a molar ratio of 0.5-10%, 0.5-5%, 1-15%, 1-10%, 1-5%, 2-15%, 2-10%, 2-5%, 5-15%, 5-10%, or 10-15% PEG lipid. In some embodiments, the lipid nanoparticle comprises a molar ratio of 0.5%, 1%, 2%, 3%, 4%, 5%, 6%, 7%, 8%, 9%, 10%, 11%, 12%, 13%, 14%, or 15% PEG- lipid. In some embodiments, the lipid nanoparticle comprises 1-5% PEG-modified lipid, optionally 1-3 mol%, for example 1.5 to 2.5 mol%, 1-2 mol%, 2-3 mol%, 3-4 mol%, or 4-5 mol%. In some embodiments, the lipid nanoparticle comprises 0.5-15 mol% PEG-modified lipid. For example, the lipid nanoparticle may comprise 0.5-10 mol%, 0.5-5 mol%, 1-15 mol%, 1-10 mol%, 1-5 mol%, 2-15 mol%, 2-10 mol%, 2-5 mol%, 5-15 mol%, 5-10 mol%, or 10-15 mol%. In some embodiments, the lipid nanoparticle comprises 0.5 mol%, 1 mol%, 2 mol%, 3 mol%, 4 mol%, 5 mol%, 6 mol%, 7 mol%, 8 mol%, 9 mol%, 10 mol%, 11 mol%, 12 mol%, 13 mol%, 14 mol%, or 15 mol% PEG-modified lipid. Some embodiments comprise adding PEG to a composition comprising an LNP encapsulating a nucleic acid (e.g., which already includes PEG in the amounts listed above). In embodiments comprise adding about 0.5mo% or more PEG to an LNP composition, such as about 1mol%, about 1.5mol%, about 2mol%, about 2.5mol%, about 3mol%, about 3.5mol%, about 4mol%, about 5mol%, or more after formation of an LNP composition (e.g., which already contains PEG in amount listed elsewhere herein). In some embodiments, the lipid nanoparticle comprises 20-60 mol% ionizable amino lipid, 5-25 mol% non-cationic lipid, 25-55 mol% sterol, and 0.5-15 mol% PEG-modified lipid. In some embodiments, a LNP of the disclosure comprises an ionizable amino lipid of Compound 1, wherein the non-cationic lipid is DSPC, the structural lipid that is cholesterol, and the PEG lipid is DMG-PEG. In some embodiments, a LNP of the disclosure comprises an ionizable amino lipid of Compound 2, wherein the non-cationic lipid is DSPC, the structural lipid that is cholesterol, and the PEG lipid is DMG-PEG. In some embodiments, a LNP comprises an ionizable amino lipid of any of Formula (AIII), (AIV), or (AV), a phospholipid comprising DSPC, a structural lipid, and a PEG lipid comprising PEG-DMG. In some embodiments, a LNP comprises an ionizable amino lipid of any of Formula (AIII), (AIV), or (AV), a phospholipid comprising DSPC, a structural lipid, and a PEG lipid comprising a compound having Formula (PII). In some embodiments, a LNP comprises an ionizable amino lipid of Formula (AIII), (AIV), or (AV), a phospholipid comprising a compound having Formula (HI), a structural lipid, and the PEG lipid comprising a compound having Formula (PI) or (PII). In some embodiments, a LNP comprises an ionizable amino lipid of Formula (AIII), (AIV), or (AV), a phospholipid comprising a compound having Formula (HI), a structural lipid, and the PEG lipid comprising a compound having Formula (PI) or (PII). In some embodiments, a LNP comprises an ionizable amino lipid of Formula (AIII), (AIV), or (AV), a phospholipid having Formula (HI), a structural lipid, and a PEG lipid comprising a compound having Formula (PII). In some embodiments, the lipid nanoparticle comprises 49 mol% ionizable amino lipid, 10 mol% DSPC, 38.5 mol% cholesterol, and 2.5 mol% DMG-PEG. In some embodiments, the lipid nanoparticle comprises 49 mol% ionizable amino lipid, 11 mol% DSPC, 38.5 mol% cholesterol, and 1.5 mol% DMG-PEG. In some embodiments, the lipid nanoparticle comprises 48 mol% ionizable amino lipid, 11 mol% DSPC, 38.5 mol% cholesterol, and 2.5 mol% DMG-PEG. In some embodiments, a LNP comprises an N:P ratio of from about 2:1 to about 30:1. In some embodiments, a LNP comprises an N:P ratio of about 6:1. In some embodiments, a LNP comprises an N:P ratio of about 3:1, 4:1, or 5:1. In some embodiments, a LNP comprises a wt/wt ratio of the ionizable amino lipid component to the RNA of from about 10:1 to about 100:1. In some embodiments, a LNP comprises a wt/wt ratio of the ionizable amino lipid component to the RNA of about 20:1. In some embodiments, a LNP comprises a wt/wt ratio of the ionizable amino lipid component to the RNA of about 10:1. Some embodiments comprise a composition having one or more LNPs having a diameter of about 150 nm or less, such as about 140 nm, 130 nm, 120 nm, 110 nm, 100 nm, 90 nm, 80 nm, 70 nm, 60 nm, 50 nm, 40 nm, 30 nm, or 20 nm or less. Some embodiments comprise a composition having a mean LNP diameter of about 150 nm or less, such as about 140 nm, 130 nm, 120 nm, 110 nm, 100 nm, 90 nm, 80 nm, 70 nm, 60 nm, 50 nm, 40 nm, 30 nm, or 20 nm or less. In some embodiments, the composition has a mean LNP diameter from about 30nm to about 150nm, or a mean diameter from about 60nm to about 120nm. A LNP may comprise or one or more types of lipids, including but not limited to amino lipids (e.g., ionizable amino lipids), neutral lipids, non-cationic lipids, charged lipids, PEG- modified lipids, phospholipids, structural lipids and sterols. In some embodiments, a LNP may further comprise one or more cargo molecules, including but not limited to nucleic acids (e.g., mRNA, plasmid DNA, DNA or RNA oligonucleotides, siRNA, shRNA, snRNA, snoRNA, lncRNA, etc.), small molecules, proteins and peptides. In some embodiments, the composition comprises a liposome. A liposome is a lipid particle comprising lipids arranged into one or more concentric lipid bilayers around a central region. The central region of a liposome may comprise an aqueous solution, suspension, or other aqueous composition. In some embodiments, the composition comprises a lipoplex. A lipoplex is a lipid particle comprising a cationic liposome and a nucleic acid (e.g., mRNA). Lipoplexes may be formed by contacting a liposome comprising a cationic lipid with a nucleic acid. A lipoplex may comprise multiple concentric lipid bilayers, each concentric bilayer separated by one or more nucleic acids. The central region of the lipoplex may comprise an aqueous solution, suspension, or other aqueous composition. In some embodiments, the composition comprises a lipopolyplex. A lipopolyplex is a lipid particle comprising a lipid bilayer surrounding a complex of a cationic polymer and a nucleic acid (e.g., mRNA). See Midoux & Pichon, Expert Rev Vaccines.2015.14(2):221–234. A lipopolyplex may be formed by contacting a cationic liposome (e.g., liposome comprising a cationic lipid) with the complex of nucleic acid and cationic polymer. The central region of the lipopolyplex may comprise an aqueous solution, suspension, or other aqueous composition. In some embodiments, the composition comprises a cationic nanoemulsion. A cationic nanoemulsion comprises a cationic lipid, hydrophilic surfactant, and hydrophobic surfactant. A liposome, lipoplex, lipopolyplex, or cationic nanoemulsion may comprise a sterol. A liposome, lipoplex, lipopolyplex, or cationic nanoemulsion may comprise a neutral lipid. A liposome, lipoplex, lipopolyplex, or cationic nanoemulsion may comprise a PEG-modified lipid. In some embodiments, a lipid nanoparticle may comprise two or more components (e.g., amino lipid and nucleic acid, PEG-lipid, phospholipid, structural lipid). For instance, a lipid nanoparticle may comprise an amino lipid and a nucleic acid. Compositions comprising the lipid nanoparticles, such as those described herein, may be used for a wide variety of applications, including the stealth delivery of therapeutic payloads with minimal adverse innate immune response. Effective in vivo delivery of nucleic acids represents a continuing medical challenge. Exogenous nucleic acids (i.e., originating from outside of a cell or organism) are readily degraded in the body, e.g., by the immune system. Accordingly, effective delivery of nucleic acids to cells often requires the use of a particulate carrier (e.g., lipid nanoparticles). The particulate carrier should be formulated to have minimal particle aggregation, be relatively stable prior to intracellular delivery, effectively deliver nucleic acids intracellularly, and illicit no or minimal immune response. To achieve minimal particle aggregation and pre-delivery stability, many conventional particulate carriers have relied on the presence and/or concentration of certain components (e.g., PEG-lipid). However, it has been discovered that certain components may decrease the stability of encapsulated nucleic acids (e.g., mRNA molecules). The reduced stability may limit the broad applicability of the particulate carriers. As such, there remains a need for methods by which to improve the stability of nucleic acid (e.g., mRNA) encapsulated within lipid nanoparticles. In some embodiments, the lipid nanoparticles comprise one or more of ionizable molecules, polynucleotides, and optional components, such as structural lipids, sterols, neutral lipids, phospholipids and a molecule capable of reducing particle aggregation (e.g., polyethylene glycol (PEG), PEG-modified lipid), such as those described above. In some embodiments, a LNP described herein may include one or more ionizable molecules (e.g., amino lipids or ionizable lipids). The ionizable molecule may comprise a charged group and may have a certain pKa. In certain embodiments, the pKa of the ionizable molecule may be greater than or equal to about 6, greater than or equal to about 6.2, greater than or equal to about 6.5, greater than or equal to about 6.8, greater than or equal to about 7, greater than or equal to about 7.2, greater than or equal to about 7.5, greater than or equal to about 7.8, greater than or equal to about 8. In some embodiments, the pKa of the ionizable molecule may be less than or equal to about 10, less than or equal to about 9.8, less than or equal to about 9.5, less than or equal to about 9.2, less than or equal to about 9.0, less than or equal to about 8.8, or less than or equal to about 8.5. Combinations of the above referenced ranges are also possible (e.g., greater than or equal to 6 and less than or equal to about 8.5). Other ranges are also possible. In embodiments in which more than one type of ionizable molecule are present in a particle, each type of ionizable molecule may independently have a pKa in one or more of the ranges described above. In general, an ionizable molecule comprises one or more charged groups. In some embodiments, an ionizable molecule may be positively charged or negatively charged. For instance, an ionizable molecule may be positively charged. For example, an ionizable molecule may comprise an amine group. As used herein, the term “ionizable molecule” has its ordinary meaning in the art and may refer to a molecule or matrix comprising one or more charged moiety. As used herein, a “charged moiety” is a chemical moiety that carries a formal electronic charge, e.g., monovalent (+1, or -1), divalent (+2, or -2), trivalent (+3, or -3), etc. The charged moiety may be anionic (i.e., negatively charged) or cationic (i.e., positively charged). Examples of positively-charged moieties include amine groups (e.g., primary, secondary, and/or tertiary amines), ammonium groups, pyridinium group, guanidine groups, and imidizolium groups. In a particular embodiment, the charged moieties comprise amine groups. Examples of negatively- charged groups or precursors thereof, include carboxylate groups, sulfonate groups, sulfate groups, phosphonate groups, phosphate groups, hydroxyl groups, and the like. The charge of the charged moiety may vary, in some cases, with the environmental conditions, for example, changes in pH may alter the charge of the moiety, and/or cause the moiety to become charged or uncharged. In general, the charge density of the molecule and/or matrix may be selected as desired. In some cases, an ionizable molecule (e.g., an amino lipid or ionizable lipid) may include one or more precursor moieties that can be converted to charged moieties. For instance, the ionizable molecule may include a neutral moiety that can be hydrolyzed to form a charged moiety, such as those described above. As a non-limiting specific example, the molecule or matrix may include an amide, which can be hydrolyzed to form an amine, respectively. Those of ordinary skill in the art will be able to determine whether a given chemical moiety carries a formal electronic charge (for example, by inspection, pH titration, ionic conductivity measurements, etc.), and/or whether a given chemical moiety can be reacted (e.g., hydrolyzed) to form a chemical moiety that carries a formal electronic charge. The ionizable molecule (e.g., amino lipid or ionizable lipid) may have any suitable molecular weight. In certain embodiments, the molecular weight of an ionizable molecule is less than or equal to about 2,500 g/mol, less than or equal to about 2,000 g/mol, less than or equal to about 1,500 g/mol, less than or equal to about 1,250 g/mol, less than or equal to about 1,000 g/mol, less than or equal to about 900 g/mol, less than or equal to about 800 g/mol, less than or equal to about 700 g/mol, less than or equal to about 600 g/mol, less than or equal to about 500 g/mol, less than or equal to about 400 g/mol, less than or equal to about 300 g/mol, less than or equal to about 200 g/mol, or less than or equal to about 100 g/mol. In some instances, the molecular weight of an ionizable molecule is greater than or equal to about 100 g/mol, greater than or equal to about 200 g/mol, greater than or equal to about 300 g/mol, greater than or equal to about 400 g/mol, greater than or equal to about 500 g/mol, greater than or equal to about 600 g/mol, greater than or equal to about 700 g/mol, greater than or equal to about 1000 g/mol, greater than or equal to about 1,250 g/mol, greater than or equal to about 1,500 g/mol, greater than or equal to about 1,750 g/mol, greater than or equal to about 2,000 g/mol, or greater than or equal to about 2,250 g/mol. Combinations of the above ranges (e.g., at least about 200 g/mol and less than or equal to about 2,500 g/mol) are also possible. In embodiments in which more than one type of ionizable molecules are present in a particle, each type of ionizable molecule may independently have a molecular weight in one or more of the ranges described above. In some embodiments, the percentage (e.g., by weight, or by mole) of a single type of ionizable molecule (e.g., amino lipid or ionizable lipid) and/or of all the ionizable molecules within a particle may be greater than or equal to about 15%, greater than or equal to about 16%, greater than or equal to about 17%, greater than or equal to about 18%, greater than or equal to about 19%, greater than or equal to about 20%, greater than or equal to about 21%, greater than or equal to about 22%, greater than or equal to about 23%, greater than or equal to about 24%, greater than or equal to about 25%, greater than or equal to about 30%, greater than or equal to about 35%, greater than or equal to about 40%, greater than or equal to about 42%, greater than or equal to about 45%, greater than or equal to about 48%, greater than or equal to about 50%, greater than or equal to about 52%, greater than or equal to about 55%, greater than or equal to about 58%, greater than or equal to about 60%, greater than or equal to about 62%, greater than or equal to about 65%, or greater than or equal to about 68%. In some instances, the percentage (e.g., by weight, or by mole) may be less than or equal to about 70%, less than or equal to about 68%, less than or equal to about 65%, less than or equal to about 62%, less than or equal to about 60%, less than or equal to about 58%, less than or equal to about 55%, less than or equal to about 52%, less than or equal to about 50%, or less than or equal to about 48%. Combinations of the above referenced ranges are also possible (e.g., greater than or equal to 20% and less than or equal to about 60%, greater than or equal to 40% and less than or equal to about 55%, etc.). In embodiments in which more than one type of ionizable molecule is present in a particle, each type of ionizable molecule may independently have a percentage (e.g., by weight, or by mole) in one or more of the ranges described above. The percentage (e.g., by weight, or by mole) may be determined by extracting the ionizable molecule(s) from the dried particles using, e.g., organic solvents, and measuring the quantity of the agent using high pressure liquid chromatography (i.e., HPLC), liquid chromatography-mass spectrometry (LC-MS), nuclear magnetic resonance (NMR), or mass spectrometry (MS). Those of ordinary skill in the art would be knowledgeable of techniques to determine the quantity of a component using the above-referenced techniques. For example, HPLC may be used to quantify the amount of a component, by, e.g., comparing the area under the curve of a HPLC chromatogram to a standard curve. It should be understood that the terms “charged” or “charged moiety” does not refer to a “partial negative charge" or “partial positive charge" on a molecule. The terms “partial negative charge" and “partial positive charge" are given their ordinary meaning in the art. A “partial negative charge" may result when a functional group comprises a bond that becomes polarized such that electron density is pulled toward one atom of the bond, creating a partial negative charge on the atom. Those of ordinary skill in the art will, in general, recognize bonds that can become polarized in this way. According to the disclosures herein, a lipid composition may comprise one or more lipids as described herein. Such lipids may include those useful in the preparation of lipid nanoparticle formulations as described above or as known in the art. Stabilizing Compounds Some embodiments of the compositions described herein are stabilized pharmaceutical compositions. Various non-viral delivery systems, including nanoparticle formulations, present attractive opportunities to overcome many challenges associated with mRNA delivery. Lipid nanoparticles (LNPs) have drawn particular attention in recent years as various LNP formulations have shown promise in a variety of pharmaceutical applications. However, lipids have been shown to degrade nucleic acids, including mRNA, and lipid nanoparticle formulations undergo rapid loss of purity when stored as refrigerated liquids. Moreover, the storage stability of mRNA encapsulated within LNPs is lower than that of unencapsulated mRNA. A class of compounds has been found to stabilize nucleic acids within a lipid carrier such as an LNP, an unexpected and unprecedented discovery which enables applications including extended refrigerated liquid shelf-life, extended in-use periods at room temperature, and extended in-use stability at physiological temperatures up to higher temperatures such as 40°C. Such stabilizing compounds solve a critical problem, as current manufacturing processes and formulations experience a 5-10% purity loss during LNP formation and processing that is typical with current large-scale LNP production. In some embodiments, the stabilized pharmaceutical composition comprises a nucleic acid formulation comprising a nucleic acid and a stabilizing compound (e.g., a compound of Formula (I), of Formula (II), or a tautomer or solvate thereof). In some embodiments, the stabilized pharmaceutical composition comprises a nucleic acid formulation comprising a nucleic acid and a lipid, and a compound of Formula (I):
. In some embodiments, the lipid composition of the pharmaceutical compositions disclosed herein does not comprise a PEG-lipid. In some embodiments, the PEG-lipids may be one or more of the PEG lipids described in U.S. Application No. US15/674,872. In some embodiments, the lipid nanoparticle comprises a molar ratio of 0.5-15% PEG lipid relative to the other lipid components. For example, the lipid nanoparticle may comprise a molar ratio of 0.5-10%, 0.5-5%, 1-15%, 1-10%, 1-5%, 2-15%, 2-10%, 2-5%, 5-15%, 5-10%, or 10-15% PEG lipid. In some embodiments, the lipid nanoparticle comprises a molar ratio of 0.5%, 1%, 2%, 3%, 4%, 5%, 6%, 7%, 8%, 9%, 10%, 11%, 12%, 13%, 14%, or 15% PEG- lipid. In some embodiments, the lipid nanoparticle comprises 1-5% PEG-modified lipid, optionally 1-3 mol%, for example 1.5 to 2.5 mol%, 1-2 mol%, 2-3 mol%, 3-4 mol%, or 4-5 mol%. In some embodiments, the lipid nanoparticle comprises 0.5-15 mol% PEG-modified lipid. For example, the lipid nanoparticle may comprise 0.5-10 mol%, 0.5-5 mol%, 1-15 mol%, 1-10 mol%, 1-5 mol%, 2-15 mol%, 2-10 mol%, 2-5 mol%, 5-15 mol%, 5-10 mol%, or 10-15 mol%. In some embodiments, the lipid nanoparticle comprises 0.5 mol%, 1 mol%, 2 mol%, 3 mol%, 4 mol%, 5 mol%, 6 mol%, 7 mol%, 8 mol%, 9 mol%, 10 mol%, 11 mol%, 12 mol%, 13 mol%, 14 mol%, or 15 mol% PEG-modified lipid. Some embodiments comprise adding PEG to a composition comprising an LNP encapsulating a nucleic acid (e.g., which already includes PEG in the amounts listed above). In embodiments comprise adding about 0.5mo% or more PEG to an LNP composition, such as about 1mol%, about 1.5mol%, about 2mol%, about 2.5mol%, about 3mol%, about 3.5mol%, about 4mol%, about 5mol%, or more after formation of an LNP composition (e.g., which already contains PEG in amount listed elsewhere herein). In some embodiments, the lipid nanoparticle comprises 20-60 mol% ionizable amino lipid, 5-25 mol% non-cationic lipid, 25-55 mol% sterol, and 0.5-15 mol% PEG-modified lipid. In some embodiments, a LNP of the disclosure comprises an ionizable amino lipid of Compound 1, wherein the non-cationic lipid is DSPC, the structural lipid that is cholesterol, and the PEG lipid is DMG-PEG. In some embodiments, a LNP of the disclosure comprises an ionizable amino lipid of Compound 2, wherein the non-cationic lipid is DSPC, the structural lipid that is cholesterol, and the PEG lipid is DMG-PEG. In some embodiments, a LNP comprises an ionizable amino lipid of any of Formula (AIII), (AIV), or (AV), a phospholipid comprising DSPC, a structural lipid, and a PEG lipid comprising PEG-DMG. In some embodiments, a LNP comprises an ionizable amino lipid of any of Formula (AIII), (AIV), or (AV), a phospholipid comprising DSPC, a structural lipid, and a PEG lipid comprising a compound having Formula (PII). In some embodiments, a LNP comprises an ionizable amino lipid of Formula (AIII), (AIV), or (AV), a phospholipid comprising a compound having Formula (HI), a structural lipid, and the PEG lipid comprising a compound having Formula (PI) or (PII). In some embodiments, a LNP comprises an ionizable amino lipid of Formula (AIII), (AIV), or (AV), a phospholipid comprising a compound having Formula (HI), a structural lipid, and the PEG lipid comprising a compound having Formula (PI) or (PII). In some embodiments, a LNP comprises an ionizable amino lipid of Formula (AIII), (AIV), or (AV), a phospholipid having Formula (HI), a structural lipid, and a PEG lipid comprising a compound having Formula (PII). In some embodiments, the lipid nanoparticle comprises 49 mol% ionizable amino lipid, 10 mol% DSPC, 38.5 mol% cholesterol, and 2.5 mol% DMG-PEG. In some embodiments, the lipid nanoparticle comprises 49 mol% ionizable amino lipid, 11 mol% DSPC, 38.5 mol% cholesterol, and 1.5 mol% DMG-PEG. In some embodiments, the lipid nanoparticle comprises 48 mol% ionizable amino lipid, 11 mol% DSPC, 38.5 mol% cholesterol, and 2.5 mol% DMG-PEG. In some embodiments, a LNP comprises an N:P ratio of from about 2:1 to about 30:1. In some embodiments, a LNP comprises an N:P ratio of about 6:1. In some embodiments, a LNP comprises an N:P ratio of about 3:1, 4:1, or 5:1. In some embodiments, a LNP comprises a wt/wt ratio of the ionizable amino lipid component to the RNA of from about 10:1 to about 100:1. In some embodiments, a LNP comprises a wt/wt ratio of the ionizable amino lipid component to the RNA of about 20:1. In some embodiments, a LNP comprises a wt/wt ratio of the ionizable amino lipid component to the RNA of about 10:1. Some embodiments comprise a composition having one or more LNPs having a diameter of about 150 nm or less, such as about 140 nm, 130 nm, 120 nm, 110 nm, 100 nm, 90 nm, 80 nm, 70 nm, 60 nm, 50 nm, 40 nm, 30 nm, or 20 nm or less. Some embodiments comprise a composition having a mean LNP diameter of about 150 nm or less, such as about 140 nm, 130 nm, 120 nm, 110 nm, 100 nm, 90 nm, 80 nm, 70 nm, 60 nm, 50 nm, 40 nm, 30 nm, or 20 nm or less. In some embodiments, the composition has a mean LNP diameter from about 30nm to about 150nm, or a mean diameter from about 60nm to about 120nm. A LNP may comprise or one or more types of lipids, including but not limited to amino lipids (e.g., ionizable amino lipids), neutral lipids, non-cationic lipids, charged lipids, PEG- modified lipids, phospholipids, structural lipids and sterols. In some embodiments, a LNP may further comprise one or more cargo molecules, including but not limited to nucleic acids (e.g., mRNA, plasmid DNA, DNA or RNA oligonucleotides, siRNA, shRNA, snRNA, snoRNA, lncRNA, etc.), small molecules, proteins and peptides. In some embodiments, the composition comprises a liposome. A liposome is a lipid particle comprising lipids arranged into one or more concentric lipid bilayers around a central region. The central region of a liposome may comprise an aqueous solution, suspension, or other aqueous composition. In some embodiments, the composition comprises a lipoplex. A lipoplex is a lipid particle comprising a cationic liposome and a nucleic acid (e.g., mRNA). Lipoplexes may be formed by contacting a liposome comprising a cationic lipid with a nucleic acid. A lipoplex may comprise multiple concentric lipid bilayers, each concentric bilayer separated by one or more nucleic acids. The central region of the lipoplex may comprise an aqueous solution, suspension, or other aqueous composition. In some embodiments, the composition comprises a lipopolyplex. A lipopolyplex is a lipid particle comprising a lipid bilayer surrounding a complex of a cationic polymer and a nucleic acid (e.g., mRNA). See Midoux & Pichon, Expert Rev Vaccines.2015.14(2):221–234. A lipopolyplex may be formed by contacting a cationic liposome (e.g., liposome comprising a cationic lipid) with the complex of nucleic acid and cationic polymer. The central region of the lipopolyplex may comprise an aqueous solution, suspension, or other aqueous composition. In some embodiments, the composition comprises a cationic nanoemulsion. A cationic nanoemulsion comprises a cationic lipid, hydrophilic surfactant, and hydrophobic surfactant. A liposome, lipoplex, lipopolyplex, or cationic nanoemulsion may comprise a sterol. A liposome, lipoplex, lipopolyplex, or cationic nanoemulsion may comprise a neutral lipid. A liposome, lipoplex, lipopolyplex, or cationic nanoemulsion may comprise a PEG-modified lipid. In some embodiments, a lipid nanoparticle may comprise two or more components (e.g., amino lipid and nucleic acid, PEG-lipid, phospholipid, structural lipid). For instance, a lipid nanoparticle may comprise an amino lipid and a nucleic acid. Compositions comprising the lipid nanoparticles, such as those described herein, may be used for a wide variety of applications, including the stealth delivery of therapeutic payloads with minimal adverse innate immune response. Effective in vivo delivery of nucleic acids represents a continuing medical challenge. Exogenous nucleic acids (i.e., originating from outside of a cell or organism) are readily degraded in the body, e.g., by the immune system. Accordingly, effective delivery of nucleic acids to cells often requires the use of a particulate carrier (e.g., lipid nanoparticles). The particulate carrier should be formulated to have minimal particle aggregation, be relatively stable prior to intracellular delivery, effectively deliver nucleic acids intracellularly, and illicit no or minimal immune response. To achieve minimal particle aggregation and pre-delivery stability, many conventional particulate carriers have relied on the presence and/or concentration of certain components (e.g., PEG-lipid). However, it has been discovered that certain components may decrease the stability of encapsulated nucleic acids (e.g., mRNA molecules). The reduced stability may limit the broad applicability of the particulate carriers. As such, there remains a need for methods by which to improve the stability of nucleic acid (e.g., mRNA) encapsulated within lipid nanoparticles. In some embodiments, the lipid nanoparticles comprise one or more of ionizable molecules, polynucleotides, and optional components, such as structural lipids, sterols, neutral lipids, phospholipids and a molecule capable of reducing particle aggregation (e.g., polyethylene glycol (PEG), PEG-modified lipid), such as those described above. In some embodiments, a LNP described herein may include one or more ionizable molecules (e.g., amino lipids or ionizable lipids). The ionizable molecule may comprise a charged group and may have a certain pKa. In certain embodiments, the pKa of the ionizable molecule may be greater than or equal to about 6, greater than or equal to about 6.2, greater than or equal to about 6.5, greater than or equal to about 6.8, greater than or equal to about 7, greater than or equal to about 7.2, greater than or equal to about 7.5, greater than or equal to about 7.8, greater than or equal to about 8. In some embodiments, the pKa of the ionizable molecule may be less than or equal to about 10, less than or equal to about 9.8, less than or equal to about 9.5, less than or equal to about 9.2, less than or equal to about 9.0, less than or equal to about 8.8, or less than or equal to about 8.5. Combinations of the above referenced ranges are also possible (e.g., greater than or equal to 6 and less than or equal to about 8.5). Other ranges are also possible. In embodiments in which more than one type of ionizable molecule are present in a particle, each type of ionizable molecule may independently have a pKa in one or more of the ranges described above. In general, an ionizable molecule comprises one or more charged groups. In some embodiments, an ionizable molecule may be positively charged or negatively charged. For instance, an ionizable molecule may be positively charged. For example, an ionizable molecule may comprise an amine group. As used herein, the term “ionizable molecule” has its ordinary meaning in the art and may refer to a molecule or matrix comprising one or more charged moiety. As used herein, a “charged moiety” is a chemical moiety that carries a formal electronic charge, e.g., monovalent (+1, or -1), divalent (+2, or -2), trivalent (+3, or -3), etc. The charged moiety may be anionic (i.e., negatively charged) or cationic (i.e., positively charged). Examples of positively-charged moieties include amine groups (e.g., primary, secondary, and/or tertiary amines), ammonium groups, pyridinium group, guanidine groups, and imidizolium groups. In a particular embodiment, the charged moieties comprise amine groups. Examples of negatively- charged groups or precursors thereof, include carboxylate groups, sulfonate groups, sulfate groups, phosphonate groups, phosphate groups, hydroxyl groups, and the like. The charge of the charged moiety may vary, in some cases, with the environmental conditions, for example, changes in pH may alter the charge of the moiety, and/or cause the moiety to become charged or uncharged. In general, the charge density of the molecule and/or matrix may be selected as desired. In some