METHODS, COMPOSITIONS, AND KITS FOR PREPARING LIBRARY AND TARGETED SEQUENCING BACKGROUND OF THE INVENTION The present invention is directed to methods and compositions for amplifying a population of population of captured target polynucleotides which are divided into portions where each portion is processed to generate (epi-)genetic information without losing a significant portion of the total information from any individual portion. An adaptor sequence is first added to the 3’ end of nucleic acids which is used for one or more cycles in one or more rounds of linear amplification. Next-generation DNA sequencing (NGS) is continuing to revolutionize clinical medicine and basic research, especially in the rapidly advancing field of liquid biopsy testing. However, while NGS has the capacity to generate hundreds of billions of nucleotides of DNA sequence in a single experiment, both the very limited mass of material available from a single patient and the inherent error rate of ∼ 1% of many sequencing technologies results in both a challenge to (i) do more than a single test on a patient sample, and (ii) overcome the potential of hundreds of millions of sequencing mistakes. The combination of limited material and scattered errors become extremely problematic when “deep sequencing” a patient sample to try and identify genetically heterogeneous mixtures, such as tumours (especially when disease stage is early), or mixed microbial populations. Cell-free DNA from liquid biopsies has the capacity to provide many different types of genetic information that can be used to provide clinical information. These includes the presence of absence of mutations (especially driver mutations), large scale chromosomal changes leading to alterations in copy number and potentially gene fusions, changes in patterns of CpG dinucleotide epigenetic marks, how DNA has fragmented in the patient with patterns of DNA ends which can change between ‘healthy’ and ‘unhealthy’ individuals. The largest hurdle to obtaining all this information from a single patient sample is the very low levels of clinical material, due to this you do not want to have to do multiple tests as every additional test you do requires the sample to be split across more and more tests reducing the sensitivity of all of them. To overcome limitations in sequencing accuracy, several methods have been reported. Duplex sequencing (Schmitt, et al PNAS 109: 14508-14513) is one of them. This approach greatly reduces errors by independently tagging and sequencing each of the two strands of a DNA duplex. As the two strands are complementary, true mutations are found at the same position in both strands. In contrast, PCR and sequencing errors result in mutations in only one strand and can thus be discounted as technical error. Another approach called Safe-Sequencing System ("Safe-SeqS) was described by Kinde et al (PNAS 2011; 108(23):9530-5). The keys to this approach are (i) assignment of a unique identifier (UID) to each template molecule, (ii) amplification of each uniquely tagged template molecule to create UID families, and (iii) redundant sequencing of the amplification products. PCR fragments with the same UID are considered mutant ("supermutants") only if ≤95% of them contain an identical mutation. US Patents US8722368B2, US8685678B2, US8742606 describe methods of sequencing polynucleotides attached with a degenerate base region to determine/estimate the number of different starting polynucleotides. However, these methods do not compare sequence information of the original two strands and involve ligating and PCR to attach degenerate base region. US Patents US8742606B2, and WO2017066592A1, and Quan Peng (Scientific Reports, 2019 Mar 18;9(1):4810. doi: 10.1038/s41598-019-41215-z) discuss methods of coupling ligation to double strand DNA together with targeted amplification to generate information on mutations from both strands of starting material. Another method, ATOM-Seq (WO2018193233A1) allows for a ligation independent method which uses polymerase based tagging of input material which allows for identification of mutations in both strands of starting material. Targeted next generation sequencing often involves the analysis of large complex fragments and this is achieved by multiplex PCR (the simultaneous amplification of different target DNA sequences in a single PCR reaction). Results obtained with multiplex PCR however are often complicated by artefacts of the amplification products. These include false negative results due to reaction failure and false-positive results (such as amplification of spurious products) due to non-specific priming events. Since the possibility of non-specific priming increases with each additional primer pair, conditions must be modified as necessary as individual primer sets are added. SUMMARY OF THE INVENTION This invention relates to methods, compositions and kits for making one or more non-specific or specific target enriched sequencing libraries from portions of an initially tagged target polynucleotide involving one or more initial steps of amplification from a universal primer binding site added to the 3’ ends of target polynucleotide. The amplified copies of the target polynucleotide are split into one or more portions where portions of the amplified polynucleotides are processed to generate a NGS library, and, the remaining tagged polynucleotides are processed using an agent to differentially convert methylcytosine and unmethylated cytosine producing converted tagged polynucleotides, the sample is amplified again and the amplified converted polynucleotides products are split into one or more portions where each portion of the amplified converted polynucleotides is processed to generate a NGS library. The NGS libraries can be uniquely indexed or dual indexed so they can be multiplex prior to sequencing. Each of the libraries can then be multiplexed and the data generated across one or more libraries can be processed using a computer to identify information present in the original target polynucleotide (i.e. patient sample) including but not limited to, single and complex nucleotide changes, insertions, deletions, copy number variation, targeted genetic information, targeted epigenetic information, whole genome genetic data, whole genome epigenetic data, high-GC genomic data, high-GC epigenetic data, patterns in DNA break points, patterns in high- GC DNA break points, chromosomal translocations and rearrangements, fusions, or any combination thereof by bioinformatic data analysis methods known to those skilled in the art (e.g. those given in WO2015100427A1, US9920366B2). Disclosed is a method of processing original target polynucleotides comprising; (a) providing a reaction mixture(s), which is designed to add an adaptor on to all 3’ and/or 5’ ends of the starting original target polynucleotide which may be a ligation based reaction, a polymerase 3’ extension based reaction (e.g. ATOM-Seq), a terminal deoxytransferase 3’ extension based reaction, or any other suitable method resulting in a universal primer binding site to produce tagged target polynucleotide ; (b) performing cycles of amplification using a primer which binds to the adaptor sequence on the tagged polynucleotide to generate amplified polynucleotides; (c) dividing the products of step (b) into one or more portions; (d) treating one portion of the tagged target polynucleotide from step (c) with an agent to differentially convert cytosine and methylcytosines to produce a converted tagged polynucleotide; (e) treating one portion of the amplified polynucleotides from step (c) to generate a NGS library which can be sequenced to obtain polynucleotide sequence information present in the original target polynucleotide; (f) (optional) performing a second round of cycles of amplification using a primer which binds to the adaptor sequence on the converted tagged polynucleotides (d) to generate amplified converted polynucleotides; and (g) further treating product (f) to generate a NGS library which can be sequenced to obtain polynucleotide sequence information present in the converted polynucleotide, for example CG methylation. Disclosed is a method for extending a population of polynucleotides comprising: (a) adding adaptor to the polynucleotides to produce tagged polynucleotides; (b) amplifying the tagged polynucleotides to produce amplified polynucleotides using universal primers capable of hybridising to the adaptor sequences; (c) dividing the products of step (b) into two or more than two portions; and (d) treating at least one portion of the tagged polynucleotides for analysing sequence information in the tagged polynucleotides and/or (e) treating at least one portion of the amplified polynucleotides for analysing sequence information in the amplified polynucleotides, wherein the sequence information in steps (d) and (e) are different, wherein the step (d) treating at least one portion of the tagged polynucleotides comprises making sequencing library to determine the epigenetic sequence information in the tagged polynucleotides. Making sequencing library may comprise: (i) converting unmethylated cytosine nucleotides into uracil nucleotides to produce converted polynucleotides, wherein said conversion is by an agent which is either enzymatic reactions or chemical treatment; (ii) amplifying the converted polynucleotide to produce amplified converted polynucleotides using one of primers capable of hybridising to the adaptor sequences, wherein the adaptor sequence in the 3’ end of the tagged polynucleotides is resistant to conversion; and (iii) further treating the product of step (ii) to produce a sequencing library. The step (iii) may comprise treating the product of step (ii) by enzymatic digestion of converted polynucleotides at uracil nucleotide sites using an enzyme having glycosylase activity. In the step (e) treating the amplified polynucleotides of at least one portion may comprise making sequencing library of the amplified polynucleotide to determine the sequence information, including but not limited to: sequence variant (SNV), end motif, fragmentation size and break point (fragmentomics), or copy number variants (CNV). Making sequencing library may comprise: (i) amplifying whole genome, or a fraction of the whole genome, or specific targets; and (ii) further treating the product of step (i) to produce a sequencing library. The step (a) may comprise adding adaptor to at least the 3’ end of single-stranded or double stranded polynucleotides. Adding adaptor may be performed by ligation of adaptor to the ends of polynucleotides.The adaptor comprises methyl cytosine nucleotides. Adding adaptor may be performed by extension of 3’ ends of the polynucleotides, wherein the extension may be performed using ATOM-seq method, wherein the extension is performed in the presence of methyl dCTP in the reaction. The amplification may be linear amplification, alternatively the amplification maybe exponential amplification, the linearl amplification is preferred. Disclosed is a method for adding adaptor or tagging or extending a population of target polynucleotides comprising: (i) incubating the target polynucleotides with adaptor template oligonucleotides (ATO) having (a) a 3’portion comprising random or/and specific motif sequence; (b) a 3’ end with a blocker, which renders ATO non-extendible; and (c) a universal sequence, 5’ to 3’ portion, The incubating buffer comprises unusual dNTPs, wherein the target polynucleotides hybridise to the 3’ portion of the ATO; (ii) performing a polymerase extension of the target polynucleotides using the ATO as a template and incorporating unusual dNTP, thereby producing tagged polynucleotides having a 3’ universal sequence, which is resistant to enzymatic or chemical cleavage; (iii) (optional) treating with an enzyme having 3’ exonuclease activity or with bisulfite; and (iv) generating a first complement sequence (CS), comprising polymerase extension of primer hybridised to the 3’ universal sequence using the target polynucleotides as templates, wherein the unusual dNTPs may be phosphorothioate dNTP (Alpha Thiol dNTP) or methy-dCTP. Disclosed is a method for adding adaptor and extending a population of target polynucleotides comprising: (i) incubating the target polynucleotides with adaptor template oligonucleotides (ATO) having (a) a 3’ portion comprising a specific motif sequence and/or random sequence; (b) a 3’ end with a blocker, which renders ATO non-extendible; (c) at least one uracil or inosine nucleotide; (d) a universal sequence, 5’ to the 3’ portion; and (e) a stem-loop structure containing a non-copiable linkage, wherein the specific motif sequence is adjacent to the double-stranded end of the stem-loop structure, wherein the target polynucleotides hybridise to the 3’ portion of the ATO; (ii) performing a polymerase extension of the target polynucleotides using the ATO as a template, thereby producing tagged polynucleotides having a 3’ universal sequence; (iii) treating with an enzyme having dU-glycosylase activity or an enzyme having inosine-specific endonuclease activity; and (iv) generating a first complement sequence (CS), comprising polymerase extension of primers hybridised to the 3’ universal sequence using the target polynucleotides as templates, wherein the specific motif sequence may be 3 to 10 nucleotides long, or 4 to 6 nucleotides which is capable of capturing