COMPOUNDS AND THEIR USE AGAINST CANCER Cross-reference to related application [0001] This application claims the benefit of priority to U.S. Provisional Patent Application No.63/464,915, filed on May 8, 2023, the entire contents of which are incorporated by reference herein. Background [0002] Ubiquitin is a small, highly conserved protein composed of 76 amino acids that is post-transcriptionally attached to target proteins, including itself, via a concerted three-step enzymatic reaction. This covalent linkage or isopeptide bond primarily occurs between the C- terminal glycine of ubiquitin and the ε-amino group of lysine residue(s) on the target protein (Pickart, C. M., Annu. Rev. Biochem., 2001 : 503-33). The functional consequence of ubiquitination is determined by the number and linkage topology of ubiquitin molecules conjugated to the target protein. For example, proteins exhibiting Lys48-linked polyubiquitin chains are generally targeted to the proteasome for degradation, while monoubiquitination or polyubiquitin chains linked through other lysines regulate several non-proteolytic functions, including cell cycle regulation (Nakayama, K. I. et al., Nat. rev. Cancer, 6(5): 369-81 (2006)), DNA repair (Bergink, S., et al., Nature 458(7237): 461 -7 (2009)), transcription (Conaway, R. C, et al., Science 296(5571): 1254-8 (2002)), and endocytosis (Mukhopadhyay, D., et al., Science 315(5809): 201 -5 (2007)). Similar to other posttranslational modifications, ubiquitination is a reversible process counteracted by a family of enzymes known as deubiquitinases (DUBs). These enzymes are cysteine proteases or metalloproteases that hydrolyze the ubiquitin isopeptide bond (Komander, D., et al., Nat. Rev. Mol. Cell Biol. 10(8): 550-63 (2007)). The human genome encodes close to 100 DUBs. [0003] DUBs and their substrate proteins are often deregulated in cancers. Targeting specific DUB family members may result in antitumor activity by enhancing the ubiquitination and subsequent degradation of oncogenic substrates, involved in tumor growth, survival, differentiation and maintenance of the tumor microenvironment. (Hussain, S. et. al., "DUBs and cancer: The role of deubiquitinating enzymes as oncogenes, non-oncogenes and tumor suppressors." Cell Cycle 8, 1688-1697 (2009)). Consequently, several members of the DUB family have been implicated in processes related to human disease, including cancer and neurodegeneration. Among them, USP1 (ubiquitin- specific peptidase 1) has gained increased interest as a novel therapeutic target given its roles in the DNA damage response. [0004] USP1 is a cysteine isopeptidase of the USP subfamily of deubiquitinases (DUBs). (Nijman, S. M. B., et al. "The deubiquitinating enzyme USP1 regulates the Fanconi anemia pathway. Mol. Cell 17, 331-339 (2005)) Full-length human USP1 is composed of 785 amino acids, including a catalytic triad composed of Cys90, His593 and Asp751. (Villamil, M. A., et al, "Serine phosphorylation is critical for the activation of ubiquitin-specific protease 1 and its interaction with WD40-repeat protein UAF1." Biochem.51, 9112-9113 (2012)). USP1 is relatively inactive on its own and full enzymatic activity is achieved only when bound in a heterodimeric complex with USP1 Associated Factor 1 (UAF1), which also binds to and regulates the activity of USP12 and USP46. (Cohn, M. A., et al, "A UAF1 -Containing Multisubunit Protein Complex Regulates the Fanconi Anemia Pathway." Mol. Cell 28, 786- 797 (2007)). [0005] USP1 deubiquitinates a variety of cellular targets involved in different processes related to cancer. For example, USP1 deubiquitinates PCNA (proliferating cell nuclear antigen), a key protein in translesion synthesis (TLS), and FANCI/FANCD2 (Fanconi anemia group complementation group D2), a key protein complex in the Fanconi anemia (FA) pathway. (Nijman, S. M. B. et al "The deubiquitinating enzyme USP1 regulates the Fanconi anemia pathway." Mol Cell 17, 331-339 (2005); Huang, T. T. et al, "Regulation of monoubiquitinated PCNA by DUB autocleavage." Nat. Cell Biol 8, 339-347 (2006)). These DNA damage response (DDR) pathways are essential for repair of DNA damage, including those induced by DNA cross-linking agents such as cisplatin, mitomycin C (MMC), diepoxybutane, ionizing radiation and ultraviolet radiation. In addition, USP1 promotes cancer cell stem maintenance by increasing inhibitor of protein binding (ID) protein stability. Thus, USP1 inhibition may antagonize cancer cell growth by inducing cell cycle arrest and decreasing cancer stem cell maintenance via a decrease in ID protein stability. (Williams, S. A. et al, “USP1 deubiquitinates ID proteins to preserve a mesenchymal stem cell program in osteosarcoma.” Cell 146: 918-30 (2011); Lee, J. K. et al, “USP1 targeting impedes GBM growth by inhibiting stem cell maintenance and radioresistance.” Neuro Oncol.18: 37-47 (2016)). [0006] The compounds GW7647 and Pimozide have been described as inactivators of USP1. However, both compounds are limited by potency and off-target pharmacology, in part because both of them have noticeable activity against unrelated targets. Another small molecule inhibitor of USP1, C527, which was reported by D’Andrea et al. in WO2011/137320, sensitizes cells to both the crosslinking agent, mitomycin C, and the topoisomerase 1 inhibitor, camptothecin. However, C527 shows low micromolar inhibition of related USPs as well as dissimilar DUBs (i.e., UCHL-1 and UCHL-3). Another small molecule USP1-UAF1 inhibitor (ML323) has been more recently disclosed (Dexheimer et al, J. Med. Chem.2014, 57, 8099-8110; Liang et al, Nature Chem. Bio.2015, 10, 298-304; US 9802904 B2). Additional USP1 inhibitors have also been described in WO2017087837, WO2020132269, WO2020139988, WO2021163530, WO2022199652, WO2022214053, WO2022228399, WO2022233263 and WO2022253188. Two applications filed by Tango Therapeutics, WO20222174184 and WO2022197892 also describe USP1 inhibitors. [0007] The foregoing shows that there exists an unmet need for new selective inhibitors of USP1, and in addition, that inhibition of USP1 with small molecule inhibitors has the potential to be a treatment for cancers and other disorders. For these reasons, there remains a considerable need for potent small molecule inhibitors of USP1. Summary [0008] In one aspect provided is a compound of Formula (I) or Formula (II) or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof; wherein:

; ; X
1 and X
2 are each independently CH or N; Ring A is selected from the group consisting of:
and
X3 wherein X
3 is CH or N and wherein the left-side attachment point connects to CR
cR
c’ and the right attachment point connects to Ring B; L is selected from the group consisting of –O– and –S–; Ring B is a 5-10 membered heteroaryl or a 3-10 membered heterocyclyl; each R
A is independently selected from the group consisting of –D, oxo, halo, – CN, –C
1–C
6 alkyl, –C
1–C
6 haloalkyl, –C
1–C
6 hydroxyalkyl, –C
3–C
10 cycloalkyl, –OR
A1 and – N(R
A1)
2; each R
A1 is independently selected from the group consisting of H, –C
1–C
6 alkyl, –C
1–C
6 haloalkyl and C
3–C
9 cycloalkyl wherein each hydrogen atom of each alkyl and cycloalkyl can be replaced by deuterium; or two instances of R
A1 on a nitrogen atom can be taken together with the nitrogen to which they are attached to form a 4-6 member monocyclic heterocycle containing one nitrogen atom and 0-1 additional heteroatoms selected from N and O; each R
B is independently selected from the group consisting of halo, –CN, –C
1–C
6 alkyl, –C
1–C
6 alkenyl, –heteroC
1-C
4 alkyl, –C
1–C
6 haloalkyl, –C
1–C
6 hydroxyalkyl, –C
3–C
10 cycloalkyl, 3-10 membered heterocyclyl, C
3-C
9 cycloalkylalkyl, –OR
B1, –N(R
B1)
2, – C(=O)R
B1, –C(=O)OR
B1, –NR
B1C(=O)R
B1, –NR
B1C(=O)OR
B1, –C(=O)N(R
B1)
2, – OC(=O)N(R
B1)
2, –S(=O)R
B1, –S(=O)
2R
B1, –SR
B1, –S(=O)(=NR
B1)R
B1, –NR
B1S(=O)
2R
B1 and –S(=O)
2N(R
B1)
2, wherein each alkyl, alkenyl, cycloalkyl, heterocyclyl and cycloalkylalkyl of R
B can be partially or fully deuterated and can be substituted at any available position with 0, 1 or 2 substituents selected from –F, –Cl, –OH– and –CH
3; each R
B1 is independently selected from the group consisting of H, –C
1–C
6 alkyl, –heteroC
1-C
4 alkyl, –C
1–C
6 haloalkyl, –C
3–C
9 cycloalkyl and 3-7 membered heterocyclyl wherein each hydrogen atom of each alkyl, cycloalkyl and heterocyclyl can be replaced by deuterium; or two instances of R
B1 on a nitrogen atom can be taken together with the nitrogen to which they are attached to form a 4-6 member monocyclic heterocycle containing one nitrogen atom and 0-1 additional heteroatoms selected from N and O; each R
C is independently selected from the group consisting of H, –D, oxo, halo, – CN, –OR
C1, –SR
C1 –NR
C12, –C
1–C
6 alkyl, –C
1–C
6 hydroxyalkyl, –C
1–C
6 haloalkyl, –C
3–C
9 cycloalkyl, 3-10 member heterocyclyl and –heteroC
1-C
4 alkyl wherein each alkyl, cycloalkyl and heterocyclyl of R
C can partially or fully deuterated and can be substituted at any available position with 0, 1 or 2 substituents selected from –F, –Cl, –OH– and –CH
3; each R
C1 is independently selected from the group consisting of H and –C
1–C
6 alkyl, wherein each hydrogen atom of the alkyl can be independently replaced by deuterium; or two instances of R
C1 on a nitrogen atom can be taken together with the nitrogen to which they are attached to form a 4-6 member monocyclic heterocycle containing one nitrogen atom and 0-1 additional heteroatoms selected from N and O; each R
D is independently selected from the group consisting of –D, halo, –CN, – C
1–C
6 alkyl, –heteroC
1-C
4 alkyl, –C
1–C
6 haloalkyl, –C
1–C
6 hydroxyalkyl, –C
3–C
10 cycloalkyl, 3-10 membered heterocyclyl, –C
6-C
10 aryl, –OR
D1, –N(R
D1)
2, –C(=O)R
D1, – C(=O)OR
D1, –NR
D1C(=O)R
D1, –NR
D1C(=O)OR
D1, –C(=O)N(R
D1)
2, –OC(=O)N(R
D1)
2, – S(=O)R
D1, –S(=O)
2R
D1, –SR
D1, –S(=O)(=NR
D1)R
D1, –NR
D1S(=O)
2R
D1 and –S(=O)
2N(R
D1)
2 , wherein each alkyl, cycloalkyl, heterocyclyl and aryl of R
D can be substituted at any available position with 0, 1 or 2 substituents selected from –F, –Cl, –OH– and –CH
3; each R
D1 is independently selected from the group consisting of H, –C
1–C
6 alkyl, –heteroC
1-C
4 alkyl, –C
1–C
6 haloalkyl, –C
3–C
9 cycloalkyl and 3-7 membered heterocyclyl, wherein each hydrogen atom of each alkyl, cycloalkyl and heterocyclyl can be replaced by deuterium; or two instances of R
D1 on a nitrogen atom can be taken together with the nitrogen to which they are attached to form a 4-6 member monocyclic heterocycle containing one nitrogen atom and 0-1 additional heteroatoms selected from N and O; R
E is selected from H, –D, halo, –CN, –C
1–C
6 alkyl, –C
1-C
6 alkynyl, –heteroC
1– C
6 alkyl, –C
1–C
6 haloalkyl, –C
1–C
6 hydroxyalkyl, –C
3–C
10 cycloalkyl, 3-10 membered heterocyclyl, –C
6-C
10 aryl, 5-10 member heteroaryl, heterocyclylalkyl, heteroarylalkyl, arylalkyl, cycloalkylalkyl, –OR
E1, –SR
E1, –N(R
E1)
2, –C
1–C
6 alkylene–NR
E1C(=O)OR
E1, – C(=O)R
E1, –C(=O)OR
E1, –NR
E1C(=O)R
E1, –NR
E1C(=O)OR
E1, –C(=O)N(R
E1)
2, and – OC(=O)N(R
E1)
2, wherein each alkyl, alkynyl, heteroalkyl, haloalkyl, cycloalkyl, heterocyclyl, aryl, heteroaryl, cycloalkylalkyl, heterocyclylalkyl, arylalkyl and heteroarylalkyl is optionally substituted at any available position; each R
E1 is independently selected from H, –C
1–C
6 alkyl (wherein each hydrogen can be independently replaced by deuterium), –heteroC
1–C
6 alkyl, –C
1–C
6 haloalkyl, –C
3–C
9 cycloalkyl, 3-7 membered heterocyclyl, cycloalkylalkyl, heterocyclylalkyl, aryl, 5-6 membered heteroaryl, arylalkyl and heteroarylalkyl wherein each alkyl, haloalkyl, cycloalkyl, heterocyclyl, aryl, heteroaryl, cycloalkylalkyl, heterocyclylalkyl, arylalkyl and heteroarylalkyl is optionally substituted at any available position; each R
c and R
c’ is independently selected from the group consisting of H, –D, – C
1–C
6 alkyl, –heteroC
1-C
4 alkyl and –C
1–C
6 haloalkyl or R
c and R
c’ can be taken together with the atom to which they are attached to form a –C
3–C
9 cycloalkyl; n is 0, 1, 2,3 or 4; m is 0, 1, 2 or 3; and p is 0, 1, 2 or 3. [0009] In an embodiment, provided are compounds selected from the compounds of Table 1, or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof. [0010] In an embodiment, provided is a pharmaceutical composition comprising a compound as described herein or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof and a pharmaceutically acceptable carrier. [0011] In an embodiment, the pharmaceutical composition comprises a second therapeutic agent. [0012] In one aspect provided is a method for treating or preventing a disease or disorder sensitive to inhibition of USP1 comprising administering to a patient in need thereof a therapeutically effective amount of a compound of Formula (I) or Formula (II) or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof. [0013] In one aspect provided is a method of treating a disease or disorder sensitive to the inhibition of USP1 comprising administering to a patient in need thereof a therapeutically effective amount of a compound of Formula (I) or Formula (II) or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof. [0014] In one aspect provided is a method for inhibiting USP1 comprising administering to a patient in need thereof an effective amount of a compound of Formula (I) or Formula (II) or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof. [0015] In one aspect provided is a method for treating or preventing cancer in a patient in need thereof comprising administering to the patient in need thereof a therapeutically effective amount of a compound of Formula (I) or Formula (II) or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof. [0016] In one aspect provided is a method for treating cancer in a patient in need thereof comprising administering to the patient in need thereof a therapeutically effective amount of a compound of Formula (I) or Formula (II) or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof. [0017] In one aspect provided is a method for treating or preventing a disease or disorder associated with DNA damage comprising administering to a patient in need of a treatment for diseases or disorders associated with DNA damage an effective amount of a compound of Formula (I) or Formula (II) or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof. [0018] In one aspect provided is a method for treating a disease or disorder associated with DNA damage comprising administering to a patient in need of a treatment for diseases or disorders associated with DNA damage an effective amount of a compound of Formula (I) or Formula (II) or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof. [0019] In one aspect provided is a method of inhibiting, modulating or reducing DNA repair activity exercised by USP1 comprising administering to a patient in need thereof an effective amount of a compound of Formula (I) or Formula (II) or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof. [0020] In one aspect provided is a compound of Formula (I) or Formula (II) or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof for use in a method for treating or preventing a disease or disorder sensitive to the inhibition of USP1 comprising administering to a patient in need thereof a therapeutically effective amount of the compound or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof. [0021] In one aspect provided is a compound of Formula (I) or Formula (II) or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof for use in a method of treating a disease or disorder sensitive to the inhibition of USP1 comprising administering to a patient in need thereof a therapeutically effective amount of the compound or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof. [0022] In one aspect provided is a compound of Formula (I) or Formula (II) or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof for use in a method for inhibiting USP1 comprising administering to a patient in need thereof an effective amount of the compound or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof. [0023] In one aspect provided is a compound of Formula (I) or Formula (II) or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof for use in a method for treating or preventing cancer in a patient in need thereof comprising administering to the patient in need thereof a therapeutically effective amount of the compound or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof. [0024] In one aspect provided is a compound of Formula (I) or Formula (II) or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof for use in a method for treating cancer in a patient in need thereof comprising administering to the patient in need thereof a therapeutically effective amount of the compound or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof. [0025] In one aspect provided is a compound of Formula (I) or Formula (II) or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof for use in a method for treating or preventing a disease or disorder associated with DNA damage. [0026] In one aspect provided is a compound of Formula (I) or Formula (II) or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof for use in a method for treating a disease or disorder associated with DNA damage. [0027] In one aspect provided is a compound of Formula (I) or Formula (II) or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof for use in a method of inhibiting, modulating or reducing DNA repair activity exercised by USP1 comprising administering to a patient in need thereof an effective amount of the compound or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof. [0028] In one aspect, provided is a use of an effective amount of a compound of Formula (I) or Formula (II) or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof in the manufacturing of a medicament for treating or preventing a disease or disorder sensitive to the inhibition of USP1. [0029] In one aspect, provided is a use of an effective amount of a compound of Formula (I) or Formula (II) or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof in the manufacturing of a medicament for treating a disease or disorder sensitive to the inhibition of USP1. [0030] In one aspect, provided is a use of an effective amount of a compound of Formula (I) or Formula (II) or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof in the manufacturing of a medicament for inhibiting USP1. [0031] In one aspect, provided is a use of an effective amount of a compound of Formula (I) or Formula (II) or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof in the manufacturing of a medicament for treating or preventing cancer in a patient in need thereof. [0032] In one aspect, provided is a use of an effective amount of a compound of Formula (I) or Formula (II) or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof in the manufacturing of a medicament for treating cancer in a patient in need thereof. [0033] In one aspect, provided is a use of an effective amount of a compound of Formula (I) or Formula (II) or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof in the manufacturing of a medicament for treating or preventing a disease or disorder associated with DNA damage. [0034] In one aspect, provided is a use of an effective amount of a compound of Formula (I) or Formula (II) or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof in the manufacturing of a medicament for treating a disease or disorder associated with DNA damage. [0035] In one aspect, provided is a use of an effective amount of a compound of Formula (I) or Formula (II) or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof for inhibiting, modulating or reducing DNA repair activity exercised by USP1. [0036] In one aspect, provided is a use of an effective amount of a compound of Formula (I) or Formula (II) or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof for treating or preventing a disease or disorder sensitive to the inhibition of USP1. [0037] In one aspect, provided is a use of an effective amount of a compound of Formula (I) or Formula (II) or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof for treating a disease or disorder sensitive to the inhibition of USP1. [0038] In one aspect, provided is a use of an effective amount of a compound of Formula (I) or Formula (II) or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof for inhibiting USP1. [0039] In one aspect, provided is a use of an effective amount of a compound of Formula (I) or Formula (II) or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof for treating or preventing cancer in a patient in need thereof. [0040] In one aspect, provided is a use of an effective amount of a compound of Formula (I) or Formula (II) or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof for treating cancer in a patient in need thereof. [0041] In one aspect, provided is a use of an effective amount of a compound of Formula (I) or Formula (II) or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof for treating or preventing a disease or disorder associated with DNA damage. [0042] In one aspect, provided is a use of an effective amount of a compound of Formula (I) or Formula (II) or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof for treating a disease or disorder associated with DNA damage. [0043] In one aspect, provided is a use of an effective amount of a compound of Formula (I) or Formula (II) or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof for inhibiting, modulating or reducing DNA repair activity exercised by USP1. Detailed Description [0044] The disclosure herein sets forth exemplary methods, parameters and the like. It should be recognized, however, that such description is not intended as a limitation on the scope of the present disclosure but is instead provided as a description of exemplary embodiments. Definitions [0045] As used in the present disclosure, the following words and phrases are generally intended to have the meanings as set forth below unless expressly indicated otherwise or the context in which they are used indicates otherwise. Chemical Definitions [0046] Definitions of specific functional groups and chemical terms are described in more detail below. The chemical elements are identified in accordance with the Periodic Table of the Elements, CAS version, Handbook of Chemistry and Physics, 75
th Ed., inside cover, and specific functional groups are generally defined as described therein. Additionally, general principles of organic chemistry, as well as specific functional moieties and reactivity, are described in Thomas Sorrell, Organic Chemistry, University Science Books, Sausalito, 1999; Smith and March, March’s Advanced Organic Chemistry, 5
th Edition, John Wiley & Sons, Inc., New York, 2001; Larock, Comprehensive Organic Transformations, VCH Publishers, Inc., New York, 1989; and Carruthers, Some Modern Methods of Organic Synthesis, 3
rd Edition, Cambridge University Press, Cambridge, 1987. [0047] Compounds described herein can comprise one or more asymmetric centers, and thus can exist in various isomeric forms, e.g., enantiomers and/or diastereomers. For example, the compounds described herein can be in the form of an individual enantiomer, diastereomer or geometric isomer, or can be in the form of a mixture of stereoisomers, including racemic mixtures and mixtures enriched in one or more stereoisomer. Unless stereochemistry is explicitly indicated in a structure, the structure is intended to embrace all possible stereoisomers of the compound depicted. If stereochemistry is explicitly indicated for one portion or portions of a molecule, but not for another portion or portions of a molecule, the structure is intended to embrace all possible stereoisomers for the portion or portions where stereochemistry is not explicitly indicated. Isomers can be isolated from mixtures by methods known to those skilled in the art, including chiral high-pressure liquid chromatography (HPLC) and the formation and crystallization of chiral salts; or preferred isomers can be prepared by asymmetric syntheses. See, for example, Jacques et al., Enantiomers, Racemates and Resolutions (Wiley Interscience, New York, 1981); Wilen et al., Tetrahedron 33:2725 (1977); Eliel, Stereochemistry of Carbon Compounds (McGraw–Hill, NY, 1962); and Wilen, Tables of Resolving Agents and Optical Resolutions p.268 (E.L. Eliel, Ed., Univ. of Notre Dame Press, Notre Dame, IN 1972). The invention additionally encompasses compounds described herein as individual isomers substantially free of other isomers, and alternatively, as mixtures of various isomers. [0048] The “enantiomeric excess” (“e.e.”) or “% enantiomeric excess” (“%e.e.”) of a composition as used herein refers to an excess of one enantiomer relative to the other enantiomer present in the composition. For example, a composition can contain 90% of one enantiomer, e.g., the S enantiomer, and 10% of the other enantiomer, i.e., the R enantiomer. e.e. = (90-10)/100 = 80%. [0049] Thus, a composition containing 90% of one enantiomer and 10% of the other enantiomer is said to have an enantiomeric excess of 80%. [0050] The “diastereomeric excess” (“d.e.”) or “% diastereomeric excess” (“%d.e.”) of a composition as used herein refers to an excess of one diastereomer relative to one or more different diastereomers present in the composition. For example, a composition can contain 90% of one diastereomer, and 10% of one or more different diastereomers. d.e. = (90-10)/100 = 80%. [0051] Thus, a composition containing 90% of one diastereomers and 10% of one or more different diastereomers is said to have a diastereomeric excess of 80%. [0052] In an alternative embodiment, compounds described herein may also comprise one or more isotopic substitutions. For example, hydrogen may be
2H (D or deuterium) or
3H (T or tritium); carbon may be, for example,
13C or
14C; oxygen may be, for example,
18O; nitrogen may be, for example,
15N, and the like. In other embodiments, a particular isotope (e.g.,
3H,
13C,
14C,
18O, or
15N) can represent at least 1%, at least 5%, at least 10%, at least 15%, at least 20%, at least 25%, at least 30%, at least 35%, at least 40%, at least 45%, at least 50%, at least 60%, at least 65%, at least 70%, at least 75%, at least 80%, at least 85%, at least 90%, at least 95%, at least 99%, or at least 99.9% of the total isotopic abundance of an element that occupies a specific site of the compound. [0053] In a formula, is a single bond where the stereochemistry of the moieties immediately attached thereto is not specified. [0054] When a range of values is listed, it is intended to encompass each value and sub– range within the range. For example, “C
1–6 alkyl” is intended to encompass, C
1, C
2, C
3, C
4, C
5, C
6, C
1–6, C
1–5, C
1–4, C
1–3, C
1–2, C
2–6, C
2–5, C
2–4, C
2–3, C
3–6, C
3–5, C
3–4, C
4–6, C
4–5, and C
5–6 alkyl. [0055] The following terms are intended to have the meanings presented therewith below and are useful in understanding the description and intended scope of the present invention. When describing the invention, which may include compounds, pharmaceutical compositions containing such compounds and methods of using such compounds and compositions, the following terms, if present, have the following meanings unless otherwise indicated. It should also be understood that when described herein any of the moieties defined forth below may be substituted with a variety of substituents, and that the respective definitions are intended to include such substituted moieties within their scope as set out below. Unless otherwise stated, the term “substituted” is to be defined as set out below. It should be further understood that the terms “groups” and “radicals” can be considered interchangeable when used herein. The articles “a” and “an” may be used herein to refer to one or to more than one (i.e. at least one) of the grammatical objects of the article. By way of example “an analogue” means one analogue or more than one analogue. [0056] The term “unsaturated bond” refers to a double or triple bond. [0057] The term “unsaturated” or “partially unsaturated” refers to a moiety that includes at least one double or triple bond. [0058] The term “saturated” refers to a moiety that does not contain a double or triple bond, i.e., the moiety only contains single bonds. [0059] Affixing the suffix “-ene” to a group indicates the group is a divalent moiety, e.g., alkylene is the divalent moiety of alkyl, alkenylene is the divalent moiety of alkenyl, alkynylene is the divalent moiety of alkynyl, heteroalkylene is the divalent moiety of heteroalkyl, heteroalkenylene is the divalent moiety of heteroalkenyl, heteroalkynylene is the divalent moiety of heteroalkynyl, carbocyclylene is the divalent moiety of carbocyclyl, heterocyclylene is the divalent moiety of heterocyclyl, arylene is the divalent moiety of aryl, and heteroarylene is the divalent moiety of heteroaryl. [0060] The term “azido” refers to the radical –N
3. [0061] “Aliphatic” refers to an alkyl, alkenyl, alkynyl, or carbocyclyl group, as defined herein. [0062] “Cycloalkylalkyl” refers to an alkyl radical in which the alkyl group is substituted with a cycloalkyl group. Typical cycloalkylalkyl groups include, but are not limited to, cyclopropylmethyl, cyclobutylmethyl, cyclopentylmethyl, cyclohexylmethyl, cycloheptylmethyl, cyclooctylmethyl, cyclopropylethyl, cyclobutylethyl, cyclopentylethyl, cyclohexylethyl, cycloheptylethyl, and cyclooctylethyl, and the like. [0063] “Heterocyclylalkyl” refers to an alkyl radical in which the alkyl group is substituted with a heterocyclyl group. Typical heterocyclylalkyl groups include, but are not limited to, pyrrolidinylmethyl, piperidinylmethyl, piperazinylmethyl, morpholinylmethyl, pyrrolidinylethyl, piperidinylethyl, piperazinylethyl, morpholinylethyl, and the like. [0064] “Aralkyl” or “arylalkyl” is a subset of alkyl and aryl, as defined herein, and refers to an optionally substituted alkyl group substituted by an optionally substituted aryl group. [0065] “Alkyl” refers to a radical of a straight–chain or branched saturated hydrocarbon group having from 1 to 20 carbon atoms (“C
1–20 alkyl”). In an embodiment, an alkyl group has 1 to 12 carbon atoms (“C
1–12 alkyl”). In an embodiment, an alkyl group has 1 to 10 carbon atoms (“C
1–10 alkyl”). In an embodiment, an alkyl group has 1 to 9 carbon atoms (“C
1–9 alkyl”). In an embodiment, an alkyl group has 1 to 8 carbon atoms (“C
1–8 alkyl”). In an embodiment, an alkyl group has 1 to 7 carbon atoms (“C
1–7 alkyl”). In an embodiment, an alkyl group has 1 to 6 carbon atoms (“C
1–6 alkyl”, also referred to herein as “lower alkyl”). In an embodiment, an alkyl group has 1 to 5 carbon atoms (“C
1–5 alkyl”). In an embodiment, an alkyl group has 1 to 4 carbon atoms (“C
1–4 alkyl”). In an embodiment, an alkyl group has 1 to 3 carbon atoms (“C
1–3 alkyl”). In an embodiment, an alkyl group has 1 to 2 carbon atoms (“C
1–2 alkyl”). In an embodiment, an alkyl group has 1 carbon atom (“C
1 alkyl”). In an embodiment, an alkyl group has 2 to 6 carbon atoms (“C
2–6 alkyl”). Examples of C
1–6 alkyl groups include methyl (C
1), ethyl (C
2), n–propyl (C
3), isopropyl (C
3), n–butyl (C
4), tert–butyl (C
4), sec–butyl (C
4), iso–butyl (C
4), n–pentyl (C
5), 3–pentanyl (C
5), amyl (C
5), neopentyl (C
5), 3–methyl–2–butanyl (C
5), tertiary amyl (C
5), and n–hexyl (C
6). Additional examples of alkyl groups include n–heptyl (C
7), n–octyl (C
8) and the like. Unless otherwise specified, each instance of an alkyl group is independently optionally substituted, i.e., unsubstituted (an “unsubstituted alkyl”) or substituted (a “substituted alkyl”) with one or more substituents; e.g., for instance from 1 to 5 substituents, 1 to 3 substituents, or 1 substituent. In an embodiment, the alkyl group is unsubstituted C
1–10 alkyl (e.g., –CH
3). In an embodiment, the alkyl group is substituted C
1–10 alkyl. Common alkyl abbreviations include Me (–CH
3), Et (– CH
2CH
3),
iPr (–CH(CH
3)
2),
nPr (–CH
2CH
2CH
3),
nBu (–CH
2CH
2CH
2CH
3), or
iBu (– CH
2CH(CH
3)
2). [0066] “Alkylene” refers to an alkyl group wherein two hydrogens are removed to provide a divalent radical, and which may be substituted or unsubstituted. Unsubstituted alkylene groups include, but are not limited to, methylene (–CH
2-), ethylene (–CH
2CH
2-), propylene (– CH
2CH
2CH
2-), butylene (–CH
2CH
2CH
2CH
2-), pentylene (–CH
2CH
2CH
2CH
2CH
2-), hexylene (–CH
2CH
2CH
2CH
2CH
2CH
2-), and the like. Exemplary substituted alkylene groups, e.g., substituted with one or more alkyl (methyl) groups, include but are not limited to, substituted methylene (–CH(CH
3)-, (–C(CH
3)
2-), substituted ethylene (–CH(CH
3)CH
2-,–CH
2CH(CH
3)-, –C(CH
3)
2CH
2-,–CH
2C(CH
3)
2-), substituted propylene (–CH(CH
3)CH
2CH
2-, – CH
2CH(CH
3)CH
2-, –CH
2CH
2CH(CH
3)-, –C(CH
3)
2CH
2CH
2-, –CH
2C(CH
3)
2CH
2-, – CH
2CH
2C(CH
3)
2-), and the like. When a range or number of carbons is provided for a particular alkylene group, it is understood that the range or number refers to the range or number of carbons in the linear carbon divalent chain. Alkylene groups may be substituted or unsubstituted with one or more substituents as described herein. [0067] “Alkenyl” refers to a radical of a straight–chain or branched hydrocarbon group having from 2 to 20 carbon atoms, one or more carbon–carbon double bonds (e.g., 1, 2, 3, or 4 carbon–carbon double bonds), and optionally one or more carbon–carbon triple bonds (e.g., 1, 2, 3, or 4 carbon–carbon triple bonds) (“C
2–20 alkenyl”). In an embodiment, alkenyl does not contain any triple bonds. In an embodiment, an alkenyl group has 2 to 10 carbon atoms (“C
2–10 alkenyl”). In an embodiment, an alkenyl group has 2 to 9 carbon atoms (“C
2–9 alkenyl”). In an embodiment, an alkenyl group has 2 to 8 carbon atoms (“C
2–8 alkenyl”). In an embodiment, an alkenyl group has 2 to 7 carbon atoms (“C
2–7 alkenyl”). In an embodiment, an alkenyl group has 2 to 6 carbon atoms (“C
2–6 alkenyl”). In an embodiment, an alkenyl group has 2 to 5 carbon atoms (“C
2–5 alkenyl”). In an embodiment, an alkenyl group has 2 to 4 carbon atoms (“C
2–4 alkenyl”). In an embodiment, an alkenyl group has 2 to 3 carbon atoms (“C
2–3 alkenyl”). In an embodiment, an alkenyl group has 2 carbon atoms (“C
2 alkenyl”). The one or more carbon–carbon double bonds can be internal (such as in 2– butenyl) or terminal (such as in 1–butenyl). Examples of C
2–4 alkenyl groups include ethenyl (C
2), 1–propenyl (C
3), 2–propenyl (C
3), 1–butenyl (C
4), 2–butenyl (C
4), butadienyl (C
4), and the like. Examples of C
2–6 alkenyl groups include the aforementioned C
2–4 alkenyl groups as well as pentenyl (C
5), pentadienyl (C
5), hexenyl (C
6), and the like. Additional examples of alkenyl include heptenyl (C
7), octenyl (C
8), octatrienyl (C
8), and the like. Unless otherwise specified, each instance of an alkenyl group is independently optionally substituted, i.e., unsubstituted (an “unsubstituted alkenyl”) or substituted (a “substituted alkenyl”) with one or more substituents e.g., for instance from 1 to 5 substituents, 1 to 3 substituents, or 1 substituent. In an embodiment, the alkenyl group is unsubstituted C
2–10 alkenyl. In an embodiment, the alkenyl group is substituted C
2–10 alkenyl. [0068] “Alkynyl” refers to a radical of a straight–chain or branched hydrocarbon group having from 2 to 20 carbon atoms, one or more carbon–carbon triple bonds (e.g., 1, 2, 3, or 4 carbon–carbon triple bonds), and optionally one or more carbon–carbon double bonds (e.g., 1, 2, 3, or 4 carbon–carbon double bonds) (“C
2–20 alkynyl”). In an embodiment, alkynyl does not contain any double bonds. In an embodiment, an alkynyl group has 2 to 10 carbon atoms (“C
2–10 alkynyl”). In an embodiment, an alkynyl group has 2 to 9 carbon atoms (“C
2–9 alkynyl”). In an embodiment, an alkynyl group has 2 to 8 carbon atoms (“C
2–8 alkynyl”). In an embodiment, an alkynyl group has 2 to 7 carbon atoms (“C
2–7 alkynyl”). In an embodiment, an alkynyl group has 2 to 6 carbon atoms (“C
2–6 alkynyl”). In an embodiment, an alkynyl group has 2 to 5 carbon atoms (“C
2–5 alkynyl”). In an embodiment, an alkynyl group has 2 to 4 carbon atoms (“C
2–4 alkynyl”). In an embodiment, an alkynyl group has 2 to 3 carbon atoms (“C
2–3 alkynyl”). In an embodiment, an alkynyl group has 2 carbon atoms (“C
2 alkynyl”). The one or more carbon–carbon triple bonds can be internal (such as in 2– butynyl) or terminal (such as in 1–butynyl). Examples of C
2–4 alkynyl groups include, without limitation, ethynyl (C
2), 1–propynyl (C
3), 2–propynyl (C
3), 1–butynyl (C
4), 2– butynyl (C
4), and the like. Examples of C
2–6 alkenyl groups include the aforementioned C
2–4 alkynyl groups as well as pentynyl (C
5), hexynyl (C
6), and the like. Additional examples of alkynyl include heptynyl (C
7), octynyl (C
8), and the like. Unless otherwise specified, each instance of an alkynyl group is independently optionally substituted, i.e., unsubstituted (an “unsubstituted alkynyl”) or substituted (a “substituted alkynyl”) with one or more substituents; e.g., for instance from 1 to 5 substituents, 1 to 3 substituents, or 1 substituent. In an embodiment, the alkynyl group is unsubstituted C
2–10 alkynyl. In an embodiment, the alkynyl group is substituted C
2–10 alkynyl. [0069] The term “heteroalkyl,” as used herein, refers to an alkyl group, as defined herein, which further comprises 1 or more (e.g., 1, 2, 3, or 4) heteroatoms (e.g., oxygen, sulfur, nitrogen, boron, silicon, phosphorus and oxidized forms thereof, e.g., SO
2) within the parent chain, wherein the one or more heteroatoms is inserted between adjacent carbon atoms within the parent carbon chain and/or one or more heteroatoms is inserted between a carbon atom and the parent molecule, i.e., between the point of attachment. In an embodiment, the heteroalkyl contains 1-2 heteroatoms independently selected from N, O and S and oxidized forms thereof (e.g., SO
2). In an embodiment, the heteroalkyl contains 1 heteroatom independently selected from N, O and S and oxidized forms thereof (e.g., SO
2). In an embodiment, the heteroalkyl contains 2 heteroatoms independently selected from N, O and S and oxidized forms thereof (e.g., SO
2). In an embodiment, a heteroalkyl group refers to a saturated group having from 1 to 10 carbon atoms and 1, 2, 3, or 4 heteroatoms (“heteroC
1–10 alkyl”). In an embodiment, a heteroalkyl group is a saturated group having 1 to 9 carbon atoms and 1, 2, 3, or 4 heteroatoms (“heteroC
1–9 alkyl”). In an embodiment, a heteroalkyl group is a saturated group having 1 to 8 carbon atoms and 1, 2, 3, or 4 heteroatoms (“heteroC
1–8 alkyl”). In an embodiment, a heteroalkyl group is a saturated group having 1 to 7 carbon atoms and 1, 2, 3, or 4 heteroatoms (“heteroC
1–7 alkyl”). In an embodiment, a heteroalkyl group is a group having 1 to 6 carbon atoms and 1, 2, or 3 heteroatoms (“heteroC
1–6 alkyl”). In an embodiment, a heteroalkyl group is a saturated group having 1 to 5 carbon atoms and 1 or 2 heteroatoms (“heteroC
1–5 alkyl”). In an embodiment, a heteroalkyl group is a saturated group having 1 to 4 carbon atoms and 1 or 2 heteroatoms (“heteroC
1–4 alkyl”). In an embodiment, a heteroalkyl group is a saturated group having 1 to 3 carbon atoms and 1 heteroatom (“heteroC
1–3 alkyl”). In an embodiment, a heteroalkyl group is a saturated group having 1 to 2 carbon atoms and 1 heteroatom (“heteroC
1–2 alkyl”). In an embodiment, a heteroalkyl group is a saturated group having 1 carbon atom and 1 heteroatom (“heteroC
1 alkyl”). In an embodiment, a heteroalkyl group is a saturated group having 2 to 6 carbon atoms and 1 or 2 heteroatoms (“heteroC
2–6 alkyl”). Unless otherwise specified, each instance of a heteroalkyl group is independently unsubstituted (an “unsubstituted heteroalkyl”) or substituted (a “substituted heteroalkyl”) with one or more substituents. In an embodiment, the heteroalkyl group is an unsubstituted heteroC
1–10 alkyl. In an embodiment, the heteroalkyl group is a substituted heteroC
1–10 alkyl. Exemplary heteroalkyl groups include: –CH
2OH, –CH
2OCH
3, –CH
2NH
2, –CH
2NH(CH
3), –CH
2N(CH
3)
2, –CH
2CH
2OH, – CH
2CH
2OCH
3, –CH
2CH
2NH
2, –CH
2CH
2NH(CH
3), –CH
2CH
2N(CH
3)
2, –CH
2CH
2S(O)
2CH
3. [0070] “Aryl” refers to a radical of a monocyclic or polycyclic (e.g., bicyclic or tricyclic) 4n+2 aromatic ring system (e.g., having 6, 10, or 14 π electrons shared in a cyclic array) having 6–14 ring carbon atoms and zero heteroatoms provided in the aromatic ring system (“C
6–14 aryl”). In an embodiment, an aryl group has six ring carbon atoms (“C
6 aryl”; e.g., phenyl). In an embodiment, an aryl group has ten ring carbon atoms (“C
10 aryl”; e.g., naphthyl such as 1–naphthyl and 2–naphthyl). In an embodiment, an aryl group has fourteen ring carbon atoms (“C
14 aryl”; e.g., anthracyl). “Aryl” also includes ring systems wherein the aryl ring, as defined above, is fused with one or more carbocyclyl or heterocyclyl groups wherein the radical or point of attachment is on the aryl ring, and in such instances, the number of carbon atoms continue to designate the number of carbon atoms in the aryl ring system. Particularly aryl groups include phenyl, naphthyl, indenyl, and tetrahydronaphthyl. Unless otherwise specified, each instance of an aryl group is independently optionally substituted, i.e., unsubstituted (an “unsubstituted aryl”) or substituted (a “substituted aryl”) with one or more substituents. In an embodiment, the aryl group is unsubstituted C
6–14 aryl. In an embodiment, the aryl group is substituted C
6–14 aryl. [0071] In an embodiment, an aryl group is substituted with one or more of groups selected from halo, C
1–C
8 alkyl, C
1–C
8 haloalkyl, cyano, hydroxy, C
1–C
8 alkoxy, and amino. [0072] Examples of representative substituted aryls include the following:
wherein one of R
56 and R
57 may be hydrogen and at least one of R
56 and R
57 is each independently selected from C
1–C
8 alkyl, C
1–C
8 haloalkyl, 4-10 membered heterocyclyl, alkanoyl, C
1–C
8 alkoxy, heteroaryloxy, alkylamino, arylamino, heteroarylamino, NR
58COR
59, NR
58SOR
59 NR
58SO
2R
59, COOalkyl, COOaryl, CONR
58R
59, CONR
58OR
59, NR
58R
59, SO
2NR
58R
59, S-alkyl, SOalkyl, SO
2alkyl, Saryl, SOaryl, SO
2aryl; or R
56 and R
57 may be joined to form a cyclic ring (saturated or unsaturated) from 5 to 8 atoms, optionally containing one or more heteroatoms selected from the group N, O, or S. R
60 and R
61 are independently hydrogen, C
1–C
8 alkyl, C
1–C
4 haloalkyl, C
3–C
10 cycloalkyl, 4-10 membered heterocyclyl, C
6–C
10 aryl, substituted C
6–C
10 aryl, 5-10 membered heteroaryl, or substituted 5-10 membered heteroaryl. [0073] “Fused aryl” refers to an aryl having two of its ring carbons in common with a second aryl or heteroaryl ring or with a carbocyclyl or heterocyclyl ring. [0074] “Heteroaryl” refers to a radical of a 5–10 membered monocyclic or bicyclic 4n+2 aromatic ring system (e.g., having 6 or 10 π electrons shared in a cyclic array) having ring carbon atoms and 1–4 ring heteroatoms provided in the aromatic ring system, wherein each heteroatom is independently selected from nitrogen, oxygen and sulfur (“5–10 membered heteroaryl”). In heteroaryl groups that contain one or more nitrogen atoms, the point of attachment can be a carbon or nitrogen atom, as valency permits. Heteroaryl bicyclic ring systems can include one or more heteroatoms in one or both rings. “Heteroaryl” includes ring systems wherein the heteroaryl ring, as defined above, is fused with one or more carbocyclyl or heterocyclyl groups wherein the point of attachment is on the heteroaryl ring, and in such instances, the number of ring members continue to designate the number of ring members in the heteroaryl ring system. “Heteroaryl” also includes ring systems wherein the heteroaryl ring, as defined above, is fused with one or more aryl groups wherein the point of attachment is either on the aryl or heteroaryl ring, and in such instances, the number of ring members designates the number of ring members in the fused (aryl/heteroaryl) ring system. Bicyclic heteroaryl groups wherein one ring does not contain a heteroatom (e.g., indolyl, quinolinyl, carbazolyl, and the like) the point of attachment can be on either ring, i.e., either the ring bearing a heteroatom (e.g., 2–indolyl) or the ring that does not contain a heteroatom (e.g., 5– indolyl). [0075] In an embodiment, a heteroaryl group is a 5–10 membered aromatic ring system having ring carbon atoms and 1–4 ring heteroatoms provided in the aromatic ring system, wherein each heteroatom is independently selected from nitrogen, oxygen, and sulfur (“5–10 membered heteroaryl”). In an embodiment, a heteroaryl group is a 5–8 membered aromatic ring system having ring carbon atoms and 1–4 ring heteroatoms provided in the aromatic ring system, wherein each heteroatom is independently selected from nitrogen, oxygen, and sulfur (“5–8 membered heteroaryl”). In an embodiment, a heteroaryl group is a 5–6 membered aromatic ring system having ring carbon atoms and 1–4 ring heteroatoms provided in the aromatic ring system, wherein each heteroatom is independently selected from nitrogen, oxygen, and sulfur (“5–6 membered heteroaryl”). In an embodiment, the 5–6 membered heteroaryl has 1–3 ring heteroatoms selected from nitrogen, oxygen, and sulfur. In an embodiment, the 5–6 membered heteroaryl has 1–2 ring heteroatoms selected from nitrogen, oxygen, and sulfur. In an embodiment, the 5–6 membered heteroaryl has 1 ring heteroatom selected from nitrogen, oxygen, and sulfur. Unless otherwise specified, each instance of a heteroaryl group is independently optionally substituted, i.e., unsubstituted (an “unsubstituted heteroaryl”) or substituted (a “substituted heteroaryl”) with one or more substituents. In an embodiment, the heteroaryl group is unsubstituted 5–14 membered heteroaryl. In an embodiment, the heteroaryl group is substituted 5–14 membered heteroaryl. In an embodiment, a heteroaryl group is a bicyclic 8-12 membered aromatic ring system having ring carbon atoms and 1-6 ring heteroatoms provided in the aromatic ring system, wherein each heteroatom is independently selected from nitrogen, oxygen, and sulfur (“8-12 membered bicyclic heteroaryl”). In an embodiment, a heteroaryl group is an 8-10 membered bicyclic aromatic ring system having ring carbon atoms and 1-6 ring heteroatoms provided in the aromatic ring system, wherein each heteroatom is independently selected from nitrogen, oxygen, and sulfur (“8-10 membered bicyclic heteroaryl”). In an embodiment, a heteroaryl group is a 9-10 membered bicyclic aromatic ring system having ring carbon atoms and 1-6 ring heteroatoms provided in the aromatic ring system, wherein each heteroatom is independently selected from nitrogen, oxygen, and sulfur (“9-10 membered bicyclic heteroaryl”). Unless otherwise specified, each instance of a heteroaryl group is independently unsubstituted (an “unsubstituted heteroaryl”) or substituted (a “substituted heteroaryl”) with one or more substituents. In an embodiment, the heteroaryl group is an unsubstituted 5-14 membered heteroaryl. In an embodiment, the heteroaryl group is a substituted 5-14 membered heteroaryl. [0076] Exemplary 5–membered heteroaryl groups containing one heteroatom include, without limitation, pyrrolyl, furanyl and thiophenyl. Exemplary 5–membered heteroaryl groups containing two heteroatoms include, without limitation, imidazolyl, pyrazolyl, oxazolyl, isoxazolyl, thiazolyl, and isothiazolyl. Exemplary 5–membered heteroaryl groups containing three heteroatoms include, without limitation, triazolyl, oxadiazolyl, and thiadiazolyl. Exemplary 5–membered heteroaryl groups containing four heteroatoms include, without limitation, tetrazolyl. Exemplary 6–membered heteroaryl groups containing one heteroatom include, without limitation, pyridinyl. Exemplary 6–membered heteroaryl groups containing two heteroatoms include, without limitation, pyridazinyl, pyrimidinyl, and pyrazinyl. Exemplary 6–membered heteroaryl groups containing three or four heteroatoms include, without limitation, triazinyl and tetrazinyl, respectively. Exemplary 7–membered heteroaryl groups containing one heteroatom include, without limitation, azepinyl, oxepinyl, and thiepinyl. Exemplary 5,6–bicyclic heteroaryl groups include, without limitation, indolyl, isoindolyl, indazolyl, benzotriazolyl, benzothiophenyl, isobenzothiophenyl, benzofuranyl, benzoisofuranyl, benzimidazolyl, benzoxazolyl, benzisoxazolyl, benzoxadiazolyl, benzthiazolyl, benzisothiazolyl, benzthiadiazolyl, indolizinyl, and purinyl. Exemplary 6,6– bicyclic heteroaryl groups include, without limitation, naphthyridinyl, pteridinyl, quinolinyl, isoquinolinyl, cinnolinyl, quinoxalinyl, phthalazinyl, and quinazolinyl. [0077] Examples of representative heteroaryls include the following:
wherein each Z is selected from carbonyl, N, NR
65, O, and S; and R
65 is independently hydrogen, C
1–C
8 alkyl, C
3–C
10 cycloalkyl, 4-10 membered heterocyclyl, C
6–C
10 aryl, and 5- 10 membered heteroaryl. [0078] In the structures described herein, a substituent attached to a polycyclic (e.g., bicyclic or tricyclic) cycloalkyl, heterocyclyl, aryl or heteroaryl with a bond that spans two or more rings is understood to mean that the substituent can be attached at any position in each of the rings. [0079] “Heteroaralkyl” or “heteroarylalkyl” is a subset of “alkyl” and refers to an alkyl group substituted by a heteroaryl group, wherein the point of attachment is on the alkyl moiety. [0080] The term “carbocyclyl” or “carbocyclic” refers to a radical of a non-aromatic monocyclic, bicyclic, or tricyclic or polycyclic hydrocarbon ring system having from 3 to 14 ring carbon atoms (“C
3-14 carbocyclyl”) and zero heteroatoms in the non-aromatic ring system. Carbocyclyl groups include fully saturated ring systems (e.g., cycloalkyls), and partially saturated ring systems. In an embodiment, a carbocyclyl group has 3 to 10 ring carbon atoms (“C
3-10 carbocyclyl”). In an embodiment, a carbocyclyl group has 3 to 8 ring carbon atoms (“C
3-8 carbocyclyl”). In an embodiment, a carbocyclyl group has 3 to 7 ring carbon atoms (“C
3-7 carbocyclyl”). In an embodiment, a carbocyclyl group has 3 to 6 ring carbon atoms (“C
3-6 carbocyclyl”). In an embodiment, a carbocyclyl group has 4 to 6 ring carbon atoms (“C
4-6 carbocyclyl”). In an embodiment, a carbocyclyl group has 5 to 6 ring carbon atoms (“C
5-6 carbocyclyl”). In an embodiment, a carbocyclyl group has 5 to 10 ring carbon atoms (“C
5-10 carbocyclyl”). Exemplary C
3-6 carbocyclyl groups include, without limitation, cyclopropyl (C
3), cyclopropenyl (C
3), cyclobutyl (C
4), cyclobutenyl (C
4), cyclopentyl (C
5), cyclopentenyl (C
5), cyclohexyl (C
6), cyclohexenyl (C
6), cyclohexadienyl (C6), and the like. Exemplary C3-8 carbocyclyl groups include, without limitation, the aforementioned C
3-6 carbocyclyl groups as well as cycloheptyl (C
7), cycloheptenyl (C
7), cycloheptadienyl (C
7), cycloheptatrienyl (C
7), cyclooctyl (C
8), cyclooctenyl (C
8), bicyclo[2.2.1]heptanyl (C
7), bicyclo[2.2.2]octanyl (C
8), and the like. Exemplary C
3-10 carbocyclyl groups include, without limitation, the aforementioned C
3-8 carbocyclyl groups as well as cyclononyl (C
9), cyclononenyl (C
9), cyclodecyl (C
10), cyclodecenyl (C
10), octahydro- 1H-indenyl (C
9), decahydronaphthalenyl (C
10), spiro[4.5]decanyl (C
10), and the like. [0081] As the foregoing examples illustrate, in an embodiment, the carbocyclyl group is either monocyclic (“monocyclic carbocyclyl”) or polycyclic (e.g., containing a fused, bridged or spiro ring system such as a bicyclic system (“bicyclic carbocyclyl”) or tricyclic system (“tricyclic carbocyclyl”)) and can be saturated or can contain one or more carbon-carbon double or triple bonds. “Carbocyclyl” also includes ring systems wherein the carbocyclyl ring, as defined above, is fused with one or more aryl or heteroaryl groups wherein the point of attachment is on the carbocyclyl ring, and in such instances, the number of carbons continue to designate the number of carbons in the carbocyclic ring system. Unless otherwise specified, each instance of a carbocyclyl group is independently unsubstituted (an “unsubstituted carbocyclyl”) or substituted (a “substituted carbocyclyl”) with one or more substituents. In an embodiment, the carbocyclyl group is an unsubstituted C
3-14 carbocyclyl. In an embodiment, the carbocyclyl group is a substituted C
3-14 carbocyclyl. [0082] The term “cycloalkyl” as employed herein includes saturated cyclic, bicyclic, tricyclic, or polycyclic hydrocarbon groups having 3 to 14 carbons containing the indicated number of rings and carbon atoms (for example a C
3–C
14 monocyclic, C
4–C
14 bicyclic, C
5– C
14 tricyclic, or C
6–C
14 polycyclic cycloalkyl). In an embodiment “cycloalkyl” is a monocyclic cycloalkyl. In an embodiment, a monocyclic cycloalkyl has 3-14 ring carbon atoms (“C
3-14 monocyclic cycloalkyl”). In an embodiment, a monocyclic cycloalkyl group has 3 to 10 ring carbon atoms (“C
3-10 monocyclic cycloalkyl”). In an embodiment, a monocyclic cycloalkyl group has 3 to 8 ring carbon atoms (“C
3-8 monocyclic cycloalkyl”). In an embodiment, a monocyclic cycloalkyl group has 3 to 6 ring carbon atoms (“C
3-6 monocyclic cycloalkyl”). In an embodiment, a monocyclic cycloalkyl group has 4 to 6 ring carbon atoms (“C
4-6 monocyclic cycloalkyl”). In an embodiment, a monocyclic cycloalkyl group has 5 to 6 ring carbon atoms (“C
5-6 monocyclic cycloalkyl”). In an embodiment, a monocyclic cycloalkyl group has 5 to 10 ring carbon atoms (“C
5-10 monocyclic cycloalkyl”). Examples of monocyclic C
5-6 cycloalkyl groups include cyclopentyl (C
5) and cyclohexyl (C
5). Examples of C
3-6 cycloalkyl groups include the aforementioned C
5-6 cycloalkyl groups as well as cyclopropyl (C
3) and cyclobutyl (C
4). Examples of C
3-8 cycloalkyl groups include the aforementioned C
3-6 cycloalkyl groups as well as cycloheptyl (C
7) and cyclooctyl (C
8). [0083] In an embodiment “cycloalkyl” is a bicyclic cycloalkyl. In an embodiment, a bicyclic cycloalkyl has 4-14 ring carbon atoms (“C
4-14 bicyclic cycloalkyl”). In an embodiment, a bicyclic cycloalkyl group has 4 to 12 ring carbon atoms (“C
4-12 bicyclic cycloalkyl”). In an embodiment, a bicyclic cycloalkyl group has 4 to 10 ring carbon atoms (“C
4-10 bicyclic cycloalkyl”). In an embodiment, a bicyclic cycloalkyl group has 5 to 10 ring carbon atoms (“C
5-10 bicyclic cycloalkyl”). In an embodiment, a bicyclic cycloalkyl group has 6 to 10 ring carbon atoms (“C
6-10 bicyclic cycloalkyl”). In an embodiment, a bicyclic cycloalkyl group has 8 to 10 ring carbon atoms (“C
8-10 bicyclic cycloalkyl”). In an embodiment, a bicyclic cycloalkyl group has 7 to 9 ring carbon atoms (“C
7-9 bicyclic cycloalkyl”). Examples of bicyclic cycloalkyls include bicyclo[1.1.0]butane (C
4), bicyclo[1.1.1]pentane (C
5), spiro[2.2] pentane (C
5), bicyclo[2.1.0]pentane (C
5), bicyclo[2.1.1]hexane (C
6), bicyclo[3.1.0]hexane (C
6), spiro[2.3] hexane (C
6), bicyclo[2.2.1]heptane (norbornane) (C
7), bicyclo[3.2.0]heptane (C
7), bicyclo[3.1.1]heptane (C
7), bicyclo[3.1.1]heptane (C
7), bicyclo[4.1.0]heptane (C
7), spiro[2.4] heptane (C
7), spiro [3.3] heptane (C
7), bicyclo[2.2.2]octane (C
8), bicyclo[4.1.1]octane (C
8)octahydropentalene (C
8), bicyclo[3.2.1]octane (C
8), bicyclo[4.2.0]octane (C
8), spiro[2.5]octane (C
8), spiro[3.4]octane (C
8), bicyclo[3.3.1]nonane (C
9), octahydro-1H-indene (C
9), bicyclo[4.2.1]nonane (C
9), spiro[3.5]nonane (C
9), spiro[4.4]nonane (C
9), bicyclo[3.3.2]decane (C
10), bicyclo[4.3.1]decane (C
10), spiro[4.5]decane (C
10), bicyclo[3.3.3]undecane (C
11), decahydronaphthalene (C
10), bicyclo[4.3.2]undecane (C
11), spiro[5.5]undecane (C
11) and bicyclo[4.3.3]dodecane (C
12). [0084] In an embodiment “cycloalkyl” is a tricyclic cycloalkyl. In an embodiment, a tricyclic cycloalkyl has 6-14 ring carbon atoms (“C
6-14 tricyclic cycloalkyl”). In an embodiment, a tricyclic cycloalkyl group has 8 to 12 ring carbon atoms (“C
8-12 tricyclic cycloalkyl”). In an embodiment, a tricyclic cycloalkyl group has 10 to 12 ring carbon atoms (“C
10-12 tricyclic cycloalkyl. Examples of tricyclic cycloalkyls include adamantine (C
12). Unless otherwise specified, each instance of a cycloalkyl group is independently unsubstituted (an “unsubstituted cycloalkyl”) or substituted (a “substituted cycloalkyl”) with one or more substituents. In an embodiment, the cycloalkyl group is an unsubstituted C
3-14 cycloalkyl. In an embodiment, the cycloalkyl group is a substituted C
3-14 cycloalkyl [0085] “Heterocyclyl” or “heterocyclic” refers to a radical of a 3– to 10–membered non– aromatic ring system having ring carbon atoms and 1 to 4 ring heteroatoms, wherein each heteroatom is independently selected from nitrogen, oxygen, sulfur, boron, phosphorus, and silicon (“3–10 membered heterocyclyl”). In heterocyclyl groups that contain one or more nitrogen atoms, the point of attachment can be a carbon or nitrogen atom, as valency permits. A heterocyclyl group can either be monocyclic (“monocyclic heterocyclyl”) or a fused, bridged or spiro ring system such as a bicyclic system (“bicyclic heterocyclyl”), and can be saturated or can be partially unsaturated. Heterocyclyl bicyclic ring systems can include one or more heteroatoms in one or both rings. “Heterocyclyl” also includes ring systems wherein the heterocyclyl ring, as defined above, is fused with one or more carbocyclyl groups wherein the point of attachment is either on the carbocyclyl or heterocyclyl ring, or ring systems wherein the heterocyclyl ring, as defined above, is fused with one or more aryl or heteroaryl groups, wherein the point of attachment is on the heterocyclyl ring, and in such instances, the number of ring members continue to designate the number of ring members in the heterocyclyl ring system. Unless otherwise specified, each instance of heterocyclyl is independently optionally substituted, i.e., unsubstituted (an “unsubstituted heterocyclyl”) or substituted (a “substituted heterocyclyl”) with one or more substituents. In an embodiment, the heterocyclyl group is unsubstituted 3–10 membered heterocyclyl. In an embodiment, the heterocyclyl group is substituted 3–10 membered heterocyclyl. [0086] In an embodiment, a heterocyclyl group is a 5–10 membered non–aromatic ring system having ring carbon atoms and 1–4 ring heteroatoms, wherein each heteroatom is independently selected from nitrogen, oxygen, sulfur, boron, phosphorus, and silicon (“5–10 membered heterocyclyl”). In an embodiment, a heterocyclyl group is a 5–8 membered non– aromatic ring system having ring carbon atoms and 1–4 ring heteroatoms, wherein each heteroatom is independently selected from nitrogen, oxygen, and sulfur (“5–8 membered heterocyclyl”). In an embodiment, a heterocyclyl group is a 5–6 membered non–aromatic ring system having ring carbon atoms and 1–4 ring heteroatoms, wherein each heteroatom is independently selected from nitrogen, oxygen, and sulfur (“5–6 membered heterocyclyl”). In an embodiment, the 5–6 membered heterocyclyl has 1–3 ring heteroatoms selected from nitrogen, oxygen, and sulfur. In an embodiment, the 5–6 membered heterocyclyl has 1–2 ring heteroatoms selected from nitrogen, oxygen, and sulfur. In an embodiment, the 5–6 membered heterocyclyl has one ring heteroatom selected from nitrogen, oxygen, and sulfur. [0087] Exemplary 3–membered heterocyclyl groups containing one heteroatom include, without limitation, aziridinyl, oxiranyl, thiorenyl. Exemplary 4–membered heterocyclyl groups containing one heteroatom include, without limitation, azetidinyl, oxetanyl and thietanyl. Exemplary 5–membered heterocyclyl groups containing one heteroatom include, without limitation, tetrahydrofuranyl, dihydrofuranyl, tetrahydrothiophenyl, dihydrothiophenyl, pyrrolidinyl, dihydropyrrolyl and pyrrolyl–2,5–dione. Exemplary 5– membered heterocyclyl groups containing two heteroatoms include, without limitation, dioxolanyl, oxasulfuranyl, disulfuranyl, and oxazolidin-2-one. Exemplary 5–membered heterocyclyl groups containing three heteroatoms include, without limitation, triazolinyl, oxadiazolinyl, and thiadiazolinyl. Exemplary 6–membered heterocyclyl groups containing one heteroatom include, without limitation, piperidinyl, tetrahydropyranyl, dihydropyridinyl, and thianyl. Exemplary 6–membered heterocyclyl groups containing two heteroatoms include, without limitation, piperazinyl, morpholinyl, dithianyl, dioxanyl. Exemplary 6– membered heterocyclyl groups containing two heteroatoms include, without limitation, triazinanyl. Exemplary 7–membered heterocyclyl groups containing one heteroatom include, without limitation, azepanyl, oxepanyl and thiepanyl. Exemplary 8–membered heterocyclyl groups containing one heteroatom include, without limitation, azocanyl, oxecanyl and thiocanyl. Exemplary 5-membered heterocyclyl groups fused to a C
6 aryl ring (also referred to herein as a 5,6-bicyclic heterocyclic ring) include, without limitation, indolinyl, isoindolinyl, dihydrobenzofuranyl, dihydrobenzothienyl, benzoxazolinonyl, and the like. Exemplary bicyclic heterocyclyl groups include, without limitation, indolinyl, isoindolinyl, dihydrobenzofuranyl, dihydrobenzothienyl, tetrahydrobenzothienyl, tetrahydrobenzofuranyl, tetrahydroindolyl, tetrahydroquinolinyl, tetrahydroisoquinolinyl, decahydroquinolinyl, decahydroisoquinolinyl, octahydrochromenyl, octahydroisochromenyl, decahydronaphthyridinyl, decahydro-1,8-naphthyridinyl, octahydropyrrolo[3,2-b]pyrrole, indolinyl, phthalimidyl, naphthalimidyl, chromanyl, chromenyl, 1H-benzo[e][1,4]diazepinyl, 1,4,5,7-tetrahydropyrano[3,4-b]pyrrolyl, 5,6-dihydro-4H-furo[3,2-b]pyrrolyl, 6,7-dihydro- 5H-furo[3,2-b]pyranyl, 5,7-dihydro-4H-thieno[2,3-c]pyranyl, 2,3-dihydro-1H-pyrrolo[2,3- b]pyridinyl, 2,3-dihydrofuro[2,3-b]pyridinyl, 4,5,6,7-tetrahydro-1H-pyrrolo[2,3-b]pyridinyl, 4,5,6,7-tetrahydrofuro[3,2-c]pyridinyl, 4,5,6,7-tetrahydrothieno[3,2-b]pyridinyl, 1,2,3,4- tetrahydro-1,6-naphthyridinyl, and the like. Exemplary 6-membered heterocyclyl groups fused to an aryl ring (also referred to herein as a 6,6-bicyclic heterocyclic ring) include, without limitation, tetrahydroquinolinyl, tetrahydroisoquinolinyl, and the like. [0088] “Nitrogen-containing heterocyclyl” group means a 4– to 7– membered non-aromatic cyclic group containing at least one nitrogen atom, for example, but without limitation, morpholine, piperidine (e.g., 2-piperidinyl, 3-piperidinyl and 4-piperidinyl), pyrrolidine (e.g., 2-pyrrolidinyl and 3-pyrrolidinyl), azetidine, pyrrolidone, imidazoline, imidazolidinone, 2- pyrazoline, pyrazolidine, piperazine, and N-alkyl piperazines such as N-methyl piperazine. Particular examples include azetidine, piperidone and piperazone. [0089] “Hetero” when used to describe a compound or a group present on a compound means that one or more carbon atoms in the compound or group have been replaced by a nitrogen, oxygen, or sulfur heteroatom. Hetero may be applied to any of the hydrocarbyl groups described above such as alkyl, e.g., heteroalkyl, cycloalkyl, e.g., heterocyclyl, aryl, e.g., heteroaryl, cycloalkenyl, e.g., cycloheteroalkenyl, and the like having from 1 to 5, and particularly from 1 to 3 heteroatoms. [0090] “Acyl” refers to a radical –C(=O)R
20, where R
20 is hydrogen, substituted or unsubstitued alkyl, substituted or unsubstitued alkenyl, substituted or unsubstitued alkynyl, substituted or unsubstitued carbocyclyl, substituted or unsubstituted heterocyclyl, substituted or unsubstituted aryl, or substituted or unsubstitued heteroaryl, as defined herein. “Alkanoyl” is an acyl group wherein R
20 is a group other than hydrogen. Representative acyl groups include, but are not limited to, formyl (–CHO), acetyl (–C(=O)CH
3), cyclohexylcarbonyl, cyclohexylmethylcarbonyl, benzoyl (–C(=O)Ph), benzylcarbonyl (–C(=O)CH
2Ph), ––C(=O)– C
1–C
8 alkyl, –C(=O)-(CH
2)
t(C
6–C
10 aryl), –C(=O)-(CH
2)
t(5-10 membered heteroaryl), – C(=O)-(CH
2)
t(C
3–C
10 cycloalkyl), and –C(=O)-(CH
2)
t(4-10 membered heterocyclyl), wherein t is an integer from 0 to 4. In an embodiment, R
21 is C
1–C
8 alkyl, substituted with halo or hydroxy; or C
3–C
10 cycloalkyl, 4-10 membered heterocyclyl, C
6–C
10 aryl, arylalkyl, 5-10 membered heteroaryl or heteroarylalkyl, each of which is substituted with unsubstituted C
1– C
4 alkyl, halo, unsubstituted C
1–C
4 alkoxy, unsubstituted C
1–C
4 haloalkyl, unsubstituted C
1– C
4 hydroxyalkyl, or unsubstituted C
1–C
4 haloalkoxy or hydroxy. [0091] The term aminoalkyl refers to a substituted alkyl group wherein one or more of the hydrogen atoms are independently replaced by an –NH
2 group. [0092] The term hydroxyalkyl refers to a substituted alkyl group wherein one or more of the hydrogen atoms are independently replaced by an –OH group. [0093] The terms “alkylamino” and “dialkylamino” refer to −NH(alkyl) and−N(alkyl)
2 radicals respectively. In an embodiment the alkylamino is a−NH(C
1−C
4 alkyl). In an embodiment the alkylamino is methylamino, ethylamino, propylamino, isopropylamino, n- butylamino, iso-butylamino, sec-butylamino or tert-butylamino. In an embodiment the dialkylamino is −N(C
1−C
6 alkyl)
2. In an embodiment the dialkylamino is a dimethylamino, a methylethylamino, a diethylamino, a methylpropylamino, a methylisopropylamino, a methylbutylamino, a methylisobutylamino or a methyltertbutylamino. [0094] The term “aryloxy” refers to an –O–aryl radical. In an embodiment the aryloxy group is phenoxy. [0095] The term “haloalkoxy” refers to alkoxy structures that are substituted with one or more halo groups or with combinations thereof. For example, the term “fluoroalkoxy” includes haloalkoxy groups, in which the halo is fluorine. In an embodiment haloalkoxy groups are difluoromethoxy and trifluoromethoxy. [0096] “Alkoxy” refers to the group –OR
29 where R
29 is substituted or unsubstituted alkyl, substituted or unsubstitued alkenyl, substituted or unsubstitued alkynyl, substituted or unsubstitued carbocyclyl, substituted or unsubstituted heterocyclyl, substituted or unsubstituted aryl, or substituted or unsubstitued heteroaryl. Particular alkoxy groups are methoxy, ethoxy, n-propoxy, isopropoxy, n-butoxy, tert-butoxy, sec-butoxy, n-pentoxy, n- hexoxy, and 1,2-dimethylbutoxy. Particular alkoxy groups are lower alkoxy, i.e. with between 1 and 6 carbon atoms. Further particular alkoxy groups have between 1 and 4 carbon atoms. [0097] In an embodiment, R
29 is a group that has 1 or more substituents, for instance from 1 to 5 substituents, and particularly from 1 to 3 substituents, in particular 1 substituent, selected from the group consisting of amino, substituted amino, C
6–C
10 aryl, aryloxy, carboxyl, cyano, C
3–C
10 cycloalkyl, 4-10 membered heterocyclyl, halogen, 5-10 membered heteroaryl, hydroxyl, nitro, thioalkoxy, thioaryloxy, thiol, alkyl-S(O)-, aryl–S(O)-, alkyl–S(O)
2– and aryl-S(O)
2-. Exemplary ’substituted alkoxy’ groups include, but are not limited to, –O– (CH
2)
t(C
6–C
10 aryl), –O–(CH
2)
t(5-10 membered heteroaryl), –O–(CH
2)
t(C
3–C
10 cycloalkyl), and –O–(CH
2)
t(4-10 membered heterocyclyl), wherein t is an integer from 0 to 4 and any aryl, heteroaryl, cycloalkyl or heterocyclyl groups present, may themselves be substituted by unsubstituted C
1–C
4 alkyl, halo, unsubstituted C
1–C
4 alkoxy, unsubstituted C
1–C
4 haloalkyl, unsubstituted C
1–C
4 hydroxyalkyl, or unsubstituted C
1–C
4 haloalkoxy or hydroxy. Particular exemplary ’substituted alkoxy’ groups are –OCF
3, –OCH
2CF
3, –OCH
2Ph, –OCH
2- cyclopropyl, –OCH
2CH
2OH, and –OCH
2CH
2NMe
2. [0098] “Amino” refers to the radical –NH
2. [0099] “Oxo group” refers to –C(=O)–. [0100] “Substituted amino” refers to an amino group of the formula –N(R
38)
2 wherein R
38 is hydrogen, substituted or unsubstituted alkyl, substituted or unsubstitued alkenyl, substituted or unsubstitued alkynyl, substituted or unsubstitued carbocyclyl, substituted or unsubstituted heterocyclyl, substituted or unsubstituted aryl, substituted or unsubstitued heteroaryl, or an amino protecting group, wherein at least one of R
38 is not a hydrogen. In an embodiment, each R
38 is independently selected from hydrogen, C
1–C
8 alkyl, C
3–C
8 alkenyl, C
3–C
8 alkynyl, C
6–C
10 aryl, 5-10 membered heteroaryl, 4-10 membered heterocyclyl, or C
3–C
10 cycloalkyl; or C
1–C
8 alkyl, substituted with halo or hydroxy; C
3–C
8 alkenyl, substituted with halo or hydroxy; C
3–C
8 alkynyl, substituted with halo or hydroxy, or -(CH
2)
t(C
6–C
10 aryl), - (CH
2)
t(5-10 membered heteroaryl), -(CH
2)
t(C
3–C
10 cycloalkyl), or -(CH
2)
t(4-10 membered heterocyclyl), wherein t is an integer between 0 and 8, each of which is substituted by unsubstituted C
1–C
4 alkyl, halo, unsubstituted C
1–C
4 alkoxy, unsubstituted C
1–C
4 haloalkyl, unsubstituted C
1–C
4 hydroxyalkyl, or unsubstituted C
1–C
4 haloalkoxy or hydroxy; or both R
38 groups are joined to form an alkylene group. [0101] Exemplary “substituted amino” groups include, but are not limited to, –NR
39–C
1–C
8 alkyl, –NR
39-(CH
2)
t(C
6–C
10 aryl), –NR
39-(CH
2)
t(5-10 membered heteroaryl), –NR
39- (CH
2)
t(C
3–C
10 cycloalkyl), and –NR
39-(CH
2)
t(4-10 membered heterocyclyl), wherein t is an integer from 0 to 4, for instance 1 or 2, each R
39 independently represents H or C
1–C
8 alkyl; and any alkyl groups present, may themselves be substituted by halo, substituted or unsubstituted amino, or hydroxy; and any aryl, heteroaryl, cycloalkyl, or heterocyclyl groups present, may themselves be substituted by unsubstituted C
1–C
4 alkyl, halo, unsubstituted C
1– C
4 alkoxy, unsubstituted C
1–C
4 haloalkyl, unsubstituted C
1–C
4 hydroxyalkyl, or unsubstituted C
1–C
4 haloalkoxy or hydroxy. For the avoidance of doubt the term ’substituted amino’ includes the groups alkylamino, substituted alkylamino, alkylarylamino, substituted alkylarylamino, arylamino, substituted arylamino, dialkylamino, and substituted dialkylamino as defined below. Substituted amino encompasses both monosubstituted amino and disubstituted amino groups. [0102] In an embodiment, the substituent present on the nitrogen atom is a nitrogen protecting group (also referred to herein as an “amino protecting group”). Nitrogen protecting groups include, but are not limited to, −OH, −OR
aa, −N(R
cc)
2, −C(=O)R
aa, −C(=O)N(R
cc)
2, −CO
2R
aa, −SO
2R
aa, −C(=NR
cc)R
aa, −C(=NR
cc)OR
aa, −C(=NR
cc)N(R
cc)
2, −SO
2N(R
cc)
2, −SO
2R
cc, −SO
2OR
cc, −SOR
aa, −C(=S)N(R
cc)
2, −C(=O)SR
cc, −C(=S)SR
cc, −C
1-10 alkyl (e.g., aralkyl, heteroaralkyl), −C
2-10 alkenyl, −C
2-10 alkynyl, heteroC
1-10 alkyl, heteroC
2-10 alkenyl, heteroC
2-10 alkynyl, C
3-10 carbocyclyl, 3-14 membered heterocyclyl, C
6-14 aryl, and 5-14 membered heteroaryl groups, wherein each alkyl, alkenyl, alkynyl, heteroalkyl, heteroalkenyl, heteroalkynyl, carbocyclyl, heterocyclyl, aralkyl, aryl, and heteroaryl is independently substituted with 0, 1, 2, 3, 4, or 5 R
dd groups, and wherein R
aa, R
bb, R
cc and R
dd are as defined herein. Nitrogen protecting groups are well known in the art and include those described in detail in Protecting Groups in Organic Synthesis, T. W. Greene and P. G. M. Wuts, 3
rd edition, John Wiley & Sons, 1999, incorporated herein by reference. In the definition of nitrogen protecting groups, each instance of R
aa is, independently, selected from −C
1-10 alkyl, −C
1-10 perhaloalkyl, −C
2-10 alkenyl, −C
2-10 alkynyl, heteroC
1-10 alkyl, heteroC
2-10 alkenyl, heteroC
2-10 alkynyl, C
3-10 carbocyclyl, 3-14 membered heterocyclyl, C
6-14 aryl, and 5-14 membered heteroaryl, or two R
aa groups are joined to form a 3-14 membered heterocyclyl or 5-14 membered heteroaryl ring, wherein each alkyl, alkenyl, alkynyl, heteroalkyl, heteroalkenyl, heteroalkynyl, carbocyclyl, heterocyclyl, aryl, and heteroaryl is independently substituted with 0, 1, 2, 3, 4, or 5 R
dd groups; each instance of R
bb is, independently, selected from hydrogen, −OH, −OR
aa, −N(R
cc)
2, −CN, −C(=O)R
aa, −C(=O)N(R
cc)
2, −CO
2R
aa, −SO
2R
aa, −C(=NR
cc)OR
aa, −C(=NR
cc)N(R
cc)
2, −SO
2N(R
cc)
2, −SO
2R
cc, −SO
2OR
cc, −SOR
aa, −C(=S)N(R
cc)
2, −C(=O)SR
cc, −C(=S)SR
cc, −P(=O)(R
aa)
2, −P(=O)(OR
cc)
2, −P(=O)(N(R
cc)
2)
2, −C
1-10 alkyl, −C
1-10 perhaloalkyl, −C
2-10 alkenyl, −C
2-10 alkynyl, heteroC
1-10 alkyl, heteroC
2-10 alkenyl, heteroC
2-10 alkynyl, C
3-10 carbocyclyl, 3-14 membered heterocyclyl, C
6-14 aryl, and 5-14 membered heteroaryl, or two R
bb groups are joined to form a 3-14 membered heterocyclyl or 5-14 membered heteroaryl ring, wherein each alkyl, alkenyl, alkynyl, heteroalkyl, heteroalkenyl, heteroalkynyl, carbocyclyl, heterocyclyl, aryl, and heteroaryl is independently substituted with 0, 1, 2, 3, 4, or 5 R
dd groups; wherein X
− is a counterion. each instance of R
cc is, independently, selected from hydrogen, −C
1-10 alkyl, −C
1-10 perhaloalkyl, −C
2-10 alkenyl, −C
2-10 alkynyl, heteroC
1-10 alkyl, heteroC
2-10 alkenyl, heteroC
2-10 alkynyl, C
3-10 carbocyclyl, 3-14 membered heterocyclyl, C
6-14 aryl, and 5-14 membered heteroaryl, or two R
cc groups are joined to form a 3-14 membered heterocyclyl or 5-14 membered heteroaryl ring, wherein each alkyl, alkenyl, alkynyl, heteroalkyl, heteroalkenyl, heteroalkynyl, carbocyclyl, heterocyclyl, aryl, and heteroaryl is independently substituted with 0, 1, 2, 3, 4, or 5 R
dd groups; each instance of R
dd is, independently, selected from halogen, −CN, −NO
2, −N
3, −SO
2H, −SO
3H, −OH, −OR
ee, −ON(R
ff)
2, −N(R
ff)
2, −N(R
ff)
3+X
−, −N(OR
ee)R
ff, −SH, −SR
ee, −SSR
ee, −C(=O)R
ee, −CO
2H, −CO
2R
ee, −OC(=O)R
ee, −OCO
2R
ee, −C(=O)N(R
ff)
2, −OC(=O)N(R
ff)
2, −NR
ffC(=O)R
ee, −NR
ffCO
2R
ee, −NR
ffC(=O)N(R
ff)
2, −C(=NR
ff)OR
ee, −OC(=NR
ff)R
ee, −OC(=NR
ff)OR
ee, −C(=NR
ff)N(R
ff)
2, −OC(=NR
ff)N(R
ff)
2, −NR
ffC(=NR
ff)N(R
ff)
2, −NR
ffSO
2R
ee, −SO
2N(R
ff)
2, −SO
2R
ee, −SO
2OR
ee, −OSO
2R
ee, −S(=O)R
ee, −Si(R
ee)
3, −OSi(R
ee)
3, −C(=S)N(R
ff)
2, −C(=O)SR
ee, −C(=S)SR
ee, −SC(=S)SR
ee, −P(=O)(OR
ee)
2, −P(=O)(R
ee)
2, −OP(=O)(R
ee)
2, −OP(=O)(OR
ee)
2, −C
1-6 alkyl, −C
1-6 perhaloalkyl, −C
2-6 alkenyl, −C
2-6 alkynyl, heteroC
1-6alkyl, heteroC
2-6alkenyl, heteroC
2-6alkynyl, C
3-10 carbocyclyl, 3-10 membered heterocyclyl, C
6-10 aryl, 5-10 membered heteroaryl, wherein each alkyl, alkenyl, alkynyl, heteroalkyl, heteroalkenyl, heteroalkynyl, carbocyclyl, heterocyclyl, aryl, and heteroaryl is independently substituted with 0, 1, 2, 3, 4, or 5 R
gg groups, or two geminal R
dd substituents can be joined to form =O or =S; wherein X
− is a counterion; each instance of R
ee is, independently, selected from −C
1-6 alkyl, −C
1-6 perhaloalkyl, −C
2-6 alkenyl, −C
2-6 alkynyl, heteroC
1-6 alkyl, heteroC
2-6alkenyl, heteroC
2-6 alkynyl, C
3-10 carbocyclyl, C
6-10 aryl, 3-10 membered heterocyclyl, and 3-10 membered heteroaryl, wherein each alkyl, alkenyl, alkynyl, heteroalkyl, heteroalkenyl, heteroalkynyl, carbocyclyl, heterocyclyl, aryl, and heteroaryl is independently substituted with 0, 1, 2, 3, 4, or 5 R
gg groups; each instance of R
ff is, independently, selected from hydrogen, −C
1-6 alkyl, −C
1-6 perhaloalkyl, −C
2-6 alkenyl, −C
2-6 alkynyl, heteroC
1-6alkyl, heteroC
2-6alkenyl, heteroC
2-6alkynyl, C
3-10 carbocyclyl, 3-10 membered heterocyclyl, C
6-10 aryl and 5-10 membered heteroaryl, or two R
ff groups are joined to form a 3-10 membered heterocyclyl or 5-10 membered heteroaryl ring, wherein each alkyl, alkenyl, alkynyl, heteroalkyl, heteroalkenyl, heteroalkynyl, carbocyclyl, heterocyclyl, aryl, and heteroaryl is independently substituted with 0, 1, 2, 3, 4, or 5 R
gg groups; and each instance of R
gg is, independently, halogen, −CN, −NO
2, −N
3, −SO
2H, −SO
3H, −OH, −OC
1-6 alkyl, −ON(C
1-6 alkyl)
2, −N(C
1-6 alkyl)
2, −N(C
1-6 alkyl)
3+X
−, −NH(C
1-6 alkyl)
2+X
−, −NH
2(C
1-6 alkyl)
+X
−, −NH
3+X
−, −N(OC
1-6 alkyl)(C
1-6 alkyl), −N(OH)(C
1-6 alkyl), −NH(OH), −SH, −SC
1-6 alkyl, −SS(C
1-6 alkyl), −C(=O)(C
1-6 alkyl), −CO
2H, −CO
2(C
1-6 alkyl), −OC(=O)(C
1-6 alkyl), −OCO
2(C
1-6 alkyl), −C(=O)NH
2, −C(=O)N(C
1-6 alkyl)
2, −OC(=O)NH(C
1-6 alkyl), −NHC(=O)(C
1-6 alkyl), −N(C
1-6 alkyl)C(=O)(C
1-6 alkyl), −NHCO
2(C
1-6 alkyl), −NHC(=O)N(C
1-6 alkyl)
2, −NHC(=O)NH(C
1-6 alkyl), −NHC(=O)NH
2, −C(=NH)O(C
1-6 alkyl), −OC(=NH)(C
1-6 alkyl), −OC(=NH)OC
1-6 alkyl, −C(=NH)N(C
1-6 alkyl)
2, −C(=NH)NH(C
1-6 alkyl), −C(=NH)NH
2, −OC(=NH)N(C
1-6 alkyl)
2, −OC(NH)NH(C
1-6 alkyl), −OC(NH)NH
2, −NHC(NH)N(C
1-6 alkyl)
2, −NHC(=NH)NH
2, −NHSO
2(C
1-6 alkyl), −SO
2N(C
1-6 alkyl)
2, −SO
2NH(C
1-6 alkyl), −SO
2NH
2, −SO
2C
1-6 alkyl, −SO
2OC
1-6 alkyl, −OSO
2C
1-6 alkyl, −SOC
1-6 alkyl, −Si(C
1-6 alkyl)
3, −OSi(C
1-6 alkyl)
3 −C(=S)N(C
1-6 alkyl)
2, −C(=S)NH(C
1-6 alkyl), −C(=S)NH
2, −C(=O)S(C
1-6 alkyl), −C(=S)SC
1-6 alkyl, −SC(=S)SC
1-6 alkyl, −P(=O)(OC
1-6 alkyl)
2, −P(=O)(C
1-6 alkyl)
2, −OP(=O)(C
1-6 alkyl)
2, −OP(=O)(OC
1-6 alkyl)
2, −C
1-6 alkyl, −C
1-6 perhaloalkyl, −C
2-6 alkenyl, −C
2-6 alkynyl, heteroC
1-6alkyl, heteroC
2-6alkenyl, heteroC
2-6alkynyl, C
3-10 carbocyclyl, C
6-10 aryl, 3-10 membered heterocyclyl, 5-10 membered heteroaryl; or two geminal R
gg substituents can be joined to form =O or =S; wherein X
− is a counterion. [0103] For example, nitrogen protecting groups such as amide groups (e.g., −C(=O)R
aa) include, but are not limited to, formamide, acetamide, chloroacetamide, trichloroacetamide, trifluoroacetamide, phenylacetamide, 3-phenylpropanamide, picolinamide, 3- pyridylcarboxamide, N-benzoylphenylalanyl derivative, benzamide, p-phenylbenzamide, o- nitrophenylacetamide, o-nitrophenoxyacetamide, acetoacetamide, (N’- dithiobenzyloxyacylamino)acetamide, 3-(p-hydroxyphenyl)propanamide, 3-(o- nitrophenyl)propanamide, 2-methyl-2-(o-nitrophenoxy)propanamide, 2-methyl-2-(o- phenylazophenoxy)propanamide, 4-chlorobutanamide, 3-methyl-3-nitrobutanamide, o- nitrocinnamide, N-acetylmethionine derivative, o-nitrobenzamide and o- (benzoyloxymethyl)benzamide. [0104] Nitrogen protecting groups such as carbamate groups (e.g., −C(=O)OR
aa) include, but are not limited to, methyl carbamate, ethyl carbamate, 9-fluorenylmethyl carbamate (Fmoc), 9-(2-sulfo)fluorenylmethyl carbamate, 9-(2,7-dibromo)fluorenylmethyl carbamate, 2,7-di-t- butyl-[9-(10,10-dioxo-10,10,10,10-tetrahydrothioxanthyl)]methyl carbamate (DBD-Tmoc), 4-methoxyphenacyl carbamate (Phenoc), 2,2,2-trichloroethyl carbamate (Troc), 2- trimethylsilylethyl carbamate (Teoc), 2-phenylethyl carbamate (hZ), 1-(1-adamantyl)-1- methylethyl carbamate (Adpoc), 1,1-dimethyl-2-haloethyl carbamate, 1,1-dimethyl-2,2- dibromoethyl carbamate (DB-t-BOC), 1,1-dimethyl-2,2,2-trichloroethyl carbamate (TCBOC), 1-methyl-1-(4-biphenylyl)ethyl carbamate (Bpoc), 1-(3,5-di-t-butylphenyl)-1- methylethyl carbamate (t-Bumeoc), 2-(2’– and 4’-pyridyl)ethyl carbamate (Pyoc), 2-(N,N- dicyclohexylcarboxamido)ethyl carbamate, t-butyl carbamate (BOC or Boc), 1-adamantyl carbamate (Adoc), vinyl carbamate (Voc), allyl carbamate (Alloc), 1-isopropylallyl carbamate (Ipaoc), cinnamyl carbamate (Coc), 4-nitrocinnamyl carbamate (Noc), 8-quinolyl carbamate, N-hydroxypiperidinyl carbamate, alkyldithio carbamate, benzyl carbamate (Cbz), p-methoxybenzyl carbamate (Moz), p-nitobenzyl carbamate, p-bromobenzyl carbamate, p- chlorobenzyl carbamate, 2,4-dichlorobenzyl carbamate, 4-methylsulfinylbenzyl carbamate (Msz), 9-anthrylmethyl carbamate, diphenylmethyl carbamate, 2-methylthioethyl carbamate, 2-methylsulfonylethyl carbamate, 2-(p-toluenesulfonyl)ethyl carbamate, [2-(1,3- dithianyl)]methyl carbamate (Dmoc), 4-methylthiophenyl carbamate (Mtpc), 2,4- dimethylthiophenyl carbamate (Bmpc), 2-phosphonioethyl carbamate (Peoc), 2- triphenylphosphonioisopropyl carbamate (Ppoc), 1,1-dimethyl-2-cyanoethyl carbamate, m- chloro-p-acyloxybenzyl carbamate, p-(dihydroxyboryl)benzyl carbamate, 5- benzisoxazolylmethyl carbamate, 2-(trifluoromethyl)-6-chromonylmethyl carbamate (Tcroc), m-nitrophenyl carbamate, 3,5-dimethoxybenzyl carbamate, o-nitrobenzyl carbamate, 3,4- dimethoxy-6-nitrobenzyl carbamate, phenyl (o-nitrophenyl)methyl carbamate, t-amyl carbamate, S-benzyl thiocarbamate, p-cyanobenzyl carbamate, cyclobutyl carbamate, cyclohexyl carbamate, cyclopentyl carbamate, cyclopropylmethyl carbamate, p- decyloxybenzyl carbamate, 2,2-dimethoxyacylvinyl carbamate, o-(N,N- dimethylcarboxamido)benzyl carbamate, 1,1-dimethyl-3-(N,N-dimethylcarboxamido)propyl carbamate, 1,1-dimethylpropynyl carbamate, di(2-pyridyl)methyl carbamate, 2-furanylmethyl carbamate, 2-iodoethyl carbamate, isobornyl carbamate, isobutyl carbamate, isonicotinyl carbamate, p-(p’-methoxyphenylazo)benzyl carbamate, 1-methylcyclobutyl carbamate, 1- methylcyclohexyl carbamate, 1-methyl-1-cyclopropylmethyl carbamate, 1-methyl-1-(3,5- dimethoxyphenyl)ethyl carbamate, 1-methyl-1-(p-phenylazophenyl)ethyl carbamate, 1- methyl-1-phenylethyl carbamate, 1-methyl-1-(4-pyridyl)ethyl carbamate, phenyl carbamate, p-(phenylazo)benzyl carbamate, 2,4,6-tri-t-butylphenyl carbamate, 4- (trimethylammonium)benzyl carbamate, and 2,4,6-trimethylbenzyl carbamate. [0105] Nitrogen protecting groups such as sulfonamide groups (e.g., −S(=O)
2R
aa) include, but are not limited to, p-toluenesulfonamide (Ts), benzenesulfonamide, 2,3,6-trimethyl-4- methoxybenzenesulfonamide (Mtr), 2,4,6-trimethoxybenzenesulfonamide (Mtb), 2,6- dimethyl-4-methoxybenzenesulfonamide (Pme), 2,3,5,6-tetramethyl-4- methoxybenzenesulfonamide (Mte), 4-methoxybenzenesulfonamide (Mbs), 2,4,6- trimethylbenzenesulfonamide (Mts), 2,6-dimethoxy-4-methylbenzenesulfonamide (iMds), 2,2,5,7,8-pentamethylchroman-6-sulfonamide (Pmc), methanesulfonamide (Ms), β- trimethylsilylethanesulfonamide (SES), 9-anthracenesulfonamide, 4-(4’,8’- dimethoxynaphthylmethyl)benzenesulfonamide (DNMBS), benzylsulfonamide, trifluoromethylsulfonamide, and phenacylsulfonamide. [0106] Other nitrogen protecting groups include, but are not limited to, phenothiazinyl-(10)- acyl derivative, N’-p-toluenesulfonylaminoacyl derivative, N’-phenylaminothioacyl derivative, N-benzoylphenylalanyl derivative, N-acetylmethionine derivative, 4,5-diphenyl-3- oxazolin-2-one, N-phthalimide, N-dithiasuccinimide (Dts), N-2,3-diphenylmaleimide, N-2,5- dimethylpyrrole, N-1,1,4,4-tetramethyldisilylazacyclopentane adduct (STABASE), 5- substituted 1,3-dimethyl-1,3,5-triazacyclohexan-2-one, 5-substituted 1,3-dibenzyl-1,3,5- triazacyclohexan-2-one, 1-substituted 3,5-dinitro-4-pyridone, N-methylamine, N-allylamine, N-[2-(trimethylsilyl)ethoxy]methylamine (SEM), N-3-acetoxypropylamine, N-(1-isopropyl- 4-nitro-2-oxo-3-pyroolin-3-yl)amine, quaternary ammonium salts, N-benzylamine, N-di(4- methoxyphenyl)methylamine, N-5-dibenzosuberylamine, N-triphenylmethylamine (Tr), N- [(4-methoxyphenyl)diphenylmethyl]amine (MMTr), N-9-phenylfluorenylamine (PhF), N- 2,7-dichloro-9-fluorenylmethyleneamine, N-ferrocenylmethylamino (Fcm), N-2- picolylamino N’-oxide, N-1,1-dimethylthiomethyleneamine, N-benzylideneamine, N-p- methoxybenzylideneamine, N-diphenylmethyleneamine, N-[(2- pyridyl)mesityl]methyleneamine, N-(N’,N’-dimethylaminomethylene)amine, N,N’- isopropylidenediamine, N-p-nitrobenzylideneamine, N-salicylideneamine, N-5- chlorosalicylideneamine, N-(5-chloro-2-hydroxyphenyl)phenylmethyleneamine, N- cyclohexylideneamine, N-(5,5-dimethyl-3-oxo-1-cyclohexenyl)amine, N-borane derivative, N-diphenylborinic acid derivative, N-[phenyl(pentaacylchromium– or tungsten)acyl]amine, N-copper chelate, N-zinc chelate, N-nitroamine, N-nitrosoamine, amine N-oxide, diphenylphosphinamide (Dpp), dimethylthiophosphinamide (Mpt), diphenylthiophosphinamide (Ppt), dialkyl phosphoramidates, dibenzyl phosphoramidate, diphenyl phosphoramidate, benzenesulfenamide, o-nitrobenzenesulfenamide (Nps), 2,4- dinitrobenzenesulfenamide, pentachlorobenzenesulfenamide, 2-nitro-4- methoxybenzenesulfenamide, triphenylmethylsulfenamide, and 3-nitropyridinesulfenamide (Npys). [0107] In an embodiment, the substituent present on an oxygen atom is an oxygen protecting group (also referred to herein as an “hydroxyl protecting group”). Oxygen protecting groups include, but are not limited to, −R
aa, −N(R
bb)
2, −C(=O)SR
aa, −C(=O)R
aa, −CO
2R
aa, −C(=O)N(R
bb)
2, −C(=NR
bb)R
aa, −C(=NR
bb)OR
aa, −C(=NR
bb)N(R
bb)
2, −S(=O)R
aa, −SO
2R
aa, −Si(R
aa)
3, −P(R
cc)
2, −P(R
cc)
3+X
−, −P(OR
cc)
2, −P(OR
cc)
3+X
−, −P(=O)(R
aa)
2, −P(=O)(OR
cc)
2, and −P(=O)(N(R
bb)
2)
2, wherein R
aa, R
bb, and R
cc are as defined herein. Oxygen protecting groups are well known in the art and include those described in detail in Protecting Groups in Organic Synthesis, T. W. Greene and P. G. M. Wuts, 3
rd edition, John Wiley & Sons, 1999, incorporated herein by reference. [0108] Exemplary oxygen protecting groups include, but are not limited to, methyl, methoxymethyl (MOM), methylthiomethyl (MTM), t-butylthiomethyl, (phenyldimethylsilyl)methoxymethyl (SMOM), benzyloxymethyl (BOM), p- methoxybenzyloxymethyl (PMBM), (4-methoxyphenoxy)methyl (p-AOM), guaiacolmethyl (GUM), t-butoxymethyl, 4-pentenyloxymethyl (POM), siloxymethyl, 2- methoxyethoxymethyl (MEM), 2,2,2-trichloroethoxymethyl, bis(2-chloroethoxy)methyl, 2- (trimethylsilyl)ethoxymethyl (SEMOR), tetrahydropyranyl (THP), 3- bromotetrahydropyranyl, tetrahydrothiopyranyl, 1-methoxycyclohexyl, 4- methoxytetrahydropyranyl (MTHP), 4-methoxytetrahydrothiopyranyl, 4- methoxytetrahydrothiopyranyl S,S-dioxide, 1-[(2-chloro-4-methyl)phenyl]-4- methoxypiperidin-4-yl (CTMP), 1,4-dioxan-2-yl, tetrahydrofuranyl, tetrahydrothiofuranyl, 2,3,3a,4,5,6,7,7a-octahydro-7,8,8-trimethyl-4,7-methanobenzofuran-2-yl, 1-ethoxyethyl, 1- (2-chloroethoxy)ethyl, 1-methyl-1-methoxyethyl, 1-methyl-1-benzyloxyethyl, 1-methyl-1- benzyloxy-2-fluoroethyl, 2,2,2-trichloroethyl, 2-trimethylsilylethyl, 2-(phenylselenyl)ethyl, t- butyl, allyl, p-chlorophenyl, p-methoxyphenyl, 2,4-dinitrophenyl, benzyl (Bn), p- methoxybenzyl, 3,4-dimethoxybenzyl, o-nitrobenzyl, p-nitrobenzyl, p-halobenzyl, 2,6- dichlorobenzyl, p-cyanobenzyl, p-phenylbenzyl, 2-picolyl, 4-picolyl, 3-methyl-2-picolyl N- oxido, diphenylmethyl, p,p’-dinitrobenzhydryl, 5-dibenzosuberyl, triphenylmethyl, α- naphthyldiphenylmethyl, p-methoxyphenyldiphenylmethyl, di(p- methoxyphenyl)phenylmethyl, tri(p-methoxyphenyl)methyl, 4-(4’- bromophenacyloxyphenyl)diphenylmethyl, 4,4′,4″-tris(4,5- dichlorophthalimidophenyl)methyl, 4,4′,4″-tris(levulinoyloxyphenyl)methyl, 4,4′,4″- tris(benzoyloxyphenyl)methyl, 3-(imidazol-1-yl)bis(4′,4″-dimethoxyphenyl)methyl, 1,1- bis(4-methoxyphenyl)-1′-pyrenylmethyl, 9-anthryl, 9-(9-phenyl)xanthenyl, 9-(9-phenyl-10- oxo)anthryl, 1,3-benzodithiolan-2-yl, benzisothiazolyl S,S-dioxido, trimethylsilyl (TMS), triethylsilyl (TES), triisopropylsilyl (TIPS), dimethylisopropylsilyl (IPDMS), diethylisopropylsilyl (DEIPS), dimethylthexylsilyl, t-butyldimethylsilyl (TBDMS), t- butyldiphenylsilyl (TBDPS), tribenzylsilyl, tri-p-xylylsilyl, triphenylsilyl, diphenylmethylsilyl (DPMS), t-butylmethoxyphenylsilyl (TBMPS), formate, benzoylformate, acetate, chloroacetate, dichloroacetate, trichloroacetate, trifluoroacetate, methoxyacetate, triphenylmethoxyacetate, phenoxyacetate, p-chlorophenoxyacetate, 3-phenylpropionate, 4- oxopentanoate (levulinate), 4,4-(ethylenedithio)pentanoate (levulinoyldithioacetal), pivaloate, adamantoate, crotonate, 4-methoxycrotonate, benzoate, p-phenylbenzoate, 2,4,6- trimethylbenzoate (mesitoate), methyl carbonate, 9-fluorenylmethyl carbonate (Fmoc), ethyl carbonate, 2,2,2-trichloroethyl carbonate (Troc), 2-(trimethylsilyl)ethyl carbonate (TMSEC), 2-(phenylsulfonyl) ethyl carbonate (Psec), 2-(triphenylphosphonio) ethyl carbonate (Peoc), isobutyl carbonate, vinyl carbonate, allyl carbonate, t-butyl carbonate (BOC or Boc), p- nitrophenyl carbonate, benzyl carbonate, p-methoxybenzyl carbonate, 3,4-dimethoxybenzyl carbonate, o-nitrobenzyl carbonate, p-nitrobenzyl carbonate, S-benzyl thiocarbonate, 4- ethoxy-1-napththyl carbonate, methyl dithiocarbonate, 2-iodobenzoate, 4-azidobutyrate, 4- nitro-4-methylpentanoate, o-(dibromomethyl)benzoate, 2-formylbenzenesulfonate, 2- (methylthiomethoxy)ethyl, 4-(methylthiomethoxy)butyrate, 2- (methylthiomethoxymethyl)benzoate, 2,6-dichloro-4-methylphenoxyacetate, 2,6-dichloro-4- (1,1,3,3-tetramethylbutyl)phenoxyacetate, 2,4-bis(1,1-dimethylpropyl)phenoxyacetate, chlorodiphenylacetate, isobutyrate, monosuccinoate, (E)-2-methyl-2-butenoate, o- (methoxyacyl)benzoate, α-naphthoate, nitrate, alkyl N,N,N’,N’- tetramethylphosphorodiamidate, alkyl N-phenylcarbamate, borate, dimethylphosphinothioyl, alkyl 2,4-dinitrophenylsulfenate, sulfate, methanesulfonate (mesylate), benzylsulfonate, and tosylate (Ts). [0109] In an embodiment, the substituent present on a sulfur atom is a sulfur protecting group (also referred to as a “thiol protecting group”). Sulfur protecting groups include, but are not limited to, −R
aa, −N(R
bb)
2, −C(=O)SR
aa, −C(=O)R
aa, −CO
2R
aa, −C(=O)N(R
bb)
2, −C(=NR
bb)R
aa, −C(=NR
bb)OR
aa, −C(=NR
bb)N(R
bb)
2, −S(=O)R
aa, −SO
2R
aa, −Si(R
aa)
3, −P(R
cc)
2, −P(R
cc)
3+X
−, −P(OR
cc)
2, −P(OR
cc)
3+X
−, −P(=O)(R
aa)
2, −P(=O)(OR
cc)
2, and −P(=O)(N(R
bb)
2)
2, wherein R
aa, R
bb, and R
cc are as defined herein. Sulfur protecting groups are well known in the art and include those described in detail in Protecting Groups in Organic Synthesis, T. W. Greene and P. G. M. Wuts, 3
rd edition, John Wiley & Sons, 1999, incorporated herein by reference. [0110] The term “leaving group” is given its ordinary meaning in the art of synthetic organic chemistry and refers to an atom or a group capable of being displaced by a nucleophile. Examples of suitable leaving groups include, but are not limited to, halogen (such as F, Cl, Br, or I (iodine)), alkoxycarbonyloxy, aryloxycarbonyloxy, alkanesulfonyloxy, arenesulfonyloxy, alkyl-carbonyloxy (e.g., acetoxy), arylcarbonyloxy, aryloxy, methoxy, N,O-dimethylhydroxylamino, pixyl, and haloformates. In an embodiment, the leaving group is halogen, alkanesulfonyloxy, arenesulfonyloxy, diazonium, alkyl diazenes, aryl diazenes, alkyl triazenes, aryl triazenes, nitro, alkyl nitrate, aryl nitrate, alkyl phosphate, aryl phosphate, alkyl carbonyl oxy, aryl carbonyl oxy, alkoxcarbonyl oxy, aryoxcarbonyl oxy ammonia, alkyl amines, aryl amines, hydroxyl group, alkyloxy group, or aryloxy. In some cases, the leaving group is a sulfonic acid ester, such as toluenesulfonate (tosylate, –OTs), methanesulfonate (mesylate, –OMs), p-bromobenzenesulfonyloxy (brosylate, –OBs), – OS(=O)
2(CF
2)
3CF
3 (nonaflate, –ONf), or trifluoromethanesulfonate (triflate, –OTf). In some cases, the leaving group is a brosylate, such as p-bromobenzenesulfonyloxy. In some cases, the leaving group is a nosylate, such as 2-nitrobenzenesulfonyloxy. In an embodiment, the leaving group is a sulfonate-containing group. In an embodiment, the leaving group is a tosylate group. The leaving group may also be a phosphineoxide (e.g., formed during a Mitsunobu reaction) or an internal leaving group such as an epoxide or cyclic sulfate. Other non-limiting examples of leaving groups are water, ammonia, alcohols, ether moieties, thioether moieties, zinc halides, magnesium moieties, diazonium salts, and copper moieties. [0111] “Carboxy” refers to the radical –C(=O)OH. [0112] “Cyano” refers to the radical –CN. [0113] “Halo” or “halogen” refers to fluoro (F), chloro (Cl), bromo (Br), and iodo (I). In an embodiment, the halo group is either fluoro or chloro. [0114] “Haloalkyl” refers to an alkyl radical in which the alkyl group is substituted with one or more halogens. Typical haloalkyl groups include, but are not limited to, trifluoromethyl (– CF
3), difluoromethyl (–CHF
2), fluoromethyl (–CH
2F), chloromethyl (–CH
2Cl), dichloromethyl (–CHCl
2), tribromomethyl (–CH
2Br), and the like. [0115] “Hydroxy” refers to the radical –OH. [0116] “Nitro” refers to the radical –NO
2. [0117] “Thioketo” refers to the group =S. [0118] Alkyl, alkenyl, alkynyl, carbocyclyl, heterocyclyl, aryl, and heteroaryl groups, as defined herein, are optionally substituted (e.g., “substituted” or “unsubstituted” alkyl, “substituted” or “unsubstituted” alkenyl, “substituted” or “unsubstituted” alkynyl, “substituted” or “unsubstituted” carbocyclyl, “substituted” or “unsubstituted” heterocyclyl, “substituted” or “unsubstituted” aryl or “substituted” or “unsubstituted” heteroaryl group). In general, the term “substituted”, whether preceded by the term “optionally” or not, means that at least one hydrogen present on a group (e.g., a carbon or nitrogen atom) is replaced with a permissible substituent, e.g., a substituent which upon substitution results in a stable compound, e.g., a compound which does not spontaneously undergo transformation such as by rearrangement, cyclization, elimination, or other reaction. Unless otherwise indicated, a “substituted” group has a substituent at one or more substitutable positions of the group, and when more than one position in any given structure is substituted, the substituent is either the same or different at each position. The term “substituted” is contemplated to include substitution with all permissible substituents of organic compounds, any of the substituents described herein that results in the formation of a stable compound. The present invention contemplates any and all such combinations in order to arrive at a stable compound. For purposes of this disclosure, heteroatoms such as nitrogen may have hydrogen substituents and/or any suitable substituent as described herein which satisfy the valencies of the heteroatoms and results in the formation of a stable moiety. [0119] Exemplary carbon atom substituents include, but are not limited to, halogen, –CN, – NO
2, –N
3, –SO
2H, –SO
3H, –OH, –OR
aa, –ON(R
bb)
2, –N(R
bb)
2, –N(R
bb)
3+X
–, –N(OR
cc)R
bb, – SH, –SR
aa, –SSR
cc, –C(=O)R
aa, –CO
2H, –CHO, –C(OR
cc)
2, –CO
2R
aa, –OC(=O)R
aa, – OCO
2R
aa, –C(=O)N(R
bb)
2, –OC(=O)N(R
bb)
2, –NR
bbC(=O)R
aa, –NR
bbCO
2R
aa, – NR
bbC(=O)N(R
bb)
2, –C(=NR
bb)R
aa, –C(=NR
bb)OR
aa, –OC(=NR
bb)R
aa, –OC(=NR
bb)OR
aa, – C(=NR
bb)N(R
bb)
2, –OC(=NR
bb)N(R
bb)
2, –NR
bbC(=NR
bb)N(R
bb)
2, –C(=O)NR
bbSO
2R
aa, – NR
bbSO
2R
aa, –SO
2N(R
bb)
2, –SO
2R
aa, –SO
2OR
aa, –OSO
2R
aa, –S(=O)R
aa, –S(=O)(=NR
bb)R
aa, – OS(=O)R
aa, –Si(R
aa)
3, –OSi(R
aa)
3 –C(=S)N(R
bb)
2, –C(=O)SR
aa, –C(=S)SR
aa, –SC(=S)SR
aa, – SC(=O)SR
aa, –OC(=O)SR
aa, –SC(=O)OR
aa, –SC(=O)R
aa, –P(=O)
2R
aa, –OP(=O)
2R
aa, – P(=O)(R
aa)
2, –OP(=O)(R
aa)
2, –OP(=O)(OR
cc)
2, –P(=O)
2N(R
bb)
2, –OP(=O)
2N(R
bb)
2, – P(=O)(NR
bb)
2, –OP(=O)(NR
bb)
2, –NR
bbP(=O)(OR
cc)
2, –NR
bbP(=O)(NR
bb)
2, –P(R
cc)
2, – P(R
cc)
3, –OP(R
cc)
2, –OP(R
cc)
3, –B(R
aa)
2, –B(OR
cc)
2, –BR
aa(OR
cc), C
1–10 alkyl, C
1–10 haloalkyl, C
2–10 alkenyl, C
2–10 alkynyl, C
3–10 carbocyclyl, 3–14 membered heterocyclyl, C
6–14 aryl, and 5–14 membered heteroaryl, wherein each alkyl, alkenyl, alkynyl, carbocyclyl, heterocyclyl, aryl, and heteroaryl is independently substituted with 0, 1, 2, 3, 4, or 5 R
dd groups; or two geminal hydrogens on a carbon atom are replaced with the group =O, =S, =NN(R
bb)
2, =NNR
bbC(=O)R
aa, =NNR
bbC(=O)OR
aa, =NNR
bbS(=O)
2R
aa, =NR
bb, or =NOR
cc; each instance of R
aa is, independently, selected from C
1–10 alkyl, C
1–10 haloalkyl, C
2–10 alkenyl, C
2–10 alkynyl, C
3–10 carbocyclyl, 3–14 membered heterocyclyl, C
6–14 aryl, and 5–14 membered heteroaryl, or two R
aa groups are joined to form a 3–14 membered heterocyclyl or 5–14 membered heteroaryl ring, wherein each alkyl, alkenyl, alkynyl, carbocyclyl, heterocyclyl, aryl, and heteroaryl is independently substituted with 0, 1, 2, 3, 4, or 5 R
dd groups; each instance of R
bb is, independently, selected from hydrogen, –OH, –OR
aa, – N(R
cc)
2, –CN, –C(=O)R
aa, –C(=O)N(R
cc)
2, –CO
2R
aa, –SO
2R
aa, –C(=NR
cc)OR
aa, – C(=NR
cc)N(R
cc)
2, –SO
2N(R
cc)
2, –SO
2R
cc, –SO
2OR
cc, –SOR
aa, –C(=S)N(R
cc)
2, –C(=O)SR
cc, – C(=S)SR
cc, –P(=O)
2R
aa, –P(=O)(R
aa)
2, –P(=O)
2N(R
cc)
2, –P(=O)(NR
cc)
2, C
1–10 alkyl, C
1–10 haloalkyl, C
2–10 alkenyl, C
2–10 alkynyl, C
3–10 carbocyclyl, 3–14 membered heterocyclyl, C
6–14 aryl, and 5–14 membered heteroaryl, or two R
bb groups are joined to form a 3–14 membered heterocyclyl or 5–14 membered heteroaryl ring, wherein each alkyl, alkenyl, alkynyl, carbocyclyl, heterocyclyl, aryl, and heteroaryl is independently substituted with 0, 1, 2, 3, 4, or 5 R
dd groups; each instance of R
cc is, independently, selected from hydrogen, C
1–10 alkyl, C
1–10 haloalkyl, C
2–10 alkenyl, C
2–10 alkynyl, C
3–10 carbocyclyl, 3–14 membered heterocyclyl, C
6–14 aryl, and 5–14 membered heteroaryl, or two R
cc groups are joined to form a 3–14 membered heterocyclyl or 5–14 membered heteroaryl ring, wherein each alkyl, alkenyl, alkynyl, carbocyclyl, heterocyclyl, aryl, and heteroaryl is independently substituted with 0, 1, 2, 3, 4, or 5 R
dd groups; each instance of R
dd is, independently, selected from halogen, –CN, –NO
2, –N
3, – SO
2H, –SO
3H, –OH, –OR
ee, –ON(R
ff)
2, –N(R
ff)
2, –N(R
ff)
3+X
–, –N(OR
ee)R
ff, –SH, –SR
ee, – SSR
ee, –C(=O)R
ee, –CO
2H, –CO
2R
ee, –OC(=O)R
ee, –OCO
2R
ee, –C(=O)N(R
ff)
2, – OC(=O)N(R
ff)
2, –NR
ffC(=O)R
ee, –NR
ffCO
2R
ee, –NR
ffC(=O)N(R
ff)
2, –C(=NR
ff)OR
ee, – OC(=NR
ff)R
ee, –OC(=NR
ff)OR
ee, –C(=NR
ff)N(R
ff)
2, –OC(=NR
ff)N(R
ff)
2, – NR
ffC(=NR
ff)N(R
ff)
2,–NR
ffSO
2R
ee, –SO
2N(R
ff)
2, –SO
2R
ee, –SO
2OR
ee, –OSO
2R
ee, –S(=O)R
ee, –Si(R
ee)
3, –OSi(R
ee)
3, –C(=S)N(R
ff)
2, –C(=O)SR
ee, –C(=S)SR
ee, –SC(=S)SR
ee, –P(=O)
2R
ee, – P(=O)(R
ee)
2, –OP(=O)(R
ee)
2, –OP(=O)(OR
ee)
2, C
1–6 alkyl, C
1–6 haloalkyl, C
2–6 alkenyl, C
2–6 alkynyl, C
3–10 carbocyclyl, 3–10 membered heterocyclyl, C
6–10 aryl, 5–10 membered heteroaryl, wherein each alkyl, alkenyl, alkynyl, carbocyclyl, heterocyclyl, aryl, and heteroaryl is independently substituted with 0, 1, 2, 3, 4, or 5 R
gg groups, or two geminal R
dd substituents can be joined to form =O or =S; each instance of R
ee is, independently, selected from C
1–6 alkyl, C
1–6 haloalkyl, C
2–6 alkenyl, C
2–6 alkynyl, C
3–10 carbocyclyl, C
6–10 aryl, 3–10 membered heterocyclyl, and 3–10 membered heteroaryl, wherein each alkyl, alkenyl, alkynyl, carbocyclyl, heterocyclyl, aryl, and heteroaryl is independently substituted with 0, 1, 2, 3, 4, or 5 R
gg groups; each instance of R
ff is, independently, selected from hydrogen, C
1–6 alkyl, C
1–6 haloalkyl, C
2–6 alkenyl, C
2–6 alkynyl, C
3–10 carbocyclyl, 3–10 membered heterocyclyl, C
6–10 aryl and 5–10 membered heteroaryl, or two R
ff groups are joined to form a 3–14 membered heterocyclyl or 5–14 membered heteroaryl ring, wherein each alkyl, alkenyl, alkynyl, carbocyclyl, heterocyclyl, aryl, and heteroaryl is independently substituted with 0, 1, 2, 3, 4, or 5 R
gg groups; and each instance of R
gg is, independently, halogen, –CN, –NO
2, –N
3, –SO
2H, –SO
3H, – OH, –OC
1–6 alkyl, –ON(C
1–6 alkyl)
2, –N(C
1–6 alkyl)
2, –N(C
1–6 alkyl)
3+X
–, –NH(C
1–6 alkyl)
2+X
–, –NH
2(C
1–6 alkyl)
+X
–, –NH
3+X
–, –N(OC
1–6 alkyl)(C
1–6 alkyl), –N(OH)(C
1–6 alkyl), –NH(OH), –SH, –SC
1–6 alkyl, –SS(C
1–6 alkyl), –C(=O)(C
1–6 alkyl), –CO
2H, –CO
2(C
1–6 alkyl), –OC(=O)(C
1–6 alkyl), –OCO
2(C
1–6 alkyl), –C(=O)NH
2, –C(=O)N(C
1–6 alkyl)
2, – OC(=O)NH(C
1–6 alkyl), –NHC(=O)(C
1–6 alkyl), –N(C
1–6 alkyl)C(=O)(C
1–6 alkyl), – NHCO
2(C
1–6 alkyl), –NHC(=O)N(C
1–6 alkyl)
2, –NHC(=O)NH(C
1–6 alkyl), –NHC(=O)NH
2, – C(=NH)O(C
1–6 alkyl),–OC(=NH)(C
1–6 alkyl), –OC(=NH)OC
1–6 alkyl, –C(=NH)N(C
1–6 alkyl)
2, –C(=NH)NH(C
1–6 alkyl), –C(=NH)NH
2, –OC(=NH)N(C
1–6 alkyl)
2, –OC(NH)NH(C
1–6 alkyl), –OC(NH)NH
2, –NHC(NH)N(C
1–6 alkyl)
2, –NHC(=NH)NH
2, –NHSO
2(C
1–6 alkyl), – SO
2N(C
1–6 alkyl)
2, –SO
2NH(C
1–6 alkyl), –SO
2NH
2,–SO
2C
1–6 alkyl, –SO
2OC
1–6 alkyl, – OSO
2C
1–6 alkyl, –SOC
1–6 alkyl, –Si(C
1–6 alkyl)
3, –OSi(C
1–6 alkyl)
3 –C(=S)N(C
1–6 alkyl)
2, C(=S)NH(C
1–6 alkyl), C(=S)NH
2, –C(=O)S(C
1–6 alkyl), –C(=S)SC
1–6 alkyl, –SC(=S)SC
1–6 alkyl, –P(=O)
2(C
1–6 alkyl), –P(=O)(C
1–6 alkyl)
2, –OP(=O)(C
1–6 alkyl)
2, –OP(=O)(OC
1–6 alkyl)
2, C
1–6 alkyl, C
1–6 haloalkyl, C
2–6 alkenyl, C
2–6 alkynyl, C
3–10 carbocyclyl, C
6–10 aryl, 3– 10 membered heterocyclyl, 5–10 membered heteroaryl; or two geminal R
gg substituents can be joined to form =O or =S; wherein X
– is a counterion. [0120] A “counterion” or “anionic counterion” is a negatively charged group associated with a cationic quaternary amino group in order to maintain electronic neutrality. Exemplary counterions include halide ions (e.g., F
–, Cl
–, Br
–, I
–), NO
3–, ClO
4–, OH
–, H
2PO
4–, HSO
4–, SO
4-2, sulfonate ions (e.g., methansulfonate, trifluoromethanesulfonate, p–toluenesulfonate, benzenesulfonate, 10–camphor sulfonate, naphthalene–2–sulfonate, naphthalene–1–sulfonic acid–5–sulfonate, ethan–1–sulfonic acid–2–sulfonate, and the like), and carboxylate ions (e.g., acetate, ethanoate, propanoate, benzoate, glycerate, lactate, tartrate, glycolate, and the like). [0121] Nitrogen atoms can be substituted or unsubstituted as valency permits, and include primary, secondary, tertiary, and quarternary nitrogen atoms. Exemplary nitrogen atom substitutents include, but are not limited to, hydrogen, –OH, –OR
aa, –N(R
cc)
2, –CN, – C(=O)R
aa, –C(=O)N(R
cc)
2, –CO
2R
aa, –SO
2R
aa, –C(=NR
bb)R
aa, –C(=NR
cc)OR
aa, – C(=NR
cc)N(R
cc)
2, –SO
2N(R
cc)
2, –SO
2R
cc, –SO
2OR
cc, –SOR
aa, –C(=S)N(R
cc)
2, –C(=O)SR
cc, – C(=S)SR
cc, –P(=O)
2R
aa, –P(=O)(R
aa)
2, –P(=O)
2N(R
cc)
2, –P(=O)(NR
cc)
2, C
1–10 alkyl, C
1–10 haloalkyl, C
2–10 alkenyl, C
2–10 alkynyl, C
3–10 carbocyclyl, 3–14 membered heterocyclyl, C
6–14 aryl, and 5–14 membered heteroaryl, or two R
cc groups attached to a nitrogen atom are joined to form a 3–14 membered heterocyclyl or 5–14 membered heteroaryl ring, wherein each alkyl, alkenyl, alkynyl, carbocyclyl, heterocyclyl, aryl, and heteroaryl is independently substituted with 0, 1, 2, 3, 4, or 5 R
dd groups, and wherein R
aa, R
bb, R
cc and R
dd are as defined above. [0122] These and other exemplary substituents are described in more detail in the Detailed Description, Examples, and Claims. The invention is not intended to be limited in any manner by the above exemplary listing of substituents. Other definitions [0123] The term "about," as used herein, includes the recited number ± 10%. Thus, "about 10" means 9 to 11. As is understood by one skilled in the art, reference to "about" a value or parameter herein includes (and describes) instances that are directed to that value or parameter per se. For example, description referring to "about X" includes description of "X." [0124] "USP1" and "ubiquitin-specific peptidase 1" as used herein refer to any native polypeptide or USP1 -encoding polynucleotide. The term "USP1" encompasses " full- length," unprocessed USP1 polypeptide as well as any forms of USP1 that result from processing within the cell (e g., removal of the signal peptide). The term also encompasses naturally occurring variants of USP1, e.g., those encoded by splice variants and allelic variants. The USP1 polypeptides described herein can be isolated from a variety of sources, such as from human tissue types or from another source, or prepared by recombinant or synthetic methods. Human USP1 sequences are known and include, for example, the sequences publicly available as UniProt No.094782 (including isoforms). As used herein, the term "human USP1 protein" refers to USP1 protein comprising the amino acid sequence as set forth in SEQ ID NO: 1 in U S. provisional patent application no.62/857,986 filed June 6, 2019. [0125] USP1 is a deubiquitinating enzyme that acts as part of a complex with UAF1. USP1’s "deubiquitinase activity" includes its ability to deubiquitinate as part of the USP1- UAF1 complex. [0126] The term "specifically binds" to a protein or domain of a protein is a term that is well understood in the art, and methods to determine such specific binding are also well known in the art. A molecule is said to exhibit "specific binding" or "preferential binding" if it reacts or associates more frequently, more rapidly, with greater duration and/or with greater affinity with a particular protein or domain of a protein than it does with alternative proteins or domains. It should be understood that a molecule that specifically or preferentially binds to a first protein or domain may or may not specifically or preferentially bind to a second protein or domain. As such, "specific binding" or "preferential binding" does not necessarily require (although it can include) exclusive binding. Generally, but not necessarily, reference to binding means preferential binding. For example, a USP1 inhibitor that specifically binds to USP1, UAF1, and/or the USP1-UAF1 complex may not bind to other deubiquitinases, other USP proteins, or other UAF1 complexes (e.g., USP46-UAF1) or may bind to other deubiquitinases, other USP proteins, or other UAF1 complexes (e.g., USP46-UAF1) with a reduced affinity as compared to binding to USP1. [0127] The terms "reduction" or "reduce" or "inhibition" or "inhibit" refer to a decrease or cessation of any phenotypic characteristic or to the decrease or cessation in the incidence, degree, or likelihood of that characteristic. To "reduce" or "inhibit" is to decrease, reduce or arrest an activity, function, and/or amount as compared to a reference. In an embodiment, by "reduce" or "inhibit" is meant the ability to cause an overall decrease of 20% or greater. In an embodiment, by "reduce" or "inhibit" is meant the ability to cause an overall decrease of 50% or greater. In an embodiment, by "reduce" or "inhibit" is meant the ability to cause an overall decrease of 75%, 85%, 90%, 95%, or greater. In an embodiment, the amount noted above is inhibited or decreased over a period of time, relative to a control over the same period of time. [0128] In an embodiment inhibiting USP1 proteins is the inhibition of one or more activities or functions of USP1 proteins. It should be appreciated that the activity or function of the one or more USP1 proteins may be inhibited in vitro or in vivo. Non limiting examples of activities and functions of USP1 include deubiquitinase activity, and formation of a complex with UAF l and are described herein. Examplary levels of inhibition of the activity of one or more USP1 proteins include at least 10% inhibiton, at least 20% inhibition, at least 30% inhibition, at least 40% inhibition, at least 50% inhibition, at least 60% inhibition, at least 70% inhibition, at least 80% inhibition, at least 90% inhibition, and up to 100% inhibition. [0129] As used herein, the term "loss of function" mutation refers to a mutation that results in the absence of a gene, decreased expression of a gene, or the production of a gene product (e.g. protein) having decreased activity or no activity. Loss of function mutations include for example, missense mutations, nucleotide insertions, nucleotide deletions, and gene deletions. Loss of function mutations also include dominant negative mutations. Thus, cancer cells with a loss of function mutation in a gene encoding BRCA1 or BRCA2 include cancer cells that contain missense mutations in a gene encoding BRCA1 or BRCA2as well as cancer cells that lack a gene encoding BRCA1 or BRCA2. [0130] As used herein, the term “salt” refers to any and all salts and encompasses pharmaceutically acceptable salts. [0131] The term “pharmaceutically acceptable salt” refers to those salts which are, within the scope of sound medical judgment, suitable for use in contact with the tissues of humans and lower animals without undue toxicity, irritation, allergic response and the like, and are commensurate with a reasonable benefit/risk ratio. Pharmaceutically acceptable salts are well known in the art. For example, Berge et al., describes pharmaceutically acceptable salts in detail in J. Pharmaceutical Sciences (1977) 66:1–19. Pharmaceutically acceptable salts of the compounds disclosed herein include those derived from suitable inorganic and organic acids and bases. Examples of pharmaceutically acceptable, nontoxic acid addition salts are salts of an amino group formed with inorganic acids such as hydrochloric acid, hydrobromic acid, phosphoric acid, sulfuric acid and perchloric acid or with organic acids such as acetic acid, oxalic acid, maleic acid, tartaric acid, citric acid, succinic acid or malonic acid or by using other methods used in the art such as ion exchange. Other pharmaceutically acceptable salts include adipate, alginate, ascorbate, aspartate, benzenesulfonate, benzoate, bisulfate, borate, butyrate, camphorate, camphorsulfonate, citrate, cyclopentanepropionate, digluconate, dodecylsulfate, ethanesulfonate, formate, fumarate, glucoheptonate, glycerophosphate, gluconate, hemisulfate, heptanoate, hexanoate, hydroiodide, 2–hydroxy–ethanesulfonate, lactobionate, lactate, laurate, lauryl sulfate, malate, maleate, malonate, methanesulfonate, 2– naphthalenesulfonate, nicotinate, nitrate, oleate, oxalate, palmitate, pamoate, pectinate, persulfate, 3–phenylpropionate, phosphate, picrate, pivalate, propionate, stearate, succinate, sulfate, tartrate, thiocyanate, p–toluenesulfonate, undecanoate, valerate salts, and the like. Pharmaceutically acceptable salts derived from appropriate bases include alkali metal, alkaline earth metal, ammonium and N
+(C
1–4alkyl)
4 salts. Representative alkali or alkaline earth metal salts include sodium, lithium, potassium, calcium, magnesium, and the like. Further pharmaceutically acceptable salts include, when appropriate, nontoxic ammonium, quaternary ammonium, and amine cations formed using counterions such as halide, hydroxide, carboxylate, sulfate, phosphate, nitrate, lower alkyl sulfonate, and aryl sulfonate. [0132] A “subject” to which administration is contemplated includes, but is not limited to, humans (i.e., a male or female of any age group, e.g., a pediatric subject (e.g., infant, child, adolescent) or adult subject (e.g., young adult, middle–aged adult or senior adult)) and/or a non-human animal, e.g., a mammal such as primates (e.g., cynomologus monkeys, rhesus monkeys), cattle, pigs, horses, sheep, goats, rodents, cats, and/or dogs. In an embodiment, the subject is a human. In an embodiment, the subject is a non-human animal. The terms “human,” “patient,” and “subject” are used interchangeably herein. [0133] Disease, disorder, and condition are used interchangeably herein. [0134] As used herein, and unless otherwise specified, the terms “treat,” “treating” and “treatment” contemplate an action that occurs while a subject is suffering from the specified disease, disorder or condition, which reduces the severity of the disease, disorder or condition, or retards or slows the progression of the disease, disorder or condition (“therapeutic treatment”), and also contemplates an action that occurs before a subject begins to suffer from the specified disease, disorder or condition (“prophylactic treatment”). In an embodiment, the compounds provided herein are contemplated to be used in methods of therapeutic treatment wherein the action occurs while a subject is suffering from the specified disease, disorder or condition and results in a reduction in the severity of the disease, disorder or condition, or retardation or slowing of the progression of the disease, disorder or condition. In an alternate embodiment, the compounds provided herein are contemplated to be used in methods of prophylactic treatment wherein the action occurs before a subject begins to suffer from the specified disease, disorder or condition and results in preventing a disease, disorder or condition, or one or more symptoms associated with the disease, disorder or condition, or preventing the recurrence of the disease, disorder or condition. [0135] In general, the “effective amount” of a compound refers to an amount sufficient to elicit the desired biological response. As will be appreciated by those of ordinary skill in this art, the effective amount of a compound disclosed herein may vary depending on such factors as the desired biological endpoint, the pharmacokinetics of the compound, the disease being treated, the mode of administration, and the age, health, and condition of the subject. An effective amount encompasses therapeutic and prophylactic treatment. An effective amount encompasses therapeutic and prophylactic treatment (i.e., encompasses a “therapeutically effective amount” and a “prophylactically effective amount”). [0136] As used herein, and unless otherwise specified, a “therapeutically effective amount” of a compound is an amount sufficient to provide a therapeutic benefit in the treatment of a disease, disorder or condition, or to delay or minimize one or more symptoms associated with the disease, disorder or condition. A therapeutically effective amount of a compound means an amount of therapeutic agent, alone or in combination with other therapies, which provides a therapeutic benefit in the treatment of the disease, disorder or condition. The term “therapeutically effective amount” can encompass an amount that improves overall therapy, reduces or avoids symptoms or causes of disease or condition, or enhances the therapeutic efficacy of another therapeutic agent. [0137] As used herein, and unless otherwise specified, a “prophylactically effective amount” of a compound is an amount sufficient to prevent a disease, disorder or condition, or one or more symptoms associated with the disease, disorder or condition, or prevent its recurrence. A prophylactically effective amount of a compound means an amount of a therapeutic agent, alone or in combination with other agents, which provides a prophylactic benefit in the prevention of the disease, disorder or condition. The term “prophylactically effective amount” can encompass an amount that improves overall prophylaxis or enhances the prophylactic efficacy of another prophylactic agent. [0138] The term "container" means any receptacle and closure therefore suitable for storing, shipping, dispensing, and/or handling a pharmaceutical product. [0139] The term "insert" or "package insert" means information accompanying a pharmaceutical product that provides a description of how to administer the product, along with the safety and efficacy data required to allow the physician, pharmacist, and patient to make an informed decision regarding use of the product. The package insert generally is regarded as the "label" for a pharmaceutical product. Compounds [0140] As generally described herein, provided are compounds (e.g., compounds of Formula (I), (II) or a compound of Table 1 or pharmaceutically acceptable salts, hydrates, solvates, prodrugs, stereoisomers or tautomers thereof) that are ubiquitin-specific peptidase 1 (USP1) inhibitors useful for treating diseases and disorders (e.g., cancers) associated with USP1. [0141] Provided herein are compounds of Formula (I) and Formula (II). Unless the context requires otherwise, reference throughout this specification to “a compound of Formula (I) and/or a compound of Formula (II)” refers to all embodiments of Formula (I), including, for example, compounds of Formula (I), Formula (I-a), Formula (I-b), Formula (I-c), Formula (I- d), Formula (I-d-1), Formula (I-e), Formula (I-f), Formula (I-f-1), Formula (II) as well as the compounds of Table 1. [0142] In an embodiment, provided are compounds of Formula (I) or Formula (II) or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof. In an embodiment, including any of the numbered embodiments described herein, the compounds are provided as free base or pharmaceutically acceptable salts. In an embodiment, including any of the numbered embodiments described herein, the compounds are provided as free base. In an embodiment, including any of the numbered embodiments described herein, the compounds are provided as pharmaceutically acceptable salts. [0143] In an embodiment, provided is a compound of Formula (I) or Formula (II) or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof; wherein:
Ring A is selected from the group consisting of:
wherein X
3 is CH or N and wherein the left-side attachment point connects to CR
cR
c’ and the right attachment point connects to Ring B; L is selected from the group consisting of –O– and –S–; Ring B is a 5-10 membered heteroaryl or a 3-10 membered heterocyclyl; each R
A is independently selected from the group consisting of –D, oxo, halo, – CN, –C
1–C
6 alkyl, –C
1–C
6 haloalkyl, –C
1–C
6 hydroxyalkyl, –C
3–C
10 cycloalkyl, –OR
A1 and – N(R
A1)
2; each R
A1 is independently selected from the group consisting of H, –C
1–C
6 alkyl, –C
1–C
6 haloalkyl and C
3–C
9 cycloalkyl wherein each hydrogen atom of each alkyl and cycloalkyl can be replaced by deuterium; or two instances of R
A1 on a nitrogen atom can be taken together with the nitrogen to which they are attached to form a 4-6 member monocyclic heterocycle containing one nitrogen atom and 0-1 additional heteroatoms selected from N and O; each R
B is independently selected from the group consisting of halo, –CN, –C
1–C
6 alkyl, –C
1–C
6 alkenyl, –heteroC
1-C
4 alkyl, –C
1–C
6 haloalkyl, –C
1–C
6 hydroxyalkyl, –C
3–C
10 cycloalkyl, 3-10 membered heterocyclyl, C
3-C
9 cycloalkylalkyl, –OR
B1, –N(R
B1)
2, – C(=O)R
B1, –C(=O)OR
B1, –NR
B1C(=O)R
B1, –NR
B1C(=O)OR
B1, –C(=O)N(R
B1)
2, – OC(=O)N(R
B1)
2, –S(=O)R
B1, –S(=O)
2R
B1, –SR
B1, –S(=O)(=NR
B1)R
B1, –NR
B1S(=O)
2R
B1 and –S(=O)
2N(R
B1)
2, wherein each alkyl, alkenyl, cycloalkyl, heterocyclyl and cycloalkylalkyl of R
B can be partially or fully deuterated and can be substituted at any available position with 0, 1 or 2 substituents selected from –F, –Cl, –OH– and –CH
3; each R
B1 is independently selected from the group consisting of H, –C
1–C
6 alkyl, –heteroC
1-C
4 alkyl, –C
1–C
6 haloalkyl, –C
3–C
9 cycloalkyl and 3-7 membered heterocyclyl wherein each hydrogen atom of each alkyl, cycloalkyl and heterocyclyl can be replaced by deuterium; or two instances of R
B1 on a nitrogen atom can be taken together with the nitrogen to which they are attached to form a 4-6 member monocyclic heterocycle containing one nitrogen atom and 0-1 additional heteroatoms selected from N and O; each R
C is independently selected from the group consisting of H, –D, oxo, halo, – CN, –OR
C1, –SR
C1 –NR
C1 2, –C
1–C
6 alkyl, –C
1–C
6 hydroxyalkyl, –C
1–C
6 haloalkyl, –C
3–C
9 cycloalkyl, 3-10 member heterocyclyl and –heteroC
1-C
4 alkyl wherein each alkyl, cycloalkyl and heterocyclyl of R
C can partially or fully deuterated and can be substituted at any available position with 0, 1 or 2 substituents selected from –F, –Cl, –OH– and –CH
3; each R
C1 is independently selected from the group consisting of H and –C
1–C
6 alkyl, wherein each hydrogen atom of the alkyl can be independently replaced by deuterium; or two instances of R
C1 on a nitrogen atom can be taken together with the nitrogen to which they are attached to form a 4-6 member monocyclic heterocycle containing one nitrogen atom and 0-1 additional heteroatoms selected from N and O; each R
D is independently selected from the group consisting of –D, halo, –CN, – C
1–C
6 alkyl, –heteroC
1-C
4 alkyl, –C
1–C
6 haloalkyl, –C
1–C
6 hydroxyalkyl, –C
3–C
10 cycloalkyl, 3-10 membered heterocyclyl, –C
6-C
10 aryl, –OR
D1, –N(R
D1)
2, –C(=O)R
D1, – C(=O)OR
D1, –NR
D1C(=O)R
D1, –NR
D1C(=O)OR
D1, –C(=O)N(R
D1)
2, –OC(=O)N(R
D1)
2, – S(=O)R
D1, –S(=O)
2R
D1, –SR
D1, –S(=O)(=NR
D1)R
D1, –NR
D1S(=O)
2R
D1 and –S(=O)
2N(R
D1)
2 , wherein each alkyl, cycloalkyl, heterocyclyl and aryl of R
D can be substituted at any available position with 0, 1 or 2 substituents selected from –F, –Cl, –OH– and –CH
3; each R
D1 is independently selected from the group consisting of H, –C
1–C
6 alkyl, –heteroC
1-C
4 alkyl, –C
1–C
6 haloalkyl, –C
3–C
9 cycloalkyl and 3-7 membered heterocyclyl, wherein each hydrogen atom of each alkyl, cycloalkyl and heterocyclyl can be replaced by deuterium; or two instances of R
D1 on a nitrogen atom can be taken together with the nitrogen to which they are attached to form a 4-6 member monocyclic heterocycle containing one nitrogen atom and 0-1 additional heteroatoms selected from N and O; R
E is selected from H, –D, halo, –CN, –C
1–C
6 alkyl, –C
1-C
6 alkynyl, –heteroC
1– C
6 alkyl, –C
1–C
6 haloalkyl, –C
1–C
6 hydroxyalkyl, –C
3–C
10 cycloalkyl, 3-10 membered heterocyclyl, –C
6-C
10 aryl, 5-10 member heteroaryl, heterocyclylalkyl, heteroarylalkyl, arylalkyl, cycloalkylalkyl, –OR
E1, –SR
E1, –N(R
E1)
2, –C
1–C
6 alkylene–NR
E1C(=O)OR
E1, – C(=O)R
E1, –C(=O)OR
E1, –NR
E1C(=O)R
E1, –NR
E1C(=O)OR
E1, –C(=O)N(R
E1)
2, and – OC(=O)N(R
E1)
2, wherein each alkyl, alkynyl, heteroalkyl, haloalkyl, cycloalkyl, heterocyclyl, aryl, heteroaryl, cycloalkylalkyl, heterocyclylalkyl, arylalkyl and heteroarylalkyl is optionally substituted at any available position; each R
E1 is independently selected from H, –C
1–C
6 alkyl (wherein each hydrogen can be independently replaced by deuterium), –heteroC
1–C
6 alkyl, –C
1–C
6 haloalkyl, –C
3–C
9 cycloalkyl, 3-7 membered heterocyclyl, cycloalkylalkyl, heterocyclylalkyl, aryl, 5-6 membered heteroaryl, arylalkyl and heteroarylalkyl wherein each alkyl, haloalkyl, cycloalkyl, heterocyclyl, aryl, heteroaryl, cycloalkylalkyl, heterocyclylalkyl, arylalkyl and heteroarylalkyl is optionally substituted at any available position; each R
c and R
c’ is independently selected from the group consisting of H, –D, – C
1–C
6 alkyl, –heteroC
1-C
4 alkyl and –C
1–C
6 haloalkyl or R
c and R
c’ can be taken together with the atom to which they are attached to form a –C
3–C
9 cycloalkyl; n is 0, 1, 2,3 or 4; m is 0, 1, 2 or 3; and p is 0, 1, 2 or 3. [0144] In an embodiment, provided is a compound of Formula (I) or Formula (II) or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof wherein X
1, X
2, Ring A, L, Ring B, R
A, R
B, R
C, R
D, R
E, R
c, R
c’, n, m and p are as defined in any of the embodiments described herein. In an embodiment, the compound is of Formula (I). In an embodiment, the compound is of Formula (II). [0145] As generally defined herein, Ring A is selected from the group consisting of:
wherein X
3 is as defined in any of the embodiments described herein, wherein the left-side attachment point connects to CR
cR
c’ and the right attachment point connects to Ring B. [0146] As generally defined herein, X
3 is CH or N. In an embodiment, X
3 is CH. In an embodiment, X
3 is N. [0147] In an embodiment, Ring A is selected from the group consisting of
and
wherein the left-side attachment point connects to CR
cR
c’ and the right attachment point connects to Ring B. [0148] In an embodiment, Ring A is
wherein the left-side attachment point connects to CR
cR
c’ and the right attachment point connects to Ring B. [0149] In an embodiment, Ring A is
wherein the left-side attachment point connects to CR
cR
c’ and the right attachment point connects to Ring B. [0150] In an embodiment, Ring A is
wherein the left-side attachment point connects to CR
cR
c’ and the right attachment point connects to Ring B. [0151] In an embodiment, Ring A is optionally substituted at any available position. In an embodiment, Ring A is substituted with 0, 1, 2, 3 or 4 instances of R
A, wherein each R
A is independently as defined in any of the embodiments described herein. In an embodiment, Ring A is substituted with 0, 1, 2 or 3 instances of R
A, wherein each R
A is independently as defined in any of the embodiments described herein. In an embodiment, Ring A is unsubstituted. In an embodiment, Ring A is substituted with 1 or 2 instances of R
A, wherein each R
A is independently as defined in any of the embodiments described herein. In an embodiment, Ring A is substituted with 1 instance of R
A, wherein each R
A is independently as defined in any of the embodiments described herein. In an embodiment, Ring A is substituted with 2 instances of R
A, wherein each R
A is independently as defined in any of the embodiments described herein. In an embodiment, Ring A is substituted with 3 instances of R
A, wherein each R
A is independently as defined in any of the embodiments described herein. [0152] In an embodiment, the moiety represented
group consisting
point connects to CR
cR
c’ and the right attachment point connects to Ring B. [0153] In an embodiment, the moiety represented
group consisting
wherein the left-side attachment point connects to CR
cR
c’ and the right attachment point connects to Ring B. [0154] In an embodiment, the moiety represented
group consisting
, wherein the left-side attachment point connects to CR
cR
c’ and the right attachment point connects to Ring B. (R
A)
n Ring A [0155] In an embodiment, the moiety represented as
is
, wherein the left-side attachment point connects to CR
cR
c’ and the right attachment point connects to Ring B. [0156] In an embodiment, the moiety represented
wherein the left-side attachment point connects to CR
cR
c’ and the right attachment point connects to Ring B. (R
A)
n [0157] In an embodiment, the moiety represented as
is
, wherein the left-side attachment point connects to CR
cR
c’ and the right attachment point connects to Ring B. [0158] In an embodiment, the moiety represented
wherein the left-side attachment point connects to CR
cR
c’ and the right attachment point connects to Ring B. [0159] In an embodiment, the moiety represented
wherein the left-side attachment point connects to CR
cR
c’ and the right attachment point connects to Ring B. [0160] In an embodiment, the moiety represented
wherein the left-side attachment point connects to CR
cR
c’ and the right attachment point connects to Ring B. [0161] In an embodiment, the moiety represented
wherein the left-side attachment point connects to CR
cR
c’ and the right attachment point connects to Ring B. (R
A)
n Ring A
[0162] In an embodiment, the moiety represented as
i
wherein the left-side attachment point connects to CR
cR
c’ and the right attachment point connects to Ring B. (R
A)
n Ring A [0163] In an embodiment, the moiety represented as
i
wherein the left-side attachment point connects to CR
cR
c’ and the right attachment point connects to Ring B. [0164] In an embodiment, the moiety represented
wherein the left-side attachment point connects to CR
cR
c’ and the right attachment point connects to Ring B. [0165] In an embodiment, the moiety represented
wherein the left-side attachment point connects to CR
cR
c’ and the right attachment point connects to Ring B.
(R
A)
n Ring A [0166] In an embodiment, the moiety represented as is
, wherein the left-side attachment point connects to CR
cR
c’ and the right attachment point connects to Ring B. [0167] In an embodiment, the moiety represented
wherein the left-side attachment point connects to CR
cR
c’ and the right attachment point connects to Ring B. [0168] In an embodiment, the moiety represented
wherein the left-side attachment point connects to CR
cR
c’ and the right attachment point connects to Ring B. [0169] In an embodiment, the moiety represented
wherein the left-side attachment point connects to CR
cR
c’ and the right attachment point connects to Ring B. [0170] In an embodiment, the moiety represented
wherein the left-side attachment point connects to CR
cR
c’ and the right attachment point connects to Ring B. [0171] In an embodiment, the moiety represented
wherein the left-side attachment point connects to CR
cR
c’ and the right attachment point connects to Ring B. [0172] In an embodiment, the moiety represented
wherein the left-side attachment point connects to CR
cR
c’ and the right attachment point connects to Ring B. [0173] In an embodiment, the moiety represented
wherein the left-side attachment point connects to CR
cR
c’ and the right attachment point connects to Ring B. [0174] In an embodiment, the moiety represented as
, wherein the left-side attachment point connects to CR
cR
c’ and the right attachment point connects to Ring B. [0175] In an embodiment, the moiety represented as
,wherein the left-side attachment point connects to CR
cR
c’ and the right attachment point connects to Ring B. [0176] As generally defined herein, X
1 is selected from N and CH. In an embodiment, X
1 is N. In an embodiment, X
1 is CH. [0177] As generally defined herein, X
2 is selected from N and CH. In an embodiment, X
2 is N. In an embodiment, X
2 is CH. [0178] In an embodiment, X
1 is N and X
2 is CH. In an embodiment, X
1 and X
2 are both N. In an embodiment, X
1 and X
2 are both CH. [0179] In an embodiment,
. In an embodiment,
. In an embodiment,
[0180] In an embodiment, the moiety represented as
is selected from the group consisting
wherein each R
1 and R
2 are as defined in any of the embodiments described herein. [0181] In an embodiment, the moiety represented as
is selected from the
[0182] In an embodiment, the moiety represented as
is selected from the
[0183] In an embodiment, the moiety represented as
is selected from the group consisting
[0184] In an embodiment, the moiety represented
embodiment, the moiety represented
embodiment, the moiety represented
embodiment, the moiety represented
embodiment, the moiety represented
embodiment, the moiety represented as