cases, an ionizable molecule (e.g., an amino lipid or ionizable lipid) may include one or more precursor moieties that can be converted to charged moieties. For instance, the ionizable molecule may include a neutral moiety that can be hydrolyzed to form a charged moiety, such as those described above. As a non-limiting specific example, the molecule or matrix may include an amide, which can be hydrolyzed to form an amine, respectively. Those of ordinary skill in the art will be able to determine whether a given chemical moiety carries a formal electronic charge (for example, by inspection, pH titration, ionic conductivity measurements, etc.), and/or whether a given chemical moiety can be reacted (e.g., hydrolyzed) to form a chemical moiety that carries a formal electronic charge. The ionizable molecule (e.g., amino lipid or ionizable lipid) may have any suitable molecular weight. In certain embodiments, the molecular weight of an ionizable molecule is less than or equal to about 2,500 g/mol, less than or equal to about 2,000 g/mol, less than or equal to about 1,500 g/mol, less than or equal to about 1,250 g/mol, less than or equal to about 1,000 g/mol, less than or equal to about 900 g/mol, less than or equal to about 800 g/mol, less than or equal to about 700 g/mol, less than or equal to about 600 g/mol, less than or equal to about 500 g/mol, less than or equal to about 400 g/mol, less than or equal to about 300 g/mol, less than or equal to about 200 g/mol, or less than or equal to about 100 g/mol. In some instances, the molecular weight of an ionizable molecule is greater than or equal to about 100 g/mol, greater than or equal to about 200 g/mol, greater than or equal to about 300 g/mol, greater than or equal to about 400 g/mol, greater than or equal to about 500 g/mol, greater than or equal to about 600 g/mol, greater than or equal to about 700 g/mol, greater than or equal to about 1000 g/mol, greater than or equal to about 1,250 g/mol, greater than or equal to about 1,500 g/mol, greater than or equal to about 1,750 g/mol, greater than or equal to about 2,000 g/mol, or greater than or equal to about 2,250 g/mol. Combinations of the above ranges (e.g., at least about 200 g/mol and less than or equal to about 2,500 g/mol) are also possible. In embodiments in which more than one type of ionizable molecules are present in a particle, each type of ionizable molecule may independently have a molecular weight in one or more of the ranges described above. In some embodiments, the percentage (e.g., by weight, or by mole) of a single type of ionizable molecule (e.g., amino lipid or ionizable lipid) and/or of all the ionizable molecules within a particle may be greater than or equal to about 15%, greater than or equal to about 16%, greater than or equal to about 17%, greater than or equal to about 18%, greater than or equal to about 19%, greater than or equal to about 20%, greater than or equal to about 21%, greater than or equal to about 22%, greater than or equal to about 23%, greater than or equal to about 24%, greater than or equal to about 25%, greater than or equal to about 30%, greater than or equal to about 35%, greater than or equal to about 40%, greater than or equal to about 42%, greater than or equal to about 45%, greater than or equal to about 48%, greater than or equal to about 50%, greater than or equal to about 52%, greater than or equal to about 55%, greater than or equal to about 58%, greater than or equal to about 60%, greater than or equal to about 62%, greater than or equal to about 65%, or greater than or equal to about 68%. In some instances, the percentage (e.g., by weight, or by mole) may be less than or equal to about 70%, less than or equal to about 68%, less than or equal to about 65%, less than or equal to about 62%, less than or equal to about 60%, less than or equal to about 58%, less than or equal to about 55%, less than or equal to about 52%, less than or equal to about 50%, or less than or equal to about 48%. Combinations of the above referenced ranges are also possible (e.g., greater than or equal to 20% and less than or equal to about 60%, greater than or equal to 40% and less than or equal to about 55%, etc.). In embodiments in which more than one type of ionizable molecule is present in a particle, each type of ionizable molecule may independently have a percentage (e.g., by weight, or by mole) in one or more of the ranges described above. The percentage (e.g., by weight, or by mole) may be determined by extracting the ionizable molecule(s) from the dried particles using, e.g., organic solvents, and measuring the quantity of the agent using high pressure liquid chromatography (i.e., HPLC), liquid chromatography-mass spectrometry (LC-MS), nuclear magnetic resonance (NMR), or mass spectrometry (MS). Those of ordinary skill in the art would be knowledgeable of techniques to determine the quantity of a component using the above-referenced techniques. For example, HPLC may be used to quantify the amount of a component, by, e.g., comparing the area under the curve of a HPLC chromatogram to a standard curve. It should be understood that the terms “charged” or “charged moiety” does not refer to a “partial negative charge" or “partial positive charge" on a molecule. The terms “partial negative charge" and “partial positive charge" are given their ordinary meaning in the art. A “partial negative charge" may result when a functional group comprises a bond that becomes polarized such that electron density is pulled toward one atom of the bond, creating a partial negative charge on the atom. Those of ordinary skill in the art will, in general, recognize bonds that can become polarized in this way. According to the disclosures herein, a lipid composition may comprise one or more lipids as described herein. Such lipids may include those useful in the preparation of lipid nanoparticle formulations as described above or as known in the art. Stabilizing Compounds Some embodiments of the compositions described herein are stabilized pharmaceutical compositions. Various non-viral delivery systems, including nanoparticle formulations, present attractive opportunities to overcome many challenges associated with mRNA delivery. Lipid nanoparticles (LNPs) have drawn particular attention in recent years as various LNP formulations have shown promise in a variety of pharmaceutical applications. However, lipids have been shown to degrade nucleic acids, including mRNA, and lipid nanoparticle formulations undergo rapid loss of purity when stored as refrigerated liquids. Moreover, the storage stability of mRNA encapsulated within LNPs is lower than that of unencapsulated mRNA. A class of compounds has been found to stabilize nucleic acids within a lipid carrier such as an LNP, an unexpected and unprecedented discovery which enables applications including extended refrigerated liquid shelf-life, extended in-use periods at room temperature, and extended in-use stability at physiological temperatures up to higher temperatures such as 40°C. Such stabilizing compounds solve a critical problem, as current manufacturing processes and formulations experience a 5-10% purity loss during LNP formation and processing that is typical with current large-scale LNP production. In some embodiments, the stabilized pharmaceutical composition comprises a nucleic acid formulation comprising a nucleic acid and a stabilizing compound (e.g., a compound of Formula (I), of Formula (II), or a tautomer or solvate thereof). In some embodiments, the stabilized pharmaceutical composition comprises a nucleic acid formulation comprising a nucleic acid and a lipid, and a compound of Formula (I):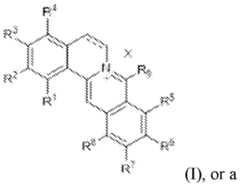 tautomer or solvate thereof, wherein: is a single bond or a double bond; R1 is H; R2 is OCH3, or together with R3 is OCH2O; R3 is OCH3, or together with R2 is OCH2O; R4 is H; R5 is H or OCH3; R6 is OCH3; R7 is H or OCH3; R8 is H; R9 is H or CH3; and X is a pharmaceutically acceptable anion, e.g., a halide such as chloride. In some embodiments, the compound of Formula (I) has the structure of:
tautomer or solvate thereof, wherein: is a single bond or a double bond; R1 is H; R2 is OCH3, or together with R3 is OCH2O; R3 is OCH3, or together with R2 is OCH2O; R4 is H; R5 is H or OCH3; R6 is OCH3; R7 is H or OCH3; R8 is H; R9 is H or CH3; and X is a pharmaceutically acceptable anion, e.g., a halide such as chloride. In some embodiments, the compound of Formula (I) has the structure of: Formula (Ia) Formula (Ib) Formula (Ic) or a tautomer or solvate thereof. In some embodiments, the stabilized pharmaceutical composition comprises a nucleic acid formulation comprising a nucleic acid and a lipid, and a compound of Formula (II):
Formula (Ia) Formula (Ib) Formula (Ic) or a tautomer or solvate thereof. In some embodiments, the stabilized pharmaceutical composition comprises a nucleic acid formulation comprising a nucleic acid and a lipid, and a compound of Formula (II): tautomer or solvate thereof, wherein: R10 is H; R11 is H; R12 together with R13 is OCH2O; R14 is H; R15 together with R16 is OCH2O; R17 is H; and X is a pharmaceutically acceptable anion, e.g., a halide such as chloride. In some embodiments, the compound of Formula (II) has the structure of:
tautomer or solvate thereof, wherein: R10 is H; R11 is H; R12 together with R13 is OCH2O; R14 is H; R15 together with R16 is OCH2O; R17 is H; and X is a pharmaceutically acceptable anion, e.g., a halide such as chloride. In some embodiments, the compound of Formula (II) has the structure of: Formula (IIa), or a tautomer or solvate thereof. Stabilizing compounds of Formulas (I), (Ia), (Ib), (Ic), (II), and (Iia) are described in International Application No. PCT/US2022/025967, which is incorporated by reference herein in its entirety. In some embodiments, the nucleic acid formulation comprises lipid nanoparticles. In some embodiments, the nucleic acid is mRNA. In some embodiments, the stabilizing compound (“the compound”) has a purity of at least 70%, 80%, 90%, 95%, or 99%. In some embodiments, the compound contains fewer than 100ppm of elemental metals. In some embodiments, the stabilized pharmaceutical composition (“the composition”) comprises a pharmaceutically acceptable metal chelator, e.g., EDTA (ethylenediaminetetraacetic acid) or DTPA (diethylenetriaminepentaacetic acid). In some embodiments, the composition is an aqueous solution. In some embodiments, the compound is present at a concentration between about 0.1mM and about 10mM in the aqueous solution. In some embodiments, the aqueous solution has a pH of or about 5 to 8, including pH of about 5, 5.5, 6, 6.5, 7, 7.5, or 8. In some embodiments, the aqueous solution does not comprise NaCl. In some embodiments, the aqueous solution comprises NaCl in a concentration of or about 150mM. In some embodiments, the aqueous solution comprises a phosphate buffer, a tris buffer, an acetate buffer, a histidine buffer, or a citrate buffer. In some embodiments, microbial growth in the composition is inhibited by the compound. In some embodiments, the composition is characterized as having a mRNA purity level of greater than 60%, greater than 70%, greater than 80%, or greater than 90% main peak mRNA purity after at least thirty days of storage. In some embodiments, the composition comprises a mRNA purity level of greater than 50% main peak mRNA purity after at least six months of storage. In some embodiments, the storage is at room temperature. In some embodiments, the composition comprises a lipid nanoparticle encapsulating a mRNA, and the composition comprises less than 50%, less than 60%, less than 70%, less than 80%, less than 90%, or less than 95% RNA fragments after at least thirty days of storage. In some embodiments, the storage temperature is greater than room temperature. In some embodiments, the storage temperature is about 4°C. In some embodiments, the compound interacts with the nucleic acid comprised within a lipid nanostructure (e.g., a lipid nanoparticle, liposome, or lipoplex), e.g., via pi-pi stacking and/or by changing backbone helicity of the nucleic acid. In some embodiments, the compound intercalates with a nucleic acid. In some embodiments, the compound binds with a nucleic acid, e.g., reversible binding, and/or binding to the stranded regions of the nucleic acid. In some embodiments, the compound self-associates, binds to nucleic acid ribose contacts, and/or binds to nucleic acid base contacts. In some embodiments, the compound does not substantially bind to nucleic acid phosphate contacts. In some embodiments, the positive charge of the compound contributes to nucleic acid binding. In some embodiments, the interacts with the nucleic acid with a binding affinity defined by an equilibrium dissociation constant of less than 10-3 M (e.g., less than 10-4 M, less than 10-5 M, less than 10-5 M, less than 10-7 M, less than 10-8 M, or less than 10-9 M). In some embodiments, the compound interacts with a nucleic acid and provides shielding from solvent, e.g., water. In some embodiments, the compound shields ribose from solvent more than the compound shields the phosphate groups of the nucleic acid. In some embodiments, the solvent exposure is measured by the solvent accessible surface area (SASA). In some embodiments, a stabilizing compound decreases the solvent accessible area of ribose to about 5- 10 nm2. In some embodiments, a stabilizing compound decreases the solvent accessible area of ribose to about 6-8 nm2. In some embodiments, a stabilizing compound decreases the solvent accessible area of phosphate to about 9-12 nm2. In some embodiments, a stabilizing compound decreases the solvent accessible area of phosphate to about 10-11 nm2. In some embodiments, a nucleic acid that is conformationally stabilized by the compound exhibits thermal unfolding temperatures (measured by circular dichroism or DSC, for example) that are higher than in the absence of the compound. In some embodiments, the compound confers increased stability, e.g., thermal stability, to the nucleic acid in a folded structure, e.g., relative to its unfolded or less folded or more linear form. In some embodiments, the compound causes compaction of the nucleic acid upon interaction with the nucleic acid. In some embodiments, the compound causes a decrease in the hydrodynamic radius of the nucleic acid molecule upon interaction with the nucleic acid. In some embodiments, a stabilizing compound causes compaction or a decrease in the hydrodynamic radius of a nucleic acid molecule by 5%, 10%, 15%, 20%, 25%, 30%, 35%, 40%, 45%, 50%, 55%, 60%, or more. In some embodiments, a stabilizing compound causes compaction or a decrease in the hydrodynamic radius of a nucleic acid molecule when the compound is in a concentration of 1 μM, 2 μM, 3 μM, 4 μM, 5 μM, 6 μM, 7 μM, 8 μM, 9 μM, 10 μM, 15 μM, 20 μM, 25 μM, 30 μM, 35 μM, 40 μM, 45 μM, 50 μM, 60 μM, 70 μM, 80 μM, 90 μM, or 100 μM. Pharmaceutical Compositions Provided herein are compositions (e.g., pharmaceutical compositions), methods, kits and reagents for prevention or treatment of coronavirus infections in humans and other mammals, for example. The compositions provided herein can be used as therapeutic or prophylactic agents. They may be used in medicine to prevent and/or treat a coronavirus infection. In some embodiments, the MERS-CoV vaccine containing RNA as described herein can be administered to a subject (e.g., a mammalian subject, such as a human subject), and the RNA polynucleotides are translated in vivo to produce an antigenic polypeptide (antigen). An “effective amount” of a composition (e.g., comprising RNA) is based, at least in part, on the target tissue, target cell type, means of administration, physical characteristics of the RNA (e.g., length, nucleotide composition, and/or extent of modified nucleosides), other components of the vaccine, and other determinants, such as age, body weight, height, sex and general health of the subject. Typically, an effective amount of a composition provides an induced or boosted immune response as a function of antigen production in the cells of the subject. In some embodiments, an effective amount of the composition containing RNA polynucleotides having at least one chemical modifications are more efficient than a composition containing a corresponding unmodified polynucleotide encoding the same antigen or a peptide antigen. Increased antigen production may be demonstrated by increased cell transfection (the percentage of cells transfected with the RNA vaccine), increased protein translation and/or expression from the polynucleotide, decreased nucleic acid degradation (as demonstrated, for example, by increased duration of protein translation from a modified polynucleotide), or altered antigen specific immune response of the host cell. The term "pharmaceutical composition" refers to the combination of an active agent with a carrier, inert or active, making the composition especially suitable for diagnostic or therapeutic use in vivo or ex vivo. A "pharmaceutically acceptable carrier," after administered to or upon a subject, does not cause undesirable physiological effects. The carrier in the pharmaceutical composition must be "acceptable" also in the sense that it is compatible with the active ingredient and can be capable of stabilizing it. One or more solubilizing agents can be utilized as pharmaceutical carriers for delivery of an active agent. Examples of a pharmaceutically acceptable carrier include, but are not limited to, biocompatible vehicles, adjuvants, additives, and diluents to achieve a composition usable as a dosage form. Examples of other carriers include colloidal silicon oxide, magnesium stearate, cellulose, and sodium lauryl sulfate. Additional suitable pharmaceutical carriers and diluents, as well as pharmaceutical necessities for their use, are described in Remington's Pharmaceutical Sciences. In some embodiments, the compositions (comprising polynucleotides and their encoded polypeptides) in accordance with the present disclosure may be used for treatment or prevention of a coronavirus infection. A composition may be administered prophylactically or therapeutically as part of an active immunization scheme to healthy individuals or early in infection during the incubation phase or during active infection after onset of symptoms. In some embodiments, the amount of RNA provided to a cell, a tissue or a subject may be an amount effective for immune prophylaxis. A composition may be administered with other prophylactic or therapeutic compounds. As a non-limiting example, a prophylactic or therapeutic compound may be an adjuvant or a booster. As used herein, when referring to a prophylactic composition, such as a vaccine, the term “booster” refers to an extra administration of the vaccine composition and may include a traditional boost, seasonal boost or a pandemic shift boost. A booster (or booster vaccine) may be given after an earlier administration of the prophylactic composition. The time of administration between the initial administration of the prophylactic composition and the booster may be, but is not limited to, 1 minute, 2 minutes, 3 minutes, 4 minutes, 5 minutes, 6 minutes, 7 minutes, 8 minutes, 9 minutes, 10 minutes, 15 minutes, 20 minutes 35 minutes, 40 minutes, 45 minutes, 50 minutes, 55 minutes, 1 hour, 2 hours, 3 hours, 4 hours, 5 hours, 6 hours, 7 hours, 8 hours, 9 hours, 10 hours, 11 hours, 12 hours, 13 hours, 14 hours, 15 hours, 16 hours, 17 hours, 18 hours, 19 hours, 20 hours, 21 hours, 22 hours, 23 hours, 1 day, 36 hours, 2 days, 3 days, 4 days, 5 days, 6 days, 1 week, 10 days, 2 weeks, 3 weeks, 1 month, 2 months, 3 months, 4 months, 5 months, or 6 months, 7 months, 8 months, 9 months, 10 months, 11 months, one year, or more. In some embodiments, the time of administration between the initial administration of the prophylactic composition and the booster is at least 6 months. In exemplary embodiments, the time of administration between the initial administration of the prophylactic composition and the booster may be, but is not limited to, 1 week, 2 weeks, 3 weeks, 1 month, 2 months, 3 months, or 6 months. As is described herein, the booster may comprise the same or different mRNAs as compared to the earlier administration of the prophylactic composition. In some embodiments, the booster may comprise a combination of the same mRNA from the earlier administration of the prophylactic composition and at least one different mRNA. In some embodiments, the ratio of the mRNA from the earlier administration of the prophylactic composition and the at least one different mRNA is 1:1, 1:2, 1:4, 4:1, or 2:1. In one embodiment, the ratio is 1:1. In some embodiments, the booster may comprise different mRNAs as compared to the earlier administration of the prophylactic compositions. In some embodiments, such a booster may comprise 1, 2, 3, 4 or more mRNAs that were not present in the prophylactic composition. In some embodiments, the ratio of two mRNA polynucleotides (none of which were in the prophylactic composition) in the booster is 1:1, 1:2, 1:4, 4:1, or 2:1. In one embodiment, the ratio is 1:1. A boost or booster dose may be administered more than once, for example 2, 3, 4, 5, 6 or more times after the initial prophylactic (prime) dose. In some embodiments, a subsequent boost is administered within weeks, e.g., within 3-4 weeks of the first (or previous) boost. In some embodiments, a second boost is administered 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, or more weeks after the first (or previous) boost. The booster, in some embodiments is monovalent (e.g., the mRNA encodes a single antigen). In some embodiments, the booster is multivalent (e.g., the mRNA encodes more than one antigen). In some embodiments, the booster dose is 5 µg-30 µg, 5 µg-25 µg, 5 µg-20 µg, 5 µg-15 µg, 5 µg-10 µg, 10 µg-30 µg, 10 µg-25 µg, 10 µg-20 µg, 10 µg-15 µg, 15 µg-30 µg, 15 µg-25 µg, 15 µg-20 µg, 20 µg-30 µg, 25 µg-30 µg, or 25 µg-300 µg. In some embodiments, the booster dose is 10 µg-60 µg, 10 µg-55 µg, 10 µg-50 µg, 10 µg-45 µg, 10 µg-40 µg, 10 µg-35 µg, 10 µg- 30 µg, 10 µg-25 µg, 10 µg-20 µg, 15 µg-60 µg, 15 µg-55 µg, 15 µg-50 µg, 15 µg-45 µg, 15 µg- 40 µg, 15 µg-35 µg, 15 µg-30 µg, 15 µg-25 µg, 15 µg-20 µg, 20 µg-60 µg, 20 µg-55 µg, 20 µg- 50 µg, 20 µg-45 µg, 20 µg-40 µg, 20 µg-35 µg, 20 µg-30 µg, 20 µg-25 µg, 25 µg-60 µg, 25 µg- 55 µg, 25 µg-50 µg, 25 µg-45 µg, 25 µg-40 µg, 25 µg-35 µg, 25 µg-30 µg, 30 µg-60 µg, 30 µg- 55 µg, 30 µg-50 µg, 30 µg-45 µg, 30 µg-40 µg, 30 µg-35 µg, 35 µg-60 µg, 35 µg-55 µg, 35 µg- 50 µg, 35 µg-45 µg, 35 µg-40 µg, 40 µg-60 µg, 40 µg-55 µg, 40 µg-50 µg, 40 µg-45 µg, 45 µg- 60 µg, 45 µg-55 µg, 45 µg-50 µg, 50 µg-60 µg, 50 µg-55 µg, or 55 µg-60 µg. In some embodiments, the booster dose is at least 10 µg and less than 25 µg of the composition. In some embodiments, the booster dose is at least 5 µg and less than 25 µg of the composition. For example, the booster dose is 5 µg, 10 µg, 15 µg, 20 µg, 25 µg, 30 µg, 35 µg, 40 µg, 45 µg, 50 µg, 55 µg, 60 µg, 65 µg, 70 µg, 75 µg, 80 µg, 85 µg, 90 µg, 95 µg, 100 µg, 110 µg, 120 µg, 130 µg, 140 µg, 150 µg, 160 µg, 170 µg, 180 µg, 190 µg, 200 µg, 250 µg, or 300 µg. In some embodiments, the booster dose is 50 μg. In some embodiments, a composition may be administered intramuscularly, intranasally or intradermally, similarly to the administration of inactivated vaccines known in the art. A composition may be utilized in various settings depending on the prevalence of the infection or the degree or level of unmet medical need. As a non-limiting example, the RNA vaccines may be utilized to treat and/or prevent a variety of infectious disease. RNA vaccines have superior properties in that they produce much larger antibody titers, better neutralizing immunity, produce more durable immune responses, and/or produce responses earlier than commercially available vaccines. Provided herein are pharmaceutical compositions including RNA and/or complexes optionally in combination with one or more pharmaceutically acceptable excipients. The RNA may be formulated or administered alone or in conjunction with one or more other components. For example, an immunizing composition may comprise other components including, but not limited to, adjuvants. In some embodiments, an immunizing composition does not include an adjuvant (it is adjuvant free). An RNA may be formulated or administered in combination with one or more pharmaceutically-acceptable excipients. In some embodiments, vaccine compositions comprise at least one additional active substances, such as, for example, a therapeutically-active substance, a prophylactically-active substance, or a combination of both. Vaccine compositions may be sterile, pyrogen-free or both sterile and pyrogen-free. General considerations in the formulation and/or manufacture of pharmaceutical agents, such as vaccine compositions, may be found, for example, in Remington: The Science and Practice of Pharmacy 21st ed., Lippincott Williams & Wilkins, 2005 (incorporated herein by reference in its entirety). In some embodiments, an immunizing composition is administered to humans, human patients or subjects. For the purposes of the present disclosure, the phrase “active ingredient” generally refers to the RNA vaccines or the polynucleotides contained therein, for example, RNA polynucleotides (e.g., mRNA polynucleotides) encoding antigens. Formulations of the vaccine compositions described herein may be prepared by any method known or hereafter developed in the art of pharmacology. In general, such preparatory methods include the step of bringing the active ingredient (e.g., mRNA polynucleotide) into association with an excipient and/or one or more other accessory ingredients, and then, if necessary and/or desirable, dividing, shaping and/or packaging the product into a desired single- or multi-dose unit. Relative amounts of the active ingredient, the pharmaceutically acceptable excipient, and/or any additional ingredients in a pharmaceutical composition in accordance with the disclosure will vary, depending upon the identity, size, and/or condition of the subject treated and further depending upon the route by which the composition is to be administered. By way of example, the composition may comprise between 0.1% and 100%, e.g., between 0.5 and 50%, between 1-30%, between 5-80%, at least 80% (w/w) active ingredient. In some embodiments, an RNA is formulated using one or more excipients to: (1) increase stability; (2) increase cell transfection; (3) permit the sustained or delayed release (e.g., from a depot formulation); (4) alter the biodistribution (e.g., target to specific tissues or cell types); (5) increase the translation of encoded protein in vivo; and/or (6) alter the release profile of encoded protein (antigen) in vivo. In addition to traditional excipients such as any and all solvents, dispersion media, diluents, or other liquid vehicles, dispersion or suspension aids, surface active agents, isotonic agents, thickening or emulsifying agents, preservatives, excipients can include, without limitation, lipidoids, liposomes, lipid nanoparticles, polymers, lipoplexes, core-shell nanoparticles, peptides, proteins, cells transfected with the RNA (e.g., for transplantation into a subject), hyaluronidase, nanoparticle mimics and combinations thereof. Administration and Dosing Provided herein are immunizing compositions (e.g., RNA vaccines), methods, kits and reagents for prevention and/or treatment of coronavirus infection in humans and other mammals. Immunizing compositions can be used as therapeutic or prophylactic agents. In some embodiments, immunizing compositions are used to provide prophylactic protection from coronavirus infection. In some embodiments, immunizing compositions are used to treat a coronavirus infection. In some embodiments, embodiments, immunizing compositions are used in the priming of immune effector cells, for example, to activate peripheral blood mononuclear cells (PBMCs) ex vivo, which are then infused (re-infused) into a subject. A subject may be any mammal, including non-human primate and human subjects. Typically, a subject is a human subject. In some embodiments, an immunizing composition (e.g., RNA vaccine) is administered to a subject (e.g., a mammalian subject, such as a human subject) in an effective amount to induce an antigen-specific immune response. The RNA encoding the coronavirus spike protein antigen is expressed and translated in vivo to produce the antigen, which then stimulates an immune response in the subject. Prophylactic protection from a coronavirus can be achieved following administration of an immunizing composition (e.g., an RNA vaccine) of the present disclosure. Immunizing compositions can be administered once, twice, three times, four times or more but it is likely sufficient to administer the vaccine once (optionally followed by a single booster). It is possible, although less desirable, to administer an immunizing composition to an infected individual to achieve a therapeutic response. Dosing may need to be adjusted accordingly. A method of eliciting an immune response in a subject against a coronavirus antigen (or multiple antigens) is provided in aspects of the present disclosure. In some embodiments, a method involves administering to the subject an immunizing composition comprising a mRNA having an open reading frame encoding a coronavirus antigen, thereby inducing in the subject an immune response specific to the coronavirus antigen, wherein anti-antigen antibody titer in the subject is increased following vaccination relative to anti-antigen antibody titer in a subject vaccinated with a prophylactically effective dose of a traditional vaccine against the antigen. An “anti-antigen antibody” is a serum antibody the binds specifically to the antigen. A prophylactically effective dose is an effective dose that prevents infection with the virus at a clinically acceptable level. In some embodiments, the effective dose is a dose listed in a package insert for the vaccine. A traditional vaccine, as used herein, refers to a vaccine other than the mRNA vaccines of the present disclosure. For instance, a traditional vaccine includes, but is not limited, to live microorganism vaccines, killed microorganism vaccines, subunit vaccines, protein antigen vaccines, DNA vaccines, virus like particle (VLP) vaccines, etc. In exemplary embodiments, a traditional vaccine is a vaccine that has achieved regulatory approval and/or is registered by a national drug regulatory body, for example the Food and Drug Administration (FDA) in the United States or the European Medicines Agency (EMA). In some embodiments, the anti-antigen antibody titer in the subject is increased 1 log to 10 log following vaccination relative to anti-antigen antibody titer in a subject vaccinated with a prophylactically effective dose of a traditional vaccine against the coronavirus or an unvaccinated subject. In some embodiments, the anti-antigen antibody titer in the subject is increased 1 log, 2 log, 3 log, 4 log, 5 log, or 10 log following vaccination relative to anti-antigen antibody titer in a subject vaccinated with a prophylactically effective dose of a traditional vaccine against the coronavirus or an unvaccinated subject. A