target polynucleotides containing the matching end motif. The ATO preferably comprises a random sequence which functions as a template for unique molecular identifiers (UMI) once copied to the target polynucleotides. Generating a first complement sequence (CS) may be a linear amplification, producing amplified products (polynucleotides) using the tagged polynucleotides as template. The methods above further comprise: (v) dividing the products of step (iv) into two or more than two portions; and (vi) treating at least one portion of the tagged polynucleotides for analysing sequence information in the tagged polynucleotides and/or (vii) treating at least one portion of the CS or amplified products for analysing sequence information in the amplified polynucleotides, wherein the sequence information in steps (vi) and (vii) are different. Sequence information in the tagged polynucleotide may be methylation information, as the tagged polynucleotide still contains the original methylation information, whereas the amplified polynucleotide has lost the methylation information. The sequence information in the amplified polynucleotide contains SNV, CNV, end motif, size, break point, fragmentomics, and fusion information and others. Disclosed is a kit for generating a library of polynucleotides comprising an adaptor template oligonucleotide (ATO), polymerases and primers compatible to an NGS platform. DETAILED DESCRIPTION While various embodiments of the compositions and methods have been shown and described herein, it will be obvious to those skilled in the art that such embodiments are provided by way of example only. Numerous variations, changes, and substitutions may occur to those skilled in the art without departing from the compositions and methods. It should be understood that various alternatives to the embodiments described herein may be employed. Accordingly, the drawings and description are to be regarded as illustrative in nature, and not as restrictive. To facilitate an understanding of the invention, a number of terms are defined below. As used herein, a "sample" refers to any substance containing or presumed to contain nucleic acids and includes a sample of tissue or fluid isolated from an individual or individuals. As used herein, the term "nucleotide sequence" refers to either a homopolymer or a heteropolymer of deoxyribonucleotides, ribonucleotides or other nucleic acids, or any combination of nucleic acids. As used herein, the term "nucleotide" generally refers to the monomer components of nucleotide sequences even though the monomers may be nucleoside and/or nucleotide analogues, and/or modified nucleosides such as amino modified nucleosides in addition to nucleotides. In addition, "nucleotide" also includes “nucleoside triphosphate” and non-naturally occurring analogue structures which may be naturally occurring or have been developed in selective or targeted approaches. The term “unusual nucleotide” and “nucleotide” may be used interchangeably with the term “unusual nucleotide” preferentially used in context of the present invention and may be used to describe any nucleotide which is in anyway functionally or chemically different from the four standard deoxynucleoside triphosphate (dNTPs) of deoxyadenosine triphosphate (dATP), deoxythymidine triphosphate (dTTP), deoxyguanosine triphosphate (dGTP) and deoxycytidine triphosphate (dCTP). As used herein, the term "nucleic acid" refers to at least two nucleotides covalently linked together. A nucleic acid of the present invention will generally contain phosphodiester bonds, although in some cases nucleic acid analogues are included that may have alternate backbones. Nucleic acids may be single-stranded or double-stranded, as specified, or contain portions of both double-stranded and single-stranded sequence. The nucleic acid may be DNA, both genomic and cDNA, RNA, DNA, DNA and RNA mixtures, or, DNA-RNA hybrids, where the nucleic acid contains any combination of deoxyribo- and ribo-nucleotides, and any combination of bases, including uracil, adenine, thymine, cytosine, guanine, inosine, xathanine, hypoxathanine, etc. Reference to a "DNA sequence" or “RNA Sequence” can include both single-stranded and double-stranded DNA or RNA. A specific sequence, unless the context indicates otherwise, refers to the single stranded DNA or RNA of such sequence, the duplex of such sequence with its complement (double stranded DNA or RNA) and/or the complement of such sequence. As used herein, the "polynucleotide" and "oligonucleotide" are types of "nucleic acid", and generally refer to primers, oligomer fragments to be detected. There is no intended distinction in length between the term "nucleic acid", "polynucleotide" and "oligonucleotide", and these terms will be used interchangeably. "Nucleic acid", "DNA" and similar terms also include nucleic acid analogues. The oligonucleotide is not necessarily physically derived from any existing or natural sequence but may be generated in any manner, including chemical synthesis, enzymatically, DNA replication, reverse transcription or any combination thereof. As used herein, the terms “original target polynucleotide”, "target sequence", "target nucleic acid", "target nucleic acid sequence", "target nucleic acid sequence" and "nucleic acids of interest" are used interchangeably and refer to a desired region which is to be either amplified, detected or both, or is the subject of hybridization with a complementary oligonucleotide, polynucleotide, e.g., a blocking oligomer, or the subject of a primer extension process. The target sequence can be composed of DNA, RNA, analogues thereof, or any combinations thereof. The target sequence can be single-stranded or double-stranded. In primer extension processes, the target nucleic acid which forms a hybridization duplex with the primer may also be referred to as a "template". A template serves as a pattern for the synthesis of a complementary polynucleotide. A target sequence for use with the present invention may be derived from any living or once living organism, including but not limited to prokaryotes, eukaryotes, plants, animals, and viruses, as well as synthetic and/or recombinant target sequences, it may also be a mixture of nucleic acids such that target nucleic acid is a subset of the total nucleic acids. "Primer" as used herein may be used interchangeably to describe, one or more than one primer or a set or plurality of multiple primers and refers to an oligonucleotide(s), whether occurring naturally or produced synthetically. The multiple primers in a set may have different sequences and hybridise to multiple different locations. The terms “first primer”, “a set of first primers” and “a first set of primers” are interchangeable, and the same applies to terms “second primer”. A “Primer” can be functionally described as a molecule capable of acting as a point of initiation of synthesis when placed under conditions in which synthesis of a primer extension product would be expected to occur, which is complementary to a nucleic acid strand is induced i.e., in the presence of nucleotides and an agent for polymerization such as DNA polymerase and at a suitable temperature and in a suitable buffer. Such conditions include the presence of one or more, two or more, three or more, or four or more different deoxyribonucleoside triphosphates which may include but is not limited to deoxyadenosine triphosphate (dATP), deoxythymidine triphosphate (dTTP), deoxyguanosine triphosphate (dGTP) and deoxycytidine triphosphate (dCTP) or suitable additional or replacement nucleotides, unusual nucleotides, and, a polymerization-inducing agent such as DNA polymerase and/or RNA polymerase and/or reverse transcriptase, in a suitable buffer ("buffer" includes substituents which are cofactors, or affect pH, ionic strength, etc.), and at a suitable temperature. The primer is preferably single-stranded for maximum efficiency in amplification. The primers herein are selected to be substantially complementary to a strand of each specific sequence to be amplified. This means that the primers must be sufficiently complementary to hybridize with their respective strands. One or more regions of non-complementary sequence may be attached to the 5' -end of the primer (5' tail portion) or in the primer (bulge portion), with the remainder of the primer sequence being complementary to the desired section of the target base sequence. Commonly, the primers are complementary, except when non-complementary nucleotides may be present at a predetermined primer terminus or middle region as described. In another expression, the primers herein are selected to be substantially identical to a strand of each specific sequence to be amplified. This means that the primers must be sufficiently identical to one strand, so that they can hybridize with their respective other strands. As used herein, the term “adaptor” is used to describe an oligonucleotide which is designed to be used in a rection as a substrate for a ligase or template for extension. An adaptor may be comprised of functional components. The term functional component may be used to interchangeable describe any position(s) or nucleotide(s) in the adaptor. As used herein, the term "complementary" refers to the ability of two nucleotide sequences, either randomly or by design, to bind in a sequence complementary dependent manor to each other by hydrogen bonding through their purine and/or pyrimidine bases according to the usual Watson- Crick rules for forming duplex nucleic acid complexes. It can also refer to the ability of nucleotide sequences that may include modified nucleotides or analogues of deoxyribonucleotides and ribonucleotides, or combinations thereof, to bind sequence-specifically to each other by other than the usual Watson Crick rules to form alternative nucleic acid duplex structures. As used herein, the term "hybridization" and "annealing" are interchangeable, and refers to the process by which two nucleotide sequences complementary to each other, either partially or fully, bind together to form a duplex sequence or segment. The terms "duplex" and "double-stranded" are interchangeable, meaning a structure formed as a result of hybridization between two complementary sequences of nucleic acids. Such duplexes can be formed by the complementary binding of two DNA segments to each other, two RNA segments to each other, or of a DNA segment to an RNA segment, or two segments composed of a mixture of RNA and DNA to one another, the latter structure being termed as a hybrid duplex. Either or both members of such duplexes can contain modified nucleotides and/or nucleotide analogues as well as nucleoside analogues. As disclosed herein, such duplexes can be formed as the result of binding of one or more blocking oligonucleotides to a sample sequence. The duplex may be partially or completely complementary and may be partially or fully double stranded. As used herein, the terms "wild-type nucleic acid", "normal nucleic acid", "nucleic acid with normal nucleotides", “wild-type”, “normal”, "wild-type DNA" and "wild-type template" are used interchangeably and refer to a polynucleotide which has a nucleotide sequence that is considered to be normal or unaltered. As used herein, the term "mutant polynucleotide", "mutant nucleic acid", "variant nucleic acid", and "nucleic acid with variant nucleotides", refers to a polynucleotide which has a nucleotide sequence that is different from the expected nucleotide sequence of the corresponding wildtype polynucleotide. The difference in the nucleotide sequence of the mutant polynucleotide as compared to the wild-type polynucleotide is referred to as the nucleotide "mutation", "variant nucleotide", “variant” or "variation." The term "variant nucleotide(s)" also refers to one or more nucleotide(s) substitution(s), deletion(s), insertion(s), methylation(s), and/or modification changes. "Amplification" as used herein denotes the use of any amplification procedures to increase the concentration or copy number of a particular nucleic acid sequence within a mixture of nucleic acid sequences. Amplification can be one or more round of linear amplification, one or more rounds of exponential amplification or a combination thereof. “Replication” or “replicate” as used herein denotes making a complementary copy of a polynucleotides which is a template for polymerase extension. Many rounds of replication result in amplification. The terms "reaction mixture", "amplification mixture" or "PCR mixture" as used herein refer to a mixture of components necessary to amplify at least one product from nucleic acid templates. The mixture may comprise one or more nucleotides (dNTPs), a polymerase (thermostable or not thermostable), primer(s), and a plurality of nucleic acid templates and other unusual nucleotide(s) necessary for the disclosed invention. The mixture may further comprise a Tris buffer, a monovalent salt and Mg