[0185] As generally defined herein, Ring B is a 5-10 membered heteroaryl or a 3-10 membered heterocyclyl. [0186] In an embodiment, Ring B is a 3-7 member monocyclic heterocyclyl containing 1-3 heteroatoms selected from O, N and S (e.g., azetidinyl, oxetanyl, tetrahydrofuranyl, pyrrolidinyl, piperidinyl, piperazinyl, morpholinyl, thiomorpholinyl). [0187] In an embodiment, Ring B is a 5-6 member monocyclic heteroaryl containing 1-3 heteroatoms selected from O, N and S (e.g., pyridinyl, pyrimidinyl, pyrazinyl, pyridazinyl, triazinyl, pyrrolyl, pyrazolyl, imidazolyl, thiazolyl, furanyl, thiophenyl, oxazolyl, thiadiazolyl, oxadiazolyl and triazolyl). In an embodiment, Ring B is selected from the group consisting of pyridinyl, pyrimidinyl, pyrazinyl, pyridazinyl, pyrrolyl, pyrazolyl, imidazolyl, thiazolyl, thiadiazolyl, oxadiazolyl and triazolyl. [0188] In an embodiment, Ring B is selected from the group consisting of pyridinyl, pyrazolyl and imidazolyl. In an embodiment, Ring B is selected from the group consisting of pyridin-2-yl, pyridin-3-yl, pyridin-4-yl, pyrimidin-2-yl, pyrimidin-4-yl, pyrimidin-5-yl, pyrimidin-6-yl, pyrazin-2-yl, pyrazin-3-yl, pyridazin-3-yl, pyridazin-4-yl, pyridazin-5-yl, pyridazin-6-yl, pyrrol-2-yl, pyrazol-1-yl, pyrazol-4-yl, pyrazol-5-yl, imidazol-2-yl, thiazol-4- yl, thiazol-5-yl, thiadiazol-3-yl, thiadiazol-5-yl, oxadiazol-3-yl, oxadiazol-5-yl, 1,2,3 triazol- 1-yl and 1,2,5 thiazol-4-yl. In an embodiment, Ring B is selected from the group consisting of pyridin-2-yl, pyrazol-1-yl and imidazol-2-yl. [0189] In an embodiment, Ring B is a 6-member monocyclic heteroaryl containing 1-3 nitrogen atoms (e.g., pyridinyl, pyrimidinyl, pyrazinyl, pyridazinyl, triazinyl). [0190] In an embodiment, Ring B is pyridinyl (e.g., pyridin-2-yl, pyridin-3-yl, pyridin-4-yl), pyrimidinyl, (e.g., pyrimidin-2-yl, pyrimidin-4-yl, pyrimidin-5-yl, pyrimidin-6-yl), pyrazinyl (e.g., pyrazin-2-yl, pyrazin-3-yl) or pyridazinyl (e.g., pyridazin-3-yl, pyridazin-4-yl, pyridazin-5-yl, pyridazin-6-yl). In an embodiment, Ring B is a 5 member monocyclic heteroaryl containing 1-3 heteroatoms selected from O, N and S. [0191] In an embodiment, Ring B is selected from pyrrolyl, pyrazolyl, imidazolyl, thiazolyl, furanyl, thiophenyl, oxazolyl, thiadiazolyl, oxadiazolyl and triazolyl. In an embodiment, Ring B is selected from pyrrolyl (e.g., pyrrol-2-yl), pyrazolyl (e.g., pyrazol-1-yl, pyrazol-4-yl, pyrazol-5-yl) , imidazolyl (e.g., imidazol-2-yl), thiazolyl (e.g., thiazol-4-yl, thiazol-5-yl), thiadiazolyl (e.g., thiadiazol-3-yl, thiadiazol-5-yl), oxadiazolyl (e.g., oxadiazol-3-yl, oxadiazol-5-yl) and triazolyl (e.g., 1,2,3 triazol-1-yl, 1,2,5 thiazol-4-yl). In an embodiment, Ring B is pyrrolyl (e.g., pyrrol-2-yl). [0192] In an embodiment, Ring B is selected from imidazolyl (e.g., imidazol-2-yl) and pyrazolyl (e.g., pyrazol-1-yl). In an embodiment, Ring B is pyrazolyl (e.g., pyrazol-1-yl, pyrazol-3-yl, pyrazol-4-yl, pyrazol-5-yl). In an embodiment, Ring B is pyrazol-1-yl. In an embodiment, Ring B is imidazolyl (e.g., imidazol-2-yl, imidazol-4-yl, imidazol-5-yl). In an embodiment, Ring B is imidazol-2-yl. In an embodiment, Ring B is thiazolyl (e.g., thiazol-2- yl, thiazol-4-yl, thiazol-5-yl). In an embodiment, Ring B is furanyl (e.g., furan-2-yl, furan-3- yl). In an embodiment, Ring B is thiophenyl (e.g., thiophen-2-yl, thiophen-3-yl). In an embodiment, Ring B is oxazolyl (e.g., oxazol-2-yl, oxazol-4-yl, oxazol-5-yl). In an embodiment, Ring B is thiadiazolyl. In an embodiment, Ring B is oxadiazolyl. [0193] In an embodiment, Ring B as defined in any of the above embodiments is optionally substituted at any available position. In an embodiment, Ring B is substituted with 0, 1, 2 or 3 instances of R
B, wherein each R
B is independently as defined in any of the embodiments described herein. In an embodiment, Ring B is substituted with 0, 1 or 2 instances of R
B, wherein each R
B is independently as defined in any of the embodiments described herein. In an embodiment, Ring B is unsubstituted. In an embodiment, Ring B is substituted with 1 or 2 instances of R
B, wherein each R
B is independently as defined in any of the embodiments described herein. In an embodiment, Ring B is substituted with 1 instance of R
B, wherein each R
B is independently as defined in any of the embodiments described herein. In an embodiment, Ring B is substituted with 2 instances of R
B, wherein each R
B is independently as defined in any of the embodiments described herein. In an embodiment, Ring B is substituted with 3 instances of R
B, wherein each R
B is independently as defined in any of the embodiments described herein.
[0195] In an embodiment, the moiety represented
selected from the group
. (R
B)p
B [0196] In an embodiment, the moiety represented by
is selected from the group consisting
(R
B)p
B [0197] In an embodiment, the moiety represented by
is selected from the group consisting
[0198] In an embodiment, the moiety represented by
embodiment, the moiety represented by
embodiment, the moiety represented by
embodiment, the moiety represented by
embodiment, the moiety represented by
embodiment, the moiety represented by
. ,
mbodiment, the moiety represented
mbodiment, the moiety represented
)p
mbodiment, the moiety represented by
is
embodiment, the moiety represented
In an embodiment, the moiety represented
embodiment, the moiety represented
the moiety represented
embodiment, the moiety
[0199] As generally defined herein, L is selected from the group consisting of –O– and –S–. [0200] In an embodiment, L is –O–. In an embodiment, L is –S–. [0201] As generally defined herein, each R
c and R
c’ is independently selected from the group consisting of H, –D, –C
1–C
6 alkyl, –heteroC
1-C
4 alkyl and –C
1–C
6 haloalkyl or R
c and R
c’ can be taken together with the atom to which they are attached to form a –C
3–C
9 cycloalkyl (e.g., cyclopropyl) [0202] In an embodiment, R
c and R
c’ are each independently selected from H, –Me, –CF
3 and –CHF
2 or are taken together to form a cyclopropyl. [0203] In an embodiment, R
c and R
c’ are each independently selected from H and –Me or are taken together to form a cyclopropyl. [0204] In an embodiment, R
c and R
c’ are each independently selected from H and –Me. [0205] In an embodiment, R
c and R
c’ are both H. [0206] In an embodiment, R
c is H and R
c’ is –Me. [0207] In an embodiment, R
c and R
c’ are each independently –C
1–C
6 alkyl (e.g., –Me, –Et, – Pr, –iPr, –
nBu, –sec-Bu, –iso-Bu, –
tBu). In an embodiment, R
c and R
c’ are each independently –Me. In an embodiment, R
c and R
c’ are each independently –Et. In an embodiment R
c and R
c’ are each independently –Pr. In an embodiment, R
c and R
c’ are each independently –iPr. [0208] In an embodiment, R
c and R
c’ are each independently –heteroC
1-C
4 alkyl. In an embodiment, R
c and R
c’ are each independently methoxymethyl (–CH
2OCH
3). In an embodiment, R
c and R
c’ are each independently hydroxymethyl (–CH
2OH). In an embodiment, R
c and R
c’ are each independently aminomethyl (e.g., –CH
2NH
2, –CH
2NHCH
3, –CH
2N(CH
3)
2. [0209] In an embodiment, R
c and R
c’ are each independently –C
1–C
6 haloalkyl. In an embodiment, R
c and R
c’ are each independently trifluoromethyl (–CF
3). In other embodiments, R
c and R
c’ are each independently difluoromethyl (–CHF
2). [0210] In an embodiment, R
c and R
c’ are taken together with the carbon to which they are attached to form a –C
3–C
9 cycloalkyl (e.g., cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl). In an embodiment, R
c and R
c’ are taken together with the carbon to which they are attached to form a cyclopropyl. In an embodiment, R
c and R
c’ are taken together with the carbon to which they are attached to form a cyclobutyl. In an embodiment, R
c and R
c’ are taken together with the carbon to which they are attached to form a cyclopentyl. In an embodiment, R
c and R
c’ are taken together with the carbon to which they are attached to form a cyclohexyl. [0211] As generally defined herein, n is 0, 1, 2, 3 or 4. In an embodiment, n is selected from 0, 1, 2 or 3. In an embodiment, n is selected from 0, 1 or 2. In an embodiment, n is selected from 0 or 1. In an embodiment n is 0. In an embodiment, n is 1 or 2. In an embodiment, n is 1. In an embodiment, n is 2. In an embodiment, n is 3. In an embodiment, n is 4 [0212] As generally defined herein, m is 0, 1, 2 or 3. In an embodiment, m is selected from 0, 1 or 2. In an embodiment, m is selected from 0 or 1. In an embodiment m is 0. In an embodiment, m is 1 or 2. In an embodiment, m is 1. In an embodiment, m is 2. [0213] As generally defined herein, p is 0, 1, 2 or 3. In an embodiment, p is selected from 1, 2 or 3. In an embodiment, p is selected from 0, 1 or 2. In an embodiment, p is selected from 0 or 1. In an embodiment p is 0. In an embodiment, p is 1 or 2. In an embodiment, p is 2 or 3. In an embodiment, p is 1. In an embodiment, p is 2. In an embodiment, p is 3. As generally defined herein, each R
A is independently selected from –D, oxo, halo, – CN, –C
1–C
6 alkyl, –C
1–C
6 haloalkyl, –C
1–C
6 hydroxyalkyl, –C
3–C
10 cycloalkyl, –OR
A1 and – N(R
A1)
2, wherein each R
A1 is as defined in any of the embodiments described herein. In an embodiment, each R
A is independently selected from –D, oxo, halo (e.g., –F, –Cl), –C
1–C
6 alkyl (e.g., –Me, –Et, –Pr, –
iPr, –
nBu, –
tBu), –OH and –O–C
1–C
6 alkyl (e.g., –OMe, –OEt, – OPr, –O
iPr, –O
nBu, –O
tBu). In an embodiment, each R
A is independently selected from In an embodiment, each R
A is independently selected from C
1-C
6 alkyl, C
1-C
6 haloalkyl, –OC
1-C
3 alkyl, and halo. [0214] In an embodiment, each R
A is independently selected from oxo and halo. In an embodiment, each R
A is independently selected from –F, –Cl, –Me, –OH and –OMe. In an embodiment, each R
A is independently selected from –F, –Cl, –CH
3, –Et
, –CH
2CF
3, –OCH
3 and –OEt. In an embodiment, each R
A is independently selected from oxo and –F. In an embodiment, each R
A is independently oxo. In an embodiment, each R
A is independently –F. [0215] As generally defined herein, each R
A1 is independently selected from the group consisting of H, –C
1–C
6 alkyl, –C
1–C
6 haloalkyl and C
3–C
9 cycloalkyl wherein each hydrogen atom of each alkyl and cycloalkyl can be replaced by deuterium; or two instances of R
A1 on a nitrogen atom can be taken together with the nitrogen to which they are attached to form a 4-6 member monocyclic heterocycle containing one nitrogen atom and 0-1 additional heteroatoms selected from N and O. In an embodiment, R
A1 is H. In an embodiment, R
A1 is –C
1–C
6 alkyl (e.g., –Me, –Et, –Pr, –
iPr, –
nBu, –
tBu). In an embodiment, R
A1 is –Me. In an embodiment R
A1 is –C
1–C
6 haloalkyl (e.g., –CF
3, –CHF
2, –CF
2CH
3, – CH
2CF
3). In an embodiment, R
A1 is –CF
3. [0216] As generally defined herein, each R
B is independently selected from the group consisting of halo, –CN, –C
1–C
6 alkyl, –C
1–C
6 alkenyl, –heteroC
1-C
4 alkyl, –C
1–C
6 haloalkyl, –C
1–C
6 hydroxyalkyl, –C
3–C
10 cycloalkyl, 3-10 membered heterocyclyl, C
3-C
9 cycloalkylalkyl, –OR
B1, –N(R
B1)
2, –C(=O)R
B1, –C(=O)OR
B1, –NR
B1C(=O)R
B1, – NR
B1C(=O)OR
B1, –C(=O)N(R
B1)
2, –OC(=O)N(R
B1)
2, –S(=O)R
B1, –S(=O)
2R
B1, –SR
B1, – S(=O)(=NR
B1)R
B1, –NR
B1S(=O)
2R
B1 and –S(=O)
2N(R
B1)
2, wherein each alkyl, alkenyl, cycloalkyl, heterocyclyl and cycloalkylalkyl of R
B can be partially or fully deuterated and can be substituted at any available position with 0, 1 or 2 substituents selected from –F, –Cl, – OH– and –CH
3, and wherein R
B1 is as defined in any of the embodiments described herein. [0217] In an embodiment, R
B is selected from halo (e.g., –F, –Cl, –Br), –CN, –OH, –C
1–C
6 alkyl including partially or fully deuterated alkyl (e.g., –Me, –CD
3, –Et, –Pr, –
iPr, –
nBu, –
tBu), –C
1–C
6 hydroxyalkyl (e.g.¸–CH
2OH), –OC
1–C
6 alkyl (e.g., –OMe, –OEt, –OPr, –O
iPr, –O
nBu, –O
tBu), –COO(C
1-C
6 alkyl) (e.g., –COOEt), –CONH
2. [0218] In an embodiment, each R
B is independently selected from the group consisting of halo, –CN, –C
1-C
6 alkyl including partially or fully deuterated alkyl, –C
1-C
6 haloalkyl, –C
1- C
6 hydroxyalkyl, C
3-C
10 cycloalkyl, OR
B1, –C(O)(NR
B1)
2 and –C(O)ONR
B1, wherein each R
B1 is as defined in any of the embodiments described herein. [0219] In an embodiment, each R
B is independently selected from the group consisting of halo, –C
1-C
6 alkyl including partially or fully deuterated alkyl, –C
1-C
6 alkenyl, –C
1-C
6 haloalkyl, -heteroC
1-C
4 alkyl, –C
3-C
10 cycloalkyl (substituted with 0, 1 or 2 instances of F), 4-7 member heterocyclyl (substituted with 0 or 1 instances of methyl), C
3-C
10 cycloalkylalkyl and OR
B1, wherein each R
B1 is as defined in any of the embodiments described herein. [0220] In an embodiment, each R
B is independently selected from the group consisting of – Cl, –CH
3, –CD
3,–Et, –
iPr, –CH=CHCH
3, –CH
2CF
3,–CH(CH
3)CF
3, –CF
3, –CHF
2, –OCH
3, – OEt, cyclopropyl, 2,2-difluorocyclopropyl, 1-Me-azetidin-3-yl, oxetan-3-yl, tetrahydropyran- 4-yl, –CH
2CH
2OCH
3, –OCH(CH
3)CH
2OCH
3, –CH
2-cyclopropyl. [0221] In an embodiment, each R
B is independently selected from the group consisting of – Cl, –CH
3, –CD
3, –Et, –
iPr , –CF
3, –OCH
3 and cyclopropyl. [0222] In an embodiment, each R
B is independently selected from the group consisting of – Cl, –CH
3, –Et, –
iPr , –CF
3, –OCH
3 and cyclopropyl. [0223] In an embodiment, each R
B is independently selected from the group consisting of – CH
3, –Et, –
iPr, –CF
3 and cyclopropyl. [0224] In an embodiment, R
B is selected from the group consisting of –Me, –Et, –
iPr, and cyclopropyl. In an embodiment, R
B is selected from the group consisting of –Me, –Et, and cyclopropyl. [0225] In an embodiment, R
B is halo (e.g., fluoro, chloro, bromo, iodo). In an embodiment, R
B is –Cl. In an embodiment, R
B is –F. In an embodiment, R
B is –Br. In an embodiment, R
B is –I. [0226] In an embodiment, R
B is –CN. [0227] In an embodiment, R
B is –C
1–C
6 alkyl including partially or fully deuterated alkyl (e.g., –Me, –CD
3, –Et, –Pr, –
iPr, –
nBu, –sec-Bu, –iso-Bu, –
tBu). In an embodiment, R
B is – Me. In an embodiment, R
B is –CD
3. In an embodiment, R
B is –Et. In an embodiment R
B is – Pr. In an embodiment, R
B is –iPr. [0228] In an embodiment, R
B is –heteroC
1-C
4 alkyl. In an embodiment, R
B is methoxymethyl (–CH
2OCH
3). In an embodiment R
B is –CH
2CH
2OMe. In an embodiment, R
B is aminomethyl (e.g., –CH
2NH
2, –CH
2NHCH
3, –CH
2N(CH
3)
2. In an embodiment, R
B is – CH
2N(CH
3)CH
2CH
3. [0229] In an embodiment, R
B is –C
1–C
6 haloalkyl (e.g., –CF
3, –CHF
2, –CH
2CH
2F, – CH
2CHF
2, –CH
2CF
3, –CF
2CH
3). In an embodiment, R
B is trifluoromethyl (–CF
3). In other embodiments, R
B is difluoromethyl (–CHF
2). In an embodiment, R
B is –CH
2CH
2F. In other embodiments, R
B is –CH
2CHF
2, [0230] In an embodiment, R
B is –C
1–C
6 hydroxyalkyl (e.g., –CH
2OH, –CH
2CH
2OH). In an embodiment, R
B is hydroxyethyl (–CH
2CH
2OH). [0231] In an embodiment, R
B is –C
3–C
10 cycloalkyl (e.g., cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl). In an embodiment, R
B is cyclopropyl. In an embodiment R
B is cyclobutyl. In an embodiment, R
B is cyclopentyl. In an embodiment, R
B is cyclohexyl. [0232] In an embodiment, R
B is 3-10 membered heterocyclyl (e.g., oxetanyl, tetrahydropyranyl, tetrahydrofuranyl, azetidinyl, pyrrolidinyl, piperidinyl, piperazinyl, morpholinyl, azepanyl, 6-oxa-1-azaspiro[3.3]heptanyl, 6-oxa-1-azaspiro[3.4]octanyl). In an embodiment, R
B is oxetanyl. In an embodiment, R
B is tetrahydropyranyl. In an embodiment, R
B is tetrahydrofuranyl. In an embodiment, R
B is azetidinyl (e.g., N-methyl azetidin-3-yl). In an embodiment, R
B is pyrrolidinyl. In an embodiment, R
B is piperidinyl. In an embodiment, R
B is piperazinyl. In an embodiment, R
B is morpholinyl. In an embodiment, R
B is azepanyl. In an embodiment, R
B is 6-oxa-1-azaspiro[3.3]heptanyl. In an embodiment, R
B is 6-oxa-1- azaspiro[3.4]octanyl. [0233] In an embodiment, R
B is –OR
B1 wherein each R
B1 is as defined in any of the embodiments described herein (e.g., hydroxy (–OH), methoxy, difluoromethoxy (–OCHF
2), trifluoromethoxy (–OCF
3), –OCH(CH
3)CF
3, –OCH
2CF
3, ethoxy, propoxy, isopropoxy, cyclopropyloxy, cyclobutyloxy, ). In an embodiment, R
B is hydroxy. In an embodiment, R
B is methoxy. In an embodiment, R
B is ethoxy. In an embodiment, R
B is propoxy. In an embodiment, R
B is isopropoxy. In an embodiment R
B is difluoromethoxy (–OCHF
2). In an embodiment, R
B is trifluoromethoxy (–OCF
3). In an embodiment, R
B is –OCH(CH
3)CF
3. In an embodiment, R
B is –OCH
2CF
3. In an embodiment, R
B is cyclopropyloxy. [0234] In an embodiment, R
B is –N(R
B1)
2 wherein each R
B1 is as defined in any of the embodiments described herein (e.g., –NH
2, –NHR
B1, –N(CH
3)R
B1). In an embodiment, R
B is –NH
2. In an embodiment, R
B is –NHR
B1 (e.g., –NHMe, –NHEt, –NHPr, –NH
iPr, – NHcyclopropyl, –NHcyclobutyl). In an embodiment, R
B is –N(CH
3)R
B1 (e.g., –NMe
2, – N(CH
3)Et, –N(CH
3)Pr, –N(CH
3)
iPr, –N(CH
3)cyclopropyl, –N(CH
3)cyclobutyl). [0235] In an embodiment, R
B is –C(=O)R
B1 or –C(=O)OR
B1 wherein each R
B1 is as defined in any of the embodiments described herein. In an embodiment, R
B is –C(=O)R
B1 wherein R
B1 is as described herein. In an embodiment, R
B is –C(=O)alkyl. In an embodiment, R
B is – C(O)CH
3, –C(O)cyclopropyl, –C(O)cyclobutyl, –C(O)
tBu, –C(O)
iPr, –C(O)Pr, –C(O)
iBu, or –C(=O)OMe. In an embodiment, R
B is acetyl (–C(=O)Me). In an embodiment, R
B is – C(=O)OR
B1. In an embodiment, R
B is –COOH. In an embodiment, R
B is COOMe. [0236] In an embodiment, R
B is –NR
B1C(=O)R
B1 wherein each R
B1 is as defined in any of the embodiments described herein. In an embodiment, R
B is –NHC(=O)R
B1 (e.g., –NHC(=O)Me, –NHC(=O)Et, –NHC(=O)Pr, –NHC(=O)
iPr, –NHC(=O)Bu, –NHC(=O)
tBu, – NHC(=O)Cyclopropyl, –NHC(=O)Cyclobutyl). In an embodiment, R
B is –N(CH
3)C(=O)R
B1 (e.g., –N(CH
3)C(=O)Me, –N(CH
3)C(=O)Et, –N(CH
3)C(=O)Pr, –N(CH
3)C(=O)
iPr, – N(CH
3)C(=O)Bu, –N(CH
3)C(=O)
tBu, –N(CH
3)C(=O)Cyclopropyl, – N(CH
3)C(=O)Cyclobutyl). [0237] In an embodiment, R
B is –NR
B1C(=O)OR
B1 wherein each R
B1 is as defined in any of the embodiments described herein. In an embodiment, R
B is –NHC(=O)OR
B1 (e.g., – NHC(=O)OMe, –NHC(=O)OEt, –NHC(=O)OPr, –NHC(=O)O
iPr, –NHC(=O)OBu, – NHC(=O)O
tBu, –NHC(=O)OCyclopropyl, –NHC(=O)OCyclobutyl). In an embodiment, R
B is –N(CH
3)C(=O)OR
B1 (e.g., –N(CH
3)C(=O)OMe, –N(CH
3)C(=O)OEt, –N(CH
3)C(=O)OPr, –N(CH
3)C(=O)O
iPr, –N(CH
3)C(=O)OBu, –N(CH
3)C(=O)O
tBu, – N(CH
3)C(=O)OCyclopropyl, –N(CH
3)C(=O)OCyclobutyl). [0238] In an embodiment, R
B is –C(=O)N(R
B1)
2 wherein each R
B1 is as defined in any of the embodiments described herein (e.g., –C(=O)NH
2, –C(=O)NHR
B1, –C(=O)N(CH
3)R
B1). In an embodiment, R
B is –C(=O)NH
2. In an embodiment, R
B is –C(=O)NHR
B1 (e.g., – C(=O)NHMe, –C(=O)NHEt, –C(=O)NHPr, –C(=O)NH
iPr, –C(=O)NHBu, –C(=O)NH
tBu, – C(=O)NHCyclopropyl, –C(=O)NHCyclobutyl). In an embodiment, R
B is –C(=O)N(CH
3)R
B1 (e.g., –C(=O)NMe
2, –C(=O)N(CH
3)Et, –C(=O)N(CH
3)Pr, –C(=O)N(CH
3)
iPr, – C(=O)N(CH
3)Bu, –C(=O)N(CH
3)
tBu, –C(=O)N(CH
3)Cyclopropyl, – C(=O)N(CH
3)Cyclobutyl). [0239] In an embodiment, R
B is –OC(=O)N(R
B1)
2 wherein each R
B1 is as defined in any of the embodiments described herein. In an embodiment, R
B is –OC(=O)NHR
B1 (e.g., – OC(=O)NHMe, –OC(=O)NHEt, –OC(=O)NHPr, –OC(=O)NH
iPr, –OC(=O)NHBu, – OC(=O)NH
tBu, –OC(=O)NHCyclopropyl, –OC(=O)NHCyclobutyl). In an embodiment, R
B is –OC(=O)N(CH
3)R
B1 (e.g., –OC(=O)NMe
2, –OC(=O)N(CH
3)Et, –OC(=O)N(CH
3)Pr, – OC(=O)N(CH
3)
iPr, –OC(=O)N(CH
3)Bu, –OC(=O)N(CH
3)
tBu, – OC(=O)N(CH
3)Cyclopropyl, –OC(=O)N(CH
3)Cyclobutyl). [0240] In an embodiment, R
B is -S(=O)R
B1 wherein each R
B1 is as defined in any of the embodiments described herein. In an embodiment, R
B is –S(=O)alkyl (e.g., –S(=O)Me, – S(=O)Et, –S(=O)Pr, –S(=O)
iPr). In an embodiment, R
B is –S(=O)cycloalkyl (e.g., – S(=O)cyclopropyl, –S(=O)cyclobutyl, –S(=O)cyclopentyl, –S(=O)cyclohexyl). [0241] In an embodiment, R
B is -S(=O)
2R
B1 wherein each R
B1 is as defined in any of the embodiments described herein. In an embodiment, R
B is –S(=O)
2alkyl (e.g., –S(=O)
2Me, – S(=O)
2Et, –S(=O)
2Pr, –S(=O)
2iPr). In an embodiment, R
B is –S(=O)
2cycloalkyl (e.g., – S(=O)
2cyclopropyl, –S(=O)
2cyclobutyl, –S(=O)
2cyclopentyl, –S(=O)
2cyclohexyl). In an embodiment, R
B is S(=O)
2aryl (e.g., S(=O)
2phenyl). [0242] In an embodiment, R
B is –SR
B1 wherein each R
B1 is as defined in any of the embodiments described herein. In an embodiment, R
B is –Salkyl (e.g., –SMe, –SEt, –SPr, – S
iPr). In an embodiment, R
B is –Scycloalkyl (e.g., –Scyclopropyl, –Scyclobutyl, – Scyclopentyl, –Scyclohexyl). In an embodiment, R
B is –Saryl (e.g., Sphenyl). [0243] In an embodiment, R
B is -S(=O)(=NR
B1)R
B1 wherein each R
B1 is as defined in any of the embodiments described herein. In an embodiment, R
B is –S(=O)(=NH)R
B1 (e.g., – S(=O)(=NH)Me, –S(=O)(=NH)Et, –S(=O)(=NH)Pr, –S(=O)(=NH)
iPr, –S(=O)(=NH)Bu, – S(=O)(=NH)
tBu, –S(=O)(=NH)Cyclopropyl, –S(=O)(=NH)Cyclobutyl). In an embodiment, R
B is –S(=O)(=NCH
3)R
B1 (e.g., –S(=O)(=NCH
3)Me, –S(=O)(=NCH
3)Et, –S(=O)(=NCH
3)Pr, –S(=O)(=NCH
3)
iPr, –S(=O)(=NCH
3)Bu, –S(=O)(=NCH
3)
tBu, –S(=O)(=NCH
3)Cyclopropyl, –S(=O)(=NCH
3)Cyclobutyl). [0244] In an embodiment, R
B is –NR
B1S(=O)
2R
B1. In an embodiment, R
B is –NHS(=O)2alkyl (e.g., –NHS(=O)
2Me, –NHS(=O)
2Et, –NHS(=O)
2Pr, –NHS(=O)
2iPr). In an embodiment, R
B is –NHS(=O)
2cycloalkyl (e.g., –NHS(=O)
2cyclopropyl, –NHS(=O)
2cyclobutyl, – NHS(=O)
2cyclopentyl, –NHS(=O)
2cyclohexyl). In an embodiment, R
B is – N(CH
3)S(=O)
2alkyl (e.g., –N(CH
3)S(=O)
2Me, –N(CH
3)S(=O)
2Et, –N(CH
3)S(=O)
2Pr, – N(CH
3)S(=O)
2iPr). In an embodiment, R
B is –N(CH
3)S(=O)
2cycloalkyl (e.g., – N(CH
3)S(=O)
2cyclopropyl, –N(CH
3)S(=O)
2cyclobutyl, –N(CH
3)S(=O)
2cyclopentyl, – N(CH
3)S(=O)
2cyclohexyl). [0245] In an embodiment, R
B is -S(=O)
2N(R
B1)
2 wherein each R
B1 is as defined in any of the embodiments described herein (e.g., –S(=O)
2NH
2, –S(=O)
2NHR
B1, –S(=O)
2N(CH
3)R
B1). In an embodiment, R
B is -S(=O)
2NH
2. In an embodiment, R
B is -S(=O)
2NHR
B1 (e.g., – S(=O)
2NHMe, –S(=O)
2NHEt, –S(=O)
2NHPr, –S(=O)
2NH
iPr, –S(=O)
2NHcyclopropyl, – S(=O)
2NHcyclobutyl). In an embodiment, R
B is -S(=O)
2N(CH
3)R
B1 (e.g., –S(=O)
2NMe
2, – S(=O)
2N(CH
3)Et, –S(=O)
2N(CH
3)Pr, –S(=O)
2N(CH
3)
iPr, –S(=O)
2N(CH
3)cyclopropyl, – S(=O)
2N(CH
3)cyclobutyl). [0246] As generally defined herein, each R
B1 is independently selected from the group consisting of H, –C
1–C
6 alkyl, –heteroC
1-C
4 alkyl, –C
1–C
6 haloalkyl, –C
3–C
9 cycloalkyl and 3-7 membered heterocyclyl wherein each hydrogen atom of each alkyl, cycloalkyl and heterocyclyl can be replaced by deuterium; or two instances of R
B1 on a nitrogen atom can be taken together with the nitrogen to which they are attached to form a 4-6 member monocyclic heterocycle containing one nitrogen atom and 0-1 additional heteroatoms selected from N and O. [0247] In an embodiment, each R
B1 is independently selected from the group consisting of H and –C
1–C
6 alkyl. [0248] In an embodiment, each R
B1 is independently selected from the group consisting of H, –Me and –Et. [0249] In an embodiment, each R
B1 is independently H. [0250] In an embodiment, each R
B1 is independently –C
1–C
6 alkyl (e.g., –Me, –Et, –Pr, –
iPr, –
nBu, –
tBu, –sec-Bu, –iso-Bu). In an embodiment, each R
B1 is independently –Me. In an embodiment, each R
B1 is independently –Et. In an embodiment, each R
B1 is independently – Pr. In an embodiment, each R
B1 is independently –
iPr. [0251] In an embodiment, each R
B1 is independently –heteroC
1-C
4 alkyl. In an embodiment, each R
B1 is independently methoxymethyl (–CH
2OCH
3). In an embodiment, each R
B1 is independently hydroxymethyl (–CH
2OH). In an embodiment, each R
B1 is independently aminomethyl (e.g., –CH
2NH
2, –CH
2NHCH
3, –CH
2N(CH
3)
2. [0252] In an embodiment, each R
B1 is independently –C
1–C
6 haloalkyl. In an embodiment, each R
B1 is independently trifluoromethyl (–CF
3). In other embodiments, each R
B1 is independently difluoromethyl (–CHF
2). In an embodiment, each R
B1 is –CH(CH
3)CF
3. In an embodiment, each R
B1 is –CH
2CF
3. [0253] In an embodiment, each R
B1 is independently –C
3–C
9 cycloalkyl (e.g., cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl). In an embodiment, each R
B1 is independently cyclopropyl. In an embodiment each R
B1 is independently cyclobutyl. In an embodiment, each R
B1 is independently cyclopentyl. In an embodiment, each R
B1 is independently cyclohexyl. [0254] In an embodiment, each R
B1 is independently 3-7 membered heterocyclyl (e.g., oxetanyl, tetrahydropyranyl, tetrahydrofuranyl, azetidinyl, pyrrolidinyl, piperidinyl, piperazinyl, morpholinyl, azepanyl). [0255] As generally defined herein, each R
C is independently selected from the group consisting of H, –D, oxo, halo, –CN, –OR
C1, –SR
C1 –NR
C1 2, –C
1–C
6 alkyl, –C
1–C
6 hydroxyalkyl, –C
1–C
6 haloalkyl, –C
3–C
9 cycloalkyl, 3-10 member heterocyclyl and – heteroC
1-C
4 alkyl wherein each alkyl, cycloalkyl and heterocyclyl of R
C can be partially or fully deuterated and can be substituted at any available position with 0, 1 or 2 substituents selected from –F, –Cl, –OH– and –CH
3, wherein each R
C1 is as defined in any of the embodiments described herein. [0256] In an embodiment, each R
C is independently selected from the group consisting of H, –OR
C1, –SR
C1, –C
1-C
6 alkyl including partially or fully deuterated alkyl, –C
1-C
6 haloalkyl – C
1-C
6 hydroxyalkyl and C
3-C
9 cycloalkyl. [0257] In an embodiment, each R
C is independently selected from the group consisting of H, –C
1-C
3 alkyl including partially or fully deuterated alkyl and –C
1-C
3 hydroxyalkyl. [0258] In an embodiment, each R
C is independently selected from the group consisting of H, –CH
3, –Et, –
iPr, –CF
3, CH
2OH, –CH(OH)CH
3, –OMe, –SMe and cyclopropyl. [0259] In an embodiment, each R
C is independently selected from the group consisting of H, –CH
3, CH
2OH and –CH(OH)CH
3. [0260] In an embodiment, each R
C is –CH
3. [0261] In an embodiment, R
C is –CH(OH)CH
3. [0262] In an embodiment, R
C is –CH
2OH. [0263] In an embodiment, R
C is H. [0264] As generally defined herein, each R
C1 is independently selected from the group consisting of H and –C
1–C
6 alkyl, wherein each hydrogen atom of the alkyl can be independently replaced by deuterium; or two instances of R
C1 on a nitrogen atom can be taken together with the nitrogen to which they are attached to form a 4-6 member monocyclic heterocycle containing one nitrogen atom and 0-1 additional heteroatoms selected from N and O. In an embodiment, R
C1 is H. In an embodiment, R
C1 is –Me. [0265] As generally defined herein, each R
D is independently selected from consisting of –D, halo, –CN, –C
1–C
6 alkyl, –heteroC
1-C
4 alkyl, –C
1–C
6 haloalkyl, –C
1–C
6 hydroxyalkyl, –C
3– C
10 cycloalkyl, 3-10 membered heterocyclyl, –C
6-C
10 aryl, –OR
D1, –N(R
D1)
2, –C(=O)R
D1, – C(=O)OR
D1, –NR
D1C(=O)R
D1, –NR
D1C(=O)OR
D1, –C(=O)N(R
D1)
2, –OC(=O)N(R
D1)
2, – S(=O)R
D1, –S(=O)
2R
D1, –SR
D1, –S(=O)(=NR
D1)R
D1, –NR
D1S(=O)
2R
D1 and –S(=O)
2N(R
D1)
2 , wherein each alkyl, cycloalkyl, heterocyclyl and aryl of R
D can be substituted at any available position with 0, 1 or 2 substituents selected from –F, –Cl, –OH– and –CH
3 and wherein each R
D1 is as defined in any of the embodiments described herein. [0266] In an embodiment, each R
D is independently selected from the group consisting of – C
1-C
6 alkyl, OR
D1 and C
3-C
10 cycloalkyl. [0267] In an embodiment, each R
C1 is independently selected from the group consisting of H, –CH
3, –Et and –CD
3. [0268] In an embodiment, each R
D is independently selected from the group consisting of – OR
D1 and C
3-C
10 cycloalkyl wherein each R
D1 is as defined in any of the embodiments described herein. [0269] In an embodiment, each R
D is independently selected from the group consisting of –n- Pr, –
iPr, –OCH
3, –OCD
3, –OEt, –OCHF
2, –OCH
2F and cyclopropyl. [0270] In an embodiment, each R
D is independently selected from the group consisting of – OCH
3, –OCD
3, –OEt, –OCHF
2, –OCH
2F and cyclopropyl. [0271] In an embodiment, each R
D is independently selected from the group consisting of – OCH
3, –OCD
3, –OCHF
2 and cyclopropyl. [0272] In an embodiment, each R
D is independently selected from the group consisting of – OCH
3 and cyclopropyl. [0273] In an embodiment R
D is –D. [0274] In an embodiment, R
D is halo (e.g., fluoro, chloro, bromo, iodo). In an embodiment, R
D is –Cl. In an embodiment, R
D is –F. In an embodiment, R
D is –Br. In an embodiment, R
D is –I. [0275] In an embodiment, R
D is –CN. [0276] In an embodiment, R
D is –C
1–C
6 alkyl (e.g., –Me, –Et, –Pr, –
iPr, –
nBu, –sec-Bu, –iso- Bu, –
tBu). In an embodiment, R
D is –Me. In an embodiment, R
D is –Et. In an embodiment R
D is –Pr. In an embodiment, R
D is –iPr. [0277] In an embodiment, R
D is –heteroC
1-C
4 alkyl. In an embodiment, R
D is methoxymethyl (–CH
2OCH
3). In an embodiment, R
D is aminomethyl (e.g., –CH
2NHCH
2CH
3, – CH
2N(CH
3)CH
2CF
3, –CH
2N(CH
3)CH
2CH
3, –CH
2N(CH
3)
2–CH
2NH
2, –CH
2NHCH
3, – CH
2N(CH
3)
2). [0278] In an embodiment, R
D is –C
1–C
6 haloalkyl (e.g., –CF
3, –CHF
2, –CF
2CH
3, –CH
2CF
3). In an embodiment, R
D is trifluoromethyl (–CF
3). In other embodiments, R
D is difluoromethyl (–CHF
2). [0279] In an embodiment, R
D is –C
1–C
6 hydroxyalkyl (e.g., –CH
2OH, –CH
2CH
2OH, – CH(OH)CH
3, –C(OH)(CH
3)
2). In an embodiment, R
D is hydroxymethyl (–CH
2OH). In an embodiment, R
D is –CH(OH)CH
3. In an embodiment, R
D is –C(OH)(CH
3)
2. [0280] In an embodiment, R
D is –C
3–C
10 cycloalkyl (e.g., cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl), each of which can be optionally substituted. In an embodiment, R
D is optionally substituted cyclopropyl (e.g., cyclopropyl substituted with 0 or 1 instances of – CN). In an embodiment R
D is cyclobutyl. In an embodiment, R
D is cyclopentyl. In an embodiment, R
D is cyclohexyl. [0281] In an embodiment, R
D is 3-10 membered heterocyclyl (e.g., oxetanyl, tetrahydropyranyl, tetrahydrofuranyl, azetidinyl, pyrrolidinyl, piperidinyl, piperazinyl, morpholinyl, azepanyl, 6-oxa-1-azaspiro[3.3]heptanyl, 6-oxa-1-azaspiro[3.4]octanyl), each of which can be optionally substituted. In an embodiment, R
D is oxetanyl. In an embodiment, R
D is tetrahydropyranyl. In an embodiment, R
D is tetrahydrofuranyl. In an embodiment, R
D is azetidinyl (e.g., azetidinyl substituted with 0 or 1 instances of halo or methyl). In an embodiment, R
D is pyrrolidinyl. In an embodiment, R
D is piperidinyl. In an embodiment, R
D is piperazinyl. In an embodiment, R
D is morpholinyl. In an embodiment, R
D is azepanyl. [0282] In an embodiment, R
D is –C
6-C
10 aryl (e.g., phenyl, naphthyl). In an embodiment, R
D is substituted phenyl (e.g., phenyl substituted with 0 or 1 instances of halo (e.g., –Cl, –F)). In an embodiment R
D is 2-chloro-phenyl. [0283] In an embodiment, R
D is –OR
D1 wherein each R
D1 is as defined in any of the embodiments described herein (e.g., hydroxy (–OH), methoxy, difluoromethoxy (–OCHF
2), trifluoromethoxy (–OCF
3), –OCH(CH
3)CF
3, –OCH
2CF
3, ethoxy, propoxy, isopropoxy, – OCH
2CH(CH
3)
3, cyclopropyloxy, cyclobutyloxy). In an embodiment, R
D is hydroxy. In an embodiment, R
D is methoxy. In an embodiment, R
D is ethoxy. In an embodiment, R
D is propoxy. In an embodiment, R
D is isopropoxy. In an embodiment R
D is fluoromethoxy (– OCH
2F).In an embodiment R
D is difluoromethoxy (–OCHF
2). In an embodiment, R
D is trifluoromethoxy (–OCF
3). In an embodiment, R
D is –OCH(CH
3)CF
3. In an embodiment, R
D is –OCH
2CF
3. In an embodiment, R
D is cyclopropyloxy. [0284] In an embodiment, R
D is –N(R
D1)
2 wherein each R
D1 is as defined in any of the embodiments described herein (e.g., –NH
2, –NHR
D1, –N(CH
3)R
D1). In an embodiment, R
D is –NH
2. In an embodiment, R
D is –NHR
D1 (e.g., –NHMe, –NHEt, –NHPr, –NH
iPr, – NHcyclopropyl, –NHcyclobutyl). In an embodiment, R
D is –N(CH
3)R
D1 (e.g., –NMe
2, – N(CH
3)Et, –N(CH
3)Pr, –N(CH
3)
iPr, –N(CH
3)cyclopropyl, –N(CH
3)cyclobutyl). [0285] In an embodiment, R
D is –C(=O)R
D1 or –C(=O)OR
D1 wherein each R
D1 is as defined in any of the embodiments described herein. In an embodiment, R
D is –C(=O)R
D1 wherein each R
D1 is as defined in any of the embodiments described herein. In an embodiment, R
D is –C(=O)alkyl. In an embodiment, R
D is –C(O)CH
3, –C(O)cyclopropyl, –C(O)cyclobutyl, – C(O)
tBu, –C(O)
iPr, –C(O)Pr, –C(O)
iBu, or –C(=O)OMe. In an embodiment, R
D is acetyl (– C(=O)Me). In an embodiment, R
D is –C(=O)OR
D1. In an embodiment, R
D is –COOH. In an embodiment, R
D is COOMe. [0286] In an embodiment, R
D is –NR
D1C(=O)R
D1 wherein each R
D1 is as defined in any of the embodiments described herein. In an embodiment, R
D is –NHC(=O)R
D1 (e.g., –NH, – NHC(=O)Me, –NH, –NHC(=O)Et, –NH, –NHC(=O)Pr, –NH, –NHC(=O)
iPr, –NH, – NHC(=O)Bu, –NH, –NHC(=O)
tBu, –NH, –NHC(=O)Cyclopropyl, –NH, – NHC(=O)Cyclobutyl). In an embodiment, R
D is –N(CH
3)C(=O)R
D1 (e.g., –N(CH
3)C(=O)Me, –N(CH
3)C(=O)Et, –N(CH
3)C(=O)Pr, –N(CH
3)C(=O)
iPr, –N(CH
3)C(=O)Bu, – N(CH
3)C(=O)
tBu, –N(CH
3)C(=O)Cyclopropyl, –N(CH
3)C(=O)Cyclobutyl). [0287] In an embodiment, R
D is –NR
D1C(=O)OR
D1 wherein each R
D1 is as defined in any of the embodiments described herein. In an embodiment, R
D is –NHC(=O)OR
D1 (e.g., –NH, – NHC(=O)OMe, –NH, –NHC(=O)OEt, –NH, –NHC(=O)OPr, –NH, –NHC(=O)O
iPr, –NH, – NHC(=O)OBu, –NH, –NHC(=O)O
tBu, –NH, –NHC(=O)OCyclopropyl, –NH, – NHC(=O)OCyclobutyl). In an embodiment, R
D is –N(CH
3)C(=O)OR
D1 (e.g., – N(CH
3)C(=O)OMe, –N(CH
3)C(=O)OEt, –N(CH
3)C(=O)OPr, –N(CH
3)C(=O)O
iPr, – N(CH
3)C(=O)OBu, –N(CH
3)C(=O)O
tBu, –N(CH
3)C(=O)OCyclopropyl, – N(CH
3)C(=O)OCyclobutyl). [0288] In an embodiment, R
D is –C(=O)N(R
D1)
2 wherein each R
D1 is as defined in any of the embodiments described herein (e.g., –C(=O)NH
2, –C(=O)NHR
D1, –C(=O)N(CH
3)R
D1). In an embodiment, R
D is –C(=O)NH
2. In an embodiment, R
D is –C(=O)NHR
D1 (e.g., – C(=O)NHMe, –C(=O)NHEt, –C(=O)NHPr, –C(=O)NH
iPr, –C(=O)NHBu, –C(=O)NH
tBu, – C(=O)NHCyclopropyl, –C(=O)NHCyclobutyl). In an embodiment, R
D is –C(=O)N(CH
3)R
D1 (e.g., –C(=O)NMe
2, –C(=O)N(CH
3)Et, –C(=O)N(CH
3)Pr, –C(=O)N(CH
3)
iPr, – C(=O)N(CH
3)Bu, –C(=O)N(CH
3)
tBu, –C(=O)N(CH
3)Cyclopropyl, – C(=O)N(CH
3)Cyclobutyl). [0289] In an embodiment, R
D is –OC(=O)N(R
D1)
2 wherein each R
D1 is as defined in any of the embodiments described herein. In an embodiment, R
D is –OC(=O)NHR
D1 (e.g., – OC(=O)NHMe, –OC(=O)NHEt, –OC(=O)NHPr, –OC(=O)NH
iPr, –OC(=O)NHBu, – OC(=O)NH
tBu, –OC(=O)NHCyclopropyl, –OC(=O)NHCyclobutyl). In an embodiment, R
D is –OC(=O)N(CH
3)R
D1 (e.g., –OC(=O)NMe
2, –OC(=O)N(CH
3)Et, –OC(=O)N(CH
3)Pr, – OC(=O)N(CH
3)
iPr, –OC(=O)N(CH
3)Bu, –OC(=O)N(CH
3)
tBu, – OC(=O)N(CH
3)Cyclopropyl, –OC(=O)N(CH
3)Cyclobutyl). [0290] In an embodiment, R
D is -S(=O)R
D1 wherein each R
D1 is as defined in any of the embodiments described herein. In an embodiment, R
D is –S(=O)alkyl (e.g., –S(=O)Me, – S(=O)Et, –S(=O)Pr, –S(=O)
iPr). In an embodiment, R
D is –S(=O)cycloalkyl (e.g., – S(=O)cyclopropyl, –S(=O)cyclobutyl, –S(=O)cyclopentyl, –S(=O)cyclohexyl). [0291] In an embodiment, R
D is -S(=O)
2R
D1 wherein each R
D1 is as defined in any of the embodiments described herein. In an embodiment, R
D is –S(=O)
2alkyl (e.g., –S(=O)
2Me, – S(=O)
2Et, –S(=O)
2Pr, –S(=O)
2iPr). In an embodiment, R
D is –S(=O)
2cycloalkyl (e.g., – S(=O)
2cyclopropyl, –S(=O)
2cyclobutyl, –S(=O)
2cyclopentyl, –S(=O)
2cyclohexyl). In an embodiment, R
D is S(=O)
2aryl (e.g., S(=O)
2phenyl). [0292] In an embodiment, R
D is –SR
D1 wherein each R
D1 is as defined in any of the embodiments described herein. In an embodiment, R
D is –Salkyl (e.g., –SMe, –SEt, –SPr, – S
iPr). In an embodiment, R
D is –Scycloalkyl (e.g., –Scyclopropyl, –Scyclobutyl, – Scyclopentyl, –Scyclohexyl). In an embodiment, R
D is –Saryl (e.g., Sphenyl). [0293] In an embodiment, R
D is -S(=O)(=NR
D1)R
D1 wherein each R
D1 is as defined in any of the embodiments described herein. In an embodiment, R
D is –S(=O)(=NH)R
D1 (e.g., – S(=O)(=NH)Me, –S(=O)(=NH)Et, –S(=O)(=NH)Pr, –S(=O)(=NH)
iPr, –S(=O)(=NH)Bu, – S(=O)(=NH)
tBu, –S(=O)(=NH)Cyclopropyl, –S(=O)(=NH)Cyclobutyl). In an embodiment, R
D is –S(=O)(=NCH
3)R
D1 (e.g., –S(=O)(=NCH
3)Me, –S(=O)(=NCH
3)Et, –S(=O)(=NCH
3)Pr, –S(=O)(=NCH
3)
iPr, –S(=O)(=NCH
3)Bu, –S(=O)(=NCH
3)
tBu, –S(=O)(=NCH
3)Cyclopropyl, –S(=O)(=NCH
3)Cyclobutyl). [0294] In an embodiment, R
D is –NR
D1S(=O)
2R
D1 wherein each R
D1 is as defined in any of the embodiments described herein. In an embodiment, R
D is –NHS(=O)
2alkyl (e.g., – NHS(=O)
2Me, –NHS(=O)
2Et, –NHS(=O)
2Pr, –NHS(=O)
2iPr). In an embodiment, R
D is – NHS(=O)
2cycloalkyl (e.g., –NHS(=O)
2cyclopropyl, –NHS(=O)
2cyclobutyl, – NHS(=O)
2cyclopentyl, –NHS(=O)
2cyclohexyl). In an embodiment, R
D is – N(CH
3)S(=O)
2alkyl (e.g., –N(CH
3)S(=O)
2Me, –N(CH
3)S(=O)
2Et, –N(CH
3)S(=O)
2Pr, – N(CH
3)S(=O)
2iPr). In an embodiment, R
D is –N(CH
3)S(=O)
2cycloalkyl (e.g., – N(CH
3)S(=O)
2cyclopropyl, –N(CH
3)S(=O)
2cyclobutyl, –N(CH
3)S(=O)
2cyclopentyl, – N(CH
3)S(=O)
2cyclohexyl). [0295] In an embodiment, R
D is -S(=O)
2N(R
D1)
2 wherein each R
D1 is as defined in any of the embodiments described herein (e.g., –S(=O)
2NH
2, –S(=O)
2NHR
D1, –S(=O)
2N(CH
3)R
D1). In an embodiment, R
D is -S(=O)
2NH
2. In an embodiment, R
D is -S(=O)
2NHR
D1 (e.g., – S(=O)
2NHMe, –S(=O)
2NHEt, –S(=O)
2NHPr, –S(=O)
2NH
iPr, –S(=O)
2NHcyclopropyl, – S(=O)
2NHcyclobutyl). In an embodiment, R
D is -S(=O)
2N(CH
3)R
D1 (e.g., –S(=O)
2NMe
2, – S(=O)
2N(CH
3)Et, –S(=O)
2N(CH
3)Pr, –S(=O)
2N(CH
3)
iPr, –S(=O)
2N(CH
3)cyclopropyl, – S(=O)
2N(CH
3)cyclobutyl). [0296] As generally defined herein, each R
D1 is independently selected from the group consisting of H, –C
1–C
6 alkyl, –heteroC
1-C
4 alkyl, –C
1–C
6 haloalkyl, –C
3–C
9 cycloalkyl and 3-7 membered heterocyclyl, wherein each hydrogen atom of each alkyl, cycloalkyl and heterocyclyl can be replaced by deuterium; or two instances of R
D1 on a nitrogen atom can be taken together with the nitrogen to which they are attached to form a 4-6 member monocyclic heterocycle containing one nitrogen atom and 0-1 additional heteroatoms selected from N and O. [0297] In an embodiment, each R
D1 is independently selected from H, –C
1–C
6 alkyl (e.g., – Me, –Et, –Pr, –
iPr, –
nBu, –sec-Bu, –iso-Bu, –
tBu), –C
1–C
6 haloalkyl (e.g., –CF
3, –CHF
2, – CH
2CF
3, –CH(CH
3)CF
3) and C
3–C
9 cycloalkyl (e.g., cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl) wherein each hydrogen atom of the alkyl can be independently replaced by deuterium. [0298] In an embodiment, each R
D1 is independently selected from the group consisting of H, –C
1-C
6 alkyl and –C
1-C
6 haloalkyl wherein each hydrogen atom of the alkyl can be independently replaced with deuterium. [0299] In an embodiment, each R
D1 is independently selected from the group consisting of H, –C
1-C
3 alkyl and –C
1-C
3 haloalkyl wherein each hydrogen atom of the alkyl can be independently replaced with deuterium. [0300] In an embodiment, each R
D1 is independently selected from the group consisting of – C
1-C
3 alkyl and –C
1-C
3 haloalkyl wherein each hydrogen atom of the alkyl can be independently replaced with deuterium. [0301] In an embodiment, each R
D1 is independently –C
1-C
3 alkyl wherein each hydrogen atom of the alkyl can be independently replaced with deuterium. [0302] In an embodiment, each R
D1 is independently selected from the group consisting of – CH
3,–Et, –CD
3, –CH
2F and –CHF
2. [0303] In an embodiment, each R
D1 is independently selected from the group consisting of – CH
3, –CD
3 and –CHF
2. [0304] In an embodiment, each R
D1 is independently selected from the group consisting of – CH
3 and –CD
3. [0305] In an embodiment, each R
D1 is independently H. [0306] In an embodiment, each R
D1 is independently –C
1–C
6 alkyl (e.g., –Me, –Et, –Pr, –
iPr, –
nBu, –
tBu, –sec-Bu, –iso-Bu) wherein each hydrogen atom of the alkyl can be independently replaced with deuterium. In an embodiment, each R
D1 is independently –Me. In an embodiment, each R
D1 is independently –CD
3. In an embodiment, each R
D1 is independently –Et. In an embodiment, each R
D1 is independently –Pr. In an embodiment, each R
D1 is independently –
iPr. [0307] In an embodiment, each R
D1 is independently –heteroC
1-C
4 alkyl. In an embodiment, each R
D1 is independently methoxymethyl (–CH
2OCH
3). In an embodiment, each R
D1 is independently aminomethyl (e.g., –CH
2NH
2, –CH
2NHCH
3, –CH
2N(CH
3)
2. [0308] In an embodiment, each R
D1 is independently –C
1–C
6 haloalkyl. In an embodiment, each R
D1 is independently trifluoromethyl (–CF
3). In other embodiments, each R
D1 is independently difluoromethyl (–CHF
2). In other embodiments, each R
D1 is independently fluoromethyl (–CH
2F). In an embodiment, each R
D1 is –CH(CH
3)CF
3. In an embodiment, each R
D1 is –CH
2CF
3. [0309] In an embodiment, each R
D1 is independently –C
3–C
9 cycloalkyl (e.g., cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl). In an embodiment, each R
D1 is independently cyclopropyl. In an embodiment each R
D1 is independently cyclobutyl. In an embodiment, each R
D1 is independently cyclopentyl. In an embodiment, each R
D1 is independently cyclohexyl. [0310] In an embodiment, each R
D1 is independently 3-7 membered heterocyclyl (e.g., oxetanyl, tetrahydropyranyl, tetrahydrofuranyl, azetidinyl, pyrrolidinyl, piperidinyl, piperazinyl, morpholinyl, azepanyl). [0311] As generally defined herein, each R
E is selected from H, –D, halo, –CN, –C
1–C
6 alkyl, –C
1-C
6 alkynyl, –heteroC
1–C
6 alkyl, –C
1–C
6 haloalkyl, –C
1–C
6 hydroxyalkyl, –C
3–C
10 cycloalkyl, 3-10 membered heterocyclyl, –C
6-C
10 aryl, 5-10 member heteroaryl, heterocyclylalkyl, heteroarylalkyl, arylalkyl, cycloalkylalkyl, –OR
E1, –SR
E1, –N(R
E1)
2, –C
1– C
6 alkylene–NR
E1C(=O)OR
E1, –C(=O)R
E1, –C(=O)OR
E1, –NR
E1C(=O)R
E1, – NR
E1C(=O)OR
E1, –C(=O)N(R
E1)
2, and –OC(=O)N(R
E1)
2, wherein each alkyl, alkynyl, heteroalkyl, haloalkyl, cycloalkyl, heterocyclyl, aryl, heteroaryl, cycloalkylalkyl, heterocyclylalkyl, arylalkyl and heteroarylalkyl is optionally substituted at any available position and wherein R
E1 is as defined in any of the embodiments described herein. [0312] In an embodiment, R
E is selected from H, –D, halo, –CN, –C
1–C
6 alkyl, –C
1-C
6 alkynyl, –heteroC
1–C
4 alkyl, –C
1–C
6 haloalkyl, –C
1–C
6 hydroxyalkyl, –C
3–C
10 cycloalkyl, 3- 10 membered heterocyclyl, –C
6-C
10 aryl, 5-10 member heteroaryl, heterocyclylalkyl, heteroarylalkyl, arylalkyl, cycloalkylalkyl, –OR
E1, –SR
E1, –N(R
E1)
2, –C
1–C
6 alkylene– NR
E1C(=O)OR
E1, –C(=O)R
E1, –C(=O)OR
E1, –NR
E1C(=O)R
E1, –NR
E1C(=O)OR
E1, – C(=O)N(R
E1)
2, and –OC(=O)N(R
E1)
2, wherein each alkyl, alkynyl, cycloalkyl, heterocyclyl, aryl, heteroaryl, cycloalkylalkyl, heterocyclylalkyl, arylalkyl and heteroarylalkyl is substituted at any available position with 0, 1 or 2 substituents independently selected from – F, –Cl–, –Me, –Et, –CN, –OH, –OMe –NCH
3, –NH
2 and –CF
3, and wherein R
E1 is as defined in any of the embodiments described herein. [0313] In an embodiment, R
E is selected from H, –D, halo, –C
1–C
6 alkyl, –heteroC
1–C
4alkyl, –C
1–C
6 haloalkyl, –C
1–C
6 hydroxyalkyl, –C
3–C
10 cycloalkyl, 3-10 membered heterocyclyl, 5-10 member heteroaryl, heterocyclylalkyl, –OR
E1, –SR
E1, –N(R
E1)
2, and –C
1–C
6 alkylene– NR
E1C(=O)OR
E1, wherein each alkyl, haloalkyl, heteroalkyl, cycloalkyl, heterocyclyl, aryl, heteroaryl and heterocyclylalkyl is substituted at any available position with 0 or 1 substituents independently selected from –F, –Cl–, –Me, –Et, –CN, –OH, –OMe –NCH
3, – NH
2 and –CF
3, and wherein R
E1 is as defined in any of the embodiments described herein
. [0314] In an embodiment, R
E is selected from H, halo, –CN, –C
1–C
6 alkyl, –C
1–C
6 haloalkyl, –C
1–C
6 hydroxyalkyl, –C
3–C
10 cycloalkyl, 3-10 membered heterocyclyl, 5-10 member heteroaryl, –OR
E1, –SR
E1, –N(R
E1)
2, –C
1–C
6 alkylene–NR
E1C(=O)OR
E1 and –C(=O)N(R
E1)
2 wherein each alkyl, haloalkyl, cycloalkyl, heterocyclyl and heteroaryl is substituted at any available position with 0 or 1 substituents independently selected from –CN, –OH, –NCH
3, – NH
2, –CF
3 and –F, and wherein R
E1 is as defined in any of the embodiments described herein
. [0315] In an embodiment, R
E is selected from H, –D, halo, –C
1–C
6 alkyl (substituted with 0 or 1 instances of –CN), –C
1–C
6 haloalkyl (substituted with 0 or 1 instances of OH, –NH
2 or – NHCH
3), heteroC
1–C
4alkyl, –C
1–C
6 hydroxyalkyl (substituted with 0 or 1 instances of –CF
3), –C
1–C
6 alkylene–NHC(=O)O(C
1-C
6alkyl), –C
3–C
10 cycloalkyl (substituted with 0 or 1 instances of –CF
3), 3-10 member heterocyclyl (substituted with 0 or 1 instances of –F), 5-10 member heteroaryl, –CH
2-heterocyclyl, –NH
2, –NH(C
1-C
6 alkyl), –N(C
1-C
6 alkyl)
2, – NH(heteroC
1–C
4alkyl), –OH, –S(C
1–C
6 alkyl), –Ocycloalkylalkyl (substituted with 0 or 1 instances of –CF
3), -Oarylalkyl, –O(C
1–C
6 haloalkyl) and –O(C
1–C
6 alkyl) (wherein each hydrogen can be independently replaced by deuterium). [0316] In an embodiment, R
E is selected from H, –D, –F, –Cl, –Me, –Et, –
iPr, –
tBu, –CHF
2, – CH
2CF
3, –CH
2CHF
2, –CF
3, –CH
2OMe, –CH(CF
3)NH
2, –CH(CF
3)NHCH
3, –CH(OH)CF
3, – C(OH)(CH
3)
2, –CH(OH)CH
3, –CH
2CN
, –C(CN)(CH
3)
2, –CH
2CH
2CH
2NHC(=O)OC(CH
3)
3, cyclopropyl, 1-CF
3-cyclopropyl, cyclobutyl, cyclopentyl, 3-F-oxetan-3-yl, tetrahydrofuran-3-
–O-tetrahydrofuran-3-yl, –OCH
2CH(NH
2)CF
3, –OCF
3, –OCH
2CHF
2, –OCH
2F, –OCHF
2, – OCH
2CF
3, –OMe, –OCD
3, –OEt and –O
iPr. [0317] In an embodiment, R
E is selected from H, –D, –CN, –F, –Cl, –Me, –Et, –Pr, –
iPr, –
nBu, –
tBu, –CF
3, –CHF
2, –C(=O)NH
2, –OH and –OMe. [0318] In an embodiment, R
E is selected from H, –F, –Cl, –Me –OCD
3 and –OMe . [0319] In an embodiment, R
E is selected from H, –Me, –OCD
3 and –OMe . [0320] In an embodiment, R
E is selected from H, –Me and –OMe. [0321] In an embodiment, R
E is H. In an embodiment, R
E is –D. [0322] In an embodiment, R
E is halo (e.g., fluoro, chloro, bromo, iodo). In an embodiment, R
E is –Cl. In an embodiment, R
E is –F. In an embodiment, R
E is –Br. In an embodiment, R
E is –I. [0323] In an embodiment, R
E is –CN. [0324] In an embodiment, R
E is –C
1–C
6 alkyl, optionally substituted (e.g., –Me, –Et, –Pr, –
iPr, –
nBu, –sec-Bu, –iso-Bu, –
tBu, -CH
2CN, –C(CN)(CH
3)
2). In an embodiment, R
E is –Me. In an embodiment, R
E is –Et. In an embodiment R
E is –Pr. In an embodiment, R
E is –
iPr. In an embodiment, R
E is –
tBu. In an embodiment, R
E is -CH
2CN. In an embodiment, R
E is – C(CN)(CH
3)
2. [0325] In an embodiment, R
E is –heteroC
1–C
6 alkyl. In an embodiment, R
E is methoxymethyl (–CH
2OCH
3). In an embodiment R
E is –CH
2CH
2OMe. In an embodiment, R
E is aminomethyl (e.g., –CH
2NH
2, –CH
2NHCH
3, –CH
2N(CH
3)
2. In an embodiment, R
E is – CH
2N(CH
3)CH
2CH
3. [0326] In an embodiment, R
E is –C
1–C
6 haloalkyl (e.g., optionally substituted, e.g., substituted at any available position with 0, 1 or 2 instances of –OH) (e.g., –CF
3, – CH(OH)CF
3, –CH(CF
3)NH
2, –CH(CF
3)NHCH
3, –CHF
2, –CH
2CH
2F, –CH
2CHF
2, –CH
2CF
3, –CF
2CH
3). In an embodiment, R
E is trifluoromethyl (–CF
3). In other embodiments, R
E is difluoromethyl (–CHF
2). In an embodiment, R
E is –CH
2CH
2F. In other embodiments, R
E is – CH
2CHF
2. In an embodiment, R
E is –CH(OH)CF
3. [0327] In an embodiment, R
E is –C
1–C
6 hydroxyalkyl, optionally substituted (e.g., with 0 or 1 instances of CF
3) (e.g., –CH
2OH, –CH
2CH
2OH, –C(OH)(CH
3)
2, –CH(OH)CH
3, – CH(OH)CF
3). In an embodiment, R
E is hydroxymethyl (–CH
2OH,). In an embodiment, R
E is hydroxyethyl (–CH
2CH
2OH). In an embodiment, R
E is –C(OH)(CH
3)
2. In an embodiment, R
E is –CH(OH)CH
3. In an embodiment, R
E is –CH(OH)CF
3. [0328] In an embodiment, R
E is optionally substituted –C
3–C
10 cycloalkyl (e.g., cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl) (e.g., substituted with 0-1 instances of -CF
3). In an embodiment, R
E is cyclopropyl. In an embodiment, R
E is 1-CF
3-cyclopropyl. In an embodiment R
E is cyclobutyl. In an embodiment, R
E is cyclopentyl. In an embodiment, R
E is cyclohexyl. [0329] In an embodiment, R
E is optionally substituted 3-10 membered heterocyclyl (e.g., oxetanyl, tetrahydropyranyl, tetrahydrofuranyl, azetidinyl, pyrrolidinyl, dihydropyrrolyl, piperidinyl, piperazinyl, morpholinyl, azepanyl, 6-oxa-1-azaspiro[3.3]heptanyl, 6-oxa-1- azaspiro[3.4]octanyl)(e.g., substituted with 0 or 1 instances of halo (e.g., –F) or –Me). In an embodiment, R
E is oxetanyl (e.g, oxetan-3-yl, 3-F-oxetan-3-yl). In an embodiment, R
E is tetrahydropyranyl. In an embodiment, R
E is tetrahydrofuranyl (e.g., tetrahydrofuran-3-yl). In an embodiment, R
E is azetidinyl (e.g., N-methyl azetidin-3-yl). In an embodiment, R
E is pyrrolidinyl (e.g.,
E . In an embodiment, R is dihydropyrrolyl (e.g.
In an embodiment, R
E is piperidinyl. In an embodiment, R
E is piperazinyl. In an embodiment, R
E is morpholinyl. In an embodiment, R
E is azepanyl. In an embodiment, R
E is 6-oxa-1- azaspiro[3.3]heptanyl. In an embodiment, R
E is 6-oxa-1-azaspiro[3.4]octanyl. [0330] In an embodiment, R
E is independently a 5-10 member heteroaryl (e.g., a 5-6 member monocyclic heteroaryl or an 8-10 member bicyclic heteroaryl containing 1-3 heteroatoms independently selected from N, O and S). In an embodiment, R
E is independently a 5-6 member monocyclic heteroaryl (e.g., a 5-member monocyclic heteroaryl containing 1-3 heteroatoms independently selected from O, N and S, a 6-member monocyclic heteroaryl containing 1-3 N heteroatoms). In an embodiment, R
E is independently a 5-member monocyclic heteroaryl (e.g., pyrazolyl, pyrolyl, thiophenyl, furyl, thiazolyl, isothiazolyl, oxazolyl, isoxazolyl, imidazolyl, triazolyl, thiadiazolyl, oxadiazolyl). In an embodiment, R
E is independently thiophenyl (e.g., thiophen-2-yl, thiophen-3-yl). In an embodiment, R
E is independently pyrolyl (e.g., pyrol-2-yl, pyrol-3-yl). In an embodiment, R
E is independently pyrazolyl (e.g. pyrazol-1-yl, pyrazol-3-yl, pyrazol-4-yl, pyrazol-5-yl). In an embodiment, R
E is independently oxazolyl (e.g., oxazol-2-yl, oxazol-4-yl, oxazol-5-yl). In an embodiment, R
E is independently thiazolyl (e.g., thiazol-2-yl, thiazol-4-yl, thiazol-5-yl). In an embodiment, R
E is independently isothiazolyl (e.g., isothiazol-3-yl, isothiazol-4-yl, isothiazol-5-yl). In an embodiment, R
E is independently a 6-member monocyclic heteroaryl (e.g., pyridyl, pyrimidinyl, triazinyl, pyrazinyl, pyridazinyl). In an embodiment, R
E is independently pyridinyl (e.g., pyridin-2-yl, pyridin-3-yl, pyridin-4-yl). In an embodiment, R
E is independently pyrimidinyl (e.g, pyrimidin-2-yl, pyrimidin-4-yl, pyrimidin-5-yl). In an embodiment, R
E is independently pyrazinyl. [0331] In an embodiment R
E is cycloalkylalkyl (e.g., cyclopropylmethyl, cyclobutylmethyl, cyclopentylmethyl, cyclohexylmethyl, cycloheptylmethyl). In an embodiment, R
E is heterocyclylalkyl (e.g., oxetanylmethyl, aziridinylmethyl, tetrahydrofuranylmethyl, pyrrolidinylmethyl, tetrahydropyranylmethyl, piperidinylmethyl, piperazinylmethyl, morpholinylmethyl, azepanylmethyl). [0332] In an embodiment, R
E is arylalkyl. In an embodiment, R
E is benzyl. [0333] In an embodiment, R
E is heteroarylalkyl (e.g., pyridinylmethyl, thiazolylmethyl, triazolylmethyl, pyrazolylmethyl). [0334] In an embodiment, R
E is –OR
E1 (e.g., hydroxy (–OH), methoxy, –OCD
3, difluoromethoxy (–OCHF
2), trifluoromethoxy (–OCF
3), –OCH(CH
3)CF
3, –OCH
2CF
3, - OCH
2F, OCH
2CHF
2, ethoxy, propoxy, isopropoxy, cyclopropyloxy, cyclobutyloxy, benzyloxy,
, –O-oxetan-3-yl, –O-tetrahydrofuran-3-yl, –OCH
2CH(NH
2)CF
3). In an embodiment, R
E is hydroxy. In an embodiment, R
E is methoxy. In an embodiment, R
E is ethoxy. In an embodiment, R
E is propoxy. In an embodiment, R
E is isopropoxy. In an embodiment R
E is fluoromethoxy (–OCH
2F).In an embodiment R
E is difluoromethoxy (– OCHF
2). In an embodiment, R
E is trifluoromethoxy (–OCF
3). In an embodiment, R
E is – OCH(CH
3)CF
3. In an embodiment, R
E is –OCH
2CF
3. In an embodiment, R
E is cyclopropyloxy. In an embodiment, R
E is
. In an embodiment, R
E is –OCD
3. In an embodiment, R
E is OCH
2F. In an embodiment, R
E is OCH
2CHF
2. In an embodiment, R
E is benzyloxy. In an embodiment, R
E is –O-oxetan-3-yl. In an embodiment, R
E is –O- tetrahydrofuran-3-yl. In an embodiment, R
E is –OCH
2CH(NH
2)CF
3). [0335] In an embodiment, R
E is –SR
E1 (e.g., –SMe). [0336] In an embodiment, R
E is –N(R
E1)
2 (e.g., –NH
2, –NHR
E1, –N(CH
3)R
E1). In an embodiment, R
E is –NH
2. In an embodiment, R
E is –NHR
E1 (e.g., –NHMe, –NHEt, –NHPr, – NH
iPr, –NHcyclopropyl, –NHcyclobutyl
, –NHCH
2CH
2OCH
3, –NHCH
2CH
2CH
2OCH
3–,
embodiment, R
E is –N(CH
3)R
E1 (e.g., –NMe
2, –N(CH
3)Et, –N(CH
3)Pr, – N(CH
3)
iPr, –N(CH
3)cyclopropyl, –N(CH
3)cyclobutyl). [0337] In an embodiment, R
E is –C(=O)R
E1 or –C(=O)OR
E1. In an embodiment, R
E is – C(=O)R
E1 wherein R
E1 is as described herein. In an embodiment, R
E is –C(=O)alkyl. In an embodiment, R
E is –C(O)CH
3, –C(O)cyclopropyl, –C(O)cyclobutyl, –C(O)
tBu, –C(O)
iPr, – C(O)Pr, –C(O)
iBu, or –C(=O)OMe. In an embodiment, R
E is acetyl (–C(=O)Me). In an embodiment, R
E is –C(=O)OR
E1. In an embodiment, R
E is –COOH. In an embodiment, R
E is COOMe. [0338] In an embodiment, R
E is –NR
E1C(=O)R
E1. In an embodiment, R
E is –NHC(=O)R
E1 (e.g., –NHC(=O)Me, –NHC(=O)Et, –NHC(=O)Pr, –NHC(=O)
iPr, –NHC(=O)Bu, – NHC(=O)
tBu, –NHC(=O)Cyclopropyl, –NHC(=O)Cyclobutyl). In an embodiment, R
E is – N(CH
3)C(=O)R
E1 (e.g., –N(CH
3)C(=O)Me, –N(CH
3)C(=O)Et, –N(CH
3)C(=O)Pr, – N(CH
3)C(=O)
iPr, –N(CH
3)C(=O)Bu, –N(CH
3)C(=O)
tBu, –N(CH
3)C(=O)Cyclopropyl, – N(CH
3)C(=O)Cyclobutyl). [0339] In an embodiment, R
E is –NR
E1C(=O)OR
E1. In an embodiment, R
E is –NHC(=O)OR
E1 (e.g., –NHC(=O)OMe, –NHC(=O)OEt, –NHC(=O)OPr, –NHC(=O)O
iPr, –NHC(=O)OBu, – NHC(=O)O
tBu, –NHC(=O)OCyclopropyl, –NHC(=O)OCyclobutyl). In an embodiment, R
E is –N(CH
3)C(=O)OR
E1 (e.g., –N(CH
3)C(=O)OMe, –N(CH
3)C(=O)OEt, –N(CH
3)C(=O)OPr, –N(CH
3)C(=O)O
iPr, –N(CH
3)C(=O)OBu, –N(CH
3)C(=O)O
tBu, – N(CH
3)C(=O)OCyclopropyl, –N(CH
3)C(=O)OCyclobutyl). [0340] In an embodiment, R
E is –C(=O)N(R
E1)
2 (e.g., –C(=O)NH
2, –C(=O)NHR
E1, – C(=O)N(CH
3)R
E1). In an embodiment, R
E is –C(=O)NH
2. In an embodiment, R
E is – C(=O)NHR
E1 (e.g., –C(=O)NHMe, –C(=O)NHEt, –C(=O)NHPr, –C(=O)NH
iPr, – C(=O)NHBu, –C(=O)NH
tBu, –C(=O)NHCyclopropyl, –C(=O)NHCyclobutyl). In an embodiment, R
E is –C(=O)N(CH
3)R
E1 (e.g., –C(=O)NMe
2, –C(=O)N(CH
3)Et, – C(=O)N(CH
3)Pr, –C(=O)N(CH
3)
iPr, –C(=O)N(CH
3)Bu, –C(=O)N(CH
3)
tBu, – C(=O)N(CH
3)Cyclopropyl, –C(=O)N(CH
3)Cyclobutyl). [0341] In an embodiment, R
E is –OC(=O)N(R
E1)
2. In an embodiment, R
E is –OC(=O)NHR
E1 (e.g., –OC(=O)NHMe, –OC(=O)NHEt, –OC(=O)NHPr, –OC(=O)NH
iPr, –OC(=O)NHBu, – OC(=O)NH
tBu, –OC(=O)NHCyclopropyl, –OC(=O)NHCyclobutyl). In an embodiment, R
E is –OC(=O)N(CH
3)R
E1 (e.g., –OC(=O)NMe
2, –OC(=O)N(CH
3)Et, –OC(=O)N(CH
3)Pr, – OC(=O)N(CH
3)
iPr, –OC(=O)N(CH
3)Bu, –OC(=O)N(CH
3)
tBu, – OC(=O)N(CH
3)Cyclopropyl, –OC(=O)N(CH
3)Cyclobutyl). [0342] –In an embodiment, R
E is –C
1–C
6 alkylene–NR
E1C(=O)OR
E1 (e.g., –C
1–C
6 alkylene– NHC(=O)O(C
1-C
6alkyl)). In an embodiment, R
E is –CH
2–NHC(=O)O(C
1-C
6alkyl) (e.g., – CH
2NHC(=O)OC(CH
3)
3). In an embodiment, R
E is –CH
2CH
2–NHC(=O)O(C
1-C
6alkyl) (e.g., –CH
2CH
2NHC(=O)OC(CH
3)
3). In an embodiment, R
E is –CH
2CH
2CH
2–NHC(=O)O(C
1- C
6alkyl) (e.g., –CH
2CH
2CH
2NHC(=O)OC(CH
3)
3) [0343] As generally defined herein, each R
E1 is independently selected from independently selected from H, –C
1–C
6 alkyl (wherein each hydrogen can be independently replaced by deuterium), –heteroC
1–C
6 alkyl, –C
1–C
6 haloalkyl, –C
3–C
9 cycloalkyl, 3-7 membered heterocyclyl, cycloalkylalkyl, heterocyclylalkyl, aryl, 5-6 membered heteroaryl, arylalkyl and heteroarylalkyl wherein each alkyl, haloalkyl, cycloalkyl, heterocyclyl, aryl, heteroaryl, cycloalkylalkyl, heterocyclylalkyl, arylalkyl and heteroarylalkyl is optionally substituted at any available position. [0344] In an embodiment, each R
E1 is independently selected from H, –C
1–C
6 alkyl (wherein each hydrogen can be independently replaced by deuterium), –heteroC
1–C
6 alkyl, –C
1–C
6 haloalkyl, –C
3–C
9 cycloalkyl, 3-7 membered heterocyclyl, cycloalkylalkyl, heterocyclylalkyl, aryl, 5-6 membered heteroaryl, arylalkyl and heteroarylalkyl wherein each alkyl, haloalkyl, cycloalkyl, heterocyclyl, aryl, heteroaryl, cycloalkylalkyl, heterocyclylalkyl, arylalkyl and heteroarylalkyl is substituted at any available position with 0, 1 or 2 substituents independently selected from –F, –Cl–, –Me, –Et, –CN, –OH, –OMe –NCH
3, –NH
2 and –CF
3. [0345] . In an embodiment, each R
E1 is independently substituted with 0 or 1 instance of CF
3. [0346] In an embodiment, each R
E1 is independently selected from –C
1–C
6 alkyl (wherein each hydrogen can be independently replaced with deuterium), –C
1–C
6 haloalkyl, –heteroC
1- C
4alkyl, –C
3-C
9 cycloalkyl, 3-7 membered heterocyclyl, cycloalkylalkyl, heterocyclylalkyl and arylalkyl, wherein each alkyl, haloalkyl, heteroalkyl, cycloalkyl, heterocyclyl, cycloalkylalkyl and arylalkyl is independently substituted at any available position with 0 or 1 substituents independently selected from –NH
2 and –CF
3. [0347] In an embodiment, each R
E1 is independently selected from –C
1–C
6 alkyl (wherein each hydrogen can be independently replaced with deuterium)(e.g., –Me, –CD
3, –Et, –
iPr), – C
1–C
6 haloalkyl (e.g., –CH
2CHF
2, –CH
2F, –CHF
2, –CH
2CF
3), –heteroC
1–C
6 alkyl (e.g., – CH
2CH
2OCH
3, –CH
2CH
2CH
2OCH
3), benzyl and CH
2Cyclopropyl (substituted with 0 or 1 instance of –CF
3). [0348] In an embodiment, each R
E1 is independently selected from H, –C
1–C
6 alkyl (e.g., – Me, –Et, –Pr, –
iPr, –
nBu, –
tBu, –sec-Bu, –iso-Bu) and –C
1–C
6 haloalkyl (e.g., –CHF
2, –CF
3, – CH(CH
3)CF
3, –CH
2CF
3). [0349] In an embodiment, each R
E1 is independently selected from the group consisting of – Me, –CD
3, –Et, –
iPr, CH
2CHF
2, –CF
3, –CHF
2, –CH
2F, Bn, cyclopropyl, oxetan-3-yl, tetrahydrofuran-3-yl, CH
2-1-CF
3-cyclopropyl, CH
2CH(NH
2)CF
3,
,– CH
2CH
2CH
2OMe and –CH
2CH
2OMe. [0350] In an embodiment, each R
E1 is independently selected from –Me, –CD
3, –Et, –
iPr, CH
2CHF
2, –CH
2F, –CHF
2, –CH
2CF
3, –CH
2CH
2OCH
3, –CH
2CH
2CH
2OCH
3, benzyl, – F
3C CH
2Cyclopropyl and (substituted with 0 or 1 instance of –CF
3). [0351] In an embodiment, each R
E1 is independently selected from –Me, –CD
3, –Et, –
iPr, CH
2CHF
2, –CH
2F, –CHF
2 and –CH
2CF
3. [0352] In an embodiment, each R
E1 is independently H. [0353] In an embodiment, each R
E1 is independently –C
1–C
6 alkyl (wherein each hydrogen can be independently replaced with deuterium)(e.g., –Me, –CD3, –Et, –Pr, –
iPr, –
nBu, –
tBu, – sec-Bu, –iso-Bu). In an embodiment, each R
E1 is independently –Me. In an embodiment each R
E1 is independently –CD
3. In an embodiment, each R
E1 is independently –Et. In an embodiment, each R
E1 is independently –Pr. In an embodiment, each R
E1 is independently –
iPr. [0354] In an embodiment, each R
E1 is independently –heteroC
1–C
6 alkyl (e.g., –CH
2OH, – CH
2OCH
3, –CH
2CH
2OCH
3, –CH
2CH
2CH
2OCH
3. In an embodiment, each R
E1 is independently methoxymethyl (–CH
2OCH
3). In an embodiment, each R
E1 is independently hydroxymethyl (–CH
2OH). In an embodiment, each R
E1 is independently –CH
2CH
2OCH
3. In an embodiment, each R
E1 is independently –CH
2CH
2CH
2OCH
3. In an embodiment, each R
E1 is independently aminomethyl (e.g., –CH
2NH
2, –CH
2NHCH
3, –CH
2N(CH
3)
2. [0355] In an embodiment, each R
E1 is independently –C
1–C
6 haloalkyl (e.g., –CF
3, – CH
2CHF
2, –CH
2F, –CHF
2, –CH
2CF
3), optionally substituted with 0-1 instances of NH
2. In an embodiment, each R
E1 is independently trifluoromethyl (–CF
3). In other embodiments, each R
E1 is independently difluoromethyl (–CHF
2). In an embodiment, each R
E1 is independently – CH
2CHF
2. In an embodiment, each R
E1 is independently –CH
2F. In an embodiment, each R
E1 is –CH(CH
3)CF
3. In an embodiment, each R
E1 is –CH
2CF
3. In an embodiment, each R
E1 is CH
2CH(NH
2)CF
3. [0356] In an embodiment, each R
E1 is independently –C
3–C
9 cycloalkyl (e.g., cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl) optionally substituted (e.g., substituted with 0, 1 or 2 instances of halo (e.g., –F, –Cl), –CN, –Me, –Et–, –
iPr, –OH, –OMe, –NH
2, –CF
3). In an embodiment, each R
E1 is independently cyclopropyl. In an embodiment each R
E1 is independently cyclobutyl. In an embodiment, each R
E1 is independently cyclopentyl. In an embodiment, each R
E1 is independently cyclohexyl. [0357] In an embodiment, each R
E1 is independently 3-10 membered heterocyclyl (e.g., oxetanyl, tetrahydropyranyl, tetrahydrofuranyl, azetidinyl, pyrrolidinyl, piperidinyl, piperazinyl, morpholinyl, azepanyl) optionally substituted (e.g., substituted with 0, 1 or 2 instances of halo (e.g., –F, –Cl), –CN, –Me, –Et–, –
iPr, –OH, –OMe, –NH
2, –CF
3). In an embodiment, each R
E1 is independently oxetan-3-yl. In an embodiment, each R
E1 is independently tetrahydrofuran-3-yl. [0358] In an embodiment, R
E1 is independently heteroaryl, optionally substituted (e.g., substituted with 0, 1 or 2 instances of halo (e.g., –F, –Cl), –CN, –Me, –Et–, –
iPr, –OH, – OMe, –NH
2, –CF
3). In an embodiment, R
E1 is independently a 5-10 member heteroaryl (e.g., a 5-6 member monocyclic heteroaryl or an 8-10 member bicyclic heteroaryl containing 1-3 heteroatoms independently selected from N, O and S). In an embodiment, R
E1 is independently a 5-6 member monocyclic heteroaryl (e.g., a 5-member monocyclic heteroaryl containing 1-3 heteroatoms independently selected from O, N and S, a 6-member monocyclic heteroaryl containing 1-3 N heteroatoms). In an embodiment, R
E1 is independently a 5- member monocyclic heteroaryl (e.g., pyrazolyl, pyrolyl, thiophenyl, furyl, thiazolyl, isothiazolyl, oxazolyl, isoxazolyl, imidazolyl, triazolyl, thiadiazolyl, oxadiazolyl). In an embodiment, R
E1 is independently thiophenyl (e.g., thiophen-2-yl, thiophen-3-yl). In an embodiment, R
E1 is independently pyrazolyl (e.g. pyrazol-1-yl, pyrazol-3-yl, pyrazol-5-yl). In an embodiment, R
E1 is independently thiazolyl (e.g., thiazol-2-yl, thiazol-4-yl, thiazol-5-yl). In an embodiment, R
E1 is independently a 6-member monocyclic heteroaryl (e.g., pyridyl, pyrimidinyl, triazinyl, pyrazinyl, pyridazinyl). In an embodiment, R
E1 is independently pyridinyl (e.g., pyridin-2-yl, pyridin-3-yl, pyridin-4-yl). In an embodiment, R
E1 is independently pyrimidinyl (e.g, pyrimidin-2-yl, pyrimidin-4-yl, pyrimidin-5-yl). [0359] In an embodiment, R
E1 is independently aryl, optionally substituted (e.g., substituted with 0, 1 or 2 instances of halo (e.g., –F, –Cl), –CN, –Me, –Et–, –
iPr, –OH, –OMe, –NH
2, – CF
3). In an embodiment, R
E1 is independently 6-10 member mono or bicyclic aryl. In an embodiment, R
E1 is independently phenyl. [0360] In an embodiment each R
E1 is independently cycloalkylalkyl (e.g., cyclopropylmethyl, cyclobutylmethyl, cyclopentylmethyl, cyclohexylmethyl, cycloheptylmethyl), optionally substituted (e.g., substituted with 0, 1 or 2 instances of halo (e.g., –F, –Cl), –CN, –Me, –Et–, –
iPr, –OH, –OMe, –NH
2, –CF
3). In an embodiment, each R
E1 is independently substituted with 0 or 1 instance of CF
3. In an embodiment, each R
E1 is independently cyclopropylmethyl (–CH
2Cyclopropyl), optionally substituted (e.g., substituted with 0, 1 or 2 instances of halo (e.g., –F, –Cl), –CN, –Me, –Et–, –
iPr, –OH, –OMe, –NH
2, –CF
3). In an embodiment, each R
E1 is independently cyclopropylmethyl (–CH
2Cyclopropyl) substituted with 0 or 1 instance of CF
3 (–CH
2-1-CF
3-cyclopropyl). In an embodiment, each R
E1 is independently heterocyclylalkyl (e.g., oxetanylmethyl, aziridinylmethyl, tetrahydrofuranylmethyl, pyrrolidinylmethyl, tetrahydropyranylmethyl, piperidinylmethyl, piperazinylmethyl, morpholinylmethyl, azepanylmethyl, thiomorpholinmethyl, thiomorpholinethyl, thiomorpholindioxide ethyl
) optionally substituted (e.g., substituted with 0, 1 or 2 instances of halo (e.g., –F, –Cl), –CN, –Me, –Et–, –
iPr, –OH, –OMe, –NH
2, –CF
3). [0361] In an embodiment, each R
E1 is independently arylalkyl optionally substituted (e.g., substituted with 0, 1 or 2 instances of halo (e.g., –F, –Cl), –CN, –Me, –Et–, –
iPr, –OH, – OMe, –NH
2, –CF
3). In an embodiment, each R
E1 is independently benzyl. [0362] In an embodiment, each R
E1 is independently heteroarylalkyl (e.g., pyridinylmethyl, thiazolylmethyl, triazolylmethyl, pyrazolylmethyl), optionally substituted (e.g., substituted with 0, 1 or 2 instances of halo (e.g., –F, –Cl), –CN, –Me, –Et–, –
iPr, –OH, –OMe, –NH
2, – CF
3). [0363] As generally defined herein, each R
1 is independently selected from the group consisting of –D, halo, –CN, –C
1–C
6 alkyl, –heteroC
1-C
4 alkyl, –C
1–C
6 haloalkyl, –C
1–C
6 hydroxyalkyl, –C
3–C
10 cycloalkyl, 3-10 membered heterocyclyl, –C
6-C
10 aryl, –OR
a1, – N(R
a1)
2, –C(=O)R
a1, –C(=O)OR
a1, –NR
a1C(=O)R
a1, –NR
a1C(=O)OR
a1, –C(=O)N(R
a1)
2, – OC(=O)N(R
a1)
2, –S(=O)R
a1, –S(=O)
2R
a1, –SR
a1, –S(=O)(=NR
a1)R
a1, –NR
a1S(=O)
2R
a1 and – S(=O)
2N(R
a1)
2 , wherein each alkyl, cycloalkyl, heterocyclyl and aryl of R
1 can be substituted at any available position with 0, 1 or 2 substituents selected from –F, –Cl, –OH– and –CH
3 wherein each R
a1 is as defined in any of the embodiments described herein. [0364] In an embodiment, each R
1 is independently selected from halo, –C
1–C
6 alkyl, phenyl substituted with 0 to 1 instances of halo, –C
1–C
6 haloalkyl, –C
1–C
6 hydroxyalkyl and –C
3– C
10 cycloalkyl. [0365] In an embodiment, each R
1 is independently selected from Cl, –Me, –
iPr, –CF
3, – CHF
2, –CF
2CH
3, 2-chlorophenyl, cyclopropyl, –CH(OH)CH
3 and –C(CH
3)
2OH. [0366] In an embodiment, each R
1 is independently selected from –
iPr, –CF
3 and cyclopropyl. [0367] In an embodiment, each R
1 is independently selected from –
iPr and cyclopropyl. [0368] In an embodiment, R
1 is H. In an embodiment R
1 is –D. [0369] In an embodiment, R
1 is halo (e.g., fluoro, chloro, bromo, iodo). In an embodiment, R
1 is –Cl. In an embodiment, R
1 is –F. In an embodiment, R
1 is –Br. In an embodiment, R
1 is –I. [0370] In an embodiment, R
1 is –CN. [0371] In an embodiment, R
1 is –C
1–C
6 alkyl (e.g., –Me, –Et, –Pr, –
iPr, –
nBu, –sec-Bu, –iso- Bu, –
tBu, –C(CH
3)CH
2CH
3,). In an embodiment, R
1 is –Me. In an embodiment, R
1 is –Et. In an embodiment R
1 is –Pr. In an embodiment, R
1 is –iPr. In an embodiment, R
1 is - C(CH
3)CH
2CH
3. [0372] In an embodiment, R
1 is –heteroC
1-C
4 alkyl. In an embodiment, R
1 is methoxymethyl (–CH
2OCH
3). In an embodiment, R
1 is aminomethyl (e.g., –CH
2NH
2, –CH
2NHCH
3, – CH
2NHCH
2CH
3, –CH
2N(CH
3)
2). In an embodiment, R
1 is –CH
2N(CH
3)CH
2CH
3. In an embodiment, R
1 is –CH
2N(CH
3)CH
2CF
3. [0373] In an embodiment, R
1 is –C
1–C
6 haloalkyl (e.g., –CF
3, –CHF
2, –CF
2CH
3, –CH
2CF
3) . In an embodiment, R
1 is trifluoromethyl (–CF
3). In other embodiments, R
1 is difluoromethyl (–CHF
2). In other embodiments, R
1 is –CH
2CF
3. In an embodiment, R
1 is –CF
2CH
3. [0374] In an embodiment, R
1 is –C
1–C
6 hydroxyalkyl (e.g., –CH
2OH, –CH(OH)CH
3, – C(OH)(CH
3)
2). In an embodiment, R
1 is hydroxymethyl (–CH
2OH). [0375] In an embodiment, R
1 is –C
3–C
10 cycloalkyl (e.g., cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl). In an embodiment, R
1 is cyclopropyl (e.g., substituted with 0 or 1 instance of –CN). In an embodiment R
1 is cyclobutyl. In an embodiment, R
1 is cyclopentyl. In an embodiment, R
1 is cyclohexyl. [0376] In an embodiment, R
1 is an optionally substituted 3-10 membered heterocyclyl (e.g., oxetanyl, tetrahydropyranyl, tetrahydrofuranyl, azetidinyl, pyrrolidinyl, piperidinyl, piperazinyl, morpholinyl, azepanyl, 6-oxa-1-azaspiro[3.3]heptanyl, 6-oxa-1- azaspiro[3.4]octanyl). In an embodiment, R
1 is oxetanyl. In an embodiment, R
1 is tetrahydropyranyl. In an embodiment, R
1 is tetrahydrofuranyl. In an embodiment, R
1 is azetidinyl. In an embodiment, the azetidinyl is optionally substituted (e.g., substituted with 0 or 1 instances of –F or –Me). In an embodiment, R
1 is pyrrolidinyl. In an embodiment, R
1 is piperidinyl. In an embodiment, R
1 is piperazinyl. In an embodiment, R
1 is morpholinyl. In an embodiment, R
1 is azepanyl. In an embodiment, R
1 is 6-oxa-1-azaspiro[3.3]heptanyl. In an embodiment, R
1 is 6-oxa-1-azaspiro[3.4]octanyl. [0377] In an embodiment, R
1 is –C
6-C
10 aryl (e.g., phenyl, naphthyl). In an embodiment, R
1 is phenyl (e.g., phenyl substituted with 0 or 1 instances of halo (e.g., –Cl, –F)). In an embodiment, R
1 is -2-Cl-phenyl. [0378] In an embodiment, R
1 is –OR
a1 wherein each R
a1 is as defined in any of the embodiments described herein (e.g., hydroxy (–OH), methoxy, difluoromethoxy (–OCHF
2), trifluoromethoxy (–OCF
3), –OCH(CH
3)CF
3, –OCH
2CF
3, ethoxy, propoxy, isopropoxy, cyclopropyloxy, cyclobutyloxy, –OCH
2CH(CH
3)
3). In an embodiment, R
1 is hydroxy. In an embodiment, R
1 is methoxy. In an embodiment, R
1 is ethoxy. In an embodiment, R
1 is propoxy. In an embodiment, R
1 is isopropoxy. In an embodiment R
1 is difluoromethoxy (– OCHF
2). In an embodiment, R
1 is trifluoromethoxy (–OCF
3). In an embodiment, R
1 is – OCH(CH
3)CF
3. In an embodiment, R
1 is –OCH
2CF
3. In an embodiment, R
1 is cyclopropyloxy. In an embodiment R
1 is –OCH
2CH(CH
3)
3. [0379] In an embodiment, R
1 is –N(R
a1)
2 wherein each R
a1 is as defined in any of the embodiments described herein (e.g., –NH
2, –NHR
a1, –N(CH
3)R
a1). In an embodiment, R
1 is – NH
2. In an embodiment, R
1 is –NHR
a1 (e.g., –NHMe, –NHEt, –NHPr, –NH
iPr, – NHcyclopropyl, –NHcyclobutyl). In an embodiment, R
1 is –N(CH
3)R
a1 (e.g., –NMe
2, – N(CH
3)Et, –N(CH
3)Pr, –N(CH
3)
iPr, –N(CH
3)cyclopropyl, –N(CH
3)cyclobutyl). [0380] In an embodiment, R
1 is –C(=O)R
a1 or –C(=O)OR
a1 wherein each R
a1 is as defined in any of the embodiments described herein. In an embodiment, R
1 is –C(=O)R
a1 wherein R
a1 is as described herein. In an embodiment, R
1 is –C(=O)alkyl. In an embodiment, R
1 is – C(O)CH
3, –C(O)cyclopropyl, –C(O)cyclobutyl, –C(O)
tBu, –C(O)
iPr, –C(O)Pr, –C(O)
iBu, or –C(=O)OMe. In an embodiment, R
1 is acetyl (–C(=O)Me). In an embodiment, R
1 is – C(=O)OR
a1. In an embodiment, R
1 is –COOH. In an embodiment, R
1 is COOMe. [0381] In an embodiment, R
1 is –NR
a1C(=O)R
a1 wherein each R
a1 is as defined in any of the embodiments described herein. In an embodiment, R
1 is –NHC(=O)R
a1 (e.g., –NH, – NHC(=O)Me, –NH, –NHC(=O)Et, –NH, –NHC(=O)Pr, –NH, –NHC(=O)
iPr, –NH, – NHC(=O)Bu, –NH, –NHC(=O)
tBu, –NH, –NHC(=O)Cyclopropyl, –NH, – NHC(=O)Cyclobutyl). In an embodiment, R
1 is –N(CH
3)C(=O)R
a1 (e.g., –N(CH
3)C(=O)Me, –N(CH
3)C(=O)Et, –N(CH
3)C(=O)Pr, –N(CH
3)C(=O)
iPr, –N(CH
3)C(=O)Bu, – N(CH
3)C(=O)
tBu, –N(CH
3)C(=O)Cyclopropyl, –N(CH
3)C(=O)Cyclobutyl). [0382] In an embodiment, R
1 is –NR
a1C(=O)OR
a1 wherein each R
a1 is as defined in any of the embodiments described herein. In an embodiment, R
1 is –NHC(=O)OR
a1 (e.g., –NH, – NHC(=O)OMe, –NH, –NHC(=O)OEt, –NH, –NHC(=O)OPr, –NHC(=O)O
iPr, – NHC(=O)OBu, –NHC(=O)O
tBu, –NHC(=O)OCyclopropyl, –NHC(=O)OCyclobutyl). In an embodiment, R
1 is –N(CH
3)C(=O)OR
a1 (e.g., –N(CH
3)C(=O)OMe, –N(CH
3)C(=O)OEt, – N(CH
3)C(=O)OPr, –N(CH
3)C(=O)O
iPr, –N(CH
3)C(=O)OBu, –N(CH
3)C(=O)O
tBu, – N(CH
3)C(=O)OCyclopropyl, –N(CH
3)C(=O)OCyclobutyl). [0383] In an embodiment, R
1 is –C(=O)N(R
a1)
2 wherein each R
a1 is as defined in any of the embodiments described herein(e.g., –C(=O)NH
2, –C(=O)NHR
a1, –C(=O)N(CH
3)R
a1). In an embodiment, R
1 is –C(=O)NH
2. In an embodiment, R
1 is –C(=O)NHR
a1 (e.g., –C(=O)NHMe, –C(=O)NHEt, –C(=O)NHPr, –C(=O)NH
iPr, –C(=O)NHBu, –C(=O)NH
tBu, – C(=O)NHCyclopropyl, –C(=O)NHCyclobutyl). In an embodiment, R
1 is –C(=O)N(CH
3)R
a1 (e.g., –C(=O)NMe
2, –C(=O)N(CH
3)Et, –C(=O)N(CH
3)Pr, –C(=O)N(CH
3)
iPr, – C(=O)N(CH
3)Bu, –C(=O)N(CH
3)
tBu, –C(=O)N(CH
3)Cyclopropyl, – C(=O)N(CH
3)Cyclobutyl). [0384] In an embodiment, R
1 is –OC(=O)N(R
a1)
2 wherein each R
a1 is as defined in any of the embodiments described herein. In an embodiment, R
1 is –OC(=O)NHR
a1 (e.g., – OC(=O)NHMe, –OC(=O)NHEt, –OC(=O)NHPr, –OC(=O)NH
iPr, –OC(=O)NHBu, – OC(=O)NH
tBu, –OC(=O)NHCyclopropyl, –OC(=O)NHCyclobutyl). In an embodiment, R
1 is –OC(=O)N(CH
3)R
a1 (e.g., –OC(=O)NMe
2, –OC(=O)N(CH
3)Et, –OC(=O)N(CH
3)Pr, – OC(=O)N(CH
3)
iPr, –OC(=O)N(CH
3)Bu, –OC(=O)N(CH
3)
tBu, – OC(=O)N(CH
3)Cyclopropyl, –OC(=O)N(CH
3)Cyclobutyl). [0385] In an embodiment, R
1 is -S(=O)R
a1 wherein each R
a1 is as defined in any of the embodiments described herein. In an embodiment, R
1 is –S(=O)alkyl (e.g., –S(=O)Me, – S(=O)Et, –S(=O)Pr, –S(=O)
iPr). In an embodiment, R
1 is –S(=O)cycloalkyl (e.g., – S(=O)cyclopropyl, –S(=O)cyclobutyl, –S(=O)cyclopentyl, –S(=O)cyclohexyl). [0386] In an embodiment, R
1 is -S(=O)
2R
a1. In an embodiment, R
1 is –S(=O)
2alkyl (e.g., – S(=O)
2Me, –S(=O)
2Et, –S(=O)
2Pr, –S(=O)
2iPr). In an embodiment, R
1 is –S(=O)
2cycloalkyl (e.g., –S(=O)
2cyclopropyl, –S(=O)
2cyclobutyl, –S(=O)
2cyclopentyl, –S(=O)
2cyclohexyl). In an embodiment, R
1 is S(=O)
2aryl (e.g., S(=O)
2phenyl). [0387] In an embodiment, R
1 is –SR
a1 wherein each R
a1 is as defined in any of the embodiments described herein. In an embodiment, R
1 is –Salkyl (e.g., –SMe, –SEt, –SPr, – S
iPr). In an embodiment, R
1 is –Scycloalkyl (e.g., –Scyclopropyl, –Scyclobutyl, – Scyclopentyl, –Scyclohexyl). In an embodiment, R
1 is –Saryl (e.g., Sphenyl). [0388] In an embodiment, R
1 is -S(=O)(=NR
a1)R
a1 wherein each R
a1 is as defined in any of the embodiments described herein. In an embodiment, R
1 is –S(=O)(=NH)R
a1 (e.g., – S(=O)(=NH)Me, –S(=O)(=NH)Et, –S(=O)(=NH)Pr, –S(=O)(=NH)
iPr, –S(=O)(=NH)Bu, – S(=O)(=NH)
tBu, –S(=O)(=NH)Cyclopropyl, –S(=O)(=NH)Cyclobutyl). In an embodiment, R
1 is –S(=O)(=NCH
3)R
a1 (e.g., –S(=O)(=NCH
3)Me, –S(=O)(=NCH
3)Et, –S(=O)(=NCH
3)Pr, –S(=O)(=NCH
3)
iPr, –S(=O)(=NCH
3)Bu, –S(=O)(=NCH
3)
tBu, –S(=O)(=NCH
3)Cyclopropyl, –S(=O)(=NCH
3)Cyclobutyl). [0389] In an embodiment, R
1 is –NR
a1S(=O)
2R
a1 wherein each R
a1 is as defined in any of the embodiments described herein. In an embodiment, R
1 is –NHS(=O)
2alkyl (e.g., – NHS(=O)
2Me, –NHS(=O)
2Et, –NHS(=O)
2Pr, –NHS(=O)
2iPr). In an embodiment, R
1 is – NHS(=O)
2cycloalkyl (e.g., –NHS(=O)
2cyclopropyl, –NHS(=O)
2cyclobutyl, – NHS(=O)
2cyclopentyl, –NHS(=O)
2cyclohexyl). In an embodiment, R
1 is – N(CH
3)S(=O)
2alkyl (e.g., –N(CH
3)S(=O)
2Me, –N(CH
3)S(=O)
2Et, –N(CH
3)S(=O)
2Pr, – N(CH
3)S(=O)
2iPr). In an embodiment, R
1 is –N(CH
3)S(=O)
2cycloalkyl (e.g., – N(CH
3)S(=O)
2cyclopropyl, –N(CH
3)S(=O)
2cyclobutyl, –N(CH
3)S(=O)
2cyclopentyl, – N(CH
3)S(=O)
2cyclohexyl). [0390] In an embodiment, R
1 is -S(=O)
2N(R
a1)
2 wherein each R
a1 is as defined in any of the embodiments described herein (e.g., –S(=O)
2NH
2, –S(=O)
2NHR
a1, –S(=O)
2N(CH
3)R
a1). In an embodiment, R
1 is -S(=O)
2NH
2. In an embodiment, R
1 is -S(=O)
2NHR
a1 (e.g., – S(=O)
2NHMe, –S(=O)
2NHEt, –S(=O)
2NHPr, –S(=O)
2NH
iPr, –S(=O)
2NHcyclopropyl, – S(=O)
2NHcyclobutyl). In an embodiment, R
1 is -S(=O)
2N(CH
3)R
a1 (e.g., –S(=O)
2NMe
2, – S(=O)
2N(CH
3)Et, –S(=O)
2N(CH
3)Pr, –S(=O)
2N(CH
3)
iPr, –S(=O)
2N(CH
3)cyclopropyl, – S(=O)
2N(CH
3)cyclobutyl). [0391] As generally defined herein, each R
a1 is independently selected from the group consisting of H, –C
1–C
6 alkyl, –heteroC
1-C
4 alkyl, –C
1–C
6 haloalkyl, –C
3–C
9 cycloalkyl and 3-7 membered heterocyclyl, wherein each hydrogen atom of each alkyl, cycloalkyl heterocyclyl can be replaced by deuterium; or two instances of R
a1 on a nitrogen atom can be taken together with the nitrogen to which they are attached to form a 4-6 member monocyclic heterocycle containing one nitrogen atom and 0-1 additional heteroatoms selected from N and O. [0392] In an embodiment, each R
a1 is independently selected from H, –C
1–C
6 alkyl (e.g., – Me, –Et, –Pr, –
iPr, –
nBu, –sec-Bu, –iso-Bu, –
tBu), –heteroC
1-C
4 alkyl, –C
1–C
6 haloalkyl (e.g., –CF
3, –CHF
2, –CH
2CF
3, –CH(CH
3)CF
3) and C
3–C
9 cycloalkyl (e.g., cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl). In an embodiment, each R
a1 is selected from H, –C
1– C
6 alkyl (e.g., –Me, –Et, –Pr, –
iPr, –
nBu, –sec-Bu, –iso-Bu, –
tBu), –C
1–C
6 haloalkyl (e.g., – CF
3, –CHF
2, –CH
2CF
3, –CH(CH
3)CF
3) and –C
3–C
9 cycloalkyl (e.g., cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl). [0393] In an embodiment, each R
a1 is independently H. [0394] In an embodiment, each R
a1 is independently –C
1–C
6 alkyl (e.g., –Me, –Et, –Pr, –
iPr, –
nBu, –
tBu, –sec-Bu, –iso-Bu). In an embodiment, each R
a1 is independently –Me. In an embodiment, each R
a1 is independently –Et. In an embodiment, each R
a1 is independently – Pr. In an embodiment, each R
a1 is independently –
iPr. [0395] In an embodiment, each R
a1 is independently –heteroC
1-C
4 alkyl. In an embodiment, each R
a1 is independently methoxymethyl (–CH
2OCH
3). In an embodiment, each R
a1 is independently aminomethyl (e.g., –CH
2NH
2, –CH
2NHCH
3, –CH
2N(CH
3)
2. [0396] In an embodiment, each R
a1 is independently –C
1–C
6 haloalkyl (e.g., –CF
3, –CHF
2, – CF
2CH
3, –CH
2CF
3). In an embodiment, each R
a1 is independently trifluoromethyl (–CF
3). In other embodiments, each R
a1 is independently difluoromethyl (–CHF
2). In an embodiment, each R
a1 is –CH(CH
3)CF
3. In an embodiment, each R
a1 is –CH
2CF
3. [0397] In an embodiment, each R
a1 is independently –C
3–C
9 cycloalkyl (e.g., cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl). In an embodiment, each R
a1 is independently cyclopropyl. In an embodiment each R
a1 is independently cyclobutyl. In an embodiment, each R
a1 is independently cyclopentyl. In an embodiment, each R
a1 is independently cyclohexyl. [0398] In an embodiment, each R
a1 is independently 3-10 membered heterocyclyl (e.g., oxetanyl, tetrahydropyranyl, tetrahydrofuranyl, azetidinyl, pyrrolidinyl, piperidinyl, piperazinyl, morpholinyl, azepanyl). [0399] As generally defined herein, each R
2 is independently selected from the group consisting of –D, halo, –CN, –C
1–C
6 alkyl, –heteroC
1-C
4 alkyl, –C
1–C
6 haloalkyl, –C
1–C
6 hydroxyalkyl, –C
3–C
10 cycloalkyl, 3-10 membered heterocyclyl, –C
6-C
10 aryl, –OR
a2, – N(R
a2)
2, –C(=O)R
a2, –C(=O)OR
a2, –NR
a2C(=O)R
a2, –NR
a2C(=O)OR
a2, –C(=O)N(R
a2)
2, – OC(=O)N(R
a2)
2, –S(=O)R
a2, –S(=O)
2R
a2, –SR
a2, –S(=O)(=NR
a2)R
a2, –NR
a2S(=O)
2R
a2 and – S(=O)
2N(R
a2)
2 , wherein each alkyl, cycloalkyl, heterocyclyl and aryl of R
2 can be substituted at any available position with 0, 1 or 2 substituents selected from –F, –Cl, –OH– and –CH
3, wherein each R
a2 is as defined in any of the embodiments described herein. [0400] In an embodiment each R
2 is independently selected from H, –D, halo, –CN, –C
1–C
6 alkyl, –heteroC
1-C
4 alkyl, –C
1–C
6 haloalkyl, –C
1–C
6 hydroxyalkyl, –C
3–C
10 cycloalkyl, 3-10 membered heterocyclyl, –C
6-C
10 aryl, –OR
a2 and –N(R
a2)
2, wherein each aryl, alkyl, cycloalkyl and heterocyclyl is substituted with 0, 1 or 2 substituents selected from –F, –Cl, – OH– and –CH
3. [0401] In an embodiment, each R
2 is independently selected from –C
1–C
6 alkyl and OR
a2. [0402] In an embodiment, each R
2 is independently selected from –CH
3, –OH, –OCH
3, – OCD
3 and –OCHF
2. [0403] In an embodiment, each R
2 is independently selected from –OCH
3, –OCD
3, –OCH
2F and –OCHF
2. [0404] In an embodiment, R
2 is H. In an embodiment R
2 is –D. [0405] In an embodiment, R
2 is halo (e.g., fluoro, chloro, bromo, iodo). In an embodiment, R
2 is –Cl. In an embodiment, R
2 is –F. In an embodiment, R
2 is –Br. In an embodiment, R
2 is –I. [0406] In an embodiment, R
2 is –CN. [0407] In an embodiment, R
2 is –C
1–C
6 alkyl (e.g., –Me, –Et, –Pr, –
iPr, –
nBu, –sec-Bu, –iso- Bu, –
tBu, –C(CH
3)CH
2CH
3,). In an embodiment, R
2 is –Me. In an embodiment, R
2 is –Et. In an embodiment R
2 is –Pr. In an embodiment, R
2 is –iPr. In an embodiment, R
2 is - C(CH
3)CH
2CH
3. [0408] In an embodiment, R
2 is –heteroC
1-C
4 alkyl. In an embodiment, R
2 is methoxymethyl (–CH
2OCH
3). In an embodiment, R
2 is aminomethyl (e.g., –CH
2NH
2, –CH
2NHCH
3, – CH
2NHCH
2CH
3, –CH
2N(CH
3)
2). In an embodiment, R
2 is –CH
2N(CH
3)CH
2CH
3. In an embodiment, R
2 is –CH
2N(CH
3)CH
2CF
3. [0409] In an embodiment, R
2 is –C
1–C
6 haloalkyl (e.g., –CF
3, –CHF
2, –CF
2CH
3, –CH
2CF
3). In an embodiment, R
2 is trifluoromethyl (–CF
3). In other embodiments, R
2 is difluoromethyl (–CHF
2). In other embodiments, R
2 is –CH
2CF
3. In an embodiment, R
2 is –CF
2CH
3. [0410] In an embodiment, R
2 is –C
1–C
6 hydroxyalkyl (e.g., –CH
2OH, –CH(OH)CH
3, – C(OH)(CH
3)
2). In an embodiment, R
2 is hydroxymethyl (–CH
2OH). [0411] In an embodiment, R
2 is optionally substituted –C
3–C
10 cycloalkyl (e.g., cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl). In an embodiment, R
2 is optionally substituted cyclopropyl (e.g., substituted with 0 or 1 instance of –CN). In an embodiment R
2 is cyclobutyl. In an embodiment, R
2 is cyclopentyl. In an embodiment, R
2 is cyclohexyl. [0412] In an embodiment, R
2 is an optionally substituted 3-10 membered heterocyclyl (e.g., oxetanyl, tetrahydropyranyl, tetrahydrofuranyl, azetidinyl, pyrrolidinyl, piperidinyl, piperazinyl, morpholinyl, azepanyl, 6-oxa-1-azaspiro[3.3]heptanyl, 6-oxa-1- azaspiro[3.4]octanyl). In an embodiment, R
2 is oxetanyl. In an embodiment, R
2 is tetrahydropyranyl. In an embodiment, R
2 is tetrahydrofuranyl. In an embodiment, R
2 is azetidinyl. In an embodiment, the azetidinyl is optionally substituted (e.g., substituted with 0 or 1 instances of –F or –Me). In an embodiment, R
2 is pyrrolidinyl. In an embodiment, R
2 is piperidinyl. In an embodiment, R
2 is piperazinyl. In an embodiment, R
2 is morpholinyl. In an embodiment, R
2 is azepanyl. In an embodiment, R
2 is 6-oxa-1-azaspiro[3.3]heptanyl. In an embodiment, R
2 is 6-oxa-1-azaspiro[3.4]octanyl. [0413] In an embodiment, R
2 is –C
6-C
10 aryl (e.g., phenyl, naphthyl). In an embodiment, R
2 is optionally substituted phenyl (e.g., phenyl substituted with 0 or 1 instances of halo (e.g., – Cl, –F)). In an embodiment, R
2 is –2-Cl-phenyl. [0414] In an embodiment, R
2 is –OR
a2 wherein each R
a2 is as defined in any of the embodiments described herein (e.g., hydroxy (–OH), methoxy, –OCD
3, difluoromethoxy (– OCHF
2), trifluoromethoxy (–OCF
3), –OCH(CH
3)CF
3, –OCH
2CF
3, ethoxy, propoxy, isopropoxy, cyclopropyloxy, cyclobutyloxy, –OCH
2CH(CH
3)
3). In an embodiment, R
2 is hydroxy. In an embodiment, each R
2 is independently selected from –OCH
3, –OCD
3, – OCH
2F and –OCHF
2. In an embodiment, each R
2 is independently selected from –OCH
3, – OCD
3 and –OCHF
2. In an embodiment, each R
2 is independently selected from –OCH
3 and – OCD
3. [0415] In an embodiment, R
2 is –OMe. In an embodiment, R
2 is –OCD
3. In an embodiment, R
2 is ethoxy. In an embodiment, R
2 is propoxy. In an embodiment, R
2 is isopropoxy. In an embodiment R
2 is difluoromethoxy (–OCHF
2). In an embodiment R
2 is fluoromethoxy (– OCH
2F). In an embodiment, R
2 is trifluoromethoxy (–OCF
3). In an embodiment, R
2 is – OCH(CH
3)CF
3. In an embodiment, R
2 is –OCH
2CF
3. In an embodiment, R
2 is cyclopropyloxy. In an embodiment R
2 is –OCH
2CH(CH
3)
3. [0416] In an embodiment, R
2 is –N(R
a2)
2 wherein each R
a2 is as defined in any of the embodiments described herein (e.g., –NH
2, –NHR
a2, –N(CH
3)R
a2). In an embodiment, R
2 is – NH
2. In an embodiment, R
2 is –NHR
a2 (e.g., –NHMe, –NHEt, –NHPr, –NH
iPr, – NHcyclopropyl, –NHcyclobutyl). In an embodiment, R
2 is –N(CH
3)R
a2 (e.g., –NMe
2, – N(CH
3)Et, –N(CH
3)Pr, –N(CH
3)
iPr, –N(CH
3)cyclopropyl, –N(CH
3)cyclobutyl). [0417] In an embodiment, R
2 is –C(=O)R
a2 or –C(=O)OR
a2 wherein each R
a2 is as defined in any of the embodiments described herein. In an embodiment, R
2 is –C(=O)R
a2 wherein R
a2 is as described herein. In an embodiment, R
2 is –C(=O)alkyl. In an embodiment, R
2 is – C(O)CH
3, –C(O)cyclopropyl, –C(O)cyclobutyl, –C(O)
tBu, –C(O)
iPr, –C(O)Pr, –C(O)
iBu, or –C(=O)OMe. In an embodiment, R
2 is acetyl (–C(=O)Me). In an embodiment, R
2 is – C(=O)OR
a2. In an embodiment, R
2 is –COOH. In an embodiment, R
2 is COOMe. [0418] In an embodiment, R
2 is –NR
a2C(=O)R
a2 wherein each R
a2 is as defined in any of the embodiments described herein. In an embodiment, R
2 is –NHC(=O)R
a2 (e.g., –NHC(=O)Me, –NHC(=O)Et, –NHC(=O)Pr, –NHC(=O)
iPr, –NHC(=O)Bu, –NHC(=O)
tBu, – NHC(=O)Cyclopropyl, –NHC(=O)Cyclobutyl). In an embodiment, R
2 is –N(CH
3)C(=O)R
a2 (e.g., –N(CH
3)C(=O)Me, –N(CH
3)C(=O)Et, –N(CH
3)C(=O)Pr, –N(CH
3)C(=O)
iPr, – N(CH
3)C(=O)Bu, –N(CH
3)C(=O)
tBu, –N(CH
3)C(=O)Cyclopropyl, – N(CH
3)C(=O)Cyclobutyl). [0419] In an embodiment, R
2 is –NR
a2C(=O)OR
a2. In an embodiment, R
2 is –NHC(=O)OR
a2 (e.g., –NHC(=O)OMe, –NHC(=O)OEt, –NHC(=O)OPr, –NHC(=O)O
iPr, –NHC(=O)OBu, – NHC(=O)O
tBu, –NHC(=O)OCyclopropyl, –NHC(=O)OCyclobutyl). In an embodiment, R
2 is –N(CH
3)C(=O)OR
a2 (e.g., –N(CH
3)C(=O)OMe, –N(CH
3)C(=O)OEt, –N(CH
3)C(=O)OPr, –N(CH
3)C(=O)O
iPr, –N(CH
3)C(=O)OBu, –N(CH
3)C(=O)O
tBu, – N(CH
3)C(=O)OCyclopropyl, –N(CH
3)C(=O)OCyclobutyl). [0420] In an embodiment, R
2 is –C(=O)N(R
a2)
2 wherein each R
a2 is as defined in any of the embodiments described herein (e.g., –C(=O)NH
2, –C(=O)NHR
a2, –C(=O)N(CH
3)R
a2). In an embodiment, R
2 is –C(=O)NH
2. In an embodiment, R
2 is –C(=O)NHR
a2 (e.g., –C(=O)NHMe, –C(=O)NHEt, –C(=O)NHPr, –C(=O)NH
iPr, –C(=O)NHBu, –C(=O)NH
tBu, – C(=O)NHCyclopropyl, –C(=O)NHCyclobutyl). In an embodiment, R
2 is –C(=O)N(CH
3)R
a2 (e.g., –C(=O)NMe
2, –C(=O)N(CH
3)Et, –C(=O)N(CH
3)Pr, –C(=O)N(CH
3)
iPr, – C(=O)N(CH
3)Bu, –C(=O)N(CH
3)
tBu, –C(=O)N(CH
3)Cyclopropyl, – C(=O)N(CH
3)Cyclobutyl). [0421] In an embodiment, R
2 is –OC(=O)N(R
a2)wherein each R
a2 is as defined in any of the embodiments described herein. In an embodiment, R
2 is –OC(=O)NHR
a2 (e.g., – OC(=O)NHMe, –OC(=O)NHEt, –OC(=O)NHPr, –OC(=O)NH
iPr, –OC(=O)NHBu, – OC(=O)NH
tBu, –OC(=O)NHCyclopropyl, –OC(=O)NHCyclobutyl). In an embodiment, R
2 is –OC(=O)N(CH
3)R
a2 (e.g., –OC(=O)NMe
2, –OC(=O)N(CH
3)Et, –OC(=O)N(CH
3)Pr, – OC(=O)N(CH
3)
iPr, –OC(=O)N(CH
3)Bu, –OC(=O)N(CH
3)
tBu, – OC(=O)N(CH
3)Cyclopropyl, –OC(=O)N(CH
3)Cyclobutyl). [0422] In an embodiment, R
2 is -S(=O)R
a2 wherein each R
a2 is as defined in any of the embodiments described herein. In an embodiment, R
2 is –S(=O)alkyl (e.g., –S(=O)Me, – S(=O)Et, –S(=O)Pr, –S(=O)
iPr). In an embodiment, R
2 is –S(=O)cycloalkyl (e.g., – S(=O)cyclopropyl, –S(=O)cyclobutyl, –S(=O)cyclopentyl, –S(=O)cyclohexyl). [0423] In an embodiment, R
2 is -S(=O)
2R
a2. In an embodiment, R
2 is –S(=O)
2alkyl (e.g., – S(=O)
2Me, –S(=O)
2Et, –S(=O)
2Pr, –S(=O)
2iPr). In an embodiment, R
2 is –S(=O)
2cycloalkyl (e.g., –S(=O)
2cyclopropyl, –S(=O)
2cyclobutyl, –S(=O)
2cyclopentyl, –S(=O)
2cyclohexyl). In an embodiment, R
2 is S(=O)
2aryl (e.g., S(=O)
2phenyl). [0424] In an embodiment, R
2 is –SR
a2 wherein each R
a2 is as defined in any of the embodiments described herein. In an embodiment, R
2 is –Salkyl (e.g., –SMe, –SEt, –SPr, – S
iPr). In an embodiment, R
2 is –Scycloalkyl (e.g., –Scyclopropyl, –Scyclobutyl, – Scyclopentyl, –Scyclohexyl). In an embodiment, R
2 is –Saryl (e.g., Sphenyl). [0425] In an embodiment, R
2 is -S(=O)(=NR
a2)R
a2 wherein each R
a2 is as defined in any of the embodiments described herein. In an embodiment, R
2 is –S(=O)(=NH)R
a2 (e.g., – S(=O)(=NH)Me, –S(=O)(=NH)Et, –S(=O)(=NH)Pr, –S(=O)(=NH)
iPr, –S(=O)(=NH)Bu, – S(=O)(=NH)
tBu, –S(=O)(=NH)Cyclopropyl, –S(=O)(=NH)Cyclobutyl). In an embodiment, R
2 is –S(=O)(=NCH
3)R
a2 (e.g., –S(=O)(=NCH
3)Me, –S(=O)(=NCH
3)Et, –S(=O)(=NCH
3)Pr, –S(=O)(=NCH
3)
iPr, –S(=O)(=NCH
3)Bu, –S(=O)(=NCH
3)
tBu, –S(=O)(=NCH
3)Cyclopropyl, –S(=O)(=NCH
3)Cyclobutyl). [0426] In an embodiment, R
2 is –NR
a2S(=O)
2R
a2 wherein each R
a2 is as defined in any of the embodiments described herein. In an embodiment, R
2 is –NHS(=O)2alkyl (e.g., – NHS(=O)
2Me, –NHS(=O)
2Et, –NHS(=O)
2Pr, –NHS(=O)
2iPr). In an embodiment, R
2 is – NHS(=O)
2cycloalkyl (e.g., –NHS(=O)
2cyclopropyl, –NHS(=O)
2cyclobutyl, – NHS(=O)
2cyclopentyl, –NHS(=O)
2cyclohexyl). In an embodiment, R
2 is – N(CH
3)S(=O)
2alkyl (e.g., –N(CH
3)S(=O)
2Me, –N(CH
3)S(=O)
2Et, –N(CH
3)S(=O)
2Pr, – N(CH
3)S(=O)
2iPr). In an embodiment, R
2 is –N(CH
3)S(=O)
2cycloalkyl (e.g., – N(CH
3)S(=O)
2cyclopropyl, –N(CH
3)S(=O)
2cyclobutyl, –N(CH
3)S(=O)
2cyclopentyl, – N(CH
3)S(=O)
2cyclohexyl). [0427] In an embodiment, R
2 is -S(=O)
2N(R
a2)
2 wherein each R
a2 is as defined in any of the embodiments described herein (e.g., –S(=O)
2NH
2, –S(=O)
2NHR
a2, –S(=O)
2N(CH
3)R
a2). In an embodiment, R
2 is -S(=O)
2NH
2. In an embodiment, R
2 is -S(=O)
2NHR
a2 (e.g., – S(=O)
2NHMe, –S(=O)
2NHEt, –S(=O)
2NHPr, –S(=O)
2NH
iPr, –S(=O)
2NHcyclopropyl, – S(=O)
2NHcyclobutyl). In an embodiment, R
2 is -S(=O)
2N(CH
3)R
a2 (e.g., –S(=O)
2NMe
2, – S(=O)
2N(CH
3)Et, –S(=O)
2N(CH
3)Pr, –S(=O)
2N(CH
3)
iPr, –S(=O)
2N(CH
3)cyclopropyl, – S(=O)
2N(CH
3)cyclobutyl). [0428] As generally defined herein, each R
a2 is independently selected from the group consisting of H, –C
1–C
6 alkyl, –heteroC
1-C
4 alkyl, –C
1–C
6 haloalkyl, –C
3–C
9 cycloalkyl and 3-7 membered heterocyclyl, wherein each hydrogen atom of each alkyl, cycloalkyl heterocyclyl can be replaced by deuterium; or two instances of R
a2 on a nitrogen atom can be taken together with the nitrogen to which they are attached to form a 4-6 member monocyclic heterocycle containing one nitrogen atom and 0-1 additional heteroatoms selected from N and O. [0429] In an embodiment, each R
a2 is independently selected from H, –C
1–C
6 alkyl (e.g., – Me, –Et, –Pr, –
iPr, –
nBu, –sec-Bu, –iso-Bu, –
tBu), –heteroC
1-C
4 alkyl, –C
1–C
6 haloalkyl (e.g., –CF
3, –CHF
2, –CH
2CF
3, –CH(CH
3)CF
3) and C
3–C
9 cycloalkyl (e.g., cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl). In an embodiment, each R
a2 is selected from H, –C
1– C
6 alkyl (e.g., –Me, –Et, –Pr, –
iPr, –
nBu, –sec-Bu, –iso-Bu, –
tBu), –C
1–C
6 haloalkyl (e.g., – CF
3, –CHF
2, –CH
2CF
3, –CH(CH
3)CF
3) and –C
3–C
9 cycloalkyl (e.g., cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl). [0430] In an embodiment, each R
a2 is independently selected from the group consisting of H, –C
1-C
6 alkyl and –C
1-C
6 haloalkyl wherein each hydrogen atom of the alkyl can be independently replaced with deuterium. [0431] In an embodiment, each R
a2 is independently selected from the group consisting of – C
1-C
6 alkyl and –C
1-C
6 haloalkyl wherein each hydrogen atom of the alkyl can be independently replaced with deuterium. [0432] In an embodiment, each R
a2 is independently selected from the group consisting of – CH
3, –CD
3,–Et, –CHF
2 and –CH
2F. [0433] In an embodiment, each R
a2 is independently selected from the group consisting of – CH
3, –CD
3 and –CHF
2. [0434] In an embodiment, each R
a2 is independently selected from the group consisting of H, –CH
3, –CD
3. [0435] In an embodiment, each R
a2 is independently H. [0436] In an embodiment, each R
a2 is independently –C
1–C
6 alkyl herein each hydrogen atom of the alkyl can be replaced by deuterium (e.g., –Me, –CD
3, –Et, –Pr, –
iPr, –
nBu, –
tBu, –sec- Bu, –iso-Bu). In an embodiment, each R
a2 is independently selected from the group consisting of –CH
3 and –CD
3. In an embodiment, each R
a2 is independently –CH
3. In an embodiment, each R
a2 is independently –CD
3. In an embodiment, each R
a2 is independently – Et. In an embodiment, each R
a2 is independently –Pr. In an embodiment, each R
a2 is independently –
iPr. [0437] In an embodiment, each R
a2 is independently –heteroC
1-C
4 alkyl. In an embodiment, each R
a2 is independently methoxymethyl (–CH
2OCH
3). In an embodiment, each R
a2 is independently aminomethyl (e.g., –CH
2NH
2, –CH
2NHCH
3, –CH
2N(CH
3)
2. [0438] In an embodiment, each R
a2 is independently –C
1–C
6 haloalkyl (e.g., –CF
3, –CHF
2, – CF
2CH
3, –CH
2CF
3). In an embodiment, each R
a2 is independently trifluoromethyl (–CF
3). In other embodiments, each R
a2 is independently difluoromethyl (–CHF
2). In other embodiments, each R
a2 is independently fluoromethyl (–CH
2F). In an embodiment, each R
a2 is –CH(CH
3)CF
3. In an embodiment, each R
a2 is –CH
2CF
3. [0439] In an embodiment, each R
a2 is independently –C
3–C
9 cycloalkyl (e.g., cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl). In an embodiment, each R
a2 is independently cyclopropyl. In an embodiment each R
a2 is independently cyclobutyl. In an embodiment, each R
a2 is independently cyclopentyl. In an embodiment, each R
a2 is independently cyclohexyl. [0440] In an embodiment, each R
a2 is independently 3-10 membered heterocyclyl (e.g., oxetanyl, tetrahydropyranyl, tetrahydrofuranyl, azetidinyl, pyrrolidinyl, piperidinyl, piperazinyl, morpholinyl, azepanyl). [0441] In an embodiment, the compound is of Formula (I-b)
Formula (I-b); wherein Ring A, Ring B, L, R
c, R
c’, R
a2, R
A, R
B, R
C, R
E, n and p are as defined in any of the embodiments described herein. [0442] In an embodiment, the compound is of Formula (I-c)
the embodiments described herein. [0443] In an embodiment, the compound is of Formula (I-d)
Formula (I-d); wherein L, R
c, R
c’, R
a2, R
A, R
C, R
E, R
B, n and p are as defined in any of the embodiments described herein. [0444] In an embodiment, the compound is of Formula (I-d-1)
Formula (I-d-1); wherein L, R
c, R
c’, R
a2, R
C, R
E, R
B, and p are as defined in any of the embodiments described herein. [0445] In an embodiment, the compound is of Formula (I-e)
wherein X
3, L, R
c, R
c’, R
a2, R
A, R
C, R
B, R
E and n are as defined in any of the embodiments described herein. [0446] In an embodiment, the compound is of Formula (I-f)
Formula (I-f); wherein L, R
c, R
c’, R
a2, R
A, R
C, R
B, R
E and n are as defined in any of the embodiments described herein. [0447] In an embodiment, the compound is of Formula (I-f-1)
Formula (I-f-1); wherein L, R
c, R
c’, R
a2, R
C, R
B and R
E are as defined in any of the embodiments described herein. [0448] In an embodiment, the compound is selected from the compounds of Table 1. [0449] In an embodiment, provided is a pharmaceutical composition comprising a compound of Formula (I), (II) or a compound of Table 1 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, tautomer or stereoisomer thereof as defined herein and a pharmaceutically acceptable carrier. In an embodiment, the pharmaceutical composition further comprises a second therapeutic agent. [0450] In various embodiments, the Compounds of the Disclosure are USP1 inhibitors that reduce the level of USP1 protein and/or inhibit or reduce at least one biological activity of USP l protein. [0451] In an embodiment, the Compounds of the Disclosure specifically bind to USP1 protein. In an embodiment, the Compounds of the Disclosure specifically bind to USP1 protein in a USP1-UAF1 complex. In an embodiment, the Compounds of the Disclosure specifically bind to USP1 mRNA. In an embodiment, the Compounds of the Disclosure specifically bind to USP1 protein (alone or in a USP1-UAF1 complex) or USP1 mRNA. In an embodiment, the Compounds of the Disclosure specifically bind to UAF1 (alone or in a USP1-UAF1 complex) and inhibit or reduces formation or activity of the USP1-UAF1 complex. [0452] In an embodiment, the Compounds of the Disclosure decrease the formation of the USP1-UAF1 complex. In an embodiment, the Compounds of the Disclosure decrease the activity of the USP1-UAF1 complex. In an embodiment, the Compounds of the Disclosure decrease the deubiquitinase activity of USP1. In an embodiment, the Compounds of the Disclosure increase mono-ubiquitinated PCNA. In an embodiment, the Compounds of the Disclosure increase mono-ubiquitinated FANCD2. In an embodiment, the Compounds of the Disclosure increase mono- ubiquitinated FANCI. [0453] In an embodiment, the Compounds of the Disclosure do not bind to other deubiquitinases, other USP proteins, or other UAF1 complexes (e.g., USP46-UAF1) or bind deubiquitinases, other USP proteins, or other UAF1 complexes (e.g., USP46-UAF1) with at least 5-fold, at least 10-fold, at least 20-fold, or at least 100-fold reduced affinity compared to the affinity for USP1 (i.e., the K
D of the USP1 inhibitor for other deubiquitinases, other USP proteins, or other UAF1 complexes (e.g., USP46-UAF1) is at least 5 -fold, at least 10-fold, at least 20 fold, least 50 fold, at least 100 fold. [0454] Certain compounds of the disclosure were assessed for USP1-UAF1 activity in a Ubiquitin Rhodamine assay as described in Biology Example 1. [0455] Table 1 indicates IC
50 values (μM) against USP1-UAF1 for exemplary compounds (column 4). For column 4, “a” indicates an IC
50 value lower than 30 nM, “b” indicates an IC
50 value equal to or greater than 30 nM and lower than 100 nM, “c” indicates an IC
50 value equal to or greater than 100 nM but lower than 5 µM, and “d” indicates an IC
50 value equal to or greater than 5 µM in the assay described in Biology Example 1 [0456] Table 1 also indicates IC
50 values in a viability assay for a non-isogenic pair of BRCA1 mutant (column 5- MDA-MB-436) and BRCA1 WT (column 6 – HCC1954) cell lines. These values indicate the effect of treatment with compound on cell survival. In columns 5 and 6, a value of “aa” and “aaa” indicates an IC
50 of less than 100 nM in the mutant and wild-type cell lines, respectively; a value of “bb” and ”bbb” indicates an IC
50 equal to or greater than 100 nM but less than 250 nM in the mutant and wild-type cell lines, respectively; a value of “cc” and “ccc” indicates an IC
50 equal to or greater than 250 nM but less than 5 µM in the mutant and wild-type cell lines, respectively; a value of “dd” and “ddd” indicates an IC
50 greater than or equal to 5 µM in the mutant and wild-type cell lines, respectively, in the assay described in Biology Example 2. [0457] Table 1 also indicates IC
50 values for exemplary compounds in an AlphaLISA assay measuring monoubiquitinated PCNA in a BRCA1 mutant cell line (MDA-MB-436; column 7). In column 7, a value of “A” indicates an IC
50 of less than 100 nM, a value of “B” indicates an IC
50 equal to or greater than 100 nM but less than 250 nM, a value of “C” indicates an IC
50 equal to or greater than 250 nM but less than 5 µM, a value of “D” indicates an IC
50 greater than or equal to 5 µM, in the assay described in Biology Example 3. [0458] Unless otherwise indicated, the absolute stereochemistry of all chiral atoms is as depicted. Compounds marked with (OR) or (rel) are single enantiomers wherein the absolute stereochemistry was arbitrarily assigned (e.g., based on chiral SFC elution as described in the Examples section). Compounds marked with (and) or (rac) are mixtures of enantiomers wherein the relative stereochemistry is as shown. Compounds marked with (abs) are single enantiomers wherein the absolute stereochemistry is as indicated. Table 1. Exemplary compounds and biological data
O N N N O N F N F 2 a aa ddd A N
F O N N N O 17 a aa ddd A
O N N N S N F 38 a aa ddd A N F
Alternative Embodiments [0459] In an alternative embodiment, compounds described herein may also comprise one or more isotopic substitutions. For example, hydrogen may be
2H (D or deuterium) or
3H (T or tritium); carbon may be, for example,
13C or
14C; oxygen may be, for example,
18O; nitrogen may be, for example,
15N, and the like. In other embodiments, a particular isotope (e.g.,
3H,
13C,
14C,
18O, or
15N) can represent at least 1%, at least 5%, at least 10%, at least 15%, at least 20%, at least 25%, at least 30%, at least 35%, at least 40%, at least 45%, at least 50%, at least 60%, at least 65%, at least 70%, at least 75%, at least 80%, at least 85%, at least 90%, at least 95%, at least 99%, or at least 99.9% of the total isotopic abundance of an element that occupies a specific site of the compound. Pharmaceutical Compositions [0460] In an embodiment, provided is a pharmaceutical composition comprising a pharmaceutically acceptable carrier and an effective amount of a compound described herein (e.g., a compound of Formula (I), (II) or a compound of Table 1), or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof. [0461] The term “pharmaceutically acceptable carrier or adjuvant” refers to a carrier or adjuvant that may be administered to a patient, together with a compound provided herewith, and which does not destroy the pharmacological activity thereof and is nontoxic when administered in doses sufficient to deliver a therapeutic amount of the compound. [0462] Pharmaceutically acceptable carriers, adjuvants and vehicles that may be used in the pharmaceutical compositions provided herewith include, but are not limited to, ion exchangers, alumina, aluminum stearate, lecithin, self-emulsifying drug delivery systems (SEDDS) such as d–α-tocopherol polyethyleneglycol 1000 succinate, surfactants used in pharmaceutical dosage forms such as Tweens or other similar polymeric delivery matrices, serum proteins, such as human serum albumin, buffer substances such as phosphates, glycine, sorbic acid, potassium sorbate, partial glyceride mixtures of saturated vegetable fatty acids, water, salts or electrolytes, such as protamine sulfate, disodium hydrogen phosphate, potassium hydrogen phosphate, sodium chloride, zinc salts, colloidal silica, magnesium trisilicate, polyvinyl pyrrolidone, cellulose based substances, polyethylene glycol, sodium carboxymethylcellulose, polyacrylates, waxes, polyethylene polyoxypropylene block polymers, polyethylene glycol and wool fat. Cyclodextrins such as α–, β–, and γ- cyclodextrin, or chemically modified derivatives such as hydroxyalkylcyclodextrins, including 2 and 3 hydroxypropyl-β-cyclodextrins, or other solubilized derivatives may also be advantageously used to enhance delivery of compounds of the formulae described herein. [0463] When employed as pharmaceuticals, the compounds provided herein are typically administered in the form of a pharmaceutical composition. Such compositions can be prepared in a manner well known in the pharmaceutical art and comprise at least one active compound. [0464] In an embodiment, with respect to the pharmaceutical composition, the carrier is a parenteral carrier, oral or topical carrier. [0465] In an embodiment, provided is a compound described herein (e.g., a compound of Formula (I), (II) or a compound of Table 1), or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof) (or pharmaceutical composition thereof) for use as a pharmaceutical or a medicament (e.g., a medicament for the treatment of a disease or disorder associated with USP1 in a subject in need thereof). In an embodiment, the disease is a proliferating disease. In a further embodiment, the disease is cancer. In an embodiment, the cancer is breast cancer (e.g., triple negative breast cancer), ovarian cancer (e.g., platinum-resistant ovarian cancer, platinum-refractory ovarian cancer), prostate cancer, pancreatic cancer or lung cancer (e.g., non-small cell lung cancer (NSCLC)). [0466] In an embodiment, provided is a compound described herein (e.g., a compound of Formula (I), (II) or a compound of Table 1), or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof) (or pharmaceutical composition thereof) for use in the treatment of a disease or disorder associated with USP1 in a subject in need thereof. In an embodiment, the disease is a proliferating disease. In a further embodiment, the disease is cancer. In an embodiment, the cancer is breast cancer (e.g., triple negative breast cancer), ovarian cancer (e.g., platinum-resistant ovarian cancer, platinum-refractory ovarian cancer), prostate cancer, pancreatic cancer or lung cancer (e.g., non-small cell lung cancer (NSCLC)). [0467] In an embodiment, provided is a compound described herein (e.g., a compound of Formula (I), (II) or a compound of Table 1), or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof) (or pharmaceutical composition thereof) for use in the manufacturing of a medicament (e.g., a medicament for the treatment of a disease or disorder associated with USP1 in a subject in need thereof). In an embodiment, the disease is a proliferating disease. In a further embodiment, the disease is cancer. In an embodiment, the cancer is breast cancer (e.g., triple negative breast cancer), ovarian cancer (e.g., platinum-resistant ovarian cancer, platinum-refractory ovarian cancer), prostate cancer, pancreatic cancer or lung cancer (e.g., non-small cell lung cancer (NSCLC)). Generally, the compounds provided herein are administered in a therapeutically effective amount. The amount of the compound actually administered will typically be determined by a physician, in the light of the relevant circumstances, including the condition to be treated, the chosen route of administration, the actual compound administered, the age, weight, and response of the individual patient, the severity of the patient’s symptoms, and the like. [0468] The pharmaceutical compositions provided herewith may be administered orally, parenterally, by inhalation spray, topically, rectally, nasally, buccally, vaginally or via an implanted reservoir, preferably by oral administration or administration by injection. The pharmaceutical compositions provided herewith may contain any conventional nontoxic pharmaceutically acceptable carriers, adjuvants or vehicles. In some cases, the pH of the formulation may be adjusted with pharmaceutically acceptable acids, bases or buffers to enhance the stability of the formulated compound or its delivery form. The term parenteral as used herein includes subcutaneous, intracutaneous, intravenous, intramuscular, intraarticular, intraarterial, intrasynovial, intrasternal, intrathecal, intralesional and intracranial injection or infusion techniques. [0469] The compositions for oral administration can take the form of bulk liquid solutions or suspensions, or bulk powders. More commonly, however, the compositions are presented in unit dosage forms to facilitate accurate dosing. The term “unit dosage forms” refers to physically discrete units suitable as unitary dosages for human subjects and other mammals, each unit containing a predetermined quantity of active material calculated to produce the desired therapeutic effect, in association with a suitable pharmaceutical excipient. Typical unit dosage forms include prefilled, premeasured ampules or syringes of the liquid compositions or pills, tablets, capsules or the like in the case of solid compositions. In such compositions, the compound is usually a minor component (from about 0.1 to about 50% by weight or preferably from about 1 to about 40% by weight) with the remainder being various vehicles or carriers and processing aids helpful for forming the desired dosing form. [0470] Liquid forms suitable for oral administration may include a suitable aqueous or nonaqueous vehicle with buffers, suspending and dispensing agents, colorants, flavors and the like. Solid forms may include, for example, any of the following ingredients, or compounds of a similar nature: a binder such as microcrystalline cellulose, gum tragacanth or gelatin; an excipient such as starch or lactose, a disintegrating agent such as alginic acid, Primogel, or corn starch; a lubricant such as magnesium stearate; a glidant such as colloidal silicon dioxide; a sweetening agent such as sucrose or saccharin; or a flavoring agent such as peppermint, methyl salicylate, or orange flavoring. [0471] Injectable compositions are typically based upon injectable sterile saline or phosphate-buffered saline or other injectable carriers known in the art. As before, the active compound in such compositions is typically a minor component, often being from about 0.05 to 10% by weight with the remainder being the injectable carrier and the like. The pharmaceutical compositions may be in the form of a sterile injectable preparation, for example, as a sterile injectable aqueous or oleaginous suspension. This suspension may be formulated according to techniques known in the art using suitable dispersing or wetting agents (such as, for example, Tween 80) and suspending agents. The sterile injectable preparation may also be a sterile injectable solution or suspension in a non-toxic parenterally acceptable diluent or solvent, for example, as a solution in 1,3–butanediol. Among the acceptable vehicles and solvents that may be employed are mannitol, water, Ringer’s solution and isotonic sodium chloride solution. In addition, sterile, fixed oils are conventionally employed as a solvent or suspending medium. For this purpose, any bland fixed oil may be employed including synthetic mono– or diglycerides. Fatty acids, such as oleic acid and its glyceride derivatives are useful in the preparation of injectables, as are natural pharmaceutically acceptable oils, such as olive oil or castor oil, especially in their polyoxyethylated versions. These oil solutions or suspensions may also contain a long chain alcohol diluent or dispersant, or carboxymethyl cellulose or similar dispersing agents which are commonly used in the formulation of pharmaceutically acceptable dosage forms such as emulsions and or suspensions. Other commonly used surfactants such as Tweens or Spans and/or other similar emulsifying agents or bioavailability enhancers which are commonly used in the manufacture of pharmaceutically acceptable solid, liquid, or other dosage forms may also be used for the purposes of formulation. [0472] Transdermal compositions are typically formulated as a topical ointment or cream containing the active ingredient(s), generally in an amount ranging from about 0.01 to about 20% by weight, preferably from about 0.1 to about 20% by weight, preferably from about 0.1 to about 10% by weight, and more preferably from about 0.5 to about 15% by weight. When formulated as an ointment, the active ingredients will typically be combined with either a paraffinic or a water-miscible ointment base. Alternatively, the active ingredients may be formulated in a cream with, for example, an oil-in-water cream base. Such transdermal formulations are well-known in the art and generally include additional ingredients to enhance the dermal penetration of stability of the active ingredients or the formulation. All such known transdermal formulations and ingredients are included within the scope provided herein. [0473] The compounds provided herein can also be administered by a transdermal device. Accordingly, transdermal administration can be accomplished using a patch either of the reservoir or porous membrane type, or of a solid matrix variety. [0474] The pharmaceutical compositions provided herewith may also be administered in the form of suppositories for rectal administration. These compositions can be prepared by mixing a compound provided herewith with a suitable non-irritating excipient which is solid at room temperature but liquid at the rectal temperature and therefore will melt in the rectum to release the active components. Such materials include, but are not limited to, cocoa butter, beeswax and polyethylene glycols. [0475] The pharmaceutical compositions provided herewith may be administered by nasal aerosol or inhalation. Such compositions are prepared according to techniques well known in the art of pharmaceutical formulation and may be prepared as solutions in saline, employing benzyl alcohol or other suitable preservatives, absorption promoters to enhance bioavailability, fluorocarbons, and/or other solubilizing or dispersing agents known in the art. [0476] The above-described components for orally administrable, injectable or topically administrable, rectally administrable and nasally administrable compositions are merely representative. Other materials as well as processing techniques and the like are set forth in Part 8 of Remington’s Pharmaceutical Sciences, 17th edition, 1985, Mack Publishing Company, Easton, Pennsylvania, which is incorporated herein by reference. [0477] The compounds disclosed herein can also be administered in sustained release forms or from sustained release drug delivery systems. A description of representative sustained release materials can be found in Remington’s Pharmaceutical Sciences. [0478] When the compositions provided herewith comprise a combination of a compound of the formulae described herein and one or more additional therapeutic or prophylactic agents, both the compound and the additional agent should be present at dosage levels of between about 1 to 100%, and more preferably between about 5 to 95% of the dosage normally administered in a monotherapy regimen. The additional agents may be administered separately, as part of a multiple dose regimen, from the compounds provided herewith. Alternatively, those agents may be part of a single dosage form, mixed together with the compounds provided herewith in a single composition. [0479] Also provided are pharmaceutically acceptable acid addition salt of a compound described herein (e.g., compound of Formula (I), (II) or a compound of Table 1). [0480] The acid which may be used to prepare the pharmaceutically acceptable salt is that which forms a non-toxic acid addition salt, i.e., a salt containing pharmacologically acceptable anions such as the hydrochloride, hydroiodide, hydrobromide, nitrate, sulfate, bisulfate, phosphate, acetate, lactate, citrate, tartrate, succinate, maleate, fumarate, benzoate, para-toluenesulfonate, and the like. [0481] The compounds described herein can, for example, be administered by injection, intravenously, intraarterially, subdermally, intraperitoneally, intramuscularly, or subcutaneously; or orally, buccally, nasally, transmucosally, topically, in an ophthalmic preparation, or by inhalation, with a dosage ranging from about 0.5 to about 100 mg/kg of body weight, alternatively dosages between 1 mg and 1000 mg/dose, every 4 to 120 hours, or according to the requirements of the particular drug. The methods herein contemplate administration of an effective amount of compound or compound composition to achieve the desired or stated effect. Typically, the pharmaceutical compositions provided herewith will be administered from about 1 to about 6 times per day or alternatively, as a continuous infusion. Such administration can be used as a chronic or acute therapy. The amount of active ingredient that may be combined with the carrier materials to produce a single dosage form will vary depending upon the host treated and the particular mode of administration. A typical preparation will contain from about 5% to about 95% active compound (w/w). Alternatively, such preparations contain from about 20% to about 80% active compound. [0482] Lower or higher doses than those recited above may be required. Specific dosage and treatment regimens for any particular patient will depend upon a variety of factors, including the activity of the specific compound employed, the age, body weight, general health status, sex, diet, time of administration, rate of excretion, drug combination, the severity and course of the disease, condition or symptoms, the patient’s disposition to the disease, condition or symptoms, and the judgment of the treating physician. [0483] Upon improvement of a patient’s condition, a maintenance dose of a compound, composition or combination provided herewith may be administered, if necessary. Subsequently, the dosage or frequency of administration, or both, may be reduced, as a function of the symptoms, to a level at which the improved condition is retained when the symptoms have been alleviated to the desired level. Patients may, however, require intermittent treatment on a long term basis upon any recurrence of disease symptoms. Methods of Treatment and Use [0484] In an embodiment, the compounds described herein can be used to inhibit the activity of a USP1 protein. For example, in an embodiment, a method of inhibiting a USP1 protein comprises contacting the USP1 protein with a compound disclosed herein. The contacting can occur in vitro or in vivo. [0485] In an embodiment, the compounds described herein can be used to treat a "USP1 protein mediated” disorder (e.g., a USP1 protein mediated cancer), a “USP1 associated” disorder (e.g., a USP1 associated cancer), or a disorder “associated with USP1” (e.g., a cancer associated with USP1). A “USP1 protein mediated”, “USP1 associated” disorder or a disorder “associated with USP1”, is any pathological condition in which a USP1 protein is known to play a role, including any cancers that require USP1 for cell proliferation and survival. In an embodiment, “USP1 protein mediated”, “USP1 associated” disorder or a disorder “associated with USP1” is a proliferative disease such as cancer. The method comprises administering to a patient in need of a treatment for a USP1 protein mediated disorder an effective amount of a compound of Formula (I), (II) or a compound of Table 1, or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof or a pharmaceutical composition comprising an effective amount of a compound of Formula (I), (II) or a compound of Table 1, or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof and a pharmaceutically acceptable excipient. [0486] In an embodiment, provided is a method of treating a disease or disorder associated with modulation of USP1. The method comprises administering to a patient in need of a treatment for diseases or disorders associated with modulation of ubiquitin specific protease 1 (USP1) an effective amount of a compound of Formula (I), (II) or a compound of Table 1, or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof or a pharmaceutical composition comprising an effective amount of a compound of Formula (I), (II) or a compound of Table 1, or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof and a pharmaceutically acceptable excipient. In an embodiment the disease or disorder is cancer. In an embodiment, the compound or composition is administered in combination with a second therapeutic agent. [0487] In an embodiment, provided is a method of treating or preventing cancer. The method comprises administering to a patient in need of a treatment for cancer an effective amount of a compound of Formula (I), (II) or a compound of Table 1 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, tautomer or stereoisomer thereof or a pharmaceutical composition comprising an effective amount of a compound of Formula (I), (II) or a compound of Table 1 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, tautomer or stereoisomer thereof and a pharmaceutically acceptable excipient. [0488] In an embodiment, provided is a method of treating cancer. The method comprises administering to a patient in need thereof of a treatment for cancer an effective amount of a compound of Formula (I), (II) or a compound of Table 1 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, tautomer or stereoisomer thereof or a pharmaceutical composition comprising an effective amount of a compound of Formula (I), (II) or a compound of Table 1 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, tautomer or stereoisomer thereof and a pharmaceutically acceptable excipient. [0489] In an embodiment, provided is a method of treating or preventing a disease or disorder associated with DNA damage. The method comprises administering to a patient in need of a treatment for diseases or disorders associated with DNA damage an effective amount of a compound of Formula (I), (II) or a compound of Table 1 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, tautomer or stereoisomer thereof or a pharmaceutical composition comprising an effective amount of a compound of Formula (I), (II) or a compound of Table 1 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, tautomer or stereoisomer thereof and a pharmaceutically acceptable excipient. In an embodiment the disease is cancer. [0490] In an embodiment, provided is a method of treating a disease or disorder associated with DNA damage. The method comprises administering to a patient in need of a treatment for diseases or disorders associated with DNA damage an effective amount of a compound of Formula (I), (II) or a compound of Table 1 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, tautomer or stereoisomer thereof or a pharmaceutical composition comprising an effective amount of a compound of Formula (I), (II) or a compound of Table 1 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, tautomer or stereoisomer thereof and a pharmaceutically acceptable excipient. [0491] In an embodiment, provided is a method of inhibiting, modulating or reducing DNA repair activity exercised by USP1. The method comprises administering to a patient in need thereof an effective amount of a compound of Formula (I), (II) or a compound of Table 1 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, tautomer or stereoisomer thereof or a pharmaceutical composition comprising an effective amount of a compound of Formula (I), (II) or a compound of Table 1 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, tautomer or stereoisomer thereof and a pharmaceutically acceptable excipient. [0492] In an embodiment, provided is (a) a compound of Formula (I), (II) or a compound of Table 1, or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof or (b) a pharmaceutical composition comprising an effective amount of a compound of Formula (I), (II) or a compound of Table 1 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, tautomer or stereoisomer thereof and a pharmaceutically acceptable excipient, for use as a medicament. [0493] In an embodiment, provided is (a) a compound of Formula (I), (II) or a compound of Table 1, or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, or (b) a pharmaceutical composition comprising an effective amount of a compound of Formula (I), (II) or a compound of Table 1 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, tautomer or stereoisomer thereof and a pharmaceutically acceptable excipient for use in the treatment or prevention of a disease sensitive to the inhibition of USP1. In an embodiment the disease is cancer. [0494] In an embodiment, provided is (a) a compound of Formula (I), (II) or a compound of Table 1 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, tautomer or stereoisomer thereof, or (b) a pharmaceutical composition comprising an effective amount of a compound of Formula (I), (II) or a compound of Table 1 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, tautomer or stereoisomer thereof and a pharmaceutically acceptable excipient for use in the treatment of a disease or disorder sensitive to the inhibition of USP1. [0495] In an embodiment, provided is (a) a compound of Formula (I), (II) or a compound of Table 1 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, tautomer or stereoisomer thereof, or (b) a pharmaceutical composition comprising an effective amount of a compound of Formula (I), (II) or a compound of Table 1 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, tautomer or stereoisomer thereof and a pharmaceutically acceptable excipient for use in the treatment or prevention of cancer. [0496] In an embodiment, provided is (a) a compound of Formula (I), (II) or a compound of Table 1 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, tautomer or stereoisomer thereof, or (b) a pharmaceutical composition comprising an effective amount of a compound of Formula (I), (II) or a compound of Table 1 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, tautomer or stereoisomer thereof and a pharmaceutically acceptable excipient for use in the treatment of cancer. [0497] In an embodiment, provided is (a) a compound of Formula (I), (II) or a compound of Table 1, or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, or (b) a pharmaceutical composition comprising an effective amount of a compound of Formula (I), (II) or a compound of Table 1, or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof and a pharmaceutically acceptable excipient for use in the treatment or prevention of a disease or disorder associated with DNA damage. In an embodiment the disease or disorder is cancer. [0498] In an embodiment, provided is (a) a compound of Formula (I), (II) or a compound of Table 1, or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, or (b) a pharmaceutical composition comprising an effective amount of a compound of Formula (I), (II) or a compound of Table 1, or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof and a pharmaceutically acceptable excipient for use in the treatment of a disease or disorder associated with DNA damage. In an embodiment the disease or disorder is cancer. [0499] In an embodiment, provided is (a) a compound of Formula (I), (II) or a compound of Table 1, or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, or (b) a pharmaceutical composition comprising an effective amount of a compound of Formula (I), (II) or a compound of Table 1, or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof and a pharmaceutically acceptable excipient for use in a method of inhibiting or reducing DNA repair activity modulated by USP1. [0500] In an embodiment, provided is a compound of Formula (I), (II) or a compound of Table 1, or a pharmaceutical composition comprising a compound of Formula (I), (II) or a compound of Table 1 and a pharmaceutically acceptable carrier used for the treatment of cancers. [0501] In an embodiment, provided is the use of (a) a compound of Formula (I), (II) or a compound of Table 1, or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, or (b) a pharmaceutical composition comprising an effective amount of a compound of Formula (I), (II) or a compound of Table 1, or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof and a pharmaceutically acceptable excipient in the manufacture of a medicament for treating or preventing a disease sensitive to the inhibition of USP1. In an embodiment the disease or disorder is cancer. [0502] In an embodiment, provided is the use of (a) a compound of Formula (I), (II) or a compound of Table 1, or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, or (b) a pharmaceutical composition comprising an effective amount of a compound of Formula (I), (II) or a compound of Table 1, or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof and a pharmaceutically acceptable excipient in the manufacture of a medicament for treating or preventing cancer. [0503] In an embodiment, provided is the use of (a) a compound of Formula (I), (II) or a compound of Table 1, or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, or (b) a pharmaceutical composition comprising an effective amount of a compound of Formula (I), (II) or a compound of Table 1, or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof and a pharmaceutically acceptable excipient in the manufacture of a medicament for treating or preventing a disease or disorder associated with DNA damage. In an embodiment, the disease or disorder is cancer. [0504] In an embodiment, provided is the use of (a) a compound of Formula (I), (II) or a compound of Table 1, or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, or (b) a pharmaceutical composition comprising an effective amount of a compound of Formula (I), (II) or a compound of Table 1, or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof and a pharmaceutically acceptable excipient in the manufacture of a medicament for treating a disease or disorder associated with DNA damage. In an embodiment, the disease or disorder is cancer. [0505] In an embodiment, provided is the use of (a) a compound of Formula (I), (II) or a compound of Table 1, or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, or (b) a pharmaceutical composition comprising an effective amount of a compound of Formula (I), (II) or a compound of Table 1, or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof and a pharmaceutically acceptable excipient in the manufacture of a medicament for inhibiting or reducing DNA repair activity modulated by USP1. [0506] In an embodiment, provided is a pharmaceutical composition comprising a compound of Formula (I), (II) or a compound of Table 1, or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof and a pharmaceutically acceptable carrier. The pharmaceutical acceptable carrier may further include an excipient, diluent, or surfactant. [0507] In an embodiment, provided are methods of treating a disease or disorder associated with modulation of USP1 including, but not limited to, cancer comprising, administering to a patient suffering from at least one of said diseases or disorder (a) an effective amount (e.g., a therapeutically effective amount) of a compound of Formula (I), (II) or a compound of Table 1, or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof or (b) a pharmaceutical composition comprising an effective amount (e.g., a therapeutically effective amount) of a compound of Formula (I), (II) or a compound of Table 1, or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof and a pharmaceutically acceptable excipient; and one or more additional anti-cancer agent(s). [0508] In an embodiment, the compound disclosed herein (e.g., a compound of Formula (I), (II) or a compound of Table 1, or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof) and the other anti-cancer agent(s) is generally administered sequentially in any order by infusion or orally. The dosing regimen may vary depending upon the stage of the disease, physical fitness of the patient, safety profiles of the individual drugs, and tolerance of the individual drugs, as well as other criteria well-known to the attending physician and medical practitioner(s) administering the combination. The compound disclosed herein and other anti-cancer agent(s) may be administered within minutes of each other, hours, days, or even weeks apart depending upon the particular cycle being used for treatment. In addition, the cycle could include administration of one drug more often than the other during the treatment cycle and at different doses per administration of the drug. [0509] In an embodiment, provided are kits that include one or more of the compounds disclosed herein (e.g., a compound of Formula (I), (II) or a compound of Table 1 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer or tautomer thereof) and a second therapeutic agent as disclosed herein are provided. Representative kits include (a) a compound disclosed herein or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof (e.g., a compound of Formula (I), (II) or a compound of Table 1 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer or tautomer thereof), (b) at least one other therapeutic agent, e.g., as indicated above, whereby such kit may comprise a package insert or other labeling including directions for administration. [0510] In an embodiment of the methods and uses described herein, the cancer is selected from adrenocortical carcinoma, AIDS-related lymphoma, AIDS-related malignancies, anal cancer, cerebellar astrocytoma, extrahepatic bile duct cancer, bladder cancer, osteosarcoma/malignant fibrous histiocytoma, brain stem glioma, ependymoma, visual pathway and hypothalamic gliomas, breast cancer, bronchial adenomas/carcinoids, carcinoid tumors, gastrointestinal carcinoid tumors, carcinoma, adrenocortical, islet cell carcinoma, primary central nervous system lymphoma, cerebellar astrocytoma, cervical cancer, chronic lymphocytic leukemia, chronic myelogenous leukemia, clear cell sarcoma of tendon sheaths, colon cancer, colorectal cancer, cutaneous t-cell lymphoma, endometrial cancer, ependymoma, esophageal cancer, Ewing’s sarcoma/family of tumors, extracranial germ cell tumors, extragonadal germ cell tumors, extrahepatic bile duct cancer, eye cancers, including intraocular melanoma, and retinoblastoma, gallbladder cancer, gastrointestinal carcinoid tumor, ovarian germ cell tumor, gestational trophoblastic tumor, hairy cell leukemia, head and neck cancer, Hodgkin’s disease, hypopharyngeal cancer, Kaposi’s sarcoma, laryngeal cancer, acute lymphoblastic leukemia, acute myeloid leukemia, liver cancer, non-small cell lung cancer, small cell lung cancer, non-Hodgkin’s lymphoma, Waldenstrom’s macroglobulinemia, malignant mesothelioma, malignant thymoma, medulloblastoma, melanoma, Merkel cell carcinoma, metastatic squamous neck cancer with occult primary, multiple endocrine neoplasia syndrome, multiple myeloma/plasma cell neoplasm, mycosis fungoides, myelodysplastic syndrome, chronic myelogenous leukemia, myeloid leukemia, multiple myeloma, myeloproliferative disorders, nasal cavity and paranasal sinus cancer, nasopharyngeal cancer, neuroblastoma, oral cancer, oral cavity and lip cancer, oropharyngeal cancer, osteosarcoma/malignant fibrous histiocytoma of bone, ovarian cancer, ovarian low malignant potential tumor, pancreatic cancer, paranasal sinus and nasal cavity cancer, parathyroid cancer, penile cancer, pheochromocytoma, pituitary tumor, pleuropulmonary blastoma, prostate cancer, rectal cancer, renal cell (kidney) cancer, transitional cell cancer (e.g., renal pelvis and ureter), retinoblastoma, rhabdomyosarcoma, salivary gland cancer, malignant fibrous histiocytoma of bone, soft tissue sarcoma, Sezary syndrome, skin cancer, small intestine cancer, stomach (gastric) cancer, supratentorial primitive neuroectodennal and pineal tumors, cutaneous t-cell lymphoma, testicular cancer, malignant thymoma, thyroid cancer, gestational trophoblastic tumor, urethral cancer, uterine sarcoma, vaginal cancer, vulvar cancer, and Wilms’ tumor. In other embodiments, the cancer is a non-small cell lung cancer. [0511] In any of the embodiments, the cancer can be any cancer in any organ, for example, a cancer is selected from the group consisting of glioma, thyroid carcinoma, breast carcinoma, small-cell lung carcinoma, non-small-cell carcinoma, gastric carcinoma, colon carcinoma, gastrointestinal stromal carcinoma, pancreatic carcinoma, bile duct carcinoma, CNS carcinoma, ovarian carcinoma, endometrial carcinoma, prostate carcinoma, renal carcinoma, anaplastic large-cell lymphoma, leukemia, multiple myeloma, mesothelioma, and melanoma, and combinations thereof. [0512] In an embodiment, the cancer to be treated with a compound disclosed herein is selected from the group consisting of bone cancer, including osteosarcoma and chondrosarcoma; brain cancer, including glioma, glioblastoma, astrocytoma, medulloblastoma, and meningioma; soft tissue cancer, including rhabdoid and sarcoma; kidney cancer; bladder cancer; skin cancer, including melanoma; and lung cancer, including non-small cell lung cancer; colon cancer, uterine cancer; nervous system cancer; head and neck cancer; pancreatic cancer; and cervical cancer. [0513] In other embodiments, the cancer is selected from liposarcoma, neuroblastoma, glioblastoma, bladder cancer, adrenocortical cancer, multiple myeloma, colorectal cancer, non- small cell lung cancer, Human Papilloma Virus-associated cervical, oropharyngeal, penis, anal, thyroid or vaginal cancer or Epstein-Barr Virus-associated nasopharyngeal carcinoma, gastric cancer, rectal cancer, thyroid cancer, Hodgkin lymphoma and diffuse large B-cell lymphoma. [0514] In an embodiment, the cancer is selected from breast cancer (e.g., triple negative breast cancer (TNBC)), ovarian cancer (e.g., platinum-resistant ovarian cancer, platinum- refractory ovarian cancer), prostate cancer, pancreatic cancer and lung cancer (e.g., non-small cell lung cancer (NSCLC)). In an embodiment, the cancer is selected from breast cancer (e.g., triple negative breast cancer (TNBC)), ovarian cancer (e.g., platinum-resistant ovarian cancer, platinum-refractory ovarian cancer), prostate cancer and lung cancer (e.g., non-small cell lung cancer (NSCLC)). In an embodiment, the cancer is breast cancer. In an embodiment the cancer is triple negative breast cancer (TNBC). In an embodiment the cancer is prostate cancer. In an embodiment the cancer is lung cancer. In an embodiment the cancer is non- small cell lung cancer (NSCLC). [0515] In an embodiment of the methods described herein, the cancer is a dedifferentiated ID-driven cancer. In other embodiments, the cancer is a cancer that is sensitive to USP1 inhibition. In yet other embodiments, the cancer is a cancer that is sensitive to USP1 inhibition due to DNA damage pathway deficiency. [0516] In an embodiment of the methods and uses described herein, the cancer is selected from the group consisting of a hematological cancer, a lymphatic cancer, and a DNA damage repair pathway deficient cancer. [0517] In an embodiment, a compound disclosed herein is used to treat a cancer, wherein the cancer is a homologous recombination deficient cancer. In an embodiment, a compound disclosed herein is used to treat a cancer that does not have a defect in the homologous recombination pathway. [0518] In an embodiment, the cancer is a DNA damage repair pathway deficient cancer. In an embodiment, the DNA damage repair pathway deficient cancer is selected from the group consisting of lung cancer, non-small cell lung cancer (NSCLC), colon cancer, bladder cancer, osteosarcoma, ovarian cancer (e.g., platinum-resistant ovarian cancer, platinum-refractory ovarian cancer), and breast cancer (e.g., triple negative breast cancer (TNBC). In an embodiment, the cancer is non-small cell lung cancer (NSCLC). In an embodiment, the cancer is colon cancer. In an embodiment, the cancer is bladder cancer. In an embodiment, the cancer is ovarian cancer or breast cancer. In an embodiment, the cancer is ovarian cancer. In an embodiment, the cancer is platinum-resistant ovarian cancer. In some embodiments, the cancer is platinum-refractory ovarian cancer. In an embodiment, the cancer is breast cancer. In an embodiment, the cancer is triple negative breast cancer. [0519] In an embodiment, the cancer is a HRR (homologous recombination repair) gene mutant cancer. In an embodiment, the cancer is a HRR (homologous recombination repair) gene mutant cancer selected from the group consisting of ATM, NBN, FANCA, BARD1, BRCA1, BRCA2, BRIP1, CDK12, CHEK1, CHEK2, FANCL, PALB2, PPP2R2A, RAD51, RAD51B, RAD51C, RAD51D, or RAD54L mutant cancer. In an embodiment, the cancer is an ATM mutant cancer. In an embodiment, the cancer is an NBN mutant cancer. In an embodiment, the cancer is a FANCA mutant cancer. In an embodiment, the cancer is an BARD1 mutant cancer. In an embodiment, the cancer is an BRCA1 mutant cancer. In an embodiment, the cancer is an BRCA2 mutant cancer. In an embodiment, the cancer is an BRIP1 mutant cancer. In an embodiment, the cancer is an CDK12 mutant cancer. In an embodiment, the cancer is an CHEK1 mutant cancer. In an embodiment, the cancer is an CHEK2 mutant cancer. In an embodiment, the cancer is an FANCL mutant cancer. In an embodiment, the cancer is an PALB2 mutant cancer. In an embodiment, the cancer is an PPP2R2A mutant cancer. In an embodiment, the cancer is an RAD51B mutant cancer. In an embodiment, the cancer is an RAD51C mutant cancer. In an embodiment, the cancer is an RAD51D mutant cancer. In an embodiment, the cancer is an RAD54L mutant cancer. [0520] In an embodiment, the cancer is a BRCA1 mutant cancer. In an embodiment, the BRCA1 mutation is a germline mutation. In an embodiment, the BRCA1 mutation is a somatic mutation. In an embodiment, the BRCA1 mutation leads to BRCA1 deficiency. In an embodiment, the cancer is a BRCA2 mutant cancer. In an embodiment, the BRCA2 mutation is a germline mutation. In an embodiment, the BRCA2 mutation is a somatic mutation. In an embodiment, the BRCA2 mutation leads to BRCA2 deficiency. In an embodiment, the cancer is a BRCA1 mutant cancer and a BRCA2 mutant cancer. In an embodiment, the cancer is a BRCA1 deficient cancer. In an embodiment, the cancer is a BRCA2 deficient cancer. In an embodiment, the cancer is a BRCA1 deficient cancer and a BRCA2 deficient cancer. In an embodiment, the cancer is not a BRCA1 mutant cancer or a BRCA2 mutant cancer. In an embodiment, the cancer is a BRCA1 deficient cancer and a BRCA2 mutant cancer. In an embodiment, the BRCA1 or BRCA2 mutant or BRCA1 or BRCA2 deficient cancer is selected from non-small cell lung cancer (NSCLC), osteosarcoma, prostate cancer, pancreatic cancer, ovarian cancer, and breast cancer. In an embodiment, the BRCA1 mutant, BRCA2 mutant, BRCA1 deficient or BRCA 2 deficient cancer as described herein is ovarian cancer, breast cancer, prostate cancer or pancreatic cancer. In an embodiment, the cancer is ovarian cancer. In an embodiment, the cancer is platinum-resistant ovarian cancer. In some embodiments, the cancer is platinum-refractory ovarian cancer. In an embodiment, the cancer is breast cancer. In an embodiment, the cancer is a triple negative breast cancer. In an embodiment, the cancer is prostate cancer. In an embodiment, the cancer is homologous recombination deficient. Homologous recombination deficiency can be measured by BRCA1/2 mutation, or genomic instability (positive homologous recombination deficiency (HRD) score) without BRCA1/2 mutations. [0521] In an embodiment, the cancer is a Poly (ADP-ribose) polymerase ("PARP") inhibitor refractory or resistant cancer. In an embodiment, the cancer is a PARP inhibitor resistant or refractory BRCA1, BRCA2, or BRCA1 and BRCA2 mutant cancer. In an embodiment, the cancer is a PARP inhibitor resistant or refractory BRCA1, BRCA2, or BRCA1 and BRCA2- deficient cancer. In an embodiment, the PARP inhibitor refractory or resistant cancer is selected from the cancers described herein. In an embodiment, the PARP inhibitor refractory or resistant cancer is selected from breast cancer (e.g., triple negative breast cancer (TNBC), ovarian cancer (e.g., platinum-resistant ovarian cancer, platinum-refractory ovarian cancer), pancreatic cancer and prostate cancer). [0522] In an embodiment, the cancer has a mutation in the gene encoding ataxia telangiectasia mutated (ATM) protein kinase or loss of ATM protein expression. In an embodiment, the cancer to be treated with a compound disclosed herein is a cancer (e.g., a cancer selected from the cancers described herein) that comprises cancer cells with a loss of function mutation in a gene encoding ATM. In an embodiment the ATM mutation is a germline mutation. In an embodiment the ATM mutation is a somatic mutation. In an embodiment, the cancer is not an ATM mutant cancer. In an embodiment the cancer is an ATM-deficient cancer. In an embodiment, the ATM-deficient cancer is selected from breast cancer (e.g., triple negative breast cancer (TNBC)), ovarian cancer (e.g., platinum-resistant ovarian cancer, platinum-refractory ovarian cancer), colorectal cancer, stomach cancer, endometrial cancer, urothelial cancer, cervical cancer, melanoma, esophageal cancer, head and neck cancer, mantle cell lymphoma, sarcoma, prostate cancer, pancreatic cancer, and lung cancer (e.g., non-small cell lung cancer (NSCLC)). [0523] In an embodiment, the cancer comprises cancer cells with elevated levels of translesion synthesis. This includes cancers that exhibit elevated PCNA monoubiquitination, with or without elevated levels of RAD18 and/or UBE2K and/or PCNA. In an embodiment, the elevated levels of RAD18 and/or UBE2K and/or PCNA are elevated RAD18 and/or UBE2K and/or PCNA protein levels. In an embodiment, the elevated levels of RAD18 and/or UBE2K and/or PCNA are elevated RAD18 and/or UBE2K and/or PCNA mRNA levels. In an embodiment, elevated levels of RAD18 and/or UBE2K and/or PCNA (e.g., RAD18 and/or UBE2K and/or PCNA protein and/or RAD18 and/or UBE2K and/or PCNA mRNA) have been detected (e.g., in a cancer sample obtained from the subject) prior to the administration. Elevated translesion synthesis can also be measured by PCNA monoubiquitination without elevated RAD18 and/or UBE2K and/or PCNA levels. In an embodiment, a subject’s cancer has been tested for RAD18 and/or UBE2K and/or PCNA levels protein or mRNA, or PCNA monoubiquitination prior to beginning treatment with a USP1 inhibitor. In an embodiment, the cancer is a breast cancer (e.g., triple negative breast cancer), an ovarian cancer, a lung cancer (e.g., non-small cell lung cancer (NSCLC)), or a prostate cancer. [0524] In an embodiment, the cancer is a BRCA1 and/or BRCA2 mutant cancer, wherein the cancer comprises cells with increased translesion synthesis, as exemplified by elevated PCNA monoubiquitination with or without elevated RAD18 and/or UBE2K and/or PCNA levels. In an embodiment, the cancer is a breast cancer (e.g., triple negative breast cancer), an ovarian cancer or a prostate cancer that is a BRCA1 and/or BRCA2 mutant cancer. [0525] In an embodiment, the cancer is selected from the group consisting of bone cancer, including osteosarcoma and chondrosarcoma; brain cancer, including glioma, glioblastoma, astrocytoma, medulloblastoma, and meningioma; soft tissue cancer, including rhabdoid and sarcoma; kidney cancer; bladder cancer; skin cancer, including melanoma; and lung cancer, including non-small cell lung cancer; colon cancer, uterine cancer; nervous system cancer; head and neck cancer; pancreatic cancer; and cervical cancer. Combination therapies [0526] In an embodiment, the compounds of the disclosure are administered in therapeutically effective amounts in a combination therapy with one or more therapeutic agents (pharmaceutical combinations) or modalities, e.g. non-drug therapies. For example, synergistic effects can occur with other anti-proliferative, anti-cancer, immunomodulatory or anti-inflammatory substances. Where the compounds of the disclosure are administered in conjunction with other therapies, dosages of the co-administered compounds will vary depending on the type of co-drug employed, on the specific drug employed, on the condition being treated and so forth. [0527] In an embodiment, provided are methods of treatment of a disease or disorder associated with the USP1 with a compound of Formula (I), (II) or a compound of Table 1 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer or tautomer thereof) in combination with a second therapeutic agent. In an embodiment, provided are methods of treatment of a disease or disorder associated with USP1 with a compound of Formula (I), (II) or a compound of Table 1 or pharmaceutically acceptable salts thereof in combination with a second therapeutic agent and a third therapeutic agent. In an embodiment, provided are methods of treatment of a disease or disorder associated with the USP1 with a compound of Formula (I), (II) or a compound of Table 1, or pharmaceutically acceptable salts thereof) in combination with a second therapeutic agent, a third therapeutic agent, and a fourth therapeutic agent. [0528] The term “Combination” refers to either a fixed combination in one dosage unit form, or a combined administration where a compound disclosed herein (e.g., a compound of Formula (I), (II) or a compound of Table 1 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer or tautomer thereof) and a combination partner (e.g., another drug as explained below, also referred to as “therapeutic agent” or “co-agent”) may be administered independently at the same time or separately within time intervals, especially where these time intervals allow that the combination partners show a cooperative, e.g., synergistic effect. The single components may be packaged in a kit or separately. One or both of the components (e.g., powders or liquids) may be reconstituted or diluted to a desired dose prior to administration. The terms “co-administration” or “combined administration” or the like as utilized herein are meant to encompass administration of the selected combination partner to a single subject in need thereof (e.g., a patient), and are intended to include treatment regimens in which the agents are not necessarily administered by the same route of administration or at the same time. The term “pharmaceutical combination” as used herein means a product that results from the mixing or combining of more than one therapeutic agent and includes both fixed and non–fixed combinations of the therapeutic agents. The term “fixed combination” means that the therapeutic agents, e.g., a compound disclosed herein (e.g., a compound of Formula (I), (II) or a compound of Table 1 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer or tautomer thereof) and a combination partner, are both administered to a patient simultaneously in the form of a single entity or dosage. The term “non-fixed combination” means that the therapeutic agents, e.g., a compound disclosed herein (e.g., a compound of Formula (I), (II) or a compound of Table 1 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer or tautomer thereof) and a combination partner, are both administered to a patient as separate entities either simultaneously, concurrently or sequentially with no specific time limits, wherein such administration provides therapeutically effective levels of the two compounds in the body of the patient. The latter also applies to cocktail therapy, e.g., the administration of three or more therapeutic agent. [0529] The term "combination therapy" refers to the administration of two or more therapeutic agents to treat a therapeutic condition or disorder described in the present disclosure. Such administration encompasses co-administration of these therapeutic agents in a substantially simultaneous manner, such as in a single capsule having a fixed ratio of active ingredients. Alternatively, such administration encompasses co-administration in multiple, or in separate containers (e.g., tablets, capsules, powders, and liquids) for each active ingredient. Powders and/or liquids may be reconstituted or diluted to a desired dose prior to administration. In addition, such administration also encompasses use of each type of therapeutic agent in a sequential manner, either at approximately the same time or at different times. [0530] In an embodiment, compounds disclosed herein are combined with other therapeutic agents, including, but not limited to, other anti-cancer agents, anti-allergic agents, anti-nausea agents (or anti-emetics), pain relievers, cytoprotective agents, and combinations thereof. [0531] In an embodiment, provided is a method of treating a disease or disorder associated with USP1 (e.g., cancer) comprising administering or coadministering, in any order, to a patient in need thereof a compound disclosed herein (e.g., a compound of Formula (I), (II) or a compound of Table 1 or pharmaceutically acceptable salts thereof) and a general chemotherapeutic agent selected from anastrozole (Arimidex®), bicalutamide (Casodex®), bleomycin sulfate (Blenoxane®), busulfan (Myleran®), busulfan injection (Busulfex®), capecitabine (Xeloda®), N4-pentoxycarbonyl-5-deoxy-5-fluorocytidine, carboplatin (Paraplatin®), carmustine (BiCNU®), chlorambucil (Leukeran®), cisplatin (Platinol®), cladribine (Leustatin®), cyclophosphamide (Cytoxan® or Neosar®), cytarabine, cytosine arabinoside (Cytosar-U®), cytarabine liposome injection (DepoCyt®), dacarbazine (DTIC- Dome®), dactinomycin (Actinomycin D, Cosmegan), daunorubicin hydrochloride (Cerubidine®), daunorubicin citrate liposome injection (DaunoXome®), dexamethasone, docetaxel (Taxotere®), doxorubicin hydrochloride (Adriamycin®, Rubex®), etoposide (Vepesid®), fludarabine phosphate (Fludara®), 5-fluorouracil (Adrucil®, Efudex®), flutamide (Eulexin®), tezacitibine, Gemcitabine (difluorodeoxycitidine), hydroxyurea (Hydrea®), Idarubicin (Idamycin®), ifosfamide (IFEX®), irinotecan (Camptosar®), L- asparaginase (ELSPAR®), leucovorin calcium, melphalan (Alkeran®), 6-mercaptopurine (Purinethol®), methotrexate (Folex®), mitoxantrone (Novantrone®), mylotarg, paclitaxel (Taxol®), nab–paclitaxel (Abraxane®), phoenix (Yttrium90/MX-DTPA), pentostatin, polifeprosan 20 with carmustine implant (Gliadel®), tamoxifen citrate (Nolvadex®), teniposide (Vumon®), 6–thioguanine, thiotepa, tirapazamine (Tirazone®), topotecan hydrochloride for injection (Hycamptin®), vinblastine (Velban®), vincristine (Oncovin®), and vinorelbine (Navelbine®). [0532] In an embodiment, provided is a method of treating a disease or disorder associated with USP1 (e.g., cancer) comprising administering or coadministering, in any order, to a patient in need thereof a compound disclosed herein (e.g., a compound of Formula (I), (II) or a compound of Table 1 or pharmaceutically acceptable salts thereof) and an EGFR- inhibitor(e.g., cetuximab, panitumimab, erlotinib, gefitinib and EGFRi NOS). In an embodiment, provided is a method of treating a disease or disorder associated with USP1 (e.g., cancer) comprising administering or coadministering, in any order, to a patient in need thereof a compound disclosed herein (e.g., a compound of Formula (I), (II) or a compound of Table 1 or pharmaceutically acceptable salts thereof) and a MAPK-pathway inhibitor (e.g., BRAFi, panRAFi, MEKi, ERKi) In an embodiment, provided is a method of treating a disease or disorder associated with USP1 (e.g., cancer) comprising administering or coadministering, in any order, to a patient in need thereof a compound disclosed herein (e.g., a compound of Formula (I), (II) or a compound of Table 1 or pharmaceutically acceptable salts thereof) and a PI3K-mTOR pathway inhibitor (e.g., alpha-specific PI3Ki, pan-class I PI3Ki and mTOR/PI3Ki, particularly everolimus and analogues thereof). [0533] In an embodiment, provided is a method of enhancing the chemotherapeutic treatment of cancer in a mammal undergoing treatment with an anti-cancer agent, which method comprises co-administering to the mammal an effective amount of a compound disclosed herein. In an embodiment, provided is a method of treating a disease or disorder associated with USP1 (e.g., cancer) comprising administering or coadministering, in any order, to a patient in need thereof a compound disclosed herein (e.g., a compound of Formula (I), (II) or a compound of Table 1 or pharmaceutically acceptable salts thereof) and a DNA damaging agent (e.g., actinomycin, amsacrine, anthracyclines, bleomycin, busulfan, camptothecin, carboplatin, chlorambucil, cisplatin, cyclophosphamide, Cytoxan, dactinomycin, daunorubicin, doxorubicin, epirubicin, hexamethylmelamineoxaliplatin, iphosphamide, melphalan, merchlorehtamine, mitomycin, mitoxantrone, nitrosourea, plicamycin, procarbazine, taxol, taxotere, tenyposide, triethylenethiophosphoramide and etoposide). In a preferred embodiment, the DNA damaging agent is cisplatin. In an embodiment, the DNA damaging agent is radiation or a biotherapeutic agent (e.g., an antibody). [0534] In an embodiment, the anti-cancer agent is selected from reversible DNA binders (e.g., topotecan hydrochloride, irinotecan (CPT11 – Camptosar), rubitecan, exatecan, nalidixic acid, TAS-103, etoposide, acridines (e.g., amsacrine, aminocrine), actinomycins (e.g., actinomycin D), anthracyclines (e.g., doxorubicin, daunorubicin), benzophenainse, XR 11576/MLN 576, benzopyridoindoles, Mitoxantrone, AQ4, Etoposide, Teniposide, epipodophyllotoxins, and bisintercalating agents such as triostin A and echinomycin), DNA alkylators (e.g., sulfur mustard, the nitrogen mustards (e.g., mechlorethamine), chlorambucil, melphalan, ethyleneimines (e.g., triethylenemelamine, carboquone, diaziquone), methyl methanesulfonate, busulfan, CC-1065, duocarmycins (e.g., duocarmycin A, duocarmycin SA), metabolically activated alkylating agents such as nitrosoureas (e.g., carmustine, lomustine, (2-chloroethyl)nitrosoureas), triazine antitumor drugs such as triazenoimidazole (e.g., dacarbazine), mitomycin C and leinamycin), DNA strand breakers (e.g., doxorubicin and daunorubicin (which are also reversible DNA binders), other anthracyclines, bleomycins, tirapazamine, enediyne antitumor antibiotics such as neocarzinostatin, esperamicins, calicheamicins, dynemicin A, hedarcidin, C-1027, N1999A2, esperamicins and zinostatin), and disruptors of DNA replication (e.g., 5-fluorodeoxyuridine). [0535] In an embodiment, the DNA damaging agent is radiation (e.g., radiation that induces a DNA cross-linking in a cell when applied to the cell, (e.g., ionizing radiation and ultraviolet (UV) radiation)). Ionizing radiation consists of subatomic particles or electromagnetic waves that are sufficiently energetic to cause ionization by detaching electrons from atoms or molecules. Ionization depends on the energy of the impinging individual particles or waves. In general, ionizing particles or photons with energies above a few electron volts can be ionizing. Non-limiting examples of ionizing particles are alpha particles, beta particles, and neutrons. The ability of photons to ionize an atom or molecule depends on its frequency. Short-wavelength radiation such as high frequency ultraviolet, x-rays, and gamma rays, is ionizing. Ionizing radiation comes from radioactive materials, x-ray tubes, and particle accelerators. [0536] In an embodiment, the anticancer agent targets a USP1 independent mechanism of DNA repair. Non-limiting examples of suitable DNA repair inhibitors are poly (ADP-ribose) polymerase (PARP) inhibitors, DNA-dependent protein kinase (DNA-PK) inhibitors, ataxia telangiectasia and Rad3-related protein (ATR) inhibitors, ataxia-telangiectasia mutated (ATM) inhibitors, checkpoint kinase 1 (CHK1) inhibitors, checkpoint kinase 2 (CHK2) inhibitors, and Wee1 inhibitors. It has been reported that BRCA1/2 status predicts the efficacy of PARP inhibitors in the clinic (Audeh et al. Lancet (2010) 376 (9737), 245-51). In general, BRCA1/2 mutant cancers have increased sensitivity to USP1 inhibitors. Accordingly, in an embodiment, a In an embodiment, provided is a method of treating a disease or disorder associated with USP1 (e.g., cancer) comprising administering or coadministering, in any order, to a patient in need thereof a compound disclosed herein (e.g., a compound of Formula (I), (II) or a compound of Table 1 or pharmaceutically acceptable salts thereof) and a PARP inhibitor (e.g., olaparib, rucaparib, niraparib, talazoparib, and veliparib). [0537] In an embodiment, the anticancer or DNA damaging agent can be a biotherapeutic. Non-limiting examples of suitable biotherapeutics include rInterferon-a2a, rlnterferon-oi2b, rInterleukin-2, rG-CSF, rGM-CSF, and rErythropoietin. [0538] In an embodiment, the anticancer agent can be an antibody, such as a monoclonal antibody. Non-limiting examples of suitable therapeutic monoclonal antibodies for use in the methods described herein include trastuzumab, an anti-ErbB2/HER2 for breast cancer, cetuximab, an anti-ErbBl/EGFR for colorectal cancer, and bevacizumab, an anti-VEGF for colorectal, breast and lung cancers (G. Adams et al., Nature Biotechnology 23: 1147-57 (2005)). Multitarget inhibitors, such as Sutent which inhibits TK activity of VEGFR, PDGFR and FGFR, are also suitable for use in the inventive method. [0539] In an embodiment, the anticancer agent can be a proteasome inhibitor, such as bortezomib. [0540] Administration of the compounds disclosed herein can be accomplished via any mode of administration of therapeutic agents including systemic or local administration such as oral, nasal, parenteral, transdermal, subcutaneous, vaginal, buccal, rectal or topical administration modes. [0541] Some patients may experience allergic reactions to the compounds disclosed herein and/or other anti-cancer agent(s) during or after administration; therefore, anti-allergic agents are often administered to minimize the risk of an allergic reaction. In an embodiment, provided is a method of treating a disease or disorder associated with USP1 (e.g., cancer) comprising administering or coadministering, in any order, to a patient in need thereof a compound disclosed herein (e.g., a compound of Formula (I), (II) or a compound of Table 1 or pharmaceutically acceptable salts thereof) and an anti-allergic agent(e.g., corticosteroids, including, but not limited to, dexamethasone (e.g., Decadron®), beclomethasone (e.g., Beclovent®), hydrocortisone (also known as cortisone, hydrocortisone sodium succinate, hydrocortisone sodium phosphate, and sold under the tradenames Ala–Cort®, hydrocortisone phosphate, Solu–Cortef®, Hydrocort Acetate® and Lanacort®), prednisolone (sold under the tradenames Delta–Cortel®, Orapred®, Pediapred® and Prelone®), prednisone (sold under the tradenames Deltasone®, Liquid Red®, Meticorten® and Orasone®), methylprednisolone (also known as 6–methylprednisolone, methylprednisolone acetate, methylprednisolone sodium succinate, sold under the tradenames Duralone®, Medralone®, Medrol®, M- Prednisol® and Solu–Medrol®); antihistamines, such as diphenhydramine (e.g., Benadryl®), hydroxyzine, and cyproheptadine; and bronchodilators, such as the beta-adrenergic receptor agonists, albuterol (e.g., Proventil®), and terbutaline (Brethine®)). [0542] Some patients may experience nausea during and after administration of the compound disclosed herein and/or other anti-cancer agent(s); therefore, anti–emetics are used in preventing nausea (upper stomach) and vomiting. In an embodiment, provided is a method of treating a disease or disorder associated with USP1 (e.g., cancer) comprising administering or coadministering, in any order, to a patient in need thereof a compound disclosed herein (e.g., a compound of Formula (I), (II) or a compound of Table 1 or pharmaceutically acceptable salts thereof) and an anti-emetic(e.g., aprepitant (Emend®), ondansetron (Zofran®), granisetron HCl (Kytril®), lorazepam (Ativan®. dexamethasone (Decadron®), prochlorperazine (Compazine®), casopitant (Rezonic® and Zunrisa®), and combinations thereof). [0543] Medication to alleviate the pain experienced during the treatment period is often prescribed to make the patient more comfortable. In an embodiment, provided is a method of treating a disease or disorder associated with USP1 (e.g., cancer) comprising administering or coadministering, in any order, to a patient in need thereof a compound disclosed herein (e.g., a compound of Formula (I), (II) or a compound of Table 1 or pharmaceutically acceptable salts thereof) and an analgesic (e.g., an over-the-counter analgesics, (e.g., Tylenol®), an opioid analgesic (e.g., hydrocodone/paracetamol or hydrocodone/acetaminophen (e.g., Vicodin®), morphine (e.g., Astramorph® or Avinza®), oxycodone (e.g., OxyContin® or Percocet®), oxymorphone hydrochloride (Opana®), and fentanyl (e.g., Duragesic®)). [0544] In an effort to protect normal cells from treatment toxicity and to limit organ toxicities, cytoprotective agents (such as neuroprotectants, free-radical scavengers, cardioprotectors, anthracycline extravasation neutralizers, nutrients and the like) may be used as an adjunct therapy. In an embodiment, provided is a method of treating a disease or disorder associated with USP1 (e.g., cancer) comprising administering or coadministering, in any order, to a patient in need thereof a compound disclosed herein (e.g., a compound of Formula (I), (II) or a compound of Table 1 or pharmaceutically acceptable salts thereof) and a cytoprotective agent (e.g., Amifostine (Ethyol®), glutamine, dimesna (Tavocept®), mesna (Mesnex®), dexrazoxane (Zinecard® or Totect®), xaliproden (Xaprila®), and leucovorin (also known as calcium leucovorin, citrovorum factor and folinic acid)). [0545] The structure of the active compounds identified by code numbers, generic or trade names may be taken from the actual edition of the standard compendium “The Merck Index” or from databases, e.g., Patents International (e.g., IMS World Publications). [0546] The above-mentioned compounds, which can be used in combination with a compound disclosed herein, can be prepared and administered as described in the art, including, but not limited to, in the documents cited above. [0547] In an embodiment, provided are pharmaceutical compositions comprising at least one compound disclosed herein (e.g., a USP1 inhibitor, e.g., a compound of Formula (I), (II) or a compound of Table 1 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer or tautomer thereof together with a pharmaceutically acceptable carrier suitable for administration to a human or animal subject, either alone or together with other anti- cancer agents. [0548] In an embodiment, provided are methods of treating human or animal subjects having or having been diagnosed with a disease or disorder associated with USP1 (e.g., cancer) comprising administering to the subject in need thereof a therapeutically effective amount of a compound disclosed herein (e.g., a compound of Formula (I), (II) or a compound of Table 1 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer or tautomer thereof in combination with a second therapeutic agent. [0549] In an embodiment, provided are methods of treating a disease or disorder associated with USP1 (e.g., cancer) in a subject in need thereof comprising administering to the subject a therapeutically effective amount of a compound disclosed herein (e.g., a compound of Formula (I), (II) or a compound of Table 1 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer or tautomer thereof in combination with a second therapeutic agent. [0550] In particular, compositions will either be formulated together as a combination therapeutic or administered separately. [0551] In combination therapy, the compound disclosed herein and other anti-cancer agent(s) may be administered either simultaneously, concurrently or sequentially with no specific time limits, wherein such administration provides therapeutically effective levels of the two compounds in the body of the patient. [0552] A compound disclosed herein (e.g., a compound of Formula (I), (II) or a compound of Table 1 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer or tautomer thereof) may also be used in combination with known therapeutic processes, for example, the administration of hormones or especially radiation. A compound disclosed herein may in particular be used as a radiosensitizer, especially for the treatment of tumors which exhibit poor sensitivity to radiotherapy. [0553] In certain instances, compounds disclosed herein are combined with other therapeutic agents, including, but not limited to, other anti-cancer agents, anti–allergic agents, anti- nausea agents (or anti-emetics), pain relievers, cytoprotective agents, and combinations thereof. Patient Selection and Monitoring Determining whether a subject will respond to treatment with USP1 inhibitors [0554] In an embodiment, provided is a method of determining if a subject having or having been diagnosed with a cancer (e.g., a cancer associated with USP1) (i.e., a cancer patient (e.g., a USP1-associated cancer patient)) will respond to therapeutic treatment with a USP1 inhibitor (e.g., a compound of Formula (I), (II) or a compound of Table 1 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer or tautomer thereof), comprising the steps of: a) detecting levels of RAD18 and/or UBE2K and/or PCNA (e.g., RAD18 and/or UBE2K and/or PCNA protein and/or RAD18 and/or UBE2K and/or PCNA mRNA) in a test cancer sample (e.g., in a cancer test sample obtained from the subject); b) comparing the test cancer sample with reference cells (e.g., a reference sample taken from a non-cancerous or normal control subject), wherein elevated levels of RAD18 and/or UBE2K and/or PCNA in said test cancer sample indicates that the subject will respond to therapeutic treatment with a USP1 inhibitor (e.g., a compound of Formula (I), (II) or a compound of Table 1 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer or tautomer thereof). [0555] In an embodiment, provided is a method of determining if a subject having or having been diagnosed with a cancer (e.g., a cancer associated with USP1) (i.e., a cancer patient (e.g., a USP1-associated cancer patient)) will respond to therapeutic treatment with a USP1 inhibitor (e.g., a compound of Formula (I), (II) or a compound of Table 1 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer or tautomer thereof), comprising the steps of: a) detecting levels of translesion synthesis (e.g., detecting PCNA monoubiquitination levels) in a test cancer sample (e.g., in a cancer test sample obtained from the subject); b) comparing the test cancer sample with reference cells (e.g., a reference sample taken from a non-cancerous or normal control subject), wherein elevated translesion synthesis (e.g., increased PCNA monoubiquitination levels) in said test cancer sample indicates that the subject will respond to therapeutic treatment with a USP1 inhibitor (e.g., a compound of Formula (I), (II) or a compound of Table 1 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer or tautomer thereof). [0556] In an embodiment, provided is a method of determining if a subject having or having been diagnosed with a cancer (e.g., a cancer associated with USP1) (i.e., a cancer patient (e.g., a USP1-associated cancer patient)) will respond to therapeutic treatment with a USP1 inhibitor (e.g., a compound of Formula (I), (II) or a compound of Table 1 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer or tautomer thereof), comprising the steps of: a) detecting mutations in a gene encoding ATM (i.e., loss function mutations) in a test cancer sample (e.g., in a cancer test sample obtained from the subject); b) wherein presence of mutations in a gene encoding ATM in said test cancer sample indicates that the subject will respond to therapeutic treatment with a USP1 inhibitor (e.g., a compound of Formula (I), (II) or a compound of Table 1 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer or tautomer thereof). [0557] In an embodiment, provided is a method of determining if a subject having or having been diagnosed with a cancer (e.g., a cancer associated with USP1) (i.e., a cancer patient (e.g., a USP1-associated cancer patient)) will respond to therapeutic treatment with a USP1 inhibitor (e.g., a compound of Formula (I), (II) or a compound of Table 1 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer or tautomer thereof), comprising the steps of: a) detecting germline or somatic mutations in a gene encoding BRCA1 (e.g., a loss of function mutation) in a subject test sample (e.g., in a cancer test sample or blood test sample obtained from the subject); b) wherein presence of mutations in a gene encoding BRCA1 in said test sample indicates that the subject will respond to therapeutic treatment with a USP1 inhibitor (e.g., a compound of Formula (I), (II) or a compound of Table 1 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer or tautomer thereof). [0558] In an embodiment, provided is a method of determining if a subject having or having been diagnosed with a cancer (e.g., a cancer patient) will respond to therapeutic treatment with a USP1 inhibitor (e.g., a compound of Formula (I), (II) or a compound of Table 1 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer or tautomer thereof), comprising the steps of: a) detecting germline or somatic mutations in a gene encoding BRCA2 (e.g., a loss of function mutation) in a subject test sample (e.g., in a cancer test sample or blood test sample obtained from the subject); b) wherein presence of mutations in a gene encoding BRCA2 in said test sample indicates that the subject will respond to therapeutic treatment with a USP1 inhibitor (e.g., a compound of Formula (I), (II) or a compound of Table 1 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer or tautomer thereof). [0559] In an embodiment, provided is a method of determining if a subject having or having been diagnosed with a cancer (e.g., a cancer associated with USP1) (i.e., a cancer patient (e.g., a USP1-associated cancer patient)) will respond to therapeutic treatment with a USP1 inhibitor (e.g., a compound of Formula (I), (II) or a compound of Table 1 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer or tautomer thereof), comprising the steps of: a) detecting deficiency in homologous recombination (e.g., as measured by a positive homologous recombination deficiency (HRD) score) in a subject test sample (e.g., in a cancer sample or blood sample obtained from the subject) b) wherein presence of homologous recombination deficiency (e.g., a positive homologous recombination deficiency (HRD) score) in said test sample indicates that the subject will respond to therapeutic treatment with a USP1 inhibitor (e.g., a compound of Formula (I), (II) or a compound of Table 1 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer or tautomer thereof). [0560] In an embodiment, the cancer is a cancer selected from the cancers disclosed herein. In an embodiment, the cancer is pancreatic cancer, breast cancer (e.g., triple negative breast cancer (TNBC)), ovarian cancer (e.g., platinum-resistant ovarian cancer, platinum-refractory ovarian cancer), prostate cancer, lung cancer (e.g., non-small cell lung cancer (NSCLC)). In an embodiment, the cancer is breast cancer (e.g., triple negative breast cancer (TNBC)), ovarian cancer (e.g., platinum-resistant ovarian cancer, platinum-refractory ovarian cancer), prostate cancer, lung cancer (e.g., non-small cell lung cancer (NSCLC)). Determining if a cancer will respond to treatment with a USP1 inhibitor [0561] In an embodiment, provided is a method of determining if a cancer (e.g., a cancer associated with USP1) will respond to therapeutic treatment with a USP1 inhibitor (e.g., a compound of Formula (I), (II) or a compound of Table 1 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer or tautomer thereof), comprising the steps of: a) detecting levels of RAD18 and/or UBE2K and/or PCNA (e.g., RAD18 and/or UBE2K and/or PCNA protein and/or RAD18 and/or UBE2K and/or PCNA mRNA) a cancer test sample (e.g., in a cancer sample obtained from the subject) b) comparing the cancer test sample with a reference (e.g., a reference sample taken from a non-cancerous or normal control subject), wherein elevated levels of RAD18 and/or UBE2K and/or PCNA in said test sample indicates that the cancer will respond to therapeutic treatment with a USP1 inhibitor (e.g., a compound of Formula (I), (II) or a compound of Table 1 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer or tautomer thereof). [0562] In an embodiment, provided is a method of determining if a cancer (e.g., a cancer associated with USP1) will respond to therapeutic treatment with a USP1 inhibitor (e.g., a compound of Formula (I), (II) or a compound of Table 1 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer or tautomer thereof), comprising the steps of: a) detecting levels of translesion synthesis (e.g., detecting PCNA monoubiquitination levels) in a test cancer sample (e.g., in a cancer sample obtained from the subject) b) comparing the test cancer sample with a reference (e.g., a reference sample taken from a non-cancerous or normal control subject), wherein elevated translesion synthesis (e.g., increased PCNA monoubiquitination levels) in said test cancer sample indicates that the cancer will respond to therapeutic treatment with a USP1 inhibitor (e.g., a compound of Formula (I), (II) or a compound of Table 1 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer or tautomer thereof). [0563] In an embodiment, provided is a method of determining if a cancer (e.g., a cancer associated with USP1) will respond to therapeutic treatment with a USP1 inhibitor (e.g., a compound of Formula (I), (II) or a compound of Table 1 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer or tautomer thereof), comprising the steps of: a) detecting mutations in a gene encoding ATM (i.e., loss function mutations) in a test cancer sample (e.g., in a cancer sample obtained from the subject) b) wherein presence of mutations in a gene encoding ATM in said cancer sample indicates that the cancer will respond to therapeutic treatment with a USP1 inhibitor (e.g., a compound of Formula (I), (II) or a compound of Table 1 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer or tautomer thereof). [0564] In an embodiment, provided is a method of determining if a cancer (e.g., a cancer associated with USP1) will respond to therapeutic treatment with a USP1 inhibitor (e.g., a compound of Formula (I), (II) or a compound of Table 1 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer or tautomer thereof), comprising the steps of: a) detecting germline or somatic mutations in a gene encoding BRCA1 (e.g., a loss of function mutation) in a cancer subject test sample (e.g., in a cancer sample or blood sample obtained from the cancer subject) b) wherein presence of mutations in a gene encoding BRCA1 in said test sample indicates that the subject’s cancer will respond to therapeutic treatment with a USP1 inhibitor (e.g., a compound of Formula (I), (II) or a compound of Table 1 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer or tautomer thereof). [0565] In an embodiment, provided is a method of determining if a cancer (e.g., a cancer associated with USP1) will respond to therapeutic treatment with a USP1 inhibitor (e.g., a compound of Formula (I), (II) or a compound of Table 1 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer or tautomer thereof), comprising the steps of: a) detecting germline or somatic mutations in a gene encoding BRCA2 (e.g., a loss of function mutation) in a cancer subject test sample (e.g., in a cancer sample or blood sample obtained from the cancer subject) b) wherein presence of mutations in a gene encoding BRCA2 in said test sample indicates that the subject’s cancer will respond to therapeutic treatment with a USP1 inhibitor (e.g., a compound of Formula (I), (II) or a compound of Table 1 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer or tautomer thereof). [0566] In an embodiment, provided is a method of determining if a cancer (e.g., a cancer associated with USP1) will respond to therapeutic treatment with a USP1 inhibitor (e.g., a compound of Formula (I), (II) or a compound of Table 1 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer or tautomer thereof), comprising the steps of: a) detecting deficiency in homologous recombination (e.g., as measured by a positive homologous recombination deficiency (HRD) score) in a cancer subject test sample (e.g., in a cancer sample or blood sample obtained from the cancer subject) b) wherein presence of homologous recombination deficiency in said test sample indicates that the subject’s cancer will respond to therapeutic treatment with a USP1 inhibitor (e.g., a compound of Formula (I), (II) or a compound of Table 1 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer or tautomer thereof). [0567] In an embodiment, the cancer is a cancer selected from the cancers disclosed herein. In an embodiment, the cancer is pancreatic cancer, breast cancer (e.g., triple negative breast cancer (TNBC)), ovarian cancer (e.g., platinum-resistant ovarian cancer, platinum-refractory ovarian cancer), prostate cancer, lung cancer (e.g., non-small cell lung cancer (NSCLC)). In an embodiment, the cancer is breast cancer (e.g., triple negative breast cancer (TNBC)), ovarian cancer (e.g., platinum-resistant ovarian cancer, platinum-refractory ovarian cancer), prostate cancer, lung cancer (e.g., non-small cell lung cancer (NSCLC)). Determining sensitivity of a cancer cell to USP1 inhibition [0568] In an embodiment, provided is a method of determining the sensitivity of a cancer cell to USP1 inhibition (e.g., inhibition with a compound of Formula (I), (II) or a compound of Table 1 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer or tautomer thereof), comprising the steps of: a) detecting levels of RAD18 and/or UBE2K and/or PCNA (e.g., RAD18 and/or UBE2K and/or PCNA protein and/or RAD18 and/or UBE2K and/or PCNA mRNA) in a cancer cell test sample (e.g., in a cancer sample obtained from the subject) b) comparing the test sample with a reference (e.g., a reference sample taken from a non-cancerous or normal control subject), wherein elevated levels of RAD18 and/or UBE2K and/or PCNA in said test sample indicates said cancer cell is sensitive to USP1 inhibition. [0569] In an embodiment, provided is a method of determining the sensitivity of a cancer cell to USP1 inhibition (e.g., inhibition with a compound of Formula (I), (II) or a compound of Table 1 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer or tautomer thereof), comprising the steps of: a) detecting levels of translesion synthesis (e.g., detecting PCNA monoubiquitination levels) in a cancer cell test sample (e.g., in a cancer sample obtained from the subject) b) comparing the test sample with a reference (e.g., a reference sample taken from a non-cancerous or normal control subject), wherein elevated translesion synthesis (e.g., increased PCNA monoubiquitination levels) in said test sample indicates said cancer cell is sensitive to USP1 inhibition. [0570] In an embodiment, provided is a method of determining the sensitivity of a cancer cell to USP1 inhibition (e.g., inhibition with a compound of Formula (I), (II) or a compound of Table 1 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer or tautomer thereof), comprising the steps of: a) detecting mutations in a gene encoding ATM (i.e., loss function mutations) in a cancer cell test sample (e.g., in a cancer sample obtained from a subject) b) wherein presence of mutations in a gene encoding ATM in said cancer cell test sample indicates said cancer cell is sensitive to USP1 inhibition. [0571] In an embodiment, provided is a method of determining the sensitivity of a cancer cell to USP1 inhibition (e.g., inhibition with a compound of Formula (I), (II) or a compound of Table 1 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer or tautomer thereof), comprising the steps of: a) detecting a mutation in a gene encoding BRCA1 (e.g., a loss of function mutation) in a cancer cell test sample (e.g., in a cancer sample obtained from a subject) b) wherein presence of mutations in a gene encoding BRCA1 in said cancer cell test sample indicates said cancer cell is sensitive to USP1 inhibition. [0572] In an embodiment, provided is a method of determining the sensitivity of a cancer cell to USP1 inhibition (e.g., inhibition with a compound of Formula (I), (II) or a compound of Table 1 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer or tautomer thereof), comprising the steps of: a) detecting a mutation in a gene encoding BRCA2 (e.g., a loss of function mutation) in a cancer cell test sample (e.g., in a cancer sample obtained from a subject); b) wherein presence of mutations in a gene encoding BRCA2 in said cancer cell test sample indicates said cancer cell is sensitive to USP1 inhibition. [0573] In an embodiment, provided is a method of determining the sensitivity of a cancer cell to USP1 inhibition (e.g., inhibition with a compound of Formula (I), (II) or a compound of Table 1 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer or tautomer thereof), comprising the steps of: a) detecting deficiency in homologous recombination (e.g., as measured by a positive homologous recombination deficiency (HRD) score) in a cancer cell test sample (e.g., in a cancer sample obtained from a subject) b) wherein presence of homologous recombination deficiency in said cancer cell test sample indicates said cancer cell is sensitive to USP1 inhibition. [0574] In an embodiment, the cancer is a cancer selected from the cancers disclosed herein. In an embodiment, the cancer is pancreatic cancer, breast cancer (e.g., triple negative breast cancer (TNBC)), ovarian cancer (e.g., platinum-resistant ovarian cancer, platinum-refractory ovarian cancer), prostate cancer, lung cancer (e.g., non-small cell lung cancer (NSCLC)). In an embodiment, the cancer is breast cancer (e.g., triple negative breast cancer (TNBC)), ovarian cancer (e.g., platinum-resistant ovarian cancer, platinum-refractory ovarian cancer), prostate cancer, lung cancer (e.g., non-small cell lung cancer (NSCLC)). Therapeutic methods for treating subjects having or having been diagnosed with cancer [0575] In an embodiment provided is a therapeutic method of treating a subject having or having been diagnosed with a cancer (e.g., a cancer associated with USP1) (i.e., a cancer patient (e.g., a USP1-associated cancer patient)) comprising the steps of: a) detecting levels of RAD18 and/or UBE2K and/or PCNA (e.g., RAD18 and/or UBE2K and/or PCNA protein and/or RAD18 and/or UBE2K and/or PCNA mRNA) in a test cancer sample (e.g., in a cancer test sample obtained from the subject); b) comparing the test cancer sample with reference cells (e.g., a reference sample taken from a non-cancerous or normal control subject), wherein elevated levels of RAD18 and/or UBE2K and/or PCNA in said test cancer sample indicates that the subject will respond to therapeutic treatment with a USP1 inhibitor (e.g., a compound of Formula (I), (II) or a compound of Table 1 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer or tautomer thereof); and c) administering a therapeutically effective amount of USP1 inhibitor (e.g., a compound of Formula (I), (II) or a compound of Table 1 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer or tautomer thereof) to the subject identified in step b). [0576] In an embodiment provided is a therapeutic method of treating a subject having or having been diagnosed with a cancer (e.g., a cancer associated with USP1) (i.e., a cancer patient (e.g., a USP1-associated cancer patient)) comprising the steps of: a) detecting levels of translesion synthesis (e.g., detecting PCNA monoubiquitination levels) in a test cancer sample (e.g., in a cancer test sample obtained from the subject); b) comparing the test cancer sample with reference cells (e.g., a reference sample taken from a non-cancerous or normal control subject), wherein elevated translesion synthesis (e.g., increased PCNA monoubiquitination levels) in said test cancer sample indicates that the subject will respond to therapeutic treatment with a USP1 inhibitor (e.g., a compound of Formula (I), (II) or a compound of Table 1 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer or tautomer thereof); and c) administering a therapeutically effective amount of USP1 inhibitor (e.g., a compound of Formula (I), (II) or a compound of Table 1 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer or tautomer thereof) to the subject identified in step b). [0577] In an embodiment provided is a therapeutic method of treating a subject having or having been diagnosed with a cancer (e.g., a cancer associated with USP1) (i.e., a cancer patient (e.g., a USP1-associated cancer patient)) comprising the steps of: a) detecting mutations in a gene encoding ATM (i.e., loss function mutations) in a test cancer sample (e.g., in a cancer test sample obtained from the subject); b) wherein presence of mutations in a gene encoding ATM in said test cancer sample indicates that the subject will respond to therapeutic treatment with a USP1 inhibitor (e.g., a compound of Formula (I), (II) or a compound of Table 1 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer or tautomer thereof); and c) administering a therapeutically effective amount of USP1 inhibitor (e.g., a compound of Formula (I), (II) or a compound of Table 1 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer or tautomer thereof) to the subject identified in step b). [0578] In an embodiment provided is a therapeutic method of treating a subject having or having been diagnosed with a cancer (e.g., a cancer associated with USP1) (i.e., a cancer patient (e.g., a USP1-associated cancer patient)) comprising the steps of: a) detecting germline or somatic mutations in a gene encoding BRCA1 (e.g., a loss of function mutation) in a subject test sample (e.g., in a cancer test sample or blood test sample obtained from the subject); b) wherein presence of mutations in a gene encoding BRCA1 in said test sample indicates that the subject will respond to therapeutic treatment with a USP1 inhibitor (e.g., a compound of Formula (I), (II) or a compound of Table 1 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer or tautomer thereof); and c) administering a therapeutically effective amount of USP1 inhibitor (e.g., a compound of Formula (I), (II) or a compound of Table 1 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer or tautomer thereof) to the subject identified in step b). [0579] In an embodiment provided is a therapeutic method of treating a subject having or having been diagnosed with a cancer (e.g., a cancer associated with USP1) (i.e., a cancer patient (e.g., a USP1-associated cancer patient)) comprising the steps of: a) detecting germline or somatic mutations in a gene encoding BRCA2 (e.g., a loss of function mutation) in a subject test sample (e.g., in a cancer test sample or blood test sample obtained from the subject); b) wherein presence of mutations in a gene encoding BRCA2 in said test sample indicates that the subject will respond to therapeutic treatment with a USP1 inhibitor (e.g., a compound of Formula (I), (II) or a compound of Table 1 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer or tautomer thereof); and c) administering a therapeutically effective amount of USP1 inhibitor (e.g., a compound of Formula (I), (II) or a compound of Table 1 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer or tautomer thereof) to the subject identified in step b). [0580] In an embodiment provided is a therapeutic method of treating a subject having or having been diagnosed with a cancer (e.g., a cancer associated with USP1) (i.e., a cancer patient (e.g., a USP1-associated cancer patient)) comprising the steps of: a) detecting deficiency in homologous recombination (e.g., as measured by a positive homologous recombination deficiency (HRD) score) in a subject test sample (e.g., in a cancer sample or blood sample obtained from the subject) b) wherein presence of homologous recombination deficiency in said test sample indicates that the subject will respond to therapeutic treatment with a USP1 inhibitor (e.g., a compound of Formula (I), (II) or a compound of Table 1 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer or tautomer thereof); and c) administering a therapeutically effective amount of USP1 inhibitor (e.g., a compound of Formula (I), (II) or a compound of Table 1 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer or tautomer thereof) to the subject identified in step b). [0581] In an embodiment, the cancer is a cancer selected from the cancers disclosed herein. In an embodiment, the cancer is pancreatic cancer, breast cancer (e.g., triple negative breast cancer (TNBC)), ovarian cancer (e.g., platinum-resistant ovarian cancer, platinum-refractory ovarian cancer), prostate cancer, lung cancer (e.g., non-small cell lung cancer (NSCLC)). In an embodiment, the cancer is breast cancer (e.g., triple negative breast cancer (TNBC)), ovarian cancer (e.g., platinum-resistant ovarian cancer, platinum-refractory ovarian cancer), prostate cancer, lung cancer (e.g., non-small cell lung cancer (NSCLC)). Therapeutic methods for treating cancer [0582] In an embodiment provided is a therapeutic method of treating a cancer (e.g., a cancer associated with USP1) in a subject in need thereof comprising the steps of: a) detecting levels of RAD18 and/or UBE2K and/or PCNA (e.g., RAD18 and/or UBE2K and/or PCNA protein and/or RAD18 and/or UBE2K and/or PCNA mRNA) a cancer test sample (e.g., in a cancer sample obtained from the subject) b) comparing the cancer test sample with a reference (e.g., a reference sample taken from a non-cancerous or normal control subject), wherein elevated levels of RAD18 and/or UBE2K and/or PCNA in said test sample indicates that the subject’s cancer will respond to therapeutic treatment with a USP1 inhibitor (e.g., a compound of Formula (I), (II) or a compound of Table 1 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer or tautomer thereof); and c) administering a therapeutically effective amount of USP1 inhibitor (e.g., a compound of Formula (I), (II) or a compound of Table 1 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer or tautomer thereof) to the subject whose cancer was identified in step b). [0583] In an embodiment provided is a therapeutic method of treating a cancer (e.g., a cancer associated with USP1) in a subject in need thereof comprising the steps of: a) detecting levels of translesion synthesis (e.g., detecting PCNA monoubiquitination levels) in a test cancer sample (e.g., in a cancer sample obtained from the subject) b) comparing the test cancer sample with a reference (e.g., a reference sample taken from a non-cancerous or normal control subject), wherein elevated translesion synthesis (e.g., increased PCNA monoubiquitination levels) in said test cancer sample indicates that the subject’s cancer will respond to therapeutic treatment with a USP1 inhibitor (e.g., a compound of Formula (I), (II) or a compound of Table 1 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer or tautomer thereof); and c) administering a therapeutically effective amount of USP1 inhibitor (e.g., a compound of Formula (I), (II) or a compound of Table 1 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer or tautomer thereof) to the subject whose cancer was identified in step b). [0584] In an embodiment provided is a therapeutic method of treating a cancer (e.g., a cancer associated with USP1) in a subject in need thereof comprising the steps of: a) detecting mutations in a gene encoding ATM (i.e., loss function mutations) in a test cancer sample (e.g., in a cancer sample obtained from the subject) b) wherein presence of mutations in a gene encoding ATM in said cancer sample indicates that the subject’s cancer will respond to therapeutic treatment with a USP1 inhibitor (e.g., a compound of Formula (I), (II) or a compound of Table 1 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer or tautomer thereof); and c) administering a therapeutically effective amount of USP1 inhibitor (e.g., a compound of Formula (I), (II) or a compound of Table 1 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer or tautomer thereof) to the subject whose cancer was identified in step b). [0585] In an embodiment provided is a therapeutic method of treating a cancer (e.g., a cancer associated with USP1) in a subject in need thereof comprising the steps of: a) detecting germline or somatic mutations in a gene encoding BRCA1 (e.g., a loss of function mutation) in a cancer subject test sample (e.g., in a cancer sample or blood sample obtained from the cancer subject) b) wherein presence of mutations in a gene encoding BRCA1 in said test sample indicates that the subject’s cancer will respond to therapeutic treatment with a USP1 inhibitor (e.g., a compound of Formula (I), (II) or a compound of Table 1 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer or tautomer thereof); and c) administering a therapeutically effective amount of USP1 inhibitor (e.g., a compound of Formula (I), (II) or a compound of Table 1 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer or tautomer thereof) to the subject whose cancer was identified in step b). [0586] In an embodiment provided is a therapeutic method of treating a cancer (e.g., a cancer associated with USP1) in a subject in need thereof comprising the steps of: a) detecting germline or somatic mutations in a gene encoding BRCA2 (e.g., a loss of function mutation) in a cancer subject test sample (e.g., in a cancer sample or blood sample obtained from the cancer subject) b) wherein presence of mutations in a gene encoding BRCA2 in said test sample indicates that the subject’s cancer will respond to therapeutic treatment with a USP1 inhibitor (e.g., a compound of Formula (I), (II) or a compound of Table 1 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer or tautomer thereof); and c) administering a therapeutically effective amount of USP1 inhibitor (e.g., a compound of Formula (I), (II) or a compound of Table 1 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer or tautomer thereof) to the subject whose cancer was identified in step b). [0587] In an embodiment provided is a therapeutic method of treating a cancer (e.g., a cancer associated with USP1) in a subject in need thereof comprising the steps of: a) detecting deficiency in homologous recombination (e.g., as measured by a positive homologous recombination deficiency (HRD) score) in a cancer subject test sample (e.g., in a cancer sample or blood sample obtained from the cancer subject) b) wherein presence of homologous recombination deficiency in said test sample indicates that the subject’s cancer will respond to therapeutic treatment with a USP1 inhibitor (e.g., a compound of Formula (I), (II) or a compound of Table 1 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer or tautomer thereof); and c) administering a therapeutically effective amount of USP1 inhibitor (e.g., a compound of Formula (I), (II) or a compound of Table 1 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer or tautomer thereof) to the subject whose cancer was identified in step b). [0588] In an embodiment, the cancer is a cancer selected from the cancers disclosed herein. In an embodiment, the cancer is pancreatic cancer, breast cancer (e.g., triple negative breast cancer (TNBC)), ovarian cancer (e.g., platinum-resistant ovarian cancer, platinum-refractory ovarian cancer), prostate cancer, lung cancer (e.g., non-small cell lung cancer (NSCLC)). In an embodiment, the cancer is breast cancer (e.g., triple negative breast cancer (TNBC)), ovarian cancer (e.g., platinum-resistant ovarian cancer, platinum-refractory ovarian cancer), prostate cancer, lung cancer (e.g., non-small cell lung cancer (NSCLC)). Sample preparation [0589] The disclosure further provides assays for the detection of levels of translesion synthesis (e.g., PCNA monoubiquitination levels, levels of RAD18, (e.g., RAD18 protein and/or RAD18 mRNA), UBE2K (e.g., UBE2K protein and/or UBE2K mRNA)). The disclosure further provides assays for detecting ATM mutations (e.g. ATM loss of function mutations), loss of ATM protein expression (e.g., as measured by immunohistochemistry), BRCA1 mutations (e.g., BRCA1 loss of function mutations), BRCA2 mutations (e.g., BRCA2 loss of function mutations), BRCA1/2 deficiency and deficiencies in homologous recombination (e.g., as measured by a positive homologous recombination deficiency (HRD) score). They detection of any of the above parameters can be performed in a patient sample, e.g., in a body fluid such as blood (e.g., serum or plasma) bone marrow, cerebral spinal fluid, peritoneal/pleural fluid, lymph fluid, ascites, serous fluid, sputum, lacrimal fluid, stool, and urine, or in a tissue such as a tumor tissue. The tumor tissue can be fresh tissue or preserved tissue (e.g., formalin fixed tissue, e.g., paraffin-embedded tissue). [0590] Body fluid samples can be obtained from a subject using any of the methods known in the art. Methods for extracting cellular DNA from body fluid samples are well known in the art. Typically, cells are lysed with detergents. After cell lysis, proteins are removed from DNA using various proteases. DNA is then extracted with phenol, precipitated in alcohol, and dissolved in an aqueous solution. Methods for extracting acellular DNA from body fluid samples are also known in the art. Commonly, a cellular DNA in a body fluid sample is separated from cells, precipitated in alcohol, and dissolved in an aqueous solution. Measurement of Gene Expression [0591] In an embodiment, elevated levels of RAD18 and/or UBE2K and/or PCNA are elevated RAD18 and/or UBE2K and/or PCNA gene expression levels. In an embodiment, elevated levels of RAD18 and/or UBE2K and/or PCNA are elevated RAD18 and/or UBE2K and/or PCNA mRNA levels. Measurement of gene expression can be performed using any method or reagent known in the art. [0592] Detection of gene expression can be by any appropriate method, including for example, detecting the quantity of mRNA transcribed from the gene or the quantity of cDNA produced from the reverse transcription of the mRNA transcribed from the gene or the quantity of the polypeptide or protein encoded by the gene. These methods can be performed on a sample by sample basis or modified for high throughput analysis. For example, using Affymetrix™ U133 microarray chips. [0593] In an embodiment, gene expression is detected and quantitated by hybridization to a probe that specifically hybridizes to the appropriate probe for that biomarker. The probes also can be attached to a solid support for use in high throughput screening assays using methods known in the art. [0594] In an embodiment, the expression level of a gene is determined through exposure of a nucleic acid sample to the probe-modified chip. Extracted nucleic acid is labeled, for example, with a fluorescent tag, preferably during an amplification step. [0595] Hybridization of the labeled sample is performed at an appropriate stringency level. The degree of probe-nucleic acid hybridization is quantitatively measured using a detection device. [0596] Alternatively, any one of gene copy number, transcription, or translation can be determined using known techniques. For example, an amplification method such as PCR may be useful. General procedures for PCR are taught in MacPherson et al., PCR: A Practical Approach, (IRL Press at Oxford University Press (1991)). However, PCR conditions used for each application reaction are empirically determined. A number of parameters influence the success of a reaction. Among them are annealing temperature and time, extension time, Mg 2+ and /or ATP concentration, pH, and the relative concentration of primers, templates, and deoxyribonucleotides. After amplification, the resulting DNA fragments can be detected by agarose gel electrophoresis followed by visualization with ethidium bromide staining and ultraviolet illumination. In an embodiment, the hybridized nucleic acids are detected by detecting one or more labels attached to the sample nucleic acids. The labels can be incorporated by any of a number of means well known to those of skill in the art. However, in an embodiment, the label is simultaneously incorporated during the amplification step in the preparation of the sample nucleic acid. Thus, for example, polymerase chain reaction (PCR) with labeled primers or labeled nucleotides will provide a labeled amplification product. In a separate embodiment, transcription amplification, as described above, using a labeled nucleotide (e.g., fluorescein-labeled UTP and/or CTP) incorporates a label in to the transcribed nucleic acids. [0597] Alternatively, a label may be added directly to the original nucleic acid sample (e.g., mRNA, polyA, mRNA, cDNA, etc.) or to the amplification product after the amplification is completed. Means of attaching labels to nucleic acids are well known to those of skill in the art and include, for example nick translation or end-labeling (e.g., with a labeled RNA) by kinasing of the nucleic acid and subsequent attachment (ligation) of a nucleic acid linker joining the sample nucleic acid to a label (e.g., a fluorophore). [0598] In one example, the gene expression can be measured through an in-situ hybridization protocol that can detect RNA molecules on a slide containing tissue sections or cells (e.g., through RNAscope®). [0599] Detectable labels suitable for use in the methods disclosed herein include any composition detectable by spectroscopic, photochemical, biochemical, immunochemical, electrical, optical or chemical means. Useful labels include biotin for staining with labeled streptavidin conjugate, magnetic beads (e.g., Dynabeads™), fluorescent dyes (e.g., fluorescein, texas red, rhodamine, green fluorescent protein, and the like), radiolabels (e.g., 3H, 125I, 35S, 14C, or 32P) enzymes (e.g., horse radish peroxidase, alkaline phosphatase and others commonly used in an ELISA), and calorimetric labels such as colloidal gold or colored glass or plastic (e.g., polystyrene, polypropylene, latex, etc.) beads. [0600] Detection of labels is well known to those of skill in the art. Thus, for example, radiolabels may be detected using photographic film or scintillation counters, fluorescent markers may be detected using a photodetector to detect emitted light. Enzymatic labels are typically detected by providing the enzyme with a substrate and detecting the reaction product produced by the action of the enzyme on the substrate, and calorimetric labels are detected by simply visualizing the colored label. The detectable label may be added to the target (sample) nucleic acid(s) prior to, or after the hybridization, such as described in WO 97/10365. These detectable labels are directly attached to or incorporated into the target (sample) nucleic acid prior to hybridization. In contrast, “indirect labels” are joined to the hybrid duplex after hybridization. Generally, the indirect label is attached to a binding moiety that has been attached to the target nucleic acid prior to the hybridization. For example, the target nucleic acid may be biotinylated before the hybridization. After hybridization, an avidin-conjugated fluorophore will bind the biotin bearing hybrid duplexes providing a label that is easily detected. For a detailed review of methods of labeling nucleic acids and detecting labeled hybridized nucleic acids see Laboratory Techniques in Biochemistry and Molecular Biology, Vol.24: Hybridization with Nucleic Acid Probes, P. Tijssen, ed. Elsevier, N.Y. (1993). [0601] In an embodiment, the detection of elevated of RAD18 and/or UBE2K and/or PCNA mRNA levels is by quantitative reverse transcriptase (RT)-polymerase chain reaction (PCR), RNA-Seq, or microarray. Detection of polypeptides [0602] Protein levels of RAD18 and/or UBE2K and/or PCNA can be determined by examining protein expression or the protein product. Determining the protein level involves measuring the amount of any immunospecific binding that occurs between an antibody that selectively recognizes and binds to the polypeptide of the biomarker in a sample obtained from a subject and comparing this to the amount of immunospecific binding of at least one biomarker in a control sample. [0603] A variety of techniques are available in the art for protein analysis. They include but are not limited to radioimmunoassays, ELISA (enzyme linked immunosorbent assays), “sandwich” immunoassays, immunoradiometric assays, in situ immunoassays (using e.g., colloidal gold, enzyme or radioisotope labels), Western blot analysis, immunoprecipitation assays, immunofluorescent assays, flow cytometry, immunohistochemistry, HPLC, mass spectrometry, confocal microscopy, enzymatic assays, surface plasmon resonance and PAGE-SDS. [0604] In an embodiment, the detection of elevated RAD18 and/or UBE2K and/or PCNA protein levels is by Western blot. In an embodiment, the detection of elevated RAD18 and/or UBE2K and/or PCNA protein levels is by fluorescence- activated cell sorting (FACS). In an embodiment, the detection of elevated RAD18 and/or UBE2K and/or PCNA protein levels is by immunohistochemistry. Other detection methods [0605] Mutations in targets of interest (e.g., BRCA1 mutations, BRCA2 mutations, ATM mutations) can be detected by methods known to those of skill in the art. [0606] For detection of germline mutation, DNA sequencing may be performed using DNA extract from body fluid such as blood (e.g., serum or plasma) bone marrow, cerebral spinal fluid, peritoneal/pleural fluid, lymph fluid, ascites, serous fluid, sputum, lacrimal fluid, stool, and urine. Alternatively, sequencing may be performed on DNA extracted from a tissue such as a tumor tissue. The tumor tissue can be fresh tissue or preserved tissue (e.g., formalin fixed tissue, e.g.paraffin-embedded tissue). Sequencing may also be performed using cell-free DNA. The coding regions and sometimes adjacent regions (e.g., introns, promoter) of genes of interest are sequenced using next generation sequencing (NGS) or Sanger sequencing (Genetic/Familial High-Risk Assessment: Breast, Ovarian, and Pancreatic, Version 2.2021, NCCN Clinical Practice Guidelines in Oncology, ESMO guideline for BRCA testing DOI: 10.1093/annonc/mdw327, Clinical testing of BRCA1 and BRCA2: a worldwide snapshot of technological practices). Loss of function mutations or gene rearrangements may be detected or validated using secondary methods such as qPCR, PCR, immunohistochemistry, Sanger sequencing, comparative genomic hybridization, or the PacBio system. [0607] Deficiencies in homologous recombination can be identified by methods known to those of skill in the art. One indicator of homologous recombination deficiencies is genomic instability (e.g., represented by a positive homologous recombination deficiency (HRD) score), which can be quantified by methods known in the art (see, e.g., Pikor L, et al., Cancer Metastasis Rev.2013;32(3-4):341-352). HRD score is measured using next generation sequencing of DNA extracted from tumor tissues (fresh or FFPE), based on genomic instability (e.g., loss of heterozygosity, telomeric allelic imbalance, and large-scale state transitions). Commercial FDA-approved assays are available for such measures (Myriad and Foundation Medicine). Kits [0608] In an embodiment kits related to methods disclosed herein are provided. [0609] In an embodiment, a kit for predicting the sensitivity of a subject having or having been diagnosed with a disease or disorder associated with USP1 for treatment with a USP1 inhibitor is provided. The kit comprises: i) reagents capable of detecting human cancer cells associated with a disease or disorder associated with USP1 (e.g., reagents capable of specifically detecting RAD18 and/or UBE2K and/or PCNA) and ii) instructions for how to use said kit. [0610] In an embodiment, the present disclosure provides kit, comprising: (a) a pharmaceutical composition comprising a USP1 inhibitor and one or more pharmaceutically acceptable excipients, and (b) a diagnostic kit comprising at least one agent capable of specifically detecting RAD18 and/or UBE2K and/or PCNA. [0611] In an embodiment, the agent capable of specifically detecting RAD18 and/or UBE2K and/or PCNA is capable of specifically hybridizing to RAD18 and/or UBE2K and/or PCNA mRNA. In an embodiment, the agent capable of specifically detecting RAD18 and/or UBE2K and/or PCNA is capable of specifically binding to RAD18 and/or UBE2K and/or PCNA protein. [0612] In another embodiment, the present disclosure provides kits which comprise a compound disclosed herein (or a composition comprising a compound disclosed herein) packaged in a manner that facilitates their use to practice methods of the present disclosure. In an embodiment, the kit includes a compound disclosed herein (or a composition comprising a compound disclosed herein) packaged in a container, such as a sealed bottle or vessel, with a label affixed to the container or included in the kit that describes use of the compound or composition to practice the method of the disclosure. In an embodiment, the compound or composition is packaged in a unit dosage form. The kit further can include a device suitable for administering the composition according to the intended route of administration. In an embodiment, the present disclosure provides a kit which comprise a compound disclosed herein, or a pharmaceutically acceptable salt or solvate thereof, and instructions for administering the compound, or a pharmaceutically acceptable salt or solvate thereof, to a patient having cancer. Selected embodiments Embodiment 1. A compound of Formula (I) or Formula (II) or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof; wherein:

; X
1 and X
2 are each independently CH or N; Ring A is selected from the group consisting of:
wherein X
3 is CH or N and wherein the left-side attachment point connects to CR
cR
c’ and the right attachment point connects to Ring B; L is selected from the group consisting of –O– and –S–; Ring B is a 5-10 membered heteroaryl or a 3-10 membered heterocyclyl; each R
A is independently selected from the group consisting of –D, oxo, halo, – CN, –C
1–C
6 alkyl, –C
1–C
6 haloalkyl, –C
1–C
6 hydroxyalkyl, –C
3–C
10 cycloalkyl, –OR
A1 and – N(R
A1)
2; each R
A1 is independently selected from the group consisting of H, –C
1–C
6 alkyl, –C
1–C
6 haloalkyl and C
3–C
9 cycloalkyl wherein each hydrogen atom of each alkyl and cycloalkyl can be replaced by deuterium; or two instances of R
A1 on a nitrogen atom can be taken together with the nitrogen to which they are attached to form a 4-6 member monocyclic heterocycle containing one nitrogen atom and 0-1 additional heteroatoms selected from N and O; each R
B is independently selected from the group consisting of halo, –CN, –C
1–C
6 alkyl, –C
1–C
6 alkenyl, –heteroC
1-C
4 alkyl, –C
1–C
6 haloalkyl, –C
1–C
6 hydroxyalkyl, –C
3–C
10 cycloalkyl, 3-10 membered heterocyclyl, C
3-C
9 cycloalkylalkyl, –OR
B1, –N(R
B1)
2, – C(=O)R
B1, –C(=O)OR
B1, –NR
B1C(=O)R
B1, –NR
B1C(=O)OR
B1, –C(=O)N(R
B1)
2, – OC(=O)N(R
B1)
2, –S(=O)R
B1, –S(=O)
2R
B1, –SR
B1, –S(=O)(=NR
B1)R
B1, –NR
B1S(=O)
2R
B1 and –S(=O)
2N(R
B1)
2, wherein each alkyl, alkenyl, cycloalkyl, heterocyclyl and cycloalkylalkyl of R
B can be partially or fully deuterated and can be substituted at any available position with 0, 1 or 2 substituents selected from –F, –Cl, –OH– and –CH
3; each R
B1 is independently selected from the group consisting of H, –C
1–C
6 alkyl, –heteroC
1-C
4 alkyl, –C
1–C
6 haloalkyl, –C
3–C
9 cycloalkyl and 3-7 membered heterocyclyl wherein each hydrogen atom of each alkyl, cycloalkyl and heterocyclyl can be replaced by deuterium; or two instances of R
B1 on a nitrogen atom can be taken together with the nitrogen to which they are attached to form a 4-6 member monocyclic heterocycle containing one nitrogen atom and 0-1 additional heteroatoms selected from N and O; each R
C is independently selected from the group consisting of H, –D, oxo, halo, – CN, –OR
C1, –SR
C1 –NR
C1 2, –C
1–C
6 alkyl, –C
1–C
6 hydroxyalkyl, –C
1–C
6 haloalkyl, –C
3–C
9 cycloalkyl, 3-10 member heterocyclyl and –heteroC
1-C
4 alkyl wherein each alkyl, cycloalkyl and heterocyclyl of R
C can partially or fully deuterated and can be substituted at any available position with 0, 1 or 2 substituents selected from –F, –Cl, –OH– and –CH
3; each R
C1 is independently selected from the group consisting of H and –C
1–C
6 alkyl, wherein each hydrogen atom of the alkyl can be independently replaced by deuterium; or two instances of R
C1 on a nitrogen atom can be taken together with the nitrogen to which they are attached to form a 4-6 member monocyclic heterocycle containing one nitrogen atom and 0-1 additional heteroatoms selected from N and O; each R
D is independently selected from the group consisting of –D, halo, –CN, – C
1–C
6 alkyl, –heteroC
1-C
4 alkyl, –C
1–C
6 haloalkyl, –C
1–C
6 hydroxyalkyl, –C
3–C
10 cycloalkyl, 3-10 membered heterocyclyl, –C
6-C
10 aryl, –OR
D1, –N(R
D1)
2, –C(=O)R
D1, – C(=O)OR
D1, –NR
D1C(=O)R
D1, –NR
D1C(=O)OR
D1, –C(=O)N(R
D1)
2, –OC(=O)N(R
D1)
2, – S(=O)R
D1, –S(=O)
2R
D1, –SR
D1, –S(=O)(=NR
D1)R
D1, –NR
D1S(=O)
2R
D1 and –S(=O)
2N(R
D1)
2 , wherein each alkyl, cycloalkyl, heterocyclyl and aryl of R
D can be substituted at any available position with 0, 1 or 2 substituents selected from –F, –Cl, –OH– and –CH
3; each R
D1 is independently selected from the group consisting of H, –C
1–C
6 alkyl, –heteroC
1-C
4 alkyl, –C
1–C
6 haloalkyl, –C
3–C
9 cycloalkyl and 3-7 membered heterocyclyl, wherein each hydrogen atom of each alkyl, cycloalkyl and heterocyclyl can be replaced by deuterium; or two instances of R
D1 on a nitrogen atom can be taken together with the nitrogen to which they are attached to form a 4-6 member monocyclic heterocycle containing one nitrogen atom and 0-1 additional heteroatoms selected from N and O; R
E is selected from H, –D, halo, –CN, –C
1–C
6 alkyl, –C
1-C
6 alkynyl, –heteroC
1– C
6 alkyl, –C
1–C
6 haloalkyl, –C
1–C
6 hydroxyalkyl, –C
3–C
10 cycloalkyl, 3-10 membered heterocyclyl, –C
6-C
10 aryl, 5-10 member heteroaryl, heterocyclylalkyl, heteroarylalkyl, arylalkyl, cycloalkylalkyl, –OR
E1, –SR
E1, –N(R
E1)
2, –C
1–C
6 alkylene–NR
E1C(=O)OR
E1, – C(=O)R
E1, –C(=O)OR
E1, –NR
E1C(=O)R
E1, –NR
E1C(=O)OR
E1, –C(=O)N(R
E1)
2, and – OC(=O)N(R
E1)
2, wherein each alkyl, alkynyl, heteroalkyl, haloalkyl, cycloalkyl, heterocyclyl, aryl, heteroaryl, cycloalkylalkyl, heterocyclylalkyl, arylalkyl and heteroarylalkyl is optionally substituted at any available position; each R
E1 is independently selected from H, –C
1–C
6 alkyl (wherein each hydrogen can be independently replaced by deuterium), –heteroC
1–C
6 alkyl, –C
1–C
6 haloalkyl, –C
3–C
9 cycloalkyl, 3-7 membered heterocyclyl, cycloalkylalkyl, heterocyclylalkyl, aryl, 5-6 membered heteroaryl, arylalkyl and heteroarylalkyl wherein each alkyl, haloalkyl, cycloalkyl, heterocyclyl, aryl, heteroaryl, cycloalkylalkyl, heterocyclylalkyl, arylalkyl and heteroarylalkyl is optionally substituted at any available position; each R
c and R
c’ is independently selected from the group consisting of H, –D, – C
1–C
6 alkyl, –heteroC
1-C
4 alkyl and –C
1–C
6 haloalkyl or R
c and R
c’ can be taken together with the atom to which they are attached to form a –C
3–C
9 cycloalkyl; n is 0, 1, 2, 3 or 4; m is 0, 1, 2 or 3; and p is 0, 1, 2 or 3. [0613] Embodiment 2. The compound of embodiment 1 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein the compound is of Formula (I). [0614] Embodiment 3. The compound of embodiment 1 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein the compound is of Formula (II). [0615] Embodiment 4. The compound of any one of embodiments 1-3 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein R
E is selected from H, –D, halo, –CN, –C
1–C
6 alkyl, –C
1-C
6 alkynyl, –heteroC
1– C
4alkyl, –C
1–C
6 haloalkyl, –C
1–C
6 hydroxyalkyl, –C
3–C
10 cycloalkyl, 3-10 membered heterocyclyl, –C
6-C
10 aryl, 5-10 member heteroaryl, heterocyclylalkyl, heteroarylalkyl, arylalkyl, cycloalkylalkyl, –OR
E1, –SR
E1, –N(R
E1)
2, –C
1–C
6 alkylene–NR
E1C(=O)OR
E1, – C(=O)R
E1, –C(=O)OR
E1, –NR
E1C(=O)R
E1, –NR
E1C(=O)OR
E1, –C(=O)N(R
E1)
2, and – OC(=O)N(R
E1)
2, wherein each alkyl, alkynyl, cycloalkyl, heterocyclyl, aryl, heteroaryl, cycloalkylalkyl, heterocyclylalkyl, arylalkyl and heteroarylalkyl is substituted at any available position with 0, 1 or 2 substituents independently selected from –F, –Cl–, –Me, –Et, –CN, –OH, –OMe –NCH
3, –NH
2 and –CF
3. [0616] Embodiment 5. The compound of any one of embodiments 1-3 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein R
E is selected from H, –D, halo, –C
1–C
6 alkyl, –heteroC
1–C
4alkyl, –C
1–C
6 haloalkyl, –C
1–C
6 hydroxyalkyl, –C
3–C
10 cycloalkyl, 3-10 membered heterocyclyl, 5-10 member heteroaryl, heterocyclylalkyl, –OR
E1, –SR
E1, –N(R
E1)
2, and –C
1–C
6 alkylene– NR
E1C(=O)OR
E1, wherein each alkyl, haloalkyl, heteroalkyl, cycloalkyl, heterocyclyl, aryl, heteroaryl and heterocyclylalkyl is substituted at any available position with 0 or 1 substituents independently selected from –F, –Cl–, –Me, –Et, –CN, –OH, –OMe –NCH
3, – NH
2 and –CF
3. [0617] Embodiment 6. The compound of any one of embodiments 1-3 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein R
E is selected from H, halo, –CN, –C
1–C
6 alkyl, –C
1–C
6 haloalkyl, –C
1–C
6 hydroxyalkyl, –C
3–C
10 cycloalkyl, 3-10 membered heterocyclyl, 5-10 member heteroaryl, – OR
E1, –SR
E1, –N(R
E1)
2, –C
1–C
6 alkylene–NR
E1C(=O)OR
E1 and –C(=O)N(R
E1)
2 wherein each alkyl, haloalkyl, cycloalkyl, heterocyclyl and heteroaryl is substituted at any available position with 0 or 1 substituents independently selected from –CN, –OH, –NCH
3, –NH
2, – CF
3 and –F
. [0618] Embodiment 7. The compound of any one of embodiments 1-6 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein each R
E1 is independently selected from H, –C
1–C
6 alkyl (wherein each hydrogen can be independently replaced by deuterium), –heteroC
1–C
6 alkyl, –C
1–C
6 haloalkyl, –C
3–C
9 cycloalkyl, 3-7 membered heterocyclyl, cycloalkylalkyl, heterocyclylalkyl, aryl, 5-6 membered heteroaryl, arylalkyl and heteroarylalkyl wherein each alkyl, haloalkyl, cycloalkyl, heterocyclyl, aryl, heteroaryl, cycloalkylalkyl, heterocyclylalkyl, arylalkyl and heteroarylalkyl is substituted at any available position with 0, 1 or 2 substituents independently selected from –F, –Cl–, –Me, –Et, –CN, –OH, –OMe –NCH
3, –NH
2 and –CF
3. [0619] Embodiment 8. The compound of any one of embodiments 1-6 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein each R
E1 is independently selected from –C
1–C
6 alkyl (wherein each hydrogen can be independently replaced with deuterium), –C
1–C
6 haloalkyl, –heteroC
1-C
4alkyl, –C
3-C
9 cycloalkyl, 3-7 membered heterocyclyl, cycloalkylalkyl, heterocyclylalkyl and arylalkyl, wherein each alkyl, haloalkyl, heteroalkyl, cycloalkyl, heterocyclyl, cycloalkylalkyl and arylalkyl is independently substituted at any available position with 0 or 1 substituents independently selected from –NH
2 and –CF
3. [0620] Embodiment 9. The compound of any one of embodiments 1-6 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein each R
E1 is independently selected from the group consisting of –Me, –CD
3, –Et, –
iPr, CH
2CHF
2, –CF
3, –CHF
2, –CH
2F, Bn, cyclopropyl, oxetan-3-yl, tetrahydrofuran-3-yl, CH
2-1-CF
3-cyclopropyl, CH
2CH(NH
2)CF
3 ,–CH
2CH
2CH
2OMe and – CH
2CH
2OMe. [0621] Embodiment 10. The compound of any one of embodiments 1-6 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein each R
E1 is –Me. [0622] Embodiment 11. The compound of any one of embodiments 1-3 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein R
E is selected from H, –D, halo, –C
1–C
6 alkyl (substituted with 0 or 1 instances of – CN), –C
1–C
6 haloalkyl (substituted with 0 or 1 instances of –NH
2 or –NHCH
3), heteroC
1– C
4alkyl, –C
1–C
6 hydroxyalkyl (substituted with 0 or 1 instances of –CF
3), –C
1–C
6 alkylene– NHC(=O)O(C
1-C
6alkyl), –C
3–C
10 cycloalkyl (substituted with 0 or 1 instances of –CF
3), 3-10 member heterocyclyl (substituted with 0 or 1 instances of –F), 5-10 member heteroaryl, – CH
2-heterocyclyl, –NH
2, –NH(C
1-C
6 alkyl), –N(C
1-C
6 alkyl)
2, –NH(heteroC
1–C
4alkyl), –OH, –S(C
1–C
6 alkyl), –Ocycloalkylalkyl (substituted with 0 or 1 instances of –CF
3), -Oarylalkyl, –O(C
1–C
6 haloalkyl) and –O(C
1–C
6 alkyl) (wherein each hydrogen can be independently replaced by deuterium). [0623] Embodiment 12. The compound of any one of embodiments 1-3 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein R
E is selected from H, –D, –F, –Cl, –Me, –Et, –
iPr, –
tBu, –CHF
2, –CH
2CF
3, – CH
2CHF
2, –CF
3, –CH
2OMe, –CH(CF
3)NH
2, –CH(CF
3)NHCH
3, –CH(OH)CF
3, – C(OH)(CH
3)
2, –CH(OH)CH
3, –CH
2CN
, –C(CN)(CH
3)
2, –CH
2CH
2CH
2NHC(=O)OC(CH
3)
3, cyclopropyl, 1-CF
3-cyclopropyl, cyclobutyl, cyclopentyl, 3-F-oxetan-3-yl, tetrahydrofuran-3-
–O-tetrahydrofuran-3-yl, –OCH
2CH(NH
2)CF
3, –OCF
3, –OCH
2CHF
2, –OCH
2F, –OCHF
2, – OCH
2CF
3, –OMe, –OCD
3, –OEt and –O
iPr. [0624] Embodiment 13. The compound of any one of embodiments 1-3 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein R
E is selected from H, –D, –CN, –F, –Cl, –Me, –Et, –Pr, –
iPr, –
nBu, –
tBu, –CF
3, – CHF
2, –C(=O)NH
2, –OH and –OMe. [0625] Embodiment 14. The compound of any one of embodiments 1-3 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein R
E is selected from H, –F, –Cl, –Me –OCD
3 and –OMe . [0626] Embodiment 15. The compound of any one of embodiments 1-3 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein R
E is selected from H, –Me, –OCD
3 and –OMe . [0627] Embodiment 16. The compound of any one of embodiments 1-3 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein R
E is selected from H, –Me and –OMe. [0628] Embodiment 17. The compound of any one of embodiments 1-3 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein R
E is –Me. [0629] Embodiment 18. The compound of any one of embodiments 1-3 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein R
E is –OMe . [0630] Embodiment 19. The compound of any one of embodiments 1-3 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein R
E is H . [0631] Embodiment 20. The compound of any one of embodiments 1-19 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein each R
C is independently selected from the group consisting of H, –OR
C1, –SR
C1, – C
1-C
6 alkyl including partially or fully deuterated alkyl, –C
1-C
6 haloalkyl –C
1-C
6 hydroxyalkyl and C
3-C
9 cycloalkyl. [0632] Embodiment 21. The compound of any one of embodiments 1-19 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein each R
C is independently selected from the group consisting of H, –C
1-C
3 alkyl including partially or fully deuterated alkyl and –C
1-C
3 hydroxyalkyl . [0633] Embodiment 22. The compound of any one of embodiments 1-21 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein each R
C1 is independently selected from the group consisting of H, –CH
3, –Et and – CD
3. [0634] Embodiment 23. The compound of any one of embodiments 1-19 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein each R
C is independently selected from the group consisting of H, –CH
3, –Et, –
iPr, – CF
3, CH
2OH, –CH(OH)CH
3, –OMe, –SMe and cyclopropyl. [0635] Embodiment 24. The compound of any one of embodiments 1-19 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein each R
C is independently selected from the group consisting of H, –CH
3, CH
2OH and –CH(OH)CH
3. [0636] Embodiment 25. The compound of any one of embodiments 1-19 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein each R
C is –CH
3. [0637] Embodiment 26. The compound of any one of embodiments 1-19 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein R
C is –CH(OH)CH
3. [0638] Embodiment 27. The compound of any one of embodiments 1-19 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein R
C is –CH
2OH. [0639] Embodiment 28. The compound of any one of embodiments 1-27 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein R
C is H. [0640] Embodiment 29. The compound of any one of embodiments 1, 2 and 4-28 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein X
1 and X
2 are both CH or are both N. [0641] Embodiment 30. The compound of any one of embodiments 1, 2 and 4-28 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein X
1 is CH and X
2 is CH or N. [0642] Embodiment 31. The compound of any one of embodiments 1, 2 and 4-28 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein X
1 and X
2 are both N. [0643] Embodiment 32. The compound of any one of embodiments 1, 2 and 4-28 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein X
1 and X
2 are both CH. [0644] Embodiment 33. The compound of any one of embodiments 1 to 32 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein each R
D is independently selected from the group consisting of –C
1-C
6 alkyl, –OR
D1 and C
3-C
10 cycloalkyl. [0645] Embodiment 34. The compound of any one of embodiments 1 to 32 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein each R
D is independently selected from the group consisting of –OR
D1 and C
3-C
10 cycloalkyl. [0646] Embodiment 35. The compound of any one of embodiments 1 to 34 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein each R
D1 is independently selected from the group consisting of H, –C
1-C
6 alkyl and –C
1-C
6 haloalkyl wherein each hydrogen atom of the alkyl can be independently replaced with deuterium. [0647] Embodiment 36. The compound of any one of embodiments 1 to 34 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein each R
D1 is independently selected from the group consisting of H, –C
1-C
3 alkyl and –C
1-C
3 haloalkyl wherein each hydrogen atom of the alkyl can be independently replaced with deuterium. [0648] Embodiment 37. The compound of any one of embodiments 1 to 34 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein each R
D1 is independently selected from the group consisting of –C
1-C
3 alkyl and – C
1-C
3 haloalkyl wherein each hydrogen atom of the alkyl can be independently replaced with deuterium. [0649] Embodiment 38. The compound of any one of embodiments 1 to 34 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein each R
D1 is independently –C
1-C
3 alkyl wherein each hydrogen atom of the alkyl can be independently replaced with deuterium. [0650] Embodiment 39. The compound of any one of embodiments 1 to 34 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein each R
D1 is independently selected from the group consisting of –CH
3, –Et, –CD
3, – CH
2F and –CHF
2. [0651] Embodiment 40. The compound of any one of embodiments 1 to 34 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein each R
D1 is independently selected from the group consisting of –CH
3 and –CD
3. [0652] Embodiment 41. The compound of any one of embodiments 1 to 32 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein each R
D is independently selected from the group consisting of –n-Pr, –
iPr, –OCH
3, –OCD
3, –OEt, –OCHF
2, –OCH
2F and cyclopropyl. [0653] Embodiment 42. The compound of any one of embodiments 1 to 32 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein each R
D is independently selected from the group consisting of –OCH
3, –OCD
3, – OEt, –OCHF
2, –OCH
2F and cyclopropyl. [0654] Embodiment 43. The compound of any one of embodiments 1 to 32 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein each R
D is independently selected from the group consisting of –OCH
3, –OCD
3, – OCHF
2 and cyclopropyl. [0655] Embodiment 44. The compound of any one of embodiments 1 to 32 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein each R
D is independently selected from the group consisting of –OCH
3 and cyclopropyl. [0656] Embodiment 45. The compound of any one of embodiments 1 to 44 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein m is 0, 1, or 2. [0657] Embodiment 46. The compound of any one of embodiments 1 to 44 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein m is 1 or 2. [0658] Embodiment 47. The compound of any one of embodiments 1 to 44 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein m is 1. [0659] Embodiment 48. The compound of any one of embodiments 1 to 44 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein m is 2. [0660] Embodiment 49. The compound of any one of embodiments 1, 2 and 4-44 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein the moiety represented as

selected from the group consisting of
each R
1 is independently selected from the group consisting of –D, halo, –CN, –C
1–C
6 alkyl, –heteroC
1-C
4 alkyl, –C
1–C
6 haloalkyl, –C
1–C
6 hydroxyalkyl, –C
3–C
10 cycloalkyl, 3-10 membered heterocyclyl, –C
6-C
10 aryl, –OR
a1, –N(R
a1)
2, –C(=O)R
a1, –C(=O)OR
a1, – NR
a1C(=O)R
a1, –NR
a1C(=O)OR
a1, –C(=O)N(R
a1)
2, –OC(=O)N(R
a1)
2, –S(=O)R
a1, – S(=O)
2R
a1, –SR
a1, –S(=O)(=NR
a1)R
a1, –NR
a1S(=O)
2R
a1 and –S(=O)
2N(R
a1)
2 , wherein each alkyl, cycloalkyl, heterocyclyl and aryl of R
1 can be substituted at any available position with 0, 1 or 2 substituents selected from –F, –Cl, –OH– and –CH
3; each R
a1 is independently selected from the group consisting of H, –C
1–C
6 alkyl, – heteroC
1-C
4 alkyl, –C
1–C
6 haloalkyl, –C
3–C
9 cycloalkyl and 3-7 membered heterocyclyl, wherein each hydrogen atom of each alkyl, cycloalkyl heterocyclyl can be replaced by deuterium; or two instances of R
a1 on a nitrogen atom can be taken together with the nitrogen to which they are attached to form a 4-6 member monocyclic heterocycle containing one nitrogen atom and 0-1 additional heteroatoms selected from N and O; each R
2 is independently selected from the group consisting of –D, halo, –CN, – C
1–C
6 alkyl, –heteroC
1-C
4 alkyl, –C
1–C
6 haloalkyl, –C
1–C
6 hydroxyalkyl, –C
3–C
10 cycloalkyl, 3-10 membered heterocyclyl, –C
6-C
10 aryl, –OR
a2, –N(R
a2)
2, –C(=O)R
a2, – C(=O)OR
a2, –NR
a2C(=O)R
a2, –NR
a2C(=O)OR
a2, –C(=O)N(R
a2)
2, –OC(=O)N(R
a2)
2, – S(=O)R
a2, –S(=O)
2R
a2, –SR
a2, –S(=O)(=NR
a2)R
a2, –NR
a2S(=O)
2R
a2 and –S(=O)
2N(R
a2)
2 , wherein each alkyl, cycloalkyl, heterocyclyl and aryl of R
2 can be substituted at any available position with 0, 1 or 2 substituents selected from –F, –Cl, –OH– and –CH
3; each R
a2 is independently selected from the group consisting of H, –C
1–C
6 alkyl, – heteroC
1-C
4 alkyl, –C
1–C
6 haloalkyl, –C
3–C
9 cycloalkyl and 3-7 membered heterocyclyl, wherein each hydrogen atom of each alkyl, cycloalkyl heterocyclyl can be replaced by deuterium; or two instances of R
a2 on a nitrogen atom can be taken together with the nitrogen to which they are attached to form a 4-6 member monocyclic heterocycle containing one nitrogen atom and 0-1 additional heteroatoms selected from N and O. [0661] Embodiment 50. The compound of embodiment 49 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein each R
1 is independently selected from halo, –C
1–C
6 alkyl, phenyl substituted with 0 to 1 instances of halo, –C
1–C
6 haloalkyl, –C
1–C
6 hydroxyalkyl and –C
3–C
10 cycloalkyl. [0662] Embodiment 51. The compound of embodiment 49 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein each R
1 is independently selected from –
iPr and cyclopropyl. [0663] Embodiment 52. The compound of embodiment 49 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein each R
1 is
iPr. [0664] Embodiment 53. The compound of embodiment 49 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein each R
1 is cyclopropyl. [0665] Embodiment 54. The compound of any one of embodiments 49-53 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein each R
2 is independently OR
a2. [0666] Embodiment 55. The compound of any one of embodiments 49-54 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein each R
a2 is independently selected from the group consisting of –C
1-C
6 alkyl and – C
1-C
6 haloalkyl wherein each hydrogen atom of the alkyl can be independently replaced with deuterium. [0667] Embodiment 56. The compound of any one of embodiments 49-54 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein each R
a2 is independently selected from the group consisting of –CH
3, –CD
3,–Et, – CHF
2 and –CH
2F. [0668] Embodiment 57. The compound of any one of embodiments 49-54 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein each R
a2 is independently selected from the group consisting of –CH
3, –CD
3 and – CHF
2. [0669] Embodiment 58. The compound of any one of embodiments 49-54 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein each R
a2 is independently selected from the group consisting of –CH
3 and –CD
3. [0670] Embodiment 59. The compound of any one of embodiments 49-53 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein each R
2 is independently selected from –OCH
3, –OCD
3, –OCH
2F and –OCHF
2. [0671] Embodiment 60. The compound of any one of embodiments 49-53 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein each R
2 is independently selected from –OCH
3, –OCD
3 and –OCHF
2. [0672] Embodiment 61. The compound of any one of embodiments 49-53 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein each R
2 is independently selected from –OCH
3 and –OCD
3. [0673] Embodiment 62. The compound of any one of embodiments 49-53 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein each R
2 is independently –OCH
3. [0674] Embodiment 63. The compound of any one of embodiments 49-53 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein each R
2 is independently –OCD
3. [0675] Embodiment 64. The compound of any one of embodiments 49-53 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein each R
2 is independently –OCHF
2. [0676] Embodiment 65. The compound of any one of embodiments 1, 2 and 4-44 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof,
[0677] Embodiment 66. The compound of any one of embodiments 1, 2 and 4-44 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof,
[0678] Embodiment 67. The compound of any one of embodiments 1, 2 and 4-44 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein the moiety represented as
is selected from the group consisting of
[0679] Embodiment 68. The compound of any one of embodiments 1, 2 and 4-44 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein the moiety represented as
. [0680] Embodiment 69. The compound of any one of embodiments 1, 2 and 4-28 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein the compound is of Formula (I-b)
Formula (I-b); wherein R
a2 is selected from the group consisting of CH
3, –CHF
2 and –CD
3. [0681] Embodiment 70. The compound of any one of embodiments 1 to 69 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein L is –O–. [0682] Embodiment 71. The compound of any one of embodiments 1 to 69 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein L is –S–. [0683] Embodiment 72. The compound of any one of embodiments 1 to 71 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein R
c and R
c’ are each independently selected from H, –Me, –CF
3 and –CHF
2 or are taken together to form a cyclopropyl. [0684] Embodiment 73. The compound of any one of embodiments 1 to 71 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein R
c and R
c’ are each independently selected from H and –Me. [0685] Embodiment 74. The compound of any one of embodiments 1 to 71 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein R
c and R
c’ are taken together to form a cyclopropyl. [0686] Embodiment 75. The compound of any one of embodiments 1 to 71 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein R
c and R
c’ are both H. [0687] Embodiment 76. The compound of any one of embodiments 1 to 75 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein Ring A is selected from the group consisting
wherein the left-side attachment point connects to CR
cR
c’ and the right attachment point connects to Ring B. [0688] Embodiment 77. The compound of any one of embodiments 1 to 75 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein Ring A is
wherein the left-side attachment point connects to CR
cR
c’ and the right attachment point connects to Ring B. [0689] Embodiment 78. The compound of any one of embodiments 1 to 75 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein Ring A is wherein the left-side attachment point connects to CR
cR
c’ and the right attachment point connects to Ring B. [0690] Embodiment 79. The compound of any one of embodiments 1 to 75 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein Ring A is
wherein the left-side attachment point connects to CR
cR
c’ and the right attachment point connects to Ring B. [0691] Embodiment 80. The compound of any one of embodiments 1 to 79 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein n is 1 or 2. [0692] Embodiment 81. The compound of any one of embodiments 1 to 79 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein n is 0 or 1. [0693] Embodiment 82. The compound of any one of embodiments 1 to 79 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein n is 1. [0694] Embodiment 83. The compound of any one of embodiments 1 to 79 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein n is 2. [0695] Embodiment 84. The compound of any one of embodiments 1 to 79 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein n is 0. [0696] Embodiment 85. The compound of any one of embodiments 1 to 84 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein each R
A is independently selected from C
1-C
6 alkyl, C
1-C
6 haloalkyl, –OC
1-C
3 alkyl, and halo. [0697] Embodiment 86. The compound of any one of embodiments 1 to 84 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein each R
A is independently halo. [0698] Embodiment 87. The compound of any one of embodiments 1 to 84 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein each R
A is independently selected from –F, –Cl, –CH
3, –Et
, –CH
2CF
3, –OCH
3 and – OEt. [0699] Embodiment 88. The compound of any one of embodiments 1 to 84 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein each R
A is independently –F. [0700] Embodiment 89. The compound of any one of embodiments 1 to 75 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof,
to CR
cR
c and the right attachment point connects to Ring B. [0701] Embodiment 90. The compound of any one of embodiments 1 to 75 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein the moiety represented
selected from the group consisting of
, wherein the left- side attachment point connects to CR
cR
c’ and the right attachment point connects to Ring B. [0702] Embodiment 91. The compound of any one of embodiments 1 to 75 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein the moiety represented
selected from the group consisting of
, wherein the left-side attachment point connects to CR
cR
c’ and the right attachment point connects to Ring B. [0703] Embodiment 92. The compound of any one of embodiments 1 to 75 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, (R
A)
n wherein the moiety represented as
is
, wherein the left-side attachment point connects to CR
cR
c’ and the right attachment point connects to Ring B. [0704] Embodiment 93. The compound of any one of embodiments 1 to 75 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, (R
A)
n wherein the moiety represented as
s , wherein the left-side attachment point connects to CR
cR
c’ and the right attachment point connects to Ring B. [0705] Embodiment 94. The compound of any one of embodiments 1 to 75 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, (R
A)
n wherein the moiety represented as
is
, wherein the left-side attachment point connects to CR
cR
c’ and the right attachment point connects to Ring B. [0706] Embodiment 95. The compound of any one of embodiments 1 to 75 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein the moiety represented
, wherein the left-side attachment point connects to CR
cR
c’ and the right attachment point connects to Ring B. [0707] Embodiment 96. The compound of any one of embodiments 1 to 75 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein the moiety represented
, wherein the left-side attachment point connects to CR
cR
c’ and the right attachment point connects to Ring B. [0708] Embodiment 97. The compound of any one of embodiments 1 to 75 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein the moiety represented as
, wherein the left-side attachment point connects to CR
cR
c’ and the right attachment point connects to Ring B. [0709] Embodiment 98. The compound of any one of embodiments 1 to 75 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein the moiety represented
, wherein the left-side attachment point connects to CR
cR
c’ and the right attachment point connects to Ring B. [0710] Embodiment 99. The compound of any one of embodiments 1 to 75 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, (R
A)
n Ring A wherein the moiety represented as i
, wherein the left-side attachment point connects to CR
cR
c’ and the right attachment point connects to Ring B. [0711] Embodiment 100. The compound of any one of embodiments 1 to 75 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, (R
A)
n Ring A wherein the moiety represented as
i
, wherein the left-side attachment point connects to CR
cR
c’ and the right attachment point connects to Ring B. [0712] Embodiment 101. The compound of any one of embodiments 1 to 75 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein the moiety represented
, wherein the left-side attachment point connects to CR
cR
c’ and the right attachment point connects to Ring B. [0713] Embodiment 102. The compound of any one of embodiments 1 to 75 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein the moiety represented
, wherein the left-side attachment point connects to CR
cR
c’ and the right attachment point connects to Ring B. [0714] Embodiment 103. The compound of any one of embodiments 1 to 75 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, (R
A)
n Ring A wherein the moiety represented as
is
, wherein the left-side attachment point connects to CR
cR
c’ and the right attachment point connects to Ring B. [0715] Embodiment 104. The compound of any one of embodiments 1 to 75 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, (R
A)
n Ring A wherein the moiety represented as
i
, wherein the left-side attachment point connects to CR
cR
c’ and the right attachment point connects to Ring B. [0716] Embodiment 105. The compound of any one of embodiments 1 to 75 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein the moiety represented
, wherein the left-side attachment point connects to CR
cR
c’ and the right attachment point connects to Ring B. [0717] Embodiment 106. The compound of any one of embodiments 1 to 75 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein the moiety represented
, wherein the left-side attachment point connects to CR
cR
c’ and the right attachment point connects to Ring B. [0718] Embodiment 107. The compound of any one of embodiments 1 to 75 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein the moiety represented
, wherein the left-side attachment point connects to CR
cR
c’ and the right attachment point connects to Ring B. [0719] Embodiment 108. The compound of any one of embodiments 1 to 75 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein the moiety represented
, wherein the left-side attachment point connects to CR
cR
c’ and the right attachment point connects to Ring B. [0720] Embodiment 109. The compound of any one of embodiments 1 to 75 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein the moiety represented
, wherein the left-side attachment point connects to CR
cR
c’ and the right attachment point connects to Ring B. [0721] Embodiment 110. The compound of any one of embodiments 1 to 75 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein the moiety represented
, wherein the left-side attachment point connects to CR
cR
c’ and the right attachment point connects to Ring B. [0722] Embodiment 111. The compound of any one of embodiments 1 to 75 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, (R
A)
n Ring A wherein the moiety represented as is
, wherein the left-side attachment point connects to CR
cR
c’ and the right attachment point connects to Ring B. [0723] Embodiment 112. The compound of any one of embodiments 1 to 75 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein the moiety represented
,wherein the left-side attachment point connects to CR
cR
c’ and the right attachment point connects to Ring B. [0724] Embodiment 113. The compound of any one of embodiments 1, 2 and 4-75 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein the compound is of Formula (I-c)
Formula (I-c); wherein R
a2 is selected from the group consisting of CH
3, –CHF
2 and –CD
3. [0725] Embodiment 114. The compound of any one of embodiments 1, 2 and 4-75 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein the compound is of Formula (I-d)
Formula (I-d); wherein R
a2 is selected from the group consisting of CH
3, –CHF
2 and –CD
3. [0726] Embodiment 115. The compound of any one of embodiments 1, 2 and 4-75 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein the compound is of Formula (I-d-1)
Formula (I-d-1); wherein R
a2 is selected from the group consisting of CH
3, –CHF
2 and –CD
3. [0727] Embodiment 116. The compound of any one of embodiments 1-113 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein Ring B is a 5-6 member monocyclic heteroaryl containing 1-3 heteroatoms selected from O, N and S. [0728] Embodiment 117. The compound of any one of embodiments 1-113 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein Ring B is a 6-member monocyclic heteroaryl containing 1-3 nitrogen atoms . [0729] Embodiment 118. The compound of any one of embodiments 1-113 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein Ring B is selected from the group consisting of pyridinyl, pyrimidinyl, pyrazinyl, pyridazinyl, pyrrolyl, pyrazolyl, imidazolyl, thiazolyl, thiadiazolyl, oxadiazolyl and triazolyl. [0730] Embodiment 119. The compound of any one of embodiments 1-113 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein Ring B is selected from the group consisting of pyridinyl, pyrazolyl and imidazolyl. [0731] Embodiment 120. The compound of any one of embodiments 1-113 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein Ring B is selected from the group consisting of pyrazolyl and imidazolyl. [0732] Embodiment 121. The compound of any one of embodiments 1-113 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein Ring B is pyrazolyl. [0733] Embodiment 122. The compound of any one of embodiments 1-113 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein Ring B is imidazolyl. [0734] Embodiment 123. The compound of any one of embodiments 1-113 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein Ring B is selected from the group consisting of pyridin-2-yl, pyridin-3-yl, pyridin-4- yl, pyrimidin-2-yl, pyrimidin-4-yl, pyrimidin-5-yl, pyrimidin-6-yl, pyrazin-2-yl, pyrazin-3-yl, pyridazin-3-yl, pyridazin-4-yl, pyridazin-5-yl, pyridazin-6-yl, pyrrol-2-yl, pyrazol-1-yl, pyrazol-4-yl, pyrazol-5-yl, imidazol-2-yl, thiazol-4-yl, thiazol-5-yl, thiadiazol-3-yl, thiadiazol-5-yl, oxadiazol-3-yl, oxadiazol-5-yl, 1,2,3 triazol-1-yl and 1,2,5 thiazol-4-yl. [0735] Embodiment 124. The compound of any one of embodiments 1-113 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein Ring B is selected from the group consisting of pyridin-2-yl, pyrazol-1-yl and imidazol-2-yl. [0736] Embodiment 125. The compound of any one of embodiments 1-113 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein Ring B is selected from the group consisting of pyrazol-1-yl and imidazol-2-yl. [0737] Embodiment 126. The compound of any one of embodiments 1-113 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein Ring B is pyrazol-1-yl. [0738] Embodiment 127. The compound of any one of embodiments 1-113 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein Ring B is imidazol-2-yl. [0739] Embodiment 128. The compound of any one of embodiments 1-127 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein p is 1, 2 or 3. [0740] Embodiment 129. The compound of any one of embodiments 1-127 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein p is 1 or 2. [0741] Embodiment 130. The compound of any one of embodiments 1-127 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein p is 2 or 3. [0742] Embodiment 131. The compound of any one of embodiments 1-127 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein p is 0. [0743] Embodiment 132. The compound of any one of embodiments 1-127 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein p is 1. [0744] Embodiment 133. The compound of any one of embodiments 1-127 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein p is 2. [0745] Embodiment 134. The compound of any one of embodiments 1-127 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein p is 3. [0746] Embodiment 135. The compound of any one of embodiments 1-134 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein each R
B is independently selected from the group consisting of halo, –CN, –C
1-C
6 alkyl (including partially or fully deuterated alkyl), –C
1-C
6 haloalkyl, –C
1-C
6 hydroxyalkyl, C
3-C
10 cycloalkyl, OR
B1, –C(O)(NR
B1)
2 and –C(O)ONR
B1. [0747] Embodiment 136. The compound of any one of embodiments 1-134 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein each R
B is independently selected from the group consisting of halo, –C
1-C
6 alkyl (including partially or fully deuterated alkyl), –C
1-C
6 alkenyl, –C
1-C
6 haloalkyl, -heteroC
1-C
4 alkyl, –C
3-C
10 cycloalkyl (substituted with 0, 1 or 2 instances of F), 4-7 member heterocyclyl (substituted with 0 or 1 instances of methyl), C
3-C
10 cycloalkylalkyl and OR
B1. [0748] Embodiment 137. The compound of any one of embodiments 1-136 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein each R
B1 is independently –C
1–C
6 alkyl. [0749] Embodiment 138. The compound of any one of embodiments 1-136 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein each R
B1 is independently selected from the group consisting of –Me and –Et. [0750] Embodiment 139. The compound of any one of embodiments 1-134 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein each R
B is independently selected from the group consisting of –Cl, –CH
3, –CD
3,–Et, –
iPr,–CH=CHCH
3, –CH
2CF
3,–CH(CH
3)CF
3, –CF
3, –CHF
2, –OCH
3, –OEt, cyclopropyl, 2,2- difluorocyclopropyl, 1-Me-azetidin-3-yl, oxetan-3-yl, tetrahydropyran-4-yl, –CH
2CH
2OCH
3, –OCH(CH
3)CH
2OCH
3, –CH
2-cyclopropyl. [0751] Embodiment 140. The compound of any one of embodiments 1-134 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein each R
B is independently selected from the group consisting of –Cl, –CD
3,–CH
3, – Et, –
iPr, –CF
3, –OCH
3 and cyclopropyl. [0752] Embodiment 141. The compound of any one of embodiments 1-134 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein each R
B is independently selected from the group consisting of –Cl, –CH
3, –Et, –
iPr, –CF
3, –OCH
3 and cyclopropyl. [0753] Embodiment 142. The compound of any one of embodiments 1-113 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein each R
B is independently selected from the group consisting of –CH
3, –Et, –
iPr, –CF
3 and cyclopropyl. [0754] Embodiment 143. The compound of any one of embodiments 1-113 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof,
[0755] Embodiment 144. The compound of any one of embodiments 1-113 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein the moiety represented
selected from the group consisting of
. [0756] Embodiment 145. The compound of any one of embodiments 1-113 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof,
Ring B wherein the moiety represented by is selected from the group consisting of
[0757] Embodiment 146. The compound of any one of embodiments 1-113 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein the moiety represented
selected from the group consisting of
[0758] Embodiment 147. The compound of any one of embodiments 1-113 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein the moiety represented
selected from the group consisting of
[0759] Embodiment 148. The compound of any one of embodiments 1-113 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein the moiety represented
[0760] Embodiment 149. The compound of any one of embodiments 1-113 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, (
B wherein the moiety represented by
[0761] Embodiment 150. The compound of any one of embodiments 1-113 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein the moiety represented
[0762] Embodiment 151. The compound of any one of embodiments 1-113 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein the moiety represented
[0763] Embodiment 152. The compound of any one of embodiments 1-113 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein the moiety represented
[0764] Embodiment 153. The compound of any one of embodiments 1-113 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein the moiety represented
[0765] Embodiment 154. The compound of any one of embodiments 1, 2, 4-75 and 80-88 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein the compound is of Formula (I-e)
Formula (I-e); wherein R
A is F; R
a2 is selected from the group consisting of CH
3,
iPr, –CHF
2 and –CD
3; and R
B is selected from the group consisting of –Me, –Et and cyclopropyl. [0766] Embodiment 155. The compound of any one of embodiments 1, 2, 4-75 and 80-88 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein the compound is of Formula (I-f)
Formula (I-f); wherein R
a2 is selected from the group consisting of CH3, –CHF2 and –CD3; and R
B is selected from the group consisting of –Me, –Et and cyclopropyl. [0767] Embodiment 156. The compound of any one of embodiments 1, 2, 4-75 and 80-88 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein the compound is of Formula (I-f-1)
Formula (I-f-1); wherein R
a2 is selected from the group consisting of CH
3, –CHF
2 and –CD
3; and R
B is selected from the group consisting of –Me, –Et and cyclopropyl. [0768] Embodiment 157. The compound of any one of embodiments 1 to 156 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein the compound is selected from the compounds of Table 1. [0769] Embodiment 158. A pharmaceutical composition comprising a compound of any one of embodiments 1 to 157 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof and a pharmaceutically acceptable carrier. [0770] Embodiment 159. The pharmaceutical composition of embodiment 158, further comprising a second therapeutic agent. [0771] Embodiment 160. A method for treating or preventing a disease or disorder associated with the inhibition of USP1 comprising administering to a patient in need thereof a therapeutically effective amount of a compound of any one of embodiments 1 to 157 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof. [0772] Embodiment 161. A method of treating a disease or disorder associated with the inhibition of USP1 comprising administering to a patient in need thereof a therapeutically effective amount of a compound of any one of embodiments 1 to 157 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof. [0773] Embodiment 162. A method for inhibiting USP1 comprising administering to a patient in need thereof an effective amount of a compound of any one of embodiments 1 to 157 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof. [0774] Embodiment 163. A method for treating or preventing cancer in a patient in need thereof comprising administering to the patient in need thereof a therapeutically effective amount of a compound of any one of embodiments 1 to 157 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof. [0775] Embodiment 164. A method for treating cancer in a patient in need thereof comprising administering to the patient in need thereof a therapeutically effective amount of a compound of any one of embodiments 1 to 157 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof. [0776] Embodiment 165. The method of embodiment 163 or 164, wherein the cancer is a dedifferentiated ID-driven cancer. [0777] Embodiment 166. The method of any one of embodiments 163 to 165, wherein the cancer is a cancer that is sensitive to USP1 inhibition. [0778] Embodiment 167. The method of any one of embodiments 163 to 166, wherein the cancer is a cancer that is sensitive to USP1 inhibition due to a dysfunctional DNA-repair pathway. [0779] Embodiment 168. The method of any one of embodiments 163 to 167, wherein the cancer is a HRR (homologous recombination repair) gene mutant cancer. [0780] Embodiment 169. The method of any one of embodiments 163 to 167, wherein the cancer is a HRR (homologous recombination repair) gene mutant cancer selected from the group consisting of ATM, NBN, FANCA, BARD1, BRCA1, BRCA2, BRIP1, CDK12, CHEK1, CHEK2, FANCL, PALB2, PPP2R2A, RAD51B, RAD51C, RAD51D, or RAD54L mutant cancer. [0781] Embodiment 170. The method of any one of embodiments 163 to 167, wherein the cancer is characterized by elevated levels of translesion synthesis (e.g., a cancer characterized by elevated levels of And/or UBE2K and/or PCNA, a cancer characterized by elevated PCNA monoubiquitination). [0782] Embodiment 171. The method of any one of embodiments 163 to 170, wherein the cancer is characterized by a deficiency in homologous recombination (e.g., a positive homologous recombination deficiency (HRD) score). [0783] Embodiment 172. The method of any one of embodiments 163 to 171, wherein the cancer is a BRCA1 and/or a BRCA2 mutant cancer. [0784] Embodiment 173. The method of any one of embodiments 163 to 172, wherein the cancer is a BRCA1 and/or a BRCA2 deficient cancer. [0785] Embodiment 174. The method of any one of embodiments 163 to 173, wherein the cancer is an ATM mutant cancer. [0786] Embodiment 175. The method of any one of embodiments 163 to 174, wherein the cancer is an BARD1 mutant cancer. [0787] Embodiment 176. The method of any one of embodiments 163 to 175, wherein the cancer is an BRIP1 mutant cancer. [0788] Embodiment 177. The method of any one of embodiments 163 to 176, wherein the cancer is an CDK12 mutant cancer. [0789] Embodiment 178. The method of any one of embodiments 163 to 177, wherein the cancer is an CHEK1 mutant cancer. [0790] Embodiment 179. The method of any one of embodiments 163 to 178, wherein the cancer is an CHEK2 mutant cancer. [0791] Embodiment 180. The method of any one of embodiments 163 to 179, wherein the cancer is an FANCL mutant cancer. [0792] Embodiment 181. The method of any one of embodiments 163 to 180, wherein the cancer is an PALB2 mutant cancer. [0793] Embodiment 182. The method of any one of embodiments 163 to 181, wherein the cancer is an PPP2R2A mutant cancer. [0794] Embodiment 183. The method of any one of embodiments 163 to 182, wherein the cancer is an RAD51B mutant cancer. [0795] Embodiment 184. The method of any one of embodiments 163 to 183, wherein the cancer is an RAD51C mutant cancer. [0796] Embodiment 185. The method of any one of embodiments 163 to 184, wherein the cancer is an RAD51D mutant cancer. [0797] Embodiment 186. The method of any one of embodiments 163 to 185, wherein the cancer is an RAD54L mutant cancer [0798] Embodiment 187. The method of any one of embodiments 163 to 186, wherein the cancer is a PARP inhibitor resistant or refractory cancer. [0799] Embodiment 188. The method of any one of embodiments 163 to 187, wherein the cancer is selected from adrenocortical carcinoma, AIDS-related lymphoma, AIDS-related malignancies, anal cancer, cerebellar astrocytoma, extrahepatic bile duct cancer, bladder cancer osteosarcoma/malignant fibrous histiocytoma, brain stem glioma, ependymoma, visual pathway and hypothalamic gliomas, breast cancer, bronchial adenomas/carcinoids, carcinoid tumors, gastrointestinal carcinoid tumors, carcinoma, adrenocortical, islet cell carcinoma, primary central nervous system lymphoma, cerebellar astrocytoma, cervical cancer, chronic lymphocytic leukemia, chronic myelogenous leukemia, clear cell sarcoma of tendon sheaths, colon cancer, colorectal cancer, cutaneous t-cell lymphoma, endometrial cancer, ependymoma, esophageal cancer, Ewing's sarcoma/family of tumors, extracranial germ cell tumors, extragonadal germ cell tumors, extrahepatic bile duct cancer, eye cancers, including intraocular melanoma, and retinoblastoma, gallbladder cancer, gastrointestinal carcinoid tumor, ovarian germ cell tumor, gestational trophoblastic tumor, hairy cell leukemia, head and neck cancer, Hodgkin's disease, hypopharyngeal cancer, hypothalamic and visual pathway glioma, intraocular melanoma, Kaposi's sarcoma, laryngeal cancer, acute lymphoblastic leukemia, acute myeloid leukemia, liver cancer, non-small cell lung cancer, small cell lung cancer, non-Hodgkin's lymphoma, Waldenstrom's macroglobulinemia, malignant mesothelioma, malignant thymoma, medulloblastoma, melanoma, intraocular melanoma, merkel cell carcinoma, metastatic squamous neck cancer with occult primary, multiple endocrine neoplasia syndrome, multiple myeloma/plasma cell neoplasm, mycosis fungoides, myelodysplastic syndrome, chronic myelogenous leukemia, myeloid leukemia, multiple myeloma, myeloproliferative disorders, nasal cavity and paranasal sinus cancer, nasopharyngeal cancer, neuroblastoma, oral cancer, oral cavity and lip cancer, oropharyngeal cancer, osteosarcoma/malignant fibrous histiocytoma of bone, ovarian cancer, ovarian low malignant potential tumor, pancreatic cancer, paranasal sinus and nasal cavity cancer, parathyroid cancer, penile cancer, pheochromocytoma, pituitary tumor, pleuropulmonary blastoma, prostate cancer, rectal cancer, renal cell (kidney) cancer, transitional cell cancer (e.g., renal pelvis and ureter), retinoblastoma, rhabdomyosarcoma, salivary gland cancer, malignant fibrous histiocytoma of bone, soft tissue sarcoma, Sezary syndrome, skin cancer, small intestine cancer, stomach (gastric) cancer, supratentorial primitive neuroectodennal and pineal tumors, cutaneous t-cell lymphoma, testicular cancer, malignant thymoma, thyroid cancer, gestational trophoblastic tumor, urethral cancer, uterine sarcoma, vaginal cancer, vulvar cancer, and Wilms' tumor. [0800] Embodiment 189. The method of any one of embodiments 163 to 187, wherein the cancer can be any cancer in any organ, for example, a cancer selected from the group consisting of glioma, thyroid carcinoma, breast carcinoma, small-cell lung carcinoma, non- small-cell carcinoma, gastric carcinoma, colon carcinoma, gastrointestinal stromal carcinoma, pancreatic carcinoma, bile duct carcinoma, CNS carcinoma, ovarian carcinoma, endometrial carcinoma, prostate carcinoma, renal carcinoma, anaplastic large-cell lymphoma, leukemia, multiple myeloma, mesothelioma, and melanoma, and combinations thereof. [0801] Embodiment 190. The method of any one of embodiments 163 to 187, wherein the cancer is selected from liposarcoma, neuroblastoma, glioblastoma, bladder cancer, adrenocortical cancer, multiple myeloma, colorectal cancer, non-small cell lung cancer, Human Papilloma Virus-associated cervical, oropharyngeal, penis, anal, thyroid or vaginal cancer or Epstein-Barr Virus-associated nasopharyngeal carcinoma, gastric cancer, rectal cancer, thyroid cancer, breast cancer, prostate cancer, ovarian cancer, pancreatic cancer, Hodgkin lymphoma and diffuse large B-cell lymphoma. [0802] Embodiment 191. The method of any one of embodiments 163 to 187, wherein the cancer is selected from breast cancer (e.g., triple negative breast cancer (TNBC)), ovarian cancer (e.g., platinum-resistant ovarian cancer, platinum-refractory ovarian cancer), pancreatic cancer, prostate cancer and lung cancer (e.g., non-small cell lung cancer (NSCLC)). [0803] Embodiment 192. The method of any one of embodiments 163 to 187, wherein the cancer is selected from breast cancer (e.g., triple negative breast cancer (TNBC)), ovarian cancer (e.g., platinum-resistant ovarian cancer, platinum-refractory ovarian cancer), prostate cancer and lung cancer (e.g., non-small cell lung cancer (NSCLC)). [0804] Embodiment 193. The method of any one of embodiments 163 to 187, wherein the cancer is breast cancer. [0805] Embodiment 194. The method of any one of embodiments 163 to 187, wherein the cancer is triple negative breast cancer (TNBC). [0806] Embodiment 195. The method of any one of embodiments 163 to 187, wherein the cancer is ovarian cancer. [0807] Embodiment 196. The method of any one of embodiments 163 to 187, wherein the cancer is platinum-resistant ovarian cancer. [0808] Embodiment 197. The method of any one of embodiments 163 to 187, wherein the cancer is platinum-refractory ovarian cancer. [0809] Embodiment 198. The method of any one of embodiments 163 to 187, wherein the cancer is prostate cancer. [0810] Embodiment 199. The method of any one of embodiments 163 to 187, wherein the cancer is lung cancer. [0811] Embodiment 200. The method of any one of embodiments 163 to 187, wherein the cancer is non-small cell lung cancer (NSCLC). [0812] Embodiment 201. A method for treating or preventing a disease or disorder associated with DNA damage comprising administering to a patient in need of a treatment for diseases or disorders associated with DNA damage an effective amount of a compound of any one of embodiments 1 to 157 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof. [0813] Embodiment 202. The method of embodiment 201, wherein the disease is cancer. [0814] Embodiment 203. A method for treating a disease or disorder associated with DNA damage comprising administering to a patient in need of a treatment for diseases or disorders associated with DNA damage an effective amount of a compound of any one of embodiments 1 to 157 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof. [0815] Embodiment 204. The method of embodiment 203, wherein the disease is cancer. [0816] Embodiment 205. A method of inhibiting, modulating or reducing DNA repair activity exercised by USP1 comprising administering to a patient in need thereof an effective amount of a compound of any one of embodiments 1 to 157 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof. [0817] Embodiment 206. A compound of any one of embodiments 1 to 157 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof for use in a method for treating or preventing a disease or disorder associated with the inhibition of USP1 comprising administering to a patient in need thereof a therapeutically effective amount of the compound or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof. [0818] Embodiment 207. A compound of any one of embodiments 1 to 157 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof for use in a method of treating a disease or disorder associated with the inhibition of USP1 comprising administering to a patient in need thereof a therapeutically effective amount of the compound or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof. [0819] Embodiment 208. A compound of any one of embodiments 1 to 157 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof for use in a method for inhibiting USP1 comprising administering to a patient in need thereof an effective amount of the compound or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof. [0820] Embodiment 209. A compound of any one of embodiments 1 to 157 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof for use in a method for treating or preventing cancer in a patient in need thereof comprising administering to the patient in need thereof a therapeutically effective amount of the compound or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof. [0821] Embodiment 210. A compound of any one of embodiments 1 to 157 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof for use in a method for treating cancer in a patient in need thereof comprising administering to the patient in need thereof a therapeutically effective amount of the compound or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof. [0822] Embodiment 211. The compound for use of embodiment 209 or 210, wherein the cancer is a dedifferentiated ID-driven cancer. [0823] Embodiment 212. The compound for use of any one of embodiments 209 to 211, wherein the cancer is a cancer that is sensitive to USP1 inhibition. [0824] Embodiment 213. The compound for use of any one of embodiments 209 to 212, wherein the cancer is a cancer that is sensitive to USP1 inhibition due to a dysfunctional DNA-repair pathway. [0825] Embodiment 214. The compound for use of any one of embodiments 209 to 213, wherein the cancer is a HRR (homologous recombination repair) gene mutant cancer. [0826] Embodiment 215. The compound for use of any one of embodiments 209 to 214, wherein the cancer is a HRR (homologous recombination repair) gene mutant cancer selected from the group consisting of ATM, NBN, FANCA, BARD1, BRCA1, BRCA2, BRIP1, CDK12, CHEK1, CHEK2, FANCL, PALB2, PPP2R2A, RAD51B, RAD51C, RAD51D, or RAD54L mutant cancer. [0827] Embodiment 216. The compound for use of any one of embodiments 209 to 215, wherein the cancer is characterized by elevated levels of translesion synthesis (e.g., a cancer characterized by elevated levels of And/or UBE2K and/or PCNA, a cancer characterized by elevated PCNA monoubiquitination). [0828] Embodiment 217. The compound for use of any one of embodiments 209 to 216, wherein the cancer is characterized by a deficiency in homologous recombination (e.g., a positive homologous recombination deficiency (HRD) score). [0829] Embodiment 218. The compound for use of any one of embodiments 209 to 217, wherein the cancer is a BRCA1 and/or a BRCA2 mutant cancer. [0830] Embodiment 219. The compound for use of any one of embodiments 209 to 218, wherein the cancer is a BRCA1 and/or a BRCA2 deficient cancer. [0831] Embodiment 220. The compound for use of any one of embodiments 209 to 219, wherein the cancer is an ATM mutant cancer. [0832] Embodiment 221. The compound for use of any one of embodiments 209 to 220, wherein the cancer is an BARD1 mutant cancer. [0833] Embodiment 222. The compound for use of any one of embodiments 209 to 221, wherein the cancer is an BRIP1 mutant cancer. [0834] Embodiment 223. The compound for use of any one of embodiments 209 to 222, wherein the cancer is an CDK12 mutant cancer. [0835] Embodiment 224. The compound for use of any one of embodiments 209 to 223, wherein the cancer is an CHEK1 mutant cancer. [0836] Embodiment 225. The compound for use of any one of embodiments 209 to 224, wherein the cancer is an CHEK2 mutant cancer. [0837] Embodiment 226. The compound for use of any one of embodiments 209 to 225, wherein the cancer is an FANCL mutant cancer. [0838] Embodiment 227. The compound for use of any one of embodiments 209 to 226, wherein the cancer is an PALB2 mutant cancer. [0839] Embodiment 228. The compound for use of any one of embodiments 209 to 227, wherein the cancer is an PPP2R2A mutant cancer. [0840] Embodiment 229. The compound for use of any one of embodiments 209 to 228, wherein the cancer is an RAD51B mutant cancer. [0841] Embodiment 230. The compound for use of any one of embodiments 209 to 229, wherein the cancer is an RAD51C mutant cancer. [0842] Embodiment 231. The compound for use of any one of embodiments 209 to 230, wherein the cancer is an RAD51D mutant cancer. [0843] Embodiment 232. The compound for use of any one of embodiments 209 to 231, wherein the cancer is an RAD54L mutant cancer. [0844] Embodiment 233. The compound for use of any one of embodiments 209 to 232, wherein the cancer is a PARP inhibitor resistant or refractory cancer. [0845] Embodiment 234. The compound for use of any one of embodiments 209 to 233, wherein the cancer is selected from adrenocortical carcinoma, AIDS-related lymphoma, AIDS-related malignancies, anal cancer, cerebellar astrocytoma, extrahepatic bile duct cancer, bladder cancer osteosarcoma/malignant fibrous histiocytoma, brain stem glioma, ependymoma, visual pathway and hypothalamic gliomas, breast cancer, bronchial adenomas/carcinoids, carcinoid tumors, gastrointestinal carcinoid tumors, carcinoma, adrenocortical, islet cell carcinoma, primary central nervous system lymphoma, cerebellar astrocytoma, cervical cancer, chronic lymphocytic leukemia, chronic myelogenous leukemia, clear cell sarcoma of tendon sheaths, colon cancer, colorectal cancer, cutaneous t-cell lymphoma, endometrial cancer, ependymoma, esophageal cancer, Ewing's sarcoma/family of tumors, extracranial germ cell tumors, extragonadal germ cell tumors, extrahepatic bile duct cancer, eye cancers, including intraocular melanoma, and retinoblastoma, gallbladder cancer, gastrointestinal carcinoid tumor, ovarian germ cell tumor, gestational trophoblastic tumor, hairy cell leukemia, head and neck cancer, Hodgkin's disease, hypopharyngeal cancer, hypothalamic and visual pathway glioma, intraocular melanoma, Kaposi's sarcoma, laryngeal cancer, acute lymphoblastic leukemia, acute myeloid leukemia, liver cancer, non-small cell lung cancer, small cell lung cancer, non-Hodgkin's lymphoma, Waldenstrom's macroglobulinemia, malignant mesothelioma, malignant thymoma, medulloblastoma, melanoma, intraocular melanoma, merkel cell carcinoma, metastatic squamous neck cancer with occult primary, multiple endocrine neoplasia syndrome, multiple myeloma/plasma cell neoplasm, mycosis fungoides, myelodysplastic syndrome, chronic myelogenous leukemia, myeloid leukemia, multiple myeloma, myeloproliferative disorders, nasal cavity and paranasal sinus cancer, nasopharyngeal cancer, neuroblastoma, oral cancer, oral cavity and lip cancer, oropharyngeal cancer, osteosarcoma/malignant fibrous histiocytoma of bone, ovarian cancer, ovarian low malignant potential tumor, pancreatic cancer, paranasal sinus and nasal cavity cancer, parathyroid cancer, penile cancer, pheochromocytoma, pituitary tumor, pleuropulmonary blastoma, prostate cancer, rectal cancer, renal cell (kidney) cancer, transitional cell cancer (e.g., renal pelvis and ureter), retinoblastoma, rhabdomyosarcoma, salivary gland cancer, malignant fibrous histiocytoma of bone, soft tissue sarcoma, Sezary syndrome, skin cancer, small intestine cancer, stomach (gastric) cancer, supratentorial primitive neuroectodennal and pineal tumors, cutaneous t-cell lymphoma, testicular cancer, malignant thymoma, thyroid cancer, gestational trophoblastic tumor, urethral cancer, uterine sarcoma, vaginal cancer, vulvar cancer, and Wilms' tumor. [0846] Embodiment 235. The compound for use of any one of embodiments 209 to 233, wherein the cancer can be any cancer in any organ, for example, a cancer selected from the group consisting of glioma, thyroid carcinoma, breast carcinoma, small-cell lung carcinoma, non-small-cell carcinoma, gastric carcinoma, colon carcinoma, gastrointestinal stromal carcinoma, pancreatic carcinoma, bile duct carcinoma, CNS carcinoma, ovarian carcinoma, endometrial carcinoma, prostate carcinoma, renal carcinoma, anaplastic large-cell lymphoma, leukemia, multiple myeloma, mesothelioma, and melanoma, and combinations thereof. [0847] Embodiment 236. The compound for use of any one of embodiments 209 to 233, wherein the cancer is selected from liposarcoma, neuroblastoma, glioblastoma, bladder cancer, adrenocortical cancer, multiple myeloma, colorectal cancer, non-small cell lung cancer, Human Papilloma Virus-associated cervical, oropharyngeal, penis, anal, thyroid or vaginal cancer or Epstein-Barr Virus-associated nasopharyngeal carcinoma, gastric cancer, rectal cancer, thyroid cancer, breast cancer, prostate cancer, ovarian cancer, pancreatic cancer, Hodgkin lymphoma and diffuse large B-cell lymphoma. [0848] Embodiment 237. The compound for use of any one of embodiments 209 to 233, wherein the cancer is selected from breast cancer (e.g., triple negative breast cancer (TNBC)), ovarian cancer (e.g., platinum-resistant ovarian cancer, platinum-refractory ovarian cancer), pancreatic cancer, prostate cancer and lung cancer (e.g., non-small cell lung cancer (NSCLC)). [0849] Embodiment 238. The compound for use of any one of embodiments 209 to 233, wherein the cancer is selected from breast cancer (e.g., triple negative breast cancer (TNBC)), ovarian cancer (e.g., platinum-resistant ovarian cancer, platinum-refractory ovarian cancer), prostate cancer and lung cancer (e.g., non-small cell lung cancer (NSCLC)). [0850] Embodiment 239. The compound for use of any one of embodiments 209 to 233, wherein the cancer is breast cancer. [0851] Embodiment 240. The compound for use of any one of embodiments 209 to 233, wherein the cancer is triple negative breast cancer (TNBC). [0852] Embodiment 241. The compound for use of any one of embodiments 209 to 233, wherein the cancer is ovarian cancer. [0853] Embodiment 242. The compound for use of any one of embodiments 209 to 233, wherein the cancer is platinum-resistant ovarian cancer. [0854] Embodiment 243. The compound for use of any one of embodiments 209 to 233, wherein the cancer is platinum-refractory ovarian cancer. [0855] Embodiment 244. The compound for use of any one of embodiments 209 to 233, wherein the cancer is prostate cancer. [0856] Embodiment 245. The compound for use of any one of embodiments 209 to 233, wherein the cancer is lung cancer. [0857] Embodiment 246. The compound for use of any one of embodiments 209 to 233, wherein the cancer is non-small cell lung cancer (NSCLC). [0858] Embodiment 247. A compound of any one of embodiments 1 to 157 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof for use in a method for treating or preventing a disease or disorder associated with DNA . [0859] Embodiment 248. The compound for use of embodiment 247, wherein the disease is cancer. [0860] Embodiment 249. A compound of any one of embodiments 1 to 157 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof for use in a method for treating a disease or disorder associated with DNA damage. [0861] Embodiment 250. The compound for use of embodiment 249, wherein the disease is cancer. [0862] Embodiment 251. A compound of any one of embodiments 1 to 157 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof for use in a method of inhibiting, modulating or reducing DNA repair activity exercised by USP1 comprising administering to a patient in need thereof an effective amount of the compound or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof. [0863] Embodiment 252. Use of an effective amount of a compound of any one of embodiments 1 to 157 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof in the manufacturing of a medicament for treating or preventing a disease or disorder associated with the inhibition of USP1. [0864] Embodiment 253. Use of an effective amount of a compound of any one of embodiments 1 to 157 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof in the manufacturing of a medicament for treating a disease or disorder associated with the inhibition of USP1. [0865] Embodiment 254. Use of an effective amount of a compound of any one of embodiments 1 to 157 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof in the manufacturing of a medicament for inhibiting USP1. [0866] Embodiment 255. Use of an effective amount of a compound of any one of embodiments 1 to 157 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof in the manufacturing of a medicament for treating or preventing cancer in a patient in need thereof. [0867] Embodiment 256. Use of an effective amount of a compound of any one of embodiments 1 to 157 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof in the manufacturing of a medicament for treating cancer in a patient in need thereof. [0868] Embodiment 257. The use of embodiment 255 or 256, wherein the cancer is a dedifferentiated ID-driven cancer. [0869] Embodiment 258. The use of any one of embodiments 255 to 257, wherein the cancer is a cancer that is sensitive to USP1 inhibition. [0870] Embodiment 259. The use of any one of embodiments 255 to 258, wherein the cancer is a cancer that is sensitive to USP1 inhibition due to a dysfunctional DNA-repair pathway. [0871] Embodiment 260. The use of any one of embodiments 255 to 259, wherein the cancer is a HRR (homologous recombination repair) gene mutant cancer. [0872] Embodiment 261. The use of any one of embodiments 255 to 260, wherein the cancer is a HRR (homologous recombination repair) gene mutant cancer selected from the group consisting of ATM, NBN, FANCA, BARD1, BRCA1, BRCA2, BRIP1, CDK12, CHEK1, CHEK2, FANCL, PALB2, PPP2R2A, RAD51B, RAD51C, RAD51D, or RAD54L mutant cancer. [0873] Embodiment 262. The use of any one of embodiments 255 to 261, wherein the cancer is characterized by elevated levels of translesion synthesis (e.g., a cancer characterized by elevated levels of And/or UBE2K and/or PCNA, a cancer characterized by elevated PCNA monoubiquitination). [0874] Embodiment 263. The use of any one of embodiments 255 to 262, wherein the cancer is characterized by a deficiency in homologous recombination (e.g., a positive homologous recombination deficiency (HRD) score). [0875] Embodiment 264. The use of any one of embodiments 255 to 263, wherein the cancer is a BRCA1 and/or a BRCA2 mutant cancer. [0876] Embodiment 265. The use of any one of embodiments 255 to 264, wherein the cancer is a BRCA1 and/or a BRCA2 deficient cancer. [0877] Embodiment 266. The use of any one of embodiments 255 to 265, wherein the cancer is an ATM mutant cancer. [0878] Embodiment 267. The use of any one of embodiments 255 to 266, wherein the cancer is an BARD1 mutant cancer. [0879] Embodiment 268. The use of any one of embodiments 255 to 267, wherein the cancer is an BRIP1 mutant cancer. [0880] Embodiment 269. The use of any one of embodiments 255 to 268, wherein the cancer is an CDK12 mutant cancer. [0881] Embodiment 270. The use of any one of embodiments 255 to 269, wherein the cancer is an CHEK1 mutant cancer. [0882] Embodiment 271. The use of any one of embodiments 255 to 270, wherein the cancer is an CHEK2 mutant cancer. [0883] Embodiment 272. The use of any one of embodiments 255 to 271, wherein the cancer is an FANCL mutant cancer. [0884] Embodiment 273. The use of any one of embodiments 255 to 272, wherein the cancer is an PALB2 mutant cancer. [0885] Embodiment 274. The use of any one of embodiments 255 to 273, wherein the cancer is an PPP2R2A mutant cancer. [0886] Embodiment 275. The use of any one of embodiments 255 to 274, wherein the cancer is an RAD51B mutant cancer. [0887] Embodiment 276. The use of any one of embodiments 255 to 275, wherein the cancer is an RAD51C mutant cancer. [0888] Embodiment 277. The use of any one of embodiments 255 to 276, wherein the cancer is an RAD51D mutant cancer. [0889] Embodiment 278. The use of any one of embodiments 255 to 277, wherein the cancer is an RAD54L mutant cancer. [0890] Embodiment 279. The use of any one of embodiments 255 to 278, wherein the cancer is a PARP inhibitor resistant or refractory cancer. [0891] Embodiment 280. The use of any one of embodiments 255 to 279, wherein the cancer is selected from adrenocortical carcinoma, AIDS-related lymphoma, AIDS-related malignancies, anal cancer, cerebellar astrocytoma, extrahepatic bile duct cancer, bladder cancer osteosarcoma/malignant fibrous histiocytoma, brain stem glioma, ependymoma, visual pathway and hypothalamic gliomas, breast cancer, bronchial adenomas/carcinoids, carcinoid tumors, gastrointestinal carcinoid tumors, carcinoma, adrenocortical, islet cell carcinoma, primary central nervous system lymphoma, cerebellar astrocytoma, cervical cancer, chronic lymphocytic leukemia, chronic myelogenous leukemia, clear cell sarcoma of tendon sheaths, colon cancer, colorectal cancer, cutaneous t-cell lymphoma, endometrial cancer, ependymoma, esophageal cancer, Ewing's sarcoma/family of tumors, extracranial germ cell tumors, extragonadal germ cell tumors, extrahepatic bile duct cancer, eye cancers, including intraocular melanoma, and retinoblastoma, gallbladder cancer, gastrointestinal carcinoid tumor, ovarian germ cell tumor, gestational trophoblastic tumor, hairy cell leukemia, head and neck cancer, Hodgkin's disease, hypopharyngeal cancer, hypothalamic and visual pathway glioma, intraocular melanoma, Kaposi's sarcoma, laryngeal cancer, acute lymphoblastic leukemia, acute myeloid leukemia, liver cancer, non-small cell lung cancer, small cell lung cancer, non-Hodgkin's lymphoma, Waldenstrom's macroglobulinemia, malignant mesothelioma, malignant thymoma, medulloblastoma, melanoma, intraocular melanoma, merkel cell carcinoma, metastatic squamous neck cancer with occult primary, multiple endocrine neoplasia syndrome, multiple myeloma/plasma cell neoplasm, mycosis fungoides, myelodysplastic syndrome, chronic myelogenous leukemia, myeloid leukemia, multiple myeloma, myeloproliferative disorders, nasal cavity and paranasal sinus cancer, nasopharyngeal cancer, neuroblastoma, oral cancer, oral cavity and lip cancer, oropharyngeal cancer, osteosarcoma/malignant fibrous histiocytoma of bone, ovarian cancer, ovarian low malignant potential tumor, pancreatic cancer, paranasal sinus and nasal cavity cancer, parathyroid cancer, penile cancer, pheochromocytoma, pituitary tumor, pleuropulmonary blastoma, prostate cancer, rectal cancer, renal cell (kidney) cancer, transitional cell cancer (e.g., renal pelvis and ureter), retinoblastoma, rhabdomyosarcoma, salivary gland cancer, malignant fibrous histiocytoma of bone, soft tissue sarcoma, Sezary syndrome, skin cancer, small intestine cancer, stomach (gastric) cancer, supratentorial primitive neuroectodennal and pineal tumors, cutaneous t-cell lymphoma, testicular cancer, malignant thymoma, thyroid cancer, gestational trophoblastic tumor, urethral cancer, uterine sarcoma, vaginal cancer, vulvar cancer, and Wilms' tumor. [0892] Embodiment 281. The use of any one of embodiments 255 to 279, wherein the cancer can be any cancer in any organ, for example, a cancer selected from the group consisting of glioma, thyroid carcinoma, breast carcinoma, small-cell lung carcinoma, non- small-cell carcinoma, gastric carcinoma, colon carcinoma, gastrointestinal stromal carcinoma, pancreatic carcinoma, bile duct carcinoma, CNS carcinoma, ovarian carcinoma, endometrial carcinoma, prostate carcinoma, renal carcinoma, anaplastic large-cell lymphoma, leukemia, multiple myeloma, mesothelioma, and melanoma, and combinations thereof. [0893] Embodiment 282. The use of any one of embodiments 255 to 279, wherein the cancer is selected from liposarcoma, neuroblastoma, glioblastoma, bladder cancer, adrenocortical cancer, multiple myeloma, colorectal cancer, non-small cell lung cancer, Human Papilloma Virus-associated cervical, oropharyngeal, penis, anal, thyroid or vaginal cancer or Epstein-Barr Virus-associated nasopharyngeal carcinoma, gastric cancer, rectal cancer, thyroid cancer, breast cancer, prostate cancer, ovarian cancer, pancreatic cancer, Hodgkin lymphoma and diffuse large B-cell lymphoma. [0894] Embodiment 283. The use of any one of embodiments 255 to 279, wherein the cancer is selected from breast cancer (e.g., triple negative breast cancer (TNBC)), ovarian cancer (e.g., platinum-resistant ovarian cancer, platinum-refractory ovarian cancer), pancreatic cancer, prostate cancer and lung cancer (e.g., non-small cell lung cancer (NSCLC)). [0895] Embodiment 284. The use of any one of embodiments 255 to 279, wherein the cancer is selected from breast cancer (e.g., triple negative breast cancer (TNBC)), ovarian cancer (e.g., platinum-resistant ovarian cancer, platinum-refractory ovarian cancer), prostate cancer and lung cancer (e.g., non-small cell lung cancer (NSCLC)). [0896] Embodiment 285. The use of any one of embodiments 255 to 279, wherein the cancer is breast cancer. [0897] Embodiment 286. The use of any one of embodiments 255 to 279, wherein the cancer is triple negative breast cancer (TNBC). [0898] Embodiment 287. The use of any one of embodiments 255 to 279, wherein the cancer is ovarian cancer. [0899] Embodiment 288. The use of any one of embodiments 255 to 279, wherein the cancer is platinum-resistant ovarian cancer. [0900] Embodiment 289. The use of any one of embodiments 255 to 279, wherein the cancer is platinum-refractory ovarian cancer. [0901] Embodiment 290. The use of any one of embodiments 255 to 279, wherein the cancer is prostate cancer. [0902] Embodiment 291. The use of any one of embodiments 255 to 279, wherein the cancer is lung cancer. [0903] Embodiment 292. The use of any one of embodiments 255 to 279, wherein the cancer is non-small cell lung cancer (NSCLC). [0904] Embodiment 293. Use of an effective amount of a compound of any one of embodiments 1 to 157 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof in the manufacturing of a medicament for treating or preventing a disease or disorder associated with DNA damage. [0905] Embodiment 294. The use of embodiment 293, wherein the disease is cancer. [0906] Embodiment 295. Use of an effective amount of a compound of any one of embodiments 1 to 157 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof in the manufacturing of a medicament for treating a disease or disorder associated with DNA damage. [0907] Embodiment 296. The use of embodiment 295, wherein the disease is cancer. [0908] Embodiment 297. Use of an effective amount of a compound of any one of embodiments 1 to 157 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof for inhibiting, modulating or reducing DNA repair activity exercised by USP1. [0909] Embodiment 298. Use of an effective amount of a compound of any one of embodiments 1 to 157 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof for treating or preventing a disease or disorder associated with the inhibition of USP1. [0910] Embodiment 299. Use of an effective amount of a compound of any one of embodiments 1 to 157 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof for treating a disease or disorder associated with the inhibition of USP1. [0911] Embodiment 300. Use of an effective amount of a compound of any one of embodiments 1 to 157 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof for inhibiting USP1. [0912] Embodiment 301. Use of an effective amount of a compound of any one of embodiments 1 to 157 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof for treating or preventing cancer in a patient in need thereof. [0913] Embodiment 302. Use of an effective amount of a compound of any one of embodiments 1 to 157 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof for treating cancer in a patient in need thereof. [0914] Embodiment 303. The use of embodiment 301 or 302, wherein the cancer is a dedifferentiated ID-driven cancer. [0915] Embodiment 304. The use of any one of embodiments 301 to 303, wherein the cancer is a cancer that is sensitive to USP1 inhibition. [0916] Embodiment 305. The use of any one of embodiments 301 to 304, wherein the cancer is a cancer that is sensitive to USP1 inhibition due to a dysfunctional DNA-repair pathway. [0917] Embodiment 306. The use of any one of embodiments 301 to 305, wherein the cancer is a HRR (homologous recombination repair) gene mutant cancer. [0918] Embodiment 307. The use of any one of embodiments 301 to 306, wherein the cancer is a HRR (homologous recombination repair) gene mutant cancer selected from the group consisting of ATM, NBN, FANCA, BARD1, BRCA1, BRCA2, BRIP1, CDK12, CHEK1, CHEK2, FANCL, PALB2, PPP2R2A, RAD51B, RAD51C, RAD51D, or RAD54L mutant cancer. [0919] Embodiment 308. The use of any one of embodiments 301 to 307, wherein the cancer is characterized by elevated levels of translesion synthesis (e.g., a cancer characterized by elevated levels of And/or UBE2K and/or PCNA, a cancer characterized by elevated PCNA monoubiquitination). [0920] Embodiment 309. The use of any one of embodiments 301 to 308, wherein the cancer is characterized by a deficiency in homologous recombination (e.g., a positive homologous recombination deficiency (HRD) score). [0921] Embodiment 310. The use of any one of embodiments 301 to 309, wherein the cancer is a BRCA1 and/or a BRCA2 mutant cancer. [0922] Embodiment 311. The use of any one of embodiments 301 to 310, wherein the cancer is a BRCA1 and/or a BRCA2 deficient cancer. [0923] Embodiment 312. The use of any one of embodiments 301 to 311, wherein the cancer is an ATM mutant cancer. [0924] Embodiment 313. The use of any one of embodiments 255 to 312, wherein the cancer is an BARD1 mutant cancer. [0925] Embodiment 314. The use of any one of embodiments 301 to 313, wherein the cancer is an BRIP1 mutant cancer. [0926] Embodiment 315. The use of any one of embodiments 301 to 314, wherein the cancer is an CDK12 mutant cancer. [0927] Embodiment 316. The use of any one of embodiments 301 to 315, wherein the cancer is an CHEK1 mutant cancer. [0928] Embodiment 317. The use of any one of embodiments 301 to 316, wherein the cancer is an CHEK2 mutant cancer. [0929] Embodiment 318. The use of any one of embodiments 301 to 317, wherein the cancer is an FANCL mutant cancer. [0930] Embodiment 319. The use of any one of embodiments 301 to 318, wherein the cancer is an PALB2 mutant cancer. [0931] Embodiment 320. The use of any one of embodiments 301 to 319, wherein the cancer is an PPP2R2A mutant cancer. [0932] Embodiment 321. The use of any one of embodiments 301 to 320, wherein the cancer is an RAD51B mutant cancer. [0933] Embodiment 322. The use of any one of embodiments 301 to 321, wherein the cancer is an RAD51C mutant cancer. [0934] Embodiment 323. The use of any one of embodiments 301 to 322, wherein the cancer is an RAD51D mutant cancer. [0935] Embodiment 324. The use of any one of embodiments 301 to 323, wherein the cancer is an RAD54L mutant cancer. [0936] Embodiment 325. The use of any one of embodiments 301 to 324, wherein the cancer is a PARP inhibitor resistant or refractory cancer. [0937] Embodiment 326. The use of any one of embodiments 301 to 325, wherein the cancer is selected from adrenocortical carcinoma, AIDS-related lymphoma, AIDS-related malignancies, anal cancer, cerebellar astrocytoma, extrahepatic bile duct cancer, bladder cancer osteosarcoma/malignant fibrous histiocytoma, brain stem glioma, ependymoma, visual pathway and hypothalamic gliomas, breast cancer, bronchial adenomas/carcinoids, carcinoid tumors, gastrointestinal carcinoid tumors, carcinoma, adrenocortical, islet cell carcinoma, primary central nervous system lymphoma, cerebellar astrocytoma, cervical cancer, chronic lymphocytic leukemia, chronic myelogenous leukemia, clear cell sarcoma of tendon sheaths, colon cancer, colorectal cancer, cutaneous t-cell lymphoma, endometrial cancer, ependymoma, esophageal cancer, Ewing's sarcoma/family of tumors, extracranial germ cell tumors, extragonadal germ cell tumors, extrahepatic bile duct cancer, eye cancers, including intraocular melanoma, and retinoblastoma, gallbladder cancer, gastrointestinal carcinoid tumor, ovarian germ cell tumor, gestational trophoblastic tumor, hairy cell leukemia, head and neck cancer, Hodgkin's disease, hypopharyngeal cancer, hypothalamic and visual pathway glioma, intraocular melanoma, Kaposi's sarcoma, laryngeal cancer, acute lymphoblastic leukemia, acute myeloid leukemia, liver cancer, non-small cell lung cancer, small cell lung cancer, non-Hodgkin's lymphoma, Waldenstrom's macroglobulinemia, malignant mesothelioma, malignant thymoma, medulloblastoma, melanoma, intraocular melanoma, merkel cell carcinoma, metastatic squamous neck cancer with occult primary, multiple endocrine neoplasia syndrome, multiple myeloma/plasma cell neoplasm, mycosis fungoides, myelodysplastic syndrome, chronic myelogenous leukemia, myeloid leukemia, multiple myeloma, myeloproliferative disorders, nasal cavity and paranasal sinus cancer, nasopharyngeal cancer, neuroblastoma, oral cancer, oral cavity and lip cancer, oropharyngeal cancer, osteosarcoma/malignant fibrous histiocytoma of bone, ovarian cancer, ovarian low malignant potential tumor, pancreatic cancer, paranasal sinus and nasal cavity cancer, parathyroid cancer, penile cancer, pheochromocytoma, pituitary tumor, pleuropulmonary blastoma, prostate cancer, rectal cancer, renal cell (kidney) cancer, transitional cell cancer (e.g., renal pelvis and ureter), retinoblastoma, rhabdomyosarcoma, salivary gland cancer, malignant fibrous histiocytoma of bone, soft tissue sarcoma, Sezary syndrome, skin cancer, small intestine cancer, stomach (gastric) cancer, supratentorial primitive neuroectodennal and pineal tumors, cutaneous t-cell lymphoma, testicular cancer, malignant thymoma, thyroid cancer, gestational trophoblastic tumor, urethral cancer, uterine sarcoma, vaginal cancer, vulvar cancer, and Wilms' tumor. [0938] Embodiment 327. The use of any one of embodiments 301 to 325, wherein the cancer can be any cancer in any organ, for example, a cancer selected from the group consisting of glioma, thyroid carcinoma, breast carcinoma, small-cell lung carcinoma, non- small-cell carcinoma, gastric carcinoma, colon carcinoma, gastrointestinal stromal carcinoma, pancreatic carcinoma, bile duct carcinoma, CNS carcinoma, ovarian carcinoma, endometrial carcinoma, prostate carcinoma, renal carcinoma, anaplastic large-cell lymphoma, leukemia, multiple myeloma, mesothelioma, and melanoma, and combinations thereof. [0939] Embodiment 328. The use of any one of embodiments 301 to 325, wherein the cancer is selected from liposarcoma, neuroblastoma, glioblastoma, bladder cancer, adrenocortical cancer, multiple myeloma, colorectal cancer, non-small cell lung cancer, Human Papilloma Virus-associated cervical, oropharyngeal, penis, anal, thyroid or vaginal cancer or Epstein-Barr Virus-associated nasopharyngeal carcinoma, gastric cancer, rectal cancer, thyroid cancer, breast cancer, prostate cancer, ovarian cancer, pancreatic cancer, Hodgkin lymphoma and diffuse large B-cell lymphoma. [0940] Embodiment 329. The use of any one of embodiments 301 to 325, wherein the cancer is selected from breast cancer (e.g., triple negative breast cancer (TNBC)), ovarian cancer (e.g., platinum-resistant ovarian cancer, platinum-refractory ovarian cancer), pancreatic cancer, prostate cancer and lung cancer (e.g., non-small cell lung cancer (NSCLC)). [0941] Embodiment 330. The use of any one of embodiments 301 to 325, wherein the cancer is selected from breast cancer (e.g., triple negative breast cancer (TNBC)), ovarian cancer (e.g., platinum-resistant ovarian cancer, platinum-refractory ovarian cancer), prostate cancer and lung cancer (e.g., non-small cell lung cancer (NSCLC)). [0942] Embodiment 331. The use of any one of embodiments 301 to 325, wherein the cancer is breast cancer. [0943] Embodiment 332. The use of any one of embodiments 301 to 325, wherein the cancer is triple negative breast cancer (TNBC). [0944] Embodiment 333. The use of any one of embodiments 301 to 325, wherein the cancer is ovarian cancer. [0945] Embodiment 334. The use of any one of embodiments 301 to 325, wherein the cancer is platinum-resistant ovarian cancer. [0946] Embodiment 335. The use of any one of embodiments 301 to 325, wherein the cancer is platinum-refractory ovarian cancer. [0947] Embodiment 336. The use of any one of embodiments 301 to 325, wherein the cancer is prostate cancer. [0948] Embodiment 337. The use of any one of embodiments 301 to 325, wherein the cancer is lung cancer. [0949] Embodiment 338. The use of any one of embodiments 301 to 325, wherein the cancer is non-small cell lung cancer (NSCLC). [0950] Embodiment 339. Use of an effective amount of a compound of any one of embodiments 1 to 157 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof for treating or preventing a disease or disorder associated with DNA damage. [0951] Embodiment 340. The use of embodiment 339, wherein the disease is cancer. [0952] Embodiment 341. Use of an effective amount of a compound of any one of embodiments 1 to 157 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof for treating a disease or disorder associated with DNA damage. [0953] Embodiment 342. The use of embodiment 341, wherein the disease is cancer. [0954] Embodiment 343. Use of an effective amount of a compound of any one of embodiments 1 to 157 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof for inhibiting, modulating or reducing DNA repair activity exercised by USP1. Examples [0955] In order that the invention described herein may be more fully understood, the following examples are set forth. The synthetic and biological examples described in this application are offered to illustrate the compounds, pharmaceutical compositions, and methods provided herein and are not to be construed in any way as limiting their scope. In the synthetic examples below, the descriptions of experimental procedures within a reaction sequence are listed in numerical order. Abbreviations General ADDP 1,1′-(azodicarbonyl)dipiperidine anhy. anhydrous aq. aqueous satd. saturated min(s) minute(s) hr(s) or h hour(s) mL milliliter mmol millimole(s) mol mole(s) MS mass spectrometry NMR nuclear magnetic resonance TLC thin layer chromatography HPLC high-performance liquid chromatography Me methyl i-Pr iso-propyl t-Bu tert-butyl
tBuXPhos 2-di-tert-butylphosphino-2′,4′,6′-triisopropylbiphenyl Ph phenyl Et ethyl Bz benzoyl RuPhos 2-dicyclohexylphosphino-2′,6′-diisopropoxybiphenyl Spectrum Hz hertz δ chemical shift J coupling constant s singlet d doublet t triplet q quartet m multiplet br broad qd quartet of doublets dquin doublet of quintets dd doublet of doublets dt doublet of triplets Solvents and Reagents DAST Diethylaminosulfurtrifluoride Cs
2CO
3 cesium carbonate CHCl
3 chloroform DCM dichloromethane DMF dimethylformamide Et
2O diethyl ether EtOH ethyl alcohol EtOAc ethyl acetate MeOH methanol MeCN or ACN acetonitrile PE petroleum ether THF tetrahydrofuran DMSO dimethyl sulfoxide t-BuOK potassium tert-butoxide 9-BBN 9-borabicyclo[3.3.1]nonane AcOH acetic acid HCl hydrochloric acid H
2SO
4 sulfuric acid NH
4Cl ammonium chloride KOH potassium hydroxide NaOH sodium hydroxide K
2CO
3 potassium carbonate Na
2CO
3 sodium carbonate TFA trifluoroacetic acid Na
2SO
4 sodium sulfate NaBH
4 sodium borohydride NaHCO
3 sodium bicarbonate LiHMDS lithium hexamethyldisilylamide NaBH
4 sodium borohydride Et
3N triethylamine Py pyridine mCPBA meta-chloroperoxybenzoic acid PCC pyridinium chlorochromate DMAP 4-(dimethylamino)pyridine DIPEA N,N-diisopropylethylamine BINAP 2,2’-bis(diphenylphosphanyl)-1,1’-binaphthyl dppf 1,1'-bis(diphenylphosphino)ferrocene PEP Phospho(enol)pyruvic acid LDH Lactate Dehydrogenase DTT DL-Dithiothreitol BSA Bovine Serum Albumin NADH β-Nicotinamide adenine dinucleotide, reduced Pd(t-Bu
3P)
2 bis(tri-tert-butylphosphine)palladium(0) AcCl acetyl chloride i-PrMgCl Isopropylmagnesium chloride TBSCl tert-Butyl(chloro)dimethylsilane (i-PrO)
4Ti titanium tetraisopropoxide BHT 2,6-di-t-butyl-4-methylphenoxide BzCl benzoyl chloride CsF cesium fluoride DCC dicyclohexylcarbodiimide DMP Dess-Martin periodinane EtMgBr ethylmagnesium bromide TEA triethylamine AlaOH alanine TBAF tetra-n-butylammonium fluoride TBS t-butyldimethylsilyl TMS trimethylsilyl TMSCF
3 (Trifluoromethyl)trimethylsilane Ts p-toluenesulfonyl Bu butyl Ti(O
iPr)
4 tetraisopropoxytitanium LAH Lithium Aluminium Hydride LDA lithium diisopropylamide LiOH.H
2O lithium hydroxide hydrates MAD methyl aluminum bis(2,6-di-t-butyl-4-methylphenoxide) NBS N-bromosuccinimide NaH sodium hydride Na
2S
2O
3 sodium thiosulfate PE PE Boc t-butoxycarbonyl MTBE methyl tert-butyl ether DIAD diisopropyl azodicarboxylate General experimental notes: [0956] In the following examples, the chemical reagents were purchased from commercial sources (such as Alfa, Acros, Sigma Aldrich, TCI and Shanghai Chemical Reagent Company), and used without further purification. Materials and Methods [0957] The compounds provided herein can be prepared from readily available starting materials using the following general methods and procedures. It will be appreciated that where typical or preferred process conditions (i.e., reaction temperatures, times, mole ratios of reactants, solvents, pressures, etc.) are given, other process conditions can also be used unless otherwise stated. Optimum reaction conditions may vary with the particular reactants or solvent used, but such conditions can be determined by one skilled in the art by routine optimization. [0958] Additionally, as will be apparent to those skilled in the art, conventional protecting groups may be necessary to prevent certain functional groups from undergoing undesired reactions. The choice of a suitable protecting group for a particular functional group as well as suitable conditions for protection and deprotection are well known in the art. For example, numerous protecting groups, and their introduction and removal, are described in T. W. Greene and P. G. M. Wuts, Protecting Groups in Organic Synthesis, Second Edition, Wiley, New York, 1991, and references cited therein. [0959] The compounds provided herein may be isolated and purified by known standard procedures. Such procedures include (but are not limited to) recrystallization, column chromatography, HPLC, or supercritical fluid chromatography (SFC). The following schemes are presented with details as to the preparation of representative pyrazoles that have been listed herein. The compounds provided herein may be prepared from known or commercially available starting materials and reagents by one skilled in the art of organic synthesis. General synthesis of compounds disclosed herein Compounds disclosed herein can be prepared according to the General Scheme. General Scheme
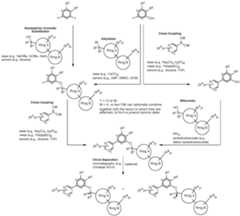
[0960] The synthesis of compounds of Formula (I) are described in the following general schemes, wherein X
1, X
2, Ring A, Ring B, R
c, R
c’, R
1, R
A, R
B, R
C, R
D, R
E, X, n, m and p are as defined herein, and each Y is independently a halogen. [0961] A general route to compounds of Formula (I), wherein R
c and R
c’ are each independently H or methyl, or R
c and R
c’ combine together with the atom to which they are attached to form a carbocyclyl, from a halo-substituted 6-membered nitrogen-containing heteroaryl (e.g., pyridine or pyrimidine) is illustrated in Scheme 1. Halo-substituted 6- membered nitrogen-containing heteroaryls are generally commercially available or synthesised according to literature procedures using general methods well known in the art of synthetic organic chemistry. Scheme 1
[0962] Reaction of the nitrogen-containing heteroaryl (1B) bearing two halide substituents with the desired alcohol building block (1A) in the presence of an alkoxide or hydride base (e.g., potassium tert-butoxide, sodium tert-butoxide, sodium hydride) at about 0 °C or room temperature in a nonpolar aprotic solvent (e.g., toluene) provides the substituted heteroaryl 1C. The left hand side aryl is introduced, for instance, by a palladium catalyzed reaction (e.g., a Suzuki reaction), with a palladium catalyst (e.g., Pd(dppf)Cl
2) and suitable boronic acid or ester derivative in the presence of a base such as a carbonate or a phosphate (e.g., sodium carbonate or potassium phosphate) in an aqueous or nonaqueous polar solvent (e.g., dioxane, THF) at about 40-120 °C to afford the compound of Formula (I). The heating in the palladium catalyst reaction is effected either by thermal heating or by microwave irradiation. Boronic acids and esters (1D) are obtained e.g., from the corresponding bromide by treatment with a base such as BuLi or similar followed by reaction with triisopropylborate or the like. Alcohol building blocks (1A) for use as shown in Scheme 1 are prepared from commercially available starting materials according to literature procedures or as described in the Chemistry Examples & Intermediates part herein below. [0963] In some compounds disclosed herein, wherein R
c and R
c’ are different (e.g., one is H and the other is methyl) and thus form a chiral center, chiral resolution is accomplished according to Scheme 2. Scheme 2
[0964] Enantiomers of a chiral center-containing compound of formula (I) are resolved by chiral chromatography/chiral HPLC (e.g., using a Chiralpak AD-H column) to afford enantiomers of formula (I) I-1 and I-2. Exemplary eluents include, but are not limited to, hexane, IPA, MeOH, MeCN, and H
2O, and mixtures thereof. [0965] In an alternative approach to compounds of Formula (I), wherein R
c and R
c’ are each independently H, the nitrogen-containing heteroaryl bears an OH substituent and a halide substituent. The heteroaryl is alkylated via reaction with a desired bromide building block, followed by reaction of an appropriate boronic acid or ester to afford compounds of formula (I). This process is shown in Scheme 3. Scheme 3
[0966] The alcohol and halide-bearing heteroaryl (2A) is treated with an appropriate bromide building block (2B) and a base, such as a carbonate base (e.g., cesium carbonate) at room temperature in a polar aprotic solvent (e.g. DMF, DMSO, DCM) to afford the substituted heteroaryl 1C. Left hand side aryl is introduced, for instance by a palladium catalyzed reaction (e.g., a Suzuki reaction), with a palladium catalyst (e.g., Pd(dppf)Cl
2) and a suitable boronic acid or ester derivative (1D) in the presence of base such as a carbonate or a phosphate (e.g., Na
2CO
3 or potassium phosphate) in an aqueous or nonaqueous polar solvent (e.g., dioxane, THF) at about 40-120 °C to afford compounds of Formula (I). [0967] In another approach to compounds of Formula (I), wherein R
c and R
c’ are each independently H, a nitrogen-containing heteroaryl bearing an alcohol and a halide substituent is first functionalized with Left hand side aryl. An appropriate alcohol building block bearing Ring A is then introduced via a Mitsunobu reaction according to Scheme 4. Scheme 4
[0968] The alcohol and halide-bearing heteroaryl (2A) is treated with an appropriate boronic acid or ester (1D), palladium catalyst (e.g., Pd(dppf)Cl
2), and a base such as a carbonate or a phosphate (e.g., Na
2CO
3 or potassium phosphate) at about 40-120 °C in an aqueous or nonaqueous polar solvent (e.g., dioxane, THF) to afford alcohol-bearing functionalized heteroaryl 4A. The heteroaryl 4A is then treated with the appropriate alcohol building block (1A), triphenylphosphine, and an azidodicarboxylate (e.g., diethyl azidodicarboxylate) at about 0 °C to room temperature in an appropriate solvent (e.g., THF, DCM) to afford compounds of Formula (I). [0969] In another approach to compounds of Formula (I), wherein R
c and R
c’ are each independently H, a nitrogen-containing heteroaryl bearing an thiomethyl ether and a halide substituent is first functionalized with Left hand side aryl. An appropriate alcohol building block bearing Ring A is then introduced via a nucleophilic substitution reaction under basic conditions according to Scheme 5. Scheme 5
Y = Cl or Br M = H, or two OM can combine, together
Scheme 5 [0970] The thiomethyl ether and halide-bearing heteroaryl (5A) is treated with an appropriate boronic acid or ester (1D), palladium catalyst (e.g., Pd(dppf)Cl
2), and a base such as a carbonate or a phosphate (e.g., Na
2CO
3 or potassium phosphate) at about 80-120 °C in an aqueous or nonaqueous polar solvent (e.g., dioxane, THF) to afford thiomethyl ether-bearing functionalized heteroaryl 5B. The heteroaryl 5B is then treated with an oxidant (e.g., mCPBA) in a nonaqueous apolar solvent (e.g., DCM) to afford the methylsulfone heteroaryl 5C. Heteroaryl 5C is then treated with an appropriate desired alcohol building block (1A) in the presence of an alkoxide or hydride base (e.g., potassium tert-butoxide, sodium tert- butoxide, sodium hydride) at about 0-100 °C in an aprotic solvent (e.g., DMF, THF, toluene) to afford compounds of Formula (I). Detailed description of the embodiments [0971] Various embodiments of the compounds and intermediates disclosed herein are illustrated by the following examples. The Examples are intended to further illustrate the invention and by no means limit the scope of the invention. Chemistry Examples & Intermediates [0972] As is well known to a person skilled in the art, reactions are performed in an inert atmosphere (including but not limited to nitrogen and argon) where necessary to protect reaction components from air or moisture. Temperatures are given in degrees Celsius (°C). Solution percentages and ratios express a volume to volume relationship, unless stated otherwise. The reactants used in the examples below may be obtained from commercial sources, or they may be prepared from commercially available starting materials as described herein or by methods known in the art. [0973] The compounds disclosed herein including intermediates are prepared as described in the Examples and in the general schemes herein. It will be apparent to a skilled person that analogous synthetic routes may be used, with appropriate modifications, to prepare the compounds disclosed herein as described herein. The progress of the reactions described herein were followed as appropriate by e.g., LC, GC or TLC, and as the skilled person will readily realize, reaction times and temperatures may be adjusted accordingly. [0974] The compound names were generated by ChemDraw Ultra software, Perkin Elmer, version 21.0.0. Intermediate 1

Step a) 4-(5-(trifluoromethyl)-1H-imidazol-2-yl)benzonitrile (I-1a) [0975] A mixture of sodium acetate (3.7 g, 44.9 mmol) and 3,3-dibromo-1,1,1- trifluoropropan-2-one (12 g, 44.03 mmol) in water (12 mL) was heated at 100 °C for 45 min, then was cooled to rt. The mixture was added to a solution of 4-formylbenzonitrile (5.8 g, 44.23 mmol) in MeOH (55 mL) followed by addition of 35% aq. NH
4OH (42 mL). The resulting reaction mixture was stirred at rt for 45 min, heated at 100 °C for 1 h, then concentrated. Water (50 mL) was added to the residue and the precipitated solid was filtered and dried, which gave the title compound (8 g) as a solid. MS (ES+) m/z 236.30 [M-H]-. The compound was taken to the next step without further purification. Step b) 4-(1-methyl-4-(trifluoromethyl)-1H-imidazol-2-yl)benzonitrile (I-1b) [0976] NaH (60%, 4.34 g, 108.5 mmol) was added at 0 °C to a solution of compound I-1a (31 g, 108.5 mmol) in THF (320 ml) and stirred at 0 °C for 1 h. CH
3I (6.8 mL, 108.5 mmol) was added at 0 °C and the mixture was stirred for 16 h at rt. Ice cold water (400 mL) was added and the mixture was extracted with EtOAc (2 x 250 mL). The combined organic layers were washed with brine, dried (Na
2SO
4), filtered and concentrated. The crude compound was purified by column chromatography on silica gel and eluted with 10-20% EtOAc in PE, which gave the title compound (12 g, 42%) as a solid. MS (ES+) 252.09 [M+H]
+. Intermediate 2
[0977] A solution of sodium hydroxide (7.01 g, 175 mmol) in water (100 mL) was added to a mixture of 4-(1-methyl-4-(trifluoromethyl)-1H-imidazol-2-yl)benzonitrile (11.0 g, 43.8 mmol) in ethanol (100 mL). The resulting mixture was stirred at 75 °C for 64 hr. The reaction mixture was cooled to room temperature and concentrated in vacuo. The residue was diluted with water (100 mL) and acidified with aqueous NaHSO
4 solution to pH < 3. The resulting precipitate was filtered, dried in air, then re-dissolved in MTBE (500 mL), dried over anhydrous Na
2SO
4 and concentrated in vacuo to afford 4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]benzoic acid (7.90 g, 29.2 mmol, 66.8% yield) as a yellow solid which was used in the next steps without further purification.
1H NMR (400 MHz, DMSO-d
6) δ 3.83 (s, 3H), 7.87 (d, 2H), 7.99 (s 1H), 8.05 (d, 2H), 13.13 (br, 1H). MS (ESI): [M+H]
+ m/z: calcd 271.07; found 271.0 Step b) [4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methanol [0978] To a suspension of lithium aluminum hydride (3.09 g, 91.04 mmol) in THF (150 mL) a solution of 4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]benzoic acid (12.3 g, 45.52 mmol) in THF (50 mL) was added dropwise at 0 °C. The resulting mixture was stirred at room temperature for 18 hr. The reaction mixture was cooled to 0 °C and quenched by dropwise addition of water (10 mL). The obtained mixture was filtered and the filtrate was evaporated to afford [4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methanol (10.5 g, 40.98 mmol, 90% yield) which was used in the next step without purification.
1H NMR (400 MHz, DMSO-d
6) δ 3.72 (s, 3H), 4.52 (d, 2H), 5.34 (t, 1H), 7.40 (d, 2H), 7.61 (d, 2H), 7.86 (s, 1H).
1H NMR (400 MHz, CDCl
3) δ 3.42 (br, 1 H), 3.69 (s, 3H), 4.66 (s, 2H), 7.26 – 7.48 (m, 5H). MS (ESI): [M+H]
+ m/z: calcd 257.1; found 257.2 Step c) 2-chloro-5-methyl-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidine and 2-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]-5-methyl-4-chloropyrimidine [0979] Potassium tert-butoxide (1.05 g, 9.37 mmol) was added portion wise to a stirred solution of [4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methanol (1.60 g, 6.24 mmol) in toluene (60 mL) in an inert atmosphere. The reaction mixture was stirred at room temperature for 30 min.2,4-Dichloro-5-methylpyrimidine (1.53 g, 9.37 mmol) was added. The resulting mixture was stirred at room temperature for 15 hr. The reaction mixture was poured into cold water (100 mL) and extracted with MTBE (3 × 40 mL). Combined organic layers were dried over anhydrous Na
2SO
4, filtered through a pad of SiO
2 and concentrated in vacuo. The residue was subjected to flash-column chromatography (SiO
2, hexanes - EtOAc) to afford 2-chloro-5-methyl-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidine (800 mg, 2.09 mmol, 33.47% yield) and 2-[[4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]-5-methyl-4-chloropyrimidine (180 mg, 470 μmol, 7.5% yield) as white solids. [0980] 2-chloro-5-methyl-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidine:
1H NMR (500 MHz, CDCl
3) δ 2.15 (s, 3H), 3.78 (s, 3H), 5.50 (s, 2H), 7.27 (s, 1H), 7.57 (d, 2H), 7.68 - 7.71 (m, 2H), 8.17 (s, 1H). MS (ESI): [M+H]
+ m/z: calcd 383.1; found 383.0. [0981] 2-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]-5-methyl-4- chloropyrimidine:
1H NMR (500 MHz, CDCl
3) δ 2.25 (s, 3H), 3.76 (s, 3H), 5.44 (s, 2H), 7.27 (s, 1H), 7.51 – 7.72 (m, 4H), 8.27 (s, 1H). MS (ESI): [M+H]
+ m/z: calcd 383.1; found 383.0. The structures were elucidated by HMBC and NOESY spectra. Intermediate 3
Step a) methyl 4-[4-(trifluoromethyl)-1H-imidazol-2-yl]benzoate [0982] 3,3-dibromo-1,1,1-trifluoro-propan-2-one (54.3 g, 201 mmol, 27.40 mL) was added to a solution of sodium acetate (34.5 g, 420 mmol) in water (200 mL). The mixture was heated at 95 °C for 1 hr. The resulting mixture was cooled to room temperature and poured into a solution of methyl 4-formylbenzoate (30.0 g, 182.75 mmol) in mixture of MeOH (1000 mL) and concentrated NH
4OH (100 mL). The reaction mixture was stirred at room temperature for 12 hr. The mixture was concentrated under reduced pressure. The residue was partitioned between EtOAc (300 ml) and water (200 ml). The organic layer was separated, dried over Na
2SO
4, and concentrated under reduced pressure. The residue was recrystallized from MTBE to afford methyl 4-[4-(trifluoromethyl)-1H-imidazol-2-yl]benzoate (38.0 g, 140 mmol, 77% yield).
1H NMR (500 MHz, DMSO-d
6) δ 3.85 (s, 3H), 7.97 (s, 1H), 8.05 (d, 2H), 8.09 (d, 2H). MS (ESI): [M+H]+ m/z: calcd 271.07; found 271.0 Step b) methyl 4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]benzoate [0983] To a stirred solution of methyl 4-[4-(trifluoromethyl)-1H-imidazol-2-yl]benzoate (38.0 g, 141 mmol) in DMF (150 mL) NaH (3.56 g, 154.70 mmol, 60% dispersion in mineral oil) was added in a few portions at 0-5°C. The resulting mixture was stirred at room temperature for 1hr. To the obtained mixture iodomethane (22.0 g, 155 mmol, 9.63 mL) was added dropwise. The reaction mixture was stirred at room temperature for 6 hr. The reaction mixture was quenched with water (200 mL) and extracted with EtOAc (2×200 mL). The combined organic layers were washed with brine (20 mL), dried over anhydrous Na
2SO
4 and evaporated under reduced pressure. The residue was recrystallized from MTBE to afford methyl 4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]benzoate (24.0 g, 84.4 mmol, 60% yield).
1H NMR (500 MHz, DMSO-d
6) δ 3.83 (s, 3H), 3.87 (s, 3H), 7.90 (d, 2H), 7.99 (s, 1H), 8.05 (d, 2H). MS (ESI): [M+H]+ m/z: calcd 285.09; found 285.2 Intermediate 4
2-chloro-5-methyl-pyrimidin-4-ol [0984] To a solution of 2,4-dichloro-5-methyl-pyrimidine (5 g, 30.67 mmol, 3.60 mL) in THF (9.8 mL) were added 1 M aqueous sodium hydroxide (1.8 g, 45.00 mmol, 45 mL) at 25 °C. The resulting mixture was stirred at 25 °C for 24 h then washed with diethyl ether. Under ice cooling, 2 M HCl aqueous solution was added to the mixture to adjust the pH to 3. The precipitated solid was collected by filtration, washed by cold water. The obtained solid was dried to give 2-chloro-5-methyl-pyrimidin-4-ol (1.8 g, 12.45 mmol, 41% yield) as a white solid.
1H NMR (400 MHz, Chloroform-d) δ 12.70 (s, 1H), 7.26 (s, 1H), 6.70 (s, 3H). Intermediate 5
Commercially available compound (e.g., PharmaBlock, BLDpharm). Intermediate 6
Step 1: The synthesis of 2-chloro-5-methoxy-4-methylsulfanyl-pyrimidine [0985] An aqueous solution of sodium thiomethoxide (58.7 mmol, 19.6 g of 21% aqueous solution) was added dropwise to a solution of 2,4-dichloro-5-methoxy-pyrimidine (10.0 g, 55.9 mmol) in THF (100 mL) at -10 °C. The reaction mixture was stirred at room temperature for 16 hr. The obtained mixture was concentrated in vacuo. The residue was diluted with water (200 mL) and extracted with MTBE (4 × 100 mL). The combined organic layers were dried over anhydrous sodium sulfate and concentrated in vacuo to afford 2-chloro-5-methoxy-4- methylsulfanyl-pyrimidine (9.00 g, 47.2 mmol, 84.5% yield) as a white solid which was used in the next steps without further purification. 1H NMR (400 MHz, CDCl3) δ 2.53 (s, 3H), 3.93 (s, 3H), 7.77 (s, 1H). LCMS(ESI): [M+H]
+ m/z: calcd 191.00; found 191.2. Step 2: The synthesis of 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-5-methoxy-4- methylsulfanyl-pyrimidine [0986] A mixture of water (4.00 mL) and dioxane (40.0 mL) was evacuated and then backfilled with argon. 2-chloro-5-methoxy-4-methylsulfanyl-pyrimidine (1.78 g, 9.34 mmol), (4- cyclopropyl-6-methoxy-pyrimidin-5-yl)boronic acid I-5 (1.99 g, 10.3 mmol), RuPhos Pd G4 (794 mg, 934 μmol) and potassium phosphate tribasic (5.95 g, 28.0 mmol) were added in an inert atmosphere at room temperature. The reaction mixture was stirred at 85 °C for 16 hr. The obtained mixture was concentrated in vacuo. The residue was diluted with H
2O (20.0 mL) and extracted with EtOAc (2×50.0 mL). The combined organic layers were dried over anhydrous sodium sulfate and concentrated in vacuo. The residue was subjected to flash-column chromatography (SiO
2; EtOAc – Hexane, 1:1) to afford 2-(4-cyclopropyl-6-methoxy- pyrimidin-5-yl)-5-methoxy-4-methylsulfanyl-pyrimidine (1.60 g, 5.26 mmol, 56.3% yield) as a brown solid. 1H NMR (400 MHz, DMSO-d
6) δ 0.86 – 0.92 (m, 2H), 1.01 – 1.06 (m, 2H), 1.67 – 1.75 (m, 1H), 2.43 (s, 3H), 3.84 (s, 3H), 4.00 (s, 3H), 8.35 (s, 1H), 8.64 (s, 1H). LCMS(ESI): [M+H]
+ m/z: calcd 305.10; found 305.2. Step 3: The synthesis of 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-5-methoxy-4- methylsulfonyl-pyrimidine I-6 [0987] mCPBA (2.54 g, 11.0 mmol, 75% purity) was added to a stirred solution of 2-(4- cyclopropyl-6-methoxy-pyrimidin-5-yl)-5-methoxy-4-methylsulfanyl-pyrimidine (1.60 g, 5.26 mmol) in Dichloromethane (30.0 mL). The resulting mixture was stirred at room temperature for 24 hr. The obtained mixture was washed with saturated aqueous NaHCO
3, dried over anhydrous sodium sulfate and concentrated in vacuo to dryness. The residue was subjected to flash-column chromatography (SiO
2; EtOAc – Hexane, 7:3) to afford 2-(4- cyclopropyl-6-methoxy-pyrimidin-5-yl)-5-methoxy-4-methylsulfonyl-pyrimidine I-6 (1.00 g, 2.97 mmol, 56.6% yield) as a light-yellow solid. 1H NMR (500 MHz, CDC
l3) δ 0.90 – 0.95 (m, 2H), 1.19 – 1.24 (m, 2H), 1.60 – 1.69 (m, 1H), 3.35 (s, 3H), 3.91 (s, 3H), 4.17 (s, 3H), 8.63 (s, 1H), 8.82 (s, 1H). LCMS(ESI): [M+H]
+ m/z: calcd 337.09; found 337.2. Intermediate 7
Intermediate 7 was prepared in a manner similar to intermediate 6 and intermediate 8 from commercially available starting materials Intermediate 8
4-cyclopropyl-6-methoxy-5-(4-methylsulfanylpyrimidin-2-yl)pyrimidine [0988] 2-Chloro-4-methylsulfanyl-pyrimidine (1.50 g, 9.34 mmol), (4-cyclopropyl-6- methoxy-pyrimidin-5-yl)boronic acid I-5 (1.99 g, 10.3 mmol), RuPhos Pd G4 (794 mg, 934 μmol) and potassium phosphate tribasic (5.95 g, 28.0 mmol) were mixed in degassed water (4 mL) and dioxane (40 mL) under argon atmosphere at room temperature. The reaction mixture was stirred at 85 °C for 16 hr. The mixture was cooled to room temperature and concentrated under reduced pressure. The residue was diluted with water (20 mL) and extracted with EtOAc (2 × 50 mL). The combined organic layers were dried over anhydrous Na
2SO
4 and concentrated under reduced pressure. The residue was subjected to flash column chromatography (SiO
2, mobile phase: EtOAc - Hexane, 1:1) to afford 4-cyclopropyl-6- methoxy-5-(4-methylsulfanylpyrimidin-2-yl)pyrimidine (2.20 g, 8.02 mmol, 85.9% yield) as a yellow solid.
1H NMR (500 MHz, DMSO-d
6) δ 0.90 – 0.95 (m, 2H), 1.02 – 1.10 (m, 2H), 1.63 – 1.71 (m, 1H), 2.53 (s, 3H), 3.86 (s, 3H), 7.47 (d, 1H), 8.62 (d, 1H), 8.68 (s, 1H). MS (ESI): [M+H]
+ m/z: calcd 275.09; found 275.0 4-cyclopropyl-6-methoxy-5-(4-methylsulfonylpyrimidin-2-yl)pyrimidine [0989] mCPBA (3.69 g, 16.0 mmol, 75% purity) was added to the stirred solution of 4- cyclopropyl-6-methoxy-5-(4-methylsulfanylpyrimidin-2-yl)pyrimidine (2.00 g, 7.29 mmol) in DCM (50 mL). The resulting mixture was stirred at room temperature for 24 hr. The mixture was washed with saturated aqueous NaHCO
3 solution (2 × 30) and dried over anhydrous Na
2SO
4. The obtained solution was concentrated under reduced pressure. The residue was subjected to flash column chromatography (SiO
2, mobile phase: EtOAc - Hexane, 7:3) to afford 4-cyclopropyl-6-methoxy-5-(4-methylsulfonylpyrimidin-2- yl)pyrimidine I-8 (1.30 g, 4.24 mmol, 58.1% yield) as a light-yellow solid.
1H NMR (500 MHz, DMSO-d
6) δ 0.88 – 0.95 (m, 2H), 1.04 – 1.12 (m, 2H), 1.74 – 1.80 (m, 1H), 3.39 (s, 3H), 3.88 (s, 3H), 8.14 (d, 1H), 8.74 (s, 1H), 9.42 (d, 1H). MS (ESI): [M+H]
+ m/z: calcd 307.08; found 307.2. Intermediate 9
The synthesis of 5-bromo-4-cyclopropyl-6-(trideuteriomethoxy)pyrimidine [0990] Sodium (325 mg, 14.1 mmol) was added portion wise to vigorously stirred trideuterio(deuteriooxy)methane (23.2 g, 642 mmol, 26.0 mL). The resulting mixture was stirred at room temperature for 1 hr, then cooled to 0°C. 5-bromo-4-chloro-6-cyclopropyl- pyrimidine (3.00 g, 12.9 mmol) was added to the mixture. The reaction mixture was stirred at room temperature for 16 hr. The reaction mixture was concentrated under reduced pressure. The residue was diluted with EtOAc (150 mL) and washed with water (30 mL). The organic layer was separated, washed with brine (20 mL), dried over anhydrous sodium sulfate and concentrated under reduced pressure to afford 5-bromo-4-cyclopropyl-6- (trideuteriomethoxy)pyrimidine (2.80 g, 12.1 mmol, 93.9% yield) as a yellow solid which was used in the next steps without further purification. [0991]
1H NMR (500 MHz, CDCl
3) δ 1.05 – 1.10 (m, 2H), 1.14 – 1.20 (m, 2H), 2.47 – 2.55 (m, 1H), 8.42 (s, 1H). LCMS(ESI): [M+H]
+ m/z: calcd 232.02; found 232.2 The synthesis of 4-cyclopropyl-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-6- (trideuteriomethoxy)pyrimidine [0992] 5-bromo-4-cyclopropyl-6-(trideuteriomethoxy)pyrimidine (2.80 g, 12.1 mmol) and 2- isopropoxy-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (3.14 g, 16.9 mmol, 3.45 mL) were mixed in THF (100 mL) under argon atmosphere. The resulting solution was cooled to -80°C. n- Butyllithium (18.1 mmol, 7.24 mL, 2.5 M in hexane) was added dropwise to the solution at - 80°C. The reaction mixture was stirred at -80°C for 3 hr, then at ambient temperature for 16 hr. The reaction mixture was quenched with a saturated aqueous solution of NH
4Cl (20 mL) and extracted with EtOAc (30 mL). The organic layer was separated, washed with water (20 mL) and brine (30 mL), dried over anhydrous sodium sulfate and concentrated under reduced pressure. The residue was subjected to flash column chromatography (SiO
2, gradient hexane - MTBE) to afford 4-cyclopropyl-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-6- (trideuteriomethoxy)pyrimidine (800 mg, 2.87 mmol, 23.8% yield) as a white solid.
1H NMR (500 MHz, CDCl
3) δ 0.93 – 1.00 (m, 2H), 1.13 – 1.19 (m, 2H), 1.38 (s, 12H), 2.04 – 2.10 (m, 1H), 8.55 (s, 1H). LCMS(ESI): [M+H]
+ m/z: calcd 280.19; found 280.2 Intermediate 10
6-cyclopropylpyrimidin-4-ol [0993] A mixture of 6-chloropyrimidin-4-ol (10 g, 76.6 mmol), cyclopropylboronic acid (32.9 g, 0.383 mol), Pd(dppf)Cl
2 (5.61 g, 7.66 mmol) and K
3PO
4 (32.5 g, 0.153 mol) in dioxane (200 mL) and H
2O (20 mL) was stirred at 110 °C for 12 hrs. The mixture was filtered through the celite. The filtrate was concentrated under reduced pressure. The residue was purified by flash chromatography (ISCO®; 120 g AgelaFlash® Silica Flash Column, DCM/MeOH with MeOH from 0-10%, 80 mL/min, 254nm) to afford a brown solid. The solid was triturated with EtOAc (20 mL). The mixture was filtered. The filter cake was dried under reduced pressure to give 6-cyclopropylpyrimidin-4-ol (1.3 g, 12.5% yield) as brown solid. The filtrate was concentrated under reduced pressure to give 6- cyclopropylpyrimidin-4-ol (1.8 g, 13.22 mmol, 17.3% yield) as solid. MS (ESI) [M+H]
+ m/z: calcd 137.1; found 137.1. 4-cyclopropyl-6-(difluoromethoxy)pyrimidine [0994] To a solution of 6-cyclopropylpyrimidin-4-ol (1.3 g, 9.55 mmol) in MeCN (13 mL) was added a solution of KOH (10.7 g, 0.191 mol) in H
2O (13 mL) dropwise at 0 °C. The mixture was stirred at 20 °C for 30 min. Then the mixture was cooled to -10 °C, and 1- [[bromo(difluoro)methyl]-ethoxy-phosphoryl]oxyethane (5.1 g, 19.1 mmol) was added dropwise. The mixture was stirred at 20 °C for 4 hrs. The resulting mixture was extracted with EtOAc (100 mL x 3). The combined organic layer was washed with brine (100 mL x 3), dried over anhydrous Na
2SO
4, filtered and concentrated under reduced pressure. The residue was purified by flash chromatography (ISCO®; 20 g AgelaFlash® Silica Flash Column, PE/EtOAc with EtOAc from 0-9%, 35 mL/min, 254nm) to afford 4-cyclopropyl-6- (difluoromethoxy)pyrimidine (890 mg, 50.1% yield) as colorless oil.
1H NMR (400 MHz, chloroform-d) δ ppm 8.62 (s, 1H), 7.68 (s, 0.25H), 7.50 (s, 0.5H), 7.32 (s, 0.25H), 6.74 (d, J = 1.1 Hz, 1H), 1.94 - 2.02 (m, 1H), 1.14 - 1.20 (m, 2H), 1.08 - 1.14 (m, 2H);
19F NMR (376 MHz, chloroform-d) δ ppm -89.80; MS (ESI) [M+H]
+ m/z: calcd 187.1; found 187.0. 5-bromo-4-cyclopropyl-6-(difluoromethoxy)pyrimidine To a solution of 4-cyclopropyl-6-(difluoromethoxy)pyrimidine (1.15 g, 6.18 mmol) in DMF (10 mL) was added Br
2 (1.6 mL, 31.2 mmol) dropwise at 0 °C. The mixture was stirred at 50 °C for 12 hrs. The resulting mixture was quenched by addition of water (100 mL) and extracted with EtOAc (100 mL x 3). The combined organic layer was washed with brine (100 mL x 3), dried over anhydrous Na
2SO
4, filtered and concentrated under reduced pressure. The residue was purified by flash chromatography (ISCO®; 20 g AgelaFlash® Silica Flash Column, PE/EtOAc with EtOAc from 0-3%, 35 mL/min, 254nm) to afford 5-bromo-4-cyclopropyl-6- (difluoromethoxy)pyrimidine (750 mg, 45.8% yield) as yellow solid. MS (ESI) [M+H]
+ m/z: calcd 267.0; found 266.9. 4-cyclopropyl-6-(difluoromethoxy)-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2- yl)pyrimidine [0995] A mixture of 5-bromo-4-cyclopropyl-6-(difluoromethoxy)pyrimidine (750 mg, 2.83 mmol), B(Pin)
2 (1.44 g, 5.66 mmol), Pd(dppf)Cl
2 (207 mg, 0.283 mmol) and KOAc (833 mg, 8.49 mmol) in dioxane (15 mL) was stirred at 90 °C for 12 hrs. The resulting mixture was filtered and concentrated under reduced pressure. The residue was purified by flash chromatography (ISCO®; 40 g AgelaFlash® Silica Flash Column, PE/EtOAc with EtOAc from 0-10%, 40 mL/min, 254nm) to afford 4-cyclopropyl-6-(difluoromethoxy)-5-(4,4,5,5- tetramethyl-1,3,2-dioxaborolan-2-yl)pyrimidine I-10 (450 mg, 51% yield) as yellow solid. MS (ESI) [M+H]
+ m/z: calcd 313.1; found 313.1. Intermediate 11
[0996] To a solution of 4-benzyloxy-2-chloro-pyrimidine (1.10 g, 5 mmol) and (4- cyclopropyl-6-methoxy-pyrimidin-5-yl) boronic acid (1.16 g, 6.00 mmol) in 1,4-dioxane (15 mL) and water (3 mL) were added potassium phosphate (2.12 g, 10.00 mmol) and 1,1'- Bis(diphenylphosphino)ferrocene-palladium(II)dichloride (408.32 mg, 500.00 μmol) at room temperature, the mixture stirred at 100 °C for 4 hours. Major target product was detected by LCMS. After cooled to room temperature, the reaction mixture was quenched by H
2O (20 mL), extracted with ethyl acetate (3 × 10 mL), the combined organic phase was washed with brine (3 × 10 mL), dried over anhydrous sodium sulfate. After filtration, the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography on silica gel, eluted with 45% ethyl acetate in petroleum ether to give 5-(4-benzyloxypyrimidin- 2-yl)-4-cyclopropyl-6-methoxy-pyrimidine (1.04 g, 3.12 mmol, 62% yield) as a brown solid. MS: m/z = 335.25 [M + H]
+.
1H NMR (400 MHz, Chloroform-d) δ 8.67 (s, 1H), 8.62 (d, J = 6.0 Hz, 1H), 7.48 - 7.44 (m, 2H), 7.42 - 7.34 (m, 3H), 6.81 (d, J = 5.6 Hz, 1H), 5.48 (s, 2H), 3.96 (s, 3H) 1.75 - 1.71 (m, 1H), 1.29 - 1.20 (m, 2H), 0.93 - 0.89 (m, 2H). 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)pyrimidin-4-ol I-11 [0997] To a solution of 5-(4-benzyloxypyrimidin-2-yl)-4-cyclopropyl-6-methoxy-pyrimidine (1.00 g, 3 mmol) in isopropyl alcohol (30 mL) was added palladium on carbon (0.25 g, 10%) at room temperature under nitrogen atmosphere. The mixture was evacuated and flushed three times with nitrogen, followed by flushing with hydrogen. The resulted mixture was stirred at 25 °C for 2 hours under an atmosphere of hydrogen. Major target product was detected by LCMS. Then the mixture was filtered. The filtrate was concentrated under reduced pressure to give 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl) pyrimidin-4-ol (700 mg, 2.87 mmol, 95% yield) as an off-white solid. MS: m/z = 245.15 [M + H]
+.
1H NMR (400 MHz, DMSO-d
6) δ 12.87 (s, 1H), 8.71 (s, 1H), 8.05 (d, J = 6.8 Hz, 1H), 6.43 - 6.25 (m, 1H), 3.92 (s, 3H), 1.90 - 1.86 (m, 1H), 1.10 - 1.00 (m, 4H). Intermediate 12
[0998] Potassium tert-butoxide (1.22 g, 10.9 mmol) was added to a solution of [4-[1-methyl- 4-(trifluoromethyl)imidazol-2-yl]phenyl]methanol I-2b (2.80 g, 10.9 mmol) in THF (100 mL). The resulting mixture was stirred at room temperature for 1 hr.2,4-dichloro-5-iodo- pyrimidine (3.00 g, 10.9 mmol) was added to the reaction mixture in one portion. The resulting mixture was stirred at room temperature for 24 hr. The reaction mixture was diluted water (100 mL) and extracted with EtOAc (2×100 mL). The combined organic layers were washed with brine (50 mL) and concentrated under reduced pressure. The residue was subjected to flash-column chromatography (SiO
2, gradient acetonitrile-chloroform) to afford 2-chloro-5-iodo-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidine I-12 (1.70 g, 3.44 mmol, 31.5% yield) as a white solid.
1H NMR (500 MHz, CDCl
3) δ 3.79 (s, 3H), 5.55 (s, 2H), 7.32 (s, 1H), 7.59 (d, 2H) 7.67 (d, 2H), 8.62 (s, 1H). LCMS(ESI): [M+H]
+ m/z: calcd 494.97; found 495.0. Example 1 (Compound 1)

2,5-dichloro-4-methyl-6-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidine [0999] To a stirred solution of [4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methanol I-2b (0.250 g, 976 μmol) in dioxane (5 mL) Potassium tert-butoxide (109 mg, 976 μmol) was added in a few portions at room temperature. The mixture was stirred for 1 h. Then 2,4,5-trichloro-6-methyl-pyrimidine (202 mg, 1.02 mmol) was added to the mixture and the resulting mixture was stirred at room temperature for 12 hr. The reaction mixture was quenched with water (10 mL). The obtained mixture was extracted with EtOAc (2 × 10 mL). The combined organic layers were washed with brine (10 mL) and concentrated under reduced pressure to afford 2,5-dichloro-4-methyl-6-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidine (300 mg, crude) as a yellow solid which was used in the next steps without further purification. MS (ESI): [M+H]
+ m/z: calcd 417.07; found 417.2. 5-chloro-2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-methyl-6-[[4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidine [1000] 2,5-dichloro-4-methyl-6-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidine (0.300 g, crude), (4-cyclopropyl-6-methoxy-pyrimidin-5- yl)boronic acid I-5 (181 mg, 935 μmol), XPhos Pd G3 (30.4 mg, 36.0 μmol), and Potassium phosphate tribasic (305 mg, 1.44 mmol) were mixed in a degassed mixture of water (3 mL) and dioxane (20 mL). The reaction vessel was evacuated and backfilled with argon. The reaction mixture was stirred at 80 °C for 18 hrs. The mixture was cooled to room temperature, diluted with water (5 mL) and extracted with EtOAc (2 × 10 mL). SiliaMetS® Dimercaptotriazine (20 mg) was added to the resulting organic layer. The resulting mixture was stirred for 30 min and filtered. The resulting filtrate concentrated under reduced pressure. The residue was subjected to HPLC (0.5-6.5 min, 52% water – ACN, flow: 30 mL/min, column: SunFire C18100 ×19 mm, 5 µm then 0.5-6.5 min, 62% water – MeOH, flow: 30 mL/min, column: SunFire C18 100 ×19 mm, 5 µm) to afford 5-chloro-2-(4-cyclopropyl-6- methoxy-pyrimidin-5-yl)-4-methyl-6-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidine 1 (11.0 mg, 20.7 μmol, 2.1% yield from [4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methanol) as an off-white solid. MS (ESI): [M+H]
+ m/z: calcd 531.15; found 531.2.
1H NMR (500 MHz, DMSO-d
6) δ 0.84 – 0.88 (m, 2H), 1.01 – 1.05 (m, 2H), 1.69 – 1.74 (m, 1H), 2.56 (s, 3H), 3.77 (s, 3H), 3.84 (s, 3H), 5.56 (s, 2H), 7.58 (d, 2H), 7.72 (d, 2H), 7.93 (s, 1H), 8.66 (s, 1H).
2-chloro-4-methylsulfanyl-6-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidine [1001] Potassium tert-butoxide (1.45 g, 12.9 mmol) was added to a solution of [4-[1-methyl- 4-(trifluoromethyl)imidazol-2-yl]phenyl]methanol I-2b (3.00 g, 11.7 mmol) and 2,4-dichloro- 6-methylsulfanyl-pyrimidine (2.51 g, 12.9 mmol) in toluene (40 mL) at 0 °C. The reaction mixture was stirred at room temperature for 15 hr. The reaction mixture was poured into water (40 mL) and extracted with EtOAc (40 mL). The organic layer was separated and concentrated under reduced pressure. The residue was subjected to flash-column chromatography (SiO
2, hexane – EtOAc 7:3) to afford 2-chloro-4-methylsulfanyl-6-[[4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidine (700 mg, 1.69 mmol, 14.4% yield) and isomeric 4-chloro-6-methylsulfanyl-2-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidine (1.50 g, 3.62 mmol, 30.9% yield) as light-yellow solids. 2-chloro-4-methylsulfanyl-6-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidine:
1H NMR (600 MHz, DMSO-d
6) δ 2.53 (s, 3H), 3.80 (s, 3H), 5.47 (s, 2H), 6.96 (s, 1H), 7.60 (d, 2H), 7.76 (d, 2H), 7.95 (s, 1H). MS (ESI): [M+H]
+ m/z: calcd 415.06; found 415.2 4-chloro-6-methylsulfanyl-2-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidine (1.50 g, 3.62 mmol, 30.9% yield):
1H NMR (600 MHz, DMSO-d
6) δ 2.55 (s, 3H), 3.80 (s, 3H), 5.48 (s, 2H), 7.32 (s, 1H), 7.60 (d, 2H), 7.76 (d, 2H), 7.94 (s, 1H). MS (ESI): [M+H]
+ m/z: calcd 415.06; found 415.0 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-methylsulfanyl-6-[[4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidine [1002] 2-Сhloro-4-methylsulfanyl-6-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidine (700 mg, 1.69 mmol), (4-cyclopropyl-6-methoxy-pyrimidin-5- yl)boronic acid I-5 (655 mg, 3.37 mmol), RuPhos Pd G4 (144 mg, 169 μmol) and potassium phosphate tribasic (1.07 g, 5.06 mmol) were mixed in degassed mixture of water (1 mL) and dioxane (10 mL) under argon atmosphere at room temperature. The reaction mixture was stirred at 85 °C for 16 hr. The reaction mixture was cooled to room temperature and concentrated under reduced pressure. The residue was diluted with water (20 mL) and extracted with EtOAc (2×50 mL). The combined organic layers were separated, dried over anhydrous Na
2SO
4 and concentrated under reduced pressure. The residue was subjected to HPLC (2-10 min, 30-50% ACN; flow: 30 mL/min, column: SunFire C18, 100 × 19 mm, 5 µm) to afford 2- (4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-methylsulfanyl-6-[[4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidine 8 (392 mg, 742 µmol, 43.9% yield) as a white solid.
1H NMR (600 MHz, DMSO-d
6) δ 0.88 – 0.92 (m, 2H), 1.02 – 1.06 (m, 2H), 1.70 – 1.76 (m, 1H), 2.53 (s, 3H), 3.78 (s, 3H), 3.87 (s, 3H), 5.49 (s, 2H), 6.94 (s, 1H), 7.58 (d, 2H), 7.74 (d, 2H), 7.94 (s, 1H), 8.67 (s, 1H). MS (ESI): [M+H]
+ m/z: calcd 529.17; found 529.2 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-methylsulfonyl-6-[[4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidine [1003] mCPBA (322 mg, 1.40 mmol, 75% purity) was added to a stirred solution of 2-(4- cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-methylsulfanyl-6-[[4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidine (370 mg, 700 μmol) in DCM (8 mL). The resulting mixture was stirred at room temperature for 24 hr. The reaction mixture was washed with saturated aqueous solution of NaHCO
3. The organic layer was separated, dried over anhydrous Na
2SO
4 and concentrated under reduced pressure. The residue was subjected to flash column chromatography (SiO
2, EtOAc – Hexane, 7:3) to afford 2-(4- cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-methylsulfonyl-6-[[4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidine (370 mg, 660 μmol, 94.3% yield) as a yellow solid. MS (ESI): [M+H]
+ m/z: calcd 561.16; found 561.2 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-methoxy-6-[[4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidine [1004] K
2CO
3 (67.8 mg, 491 μmol) was added to a stirred solution of 2-(4-cyclopropyl-6- methoxy-pyrimidin-5-yl)-4-methylsulfonyl-6-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidine (110 mg, 196 μmol) in MeOH (5.0 mL) and Dioxane (5.0 mL). The resulting mixture was stirred at room temperature for 12 hr. The reaction mixture was filtered and concentrated under reduced pressure. The residue was subjected to HPLC (2-10 min, 30-70% ACN; flow: 30 mL/min, column: SunFire C18, 100×19 mm, 5 µm) to afford 2- (4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-methoxy-6-[[4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidine 49 (42.0 mg, 82.0 μmol, 41.8% yield) as a brown solid.
1H NMR (600 MHz, DMSO-d
6) δ 0.87 – 0.92 (m, 2H), 1.02 – 1.06 (m, 2H), 1.73 – 1.79 (m, 1H), 3.78 (s, 3H), 3.87 (s, 3H), 3.90 (s, 3H), 5.47 (s, 2H), 6.41 (s, 1H), 7.57 (d, 2H), 7.73 (d, 2H), 7.94 (s, 1H), 8.67 (s, 1H). MS (ESI): [M+H]
+ m/z: calcd 513.21; found 513.2
Methyl 3-fluoro-2-methoxy-4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]benzoate [1005] 2-Bromo-1-methyl-4-(trifluoromethyl)imidazole (591 mg, 2.58 mmol) and Na
2CO
3 (820 mg, 7.74 mmol) were added to a stirred solution of methyl 3-fluoro-2-methoxy-4-(4,4,5,5- tetramethyl-1,3,2-dioxaborolan-2-yl)benzoate (800 mg, 2.58 mmol) in degassed mixture of dioxane (16 mL) and water (4.0 mL) under argon atmosphere. Bis(diphenylphosphino)ferrocene]dichloropalladium(II)-DCM (105 mg, 129 μmol) was added to the mixture. The resulting mixture was stirred at 90°C for 16 hr under argon atmosphere. The reaction mixture was cooled to room temperature and concentrated under reduced pressure. The residue was dissolved in EtOAc (40 mL), then solids were filtered off. The filtrate was concentrated under reduced pressure to afford methyl 3-fluoro-2-methoxy-4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]benzoate (590 mg, crude) as a brown gum. MS (ESI): [M+H]
+ m/z: calcd 333.09; found 333.0 [3-fluoro-2-methoxy-4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methanol [1006] A solution of ethyl 3-fluoro-2-methoxy-4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]benzoate (590 mg, 1.78 mmol) in THF (10 mL) was added dropwise to a stirred suspension of LiAlH
4 (101 mg, 2.66 mmol) in THF (25 mL) at -10 °C under argon atmosphere. The reaction mixture was allowed to warm to room temperature and stirred at room temperature overnight. Water (300 µL) in THF (5.0 mL) was added dropwise to the reaction mixture at 0°C. The resulting mixture was stirred for 15 min. K
2CO
3 (3.0 g) and Na
2SO
4 (3.0 g) were added to the mixture. The resulting mixture was stirred for 20 min. The resulting mixture was filtered, the filter cake was washed with THF (2 × 15 mL). The combined filtrate was concentrated under reduced pressure to afford [3-fluoro-2-methoxy-4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methanol (330 mg, crude) as a brown gum which was used in the next step without further purification. MS (ESI): [M+H]
+ m/z: calcd 305.09; found 305.0 4'-cyclopropyl-4-((3-fluoro-2-methoxy-4-(1-methyl-4-(trifluoromethyl)-1H-imidazol-2- yl)benzyl)oxy)-5,6'-dimethoxy-2,5'-bipyrimidine [1007] NaH (45.6 mg, 1.14 mmol, 60% dispersion in mineral oil) was added to a stirred solution of [3-fluoro-2-methoxy-4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methanol (330 mg, 759 μmol) in DMF (7 mL). The resulting mixture was stirred at room temperature for 30 min. 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-5-methoxy-4- methylsulfonyl-pyrimidine I-6 (255 mg, 759 μmol) was added to the mixture. The reaction mixture was stirred at room temperature for 16 hr. The reaction mixture was diluted with saturated aqueous NH
4Cl solution (8 mL) and extracted with EtOAc (3 × 15 mL). The combined organic layers were separated, washed with brine (10 mL), dried over anhydrous Na
2SO
4 and filtered. The filtrate was concentrated under reduced pressure. The residue was subjected to HPLC (0-1-5 min, 20-20-70% water+FA (0.1% vol.) - MeOH+FA (0.1% vol.); flow: 30 mL/min, column: Chromatorex 18 SMB100-5T, 100×19 mm, 5 µm), then repurified by HPLC (0-5 min., 40-60% water+FA (0.1% vol.) - ACN+FA (0.1% vol.); flow: 30 mL/min, column: Chromatorex 18 SMB100-5T, 100×19 mm, 5 µm), then repurified by HPLC (0-1.5-5 min, 40-40-45% water - MeOH; flow: 30 mL/min, column: Kinetex PFP, 100×21 mm, 5 µm) to afford 4'-cyclopropyl-4-((3-fluoro-2-methoxy-4-(1-methyl-4-(trifluoromethyl)-1H- imidazol-2-yl)benzyl)oxy)-5,6'-dimethoxy-2,5'-bipyrimidine 15 (18.6 mg, 33.2 μmol, 4.4% yield) as a beige solid.
1H NMR (600 MHz, DMSO-d
6) δ 0.85 – 0.92 (m, 2H), 1.01 – 1.07 (m, 2H), 1.68 – 1.75 (m, 1H), 3.63 (s, 3H), 3.85 (s, 3H), 3.89 (s, 3H), 3.95 (s, 3H), 5.50 (s, 2H), 7.30 – 7.41 (m, 2H), 8.03 (s, 1H), 8.44 (s, 1H), 8.65 (s, 1H). MS (ESI): [M+H]
+ m/z: calcd 561.19; found 561.2 Example 4 (Compound 21)
4-cyclopropyl-5-[4-[[4-[1-cyclopropyl-4-(trifluoromethyl)imidazol-2-yl]-3-fluoro- phenyl]methoxy]pyrimidin-2-yl]-6-methoxy-pyrimidine Synthesis of the starting (4-(1-cyclopropyl-4-(trifluoromethyl)-1H-imidazol-2-yl)-3- fluorophenyl)methanol is described for compound 154. [1008] NaH (7.33 mg, 183 μmol, 60% dispersion in mineral oil) was added to a solution of [4-[1-cyclopropyl-4-(trifluoromethyl)imidazol-2-yl]-3-fluoro-phenyl]methanol (50.0 mg, 167 μmol) in THF (500 µL) under argon atmosphere. The reaction mixture was stirred at room temperature for 1 hr.4-cyclopropyl-6-methoxy-5-(4-methylsulfonylpyrimidin-2- yl)pyrimidine I-8 (56.1 mg, 183 μmol) was added to the reaction mixture. The resulting mixture was stirred at room temperature for 48 hr. The reaction mixture was diluted with EtOAc (20 mL) and washed with brine (20 mL). The organic layer was separated, washed with brine (2 × 15 mL), dried over anhydrous Na
2SO
4 and filtered. SiliaMetS
® Dimercaptotriazine (100 mg) was added to the resulting organic layer. The resulting mixture was stirred for 30 min and filtered. The filtrate was concentrated under reduced pressure. The residue was subjected to HPLC (0-2-10 min., 43-50-60% water – ACN, +0.1% vol. of 25% aq. NH
3, flow: 30 mL/min, column: Chromatorex C18 SMB100-5T 100×19 mm, 5 µm) to afford 4-cyclopropyl-5-[4-[[4-[1-cyclopropyl-4-(trifluoromethyl)imidazol-2-yl]-3-fluoro- phenyl]methoxy]pyrimidin-2-yl]-6-methoxy-pyrimidine 21 (40.0 mg, 76.0 μmol, 45.6% yield) as a yellow solid.
1H NMR (500 MHz, DMSO-d
6) δ 0.79 – 0.93 (m, 6H), 1.02 – 1.07 (m, 2H), 1.66 – 1.74 (m, 1H), 3.44 – 3.51 (m, 1H), 3.85 (s, 3H), 5.53 (s, 2H), 7.12 (d, 1H), 7.45 (d, 1H), 7.51 (d, 1H), 7.65 (t, 1H), 8.01 (s, 1H), 8.68 (s, 1H), 8.73 (d, 1H). MS (ESI): [M+H]
+ m/z: calcd 527.21; found 527.2 Example 5 (Compound 24)
2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[5-fluoro-6-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]-3-pyridyl]methoxy]-5-methyl-pyrimidine Synthesis of the starting [5-fluoro-6-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]-3- pyridyl]methanol is described for compound 48. [1009] NaH (21.8 mg, 545 μmol, 60% dispersion in mineral oil) was added to a solution of 2- (4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-5-methyl-4-methylsulfonyl-pyrimidine I-7 (87.3 mg, 273 μmol) and [5-fluoro-6-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]-3- pyridyl]methanol (75.0 mg, 273 μmol) in THF (5.0 mL). The resulting mixture was stirred at room temperature for 13 hr. The reaction mixture was poured into a saturated aqueous solution of NH
4Cl (15 mL) and extracted with MTBE (3×15 mL). The combined organic layers were dried over anhydrous Na
2SO
4 and concentrated under reduced pressure. The residue was dissolved in ACN (6.0 mL). Metal scavenger SiliaMetS
® Dimercaptotriazine (100 mg) was added to the solution. The resulting suspension was stirred at room temperature for 7 hr. The suspension was filtered. The filtrate was subjected to HPLC (0-1.3-5.5 min., 35- 40-40% water – ACN, flow: 30 mL/min, column: Chromatorex 100×19 mm, 5 µm) to afford 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[5-fluoro-6-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]-3-pyridyl]methoxy]-5-methyl-pyrimidine 24 (44.0 mg, 85.4 μmol, 31.3% yield) as a yellow solid.
1H NMR (600 MHz, DMSO-d
6) δ 0.86 – 0.90 (m, 2H), 1.02 – 1.06 (m, 2H), 1.65 – 1.71 (m, 1H), 2.25 (s, 3H), 3.84 (s, 3H), 3.86 (s, 3H), 5.59 (s, 2H), 8.04 (d, 1H), 8.06 (s, 2H), 8.58 (s, 1H), 8.67 (s, 1H), 8.69 (s, 1H). MS (ESI): [M+H]
+ m/z: calcd 516.18; found 516.2
2,6-dichloro-N-methoxy-N-methylpyrimidine-4-carboxamide [1010] TEA (957 mg, 9.46 mmol, 1.32 mL) was added dropwise to a stirred mixture of 2,6- dichloropyrimidine-4-carbonyl chloride (800 mg, 3.78 mmol) and N-methoxymethanamine (369 mg, 3.78 mmol, HCl) in DCM (20 mL) at -20 °C. The reaction mixture was allowed to warm to room temperature and stirred for 3 hr. The reaction mixture was washed with an aqueous solution of citric acid (25 mL, 10% wt.) and water (2×25 mL). The organic layer was separated, dried over anhydrous Na
2SO
4 and concentrated under reduced pressure to afford 2,6-dichloro-N-methoxy-N-methylpyrimidine-4-carboxamide (760 mg, 3.22 mmol, 85.1% yield) as a light-yellow solid which was used in the next step without further purification.
1H NMR (500 MHz, DMSO-d6) δ 3.32 (s, 3H), 3.76 (s, 3H), 7.48 (s, 1H). 2-chloro-6-[[3-fluoro-4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]-N- methoxy-N-methyl-pyrimidine-4-carboxamide Synthesis of the starting [3-fluoro-4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methanol is described for compound 97. [1011] Cs
2CO
3 (1.05 g, 3.22 mmol) was suspended in THF (5.0 mL) at 0 °C. [3-fluoro-4-[1- methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methanol (441 mg, 1.61 mmol) was added to the mixture at the same temperature, followed by 2,6-dichloro-N-methoxy-N-methyl- pyrimidine-4-carboxamide (760 mg, 3.22 mmol) in an hour. The reaction mixture was stirred at room temperature for 12 hr. The reaction mixture was concentrated under reduced pressure. The residue was dissolved in EtOAc (15 mL) and washed with brine (2×10 mL). The organic layer was separated, dried over anhydrous Na
2SO
4 and concentrated under reduced pressure. The residue was subjected to flash column chromatography (silica, gradient hexane - MTBE) to afford of 2-chloro-6-[[3-fluoro-4-[1-methyl-4-(trifluoromethyl)imidazol- 2-yl]phenyl]methoxy]-N-methoxy-N-methyl-pyrimidine-4-carboxamide (560 mg, 1.18 mmol, 36.7% yield) as a yellow oil.
1H NMR (500 MHz, CDCl
3) δ 3.33 (br. s., 3H), 3.66 (s, 3H), 3.77 (br. s., 3H), 5.52 (s, 2H), 6.94 (br. s., 1H), 7.28 (d, 1H), 7.33 – 7.38 (m, 2H), 7.64 (t, 1H). MS (ESI): [M+H]
+ m/z: calcd 474.10; found 474.0 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-6-[[3-fluoro-4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]-N-methoxy-N-methyl-pyrimidine-4- carboxamide [1012] 2-Chloro-6-[[3-fluoro-4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]-N-methoxy-N-methyl-pyrimidine-4-carboxamide (400 mg, 844 μmol) was added to the mixture of (4-cyclopropyl-6-methoxy-pyrimidin-5-yl)boronic acid I-5 (213 mg, 1.10 mmol), DIPEA (327 mg, 2.53 mmol, 441 μL) and XPhos Pd G3 (71.5 mg, 84.4 μmol) in a degassed mixture of dioxane (600 mL) and water (600 µL). The resulting mixture was stirred at 70 °C for 12 hr. under argon atmosphere. The reaction mixture was cooled to room temperature. SiliaMetS
® Dimercaptotriazine (300 mg) was added to the mixture and the resulting mixture was stirred for 30 min. The mixture was filtered. The filtrate was concentrated under reduced pressure. The residue was subjected to HPLC (0-1-6 min., 40- 60% water – ACN, +0.1% vol. of 25% aq. NH
3, flow: 30 mL/min, column: XBridge OBD C18100×19 mm, 5 µm) to afford 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-6-[[3-fluoro- 4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]-N-methoxy-N-methyl- pyrimidine-4-carboxamide (64.0 mg, 109 μmol, 12.9% yield) as a red oil. MS (ESI): [M+H]
+ m/z: calcd 588.22; found 588.2 1-[2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-6-[[3-fluoro-4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidin-4-yl]ethanone [1013] A solution of chloro(methyl)magnesium (102 μL, 3 M in THF) was added to a stirred solution of 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-6-[[3-fluoro-4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]-N-methoxy-N-methyl-pyrimidine-4- carboxamide (45.0 mg, 76.6 μmol) in THF (2 mL) at -20 °C under argon atmosphere. The reaction mixture was stirred at -20 °C for 2 hr. The reaction mixture was diluted with MeOH (3.0 mL). The resulting solution was used in the next step as is. MS (ESI): [M+H]
+ m/z: calcd 543.18; found 543.1 1-[2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-6-[[3-fluoro-4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidin-4-yl]ethanol [1014] Sodium Borohydride (9.41 mg, 249 μmol) was added to a stirred solution of 1-[2-(4- cyclopropyl-6-methoxy-pyrimidin-5-yl)-6-[[3-fluoro-4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidin-4-yl]ethanone (45.0 mg, 83.0 μmol) in MeOH (3 mL) and THF (2 mL) at 0 °C (solution from previous step). The reaction mixture was stirred at 0 °C for 3 hr. The reaction mixture was allowed to warm to room temperature and directly subjected to HPLC (0-5 min, 25-75% water+FA (0.1% vol.) - ACN+FA (0.1% vol.); flow: 30 mL/min, column: XBridge C18 OBD, 100×19 mm, 5 µm) to afford 1-[2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-6-[[3-fluoro-4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidin-4-yl]ethanol (5.20 mg, 9.55 μmol, 11.5% yield) as a light-yellow oil. MS (ESI): [M+H]
+ m/z: calcd 545.22; found 545.2 rel-(R)-1-(4'-cyclopropyl-6-((3-fluoro-4-(1-methyl-4-(trifluoromethyl)-1H-imidazol-2- yl)benzyl)oxy)-6'-methoxy-[2,5'-bipyrimidin]-4-yl)ethanol and rel-(S)-1-(4'-cyclopropyl-6-((3-fluoro-4-(1-methyl-4-(trifluoromethyl)-1H-imidazol-2- yl)benzyl)oxy)-6'-methoxy-[2,5'-bipyrimidin]-4-yl)ethanol [1015] 1-[2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-6-[[3-fluoro-4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidin-4-yl]ethanol (5.20 mg, 9.55 μmol) was subjected to chiral HPLC (Column: Chiralcel OZ-H (250×4.6 mm, 5 µm)-7, mobile phase: Hexane:IPA:MeOH, 90:5:5, flow rate: 0.6 mL/min) to afford rel-(S)-1-(4'- cyclopropyl-6-((3-fluoro-4-(1-methyl-4-(trifluoromethyl)-1H-imidazol-2-yl)benzyl)oxy)-6'- methoxy-[2,5'-bipyrimidin]-4-yl)ethanol (2.71 mg, 4.98 μmol, 94.8% yield) and rel-(R)-1-(4'- cyclopropyl-6-((3-fluoro-4-(1-methyl-4-(trifluoromethyl)-1H-imidazol-2-yl)benzyl)oxy)-6'- methoxy-[2,5'-bipyrimidin]-4-yl)ethanol (1.58 mg, 2.90 μmol, 67.5% yield) as light-yellow solids. rel-(R)-1-(4'-cyclopropyl-6-((3-fluoro-4-(1-methyl-4-(trifluoromethyl)-1H-imidazol- 2-yl)benzyl)oxy)-6'-methoxy-[2,5'-bipyrimidin]-4-yl)ethanol (25): MS (ESI): [M+H]
+ m/z: calcd 545.22; found 545.4 Enantiopurity: >99% (column: Chiralcel OZ-H, 250 × 4.6 mm, 5 μm; mobile phase: Hexane-IPA-MeOH, 90-5-5; flow: 0.6 mL/min; RT=36.0 min). rel-(S)-1-(4'-cyclopropyl-6-((3-fluoro-4-(1-methyl-4-(trifluoromethyl)-1H-imidazol- 2-yl)benzyl)oxy)-6'-methoxy-[2,5'-bipyrimidin]-4-yl)ethanol (ent-25): MS (ESI): [M+H]
+ m/z: calcd 545.22; found 545.4 Enantiopurity: 90.4% (column: Chiralcel OZ-H, 250 × 4.6 mm, 5 μm; mobile phase: Hexane-IPA-MeOH, 90-5-5; flow: 0.6 mL/min; RT=45.0 min). Example 7 (Compound 26)