method of eliciting an immune response in a subject against a coronavirus is provided in other aspects of the disclosure. The method involves administering to the subject a composition comprising an mRNA comprising an open reading frame encoding a coronavirus antigen, thereby inducing in the subject an immune response specific to the coronavirus, wherein the immune response in the subject is equivalent to an immune response in a subject vaccinated with a traditional vaccine against the coronavirus at 2 times to 100 times the dosage level relative to the composition. In some embodiments, the immune response in the subject is equivalent to an immune response in a subject vaccinated with a traditional vaccine at twice the dosage level relative to a composition of the present disclosure. In some embodiments, the immune response in the subject is equivalent to an immune response in a subject vaccinated with a traditional vaccine at three times the dosage level relative to a composition of the present disclosure. In some embodiments, the immune response in the subject is equivalent to an immune response in a subject vaccinated with a traditional vaccine at 4 times, 5 times, 10 times, 50 times, or 100 times the dosage level relative to a composition of the present disclosure. In some embodiments, the immune response in the subject is equivalent to an immune response in a subject vaccinated with a traditional vaccine at 10 times to 1000 times the dosage level relative to a composition of the present disclosure. In some embodiments, the immune response in the subject is equivalent to an immune response in a subject vaccinated with a traditional vaccine at 100 times to 1000 times the dosage level relative to a composition of the present disclosure. In other embodiments, the immune response is assessed by determining [protein] antibody titer in the subject. In other embodiments, the ability of serum or antibody from an immunized subject is tested for its ability to neutralize viral uptake or reduce coronavirus transformation of human B lymphocytes. In other embodiments, the ability to promote a robust T cell response(s) is measured using art recognized techniques. Other aspects the disclosure provide methods of eliciting an immune response in a subject against a coronavirus by administering to the subject composition comprising an mRNA having an open reading frame encoding a coronavirus antigen, thereby inducing in the subject an immune response specific to the coronavirus antigen, wherein the immune response in the subject is induced 2 days to 10 weeks earlier relative to an immune response induced in a subject vaccinated with a prophylactically effective dose of a traditional vaccine against the coronavirus. In some embodiments, the immune response in the subject is induced in a subject vaccinated with a prophylactically effective dose of a traditional vaccine at 2 times to 100 times the dosage level relative to a composition of the present disclosure. In some embodiments, the immune response in the subject is induced 2 days, 3 days, 1 week, 2 weeks, 3 weeks, 5 weeks, or 10 weeks earlier relative to an immune response induced in a subject vaccinated with a prophylactically effective dose of a traditional vaccine. Also provided herein are methods of eliciting an immune response in a subject against a coronavirus by administering to the subject an mRNA having an open reading frame encoding a first antigen, wherein the RNA does not include a stabilization element, and wherein an adjuvant is not co-formulated or co-administered with the vaccine. A composition may be administered by any route that results in a therapeutically effective outcome. These include, but are not limited, to intradermal, intramuscular, intranasal, and/or subcutaneous administration. The present disclosure provides methods comprising administering RNA vaccines to a subject in need thereof. The exact amount required will vary from subject to subject, depending on the species, age, and general condition of the subject, the severity of the disease, the particular composition, its mode of administration, its mode of activity, and the like. The RNA is typically formulated in dosage unit form for ease of administration and uniformity of dosage. It will be understood, however, that the total daily usage of the RNA may be decided by the attending physician within the scope of sound medical judgment. The specific therapeutically effective, prophylactically effective, or appropriate imaging dose level for any particular patient will depend upon a variety of factors including the disorder being treated and the severity of the disorder; the activity of the specific compound employed; the specific composition employed; the age, body weight, general health, sex and diet of the patient; the time of administration, route of administration, and rate of excretion of the specific compound employed; the duration of the treatment; drugs used in combination or coincidental with the specific compound employed; and like factors well known in the medical arts. The effective amount (e.g., effective dose) of the RNA, as provided herein, may be as low as 20 µg, administered for example as a single dose or as two 10 µg doses. In some embodiments, the effective amount (e.g., effective dose) is a total dose of 20 µg-300 µg5 µg-30 µg, 5 µg -25 µg, 5 µg -20 µg, 5 µg -15 µg, 5 µg -10 µg, 10 µg -30 µg, 10 µg -25 µg, 10 µg-20 µg, 10 µg -15 µg, 15 µg -30 µg, 15 µg -25 µg, 15 µg -20 µg, 20 µg -30 µg, 25 µg -30 µg, or 25 µg-300 µg. In some embodiments, the effective dose (e.g., effective amount) is at least 10 µg and less than 25 µg of the composition. In some embodiments, the effective dose (e.g., effective amount) is at least 5 µg and less than 25 µg of the composition. For example, the effective amount may be a total dose of 5 µg, 10 µg, 15 µg, 20 µg, 25 µg, 30 µg, 35 µg, 40 µg, 45 µg, 50 µg, 55 µg, 60 µg, 65 µg, 70 µg, 75 µg, 80 µg, 85 µg, 90 µg, 95 µg, 100 µg, 110 µg, 120 µg, 130 µg, 140 µg, 150 µg, 160 µg, 170 µg, 180 µg, 190 µg, 200 µg, 250 µg, or 300 µg. In some embodiments, the effective amount (e.g., effective dose) is a total dose of 10 μg. In some embodiments, the effective amount is a total dose of 20 μg (e.g., two 10 μg doses). In some embodiments, the effective amount is a total dose of 25 μg. In some embodiments, the effective amount is a total dose of 30 μg. In some embodiments, the effective amount is a total dose of 50 μg. In some embodiments, the effective amount is a total dose of 60 μg (e.g., two 30 μg doses). In some embodiments, the effective amount is a total dose of 75 μg. In some embodiments, the effective amount is a total dose of 100 μg. In some embodiments, the effective amount is a total dose of 150 μg. In some embodiments, the effective amount is a total dose of 200 μg. In some embodiments, the effective amount is a total dose of 250 μg. In some embodiments, the effective amount is a total dose of 300 μg. Any of the doses provided above may be an effective amount for a booster dose; for example, in some embodiments, the booster dose is a total dose of 50 μg. In some embodiments, the composition comprises two or more mRNA polynucleotides and effective amount is a total dose of 20 μg (e.g., 10 μg of a first mRNA and 10 μg of a second mRNA). In some embodiments, the composition comprises two or more mRNA polynucleotides and effective amount is a total dose of 50 μg (e.g., 25 μg of a first mRNA and 25 μg of a second mRNA). In some embodiments, the composition comprises two or more mRNA polynucleotides and effective amount is a total dose of 100 μg (e.g., 50 μg of a first mRNA and 50 μg of a second mRNA). The RNA described herein can be formulated into a dosage form described herein, such as an intranasal, intratracheal, or injectable (e.g., intravenous, intraocular, intravitreal, intramuscular, intradermal, intracardiac, intraperitoneal, and subcutaneous). Vaccine Efficacy Some aspects of the present disclosure provide formulations of the compositions (e.g., RNA vaccines), wherein the RNA is formulated in an effective amount to produce an antigen specific immune response in a subject (e.g., production of antibodies specific to a coronavirus antigen). “An effective amount” is a dose of the RNA effective to produce an antigen-specific immune response. Also provided herein are methods of inducing an antigen-specific immune response in a subject. As used herein, an immune response to a vaccine or LNP of the present disclosure is the development in a subject of a humoral and/or a cellular immune response to a (one or more) coronavirus protein(s) present in the vaccine. For purposes of the present disclosure, a “humoral” immune response refers to an immune response mediated by antibody molecules, including, e.g., secretory (IgA) or IgG molecules, while a “cellular” immune response is one mediated by T- lymphocytes (e.g., CD4+ helper and/or CD8+ T cells (e.g., CTLs) and/or other white blood cells. One important aspect of cellular immunity involves an antigen-specific response by cytolytic T- cells (CTLs). CTLs have specificity for peptide antigens that are presented in association with proteins encoded by the major histocompatibility complex (MHC) and expressed on the surfaces of cells. CTLs help induce and promote the destruction of intracellular microbes or the lysis of cells infected with such microbes. Another aspect of cellular immunity involves and antigen- specific response by helper T-cells. Helper T-cells act to help stimulate the function and focus the activity nonspecific effector cells against cells displaying peptide antigens in association with MHC molecules on their surface. A cellular immune response also leads to the production of cytokines, chemokines, and other such molecules produced by activated T-cells and/or other white blood cells including those derived from CD4+ and CD8+ T-cells. In some embodiments, the antigen-specific immune response is characterized by measuring an anti-coronavirus antigen antibody titer produced in a subject administered a composition as provided herein. An antibody titer is a measurement of the amount of antibodies within a subject, for example, antibodies that are specific to a particular antigen or epitope of an antigen. Antibody titer is typically expressed as the inverse of the greatest dilution that provides a positive result. Enzyme-linked immunosorbent assay (ELISA) is a common assay for determining antibody titers, for example. A variety of serological tests can be used to measure antibody against encoded antigen of interest, for example, SAR-CoV-2 virus or SAR-CoV-2 viral antigen, e.g., SAR-CoV-2 spike or S protein, of domain thereof. These tests include the hemagglutination-inhibition test, complement fixation test, fluorescent antibody test, enzyme-linked immunosorbent assay (ELISA), and plaque reduction neutralization test (PRNT). Each of these tests measures different antibody activities. In exemplary embodiments, A plaque reduction neutralization test, or PRNT (e.g., PRNT50 or PRNT90) is used as a serological correlate of protection. PRNT measures the biological parameter of in vitro virus neutralization and is the most serologically virus-specific test among certain classes of viruses, correlating well to serum levels of protection from virus infection. The basic design of the PRNT allows for virus-antibody interaction to occur in a test tube or microtiter plate, and then measuring antibody effects on viral infectivity by plating the mixture on virus-susceptible cells, preferably cells of mammalian origin. The cells are overlaid with a semi-solid media that restricts spread of progeny virus. Each virus that initiates a productive infection produces a localized area of infection (a plaque), that can be detected in a variety of ways. Plaques are counted and compared back to the starting concentration of virus to determine the percent reduction in total virus infectivity. In PRNT, the serum sample being tested is usually subjected to serial dilutions prior to mixing with a standardized amount of virus. The concentration of virus is held constant such that, when added to susceptible cells and overlaid with semi-solid media, individual plaques can be discerned and counted. In this way, PRNT end- point titers can be calculated for each serum sample at any selected percent reduction of virus activity. In functional assays intended to assess vaccinal immunogenicity, the serum sample dilution series for antibody titration should ideally start below the “seroprotective” threshold titer. Regarding MERS-CoV neutralizing antibodies, the “seroprotective” threshold titer remains unknown; but a seropositivity threshold of 1:10 can be considered a seroprotection threshold in certain embodiments. In some embodiments a neutralizing immune response is an immune response that produces a level of antibodies that meet or exceed a seroprotection threshold. PRNT end-point titers are expressed as the reciprocal of the last serum dilution showing the desired percent reduction in plaque counts. The PRNT titer can be calculated based on a 50% or greater reduction in plaque counts (PRNT50). A PRNT50 titer is preferred over titers using higher cut-offs (e.g., PRNT90) for vaccine sera, providing more accurate results from the linear portion of the titration curve. There are several ways to calculate PRNT titers. The simplest and most widely used way to calculate titers is to count plaques and report the titer as the reciprocal of the last serum dilution to show >50% reduction of the input plaque count as based on the back-titration of input plaques. Use of curve fitting methods from several serum dilutions may permit calculation of a more precise result. There are a variety of computer analysis programs available for this (e.g., SPSS or GraphPad Prism). In some embodiments, an antibody titer is used to assess whether a subject has had an infection or to determine whether immunizations are required. In some embodiments, an antibody titer is used to determine the strength of an autoimmune response, to determine whether a booster immunization is needed, to determine whether a previous vaccine was effective, and to identify any recent or prior infections. In accordance with the present disclosure, an antibody titer may be used to determine the strength of an immune response induced in a subject by a composition (e.g., RNA vaccine). In some embodiments, an anti-coronavirus antigen antibody titer produced in a subject is increased by at least 1 log relative to a control. For example, anti-coronavirus antigen antibody titer produced in a subject may be increased by at least 1.5, at least 2, at least 2.5, or at least 3 log relative to a control. In some embodiments, the anti-coronavirus antigen antibody titer produced in the subject is increased by 1, 1.5, 2, 2.5 or 3 log relative to a control. In some embodiments, the anti-coronavirus antigen antibody titer produced in the subject is increased by 1-3 log relative to a control. For example, the anti-coronavirus antigen antibody titer produced in a subject may be increased by 1-1.5, 1-2, 1-2.5, 1-3, 1.5-2, 1.5-2.5, 1.5-3, 2-2.5, 2-3, or 2.5-3 log relative to a control. In some embodiments, the anti-coronavirus antigen antibody titer produced in a subject is increased at least 2 times relative to a control. For example, the anti-coronavirus antigen n antibody titer produced in a subject may be increased at least 3 times, at least 4 times, at least 5 times, at least 6 times, at least 7 times, at least 8 times, at least 9 times, or at least 10 times relative to a control. In some embodiments, the anti-coronavirus antigen antibody titer produced in the subject is increased 2, 3, 4, 5, 6, 7, 8, 9, or 10 times relative to a control. In some embodiments, the anti-coronavirus antigen antibody titer produced in a subject is increased 2-10 times relative to a control. For example, the anti-coronavirus antigen antibody titer produced in a subject may be increased 2-10, 2-9, 2-8, 2-7, 2-6, 2-5, 2-4, 2-3, 3-10, 3-9, 3-8, 3-7, 3-6, 3-5, 3-4, 4-10, 4-9, 4-8, 4-7, 4-6, 4-5, 5-10, 5-9, 5-8, 5-7, 5-6, 6-10, 6-9, 6-8, 6-7, 7-10, 7-9, 7-8, 8-10, 8-9, or 9-10 times relative to a control. In some embodiments, an antigen-specific immune response is measured as a ratio of geometric mean titer (GMT), referred to as a geometric mean ratio (GMR), of serum neutralizing antibody titers to coronavirus. A geometric mean titer (GMT) is the average antibody titer for a group of subjects calculated by multiplying all values and taking the nth root of the number, where n is the number of subjects with available data. A control, in some embodiments, is an anti-coronavirus antigen antibody titer produced in a subject who has not been administered a composition (e.g., RNA vaccine). In some embodiments, a control is an anti-coronavirus antigen antibody titer produced in a subject administered a recombinant or purified protein vaccine. Recombinant protein vaccines typically include protein antigens that either have been produced in a heterologous expression system (e.g., bacteria or yeast) or purified from large amounts of the pathogenic organism. In some embodiments, the ability of a composition (e.g., RNA vaccine) to be effective is measured in a murine model. For example, a composition may be administered to a murine model and the murine model assayed for induction of neutralizing antibody titers. Viral challenge studies may also be used to assess the efficacy of a vaccine of the present disclosure. For example, a composition may be administered to a murine model, the murine model challenged with virus, and the murine model assayed for survival and/or immune response (e.g., neutralizing antibody response, T cell response (e.g., cytokine response)). In some embodiments, an effective amount of a composition (e.g., RNA vaccine) is a dose that is reduced compared to the standard of care dose of a recombinant protein vaccine. A “standard of care,” as provided herein, refers to a medical or psychological treatment guideline and can be general or specific. “Standard of care” specifies appropriate treatment based on scientific evidence and collaboration between medical professionals involved in the treatment of a given condition. It is the diagnostic and treatment process that a physician/ clinician should follow for a certain type of patient, illness or clinical circumstance. A “standard of care dose,” as provided herein, refers to the dose of a recombinant or purified protein vaccine, or a live attenuated or inactivated vaccine, or a VLP vaccine, that a physician/clinician or other medical professional would administer to a subject to treat or prevent coronavirus infection or a related condition, while following the standard of care guideline for treating or preventing coronavirus infection or a related condition. In some embodiments, the anti-coronavirus antigen antibody titer produced in a subject administered an effective amount of an composition is equivalent to an anti-coronavirus antigen antibody titer produced in a control subject administered a standard of care dose of a recombinant or purified protein vaccine, or a live attenuated or inactivated vaccine, or a VLP vaccine. Vaccine efficacy may be assessed using standard analyses (see, e.g., Weinberg et al., J Infect Dis.2010 Jun 1;201(11):1607-10). For example, vaccine efficacy may be measured by double-blind, randomized, clinical controlled trials. Vaccine efficacy may be expressed as a proportionate reduction in disease attack rate (AR) between the unvaccinated (ARU) and vaccinated (ARV) study cohorts and can be calculated from the relative risk (RR) of disease among the vaccinated group with use of the following formulas: Efficacy = (ARU – ARV)/ARU x 100; and Efficacy = (1 – RR) x 100. Likewise, vaccine effectiveness may be assessed using standard analyses (see, e.g., Weinberg et al., J Infect Dis.2010 Jun 1;201(11):1607-10). Vaccine effectiveness is an assessment of how a vaccine (which may have already proven to have high vaccine efficacy) reduces disease in a population. This measure can assess the net balance of benefits and adverse effects of a vaccination program, not just the vaccine itself, under natural field conditions rather than in a controlled clinical trial. Vaccine effectiveness is proportional to vaccine efficacy (potency) but is also affected by how well target groups in the population are immunized, as well as by other non-vaccine-related factors that influence the ‘real-world’ outcomes of hospitalizations, ambulatory visits, or costs. For example, a retrospective case control analysis may be used, in which the rates of vaccination among a set of infected cases and appropriate controls are compared. Vaccine effectiveness may be expressed as a rate difference, with use of the odds ratio (OR) for developing infection despite vaccination: Effectiveness = (1 – OR) x 100. In some embodiments, efficacy of the composition (e.g., RNA vaccine) is at least 60% relative to unvaccinated control subjects. For example, efficacy of the composition may be at least 65%, at least 70%, at least 75%, at least 80%, at least 85%, at least 95%, at least 98%, or 100% relative to unvaccinated control subjects. Sterilizing Immunity. Sterilizing immunity refers to a unique immune status that prevents effective pathogen infection into the host. In some embodiments, the effective amount of a composition of the present disclosure is sufficient to provide sterilizing immunity in the subject for at least 1 year. For example, the effective amount of a composition of the present disclosure is sufficient to provide sterilizing immunity in the subject for at least 2 years, at least 3 years, at least 4 years, or at least 5 years. In some embodiments, the effective amount of a composition of the present disclosure is sufficient to provide sterilizing immunity in the subject at an at least 5-fold lower dose relative to control. For example, the effective amount may be sufficient to provide sterilizing immunity in the subject at an at least 10-fold lower, 15-fold, or 20-fold lower dose relative to a control. Detectable Antigen. In some embodiments, the effective amount of a composition of the present disclosure is sufficient to produce detectable levels of coronavirus antigen as measured in serum of the subject at 1-72 hours post administration. Titer. An antibody titer is a measurement of the number of antibodies within a subject, for example, antibodies that are specific to a particular antigen (e.g., an anti-coronavirus antigen). Antibody titer is typically expressed as the inverse of the greatest dilution that provides a positive result. Enzyme-linked immunosorbent assay (ELISA) is a common assay for determining antibody titers, for example. A neutralizing immune response is an immune response that is a neutralizing antibody response and/or an effective neutralizing T cell response. In some embodiments a neutralizing antibody response produces a level of antibodies that meet or exceed a seroprotection threshold. An effective T cell response is a response which produces a baseline level of viral activated or viral specific T cells including CD8+ and CD4+ T helper type 1 cells. CD8+ cytotoxic T lymphocytes typically clear the intracellular virus compartment and CD4+ T cells exert various functions in the body such as helping B and other T cells, promoting memory generation and indirect or direct cytotoxic activity. In some embodiments the effective T cells comprises a high proportion of CD8+ T cells and/or CD4+ T cells, relative to a baseline level (in a naïve subject). In some embodiments these T cells are differentiated towards an early- differentiated memory phenotype with co-expression of CD27 and CD28. In some embodiments, the effective amount of a composition of the present disclosure is sufficient to produce a 1,000-10,000 neutralizing antibody titer produced by neutralizing antibody against the coronavirus antigen as measured in serum of the subject at 1-72 hours post administration. In some embodiments, the effective amount is sufficient to produce a 1,000-5,000 neutralizing antibody titer produced by neutralizing antibody against the coronavirus antigen as measured in serum of the subject at 1-72 hours post administration. In some embodiments, the effective amount is sufficient to produce a 5,000-10,000 neutralizing antibody titer produced by neutralizing antibody against the coronavirus antigen as measured in serum of the subject at 1-72 hours post administration. In some embodiments, the neutralizing antibody titer is at least 100 NT50. For example, the neutralizing antibody titer may be at least 200, 300, 400, 500, 600, 700, 800, 900 or 1000 NT50. In some embodiments, the neutralizing antibody titer is at least 10,000 NT50. In some embodiments, the neutralizing antibody titer is at least 100 neutralizing units per milliliter (NU/mL). For example, the neutralizing antibody titer may be at least 200, 300, 400, 500, 600, 700, 800, 900 or 1000 NU/mL. In some embodiments, the neutralizing antibody titer is at least 10,000 NU/mL. In some embodiments, an anti-coronavirus antigen antibody titer produced in the subject is increased by at least 1 log relative to a control. For example, an anti-coronavirus antigen antibody titer produced in the subject may be increased by at least 2, 3, 4, 5, 6, 7, 8, 9 or 10 log relative to a control. In some embodiments, an anti-coronavirus antigen antibody titer produced in the subject is increased at least 2 times relative to a control. For example, an anti-coronavirus antigen antibody titer produced in the subject is increased by at least 3, 4, 5, 6, 7, 8, 9 or 10 times relative to a control. In some embodiments, a geometric mean, which is the nth root of the product of n numbers, is generally used to describe proportional growth. Geometric mean, in some embodiments, is used to characterize antibody titer produced in a subject. A control may be, for example, an unvaccinated subject, or a subject administered a live attenuated viral vaccine, an inactivated viral vaccine, or a protein subunit vaccine. EXAMPLES Methods Manufacture of polynucleotides described herein, and/or parts or regions thereof, may be accomplished using the methods taught in PCT Publication No. WO 2014/152027, entitled “Manufacturing Methods for Production of RNA Transcripts,” the contents of which are incorporated herein by reference in their entirety. Purification methods may include those described in PCT Publication No. WO 2014/152030 and PCT Publication No. WO 2014/152031, the contents of each of which are incorporated herein by reference in their entirety. Detection and characterization methods of the polynucleotides may be performed as described in PCT Publication No. WO 2014/144039, the contents of which are incorporated herein by reference in their entirety. Characterization of the polynucleotides of the disclosure may be accomplished using polynucleotide mapping, reverse transcriptase sequencing, charge distribution analysis, detection of RNA impurities, or any combination of two or more of the foregoing. “Characterizing” comprises determining the RNA transcript sequence, determining the purity of the RNA transcript, or determining the charge heterogeneity of the RNA transcript, for example. Such methods are taught in, for example, PCT Publication No. WO 2014/144711 and PCT Publication No. WO 2014/144767, the contents of each of which are incorporated herein by reference in their entirety. In experiments where a lipid nanoparticle (LNP) formulation was or is used, the formulation included or includes 48 mol% lipid of Compound 1, 11 mol% 1,2 distearoyl-sn- glycero-3-phosphocholine (DSPC), 38.5 mol% cholesterol, and 2.5 mol% PEG-modified 1,2 dimyristoyl-sn-glycerol, methoxypolyethyleneglycol (PEG2000 DMG). Immunization Methods Vaccine compositions of lipid nanoparticles containing mRNAs are administered to mice according to the following administration schedule. C57BL/6 mice are immunized with two doses of a given composition, receiving the first dose on day 0, and the second dose on day 22. Sera are collected on day 21, three weeks after the first (prime) dose but before administration of the second (boost) dose, and day 36, two weeks after administration of the second (boost) dose. Where T cells are evaluated, mice are euthanized on day 36, and spleens are collected and processed to harvest splenocytes. Splenocytes are stimulated with one of a panel of peptide pools, each pool containing peptides from a single MERS-CoV antigen, in the presence of a Golgi blocker, so that cells producing cytokines in response to stimulation retain cytokines instead of secreting them. Cell surfaces are stained for lymphocyte markers, including CD3, CD4, CD8, and/or CD107a, and cells are permeabilized and stained for multiple cytokines, including TNF-α, IFN-γ, IL-2, IL-4, IL-5, IL-10, and/or IL-13. Stained cells are incubated with a viability dye and analyzed by flow cytometry. Neutralization Assays Antibodies in serum, when bound to a viral surface protein that is essential for infection, can prevent a virus from infecting a target cell, an activity referred to as “neutralization.” To determine the ability of mRNA compositions to generate neutralizing antibodies against MERS- CoV, the neutralization activity of sera is quantified using a neutralization assay. For each assay, ARPE-19 cells are plated in 96-well plates, at a density of 2*104 cells/well and incubated for 20– 24 hours. Then, serial 3-fold dilutions of each serum sample are prepared in phenol red-free cDMEM. A consistent amount of MERS-CoV reporter virus, containing a gene encoding GFP, is incubated with each serum dilution sample, to allow for binding of any MERS-CoV-specific antibodies to the virus. After incubation, cells are washed, and incubated with MERS-CoV /serum mixtures.24 hours after incubation, GFP fluorescence in each well is measured to determine the extent of infection. For a given serum sample, the 50% neutralization titer (NT50) is calculated as the reciprocal of the serum dilution at which 50% of GFP+ cells are observed. Recombinant VSV-based Pseudovirus Neutralization. Codon-optimized nucleotide sequences encoding MERS-CoV Spike proteins are cloned into pCAGGS vectors. To make MERS-CoV Spike proteinpseudotyped recombinant VSV-ΔG-firefly luciferase virus, BHK- 21/WI-2 cells (Kerafast, EH1011) are transfected with the Spike protein expression plasmid, and subsequently infected with VSV∆G-firefly-luciferase as previously described (Whitt, 2010, Journal of Virological Methods 169, 365–374). For the neutralization assay, serially diluted serum samples are mixed with pseudovirus and incubated at 37°C for 45 minutes. The virus- serum mix is subsequently used to infect Vero-81 or Huh7.5s cells for 18 hours at 37°C before adding ONE-Glo reagent (Promega E6120) for measurement of luciferase signal (relative luminescence unit; RLU). The percentage of neutralization is calculated based on RLU of the virus only control, and subsequently analyzed using four-parameter logistic curve (Prism 8). Antibody-dependent cell-mediated cytotoxicity assays Antibodies in serum, when bound to viral protein expressed on the surface of an infected cell, can be recognized by effector cells, such as natural killer (NK) cells. Effector cells recognize the constant (Fc) region of antibodies bound to the target cells, and following recognition, release cytotoxic granules that induce apoptosis in the infected target cells. This process is referred to as “antibody-dependent cell-mediated cytotoxicity” (ADCC). To determine the ability of mRNA compositions to generate antibodies that can facilitate ADCC, the ADCC activity of sera is quantified using an ADCC assay. This assay uses Jurkat cells that constitutively express mouse FcγRIV, allowing for recognition of antibody Fc regions, and luciferase under the control of the NFAT pathway, which is activated following Fc recognition. In each assay, Vero cells are plated in 96-well plates, at a density of 2.5*104 cells/well, incubated for 20–24 hours, then inoculated with MERS-CoV at a multiplicity of infection (MOI) of 5 plaque-forming units (PFU) per cell.16 hours after inoculation, serial 3-fold dilutions of serum samples are prepared in RPMI + 4% fetal bovine serum (FBS) containing only minimal amounts of IgG, to reduce background. Serum samples are added to each well, to allow antibodies to bind to infected Vero cells expressing viral surface proteins. Then, reporter effector cells are serially diluted in RPMI + 4% low-IgG FBS, added to wells, and incubated for 6 hours to allow for recognition of surface- bound antibodies and expression of luciferase. After the 6 hours of incubation, a luciferase substrate is added to wells, so that any luciferase present can react with the substrate to produce light. Light emitted from wells is measured to quantify the amount of luciferase activity as a measurement of ADCC activity. Serum-depletion Assays Epitope- or domain-level specificity of antibodies in serum is evaluated by depleting antibodies with a given specificity from serum, and comparing such antigen-depleted (Ag- depleted) serum with mock-depleted serum. Antibodies specific to a given epitope or antigen may be depleted by incubating serum with a solid support (e.g., surface of a 96-well plate) on which the epitope or antigen is immobilized. Incubation allows antibodies with the desired specificity to bind to the immobilized epitope or antigen, such that the serum collected after incubation does not contain the surface-bound antibodies. Mock-depleted serum is prepared by incubating serum under similar conditions, where the solid support does not contain an immobilized epitope or antigen. Antigen-depleted sera may be compared to mock-depleted sera in multiple in vitro assays, such as ELISA, neutralization assays, ADCC assays, and/or in vivo, such as by passive transfer of sera before, during, or after inoculation with a virus. Example 1: Design of mRNA encoding MERS-CoV S proteins, fusion proteins, and variants thereof. MERS-CoV S protein sequences were modified with one or more changes as described in Table E1-1. Modifications included (i) introduction of two proline substitutions to stabilize the S protein in a prefusion conformation (S2P); (ii) inactivation of a furin cleavage site; (iii) deletion of an endoplasmic reticulum (ER) retention signal; and/or truncation of a cytoplasmic tail Fusion proteins comprising one or more domains of MERS-CoV S protein, and optionally one or more linkers and/or a transmembrane domain, were designed as described in Table E1-2. Table E1-1. MERS-CoV S protein modifications