2+. The concentration of each component, apart from the unusual nucleotide as necessary for the disclosed invention, is well known in the art and can be further optimized by an ordinary skilled artisan. The terms “amplified product” or “amplicon” refer to a fragment of DNA or RNA amplified by a polymerase a primer, pool of primer, a pair of primers, a pool of pairs of primers or any combination thereof in an amplification method. The terms “tagged target polynucleotide”, “amplified copies of the target polynucleotide”, “amplified polynucleotide”, “tagged amplified polynucleotide”, “converted tagged polynucleotide”, “converted polynucleotides”, “amplified polynucleotides”, “amplified converted polynucleotide” and “tagged amplified converted polynucleotide” are used in way of an example of types of polynucleotide which may be used in the context of their mention, this does not limit the polynucleotide to just that which is mentioned. The terms “primer extension product” refer to a fragment of DNA or RNA extended by a polymerase using one or a pair of primers in a reaction, which may involve one pass extension, for example first strand cDNA synthesis, or two pass extension, for example double strand cDNA syntheses, or many cycles of extension, which may be a PCR. The term “compatible” refers to a primer sequence or a portion of primer sequence which is identical, or substantially identical, complementary, substantially complementary or similar to a PCR primer sequence/sequencing primer sequence used in a massive parallel sequencing platform. The practice of the present invention will employ, unless otherwise indicated, conventional techniques of molecular biology, microbiology, recombinant DNA, and next generation sequencing techniques, bioinformatics, data analysis, which are within the skill of a person skilled in the art. All patents, patent applications, and publications mentioned herein, both supra and infra, are hereby incorporated by reference as if each individual publication, patent, or patent application was specifically and individually indicated to be incorporated by reference. Disclosed herein are methods, systems, and compositions that can significantly increase the amount of information which can be obtained from a single patient sample compared to other technologies, especially those trying to combine detection of both genetic and epigenetic information as the invention allows for testing of one sample with multiple methods while minimising the loss of sensitivity of having to divide the starting material between different tests. DNA methylation is an epigenetic modification that plays a role in regulating gene expression and, consequently, can influence a variety of biological processes and diseases. The addition of a methyl group to a base present in a nucleotide of a polynucleotide, for example at the 5' position of a cytosine residue, can be a mechanism in gene expression, chromatin structure regulation, or both. The functional presence of this methylated nucleotide, e.g., 5mC (5- methylcytosine), in gene promoters can be associated with transcriptional repression, in some cases due to structural chromatin alterations, while the absence of 5mC can be linked with transcriptional activity. Methylation of cytosines to form 5-methylcytosine (5mC or mC), e.g., at cytosines followed by guanine residues (e.g., cytosine-phosphate-guanine motifs, or CpGs), can be an epigenetic mark with important roles in development and tissue specificity, genomic imprinting, and environmental responses. Dysregulation of 5mC can cause aberrant gene expression, and in some cases can affect cancer risk, progression or treatment response.5- hydroxymethylcytosine (5hmC or hmC) can be an intermediate in the cell’s active DNA demethylation pathway with tissue-specific distribution affecting gene expression and carcinogenesis Gene body DNA methylation (as used herein, methylation can mean addition of or the presence of a methyl group on a base of a nucleic acid; the methyl group can be in an oxygenated or unoxygenated state; an unoxygenated methyl group can be e.g., methyl; an oxygenated methyl group can be a hydroxymethyl, a formyl group, a carboxylic acid group, or a salt of carboxylic acid) can play a role in repetitive DNA elements’ silencing and alternative splicing. DNA methylation can be associated with several biological processes such as genomic imprinting, transposon inactivation, stem cell differentiation, transcription repression, and inflammation. DNA methylation profiles can in some cases be inherited through cell division and sometimes through generations. Since methyl marks can play a very relevant role in both physiologic and pathologic conditions, there may be significant application for profiling DNA methylation to answer biological questions. Moreover, uncovering of DNA methylation genomic regions can be appealing to translational research because methyl sites can be modifiable by pharmacologic intervention. The method presented herein can be used to determine and identify a genetic change and/or an epigenetic modification and/or a gene fusion/chromosomal rearrangement event present in an original target polynucleotide. In some cases, the target polynucleotide is amplified and the amplified polynucleotides are divided into two or more portions where one or more portion is processed to by one or more workflows to generate one or more different final NGS libraries using the amplified polynucleotide as a starting point, where a second portion is processed and treated by an agent to differentially convert cytosine and methylated cytosines of the tagged target polynucleotide where the converted tagged polynucleotide is amplified to generate amplified converted polynucleotides which are divided into one or more portions where one or more portion is processed to by one or more workflows to generate one or more different final NGS libraries using the amplified converted polynucleotide as a starting point. All the different final libraries derived from the original target polynucleotide are pooled and sequenced with any suitable method which includes but is not limited to Pacific Biosciences, Ion Torrent sequencing, Illumina, Nanopore, GenapSys, MGI, and/or Element Biosciences. The pooled sequencing data, depending on how it has been processed, may be analysed to produce some or all of the following disease associated changes which include: • Single nucleotide changes • Insertions • Deletions • Copy number variation • Chromosomal rearrangements • Including gene fusions • Epigenetic changes • Changes in relative or absolute DNA break points (fragmentomics) • From whole genome • From GC rich DNA • Of specific DNA break point ends In some cases, these data are further enhanced by the use of unique molecule identifiers (UMI), or, molecular barcodes (MB). This enhanced error correction allows for higher sensitivities for identification of ultra-low level variants when compared to that necessary for approaches not utilising UMIs, such as when original target polynucleotides are tumour DNA molecules or fragments thereof derived from a liquid biopsy sample obtained from a subject. In some cases, genetic mutations, epigenetic mutations, or fusions can be determined using a computer program (e.g., comprising instructions for the analysis of sequencing data and/or for performing one or more operations of a method presented herein). In some cases, such a computer program can be stored on a memory of a computer. In many cases, methods and systems presented herein comprise tagging step for the generation of a tagged molecule comprising an original target polynucleotide, which in many cases, has a 3’ extension forming a tagged target polynucleotide. This tag allows for the amplification of the tagged target polynucleotide, by producing many amplified copies it is therefore possible to divide the copies in to portions where each portion can be processed as replicates and/or differently, where each portion has representatives of many or all the original target polynucleotide. As portions may take a small part of the amplification reaction, a large portion may be taken which contains the majority of the tagged target polynucleotide allowing it to be further processed, in many cases by an agent, to differentially convert cytosine and methylated cytosine where the produced converted tagged polynucleotide is further amplified, by producing many amplified converted polynucleotides copies it is there for possible to divide the copies in to portions where each portion can be processed differently, where each portion has representative of all the original target polynucleotide which has been converted by an agent. In many cases, the portion of the first amplified polynucleotides of the tagged polynucleotide may be preferentially used to generate sequencing data for identifying; • Single nucleotide changes (SNV) • Insertions • Deletions • Copy number variation (CNV) • Chromosomal rearrangements • Including gene fusions • Changes in relative or absolute DNA break points (fragmentomics) • From whole genome • From GC rich DNA • Of specific DNA break point end In many cases, the portion of the amplified converted polynucleotides of the converted tagged polynucleotide may be preferentially used to generate sequencing data for identifying; • Epigenetic changes In some cases, portions of the amplified polynucleotides and amplified converted polynucleotides or any portions thereof may be used to identify some or all or any combination of the above detailed genetic sequencing information. In some cases, not all of the above information is obtained. In some cases, different or additional information is obtained. Methods and systems presented herein can comprise providing and/or (e.g., chemical or enzymatic) processing a single- or double-strand DNA polynucleotide. A single- or double-strand DNA polynucleotide can comprise an original target polynucleotide, described herein. In some cases, an original target polynucleotide is captured or tagged to (e.g., via an addition to its 3’, its 5’, or 3’ and 5’) preferentially generate a 3’ tagged polynucleotide where the 3’ tagged with contain a universal priming site. Preferentially, the method for generating a 3’ extension is an ATO-Reaction which is provided in WO2018/193233 A1, which is incorporated by reference herein in its entirety. In many cases, a 3’ extension reaction may comprise the use of one or more unusual nucleotides which may include any combination or mixture of, 5-Methyl-2'-deoxycytidine-5'- Triphosphate, 5-hydroxyMethyl-2'-deoxycytidine-5'-Triphosphate, 5-formyl-2'-deoxycytidine-5'- Triphosphate, 5-Carboxy-2'-deoxycytidine-5'-Triphosphate, 2´-Deoxyuridine, 5´-Triphosphate, 2´- Deoxyuridine, 5´-Triphosphate and/or 2'-deoxyinosine 5'-triphosphate. In some cases, unusual nucleotides may comprise an (α-thio)-triphosphate in place of a triphosphate to produce polymerase extension products which are resistant to exonucleases due to the substitution of an Oxygen atom with a Sulphur atom. In some cases, a method for extending a population of target polynucleotides comprises: (i) incubating the target polynucleotides with adaptor template oligonucleotides (ATO) having (a) a 3’portion comprising random or/and specific motif sequence; (b) a 3’ end with a blocker, which renders ATO non-extendible; and (c) a universal sequence, 5’ to the 3’ portion, The incubating buffer comprises unusual dNTPs, wherein the target polynucleotides hybridise to the 3’ portion of the ATO; (ii) performing a polymerase extension of the original target polynucleotides using the ATO as a template and incorporating unusual dNTP, thereby producing tagged target polynucleotides having a 3’ universal sequence, which is resistant to enzymatic or chemical cleavage; (iii) treating with an enzyme having 3’ exonuclease activity or with bisulfite; and (iv) generating a first amplified polynucleotides, wherein generating the first amplified polynucleotides comprises polymerase extension from primers hybridised to the 3’ universal sequence using the target polynucleotides as templates. In some cases, the ATO has a hairpin structure. In some cases, the ATO has a region which when copied to the tagged polynucleotides has the function of a unique molecular identifies (also known as a molecular barcode). In some cases, the ATO contains nucleotides which allow it to be acted on by an agent leading to it degradation or otherwise rendering it non-functional. In some cases, a method for tagging or extending a population of target polynucleotides comprising: (i) incubating the target polynucleotides with adaptor template oligonucleotides (ATO) having (a) a 3’portion comprising random or/and specific motif sequence; (b) a 3’ end with a blocker, which renders ATO non-extendible; and (c) a universal sequence, 5’ to 3’ portion, The incubating buffer comprises unusual dNTPs, wherein the target polynucleotides hybridise to the 3’ portion of the ATO; (ii) performing a polymerase extension of the target polynucleotides using the ATO as a template and incorporating unusual dNTP, thereby producing tagged polynucleotides having a 3’ universal sequence, which is resistant to enzymatic or chemical cleavage; (iii) treating with an enzyme having 3’ exonuclease activity or with bisulfite; and (iv) generating a first complement sequence (CS), comprising polymerase extension of primer hybridised to the 3’ universal sequence using the target polynucleotides as templates. The unusual dNTPs may be phosphorothioate dNTP (Alpha Thiol dNTP), or methyl-dCTP. In some cases, a method for extending a population of target polynucleotides comprising: (i) incubating the target polynucleotides with adaptor template oligonucleotides (ATO) having (a) a 3’ portion comprising a specific motif sequence and/or random sequence; (b) a 3’ end with a blocker, which