5-chloro-2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[5-fluoro-6-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]-3-pyridyl]methoxy]pyrimidine Synthesis of the starting 5-chloro-2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4- methylsulfonyl-pyrimidine is described for Compound 123. Synthesis of the starting [5-fluoro-6-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]-3- pyridyl]methanol is described for compound 48. [1016] Potassium tert-butoxide (11.0 mg, 98.0 μmol) was added to a stirred solution of [5- fluoro-6-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]-3-pyridyl]methanol (50.0 mg, 182 μmol) and 5-chloro-2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-methylsulfonyl- pyrimidine (61.9 mg, 182 μmol) in THF (1 mL) at 0 °C. The reaction mixture was stirred at 0°C for 1 hr. The reaction was quenched by addition of water (0.1 mL). The resulting mixture was directly subjected to HPLC (0-2-9 min., 43-50-65% water – ACN, flow: 30 mL/min, column: Chromatorex C18 SMB100-5T 100×19 mm, 5 µm) to afford 5-chloro-2-(4- cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[5-fluoro-6-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]-3-pyridyl]methoxy]pyrimidine 26 (11.9 mg, 22.2 μmol, 12.2% yield) as a white solid.
1H NMR (500 MHz, DMSO-d
6) δ 0.89 – 0.94 (m, 2H), 1.04 – 1.08 (m, 2H), 1.77 – 1.83 (m, 1H), 3.86 (s, 3H), 3.87 (s, 3H), 5.66 (s, 2H), 8.03 – 8.08 (m, 2H), 8.69 – 8.72 (m, 2H), 8.92 (s, 1H). MS (ESI): [M+H]
+ m/z: calcd 536.14; found 536.2 Example 8 (Compound 27)
5-bromo-2-chloro-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidine [1017] 5-Bromo-2,4-dichloro-pyrimidine (680 mg, 2.98 mmol) was added to a stirred mixture of [4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methanol I-2b (497 mg, 1.94 mmol) and Cs
2CO
3 (1.17 g, 3.58 mmol) in ACN (10 mL). The resulting mixture was stirred at 65 °C for 12 hr. The reaction mixture was cooled to room temperature and concentrated under reduced pressure. The residue was diluted with water (30 mL) and extracted with EtOAc (40 mL). The organic layer was separated, washed with brine (30 mL), dried over anhydrous Na
2SO
4 and concentrated under reduced pressure to afford 5-bromo-2- chloro-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidine (800 g, 1.79 mmol, 59.9% yield) as a yellow solid which was used in the next steps without further purification.
1H NMR (500 MHz, DMSO-d
6) 3.80 (s, 3H), 5.57 (s, 2H), 7.63 (d, 2H), 7.77 (d, 2H), 7.94 (s, 1H), 8.77 (s, 1H). Tert-butyl 2-[2-chloro-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidin-5-yl]pyrrole-1-carboxylate [1018] (1-Tert-butoxycarbonylpyrrol-2-yl)boronic acid (191 mg, 905 μmol), 5-bromo-2- chloro-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidine (300 mg, 670 μmol), bis(diphenylphosphino)ferrocene]dichloropalladium(II)-DCM (38.3 mg, 46.9 μmol) and tripotassium phosphate (213 mg, 1.01 mmol) were mixed in a degassed mixture dioxane (7.2 mL) and water (2.1 mL) under argon atmosphere at room temperature. The reaction mixture was stirred at 70 °C for 12 hr. Additional portion of (1-tert- butoxycarbonylpyrrol-2-yl)boronic acid (191 mg, 905 μmol) and tripotassium phosphate (213 mg, 1.01 mmol) were added to the reaction mixture. The resulting mixture was stirred at 90 °C for 12 hr. The reaction mixture was cooled to room temperature and filtered. The filtrate was concentrated under reduced pressure. The residue was subjected to flash column chromatography (SiO
2, gradient hexane - EtOAc) to afford tert-butyl 2-[2-chloro-4-[[4-[1- methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidin-5-yl]pyrrole-1- carboxylate (130 mg, 244 μmol, 36.3% yield) as a brown solid. MS (ESI): [M+H]
+ m/z: calcd 534.15; found 534.0 Tert-butyl 2-[2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidin-5-yl]pyrrole-1-carboxylate Tert-butyl 2-[2-chloro-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidin-5-yl]pyrrole-1-carboxylate (132 mg, 247 μmol), (4- cyclopropyl-6-methoxy-pyrimidin-5-yl)boronic acid I-5 (95.9 mg, 494 μmol), potassium phosphate tribasic (157 mg, 741 μmol) and RuPhos Pd G3 (10.5 mg, 12.4 μmol) were mixed in a degassed mixture of dioxane (2 mL) and water (200 µL) under argon atmosphere. The reaction mixture was stirred at room temperature for 20 hr. The organic layer was separated and subjected to HPLC (2-10 min., 40-65% water – ACN, flow: 30 mL/min, column: Chromatorex SMB100-5T 100×19 mm, 5 µm) to afford tert-butyl 2-[2-(4-cyclopropyl-6- methoxy-pyrimidin-5-yl)-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidin-5-yl]pyrrole-1-carboxylate (20.0 mg, 30.9 μmol, 12.5% yield) as a white solid. MS (ESI): [M+H]
+ m/z: calcd 648.26; found 648.2 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]-5-(1H-pyrrol-2-yl)pyrimidine Tert-butyl 2-[2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidin-5-yl]pyrrole-1-carboxylate (20.0 mg, 31.0 μmol) was dissolved in a mixture of DCM (100 µL) and TFA (100 μL). The reaction mixture was stirred at room temperature for 2 hr. The reaction mixture was concentrated under reduced pressure. The residue was subjected to HPLC (0-2-10 min., 30-40-65% water – ACN, flow: 30 mL/min, column: Chromatorex SMB100-5T 100×19 mm, 5 µm) to afford 2-(4- cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]-5-(1H-pyrrol-2-yl)pyrimidine 27 (7.00 mg, 12.8 μmol, 41.4% yield) as a yellow solid.
1H NMR (600 MHz, DMSO-d
6) δ 0.84 – 0.89 (m, 2H), 1.02 – 1.06 (m, 2H), 1.74 – 1.79 (m, 1H), 3.79 (s, 3H), 3.86 (s, 3H), 5.65 (s, 2H), 6.21 (s, 1H), 6.83 (s, 1H), 7.02 (s, 1H), 7.63 (d, 2H), 7.75 (d, 2H), 7.94 (s, 1H), 8.67 (s, 1H), 9.02 (s, 1H), 11.45 (s, 1H). MS (ESI): [M+H]
+ m/z: calcd 548.23; found 548.0 Example 9 (Compound 28)

2-[4-cyclopropyl-6-(difluoromethoxy)pyrimidin-5-yl]-4-[[4-[1-ethyl-4- (trifluoromethyl)imidazol-2-yl]-3-fluoro-phenyl]methoxy]-6-methyl-pyrimidine Synthesis of the starting [4-[1-ethyl-4-(trifluoromethyl)imidazol-2-yl]-3-fluoro- phenyl]methanol is described for Compound 131. Synthesis of the starting 2-[4-cyclopropyl-6-(difluoromethoxy)pyrimidin-5-yl]-4- methyl-6-methylsulfonyl-pyrimidine is described for Compound 105. [1019] The mixture of 2-[4-cyclopropyl-6-(difluoromethoxy)pyrimidin-5-yl]-4-methyl-6- methylsulfonyl-pyrimidine (100 mg, 281 μmol), [4-[1-ethyl-4-(trifluoromethyl)imidazol-2- yl]-3-fluoro-phenyl]methanol (80.9 mg, 281 μmol) and NaH (11.2 mg, 281 μmol, 60% dispersion in mineral oil) in THF (2 mL) was stirred at room temperature for 12 hr. The reaction mixture was directly subjected to HPLC (0.6-8.6 min., 42-50-80% water – ACN, flow: 30 mL/min, column: Chromatorex C18 SMB100-5T 100×19 mm, 5 µm) to afford 2-[4- cyclopropyl-6-(difluoromethoxy)pyrimidin-5-yl]-4-[[4-[1-ethyl-4-(trifluoromethyl)imidazol- 2-yl]-3-fluoro-phenyl]methoxy]-6-methyl-pyrimidine 28 (27.0 mg, 47.8 μmol, 17% yield) as an off-white solid.
1H NMR (600 MHz, DMSO-d
6) δ 0.96 – 1.01 (m, 2H), 1.09 – 1.13 (m, 2H), 1.27 (t, 3H), 1.81 – 1.87 (m, 1H), 2.52 (s, 3H), 3.88 (q, 2H), 5.53 (s, 2H), 7.04 (s, 1H), 7.43 (d, 1H), 7.49 (d, 1H), 7.57 (t, 1 H), 7.80 (t, 1H, CHF2), 8.11 (s, 1H), 8.80 (s, 1H). MS (ESI): [M+H]
+ m/z: calcd 565.21; found 565.2 Example 10 (Compounds 229 & 29)
1-[2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-6-[[6-[1-cyclopropyl-4- (trifluoromethyl)imidazol-2-yl]-5-fluoro-3-pyridyl]methoxy]pyrimidin-4-yl]ethanone Synthesis of the starting [6-[1-cyclopropyl-4-(trifluoromethyl)imidazol-2-yl]-5-fluoro- 3-pyridyl]methanol is described for Compound 43. Synthesis of the 1-[2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-6-methylsulfonyl- pyrimidin-4-yl]ethanone is described for Compound 36 and 233. [1020] Potassium tert-butoxide (30.9 mg, 276 μmol) was added to a stirred solution of 1-[2- (4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-6-methylsulfonyl-pyrimidin-4-yl]ethanone (80.0 mg, 230 μmol) and [6-[1-cyclopropyl-4-(trifluoromethyl)imidazol-2-yl]-5-fluoro-3- pyridyl]methanol (69.2 mg, 230 μmol) in THF (1 mL). The resulting mixture was stirred at room temperature for 10 hr. The reaction mixture was poured into a saturated aqueous solution NH
4Cl (2.0 mL) and extracted with EtOAc (3×3 mL). The combined organic layers were dried over anhydrous Na
2SO
4 and concentrated under reduced pressure to afford 1-[2- (4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-6-[[6-[1-cyclopropyl-4-(trifluoromethyl)imidazol- 2-yl]-5-fluoro-3-pyridyl]methoxy]pyrimidin-4-yl]ethanone (115 mg, crude) as a yellow oil which was used in the next step without further purification. MS (ESI): [M+H]
+ m/z: calcd 570.19; found 570.6 1-[2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-6-[[6-[1-cyclopropyl-4- (trifluoromethyl)imidazol-2-yl]-5-fluoro-3-pyridyl]methoxy]pyrimidin-4-yl]ethanol [1021] Sodium borohydride (7.79 mg, 206 μmol, 7.3 μL) was added to a stirred solution of 1- [2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-6-[[6-[1-cyclopropyl-4- (trifluoromethyl)imidazol-2-yl]-5-fluoro-3-pyridyl]methoxy]pyrimidin-4-yl]ethanone (115 mg, crude) in MeOH (3.0 mL) at 0 °C. The reaction mixture was stirred at 0 °C for 3 hr. The mixture was allowed to warm to room temperature. The reaction mixture was subjected to HPLC (0-1-6 min, 40-40-70% water+FA (0.1% vol.) - ACN+FA (0.1% vol.); flow: 60 mL/min, column: XBridge OBD C18, 100×30 mm, 5 µm) to afford 1-[2-(4-cyclopropyl-6- methoxy-pyrimidin-5-yl)-6-[[6-[1-cyclopropyl-4-(trifluoromethyl)imidazol-2-yl]-5-fluoro-3- pyridyl]methoxy]pyrimidin-4-yl]ethanol (19.7 mg, 34.5 μmol, 15.0% yield from 1-[2-(4- cyclopropyl-6-methoxy-pyrimidin-5-yl)-6-methylsulfonyl-pyrimidin-4-yl]ethanone) as a light-yellow solid.
1H NMR (600 MHz, DMSO-d
6) δ 0.84 – 0.92 (m, 6H), 1.03 – 1.08 (m, 2H), 1.37 (d, 3H), 1.67 – 1.72 (m, 1H), 3.73 – 3.78 (m, 1H), 3.85 (s, 3H), 4.66 – 4.71 (m, 1H), 5.55 – 5.63 (m, 3H), 7.08 (s, 1H), 8.05 – 8.09 (m, 2H), 8.68 (s, 1H), 8.71 (s, 1H). MS (ESI): [M+H]
+ m/z: calcd 572.23; found 572.2 rel-(R)-1-(4'-cyclopropyl-6-((6-(1-cyclopropyl-4-(trifluoromethyl)-1H-imidazol-2-yl)-5- fluoropyridin-3-yl)methoxy)-6'-methoxy-[2,5'-bipyrimidin]-4-yl)ethanol & rel-(S)-1-(4'- cyclopropyl-6-((6-(1-cyclopropyl-4-(trifluoromethyl)-1H-imidazol-2-yl)-5-fluoropyridin-3- yl)methoxy)-6'-methoxy-[2,5'-bipyrimidin]-4-yl)ethanol [1022] 1-[2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-6-[[6-[1-cyclopropyl-4- (trifluoromethyl)imidazol-2-yl]-5-fluoro-3-pyridyl]methoxy]pyrimidin-4-yl]ethanol (19.7 mg, 34.5 μmol) was subjected to chiral HPLC (Column: Chiralcel OD-H (250×20 mm, 5 µm); mobile phase: Hexane:IPA:MeOH, 90:5:5, flow rate: 12 mL/min) to afford rel-(S)-1- (4'-cyclopropyl-6-((6-(1-cyclopropyl-4-(trifluoromethyl)-1H-imidazol-2-yl)-5-fluoropyridin- 3-yl)methoxy)-6'-methoxy-[2,5'-bipyrimidin]-4-yl)ethanol (7.85 mg, 13.7 μmol, 79.7% yield) and rel-(R)-1-(4'-cyclopropyl-6-((6-(1-cyclopropyl-4-(trifluoromethyl)-1H-imidazol-2-yl)-5- fluoropyridin-3-yl)methoxy)-6'-methoxy-[2,5'-bipyrimidin]-4-yl)ethanol (7.55 mg, 13.2 μmol, 76.6% yield) as light-yellow solids. rel-(R)-1-(4'-cyclopropyl-6-((4-(1-cyclopropyl-4-(trifluoromethyl)-1H-imidazol-2-yl)- 3-fluorobenzyl)oxy)-6'-methoxy-[2,5'-bipyrimidin]-4-yl)ethanol (229): 1H NMR (600 MHz, DMSO-d
6) δ 0.84 – 0.92 (m, 6H), 1.03 – 1.08 (m, 2H), 1.37 (d, 3H), 1.67 – 1.72 (m, 1H), 3.73 – 3.78 (m, 1H), 3.85 (s, 3H), 4.66 – 4.71 (m, 1H), 5.55 – 5.63 (m, 3H), 7.08 (s, 1H), 8.05 – 8.09 (m, 2H), 8.68 (s, 1H), 8.71 (s, 1H). MS (ESI): [M+H]
+ m/z: calcd 572.23; found 572.2 Enantiopurity: 99% (column: Chiralcel OD-H, 250 × 4.6 mm, 5 μm; mobile phase: Hexane-IPA-MeOH, 90-5-5; flow: 0.6 mL/min; RT=34.1 min). rel-(S)-1-(4'-cyclopropyl-6-((4-(1-cyclopropyl-4-(trifluoromethyl)-1H-imidazol-2-yl)- 3-fluorobenzyl)oxy)-6'-methoxy-[2,5'-bipyrimidin]-4-yl)ethanol (29): 1H NMR (600 MHz, DMSO-d
6) δ 0.84 – 0.92 (m, 6H), 1.03 – 1.08 (m, 2H), 1.37 (d, 3H), 1.67 – 1.72 (m, 1H), 3.73 – 3.78 (m, 1H), 3.85 (s, 3H), 4.66 – 4.71 (m, 1H), 5.55 – 5.63 (m, 3H), 7.08 (s, 1H), 8.05 – 8.09 (m, 2H), 8.68 (s, 1H), 8.71 (s, 1H). MS (ESI): [M+H]
+ m/z: calcd 572.23; found 572.2 Enantiopurity: 97% (column: Chiralcel OD-H, 250 × 4.6 mm, 5 μm; mobile phase: Hexane-IPA-MeOH, 90-5-5; flow: 0.6 mL/min; RT=29.8 min).
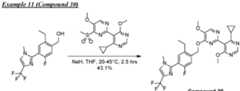
Compound 30 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[2-ethyl-5-fluoro-4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]-5-methoxy-pyrimidine (30) The synthesis of [2-ethyl-5-fluoro-4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methanol is described for Compound 83. [1023] To a solution of [2-ethyl-5-fluoro-4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methanol (70 mg, 0.232 mmol) in THF (2 mL) was added NaH (15 mg, 0.375 mmol, 60 wt% in mineral oil). The mixture was stirred at 20 °C for 30 min.2-(4-cyclopropyl- 6-methoxy-pyrimidin-5-yl)-5-methoxy-4-methylsulfonyl-pyrimidine I-6 (200 mg, 0.595 mmol) was added, and the mixture was stirred at 45 °C for 2 hrs. The resulting mixture was quenched by addition of water (5 mL) and extracted with EtOAc (50 mL x 3). The combined organic layer was washed with brine (40 mL x 2), dried over anhydrous Na
2SO
4, filtered and concentrated under reduced pressure. The residue was purified by preparative HPLC (Instrument: Gilson GX-281 Liquid Handler, Gilson 322 Pump, Gilson 156 UV Detector; Column: 2_Phenomenex Gemini C1875 x 40 mm x 3 μm; Mobile phase A: H
2O with NH
3- H
2O (v%); Mobile phase B: MeCN; Gradient: B from 50% to 80% in 7.8 min, hold 100% B for 2 min; Flow Rate: 30 mL/min; Column Temperature: 30 °C; Wavelength: 220 nm, 254 nm). The fraction was concentrated under reduced pressure and then lyophilized for overnight to afford 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[2-ethyl-5-fluoro-4-[1- methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]-5-methoxy-pyrimidine (55.8 mg, 43.1% yield) as white solid.
1H NMR (400 MHz, DMSO-d
6) δ ppm 8.65 (s, 1H), 8.44 (s, 1H), 8.01 (d, J = 1.0 Hz, 1H), 7.38 - 7.49 (m, 2H), 5.52 (s, 2H), 3.95 (s, 3H), 3.84 (s, 3H), 3.61 (d, J = 1.3 Hz, 3H), 2.70 (q, J = 7.5 Hz, 2H), 1.70 - 1.78 (m, 1H), 1.16 (t, J = 7.6 Hz, 3H), 1.00 - 1.06 (m, 2H), 0.84 - 0.92 (m, 2H);
19F NMR (376 MHz, DMSO-d
6) δ ppm -60.82, -118.17; MS (ESI) [M+H]
+ m/z: calcd 559.2, found 559.3. Example 12 (Compound 7)
Step a) [4-[5-methyl-3-(trifluoromethyl)pyrazol-1-yl]phenyl]methanol [1024] A mixture of (4-iodophenyl)methanol (1.00 g, 4.27 mmol), 5-methyl-3- (trifluoromethyl)-1H-pyrazole (693 mg, 4.61 mmol), Cs
2CO
3 (2.92 g, 8.97 mmol), copper (I) iodide (97.6 mg, 513 μmol) and trans-N,N′-Dimethylcyclohexane-1,2-diamine (334 mg, 2.35 mmol) in DMF (5 mL) was stirred in an inert atmosphere at 110 °C for 12 hr. The reaction mixture was cooled to room temperature and poured into water (10 mL). The obtained mixture was extracted with EtOAc (2 × 15 mL). The combined organic layers were washed with brine (15 mL), dried over anhydrous Na
2SO
4 and concentrated in vacuo. The residue was subjected to flash-column chromatography (SiO
2, gradient hexane - EtOAc) to afford [4- [5-methyl-3-(trifluoromethyl)pyrazol-1-yl]phenyl]methanol (530 mg, 2.07 mmol, 48.4% yield) as a yellow solid.
1H NMR (400 MHz, CDCl
3) δ 2.33 (s, 3H), 4.77 (s, 2H), 6.44 (s, 1H), 7.42 (d, 2H), 7.48 (d, 2H). MS (ESI): [M+H]+ m/z: calcd 257.1; found 257.0. Step b) 2-chloro-5-methyl-4-[[4-[5-methyl-3-(trifluoromethyl)pyrazol-1- yl]phenyl]methoxy]pyrimidine [1025] [4-[5-methyl-3-(trifluoromethyl)pyrazol-1-yl]phenyl]methanol (530 mg, 2.07 mmol) was added to a suspension of NaH (124 mg, 3.10 mmol, 60% dispersion in mineral oil) in DMF (10 mL) at room temperature, followed by the addition of 2,4-dichloro-5-methyl- pyrimidine (506 mg, 3.10 mmol). The resulting mixture was stirred at room temperature for 12 hr. The reaction mixture was poured into cold aqueous NH
4Cl solution (25 mL). The resulting mixture was extracted with EtOAc (2 × 30 mL). The combined organic layers were dried over anhydrous Na
2SO
4 and concentrated in vacuo. The residue was subjected to HPLC (0.6-7 min 45-70% water - ACN; flow: 30 mL/min, column: Waters SunFire C18, 100 x 19 mm, 5 µm) to afford 2-chloro-5-methyl-4-[[4-[5-methyl-3-(trifluoromethyl)pyrazol-1- yl]phenyl]methoxy]pyrimidine (156 mg, 408 μmol, 19.7% yield) as a white solid.
1H NMR (400 MHz, DMSO-d
6) δ 2.13 (s, 3H), 2.36 (s, 3H), 5.53 (s, 2H), 6.77 (s, 1H), 7.62 (d, 2H), 7.67 (d, 2H), 8.36 (s, 1H). MS (ESI): [M+H]
+ m/z: calcd 383.1; found 383.0. Step c) 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-5-methyl-4-[[4-[5-methyl-3- (trifluoromethyl)pyrazol-1-yl]phenyl]methoxy]pyrimidine [1026] A mixture of 2-chloro-5-methyl-4-[[4-[5-methyl-3-(trifluoromethyl)pyrazol-1- yl]phenyl]methoxy]pyrimidine (60.0 mg, 157 μmol), 4-cyclopropyl-6-methoxy-5-(4,4,5,5- tetramethyl-1,3,2-dioxaborolan-2-yl)pyrimidine I-5 (47.6 mg, 172 μmol), RuPhos Pd G4 (13.3 mg, 15.7 μmol) and potassium phosphate (99.8 mg, 470 μmol) in dioxane (1 mL) was stirred at 95 °C in an inert atmosphere for 12 hr The reaction mixture was cooled to room temperature and diluted with EtOAc (5 mL). Anhydrous Na
2SO
4 and SiliaMetS
® Dimercaptotriazine (50 mg) were added and the mixture was stirred for 1 hr. The solid was filtered out and washed with EtOAc (5 mL). The filtrate was concentrated in vacuo. The residue was subjected to HPLC (0.6-8.6 min 40-60% water – ACN, +0.1% vol. of 25% aq. NH
3, 30 mL/min, column: YMC-Actus Triart C18, 100x20mm, 5 µm) to afford 2-(4- cyclopropyl-6-methoxy-pyrimidin-5-yl)-5-methyl-4-[[4-[5-methyl-3- (trifluoromethyl)pyrazol-1-yl]phenyl]methoxy]pyrimidine (Compound 7)(18.2 mg, 36.7 μmol, 23.4% yield) as an off-white solid.
1H NMR (600 MHz, DMSO-d
6) δ 0.83 – 8.87 (m, 2H), 0.99 – 1.03 (m, 2H), 1.63 – 1.67 (m, 1H), 2.22 (s, 3H), 2.33 (s, 3H), 3.82 (s, 3H), 5.52 (s, 2H), 6.75 (s, 1H), 7.58 (d, 2H), 7.63 (d, 2H), 8.55 (s, 1H), 8.64 (s, 1H). MS (ESI): [M+H]
+ m/z: calcd 497.21; found 497.2. Example 13 (Compound 32)

Methyl 2-[4-cyclopropyl-6-(difluoromethoxy)pyrimidin-5-yl]-6-[[4-[1-cyclopropyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidine-4-carboxylate Synthesis of the starting methyl 2-[4-cyclopropyl-6-(difluoromethoxy)pyrimidin-5-yl]- 6-methylsulfonyl-pyrimidine-4-carboxylate is described for Compound 44. Synthesis of the starting [4-[1-cyclopropyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methanol is described for compound 2. [1027] DBU (26.6 mg, 175 μmol, 26 μL) was added to a stirred solution of [4-[1- cyclopropyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methanol (35.3 mg, 125 μmol) and methyl 2-[4-cyclopropyl-6-(difluoromethoxy)pyrimidin-5-yl]-6-methylsulfonyl-pyrimidine- 4-carboxylate (50 mg, 125 μmol) in THF (2 mL). The reaction mixture was stirred at room temperature for 10 hr. The reaction was quenched by addition of water (5 mL) and extracted with EtOAc (2×5 mL). The combined organic layers were washed with brine (5 mL) and concentrated under reduced pressure to afford 2-[4-cyclopropyl-6- (difluoromethoxy)pyrimidin-5-yl]-6-[[4-[1-cyclopropyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidine-4-carboxylate (70 mg, crude) as a yellow oil which was used in the next step without further purification. MS (ESI): [M+H]
+ m/z: calcd 603.18; found 603.0 [2-[4-cyclopropyl-6-(difluoromethoxy)pyrimidin-5-yl]-6-[[4-[1-cyclopropyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidin-4-yl]methanol [1028] DIBAL (49.6 mg, 349 μmol, 74 μL) was added to a stirred solution of methyl 2-[4- cyclopropyl-6-(difluoromethoxy)pyrimidin-5-yl]-6-[[4-[1-cyclopropyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidine-4-carboxylate (35 mg, crude from previous step) in THF (1 mL). The reaction mixture was stirred at 0 °C for 1 hr. The reaction mixture was quenched by addition of MeOH (1 mL) and filtered. The filtrate was subjected to HPLC (0-2-9 min., 38-45-55% water – ACN, flow: 30 mL/min, column: Chromatorex C18 SMB100-5T, 100×19 mm, 5 µm) to afford [2-[4-cyclopropyl-6- (difluoromethoxy)pyrimidin-5-yl]-6-[[4-[1-cyclopropyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidin-4-yl]methanol (3.80 mg, 6.61 μmol, 10.6% yield from [4-[1- cyclopropyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methanol) as a yellow solid.
1H NMR (600 MHz, DMSO-d
6) δ 0.89 – 0.90 (m, 2H), 0.96 – 1.02 (m, 4H), 1.09 – 1.13 (m, 2H), 1.83 – 1.89 (m, 1H), 3.72 – 3.77 (m, 1H), 4.58 (d, 2H), 5.54 (s, 2H), 5.69 (t, 1H), 7.06 (s, 1H), 7.59 (d, 2H), 7.81 (t, 1H, CHF
2), 7.91 (d, 2H), 7.95 (s, 1H), 8.80 (s, 1H). MS (ESI): [M+H]
+ m/z: calcd 575.21; found 575.2. Example 14 (Compound 23)
Step a) 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-5-methyl-pyrimidin-4-ol [1029] To a solution of 2-chloro-5-methyl-pyrimidin-4-ol (0.3 g, 2.08 mmol) and (4- cyclopropyl-6-methoxy-pyrimidin-5-yl)boronic acid I-5 (483.11 mg, 2.49 mmol) in 1, 4- dioxane (2 mL) were added potassium phosphate (881.02 mg, 4.15 mmol) and [1,1- bis(diphenylphosphino)ferrocene]dichloropalladium(II) (169.89 mg, 207.53 µmol). After stirring at 110 °C for 16 hrs under nitrogen, the resulting solution was diluted with water (50 mL) and extracted with EtOAc (3 x 100 mL). The combined organic layers were dried over anhydrous Na2SO4. After filtration, the filtrate was concentrated under reduced pressure. The residue was purified by silica gel column chromatography, eluting with EA in PE (0% - 55%) to afford 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-5-methyl-pyrimidin-4-ol (55 mg, 212.95 µmol, 10% yield) as a yellow solid. MS: m/z = 259.15 [M + H]
+.
1H NMR (400 MHz, Methanol-d
4) δ 8.65 (s, 1H), 8.00 (s, 1H), 4.01 (s, 3H), 2.11 (s, 3H), 1.96 - 1.86 (m, 1H), 1.25 - 1.17 (m, 2H), 1.09 - 1.03 (m, 2H). Step b) 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-5-methyl-4-[[4-[1-(1-methylazetidin-3- yl)-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidine & 2-(4-cyclopropyl-6- methoxy-pyrimidin-5-yl)-5-methyl-3-[[4-[1-(1-methylazetidin-3-yl)-4- (trifluoromethyl)imidazol-2-yl]phenyl]methyl]pyrimidin-4-one [1030] To a solution of 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-5-methyl-pyrimidin-4- ol (0.045 g, 174.23 µmol), [4-[1-(1-methylazetidin-3-yl)-4-(trifluoromethyl)imidazol-2- yl]phenyl]methanol (54.24 mg, 174.23 µmol) and triphenylphosphine (68.55 mg, 261.35 µmol) in THF (2 mL) was added diethyl (E)-1,2-diazenedicarboxylate (45.52 mg, 261.35 µmol) dropwise. The resulting solution was stirred at room temperature for 2 h. The residue was purified by prep-TLC (DCM : MeOH = 15 : 1) to afford a mixture including two isomers (yellow solid, 100 mg). The residue was purified by reverse phase chromatography with the following conditions: Column: Spherical C18, 20-40 um, 40 g; Mobile Phase A: Water (10 mM NH
4HCO
3), Mobile Phase B: ACN; Flow rate: 40 mL/min; Gradient (B %): 0% hold 5 min, 5% - 30% within 10 min; 30% - 50% within 20 min, 50% - 95% within 30 min, 95% hold 5 min; Detector: UV 254 & 220 nm. The collected fractions were combined and concentrated under reduced pressure to afford 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)- 5-methyl-3-[[4-[1-(1-methylazetidin-3-yl)-4-(trifluoromethyl)imidazol-2- yl]phenyl]methyl]pyrimidin-4-one (20.1 mg, 36.44 µmol, 20% yield) as a white solid. MS: m/z = 552.30 [M + H]
+.
1H NMR (300 MHz, Methanol-d
4) δ 8.66 (s, 1H), 8.16 (s, 1H), 8.01 (s, 1H), 7.42 (d, J = 6.0 Hz, 2H), 7.09 (d, J = 6.0 Hz, 2H), 5.34 (d, J = 12.0 Hz, 1H), 5.14 (d, J = 15.6 Hz, 1H), 4.98 - 4.91 (m, 1H), 3.84 (s, 3H), 3.81 - 3.71 (m, 2H), 3.44 (t, J = 7.8 Hz, 2H), 2.43 (s, 3H), 2.21 (s, 3H), 1.57 - 1.44 (m, 1H), 1.23 - 1.12 (m, 1H), 1.04 - 0.90 (m, 2H), 0.86 - 0.73 (m, 1H).
19F NMR (282 MHz, Methanol-d
4) δ -64.07 [1031] And afford 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-5-methyl-4-[[4-[1-(1- methylazetidin-3-yl)-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidine (Compound 23)(10 mg, 18.13 µmol, 10% yield) as a white solid. MS: m/z = 552.30 [M + H]
+1H NMR (300 MHz, Methanol-d
4) δ 8.61 (s, 1H), 8.48 (s, 1H), 8.17 (s, 1H), 7.65 (d, J = 8.4 Hz, 2H), 7.56 (d, J = 8.4 Hz, 2H), 5.62 (s, 2H), 5.01 - 4.92 (m, 1H), 3.93 (s, 3H), 3.80 - 3.69 (m, 2H), 3.50 - 3.37 (m, 2H), 2.42 (s, 3H), 2.32 (s, 3H), 1.77 - 1.63 (m, 1H), 1.20 - 1.09 (m, 2H), 0.97 - 0.85 (m, 2H).
19F NMR (282 MHz, Methanol-d
4) δ -64.07. Example 15 (Compound 33)
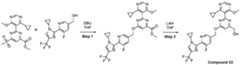
Methyl 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-6-[[6-[1-cyclopropyl-4- (trifluoromethyl)imidazol-2-yl]-5-fluoro-3-pyridyl]methoxy]pyrimidine-4-carboxylate Synthesis of the starting methyl 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-6- methylsulfonyl-pyrimidine-4-carboxylate is described for compound 58 Synthesis of the starting [6-[1-cyclopropyl-4-(trifluoromethyl)imidazol-2-yl]-5-fluoro- 3-pyridyl]methanol is described for compound 43. [1032] DBU (50.0 mg, 325 μmol, 48.7 μL) was added to a stirred solution of [6-[1- cyclopropyl-4-(trifluoromethyl)imidazol-2-yl]-5-fluoro-3-pyridyl]methanol (70.0 mg, 232 μmol) and methyl 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-6-methylsulfonyl- pyrimidine-4-carboxylate (84.7 mg, 232 μmol) in THF (2 mL). The reaction mixture was stirred at room temperature for 10 hr. The reaction mixture was quenched by addition of water (5 mL) and extracted with EtOAc (2×5 mL). The combined organic layers were washed with brine (5 mL) and concentrated under reduced pressure to afford methyl 2-(4- cyclopropyl-6-methoxy-pyrimidin-5-yl)-6-[[6-[1-cyclopropyl-4-(trifluoromethyl)imidazol-2- yl]-5-fluoro-3-pyridyl]methoxy]pyrimidine-4-carboxylate (100 mg, 171 μmol, 73.5% yield) as a yellow oil which was used in the next step without further purification. MS (ESI): [M+H]
+ m/z: calcd 586.18; found 586.1 [2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-6-[[6-[1-cyclopropyl-4- (trifluoromethyl)imidazol-2-yl]-5-fluoro-3-pyridyl]methoxy]pyrimidin-4-yl]methanol [1033] A solution of methyl 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-6-[[6-[1- cyclopropyl-4-(trifluoromethyl)imidazol-2-yl]-5-fluoro-3-pyridyl]methoxy]pyrimidine-4- carboxylate (70.0 mg, 120 μmol) in THF (0.5 mL) was added dropwise to a stirred suspension of LAH (10.5 mg, 263 μmol) in THF (1 mL) at 0 °C. The reaction mixture was stirred at 0 °C for 1 hr. The reaction mixture was quenched by addition of water (0.5 mL). The organic layer was separated, dried over anhydrous magnesium sulfate and filtered. The filtrate was subjected to HPLC (0-2-9 min., 23-30-40% water – ACN, flow: 30 mL/min, column: Chromatorex C18 SMB100-5T, 100×19 mm, 5 µm) to afford [2-(4-cyclopropyl-6- methoxy-pyrimidin-5-yl)-6-[[6-[1-cyclopropyl-4-(trifluoromethyl)imidazol-2-yl]-5-fluoro-3- pyridyl]methoxy]pyrimidin-4-yl]methanol (12.0 mg, 21.5 μmol, 18% yield) as a white solid.
1H NMR (500 MHz, DMSO-d
6) δ 0.83 – 0.92 (m, 6H), 1.02 – 1.07 (m, 2H), 1.63 – 1.70 (m, 1H), 3.72 – 3.78 (m, 1H), 3.85 (s, 3H), 4.56 (s, 2H), 5.59 (s, 2H), 5.69 (br. s., 1H), 7.06 (s, 1H), 8.04 – 8.09 (m, 2H), 8.67 (s, 1H), 8.71 (s, 1H). MS (ESI): [M+H]
+ m/z: calcd 558.21; found 558.2. Example 16 (Compound 34)
2-chloro-4-methylsulfanyl-pyrimidine-5-carbaldehyde [1034] To a mixture of 2,4-dichloropyrimidine-5-carbaldehyde (8.5 g, 48.0 mmol) in THF (70 mL) was added NaSMe (17.99 g, 51.3 mmol, 20 wt% in H
2O) at -10 °C. The mixture was stirred at 20°C for 12 hrs. The resulting mixture was diluted by addition of water (40 mL) and extracted with EtOAc (200 mL x 3). The combined organic layer was washed with brine (100 mL x 2), dried over anhydrous Na
2SO
4, filtered and concentrated under reduced pressure. The residue was purified by flash chromatography (ISCO®; 20 g AgelaFlash® Silica Flash Column, PE/EtOAc with EtOAc from 0-5%, flow rate = 45 mL/min, 254 nm) to afford 2- chloro-4-methylsulfanyl-pyrimidine-5-carbaldehyde (1.8 g, 20% yield) as white solid. MS (ESI) [M+H]
+ m/z: calcd 189.0, found 189.0. 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-methylsulfanyl-pyrimidine-5-carbaldehyde [1035] A mixture of 2-chloro-4-methylsulfanyl-pyrimidine-5-carbaldehyde (1.1 g, 5.83 mmol), (4-cyclopropyl-6-methoxy-pyrimidin-5-yl)boronic acid I-5 (1.2 g, 6.19 mmol), RuPhos Pd G
3 (260 mg, 0.310 mmol), XPhos (165 mg, 0.346 mmol), K
3PO
4 (3.85 g, 18.1 mmol) in dioxane (10 mL) and H
2O (2 mL) was stirred at 80°C for 12 hrs under N
2. The resulting mixture was diluted by addition of water (10 mL) and extracted with EtOAc (70 mL x 3). The combined organic layer was washed with brine (50 mL x 2), dried over anhydrous Na
2SO
4, filtered and concentrated under reduced pressure. The residue was purified by flash chromatography (ISCO®; 12 g AgelaFlash® Silica Flash Column, PE/EtOAc with EtOAc from 0-55%, flow rate = 35 mL/min, 254 nm) to afford 2-(4-cyclopropyl-6-methoxy- pyrimidin-5-yl)-4-methylsulfanyl-pyrimidine-5-carbaldehyde (580 mg, 32.9% yield) as yellow oil. MS (ESI) [M+H]
+ m/z: calcd 303.1, found 303.0. [2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-methylsulfanyl-pyrimidin-5-yl]methanol [1036] To a mixture of 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-methylsulfanyl- pyrimidine-5-carbaldehyde (180 mg, 0.595 mmol), NaBH
4 (60 mg, 1.59 mmol) in MeOH (2 mL) at 0 °C slowly. The mixture was stirred at 20 °C for 1 hr. The resulting mixture quenched was quenched by addition of water (10 mL) and extracted with EtOAc (80 mL x 3). The combined organic layer was washed with brine (40 mL x 2), dried over anhydrous Na
2SO
4, filtered and concentrated under reduced pressure to afford [2-(4-cyclopropyl-6- methoxy-pyrimidin-5-yl)-4-methylsulfanyl-pyrimidin-5-yl]methanol (180 mg, 99.3% yield) was brown solid. MS (ESI) [M+H]
+ m/z: calcd 305.1, found 305.1. 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-5-(methoxymethyl)-4-methylsulfanyl-pyrimidine [1037] To a solution of [2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-methylsulfanyl- pyrimidin-5-yl]methanol (180 mg, 0.591 mmol) in THF (2 mL) was added NaH (40 mg, 1.00 mmol, 60 wt% in mineral oil). The mixture was stirred at 0 °C for 30 minutes. MeI (120 mg, 0.845 mmol) was added, and the mixture was stirred at 20 °C for 1 hr. The resulting mixture was quenched by addition of water (10 mL) and extracted with EtOAc (60 mL x 3). The combined organic layer was washed with brine (40 mL x 2), dried over anhydrous Na
2SO
4, filtered and concentrated under reduced pressure to afford 2-(4-cyclopropyl-6-methoxy- pyrimidin-5-yl)-5-(methoxymethyl)-4-methylsulfanyl-pyrimidine (100 mg, 53.1% yield) as yellow solid. MS (ESI) [M+H]
+ m/z: calcd 319.1, found 319.1. 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-5-(methoxymethyl)-4-methylsulfonyl-pyrimidine [1038] A mixture of 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-5-(methoxymethyl)-4- methylsulfanyl-pyrimidine (80 mg, 0.251 mmol), 3-chlorobenzenecarboperoxoic acid (120 mg, 0.591 mmol, 85 wt%) in DCM (3 mL) was stirred at 20 °C for 12 hrs. The resulting mixture was diluted by addition of water (10 mL) and extracted with EtOAc (60 mL x 3). The combined organic layer was washed with saturated Na
2SO
3 aqueous solution (30 mL x 2), NaHCO
3 aqueous solution (30 mL x 2), brine (50 mL x 2), dried over anhydrous Na
2SO
4, filtered and concentrated under reduced pressure to afford 2-(4-cyclopropyl-6-methoxy- pyrimidin-5-yl)-5-(methoxymethyl)-4-methylsulfonyl-pyrimidine (84 mg, 95.4% yield) as yellow oil. MS (ESI) [M+H]
+ m/z: calcd 351.1, found 351.0. 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-5-(methoxymethyl)-4-[[4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidine (34) [1039] To a solution of [4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methanol I-2b (50 mg, 0.195 mmol) in THF (3 mL) was added NaH (25 mg, 0.625 mmol, 60 wt% in mineral oil). The mixture was stirred at 20 °C for 30 minutes.2-(4-cyclopropyl-6-methoxy- pyrimidin-5-yl)-5-(methoxymethyl)-4-methylsulfonyl-pyrimidine (64 mg, 0.183 mmol) was added, and the mixture was stirred at 45°C for 2 hrs. The resulting mixture was quenched by addition of water (10 mL) and extracted with EtOAc (60 mL x 3). The combined organic layer was washed with brine (40 mL x 2), dried over anhydrous Na
2SO
4, filtered and concentrated under reduced pressure. The residue was purified by preparative HPLC (Instrument: Gilson GX-281 Liquid Handler, Gilson 322 Pump, Gilson 156 UV Detector; Column: Xtimate C1875 x 40 mm x 3 μm; Mobile phase A: H
2O with NH
3-H
2O and NH
4HCO
3 (v%); Mobile phase B: MeCN; Gradient: B from 46% to 76% in 7.8 min, hold 100% B for 2 min; Flow Rate: 30mL/min; Column Temperature: 30 °C; Wavelength: 220 nm, 254 nm). The fraction was concentrated under reduced pressure and then lyophilized for overnight to afford 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-5-(methoxymethyl)-4-[[4- [1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidine (20.4 mg, 21.2% yield) as white solid.
1H NMR (400 MHz, chloroform-d) δ ppm 8.66 (d, J = 10.5 Hz, 2H), 7.66 (d, J = 8.3 Hz, 2H), 7.55 (d, J = 8.3 Hz, 2H), 7.33 (d, J = 0.9 Hz, 1H), 5.55 (s, 2H), 4.55 (s, 2H), 3.93 (s, 3H), 3.79 (s, 3H), 3.52 (s, 3H), 1.68 - 1.72 (m, 1H), 1.15 - 1.25 (m, 2H), 0.84 - 0.92 (m, 2H);
19F NMR (376 MHz, chloroform-d) δ ppm -62.75; MS (ESI) [M+H]
+ m/z: calcd 527.2, found 527.1. Example 17 (Compound 6)
4-(2-isopropylphenyl)-5-methyl-2-((4-(1-methyl-4-(trifluoromethyl)-1H-imidazol-2- yl)benzyl)oxy)pyrimidine [1040] To a solution of 2-chloro-5-methyl-4-((4-(1-methyl-4-(trifluoromethyl)-1H-imidazol- 2-yl)benzyl)oxy)pyrimidine (0.60 g, 1.57 mmol) in a mixture of 1,4-dioxane (10 mL) and water (3 mL) Na
2CO
3 (0.50 g, 4.71 mmol) and (2-isopropylphenyl)boronic acid (0.386 g, 2.35 mmol) were added. The resulting mixture was evacuated and then backfilled with argon. Bis(diphenylphosphino)ferrocene]dichloropalladium(II)-DCM (64 mg, 78.5 μmol) was added to the obtained mixture. The resulting mixture was stirred at 80 °C for 12 hr. The reaction mixture was cooled to room temperature and filtered. The filter cake was washed with 1,4- dioxane (2×5 mL) and discarded. The filtrate was concentrated in vacuo. The residue was subjected to HPLC (0-5 min 60-85% water - ACN; flow: 30 mL/min, column: Waters SunFire C18, 100x19 mm, 5 µm) to afford 4-(2-isopropylphenyl)-5-methyl-2-((4-(1-methyl- 4-(trifluoromethyl)-1H-imidazol-2-yl)benzyl)oxy)pyrimidine (Compound 6) (37.0 mg, 79.3 μmol) as a brownish solid.
1H NMR (600 MHz, DMSO-d
6) δ 1.10 (d, 6H), 2.20 (s, 3H), 3.50 (hept, 1H), 3.76 (s, 3H), 5.55 (s, 2H), 7.23 (td, 1H), 7.36 – 7.44 (m, 2H), 7.51 (dd, 1H), 7.56 (d, 2H), 7.70 – 7.74 (m, 2H), 7.91 (s, 1H), 8.51 (s, 1H). MS (ESI): [M+H]+ m/z: calcd 467.21; found 467.2 Example 18 (Compound 19)
Compound 19 4'-cyclopropyl-6'-methoxy-5-methyl-4-((4-(1-methyl-4-(trifluoromethyl)-1H-imidazol-2- yl)benzyl)oxy)-2,5'-bipyrimidine [1041] A mixture of 2-chloro-5-methyl-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidine I-2c (110 mg, 287 μmol), 4-cyclopropyl-6-methoxy-5- (4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyrimidine (103 mg, 374 μmol), K
2CO
3 (79 mg, 575 μmol) and Xphos (8.2 mg, 17 μmol) in dioxane (10 mL) and water (2 mL) was evacuated and backfilled with argon. Xphos Pd G3 (15 mg, 17 μmol) was added to the mixture. The reaction mixture was stirred at 95 °C for 15 hr. The reaction mixture was cooled to room temperature and concentrated under reduced pressure. The residue was diluted with EtOAc (50 mL), filtered through a short pad of SiO
2 and concentrated under reduced pressure. The residue was dissolved in MeOH (10 mL) and treated with SiliaMetS® Dimercaptotriazine for 7 hr at room temperature. The obtained mixture was filtered and the filtrate was subjected to HPLC (2-10 min.50-80% water+FA (0.1% vol.) - ACN+FA (0.1% vol.); flow: 30 mL/min, column: Waters SunFire C18, 100 x 19 mm, 5 µm) affording 2 portions with 70% and 92.9% purity. The second portion was re-purified by HPLC (2-10 min.30-80% water+FA (0.1% vol.) - ACN+FA (0.1% vol.); flow: 30 mL/min, column: Waters SunFire C18, 100x19 mm, 5 µm) to afford the 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-5-methyl-4-[[4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidine (Compound 19)(7.0 mg, 14.1 μmol, 4.9% yield).
1H NMR (600 MHz, DMSO-d
6) δ 0.87 – 0.79 (m, 2H), 1.04 – 0.97 (m, 2H), 1.70 – 1.60 (m, 1H), 2.22 (s, 3H), 3.77 (s, 3H), 3.82 (s, 3H), 5.51 (s, 2H), 7.58 (d, 2H), 7.72 (d, 2H), 7.92 (s, 1H), 8.54 (s, 1H), 8.64 (s, 1H). MS (ESI): [M+H]+ m/z: calcd 497.19; found 497.2
Step a) 2,4-dichloro-5-methoxypyrimidine [1042] Dimethylaniline (1.94 g, 16.0 mmol, 2.03 mL) was slowly added to a stirred mixture of 5-methoxypyrimidine-2,4-diol (5.00 g, 35.2 mmol) and phosphoryl chloride (19.4 g, 127 mmol). The reaction mixture was stirred at 101 °C for 12 hr. The reaction mixture was cooled and concentrated under reduced pressure. The residue was diluted with ice-cooled saturated aqueous NaHCO
3 solution (300 mL). To the obtained mixture NaHCO
3 was added in small portion until pH=6-8. The resulting mixture was stirred for 40 min. The precipitate formed was filtered off, washed with water and hexane and dried on air to afford 2,4-dichloro-5- methoxypyrimidine (5.0 g, 27.9 mmol, 79.4% yield) which was used in the next step without further purification.
1H NMR (400 MHz, DMSO-d
6) δ 4.01 (s, 3H), 8.64 (s, 1H). MS (ESI): [M+H]+ m/z: calcd 178.98; found 178.8 Step b) 2-chloro-5-methoxy-4-((4-(1-methyl-4-(trifluoromethyl)-1H-imidazol-2- yl)benzyl)oxy)pyrimidine [1043] [4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methanol I-2b(0.75 g, 2.93 mmol) was dissolved in THF (20 mL) and the mixture was cooled to 0 °C. To the obtained mixture NaH (140 mg, 2.93 mmol, 50% dispersion in mineral oil) was added. The mixture was stirred at room temperature for 30 minutes and cooled to 0 °C again.2,4-dichloro-5- methoxypyrimidine (524 mg, 2.93 mmol) dissolved in THF (3 mL) was added dropwise to the mixture. The resulting mixture was stirred at room temperature for 3 hr. An aqueous solution of HCl (prepared with 0.4 mL HCl (36% wt.) and 4 mL H
2O) was added to the mixture. The obtained mixture was extracted with EtOAc. The organic layer was separated, washed with saturated aqueous solution of sodium hydrogen carbonate and brine and dried over anhydrous Na
2SO
4. The solvent was evaporated under reduced pressure. The residue was subjected to flash column chromatography (SiO
2, gradient chloroform - acetonitrile) to afford 2-chloro-5-methoxy-4-((4-(1-methyl-4-(trifluoromethyl)-1H-imidazol-2- yl)benzyl)oxy)pyrimidine (500 mg, 1.25 mmol, 42.8% yield).
1H NMR (600 MHz, CDCl
3) δ 3.77 (s, 3H), 3.92 (s, 3H), 5.54 (s, 2H), 7.31 (s, 1H), 7.59 (d, 2H), 7.66 (d, 2H), 7.92 (s, 1H). MS (ESI): [M+H]
+ m/z: calcd 399.08; found 399.0 Step c) 4'-cyclopropyl-5,6'-dimethoxy-4-((4-(1-methyl-4-(trifluoromethyl)-1H-imidazol-2- yl)benzyl)oxy)-2,5'-bipyrimidine [1044] 2-chloro-5-methoxy-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidine (100 mg, 251 μmol), 4-cyclopropyl-6-methoxy-5-(4,4,5,5- tetramethyl-1,3,2-dioxaborolan-2-yl)pyrimidine (104 mg, 377 μmol), XPhos Pd G3 (10.6 mg, 12.5 μmol) and potassium phosphate tribasic anhydrous (156 mg, 752 μmol) were sequentially added to a degassed mixture of dioxane (5 mL) and water (1 mL). The resulting mixture was stirred at 100 °C under argon for 24 hr. The reaction mixture was cooled, diluted with EtOAc (5 mL), washed with water (3 mL) and brine (3 mL) and dried over anhydrous Na
2SO
4. To the obtained organic phase SiliaMetS® Dimercaptotriazine (50 mg) was added and the mixture was stirred for 30 min at room temperature. The mixture was filtered and the filtrate was concentrated under reduce pressure. The residue was subjected to HPLC to afford 4'-cyclopropyl-5,6'-dimethoxy-4-((4-(1-methyl-4-(trifluoromethyl)-1H-imidazol-2- yl)benzyl)oxy)-2,5'-bipyrimidine (Compound 12)(37.0 mg, 72.2 μmol, 28.8% yield).
1H NMR (600 MHz, CDCl
3) δ 0.84 – 0.90 (m, 2H), 1.00 – 1.05 (m, 2H), 1.68 – 1.74 (m, 1H), 3.79 (s, 3H), 3.85 (s, 3H), 3.95 (s, 3H), 5.50 (s, 2H), 7.59 (d, 2H), 7.74 (d, 2H), 7.95 (s, 1H), 8.43 (s, 1H), 8.65 (s, 1H). MS (ESI): [M+H]
+ m/z: calcd 513.19; found 513.2
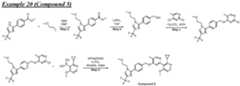
Step a): The synthesis of methyl 4-(1-(2-methoxyethyl)-4-(trifluoromethyl)-1H-imidazol-2- yl)benzoate [1045] To a solution methyl 4-(4-(trifluoromethyl)-1H-imidazol-2-yl)benzoate (500 mg, 1.85 mmol) in Dimethylformamide (10 mL) NaH (88.8 mg, 2.22 mmol, 60% in mineral oil) was added at room temperature followed by a solution of 1-iodo-2-methoxyethane (516 mg, 2.78 mmol). The reaction mixture was stirred at 95 °C for 12 hr. The mixture was concentrated in vacuo. The residue was diluted with water (20 mL). The obtained mixture was extracted with MTBE (35 mL) and EtOAc (35 mL). The combined organic layers were washed with brine (40 mL), dried over anhydrous Na2SO4 and concentrated in vacuo to afford methyl 4-(1-(2- methoxyethyl)-4-(trifluoromethyl)-1H-imidazol-2-yl)benzoate (580 mg, 1.77 mmol, 95.5% yield) as a brown viscous oil which was used in the next steps without further purification. MS (ESI): [M+H]+ m/z: calcd 329.12; found 329.1 Step b): The synthesis of (4-(1-methoxyethyl-4-(trifluoromethyl)-1H-imidazol-2- yl)phenyl)methanol [1046] Lithium aluminum hydride (134 mg, 3.53 mmol) was suspended in THF (15 mL). The resulting mixture was cooled to 0 °C and then a solution of methyl 4-(1-(2-methoxyethyl)-4- (trifluoromethyl)-1H-imidazol-2-yl)benzoate (580 mg, 1.77 mmol) in THF (5 mL) was added dropwise. The reaction mixture was stirred at ambient temperature for 2 hr. The reaction was quenched by the dropwise addition of water (1 mL), followed 15% aqueous NaOH (1 mL) then water (3 mL) with rapid stirring. The resulting solid was filtered out and the filter cake was rinsed with THF. Combined filtrate was concentrated in vacuo to afford (4-(1-(2- methoxyethyl)-4-(trifluoromethyl)-1H-imidazol-2-yl)phenyl)methanol (380 mg, 1.27 mmol, 71.6% yield) as a light-yellow viscous oil which was used in the next steps without further purification.
1H NMR (500 MHz, DMSO-d
6) δ 3.19 (s, 3H), 3.63 (t, 2H), 4.21 (t, 2H), 4.58 (s, 2H), 5.27 (br, 1 H), 7.45 (d, 2H), 7.61 (d, 2H), 7.94 (s, 1H). MS (ESI): [M+H]
+ m/z: calcd 311.13; found 301.2 Step c): The synthesis of 2-chloro-4-((4-(1-(2-methoxyethyl)-4-(trifluoromethyl)-1H-imidazol- 2-yl)benzyl)oxy)-5-methylpyrimidine [1047] (4-(1-(2-Methoxyethyl)-4-(trifluoromethyl)-1H-imidazol-2-yl)phenyl)methanol (380 mg, 1.27 mmol), 2,4-dichloro-5-methylpyrimidine (206 mg, 1.27 mmol) and Cs
2CO
3 (412 mg, 1.27 mmol) were mixed in Acetonitrile (5 mL). The reaction mixture was stirred at 80 °C for 6 hr. The reaction mixture was cooled to room temperature and filtered. The filtrate was concentrated under reduced pressure to give 2-chloro-4-((4-(1-(2-methoxyethyl)-4- (trifluoromethyl)-1H-imidazol-2-yl)benzyl)oxy)-5-methylpyrimidine (0.50 g, 1.17 mmol, 92.6% yield) which was used directly in the next steps without further purification. MS (ESI): [M+H]
+ m/z: calcd 427.13; found 427.2 Step d): The synthesis of 4'-cyclopropyl-6'-methoxy-4-((4-(1-(2-methoxyethyl)-4- (trifluoromethyl)-1H-imidazol-2-yl)benzyl)oxy)-5-methyl-2,5'-bipyrimidine [1048] 2-Chloro-4-((4-(1-(2-methoxyethyl)-4-(trifluoromethyl)-1H-imidazol-2- yl)benzyl)oxy)-5-methylpyrimidine (0.50 g, 1.17 mmol), (4-cyclopropyl-6- methoxypyrimidin-5-yl)boronic acid I-5 (341 mg, 1.76 mmol), XPhos Pd G3 (49.6 mg, 58.6 μmol) and Potassium phosphate tribasic anhydrous (746 mg, 3.51 mmol) were mixed in degassed dioxane (5 mL) and water (1 mL). The reaction mixture was stirred at 95 °C under argon atmosphere for 20 hr. The reaction mixture was cooled to room temperature and diluted with EtOAc (5 mL). The resulting solution was washed with water (3 mL) and brine (3 mL). SiliaMetS® Dimercaptotriazine (100 mg) was added to the obtained solution and the mixture was stirred for 30 min and then filtered. The filtrate was concentrated under reduce pressure. The residue was subjected to HPLC (0-1-6 min 30-30-80% water – methanol, +0.1% vol. of 25% aq. NH
3, 30 mL/min, column: YMC-Actus Triart C18, 100x20mm, 5 µm, then 0-1-6 min 40-40-75% water – methanol, +0.1% vol. of 25% aq. NH
3, 30 mL/min, column: YMC-Actus Triart C18, 100x20mm, 5 µm) to afford 4'-cyclopropyl-6'-methoxy-4- ((4-(1-(2-methoxyethyl)-4-(trifluoromethyl)-1H-imidazol-2-yl)benzyl)oxy)-5-methyl-2,5'- bipyrimidine (Compound 5)(94.0 mg, 173.90 μmol, 14.8% yield) as a light-yellow solid.
1H NMR (600 MHz, DMSO-d
6) δ 0.85 – 0.89 (m, 2H), 1.01 – 1.03 (m, 2H), 1.66 – 1.70 (m, 1H), 2.23 (s, 3H), 3.19 (s, 3H), 3.64 (t, 2H), 3.84 (s, 3H), 4.22 (t, 2H), 5.53 (s, 2H), 7.60 (d, 2H), 7.68 (d, 2H), 7.99 (s, 1H), 8.56 (s, 1H), 8.66 (s, 1H). MS (ESI): [M+H]
+ m/z: calcd 541.24; found 541.2 Example 21 (Compound 188)
Step 1: The synthesis of 3,5-difluoro-4-[4-(trifluoromethyl)-1H-imidazol-2-yl]benzoic acid [1049] A solution of 3,3-dibromo-1,1,1-trifluoro-propan-2-one (8.09 g, 30,0 mmol) and sodium acetate (4.71 g, 57.5 mmol) in water (76 mL) was stirred at 100 °C for 45 min. The obtained mixture was cooled to room temperature and poured into a mixture of methyl 3,5- difluoro-4-formyl-benzoate (5.0 g, 25.0 mmol) and an aqueous ammonium hydroxide (8.76 g, 250 mmol, 9.73 mL, 28% wt.) in MeOH (287 mL). The reaction mixture was stirred at room temperature for 12 hr. The reaction mixture was concentrated under reduced pressure to a half volume. The residue was extracted with EtOAc (2×40 mL). The combined organic layers were washed with brine (50 mL), dried over anhydrous Na
2SO
4 and filtered. The filtrate was concentrated under reduced pressure to afford crude 3,5-difluoro-4-[4-(trifluoromethyl)-1H- imidazol-2-yl]benzoic acid (4.00 g, 13.7 mmol, 54.8% yield) a light-yellow solid which was used in the next steps without further purification. MS (ESI): [M+H]+ m/z: calcd 293.04; found 293.0 Step 2: The synthesis of isopropyl 3,5-difluoro-4-[1-isopropyl-4-(trifluoromethyl)imidazol-2- yl]benzoate [1050] Cs
2CO
3 (22.3 g, 68.5 mmol) and 2-iodopropane (23.3 g, 137 mmol) were added to a solution of 3,5-difluoro-4-[4-(trifluoromethyl)-1H-imidazol-2-yl]benzoic acid (4.00 g, 13.7 mmol) in ACN (200 mL). The reaction mixture was stirred at room temperature for 48 hr. then an additional portion of 2-iodopropane (12.0 g) and Cs
2CO
3 (10 g) were added to the reaction mixture. The reaction mixture was stirred at 45 °C for 24 hr.2-iodopropane (12.0 g) and Cs
2CO
3 (10 g) were added to the reaction mixture. The reaction mixture was stirred at 85 °C for 24 hr. The reaction mixture was cooled and filtered. The filtrate was concentrated under reduced pressure. The residue was dissolved in EtOAc (150 mL). The obtained solution was washed with water (50 mL) and brine (30 mL), dried over anhydrous Na
2SO
4 and filtered. The filtrate was concentrated under reduced pressure to afford isopropyl 3,5- difluoro-4-[1-isopropyl-4-(trifluoromethyl)imidazol-2-yl]benzoate (5.0 g, crude) as a red oil which was used in the next steps without further purification. MS (ESI): [M+H]+ m/z: calcd 377.13; found 377.0 Step 3: The synthesis of [3,5-difluoro-4-[1-isopropyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methanol [1051] A solution of isopropyl 3,5-difluoro-4-[1-isopropyl-4-(trifluoromethyl)imidazol-2- yl]benzoate (5.00 g, crude) in THF (50 mL) was added dropwise to a vigorously stirred suspension of LAH (196 mg, 5.78 mmol) in THF (150 mL). The reaction mixture was stirred at room temperature for 2 hr. An aqueous NaOH (30 mL, 10% wt.) was added dropwise to the reaction mixture followed by granular NaOH (10.0 g). The resulting mixture was filtered. The filtrate was concentrated under reduced pressure. The residue was subjected to flash column chromatography (SiO
2, gradient Hexane-EtOAc) to afford [3,5-difluoro-4-[1- isopropyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methanol (280 mg, 874 μmol, 6.3% yield from 3,5-difluoro-4-[4-(trifluoromethyl)-1H-imidazol-2-yl]benzoic acid) as an yellow solid.
1H NMR (400 MHz, CDCl
3) δ 1.40 (d, 6H), 4.08 – 4.16 (m, 1H), 4.71 (s, 2H), 6.94 (d, 2H), 7.48 (s, 1H). MS (ESI): [M+H]+ m/z: calcd 321.10; found 321.2. Step 4: The synthesis of 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[3,5-difluoro-4-[1- isopropyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]-5-methoxy-pyrimidine [1052] NaH (13.9 mg, 347 μmol, 60% dispersion in mineral oil) was added to a solution of [3,5-difluoro-4-[1-isopropyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methanol (111 mg, 347 μmol) in DMF (2.0 mL). The reaction mixture was stirred at room temperature for 15 min.2- (4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-5-methoxy-4-methylsulfonyl-pyrimidine I-6 (117 mg, 347 μmol) was added to the reaction mixture. The reaction mixture was stirred at room temperature for 12 hr. The reaction mixture was diluted with water (5 mL) and extracted with EtOAc (5 mL). The organic layer was concentrated under reduced pressure. The residue was subjected to HPLC (0.5-6.5 min, 48-63% water – MeOH, +0.1% vol. of 25% aq. NH
3, flow: 30 mL/min, column: Chromatorex Phenyl SMB 100-5100 ×19 mm, 5 µm) to afford 2-(4- cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[3,5-difluoro-4-[1-isopropyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]-5-methoxy-pyrimidine (38.0 mg, 65.9 µmol, 19.0% yield) as a light-yellow solid.
1H NMR (600 MHz, DMSO-d6) δ 0.83 – 0.88 (m, 2H), 0.99 – 1.03 (m, 2H), 1.34 (d, 6H), 1.66 – 1.73 (m, 1H), 3.82 (s, 3H), 3.96 (s, 3H), 4.06 – 4.12 (m, 1H), 5.51 (s, 2H), 7.41 (d, 2H), 8.32 (s, 1H), 8.45 (s, 1H), 8.64 (s, 1H). MS (ESI): [M+H]+ m/z: calcd 577.23; found 577.0. Example 22 (Compound 131)
Step 1: The synthesis of methyl 3-fluoro-4-(4-(trifluoromethyl)-1H-imidazol-2-yl)benzoate [1053] 3,3-dibromo-1,1,1-trifluoro-propan-2-one (8.15 g, 30.2 mmol) was added to a solution of sodium acetate (4.95 g, 60.4 mmol) in water (35 mL). The reaction mixture was stirred at 95°C for 1 hr. The reaction mixture was cooled to room temperature and poured into a solution of methyl 3-fluoro-4-formyl-benzoate (5.00 g, 27.5 mmol) in MeOH (170 mL) and an aqueous NH
4OH (50 mL, 25% wt.). The resulting mixture was stirred at room temperature for 12 hr. The reaction mixture was concentrated under reduced pressure. The residue was diluted in EtOAc (100 mL) and washed with water (100 mL) and brine (50 mL). The organic layer was dried over anhydrous Na
2SO
4 and filtered. The filtrate was concentrated under reduced pressure to afford methyl 3-fluoro-4-(4-(trifluoromethyl)-1H-imidazol-2-yl)benzoate (6.90 g, 23.9 mmol, 87.2% yield) as a yellow solid which was used in the next steps without further purification.
1H NMR (500 MHz, DMSO-d6) δ 3.87 (s, 3H), 7.80 – 7.90 (m, 2H), 7.95 (s, 1H), 8.11 – 8.16 (m, 1H). MS (ESI): [M+H]+ m/z: calcd 289.06; found 289.0 Step 2: The synthesis of methyl 4-(1-ethyl-4-(trifluoromethyl)-1H-imidazol-2-yl)-3- fluorobenzoate [1054] Ethyl iodide (3.97 g, 25.5 mmol, 2.05 mL) was added to a stirred solution of methyl 3-fluoro-4-(4-(trifluoromethyl)-1H-imidazol-2-yl)benzoate (3.67 g, 12.7 mmol) and K
2CO
3 (3.52 g, 25.5 mmol) in ACN (75 mL) at room temperature. The reaction mixture was stirred at 55 °C for 16 hr. The reaction mixture was cooled to room temperature and concentrated under reduced pressure. The residue was diluted with DCM (100 mL) and washed with water (100 mL) and brine (50 mL). The organic layer was dried over anhydrous Na
2SO
4 and filtered. The filtrate was concentrated under reduced pressure to afford methyl 4-(1-ethyl-4- (trifluoromethyl)-1H-imidazol-2-yl)-3-fluorobenzoate (3.50 g, 11.1 mmol, 86.9% yield) as a yellow oil which was used in the next steps without further purification.
1H NMR (400 MHz, CDCl
3) δ 1.39 (t, 3H), 3.85 – 3.97 (m, 5H), 7.42 (s, 1H), 7.63 – 7.70 (m, 1H), 7.82 (d, 1H), 7.92 (d, 1H). MS (ESI): [M+H]+ m/z: calcd 317.09; found 317.0 Step 3: The synthesis of [4-[1-ethyl-4-(trifluoromethyl)imidazol-2-yl]-3-fluoro- phenyl]methanol [1055] A solution of methyl 4-[1-ethyl-4-(trifluoromethyl)imidazol-2-yl]-3-fluoro-benzoate (3.50 g, 11.1 mmol) in THF (10 mL) was added dropwise to a precooled to 0 °C suspension of lithium aluminium hydride (563 mg, 14.8 mmol) in THF (30 mL) at vigorous stirring. The reaction mixture was stirred at room temperature for 2 hr. The reaction mixture was cooled to 0 °C and quenched by dropwise addition of aqueous solution of KOH (5 mL, 30%). The solids were filtered out. The filtrate was concentrated under reduced pressure to afford [4-[1- ethyl-4-(trifluoromethyl)imidazol-2-yl]-3-fluoro-phenyl]methanol (2.90 g, 10.1 mmol, 90.9% yield) as a yellow oil which was used in the next steps without further purification.
1H NMR (400 MHz, CDCl
3) δ 1.35 (t, 3H), 3.89 (q, 2H), 4.70 (s, 3H), 7.08 – 7.16 (m, 2H), 7.36 – 7.45 (m, 2H). MS (ESI): [M+H]+ m/z: calcd 289.10; found 289.2 Step 4: The synthesis of 4-cyclopropyl-5-[4-[[4-[1-ethyl-4-(trifluoromethyl)imidazol-2-yl]-3- fluoro-phenyl]methoxy]pyrimidin-2-yl]-6-methoxy-pyrimidine [1056] NaH (43.7 mg, 1.09 mmol, 60% dispersion in mineral oil) was added to a stirred solution of [4-[1-ethyl-4-(trifluoromethyl)imidazol-2-yl]-3-fluoro-phenyl]methanol (286 mg, 992 μmol) in DMF (2.0 mL). The reaction mixture was stirred at room temperature for 30 min.4-cyclopropyl-6-methoxy-5-(4-methylsulfonylpyrimidin-2-yl)pyrimidine I-8 (304 mg, 992 μmol) was added to the reaction mixture. The resulting mixture was stirred at room temperature for 16 hr. The reaction mixture was diluted with a saturated aqueous solution of NH
4Cl (15 mL) and extracted with EtOAc (2×20 mL). The combined organic layers were washed with brine (10 mL), dried over anhydrous Na
2SO
4 and filtered. The filtrate was concentrated under reduced pressure. The residue was subjected to HPLC (0-5 min., 40-40- 80% water - ACN, +0.1% vol. of 25% aq. NH
3, flow: 30 mL/min, column: XBridge C18 OBD 100×19 mm, 5 µm) to afford 4-cyclopropyl-5-[4-[[4-[1-ethyl-4- (trifluoromethyl)imidazol-2-yl]-3-fluoro-phenyl]methoxy]pyrimidin-2-yl]-6-methoxy- pyrimidine (154 mg, 299 μmol, 30.2% yield) as a light-yellow solid.
1H NMR (600 MHz, DMSO-d6) δ 0.85 – 0.89 (m, 2H), 1.01 – 1.04 (m, 2H), 1.26 (t, 3H), 1.64 – 1.70 (m, 1H), 3.83 (s, 3H), 3.87 (q, 2H), 5.51 (s, 2H), 7.09 (d, 1H), 7.43 (d, 1H), 7.50 (d, 1H), 7.57 (t, 1H), 8.09 (s, 1H), 8.66 (s, 1H), 8.71 (d, 1H). MS (ESI): [M+H]+ m/z: calcd 515.21; found 515.2.

Step 1: The synthesis of 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[4-[1-ethyl-4- (trifluoromethyl)imidazol-2-yl]-3-fluoro-phenyl]methoxy]-5-methoxy-pyrimidine Synthesis of the starting [4-[1-ethyl-4-(trifluoromethyl)imidazol-2-yl]-3-fluoro- phenyl]methanol is described for Compound 131. [1057] NaH (36.9 mg, 924 μmol, 60% dispersion in mineral oil) was added to a stirred solution of [4-[1-ethyl-4-(trifluoromethyl)imidazol-2-yl]-3-fluoro-phenyl]methanol (242 mg, 840 μmol) in DMF (2 mL). The reaction mixture was stirred at room temperature for 30 min. 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-5-methoxy-4-methylsulfonyl-pyrimidine I-6 (282 mg, 840 μmol) was added to the reaction mixture. The resulting mixture was stirred at room temperature for 16 hr. The reaction mixture was diluted with a saturated aqueous solution of NH
4Cl (15 mL) and extracted with EtOAc (2×20 mL). The combined organic layers were washed with brine (10 mL), dried over anhydrous Na
2SO
4 and filtered. The filtrate was concentrated under reduced pressure. The residue was subjected to HPLC (0-5 min., 40-90% water - ACN, +0.1% vol. of 25% aq. NH
3, flow: 30 mL/min, column: XBridge C18 OBD 100×19 mm, 5 µm) to afford 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[4- [1-ethyl-4-(trifluoromethyl)imidazol-2-yl]-3-fluoro-phenyl]methoxy]-5-methoxy-pyrimidine (164 mg, 301 μmol, 35.9% yield) as a light-yellow solid.
1H NMR (600 MHz, DMSO-d
6) δ 0.83 – 0.88 (m, 2H), 0.99 – 1.03 (m, 2H), 1.26 (t, 3H), 1.66 – 1.72 (m, 1H), 3.82 (s, 3H), 3.87 (q, 2H), 3.95 (s, 3H), 5.51 (s, 2H), 7.43 (d, 1H), 7.48 (d, 1H), 7.58 (t, 1H), 8.09 (s, 1H), 8.43 (s, 1H), 8.63 (s, 1H). MS (ESI): [M+H]
+ m/z: calcd 545.22; found 545.2. Example 24 (Compound 4)
Step 1: Synthesis of 4-[5-hydroxy-3-(trifluoromethyl)pyrazol-1-yl]benzonitrile [1058] To a solution of tert-butyl-dimethyl-[[4-[4-(trifluoromethyl)-1H-imidazol-2- yl]phenyl]methoxy]silane (700 mg, 1.96 mmol) in DMF (10 mL) NaH (45.2 mg, 1.96 mmol, 60% dispersion in mineral oil) was added at room temperature. The mixture was stirred for 1 hr., and then iodoethane (337 mg, 2.16 mmol, 174 μL) was added. The solution was stirred at room temperature for 24 hr. The resulting mixture was diluted with water (20 mL) and extracted with DCM (20 mL). The organic layer was washed with water (20 mL × 2) and concentrated under reduced pressure to afford tert-butyl-[[4-[1-ethyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]-dimethyl-silane (580 mg, 1.51 mmol, 76.8% yield) as an yellow oil which was used in the next steps without further purification.
1H NMR (400 MHz, DMSO-d6) δ 0.10 (s, 6H), 0.92 (s, 9H), 1.31 (t, 3H), 4.08 (q, 2H), 4.79 (s, 2H), 7.34 (d, 2H), 7.61 (d, 2H), 8.00 (s, 1H). MS (ESI): [M+H]+ m/z: calcd 385.25; found 385.2. Step 2: Synthesis of [4-[1-ethyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methanol [1059] TBAF (1 M in THF, 1.54 mL, 1.54 mmol) was added to a solution of tert-butyl-[[4- [1-ethyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]-dimethyl-silane (480 mg, 1.25 mmol) in THF (20 mL) at room temperature. The resulting mixture was stirred at room temperature for 24 hr. The mixture was concentrated under reduced pressure. The residue was diluted with EtOAc (30 mL) and washed with water (20 mL × 2). Organic layer was dried over anhydrous Na
2SO
4 concentrated under reduced pressure to afford [4-[1-ethyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methanol (450 mg, crude) as an yellow oil which was used in the next steps without further purification. MS (ESI): [M+H]+ m/z: calcd 271.13; found 271.0. Step 3: Synthesis of 2-chloro-4-[[4-[1-ethyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]-5-methyl-pyrimidine [1060] To a solution of [4-[1-ethyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methanol (450 mg, crude) in DMF (5 mL), Potassium tert-butoxide (139 mg, 1.24 mmol) was added. The resulting solution was stirred at room temperature for 20 min and then was added dropwise into a solution of 2,4-dichloro-5-methyl-pyrimidine (212 mg, 1.30 mmol,) in DMF (5 mL) at room temperature. The resulting mixture was stirred for 24. The mixture was diluted with water (10 mL) and extracted with CH2Cl2 (20 mL). The organic layer was washed with water (10 mL × 2) and concentrated under reduced pressure. The residue was subjected to HPLC (0.5-6.5 min, 51% water - ACN; flow: 30 mL/min, column: Waters SunFire C18, 100 x 19 mm, 5 µm) to afford 2-chloro-4-[[4-[1-ethyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]-5-methyl-pyrimidine (190 mg, 479 μmol, 38.3% yield from tert-butyl- [[4-[1-ethyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]-dimethyl-silane) as a white solid. MS (ESI): [M+H]+ m/z: calcd 397.13; found 397.2. Step 4: Synthesis of 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[4-[1-ethyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]-5-methyl-pyrimidine [1061] 2-Chloro-4-[[4-[1-ethyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]-5-methyl- pyrimidine (82.0 mg, 207 μmol), (4-cyclopropyl-6-methoxy-pyrimidin-5-yl)boronic acid I-5 (56.1 mg, 289 μmol), XPhos Pd G3 (8.75 mg, 10.3 μmol) and potassium phosphate tribasic (132 mg, 620 μmol) were sequentially added to degassed dioxane (3 mL) with 5% of water under argon atmosphere. The resulting mixture was stirred at 100 °C for 24 hr. The mixture was cooled to room temperature, diluted with EtOAc (10 mL) and washed with water (10 mL × 2). To the organic layer SiliaMetS® Dimercaptotriazine was added. The resulting mixture was stirred for 30 min. The mixture was filtrated. The filtrate was concentrated under reduced pressure. The residue was subjected to HPLC (0.5-6.5 min, 40-90% water - ACN; flow: 30 mL/min, column: Waters SunFire C18, 100 x 19 mm, 5 µm) to afford 2-(4-cyclopropyl-6- methoxy-pyrimidin-5-yl)-4-[[4-[1-ethyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]- 5-methyl-pyrimidine (11.2 mg, 21.9 μmol, 10.6% yield) as an yellow solid. In another HPLC-fraction a side product 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-3-[[4- [1-ethyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methyl]-5-methyl-pyrimidin-4-one (13.2 mg, 25.9 μmol, 12.5% yield) was obtained as an yellow solid.
1H NMR (600 MHz, DMSO- d6) δ 0.81 – 0.87 (m, 2H), 0.98 – 1.03 (m, 2H), 1.31 (t, 3H), 1.63 – 1.69 (m, 1H), 2.22 (s, 3H), 3.82 (s, 3H), 4.08 (q, 2H), 5.51 (s, 2H), 7.58 (d, 2H), 7.64 (d, 2H), 8.02 (s, 1H), 8.54 (s, 1H), 8.64 (s, 1H). MS (ESI): [M+H]+ m/z: calcd 511.24; found 511.2. Example 25 (Compound 167)
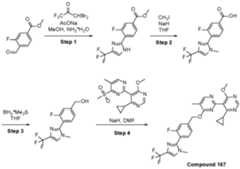
Step 1: The synthesis methyl 3-fluoro-4-[4-(trifluoromethyl)-1H-imidazol-2-yl]benzoate [1062] A mixture of 3,3-dibromo-1,1,1-trifluoro-propan-2-one (8.52 g, 31.6 mmol), sodium acetate (5.18 g, 63.1 mmol) in water (6.00 mL) was stirred at 100 °C for 45 min. The reaction mixture was cooled to room temperature. A solution of methyl 3-fluoro-4-formyl-benzoate (5.00 g, 27.5 mmol) in MeOH (150 mL) and aqueous ammonium hydroxide (30 mL, 25% wt.) were added to the reaction mixture. The resulting mixture was stirred at room temperature for 12 hr. The reaction mixture was concentrated under reduced pressure. The residue was diluted with water (100 mL) and extracted with EtOAc (3×150 mL). The combined organic layers were washed with brine (150 mL), dried over anhydrous Na2SO4 and concentrated under reduced pressure. The residue was subjected to flash-column chromatography (SiO
2, gradient chloroform - MTBE) to afford methyl 3-fluoro-4-[4- (trifluoromethyl)-1H-imidazol-2-yl]benzoate (1.00 g, 3.47 mmol, 12.6% yield) as a light- yellow solid.
1H NMR (500 MHz, CDCl
3) δ 3.95 (s, 3H), 7.52 (s, 1H), 7.85 (d, 1H), 7.94 (d, 1H), 8.40 (t, 1H), 10.09 (br., 1H). MS (ESI): [M+H]+ m/z: calcd 289.06; found 289.0. Step 2: The synthesis of 3-fluoro-4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]benzoic acid [1063] NaH (199 mg, 5.20 mmol, 60% dispersion in mineral oil) was added to a solution of methyl 3-fluoro-4-[4-(trifluoromethyl)-1H-imidazol-2-yl]benzoate (1.00 g, 3.47 mmol) in THF (30 mL). The reaction mixture was stirred at room temperature for 30 min. Iodomethane (640 mg, 4.51 mmol, 281 μL) was added to the reaction mixture. The resulting mixture was stirred for at room temperature for 15 hr. An additional portion of NaH (0.3 eq.) and MeI (0.5 eq.) were added to the reaction mixture. The resulting mixture was stirred at room temperature for 13 hr. The mixture was diluted with water (20 mL) and washed with EtOAc (50 mL). The water layer was separated, acidified with 6 N HCl to pH≈2 and extracted with EtOAc (50 mL). The organic layer was dried over anhydrous Na
2SO
4 and concentrated under reduced pressure to afford 3-fluoro-4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]benzoic acid (700 mg, 2.43 mmol, 70% yield) as a brown solid which was used in the next steps without further purification. MS (ESI): [M+H]+ m/z: calcd 289.06; found 289.0. Step 3: The synthesis of [3-fluoro-4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methanol [1064] Borane dimethyl sulfide complex (79.1 mg, 1.04 mmol, 98.7 μL) was added dropwise to a stirred solution of 3-fluoro-4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]benzoic acid (100 mg, 347 μmol) in THF (10 mL). The reaction mixture was stirred at room temperature for 15 hr. Methanol (10 mL) was added dropwise to the reaction mixture. The resulting mixture was heated at reflux for 5 hr. The resulting mixture was cooled to room temperature and concentrated under reduced pressure. The residue was diluted with saturated aqueous NaHCO
3 (10 mL) and extracted with EtOAc (40 mL). The organic layer was separated, dried over anhydrous Na
2SO
4 and concentrated under reduced pressure to afford [3-fluoro-4-[1- methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methanol (90.0 mg, 328 μmol, 94.6% yield) as an yellow solid which was used in the next steps without further purification.
1H NMR (400 MHz, CDCl
3) δ 3.60 (s, 3H), 4.60 (d, 2H), 5.45 (t, 1H), 7.28 – 7.35 (m, 2H), 7.54 (t, 1H), 7.99 (s, 1H). MS (ESI): [M+H]+ m/z: calcd 275.08; found 275.0. Step 4: The synthesis of 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[3-fluoro-4-[1- methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]-5-methyl-pyrimidine [1065] NaH (10.9 mg, 285 μmol, 60% dispersion in mineral oil) was added to a solution of [3-fluoro-4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methanol (65 mg, 237 μmol) in DMF (4 mL). The reaction mixture was stirred at room temperature for 30 min.2-(4- cyclopropyl-6-methoxy-pyrimidin-5-yl)-5-methyl-4-methylsulfonyl-pyrimidine I-7 (75.9 mg, 237 μmol) was added to the reaction mixture. The resulting mixture was stirred at room temperature for 15 hr. The mixture was diluted with water (20 mL) and extracted with EtOAc (30 mL). The organic layer was separated, dried over anhydrous Na
2SO
4 and concentrated under reduced pressure. The residue was subjected by HPLC (2-10 min, 20 - 50% water+FA (0.1% vol.) - ACN+FA (0.1% vol.); flow: 30 mL/min, column: Waters SunFire C18, 100 x 19 mm, 5 µm) to afford 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[3-fluoro-4-[1- methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]-5-methyl-pyrimidine (51.2 mg, 99.5 μmol, 42.0% yield) as a white solid.
1H NMR (400 MHz, DMSO-d6) δ 0.82 – 0.89 (m, 2H), 0.99 - 1.06 (m, 2H), 1.64 – 1.72 (m, 1H), 2.25 (s, 3H), 3.60 (s, 3H), 3.83 (s, 3H), 5.53 (s, 2H), 7.43 – 7.54 (m, 2H), 7.61 (t, 1H), 8.00 (s, 1H), 8.57 (s, 1H), 8.66 (s, 1H). MS (ESI): [M+H]+ m/z: calcd 515.21; found 515.2.
Step 1: The synthesis of 2-[4-(chloromethyl)phenyl]-1-methyl-4-(trifluoromethyl)imidazole [1066] Mesylchloride (11.7 g, 103 mmol, 7.93 mL) was added dropwise to a stirred solution of [4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methanol I-2b (15.0 g, 58.6 mmol) and TEA (11.9 g, 117 mmol, 16.3 mL) in DCM (400 mL) at 0 °C. The reaction mixture was stirred at room temperature for 12 hr. The reaction mixture was washed with a saturated aqueous solution of NaHCO3 (3×100 mL). The organic layer was dried over anhydrous Na
2SO
4 and filtered through a thin pad of silica gel. The filtrate was concentrated under reduced pressure. The residue was triturated with Hexane (100 mL) to afford 2-[4- (chloromethyl)phenyl]-1-methyl-4-(trifluoromethyl)imidazole (13.8 g, 50.1 mmol, 85.5% yield) as a white solid.
1H NMR (500 MHz, DMSO-d6) δ 3.78 (s, 3H), 4.83 (s, 2H), 7.56 (d, 2H), 7.72 (d, 2H), 7.93 (s, 1H). MS (ESI): [M+H]+ m/z: calcd 275.06; found 275.2 Step 2: The synthesis of S-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methyl]ethanethioate [1067] 2-[4-(Chloromethyl)phenyl]-1-methyl-4-(trifluoromethyl)imidazole (450 mg, 1.64 mmol) and potassium thioacetate (225 mg, 1.97 mmol) were mixed in DMF (15 mL). The reaction mixture was stirred at room temperature for 10 hr. The reaction mixture was poured into ice-cold water (20 mL) and extracted with EtOAc (40 mL). The organic layer was washed with water (20 mL) and brine (3×20 mL) and concentrated under reduced pressure to afford S-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methyl]ethanethioate (400 mg, 1.27 mmol, 77.7% yield) as a brown solid which was used in the next steps without further purification. MS (ESI): [M+H]+ m/z: calcd 315.10; found 315.2 Step 3: The synthesis of 2-chloro-5-methyl-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methylsulfanyl]pyrimidine [1068] S-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methyl]ethanethioate (400 mg, 1.27 mmol) and 2,4-dichloro-5-methyl-pyrimidine (189 mg, 1.16 mmol) were mixed in THF (4.0 mL) and MeOH (15 mL). The resulting mixture was cooled to 0°C. K
2CO
3 (176 mg, 1.27 mmol) was added to the mixture. The reaction mixture was stirred at 0°C for 3 hr. The reaction mixture was allowed to warm to room temperature, diluted with EtOAc (40 mL) and washed with water (30 mL) an brine (3×20 mL). The organic layer was concentrated under reduced pressure to afford 2-chloro-5-methyl-4-[[4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methylsulfanyl]pyrimidine (500 mg, crude) as a light- yellow solid which was used in the next step without further purification. MS (ESI): [M+H]+ m/z: calcd 399.09; found 399.0 Step 4: The synthesis of 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-5-methyl-4-[[4-[1- methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methylsulfanyl]pyrimidine [1069] (4-cyclopropyl-6-methoxy-pyrimidin-5-yl)boronic acid I-5 (523 mg, 2.70 mmol), 2- chloro-5-methyl-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methylsulfanyl]pyrimidine (500 mg, 1.08 mmol) and potassium phosphate tribasic (572 mg, 2.70 mmol) were mixed in a degassed mixture of water (6.0 mL) and dioxane (30 mL). RuPhos Pd G4 (91.7 mg, 108 μmol) was added to the mixture. The reaction mixture was stirred at 90 °C for 15 hr. The reaction mixture was cooled to room temperature and concentrated under reduced pressure. The residue was diluted with water (10 mL) and extracted with EtOAc (4×20 mL). The combined organic layers were filtered through a thin pad of silica gel. The filtrate was dried over anhydrous Na
2SO
4 and concentrated under reduced pressure. The residue was subjected to column chromatography (Interchim 40 g SiO
2, HEX-EtOAc from 0-100%, flow rate = 50 mL/min, cv=56.7) to afford 2-(4- cyclopropyl-6-methoxy-pyrimidin-5-yl)-5-methyl-4-[[4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methylsulfanyl]pyrimidine (325 mg, 634 μmol, 58.8% yield) as a white solid.
1H NMR (600 MHz, DMSO-d6) δ 0.87 – 0.92 (m, 2H), 1.04 – 1.08 (m, 2H), 1.66 – 1.72 (m, 1H), 2.22 (s, 3H), 3.76 (s, 3H), 3.87 (s, 3H), 4.55 (s, 2H), 7.50 (d, 2H), 7.62 (d, 2H), 7.92 (s, 1H), 8.51 (s, 1H), 8.69 (s, 1H). MS (ESI): [M+H]+ m/z: calcd 513.20; found 513.2.

Step 1: Synthesis of 2-chloro-4-[[4-[1-ethyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]-5-methoxy-pyrimidine Synthesis of the starting [4-[1-ethyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methanol is described for Compound 4. [1070] To a solution of [4-[1-ethyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methanol (250 mg, 833 μmol) in DMF (2 mL) potassium tert-butoxide (98.1 mg, 874 μmol) was added at room temperature. The mixture was stirred for 20 min and added dropwise into solution of 2,4-dichloro-5-methoxy-pyrimidine (164 mg, 916 μmol) in DMF (2 mL) at room temperature. The resulting mixture was stirred for 24 hr. The mixture was diluted with water (10 mL) and extracted with CH2Cl2 (20 ml). The organic layer was washed with water (10 mL × 2), dried over anhydrous Na
2SO
4 and concentrated under reduced pressure to afford 2- chloro-4-[[4-[1-ethyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]-5-methoxy- pyrimidine (320 mg, crude) as an yellow oil which was used in the next steps without further purification. MS (ESI): [M+H]+ m/z: calcd 413.12; found 413.0. Step 2: Synthesis of 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[4-[1-ethyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]-5-methoxy-pyrimidine [1071] 2-chloro-4-[[4-[1-ethyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]-5- methoxy-pyrimidine (320 mg, crude), (4-cyclopropyl-6-methoxy-pyrimidin-5-yl)boronic acid I-5 (60.2 mg, 310 μmol), potassium phosphate tribasic (197 mg, 930 μmol) and XPhos Pd G3 (13.12 mg, 15.5 μmol) were mixed in a degassed mixture of water (0.25 mL) and dioxane (5 mL). The reaction mixture was stirred under argon atmosphere at 100 °C for 24 hr. The reaction mixture was cooled room temperature, diluted with EtOAc (10 mL) and washed with water (2 × 10 mL) and brine (10 mL). To the obtained organic phase SiliaMetS® Dimercaptotriazine (20 mg) was added. The resulting mixture was stirred for 30 min and filtered. The filtrate was evaporated under reduced pressure. The residue was subjected to HPLC (0.5-6.5 min, 45% water - ACN; flow: 30 mL/min, column: Waters SunFire C18, 100 x 19 mm, 5 µm) to afford 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[4-[1-ethyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]-5-methoxy-pyrimidine (56.0 mg, 106 μmol, 12.7% yield from [4-[1-ethyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methanol) as a white solid.
1H NMR (600 MHz, DMSO-d6) δ 0.82 – 0.88 (m, 2H), 0.98 – 1.03 (m, 2H), 1.31 (t, 3H), 1.66 – 1.73 (m, 1H), 3.83 (s, 3H), 3.93 (s, 3H), 4.08 (q, 2H), 5.49 (s, 2H), 7.57 (d, 2H), 7.64 (d, 2H), 8.02 (s, 1H), 8.42 (s, 1H), 8.63 (s, 1H). MS (ESI): [M+H]+ m/z: calcd 527.23; found 527.0.
Step 1: Synthesis of 2-chloro-5-ethyl-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidine [1072] 2,4-dichloro-5-ethyl-pyrimidine (155 mg, 878 μmol) and [4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methanol I-2b (150 mg, 585 μmol) were mixed in toluene (10 mL). The mixture was cooled to 0 °C then potassium tert-butoxide (98.5 mg, 878 μmol) was added portion wise. The resulting mixture was stirred at room temperature for 16 hr. The reaction mixture was diluted with water (30 mL) and extracted with MTBE (20 mL). The organic layer was washed with brine (30 mL), dried over anhydrous Na
2SO
4 and concentrated under reduced pressure to afford 2-chloro-5-ethyl-4-[[4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidine (230 mg, 580 μmol, 99.0% yield) as a light-yellow solid which was used in the next steps without further purification. MS (ESI): [M+H]+ m/z: calcd 397.13; found 397.2. Step 2: Synthesis of 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-5-ethyl-4-[[4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidine [1073] 2-chloro-5-ethyl-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidine (130 mg, 328 μmol), 4-cyclopropyl-6-methoxy-5-(4,4,5,5- tetramethyl-1,3,2-dioxaborolan-2-yl)pyrimidine (181 mg, 655 μmol), Cs
2CO
3 (320 mg, 983 μmol) and XPhos Pd G3 (13.9 mg, 16.4 μmol) were mixed in degassed dioxane (4 mL) and water (1 mL) under inert atmosphere of argon. The reaction mixture was stirred at 90 °C and for 16 hr. The reaction mixture was cooled to room temperature and concentrated under reduced pressure, the residue was diluted with EtOAc (20 mL), washed with brine (10 mL), dried over anhydrous Na
2SO
4 and concentrated under reduced pressure. The residue was subjected to HPLC purification (0-5 min., 40-90% water – ACN, +0.1% vol. of 25% aq. NH
3, flow: 30 mL/min, column: YMC Triart C18100 × 20 mm, 5 μm) to afford 2-(4-cyclopropyl- 6-methoxy-pyrimidin-5-yl)-5-ethyl-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidine (40.0 mg, 78.4 μmol, 23.9% yield) as a light-yellow solid.
1H NMR (600 MHz, DMSO-d6) δ 0.83 – 0.87 (m, 2H), 0.99 – 1.03 (m, 2H), 1.23 (t, 3H), 1.63 – 1.69 (m, 1H), 2.65 (q, 2H), 3.77 (s, 3H), 3.82 (s, 3H), 5.51 (s, 2H), 7.57 (d, 2H), 7.73 (d, 2H), 7.92 (s, 1H), 8.55 (s, 1H), 8.64 (s, 1H). MS (ESI): [M+H]+ m/z: calcd 511.24; found 511.2. Example 29 (Compound 86)
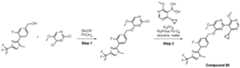
Step 1: The synthesis 2-chloro-4-[[3-fluoro-4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]-5-methoxy-pyrimidine Synthesis of the starting [3-fluoro-4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methanol is described for compound 97. [1074] Potassium tert-butoxide (135 mg, 1.20 mmol) was added to a solution of [3-fluoro-4- [1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methanol (300 mg, 1.09 mmol) in toluene (4 mL) and the reaction mixture was stirred at room temperature for 30 min.2,4-Dichloro-5- methoxy-pyrimidine (196 mg, 1.09 mmol) was added to the reaction mixture. The reaction mixture was stirred at room temperature for 15 hr. The reaction mixture was diluted with water (20 mL) and extracted with EtOAc (30 mL). The organic layer was separated, dried over anhydrous Na
2SO
4 and concentrated under reduced pressure. The residue was subjected to flash-column chromatography (SiO
2, gradient chloroform - MTBE) to afford 2-chloro-4- [[3-fluoro-4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]-5-methoxy- pyrimidine (170 mg, 0.408 mmol, 37.2% yield) as a white solid. MS (ESI): [M+H]+ m/z: calcd 417.08; found 417.0 Step 2: the synthesis of 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[3-fluoro-4-[1- methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]-5-methoxy-pyrimidine [1075] 2-chloro-4-[[3-fluoro-4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]- 5-methoxy-pyrimidine (100 mg, 240 μmol), (4-cyclopropyl-6-methoxy-pyrimidin-5- yl)boronic acid I-5 (69.8 mg, 360 μmol), potassium phosphate tribasic (153 mg, 720 μmol) and RuPhosPdG4 (15.3 mg, 18.0 μmol) were mixed in degassed dioxane (5 mL) and water (1 mL) under argon atmosphere at room temperature. The reaction mixture was stirred at 90 °C for 15 hr. The reaction mixture was cooled to room temperature and concentrated under reduced temperature. The residue was diluted with water (10 mL) and extracted with EtOAc (3×20 mL). The combined organic layers were dried over anhydrous Na
2SO
4 and concentrated under reduced pressure. The residue was subjected to HPLC (2-10 min, 30% water+FA (0.1% vol.) - ACN+FA (0.1% vol.); flow: 30 mL/min, column: Waters SunFire C18, 100 x 19 mm, 5 µm), then repurified by SFC (column: SiO
2, 19×100 mm, 5 µm; eluent: CO2-IPA 70-30; flow: 50 mL/min) to afford 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4- [[3-fluoro-4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]-5-methoxy- pyrimidine (9.6 mg, 18.1 μmol, 7.5% yield) as an off-white solid.
1H NMR (600 MHz, DMSO-d6) δ 0.83 – 0.87 (m, 2H), 0.99 - 1.02 (m, 2H), 1.67 – 1.71 (m, 1H), 3.59 (s, 3H), 3.82 (s, 3H), 3.95 (s, 3H), 5.50 (s, 2H), 7.43 (d, 1H), 7.47 (d, 1H), 7.60 (t, 1H), 7.99 (s, 1H), 8.43 (s, 1H), 8.64 (s, 1H). MS (ESI): [M+H]+ m/z: calcd 531.18; found 531.2.
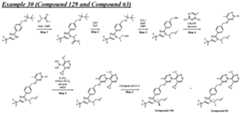
Step 1: The synthesis of methyl 2-[2-[4-[[tert-butyl(dimethyl)silyl]oxymethyl]phenyl]-4- (trifluoromethyl)imidazol-1-yl]propanoate [1076] NaH (185 mg, 4.63 mmol, 60% dispersion in mineral oil) was added to a solution of tert-butyl-dimethyl-[[4-[4-(trifluoromethyl)-1H-imidazol-2-yl]phenyl]methoxy]silane (1.50 g, 4.21 mmol) in DMF (5.0 mL). The reaction mixture was stirred at room temperature for 15 min. Methyl 2-chloropropanoate (670 mg, 5.47 mmol) was added to the reaction mixture. The resulting mixture was stirred at room temperature overnight. The reaction mixture was diluted with water (10 mL) and extracted with EtOAc (10 mL). The organic layer was washed with water (5 mL) and brine (5 mL) and concentrated under reduced pressure to afford methyl 2-[2-[4-[[tert-butyl(dimethyl)silyl]oxymethyl]phenyl]-4- (trifluoromethyl)imidazol-1-yl]propanoate (1.80 g, 4.07 mmol, 96.7% yield) as a light-yellow oil which was used in the next steps without further purification. MS (ESI): [M+H]+ m/z: calcd 443.25; found 443.0 Step 2: The synthesis of 2-[2-[4-[[tert-butyl(dimethyl)silyl]oxymethyl]phenyl]-4- (trifluoromethyl)imidazol-1-yl]propan-1-ol [1077] A solution of methyl 2-[2-[4-[[tert-butyl(dimethyl)silyl]oxymethyl]phenyl]-4- (trifluoromethyl)imidazol-1-yl]propanoate (1.80 g, 4.07 mmol) in THF (20 mL) was added dropwise to a vigorously stirred suspension of LAH (168 mg, 4.94 mmol) in THF (50 mL) at 0°C. The reaction mixture was stirred at 0 °C for 2 hr. An aqueous NaOH (1 mL, 10% wt.) was added dropwise to the reaction mixture. The resulting mixture was stirred for 30 min then solids were filtered out. The filtrate was concentrated under reduced pressure to afford 2-[2-[4-[[tert-butyl(dimethyl)silyl]oxymethyl]phenyl]-4-(trifluoromethyl)imidazol-1- yl]propan-1-ol (1.00 g, 2.41 mmol, 73.2% yield) as a light-yellow oil which was used in the next steps without further purification.
1H NMR (500 MHz, CDCl
3) δ 0.10 (s, 6H), 0.94 (s, 9H), 1.40 (d, 3H), 3.77 (d, 2H), 4.50 – 4.58 (m, 1H), 4.77 (s, 2H), 7.40 (d, 2H), 7.45 (s, 1H) 7.54 (d, 2H). Step 3: The synthesis of [4-[1-(2-methoxy-1-methyl-ethyl)-4-(trifluoromethyl)imidazol-2- yl]phenyl]methanol [1078] NaH (191 mg, 4.78 mmol, 60% dispersion in mineral oil) was added to a solution of 2-[2-[4-[[tert-butyl(dimethyl)silyl]oxymethyl]phenyl]-4-(trifluoromethyl)imidazol-1- yl]propan-1-ol (1.80 g, 4.34 mmol) in DMF (10 mL). The reaction mixture was stirred at room temperature for 30 min. Iodomethane (801 mg, 5.64 mmol, 351 μL) was added to the reaction mixture. The resulting mixture was stirred at room temperature overnight. The reaction mixture was diluted with water (20 mL) and extracted with EtOAc (2×10 mL). Combined organic layers were washed with water (10 mL) and brine (10 mL) and concentrated under reduced pressure. The residue was subjected to flash column chromatography (SiO
2, gradient DCM - EtOAc) to afford [4-[1-(2-methoxy-1-methyl-ethyl)- 4-(trifluoromethyl)imidazol-2-yl]phenyl]methanol (240 mg, 764 μmol, 17.7% yield) as an yellowish oil. MS (ESI): [M+H]+ m/z: calcd 315.16; found 315.0. Step 4: The synthesis of 2-chloro-4-[[4-[1-(2-methoxy-1-methyl-ethyl)-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]-5-methyl-pyrimidine [1079] Potassium tert-butoxide (66.0 mg, 588 μmol) was added to a solution of [4-[1-(2- methoxy-1-methyl-ethyl)-4-(trifluoromethyl)imidazol-2-yl]phenyl]methanol (240 mg, 764 μmol) in dioxane (10 mL). The reaction mixture was stirred at room temperature for 15 min. 2,4-dichloro-5-methyl-pyrimidine (87.1 mg, 535 μmol) was added to the reaction mixture. The resulting mixture was stirred at room temperature for 12 hr. The resulting mixture was diluted with water (15 mL) and extracted with EtOAc (20 mL). The resulting organic layer was washed with brine (5.0 mL), dried over anhydrous Na
2SO
4 and filtered. The filtrate was concentrated under reduced pressure. The residue was subjected to HPLC (0.5-5.5-9.0 min, 45-45-48% water - ACN; flow: 30 mL/min, column: Waters SunFire C18, 100 x 19 mm, 5 µm) to afford 2-chloro-4-[[4-[1-(2-methoxy-1-methyl-ethyl)-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]-5-methyl-pyrimidine (94.0 mg, 213 μmol, 39.9% yield) as a light-yellow solid. MS (ESI): [M+H]+ m/z: calcd 441.16; found 441.2. Step 5: The synthesis of 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[4-[1-(2-methoxy-1- methyl-ethyl)-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]-5-methyl-pyrimidine [1080] (4-cyclopropyl-6-methoxy-pyrimidin-5-yl)boronic acid I-5 (33.0 mg, 170 μmol), 2- chloro-4-[[4-[1-(2-methoxy-1-methyl-ethyl)-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]-5-methyl-pyrimidine (50.0 mg, 113 μmol), potassium phosphate tribasic (72.2 mg, 340 μmol) and XPhos Pd G3 (1.13 μmol) were mixed in degassed dioxane (2.0 mL) and water (300 μL) under argon atmosphere. The reaction mixture was stirred at 95 °C overnight. The reaction mixture was cooled to room temperature and concentrated under reduced pressure. The residue was diluted with EtOAc (20 mL) and washed with brine (10 mL). The organic layer was dried over anhydrous Na
2SO
4 and filtered. SiliaMetS® Dimercaptotriazine (30.0 mg) was added to the filtrate. The resulting mixture was stirred at room temperature for 15 min, then solids were filtered out. The filtrate was concentrated under reduced pressure. The residue was subjected to HPLC (0.5-6.5min, 53%, water - ACN; flow 30ml/min; column SunFireC18100x19 mm 5 μm) to afford 2-(4-cyclopropyl-6- methoxy-pyrimidin-5-yl)-4-[[4-[1-(2-methoxy-1-methyl-ethyl)-4-(trifluoromethyl)imidazol- 2-yl]phenyl]methoxy]-5-methyl-pyrimidine (20.0 mg, 36.1 μmol, 31.8% yield) as a brown solid. MS (ESI): [M+H]+ m/z: calcd 555.27; found 555.2. Step 6: The separation of rel-(R)-4'-cyclopropyl-6'-methoxy-4-((4-(1-(1-methoxypropan-2- yl)-4-(trifluoromethyl)-1H-imidazol-2-yl)benzyl)oxy)-5-methyl-2,5'-bipyrimidine (129) and rel-(S)-4'-cyclopropyl-6'-methoxy-4-((4-(1-(1-methoxypropan-2-yl)-4-(trifluoromethyl)-1H- imidazol-2-yl)benzyl)oxy)-5-methyl-2,5'-bipyrimidine (63) [1081] Racemic 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[4-[1-(2-methoxy-1-methyl- ethyl)-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]-5-methyl-pyrimidine (20.0 mg, 36.1 μmol) was subjected to chiral HPLC (column: Chiralpak AD-H V, 250×20 mm, 5 μm; mobile phase: Hexane-IPA-MeOH, 90-5-5; flow: 13 mL/min) to afford rel-(R)-4'- cyclopropyl-6'-methoxy-4-((4-(1-(1-methoxypropan-2-yl)-4-(trifluoromethyl)-1H-imidazol- 2-yl)benzyl)oxy)-5-methyl-2,5'-bipyrimidine (5.0 mg, 9.02 μmol, 25.0% yield) and rel-(S)-4'- cyclopropyl-6'-methoxy-4-((4-(1-(1-methoxypropan-2-yl)-4-(trifluoromethyl)-1H-imidazol- 2-yl)benzyl)oxy)-5-methyl-2,5'-bipyrimidine (5.0 mg, 9.02 μmol, 25% yield) as a white solids. rel-(R)-4'-cyclopropyl-6'-methoxy-4-((4-(1-(1-methoxypropan-2-yl)-4-(trifluoromethyl)-1H- imidazol-2-yl)benzyl)oxy)-5-methyl-2,5'-bipyrimidine (129):
1H NMR (600 MHz, DMSO-d6) δ 0.82 – 0.86 (m, 2H), 0.99 – 1.03 (m, 2H), 1.35 (d, 3H), 1.63 – 1.69 (m, 1H), 2.22 (s, 3H), 3.12 (s, 3H), 3.50 – 3.55 (m, 1H), 3.59 – 3.65 (m, 1H), 3.82 (s, 3H), 4.47 – 4.53 (m, 1H), 5.51 (s, 2H), 7.57 – 7.61 (m, 4H), 8.15 (s, 1H), 8.54 (s, 1H), 8.64 (s, 1H). MS (ESI): [M+H]+ m/z: calcd 555.27; found 555.2 Enantiopurity: 99% (column: Chiralpak AD-H, 250 × 4.6 mm, 5 μm; mobile phase: Hexane- IPA-MeOH, 90-5-5; flow: 0.6 mL/min; RT=47.3 min) rel-(S)-4'-cyclopropyl-6'-methoxy-4-((4-(1-(1-methoxypropan-2-yl)-4-(trifluoromethyl)-1H- imidazol-2-yl)benzyl)oxy)-5-methyl-2,5'-bipyrimidine (63):
1H NMR (600 MHz, DMSO-d6) δ 0.82 – 0.86 (m, 2H), 0.99 – 1.03 (m, 2H), 1.35 (d, 3H), 1.63 – 1.69 (m, 1H), 2.22 (s, 3H), 3.12 (s, 3H), 3.50 – 3.55 (m, 1H), 3.59 – 3.65 (m, 1H), 3.82 (s, 3H), 4.47 – 4.53 (m, 1H), 5.51 (s, 2H), 7.57 – 7.61 (m, 4H), 8.15 (s, 1H), 8.54 (s, 1H), 8.64 (s, 1H). MS (ESI): [M+H]+ m/z: calcd 555.27; found 555.2. Enantiopurity: 100% (column: Chiralpak AD-H, 250 × 4.6 mm, 5 μm; mobile phase: Hexane- IPA-MeOH, 90-5-5; flow: 0.6 mL/min; RT=39.1 min). Example 31 (Compound 168)
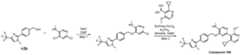
Step 1: Synthesis of 2-chloro-N-methyl-4-((4-(1-methyl-4-(trifluoromethyl)-1H-imidazol-2- yl)benzyl)oxy)pyrimidin-5-amine [1082] To a solution of [4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methanol I-2b (1.00 g, 3.90 mmol) in DMF (50 mL), NaH (98.7 mg, 4.11 mmol, 60% dispersion in mineral oil) was added. The mixture was stirred for 1 hr. then 2,4-dichloro-N-methyl-pyrimidin-5- amine (764 mg, 4.29 mmol) was added. The resulting mixture was stirred at 70 °C for 16 hr. The mixture was cooled to room temperature, quenched with water (50 mL), diluted with EtOAc (75 mL) and washed with water (50 mL × 2) and brine (50 mL). The organic layer was concentrated under reduced pressure to afford 2-chloro-N-methyl-4-((4-(1-methyl-4- (trifluoromethyl)-1H-imidazol-2-yl)benzyl)oxy)pyrimidin-5-amine (1.8 g, crude) which was used in the next steps without further purification. MS (ESI): [M+H]+ m/z: calcd 398.1; found 398.0. Step 2: Synthesis of 4'-cyclopropyl-6'-methoxy-N-methyl-4-((4-(1-methyl-4-(trifluoromethyl)- 1H-imidazol-2-yl)benzyl)oxy)-[2,5'-bipyrimidin]-5-amine [1083] 2-Chloro-N-methyl-4-((4-(1-methyl-4-(trifluoromethyl)-1H-imidazol-2- yl)benzyl)oxy)pyrimidin-5-amine (500 mg, 777 μmol), (4-cyclopropyl-6-methoxy-pyrimidin- 5-yl)boronic acid I-5 (211 mg, 1.09 mmol), Potassium phosphate tribasic (495 mg, 2.33 mmol) and RuPhos Pd G4 (6.61 mg, 7.77 μmol) were mixed in degassed dioxane (100 mL) with 5% of water under argon atmosphere. The resulting mixture was stirred at 60 °C for 16 hr. The mixture was cooled to room temperature, diluted with EtOAc (80 mL) and washed with water (50 mL × 2). To the obtained solution SiliaMetS® Dimercaptotriazine (300 mg) was added, and the mixture was stirred for 1 hr. The mixture was filtered. The filtrate was concentrated under reduced pressure. The residue was subjected to HPLC (0.5-6.5 min., 40- 90% water – MeOH, +0.1% vol. of 25% aq. NH
3, flow: 30 mL/min, column: XBridge 100 ×19 mm, 5 µm) to afford 4'-cyclopropyl-6'-methoxy-N-methyl-4-((4-(1-methyl-4- (trifluoromethyl)-1H-imidazol-2-yl)benzyl)oxy)-[2,5'-bipyrimidin]-5-amine (59.0 mg, 115 μmol, 9.69% yield from 2,4-dichloro-N-methyl-pyrimidin-5-amine) as a light yellow solid.
1H NMR (600 MHz, DMSO-d6) δ 0.79 – 0.85 (m, 2H), 0.96 – 1.01 (m, 2H), 1.68 – 1.76 (m, 1H), 2.80 (d, 3H), 3.76 (s, 3H), 3.81 (s, 3H), 5.50 (s, 2H), 5.72 (q, 1H), 7.62 (d, 2H), 7.71 (d, 2H), 7.86 (s, 1H), 7.92 (s, 1H), 8.59 (s, 1H). MS (ESI): [M+H]+ m/z: calcd 512.23; found 512.2.
Step 1: The synthesis of methyl 4-[1-cyclopropyl-4-(trifluoromethyl)imidazol-2-yl]benzoate [1084] 2,2′-Bipyridine (751 mg, 4.81 mmol), pyridine (293 mg, 3.70 mmol, 299 μL), Na
2CO
3 (785 mg, 7.40 mmol) and copper acetate (874 mg, 4.81 mmol) were added sequentially to a solution of methyl 4-[4-(trifluoromethyl)-1H-imidazol-2-yl]benzoate I-3a (1.00 g, 3.70 mmol) and cyclopropylboronic acid (477 mg, 5.55 mmol) in THF (10 mL) and DCM (10 mL). The reaction mixture was stirred at room temperature for 48 hr. Additional portion of cyclopropylboronic acid (954 mg, 11.1 mmol) was added to the reaction mixture. The mixture was stirred at room temperature for 48 hr. The mixture was quenched with water (20 mL) and extracted with EtOAc (2×20 mL). The combined organic layers were washed with water (20 mL) and brine (2×10 mL) and concentrated under reduced pressure to afford methyl 4-[1-cyclopropyl-4-(trifluoromethyl)imidazol-2-yl]benzoate (800 mg, 2.58 mmol, 69.7% yield) as a white solid which was used in the next steps without further purification. MS (ESI): [M+H]+ m/z: calcd 311.12; found 311.2 Step 2: The synthesis of [4-[1-cyclopropyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methanol [1085] A solution of methyl 4-[1-cyclopropyl-4-(trifluoromethyl)imidazol-2-yl]benzoate (500 mg, 1.61 mmol) in THF (2 mL) was added dropwise to a vigorously stirred suspension of LAH (184 mg, 4.83 mmol) in THF (10 mL) under argon atmosphere at 0 °C. The reaction mixture was stirred at 0 °C for 3 hr. The mixture was quenched by dropwise addition of a mixture of water and THF (1:1, 5 mL) at 0 °C. The resulting mixture was stirred at room temperature for 30 min and then solids were filtered out. The filtrate was concentrated under reduced pressure to afford [4-[1-cyclopropyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methanol (400 mg, 1.42 mmol, 87.9% yield) as a light-yellow solid which was used in the next steps without further purification.
1H NMR (500 MHz, CDCl
3) δ 0.83 – 0.91 (m, 2H), 1.01 – 1.09 (m, 2H), 2.02 (br, 1H), 3.45 – 3.55 (m, 1H), 4.76 (s, 2H), 7.34 (s, 1H), 7.45 (d, 2H), 7.80 (d, 2H). MS (ESI): [M+H]+ m/z: calcd 283.13; found 283.0 Step 3: The synthesis of 2-bromo-4-[[4-[1-cyclopropyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]-5-methyl-pyrimidine [1086] Potassium tert-butoxide (167 mg, 1.49 mmol) was added to a solution of [4-[1- cyclopropyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methanol (400 mg, 1.42 mmol) in THF (5 mL). The reaction mixture was stirred at room temperature for 1 hr. The obtained reaction mixture was added dropwise to a solution of 2,4-dibromo-5-methyl-pyrimidine (375 mg, 1.49 mmol) in THF (5.00 mL). The resulting mixture was stirred at room temperature for 12 hr. The mixture was diluted with EtOAc (10.0 mL) and washed with water (2×5 mL) and brine (10 mL). The organic layer was dried over anhydrous Na
2SO
4 and concentrated under reduced pressure to afford 2-bromo-4-[[4-[1-cyclopropyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]-5-methyl-pyrimidine (700 g, crude) as a light-yellow solid which was used in the next steps without further purification. MS (ESI): [M+H]+ m/z: calcd 453.08; found 453.0 Step 4: The synthesis of 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[4-[1-cyclopropyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]-5-methyl-pyrimidine [1087] 2-Bromo-4-[[4-[1-cyclopropyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]-5- methyl-pyrimidine (200 mg, crude), (4-cyclopropyl-6-methoxy-pyrimidin-5-yl)boronic acid I-5 (41.1 mg, 212 μmol), potassium phosphate tribasic (74.9 mg, 353 μmol) and XPhos Pd G3 (7.47 mg, 8.83 μmol) were mixed in degassed dioxane (5 mL) under argon atmosphere at room temperature. The reaction mixture was stirred at 90 °C for 6 hr. The reaction mixture was cooled to room temperature and concentrated under reduced pressure. The residue was diluted with H2O (10 mL) and extracted with EtOAc (2 × 10 mL). The combined organic layers were washed with water (10 mL) and brine (10 mL). The organic layer was dried over anhydrous Na
2SO
4 and concentrated under reduced pressure. The residue was subjected to HPLC (0.5 - 6.5 min, 52% water - ACN; flow: 30 mL/min, column: Waters SunFire C18, 100 x 19 mm, 5 µm then 0.5 - 6.5 min, 63% water - methanol; flow: 30 mL/min, column: Waters SunFire C18, 100 x 19 mm, 5 µm) to afford (4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4- [[4-[1-cyclopropyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]-5-methyl-pyrimidine (4.50 mg, 8.61 μmol, 2.1% yield from [4-[1-cyclopropyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methanol) as a white solid.
1H NMR (600 MHz, DMSO-d6) δ 0.83 – 0.86 (m, 2H), 0.88 – 0.91 (m, 2H), 0.95 – 1.03 (m, 4H), 1.62 – 1.72 (m, 1H), 2.22 (s, 3H), 3.63 – 3.73 (m, 1H), 3.82 (s, 3H), 5.51 (s, 2H), 7.57 (d, 2H), 7.90 (d, 2H), 7.93 (s, 1H), 8.55 (s, 1H), 8.64 (s, 1H). MS (ESI): [M+H]+ m/z: calcd 523.24; found 524.2. Example 33 (Compound 130)

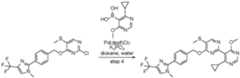

Step 1: The synthesis of 5-(methylthio)pyrimidine-2,4-diol [1088] 5-bromopyrimidine-2,4-diol (3.00 g, 15.7 mmol) was refluxed in aqueous sodium methanethiolate (30.0 g, 89.9 mmol, 21% wt. in water) for 5 hr. The mixture was cooled to 10 °C and slowly adjusted to pH ≥ 5 with 12 mL of conc. HCl. The precipitate formed was filtered off and dried on air to afford 5-(methylthio)pyrimidine-2,4-diol (2.00 g, 12.6 mmol, 80.6% yield) as white solid which was used in the next steps without further purification. MS (ESI): [M+H]+ m/z: calcd 159.02; found 159.0. Step 2: The synthesis of 2,4-dichloro-5-(methylthio)pyrimidine [1089] 5-methylsulfanylpyrimidine-2,4-diol (1.25 g, 7.90 mmol) was mixed with phosphoryl chloride (25.0 g, 163 mmol) and dimethylaniline (250 mg, 2.06 mmol, 262 μL). The reaction mixture was stirred at 100 °C for 20 hr. The reaction mixture was cooled to room temperature and concentrated in vacuo. The residue was quenched with ice. The precipitate formed was filtered off and dissolved in DCM (50 mL). The solution was dried over anhydrous Na
2SO
4 and the solvent was evaporated in vacuo to afford 2,4-dichloro-5-(methylthio)pyrimidine (1.20 g, 6.15 mmol, 77.9% yield) as a grey solid which was used in the next steps without further purification.
1H NMR (400 MHz, CDCl
3) δ 2.55 (s, 3H), 8.29 (s, 1H). GCMS: [M]+ m/z: calcd 193.95; found 194. Step 3: The synthesis of 2-chloro-4-((4-(1-methyl-4-(trifluoromethyl)-1H-imidazol-2- yl)benzyl)oxy)-5-(methylthio)pyrimidine [1090] 2,4-dichloro-5-methylsulfanyl-pyrimidine (0.500 g, 2.56 mmol), [4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methanol I-2b (657 mg, 2.56 mmol) and Cs
2CO
3 (919 mg, 2.82 mmol) were mixed in Acetonitrile (15 mL). The reaction mixture was stirred at 80 °C for 4 hr. The reaction mixture was cooled to room temperature and filtered. The filtrate was concentrated under reduced pressure. The residue was subjected to flash-column chromatography (SiO
2, gradient MTBE-chloroform) to afford 2-chloro-4-((4-(1-methyl-4- (trifluoromethyl)-1H-imidazol-2-yl)benzyl)oxy)-5-(methylthio)pyrimidine (90.0 mg, 217 μmol, 8.46% yield) as a yellow solid.
1H NMR (500 MHz, CDCl
3) δ 2.45 (s, 3H), 3.79 (s, 3H), 5.57 (s, 3H), 7.32 (s, 1H), 7.58 (d, 2H), 7.68 (d, 2H), 8.13 (s, 1H). MS (ESI): [M+H]+ m/z: calcd 415.08; found 415.0. Step 4: The synthesis of 4'-cyclopropyl-6'-methoxy-4-((4-(1-methyl-4-(trifluoromethyl)-1H- imidazol-2-yl)benzyl)oxy)-5-(methylthio)-2,5'-bipyrimidine [1091] 2-chloro-5-methylsulfanyl-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidine (90.0 mg, 217 μmol), (4-cyclopropyl-6-methoxy-pyrimidin-5- yl)boronic acid I-5 (63.1 mg, 325 μmol), bis(diphenylphosphino)ferrocene]dichloropalladium(II)-DCM (18.5 mg, 21.7 μmol) and potassium phosphate tribasic (138 mg, 651 μmol) were mixed in degassed Dioxane (2.0 mL) and Water (0.15 mL) under atmosphere of argon. The resulting mixture was stirred at room temperature for 12 hr. To the obtained mixture SiliaMetS® Dimercaptotriazine (100 mg) was added, and the mixture was stirred 30 min at room temperature. The mixture was filtered. The filtrate was concentrated under reduced pressure. The residue was subjected to HPLC (0-5 min., 40-90% water – MeOH, +0.1% vol. of 25% aq. NH
3, flow: 30 mL/min, column: XBridge 100 ×19 mm, 5 µm) to afford 4'-cyclopropyl-6'-methoxy-4-((4-(1-methyl-4- (trifluoromethyl)-1H-imidazol-2-yl)benzyl)oxy)-5-(methylthio)-2,5'-bipyrimidine (7.0 mg, 13 μmol, 6.1% yield) as a light-yellow solid.
1H NMR (600 MHz, DMSO-d6) δ 0.83 – 0.88 (m, 2H), 1.00 – 1.04 (m, 2H), 1.69 – 1.74 (m, 1H), 2.56 (s, 3H), 3.77 (s, 3H), 3.83 (s, 3H), 5.56 (s, 2H), 7.57 (d, 2H), 7.73 (d, 2H), 7.92 (s, 1H), 8.52 (s, 1H), 8.65 (s, 1H). MS (ESI): [M+H]+ m/z: calcd 529.19; found 529.2. Example 34 (Compound 186)

Compound 186 Step 1: The synthesis of 3,5-difluoro-4-[4-(trifluoromethyl)-1H-imidazol-2-yl]benzonitrile [1092] A mixture of 3,3-dibromo-1,1,1-trifluoro-propan-2-one (26.6 g, 98.7 mmol), sodium acetate (16.2 g, 197 mmol) and water (25 mL) was stirred at 95 °C for 1 hr. The mixture was cooled to ambient temperature and poured into a solution 3,5-difluoro-4-formyl-benzonitrile (15.0 g, 89.8 mmol) and aqueous ammonium hydroxide (75 mL, 25% wt.) in MeOH (600 mL). The resulting mixture was stirred for 40 min at ambient temperature, then heated to 100 °C and stirred at this temperature for 1 hr. The mixture was cooled to ambient temperature and concentrated under reduced pressure to approximately 100 mL. The precipitate formed was filtered off and dissolved in EtOAc (400 mL). The organic layer was washed with water (50 mL) and brine (2 × 100 mL) and concentrated under reduced pressure. The residue was subjected to flash-column chromatography (SiO
2, gradient chloroform - ACN) to afford 3,5- difluoro-4-[4-(trifluoromethyl)-1H-imidazol-2-yl]benzonitrile (10.0 g, 36.6 mmol, 40.8% yield) as a light yellow solid.
1H NMR (500 MHz, DMSO-d6) δ 7.99 (d, 2H), 8.07 (s, 1H), 13.35 (br., 1H). MS (ESI): [M+H]+ m/z: calcd 274.04; found 274.0 Step 2: The synthesis of 3,5-difluoro-4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]benzonitrile [1093] To a solution of 3,5-difluoro-4-[4-(trifluoromethyl)-1H-imidazol-2-yl]benzonitrile (10.0 g, 36.6 mmol) in DMF (100 mL) NaH (1.61 mg, 40.27 mmol, 60% dispersion in mineral oil) was added portion wise at 0 °C. The reaction mixture was stirred at ambient temperature for 2 hr. The mixture was cooled to 0 °C and methyl iodide (5.98 g, 42.1 mmol, 2.62 mL) was added in one portion. The solution was stirred at ambient temperature for 16 hr. The resulting mixture was poured into ice-water mixture (700 mL). The solid precipitate formed was filtered off and dissolved in EtOAc (400 mL). The resulting solution was washed with brine (150 mL). The organic layer was separated, dried over anhydrous Na
2SO
4 and concentrated under reduced pressure to afford 3,5-difluoro-4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]benzonitrile (9.00 g, 31.3 mmol, 85.6% yield) as a light- yellow solid which was used in the next steps without further purification.
1H NMR (500 MHz, DMSO-d6) δ 3.60 (s, 3H), 8.04 (d, 2H), 8.12 (s, 1H). MS (ESI): [M+H]+ m/z: calcd 288.06; found 288.2 Step 3: The synthesis of 3,5-difluoro-4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]benzoic acid [1094] Aqueous sodium hydroxide soln. (5 mL, 20% wt.) was added to a solution of 3,5- difluoro-4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]benzonitrile (2.00 g, 6.96 mmol) in EtOH (10 mL). The reaction mixture was stirred at 80 °C for 16 hr. The reaction mixture was cooled to room temperature and concentrated under reduced pressure. The residue was diluted with MTBE (20 mL) and water (40 mL). The water layer was separated and acidified with saturated solution of citric acid (10 mL). The solid precipitate formed was filtered off and dissolved in EtOAc (40 mL). The obtained solution was washed with brine (2×20 mL), dried over anhydrous Na
2SO
4 and concentrated under reduced pressure to afford 3,5-difluoro- 4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]benzoic acid (1.70 g, 5.55 mmol, 79.7% yield) as a yellow solid which was used in the next steps without further purification. MS (ESI): [M+H]+ m/z: calcd 307.05; found 307.0 Step 4: The synthesis of [3,5-difluoro-4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methanol [1095] Borane dimethyl sulfide complex (1.27 g, 16.7 mmol, 1.58 mL) was added dropwise to a vigorously stirred solution of 3,5-difluoro-4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]benzoic acid (1.70 g, 5.55 mmol) in THF (100 mL) under argon atmosphere. The reaction mixture was stirred at room temperature for 16 hr. The reaction mixture was quenched by dropwise addition of water (10 mL) and stirred at room temperature overnight. The reaction mixture was concentrated under reduced pressure. The residue was diluted with EtOAc (80 mL) and washed with water (30 mL). The organic layer was separated, washed with brine (2×20 mL), dried over anhydrous Na
2SO
4 and concentrated under reduced pressure to afford [3,5-difluoro-4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methanol (1.10 g, 3.76 mmol, 67.8% yield) as a yellow solid which was used in the next steps without further purification.
1H NMR (400 MHz, CDCl
3) δ 3.56 (s, 3H), 4.60 (d, 2H), 5.59 (t, 1H), 7.23 (d, 2H), 8.06 (s, 1H). MS (ESI): [M+H]+ m/z: calcd 293.07; found 293.2. Step 5: The synthesis of 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[3,5-difluoro-4-[1- methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]-5-methyl-pyrimidine [1096] NaH (15.0 mg, 376 μmol, 60% dispersion in mineral oil) was added to a solution of [3,5-difluoro-4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methanol (100 mg, 342 μmol) in DMF (3.0 mL) under argon atmosphere. The reaction mixture was stirred at room temperature for 1 hr.2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-5-methyl-4- methylsulfonyl-pyrimidine I-7 (110 mg, 342 μmol) was added to the reaction mixture. The resulting mixture was stirred at room temperature for 48 hr. The reaction mixture was diluted with EtOAc (20 mL) and brine (20 mL). The organic layer was separated, washed with brine (2×15 mL) and dried over anhydrous Na
2SO
4. To the resulting solution SiliaMetS® Dimercaptotriazine (100 mg) was added, and the mixture was stirred for 30 min. The mixture was filtered. The filtrate was concentrated under reduced pressure. The residue was subjected to HPLC (2-10 min., 35-50% water – ACN, +0.1% vol. of 25% aq. NH
3, flow: 30 mL/min, column: SunFire 100 ×19 mm, 5 µm) to afford 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)- 4-[[3,5-difluoro-4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]-5-methyl- pyrimidine (57.0 mg, 107 μmol, 31.3% yield) as a white solid.
1H NMR (600 MHz, DMSO- d6) δ 0.83 – 0.88 (m, 2H), 0.99 – 1.03 (m, 2H), 1.63 – 1.69 (m, 1H), 2.25 (s, 3H), 3.56 (s, 3H), 3.81 (s, 3H), 5.52 (s, 2H), 7.43 (d, 2H), 8.06 (s, 1H), 8.57 (s, 1H), 8.65 (s, 1H). MS (ESI): [M+H]+ m/z: calcd 533.20; found 533.0. Example 35 (Compound 138)
[1097] NaH (13.2 mg, 329 μmol, 60% dispersion in mineral oil) was added to a stirred solution of [5-fluoro-2-methoxy-4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methanol (130 mg, 299 μmol) in DMF (5 mL). The resulting mixture was stirred at room temperature for 30 min.2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-5-methoxy-4- methylsulfonyl-pyrimidine I-6 (101 mg, 299 μmol) was added to the mixture. The reaction mixture was stirred at room temperature for 16 hr. The reaction mixture was diluted with an aqueous solution of NH
4Cl (10 mL) and extracted with EtOAc (3×20 mL). The combined organic layers were washed with brine (10 mL), dried over anhydrous Na
2SO
4 and filtered. The filtrate was concentrated under reduced pressure. The residue was subjected to HPLC (0- 5 min, 55-80% water+FA (0.1% vol.) - MeOH+FA (0.1% vol.); flow: 30 mL/min, column: Chromatorex 18 SMB100-5T 100×19 mm, 5 µm) to afford 2-(4-cyclopropyl-6-methoxy- pyrimidin-5-yl)-4-[[5-fluoro-2-methoxy-4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]-5-methoxy-pyrimidine (52.0 mg, 92.8 μmol, 31% yield) as an off-white solid.
1H NMR (DMSO-d6, 600 MHz) δ 0.85 – 0.90 (m, 2H), 1.02 – 1.06 (m, 2H), 1.70 – 1.75 (m, 1H), 3.62 (s, 3H), 3.84 (s, 3H), 3.85 (s, 3H), 3.96 (s, 3H), 5.44 (s, 2H), 7.18 (d, 1H), 7.40 (d, 1H), 8.02 (s, 1H), 8.44 (s, 1H), 8.66 (s, 1H). MS (ESI): [M+H]+ m/z: calcd 561.21; found 561.2. Example 36 (Compound 183)
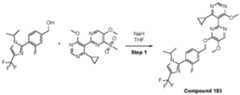
The synthesis of the starting [3-fluoro-4-[1-isopropyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methanol was described in Example 76 (Compound 149). [1098] NaH (12.2 mg, 305 μmol, 60% dispersion in mineral oil) was added to a stirred solution of [3-fluoro-4-[1-isopropyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methanol (80.0 mg, 265 μmol) in THF (3.0 mL). The reaction mixture was stirred at room temperature for 15 min.2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-5-methoxy-4-methylsulfonyl-pyrimidine I-6 (89.0 mg, 265 μmol) was added to the reaction mixture. The resulting mixture was stirred at room temperature for 12 hr. The reaction mixture was diluted with a saturated aqueous solution of NH
4Cl (2.0 mL). The organic layer was separated and subjected to HPLC (0.5-6.5 min, 49-63% water – ACN; flow: 30 mL/min, column: SunFire C18, 100 x 19 mm, 5 µm) to afford 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[3-fluoro-4-[1-isopropyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]-5-methoxy-pyrimidine (80.0 mg, 143 μmol, 54.1% yield) as a yellow solid.
1H NMR (600 MHz, DMSO-d6) δ 0.82 – 0.87 (m, 2H), 0.99 – 1.03 (m, 2H), 1.35 (d, 6H), 1.66 – 1.72 (m, 1H), 3.82 (s, 3H), 3.95 (s, 3H), 4.10 – 4.17 (m, 1H), 5.51 (s, 2H), 7.43 (d, 1H), 7.48 (d, 1H), 7.56 (t, 1H), 8.24 (s, 1H), 8.44 (s, 1H), 8.64 (s, 1H). MS (ESI): [M+H]+ m/z: calcd 559.24; found 559.2.
Step 1: Synthesis of 4-(1-ethyl-4-(trifluoromethyl)-1H-imidazol-2-yl)benzonitrile [1099] The solution of Ethyl iodide (5.43 g, 34.8 mmol) in Acetonitrile (10 mL) was added dropwise to a mixture of 4-[4-(trifluoromethyl)-1H-imidazol-2-yl]benzonitrile I-1a (7.50 g, 31.6 mmol) and Cs
2CO
3 (20.6 g, 63.2 mmol) in ACN (100 mL). The resulting mixture was stirred at 80 °C for 16 hr. The reaction mixture was cooled to room temperature and filtered. The filtrate was concentrated under reduced pressure. The residue was diluted with DCM (30 mL) and washed with water (2 × 10 mL) and brine (10 mL). The organic layer was concentrated under reduced pressure to afford 4-(1-ethyl-4-(trifluoromethyl)-1H-imidazol-2- yl)benzonitrile (8.00 g, 30.6 mmol, 96.8% yield) as a yellow solid which was used in the next steps without further purification.
1H NMR (500 MHz, DMSO-d6) δ 1.34 (t, 3H), 4.15 (q, 2H), 7.86 (d, 2H), 7.99 (d, 2H), 8.12 (s, 1H). MS (ESI): [M+H]+ m/z: calcd 266.11; found 266.0. Step 2: Synthesis of 4-(1-ethyl-4-(trifluoromethyl)-1H-imidazol-2-yl)benzaldehyde [1100] Diisobutylaluminum hydride (13.6 mmol, 13.6 mL, 1M solution in hexane) was added dropwise to a stirred solution of 4-[1-ethyl-4-(trifluoromethyl)imidazol-2- yl]benzonitrile (3.00 g, 11.3 mmol) in DCM (100 mL) at -78 °C under argon atmosphere. The resulting mixture was stirred overnight, during this time it was allowed to warm to room temperature. The reaction mixture was poured into ice-cooled aqueous solution of sulfuric acid (100 mL, 3% wt.). The organic layer was separated, washed with brine (20 mL), dried over anhydrous Na
2SO
4 and concentrated under reduced pressure to afford 4-(1-ethyl-4- (trifluoromethyl)-1H-imidazol-2-yl)benzaldehyde (2.50 g, 9.32 mmol, 82.5% yield) as a yellow oil which was used in the next steps without further purification.
1H NMR (500 MHz, DMSO-d6) δ 1.34 (t, 3H), 4.17 (q, 2H), 7.90 (d, 2H), 8.40 (d, 2H), 8.12 (s, 1H), 10.10 (s, 1H). MS (ESI): [M+H]+ m/z: calcd 269.11; found 269.2. Step 3: The synthesis of 1-(4-(1-ethyl-4-(trifluoromethyl)-1H-imidazol-2-yl)phenyl)ethanol [1101] Methylmagnesium bromide (14.0 mmol, 4.70 mL, 3M in ether) was added dropwise to a solution of 4-[1-ethyl-4-(trifluoromethyl)imidazol-2-yl]benzaldehyde (2.50 g, 9.32 mmol) in THF (50 mL) at -80 °C under argon atmosphere. The resulting mixture was stirred for 2 hr., during this time it was allowed to warm to -20 °C. The mixture was quenched by dropwise addition of aqueous saturated solution of NH
4Cl (50 mL) and stirred for 15 min. The resulting mixture was extracted with EtOAc (50 mL). Organic layer was washed brine (50 mL) and concentrated under reduced pressure. The residue was subjected to flash-column chromatography (SiO
2, gradient CHCl3 - MTBE) to afford 1-(4-(1-ethyl-4-(trifluoromethyl)- 1H-imidazol-2-yl)phenyl)ethanol (1.00 g, 3.52 mmol, 37.7% yield) as a yellow oil. MS (ESI): [M+H]+ m/z: calcd 285.14; found 285.0. Step 4: The synthesis of rel-(R)-1-(4-(1-ethyl-4-(trifluoromethyl)-1H-imidazol-2- yl)phenyl)ethanol and rel-(S)-1-(4-(1-ethyl-4-(trifluoromethyl)-1H-imidazol-2- yl)phenyl)ethanol [1102] Racemic 1-[4-[1-ethyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]ethanol (1.00 g, 3.52 mmol) was subjected to chiral HPLC (column: Chiralcel OJ-H-I (250×20 mm, 5 μm), Hexane-IPA-MeOH, 80-10-10, 12 mL/min) to afford rel-(R)-1-(4-(1-ethyl-4- (trifluoromethyl)-1H-imidazol-2-yl)phenyl)ethanol (0.263 g, 925 μmol, 26.3% yield) and rel- (S)-1-(4-(1-ethyl-4-(trifluoromethyl)-1H-imidazol-2-yl)phenyl)ethanol (0.256 g, 901 μmol, 25.6% yield) as a white solids. rel-(R)-1-(4-(1-ethyl-4-(trifluoromethyl)-1H-imidazol-2-yl)phenyl)ethanol: MS (ESI): [M+H]+ m/z: calcd 285.15; found 285.2 Enantiopurity: 100% (column: Chiralcel OJ-H-I, 250 × 4.6 mm, 5 μm; mobile phase: Hexane-IPA-MeOH, 80-10-10; flow: 0.6 mL/min; RT=12.6 min) rel-(S)-1-(4-(1-ethyl-4-(trifluoromethyl)-1H-imidazol-2-yl)phenyl)ethanol: MS (ESI): [M+H]+ m/z: calcd 285.15; found 285.2 Enantiopurity: 98% (column: Chiralcel OJ-H-I, 250 × 4.6 mm, 5 μm; mobile phase: Hexane- IPA-MeOH, 50-25-25; flow: 0.6 mL/min; RT=16.2 min) Step 5: Synthesis of 2-chloro-5-methoxy-4-[(1S)-1-[4-[1-ethyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]ethoxy]pyrimidine [1103] rel-(1S)-1-[4-[1-ethyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]ethanol (163 mg, 573 μmol), 2,4-dichloro-5-methoxy-pyrimidine (108 mg, 602 μmol) and Cs
2CO
3 (206 mg, 631 μmol) were mixed in ACN (3 mL). The resulting mixture was stirred at 80 °C for 12 hr. The reaction mixture was cooled to room temperature and filtered. The filtrate was concentrated under reduced pressure. The residue was subjected to HPLC (0-6 min., 40-65% water – acetonitrile, +0.1% vol. of 25% aq. NH
3, flow: 30 mL/min, column: XBridge 100×19 mm, 5 µm) to afford rel-2-chloro-5-methoxy-4-[(1S)-1-[4-[1-ethyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]ethoxy]pyrimidine (140 mg, 328 μmol, 57.2% yield) as a yellow solid. MS (ESI): [M+H]+ m/z: calcd 427.13; found 428.0. Step 6: The synthesis of rel-2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-5-methoxy-4-[(1S)- 1-[4-[1-ethyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]ethoxy]pyrimidine. [1104] Rel-2-chloro-5-methoxy-4-[(1S)-1-[4-[1-ethyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]ethoxy]pyrimidine (140 mg, 328 μmol), (4-cyclopropyl-6-methoxy-pyrimidin-5- yl)boronic acid I-5 (255 mg, 1.31 mmol), N,N-Diisopropylethylamine (209 mg, 1.62 mmol) and RuPhos Pd G3 (27.4 mg, 32.8 μmol) were mixed in a degassed mixture of dioxane (0.8 mL) and water (0.2 mL). The reaction mixture was stirred at 100 °C for 14 hr. The reaction mixture was cooled to room temperature. The reaction mixture was diluted with EtOAc (10 mL) and washed with water (5 mL) and brine (5 mL). To the obtained organic phase Dimercaptotriazine (20 mg) was added, and the mixture was stirred for 30 min. The mixture was filtered. The filtrate was concentrated under reduced pressure. The residue was subjected to HPLC (0- 5 min., 40-85% water – MeOH, +0.1% vol. of 25% aq. NH
3, flow: 30 mL/min, column: XBridge 100 ×19 mm, 5 µm) to afford rel-2-(4-cyclopropyl-6-methoxy-pyrimidin-5- yl)-5-methoxy-4-[(1S)-1-[4-[1-ethyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]ethoxy]pyrimidine (61.0 mg, 113 μmol, 34.4% yield) as a white solid.
1H NMR (600 MHz, DMSO-d6) δ 0.70 – 0.76 (m, 1H), 0.78 – 0.84 (m, 1H), 0.92 – 1.01 (m, 2H), 1.32 (t, 3H), 1.55 – 1.61 (m, 1H), 1.65 (d, 3H), 3.78 (s, 3H), 3.96 (s, 3H), 4.08 (q, 2H), 6.31 (q, 1H), 7.55 (d, 2H), 7.62 (d, 2H), 8.02 (s, 1H), 8.40 (s, 1H), 8.62 (s, 1H). MS (ESI): [M+H]+ m/z: calcd 541.25; found 541.24. The synthesis of rel-2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-5-methoxy-4-[(1R)-1-[4- [1-ethyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]ethoxy]pyrimidine (Compound 172) Step 7: Synthesis of rel-2-chloro-5-methoxy-4-[(1R)-1-[4-[1-ethyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]ethoxy]pyrimidine. [1105] rel-(1R)-1-[4-[1-ethyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]ethanol (164 mg, 577 μmol), 2,4-dichloro-5-methoxy-pyrimidine (108 mg, 606 μmol) and Cs
2CO
3 (207 mg, 635 μmol) were mixed in ACN (3 mL). The resulting mixture was stirred at 80 °C for 12 hr. The reaction mixture was cooled to room temperature and filtered. The filtrate was concentrated under reduced pressure. The residue was subjected to HPLC (0-6 min., 45-65% water – acetonitrile, flow: 30 mL/min, column: XBridge 100×19 mm, 5 µm) to afford rel-2-chloro-5- methoxy-4-[(1R)-1-[4-[1-ethyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]ethoxy]pyrimidine (105 mg, 246 μmol) as a yellow solid. MS (ESI): [M+H]+ m/z: calcd 427.14; found 427.0. Step 8: The synthesis of rel-2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-5-methoxy-4-[(1R)- 1-[4-[1-ethyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]ethoxy]pyrimidine. [1106] rel-2-Chloro-5-methoxy-4-[(1R)-1-[4-[1-ethyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]ethoxy]pyrimidine (105 mg, 246 μmol), (4-cyclopropyl-6-methoxy-pyrimidin-5- yl)boronic acid I-5 (191 mg, 984 μmol), N,N-Diisopropylethylamine (157 mg, 1.21 mmol) and RuPhos Pd G3 (20.6 mg, 24.6 μmol) were mixed in a degassed mixture of dioxane (0.8 mL) and water (0.2 mL). The reaction mixture was stirred at 100 °C for 14 hr. The reaction mixture was cooled to room temperature. The reaction mixture was diluted with EtOAc (10 mL) and washed with water and brine (2 × 5 mL). To the obtained organic phase Dimercaptotriazine (20 mg) was added, and the mixture was stirred for 30 min. The mixture was filtered. The filtrate was concentrated under reduced pressure. The residue was subjected to HPLC (0-5 min., 45-80% water – MeOH, +0.1% vol. of 25% aq. NH
3, flow: 30 mL/min, column: XBridge 100 ×19 mm, 5 µm) to afford rel-2-(4-cyclopropyl-6-methoxy-pyrimidin-5- yl)-5-methoxy-4-[(1R)-1-[4-[1-ethyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]ethoxy]pyrimidine (17.5 mg, 32.4 μmol, 13.2% yield) as a white solid.
1H NMR (600 MHz, DMSO-d6) δ 0.70 – 0.76 (m, 1H), 0.78 – 0.84 (m, 1H), 0.92 – 1.01 (m, 2H), 1.32 (t, 3H), 1.55 – 1.61 (m, 1H), 1.65 (d, 3H), 3.78 (s, 3H), 3.96 (s, 3H), 4.08 (q, 2H), 6.31 (q, 1H), 7.55 (d, 2H), 7.62 (d, 2H), 8.02 (s, 1H), 8.40 (s, 1H), 8.62 (s, 1H). MS (ESI): [M+H]+ m/z: calcd 541.25; found 541.24. Example 38 (Compound 90)
Step 1: The synthesis of 2-chloro-5-cyclopropyl-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol- 2-yl]phenyl]methoxy]pyrimidine [1107] Cs
2CO
3 (270 mg, 828 μmol) was added to a solution [4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methanol I-2b (177 mg, 690 μmol) in ACN (10 mL). 2,4-Dichloro-5-cyclopropyl-pyrimidine (150 mg, 794 μmol) was added to the mixture at the same temperature. The resulting reaction mixture was stirred at 65 °C for 12 hr. The mixture was cooled to room temperature and filtered. The filtrate was evaporated in vacuo to afford 2- chloro-5-cyclopropyl-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidine (100 mg, crude) as a white solid which was used in the next steps without further purification. MS (ESI): [M+H]+ m/z: calcd 409.11; found 409.00. Step 2: The synthesis of 4',5-dicyclopropyl-6'-methoxy-4-({4-[1-methyl-4-(trifluoromethyl)- 1H-imidazol-2-yl]phenyl}methoxy)-2,5'-bipyrimidine [1108] 2-Chloro-5-cyclopropyl-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidine (100 mg, crude), (4-cyclopropyl-6-methoxy-pyrimidin-5- yl)boronic acid I-5 (142 mg, 734 μmol), K
2CO
3 (67.6 mg, 489 μmol), RuPhos Pd G4 (10.4 mg, 12.2 μmol) and RuPhos (5.71 mg, 12.2 μmol) were mixed in a degassed mixture of dioxane (5 mL) and water (0.5 mL). The mixture was stirred at 100 °C under argon atmosphere for 12 hr. The mixture was cooled to room temperature and filtered. The filtrate was subjected to HPLC (0-5 min, 40-65% +0.1% water-ACN +0.1% vol. of 25% aq. NH
3, flow: 30 mL/min, column: XBridge C18100x19 mm, 5 μm) to afford 4-cyclopropyl-5-[5- cyclopropyl-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidin-2- yl]-6-methoxy-pyrimidine (29.9 mg, 57.2 μmol, 8.3% yield from [4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methanol) as a white solid.
1H NMR (600 MHz, DMSO-d6) δ 0.82 – 0.86 (m, 2H), 0.88 – 0.91 (m, 2H), 0.96 – 1.02 (m, 4H), 1.65 – 1.69 (m, 1H), 1.99 – 2.04 (m, 1H), 3.77 (s, 3H), 3.82 (s, 3H), 5.52 (s, 2H), 7.59 (d, 2H), 7.73 (d, 2H), 7.92 (s, 1H), 8.35 (s, 1H), 8.64 (s, 1H). MS (ESI): [M+H]+ m/z: calcd 523.24; found 523.00. Example 39 (Compound 144)
Step 1: The synthesis of [3-methyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2- yl)phenyl]methanol [1109] (4-bromo-3-methyl-phenyl)methanol (1.50 g, 7.46 mmol), 4,4,5,5-tetramethyl-2- (4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1,3,2-dioxaborolane (2.84 g, 11.2 mmol) and bis(diphenylphosphino)ferrocene]dichloropalladium(II)-DCM (305 mg, 0.373 mmol) were mixed in degassed dioxane (50 mL). The reaction mixture was stirred under argon atmosphere at 100 °C for 24 hr. The reaction mixture was cooled to room temperature, diluted with EtOAc (100 mL) and washed with water (2×50 mL). The organic layer was dried over anhydrous Na
2SO
4 and concentrated under reduced pressure to afford [3-methyl-4- (4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl]methanol (2.20 g, crude) as a solid which was used in the next steps without further purification. MS (ESI): [M-OH]+ m/z: calcd 231.16; found 231.2. Step 2: The synthesis of [3-methyl-4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methanol [1110] [3-methyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl]methanol (2.20 g, crude), 2-bromo-1-methyl-4-(trifluoromethyl)imidazole (1.60 g, 7.00 mmol), Cs
2CO
3 (5.71 g, 17.5 mmol) and bis(diphenylphosphino)ferrocene]dichloropalladium(II)-DCM (286 mg, 350 μmol) were mixed in degassed dioxane (50 mL) and water (2.5 mL). The reaction mixture was stirred under argon atmosphere at 90 °C for 16 hr. The reaction mixture was cooled to room temperature, diluted with EtOAc (100 mL) and washed with water (2×50 mL). The organic layer was dried over anhydrous Na
2SO
4 and concentrated under reduced pressure. The residue was subjected to flash-column chromatography (SiO
2, gradient chloroform - acetonitrile) to afford [3-methyl-4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methanol (760 mg, 2.81 mmol, 37.7% yield from (4-bromo-3-methyl- phenyl)methanol) as a light-yellow solid.
1H NMR (400 MHz, CDCl
3) δ 2.22 (s, 3H), 3.51 (s, 3H), 4.71 (s, 2H), 7.24 – 7.36 (m, 4H). MS (ESI): [M+H]+ m/z: calcd 271.11; found 271.2. Step 3: The synthesis of 2-chloro-5-methoxy-4-[[3-methyl-4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidine [1111] 2,4-dichloro-5-methoxy-pyrimidine (255 mg, 1.41 mmol) was added to a mixture of [3-methyl-4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methanol (380 mg, 1.41 mmol) and potassium tert-butoxide (158 mg, 1.41 mmol) in dioxane (10 mL). The reaction mixture was stirred at 80 °C for 32 hr. The reaction mixture was cooled to room temperature, diluted with EtOAc (30 mL) and washed with water (2×10 mL) and brine (10 mL). The organic layer was dried over anhydrous Na
2SO
4 and concentrated under reduced pressure. The residue was subjected to HPLC (0.5-6.5 min, 45 - 70% water - ACN; flow: 30 mL/min, column: Waters SunFire C18, 100 x 19 mm, 5 µm) to afford 2-chloro-5-methoxy-4-[[3- methyl-4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidine (180 mg, 436 μmol, 31.0% yield) as a white solid. MS (ESI): [M+H]+ m/z: calcd 413.12; found 413.2. Step 4: The synthesis of 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-5-methoxy-4-[[3- methyl-4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidine [1112] 2-chloro-5-methoxy-4-[[3-methyl-4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidine (90.0 mg, 218 μmol), (4-cyclopropyl-6-methoxy-pyrimidin-5- yl)boronic acid I-5 (59.2 mg, 305 μmol) and DIPEA (84.5 mg, 654 μmol, 114 μL) were mixed in degassed dioxane (3 mL) and water (150 µL). RuPhos Pd G4 (1.85 mg, 2.18 μmol) was added to the reaction mixture. The reaction mixture was stirred under argon atmosphere at 60 °C for 16 hr. The reaction mixture was cooled to room temperature, diluted with EtOAc (20 mL) and washed with water (2×10 mL). To the resulting organic layer SiliaMetS® Dimercaptotriazine (50 mg) was added, and the resulting mixture was stirred for 30 min. The mixture was filtered. The filtrate was concentrated under reduced pressure. The residue was subjected to HPLC (0.5-6.5 min, 40 - 65% water - ACN; flow: 30 mL/min, column: Waters SunFire C18, 100 x 19 mm, 5 µm, then repurified 0.5-6.5 min., 40-90% water – MeOH, +0.1% vol. of 25% aq. NH
3, flow: 30 mL/min, column: XBridge 100 ×19 mm, 5 µm) to afford 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-5-methoxy-4-[[3-methyl-4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidine (13.0 mg, 24.7 μmol, 11.3% yield) as a white solid.
1H NMR (400 MHz, DMSO-d6) δ 0.84 – 0.88 (m, 2H), 0.99 – 1.03 (m, 2H), 1.68 – 1.74 (m, 1H), 2.15 (s, 3H), 3.49 (s, 3H), 3.83 (s, 3H), 3.93 (s, 3H), 5.44 (s, 2H), 7.37 – 7.39 (m, 2H), 7.43 (s, 1H), 7.92 (s, 1H), 8.41 (s, 1H), 8.64 (s, 1H). MS (ESI): [M+H]+ m/z: calcd 527.23; found 527.2. Example 40 (Compound 79)

2-[4-cyclopropyl-6-(trideuteriomethoxy)pyrimidin-5-yl]-4-[[4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]-5-(trideuteriomethoxy)pyrimidine [1113] To a stirred solution of 2-chloro-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]-5-(trideuteriomethoxy)pyrimidine 171a (200 mg, 497.78 μmol) and 4- cyclopropyl-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-6- (trideuteriomethoxy)pyrimidine I-9 (308.49 mg, 1.11 mmol) in THF (2 mL) and water (0.4 mL) were added [1,1’-Bis(dicyclohexylphosphino)ferrocene]dichloropalladium(II) (41.95 mg, 49.78 μmol) and potassium phosphate (316.98 mg, 1.49 mmol) at 25 °C under nitrogen atmosphere. The resulting mixture was stirred for 16 hrs at 40 °C under nitrogen atmosphere. The resulting mixture was concentrated under reduced pressure. The residue was purified by Prep-TLC (100% EtOAc) to afford crude product (115 mg). The obtained product was further purified by RP-Flash with the following conditions: Column: C18 spherical Column, 20-35 um, 100A, 25 g; Mobile Phase A: Water (5 mM aq. NH4HCO3), Mobile Phase B: MeCN; Flow rate: 35 mL/min; Gradient: 5% B to 5% B in 5 min, 5% B to 60% B in 20 min, 60% B to 60% B in 1 min, 60% B to 95% B in 5 min; Detector: UV 254 & 210 nm. The collected fractions were combined, concentrated under reduced pressure and then lyophilized to give 2- [4-cyclopropyl-6-(trideuteriomethoxy)pyrimidin-5-yl]-4-[[4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]-5-(trideuteriomethoxy)pyrimidine (100.7 mg, 194.21 μmol, 39% yield) as a white solid. MS: m/z = 519.20 [M + H]+.
1H NMR (400 MHz, Chloroform-d) δ 8.68 (s, 1H), 8.26 (s, 1H), 7.66 (d, J = 8.0 Hz, 2H), 7.60 (d, J = 8.4 Hz, 2H), 7.34 (s, 1H), 5.60 (s, 2H), 3.79 (s, 3H), 1.79 - 1.72 (m, 1H), 1.29 - 1.24 (m, 2H), 0.98 - 0.89 (m, 2H).
19F NMR (377 MHz, Chloroform-d) δ -62.77. Example 41 (Compound 3)
Step 1: Synthesis of methyl 4-[4-(trifluoromethyl)-1H-imidazol-2-yl]benzoate [1114] Sodium acetate (34.5 g, 420 mmol) was dissolved in H2O (200 mL) then 3,3-dibromo- 1,1,1-trifluoro-propan-2-one (54.3 g, 201 mmol) was added and the reaction mixture was stirred at 95 °C for 1hr. The resulting mixture was cooled to room temperature and poured into a solution of methyl 4-formylbenzoate (30.0 g, 183 mmol) and NH
4OH (100 mL, 25% wt.) in MeOH (1000 mL). The obtained mixture was stirred at room temperature for 12 hr. The mixture was concentrated under reduced pressure. The residue was distributed between EtOAc (300 mL) and water (200 mL). The organic layer was separated, dried over anhydrous Na
2SO
4, filtered and concentrated under reduced pressure. The residue was recrystallized from MTBE to afford methyl 4-[4-(trifluoromethyl)-1H-imidazol-2-yl]benzoate (38.0 g, 141 mmol, 77% yield) as a light-yellow solid which was used in the next steps without further purification.
1H NMR (400 MHz, CDCl3) δ 3.85 (s, 3H), 7.98 (s, 1H), 8.04 (d, 2H), 8.10 (d, 2H). MS (ESI): [M+H]
+ m/z: calcd 271.08; found 271.0. Step 2: Synthesis of methyl 4-[1-isopropyl-4-(trifluoromethyl)imidazol-2-yl]benzoate [1115] Methyl 4-[4-(trifluoromethyl)-1H-imidazol-2-yl]benzoate (1.00 g, 3.70 mmol), 2- iodopropane (1.89 g, 11.1 mmol, 1.11 mL) and Cs
2CO
3 (3.62 g, 11.1 mmol) were mixed in DMF (3 mL). The mixture was stirred at 80 °C for 36 hr. The mixture was cooled to room temperature and diluted with water (10 mL). The resulting mixture was extracted with MTBE (2 × 10 mL). Combined organic layers were washed with brine (10mL) and concentrated under reduced pressure to afford methyl 4-[1-isopropyl-4-(trifluoromethyl)imidazol-2- yl]benzoate (1.00 g, 3.20 mmol, 86.5% yield) as an yellow oil which was used in the next steps without further purification.
1H NMR (500 MHz, CDCl3) δ 1.44 (d, 6H), 3.91 (s, 3H), 4.47 – 4.60 (m, 1H), 7.44 (s, 1H), 7.60 (d, 2H), 8.10 (d, 2H). Step 3: Synthesis of [4-[1-isopropyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methanol [1116] A solution of methyl 4-[1-isopropyl-4-(trifluoromethyl)imidazol-2-yl]benzoate (1.15 g, 3.68 mmol) in THF (5 mL) was added dropwise to a vigorously stirred suspension of lithium aluminum hydride (375 mg, 11.1 mmol) in THF (20 mL) under argon atmosphere at +5°C. The resulting mixture was stirred at ambient temperature for 12 hr. The mixture was carefully quenched by dropwise addition of aqueous NaOH (10 mL, 10% wt.). The resulting mixture was stirred for 15 min and filtered. The filter cake was washed with THF (10 mL). The combined filtrate was dried over anhydrous Na
2SO
4 and concentrated under reduced pressure to afford [4-[1-isopropyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methanol (700 mg, 2.46 mmol, 73.5% yield) as a red gum which was used in the next steps without further purification. MS (ESI): [M+H]
+ m/z: calcd 285.15; found 285.2. Step 4: Synthesis of 2-chloro-4-[[4-[1-isopropyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]-5-methoxy-pyrimidine [1117] Potassium tert-butoxide (158 mg, 1.41 mmol) was added to a solution of [4-[1- isopropyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methanol (400 mg, 1.41 mmol) in dioxane (10 mL) at room temperature. The resulting mixture was stirred for 1 hr. To the obtained mixture 2,4-dichloro-5-methoxy-pyrimidine (252 mg, 1.41 mmol) was added in one portion. The resulting mixture was stirred at 80 °C for 12 hr. The reaction mixture was cooled to room temperature, diluted with water (10 mL) and extracted with EtOAc (2×10 mL). The organic layer was washed with brine (10 mL) and concentrated under reduced pressure. The residue was subjected to HPLC (0.5-6 min, 55-63% water – ACN, +0.1% vol. of 25% aq. NH
3, flow: 30 mL/min, column: XBridge C18, 100 ×19 mm, 5 µm) to afford 2-chloro-4-[[4-[1- isopropyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]-5-methoxy-pyrimidine (130 mg, 305 μmol, 21.7% yield) as an yellow solid. MS (ESI): [M+H]+ m/z: calcd 427.14; found 427.0. Step 5: Synthesis of 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[4-[1-isopropyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]-5-methoxy-pyrimidine [1118] 2-chloro-4-[[4-[1-isopropyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]-5- methoxy-pyrimidine (130 mg, 305 μmol), (4-cyclopropyl-6-methoxy-pyrimidin-5-yl)boronic acid I-5 (88.6 mg, 457 μmol), DIPEA (118 mg, 914 μmol, 159 μL) and XPhos Pd G3 (12.9 mg, 15.2 μmol) were mixed in a degassed mixture of water (0.5 mL) and dioxane (5 mL). The reaction mixture was stirred under argon atmosphere at 80 °C for 12 hr. The reaction mixture was cooled room temperature, diluted with water (5 mL) and extracted with EtOAc (2 × 10 mL). The combined organic layers were washed with water (2 × 10 mL) and brine (10 mL). To the obtained organic phase SiliaMetS® Dimercaptotriazine (20 mg) was added. The resulting mixture was stirred for 30 min and filtered. The filtrate was evaporated under reduced pressure. The residue was subjected to HPLC (0.5-6.5 min, 49-64% water - ACN; flow: 30 mL/min, column: Waters SunFire C18, 100 x 19 mm, 5 µm, then re-purified 0.5-6 min, 55-71% water – MeOH, +0.1% vol. of 25% aq. NH
3, flow: 30 mL/min, column: XBridge C18100 ×19 mm, 5 µm) to afford 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4- [[4-[1-isopropyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]-5-methoxy-pyrimidine (22.0 mg, 40.7 μmol, 13.4% yield) as a light-yellow solid.
1H NMR (600 MHz, DMSO-d6) δ 0.82 – 0.87 (m, 2H), 0.99 – 1.03 (m, 2H), 1.39 (d, 6H), 1.67 – 1.73 (m, 1H), 3.83 (s, 3H), 3.93 (s, 3H), 4.43 – 4.50 (m, 1H), 5.50 (s, 2H), 7.55 – 7.60 (m, 4H), 8.17 (s, 1H), 8.42 (s, 1H), 8.63 (s, 1H). MS (ESI): [M+H]+ m/z: calcd 541.25; found 541.2.
Compound 9 Step 1: The synthesis methyl 4-[1-(trideuteriomethyl)-4-(trifluoromethyl)imidazol-2- yl]benzoate [1119] Cs
2CO
3 (1.27 g, 3.89 mmol) was added to a stirred solution of methyl 4-[4- (trifluoromethyl)-1H-imidazol-2-yl]benzoate I-3a (700 mg, 2.59 mmol) in acetonitrile (15 mL) under argon atmosphere at room temperature. The mixture was stirred at room temperature for 5 min. Methyliodide-d3 (432 mg, 2.98 mmol, 185 μL) was added to the reaction mixture. The reaction mixture was stirred at room temperature for 16 hr. The reaction was diluted with water (30 mL) and extracted with DCM (2×15 mL). The combined organic layers were dried over anhydrous Na
2SO
4 and filtered. The filtrate was concentrated under reduced pressure to afford methyl 4-[1-(trideuteriomethyl)-4- (trifluoromethyl)imidazol-2-yl]benzoate (659 mg, 2.29 mmol, 88.6% yield) as a light-yellow liquid which was used in the next steps without further purification. MS (ESI): [M+H]+ m/z: calcd 288.11; found 288.1. Step 2: The synthesis of [4-[1-(trideuteriomethyl)-4-(trifluoromethyl)imidazol-2- yl]phenyl]methanol [1120] To a vigorously stirred solution of methyl 4-[1-(trideuteriomethyl)-4- (trifluoromethyl)imidazol-2-yl]benzoate (659 mg, 2.11 mmol) in THF (15 mL) a suspension of LAH (81.0 mg, 2.13 mmol) in THF (3 mL) was added dropwise at 0 °C. The reaction mixture was stirred at room temperature for 3 hr. The mixture was quenched by dropwise addition of water (1 mL) and 5 N aqueous NaOH (1 mL). The resulting suspension was diluted with CHCl
3 (30 mL) and filtered through a pad of Celite. The filtrate was concentrated under reduced pressure to afford [4-[1-(trideuteriomethyl)-4- (trifluoromethyl)imidazol-2-yl]phenyl]methanol (586 mg, crude) as a light-yellow oil which was used in the next steps without further purification. MS (ESI): [M+H]+ m/z: calcd 260.11; found 260.1 Step 3: The synthesis of 2-chloro-5-methoxy-4-[[4-[1-(trideuteriomethyl)-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidine [1121] Potassium tert-butoxide (334 mg, 2.97 mmol) was added to a stirred solution of [4-[1- (trideuteriomethyl)-4-(trifluoromethyl)imidazol-2-yl]phenyl]methanol (586 mg, crude) in acetonitrile (10 mL) under argon atmosphere at room temperature. The reaction mixture was stirred at room temperature for 5 min.2,4-dichloro-5-methoxy-pyrimidine (450 mg, 2.52 mmol) was added to the reaction mixture. The resulting mixture was stirred at room temperature for 17 hr. The reaction mixture was diluted with water (15 mL) and extracted with DCM (2×10 mL). The combined organic layers were separated, dried over anhydrous Na
2SO
4 and filtered. The filtrate was concentrated under reduced pressure to afford 2-chloro- 5-methoxy-4-[[4-[1-(trideuteriomethyl)-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidine (528 mg, crude) as an yellow oil which was used in the next steps without further purification. MS (ESI): [M+H]+ m/z: calcd 402.10; found 402.1 Step 4: The synthesis of 4,6-dimethoxy-5-[5-methoxy-4-[[4-[1-(trideuteriomethyl)-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidin-2-yl]pyrimidine [1122] 2-chloro-5-methoxy-4-[[4-[1-(trideuteriomethyl)-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidine (100 mg, crude), (4-cyclopropyl-6-methoxy-pyrimidin-5- yl)boronic acid I-5 (64.9 mg, 335 μmol), potassium phosphate tribasic (152 mg, 717 μmol) and RuPhos Pd G4 (10.2 mg, 12.0 μmol) were mixed in degassed dioxane (6.0 mL) and water (1.0 mL) under argon atmosphere. The reaction mixture was stirred at 85 °C for 16 hr. The reaction mixture was cooled to room temperature and concentrated under reduced pressure. The residue was diluted with EtOAc (20 mL) and washed with brine (10 mL). SiliaMetS® Dimercaptotriazine (50 mg) and anhydrous Na
2SO
4 were added to the organic layer. The resulting mixture was stirred at room temperature for 1 hr and then the solids were filtered out. The filtrate was concentrated under reduced pressure. The residue was subjected to HPLC (2-10 min, 25-45% ACN, +0.1% vol. of 25% aq. NH
3, flow: 30 mL/min, column: SunFire 100 ×19 mm, 5 µm) to afford 4,6-dimethoxy-5-[5-methoxy-4-[[4-[1- (trideuteriomethyl)-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidin-2- yl]pyrimidine (15.7 mg, 30.5 μmol, 7.63% yield from 4-[1-(trideuteriomethyl)-4- (trifluoromethyl)imidazol-2-yl]benzoate) as an yellow solid.
1H NMR (600 MHz, DMSO-d6) δ 0.83 – 0.88 (m, 2H), 0.99 – 1.03 (m, 2H), 1.67 – 1.72 (m, 1H), 3.83 (s, 3H), 3.93 (s, 3H), 5.48 (s, 2H), 7.57 (d, 2H), 7.72 (d, 2H), 7.92 (s, 1H), 8.41 (s, 1H), 8.63 (s, 1H). MS (ESI): [M+H]+ m/z: calcd 516.21; found 516.2. Example 43 (Compound 89)
Step 1: The synthesis of 2-chloro-5-isopropyl-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidine [1123] Potassium tert-butoxide (214 mg, 1.90 mmol) was added to a stirred solution of [4-[1- methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methanol I-2b (244 mg, 952 μmol) in toluene (10 mL) at room temperature. The reaction mixture was stirred at room temperature for 30 min. A solution of 2,4-dichloro-5-isopropyl-pyrimidine (200 mg, 1.05 mmol) in toluene (5.0 mL) was added to the reaction mixture. The resulting mixture was stirred at room temperature for 16 hr. The reaction mixture was diluted with a saturated aqueous solution of NH
4Cl (10 mL) and extracted with EtOAc (3×20 mL). The combined organic layers were washed with brine, dried over anhydrous Na
2SO
4 and filtered. The filtrate was concentrated under reduced pressure to afford 2-chloro-5-isopropyl-4-[[4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidine (300 mg, crude) as a brown oil which was used in the next steps without further purification. MS: [M+H]+ m/z: calcd 411.12; found 411.0 Step 2: The synthesis of 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-5-isopropyl-4-[[4-[1- methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidine [1124] (4-Сyclopropyl-6-methoxy-pyrimidin-5-yl)boronic acid (61.4 mg, 316 μmol), 2- chloro-5-isopropyl-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidine (130 mg, crude) and potassium phosphate tribasic anhydrous (202 mg, 949 μmol) were mixed in degassed dioxane (8.0 mL) and water (2.0 mL) under argon atmosphere. RuPhos Pd G4 (13.5 mg, 15.8 μmol) was added to the reaction mixture. The resulting mixture was stirred at 90°C for 16 hr. under argon atmosphere. The reaction mixture was cooled to room temperature and concentrated under reduced pressure. The residue was diluted with water (10 mL) and extracted with EtOAc (2×20 mL). The combined organic layers were dried over anhydrous Na
2SO
4 and filtered. The filtrate was concentrated under reduced pressure. The residue was subjected to HPLC (0-5 min, 40-90% water+FA (0.1% vol.) - MeOH+FA (0.1% vol.); flow: 30 mL/min, column: Chromatorex 18 SMB100- 5T, 100 x 19 mm, 5 µm), then repurified by HPLC (0-5 min., 40-65% water – ACN, +0.1% vol. of 25% aq. NH
3, flow: 30 mL/min, column: XBridge C18100 ×19 mm, 5 µm) to afford 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-5-isopropyl-4-[[4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidine (6.90 mg, 13.2 μmol, 3.2% yield from [4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methanol) as an off-white solid.
1H NMR (600 MHz, DMSO-d6) δ 0.83 – 0.88 (m, 2H), 0.99 – 1.03 (m, 2H), 1.29 (d, 6H), 1.64 – 1.70 (m, 1H), 3.12 – 3.18 (m, 1H), 3.77 (s, 3H), 3.83 (s, 3H), 5.51 (s, 2H), 7.58 (d, 2H), 7.73 (d, 2H), 7.92 (s, 1H), 8.58 (s, 1H), 8.65 (s, 1H). MS (ESI): [M+H]+ m/z: calcd 525.26; found 525.2. Example 44 (Compound 108)
Step 1: The synthesis of 3,5-difluoro-4-hydrazino-benzonitrile [1125] A solution of 3,4,5-trifluorobenzonitrile (10.4 g, 66.2 mmol) and hydrazine monohydrate (4.64 g, 92.7 mmol, 4.52 mL) in dioxane (150 mL) was stirred at 60 °C for 64 hr. The reaction mixture was cooled to room temperature and concentrated under reduced pressure. The residue was triturated with cold MTBE (75 mL). The solid precipitate formed was filtered off and dried on air to afford 3,5-difluoro-4-hydrazino-benzonitrile (7.50 g, 44.3 mmol, 67.0% yield) as a light-yellow solid which was used in the next steps without further purification.
1H NMR (400 MHz, DMSO-d6) δ 4.52 (s, 2H), 7.29 (s, 1H), 7.43 – 7.54 (m, 2H). GCMS: [M]+ m/z: calcd 169.05; found 169.0 Step 2: The synthesis of 3,5-difluoro-4-[5-methyl-3-(trifluoromethyl)pyrazol-1-yl]benzonitrile A mixture of 3,5-difluoro-4-hydrazino-benzonitrile (5.00 g, 29.6 mmol) and 1,1,1- trifluoropentane-2,4-dione (4.78 g, 31.0 mmol, 3.75 mL) in AcOH (75 mL) was stirred at 110°C for 16 hr. The reaction mixture was cooled to room temperature and concentrated under reduced pressure. The residue was subjected to FCC (SiO
2, gradient Hexanes - EtOAc) to afford 3,5-difluoro-4-[5-methyl-3-(trifluoromethyl)pyrazol-1-yl]benzonitrile (2.30 g, 8.01 mmol, 27.1% yield) as a light-yellow solid.
1H NMR (400 MHz, DMSO-d6) δ 2.22 (s, 3H), 6.89 (s, 1H), 8.19 (d, 2H). MS (ESI): [M+H]+ m/z: calcd 288.06; found 288.0. Step 3: The synthesis of 3,5-difluoro-4-[5-methyl-3-(trifluoromethyl)pyrazol-1-yl]benzoic acid [1126] A solution of sodium hydroxide (961 mg, 24.0 mmol) in water (20 mL) was added to a solution of 3,5-difluoro-4-[5-methyl-3-(trifluoromethyl)pyrazol-1-yl]benzonitrile (2.30 g, 8.01 mmol) in EtOH (20 mL). The resulting mixture was stirred at 70 °C for 30 hr. The reaction mixture was cooled to room temperature and concentrated under reduced pressure. The residue was diluted with water (125 mL) and acidified with an aqueous 2 N HCl to pH<3. The resulting precipitate was filtered off to afford 3,5-difluoro-4-[5-methyl-3- (trifluoromethyl)pyrazol-1-yl]benzoic acid (2.00 g, 6.53 mmol, 81.6% yield) as a yellow solid which was used in the next steps without further purification.
1H NMR (500 MHz, DMSO- d6) δ 2.22 (s, 3H), 6.86 (s, 1H), 7.84 (d, 2H). MS (ESI): [M+H]+ m/z: calcd 307.05; found 307.0. Step 4: The synthesis of [3,5-difluoro-4-[5-methyl-3-(trifluoromethyl)pyrazol-1- yl]phenyl]methanol [1127] Borane dimethyl sulfide complex (2.48 g, 32.7 mmol, 3.10 mL) was added dropwise to a solution of 3,5-difluoro-4-[5-methyl-3-(trifluoromethyl)pyrazol-1-yl]benzoic acid (2.00 g, 6.53 mmol) in THF (125 mL). The resulting mixture was stirred at room temperature for 48 hr. The reaction mixture was cooled to 0 °C. MeOH (100 mL) was added dropwise to the reaction mixture. The resulting mixture was stirred at 0 °C for 4 hr. The mixture was concentrated under reduced pressure. The residue was redissolved in MeOH (100 mL) and the mixture was concentrated under reduced pressure. The residue was dissolved in MTBE (100 mL). The organic layer was washed with aqueous NaOH (3×25 mL, 10%), dried over anhydrous Na
2SO
4 and concentrated under reduced pressure to afford [3,5-difluoro-4-[5- methyl-3-(trifluoromethyl)pyrazol-1-yl]phenyl]methanol (1.30 g, 4.45 mmol, 68.1% yield) as a white solid which was used in the next step without further purification.
1H NMR (500 MHz, DMSO-d6) δ 2.18 (s, 3H), 4.62 (d, 2H), 5.64 (t, 1H), 6.83 (s, 1H), 7.34 (d, 2H). MS (ESI): [M+H]+ m/z: calcd 293.07; found 293.0. Step 5: The synthesis of 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[3,5-difluoro-4-[5- methyl-3-(trifluoromethyl)pyrazol-1-yl]phenyl]methoxy]-5-methoxy-pyrimidine [1128] NaH (19.3 mg, 483 μmol, 60% dispersion in mineral oil) was added to a solution of [3,5-difluoro-4-[5-methyl-3-(trifluoromethyl)pyrazol-1-yl]phenyl]methanol (160 mg, 438 μmol) and 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-5-methoxy-4-methylsulfonyl- pyrimidine I-6 (147 mg, 438 μmol) in DMF (10 mL). The reaction mixture was stirred at room temperature for 15 hr. The reaction mixture was diluted with water (25 mL). The resulting precipitate was filtered off and re-dissolved in ACN (20 mL) and DMF (3 mL). Metal scavenger SiliaMetS® Dimercaptotriazine (50 mg) was added to this solution. The resulting suspension was stirred at room temperature for 10 hr. The mixture was filtered. The filtrate was subjected to HPLC (2-10 min, 10-50% water+FA (0.1% vol.) - ACN+FA (0.1% vol.); flow: 30 mL/min, column: Waters SunFire C18, 100 x 19 mm, 5 µm) to afford 2-(4- cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[3,5-difluoro-4-[5-methyl-3- (trifluoromethyl)pyrazol-1-yl]phenyl]methoxy]-5-methoxy-pyrimidine (85.0 mg, 155 μmol, 35.4% yield) as a white solid.
1H NMR (600 MHz, DMSO-d6) δ 0.83 – 0.89 (m, 2H), 1.00 – 1.04 (m, 2H), 1.66 – 1.72 (m, 1H), 2.19 (s, 3H), 3.82 (s, 3H), 3.96 (s, 3H), 5.52 (s, 2H), 6.83 (s, 1H), 7.53 (d, 2H), 8.45 (s, 1H), 8.64 (s, 1H). MS (ESI): [M+H]+ m/z: calcd 549.19; found 549.4. Example 45 (Compound 40)

The synthesis of starting 2-chloro-5-methoxy-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidine is described for Compound 12 and compound 132. Step 1: The synthesis of 5-bromo-6-cyclopropyl-pyrimidin-4-ol [1129] N-Bromosuccinimide (2.75 g, 15.4 mmol) was added portion wise to a solution of 6- cyclopropylpyrimidin-4-ol (2.00 g, 14.7 mmol) in ACN (14.7 mL) at room temperature. The reaction mixture was stirred at room temperature for 1 hr. The reaction mixture was concentrated under reduced pressure. The residue was diluted with aqueous thiosulfate (5 % wt.). The resulting mixture was stirred for 10 min. The solid precipitate was filtered off, washed with water and dried on air to afford 5-bromo-6-cyclopropyl-pyrimidin-4-ol (2.60 g, 12.1 mmol, 82.3% yield) as a light-yellow solid which was used in the next steps without further purification.
1H NMR (500 MHz, DMSO-d6) δ 0.95 – 1.05 (m, 4H), 2.31 – 2.35 (m, 1H), 8.04 (s, 1H), 12.75 (br, 1H). MS (ESI): [M+H]+ m/z: calcd 214.98; found 215.0. Step 2: The synthesis of 5-bromo-4-cyclopropyl-6-(fluoromethoxy)pyrimidine 5-bromo-6-cyclopropyl-pyrimidin-4-ol (900 mg, 4.19 mmol), fluoroiodomethane (3.00 g, 18.8 mmol) and silver carbonate (1.05 g, 6.28 mmol) were mixed in chloroform (5.0 mL). The reaction mixture was stirred at 50 °C for 72 hr. The reaction mixture was cooled to room temperature and filtered. The filtrate was concentrated under reduced pressure. The residue was subjected to flash column chromatography (SiO
2, gradient DCM - EtOAc) to afford 5- bromo-4-cyclopropyl-6-(fluoromethoxy)pyrimidine (350 mg, 1.42 mmol, 33.9% yield) as a yellow solid.
1H NMR (500 MHz, CDCl
3) δ 1.12 – 1.16 (m, 2H), 1.19 – 1.23 (m, 2H), 2.54 – 2.60 (m, 1H), 6.11 (d, 2H, CH2F), 8.49 (s, 1H). MS (ESI): [M+H]+ m/z: calcd 247.00; found 247.0. Step 3: The synthesis of 4-cyclopropyl-6-(fluoromethoxy)-5-(4,4,5,5-tetramethyl-1,3,2- dioxaborolan-2-yl)pyrimidine [1130] Bis(diphenylphosphino)ferrocene]dichloropalladium(II)-DCM (33.0 mg, 40.5 μmol) was added to a mixture of 5-bromo-4-cyclopropyl-6-(fluoromethoxy)pyrimidine (200 mg, 810 μmol), cesium pivalate (322 mg, 1.38 mmol) and bis(pinacolato)diboron (308 mg, 1.21 mmol) in degassed dioxane (3.0 mL) under argon atmosphere. The reaction mixture was stirred at 75 °C for 12 hr. The reaction mixture was cooled to room temperature and filtered through a pad of silica gel. The filtrate was concentrated under reduced pressure to afford 4- cyclopropyl-6-(fluoromethoxy)-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyrimidine (700 mg, crude) as a yellow oil which as used in the next step without further purification. MS (ESI): [M+H]+ m/z: calcd 295.17; found 295.2. Step 4: The synthesis of 2-[4-cyclopropyl-6-(fluoromethoxy)pyrimidin-5-yl]-5-methoxy-4-[[4- [1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidine [1131] 4-cyclopropyl-6-(fluoromethoxy)-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2- yl)pyrimidine (363 mg, 1.23 mmol), 2-chloro-5-methoxy-4-[[4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidine (176 mg, 440 μmol), potassium phosphate tribasic (281 mg, 1.32 mmol) and XPhos Pd G3 (22.4 mg, 26.4 μmol) were mixed in degassed dioxane (9 mL) and water (1 mL) under argon atmosphere. The reaction mixture was stirred at 75 °C for 12 hr. The reaction mixture was cooled to room temperature and subjected to HPLC (2-10 min., 35-50% ACN - water +0.1% vol. of 25% aq. NH
3, flow: 30 mL/min, column: YMC-Actus Triar 100 ×19 mm, 5 µm) to afford 2-[4-cyclopropyl-6- (fluoromethoxy)pyrimidin-5-yl]-5-methoxy-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidine (17.0 mg, 32.1 μmol, 7.28% yield) as a light-yellow solid.
1H NMR (600 MHz, DMSO-d6) δ 0.90 – 0.94 (m, 2H), 1.04 – 1.08 (m, 2H), 1.77 – 1.82 (m, 1H), 3.77 (s, 3H), 3.94 (s, 3H), 5.49 (s, 2H), 6.05 (d, 2H, CH2F), 7.57 (d, 2H), 7.72 (d, 2H), 7.93 (s, 1H), 8.45 (s, 1H), 8.73 (s, 1H). MS (ESI): [M+H]+ m/z: calcd 531.20; found 531.0. Example 46 (Compound 17)

Step 1: The synthesis of 5-tert-butyl-2-chloro-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidine [1132] Potassium tert-butoxide (98.1 mg, 874 μmol) was added to a solution of [4-[1-methyl- 4-(trifluoromethyl)imidazol-2-yl]phenyl]methanol I-2b (150 mg, 585 μmol) in dioxane (5 mL). The reaction mixture was stirred at room temperature for 20 min. The reaction mixture was added to a solution of 5-tert-butyl-2,4-dichloro-pyrimidine (120 mg, 585 μmol) in dioxane (5 mL). The resulting mixture was stirred at 50 °C for 16 hr. The reaction mixture was cooled to room temperature, diluted with water (10 mL) and extracted with EtOAc (20 mL). The organic layer was washed with water (10 mL) and brine (10 mL) and concentrated under reduced pressure to afford 5-tert-butyl-2-chloro-4-[[4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidine (276 mg, crude) as a yellow oil which was used in the next steps without further purification. MS (ESI): [M+H]+ m/z: calcd 425.14; found 425.0. Step 2: The synthesis of 5-tert-butyl-2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[4-[1- methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidine [1133] 5-tert-butyl-2-chloro-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidine (276 mg, crude), (4-cyclopropyl-6-methoxy-pyrimidin-5- yl)boronic acid I-5 (163 mg, 838 μmol) and potassium phosphate tribasic (356 mg, 1.68 mmol) were mixed in a degassed mixture of dioxane (3 mL) and water (150 μL) under argon atmosphere. XPhosPdG3 (23.7 mg, 27.9 μmol) was added to the mixture. The reaction mixture was degassed ones more and stirred at 90 °C for 16 hr. under argon atmosphere. The reaction mixture was cooled to room temperature, diluted with EtOAc (10 mL) and washed with water (2×5 mL). SiliaMetS® Dimercaptotriazine (100 mg) was added to the organic layer. The resulting mixture was stirred for 30 min and filtered. The filtrate was concentrated under reduced pressure. The residue was subjected to HPLC (0.5-6.5 min., 50-100% water - ACN, column: SunFire C18100×19 mm, 5 µm) to afford 5-tert-butyl-2-(4-cyclopropyl-6- methoxy-pyrimidin-5-yl)-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidine (11.0 mg, 20.4 μmol, 3.7% yield from 5-tert-butyl-2,4- dichloro-pyrimidine) as a yellow solid.
1H NMR (600 MHz, DMSO-d6) δ 0.84 – 0.88 (m, 2H), 0.99 – 1.03 (m, 2H), 1.40 (s, 9H), 1.66 – 1.71 (m, 1H), 3.78 (s, 3H), 3.83 (s, 3H), 5.54 (s, 2H), 7.58 (d, 2H), 7.74 (d, 2H), 7.92 (s, 1H), 8.57 (s, 1H), 8.65 (s, 1H). MS (ESI): [M+H]+ m/z: calcd 539.28; found 539.2. Example 47 (Compound 73)
Step 1: The synthesis of 2,5-dichloro-4-((4-(1-methyl-4-(trifluoromethyl)-1H-imidazol-2- yl)benzyl)oxy)pyrimidine [1134] (4-(1-methyl-4-(trifluoromethyl)-1H-imidazol-2-yl)phenyl)methanol I-2b (0.25 g, 976 μmol), 2,4,5-trichloropyrimidine (179 mg, 976 μmol) and Cs
2CO
3 (318 mg, 976 μmol) were mixed in Acetonitrile (5 mL). The reaction mixture was stirred at 80 °C for 3 hr. The reaction mixture was cooled to room temperature and filtered. The filtrate was concentrated under reduced pressure. The residue was subjected to flash-column chromatography (SiO
2, gradient chloroform – MTBE) to afford 2,5-dichloro-4-((4-(1-methyl-4-(trifluoromethyl)-1H- imidazol-2-yl)benzyl)oxy)pyrimidine (320 mg, 0.793 mmol, 81.4% yield) as white solid. MS (ESI): [M+H]+ m/z: calcd 403.05; found 403.0. Step 2: The synthesis of 5-chloro-4'-cyclopropyl-6'-methoxy-4-((4-(1-methyl-4- (trifluoromethyl)-1H-imidazol-2-yl)benzyl)oxy)-2,5'-bipyrimidine [1135] The 2,5-dichloro-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidin (100 mg, 248 μmol), (4-cyclopropyl-6-methoxypyrimidin-5- yl)boronic acid I-5 (57.7 mg, 298 μmol), XPhos Pd G3 (10.5 mg, 12.4 μmol) and Potassium phosphate tribasic anhydrous (158 mg, 744 μmol) were mixed in degassed dioxane (5 mL) and water (1 mL). The reaction mixture was stirred at 75 °C for 24 hr. under argon atmosphere. The reaction mixture was cooled, diluted with EtOAc (5 mL), washed water (3 mL) and brine (3 mL). To the obtained solution SiliaMetS® Dimercaptotriazine (10 mg) was added. The mixture was stirred for 30 min and filtered. The filtrate was evaporated under reduce pressure. The residue was subjected to HPLC (0-5 min, 40-90% water – MeOH, flow: 30 mL/min, column: Kinetex PFP 100 × 21.2 mm, 5 µm) to afford 5-chloro-4'-cyclopropyl- 6'-methoxy-4-((4-(1-methyl-4-(trifluoromethyl)-1H-imidazol-2-yl)benzyl)oxy)-2,5'- bipyrimidine (2.0 mg, 3.9 μmol, 1.6% yield) as a light yellow solid.
1H NMR (600 MHz, DMSO-d6) δ 0.85 – 0.90 (m, 2H), 1.02 – 1.06 (m, 2H), 1.74 – 1.80 (m, 1H), 3.78 (s, 3H), 3.84 (s, 3H), 5.58 (s, 2H), 7.60 (d, 2H), 7.74 (d, 2H), 7.93 (s, 1H), 8.67 (s, 1H), 8.88 (s, 1H). MS (ESI): [M+H]+ m/z: calcd 517.16; found 517.2.