Formula (IIa), or a tautomer or solvate thereof. Stabilizing compounds of Formulas (I), (Ia), (Ib), (Ic), (II), and (Iia) are described in International Application No. PCT/US2022/025967, which is incorporated by reference herein in its entirety. In some embodiments, the nucleic acid formulation comprises lipid nanoparticles. In some embodiments, the nucleic acid is mRNA. In some embodiments, the stabilizing compound (“the compound”) has a purity of at least 70%, 80%, 90%, 95%, or 99%. In some embodiments, the compound contains fewer than 100ppm of elemental metals. In some embodiments, the stabilized pharmaceutical composition (“the composition”) comprises a pharmaceutically acceptable metal chelator, e.g., EDTA (ethylenediaminetetraacetic acid) or DTPA (diethylenetriaminepentaacetic acid). In some embodiments, the composition is an aqueous solution. In some embodiments, the compound is present at a concentration between about 0.1mM and about 10mM in the aqueous solution. In some embodiments, the aqueous solution has a pH of or about 5 to 8, including pH of about 5, 5.5, 6, 6.5, 7, 7.5, or 8. In some embodiments, the aqueous solution does not comprise NaCl. In some embodiments, the aqueous solution comprises NaCl in a concentration of or about 150mM. In some embodiments, the aqueous solution comprises a phosphate buffer, a tris buffer, an acetate buffer, a histidine buffer, or a citrate buffer. In some embodiments, microbial growth in the composition is inhibited by the compound. In some embodiments, the composition is characterized as having a mRNA purity level of greater than 60%, greater than 70%, greater than 80%, or greater than 90% main peak mRNA purity after at least thirty days of storage. In some embodiments, the composition comprises a mRNA purity level of greater than 50% main peak mRNA purity after at least six months of storage. In some embodiments, the storage is at room temperature. In some embodiments, the composition comprises a lipid nanoparticle encapsulating a mRNA, and the composition comprises less than 50%, less than 60%, less than 70%, less than 80%, less than 90%, or less than 95% RNA fragments after at least thirty days of storage. In some embodiments, the storage temperature is greater than room temperature. In some embodiments, the storage temperature is about 4°C. In some embodiments, the compound interacts with the nucleic acid comprised within a lipid nanostructure (e.g., a lipid nanoparticle, liposome, or lipoplex), e.g., via pi-pi stacking and/or by changing backbone helicity of the nucleic acid. In some embodiments, the compound intercalates with a nucleic acid. In some embodiments, the compound binds with a nucleic acid, e.g., reversible binding, and/or binding to the stranded regions of the nucleic acid. In some embodiments, the compound self-associates, binds to nucleic acid ribose contacts, and/or binds to nucleic acid base contacts. In some embodiments, the compound does not substantially bind to nucleic acid phosphate contacts. In some embodiments, the positive charge of the compound contributes to nucleic acid binding. In some embodiments, the interacts with the nucleic acid with a binding affinity defined by an equilibrium dissociation constant of less than 10-3 M (e.g., less than 10-4 M, less than 10-5 M, less than 10-5 M, less than 10-7 M, less than 10-8 M, or less than 10-9 M). In some embodiments, the compound interacts with a nucleic acid and provides shielding from solvent, e.g., water. In some embodiments, the compound shields ribose from solvent more than the compound shields the phosphate groups of the nucleic acid. In some embodiments, the solvent exposure is measured by the solvent accessible surface area (SASA). In some embodiments, a stabilizing compound decreases the solvent accessible area of ribose to about 5- 10 nm2. In some embodiments, a stabilizing compound decreases the solvent accessible area of ribose to about 6-8 nm2. In some embodiments, a stabilizing compound decreases the solvent accessible area of phosphate to about 9-12 nm2. In some embodiments, a stabilizing compound decreases the solvent accessible area of phosphate to about 10-11 nm2. In some embodiments, a nucleic acid that is conformationally stabilized by the compound exhibits thermal unfolding temperatures (measured by circular dichroism or DSC, for example) that are higher than in the absence of the compound. In some embodiments, the compound confers increased stability, e.g., thermal stability, to the nucleic acid in a folded structure, e.g., relative to its unfolded or less folded or more linear form. In some embodiments, the compound causes compaction of the nucleic acid upon interaction with the nucleic acid. In some embodiments, the compound causes a decrease in the hydrodynamic radius of the nucleic acid molecule upon interaction with the nucleic acid. In some embodiments, a stabilizing compound causes compaction or a decrease in the hydrodynamic radius of a nucleic acid molecule by 5%, 10%, 15%, 20%, 25%, 30%, 35%, 40%, 45%, 50%, 55%, 60%, or more. In some embodiments, a stabilizing compound causes compaction or a decrease in the hydrodynamic radius of a nucleic acid molecule when the compound is in a concentration of 1 μM, 2 μM, 3 μM, 4 μM, 5 μM, 6 μM, 7 μM, 8 μM, 9 μM, 10 μM, 15 μM, 20 μM, 25 μM, 30 μM, 35 μM, 40 μM, 45 μM, 50 μM, 60 μM, 70 μM, 80 μM, 90 μM, or 100 μM. Pharmaceutical Compositions Provided herein are compositions (e.g., pharmaceutical compositions), methods, kits and reagents for prevention or treatment of coronavirus infections in humans and other mammals, for example. The compositions provided herein can be used as therapeutic or prophylactic agents. They may be used in medicine to prevent and/or treat a coronavirus infection. In some embodiments, the MERS-CoV vaccine containing RNA as described herein can be administered to a subject (e.g., a mammalian subject, such as a human subject), and the RNA polynucleotides are translated in vivo to produce an antigenic polypeptide (antigen). An “effective amount” of a composition (e.g., comprising RNA) is based, at least in part, on the target tissue, target cell type, means of administration, physical characteristics of the RNA (e.g., length, nucleotide composition, and/or extent of modified nucleosides), other components of the vaccine, and other determinants, such as age, body weight, height, sex and general health of the subject. Typically, an effective amount of a composition provides an induced or boosted immune response as a function of antigen production in the cells of the subject. In some embodiments, an effective amount of the composition containing RNA polynucleotides having at least one chemical modifications are more efficient than a composition containing a corresponding unmodified polynucleotide encoding the same antigen or a peptide antigen. Increased antigen production may be demonstrated by increased cell transfection (the percentage of cells transfected with the RNA vaccine), increased protein translation and/or expression from the polynucleotide, decreased nucleic acid degradation (as demonstrated, for example, by increased duration of protein translation from a modified polynucleotide), or altered antigen specific immune response of the host cell. The term "pharmaceutical composition" refers to the combination of an active agent with a carrier, inert or active, making the composition especially suitable for diagnostic or therapeutic use in vivo or ex vivo. A "pharmaceutically acceptable carrier," after administered to or upon a subject, does not cause undesirable physiological effects. The carrier in the pharmaceutical composition must be "acceptable" also in the sense that it is compatible with the active ingredient and can be capable of stabilizing it. One or more solubilizing agents can be utilized as pharmaceutical carriers for delivery of an active agent. Examples of a pharmaceutically acceptable carrier include, but are not limited to, biocompatible vehicles, adjuvants, additives, and diluents to achieve a composition usable as a dosage form. Examples of other carriers include colloidal silicon oxide, magnesium stearate, cellulose, and sodium lauryl sulfate. Additional suitable pharmaceutical carriers and diluents, as well as pharmaceutical necessities for their use, are described in Remington's Pharmaceutical Sciences. In some embodiments, the compositions (comprising polynucleotides and their encoded polypeptides) in accordance with the present disclosure may be used for treatment or prevention of a coronavirus infection. A composition may be administered prophylactically or therapeutically as part of an active immunization scheme to healthy individuals or early in infection during the incubation phase or during active infection after onset of symptoms. In some embodiments, the amount of RNA provided to a cell, a tissue or a subject may be an amount effective for immune prophylaxis. A composition may be administered with other prophylactic or therapeutic compounds. As a non-limiting example, a prophylactic or therapeutic compound may be an adjuvant or a booster. As used herein, when referring to a prophylactic composition, such as a vaccine, the term “booster” refers to an extra administration of the vaccine composition and may include a traditional boost, seasonal boost or a pandemic shift boost. A booster (or booster vaccine) may be given after an earlier administration of the prophylactic composition. The time of administration between the initial administration of the prophylactic composition and the booster may be, but is not limited to, 1 minute, 2 minutes, 3 minutes, 4 minutes, 5 minutes, 6 minutes, 7 minutes, 8 minutes, 9 minutes, 10 minutes, 15 minutes, 20 minutes 35 minutes, 40 minutes, 45 minutes, 50 minutes, 55 minutes, 1 hour, 2 hours, 3 hours, 4 hours, 5 hours, 6 hours, 7 hours, 8 hours, 9 hours, 10 hours, 11 hours, 12 hours, 13 hours, 14 hours, 15 hours, 16 hours, 17 hours, 18 hours, 19 hours, 20 hours, 21 hours, 22 hours, 23 hours, 1 day, 36 hours, 2 days, 3 days, 4 days, 5 days, 6 days, 1 week, 10 days, 2 weeks, 3 weeks, 1 month, 2 months, 3 months, 4 months, 5 months, or 6 months, 7 months, 8 months, 9 months, 10 months, 11 months, one year, or more. In some embodiments, the time of administration between the initial administration of the prophylactic composition and the booster is at least 6 months. In exemplary embodiments, the time of administration between the initial administration of the prophylactic composition and the booster may be, but is not limited to, 1 week, 2 weeks, 3 weeks, 1 month, 2 months, 3 months, or 6 months. As is described herein, the booster may comprise the same or different mRNAs as compared to the earlier administration of the prophylactic composition. In some embodiments, the booster may comprise a combination of the same mRNA from the earlier administration of the prophylactic composition and at least one different mRNA. In some embodiments, the ratio of the mRNA from the earlier administration of the prophylactic composition and the at least one different mRNA is 1:1, 1:2, 1:4, 4:1, or 2:1. In one embodiment, the ratio is 1:1. In some embodiments, the booster may comprise different mRNAs as compared to the earlier administration of the prophylactic compositions. In some embodiments, such a booster may comprise 1, 2, 3, 4 or more mRNAs that were not present in the prophylactic composition. In some embodiments, the ratio of two mRNA polynucleotides (none of which were in the prophylactic composition) in the booster is 1:1, 1:2, 1:4, 4:1, or 2:1. In one embodiment, the ratio is 1:1. A boost or booster dose may be administered more than once, for example 2, 3, 4, 5, 6 or more times after the initial prophylactic (prime) dose. In some embodiments, a subsequent boost is administered within weeks, e.g., within 3-4 weeks of the first (or previous) boost. In some embodiments, a second boost is administered 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, or more weeks after the first (or previous) boost. The booster, in some embodiments is monovalent (e.g., the mRNA encodes a single antigen). In some embodiments, the booster is multivalent (e.g., the mRNA encodes more than one antigen). In some embodiments, the booster dose is 5 µg-30 µg, 5 µg-25 µg, 5 µg-20 µg, 5 µg-15 µg, 5 µg-10 µg, 10 µg-30 µg, 10 µg-25 µg, 10 µg-20 µg, 10 µg-15 µg, 15 µg-30 µg, 15 µg-25 µg, 15 µg-20 µg, 20 µg-30 µg, 25 µg-30 µg, or 25 µg-300 µg. In some embodiments, the booster dose is 10 µg-60 µg, 10 µg-55 µg, 10 µg-50 µg, 10 µg-45 µg, 10 µg-40 µg, 10 µg-35 µg, 10 µg- 30 µg, 10 µg-25 µg, 10 µg-20 µg, 15 µg-60 µg, 15 µg-55 µg, 15 µg-50 µg, 15 µg-45 µg, 15 µg- 40 µg, 15 µg-35 µg, 15 µg-30 µg, 15 µg-25 µg, 15 µg-20 µg, 20 µg-60 µg, 20 µg-55 µg, 20 µg- 50 µg, 20 µg-45 µg, 20 µg-40 µg, 20 µg-35 µg, 20 µg-30 µg, 20 µg-25 µg, 25 µg-60 µg, 25 µg- 55 µg, 25 µg-50 µg, 25 µg-45 µg, 25 µg-40 µg, 25 µg-35 µg, 25 µg-30 µg, 30 µg-60 µg, 30 µg- 55 µg, 30 µg-50 µg, 30 µg-45 µg, 30 µg-40 µg, 30 µg-35 µg, 35 µg-60 µg, 35 µg-55 µg, 35 µg- 50 µg, 35 µg-45 µg, 35 µg-40 µg, 40 µg-60 µg, 40 µg-55 µg, 40 µg-50 µg, 40 µg-45 µg, 45 µg- 60 µg, 45 µg-55 µg, 45 µg-50 µg, 50 µg-60 µg, 50 µg-55 µg, or 55 µg-60 µg. In some embodiments, the booster dose is at least 10 µg and less than 25 µg of the composition. In some embodiments, the booster dose is at least 5 µg and less than 25 µg of the composition. For example, the booster dose is 5 µg, 10 µg, 15 µg, 20 µg, 25 µg, 30 µg, 35 µg, 40 µg, 45 µg, 50 µg, 55 µg, 60 µg, 65 µg, 70 µg, 75 µg, 80 µg, 85 µg, 90 µg, 95 µg, 100 µg, 110 µg, 120 µg, 130 µg, 140 µg, 150 µg, 160 µg, 170 µg, 180 µg, 190 µg, 200 µg, 250 µg, or 300 µg. In some embodiments, the booster dose is 50 μg. In some embodiments, a composition may be administered intramuscularly, intranasally or intradermally, similarly to the administration of inactivated vaccines known in the art. A composition may be utilized in various settings depending on the prevalence of the infection or the degree or level of unmet medical need. As a non-limiting example, the RNA vaccines may be utilized to treat and/or prevent a variety of infectious disease. RNA vaccines have superior properties in that they produce much larger antibody titers, better neutralizing immunity, produce more durable immune responses, and/or produce responses earlier than commercially available vaccines. Provided herein are pharmaceutical compositions including RNA and/or complexes optionally in combination with one or more pharmaceutically acceptable excipients. The RNA may be formulated or administered alone or in conjunction with one or more other components. For example, an immunizing composition may comprise other components including, but not limited to, adjuvants. In some embodiments, an immunizing composition does not include an adjuvant (it is adjuvant free). An RNA may be formulated or administered in combination with one or more pharmaceutically-acceptable excipients. In some embodiments, vaccine compositions comprise at least one additional active substances, such as, for example, a therapeutically-active substance, a prophylactically-active substance, or a combination of both. Vaccine compositions may be sterile, pyrogen-free or both sterile and pyrogen-free. General considerations in the formulation and/or manufacture of pharmaceutical agents, such as vaccine compositions, may be found, for example, in Remington: The Science and Practice of Pharmacy 21st ed., Lippincott Williams & Wilkins, 2005 (incorporated herein by reference in its entirety). In some embodiments, an immunizing composition is administered to humans, human patients or subjects. For the purposes of the present disclosure, the phrase “active ingredient” generally refers to the RNA vaccines or the polynucleotides contained therein, for example, RNA polynucleotides (e.g., mRNA polynucleotides) encoding antigens. Formulations of the vaccine compositions described herein may be prepared by any method known or hereafter developed in the art of pharmacology. In general, such preparatory methods include the step of bringing the active ingredient (e.g., mRNA polynucleotide) into association with an excipient and/or one or more other accessory ingredients, and then, if necessary and/or desirable, dividing, shaping and/or packaging the product into a desired single- or multi-dose unit. Relative amounts of the active ingredient, the pharmaceutically acceptable excipient, and/or any additional ingredients in a pharmaceutical composition in accordance with the disclosure will vary, depending upon the identity, size, and/or condition of the subject treated and further depending upon the route by which the composition is to be administered. By way of example, the composition may comprise between 0.1% and 100%, e.g., between 0.5 and 50%, between 1-30%, between 5-80%, at least 80% (w/w) active ingredient. In some embodiments, an RNA is formulated using one or more excipients to: (1) increase stability; (2) increase cell transfection; (3) permit the sustained or delayed release (e.g., from a depot formulation); (4) alter the biodistribution (e.g., target to specific tissues or cell types); (5) increase the translation of encoded protein in vivo; and/or (6) alter the release profile of encoded protein (antigen) in vivo. In addition to traditional excipients such as any and all solvents, dispersion media, diluents, or other liquid vehicles, dispersion or suspension aids, surface active agents, isotonic agents, thickening or emulsifying agents, preservatives, excipients can include, without limitation, lipidoids, liposomes, lipid nanoparticles, polymers, lipoplexes, core-shell nanoparticles, peptides, proteins, cells transfected with the RNA (e.g., for transplantation into a subject), hyaluronidase, nanoparticle mimics and combinations thereof. Administration and Dosing Provided herein are immunizing compositions (e.g., RNA vaccines), methods, kits and reagents for prevention and/or treatment of coronavirus infection in humans and other mammals. Immunizing compositions can be used as therapeutic or prophylactic agents. In some embodiments, immunizing compositions are used to provide prophylactic protection from coronavirus infection. In some embodiments, immunizing compositions are used to treat a coronavirus infection. In some embodiments, embodiments, immunizing compositions are used in the priming of immune effector cells, for example, to activate peripheral blood mononuclear cells (PBMCs) ex vivo, which are then infused (re-infused) into a subject. A subject may be any mammal, including non-human primate and human subjects. Typically, a subject is a human subject. In some embodiments, an immunizing composition (e.g., RNA vaccine) is administered to a subject (e.g., a mammalian subject, such as a human subject) in an effective amount to induce an antigen-specific immune response. The RNA encoding the coronavirus spike protein antigen is expressed and translated in vivo to produce the antigen, which then stimulates an immune response in the subject. Prophylactic protection from a coronavirus can be achieved following administration of an immunizing composition (e.g., an RNA vaccine) of the present disclosure. Immunizing compositions can be administered once, twice, three times, four times or more but it is likely sufficient to administer the vaccine once (optionally followed by a single booster). It is possible, although less desirable, to administer an immunizing composition to an infected individual to achieve a therapeutic response. Dosing may need to be adjusted accordingly. A method of eliciting an immune response in a subject against a coronavirus antigen (or multiple antigens) is provided in aspects of the present disclosure. In some embodiments, a method involves administering to the subject an immunizing composition comprising a mRNA having an open reading frame encoding a coronavirus antigen, thereby inducing in the subject an immune response specific to the coronavirus antigen, wherein anti-antigen antibody titer in the subject is increased following vaccination relative to anti-antigen antibody titer in a subject vaccinated with a prophylactically effective dose of a traditional vaccine against the antigen. An “anti-antigen antibody” is a serum antibody the binds specifically to the antigen. A prophylactically effective dose is an effective dose that prevents infection with the virus at a clinically acceptable level. In some embodiments, the effective dose is a dose listed in a package insert for the vaccine. A traditional vaccine, as used herein, refers to a vaccine other than the mRNA vaccines of the present disclosure. For instance, a traditional vaccine includes, but is not limited, to live microorganism vaccines, killed microorganism vaccines, subunit vaccines, protein antigen vaccines, DNA vaccines, virus like particle (VLP) vaccines, etc. In exemplary embodiments, a traditional vaccine is a vaccine that has achieved regulatory approval and/or is registered by a national drug regulatory body, for example the Food and Drug Administration (FDA) in the United States or the European Medicines Agency (EMA). In some embodiments, the anti-antigen antibody titer in the subject is increased 1 log to 10 log following vaccination relative to anti-antigen antibody titer in a subject vaccinated with a prophylactically effective dose of a traditional vaccine against the coronavirus or an unvaccinated subject. In some embodiments, the anti-antigen antibody titer in the subject is increased 1 log, 2 log, 3 log, 4 log, 5 log, or 10 log following vaccination relative to anti-antigen antibody titer in a subject vaccinated with a prophylactically effective dose of a traditional vaccine against the coronavirus or an unvaccinated subject. A method of eliciting an immune response in a subject against a coronavirus is provided in other aspects of the disclosure. The method involves administering to the subject a composition comprising an mRNA comprising an open reading frame encoding a coronavirus antigen, thereby inducing in the subject an immune response specific to the coronavirus, wherein the immune response in the subject is equivalent to an immune response in a subject vaccinated with a traditional vaccine against the coronavirus at 2 times to 100 times the dosage level relative to the composition. In some embodiments, the immune response in the subject is equivalent to an immune response in a subject vaccinated with a traditional vaccine at twice the dosage level relative to a composition of the present disclosure. In some embodiments, the immune response in the subject is equivalent to an immune response in a subject vaccinated with a traditional vaccine at three times the dosage level relative to a composition of the present disclosure. In some embodiments, the immune response in the subject is equivalent to an immune response in a subject vaccinated with a traditional vaccine at 4 times, 5 times, 10 times, 50 times, or 100 times the dosage level relative to a composition of the present disclosure. In some embodiments, the immune response in the subject is equivalent to an immune response in a subject vaccinated with a traditional vaccine at 10 times to 1000 times the dosage level relative to a composition of the present disclosure. In some embodiments, the immune response in the subject is equivalent to an immune response in a subject vaccinated with a traditional vaccine at 100 times to 1000 times the dosage level relative to a composition of the present disclosure. In other embodiments, the immune response is assessed by determining [protein] antibody titer in the subject. In other embodiments, the ability of serum or antibody from an immunized subject is tested for its ability to neutralize viral uptake or reduce coronavirus transformation of human B lymphocytes. In other embodiments, the ability to promote a robust T cell response(s) is measured using art recognized techniques. Other aspects the disclosure provide methods of eliciting an immune response in a subject against a coronavirus by administering to the subject composition comprising an mRNA having an open reading frame encoding a coronavirus antigen, thereby inducing in the subject an immune response specific to the coronavirus antigen, wherein the immune response in the subject is induced 2 days to 10 weeks earlier relative to an immune response induced in a subject vaccinated with a prophylactically effective dose of a traditional vaccine against the coronavirus. In some embodiments, the immune response in the subject is induced in a subject vaccinated with a prophylactically effective dose of a traditional vaccine at 2 times to 100 times the dosage level relative to a composition of the present disclosure. In some embodiments, the immune response in the subject is induced 2 days, 3 days, 1 week, 2 weeks, 3 weeks, 5 weeks, or 10 weeks earlier relative to an immune response induced in a subject vaccinated with a prophylactically effective dose of a traditional vaccine. Also provided herein are methods of eliciting an immune response in a subject against a coronavirus by administering to the subject an mRNA having an open reading frame encoding a first antigen, wherein the RNA does not include a stabilization element, and wherein an adjuvant is not co-formulated or co-administered with the vaccine. A composition may be administered by any route that results in a therapeutically effective outcome. These include, but are not limited, to intradermal, intramuscular, intranasal, and/or subcutaneous administration. The present disclosure provides methods comprising administering RNA vaccines to a subject in need thereof. The exact amount required will vary from subject to subject, depending on the species, age, and general condition of the subject, the severity of the disease, the particular composition, its mode of administration, its mode of activity, and the like. The RNA is typically formulated in dosage unit form for ease of administration and uniformity of dosage. It will be understood, however, that the total daily usage of the RNA may be decided by the attending physician within the scope of sound medical judgment. The specific therapeutically effective, prophylactically effective, or appropriate imaging dose level for any particular patient will depend upon a variety of factors including the disorder being treated and the severity of the disorder; the activity of the specific compound employed; the specific composition employed; the age, body weight, general health, sex and diet of the patient; the time of administration, route of administration, and rate of excretion of the specific compound employed; the duration of the treatment; drugs used in combination or coincidental with the specific compound employed; and like factors well known in the medical arts. The effective amount (e.g., effective dose) of the RNA, as provided herein, may be as low as 20 µg, administered for example as a single dose or as two 10 µg doses. In some embodiments, the effective amount (e.g., effective dose) is a total dose of 20 µg-300 µg5 µg-30 µg, 5 µg -25 µg, 5 µg -20 µg, 5 µg -15 µg, 5 µg -10 µg, 10 µg -30 µg, 10 µg -25 µg, 10 µg-20 µg, 10 µg -15 µg, 15 µg -30 µg, 15 µg -25 µg, 15 µg -20 µg, 20 µg -30 µg, 25 µg -30 µg, or 25 µg-300 µg. In some embodiments, the effective dose (e.g., effective amount) is at least 10 µg and less than 25 µg of the composition. In some embodiments, the effective dose (e.g., effective amount) is at least 5 µg and less than 25 µg of the composition. For example, the effective amount may be a total dose of 5 µg, 10 µg, 15 µg, 20 µg, 25 µg, 30 µg, 35 µg, 40 µg, 45 µg, 50 µg, 55 µg, 60 µg, 65 µg, 70 µg, 75 µg, 80 µg, 85 µg, 90 µg, 95 µg, 100 µg, 110 µg, 120 µg, 130 µg, 140 µg, 150 µg, 160 µg, 170 µg, 180 µg, 190 µg, 200 µg, 250 µg, or 300 µg. In some embodiments, the effective amount (e.g., effective dose) is a total dose of 10 μg. In some embodiments, the effective amount