renders ATO non-extendible; (c) at least one uracil or inosine nucleotide; (d) a universal sequence, 5’ to the 3’ portion; and (e) a stem-loop structure containing a non-copiable linkage, wherein the specific motif sequence is adjacent to the double-stranded end of the stem-loop structure, wherein the target polynucleotides hybridise to the 3’ portion of the ATO; (ii) performing a polymerase extension of the original target polynucleotides using the ATO as a template, thereby producing tagged target polynucleotides having a 3’ universal sequence; (iii) treating with an enzyme having dU-glycosylase activity or an enzyme having inosine-specific endonuclease activity; and (iv) generating a first complement sequence (CS), comprising polymerase extension of primers hybridised to the 3’ universal sequence using the target polynucleotides as templates. wherein the specific motif sequence is 3 to 10 nucleotides long. wherein the specific motif sequence is 4 to 6 nucleotides which is capable of capturing target polynucleotides containing the matching end motif. wherein the ATO comprises a random sequence which functions as a template for unique molecular identifiers (UMI) once copied to the target polynucleotides. In many cases the ATO design is any combinations of designs or function by any method here in, and/or, any combination of designs or functions or methods as found in WO2018/193233 A1. In many cases the ATO will have the following design: (a) a 3’ end with a blocker, which renders ATO non-extendible; (b) 5’ to (a) one of the following or any combination or components thereof (5’ to 3’) (i) a random sequence of 3 to 36 ‘N’ bases; (ii) a target specific region of 4 to 10 bases followed by a random region of 3 to 32 bases (c) a universal sequence, 5’ (b); (d) one or more moieties which renders the ATO degradable. In one embodiment, the moieties are uracil nucleotides, wherein the agent is a dU-glycosylase, or a dU-glycosylase and apurinic/apyrimidinic endonuclease, which is capable of digesting/removing the ATO following the first extension reaction. In another embodiment, the moieties are ribonucleotides, wherein ribonucleotides are incorporated during oligo synthesis into the ATO in the place of any nucleotides or all nucleotides; wherein the agent is a ribonuclease, which is capable of digesting/removing the ATO following the first extension reaction. In another embodiment, the moieties are deoxyinosine, wherein deoxyinosine are incorporated during oligo synthesis into the ATO in the place of any nucleotides or all nucleotides; wherein the agent is an enzyme, which is capable of digesting/removing the ATO following the first extension reaction. The enzyme may be an endocucleases which may be Endonuclease V, the enzyme may be a glycosylase which may be human alkyl adenine DNA glycosylase. The ATO may be an RNA oligo, or a DNA oligo, or a combination of DNA and RNA oligos and may have other non-canonical nucleotides. The ATO may be a combination of one or more different ATO. The combination of ATO may vary in sequence. The combined ATOs may vary in design. The combined ATO may vary in function. Herein the term ‘ATO’ may refer to a combination of one or more ATOs, to any sequence of ATO, to any designed of ATO with any combination of ATO design features, to any combination of ATO with any combination of functions. When using a combination of one or more ATO there may be variation within the universal sequence with the ATO used, the term universal sequence is used in these cases as well. The universal sequence may be double-stranded or partially double-stranded. Having the universal sequence protected as a double stranded region prevents hybridisation with the randomised 3’-ends of the original target polynucleotide and the ATO. In one embodiment, the ATO comprises a 5’ stem portion sequence which is complementary or partially complementary to all or part of the universal sequence, which are capable of forming a stem-loop structure or split stem-loop structure. Alternatively, the loop part may not comprise a non-copiable linkage. Alternatively, the stem part may comprise a non-copiable linkage. If the 5’ of the stem portion comprises an additional sequence, a non- copiable linkage may be present between the stem portion and the additional sequence. Alternatively, the stem part may not comprise a non-copiable linkage. In another embodiment, the 5’ stem portion comprises a non-copiable linkage. The non-copiable linkage may be selected from group but not limited to C3 Spacer phosphoramidite, or a triethylene glycol spacer, or an 18-atom hexa- ethyleneglycol spacer, or 1’,2’-Dideoxyribose (dSpacer). The double-stranded stem part may comprise non-complementary region(s), wherein the non- complementary region(s) in the universal sequence strand comprises a random, a degenerate sequence(s), or a specially designed mismatch. The stem portion may form two or more split sections separated by one or more non-copiable linkage(s). The stem portion may form two or more split sections separated by one or region of mismatches base pairs. The ATO may further comprise a specific sequence 5’ and/or 3’of the random sequence, wherein the specific sequence is capable of hybridising to a specific place of the target polynucleotide, or a specific sequence which is not designed for a specific target, and part of the 3’ random/degenerated sequence serves as templates on which the polynucleotide is extended by a polymerase. The 3’ random sequence of the ATO may be separated into two or more portions by specially designed specific sequences, and part of the 3’ random/degenerated/target specific sequence serves as templates on which the target polynucleotide is extended by a polymerase. In some cases ligation is used, the 5’ end of the ATO may comprise a phosphate group, the 3’ end of the upper separate strand may comprise a biotin. In the embodiments where the extension is used without ligation, the 5’ end of the upper separate strand does not comprise a phosphate group, the 3’ end of the upper separate strand does not comprise a biotin, but the upper separate strand may comprise nucleotides which can be digested such as uracil nucleotide. A original target polynucleotide may be randomly or specifically or a combination of randomly and specifically fragmented either naturally or artificially. The 3’ end of the fragmented target polynucleotide is tagged, for example by extending after hybridisation with the random and/or target specific sequence of an ATO. The combination of the random 3’ end sequence of the target polynucleotide and extended part on random template provides a unique identification (UMI, UID, molecular barcode) sequence which can be used for grouping sequencing reads into family after the generation of sequencing data. In addition, ATO may comprise one or more additional UID(s) located in the universal sequence. Fragmentation may have happened naturally prior to or after collection and extraction of the original target polynucleotide, for example by nucleases present in liquid biopsy samples. It may be accomplished by the use of physical shearing such as a sonicator, enzymatically (e.g using nucleaseses, DNAses, exonucleases, endonucleases, transposases), targeted fragmentation using in-vitro or in-vivo crisper systems. In some cases, the target polynucleotide may be tagged generating a tagged target polynucleotide by any suitable method which may generate tagging at the 3’ and/or 5’ ends of original target polynucleotide including ligating single strand adaptor(s), ligating double strand adaptor(s), an ATOM-Seq reaction, a Terminal deoxynucleotidyl transferase (TdT) reaction (following any method of US9896709B2), whole genome amplification of any combination of the original polynucleotide or any polynucleotide generated by any methods detailed herein. In some cases, the ligation-based reactions may or may not include end repair steps which may including one or more of filling in 5’ overhangs, trimming 3’ overhangs, unblocking 3’ ends, generating double-stand blunt ends, generating ‘A-tailed’ double strand ends, (de-)phosphorylating 5’ and/or 3’ ends. In some cases, the adapters to be ligated may be fully or partially single strand, double strand, have hairpins, have a DNA or RNA splint, the split may be target specific or random, have a 5’ overhang, have a 3’ overhang, be comprised of RNA and/or DNA, have phosphorylated 3’/5’ ends or have 3’/5’ -OH groups, have non-canonical nucleotides including uracil, methyl-cytosine, inosine, a- basic sites, linkers (e.g. C3), affinity moieties (e.g. biotin), be blunt ended or have a T nucleotide overhang, require nick extension prior to ligation, may follow Golden Gate Assembly methods to concatenate nucleotides, may be “Y” shaped, may incorporate single end duplex-UMIs (WO2018013598A1). In some cases, the ligases that may be used to practice the methods of the disclosure include but are not limited to T3 DNA ligase, T4 DNA ligase, SplintR® Ligase, Taq DNA ligase, HiFi Taq DNA Ligase, T4 RNA ligase, E. coli DNA ligase, Instant Sticky-end Ligase, and E. coli RNA ligase. In one embodiment, generating amplified polynucleotide or amplified converted polynucleotide involves one or more first primers. A first primer may anneal to the 3’ universal region of the tagged target polynucleotides and a second first primer may be capable of annealing to copies of the 5’ end of tagged target polynucleotides. These primers may be used to generate linear and or PCR amplified polynucleotide and/or linear and or PCR amplified converted polynucleotide. First primers may be a mixture of universal primers and target specific primer, which may have a 3’ target specific region and a 5’ tail, the tail on the target specific primer may be different to the universal primers so that you can differentially non-specifically amplify the tagged target polynucleotide and selectively amply the regions of interest. In one embodiment, generating amplified polynucleotides comprises extension using a first primer, and using the tagged target polynucleotides as template, wherein the first primer is hybridised to the tagged target polynucleotides and is extended by a polymerase. The first primer anneals to the 3’ universal region of the tagged target polynucleotides. The first primer may comprise additional sequence compatible to a NGS platform, for example a 5’ tail containing necessary sequences. The first primer may comprise unusual nucleotides such as uracil nucleotide or inosine (dinosine) nucleotide, which make the first primer uncopiable by a polymerase. The polymerase may be enzyme with 3’ exonuclease activity such as Phusion, Pfu, Kod, Q5 polymerases etc. The uncopiable first primers ensure the amplification is linear amplification. In yet another embodiment, generating amplified polynucleotides comprises heat denaturing the tagged target polynucleotide, self-annealing a 3’ stem-loop structure generated during the 3’ extension reaction of the tagged polynucleotide and self-priming to extend to form the amplified polynucleotides. In yet another embodiment, said generating amplified polynucleotides comprises target specific primer annealing to the tagged target polynucleotide and extension by a polymerase. The target- specific primer anneals to the target sequence of interest. The double-stranded end of the amplified polynucleotides can be ligated to an adaptor by any suitable method such as “A-tail” or blunt-end ligation methods. The method may further comprise digesting ATO before or after generating a amplified polynucleotides and or amplified converted polynucleotides of the converted target polynucleotide. The method may further comprise affinity capturing before or after generating a amplified polynucleotides and or amplified converted polynucleotides of the tagged target polynucleotide or converted tagged polynucleotide. In the first ATO reaction, the method may comprise ligation, with or without an extension, wherein if the 3’ ends are extended a polymerase extends the 3’ end of the target, and a ligase ligates the extended target sequence to the 5’ stem portion of ATO or upper separate strand of ATO. In some cases, the 3’ ends of the DNA anneal with the ATO in a way which can be immediately ligated to the ATO without the need for an extension reaction. The method may further comprise generating a tagged target polynucleotides or tagged amplified polynucleotides or tagged amplified converted polynucleotides by incubating amplified polynucleotides or amplified converted polynucleotides with any of the compositions or methods described, wherein the 3’ end of the amplified polynucleotides or amplified converted polynucleotides hybridises to the 3’ random sequence of a second adaptor template oligonucleotide (second ATO) in an enzymatic second ATO reaction, in which the 3’ end of the amplified polynucleotides or amplified converted polynucleotides is extended using an ATO as a template, wherein the second ATO comprises a different 5’ universal sequence of the first ATO. The 5’ and 3’ ends of the tagged amplified polynucleotide and tagged amplified converted polynucleotide therefore have different tagged sequences which can be used for whole sample amplification. The method may further comprise extending a second primer hybridized to the tagged or not tagged, amplified polynucleotides or amplified converted polynucleotides, thereby generating a