Step 1: The synthesis methyl 3-chloro-4-[4-(trifluoromethyl)-1H-imidazol-2-yl]benzoate [1136] 3,3-dibromo-1,1,1-trifluoro-propan-2-one (15.0 g, 55.4 mmol) was added to a solution of sodium acetate (9.09 g, 111 mmol) in water (60 mL). The reaction mixture was stirred at 95°C for 1 hr. The reaction mixture was cooled to room temperature. A mixture of methyl 3- chloro-4-formyl-benzoate (10.0 g, 50.4 mmol) and aqueous ammonium hydroxide (100 mL, 25% wt.) in MeOH (260 mL) was added to the reaction mixture. The resulting mixture was stirred at room temperature for 14 hr. The mixture was concentrated under reduced pressure. The residue was diluted with water (300 mL) and extracted with CHCl3 (3×100 mL). The combined organic layers were washed with brine (100 mL), dried over anhydrous Na
2SO
4 and filtered through the pad of silica gel. The filtrate was concentrated under reduced pressure to afford methyl 3-chloro-4-[4-(trifluoromethyl)-1H-imidazol-2-yl]benzoate (11.0 g, 36.1 mmol, 71.7% yield) as an yellow solid which was used in the next steps without further purification. MS (ESI): [M+H]+ m/z: calcd 305.03; found 305.0. Step 2: The synthesis of methyl 3-chloro-4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]benzoate [1137] Methyliodide (8.67 g, 61.1 mmol, 3.80 mL) was added to a stirred mixture of methyl 3-chloro-4-[4-(trifluoromethyl)-1H-imidazol-2-yl]benzoate (7.75 g, 20.4 mmol), K
2CO
3 (4.22 g, 30.5 mmol) and Cs
2CO
3 (6.63 g, 20.4 mmol) in DMF (150 mL). The reaction mixture was stirred at 50 °C for 48 hr. The reaction mixture was cooled to room temperature, diluted with water (300 mL) and extracted with EtOAc (4×120 mL). The combined organic layers were washed with brine (150 mL) and filtered through the pad of silica gel. The filtrate was dried over anhydrous Na
2SO
4 and concentrated under reduced pressure. The residue was subjected to flash column chromatography (SiO
2, gradient Hexanes - EtOAc) to afford methyl 3- chloro-4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]benzoate (3.00 g, 9.41 mmol, 46.3% yield) as a light-yellow solid which was used in the next steps.
1H NMR (500 MHz, CDCl
3) δ 3.58 (s, 3H), 3.95 (s, 3H), 7.36 (s, 1H), 7.59 (d, 1H), 8.02 (d, 1H), 8.15 (s, 1H). MS (ESI): [M+H]+ m/z: calcd 319.05; found 319.2. Step 3: The synthesis of [3-chloro-4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methanol [1138] Diisobutylaluminum hydride (3.35 g, 23.5 mmol, 4.19 mL) was added dropwise to the solution of methyl 3-chloro-4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]benzoate (3.00 g, 9.41 mmol) in DCM (30 mL) at -78 °C under argon atmosphere. The reaction mixture was stirred at -78 °C for 5 hr. The reaction mixture was allowed to warm to 0 °C, quenched by addition of aqueous citric acid solution (50 mL, 10% wt.) and stirred at room temperature overnight. The resulting mixture was filtered. The filtrate was extracted with DCM (2×30 mL). The combined organic layers were dried over anhydrous Na
2SO
4 and concentrated under reduced pressure to afford [3-chloro-4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methanol (1.50 g, 5.16 mmol, 54.8% yield) as an yellow gum which was used in the next steps without further purification. MS (ESI): [M+H]+ m/z: calcd 291.05; found 291.0. Step 4: The synthesis of 4-[[3-chloro-4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]-2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-5-methyl-pyrimidine [1139] NaH (12.3 mg, 322 μmol, 60% dispersion in mineral oil) was added to a solution of [3-chloro-4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methanol (85.0 mg, 292 μmol) in DMF (10 mL). The reaction mixture was stirred at room temperature for 30 min.2- (4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-5-methyl-4-methylsulfonyl-pyrimidine I-7 (93.7 mg, 292 μmol) was added to the reaction mixture. The resulting mixture was stirred at room temperature for 15 hr. The mixture was diluted with water (10 mL) and extracted with EtOAc (3×10 mL). The combined organic layers were dried over anhydrous Na
2SO
4 and concentrated under reduced pressure. The residue was subjected to HPLC (2-10 min, 40 - 60% water - ACN; flow: 30 mL/min, column: Waters SunFire C18, 100 x 19 mm, 5 µm) to afford 4-[[3-chloro-4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]-2-(4- cyclopropyl-6-methoxy-pyrimidin-5-yl)-5-methyl-pyrimidine (64.0 mg, 121 μmol, 41.2% yield) as a solid.
1H NMR (600 MHz, DMSO-d6) δ 0.83 – 0.88 (m, 2H), 0.99 – 1.03 (m, 2H), 1.63 – 1.69 (m, 1H), 2.23 (s, 3H), 3.50 (s, 3H), 3.82 (s, 3H), 5.52 (s, 2H), 7.56 (s, 2H), 7.72 (s, 1H), 7.97 (s, 1H), 7.56 (s, 1H), 8.64 (s, 1H). MS (ESI): [M+H]+ m/z: calcd 531.18; found 531.2.
[1140] NaH (12.3 mg, 322 μmol, 60% dispersion in mineral oil) was added to a solution of [3-chloro-4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methanol (85.0 mg, 292 μmol) in DMF (10 mL). The reaction mixture was stirred at room temperature for 30 min.2- (4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-5-methoxy-4-methylsulfonyl-pyrimidine I-6 (98.4 mg, 292 μmol) was added to the reaction mixture. The resulting mixture was stirred at room temperature for 15 hr. The mixture was diluted with water (10 mL) and extracted with EtOAc (3×10 mL). The combined organic layers were dried over anhydrous Na
2SO
4 and concentrated under reduced pressure. The residue was subjected to HPLC (2-10 min, 30% water+FA (0.1% vol.) - ACN+FA (0.1% vol.); flow: 30 mL/min, column: Waters SunFire C18, 100 x 19 mm, 5 µm) to afford 4-[[3-chloro-4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]-2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-5-methoxy-pyrimidine (63.0 mg, 115 μmol, 39.4% yield) as a light-yellow gum.
1H NMR (600 MHz, DMSO-d6) δ 0.84 – 0.91 (m, 2H), 1.00 – 1.04 (m, 2H), 1.66 – 1.73 (m, 1H), 3.51 (s, 3H), 3.83 (s, 3H), 3.95 (s, 3H), 5.50 (s, 2H), 7.52 – 7.59 (m, 2H), 7.71 (s, 1H), 7.98 (s, 1H), 8.43 (s, 1H), 8.63 (s, 1H). MS (ESI): [M+H]+ m/z: calcd 547.17; found 547.4. Example 50 (Compound 143)
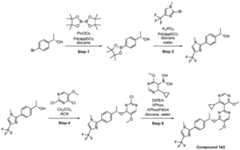
Step 1: The synthesis of (S)-1-(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)ethanol [1141] (S)-1-(4-bromophenyl)ethanol (5.00 g, 24.9 mmol), 4,4,5,5-tetramethyl-2-(4,4,5,5- tetramethyl-1,3,2-dioxaborolan-2-yl)-1,3,2-dioxaborolane (6.95 g, 27.4 mmol), cesium pivalate (11.6 g, 49.7 mmol) and bis(diphenylphosphino)ferrocene]dichloropalladium(II)- DCM (1.02 g, 1.24 mmol) were mixed in degassed dioxane (100 mL). The resulting mixture was stirred at 100 °C for 3 hr. The reaction mixture was cooled to room temperature, filtered and concentrated under reduced pressure to afford (S)-1-(4-(4,4,5,5-tetramethyl-1,3,2- dioxaborolan-2-yl)phenyl)ethanol (10.0 g, crude) which was used in the next steps without further purification. Step 2: The synthesis of (S)-1-(4-(1-methyl-4-(trifluoromethyl)-1H-imidazol-2- yl)phenyl)ethanol [1142] (1S)-1-[4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl]ethanol (1.30 g, crude), 2-bromo-1-methyl-4-(trifluoromethyl)-1H-imidazole (1.00 g, 4.37 mmol), N,N- Diisopropylethylamine (1.41 g, 10.92 mmol, 1.90 mL), XPhos Pd G4 (188 mg, 218 μmol), and XPhos (104 mg, 218 μmol) were mixed in a degassed mixture of dioxane (40 mL) and water (9 mL) under argon atmosphere. The reaction mixture was stirred at 95 °C for 12 hr. The mixture was cooled to room temperature and concentrated under reduced pressure. The residue was redissolved in EtOAc (100 mL), washed with water (50 mL) and brine (50 mL) and concentrated under reduced pressure. The residue was subjected to flash-column chromatography (SiO
2, gradient chloroform - MTBE) to afford (S)-1-(4-(1-methyl-4- (trifluoromethyl)-1H-imidazol-2-yl)phenyl)ethanol (170 mg, 629 μmol, 19.4% yield from (S)-1-(4-bromophenyl)ethanol) as a light yellow oil. MS (ESI): [M+H]+ m/z: calcd 271.13; found 271.2. Step 3: The synthesis of (S)-2-chloro-5-methoxy-4-(1-(4-(1-methyl-4-(trifluoromethyl)-1H- imidazol-2-yl)phenyl)ethoxy)pyrimidine [1143] (1S)-1-[4-[1-Methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]ethanol (170 mg, 629 μmol), 2,4-dichloro-5-methoxypyrimidine (113 mg, 629 μmol) and Cs
2CO
3 (205 mg, 629 μmol) were mixed in ACN (5.0 mL). The reaction mixture was stirred at 80 °C for 20 hr. The reaction mixture was cooled to room temperature and filtered. The filtrate was concentrated under reduced pressure to afford (S)-2-chloro-5-methoxy-4-(1-(4-(1-methyl-4- (trifluoromethyl)-1H-imidazol-2-yl)phenyl)ethoxy)pyrimidine (250 mg, 606 μmol, 96.2% yield) as a light-yellow gum which was used in the next step without further purification. MS (ESI): [M+H]+ m/z: calcd 413.10; found 413.0. Step 4: The synthesis of (S)-4'-cyclopropyl-5,6'-dimethoxy-4-(1-(4-(1-methyl-4- (trifluoromethyl)-1H-imidazol-2-yl)phenyl)ethoxy)-2,5'-bipyrimidine [1144] (S)-2-Chloro-5-methoxy-4-(1-(4-(1-methyl-4-(trifluoromethyl)-1H-imidazol-2- yl)phenyl)ethoxy)pyrimidine (250 mg, 606 μmol), (4-cyclopropyl-6-methoxy-pyrimidin-5- yl)boronic acid I-5 (141 mg, 727 μmol), XPhos Pd G4 (39.1 mg, 45.4 μmol), DIPEA (235 mg, 1.82 mmol, 317 μL) and XPhos (21.7 mg, 45.4 μmol) were mixed in a degassed mixture of dioxane (8 mL) and water (2 mL) under argon atmosphere at room temperature. The reaction mixture was stirred at 95 °C for 12 hr. The reaction mixture was cooled to room temperature. Pd scavenger (100 mg) was added to the reaction mixture. The resulting mixture was stirred at room temperature for 30 min, then solids were filtered out. The filtrate was concentrated under reduced pressure. The residue was subjected to HPLC (0-1-6 min., 50-50- 80% water – MeOH, +0.1% vol. of 25% aq. NH
3, flow: 30 mL/min, column: YMC Triart C18100×20 mm, 5 µm) to afford (S)-4'-cyclopropyl-5,6'-dimethoxy-4-(1-(4-(1-methyl-4- (trifluoromethyl)-1H-imidazol-2-yl)phenyl)ethoxy)-2,5'-bipyrimidine (65.0 mg, 124 μmol, 20.4% yield) as a yellow solid.
1H NMR (600 MHz, DMSO-d6) δ 0.70 – 0.76 (m, 1H), 0.79 – 0.84 (m, 1H), 0.93 – 1.01 (m, 2H), 1.55 – 1.61 (m, 1H), 1.64 (d, 3H), 3.77 (s, 3H), 3.78 (s, 3H), 3.96 (s, 3H), 6.31 (q, 1H), 7.54 (d, 2H), 7.70 (d, 2H), 7.93 (s, 1H), 8.40 (s, 1H), 8.62 (s, 1H). MS (ESI): [M+H]+ m/z: calcd 527.23; found 527.2. Enantiopurity: 99.8% (column: Chiralpak IC, 250 × 4.6 mm, 5 μm; mobile phase: Hexane- IPA-MeOH, 70-15-15; flow: 0.8 mL/min; RT=16.5 min). Example 51 (Compound 140)

Step 1: Synthesis of 2-chloro-5-cyclobutyl-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidine Potassium 2-methylpropan-2-olate (112 mg, 1.01 mmol) was added to a stirred solution of [4- [1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methanol I-2b (172 mg, 672 μmol) in THF (6 mL) under argon atmosphere at room temperature. The mixture was stirred for 5 mins, then 2,4-dichloro-5-cyclobutyl-pyrimidine (150 mg, 739 μmol) was added to the reaction mixture. The resulting mixture was stirred for 14 hr. The reaction mixture was diluted with water (10 mL) and extracted with EtOAc (2 × 15 mL). The combined organic layers were washed with brine (10 mL), dried over anhydrous Na
2SO
4 and concentrated under reduced pressure. The residue was subjected to HPLC (2-10 min, 20-45% water – MeOH, +0.1% vol. of 25% aq. NH
3, flow: 30 mL/min, column: SunFire C18100 ×19 mm, 5 µm) to afford 2-chloro-5-cyclobutyl-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidine (102 mg, 241 μmol, 35.9% yield) as yellow solid. MS (ESI): [M+H]+ m/z: calcd 423.15; found 423.0. Step 2: Synthesis of 5-cyclobutyl-2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[4-[1- methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidine [1145] To a stirred solution of 2-chloro-5-cyclobutyl-4-[[4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidine (102 mg, 241 μmol) in a degassed mixture of dioxane (7 mL) and water (2 mL) (4-cyclopropyl-6-methoxy-pyrimidin- 5-yl) boronic acid (60.0 mg, 314 μmol), DIPEA (153 mg, 1.19 mmol, 207 μL) and RuPhos Pd G4 (10.3 mg, 12.1 μmol) were added under argon atmosphere. The resulting mixture was stirred under argon atmosphere at 90 °C for 16 hr. The reaction mixture was cooled to room temperature and concentrated under reduced pressure. The residue was diluted with water (10 mL) and EtOAc (25 mL). The organic layer was separated, washed with brine (10 mL), dried over anhydrous Na
2SO
4 and concentrated under reduced pressure. The residue was subjected to HPLC (2-10 min, 20-45% water – ACN, flow: 30 mL/min, column: SunFire C18100 ×19 mm, 5 µm) to afford 5-cyclobutyl-2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[4-[1- methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidine (48.0 mg, 89.5 μmol, 37.1% yield) as light-yellow solid.
1H NMR (600 MHz, DMSO-d6) δ 0.82 – 0.87 (m, 2H), 0.99 – 1.03 (m, 2H), 1.64 – 1.70 (m, 1H), 1.90 – 1.86 (m, 1H), 1.98 – 2.08 (m, 1H), 2.20 – 2.27 (m, 2H), 2.28 – 2.34 (m, 2H), 3.61 – 3.69 (m, 1H), 3.77 (s, 3H), 3.82 (s, 3H), 5.49 (s, 2H), 7.56 (d, 2H), 7.72 (d, 2H), 7.93 (s, 1H), 8.56 (s, 1H), 8.64 (s, 1H). MS (ESI): [M+H]+ m/z: calcd 537.26; found 537.2.

Step 1: The synthesis of 2-chloro-5-methoxy-4-[1-[4-[1-methyl-4-(trifluoromethyl)imidazol- 2-yl]phenyl]ethoxy]pyrimidine The synthesis of the starting 1-[4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]ethanol is described in Example 55 (Compound 100, 74, and 205). [1146] NaH (37.0 mg, 925 μmol, 60% dispersion in mineral oil) was added to a solution 1- [4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]ethanol (0.250 g, 925 μmol) in DMF (3 mL) at 0 °C. The reaction mixture was stirred at 0 °C for 1 hr.2,4-Dichloro-5-methoxy- pyrimidine (166 mg, 925 μmol) was added to the reaction mixture. The resulting mixture was stirred at room temperature for 16 hr. The reaction mixture was diluted with water (10 mL) and extracted with EtOAc (2×10 mL). The combined organic layers were washed with brine (10 mL) and concentrated under reduced pressure to afford 2-chloro-5-methoxy-4-[1-[4-[1- methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]ethoxy]pyrimidine (0.380 g, 920 μmol, 99.5% yield) as an yellow solid which was used in the next steps without further purification. MS (ESI): [M+H]+ m/z: calcd 413.12; found 413.0. Step 2: The synthesis of 4'-cyclopropyl-5,6'-dimethoxy-4-(1-(4-(1-methyl-4-(trifluoromethyl)- 1H-imidazol-2-yl)phenyl)ethoxy)-2,5'-bipyrimidine 2-Chloro-5-methoxy-4-[1-[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]ethoxy]pyrimidine (0.200 g, 392 μmol), 4-cyclopropyl-6-methoxy-5-(4,4,5,5- tetramethyl-1,3,2-dioxaborolan-2-yl)pyrimidine (141 mg, 510 μmol), potassium phosphate tribasic (250 mg, 1.18 mmol) and XPhos Pd G3 (16.6 mg, 19.6 μmol) were mixed in a degassed mixture of dioxane (3 mL) and water (300 µL). The reaction mixture was stirred at 95 °C for 14 hr. The reaction mixture was cooled to room temperature. The reaction mixture was diluted with EtOAc (10 mL) and washed with water and brine (2 × 5 mL). To the obtained organic phase SiliaMetS® Dimercaptotriazine (20 mg) was added, and the mixture was stirred for 30 min. The mixture was filtered. The filtrate was concentrated under reduced pressure. The residue was subjected to HPLC (0.5-6.5 min, 47% water – acetonitrile, flow: 30 mL/min, column: SunFire C18100 ×19 mm, 5 µm) to afford 4'-cyclopropyl-5,6'-dimethoxy- 4-(1-(4-(1-methyl-4-(trifluoromethyl)-1H-imidazol-2-yl)phenyl)ethoxy)-2,5'-bipyrimidine (90.0 mg, 159 μmol, 43.5% yield) as a yellow solid.
1H NMR (600 MHz, DMSO-d6) δ 0.68 – 0.75 (m, 1H), 0.77 – 0.82 (m, 1H), 0.91 – 1.00 (m, 2H), 1.54 – 1.59 (m, 1H), 1.63 (d, 3H), 3.74 – 3.78 (m, 6H), 3.95 (s, 3H), 6.29 (q, 1H), 7.53 (d, 2H), 7.69 (d, 2H), 7.91 (s, 1H), 8.38 (s, 1H), 8.61 (s, 1H). MS (ESI): [M+H]+ m/z: calcd 527.23; found 527.2. Step 3: The synthesis of rel-2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-5-methoxy-4-[(1R)- 1-[4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]ethoxy]pyrimidine 111 and rel-2-(4- cyclopropyl-6-methoxy-pyrimidin-5-yl)-5-methoxy-4-[(1S)-1-[4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]ethoxy]pyrimidine 200 [1147] Racemic 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-5-methoxy-4-[1-[4-[1-methyl- 4-(trifluoromethyl)imidazol-2-yl]phenyl]ethoxy]pyrimidine (90.0 mg, 159 μmol) was subjected to chiral HPLC (column: Chiralpak IC (250 × 20 mm, 5 mkm); Mobile phase : Hexane-IPA-MeOH, 70-15-15.; flow: 15 mL/min) to afford 2-(4-cyclopropyl-6-methoxy- pyrimidin-5-yl)-5-methoxy-4-[rac-(1R)-1-[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]ethoxy]pyrimidine (15.0 mg, 28.5 μmol, 17.9% yield) and 2-(4-cyclopropyl-6- methoxy-pyrimidin-5-yl)-5-methoxy-4-[rac-(1S)-1-[4-[1-methyl-4-(trifluoromethyl)imidazol- 2-yl]phenyl]ethoxy]pyrimidine (17.0 mg, 32.3 μmol, 20.3% yield) as a white solids. Rel-2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-5-methoxy-4-[(1R)-1-[4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]ethoxy]pyrimidine (111):
1H NMR (600 MHz, DMSO-d6) δ 0.68 – 0.75 (m, 1H), 0.77 – 0.82 (m, 1H), 0.91 – 1.00 (m, 2H), 1.54 – 1.59 (m, 1H), 1.63 (d, 3H), 3.74 – 3.78 (m, 6H), 3.95 (s, 3H), 6.29 (q, 1H), 7.53 (d, 2H), 7.69 (d, 2H), 7.91 (s, 1H), 8.38 (s, 1H), 8.61 (s, 1H). MS (ESI): [M+H]+ m/z: calcd 527.23; found 527.4. Enantiopurity: 100% (column: Chiralpak IC, 250 × 4.6 mm, 5 μm; mobile phase: Hexane- IPA-MeOH, 70-15-15; flow: 0.6 mL/min; RT=21.7 min) Rel-2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-5-methoxy-4-[(1S)-1-[4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]ethoxy]pyrimidine (200):
1H NMR (600 MHz, DMSO-d6) δ 0.68 – 0.75 (m, 1H), 0.77 – 0.82 (m, 1H), 0.91 – 1.00 (m, 2H), 1.54 – 1.59 (m, 1H), 1.63 (d, 3H), 3.74 – 3.78 (m, 6H), 3.95 (s, 3H), 6.29 (q, 1H), 7.53 (d, 2H), 7.69 (d, 2H), 7.91 (s, 1H), 8.38 (s, 1H), 8.61 (s, 1H). MS (ESI): [M+H]+ m/z: calcd 527.23; found 527.4. Enantiopurity: 100% (column: Chiralpak IC, 250 × 4.6 mm, 5 μm; mobile phase: Hexane- IPA-MeOH, 70-15-15; flow: 0.6 mL/min; RT=25.4 min).
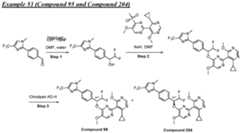
Step 1: Synthesis of 2,2-difluoro-1-(4-(1-methyl-4-(trifluoromethyl)-1H-imidazol-2- yl)phenyl)ethanol [1148] The difluoromethyl(trimethyl)silane (586 mg, 4.72 mmol) was added to mixture of 4- [1-methyl-4-(trifluoromethyl)imidazol-2-yl]benzaldehyde (0.600 g, 2.36 mmol) and CsF (35.9 mg, 236 μmol) in DMF (6 mL) at 0 °C. The reaction mixture was stirred at room temperature for 12 hr. Tetrabutylammonium fluoride (472 μmol, 472 μL, 1M solution in THF) was added to reaction mixture. The resulting mixture was stirred for 2 hr. The mixture was diluted with saturated aqueous solution of NaHCO
3 (10 mL). The resulting mixture was extracted EtOAc (20 mL). Organic layer was separated, dried over anhydrous Na
2SO
4 and concentrated under reduced pressure. The residue was subjected to HPLC (0.5-6.5 min., 25- 50% water – ACN, flow: 30 mL/min, column: SunFire C18100 ×19 mm, 5 µm) to afford 2,2-difluoro-1-(4-(1-methyl-4-(trifluoromethyl)-1H-imidazol-2-yl)phenyl)ethanol (268 mg, 875 μmol, 37.1% yield) as a white solid. MS (ESI): [M+H]+ m/z: calcd 307.11; found 307.2. Step 2: The synthesis of 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[2,2-difluoro-1-[4-[1- methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]ethoxy]-5-methoxy-pyrimidine [1149] NaH (5.88 mg, 147 μmol 60% dispersion in mineral oil) was added to a stirred solution of 2,2-difluoro-1-[4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]ethanol (54.0 mg, 176 μmol) in DMF (2 mL). The reaction mixture was stirred at ambient temperature for 0.5 hr.2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-5-methoxy-4- methylsulfonyl-pyrimidine I-6 (49.4 mg, 147 μmol) was added to the reaction mixture. The resulting mixture was stirred at room temperature for 12 hr. The reaction mixture was diluted with water (10 mL) and extracted with EtOAc (2×10 mL). The combined organic layers were washed with brine (10 mL) and concentrated under reduced pressure. The residue was subjected to HPLC (0.5-6.5 min., 50-75% water – ACN, +0.1% vol. of 25% aq. NH
3, flow: 30 mL/min, column: XBridge 100 ×19 mm, 5 µm) to afford 2-(4-cyclopropyl-6-methoxy- pyrimidin-5-yl)-4-[2,2-difluoro-1-[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]ethoxy]-5-methoxy-pyrimidine (49.0 mg, 87.1 μmol, 59.3% yield) as a white solid.
1H NMR (600 MHz, DMSO-d6) δ 0.64 – 0.70 (m, 1H), 0.74 – 0.80 (m, 1H), 0.90 – 0.99 (m, 2H), 1.49 – 1.55 (m, 1H), 3.73 (s, 3H), 3.78 (s, 3H), 4.02 (s, 3H), 6.46 – 6.68 (m, 2H), 7.60 (d, 2H), 7.77 (d, 2H), 7.94 (s, 1H), 8.51 (s, 1H), 8.62 (s, 1H). MS (ESI): [M+H]+ m/z: calcd 563.21; found 563.0. Step 3: Chiral resolution of 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-5-methoxy-4-[rel- (1R)-2,2-difluoro-1-[4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]ethoxy]pyrimidine (95) and 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-5-methoxy-4-[rel-(1S)-2,2-difluoro-1- [4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]ethoxy]pyrimidine (204) [1150] Racemic 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[2,2-difluoro-1-[4-[1- methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]ethoxy]-5-methoxy-pyrimidine (47.0 mg, 83.6 μmol) was subjected to chiral HPLC (column: CHIRALPAK AD-H (250×20 mm, 5 μm), Mobile Phase: Hexane-IPA-MeOH, 70-15-15, Flow Rate: 15 mL/min) to afford 2-(4- cyclopropyl-6-methoxy-pyrimidin-5-yl)-5-methoxy-4-[rel-(1R)-2,2-difluoro-1-[4-[1-methyl- 4-(trifluoromethyl)imidazol-2-yl]phenyl]ethoxy]pyrimidine (19.0 mg, 33.8 μmol, 40.4% yield) and 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-5-methoxy-4-[rel-(1S)-2,2-difluoro- 1-[4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]ethoxy]pyrimidine (19.0 mg, 33.8 μmol, 40.4% yield) as a white solids. 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-5-methoxy-4-[rel-(1R)-2,2-difluoro-1-[4-[1- methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]ethoxy]pyrimidine (95):
1H NMR (600 MHz, DMSO-d6) δ 0.64 – 0.70 (m, 1H), 0.74 – 0.80 (m, 1H), 0.90 – 0.99 (m, 2H), 1.49 – 1.55 (m, 1H), 3.73 (s, 3H), 3.78 (s, 3H), 4.02 (s, 3H), 6.46 – 6.68 (m, 2H), 7.60 (d, 2H), 7.77 (d, 2H), 7.94 (s, 1H), 8.51 (s, 1H), 8.62 (s, 1H). MS (ESI): [M+H]+ m/z: calcd 563.21; found 563.0 Enantiopurity: 100% (column: CHIRALPAK AD-H, 250 × 4.6 mm, 5 μm; mobile phase: Hexane-IPA-MeOH, 70-15-15; flow: 0.6 mL/min; RT=12.4 min) 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-5-methoxy-4-[rel-(1S)-2,2-difluoro-1-[4-[1- methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]ethoxy]pyrimidine (204):
1H NMR (600 MHz, DMSO-d6) δ 0.64 – 0.70 (m, 1H), 0.74 – 0.80 (m, 1H), 0.90 – 0.99 (m, 2H), 1.49 – 1.55 (m, 1H), 3.73 (s, 3H), 3.78 (s, 3H), 4.02 (s, 3H), 6.46 – 6.68 (m, 2H), 7.60 (d, 2H), 7.77 (d, 2H), 7.94 (s, 1H), 8.51 (s, 1H), 8.62 (s, 1H). MS (ESI): [M+H]+ m/z: calcd 563.21; found 563.4. Enantiopurity: 100% (column: CHIRALPAK AD-H, 250 × 4.6 mm, 5 μm; mobile phase: Hexane-IPA-MeOH, 70-15-15; flow: 0.6 mL/min; RT=6.2 min). Example 54 (Compound 141)

Step 1: The synthesis of 2-chloro-N-methyl-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidin-5-amine [1151] Potassium tert-butoxide (241 mg, 2.15 mmol) was added to a solution of [4-[1- methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methanol I-2b (500 mg, 1.95 mmol) in THF (10 mL) at 0°C. The reaction mixture was stirred at 0 °C for 10 min.2,4-dichloro-N-methyl- pyrimidin-5-amine (347 mg, 1.95 mmol) was added to the mixture. The resulting mixture was stirred at room temperature for 15 hr. The reaction mixture was poured into water (15 mL) and extracted with EtOAc (20 mL). The organic layer was dried over anhydrous Na
2SO
4 and concentrated under reduced pressure to afford 2-chloro-N-methyl-4-[[4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidin-5-amine (750 mg, 1.89 mmol, 96.6% yield) as a yellow solid which was used in the next steps without further purification.
1H NMR (500 MHz, CDCl
3) δ 2.88 (s, 3H), 3.79 (s, 3H), 4.04 (br., 1H), 5.50 (s, 2H), 7.32 (s, 1H), 7.54 (d, 2H), 7.60 (s, 1H), 7.66 (d, 2H). MS (ESI): [M+H]+ m/z: calcd 398.10; found 398.0 Step 2: The synthesis of 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-N-methyl-4-[[4-[1- methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidin-5-amine [1152] 2-chloro-N-methyl-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidin-5-amine (800 mg, 1.51 mmol), (4-cyclopropyl-6-methoxy- pyrimidin-5-yl)boronic acid I-5 (380 mg, 1.96 mmol), RuPhos Pd G4 (96.2 mg, 113 μmol) and potassium phosphate tribasic anhydrous (801 mg, 3.77 mmol) were mixed in degassed mixture of water (1 mL) and dioxane (10 mL) at room temperature under argon atmosphere. The reaction mixture was stirred at 90 °C for 15 hr. The reaction mixture was cooled to room temperature and concentrated under reduced pressure. The residue was diluted with water (10 mL) and extracted with EtOAc (2×50 mL). The combined organic layers were dried over anhydrous Na
2SO
4 and concentrated under reduced pressure. The residue was subjected to HPLC (2-10 min, 30-60% ACN+FA (0.1% vol.); flow: 30 mL/min, column: Waters SunFire C18, 100 x 19 mm, 5 µm) to afford 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-N-methyl- 4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidin-5-amine (180 mg, 352 μmol, 23.3% yield) as a yellow solid. MS (ESI): [M+H]+ m/z: calcd 512.23; found 512.0. Step 3: The synthesis of 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-N,N-dimethyl-4-[[4-[1- methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidin-5-amine [1153] Formaldehyde (440 μmol, 33.0 µL, 37% in water, stab. with 10-15% MeOH) was added to a solution of 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-N-methyl-4-[[4-[1- methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidin-5-amine (75.0 mg, 147 μmol) in AcOH (1.0 mL). The reaction mixture was stirred at room temperature for 30 min. Sodium cyanoborohydride (12.0 mg, 191 μmol) was added to the reaction mixture. The resulting mixture was stirred at room temperature for 20 hr. The reaction mixture was subjected to HPLC (2-10 min, 30-50% ACN+FA (0.1% vol.); flow: 30 mL/min, column: Waters SunFire C18, 100 x 19 mm, 5 µm) to afford 2-(4-cyclopropyl-6-methoxy-pyrimidin- 5-yl)-N,N-dimethyl-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidin-5-amine (51.2 mg, 97.4 μmol, 66.4% yield) as an off-white solid.
1H NMR (600 MHz, DMSO-d6) δ 0.82 – 0.88 (m, 2H), 0.98 – 1.03 (m, 2H), 1.70 – 1.75 (m, 1H), 2.86 (s, 6H), 3.77 (s, 3H), 3.83 (s, 3H), 5.51 (s, 2H), 7.59 (d, 2H), 7.73 (d, 2H), 7.92 (s, 1H), 8.18 (s, 1H), 8.62 (s, 1H). MS (ESI): [M+H]+ m/z: calcd 526.22; found 526.2.

Step 1: The synthesis of 1-[4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]ethanol [1154] Methylmagnesium bromide (17.1 mmol, 5.83 mL, 3M in ether) was added dropwise to a solution of 4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]benzaldehyde (2.90 g, 11.4 mmol) in THF (50 mL) at -78 °C under argon atmosphere. The resulting mixture was stirred for 3 hr, during which time the mixture was allowed to warm to -20 °C. At this temperature the mixture was quenched with of 1 N aqueous HCl solution (10 mL). The resulting mixture was stirred for 15 min. The mixture was diluted with water (50 mL) and extracted with EtOAc (2×100 mL). The combined organic layers were washed with brine (10 mL) and concentrated under reduced pressure to afford 1-[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]ethanol (2.45 g, 9.07 mmol, 79.5% yield) as a brown solid which was used in the next steps without further purification.
1H NMR (400 MHz, CDCl
3) δ 1.45 (d, 3H), 2.88 (br., 1H), 3.70 (s, 3H), 4.88 (q, 1H), 7.25 (s, 1H), 7.38 (d, 2H), 7.50 (d, 2H). MS (ESI): [M+H]+ m/z: calcd 271.13; found 271.0. Step 2: The synthesis of 2-chloro-5-methyl-4-[1-[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]ethoxy]pyrimidine [1155] NaH (29.6 mg, 740 μmol, 60% dispersion in mineral oil) was added to a solution of 1- [4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]ethanol (200 mg, 740 μmol) in DMF (5 mL) at 0°C. The reaction mixture was stirred at 0 °C for 1 hr.2,4-dichloro-5-methyl- pyrimidine (121 mg, 740 μmol) was added to the reaction mixture. The resulting mixture was stirred at room temperature for 16 hr. The reaction mixture was diluted with water (10 mL) and extracted with EtOAc (2×10 mL). The combined organic layers were washed with brine (10 mL) and concentrated under reduced pressure to afford 2-chloro-5-methyl-4-[1-[4-[1- methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]ethoxy]pyrimidine (310 mg, crude) as an yellow solid which was used in the next steps without further purification.
1H NMR (400 MHz, DMSO-d6) δ 1.64 (d, 3H), 2.14 (s, 3H), 3.77 (s, 3H), 6.27 (q, 1H), 7.58 (d, 2H), 7.71 (d, 2H), 7.91 (s, 1H), 8.32 (s, 1H). MS (ESI): [M+H]+ m/z: calcd 397.13; found 397.2. Step 3: The synthesis of 4'-cyclopropyl-6'-methoxy-5-methyl-4-(1-(4-(1-methyl-4- (trifluoromethyl)-1H-imidazol-2-yl)phenyl)ethoxy)-2,5'-bipyrimidine [1156] 2-Chloro-5-methyl-4-[1-[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]ethoxy]pyrimidine (150 mg, 378 μmol), (4-cyclopropyl-6-methoxy-pyrimidin-5- yl)boronic acid I-5 (103 mg, 529 μmol), potassium phosphate tribasic (241 mg, 1.13 mmol) and XPhos Pd G3 (16.0 mg, 18.9 μmol) were mixed in a degassed mixture of dioxane (3 mL) and water (100 µL). The reaction mixture was stirred at 100 °C for 14 hr. The reaction mixture was cooled to room temperature. The reaction mixture was diluted with EtOAc (10 mL) and washed with water (5 mL) and brine (5 mL). SiliaMetS® Dimercaptotriazine (20 mg) was added to the organic layer. The resulting mixture was stirred for 30 min and filtered. The filtrate was concentrated under reduced pressure. The residue was subjected to HPLC (0.5-6.5 min., 50-100% water – MeOH, +0.1% vol. of 25% aq. NH
3, flow: 30 mL/min, column: XBridge 100 ×19 mm, 5 µm) to afford 4'-cyclopropyl-6'-methoxy-5-methyl-4-(1-(4- (1-methyl-4-(trifluoromethyl)-1H-imidazol-2-yl)phenyl)ethoxy)-2,5'-bipyrimidine (69.0 mg, 135 μmol, 35.7% yield) as a yellow solid.
1H NMR (600 MHz, DMSO-d6) δ 0.68 – 0.74 (m, 1H), 0.77 – 0.82 (m, 1H), 0.91 – 1.00 (m, 2H), 1.51 – 1.56 (m, 1H), 1.62 (d, 3H), 2.23 (s, 3H), 3.75 (s, 3H), 3.76 (s, 3H), 6.30 (q, 1H), 7.54 (d, 2H), 7.68 (d, 2H), 7.91 (s, 1H), 8.51 (s, 1H), 8.62 (s, 1H). MS (ESI): [M+H]+ m/z: calcd 511.24; found 511.2. Step 4. The synthesis of rel-2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-5-methyl-4-[(1S)-1- [4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]ethoxy]pyrimidine (100) and rel-2-(4- cyclopropyl-6-methoxy-pyrimidin-5-yl)-5-methyl-4-[(1R)-1-[4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]ethoxy]pyrimidine (205). [1157] Racemic 4'-cyclopropyl-6'-methoxy-5-methyl-4-(1-(4-(1-methyl-4-(trifluoromethyl)- 1H-imidazol-2-yl)phenyl)ethoxy)-2,5'-bipyrimidine (69.0 mg, 135 μmol) was subjected to chiral HPLC (column: YMC CHIRAL ART AD-H V, 250×20 mm, 5 μm; mobile phase: Hexane-IPA-MeOH, 70-15-15; flow: 12 mL/min) to afford rel-2-(4-cyclopropyl-6-methoxy- pyrimidin-5-yl)-5-methyl-4-[(1S)-1-[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]ethoxy]pyrimidine (8.40 mg, 16.5 μmol, 12.2% yield) and rel-2-(4-cyclopropyl-6- methoxy-pyrimidin-5-yl)-5-methyl-4-[(1R)-1-[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]ethoxy]pyrimidine (9.60 mg, 18.8 μmol, 13.9% yield) as a white solids. rel-2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-5-methyl-4-[(1S)-1-[4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]ethoxy]pyrimidine (100):
1H NMR (600 MHz, DMSO-d6) δ 0.68 – 0.74 (m, 1H), 0.77 – 0.82 (m, 1H), 0.91 – 1.00 (m, 2H), 1.51 – 1.56 (m, 1H), 1.62 (d, 3H), 2.23 (s, 3H), 3.75 (s, 3H), 3.76 (s, 3H), 6.30 (q, 1H), 7.54 (d, 2H), 7.68 (d, 2H), 7.91 (s, 1H), 8.51 (s, 1H), 8.62 (s, 1H). MS (ESI): [M+H]+ m/z: calcd 511.24; found 511.2. Enantiopurity: 100% (column: Chiralpak IC, 250 × 4.6 mm, 5 μm; mobile phase: Hexane- IPA-MeOH, 50-25-25; flow: 0.6 mL/min; RT=13.5 min) rel-2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-5-methyl-4-[(1R)-1-[4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]ethoxy]pyrimidine (205):
1H NMR (600 MHz, DMSO-d6) δ 0.68 – 0.74 (m, 1H), 0.77 – 0.82 (m, 1H), 0.91 – 1.00 (m, 2H), 1.51 – 1.56 (m, 1H), 1.62 (d, 3H), 2.23 (s, 3H), 3.75 (s, 3H), 3.76 (s, 3H), 6.30 (q, 1H), 7.54 (d, 2H), 7.68 (d, 2H), 7.91 (s, 1H), 8.51 (s, 1H), 8.62 (s, 1H). MS (ESI): [M+H]+ m/z: calcd 511.24; found 511.2. Enantiopurity: 100% (column: Chiralpak IC, 250 × 4.6 mm, 5 μm; mobile phase: Hexane- IPA-MeOH, 50-25-25; flow: 0.6 mL/min; RT=11.6 min).
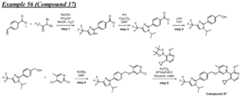
Step 1: Synthesis of methyl 4-[4-(trifluoromethyl)-1H-imidazol-2-yl]benzoate [1158] Sodium acetate (34.5 g, 420 mmol) was dissolved in water (200 mL) then 3,3- dibromo-1,1,1-trifluoro-propan-2-one (54.3 g, 201 mmol) was added and the reaction mixture was stirred at 95 °C for 1hr. The resulting mixture was cooled to room temperature and poured into a solution of methyl 4-formylbenzoate (30.0 g, 183 mmol) and NH4OH (100 mL, 25% wt.) in MeOH (1000 mL). The obtained mixture was stirred at room temperature for 12 hr. The mixture was concentrated under reduced pressure. The residue was distributed between EtOAc (300 mL) and water (200 mL). The organic layer was separated, dried over anhydrous Na
2SO
4, filtered and concentrated under reduced pressure. The residue was recrystallized from MTBE to afford methyl 4-[4-(trifluoromethyl)-1H-imidazol-2-yl]benzoate (38.0 g, 141 mmol, 77.0% yield) as a light-yellow solid which was used in the next steps without further purification.
1H NMR (400 MHz, CDCl
3) δ 3.85 (s, 3H), 7.98 (s, 1H), 8.04 (d, 2H), 8.10 (d, 2H). MS (ESI): [M+H]+ m/z: calcd 271.08; found 271.0. Step 2: Synthesis of methyl 4-[1-isopropyl-4-(trifluoromethyl)imidazol-2-yl]benzoate [1159] Methyl 4-[4-(trifluoromethyl)-1H-imidazol-2-yl]benzoate (1.00 g, 3.70 mmol), 2- iodopropane (1.89 g, 11.1 mmol, 1.11 mL) and Cs
2CO
3 (3.62 g, 11.1 mmol) were mixed in DMF (3 mL). The mixture was stirred at 80 °C for 36 hr. The mixture was cooled to room temperature and diluted with water (10 mL). The resulting mixture was extracted with MTBE (2 × 10 mL). Combined organic layers were washed with brine (10mL) and concentrated under reduced pressure to afford methyl 4-[1-isopropyl-4-(trifluoromethyl)imidazol-2- yl]benzoate (1.00 g, 3.20 mmol, 86.5% yield) as an yellow oil which was used in the next steps without further purification.
1H NMR (500 MHz, CDCl
3) δ 1.44 (d, 6H), 3.91 (s, 3H), 4.47 – 4.60 (m, 1H), 7.44 (s, 1H), 7.60 (d, 2H), 8.10 (d, 2H). Step 3: Synthesis of [4-[1-isopropyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methanol [1160] A solution of methyl 4-[1-isopropyl-4-(trifluoromethyl)imidazol-2-yl]benzoate (1.15 g, 3.68 mmol) in THF (5 mL) was added dropwise to a vigorously stirred suspension of lithium aluminum hydride (375 mg, 11.1 mmol) in THF (20 mL) under argon atmosphere at +5 °C. The resulting mixture was stirred at ambient temperature for 12 hr. The mixture was carefully quenched by dropwise addition of aqueous NaOH (10 mL, 10% wt.). The resulting mixture was stirred for 15 min and filtered. The filter cake was washed with THF (10 mL). The combined filtrate was dried over anhydrous Na
2SO
4 and concentrated under reduced pressure to afford [4-[1-isopropyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methanol (700 mg, 2.46 mmol, 73.5% yield) as a red gum which was used in the next steps without further purification. MS (ESI): [M+H]+ m/z: calcd 285.15; found 285.2. Step 4: Synthesis of 2-chloro-4-[[4-[1-isopropyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]-5-methyl-pyrimidine [1161] Potassium tert-butoxide (101 mg, 904 µmol) was added to a solution of [4-[1- isopropyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methanol (245 mg, 861.13 μmol) in DMF (5 mL). The mixture was stirred for 20 min at room temperature and added dropwise to a solution of 2,4-dichloro-5-methyl-pyrimidine (154 mg, 947 μmol) in DMF (5 mL) at room temperature. The resulting mixture was stirred for 24 hr. The mixture was diluted with water (5 mL) and extracted with DCM (20 mL). The organic layer was washed with water (10 mL × 2), dried over anhydrous Na
2SO
4 and concentrated under reduced pressure. The residue was subjected to HPLC (0.5-6.5 min, 29% water - ACN; flow: 30 mL/min, column: Waters SunFire C18, 100 x 19 mm, 5 µm) to afford 2-chloro-4-[[4-[1-isopropyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]-5-methyl-pyrimidine (83.0 mg, 202 μmol, 23.5% yield) as a yellow solid. MS (ESI): [M+H]+ m/z: calcd 397.13; found 397.2. Step 5: Synthesis of 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[4-[1-isopropyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]-5-methyl-pyrimidine [1162] 2-Chloro-4-[[4-[1-isopropyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]-5- methyl-pyrimidine (32.0 mg, 77.9 μmol), (4-cyclopropyl-6-methoxy-pyrimidin-5-yl)boronic acid I-5 (21.2 mg, 109 μmol), XPhos Pd G3 (3.30 mg, 3.89 μmol) and potassium phosphate tribasic (49.6 mg, 234 μmol) were mixed in a degassed mixture of dioxane (3 mL) and water (0.15 mL) under argon atmosphere. The mixture was stirred at 100 °C for 24 hr. The mixture was cooled to room temperature, diluted with EtOAc (10 mL) and washed with water (10 mL × 2). To the obtained organic phase SiliaMetS® Dimercaptotriazine (30 mg) was added, and the mixture was stirred for 30 min. The mixture was filtered. The filtrate was concentrated under reduced pressure. The residue was subjected HPLC (0.5-6.5 min, 40-90%% water - ACN; flow: 30 mL/min, column: Waters SunFire C18, 100 x 19 mm, 5 µm) to afford 2-(4- cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[4-[1-isopropyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]-5-methyl-pyrimidine (4.90 mg, 9.34 μmol, 12.0% yield) as a yellow solid.
1H NMR (600 MHz, DMSO-d6) δ 0.81 – 0.86 (m, 2H), 0.98 – 1.03 (m, 2H), 1.39 (d, 6H), 1.62 – 1.69 (m, 1H), 2.22 (s, 3H), 3.82 (s, 3H), 4.42 – 4.50 (m, 1H), 5.52 (s, 2H), 7.54 – 7.61 (m, 4H), 8.17 (s, 1H), 8.55 (s, 1H), 8.64 (s, 1H). MS (ESI): [M+H]+ m/z: calcd 525.26; found 525.0. Example 57 (Compound 192)
The synthesis of the starting [3-fluoro-4-[1-isopropyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methanol was described in Example 76 (Compound 149). The synthesis of 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[3-fluoro-4-[1-isopropyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]-5-methyl-pyrimidine [1163] NaH (15.2 mg, 380 μmol, 60% dispersion in mineral oil) was added to a stirred solution of [3-fluoro-4-[1-isopropyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methanol (100 mg, 331 μmol) in THF (4.0 mL). The reaction mixture was stirred at room temperature for 15 min.2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-5-methyl-4-[[4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methylsulfonyl]pyrimidine (198 mg, 364 μmol) was added to the reaction mixture. The resulting mixture was stirred at room temperature for 12 hr. The reaction mixture was diluted with a saturated aqueous solution of NH
4Cl (2.0 mL). The organic layer was separated and subjected to HPLC (0.5-6.5 min, 55-65% water – ACN; flow: 30 mL/min, column: SunFire C18, 100 x 19 mm, 5 µm) to afford 2-(4-cyclopropyl-6- methoxy-pyrimidin-5-yl)-4-[[3-fluoro-4-[1-isopropyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]-5-methyl-pyrimidine (52.0 mg, 95.9 μmol, 29.0% yield) as a yellow solid.
1H NMR (600 MHz, DMSO-d6) δ 0.81 – 0.87 (m, 2H), 0.98 – 1.03 (m, 2H), 1.35 (d, 6H), 1.62 – 1.69 (m, 1H), 2.24 (s, 3H), 3.82 (s, 3H), 4.10 – 4.17 (m, 1H), 5.53 (s, 2H), 7.44 (d, 1H), 7.49 (d, 1H), 7.56 (t, 1H), 8.23 (s, 1H), 8.56 (s, 1H), 8.64 (s, 1H). MS (ESI): [M+H]+ m/z: calcd 543.25; found 543.2.

Step 1: The synthesis of 5-bromo-2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[4-[1- methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidine The synthesis of the starting 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidin-5-amine is described in Example 67 (Compound 142). [1164] A solution of 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidin-5-amine (110 mg, 221 μmol) in ACN (17 mL) was added dropwise to a mixture of copper (II) bromide (74.1 mg, 332 μmol) and tertbutyl nitrite (68.4 mg, 663 μmol, 78.9 μL) in ACN (17 mL) at 0 °C. The reaction mixture was stirred at room temperature for 12 hr. The reaction mixture was diluted with water (25 mL) and extracted with EtOAc (30 mL). The organic layer was separated, washed with water (2×20 mL), brine (20 mL), dried over anhydrous Na
2SO
4 and concentrated under reduced pressure to afford 5-bromo-2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[4-[1- methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidine (140 mg) as a yellow solid which was used in the next steps without further purification. MS (ESI): [M+H]+ m/z: calcd 561.09; found 561.2. Step 2: The synthesis of 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]-5-(1-tetrahydropyran-2-ylpyrazol-4- yl)pyrimidine [1165] 5-bromo-2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidine (140 mg, crude), 1- tetrahydropyran-2-yl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyrazole (104 mg, 374 μmol), tripotassium phosphate (106 mg, 499 μmol) and bis(diphenylphosphino)ferrocene]dichloropalladium(II)-DCM (20.4 mg, 24.9 μmol) were mixed in degassed dioxane (10 mL) and water (500 µL). The reaction mixture was stirred at 90 °C for 12 hr under argon atmosphere. The reaction mixture was cooled to room temperature and filtered. The filtrate was concentrated under reduced pressure to afford 2-(4- cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]-5-(1-tetrahydropyran-2-ylpyrazol-4-yl)pyrimidine (150 mg, crude) as a brown solid which was used in the next steps without further purification. MS (ESI): [M+H]+ m/z: calcd 633.26; found 633.4. Step 3: The synthesis of 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]-5-(1H-pyrazol-4-yl)pyrimidine [1166] Hydrogen chloride solution in dioxane (110 μL, 4.0 M) was added to a solution of 2- (4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]-5-(1-tetrahydropyran-2-ylpyrazol-4-yl)pyrimidine (150 mg, crude) in MeOH (3 mL). The reaction mixture was stirred at room temperature for 24 hr. The reaction mixture was concentrated under reduced pressure. The residue was subjected to HPLC (0-5 min, 10-60% water+FA (0.1% vol.) - ACN+FA (0.1% vol.); flow: 30 mL/min, column: Chromatorex 18 SMB100-5T, 100 x 19 mm, 5 µm) to afford 2-(4-cyclopropyl-6-methoxy- pyrimidin-5-yl)-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]-5-(1H- pyrazol-4-yl)pyrimidine (4.20 mg, 7.66 μmol, 3.46% yield from 2-(4-cyclopropyl-6- methoxy-pyrimidin-5-yl)-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidin-5-amine) as a yellow solid.
1H NMR (600 MHz, DMSO-d6) δ 0.84 – 0.89 (m, 2H), 1.01 – 1.06 (m, 2H), 1.74 – 1.79 (m, 1H), 3.77 (s, 3H), 3.84 (s, 3H), 5.61 (s, 2H), 7.62 (d, 2H), 7.74 (d, 2H), 7.93 (s, 1H), 8.09 – 8.36 (m, 2H), 8.66 (s, 1H), 9.07 (s, 1H), 13.14 (br., 1H). MS (ESI): [M+H]+ m/z: calcd 549.22; found 549.2.
Step 1: Synthesis of ethyl 4-[5-oxo-3-(trifluoromethyl)-4H-pyrazol-1-yl]benzoate [1167] To a stirred solution of 4-[5-oxo-3-(trifluoromethyl)-4H-pyrazol-1-yl]benzoic acid (1.20 g, 4.41 mmol) in EtOH (25 mL) thionyl chloride (1.31 g, 11.0 mmol, 804 μL) was added dropwise at room temperature. The resulting reaction mixture was stirred at 75 °C for 16 hr. The reaction mixture was cooled to room temperature and concentrated under reduced pressure to afford ethyl 4-[5-oxo-3-(trifluoromethyl)-4H-pyrazol-1-yl]benzoate (1.32 g, 4.40 mmol, 100% yield) as a yellow solid which was used in the next steps without further purification.
1H NMR (500 MHz, DMSO-d6) δ 1.32 (t, 3H), 4.32 (q, 2H), 5.96 (s, 1H), 7.92 (d, 2H), 8.07 (d, 2H), 12.86 (br., 1H). MS (ESI): [M+H]+ m/z: calcd 301.09; found 301.0. Step 2: Synthesis of ethyl 4-[5-ethoxy-3-(trifluoromethyl)pyrazol-1-yl]benzoate [1168] A stirred solution of ethyl 4-[5-oxo-3-(trifluoromethyl)-4H-pyrazol-1-yl]benzoate (1.32 g, 4.40 mmol) in DMF (15 mL) was cooled to 0 °C. To the obtained solution NaH (202 mg, 5.28 mmol, 60% dispersion in mineral oil) was added in a few portions. The resulting mixture was stirred at room temperature for 2 hr. To the obtained mixture ethyliodide (891 mg, 5.72 mmol, 460 μL) was added dropwise and the solution was stirred at room temperature for 16 hr. The reaction mixture was poured into ice-water (75 mL). The resulting mixture was extracted with MTBE (3 × 75 mL). The combined organic layers were washed with water (100 mL) and brine (100 mL), dried over anhydrous Na
2SO
4, filtered and concentrated under reduced pressure to afford ethyl 4-[5-ethoxy-3-(trifluoromethyl)pyrazol- 1-yl]benzoate (1.33 g, 4.05 mmol, 92.4% yield) as a yellow solid which was used in the next steps without further purification.
1H NMR (400 MHz, CDCl
3) δ 1.41 (t, 3H), 1.50 (t, 3H), 4.25 (q, 2H), 4.40 (q, 2H), 5.94 (s, 1H), 7.86 (d, 2H), 8.13 (d, 2H). MS (ESI): [M+H]+ m/z: calcd 328.13; found 329.2. Step 3: Synthesis of [4-[5-ethoxy-3-(trifluoromethyl)pyrazol-1-yl]phenyl]methanol [1169] A stirred suspension of LAH (277 mg, 7.29 mmol) in THF (80 mL) was cooled to -10 °C under argon atmosphere. To the obtained mixture ethyl 4-[5-ethoxy-3- (trifluoromethyl)pyrazol-1-yl]benzoate (1.33 g, 4.05 mmol) in THF (20 mL) was added dropwise at vigorous stirring. The resulting mixture was allowed to warm to room temperature and stirred overnight. The reaction mixture was quenched by dropwise addition of water (1 mL) in THF (5 mL) at 0 °C. The mixture was stirred for 15 minutes and then anhydrous K
2CO
3 and Na
2SO
4 were added. The resulting mixture was stirred for 15 minutes. The resulting solids were filtered out and washed with THF (2 × 10 mL). The combined filtrate was concentrated under reduced pressure to afford [4-[5-ethoxy-3- (trifluoromethyl)pyrazol-1-yl]phenyl]methanol (950 mg, 3.32 mmol, 81.9% yield) as a yellow oil which was used in the next steps without further purification.
1H NMR (400 MHz, CDCl
3) δ 1.47 (t, 3H), 4.21 (q, 2H), 4.72 (s, 2H), 5.92 (s, 1H), 7.43 (d, 2H), 7.69 (d, 2H). MS (ESI): [M+H]+ m/z: calcd 287.12; found 287.0. Step 4: Synthesis of 2-chloro-4-[[4-[5-ethoxy-3-(trifluoromethyl)pyrazol-1- yl]phenyl]methoxy]-5-methyl-pyrimidine [1170] [4-[5-Ethoxy-3-(trifluoromethyl)pyrazol-1-yl]phenyl]methanol (250 mg, 873 μmol) was added to a stirred solution of NaH (50.2 mg, 1.31 mmol, 60% dispersion in mineral oil) in DMF (6 mL) at room temperature. The mixture was stirred for 30 min. To the resulting solution 2,4-dichloro-5-methyl-pyrimidine (214 mg, 1.31 mmol) was added. The reaction mixture was stirred at room temperature for 16 hr. The reaction mixture was poured into saturated aqueous NH
4Cl solution (15 mL). The obtained mixture was stirred for 10 minutes. The solid precipitate was filtered off and washed with water (10 mL) and n-Hexane (10 mL) and dried under reduced pressure to afford 2-chloro-4-[[4-[5-ethoxy-3- (trifluoromethyl)pyrazol-1-yl]phenyl]methoxy]-5-methyl-pyrimidine (200 mg, 485 μmol, 55.4% yield) as a yellow solid which was used in the next steps without further purification. MS (ESI): [M+H]+ m/z: calcd 413.12; found 413.1. Step 5: Synthesis of 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[4-[5-ethoxy-3- (trifluoromethyl)pyrazol-1-yl]phenyl]methoxy]-5-methyl-pyrimidine [1171] 2-Chloro-4-[[4-[5-ethoxy-3-(trifluoromethyl)pyrazol-1-yl]phenyl]methoxy]-5-methyl- pyrimidine (80.0 mg, 194 μmol), 4-cyclopropyl-6-methoxy-5-(4,4,5,5-tetramethyl-1,3,2- dioxaborolan-2-yl)pyrimidine (58.9 mg, 213 μmol), Na
2CO
3 (61.6 mg, 581 μmol) and XPhos Pd G3 (8.20 mg, 9.69 μmol) were mixed in a degassed mixture of water (2 mL) and dioxane (6 mL) under argon atmosphere. The resulting mixture was stirred under argon atmosphere at 95 °C for 16 hr. The reaction mixture was cooled to room temperature and filtered through a short pad of SiO
2. The filtrate was concentrated under reduced pressure. The residue was subjected to HPLC (0-5 min, 60-80% water – MeOH, +0.1% vol. of 25% aq. NH
3, flow: 30 mL/min, column: XBridge C18100 ×19 mm, 5 µm) to afford 2-(4-cyclopropyl-6-methoxy- pyrimidin-5-yl)-4-[[4-[5-ethoxy-3-(trifluoromethyl)pyrazol-1-yl]phenyl]methoxy]-5-methyl- pyrimidine (21.6 mg, 41.0 μmol, 21.2% yield) as an off-white solid. In another HPLC- fraction a side product 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-3-[[4-[5-ethoxy-3- (trifluoromethyl)pyrazol-1-yl]phenyl]methyl]-5-methyl-pyrimidin-4-one (16.4 mg, 31.2 μmol, 16.1% yield) was obtained as an off-white solid.
1H NMR (600 MHz, DMSO-d6) δ 0.81 – 0.85 (m, 2H), 0.98 – 1.02 (m, 2H), 1.34 (t, 3H), 1.61 – 1.67 (m, 1H), 2.21 (s, 3H), 3.81 (s, 3H), 4.28 (q, 2H), 5.49 (s, 2H), 6.45 (s, 1H), 7.58 (d, 2H), 7.66 (d, 2H), 8.54 (s, 1H), 8.64 (s, 1H). MS (ESI): [M+H]+ m/z: calcd 527.23; found 527.2. Example 60 (Compound 52)

Step 1: Synthesis of 4-[5-hydroxy-3-(trifluoromethyl)pyrazol-1-yl]benzonitrile [1172] 4-hydrazinobenzonitrile (30.5 g, 180 mmol, HCl) was dissolved in EtOH (200 mL) then Sodium hydroxide (7.19 g, 180 mmol) was added. The reaction mixture was stirred at room temperature for 40 min. To the resulting mixture ethyl 4,4,4-trifluoro-3-oxo-butanoate (39.7 g, 216 mmol, 31.53 mL) in EtOH (20 ml) was added in one portion. The resulting mixture was stirred under reflux for 24 h. The mixture was cooled to room temperature and filtered. The filtrate was concentrated under reduced pressure. The residue was dissolved in toluene (200 mL) then p-toluenesulfonic acid (a catalytic amount) was added. The resulting solution was stirred at 120 °C for 6 hr. The mixture was cooled to room temperature and concentrated under reduced pressure to afford 4-[5-hydroxy-3-(trifluoromethyl)pyrazol-1- yl]benzonitrile (45.0 g, 178 mmol, 98.8% yield) as a yellow solid which was used in the next steps without further purification.
1H NMR (500 MHz, DMSO-d6) δ 5.96 (s, 1H), 7.94 – 8.02 (m, 4H). MS (ESI): [M+H]+ m/z: calcd 254.06; found 254.0. Step 2: Synthesis of 4-[5-methoxy-3-(trifluoromethyl)pyrazol-1-yl]benzonitrile 4-[5-hydroxy-3-(trifluoromethyl)pyrazol-1-yl]benzonitrile (23.0 g, 90.8 mmol) was dissolved in DMF (100 mL) and the solution was cooled to 0 °C. To the solution NaH (2.40 g, 99.9 mmol, 60% dispersion in mineral oil) was added in a few portions. The reaction mixture was stirred at ambient temperature for 30 min. To the obtained mixture methyliodide (15.5 g, 109 mmol, 6.79 mL) was added dropwise. The reaction mixture was stirred at ambient temperature for 16 hr. The reaction mixture was poured into ice-water mixture (200 mL). The solid precipitate formed was filtered off and dissolved in EtOAc (200 mL). The obtained solution was washed with water (150 mL) and brine (100 mL) and dried over anhydrous Na
2SO
4. The mixture was concentrated under reduced pressure to afford 4-[5-methoxy-3- (trifluoromethyl)pyrazol-1-yl]benzonitrile (21.0 g, 78.6 mmol, 86.5% yield) as yellow solid which was used in the next steps without further purification.
1H NMR (500 MHz, CDCl
3) δ 4.03 (s, 3H), 5.97 (s, 1H), 7.72 (d, 2H), 7.93 (d, 2H). MS (ESI): [M+H]+ m/z: calcd 268.08; found 268.0 Step 3: Synthesis of 4-[5-methoxy-3-(trifluoromethyl)pyrazol-1-yl]benzoic acid [1173] A solution of sodium hydroxide (1.80 g, 44.9 mmol) in water (35 mL) was added to a solution of 4-[5-methoxy-3-(trifluoromethyl)pyrazol-1-yl]benzonitrile (3.00 g, 11.2 mmol) in MeOH (60 mL). The resulting mixture was stirred at 65 °C for 80 hr. The mixture was cooled to room temperature and concentrated under reduced pressure. The residue was diluted with water (100 mL). The resulting mixture washed with MTBE (2 × 30 mL). The aqueous layer was acidified with aqueous citric acid solution (10% wt.). The resulting precipitate was filtered off and dissolved in MTBE (300 mL). The obtained solution was dried over anhydrous Na
2SO
4 and concentrated under reduced pressure to afford 4-[5-methoxy-3- (trifluoromethyl)pyrazol-1-yl]benzoic acid (2.70 g, 9.43 mmol, 84.1% yield) as a light yellow solid which was used in the next steps without further purification.
1H NMR (500 MHz, CDCl
3) δ 4.03 (s, 3H), 6.49 (s, 1H), 7.83 (d, 2H), 8.06 (d, 2H). MS (ESI): [M+H]+ m/z: calcd 287.07; found 287.2. Step 4: Synthesis of [4-[5-methoxy-3-(trifluoromethyl)pyrazol-1-yl]phenyl]methanol Borane dimethyl sulfide complex (3.19 g, 41.9 mmol, 3.98 mL) was added dropwise to a stirred solution of 4-[5-methoxy-3-(trifluoromethyl)pyrazol-1-yl]benzoic acid (1.50 g, 4.19 mmol) in THF (40 mL) at ambient temperature. The mixture was stirred at room temperature for 60 hr. The mixture was quenched by dropwise addition of MeOH (50 mL). The resulting mixture was stirred at room temperature for 2 hr. and then stirred under reflux for 5 hr. The mixture was cooled to room temperature and concentrated under reduced pressure. The residue was redissolved in MeOH (100 mL). The resulting mixture was stirred under reflux for 3 hr. The mixture was cooled to room temperature and concentrated under reduced pressure. The residue was redissolved in MTBE (50 mL). The resulting solution was washed with saturated aqueous NaHCO
3 solution (2 × 15 mL), dried over dried over anhydrous Na
2SO
4 and concentrated under reduced pressure to afford [4-[5-methoxy-3- (trifluoromethyl)pyrazol-1-yl]phenyl]methanol (1.10 g, 4.04 mmol, 96.5% yield) as a light- yellow gum which was used in the next steps without further purification. MS (ESI): [M+H]+ m/z: calcd 273.10; found 273.2. Step 5: Synthesis of 2-chloro-4-[[4-[5-methoxy-3-(trifluoromethyl)pyrazol-1- yl]phenyl]methoxy]-5-methyl-pyrimidine [1174] Potassium tert-butoxide (510 mg, 4.55 mmol) was added portion wise to a stirred solution of [4-[5-methoxy-3-(trifluoromethyl)pyrazol-1-yl]phenyl]methanol (1.10 g, 4.04 mmol) and 2,4-Dichloro-5-methylpyrimidine (741 mg, 4.55 mmol) in toluene (50 mL) at 0 °C. The resulting solution was stirred at ambient temperature for 15 hr. The mixture was poured into cold water (50 mL). The resulting mixture was extracted with MTBE (3 × 15 mL). The combined organic layers were dried over anhydrous Na
2SO
4, filtered through a short silica pad and concentrated under reduced pressure. The residue was subjected to column chromatography (SiO
2, two runs: gradient Hexanes - EtOAc then gradient Hexanes - IPA) to afford 2-chloro-4-[[4-[5-methoxy-3-(trifluoromethyl)pyrazol-1-yl]phenyl]methoxy]- 5-methyl-pyrimidine (750 mg, 1.88 mmol, 62.1% yield) as a light-yellow solid. MS (ESI): [M+H]+ m/z: calcd 399.10; found 399.0. Step 6: Synthesis of 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[4-[5-methoxy-3- (trifluoromethyl)pyrazol-1-yl]phenyl]methoxy]-5-methyl-pyrimidine [1175] 2-chloro-4-[[4-[5-methoxy-3-(trifluoromethyl)pyrazol-1-yl]phenyl]methoxy]-5- methyl-pyrimidine (120 mg, 301 μmol), (4-cyclopropyl-6-methoxy-pyrimidin-5-yl)boronic acid I-5 (117 mg, 602 μmol), XPhos (7.17 mg, 15.1 μmol), XPhos Pd G3 (25.5 mg, 30.1 μmol) and K
2CO
3 (104 mg, 752 μmol) were mixed in a degassed mixture of dioxane (15 mL) and Water (3 mL). The resulting mixture was stirred under argon atmosphere at 90 °C for 15 hr. The mixture was cooled to room temperature and concentrated under reduced pressure. The residue was diluted with water (5 mL) and extracted with EtOAc (4 × 10 mL). The combined organic layers were dried over anhydrous Na
2SO
4 and evaporated to dryness. The residue was redissolved in MeCN (10 mL), then SiliaMetS® Dimercaptotriazine was added. The resulting mixture was stirred for 10 hr. The mixture was filtered. The filtrate was subjected to HPLC (2-10 min, 10-50% water+FA (0.1% vol.) - ACN+FA (0.1% vol.); flow: 30 mL/min, column: Waters SunFire C18, 100 x 19 mm, 5 µm) to afford to afford a residue. The residue was subjected to preparative SFC (0.5-10.5 min, 5-50% MeOH in CO
2, flow: 50 ml/min, column: SiO
2, 19 × 100 mm, 5 µm) to afford 2-(4-cyclopropyl-6-methoxy- pyrimidin-5-yl)-4-[[4-[5-methoxy-3-(trifluoromethyl)pyrazol-1-yl]phenyl]methoxy]-5- methyl-pyrimidine (10.0 mg, 19.5 μmol, 6.5% yield) as a light-yellow solid.
1H NMR (600 MHz, DMSO-d6) δ 0.81 – 0.86 (m, 2H), 0.98 – 1.02 (m, 2H), 1.60 – 1.66 (m, 1H), 2.21 (s, 3H), 3.81 (s, 3H), 3.98 (s, 3H), 5.48 (s, 2H), 6.46 (s, 1H), 7.59 (d, 2H), 7.63 (d, 2H), 8.54 (s, 1H), 8.64 (s, 1H). MS (ESI): [M+H]+ m/z: calcd 513.21; found 513.2. Example 61 (Compound 128)
Step 1: The synthesis of 2-chloro-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methylsulfanyl]pyrimidine [1176] S-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methyl]ethanethioate (1.50 g, 4.77 mmol) and 2,4-dichloropyrimidine (646 mg, 4.34 mmol) were mixed in THF (4.0 mL) and MeOH (15 mL). The resulting mixture was cooled to 0 °C. K
2CO
3 (660 mg, 4.77 mmol) was added to the mixture. The reaction mixture was stirred at 0 °C for 3 hr and then at room temperature for 8 hr. The reaction mixture was diluted with EtOAc (50 mL) and washed with water (50 mL) and brine (3×40 mL). The organic layer was concentrated under reduced pressure. The residue was subjected to flash column chromatography (SiO
2, gradient hexane - EtOAc) to afford 2-chloro-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methylsulfanyl]pyrimidine (800 mg, 2.08 mmol, 47.9% yield) as a light-yellow solid which was used in the next step without further purification. MS (ESI): [M+H]+ m/z: calcd 385.07; found 385.2 Step 2: The synthesis of 4-cyclopropyl-6-methoxy-5-[4-[[4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methylsulfanyl]pyrimidin-2-yl]pyrimidine [1177] 2-Chloro-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methylsulfanyl]pyrimidine (90 mg, 234 μmol), (4-cyclopropyl-6-methoxy- pyrimidin-5-yl)boronic acid I-5 (90.7 mg, 468 μmol) and K
2CO
3 (64.7 mg, 468 μmol) were mixed in a degassed mixture of dioxane (5 mL) and water (1 mL). The resulting mixture was degassed thrice. RuPhos Pd G4 (19.9 mg, 23.4 μmol) was added to the mixture. The reaction mixture was stirred at 90°C for 14 hr. The reaction mixture was cooled to room temperature and concentrated under reduced pressure. The residue was diluted with water (5 mL) and extracted with MTBE (4×10 mL). The combined organic layers were dried over anhydrous Na
2SO
4 and concentrated under reduced pressure. The residue was subjected to HPLC (2-10 min, 0-55% water – ACN, flow: 30 mL/min, column: SunFire C18100 ×19 mm, 5 µm) to afford 4-cyclopropyl-6-methoxy-5-[4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methylsulfanyl]pyrimidin-2-yl]pyrimidine (76.0 mg, 152 μmol, 65.2% yield) as a yellow solid.
1H NMR (400 MHz, DMSO-d6) δ 0.88 – 0.94 (m, 2H), 1.03 – 1.09 (m, 2H), 1.62 – 1.70 (m, 1H), 3.77 (s, 3H), 3.87 (s, 3H), 4.54 (s, 2H), 7.50 – 7.55 (m, 3H), 7.66 (d, 2H), 7.93 (s, 1H), 8.64 (d, 1H), 8.69 (s, 1H). MS (ESI): [M+H]+ m/z: calcd 499.15; found 499.2.

Compound 113 Step 1: Synthesis of 2-chloro-5-methoxy-4-[[4-[5-methoxy-3-(trifluoromethyl)pyrazol-1- yl]phenyl]methoxy]pyrimidine Starting [4-[5-methoxy-3-(trifluoromethyl)pyrazol-1-yl]phenyl]methanol was prepared as described for Compound 52 [1178] Potassium tert-butoxide (227 mg, 2.02 mmol) was added to a stirred solution of [4-[5- methoxy-3-(trifluoromethyl)pyrazol-1-yl]phenyl]methanol (500 mg, 1.84 mmol) in dioxane (10 mL) at room temperature and the mixture was stirred for 1 hr. To the obtained mixture 2,4-dichloro-5-methoxy-pyrimidine (329 mg, 1.84 mmol) was added in one portion. The resulting mixture was stirred at 80 °C for 12 hr. The reaction mixture was cooled to room temperature, quenched with water (10 mL) and extracted with EtOAc (2 × 10 mL). The combined organic layers were washed with brine (10 mL) and concentrated under reduced pressure to afford 2-chloro-5-methoxy-4-[[4-[5-methoxy-3-(trifluoromethyl)pyrazol-1- yl]phenyl]methoxy]pyrimidine (0.7 g, crude) as a light-yellow solid which was used in the next steps without further purification. MS (ESI): [M+H]+ m/z: calcd 415.08; found 415.0. Step 2: Synthesis of 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[4-[5-methoxy-3- (trifluoromethyl)pyrazol-1-yl]phenyl]methoxy]-5-methoxy-pyrimidine 2-Chloro-5-methoxy-4-[[4-[5-methoxy-3-(trifluoromethyl)pyrazol-1- yl]phenyl]methoxy]pyrimidine (200 mg, 482 μmol), (4-cyclopropyl-6-methoxy-pyrimidin-5- yl)boronic acid I-5 (112 mg, 579 μmol), potassium phosphate tribasic (256 mg, 1.21 mmol) and XPhos Pd G3 (20.4 mg, 24.1 μmol) were mixed in a degassed mixture of water (0.5 mL) and dioxane (5 mL). The reaction mixture was stirred under argon atmosphere at 80 °C for 12 hr. The reaction mixture was cooled room temperature, diluted with water (5 mL) and extracted with EtOAc (2 × 10 mL). The combined organic layers were washed with water (10 mL) and brine (2 × 10 mL). To the obtained organic phase SiliaMetS® Dimercaptotriazine (20 mg) was added. The resulting mixture was stirred for 30 min and filtered. The filtrate was evaporated under reduced pressure. The residue was subjected to HPLC (0.5-6.5min, 61% water – MeOH, +0.1% vol. of 25% aq. NH
3, flow: 30 mL/min, column: XBridge C18100 ×19 mm, 5 µm) to afford 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[4-[5-methoxy-3- (trifluoromethyl)pyrazol-1-yl]phenyl]methoxy]-5-methoxy-pyrimidine (27.0 mg, 51.3 μmol, 9.8% yield from [4-[5-methoxy-3-(trifluoromethyl)pyrazol-1-yl]phenyl]methanol) as a light yellow solid.
1H NMR (600 MHz, DMSO-d6) δ 0.81 – 0.87 (m, 2H), 1.03 – 0.99 (m, 2H), 1.63 – 1.71 (m, 1H), 3.82 (s, 3H), 3.93 (s, 3H), 3.99 (s, 3H), 5.46 (s, 2H), 6.46 (s, 1H), 7.57 (d, 2H), 7.65 (d, 2H), 8.41 (s, 1H), 8.63 (s, 1H). MS (ESI): [M+H]+ m/z: calcd 529.20; found 529.0. Example 63 (Compound 148)
Step 1: The synthesis of [4-[[tert-butyl(dimethyl)silyl]oxymethyl]-2-fluoro-6-methyl- phenyl]methanol [1179] Tert-butyl(chloro)dimethylsilane (2.66 g, 17.6 mmol, 3.28 mL) was added to a solution of [3-fluoro-4-(hydroxymethyl)-5-methyl-phenyl]methanol (3.00 g, 17.6 mmol) and 1H-imidazole (1.32 g, 19.4 mmol) in THF (60 mL) at 0 °C. The reaction mixture was stirred at 0 °C for 16 hr. The reaction mixture was diluted with EtOAc (60 ml) and washed with water (2×30 mL). The organic layer was concentrated under reduced pressure. The residue was subjected to flash column chromatography (SiO
2, gradient hexane – MTBE) to afford [4- [[tert-butyl(dimethyl)silyl]oxymethyl]-2-fluoro-6-methyl-phenyl]methanol (1.80 g, 6.33 mmol, 35.9% yield) as a white solid.
1H NMR (500 MHz, CDCl
3) δ 0.09 (s, 6H), 0.93 (s, 9H), 2.41 (s, 3H), 4.66 (s, 2H), 4.72 (s, 2H), 6.87 – 6.91 (m, 2H). MS (ESI): [M-OH]+ m/z: calcd 267.17; found 267.2. Step 2: The synthesis of 4-[[tert-butyl(dimethyl)silyl]oxymethyl]-2-fluoro-6-methyl- benzaldehyde [1180] DMSO (1.38 g, 17.7 mmol, 1.26 mL) in DCM (5 mL) was added dropwise to a solution of oxalyl chloride (1.04 g, 8.23 mmol, 718 μL) in DCM (30 mL) at -78°C. The reaction mixture was stirred at -78°C for 40 min. A solution of [4-[[tert- butyl(dimethyl)silyl]oxymethyl]-2-fluoro-6-methyl-phenyl]methanol (1.80 g, 6.33 mmol) in DCM (10 mL) was added dropwise to the reaction mixture. The resulting mixture was stirred at -75 °C for 30 min. TEA (3.20 g, 31.6 mmol, 4.41 mL) was added dropwise to the mixture at -65 °C. The reaction mixture was allowed to warm to 0 °C and quenched by dropwise addition of water (30 mL). The organic layer was separated, washed with saturated citric acid solution (30 mL), water (20 mL) and brine (20 mL), dried over anhydrous Na
2SO
4 and concentrated under reduced pressure to afford 4-[[tert-butyl(dimethyl)silyl]oxymethyl]-2- fluoro-6-methyl-benzaldehyde (1.40 g, 4.96 mmol, 78.2% yield) as a light-yellow oil which was used in the next steps without further purification.
1H NMR (500 MHz, CDCl
3) δ 0.10 (s, 6H), 0.94 (s, 9H), 2.60 (s, 3H), 4.71 (s, 2H), 6.92 (s, 1H), 7.00 (d, 1H), 10.48 (s, 1H). MS (ESI): [M+H]+ m/z: calcd 283.16; found 283.2. Step 3: The synthesis of tert-butyl-[[3-fluoro-5-methyl-4-[4-(trifluoromethyl)-1H-imidazol-2- yl]phenyl]methoxy]-dimethyl-silane [1181] 3,3-dibromo-1,1,1-trifluoro-propan-2-one (562 mg, 2.08 mmol) was added to a solution of anhydrous sodium acetate (328 mg, 3.99 mmol) in water (20 mL). The resulting mixture was stirred at 100 °C for 1 hr. The mixture was cooled to 5 °C and poured into a cooled to 0 °C stirred solution of 4-[[tert-butyl(dimethyl)silyl]oxymethyl]-2-fluoro-6-methyl- benzaldehyde (700 mg, 1.73 mmol, 70% purity) in MeOH (80 mL). An aqueous ammonium hydroxide (296 mg, 17.4 mmol, 25% wt.) was added to this mixture. The resulting mixture was stirred at 0 °C for 16 hr. The reaction mixture was concentrated under reduced pressure to 1/4 of its volume. The precipitate formed was filtered off and dried on air to afford tert- butyl-[[3-fluoro-5-methyl-4-[4-(trifluoromethyl)-1H-imidazol-2-yl]phenyl]methoxy]- dimethyl-silane (1.00 g, crude) as a yellow solid which was used in the next steps without further purification. MS (ESI): [M+H]+ m/z: calcd 389.17; found 389.0. Step 4: The synthesis of tert-butyl-[[3-fluoro-5-methyl-4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]-dimethyl-silane [1182] Methyliodide (135 mg, 950 μmol, 59.1 μL) and Cs
2CO
3 (688 mg, 2.11 mmol) were added to a solution of tert-butyl-[[3-fluoro-5-methyl-4-[4-(trifluoromethyl)-1H-imidazol-2- yl]phenyl]methoxy]-dimethyl-silane (1.00 g, crude) in ACN (10 mL). The reaction mixture was stirred at 40 °C for 16 hr. The reaction mixture was cooled to room temperature and concentrated under reduced pressure. The residue was diluted with EtOAc (20 mL) and washed with water (2×20 mL). The organic layer was dried over anhydrous Na
2SO
4 and concentrated under reduced pressure to afford tert-butyl-[[3-fluoro-5-methyl-4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]-dimethyl-silane (760 mg, crude) as a yellow solid which was used in the next steps without further purification. MS (ESI): [M+H]+ m/z: calcd 403.19; found 403.2. Step 5: The synthesis of [3-fluoro-5-methyl-4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methanol [1183] A solution of TBAF (520 mg, 1.99 mmol) in THF (5.0 mL) was added to a solution of tert-butyl-[[3-fluoro-5-methyl-4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]-dimethyl-silane (760 mg, crude) in THF (20 mL). The reaction mixture was stirred at room temperature for 16 hr. The reaction mixture was diluted with EtOAc (20 mL) and washed with water (2×20 mL) and brine (20 mL). The organic layer was dried over anhydrous Na
2SO
4 and concentrated under reduced pressure. The residue was subjected to HPLC (0.5-6.5 min, 20-70% water - ACN; flow: 4 mL/min, column: SunFire C18, 100 x 19 mm, 5 µm) to afford [3-fluoro-5-methyl-4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methanol (168 mg, 583 μmol, 23.5% yield from 4-[[tert- butyl(dimethyl)silyl]oxymethyl]-2-fluoro-6-methyl-benzaldehyde) as a light-yellow solid. MS (ESI): [M+H]+ m/z: calcd 289.12; found 289.2. Step 6: The synthesis of 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[3-fluoro-5-methyl-4- [1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]-5-methoxy-pyrimidine [1184] NaH (11.0 mg, 274 μmol, 60% dispersion in mineral oil) was added to a solution of [3-fluoro-5-methyl-4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methanol (75.0 mg, 260 μmol) in DMF (1 mL). The reaction mixture was stirred at room temperature for 30 min. 4-cyclopropyl-6-methoxy-5-(4-methylsulfonylpyrimidin-2-yl)pyrimidine I-8 (79.7 mg, 260 μmol) was added to the reaction mixture. The resulting mixture was stirred at room temperature for 16 hr. The reaction mixture was quenched by addition of water (1 mL). The resulting mixture was diluted with EtOAc (2.0 mL) and washed with water (3 × 1 mL). The organic layer was separated, dried over anhydrous Na
2SO
4 and concentrated under reduced pressure. The residue was subjected to HPLC (0.5-6.5 min, 30-80% water - ACN; flow: 4 mL/min, column: SunFire C18, 100 x 19 mm, 5 µm) to afford 2-(4-cyclopropyl-6-methoxy- pyrimidin-5-yl)-4-[[3-fluoro-5-methyl-4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]-5-methoxy-pyrimidine (51.0 mg, 93.7 μmol, 36.0% yield) as a white solid.
1H NMR (500 MHz, DMSO-d6) δ 0.84 – 0.88 (m, 2H), 100 – 1.04 (m, 2H), 1.67 – 1.73 (m, 1H), 2.11 (s, 3H), 3.47 (s, 3H), 3.83 (s, 3H), 3.94 (s, 3H), 5.45 (s, 2H), 7.28 (d, 1H), 7.32 (s, 1H), 8.00 (s, 1H), 8.43 (s, 1H), 8.64 (s, 1H). MS (ESI): [M+H]+ m/z: calcd 545.22; found 545.2. Example 64 (Compound 198)
Step 1: The synthesis of 2,4-dichloro-5-(fluoromethoxy)pyrimidine [1185] 2,4-dichloropyrimidin-5-ol (450 mg, 2.73 mmol), Cs
2CO
3 (1.16 g, 3.55 mmol) and fluoroiodomethane (873 mg, 5.46 mmol, 368 μL) were mixed in DMF (2 mL). The reaction mixture was stirred at 65 °C 12 hr. The reaction mixture was cooled to room temperature, diluted with water (10 mL) and extracted with MTBE (2×10 mL). The combined organic layers were washed with water (10 mL), dried over anhydrous Na
2SO
4 and concentrated under reduced pressure to afford 2,4-dichloro-5-(fluoromethoxy)pyrimidine (300 mg, 1.52 mmol, 55.8% yield) as a colorless oil which was used in the next steps without further purification. GCMS: [M]+ m/z: calcd 195.96; found 195.9. Step 2: The synthesis of 2-chloro-5-(fluoromethoxy)-4-((4-(1-methyl-4-(trifluoromethyl)-1H- imidazol-2-yl)benzyl)oxy)pyrimidine [1186] [4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methanol I-2b (273 mg, 1.07 mmol), 2,4-dichloro-5-(fluoromethoxy)pyrimidine (300 mg, 1.52 mmol) and Cs
2CO
3 (496 mg, 1.52 mmol) were mixed in THF (4.0 mL). The resulting mixture was stirred under reflux for 12 hr. The reaction mixture was cooled to room temperature and concentrated under reduced pressure. The residue was subjected to flash-column chromatography (SiO
2, gradient chloroform - acetonitrile) to afford 2-chloro-5-(fluoromethoxy)-4-((4-(1-methyl-4- (trifluoromethyl)-1H-imidazol-2-yl)benzyl)oxy)pyrimidine (150 mg, 360 μmol, 23.6% yield) as a yellow oil.
1H NMR (400 MHz, CDCl
3) δ 3.79 (s, 3H), 5.55 (s, 2H), 5.68 (d, 2H, CH2F), 7.32 (s, 1H), 7.58 (d, 2H), 7.68 (d, 2H), 8.20 (s, 1H). MS (ESI): [M+H]+ m/z: calcd 417.08; found 417.0. Step 3: The synthesis of 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-5-(fluoromethoxy)-4- [[4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidine [1187] 2-Chloro-5-(fluoromethoxy)-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidine (80.0 mg, 192 μmol), (4-cyclopropyl-6-methoxy-pyrimidin-5- yl)boronic acid I-5 (112 mg, 576 μmol), potassium phosphate tribasic (122 mg, 576 μmol), XPhos Pd G3 (8.06 mg, 9.60 μmol) were mixed in degassed mixture of dioxane (4 mL) and water (400 µL) under argon atmosphere. The reaction mixture was stirred at room temperature for 24 hr. The mixture was filtered and subjected to HPLC (2-10 min, 50-90% MeOH; flow: 30 mL/min, column: SunFire C18, 100×19 mm, 5 µm) to afford 2-(4- cyclopropyl-6-methoxy-pyrimidin-5-yl)-5-(fluoromethoxy)-4-[[4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidine (8.00 mg, 15.1 μmol, 7.9% yield) as a yellow solid.
1H NMR (600 MHz, DMSO-d6) δ 0.86 – 0.91 (m, 2H), 1.02 – 1.06 (m, 2H), 1.71 – 1.76 (m, 1H), 3.79 (s, 3H), 3.86 (s, 3H), 5.54 (s, 2H), 6.00 (d, 2H, CH2F), 7.60 (d, 2H), 7.74 (d, 2H), 7.95 (s, 1H), 8.60 (s, 1H), 8.67 (s, 1H). MS (ESI): [M+H]+ m/z: calcd 531.20; found 531.2. Example 65 (Compound 77)
Step 1: Synthesis of 4-cyclopropyl-6-methoxy-2-methyl-pyrimidine [1188] Potassium tert-butoxide (1.49 g, 13.3 mmol) was added portion wise to a solution of 4-bromo-6-cyclopropyl-2-methyl-pyrimidine (2.70 g, 12.7 mmol) in MeOH (25 mL) at 0 °C. The cooling bath was removed, and the resulting mixture was stirred at ambient temperature for 12 hr. The reaction mixture was concentrated under reduced pressure. The residue was dissolved in DCM (200 mL). The obtained solution was washed with water (2×100 mL) and brine (150 mL), dried over anhydrous Na
2SO
4, filtered and concentrated under reduced pressure to afford 4-cyclopropyl-6-methoxy-2-methyl-pyrimidine (1.50 g, 9.13 mmol, 72.1% yield), as yellow liquid which was used in the next steps without further purification.
1H NMR (400 MHz, CDCl
3) δ 0.94 – 1.06 (m, 4H), 1.84 – 1.93 (m, 1H), 2.52 (s, 3H), 3.91 (s, 3H), 6.28 (s, 1H). MS (ESI): [M+H]+ m/z: calcd 165.10; found 165.2. Step 2: Synthesis of 5-bromo-4-cyclopropyl-6-methoxy-2-methyl-pyrimidine [1189] To a stirred solution of 4-cyclopropyl-6-methoxy-2-methyl-pyrimidine (1.50 g, 9.13 mmol) in acetic acid (15 mL) N-Bromosuccinimide (1.63 g, 9.13 mmol) was added at ambient temperature. The resulting mixture was stirred at 60 °C for 12 hr. The resulting mixture was cooled to room temperature and concentrated under reduced pressure. The residue was subjected to flash chromatography (SiO
2, gradient hexane-chloroform) to afford 5-bromo-4-cyclopropyl-6-methoxy-2-methyl-pyrimidine (1.00 g, 4.11 mmol, 45.0% yield) as white solid.
1H NMR (500 MHz, CDCl
3) δ 0.98 – 1.05 (m, 2H), 1.10 – 1.18 (m, 2H), 1.58 (s, 1H), 2.46 (s, 3H), 4.00 (s, 3H). MS (ESI): [M+H]+ m/z: calcd 243.01 and 245.01; found 243.0 and 245.0. Step 3: Synthesis of 4-cyclopropyl-6-methoxy-2-methyl-5-(4,4,5,5-tetramethyl-1,3,2- dioxaborolan-2-yl)pyrimidine [1190] 5-bromo-4-cyclopropyl-6-methoxy-2-methyl-pyrimidine (500 mg, 2.06 mmol) was added to a mixture of Bis(pinacolato)diboron (575 mg, 2.26 mmol), Cesium pivalate (967 mg, 4.11 mmol) and bis(diphenylphosphino)ferrocene]dichloropalladium(II)-DCM (168 mg, 206 μmol) in degassed dioxane (20 mL). The resulting mixture was stirred under argon atmosphere at 90 °C for 12 hr. The resulting mixture was cooled to room temperature and filtered. The filtrate was concentrated under reduced pressure to afford 4-cyclopropyl-6- methoxy-2-methyl-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyrimidine (600 mg, crude) as brown solid which was used in the next steps without further purification. MS (ESI): [M+H]+ m/z: calcd 291.19; found 291.4. Step 4: Synthesis of 4-cyclopropyl-6-methoxy-2-methyl-5-[5-methyl-4-[[4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidin-2-yl]pyrimidine [1191] 2-Chloro-5-methyl-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidine I-2c (150 mg, 392 μmol), 4-cyclopropyl-6-methoxy-2- methyl-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyrimidine (341 mg, 1.18 mmol), RuPhos Pd G4 (33.3 mg, 39.2 μmol) and potassium phosphate tribasic (101 mg, 477 μmol) were mixed in a degassed mixture of dioxane (20 mL) and water (0.5 mL) under argon atmosphere. The reaction mixture was stirred at 100 °C for 12 hr. The reaction mixture was cooled to room temperature and filtered. The filtrate was concentrated in vacuo. The residue was subjected to HPLC (0-5 min, 50-80% water+FA (0.1% vol.) - ACN+FA (0.1% vol.); flow: 30 mL/min, column: Chromatorex 18 SMB100-5T, 100 x 19 mm, 5 µm) to afford 4- cyclopropyl-6-methoxy-2-methyl-5-[5-methyl-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol- 2-yl]phenyl]methoxy]pyrimidin-2-yl]pyrimidine (46.0 mg, 90.1 μmol, 23.0% yield) as an off- white solid.
1H NMR (600 MHz, DMSO-d6) δ 0.76 – 0.79 (m, 2H), 0.95 – 1.01 (m, 2H), 1.60 – 1.67 (m, 1H), 2.20 (s, 3H), 2.46 (s, 3H), 3.76 (s, 3H), 3.79 (s, 3H), 5.50 (s, 2H), 7.57 (d, 2H), 7.71 (d, 2H), 7.92 (s, 1H), 8.52 (s, 1H). MS (ESI): [M+H]+ m/z: calcd 511.24; found 511.2. Example 66 (Compound 88)

The synthesis of starting 2-chloro-5-methoxy-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidine is described for Compound 12 and compound 132. Synthesis of 4-cyclopropyl-6-methoxy-5-[5-methoxy-4-[[4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidin-2-yl]-2-methyl-pyrimidine [1192] 2-Chloro-5-methoxy-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidine (100 mg, 251 μmol), 4-cyclopropyl-6-methoxy-2-methyl-5- (4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyrimidine (153 mg, 527 μmol), RuPhos Pd G4 (21.3 mg, 25.1 μmol) and potassium phosphate tribasic (107 mg, 502 μmol) were mixed in a degassed mixture of dioxane (20 mL) and water (0.5 mL) under argon atmosphere. The reaction mixture was stirred at 100 °C for 12 hr. The reaction mixture was cooled to room temperature and filtered. The filtrate was concentrated in vacuo. The residue was subjected to HPLC (0-5 min., 40-90% water – MeOH, +0.1% vol. of 25% aq. NH
3, flow: 30 mL/min, column: XBridge C18100 ×19 mm, 5 µm) to afford 4-cyclopropyl-6-methoxy-5-[5-methoxy- 4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidin-2-yl]-2-methyl- pyrimidine (15.9 mg, 30.2 μmol, 12.0% yield) as yellow solid.
1H NMR (600 MHz, DMSO- d6) δ 0.78 – 0.83 (m, 2H), 0.95 – 0.99 (m, 2H), 1.64 – 1.69 (m, 1H), 2.46 (s, 3H), 3.77 (s, 3H), 3.80 (s, 3H), 3.92 (s, 3H), 5.47 (s, 2H), 7.56 (d, 2H), 7.72 (d, 2H), 7.93 (s, 1H), 8.39 (s, 1H). MS (ESI): [M+H]+ m/z: calcd 527.23; found 527.2. Example 67 (Compound 142)
Step 1: The synthesis of 2-chloro-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidin-5-amine [1193] Potassium tert-butoxide (72.3 mg, 644 μmol) was added to a solution of [4-[1-methyl- 4-(trifluoromethyl)imidazol-2-yl]phenyl]methanol I-2b (150 mg, 585 μmol) in THF (5 mL) at 0 °C. The reaction mixture was stirred at 0 °C for 10 min.2,4-dichloropyrimidin-5-amine (96.0 mg, 585 μmol) was added to the mixture. The resulting mixture was stirred at room temperature for 14 hr. The reaction mixture was poured into water (10 mL) and extracted with EtOAc (10 mL). The organic layer was dried over anhydrous Na
2SO
4 and concentrated under reduced pressure to afford 2-chloro-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidin-5-amine (200 mg, 521 μmol, 88.9% yield) as a brown solid which was used in the next step without further purification.
1H NMR (500 MHz, CDCl
3) δ 3.69 – 3.84 (m, 5H), 5.51 (s, 2H), 7.32 (s, 1H), 7.56 (d, 2H), 7.67 (d, 2H), 7.82 (s, 1H). MS (ESI): [M+H]+ m/z: calcd 384.09; found 384.0 Step 2: The synthesis of 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidin-5-amine [1194] 2-chloro-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidin-5-amine (200 mg, 469 μmol), (4-cyclopropyl-6-methoxy- pyrimidin-5-yl)boronic acid I-5 (137 mg, 704 μmol), RuPhos Pd G4 (29.9 mg, 35.2 μmol) and potassium phosphate tribasic anhydrous (249 mg, 1.17 mmol) were mixed in a degassed mixture of water (1 mL) and dioxane (4 mL) under argon atmosphere at room temperature. The reaction mixture was stirred at 90 °C for 15 hr. The reaction mixture was cooled to room temperature and concentrated under reduced pressure. The residue was diluted with water (10 mL) and extracted with EtOAc (3×20 mL). The combined organic layers were dried over anhydrous Na
2SO
4 and concentrated under reduced pressure. The residue was subjected to HPLC (2-10 min, 40-95% water+FA (0.1% vol.) - ACN+FA (0.1% vol.); flow: 30 mL/min, column: Waters SunFire C18, 100 x 19 mm, 5 µm), then repurified by SFC to afford 2-(4- cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidin-5-amine (3.60 mg, 7.24 μmol, 1.5% yield) as a brown solid.
1H NMR (600 MHz, DMSO-d6) δ 0.78 – 0.84 (m, 2H), 0.96 – 1.00 (m, 2H), 1.68 – 1.73 (m, 1H), 3.76 (s, 3H), 3.80 (s, 3H), 5.37 (s, 2H), 5.49 (s, 2H), 7.61 (d, 2H), 7.71 (d, 2H), 7.92 (s, 1H), 8.01 (s, 1H), 8.59 (s, 1H). MS (ESI): [M+H]+ m/z: calcd 498.20; found 498.2.

Step 1: The synthesis of ethyl 3-[(4-cyanophenyl)hydrazono]-4,4-difluoro-butanoate [1195] Sodium hydroxide (590 mg, 14.7 mmol) was added to a solution of 4- hydrazinobenzonitrile (2.50 g, 14.7 mmol, HCl) in EtOH (30 mL). The reaction mixture was stirred at room temperature for 40 min. A solution of ethyl 4,4-difluoro-3-oxo-butanoate (2.45 g, 14.7 mmol) in EtOH (20 mL) was added to the reaction mixture. The resulting mixture was stirred at 80°C for 12 hr. The reaction mixture was cooled to room temperature and filtered. The filtrate was concentrated under reduced pressure to afford ethyl 3-[(4- cyanophenyl)hydrazono]-4,4-difluoro-butanoate (2.50 g, 8.89 mmol, 60.2% yield) as an orange solid which was used in the next step without further purification. MS (ESI): [M+H]+ m/z: calcd 282.11; found 282.0 Step 2: The synthesis of 4-[3-(difluoromethyl)-5-hydroxy-pyrazol-1-yl]benzonitrile [1196] A solution of ethyl 3-[(4-cyanophenyl)hydrazono]-4,4-difluoro-butanoate (2.50 g, 8.89 mmol) in toluene (50 mL) was stirred under reflux for 12 hr. The reaction mixture was cooled to room temperature and concentrated under reduced pressure. The residue was subjected to flash column chromatography (SiO
2, hexane – MTBE) to afford 4-[3- (difluoromethyl)-5-hydroxy-pyrazol-1-yl]benzonitrile (900 mg, 3.83 mmol, 43.1% yield) as an orange solid.
1H NMR (500 MHz, DMSO-d6) δ 5.78 (s, 1H), 6.88 (t, 1H, CHF2), 7.94 – 8.00 (m, 4H), 12.75 (s, 2H). MS (ESI): [M+H]+ m/z: calcd 236.2; found 236.0. Step 3: The synthesis of 4-[3-(difluoromethyl)-5-methoxy-pyrazol-1-yl]benzonitrile [1197] 4-[3-(difluoromethyl)-5-hydroxy-pyrazol-1-yl]benzonitrile (800 mg, 3.40 mmol) was added to a stirred suspension of NaH (136 mg, 3.40 mmol, 60% dispersion in mineral oil) in DMF (10 mL) at 0 °C. The reaction mixture was stirred at 0 °C for 1 hr. Iodomethane (483 mg, 3.40 mmol, 212 μL) was added to the reaction mixture. The resulting reaction mixture was stirred at room temperature for 12 hr. The reaction mixture was diluted with water (30 mL) and extracted with EtOAc (40 mL). The organic layer was washed with brine (30 mL), dried over anhydrous Na
2SO
4 and concentrated under reduced pressure to afford 4-[3- (difluoromethyl)-5-methoxy-pyrazol-1-yl]benzonitrile (620 mg, 2.49 mmol, 73.2 % yield) as a red oil which was used in the next steps without further purification. MS (ESI): [M+H]+ m/z: calcd 250.08; found 250.0 Step 4: The synthesis of 4-[3-(difluoromethyl)-5-methoxy-pyrazol-1-yl]benzoic acid [1198] A solution of potassium hydroxide (141 mg, 2.51 mmol) in water (2.0 mL) was added to a solution of 4-[3-(difluoromethyl)-5-methoxy-pyrazol-1-yl]benzonitrile (250 mg, 1.00 mmol) in EtOH (5 mL). The resulting mixture was stirred at 100 °C for 12 hr. The reaction mixture was cooled to room temperature and concentrated under reduced pressure. The residue was diluted with water (30 mL), quenched with AcOH and extracted with EtOAc (40 mL). The organic layer was washed with brine (30 mL), dried over anhydrous Na
2SO
4 and concentrated under reduced pressure to afford 4-[3-(difluoromethyl)-5-methoxy-pyrazol-1- yl]benzoic acid (150 mg, 559 μmol, 55.8% yield) as a red solid which was used in the next steps without further purification. MS (ESI): [M+H]+ m/z: calcd 269.08; found 269.0. Step 5: The synthesis of [4-[3-(difluoromethyl)-5-methoxy-pyrazol-1-yl]phenyl]methanol [1199] Borane dimethyl sulfide complex (93.5 mg, 1.23 mmol, 117 μL) was added to a solution of 4-[3-(difluoromethyl)-5-methoxy-pyrazol-1-yl]benzoic acid (110 mg, 410 μmol) in THF (7 mL) under argon atmosphere at 0 °C. The reaction mixture was stirred at room temperature for 12 hr. The reaction mixture was quenched by dropwise addition of MeOH (2 mL). The resulting mixture was concentrated under reduced pressure. The residue was diluted with water (7 mL) and extracted with EtOAc (20 mL). The organic layer was washed with brine (10 mL), dried over anhydrous Na
2SO
4 and concentrated under reduced pressure to afford [4-[3-(difluoromethyl)-5-methoxy-pyrazol-1-yl]phenyl]methanol (100 mg, 393 μmol, 95.9% yield) as a yellow solid which was used in the next steps without further purification. MS (ESI): [M+H]+ m/z: calcd 255.10; found 255.0 Step 6: The synthesis of 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[4-[3- (difluoromethyl)-5-methoxy-pyrazol-1-yl]phenyl]methoxy]-5-methoxy-pyrimidine [1200] [4-[3-(difluoromethyl)-5-methoxy-pyrazol-1-yl]phenyl]methanol (100 mg, 393 μmol) was added to a solution of NaH (17.3 mg, 433 μmol, 60% dispersion in mineral oil) in THF (5 mL) at 0 °C. The reaction mixture was stirred at 0 °C for 1 hr.2-(4-cyclopropyl-6- methoxy-pyrimidin-5-yl)-5-methoxy-4-methylsulfonyl-pyrimidine I-6 (132 mg, 393 μmol) was added to the reaction mixture. The resulting mixture was stirred at 50 °C for 12 hr. The reaction mixture was cooled to room temperature and concentrated under reduced pressure. The residue was subjected to HPLC (0-1.5-6 min., 30-30-80% water – MeOH, +0.1% vol. of 25% aq. NH
3, flow: 40 mL/min, column: YMC Triart C18100 ×19 mm, 5 µm) to afford 2- (4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[4-[3-(difluoromethyl)-5-methoxy-pyrazol-1- yl]phenyl]methoxy]-5-methoxy-pyrimidine (45.8 mg, 89.7 μmol, 20.7% yield) as a white solid.
1H NMR (600 MHz, DMSO-d6) δ 0.82 – 0.87 (m, 2H), 0.99 – 1.03 (m, 2H), 1.64 – 1.70 (m, 1H), 3.82 (s, 3H), 3.92 (s, 3H), 3.97 (s, 3H), 5.45 (s, 2H), 6.22 (s, 1H), 6.92 (t, 1H, CHF2) 7.56 (d, 2H), 7.64 (d, 2H), 8.40 (s, 1H), 8.63 (s, 1H). MS (ESI): [M+H]+ m/z: calcd 511.21; found 511.0.
Step 1: Synthesis of 2,2,2-trifluoro-1-[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]ethanol [1201] The trimethyl(trifluoromethyl)silane (336 mg, 2.36 mmol) was added to solution of 4- [1-methyl-4-(trifluoromethyl)imidazol-2-yl]benzaldehyde (500 mg, 1.97 mmol) and Tetrabutylammonium fluoride (98.34 μmol, 98.3 μL, 1 M solution in THF) in THF (25 mL) . The reaction mixture was stirred at 25 °C for 12 hr. The reaction mixture was diluted with HCl, 36% w/w aq. solution (143.43 mg, 3.93 mmol, 179.28 μL). Resulting mixture was stirred for 2 hr, then diluted with saturated aqueous solution of NaHCO
3. Resulting mixture was extracted EtOAc (20 mL). Organic layer was concentrated under reduced pressure to afford 2,2,2-trifluoro-1-[4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]ethanol (460 mg, 1.42 mmol, 72.1% yield) as a yellow solid which was used in the next steps without further purification.
1H NMR (500 MHz, DMSO-d6) δ 3.76 (s, 3H), 4.90 – 4.93 (m, 1H), 7.34 (s, 1H), 7.42 (d, 2H), 7.52 (d, 2H). MS (ESI): [M+H]+ m/z: calcd 325.07; found 325.2. Step 2: The synthesis of 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-5-methoxy-4-[2,2,2- trifluoro-1-[4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]ethoxy]pyrimidine. (151). [1202] NaH (6.28 mg, 157 μmol, 60% dispersion in mineral oil) was added to a solution of 2,2,2-trifluoro-1-[4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]ethanol (50.9 mg, 157 μmol) in DMF (2 mL). The reaction mixture was stirred at ambient temperature for 0.5 hr.2- (4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-5-methoxy-4-methylsulfonyl-pyrimidine I-6 (44.0 mg, 131 μmol) was added to the reaction mixture. The resulting mixture was stirred at room temperature for 12 hr. The reaction mixture was diluted with water (10 mL) and extracted with EtOAc (2×10 mL). The combined organic layers were washed with brine (10 mL) and concentrated under reduced pressure. The residue was subjected to HPLC (0.5-6.5 min., 54% water – ACN, +0.1% vol. of 25% aq. NH
3, flow: 30 mL/min, column: SunFire C18100 ×19 mm, 5 µm) to afford 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-5-methoxy-4-[2,2,2- trifluoro-1-[4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]ethoxy]pyrimidine (40.0 mg, 68.9 μmol, 52.7% yield) as an white solid.
1H NMR (600 MHz, DMSO-d6) δ 0.62 – 0.66 (m, 1H), 0.71 – 0.75 (m, 1H), 0.87 – 0.96 (m, 2H), 1.42 – 1.47 (m, 1H), 3.67 (s, 3H), 3.77 (s, 3H), 4.03 (s, 3H), 6.93 – 6.99 (m, 1H), 7.66 (d, 2H), 7.79 (d, 2H), 7.94 (s, 1H), 8.54 (s, 1H), 8.60 (s, 1H). MS (ESI): [M+H]+ m/z: calcd 581.19; found 581.2. Step 3: The synthesis of 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-5-methoxy-4-[rac-(1R)- 2,2,2-trifluoro-1-[4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]ethoxy]pyrimidine (11) and 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-5-methoxy-4-[rac-(1S)-2,2,2-trifluoro- 1-[4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]ethoxy]pyrimidine (203). [1203] Racemic 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-5-methoxy-4-[2,2,2-trifluoro- 1-[4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]ethoxy]pyrimidine (0.037 g, 63.7 μmol) was subjected to chiral HPLC (column: CHIRALPAK AD-H (250x21 mm, 5 μm), Mobile Phase: Hexane:IPA-MeOH, 90:5:5, Flow Rate: 12 ml/min) to afford 2-(4- cyclopropyl-6-methoxy-pyrimidin-5-yl)-5-methoxy-4-[rac-(1R)-2,2,2-trifluoro-1-[4-[1- methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]ethoxy]pyrimidine (14 mg, 24.12 μmol, 37.84% yield) and 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-5-methoxy-4-[rac-(1S)- 2,2,2-trifluoro-1-[4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]ethoxy]pyrimidine (13.0 mg, 22.40 μmol, 35.1% yield) as a white solids. 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-5-methoxy-4-[rac-(1R)-2,2,2-trifluoro-1-[4-[1- methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]ethoxy]pyrimidine (14.0 mg, 24.1 μmol, 37.8% yield) (11):
1H NMR (600 MHz, DMSO-d6) δ 0.62 – 0.66 (m, 1H), 0.71 – 0.75 (m, 1H), 0.87 – 0.96 (m, 2H), 1.42 – 1.47 (m, 1H), 3.67 (s, 3H), 3.77 (s, 3H), 4.03 (s, 3H), 6.93 – 6.99 (m, 1H), 7.66 (d, 2H), 7.79 (d, 2H), 7.94 (s, 1H), 8.54 (s, 1H), 8.60 (s, 1H). MS (ESI): [M+H]+ m/z: calcd 563.2; found 563.2. Enantiopurity: 100% (column: CHIRALPAK AD-H, 250 × 4.6 mm, 5 μm; mobile phase: Hexane-IPA-MeOH, 80-10-10; flow: 0.6 mL/min; RT=18.2 min) 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-5-methoxy-4-[rac-(1S)-2,2,2-trifluoro-1-[4-[1- methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]ethoxy]pyrimidine (13.0 mg, 22.4 μmol, 35.1% yield) (203):
1H NMR (600 MHz, DMSO-d6) δ 0.62 – 0.65 (m, 1H), 0.71 – 0.75 (m, 1H), 0.87 – 0.96 (m, 2H), 1.41 – 1.46 (m, 1H), 3.67 (s, 3H), 3.77 (s, 3H), 4.03 (s, 3H), 6.93 – 6.99 (m, 1H), 7.66 (d, 2H), 7.79 (d, 2H), 7.94 (s, 1H), 8.54 (s, 1H), 8.60 (s, 1H). MS (ESI): [M+H]+ m/z: calcd 563.2; found 563.4. Enantiopurity: 100% (column: CHIRALPAK AD-H, 250 × 4.6 mm, 5 μm; mobile phase: Hexane-IPA-MeOH, 80-10-10; flow: 0.6 mL/min; RT=9.3 min). Example 70 (Compound 96 and 231)

Step 1: The synthesis of 4-cyclopropyl-5-[4-[2,2-difluoro-1-[4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]ethoxy]pyrimidin-2-yl]-6-methoxy-pyrimidine The synthesis of the starting 2,2-difluoro-1-[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]ethanol is described in Example 53 (Compound 95 and Compound 204). [1204] NaH (5.88 mg, 147 μmol, 60% dispersion in mineral oil) was added to a solution of 2,2-difluoro-1-[4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]ethanol (54.0 mg, 176 μmol) in DMF (2 mL). The reaction mixture was stirred at ambient temperature for 0.5 hr. The 4-cyclopropyl-6-methoxy-5-(4-methylsulfonylpyrimidin-2-yl)pyrimidine I-8 (45.0 mg, 147 μmol) was added to the reaction mixture. The resulting mixture was stirred at room temperature for 12 hr. The reaction mixture was diluted with water (10 mL) and extracted with EtOAc (2 × 10 mL). The combined organic layers were washed with brine (10 mL) and concentrated under reduced pressure. The residue was subjected to HPLC (0.5-6.5 min., 45- 65% water – ACN, +0.1% vol. of 25% aq. NH
3, flow: 30 mL/min, column: XBridge 100 ×19 mm, 5 µm) to afford 4-cyclopropyl-5-[4-[2,2-difluoro-1-[4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]ethoxy]pyrimidin-2-yl]-6-methoxy-pyrimidine (28.0 mg, 52.6 μmol, 35.8% yield) as an white solid.
1H NMR (500 MHz, DMSO-d6) δ 0.65 – 0.72 (m, 1H), 0.76 – 0.82 (m, 1H), 0.91 – 1.01 (m, 2H), 1.47 – 1.53 (m, 1H), 3.74 (s, 3H), 3.78 (s, 3H), 6.43 – 6.67 (m, 2H), 7.23 (d, 1H), 7.62 (d, 2H), 7.77 (d, 2H), 7.95 (s, 1H), 8.65 (s, 1H), 8.77 (d, 1H). MS (ESI): [M+H]+ m/z: calcd 533.17; found 533.2. Step 2: Chiral resolution of 4-cyclopropyl-6-methoxy-5-[4-[rel-(1S)-2,2-difluoro-1-[4-[1- methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]ethoxy]pyrimidin-2-yl]pyrimidine (231) and 4-cyclopropyl-6-methoxy-5-[4-[rel-(1R)-2,2-difluoro-1-[4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]ethoxy]pyrimidin-2-yl]pyrimidine (96) [1205] Racemic 4-cyclopropyl-5-[4-[2,2-difluoro-1-[4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]ethoxy]pyrimidin-2-yl]-6-methoxy-pyrimidine (27.0 mg, 50.7 μmol) was subjected to chiral HPLC (column: CHIRALPAK AD-H (250×20 mm, 5 μm), Mobile Phase: Hexane-IPA-MeOH, 70-15-15, Flow Rate: 15 ml/min) to afford 4- cyclopropyl-6-methoxy-5-[4-[rel-(1S)-2,2-difluoro-1-[4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]ethoxy]pyrimidin-2-yl]pyrimidine (10.0 mg, 18.8 μmol, 37.0% yield) and 4-cyclopropyl-6-methoxy-5-[4-[rel-(1R)-2,2-difluoro-1-[4-[1-methyl- 4-(trifluoromethyl)imidazol-2-yl]phenyl]ethoxy]pyrimidin-2-yl]pyrimidine (10.0 mg, 18.8 μmol, 37.0% yield) as a white solids. 4-cyclopropyl-6-methoxy-5-[4-[rel-(1S)-2,2-difluoro-1-[4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]ethoxy]pyrimidin-2-yl]pyrimidine (231):
1H NMR (600 MHz, DMSO-d6) δ 0.66 – 0.72 (m, 1H), 0.76 – 0.82 (m, 1H), 0.92 – 1.01 (m, 2H), 1.47 – 1.53 (m, 1H), 3.74 (s, 3H), 3.78 (s, 3H), 6.45 – 6.66 (m, 2H), 7.23 (d, 1H), 7.62 (d, 2H), 7.77 (d, 2H), 7.95 (s, 1H), 8.65 (s, 1H), 8.77 (d, 1H). MS (ESI): [M+H]+ m/z: calcd 533.20; found 533.2. Enantiopurity: 100% (column: CHIRALPAK AD-H, 250 × 4.6 mm, 5 μm; mobile phase: Hexane-IPA-MeOH, 70-15-15; flow: 0.8 mL/min; RT=5.98 min) 4-cyclopropyl-6-methoxy-5-[4-[rel-(1R)-2,2-difluoro-1-[4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]ethoxy]pyrimidin-2-yl]pyrimidine (96):
1H NMR (600 MHz, DMSO-d6) δ 0.66 – 0.72 (m, 1H), 0.76 – 0.82 (m, 1H), 0.92 – 1.01 (m, 2H), 1.47 – 1.53 (m, 1H), 3.74 (s, 3H), 3.78 (s, 3H), 6.45 – 6.66 (m, 2H), 7.23 (d, 1H), 7.62 (d, 2H), 7.77 (d, 2H), 7.95 (s, 1H), 8.65 (s, 1H), 8.77 (d, 1H). MS (ESI): [M+H]+ m/z: calcd 533.20; found 533.2. Enantiopurity: 100% (column: CHIRALPAK AD-H, 250 × 4.6 mm, 5 μm; mobile phase: Hexane-IPA-MeOH, 70-15-15; flow: 0.8 mL/min; RT=11.8 min). Example 71 (Compound 177)
Step 1: The synthesis of 2-bromo-3-methyl-6-(trifluoromethyl)pyridine [1206] 6-bromo-5-methyl-pyridine-2-carboxylic acid (4.00 g, 18.5 mmol), HF (7.41 g, 370 mmol) and sulfur tetrafluoride (6.00 g, 55.6 mmol) were mixed in a corrosion-resistant steel autoclave. The autoclave was sealed and the reaction mixture was stirred at 85 °C for 12 hr. The reaction mixture was cooled to room temperature and concentrated under reduced pressure. The residue was distilled in vacuo to afford 2-bromo-3-methyl-6- (trifluoromethyl)pyridine (3.50 g, 14.6 mmol, 78.8% yield) as a light-yellow solid which was used in the next steps without further purification.
1H NMR (500 MHz, CDCl3) δ 2.47 (s, 3H), 7.55 (d, 1H), 7.68 (d, 1H). MS (ESI): [M+H]+ m/z: calcd 239.97; found 239.8. Step 2: The synthesis of [4-[3-methyl-6-(trifluoromethyl)-2-pyridyl]phenyl]methanol [1207] 2-bromo-3-methyl-6-(trifluoromethyl)pyridine (500 mg, 2.08 mmol), [4- (hydroxymethyl)phenyl]methanediol (450 mg, 2.92 mmol), potassium phosphate tribasic (1.33 g, 6.25 mmol) and bis(diphenylphosphino)ferrocene]dichloropalladium(II)-DCM (85.1 mg, 104 μmol) were mixed in degassed dioxane (25 mL) and water (1.25 mL). The reaction mixture was stirred at 90 °C for 16 hr under argon atmosphere. The reaction mixture was cooled to room temperature, diluted with EtOAc (100 mL) and washed with water (2×50 mL). The organic layer was concentrated under reduced pressure to afford [4-[3-methyl-6- (trifluoromethyl)-2-pyridyl]phenyl]methanol (700 mg, crude) which was used in the next step without further purification. MS (ESI): [M+H]+ m/z: calcd 268.10; found 268.2 Step 3: The synthesis of 4-cyclopropyl-6-methoxy-5-[4-[[4-[3-methyl-6-(trifluoromethyl)-2- pyridyl]phenyl]methoxy]pyrimidin-2-yl]pyrimidine [1208] NaH (12.0 mg, 300 μmol, 60% dispersion in mineral oil) was added to the stirred solution of [4-[3-methyl-6-(trifluoromethyl)-2-pyridyl]phenyl]methanol (90.0 mg, 273 μmol) in DMF (1.0 mL). The reaction mixture was stirred at room temperature for 1 hr.4- cyclopropyl-6-methoxy-5-(4-methylsulfonylpyrimidin-2-yl)pyrimidine I-8 (83.6 mg, 273 μmol) was added to the reaction mixture. The resulting mixture was stirred at room temperature for 16 hr. The reaction mixture was diluted with water (5 mL) and extracted with EtOAc (20 mL). The organic layer was washed with water (20 mL) and concentrated under reduced pressure. The residue was subjected to HPLC (0.5-6.5 min., 50-100% water – MeOH, +0.1% vol. of 25% aq. NH
3, flow: 30 mL/min, column: XBridge 100 ×19 mm, 5 µm) to afford 4-cyclopropyl-6-methoxy-5-[4-[[4-[3-methyl-6-(trifluoromethyl)-2- pyridyl]phenyl]methoxy]pyrimidin-2-yl]pyrimidine (45.0 mg, 91.2 μmol, 33.4% yield) as a white solid.
1H NMR (600 MHz, DMSO-d6) δ 0.84 – 0.89 (m, 2H), 1.00 – 1.04 (m, 2H), 1.63 – 1.69 (m, 1H), 2.40 (s, 3H), 3.84 (s, 3H), 5.50 (s, 2H), 7.06 (d, 1H), 7.53 – 7.60 (m, 4H), 7.79 (d, 1H), 8.01 (d, 1H), 8.66 (s, 1H), 8.69 (d, 1H). MS (ESI): [M+H]+ m/z: calcd 494.21; found 494.0. Example 72 (Compound 193)
Compound 193 Step 1: Synthesis of methyl 2-chloro-4-[4-(trifluoromethyl)-1H-imidazol-2-yl]benzoate Sodium acetate (3.80 g, 46.3 mmol) was dissolved in H2O (25 mL) then 3,3-dibromo-1,1,1- trifluoro-propan-2-one (6.00 g, 22.2 mmol) was added, and the reaction mixture was stirred at 95 °C for 1hr. The resulting mixture was cooled to room temperature and poured into a solution of methyl 2-chloro-4-formyl-benzoate (4.00 g, 20.2 mmol) and NH
4OH (30 mL, 25% wt.) in MeOH (100 mL). The obtained mixture was stirred at room temperature for 12 hr. The mixture was concentrated under reduced pressure. The residue was distributed between EtOAc (30 mL) and water (20 mL). The organic layer was separated, dried over anhydrous Na
2SO
4, filtered and concentrated under reduced pressure to afford methyl 2- chloro-4-[4-(trifluoromethyl)-1H-imidazol-2-yl]benzoate (5.93 g, 19.5 mmol, 96.6% yield) as a yellow oil which was used in the next steps without further purification.
1H NMR (400 MHz, CDCl
3) δ 3.93 (s, 3H), 7.48 (s, 1H), 7.77 (d, 1H), 7.86 (d, 1H), 7.93 (s, 1H). MS (ESI): [M+H]+ m/z: calcd 305.04; found 305.0. Step 2: Synthesis of methyl 2-chloro-4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]benzoate [1209] NaH (441 mg, 11.0 mmol, 60% in mineral oil) was added in a few portions to a solution of methyl 2-chloro-4-[4-(trifluoromethyl)-1H-imidazol-2-yl]benzoate (5.93 g, 10.5 mmol) in DMF (10 mL) under argon atmosphere at 0 °C. The reaction mixture was stirred at ambient temperature for 1 hr. Methyl iodide (1.64 g, 11.6 mmol) was added dropwise. The resulting mixture was stirred at ambient temperature for 12 hr. The reaction mixture was diluted with EtOAc (50 mL) and brine (30 mL). The organic layer was separated, washed with brine (2 × 50 mL) and concentrated under reduced pressure to afford methyl 2-chloro-4- [1-methyl-4-(trifluoromethyl)imidazol-2-yl]benzoate (2.50 g, 7.84 mmol, 74.7% yield) as a red oil which was used in the next steps without further purification. MS (ESI): [M+H]+m/z: calcd 319.06; found 319.0. Step 3: Synthesis [2-chloro-4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methanol [1210] Borane dimethyl sulfide complex (942 mg, 12.4 mmol,) was added dropwise to a solution of methyl 2-chloro-4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]benzoate (2.50 g, 6.20 mmol) in THF (50 mL) under argon atmosphere. The reaction mixture was stirred at 40 °C for 12 hr. The mixture was cooled to room temperature and carefully quenched by dropwise addition of water (10 mL) under argon atmosphere. The resulting mixture was stirred at 25 °C for 3 hr. The resulting mixture was concentrated under reduced pressure to half volume. The residue was extracted with EtOAc (2 × 50 mL). The combined organic layers were dried over anhydrous Na
2SO
4 and concentrated under reduced pressure to afford [2-chloro-4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methanol (1.80 g, 6.19 mmol, 99.9% yield) as a yellow solid which was used in the next steps without further purification.
1H NMR (400 MHz, DMSO-d6) δ 3.76 (s, 3H), 4.57 – 4.61 (m, 2H), 5.46 – 5.50 (m, 1H), 7.67 – 7.74 (m, 3H), 7.93 (s, 1H). MS (ESI): [M+H]+m/z: calcd 291.07; found 291.0. Step 4: Synthesis of 4-[[2-chloro-4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]-2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-5-methoxy-pyrimidine [1211] NaH (10.7 mg, 266 μmol, 60% dispersion in mineral oil) was added to a solution of [2-chloro-4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methanol (80.0 mg, 275 μmol) in DMF (3 mL) under argon atmosphere. The reaction mixture was stirred at ambient temperature for 1 hr.2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-5-methoxy-4- methylsulfonyl-pyrimidine I-6 (92.6 mg, 275 μmol) was added to the mixture. The resulting mixture was stirred at ambient temperature for 48 hr. The reaction mixture was diluted with EtOAc (20 mL) and brine (10 mL). The organic layer was separated, washed with brine (2 × 10 mL) and dried over anhydrous Na
2SO
4. To the obtained solution SiliaMetS® Dimercaptotriazine (100 mg) was added. The mixture was stirred for 30 min. The mixture was filtered. The filtrate was concentrated under reduced pressure. The residue was subjected to HPLC (0.5-6.5 min, 48-63% water - ACN; flow: 30 mL/min, column: SunFire C18, 100 x 19 mm, 5 µm) to afford 4-[[2-chloro-4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]-2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-5-methoxy-pyrimidine (40.0 mg, 73.1 μmol, 26.5% yield) as a light-yellow solid.
1H NMR (600 MHz, DMSO-d6) δ 0.82 – 0.86 (m, 2H), 1.00 – 1.03 (m, 2H), 1.69 – 1.74 (m, 1H), 3.80 (s, 3H), 3.82 (s, 3H), 3.94 (s, 3H), 5.54 (s, 2H), 7.67 (d, 1H), 7.73 (d, 1H), 7.84 (s, 1H), 7.97 (s, 1H), 8.44 (s, 1H), 8.64 (s, 1H). MS (ESI): [M+H]+ m/z: calcd 547.17; found 547.0. Example 73 (Compound 55)
C
ompound 55 Step 1: The synthesis of 2-chloro-5-iodo-4-methylsulfanyl-pyrimidine [1212] Sodium thiomethoxide (5.34 g, 16.0 mmol, 21% aqueous solution) was added dropwise to a cooled to 5 °C solution of 2,4-dichloro-5-iodo-pyrimidine (4.00 g, 14.6 mmol) in THF (50 mL). The reaction mixture was stirred at room temperature for 13 h. The mixture was concentrated under reduced pressure. The residue was diluted with MTBE (150 mL), washed with water (2 × 25 mL) and brine (2 × 15 mL), dried over anhydrous Na
2SO
4, filtered through a thin pad of silica and concentrated under reduced pressure to afford 2-chloro-5- iodo-4-methylsulfanyl-pyrimidine (3.90 g, 13.6 mmol, 93.5 % yield) as a white solid which was used in the next step without further purification.
1H NMR (400 MHz, CDCl
3) δ 2.54 (s, 3H), 8.45 (s, 1H). GCMS: [M]+ m/z: calcd 285.88; found 286.0. Step 2: The synthesis of tert-butyl N-[3-(2-chloro-4-methylsulfanyl-pyrimidin-5- yl)allyl]carbamate [1213] A mixture of tert-butyl N-[(E)-3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2- yl)allyl]carbamate (1.63 g, 5.76 mmol), 2-chloro-5-iodo-4-methylsulfanyl-pyrimidine (1.50 g, 5.24 mmol) and K
2CO
3 (1.81 g, 13.1 mmol) were mixed in a degassed mixture of water (8 mL) and dioxane (60 mL). Bis(diphenylphosphino)ferrocene]dichloropalladium(II)-DCM (428 mg, 524 μmol) was added to the reaction mixture. The mixture was heated at 90 °C for 15 hr. under argon atmosphere. The reaction mixture was cooled to room temperature and concentrated under reduced pressure. The residue was diluted with water (50 mL) and extracted with EtOAc (5 × 50 mL). The combined organic layers were dried over anhydrous Na
2SO
4 and concentrated under reduced pressure. The residue was subjected to flash column chromatography (SiO
2, gradient Hexanes – EtOAc) to afford tert-butyl N-[3-(2-chloro-4- methylsulfanyl-pyrimidin-5-yl)allyl]carbamate (1.17 g, 3.70 mmol, 70.8% yield) as a white solid.
1H NMR (400 MHz, CDCl
3) δ 1.46 (s, 9H), 2.58 (s, 3H), 3.88 – 3.99 (m, 2H), 4.77 (br., 1H), 6.18 – 6.29 (m, 1H), 6.39 – 6.47 (m, 1H), 8.22 (s, 1H). MS (ESI): [M+H]+ m/z: calcd 316.09; found 316.0. Step 3: The synthesis of tert-butyl N-[3-[2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4- methylsulfanyl-pyrimidin-5-yl]allyl]carbamate [1214] (4-cyclopropyl-6-methoxy-pyrimidin-5-yl)boronic acid I-5 (1.14 g, 5.89 mmol), tert- butyl N-[3-(2-chloro-4-methylsulfanyl-pyrimidin-5-yl)allyl]carbamate (1.20 g, 3.80 mmol) and tripotassium phosphate (2.02 g, 9.50 mmol) were mixed in a degassed mixture of water (6 mL) and dioxane (50 mL). RuPhos Pd G4 (323 mg, 380 μmol) was added to the reaction mixture. The mixture was stirred at 90 °C for 15 hr. The mixture was cooled to room temperature and concentrated under reduced pressure. The residue was diluted with water (50 mL) and extracted with EtOAc (5 × 50 mL). The combined organic layers were dried over anhydrous Na
2SO
4 and concentrated under reduced pressure. The residue was The residue was subjected to flash column chromatography (SiO
2, gradient Hexanes – EtOAc) to afford tert-butyl N-[3-[2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-methylsulfanyl-pyrimidin-5- yl]allyl]carbamate (1.20 g, 2.79 mmol, 73.5 % yield) as a light-yellow solid.
1H NMR (600 MHz, DMSO-d6) δ 0.88 – 0.93 (m, 2H), 1.02 – 1.07 (m, 2H), 1.40 (s, 9H), 1.72 – 1.80 (m, 1H), 2.49 (s, 3H), 3.72 – 3.81 (m, 2H), 3.85 (s, 3H), 6.40 – 6.52 (m, 2H), 7.20 (br., 1H), 8.66 (s, 1H), 8.71 (s, 1H). MS (ESI): [M+H]+ m/z: calcd 430.22; found 430.2. Step 4: The synthesis of tert-butyl N-[3-[2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4- methylsulfanyl-pyrimidin-5-yl]propyl]carbamate [1215] A solution of tert-butyl N-[3-[2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4- methylsulfanyl-pyrimidin-5-yl]allyl]carbamate (1.15 g, 2.68 mmol) in EtOAc (75 mL) was subjected to hydrogenation at 1 bar at room temperature for 60 hr with 5% palladium on carbon (150 mg) as a catalyst. The reaction mixture was filtered and concentrated under reduced pressure to afford tert-butyl N-[3-[2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4- methylsulfanyl-pyrimidin-5-yl]propyl]carbamate (1.10 g, 2.55 mmol, 95.2 % yield) as an off- white foam which was used in the next step without further purification.
1H NMR (400 MHz, CDCl
3) δ 0.86 – 0.98 (m, 2H), 1.18 – 1.26 (m, 2H), 1.45 (s, 9H), 1.74 – 1.84 (m, 1H), 1.86 – 1.97 (m, 2H), 2.55 (s, 3H), 2.65 (t, 2H), 3.14 – 3.31 (m, 2H), 3.93 (s, 3H), 4.61 – 4.75 (m, 1H), 8.33 (s, 1H), 8.64 (s, 1H). MS (ESI): [M+H]+ m/z: calcd 432.21; found 432.2. Step 5: The synthesis of tert-butyl N-[3-[2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4- methylsulfonyl-pyrimidin-5-yl]propyl]carbamate [1216] mCPBA (494 mg, 2.43 mmol, 85% purity) was added to a stirred solution of tert- butyl N-[3-[2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-methylsulfanyl-pyrimidin-5- yl]propyl]carbamate (500 mg, 1.16 mmol) in DCM (100 mL). The resulting mixture was stirred at room temperature for 12 hr. The mixture was washed with saturated aqueous solution of NaHCO
3 (3 × 30 mL), dried over anhydrous Na
2SO
4 and concentrated under reduced pressure to afford tert-butyl N-[3-[2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4- methylsulfonyl-pyrimidin-5-yl]propyl]carbamate (525 mg, crude) as a yellow gum which was used in the next step without further purification. MS (ESI): [M+H]+ m/z: calcd 464.20; found 464.2. Step 6: The synthesis of tert-butyl N-[3-[2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[4- [1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidin-5- yl]propyl]carbamate [1217] NaH (46.7 mg, 1.17 mmol, 60% dispersion in mineral oil) was added to a mixture of tert-butyl N-[3-[2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-methylsulfonyl-pyrimidin-5- yl]propyl]carbamate (525 mg, crude) and [4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methanol I-2b (224 mg, 875 μmol) in DMF (25 mL). The resulting mixture was stirred at room temperature for 13 hr. The reaction mixture was poured into water (30 mL). The resulting precipitate was filtered off. The precipitate was redissolved in ACN (8 mL). To the obtained solution SiliaMetS® Dimercaptotriazine (200 mg) was added and the resulting mixture was stirred for 10 hr. The mixture was filtered. The filtrate was subjected to HPLC (2 -10 min, 30-60 % water – ACN, flow: 30 mL/min, column: SunFire C18100 ×19 mm, 5 µm) to afford tert-butyl N-[3-[2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidin-5-yl]propyl]carbamate (220 mg, 344 μmol, 29.7% total yield from tert-butyl N-[3-[2-(4-cyclopropyl-6-methoxy-pyrimidin-5- yl)-4-methylsulfanyl-pyrimidin-5-yl]propyl]carbamate) as a light-yellow solid.
1H NMR (600 MHz, DMSO-d6) δ 0.84 – 0.90 (m, 2H), 1.01 – 1.06 (m, 2H), 1.37 (s, 9H), 1.65 – 1.71 (m, 1H), 1.73 – 1.80 (m, 2H), 2.60 – 2.66 (m, 2H), 2.96 – 3.03 (m, 2H), 3.79 (s, 3H), 3.84 (s, 3H), 5.53 (s, 2H), 6.89 – 6.95 (m, 1H), 7.58 (d, 2H), 7.74 (d, 2H), 7.94 (s, 1H), 8.57 (s, 1H), 8.66 (s, 1H). MS (ESI): [M+H]+ m/z: calcd 640.29; found 640.4. Example 74 (Compound 139)
Compound 139 Step 1: The synthesis of 5-bromo-2-chloro-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidine [1218] 5-Bromo-2,4-dichloro-pyrimidine (300 mg, 1.32 mmol), [4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methanol I-2b (337 mg, 1.32 mmol) and Cs
2CO
3 (858 mg, 2.63 mmol) were mixed in ACN (20 mL). The resulting mixture was stirred at 65 °C for 12 hr. The mixture was cooled to room temperature and filtered. The filtrate concentrated under reduced pressure to afford 5-bromo-2-chloro-4-[[4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidine (400 mg, 894 μmol, 67.9% yield) as an yellow oil which was used in the next steps without further purification. MS (ESI): [M+H]+ m/z: calcd 448.98; found 449.0. Step 2: The synthesis of 2-chloro-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]-5-(1H-pyrazol-5-yl)pyrimidine [1219] 5-Bromo-2-chloro-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidine (400 mg, 894 μmol), 1-tetrahydropyran-2-yl-5-(4,4,5,5- tetramethyl-1,3,2-dioxaborolan-2-yl)pyrazole (268 mg, 965 μmol), potassium phosphate tribasic (427 mg, 2.01 mmol) and bis(diphenylphosphino)ferrocene]dichloropalladium(II)- DCM (132 mg, 161 μmol) were mixed in degassed mixture of dioxane (9 mL) and water (1 mL). The reaction mixture was stirred at 90 °C for 12 hr. To the reaction mixture was added 1-tetrahydropyran-2-yl-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyrazole (268. mg, 965 μmol) and the mixture was stirred at 90 °C for 12 hr. The reaction mixture was cooled to room temperature, diluted with water (5 mL) and extracted with EtOAc (2 × 5 mL). The organic layer was dried over anhydrous Na
2SO
4 and concentrated under reduced pressure. The residue was subjected to HPLC (0.5-6.5 min, 43% water – ACN, flow: 30 mL/min, column: SunFire C18100 ×19 mm, 5 µm) to afford 2-chloro-4-[[4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]-5-(1H-pyrazol-5-yl)pyrimidine (207 mg, 476 μmol, 59.1% yield) as an yellow solid. MS (ESI): [M+H]+ m/z: calcd 435.11; found 435.0. Step 3: The synthesis of 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]-5-(1H-pyrazol-5-yl)pyrimidine [1220] 2-Chloro-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]-5-(1H- pyrazol-5-yl)pyrimidine (59.1 mg, 136 μmol), (4-cyclopropyl-6-methoxy-pyrimidin-5- yl)boronic acid I-5 (29.0 mg, 149 μmol), potassium phosphate tribasic (72.1 mg, 340 μmol) and XPhos Pd G3 (5.74 mg, 6.79 μmol) were mixed in a degassed mixture of dioxane (3.6 mL) and water (0.4 mL). The reaction mixture was stirred at 33 °C under argon atmosphere for 12 hr. To the reaction mixture an additional portion of (4-cyclopropyl-6-methoxy- pyrimidin-5-yl)boronic acid I-5 (29.0 mg, 149 μmol) was added and the mixture was stirred at 33 °C for 12 hr. The mixture was concentrated under reduced pressure. The residue was subjected to HPLC (0.5-6.5 min, 25-80% water – ACN, +0.1% vol. of 25% aq. NH
3, flow: 30 mL/min, column: SunFire C18100 ×19 mm, 5 µm) to afford a residue which was re-purified by HPLC (0.5-6.5 min, 52% water – MeOH, +0.1% vol. of 25% aq. NH
3, flow: 30 mL/min, column: SunFire C18, 100 ×19 mm, 5 µm) to afford 2-(4-cyclopropyl-6-methoxy-pyrimidin- 5-yl)-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]-5-(1H-pyrazol-5- yl)pyrimidine (3.50 mg, 6.38 μmol, 4.7% yield) as a beige solid.
1H NMR (400 MHz, DMSO-d6) δ 0.85 – 0.91 (m, 2H), 1.02 – 1.06 (m, 2H), 1.73 – 1.81 (m, 1H), 3.78 (s, 3H), 3.85 (s, 3H), 5.61 (s, 2H), 6.80 (s, 1H), 7.63 (d, 2H), 7.74 (d, 2H), 7.86 (s, 1H), 7.92 (s, 1H), 8.67 (s, 1H), 9.19 (s, 1H), 13.21 (s, 1H). MS (ESI): [M+H]+ m/z: calcd 549.22; found 549.2. Example 75 (Compound 13)
Step 1: Synthesis of 2-[4-bromo-3-(trifluoromethyl)phenyl]-4-(trifluoromethyl)-1H-imidazole [1221] Sodium acetate (14.9 g, 182 mmol) was dissolved in H
2O (300 mL) then 3,3- dibromo-1,1,1-trifluoro-propan-2-one (23.5 g, 86.9 mmol) was added and the resulting mixture was stirred at 95 °C for 1hr. The resulting mixture was cooled to room temperature and poured into a solution of 4-bromo-3-(trifluoromethyl)benzaldehyde (20.0 g, 79.1 mmol) and aqueous NH
4OH (100 mL, 25% wt.) in MeOH (1000 mL). The obtained mixture was stirred at room temperature for 12 hr. The mixture was concentrated under reduced pressure. The residue was distributed between EtOAc (300 mL) and water (200 mL). The organic layer was separated, dried over anhydrous Na
2SO
4, filtered and concentrated under reduced pressure to afford 2-[4-bromo-3-(trifluoromethyl)phenyl]-4-(trifluoromethyl)-1H-imidazole (21.0 g, 58.5 mmol, 74% yield) as a yellow solid which was used in the next steps without further purification.
1H NMR (500 MHz, DMSO-d6) δ 7.96 – 8.01 (m, 2H), 8.13 (d, 2H), 8.38 (s, 1H). MS (ESI): [M+H]+ m/z: calcd 358.96; found 359.0. Step 2: Synthesis of methyl 2-(trifluoromethyl)-4-[4-(trifluoromethyl)-1H-imidazol-2- yl]benzoate [1222] A mixture of 2-[4-bromo-3-(trifluoromethyl)phenyl]-4-(trifluoromethyl)-1H- imidazole (15.0 g, 41.8 mmol), TEA (8.45 g, 83.6 mmol) and bis(diphenylphosphino)ferrocene]dichloropalladium(II)-DCM (1.73 g, 2.09 mmol) in MeOH (400 mL) was placed in high pressure stainless steel vessel. The reaction mixture was stirred under CO atmosphere (20 bar) at 120 °C for 12 hr. The reaction mixture was cooled to room temperature and concentrated under reduced pressure to 1/3 volume. The residue was diluted EtOAc (200 mL) and washed water (50 mL) and brine (50 mL). The organic layer was filtered through short pad of silicagel. The filtrate was concentrated under reduced pressure to afford methyl 2-(trifluoromethyl)-4-[4-(trifluoromethyl)-1H-imidazol-2-yl]benzoate (11.0 g, 32.5 mmol, 77.9% yield) as a red oil which was used in the next steps without further purification.
1H NMR (500 MHz, DMSO-d6) δ 3.88 (s, 3H), 7.98 (d, 1H), 8.06 (s, 1H), 8.34 (d, 1H), 8.43 (s, 1H). MS (ESI): [M+H]+ m/z: calcd 339.06; found 339.0. Step 3: Synthesis of methyl 4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]-2- (trifluoromethyl)benzoate [1223] Methyl iodide (923 mg, 6.50 mmol) was added dropwise to a mixture of methyl 2- (trifluoromethyl)-4-[4-(trifluoromethyl)-1H-imidazol-2-yl]benzoate (2.00 g, 5.91 mmol) and Cs
2CO
3 (2.31 g, 7.10 mmol) in DMF (10 mL). The resulting mixture was stirred 40 °C for 12 hr. The reaction mixture was cooled to room temperature and diluted with EtOAc (30 mL) and water (20 mL). The organic layer was separated, washed with brine (2 × 10 mL) and concentrated under reduced pressure to afford methyl 4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]-2-(trifluoromethyl)benzoate (2.00 g, 5.68 mmol, 96.0% yield) as a yellow solid which was used in the next steps without further purification.
1H NMR (400 MHz, DMSO-d6) δ 3.87 (s, 3H), 3.91 (s, 3H), 8.00 (d, 1H), 8.06 (s, 1H), 8.16 – 8.22 (m, 2H). MS (ESI): [M+H]+ m/z: calcd 353.07; found 353.0. Step 4: Synthesis of [4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]-2- (trifluoromethyl)phenyl]methanol and [2-methyl-4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methanol [1224] A solution of methyl 4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]-2- (trifluoromethyl)benzoate (2.00 g, 5.68 mmol) in THF (5 mL) was added dropwise to a vigorously stirred suspension of lithium aluminium hydride (578 mg, 17.0 mmol) in THF (20 mL) under argon atmosphere at 5 °C. The reaction mixture was stirred at ambient temperature for 8 hr. The mixture was cooled to 0 °C and quenched by dropwise addition of water (5 mL). The resulting mixture was stirred for 15 minutes and filtered. The filter cake was washed with THF (10 mL). The combined filtrate was dried over anhydrous Na
2SO
4 and concentrated under reduced pressure. The residue was subjected to flash chromatography (SiO
2, gradient hexane - EtOAc) to afford [4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]-2- (trifluoromethyl)phenyl]methanol (0.140 g, 432 μmol, 7.60% yield) and [4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]-2-(trifluoromethyl)phenyl]methanol (0.250 g, 925 μmol, 16.3% yield) as light-yellow solids. [4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]-2-(trifluoromethyl)phenyl]methanol:
1H NMR (400 MHz, CDCl
3) δ 3.41 (br., 1H), 3.73 (s, 3H), 4.85 (s, 2H), 7.33 (s, 1H), 7.66 (d, 1H), 7.76 (d, 1H), 7.79 (s, 1H). MS (ESI): [M+H]+ m/z: calcd 325.08; found 325.2. [2-methyl-4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methanol:
1H NMR (400 MHz, DMSO-d6) δ 2.30 (s, 3H), 3.76 (s, 3H), 4.52 – 4.56 (m, 2H), 5.14 (t, 1H), 7.46 – 7.51 (m, 3H), 7.88 (s, 1H). MS (ESI): [M+H]+ m/z: calcd 271.11; found 271.2. Step 5: The synthesis of 4-cyclopropyl-6-methoxy-5-[4-[[2-methyl-4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidin-2-yl]pyrimidine [1225] NaH (16.3 mg, 407 μmol, 60% dispersion in mineral oil) was added to a solution [2- methyl-4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methanol (0.120 g, 444 μmol) in THF (5 mL) at 0°C. The reaction mixture was stirred at 0 °C for 1 hr. The 4-cyclopropyl- 6-methoxy-5-(4-methylsulfonylpyrimidin-2-yl)pyrimidine I-8 (136 mg, 444 μmol) was added to the reaction mixture. The resulting mixture was stirred at room temperature for 6 hr. The reaction mixture was diluted with water (10 mL) and extracted with EtOAc (2×10 mL). The combined organic layers were washed with brine (10 mL) and concentrated under reduced pressure. The residue was subjected to HPLC (0.5-6.5 min., 45-60% water – ACN, flow: 30 mL/min, column: SunFire C18100 ×19 mm, 5 µm) to afford 4-cyclopropyl-6-methoxy-5-[4- [[2-methyl-4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidin-2- yl]pyrimidine (0.110 g, 222 μmol, 49.9% yield) as a light-yellow solid.
1H NMR (600 MHz, DMSO-d6) δ 0.85 – 0.90 (m, 2H), 1.02 – 1.05 (m, 2H), 1.67 – 1.72 (m, 1H), 2.38 (s, 3H), 3.77 (s, 3H) 3.84 (s, 3H), 5.48 (s, 2H), 7.05 (d, 1H), 7.49 – 7.54 (m, 2H), 7.58 (s, 1H), 7.91 (s, 1H), 8.66 (s, 1H), 8.69 (d, 1H). MS (ESI): [M+H]+ m/z: calcd 497.22; found 497.4. Example 76 (Compound 149)

Compound 149 Step 1: The synthesis of methyl 3-fluoro-4-[4-(trifluoromethyl)-1H-imidazol-2-yl]benzoate [1226] 3,3-dibromo-1,1,1-trifluoro-propan-2-one (8.89 g, 32.9 mmol, 4.49 mL) and sodium acetate (5.18 g, 63.1 mmol) were mixed in water (76 mL). The reaction mixture was stirred at 100°C for 45 min. The reaction mixture was cooled to room temperature. A solution of methyl 3-fluoro-4-formyl-benzoate (5.00 g, 27.5 mmol) and an aqueous ammonium hydroxide (9.62 g, 275 mmol, 10.7 mL, 28% wt.) in MeOH (286 mL) was added to the reaction mixture. The resulting mixture was stirred at room temperature for 12 hr. The reaction mixture was concentrated to half volume and extracted with EtOAc (2×80 mL). The combined organic layers were washed with brine (50 mL), dried over anhydrous Na
2SO
4 and filtered. The filtrate was concentrated under reduced pressure to afford methyl 3-fluoro-4-[4- (trifluoromethyl)-1H-imidazol-2-yl]benzoate (5.00 g, 17.4 mmol, 63.2% yield) as a brown solid which was used in the next steps without further purification.
1H NMR (500 MHz, DMSO-d6) δ 3.88 (s, 3H), 7.82 – 7.91 (m, 2H), 8.11 (s, 1H), 8.17 (t, 1H), 13.08 (br., 1H). MS (ESI): [M+H]+ m/z: calcd 289.06; found 289.2. Step 2: The synthesis of methyl 3-fluoro-4-[1-isopropyl-4-(trifluoromethyl)imidazol-2- yl]benzoate [1227] NMP (30 mL), Cs
2CO
3 (8.82 g, 27.1 mmol), methyl 3-fluoro-4-[4-(trifluoromethyl)- 1H-imidazol-2-yl]benzoate (2.60 g, 9.02 mmol), 2-iodopropane (7.67 g, 45.1 mmol, 4.51 mL) were mixed in a sealed tube. The reaction mixture was stirred at 110°C for 48 hr. The reaction mixture was cooled to room temperature, diluted with water (100 mL) and extracted with EtOAc (2×20 mL). The combined organic layers were washed with water (50 mL) and brine (20 mL), dried over anhydrous Na
2SO
4 and filtered. The filtrate was concentrated under reduced pressure to afford methyl 3-fluoro-4-[1-isopropyl-4-(trifluoromethyl)imidazol-2- yl]benzoate (3.0 g, crude) as a gum which was used in the next steps without purification. MS (ESI): [M+H]+ m/z: calcd 331.11; found 331.2. Step 3: The synthesis of [3-fluoro-4-[1-isopropyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methanol [1228] A solution of methyl 3-fluoro-4-[1-isopropyl-4-(trifluoromethyl)imidazol-2- yl]benzoate (3.00 g, crude) in THF (50 mL) was added dropwise to a vigorously stirred suspension of LAH (629 mg, 18.5 mmol) in THF (100 mL). The reaction mixture was stirred at room temperature for 3 hr. The reaction mixture was quenched by addition of an aqueous NaOH (30 mL, 10% wt.) followed by granular NaOH (10.0 g). The resulting mixture was filtered. The filtrate was concentrated under reduced pressure to give 2.00 g of a crude product.1.00 of the crude was subjected to HPLC (0.5-6.5 min, 27-43% water - ACN; flow: 30 mL/min, column: Waters SunFire C18, 100 x 19 mm, 5 µm) to afford [3-fluoro-4-[1- isopropyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methanol (300 mg, 992 µmol, 22.0% yield from methyl 3-fluoro-4-[4-(trifluoromethyl)-1H-imidazol-2-yl]benzoate) as a yellow oil. MS (ESI): [M+H]+ m/z: calcd 303.14; found 303.2. Step 4: The synthesis of 4-cyclopropyl-5-[4-[[3-fluoro-4-[1-isopropyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidin-2-yl]-6-methoxy-pyrimidine [1229] NaH (17.5 mg, 437 μmol, 60% dispersion in mineral oil) was added to a solution of [3-fluoro-4-[1-isopropyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methanol (120 mg, 397 μmol) in DMF (2 mL). The reaction mixture was stirred at room temperature for 30 min.4- cyclopropyl-6-methoxy-5-(4-methylsulfonylpyrimidin-2-yl)pyrimidine I-8 (122 mg, 397 μmol) was added to the reaction mixture. The reaction mixture was stirred at room temperature for 12 hr. The reaction mixture was diluted with water (5.0 mL) and extracted with EtOAc (5.0 mL). The organic layer was separated and concentrated under reduced pressure. The residue was subjected to HPLC (0.5 - 6.5 min, 50-65% water - ACN; flow: 4 mL/min, column: Waters SunFire C18, 100 x 19 mm, 5 µm) to afford 4-cyclopropyl-5-[4-[[3- fluoro-4-[1-isopropyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidin-2-yl]-6- methoxy-pyrimidine (51.0 mg, 96.5 μmol, 24.3% yield) as a yellow gum.
1H NMR (600 MHz, DMSO-d6) δ 0.83 – 0.89 (m, 2H), 1.00 – 1.05 (m, 2H), 1.35 (d, 6H), 1.64 – 1.70 (m, 1H), 3.83 (s, 3H), 4.09 – 4.16 (m, 1H), 5.51 (s, 2H), 7.10 (d, 1H), 7.44 (d, 1H), 7.50 (d, 1H), 7.56 (t, 1H), 8.24 (s, 1H), 8.66 (s, 1H), 8.72 (s, 1H). MS (ESI): [M+H]+ m/z: calcd 529.23; found 529.4. Example 77 (Compound 97)
Step 1: The synthesis of methyl 3-fluoro-4-[4-(trifluoromethyl)-1H-imidazol-2-yl]benzoate [1230] A solution of 3,3-dibromo-1,1,1-trifluoro-propan-2-one (8.89 g, 32.9 mmol, 4.49 mL) and sodium acetate (5.18 g, 63.1 mmol) in water (76 mL) was stirred at 100 °C for 45 min. The reaction mixture was cooled to room temperature. To the reaction mixture was added a solution of methyl 3-fluoro-4-formyl-benzoate (5.00 g, 27.5 mmol) followed by a solution of aqueous ammonium hydroxide (10.7 mL, 28% wt.) in MeOH (286 mL). The reaction mixture was stirred at room temperature for 12 hr. The mixture was concentrated under reduced pressure to a half of a volume. The residue was extracted with EtOAc (2×80 mL). The combined organic layers were washed with brine (50 mL), dried over anhydrous Na
2SO
4 and concentrated under reduce pressure to afford methyl 3-fluoro-4-[4-(trifluoromethyl)-1H- imidazol-2-yl]benzoate (5.00 g, 17.4 mmol, 63.2% yield) as a brown solid which was used in the next step without further purification.
1H NMR (500 MHz, DMSO-d6) δ 3.88 (s, 3H), 7.81 – 7.95 (m, 2H), 7.96 (s, 1H), 8.14 (t, 1H), 13.04 (br, 1H). MS (ESI): [M+H]+ m/z: calcd 289.06; found 289.2. Step 2: The synthesis of methyl 3-fluoro-4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]benzoate [1231] Methyl 3-fluoro-4-[4-(trifluoromethyl)-1H-imidazol-2-yl]benzoate (2.40 g, 8.33 mmol), iodomethane (2.36 g, 16.7 mmol, 1.04 mL) and Cs2CO3 (2.71 g, 8.33 mmol) were mixed in acetonitrile (49.0 mL). The reaction mixture was stirred at 40 °C for 12 hr. The reaction mixture was cooled to room temperature. The reaction mixture was diluted with water (50 mL) and extracted with EtOAc (2×20 mL). The combined organic layers were washed with water (50 mL) and brine (20 mL), dried over anhydrous Na
2SO
4 and concentrated under reduce pressure to afford methyl 3-fluoro-4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]benzoate (2.00 g, 6.62 mmol, 79.5% yield) as a red gum which was used in the next step without further purification. MS (ESI): [M+H]+ m/z: calcd 303.08; found 303.2. Step 3: The synthesis of [3-fluoro-4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methanol [1232] А solution of methyl 3-fluoro-4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]benzoate (2.00 g, 6.62 mmol) in THF (50.0 mL) was added dropwise to a vigorously stirred suspension of LAH (561 mg, 16.5 mmol) in THF (100 mL) at room temperature. The reaction mixture was stirred at room temperature for 3 hr. The mixture was quenched by dropwise addition of aqueous NaOH (30 mL, 28% wt.). To the obtained mixture solid granulated NaOH (10.0 g, 250 mmol) was added. The mixture was stirred for 10 min. and then solids were filtered out. The filtrate was concentrated under reduced pressure to afford [3-fluoro-4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methanol (1.50 g, 5.47 mmol, 82.7% yield) as a red oil which was used in the next steps without further purification.
1H NMR (500 MHz, DMSO- d6) δ 3.57 (s, 3H), 4.58 (d, 2H), 5.45 (t, 1H), 7.25 – 7.33 (m, 2H), 7.52 (t, 1H), 7.97 (s, 1H). MS (ESI): [M+H]+ m/z: calcd 275.1; found 275.2. Step 4: The synthesis of 4-cyclopropyl-5-[4-[[3-fluoro-4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidin-2-yl]-6-methoxy-pyrimidine [1233] NaH (19.3 mg, 303 μmol, 60% dispersion in mineral oil) was added to a solution of [3-fluoro-4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methanol (120 mg, 276 μmol) in DMF (2.00 mL). The reaction mixture was stirred at room temperature for 15 min.4- cyclopropyl-6-methoxy-5-(4-methylsulfonylpyrimidin-2-yl)pyrimidine I-8 (84.5 mg, 276 μmol) was added to the reaction mixture. The reaction mixture was stirred at room temperature for 12 hr. The reaction mixture was diluted with water (5 mL) and extracted with EtOAc (5 mL). The organic layer was separated and concentrated under reduce pressure. The residue was subjected to HPLC (0.5 - 6.5 min, 42 - 57% water - ACN; flow: 30 mL/min, column: Waters SunFire C18, 100 x 19 mm, 5 µm) to afford 4-cyclopropyl-5-[4-[[3-fluoro-4- [1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidin-2-yl]-6-methoxy- pyrimidine (110 mg, 220 μmol, 80.0% yield) as a yellow gum which solidified upon freeze- drying.
1H NMR (600 MHz, DMSO-d6) δ 0.85 – 0.90 (m, 2H), 1.01 – 1.05 (m, 2H), 1.64 – 1.70 (m, 1H), 3.59 (s, 3H), 3.83 (s, 3H), 5.50 (s, 2H), 7.10 (d, 1H), 7.44 (d, 1H), 7.50 (d, 1H), 7.60 (t, 1H), 7.99 (s, 1H), 8.66 (s, 1H), 8.72 (d, 1H). MS (ESI): [M+H]+ m/z: calcd 501.19; found 501.0. Example 78 (Compound 159)
Step 1: The synthesis of 3,5-difluoro-4-hydrazino-benzonitrile [1234] A solution of 3,4,5-trifluorobenzonitrile (10.4 g, 66.2 mmol) and hydrazine monohydrate (4.64 g, 92.7 mmol, 4.52 mL) in dioxane (150 mL) was stirred at 60 °C for 64 hr. The reaction mixture was cooled to room temperature and concentrated under reduced pressure. The residue was triturated with cold MTBE (75 mL). The solid precipitate formed was filtered off and dried on air to afford 3,5-difluoro-4-hydrazino-benzonitrile (7.50 g, 44.3 mmol, 67.0% yield) as a light-yellow solid which was used in the next steps without further purification.
1H NMR (400 MHz, DMSO-d6) δ 4.52 (s, 2H), 7.29 (s, 1H), 7.43 – 7.54 (m, 2H). GCMS: [M]+ m/z: calcd 169.05; found 169.0. Step 2: The synthesis of 3,5-difluoro-4-[5-methyl-3-(trifluoromethyl)pyrazol-1-yl]benzonitrile A mixture of 3,5-difluoro-4-hydrazino-benzonitrile (5.00 g, 29.6 mmol) and 1,1,1- trifluoropentane-2,4-dione (4.78 g, 31.0 mmol, 3.75 mL) in AcOH (75 mL) was stirred at 110 °C for 16 hr. The reaction mixture was cooled to room temperature and concentrated under reduced pressure. The residue was subjected to FCC (SiO
2, gradient Hexanes - EtOAc) to afford 3,5-difluoro-4-[5-methyl-3-(trifluoromethyl)pyrazol-1-yl]benzonitrile (2.30 g, 8.01 mmol, 27.1% yield) as a light-yellow solid.
1H NMR (400 MHz, DMSO-d6) δ 2.22 (s, 3H), 6.89 (s, 1H), 8.19 (d, 2H). MS (ESI): [M+H]+ m/z: calcd 288.06; found 288.0. Step 3: The synthesis of 3-ethoxy-5-fluoro-4-(5-methyl-3-(trifluoromethyl)-1H-pyrazol-1- yl)benzoic acid [1235] A solution of sodium hydroxide (961 mg, 24.0 mmol) in water (20.0 mL) was added to a solution of 3,5-difluoro-4-[5-methyl-3-(trifluoromethyl)pyrazol-1-yl]benzonitrile (2.30 g, 8.01 mmol) in EtOH (20 mL). The resulting mixture was stirred at 70 °C for 30 hr. The reaction mixture was cooled to room temperature and concentrated under reduced pressure. The residue was diluted with water (125 mL) and acidified with an aqueous 2 N HCl to pH<3. The resulting precipitate was filtered off to afford 3-ethoxy-5-fluoro-4-(5-methyl-3- (trifluoromethyl)-1H-pyrazol-1-yl)benzoic acid in mixture with 3,5-difluoro-4-[5-methyl-3- (trifluoromethyl)pyrazol-1-yl]benzoic acid in a ratio 12% : 88% (2.00 g) as a yellow solid which was used in the next steps without further purification.
1H NMR (500 MHz, DMSO- d6) δ 1.20 (t, 3H), 2.13 (s, 3H), 4.20 (q, 2H), 6.75 (s, 1H), 7.51 (d, 1H), 7.57 (s, 1H). MS (ESI): [M+H]+ m/z: calcd 333.09; found 333.2. Step 4: The synthesis of (3-ethoxy-5-fluoro-4-(5-methyl-3-(trifluoromethyl)-1H-pyrazol-1- yl)phenyl)methanol [1236] Borane dimethyl sulfide complex (2.48 g, 32.7 mmol, 3.10 mL) was added dropwise to a solution of 3-ethoxy-5-fluoro-4-(5-methyl-3-(trifluoromethyl)-1H-pyrazol-1-yl)benzoic acid (2.00 g, сrude, mixture with 3,5-difluoro-4-(5-methyl-3-(trifluoromethyl)-1H-pyrazol-1- yl)benzoic acid) in THF (125 mL). The resulting mixture was stirred at room temperature for 48 hr. The reaction mixture was cooled to 0 °C. MeOH (100 mL) was added dropwise to the reaction mixture. The resulting mixture was stirred at 0 °C for 4 hr. The mixture was concentrated under reduced pressure. The residue was redissolved in MeOH (100 mL) and the mixture was concentrated under reduced pressure. The residue was dissolved in MTBE (100 mL). The organic layer was washed with aqueous NaOH (3×25 mL, 10% wt.), dried over anhydrous Na
2SO
4 and concentrated under reduced pressure to afford (3-ethoxy-5- fluoro-4-(5-methyl-3-(trifluoromethyl)-1H-pyrazol-1-yl)phenyl)methanol as in mixture with [3,5-difluoro-4-[5-methyl-3-(trifluoromethyl)pyrazol-1-yl]phenyl]methanol in ratio 12% : 88% (1.30 g, crude) as a white solid which was used in the next steps without further purification.
1H NMR (500 MHz, DMSO-d6) δ 1.18 (t, 3H), 2.09 (s, 3H), 4.11 (q, 2H), 4.59 (d, 2H), 5.50 (t, 1H), 6.70 (s, 1H), 6.98 (d, 1H), 7.05 (s, 1H). MS (ESI): [M+H]+ m/z: calcd 319.11; found 319.0. Step 5: The synthesis of 4'-cyclopropyl-4-((3-ethoxy-5-fluoro-4-(5-methyl-3-(trifluoromethyl)- 1H-pyrazol-1-yl)benzyl)oxy)-5,6'-dimethoxy-2,5'-bipyrimidine [1237] NaH (19.3 mg, 483 μmol, 60% dispersion in mineral oil) was added to a solution of (3-ethoxy-5-fluoro-4-(5-methyl-3-(trifluoromethyl)-1H-pyrazol-1-yl)phenyl)methanol (160 mg, crude, mixture with [3,5-difluoro-4-[5-methyl-3-(trifluoromethyl)pyrazol-1- yl]phenyl]methanol) and 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-5-methoxy-4- methylsulfonyl-pyrimidine I-6 (147 mg, 438 μmol) in DMF (10 mL). The reaction mixture was stirred at room temperature for 15 hr. The reaction mixture was diluted with water (25 mL). The resulting precipitate was filtered off and re-dissolved in ACN (20 mL) and DMF (3.0 mL). SiliaMetS® Dimercaptotriazine (50 mg) was added to the solution. The resulting mixture was stirred at room temperature for 10 hr. The mixture was filtered. The filtrate was subjected to HPLC (2-10 min, 10-50% water+FA (0.1% vol.) - ACN+FA (0.1% vol.); flow: 30 mL/min, column: Waters SunFire C18, 100 × 19 mm, 5 µm) and then re-purified with SFC (Chiralpak IC I (250 × 20 mm, 5 mkm); CO2:MeOH, 85%:15%; 50.0 mL/min) to afford 4'-cyclopropyl-4-((3-ethoxy-5-fluoro-4-(5-methyl-3-(trifluoromethyl)-1H-pyrazol-1- yl)benzyl)oxy)-5,6'-dimethoxy-2,5'-bipyrimidine (4.2 mg, 7.31 μmol, 0.7% yield from 3,5- difluoro-4-[5-methyl-3-(trifluoromethyl)pyrazol-1-yl]benzonitrile) as an off-white solid.
1H NMR (600 MHz, DMSO-d6) δ 0.86 – 0.90 (m, 2H), 1.01 – 1.05 (m, 2H), 1.17 (t, 3H), 1.67 – 1.73 (m, 1H), 2.11 (s, 3H), 3.84 (s, 3H), 3.97 (s, 3H), 4.05 – 4.13 (m, 2H), 5.49 (s, 2H), 6.73 (s, 1H), 7.14 (d, 1H), 7.25 (s, 1H), 8.46 (s, 1H), 8.65 (s, 1H). MS (ESI): [M+H]+ m/z: calcd 575.23; found 575.0. Example 79 (Compound 173)

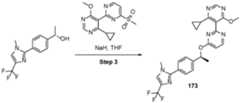
Step 1: The synthesis of (1S)-1-[4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2- yl)phenyl]ethanol [1238] (1S)-1-(4-bromophenyl)ethanol (1.00 g, 4.97 mmol), 4,4,5,5-tetramethyl-2-(4,4,5,5- tetramethyl-1,3,2-dioxaborolan-2-yl)-1,3,2-dioxaborolane (1.39 g, 5.47 mmol), potassium acetate (1.22 g, 12.4 mmol) and bis(diphenylphosphino)ferrocene]dichloropalladium(II)- DCM (75.0 mg, 103 μmol) were mixed in degassed dioxane (80 mL) under argon atmosphere. The reaction mixture was stirred at 85 °C for 16 hr. The reaction mixture was cooled to room temperature, then solids were filtered out. The filtrate was used directly in the next step. MS (ESI): [M-OH]+ m/z: calcd 231.16; found 231.2. Step 2: The synthesis of (1S)-1-[4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]ethanol [1239] 2-bromo-1-methyl-4-(trifluoromethyl)imidazole (430 mg, 1.88 mmol), Cs
2CO
3 (1.84 g, 5.63 mmol), bis(diphenylphosphino)ferrocene]dichloropalladium(II)-DCM (300 mg, 18.8 μmol) and degassed water (10 mL) were added to a solution of (1S)-1-[4-(4,4,5,5- tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl]ethanol (466 mg, 1.88 mmol) in dioxane (80 mL) obtained in the previous step under argon atmosphere. The reaction mixture was stirred at 85 °C for 24 hr. The reaction mixture was cooled to room temperature and concentrated under reduced pressure. The residue was diluted with EtOAc (200 mL) and washed with water (50 mL). The organic layer was separated, washed with brine (2×50 mL), dried over anhydrous Na
2SO
4 and concentrated under reduced pressure. The residue was subjected to flash column chromatography (SiO
2, gradient chloroform - MTBE) to afford (1S)-1-[4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]ethanol (81.8 mg, 303 μmol, 16.1% yield) as a white solid. MS (ESI): [M+H]+ m/z: calcd 271.13; found 271.2. Step 3: The synthesis of 4-cyclopropyl-6-methoxy-5-[4-[(1S)-1-[4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]ethoxy]pyrimidin-2-yl]pyrimidine [1240] NaH (13.2 mg, 333 μmol, 60% dispersion in mineral oil), (1S)-1-[4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]ethanol (81.8 mg, 303 μmol) and 4-cyclopropyl-6- methoxy-5-(4-methylsulfonylpyrimidin-2-yl)pyrimidine I-8 (92.7 mg, 303 μmol) were mixed in THF (5 mL) at 0 °C. The resulting mixture was stirred at 50 °C for 12 hr. The reaction mixture was cooled to room temperature and concentrated under reduced pressure. The residue was subjected to HPLC (0-5 min., 30-65% water – ACN, +0.1% vol. of 25% aq. NH
3, flow: 30 mL/min, column: XBridge BEH C18100×19 mm, 5 µm) to afford 4-cyclopropyl-6- methoxy-5-[4-[(1S)-1-[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]ethoxy]pyrimidin-2-yl]pyrimidine (29.0 mg, 58.4 μmol, 19.3% yield) as a yellow solid.
1H NMR (600 MHz, DMSO-d6) δ 0.72 – 0.78 (m, 1H), 0.80 – 0.86 (m, 1H), 0.95 – 1.03 (m, 2H), 1.53 – 1.59 (m, 1H), 1.64 (d, 3H), 3.77 (s, 3H), 3.79 (s, 3H), 6.29 (q, 1H), 7.04 (d, 1H), 7.56 (d, 2H), 7.71 (d, 2H), 7.93 (s, 1H), 8.65 (s, 1H), 8.67 (d, 1H). MS (ESI): [M+H]+ m/z: calcd 497.22; found 497.2. Example 80 (Compound 84)