is a total dose of 20 μg (e.g., two 10 μg doses). In some embodiments, the effective amount is a total dose of 25 μg. In some embodiments, the effective amount is a total dose of 30 μg. In some embodiments, the effective amount is a total dose of 50 μg. In some embodiments, the effective amount is a total dose of 60 μg (e.g., two 30 μg doses). In some embodiments, the effective amount is a total dose of 75 μg. In some embodiments, the effective amount is a total dose of 100 μg. In some embodiments, the effective amount is a total dose of 150 μg. In some embodiments, the effective amount is a total dose of 200 μg. In some embodiments, the effective amount is a total dose of 250 μg. In some embodiments, the effective amount is a total dose of 300 μg. Any of the doses provided above may be an effective amount for a booster dose; for example, in some embodiments, the booster dose is a total dose of 50 μg. In some embodiments, the composition comprises two or more mRNA polynucleotides and effective amount is a total dose of 20 μg (e.g., 10 μg of a first mRNA and 10 μg of a second mRNA). In some embodiments, the composition comprises two or more mRNA polynucleotides and effective amount is a total dose of 50 μg (e.g., 25 μg of a first mRNA and 25 μg of a second mRNA). In some embodiments, the composition comprises two or more mRNA polynucleotides and effective amount is a total dose of 100 μg (e.g., 50 μg of a first mRNA and 50 μg of a second mRNA). The RNA described herein can be formulated into a dosage form described herein, such as an intranasal, intratracheal, or injectable (e.g., intravenous, intraocular, intravitreal, intramuscular, intradermal, intracardiac, intraperitoneal, and subcutaneous). Vaccine Efficacy Some aspects of the present disclosure provide formulations of the compositions (e.g., RNA vaccines), wherein the RNA is formulated in an effective amount to produce an antigen specific immune response in a subject (e.g., production of antibodies specific to a coronavirus antigen). “An effective amount” is a dose of the RNA effective to produce an antigen-specific immune response. Also provided herein are methods of inducing an antigen-specific immune response in a subject. As used herein, an immune response to a vaccine or LNP of the present disclosure is the development in a subject of a humoral and/or a cellular immune response to a (one or more) coronavirus protein(s) present in the vaccine. For purposes of the present disclosure, a “humoral” immune response refers to an immune response mediated by antibody molecules, including, e.g., secretory (IgA) or IgG molecules, while a “cellular” immune response is one mediated by T- lymphocytes (e.g., CD4+ helper and/or CD8+ T cells (e.g., CTLs) and/or other white blood cells. One important aspect of cellular immunity involves an antigen-specific response by cytolytic T- cells (CTLs). CTLs have specificity for peptide antigens that are presented in association with proteins encoded by the major histocompatibility complex (MHC) and expressed on the surfaces of cells. CTLs help induce and promote the destruction of intracellular microbes or the lysis of cells infected with such microbes. Another aspect of cellular immunity involves and antigen- specific response by helper T-cells. Helper T-cells act to help stimulate the function and focus the activity nonspecific effector cells against cells displaying peptide antigens in association with MHC molecules on their surface. A cellular immune response also leads to the production of cytokines, chemokines, and other such molecules produced by activated T-cells and/or other white blood cells including those derived from CD4+ and CD8+ T-cells. In some embodiments, the antigen-specific immune response is characterized by measuring an anti-coronavirus antigen antibody titer produced in a subject administered a composition as provided herein. An antibody titer is a measurement of the amount of antibodies within a subject, for example, antibodies that are specific to a particular antigen or epitope of an antigen. Antibody titer is typically expressed as the inverse of the greatest dilution that provides a positive result. Enzyme-linked immunosorbent assay (ELISA) is a common assay for determining antibody titers, for example. A variety of serological tests can be used to measure antibody against encoded antigen of interest, for example, SAR-CoV-2 virus or SAR-CoV-2 viral antigen, e.g., SAR-CoV-2 spike or S protein, of domain thereof. These tests include the hemagglutination-inhibition test, complement fixation test, fluorescent antibody test, enzyme-linked immunosorbent assay (ELISA), and plaque reduction neutralization test (PRNT). Each of these tests measures different antibody activities. In exemplary embodiments, A plaque reduction neutralization test, or PRNT (e.g., PRNT50 or PRNT90) is used as a serological correlate of protection. PRNT measures the biological parameter of in vitro virus neutralization and is the most serologically virus-specific test among certain classes of viruses, correlating well to serum levels of protection from virus infection. The basic design of the PRNT allows for virus-antibody interaction to occur in a test tube or microtiter plate, and then measuring antibody effects on viral infectivity by plating the mixture on virus-susceptible cells, preferably cells of mammalian origin. The cells are overlaid with a semi-solid media that restricts spread of progeny virus. Each virus that initiates a productive infection produces a localized area of infection (a plaque), that can be detected in a variety of ways. Plaques are counted and compared back to the starting concentration of virus to determine the percent reduction in total virus infectivity. In PRNT, the serum sample being tested is usually subjected to serial dilutions prior to mixing with a standardized amount of virus. The concentration of virus is held constant such that, when added to susceptible cells and overlaid with semi-solid media, individual plaques can be discerned and counted. In this way, PRNT end- point titers can be calculated for each serum sample at any selected percent reduction of virus activity. In functional assays intended to assess vaccinal immunogenicity, the serum sample dilution series for antibody titration should ideally start below the “seroprotective” threshold titer. Regarding MERS-CoV neutralizing antibodies, the “seroprotective” threshold titer remains unknown; but a seropositivity threshold of 1:10 can be considered a seroprotection threshold in certain embodiments. In some embodiments a neutralizing immune response is an immune response that produces a level of antibodies that meet or exceed a seroprotection threshold. PRNT end-point titers are expressed as the reciprocal of the last serum dilution showing the desired percent reduction in plaque counts. The PRNT titer can be calculated based on a 50% or greater reduction in plaque counts (PRNT50). A PRNT50 titer is preferred over titers using higher cut-offs (e.g., PRNT90) for vaccine sera, providing more accurate results from the linear portion of the titration curve. There are several ways to calculate PRNT titers. The simplest and most widely used way to calculate titers is to count plaques and report the titer as the reciprocal of the last serum dilution to show >50% reduction of the input plaque count as based on the back-titration of input plaques. Use of curve fitting methods from several serum dilutions may permit calculation of a more precise result. There are a variety of computer analysis programs available for this (e.g., SPSS or GraphPad Prism). In some embodiments, an antibody titer is used to assess whether a subject has had an infection or to determine whether immunizations are required. In some embodiments, an antibody titer is used to determine the strength of an autoimmune response, to determine whether a booster immunization is needed, to determine whether a previous vaccine was effective, and to identify any recent or prior infections. In accordance with the present disclosure, an antibody titer may be used to determine the strength of an immune response induced in a subject by a composition (e.g., RNA vaccine). In some embodiments, an anti-coronavirus antigen antibody titer produced in a subject is increased by at least 1 log relative to a control. For example, anti-coronavirus antigen antibody titer produced in a subject may be increased by at least 1.5, at least 2, at least 2.5, or at least 3 log relative to a control. In some embodiments, the anti-coronavirus antigen antibody titer produced in the subject is increased by 1, 1.5, 2, 2.5 or 3 log relative to a control. In some embodiments, the anti-coronavirus antigen antibody titer produced in the subject is increased by 1-3 log relative to a control. For example, the anti-coronavirus antigen antibody titer produced in a subject may be increased by 1-1.5, 1-2, 1-2.5, 1-3, 1.5-2, 1.5-2.5, 1.5-3, 2-2.5, 2-3, or 2.5-3 log relative to a control. In some embodiments, the anti-coronavirus antigen antibody titer produced in a subject is increased at least 2 times relative to a control. For example, the anti-coronavirus antigen n antibody titer produced in a subject may be increased at least 3 times, at least 4 times, at least 5 times, at least 6 times, at least 7 times, at least 8 times, at least 9 times, or at least 10 times relative to a control. In some embodiments, the anti-coronavirus antigen antibody titer produced in the subject is increased 2, 3, 4, 5, 6, 7, 8, 9, or 10 times relative to a control. In some embodiments, the anti-coronavirus antigen antibody titer produced in a subject is increased 2-10 times relative to a control. For example, the anti-coronavirus antigen antibody titer produced in a subject may be increased 2-10, 2-9, 2-8, 2-7, 2-6, 2-5, 2-4, 2-3, 3-10, 3-9, 3-8, 3-7, 3-6, 3-5, 3-4, 4-10, 4-9, 4-8, 4-7, 4-6, 4-5, 5-10, 5-9, 5-8, 5-7, 5-6, 6-10, 6-9, 6-8, 6-7, 7-10, 7-9, 7-8, 8-10, 8-9, or 9-10 times relative to a control. In some embodiments, an antigen-specific immune response is measured as a ratio of geometric mean titer (GMT), referred to as a geometric mean ratio (GMR), of serum neutralizing antibody titers to coronavirus. A geometric mean titer (GMT) is the average antibody titer for a group of subjects calculated by multiplying all values and taking the nth root of the number, where n is the number of subjects with available data. A control, in some embodiments, is an anti-coronavirus antigen antibody titer produced in a subject who has not been administered a composition (e.g., RNA vaccine). In some embodiments, a control is an anti-coronavirus antigen antibody titer produced in a subject administered a recombinant or purified protein vaccine. Recombinant protein vaccines typically include protein antigens that either have been produced in a heterologous expression system (e.g., bacteria or yeast) or purified from large amounts of the pathogenic organism. In some embodiments, the ability of a composition (e.g., RNA vaccine) to be effective is measured in a murine model. For example, a composition may be administered to a murine model and the murine model assayed for induction of neutralizing antibody titers. Viral challenge studies may also be used to assess the efficacy of a vaccine of the present disclosure. For example, a composition may be administered to a murine model, the murine model challenged with virus, and the murine model assayed for survival and/or immune response (e.g., neutralizing antibody response, T cell response (e.g., cytokine response)). In some embodiments, an effective amount of a composition (e.g., RNA vaccine) is a dose that is reduced compared to the standard of care dose of a recombinant protein vaccine. A “standard of care,” as provided herein, refers to a medical or psychological treatment guideline and can be general or specific. “Standard of care” specifies appropriate treatment based on scientific evidence and collaboration between medical professionals involved in the treatment of a given condition. It is the diagnostic and treatment process that a physician/ clinician should follow for a certain type of patient, illness or clinical circumstance. A “standard of care dose,” as provided herein, refers to the dose of a recombinant or purified protein vaccine, or a live attenuated or inactivated vaccine, or a VLP vaccine, that a physician/clinician or other medical professional would administer to a subject to treat or prevent coronavirus infection or a related condition, while following the standard of care guideline for treating or preventing coronavirus infection or a related condition. In some embodiments, the anti-coronavirus antigen antibody titer produced in a subject administered an effective amount of an composition is equivalent to an anti-coronavirus antigen antibody titer produced in a control subject administered a standard of care dose of a recombinant or purified protein vaccine, or a live attenuated or inactivated vaccine, or a VLP vaccine. Vaccine efficacy may be assessed using standard analyses (see, e.g., Weinberg et al., J Infect Dis.2010 Jun 1;201(11):1607-10). For example, vaccine efficacy may be measured by double-blind, randomized, clinical controlled trials. Vaccine efficacy may be expressed as a proportionate reduction in disease attack rate (AR) between the unvaccinated (ARU) and vaccinated (ARV) study cohorts and can be calculated from the relative risk (RR) of disease among the vaccinated group with use of the following formulas: Efficacy = (ARU – ARV)/ARU x 100; and Efficacy = (1 – RR) x 100. Likewise, vaccine effectiveness may be assessed using standard analyses (see, e.g., Weinberg et al., J Infect Dis.2010 Jun 1;201(11):1607-10). Vaccine effectiveness is an assessment of how a vaccine (which may have already proven to have high vaccine efficacy) reduces disease in a population. This measure can assess the net balance of benefits and adverse effects of a vaccination program, not just the vaccine itself, under natural field conditions rather than in a controlled clinical trial. Vaccine effectiveness is proportional to vaccine efficacy (potency) but is also affected by how well target groups in the population are immunized, as well as by other non-vaccine-related factors that influence the ‘real-world’ outcomes of hospitalizations, ambulatory visits, or costs. For example, a retrospective case control analysis may be used, in which the rates of vaccination among a set of infected cases and appropriate controls are compared. Vaccine effectiveness may be expressed as a rate difference, with use of the odds ratio (OR) for developing infection despite vaccination: Effectiveness = (1 – OR) x 100. In some embodiments, efficacy of the composition (e.g., RNA vaccine) is at least 60% relative to unvaccinated control subjects. For example, efficacy of the composition may be at least 65%, at least 70%, at least 75%, at least 80%, at least 85%, at least 95%, at least 98%, or 100% relative to unvaccinated control subjects. Sterilizing Immunity. Sterilizing immunity refers to a unique immune status that prevents effective pathogen infection into the host. In some embodiments, the effective amount of a composition of the present disclosure is sufficient to provide sterilizing immunity in the subject for at least 1 year. For example, the effective amount of a composition of the present disclosure is sufficient to provide sterilizing immunity in the subject for at least 2 years, at least 3 years, at least 4 years, or at least 5 years. In some embodiments, the effective amount of a composition of the present disclosure is sufficient to provide sterilizing immunity in the subject at an at least 5-fold lower dose relative to control. For example, the effective amount may be sufficient to provide sterilizing immunity in the subject at an at least 10-fold lower, 15-fold, or 20-fold lower dose relative to a control. Detectable Antigen. In some embodiments, the effective amount of a composition of the present disclosure is sufficient to produce detectable levels of coronavirus antigen as measured in serum of the subject at 1-72 hours post administration. Titer. An antibody titer is a measurement of the number of antibodies within a subject, for example, antibodies that are specific to a particular antigen (e.g., an anti-coronavirus antigen). Antibody titer is typically expressed as the inverse of the greatest dilution that provides a positive result. Enzyme-linked immunosorbent assay (ELISA) is a common assay for determining antibody titers, for example. A neutralizing immune response is an immune response that is a neutralizing antibody response and/or an effective neutralizing T cell response. In some embodiments a neutralizing antibody response produces a level of antibodies that meet or exceed a seroprotection threshold. An effective T cell response is a response which produces a baseline level of viral activated or viral specific T cells including CD8+ and CD4+ T helper type 1 cells. CD8+ cytotoxic T lymphocytes typically clear the intracellular virus compartment and CD4+ T cells exert various functions in the body such as helping B and other T cells, promoting memory generation and indirect or direct cytotoxic activity. In some embodiments the effective T cells comprises a high proportion of CD8+ T cells and/or CD4+ T cells, relative to a baseline level (in a naïve subject). In some embodiments these T cells are differentiated towards an early- differentiated memory phenotype with co-expression of CD27 and CD28. In some embodiments, the effective amount of a composition of the present disclosure is sufficient to produce a 1,000-10,000 neutralizing antibody titer produced by neutralizing antibody against the coronavirus antigen as measured in serum of the subject at 1-72 hours post administration. In some embodiments, the effective amount is sufficient to produce a 1,000-5,000 neutralizing antibody titer produced by neutralizing antibody against the coronavirus antigen as measured in serum of the subject at 1-72 hours post administration. In some embodiments, the effective amount is sufficient to produce a 5,000-10,000 neutralizing antibody titer produced by neutralizing antibody against the coronavirus antigen as measured in serum of the subject at 1-72 hours post administration. In some embodiments, the neutralizing antibody titer is at least 100 NT50. For example, the neutralizing antibody titer may be at least 200, 300, 400, 500, 600, 700, 800, 900 or 1000 NT50. In some embodiments, the neutralizing antibody titer is at least 10,000 NT50. In some embodiments, the neutralizing antibody titer is at least 100 neutralizing units per milliliter (NU/mL). For example, the neutralizing antibody titer may be at least 200, 300, 400, 500, 600, 700, 800, 900 or 1000 NU/mL. In some embodiments, the neutralizing antibody titer is at least 10,000 NU/mL. In some embodiments, an anti-coronavirus antigen antibody titer produced in the subject is increased by at least 1 log relative to a control. For example, an anti-coronavirus antigen antibody titer produced in the subject may be increased by at least 2, 3, 4, 5, 6, 7, 8, 9 or 10 log relative to a control. In some embodiments, an anti-coronavirus antigen antibody titer produced in the subject is increased at least 2 times relative to a control. For example, an anti-coronavirus antigen antibody titer produced in the subject is increased by at least 3, 4, 5, 6, 7, 8, 9 or 10 times relative to a control. In some embodiments, a geometric mean, which is the nth root of the product of n numbers, is generally used to describe proportional growth. Geometric mean, in some embodiments, is used to characterize antibody titer produced in a subject. A control may be, for example, an unvaccinated subject, or a subject administered a live attenuated viral vaccine, an inactivated viral vaccine, or a protein subunit vaccine. EXAMPLES Methods Manufacture of polynucleotides described herein, and/or parts or regions thereof, may be accomplished using the methods taught in PCT Publication No. WO 2014/152027, entitled “Manufacturing Methods for Production of RNA Transcripts,” the contents of which are incorporated herein by reference in their entirety. Purification methods may include those described in PCT Publication No. WO 2014/152030 and PCT Publication No. WO 2014/152031, the contents of each of which are incorporated herein by reference in their entirety. Detection and characterization methods of the polynucleotides may be performed as described in PCT Publication No. WO 2014/144039, the contents of which are incorporated herein by reference in their entirety. Characterization of the polynucleotides of the disclosure may be accomplished using polynucleotide mapping, reverse transcriptase sequencing, charge distribution analysis, detection of RNA impurities, or any combination of two or more of the foregoing. “Characterizing” comprises determining the RNA transcript sequence, determining the purity of the RNA transcript, or determining the charge heterogeneity of the RNA transcript, for example. Such methods are taught in, for example, PCT Publication No. WO 2014/144711 and PCT Publication No. WO 2014/144767, the contents of each of which are incorporated herein by reference in their entirety. In experiments where a lipid nanoparticle (LNP) formulation was or is used, the formulation included or includes 48 mol% lipid of Compound 1, 11 mol% 1,2 distearoyl-sn- glycero-3-phosphocholine (DSPC), 38.5 mol% cholesterol, and 2.5 mol% PEG-modified 1,2 dimyristoyl-sn-glycerol, methoxypolyethyleneglycol (PEG2000 DMG). Immunization Methods Vaccine compositions of lipid nanoparticles containing mRNAs are administered to mice according to the following administration schedule. C57BL/6 mice are immunized with two doses of a given composition, receiving the first dose on day 0, and the second dose on day 22. Sera are collected on day 21, three weeks after the first (prime) dose but before administration of the second (boost) dose, and day 36, two weeks after administration of the second (boost) dose. Where T cells are evaluated, mice are euthanized on day 36, and spleens are collected and processed to harvest splenocytes. Splenocytes are stimulated with one of a panel of peptide pools, each pool containing peptides from a single MERS-CoV antigen, in the presence of a Golgi blocker, so that cells producing cytokines in response to stimulation retain cytokines instead of secreting them. Cell surfaces are stained for lymphocyte markers, including CD3, CD4, CD8, and/or CD107a, and cells are permeabilized and stained for multiple cytokines, including TNF-α, IFN-γ, IL-2, IL-4, IL-5, IL-10, and/or IL-13. Stained cells are incubated with a viability dye and analyzed by flow cytometry. Neutralization Assays Antibodies in serum, when bound to a viral surface protein that is essential for infection, can prevent a virus from infecting a target cell, an activity referred to as “neutralization.” To determine the ability of mRNA compositions to generate neutralizing antibodies against MERS- CoV, the neutralization activity of sera is quantified using a neutralization assay. For each assay, ARPE-19 cells are plated in 96-well plates, at a density of 2*104 cells/well and incubated for 20– 24 hours. Then, serial 3-fold dilutions of each serum sample are prepared in phenol red-free cDMEM. A consistent amount of MERS-CoV reporter virus, containing a gene encoding GFP, is incubated with each serum dilution sample, to allow for binding of any MERS-CoV-specific antibodies to the virus. After incubation, cells are washed, and incubated with MERS-CoV /serum mixtures.24 hours after incubation, GFP fluorescence in each well is measured to determine the extent of infection. For a given serum sample, the 50% neutralization titer (NT50) is calculated as the reciprocal of the serum dilution at which 50% of GFP+ cells are observed. Recombinant VSV-based Pseudovirus Neutralization. Codon-optimized nucleotide sequences encoding MERS-CoV Spike proteins are cloned into pCAGGS vectors. To make MERS-CoV Spike proteinpseudotyped recombinant VSV-ΔG-firefly luciferase virus, BHK- 21/WI-2 cells (Kerafast, EH1011) are transfected with the Spike protein expression plasmid, and subsequently infected with VSV∆G-firefly-luciferase as previously described (Whitt, 2010, Journal of Virological Methods 169, 365–374). For the neutralization assay, serially diluted serum samples are mixed with pseudovirus and incubated at 37°C for 45 minutes. The virus- serum mix is subsequently used to infect Vero-81 or Huh7.5s cells for 18 hours at 37°C before adding ONE-Glo reagent (Promega E6120) for measurement of luciferase signal (relative luminescence unit; RLU). The percentage of neutralization is calculated based on RLU of the virus only control, and subsequently analyzed using four-parameter logistic curve (Prism 8). Antibody-dependent cell-mediated cytotoxicity assays Antibodies in serum, when bound to viral protein expressed on the surface of an infected cell, can be recognized by effector cells, such as natural killer (NK) cells. Effector cells recognize the constant (Fc) region of antibodies bound to the target cells, and following recognition, release cytotoxic granules that induce apoptosis in the infected target cells. This process is referred to as “antibody-dependent cell-mediated cytotoxicity” (ADCC). To determine the ability of mRNA compositions to generate antibodies that can facilitate ADCC, the ADCC activity of sera is quantified using an ADCC assay. This assay uses Jurkat cells that constitutively express mouse FcγRIV, allowing for recognition of antibody Fc regions, and luciferase under the control of the NFAT pathway, which is activated following Fc recognition. In each assay, Vero cells are plated in 96-well plates, at a density of 2.5*104 cells/well, incubated for 20–24 hours, then inoculated with MERS-CoV at a multiplicity of infection (MOI) of 5 plaque-forming units (PFU) per cell.16 hours after inoculation, serial 3-fold dilutions of serum samples are prepared in RPMI + 4% fetal bovine serum (FBS) containing only minimal amounts of IgG, to reduce background. Serum samples are added to each well, to allow antibodies to bind to infected Vero cells expressing viral surface proteins. Then, reporter effector cells are serially diluted in RPMI + 4% low-IgG FBS, added to wells, and incubated for 6 hours to allow for recognition of surface- bound antibodies and expression of luciferase. After the 6 hours of incubation, a luciferase substrate is added to wells, so that any luciferase present can react with the substrate to produce light. Light emitted from wells is measured to quantify the amount of luciferase activity as a measurement of ADCC activity. Serum-depletion Assays Epitope- or domain-level specificity of antibodies in serum is evaluated by depleting antibodies with a given specificity from serum, and comparing such antigen-depleted (Ag- depleted) serum with mock-depleted serum. Antibodies specific to a given epitope or antigen may be depleted by incubating serum with a solid support (e.g., surface of a 96-well plate) on which the epitope or antigen is immobilized. Incubation allows antibodies with the desired specificity to bind to the immobilized epitope or antigen, such that the serum collected after incubation does not contain the surface-bound antibodies. Mock-depleted serum is prepared by incubating serum under similar conditions, where the solid support does not contain an immobilized epitope or antigen. Antigen-depleted sera may be compared to mock-depleted sera in multiple in vitro assays, such as ELISA, neutralization assays, ADCC assays, and/or in vivo, such as by passive transfer of sera before, during, or after inoculation with a virus. Example 1: Design of mRNA encoding MERS-CoV S proteins, fusion proteins, and variants thereof. MERS-CoV S protein sequences were modified with one or more changes as described in Table E1-1. Modifications included (i) introduction of two proline substitutions to stabilize the S protein in a prefusion conformation (S2P); (ii) inactivation of a furin cleavage site; (iii) deletion of an endoplasmic reticulum (ER) retention signal; and/or truncation of a cytoplasmic tail Fusion proteins comprising one or more domains of MERS-CoV S protein, and optionally one or more linkers and/or a transmembrane domain, were designed as described in Table E1-2. Table E1-1. MERS-CoV S protein modifications Table E1-2. MERS-CoV S protein domains and combinations
Table E1-2. MERS-CoV S protein domains and combinations
 Relative positions of N-terminal domain (NTD) and receptor-binding domain (RBD) on betacoronavirus S proteins, and exemplary arrangement of an NTD-RBD-TM fusion protein, are shown in FIG.1. To evaluate immunogenicity of such fusion proteins, lipid nanoparticles (LNPs) containing mRNAs encoding a 2P-stabilized betacoronavirus S protein having two proline substitutions (S-2P) or a fusion protein comprising an NTD and RBD of the same betacoronavirus S protein, linked to a transmembrane (TM) domain (NTD-RBD-TM), were administered to mice in one dose containing 1 ug mRNA, or in two doses, each dose containing the same amount of mRNA, at either 0.001, 0.01 ug, 0.1 ug, or 1 ug. The first dose was administered on day 0, and second dose was administered on day 22. In two-dose administration experiments, sera were collected at day 36, and evaluated for neutralizing antibody titers as described above in “Neutralization Assays.” Compositions containing mRNA encoding the NTD- RBD fusion protein (NTD-RBD-TM) elicited significantly higher neutralizing antibody titers, relative to those containing mRNA encoding the 2P-stabilized prefusion S protein (S-2P) (FIG. 2). Example 2: Immunization of mice with compositions containing mRNAs encoding MERS- CoV proteins. Mice are administered lipid nanoparticles (LNPs) containing the mRNAs encoding MERS-CoV Spike proteins and/or fusion proteins. The proteins encoded by the mRNAs of each composition are shown with their sequences in Table E2-1. A first dose (prime) is administered on day 0, and a second dose (booster) is administered on day 22. Serum is collected (i) on day 21, three weeks after administration of the first dose, but before booster dose administration, and (ii) on day 36, two weeks after the administration of the booster dose. At day 36, mice are also euthanized to collect spleens for analysis of T cells by cell surface marker and intracellular cytokine staining. T cell phenotypes and epitope-specificity is evaluated as described above in “Immunization Methods.” T cells are incubated with peptides from the MERS-CoV S protein NTD, RBD, S1 subunit, S2 subunit, and/or full-length S protein to evaluate specificity to different regions of the S protein. Sera are evaluated for antiviral activities such as neutralization, ADCC activity, and/or prevention of cell-cell spread by MERS-CoV. Sera are incubated with immobilized MERS-CoV S protein NTD, RBD, S1 subunit, S2 subunit, and/or full-length S protein to generate sera depleted of NTD-specific, RBD-specific, S1 subunit-specific, S2 subunit-specific, and/or full- length S protein-specific antibodies, and depleted sera are compared to mock-depleted sera to evaluate the contribution of antibody specificity to neutralization, ADCC activity, and/or prevention of cell-cell spread by MERS-CoV. Table E2-1. MERS-CoV Proteins