copied amplified polynucleotides, wherein the second primer comprises a target-specific portion or universal sequence, or both 3’ target specific and 5’ universal sequence. The method may further comprise hybridising a primer complementary to the universal region of the tagged target polynucleotide, followed by one or more rounds of linear amplification to generate amplified polynucleotides. The linear amplification reaction product may be separated into two or more separate portions, where such portions are used in a reaction which contains a primer complementary to the universal region of the tagged target polynucleotide, with a target-specific second primer, or pool of target-specific primers, complementary to forward strand of a target sequence, and a second reaction comprising a target-specific second primer, or pool of target-specific primers, complementary to reverse strand of the target sequence, wherein forward strand and reverse strand of the target sequence are complementary. In another embodiment, the pools of target-specific primers can contain a mixture of primers, where individual primers can target either the forward or reverse strand for different targets, such that the final pool can target different regions of both the forward and reverse strands. Wherein the forward strand and reverse strand of the target sequence are complementary, and the forward and reverse target specific primers are separated into different pools of primers as long as consideration is taken that no two primers which target the forward and reverse strands are added to the same pool if they are capable of together acting as primers for PCR resulting in the generation of unwanted PCR products. Further, all references to ‘forward’ and/or ‘reverse’ pools allows for each pool to contain primers which target both the forward and reverse strands, as mentioned above. The method may further comprise an end repair step of the double strand target polynucleotide followed by ligation of a double strand adaptor. The ligation product is then hybridised with a primer complementary to the universal region of the target polynucleotide ligation product, followed by one or more rounds of linear amplification to generate amplified polynucleotides. The amplified polynucleotides are separated into two or more portions or two reactions, where each reaction contains a primer complementary to the universal region of the tagged target polynucleotide, with a reaction comprising a target-specific second primer, or pool of target-specific primers, complementary to forward strand of a target sequence, and a second reaction comprising a target- specific second primer, or pool of target-specific primers, complementary to reverse strand of the target sequence, wherein forward strand and reverse strand of the target sequence are complementary. In one aspect, generating the amplified polynucleotides comprises one pass extension or linear amplification or PCR using the first primer, which is a universal primer targeting the 3’ ends of the tagged polynucleotides or 3’ and 5’ ends of the tagged polynucleotides. The amplification may have 1- 30 cycles, 2-25cycles, 3-24 cycles, 4-23 cycles, 5-22 cycles, 6-21 cycles, 7-20 cycles 8-19 cycles 9-18 cycles 10-17 cycles, 31-40 cycles, 41-50 cycles, 51-100 cycles, or more cycles of amplification. The method may further comprise exponential amplification using first primer and second primer. The first primer may be a universal primer targeting the 3’ extended universal part of the tagged target polynucleotide; the second primer may be a universal primer targeting the 3’ extended universal part of the first amplified product. Alternatively, the second primer is a target specific primer annealing to a specific region of interest of the amplified product. The second primer may be a set of multiple primers targeting multiple sequence regions of interest. When the second primer is a target- specific primer, after linear or exponential amplification using the second primer, a nested target- specific third primer is used for a further amplification. The first primer, second primer, or third primer may comprise a sample barcode (SBC) sequence and additional universal sequence(s) necessary for compatibility with an NGS platform. The original target polynucleotide is preferably fragmented either naturally or artificially. The target polynucleotide may be any nucleic acids such as DNA, cDNA, RNA, mRNA, small RNA, or microRNA, or any combination thereof. The original target polynucleotide may comprise a plurality of target polynucleotides. Each of the target polynucleotides of the plurality may comprise different sequences or the same sequence. One or more of the target polynucleotides or plurality of target polynucleotides may comprise a variant sequence. Depending on the type of original target polynucleotide and ATO which are either DNA and/or RNA, the methods can utilize reverse transcription or primer extension. A primer extension reaction can be a single primer extension step. A primer extension reaction can comprise extending one or more individual primers once. A primer extension reaction can comprise extending one or more individual primers in one step. In one step the 3’ end, or trimmed 3’ end, of the original target polynucleotides act as primer, which is extended using ATO as template, the extension or amplification primers are the first primer which anneal to the 3’ extended part of the target polynucleotide. In one embodiment in the 3’ end of the original target polynucleotides acting as primers are hybridised to the ATO through the 3’ end of the sequence which may be randomly fragmented or specifically fragmented, are trimmed or not trimmed to remove 3’ overhang, if any is present, by the 3’ to 5’ exonuclease activity of a polymerase, and is extended using ATO as template to generate tagged target polynucleotides. Because of the nature of randomness of ATO’s 3’ random sequence portion, the perfect hybridisation between the 3’ end of the target polynucleotides and a ATO may not be readily obtained, less stringent conditions are applied. For example, high concentration of ATO may be used, low temperature of hybridization such as 4 degree C may be used, and/or multiple cycle of extension may be used, and/or longer hybridization times. Extension may be carried out by any polymerase and/or any reverse transcriptase, or mix of different polymerases. The DNA polymerase may have a 3’ to 5’ exonuclease activity, so that any 3’ overhang if present is digested (trimmed) and extension can take place. The DNA polymerase may contain strand displacement activity, so that the stem-loop structure and the double-stranded universal portion of the ATO molecules can be opened and copied. Alternatively, the DNA polymerase may contain 5’ to 3’ exonuclease activity, so that the 5’ end of stem-loop structure and the double-stranded universal portion of the ATO molecules can be digested and the lower strand of ATO is copied. The DNA polymerase is preferably active at low temperature. The polymerase may contain a mix of different polymerases which may have 3’ to 5’ exonuclease activity, 5’ to 3’ exonuclease activity and/or strand displacement activity. The polymerases that may be used to practice the methods disclosed herein include but are not limited to Deep VentR™ DNA Polymerase, LongAmp™ Taq DNA Polymerase, Phusion™ High-Fidelity DNA Polymerase, Phusion™ Hot Start High-Fidelity DNA Polymerase, VentR® DNA Polymerase, DyNAzyme™ II Hot Start DNA Polymerase, Phire™ Hot Start DNA Polymerase, Crimson LongAmp™ Taq DNA Polymerase, DyNAzyme™ EXT DNA Polymerase, LongAmp™ Taq DNA Polymerase, Taq DNA Polymerase with Standard Taq (Mg- free) Buffer, Taq DNA Polymerase with Standard Taq Buffer, Taq DNA Polymerase with ThermoPol II (Mg-free) Buffer, Taq DNA Polymerase with ThermoPol Buffer, Crimson Taq™ DNA Polymerase, Crimson Taq™ DNA Polymerase with (Mg-free) Buffer, VentR® (exo-) DNA Polymerase, Hemo KlenTaq™, Deep VentR™ (exo-) DNA Polymerase, ProtoScript® AMV First Strand cDNA Synthesis Kit, ProtoScript® M-MuLV First Strand cDNA Synthesis Kit, Bst DNA Polymerase, Full Length, Bst DNA Polymerase, Large Fragment, Taq DNA Polymerase with ThermoPol Buffer, 9° Nm DNA Polymerase, Crimson Taq™ DNA Polymerase, Crimson Taq™ DNA Polymerase with (Mg-free) Buffer, Deep VentR™ (exo-) DNA Polymerase, Deep VentR™ DNA Polymerase, DyNAzyme™ EXT DNA Polymerase, DyNAzyme™ II Hot Start DNA Polymerase, Hemo KlenTaq™, Phusion™ High-Fidelity DNA Polymerase, Phusion™ Hot Start High-Fidelity DNA Polymerase, Sulfolobus DNA Polymerase IV, Therminator™ γ DNA Polymerase, Therminator™ DNA Polymerase, Therminator™ II DNA Polymerase, Therminator™ III DNA Polymerase, VentR® DNA Polymerase, VentR® (exo-) DNA Polymerase, Bsu DNA Polymerase, Large Fragment, Bst DNA Polymerase, Large Fragment, DNA Polymerase I (E. coli), DNA Polymerase I, Large (Klenow) Fragment, Klenow Fragment (3ʹ→5ʹ exo-), phi29 DNA Polymerase, T4 DNA Polymerase, T7 DNA Polymerase (unmodified), Reverse Transcriptases and RNA Polymerases, AMV Reverse Transcriptase, M-MuLV Reverse Transcriptase, phi6 RNA Polymerase (RdRP), SP6 RNA Polymerase, and T7 RNA Polymerase. Ligases that may be used to practice the methods of the disclosure include but are not limited to T4 DNA ligase, T4 RNA ligase, E. coli DNA ligase, and E. coli RNA ligase. Multiple cycles of extension may be carried out through repeated cycling of temperatures: annealing, extension, and denaturing. The tagged target polynucleotide has extended 3’ part, which may comprise some random sequence and a universal sequence providing a binding site for a primer. The extended 3’ part may also comprise additional UID. In one embodiment, in the universal portion of ATO comprises weak pairing nucleotides such as Inosine, the removal of ATO after first extension reaction may be not needed. Otherwise the removal or digestion of ATO from the reaction mix may be performed by any means. For example, if the ATO comprises Uracil residues, the ATO is digested/removed by UNG digestion; if the ATO comprises RNA, the ATO is digested/removed by RNase digestion; if the ATO comprises restriction enzyme site, the ATO is digested/removed by restriction enzyme digestion; or if the ATO comprises biotin, the ATO is removed by capturing on streptavidin beads. In another embodiment, the ATO may not need to be digested or removed from the reaction mix if the ATO comprises the hairpin/stem-loop structure (Fig. 3B and 3C), as the hairpin structure of ATO makes the hybridisation of ATO to the 3’ extended part of the tagged target polynucleotide impossible. In one embodiment, the first ATO reaction is a primer extension reaction, wherein the target polynucleotide serves as primer is extended on the ATO template by a DNA polymerase. The DNA polymerase may comprise a strand displacement activity or 5’ to 3’ exonuclease activity, wherein during the extension the stem-loop structure is opened or the upper ATO strand is displaced or digested. Any polymerase can be use, for example Klenow exo-, Bst polymerase, or T4 DNA polymerase. In another embodiment, the first reaction is an extension-ligation reaction, wherein a DNA polymerase extends the original target polynucleotide and a DNA ligase ligates the extended target sequence to the 5’ stem portion of ATO or upper strand of ATO. Any DNA polymerase and DNA ligase can be used, for example, Klenow large fragment, T4 DNA ligase. In another embodiment, the first reaction is a ligation reaction, wherein a DNA ligase ligates the target polynucleotide to the 5’ stem portion of ATO or upper strand of ATO. Any DNA ligase can be used, for example T4 DNA ligase. After the first reaction, the method may comprise digesting part of ATO or removing part of ATO by affinity capturing. Tagged target polynucleotides and/or amplified polynucleotide may incorporate affinity moieties (e.g. biotin) or be purified by a probe using an affinity moiety (i.e. Hybrid capture). Amplified polynucleotide may be generated using polynucleotide and this may form a portion which is collect by affinity capture. The tagged target polynucleotide may be bound to magnetic bead by affinity before or after generation of the amplified polynucleotide, it may be purified out of the reaction solution and used in a second or more step whereas portions of the amplified polynucleotide are processed (e.g. WO2023018944A1). A second or additional amplified polynucleotide may be generated using polynucleotide again as the template, each of which is a portion which can be individually collect by affinity capture. Each affinity captured portion may then be used to may be preferentially used to generate one or more type of previously mentioned sequencing data. Multiple portions of amplified polynucleotide may be generated in a single reaction by use multiple affinity moieties including biotin:avdin, Glutathione:Glutathione S-transferase, Maltose:Maltose-binding Protein and Chitin, Chitin-binding Protein In one embodiment, the method further comprises: using a portion of the first amplified polynucleotides, or, a portion of the amplified converted polynucleotides, in a second ATOM-Seq reaction using a second adaptor template oligonucleotide (ATO) of any one of above-mentioned oligo as template. The 3’ ends of the amplified polynucleotides or amplified converted polynucleotide in a portion are extended on an ATO template to produce