The synthesis of 5-[4-[[3-chloro-4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidin-2-yl]-4-cyclopropyl-6-methoxy-pyrimidine The synthesis of the starting [3-chloro-4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methanol is described in Example 49 (Compound 75). [1241] NaH (31.6 mg, 826 μmol, 60% dispersion in mineral oil) was suspended in THF (5.0 mL) at 0°C. [3-chloro-4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methanol (200 mg, 688 μmol) was added to the suspension at the same temperature. The resulting mixture was stirred at 0°C for 1 hr.4-cyclopropyl-6-methoxy-5-(4-methylsulfonylpyrimidin-2- yl)pyrimidine I-8 (211 mg, 688 μmol) was added to the mixture. The reaction mixture was stirred at 40 °C for 12 hr. The reaction mixture was cooled to room temperature, quenched by addition of water (10 mL) and extracted with MTBE (20 mL). The organic layer was washed with brine (10 mL), dried over anhydrous Na
2SO
4 and concentrated under reduced pressure. The residue was subjected to HPLC (0-5 min., 45-70% water+FA (0.1% vol.) - ACN+FA (0.1% vol.); flow: 30 mL/min, column: Chromatorex 18 SMB100-5T 100×19 mm, 5 µm) to afford 5-[4-[[3-chloro-4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidin-2-yl]-4-cyclopropyl-6-methoxy-pyrimidine (45.7 mg, 88.4 μmol, 12.9% yield) as a white solid.
1H NMR (600 MHz, DMSO-d6) δ 0.99 – 0.92 (m, 2H), 1.03 – 1.06 (m, 2H), 1.67 – 1.72 (m, 1H), 3.52 (s, 3H), 3.85 (s, 3H), 5.52 (s, 2H), 7.12 (d, 1H), 7.55 – 7.60 (m, 2H), 7.75 (s, 1H), 8.00 (s, 1H), 8.68 (s, 1H), 8.73 (d, 1H). MS (ESI): [M+H]+ m/z: calcd 517.16; found 517.2. Example 81 (Compound 91)
Step 1: The synthesis of 2-chloro-5-cyclopentyl-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol- 2-yl]phenyl]methoxy]pyrimidine [1242] Potassium tert-butoxide (71.5 mg, 637 μmol) was added portion wise to a stirred mixture of 2,4-dichloro-5-cyclopentyl-pyrimidine (115 mg, 531 μmol) and [4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methanol I-2b(136 mg, 531 μmol) in toluene (10 mL). The reaction mixture was stirred at room temperature for 16 hr. The reaction mixture was diluted with water (30 mL) and extracted with MTBE (20 mL). The organic layer was washed with brine (30 mL), dried over anhydrous Na
2SO
4 and concentrated under reduced pressure to afford 2-chloro-5-cyclopentyl-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidine (240 mg, crude) as a light-yellow gum which was used in the next steps without further purification. MS (ESI): [M+H]+ m/z: calcd 437.14; found 437.2. Step 2: The synthesis of 5-cyclopentyl-2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[4-[1- methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidine [1243] (4-cyclopropyl-6-methoxy-pyrimidin-5-yl)boronic acid I-5 (213 mg, 1.10 mmol), 2- chloro-5-cyclopentyl-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidine (240 mg, crude), potassium phosphate tribasic (117 mg, 549 μmol) and XPhos Pd G3 (23.3 mg, 27.5 μmol) were mixed in a degassed mixture of dioxane (4 mL) and water (1 mL) under argon atmosphere. The reaction mixture was stirred at room temperature for 72 hr. The reaction mixture was concentrated under reduced pressure. The residue was diluted with EtOAc (20 mL) and washed with brine (10 mL). The organic layer was dried over anhydrous Na
2SO
4 and filtered. The filtrate was concentrated under reduced pressure. The residue was subjected to HPLC (0-1-6 min., 38-38-50% water – MeOH, +0.1% vol. of 25% aq. NH
3, flow: 30 mL/min, column: XBridge BEH C18100×19mm, 5 µm) to afford 5-cyclopentyl-2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidine (18.8 mg, 34.2 μmol, 6.44% yield from 2,4-dichloro-5-cyclopentyl-pyrimidine) as a yellow solid.
1H NMR (600 MHz, DMSO-d6) δ 0.83 – 0.88 (m, 2H), 0.99 – 1.03 (m, 2H), 1.59 – 1.80 (m, 7H), 1.99 – 2.06 (m, 2H), 3.13 – 3.20 (m, 1H), 3.77 (s, 3H), 3.82 (s, 3H), 5.51 (s, 2H), 7.56 (d, 2H), 7.73 (d, 2H), 7.93 (s, 1H), 8.58 (s, 1H), 8.64 (s, 1H). MS (ESI): [M+H]+ m/z: calcd 551.28; found 551.2.
Step 1: The synthesis of 2,2,2-trifluoro-1-[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]ethanol [1244] Trimethyl(trifluoromethyl)silane (336 mg, 2.36 mmol, 375 μL) was added to a stirred solution of 4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]benzaldehyde (500 mg, 1.97 mmol) in THF (25 mL) followed by a catalytic amount of tetrabutylammonium fluoride (98.3 μL, 1 M in THF). The reaction mixture was stirred at room temperature for 12 hr. The reaction mixture was diluted with aqueous HCl (180 μL, 36% wt.) and stirred at room temperature for 2 hr. The reaction mixture was diluted with saturated aqueous NaHCO
3 solution (30 mL). The resulting mixture was extracted with EtOAc (25 mL). The organic layer was dried over anhydrous Na
2SO
4 and concentrated under reduced pressure to afford 2,2,2-trifluoro-1-[4-[1- methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]ethanol (460 mg, 1.42 mmol, 72.13% yield) as a yellow solid which was used in the next steps without further purification.
1H NMR (500 MHz, CDCl
3) δ 3.76 (s, 3H), 4.92 (q, 1H), 7.34 (s, 1H), 7.42 (d, 2H), 7.52 (d, 2H). MS (ESI): [M+H]+ m/z: calcd 325.08; found 325.2. Step 2: The synthesis of 4-cyclopropyl-6-methoxy-5-[4-[2,2,2-trifluoro-1-[4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]ethoxy]pyrimidin-2-yl]pyrimidin [1245] 2,2,2-trifluoro-1-[4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]ethanol (69.9 mg, 215 μmol) was added to a suspension of NaH (8.62 mg, 215 μmol, 60% in mineral oil) in DMF (2 mL). The resulting mixture was stirred at room temperature for 30 min.4- cyclopropyl-6-methoxy-5-(4-methylsulfonylpyrimidin-2-yl)pyrimidine I-8 (55.0 mg, 180 μmol) was added to the mixture. The mixture was stirred at room temperature for 12 hr. The reaction mixture was directly subjected to HPLC(0.5-6.5 min, 40-65% water – ACN, +0.1% vol. of 25% aq. NH
3, flow: 30 mL/min, column: YMC-Actus Triart C18100×20 mm, 5 μm) to afford 4-cyclopropyl-6-methoxy-5-[4-[2,2,2-trifluoro-1-[4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]ethoxy]pyrimidin-2-yl]pyrimidine (47.0 mg, 85.4 μmol, 47.6% yield) as white solid. MS (ESI): [M+H]+ m/z: calcd 551.19; found 551.0. Step 3: The synthesis of rel-(S)-4'-cyclopropyl-6'-methoxy-4-(2,2,2-trifluoro-1-(4-(1-methyl- 4-(trifluoromethyl)-1H-imidazol-2-yl)phenyl)ethoxy)-2,5'-bipyrimidine (64) and rel-(R)-4'- cyclopropyl-6'-methoxy-4-(2,2,2-trifluoro-1-(4-(1-methyl-4-(trifluoromethyl)-1H-imidazol-2- yl)phenyl)ethoxy)-2,5'-bipyrimidine (230) [1246] Racemic 4-cyclopropyl-6-methoxy-5-[4-[2,2,2-trifluoro-1-[4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]ethoxy]pyrimidin-2-yl]pyrimidine (30 mg, 54.5 μmol) was subjected to chiral HPLC (column: Chiralpak IC, 250×20 mm, 5 μm; mobile phase: Hexane-IPA-MeOH, 80-10-10; flow: 15 mL/min) to afford rel-(S)-4'-cyclopropyl-6'- methoxy-4-(2,2,2-trifluoro-1-(4-(1-methyl-4-(trifluoromethyl)-1H-imidazol-2- yl)phenyl)ethoxy)-2,5'-bipyrimidine (12.0 mg, 21.8 μmol, 40.0% yield) and rel-(R)-4'- cyclopropyl-6'-methoxy-4-(2,2,2-trifluoro-1-(4-(1-methyl-4-(trifluoromethyl)-1H-imidazol-2- yl)phenyl)ethoxy)-2,5'-bipyrimidine (12.0 mg, 21.8 μmol, 40.0% yield) as a white solids. rel-(R)-4'-cyclopropyl-6'-methoxy-4-(2,2,2-trifluoro-1-(4-(1-methyl-4-(trifluoromethyl)-1H- imidazol-2-yl)phenyl)ethoxy)-2,5'-bipyrimidine (230):
1H NMR (600 MHz, DMSO-d6) δ 0.63 – 0.69 (m, 1H), 0.72 – 0.78 (m, 1H), 0.89 – 0.99 (m, 2H), 1.40 – 1.46 (m, 1H), 3.69 (s, 3H), 3.77 (s, 3H), 6.99 (q, 1H), 7.30 (d, 1H), 7.70 (d, 2H), 7.80 (d, 2H), 7.94 (s, 1H), 8.63 (s, 1H), 8.80 (d, 1H). MS (ESI): [M+H]+ m/z: calcd 551.19; found 551.2. Enantiopurity: >99% (column: Chiralpak IC, 250 × 4.6 mm, 5 μm; mobile phase: Hexane- IPA-MeOH, 70-15-15; flow: 0.6 mL/min; RT=13.4 min) rel-(S)-4'-cyclopropyl-6'-methoxy-4-(2,2,2-trifluoro-1-(4-(1-methyl-4-(trifluoromethyl)-1H- imidazol-2-yl)phenyl)ethoxy)-2,5'-bipyrimidine (64):
1H NMR (600 MHz, DMSO-d6) δ 0.63 – 0.69 (m, 1H), 0.72 – 0.78 (m, 1H), 0.89 – 0.99 (m, 2H), 1.40 – 1.46 (m, 1H), 3.69 (s, 3H), 3.77 (s, 3H), 6.99 (q, 1H), 7.30 (d, 1H), 7.70 (d, 2H), 7.80 (d, 2H), 7.94 (s, 1H), 8.63 (s, 1H), 8.80 (d, 1H). MS (ESI): [M+H]+ m/z: calcd 551.19; found 551.2. Enantiopurity: >99% (column: Chiralpak IC, 250 × 4.6 mm, 5 μm; mobile phase: Hexane- IPA-MeOH, 70-15-15; flow: 0.6 mL/min; RT=11.5 min). Example 83 (Compound 87)

Step 1: The synthesis of 4-[3-(difluoromethyl)-5-methyl-pyrazol-1-yl]benzonitrile [1247] 4-hydrazinobenzonitrile (5.00 g, 29.5 mmol, HCl) and 1,1-difluoropentane-2,4-dione (4.81 g, 35.4 mmol) were mixed in HFIP (60 mL). The resulting mixture was cooled to -20 °C and then TEA (7.76 g, 76.7 mmol, 11.0 mL) was added dropwise to the mixture. The reaction mixture was stirred at ambient temperature for 24 hr. The reaction mixture was concentrated under reduced pressure. The residue was dissolved in DCM (300 mL) and washed with brine (100 mL). The organic layer was dried over anhydrous Na
2SO
4 and concentrated under reduced pressure. The residue was subjected to flash column chromatography (SiO
2, gradient chloroform – acetonitrile) to afford 4-[3-(difluoromethyl)-5- methyl-pyrazol-1-yl]benzonitrile (5.00 g, 21.4 mmol, 72.7% yield) as a yellow solid.
1H NMR (400 MHz, DMSO-d6) δ 2.43 (s, 3H), 6.64 (s, 1H), 7.04 (t, 1H, CHF2), 7.82 (d, 2H), 8.04 (d, 2H). MS (ESI): [M+H]+ m/z: calcd 234.09; found 234.0. Step 2: The synthesis of 4-[3-(difluoromethyl)-5-methyl-pyrazol-1-yl]benzoic acid [1248] An aqueous NaOH (1.54 g, 38.6 mmol in 8 mL of water) was added to a solution of 4-[3-(difluoromethyl)-5-methyl-pyrazol-1-yl]benzonitrile (3.00 g, 12.9 mmol) in EtOH (30 mL). The reaction mixture was stirred at 80 °C for 24 hr. The reaction mixture was cooled to room temperature and concentrated under reduced pressure. The residue was diluted with MTBE (20 mL) and washed with water (50 mL). The water layer was acidified with saturated solution of citric acid (10 mL). The solids were filtered off and dissolved in EtOAc (50 mL). The resulting solution was washed with brine (2×20 mL), dried over anhydrous Na
2SO
4 and concentrated under reduced pressure to afford 4-[3-(difluoromethyl)-5-methyl-pyrazol-1- yl]benzoic acid (2.50 g, 9.91 mmol, 77.1% yield) as a yellow solid which was used in the next steps without further purification. MS (ESI): [M+H]+ m/z: calcd 253.08; found 253.1. Step 3: The synthesis of [4-[3-(difluoromethyl)-5-methyl-pyrazol-1-yl]phenyl]methanol [1249] Borane dimethyl sulfide complex (2.26 g, 29.7 mmol, 2.82 mL) was added dropwise to a stirred solution of 4-[3-(difluoromethyl)-5-methyl-pyrazol-1-yl]benzoic acid (2.50 g, 9.91 mmol) in THF (100 mL) under argon atmosphere. The reaction mixture was stirred at room temperature for 48 hr. The reaction mixture was quenched by dropwise addition of water (20 mL). The resulting mixture was stirred at room temperature overnight. The reaction mixture was concentrated under reduced pressure. The residue was diluted with EtOAc (150 mL) and washed with water (50 mL) and brine (2×50 mL), dried over anhydrous Na
2SO
4 and concentrated under reduced pressure to afford [4-[3-(difluoromethyl)-5-methyl-pyrazol-1- yl]phenyl]methanol (2.00 g, 8.40 mmol, 84.7% yield) as a light-yellow oil which was used in the next steps without further purification.
1H NMR (400 MHz, DMSO-d6) δ 2.32 (s, 3H), 4.56 (d, 2H), 5.32 (t, 1H), 6.53 (s, 1H), 6.99 (t, 1H, CHF2), 7.44 – 7.52 (m, 4H). MS (ESI): [M+H]+ m/z: calcd 239.10; found 239.2. Step 4: The synthesis of 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[4-[3- (difluoromethyl)-5-methyl-pyrazol-1-yl]phenyl]methoxy]-5-methoxy-pyrimidine [1250] NaH (18.5 mg, 462 μmol, 60% dispersion in mineral oil) was added to a solution of [4-[3-(difluoromethyl)-5-methyl-pyrazol-1-yl]phenyl]methanol (100 mg, 420 μmol) in DMF (4.0 mL) under argon atmosphere. The reaction mixture was stirred at room temperature for 1 hr.2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-5-methoxy-4-methylsulfonyl-pyrimidine I-6 (141 mg, 420 μmol) was added to the reaction mixture. The resulting mixture was stirred at room temperature for 16 hr. The reaction mixture was diluted with EtOAc (20 mL) and washed with brine (20 mL). The organic layer was separated, washed with brine (15 mL), dried over anhydrous Na
2SO
4 and concentrated under reduced pressure. The residue was subjected to HPLC (2-10 min., 10-40% ACN, +0.1% vol. of 25% aq. NH
3, flow: 30 mL/min, column: SunFire 100 ×19 mm, 5 µm) to afford 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)- 4-[[4-[3-(difluoromethyl)-5-methyl-pyrazol-1-yl]phenyl]methoxy]-5-methoxy-pyrimidine (59.0 mg, 119 μmol, 28.4% yield) as a white solid.
1H NMR (600 MHz, DMSO-d6) δ 0.83 – 0.87 (m, 2H), 0.99 – 1.03 (m, 2H), 1.66 – 1.72 (m, 1H), 2.33 (s, 3H), 3.83 (s, 3H), 3.93 (s, 3H), 5.49 (s, 2H), 6.54 (s, 1H), 6.98 (t, 1H, CHF2), 7.56 (d, 2H), 7.61 (d, 2H), 8.42 (s, 1H), 8.64 (s, 1H). MS (ESI): [M+H]+ m/z: calcd 495.22; found 495.2.
Compound 51 Step 1: The synthesis of [4-[1-(oxetan-3-yl)-4-(trifluoromethyl)imidazol-2- yl]phenyl]methanol [1251] Tert-butyl-dimethyl-[[4-[4-(trifluoromethyl)-1H-imidazol-2-yl]phenyl]methoxy]silane (1.00 g, 2.81 mmol), 3-iodooxetane (1.55 g, 8.42 mmol, 724 μL) and Cs
2CO
3 (2.74 g, 8.42 mmol) were mixed in DMSO (3.5 mL). The reaction mixture was stirred at 110 °C for 12 hr. The reaction mixture was cooled to room temperature and diluted with cold water (10 mL). Th resulting mixture was extracted with EtOAc (15 mL). The organic layer was washed with brine (10 mL), dried over anhydrous Na
2SO
4 and concentrated under reduced pressure. The residue was subjected to HPLC (0.5-6.5 min, 15% water - ACN; flow: 30 mL/min, column: Waters SunFire C18, 100 x 19 mm, 5 µm) to afford [4-[1-(oxetan-3-yl)-4- (trifluoromethyl)imidazol-2-yl]phenyl]methanol (80.0 mg, 268 μmol, 9.56% yield) as a white solid. MS (ESI): [M+H]+ m/z: calcd 299.12; found 299.2. Step 2: The synthesis of 2-chloro-5-methyl-4-[[4-[1-(oxetan-3-yl)-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidine [1252] NaH (107 mg, 2.68 mmol, 60% dispersion in mineral oil) was added to a solution of [4-[1-(oxetan-3-yl)-4-(trifluoromethyl)imidazol-2-yl]phenyl]methanol (79.9 mg, 0.268 mmol) in DMF (5.00 mL). The reaction mixture was stirred at room temperature for 1 hr.2,4- Dichloro-5-methyl-pyrimidine (437 mg, 2.68 mmol) was added to the reaction mixture. The mixture was stirred at room temperature for 12 hr. The reaction mixture was diluted with aqueous NH
4Cl (10 mL) and extracted with EtOAc (2 × 10 mL). The combined organic layers were washed with brine (10 mL), dried over anhydrous Na
2SO
4 and concentrated under reduced pressure. The residue was subjected to HPLC (0.5-6.5 min, 40-65% water - ACN; flow: 30 mL/min, column: Waters SunFire C18, 100 x 19 mm, 5 µm) to afford 2- chloro-5-methyl-4-[[4-[1-(oxetan-3-yl)-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidine (43.0 mg, 101 μmol, 37.8% yield) as an off-white solid. Step 3: The synthesis of 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-5-methyl-4-[[4-[1- (oxetan-3-yl)-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidine [1253] 2-Chloro-5-methyl-4-[[4-[1-(oxetan-3-yl)-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidine (100 mg, 235 μmol), (4-cyclopropyl-6-methoxy-pyrimidin-5- yl)boronic acid I-5 (68.5 mg, 353 μmol), bis(diphenylphosphino)ferrocene]dichloropalladium(II)-DCM (9.60 mg, 11.8 μmol) and potassium phosphate tribasic (150 mg, 706 μmol) were mixed in degassed dioxane (5 mL) and water (150 µL) under argon atmosphere. The reaction mixture was stirred at 75 °C for 12 hr. The reaction mixture was cooled to room temperature and concentrated under reduced pressure. The residue was diluted with EtOAc. An anhydrous Na
2SO
4 and SiliaMetS® Dimercaptotriazine (50 mg) were added to the solution. The mixture was stirred at room temperature for 1 hr. and then solids were filtered out. The filtrate was concentrated under reduced pressure. The residue was subjected to HPLC (2-7 min., 42% water – ACN, +0.1% vol. of 25% aq. NH
3, flow: 30 mL/min, column: SunFire 100 ×19 mm, 5 µm) to afford 2-(4- cyclopropyl-6-methoxy-pyrimidin-5-yl)-5-methyl-4-[[4-[1-(oxetan-3-yl)-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidine (30 mg, 55.7 μmol, 23.7% yield) as an off-white solid. MS (ESI): [M+H]+ m/z: calcd 539.23; found 539.0.
1H NMR (600 MHz, DMSO-d6) δ 0.82 – 0.88 (m, 2H), 0.99 – 1.03 (m, 2H), 1.64 – 1.70 (m, 1H), 2.22 (s, 3H), 3.82 (s, 3H), 4.79 (t, 2H), 4.86 (t, 2H), 5.51 (s, 2H), 7.53 (d, 2H), 7.59 (d, 2H), 8.48 (s, 1H), 8.55 (s, 1H), 8.65 (s, 1H). Example 85 (Compound 164)
Compound 164 The synthesis of starting 2-chloro-5-methoxy-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidine is described for Compound 12 and compound 132. Step 1: Synthesis of 5-bromo-4-cyclopropyl-6-ethoxy-pyrimidine [1254] NaH (295 mg, 12.9 mmol, 60% dispersion in mineral oi) was added in few portions to stirred EtOH (150 mL) under argon atmosphere. The solution was stirred at room temperature for 20 minutes and then cooled to -20 oC.5-bromo-4-chloro-6-cyclopropyl-pyrimidine (3.00 g, 12.9 mmol) was added in one portion. The reaction mixture was stirred at room temperature for 16 hr. The mixture was concentrated under reduced pressure. The residue was poured into ice-water (100 mL). The solid precipitate formed was filtered off and dissolved in EtOAc (120 mL). The obtained solution was washed with brine (2 × 30 mL), dried over anhydrous Na
2SO
4 and concentrated under reduced pressure to afford 5-bromo-4- cyclopropyl-6-ethoxy-pyrimidine (2.80 g, 11.5 mmol, 89.6% yield) as light yellow solid which was used in the next steps without further purification.
1H NMR (400 MHz, CDCl
3) δ 1.05 – 1.11 (m, 2H), 1.13 – 1.19 (m, 2H), 1.44 (t, 3H), 2.49 – 2.56 (m, 1H), 4.47 (q, 2H), 8.41 (s, 1H). MS (ESI): [M+H]+ m/z: calcd 243.02; found 243.0. Step 2: Synthesis of 4-cyclopropyl-6-ethoxy-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2- yl)pyrimidine [1255] 5-bromo-4-cyclopropyl-6-ethoxy-pyrimidine (1.80 g, 7.40 mmol) and 2-isopropoxy- 4,4,5,5-tetramethyl-1,3,2-dioxaborolane (2.07 g, 11.1 mmol, 2.27 mL) were dissolved in THF (100 mL) under argon atmosphere. The solution was cooled to -78 °C and then n- Butyllithium, 2.5 M in hexane (5.32 mL, 13.3 mmol) was added dropwise. The reaction mixture was stirred at -70 - -65 °C for 3 hr. The mixture was allowed to warm to room temperature and quenched with saturated aqueous NH
4Cl solution (20 mL). The obtained mixture was extracted with EtOAc (50 mL). The organic layer was separated, washed with brine (2 × 20 mL), dried over anhydrous Na
2SO
4 and concentrated under reduced pressure. The residue was subjected to flash-column chromatography to (SiO
2, gradient hexane- MTBE) to afford 4-cyclopropyl-6-ethoxy-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2- yl)pyrimidine (1.36 g, 4.70 mmol, 63.5% yield) as a light-yellow solid.
1H NMR (400 MHz, CDCl
3) δ 0.96 – 1.02 (m, 2H), 1.15 – 1.21 (m, 2H), 1.37 (t, 3H), 1.40 (s, 12H), 1.99 – 2.07 (m, 1H), 4.36 (q, 2H), 8.55 (s, 1H). MS (ESI): [M+H]+ m/z: calcd 291.17; found 291.0. Step 3: Synthesis of 2-(4-cyclopropyl-6-ethoxy-pyrimidin-5-yl)-5-methoxy-4-[[4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidine [1256] 2-chloro-5-methoxy-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidine (400 mg, 1.00 mmol), 4-cyclopropyl-6-ethoxy-5-(4,4,5,5- tetramethyl-1,3,2-dioxaborolan-2-yl)pyrimidine (582 mg, 2.01 mmol), Potassium phosphate tribasic (639 mg, 3.01 mmol) and XPhos Pd G3 (42.5 mg, 50.2 μmol) were mixed in degassed dioxane (12 mL) and water (2 mL) under argon atmosphere. The reaction mixture was stirred at 60 °C for 6 hr. The mixture was cooled to room temperature and concentrated under reduced pressure. The residue was diluted with EtOAc (25 mL) and water (10 mL). The organic layer was separated, washed with brine (2 × 15 mL), dried over anhydrous Na
2SO
4 and treated with SiliaMetS® Dimercaptotriazine (300 mg) for 30 min. The resulting mixture was filtered. The filtrate was concentrated under reduced pressure. The residue was subjected to HPLC (2-10 min, 20 - 50% water - ACN; flow: 30 mL/min, column: Waters SunFire C18, 100 x 19 mm, 5 µm) to afford 2-(4-cyclopropyl-6-ethoxy-pyrimidin-5-yl)-5- methoxy-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidine (96.6 mg, 183 μmol, 18.3% yield) as a light-yellow solid.
1H NMR (600 MHz, DMSO-d6) δ 0.83 – 0.88 (m, 2H), 0.98 – 1.03 (m, 2H), 1.16 (t, 3H), 1.67 – 1.73 (m, 1H), 3.77 (s, 3H), 3.93 (s, 3H), 4.34 (q, 2H), 5.49 (s, 2H), 7.57 (d, 2H), 7.72 (d, 2H), 7.92 (s, 1H), 8.42 (s, 1H), 8.60 (s, 1H). MS (ESI): [M+H]+ m/z: calcd 527.23; found 527.2. Example 86 (Compound 127)
Compound 127 The synthesis of starting 2-chloro-5-methoxy-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidine is described for Compound 12 and compound 132. Step 1: The synthesis of 3-bromo-2-cyclopropyl-pyridine 2-cyclopropylpyridin-3-amine (11.1 g, 82.7 mmol) was suspended in Hydrobromic acid (35.9 mL, 48% wt. in water) and the resulting mixture was cooled to -10 °C. Bromine (17.2 g, 107 mmol) followed by Sodium Nitrite (11.4 g, 165 mmol) in water (90 mL) were added dropwise to the mixture maintaining internal temperature below -10 °C. The reaction mixture was stirred for 1 hr. at ambient temperature. The mixture was diluted with water (300 mL) and quenched with NaOH to pH≈9. The resulting mixture was extracted with MTBE (2×200 mL). the combined organic layers were washed with water (150 mL), dried over anhydrous Na
2SO
4 and concentrated under reduced pressure. The residue was distilled under reduce pressure (0.3 mm Hg, b.p. approximately 45 °C) to afford 3-bromo-2-cyclopropyl-pyridine (12.0 g, 60.6 mmol, 73.2% yield) as a light-yellow oil which was used in the next steps without further purification.
1H NMR (500 MHz, CDCl
3) δ 0.99 – 1.14 (m, 4H), 2.48 – 2.57 (m, 1H), 6.87 – 6.94 (m, 1H), 7.78 (d, 1H), 8.36 (d, 1H). MS (ESI): [M+H]+ m/z: calcd 197.99; found 198. Step 2: The synthesis of 2-cyclopropyl-3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2- yl)pyridine [1257] 3-bromo-2-cyclopropyl-pyridine (5.00 g, 25.2 mmol), 4,4,5,5-tetramethyl-2-(4,4,5,5- tetramethyl-1,3,2-dioxaborolan-2-yl)-1,3,2-dioxaborolane (9.62 g, 37.9 mmol), Potassium Acetate (6.19 g, 63.11 mmol) and bis(diphenylphosphino)ferrocene]dichloropalladium(II)- DCM (2.06 g, 2.52 mmol) were mixed in degassed dioxane (75 mL). The reaction mixture was stirred at 95 °C for 16 hr. The mixture was cooled to room temperature and concentrated under reduced pressure. The residue was diluted with MTBE (200 mL) and washed with brine (100 mL). The organic layer was dried over Na
2SO
4, filtered and concentrated under reduced pressure to afford 2-cyclopropyl-3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2- yl)pyridine (12 g, crude) which was used in the next steps without further purification.
1H NMR (500 MHz, CDCl
3) δ 0.95 – 1.03 (m, 2H), 1.10 – 1.17 (m, 2H), 1.35 (s, 12H), 2.83 – 2.90 (m, 1H), 6.98 – 7.05 (m, 1H), 8.01 (d, 1H), 8.47 (d, 1H). Step 3: The synthesis of 2-(2-cyclopropyl-3-pyridyl)-5-methoxy-4-[[4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidine [1258] 2-chloro-5-methoxy-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidine (400 mg, 512 μmol), 2-cyclopropyl-3-(4,4,5,5-tetramethyl- 1,3,2-dioxaborolan-2-yl)pyridine (752 mg, crude), potassium phosphate tribasic (326 mg, 1.53 mmol) were mixed in a degassed mixture of dioxane (9 mL) and water (1 mL). XPhosPdG3 (21.7 mg, 25.6 μmol) was added to the mixture. The reaction mixture was stirred at 60°C for 4 hr. The reaction mixture was diluted with EtOAc (20 mL) and washed with water (2×20 mL). SiliaMetS® Dimercaptotriazine (150 mg) was added to the organic layer. The resulting mixture was stirred for 30 min and filtered. The filtrate was concentrated under reduced pressure. The residue was subjected to HPLC (0.5-6.5 min, 40-90% water - ACN, flow 30ml/min; column SunFire C18100×19mm, 5μm), then repurified by HPLC (0.5-6.5 min, 55% water – ACN - MeOH, flow 30ml/min; column SunFire C18100×19 mm, 5μm) to afford 2-(2-cyclopropyl-3-pyridyl)-5-methoxy-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol- 2-yl]phenyl]methoxy]pyrimidine (33.0 mg, 68.5 μmol, 13.4% yield) as a light-yellow solid.
1H NMR (600 MHz, DMSO-d6) δ 0.79 – 0.83 (m, 2H), 0.96 – 0.99 (m, 2H), 2.81 – 2.87 (m, 1H), 3.77 (s, 3H), 3.94 (s, 3H), 5.58 (s, 2H), 7.20 – 7.23 (m, 1H), 7.58 (d, 2H), 7.73 (d, 2H), 7.92 (s, 1H), 8.01(d, 1H), 8.43 – 8.46 (m, 2H). MS (ESI): [M+H]+ m/z: calcd 482.21; found 482.0. Example 87 (Compound 165)
Step 1: The synthesis of 6-cyclopropyl-2-propyl-pyrimidin-4-ol [1259] NaH (4.01 g, 100 mmol, 60% dispersion in mineral oil) was added portion wise to a vigorously stirred MeOH (300 mL) at 0 °C. The resulting solution was stirred at room temperature for 1 hr. then methyl 3-cyclopropyl-3-oxo-propanoate (8.00 g, 56.3 mmol) was added dropwise to the solution. The resulting mixture was stirred at room temperature for 15 min. The mixture was cooled to 0 °C then butanamidine (6.90 g, 56.3 mmol, HCl) was added. The reaction mixture was stirred at room temperature for 72 hr. The reaction mixture was concentrated under reduced pressure. The residue was diluted with water (200 mL) and acidified with an aqueous solution of HCl (5% wt.) to pH = 3. The solid precipitate formed was filtered off, washed with water (30 mL) and dissolved in EtOAc (200 mL). The resulting solution was washed with brine (2×50 mL), dried over anhydrous Na
2SO
4 and concentrated under reduced pressure to afford 6-cyclopropyl-2-propyl-pyrimidin-4-ol (3.00 g, 16.8 mmol, 29.9% yield) as a light-yellow solid which was used in the next steps without further purification.
1H NMR (500 MHz, DMSO-d6) δ 0.77 – 0.90 (m, 7H), 1.53 – 1.65 (m, 2H), 1.77 – 1.81 (m, 1H), 2.40 (t, 2H), 6.04 (s, 1H), 12.07 (br., 1H). MS: [M+H]+ m/z: calcd 179.12; found 179.2. Step 2: The synthesis of 5-bromo-6-cyclopropyl-2-propyl-pyrimidin-4-ol [1260] Bromine (3.40 g, 42.1 mmol) was added dropwise to a vigorously stirred solution of 6-cyclopropyl-2-propyl-pyrimidin-4-ol (3.00 g, 16.8 mmol) in EtOH (150 mL) at room temperature. The reaction mixture was stirred at room temperature for 48 hr. The reaction mixture was concentrated under reduced pressure. The residue was diluted with a saturated aqueous NaHCO
3 solution (80 mL) and extracted with EtOAc (300 mL). The organic layer was separated, washed with water (50 mL), an aqueous solution of Na2S2O3 (100 mL), water (100 mL) and brine (200 mL), dried over anhydrous Na
2SO
4 and concentrated under reduced pressure to afford 5-bromo-6-cyclopropyl-2-propyl-pyrimidin-4-ol (3.10 g, 12.1 mmol, 71.6% yield) as a light-yellow solid which was used in the next steps without further purification. MS (ESI): [M+H]+ m/z: calcd 257.03; found 257.0. Step 3: The synthesis of 5-bromo-4-chloro-6-cyclopropyl-2-propyl-pyrimidine Phosphorus oxychloride (4.77 g, 31.1 mmol) was added dropwise to a solution of 5-bromo-6- cyclopropyl-2-propyl-pyrimidin-4-ol (4.00 g, 15.6 mmol) and DIPEA (4.02 g, 31.1 mmol, 5.42 mL) in ACN (150 mL). The reaction mixture was stirred at 65 °C for 72 hr. The reaction mixture was cooled to room temperature and concentrated under reduced pressure. The residue was subjected to flash column chromatography (SiO
2, gradient hexane – MTBE) to afford 5-bromo-4-chloro-6-cyclopropyl-2-propyl-pyrimidine (1.50 g, 5.44 mmol, 35.0% yield) as a light-yellow oil.
1H NMR (400 MHz, CDCl
3) δ 0.93 (t, 3H), 1.06 – 1.21 (m, 4H), 1.69 – 1.80 (m, 2H), 2.47 – 2.58 (m, 1H), 2.72 (t, 2H). MS (ESI): [M+H]+ m/z: calcd 275.00, 276.99; found 275.0, 277.0. Step 4: The synthesis of 5-bromo-4-cyclopropyl-6-methoxy-2-propyl-pyrimidine [1261] NaH (229 mg, 5.72 mmol, 60% dispersion in mineral oil) was added portion wise to a vigorously stirred MeOH (50 mL) at 0 °C. The solution was stirred at room temperature for 1 hr., then cooled to -20 °C. A solution of 5-bromo-4-chloro-6-cyclopropyl-2-propyl- pyrimidine (1.50 g, 5.44 mmol) in MeOH (5.0 mL) was added to the mixture. The reaction mixture was stirred at room temperature for 16 hr. The reaction mixture was concentrated under reduced pressure. The residue was diluted with EtOAc (30 mL) and washed with water (15 mL). The organic layer was separated, washed with brine (2×20 mL), dried over anhydrous Na
2SO
4 and concentrated under reduced pressure to afford 5-bromo-4- cyclopropyl-6-methoxy-2-propyl-pyrimidine (1.30 g, 4.79 mmol, 88.1% yield) as a light- yellow oil which was used in the next steps without further purification.
1H NMR (400 MHz, CDCl
3) δ 0.94 (t, 3H), 0.98 – 1.04 (m, 2H), 1.12 – 1.18 (m, 2H), 1.70 – 1.80 (m, 2H), 2.43 – 2.50 (m, 1H), 2.67 (t, 2H), 4.00 (s, 3H). MS (ESI): [M+H]+ m/z: calcd 271.04, 273.04; found 271.2, 273.2 Step 5: The synthesis of 4-cyclopropyl-6-methoxy-2-propyl-5-(4,4,5,5-tetramethyl-1,3,2- dioxaborolan-2-yl)pyrimidine [1262] A solution of 5-bromo-4-cyclopropyl-6-methoxy-2-propyl-pyrimidine (600 mg, 2.21 mmol) and 2-isopropoxy-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (618 mg, 3.32 mmol, 677 μL) in THF (30 mL) was cooled to -80 °C under argon atmosphere. n-Butyllithium, 2.5 M in hexane (2.50 mmol, 1.00 mL) was added dropwise to the solution. The reaction mixture was stirred at -65 to -78 °C for 3 hr. The reaction mixture was allowed to warm to room temperature and quenched by dropwise addition of a saturated aqueous solution of NH
4Cl (15 mL). The resulting mixture was extracted with MTBE (20 mL). The organic layer was separated, washed with water (10 mL) and brine (20 mL), dried over anhydrous Na
2SO
4 and concentrated under reduced pressure to afford 4-cyclopropyl-6-methoxy-2-propyl-5-(4,4,5,5- tetramethyl-1,3,2-dioxaborolan-2-yl)pyrimidine (700 mg, 2.20 mmol, 99.4% yield) as a light- yellow oil which was used in the next steps without further purification. MS (ESI): [M+H]+ m/z: calcd 319.22; found 319.2. Step 6: The synthesis of 4-cyclopropyl-6-methoxy-5-[4-[[4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidin-2-yl]-2-propyl-pyrimidine [1263] 4-Cyclopropyl-6-methoxy-2-propyl-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2- yl)pyrimidine (300 mg, 943 μmol), 2-chloro-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidine (116 mg, 314 μmol), RuPhos Pd G4 (13.3 mg, 15.6 μmol) and potassium phosphate tribasic (200 mg, 943 μmol) were mixed in a degassed mixture of dioxane (5 mL) and water (800 µL) under argon atmosphere. The reaction mixture was stirred at 65 °C for 5 hr. The reaction mixture was cooled to room temperature and concentrated under reduced pressure. The residue was diluted with EtOAc (20 mL) and washed with water (10 mL). The organic layer was separated, washed with brine (20 mL), dried over anhydrous Na
2SO
4 and filtered. To the obtained solution SiliaMetS® Dimercaptotriazine (200 mg) was added, and the mixture was stirred for 1 hr. The mixture was filtered. The filtrate was concentrated under reduced pressure. The residue was subjected to HPLC to afford 4-cyclopropyl-6-methoxy-5-[4-[[4-[1-methyl-4-(trifluoromethyl)imidazol- 2-yl]phenyl]methoxy]pyrimidin-2-yl]-2-propyl-pyrimidine (75.0 mg, 143 μmol, 45.5% yield) as a light-yellow solid.
1H NMR (600 MHz, DMSO-d6) δ 0.80 – 0.85 (m, 2H), 0.93 (t, 3H), 0.98 – 1.03 (m, 2H), 1.63 – 1.69 (m, 1H), 1.70 – 1.78 (m, 2H), 2.69 (t, 3H), 3.77 (s, 3H), 3.81 (s, 3H), 5.48 (s, 2H), 7.04 (d, 1H), 7.57 (d, 2H), 7.72 (d, 2H), 7.92 (s, 1H), 8.68 (d, 1H). MS (ESI): [M+H]+ m/z: calcd 525.26; found 525.2. Example 88 (Compound 18)
Step 1: The synthesis of [1-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]-4-piperidyl]methanol [1264] 2-bromo-1-methyl-4-(trifluoromethyl)imidazole (600 mg, 2.62 mmol), tert-butyl- dimethyl-(4-piperidylmethoxy)silane (631 mg, 2.75 mmol) and KF (304 mg, 5.24 mmol) were mixed in DMSO (2.0 mL) under argon atmosphere. The reaction mixture was stirred at 125 °C for 72 hr. The reaction mixture was cooled to room temperature, diluted with water (5.0 mL) and extracted with EtOAc (20 mL). The organic layer was separated, washed with water (10 mL), dried over anhydrous Na
2SO
4 and concentrated under reduced pressure to afford [1-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]-4-piperidyl]methanol (650 mg, 2.47 mmol, 94.2% yield) as an yellow oil which was used in the next steps without further purification. MS (ESI): [M+H]+ m/z: calcd 378.28; found 378.4. Step 2: The synthesis of 2-chloro-5-methyl-4-[[1-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]- 4-piperidyl]methoxy]pyrimidine [1265] 2,4-Dichloro-5-methyl-pyrimidine (232 mg, 1.42 mmol) was added to a vigorously stirred solution of [1-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]-4-piperidyl]methanol (250 mg, 950 μmol) and potassium tert-butoxide (192 mg, 1.71 mmol) in THF (2 mL) at 0 °C. The reaction mixture was stirred at room temperature for 72 hr. The reaction mixture was concentrated under reduced pressure to afford 2-bromo-4-[[4-[1-cyclopropyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]-5-methyl-pyrimidine (330 mg, crude) as an yellow oil which was used in the next steps without further purification. MS (ESI): [M+H]+ m/z: calcd 390.16; found 390.0. Step 3: The synthesis of 4'-cyclopropyl-6'-methoxy-5-methyl-4-((1-(1-methyl-4- (trifluoromethyl)-1H-imidazol-2-yl)piperidin-4-yl)methoxy)-2,5'-bipyrimidine [1266] 2-chloro-5-methyl-4-[[1-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]-4- piperidyl]methoxy]pyrimidine (110 mg, 282 μmol), (4-cyclopropyl-6-methoxy-pyrimidin-5- yl)boronic acid I-5 (110 mg, 564 μmol), potassium phosphate tribasic (150 mg, 706 μmol), RuPhos Pd G4 (12.0 mg, 14.1 μmol) and RuPhos (6.59 mg, 14.1 μmol) were mixed in degassed dioxane (5 mL) and water (1 mL) under argon atmosphere. The mixture was stirred at 90 °C for 12 hr. The reaction mixture was cooled to room temperature. SiliaMetS® Dimercaptotriazine (100 mg) was added to the reaction mixture. The resulting mixture was stirred at room temperature for 3 hr. The mixture was diluted with MTBE (5.0 mL) and filtered through pad of silica gel. The filtrate was concentrated under reduced pressure. The residue was subjected to HPLC (2-12 min, 30 - 40% water - ACN; flow: 30 mL/min, column: Waters SunFire C18, 100 × 19 mm, 5 µm) to afford 4'-cyclopropyl-6'-methoxy-5-methyl-4- ((1-(1-methyl-4-(trifluoromethyl)-1H-imidazol-2-yl)piperidin-4-yl)methoxy)-2,5'- bipyrimidine (20.0 mg, 51.3 μmol, 18.2% yield) as an yellow solid.
1H NMR (600 MHz, DMSO-d6) δ 0.87 – 0.92 (m, 2H), 1.01 – 1.05 (m, 2H), 1.41 – 1.50 (m, 2H), 1.66 – 1.72 (m, 1H), 1.77 – 1.83 (m, 2H), 1.90 – 1.98 (m, 1H), 2.17 (s, 3H), 2.74 (t, 2H), 3.24 – 3.30 (m, 2H), 3.47 (s, 3H), 3.83 (s, 3H), 4.24 (d, 2H), 7.51 (s, 1H), 8.49 (s, 1H), 8.64 (s, 1H). MS (ESI): [M+H]+ m/z: calcd 504.27; found 504.4.

Step 1: 5-benzyloxy-2-chloro-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidine [1267] To a stirred solution of [4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methanol I-2b (180.79 mg, 705.61 μmol) in THF (4 mL) was added NaH (28.22 mg, 705.61 μmol, 60% purity) at 25 °C under nitrogen atmosphere. The resulting mixture was stirred at 0 °C for 30 minutes under nitrogen atmosphere. To the resulting mixture was added 5-benzyloxy-2,4-dichloro-pyrimidine (200 mg, 784.01 μmol) at 0 °C under nitrogen atmosphere. The resulting mixture was stirred for 16 hrs at 25 °C under nitrogen atmosphere. The reaction was quenched by the addition of saturated NH
4Cl aqueous solution (10 mL) at 0 °C. The resulting mixture was extracted with EtOAc (3 x 5 mL). The combined organic layers were washed with brine (5 mL), dried over anhydrous Na
2SO
4. After filtration, the filtrate was concentrated under reduced pressure. The residue was purified by Prep-TLC (30% EtOAc in PE) to afford 5-benzyloxy-2-chloro-4-[[4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidine (300 mg, 631.76 μmol, 80% yield) as a light yellow solid. MS: m/z = 475.05 [M + H]+.
1H NMR (400 MHz, Chloroform- d) δ 7.96 (s, 1H), 7.69 (d, J = 8.4 Hz, 2H), 7.60 (d, J = 8.0 Hz, 2H), 7.43 - 7.31 (m, 6H), 5.57 (s, 2H), 5.17 (s, 2H), 3.80 (s, 3H). Step 2: 5-benzyloxy-2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidine [1268] To a stirred solution of 5-benzyloxy-2-chloro-4-[[4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidine (200 mg, 421.17 μmol) and 4- cyclopropyl-6-methoxy-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyrimidine I-5 (116.30 mg, 421.17 μmol) in THF (3 mL) and water (0.6 mL) were added potassium phosphate (268.20 mg, 1.26 mmol) and 1,1μ-Bis(di-cyclohexylphosphino)ferrocene palladium dichloride (35.49 mg, 42.12 μmol) at 25 °C under nitrogen atmosphere. The resulting mixture was stirred at 40 °C for 16 hrs under nitrogen atmosphere. The mixture was allowed to cool down to 25 °C, filtered and the filtrate was concentrated under reduced pressure. The residue was subject to Prep-TLC (10% MeOH in DCM) to afford crude product. The obtained crude product was purified by RP-Flash with the following conditions: Column: C18 spherical Column, 20-35 um, 100A, 25 g; Mobile Phase A: Water (5 mM aq. NH
4HCO
3), Mobile Phase B: MeCN; Flow rate: 35 mL/min; Gradient: 5% B to 5% B in 5 min, 5% B to 48% B in 15 min, 48% B to 48% B in 2 min, 48% B to 95% B in 5 min; Detector: UV 254 & 210 nm. The collected fractions were combined and lyophilized to give 5-benzyloxy-2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidine (187.5 mg, 318.56 μmol, 75% yield) as a white solid. MS: m/z = 589.20 [M + H]+.
1H NMR (400 MHz, Chloroform-d) δ 8.67 (s, 1H), 8.30 (s, 1H), 7.67 (d, J = 8.4 Hz, 2H), 7.60 (d, J = 8.0 Hz, 2H), 7.52 - 7.36 (m, 5H), 7.34 (s, 1H), 5.61 (s, 2H), 5.27 (s, 2H), 3.96 (s, 3H), 3.80 (s, 3H), 1.78 - 1.72 (m, 1H), 1.29 - 1.23 (m, 2H), 0.94 - 0.90 (m, 2H).
19F NMR (377 MHz, Chloroform-d) δ -62.77. Example 90 (Compound 122)
Step 1: The synthesis of 2,4-dichloropyrimidin-5-ol [1269] Boron tribromide (35.0 g, 140 mmol, 13.3 mL) was slowly added to a solution of 2,4- dichloro-5-methoxy-pyrimidine (5.00 g, 27.9 mmol) in dichloroethane (50 mL) at 0 °C. The reaction mixture was stirred at 82 °C for 48 hr. The reaction mixture was cooled to 0°C and quenched with a 1 N aqueous NaOH solution to pH≈9-10. The mixture was stirred at room temperature for 1 hr and then pH was adjusted to 6-7 with AcOH. The layers were separated, and the aqueous layer was extracted with EtOAc (3×50 mL). The combined organic layers were dried over dried over anhydrous Na
2SO
4, filtered and concentrated in vacuo to afford 2,4-dichloropyrimidin-5-ol (3.20 g, 19.4 mmol, 69.4% yield) as a light-yellow solid which was used in the next steps without further purification.
1H NMR (500 MHz, DMSO-d6) δ 8.23 (s, 1H), 11.71 (s, 1H). MS (ESI): [M+H]+ m/z: calcd 164.96; found 165.0. Step 2: The synthesis of 2-chloro-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidin-5-ol [1270] Potassium tert-butoxide (425 mg, 3.79 mmol) was added portion wise to a stirred mixture of [4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methanol I-2b (388 mg, 1.52 mmol) and 2,4-dichloropyrimidin-5-ol (250 mg, 1.52 mmol) in toluene (35 mL) at room temperature. The resulting mixture was stirred at room temperature for 12 hr. The resulting mixture was poured into saturated aqueous NH
4Cl solution (50 mL) and extracted with MTBE (4 × 20 mL). The combined organic layers were dried over anhydrous Na
2SO
4 and concentrated under reduced pressure. The residue was subjected to HPLC (2-10 min, 30-50% water – ACN, flow: 30 mL/min, column: SunFire C18100 ×19 mm, 5 µm) to afford 2- chloro-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidin-5-ol (112 mg, 291 μmol, 19.2% yield) as a yellow gum.
1H NMR (600 MHz, DMSO-d6) δ 3.78 (s, 3H), 5.47 (s, 3H), 7.61 (d, 2H), 7.74 (d, 2H), 7.97 (s, 1H), 8.11 (s, 1H), 10.46 (s, 1H). MS (ESI): [M+H]+ m/z: calcd 385.08; found 385.2. Step 3: The synthesis of 2-chloro-5-isopropoxy-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol- 2-yl]phenyl]methoxy]pyrimidine [1271] 2-Iodopropane (74.2 mg, 437 μmol, 43.6 μL) was added to a stirred mixture of 2- chloro-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidin-5-ol (112 mg, 291 μmol) and K
2CO
3 (80.5 mg, 582 μmol) in DMF (7 mL). The resulting mixture was stirred at 40 °C for 14 hr. The mixture was cooled to room temperature and poured into water (25 mL). The resulting mixture was extracted with EtOAc (4 × 15 mL). The combined organic layers were washed with water (2 × 10 mL) and brine (2 × 5 mL), dried over anhydrous Na
2SO
4 and concentrated under reduced pressure to afford 2-chloro-5-isopropoxy- 4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidine (100 mg, 234 μmol, 80.5% yield) as a light-yellow gum which was used in the next steps without further purification. MS (ESI): [M+H]+ m/z: calcd 427.12; found 427.2 Step 4: The synthesis of 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-5-isopropoxy-4-[[4-[1- methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidine [1272] 2-Chloro-5-isopropoxy-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidine (250 mg, 586 μmol), (4-cyclopropyl-6-methoxy-pyrimidin-5- yl)boronic acid I-5 (170 mg, 879 μmol), potassium phosphate tribasic (373 mg, 1.76 mmol) and XPhos Pd G3 (24.8 mg, 29.3 μmol) were mixed in a degassed mixture of dioxane (9 mL) and water (2 mL) under argon atmosphere. The reaction mixture was stirred at 60 °C for 6 hr. The reaction mixture was cooled to room temperature and concentrated under reduced pressure. The residue was diluted with EtOAc (25 mL) and washed with water (10 mL). The organic layer was separated, washed with brine (2×20 mL), dried over anhydrous Na
2SO
4 and concentrated under reduced pressure. The residue was subjected to HPLC (2-10 min, 30-90% ACN; flow: 30 mL/min, column: Chromatorex, 100×19 mm, 5 µm) to afford 2-(4- cyclopropyl-6-methoxy-pyrimidin-5-yl)-5-isopropoxy-4-[[4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidine (70.0 mg, 130 μmol, 22.1% yield) as a light-yellow solid.
1H NMR (600 MHz, DMSO-d6) δ 0.84 – 0.88 (m, 2H), 1.00 – 1.04 (m, 2H), 1.34 (d, 6H), 1.69 – 1.75 (m, 1H), 3.77 (s, 3H), 3.84 (s, 3H), 4.75 – 4.81 (m, 1H), 5.51 (s, 2H), 7.58 (d, 2H), 7.74 (d, 2H), 7.94 (s, 1H), 8.44 (s, 1H), 8.65 (s, 1H). MS (ESI): [M+H]+ m/z: calcd 541.25; found 541.2.
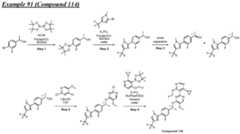
Step 1: The synthesis of 1-[3-fluoro-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2- yl)phenyl]ethanol [1273] 1-(4-bromo-3-fluoro-phenyl)ethanol (5.40 g, 24.7 mmol) was added to a mixture of 4,4,5,5-tetramethyl-2-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1,3,2-dioxaborolane (6.89 g, 27.1 mmol), potassium acetate (4.84 g, 49.3 mmol) and bis(diphenylphosphino)ferrocene]dichloropalladium(II)-DCM (1.01 g, 1.23 mmol) in degassed dioxane (60 mL). The resulting mixture was stirred at 100 °C for 4 hr. under argon atmosphere. The reaction mixture was cooled to room temperature and filtered. The filtrate was concentrated under reduced pressure to afford 1-[3-fluoro-4-(4,4,5,5-tetramethyl-1,3,2- dioxaborolan-2-yl)phenyl]ethanol (8.00 g, crude) as a brown oil which was used in the next steps without further purification. MS (ESI): [M+H]+ m/z: calcd 267.16; found 267.0. Step 2: The synthesis of 1-[3-fluoro-4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]ethanol [1274] 1-[3-fluoro-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl]ethanol (1.10 g, 4.13 mmol), 2-bromo-1-methyl-4-(trifluoromethyl)imidazole (947 mg, 4.13 mmol), potassium phosphate tribasic (1.75 g, 8.27 mmol) and bis(diphenylphosphino)ferrocene]dichloropalladium(II)-DCM (169 mg, 207 μmol) were mixed in a degassed mixture of dioxane (9 mL) and water (1 mL) under argon atmosphere. The reaction mixture was stirred at 95 °C for 16 hr. The reaction mixture was cooled to room temperature and filtered. The filtrate was concentrated under reduced pressure. The residue was subjected to flash column chromatography (SiO
2, gradient hexane - EtOAc) to afford 1- [3-fluoro-4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]ethanol (300 mg, 1.04 mmol, 25.2% yield) as a colorless oil.
1H NMR (400 MHz, CDCl
3) δ 1.49 (d, 3H), 3.64 (s, 3H), 4.94 (q, 1H), 7.18 – 7.26 (m, 2H), 7.35 (s, 1H), 7.49 – 7.57 (m, 1H). MS (ESI): [M+H]+ m/z: calcd 289.10; found 289.0. Step 3: Chiral resolution of rel-(1R)-1-[3-fluoro-4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]ethanol and rel-(1S)-1-[3-fluoro-4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]ethanol [1275] Racemic 1-[3-fluoro-4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]ethanol (300 mg, 1.04 mmol) was subjected to chiral HPLC (column: CHIRALCEL OJ-H (250×20 mm, 5 μm), Mobile Phase: Hexane-IPA-MeOH, 80-10-10, Flow Rate: 20 mL/min) to afford rel-(1S)-1-[3-fluoro-4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]ethanol (140 mg, 486 μmol, 46.7% yield) and rel-(1R)-1-[3-fluoro-4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]ethanol (140 mg, 486 μmol, 46.7% yield) as a colorless oil. rel-(1S)-1-[3-fluoro-4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]ethanol: Enantiopurity: 100% (column Chiralcel OJ-H, 250 × 4.6 mm, 5 μm; mobile phase: Hexane- IPA-MeOH, 80-10-10; flow: 0.6 mL/min; RT=14.7 min). rel-(1R)-1-[3-fluoro-4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]ethanol: Enantiopurity: 99.9% (column Chiralcel OJ-H, 250 × 4.6 mm, 5 μm; mobile phase: Hexane- IPA-MeOH, 80-10-10; flow: 0.6 mL/min). Step 4: The synthesis of 2-chloro-5-methoxy-4-[rel-(1R)-1-[3-fluoro-4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]ethoxy]pyrimidine rel-(1R)-1-[3-fluoro-4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]ethanol (50.0 mg, 174 μmol) and potassium tert-butoxide (27.3 mg, 243 μmol) were mixed in THF (2.0 mL) under argon atmosphere. The solution was stirred at room temperature for 2 hr.2,4-dichloro- 5-methoxy-pyrimidine (40.4 mg, 226 μmol) was added at -20 °C. The reaction mixture was stirred at room temperature for 16 hr. The reaction mixture was concentrated under reduced pressure. The residue was diluted with MTBE (30 mL) and washed with water (15 mL). The organic layer was separated, washed with brine (20 mL), dried over anhydrous Na
2SO
4 and concentrated under reduced pressure to afford 2-chloro-5-methoxy-4-[rel-(1R)-1-[3-fluoro-4- [1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]ethoxy]pyrimidine (70.0 mg, 163 μmol, 93.7% yield) as a white solid which was used in the next steps without further purification. MS (ESI): [M+H]+ m/z: calcd 431.09; found 431.1. Step 5: The synthesis of 2-[4-cyclopropyl-6-(fluoromethoxy)pyrimidin-5-yl]-5-methoxy-4- [rel-(1R)-1-[3-fluoro-4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]ethoxy]pyrimidine The synthesis of the starting 4-cyclopropyl-6-(fluoromethoxy)-5-(4,4,5,5-tetramethyl-1,3,2- dioxaborolan-2-yl)pyrimidine was described in Example 45 (Compound 40). [1276] 2-chloro-5-methoxy-4-[rel-(1R)-1-[3-fluoro-4-[1-methyl-4-(trifluoromethyl)imidazol- 2-yl]phenyl]ethoxy]pyrimidine (65.0 mg, 151 μmol), 4-cyclopropyl-6-(fluoromethoxy)-5- (4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyrimidine (88.8 mg, 302 μmol), potassium phosphate tribasic anhydrous (96.1 mg, 453 μmol), RuPhos Pd G3 (6.41 mg, 7.54 μmol) were mixed in a degassed mixture of dioxane (2 mL) and water (200 µL) under argon atmosphere. The reaction mixture was stirred at room temperature for 20 hr. The mixture was filtered. The filtrate was subjected to HPLC (2-10 min, 40-65% ACN; flow: 30 mL/min, column: Chromatorex C18, 100 x 19 mm, 5 µm) to afford 2-[4-cyclopropyl-6- (fluoromethoxy)pyrimidin-5-yl]-5-methoxy-4-[rel-(1R)-1-[3-fluoro-4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]ethoxy]pyrimidine (16.0 mg, 28.4 μmol, 18.9% yield) as a yellow solid.
1H NMR (600 MHz, DMSO-d6) δ 0.80 – 0.86 (m, 1H), 0.88 – 0.94 (m, 1H), 1.00 – 1.08 (m, 2H), 1.63 – 1.70 (m, 4H), 3.59 (s, 3H), 3.99 (s, 3H), 5.91 – 6.08 (m, 2H), 6.32 (q, 1H), 7.41 (d, 1H), 7.46 (d, 1H), 7.59 (t, 1H), 8.00 (s, 1H), 8.46 (s, 1H), 8.73 (s, 1H). MS (ESI): [M+H]+ m/z: calcd 563.21; found 563.2.
Step 1: The synthesis of 5-bromo-2-chloro-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidine [1277] The [4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methanol I-2b (2.00 g, 7.81 mmol), 5-bromo-2,4-dichloro-pyrimidine (1.78 g, 7.81 mmol) and Cs
2CO
3 (3.81 g, 11.7 mmol) were mixed in CH3CN (40 mL). The resulting mixture was stirred at room temperature for 16 hr. The reaction mixture was filtered. The filtrate was concentrated under reduced pressure. The residue was diluted with EtOAc (50 mL) and washed with water (20 mL × 2). The organic layer was concentrated under reduced pressure to afford 5-bromo-2- chloro-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidine (3.20 g, 7.15 mmol, 91.6% yield) as a yellow solid which was used in the next steps without further purification.
1H NMR (500 MHz, DMSO-d6) δ 3.79 (s, 3H), 5.56 (s, 2H), 7.62 (d, 2H), 7.76 (d, 2H), 7.94 (s, 1H), 8.77 (s, 1H). MS (ESI): [M+H]+ m/z: calcd 446.99; found 447.0. Step 2: The synthesis of tert-butyl 3-[2-chloro-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidin-5-yl]-2,5-dihydropyrrole-1-carboxylate [1278] 5-bromo-2-chloro-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidine (1.00 g, 1.92 mmol), tert-butyl 3-(4,4,5,5-tetramethyl-1,3,2- dioxaborolan-2-yl)-2,5-dihydropyrrole-1-carboxylate (681 mg, 2.31 mmol), Cs
2CO
3 (1.88 g, 5.76 mmol) and bis(diphenylphosphino)ferrocene]dichloropalladium(II)-DCM (70.3 mg, 96.1 μmol) were mixed in a degassed mixture of dioxane (30 mL) and water (3 mL). The reaction mixture was stirred at 90 °C for 16 hr. The reaction mixture was cooled to room temperature and diluted water (10 mL). The obtained mixture was extracted with EtOAc (2×10 mL). The combined organic layers were washed with brine (10 mL) and concentrated under reduced pressure to afford tert-butyl 3-[2-chloro-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidin-5-yl]-2,5-dihydropyrrole-1-carboxylate (1.56 g, crude) as a black solid which was used in the next steps without further purification. MS (ESI): [M+H]+ m/z: calcd 536.17; found 536.2. Step 3: Synthesis of tert-butyl 3-[2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[4-[1- methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidin-5-yl]-2,5-dihydropyrrole- 1-carboxylate [1279] Tert-butyl 3-[2-chloro-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidin-5-yl]-2,5-dihydropyrrole-1-carboxylate (720 mg, crude), (4- cyclopropyl-6-methoxy-pyrimidin-5-yl)boronic acid I-5 (131 mg, 677 μmol), potassium phosphate tribasic anhydrous (308 mg, 1.45 mmol) and XPhos Pd G3 (20.5 mg, 24.2 μmol) were mixed in a degassed mixture of dioxane (18 mL) and water (2 mL). The reaction mixture was stirred at 60 °C for 12 hr . The reaction mixture was cooled to room temperature. The reaction mixture was diluted with EtOAc (20 mL) and washed with water (10 mL) and brine (10 mL). To the obtained organic phase SiliaMetS® Dimercaptotriazine (20 mg) was added, and the resulting mixture was stirred for 30 min. The mixture was filtered through a short pad of silicagel. The filtrate was concentrated under reduced pressure to afford tert- butyl 3-[2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidin-5-yl]-2,5-dihydropyrrole-1- carboxylate (104 mg) as an yellow solid which was used in the next steps without further purification. MS (ESI): [M+H]+ m/z: calcd 650.27; found 650.2. Step 4: The synthesis of 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-5-(2,5-dihydro-1H- pyrrol-3-yl)-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidine [1280] Trifluoroacetic acid (42.0 mg, 368 μmol) was added dropwise to a solution of tert- butyl 3-[2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidin-5-yl]-2,5-dihydropyrrole-1- carboxylate (104 mg, crude) in DCM (1 mL) at 0°C. The resulting mixture was stirred at room temperature for 24 hr. The reaction mixture was diluted DCM (5 mL) and aqueous NH4OH solution (5 mL, 25% wt.). The organic layer was separated and concentrated under reduced pressure. The residue was subjected to HPLC (0.5-6.5 min, 30-80% water - ACN, +0.1% vol. of TFA; flow: 30 mL/min, column: SunFire C18, 100 x 19 mm, 5 µm) to afford 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-5-(2,5-dihydro-1H-pyrrol-3-yl)-4-[[4-[1- methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidine (6.00 mg, 10.9 μmol, 1.54% yield from 5-bromo-2-chloro-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidine, TFA-salt) as a colorless gum.
1H NMR (600 MHz, DMSO- d6) δ 0.84 – 0.89 (m, 2H), 1.02 – 1.08 (m, 2H), 1.72 – 1.77 (m, 1H), 3.77 (s, 3H), 3.84 (s, 3H), 4.15 – 4.19 (m, 2H), 4.42 – 4.47 (m, 2H), 5.61 (s, 2H), 6.67 (s, 1H), 7.60 (d, 2H), 7.74 (d, 2H), 7.94 (s, 1H), 8.68 (s, 1H) 8.76 (s, 1H), 9.28 (br., 2H). MS (ESI): [M+H]+ m/z: calcd 550.25; found 550.2. Example 93 (Compound 78)

The starting material [4-[1-isopropyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methanol was prepared as described for Compound 3 Synthesis of 4-cyclopropyl-5-[4-[[4-[1-isopropyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidin-2-yl]-6-methoxy-pyrimidine [1281] NaH (16.9 mg, 422 μmol, 60% dispersion in mineral oil) was added to a solution of [4-[1-isopropyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methanol (100 mg, 352 μmol) in DMF (0.5 mL) under argon atmosphere. The reaction mixture was stirred at ambient temperature for 1 hr. and then 4-cyclopropyl-6-methoxy-5-(4-methylsulfonylpyrimidin-2- yl)pyrimidine I-8 (108 mg, 352 μmol) was added in one portion. The resulting mixture was stirred at ambient temperature for 48 hr. The reaction mixture was diluted with EtOAc (20 mL) and brine (20 mL). The organic layer was separated, washed with brine (2 × 15 mL) and dried over anhydrous Na
2SO
4. To the obtained solution SiliaMetS® Dimercaptotriazine (100 mg) was added. The mixture was stirred for 30 min. The mixture was filtered. The filtrate was concentrated under reduced pressure. The residue was subjected to HPLC (2-10 min, 40- 65% water – MeOH, +0.1% vol. of 25% aq. NH
3, flow: 30 mL/min, column: YMC-Actus Triart C18100×20 mm, 5 µM ) to afford 4-cyclopropyl-5-[4-[[4-[1-isopropyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidin-2-yl]-6-methoxy-pyrimidine (80.0 mg, 157 μmol, 44.6% yield) as a white solid.
1H NMR (600 MHz, DMSO-d6) δ 0.83 – 0.89 (m, 2H), 1.00 – 1.05 (m, 2H), 1.39 (d, 6H), 1.64 – 1.70 (m, 1H), 3.84 (s, 3H), 4.42 – 4.50 (m, 1H), 5.50 (s, 2H), 7.06 (d, 1H), 7.55 – 7.62 (m, 4H), 8.17 (s, 1H), 8.66 (s, 1H), 8.70 (d, 1H). MS (ESI): [M+H]+ m/z: calcd 511.24; found 511.4. Example 94 (Compound 126)

Compound 126 Synthesis of 4-cyclopropyl-6-(fluoromethoxy)-5-[4-[[4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidin-2-yl]pyrimidine [1282] 4-cyclopropyl-6-(fluoromethoxy)-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2- yl)pyrimidine (340 mg, 1.16 mmol), 2-chloro-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidine (142 mg, 385 μmol), potassium phosphate tribasic (286 mg, 1.35 mmol) and XPhos Pd G3 (19.6 mg, 23.1 μmol) were mixed in degassed dioxane (6.3 mL) and water (0.7 mL) under argon atmosphere. The reaction mixture was stirred at 20°C for 96 hr. The reaction mixture was filtered and the filtrate was subjected to HPLC (2-10 min, 30-55% ACN - water flow: 30 mL/min, column: SunFire C18, 100 ×19 mm, 5 µm) to afford 4-cyclopropyl-6-(fluoromethoxy)-5-[4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidin-2-yl]pyrimidine (65.0 mg, 130 μmol, 33.7% yield) as an yellow solid.
1H NMR (600 MHz, DMSO-d6) δ 0.91 – 0.96 (m, 2H), 1.06 – 1.09 (m, 2H), 1.74 – 1.80 (m, 1H), 3.77 (s, 3H), 5.49 (s, 2H), 6.05 (d, 2H, CH
2F), 7.10 (d, 1H), 7.57 (d, 2H), 7.72 (d, 2H), 7.92 (s, 1H), 8.73 (d, 1H), 8.76 (s, 1H). MS (ESI): [M+H]
+ m/z: calcd 501.19; found 501.2.
Compound 20 Step 1: The synthesis of methyl 4-[1-(2,2,2-trifluoroethyl)-4-(trifluoromethyl)imidazol-2- yl]benzoate [1283] NaH (100 mg, 4.16 mmol, 60% dispersion in mineral oil) was added to a stirred suspension of methyl 4-[4-(trifluoromethyl)-1H-imidazol-2-yl]benzoate I-3a (1.07 g, 3.95 mmol) in DMF (30.0 mL). The reaction mixture was stirred at room temperature for 1 hr. 2,2,2-Trifluoroethyl trifluoromethanesulfonate (1.10 g, 4.74 mmol) was added to the reaction mixture. The reaction mixture was stirred at room temperature for 12 hr. The reaction mixture was diluted with water (50.0 mL) and extracted with EtOAc (100 mL). The organic layer was washed with water (50 mL) and brine (50 mL), dried over anhydrous Na
2SO
4 and concentrated under reduced pressure to afford 4-[1-(2,2,2-trifluoroethyl)-4- (trifluoromethyl)imidazol-2-yl]benzoate (2.00 g, crude) as a yellow oil which was used in the next steps without further purification. MS (ESI): [M+H]+ m/z: calcd 353.09; found 353.0. Step 2: The synthesis of [4-[1-(2,2,2-trifluoroethyl)-4-(trifluoromethyl)imidazol-2- yl]phenyl]methanol [1284] A solution of 4-[1-(2,2,2-trifluoroethyl)-4-(trifluoromethyl)imidazol-2-yl]benzoate (2.00 g, crude) in THF (10 mL) was added dropwise to a vigorously stirred suspension of LAH (290 mg, 7.63 μmol) in THF (50 mL) under argon atmosphere at 0 °C. The reaction mixture was stirred at 0 °C for 2 hr. The mixture was quenched by dropwise addition of aqueous NaOH (5.00 mL, 10% wt.). The resulting mixture was stirred at room temperature for 30 min and then solids were filtered out. The filtrate was concentrated under reduced pressure. The residue was subjected to flash-column chromatography (SiO
2, gradient hexane- ethyl acetate) to afford [4-[1-(2,2,2-trifluoroethyl)-4-(trifluoromethyl)imidazol-2- yl]phenyl]methanol (0.47 g, 1.45 mmol, 36.7% yield from methyl 4-[4-(trifluoromethyl)-1H- imidazol-2-yl]benzoate) as a light-yellow oil. MS (ESI): [M+H]+ m/z: calcd 325.23; found 325.2. Step 3: The synthesis of 2-chloro-5-methyl-4-[[4-[1-(2,2,2-trifluoroethyl)-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidine [1285] Potassium tert-butoxide (69.8 mg, 622 μmol) was added dropwise to a solution of 2,4- dichloro-5-methyl-pyrimidine (101 mg, 622 μmol) and [4-[1-(2,2,2-trifluoroethyl)-4- (trifluoromethyl)imidazol-2-yl]phenyl]methanol (210 mg, 622 μmol,) in toluene (10.0 mL). The reaction mixture was stirred at room temperature for 12 hr. The reaction mixture was diluted with water (10 mL) and extracted with EtOAc (10 mL). The organic layer was separated and washed with brine (5 mL). The organic layer was dried over anhydrous Na
2SO
4 and concentrated under reduced pressure to afford 2-chloro-5-methyl-4-[[4-[1-(2,2,2- trifluoroethyl)-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidine (250 mg, 555 µmol, 89.2% yield) as a red gum which was used in the next steps without further purification. MS (ESI): [M+H]+ m/z: calcd 451.10; found 451.0. Step 4: The synthesis of 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-5-methyl-4-[[4-[1- (2,2,2-trifluoroethyl)-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidine [1286] 2-Chloro-5-methyl-4-[[4-[1-(2,2,2-trifluoroethyl)-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidine (250 mg, 438 μmol), (4-cyclopropyl-6-methoxy-pyrimidin-5- yl)boronic acid I-5 (128 mg, 657 μmol), XPhos Pd G3 (21.9 μmol) and potassium phosphate tribasic (279 mg, 1.31 mmol) were mixed in degassed dioxane (5 mL) under argon atmosphere at room temperature. The reaction mixture was stirred at 90 °C for 48 hr. The reaction mixture was cooled to room temperature and concentrated under reduced pressure. The residue was diluted with water (5 mL) and extracted with EtOAc (5 mL). The organic layer was dried over anhydrous Na
2SO
4 and concentrated under reduced pressure. The residue was subjected to HPLC (0.5 - 6.5 min, 51% water - ACN; flow: 30 mL/min, column: Waters SunFire C18, 100 x 19 mm, 5 µm) to give a residue which was purified by HPLC (column: ChiralpakAD-HV, 250 x 20 mm, 5 µm; eluent: Hexane-IPA-MeOH 90-5-5; flow: 12 mL/min,) to afford 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-5-methyl-4-[[4-[1-(2,2,2- trifluoroethyl)-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidine (7.00 mg, 12.4 μmol, 2.8% yield) as a light-yellow solid.
1H NMR (600 MHz, DMSO-d6) δ 0.82 – 0.86 (m, 2H), 0.98 – 1.02 (m, 2H), 1.64 – 1.70 (m, 1H), 2.22 (s, 3H), 3.82 (s, 3H), 5.08 – 5.14 (m, 2H), 5.53 (s, 2H), 7.60 (d, 2H), 7.65 (d, 2H), 8.06 (s, 1H), 8.55 (s, 1H), 8.64 (s, 1H). MS (ESI): [M+H]+m/z: calcd 565.21; found 565.0.
Step 1: Synthesis of 6-[5-methoxy-3-(trifluoromethyl)pyrazol-1-yl]pyridine-3-carbonitrile [1287] Potassium tert-butoxide (3.65 g, 32.5 mmol) was added to a solution of 5-methoxy-3- (trifluoromethyl)-1H-pyrazole (5.40 g, 32.5 mmol) in DMF (30 mL) under argon atmosphere. The reaction mixture was stirred at ambient temperature for 1 hr.6-Fluoropyridine-3- carbonitrile (3.97 g, 32.5 mmol) was added to the mixture. The resulting mixture was stirred at 60°C for 6 hr. The reaction mixture was cooled to room temperature and diluted with EtOAc (200 mL) and water (50 mL). The organic layer was separated, washed with brine (2 × 100 mL), dried over anhydrous Na
2SO
4 and concentrated under reduced pressure to afford 6-[5-methoxy-3-(trifluoromethyl)pyrazol-1-yl]pyridine-3-carbonitrile (7.00 g, 26.1 mmol, 80.3% yield) as a yellow solid which was used in the next steps without further purification.
1H NMR (600 MHz, DMSO-d6) δ 4.02 (s, 3H), 6.54 (s, 1H), 7.89 – 7.95 (m, 2H), 8.49 (dd, 1H), 9.02 (d, 1H). MS (ESI): [M+H]+ m/z: calcd 269.07; found 269.0. Step 2: The synthesis of 6-[5-methoxy-3-(trifluoromethyl)pyrazol-1-yl]pyridine-3-carboxylic acid [1288] A solution of sodium hydroxide (2.98 g, 74.6 mmol) in water (10 mL) was added to a solution of 6-[5-methoxy-3-(trifluoromethyl)pyrazol-1-yl]pyridine-3-carbonitrile (5.00 g, 18.6 mmol) in ethanol (50 mL). The reaction mixture was stirred at 80 °C for 48 hr. The reaction mixture was cooled to room temperature, diluted with water (10 mL) and concentrated under reduced pressure to half volume. This residue was acidified with saturated solution of citric acid to pH≈5. The resulting mixture was extracted with EtOAc (2×20 mL). The combined organic layers were washed with water (10 mL), dried over anhydrous Na
2SO
4 and concentrated under reduced pressure to afford 6-[5-methoxy-3-(trifluoromethyl)pyrazol- 1-yl]pyridine-3-carboxylic acid (4.50 g, 15.7 mmol, 84.1% yield) as a yellow solid which was used in the next steps without further purification. MS (ESI): [M+H]+ m/z: calcd 288.06; found 288.0 Step 3: Synthesis of 2-[5-(hydroxymethyl)-2-pyridyl]-5-(trifluoromethyl)pyrazol-3-ol [1289] Borane dimethyl sulfide complex (3.57 g, 47.0 mmol) was added dropwise to a stirred solution of 6-[5-methoxy-3-(trifluoromethyl)pyrazol-1-yl]pyridine-3-carboxylic acid (4.50 g, 15.7 mmol) in THF (100 mL) under argon atmosphere. The reaction mixture was stirred at room temperature for 12 hr. The mixture was carefully quenched by dropwise addition of water (10 mL) under argon atmosphere. The resulting mixture was stirred at room temperature for 3 hr. The resulting mixture was concentrated under reduced pressure to half volume and extracted with EtOAc (2 × 50 mL). The combined organic layers were dried over anhydrous Na
2SO
4 and concentrated under reduced pressure. The residue was subjected to flash column chromatography (SiO
2, gradient hexane - EtOAc) to afford 2-[5- (hydroxymethyl)-2-pyridyl]-5-(trifluoromethyl)pyrazol-3-ol (0.350 g, 1.35 mmol, 8.62% yield) as light-yellow oil.
1H NMR (400 MHz, DMSO-d6) δ 4.59 (s, 2H), 5.46 (br., 1H), 6.02 (s, 1H), 7.73 (d, 1H), 7.99 (d, 1H), 8.46 (s, 1H). MS (ESI): [M+H]+ m/z: calcd 260.07; found 260.0. Step 4: Synthesis of [6-[5-methoxy-3-(trifluoromethyl)pyrazol-1-yl]-3-pyridyl]methanol [1290] Methyl iodide (211 mg, 1.49 mmol) was added dropwise to a mixture of 2-[5- (hydroxymethyl)-2-pyridyl]-5-(trifluoromethyl)pyrazol-3-ol (0.350 g, 1.35 mmol) and K
2CO
3 (280 mg, 2.03 mmol) in DMF (5 mL). The resulting mixture was stirred at ambient temperature for 12 hr. The reaction mixture was diluted with EtOAc (20 mL) and brine (10 mL). The organic layer was separated, washed with brine (2 × 10 mL) and concentrated under reduced pressure to afford [6-[5-methoxy-3-(trifluoromethyl)pyrazol-1-yl]-3- pyridyl]methanol (0.200 g, 732 μmol, 54.2% yield) as a yellow oil which was used in the next steps without further purification. MS (ESI): [M+H]+ m/z: calcd 274.08; found 274.2. Step 5: Synthesis of 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-5-methoxy-4-[[6-[5- methoxy-3-(trifluoromethyl)pyrazol-1-yl]-3-pyridyl]methoxy]pyrimidine [1291] NaH (29.6 mg, 739 μmol, 60% dispersion in mineral oil) was added to a solution of [6-[5-methoxy-3-(trifluoromethyl)pyrazol-1-yl]-3-pyridyl]methanol (0.200 g, 732 μmol) in DMF (5 mL) under argon atmosphere. The reaction mixture was stirred at ambient temperature for 1 hr.2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-5-methoxy-4- methylsulfonyl-pyrimidine I-6 (246 mg, 732 μmol) was added to the mixture. The resulting mixture was stirred at ambient temperature for 6 hr. The reaction mixture was diluted with EtOAc (20 mL) and brine (10 mL). The organic layer was separated, washed with brine (2 × 10 mL), dried over anhydrous Na
2SO
4 and concentrated under reduced pressure. The residue was subjected to HPLC (0.5-6.5 min, 42-57% water - ACN; flow: 30 mL/min, column: SunFire C18, 100 x 19 mm, 5 µm) to afford 4-[[2-chloro-4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]-2-(4-cyclopropyl-6-methoxy-pyrimidin-5- yl)-5-methoxy-pyrimidine (40.0 mg, 73.1 μmol, 26.5% yield) as a white solid.
1H NMR (600 MHz, DMSO-d6) δ 0.85 – 0.89 (m, 2H), 0.99 – 1.05 (m, 2H), 1.64 – 1.70 (m, 1H), 3.82 (s, 3H), 3.93 (s, 3H), 3.97 (s, 3H), 5.52 (s, 2H), 6.47 (s, 1H), 7.69 (d, 1H), 8.08 (d, 1H), 8.42 (s, 1H), 8.63 – 8.65 (m, 2H). MS (ESI): [M+H]+ m/z: calcd 530.19; found 530.2. Example 97 (Compound 99)
Step 1: 2-chloro-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]-5- (trifluoromethyl)pyrimidine & 4-chloro-2-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]-5-(trifluoromethyl)pyrimidine [1292] To a solution of [4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methanol I-2b (389.69 mg, 1.52 mmol) in THF (12 mL) was added NaH (60% dispersion in mineral oil, 58.28 mg, 1.52 mmol) at 0 °C. The resulted reaction was stirred at 15 °C for 1 hour. The above resulted solution was added dropwise 2,4-dichloro-5-(trifluoromethyl)pyrimidine (300 mg, 1.38 mmol) in THF (2 mL). The reaction mixture was stirred at 15 °C for 2 hrs. The resulted solution was quenched by saturated NH
4Cl aqueous solution (100 mL), extracted by EtOAc (3 x 50 mL). The combined organic layers were dried over anhydrous Na
2SO
4. After filtration, the filtrate was concentrated under reduced pressure. The residue was purified by Prep-TLC (PE : EtOAc = 2 : 1) to give two isomers. The less polar isomer fractions was concentrated under reduced pressure to give 2-chloro-4-[[4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]-5-(trifluoromethyl)pyrimidine (150 mg, 343.46 µmol, 24% yield) as a white solid. MS: m/z = 437.15 [M + H]+.
1H NMR (400 MHz, Chloroform-d) δ 8.63 (s, 1H), 7.79 - 7.72 (m, 2H), 7.64 - 7.57 (m, 2H), 7.38 (s, 1H), 5.67 (s, 2H), 3.85 (s, 3H). The more polar isomer fraction was concentrated under reduced pressure to give 4-chloro-2- [[4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]-5- (trifluoromethyl)pyrimidine (310 mg, 709.81 μmol, 51% yield) as a white solid. MS: m/z = 437.15 [M + H]+.
1H NMR (400 MHz, Chloroform-d) δ 8.76 (s, 1H), 7.70 (d, J = 7.6 Hz, 2H), 7.62 (d, J = 8.0 Hz, 2H), 7.36 (s, 1H), 5.59 (s, 2H), 3.81 (s, 3H). Step 2: 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]-5-(trifluoromethyl)pyrimidine [1293] To a solution of 2-chloro-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]-5-(trifluoromethyl)pyrimidine (130 mg, 297.66 µmol) in 1,4-dioxane (4 mL) and water (0.8 mL) were added (4-cyclopropyl-6-methoxy-pyrimidin-5-yl)boronic acid I-5 (57.74 mg, 297.66 µmol), bis(triphenylphosphine)palladium (II) chloride (20.89 mg, 29.77 µmol) and Cs
2CO
3 (193.97 mg, 595.32 µmol) at 15 °C. The resulted reaction was stirred at 90 °C for 16 hrs. The mixture solution was filtrated. The filtrate was concentrated under reduced pressure. The residue was purified by Prep-TLC (DCM : EtOAc = 8 : 1) to give 45 mg crude product. The crude product was further purified by RP-Flash chromatography with the following conditions: Column: C18 spherical, 20-35 um, 100A, 20 g; Mobile Phase A: 10 mM aq. ammonium hydrogen carbonate, Mobile Phase B: acetonitrile; Flow rate: 40 mL/min; Gradient: 5% B to 5% B in 5 min, 5% B to 40% B in 5 min, 40% B to 60% B in 25 min, 60% B to 95% B in 5 min; Detector: UV 254 & 220 nm; to give 2-(4- cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]-5-(trifluoromethyl)pyrimidine (17.3 mg, 31.43 µmol, 10% yield) as a white solid. MS: m/z = 551.30 [M + H]+.
1H NMR (400 MHz, Chloroform-d) δ 8.91 (s, 1H), 8.78 (s, 1H), 7.70 (d, J = 8.0 Hz, 2H), 7.57 (d, J = 8.0 Hz, 2H), 7.35 (s, 1H), 5.67 (s, 2H), 4.00 (s, 3H), 3.82 (s, 3H), 1.78 - 1.72 (m, 1H), 1.44 - 1.40 (s, 2H), 1.08 - 1.01 (s, 2H).
19F NMR (376 MHz, Chloroform-d) δ -62.77, -63.43.
Step 1: The synthesis of methyl 3-fluoro-4-[5-methyl-3-(trifluoromethyl)pyrazol-1- yl]benzoate [1294] Acetic acid (359 mg, 5.97 mmol, 342 μL) and 1,1,1-trifluoropentane-2,4-dione (837 mg, 5.43 mmol, 657 μL) were added to a stirred solution of methyl 3-fluoro-4-hydrazino- benzoate (1.00 g, 5.43 mmol) in EtOH (50 mL) at room temperature. The reaction mixture was stirred at room temperature for 16 hr. The reaction mixture was concentrated under reduced pressure. The residue was diluted with DCM (80 mL) and washed with brine (50 mL). The organic layer was dried over anhydrous Na
2SO
4 and filtered. The filtrate was concentrated under reduced pressure. The residue was subjected to column chromatography (SiO
2, gradient hexane - MTBE) to afford methyl 3-fluoro-4-[5-methyl-3- (trifluoromethyl)pyrazol-1-yl]benzoate (460 mg, 1.52 mmol, 28.0% yield) as a light-yellow solid.
1H NMR (500 MHz, CDCl
3) δ 2.27 (s, 3H), 3.97 (s, 3H), 6.49 (s, 1H), 7.58 (t, 1H), 7.93 (d, 1H), 7.98 (d, 1H). MS (ESI): [M+H]+ m/z: calcd 303.08; found 303.0. Step 2: The synthesis of [3-fluoro-4-[5-methyl-3-(trifluoromethyl)pyrazol-1- yl]phenyl]methanol A solution of methyl 3-fluoro-4-[5-methyl-3-(trifluoromethyl)pyrazol-1-yl]benzoate (468 mg, 1.55 mmol) in THF (10 mL) was added dropwise to a vigorously stirred suspension of lithium aluminium hydride (118 mg, 3.10 mmol) in THF (25 mL) at -10°C under argon atmosphere. The reaction mixture was allowed to warm to room temperature and stirred at room temperature overnight. The reaction mixture was quenched by dropwise addition of water (200 µL) in THF (5.0 mL) at 0 °C and stirred for 15 min. K
2CO
3 (500 mg) and anhydrous Na
2SO
4 (1.00 g) were added to the mixture. The resulting mixture was stirred at room temperature for 10 min and filtered. The filtrate was concentrated under reduced pressure to afford [3-fluoro-4-[5-methyl-3-(trifluoromethyl)pyrazol-1-yl]phenyl]methanol (300 mg, 1.09 mmol, 70.7% yield) as a colorless oil which was used in the next step without further purification.
1H NMR (500 MHz, CDCl
3) δ 2.23 (s, 3H), 4.76 (s, 2H), 6.46 (s, 1H), 7.21 – 7.30 (m, 2H), 7.39 - 7.46 (m, 1H). MS (ESI): [M+H]+ m/z: calcd 275.08; found 275.0. Step 3: The synthesis of 4'-cyclopropyl-4-((3-fluoro-4-(5-methyl-3-(trifluoromethyl)-1H- pyrazol-1-yl)benzyl)oxy)-6'-methoxy-2,5'-bipyrimidine [1295] NaH (25.2 mg, 629 μmol, 60% dispersion in mineral oil) was added to a stirred solution of [3-fluoro-4-[5-methyl-3-(trifluoromethyl)pyrazol-1-yl]phenyl]methanol (150 mg, 547 μmol) in DMF (5.0 mL). The reaction mixture was stirred at room temperature for 30 min.4-cyclopropyl-6-methoxy-5-(4-methylsulfonylpyrimidin-2-yl)pyrimidine I-8 (168 mg, 547 μmol) was added to the reaction mixture. The resulting mixture was stirred at room temperature for 16 hr. The reaction mixture was diluted with a saturated aqueous solution of NH
4Cl (10 mL) and extracted with EtOAc (3×20 mL). The combined organic layers were dried over anhydrous Na
2SO
4 and filtered. The filtrate was concentrated under reduced pressure. The residue was subjected to HPLC (0-5 min., 40-65% water – ACN, +0.1% vol. of 25% aq. NH
3, flow: 30 mL/min, column: XBridge BEH C18100 ×19 mm, 5 µm), then repurified by HPLC (0-5 min., 50-55% water – ACN, +0.1% vol. of 25% aq. NH
3, flow: 30 mL/min, column: XBridge BEH C18100 ×19 mm, 5 µm) to afford 4-cyclopropyl-5-[4-[[2- fluoro-4-[5-methyl-3-(trifluoromethyl)pyrazol-1-yl]phenyl]methoxy]pyrimidin-2-yl]-6- methoxy-pyrimidine (39.4 mg, 78.7 μmol, 14.4% yield) as an off-white solid.
1H NMR (400 MHz, DMSO-d6) δ 0.87 – 0.91 (m, 2H), 1.02 – 1.06 (m, 2H), 1.65 – 1.71 (m, 1H), 2.21 (s, 3H), 3.85 (s, 3H), 5.53 (s, 2H), 6.80 (s, 1H), 7.12 (d, 1H), 7.50 (d, 1H), 7.59 – 7.69 (m, 2H), 8.68 (s, 1H), 8.74 (d, 1H). MS (ESI): [M+H]+ m/z: calcd 501.19; found 501.2. Example 99 (Compound 191)
Compound 191 Step 1: The synthesis of 5-bromo-2-chloro-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidine [1296] 5-Bromo-2,4-dichloro-pyrimidine (1.78 g, 7.81 mmol) was added to a stirred mixture of [4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methanol I-2b (2.00 g, 7.81 mmol) and Cs
2CO
3 (3.81 g, 11.7 mmol) in ACN (40 mL). The resulting mixture was stirred at 60 °C for 16 hr. The mixture was cooled to room temperature and concentrated under reduced pressure. The residue was diluted with EtOAc (50 mL), washed with water (2×20 mL), dried over anhydrous Na
2SO
4 and concentrated under reduced pressure to afford 5-bromo-2-chloro- 4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidine (3.20 g, 7.15 mmol, 91.6% yield) as a yellow solid.
1H NMR (500 MHz, DMSO-d6) δ 3.79 (s, 3H), 5.56 (s, 2H), 7.62 (d, 2H), 7.77 (d, 2H), 7.94 (s, 1H), 8.77 (s, 1H). MS (ESI): [M+H]+ m/z: calcd 448.98, 446.98; found 449.0, 447.0. Step 2: The synthesis of 1-[2-chloro-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidin-5-yl]ethanol [1297] Isopropyl magnesium chloride (50.6 mg, 492 μmol) in THF (1 mL) was added dropwise to a solution of 5-bromo-2-chloro-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidine (200 mg, 447 μmol) in THF (1 mL) at -30 °C. The reaction mixture was allowed to warm to 0 °C during 30 min. The mixture was cooled to -30 °C again and acetaldehyde (78.7 mg, 1.79 mmol) was added to the mixture. The resulting mixture was stirred at ambient temperature for 1 hr. The reaction mixture was quenched by addition of water (5 mL) and extracted with MTBE (3×10 mL). The combined organic layers were washed with brine (10 mL) and concentrated under reduced pressure to afford 1-[2-chloro-4- [[4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidin-5-yl]ethanol (160 mg, crude) as a yellow oil which was used in the next steps without further purification. MS (ESI): [M+H]+ m/z: calcd 413.10; found 413.0. Step 3: The synthesis of 1-[2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidin-5-yl]ethanol [1298] 1-[2-Chloro-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidin-5-yl]ethanol (160 mg, 388 μmol), (4-cyclopropyl-6-methoxy- pyrimidin-5-yl)boronic acid I-5 (190 mg, 979 μmol), potassium phosphate tribasic (247 mg, 1.16 mmol) and XPhos Pd G3 (19.7 mg, 23.3 μmol) were mixed in degassed mixture of dioxane (5.0 mL) and water (500 µL) under argon atmosphere. The reaction mixture was stirred at room temperature for 12 hr, then at 50°C for 12 hr. The reaction mixture was cooled to room temperature and filtered. The filtrate was subjected to HPLC (2-10 min, 10-40% ACN+FA (0.1% vol.); flow: 30 mL/min, column: SunFire C18, 100×19 mm, 5 µm) to afford 1-[2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol- 2-yl]phenyl]methoxy]pyrimidin-5-yl]ethanol (6.00 mg, 11.4 μmol, 2.55% yield from 5- bromo-2-chloro-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidine) as a yellow solid.
1H NMR (600 MHz, DMSO-d6) δ 0.85 – 0.89 (m, 2H), 1.01 – 1.06 (m, 2H), 1.43 (d, 3H), 1.64 – 1.69 (m, 1H), 3.79 (s, 3H), 3.84 (s, 3H), 4.97 – 5.02 (m, 2H), 5.45 (d, 1H), 5.54 (s, 2H), 7.58 (d, 2H), 7.74 (d, 2H), 7.94 (s, 1H), 8.67 (s, 1H), 8.72 (s, 1H). MS (ESI): [M+H]+ m/z: calcd 527.23; found 527.0. Example 100 (Compound 150)

Compound 150 The synthesis of starting 2-chloro-5-methoxy-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidine is described for Compound 12 and compound 132. Step 1: The synthesis of 2-(3-bromo-2-pyridyl)-2-methyl-propanoic acid [1299] A solution of sodium hydroxide (14.0 g, 349 mmol) in water (40 mL) was added to a solution of methyl 2-(3-bromo-2-pyridyl)-2-methyl-propanoate (30.0 g, 116.23 mmol) in ethanol (200 mL). The resulting mixture was stirred at 80 °C for 12 hr. The mixture was cooled too room temperature and dilute with water (200 mL). The resulting mixture was concentrated under reduced pressure to half volume. The residue was washed with MTBE (100 mL). The organic layer was discarded. Water layer was quenched with 1 M aqueous HCl to pH≈5-6. The resulting mixture was extracted with EtOAc (2×200 mL). The combined organic layers were dried over anhydrous MgS04 and concentrated under reduced pressure to afford 2-(3-bromo-2-pyridyl)-2-methyl-propanoic acid (24.0 g, 98.3 mmol, 84.6% yield) as yellow solid which was used in the next steps without further purification.
1H NMR (400 MHz, CDCl
3) δ 1.51 (s, 6H), 7.21 (dd, 1H), 8.00 (d, 1H), 8.48 (d, 1H), 12.43 (br, 1H). MS (ESI): [M+H]+ m/z: calcd 244.01; found 244.2. Step 2: The synthesis of 3-bromo-2-isopropyl-pyridine [1300] 2-(3-bromo-2-pyridyl)-2-methyl-propanoic acid (24.0 g, 98.3 mmol) was heated in neat at 130 °C until gas evolution ceased, which took approximately 1 hr. The melt was cooled to room temperature and distilled under reduce pressure (5 mmHg, b.p. approximately 65-70 °C) to afford 3-bromo-2-isopropyl-pyridine (15.0 g, 75.0 mmol, 76.3% yield) as a colorless liquid.
1H NMR (500 MHz, CDCl
3,) δ 1.29 (d, 6H), 3.51 – 3.60 (m, 1H), 6.98 (dd, 1H), 7.81 (d, 1H), 8.51 (d, 1H). Step 3: The synthesis of (2-isopropyl-3-pyridyl)boronic acid [1301] To a solution 3-bromo-2-isopropyl-pyridine (10.0 g, 50.0 mmol) and Triisopropyl borate (14.1 g, 75.0 mmol, 17.30 mL) in dry THF (200 mL) n-Butyllithium (75.0 mmol, 30 mL, 2.5 M soln. in hexane) was added dropwise at -78 °C under argon atmosphere. The reaction mixture was stirred at -78 °C for 4 hr. The mixture was quenched with water (100 mL) and warmed to room temperature. The mixture was concentrated under reduced pressure to half a volume. The residue was washed with MTBE (50 mL). The organic layer was discarded. The aqueous layer was acidified to pH 2-3 with conc. HCl and concentrated under reduced pressure to afford (2-isopropyl-3-pyridyl)boronic acid (11.0 g, crude, HCl) as a white solid which was used in the next steps without further purification. MS (ESI): [M+H]+ m/z: calcd 166.12; found 166.0. Step 4: The synthesis of 2-(2-isopropyl-3-pyridyl)-5-methoxy-4-[[4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidine [1302] 2-chloro-5-methoxy-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidine (200 mg, 256 μmol), (2-isopropyl-3-pyridyl)boronic acid (147 mg, 512 μmol, HCl) and potassium phosphate tribasic anhydrous (163 mg, 767 μmol) were mixed in a degassed mixture of dioxane (9 mL) and water (1 mL). XPhos Pd G3 (10.8 mg, 12.8 μmol) was added to the mixture. The reaction mixture was stirred at 60 °C for 4 hr. XPhos Pd G3 (10.8 mg, 12.8 μmol) and (2-isopropyl-3-pyridyl)boronic acid (147 mg, 512 μmol, HCl) were added to the mixture. The reaction mixture was stirred at 80°C for 16 hr. The reaction mixture was cooled to room temperature and diluted with EtOAc (20 mL). The resulting mixture was washed with water (2×20 mL). SiliaMetS® Dimercaptotriazine (100 mg) was added to the organic layer. The resulting mixture was stirred for 30 min and filtered. The filtrate was concentrated under reduced pressure. The residue was subjected to HPLC (0.5-6.5 min, 40-90% water - ACN, flow 30ml/min; column SunFire C18100×19 mm, 5 μm) to afford 2-(2-isopropyl-3-pyridyl)-5-methoxy-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol- 2-yl]phenyl]methoxy]pyrimidine (8.80 mg, 18.2 μmol, 7.12% yield) as a light-yellow solid.
1H NMR (600 MHz, DMSO-d6) δ 1.16 (d, 6H), 3.69 – 3.73 (m, 1H), 3.77 (s, 3H), 3.94 (s, 3H), 5.56 (s, 2H), 7.30 (dd, 1H), 7.57 (d, 2H), 7.74 (d, 2H), 7.93 (s, 1H), 7.98 (d, 1H), 8.44 (s, 1H), 8.58 (d, 1H). MS (ESI): [M+H]+ m/z: calcd 484.23; found 484.2. Example 101 (Compound 110 and Compound 235)
Step 1: The synthesis of methyl 2-[2-(4-cyanophenyl)-4-(trifluoromethyl)imidazol-1- yl]propanoate [1303] K
2CO
3 (1.75 g, 12.7 mmol) and 4-[4-(trifluoromethyl)-1H-imidazol-2-yl]benzonitrile I-1a (3.00 g, 12.7 mmol) were added to a solution of methyl 2-bromopropanoate (2.53 g, 15.2 mmol, 1.69 mL) in DMF (30 mL). The reaction mixture was stirred at 100 °C for 8 hr. The reaction mixture was cooled to room temperature and poured into ice-cold water (100 mL). The obtained mixture was extracted with EtOAc (70 mL). The organic layer was washed with brine (3×50 mL) and concentrated under reduced pressure to afford methyl 2-[2-(4- cyanophenyl)-4-(trifluoromethyl)imidazol-1-yl]propanoate (3.40 g, 10.5 mmol, 83.2% yield) as a yellow solid which was used in the next steps without further purification.
1H NMR (500 MHz, DMSO-d6) δ 1.72 (d, 3H), 3.64 (s, 3H), 5.23 (q, 1H), 7.74 (d, 2H), 7.98 (d, 2H), 8.23 (s, 1H). MS (ESI): [M+H]+ m/z: calcd 324.11; found 324.2. Step 2: The synthesis of 2-[2-(4-cyanophenyl)-4-(trifluoromethyl)imidazol-1-yl]propanoic acid A solution of lithium hydroxide monohydrate (883 mg, 21.0 mmol) in water (2.0 mL) was added to a solution of methyl 2-[2-(4-cyanophenyl)-4-(trifluoromethyl)imidazol-1- yl]propanoate (3.40 g, 10.5 mmol) in the mixture of methanol (10 mL) and THF (4 mL). The reaction mixture was stirred at room temperature for 3 hr. The reaction mixture was diluted with water (30 mL) and washed with MTBE (20 mL). The water layer was acidified by addition of 1N HCl to pH≈2 and extracted with EtOAc (3×15 mL). The combined organic layers were dried over anhydrous Na
2SO
4 and concentrated under reduced pressure to afford 2-[2-(4-cyanophenyl)-4-(trifluoromethyl)imidazol-1-yl]propanoic acid (2.05 g, 6.63 mmol, 63.0% yield) as a light-yellow solid which was used in the next steps without further purification.
1H NMR (500 MHz, DMSO-d6) δ 1.71 (d, 3H), 5.08 – 5.14 (m, 1H), 7.74 (d, 2H), 7.99 (d, 2H), 8.23 (s, 1H), 13.49 (br., 1H). MS (ESI): [M+H]+ m/z: calcd 310.09; found 310.2. Step 3: The synthesis of 4-[4-(trifluoromethyl)-1-(2,2,2-trifluoro-1-methyl-ethyl)imidazol-2- yl]benzonitrile [1304] Sulfur tetrafluoride (2.45 g, 22.6 mmol), hydrogen fluoride (1.13 g, 56.6 mmol) and 2-[2-(4-cyanophenyl)-4-(trifluoromethyl)imidazol-1-yl]propanoic acid (1.75 g, 5.66 mmol) were mixed in a stainless steel reaction vessel. The vessel was sealed, and the reaction mixture was stirred at room temperature for 8 hr. The reaction mixture was poured into saturated K
2CO
3 solution (20 mL). The resulting mixture was extracted with MTBE (3×10 mL). The combined organic layers were dried over anhydrous Na
2SO
4 and concentrated under reduced pressure to afford 4-[4-(trifluoromethyl)-1-(2,2,2-trifluoro-1-methyl- ethyl)imidazol-2-yl]benzonitrile (1.80 g, 5.40 mmol, 95.5% yield) as a yellow solid which was used in the next steps without further purification. MS (ESI): [M+H]+ m/z: calcd 334.10; found 334.0. Step 4: The synthesis of 4-[4-(trifluoromethyl)-1-(2,2,2-trifluoro-1-methyl-ethyl)imidazol-2- yl]benzoic acid [1305] A solution of potassium hydroxide (1.35 g, 24.0 mmol) in water (10 mL) was added to a solution of 4-[4-(trifluoromethyl)-1-(2,2,2-trifluoro-1-methyl-ethyl)imidazol-2- yl]benzonitrile (2.00 g, 6.00 mmol) in EtOH (50 mL). The reaction mixture was stirred at 60 °C for 18 hr. The reaction mixture was cooled to room temperature, diluted with water (50 mL) and washed with MTBE (20 mL). The water layer was acidified by addition of 1N HCl to pH≈2 and extracted with EtOAc (3×30 mL). The combined organic layers were dried over anhydrous Na
2SO
4 and concentrated under reduced pressure to afford 4-[4-(trifluoromethyl)- 1-(2,2,2-trifluoro-1-methyl-ethyl)imidazol-2-yl]benzoic acid (1.50 g, 4.26 mmol, 71.0% yield) as a yellow solid which was used in the next steps without further purification.
1H NMR (500 MHz, DMSO-d6) δ 1.77 (d, 3H), 5.23 – 5.29 (m, 1H), 7.71 (d, 2H), 8.07 (d, 2H), 8.41 (s, 1H). MS (ESI): [M+H]+ m/z: calcd 353.09; found 353.2. Step 5: The synthesis of [4-[4-(trifluoromethyl)-1-(2,2,2-trifluoro-1-methyl-ethyl)imidazol-2- yl]phenyl]methanol [1306] Borane dimethyl sulfide complex (518 mg, 6.81 mmol, 646 μL) was added dropwise to a solution of 4-[4-(trifluoromethyl)-1-(2,2,2-trifluoro-1-methyl-ethyl)imidazol-2- yl]benzoic acid (1.20 g, 3.41 mmol) in THF (100 mL) under argon atmosphere. The reaction mixture was stirred at 65 °C for 18 hr. The reaction mixture was cooled to room temperature and quenched by dropwise addition of MeOH (20 mL) under argon atmosphere. The resulting mixture was stirred at 65 °C for 3 hr. The reaction mixture was cooled to room temperature and concentrated under reduced pressure to afford [4-[4-(trifluoromethyl)-1-(2,2,2-trifluoro- 1-methyl-ethyl)imidazol-2-yl]phenyl]methanol (1.10 g, 3.25 mmol, 95.5% yield) as a light- yellow oil which was used in the next steps without further purification.
1H NMR (400 MHz, DMSO-d6) δ 1.76 (d, 3H), 4.58 (s, 2H), 5.13 – 5.20 (m, 1H), 7.48 (d, 2H), 7.53 (d, 2H), 8.34 (s, 1H). MS (ESI): [M+H]+ m/z: calcd 339.12; found 339.0. Step 6: The synthesis of 2-chloro-5-methyl-4-[[4-[4-(trifluoromethyl)-1-(2,2,2-trifluoro-1- methyl-ethyl)imidazol-2-yl]phenyl]methoxy]pyrimidine [1307] 2,4-dichloro-5-methyl-pyrimidine (627 mg, 3.84 mmol) and potassium tert-butoxide (500 mg, 4.46 mmol) were added to a solution of [4-[4-(trifluoromethyl)-1-(2,2,2-trifluoro-1- methyl-ethyl)imidazol-2-yl]phenyl]methanol (1.00 g, 2.96 mmol) in toluene (100 mL). The reaction mixture was stirred at room temperature for 40 hr. The reaction mixture was cooled to room temperature, diluted with water (200 mL) and extracted with EtOAc (3×50 mL). The combined organic layers were dried over anhydrous Na
2SO
4 and concentrated under reduced pressure. The residue was subjected to flash-column chromatography (SiO
2, hexane - EtOAc 7:3) to afford 2-chloro-5-methyl-4-[[4-[4-(trifluoromethyl)-1-(2,2,2-trifluoro-1-methyl- ethyl)imidazol-2-yl]phenyl]methoxy]pyrimidine (310 mg, 667 μmol, 22.6% yield) as a colorless oil. MS (ESI): [M+H]+ m/z: calcd 465.12; found 465.0. Step 7: The synthesis of 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-5-methyl-4-[[4-[4- (trifluoromethyl)-1-(2,2,2-trifluoro-1-methyl-ethyl)imidazol-2-yl]phenyl]methoxy]pyrimidine [1308] 2-chloro-5-methyl-4-[[4-[4-(trifluoromethyl)-1-(2,2,2-trifluoro-1-methyl- ethyl)imidazol-2-yl]phenyl]methoxy]pyrimidine (150 mg, 322.73 μmol), (4-cyclopropyl-6- methoxy-pyrimidin-5-yl)boronic acid I-5 (100 mg, 515.48 μmol), potassium phosphate tribasic (205 mg, 966 μmol) and XPhos Pd G3 (30.0 mg, 35.4 μmol) were mixed in degassed dioxane (3.0 mL) and water (100 µL). The reaction mixture was stirred at 85 °C for 14 hr. The reaction mixture was cooled to room temperature and diluted with MeOH (10 mL) To the obtained mixture SiliaMetS® Dimercaptotriazine (100 mg) was added and the resulting mixture was stirred for 8 hr. The mixture was filtered. The filtrated was concentrated under reduced pressure. The residue was subjected to HPLC (0.5-6.5 min., 40-80% water – ACN, +0.1% vol. of 25% aq. NH
3, flow: 30 mL/min, column: XBridge 100 ×19 mm, 5 µm) to afford 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-5-methyl-4-[[4-[4-(trifluoromethyl)-1- (2,2,2-trifluoro-1-methyl-ethyl)imidazol-2-yl]phenyl]methoxy]pyrimidine (75.0 mg, 130 μmol, 40.2% yield) as a white solid. MS (ESI): [M+H]+ m/z: calcd 579.23; found 579.4. Step 8: The synthesis of rel-(S)-4'-cyclopropyl-6'-methoxy-5-methyl-4-((4-(4- (trifluoromethyl)-1-(1,1,1-trifluoropropan-2-yl)-1H-imidazol-2-yl)benzyl)oxy)-2,5'- bipyrimidine (110) and rel-(R)-4'-cyclopropyl-6'-methoxy-5-methyl-4-((4-(4- (trifluoromethyl)-1-(1,1,1-trifluoropropan-2-yl)-1H-imidazol-2-yl)benzyl)oxy)-2,5'- bipyrimidine (Compound 235) [1309] Racemic 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-5-methyl-4-[[4-[4- (trifluoromethyl)-1-(2,2,2-trifluoro-1-methyl-ethyl)imidazol-2-yl]phenyl]methoxy]pyrimidine (75 mg, 129.64 μmol) was subjected to chiral HPLC (column: Chiralpak AD-H V, 250×30 mm, 5 μm; mobile phase: Hexane-IPA-MeOH, 90-5-5; flow: 12 mL/min) to afford rel-(S)-4'- cyclopropyl-6'-methoxy-5-methyl-4-((4-(4-(trifluoromethyl)-1-(1,1,1-trifluoropropan-2-yl)- 1H-imidazol-2-yl)benzyl)oxy)-2,5'-bipyrimidine (28.0 mg, 48.4 μmol, 37.3% yield) and rel- (R)-4'-cyclopropyl-6'-methoxy-5-methyl-4-((4-(4-(trifluoromethyl)-1-(1,1,1-trifluoropropan- 2-yl)-1H-imidazol-2-yl)benzyl)oxy)-2,5'-bipyrimidine (27.0 mg, 46.7 μmol, 36.0% yield) as a white solids. rel-(R)-4'-cyclopropyl-6'-methoxy-5-methyl-4-((4-(4-(trifluoromethyl)-1-(1,1,1- trifluoropropan-2-yl)-1H-imidazol-2-yl)benzyl)oxy)-2,5'-bipyrimidine (Compound 235):
1H NMR (600 MHz, DMSO-d6) δ 0.82 – 0.86 (m, 2H), 0.99 – 1.02 (m, 2H), 1.63 – 1.69 (m, 1H), 1.75 (d, 3H), 2.23 (s, 3H), 3.82 (s, 3H), 5.15 – 5.22 (m, 1H), 5.53 (s, 2H), 7.58 (d, 2H), 7.62 (d, 2H), 8.35 (s, 1H), 8.55 (s, 1H), 8.64 (s, 1H). MS (ESI): [M+H]+ m/z: calcd 579.23; found 579.2 Enantiopurity: 100% (column: Chiralpak AD-H, 250 × 4.6 mm, 5 μm; mobile phase: Hexane- IPA-MeOH, 90-5-5; flow: 0.6 mL/min; RT=28.5 min) rel-(S)-4'-cyclopropyl-6'-methoxy-5-methyl-4-((4-(4-(trifluoromethyl)-1-(1,1,1- trifluoropropan-2-yl)-1H-imidazol-2-yl)benzyl)oxy)-2,5'-bipyrimidine (110):
1H NMR (600 MHz, DMSO-d6) δ 0.82 – 0.86 (m, 2H), 0.99 – 1.02 (m, 2H), 1.63 – 1.69 (m, 1H), 1.75 (d, 3H), 2.23 (s, 3H), 3.82 (s, 3H), 5.15 – 5.22 (m, 1H), 5.53 (s, 2H), 7.58 (d, 2H), 7.62 (d, 2H), 8.35 (s, 1H), 8.55 (s, 1H), 8.64 (s, 1H). MS (ESI): [M+H]+ m/z: calcd 579.23; found 579.4. Enantiopurity: 100% (column: Chiralpak AD-H, 250 × 4.6 mm, 5 μm; mobile phase: Hexane- IPA-MeOH, 90-5-5; flow: 0.6 mL/min; RT=15.9 min). Example 102 (Compound 22)

Step 1: The synthesis of 2-chloro-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidin-5-amine [1310] Potassium tert-butoxide (1.49 g, 13.3 mmol) was added to a solution of [4-[1-methyl- 4-(trifluoromethyl)imidazol-2-yl]phenyl]methanol I-2b (3.00 g, 11.7 mmol) in THF (5 mL) at 0 °C. The reaction mixture was stirred at 0 °C for 10 min.2,4-dichloropyrimidin-5-amine (1.75 g, 10.6 mmol) was added to the mixture. The resulting mixture was stirred at room temperature for 14 hr. The reaction mixture was poured into water (10 mL) and extracted with EtOAc (90 mL). The organic layer was dried over anhydrous Na
2SO
4 and concentrated under reduced pressure. The residue was subjected to flash column chromatography (SiO
2, gradient chloroform – acetonitrile) to afford 2-chloro-4-[[4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidin-5-amine (2.50 g, 6.51 mmol, 61.2% yield) as a brown solid.
1H NMR (400 MHz, DMSO-d6) δ 3.79 (s, 3H), 5.40 (s, 2H), 5.50 (s, 2H), 7.65 (d, 2H), 7.74 (d, 2H), 7.79 (s, 1H), 7.95 (s, 1H). MS (ESI): [M+H]+ m/z: calcd 384.09; found 384.2. Step 2: The synthesis of 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidin-5-amine [1311] 2-chloro-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidin-5-amine (2.00 g, 5.21 mmol), (4-cyclopropyl-6-methoxy- pyrimidin-5-yl)boronic acid I-5 (1.52 g, 7.82 mmol), potassium phosphate tribasic anhydrous (2.77 g, 13.0 mmol) and XPhos Pd G3 (331 mg, 391 μmol) were mixed in degassed water (4 mL) and dioxane (20 mL) at room temperature under argon atmosphere. The reaction mixture was stirred at 55°C for 15 hr. The reaction mixture was cooled to room temperature and concentrated under reduced pressure. The residue was diluted with water (40 mL) and extracted with EtOAc (2×50 mL). The combined organic layers were dried over anhydrous Na
2SO
4 and concentrated under reduced pressure. The residue was subjected to flash column chromatography (SiO
2, gradient chloroform – acetonitrile) to afford 2-(4-cyclopropyl-6- methoxy-pyrimidin-5-yl)-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidin-5-amine (1.00 g, 2.01 mmol, 38.6% yield) as a yellow solid. MS (ESI): [M+H]+ m/z: calcd 498.19; found 498.2. Step 3: The synthesis of 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-N-(3-methoxypropyl)-4- [[4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidin-5-amine (22) [1312] 3-methoxypropanal (9.74 mg, 111 μmol) was added to a solution of 2-(4-cyclopropyl- 6-methoxy-pyrimidin-5-yl)-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidin-5-amine (50.0 mg, 101 μmol) in EtOH (2 mL) and AcOH (2 mL). The reaction mixture was stirred at room temperature for 3 hr. Sodium cyanoborohydride (7.90 mg, 126 μmol) was added to the reaction mixture. The resulting mixture was stirred at room temperature for 15 hr. The reaction mixture was subjected to HPLC (2-10 min., 30-60% ACN, +0.1% vol. of 25% aq. NH
3, flow: 30 mL/min, column: SunFire 100 ×19 mm, 5 µm) to afford 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-N-(3- methoxypropyl)-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidin-5-amine (24.1 mg, 42.3 μmol, 42.1% yield) as an-off-white solid.
1H NMR (600 MHz, DMSO-d6) δ 0.80 – 0.84 (m, 2H), 0.96 – 0.99 (m, 2H), 1.70 – 1.76 (m, 1H), 1.80 – 1.84 (m, 2H), 3.19 – 3.25 (m, 5H), 3.42 (t, 2H), 3.76 (s, 3H), 3.81 (s, 3H), 5.50 (s, 2H), 5.60 (t, 1H), 7.60 (d, 2H), 7.71 (d, 2H), 7.92 (s, 2H), 8.59 (s, 1H). MS (ESI): [M+H]+ m/z: calcd 570.25; found 570.4.

The synthesis of the starting 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidin-5-amine is described in Example 67 (Compound 142). Step 1: The synthesis of N-[2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidin-5-yl]-2-methoxy-acetamide [1313] 2-Methoxyacetyl chloride (24.5 mg, 226 μmol) was added to a solution of 2-(4- cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidin-5-amine (90.0 mg, 181 μmol) and TEA (32.0 mg, 317 μmol, 44.1 μL) in DCM (4 mL). The reaction mixture was stirred at room temperature for 10 hr. The reaction mixture was washed with water (3 × 3 mL). The organic layer was dried over anhydrous Na
2SO
4 and concentrated under reduced pressure to afford N-[2-(4-cyclopropyl-6- methoxy-pyrimidin-5-yl)-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidin-5-yl]-2-methoxy-acetamide (80.0 mg, 141 μmol, 77.6% yield) as a yellow solid which was used in the next step without further purification. MS (ESI): [M+H]+ m/z: calcd 570.21; found 570.0. Step 2: The synthesis of 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-N-(2-methoxyethyl)-4- [[4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidin-5-amine [1314] Borane dimethyl sulfide complex (21.3 mg, 281 μmol, 26.6 μL) was added to a solution of N-[2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidin-5-yl]-2-methoxy-acetamide (80.0 mg, 141 μmol) in THF (3 mL). The reaction mixture was stirred at 50 °C for 15 hr. The reaction mixture was cooled to room temperature and quenched by dropwise addition of MeOH (3.0 mL). The resulting mixture was stirred at 100 °C for 4 hr. The reaction mixture was cooled to room temperature and concentrated under reduced pressure. The residue was subjected to HPLC (2-10 min, 20-35% ACN+FA, flow 30ml/min; column SunFireC18 100×19mm 5μm) to afford 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-N-(2- methoxyethyl)-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidin- 5-amine (7.4 mg, 13.32 μmol, 9.5% yield) as an off-white solid.
1H NMR (DMSO-d6, 600 MHz) δ 0.80 – 0.84 (m, 2H), 0.96 – 1.00 (m, 2H), 1.69 – 1.74 (m, 1H), 3.32 – 3.38 (m, 2H), 3.53 (t, 1H), 3.77 (s, 3H), 3.81 (s, 3H), 5.43 (t, 1H), 5.51 (s, 2H), 7.60 (d, 1H), 7.71 (d, 1H), 7.92 (s, 1H), 8.01 (s, 1H), 8.59 (s, 1H). MS (ESI): [M+H]+ m/z: calcd 556.26; found 556.2.
Step 1: The synthesis of 5-fluoro-2-methoxy-4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]benzaldehyde [1315] 2-bromo-1-methyl-4-(trifluoromethyl)imidazole (245 mg, 1.07 mmol) and Na
2CO
3 (341 mg, 3.21 mmol) was added to a stirred solution of 5-fluoro-2-methoxy-4-(4,4,5,5- tetramethyl-1,3,2-dioxaborolan-2-yl)benzaldehyde (300 mg, 1.07 mmol) in a degassed mixture of dioxane (12 mL) and water (3 mL) under argon atmosphere.is(diphenylphosphino)ferrocene] dichloropalladium(II)-DCM (43.7 mg, 53.6 μmol) was added to the mixture. The reaction mixture was stirred at 90 °C for 16 hr. under argon atmosphere. The reaction mixture was cooled to room temperature and concentrated under reduced pressure. The residue was diluted with EtOAc (50 mL) and filtered. The filtrate was concentrated under reduced pressure to afford 5-fluoro-2-methoxy-4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]benzaldehyde (290 mg, 960 μmol, 89.5% yield) as a yellow solid which was used in the next steps without further purification. MS (ESI): [M+H]+ m/z: calcd 303.08; found 303.0. Step 2: The synthesis of [5-fluoro-2-methoxy-4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methanol Sodium borohydride (36.3 mg, 960 μmol) was added to a stirred solution of 5-fluoro-2- methoxy-4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]benzaldehyde (290 mg, 960 μmol) in MeOH (15 mL). The reaction mixture was stirred at room temperature for 16 hr. The reaction mixture was concentrated under reduced pressure. The residue was diluted with water (10 mL) and extracted with EtOAc (30 mL). The organic layer was separated, washed with brine (5 mL), dried over anhydrous Na
2SO
4 and filtered. The filtrate was concentrated under reduced pressure to afford [5-fluoro-2-methoxy-4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methanol (260 mg, 855 μmol, 89.0% yield) as a brown gum which was used in the next steps without further purification. MS (ESI): [M+H]+ m/z: calcd 305.09; found 305.0. Step 3: The synthesis of 4-cyclopropyl-5-[4-[[5-fluoro-2-methoxy-4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidin-2-yl]-6-methoxy-pyrimidine [1316] NaH (13.2 mg, 344 μmol, 60% dispersion in mineral oil) was added to a stirred solution of [5-fluoro-2-methoxy-4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methanol (130 mg, 299 μmol) in DMF (5 mL). The resulting mixture was stirred at room temperature for 30 min.4-cyclopropyl-6-methoxy-5-(4-methylsulfonylpyrimidin-2- yl)pyrimidine I-8 (91.6 mg, 299 μmol) was added to the mixture. The reaction mixture was stirred at room temperature for 16 hr. The reaction mixture was diluted with a saturated aqueous solution of NH
4Cl (10 mL) and extracted with EtOAc (3×20 mL). The combined organic layers were washed with brine (10 mL), dried over anhydrous Na
2SO
4 and filtered. The filtrate was concentrated under reduced pressure. The residue was subjected to HPLC (0- 5 min., 45-70% water - ACN, +0.1% vol. of 25% aq. NH
3, flow: 30 mL/min, column: XBridge BEH C18100×19 mm, 5 µm), then repurified by HPLC (0-5 min, 55-90% water+FA (0.1% vol.) - ACN+FA (0.1% vol.); flow: 30 mL/min, column: Chromatorex 18 SMB100-5T, 100×19 mm, 5 µm) to afford 4-cyclopropyl-5-[4-[[5-fluoro-2-methoxy-4-[1- methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidin-2-yl]-6-methoxy- pyrimidine (72.8 mg, 137 μmol, 45.9% yield) as an off-white solid.
1H NMR (DMSO-d6, 600 MHz) δ 0.87 – 0.92 (m, 2H), 1.03 – 1.08 (m, 2H), 1.68 – 1.73 (m, 1H), 3.62 (s, 3H), 3.84 (s, 3H), 3.86 (s, 3H), 5.44 (s, 2H), 7.10 (d, 1H), 7.18 (d, 1H), 7.45 (d, 1H), 8.02 (s, 1H), 8.68 (s, 1H), 8.72 (d, 1H). MS (ESI): [M+H]+ m/z: calcd 531.20; found 531.2. Example 105 (Compound 133)
Compound 133 Step 1: Synthesis of 4-(difluoromethyl)-1-isopropyl-imidazole. [1317] A mixture of 1-isopropylimidazole-4-carbaldehyde (5.00 g, 36.2 mmol) and water (0.7 mL) was placed in high pressure reaction vessel. The vessel was sealed and cooled to -50 °C. Sulfur tetrafluoride (16 g, 14 mmol, approximate amount) was pumped into the vessel. The reaction mixture was allowed to warm to room temperature and stirred at 95 °C for 18 hr. The reaction mixture was cooled to room temperature and poured onto crashed ice (50 g). The resulting mixture was neutralized with sat. aqueous solution of NaHCO
3 to pH≈7 and extracted with DCM (2×50 mL). The combined organic layers were concentrated under reduced pressure. The residue was distilled under reduced pressure (b.p. approximately 50-55 °C at 0.5 mbar) to afford 4-(difluoromethyl)-1-isopropyl-imidazole (2.94 g, 18.4 mmol, 50.7% yield) as a colorless oil which was used in the next steps without further purification.
1H NMR (400 MHz, CDCl
3) δ 1.45 (d, 6H), 4.26 – 4.37 (m, 1H), 6.61 (t, 1H, CHF2), 7.17 (s, 1H), 7.50 (s, 1H). MS (ESI): [M+H]+ m/z: calcd 161.09; found 161.4. Step 2: The synthesis of 2-bromo-4-(difluoromethyl)-1-isopropyl-1H-imidazole [1318] Butyl lithium (10.0 mmol, 4.00 mL, 2.5M in hexane) was added dropwise to a solution of 4-(difluoromethyl)-1-isopropyl-imidazole (1.46 g, 9.12 mmol) in THF (40 mL) at -78 °C. The resulting mixture was stirred at -78 °C for 40 min.1,2-Dibromotetrafluoroethane (2.61 g, 10.0 mmol, 1.20 mL) was added dropwise to the resulting mixture at -78 °C. The reaction mixture was stirred for 1 hr, during this time it was allowed to warm to room temperature. The mixture was poured into sat. aqueous NH
4Cl solution (30 mL). The obtained mixture was extracted with EtOAc (2 × 30 mL). The combined organic layers were concentrated under reduced pressure to afford 2-bromo-4-(difluoromethyl)-1-isopropyl-1H- imidazole (2.00 mg, 8.37 mmol, 91.8% yield) as a yellow oil which was used in the next steps without further purification.
1H NMR (400 MHz, CDCl
3) δ 1.44 (d, 6H), 4.44 – 4.55 (m, 1H), 6.57 (t, 1H), 7.25 (s, 1H). Step 3: The synthesis of [4-[4-(difluoromethyl)-1-methyl-imidazol-2-yl]phenyl]methanol [1319] 2-bromo-4-(difluoromethyl)-1-isopropyl-imidazole (0.400 g, 1.67 mmol), [4- (hydroxymethyl)phenyl]boronic acid (305 mg, 2.01 mmol), Potassium phosphate tribasic (1.07 g, 5.02 mmol) and bis(diphenylphosphino)ferrocene]dichloropalladium(II)-DCM (137 mg, 167 μmol) were mixed in a degassed mixture of dioxane (11 mL) and water (1 mL). The reaction mixture was stirred at 75°C for 16 hr. The reaction mixture was cooled to room temperature and diluted water (10 mL). The obtained mixture was extracted with EtOAc (2×10 mL). To combined organic layers SiliaMetS® Dimercaptotriazine (100 mg) was added and the resulting mixture was stirred for 8 hr. The mixture was filtered. The filtrated was concentrated under reduced pressure. The residue was subjected to HPLC (2-7 min., 25-50%, water – ACN, +0.1% vol. of 25% aq. NH
3, flow: 30 mL/min, column: XBridge 150 ×30 mm, 5 µm) to afford [4-[4-(difluoromethyl)-1-methyl-imidazol-2-yl]phenyl]methanol (352 mg, 1.48 mmol, 88.3% yield) as a white solid.
1H NMR (400 MHz, CDCl
3) δ 1.41 (d, 6H), 4.45 – 4.55 (m, 1H), 4.71 (s, 2H), 6.71 (t, 1H), 7.32 (s, 1H), 7.30 – 7.45 (m, 4H). MS (ESI): [M+H]+ m/z: calcd 267.13; found 267.2. Step 4: The synthesis of 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[4-[4- (difluoromethyl)-1-isopropyl-imidazol-2-yl]phenyl]methoxy]-5-methoxy-pyrimidine [1320] NaH (3.57 mg, 89.2 μmol, 60% dispersion in mineral oil) was added to a solution of [4-[4-(difluoromethyl)-1-isopropyl-imidazol-2-yl]phenyl]methanol (21.0 mg, 78.9 μmol) in DMF (1 mL). The reaction mixture was stirred at ambient temperature for 0.5 hr. The 2-(4- cyclopropyl-6-methoxy-pyrimidin-5-yl)-5-methoxy-4-methylsulfonyl-pyrimidine I-6 (25.0 mg, 74.3 μmol) was added to the reaction mixture. The resulting mixture was stirred at room temperature for 6 hr. The reaction mixture was diluted with water (10 mL) and extracted with EtOAc (2×10 mL). The combined organic layers were washed with brine (10 mL) and concentrated under reduced pressure. The residue was subjected to HPLC (0.5-6.5 min., 30- 60 water – ACN, +0.1% vol. of 25% aq. NH
3, flow: 30 mL/min, column: SunFire C18100 ×19 mm, 5 µm) to afford 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[4-[4- (difluoromethyl)-1-isopropyl-imidazol-2-yl]phenyl]methoxy]-5-methoxy-pyrimidine (21.0 mg, 40.2 μmol, 54.1% yield) as a white solid.
1H NMR (400 MHz, DMSO-d6) δ 0.84 – 0.89 (m, 2H), 1.01 – 1.05 (m, 2H), 1.40 (d, 3H), 1.68 – 1.74 (m, 1H), 3.85 (s, 3H) 3.95 (s, 3H), 4.44 – 4.51 (m, 1H), 5.51 (s, 2H), 6.90 (t, 1H, CHF2), 7.54 – 7.61 (m, 4H), 7.86 (s, 1H), 8.43 (s, 1H), 8.65 (s, 1H). MS (ESI): [M+H]+ m/z: calcd 523.25; found 523.2.

Step 1: Synthesis of 2-chloro-5-fluoro-4-[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl-methoxy]pyrimidine [1321] To a solution of [4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methanol I-2b (100 mg, 390 μmol) in toluene (10 mL), Potassium tert-butoxide (43.8 mg, 390 μmol) was added. The resulting mixture was stirred at room temperature for 1 hr. then 2,4-dichloro-5- fluoro-pyrimidine (71.7 mg, 429 μmol) was added and stirring was continued for 16 hr. at room temperature. The mixture was concentrated under reduced pressure. The residue was diluted with EtOAc (20 mL) and washed with water (10 mL × 2) and brine (10 mL). The organic layer was concentrated under reduced pressure to afford 2-chloro-5-fluoro-4-[4-[1- methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl-methoxy]pyrimidine (140 mg, crude) as a brown solid which was used in the next steps without further purification. MS (ESI): [M+H]+ m/z: calcd 387.07; found 387.0. Step 2: Synthesis of 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-5-fluoro-4-[4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl-methoxy]pyrimidine [1322] 2-chloro-5-fluoro-4-[4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl- methoxy]pyrimidine (140 mg, 161 μmol), (4-cyclopropyl-6-methoxy-pyrimidin-5-yl)boronic acid I-5 (43.6 mg, 225 μmol), Potassium phosphate tribasic (102 mg, 482 μmol) and RuPhos Pd G4 (1.37 mg, 1.61 μmol) were mixed in degassed dioxane (2 mL) with 5% of water under inert atmosphere of argon. The resulting mixture was stirred at 90 °C for 16 hr. The mixture was cooled to room temperature, diluted with EtOAc (10 mL) and washed with water (5 mL × 2). To the obtained solution SiliaMetS® Dimercaptotriazine (40 mg) was added, and the mixture was stirred for 1hr. The mixture was filtered. The filtrate was evaporated under reduced pressure. The residue was subjected to HPLC (0.5-6.5 min., 54% water – MeOH, +0.1% vol. of 25% aq. NH
3, flow: 30 mL/min, column: XBridge 100 ×19 mm, 5 µm) to afford 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-5-fluoro-4-[4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl-methoxy]pyrimidine (36.0 mg, 72.0 μmol, 16.9% yield from 2,4-dichloro-5-fluoro-pyrimidine) as a light-yellow solid.
1H NMR (600 MHz, DMSO-d6) δ 0.85 – 0.90 (m, 2H), 1.01 – 1.06 (m, 2H), 1.70 – 1.76 (m, 1H), 3.78 (s, 3H), 3.84 (s, 3H), 5.57 (s, 2H), 7.61 (d, 2H), 7.74 (d, 2H), 7.93 (s, 1H), 8.66 (s, 1H), 8.80 (s, 1H). MS (ESI): [M+H]+ m/z: calcd 501.19; found 501.2.

Step 1: The synthesis of [2,5-difluoro-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2- yl)phenyl]methanol [1323] Bis(pinacolato) diboron (2.28 g, 8.97 mmol) and potassium acetate (2.64 g, 26.9 mmol, 1.68 mL) were added to a stirred solution of (4-bromo-2,5-difluoro-phenyl)methanol (2.00 g, 8.97 mmol) in degassed dioxane (80 mL) under argon atmosphere. Bis(diphenylphosphino)ferrocene]dichloropalladium(II)-DCM (220 mg, 269 μmol) was added to the mixture. The reaction mixture was stirred at 100 °C for 16 hr under argon atmosphere. The reaction mixture was cooled to room temperature and concentrated under reduced pressure. The residue was diluted with EtOAc (15 mL) and filtered. The filtrate was concentrated under reduced pressure. The residue was subjected to column chromatography (SiO
2, 0-100% MTBE in n-Hexane) to afford [2,5-difluoro-4-(4,4,5,5-tetramethyl-1,3,2- dioxaborolan-2-yl)phenyl]methanol (1.50 g, 5.55 mmol, 61.9% yield) as a colorless gum.
1H NMR (CDCl
3, 500 MHz) δ 1.35 (s, 12H), 1.76 (br, 1H), 4.76 (s, 2H), 7.14 (dd, 1H), 7.36 (dd, 1H). Step 2: The synthesis of [2,5-difluoro-4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methanol [1324] [2,5-difluoro-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl]methanol (200 mg, 741 μmol), 2-bromo-1-methyl-4-(trifluoromethyl)imidazole (170 mg, 741 μmol), Na
2CO
3 (236 mg, 2.22 mmol) and Bis(diphenylphosphino)ferrocene]dichloropalladium(II)- DCM (30.2 mg, 37.0 μmol) were mixed in a degassed mixture of dioxane (8 mL) and water (2 mL) under argon atmosphere. The reaction mixture was stirred at 90 °C for 16 hr. under argon atmosphere. The reaction mixture was cooled to room temperature and concentrated under reduced pressure. The residue was diluted with EtOAc (50 mL) and filtered. The filtrate was concentrated under reduced pressure to afford [2,5-difluoro-4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methanol (200 mg) as a gum which was used in the next steps without further purification. MS (ESI): [M+H]+ m/z: calcd 293.07; found 293.0. Step 3: The synthesis of 4-cyclopropyl-5-[4-[[2,5-difluoro-4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidin-2-yl]-6-methoxy-pyrimidine [1325] NaH (16.4 mg, 411 μmol, 60% dispersion in mineral oil) was added to a stirred solution of [2,5-difluoro-4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methanol (200 mg, 342 μmol) in DMF (5 mL). The resulting mixture was stirred at room temperature for 30 min.4-cyclopropyl-6-methoxy-5-(4-methylsulfonylpyrimidin-2-yl)pyrimidine I-8 (105 mg, 342 μmol) was added to the mixture. The reaction mixture was stirred at room temperature for 16 hr. The reaction mixture was diluted with a saturated aqueous solution of NH
4Cl (10 mL) and extracted with EtOAc (3×20 mL). The combined organic layers were washed with brine (10 mL), dried over anhydrous Na
2SO
4 and filtered. The filtrate was concentrated under reduced pressure. The residue was subjected to HPLC (0-5 min, 40-65% water+FA (0.1% vol.) - ACN+FA (0.1% vol.); flow: 30 mL/min, column: Chromatorex 18 SMB100-5T, 100×19 mm, 5 µm) to afford 4-cyclopropyl-5-[4-[[2,5-difluoro-4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidin-2-yl]-6-methoxy-pyrimidine (32.6 mg, 62.9 μmol, 18.4% yield) as an off-white solid.
1H NMR (DMSO-d6, 600 MHz) δ 0.88 – 0.93 (m, 2H), 1.03 – 1.07 (m, 2H), 1.68 – 1.74 (m, 1H), 3.64 (s, 3H), 3.85 (s, 3H), 5.52 (s, 2H), 7.11 (d, 1H), 7.58 (dd, 1H), 7.63 (dd, 1H), 8.04 (s, 1H), 8.68 (s, 1H), 8.73 (d, 1H). MS (ESI): [M+H]+ m/z: calcd 519.18; found 519.2. Example 109 (Compound 35)
2-chloro-4-[[4-[1-cyclopropyl-4-(trifluoromethyl)imidazol-2-yl]-3-fluoro- phenyl]methoxy]pyrimidine Synthesis of the starting [4-[1-cyclopropyl-4-(trifluoromethyl)imidazol-2-yl]-3-fluoro- phenyl]methanol is described for compound 154. [4-[1-Cyclopropyl-4-(trifluoromethyl)imidazol-2-yl]-3-fluoro-phenyl]methanol (150 mg, 500 μmol) and potassium tert-butoxide (84.1 mg, 749 μmol) were mixed in THF (2.0 mL) under argon atmosphere. The solution was stirred at room temperature for 1 hr. 2,4- dichloropyrimidine (149 mg, 999 μmol) was added to the solution at -20 °C. The reaction mixture was stirred at -20 °C for 1 hr. The reaction mixture was allowed to warm to room temperature and concentrated under reduced pressure. The residue was diluted with MTBE (30 mL) and washed with water (15 mL). The organic layer was separated, washed with brine (20 mL), dried over anhydrous Na
2SO
4 and concentrated under reduced pressure. The residue was subjected to flash column chromatography (silica, gradient hexane - EtOAc) to afford 2-chloro- 4-[[4-[1-cyclopropyl-4-(trifluoromethyl)imidazol-2-yl]-3-fluoro-phenyl]methoxy]pyrimidine (75.0 mg, 182 μmol, 36.4% yield) as a white solid. MS (ESI): [M+H]
+ m/z: calcd 413.08; found 413.0 4-cyclopropyl-5-[4-[[4-[1-cyclopropyl-4-(trifluoromethyl)imidazol-2-yl]-3-fluoro- phenyl]methoxy]pyrimidin-2-yl]-6-(difluoromethoxy)pyrimidine 2-Сhloro-4-[[4-[1-cyclopropyl-4-(trifluoromethyl)imidazol-2-yl]-3-fluoro- phenyl]methoxy]pyrimidine (75.0 mg, 182 μmol), 4-cyclopropyl-6-(difluoromethoxy)-5- (4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyrimidine I-10 (85.1 mg, 273 μmol), potassium phosphate tribasic (77.1 mg, 363 μmol) and XPhos Pd G3 (7.69 mg, 9.09 μmol) were mixed in degassed mixture of dioxane (2 mL) and water (200 µL). The reaction mixture was stirred at 90 °C for 5 hr. The reaction mixture was cooled to room temperature, filtered and subjected to HPLC (0-2-9 min., 43-50-60% water – ACN, +0.1% vol. of 25% aq. NH
3, flow: 30 mL/min, column: XBridge BEH C18100×19 mm, 5 µm; then Hexane:IPA:MeOH, 80:10:10, Flow Rate: 16 mL/min, Column: CHIRALPAK AD-H 250×20 mm, 5 µm) to afford 4-cyclopropyl-5-[4- [[4-[1-cyclopropyl-4-(trifluoromethyl)imidazol-2-yl]-3-fluoro-phenyl]methoxy]pyrimidin-2- yl]-6-(difluoromethoxy)pyrimidine (10.0 mg, 17.8 μmol, 9.8% yield) as a white solid.
1H NMR (500 MHz, DMSO-d
6) δ 0.78 – 0.83 (m, 4H), 0.96 – 1.02 (m, 2H), 1.08 – 1.13 (m, 2H), 1.83 – 1.89 (m, 1H), 3.42 – 3.49 (m, 1H), 5.52 (s, 2H), 7.17 (d, 1H), 7.43 (d, 1H), 7.50 (d, 1H), 7.63 (t, 1H), 7.80 (t, 1H, CHF
2), 8.00 (s, 1H), 8.77 (d, 1H), 8.80 (s, 1H). MS (ESI): [M+H]
+ m/z: calcd 563.19; found 563.0. Example 110 (Compounds 36 and 233)
1-(2,6-dichloropyrimidin-4-yl)ethanone [1326] A solution of chloro(methyl)magnesium (3 M, 12.1 mL) in THF (8.7 mL) was added dropwise to a solution of methyl 2,6-dichloropyrimidine-4-carboxylate (5.00 g, 24.2 mmol) in THF (8.7 mL) at -78°C under argon atmosphere. The reaction mixture was stirred at -78°C for 2 hr. The reaction mixture was allowed to warm to 0°C and quenched by dropwise addition of water (5.0 mL). The organic layer was separated, washed with brine, dried over anhydrous Na
2SO
4 and concentrated under reduced pressure. The residue was subjected to flash column chromatography (silica, gradient CHCl
3 - EtOAc) to afford 1-(2,6- dichloropyrimidin-4-yl)ethanone (900 mg, 4.71 mmol, 19.5% yield) as a light-yellow oil.
1H NMR (400 MHz, CDCl
3) δ 2.71 (s, 3H), 7.85 (s, 1H). 1-(2-chloro-6-methylsulfanyl-pyrimidin-4-yl)ethanone [1327] Sodium methanethiolate (4.95 mmol, 1.65 mL, 21% wt. in water) was added to a stirred solution of 1-(2,6-dichloropyrimidin-4-yl)ethanone (900 mg, 4.71 mmol) in THF (120 mL) at 5°C. The resulting mixture was stirred at room temperature for 16 hr. The reaction mixture was concentrated under reduced pressure. The residue was diluted with MTBE (80 mL) and washed with water (3×20 mL). The organic layer was washed with brine, dried over anhydrous Na
2SO
4 and filtered through a short pad of silica. The filtrate was concentrated under reduced pressure to afford 1-(2-chloro-6-methylsulfanyl-pyrimidin-4-yl)ethanone (890 mg, 4.39 mmol, 93.2% yield) as a white solid which was used in the next steps without further purification. MS (ESI): [M+H]
+ m/z: calcd 203.01; found 203.0 1-[2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-6-methylsulfanyl-pyrimidin-4-yl]ethanone [1328] 1-(2-Chloro-6-methylsulfanyl-pyrimidin-4-yl)ethanone (890 mg, 4.39 mmol), (4- cyclopropyl-6-methoxy-pyrimidin-5-yl)boronic acid I-5 (1.28 g, 6.59 mmol), XPhos Pd G3 (372 mg, 439 μmol) and DIPEA (1.70 g, 13.2 mmol, 2.3 mL) were mixed in a degassed mixture of dioxane (18 mL) and water (2.0 mL). The resulting mixture was stirred at 65°C for 12 hr under argon atmosphere. The reaction mixture was cooled to room temperature and diluted in EtOAc (50 mL). The organic layer was separated, washed with brine (10 mL) and concentrated under reduced pressure. The residue was subjected to flash column chromatography (silica, gradient hexane - EtOAc) to afford 1-[2-(4-cyclopropyl-6-methoxy- pyrimidin-5-yl)-6-methylsulfanyl-pyrimidin-4-yl]ethanone (600 mg, 1.90 mmol, 43.2% yield) as a yellow oil.
1H NMR (400 MHz, CDCl
3) δ 0.89 – 0.97 (m, 2H), 1.20 – 1.29 (m, 2H), 1.75 – 1.83 (m, 1H), 2.59 (s, 3H), 2.68 (s, 3H), 3.93 (s, 3H), 7.69 (s, 1H), 8.66 (s, 1H). MS (ESI): [M+H]
+ m/z: calcd 317.11; found 317.0 1-[2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-6-methylsulfonyl-pyrimidin-4-yl]ethanone [1329] 3-Chlorobenzenecarboperoxoic acid (1.05 g, 4.55 mmol) was added to a stirred solution of 1-[2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-6-methylsulfanyl-pyrimidin-4- yl]ethanone (600 mg, 1.90 mmol) in DCM (25 mL). The reaction mixture was stirred at room temperature for 12 hr. The reaction was diluted with a saturated aqueous solution of NaHCO
3 (20 mL) and extracted with DCM (2×20 mL). The combined organic layers were separated, dried over anhydrous Na
2SO
4 and filtered. The filtrate was concentrated under reduced pressure to afford 1-[2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-6-methylsulfonyl- pyrimidin-4-yl]ethanone (490 mg, 1.41 mmol, 74.2% yield) as a yellow solid which was used in the next step without further purification. MS (ESI): [M+H]
+ m/z: calcd 349.1; found 349.2 1-[2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-6-[[4-[1-cyclopropyl-4- (trifluoromethyl)imidazol-2-yl]-3-fluoro-phenyl]methoxy]pyrimidin-4-yl]ethanone Synthesis of the starting [4-[1-cyclopropyl-4-(trifluoromethyl)imidazol-2-yl]-3-fluoro- phenyl]methanol is described for Compound 154. [1330] Potassium tert-butoxide (30.9 mg, 276 μmol) was added to a stirred solution of 1-[2- (4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-6-methylsulfonyl-pyrimidin-4-yl]ethanone (80.0 mg, 230 μmol) and [4-[1-cyclopropyl-4-(trifluoromethyl)imidazol-2-yl]-3-fluoro- phenyl]methanol (69.0 mg, 230 μmol) in THF (1 mL). The resulting mixture was stirred at room temperature for 10 hr. The reaction mixture was poured into a saturated aqueous solution NH
4Cl (2 mL) and extracted with EtOAc (3×3 mL). The combined organic layers were dried over anhydrous Na
2SO
4 and concentrated under reduced pressure to afford 1-[2- (4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-6-[[4-[1-cyclopropyl-4-(trifluoromethyl)imidazol- 2-yl]-3-fluoro-phenyl]methoxy]pyrimidin-4-yl]ethanone (110 mg, crude) as a yellow oil which was used in the next step without further purification. MS (ESI): [M+H]
+ m/z: calcd 569.19; found 569.6 1-[2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-6-[[4-[1-cyclopropyl-4- (trifluoromethyl)imidazol-2-yl]-3-fluoro-phenyl]methoxy]pyrimidin-4-yl]ethanol Sodium borohydride (22.0 mg, 581 μmol) was added to a stirred solution of 1-[2-(4- cyclopropyl-6-methoxy-pyrimidin-5-yl)-6-[[4-[1-cyclopropyl-4-(trifluoromethyl)imidazol-2- yl]-3-fluoro-phenyl]methoxy]pyrimidin-4-yl]ethanone (110 mg, crude) in MeOH (3.0 mL) at 0°C. The reaction mixture was stirred at 0 °C for 3 hr. The mixture was allowed to warm to room temperature. The reaction mixture was subjected to HPLC (0-1-6 min, 35-35-75% water+FA (0.1% vol.) - ACN+FA (0.1% vol.); flow: 60 mL/min, column: XBridge OBD C18, 100×30 mm, 5 µm) to afford 1-[2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-6-[[4-[1- cyclopropyl-4-(trifluoromethyl)imidazol-2-yl]-3-fluoro-phenyl]methoxy]pyrimidin-4- yl]ethanol (28.3 mg, 49.6 μmol, 21.6% yield from 1-[2-(4-cyclopropyl-6-methoxy-pyrimidin- 5-yl)-6-methylsulfonyl-pyrimidin-4-yl]ethanone) as a light-yellow solid.
1H NMR (600 MHz, DMSO-d
6) δ 0.81 – 0.85 (m, 4H), 0.87 – 0.91 (m, 2H), 1.02 – 1.06 (m, 2H), 1.38 (d, 3H), 1.67 – 1.72 (m, 1H), 3.45 – 3.50 (m, 1H), 3.85 (s, 3H), 4.66 – 4.71 (m, 1H), 5.53 (q, 2H), 5.61 (d, 1H), 7.06 (s, 1H), 7.44 (d, 1H), 7.51 (d, 1H), 7.65 (t, 1H), 8.01 (s, 1H), 8.67 (s, 1H). MS (ESI): [M+H]
+ m/z: calcd 571.24; found 571.4 rel-(R)-1-(4'-cyclopropyl-6-((4-(1-cyclopropyl-4-(trifluoromethyl)-1H-imidazol-2-yl)-3- fluorobenzyl)oxy)-6'-methoxy-[2,5'-bipyrimidin]-4-yl)ethanol & rel-(S)-1-(4'-cyclopropyl-6- ((4-(1-cyclopropyl-4-(trifluoromethyl)-1H-imidazol-2-yl)-3-fluorobenzyl)oxy)-6'-methoxy- [2,5'-bipyrimidin]-4-yl)ethanol [1331] 1-[2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-6-[[4-[1-cyclopropyl-4- (trifluoromethyl)imidazol-2-yl]-3-fluoro-phenyl]methoxy]pyrimidin-4-yl]ethanol (28.3 mg, 49.6 μmol) was subjected to chiral HPLC (Column: Chiralcel OZ-H (250×20 mm, 5 µm); mobile phase: Hexane:IPA:MeOH, 90:5:5, flow rate: 18 mL/min) to afford rel-(S)-1-(4'- cyclopropyl-6-((4-(1-cyclopropyl-4-(trifluoromethyl)-1H-imidazol-2-yl)-3- fluorobenzyl)oxy)-6'-methoxy-[2,5'-bipyrimidin]-4-yl)ethanol (9.22 mg, 16.2 μmol, 65.2% yield) and rel-(R)-1-(4'-cyclopropyl-6-((4-(1-cyclopropyl-4-(trifluoromethyl)-1H-imidazol-2- yl)-3-fluorobenzyl)oxy)-6'-methoxy-[2,5'-bipyrimidin]-4-yl)ethanol (4.69 mg, 8.22 μmol, 33.1% yield) as light-yellow solids. rel-(R)-1-(4'-cyclopropyl-6-((4-(1-cyclopropyl-4-(trifluoromethyl)-1H-imidazol-2-yl)- 3-fluorobenzyl)oxy)-6'-methoxy-[2,5'-bipyrimidin]-4-yl)ethanol (36): 1H NMR (600 MHz, DMSO-d
6) δ 0.81 – 0.85 (m, 4H), 0.87 – 0.91 (m, 2H), 1.02 – 1.06 (m, 2H), 1.38 (d, 3H), 1.67 – 1.72 (m, 1H), 3.45 – 3.50 (m, 1H), 3.85 (s, 3H), 4.66 – 4.71 (m, 1H), 5.53 (q, 2H), 5.61 (d, 1H), 7.06 (s, 1H), 7.44 (d, 1H), 7.51 (d, 1H), 7.65 (t, 1H), 8.01 (s, 1H), 8.67 (s, 1H). MS (ESI): [M+H]
+ m/z: calcd 571.24; found 571.4 Enantiopurity: 98.5% (column: Chiralcel OZ-H, 250 × 4.6 mm, 5 μm; mobile phase: Hexane-IPA-MeOH, 90-5-5; flow: 0.6 mL/min; RT=39.1 min). rel-(S)-1-(4'-cyclopropyl-6-((4-(1-cyclopropyl-4-(trifluoromethyl)-1H-imidazol-2-yl)- 3-fluorobenzyl)oxy)-6'-methoxy-[2,5'-bipyrimidin]-4-yl)ethanol (233): 1H NMR (600 MHz, DMSO-d
6) δ 0.81 – 0.85 (m, 4H), 0.87 – 0.91 (m, 2H), 1.02 – 1.06 (m, 2H), 1.38 (d, 3H), 1.67 – 1.72 (m, 1H), 3.45 – 3.50 (m, 1H), 3.85 (s, 3H), 4.66 – 4.71 (m, 1H), 5.53 (q, 2H), 5.61 (d, 1H), 7.06 (s, 1H), 7.44 (d, 1H), 7.51 (d, 1H), 7.65 (t, 1H), 8.01 (s, 1H), 8.67 (s, 1H). MS (ESI): [M+H]
+ m/z: calcd 571.24; found 571.4 Enantiopurity: >99% (column: Chiralcel OZ-H, 250 × 4.6 mm, 5 μm; mobile phase: Hexane-IPA-MeOH, 90-5-5; flow: 0.6 mL/min; RT=29.8 min).

1-[2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-6-[[4-[1-cyclopropyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidin-4-yl]ethanone Synthesis of the starting [4-[1-cyclopropyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methanol is described for compound 2. Synthesis of the starting 1-[2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-6- methylsulfonyl-pyrimidin-4-yl]ethanone is described for compound 36 and 233. [1332] Potassium tert-butoxide (30.9 mg, 276 μmol) was added to a stirred solution of 1-[2- (4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-6-methylsulfonyl-pyrimidin-4-yl]ethanone (80.0 mg, 230 μmol) and [4-[1-cyclopropyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methanol (64.8 mg, 230 μmol) in THF (1 mL). The resulting mixture was stirred at room temperature for 10 hr. The reaction mixture was poured into a saturated aqueous solution of NH
4Cl (2 mL) and extracted with EtOAc (3×3 mL). The combined organic layers were dried over anhydrous Na
2SO
4 and concentrated under reduced pressure to afford 1-[2-(4-cyclopropyl-6- methoxy-pyrimidin-5-yl)-6-[[4-[1-cyclopropyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidin-4-yl]ethanone (115 mg, crude) as a yellow oil which was used in the next step without further purification. MS (ESI): [M+H]
+ m/z: calcd 551.20; found 551.6 1-[2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-6-[[4-[1-cyclopropyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidin-4-yl]ethanol Sodium borohydride (9.74 mg, 258 μmol) was added to a stirred solution of 1-[2-(4- cyclopropyl-6-methoxy-pyrimidin-5-yl)-6-[[4-[1-cyclopropyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidin-4-yl]ethanone (115 mg, crude) in MeOH (3.0 mL) at 0°C. The reaction mixture was stirred at 0 °C for 3 hr. The mixture was allowed to warm to room temperature. The reaction mixture was subjected to HPLC (0-1-6 min, 35-35-75% water+FA (0.1% vol.) - ACN+FA (0.1% vol.); flow: 60 mL/min, column: XBridge OBD C18, 100×30 mm, 5 µm) to afford 1-[2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-6-[[4-[1-cyclopropyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidin-4-yl]ethanol (45.7 mg, 82.7 μmol, 36.0% yield from 1-[2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-6-methylsulfonyl- pyrimidin-4-yl]ethanone) as a light-yellow solid.
1H NMR (600 MHz, DMSO-d
6) δ 0.86 – 0.93 (m, 4H), 0.97 – 1.02 (m, 2H), 1.02 – 1.06 (m, 2H), 1.37 (d, 3H), 1.67 – 1.72 (m, 1H), 3.73 – 3.78 (m, 1H), 3.85 (s, 3H), 4.65 – 4.70 (m, 1H), 5.51 (q, 2H), 5.59 (d, 1H), 7.03 (s, 1H), 7.58 (d, 2H), 7.91 (d, 2H), 7.94 (s, 1H), 8.67 (s, 1H). MS (ESI): [M+H]
+ m/z: calcd 553.25; found 553.2 rel-(R)-1-(4'-cyclopropyl-6-((4-(1-cyclopropyl-4-(trifluoromethyl)-1H-imidazol-2- yl)benzyl)oxy)-6'-methoxy-[2,5'-bipyrimidin]-4-yl)ethanol and rel-(S)-1-(4'-cyclopropyl-6- ((4-(1-cyclopropyl-4-(trifluoromethyl)-1H-imidazol-2-yl)benzyl)oxy)-6'-methoxy-[2,5'- bipyrimidin]-4-yl)ethanol [1333] 1-[2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-6-[[4-[1-cyclopropyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidin-4-yl]ethanol (45.6 mg, 82.5 μmol) was subjected to chiral HPLC (Column: CHIRALPAK IС (250×21 mm, 5 µm); mobile phase: Hexane:IPA:MeOH, 80:10:10, flow rate: 20 mL/min) to afford rel-(S)-1-(4'- cyclopropyl-6-((4-(1-cyclopropyl-4-(trifluoromethyl)-1H-imidazol-2-yl)benzyl)oxy)-6'- methoxy-[2,5'-bipyrimidin]-4-yl)ethanol (20.5 mg, 37.1 μmol, 89.9% yield) and rel-(R)-1-(4'- cyclopropyl-6-((4-(1-cyclopropyl-4-(trifluoromethyl)-1H-imidazol-2-yl)benzyl)oxy)-6'- methoxy-[2,5'-bipyrimidin]-4-yl)ethanol (12.0 mg, 21.7 μmol, 52.6% yield) as light-yellow solids. rel-(S)-1-(4'-cyclopropyl-6-((4-(1-cyclopropyl-4-(trifluoromethyl)-1H-imidazol-2- yl)benzyl)oxy)-6'-methoxy-[2,5'-bipyrimidin]-4-yl)ethanol (234):
1H NMR (600 MHz, DMSO-d
6) δ 0.86 – 0.93 (m, 4H), 0.97 – 1.02 (m, 2H), 1.02 – 1.06 (m, 2H), 1.37 (d, 3H), 1.67 – 1.72 (m, 1H), 3.73 – 3.78 (m, 1H), 3.85 (s, 3H), 4.65 – 4.70 (m, 1H), 5.51 (q, 2H), 5.59 (d, 1H), 7.03 (s, 1H), 7.58 (d, 2H), 7.91 (d, 2H), 7.94 (s, 1H), 8.67 (s, 1H). MS (ESI): [M+H]
+ m/z: calcd 553.25; found 553.2 Enantiopurity: >99% (column: CHIRALPAK IС, 250 × 4.6 mm, 5 μm; mobile phase: Hexane-IPA-MeOH, 70-15-15; flow: 0.6 mL/min; RT=8.2 min). rel-(R)-1-(4'-cyclopropyl-6-((4-(1-cyclopropyl-4-(trifluoromethyl)-1H-imidazol-2- yl)benzyl)oxy)-6'-methoxy-[2,5'-bipyrimidin]-4-yl)ethanol (59): 1H NMR (600 MHz, DMSO-d
6) δ 0.86 – 0.93 (m, 4H), 0.97 – 1.02 (m, 2H), 1.02 – 1.06 (m, 2H), 1.37 (d, 3H), 1.67 – 1.72 (m, 1H), 3.73 – 3.78 (m, 1H), 3.85 (s, 3H), 4.65 – 4.70 (m, 1H), 5.51 (q, 2H), 5.59 (d, 1H), 7.03 (s, 1H), 7.58 (d, 2H), 7.91 (d, 2H), 7.94 (s, 1H), 8.67 (s, 1H). MS (ESI): [M+H]
+ m/z: calcd 553.25; found 553.2 Enantiopurity: 98% (column: CHIRALPAK IС, 250 × 4.6 mm, 5 μm; mobile phase: Hexane-IPA-MeOH, 70-15-15; flow: 0.6 mL/min; RT=9.1 min). Example 112 (Compound 42)
1-[4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]cyclopropanol [1334] Bromo(ethyl)magnesium (6.53 mL, 22.2 mmol, 3.4 M solution in THF) was added dropwise to a stirred solution of methyl 4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]benzoate I-3b (3.00 g, 10.6 mmol) and titanium(IV) isopropoxide (600 mg, 2.11 mmol, 628 μL) in THF (150 mL) at 0 °C. The reaction mixture was stirred at room temperature for 6 hr. The reaction mixture was poured into cooled (5 °C) water (50 mL) and extracted with Et
2O (2×50 mL). The combined organic layers were separated, washed with brine (30 mL) and concentrated under reduced pressure. The residue was subjected to flash column chromatography (SiO
2, gradient chloroform – MTBE) to afford 1-[4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]cyclopropanol (460 mg, 1.63 mmol, 15.4% yield) as a yellow solid. MS (ESI): [M+H]
+ m/z: calcd 283.11; found 283.0 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-5-methoxy-4-[1-[4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]cyclopropoxy]pyrimidine [1335] NaH (6.31 mg, 158 μmol, 60% dispersion in mineral oil) was added to a stirring solution of 1-[4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]cyclopropanol (60.0 mg, 196 μmol) in DMF (2.0 mL). The resulting mixture was stirred at room temperature for 1 hr. 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-5-methoxy-4-methylsulfonyl-pyrimidine I-6 (65.8 mg, 196 μmol) was added to the mixture. The reaction mixture was stirred at room temperature for 16 hr. The reaction mixture was quenched by addition of water (2 mL), extracted with EtOAc (8 mL). The organic layer was separated, washed with water (10 mL) and concentrated under reduced pressure. The residue was subjected to HPLC (0.5-6.5 min., 40-90% water – ACN, column: SunFire C18100×19 mm, 5 µm) to afford 2-(4-cyclopropyl- 6-methoxy-pyrimidin-5-yl)-5-methoxy-4-[1-[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]cyclopropoxy]pyrimidine (53.9 mg, 100 μmol, 51.2% yield) as a white solid.
1H NMR (600 MHz, DMSO-d
6) δ 0.61 – 0.65 (m, 2H), 0.85 – 0.89 (m, 2H), 1.42 – 1.51 (m, 4H), 1.51 – 1.57 (m, 1H), 3.73 (s, 3H), 3.74 (s, 3H), 3.99 (s, 3H), 7.25 (d, 2H), 7.61 (d, 2H), 7.90 (s, 1H), 8.44 (s, 1H), 8.56 (s, 1H). MS (ESI): [M+H]
+ m/z: calcd 539.23; found 539.2
methyl 5-fluoro-6-[4-(trifluoromethyl)-1H-imidazol-2-yl]pyridine-3-carboxylate Synthesis of the starting 5-fluoro-6-[4-(trifluoromethyl)-1H-imidazol-2-yl]pyridine-3- carboxylic acid was described for Compound 48. [1336] Thionyl chloride (1.23 g, 10.4 mmol, 756 μL) was added dropwise to a solution of 5- fluoro-6-[4-(trifluoromethyl)-1H-imidazol-2-yl]pyridine-3-carboxylic acid (1.90 g, 6.91 mmol) in MeOH (100 mL). The reaction mixture was stirred at room temperature for 12 hr. The reaction mixture was concentrated under reduced pressure to afford methyl 5-fluoro-6- [4-(trifluoromethyl)-1H-imidazol-2-yl]pyridine-3-carboxylate (2.00 g, 6.92 mmol, 100% yield) which was used in the next step without further purification. MS (ESI): [M+H]
+ m/z: calcd 290.06; found 290.0 methyl 6-[1-cyclopropyl-4-(trifluoromethyl)imidazol-2-yl]-5-fluoro-pyridine-3-carboxylate [1337] Methyl 5-fluoro-6-[4-(trifluoromethyl)-1H-imidazol-2-yl]pyridine-3-carboxylate (1.50 g, 5.19 mmol), cyclopropylboronic acid (4.46 g, 51.9 mmol), copper (I) bromide (744 mg, 5.19 mmol, 158 μL), pyridine (5.74 g, 72.6 mmol, 5.85 mL), bipyridine (810 mg, 5.19 mmol) and K
2CO
3 (860 mg, 6.22 mmol) were mixed in dioxane (150 mL). The reaction mixture was stirred at 65 °C for 12 hr with air access. The reaction mixture was cooled to room temperature and concentrated under reduced pressure. The residue was subjected to flash column chromatography (SiO
2, gradient hexane - EtOAc) to afford methyl 6-[1- cyclopropyl-4-(trifluoromethyl)imidazol-2-yl]-5-fluoro-pyridine-3-carboxylate (750 mg, 2.28 mmol, 43.9% yield) as a colorless oil.
1H NMR (400 MHz, CDCl
3) δ 0.74 – 0.80 (m, 2H), 0.91 – 0.98 (m, 2H), 3.76 – 3.84 (m, 1H), 3.98 (s, 3H), 7.43 (s, 1H), 8.12 (d, 2H), 9.09 (s, 1H). MS (ESI): [M+H]
+ m/z: calcd 330.09; found 330.2 [6-[1-cyclopropyl-4-(trifluoromethyl)imidazol-2-yl]-5-fluoro-3-pyridyl]methanol [1338] A solution of methyl 6-[1-cyclopropyl-4-(trifluoromethyl)imidazol-2-yl]-5-fluoro- pyridine-3-carboxylate (420 mg, 1.28 mmol) in diethyl ether (5 mL) was added dropwise to a stirred suspension of LAH (97.0 mg, 2.55 mmol) in diethyl ether (20 mL) at 0°C. The reaction mixture was stirred at 0 °C for 20 min, quenched by dropwise addition of 30% aqueous potassium hydroxide (500 mL) and filtered. The filtrate was concentrated under reduced pressure to afford [6-[1-cyclopropyl-4-(trifluoromethyl)imidazol-2-yl]-5-fluoro-3- pyridyl]methanol (300 mg, 996 μmol, 78.1% yield) as a yellow oil which was used in the next step without further purification. MS (ESI): [M+H]
+ m/z: calcd 302.09; found 302.0 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[6-[1-cyclopropyl-4- (trifluoromethyl)imidazol-2-yl]-5-fluoro-3-pyridyl]methoxy]-5-methoxy-pyrimidine [1339] Potassium tert-butoxide (14.9 mg, 133 μmol) was added to a solution of 2-(4- cyclopropyl-6-methoxy-pyrimidin-5-yl)-5-methoxy-4-methylsulfonyl-pyrimidine I-6 (44.7 mg, 133 μmol) and [6-[1-cyclopropyl-4-(trifluoromethyl)imidazol-2-yl]-5-fluoro-3- pyridyl]methanol (40.0 mg, 133 μmol) in THF (6 mL). The reaction mixture was stirred at room temperature for 13 hr. The reaction mixture was poured into a saturated aqueous solution of NH
4Cl (15 mL). The resulting mixture was extracted with MTBE (3×15 mL). The combined organic layers were dried over anhydrous Na
2SO
4 and concentrated under reduced pressure. The residue was dissolved in ACN (6.0 mL). Metal scavenger SiliaMetS
® Dimercaptotriazine (100 mg) was added to the solution. The resulting suspension was stirred at ambient temperature for 20 hr, then solids were filtered out. The filtrate was subjected to HPLC (0-6 min., 40-60% water – ACN, +0.1% vol. of 25% aq. NH
3, flow: 60 mL/min, column: XBridge OBD C18100×30 mm, 5 µm) to afford 2-(4-cyclopropyl-6-methoxy- pyrimidin-5-yl)-4-[[6-[1-cyclopropyl-4-(trifluoromethyl)imidazol-2-yl]-5-fluoro-3- pyridyl]methoxy]-5-methoxy-pyrimidine (46.0 mg, 82.5 μmol, 62.1% yield) as yellow solid.
1H NMR (600 MHz, DMSO-d
6) δ 0.84 – 0.92 (m, 6H), 1.01 – 1.06 (m, 2H), 1.69 – 1.76 (m, 1H), 3.73 – 3.79 (m, 1H), 3.85 (s, 3H), 3.96 (s, 3H), 5.59 (s, 2H), 8.03 (d, 1H), 8.08 (s, 1H), 8.46 (s, 1H), 8.66 (s, 1H), 8.70 (s, 1H). MS (ESI): [M+H]
+ m/z: calcd 558.21; found 558.2

methyl 2-[4-cyclopropyl-6-(difluoromethoxy)pyrimidin-5-yl]-6-methylsulfanyl-pyrimidine-4- carboxylate Synthesis of the starting 2-chloro-6-methylsulfanyl-pyrimidine-4-carboxylate is described for Compound 58. [1340] Methyl 2-chloro-6-methylsulfanyl-pyrimidine-4-carboxylate (200 mg, 915 μmol), 4- cyclopropyl-6-(difluoromethoxy)-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyrimidine I-10 (314 mg, 1.01 mmol), potassium phosphate tribasic (175 mg, 823 μmol) and XPhos Pd G3 (38.9 mg, 45.7 μmol) were mixed in a degassed mixture of dioxane (9 mL) and water (2 mL) under argon atmosphere. The reaction mixture was stirred at 70 °C for 16 hr. The reaction mixture was cooled to room temperature and concentrated under reduced pressure. The residue was dissolved in DCM (20 mL) and filtered through a shot pad of silica. The filtrate was concentrated under reduced pressure to afford methyl 2-[4-cyclopropyl-6- (difluoromethoxy)pyrimidin-5-yl]-6-methylsulfanyl-pyrimidine-4-carboxylate (140 mg, 380 μmol, 41.6% yield) as a white solid.
1H NMR (400 MHz, CDCl
3) δ 0.95 – 1.03 (m, 2H), 1.23 – 1.30 (m, 2H), 1.81 – 1.89 (m, 1H), 2.60 (s, 3H), 4.01 (s, 3H), 7.47 (t, 1H, CHF
2), 7.88 (s, 1H), 8.64 (s, 1H). MS (ESI): [M+H]
+ m/z: calcd 369.09; found 369.0 methyl 2-[4-cyclopropyl-6-(difluoromethoxy)pyrimidin-5-yl]-6-methylsulfonyl-pyrimidine-4- carboxylate [1341] mCPBA (192 mg, 836 μmol, 75% purity) was added to a stirred solution of methyl 2- [4-cyclopropyl-6-(difluoromethoxy)pyrimidin-5-yl]-6-methylsulfanyl-pyrimidine-4- carboxylate (140 mg, 380 μmol) in DCM (5 mL). The resulting mixture was stirred at room temperature for 24 hr. The reaction mixture was washed with a saturated aqueous solution of NaHCO
3 (30 mL), dried over anhydrous Na
2SO
4 and concentrated under reduced pressure to afford methyl 2-[4-cyclopropyl-6-(difluoromethoxy)pyrimidin-5-yl]-6-methylsulfonyl- pyrimidine-4-carboxylate (150 mg, 375 μmol, 98.6% yield) as a light-yellow solid which was used in the next step without further purification.
1H NMR (400 MHz, CDCl
3) δ 1.01 – 1.10 (m, 2H), 1.36 – 1.28 (m, 2H), 1.88 – 1.99 (m, 1H), 3.32 (s, 3H), 4.08 (s, 3H), 7.49 (t, 1H, CHF
2), 8.64 (s, 1H), 8.68 (s, 1H). MS (ESI): [M+H]
+ m/z: calcd 401.08; found 401.0 methyl 2-[4-cyclopropyl-6-(difluoromethoxy)pyrimidin-5-yl]-6-[[4-[1-cyclopropyl-4- (trifluoromethyl)imidazol-2-yl]-3-fluoro-phenyl]methoxy]pyrimidine-4-carboxylate Synthesis of the starting [4-[1-cyclopropyl-4-(trifluoromethyl)imidazol-2-yl]-3-fluoro- phenyl]methanol is described for compound 154. [1342] DBU (26.6 mg, 175 μmol, 26 μL) was added to a stirred solution of [4-[1- cyclopropyl-4-(trifluoromethyl)imidazol-2-yl]-3-fluoro-phenyl]methanol (37.5 mg, 125 μmol) and methyl 2-[4-cyclopropyl-6-(difluoromethoxy)pyrimidin-5-yl]-6-methylsulfonyl- pyrimidine-4-carboxylate (50.0 mg, 125 μmol) in THF (2 mL). The reaction mixture was stirred at room temperature for 10 hr. The reaction was quenched by addition of water (5 mL) and extracted with EtOAc (2×5 mL). The combined organic layers were dried over anhydrous Na
2SO
4 and concentrated under reduced pressure to afford methyl 2-[4-cyclopropyl-6- (difluoromethoxy)pyrimidin-5-yl]-6-[[4-[1-cyclopropyl-4-(trifluoromethyl)imidazol-2-yl]-3- fluoro-phenyl]methoxy]pyrimidine-4-carboxylate (70.0 mg, 113 μmol, 90.3% yield) as a yellow oil which was used in the next step without further purification. MS (ESI): [M+H]
+ m/z: calcd 621.17; found 621.0 [2-[4-cyclopropyl-6-(difluoromethoxy)pyrimidin-5-yl]-6-[[4-[1-cyclopropyl-4- (trifluoromethyl)imidazol-2-yl]-3-fluoro-phenyl]methoxy]pyrimidin-4-yl]methanol [1343] DIBAL (10.4 mg, 72.9 μmol, 15.6 μL) was added to a stirred solution of methyl 2-[4- cyclopropyl-6-(difluoromethoxy)pyrimidin-5-yl]-6-[[4-[1-cyclopropyl-4- (trifluoromethyl)imidazol-2-yl]-3-fluoro-phenyl]methoxy]pyrimidine-4-carboxylate (36.9 mg, 59.5 μmol) in THF (1 mL) at 0 °C. The reaction mixture was stirred at 0 °C for 1 hr. the reaction mixture was quenched by addition of MeOH (1 mL) and subjected to HPLC (0-2-9 min., 38-45-55% water – ACN, flow: 30 mL/min, column: Chromatorex C18 SMB100-5T, 100×19 mm, 5 µm) to afford [2-[4-cyclopropyl-6-(difluoromethoxy)pyrimidin-5-yl]-6-[[4-[1- cyclopropyl-4-(trifluoromethyl)imidazol-2-yl]-3-fluoro-phenyl]methoxy]pyrimidin-4- yl]methanol (3.00 mg, 5.06 μmol, 8.5% yield) as a yellow solid.
1H NMR (600 MHz, DMSO- d
6) δ 0.80 – 0.85 (m, 4H), 0.97 – 1.02 (m, 2H), 1.09 – 1.13 (m, 2H), 1.83 – 1.88 (m, 1H), 3.44 – 3.49 (m, 1H), 4.59 (d, 2H), 5.56 (s, 2H), 5.71 (t, 1H), 7.10 (s, 1H), 7.44 (d, 1H), 7.50 (d, 1H), 7.64 (t, 1H), 7.80 (t, 1H, CHF
2), 8.01 (s, 1H), 8.80 (s, 1H). MS (ESI): [M+H]
+ m/z: calcd 593.20; found 593.2.
2-chloro-4-[[5-fluoro-6-[1-isopropyl-4-(trifluoromethyl)imidazol-2-yl]-3-pyridyl]methoxy]- 5-methoxy-pyrimidine Synthesis of the starting [5-fluoro-6-[1-isopropyl-4-(trifluoromethyl)imidazol-2-yl]-3- pyridyl]methanol is described for Compound 119. [1344] Potassium tert-butoxide (50.0 mg, 445 μmol) was added to a stirred solution of 2,4- dichloro-5-methoxy-pyrimidine (79.7 mg, 445 μmol) and [5-fluoro-6-[1-isopropyl-4- (trifluoromethyl)imidazol-2-yl]-3-pyridyl]methanol (135 mg, 445 μmol) in THF (6 mL). The resulting mixture was stirred at room temperature for 13 hr. The reaction mixture was poured into a saturated aqueous solution of NH
4Cl (15 mL) and extracted with MTBE (3×15 mL). The combined organic layers were dried over anhydrous Na
2SO
4 and concentrated under reduced pressure to afford 2-chloro-4-[[5-fluoro-6-[1-isopropyl-4-(trifluoromethyl)imidazol- 2-yl]-3-pyridyl]methoxy]-5-methoxy-pyrimidine (142 mg, 319 μmol, 71.6% yield) as a yellow solid which was used in the next step without further purification. MS (ESI): [M+H]
+ m/z: calcd 446.10; found 446.1 2-[4-cyclopropyl-6-(difluoromethoxy)pyrimidin-5-yl]-4-[[5-fluoro-6-[1-isopropyl-4- (trifluoromethyl)imidazol-2-yl]-3-pyridyl]methoxy]-5-methoxy-pyrimidine [1345] 2-Chloro-4-[[5-fluoro-6-[1-isopropyl-4-(trifluoromethyl)imidazol-2-yl]-3- pyridyl]methoxy]-5-methoxy-pyrimidine (142 mg, 163 μmol), 4-cyclopropyl-6- (difluoromethoxy)-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyrimidine I-10 (50.7 mg, 163 μmol), XPhosPdG3 (13.8 mg, 16.3 μmol) and DIPEA (63.0 mg, 487 μmol, 85 μL) were mixed in in a degassed mixture of dioxane (7 mL) and water (100 µL). The resulting mixture was stirred at 65 °C for 12 hr. under argon atmosphere. The reaction mixture was cooled to room temperature. SiliaMetS
® Dimercaptotriazine was added to the reaction mixture and the mixture was stirred for 30 min. The mixture was filtered. The filtrate was concentrated under reduced pressure. The residue was subjected to HPLC (0-1-5 min., 40-40-80% water – ACN, flow: 60 mL/min, column: XBridge BEH C18100×30 mm, 5 µm) to afford 2-[4-cyclopropyl- 6-(difluoromethoxy)pyrimidin-5-yl]-4-[[5-fluoro-6-[1-isopropyl-4-(trifluoromethyl)imidazol- 2-yl]-3-pyridyl]methoxy]-5-methoxy-pyrimidine (4.00 mg, 6.72 μmol, 4.1% yield) as a yellow solid.
1H NMR (600 MHz, DMSO-d
6) δ 0.97 – 1.02 (m, 2H), 1.09 – 1.13 (m, 2H), 1.42 (d, 6H), 1.87 – 1.92 (m, 1H), 3.98 (s, 3H), 4.76 – 4.82 (m, 1H), 5.59 (s, 2H), 7.80 (t, 1H, CHF
2), 8.03 (d, 1H), 8.31 (s, 1H), 8.52 (s, 1H), 8.68 (s, 1H), 8.79 (s, 1H). MS (ESI): [M+H]
+ m/z: calcd 596.21; found 596.2. Example 116 (Compound 46)