Relative positions of N-terminal domain (NTD) and receptor-binding domain (RBD) on betacoronavirus S proteins, and exemplary arrangement of an NTD-RBD-TM fusion protein, are shown in FIG.1. To evaluate immunogenicity of such fusion proteins, lipid nanoparticles (LNPs) containing mRNAs encoding a 2P-stabilized betacoronavirus S protein having two proline substitutions (S-2P) or a fusion protein comprising an NTD and RBD of the same betacoronavirus S protein, linked to a transmembrane (TM) domain (NTD-RBD-TM), were administered to mice in one dose containing 1 ug mRNA, or in two doses, each dose containing the same amount of mRNA, at either 0.001, 0.01 ug, 0.1 ug, or 1 ug. The first dose was administered on day 0, and second dose was administered on day 22. In two-dose administration experiments, sera were collected at day 36, and evaluated for neutralizing antibody titers as described above in “Neutralization Assays.” Compositions containing mRNA encoding the NTD- RBD fusion protein (NTD-RBD-TM) elicited significantly higher neutralizing antibody titers, relative to those containing mRNA encoding the 2P-stabilized prefusion S protein (S-2P) (FIG. 2). Example 2: Immunization of mice with compositions containing mRNAs encoding MERS- CoV proteins. Mice are administered lipid nanoparticles (LNPs) containing the mRNAs encoding MERS-CoV Spike proteins and/or fusion proteins. The proteins encoded by the mRNAs of each composition are shown with their sequences in Table E2-1. A first dose (prime) is administered on day 0, and a second dose (booster) is administered on day 22. Serum is collected (i) on day 21, three weeks after administration of the first dose, but before booster dose administration, and (ii) on day 36, two weeks after the administration of the booster dose. At day 36, mice are also euthanized to collect spleens for analysis of T cells by cell surface marker and intracellular cytokine staining. T cell phenotypes and epitope-specificity is evaluated as described above in “Immunization Methods.” T cells are incubated with peptides from the MERS-CoV S protein NTD, RBD, S1 subunit, S2 subunit, and/or full-length S protein to evaluate specificity to different regions of the S protein. Sera are evaluated for antiviral activities such as neutralization, ADCC activity, and/or prevention of cell-cell spread by MERS-CoV. Sera are incubated with immobilized MERS-CoV S protein NTD, RBD, S1 subunit, S2 subunit, and/or full-length S protein to generate sera depleted of NTD-specific, RBD-specific, S1 subunit-specific, S2 subunit-specific, and/or full- length S protein-specific antibodies, and depleted sera are compared to mock-depleted sera to evaluate the contribution of antibody specificity to neutralization, ADCC activity, and/or prevention of cell-cell spread by MERS-CoV. Table E2-1. MERS-CoV Proteins Example 3. Immunogenicity and Neutralization Assay at Day 21 Following a Single Dose Mice are administered lipid nanoparticles (LNPs) containing mRNAs encoding MERS- CoV Spike proteins and/or fusion proteins (Table E2-1). MERS-CoV S protein-specific IgG titers are measured by ELISA at Day 21 post-vaccination. Sera are evaluated for antiviral activities such as neutralization, ADCC activity, and/or prevention of cell-cell spread by MERS- CoV. Sera depleted of specific antibodies are prepared and compared to mock-depleted sera to evaluate the contribution of specific antibodies to neutralization, ADCC activity, and/or prevention of cell-cell spread by MERS-CoV. Example 4. Immunogenicity and Neutralization Assay at Day 36 Following Two Doses The same LNP-mRNA compositions described in Example 3 are again administered to mice as booster doses on Day 22 post-vaccination with the first dose. The titers of antibodies generated after the booster dose S protein are measured by ELISA from day 36 serum. Sera are evaluated for antiviral activities such as neutralization, ADCC activity, and/or prevention of cell- cell spread by MERS-CoV. Sera depleted of specific antibodies are prepared and compared to mock-depleted sera to evaluate the contribution of specific antibodies to neutralization, ADCC activity, and/or prevention of cell-cell spread by MERS-CoV. EXEMPLARY SEQUENCES It should be understood that any of the mRNA sequences described herein may include a 5’ UTR and/or a 3’ UTR. The UTR sequences may be selected from the following sequences, or other known UTR sequences may be used. It should also be understood that any of the mRNA constructs described herein may further comprise a poly(A) tail and/or cap (e.g., 7mG(5’)ppp(5’)NlmpNp). Further, while many of the mRNAs and encoded antigen sequences described herein include a signal peptide and/or a peptide tag (e.g., C-terminal His tag), it should be understood that the indicated signal peptide and/or peptide tag may be substituted for a different signal peptide and/or peptide tag, or the signal peptide and/or peptide tag may be omitted. 5’ UTR: GGGAAAUAAGAGAGAAAAGAAGAGUAAGAAGAAAUAUAAGAGCCACC (SEQ ID NO: 1) 5’ UTR: GGGAAAUAAGAGAGAAAAGAAGAGUAAGAAGAAAUAUAAGACCCCGGCGCCGCCACC (SEQ ID NO: 2) 3’ UTR: UGAUAAUAGGCUGGAGCCUCGGUGGCCAUGCUUCUUGCCCCUUGGGCCUCCCCCCAGCCCCUCCUCCCCUUCCUGCAC CCGUACCCCCGUGGUCUUUGAAUAAAGUCUGAGUGGGCGGC (SEQ ID NO: 3) 3’ UTR: UGAUAAUAGGCUGGAGCCUCGGUGGCCUAGCUUCUUGCCCCUUGGGCCUCCCCCCAGCCCCUCCUCCCCUUCCUGCAC CCGUACCCCCGUGGUCUUUGAAUAAAGUCUGAGUGGGCGGC (SEQ ID NO: 4) Table 1.5′ UTR sequences
Example 3. Immunogenicity and Neutralization Assay at Day 21 Following a Single Dose Mice are administered lipid nanoparticles (LNPs) containing mRNAs encoding MERS- CoV Spike proteins and/or fusion proteins (Table E2-1). MERS-CoV S protein-specific IgG titers are measured by ELISA at Day 21 post-vaccination. Sera are evaluated for antiviral activities such as neutralization, ADCC activity, and/or prevention of cell-cell spread by MERS- CoV. Sera depleted of specific antibodies are prepared and compared to mock-depleted sera to evaluate the contribution of specific antibodies to neutralization, ADCC activity, and/or prevention of cell-cell spread by MERS-CoV. Example 4. Immunogenicity and Neutralization Assay at Day 36 Following Two Doses The same LNP-mRNA compositions described in Example 3 are again administered to mice as booster doses on Day 22 post-vaccination with the first dose. The titers of antibodies generated after the booster dose S protein are measured by ELISA from day 36 serum. Sera are evaluated for antiviral activities such as neutralization, ADCC activity, and/or prevention of cell- cell spread by MERS-CoV. Sera depleted of specific antibodies are prepared and compared to mock-depleted sera to evaluate the contribution of specific antibodies to neutralization, ADCC activity, and/or prevention of cell-cell spread by MERS-CoV. EXEMPLARY SEQUENCES It should be understood that any of the mRNA sequences described herein may include a 5’ UTR and/or a 3’ UTR. The UTR sequences may be selected from the following sequences, or other known UTR sequences may be used. It should also be understood that any of the mRNA constructs described herein may further comprise a poly(A) tail and/or cap (e.g., 7mG(5’)ppp(5’)NlmpNp). Further, while many of the mRNAs and encoded antigen sequences described herein include a signal peptide and/or a peptide tag (e.g., C-terminal His tag), it should be understood that the indicated signal peptide and/or peptide tag may be substituted for a different signal peptide and/or peptide tag, or the signal peptide and/or peptide tag may be omitted. 5’ UTR: GGGAAAUAAGAGAGAAAAGAAGAGUAAGAAGAAAUAUAAGAGCCACC (SEQ ID NO: 1) 5’ UTR: GGGAAAUAAGAGAGAAAAGAAGAGUAAGAAGAAAUAUAAGACCCCGGCGCCGCCACC (SEQ ID NO: 2) 3’ UTR: UGAUAAUAGGCUGGAGCCUCGGUGGCCAUGCUUCUUGCCCCUUGGGCCUCCCCCCAGCCCCUCCUCCCCUUCCUGCAC CCGUACCCCCGUGGUCUUUGAAUAAAGUCUGAGUGGGCGGC (SEQ ID NO: 3) 3’ UTR: UGAUAAUAGGCUGGAGCCUCGGUGGCCUAGCUUCUUGCCCCUUGGGCCUCCCCCCAGCCCCUCCUCCCCUUCCUGCAC CCGUACCCCCGUGGUCUUUGAAUAAAGUCUGAGUGGGCGGC (SEQ ID NO: 4) Table 1.5′ UTR sequences
 Table 2.3′ UTR sequences (stop cassette is italicized; miR binding sites are boldened)
Table 2.3′ UTR sequences (stop cassette is italicized; miR binding sites are boldened)
 Table 3. Signal Peptides
Table 3. Signal Peptides Wild-type MERS-CoV (Kingdom of Saudi Arabia (KSA) 2019) Spike (S) Protein Sequence (SEQ ID NO: 84) (GenBank Accession No. QEJ82215.1) MIHSVFLLMFLLTPTESYVDVGPDSLKSACIEVDIQQTFFDKTWPRPIDVSKADGIIYPQGRTYSNITITYQGLFPYQ GDHGDMYVYSAGHATGTTPQKLFVANYSQDVKQFANGFVVRIGAAANSTGTVIISPSTSATIRKIYPAFMLGSSVGNF SDGKMGRFFNHTLVLLPDGCGTLLRAFYCILEPRSGNHCPAGNSYTSFATYHTPATDCSDGNYNRNASLNSFKEYFNL RNCTFMYTYNITEDEILEWFGITQTAQGVHLFSSRYVDLYGGNMFQFATLPVYDTIKYYSIIPHSIRSIQSDRKAWAA FYVYKLQPLTFLLDFSVDGYIRRAIDCGFNDLSQLHCSYESFDVESGVYSVSSFEAKPSGSVVEQAEGVECDFSPLLS GTPPQVYNFKRLVFTNCNYNLTKLLSLFSVNDFTCSQISPAAIASNCYSSLILDYFSYPLSMKSDLSVSSAGPISQFN YKQSFSNPTCLILATVPHNLTTITKPLKYSYINKCSRLLSDDRTEVPQLVNANQYSPCVSIAPSTVWEDGDYYRKQLS PLEGGGWLVASGSTVAMTEQLQMGFGITVQYGTDTNSVCPKLEFANDTKIASQLGNCVEYSLYGVSGRGVFQNCTAVG VRQQRFVYDAYQNLVGYYSDDGNYYCLRACVSVPVSVIYDKETKTHATLFGSVACEHISSTMSQYSRSTRSMLKRRDS TYGPLQTPVGCVLGLVNSSLFVEDCKLPLGQSLCALPDTPSTLTPRSVRSVPGEMRLASIAFNHPIQVDQLNSSYFKL SIPTNFSFGVTQEYIQTTIQKVTVDCKQYVCNGFQKCEQLLREYGQFCSKINQALHGANLRQDDSVRNLFASVKSSQS SPIIPGFGGDFNLTLLEPVSISTGSRSARSAIEDLLFDKVTIADPGYMQGYDDCMKQGPASARDLICAQYVAGYKVLP PLMDVNMEAAYTSSLLGSIAGVGWTAGLSSFAAIPFAQSIFYRLNGVGITQQVLSENQKLIANKFNQALGAMQTGFTT TNEAFRKVQDAVNNNAQALSKLASELSNTFGAISASIGDIIQRLDVLEQDAQIDRLINGRLTTLNAFVAQQLVRSESA ALSAQLAKDKVNECVKAQSKRSGFCGQGTHIVSFVVNAPNGLYFMHVGYYPSNHIEVVSAYGLCDAANPTNCIAPVNG YFIKTNNTRIVDEWSYTGSSFYAPEPITSLNTKYVAPQVTYQNISTNLPPPLLGNSTGIDFQDELDEFFKNVSTSIPN FGSLTQINTTLLDLTYEMLSLQQVVKALNESYIDLKELGNYTYYNKWPWYIWLGFIAGLVALALCVFFILCCTGCGTN CMGKLKCNRCCDRYEEYDLEPHKVHVH Wild-type MERS-CoV (Riyadh 2013) Spike (S) Protein Sequence (SEQ ID NO: 85) MIHSVFLLMFLLTPTESYVDVGPDSVKSACIEVDIQQTFFDKTWPRPIDVSKADGIIYPQGRTYSNITITYQGLFPYQ GDHGDMYVYSAGHATGTTPQKLFVANYSQDVKQFANGFVVRIGAAANSTGTVIISPSTSATIRKIYPAFMLGSSVGNF SDGKMGRFFNHTLVLLPDGCGTLLRAFYCILEPRSGNHCPAGNSYTSFATYHTPATDCSDGNYNRNASLNSFKEYFNL RNCTFMYTYNITEDEILEWFGITQTAQGVHLFSSRYVDLYGGNMFQFATLPVYDTIKYYSIIPHSIRSIQSDRKAWAA FYVYKLQPLTFLLDFSVDGYIRRAIDCGFNDLSQLHCSYESFDVESGVYSVSSFEAKPSGSVVEQAEGVECDFSPLLS GTPPQVYNFKRLVFTNCNYNLTKLLSLFSVNDFTCSQISPAAIASNCYSSLILDYFSYPLSMKSDLSVSSAGPISQFN YKQSFSNPTCLILATVPHNLTTITKPLKYSYINKCSRLLSDDRTEVPQLVNANQYSPCVSIVPSTVWEDGDYYRKQLS PLEGGGWLVASGSTVAMTEQLQMGFGITVQYGTDTNSVCPKLEFANDTKIASQLGNCVEYSLYGVSGRGVFQNCTAVG VRQQRFVYDAYQNLVGYYSDDGNYYCLRACVSVPVSVIYDKETKTHATLFGSVACEHISSTMSQYSRSTRSMLKRRDS TYGPLQTPVGCVLGLVNSSLFVEDCKLPLGQSLCALPDTPSTLTPRSVRSVPGEMRLASIAFNHPIQVDQLNSSYFKL SIPTNFSFGVTQEYIQTTIQKVTVDCKQYVCNGFQKCEQLLREYGQFCSKINQALHGANLRQDDSVRNLFASVKSSQS SPIIPGFGGDFNLTLLEPVSISTGSRSARSAIEDLLFDKVTIADPGYMQGYDDCMQQGPASARDLICAQYVAGYKVLP PLMDVNMEAAYTSSLLGSIAGVGWTAGLSSFAAIPFAQSIFYRLNGVGITQQVLSENQKLIANKFNQALGAMQTGFTT TNEAFRKVQDAVNNNAQALSKLASELSNTFGAISASIGDIIQRLDVLEQDAQIDRLINGRLTTLNAFVAQQLVRSESA ALSAQLAKDKVNECVKAQSKRSGFCGQGTHIVSFVVNAPNGLYFMHVGYYPSNHIEVVSAYGLCDAANPTNCIAPVNG YFIKTNNTRIVDEWSYTGSSFYAPEPITSLNTKYVAPQVTYQNISTNLPPPLLGNSTGIDFQDELDEFFKNVSTSIPN FGSLTQINTTLLDLTYEMLSLQQVVKALNESYIDLKELGNYTYYNKWPWYIWLGFIAGLVALALCVFFILCCTGCGTN CMGKLKCNRCCDRYEEYDLEPHKVHVH Wild-type MERS-CoV (England 2012) Spike (S) Protein Sequence (SEQ ID NO: 86) (GenBank Accession No. YP_007188579.1) MIHSVFLLMFLLTPTESYVDVGPDSVKSACIEVDIQQTFFDKTWPRPIDVSKADGIIYPQGRTYSNITITYQGLFPYQ GDHGDMYVYSAGHATGTTPQKLFVANYSQDVKQFANGFVVRIGAAANSTGTVIISPSTSATIRKIYPAFMLGSSVGNF SDGKMGRFFNHTLVLLPDGCGTLLRAFYCILEPRSGNHCPAGNSYTSFATYHTPATDCSDGNYNRNASLNSFKEYFNL RNCTFMYTYNITEDEILEWFGITQTAQGVHLFSSRYVDLYGGNMFQFATLPVYDTIKYYSIIPHSIRSIQSDRKAWAA FYVYKLQPLTFLLDFSVDGYIRRAIDCGFNDLSQLHCSYESFDVESGVYSVSSFEAKPSGSVVEQAEGVECDFSPLLS GTPPQVYNFKRLVFTNCNYNLTKLLSLFSVNDFTCSQISPAAIASNCYSSLILDYFSYPLSMKSDLSVSSAGPISQFN YKQSFSNPTCLILATVPHNLTTITKPLKYSYINKCSRFLSDDRTEVPQLVNANQYSPCVSIVPSTVWEDGDYYRKQLS PLEGGGWLVASGSTVAMTEQLQMGFGITVQYGTDTNSVCPKLEFANDTKIASQLGNCVEYSLYGVSGRGVFQNCTAVG VRQQRFVYDAYQNLVGYYSDDGNYYCLRACVSVPVSVIYDKETKTHATLFGSVACEHISSTMSQYSRSTRSMLKRRDS TYGPLQTPVGCVLGLVNSSLFVEDCKLPLGQSLCALPDTPSTLTPRSVRSVPGEMRLASIAFNHPIQVDQLNSSYFKL SIPTNFSFGVTQEYIQTTIQKVTVDCKQYVCNGFQKCEQLLREYGQFCSKINQALHGANLRQDDSVRNLFASVKSSQS SPIIPGFGGDFNLTLLEPVSISTGSRSARSAIEDLLFDKVTIADPGYMQGYDDCMQQGPASARDLICAQYVAGYKVLP PLMDVNMEAAYTSSLLGSIAGVGWTAGLSSFAAIPFAQSIFYRLNGVGITQQVLSENQKLIANKFNQALGAMQTGFTT TNEAFHKVQDAVNNNAQALSKLASELSNTFGAISASIGDIIQRLDVLEQDAQIDRLINGRLTTLNAFVAQQLVRSESA ALSAQLAKDKVNECVKAQSKRSGFCGQGTHIVSFVVNAPNGLYFMHVGYYPSNHIEVVSAYGLCDAANPTNCIAPVNG YFIKTNNTRIVDEWSYTGSSFYAPEPITSLNTKYVAPQVTYQNISTNLPPPLLGNSTGIDFQDELDEFFKNVSTSIPN FGSLTQINTTLLDLTYEMLSLQQVVKALNESYIDLKELGNYTYYNKWPWYIWLGFIAGLVALALCVFFILCCTGCGTN CMGKLKCNRCCDRYEEYDLEPHKVHVH MERS-CoV (KSA 2019) Spike (S) Protein N-terminal Domain (NTD) (SEQ ID NO: 87) YVDVGPDSLKSACIEVDIQQTFFDKTWPRPIDVSKADGIIYPQGRTYSNITITYQGLFPYQGDHGDMYVYSAGHATGT TPQKLFVANYSQDVKQFANGFVVRIGAAANSTGTVIISPSTSATIRKIYPAFMLGSSVGNFSDGKMGRFFNHTLVLLP DGCGTLLRAFYCILEPRSGNHCPAGNSYTSFATYHTPATDCSDGNYNRNASLNSFKEYFNLRNCTFMYTYNITEDEIL EWFGITQTAQGVHLFSSRYVDLYGGNMFQFATLPVYDTIKYYSIIPHSIRSIQSDRKAWAAFYVYKLQPLTFLLDFSV DGYIRRAID MERS-CoV (KSA 2019) Spike (S) Protein Receptor-binding Domain (RBD) (SEQ ID NO: 88) EQAEGVECDFSPLLSGTPPQVYNFKRLVFTNCNYNLTKLLSLFSVNDFTCSQISPAAIASNCYSSLILDYFSYPLSMK SDLSVSSAGPISQFNYKQSFSNPTCLILATVPHNLTTITKPLKYSYINKCSRLLSDDRTEVPQLVNANQYSPCVSIAP STVWEDGDYYRKQLSPLEGGGWLVASGSTVAMTEQLQMGFGITVQYGTDTNSVCPKLE 2P-Stabilized MERS-CoV S Protein Sequence (SEQ ID NO: 89) MIHSVFLLMFLLTPTESYVDVGPDSLKSACIEVDIQQTFFDKTWPRPIDVSKADGIIYPQGRTYSNITITYQGLFPYQ GDHGDMYVYSAGHATGTTPQKLFVANYSQDVKQFANGFVVRIGAAANSTGTVIISPSTSATIRKIYPAFMLGSSVGNF SDGKMGRFFNHTLVLLPDGCGTLLRAFYCILEPRSGNHCPAGNSYTSFATYHTPATDCSDGNYNRNASLNSFKEYFNL RNCTFMYTYNITEDEILEWFGITQTAQGVHLFSSRYVDLYGGNMFQFATLPVYDTIKYYSIIPHSIRSIQSDRKAWAA FYVYKLQPLTFLLDFSVDGYIRRAIDCGFNDLSQLHCSYESFDVESGVYSVSSFEAKPSGSVVEQAEGVECDFSPLLS GTPPQVYNFKRLVFTNCNYNLTKLLSLFSVNDFTCSQISPAAIASNCYSSLILDYFSYPLSMKSDLSVSSAGPISQFN YKQSFSNPTCLILATVPHNLTTITKPLKYSYINKCSRLLSDDRTEVPQLVNANQYSPCVSIAPSTVWEDGDYYRKQLS PLEGGGWLVASGSTVAMTEQLQMGFGITVQYGTDTNSVCPKLEFANDTKIASQLGNCVEYSLYGVSGRGVFQNCTAVG VRQQRFVYDAYQNLVGYYSDDGNYYCLRACVSVPVSVIYDKETKTHATLFGSVACEHISSTMSQYSRSTRSMLKRRDS TYGPLQTPVGCVLGLVNSSLFVEDCKLPLGQSLCALPDTPSTLTPRSVRSVPGEMRLASIAFNHPIQVDQLNSSYFKL SIPTNFSFGVTQEYIQTTIQKVTVDCKQYVCNGFQKCEQLLREYGQFCSKINQALHGANLRQDDSVRNLFASVKSSQS SPIIPGFGGDFNLTLLEPVSISTGSRSARSAIEDLLFDKVTIADPGYMQGYDDCMKQGPASARDLICAQYVAGYKVLP PLMDVNMEAAYTSSLLGSIAGVGWTAGLSSFAAIPFAQSIFYRLNGVGITQQVLSENQKLIANKFNQALGAMQTGFTT TNEAFRKVQDAVNNNAQALSKLASELSNTFGAISASIGDIIQRLDPPEQDAQIDRLINGRLTTLNAFVAQQLVRSESA ALSAQLAKDKVNECVKAQSKRSGFCGQGTHIVSFVVNAPNGLYFMHVGYYPSNHIEVVSAYGLCDAANPTNCIAPVNG YFIKTNNTRIVDEWSYTGSSFYAPEPITSLNTKYVAPQVTYQNISTNLPPPLLGNSTGIDFQDELDEFFKNVSTSIPN FGSLTQINTTLLDLTYEMLSLQQVVKALNESYIDLKELGNYTYYNKWPWYIWLGFIAGLVALALCVFFILCCTGCGTN CMGKLKCNRCCDRYEEYDLEPHKVHVH MERS-CoV S Protein with Inactive Furin Cleavage Site (SEQ ID NO: 90) MIHSVFLLMFLLTPTESYVDVGPDSLKSACIEVDIQQTFFDKTWPRPIDVSKADGIIYPQGRTYSNITITYQGLFPYQ GDHGDMYVYSAGHATGTTPQKLFVANYSQDVKQFANGFVVRIGAAANSTGTVIISPSTSATIRKIYPAFMLGSSVGNF SDGKMGRFFNHTLVLLPDGCGTLLRAFYCILEPRSGNHCPAGNSYTSFATYHTPATDCSDGNYNRNASLNSFKEYFNL RNCTFMYTYNITEDEILEWFGITQTAQGVHLFSSRYVDLYGGNMFQFATLPVYDTIKYYSIIPHSIRSIQSDRKAWAA FYVYKLQPLTFLLDFSVDGYIRRAIDCGFNDLSQLHCSYESFDVESGVYSVSSFEAKPSGSVVEQAEGVECDFSPLLS GTPPQVYNFKRLVFTNCNYNLTKLLSLFSVNDFTCSQISPAAIASNCYSSLILDYFSYPLSMKSDLSVSSAGPISQFN YKQSFSNPTCLILATVPHNLTTITKPLKYSYINKCSRLLSDDRTEVPQLVNANQYSPCVSIAPSTVWEDGDYYRKQLS PLEGGGWLVASGSTVAMTEQLQMGFGITVQYGTDTNSVCPKLEFANDTKIASQLGNCVEYSLYGVSGRGVFQNCTAVG VRQQRFVYDAYQNLVGYYSDDGNYYCLRACVSVPVSVIYDKETKTHATLFGSVACEHISSTMSQYSRSTRSMLKRRDS TYGPLQTPVGCVLGLVNSSLFVEDCKLPLGQSLCALPDTPSTLTPASVGSVPGEMRLASIAFNHPIQVDQLNSSYFKL SIPTNFSFGVTQEYIQTTIQKVTVDCKQYVCNGFQKCEQLLREYGQFCSKINQALHGANLRQDDSVRNLFASVKSSQS SPIIPGFGGDFNLTLLEPVSISTGSRSARSAIEDLLFDKVTIADPGYMQGYDDCMKQGPASARDLICAQYVAGYKVLP PLMDVNMEAAYTSSLLGSIAGVGWTAGLSSFAAIPFAQSIFYRLNGVGITQQVLSENQKLIANKFNQALGAMQTGFTT TNEAFRKVQDAVNNNAQALSKLASELSNTFGAISASIGDIIQRLDVLEQDAQIDRLINGRLTTLNAFVAQQLVRSESA ALSAQLAKDKVNECVKAQSKRSGFCGQGTHIVSFVVNAPNGLYFMHVGYYPSNHIEVVSAYGLCDAANPTNCIAPVNG YFIKTNNTRIVDEWSYTGSSFYAPEPITSLNTKYVAPQVTYQNISTNLPPPLLGNSTGIDFQDELDEFFKNVSTSIPN FGSLTQINTTLLDLTYEMLSLQQVVKALNESYIDLKELGNYTYYNKWPWYIWLGFIAGLVALALCVFFILCCTGCGTN CMGKLKCNRCCDRYEEYDLEPHKVHVH MERS-CoV S Protein Lacking Endoplasmic Reticulum (ER) Retention Signal (SEQ ID NO: 91) MIHSVFLLMFLLTPTESYVDVGPDSLKSACIEVDIQQTFFDKTWPRPIDVSKADGIIYPQGRTYSNITITYQGLFPYQ GDHGDMYVYSAGHATGTTPQKLFVANYSQDVKQFANGFVVRIGAAANSTGTVIISPSTSATIRKIYPAFMLGSSVGNF SDGKMGRFFNHTLVLLPDGCGTLLRAFYCILEPRSGNHCPAGNSYTSFATYHTPATDCSDGNYNRNASLNSFKEYFNL RNCTFMYTYNITEDEILEWFGITQTAQGVHLFSSRYVDLYGGNMFQFATLPVYDTIKYYSIIPHSIRSIQSDRKAWAA FYVYKLQPLTFLLDFSVDGYIRRAIDCGFNDLSQLHCSYESFDVESGVYSVSSFEAKPSGSVVEQAEGVECDFSPLLS GTPPQVYNFKRLVFTNCNYNLTKLLSLFSVNDFTCSQISPAAIASNCYSSLILDYFSYPLSMKSDLSVSSAGPISQFN YKQSFSNPTCLILATVPHNLTTITKPLKYSYINKCSRLLSDDRTEVPQLVNANQYSPCVSIAPSTVWEDGDYYRKQLS PLEGGGWLVASGSTVAMTEQLQMGFGITVQYGTDTNSVCPKLEFANDTKIASQLGNCVEYSLYGVSGRGVFQNCTAVG VRQQRFVYDAYQNLVGYYSDDGNYYCLRACVSVPVSVIYDKETKTHATLFGSVACEHISSTMSQYSRSTRSMLKRRDS TYGPLQTPVGCVLGLVNSSLFVEDCKLPLGQSLCALPDTPSTLTPRSVRSVPGEMRLASIAFNHPIQVDQLNSSYFKL SIPTNFSFGVTQEYIQTTIQKVTVDCKQYVCNGFQKCEQLLREYGQFCSKINQALHGANLRQDDSVRNLFASVKSSQS SPIIPGFGGDFNLTLLEPVSISTGSRSARSAIEDLLFDKVTIADPGYMQGYDDCMKQGPASARDLICAQYVAGYKVLP PLMDVNMEAAYTSSLLGSIAGVGWTAGLSSFAAIPFAQSIFYRLNGVGITQQVLSENQKLIANKFNQALGAMQTGFTT TNEAFRKVQDAVNNNAQALSKLASELSNTFGAISASIGDIIQRLDVLEQDAQIDRLINGRLTTLNAFVAQQLVRSESA ALSAQLAKDKVNECVKAQSKRSGFCGQGTHIVSFVVNAPNGLYFMHVGYYPSNHIEVVSAYGLCDAANPTNCIAPVNG YFIKTNNTRIVDEWSYTGSSFYAPEPITSLNTKYVAPQVTYQNISTNLPPPLLGNSTGIDFQDELDEFFKNVSTSIPN FGSLTQINTTLLDLTYEMLSLQQVVKALNESYIDLKELGNYTYYNKWPWYIWLGFIAGLVALALCVFFILCCTGCGTN CMGKLKCNRCCDRYEEYDLEP MERS-CoV S Protein with Truncated Cytoplasmic Tail (SEQ ID NO: 92) MIHSVFLLMFLLTPTESYVDVGPDSLKSACIEVDIQQTFFDKTWPRPIDVSKADGIIYPQGRTYSNITITYQGLFPYQ GDHGDMYVYSAGHATGTTPQKLFVANYSQDVKQFANGFVVRIGAAANSTGTVIISPSTSATIRKIYPAFMLGSSVGNF SDGKMGRFFNHTLVLLPDGCGTLLRAFYCILEPRSGNHCPAGNSYTSFATYHTPATDCSDGNYNRNASLNSFKEYFNL RNCTFMYTYNITEDEILEWFGITQTAQGVHLFSSRYVDLYGGNMFQFATLPVYDTIKYYSIIPHSIRSIQSDRKAWAA FYVYKLQPLTFLLDFSVDGYIRRAIDCGFNDLSQLHCSYESFDVESGVYSVSSFEAKPSGSVVEQAEGVECDFSPLLS GTPPQVYNFKRLVFTNCNYNLTKLLSLFSVNDFTCSQISPAAIASNCYSSLILDYFSYPLSMKSDLSVSSAGPISQFN YKQSFSNPTCLILATVPHNLTTITKPLKYSYINKCSRLLSDDRTEVPQLVNANQYSPCVSIAPSTVWEDGDYYRKQLS PLEGGGWLVASGSTVAMTEQLQMGFGITVQYGTDTNSVCPKLEFANDTKIASQLGNCVEYSLYGVSGRGVFQNCTAVG VRQQRFVYDAYQNLVGYYSDDGNYYCLRACVSVPVSVIYDKETKTHATLFGSVACEHISSTMSQYSRSTRSMLKRRDS TYGPLQTPVGCVLGLVNSSLFVEDCKLPLGQSLCALPDTPSTLTPRSVRSVPGEMRLASIAFNHPIQVDQLNSSYFKL SIPTNFSFGVTQEYIQTTIQKVTVDCKQYVCNGFQKCEQLLREYGQFCSKINQALHGANLRQDDSVRNLFASVKSSQS SPIIPGFGGDFNLTLLEPVSISTGSRSARSAIEDLLFDKVTIADPGYMQGYDDCMKQGPASARDLICAQYVAGYKVLP PLMDVNMEAAYTSSLLGSIAGVGWTAGLSSFAAIPFAQSIFYRLNGVGITQQVLSENQKLIANKFNQALGAMQTGFTT TNEAFRKVQDAVNNNAQALSKLASELSNTFGAISASIGDIIQRLDVLEQDAQIDRLINGRLTTLNAFVAQQLVRSESA ALSAQLAKDKVNECVKAQSKRSGFCGQGTHIVSFVVNAPNGLYFMHVGYYPSNHIEVVSAYGLCDAANPTNCIAPVNG YFIKTNNTRIVDEWSYTGSSFYAPEPITSLNTKYVAPQVTYQNISTNLPPPLLGNSTGIDFQDELDEFFKNVSTSIPN FGSLTQINTTLLDLTYEMLSLQQVVKALNESYIDLKELGNYTYYNKWPWYIWLGFIAGLVALALCVFFILCCTGCGTN CMGKLKCNRCCD 2P-Stabilized MERS-CoV S Protein with Inactive Furin Cleavage Site (SEQ ID NO: 93) MIHSVFLLMFLLTPTESYVDVGPDSLKSACIEVDIQQTFFDKTWPRPIDVSKADGIIYPQGRTYSNITITYQGLFPYQ GDHGDMYVYSAGHATGTTPQKLFVANYSQDVKQFANGFVVRIGAAANSTGTVIISPSTSATIRKIYPAFMLGSSVGNF SDGKMGRFFNHTLVLLPDGCGTLLRAFYCILEPRSGNHCPAGNSYTSFATYHTPATDCSDGNYNRNASLNSFKEYFNL RNCTFMYTYNITEDEILEWFGITQTAQGVHLFSSRYVDLYGGNMFQFATLPVYDTIKYYSIIPHSIRSIQSDRKAWAA FYVYKLQPLTFLLDFSVDGYIRRAIDCGFNDLSQLHCSYESFDVESGVYSVSSFEAKPSGSVVEQAEGVECDFSPLLS GTPPQVYNFKRLVFTNCNYNLTKLLSLFSVNDFTCSQISPAAIASNCYSSLILDYFSYPLSMKSDLSVSSAGPISQFN YKQSFSNPTCLILATVPHNLTTITKPLKYSYINKCSRLLSDDRTEVPQLVNANQYSPCVSIAPSTVWEDGDYYRKQLS PLEGGGWLVASGSTVAMTEQLQMGFGITVQYGTDTNSVCPKLEFANDTKIASQLGNCVEYSLYGVSGRGVFQNCTAVG VRQQRFVYDAYQNLVGYYSDDGNYYCLRACVSVPVSVIYDKETKTHATLFGSVACEHISSTMSQYSRSTRSMLKRRDS TYGPLQTPVGCVLGLVNSSLFVEDCKLPLGQSLCALPDTPSTLTPASVGSVPGEMRLASIAFNHPIQVDQLNSSYFKL SIPTNFSFGVTQEYIQTTIQKVTVDCKQYVCNGFQKCEQLLREYGQFCSKINQALHGANLRQDDSVRNLFASVKSSQS SPIIPGFGGDFNLTLLEPVSISTGSRSARSAIEDLLFDKVTIADPGYMQGYDDCMKQGPASARDLICAQYVAGYKVLP PLMDVNMEAAYTSSLLGSIAGVGWTAGLSSFAAIPFAQSIFYRLNGVGITQQVLSENQKLIANKFNQALGAMQTGFTT TNEAFRKVQDAVNNNAQALSKLASELSNTFGAISASIGDIIQRLDPPEQDAQIDRLINGRLTTLNAFVAQQLVRSESA ALSAQLAKDKVNECVKAQSKRSGFCGQGTHIVSFVVNAPNGLYFMHVGYYPSNHIEVVSAYGLCDAANPTNCIAPVNG YFIKTNNTRIVDEWSYTGSSFYAPEPITSLNTKYVAPQVTYQNISTNLPPPLLGNSTGIDFQDELDEFFKNVSTSIPN FGSLTQINTTLLDLTYEMLSLQQVVKALNESYIDLKELGNYTYYNKWPWYIWLGFIAGLVALALCVFFILCCTGCGTN CMGKLKCNRCCDRYEEYDLEPHKVHVH MERS-CoV S Protein with Inactive Furin Cleavage Site and Lacking Endoplasmic Reticulum (ER) Retention Signal (SEQ ID NO: 94) MIHSVFLLMFLLTPTESYVDVGPDSLKSACIEVDIQQTFFDKTWPRPIDVSKADGIIYPQGRTYSNITITYQGLFPYQ GDHGDMYVYSAGHATGTTPQKLFVANYSQDVKQFANGFVVRIGAAANSTGTVIISPSTSATIRKIYPAFMLGSSVGNF SDGKMGRFFNHTLVLLPDGCGTLLRAFYCILEPRSGNHCPAGNSYTSFATYHTPATDCSDGNYNRNASLNSFKEYFNL RNCTFMYTYNITEDEILEWFGITQTAQGVHLFSSRYVDLYGGNMFQFATLPVYDTIKYYSIIPHSIRSIQSDRKAWAA FYVYKLQPLTFLLDFSVDGYIRRAIDCGFNDLSQLHCSYESFDVESGVYSVSSFEAKPSGSVVEQAEGVECDFSPLLS GTPPQVYNFKRLVFTNCNYNLTKLLSLFSVNDFTCSQISPAAIASNCYSSLILDYFSYPLSMKSDLSVSSAGPISQFN YKQSFSNPTCLILATVPHNLTTITKPLKYSYINKCSRLLSDDRTEVPQLVNANQYSPCVSIAPSTVWEDGDYYRKQLS PLEGGGWLVASGSTVAMTEQLQMGFGITVQYGTDTNSVCPKLEFANDTKIASQLGNCVEYSLYGVSGRGVFQNCTAVG VRQQRFVYDAYQNLVGYYSDDGNYYCLRACVSVPVSVIYDKETKTHATLFGSVACEHISSTMSQYSRSTRSMLKRRDS TYGPLQTPVGCVLGLVNSSLFVEDCKLPLGQSLCALPDTPSTLTPASVGSVPGEMRLASIAFNHPIQVDQLNSSYFKL SIPTNFSFGVTQEYIQTTIQKVTVDCKQYVCNGFQKCEQLLREYGQFCSKINQALHGANLRQDDSVRNLFASVKSSQS SPIIPGFGGDFNLTLLEPVSISTGSRSARSAIEDLLFDKVTIADPGYMQGYDDCMKQGPASARDLICAQYVAGYKVLP PLMDVNMEAAYTSSLLGSIAGVGWTAGLSSFAAIPFAQSIFYRLNGVGITQQVLSENQKLIANKFNQALGAMQTGFTT TNEAFRKVQDAVNNNAQALSKLASELSNTFGAISASIGDIIQRLDPPEQDAQIDRLINGRLTTLNAFVAQQLVRSESA ALSAQLAKDKVNECVKAQSKRSGFCGQGTHIVSFVVNAPNGLYFMHVGYYPSNHIEVVSAYGLCDAANPTNCIAPVNG YFIKTNNTRIVDEWSYTGSSFYAPEPITSLNTKYVAPQVTYQNISTNLPPPLLGNSTGIDFQDELDEFFKNVSTSIPN FGSLTQINTTLLDLTYEMLSLQQVVKALNESYIDLKELGNYTYYNKWPWYIWLGFIAGLVALALCVFFILCCTGCGTN CMGKLKCNRCCDRYEEYDLEP MERS-CoV S Protein with Inactive Furin Cleavage Site and Truncated Cytoplasmic Tail (SEQ ID NO: 95) MIHSVFLLMFLLTPTESYVDVGPDSLKSACIEVDIQQTFFDKTWPRPIDVSKADGIIYPQGRTYSNITITYQGLFPYQ GDHGDMYVYSAGHATGTTPQKLFVANYSQDVKQFANGFVVRIGAAANSTGTVIISPSTSATIRKIYPAFMLGSSVGNF SDGKMGRFFNHTLVLLPDGCGTLLRAFYCILEPRSGNHCPAGNSYTSFATYHTPATDCSDGNYNRNASLNSFKEYFNL RNCTFMYTYNITEDEILEWFGITQTAQGVHLFSSRYVDLYGGNMFQFATLPVYDTIKYYSIIPHSIRSIQSDRKAWAA FYVYKLQPLTFLLDFSVDGYIRRAIDCGFNDLSQLHCSYESFDVESGVYSVSSFEAKPSGSVVEQAEGVECDFSPLLS GTPPQVYNFKRLVFTNCNYNLTKLLSLFSVNDFTCSQISPAAIASNCYSSLILDYFSYPLSMKSDLSVSSAGPISQFN YKQSFSNPTCLILATVPHNLTTITKPLKYSYINKCSRLLSDDRTEVPQLVNANQYSPCVSIAPSTVWEDGDYYRKQLS PLEGGGWLVASGSTVAMTEQLQMGFGITVQYGTDTNSVCPKLEFANDTKIASQLGNCVEYSLYGVSGRGVFQNCTAVG VRQQRFVYDAYQNLVGYYSDDGNYYCLRACVSVPVSVIYDKETKTHATLFGSVACEHISSTMSQYSRSTRSMLKRRDS TYGPLQTPVGCVLGLVNSSLFVEDCKLPLGQSLCALPDTPSTLTPASVGSVPGEMRLASIAFNHPIQVDQLNSSYFKL SIPTNFSFGVTQEYIQTTIQKVTVDCKQYVCNGFQKCEQLLREYGQFCSKINQALHGANLRQDDSVRNLFASVKSSQS SPIIPGFGGDFNLTLLEPVSISTGSRSARSAIEDLLFDKVTIADPGYMQGYDDCMKQGPASARDLICAQYVAGYKVLP PLMDVNMEAAYTSSLLGSIAGVGWTAGLSSFAAIPFAQSIFYRLNGVGITQQVLSENQKLIANKFNQALGAMQTGFTT TNEAFRKVQDAVNNNAQALSKLASELSNTFGAISASIGDIIQRLDPPEQDAQIDRLINGRLTTLNAFVAQQLVRSESA ALSAQLAKDKVNECVKAQSKRSGFCGQGTHIVSFVVNAPNGLYFMHVGYYPSNHIEVVSAYGLCDAANPTNCIAPVNG YFIKTNNTRIVDEWSYTGSSFYAPEPITSLNTKYVAPQVTYQNISTNLPPPLLGNSTGIDFQDELDEFFKNVSTSIPN FGSLTQINTTLLDLTYEMLSLQQVVKALNESYIDLKELGNYTYYNKWPWYIWLGFIAGLVALALCVFFILCCTGCGTN CMGKLKCNRCCD MERS-CoV NTD-RBD-TM Protein (SEQ ID NO: 96) MIHSVFLLMFLLTPTESYVDVGPDSLKSACIEVDIQQTFFDKTWPRPIDVSKADGIIYPQGRTYSNITITYQGLFPYQ GDHGDMYVYSAGHATGTTPQKLFVANYSQDVKQFANGFVVRIGAAANSTGTVIISPSTSATIRKIYPAFMLGSSVGNF SDGKMGRFFNHTLVLLPDGCGTLLRAFYCILEPRSGNHCPAGNSYTSFATYHTPATDCSDGNYNRNASLNSFKEYFNL RNCTFMYTYNITEDEILEWFGITQTAQGVHLFSSRYVDLYGGNMFQFATLPVYDTIKYYSIIPHSIRSIQSDRKAWAA FYVYKLQPLTFLLDFSVDGYIRRAIDGGGSGGGEQAEGVECDFSPLLSGTPPQVYNFKRLVFTNCNYNLTKLLSLFSV NDFTCSQISPAAIASNCYSSLILDYFSYPLSMKSDLSVSSAGPISQFNYKQSFSNPTCLILATVPHNLTTITKPLKYS YINKCSRLLSDDRTEVPQLVNANQYSPCVSIAPSTVWEDGDYYRKQLSPLEGGGWLVASGSTVAMTEQLQMGFGITVQ YGTDTNSVCPKLESGGGSILAIYSTVASSLVLLVSLGAISF MERS-CoV NTD-TM Protein (SEQ ID NO: 97) MIHSVFLLMFLLTPTESYVDVGPDSLKSACIEVDIQQTFFDKTWPRPIDVSKADGIIYPQGRTYSNITITYQGLFPYQ GDHGDMYVYSAGHATGTTPQKLFVANYSQDVKQFANGFVVRIGAAANSTGTVIISPSTSATIRKIYPAFMLGSSVGNF SDGKMGRFFNHTLVLLPDGCGTLLRAFYCILEPRSGNHCPAGNSYTSFATYHTPATDCSDGNYNRNASLNSFKEYFNL RNCTFMYTYNITEDEILEWFGITQTAQGVHLFSSRYVDLYGGNMFQFATLPVYDTIKYYSIIPHSIRSIQSDRKAWAA FYVYKLQPLTFLLDFSVDGYIRRAIDSGGGSILAIYSTVASSLVLLVSLGAISF MERS-CoV RBD-TM Protein (SEQ ID NO: 98) MYSMQLASCVTLTLVLLVNSQEQAEGVECDFSPLLSGTPPQVYNFKRLVFTNCNYNLTKLLSLFSVNDFTCSQISPAA IASNCYSSLILDYFSYPLSMKSDLSVSSAGPISQFNYKQSFSNPTCLILATVPHNLTTITKPLKYSYINKCSRLLSDD RTEVPQLVNANQYSPCVSIAPSTVWEDGDYYRKQLSPLEGGGWLVASGSTVAMTEQLQMGFGITVQYGTDTNSVCPKL ESGGGSILAIYSTVASSLVLLVSLGAISF MERS-CoV S Protein ER Retention Signal (SEQ ID NO: 99) KVHVH MERS-CoV S Protein Cytoplasmic Tail (SEQ ID NO: 100) LCCTGCGTNCMGKLKCNRCCDRYEEYDLEPHKVHVH MERS-CoV S Protein transmembrane (TM) domain (SEQ ID NO: 101) WYIWLGFIAGLVALALCVFFI Influenza A virus hemagglutinin (HA) transmembrane (TM) domain (SEQ ID NO: 102) ILAIYSTVASSLVLLVSLGAISF EQUIVALENTS AND SCOPE While several inventive embodiments have been described and illustrated herein, those of ordinary skill in the art will readily envision a variety of other means and/or structures for performing the function and/or obtaining the results and/or one or more of the advantages described herein, and each of such variations and/or modifications is deemed to be within the scope of the inventive embodiments described herein. More generally, those skilled in the art will readily appreciate that all parameters, dimensions, materials, and configurations described herein are meant to be exemplary and that the actual parameters, dimensions, materials, and/or configurations will depend upon the specific application or applications for which the inventive teachings is/are used. Those skilled in the art will recognize, or be able to ascertain using no more than routine experimentation, many equivalents to the specific inventive embodiments described herein. It is, therefore, to be understood that the foregoing embodiments are presented by way of example only and that, within the scope of the appended claims and equivalents thereto, inventive embodiments may be practiced otherwise than as specifically described and claimed. Inventive embodiments of the present disclosure are directed to each individual feature, system, article, material, kit, and/or method described herein. In addition, any combination of two or more such features, systems, articles, materials, kits, and/or methods, if such features, systems, articles, materials, kits, and/or methods are not mutually inconsistent, is included within the inventive scope of the present disclosure. All definitions, as defined and used herein, should be understood to control over dictionary definitions, definitions in documents incorporated by reference, and/or ordinary meanings of the defined terms. All references, patents and patent applications disclosed herein are incorporated by reference with respect to the subject matter for which each is cited, which in some cases may encompass the entirety of the document. The indefinite articles “a” and “an,” as used herein in the specification and in the claims, unless clearly indicated to the contrary, should be understood to mean “at least one.” The phrase “and/or,” as used herein in the specification and in the claims, should be understood to mean “either or both” of the elements so conjoined, i.e., elements that are conjunctively present in some cases and disjunctively present in other cases. Multiple elements listed with “and/or” should be construed in the same fashion, i.e., “one or more” of the elements so conjoined. Other elements may optionally be present other than the elements specifically identified by the “and/or” clause, whether related or unrelated to those elements specifically identified. Thus, as a non-limiting example, a reference to “A and/or B”, when used in conjunction with open-ended language such as “comprising” can refer, in some embodiments, to A only (optionally including elements other than B); in another embodiment, to B only (optionally including elements other than A); in yet another embodiment, to both A and B (optionally including other elements); etc. As used herein in the specification and in the claims, “or” should be understood to have the same meaning as “and/or” as defined above. For example, when separating items in a list, “or” or “and/or” shall be interpreted as being inclusive, i.e., the inclusion of at least one, but also including more than one, of a number or list of elements, and, optionally, additional unlisted items. Only terms clearly indicated to the contrary, such as “only one of” or “exactly one of,” or, when used in the claims, “consisting of,” will refer to the inclusion of exactly one element of a number or list of elements. In general, the term “or” as used herein shall only be interpreted as indicating exclusive alternatives (i.e. “one or the other but not both”) when preceded by terms of exclusivity, such as “either,” “one of,” “only one of,” or “exactly one of.” “Consisting essentially of,” when used in the claims, shall have its ordinary meaning as used in the field of patent law. As used herein in the specification and in the claims, the phrase “at least one,” in reference to a list of one or more elements, should be understood to mean at least one element selected from any one or more of the elements in the list of elements, but not necessarily including at least one of each and every element specifically listed within the list of elements and not excluding any combinations of elements in the list of elements. This definition also allows that elements may optionally be present other than the elements specifically identified within the list of elements to which the phrase “at least one” refers, whether related or unrelated to those elements specifically identified. Thus, as a non-limiting example, “at least one of A and B” (or, equivalently, “at least one of A or B,” or, equivalently “at least one of A and/or B”) can refer, in some embodiments, to at least one, optionally including more than one, A, with no B present (and optionally including elements other than B); in another embodiment, to at least one, optionally including more than one, B, with no A present (and optionally including elements other than A); in yet another embodiment, to at least one, optionally including more than one, A, and at least one, optionally including more than one, B (and optionally including other elements); etc. Each possibility represents a separate embodiment of the present invention. It should be understood that, unless clearly indicated to the contrary, the disclosure of numerical values and ranges of numerical values in the specification includes both i) the exact value(s) or range specified, and ii) values that are “about” the value(s) or ranges specified (e.g., values or ranges falling within a reasonable range (e.g., about 10% similar)) as would be understood by a person of ordinary skill in the art. It should also be understood that, unless clearly indicated to the contrary, in any methods disclosed herein that include more than one step or act, the order of the steps or acts of the method is not necessarily limited to the order in which the steps or acts of the method are disclosed. In the claims, as well as in the specification above, all transitional phrases such as “comprising,” “including,” “carrying,” “having,” “containing,” “involving,” “holding,” “composed of,” and the like are to be understood to be open-ended, i.e., to mean including but not limited to. Only the transitional phrases “consisting of” and “consisting essentially of” shall be closed or semi-closed transitional phrases, respectively, as set forth in the United States Patent Office Manual of Patent Examining Procedures, Section 2111.03.