tagged amplified polynucleotide and/or tagged amplified converted polynucleotide which comprises a second universal sequence in the 3’ end. Following removal of second ATO, the tagged amplified converted polynucleotides and/or tagged converted polynucleotides may be linear or PCR amplified using one or two universal primers or other primers mention here in. In some embodiments, multiple different portions may be used in multiple different reactions to generate multiple populations of tagged amplified polynucleotides and/or tagged amplified converted polynucleotides. Second ATOs may be; sequence specific at the 3’ end to allow for capturing of amplified polynucleotides and/or amplified converted polynucleotides with a specific sequence of nucleotides at their 3’ end; enriched in GC or AT nucleotides at their 3’ ends to allow for capturing of amplified polynucleotides and/or amplified converted polynucleotides which are GC, or AT rich; random at their 3’ ends to capture all of the amplified polynucleotides and/or amplified converted polynucleotides, or any combination thereof. In another embodiment, the method further comprises: extending a second primer hybridized to the amplified polynucleotides, converted tagged polynucleotides, amplified converted polynucleotides, tagged amplified converted polynucleotides and/or thereby forming a second amplified polynucleotide, wherein the second primer comprises a target-specific portion or universal sequence, or both 3’ target specific and 5’ universal sequence. In one aspect, when second primer is a target specific primer, which may comprise a 3’ target specific portion with or without a 5’ universal portion. The extending using second primer may be one pass extension, or multiple cycles of linear amplification. Alternatively, steps are combined into one single PCR reaction, which uses first primer to generate amplified polynucleotides and uses second primer to form second amplified polynucleotides, where the amplified polynucleotides and second amplified polynucleotides are generated simultaneously after first PCR cycle. After PCR reaction or linear amplification, the product may be purified, or primers are removed by single-strand specific nuclease digestion. If the first primer contains a sample barcode, the purified PCR products from multiple samples may be pooled together. The (pooled) purified PCR or linear amplification product is further PCR amplified using nested target- specific third primers for amplified polynucleotides and a universal primer for second amplified polynucleotides. The nested target-specific third primers for amplified polynucleotides comprises 3’ target specific portion and 5’ universal sequence portion, wherein the 5’ universal sequence portion is compatible to a NGS platform. The PCR product is then purified ready for sequencing. In another aspect, when second primer is a target specific primer, which may comprise a 3’ target specific portion and a 5’ universal portion, wherein the 5’ universal portion is compatible with a NGS platform. The steps are combined into one single PCR reaction, which uses first primer to generate amplified polynucleotides and uses second primer to form second amplified polynucleotides, where the amplified polynucleotides and second amplified polynucleotides are generated simultaneously after first PCR cycle. The PCR product is then purified ready for sequencing. The first primer may comprise a sample barcode (SBC) sequence and additional 5’ universal sequence compatible for a NGS platform. The present disclosure further provides a method of accurately determining the sequence of amplified polynucleotides from a portion of amplified polynucleotides or amplified converted polynucleotide: (i) sequencing at least one of the amplified polynucleotides of any one of the above-mentioned methods; (ii) aligning at least two sequences containing the same UID from (i) and/or aligning same target sequences of two reactions two or more portions, each reaction generates sequence information of one strand and/or complementary strand of a duplex target sequence; and (iii) determining a consensus sequence and/or identical variant sequence of two reactions based on (ii), wherein the consensus sequence and/or variant sequence accurately represents the target polynucleotide sequence. The methods can be used to quantitate the starting molecules, the counting of UID families of a target sequence in comparison with other samples or comparing between forward reaction and reverse reaction may provide accurate counting information. The purpose of UID is twofold. First is the assignment of a unique UID to each original target polynucleotide. The second is the amplification of each uniquely tagged template, so that many daughter molecules with the identical UID sequence are generated (defined as a UID family). If a mutation pre-existed in the template molecule used for amplification, that mutation should be present in every daughter molecule containing that UID. The present disclosure further provides a kit for generating a one or more libraries of polynucleotides from one or more portions where each library uses a portion of amplified polynucleotides comprising an adaptor template oligonucleotide (ATO) of any one of the above- mentioned ATO, and primers compatible to a NGS platform. A kit comprises the composition described above. A kit for generating a library of polynucleotides comprises an adaptor template oligonucleotide (ATO) described above, polymerase and primers compatible to NGS platform. A target polynucleotide is a polynucleotide, tagged polynucleotide, amplified polynucleotides, converted tagged polynucleotides, amplified converted polynucleotide, tagged amplified converted polynucleotide or combination thereof as described herein. The target polynucleotide is, in various embodiments, DNA, RNA, cDNA, or a combination thereof. In another embodiment, the target polynucleotide is chemically treated nucleic acid, including but not limited to embodiments wherein the substrate polynucleotide is bisulfite-treated DNA to detect methylation status by NGS. The target polynucleotides are obtained from naturally occurring sources or they can be synthetic. The naturally occurring sources are RNA and/or genomic DNA from a prokaryote or a eukaryote. For example and without limitation, the source can be a human, mouse, virus, plant or bacteria. In various aspects, the target polynucleotide is extended at the 3’ end with an adaptor sequence for use in assays involving microarrays and creating libraries for next generation nucleic acid sequencing. If the source of the target polynucleotide is genomic DNA or RNA or both, in some embodiments the genomic DNA or RNA or both is fragmented prior to its being extended. Fragmenting of genomic DNA/RNA is a general procedure known to those of skill in the art and is performed, for example and without limitation in vitro by shearing (nebulizing) the DNA/RNA, cleaving the DNA/RNA with an endonuclease, sonicating the DNA/RNA, by heating the DNA/RNA, by irradiation of DNA/RNA using alpha, beta, gamma or other radioactive sources, by light, by chemical cleavage of DNA/RNA in the presence of metal ions, by radical cleavage and combinations thereof. Fragmenting of genomic DNA/RNA can also occur in vivo, for example and without limitation due to apoptosis, radiation and/or exposure to asbestos. According to the methods provided herein, a population of target polynucleotides are not required to be of a uniform size. Thus, the methods of the disclosure are effective for use with a population of differently-sized target polynucleotide fragments. The universal primers may contain one, or two, or more terminal phosphorothioates to make them resistant to any exonuclease activity. They may also contain 5’-grafting sequences necessary for hybridization to NGS flow cell, for example the Illumina GA IIx flow cell. Finally, they may contain an index sequence between the grafting sequence and the universal tag sequence. This index sequence enables the PCR products from multiple different individuals to be simultaneously analysed in the same flow cell compartment of the sequencer. DNA methylation is an important epigenetic modification of the genome. Abnormal DNA methylation may result in silencing of tumour suppressor genes and is common in a variety of human cancer cells. In some embodiments, in order to detect the presence of any abnormal methylation in the target polynucleotide the tag of the tagged polynucleotide contains methylated cytosine. In some embodiments, a portion, preferably containing mC tagged polynucleotide, is modified by an agent, producing converted tagged polynucleotide. The agent chemically and/or enzymatically modifies the tagged polynucleotide to allow for the discrimination of methylated and unmethylated cytosines. The agent may be a bisulphite treatment, which will convert cytosine to uracil but not the methylated cytosine (i.e., 5-methylcytosine, which is resistant to this treatment and remains as cytosine), an enzymatic treatment such as the combination of a TET family member with APOBEC which results in the conversion of unmethylated C to U but not the methylated cytosine, or chemical conversion by ‘TAPS chemistry’ where the 5mC or 5hmC present in original target polynucleotids are converted to a uracil derivate in a two-step process by first converting the 5mC and/or 5hmC to 5caC and/or 5fC comprises contacting a polynucleotide with a ten eleven translocation (TET) enzyme followed by conversion to dihydrouracil (DHU) by contacting the nucleic acid sample with a reducing agent. In some embodiments, converted tagged polynucleotide is used as a template with any of the included methods to generate amplified converted polynucleotides. One of more portions of the amplified converted polynucleotides may then be taken. In one embodiment, the method further comprises: using a portion of the amplified converted polynucleotides as primer and using a second adaptor template oligonucleotide (ATO) of any one of above-mentioned oligo as template. The 3’ ends of the amplified polynucleotides in a portion are extended on an ATO template to generate the tagged amplified converted polynucleotides which comprises a second universal sequence in the 3’ end. Following removal of second ATO, the extended amplified polynucleotides may be PCR amplified using two universal primers. In some embodiments, multiple different portions may be used in multiple different reactions to generate multiple populations of extended amplified converted polynucleotides. Second ATOs may be; sequence specific at the 3’ end to allow for capturing of amplified polynucleotides with a specific sequence of nucleotides at their 3’ end; enriched in GC or AT nucleotides at their 3’ ends to allow for capturing of amplified converted polynucleotides which are GC rich, or, AT; random at their 3’ ends to capture all of the amplified converted polynucleotides. With these modifications, the method can be applied to the detection of abnormal methylation(s) in the target nucleic acid. The present disclosure provides a method of analysing a biological sample for the presence and/or the amount and/or frequency of mutations or polymorphisms or other information at multiple loci of different target nucleic acid sequences by linear amplifying the tagged polynucleotides and taking one or more portions to obtain different information. In another aspect, the present disclosure provides a method of analysing a biological sample for chromosomes abnormality of, for example trisomy. The ATO reaction may be followed by next generation sequencing, digital PCR, microarray, or other high throughput analysis. The number of multiplexing of target loci may be more than 5, or more than 10, or more than 30, or more than 50, or more than 100, or more than 500, more than 1000, even more than 2000. When a mutant is in very low concentration in a sample, for example one or two mutants are present in the sample, with sufficient amplified polynucleotide copies, these rare molecules may be represented in all portions. In some embodiments, the reaction generating the amplified polynucleotides contains template, polymerase, buffer, nucleotides, primer, in a volume of 5 µl, 6-10 µl, 11-20 µl, 21-30 µl, 31- 40 µl, 41-50 µl, or larger than 50 µl. Preferentially the reaction volume is 50 µl. In some embodiments a portion may be taken from any step with the volume of a portion is less than 1 µl, 1-5 µl, 6-10 µl, 11-20 µl, 21-30 µl, 31-40 µl, 41-50ul, or larger then 50 µl. Preferentially the portion volume is 1-10 µl. In some embodiments 1 portion may be taken, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21-30, 31-40, 41-50, or more than 50 portions may be taken. Hypergeometirc distributions can be used to estimate the average number of amplified polynucleotides which will be present in a portion of volume of 50 µl when different numbers of copies have been generated. When 0 amplification cycles are used then a 1 µl portion will contain 2% of the tagged polynucleotide and on average 0 amplified polynucleotides. When 5 amplification cycles are used then a 1 µl portion will contain 2% of the tagged polynucleotide and a on average 10% chance to contain an amplified polynucleotide copy of each tagged polynucleotide. When 25 amplification cycles are used then a 1 µl portion will contain 2% of the tagged polynucleotide