Compound 46 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[6-[1-(cyclopropylmethyl)-4- (trifluoromethyl)imidazol-2-yl]-5-fluoro-3-pyridyl]methoxy]-5-methyl-pyrimidine Synthesis of the starting [6-[1-(cyclopropylmethyl)-4-(trifluoromethyl)imidazol-2-yl]- 5-fluoro-3-pyridyl]methanol is described for Compound 82. [1346] A mixture of [6-[1-(cyclopropylmethyl)-4-(trifluoromethyl)imidazol-2-yl]-5-fluoro-3- pyridyl]methanol (16.0 mg, 50.8 μmol), 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-5- methyl-4-methylsulfonyl-pyrimidine I-7 (17.9 mg, 55.8 μmol) and NaH (2.03 mg, 50.8 μmol, 60% dispersion in mineral oil) in THF (2 mL) was stirred at room temperature for 12 hr. The reaction mixture was directly subjected to HPLC (0.5-6.5 min., 50-75% water – ACN, flow: 30 mL/min, column: Chromatorex C18 SMB100-5T 100×19 mm, 5 µm) to afford 2-(4- cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[6-[1-(cyclopropylmethyl)-4- (trifluoromethyl)imidazol-2-yl]-5-fluoro-3-pyridyl]methoxy]-5-methyl-pyrimidine (13.0 mg, 23.4 μmol, 46.1% yield) as an off-white solid.
1H NMR (600 MHz, DMSO-d
6) δ 0.29 – 0.33 (m, 2H), 0.46 – 0.51 (m, 2H), 0.86 – 0.90 (m, 2H), 1.02 – 1.06 (m, 2H), 1.20 – 1.27 (m, 1H), 1.65 – 1.71 (m, 1H), 2.25 (s, 3H), 3.84 (s, 3H), 4.11 (d, 2H), 5.59 (s, 2H), 8.05 (d, 1H), 8.17 (s, 1H), 8.58 (s, 1H), 8.67 (s, 1H), 8.69 (s, 1H). MS (ESI): [M+H]
+ m/z: calcd 556.24; found 556.2.
2-chloro-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]-5-(2,2,2- trifluoroethoxy)pyrimidine Synthesis of the starting 2-chloro-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidin-5-ol is described in for Compound 122. [1347] 2-Chloro-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidin-5-ol (200 mg, 520 μmol), 2,2,2-trifluoroethyl trifluoromethanesulfonate (133 mg, 572 μmol, 82.4 μL) and K
2CO
3 (93.4 mg, 676 μmol) were mixed in DMF (4.0 mL). The reaction mixture was stirred at 40°C for 12 hr. The reaction mixture was cooled to room temperature, diluted with EtOAc (20 mL) and washed with brine (10 mL). The organic layer was separated, washed with brine (2×10 mL), dried over anhydrous Na
2SO
4 and concentrated under reduced pressure to afford 2-chloro-4-[[4-[1- methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]-5-(2,2,2- trifluoroethoxy)pyrimidine (205 mg, 439 μmol, 84.5% yield) as a yellow solid which was used in the next step without further purification. MS (ESI): [M+H]
+ m/z: calcd 467.07; found 467.0 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]-5-(2,2,2-trifluoroethoxy)pyrimidine [1348] 2-Chloro-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]-5- (2,2,2-trifluoroethoxy)pyrimidine (200 mg, 429 μmol), (4-cyclopropyl-6-methoxy-pyrimidin- 5-yl)boronic acid I-5 (125 mg, 643 μmol), potassium phosphate tribasic (273 mg, 1.29 mmol) and XPhos Pd G3 (18.1 mg, 21.4 μmol) were mixed in degassed mixture of dioxane (10 mL) and water (1.5 mL). The resulting mixture was evacuated and backfilled with argon. The reaction mixture was stirred at 60 °C for 6 hr. The reaction mixture was cooled to room temperature and concentrated under reduced pressure. The residue was diluted with EtOAc (20 mL) and washed with water (5 mL). The organic layer was separated, washed with brine (2×20 mL), dried over anhydrous Na
2SO
4 and concentrated under reduced pressure. The residue was subjected to HPLC (2-10 min., 30-55% ACN, +0.1% vol. of 25% aq. NH
3, flow: 30 mL/min, column: XBridge BEH C18100×20 mm, 5 µm) to afford 2-(4-cyclopropyl-6- methoxy-pyrimidin-5-yl)-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]- 5-(2,2,2-trifluoroethoxy)pyrimidine (33.0 mg, 56.9 μmol, 13.3% yield) as a light-yellow solid.
1H NMR (600 MHz, DMSO-d
6) δ 0.84 – 0.90 (m, 2H), 1.01 – 1.06 (m, 2H), 1.67 – 1.74 (m, 1H), 3.79 (s, 3H), 3.85 (s, 3H), 5.00 (q, 2H), 5.56 (s, 1H), 7.59 (d, 2H), 7.74 (d, 2H), 7.94 (s, 1H), 8.57 (s, 1H), 8.66 (s, 1H). MS (ESI): [M+H]
+ m/z: calcd 581.20; found 581.2. Example 118 (Compound 48)
5-fluoro-6-[4-(trifluoromethyl)-1H-imidazol-2-yl]pyridine-3-carboxylic acid [1349] A mixture of 3,3-dibromo-1,1,1-trifluoro-propan-2-one (3.99 g, 14.8 mmol) and sodium acetate, anhydrous (1.21 g, 14.8 mmol) in water (50 mL) was stirred at 100 °C for 1 hr. The reaction mixture was cooled to room temperature and poured into a precooled to 0 °C solution of 5-fluoro-6-formyl-pyridine-3-carboxylic acid (2.50 g, 14.8 mmol) and ammonium hydroxide (10.4 g, 296 mmol, 11.5 mL, 28% wt. NH
3) in MeOH (100 mL). The resulting mixture was stirred at ambient temperature for 24 hr. The reaction mixture was concentrated under reduced pressure to a half volume. The precipitate formed was filtered off, air-dried and recrystallized from MTBE to afford 5-fluoro-6-[4-(trifluoromethyl)-1H-imidazol-2- yl]pyridine-3-carboxylic acid (1.60 g, 5.81 mmol, 39.3% yield) as a white solid which was used in the next steps without further purification.
1H NMR (400 MHz, DMSO-d
6) δ 7.90 (s, 1H), 7.99 (d, 1H), 8.89 (s, 1H). MS (ESI): [M+H]
+ m/z: calcd 276.04; found 276.0 methyl 5-fluoro-6-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]pyridine-3-carboxylate Methyl iodide (645 mg, 4.54 mmol, 283 μL) was added to a mixture of 5-fluoro-6-[4- (trifluoromethyl)-1H-imidazol-2-yl]pyridine-3-carboxylic acid (500 mg, 1.82 mmol) and Cs
2CO
3 (1.18 g, 3.63 mmol) in DMF (10 mL). The reaction mixture was stirred at 40 °C for 16 hr. The reaction mixture was cooled to room temperature, diluted with EtOAc (20 mL) and washed with water (2×20 mL) and brine (20 mL). The organic layer was separated and concentrated under reduced pressure to afford methyl 5-fluoro-6-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]pyridine-3-carboxylate (380 mg, 1.25 mmol, 69.0% yield) as a white solid which was used in the next step without further purification.
1H NMR (400 MHz, DMSO-d
6) δ 3.92 (s, 3H), 3.95 (s, 3H), 8.13 (s, 1H), 8.34 (d, 1H), 9.03 (s, 1H). MS (ESI): [M+H]
+ m/z: calcd 304.07; found 304.2 [5-fluoro-6-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]-3-pyridyl]methanol [1350] A solution of methyl 5-fluoro-6-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]pyridine- 3-carboxylate (380 mg, 1.13 mmol) in THF (5 mL) was added dropwise to a stirred suspension of LAH (64.2 mg, 1.69 mmol) in THF (30 mL) at -10 °C. The reaction mixture was stirred of at 0 °C for 2 hr. The reaction mixture was cooled to -10 °Cand quenched by the dropwise addition of aqueous NaOH (500 µl, 20%). To the resulting mixture NaOH (solid) was added and the mixture was stirred for 15 min. The solids were filtered out. The filtrate was concentrated under reduced pressure to afford [5-fluoro-6-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]-3-pyridyl]methanol (330 mg, crude) as a yellow oil which was used in the next step without further purification. MS (ESI): [M+H]
+ m/z: calcd 276.08; found 276.0 4'-cyclopropyl-4-((5-fluoro-6-(1-methyl-4-(trifluoromethyl)-1H-imidazol-2-yl)pyridin-3- yl)methoxy)-5,6'-dimethoxy-2,5'-bipyrimidine [1351] NaH (9.88 mg, 247 μmol, 60% dispersion in mineral oil) was added to a stirred solution of [5-fluoro-6-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]-3-pyridyl]methanol (165 mg, 258 μmol) in DMF (2 mL). The resulting mixture was stirred 10 min at room temperature then 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-5-methoxy-4-methylsulfonyl- pyrimidine I-6 (86.7 mg, 258 μmol) was added. The reaction mixture was stirred at room temperature for 16 hr. The reaction mixture was diluted with water (4 mL) and extracted with EtOAc (10 mL). The organic layer was separated, washed with water (5 mL) and concentrated under reduced pressure. The residue was subjected to HPLC (0.5-6.5 min., 30- 55% water – ACN, +0.1% vol. of 25% aq. NH
3, flow: 30 mL/min, column: XBridge 100×19 mm, 5 µm), then repurified by HPLC (0.5-6.5 min., 40-60% water – MeOH, flow: 30 mL/min, column: SunFire C18100×19 mm, 5 µm) to afford 4'-cyclopropyl-4-((5-fluoro-6-(1- methyl-4-(trifluoromethyl)-1H-imidazol-2-yl)pyridin-3-yl)methoxy)-5,6'-dimethoxy-2,5'- bipyrimidine (14.0 mg, 26.3 μmol, 10.2% yield) as a light-yellow solid.
1H NMR (600 MHz, DMSO-d
6) δ 0.87 – 0.91 (m, 2H), 1.02 – 1.05 (m, 2H), 1.69 – 1.75 (m, 1H), 3.85 (s, 3H), 3.86 (s, 3H), 3.96 (s, 3H), 5.57 (s, 2H), 8.02 (d, 1H), 8.06 (s, 1H), 8.46 (s, 1H), 8.66 (s, 1H), 8.68 (s, 1H). MS (ESI): [M+H]
+ m/z: calcd 532.19; found 532.2. Example 119 (Compound 56)
Compound 56 2-(4-bromo-2-fluoro-6-methoxy-phenyl)-4-(trifluoromethyl)-1H-imidazole [1352] 3,3-Dibromo-1,1,1-trifluoro-propan-2-one (5.56 g, 20.6 mmol) and sodium acetate (3.24 g, 39.5 mmol) were mixed in water (100 mL). The resulting mixture was stirred at 100 °C for 45 min. The mixture was cooled to room temperature. The solution of 4-bromo-2- fluoro-6-methoxy-benzaldehyde (4.00 g, 17.2 mmol) and ammonium hydroxide (6.02 g, 172 mmol, 6.68 mL, 28% NH
3) in MeOH (400 mL) was added to the mixture. The reaction mixture was stirred at room temperature for 12 hr. The reaction mixture was concentrated under reduced pressure to half volume. The solid precipitate formed weas filtered off to afford 2-(4-bromo-2-fluoro-6-methoxy-phenyl)-4-(trifluoromethyl)-1H-imidazole (4.00 g, 11.8 mmol, 68.7% yield) as a yellow solid which was used in the next step without further purification. MS (ESI): [M+H]
+ m/z: calcd 338.98, 340.97; found 339.0, 341.0 2-(4-bromo-2-fluoro-6-methoxy-phenyl)-1-methyl-4-(trifluoromethyl)imidazole [1353] Cs
2CO
3 (7.69 g, 23.6 mmol) and iodomethane (2.51 g, 17.7 mmol, 1.10 mL) were added to a solution of 2-(4-bromo-2-fluoro-6-methoxy-phenyl)-4-(trifluoromethyl)-1H- imidazole (4.00 g, 11.8 mmol) in ACN (100 mL). The reaction mixture was stirred at room temperature for 24 hr. The reaction mixture was filtered. The filtrate was concentrated under reduced pressure to afford 2-(4-bromo-2-fluoro-6-methoxy-phenyl)-1-methyl-4- (trifluoromethyl)imidazole (4.00 g, 11.3 mmol, 95.9% yield) as a yellow solid which was used in the next step without further purification.
1H NMR (500 MHz, DMSO-d
6) δ 3.46 (s, 3H), 3.76 (s, 3H), 7.28 (s, 1H), 7.33 (d, 1H), 7.96 (s, 1H). MS (ESI): [M+H]
+ m/z: calcd 352.99, 354.99; found 353.0, 355.0 methyl 3-fluoro-5-methoxy-4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]benzoate [1354] A mixture of 2-(4-bromo-2-fluoro-6-methoxy-phenyl)-1-methyl-4- (trifluoromethyl)imidazole (4.00 g, 11.3 mmol), TEA (2.29 g, 22.7 mmol, 3.16 mL) and bis(diphenylphosphino)ferrocene]dichloropalladium(II)-DCM (925 mg, 1.13 mmol) in MeOH (96 mL) was stirred under 20 bar CO pressure at 120 °C for 12 hr. The reaction mixture was cooled to room temperature and concentrated under reduced pressure. The residue was dissolved in THF (20 mL), then solids were filtered out. The filtrate was concentrated under reduced pressure to afford methyl 3-fluoro-5-methoxy-4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]benzoate (2.50 g, 7.52 mmol, 66.5% yield) as a black gum which was used in the next step without further purification. MS (ESI): [M+H]
+ m/z: calcd 333.09; found 333.0 [3-fluoro-5-methoxy-4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methanol [1355] DIBAL (1.87 g, 13.2 mmol, 2.67 mL) was added dropwise to a stirred solution of methyl 3-fluoro-5-methoxy-4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]benzoate (2.50 g, 5.27 mmol) in THF (80 mL) at -40 °C. The reaction mixture was stirred at room temperature overnight. The reaction mixture was quenched with water (5 mL). An aqueous NaOH (10 mL, 10% wt.) was added to the resulting mixture. The resulting mixture was stirred at room temperature until solid precipitated dissolved. EtOAc (30 mL) was added to the mixture after. The organic layer was separated, washed with brine (10 mL), dried over anhydrous Na
2SO
4 and concentrated under reduced pressure. The residue was subjected to flash column chromatography (SiO
2, gradient DCM - ACN) to afford [3-fluoro-5-methoxy-4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methanol (1.20 g, 3.94 mmol, 74.9% yield) as a beige solid.
1H NMR (400 MHz, CDCl
3) δ 3.51 (s, 3H), 3.77 (s, 3H), 4.70 (s, 2H), 6.69 (d, 1H), 6.77 (s, 1H), 7.37 (s, 1H). MS (ESI): [M+H]
+ m/z: calcd 305.09; found 305.0 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[3-fluoro-5-methoxy-4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]-5-methoxy-pyrimidine [1356] NaH (11.1 mg, 277 μmol, 60% dispersion in mineral oil) was added to the stirred solution of [3-fluoro-5-methoxy-4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methanol (80.0 mg, 263 μmol) in THF (4 mL). The resulting mixture was stirred at room temperature for 30 min.2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-5-methoxy-4- methylsulfonyl-pyrimidine I-6 (88.5 mg, 263 μmol) was added to the mixture. The reaction mixture was stirred at room temperature for 12 hr. The reaction mixture was diluted with saturated aqueous NH
4Cl solution (2 mL) and extracted with EtOAc (2× 2 mL). The combined organic layers were washed with brine (1.0 mL), dried over anhydrous Na
2SO
4 and filtered. The filtrate was concentrated under reduced pressure. The residue was subjected to HPLC (0.5-6.5 min., 38-46% water – ACN, flow: 30 mL/min, column: SunFire C18 (L) 100×19 mm, 5 µm) to afford 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[3-fluoro-5- methoxy-4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]-5-methoxy- pyrimidine (56.0 mg, 99.9 μmol, 38.0% yield) as a yellow gum.
1H NMR (600 MHz, DMSO- d
6) δ 0.86 – 0.91 (m, 2H), 1.01 – 1.06 (m, 2H), 1.69 – 1.74 (m, 1H), 3.47 (s, 3H), 3.78 (s, 3H), 3.84 (s, 3H), 3.97 (s, 3H), 5.49 (s, 2H), 7.06 (d, 1H), 7.18 (s, 1H), 7.97 (s, 1H), 8.46 (s, 1H), 8.66 (s, 1H). MS (ESI): [M+H]
+ m/z: calcd 561.21; found 561.2. Example 120 (Compound 57)
Compound 57 2-chloro-4-[[6-[1-cyclopropyl-4-(trifluoromethyl)imidazol-2-yl]-5-fluoro-3- pyridyl]methoxy]-5-methoxy-pyrimidine Synthesis of the starting [6-[1-cyclopropyl-4-(trifluoromethyl)imidazol-2-yl]-5-fluoro- 3-pyridyl]methanol is described for compound 43. [1357] To a solution of [6-[1-cyclopropyl-4-(trifluoromethyl)imidazol-2-yl]-5-fluoro-3- pyridyl]methanol (80 mg, 265.57 μmol) in THF (1 mL) was added NaH (9.56 mg, 398.35 μmol, 60% purity) at 0 °C under nitrogen atmosphere. The reaction mixture was stirred at 0 °C for 1 hr. Then to the mixture was added 2,4-dichloro-5-methoxy-pyrimidine (57.05 mg, 318.68 μmol) at 0 °C under nitrogen atmosphere. The reaction mixture was stirred at 25 °C for 16 hrs. then was quenched by the addition of NH4Cl aqueous solution (15 mL). The resulting mixture was extracted with EtOAc (3 x 10 mL). The combined organic layers were washed with brine (15 mL), dried over anhydrous Na
2SO
4. After filtration, the filtrate was concentrated under reduced pressure. The residue was purified by Prep-TLC to give 2- chloro-4-[[6-[1-cyclopropyl-4-(trifluoromethyl)imidazol-2-yl]-5-fluoro-3-pyridyl]methoxy]- 5-methoxy-pyrimidine (60 mg, 135.20 μmol, 50% yield) as an off-white solid. MS: m/z = 444.00 [M + H]
+.
1H NMR (300 MHz, Chloroform-d) δ 8.65 (s, 1H), 7.97 (s, 1H), 7.73 (dd, J = 9.9, 1.8 Hz, 1H), 7.42 (s, 1H), 5.60 (s, 2H), 3.95 (s, 3H), 3.80 - 3.74 (m, 1H), 1.33 - 1.21 (m, 1H), 1.02 - 0.70 (m, 4H).
19F NMR (282 MHz, Chloroform-d) δ -62.69, -119.29. 2-[4-cyclopropyl-6-(trideuteriomethoxy)pyrimidin-5-yl]-4-[[6-[1-cyclopropyl-4- (trifluoromethyl)imidazol-2-yl]-5-fluoro-3-pyridyl]methoxy]-5-methoxy-pyrimidine [1358] To a solution of 2-chloro-4-[[6-[1-cyclopropyl-4-(trifluoromethyl)imidazol-2-yl]-5- fluoro-3-pyridyl]methoxy]-5-methoxy-pyrimidine (40 mg, 90.13 μmol) and [4-cyclopropyl- 6-(trideuteriomethoxy)pyrimidin-5-yl]boronic acid (17.76 mg, 90.13 μmol) in water (0.2 mL) and 1,4-dioxane (1 mL) were added 2-(Dicyclohexylphosphino)-2',4',6'-Triisopropylbiphenyl (4.30 mg, 9.01 μmol), Methanesulfonato(2-dicyclohexylphosphino-2',4',6'-tri-i-propyl-1,1'- biphenyl)(2'-amino-1,1'-biphenyl-2-yl)palladium(II) (7.63 mg, 9.01 μmol) and potassium phosphate tribasic (38.26 mg, 180.27 μmol) at 20 °C under nitrogen atmosphere. The reaction mixture was stirred at 50 °C for 16 hrs. The mixture was cooled down to room temperature and was quenched by the addition of NH
4Cl aqueous solution (30 mL). The resulting mixture was extracted with EtOAc (3 x 20 mL). The combined organic layers were washed with brine (20 mL), dried over anhydrous Na
2SO
4. After filtration, the filtrate was concentrated under reduced pressure. The residue was purified by Prep-TLC to give a crude product. The crude product was further purified by RP-Flash with the following conditions: Column: C18 spherical Column, 20-35 um, 100A, 40 g; Mobile Phase A: Water (5 mM aq. NH
4HCO
3), Mobile Phase B: MeCN; Flow rate: 40 mL/min; Gradient: 5% B to 5% B in 3 min, 5% B to 60% B in 15 min, 60% B to 60% B in 2 min, 60% B to 95% B in 8 min; Detector: UV 254 & 220 nm. The collected fractions were combined, concentrated and then lyophilized to give 2- [4-cyclopropyl-6-(trideuteriomethoxy)pyrimidin-5-yl]-4-[[6-[1-cyclopropyl-4- (trifluoromethyl)imidazol-2-yl]-5-fluoro-3-pyridyl]methoxy]-5-methoxy-pyrimidine (9.1 mg, 16.24 μmol, 18% yield) as an off-white solid. MS: m/z = 561.30 [M + H]
+.
1H NMR (300 MHz, Chloroform-d) δ 8.66 (s, 1H), 8.64 (s, 1H), 8.27 (s, 1H), 7.75 (d, J = 9.0 Hz, 1H), 7.41 (s, 1H), 5.64 (s, 2H), 4.03 (s, 3H), 3.81 - 3.72 (m, 1H), 1.78 - 1.69 (m, 1H), 1.28 - 1.20 (m, 2H), 0.99 - 0.89 (m, 4H), 0.83 - 0.77 (m, 2H).
19F NMR (282 MHz, Chloroform-d) δ -62.71, - 119.42. Example 121 (Compound 58)
methyl 2-chloro-6-methylsulfanyl-pyrimidine-4-carboxylate [1359] Sodium methanethiolate (15.2 mmol, 5.08 mL, 21% wt. in water) was added to a stirred solution of methyl 2,6-dichloropyrimidine-4-carboxylate (3.00 g, 14.5 mmol) in THF (30 mL) at 5 °C. The resulting mixture was stirred at room temperature for 16 hr. The reaction mixture was concentrated under reduced pressure. The residue was diluted with water (3 mL). The precipitate formed was filtered off and air-dried to afford methyl 2-chloro- 6-methylsulfanyl-pyrimidine-4-carboxylate (2.40 g, 11.0 mmol, 75.7% yield) as a white solid which was used in the next step without further purification.
1H NMR (400 MHz, CDCl
3) δ 2.64 (s, 3H), 4.01 (s, 3H), 7.79 (s, 1H). MS (ESI): [M+H]
+ m/z: calcd 219.0; found 219.0 methyl 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-6-methylsulfanyl-pyrimidine-4- carboxylate [1360] Methyl 2-chloro-6-methylsulfanyl-pyrimidine-4-carboxylate (1.19 g, 5.03 mmol), (4- cyclopropyl-6-methoxy-pyrimidin-5-yl)boronic acid I-5 (1.17 g, 6.04 mmol), potassium phosphate tribasic (1.07 g, 5.03 mmol) and XPhos Pd G3 (214 mg, 252 μmol) were mixed in a degassed mixture of dioxane (9 mL) and water (2 mL) under argon atmosphere. The reaction mixture was stirred at 70 °C for 16 hr. The reaction mixture was cooled to room temperature and concentrated under reduced pressure. The residue was subjected to flash column chromatography (silica, gradient hexane - MTBE) to afford methyl 2-(4-cyclopropyl- 6-methoxy-pyrimidin-5-yl)-6-methylsulfanyl-pyrimidine-4-carboxylate (1.30 g, 3.91 mmol, 77.8% yield) as a white solid.
1H NMR (400 MHz, CDCl
3) δ 0.87 – 0.97 (m, 2H), 1.18 – 1.27 (m, 2H), 1.63 – 1.71 (m, 1H), 2.61 (s, 3H), 3.91 (s, 3H), 4.02 (s, 3H), 7.86 (s, 1H), 8.63 (s, 1H). MS (ESI): [M+H]
+ m/z: calcd 333.1; found 333.1 methyl 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-6-methylsulfonyl-pyrimidine-4- carboxylate [1361] mCPBA (1.22 g, 5.30 mmol, 75% purity) was added to a stirred solution of methyl 2- (4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-6-methylsulfanyl-pyrimidine-4-carboxylate (800 mg, 2.41 mmol) in DCM (20 mL). The resulting mixture was stirred at room temperature for 24 hr. The reaction mixture was washed with a saturated aqueous solution of NaHCO
3 (30 mL), dried over anhydrous Na
2SO
4 and concentrated under reduced pressure to afford methyl 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-6-methylsulfonyl-pyrimidine-4-carboxylate (500 mg, 1.37 mmol, 57.0% yield) as a light-yellow solid which was used in the next step without further purification.
1H NMR (400 MHz, CDCl
3) δ 0.87 – 0.95 (m, 2H), 1.21 – 1.27 (m, 2H), 1.56 – 1.65 (m, 1H), 3.31 (s, 3H), 3.88 (s, 3H), 4.07 (s, 3H), 8.59 (s, 1H), 8.64 (s, 1H). MS (ESI): [M+H]
+ m/z: calcd 365.09; found 365.0 methyl 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-6-[[4-[1-cyclopropyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidine-4-carboxylate Synthesis of the starting [4-[1-cyclopropyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methanol is described for compound 2. [1362] DBU (52.6 mg, 345 μmol, 52.0 μL) was added to a stirred solution of [4-[1- cyclopropyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methanol (75.0 mg, 266 μmol) and methyl 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-6-methylsulfonyl-pyrimidine-4- carboxylate (96.8 mg, 266 μmol) in THF (1 mL). The reaction mixture was stirred at room temperature for 1 hr. The reaction was quenched by addition of water (5 mL) and extracted with EtOAc (2×5 mL). The combined organic layers were dried over anhydrous Na
2SO
4 and concentrated under reduced pressure to afford methyl 2-(4-cyclopropyl-6-methoxy- pyrimidin-5-yl)-6-[[4-[1-cyclopropyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidine-4-carboxylate (110 mg, 194 μmol, 73.1% yield) as a yellow oil which was used in the next step without further purification. MS (ESI): [M+H]
+ m/z: calcd 567.20; found 567.2 [2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-6-[[4-[1-cyclopropyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidin-4-yl]methanol [1363] A solution of methyl 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-6-[[4-[1- cyclopropyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidine-4-carboxylate (110 mg, 194 μmol) in THF (0.5 mL) was added dropwise to a suspension if LAH (18.3 mg, 483 μmol) in THF (2 mL). The reaction mixture was stirred at 0 °C for 30 min. The reaction mixture was quenched by addition of water (0.1 mL). The mixture was dried over anhydrous Na
2SO
4 and filtered. The filtrate subjected to HPLC (0-2-9 min., 33-40-45% water – ACN, flow: 30 mL/min, column: Chromatorex C18 SMB100-5T, 100×19 mm, 5 µm) to afford [2- (4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-6-[[4-[1-cyclopropyl-4-(trifluoromethyl)imidazol- 2-yl]phenyl]methoxy]pyrimidin-4-yl]methanol (15.0 mg, 27.9 μmol, 14.4% yield) as a yellow solid.
1H NMR (500 MHz, DMSO-d
6) δ 0.85 – 0.93 (m, 4H), 0.96 – 1.05 (m, 4H), 1.63 – 1.70 (m, 1H), 3.72 – 3.79 (m, 1H), 3.85 (s, 3H), 4.55 (s, 2H), 5.51 (s, 2H), 5.66 (br. s., 1H), 7.01 (s, 1H), 7.58 (d, 2H), 7.91 (d, 2H), 7.95 (s, 1H), 8.67 (s, 1H). MS (ESI): [M+H]
+ m/z: calcd 539.23; found 539.2. Example 122 (Compound 60)
2,4-dichloro-6-methylsulfanyl-pyrimidine [1364] To a solution of 2,4,6-trichloropyrimidine (4.8 g, 26.17 mmol, 3.01 mL) in THF (67 mL) and H2O (12 mL) was added sodium methanethiolate (1.93 g, 27.48 mmol) at 0 °C. The mixture was stirred at 25 °C for 4.5 hrs. The reaction mixture was quenched with saturated NH
4Cl aqueous solution (50 mL), extracted with diethyl ether (50 mL x 3). The organic layers were combined, dried over anhydrous Na
2SO
4. After filtration, the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography on silica gel, eluted with 18% EtOAc in PE to give 2,4-dichloro-6-methylsulfanyl-pyrimidine (3.4 g, 17.43 mmol, 67% yield) as a white solid. MS: m/z = 194.90 [M + H]+.
1H NMR (300 MHz, Chloroform-d) δ 7.13 (s, 1H), 2.59 (s, 3H). 2-chloro-4-[[6-[1-cyclopropyl-4-(trifluoromethyl)imidazol-2-yl]-5-fluoro-3- pyridyl]methoxy]-6-methylsulfanyl-pyrimidine & 4-chloro-2-[[6-[1-cyclopropyl-4-(1,1- difluoroethyl)imidazol-2-yl]-5-fluoro-3-pyridyl]methoxy]-6-methylsulfanyl-pyrimidine Synthesis of the starting [6-[1-cyclopropyl-4-(trifluoromethyl)imidazol-2-yl]-5-fluoro- 3-pyridyl]methanol is described for compound 43. [1365] To a solution of [6-[1-cyclopropyl-4-(trifluoromethyl)imidazol-2-yl]-5-fluoro-3- pyridyl]methanol (120 mg, 398.35 μmol) in THF (3 mL) was added NaH (19.12 mg, 478.03 μmol, 60% purity) under nitrogen atmosphere at 0 °C. The resulting mixture was stirred for 1 hour at 0 °C. To the above mixture was added 2,4-dichloro-6-methylsulfanyl-pyrimidine (93.25 mg, 478.03 μmol) under nitrogen atmosphere at 25 °C. The resulting mixture was stirred at 25 °C for 16 hrs. The reaction was quenched by the addition of NH
4Cl aqueous solution (30 mL). The resulting mixture was extracted with EtOAc (3 x 10 mL). The combined organic layers were washed with brine (3 x 10 mL), dried over anhydrous Na
2SO
4. After filtration, the filtrate was concentrated under reduced pressure. The residue was purified by Prep-TLC, eluted with 10% MeOH in DCM to give a mixture of 2-chloro-4-[[6-[1- cyclopropyl-4-(trifluoromethyl)imidazol-2-yl]-5-fluoro-3-pyridyl]methoxy]-6- methylsulfanyl-pyrimidine & 4-chloro-2-[[6-[1-cyclopropyl-4-(1,1-difluoroethyl)imidazol-2- yl]-5-fluoro-3-pyridyl]methoxy]-6-methylsulfanyl-pyrimidine (120 mg, 263.23 μmol, 66% yield) as an off-white solid. MS: m/z = 460.00 [M + H]
+.
1H NMR (300 MHz, Chloroform-d) δ 8.64 - 8.59 (m, 1H), 7.75 - 7.63 (m, 1H), 7.42 (s, 1H), 6.91 (s, 1H), 5.54 - 5.51 (m, 2H), 3.76 - 3.73 (m, 1H), 2.57 - 2.55 (m, 3H), 0.97 - 0.91 (m, 2H), 0.82 - 0.76 (m, 2H). 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[6-[1-cyclopropyl-4- (trifluoromethyl)imidazol-2-yl]-5-fluoro-3-pyridyl]methoxy]-6-methylsulfanyl-pyrimidine) [1366] To a stirred mixture of 2-chloro-4-[[6-[1-cyclopropyl-4-(trifluoromethyl)imidazol-2- yl]-5-fluoro-3-pyridyl]methoxy]-6-methylsulfanyl-pyrimidine & 4-chloro-2-[[6-[1- cyclopropyl-4-(1,1-difluoroethyl)imidazol-2-yl]-5-fluoro-3-pyridyl]methoxy]-6- methylsulfanyl-pyrimidine (160.00 mg, 347.94 μmol) in 1,4-dioxane (5 mL) and water (1 mL) were added (4-cyclopropyl-6-methoxypyrimidin-5-yl)boronic acid I-5 (67.5 mg, 347.94 μmol), 1,1'-bis(diphenylphosphino)ferrocene-palladium(II)dichloride (28.41 mg, 34.79 μmol) and potassium phosphate (147.71 mg, 695.88 μmol) under nitrogen atmosphere at 25 °C. The resulting mixture was stirred under nitrogen atmosphere for 16 hrs at 40 °C. The reaction mixture was cooled to room temperature, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by Prep-TLC (EtOAc : PE = 2 : 1) to give crude product (150 mg, mixture of two isomers). The crude product was separated by Prep- CHIRAL-HPLC with the following conditions: CHIRALPAK IE 2x25 cm, 5 μm; Mobile Phase A: Hex : DCM = 3 : 1 (0.5% 2 M NH
3-MeOH), Mobile Phase B: EtOH; Flow rate: 20 mL/min; Gradient: isocratic 5; Wavelength: 220/254 nm. [1367] The first eluting isomer peak was combined, concentrated under reduced pressure and then lyophilized to give 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[6-[1-cyclopropyl-4- (trifluoromethyl)imidazol-2-yl]-5-fluoro-3-pyridyl]methoxy]-6-methylsulfanyl-pyrimidine (19.4 mg, 33.82 μmol, 10% yield, as an off-white solid. MS: m/z = 574.05 [M + H]
+.
1H NMR (300 MHz, Chloroform-d) δ 8.65 (s, 1H), 8.59 (s, 1H), 7.70 (d, J = 9.6 Hz, 1H), 7.42 (s, 1H), 6.65 (s, 1H), 5.55 (s, 2H), 3.94 (s, 3H), 3.79 - 3.71 (m, 1H), 2.55 (s, 3H), 1.84 - 1.75 (m, 1H), 1.26 - 1.21 (m, 2H), 0.99 - 0.90 (m, 4H), 0.83 - 0.76 (m, 2H).
19F NMR (282 MHz, Chloroform-d) δ -62.69, -119.55.
2-chloro-4-[[6-[1-cyclopropyl-4-(trifluoromethyl)imidazol-2-yl]-5-fluoro-3- pyridyl]methoxy]-5-methoxy-pyrimidine Synthesis of the methyl 6-[1-cyclopropyl-4-(trifluoromethyl)imidazol-2-yl]-5-fluoro- pyridine-3-carboxylate is described for compound 43. [1368] Sodium tert-butoxide (43.1 mg, 448 μmol) was added to a solution of [6-[1- cyclopropyl-4-(trifluoromethyl)imidazol-2-yl]-5-fluoro-3-pyridyl]methanol (90.0 mg, 299 μmol) and 2,4-dichloro-5-methoxy-pyrimidine (64.2 mg, 359 μmol) in THF (2 mL) at room temperature. The resulting mixture was stirred at room temperature for 13 hr. The reaction mixture was diluted with water (5 mL) and extracted with EtOAc (2×5 mL). The combined organic layers were washed with brine (5 mL), dried over anhydrous Na
2SO
4 and filtered. The filtrate was concentered under reduced pressure to afford 2-chloro-4-[[6-[1-cyclopropyl- 4-(trifluoromethyl)imidazol-2-yl]-5-fluoro-3-pyridyl]methoxy]-5-methoxy-pyrimidine (120 mg) as light-yellow gum which was used in the next step without further purification. MS (ESI): [M+H]
+ m/z: calcd 444.09; found 444.3 2-[4-cyclopropyl-6-(difluoromethoxy)pyrimidin-5-yl]-4-[[6-[1-cyclopropyl-4- (trifluoromethyl)imidazol-2-yl]-5-fluoro-3-pyridyl]methoxy]-5-methoxy-pyrimidine [1369] 2-Chloro-4-[[6-[1-cyclopropyl-4-(trifluoromethyl)imidazol-2-yl]-5-fluoro-3- pyridyl]methoxy]-5-methoxy-pyrimidine (120 mg, crude), 4-cyclopropyl-6- (difluoromethoxy)-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyrimidine I-10 (84.4 mg, 270 μmol), XPhos Pd G3 (11.4 mg, 13.5 μmol) and potassium phosphate tribasic (115 mg, 541 μmol) were mixed in a degassed mixture of dioxane (20 mL) and water (1 mL) under argon atmosphere. The reaction mixture was stirred at 90 °C for 2 hr. The reaction mixture was cooled to room temperature and filtered. The filtrate was concentrated under reduced pressure. The residue was subjected to HPLC (0-5 min, 60-85% water+FA (0.1% vol.) - MeOH+FA (0.1% vol.); flow: 30 mL/min, column: XSelect CSH PFP, 100×19 mm, 5 µm) to afford 2-[4-cyclopropyl-6-(difluoromethoxy)pyrimidin-5-yl]-4-[[6-[1-cyclopropyl-4- (trifluoromethyl)imidazol-2-yl]-5-fluoro-3-pyridyl]methoxy]-5-methoxy-pyrimidine (18.0 mg, 30.3 μmol, 11.2% yield) as a beige solid.
1H NMR (500 MHz, DMSO-d
6) δ 0.83 – 0.90 (m, 4H), 0.97 – 1.03 (m, 2H), 1.08 – 1.14 (m, 2H), 1.87 – 1.94 (m, 1H), 3.73 – 3.79 (m, 1H), 3.98 (s, 3H), 5.60 (s, 2H), 7.81 (t, 1H, CHF
2), 8.03 (d, 1H), 8.07 (s, 1H), 8.53 (s, 1H), 8.70 (s, 1H), 8.79 (s, 1H). MS (ESI): [M+H]
+ m/z: calcd 594.19; found 594.2. Example 124 (Compound 62)
2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[6-[1-ethyl-4-(trifluoromethyl)imidazol-2-yl]- 5-fluoro-3-pyridyl]methoxy]-5-methyl-pyrimidine Synthesis of the starting [6-[1-ethyl-4-(trifluoromethyl)imidazol-2-yl]-5-fluoro-3- pyridyl]methanol is described for Compound 118. [1370] NaH (31.1 mg, 778 μmol, 60% dispersion in mineral oil) was added to a solution of [6-[1-ethyl-4-(trifluoromethyl)imidazol-2-yl]-5-fluoro-3-pyridyl]methanol (150 mg, 519 μmol) and 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-5-methyl-4-methylsulfonyl- pyrimidine I-7 (175 mg, 545 μmol) in THF (7 mL). The resulting mixture was stirred at room temperature for 13 hr. The reaction mixture was poured into a saturated aqueous solution of NH
4Cl (15 mL) and extracted with MTBE (3×15 mL). The combined organic layers were dried over anhydrous Na
2SO
4 and concentrated under reduced pressure. The residue was dissolved in ACN (6.0 mL). Metal scavenger SiliaMetS
® Dimercaptotriazine (100 mg) was added to the solution. The resulting suspension was stirred at room temperature for 20 hr. The suspension was filtered. The filtrate was subjected to HPLC (0-1.3-5.5 min., 35-40-50% water – ACN, flow: 30 mL/min, column: Chromatorex 100×19 mm, 5 µm) to afford 2-(4- cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[6-[1-ethyl-4-(trifluoromethyl)imidazol-2-yl]-5- fluoro-3-pyridyl]methoxy]-5-methyl-pyrimidine (27.0 mg, 51.0 μmol, 9.8% yield) as a yellow solid.
1H NMR (500 MHz, DMSO-d
6) δ 0.85 – 0.90 (m, 2H), 1.01 – 1.06 (m, 2H), 1.34 (t, 3H), 1.64 – 1.71 (m, 1H), 2.25 (s, 3H), 3.83 (s, 3H), 4.25 (q, 2H), 5.59 (s, 2H), 8.05 (d, 1H), 8.16 (s, 1H), 8.58 (s, 1H), 8.67 (s, 1H), 8.70 (s, 1H). MS (ESI): [M+H]
+ m/z: calcd 530.22; found 530.2. Example 125 (Compound 66)
2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-5-(2,2-difluorovinyl)-4-[[4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidine [1371] 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidine-5-carbaldehyde 68a (80 mg, 156.72 μmol) and 2,2-difluoro-2-triphenylphosphaniumyl-acetate (111.68 mg, 313.44 μmol) were added to the reaction flask and then backfilled with nitrogen. Dimethylformamide (0.8 mL) was added via syringe. The resulting mixture was stirred at 60 °C for 40 minutes under nitrogen atmosphere. SM was disappeared by TLC. The mixture was allowed to cool down to room temperature. The mixture was purified directly by RP-Flash with the following conditions: Column: C18 spherical Column, 20-35 um, 100A, 40 g; Mobile Phase A: Water (5 mM aq. NH
4HCO
3), Mobile Phase B: MeCN; Flow rate: 60 mL/min; Gradient: 5% B to 5% B in 5 min, 5% B to 62% B in 20 min, 62% B to 62% B in 3 min, 62% B to 95% B in 5 min; Detector: UV 254 & 220 nm. The collected fractions were combined, concentrated and then lyophilized to give 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-5-(2,2-difluorovinyl)-4- [[4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidine (68 mg, 124.89 μmol, 80% yield) as a yellow solid. MS: m/z = 567.15 [M + Na]
+.
1H NMR (400 MHz, Chloroform-d) δ 8.81 (s, 2H), 7.70 (d, J = 8.0 Hz, 2H), 7.56 (d, J = 8.0 Hz, 2H), 7.36 (s, 1H), 5.61 - 5.50 (m, 2H), 4.04 (s, 3H), 3.82 (s, 3H), 1.88 - 1.81 (m, 1H), 1.46 - 1.32 - 1.28 (m, 2H), 1.09 - 1.00 (m, 2H). 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-5-(2,2-difluoroethyl)-4-[[4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidine [1372] To a stirred solution of 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-5-(2,2- difluorovinyl)-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidine (68 mg, 124.89 μmol) in EtOAc (8 mL) was added 10% Palladium on carbon (45 mg) at room temperature. The resulting mixture was stirred for 7 hrs at room temperature under hydrogen atmosphere (1.5 atm). The reaction was filtered and the filtrate was concentrated under reduced pressure. The residue was purified by Prep-TLC (EtOAc / PE = 2 / 1) to afford the residue. The residue was further purified by RP-Flash with the following conditions: Column: C18 spherical Column, 20-35 um, 100A, 40 g; Mobile Phase A: Water (5 mM aq. NH
4HCO
3), Mobile Phase B: MeCN; Flow rate: 60 mL/min; Gradient: 5% B to 5% B in 5 min, 5% B to 55% B in 20 min, 55% B to 55% B in 1 min, 59% B to 95% B in 5 min; Detector: UV 254 & 220 nm. The collected fractions were combined, concentrated and then lyophilized to give 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-5-(2,2-difluoroethyl)-4-[[4- [1-methyl-4-(trifluoromethyl)imidazol-2yl]phenyl]methoxy]pyrimidine (32.6 mg, 59.65 μmol, 36% yield) as a white solid. MS: m/z = 547.20 [M + H]
+; 569.20 [M + Na]
+.
1H NMR (400 MHz, Chloroform-d) δ 8.82 (s, 1H), 8.59 (s, 1H), 7.70 (d, J = 8.4 Hz, 2H), 7.55 (d, J = 8.4 Hz, 2H), 7.36 (s, 1H), 6.29 - 5.98 (m, 1H), 5.58 (s, 2H), 4.05 (s, 3H), 3.86 (s, 3H), 3.32 - 3.21 (m, 2H), 1.86 - 1.79 (m, 1H), 1.58 - 1.52 (m, 2H), 1.16 - 1.07 (m, 2H).
19F NMR (376 MHz, Chloroform-d) δ -62.78, -114.93. Example 126 (Compound 67)

2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[4-[1-(cyclopropylmethyl)-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]-5-methoxy-pyrimidine (67) [1373] To a mixture of 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-5-methoxy-4- methylsulfonyl-pyrimidine I-6 (171 mg, 0.508 mmol) in THF (2 mL) was added NaH (24.3 mg, 0.608 mmol, 60 wt% in mineral oil). The mixture was stirred at 25 °C for 30 min. To the mixture was added [4-[1-(cyclopropylmethyl)-4-(trifluoromethyl)imidazol-2- yl]phenyl]methanol (150 mg, 0.506 mmol). The mixture was stirred at 45 °C for 2.5 hrs. The mixture was quenched by addition of water (1 mL) and concentrated. The residue was purified by preparative HPLC (Instrument: Gilson GX-281 Liquid Handler, Gilson 322 Pump, Gilson 156 UV Detector; Column: 2_Phenomenex Gemini C1875 x 40 mm x 3 µm; Mobile phase A: H2O with 0.05% NH4HCO3 (v%); Mobile phase B: MeCN; Gradient: B from 53% to 83% in 7.8 min, hold 100% B for 1 min; Flow Rate: 30 mL/min; Column Temperature: 30 °C; Wavelength: 220 nm, 254 nm) to afford 2-(4-cyclopropyl-6-methoxy- pyrimidin-5-yl)-4-[[4-[1-(cyclopropylmethyl)-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]-5-methoxy-pyrimidine (60.3 mg, 21.6% yield) as white solid.
1H NMR (400 MHz, DMSO-d
6) δ ppm 8.65 (s, 1H), 8.43 (s, 1H), 8.05 (d, J = 1.3 Hz, 1H), 7.65 - 7.72 (m, 2H), 7.56 - 7.61 (m, 2H), 5.50 (s, 2H), 3.93 - 3.96 (m, 5H), 3.85 (s, 3H), 1.71 (s, 1H), 1.15 (t, J = 7.5 Hz, 1H), 0.99 - 1.06 (m, 2H), 0.87 (dd, J = 7.9, 3.1 Hz, 2H), 0.46 - 0.54 (m, 2H), 0.25 - 0.34 (m, 2H);
19FNMR (376 MHz, DMSO-d
6) δ ppm -60.71; MS (ESI) [M+H]
+ m/z: calcd 553.2, found 553.2 Example 127 (Compounds 222 & 68)
5-chloro-7-((3-fluoro-4-(1-methyl-4-(trifluoromethyl)-1H-imidazol-2-yl)benzyl)oxy)-2- (tetrahydro-2H-pyran-2-yl)-2H-pyrazolo[4,3-d]pyrimidine [1374] To a stirred solution of [4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methanol I-2b (1.5 g, 5.85 mmol) in THF (15 mL) was added NaH (168.59 mg, 7.03 mmol) at 0 °C under nitrogen atmosphere. After stirring at 0 °C for 0.5 hr, ethyl 2,4- dichloropyrimidine-5-carboxylate (1.55 g, 7.03 mmol) was added at 0 °C. The resulting mixture was stirred at 25 °C for 16 hrs. The reaction was quenched by the addition of saturated NH
4Cl aqueous solution (30 mL) at 0 °C. The resulting mixture was extracted with EtOAc (3 x 60 mL). The combined organic layers were dried over anhydrous Na
2SO
4. After filtration, the filtrate was concentrated under reduced pressure. The residue was purified by silica gel column chromatography, eluted with 36% EtOAc in PE to give desired isomer ethyl 2-chloro-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidine-5- carboxylate (1.6 g, 3.63 mmol, 62% yield, smaller polarity) as an off-white solid and undesired isomer ethyl 4-chloro-2-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidine-5-carboxylate (140 mg, 317.60 μmol, 5% yield, higher polarity) as an off-white solid. MS: m/z = 441.05 [M + H]
+.
1H NMR (400 MHz, Chloroform- d) δ 8.93 (s, 1H), 7.70 (d, J = 8.4 Hz, 2H), 7.64 (d, J = 8.4 Hz, 2H), 7.34 (s, 1H), 5.64 (s, 2H), 4,41 (q, J = 7.2 Hz, 2H), 3.80 (s, 3H), 1.39 (t, J = 7.2 Hz, 3H). ethyl 4-cyclopropyl-6-methoxy-4-((4-(1-methyl-4-(trifluoromethyl)-1H-imidazol-2- yl)benzyl)oxy)-[2,5-bipyrimidine]-5-carboxylate [1375] To a mixture of ethyl 2-chloro-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidine-5-carboxylate (1.2 g, 2.72 mmol) and (4-cyclopropyl-6- methoxy-pyrimidin-5-yl)boronic acid I-5 (792.17 mg, 4.08 mmol) in 1,4-dioxane (20 mL) and water (4 mL) under nitrogen atmosphere were added potassium phosphate (1.16 g, 5.44 mmol) and [1,1-Bis(diphenylphosphino)ferrocene]dichloropalladium(II) (222.31 mg, 272.23 μmol) at 25 °C. The resulting mixture was stirred at 25 °C for 3 hrs then filtered. The filtrate was concentrated under reduced pressure. The residue was purified by silica gel column chromatography, eluted with 60% EtOAc in PE to give ethyl 4-cyclopropyl-6-methoxy-4-((4- (1-methyl-4-(trifluoromethyl)-1H-imidazol-2-yl)benzyl)oxy)-[2,5-bipyrimidine]-5- carboxylate (970 mg, 1.75 mmol, 64% yield) as a yellow solid. MS: m/z = 555.35 [M + H]
+.
1H NMR (400 MHz, Chloroform-d) δ 9.20 (s, 1H), 8.68 (s, 1H), 7.71 - 7.62 (m, 4H), 7.34 (s, 1H), 5.66 (s, 2H), 4.46 (q, J = 7.2 Hz, 2H), 3.95 (s, 3H), 3.80 (s, 3H), 1.79 - 1.71 (m, 1H), 1.44 (t, J = 7.6 Hz, 3H), 1.29 - 1.23 (m, 2H), 0.96 - 0.90 (m, 2H).
19F NMR (376 MHz, Chloroform-d) δ -62.74. [2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidin-5-yl]methanol [1376] To a stirred solution of ethyl 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[4-[1- methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidine-5-carboxylate (600 mg, 1.08 mmol) in THF (10 mL) was added lithium aluminum hydride (20.53 mg, 541.01 μmol) at 0 °C under nitrogen atmosphere. The resulting mixture was stirred for 3 hrs at 0 °C then quenched with saturated NH
4Cl aqueous solution (100 mL) at 0 °C. The resulting mixture was extracted with EtOAc (3 x 100 mL). The combined organic layers were washed with brine (200 mL), dried over anhydrous Na
2SO
4. After filtration, the filtrate was concentrated under reduced pressure. The residue was purified by silica gel column chromatography, eluted with 80% EtOAc in PE to give [2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[4-[1- methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidin-5-yl]methanol (295 mg, 575.63 μmol, 53% yield) as a yellow solid. MS: m/z = 513.30 [M + H]
+.
1H NMR (400 MHz, Chloroform-d) δ 8.68 (s, 1H), 8.66 (s, 1H), 7.66 (d, J = 8.4 Hz, 2H), 7.56 (d, J = 8.4 Hz, 2H), 7.34 (s, 1H), 5.57 (s, 2H), 4.78 (s, 2H), 3.94 (s, 3H), 3.80 (s, 3H), 2.79 (s, 1H), 1.77 - 1.71 (m, 1H), 1.26 - 1.20 (m, 2H), 0.93 - 0.86 (m, 2H).
19F NMR (376 MHz, Chloroform-d) δ -62.74. 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidine-5-carbaldehyde [1377] To a stirred solution of [2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[4-[1- methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidin-5-yl]methanol (900 mg, 1.76 mmol) in DCM (10 mL) was added Dess-Martin periodinane (893.83 mg, 2.11 mmol) at 0 °C under nitrogen atmosphere. The resulting mixture was stirred for 1 hour at 0 °C under nitrogen atmosphere. The reaction mixture was quenched by saturated NaHCO
3 aqueous solution (200 mL) at 0 °C. The resulting mixture was extracted with EtOAc (3 x 150 mL). The combined organic layers were washed with brine (200 mL), dried over anhydrous Na
2SO
4. After filtration, the filtrate was concentrated under reduced pressure. The residue was purified by silica gel column chromatography, eluted with 40% EtOAc in PE to give 2- (4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidine-5-carbaldehyde (500 mg, 979.50 μmol, 56% yield) as a yellow solid. MS: m/z = 511.10 [M + H]
+.
1H NMR (400 MHz, Chloroform-d) δ 10.46 (s, 1H), 9.12 (s, 1H), 8.69 (s, 1H), 7.70 (d, J = 8.0 Hz, 2H), 7.61 (d, J = 8.0 Hz, 2H), 7.34 (d, J = 1.6 Hz, 1H), 5.69 (s, 2H), 3.96 (s, 3H), 3.81 (s, 3H), 1.80 - 1.72 (m, 2H), 1.31 - 1.23 (m, 2H), 0.98 - 0.92 (m, 2H). 1-[2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol- 2-yl]phenyl]methoxy]pyrimidin-5-yl]-2,2,2-trifluoro-ethanol [1378] To a solution of 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidine-5-carbaldehyde 68a (100 mg, 195.90 μmol) and K
2CO
3 (2.71 mg, 19.59 μmol) in DMF (2 mL) was added (trifluoromethyl)trimethylsilane (83.57 mg, 587.70 μmol, 93.37 μL) at 25 °C. The reaction was stirred at room temperature for 2 hrs. Then tetrabutylammonium fluoride (0.2 mL, 0.2 mmol, 1mol/L in THF) was added at 0 °C. The reaction solution was stirred at 0 °C for 30 minutes then was quenched by saturated NH
4Cl aqueous solution (50 mL) at 0 °C. The resulting mixture was extracted with EtOAc (3 x 50 mL). The combined organic layers were washed with brine (100 mL), dried over anhydrous Na
2SO
4. After filtration, the filtrate was concentrated under reduced pressure. The residue was purified by silica gel column chromatography, eluted with 80% EtOAc in PE to give 85 mg crude product. The crude product was re-purified by RP-Flash with the following conditions: Column: C18 spherical, 20-30 um, 100A, 40 g; Mobile Phase A: Water (5 mM aq. NH
4HCO
3), Mobile Phase B: MeCN; Flow rate: 60 mL/min; Gradient: 2% B to 2% B in 5 min, 5% B to 50% B in 17 min, 50% B to 50% B in 2 min, 50% B to 95% B in 5 min; Detector: UV 254 & 210 nm. The collected fractions were combined, concentrated and then lyophilized to give 1-[2-(4- cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidin-5-yl]-2,2,2-trifluoro-ethanol (80 mg, 137.82 μmol, 70% yield) as an off-white solid. MS: m/z = 581.15 [M + H]
+.
1H NMR (400 MHz, Chloroform-d) δ 8.81 (s, 1H), 8.65 (s, 1H), 7.59 (d, J = 8.0 Hz, 2H), 7.47 (d, J = 8.0 Hz, 2H), 7.35 (s, 1H), 5.92 (s, 1H), 5.59 (d, J = 12.8 Hz, 1H), 5.50 (d, J = 12.8 Hz, 1H), 5.41 - 5.35 (m, 1H), 3.94 (s, 3H), 3.79 (s, 3H), 1.74 - 1.66 (m, 1H), 1.32 - 1.13 (m, 2H), 0.97 - 0.85 (m, 2H).
19F NMR (376 MHz, Chloroform-d) δ -62.731, -78.01. [1-[2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidin-5-yl]-2,2,2-trifluoro-ethyl] 4- methylbenzenesulfonate To a stirred solution of 1-[2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidin-5-yl]-2,2,2-trifluoro-ethanol (110 mg, 189.50 μmol) in THF (2.5 mL) was added NaH (8.34 mg, 208.45 μmol, 60% dispersion in oil) at 0 °C under nitrogen atmosphere. The resulting mixture was stirred for 1 hour at 0 °C under nitrogen atmosphere. To the above mixture was added 4-methylbenzenesulfonyl chloride (43.35 mg, 227.40 μmol) at 0 °C. The resulting mixture was stirred at 25 °C for 2 hrs. The reaction mixture was quenched by saturated NH
4Cl aqueous solution (80 mL) at 0 °C. The resulting mixture was extracted with EtOAc (60 mL x 2). The combined organic layers were washed with brine (100 mL), dried over anhydrous Na
2SO
4. After filtration, the filtrate was concentrated under reduced pressure. The residue was purified by Prep-TLC, eluted with 40% EtOAc in PE to give [1-[2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidin-5-yl]-2,2,2-trifluoro-ethyl] 4- methylbenzenesulfonate (81 mg, 110.25 μmol, 58% yield) as an off-white solid. MS: m/z = 735.35 [M + H]
+.
1H NMR (400 MHz, Chloroform-d) δ 8.73 (s, 1H), 8.68 (s, 1H), 7.75 - 7.70 (m, 4H), 7.57 (d, J = 8.0 Hz, 2H), 7.35 (s, 1H), 7.31 (d, J = 8.0 Hz, 2H), 6.22 - 6.18 (m, 1H), 5.58 (d, J = 2.0 Hz, 2H), 3.97 (s, 3H), 3.82 (s, 3H), 2.44 (s, 3H), 1.72 - 1.66 (m, 1H), 1.31 - 1.23 (m, 2H), 0.98 - 0.92 (m, 2H).
19F NMR (376 MHz, Chloroform-d) δ -62.78, -75.96. 1-[2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol- 2-yl]phenyl]methoxy]pyrimidin-5-yl]-2,2,2-trifluoro-ethanamine [1379] To a solution of [1-[2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidin-5-yl]-2,2,2-trifluoro-ethyl] 4- methylbenzenesulfonate 68b (40 mg, 54.45 μmol) in THF (0.5 mL) was added Ammonium hydroxide (0.5 mL, 28% aqueous solution) at 25 °C under nitrogen atmosphere. The resulting mixture was stirred for 16 hrs at 80 °C then concentrated under reduced pressure. The residue was purified by Prep-TLC, eluted with 75% EtOAc in PE to give the less polar undesired isomer (6.6 mg) and the more polar desired isomer (1.3 mg). The desired isomer was re-purified by RP-Flash with the following conditions: Column: C18 spherical, 20-30 um, 100A, 20 g; Mobile Phase A: Water (5 mM aq. NH
4HCO
3), Mobile Phase B: MeCN; Flow rate: 25 mL/min; Gradient: 2% B to 2% B in 5 min, 5% B to 40% B in 15 min, 40% B to 40% B in 2 min, 40% B to 95% B in 5 min; Detector: UV 254 & 210 nm. The collected fractions were combined, concentrated and then lyophilized to give 1-[2-(4- cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidin-5-yl]-2,2,2-trifluoro-ethanamine (0.7 mg, 1.21 μmol, 2% yield). MS: m/z = 580.20 [M + H]
+.
1H NMR (400 MHz, DMSO-d
6) δ 8.96 (s, 1H), 8.69 (s, 1H), 7.95 (s, 1H), 7.75 (d, J = 8.0 Hz, 2H), 7.58 (d, J = 8.0 Hz, 2H), 5.62 – 5.52 (m, 2H), 4.88 – 4.78 (m, 1H), 3.86 (s, 3H), 3.80 (s, 3H), 2.78 (d, J = 8.0 Hz, 2H), 1.69 – 1.65 (m, 1H), 1.11 – 1.02 (m, 2H), 0.93 – 0.87 (m, 2H).
19F NMR (376 MHz, DMSO-d
6) δ -60.82, -74.57.
carboxylate Starting material ethyl 6-[1-cyclopropyl-4-(trifluoromethyl)imidazol-2-yl]-5-fluoro-pyridine- 3-carboxylate was prepared in a similar manner to the synthesis of methyl 6-[1-cyclopropyl-4- (trifluoromethyl)imidazol-2-yl]-5-fluoro-pyridine-3-carboxylate described for Compound 43. [1380] To a solution of ethyl 6-[1-cyclopropyl-4-(trifluoromethyl)imidazol-2-yl]-5-fluoro- pyridine-3-carboxylate (197 mg, 573.88 μmol) in DMF (3 mL) was added N- Chlorosuccinimide (229.90 mg, 1.72 mmol) at room temperature. The mixture was stirred at 45 °C for 5 hrs. The resulted mixture was cooled down to room temperature and the reaction mixture was diluted with EtOAc (50 mL). The organic layers were washed with brine (30 mL x 3), dried over anhydrous Na
2SO
4. After filtration, the filtrate was concentrated under reduced pressure. The resulted residue was purified by column chromatography on silica gel, eluted with 25% EtOAc in PE to give ethyl 6-[5-chloro-1-cyclopropyl-4- (trifluoromethyl)imidazol-2-yl]-5-fluoro-pyridine-3-carboxylate (187 mg, 495.07 μmol, 86% yield) as a yellow oil. MS: m/z = 378.05 [M + H]
+.
1H NMR (300 MHz, Chloroform-d) δ 9.14 (t, J = 1.5 Hz, 1H), 8.15 (dd, J = 9.6, 1.8 Hz, 1H), 4.47 (q, J = 7.2 Hz, 2H), 3.44 – 3.37 (m, 1H), 1.45 (t, J = 7.2 Hz, 3H), 1.10 – 0.99 (m, 2H), 0.72 – 0.61 (m, 2H). [6-[5-chloro-1-cyclopropyl-4-(trifluoromethyl)imidazol-2-yl]-5-fluoro-3-pyridyl]methanol [1381] To a solution of ethyl 6-[5-chloro-1-cyclopropyl-4-(trifluoromethyl)imidazol-2-yl]-5- fluoro-pyridine-3-carboxylate (180 mg, 476.54 μmol) in methanol (2 mL) was added sodium borohydride (36.06 mg, 953.08 μmol) at 0 °C under nitrogen atmosphere. The reaction mixture was stirred at room temperature for 16 hrs then was concentrated under reduced pressure. The residue was purified by Prep-TLC, eluted with 33% EtOAc in PE to give [6-[5- chloro-1-cyclopropyl-4-(trifluoromethyl)imidazol-2-yl]-5-fluoro-3-pyridyl]methanol (60 mg, 175.76 μmol, 36% yield) as a yellow oil. MS: m/z = 336.15 [M + H]
+.
1H NMR (400 MHz, Chloroform-d) δ 8.44 (s, 1H), 7.59 (dd, J = 10.0, 1.6 Hz, 1H), 4.84 (s, 2H), 3.37 – 3.29 (m, 1H), 2.20 (b, 1H), 1.02 – 0.96 (m, 2H), 0.68 – 0.63 (m, 2H). 5-[4-[[6-[5-chloro-1-cyclopropyl-4-(trifluoromethyl)imidazol-2-yl]-5-fluoro-3- pyridyl]methoxy]pyrimidin-2-yl]-4-cyclopropyl-6-methoxy-pyrimidine [1382] To a solution of [6-[5-chloro-1-cyclopropyl-4-(trifluoromethyl)imidazol-2-yl]-5- fluoro-3-pyridyl]methanol (60 mg, 178.74 μmol) and 2-(4-cyclopropyl-6-methoxy-pyrimidin- 5-yl)pyrimidin-4-ol I-11 (52.39 mg, 214.49 μmol) in THF (3 mL) was added triphenylphosphine (140.64 mg, 536.22 μmol) at room temperature. Then the mixture was added diisopropyl azodicarboxylate (108.43 mg, 536.22 μmol, 105.6 μL) at room temperature. The mixture was stirred at room temperature for 16 hrs then concentrated under reduced pressure. The resulting residue was purified by Prep-TLC (EtOAc/PE = 2/1) to give two isomers. The less polar isomer fraction was concentrated under reduced pressure to give crude product, then the residue was further purified by reverse phase chromatography with the following conditions: Column: C18 gel column 40 g, 20-35 μm; Mobile Phase A: 5 mM aq. NH
4HCO
3, Mobile Phase B: MeCN; Gradient: 5% B to 5% B in 5 min, 5% B to 65% B in 20 min, 65% B to 65% B in 5 min, 65% B to 95% B in 5 min, 95% B hold 3 min; Flow rate: 40 mL/min; Detector: UV 254 & 220 nm. The product-containing fractions were collected and evaporated in vacuo and then lyophilized to give 5-[4-[[6-[5-chloro-1-cyclopropyl-4- (trifluoromethyl)imidazol-2-yl]-5-fluoro-3-pyridyl]methoxy]pyrimidin-2-yl]-4-cyclopropyl- 6-methoxy-pyrimidine (22.1 mg, 39.33 μmol, 22% yield) as a white solid. MS: m/z = 562.05 [M + H]
+.
1H NMR (300 MHz, Chloroform-d) δ 8.69 – 8.63 (m, 3H), 7.72 (d, J = 9.9 Hz, 1H), 6.86 (d, J = 5.7 Hz, 1H), 5.59 (s, 2H), 3.94 (s, 3H), 3.40 – 3.31 (m, 1H), 1.76 – 1.65 (m, 1H), 1.31 – 1.22 (m, 2H), 1.06 – 0.99 (m, 2H), 0.98 – 0.90 (m, 2H), 0.69 – 0.60 (m, 2H).
19F NMR (282 MHz, Chloroform-d) δ -61.92, -119.98.
2-chloro-4-[[4-[1-cyclopropyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidine Synthesis of the starting [4-[1-cyclopropyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methanol is described in for Compound 2. [1383] [4-[1-Cyclopropyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methanol (200 mg, 709 μmol), 2,4-dichloropyrimidine (127 mg, 850 μmol) and Cs
2CO
3 (346 mg, 1.06 mmol) were mixed in ACN (10 mL). The reaction mixture was stirred at room temperature for 12 hr. The reaction mixture was filtered. The filtrate was concentrated under reduced pressure to afford 2-chloro-4-[[4-[1-cyclopropyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidine (200 mg, 507 μmol, 71.5% yield) as a brown solid. MS (ESI): [M+H]
+ m/z: calcd 395.09; found 395.0 4-cyclopropyl-5-[4-[[4-[1-cyclopropyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidin-2-yl]-6-(difluoromethoxy)pyrimidine [1384] 2-Chloro-4-[[4-[1-cyclopropyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidine (73.0 mg, 185 μmol), 4-cyclopropyl-6-(difluoromethoxy)-5- (4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyrimidine I-10 (69.3 mg, 222 μmol), potassium phosphate tribasic (58.9 mg, 277 μmol) and Xphos Pd G3 (7.83 mg, 9.25 μmol) were mixed in a degassed mixture of dioxane (5 mL) and water (500 µL). The reaction mixture was stirred at 90 °C for 2 hr. The reaction mixture was cooled to room temperature. To the reaction mixture SiliaMetS
® Dimercaptotriazine (50 mg) was added, and the resulting mixture was stirred for 30 min. The mixture was filtered. The filtrate was subjected to HPLC (0.5-6.5 min., 40-90% water – ACN, flow: 30 mL/min, column: SunFire 100×19 mm, 5 µm; then Hexane:IPA 10:60, Flow Rate: 12 mL/min, Column: Chromatorex EP4 (100×19 mm, 5 µm) to afford 4-cyclopropyl-5-[4-[[4-[1-cyclopropyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidin-2-yl]-6-(difluoromethoxy)pyrimidine (4.50 mg, 8.26 μmol, 4.47% yield) as a white solid.
1H NMR (500 MHz, CD
3OD) δ 0.85 – 0.89 (m, 2H), 0.97 – 1.06 (m, 4H), 1.20 – 1.25 (m, 2H), 1.80 – 1.86 (m, 1H), 3.62 – 3.69 (m, 1H), 5.59 (s, 2H), 7.05 (d, 1H), 7.62 (d, 2H), 7.66 (t, 1H, CHF
2), 7.74 (s, 1H), 7.84 (d, 2H), 8.67 (d, 1H), 8.69 (s, 1H). MS (ESI): [M+H]
+ m/z: calcd 545.20; found 545.0. Example 130 (Compound 71)
yl)benzyl)oxy)pyrimidine [1385] To a stirred solution of 2-chloro-4-((4-(1-methyl-4-(trifluoromethyl)-1H-imidazol-2- yl)benzyl)oxy)pyrimidin-5-ol 171b (150 mg, 390.56 μmol) in THF (3 mL) was added NaH (18.75 mg, 468.68 μmol, 60% purity) at 0 °C under nitrogen atmosphere. Then dibromo(difluoro)methane (239.46 mg, 1.15 mmol) was added at 0 °C. The resulting mixture was stirred for 16 hrs at 25 °C under nitrogen atmosphere then was quenched by addition of saturated NH
4Cl aqueous solution (5 mL) at 0 °C. The resulting mixture was extracted with EtOAc (3 x 10 mL). The combined organic layers were washed with brine (5 mL), dried over anhydrous Na
2SO
4. After filtration, the filtrate was concentrated under reduced pressure. The residue was purified by Prep-TLC (50% EtOAc in PE) to afford 5-(bromodifluoromethoxy)- 2-chloro-4-((4-(1-methyl-4-(trifluoromethyl)-1H-imidazol-2-yl)benzyl)oxy)pyrimidine (90 mg, 175.79 μmol, 44% yield) as a yellow solid. MS: m/z = 512.95, 514.95 [M + H]
+.
1H NMR (400 MHz, Chloroform-d) δ 8.39 (s, 1H), 7.71 (d, J = 8.0 Hz, 2H), 7.60 (d, J = 8.0 Hz, 2H), 7.35 (s, 1H), 5.60 (s, 2H), 3.82 (s, 3H). 2-chloro-4-((4-(1-methyl-4-(trifluoromethyl)-1H-imidazol-2-yl)benzyl)oxy)-5- (trifluoromethoxy)pyrimidine [1386] To a stirred solution of 5-(bromodifluoromethoxy)-2-chloro-4-((4-(1-methyl-4- (trifluoromethyl)-1H-imidazol-2-yl)benzyl)oxy)pyrimidine (90 mg, 175.22 μmol) in DCM (1 mL) was added silver tetrafluoroborate (102.33 mg, 525.66 μmol) at -60 °C under nitrogen atmosphere. The resulting mixture was stirred for 16 hrs at 0 °C under nitrogen atmosphere then was quenched by the addition of saturated NaHCO
3 aqueous solution (5 mL) at 0 °C. The resulting mixture was extracted with EtOAc (3 x 10 mL). The combined organic layers were washed with brine (5 mL), dried over anhydrous Na
2SO
4. After filtration, the filtrate was concentrated under reduced pressure. The residue was purified by Prep-TLC (50% EtOAc in PE) to afford 2-chloro-4-((4-(1-methyl-4-(trifluoromethyl)-1H-imidazol-2- yl)benzyl)oxy)-5-(trifluoromethoxy)pyrimidine (50 mg, 110.44 μmol, 63% yield) as a yellow solid. MS: m/z = 453.00 [M + H]
+.
1H NMR (400 MHz, Chloroform-d) δ 8.34 (s, 1H), 7.72 (d, J = 8.0 Hz, 2H), 7.59 (d, J = 8.0 Hz, 2H), 7.35 (s, 1H), 5.61 (s, 2H), 3.82 (s, 3H). 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]-5-(trifluoromethoxy)pyrimidine [1387] To a stirred solution of 2-chloro-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]-5-(trifluoromethoxy)pyrimidine (50 mg, 110.44 μmol) and (4- cyclopropyl-6-methoxy-pyrimidin-5-yl)boronic acid I-5 (32.14 mg, 165.66 μmol) in THF (1 mL) and H
2O (0.2 mL) were added potassium phosphate (70.33 mg, 331.32 μmol) and [1,1’- Bis(dicyclohexylphosphino)ferrocene]dichloropalladium(II) (9.31 mg, 11.04 μmol) at 25 °C under nitrogen atmosphere. The resulting mixture was stirred for 16 hrs at 40 °C under nitrogen atmosphere then was diluted with water (5 mL). The resulting mixture was extracted with EtOAc (3 x 10 mL). The combined organic layers were washed with brine (5 mL), dried over anhydrous Na
2SO
4. After filtration, the filtrate was concentrated under reduced pressure. The residue was subject to Prep-TLC (50% EtOAc in PE) to afford crude product. The crude product was purified by RP-Flash with the following conditions: Column: C18 spherical Column, 20-35 um, 100A, 25 g; Mobile Phase A: Water (5 mM aq. NH
4HCO
3), Mobile Phase B: MeCN; Flow rate: 35 mL/min; Gradient: 5% B to 5% B in 5 min, 5% B to 45% B in 12 min, 45% B to 45% B in 2 min, 45% B to 95% B in 5 min; Detector: UV 254 & 210 nm. The collected fractions were combined and lyophilized to give 2-(4-cyclopropyl-6-methoxy- pyrimidin-5-yl)-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]-5- (trifluoromethoxy)pyrimidine (11.1 mg, 19.61 μmol, 18% yield) as a light yellow solid. MS: m/z = 567.20 [M + H]
+.
1H NMR (400 MHz, Chloroform-d) δ 8.78 (s, 1H), 8.62 (s, 1H), 7.70 (d, J = 8.0 Hz, 2H), 7.57 (d, J = 8.0 Hz, 2H), 7.35 (s, 1H), 5.61 (s, 2H), 4.01 (s, 3H), 3.82 (s, 3H), 1.91 – 1.73 (m, 1H), 1.45 – 1.40 (m, 2H), 1.08 – 1.02 (m, 2H).
19F NMR (377 MHz, Chloroform-d) δ -58.44, -62.78. Example 131 (Compound 73)
2,5-dichloro-4-((4-(1-methyl-4-(trifluoromethyl)-1H-imidazol-2-yl)benzyl)oxy)pyrimidine [1388] (4-(1-Methyl-4-(trifluoromethyl)-1H-imidazol-2-yl)phenyl)methanol I-2b (0.25 g, 976 μmol), 2,4,5-trichloropyrimidine (179 mg, 976 μmol) and Cs
2CO
3 (318 mg, 976 μmol) were mixed in Acetonitrile (5 mL). The reaction mixture was stirred at 80 °C for 3 hr. The reaction mixture was cooled to room temperature and filtered. The filtrate was concentrated under reduced pressure. The residue was subjected to flash-column chromatography (SiO
2, gradient chloroform – MTBE) to afford 2,5-dichloro-4-((4-(1-methyl-4-(trifluoromethyl)-1H- imidazol-2-yl)benzyl)oxy)pyrimidine (320 mg, 0.793 mmol, 81.4% yield) as white solid.
1H NMR (400 MHz, CDCl
3) δ 3.80 (s, 3H), 5.58 (s, 2H), 7.33 (s, 1H), 7.59 (d, 2H), 7.69 (d, 2H), 8.36 (s, 1H). MS (ESI): [M+H]
+ m/z: calcd 403.03, 405.03; found 403.0, 405.0. 5-chloro-4’-cyclopropyl-6’-methoxy-4-((4-(1-methyl-4-(trifluoromethyl)-1H-imidazol-2- yl)benzyl)oxy)-2,5’-bipyrimidine (73) [1389] 2,5-Dichloro-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidine (0.300 g, 744 μmol), (4-cyclopropyl-6-methoxypyrimidin-5- yl)boronic acid I-5 (144 mg, 744 μmol), Xphos Pd G3 (63.0 mg, 74.4 μmol) and Potassium phosphate tribasic (473 mg, 2.23 mmol) were mixed in a degassed mixture of dioxane (5 mL) and water (1 mL). The reaction mixture was stirred at 75 °C for 24 hr under argon atmosphere. The reaction mixture was cooled, diluted with EtOAc (5 mL), washed water (3 mL) and brine (3 mL). To the obtained solution SiliaMetS
® Dimercaptotriazine (100 mg) was added. The mixture was stirred for 30 min and filtered. The filtrate was concentrated under reduce pressure. The residue was subjected to HPLC (0-5 min., 40-90% water – MeOH, flow: 30 mL/min, column: Kinetex PFP 100 × 21.2 mm, 5 µm) to afford 5-chloro-4’-cyclopropyl- 6’-methoxy-4-((4-(1-methyl-4-(trifluoromethyl)-1H-imidazol-2-yl)benzyl)oxy)-2,5’- bipyrimidine (23.0 mg, 44.5 μmol, 5.98% yield) and by-product 5-chloro-4’-cyclopropyl-6’- methoxy-3-(4-(1-methyl-4-(trifluoromethyl)-1H-imidazol-2-yl)benzyl)-[2,5’-bipyrimidin]- 4(3H)-one (29.0 mg, 56.1 μmol, 7.54% yield) as a light yellow solids. 5-Chloro-4’-cyclopropyl-6’-methoxy-4-((4-(1-methyl-4-(trifluoromethyl)-1H- imidazol-2-yl)benzyl)oxy)-2,5’-bipyrimidine (73): 1H NMR (600 MHz, DMSO-d
6) δ 0.87 – 0.91 (m, 2H), 1.03 – 1.08 (m, 2H), 1.76 – 1.82 (m, 1H), 3.79 (s, 3H), 3.86 (s, 3H), 5.60 (s, 2H), 7.61 (d, 2H), 7.76 (d, 2H), 7.95 (s, 1H), 8.69 (s, 1H), 8.89 (s, 1H). MS (ESI): [M+H]
+ m/z: calcd 517.16; found 517.0. 5-Chloro-4’-cyclopropyl-6’-methoxy-3-(4-(1-methyl-4-(trifluoromethyl)-1H- imidazol-2-yl)benzyl)-[2,5’-bipyrimidin]-4(3H)-one: 1H NMR (600 MHz, DMSO-d
6) δ 0.91 – 0.96 (m, 2H), 1.02 – 1.07 (m, 1H), 1.08 – 1.13 (m, 1H), 1.63 – 1.68 (m, 1H), 3.79 (s, 3H), 3.89 (s, 3H), 5.49 (q, 2H), 7.61 (d, 2H), 7.75 (d, 2H), 7.95 (s, 1H), 8.74 (s, 1H), 8.92 (s, 1H). MS (ESI): [M+H]
+ m/z: calcd 517.16; found 517.0. Example 132 (Compound 80)
K
3PO4, THF, H2O Compound 80 2-chloro-4-[[4-[1-cyclopropyl-4-(trifluoromethyl)imidazol-2-yl]-3-fluoro-phenyl]methoxy]-5- methoxy-pyrimidine Synthesis of the starting (4-(1-cyclopropyl-4-(trifluoromethyl)-1H-imidazol-2-yl)-3- fluorophenyl)methanol is described for compound 154. [1390] To a solution of [4-[1-cyclopropyl-4-(trifluoromethyl)imidazol-2-yl]-3-fluoro- phenyl]methanol (100 mg, 333.05 μmol) in THF (2 mL) was added NaH (11.99 mg, 499.58 μmol 60% purity) at 0 °C under nitrogen atmosphere. The reaction mixture was stirred at 0 °C for 1 hour. Then to the mixture was added 2,4-dichloro-5-methoxy-pyrimidine (71.54 mg, 399.67 μmol) at 0 °C under nitrogen atmosphere. The reaction mixture was stirred at 25 °C for 16 hrs then was quenched by the addition of NH
4Cl aqueous solution (15 mL). The resulting mixture was extracted with EtOAc (3 x 10 mL). The combined organic layers were washed with brine (15 mL), dried over anhydrous Na
2SO
4. After filtration, the filtrate was concentrated under reduced pressure. The residue was purified by Prep-TLC to give 2- chloro-4-[[4-[1-cyclopropyl-4-(trifluoromethyl)imidazol-2-yl]-3-fluoro-phenyl]methoxy]-5- methoxy-pyrimidine (90 mg, 203.25 μmol, 61% yield) as an off-white solid. MS: m/z = 443.10 [M + H]
+.
1H NMR (300 MHz, Chloroform-d) δ 7.95 (s, 1H), 7.63 (t, J = 7.6 Hz, 1H), 7.42 – 7.27 (m, 3H), 5.55 (s, 2H), 3.95 (s, 3H), 3.52 – 3.44 (m, 1H), 0.96 – 0.80 (m, 2H), 0.77 – 0.71 (m, 2H).
19F NMR (282 MHz, Chloroform-d) δ -62.77, -111.55. 2-[4-cyclopropyl-6-(trideuteriomethoxy)pyrimidin-5-yl]-4-[[4-[1-cyclopropyl-4- (trifluoromethyl)imidazol-2-yl]-3-fluoro-phenyl]methoxy]-5-methoxy-pyrimidine [1391] To a solution of 2-chloro-4-[[4-[1-cyclopropyl-4-(trifluoromethyl)imidazol-2-yl]-3- fluoro-phenyl]methoxy]-5-methoxy-pyrimidine (40 mg, 90.34 μmol) and 4-cyclopropyl-5- (4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-6-(trideuteriomethoxy)pyrimidine I-9 (25.22 mg, 90.34 μmol) in THF (1 mL) and water (0.2 mL) were added potassium phosphate (38.35 mg, 180.67 μmol), 2-(Dicyclohexylphosphino)-2’,4’,6’-Triisopropylbiphenyl (4.31 mg, 9.03 μmol) and Methanesulfonato(2-dicyclohexylphosphino-2’,4’,6’-tri-i-propyl-1,1’- biphenyl)(2’-amino-1,1’-biphenyl-2-yl)palladium(II) (7.65 mg, 9.03 μmol) at 20 °C under nitrogen atmosphere. The reaction mixture was stirred at 60 °C for 16 hrs then was quenched by the addition of NH
4Cl aqueous solution (30 mL). The resulting mixture was extracted with EtOAc (3 x 20 mL). The combined organic layers were washed with brine (20 mL), dried over anhydrous Na
2SO
4. After filtration, the filtrate was concentrated under reduced pressure. The residue was purified by Prep-TLC to give a crude product. The crude product was further purified by RP-Flash with the following conditions: Column: C18 spherical Column, 20-35 um, 100A, 40 g; Mobile Phase A: Water (5 mM aq. NH
4HCO
3), Mobile Phase B: MeCN; Flow rate: 40 mL/min; Gradient: 5% B to 5% B in 3 min, 5% B to 70% B in 14 min, 70% B to 70% B in 1 min, 70% B to 95% B in 8 min; Detector: UV 254 & 220 nm. The collected fractions were combined, concentrated under reduced pressure and then lyophilized to give 2-[4-cyclopropyl-6-(trideuteriomethoxy)pyrimidin-5-yl]-4-[[4-[1-cyclopropyl-4- (trifluoromethyl)imidazol-2-yl]-3-fluoro-phenyl]methoxy]-5-methoxy-pyrimidine (7.1 mg, 12.69 μmol, 14% yield) as an off-white solid. MS: m/z = 560.20 [M + H]
+.
1H NMR (400 MHz, Chloroform-d) δ 8.68 (s, 1H), 8.26 (s, 1H), 7.64 – 7.58 (m, 1H), 7.43 – 7.32 (m, 3H), 5.58 (s, 2H), 4.03 (s, 3H), 3.51 – 3.43 (m, 1H), 1.80 – 1.70 (m, 1H), 1.29 – 1.23 (m, 2H), 0.99 – 0.88 (m, 4H), 0.80 – 0.71 (m, 2H).
19F NMR (377 MHz, Chloroform-d) δ -62.80, -111.68. Example 133 (Compound 81)
2-chloro-4-[[4-[1-cyclopropyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]-5-methoxy- pyrimidine Synthesis of the starting [4-[1-cyclopropyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methanol is described for compound 2. [1392] [4-[1-cyclopropyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methanol (200 mg, 709 μmol), 2,4-dichloro-5-methoxy-pyrimidine (133 mg, 744 μmol) and Cs
2CO
3 (346 mg, 1.06 mmol) were mixed in ACN (10 mL). The reaction mixture was stirred at room temperature for 12 hr. The reaction mixture was filtered. The filtrate was concentrated under reduced pressure. The residue was subjected to flash column chromatography (silica, gradient DCM – EtOAc) to afford 2-chloro-4-[[4-[1-cyclopropyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]-5-methoxy-pyrimidine (180 mg, 424 μmol, 59.8% yield) as a white solid. MS (ESI): [M+H]
+ m/z: calcd 425.10; found 425.0 2-[4-cyclopropyl-6-(difluoromethoxy)pyrimidin-5-yl]-4-[[4-[1-cyclopropyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]-5-methoxy-pyrimidine [1393] 2-Chloro-4-[[4-[1-cyclopropyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]-5- methoxy-pyrimidine (100 mg, 235 μmol), 4-cyclopropyl-6-(difluoromethoxy)-5-(4,4,5,5- tetramethyl-1,3,2-dioxaborolan-2-yl)pyrimidine I-10 (88.2 mg, 283 μmol), potassium phosphate tribasic (99.9 mg, 471 μmol) and Xphos Pd G3 (9.96 mg, 11.8 μmol) were mixed in degassed mixture of dioxane (5.0 mL) and water (500 µL). The reaction mixture was stirred at 100 °C for 2 hr. The reaction mixture was cooled to room temperature. To the reaction mixture SiliaMetS
® Dimercaptotriazine (50 mg) was added, and the resulting mixture was stirred for 30 min. The mixture was filtered. The filtrate was subjected to HPLC (0.5-6.5 min., 40-90% water – ACN, flow: 30 mL/min, column: SunFire C18 can 100×19 mm, 5 µm) to afford 2-[4-cyclopropyl-6-(difluoromethoxy)pyrimidin-5-yl]-4-[[4-[1- cyclopropyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]-5-methoxy-pyrimidine (25.0 mg, 43.5 μmol, 18.5% yield) as a beige solid.
1H NMR (500 MHz, DMSO-d
6) δ 0.89 – 0.94 (m, 2H), 0.95 – 1.03 (m, 4H), 1.08 – 1.13 (m, 2H), 1.88 – 1.94 (m, 1H), 3.72 – 3.78 (m, 1H), 3.97 (s, 3H), 5.52 (s, 2H), 7.59 (d, 2H), 7.82 (t, 1H, CHF
2), 7.91 (d, 2H), 7.95 (s, 1H), 8.50 (s, 1H), 8.79 (s, 1H). MS (ESI): [M+H]
+ m/z: calcd 575.21; found 575.2. Example 134 (Compound 82)
Compound 82 Methyl 6-[1-(cyclopropylmethyl)-4-(trifluoromethyl)imidazol-2-yl]-5-fluoro-pyridine-3- carboxylate Synthesis of the starting methyl 5-fluoro-6-[4-(trifluoromethyl)-1H-imidazol-2- yl]pyridine-3-carboxylate is described for Compound 43. [1394] A mixture of methyl 5-fluoro-6-[4-(trifluoromethyl)-1H-imidazol-2-yl]pyridine-3- carboxylate (440 mg, 1.52 mmol), iodomethylcyclopropane (554 mg, 3.04 mmol, 283 μL) and K
2CO
3 (421 mg, 3.04 mmol) in DMF (5 mL) was stirred at room temperature for 12 hr. The reaction mixture was poured into cold water (50 mL) and extracted with EtOAc (2×30 mL). The combined extracts were washed with brine, dried over anhydrous Na
2SO
4 and concentrated under reduced pressure to afford methyl 6-[1-(cyclopropylmethyl)-4- (trifluoromethyl)imidazol-2-yl]-5-fluoro-pyridine-3-carboxylate (522 mg, 1.52 mmol, 99.9% yield) as a yellow solid which was used in the next step without further purification.
1H NMR (400 MHz, CDCl
3) δ 0.31 – 0.40 (m, 2H), 0.65 – 0.73 (m, 2H), 1.23 – 1.31 (m, 1H), 4.02 (s, 3H), 4.21 (d, 2H), 7.62 (s, 1H), 8.16 (d, 1H), 9.09 (s, 1H). MS (ESI): [M+H]
+ m/z: calcd 344.10; found 344.2 [6-[1-(cyclopropylmethyl)-4-(trifluoromethyl)imidazol-2-yl]-5-fluoro-3-pyridyl]methanol [1395] A solution of DIBAL (1.80 mL, 1.07 M in toluene, 1.92 mmol) was diluted with diethyl ether (18 mL) at 0 °C. To the resulting solution a solution of methyl 6-[1- (cyclopropylmethyl)-4-(trifluoromethyl)imidazol-2-yl]-5-fluoro-pyridine-3-carboxylate (300 mg, 874 μmol) in diethyl ether (2 mL) was added dropwise at 0 °C. The reaction mixture was stirred at 0 °C for 30 min. Sodium hydroxide (28.5% wt. aqueous solution, 140 µL) was added to the reaction mixture at 0 °C. The solids formed were filtered out. The filtrate was concentrated under reduced pressure. The residue was subjected to HPLC (0.5-6.5 min., 30- 45% water – ACN, flow: 30 mL/min, column: CROMATOREX SMB C18100×20 mm, 5 µm) to afford [6-[1-(cyclopropylmethyl)-4-(trifluoromethyl)imidazol-2-yl]-5-fluoro-3- pyridyl]methanol (71.0 mg, 225 μmol, 25.8% yield) as a white solid. MS (ESI): [M+H]
+ m/z: calcd 316.13; found 315.9 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[6-[1-(cyclopropylmethyl)-4- (trifluoromethyl)imidazol-2-yl]-5-fluoro-3-pyridyl]methoxy]-6-methyl-pyrimidine Synthesis of the starting 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-methyl-6- methylsulfonyl-pyrimidine is described for Compound 120. [1396] A mixture of [6-[1-(cyclopropylmethyl)-4-(trifluoromethyl)imidazol-2-yl]-5-fluoro-3- pyridyl]methanol (16.0 mg, 50.8 μmol), 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4- methyl-6-methylsulfonyl-pyrimidine (17.9 mg, 55.8 μmol) and NaH (2.03 mg, 50.8 μmol, 60% dispersion in mineral oil) in THF (2.0 mL) was stirred at room temperature for 12 hr. The reaction mixture was directly subjected to HPLC (0.5-6.5 min., 60% water – MeOH, +0.1% vol. of 25% aq. NH
3, flow: 30 mL/min, column: YMC-ACTUS TRIART C18100×20 mm, 5 µm) to afford 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[6-[1- (cyclopropylmethyl)-4-(trifluoromethyl)imidazol-2-yl]-5-fluoro-3-pyridyl]methoxy]-6- methyl-pyrimidine (8.20 mg, 14.8 μmol, 29.1% yield) as a yellow gum.
1H NMR (500 MHz, DMSO-d
6) δ 0.29 – 0.33 (m, 2H), 0.45 – 0.50 (m, 2H), 0.87 – 0.92 (m, 2H), 1.02 – 1.06 (m, 2H), 1.20 – 1.26 (m, 1H), 1.63 – 1.68 (m, 1H), 2.47 (s, 3H), 3.85 (s, 3H), 4.10 (d, 2H), 5.55 (s, 2H), 6.97 (s, 1H), 8.03 (d, 1H), 8.17 (s, 1H), 8.67 (s, 2H). MS (ESI): [M+H]
+ m/z: calcd 556.24; found 556.2. Example 135 (Compound 83)
Methyl 2-bromo-4-(dibromomethyl)-5-fluoro-benzoate [1397] A mixture of methyl 2-bromo-5-fluoro-4-methyl-benzoate (7.5 g, 30.4 mmol), NBS (17 g, 95.5 mmol), BPO (750 mg, 3.10 mmol) in CCl
4 (50 mL) was stirred at 90 °C for 18 hrs. The resulting mixture was concentrated under reduced pressure. The resulting mixture was diluted by addition of water (30 mL) and washed with TBME (60 mL x 2). The aqueous phase was then extracted with EtOAc (200 mL x 3). The combined organic layer was washed with brine (70 mL x 2), dried over anhydrous Na
2SO
4, filtered and concentrated under reduced pressure. The residue was purified by flash chromatography (ISCO®; 80 g AgelaFlash® Silica Flash Column, PE/EtOAc with EtOAc from 0-9%, flow rate = 100 mL/min, 254 nm) to afford methyl 2-bromo-4-(dibromomethyl)-5-fluoro-benzoate (11.5 g, crude) as yellow oil.
1H NMR (400 MHz, chloroform-d) δ ppm 8.10 (d, J = 6.8 Hz, 1H), 7.52 (d, J = 10.0 Hz, 1H), 6.82 (s, 1H), 3.96 (s, 3H).
19F NMR (376 MHz, chloroform-d) δ ppm - 117.44. Methyl 2-bromo-5-fluoro-4-formyl-benzoate [1398] To a mixture of methyl 2-bromo-4-(dibromomethyl)-5-fluoro-benzoate (11.5 g, 28.4 mmol) in THF (75 mL) and H
2O (25 mL) was added AgNO
3 (34 g, 0.140 mol). The mixture was stirred at 80°C for 2 hrs. The reaction mixture was diluted by addition of water (20 mL) and extracted with EtOAc (150 mL x 3). The combined organic layer was washed with brine (60 mL x 8), dried over anhydrous Na
2SO
4, filtered and concentrated under reduced pressure. The residue was purified by flash chromatography (ISCO®; 120 g AgelaFlash® Silica Flash Column, PE/EtOAc with EtOAc from 0-6%, flow rate = 80 mL/min, 254 nm) to afford methyl 2-bromo-5-fluoro-4-formyl-benzoate (4.7 g, crude) as light yellow oil.
1H NMR (400 MHz, DMSO-d
6) δ ppm 10.14 (s, 1H), 8.10 (d, J = 6.3 Hz, 1H), 7.83 (d, J = 10.3 Hz, 1H), 3.90 (s, 3H);
19F NMR (376 MHz, DMSO-d
6) δ ppm -120.52. Methyl 2-bromo-5-fluoro-4-[4-(trifluoromethyl)-1H-imidazol-2-yl]benzoate [1399] To a solution of 3,3-dibromo-1,1,1-trifluoro-propan-2-one (4 mL, 29.7 mmol) in H
2O (30 mL) was added NaOAc (4.5 g, 54.9 mmol). The mixture was stirred at 95°C for 30 minutes, then cooled the solution in an ice bath. To the mixture was added methyl 2-bromo- 5-fluoro-4-formyl-benzoate (4.7 g, 18.0 mmol), MeOH (30 mL) and ammonium hydroxide (30 mL, 0.193 mol, 25 wt%). The mixture was stirred at 20 °C to for 4 hrs. The reaction solution was diluted by addition of water (20 mL) and extracted with EtOAc (150 mL x 3). The combined organic layer was washed with brine (100 mL x 2), dried over anhydrous Na
2SO
4, filtered and concentrated under reduced pressure. The residue was purified by flash chromatography (ISCO®; 120 g AgelaFlash® Silica Flash Column, PE/EtOAc with EtOAc from 0-19%, flow rate = 100 mL/min, 254 nm) to afford methyl 2-bromo-5-fluoro-4-[4- (trifluoromethyl)-1H-imidazol-2-yl]benzoate (1.45 g, 21.9% yield) as light yellow solid.
1H NMR (400 MHz, DMSO-d
6) δ ppm 13.23 (brs, 1H), 8.27 (d, J = 6.5 Hz, 1H), 8.01 (s, 1H), 7.85 (d, J = 10.8 Hz, 1H), 3.89 (s, 3H);
19F NMR (376 MHz, DMSO-d
6) δ ppm -60.75, - 115.64; MS (ESI) [M+H]
+ m/z: calcd 369.0, found 369.0. Methyl 2-bromo-5-fluoro-4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]benzoate [1400] To a solution of methyl 2-bromo-5-fluoro-4-[4-(trifluoromethyl)-1H-imidazol-2- yl]benzoate (1.45 g, 3.95 mmol) in THF (10 mL) was added t-BuOK (700 mg, 6.24 mmol) at 0°C. The mixture was stirred at 0°C for 1.5 hrs. MeI (660 mg, 4.65 mmol) was added. The mixture was stirred at 20°C for 3.5 hrs. The resulting mixture was diluted by addition of water (10 mL) and extracted with EtOAc (70 mL x 3). The combined organic layer was washed with brine (30 mL x 2), dried over anhydrous Na
2SO
4, filtered and concentrated under reduced pressure to afford methyl 2-bromo-5-fluoro-4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]benzoate (760 mg, 50.5% yield) as light yellow solid. The aqueous phase was adjusted pH to 4-5 with 1N HCl aqueous solution. The aqueous phase was extracted with EtOAc (80 mL x 3). The combined organic layer was washed with brine (50 mL x 2), dried over anhydrous Na
2SO
4, filtered and concentrated under reduced pressure to afford 2-bromo-5-fluoro-4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]benzoic acid (640 mg, 44.1% yield) as yellow solid. MS (ESI) [M+H]
+ m/z: calcd 381.0, found 381.0. Methyl 5-fluoro-4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]-2-vinyl-benzoate [1401] A mixture of methyl 2-bromo-5-fluoro-4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]benzoate (760 mg, 1.99 mmol), potassium;trifluoro(vinyl)boranuide (350 mg, 2.61 mmol), cyclopentyl(diphenyl)phosphane;dichloropalladium;iron (150 mg, 0.205 mmol), tripotassium;carbonate (840 mg, 6.08 mmol) in dioxane (5 mL) and H
2O (1 mL) was stirred at 100 °C for 12 hrs under N
2. The resulting mixture was diluted by addition of water (5 mL) and extracted with EtOAc (80 mL x 3). The combined organic layer was washed with brine (50 mL x 2), dried over anhydrous Na
2SO
4, filtered and concentrated under reduced pressure. The residue was purified by flash chromatography (ISCO®; 20 g AgelaFlash® Silica Flash Column, PE/EtOAc with EtOAc from 0-15%, flow rate = 35 mL/min, 254 nm) to afford methyl 5-fluoro-4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]-2-vinyl-benzoate (230 mg, 35.1% yield) as light yellow solid. MS (ESI) [M+H]
+ m/z: calcd 329.1, found 329.1. Methyl 2-ethyl-5-fluoro-4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]benzoate [1402] To a mixture of methyl 5-fluoro-4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]-2- vinyl-benzoate (230 mg, 0.701 mmol) in MeOH (3 mL) was added Pd/C (50 mg, 10 wt% Pd with 50 wt% water) under N
2 atmosphere. The suspension was degassed and purged with hydrogen for 3 times. The mixture was stirred under H
2 (in balloon) at 20°C for 12 hrs. The resulting mixture was filtered and concentrated under reduced pressure to afford methyl 2- ethyl-5-fluoro-4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]benzoate (220 mg, 95.1% yield) as light yellow solid. MS (ESI) [M+H]
+ m/z: calcd 331.1, found 331.1. [2-ethyl-5-fluoro-4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methanol [1403] To a solution of methyl 2-ethyl-5-fluoro-4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]benzoate (200 mg, 0.606 mmol) in THF (3 mL) was added LAH (40 mg, 1.05 mmol) at 0 °C under N
2. The mixture was stirred at 20°C for 1 hour. The resulting mixture was added water (0.1 mL), NaOH aqueous solution (0.1 mL, 15 wt%), water (0.3 mL). Then to the mixture was added excessive MgSO
4 solid. The mixture was filtered and concentered under pressure to afford [2-ethyl-5-fluoro-4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methanol (180 mg, 98.3% yield) as white solid. MS (ESI) [M+H]
+ m/z: calcd 303.1, found 303.0. 4-cyclopropyl-5-[4-[[2-ethyl-5-fluoro-4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidin-2-yl]-6-methoxy-pyrimidine (83) [1404] To a solution of [2-ethyl-5-fluoro-4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methanol (70 mg, 0.232 mmol) in THF (2 mL) was added NaH (15 mg, 0.375 mmol, 60 wt% in mineral oil). The mixture was stirred at 20 °C for 30 min.4-cyclopropyl-6- methoxy-5-(4-methylsulfonylpyrimidin-2-yl)pyrimidine I-8 (150 mg, 0.490 mmol) was added, and the mixture was stirred at 45 °C for 2 hrs. The resulting mixture was quenched by addition of water (5 mL) and extracted with EtOAc (50 mL x 3). The combined organic layer was washed with brine (40 mL x 2), dried over anhydrous Na
2SO
4, filtered and concentrated under reduced pressure. The residue was purified by preparative HPLC (Instrument: Gilson GX-281 Liquid Handler, Gilson 322 Pump, Gilson 156 UV Detector; Column: 2_Phenomenex Gemini C1875 x 40 mm x 3 μm; Mobile phase A: H
2O with NH
3-H
2O (v%); Mobile phase B: MeCN; Gradient: B from 48% to 78% in 9.5 min, hold 100% B for 1 min; Flow Rate: 30 mL/min; Column Temperature: 30 °C; Wavelength: 220 nm, 254 nm). The fraction was concentrated under reduced pressure and then lyophilized for overnight to afford 4-cyclopropyl-5-[4-[[2-ethyl-5-fluoro-4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidin-2-yl]-6-methoxy-pyrimidine (41.3 mg, 33.7% yield) as white solid.
1H NMR (400 MHz, DMSO-d
6) δ ppm 8.72 (d, J = 5.8 Hz, 1H), 8.68 (s, 1H), 8.01 (d, J = 0.8 Hz, 1H), 7.41 - 7.49 (m, 2H), 7.10 (d, J = 5.8 Hz, 1H), 5.53 (s, 2H), 3.85 (s, 3H), 3.61 (d, J = 1.0 Hz, 3H), 2.71 (q, J = 7.4 Hz, 2H), 1.66 - 1.79 (m, 1H), 1.17 (t, J = 7.5 Hz, 3H), 1.01 - 1.10 (m, 2H), 0.85 - 0.95 (m, 2H);
19F NMR (376 MHz, DMSO-d
6) δ ppm -60.83, - 118.15; MS (ESI) [M+H]
+ m/z: calcd 529.2, found 529.3. Example 136 (Compound 228)
Compound 228 2-chloro-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]-5-pyrazin-2-yl- pyrimidine [1405] To a solution of [2-chloro-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidin-5-yl]boronic acid (100 mg, 242.39 μmol) and 2-bromopyrazine (38.54 mg, 242.39 μmol) in water (0.4 mL) and 1,4-dioxane (2 mL) were added potassium phosphate tribasic (102.90 mg, 484.78 μmol) (5-diphenylphosphanyl-9,9-dimethyl-xanthen- 4-yl)-diphenyl-phosphane (14.03 mg, 24.24 μmol) and [4,5-Bis(diphenylphosphino)-9,9- dimethylxanthene][2’-amino-2-biphenylyl][(methylsulfonyl)oxy]palladium(II) (23 mg, 24.24 μmol) at 20 °C under nitrogen atmosphere. The reaction mixture was stirred at 40 °C for 16 hrs, cooled to room temperature and quenched by the addition of NH
4Cl aqueous solution (30 mL). The resulting mixture was extracted with EtOAc (3 x 20 mL). The combined organic layers were washed with brine (30 mL), dried over anhydrous Na
2SO
4. After filtration, the filtrate was concentrated under reduced pressure. The residue was purified by Prep-TLC to afford 2-chloro-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]-5- pyrazin-2-yl-pyrimidine (60 mg, 134.28 μmol, 55% yield) as an off-white solid. MS: m/z = 447.15 [M + H]
+.
1H NMR (400 MHz, Chloroform-d) δ 9.26 (s, 1H), 9.14 (s, 1H), 8.73 - 8.71 (m, 1H), 8.60 (d, J = 2.4 Hz, 1H), 7.74 (d, J = 8.0 Hz, 2H), 7.63 (d, J = 8.0 Hz, 2H), 7.37 (s, 1H), 5.71 (s, 2H), 3.84 (s, 3H).
19F NMR (377 MHz, Chloroform-d) δ -62.56. 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]-5-pyrazin-2-yl-pyrimidine To a solution of 2-chloro-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]- 5-pyrazin-2-yl-pyrimidine (60 mg, 134.28 μmol) and (4-cyclopropyl-6-methoxy-pyrimidin-5- yl)boronic acid I-5 (26.05 mg, 134.28 μmol) in 1,4-dioxane (2 mL) and water (0.4 mL) were added 2-(Dicyclohexylphosphino)-2',4',6'-Triisopropylbiphenyl (6.40 mg, 13.43 μmol), Methanesulfonato(2-dicyclohexylphosphino-2',4',6'-tri-i-propyl-1,1'-biphenyl)(2'-amino-1,1'- biphenyl-2-yl)palladium(II) (11.37 mg, 13.43 μmol) and potassium phosphate tribasic (57.01 mg, 268.57 μmol) at 20 °C under nitrogen atmosphere. The reaction mixture was stirred at 40 °C for 16 hrs, was cooled to room temperature and quenched by the addition of NH
4Cl aqueous solution (30 mL). The resulting mixture was extracted with EtOAc (3 x 20 mL). The combined organic layers were washed with brine (30 mL), dried over anhydrous Na
2SO
4. After filtration, the filtrate was concentrated under reduced pressure. The residue was purified by Prep-TLC to give a crude product. The crude product was further purified by Prep-HPLC with the following conditions: Column: XBridge Prep OBD C18 Column, 19x250 mm, 5μm; Mobile Phase A: Water (10 mM aq. NH
4HCO
3), Mobile Phase B: ACN; Flow rate: 25 mL/min; Gradient: 50% B to 55% B in 10 min, 55% B; Wavelength: 254 nm. The collected fractions were combined, concentrated and then lyophilized to give 2-(4-cyclopropyl-6-methoxy- pyrimidin-5-yl)-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]-5- pyrazin-2-yl-pyrimidine (4.1 mg, 7.31 μmol, 5% yield) as an off-white solid. MS: m/z = 561.20 [M + H]
+.
1H NMR (400 MHz, Chloroform-d) δ 9.38 - 9.36 (m, 2H), 8.80 - 8.54 (m, 3H), 7.68 (d, J = 8.0 Hz, 2H), 7.59 (d, J = 8.0 Hz, 2H), 7.34 (s, 1H), 5.71 (s, 2H), 3.98 (s, 3H), 3.81 (s, 3H), 1.88 - 1.80 (m, 1H), 1.33 - 1.23 (m, 2H), 0.9 - 0.91 (m, 2H).
19F NMR (377 MHz, Chloroform-d) δ -62.79.
pyrimidine & 4-chloro-2-[[3-fluoro-4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]-5-iodo-pyrimidine Synthesis of the starting [3-fluoro-4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methanol is described for compound 97. [1406] To a solution of [3-fluoro-4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methanol (500 mg, 1.82 mmol) in THF (8 mL) was added NaH (72.93 mg, 1.82 mmol, 60% purity) at 0 °C. The mixture was stirred at 0 °C for 30 minutes. Then the mixture was added 2,4-dichloro-5-iodo-pyrimidine (601.44 mg, 2.19 mmol) at 0 °C. Then the mixture was stirred at room temperature for 16 hrs. The reaction mixture was diluted with EtOAc (50 mL), washed with NH
4Cl aqueous solution (30 mL x 3), dried over anhydrous Na
2SO
4. Then the mixture was concentrated under reduced pressure. The resulted residue was subject to column chromatography on silica gel, eluted with 28% EtOAc in PE, to give two isomers. The less polar isomer was purified by Prep-TLC, eluted with 33% EtOAc in PE to give 2-chloro-4-[[3-fluoro-4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]-5-iodo- pyrimidine (200 mg, 390.15 μmol, 21% yield) as a yellow solid. MS: m/z = 513.00 [M + H]
+.
1H NMR (400 MHz, Chloroform-d) δ 8.67 (s, 1H), 7.74 - 7.65 (m, 1H), 7.43 - 7.34 (m, 3H), 5.56 (s, 2H), 3.69 (s, 3H). The more polar isomer was purified by Prep-TLC, eluted with 33% EtOAc in PE to give 4-chloro-2-[[3-fluoro-4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]-5-iodo- pyrimidine (137 mg, 267.25 μmol, 14% yield) as a yellow solid. MS: m/z = 513.00 [M + H]
+.
1H NMR (400 MHz, Chloroform-d) δ 8.74 (s, 1H), 7.68 - 7.61 (m, 1H), 7.41 - 7.32 (m, 3H), 5.49 (s, 2H), 3.67 (s, 3H). 2-chloro-N-[2-(1,1-dioxo-1,4-thiazinan-4-yl)ethyl]-4-[[3-fluoro-4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidin-5-amine [1407] To a solution of 2-chloro-4-[[3-fluoro-4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]-5-iodo-pyrimidine (150 mg, 292.61 μmol) and 2-(1,1-dioxo-1,4- thiazinan-4-yl)ethanamine (62.59 mg, 351.13 μmol) in 1,4-dioxane (5 mL) were added (SP- 4-1)-[1,3-BIs[2,6-bis(1-ethylpropyl)phenyl]-4,5-dichloro-1,3-dihydro-2H-imidazol-2- ylidene]dichloro(2-methylpyridine)palladium (24.58 mg, 29.26 μmol) and Cs
2CO
3 (190.68 mg, 585.22 μmol) at room temperature, the mixture was stirred at 70 °C for 16 hrs. The reaction mixture was diluted with EtOAc (50 mL), washed with NH
4Cl aqueous solution (25 mL x 3), dried over anhydrous Na
2SO
4. Then the mixture was concentrated under reduced pressure. The resulted residue was purified by Prep-TLC, eluted with 7% methanol in DCM give 2-chloro-N-[2-(1,1-dioxo-1,4-thiazinan-4-yl)ethyl]-4-[[3-fluoro-4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidin-5-amine (38 mg, 67.50 μmol, 23% yield) as a white solid. MS: m/z = 563.15 [M + H]
+.
1H NMR (400 MHz, Chloroform-d) δ 7.73 - 7.67 (m, 2H), 7.41 - 7.37 (m, 2H), 7.31 (s, 1H), 5.56 (s, 2H), 3.70 (s, 3H), 3.29 (br., 2H), 3.14 (br., 8H), 2.93 (br., 2H). 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-N-[2-(1,1-dioxo-1,4-thiazinan-4-yl)ethyl]-4-[[3- fluoro-4-[1-methyl-4-(trifluoromethyl)imidazol-2yl]phenyl]methoxy]pyrimidin-5-amine [1408] To a solution of 2-chloro-N-[2-(1,1-dioxo-1,4-thiazinan-4-yl)ethyl]-4-[[3-fluoro-4-[1- methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidin-5-amine (25 mg, 44.41 μmol) and (4-cyclopropyl-6-methoxy-pyrimidin-5-yl)boronic acid I-5 (12.92 mg, 66.61 μmol) in 1,4-dioxane (1 mL) and H
2O (0.2 mL) were added Methanesulfonato(2- dicyclohexylphosphino-2',4',6'-tri-i-propyl-1,1'-biphenyl)(2'-amino-1,1'-biphenyl-2- yl)palladium(II) (3.76 mg, 4.44 μmol), 2-(Dicyclohexylphosphino)-2',4',6'- Triisopropylbiphenyl (2.12 mg, 4.44 μmol) and potassium phosphate (18.85 mg, 88.82 μmol) at room temperature, the mixture was stirred at 40 °C for 16 hrs then was concentrated under reduced pressure. The resulted residue was purified by Prep-TLC and then was further purified by Prep-HPLC with the following conditions (Column: XSelect CSH Prep C18 OBD Column, 19 x 250 mm, 5μm; Mobile Phase A: Water (10 mM aq. NH
4HCO
3), Mobile Phase B: ACN; Flow rate: 20 mL/min; Gradient: 50% B to 55% B in 8 min, 55% B; Wavelength: 254 nm) to afford 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-N-[2-(1,1- dioxo-1,4-thiazinan-4-yl)ethyl]-4-[[3-fluoro-4-[1-methyl-4-(trifluoromethyl)imidazol- 2yl]phenyl]methoxy]pyrimidin-5-amine (2.5 mg, 3.69 μmol, 8% yield) as a white solid. MS: m/z = 677.25 [M + H]
+.
1H NMR (400 MHz, Chloroform-d) δ 8.69 (s, 1H), 8.01 (s, 1H), 7.68 (t, J = 7.6 Hz, 1H), 7.40 - 7.30 (m, 3H), 5.60 (s, 2H), 4.00 (s, 3H), 3.70 (s, 3H), 3.39 (br., 2H), 3.32 - 3.17 (m, 8H), 2.99 (br., 2H), 1.84 - 1.72 (m, 1H), 1.30 - 1.22 (m, 2H), 0.96 - 0.88 (m, 2H).
19F NMR (376 MHz, Chloroform-d) δ -62.75, -112.41. Example 138 (Compound 226)
doxane,
2O Compound 226 2-chloro-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]-5-(2- pyridyl)pyrimidine [1409] To a stirred mixture of [2-chloro-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidin-5-yl]boronic acid (100 mg, 242.39 μmol) in 1,4-Dioxane (1 mL) and water (0.2 mL) were added 2-bromopyridine (38.30 mg, 242.39 μmol), potassium phosphate (102.90 mg, 484.78 μmol) and Dichloro[9,9-dimethyl-4,5- bis(diphenylphosphino)xanthene]palladium(II) (18.3 mg, 24.24 μmol) under nitrogen atmosphere at 25 °C. The resulting mixture was stirred under nitrogen atmosphere for 16 hrs at 40 °C. The reaction mixture was cooled down to room temperature. The reaction filtered and the filtrate was concentrated under reduced pressure. The residue was purified by Prep- TLC (EtOAc : PE = 2 : 1) to give 2-chloro-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]-5-(2-pyridyl)pyrimidine (45 mg, 100.94 μmol, 41% yield) as an off- white solid. MS: m/z = 466.00 [M + H]
+.
1H NMR (400 MHz, Chloroform-d) δ 9.06 (s, 1H), 8.82 (d, J = 5.2 Hz, 1H), 8.02 -7.93 (m, 2H), 7.69 (d, J = 8.0 Hz, 2H), 7.61 (d, J = 8.0 Hz, 2H), 7.53 (s, 1H), 7.35 (s, 1H), 5.68 (s, 2H), 3.81 (s, 3H). 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]-5-phenyl-pyrimidine [1410] To a stirred mixture of 2-chloro-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]-5-phenyl-pyrimidine (45 mg, 101.16 μmol) in 1,4-Dioxane (1 mL) and water (0.2 mL) were added (4-cyclopropyl-6-methoxy-pyrimidin-5-yl)boronic acid I-5 (23.55 mg, 121.39 μmol), dicyclohexyl-[2-(2,4,6-triisopropylphenyl)phenyl]phosphane (4.82 mg, 10.12 μmol), Methanesulfonato(2-dicyclohexylphosphino-2',4',6'-tri-i-propyl-1,1'- biphenyl)(2'-amino-1,1'-biphenyl-2-yl)palladium(II) (8.56 mg, 10.12 μmol) and potassium phosphate tribasic (42.95 mg, 202.32 μmol) under nitrogen atmosphere at 25 °C. The resulting mixture was stirred under nitrogen atmosphere for 16 hrs at 40 °C. The reaction mixture was cooled down to room temperature and concentrated. The residue was purified by Prep-TLC (EtOAc : PE = 1 : 1) to give 30 mg crude product. The crude product was purified by reverse phase chromatography with the following conditions: Column: C18 spherical Column, 20-35 um, 100A, 40 g; Mobile Phase A: Water (5 mM aq. NH
4HCO
3), Mobile Phase B: MeCN; Flow rate: 35 mL/min; Gradient: 5% B to 5% B in 5 min, 5% B to 73% B in 2 min, 73% B to 73% B in 1 min, 73% B to 95% B in 10 min; Detector: UV 254 & 210 nm. The product-containing fractions were collected and evaporated in vacuo and then lyophilized to give 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]-5-phenyl-pyrimidine (8.2 mg, 14.68 μmol, 14% yield) as an off-white solid. MS: m/z = 560.30 [M + H]
+.
1H NMR (400 MHz, Chloroform-d) δ 9.32 (s, 1H), 8.80 (d, J = 4.0 Hz, 1H), 8.70 (s, 1H), 8.02 (d, J = 8.0 Hz, 1H), 7.82 (t, J = 7.2 Hz, 1H), 7.67 (d, J = 8.0 Hz, 2H), 7.57 (d, J = 8.0 Hz, 2H), 7.38 - 7.34 (m, 2H), 5.67 (s, 2H), 3.99 (s, 3H), 3.80 (s, 3H), 1.87 - 1.81 (m, 1H), 1.31 - 1.28 (m, 2H), 0.97 - 0.92 (m, 2H).
19F NMR (377 MHz, Chloroform-d) δ -62.79. Example 139 (Compound 225)
XPhosPdG
3, XPhos, K
3PO
4 1
,4-dioxane, H2O Compound 225 4-[2-chloro-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidin-5- yl]oxazole [1411] To a solution of 2-chloro-5-iodo-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidine I-12 (80 mg, 161.73 μmol) and 4-(4,4,5,5-tetramethyl-1,3,2- dioxaborolan-2-yl)oxazole (37.9 mg, 194.08 μmol) in 1,4-dioxane (3 mL) and H
2O (0.5 mL) were added dichloro[9,9-dimethyl-4,5-bis(diphenylphosphino)xanthene]palladium(II) (12.26 mg, 16.17 μmol) and potassium phosphate (68.7 mg, 323.47 μmol) at room temperature, the mixture was stirred at 40 °C for 16 hrs. The resulting reaction was diluted with EtOAc (20 mL), washed with NH
4Cl aqueous solution (10 mL x 3), dried over anhydrous Na
2SO
4. After filtration, the filtrate was concentrated under reduced pressure. Then the mixture was concentrated under reduced pressure and the residue was purified by column chromatography on silica gel, eluted with 15% EtOAc in PE to afford 4-[2-chloro-4-[[4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidin-5-yl]oxazole (56 mg, 128.50 μmol, 79% yield) as a yellow solid. MS: m/z = 436.15 [M + H]
+.
1H NMR (400 MHz, Chloroform-d) δ 9.14 (s, 1H), 8.08 (s, 1H), 7.99 (s, 1H), 7.76 (d, J = 8.0 Hz, 2H), 7.64 (d, J = 8.0 Hz, 2H), 7.38 (s, 1H), 5.68 (s, 2H), 3.85 (s, 3H). 4-[2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol- 2-yl]phenyl]methoxy]pyrimidin-5-yl]oxazole [1412] To a solution of 4-[2-chloro-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidin-5-yl]oxazole (45 mg, 103.26 μmol) and 2-(4-cyclopropyl-6- methoxy-pyrimidin-5-yl)-1,3,6,2-dioxazaborocane (54.3 mg, 206.52 μmol) in 1,4-dioxane (3 mL) and H
2O (0.6 mL) were added methanesulfonato(2-dicyclohexylphosphino-2',4',6'-tri-i- propyl-1,1'-biphenyl)(2'-amino-1,1'-biphenyl-2-yl)palladium(II) (8.7 mg, 10.33 μmol), 2- (dicyclohexylphosphino)-2',4',6'-triisopropylbiphenyl (4.9 mg, 10.33 μmol) and potassium phosphate (43.8 mg, 206.52 μmol) at room temperature, the mixture was stirred at 40 °C for 7 hrs. The resulting reaction was diluted with EtOAc (50 mL), washed NH
4Cl aqueous solution (15 mL x 3), dried over anhydrous Na
2SO
4 and filtered. The filtrate was concentrated under reduced pressure. The residue was purified by RP-Flash with the following conditions: Column: C18 gel column 40 g, 20-35 µm; Mobile Phase A: Water (5 mM aq. NH
4HCO
3), Mobile Phase B: MeCN; Gradient: 5% B hold 5 min, up to 61% B within 18 min, 61% B hold 5 min, up to 95% B within 5 min; Flow rate: 40 mL/min; Detector: UV 254 & 220 nm. The product-containing fractions were collected, concentrated and then lyophilized to give 4- [2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidin-5-yl]oxazole (12.3 mg, 22.38 μmol, 21% yield) as a white solid. MS: m/z = 550.20 [M + H]
+.
1H NMR (400 MHz, Chloroform-d) δ 9.44 (s, 1H), 8.80 (s, 1H), 8.18 (s, 1H), 8.03 (s, 1H), 7.74 (d, J = 8.0 Hz, 2H), 7.61 (d, J = 8.0 Hz, 2H), 7.36 (s, 1H), 5.69 (s, 2H), 4.04 (s, 3H), 3.86 (s, 3H), 1.89 - 1.86 (m, 1H), 1.47 - 1.45 (m, 2H), 1.07 - 1.05 (m, 2H).
19F NMR (376 MHz, Chloroform-d) δ -62.79. Example 140 (Compound 224)
4-[2-chloro-4-[[4
yl]thiazole [1413] To a stirred mixture of 2-chloro-5-iodo-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol- 2-yl]phenyl]methoxy]pyrimidine I-12 (210 mg, 424.55 μmol) in 1,4-dioxane (2 mL) and water (0.4 mL) were added thiazol-4-ylboronic acid (54.74 mg, 424.55 μmol), potassium phosphate (180.2 mg, 849.11 μmol) and Dichloro[9,9-dimethyl-4,5- bis(diphenylphosphino)xanthene]palladium(II) (32 mg, 42.46 μmol) under nitrogen atmosphere at 25 °C. The resulting mixture was stirred under nitrogen atmosphere for 16 hrs at 40 °C. The reaction mixture was cooled to room temperature, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by Prep-TLC (EtOAc : PE = 2 : 1) to give 4-[2-chloro-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidin-5-yl]thiazole (60 mg, 132.79 μmol, 31% yield) as a yellow solid. MS: m/z = 452.10 [M + H]
+. 4-[2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol- 2-yl]phenyl]methoxy]pyrimidin-5-yl]thiazole [1414] To a stirred mixture of 4-[2-chloro-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidin-5-yl]thiazole (60 mg, 132.79 μmol) in 1,4-dioxane (1.5 mL) and water (0.3 mL) were added 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-1,3,6,2- dioxazaborocane (41.9 mg, 159.34 μmol), dicyclohexyl-[2-(2,4,6- triisopropylphenyl)phenyl]phosphane (6.3 mg, 13.28 μmol), Methanesulfonato(2- dicyclohexylphosphino-2',4',6'-tri-i-propyl-1,1'-biphenyl)(2'-amino-1,1'-biphenyl-2- yl)palladium(II) (11.24 mg, 13.28 μmol) and potassium phosphate (56.37 mg, 265.57 μmol) under nitrogen atmosphere at 25 °C. The resulting mixture was stirred under nitrogen atmosphere for 16 hrs at 40 °C. The reaction mixture was cooled to room temperature and concentrated under reduced pressure. The resulting residue was purified by Prep-TLC (EtOAc : PE = 2 : 1) to give 35 mg crude product. The crude product was purified by reverse phase chromatography with the following conditions: Column: C18 spherical Column, 20-35 um, 100A, 40 g; Mobile Phase A: Water (5 mM aq. NH
4HCO
3), Mobile Phase B: MeCN; Flow rate: 35 mL/min; Gradient: 5% B to 5% B in 5 min, 5% B to 73% B in 5 min, 73% B to 73% B in 1 min, 73% B to 95% B in 10 min; Detector: UV 254 & 210 nm. The product- containing fractions were collected and evaporated in vacuo and then lyophilized to give 4- [2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidin-5-yl]thiazole (29.2 mg, 51.63 μmol, 38% yield) as an off-white solid. MS: m/z = 566.30 [M + H]
+.
1H NMR (400 MHz, Chloroform-d) δ 9.65 (s, 1H), 8.94 (d, J = 2.0 Hz, 1H), 8.71 (s, 1H), 8.06 (d, J = 2.0 Hz, 1H), 7.72 (d, J = 8.4 Hz, 2H), 7.64 (d, J = 8.4 Hz, 2H), 7.35 (s, 1H), 5.71 (s, 2H), 3.99 (s, 3H), 3.83 (s, 3H), 1.88 - 1.81 (m, 1H), 1.32 - 1.28 (m, 2H), 0.98 - 0.93 (m, 2H).
19F NMR (376 MHz, Chloroform-d) δ -62.79. Example 141 (Compound 223)
[1415] To a solution of 5-bromo-3-fluoro-2-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]pyridine (400 mg, 1.23 mmol) in ethanol (10 mL) were added sodium acetate trihydrate (335.9 mg, 2.47 mmol) and 1,1'-bis(diphenylphosphino)ferrocene-palladium(II)dichloride (201.6 mg, 246.86 μmol) at 25 °C under carbon monoxide atmosphere (10 atm) in a High pressure reactor. The resulting mixture was stirred at 110 °C for 16 hrs. The reaction mixture was cooled down to room temperature and filtered. The filtrate was concentrated under reduced pressure. The residue was purified by silica gel column chromatography, eluted with 60% EtOAc in PE to give ethyl 5-fluoro-6-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]pyridine-3-carboxylate (300 mg, 945.66 μmol, 77% yield) as a yellow solid. MS: m/z = 318.10 [M + H]
+.
1H NMR (300 MHz, Chloroform-d) δ 9.09 - 9.07 (m, 1H), 8.14 (dd, J = 9.9, 1.8 Hz, 1H), 7.40 (d, J = 1.5 Hz, 1H), 4.46 (q, J = 7.2 Hz, 2H), 3.99 (s, 3H), 1.44 (t, J = 7.2 Hz, 3H). Ethyl 6-[5-chloro-1-methyl-4-(trifluoromethyl)imidazol-2-yl]-5-fluoro-pyridine-3- carboxylate [1416] To a solution of ethyl 5-fluoro-6-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]pyridine-3-carboxylate (500 mg, 1.58 mmol) in DMF was added N-chlorosuccinimide (631.4 mg, 4.73 mmol) at 25 °C, then stirred at 45 °C for 16 hrs under nitrogen atmosphere. The reaction was cooled down to room temperature, then diluted with water (50 mL), extracted with DCM (3 x 30 mL). The combined organic layers were washed with brine (3 x 30 mL), dried over anhydrous Na
2SO
4. After filtration, the filtrate was concentrated under reduced pressure. The residue was purified by silica gel column chromatography, eluted with 45% - 50% EtOAc in PE. The collected fractions were combined and concentrated under reduced pressure to give ethyl 6-[5-chloro-1-methyl-4-(trifluoromethyl)imidazol-2-yl]-5- fluoro-pyridine-3-carboxylate (400 mg, 1.14 mmol, 72% yield) as a yellow solid. MS: m/z = 352.10 [M + H]
+.
1H NMR (300 MHz, Chloroform-d) δ 9.01 (s, 1H), 8.07 (dd, J = 9.9, 1.5 Hz, 1H), 4.39 (q, J = 7.2 Hz, 2H), 3.84 (s, 3H), 1.37 (t, J = 7.2 Hz, 3H). [6-[5-chloro-1-methyl-4-(trifluoromethyl)imidazol-2-yl]-5-fluoro-3-pyridyl]methanol [1417] To a solution of ethyl 6-[5-chloro-1-methyl-4-(trifluoromethyl)imidazol-2-yl]-5- fluoro-pyridine-3-carboxylate (500 mg, 1.42 mmol) in methanol (10 mL) was added sodium borohydride (215.1 mg, 5.69 mmol) at 0 °C under nitrogen atmosphere. The reaction mixture was stirred at 25 °C for 16 hrs. The resulting mixture was concentrated under reduced pressure. The residue was purified by Prep-TLC, eluted with 7% methanol in DCM to give [6-[5-chloro-1-methyl-4-(trifluoromethyl)imidazol-2-yl]-5-fluoro-3-pyridyl]methanol (200 mg, 645.90 μmol, 45% yield) as an off-white solid. MS: m/z = 310.00 [M + H]
+.
1H NMR (300 MHz, Chloroform-d) δ 8.34 (d, J = 1.5 Hz, 1H), 7.52 - 7.47 (m, 1H), 4.73 (s, 2H), 3.71 (s, 3H). 5-[4-[[6-[5-chloro-1-methyl-4-(trifluoromethyl)imidazol-2-yl]-5-fluoro-3- pyridyl]methoxy]pyrimidin-2-yl]-4-cyclopropyl-6-methoxy-pyrimidine [1418] To a stirring mixture of 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)pyrimidin-4-ol I- 11(130 mg, 532.24 μmol) and [6-[5-chloro-1-methyl-4-(trifluoromethyl)imidazol-2-yl]-5- fluoro-3-pyridyl]methanol (164.8 mg, 532.24 μmol) in THF (5 mL) were added trimethylphosphine (121.5 mg, 1.60 mmol) and 1,1'-(azodicarbonyl)-dipiperidine (402.9 mg, 1.60 mmol) under nitrogen atmosphere at 25 °C. The resulting mixture was stirred for 16 hrs at 25 °C then concentrated under reduced pressure. The residue was purified by Prep-TLC, eluted with 10% MeOH in DCM to give 60 mg of a more polar isomer and 70 mg of a less polar isomer. The less polar isomer was further purified by RP-Flash with the following conditions: Column: C18 spherical Column, 20-35 um, 100A, 20 g; Mobile Phase A: Water (5 mM aq. NH
4HCO
3), Mobile Phase B: MeCN; Flow rate: 60 mL/min; Gradient: 5% B to 5% B in 5 min, 5% B to 65% B in 10 min, 65% B to 65% B in 2 min, 65% B to 95% B in 5 min; Detector: UV 254 & 210 nm. The collected fractions were combined, concentrated and then lyophilized to give 5-[4-[[6-[5-chloro-1-methyl-4-(trifluoromethyl)imidazol-2-yl]-5-fluoro-3- pyridyl]methoxy]pyrimidin-2-yl]-4-cyclopropyl-6-methoxy-pyrimidine (57.3 mg, 106.93 μmol, 20% yield) as an off-white solid. MS: m/z = 536.20 [M + H]
+.
1H NMR (300 MHz, Chloroform-d) δ 8.71 - 8.63 (m, 2H), 8.59 (s, 1H), 7.72 (d, J = 10.2 Hz, 1H), 7.26 (s, 1H), 6.85 (d, J = 5.7 Hz, 1H), 5.57 (s, 2H), 3.95 (s, 3H), 3.85 (s, 3H), 1.74 - 1.65 (m, 1H), 1.32 - 1.20 (m, 2H), 0.99 - 0.86 (m, 2H).
19F NMR (282 MHz, Chloroform-d) δ -61.91, -118.60. Example 142 (Compound 221)
[4-(4-chloro-1-methyl-imidazol-2-yl)phenyl]methanol [1419] [4-(Hydroxymethyl)phenyl]boronic acid (233 mg, 1.53 mmol), 2-bromo-4-chloro-1- methyl-1H-imidazole (250 mg, 1.28 mmol), bis(diphenylphosphino)ferrocene]dichloropalladium(II)-DCM (105 mg, 128 μmol) and K
2CO
3 (354 mg, 2.56 mmol) were mixed in a degassed mixture of dioxane (5 mL) and water (500 μL) under argon atmosphere. The reaction mixture was stirred at 70 °C for 12 hr. The reaction mixture was cooled to room temperature and filtered. The filtrate was concentrated under reduced pressure to afford [4-(4-chloro-1-methyl-imidazol-2-yl)phenyl]methanol (300 mg, crude) as a brown oil which was used in the next step without further purification. MS (ESI): [M+H]
+ m/z: calcd 223.07; found 223.0 5-[4-[[4-(4-chloro-1-methyl-imidazol-2-yl)phenyl]methoxy]pyrimidin-2-yl]-4-cyclopropyl-6- methoxy-pyrimidine [1420] NaH (19.3 mg, 503 μmol, 60% dispersion in mineral oil) was suspended in THF (10 mL) at 0 °C. [4-(4-Chloro-1-methyl-imidazol-2-yl)phenyl]methanol (102 mg, crude from previous step) was added to the mixture followed by 4-cyclopropyl-6-methoxy-5-(4- methylsulfonylpyrimidin-2-yl)pyrimidine I-8 (140 mg, 457 μmol) at 0 °C. The reaction mixture was stirred at 40 °C for 12 hr. The reaction mixture was cooled to room temperature and filtered. The filtrate was concentrated under reduced pressure. The residue was subjected to HPLC (0-5 min, 35-60% water+FA (0.1% vol) - ACN+FA (0.1% vol.); flow: 30 mL/min, column: Chromatorex 18 SMB100-5T, 100 × 19 mm, 5 µm) to afford 5-[4-[[4-(4-chloro-1- methyl-imidazol-2-yl)phenyl]methoxy]pyrimidin-2-yl]-4-cyclopropyl-6-methoxy-pyrimidine (19.2 mg, 42.8 μmol, 9.4% yield) as a white solid.
1H NMR (600 MHz, DMSO-d
6) δ 0.86 – 0.90 (m, 2H), 1.02 – 1.06 (m, 2H), 1.66 – 1.71 (m, 1H), 3.72 (s, 3H), 3.85 (s, 3H), 5.49 (s, 2H), 7.07 (d, 1H), 7.38 (s, 1H), 7.56 (d, 2H), 7.69 (d, 2H), 8.68 (s, 1H), 8.71 (d, 1H). MS (ESI): [M+H]
+ m/z: calcd 449.17; found 449.2. Example 143 (Compound 220)

(R)-tert-butyl (3-((2-chloro-4-((4-(1-methyl-4-(trifluoromethyl)-1H-imidazol-2- yl)benzyl)oxy)pyrimidin-5-yl)oxy)-1,1,1-trifluoropropan-2-yl)carbamate Synthesis of the starting 2-chloro-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidin-5-ol is described for Compound 122. [1421] Triphenyl phosphine (1.06 g, 4.05 mmol) and DIAD (820 mg, 4.05 mmol, 798 μL) were added to a stirred solution of 2-chloro-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidin-5-ol (1.30 g, 3.38 mmol) and (R)-tert-butyl (1,1,1-trifluoro-3- hydroxypropan-2-yl)carbamate (929 mg, 4.05 mmol) in THF (20 mL). The reaction mixture was stirred at room temperature for 16 hr. The reaction mixture was concentrated under reduced pressure. The residue was subjected to flash column chromatography (silica, gradient hexane-EtOAc) to afford (R)-tert-butyl (3-((2-chloro-4-((4-(1-methyl-4-(trifluoromethyl)-1H- imidazol-2-yl)benzyl)oxy)pyrimidin-5-yl)oxy)-1,1,1-trifluoropropan-2-yl)carbamate (1.00 g, 1.68 mmol, 49.7% yield) as a white solid. MS (ESI): [M+H]
+ m/z: calcd 596.15; found 596.2 tert-butyl N-[(1R)-1-[[2-chloro-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidin-5-yl]oxymethyl]-2,2,2-trifluoro-ethyl]carbamate [1422] (R)-tert-butyl (3-((2-chloro-4-((4-(1-methyl-4-(trifluoromethyl)-1H-imidazol-2- yl)benzyl)oxy)pyrimidin-5-yl)oxy)-1,1,1-trifluoropropan-2-yl)carbamate (1.00 g, 1.68 mmol), (4-cyclopropyl-6-methoxy-pyrimidin-5-yl)boronic acid I-5 (456 mg, 2.35 mmol) and potassium phosphate tribasic (1.07 g, 5.03 mmol) were mixed in a degassed mixture of dioxane (70 mL) and water (3.5 mL). XPhos Pd G3 (71 mg, 83.9 μmol) was added to the mixture. The reaction mixture was stirred at 60°C for 16 hr under reduced pressure. Additional portions of (4-cyclopropyl-6-methoxy-pyrimidin-5-yl)boronic acid I-5 (456 mg, 2.35 mmol) and XPhos Pd G3 (71.0 mg, 83.9 μmol) were added to the mixture. The reaction mixture was stirred at 60°C for 12 hr. The reaction mixture was cooled to room temperature, diluted with EtOAc (50 mL) and washed with water (2×50 mL). The combined organic layers were separated and concentrated under reduced pressure. The residue was subjected to flash column chromatography (silica, gradient CHCl
3 - MTBE) to afford tert-butyl N-[(1R)-1-[[2- chloro-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidin-5- yl]oxymethyl]-2,2,2-trifluoro-ethyl]carbamate (100 mg, 141 μmol, 8.4% yield) as a yellow solid. MS (ESI): [M+H]
+ m/z: calcd 710.25; found 710.2 (2R)-3-[2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidin-5-yl]oxy-1,1,1-trifluoro-propan-2- amine [1423] TFA (129 mg, 1.13 mmol, 87 μL) was added to a stirred solution of tert-butyl N- [(1R)-1-[[2-chloro-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidin-5-yl]oxymethyl]-2,2,2-trifluoro-ethyl]carbamate (80.0 mg, 113 μmol) in DCM (5 mL). The reaction mixture was stirred at room temperature for 16 hr. The reaction mixture was concentrated under reduced pressure. The residue was subjected to HPLC (0.5-6.5 min., 20-70% water – ACN, flow: 30 mL/min, column: SunFire C18 (R),100×19 mm, 5 µm) to afford (2R)-3-[2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[4- [1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidin-5-yl]oxy-1,1,1- trifluoro-propan-2-amine (9.00 mg, 14.8 μmol, 13.1% yield) were obtained as a white solid.
1H NMR (600 MHz, DMSO-d
6) δ 0.84 – 0.88 (m, 2H), 1.01 – 1.05 (m, 2H), 1.68 – 1.74 (m, 1H), 2.20 (d, 2H), 3.75 – 3.80 (m, 4H), 3.84 (s, 3H), 4.26 – 4.36 (m, 2H), 5.54 (s, 2H), 7.58 (d, 2H), 7.73 (d, 2H), 7.94 (s, 1H), 8.51 (s, 1H), 8.65 (s, 1H). MS (ESI): [M+H]
+ m/z: calcd 610.23; found 610.2. Example 144 (Compound 218)
Ethyl 2-chloro-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidine- 5-carboxylate [1424] A mixture of NaH (156 mg, 3.90 mmol, 60% dispersion in mineral oil) and [4-[1- methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methanol I-2b (1.00 g, 3.90 mmol) in THF (40 mL) was stirred at room temperature for 30 min. Ethyl 2,4-dichloropyrimidine-5- carboxylate (863 mg, 3.90 mmol) was added to the mixture. The resulting mixture was stirred at room temperature for 12 hr. The reaction mixture was poured into cold water and extracted with EtOAc (2×50 mL). The combined organic layers were washed with brine, dried over anhydrous Na
2SO
4 and concentrated under reduced pressure. The residue was subjected to flash column chromatography (silica, gradient MTBE - CHCl
3) to afford ethyl 2-chloro-4-[[4- [1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidine-5-carboxylate (1.10 g, 2.50 mmol, 63.9% yield) as a white solid.
1H NMR (600 MHz, DMSO-d
6) δ 1.29 (t, 3H), 3.80 (s, 3H), 4.33 (q, 2H), 5.61 (s, 2H), 7.66 (d, 2H), 7.78 (d, 2H), 7.95 (s, 1H), 8.93 (s, 1H). MS (ESI): [M+H]
+ m/z: calcd 441.10; found 441.2 Ethyl 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidine-5-carboxylate [1425] A mixture of ethyl 2-chloro-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidine-5-carboxylate (800 mg, 1.81 mmol), (4-cyclopropyl-6- methoxy-pyrimidin-5-yl)boronic acid I-5 (880 mg, 4.54 mmol), potassium phosphate tribasic (2.31 g, 10.9 mmol), RuPhos (84.7 mg, 182 μmol) and RuPhos Pd G4 (154 mg, 182 μmol) in a degassed mixture of dioxane (25 mL) and water (2.5 mL) was stirred at room temperature for 12 hr under inert atmosphere. The reaction mixture was diluted with EtOAc (50 mL). Anhydrous Na
2SO
4 (10 g) was added to the mixture. The resulting mixture was stirred for 30 min, then solids were filtered out and washed with EtOAc. The filtrate was concentrated under reduced pressure to afford ethyl 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[4-[1- methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidine-5-carboxylate (1.50 g, crude) as a yellow gum which was used in the next step without further purification. MS (ESI): [M+H]
+ m/z: calcd 555.20; found 555.2 [2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidin-5-yl]methanol [1426] A solution of ethyl 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidine-5-carboxylate (1.50 g, crude from previous step) in THF (5 mL) was added dropwise to a vigorously stirred suspension of LAH (348 mg, 9.17 mmol) in THF (20 mL) at -45 °C. The reaction mixture was stirred at -45 °C for 2 hr. Water (350 µL) was added dropwise to the reaction mixture followed by an aqueous solution of NaOH (400 µL, 28.5%) and water (450 µL). The resulting mixture was allowed to warm to room temperature at stirring, then solids were filtered out and washed with THF. The filtrate was concentrated under reduced pressure. The residue was subjected to flash column chromatography (silica, gradient MeOH - CHCl
3) to afford [2-(4- cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidin-5-yl]methanol (720 mg, 1.40 mmol, 77.3% yield from ethyl 2- chloro-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidine-5- carboxylate) as a yellow gum.
1H NMR (400 MHz, CDCl
3) δ 0.88 – 0.94 (m, 2H), 1.21 – 1.27 (m, 2H), 1.71 – 1.79 (m, 1H), 3.71 (t, 1H), 3.80 (s, 3H), 3.95 (s, 3H), 4.81 (s, 2H), 5.59 (s, 2H), 7.34 (s, 1H), 7.56 (d, 2H), 7.67 (d, 2H), 8.67 (s, 1H), 8.69 (s, 1H). MS (ESI): [M+H]
+ m/z: calcd 513.19; found 513.2 [2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidin-5-yl]methyl methanesulfonate [1427] Methanesulfonyl chloride (33.5 mg, 293 μmol, 22.7 μL) was added to a stirred solution of [2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidin-5-yl]methanol (100 mg, 195 μmol) and TEA (50.4 mg, 498 μmol, 69.5 μL) in DCM (5 mL) at 0 °C. The reaction mixture was stirred at 0 °C for 2 hr. The reaction mixture was poured into cold water. The organic layer was separated, washed with an aqueous solution of NaHCO
3 (20 mL), dried over anhydrous Na
2SO
4 and concentrated under reduced pressure to afford [2-(4-cyclopropyl-6- methoxy-pyrimidin-5-yl)-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidin-5-yl]methyl methanesulfonate (100 mg) as a yellow gum which was used in the next step without further purification. MS (ESI): [M+H]
+ m/z: calcd 591.17; found 591.2 4-[[2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol- 2-yl]phenyl]methoxy]pyrimidin-5-yl]methyl]morpholine [1428] A mixture of [2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidin-5-yl]methyl methanesulfonate (50.0 mg, crude from previous step) and morpholine (22.1 mg, 254 μmol, 22.2 μL) in DMF (1.0 mL) was stirred at room temperature for 12 hr. The reaction mixture was subjected to HPLC (2-7 min., 40-65% water – ACN, +0.1% vol. of 25% aq. NH
3, flow: 30 mL/min, column: XBridge C18 (L), 100×19 mm, 5 µm) to afford 4-[[2-(4-cyclopropyl-6-methoxy- pyrimidin-5-yl)-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidin-5-yl]methyl]morpholine (2.60 mg, 4.47 μmol, 4.58% yield) as a yellow gum.
1H NMR (600 MHz, DMSO-d
6) δ 0.84 – 0.89 (m, 2H), 1.01 – 1.05 (m, 2H), 1.67 – 1.72 (m, 1H), 2.45 – 2.49 (m, 4H), 3.57 (s, 2H), 3.59 – 3.63 (m, 4H), 3.79 (s, 3H), 3.84 (s, 3H), 5.54 (s, 2H), 7.61 (d, 2H), 7.74 (d, 2H), 7.94 (s, 1H), 8.65 (s, 1H), 8.67 (s, 1H). MS (ESI): [M+H]
+ m/z: calcd 582.28; found 582.2.

2-chloro-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]-5-[[1- (trifluoromethyl)cyclopropyl]methoxy]pyrimidine [1429] Synthesis of the starting 2-chloro-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidin-5-ol is described in the document “122”. DIAD (60.6 mg, 299 μmol) was added to a stirred solution of 2-chloro-4-[[4-[1-methyl- 4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidin-5-ol (96.0 mg, 250 μmol), [1- (trifluoromethyl)cyclopropyl]methanol (42.0 mg, 299 μmol) and triphenylphosphine (72.0 mg, 275 μmol) in THF (3 mL). The reaction mixture was stirred at room temperature for 12 hr. The reaction mixture was diluted with water (500 µL) and ACN (2 mL). The resulting mixture was subjected to HPLC (0.5-6.5 min., 45-70% water – ACN, flow: 30 mL/min, column: SunFire C18 (R), 100×19 mm, 5 µm) to afford 2-chloro-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol- 2-yl]phenyl]methoxy]-5-[[1-(trifluoromethyl)cyclopropyl]methoxy]pyrimidine (80.0 mg, 158 μmol, 63.3% yield) as a white solid. MS (ESI): [M+H]
+ m/z: calcd 507.13; found 507.0 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]-5-[[1-(trifluoromethyl)cyclopropyl]methoxy]pyrimidine 2-Chloro-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]-5-[[1- (trifluoromethyl)cyclopropyl]methoxy]pyrimidine (80.0 mg, 158 μmol), (4-cyclopropyl-6- methoxy-pyrimidin-5-yl)boronic acid I-5 (45.9 mg, 237 μmol), potassium phosphate tribasic (67.0 mg, 316 μmol) and XPhos Pd G3 (6.68 mg, 7.89 μmol) were mixed in a degassed mixture of dioxane (3.0 mL) and water (300 µL). The reaction mixture was stirred at 60 °C for 4 hr under argon atmosphere. The reaction mixture was cooled to room temperature, diluted with water (5.0 mL), and extracted with EtOAc (2×10 mL). The combined organic layers were washed with water (2×10 mL) and brine (10 mL). SiliaMetS
® Dimercaptotriazine (20 mg) was added to the organic layer. The resulting mixture was stirred for 30 min, then solids were filtered out. The filtrate was concentrated under reduced pressure. The residue was subjected to HPLC (2-7 min, 50-75% water – ACN, flow: 30 mL/min, column: SunFire C18 (R), 100×19 mm, 5 µm) to afford 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]-5-[[1- (trifluoromethyl)cyclopropyl]methoxy]pyrimidine (24.0 mg, 38.7 μmol, 24.5% yield) as a beige solid.
1H NMR (600 MHz, DMSO-d
6) δ 0.83 – 0.87 (m, 2H), 1.00 – 1.06 (m, 4H), 1.13 – 1.16 (m, 2H), 1.67 – 1.72 (m, 1H), 3.79 (s, 3H), 3.83 (s, 3H), 4.34 (s, 2H), 5.55 (s, 2H), 7.58 (d, 2H), 7.73 (d, 2H), 7.94 (s, 1H), 8.43 (s, 1H), 8.65 (s, 1H). MS (ESI): [M+H]
+ m/z: calcd 621.24; found 621.2.
5-bromo-2-chloro-4-[[3-fluoro-4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidine Synthesis of the starting [3-fluoro-4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methanol is described for compound 97. [1430] 5-Bromo-2,4-dichloro-pyrimidine (400 mg, 1.76 mmol) was added to a stirred mixture of [3-fluoro-4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methanol (313 mg, 1.14 mmol) and Cs
2CO
3 (686 mg, 2.11 mmol) in ACN (10 mL). The reaction mixture was stirred at 65 °C for 12 hr. The reaction mixture was cooled to room temperature and concentrated under reduced pressure. The residue was diluted with water (30 mL) and extracted with DCM (40 mL). The organic layer was separated, washed with brine (30 mL), dried over anhydrous Na
2SO
4 and concentrated under reduced pressure to afford 5-bromo-2- chloro-4-[[3-fluoro-4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidine (400 mg, 859 μmol, 48.9% yield) as a red solid which was used in the next step without further purification.
1H NMR (500 MHz, DMSO-d
6) δ 3.63 (s, 3H), 5.60 (s, 2H), 7.49 (d, 1H), 7.55 (d, 1H), 7.66 (d, 1H), 8.01 (s, 1H), 8.79 (s, 1H). MS (ESI): [M+H]
+ m/z: calcd 466.97; found 467.0 2-[2-chloro-4-[[3-fluoro-4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidin-5-yl]propan-2-ol [1431] 5-Bromo-2-chloro-4-[[3-fluoro-4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidine (200 mg, 430 μmol) was dissolved in THF (2 mL) and cooled to -100 °C. n-Butyl lithium (644 μmol, 270 μL, 2.4 M in hexane) was added to the solution and the mixture was stirred for 5 min at -90 °C. Acetone (99.8 mg, 1.72 mmol, 126 μL) was added to the mixture at -90 °C. The rection mixture was stirred for 5 min at -90 °C. The resulting mixture was quenched with water (0.2 mL) and diluted with MTBE (10 mL). The organic layer was washed with brine (2 mL) and concentrated under reduced pressure to afford 2-[2-chloro-4-[[3-fluoro-4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidin-5-yl]propan-2-ol (250 mg, crude) as a yellow oil which was used in the next step without further purification. MS (ESI): [M+H]
+ m/z: calcd 445.11; found 445.0 2-[2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[3-fluoro-4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidin-5-yl]propan-2-ol [1432] 2-[2-Сhloro-4-[[3-fluoro-4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidin-5-yl]propan-2-ol (20.0 mg, crude from previous step), (4- cyclopropyl-6-methoxy-pyrimidin-5-yl)boronic acid I-5 (17.5 mg, 89.9 μmol), potassium phosphate tribasic (28.6 mg, 135 μmol) and RuPhos Pd G3 (1.9 mg, 2.25 μmol) were mixed in a degassed mixture of dioxane (2.0 mL) and water (200 µL) under argon atmosphere. The reaction mixture was stirred at room temperature for 20 hr. Additional portions of (4- cyclopropyl-6-methoxy-pyrimidin-5-yl)boronic acid I-5 (17.5 mg, 89.9 μmol) and RuPhos Pd G3 (1.91 mg, 2.25 μmol) were added to the mixture. The resulting mixture was stirred at 70 °C for 14 hr. The organic layer was separated, filtered and subjected to HPLC (0-2-12 min., 20-20-40% water – ACN, flow: 30 mL/min, column: Chromatorex SMB100-5T 100×19 mm, 5 µm) to afford 2-[2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[3-fluoro-4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidin-5-yl]propan-2-ol (4.00 mg, 7.16 μmol, 20.8% yield from 5-bromo-2-chloro-4-[[3-fluoro-4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidine) as a white solid.
1H NMR (600 MHz, DMSO-d
6) δ 0.91 – 0.95 (m, 2H), 1.06 – 1.10 (m, 2H), 1.61 (s, 6H), 1.69 – 1.75 (m, 1H), 3.65 (s, 3H), 3.88 (s, 3H), 5.46 (br. s., 1H), 5.60 (s, 2H), 7.48 (d, 1H), 7.53 (d, 1H), 7.67 (t, 1H), 8.05 (s, 1H), 8.71 (s, 1H), 8.87 (s, 1H). MS (ESI): [M+H]
+ m/z: calcd 559.24; found 559.2.
3-chloro-5-fluoro-4-formyl-benzonitrile [1433] Butyllithium (2.5 M, 15.4 mL) was added dropwise to a solution of diisopropylamine (3.90 g, 38.6 mmol, 5.44 mL) in THF (50 mL) at -20 °C. The resulting mixture was stirred at -20°C for 15 min. The mixture was cooled to -78 °C.3-chloro-5-fluoro-benzonitrile (5.00 g, 32.1 mmol) was added to the mixture. The resulting mixture was stirred for 30 min at -78 °C. Ethyl formate (3.33 g, 45.0 mmol, 3.62 mL) was added to the mixture dropwise at -78 °C. The reaction mixture was allowed to warm to room temperature and poured into cold saturated aqueous solution of NH
4Cl (100 mL). The resulting mixture was extracted with EtOAc (2×50 mL). The combined organic layers were washed with brine (50 mL), dried over anhydrous Na
2SO
4 and concentrated under reduced pressure. The residue was subjected to flash column chromatography (silica, gradient Hexane - EtOAc) to afford 3-chloro-5-fluoro- 4-formyl-benzonitrile (1.20 g, 6.54 mmol, 20.3% yield) as a white solid.
1H NMR (400 MHz, CDCl
3) δ 7.40 (d, 1H), 7.58 (s, 1H), 10.42 (s, 1H). MS (ESI): [M+H]
+ m/z: calcd 184.00; found 184.2 3-chloro-5-fluoro-4-[4-(trifluoromethyl)-1H-imidazol-2-yl]benzonitrile [1434] A mixture of 3,3-dibromo-1,1,1-trifluoro-propan-2-one (1.94 g, 7.19 mmol) and sodium acetate (1.18 g, 14.4 mmol) in water (2 mL) was stirred at 100 °C for 2 hr. The mixture was cooled to room temperature. A solution of 3-chloro-5-fluoro-4-formyl- benzonitrile (1.20 g, 6.54 mmol) and ammonium hydroxide (5.90 mL, 25% wt. in water) in MeOH (39.4 mL) was added to the mixture. The resulting mixture was stirred at 100 °C for 1 hr. The reaction mixture was cooled to room temperature and concentrated under reduced pressure. The residue was diluted with water (50 mL) and extracted with EtOAc (2×50 mL). The combined organic layers were washed with brine (40 mL), dried over anhydrous Na
2SO
4 and concentrated under reduced pressure to afford 3-chloro-5-fluoro-4-[4-(trifluoromethyl)- 1H-imidazol-2-yl]benzonitrile (1.50 g, 5.18 mmol, 79.4% yield) as a yellow solid which was used in the next step without further purification. MS (ESI): [M+H]
+ m/z: calcd 290.01; found 290.0 3-chloro-5-fluoro-4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]benzonitrile [1435] A mixture of 3-chloro-5-fluoro-4-[4-(trifluoromethyl)-1H-imidazol-2-yl]benzonitrile (12.0 g, 41.4 mmol), methyl iodide (6.47 g, 45.6 mmol, 2.84 mL) and Cs
2CO
3 (27.0 g, 82.9 mmol) in acetone (240 mL) was stirred under reflux for 12 hr. The mixture was cooled to room temperature and filtered. The filtrate was concentrated under reduced pressure. The residue was diluted with water (100 mL) and extracted with EtOAc (2×250 mL). The combined organic layers were washed with brine (200 mL), dried over anhydrous Na
2SO
4 and concentrated under reduced pressure to afford 3-chloro-5-fluoro-4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]benzonitrile (12.0 g, 39.5 mmol, 95.2% yield) as a yellow solid which was used in the next step without further purification. MS (ESI): [M+H]
+ m/z: calcd 304.03; found 304.0 3-chloro-5-fluoro-4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]benzoic acid [1436] A mixture of 3-chloro-5-fluoro-4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]benzonitrile (12.0 g, 39.5 mmol) and sodium hydroxide (4.74 g, 119 mmol) in MeOH (120 mL) was stirred under reflux for 12 hr. The reaction mixture was cooled to room temperature and concentrated under reduced pressure. The residue was distributed between water (40 mL) and MTBE (50 mL). The aqueous layer was separated, acidified with aq. HCl to pH = 2 and extracted with CHCl
3 (30 mL). The organic layer was washed with water (30 mL), dried over anhydrous Na
2SO
4 and concentrated under reduced pressure to afford 3-chloro-5-fluoro-4-[1- methyl-4-(trifluoromethyl)imidazol-2-yl]benzoic acid (5.50 g, 17.1 mmol, 43.0% yield) as an orange solid which was used in the next step without further purification.
1H NMR (400 MHz, CDCl
3) δ 3.38 (s, 3H), 7.31 (s, 1H), 7.57 (d, 1H), 7.79 (s, 1H). MS (ESI): [M+H]
+ m/z: calcd 323.02; found 323.0 [3-chloro-5-fluoro-4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methanol [1437] Borane dimethyl sulfide complex (1.04 g, 13.7 mmol, 1.30 mL) was added to a stirred solution of 3-chloro-5-fluoro-4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]benzoic acid (1.47 g, crude) in THF (50 mL). The resulting mixture was stirred under refluxed for 12 hr. The reaction mixture was cooled to room temperature and quenched by dropwise addition of MeOH (50 mL). The resulting mixture was concentrated under reduced pressure. The residue was distributed between EtOAc (400 mL) and saturated aqueous solution of NH
4Cl (200 mL). The organic layer was separated, washed with water (200 mL), dried over anhydrous Na
2SO
4 and concentrated under reduced pressure. The residue was subjected to flash column chromatography (silica, DCM) to afford [3-chloro-5-fluoro-4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methanol (1.00 g, 3.24 mmol, 71.1% yield) as a yellow solid.
1H NMR (400 MHz, CDCl
3) δ 3.51 (s, 3H), 4.59 (d, 2H), 5.59 (t, 1H), 7.35 (d, 1H), 7.47 (s, 1H), 8.05 (s, 1H). MS (ESI): [M+H]
+ m/z: calcd 309.04; found 309.0 5-[4-[[3-chloro-5-fluoro-4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidin-2-yl]-4-cyclopropyl-6-methoxy-pyrimidine [1438] [3-Chloro-5-fluoro-4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methanol (111 mg, 359 μmol) was added to a stirred mixture of NaH (14.4 mg, 359 μmol, 60% dispersion in mineral oil) in DMF (2 mL) at room temperature. The resulting mixture was stirred for 15 min.4-cyclopropyl-6-methoxy-5-(4-methylsulfonylpyrimidin-2-yl)pyrimidine I-8 (100 mg, 326 μmol) was added to the mixture. The resulting mixture was stirred at room temperature for 12 hr. The reaction mixture was subjected to HPLC (0.5-6.5 min., 45-70% water – ACN, +0.1% vol. of 25% aq. NH
3, flow: 30 mL/min, column: XBridge, 100 × 19 mm, 5 µm) to a residue which was subjected to SFC (0.5-10.5 min., 5-30% CO
2-MeOH; column: Chromatorex PEI, 100 × 19 mm, 5 µm; flow; 50 mL/min) to afford 5-[4-[[3-chloro- 5-fluoro-4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidin-2-yl]-4- cyclopropyl-6-methoxy-pyrimidine (4.30 mg, 8.04 μmol, 2.5% yield) as an off-white solid.
1H NMR (600 MHz, DMSO-d
6) δ 0.87 – 0.93 (m, 2H), 1.02 – 1.07 (m, 2H), 1.66 – 1.73 (m, 1H), 3.53 (s, 3H), 3.85 (s, 3H), 5.52 (s, 2H), 7.14 (d, 1H), 7.57 (d, 1H), 7.67 (s, 1H), 8.07 (s, 1H), 8.68 (s, 1H), 7.74 (d, 1H). MS (ESI): [M+H]
+ m/z: calcd 535.15; found 535.2.
2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidin-5-ol Synthesis of the starting 2-chloro-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidin-5-ol is described for compound 122. [1439] 2-Chloro-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidin-5-ol (200 mg, 520 μmol), (4-cyclopropyl-6-methoxy- pyrimidin-5-yl)boronic acid I-5 (252 mg, 1.30 mmol) and DIPEA (336 mg, 2.60 mmol, 453 μL) were mixed in a degassed mixture of dioxane (12 mL) and water (1 mL). RuPhos Pd G4 (88.4 mg, 104 μmol) was added to the mixture and the reaction mixture was stirred at 80 °C for 24 h. The mixture was cooled to room temperature and concentrated under reduced pressure. The crude residue was dissolved in DMF (5 mL). SiliaMetS
® Dimercaptotriazine (400 mg) was added to the mixture and the resulting suspension was stirred for 10 hr. The suspension was filtered. The filtrate was subjected to HPLC (0.5-6.5 min, 15-40% water – ACN, flow: 30 mL/min, column: SunFire C18100 ×19 mm, 5 µm) to afford 160 mg of mixture of desired product (29%) and starting chloride (71%). This mixture was subjected to preparative SFC purification (0-0.5-10.5-12.5 min, 5-5-50-50% IPA-ACN; flow: 50 mL/min; column: Chromatorex_Diol, 100×19 mm, 5 µm, ) to afford 2-(4-cyclopropyl-6-methoxy- pyrimidin-5-yl)-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidin-5-ol (32 mg, 64.20 μmol, 12.4% yield) as a light-yellow solid.
1H NMR (600 MHz, DMSO-d
6) δ 0.83 – 0.88 (m, 2H), 0.99 – 1.02 (m, 2H), 1.68 – 1.73 (m, 1H), 3.79 (s, 3H), 3.84 (s, 3H), 5.50 (s, 2H), 7.60 (d, 2H), 7.73 (d, 2H), 7.94 (s, 1H), 8.22 (s, 1H), 8.63 (s, 1H), 10.34 (br. s, 1H). MS (ESI): [M+H]
+ m/z: calcd 499.19; found 499.2 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]-5-tetrahydrofuran-3-yloxy-pyrimidine [1440] Diisopropyl azodicarboxylate (38.1 mg, 189 μmol, 37.1 μL) was added to a stirred solution of 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidin-5-ol (47.0 mg, 94.3 μmol), 3- hydroxytetrahydrofuran (16.6 mg, 189 μmol) and triphenylphosphine (49.5 mg, 189 μmol) in THF (2 mL) at 0 °C. The reaction mixture was stirred at ambient temperature for 12 hr. The reaction mixture was subjected to HPLC (0.5-6.5 min, 40-65% water - ACN; flow: 30 mL/min, column: Chromatorex SMB 100-5, 100×19 mm, 5 µm) to afford 2-(4-cyclopropyl- 6-methoxy-pyrimidin-5-yl)-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]-5-tetrahydrofuran-3-yloxy-pyrimidine (24.0 mg, 42.2 μmol, 44.8% yield) as a yellow gum.
1H NMR (600 MHz, DMSO-d
6) δ 0.84 – 0.89 (m, 2H), 1.01 – 1.05 (m, 2H), 1.70 – 1.77 (m, 1H), 2.02 – 2.11 (m, 1H), 2.24 – 2.33 (m, 1H), 3.74 – 3.81 (m, 4H), 3.83 – 3.93 (m, 6H), 5.19 – 5.25 (m, 1H), 5.53 (s, 2H), 7.58 (d, 2H), 7.74 (d, 2H), 7.95 (s, 1H), 8.43 (s, 1H), 8.65 (s, 1H). MS (ESI): [M+H]
+ m/z: calcd 569.24; found 569.1.
2-chloro-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]-6- (trifluoromethyl)pyrimidine [1441] Potassium tert-butoxide (241 mg, 2.15 mmol) was added to a solution of [4-[1- methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methanol I-2b (500 mg, 1.95 mmol) and 2,4- dichloro-6-(trifluoromethyl)pyrimidine (466 mg, 2.15 mmol) in toluene (20 mL) at 0 °C. The reaction mixture was stirred at room temperature for 15 hr. The reaction mixture was poured into water (40 mL) and extracted with EtOAc (40 mL). The organic layer was concentrated under reduced pressure. The residue was subjected to flash column chromatography (silica, hexane – EtOAc 7/3) to afford 2-chloro-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]-6-(trifluoromethyl)pyrimidine (100 mg, 229 μmol, 11.7% yield) as a light-yellow solid.
1H NMR (600 MHz, DMSO-d
6) 3.80 (s, 3H), 5.59 (s, 2H), 7.65 (d, 2H), 7.72 (s, 1H), 7.78 (d, 2H), 7.96 (s, 1H), 8.61 (s, 1H). MS (ESI): [M+H]
+ m/z: calcd 437.06; found 437.2 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]-6-(trifluoromethyl)pyrimidine [1442] 2-Chloro-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]-6- (trifluoromethyl)pyrimidine (90.0 mg, 206 μmol), (4-cyclopropyl-6-methoxy-pyrimidin-5- yl)boronic acid I-5 (60.0 mg, 309 μmol), RuPhos Pd G4 (17.5 mg, 20.6 μmol) and potassium phosphate tribasic (131 mg, 618 μmol) were mixed in a degassed mixture of water (1 mL) and dioxane (10 mL). The reaction mixture was stirred at 30 °C for 12 hr under argon atmosphere. The reaction mixture was cooled to room temperature and concentrated under reduced pressure. The residue was diluted with water (10 mL) and extracted with EtOAc (2×10 mL). The combined organic layers were separated, dried over anhydrous Na
2SO
4 and concentrated under reduced pressure. The residue was subjected to HPLC (0.5-6.5 min, 20- 35% water+FA (0.1% vol.) - ACN+FA (0.1% vol.); flow: 30 mL/min, column: SunFire C18, 100×19 mm, 5 µm) to afford 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]-6-(trifluoromethyl)pyrimidine (16.0 mg, 29.1 μmol, 14.1% yield) as a yellow solid.
1H NMR (600 MHz, DMSO-d
6) δ 0.89 – 0.93 (m, 2H), 1.06 – 1.09 (m, 2H), 1.74 – 1.79 (m, 1H), 3.79 (s, 3H), 3.88 (s, 3H), 5.60 (s, 2H), 7.62 (d, 2H), 7.66 (s, 1H), 7.76 (d, 2H), 7.95 (s, 1H), 8.72 (s, 1H). MS (ESI): [M+H]
+ m/z: calcd 551.19; found 551.0. Example 150 (Compound 211)
Compound 211 3-[2-chloro-4-[[3-fluoro-4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidin-5-yl]oxetan-3-ol Synthesis of the starting 5-bromo-2-chloro-4-[[3-fluoro-4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidine is described for compound 215. [1443] 5-Bromo-2-chloro-4-[[3-fluoro-4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidine (830 mg, 1.78 mmol) was dissolved in THF (10 mL) and cooled to -85 °C. To the obtained solution butyl lithium (2.67 mmol, 1.1 mL, 2.4 M in hexane) was added at stirring. The clear solution was formed after 5 min. Oxetan-3-one (514 mg, 7.13 mmol, 457 μL) was added to the mixture at -95 °C. The resulting mixture was stirred at -85 °C for 5 min. The reaction mixture was quenched with water (5.0 mL) at -70 °C. The resulting mixture was extracted with EtOAc (10 mL). The organic layer was separated, dried over anhydrous Na
2SO
4 and concentrated under reduced pressure to afford 3-[2-chloro- 4-[[3-fluoro-4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidin-5- yl]oxetan-3-ol (800 mg, 1.74 mmol, 97.8% yield) as a yellow oil which was used in the next step without further purification. MS (ESI): [M+H]
+ m/z: calcd 459.08; found 459.0. 2-chloro-4-[[3-fluoro-4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]-5-(3- fluorooxetan-3-yl)pyrimidine [1444] 3-[2-Chloro-4-[[3-fluoro-4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidin-5-yl]oxetan-3-ol (300 mg, 654 μmol) was added to a stirred solution of morpholinosulfur trifluoride (229 mg, 1.31 mmol) in DCM (7 mL) at -70 °C. The resulting mixture was allowed to warm to 0 °C over 1 hr. The reaction mixture was quenched by addition of an aqueous solution of NaHCO
3 (20 mL). The organic layer was separated, dried over anhydrous Na
2SO
4 and subjected to flash column chromatography (silica, gradient EtOAc - DCM) to afford 2-chloro-4-[[3-fluoro-4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]-5-(3-fluorooxetan-3-yl)pyrimidine (100 mg, 217 μmol, 33.2% yield) as a yellow oil. MS (ESI): [M+H]
+ m/z: calcd 461.08; found 461.0 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[3-fluoro-4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]-5-(3-fluorooxetan-3-yl)pyrimidine 2-Chloro-4-[[3-fluoro-4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]-5-(3-fluorooxetan-3-yl)pyrimidine (40.0 mg, 86.8 μmol), (4-cyclopropyl- 6-methoxy-pyrimidin-5-yl)boronic acid I-5 (67.4 mg, 347 μmol), potassium phosphate tribasic (55.3 mg, 260 μmol) and RuPhos Pd G3 (5.17 mg, 6.08 μmol) were mixed in dioxane (5 mL) and water (500 µL) under argon atmosphere. The reaction mixture was stirred at room temperature for 4 hr, then at 70 °C for 12 hr. The reaction mixture was cooled to room temperature. SiliaMetS
® Dimercaptotriazine (100 mg) was added to the resulting mixture. The resulting mixture was stirred for 3 hr. The mixture was diluted with MTBE (5 mL) and filtered through a thin pad of silica. The filtrate was concentrated under reduced pressure. The residue was subjected to HPLC (0-2-9 min., 28-35-50% water – ACN, +0.1% vol. of 25% aq. NH
3, flow: 30 mL/min, column: XBrіdge BEH C18, 100×19 mm, 5 µm) to afford 2-(4-cyclopropyl- 6-methoxy-pyrimidin-5-yl)-4-[[3-fluoro-4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]-5-(3-fluorooxetan-3-yl)pyrimidine (6.00 mg, 10.4 μmol, 12.0% yield) as a yellow oil.
1H NMR (600 MHz, DMSO-d
6) δ 0.88 – 0.93 (m, 2H), 1.04 – 1.08 (m, 2H), 1.73 – 1.79 (m, 1H), 3.61 (s, 3H), 3.86 (s, 3H), 4.96 (dd, 2H), 5.30 (dd, 2H), 5.59 (s, 2H), 7.45 (d, 1H), 7.51 (d, 1H), 7.63 (t, 1H), 8.01 (s, 1H), 8.70 (s, 1H), 8.90 (s, 1H). MS (ESI): [M+H]
+ m/z: calcd 575.21; found 575.2. Example 152 (Compound 219) and (Ent-219)
Step 1: The synthesis of 3,5-difluoro-4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]benzaldehyde [1445] DIBAL (12.5 mmol, 12.5 mL, 1M solution in hexane) was added dropwise to a stirred solution of 3,5-difluoro-4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]benzonitrile (3.00 g, 10.5 mmol) in DCM (100 mL) under argon atmosphere at -60 °C. The reaction mixture was stirred at -60 °C for 2 hr. i-PrOH (20 mL, 50% wt. in water) was added dropwise at -20 °C. The resulting mixture was stirred at -20 °C for 1 hr, then allowed to warm to room temperature and filtered. The filtrate was concentrated under reduced pressure to afford 3,5- difluoro-4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]benzaldehyde (2.10 g, 7.24 mmol, 69.3% yield) as a yellow oil which was used in the next step without further purification. MS (ESI): [M+H]
+ m/z: calcd 291.06; found 291.0 Step 2: The synthesis of 1-[3,5-difluoro-4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]ethanol [1446] Methylmagnesium bromide (10.9 mmol, 3.63 mL, 3M in ether) was added dropwise to a stirred solution of 3,5-difluoro-4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]benzaldehyde (2.10 g, 7.24 mmol) in THF (49 mL) under argon atmosphere at -80 °C. The resulting mixture was stirred at -80 °C for 4 hr. The reaction mixture was allowed to warm to 0 °C. An aqueous solution of NH
4Cl (50 mL) was added dropwise to the reaction mixture. The resulting mixture was stirred for 15 min and extracted with EtOAc (2×100 mL). The combined organic layers were dried over anhydrous Na
2SO
4 and concentrated under reduced pressure. The residue was subjected to flash column chromatography (silica: gradient MTBE/MeOH) to afford partially purified product which was then recrystallized from MTBE to afford 1-[3,5-difluoro-4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]ethanol (160 mg, 523 μmol, 7.2% yield) as a yellow oil. MS (ESI): [M+H]
+ m/z: calcd 307.09; found 307.2 Step 3: The synthesis of 4'-cyclopropyl-4-(1-(3,5-difluoro-4-(1-methyl-4-(trifluoromethyl)- 1H-imidazol-2-yl)phenyl)ethoxy)-6'-methoxy-2,5'-bipyrimidine [1447] NaH (20.9 mg, 523 μmol, 60% dispersion in mineral oil) was added to a stirred solution of 1-[3,5-difluoro-4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]ethanol (160 mg, 523 μmol) in DMF (5.0 mL). The resulting mixture was stirred at 0 °C for 1 hr.4- cyclopropyl-6-methoxy-5-(4-methylsulfonylpyrimidin-2-yl)pyrimidine I-8 (160 mg, 523 μmol) was added to the mixture. The reaction mixture was stirred at room temperature for 18 hr. The reaction mixture was poured into water (10 mL) and extracted with EtOAc (2×10 mL). The combined organic layers were separated, washed with brine (10 mL) and concentrated under reduced pressure. The residue was subjected to HPLC (0.5-6.5 min., 49% water – ACN, +0.1% vol. of 25% aq. NH
3, flow: 30 mL/min, column: SunFire 100×19 mm, 5 µm) to afford 4'-cyclopropyl-4-(1-(3,5-difluoro-4-(1-methyl-4-(trifluoromethyl)-1H- imidazol-2-yl)phenyl)ethoxy)-6'-methoxy-2,5'-bipyrimidine (27.0 mg, 50.7 μmol, 9.7% yield) as a yellow oil. MS (ESI): [M+H]
+ m/z: calcd 533.20; found 533.2 Step 4: The synthesis of rel-(R)-4'-cyclopropyl-4-(1-(3,5-difluoro-4-(1-methyl-4- (trifluoromethyl)-1H-imidazol-2-yl)phenyl)ethoxy)-6'-methoxy-2,5'-bipyrimidine (ent-219) and rel-(S)-4'-cyclopropyl-4-(1-(3,5-difluoro-4-(1-methyl-4-(trifluoromethyl)-1H-imidazol-2- yl)phenyl)ethoxy)-6'-methoxy-2,5'-bipyrimidine (219) 4'-Cyclopropyl-4-(1-(3,5-difluoro-4-(1-methyl-4-(trifluoromethyl)-1H-imidazol-2- yl)phenyl)ethoxy)-6'-methoxy-2,5'-bipyrimidine (27.0 mg, 50.7 μmol) was subjected to chiral HPLC (Column: Chiralpak AD-H-V (250×20 mm, 5 µm); Mobile phase: Hexane-IPA-MeOH, 70-15-15, flow rate: 12 mL/min; Column Temperature: 24 °C; Wavelength: 210 nm, 248 nm), RetTime (isomer A) = 7.57 min; RetTime (isomer B) = 14.16 min) to afford rel-(R)-4'- cyclopropyl-4-(1-(3,5-difluoro-4-(1-methyl-4-(trifluoromethyl)-1H-imidazol-2- yl)phenyl)ethoxy)-6'-methoxy-2,5'-bipyrimidine (10.0 mg, 18.8 μmol, 74.1% yield) and rel- (S)-4'-cyclopropyl-4-(1-(3,5-difluoro-4-(1-methyl-4-(trifluoromethyl)-1H-imidazol-2- yl)phenyl)ethoxy)-6'-methoxy-2,5'-bipyrimidine (7.00 mg, 13.1 μmol, 51.9% yield) as yellow solids. rel-(R)-4'-cyclopropyl-4-(1-(3,5-difluoro-4-(1-methyl-4-(trifluoromethyl)-1H- imidazol-2-yl)phenyl)ethoxy)-6'-methoxy-2,5'-bipyrimidine (Ent-219): 1H NMR (600 MHz, DMSO-d
6) δ 0.79 – 0.90 (m, 2H), 0.97 – 1.05 (m, 2H), 1.57 – 1.63 (m, 1H), 1.65 (d, 3H), 3.57 (s, 3H), 3.80 (s, 3H), 6.30 (q, 1H), 7.09 (d, 1H), 7.43 (d, 2H), 8.07 (s, 1H), 8.66 (s, 1H), 8.71 (d, 1H). MS (ESI): [M+H]
+ m/z: calcd 533.20; found 533.0 Enantiopurity: >99% (column: Chiralpak AD-H, 250 × 4.6 mm, 5 μm; mobile phase: Hexane-IPA-MeOH, 50-25-25; flow: 0.6 mL/min; RT=8.4 min). rel-(S)-4'-cyclopropyl-4-(1-(3,5-difluoro-4-(1-methyl-4-(trifluoromethyl)-1H- imidazol-2-yl)phenyl)ethoxy)-6'-methoxy-2,5'-bipyrimidine (219): 1H NMR (600 MHz, DMSO-d
6) δ 0.79 – 0.90 (m, 2H), 0.97 – 1.05 (m, 2H), 1.57 – 1.63 (m, 1H), 1.65 (d, 3H), 3.57 (s, 3H), 3.80 (s, 3H), 6.30 (q, 1H), 7.09 (d, 1H), 7.43 (d, 2H), 8.07 (s, 1H), 8.66 (s, 1H), 8.71 (d, 1H). MS (ESI): [M+H]
+ m/z: calcd 533.20; found 533.0 Enantiopurity: >99% (column: Chiralpak AD-H, 250 × 4.6 mm, 5 μm; mobile phase: Hexane-IPA-MeOH, 50-25-25; flow: 0.6 mL/min; RT=6.0 min).
2-chloro-4-[[3-fluoro-4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]-6- isopropyl-pyrimidine Synthesis of the starting [3-fluoro-4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methanol is described for compound 97. [1448] 2,4-Dichloro-6-isopropyl-pyrimidine (378 mg, 1.98 mmol) was added to a stirred mixture of [3-fluoro-4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methanol (314 mg, 1.15 mmol) and Cs
2CO
3 (773 mg, 2.37 mmol) in ACN (10 mL). The resulting mixture was stirred at 65 °C for 12 hr. The reaction mixture was cooled to room temperature and concentrated under reduced pressure. The residue was diluted with water (30 mL) and extracted with DCM (40 mL). The organic layer was washed with brine (30 mL), dried over anhydrous Na
2SO
4 and concentrated under reduced pressure. The residue was subjected to flash column chromatography (silica, gradient DCM - EtOAc) to afford 2-chloro-4-[[3- fluoro-4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]-6-isopropyl- pyrimidine (100 mg, 233 μmol, 11.8% yield) as a white solid. MS (ESI): [M-H]- m/z: calcd 429.11; found 429.0 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[3-fluoro-4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]-6-isopropyl-pyrimidine [1449] 2-Chloro-4-[[3-fluoro-4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]-6-isopropyl-pyrimidine (100 mg, 233 μmol), (4-cyclopropyl-6-methoxy- pyrimidin-5-yl)boronic acid I-5 (113 mg, 583 μmol), potassium phosphate tribasic (149 mg, 700 μmol) and RuPhos Pd G3 (9.9 mg, 11.7 μmol) were mixed in a degassed mixture of dioxane (4.0 mL) and water (400 µL) under argon atmosphere. The reaction mixture was stirred at 70 °C for 20 hr. Additional portions of (4-cyclopropyl-6-methoxy-pyrimidin-5- yl)boronic acid I-5 (113 mg, 583 μmol) and RuPhos Pd G3 (9.91 mg, 11.7 μmol) were added to the mixture. The resulting mixture was stirred at 80 °C for 5 hr. The reaction mixture was cooled to room temperature. The organic layer was separated. SiliaMetS
® Dimercaptotriazine (100 mg) was added to the organic layer. The resulting mixture was stirred for 3 hr. The mixture was filtered. The filtrate was subjected to HPLC (0-2-10 min., 48-55-60% water – ACN, +0.1% vol. of 25% aq. NH
3, flow: 30 mL/min, column: Chromatorex SMB100-51, 100×19 mm, 5 µm) to afford 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[3-fluoro-4-[1- methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]-6-isopropyl-pyrimidine (44.0 mg, 81.1 μmol, 34.8% yield) as a yellow solid.
1H NMR (600 MHz, CDCl
3) δ 0.86 – 0.91 (m, 2H), 1.19 – 1.23 (m, 2H), 1.33 (d, 6H), 1.69 – 1.74 (m, 1H), 3.00 – 3.07 (m, 1H), 3.66 (s, 3H), 3.92 (s, 3H), 5.49 (s, 2H), 6.67 (s, 1H), 7.29 – 7.37 (m, 3H), 7.62 (t, 1H), 8.63 (s, 1H). MS (ESI): [M+H]
+ m/z: calcd 543.25; found 543.2. Example 153 (Compound 208)
Compound 208 5-bromo-2-chloro-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidine [1450] 5-Bromo-2,4-dichloro-pyrimidine (1.78 g, 7.81 mmol) and Cs
2CO
3 (3.81 g, 11.7 mmol) were added to a stirred solution of [4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methanol I-2b (2.00 g, 7.81 mmol) in ACN (40 mL). The reaction mixture was stirred at 60 °C for 16 hr. The reaction mixture was cooled to room temperature and concentrated under reduced pressure. The residue was diluted with EtOAc (50 mL) and washed with water (2×20 mL). The combined organic layers were washed with brine (20 mL), dried over anhydrous Na
2SO
4 and concentrated under reduced pressure to afford 5- bromo-2-chloro-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidine (3.20 g, 7.15 mmol, 91.6% yield) as a yellow solid which was used in the next step without further purification.
1H NMR (500 MHz, DMSO-d
6) δ 3.79 (s, 3H), 5.56 (s, 2H), 7.62 (d, 2H), 7.77 (d, 2H), 7.94 (s, 1H), 8.77 (s, 1H). MS (ESI): [M+H]
+ m/z: calcd 448.98, 446.98; found 449.0, 447.0 2-chloro-5-(2,5-dihydrofuran-3-yl)-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidine [1451] 5-Bromo-2-chloro-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidine (460 mg, 884 μmol), 2-(2,5-dihydrofuran-3-yl)-4,4,5,5- tetramethyl-1,3,2-dioxaborolane (208 mg, 1.06 mmol), bis(diphenylphosphino)ferrocene]dichloropalladium(II)-DCM (32.3 mg, 44.2 μmol) and Cs
2CO
3 (864 mg, 2.65 mmol) were mixed in a degassed mixture of dioxane (10 mL) and water (1.0 mL). The mixture was stirred at 90 °C for 16 hr. Additional portions of 2-(2,5- dihydrofuran-3-yl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (208 mg, 1.06 mmol) and bis(diphenylphosphino)ferrocene]dichloropalladium(II)-DCM (32.3 mg, 44.2 μmol) were added to the mixture. The resulting mixture was stirred for 16 hr. The mixture was cooled to room temperature and filtered. The filtrate was concentrated under reduced pressure. The residue was dissolved in EtOAc-hexane (50:50) mixture. The resulting solution was filtered through a thin pad of silica. The filtrate was concentrated under reduced pressure to afford 2- chloro-5-(2,5-dihydrofuran-3-yl)-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidine (390 mg, crude) as yellow oil which was used in the next steps without further purification. MS (ESI): [M+H]
+ m/z: calcd 437.1; found 437.2 2-chloro-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]-5- tetrahydrofuran-3-yl-pyrimidine [1452] Platinum, 5% on carbon (40 mg,) was added to a solution of 2-chloro-5-(2,5- dihydrofuran-3-yl)-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidine (390 mg, crude from previous step) in degassed MeOH (10 mL). The resulting mixture was stirred under hydrogen atmosphere (1 bar) at room temperature for 16 hr. An additional portion of platinum, 5% on carbon (40 mg,) was added to the mixture. The reaction mixture was stirred for 16 hr. The reaction mixture was filtered. The filtrate was concentrated under reduced pressure to afford 2-chloro-4-[[4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]-5-tetrahydrofuran-3-yl-pyrimidine (260 mg, crude) as a brown oil which was used in the next step without further purification. MS (ESI): [M+H]
+ m/z: calcd 439.12; found 439.0 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]-5-tetrahydrofuran-3-yl-pyrimidine [1453] 2-Chloro-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]-5- tetrahydrofuran-3-yl-pyrimidine (260 mg, 172 μmol), (4-cyclopropyl-6-methoxy-pyrimidin- 5-yl)boronic acid I-5 (33.3 mg, 172 μmol), potassium phosphate tribasic (109 mg, 516 μmol) and XPhos Pd G3 (1.5 mg, 1.72 μmol) were mixed in a degassed mixture of dioxane (3 mL) and water (150 µL). The reaction mixture was stirred at 60 °C for 16 hr under argon atmosphere. The reaction mixture was cooled to room temperature, diluted with EtOAc (10 mL) and washed with water (2×5 mL). SiliaMetS
® Dimercaptotriazine (50 mg) was added to the resulting organic layer. The resulting mixture was stirred for 30 min. and filtered. The filtrate was concentrated under reduced pressure. The residue was subjected to HPLC (0.5- 6.5 min., 30-80% water – ACN, flow: 30 mL/min, column: SunFire C18 (L), 100×19 mm, 5 µm) to afford 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]-5-tetrahydrofuran-3-yl-pyrimidine (11.0 mg, 19.9 μmol, 2.25% yield from 5-bromo-2-chloro-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol- 2-yl]phenyl]methoxy]pyrimidine) as a light-yellow solid.
1H NMR (600 MHz, DMSO-d
6) δ 0.85 – 0.90 (m, 2H), 1.02 – 1.05 (m, 2H), 1.68 – 1.74 (m, 1H), 2.12 – 2.19 (m, 1H), 2.26 – 2.33 (m, 1H), 3.55 – 3.61 (m, 1H), 3.73 (t, 1H), 3.78 – 3.86 (m, 7H), 3.90 – 3.94 (m, 1H), 4.05 (t, 1H), 5.53 (s, 2H), 7.60 (d, 2H), 7.75 (d, 2H), 7.94 (s, 1H), 8.62 (s, 1H), 8.67 (s, 1H). MS (ESI): [M+H]
+ m/z: calcd 553.25; found 553.2.
pyridyl]methoxy]pyrimidine & 4-chloro-6-cyclopropyl-2-[[5-fluoro-6-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]-3- pyridyl]methoxy]pyrimidine Synthesis of the starting [5-fluoro-6-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]-3- pyridyl]methanol is described for compound 48. [1454] To a solution of [5-fluoro-6-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]-3- pyridyl]methanol (150 mg, 545.05 μmol) in THF (3 mL) was added NaH (26.2 mg, 654.07 μmol, 60% purity) at 0 °C. The mixture was stirred at 0 °C for 1 hour. Then the mixture was added 2,4-dichloro-6-cyclopropyl-pyrimidine (123.7 mg, 654.07 μmol) at 0 °C. Then the mixture was stirred at room temperature for 16 hrs. The reaction mixture was diluted with EtOAc (30 mL and washed with saturated NH
4Cl aqueous solution (20 mL x 3), dried over anhydrous Na
2SO
4. After filtration, the filtrate was concentrated under reduced pressure. The resulted residue was purified by Prep-TLC, eluted with 33% EtOAc in PE to give 2-chloro-4- cyclopropyl-6-[[5-fluoro-6-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]-3- pyridyl]methoxy]pyrimidine and 4-chloro-6-cyclopropyl-2-[[5-fluoro-6-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]-3-pyridyl]methoxy]pyrimidine (188 mg, 439.48 μmol, 80% yield, mixture of two isomers) as a yellow oil. MS: m/z = 428.15 [M + H]
+. 4-cyclopropyl-2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-6-[[5-fluoro-6-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]-3-pyridyl]methoxy]pyrimidine [1455] To a solution of 2-chloro-4-cyclopropyl-6-[[5-fluoro-6-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]-3-pyridyl]methoxy]pyrimidine , 4-chloro-6-cyclopropyl-2- [[5-fluoro-6-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]-3-pyridyl]methoxy]pyrimidine (188.00 mg, 439.48 μmol, mixture of two isomers) and (4-cyclopropyl-6-methoxy-pyrimidin- 5-yl)boronic acid I-5 (127.88 mg, 659.21 μmol) in 1,4-dioxane (4 mL) and H
2O (0.8 mL) were added methanesulfonato(2-dicyclohexylphosphino-2',4',6'-tri-i-propyl-1,1'-biphenyl)(2'- amino-1,1'-biphenyl-2-yl)palladium(II) (37.20 mg, 43.95 μmol), 2-(Dicyclohexylphosphino)- 2',4',6'-Triisopropylbiphenyl (20.95 mg, 43.95 μmol) and potassium phosphate (186.57 mg, 878.95 μmol) at room temperature, the mixture was stirred at 40 °C for 16 hrs. The resulted mixture was cooled to room temperature and was diluted with EtOAc (50 mL). The organic layers were washed with saturated NH
4Cl aqueous solution (20 mL x 3), dried over anhydrous Na
2SO
4. Then the mixture was concentrated under reduced pressure. The resulting residue was purified by Prep-TLC to give two isomers. [1456] The less polar isomer fraction was concentrated under reduced pressure to give crude product, then was further purified by reverse phase chromatography with the following conditions: Column: C18 gel column 40g, 20-35 μm; Mobile Phase A: 5 mM aq. NH
4HCO
3, Mobile Phase B: MeCN; Gradient: 5% B to 5% B in 5 min, 5% B to 65% B in 20 min, 65% B to 65% B in 5 min, 65% B to 95% B in 5 min, 95% B hold 3 min; Flow rate: 40 mL/min; Detector: UV 254 & 220 nm. The product-containing fractions were collected and evaporated in vacuo and then lyophilized to give 4-cyclopropyl-2-(4-cyclopropyl-6-methoxy-pyrimidin- 5-yl)-6-[[5-fluoro-6-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]-3- pyridyl]methoxy]pyrimidine (60.4 mg, 111.54 μmol, 25% yield) as a white solid. MS: m/z = 542.15 [M + H]
+.
1H NMR (400 MHz, Chloroform-d) δ 8.63 (s, 1H), 8.57 (s, 1H), 7.72 - 7.69 (m, 1H), 7.37 (s, 1H), 6.60 (s, 1H), 5.53 (s, 2H), 3.92 (s, 6H), 2.05 - 1.98 (m, 1H), 1.74 - 1.67 (m, 1H), 1.26 - 1.18 (m, 2H), 1.16 - 1.06 (m, 4H), 0.91 - 0.85 (m, 2H).
19F NMR (376 MHz, Chloroform-d) δ -62.62, -118.62. Example 155 (Compound 206)
2-chloro-5-methoxy-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]-2-(2,2,2- trifluoroethyl)phenyl]methoxy]pyrimidine [1457] To a stirred solution of [4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]-2-(2,2,2- trifluoroethyl)phenyl]methanol (130 mg, 384.33 μmol) in THF (5 mL) was added NaH (15.4 mg, 384.33 μmol, 60% purity) at 0 °C under nitrogen atmosphere. The resulting mixture was stirred for 1 hour at 0 °C under nitrogen atmosphere. To the above mixture was added 2,4- dichloro-5-methoxy-pyrimidine (82.6 mg, 461.20 μmol) at 0 °C. The resulting mixture was stirred at 25 °C for 16 hrs. The reaction was quenched with sat. NH
4Cl (aq.) (100 mL) at 0 °C. The resulting mixture was extracted with EtOAc (2 x 100 mL). The combined organic layers were washed with brine (100 mL), dried over anhydrous Na
2SO
4. After filtration, the filtrate was concentrated under reduced pressure. The residue was purified by silica gel column chromatography, eluted with 60% EtOAc in PE to give 2-chloro-5-methoxy-4-[[4-[1- methyl-4-(trifluoromethyl)imidazol-2-yl]-2-(2,2,2-trifluoroethyl)phenyl]methoxy]pyrimidine (154 mg, 320.31 μmol, 83% yield) as a yellow solid. MS: m/z = 481.10 [M + H]
+.
1H NMR (400 MHz, Chloroform-d) δ 7.93 (s, 1H), 7.74 - 7.64 (m, 3H), 7.36 (s, 1H), 5.59 (s, 2H), 3.93 (s, 3H), 3.82 (s, 3H), 7.77 (t, J = 10.6 Hz, 2H).
19F NMR (376 MHz, Chloroform-d) δ -62.74, -64.93. 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-5-methoxy-4-[[4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]-2-(2,2,2-trifluoroethyl)phenyl]methoxy]pyrimidine [1458] To a mixture of 2-chloro-5-methoxy-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]-2-(2,2,2-trifluoroethyl)phenyl]methoxy]pyrimidine (200 mg, 415.98 μmol) and (4- cyclopropyl-6-methoxy-pyrimidin-5-yl)boronic acid I-5 (121 mg, 623.97 μmol) in dioxane (2.5 mL) and water (0.5 mL) under nitrogen atmosphere were added Methanesulfonato(2- dicyclohexylphosphino-2',4',6'-tri-i-propyl-1,1'-biphenyl)(2'-amino-1,1'-biphenyl-2- yl)palladium(II) (35.2 mg, 41.60 μmol), 2-(Dicyclohexylphosphino)-2',4',6'-tri-i-propyl-1,1'- biphenyl (19.8 mg, 41.60 μmol) and potassium phosphate (176.6 mg, 831.96 μmol) at 25 °C. The resulting mixture was stirred for 16 hrs at 40 °C, cooled to room temperature and filtered. The filtrate was concentrated under reduced pressure. The residue was subject to silica gel column chromatography, eluted with 70% EtOAc in PE to give product a residue. The residue was purified by RP-Flash with the following conditions: Column: C18 spherical, 20-30 um, 100A, 20 g; Mobile Phase A: Water (5 mM aq. NH
4HCO
3), Mobile Phase B: MeCN; Flow rate: 35 mL/min; Gradient: 2% B to 2% B in 5 min, 5% B to 70% B in 15 min, 70% B to 70% B in 2 min, 70% B to 95% B in 5 min; Detector: UV 254 & 210 nm. The collected fractions were combined, concentrated and then lyophilized to give 2-(4- cyclopropyl-6-methoxy-pyrimidin-5-yl)-5-methoxy-4-[[4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]-2-(2,2,2-trifluoroethyl)phenyl]methoxy]pyrimidine (52 mg, 87.47 μmol, 21% yield) as an off-white solid. MS: m/z = 595.15 [M + H]
+.
1H NMR (400 MHz, Chloroform-d) δ 8.67 (s, 1H), 8.25 (s, 1H), 7.71 - 7.62 (m, 3H), 7.34 (s, 1H), 5.61 (s, 2H), 4.01 (s, 3H), 3.94 (s, 3H), 3.79 (s, 3H), 3.67 (q, J = 10.6 Hz, 2H), 1.75 - 1.71 (s, 1H), 1.27 - 1.21 (m, 2H), 0.93 - 0.88 (m, 2H).
19F NMR (376 MHz, Chloroform-d) δ -62.80, - 64.94. Example 156 (Compound 196)
2-chloro-5-(2,2-difluoroethoxy)-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidine Synthesis of the starting 2-chloro-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidin-5-ol is described for Compound 122. [1459] 2-Chloro-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidin-5-ol (129 mg, 335 μmol), 2,2-difluoroethanol (33.0 mg, 402 μmol, 25.5 μL), triphenylphosphine (96.7 mg, 369 μmol) were mixed in THF (3 mL). DIAD (81.4 mg, 402 μmol) was added to the obtained mixture. The reaction mixture was stirred at room temperature for 12 hr. The reaction mixture was diluted with water (500 µL) and ACN (2.0 mL). The resulting solution was subjected to HPLC (0.5-6.5 min., 40-65% water – ACN, +0.1% vol. of 25% aq. NH
3, flow: 30 mL/min, column: XBridge 100×19 mm, 5 µm) to afford 2-chloro-5-(2,2-difluoroethoxy)-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidine (57.0 mg, 127 μmol, 37.9% yield) as a white solid. MS (ESI): [M+H]
+ m/z: calcd 449.10; found 449.0 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-5-(2,2-difluoroethoxy)-4-[[4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidine [1460] 2-Chloro-5-(2,2-difluoroethoxy)-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidine (57 mg, 127 μmol), 5-(4,4,5,5-tetramethyl-1,3,2- dioxaborolan-2-yl)pyrimidine (45.6 mg, 165 μmol), potassium phosphate tribasic (53.9 mg, 254 μmol) and XPhos Pd G3 (5.38 mg, 6.35 μmol) were mixed in degassed mixture of dioxane (2.7 mL) and water (0.3 mL). The reaction mixture was stirred at 60 °C for 4 hr under argon atmosphere. The reaction mixture was cooled to room temperature, diluted with water (5 mL), and extracted with EtOAc (2×10 mL). The combined organic layers were separated, washed with water (2×10 mL) and brine (10 mL). SiliaMetS
® Dimercaptotriazine (20 mg) was added to the obtained organic layer. The resulting mixture was stirred at room temperature for 30 mi and filtered. The filtrate was concentrated under reduced pressure. The residue was subjected to HPLC (0.8-12 min., 52% water – MeOH, flow: 30 mL/min, column: SunFire C18 (R) 100×19 mm, 5 µm) to afford 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)- 5-(2,2-difluoroethoxy)-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidine (9.00 mg, 16.0 μmol, 12.6% yield) as a yellow solid.
1H NMR (600 MHz, DMSO-d
6) δ 0.84 – 0.89 (m, 2H), 1.01 – 1.05 (m, 2H), 1.68 – 1.73 (m, 1H), 3.79 (s, 3H), 3.85 (s, 3H), 4.55 (dt, 1H), 5.55 (s, 2H), 6.46 (tt, 1H, CHF
2), 7.59 (d, 2H), 7.74 (d, 2H), 7.94 (s, 1H), 8.53 (s, 1H), 8.66 (s, 1H). MS (ESI): [M+H]
+ m/z: calcd 563.21; found 563.2. Example 157 (Compound 194)
2-chloro-5-(cyclopropoxy)-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidine Synthesis of the starting 2-chloro-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidin-5-ol is described for Compound 122. [1461] Cyclopropyl trifluoromethanesulfonate (494 mg, 2.60 mmol) was added to a stirred mixture of 2-chloro-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidin-5-ol (250 mg, 650 μmol) and Cs
2CO
3 (318 mg, 975 μmol) in DMF (7.0 mL). The reaction mixture was stirred at 40°C for 24 hr. The reaction mixture was cooled to room temperature, poured into water (30 mL) and extracted with MTBE (4×15 mL). The combined organic layers were separated, washed with water (20 mL) and brine (20 mL), dried over anhydrous Na
2SO
4 and filtered through a thin pad of silica gel. The filtrate was concentrated under reduced pressure to afford 2-chloro-5-(cyclopropoxy)-4-[[4-[1- methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidine (240 mg, 565 μmol, 87% yield) as a light-yellow solid which was used in the next steps without further purification. MS (ESI): [M+H]
+ m/z: calcd 425.10; found 425.2 5-(cyclopropoxy)-2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidine [1462] 2-Chloro-5-(cyclopropoxy)-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidine (110 mg, 259 μmol), (4-cyclopropyl-6-methoxy-pyrimidin-5- yl)boronic acid I-5 (151 mg, 777 μmol), potassium phosphate tribasic (330 mg, 1.55 mmol) and bis(diphenylphosphino)ferrocene]dichloropalladium(II)-DCM (42.3 mg, 51.8 μmol) were mixed in degassed dioxane (4 mL). Water (400 µL) was added to the mixture. The reaction mixture was stirred at room temperature for 24 hr. The reaction mixture was diluted with EtOAc (5.0 mL). Na
2SO
4 (1.0 g) and SiliaMetS
® Dimercaptotriazine (400 mg) were added to the mixture and the resulting mixture was stirred at room temperature for 12 hr. Then solids were filtered out and washed with EtOAc (2 mL). The combined filtrate was concentrated under reduced pressure. The residue was subjected to HPLC (0-2-6 min., 40-50-100% water – MeOH, +0.1% vol. of 25% aq. NH
3, flow: 30 mL/min, column: Chromatorex C18100×19 mm, 5 µm) to afford 5-(cyclopropoxy)-2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[4- [1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidine (58.0 mg, 108 μmol, 41.6% yield) as a light-yellow solid. MS (ESI): [M+H]
+ m/z: calcd 539.23; found 539.2.
1H NMR (600 MHz, DMSO-d
6) δ 0.77 – 0.90 (m, 6H), 1.00 – 1.05 (m, 2H), 1.69 – 1.76 (m, 1H), 3.79 (s, 3H), 3.85 (s, 3H), 4.06 – 4.12 (m, 1H), 5.49 (s, 2H), 7.57 (d, 2H), 7.73 (d, 2H), 7.94 (s, 1H), 8.65 (s, 2H). Example 158 (Compound 190)