Wild-type MERS-CoV (Kingdom of Saudi Arabia (KSA) 2019) Spike (S) Protein Sequence (SEQ ID NO: 84) (GenBank Accession No. QEJ82215.1) MIHSVFLLMFLLTPTESYVDVGPDSLKSACIEVDIQQTFFDKTWPRPIDVSKADGIIYPQGRTYSNITITYQGLFPYQ GDHGDMYVYSAGHATGTTPQKLFVANYSQDVKQFANGFVVRIGAAANSTGTVIISPSTSATIRKIYPAFMLGSSVGNF SDGKMGRFFNHTLVLLPDGCGTLLRAFYCILEPRSGNHCPAGNSYTSFATYHTPATDCSDGNYNRNASLNSFKEYFNL RNCTFMYTYNITEDEILEWFGITQTAQGVHLFSSRYVDLYGGNMFQFATLPVYDTIKYYSIIPHSIRSIQSDRKAWAA FYVYKLQPLTFLLDFSVDGYIRRAIDCGFNDLSQLHCSYESFDVESGVYSVSSFEAKPSGSVVEQAEGVECDFSPLLS GTPPQVYNFKRLVFTNCNYNLTKLLSLFSVNDFTCSQISPAAIASNCYSSLILDYFSYPLSMKSDLSVSSAGPISQFN YKQSFSNPTCLILATVPHNLTTITKPLKYSYINKCSRLLSDDRTEVPQLVNANQYSPCVSIAPSTVWEDGDYYRKQLS PLEGGGWLVASGSTVAMTEQLQMGFGITVQYGTDTNSVCPKLEFANDTKIASQLGNCVEYSLYGVSGRGVFQNCTAVG VRQQRFVYDAYQNLVGYYSDDGNYYCLRACVSVPVSVIYDKETKTHATLFGSVACEHISSTMSQYSRSTRSMLKRRDS TYGPLQTPVGCVLGLVNSSLFVEDCKLPLGQSLCALPDTPSTLTPRSVRSVPGEMRLASIAFNHPIQVDQLNSSYFKL SIPTNFSFGVTQEYIQTTIQKVTVDCKQYVCNGFQKCEQLLREYGQFCSKINQALHGANLRQDDSVRNLFASVKSSQS SPIIPGFGGDFNLTLLEPVSISTGSRSARSAIEDLLFDKVTIADPGYMQGYDDCMKQGPASARDLICAQYVAGYKVLP PLMDVNMEAAYTSSLLGSIAGVGWTAGLSSFAAIPFAQSIFYRLNGVGITQQVLSENQKLIANKFNQALGAMQTGFTT TNEAFRKVQDAVNNNAQALSKLASELSNTFGAISASIGDIIQRLDVLEQDAQIDRLINGRLTTLNAFVAQQLVRSESA ALSAQLAKDKVNECVKAQSKRSGFCGQGTHIVSFVVNAPNGLYFMHVGYYPSNHIEVVSAYGLCDAANPTNCIAPVNG YFIKTNNTRIVDEWSYTGSSFYAPEPITSLNTKYVAPQVTYQNISTNLPPPLLGNSTGIDFQDELDEFFKNVSTSIPN FGSLTQINTTLLDLTYEMLSLQQVVKALNESYIDLKELGNYTYYNKWPWYIWLGFIAGLVALALCVFFILCCTGCGTN CMGKLKCNRCCDRYEEYDLEPHKVHVH Wild-type MERS-CoV (Riyadh 2013) Spike (S) Protein Sequence (SEQ ID NO: 85) MIHSVFLLMFLLTPTESYVDVGPDSVKSACIEVDIQQTFFDKTWPRPIDVSKADGIIYPQGRTYSNITITYQGLFPYQ GDHGDMYVYSAGHATGTTPQKLFVANYSQDVKQFANGFVVRIGAAANSTGTVIISPSTSATIRKIYPAFMLGSSVGNF SDGKMGRFFNHTLVLLPDGCGTLLRAFYCILEPRSGNHCPAGNSYTSFATYHTPATDCSDGNYNRNASLNSFKEYFNL RNCTFMYTYNITEDEILEWFGITQTAQGVHLFSSRYVDLYGGNMFQFATLPVYDTIKYYSIIPHSIRSIQSDRKAWAA FYVYKLQPLTFLLDFSVDGYIRRAIDCGFNDLSQLHCSYESFDVESGVYSVSSFEAKPSGSVVEQAEGVECDFSPLLS GTPPQVYNFKRLVFTNCNYNLTKLLSLFSVNDFTCSQISPAAIASNCYSSLILDYFSYPLSMKSDLSVSSAGPISQFN YKQSFSNPTCLILATVPHNLTTITKPLKYSYINKCSRLLSDDRTEVPQLVNANQYSPCVSIVPSTVWEDGDYYRKQLS PLEGGGWLVASGSTVAMTEQLQMGFGITVQYGTDTNSVCPKLEFANDTKIASQLGNCVEYSLYGVSGRGVFQNCTAVG VRQQRFVYDAYQNLVGYYSDDGNYYCLRACVSVPVSVIYDKETKTHATLFGSVACEHISSTMSQYSRSTRSMLKRRDS TYGPLQTPVGCVLGLVNSSLFVEDCKLPLGQSLCALPDTPSTLTPRSVRSVPGEMRLASIAFNHPIQVDQLNSSYFKL SIPTNFSFGVTQEYIQTTIQKVTVDCKQYVCNGFQKCEQLLREYGQFCSKINQALHGANLRQDDSVRNLFASVKSSQS SPIIPGFGGDFNLTLLEPVSISTGSRSARSAIEDLLFDKVTIADPGYMQGYDDCMQQGPASARDLICAQYVAGYKVLP PLMDVNMEAAYTSSLLGSIAGVGWTAGLSSFAAIPFAQSIFYRLNGVGITQQVLSENQKLIANKFNQALGAMQTGFTT TNEAFRKVQDAVNNNAQALSKLASELSNTFGAISASIGDIIQRLDVLEQDAQIDRLINGRLTTLNAFVAQQLVRSESA ALSAQLAKDKVNECVKAQSKRSGFCGQGTHIVSFVVNAPNGLYFMHVGYYPSNHIEVVSAYGLCDAANPTNCIAPVNG YFIKTNNTRIVDEWSYTGSSFYAPEPITSLNTKYVAPQVTYQNISTNLPPPLLGNSTGIDFQDELDEFFKNVSTSIPN FGSLTQINTTLLDLTYEMLSLQQVVKALNESYIDLKELGNYTYYNKWPWYIWLGFIAGLVALALCVFFILCCTGCGTN CMGKLKCNRCCDRYEEYDLEPHKVHVH Wild-type MERS-CoV (England 2012) Spike (S) Protein Sequence (SEQ ID NO: 86) (GenBank Accession No. YP_007188579.1) MIHSVFLLMFLLTPTESYVDVGPDSVKSACIEVDIQQTFFDKTWPRPIDVSKADGIIYPQGRTYSNITITYQGLFPYQ GDHGDMYVYSAGHATGTTPQKLFVANYSQDVKQFANGFVVRIGAAANSTGTVIISPSTSATIRKIYPAFMLGSSVGNF SDGKMGRFFNHTLVLLPDGCGTLLRAFYCILEPRSGNHCPAGNSYTSFATYHTPATDCSDGNYNRNASLNSFKEYFNL RNCTFMYTYNITEDEILEWFGITQTAQGVHLFSSRYVDLYGGNMFQFATLPVYDTIKYYSIIPHSIRSIQSDRKAWAA FYVYKLQPLTFLLDFSVDGYIRRAIDCGFNDLSQLHCSYESFDVESGVYSVSSFEAKPSGSVVEQAEGVECDFSPLLS GTPPQVYNFKRLVFTNCNYNLTKLLSLFSVNDFTCSQISPAAIASNCYSSLILDYFSYPLSMKSDLSVSSAGPISQFN YKQSFSNPTCLILATVPHNLTTITKPLKYSYINKCSRFLSDDRTEVPQLVNANQYSPCVSIVPSTVWEDGDYYRKQLS PLEGGGWLVASGSTVAMTEQLQMGFGITVQYGTDTNSVCPKLEFANDTKIASQLGNCVEYSLYGVSGRGVFQNCTAVG VRQQRFVYDAYQNLVGYYSDDGNYYCLRACVSVPVSVIYDKETKTHATLFGSVACEHISSTMSQYSRSTRSMLKRRDS TYGPLQTPVGCVLGLVNSSLFVEDCKLPLGQSLCALPDTPSTLTPRSVRSVPGEMRLASIAFNHPIQVDQLNSSYFKL SIPTNFSFGVTQEYIQTTIQKVTVDCKQYVCNGFQKCEQLLREYGQFCSKINQALHGANLRQDDSVRNLFASVKSSQS SPIIPGFGGDFNLTLLEPVSISTGSRSARSAIEDLLFDKVTIADPGYMQGYDDCMQQGPASARDLICAQYVAGYKVLP PLMDVNMEAAYTSSLLGSIAGVGWTAGLSSFAAIPFAQSIFYRLNGVGITQQVLSENQKLIANKFNQALGAMQTGFTT TNEAFHKVQDAVNNNAQALSKLASELSNTFGAISASIGDIIQRLDVLEQDAQIDRLINGRLTTLNAFVAQQLVRSESA ALSAQLAKDKVNECVKAQSKRSGFCGQGTHIVSFVVNAPNGLYFMHVGYYPSNHIEVVSAYGLCDAANPTNCIAPVNG YFIKTNNTRIVDEWSYTGSSFYAPEPITSLNTKYVAPQVTYQNISTNLPPPLLGNSTGIDFQDELDEFFKNVSTSIPN FGSLTQINTTLLDLTYEMLSLQQVVKALNESYIDLKELGNYTYYNKWPWYIWLGFIAGLVALALCVFFILCCTGCGTN CMGKLKCNRCCDRYEEYDLEPHKVHVH MERS-CoV (KSA 2019) Spike (S) Protein N-terminal Domain (NTD) (SEQ ID NO: 87) YVDVGPDSLKSACIEVDIQQTFFDKTWPRPIDVSKADGIIYPQGRTYSNITITYQGLFPYQGDHGDMYVYSAGHATGT TPQKLFVANYSQDVKQFANGFVVRIGAAANSTGTVIISPSTSATIRKIYPAFMLGSSVGNFSDGKMGRFFNHTLVLLP DGCGTLLRAFYCILEPRSGNHCPAGNSYTSFATYHTPATDCSDGNYNRNASLNSFKEYFNLRNCTFMYTYNITEDEIL EWFGITQTAQGVHLFSSRYVDLYGGNMFQFATLPVYDTIKYYSIIPHSIRSIQSDRKAWAAFYVYKLQPLTFLLDFSV DGYIRRAID MERS-CoV (KSA 2019) Spike (S) Protein Receptor-binding Domain (RBD) (SEQ ID NO: 88) EQAEGVECDFSPLLSGTPPQVYNFKRLVFTNCNYNLTKLLSLFSVNDFTCSQISPAAIASNCYSSLILDYFSYPLSMK SDLSVSSAGPISQFNYKQSFSNPTCLILATVPHNLTTITKPLKYSYINKCSRLLSDDRTEVPQLVNANQYSPCVSIAP STVWEDGDYYRKQLSPLEGGGWLVASGSTVAMTEQLQMGFGITVQYGTDTNSVCPKLE 2P-Stabilized MERS-CoV S Protein Sequence (SEQ ID NO: 89) MIHSVFLLMFLLTPTESYVDVGPDSLKSACIEVDIQQTFFDKTWPRPIDVSKADGIIYPQGRTYSNITITYQGLFPYQ GDHGDMYVYSAGHATGTTPQKLFVANYSQDVKQFANGFVVRIGAAANSTGTVIISPSTSATIRKIYPAFMLGSSVGNF SDGKMGRFFNHTLVLLPDGCGTLLRAFYCILEPRSGNHCPAGNSYTSFATYHTPATDCSDGNYNRNASLNSFKEYFNL RNCTFMYTYNITEDEILEWFGITQTAQGVHLFSSRYVDLYGGNMFQFATLPVYDTIKYYSIIPHSIRSIQSDRKAWAA FYVYKLQPLTFLLDFSVDGYIRRAIDCGFNDLSQLHCSYESFDVESGVYSVSSFEAKPSGSVVEQAEGVECDFSPLLS GTPPQVYNFKRLVFTNCNYNLTKLLSLFSVNDFTCSQISPAAIASNCYSSLILDYFSYPLSMKSDLSVSSAGPISQFN YKQSFSNPTCLILATVPHNLTTITKPLKYSYINKCSRLLSDDRTEVPQLVNANQYSPCVSIAPSTVWEDGDYYRKQLS PLEGGGWLVASGSTVAMTEQLQMGFGITVQYGTDTNSVCPKLEFANDTKIASQLGNCVEYSLYGVSGRGVFQNCTAVG VRQQRFVYDAYQNLVGYYSDDGNYYCLRACVSVPVSVIYDKETKTHATLFGSVACEHISSTMSQYSRSTRSMLKRRDS TYGPLQTPVGCVLGLVNSSLFVEDCKLPLGQSLCALPDTPSTLTPRSVRSVPGEMRLASIAFNHPIQVDQLNSSYFKL SIPTNFSFGVTQEYIQTTIQKVTVDCKQYVCNGFQKCEQLLREYGQFCSKINQALHGANLRQDDSVRNLFASVKSSQS SPIIPGFGGDFNLTLLEPVSISTGSRSARSAIEDLLFDKVTIADPGYMQGYDDCMKQGPASARDLICAQYVAGYKVLP PLMDVNMEAAYTSSLLGSIAGVGWTAGLSSFAAIPFAQSIFYRLNGVGITQQVLSENQKLIANKFNQALGAMQTGFTT TNEAFRKVQDAVNNNAQALSKLASELSNTFGAISASIGDIIQRLDPPEQDAQIDRLINGRLTTLNAFVAQQLVRSESA ALSAQLAKDKVNECVKAQSKRSGFCGQGTHIVSFVVNAPNGLYFMHVGYYPSNHIEVVSAYGLCDAANPTNCIAPVNG YFIKTNNTRIVDEWSYTGSSFYAPEPITSLNTKYVAPQVTYQNISTNLPPPLLGNSTGIDFQDELDEFFKNVSTSIPN FGSLTQINTTLLDLTYEMLSLQQVVKALNESYIDLKELGNYTYYNKWPWYIWLGFIAGLVALALCVFFILCCTGCGTN CMGKLKCNRCCDRYEEYDLEPHKVHVH MERS-CoV S Protein with Inactive Furin Cleavage Site (SEQ ID NO: 90) MIHSVFLLMFLLTPTESYVDVGPDSLKSACIEVDIQQTFFDKTWPRPIDVSKADGIIYPQGRTYSNITITYQGLFPYQ GDHGDMYVYSAGHATGTTPQKLFVANYSQDVKQFANGFVVRIGAAANSTGTVIISPSTSATIRKIYPAFMLGSSVGNF SDGKMGRFFNHTLVLLPDGCGTLLRAFYCILEPRSGNHCPAGNSYTSFATYHTPATDCSDGNYNRNASLNSFKEYFNL RNCTFMYTYNITEDEILEWFGITQTAQGVHLFSSRYVDLYGGNMFQFATLPVYDTIKYYSIIPHSIRSIQSDRKAWAA FYVYKLQPLTFLLDFSVDGYIRRAIDCGFNDLSQLHCSYESFDVESGVYSVSSFEAKPSGSVVEQAEGVECDFSPLLS GTPPQVYNFKRLVFTNCNYNLTKLLSLFSVNDFTCSQISPAAIASNCYSSLILDYFSYPLSMKSDLSVSSAGPISQFN YKQSFSNPTCLILATVPHNLTTITKPLKYSYINKCSRLLSDDRTEVPQLVNANQYSPCVSIAPSTVWEDGDYYRKQLS PLEGGGWLVASGSTVAMTEQLQMGFGITVQYGTDTNSVCPKLEFANDTKIASQLGNCVEYSLYGVSGRGVFQNCTAVG VRQQRFVYDAYQNLVGYYSDDGNYYCLRACVSVPVSVIYDKETKTHATLFGSVACEHISSTMSQYSRSTRSMLKRRDS TYGPLQTPVGCVLGLVNSSLFVEDCKLPLGQSLCALPDTPSTLTPASVGSVPGEMRLASIAFNHPIQVDQLNSSYFKL SIPTNFSFGVTQEYIQTTIQKVTVDCKQYVCNGFQKCEQLLREYGQFCSKINQALHGANLRQDDSVRNLFASVKSSQS SPIIPGFGGDFNLTLLEPVSISTGSRSARSAIEDLLFDKVTIADPGYMQGYDDCMKQGPASARDLICAQYVAGYKVLP PLMDVNMEAAYTSSLLGSIAGVGWTAGLSSFAAIPFAQSIFYRLNGVGITQQVLSENQKLIANKFNQALGAMQTGFTT TNEAFRKVQDAVNNNAQALSKLASELSNTFGAISASIGDIIQRLDVLEQDAQIDRLINGRLTTLNAFVAQQLVRSESA ALSAQLAKDKVNECVKAQSKRSGFCGQGTHIVSFVVNAPNGLYFMHVGYYPSNHIEVVSAYGLCDAANPTNCIAPVNG YFIKTNNTRIVDEWSYTGSSFYAPEPITSLNTKYVAPQVTYQNISTNLPPPLLGNSTGIDFQDELDEFFKNVSTSIPN FGSLTQINTTLLDLTYEMLSLQQVVKALNESYIDLKELGNYTYYNKWPWYIWLGFIAGLVALALCVFFILCCTGCGTN CMGKLKCNRCCDRYEEYDLEPHKVHVH MERS-CoV S Protein Lacking Endoplasmic Reticulum (ER) Retention Signal (SEQ ID NO: 91) MIHSVFLLMFLLTPTESYVDVGPDSLKSACIEVDIQQTFFDKTWPRPIDVSKADGIIYPQGRTYSNITITYQGLFPYQ GDHGDMYVYSAGHATGTTPQKLFVANYSQDVKQFANGFVVRIGAAANSTGTVIISPSTSATIRKIYPAFMLGSSVGNF SDGKMGRFFNHTLVLLPDGCGTLLRAFYCILEPRSGNHCPAGNSYTSFATYHTPATDCSDGNYNRNASLNSFKEYFNL RNCTFMYTYNITEDEILEWFGITQTAQGVHLFSSRYVDLYGGNMFQFATLPVYDTIKYYSIIPHSIRSIQSDRKAWAA FYVYKLQPLTFLLDFSVDGYIRRAIDCGFNDLSQLHCSYESFDVESGVYSVSSFEAKPSGSVVEQAEGVECDFSPLLS GTPPQVYNFKRLVFTNCNYNLTKLLSLFSVNDFTCSQISPAAIASNCYSSLILDYFSYPLSMKSDLSVSSAGPISQFN YKQSFSNPTCLILATVPHNLTTITKPLKYSYINKCSRLLSDDRTEVPQLVNANQYSPCVSIAPSTVWEDGDYYRKQLS PLEGGGWLVASGSTVAMTEQLQMGFGITVQYGTDTNSVCPKLEFANDTKIASQLGNCVEYSLYGVSGRGVFQNCTAVG VRQQRFVYDAYQNLVGYYSDDGNYYCLRACVSVPVSVIYDKETKTHATLFGSVACEHISSTMSQYSRSTRSMLKRRDS TYGPLQTPVGCVLGLVNSSLFVEDCKLPLGQSLCALPDTPSTLTPRSVRSVPGEMRLASIAFNHPIQVDQLNSSYFKL SIPTNFSFGVTQEYIQTTIQKVTVDCKQYVCNGFQKCEQLLREYGQFCSKINQALHGANLRQDDSVRNLFASVKSSQS SPIIPGFGGDFNLTLLEPVSISTGSRSARSAIEDLLFDKVTIADPGYMQGYDDCMKQGPASARDLICAQYVAGYKVLP PLMDVNMEAAYTSSLLGSIAGVGWTAGLSSFAAIPFAQSIFYRLNGVGITQQVLSENQKLIANKFNQALGAMQTGFTT TNEAFRKVQDAVNNNAQALSKLASELSNTFGAISASIGDIIQRLDVLEQDAQIDRLINGRLTTLNAFVAQQLVRSESA ALSAQLAKDKVNECVKAQSKRSGFCGQGTHIVSFVVNAPNGLYFMHVGYYPSNHIEVVSAYGLCDAANPTNCIAPVNG YFIKTNNTRIVDEWSYTGSSFYAPEPITSLNTKYVAPQVTYQNISTNLPPPLLGNSTGIDFQDELDEFFKNVSTSIPN FGSLTQINTTLLDLTYEMLSLQQVVKALNESYIDLKELGNYTYYNKWPWYIWLGFIAGLVALALCVFFILCCTGCGTN CMGKLKCNRCCDRYEEYDLEP MERS-CoV S Protein with Truncated Cytoplasmic Tail (SEQ ID NO: 92) MIHSVFLLMFLLTPTESYVDVGPDSLKSACIEVDIQQTFFDKTWPRPIDVSKADGIIYPQGRTYSNITITYQGLFPYQ GDHGDMYVYSAGHATGTTPQKLFVANYSQDVKQFANGFVVRIGAAANSTGTVIISPSTSATIRKIYPAFMLGSSVGNF SDGKMGRFFNHTLVLLPDGCGTLLRAFYCILEPRSGNHCPAGNSYTSFATYHTPATDCSDGNYNRNASLNSFKEYFNL RNCTFMYTYNITEDEILEWFGITQTAQGVHLFSSRYVDLYGGNMFQFATLPVYDTIKYYSIIPHSIRSIQSDRKAWAA FYVYKLQPLTFLLDFSVDGYIRRAIDCGFNDLSQLHCSYESFDVESGVYSVSSFEAKPSGSVVEQAEGVECDFSPLLS GTPPQVYNFKRLVFTNCNYNLTKLLSLFSVNDFTCSQISPAAIASNCYSSLILDYFSYPLSMKSDLSVSSAGPISQFN YKQSFSNPTCLILATVPHNLTTITKPLKYSYINKCSRLLSDDRTEVPQLVNANQYSPCVSIAPSTVWEDGDYYRKQLS PLEGGGWLVASGSTVAMTEQLQMGFGITVQYGTDTNSVCPKLEFANDTKIASQLGNCVEYSLYGVSGRGVFQNCTAVG VRQQRFVYDAYQNLVGYYSDDGNYYCLRACVSVPVSVIYDKETKTHATLFGSVACEHISSTMSQYSRSTRSMLKRRDS TYGPLQTPVGCVLGLVNSSLFVEDCKLPLGQSLCALPDTPSTLTPRSVRSVPGEMRLASIAFNHPIQVDQLNSSYFKL SIPTNFSFGVTQEYIQTTIQKVTVDCKQYVCNGFQKCEQLLREYGQFCSKINQALHGANLRQDDSVRNLFASVKSSQS SPIIPGFGGDFNLTLLEPVSISTGSRSARSAIEDLLFDKVTIADPGYMQGYDDCMKQGPASARDLICAQYVAGYKVLP PLMDVNMEAAYTSSLLGSIAGVGWTAGLSSFAAIPFAQSIFYRLNGVGITQQVLSENQKLIANKFNQALGAMQTGFTT TNEAFRKVQDAVNNNAQALSKLASELSNTFGAISASIGDIIQRLDVLEQDAQIDRLINGRLTTLNAFVAQQLVRSESA ALSAQLAKDKVNECVKAQSKRSGFCGQGTHIVSFVVNAPNGLYFMHVGYYPSNHIEVVSAYGLCDAANPTNCIAPVNG YFIKTNNTRIVDEWSYTGSSFYAPEPITSLNTKYVAPQVTYQNISTNLPPPLLGNSTGIDFQDELDEFFKNVSTSIPN FGSLTQINTTLLDLTYEMLSLQQVVKALNESYIDLKELGNYTYYNKWPWYIWLGFIAGLVALALCVFFILCCTGCGTN CMGKLKCNRCCD 2P-Stabilized MERS-CoV S Protein with Inactive Furin Cleavage Site (SEQ ID NO: 93) MIHSVFLLMFLLTPTESYVDVGPDSLKSACIEVDIQQTFFDKTWPRPIDVSKADGIIYPQGRTYSNITITYQGLFPYQ GDHGDMYVYSAGHATGTTPQKLFVANYSQDVKQFANGFVVRIGAAANSTGTVIISPSTSATIRKIYPAFMLGSSVGNF SDGKMGRFFNHTLVLLPDGCGTLLRAFYCILEPRSGNHCPAGNSYTSFATYHTPATDCSDGNYNRNASLNSFKEYFNL RNCTFMYTYNITEDEILEWFGITQTAQGVHLFSSRYVDLYGGNMFQFATLPVYDTIKYYSIIPHSIRSIQSDRKAWAA FYVYKLQPLTFLLDFSVDGYIRRAIDCGFNDLSQLHCSYESFDVESGVYSVSSFEAKPSGSVVEQAEGVECDFSPLLS GTPPQVYNFKRLVFTNCNYNLTKLLSLFSVNDFTCSQISPAAIASNCYSSLILDYFSYPLSMKSDLSVSSAGPISQFN YKQSFSNPTCLILATVPHNLTTITKPLKYSYINKCSRLLSDDRTEVPQLVNANQYSPCVSIAPSTVWEDGDYYRKQLS PLEGGGWLVASGSTVAMTEQLQMGFGITVQYGTDTNSVCPKLEFANDTKIASQLGNCVEYSLYGVSGRGVFQNCTAVG VRQQRFVYDAYQNLVGYYSDDGNYYCLRACVSVPVSVIYDKETKTHATLFGSVACEHISSTMSQYSRSTRSMLKRRDS TYGPLQTPVGCVLGLVNSSLFVEDCKLPLGQSLCALPDTPSTLTPASVGSVPGEMRLASIAFNHPIQVDQLNSSYFKL SIPTNFSFGVTQEYIQTTIQKVTVDCKQYVCNGFQKCEQLLREYGQFCSKINQALHGANLRQDDSVRNLFASVKSSQS SPIIPGFGGDFNLTLLEPVSISTGSRSARSAIEDLLFDKVTIADPGYMQGYDDCMKQGPASARDLICAQYVAGYKVLP PLMDVNMEAAYTSSLLGSIAGVGWTAGLSSFAAIPFAQSIFYRLNGVGITQQVLSENQKLIANKFNQALGAMQTGFTT TNEAFRKVQDAVNNNAQALSKLASELSNTFGAISASIGDIIQRLDPPEQDAQIDRLINGRLTTLNAFVAQQLVRSESA ALSAQLAKDKVNECVKAQSKRSGFCGQGTHIVSFVVNAPNGLYFMHVGYYPSNHIEVVSAYGLCDAANPTNCIAPVNG YFIKTNNTRIVDEWSYTGSSFYAPEPITSLNTKYVAPQVTYQNISTNLPPPLLGNSTGIDFQDELDEFFKNVSTSIPN FGSLTQINTTLLDLTYEMLSLQQVVKALNESYIDLKELGNYTYYNKWPWYIWLGFIAGLVALALCVFFILCCTGCGTN CMGKLKCNRCCDRYEEYDLEPHKVHVH MERS-CoV S Protein with Inactive Furin Cleavage Site and Lacking Endoplasmic Reticulum (ER) Retention Signal (SEQ ID NO: 94) MIHSVFLLMFLLTPTESYVDVGPDSLKSACIEVDIQQTFFDKTWPRPIDVSKADGIIYPQGRTYSNITITYQGLFPYQ GDHGDMYVYSAGHATGTTPQKLFVANYSQDVKQFANGFVVRIGAAANSTGTVIISPSTSATIRKIYPAFMLGSSVGNF SDGKMGRFFNHTLVLLPDGCGTLLRAFYCILEPRSGNHCPAGNSYTSFATYHTPATDCSDGNYNRNASLNSFKEYFNL RNCTFMYTYNITEDEILEWFGITQTAQGVHLFSSRYVDLYGGNMFQFATLPVYDTIKYYSIIPHSIRSIQSDRKAWAA FYVYKLQPLTFLLDFSVDGYIRRAIDCGFNDLSQLHCSYESFDVESGVYSVSSFEAKPSGSVVEQAEGVECDFSPLLS GTPPQVYNFKRLVFTNCNYNLTKLLSLFSVNDFTCSQISPAAIASNCYSSLILDYFSYPLSMKSDLSVSSAGPISQFN YKQSFSNPTCLILATVPHNLTTITKPLKYSYINKCSRLLSDDRTEVPQLVNANQYSPCVSIAPSTVWEDGDYYRKQLS PLEGGGWLVASGSTVAMTEQLQMGFGITVQYGTDTNSVCPKLEFANDTKIASQLGNCVEYSLYGVSGRGVFQNCTAVG VRQQRFVYDAYQNLVGYYSDDGNYYCLRACVSVPVSVIYDKETKTHATLFGSVACEHISSTMSQYSRSTRSMLKRRDS TYGPLQTPVGCVLGLVNSSLFVEDCKLPLGQSLCALPDTPSTLTPASVGSVPGEMRLASIAFNHPIQVDQLNSSYFKL SIPTNFSFGVTQEYIQTTIQKVTVDCKQYVCNGFQKCEQLLREYGQFCSKINQALHGANLRQDDSVRNLFASVKSSQS SPIIPGFGGDFNLTLLEPVSISTGSRSARSAIEDLLFDKVTIADPGYMQGYDDCMKQGPASARDLICAQYVAGYKVLP PLMDVNMEAAYTSSLLGSIAGVGWTAGLSSFAAIPFAQSIFYRLNGVGITQQVLSENQKLIANKFNQALGAMQTGFTT TNEAFRKVQDAVNNNAQALSKLASELSNTFGAISASIGDIIQRLDPPEQDAQIDRLINGRLTTLNAFVAQQLVRSESA ALSAQLAKDKVNECVKAQSKRSGFCGQGTHIVSFVVNAPNGLYFMHVGYYPSNHIEVVSAYGLCDAANPTNCIAPVNG YFIKTNNTRIVDEWSYTGSSFYAPEPITSLNTKYVAPQVTYQNISTNLPPPLLGNSTGIDFQDELDEFFKNVSTSIPN FGSLTQINTTLLDLTYEMLSLQQVVKALNESYIDLKELGNYTYYNKWPWYIWLGFIAGLVALALCVFFILCCTGCGTN CMGKLKCNRCCDRYEEYDLEP MERS-CoV S Protein with Inactive Furin Cleavage Site and Truncated Cytoplasmic Tail (SEQ ID NO: 95) MIHSVFLLMFLLTPTESYVDVGPDSLKSACIEVDIQQTFFDKTWPRPIDVSKADGIIYPQGRTYSNITITYQGLFPYQ GDHGDMYVYSAGHATGTTPQKLFVANYSQDVKQFANGFVVRIGAAANSTGTVIISPSTSATIRKIYPAFMLGSSVGNF SDGKMGRFFNHTLVLLPDGCGTLLRAFYCILEPRSGNHCPAGNSYTSFATYHTPATDCSDGNYNRNASLNSFKEYFNL RNCTFMYTYNITEDEILEWFGITQTAQGVHLFSSRYVDLYGGNMFQFATLPVYDTIKYYSIIPHSIRSIQSDRKAWAA FYVYKLQPLTFLLDFSVDGYIRRAIDCGFNDLSQLHCSYESFDVESGVYSVSSFEAKPSGSVVEQAEGVECDFSPLLS GTPPQVYNFKRLVFTNCNYNLTKLLSLFSVNDFTCSQISPAAIASNCYSSLILDYFSYPLSMKSDLSVSSAGPISQFN YKQSFSNPTCLILATVPHNLTTITKPLKYSYINKCSRLLSDDRTEVPQLVNANQYSPCVSIAPSTVWEDGDYYRKQLS PLEGGGWLVASGSTVAMTEQLQMGFGITVQYGTDTNSVCPKLEFANDTKIASQLGNCVEYSLYGVSGRGVFQNCTAVG VRQQRFVYDAYQNLVGYYSDDGNYYCLRACVSVPVSVIYDKETKTHATLFGSVACEHISSTMSQYSRSTRSMLKRRDS TYGPLQTPVGCVLGLVNSSLFVEDCKLPLGQSLCALPDTPSTLTPASVGSVPGEMRLASIAFNHPIQVDQLNSSYFKL SIPTNFSFGVTQEYIQTTIQKVTVDCKQYVCNGFQKCEQLLREYGQFCSKINQALHGANLRQDDSVRNLFASVKSSQS SPIIPGFGGDFNLTLLEPVSISTGSRSARSAIEDLLFDKVTIADPGYMQGYDDCMKQGPASARDLICAQYVAGYKVLP PLMDVNMEAAYTSSLLGSIAGVGWTAGLSSFAAIPFAQSIFYRLNGVGITQQVLSENQKLIANKFNQALGAMQTGFTT TNEAFRKVQDAVNNNAQALSKLASELSNTFGAISASIGDIIQRLDPPEQDAQIDRLINGRLTTLNAFVAQQLVRSESA ALSAQLAKDKVNECVKAQSKRSGFCGQGTHIVSFVVNAPNGLYFMHVGYYPSNHIEVVSAYGLCDAANPTNCIAPVNG YFIKTNNTRIVDEWSYTGSSFYAPEPITSLNTKYVAPQVTYQNISTNLPPPLLGNSTGIDFQDELDEFFKNVSTSIPN FGSLTQINTTLLDLTYEMLSLQQVVKALNESYIDLKELGNYTYYNKWPWYIWLGFIAGLVALALCVFFILCCTGCGTN CMGKLKCNRCCD MERS-CoV NTD-RBD-TM Protein (SEQ ID NO: 96) MIHSVFLLMFLLTPTESYVDVGPDSLKSACIEVDIQQTFFDKTWPRPIDVSKADGIIYPQGRTYSNITITYQGLFPYQ GDHGDMYVYSAGHATGTTPQKLFVANYSQDVKQFANGFVVRIGAAANSTGTVIISPSTSATIRKIYPAFMLGSSVGNF SDGKMGRFFNHTLVLLPDGCGTLLRAFYCILEPRSGNHCPAGNSYTSFATYHTPATDCSDGNYNRNASLNSFKEYFNL RNCTFMYTYNITEDEILEWFGITQTAQGVHLFSSRYVDLYGGNMFQFATLPVYDTIKYYSIIPHSIRSIQSDRKAWAA FYVYKLQPLTFLLDFSVDGYIRRAIDGGGSGGGEQAEGVECDFSPLLSGTPPQVYNFKRLVFTNCNYNLTKLLSLFSV NDFTCSQISPAAIASNCYSSLILDYFSYPLSMKSDLSVSSAGPISQFNYKQSFSNPTCLILATVPHNLTTITKPLKYS YINKCSRLLSDDRTEVPQLVNANQYSPCVSIAPSTVWEDGDYYRKQLSPLEGGGWLVASGSTVAMTEQLQMGFGITVQ YGTDTNSVCPKLESGGGSILAIYSTVASSLVLLVSLGAISF MERS-CoV NTD-TM Protein (SEQ ID NO: 97) MIHSVFLLMFLLTPTESYVDVGPDSLKSACIEVDIQQTFFDKTWPRPIDVSKADGIIYPQGRTYSNITITYQGLFPYQ GDHGDMYVYSAGHATGTTPQKLFVANYSQDVKQFANGFVVRIGAAANSTGTVIISPSTSATIRKIYPAFMLGSSVGNF SDGKMGRFFNHTLVLLPDGCGTLLRAFYCILEPRSGNHCPAGNSYTSFATYHTPATDCSDGNYNRNASLNSFKEYFNL RNCTFMYTYNITEDEILEWFGITQTAQGVHLFSSRYVDLYGGNMFQFATLPVYDTIKYYSIIPHSIRSIQSDRKAWAA FYVYKLQPLTFLLDFSVDGYIRRAIDSGGGSILAIYSTVASSLVLLVSLGAISF MERS-CoV RBD-TM Protein (SEQ ID NO: 98) MYSMQLASCVTLTLVLLVNSQEQAEGVECDFSPLLSGTPPQVYNFKRLVFTNCNYNLTKLLSLFSVNDFTCSQISPAA IASNCYSSLILDYFSYPLSMKSDLSVSSAGPISQFNYKQSFSNPTCLILATVPHNLTTITKPLKYSYINKCSRLLSDD RTEVPQLVNANQYSPCVSIAPSTVWEDGDYYRKQLSPLEGGGWLVASGSTVAMTEQLQMGFGITVQYGTDTNSVCPKL ESGGGSILAIYSTVASSLVLLVSLGAISF MERS-CoV S Protein ER Retention Signal (SEQ ID NO: 99) KVHVH MERS-CoV S Protein Cytoplasmic Tail (SEQ ID NO: 100) LCCTGCGTNCMGKLKCNRCCDRYEEYDLEPHKVHVH MERS-CoV S Protein transmembrane (TM) domain (SEQ ID NO: 101) WYIWLGFIAGLVALALCVFFI Influenza A virus hemagglutinin (HA) transmembrane (TM) domain (SEQ ID NO: 102) ILAIYSTVASSLVLLVSLGAISF EQUIVALENTS AND SCOPE While several inventive embodiments have been described and illustrated herein, those of ordinary skill in the art will readily envision a variety of other means and/or structures for performing the function and/or obtaining the results and/or one or more of the advantages described herein, and each of such variations and/or modifications is deemed to be within the scope of the inventive embodiments described herein. More generally, those skilled in the art will readily appreciate that all parameters, dimensions, materials, and configurations described herein are meant to be exemplary and that the actual parameters, dimensions, materials, and/or configurations will depend upon the specific application or applications for which the inventive teachings is/are used. Those skilled in the art will recognize, or be able to ascertain using no more than routine experimentation, many equivalents to the specific inventive embodiments described herein. It is, therefore, to be understood that the foregoing embodiments are presented by way of example only and that, within the scope of the appended claims and equivalents thereto, inventive embodiments may be practiced otherwise than as specifically described and claimed. Inventive embodiments of the present disclosure are directed to each individual feature, system, article, material, kit, and/or method described herein. In addition, any combination of two or more such features, systems, articles, materials, kits, and/or methods, if such features, systems, articles, materials, kits, and/or methods are not mutually inconsistent, is included within the inventive scope of the present disclosure. All definitions, as defined and used herein, should be understood to control over dictionary definitions, definitions in documents incorporated by reference, and/or ordinary meanings of the defined terms. All references, patents and patent applications disclosed herein are incorporated by reference with respect to the subject matter for which each is cited, which in some cases may encompass the entirety of the document. The indefinite articles “a” and “an,” as used herein in the specification and in the claims, unless clearly indicated to the contrary, should be understood to mean “at least one.” The phrase “and/or,” as used herein in the specification and in the claims, should be understood to mean “either or both” of the elements so conjoined, i.e., elements that are conjunctively present in some cases and disjunctively present in other cases. Multiple elements listed with “and/or” should be construed in the same fashion, i.e., “one or more” of the elements so conjoined. Other elements may optionally be present other than the elements specifically identified by the “and/or” clause, whether related or unrelated to those elements specifically identified. Thus, as a non-limiting example, a reference to “A and/or B”, when used in conjunction with open-ended language such as “comprising” can refer, in some embodiments, to A only (optionally including elements other than B); in another embodiment, to B only (optionally including elements other than A); in yet another embodiment, to both A and B (optionally including other elements); etc. As used herein in the specification and in the claims, “or” should be understood to have the same meaning as “and/or” as defined above. For example, when separating items in a list, “or” or “and/or” shall be interpreted as being inclusive, i.e., the inclusion of at least one, but also including more than one, of a number or list of elements, and, optionally, additional unlisted items. Only terms clearly indicated to the contrary, such as “only one of” or “exactly one of,” or, when used in the claims, “consisting of,” will refer to the inclusion of exactly one element of a number or list of elements. In general, the term “or” as used herein shall only be interpreted as indicating exclusive alternatives (i.e. “one or the other but not both”) when preceded by terms of exclusivity, such as “either,” “one of,” “only one of,” or “exactly one of.” “Consisting essentially of,” when used in the claims, shall have its ordinary meaning as used in the field of patent law. As used herein in the specification and in the claims, the phrase “at least one,” in reference to a list of one or more elements, should be understood to mean at least one element selected from any one or more of the elements in the list of elements, but not necessarily including at least one of each and every element specifically listed within the list of elements and not excluding any combinations of elements in the list of elements. This definition also allows that elements may optionally be present other than the elements specifically identified within the list of elements to which the phrase “at least one” refers, whether related or unrelated to those elements specifically identified. Thus, as a non-limiting example, “at least one of A and B” (or, equivalently, “at least one of A or B,” or, equivalently “at least one of A and/or B”) can refer, in some embodiments, to at least one, optionally including more than one, A, with no B present (and optionally including elements other than B); in another embodiment, to at least one, optionally including more than one, B, with no A present (and optionally including elements other than A); in yet another embodiment, to at least one, optionally including more than one, A, and at least one, optionally including more than one, B (and optionally including other elements); etc. Each possibility represents a separate embodiment of the present invention. It should be understood that, unless clearly indicated to the contrary, the disclosure of numerical values and ranges of numerical values in the specification includes both i) the exact value(s) or range specified, and ii) values that are “about” the value(s) or ranges specified (e.g., values or ranges falling within a reasonable range (e.g., about 10% similar)) as would be understood by a person of ordinary skill in the art. It should also be understood that, unless clearly indicated to the contrary, in any methods disclosed herein that include more than one step or act, the order of the steps or acts of the method is not necessarily limited to the order in which the steps or acts of the method are disclosed. In the claims, as well as in the specification above, all transitional phrases such as “comprising,” “including,” “carrying,” “having,” “containing,” “involving,” “holding,” “composed of,” and the like are to be understood to be open-ended, i.e., to mean including but not limited to. Only the transitional phrases “consisting of” and “consisting essentially of” shall be closed or semi-closed transitional phrases, respectively, as set forth in the United States Patent Office Manual of Patent Examining Procedures, Section 2111.03.





































































































































Claims
CLAIMS What is claimed is: 1. A composition comprising a lipid delivery vehicle and a messenger ribonucleic acid (mRNA) comprising an open reading frame encoding a Middle East Respiratory Syndrome (MERS)-CoV fusion protein comprising: (a) an N-terminal domain (NTD) of a full-length MERS-CoV Spike (S) protein; and (b) a receptor-binding domain (RBD) of the full-length MERS-CoV-S protein, wherein the MERS-CoV fusion protein does not comprise an NTD or an RBD of an S protein of a second coronavirus that is not MERS-CoV.
2. The composition of claim 1, wherein the NTD and RBD are connected by a linker.
3. The composition of claim 2, wherein the linker is a glycine linker or a glycine-serine linker.
4. The composition of claim 2, wherein the linker comprises a pan-HLA-DR-binding epitope (PADRE).
5. The composition of any one of claims 1–4, wherein the MERS-CoV fusion protein further comprises a transmembrane (TM) domain.
6. The composition of claim 5, wherein the TM domain is a MERS-CoV S protein TM domain or derivative thereof.
7. The composition of claim 5, wherein the TM domain is heterologous to MERS-CoV.
8. The composition of claim 5 or 7, wherein the TM domain is an influenza virus hemagglutinin (HA) TM domain or derivative thereof.
9. The composition of any one of claims 5–8, wherein the MERS-CoV fusion protein comprises, in N-to-C-terminal order, the NTD, the RBD, and the TM domain.
10. The composition of any one of claim 1–4, wherein the MERS-CoV fusion protein does not comprise a transmembrane (TM) domain.
11. The composition of any one of claims 1–4 or 10, wherein the MERS-CoV fusion protein is soluble.
12. The composition of any one of claims 1–11, wherein the MERS-CoV fusion protein further comprises a signal peptide.
13. The composition of claim 12, wherein the signal peptide is a MERS-CoV S protein signal peptide.
14. The composition of claim 12, wherein the signal peptide is heterologous to MERS-CoV.
15. A composition comprising a lipid delivery vehicle and a messenger ribonucleic acid (mRNA) comprising an open reading frame encoding a mutant Middle East Respiratory Syndrome (MERS)-CoV S protein, wherein the mutant MERS-CoV S protein comprises (a) one or more proline substitutions or insertions, and (b) one or more substitutions or deletions in a furin cleavage site, relative to a full-length MERS-CoV S protein.
16 The composition of claim 15, wherein the mutant MERS-CoV S protein comprises prolines at positions corresponding to residues 1061 and 1061 of the full-length MERS-CoV S protein.
17. A composition comprising a lipid delivery vehicle and a messenger ribonucleic acid (mRNA) comprising an open reading frame encoding a mutant Middle East Respiratory Syndrome (MERS)-CoV S protein, wherein the mutant MERS-CoV S protein comprises (a) one or more substitutions or deletions in a furin cleavage site, and (b) a truncated endoplasmic reticulum (ER) retention signal, relative to a full-length MERS-CoV S protein.
18 The composition of claim 17, wherein the mutant MERS-CoV S protein does not comprise an ER retention signal.
19. A composition comprising a lipid delivery vehicle and a messenger ribonucleic acid (mRNA) comprising an open reading frame encoding a mutant Middle East Respiratory Syndrome (MERS)-CoV S protein, wherein the mutant MERS-CoV S protein comprises (a) one or more substitutions or deletions in a furin cleavage site, and (b) a truncated cytoplasmic tail, relative to a full-length MERS-CoV S protein.
20. The composition of claim 19, wherein the mutant MERS-CoV S protein does not comprise a cytoplasmic tail, or comprises a cytoplasmic tail comprising 21 or fewer amino acids.
21. A composition comprising a lipid delivery vehicle and a messenger ribonucleic acid (mRNA) comprising an open reading frame encoding a Middle East Respiratory Syndrome (MERS)-CoV fusion protein, wherein the MERS-CoV fusion protein consists essentially of (a) an N-terminal domain (NTD) of a full-length MERS-CoV S protein, (b) a receptor-binding domain of the full-length MERS-CoV protein, and (c) a transmembrane (TM) domain.
22. A composition comprising a lipid delivery vehicle and a messenger ribonucleic acid (mRNA) comprising an open reading frame encoding a Middle East Respiratory Syndrome (MERS)-CoV fusion protein, wherein the MERS-CoV fusion protein consists essentially of (a) an N-terminal domain (NTD) of a full-length MERS-CoV S protein, and (b) a transmembrane (TM) domain.
23. The composition of claim 21 or 22, wherein the NTD comprises amino acids 18–338 of a full-length MERS-CoV S protein.
24. A composition comprising a lipid delivery vehicle and a messenger ribonucleic acid (mRNA) comprising an open reading frame encoding a Middle East Respiratory Syndrome (MERS)-CoV fusion protein, wherein the MERS-CoV fusion protein consists essentially of (a) a receptor-binding domain (RBD) of a full-length MERS-CoV S protein, and (b) a transmembrane (TM) domain.
25. The composition of claim 22 or 24, wherein the RBD comprises amino acids 376–589 of a full-length MERS-CoV S protein.
26. The composition of any one of claims 22–25, wherein the TM domain is heterologous to MERS-CoV.
27. The composition of claim 26, wherein the TM domain is an influenza virus hemagglutinin (HA) TM domain.
28. The composition of any one of claim 1–27, wherein the full-length MERS-CoV- S protein comprises the amino acid sequence of SEQ ID NO: 84.
29. A composition comprising a lipid delivery vehicle and a messenger ribonucleic acid (mRNA) comprising an open reading frame encoding a protein comprising an amino acid sequence with at least 90%, at least 95%, at least 97%, at least 98%, at least 99%, or 100% sequence identity to the amino acid sequence of any one of SEQ ID NOs: 89–98.
30. The composition of any one of claims 1–29, wherein the mRNA comprises a 5′ untranslated region (UTR), wherein the 5′ UTR comprises a nucleotide sequence with at least 90% sequence identity to a nucleotide sequence selected from SEQ ID NOs: 1, 2, 5–35, 66, 67, 70–72, 75, 76, and 81.
31. The composition of claim 30, wherein the 5′ UTR comprises a nucleotide sequence selected from SEQ ID NOs: 1, 2, 5–35, 66, 67, 70–72, 75, 76, and 81.
32. The composition of any one of claims 1–31, wherein the mRNA comprises a 3′ untranslated region (UTR), wherein the 5′ UTR comprises a nucleotide sequence with at least 90% sequence identity to a nucleotide sequence selected from SEQ ID NOs: 3–4, 36–44, 68, 69, 73, 74, 77–79, and 82.
33. The composition of claim 32, wherein the 3′ UTR comprises a nucleotide sequence selected from SEQ ID NOs: 3–4, 36–44, 68, 69, 73, 74, 77–79, and 82.
34. The composition of any one of claims 1–33, wherein the mRNA comprises one or more stop codons immediately following the last amino acid-encoding codon.