and a on average 50% chance to contain an amplified polynucleotide copy of each tagged polynucleotide. When 50 amplification cycles are used then a 1 µl portion will contain 2% of the tagged polynucleotide and a on average 100% chance to contain an amplified polynucleotide copy of each tagged polynucleotide. In some embodiments the volume may be larger or small and/or the cycle numbers may be higher or lower and/or the volumes of the portions may be high or lower, these will all alter the proportion of tagged polynucleotide and/or amplified polynucleotides present in all volumes used. In some embodiments, four 1 µl portions (a,b,c,d) of the amplified polynucleotides are taken. Portion a) may be used for target specific enrichment of target regions. Portion b) may be used for whole sample capture. Portion c) may be used for High-GC whole sample capture. Portion d) may be used for targeted DNA end capture. In some embodiments, the remaining 46 µl containing 92% of the total tagged polynucleotide is processed by an agent by any of the methods here in. A second 50 µl amplification reaction generates amplified converted polynucleotides. When 0 amplification cycles are used then a 5 µl portion will contain 9.2% of the tagged converted polynucleotide and on average 0 copies of each amplified converted polynucleotides. When 5 amplification cycles are used then a 5 µl portion will contain 9.2% of the tagged converted polynucleotide and on average <1 amplified converted polynucleotides. When 10 amplification cycles are used then a 5 µl portion will contain 9.2% of the tagged converted polynucleotide and on average 1 copy of each amplified converted polynucleotides. When 25 amplification cycles are used then a 5 µl portion will contain 9.2% of the tagged converted polynucleotide and on average 2.5 copies of each amplified converted polynucleotides. When 50 amplification cycles are used then a 5 µl portion will contain 9.2% of the tagged converted polynucleotide and on average 5 copies of each amplified converted polynucleotides. In some embodiments, after the generation of the amplified converted polynucleotides, the unmethylated cytosines will be converted to uracil in the converted tagged polynucleotide, is used as a template to make amplified converted tagged polynucleotide and converted amplified polynucleotides may be acted upon by a second agent. The second agent may be a dU-glycosylase, or a dU-glycosylase and apurinic/apyrimidinic endonuclease, which is capable of digesting/removing the converted tagged polynucleotide and converted amplified polynucleotided. The release of cell-free DNA into the bloodstream from dying tumour cells has been well documented in patients with various types of cancer. Research has shown that circulating tumour DNA can be used as a non-invasive biomarker to detect the presence of malignancy, follow treatment response, or monitor for recurrence. However, current methods of detection have significant limitations. Next Generation Sequencing (NGS) methods have revolutionised genomic exploration by allowing simultaneous sequencing of hundreds of billions of base pairs at a small fraction of the time and cost of traditional methods. However, the error rate of ∼1% results in hundreds of millions of sequencing mistakes, which is unacceptable when aiming to identify rare mutants in genetically heterogeneous mixtures, such as tumours and plasma. The methods of this invention overcome these limitations in sequencing accuracy. Mutation-harbouring cell-free DNA (cfDNA) can be obscured by a relative excess of background wild-type DNA; detection has proven to be challenging. The method greatly reduces errors by independently tagging and sequencing each original DNA duplex. The methods of the present invention can substantially improve both the accuracy and sensitivity of testing of a single patient sample, by allowing for serial testing workflows without dividing the initial input material. The approach allows for identifying rare mutants in a population of DNA templates. The two strands of one target template in sample, each is uniquely tagged and independently sequenced. Comparing the sequences of the two strands results in either agreement to each other or disagreement. The agreement gives the confidence to score a mutation as true positive. After sequencing, members of each read family are identified and grouped by virtue of sharing the identical UID tag sequence. The sequences of uniquely UID tagged family and one or two strands of target sequences are then compared to create a consensus sequence. This step filters out random errors introduced during sequencing or PCR to yield a set of sequences, each of which derives from an individual molecule of single-stranded DNA. In addition to their application for high sensitivity detection of rare DNA variants, the barcoded random sequence identifier in the target specific primer can also be used for single-molecule counting to precisely determine relative or absolute DNA and/or RNA copy numbers. Because tagging occurs before major amplification, the relative abundance of variants in a population can be accurately assessed given that proportional representation is not subject to skewing by amplification biases. The methods of the present invention greatly reduce errors by tagging each target sequence with random sequence identifier and sequencing the two strands. Analysis provides error-corrected consensus sequences by grouping the sequenced uniquely tagged sequences; removing the target sequences of the same family having one or more nucleotide positions where the target sequence disagrees with majority members in a family; and same mutations appearing in the two populations would be the true mutations. The method can be used for detecting mutation in any sample such as FFPE or blood. The accurate counting of sequencing reads which reflect the original molecules present in a sample provides information for copy number variations or for prenatal test for chromosome abnormality. Reagents employed in the methods of the invention can be packaged into kits. Kits include ATO(s), polymerase(s), the primer(s), in separate containers or in a single master mixture container. The kit may also contain other suitably packaged reagents and materials needed for extension, amplification, enrichment, for example, buffers, dNTPs, and/or polymerizing means; and for detection analysis, for example, and enzymes, as well as instructions for conducting the assay. BRIEF DESCRIPTION OF THE DRAWINGS FIG.1 depicts a schematic of an illustrative embodiment of the present invention. A polynucleotide population (PCR product(s), or gDNA, or RNA, or any mixture thereof may be double-stranded, or single-stranded. An adaptor is added to at least 3’ end of the polynucleotides to produced tagged polynucleotides. The addition of adaptors may be achieved by ligation or extension by a polymerase or transferase. After addition of adaptors, the tagged polynucleotides are amplified which is either linear amplification or exponential amplification. After first amplification, the amplified products are divided into several sub-portion. At least one sub-portion was treated for making sequencing library. For example, treating the tagged polynucleotides of at least one portion comprises making sequencing library to determine the epigenetic sequence information in the tagged polynucleotides. Another example is treating the amplified polynucleotides of at least one portion comprises making sequencing library of the amplified polynucleotide to determine the sequence information, including but not limited to: sequence variant (SNV), end motif, fragmentation size and break point (fragmentomics), or CNV. FIG.2 depicts a schematic of an adaptor template oligonucleotides (ATO) having (a) a 3’ portion comprising a specific motif sequence and/or random sequence; (b) a 3’ end with a blocker, which renders ATO non-extendible; (c) at least one uracil or inosine nucleotide; (d) a universal sequence, 5’ to the 3’ portion; and (e) a stem-loop structure containing a non-copiable linkage, wherein the specific motif sequence is adjacent to the double-stranded end of the stem-loop structure, wherein the target polynucleotides hybridise to the 3’ portion of the ATO. FIG.3 depicts a schematic of an illustrative embodiment of the present invention. A tagged polynucleotide is generated and amplified, and multiple portions of amplified polynucleotides are taken to multiple down-stream workflows. The first portion is taken for use in targeted enrichment for regions of interest in the original target polynucleotide. A second portion is taken for use in whole genome capture workflows. A third portion is used to enrich for high GC content DNA. A fourth portion is taken and used for targeted end capture to enrich amplified polynucleotides with specific ‘3 end sequences. Additional portions may be taken for other analysis pathways. The remaining volume of the amplified tagged polynucleotide (which contains tagged target polynucleotide) is treated with an agent to selectively convert unmethylated cytosines to uracil (or selectively convert methylated cytosines to uracil). The converted tagged polynucleotide are then amplified to generate amplified converted polynucleotide. The first portion is taken for use in targeted enrichment for regions of interest in the original target polynucleotide. A second portion is taken for use in whole genome capture workflows. A third portion is used to enrich for high GC content DNA. A fourth portion is taken and used for targeted end capture to enrich amplified polynucleotides with specific ‘3 end sequences. Additional portions may be taken for other analysis pathways. All of the NGS libraries generated from all of the portions is sequenced and the generated data is used to detect disease associated changes. FIG.4 depicts results from an example implementation of the present invention. A cfDNA sample, half of which was invitro methylated, was processed as described in example 2. The results demonstrate that a single cfDNA sample can successfully generate both a targeted methylation enrichment library and a whole genome library from a single cfDNA sample. FIG.5 depicts results from an example implementation of the present invention. Three independent FFPE samples were processed as described in example 3. The results demonstrate that a FFPE samples can successfully generate both a targeted methylation enrichment library and a targeted mutation enrichment library. EXAMPLES Example 1 A method for enriching multiple portions from a tagged cell-free DNA samples for multiple target regions for two forms of genetic information including target enrichment for cancer associated mutation hot spots, and whole sample enrichment for fragmentomics. Materials Cell-free DNA extracted from healthy plasma, Adaptor template oligos (ATO), 1-001, 1-002, 1-003, 1- 004, 1-006, 1-007, 1-008, 1-009 (Table 1), TAQ DNA polymerase, DNA Polymerase mix, Deoxynucleotide (dNTP) Solution Set, 5-methyl-dCTP, USER® Enzyme, Phusion® High-Fidelity DNA Polymerase, Phusion buffer, NEBNext® Q5U® Master Mix, Primers, 1-005, 1-010, 1-011 (Table 1), AMPure XP Beads, Bioanalyzer high sensitivity kit, EpiTect Fast Bisulfite Kit, Phusion U Hot Start DNA Polymerase, CIP, CpG Methyltransferase Method ATOM-Seq reaction In this example two 20 ng of cfDNA samples were processed. Both extracted from healthy donor plasma, one was invitro methylated following manufactures instruction using CpG Methyltransferase. Both were mixed with each of four ATOs (1-001, 1-002, 1-003, 1-004), 2 µl of 10x TAQ DNA polymerase buffer, and 10 nmole of each dNTP (dATP/dTTP/5-methyl-dCTP/dGTP), in H
2O to a final volume of 20 µl. The mixture is heated at 98
oC, for 2 minutes followed by 4
oC for 2 minutes. To this 2.5 units of polymerase mix and 2.5 units of TAQ DNA polymerase are added and thermocycled as follows: 10
oC for 1 min, 26
oC for 12 min, 30
oC for 20 min, and two cycles of 65
oC for 1 min, 10
oC for 1 min, 26
oC for 12 min, and 30
oC for 20 min. To this 2 units of USER and 5 units of Quick CIP were added followed by sequential incubations of 37
oC for 30 minutes, 25
oC for 15 minutes and then 80
oC for 2 minutes. Whole Sample Linear Amplification The treated tagged target polynucleotide is combined with 50 pmoles of a primer complementary to the universal sequence now present on 3’ end of tagged target polynucleotide (1-005), 2 units of Phusion DNA polymerase, 10 µl of 5x Phusion DNA polymerase buffer, and 12 nmole of each dNTP (dATP/dTTP/dCTP/dGTP) and H
2O to a final volume of 50 µl. The mixture is then thermocycled to linearly amplify the target nucleic acid extension product to generate amplified polynucleotide, in this example 98
oC for 30 seconds, 15 cycles of 98
oC for 5 seconds, 60
oC for 30 seconds, and 72
oC for 30 seconds, followed by 72
oC for 2 minutes. The amplified polynucleotide product can then be optionally purified by any suitable method, for example using magnetic beads. Bead purification of portion one of amplified polynucleotide A 10 µl portion of the amplified polynucleotide is taken and 40 µl of molecular biology grade water is added to increase the volume to 50 µl. The sample is then bead purified by adding 90 µl of amp pure beads, mixing well and incubated at RT for 20 minutes. The sample bead mixture is then place on magnets for 3 minutes and the supernatant discarded. The beads are washed twice with 180 µl fresh 70% EtOH and then left, lid off, at RT for 3 minutes. The samples are spun briefly to collect residual EtOH and remove it before eluting the sample in 15 of molecular biology grade water at RT for 5 minutes. The samples are left on magnets for 3 minutes before transferring 13 µl of the eluent into a clean tube. ATOM-Seq reaction 2 on portion 1. The whole of the bead purified sample was used for a second ATO2 reaction as detailed previously, where dCTP was used in place of 5-methyl-dCTP, generating tagged amplified polynucleotide with 1- 006, 1-007, 1-008, and 1-009. Global PCR To generate the final library, 25 pmol of 1-010 and 25 pmol 1-011, the 22 µl of the ATOM-Seq reaction 2 product, 2 units of Phusion DNA Polymerase, 10 µl of 5x Phusion DNA polymerase buffer are combine with H
2O up to 50 µl. The mixture was then cycled as follows, 98
oC for 30 seconds, then 11 cycles of 98
oC for 10 seconds 60
oC for 30 seconds, and 72
oC for 60 seconds, followed by 72
oC for 2 minutes. The sample is then bead purified as before. Bisulfite Conversion of portion two The 40 µl second portion of the amplified polynucleotide (containing tagged polynucleotide) was bisulfite converted following the manufacture’s recommended protocol with a final elution volume of 15 µl. Converted tagged polynucleotide linear amplification A total of 13 µl of converted tagged polynucleotide is combined with 12.5 of NEBNext Q5U Master Mix and H
2O to a final volume of 25 µl and thermocycled as follows 98
oC for 30 seconds, 15 cycles of 98
oC for 5 seconds, 60
oC for 30 seconds, and 65
oC for 30 seconds, followed by 65
oC for 2 minutes. To this 2 units of USER was added followed by sequential incubations of 37
oC for 30 minutes and then 25
oC for 15 minutes. First target enrichment PCR for differentially methylated target regions A pool of differentially methylated target region specific primers was added to the 25 µl of amplified converted polynucleotide 1 unit of Phusion DNA polymerase, 10 µl of 5x Phusion DNA polymerase buffer, H
2O to 50 µl and thermocycled as follows 98
oC for 30 seconds, 65
oC for 3 minutes, 72
oC for 30 seconds, followed by 14 cycles of 98
oC for 5 seconds, 72
oC for 30 seconds, 65
oC for 3 seconds, and 72
oC for 30 seconds, followed by 72
oC for 2 minutes. The sample is then bead purified as before, except using 90 µl of AMPure XP beads and eluting in 23 µl of H2O. Second target enrichment PCR for differentially methylated target regions The product from the first PCR is used as template for a nested PCR.22 µl of the previous product was combined with a pool of differentially methylated target region specific primers, each of two universal primers (1-010,1-011) containing sequences necessary for NGS, 2 units of Phusion DNA polymerase, 10 µl of 5x Phusion DNA polymerase buffer, and 12 nmole of each dNTP (dATP/dTTP/dCTP/dGTP) and H
2O to a final volume of 50 µl. The sample is then bead purified as before, except using 60 µl of AMPure XP beads and eluting in 30 µl of 1x TE (1mM EDTA, 10 mM Tris pH 8.0). Final libraries were visualised using a Bioanalyzer high sensitivity kit from Agilent Example 2 A method for enriching multiple portions from a tagged FFPE DNA samples for multiple target regions for two forms of genetic information including target enrichment for cancer associated mutation hot spots, and cancer associated methylation hot spots. Materials Adaptor template oligos (ATO), 1-001, 1-002, 1-003, 1-004, (Table 1), TAQ DNA polymerase, DNA Polymerase mix, Deoxynucleotide (dNTP) Solution Set, 5-methyl-dCTP, USER® Enzyme, Phusion® High- Fidelity DNA Polymerase, Phusion buffer, NEBNext Q5U Master Mix, Primers, 1-005, 1-010, 1-011 (Table 1), AMPure XP Beads, Bioanalyzer high sensitivity kit, EpiTect Fast Bisulfite Kit, CIP, CpG Methyltransferase . Method DNA Fragmentation For each of the 10 FFPE samples, 80 ng of FFPE DNA were mixed with 2.0 µl of 5X SureSelect Fragmentation Buffer and 1.0 µl of SureSelect Fragmentation Enzyme and incubated at 37
oC for 20 minutes and 98
oC for 2 minutes. ATOM-Seq reaction The whole of the fragmented DNA sample was used as starting material and followed the protocol in example 1. Whole Sample Linear Amplification As in example 1. First target enrichment PCR for mutation hotspots. As in example 1, except using a 10 µl portion of the amplified polynucleotide was used as starting material and with a pool of primers designed to target mutation hotspots associated with bowel cancer. Second target enrichment PCR for mutation hotspots As in example 1, except using a pool of primers designed to target mutation hotspots associated with bowel cancer. Bisulfite Conversion of tagged target polynucleotide As in example 1. Converted tagged polynucleotide linear amplification As in example 1. First target enrichment PCR for differentially methylated target regions As in example 1. Second target enrichment PCR for differentially methylated target regions As in example 1. Example 3 A method for enriching multiple portions from a 3’ extension based tagged cfDNA samples for multiple target regions for four forms of genetic information including target enrichment for cancer associated mutation hot spots, whole sample capture for fragmentomics, cancer associated methylation hot spots, and whole genome methylation. Materials Cell-free DNA extracted from healthy plasma, Adaptor template oligos (ATO), 1-001, 1-002, 1-003, 1- 004, 1-006, 1-007, 1-008, 1-009 (Table 1), TAQ DNA polymerase, DNA Polymerase mix, Deoxynucleotide (dNTP) Solution Set, 5-methyl-dCTP, USER® Enzyme, Phusion® High-Fidelity DNA Polymerase, Phusion buffer, NEBNext Q5U Master Mix, Primers, 1-005, 1-010, 1-011 (Table 1), Agencourt AMPure XP Beads, EpiTect Fast Bisulfite Kit, Phusion U Hot Start DNA Polymerase, CIP, CpG Methyltransferase Method ATOM-Seq reaction In this example 20 ng of cfDNA was used as starting material and followed the protocol in example 1. Whole Sample Linear Amplification As in example 1 except with 30 cycles. Portion 1: Bead purification of amplified polynucleotide As in example 1, except a 5 µl portion was diluted to 50 µl. Portion 1: First ATOM-Seq reaction 2 As in example 1. Portion 1: Global PCR As in example 1. Portion 2: First target enrichment PCR for mutation hotspots. As in example 2, except a 5 µl portion was used. Portion 2: Second target enrichment PCR for mutation hotspots As in example 2. Bisulfite Conversion of tagged target polynucleotide As in example 1. Converted tagged polynucleotide linear amplification As in example 1. Portion 3: Bead purification of amplified polynucleotide As in example 1, except a 10 µl portion of amplified converted polynucleotide was diluted to 50 µl. Portion 3: Second ATOM-Seq reaction 2 As in example 1 except using ATO 1-012. Portion 3: Global PCR As above. Portion 4: First target enrichment PCR for differentially methylated target regions The remainder of the amplified converted polynucleotide is used as in example 1. Portion 4: Second target enrichment PCR for differentially methylated target regions As in example 1. Example 4 A method for enriching multiple portions from a ligation based tagged cfDNA samples for multiple target regions for four forms of genetic information including target enrichment for cancer associated mutation hot spots, whole sample capture for fragmentomics, cancer associated methylation hot spots, and whole genome methylation. Materials Cell-free DNA extracted from healthy plasma, NEBNext® Ultra™ II DNA Library Prep Kit for Illumina®, Adaptor Oligo 1-013, 1-014 (Table 1), TAQ DNA polymerase, DNA Polymerase mix, Deoxynucleotide (dNTP) Solution Set, USER® Enzyme, Phusion® High-Fidelity DNA Polymerase, Phusion buffer, NEBNext Q5U Master Mix, Primers, 1-005, 1-010, 1-011 (Table 1), Agencourt AMPure XP Beads, EpiTect Fast Bisulfite Kit, CIP, CpG Methyltransferase Adaptor annealing. To generate double strand adaptor, 50 µl of 100 µM of 1-013 and 1-014 were combined, heated to 98
oC for 2 minutes and allowed to cool to room temperature. These were then diluted to 1.5 µM in dilution 10 mM Tris-HCl, pH 8.0 with 10 mM NaCl. cfDNA End repair, A-tail and ligation. The recommended protocol for 5-100 ng of material for the NEBNext Ultra™ II DNA Library Prep Kit for Illumina was followed from step 1 to step 3, except that the previously made 1.5 µM adaptor was used and the sample was eluted in 24 µl of Low TE (10 mM Tris-HCl, pH 8.00.1mM EDTA). Whole Sample Linear Amplification As in example 1 except with 30 cycles. Portion 1: Bead purification of amplified polynucleotide As in example 1, except a 5 µl portion was diluted to 50 µl. Portion 1: First ATOM-Seq reaction 2 As in example 1. Portion 1: Global PCR As in example 1. Portion 2: First target enrichment PCR for mutation hotspots. As in example 2, except a 5 µl portion was used. Portion 2: Second target enrichment PCR for mutation hotspots As in example 2. Bisulfite Conversion of tagged target polynucleotide As in example 1. Converted tagged polynucleotide linear amplification As in example 1. Portion 3: Bead purification of amplified polynucleotide As in example 1, except a 10 µl portion of amplified converted polynucleotide was diluted to 50 µl. Portion 3: Second ATOM-Seq reaction 2 As in example 1 except using ATO 1-012. Portion 3: Global PCR As above. Portion 4: First target enrichment PCR for differentially methylated target regions The remainder of the amplified converted polynucleotide is used as in example 1. Portion 4: Second target enrichment PCR for differentially methylated target regions As in example 1. Example 5 A method for enriching multiple exponential amplified portions from a ligation based tagged cfDNA samples for multiple target regions for multiple forms of genetic information including whole sample capture for fragmentomics, cancer associated methylation hot spots, and whole genome methylation. Materials Cell-free DNA extracted from healthy plasma, NEBNext® Ultra™ II DNA Library Prep Kit for Illumina®, NEBNext® Multiplex Oligos for Illumina® (Methylated Adaptor, Index Primers Set 1), NEBNext® Ultra™ II Q5® Master Mix, TAQ DNA polymerase, DNA Polymerase mix, Deoxynucleotide (dNTP) Solution Set, USER® Enzyme, Phusion® High-Fidelity DNA Polymerase, Phusion buffer, Primers, 1-005, 1-010, 1-011 (Table 1), Agencourt AMPure XP Beads, Bioanalyzer high sensitivity kit from Agilent, EpiTect Fast Bisulfite Kit, Phusion U Hot Start DNA Polymerase, CIP, CpG Methyltransferase cfDNA End repair, A-tail, ligation, clean up, and PCR. The recommended protocol for 5-100 ng of material for the NEBNext Ultra™ II DNA Library Prep Kit for Illumina was followed with 10 ng of cfDNA from step 1 to step 4.1.3, except that NEB Methylated Adaptor at 1.5 µM was used, and only 3 PCR cycles were used. Portion 1: PCR of PCR amplified polynucleotide A 10 µl portion of the PCR amplified polynucleotide was combined with 25 µl of NEBNext Ultra II Q5 Master Mix, 25 pmole of 1-010 and 1-011, and H
2O to a final volume of 50 µl. The mix was thermocycled as followed, 98
oC for 30 seconds, then 10 cycles of 98
oC for 5 seconds, 65
oC for 30 seconds, and 72
oC for 30 seconds, followed by 72
oC for 2 minutes. The sample was then bead purified as in example 1. Bisulfite Conversion of ligation tagged target polynucleotide As in example 1. Portion 2: PCR of converted tagged polynucleotide A 3 µl portion of the converted tagged polynucleotide was combined with 25 µl of NEBNext Ultra II Q5 Master Mix, 25 pmole of 1-010 and 1-011, and H
2O to a final volume of 50 µl. The mix was thermocycled as followed, 98
oC for 30 seconds, then 10 cycles of 98
oC for 5 seconds, 65
oC for 30 seconds, and 72
oC for 30 seconds, followed by 72
oC for 2 minutes. The sample was then bead purified as in example 1. Portion 3: Converted tagged polynucleotide linear amplification As in example 1 except a pool of target specific primers are used. Portion 3: First target enrichment PCR for differentially methylated target regions As in example 1 using the targeted specific amplified converted polynucleotide. Portion 3: Second target enrichment PCR for differentially methylated target regions As in example 1. Table 1 ID Seq-ID Sequence NCGNNNAGA[U]CGGA[U]GAGC[SpC18]TCAGACGTGTGCTC[U]TCCGA[U 1-001 1,2 ]C[U]NNNCGNNNNNNNNNNNNNNNN*T[PHO] NCANNNAGA[U]CGGA[U]GAGC[SpC18]TCAGACGTGTGCTC[U]TCCGA[U] 1-002 3,4 C[U] [PHO]
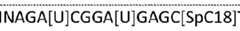
NCCNNNAGA[U]CGGA[U]GAGC[SpC18]TCAGACGTGTGCTC[U]TCCGA[U] 1-003 5,6 C[U]NNNGGNNNNNNNNNNNNNNNN*C[PHO] NGGNNNAGA[U]CGGA[U]GAGC[SpC18]TCAGACGTGTGCTC[U]TCCGA[U 1-004 7,8 ]C[U]NNNCCNNNNNNNNNNNNNNNN*G[PHO] 1-005 9 GTGACTGGAGTTCAGACGTGTGCTCTTCCGATC*T AGA[U]CGGA[U]GAGC[SpC18]CTACACGACGCTCTTCCGATC[U]CCCANN 1-006 10,11 NN*G[PHO] AGA[U]CGGA[U]GAGC[SpC18]CTACACGACGCTCTTCCGATC[U]CCAGNN-007 12,13 NN*G[PHO] AGA[U]CGGA[U]GAGC[SpC18]CTACACGACGCTCTTCCGATC[U]CCTGNN-008 14,15 NN*G[PHO] AGA[U]CGGA[U]GAGC[SpC18]CTACACGACGCTCTTCCGATC[U]TAAANN-009 16,17 NN*G[PHO] -010
-011 19 TGTGCTCTTCCGAT*C*T AGA[U]CGGA -012 20,21
NNNNNNN[U]*G[PHO] -013 22 CGTGTGCTCTTCCGATCT -014 23 [PHO]GAT[5mC]GGAAGAG[5mC]A[5mC]A[5mC]GA[5mC]AGA
 -011 19 TGTGCTCTTCCGAT*C*T AGA[U]CGGA -012 20,21
-011 19 TGTGCTCTTCCGAT*C*T AGA[U]CGGA -012 20,21 NNNNNNN[U]*G[PHO] -013 22 CGTGTGCTCTTCCGATCT -014 23 [PHO]GAT[5mC]GGAAGAG[5mC]A[5mC]A[5mC]GA[5mC]AGA
NNNNNNN[U]*G[PHO] -013 22 CGTGTGCTCTTCCGATCT -014 23 [PHO]GAT[5mC]GGAAGAG[5mC]A[5mC]A[5mC]GA[5mC]AGA