35. The composition of claim 34, wherein the one or more stop codons comprise the nucleotide sequence UGAUGA.
36. The composition of claim 34, wherein the one or more stop codons comprise the nucleotide sequence UGAUAAUAG.
37. The composition of any one of claims 1–36, wherein the mRNA comprises a polyadenosine (polyA) sequence comprising 20 or more consecutive adenosine nucleotides.
38. The composition of claim 37, wherein the polyA sequence comprises 100 consecutive adenosine nucleotides.
39. The composition of claim 37, wherein the polyA sequence comprises, in 5′-to-3′ order, a first nucleotide sequence comprising 30 consecutive adenosine nucleotides, an intervening sequence comprising no more than three adenosine nucleotides, and a second nucleotide sequence comprising 70 consecutive adenosine nucleotides.
40. The composition of claim 37 or 39, wherein the polyA sequence comprises the nucleotide sequence of SEQ ID NO: 80.
41. The composition of claim 37, wherein the mRNA further comprises a polycytidine (polyC) sequence comprising 20 or more consecutive cytidine nucleotides.
42. The composition of claim 41, wherein the polyC sequence comprises 30 consecutive cytidine nucleotides.
43. The composition of claim 41 or 42, wherein the polyC sequence is downstream from the polyA sequence, wherein the polyA sequence comprises 64 consecutive adenosine nucleotides.
44. The composition of claim 37, wherein the polyA sequence comprises 109 consecutive adenosine nucleotides.
45. The composition of any one of claims 1–44, wherein the mRNA comprises a 5′ cap analog.
46. The composition of claim 45, wherein the 5′ cap analog comprises a 7mG(5′)ppp(5′)NlmpNp cap.
47. The composition of claim 46, wherein the 5′ cap analog comprises an m23′O,7 G+(5′)ppp(5′)Am cap having the structure: .
G+(5′)ppp(5′)Am cap having the structure: .

48. The composition of any one of claims 1–47, wherein the open reading frame comprises one or more chemically modified nucleotides.
49. The composition of any one of claims 1–48, wherein the open reading frame comprises N1-methylpseudouridine.
50. The composition of any one of claims 1–49, wherein at least 80% of uracil nucleotides in the open reading frame comprise N1-methylpseudouridine.
51. The composition of any one of claims 1–50, wherein 100% of uracil nucleotides in the open reading frame comprise N1-methylpseudouridine.
52. The composition of any one of claims 1–51, wherein the open reading frame comprises 5- methylcytidine.
53. The composition of any one of claims 1–52, wherein at least 80% of cytosine nucleotides in the open reading frame comprise 5-methylcytidine.
54. The composition of any one of claims 1–53, wherein 100% of cytosine nucleotides in the open reading frame comprise 5-methylcytidine.
55. The composition of any one of claims 1–48 or 52–54, wherein the open reading frame comprises 5-methyluridine.
56. The composition of any one of claims 1– 48 or 52–55, wherein at least 80% of uracil nucleotides in the open reading frame comprise 5-methyluridine.
57. The composition of any one of claims 1– 48 or 52–56, wherein 100% of uracil nucleotides in the open reading frame comprise 5-methyluridine.
58. The composition of any one of claims 1–57, wherein the lipid delivery vehicle is a lipid nanoparticle (LNP).
59. The composition of claim 58, wherein the lipid nanoparticle comprises 40-55 mol% of an amino lipid, 30-45 mol% of a sterol, 5-15 mol% of a neutral lipid, and 1-5 mol% of a PEG- modified lipid.
60. The composition of claim 59, wherein the amino lipid comprises a compound of Formula (I): or a salt or isomer thereof, wherein: R1 is R”M’R’ or C5-20 alkenyl; R2 and R3 are each independently selected from C1-14 alkyl and C2-14 alkenyl; R4 is -(CH2)nQ, wherein Q is OH and n is selected from 3, 4, and 5; M and M’ are each independently -OC(O)- or -C(O)O-; R5, R6, and R7 are each H; R’ is a linear C1-12 alkyl, or C1-12 alkyl substituted with C6-9 alkyl; R” is C3-14 alkyl; m is selected from 5, 6, 7, 8, 9, 10, 11, 12, and 13.
or a salt or isomer thereof, wherein: R1 is R”M’R’ or C5-20 alkenyl; R2 and R3 are each independently selected from C1-14 alkyl and C2-14 alkenyl; R4 is -(CH2)nQ, wherein Q is OH and n is selected from 3, 4, and 5; M and M’ are each independently -OC(O)- or -C(O)O-; R5, R6, and R7 are each H; R’ is a linear C1-12 alkyl, or C1-12 alkyl substituted with C6-9 alkyl; R” is C3-14 alkyl; m is selected from 5, 6, 7, 8, 9, 10, 11, 12, and 13.

61. The composition of claim 59 or 60, wherein the amino lipid comprises Compound 1: (Compound 1).
(Compound 1).

62. The composition of claim 59 or 60, wherein the amino lipid comprises a compound of the structure A7: (A7).
structure A7: (A7).

63. The composition of any one of claims 59–62, wherein the neutral lipid is 1,2 distearoyl- sn-glycero-3-phosphocholine (DSPC).
64. The composition of any one of claims 59–63, wherein the sterol is cholesterol.
65. The composition of any one of claims 59–64, wherein the PEG-modified lipid is PEG2000-DMG.
66. The composition of any one of claims 1–57, wherein the lipid delivery vehicle is a liposome.
67. The composition of any one of claims 1–57, wherein the lipid delivery vehicle is a lipoplex.
68. The composition of any one of claims 1–57, wherein the lipid delivery vehicle is a lipopolyplex.
69. The composition of any one of claims 1–57, wherein the lipid delivery vehicle is a lipidoid.
70. A pharmaceutical composition comprising the composition of any one of claims 1–69, and a pharmaceutically acceptable excipient.
71. A method comprising administering to a subject the composition of any one of claims 1– 70.
72. The method of claim 71, wherein the composition is administered intramuscularly.
73. The method of claim 71 or 72, wherein the composition is effective to elicit, in the subject, CD4+ and/or CD8+ T cells specific to one or more epitopes of the protein.
74. The method of any one of claims 71–3, wherein the composition is effective to elicit, in the subject, neutralizing antibodies to MERS-CoV.
75. The method of any one of claims 71–74, wherein the composition is effective to elicit, in the subject, antibodies that mediate antibody-dependent cell-mediated cytotoxicity (ADCC) against MERS-CoV-infected cells.
76. The method of any one of claims 71–75, the method comprising administering a first dose and a second dose of the composition.
77. A MERS-CoV fusion protein comprising: (a) an N-terminal domain (NTD) of a MERS-CoV Spike (S) protein; and (b) a receptor-binding domain (RBD) of the MERS-CoV-S protein, wherein the MERS-CoV fusion protein does not comprise an NTD or an RBD of an S protein of a coronavirus that is not MERS-CoV.
78. The MERS-CoV fusion protein of claim 77, wherein the NTD and RBD are connected by a linker.
79. The MERS-CoV fusion protein of claim 78, wherein the linker is a glycine linker or a glycine-serine linker.
80. The MERS-CoV fusion protein of claim 78, wherein the linker comprises a pan-HLA- DR-binding epitope (PADRE).
81. The MERS-CoV fusion protein of any one of claims 71–80, wherein the MERS-CoV fusion protein further comprises a transmembrane (TM) domain.
82. The MERS-CoV fusion protein of claim 81, wherein the TM domain is a MERS-CoV S protein TM domain or derivative thereof.
83. The MERS-CoV fusion protein of claim 81, wherein the TM domain is heterologous to MERS-CoV.
84. The MERS-CoV fusion protein of claim 81 or 83, wherein the TM domain is an influenza virus hemagglutinin (HA) TM domain or derivative thereof.
85. The MERS-CoV fusion protein of any one of claims 81–84, wherein the MERS-CoV fusion protein comprises, in N-to-C-terminal order, the NTD, the RBD, and the TM domain.
86. The MERS-CoV fusion protein of any one of claim 84–80, wherein the MERS-CoV fusion protein does not comprise a transmembrane (TM) domain.
87. The MERS-CoV fusion protein of any one of claims 84–80 or 86, wherein the MERS- CoV fusion protein is soluble.
88. The MERS-CoV fusion protein of any one of claims 84–87, wherein the MERS-CoV fusion protein further comprises a signal peptide.
89. The MERS-CoV fusion protein of claim 88, wherein the signal peptide is a MERS-CoV S protein signal peptide.
90. The MERS-CoV fusion protein of claim 88, wherein the signal peptide is heterologous to MERS-CoV.
91. A mutant Middle East Respiratory Syndrome (MERS)-CoV S protein, comprising (a) one or more proline substitutions or insertions, and (b) one or more substitutions or deletions in a furin cleavage site, relative to a full-length MERS-CoV S protein.
92. The mutant MERS-CoV S protein of claim 91, wherein the mutant MERS-CoV S protein comprises prolines at positions corresponding to residues 1060 and 1061 of the full-length MERS-CoV S protein.
93. A mutant Middle East Respiratory Syndrome (MERS)-CoV S protein comprising (a) one or more substitutions or deletions in a furin cleavage site, and (b) a truncated endoplasmic reticulum (ER) retention signal, relative to a full-length MERS-CoV S protein.
94. The mutant MERS-CoV S protein of claim 93, wherein the mutant MERS-CoV S protein does not comprise an ER retention signal.
95. A mutant MERS-CoV S protein comprising (a) one or more substitutions or deletions in a furin cleavage site, and (b) a truncated cytoplasmic tail, relative to a full-length MERS-CoV S protein.
96. The mutant MERS-CoV S protein of claim 95, wherein the mutant MERS-CoV S protein does not comprise a cytoplasmic tail, or comprises a cytoplasmic tail comprising 21 or fewer amino acids.
97. A MERS-CoV fusion protein consisting essentially of (a) an N-terminal domain (NTD) of a full-length MERS-CoV S protein, (b) a receptor-binding domain of the full-length MERS-CoV protein, and (c) a transmembrane (TM) domain.
98. A Middle East Respiratory Syndrome (MERS)-CoV fusion proteinconsisting essentially of (a) an N-terminal domain (NTD) of a full-length MERS-CoV S protein, and (b) a transmembrane (TM) domain.
99. The MERS-CoV fusion protein of claim 97 or 98, wherein the NTD comprises amino acids 18–338 of a full-length MERS-CoV S protein.
100. A MERS-CoV fusion protein consisting essentially of (a) a receptor-binding domain (RBD) of a full-length MERS-CoV S protein, and (b) a transmembrane (TM) domain.
101. The MERS-CoV fusion protein of claim 97 or 100, wherein the RBD comprises amino acids 376–589 of a full-length MERS-CoV S protein.
102. The MERS-CoV fusion protein of any one of claims 97–101, wherein the TM domain is heterologous to MERS-CoV.
103. The MERS-CoV fusion protein of claim 102, wherein the TM domain is an influenza virus hemagglutinin (HA) TM domain.
104. The protein of any one of claim 77–103, wherein the full-length MERS-CoV- S protein comprises the amino acid sequence of SEQ ID NO: 84.
105. A protein comprising an amino acid sequence with at least 95%, at least 97%, at least 98%, at least 99%, or 100% sequence identity to the amino acid sequence of any one of SEQ ID NOs: 87–98.
106. A ribonucleic acid (RNA) comprising an open reading frame encoding the protein of any one of claims 77–105.
107. A messenger ribonucleic acid comprising an open reading frame encoding the protein of any one of claims 77–105.
108. The mRNA of claim 107, wherein the mRNA comprises one or more chemically modified nucleotides.
109. The mRNA of claim 107 or 108, wherein 100% of the uracil nucleotides of the mRNA comprise are chemically modified nucleotides.
110. The mRNA of any one of claims 107–109, wherein 100% of uracil nucleotides of the mRNA comprise N1-methylpseudouridine.
111. A self-amplifying ribonucleic acid (saRNA) comprising an open reading frame encoding a protein comprising: (a) an N-terminal domain (NTD) of a MERS-CoV Spike (S) protein; and (b) a receptor-binding domain (RBD) of the MERS-CoV-S protein, wherein the MERS-CoV fusion protein does not comprise an NTD or an RBD of an S protein of a coronavirus that is not MERS-CoV.
112. The saRNA of claim 111, wherein the NTD and RBD are connected by a linker.
113. The saRNA of claim 113, wherein the linker is a glycine linker or a glycine-serine linker.
114. The saRNA of claim 113, wherein the linker comprises a pan-HLA-DR-binding epitope (PADRE).
115. The saRNA of any one of claims 111–114, wherein the MERS-CoV fusion protein further comprises a transmembrane (TM) domain.
116. The saRNA of claim 115, wherein the TM domain is a MERS-CoV S protein TM domain or derivative thereof.
117. The saRNA of claim 115, wherein the TM domain is heterologous to MERS-CoV.
118. The saRNA of claim 115 or 117, wherein the TM domain is an influenza virus hemagglutinin (HA) TM domain or derivative thereof.
119. The saRNA of any one of claims 115–118, wherein the MERS-CoV fusion protein comprises, in N-to-C-terminal order, the NTD, the RBD, and the TM domain.
120. The saRNA of any one of claim 111–114, wherein the MERS-CoV fusion protein does not comprise a transmembrane (TM) domain.
121. The saRNA of any one of claims 111–114 or 120, wherein the MERS-CoV fusion protein is soluble.
122. The saRNA of any one of claims 111–121, wherein the MERS-CoV fusion protein further comprises a signal peptide.
123. The saRNA of claim 122, wherein the signal peptide is a MERS-CoV S protein signal peptide.
124. The saRNA of claim 122, wherein the signal peptide is heterologous to MERS-CoV.
125. A self-amplifying ribonucleic acid (saRNA) comprising an open reading frame encoding a mutant Middle East Respiratory Syndrome (MERS)-CoV S protein, wherein the mutant MERS-CoV S protein comprises (a) one or more proline substitutions or insertions, and (b) one or more substitutions or deletions in a furin cleavage site, relative to a full-length MERS-CoV S protein.
126. The saRNA of claim 125, wherein the mutant MERS-CoV S protein comprises prolines at positions corresponding to residues 1060 and 1061 of the full-length MERS-CoV S protein.
127. A self-amplifying ribonucleic acid (saRNA) comprising an open reading frame encoding a mutant Middle East Respiratory Syndrome (MERS)-CoV S protein, wherein the mutant MERS-CoV S protein comprises (a) one or more substitutions or deletions in a furin cleavage site, and (b) a truncated endoplasmic reticulum (ER) retention signal, relative to a full-length MERS-CoV S protein.
128. The saRNA of claim 127, wherein the mutant MERS-CoV S protein does not comprise an ER retention signal.
129. A self-amplifying ribonucleic acid (saRNA) comprising an open reading frame encoding a mutant Middle East Respiratory Syndrome (MERS)-CoV S protein, wherein the mutant MERS-CoV S protein comprises (a) one or more substitutions or deletions in a furin cleavage site, and (b) a truncated cytoplasmic tail, relative to a full-length MERS-CoV S protein.
130. The saRNA of claim 129, wherein the mutant MERS-CoV S protein does not comprise a cytoplasmic tail, or comprises a cytoplasmic tail comprising 21 or fewer amino acids.
131. A self-amplifying ribonucleic acid (saRNA) comprising an open reading frame encoding a Middle East Respiratory Syndrome (MERS)-CoV fusion protein, wherein the MERS-CoV fusion protein consists essentially of (a) an N-terminal domain (NTD) of a full-length MERS-CoV S protein, (b) a receptor-binding domain of the full-length MERS-CoV protein, and (c) a transmembrane (TM) domain.
132. A self-amplifying ribonucleic acid (saRNA) comprising an open reading frame encoding a Middle East Respiratory Syndrome (MERS)-CoV fusion protein, wherein the MERS-CoV fusion protein consists essentially of (a) an N-terminal domain (NTD) of a full-length MERS-CoV S protein, and (b) a transmembrane (TM) domain.
133. The saRNA of claim 131 or 132, wherein the NTD comprises amino acids 18–338 of a full-length MERS-CoV S protein.
134. A self-amplifying ribonucleic acid (saRNA) comprising an open reading frame encoding a Middle East Respiratory Syndrome (MERS)-CoV fusion protein, wherein the MERS-CoV fusion protein consists essentially of (a) a receptor-binding domain (RBD) of a full-length MERS-CoV S protein, and (b) a transmembrane (TM) domain.
135. The saRNA of claim 131 or 134, wherein the RBD comprises amino acids 376–589 of a full-length MERS-CoV S protein.
136. The saRNA of any one of claims 131–135, wherein the TM domain is heterologous to MERS-CoV.
137. The saRNA of claim 136, wherein the TM domain is an influenza virus hemagglutinin (HA) TM domain.
138. The saRNA of any one of claims 111–137, wherein the full-length MERS-CoV- S protein comprises the amino acid sequence of SEQ ID NO: 84.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US202363513614P | 2023-07-14 | 2023-07-14 | |
| US63/513,614 | 2023-07-14 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2025019352A2true WO2025019352A2 (en) | 2025-01-23 |
| WO2025019352A3 WO2025019352A3 (en) | 2025-04-24 |
Family
ID=92214438
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2024/037879PendingWO2025019352A2 (en) | 2023-07-14 | 2024-07-12 | Mers-cov mrna vaccines |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2025019352A2 (en) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US12428577B2 (en) | 2021-05-14 | 2025-09-30 | Modernatx, Inc. | Methods of monitoring in vitro transcription of mRNA and/or post-in vitro transcription processes |
Citations (35)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2002098443A2 (en) | 2001-06-05 | 2002-12-12 | Curevac Gmbh | Stabilised mrna with an increased g/c content and optimised codon for use in gene therapy |
| US20090226470A1 (en) | 2007-12-11 | 2009-09-10 | Mauro Vincent P | Compositions and methods related to mRNA translational enhancer elements |
| US20100129877A1 (en) | 2005-09-28 | 2010-05-27 | Ugur Sahin | Modification of RNA, Producing an Increased Transcript Stability and Translation Efficiency |
| US20100293625A1 (en) | 2007-09-26 | 2010-11-18 | Interexon Corporation | Synthetic 5'UTRs, Expression Vectors, and Methods for Increasing Transgene Expression |
| US8158601B2 (en) | 2009-06-10 | 2012-04-17 | Alnylam Pharmaceuticals, Inc. | Lipid formulation |
| WO2012099755A1 (en) | 2011-01-11 | 2012-07-26 | Alnylam Pharmaceuticals, Inc. | Pegylated lipids and their use for drug delivery |
| US8278063B2 (en) | 2007-06-29 | 2012-10-02 | Commonwealth Scientific And Industrial Research Organisation | Methods for degrading toxic compounds |
| WO2013052523A1 (en) | 2011-10-03 | 2013-04-11 | modeRNA Therapeutics | Modified nucleosides, nucleotides, and nucleic acids, and uses thereof |
| WO2013185069A1 (en) | 2012-06-08 | 2013-12-12 | Shire Human Genetic Therapies, Inc. | Pulmonary delivery of mrna to non-lung target cells |
| WO2014093924A1 (en) | 2012-12-13 | 2014-06-19 | Moderna Therapeutics, Inc. | Modified nucleic acid molecules and uses thereof |
| US20140206753A1 (en) | 2011-06-08 | 2014-07-24 | Shire Human Genetic Therapies, Inc. | Lipid nanoparticle compositions and methods for mrna delivery |
| WO2014144039A1 (en) | 2013-03-15 | 2014-09-18 | Moderna Therapeutics, Inc. | Characterization of mrna molecules |
| WO2014144711A1 (en) | 2013-03-15 | 2014-09-18 | Moderna Therapeutics, Inc. | Analysis of mrna heterogeneity and stability |
| WO2014144767A1 (en) | 2013-03-15 | 2014-09-18 | Moderna Therapeutics, Inc. | Ion exchange purification of mrna |
| WO2014144196A1 (en) | 2013-03-15 | 2014-09-18 | Shire Human Genetic Therapies, Inc. | Synergistic enhancement of the delivery of nucleic acids via blended formulations |
| WO2014152027A1 (en) | 2013-03-15 | 2014-09-25 | Moderna Therapeutics, Inc. | Manufacturing methods for production of rna transcripts |
| WO2014152031A1 (en) | 2013-03-15 | 2014-09-25 | Moderna Therapeutics, Inc. | Ribonucleic acid purification |
| WO2014152030A1 (en) | 2013-03-15 | 2014-09-25 | Moderna Therapeutics, Inc. | Removal of dna fragments in mrna production process |
| WO2015024667A1 (en) | 2013-08-21 | 2015-02-26 | Curevac Gmbh | Method for increasing expression of rna-encoded proteins |
| WO2015051173A2 (en) | 2013-10-02 | 2015-04-09 | Moderna Therapeutics, Inc | Polynucleotide molecules and uses thereof |
| WO2015051169A2 (en) | 2013-10-02 | 2015-04-09 | Moderna Therapeutics, Inc. | Polynucleotide molecules and uses thereof |
| US9012219B2 (en) | 2005-08-23 | 2015-04-21 | The Trustees Of The University Of Pennsylvania | RNA preparations comprising purified modified RNA for reprogramming cells |
| WO2015062738A1 (en) | 2013-11-01 | 2015-05-07 | Curevac Gmbh | Modified rna with decreased immunostimulatory properties |
| WO2015085318A2 (en) | 2013-12-06 | 2015-06-11 | Moderna Therapeutics, Inc. | Targeted adaptive vaccines |
| WO2015089511A2 (en) | 2013-12-13 | 2015-06-18 | Moderna Therapeutics, Inc. | Modified nucleic acid molecules and uses thereof |
| WO2015101415A1 (en) | 2013-12-30 | 2015-07-09 | Curevac Gmbh | Artificial nucleic acid molecules |
| WO2015101414A2 (en) | 2013-12-30 | 2015-07-09 | Curevac Gmbh | Artificial nucleic acid molecules |
| WO2015130584A2 (en) | 2014-02-25 | 2015-09-03 | Merck Sharp & Dohme Corp. | Lipid nanoparticle vaccine adjuvants and antigen delivery systems |
| WO2016040359A1 (en) | 2014-09-08 | 2016-03-17 | WebMD Health Corporation | Structuring multi-sourced medical information into a collaborative health record |
| WO2017066797A1 (en) | 2015-10-16 | 2017-04-20 | Modernatx, Inc. | Trinucleotide mrna cap analogs |
| WO2017127750A1 (en) | 2016-01-22 | 2017-07-27 | Modernatx, Inc. | Messenger ribonucleic acids for the production of intracellular binding polypeptides and methods of use thereof |
| WO2017153936A1 (en) | 2016-03-10 | 2017-09-14 | Novartis Ag | Chemically modified messenger rna's |
| WO2018053209A1 (en) | 2016-09-14 | 2018-03-22 | Modernatx, Inc. | High purity rna compositions and methods for preparation thereof |
| WO2019036682A1 (en) | 2017-08-18 | 2019-02-21 | Modernatx, Inc. | Rna polymerase variants |
| WO2020172239A1 (en) | 2019-02-20 | 2020-08-27 | Modernatx, Inc. | Rna polymerase variants for co-transcriptional capping |
Family Cites Families (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2005027963A2 (en)* | 2003-09-15 | 2005-03-31 | The United States Of America As Represented By Thesecretary Of Health And Human Services, Nih | METHODS AND COMPOSITIONS FOR THE GENERATION OF A PROTECTIVE IMMUNE RESPONSE AGAINTS SARS-CoV |
| RS63329B1 (en)* | 2010-08-31 | 2022-07-29 | Glaxosmithkline Biologicals Sa | Pegylated liposomes for delivery of immunogen-encoding rna |
| CA3018046A1 (en)* | 2011-12-16 | 2013-06-20 | Moderna Therapeutics, Inc. | Modified nucleoside, nucleotide, and nucleic acid compositions |
| EP3628335B1 (en)* | 2012-12-07 | 2023-11-08 | Translate Bio, Inc. | Lipidic nanoparticles for mrna delivery in the lungs |
| PT3083556T (en)* | 2013-12-19 | 2020-03-05 | Novartis Ag | Lipids and lipid compositions for the delivery of active agents |
| EP4349405A3 (en)* | 2015-10-22 | 2024-06-19 | ModernaTX, Inc. | Respiratory virus vaccines |
| EP3558356A2 (en)* | 2016-12-23 | 2019-10-30 | CureVac AG | Mers coronavirus vaccine |
| WO2023064907A1 (en)* | 2021-10-15 | 2023-04-20 | Yale University | Compositions and methods for vaccination against pathogenic coronavirus species and variants |
- 2024
- 2024-07-12WOPCT/US2024/037879patent/WO2025019352A2/enactivePending
Patent Citations (35)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2002098443A2 (en) | 2001-06-05 | 2002-12-12 | Curevac Gmbh | Stabilised mrna with an increased g/c content and optimised codon for use in gene therapy |
| US9012219B2 (en) | 2005-08-23 | 2015-04-21 | The Trustees Of The University Of Pennsylvania | RNA preparations comprising purified modified RNA for reprogramming cells |
| US20100129877A1 (en) | 2005-09-28 | 2010-05-27 | Ugur Sahin | Modification of RNA, Producing an Increased Transcript Stability and Translation Efficiency |
| US8278063B2 (en) | 2007-06-29 | 2012-10-02 | Commonwealth Scientific And Industrial Research Organisation | Methods for degrading toxic compounds |
| US20100293625A1 (en) | 2007-09-26 | 2010-11-18 | Interexon Corporation | Synthetic 5'UTRs, Expression Vectors, and Methods for Increasing Transgene Expression |
| US20090226470A1 (en) | 2007-12-11 | 2009-09-10 | Mauro Vincent P | Compositions and methods related to mRNA translational enhancer elements |
| US8158601B2 (en) | 2009-06-10 | 2012-04-17 | Alnylam Pharmaceuticals, Inc. | Lipid formulation |
| WO2012099755A1 (en) | 2011-01-11 | 2012-07-26 | Alnylam Pharmaceuticals, Inc. | Pegylated lipids and their use for drug delivery |
| US20140206753A1 (en) | 2011-06-08 | 2014-07-24 | Shire Human Genetic Therapies, Inc. | Lipid nanoparticle compositions and methods for mrna delivery |
| WO2013052523A1 (en) | 2011-10-03 | 2013-04-11 | modeRNA Therapeutics | Modified nucleosides, nucleotides, and nucleic acids, and uses thereof |
| WO2013185069A1 (en) | 2012-06-08 | 2013-12-12 | Shire Human Genetic Therapies, Inc. | Pulmonary delivery of mrna to non-lung target cells |
| WO2014093924A1 (en) | 2012-12-13 | 2014-06-19 | Moderna Therapeutics, Inc. | Modified nucleic acid molecules and uses thereof |
| WO2014152031A1 (en) | 2013-03-15 | 2014-09-25 | Moderna Therapeutics, Inc. | Ribonucleic acid purification |
| WO2014144767A1 (en) | 2013-03-15 | 2014-09-18 | Moderna Therapeutics, Inc. | Ion exchange purification of mrna |
| WO2014144196A1 (en) | 2013-03-15 | 2014-09-18 | Shire Human Genetic Therapies, Inc. | Synergistic enhancement of the delivery of nucleic acids via blended formulations |
| WO2014152027A1 (en) | 2013-03-15 | 2014-09-25 | Moderna Therapeutics, Inc. | Manufacturing methods for production of rna transcripts |
| WO2014144711A1 (en) | 2013-03-15 | 2014-09-18 | Moderna Therapeutics, Inc. | Analysis of mrna heterogeneity and stability |
| WO2014152030A1 (en) | 2013-03-15 | 2014-09-25 | Moderna Therapeutics, Inc. | Removal of dna fragments in mrna production process |
| WO2014144039A1 (en) | 2013-03-15 | 2014-09-18 | Moderna Therapeutics, Inc. | Characterization of mrna molecules |
| WO2015024667A1 (en) | 2013-08-21 | 2015-02-26 | Curevac Gmbh | Method for increasing expression of rna-encoded proteins |
| WO2015051173A2 (en) | 2013-10-02 | 2015-04-09 | Moderna Therapeutics, Inc | Polynucleotide molecules and uses thereof |
| WO2015051169A2 (en) | 2013-10-02 | 2015-04-09 | Moderna Therapeutics, Inc. | Polynucleotide molecules and uses thereof |
| WO2015062738A1 (en) | 2013-11-01 | 2015-05-07 | Curevac Gmbh | Modified rna with decreased immunostimulatory properties |
| WO2015085318A2 (en) | 2013-12-06 | 2015-06-11 | Moderna Therapeutics, Inc. | Targeted adaptive vaccines |
| WO2015089511A2 (en) | 2013-12-13 | 2015-06-18 | Moderna Therapeutics, Inc. | Modified nucleic acid molecules and uses thereof |
| WO2015101415A1 (en) | 2013-12-30 | 2015-07-09 | Curevac Gmbh | Artificial nucleic acid molecules |
| WO2015101414A2 (en) | 2013-12-30 | 2015-07-09 | Curevac Gmbh | Artificial nucleic acid molecules |
| WO2015130584A2 (en) | 2014-02-25 | 2015-09-03 | Merck Sharp & Dohme Corp. | Lipid nanoparticle vaccine adjuvants and antigen delivery systems |
| WO2016040359A1 (en) | 2014-09-08 | 2016-03-17 | WebMD Health Corporation | Structuring multi-sourced medical information into a collaborative health record |
| WO2017066797A1 (en) | 2015-10-16 | 2017-04-20 | Modernatx, Inc. | Trinucleotide mrna cap analogs |
| WO2017127750A1 (en) | 2016-01-22 | 2017-07-27 | Modernatx, Inc. | Messenger ribonucleic acids for the production of intracellular binding polypeptides and methods of use thereof |
| WO2017153936A1 (en) | 2016-03-10 | 2017-09-14 | Novartis Ag | Chemically modified messenger rna's |
| WO2018053209A1 (en) | 2016-09-14 | 2018-03-22 | Modernatx, Inc. | High purity rna compositions and methods for preparation thereof |
| WO2019036682A1 (en) | 2017-08-18 | 2019-02-21 | Modernatx, Inc. | Rna polymerase variants |
| WO2020172239A1 (en) | 2019-02-20 | 2020-08-27 | Modernatx, Inc. | Rna polymerase variants for co-transcriptional capping |
Non-Patent Citations (16)
| Title |
|---|
| "Remington: The Science and Practice of Pharmacy", 2005, LIPPINCOTT WILLIAMS & WILKINS |
| ALEXANDER ET AL., J IMMUNOL., vol. 164, no. 3, 2000, pages 1625 - 1633 |
| BLOOM K ET AL., GENE THERAPY, vol. 28, 2021, pages 117 - 129 |
| CUI ET AL., NAT. REV. MICROBIOL.,, vol. 17, no. 3, pages 181 - 192 |
| KIM, J.H. ET AL., PLOS ONE, vol. 6, 2011, pages 18556 |
| MIDOUXPICHON, EXPERT REV VACCINES., vol. 14, no. 2, 2015, pages 221 - 234 |
| MILLETWHITTAKER, PROC NATL ACAD SCI U S A., vol. 111, no. 42, 2014, pages 15214 - 15219 |
| NEEDLEMAN, S.B.WUNSCH, C.D.: "A general method applicable to the search for similarities in the amino acid sequences of two proteins", J. MOL. BIOL., vol. 48, 1970, pages 443 - 453 |
| RAJ ET AL., NATURE, vol. 495, no. 7440, 2013, pages 251 - 254 |
| RAMADAN ET AL., GERMS, vol. 9, no. 1, 2019, pages 35 - 42 |
| SMITH, T.FWATERMAN, M.S.: "Identification of common molecular subsequences", J. MOL. BIOL., vol. 147, 1981, pages 195 - 197, XP024015032, DOI: 10.1016/0022-2836(81)90087-5 |
| SONG ET AL., VIRUSES, vol. 11, no. 1, 2019, pages 59 |
| STEPHEN F. ALTSCHUL ET AL.: "Gapped BLAST and PSI-BLAST: a new generation of protein database search programs", NUCLEIC ACIDS RES., vol. 25, 1997, pages 3389 - 3402, XP002905950, DOI: 10.1093/nar/25.17.3389 |
| WAGNER ET AL., NATURE CHEMICAL BIOLOGY, 2018 |
| WEINBERG ET AL., J INFECT DIS., vol. 201, no. 11, 1 June 2010 (2010-06-01), pages 1607 - 10 |
| WHITT, JOURNAL OF VIROLOGICAL METHODS, vol. 169, 2010, pages 365 - 374 |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US12428577B2 (en) | 2021-05-14 | 2025-09-30 | Modernatx, Inc. | Methods of monitoring in vitro transcription of mRNA and/or post-in vitro transcription processes |
Also Published As
| Publication number | Publication date |
|---|---|
| WO2025019352A3 (en) | 2025-04-24 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US20230346914A1 (en) | Sars-cov-2 mrna domain vaccines | |
| US20240100151A1 (en) | Variant strain-based coronavirus vaccines | |
| US20240139309A1 (en) | Variant strain-based coronavirus vaccines | |
| US20230355743A1 (en) | Multi-proline-substituted coronavirus spike protein vaccines | |
| US20240382581A1 (en) | Pan-human coronavirus vaccines | |
| US20240299531A1 (en) | Therapeutic use of sars-cov-2 mrna domain vaccines | |
| US20240285754A1 (en) | Mrna vaccines encoding flexible coronavirus spike proteins | |
| AU2021213108A1 (en) | Coronavirus RNA vaccines | |
| JP2024514182A (en) | Respiratory virus combination vaccine | |
| WO2023283642A2 (en) | Pan-human coronavirus concatemeric vaccines | |
| EP4355891A1 (en) | Coronavirus glycosylation variant vaccines | |
| WO2023283645A1 (en) | Pan-human coronavirus domain vaccines | |
| WO2024050483A1 (en) | Variant strain-based coronavirus vaccines and uses thereof | |
| WO2024163465A1 (en) | Epstein-barr virus mrna vaccines | |
| WO2025019352A2 (en) | Mers-cov mrna vaccines | |
| WO2024263826A1 (en) | Sars-cov-2 t cell vaccines | |
| WO2025034612A1 (en) | Varicella-zoster virus mrna vaccine |