3-fluoro-4-[4-(trifluoromethyl)-1H-imidazol-2-yl]benzonitrile [1463] A solution of 3,3-dibromo-1,1,1-trifluoro-propan-2-one (9.95 g, 36.9 mmol, 5.03 mL) and sodium acetate (6.05 g, 73.8 mmol) in water (20 mL) was stirred at 95°C for 45 min. The reaction mixture was cooled to room temperature. To the reaction mixture was added a solution of 3-fluoro-4-formyl-benzonitrile (5.00 g, 33.5 mmol) followed by a solution of aqueous ammonium hydroxide (15 mL, 25% wt.) in MeOH (80 mL). The reaction mixture was stirred at room temperature for 12 hr. The mixture was concentrated under reduced pressure. The residue was diluted with water. The solid formed was filtered off and dried on air to afford 3-fluoro-4-[4-(trifluoromethyl)-1H-imidazol-2-yl]benzonitrile (6.00 g, 23.5 mmol, 70.1% yield) as a yellow solid which was used in the next steps without further purification. 3-fluoro-4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]benzonitrile [1464] To a solution of 3-fluoro-4-[4-(trifluoromethyl)-1H-imidazol-2-yl]benzonitrile (500 mg, 1.96 mmol) in THF (5 mL) was added t-BuOK (350 mg, 3.12 mmol) and the mixture was stirred at 0 °C for 30 minutes. To the mixture was added MeI (420 mg, 2.96 mmol) and the mixture stirred at 20 °C for 2 hrs. The resulting mixture was diluted by addition of water (10 mL) and extracted with EtOAc (100 mL x 3). The combined organic layer was washed with brine (100 mL x 2), dried over anhydrous Na
2SO
4, filtered and concentrated under reduced pressure. The residue was purified by flash chromatography (ISCO®; 12 g AgelaFlash® Silica Flash Column, PE/EtOAc with EtOAc from 0-20%, flow rate = 35 mL/min, 254 nm) to afford 3-fluoro-4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]benzonitrile (520 mg, 98.6% yield) as light yellow solid. MS (ESI) [M+H]
+ m/z: calcd 270.1, found 270.0. 4-[5-chloro-1-methyl-4-(trifluoromethyl)imidazol-2-yl]-3-fluoro-benzonitrile A mixture of 3-fluoro-4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]benzonitrile (520 mg, 1.93 mmol), NCS (530 mg, 3.97 mmol) in AcOH (4 mL) was stirred at 45 °C for 2.5 hrs. The mixture was adjusted pH to 7-8 with saturated NaHCO
3 aqueous solution. The resulting mixture was diluted by addition of water (10 mL) and extracted with EtOAc (100 mL x 3). The combined organic layer was washed with brine (40 mL x 2), dried over anhydrous Na
2SO
4, filtered and concentrated under reduced pressure. The residue was purified by flash chromatography (ISCO®; 12 g AgelaFlash® Silica Flash Column, PE/EtOAc with EtOAc from 0-10%, flow rate = 30 mL/min, 254 nm) to afford 4-[5-chloro-1-methyl-4- (trifluoromethyl)imidazol-2-yl]-3-fluoro-benzonitrile (400 mg, 68.2% yield) as white solid. MS (ESI) [M+H]
+ m/z: calcd 304.0, found 304.0. 4-[5-chloro-1-methyl-4-(trifluoromethyl)imidazol-2-yl]-3-fluoro-benzoic acid [1465] To a solution of 4-[5-chloro-1-methyl-4-(trifluoromethyl)imidazol-2-yl]-3-fluoro- benzonitrile (400 mg, 1.32 mmol) in EtOH (4 mL) and H
2O (1 mL) was added KOH (400 mg, 7.13 mmol) and the mixture was stirred at 80 °C for 12 hrs. The resulting mixture was concentrated under reduced pressure. The mixture was adjusted pH to 4-5 with 1N HCl aqueous solution. The resulting mixture was diluted by addition of water (5 mL) and extracted with EtOAc (30 mL x 3). The combined organic layer was washed with brine (20 mL x 2), dried over anhydrous Na
2SO
4, filtered and concentrated under reduced pressure to afford 4-[5-chloro-1-methyl-4-(trifluoromethyl)imidazol-2-yl]-3-fluoro-benzoic acid (340 mg, 80% yield) as light-yellow solid.
1H NMR (400 MHz, DMSO-d
6) δ ppm 7.92 (dd, J = 8.0, 1.5 Hz, 1H), 7.86 (dd, J = 10.4, 1.3 Hz, 1H), 7.76 (t, J = 7.6 Hz, 1H), 3.56 (d, J = 1.6 Hz, 3H);
19F NMR (376 MHz, DMSO-d
6) δ ppm -66.43, -112.91; MS (ESI) [M+H]
+ m/z: calcd 323.0, found 323.0. [4-[5-chloro-1-methyl-4-(trifluoromethyl)imidazol-2-yl]-3-fluoro-phenyl]methanol To a solution of 4-[5-chloro-1-methyl-4-(trifluoromethyl)imidazol-2-yl]-3-fluoro-benzoic acid (340 mg, 1.05 mmol) in THF (3 mL) was added 1 M BH
3-THF (7 mL, 7.00 mmol) at 0°C. The mixture was stirred at 20°C for 3 hrs. The resulting mixture was quenched by addition of MeOH (5 mL), filtered and concentrated under reduced pressure. The residue was purified by flash chromatography (ISCO®; 12 g AgelaFlash® Silica Flash Column, PE/EtOAc with EtOAc from 0-50%, flow rate = 30 mL/min, 254 nm) to afford [4-[5-chloro-1-methyl-4- (trifluoromethyl)imidazol-2-yl]-3-fluoro-phenyl]methanol (150 mg, 46.1% yield) as colorless oil. MS (ESI) [M+H]
+ m/z: calcd 309.0, found 309.0. 5-[4-[[4-[5-chloro-1-methyl-4-(trifluoromethyl)imidazol-2-yl]-3-fluoro- phenyl]methoxy]pyrimidin-2-yl]-4-cyclopropyl-6-methoxy-pyrimidine (190) To a solution of [4-[5-chloro-1-methyl-4-(trifluoromethyl)imidazol-2-yl]-3-fluoro- phenyl]methanol (150 mg, 0.486 mmol) in THF (3 mL) was slowly added NaH (60 mg, 1.50 mmol, 60 wt% in mineral oil). The mixture was stirred at 20 °C for 30 min. Then to the mixture was added 4-cyclopropyl-6-methoxy-5-(4-methylsulfonylpyrimidin-2-yl)pyrimidine I-8 (180 mg, 0.588 mmol) and the mixture was stirred at 45 °C for 1 hr. The resulting mixture was quenched by addition of water (10 mL) and extracted with EtOAc (50 mL x 3). The combined organic layer was washed with brine (40 mL x 2), dried over anhydrous Na
2SO
4, filtered and concentrated under reduced pressure. The residue was purified by flash chromatography (Biotage®, Column: SepaFlash® Sphercial C18, 40 g, 40-60 μm, 120Å; MeCN/water (0.05%NH
3-H
2O) with MeCN from 0-75%, 35 mL/min, 254 nm). The fraction was concentrated under reduced pressure and then lyophilized for overnight to afford 5-[4-[[4-[5- chloro-1-methyl-4-(trifluoromethyl)imidazol-2-yl]-3-fluoro-phenyl]methoxy]pyrimidin-2-yl]- 4-cyclopropyl-6-methoxy-pyrimidine (23.0 mg, 8.9% yield) as white solid.
1H NMR (400 MHz, DMSO-d
6) δ ppm 8.73 (d, J = 5.9 Hz, 1H), 8.68 (s, 1H), 7.64 (t, J = 7.7 Hz, 1H), 7.54 (d, J = 10.9 Hz, 1H), 7.48 (d, J = 7.9 Hz, 1H), 7.11 (d, J = 5.9 Hz, 1H), 5.53 (s, 2H), 3.84 (s, 3H), 3.53 (d, J = 1.1 Hz, 3H), 1.63 - 1.74 (m, 1H), 1.04 (dt, J = 7.2, 3.5 Hz, 2H), 0.84 - 0.95 (m, 2H);
19F NMR (376 MHz, DMSO-d
6) δ ppm -60.31, -113.71; MS (ESI) [M+H]
+ m/z: calcd 535.1, found 535.2.
4-(1-(tetrahydro-2H-pyran-4-yl)-4-(trifluoromethyl)-1H-imidazol-2-yl)benzonitrile [1466] Cs
2CO
3 (8.24 g, 25.3 mmol) followed by tetrahydropyran-4-yl trifluoromethanesulfonate (5.92 g, 25.3 mmol) were added to a stirred solution of 4-[4- (trifluoromethyl)-1H-imidazol-2-yl]benzonitrile I-1a (2.00 g, 8.43 mmol) in ACN (10 mL). The resulting mixture was stirred at 50 °C for 16 hr. The reaction mixture was cooled to room temperature and filtered. The filtrate was concentrated under reduced pressure. The residue was subjected by flash column chromatography (SiO
2, gradient chloroform – acetonitrile) to afford 4-(1-(tetrahydro-2H-pyran-4-yl)-4-(trifluoromethyl)-1H-imidazol-2-yl)benzonitrile (1.00 g, 3.11 mmol, 37.0% yield) as a yellow oil.
1H NMR (400 MHz, DMSO-d
6) δ 1.89 – 1.97 (m, 2H), 1.97 – 2.10 (m, 2H), 3.37 (t, 2H), 3.87 – 3.96 (m, 2H), 4.30 – 4.40 (m, 1H), 7.82 (d, 2H), 8.01 (d, 2H), 8.33 (s, 1H). MS (ESI): [M+H]
+ m/z: calcd 322.12; found 322.2 4-(1-(tetrahydro-2H-pyran-4-yl)-4-(trifluoromethyl)-1H-imidazol-2-yl)benzoic acid [1467] 4-(1-(Tetrahydro-2H-pyran-4-yl)-4-(trifluoromethyl)-1H-imidazol-2-yl)benzonitrile (1.00 g, 3.11 mmol) was added to a solution of sodium hydroxide (376 mg, 9.39 mmol) in water (20 mL) and MeOH (20 mL). The reaction mixture was stirred at 60 °C for 12 hr. The reaction mixture was cooled to room temperature and concentrated under reduced pressure. The residue was diluted with an aqueous HCl (2 mL, 5N). The precipitate formed was filtered off, washed with water (2 mL) and air-dried to afford 4-(1-(tetrahydro-2H-pyran-4-yl)-4- (trifluoromethyl)-1H-imidazol-2-yl)benzoic acid (500 mg, 1.47 mmol, 47.2% yield) as a white solid which was used in the next step without further purification. MS (ESI): [M+H]
+ m/z: calcd 341.11; found 341.0 (4-(1-(tetrahydro-2H-pyran-4-yl)-4-(trifluoromethyl)-1H-imidazol-2-yl)phenyl)methanol [1468] Borane dimethyl sulfide complex (357 mg, 4.69 mmol, 445 μL) was added dropwise to a stirred solution of 4-(1-(tetrahydro-2H-pyran-4-yl)-4-(trifluoromethyl)-1H-imidazol-2- yl)benzoic acid (500 g, 1.47 mmol) in THF (20 mL). The reaction mixture was stirred at room temperature for 12 hr. The reaction mixture was quenched by dropwise addition of water (20 mL). The resulting mixture was stirred at room temperature for 30 min, then an aqueous NaOH (5 mL, 20%) was added. The resulting mixture was stirred at room temperature for 30 min. The resulting mixture was extracted with EtOAc (2×10 mL). The combined organic layers were separated, washed with brine (5 mL), dried over anhydrous Na
2SO
4 and concentrated under reduced pressure. The residue was subjected to HPLC (2-10 min, 30-69% ACN+FA (0.1% vol.); flow: 30 mL/min, column: SunFire C18, 100×19 mm, 5 µm) to afford (4-(1-(tetrahydro-2H-pyran-4-yl)-4-(trifluoromethyl)-1H-imidazol-2- yl)phenyl)methanol (110 mg, 336 μmol, 22.9% yield) as a white solid. MS (ESI): [M+H]
+ m/z: calcd 327.16; found 327.2 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-5-methyl-4-[[4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methylsulfonyl]pyrimidine Synthesis of the starting 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-5-methyl-4-[[4- [1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methylsulfanyl]pyrimidine is described for Compound 38. [1469] mCPBA (999 mg, 4.92 mmol, 85% purity) was added to a stirred solution of 2-(4- cyclopropyl-6-methoxy-pyrimidin-5-yl)-5-methyl-4-[[4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methylsulfanyl]pyrimidine (1.2 g, 2.34 mmol) in DCM (80 mL) and the resulting mixture was stirred at room temperature for 48 hr. The mixture was washed with saturated aqueous NaHCO
3 solution (3 × 25 mL), dried over anhydrous Na
2SO
4 and concentrated under reduced pressure. The residue was dissolved in EtOAc (50 mL). SiliaMetS
® Dimercaptotriazine (500 mg) was added to the resulting organic layer. The resulting mixture was stirred for 10 hr. and filtered. The filtrate was concentrated under reduced pressure. The residue was subjected to flash-column chromatography (SiO
2, Hexanes - EtOAc, gradient from 40% to 80% EtOAc) to afford 2-(4-cyclopropyl-6-methoxy- pyrimidin-5-yl)-5-methyl-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methylsulfonyl]pyrimidine (620 mg, 1.14 mmol, 48.6% yield) as a light-yellow solid which was used in the next steps without further purification. MS (ESI): [M+H]
+ m/z: calcd 545.17; found 545.0 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-5-methyl-4-[[4-[1-tetrahydropyran-4-yl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidine [1470] NaH (6.76 mg, 169 μmol, 60% dispersion in mineral oil) followed by 2-(4- cyclopropyl-6-methoxy-pyrimidin-5-yl)-5-methyl-4-[[4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methylsulfonyl]pyrimidine (83.4 mg, 153 μmol) were added to a solution of [4-[1-tetrahydropyran-4-yl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methanol (50 mg, 153 μmol) in THF (5.0 mL). The reaction mixture was stirred at room temperature for 36 hr. The reaction mixture was poured into water (10 mL) and extracted with EtOAc (10 mL). The organic layer was separated and concentrated under reduced pressure. The residue was subjected to HPLC (2-10 min, 0-35% ACN+FA (0.1% vol.); flow: 30 mL/min, column: SunFire C18, 100×19 mm, 5 µm) to afford 2-(4- cyclopropyl-6-methoxy-pyrimidin-5-yl)-5-methyl-4-[[4-[1-tetrahydropyran-4-yl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidine (4.00 mg, 7.06 μmol, 4.6% yield) as a yellow solid.
1H NMR (600 MHz, DMSO-d
6) δ 0.83 – 0.88 (m, 2H), 1.00 – 1.05 (m, 2H), 1.66 – 1.71 (m, 1H), 1.88 – 1.94 (m, 2H), 1.99 – 2.08 (m, 2H), 2.24 (s, 3H), 3.30 – 3.38 (m, 2H), 3.84 (s, 3H), 3.89 – 3.94 (m, 2H), 4.30 – 4.37 (m, 1H), 5.55 (s, 2H), 7.61 (s, 4H), 8.25 (s, 1H), 8.57 (s, 1H), 8.66 (s, 1H). MS (ESI): [M+H]
+ m/z: calcd 567.27; found 567.0.
3,5-difluoro-4-[4-(trifluoromethyl)-1H-imidazol-2-yl]benzoic acid [1471] A solution of 3,3-dibromo-1,1,1-trifluoro-propan-2-one (8.09 g, 30,0 mmol) and sodium acetate (4.71 g, 57.5 mmol, 3.08 mL) in water (76 mL) was stirred at 100 °C for 45 min. The obtained mixture was cooled to room temperature and poured into a mixture of methyl 3,5-difluoro-4-formyl-benzoate (5.00 g, 25.0 mmol) and an aqueous ammonium hydroxide (8.76 g, 250 mmol, 9.73 mL, 28% wt.) in MeOH (287 mL). The reaction mixture was stirred at room temperature for 12 hr. The reaction mixture was concentrated under reduced pressure to a half volume. The residue was extracted with EtOAc (2×40 mL). The combined organic layers were washed with brine (50 mL), dried over anhydrous Na
2SO
4 and filtered. The filtrate was concentrated under reduced pressure to afford crude 3,5-difluoro-4- [4-(trifluoromethyl)-1H-imidazol-2-yl]benzoic acid (4.00 g, 13.7 mmol, 54.8% yield) a light- yellow solid which was used in the next steps without further purification. MS (ESI): [M+H]
+ m/z: calcd 293.04; found 293.0 isopropyl 3,5-difluoro-4-[1-isopropyl-4-(trifluoromethyl)imidazol-2-yl]benzoate [1472] Cs
2CO
3 (22.3 g, 68.5 mmol) and 2-iodopropane (23.3 g, 137 mmol) were added to a solution of 3,5-difluoro-4-[4-(trifluoromethyl)-1H-imidazol-2-yl]benzoic acid (4.00 g, 13.7 mmol) in ACN (200 mL). The reaction mixture was stirred at room temperature for 48 hr. then an additional portion of 2-iodopropane (12.0 g) and Cs
2CO
3 (10.0 g) were added to the reaction mixture. The reaction mixture was stirred at 45 °C for 24 hr.2-iodopropane (12.0 g) and Cs
2CO
3 (10.0 g) were added to the reaction mixture. The reaction mixture was stirred at 85 °C for 24 hr. The reaction mixture was cooled and filtered. The filtrate was concentrated under reduced pressure. The residue was dissolved in EtOAc (150 mL). The obtained solution was washed with water (50 mL) and brine (30 mL), dried over anhydrous Na
2SO
4 and filtered. The filtrate was concentrated under reduced pressure to afford isopropyl 3,5- difluoro-4-[1-isopropyl-4-(trifluoromethyl)imidazol-2-yl]benzoate (5.00 g, crude) as a red oil which was used in the next steps without further purification. MS (ESI): [M+H]
+ m/z: calcd 377.13; found 377.0 [3,5-difluoro-4-[1-isopropyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methanol [1473] A solution of isopropyl 3,5-difluoro-4-[1-isopropyl-4-(trifluoromethyl)imidazol-2- yl]benzoate (5.00 g, crude) in THF (50 mL) was added dropwise to a vigorously stirred suspension of LAH (196 mg, 5.78 mmol) in THF (150 mL). The reaction mixture was stirred at room temperature for 2 hr. An aqueous NaOH (30 mL, 10% wt.) was added dropwise to the reaction mixture followed by granular NaOH (10.0 g). The resulting mixture was filtered. The filtrate was concentrated under reduced pressure. The residue was subjected to flash column chromatography (SiO
2, gradient Hexane-EtOAc) to afford [3,5-difluoro-4-[1- isopropyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methanol (280 mg, 874 μmol, 6.34% yield from 3,5-difluoro-4-[4-(trifluoromethyl)-1H-imidazol-2-yl]benzoic acid) as an yellow solid.
1H NMR (400 MHz, CDCl
3) δ 1.40 (d, 6H), 4.08 – 4.16 (m, 1H), 4.71 (s, 2H), 6.94 (d, 2H), 7.48 (s, 1H). MS (ESI): [M+H]
+ m/z: calcd 321.10; found 321.2. 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[3,5-difluoro-4-[1-isopropyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]-5-methoxy-pyrimidine NaH (13.9 mg, 347 μmol, 60% dispersion in mineral oil) was added to a solution of [3,5-difluoro-4-[1-isopropyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methanol (111 mg, 347 μmol) in DMF (2 mL). The reaction mixture was stirred at room temperature for 15 min.2-(4- cyclopropyl-6-methoxy-pyrimidin-5-yl)-5-methoxy-4-methylsulfonyl-pyrimidine I-6 (117 mg, 347 μmol) was added to the reaction mixture. The reaction mixture was stirred at room temperature for 12 hr. The reaction mixture was diluted with water (5.0 mL) and extracted with EtOAc (5 mL). The organic layer was concentrated under reduced pressure. The residue was subjected to HPLC (0.5-6.5 min, 48-63% water – MeOH, +0.1% vol. of 25% aq. NH
3, flow: 30 mL/min, column: Chromatorex Phenyl SMB 100-5100 ×19 mm, 5 µm) to afford 2-(4- cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[3,5-difluoro-4-[1-isopropyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]-5-methoxy-pyrimidine (38.0 mg, 65.9 µmol, 19% yield) as a light-yellow solid.
1H NMR (600 MHz, DMSO-d
6) δ 0.83 – 0.88 (m, 2H), 0.99 – 1.03 (m, 2H), 1.34 (d, 6H), 1.66 – 1.73 (m, 1H), 3.82 (s, 3H), 3.96 (s, 3H), 4.06 – 4.12 (m, 1H), 5.51 (s, 2H), 7.41 (d, 2H), 8.32 (s, 1H), 8.45 (s, 1H), 8.64 (s, 1H). MS (ESI): [M+H]
+ m/z: calcd 577.23; found 577.0 The synthesis of 4-cyclopropyl-5-[4-[[3,5-difluoro-4-[1-isopropyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidin-2-yl]-6-methoxy-pyrimidine NaH (11.3 mg, 282 μmol, 60% dispersion in mineral oil) was added to a solution of [3,5-difluoro-4-[1-isopropyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methanol (129 mg, 282 μmol) in DMF (2 mL). The reaction mixture was stirred at room temperature for 15 min. 4- cyclopropyl-6-methoxy-5-(4-methylsulfonylpyrimidin-2-yl)pyrimidine I-8 (86.4 mg, 282 μmol) was added to the reaction mixture. The reaction mixture was stirred at room temperature for 12 hr. The reaction mixture was diluted with water (2 mL), extracted with EtOAc (2 × 2 mL). The combined organic layers were washed with brine (3 mL) and concentrated under reduced pressure. The residue was subjected to HPLC (0.5-6.5 min., 50-75% water – MeOH, +0.1% vol. of 25% aq. NH
3, flow: 30 mL/min, column: Chromatorex Phenyl SMB 100-5100 ×19 mm, 5 µm) to afford 4-cyclopropyl-5-[4-[[3,5-difluoro-4-[1-isopropyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidin-2-yl]-6-methoxy-pyrimidine (40.0 mg, 73.2 μmol, 26% yield) as a light-yellow solid.
1H NMR (600 MHz, DMSO-d
6) δ 0.85 – 0.90 (m, 2H), 1.00 – 1.05 (m, 2H), 1.34 (d, 6H), 1.64 – 1.71 (m, 1H), 3.83 (s, 3H), 4.05 – 4.11 (m, 1H), 5.51 (s, 2H), 7.11 (d, 1H), 7.44 (d, 2H), 8.32 (s, 1H), 8.66 (s, 1H), 8.72 (d, 1H). MS (ESI): [M+H]
+ m/z: calcd 547.22; found 547.4.
2-chloro-4-methylsulfanyl-5-[1-(trifluoromethyl)vinyl]pyrimidine [1474] To a solution of 5-bromo-2-chloro-4-methylsulfanyl-pyrimidine (2 g, 8.35 mmol), 4,4,6-trimethyl-2-[1-(trifluoromethyl)vinyl]-1,3,2-dioxaborinane (3.6 g, 16.2 mmol), K
2CO
3 (4.6 g, 33.3 mmol) in H
2O (15 mL) and 1,2-dimethoxyethane (30 mL) was added palladium;triphenylphosphane (480 mg, 0.415 mmol). The mixture was stirred at 80 °C for 12 hrs under N
2. The mixture was filtered. The filtrate was extracted with water (50 mL) and EtOAc (60 mL x 3). The combined organic layer was dried over anhydrous Na
2SO
4, filtered and concentrated under reduced pressure. The residue was purified by flash chromatography (ISCO
®; 20 g AgelaFlash
® Silica Flash Column, PE/EtOAc with EtOAc from 0-10%, flow rate = 40 mL/min, 254 nm) to afford 2-chloro-4-methylsulfanyl-5-[1- (trifluoromethyl)vinyl]pyrimidine (1.6 g, 75.2% yield) as yellow oil.
1H NMR (400 MHz, chloroform-d) δ ppm 8.11 (s, 1H), 6.34 (d, J = 1.2 Hz, 1H), 5.86 (d, J = 1.2 Hz, 1H), 2.57 (s, 3H);
19F NMR (377 MHz, chloroform-d) δ ppm -66.92; MS (ESI) [M+H]
+ m/z: calcd 255.0, found 255.0. 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-methylsulfanyl-5-[1- (trifluoromethyl)vinyl]pyrimidine [1475] A mixture of 2-chloro-4-methylsulfanyl-5-[1-(trifluoromethyl)vinyl]pyrimidine (500 mg, 1.96 mmol), (4-cyclopropyl-6-methoxy-pyrimidin-5-yl)boronic acid I-5 (400 mg, 2.06 mmol), XPhos Pd G
3 (90 mg, 0.106 mmol), XPhos (50 mg, 0.105 mmol), K
3PO
4 (1.05 g, 4.95 mmol) in dioxane (10 mL) and H
2O (2 mL) was stirred at 90°C for 2 hrs. The mixture was filtered. The resulting mixture was extracted with water (50 mL) and EtOAc (60 mL x 3). The combined organic layer was dried over anhydrous Na
2SO
4, filtered and concentrated under reduced pressure. The residue was purified by flash chromatography (ISCO
®; 12 g AgelaFlash
® Silica Flash Column, PE/EtOAc with EtOAc from 0-12%, flow rate = 40 mL/min, 254 nm) to afford 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4- methylsulfanyl-5-[1-(trifluoromethyl)vinyl]pyrimidine (530 mg, 73.3% yield) as yellow oil.
1H NMR (400 MHz, chloroform-d) δ ppm 8.66 (s, 1H), 8.40 (s, 1H), 6.38 (d, J = 1.3 Hz, 1H), 5.96 (d, J = 1.0 Hz, 1H), 3.96 (s, 3H), 2.53 (s, 3H), 1.80 - 1.87 (m, 1H), 1.25 (s, 2H), 0.96 (dd, J = 8.0, 3.0 Hz, 2H);
19F NMR (377 MHz, chloroform-d) δ ppm -66.56; MS (ESI) [M+H]
+ m/z: calcd 369.1, found 369.0. 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-methylsulfanyl-5-[1- (trifluoromethyl)cyclopropyl]pyrimidine [1476] A mixture of 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-methylsulfanyl-5-[1- (trifluoromethyl)vinyl]pyrimidine (430 mg, 1.17 mmol), Methyldiphenylsulfonium tetrafluoroborate (430 mg, 1.42 mmol, 95 wt%) and THF (10 mL) was stirred at 20 °C for 30 minutes.1M NaHMDS/THF (1.9 mL, 1.90 mmol) was added at 0 °C and the mixture was stirred at 20 °C for 1 hour. The resulting mixture was quenched by addition of water (30 mL) and extracted with EtOAc (30 mL x 3). The combined organic layer was washed with dried over anhydrous Na
2SO
4, filtered and concentrated under reduced pressure. The residue was purified by flash chromatography (ISCO
®; 12 g AgelaFlash
® Silica Flash Column, PE/EtOAc with EtOAc from 0-25%, flow rate = 40 mL/min, 254 nm) to afford 2-(4-cyclopropyl-6- methoxy-pyrimidin-5-yl)-4-methylsulfanyl-5-[1-(trifluoromethyl)cyclopropyl]pyrimidine (200 mg, 44.8% yield) as yellow oil.
1H NMR (400 MHz, chloroform-d) δ ppm 8.65 (s, 1H), 8.58 (s, 1H), 3.95 (s, 3H), 2.56 (s, 3H), 1.77 - 1.84 (m, 1H), 1.25 - 1.29 (m, 2H), 1.22 - 1.25 (m, 4H), 0.95 (dd, J = 8.1, 2.9 Hz, 2H),
19F NMR (377 MHz, chloroform-d) δ ppm -69.23; MS (ESI) [M+H]
+ m/z: calcd 383.1, found 383.0. 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-methylsulfonyl-5-[1- (trifluoromethyl)cyclopropyl]pyrimidine [1477] To a solution of 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-methylsulfanyl-5-[1- (trifluoromethyl)cyclopropyl]pyrimidine (110 mg, 0.288 mmol) in DCM (5 mL) was added mCPBA (120 mg, 0.591 mmol, 85 wt%) at 0 °C. The mixture was stirred at 20 °C for 2 hrs. The resulting mixture was quenched by addition of saturated Na
2SO
3 aqueous solution (20 mL) and extracted with DCM (30 mL x 3). The combined organic layer was dried over anhydrous Na
2SO
4, filtered and concentrated under reduced pressure to give 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-methylsulfonyl-5-[1- (trifluoromethyl)cyclopropyl]pyrimidine (160 mg, crude) as colorless oil. MS (ESI) [M+H]
+ m/z: calcd 415.1, found 415.0. 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]-5-[1-(trifluoromethyl)cyclopropyl]pyrimidine (187) [1478] To a solution of [4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methanol I-2b (70 mg, 0.273 mmol) in THF (3 mL) was added NaH (13 mg, 0.325 mmol, 60 wt% in mineral oil). The mixture was stirred at 20 °C for 30 minutes.2-(4-cyclopropyl-6-methoxy- pyrimidin-5-yl)-4-methylsulfonyl-5-[1-(trifluoromethyl)cyclopropyl]pyrimidine (110 mg, 0.265 mmol) was added and the mixture was stirred at 45 °C for 2 hrs. The resulting mixture was quenched by addition of water (20 mL) extracted with EtOAc (30 mL x 3). The combined organic layer was dried over anhydrous Na
2SO
4, filtered and concentrated under reduced pressure. The residue was purified by preparative HPLC (Instrument: Gilson GX-281 Liquid Handler, Gilson 322 Pump, Gilson 156 UV Detector; Column: 2_Phenomenex Gemini C1875 × 40 mm × 3 μm; Mobile phase A: H
2O with 10 mmol NH
4HCO
3 (v%); Mobile phase B: MeCN; Gradient: B from 56% to 86% in 7.8 min, hold 100% B for 1 min; Flow Rate: 25 mL/min; Column Temperature: 30 °C; Wavelength: 220 nm, 254 nm) to afford 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]-5-[1- (trifluoromethyl)cyclopropyl]pyrimidine (42.3 mg, 27% yield) as white solid.
1H NMR (400 MHz, chloroform-d) δ ppm 8.69 (s, 1H), 8.66 (s, 1H), 7.67 (d, J = 8.3 Hz, 2H), 7.56 (d, J = 8.3 Hz, 2H), 7.33 (d, J = 0.9 Hz, 1H), 5.56 (s, 2H), 3.94 (s, 3H), 3.80 (s, 3H), 1.68 - 1.75 (m, 1H), 1.49 - 1.52 (m, 2H), 1.24 (dd, J = 4.5, 2.9 Hz, 2H), 1.11 (s, 2H), 0.93 (dd, J = 8.0, 3.0 Hz, 2H),
19F NMR (377 MHz, chloroform-d) δ ppm -62.77, -69.97; MS (ESI) [M+H]
+ m/z: calcd 591.2, found 591.1. Example 162 (Compound 185)
T
HF, H2O Compound 185 2-chloro-4-[[3-fluoro-4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]-5- methoxy-pyrimidine Synthesis of the starting [3-fluoro-4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methanol is described for compound 97. [1479] To a stirred solution of [3-fluoro-4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methanol (150 mg, 547.02 μmol) in THF (1 mL) was added NaH (21.88 mg, 547.02 μmol, 60% purity) at 0 °C under nitrogen atmosphere. After 30 minutes, 2,4-dichloro- 5-methoxy-pyrimidine (117.5 mg, 656.42 μmol) was added. The resulting mixture was stirred for 2 hrs at 0 °C under nitrogen atmosphere. The reaction was quenched by the addition of saturated NH
4Cl aqueous solution (5 mL) at 0 °C. The resulting mixture was extracted with EtOAc (3 x 10 mL). The combined organic layers were washed with brine (5 mL), dried over anhydrous Na
2SO
4. After filtration, the filtrate was concentrated under reduced pressure. The residue was purified by Prep-TLC (50% EtOAc in PE) to afford 2-chloro-4-[[3-fluoro-4-[1- methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]-5-methoxy-pyrimidine (200 mg, 479.90 μmol, 87% yield) as a yellow solid. MS: m/z = 417.10, 419.10 [M + H]
+.
1H NMR (400 MHz, Chloroform-d) δ 7.96 (d, J = 1.8 Hz, 1H), 7.69 - 7.61 (m, 1H), 7.42 - 7.30 (m, 3H), 5.55 (s, 2H), 3.95 (s, 3H), 3.67 (s, 3H). 2-[4-cyclopropyl-6-(trideuteriomethoxy)pyrimidin-5-yl]-4-[[3-fluoro-4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]-5-methoxy-pyrimidine [1480] To a stirred solution of 2-chloro-4-[[3-fluoro-4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]-5-methoxy-pyrimidine (100 mg, 239.95 μmol) and 4-cyclopropyl-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-6- (trideuteriomethoxy)pyrimidine I-9 (67 mg, 239.95 μmol) in THF (2 mL) and H
2O (0.4 mL) were added potassium phosphate (101.9 mg, 479.90 μmol) and [1,1’- Bis(dicyclohexylphosphino)ferrocene]dichloropalladium(II) (20.22 mg, 23.99 μmol) at 25 °C under nitrogen atmosphere. The resulting mixture was stirred for 16 hrs at 40 °C under nitrogen atmosphere. After filtration, the filtrate was concentrated under reduced pressure. The residue was purified by Prep-TLC (10% MeOH in DCM) to afford crude product. The obtained crude product was purified by RP-Flash with the following conditions: Column: C18 spherical Column, 20-35 um, 100A, 25 g; Mobile Phase A: Water (5 mM aq. NH
4HCO
3), Mobile Phase B: MeCN; Flow rate: 35 mL/min; Gradient: 5% B to 5% B in 5 min, 5% B to 61% B in 12 min, 61% B to 95% B in 5min; Detector: UV 254 & 210 nm. The collected fractions were combined and lyophilized to give 2-[4-cyclopropyl-6- (trideuteriomethoxy)pyrimidin-5-yl]-4-[[3-fluoro-4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]-5-methoxy-pyrimidine (14 mg, 26.24 μmol, 11% yield) as a white solid. MS: m/z = 534.65 [M + H]
+.
1H NMR (400 MHz, Chloroform-d) δ 8.67 (s, 1H), 8.26 (s, 1H), 7.62 (t, J = 7.6 Hz, 1H), 7.40 - 7.30 (m, 3H), 5.57 (s, 2H), 4.02 (s, 3H), 3.65 (s, 3H), 1.76 - 1.70 (m, 1H), 1.30 - 1.22 (m, 2H), 0.98 - 0.89 (m, 2H).
19F NMR (376 MHz, Chloroform-d) δ
1-[2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol- 2-yl]phenyl]methoxy]pyrimidin-5-yl]-2,2,2-trifluoro-N-methyl-ethanamine [1481] A solution of [1-[2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidin-5-yl]-2,2,2-trifluoro-ethyl] 4- methylbenzenesulfonate 68b (56 mg, 76.23 μmol) in Methylamine solution (0.5 mL, 1 mmol, 2 M in THF) was stirred at 25 °C under nitrogen atmosphere. The resulting mixture was stirred for 16 hrs at 80 °C then concentrated under reduced pressure. The residue was subject to by Prep-TLC, eluted with 50% EtOAc in PE, to give a mixture of two isomers. The more polar isomer was purified by RP-Flash with the following conditions: Column: C18 spherical, 20-30 um, 100A, 20 g; Mobile Phase A: Water (5 mM aq. NH
4HCO
3), Mobile Phase B: MeCN; Flow rate: 35 mL/min; Gradient: 2% B to 2% B in 5 min, 2% B to 55% B in 15 min, 55% B to 55% B in 2 min, 55% B to 95% B in 5 min; Detector: UV 254 & 210 nm. The collected fractions were combined, concentrated and then lyophilized to give 1-[2-(4- cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidin-5-yl]-2,2,2-trifluoro-N-methyl-ethanamine (12.4 mg, 20.89 μmol, 27% yield). MS: m/z = 594.25 [M + H]
+.
1H NMR (400 MHz, Chloroform-d) δ 8.75 (s, 1H), 8.67 (s, 1H), 7.69 (d, J = 8.0 Hz, 2H), 7.55 (d, J = 8.0 Hz, 2H), 7.35 (s, 1H), 5.63 - 5.53 (m, 2H), 4.57 - 4.51 (m, 1H), 3.96 (s, 3H), 3.81 (s, 3H), 2.53 (s, 3H), 1.75 - 1.69 (m, 1H), 1.28 - 1.23 (m, 2H), 0.95 - 0.91 (m, 2H).
19F NMR (376 MHz, Chloroform-d) δ -62.78, - 73.04.
2,2-difluoro-2-triphenylphosphaniumylacetate [1482] To a solution of (2-bromo-2,2-difluoro-acetyl)oxypotassium (1.02 g, 4.77 mmol) in DMF (6 mL) was added triphenylphosphine (1.25 g, 4.77 mmol) at 25 °C under nitrogen atmosphere. The reaction mixture was stirred at 25 °C for 16 hrs. The precipitated solid was collected by filtration, then washed with cold DMF (2 mL), water (2 × 3 mL) and ethyl ether (5 mL). The solid was dried under reduced pressure to give 2,2-difluoro-2- triphenylphosphaniumylacetate (1.2 g, 3.37 mmol, 70% yield) as a white solid. MS: m/z = 357.00 [M + H]
+.
1H NMR (400 MHz, Chloroform-d) δ 7.88 - 7.76 (m, 8H), 7.68 - 7.61 (m, 6H), 7.51 - 7.45 (m, 1H).
19F NMR (377 MHz, Chloroform-d) δ -92.62, -92.85. 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]-5-(2,2,2-trifluoroethyl)pyrimidine Starting material 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidine-5-carbaldehyde was prepared as described for Compound 68 [1483] 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidine-5-carbaldehyde 68a(50 mg, 97.95 μmol) and 2,2-difluoro-2-triphenylphosphaniumylacetate (69.8 mg, 195.90 μmol) were added to the reaction flask and then backfilled with nitrogen. DMF (0.5 mL) was added dropwise via syringe. The resulting mixture was stirred for 40 min at 60 °C under nitrogen atmosphere. Tetrabutylammonium fluoride (0.3 mL, 0.3 mmol, 1 M in THF) was added dropwise via syringe. The resulting mixture was stirred for additional 30 min at 60 °C under nitrogen atmosphere. The reaction mixture was allowed to cool down to room temperature. Water (10 mL) was added to the mixture. The resulting mixture was extracted with EtOAc (3 x 20 mL). The combined organic layers were washed with brine (2 x 10 mL), dried over anhydrous Na
2SO
4. After filtration, the filtrate was concentrated under reduced pressure to give a residue. The residue was purified by Prep-TLC (EtOAc / PE = 2 / 1) to afford 10 mg crude product. The crude product was further purified by RP-Flash with the following conditions: Column: C18 spherical Column, 20-35 um, 100A, 40 g; Mobile Phase A: Water (5 mM aq. NH
4HCO
3), Mobile Phase B: MeCN; Flow rate: 60 mL/min; Gradient: 5% B to 5% B in 5 min, 5% B to 59% B in 20 min, 59% B to 59% B in 1 min, 59% B to 95% B in 5 min; Detector: UV 254 & 220 nm. The collected fractions were combined, concentrated and then lyophilized to give 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[4-[1-methyl-4- (trfluoromeithyl)imidazol-2-yl]phenyl]methoxy]-5-(2,2,2-trifluoroethyl)pyrimidine (3.2 mg, 5.67 μmol, 6% yield) as an off-white solid. MS: m/z = 565.30 [M + H]
+; 587.10 [M + Na]
+.
1H NMR (400 MHz, Chloroform-d) δ 8.74 (s, 1H), 8.68 (s, 1H), 7.68 (d, J = 8.0 Hz, 2H), 7.55 (d, J = 8.0 Hz, 2H), 7.37 (s, 1H), 5.57 (s, 2H), 3.99 (s, 3H), 3.81 (s, 3H), 3.63 - 3.48 (m, 2H), 1.79 - 1.70 (m, 1H), 1.29 - 1.24 (m, 2H), 0.98 - 0.87 (m, 2H).
19F NMR (377 MHz, Chloroform-d) δ -62.78, -65.04.
2-chloro-5-(difluoromethoxy)-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidine Synthesis of the starting 2-chloro-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidin-5-ol is described for Compound 122. 2-Chloro-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidin-5-ol (434 mg, 1.13 mmol), K
2CO
3 (624 mg, 4.51 mmol) and (2- chloro-2,2-difluoro-acetyl)oxysodium (688 mg, 4.51 mmol) were mixed in DMF (10 mL) and Water (2.5 mL). The reaction mixture was stirred at 85 °C for 12 hr. Additional portion of (2- chloro-2,2-difluoro-acetyl)oxysodium (689 mg, 4.51 mmol) was added to the reaction mixture. The resulting mixture was stirred at 85 °C for 36 hr. The reaction mixture was cooled to room temperature, poured into cold water (40 mL) and extracted with EtOAc (2×20 mL). The combined organic layers were separated, washed with brine (3×40 mL), dried over anhydrous Na
2SO
4 and concentrated under reduced pressure. The residue was subjected to HPLC (0.5-6.5 min, 40-80% water - ACN; flow: 30 mL/min, column: SunFire C18, 100×19 mm, 5 µm) to afford 2-chloro-5-(difluoromethoxy)-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidine (83.0 mg, 191 μmol, 16.9% yield) as a white solid. MS (ESI): [M+H]
+ m/z: calcd 435.08; found 435.0 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-5-(difluoromethoxy)-4-[[4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidine [1484] 2-Chloro-5-(difluoromethoxy)-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidine (83.0 mg, 191 μmol), (4-cyclopropyl-6-methoxy-pyrimidin-5- yl)boronic acid I-5 (111 mg, 573 μmol), potassium phosphate tribasic (243 mg, 1.15 mmol) and bis(diphenylphosphino)ferrocene]dichloropalladium(II)-DCM (31.2 mg, 38.2 μmol) were mixed in a degassed mixture of dioxane (3 mL) and water (300 µL). The reaction mixture was stirred at room temperature for 12 hr under argon atmosphere. The reaction mixture was diluted with EtOAc (6.0 mL). Na
2SO
4 (1 g) and SiliaMetS
® Dimercaptotriazine (300 mg) were added to the mixture. The resulting mixture was stirred at room temperature for 1 hr, then solids were filtered out and washed with EtOAc. The filtrate was concentrated under reduced pressure. The residue was subjected to HPLC (0.5-6.5 min., 48% water – ACN, +0.1% vol. of 25% aq. NH
3, flow: 30 mL/min, column: Cromatotex C18100×19 mm, 5 µm) to afford 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-5-(difluoromethoxy)-4-[[4-[1-methyl- 4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidine (51.0 mg, 93.0 μmol, 48.7% yield) as a white solid.
1H NMR (600 MHz, DMSO-d
6) δ 0.87 – 0.92 (m, 2H), 1.02 – 1.07 (m, 2H), 1.72 – 1.78 (m, 1H), 3.79 (s, 3H), 3.86 (s, 3H), 5.57 (s, 2H), 7.36 (t, 1H, CHF
2), 7.60 (d, 2H), 7.75 (d, 2H), 7.95 (s, 1H), 8.68 (s, 1H), 8.69 (s, 1H). MS (ESI): [M+H]
+ m/z: calcd 549.19; found 549.2. Example 166 (Compound 179)
2-chloro-4-[[4-
methoxy-pyrimidine [1485] To a stirred solution of [4-fluoro-1-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]-4- piperidyl]methanol (145 mg, 515.56 μmol) in THF (3 mL) was added NaH (24.74 mg, 618.67 μmol, 60% purity) at 25 °C under nitrogen atmosphere. After 30 min, 2,4-dichloro-5- methoxy-pyrimidine (184.57 mg, 1.03 mmol) was added. The resulting mixture was stirred for 16 hrs at 25 °C under nitrogen atmosphere. The reaction was quenched by the addition of saturated NH
4Cl aqueous solution (5 mL) at 0 °C. The resulting mixture was extracted with EtOAc (3 x 10 mL). The combined organic layers were washed with brine (5 mL), dried over anhydrous Na
2SO
4. After filtration, the filtrate was concentrated under reduced pressure. The residue was purified by Prep-TLC (50% EtOAc in PE) to afford 2-chloro-4-[[4-fluoro-1-[1- methyl-4-(trifluoromethyl)imidazol-2-yl]-4-piperidyl]methoxy]-5-methoxy-pyrimidine (120 mg, 283.16 μmol, 54% yield) as a yellow solid. MS: m/z = 424.20 [M + H]
+.
1H NMR (400 MHz, Chloroform-d) δ 7.94 (s, 1H), 7.05 (s, 1H), 4.56 (s, 1H), 4.51 (s, 1H), 3.93 (s, 3H), 3.54 (s, 3H), 3.35 - 3.13 (m, 4H), 2.17 - 1.96 (m, 4H). 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[4-fluoro-1-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]-4-piperidyl]methoxy]-5-methoxy-pyrimidine [1486] To a stirred solution of 2-chloro-4-[[4-fluoro-1-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]-4-piperidyl]methoxy]-5-methoxy-pyrimidine (100 mg, 235.96 μmol) and (4-cyclopropyl-6-methoxy-pyrimidin-5-yl)boronic acid I-5 (45.8 mg, 235.96 μmol) in 1,4-dioxane (2 mL) and water (0.4 mL) were added potassium phosphate (50.09 mg, 235.96 μmol), dicyclohexyl-[2-(2,4,6-triisopropylphenyl)phenyl]phosphane (11.3 mg, 23.60 μmol) and Methanesulfonato(2-dicyclohexylphosphino-2',4',6'-tri-i-propyl-1,1'- biphenyl)(2'-amino-1,1'-biphenyl-2-yl)palladium(II) (20 mg, 23.60 μmol) at 25 °C under nitrogen atmosphere. The resulting mixture was stirred for 16 hrs at 40 °C under nitrogen atmosphere. The resulting mixture was diluted with water (2 mL) and extracted with EtOAc (3 x 10 mL). The combined organic layers were washed with brine (5 mL), dried over anhydrous Na
2SO
4. After filtration, the filtrate was concentrated under reduced pressure. The residue was subject to Prep-TLC (50% EtOAc in PE) to afford crude product. The obtained crude product was purified by RP-Flash with the following conditions: Column: C18 spherical Column, 20-35 um, 100A, 25 g; Mobile Phase A: Water (5 mM aq. NH
4HCO
3), Mobile Phase B: MeCN; Flow rate: 35 mL/min; Gradient: 5% B to 5% B in 5 min, 5% B to 36% B in 9 min, 36% B to 36% B in 1 min, 36% B to 95% B in 5 min; Detector: UV 254 & 210 nm. The collected fractions were combined, concentrated and then lyophilized to give 2- (4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[4-fluoro-1-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]-4-piperidyl]methoxy]-5-methoxy-pyrimidine (17.9 mg, 33.30 μmol, 14% yield) as a white solid. MS: m/z = 538.25 [M + H]
+.
1H NMR (400 MHz, Chloroform-d) δ 8.62 (s, 1H), 8.23 (s, 1H), 7.02 (s, 1H), 4.57 (s, 1H), 4.52 (s, 1H), 3.99 (s, 3H), 3.93 (s, 3H), 3.52 (s, 3H), 3.35 - 3.28 (m, 2H), 3.20 - 3.15 (m, 2H), 2.19 - 1.91 (m, 4H), 1.76 - 1.69 (m, 1H), 1.26 - 1.18 (m, 2H), 0.93 - 0.89 (m, 2H).
19F NMR (376 MHz, Chloroform-d) δ -63.05, -165.10. Example 167 (Compound 178)
Compound 178 Methyl 4-[5-methyl-3-(trifluoromethyl)pyrazol-1-yl]benzoate [1487] To a solution of methyl 4-hydrazinobenzoate hydrochloride (700 mg, 3.45 mmol) and anhydrous sodium acetate (298 mg, 3.63 mmol) in EtOH (50 mL) 1,1,1-trifluoropentane-2,4- dione (532 mg, 3.45 mmol, 418 μL) was added at room temperature. The resulting mixture was stirred at room temperature for 16 hr. The mixture was concentrated under reduced pressure. The residue was distributed between DCM (80 mL) and brine (50 mL). The organic layer was separated, dried over anhydrous Na
2SO
4 and concentrated under reduced pressure. The crude was subjected to flash-column chromatography (SiO
2, gradient hexane - MTBE with MTBE from 0% to 50%) to afford methyl 4-[5-methyl-3-(trifluoromethyl)pyrazol-1- yl]benzoate (200 mg, 704 μmol, 20.4% yield) as a light-yellow solid.
1H NMR (500 MHz, CDCl
3) δ 2.40 (s, 3H), 3.95 (s, 3H), 6.48 (s, 1H), 7.55 (d, 2H), 8.17 (d, 2H). MS (ESI): [M+H]
+ m/z: calcd 285.08; found 285.0. [4-[5-methyl-3-(trifluoromethyl)pyrazol-1-yl]phenyl]methanol [1488] Lithium aluminium hydride (47.7 mg, 1.41 mmol) was suspended in THF (25 mL). The resulting mixture was cooled to 0 °C. To the obtained mixture a solution of methyl 4-[5- methyl-3-(trifluoromethyl)pyrazol-1-yl]benzoate (200 mg, 704 μmol) in THF (10 mL) was added dropwise. The reaction mixture was stirred at room temperature for 2 hr. The reaction mixture was cooled to 0 °C and quenched by dropwise addition of water (2 mL). The solid was filtered and off and the filtrate was evaporated under reduced pressure to afford [4-[5- methyl-3-(trifluoromethyl)pyrazol-1-yl]phenyl]methanol (195 mg, crude) as a yellow oil which was used in the next steps without further purification.
1H NMR (400 MHz, CDCl
3) δ 2.31 (s, 3H), 4.74 (s, 2H), 6.43 (s, 1H), 7.41 (d, 2H), 7.46 (d, 2H). MS (ESI): [M+H]
+ m/z: calcd 257.08; found 257.2. 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-5-methoxy-4-[[4-[5-methyl-3- (trifluoromethyl)pyrazol-1-yl]phenyl]methoxy]pyrimidine [1489] NaH (29.2 mg, 761 μmol, 60% dispersion in mineral oil) was suspended in DMF (3 mL). The resulting mixture was cooled to 0 °C. To the obtained mixture [4-[5-methyl-3- (trifluoromethyl)pyrazol-1-yl]phenyl]methanol (195 mg, 761 μmol) was added at 0 °C. The obtained mixture was stirred at 0 °C for 15 min and then 2-(4-cyclopropyl-6-methoxy- pyrimidin-5-yl)-5-methoxy-4-methylsulfonyl-pyrimidine I-6 (256 mg, 761 μmol) was added. The mixture was stirred at room temperature for 16 hr. The mixture was poured into ice- water (5 mL) and extracted with EtOAc (2 × 5 mL). Combined organic layers were dried over anhydrous Na
2SO
4 and evaporated under reduced pressure The crude subjected to HPLC (0-1-5 min., 40-80% water – ACN, +0.1% vol. of 25% aq. NH
3; flow: 30 mL/min, column: XBridge BEH C18, 200 × 19 mm, 5 μm) to afford 2-(4-cyclopropyl-6-methoxy-pyrimidin-5- yl)-5-methoxy-4-[[4-[5-methyl-3-(trifluoromethyl)pyrazol-1-yl]phenyl]methoxy]pyrimidine (61.3 mg, 120 μmol, 17% yield from methyl 4-[5-methyl-3-(trifluoromethyl)pyrazol-1- yl]benzoate) as a white solid.
1H NMR (600 MHz, DMSO-d
6) δ 0.83 – 0.88 (m, 2H), 0.99 – 1.03 (m, 2H), 1.66 – 1.72 (m, 1H), 2.33 (s, 3H), 3.83 (s, 3H), 3.93 (s, 3H), 5.50 (s, 2H), 6.75 (s, 1H), 7.58 (d, 2H), 7.62 (d, 2H), 8.41 (s, 1H), 8.63 (s, 1H). MS (ESI): [M+H]
+ m/z: calcd 513.20; found 513.4. Example 168 (Compound 176)
2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[5-fluoro-6-[1-isopropyl-4- (trifluoromethyl)imidazol-2-yl]-3-pyridyl]methoxy]-5-methoxy-pyrimidine (176) Synthesis of the starting [5-fluoro-6-[1-isopropyl-4-(trifluoromethyl)imidazol-2-yl]-3- pyridyl]methanol is described for Compound 119. [1490] To a solution of [5-fluoro-6-[1-isopropyl-4-(trifluoromethyl)imidazol-2-yl]-3- pyridyl]methanol (100 mg, 0.330 mmol) in THF (3 mL) was added NaH (16 mg, 0.400 mmol, 60 wt% in mineral oil). The mixture was stirred at 20° C for 30 minutes.2-(4- cyclopropyl-6-methoxy-pyrimidin-5-yl)-5-methoxy-4-methylsulfonyl-pyrimidine I-6 (110 mg, 0.327 mmol) was added and the mixture was stirred at 45 °C for 2 hrs. The resulting mixture was quenched by addition of water (20 mL) and extracted with EtOAc (30 mL x 3). The combined organic layer was dried over anhydrous Na
2SO
4, filtered and concentrated under reduced pressure. The residue was purified by preparative HPLC (Instrument: Gilson GX-281 Liquid Handler, Gilson 322 Pump, Gilson 156 UV Detector; Column: 2_Phenomenex Gemini C1875 × 40 mm × 3 μm; Mobile phase A: H
2O with 0.05% NH
3-H
2O + NH
4HCO
3(v%); Mobile phase B: MeCN; Gradient: B from 49% to 79% in 7.8 min, hold 100% B for 2 min; Flow Rate: 30 mL/min; Column Temperature: 30 °C; Wavelength: 220 nm, 254 nm) to afford 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[5- fluoro-6-[1-isopropyl-4-(trifluoromethyl)imidazol-2-yl]-3-pyridyl]methoxy]-5-methoxy- pyrimidine (16 mg, 8.7% yield) as white solid.
1H NMR (400 MHz, DMSO-d
6) δ ppm 8.69 (s, 1H), 8.66 (s, 1H), 8.46 (s, 1H), 8.31 (d, J = 1.2 Hz, 1H), 8.03 (dd, J = 10.4, 1.6 Hz, 1H), 5.57 (s, 2H), 4.79 (dt, J = 13.2, 6.8 Hz, 1H), 3.96 (s, 3H), 3.84 (s, 3H), 1.71 (dq, J = 8.4, 4.0 Hz, 1H), 1.42 (d, J = 6.4 Hz, 6H), 1.01 - 1.06 (m, 2H), 0.86 - 0.91 (m, 2H),
19F NMR (376 MHz, DMSO-d
6) δ ppm -60.73, -120.53; MS (ESI) [M+H]
+ m/z: calcd 560.2, found 560.1. Example 169 (Compound 175)
Synthesis of the starting [5-fluoro-6-[1-isopropyl-4-(trifluoromethyl)imidazol-2-yl]-3- pyridyl]methanol is described for Compound 119. Synthesis of the starting 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-methyl-6- methylsulfonyl-pyrimidine is described for Compound 120. [1491] To a solution of [5-fluoro-6-[1-isopropyl-4-(trifluoromethyl)imidazol-2-yl]-3- pyridyl]methanol (160 mg, 0.528 mmol) in THF (5 mL) was added NaH (25 mg, 0.625 mmol, 60 wt% in mineral oil) at 20°C. The mixture was stirred at 20 °C for 30 minures.2-(4- cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-methyl-6-methylsulfonyl-pyrimidine (170 mg, 0.530 mmol) was added and stirred at 45 °C for 2 hrs. The resulting mixture was quenched by addition of water (20 mL) and extracted with EtOAc (30 mL x 3). The combined organic layer was dried over anhydrous Na
2SO
4, filtered and concentrated under reduced pressure. The residue was purified by preparative HPLC (Instrument: Gilson GX-281 Liquid Handler, Gilson 322 Pump, Gilson 156 UV Detector; Column: 2_Phenomenex Gemini C1875 × 40 mm × 3 μm; Mobile phase A: H
2O with 0.05% NH
3-H
2O + NH
4HCO
3(v%); Mobile phase B: MeCN; Gradient: B from 46% to 76% in 9.5 min, hold 100% B for 2 min; Flow Rate: 30 mL/min; Column Temperature: 30 °C; Wavelength: 220 nm, 254 nm) to 2-(4-cyclopropyl- 6-methoxy-pyrimidin-5-yl)-4-[[5-fluoro-6-[1-isopropyl-4-(trifluoromethyl)imidazol-2-yl]-3- pyridyl]methoxy]-6-methyl-pyrimidine (33.8 mg, 11.8% yield) as yellow solid.
1H NMR (400 MHz, methanol-d
4) δ ppm 8.66 (s, 1H), 8.60 (s, 1H), 8.03 (d, J = 0.8 Hz, 1H), 7.93 (dd, J = 10.4, 1.6 Hz, 1H), 6.91 (s, 1H), 5.63 (s, 2H), 4.76 (dt, J = 13.2, 6.8 Hz, 1H), 3.91 (s, 3H), 2.52 (s, 3H), 1.61 - 1.68 (m, 1H), 1.47 (d, J = 6.8 Hz, 6H), 1.12 - 1.16 (m, 2H), 0.90 - 0.95 (m, 2H),
19F NMR (376 MHz, methanol-d
4) δ ppm -63.87, -122.06; MS (ESI) [M+H]
+ m/z: calcd 544.2, found 544.3. Example 170 (Compound 174)
[1492] To a stirred solution of 2-chloro-4-methyl-6-methylsulfanyl-pyrimidine (1.5 g, 8.59 mmol) and (4-cyclopropyl-6-methoxy-pyrimidin-5-yl)boronic acid I-5 (2.50 g, 12.88 mmol) in 1,4-dioxane (25 mL) and water (5 mL) were added potassium phosphate (5.47 g, 25.77 mmol) and Methanesulfonato(2-dicyclohexylphosphino-2’,6’-di-isopropoxy-1,1’- biphenyl)[2’-(methylamino)-2-biphenylyl]palladium(II) (730.9 mg, 858.86 μmol) at 25 °C under nitrogen atmosphere. The resulting mixture was stirred for 16 hrs at 90 °C under nitrogen atmosphere. The mixture was allowed to cool down to 25 °C. The resulting mixture was diluted with water (50 mL). The resulting mixture was extracted with EtOAc (3 x 10 mL). The combined organic layers were washed with brine (50 mL), dried over anhydrous Na
2SO
4. After filtration, the filtrate was concentrated under reduced pressure. The residue was purified by Prep-TLC (30% EtOAc in PE) to afford 2-(4-cyclopropyl-6-methoxy- pyrimidin-5-yl)-4-methyl-6-methylsulfanyl-pyrimidine (485 mg, 1.68 mmol, 19% yield) as a yellow solid. MS: m/z = 289.10 [M + H]
+.
1H NMR (400 MHz, Chloroform-d) δ 8.65 (s, 1H), 7.04 (s, 1H), 3.95 (s, 3H), 2.60 (s, 3H), 2.54 (s, 3H), 1.78 - 1.70 (m, 1H), 1.30 - 1.16 (m, 2H), 1.00 - 0.89 (m, 2H). 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-methyl-6-methylsulfonyl-pyrimidine [1493] To a stirred solution of 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-methyl-6- methylsulfanyl-pyrimidine (485 mg, 1.68 mmol) in DCM (5 mL) was added 3- chlorobenzenecarboperoxoic acid (870.7 mg, 5.05 mmol) at 25 °C under nitrogen atmosphere. The resulting mixture was stirred for 16 hrs at 25 °C under nitrogen atmosphere. The resulting mixture was directly concentrated under reduced pressure. The residue was purified by Prep-TLC (50% EtOAc in PE) to afford 2-(4-cyclopropyl-6-methoxy-pyrimidin- 5-yl)-4-methyl-6-methylsulfonyl-pyrimidine (360 mg, 1.12 mmol, 66 % yield) as a white solid. MS: m/z = 320.90 [M + H]
+. 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-methyl-6-[[4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidine [1494] To a stirred solution of [4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methanol I-2b (345.51 mg, 1.35 mmol) in DMF (5 mL) was added NaH (53.94 mg, 1.35 mmol, 60% purity) at 0 °C under nitrogen atmosphere. After 30 min, 2-(4- cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-methyl-6-methylsulfonyl-pyrimidine (360 mg, 1.12 mmol) was added at 0 °C. The resulting mixture was stirred for 16 hrs at 25 °C under nitrogen atmosphere. The reaction was quenched by the addition of saturated NH
4Cl aqueous solution (15 mL) at 0 °C. The mixture was diluted with EtOAc (200 mL) and washed with brine (3 x 100 mL), the organic layer was dried over anhydrous Na
2SO
4. After filtration, the filtrate was concentrated under reduced pressure. The residue was subject to silica gel column chromatography, eluted with 0 - 100% EtOAc in PE, to afford a residue. The residue was purified by RP-Flash with the following conditions: Column: C18 spherical Column, 20-35 um, 100A, 80 g; Mobile Phase A: Water (5 mM aq. NH
4HCO
3), Mobile Phase B: MeCN; Flow rate: 50 mL/min; Gradient: 5% B to 5% B in 5 min, 5% B to 60% B in 15 min, 60% B to 60% B in 1 min; Detector: UV 254 & 210 nm. The collected fractions were combined and lyophilized to give 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-methyl-6-[[4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidine (401.9 mg, 809.49 μmol, 72% yield) as a yellow solid. MS: m/z = 497.30 [M + H]
+.
1H NMR (400 MHz, DMSO-d
6) δ 8.67 (s, 1H), 7.95 (s, 1H), 7.74 (d, J = 8.0 Hz, 2H), 7.58 (d, J = 8.0 Hz, 2H), 6.94 (s, 1H), 5.49 (s, 2H), 3.86 (s, 3H), 3.79 (s, 3H), 2.47 (s, 3H), 1.70 - 1.63 (m, 1H), 1.06 - 1.02 (m, 2H), 0.91 - 0.86 (m, 2H).
19F NMR (377 MHz, DMSO-d
6) δ -60.82.
[1495] To a stirred solution of [4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methanol I-2b (1.00 g, 3.90 mmol) in THF (15 mL) was added NaH (156.13 mg, 3.90 mmol, 60% dispersion in mineral oil) at 0 °C under nitrogen atmosphere. After stirred at 0 °C for 1 hour, a solution of 2,4-dichloropyrimidin-5-ol (322 mg, 1.95 mmol) in THF (2 mL) was added dropwise with stirring at 0 °C. The resulting mixture was stirred at 25 °C for 16 hrs. The reaction was quenched by the addition of saturated NH
4Cl aqueous solution (100 mL) at 0 °C. The resulting mixture was extracted with EtOAc (3 x 80 mL). The combined organic layers were dried over anhydrous Na
2SO
4. After filtration, the filtrate was concentrated under reduced pressure. The residue was purified by silica gel column chromatography, eluted with 59% EtOAc in PE to give 2-chloro-4-[[4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidin-5-ol (644 mg, 1.67 mmol, 85% yield) as a light yellow solid. MS: m/z = 385.05, 387.05 [M + H]
+.
1H NMR (400 MHz, DMSO-d
6) δ 10.38 (br., 1H), 7.99 (s, 1H), 7.95 (s, 1H), 7.77 (d, J = 8.0 Hz, 2H), 7.64 (d, J = 8.0 Hz, 2H), 5.50 (s, 2H), 3.81 (s, 3H). 2-chloro-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]-5- (trideuteriomethoxy)pyrimidine [1496] To a mixture of 2-chloro-4-[[4-[4-(1,1-difluoroethyl)-1-methyl-imidazol-2- yl]phenyl]methoxy]pyrimidin-5-ol 171b (644 mg, 1.69 mmol), trimethylphosphane (5.07 mL, 1 M in THF) and methanol-d
4 (183.00 mg, 5.07 mmol, 205.6 μL) in THF (20 mL) under nitrogen atmosphere was added diisopropyl (E)-diazene-1,2-dicarboxylate (1.03 g, 5.07 mmol) dropwise with stirring at 0 °C. The resulting mixture was stirred at 25 °C for 16 hrs. The resulted mixture was concentrated under reduced pressure. The residue was purified by silica gel column chromatography, eluted with 53% EtOAc in PE to give 2-chloro-4-[[4-[1- methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]-5-(trideuteriomethoxy)pyrimidine (604 mg, 1.50 mmol, 88% yield) as an off-white solid. MS: m/z = 402.10, 404.10 [M + H]
+.
1H NMR (400 MHz, Chloroform-d) δ 7.94 (s, 1H), 7.70 (d, J = 8.4 Hz, 2H), 7.62 (d, J = 8.0 Hz, 2H), 7.36 (s, 1H), 5.57 (s, 2H), 3.81 (s, 3H).
19F NMR (376 MHz, Chloroform-d) δ - 62.63. 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]-5-(trideuteriomethoxy)pyrimidine [1497] To a mixture of 2-chloro-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]-5-(trideuteriomethoxy)pyrimidine 171a (300 mg, 746.67 μmol) and (4- cyclopropyl-6-methoxy-pyrimidin-5-yl)boronic acid I-5 (217.3 mg, 1.12 mmol) in THF (5 mL) and water (1 mL) under nitrogen atmosphere were added dichloro[1,1'- bis(dicyclohexylphosphino)ferrocene]palladium (II) (56.4 mg, 74.67 μmol) and potassium phosphate (475.5 mg, 2.24 mmol) at 25 °C. The resulting mixture was stirred at 40 °C for 16 hrs then cooled to room temperature and filtered. The filtrate was concentrated under reduced pressure. The residue was subject to silica gel column chromatography, eluted with 90% EtOAc in PE, to give the crude product. The crude product was purified by RP-Flash with the following conditions: Column: C18 spherical, 20-30 um, 100A, 40 g; Mobile Phase A: Water (5 mM aq. NH
4HCO
3), Mobile Phase B: MeCN; Flow rate: 40 mL/min; Gradient: 2% B to 2% B in 5 min, 2% B to 56% B in 17 min, 56% B to 56% B in 5 min, 56% B to 95% B in 5 min; Detector: UV 254 & 210 nm. The collected fractions were combined, concentrated and then lyophilized to give 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]-5-(trideuteriomethoxy)pyrimidine (251.6 mg, 488.07 μmol, 65% yield) as a white solid. MS: m/z = 516.15 [M + H]
+.
1H NMR (400 MHz, Chloroform-d) δ 8.67 (s, 1H), 8.26 (s, 1H), 7.66 (d, J = 8.4 Hz, 2H), 7.60 (d, J = 8.0 Hz, 2H), 7.34 (s, 1H), 5.60 (s, 2H), 3.96 (s, 3H), 3.79 (s, 3H), 1.78 - 1.71 (m, 1H), 1.27 - 1.23 (m, 2H), 0.94 - 0.88 (m, 2H).
19F NMR (376 MHz, Chloroform-d) δ -62.77.
5-bromo-2-chloro-4-[[3-fluoro-4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidine Synthesis of the starting [3-fluoro-4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methanol is described for compound 97. [1498] 5-Bromo-2,4-dichloro-pyrimidine (1.28 g, 5.61 mmol) was added to a stirred mixture of [3-fluoro-4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methanol (1.00 g, 3.65 mmol) and Cs
2CO
3 (2.19 g, 6.73 mmol) in ACN (9.5 mL). The reaction mixture was stirred at 65 °C for 12 hr. The reaction mixture was cooled to room temperature and concentrated under reduced pressure. The residue was diluted with water (30 mL) and extracted with DCM (40 mL). The organic layer was separated, washed with brine (30 mL), dried over anhydrous Na
2SO
4 and concentrated under reduced pressure. The residue was subjected to flash column chromatography (SiO
2, gradient hexane - EtOAc) to afford 5-bromo-2-chloro-4-[[3-fluoro-4- [1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidine (1.10 g, 2.36 mmol, 42.1% yield) as a white solid. MS (ESI): [M+H]
+ m/z: calcd 466.97, 464.97; found 467.0, 465.0 1-[2-chloro-4-[[3-fluoro-4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidin-5-yl]ethanol [1499] A solution of 5-Bromo-2-chloro-4-[[3-fluoro-4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidine (350 mg, 752 μmol) in THF (5 mL) was cooled to -90 °C. n-Butyl lithium (500 µL, 1.20 mmol, 2.4 M solution in hexane) was added dropwise. The mixture was stirred for 5 min at -90 °C then acetaldehyde (133 mg, 3.01 mmol, 170 μL) was added. The mixture was stirred at -90 °C for 5 min. The mixture was quenched with water (500 µL). The reaction mixture was allowed to warm to the room temperature, diluted with water (5 mL) and extracted with EtOAc (10 mL). The organic layer was separated, dried over anhydrous Na
2SO
4 and concentrated under reduced pressure to afford 1-[2-chloro-4-[[3-fluoro-4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidin-5-yl]ethanol (150 mg, 348 μmol, 46.3% yield) as a yellow oil which was used in the next step without further purification. MS (ESI): [M+H]
+ m/z: calcd 431.09; found 431.0 1-[2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[3-fluoro-4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidin-5-yl]ethanol [1500] 1-[2-Chloro-4-[[3-fluoro-4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidin-5-yl]ethanol (150 mg, 348 μmol), (4-cyclopropyl-6-methoxy- pyrimidin-5-yl)boronic acid I-5 (135 mg, 696 µmol), potassium phosphate tribasic (222 mg, 1.05 mmol) and RuPhos Pd G3 (14.8 mg, 17.4 μmol) were mixed in degassed mixture of dioxane (5 mL) and water (500 µL) under argon atmosphere. The reaction mixture was stirred at room temperature for 20 hr. Additional portions of (4-cyclopropyl-6-methoxy- pyrimidin-5-yl)boronic acid I-5 (135 mg, 696 µmol) and RuPhos Pd G3 (14.8 mg, 17.4 μmol) were added to the reaction mixture. The resulting mixture was stirred at 70°C for 12 hr. The reaction mixture was cooled to room temperature, dried over anhydrous Na
2SO
4 and filtered. The filtrate was subjected to HPLC (0-2-10 min., 30-60% water – ACN, +0.1% vol. of 25% aq. NH
3, flow: 30 mL/min, column: Chromatorex C18 SMB100-5T 100×19 mm, 5 µm) to afford 1-[2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[3-fluoro-4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidin-5-yl]ethanol (19.0 mg, 34.9 μmol, 10% yield) as a yellow oil. MS (ESI): [M+H]
+ m/z: calcd 545.22; found 545.2 rel-(S)-1-(4'-cyclopropyl-4-((3-fluoro-4-(1-methyl-4-(trifluoromethyl)-1H-imidazol-2- yl)benzyl)oxy)-6'-methoxy-[2,5'-bipyrimidin]-5-yl)ethanol (170) and rel-(R)-1-(4'-cyclopropyl-4-((3-fluoro-4-(1-methyl-4-(trifluoromethyl)-1H-imidazol-2- yl)benzyl)oxy)-6'-methoxy-[2,5'-bipyrimidin]-5-yl)ethanol (92) [1501] 1-[2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[3-fluoro-4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidin-5-yl]ethanol (15.0 mg, 27.6 μmol) was subjected to chiral HPLC (Injection Volume: 500 µL, Column: CHIRALCEL OD-H (250×20 mm, 5 µm)-I, Mobile Phase: Hexane:IPA:MeOH, 90:5:5, flow rate: 15 mL/min) to afford rel-(S)-1-(4'-cyclopropyl-4-((3-fluoro-4-(1-methyl-4-(trifluoromethyl)-1H-imidazol-2- yl)benzyl)oxy)-6'-methoxy-[2,5'-bipyrimidin]-5-yl)ethanol (6.00 mg, 11.0 μmol, 40.0% yield) and rel-(R)-1-(4'-cyclopropyl-4-((3-fluoro-4-(1-methyl-4-(trifluoromethyl)-1H- imidazol-2-yl)benzyl)oxy)-6'-methoxy-[2,5'-bipyrimidin]-5-yl)ethanol (6.00 mg, 11.0 μmol, 40.0% yield) as yellow oils. rel-(S)-1-(4'-cyclopropyl-4-((3-fluoro-4-(1-methyl-4-(trifluoromethyl)-1H-imidazol- 2-yl)benzyl)oxy)-6'-methoxy-[2,5'-bipyrimidin]-5-yl)ethanol (170): 1H NMR (600 MHz, DMSO-d
6) δ 0.85 – 0.90 (m, 2H), 1.01 – 1.06 (m, 2H), 1.45 (d, 3H), 1.63 – 1.69 (m, 1H), 3.61 (s, 3H), 3.84 (s, 3H), 4.98 – 5.05 (m, 1H), 5.46 (d, 1H), 5.55 (s, 2H), 7.44 (d, 1H), 7.50 (d, 1H), 7.62 (t, 1H), 8.01 (s, 1H), 8.67 (s, 1H), 8.74 (s, 1H). MS (ESI): [M+H]
+ m/z: calcd 545.22; found 545.0 Enantiopurity: >99% (column: Chiralcel OD-H, 250 × 4.6 mm, 5 μm; mobile phase: Hexane-IPA-MeOH, 70-15-15; flow: 0.6 mL/min; RT=7.5 min). rel-(R)-1-(4'-cyclopropyl-4-((3-fluoro-4-(1-methyl-4-(trifluoromethyl)-1H-imidazol- 2-yl)benzyl)oxy)-6'-methoxy-[2,5'-bipyrimidin]-5-yl)ethanol (92): 1H NMR (600 MHz, DMSO-d
6) δ 0.85 – 0.90 (m, 2H), 1.01 – 1.06 (m, 2H), 1.45 (d, 3H), 1.63 – 1.69 (m, 1H), 3.61 (s, 3H), 3.84 (s, 3H), 4.98 – 5.05 (m, 1H), 5.46 (d, 1H), 5.55 (s, 2H), 7.44 (d, 1H), 7.50 (d, 1H), 7.62 (t, 1H), 8.01 (s, 1H), 8.67 (s, 1H), 8.74 (s, 1H). MS (ESI): [M+H]
+ m/z: calcd 545.22; found 545.0 Enantiopurity: >99% (column: Chiralcel OD-H, 250 × 4.6 mm, 5 μm; mobile phase: Hexane-IPA-MeOH, 70-15-15; flow: 0.6 mL/min; RT=9.3 min). Example 173 (Compound 169)
2,4-dichloro-5-(difluoromethoxy)pyrimidine Synthesis of the starting 2,4-dichloropyrimidin-5-ol is described for Compound 122 [1502] 2,2-Difluoro-2-fluorosulfonyl-acetic acid (533 mg, 2.99 mmol, 310 μL) and K2CO3 (1.09 g, 4.74 mmol) were added to a stirred solution of 2,4-dichloropyrimidin-5-ol (380 mg, 2.30 mmol) in anhydrous acetonitrile (5.0 mL). The reaction mixture was stirred at room temperature for 12 hr under nitrogen atmosphere. The reaction mixture was quenched by addition of water (10 mL) and extracted with EtOAc (30mL). The organic layer was separated, dried over anhydrous Na
2SO
4 and concentrated under reduced pressure to afford 2,4-dichloro-5-(difluoromethoxy)pyrimidine (222 mg, 1.03 mmol, 44.8% yield) as a light- yellow solid which was used in the next step without further purification. 2-chloro-5-(difluoromethoxy)-4-[[4-[1-isopropyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidine The synthesis of the starting [4-[1-isopropyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methanol is described for Compound 37. [1503] [4-[1-isopropyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methanol (150 mg, 528 μmol), 2,4-dichloro-5-(difluoromethoxy)pyrimidine (227 mg, 1.06 mmol) and Cs
2CO
3 (344 mg, 1.06 mmol) were mixed in ACN (3 mL). The reaction mixture was stirred at 65 °C for 2 hr. then at 70 °C for 12 hr. The reaction mixture was cooled to room temperature, diluted with MTBE (30 mL) and washed with water (15 mL). The organic layer was separated, washed with brine (20 mL), dried over anhydrous Na
2SO
4 and concentrated under reduced pressure. The residue was subjected to flash column chromatography (SiO
2, gradient hexane- EtOAc) to afford 2-chloro-5-(difluoromethoxy)-4-[[4-[1-isopropyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidine (40.0 mg, 86.4 μmol, 16.4% yield) as a white solid. MS (ESI): [M+H]
+ m/z: calcd 463.1; found 463.0 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-5-(difluoromethoxy)-4-[[4-[1-isopropyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidine [1504] 2-Chloro-5-(difluoromethoxy)-4-[[4-[1-isopropyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidine (40.0 mg, 86.4 μmol), (4-cyclopropyl-6-methoxy-pyrimidin- 5-yl)boronic acid I-5 (50.3 mg, 259 μmol), potassium phosphate tribasic anhydrous (55.0 mg, 259 μmol), RuPhos Pd G3 (5.1 mg, 6.05 μmol) were mixed in a degassed mixture of dioxane (450 µL) and water (50 µL) under argon atmosphere. The reaction mixture was stirred at room temperature for 24 hr and then at 70 °C for 12 hr. SiliaMetS
® Dimercaptotriazine (100 mg) was added to the reaction mixture. The resulting mixture was stirred for 3 hr. The mixture was diluted with MTBE (5.0 mL) and filtered through a thin pad of silica gel. The filtrate was concentrated under reduced pressure. The residue was subjected to HPLC (0-2-10 min., 43-50-60% water – ACN, +0.1% vol. of 25% aq. NH
3, flow: 30 mL/min, column: Chromatorex C18 SMB100-5T 100×19 mm, 5 µm, and then 0-2-10 min., 43-50-60% water – ACN, +0.1% vol. of 25% aq. NH
3, flow: 30 mL/min, column: Chromatorex C18 SMB100-5T 100×19 mm, 5 µm) to afford 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-5- (difluoromethoxy)-4-[[4-[1-isopropyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidine (4.00 mg, 6.94 μmol, 8% yield) as a white solid.
1H NMR (600 MHz, DMSO-d
6) δ 0.86 – 0.91 (m, 2H), 1.03 – 1.07 (m, 2H), 1.41 (d, 6H), 1.71 – 1.78 (m, 1H), 3.86 (s, 3H), 4.45 – 4.51 (m, 1H), 5.58 (s, 2H), 7.37 (t, 1H, CHF
2), 7.61 (dd, 4H), 8.19 (s, 1H), 8.68 (s, 1H), 8.70 (s, 1H). MS (ESI): [M+H]
+ m/z: calcd 577.23; found 577.2. Example 174 (Compound 166)
2-chloro-5-(difluoromethyl)-4-methylsulfanyl-pyrimidine [1505] An aqueous solution of sodium methanethiolate (440 mg, 21% wt.) was added to a stirred solution of 2,4-dichloro-5-(difluoromethyl)pyrimidine (250 mg, 1.26 mmol) in THF (5 mL) at 5 °C. The reaction mixture was stirred at room temperature for 16 hr. The reaction mixture was concentrated under reduced pressure. The residue was diluted with MTBE (15 mL) and washed with water (5 mL) and brine (5 mL), dried over anhydrous Na
2SO
4 and filtered. The filtrate was concentrated under reduced pressure to afford 2-chloro-5- (difluoromethyl)-4-methylsulfanyl-pyrimidine (240 mg, 1.14 mmol, 90.6% yield) as a yellow liquid which was used in the next steps without further purification.
1H NMR (500 MHz, CDCl
3) δ 2.66 (s, 3H), 6.73 (t, 1H, CHF
2), 8.45 (s, 1H). MS: [M+H]
+ m/z: calcd 211.00; found 211.0 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-5-(difluoromethyl)-4-methylsulfanyl-pyrimidine [1506] (4-cyclopropyl-6-methoxy-pyrimidin-5-yl)boronic acid I-5 (212 mg, 1.09 mmol) and potassium phosphate tribasic (696 mg, 3.28 mmol) was added to a stirred solution of 2- chloro-5-(difluoromethyl)-4-methylsulfanyl-pyrimidine (230 mg, 1.09 mmol) in a degassed mixture dioxane (8 mL) and water (2 mL) under argon atmosphere. To the resulting mixture RuPhos Pd G4 (46.4 mg, 54.6 μmol) was added. The resulting mixture was stirred at 90 °C for 16 hr. under argon atmosphere. The reaction mixture was cooled to room temperature and concentrated under reduced pressure. The residue was diluted with water (10 mL) and extracted with EtOAc (35 mL). The organic layer was separated, washed with water (5.0 mL) and brine (5.0 mL), dried over anhydrous Na
2SO
4 and filtered. The filtrate was concentrated under reduced pressure to afford 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-5- (difluoromethyl)-4-methylsulfanyl-pyrimidine (290 mg, 894 μmol, 81.9% yield) as an orange gum which was used in the next steps without further purification. MS (ESI): [M+H]
+ m/z: calcd 325.1; found 325.0 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-5-(difluoromethyl)-4-methylsulfonyl-pyrimidine [1507] mCPBA (613 mg, 3.02 mmol, 85% purity) was added to a stirred solution of 2-(4- cyclopropyl-6-methoxy-pyrimidin-5-yl)-5-(difluoromethyl)-4-methylsulfanyl-pyrimidine (326 mg, 1.01 mmol) in DCM (15 mL). The reaction mixture was stirred at room temperature for 16 hr. The reaction mixture was washed with a saturated aqueous solution NaHCO
3 (15 mL). The organic layer was washed with water (5 mL), brine (5 mL), dried over anhydrous Na
2SO
4 and filtered. The filtrate was concentrated under reduced pressure. The residue was subjected to column chromatography (SiO
2, gradient CHCl
3 - MeOH) to afford 2-(4- cyclopropyl-6-methoxy-pyrimidin-5-yl)-5-(difluoromethyl)-4-methylsulfonyl-pyrimidine (140 mg, 393 μmol, 31.2% yield) as a light-yellow solid. MS (ESI): [M+H]
+ m/z: calcd 357.09; found 357.0 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-5-(difluoromethyl)-4-[[4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidine [1508] NaH (17.3 mg, 433 μmol, 60% dispersion in mineral oil) was added to a stirred solution of [4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methanol I-2b (101 mg, 393 μmol) in DMF (5 mL). The resulting mixture was stirred at room temperature for 30 min. 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-5-(difluoromethyl)-4-methylsulfonyl- pyrimidine (140 mg, 393 μmol) was added to the mixture. The reaction mixture was stirred at room temperature for 16 hr. The reaction mixture was diluted with a saturated aqueous solution of NH
4Cl (5 mL) and extracted with EtOAc (3×20 mL). The combined organic layers were washed with brine (5 mL), dried over anhydrous Na
2SO
4 and filtered. The filtrate was concentrated under reduced pressure. The residue was subjected to HPLC (0-5 min, 40- 65% water+FA (0.1% vol.) - ACN+FA (0.1% vol.); flow: 30 mL/min, column: Chromatorex 18 SMB100-5T, 100 x 19 mm, 5 µm) to afford 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)- 5-(difluoromethyl)-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidine (17.2 mg, 32.3 μmol, 8.2% yield) as an off-white solid.
1H NMR (500 MHz, DMSO-d
6) δ 0.84 – 0.90 (m, 2H), 1.01 – 1.06 (m, 2H), 1.69 – 1.75 (m, 1H), 3.77 (s, 3H), 3.84 (s, 3H), 5.60 (s, 2H), 7.25 (t, 1H, CHF
2), 7.58 (d, 2H), 7.73 (d, 2H), 7.93 (s, 1H), 8.68 (s, 1H), 8.93 (s, 1H). MS (ESI): [M+H]
+ m/z: calcd 533.20; found 533.2. Example 175 (Compound 163)

5-chloro-2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[6-[1-cyclopropyl-4- (trifluoromethyl)imidazol-2-yl]-5-fluoro-3-pyridyl]methoxy]pyrimidine Synthesis of the starting 5-chloro-2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4- methylsulfonyl-pyrimidine is described for Compound 123. Synthesis of the starting [6-[1-cyclopropyl-4-(trifluoromethyl)imidazol-2-yl]-5-fluoro- 3-pyridyl]methanol is described for Compound 43. [1509] Potassium tert-butoxide (28.0 mg, 249 μmol) was added to a stirred solution of [6-[1- cyclopropyl-4-(trifluoromethyl)imidazol-2-yl]-5-fluoro-3-pyridyl]methanol (50.0 mg, 166 μmol) and 5-chloro-2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-methylsulfonyl- pyrimidine (56.6 mg, 156 μmol) in THF (1 mL) at 0 °C. The reaction mixture was stirred at 0 °C for 1 hr. The reaction was quenched by addition of water (0.1 mL). The resulting mixture was directly subjected to HPLC (0-2-9 min., 43-50-65% water – ACN, +0.1% vol. of 25% aq. NH
3, flow: 30 mL/min, column: XBridge BEH 100×19 mm, 5 µm) to afford 5-chloro-2- (4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[6-[1-cyclopropyl-4-(trifluoromethyl)imidazol- 2-yl]-5-fluoro-3-pyridyl]methoxy]pyrimidine (30.0 mg, 53.4 μmol, 32.2% yield) as a yellow solid.
1H NMR (500 MHz, DMSO-d
6) δ 0.84 – 0.94 (m, 6H), 1.04 – 1.08 (m, 2H), 1.78 – 1.84 (m, 1H), 3.74 – 3.80 (m, 1H), 3.86 (s, 3H), 5.68 (s, 2H), 8.04 – 8.09 (m, 2H), 8.69 (s, 1H), 8.73 (s, 1H), 8.92 (s, 1H). MS (ESI): [M+H]
+ m/z: calcd 562.16; found 562.0. Example 176 (Compound 162)
oxetan-3-yl trifluoromethanesulfonate [1510] Trifluoromethanesulfonic anhydride (2.00 g, 7.09 mmol, 1.19 mL) was added to the mixture of oxetan-3-ol (500 mg, 6.75 mmol, 429 μL) and pyridine (1.07 g, 13.5 mmol, 1.09 mL) in DCM (14.5 mL) at -20 °C. The reaction mixture was stirred at room temperature for 12 hr. The reaction mixture was poured into ice cold water. The organic layer was separated, dried over anhydrous Na
2SO
4 and concentrated under reduced pressure to afford oxetan-3-yl trifluoromethanesulfonate (1.00 g, crude, contaminated with pyridine) as a yellow liquid which was used in the nest step without further purification.
1H NMR (400 MHz, CDCl
3) δ 4.81 – 4.95 (m, 4H), 5.67 – 5.74 (m, 1H). 2-chloro-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]-5-(oxetan-3- yloxy)pyrimidine Synthesis of the starting 2-chloro-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidin-5-ol is described for Compound 122. [1511] Oxetan-3-yl trifluoromethanesulfonate (557 mg, crude from previous step) was added to a stirred mixture of 2-chloro-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidin-5-ol (130 mg, 338 μmol) and Cs
2CO
3 (221 mg, 676 μmol) in DMF (10 mL). The resulting mixture was stirred at 40°C for 12 hr. The reaction mixture was cooled to room temperature, poured into water (25 mL) and extracted with MTBE (3×15 mL). The combined organic layers were washed with water (2×10 mL) and brine (2×5 mL), dried over anhydrous Na
2SO
4 and concentrated under reduced pressure. The residue was subjected to column chromatography (SiO
2, Hexanes - EtOAc, gradient from 50% to 80% EtOAc) to afford 2-chloro-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]-5-(oxetan-3-yloxy)pyrimidine (85.0 mg, 193 μmol, 57.1% yield) as a white solid. MS (ESI): [M+H]
+ m/z: calcd 441.10; found 441.0 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]-5-(oxetan-3-yloxy)pyrimidine [1512] 2-Сhloro-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]-5- (oxetan-3-yloxy)pyrimidine (66.0 mg, 150 μmol), (4-cyclopropyl-6-methoxy-pyrimidin-5- yl)boronic acid I-5 (87.1 mg, 449 μmol), potassium phosphate tribasic (191 mg, 898 μmol) and bis(diphenylphosphino)ferrocene]dichloropalladium(II)-DCM (24.4 mg, 30.0 μmol) were mixed in a degassed dioxane (2 mL). The resulting mixture was degassed by three cycles of vacuo and argon replacement. Then water (200 µL) was added and the reaction mixture was stirred at room temperature for 24 hr. The reaction mixture was diluted with EtOAc (5 mL). Anhydrous Na
2SO
4 (1 g) and SiliaMetS
® Dimercaptotriazine (400 mg) were added to the mixture. The resulting mixture was stirred at room temperature for 12 hr. Then solids were filtered out and washed with EtOAc (2 mL). The resulting organic solution was filtered through a thin pad of silica gel. The filtrate was concentrated under reduced pressure. The residue was subjected to HPLC (0.5-6.5 min., 25-35-55% water - ACN, +0.1% vol. of 25% aq. NH
3, flow: 30 mL/min, column: Chromatorex C18 Phenil 100×19 mm, 5 µm), then subjected to SFC repurification (Chromatorex EP-2 (19×100, 5µm), IPA-CO
23:7) to afford 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]-5-(oxetan-3-yloxy)pyrimidine (8.0 mg, 14.4 μmol, 9.6% yield) as a white solid.
1H NMR (600 MHz, DMSO-d
6) δ 0.84 – 0.88 (m, 2H), 1.01 – 1.04 (m, 2H), 1.69 – 1.75 (m, 1H), 3.79 (s, 3H), 3.84 (s, 3H), 4.64 (dd, 2H), 4.94 (t, 2H), 5.43 – 5.48 (m, 1H), 5.54 (s, 2H), 7.60 (d, 2H), 7.75 (d, 2H), 7.95 (s, 1H), 8.14 (s, 1H), 8.65 (s, 1H). MS (ESI): [M+H]
+ m/z: calcd 555.22; found 555.0. Example 177 (Compound 161)
Compound 161 4-[4-(trifluoromethyl)-1-vinyl-imidazol-2-yl]benzonitrile [1513] K
2CO
3, anhydrous (4.66 g, 33.7 mmol), 4-[4-(trifluoromethyl)-1H-imidazol-2- yl]benzonitrile I-1a (2.00 g, 8.43 mmol) and 1,2-dibromoethane (7.92 g, 42.2 mmol, 3.63 mL) were mixed in DMF (10 mL). The resulting mixture was stirred at 50 °C for 14 hr. The reaction mixture was cooled to room temperature, diluted with water (10 mL) and extracted with EtOAc (2×20 mL). The combined organic layers were separated, washed with water (20 mL) and brine (20 mL), dried over anhydrous Na
2SO
4 and filtered. The filtrate was concentrated under reduced pressure to afford 4-[4-(trifluoromethyl)-1-vinyl-imidazol-2- yl]benzonitrile (1.70 g, 6.46 mmol, 76.6% yield) as a yellow oil which was used in the next step without further purification. MS (ESI): [M+H]
+ m/z: calcd 264.07; found 264.0 4-[1-(2,2-difluorocyclopropyl)-4-(trifluoromethyl)imidazol-2-yl]benzonitrile [1514] Sodium iodide (1.71 mg, 11.4 mmol) was added to a stirred solution of 4-[4- (trifluoromethyl)-1-vinyl-imidazol-2-yl]benzonitrile (2.00 g, 7.60 mmol) in THF (3.0 mL) at 65 °C under argon atmosphere. Trimethyl(trifluoromethyl)silane (4.32 g, 30.4 mmol, 4.82 mL) was added to the mixture at room temperature. The reaction mixture was stirred at room temperature for 15 hr. The reaction was diluted with water (20 mL) and extracted with EtOAc (2×30 mL). The combined organic layers were separated, dried over anhydrous Na
2SO
4 and filtered. The filtrate was concentrated under reduced pressure. The residue was subjected to flash column chromatography (SiO
2, gradient Hexane – EtOAc) to afford 4-[1-(2,2- difluorocyclopropyl)-4-(trifluoromethyl)imidazol-2-yl]benzonitrile (1.19 g, 3.80 mmol, 100% yield) as a light yellow solid.
1H NMR (400 MHz, CDCl
3) δ 1.73 – 1.84 (m, 1H), 2.10 – 2.22 (m, 1H), 4.02 – 4.11 (m, 1H), 7.43 (s, 1H), 7.76 (d, 2H), 7.90 (d, 2H). MS (ESI): [M+H]
+ m/z: calcd 314.07; found 314.0 4-[1-(2,2-difluorocyclopropyl)-4-(trifluoromethyl)imidazol-2-yl]benzaldehyde [1515] Diisobutylaluminum hydride (1 M in THF, 3.67 mmol, 3.67 mL) was added dropwise to a precooled to -30 °C solution of 4-[1-(2,2-difluorocyclopropyl)-4- (trifluoromethyl)imidazol-2-yl]benzonitrile (1.00 g, 3.19 mmol) in DCM (10 mL). The reaction mixture was stirred at ambient temperature for 18 hr. The reaction mixture was quenched by dropwise addition of aqueous citric acid (3 mL, 10% wt.). The resulting suspension was diluted with CHCl
3 (10 mL) and filtered through a thin pad of celite. The filtrate was concentrated under reduced pressure to afford 4-[1-(2,2-difluorocyclopropyl)-4- (trifluoromethyl)imidazol-2-yl]benzaldehyde (860 mg, 2.72 mmol, 85.2% yield) as light yellow oil which was used in the next step without further purification.
1H NMR (400 MHz, CDCl
3) δ 1.68 – 1.77 (m, 1H), 2.06 – 2.16 (m, 1H), 4.04 – 4.14 (m, 1H), 7.41 (s, 1H), 7.90 (d, 2H), 7.98 (d, 2H), 10.05 (s, 1H). MS (ESI): [M+H]
+ m/z: calcd 317.07; found 317.1 [4-[1-(2,2-difluorocyclopropyl)-4-(trifluoromethyl)imidazol-2-yl]phenyl]methanol [1516] Sodium Borohydride (115 mg, 3.04 mmol) was added to a precooled to 0 °C solution of 4-[1-(2,2-difluorocyclopropyl)-4-(trifluoromethyl)imidazol-2-yl]benzaldehyde (960 mg, 3.04 mmol) in MeOH (25 mL). The reaction mixture was stirred at room temperature for 18 hr. The reaction mixture was quenched with water (30 mL) and an aqueous 5 N NaOH (10 mL). The resulting suspension was extracted with CHCl
3 (2 × 30 mL). The combined organic layers were washed with water (20 mL), dried over anhydrous Na
2SO
4 and concentrated under reduced pressure to afford [4-[1-(2,2-difluorocyclopropyl)-4-(trifluoromethyl)imidazol- 2-yl]phenyl]methanol (760 mg, 2.39 mmol, 78.7% yield) as a light yellow oil which was used in the next step without further purification. MS (ESI): [M+H]
+ m/z: calcd 319.09; found 319.1 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[4-[1-(2,2-difluorocyclopropyl)-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]-5-methoxy-pyrimidine [1517] NaH (20.7 mg, 518 μmol, 60% dispersion in mineral oil) was suspended in THF (5 mL) at 0 °C. [4-[1-(2,2-Difluorocyclopropyl)-4-(trifluoromethyl)imidazol-2- yl]phenyl]methanol (150 mg, 471 μmol) was added to the suspension at the same temperature followed by 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-5-methoxy-4-methylsulfonyl- pyrimidine I-6 (159 mg, 47 μmol). The reaction mixture was stirred at 50 °C for 12 hr. The reaction mixture was cooled to room temperature and concentrated under reduced pressure. The residue was subjected to HPLC (0-2-10 min., 30-40-65% water – ACN, flow: 30 mL/min, column: Chromatorex C18 SMB100-5T, 100×19 mm, 5 µm) to afford 2-(4- cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[4-[1-(2,2-difluorocyclopropyl)-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]-5-methoxy-pyrimidine (78.0 mg, 136 μmol, 28.8% yield) as a light-yellow solid.
1H NMR (600 MHz, DMSO-d
6) δ 0.85 – 0.91 (m, 2H), 1.00 – 1.06 (m, 2H), 1.70 – 1.76 (m, 1H), 2.19 – 2.27 (m, 1H), 2.31 – 2.39 (m, 1H), 3.85 (s, 3H), 3.95 (s, 3H), 4.72 – 4.79 (m, 1H), 5.51 (s, 2H), 7.60 (d, 2H), 7.83 (d, 2H), 8.10 (s, 1H), 8.44 (s, 1H), 8.65 (s, 1H). MS (ESI): [M+H]
+ m/z: calcd 575.21; found 575.0. Example 178 (Compound 157)
4-cyclopropyl-6-methoxy-5-[4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidin-2-yl]pyrimidine [1518] To a stirred solution of [4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methanol I-2b (75.3 mg, 294 μmol) in DMF (5 mL) NaH (13.5 mg, 338 μmol, 60% dispersion in mineral oil) was added. The resulting mixture was stirred for 30 minutes at room temperature. To the obtained mixture 4-cyclopropyl-6-methoxy-5-(4- methylsulfonylpyrimidin-2-yl)pyrimidine I-8 (90.0 mg, 294 μmol) was added and the obtained reaction mixture was stirred at room temperature for 16 hrs. The reaction mixture was quenched with saturated aqueous NH
4Cl solution (20 mL). The obtained mixture was extracted with EtOAc (3 × 20 mL). The combined organic layers were washed with brine (20 mL), dried over anhydrous Na
2SO
4, filtered and concentrated under reduced pressure. The residue was submitted to HPLC (0 - 5 min., 30 - 60% water – acetonitrile, +0.1% vol. of 25% aq. NH
3, flow rate: 30 mL/min; column: XBridge C18, 100 x 19mm, 5 µm) to afford 4- cyclopropyl-6-methoxy-5-[4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidin-2-yl]pyrimidine (50.2 mg, 104.1 μmol, 35.4% yield) as a white solid.
1H NMR (600 MHz, DMSO-d
6) δ 0.84 – 0.89 (m, 2H), 1.00 – 1.05 (m, 2H), 1.65 – 1.71 (m, 1H), 3.77 (s, 3H), 3.84 (s, 3H), 5.48 (s, 2H), 7.06 (d, 1H), 7.57 (d, 2H), 7.72 (d, 2H), 7.93 (s, 1H), 8.66 (s, 1H), 8.69 (d, 1H). MS (ESI): [M+H]
+ m/z: calcd 483.18; found 483.2. Example 179 (Compound 156)
3-fluoro-4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]benzonitrile Starting material 3-fluoro-4-[4-(trifluoromethyl)-1H-imidazol-2-yl]benzonitrile was prepared as described for Compound 190. To a solution of 3-fluoro-4-[4-(trifluoromethyl)-1H-imidazol-2-yl]benzonitrile (1 g, 3.92 mmol) in THF (10 mL) was added potassium tert-butoxide (700 mg, 6.24 mmol) and The mixture was stirred at 0 °C for 30 minutes. To the mixture was added MeI (840 mg, 5.92 mmol) and the mixture stirred at 20 °C for 2 hrs. The resulting mixture was diluted by addition of water (10 mL) and extracted with EtOAc (100 mL x 3). The combined organic layer was washed with brine (100 mL x 2), dried over anhydrous Na
2SO
4, filtered and concentrated under reduced pressure. The residue was purified by flash chromatography (ISCO®; 12 g AgelaFlash® Silica Flash Column, PE/EtOAc with EtOAc from 0-20%, flow rate = 35 mL/min, 254 nm) to afford 3-fluoro-4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]benzonitrile (930 mg, 88.2% yield) as light yellow solid.
1H NMR (400 MHz, DMSO-d
6) δ ppm 8.06 - 8.17 (m, 2H), 7.81 - 7.93 (m, 2H), 3.65 (d, J = 1.5 Hz, 3H);
19F NMR (376 MHz, DMSO-d
6) δ ppm -66.91, -111.24; MS (ESI) [M+H]
+ m/z: calcd 270.1, found 270.0. 4-[5-chloro-1-methyl-4-(trifluoromethyl)imidazol-2-yl]-3-fluoro-benzonitrile [1519] A mixture of 3-fluoro-4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]benzonitrile (930 mg, 3.45 mmol), NCS (950 mg, 7.11 mmol) in AcOH (5 mL) was stirred at 45 °C for 2.5 hrs. The mixture was adjusted pH to 7-8 with saturated NaHCO
3 aqueous solution. The resulting mixture was diluted by addition of water (10 mL) and extracted with EtOAc (100 mL x 3). The combined organic layer was washed with brine (40 mL x 2), dried over anhydrous Na
2SO
4, filtered and concentrated under reduced pressure. The residue was purified by flash chromatography (ISCO®; 12 g AgelaFlash® Silica Flash Column, PE/EtOAc with EtOAc from 0-10%, flow rate = 30 mL/min, 254 nm) to afford 4-[5-chloro-1-methyl-4- (trifluoromethyl)imidazol-2-yl]-3-fluoro-benzonitrile (1.04 g, 99.1% yield) as white solid. MS (ESI) [M+H]
+ m/z: calcd 304.0, found 304.0. 4-[5-chloro-1-methyl-4-(trifluoromethyl)imidazol-2-yl]-3-fluoro-benzoic acid [1520] To a solution of 4-[5-chloro-1-methyl-4-(trifluoromethyl)imidazol-2-yl]-3-fluoro- benzonitrile (680 mg, 2.24 mmol) in EtOH (5 mL) and H
2O (1 mL) was added KOH (680 mg, 12.1 mmol) and the mixture was stirred at 80 °C for 12 hrs. The resulting mixture was concentrated under reduced pressure. The mixture was adjusted pH to 4-5 with 1 N HCl aqueous solution. The resulting mixture was diluted by addition of water (5 mL) and extracted with EtOAc (30 mL x 3). The combined organic layer was washed with brine (20 mL x 2), dried over anhydrous Na
2SO
4, filtered and concentrated under reduced pressure to afford 4-[5-chloro-1-methyl-4-(trifluoromethyl)imidazol-2-yl]-3-fluoro-benzoic acid (670 mg, 92.7% yield) as light yellow solid. MS (ESI) [M+H]
+ m/z: calcd 323.0, found 322.9. [4-[5-chloro-1-methyl-4-(trifluoromethyl)imidazol-2-yl]-3-fluoro-phenyl]methanol [1521] To a solution of 4-[5-chloro-1-methyl-4-(trifluoromethyl)imidazol-2-yl]-3-fluoro- benzoic acid (600 mg, 1.86 mmol) in THF (5 mL) was added 1 M BH
3-THF (12 mL, 12.0 mmol) at 0 °C. The mixture was stirred at 20 °C for 3 hrs. The resulting mixture was quenched by addition of MeOH (5 mL), filtered and concentrated under reduced pressure. The residue was purified by flash chromatography (ISCO®; 12 g AgelaFlash® Silica Flash Column, PE/EtOAc with EtOAc from 0-50%, flow rate = 30 mL/min, 254 nm) to afford [4- [5-chloro-1-methyl-4-(trifluoromethyl)imidazol-2-yl]-3-fluoro-phenyl]methanol (430 mg, 74.9% yield) as colorless oil.
1H NMR (400 MHz, DMSO-d
6) δ ppm 7.56 (t, J = 7.8 Hz, 1H), 7.27 - 7.39 (m, 2H), 5.48 (t, J = 5.8 Hz, 1H), 4.61 (d, J = 5.8 Hz, 2H), 3.53 (d, J = 1.5 Hz, 3H);
19F NMR (376 MHz, DMSO-d
6) δ ppm -60.32, -114.42, MS (ESI) [M+H]
+ m/z: calcd 309.0, found 309.0. 4-[[4-[5-chloro-1-methyl-4-(trifluoromethyl)imidazol-2-yl]-3-fluoro-phenyl]methoxy]-2-(4- cyclopropyl-6-methoxy-pyrimidin-5-yl)-5-methoxy-pyrimidine (156) [1522] To a solution of [4-[5-chloro-1-methyl-4-(trifluoromethyl)imidazol-2-yl]-3-fluoro- phenyl]methanol (150 mg, 0.486 mmol) in THF (3 mL) was slowly added NaH (60 mg, 1.50 mmol, 60 wt% in mineral oil). The mixture was stirred at 20 °C for 30 min. Then to the mixture was added 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-5-methoxy-4- methylsulfonyl-pyrimidine I-6 (180 mg, 0.535 mmol) and the mixture was stirred at 45 °C for 1.5 hrs. The resulting mixture was quenched by addition of water (10 mL) and extracted with EtOAc (50 mL x 3). The combined organic layer was washed with brine (40 mL x 2), dried over anhydrous Na
2SO
4, filtered and concentrated under reduced pressure. The residue was purified by preparative HPLC (Instrument: Gilson GX-281 Liquid Handler, Gilson 322 Pump, Gilson 156 UV Detector; Column: Xtimate C1875 x 40 mm x 3 μm; Mobile phase A: H
2O with NH
3-H
2O (v%); Mobile phase B: MeCN; Gradient: B from 54% to 84% in 7.8 min, hold 100% B for 1 min; Flow Rate: 30 mL/min; Column Temperature: 30 °C; Wavelength: 220 nm, 254 nm). The fraction was concentrated under reduced pressure and then lyophilized for overnight to afford 4-[[4-[5-chloro-1-methyl-4-(trifluoromethyl)imidazol-2-yl]-3-fluoro- phenyl]methoxy]-2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-5-methoxy-pyrimidine (49.2 mg, 17.9% yield) as white solid.
1H NMR (400 MHz, DMSO-d
6) δ ppm 8.65 (s, 1H), 8.45 (s, 1H), 7.64 (t, J = 7.7 Hz, 1H), 7.43 - 7.55 (m, 2H), 5.53 (s, 2H), 3.96 (s, 3H), 3.84 (s, 3H), 3.53 (d, J = 1.3 Hz, 3H), 1.65 - 1.77 (m, 1H), 1.03 (quin, J = 3.6 Hz, 2H), 0.82 - 0.91 (m, 2H);
19F NMR (376 MHz, DMSO-d
6) δ ppm -66.36, -113.71; MS (ESI) [M+H]
+ m/z: calcd 565.1, found 565.1. Example 180 (Compounds 155 & 135)
Ethyl 2-chloro-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidine- 5-carboxylate [1523] A mixture of NaH (156 mg, 3.90 mmol, 60% dispersion in mineral oil) and [4-[1- methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methanol I-2b (1.00 g, 3.90 mmol) in THF (40 mL) was stirred at room temperature for 30 min. Ethyl 2,4-dichloropyrimidine-5- carboxylate (863 mg, 3.90 mmol) was added to the mixture. The reaction mixture was stirred at room temperature for 12 hr. The reaction mixture was poured into cold water and extracted with EtOAc (2×50 mL). The combined organic layers were separated, washed with brine (30 mL), dried over anhydrous Na
2SO
4 and concentrated under reduced pressure. The residue was subjected to flash column chromatography (silica, gradient MTBE - chloroform) to afford ethyl 2-chloro-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidine-5-carboxylate (0.917 g, 2.08 mmol, 53.3% yield) as a white solid.
1H NMR (500 MHz, DMSO-d
6) 1.27 (t, 3H), 3.78 (s, 3H), 4.30 (q, 2H), 5.59 (s, 2H), 7.65 (d, 2H), 7.56 (d, 2H), 7.93 (s, 1H), 8.91 (s, 1H). MS (ESI): [M+H]
+ m/z: calcd 441.10; found 441.2 Ethyl 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidine-5-carboxylate [1524] Ethyl 2-chloro-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidine-5-carboxylate (800 mg, 1.81 mmol), (4-cyclopropyl-6- methoxy-pyrimidin-5-yl)boronic acid I-5 (880 mg, 4.54 mmol), potassium phosphate tribasic (2.31 g, 10.9 mmol), RuPhos (84.7 mg, 182 μmol) and RuPhos Pd G4 (154 mg, 182 μmol) were mixed in degassed mixture of dioxane (25 mL) and water (2.5 mL). The mixture was stirred at room temperature for 12 hr under argon atmosphere. The reaction mixture was diluted with EtOAc (50 mL). Anhydrous Na
2SO
4 (10 g) was added to the mixture. The resulting mixture was stirred at room temperature for 30 min, then solids were filtered out. The filtrate was concentrated under reduced pressure to afford ethyl 2-(4-cyclopropyl-6- methoxy-pyrimidin-5-yl)-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidine-5-carboxylate (1.50 g) as a yellow gum which was used in the next step without further purification. MS (ESI): [M+H]
+ m/z: calcd 555.20; found 555.2 [2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidin-5-yl]methanol [1525] A solution of ethyl 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidine-5-carboxylate (1.50 g, crude) in THF (5 mL) was added dropwise to a stirred suspension of LAH (348 mg, 9.17 mmol) in THF (20 mL) at -45 °C. The reaction mixture was stirred at -45 °C for 2 hr. The reaction mixture was quenched by dropwise addition of water (350 µL) followed by an aqueous NaOH (400 µL, 30% wt.) and then water (450 µL). The resulting mixture was allowed to warm to room temperature under constant stirring, then the solids were filtered out. The filtrate was concentrated under reduced pressure. The residue was subjected to flash column chromatography (silica, gradient CHCl
3 - MeOH) to afford [2-(4-cyclopropyl-6-methoxy- pyrimidin-5-yl)-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidin-5-yl]methanol (720 mg, 1.40 mmol, 77.4% yield from ethyl 2- chloro-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidine-5- carboxylate) as a yellow gum which was used in the next steps without further purification.
1H NMR (400 MHz, CDCl
3) δ 0.88 – 0.94 (m, 2H), 1.21 – 1.27 (m, 2H), 1.67 – 1.74 (m, 1H), 3.80 (s, 3H), 3.95 (s, 3H), 4.81 (s, 2H), 5.59 (s, 2H), 7.34 (s, 1H), 7.56 (d, 2H), 7.67 (d, 2H), 8.67 (s, 1H), 8.69 (s, 1H). MS (ESI): [M+H]
+ m/z: calcd 513.19; found 513.2 [2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidin-5-yl]methyl methanesulfonate [1526] A solution of methanesulfonyl chloride (117 mg, 1.02 mmol, 79.5 μL) in DCM (1 mL) was added dropwise to a mixture of [2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[4- [1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidin-5-yl]methanol (350 mg, 683 μmol) and TEA (177 mg, 1.74 mmol, 243 μL) in DCM (9 mL) at 0 °C. The reaction mixture was stirred at 0°C for 2 hr. The reaction mixture was poured into cold water (20 mL) and extracted with DCM (30 mL). The organic layer was separated, washed with saturated aqueous NaHCO
3 solution (20 mL), dried over anhydrous Na
2SO
4 and concentrated under reduced pressure to afford [2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidin-5-yl]methyl methanesulfonate (400 mg) as a gum which was used in the next step without further purification. MS (ESI): [M+H]
+ m/z: calcd 591.16; found 591.0 2-[2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol- 2-yl]phenyl]methoxy]pyrimidin-5-yl]acetonitrile (Compound 135) [1527] A mixture of [2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidin-5-yl]methyl methanesulfonate (200 mg, crude) and sodium cyanide (33.2 mg, 677 μmol) in DMF (3 mL) was stirred at room temperature for 12 hr. The reaction mixture was filtered and subjected to HPLC (2-7 min., 50-100% water – MeOH, +0.1% vol. of 25% aq. NH
3, flow: 30 mL/min, column: XBridge (R) 100×19 mm, 5 µm) to afford 2-[2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4- [[4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidin-5-yl]acetonitrile (11.6 mg, 22.2 μmol, 6.50% yield from [2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[4- [1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidin-5-yl]methanol) as a white solid.
1H NMR (500 MHz, DMSO-d
6) δ 0.84 – 0.91 (m, 2H), 1.01 – 1.07 (m, 2H), 1.66 – 1.73 (m, 1H), 3.79 (s, 3H), 3.84 (s, 3H), 4.07 (s, 2H), 5.59 (s, 2H), 7.64 (d, 2H), 7.74 (d, 2H), 7.94 (s, 1H), 8.68 (s, 1H), 8.72 (s, 1H). MS (ESI): [M+H]
+ m/z: calcd 522.19; found 522.2 2-[2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol- 2-yl]phenyl]methoxy]pyrimidin-5-yl]-2-methyl-propanenitrile [1528] A mixture of 2-[2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidin-5-yl]acetonitrile (8.00 mg, 15.3 μmol) and NaH (4.00 mg, 100 μmol, 60% dispersion in mineral oil) in THF (1 mL) was stirred at room temperature for 1 hr. Methyl iodide (14.2 mg, 100 μmol, 6.2 μL) was added to the mixture. The reaction mixture was stirred at room temperature for 12 hr. The reaction mixture was subjected to HPLC (0.5-6.5 min., 40-65% water – ACN, +0.1% vol. of 25% aq. NH
3, flow: 30 mL/min, column: Chromatorex Phen1l SMB100-5T 100×19 mm, 5 µm) to afford 2-[2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidin-5-yl]-2-methyl-propanenitrile (1.40 mg, 2.55 μmol, 16.6% yield) as a yellow gum.
1H NMR (500 MHz, CD
3OD) δ 0.89 – 0.94 (m, 2H), 1.13 – 1.17 (m, 2H), 1.69 – 1.75 (m, 1H), 1.87 (s, 6H), 3.80 (s, 3H), 3.92 (s, 3H), 5.71 (s, 2H), 7.67 – 7.74 (m, 4H), 8.61 (s, 1H), 8.71 (s, 1H). MS (ESI): [M+H]
+ m/z: calcd 550.25; found 550.2.
Methyl 3-fluoro-4-[4-(trifluoromethyl)-1H-imidazol-2-yl]benzoate [1529] A mixture of 3,3-dibromo-1,1,1-trifluoro-propan-2-one (21.5 g, 79.6 mmol) and sodium acetate (7.18 g, 87.6 mmol) in water (20 mL) was stirred at 100 °C for 45 min. The mixture was cooled to room temperature. The solution of methyl 3-fluoro-4-formyl-benzoate (14.5 g, 79.6 mmol) and ammonium hydroxide (27.9 g, 796 mmol, 31.0 mL, 28% NH
3) in MeOH (240 mL) was added to the mixture. The resulting mixture was stirred at room temperature for 12 hr. The reaction mixture was concentrated under reduced pressure to half volume and extracted with EtOAc (2×300 mL). The combined organic layers were separated, washed with brine (300 mL), dried over anhydrous Na
2SO
4 and filtered. The filtrate was concentrated under reduced pressure. The residue was subjected to flash column chromatography (SiO
2, gradient chloroform – EtOAc) to afford methyl 3-fluoro-4-[4- (trifluoromethyl)-1H-imidazol-2-yl]benzoate (7.80 g, 27.1 mmol, 34.0% yield) as a light- yellow solid. MS (ESI): [M+H]
+ m/z: calcd 289.06; found 289.0 Methyl 4-[1-cyclopropyl-4-(trifluoromethyl)imidazol-2-yl]-3-fluoro-benzoate [1530] Methyl 3-fluoro-4-[4-(trifluoromethyl)-1H-imidazol-2-yl]benzoate (500 mg, 1.73 mmol), cyclopropylboronic acid (596 mg, 6.94 mmol), copper bromide (194 mg, 868 μmol) and pyridine (1.37 g, 17.4 mmol, 1.40 mL) were mixed in dioxane (14 mL). The reaction mixture was stirred at room temperature for 24 hr with air access. The mixture was concentrated under reduced pressure. The residue was subjected to flash column chromatography (SiO
2, gradient DCM – EtOAc) to afford methyl 4-[1-cyclopropyl-4- (trifluoromethyl)imidazol-2-yl]-3-fluoro-benzoate (120 mg, 366 μmol, 21.1% yield) as a colorless oil.
1H NMR (400 MHz, CDCl
3) δ 0.67 – 0.73 (m, 2H), 0.86 – 0.94 (m, 2H), 3.45 – 3.53 (m, 1H), 3.95 (s, 3H), 7.41 (s, 1H), 7.59 (t, 1H), 7.83 (d, 1H), 7.92 (d, 1H). MS (ESI): [M+H]
+ m/z: calcd 329.09; found 329.0 [4-[1-cyclopropyl-4-(trifluoromethyl)imidazol-2-yl]-3-fluoro-phenyl]methanol [1531] A solution of methyl 4-[1-cyclopropyl-4-(trifluoromethyl)imidazol-2-yl]-3-fluoro- benzoate (120 mg, 366 μmol) in THF (0.5 mL) was added dropwise to a suspension of LiAlH
4 (55.6 mg, 1.46 mmol) in diethyl ether (3.0 mL) at 0 °C. The resulting mixture was stirred at 0 °C for 20 min. The reaction mixture was quenched by addition of aqueous potassium hydroxide (500 µl, 30% wt.). The solid precipitate formed was filtered out. The filtrate was concentrated under reduced pressure to afford [4-[1-cyclopropyl-4- (trifluoromethyl)imidazol-2-yl]-3-fluoro-phenyl]methanol (109 mg, 363 μmol, 99.3% yield) as a yellow oil which was used in the next steps without further purification. MS (ESI): [M+H]
+ m/z: calcd 301.10; found 301.2 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[4-[1-cyclopropyl-4- (trifluoromethyl)imidazol-2-yl]-3-fluoro-phenyl]methoxy]-5-methoxy-pyrimidine [1532] NaH (7.33 mg, 183 μmol, 60% dispersion in mineral oil) was added to a solution of [4-[1-cyclopropyl-4-(trifluoromethyl)imidazol-2-yl]-3-fluoro-phenyl]methanol (50.0 mg, 167 μmol) in THF (500 µL) under argon atmosphere. The reaction mixture was stirred at room atmosphere for 1 hr.2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-5-methoxy-4- methylsulfonyl-pyrimidine I-6 (61.6 mg, 183 μmol) was added to the reaction mixture. The resulting mixture was stirred at room temperature for 48 hr. The reaction mixture was diluted with EtOAc (20 mL) and washed with brine (20 mL). The organic layer was separated, washed with brine (2×15 mL), dried over anhydrous Na
2SO
4 and filtered. SiliaMetS
® Dimercaptotriazine (100 mg) was added to the resulting organic layer. The resulting mixture was stirred for 30 min and filtered. The filtrate was concentrated under reduced pressure. The residue was subjected to HPLC (0-2-10 min., 38-45-60% water – ACN, +0.1% vol. of 25% aq. NH
3, flow: 30 mL/min, column: Chromatorex C18 SMB100-5T 100×19 mm, 5 µm) to afford 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[4-[1-cyclopropyl-4- (trifluoromethyl)imidazol-2-yl]-3-fluoro-phenyl]methoxy]-5-methoxy-pyrimidine (37.0 mg, 66.5 μmol, 39.9% yield) as a yellow solid.
1H NMR (500 MHz, DMSO-d
6) δ 0.79 – 0.91 (m, 6H), 1.00 – 1.06 (m, 2H), 1.68 – 1.76 (m, 1H), 3.44 – 3.51 (m, 1H), 3.84 (s, 3H), 3.97 (s, 3H), 5.53 (s, 2H), 7.44 (d, 1H), 7.49 (d, 1H), 7.66 (t, 1H), 8.01 (s, 1H), 8.45 (s, 1H), 8.65 (s, 1H). MS (ESI): [M+H]
+ m/z: calcd 557.22; found 557.2.
Methyl 3-fluoro-4-[1-[prop-1-enyl]-4-(trifluoromethyl)imidazol-2-yl]benzoate Synthesis of the starting methyl 3-fluoro-4-[4-(trifluoromethyl)-1H-imidazol-2- yl]benzoate is described for Compound 149. [1533] Methyl 3-fluoro-4-[4-(trifluoromethyl)-1H-imidazol-2-yl]benzoate (300 mg, 1.04 mmol), cyclopropyl trifluoromethanesulfonate (594 mg, 3.12 mmol), NaH (75.0 mg, 3.12 mmol) were mixed in DMF (1 mL). The reaction mixture was stirred at 80 °C for 12 hr. The reaction mixture was cooled to room temperature, diluted with EtOAc (25 mL) and water (25 mL). The organic layer was separated, washed with water (2×20 mL), dried over anhydrous Na
2SO
4 and concentrated under reduced pressure to afford methyl 3-fluoro-4-[1-[prop-1- enyl]-4-(trifluoromethyl)imidazol-2-yl]benzoate (400 mg, crude) as a brown oil which was used in the next step without further purification. MS (ESI): [M+H]
+ m/z: calcd 329.09; found 329.1 [3-fluoro-4-[1-[prop-1-enyl]-4-(trifluoromethyl)imidazol-2-yl]phenyl]methanol [1534] A solution of methyl 3-fluoro-4-[1-[prop-1-enyl]-4-(trifluoromethyl)imidazol-2- yl]benzoate (400 mg, crude) in diethyl ether (2 mL) was added dropwise to a suspension of LAH (139 mg, 3.66 mmol) in diethyl ether (10 mL) at 0 °C. The reaction mixture was stirred at 0°C for 20 min. The reaction mixture was quenched by addition of aqueous KOH (1.5 mL, 30%), then solids were filtered out. The filtrate was concentrated under reduced pressure to afford [3-fluoro-4-[1-[prop-1-enyl]-4-(trifluoromethyl)imidazol-2-yl]phenyl]methanol (300 mg, crude) as a yellow oil which was used in the next step without further purification. MS (ESI): [M+H]
+ m/z: calcd 301.10; found 301.1 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[3-fluoro-4-[1-[prop-1-enyl]-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]-5-methoxy-pyrimidine [1535] NaH (10.0 mg, 250 μmol, 60% dispersion in mineral oil) was added to a solution of [3-fluoro-4-[1-[prop-1-enyl]-4-(trifluoromethyl)imidazol-2-yl]phenyl]methanol (50 mg, 167 μmol) in THF (500 µL) under argon atmosphere. The resulting mixture was stirred at room temperature for 1 hr.2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-5-methoxy-4- methylsulfonyl-pyrimidine I-6 (61.6 mg, 183 μmol) was added to the mixture. The reaction mixture was stirred at room temperature for 12 hr. The reaction mixture was diluted with EtOAc (20 mL) and washed with brine (20 mL). The organic layer was separated and dried over anhydrous Na
2SO
4. SiliaMetS
® Dimercaptotriazine (100 mg) was added to the resulting solution. The resulting mixture was stirred for 30 min. and filtered. The filtrate was concentrated under reduced pressure. The residue was subjected to HPLC (0-2-10 min, 30- 40-50% water - ACN; flow: 30 mL/min, column: Chromatorex SMB100-5T, 100×19 mm, 5 µm) to afford 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[3-fluoro-4-[1-[prop-1-enyl]- 4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]-5-methoxy-pyrimidine (8.00 mg, 14.4 μmol, 8.6% yield) as a yellow solid.
1H NMR (600 MHz, DMSO-d
6) δ 0.85 – 0.89 (m, 2H), 1.00 – 1.04 (m, 2H), 1.49 (d, 3H), 1.68 – 1.73 (m, 1H), 3.83 (s, 3H), 3.96 (s, 3H), 5.50 (s, 2H), 5.74 – 5.81 (m, 1H), 6.61 (d, 1H), 7.39 – 7.46 (m, 2H), 7.60 (t, 1H), 8.09 (s, 1H), 8.45 (s, 1H), 8.65 (s, 1H). MS (ESI): [M+H]
+ m/z: calcd 557.22; found 557.2. Example 183 (Compound 152)
Methyl 2-bromo-4-[4-(trifluoromethyl)-1H-imidazol-2-yl]benzoate [1536] 3,3-Вibromo-1,1,1-trifluoro-propan-2-one (9.04 g, 33.5 mmol) was added to a solution of sodium acetate (5.49 g, 67.0 mmol,) in water (40 mL). The reaction mixture was stirred at 95 °C for 1 hr. The reaction mixture was cooled to room temperature and poured into a solution of methyl 2-bromo-4-formyl-benzoate (7.40 g, 30.5 mmol) and an aqueous NH
3 (30 mL) in MeOH (300 mL). The resulting mixture was stirred at room temperature for 14 hr. The mixture was concentrated under reduced pressure. The residue was diluted with water (300 mL) and extracted with CHCl
3 (3×100 mL). The combined organic layers were separated, dried over anhydrous Na
2SO
4 and filtered through a thin pad of silica gel. The filtrate was concentrated under reduced pressure to afford methyl 2-bromo-4-[4- (trifluoromethyl)-1H-imidazol-2-yl]benzoate (10.0 g, 28.6 mmol, 94.1% yield) as a yellow gum which was used in the next steps without further purification.
1H NMR (400 MHz, DMSO-d
6) 3.89 (s, 3H), 7.92 (d, 1H), 8.05 (s, 1H), 8.11 (d, 1H), 8.35 (s, 1H), 13.53 (br. s, 1H). MS (ESI): [M+H]
+ m/z: calcd 348.98, 350.98; found 349.0, 351.0 Methyl 2-bromo-4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]benzoate [1537] Cs
2CO
3 (18.7 g, 57.3 mmol) and iodomethane (5.29 g, 37.2 mmol, 2.32 mL) were added to a solution of methyl 2-bromo-4-[4-(trifluoromethyl)-1H-imidazol-2-yl]benzoate (10.0 g, 28.6 mmol) in ACN (300 mL). The reaction mixture was stirred at 40 °C for 12 hr. The reaction mixture was cooled to room temperature, poured into water (300 mL) and extracted with EtOAc (4×120 mL). The combined organic layers were dried over anhydrous Na2SO4 and filtered through a thin pad of silica gel. The filtrate was concentrated under reduced pressure to afford methyl 2-bromo-4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]benzoate (4.00 g, 11.0 mmol, 38.5% yield) as a yellow solid.
1H NMR (400 MHz, CDCl
3) 3.80 (s, 3H), 3.94 (s, 3H), 7.33 (s, 1H), 7.64 (d, 1H), 7.88 (d, 1H), 7.97 (s, 1H). MS (ESI): [M+H]
+ m/z: calcd 363.00, 364.99; found 363.0, 365.0 methyl 4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]-2-vinyl-benzoate [1538] Methyl 2-bromo-4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]benzoate (2.00 g, 5.51 mmol), potassium trifluoro(vinyl)boron (1.10 g, 8.26 mmol), K
2CO
3 (1.52 g, 11.0 mmol) and bis(diphenylphosphino)ferrocene]dichloropalladium(II)-DCM (202 mg, 275 μmol) were mixed in degassed DMSO (20 mL). The reaction mixture was stirred at 100 °C for 3 hr. The reaction mixture was cooled to room temperature, diluted with water (50 mL) and extracted with EtOAc (2×30 mL). The combined organic layers were separated, washed with water (20 mL) and brine (20 mL), dried over anhydrous Na
2SO
4 and concentrated under reduced pressure to afford methyl 4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]-2-vinyl-benzoate (1.50 g) as a brown solid which was used next step without further purification. MS (ESI): [M+H]
+ m/z: calcd 311.10; found 311.0 Methyl 2-ethyl-4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]benzoate [1539] 10% Pd on carbon (300 mg) was added to a solution of methyl 4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]-2-vinyl-benzoate (1.50 g) in MeOH (60 mL). The reaction mixture was evacuated and backfilled with hydrogen. The reaction mixture was stirred at room temperature under hydrogen atmosphere (atmospheric pressure) for 12 hr. The reaction mixture was filtered. The filtrate was concentrated under reduced pressure to afford methyl 2- ethyl-4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]benzoate (1.50 g, crude) as a brown gum which was used in the next step without further purification. MS (ESI): [M+H]
+ m/z: calcd 313.12; found 313.0 [2-ethyl-4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methanol [1540] DIBAL (2.05 g, 14.4 mmol, 2.92 mL) was added dropwise to a solution of methyl 2- ethyl-4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]benzoate (1.50 g, crude) in THF (30 mL) at -78 °C under argon atmosphere. The reaction mixture was stirred at room temperature for 12 hr. The reaction mixture was cooled to -20 °C and quenched by dropwise addition of an aqueous solution of 10 % NaOH (3.0 mL). NaOH (5.0 g, solid) was added to the reaction mixture. The resulting mixture was stirred at room temperature for 20 min. Then organic solvent was decanted and concentrated under reduced pressure. The residue was subjected to flash column chromatography (SiO
2, gradient hexane – EtOAc) to afford [2-ethyl-4-[1- methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methanol (450 mg, 1.58 mmol, 28.7% yield from 2-bromo-4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]benzoate) as a beige solid.
1H NMR (400 MHz, CDCl
3) 1.23 (t, 3H), 2.70 (q, 2H), 3.74 (s, 3H), 4.64 – 4.78 (m, 3H), 7.29 (s, 1H), 7.38 (d, 1H), 7.44 – 7.49 (m, 2H). MS (ESI): [M+H]
+ m/z: calcd 285.12; found 285.2 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[2-ethyl-4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]-5-methoxy-pyrimidine [1541] [2-Ethyl-4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methanol (80.0 mg, 281 μmol) was added to a suspension of NaH (13.5 mg, 338 μmol, 60% dispersion in mineral oil) in THF (4 mL) at room temperature. The reaction mixture was stirred at room temperature for 15 min.2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-5-methoxy-4- methylsulfonyl-pyrimidine I-6 (94.7 mg, 281 μmol) was added to the reaction mixture. The resulting mixture was stirred at room temperature for 12 hr. The reaction mixture was diluted with water (5 mL) and extracted with EtOAc (2×5 mL). The combined organic layers were separated, washed with brine (5 mL) and concentrated under reduced pressure. The residue was subjected to HPLC (1.5-9 min., 45-53% water – ACN, flow: 30 mL/min, column: SunFire C18100×19 mm, 5 µm) to afford 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4- [[2-ethyl-4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]-5-methoxy- pyrimidine (48.0 mg, 88.8 μmol, 31.6% yield) as a beige solid.
1H NMR (600 MHz, DMSO- d
6) δ 0.84 – 0.90 (m, 2H), 1.01 – 1.06 (m, 2H), 1.19 (t, 3H), 1.71 – 1.77 (m, 1H), 2.74 (q, 2H), 3.79 (s, 3H), 3.85 (s, 3H), 3.93 (s, 3H), 5.52 (s, 2H), 7.50 – 7.57 (m, 2H), 7.59 (s, 1H), 7.94 (s, 1H), 8.42 (s, 1H), 8.65 (s, 1H). MS (ESI): [M+H]
+ m/z: calcd 541.25; found 541.2. Example 184 (Compound 147)
2-chloro-5-methoxy-4-methylsulfanyl-pyrimidine [1542] To a solution of 2,4-dichloro-5-methoxy-pyrimidine (2 g, 11.2 mmol) in THF (20 mL) was added MeSNa (4.04 g, 11.53 mmol, 20 wt% in H
2O) at -10 °C. The mixture was stirred at 20 °C for 16 hrs. The resulting mixture was quenched by addition of water (100 mL) and extracted with EtOAc (100 mL x 3). The combined organic layer was washed with brine (100 mL x 2), dried over anhydrous Na
2SO
4, filtered and concentrated under reduced pressure to afford 2-chloro-5-methoxy-4-methylsulfanyl-pyrimidine (2.15 g, crude) as white solid.
1H NMR (400 MHz, chloroform-d) δ ppm 7.79 (s, 1H), 3.95 (s, 3H), 2.55 (s, 3H); MS (ESI) [M+H]
+ m/z: calcd 191.0, found 191.0. 2-[4-cyclopropyl-6-(difluoromethoxy)pyrimidin-5-yl]-5-methoxy-4-methylsulfanyl-pyrimidine [1543] A mixture of 2-chloro-5-methoxy-4-methylsulfanyl-pyrimidine (150 mg, 0.787 mmol), 4-cyclopropyl-6-(difluoromethoxy)-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2- yl)pyrimidine I-10 (450 mg, 1.44 mmol), XPhos Pd G
3 (33 mg, 0.0390 mmol), XPhos (19 mg, 0.0399 mmol), K
3PO
4 (418 mg, 1.97 mmol) in dioxane (15 mL) and H
2O (3 mL) was stirred at 90 °C for 12 hrs. The resulting mixture was quenched by addition of water (20 mL) and extracted with EtOAc (20 mL x 3). The combined organic layer was washed with brine (50 mL), dried over anhydrous Na
2SO
4, filtered and concentrated under reduced pressure. The residue was purified by flash chromatography (ISCO®; 12 g AgelaFlash® Silica Flash Column, PE/EtOAc with EtOAc from 0-40%, 30 mL/min, 254nm) to afford 2-[4- cyclopropyl-6-(difluoromethoxy)pyrimidin-5-yl]-5-methoxy-4-methylsulfanyl-pyrimidine (255 mg, 95.2% yield) as yellow solid. MS (ESI) [M+H]
+ m/z: calcd 341.1; found 341.1. 2-[4-cyclopropyl-6-(difluoromethoxy)pyrimidin-5-yl]-5-methoxy-4-methylsulfonyl-pyrimidine [1544] To a solution of 2-[4-cyclopropyl-6-(difluoromethoxy)pyrimidin-5-yl]-5-methoxy-4- methylsulfanyl-pyrimidine (255 mg, 0.749 mmol) in DCM (10 mL) was added mCPBA (319 mg, 1.57 mmol, 85 wt%). The mixture was stirred at 20 °C for 12 hrs. The resulting mixture was quenched by addition of saturated Na
2SO
3 aqueous solution (10 mL), saturated NaHCO
3 aqueous solution (10 mL), and extracted with EtOAc (20 mL x 3). The combined organic layer was dried over anhydrous Na
2SO
4, filtered and concentrated under reduced pressure to give 2-[4-cyclopropyl-6-(difluoromethoxy)pyrimidin-5-yl]-5-methoxy-4-methylsulfonyl- pyrimidine (300 mg, crude) as colorless oil, which was directly used without further purification. MS (ESI) [M+H]
+ m/z: calcd 373.1; found 373.0. 2-[4-cyclopropyl-6-(difluoromethoxy)pyrimidin-5-yl]-4-[[4-[1-isopropyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]-5-methoxy-pyrimidine (147) [1545] To a solution of [4-[1-isopropyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methanol (115 mg, 0.405 mmol) in THF (5 mL) was added NaH (19 mg, 0.475 mmol, 60 wt%). The mixture was stirred at 20 °C for 30 min.2-[4-cyclopropyl-6-(difluoromethoxy)pyrimidin-5- yl]-5-methoxy-4-methylsulfonyl-pyrimidine (150 mg, 0.403 mmol) was added, and the mixture was stirred at 45 °C for 2 hrs. The resulting mixture was quenched by addition of water (20 mL) and extracted with EtOAc (20 mL x 3). The combined organic layer was dried over anhydrous Na
2SO
4, filtered and concentrated under reduced pressure. The residue was purified by preparative HPLC (Instrument: Gilson 281, Gilson 333 and 334 Pumps, Gilson 159 UV Detector; Column: Xtimate C18150 × 40 mm × 5 μm; Mobile phase A: H
2O with 0.05% NH
3-H
2O + NH
4HCO
3 (v%); Mobile phase B: MeCN; Gradient: B from 55% to 80% in 18 min, hold 100% B for 5 min; Flow Rate: 30 mL/min; Column Temperature: 30 °C; Wavelength: 220 nm, 254 nm) to give 2-[4-cyclopropyl-6-(difluoromethoxy)pyrimidin-5-yl]- 4-[[4-[1-isopropyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]-5-methoxy-pyrimidine (32 mg, 13.8% yield) as white dry powder.
1H NMR (400 MHz, methanol-d
4) δ ppm 8.67 (s, 1H), 8.35 (s, 1H), 7.92 (d, J = 1.1 Hz, 1H), 7.82 (s, 0.25H), 7.62 - 7.68 (m, 2.5H), 7.54 - 7.59 (m, 2H), 7.46 (s, 0.25H), 5.61 (s, 2H), 4.55 (dt, J = 13.4, 6.7 Hz, 1H), 4.02 (s, 3H), 1.80 - 1.87 (m, 1H), 1.46 (d, J = 6.7 Hz, 6H), 1.18 - 1.22 (m, 2H), 0.95 - 1.01 (m, 2H);
19F NMR (376 MHz, methanol-d
4) δ ppm -91.20, -63.86; MS (ESI) [M+H]
+ m/z: calcd 577.2; found 577.1. Example 185 (Compound 146)
2-[4-cyclopropyl-6-(difluoromethoxy)pyrimidin-5-yl]-5-methoxy-4-[[4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidine (146) [1546] To a solution of [4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methanol I-2b (103 mg, 0.402 mmol) in THF (5 mL) was added NaH (19 mg, 0.475 mmol, 60 wt%). The mixture was stirred at 20 °C for 30 minutes.2-[4-cyclopropyl-6-(difluoromethoxy)pyrimidin- 5-yl]-5-methoxy-4-methylsulfonyl-pyrimidine (150 mg, 0.403 mmol) was added, and the mixture was stirred at 45 °C for 2 hrs. The resulting mixture was quenched by addition of water (20 mL) and extracted with EtOAc (20 mL x 3). The combined organic layer was dried over anhydrous Na
2SO
4, filtered and concentrated under reduced pressure. The residue was purified by preparative HPLC (Instrument: Gilson 281, Gilson 333 and 334 Pumps, Gilson 159 UV Detector; Column: Xtimate C18150 × 40 mm × 5 μm; Mobile phase A: H
2O with 0.05% NH
3-H
2O + NH
4HCO
3 (v%); Mobile phase B: MeCN; Gradient: B from 46% to 76% in 20 min, hold 100% B for 5 min; Flow Rate: 30 mL/min; Column Temperature: 30 °C; Wavelength: 220 nm, 254 nm) to give 2-[4-cyclopropyl-6-(difluoromethoxy)pyrimidin-5-yl]- 5-methoxy-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidine (32 mg, 14.5% yield) as white dry powder.
1H NMR (400 MHz, methanol-d
4) δ ppm 8.67 (s, 1H), 8.35 (s, 1H), 7.82 (s, 1H), 7.70 (d, J = 0.8 Hz, 1H), 7.61 - 7.68 (m, 5H), 7.46 (s, 1H), 5.60 (s, 2H), 4.02 (s, 3H), 3.78 (s, 3H), 1.79 - 1.87 (m, 1H), 1.17 - 1.22 (m, 2H), 0.95 - 1.01 (m, 2H);
19F NMR (376 MHz, methanol-d
4) δ ppm -91.19, -63.92; MS (ESI) [M+H]
+ m/z: calcd 549.2; found 549.1 Example 186 (Compound 137)
yl]isothiazole [1547] To a stirred mixture of 2-chloro-5-iodo-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol- 2-yl]phenyl]methoxy]pyrimidine I-12 (120 mg, 242.60 μmol) in 1,4-dioxane (2 mL) and water (0.4 mL) were added 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)isothiazole (51.21 mg, 242.60 μmol), potassium phosphate tribasic (102.99 mg, 485.20 μmol) and Dichloro[9,9- dimethyl-4,5-bis(diphenylphosphino)xanthene]palladium(II) (18.3 mg, 24.26 μmol) under nitrogen atmosphere at 25 °C. The resulting mixture was stirred under nitrogen atmosphere for 16 hrs at 40 °C. The reaction mixture was cooled down to room temperature and filtered. The filtrate was concentrated under reduced pressure. The residue was purified by Prep-TLC (EtOAc : PE = 2 : 1) to give 4-[2-chloro-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidin-5-yl]isothiazole (90 mg, 199.18 μmol, 82% yield) as a yellow solid. MS: m/z = 452.00 [M + H]
+. 4-[2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol- 2-yl]phenyl]methoxy]pyrimidin-5-yl]isothiazole [1548] To a stirred mixture of 4-[2-chloro-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidin-5-yl]isothiazole (75 mg, 165.98 μmol) in 1,4-dioxane (2 mL) and water (0.4 mL) were added (4-cyclopropyl-6-methoxy-pyrimidin-5-yl)boronic acid I-5 (38.6 mg, 199.18 μmol), dicyclohexyl-[2-(2,4,6-triisopropylphenyl)phenyl]phosphane (7.9 mg, 16.60 μmol), Methanesulfonato(2-dicyclohexylphosphino-2',4',6'-tri-i-propyl-1,1'- biphenyl)(2'-amino-1,1'-biphenyl-2-yl)palladium(II) (14.1 mg, 16.60 μmol) and potassium phosphate tribasic (70.5 mg, 331.97 μmol) under nitrogen atmosphere at 25 °C. The resulting mixture was stirred under nitrogen atmosphere for 16 hrs at 40 °C. The reaction mixture was cooled down to room temperature, concentrated and the residue was subject to Prep-TLC (EtOAc : PE = 2 : 1). The crude product was purified by reverse phase chromatography with the following conditions: Column: C18 spherical Column, 20-35 um, 100A, 40 g; Mobile Phase A: Water (5 mM aq. NH
4HCO
3), Mobile Phase B: MeCN; Flow rate: 35 mL/min; Gradient: 5% B to 5% B in 5 min, 5% B to 65% B in 5 min, 65% B to 65% B in 1 min, 65% B to 95% B in 10 min; Detector: UV 254 & 210 nm. The product-containing fractions were collected and evaporated in vacuo and then lyophilized to give 4-[2-(4-cyclopropyl-6- methoxy-pyrimidin-5-yl)-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidin-5-yl]isothiazole (13.7 mg, 24.22 μmol, 14% yield) as an off- white solid. MS: m/z = 566.25 [M + H]
+.
1H NMR (400 MHz, Chloroform-d) δ 9.07 (s, 1H), 8.99 (s, 1H), 8.96 (s, 1H), 8.73 (s, 1H), 7.70 (d, J = 8.0 Hz, 2H), 7.58 (d, J = 8.0 Hz, 2H), 7.35 (s, 1H), 5.67 (s, 2H), 4.01 (s, 3H), 3.82 (s, 3H), 1.89 - 1.83 (m, 1H), 1.36 - 1.32 (m, 2H), 1.02 - 0.97 (m, 2H).
19F NMR (376 MHz, Chloroform-d) δ -62.79. Example 187 (Compound 136)
2-chloro-4-ethyl-6-[[3-fluoro-4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidine Synthesis of the starting [3-fluoro-4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methanol is described for compound 97. [1549] 2,4-Dichloro-6-ethyl-pyrimidine (350 mg, 1.98 mmol) was added to a stirred suspension of [3-fluoro-4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methanol (314 mg, 1.15 mmol) and Cs
2CO
3 (773 mg, 2.37 mmol) in ACN (9.5 mL). The reaction mixture was stirred at 65 °C for 12 hr. The reaction mixture was cooled to room temperature and concentrated under reduced pressure. The residue was diluted with water (30 mL) and extracted with DCM (40 mL). The organic layer was separated, washed with brine (30 mL), dried over anhydrous Na2SO4 and concentrated under reduced pressure. The residue was subjected to flash column chromatography (SiO
2, gradient DCM - EtOAc) to afford 2-chloro- 4-ethyl-6-[[3-fluoro-4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidine (150 mg, 362 μmol, 18.3% yield) as a white solid. MS (ESI): [M+H]
+ m/z: calcd 415.10; found 415.0 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-ethyl-6-[[3-fluoro-4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidine [1550] 2-Chloro-4-ethyl-6-[[3-fluoro-4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidine (150 mg, 362 μmol), (4-cyclopropyl-6-methoxy-pyrimidin-5- yl)boronic acid I-5 (175 mg, 904 μmol), potassium phosphate tribasic (230 mg, 1.08 mmol), and RuPhos Pd G3 (15.4 mg, 18.1 μmol) were mixed in a mixture of dioxane and water (4 mL, 9:1) under argon atmosphere. The resulting mixture was stirred at 70 °C for 20 hr. Additional portions of (4-cyclopropyl-6-methoxy-pyrimidin-5-yl)boronic acid I-5 (175 mg, 904 μmol) and RuPhos Pd G3 (15.4 mg, 18.1 μmol) were added to the mixture. The reaction mixture was stirred at 80 °C for 5 hr. The reaction mixture was cooled to room temperature and diluted with EtOAc (10 mL) and water (5 mL). The organic layer was separated. SiliaMetS
® Dimercaptotriazine (100 mg) was added to the organic layer. The resulting mixture was stirred at room temperature for 3 hr. The mixture was filtered. The filtrate was subjected to HPLC (0-2-10 min., 43-50-65% water – ACN, +0.1% vol. of 25% aq. NH
3, flow: 30 mL/min, column: Chromatorex SMB100-51100×19 mm, 5 µm) to afford 2-(4- cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-ethyl-6-[[3-fluoro-4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidine (30.0 mg, 56.8 μmol, 15.7% yield) as a yellow solid.
1H NMR (600 MHz, CDCl
3) δ 0.86 – 0.91 (m, 2H), 1.19 – 1.23 (m, 2H), 1.33 (t, 3H), 1.65 – 1.72 (m, 1H), 2.82 (q, 2H), 3.65 (s, 3H), 3.92 (s, 3H), 5.50 (s, 2H), 6.68 (s, 1H), 7.29 – 7.37 (m, 3H), 7.62 (t, 2H), 8.63 (s, 1H). MS (ESI): [M+H]
+ m/z: calcd 529.23; found 529.2. Example 188 (Compound 134)
2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[6-[1-cyclopropyl-4- (trifluoromethyl)imidazol-2-yl]-5-fluoro-3-pyridyl]methoxy]-5-methyl-pyrimidine Synthesis of the starting [6-[1-cyclopropyl-4-(trifluoromethyl)imidazol-2-yl]-5-fluoro- 3-pyridyl]methanol is described for Compound 43. [1551] NaH (26.6 mg, 664 μmol, 60% dispersion in mineral oil) was added to a stirred solution of 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-5-methyl-4-methylsulfonyl- pyrimidine I-7 (106 mg, 332 μmol) and [6-[1-cyclopropyl-4-(trifluoromethyl)imidazol-2-yl]- 5-fluoro-3-pyridyl]methanol (100 mg, 332 μmol) in THF (6 mL). The reaction mixture was stirred at room temperature for 13 hr. The reaction mixture was poured into an aqueous solution of NH
4Cl (15 mL). the resulting mixture was extracted with MTBE (3×15 mL). The combined organic layers were dried over anhydrous Na
2SO
4 and concentrated under reduced pressure. The residue was dissolved in ACN (6 mL). SiliaMetS
® Dimercaptotriazine (100 mg) was added to the mixture. The resulting suspension was stirred at room temperature for 20 hr. The suspension was filtered. The filtrate was subjected to HPLC (0-0.5-6 min., 20-30- 30% water – ACN, flow: 30 mL/min, column: Chromatorex 100×19 mm, 5 µm) to afford 2- (4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[6-[1-cyclopropyl-4-(trifluoromethyl)imidazol- 2-yl]-5-fluoro-3-pyridyl]methoxy]-5-methyl-pyrimidine (66.0 mg, 122 μmol, 36.7% yield) as a yellow solid.
1H NMR (600 MHz, DMSO-d
6) δ 0.84 – 0.92 (m, 6H), 1.02 – 1.06 (m, 2H), 1.66 – 1.72 (m, 1H), 2.26 (s, 3H), 3.74 – 3.79 (m, 1H), 3.84 (s, 3H), 5.61 (s, 2H), 8.04 - 8.08 (m, 2H), 8.59 (s, 1H), 8.67 (s, 1H), 8.72 (s, 1H). MS (ESI): [M+H]
+ m/z: calcd 542.22; found 542.6. Example 189 (Compound 132)
Compound 132 2-chloro-5-methoxy-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidine [1552] To a stirred solution of 2,4-dichloro-5-methoxy-pyrimidine (200 mg, 1.12 mmol) in THF (2 mL) was added NaH (26.81 mg, 1.12 mmol, 60% dispersion in mineral oil) at 0 °C under nitrogen atmosphere. The resulting mixture was stirred for 30 min at 0 °C. To the resulting mixture added [4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methanol I-2b (286.28 mg, 1.12 mmol) at 0 °C under nitrogen atmosphere. The resulting mixture was stirred at 25 °C for 16 hrs then was quenched by the addition of saturated NH
4Cl aqueous solution (5 mL) at 0 °C. The resulting mixture was extracted with EtOAc (3 x 10 mL). The combined organic layers were washed with brine (5 mL), dried over anhydrous Na
2SO
4. After filtration, the filtrate was concentrated under reduced pressure. The residue was purified by Prep-TLC (10% MeOH in DCM) to afford 2-chloro-5-methoxy-4-[[4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidine (390 mg, 978.02 μmol, 87% yield) as a white solid. MS: m/z = 399.00 [M + H]
+.
1H NMR (400 MHz, DMSO-d
6) δ 8.23 (s, 1H), 7.95 (s, 1H), 7.78 (d, J = 8.0 Hz, 2H), 7.62 (d, J = 8.0 Hz, 2H), 5.50 (s, 2H), 3.88 (s, 3H), 3.81 (s, 3H). 2-[4-cyclopropyl-6-(trideuteriomethoxy)pyrimidin-5-yl]-5-methoxy-4-[[4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidine [1553] To a stirred solution of 2-chloro-5-methoxy-4-[[4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidine (100 mg, 250.77 μmol) and 4- cyclopropyl-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-6- (trideuteriomethoxy)pyrimidine I-9 (105.01 mg, 376.16 μmol) in 1,4-dioxane (2 mL) and water (0.4 mL) were added dicyclohexyl-[2-(2,4,6-triisopropylphenyl)phenyl]phosphane (12 mg, 25.08 μmol), Methanesulfonato(2-dicyclohexylphosphino-2',4',6'-tri-i-propyl-1,1'- biphenyl)(2'-amino-1,1'-biphenyl-2-yl)palladium(II) (21.2 mg, 25.08 μmol) and potassium phosphate (106.5 mg, 501.55 μmol) in portions at 25 °C under nitrogen atmosphere. The resulting mixture was stirred for 16 hrs at 90 °C under nitrogen atmosphere. The mixture was allowed to cool down to 25 °C. The resulting mixture was concentrated under reduced pressure. The residue was purified by Prep-TLC (100% EtOAc) to afford a less polar isomer and a more polar isomer. The less polar isomer was purified by RP-Flash with the following conditions: Column: C18 spherical Column, 20-35 um, 100A, 40 g; Mobile Phase A: Water (5 mM aq. NH
4HCO
3), Mobile Phase B: MeCN; Flow rate: 35 mL/min; Gradient: 5% B to 5% B in 5 min, 5% B to 68% B in 19 min, 68% B to 68% B in 1 min, 68% B to 95% B in 5 min; Detector: UV 254 & 210 nm. The collected fractions were combined and lyophilized to give 2-[4-cyclopropyl-6- (trideuteriomethoxy)pyrimidin-5-yl]-5-methoxy-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol- 2-yl]phenyl]methoxy]pyrimidine, 22.6 mg, 43.84 μmol, 17% yield) as a white solid. MS: m/z = 516.15 [M + H]
+.
1H NMR (400 MHz, Chloroform-d) δ 8.65 (s, 1H), 8.25 (s, 1H), 7.65 (d, J = 8.0 Hz, 2H), 7.59 (d, J = 8.4 Hz, 2H), 7.33 (s, 1H), 5.59 (s, 2H), 4.02 (s, 3H), 3.78 (s, 3H), 1.78 - 1.69 (m, 1H), 1.28 - 1.19 (m, 2H), 0.95 - 0.88 (m, 2H).
19F NMR (377 MHz, Chloroform- d) δ -62.73. 2-[4-cyclopropyl-6-(trideuteriomethoxy)pyrimidin-5-yl]-5-methoxy-4-[[4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidine [1554] To a stirred solution of 2-chloro-5-methoxy-4-[[4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidine (220 mg, 551.70 μmol) and 4- cyclopropyl-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-6- (trideuteriomethoxy)pyrimidine I-9 (231 mg, 827.55 μmol) in THF (1 mL) and water (0.2 mL) were added potassium phosphate (351.3 mg, 1.66 mmol) and [1,1’- Bis(dicyclohexylphosphino)ferrocene]dichloropalladium(II) (46.5 mg, 55.17 μmol) at 25 °C. The resulting mixture was stirred for 16 hrs at 40 °C under nitrogen atmosphere. The mixture was allowed to cool down to 25 °C and was concentrated under reduced pressure. The residue was purified by silica gel column chromatography, eluted with 1% - 100% EtOAc in PE to afford crude product. The obtained crude product was purified by RP-Flash with the following conditions: Column: C18 spherical Column, 20-35 um, 100A, 40 g; Mobile Phase A: Water (5 mM aq. NH
4HCO
3), Mobile Phase B: MeCN; Flow rate: 35 mL/min; Gradient: 5% B to 5% B in 5 min, 5% B to 68% B in 19 min, 68% B to 68% B in 2 min, 95% B to 95% B in 5 min; Detector: UV 254 & 210 nm. The collected fractions were combined and lyophilized to give 2-[4-cyclopropyl-6-(trideuteriomethoxy)pyrimidin-5-yl]-5-methoxy-4-[[4- [1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidine, 148.6 mg, 288.26 μmol, 52% yield) as a white solid. MS: m/z = 516.35 [M + H]
+.
1H NMR (400 MHz, Chloroform-d) δ 8.67 (s, 1H), 8.26 (s, 1H), 7.66 (d, J = 8.4 Hz, 2H), 7.60 (d, J = 8.0 Hz, 2H), 7.34 (s, 1H), 5.60 (s, 2H), 4.03 (s, 3H), 3.79 (s, 3H), 1.78 - 1.70 (m, 1H), 1.26 - 1.23 (m, 2H), 0.94 - 0.89 (m, 2H).
19F NMR (377 MHz, Chloroform-d) δ -62.76. Example 190 (Compound 125)
Compound 125 4-cyclopropyl-5-[4-[[6-[1-cyclopropyl-4-(trifluoromethyl)imidazol-2-yl]-5-fluoro-3- pyridyl]methoxy]pyrimidin-2-yl]-6-methoxy-pyrimidine Synthesis of the [6-[1-cyclopropyl-4-(trifluoromethyl)imidazol-2-yl]-5-fluoro-3- pyridyl]methanol is described for Compound 43. [1555] Potassium tert-butoxide (14.9 mg, 133 μmol) was added to a solution of 4- cyclopropyl-6-methoxy-5-(4-methylsulfonylpyrimidin-2-yl)pyrimidine I-8 (40.7 mg, 133 μmol) and [6-[1-cyclopropyl-4-(trifluoromethyl)imidazol-2-yl]-5-fluoro-3-pyridyl]methanol (40 mg, 133 μmol) in THF (6 mL). The resulting mixture was stirred at room temperature for 13 hr. The reaction mixture was poured into a saturated aqueous solution of NH
4Cl (15 mL) and extracted with MTBE (3×15 mL). The combined organic layers were dried over anhydrous Na
2SO
4 and concentrated under reduced pressure. The residue was dissolved in ACN (6 mL). SiliaMetS
® Dimercaptotriazine (100 mg) was added to the solution. The resulting suspension was stirred for 20 hr. at room temperature and filtered. The filtrate was subjected to HPLC (0-6 min., 40-60% water – ACN, +0.1% vol. of 25% aq. NH
3, flow: 60 mL/min, column: XBridge OBD C18100×30 mm, 5 µm) to afford 4-cyclopropyl-5-[4-[[6-[1- cyclopropyl-4-(trifluoromethyl)imidazol-2-yl]-5-fluoro-3-pyridyl]methoxy]pyrimidin-2-yl]- 6-methoxy-pyrimidine (31.0 mg, 58.8 μmol, 44.3% yield) as a yellow solid.
1H NMR (600 MHz, DMSO-d
6) δ 0.84 – 0.94 (m, 6H), 1.03 – 1.07 (m, 2H), 1.67 – 1.73 (m, 1H), 3.72 – 3.78 (m, 1H), 3.85 (s, 3H), 5.59 (s, 2H), 7.12 (d, 1H), 8.04 – 8.09 (m, 2H), 8.68 (s, 1H), 8.71 (s, 1H), 8.74 (d, 1H). MS (ESI): [M+H]
+ m/z: calcd 528.20; found 528.0.
Methyl 2-bromo-4-[4-(trifluoromethyl)-1H-imidazol-2-yl]benzoate [1556] 3,3-Dibromo-1,1,1-trifluoro-propan-2-one (9.04 g, 33.5 mmol, 4.6 mL) was added to a solution of sodium acetate (5.49 g, 67.0 mmol) in water (40 mL). The reaction mixture was stirred at 95 °C for 1 hr. The reaction mixture was cooled to room temperature and poured into a solution of methyl 2-bromo-4-formyl-benzoate (7.40 g, 30.5 mmol) in MeOH (300 mL) and concentrated aqueous NH
4OH (30 mL). The resulting mixture was stirred at room temperature for 14 hr. The reaction mixture was concentrated under reduced pressure. The residue was diluted with water (300 mL) and extracted with CHCl
3 (3×100 mL). The combined organic layers were dried over anhydrous Na
2SO
4 and filtered through a thin pad of silica gel. The filtrate was concentrated under reduced pressure to afford methyl 2-bromo-4- [4-(trifluoromethyl)-1H-imidazol-2-yl]benzoate (10.0 g, 28.6 mmol, 94.1% yield) as a yellow gum which was used in the next steps without further purification.
1H NMR (400 MHz, DMSO-d
6) δ 3.88 (s, 3H), 7.91 (d, 1H), 8.04 (s, 1H), 8.08 (d, 1H), 8.33 (s, 1H), 13.52 (br. s., 1H). MS (ESI): [M+H]
+ m/z: calcd 348.98, 350.98; found 349.0, 351.0 Methyl 2-bromo-4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]benzoate [1557] Cs
2CO
3 (18.7 g, 57.3 mmol) and iodomethane (5.29 g, 37.2 mmol, 2.32 mL) were added to a solution of methyl 2-bromo-4-[4-(trifluoromethyl)-1H-imidazol-2-yl]benzoate (10.0 g, 28.6 mmol) in ACN (300 mL). The reaction mixture was stirred at 40 °C for 12 hr. The reaction mixture was cooled to room temperature, poured into water (300 mL) and extracted with EtOAc (4×120 mL). The combined organic layers were dried over anhydrous Na
2SO
4 and filtered through a thin pad of silica gel. The filtrate was concentrated under reduced pressure to afford methyl 2-bromo-4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]benzoate (4.00 g, 11.0 mmol, 38.5% yield) as a yellow solid which was used in the next step without further purification.
1H NMR (400 MHz, CDCl
3) δ 3.80 (s, 3H), 3.94 (s, 3H), 7.33 (s, 1H), 7.64 (d, 1H), 7.88 (d, 1H), 7.97 (s, 1H). MS (ESI): [M+H]
+ m/z: calcd 363.00, 364.99; found 363.0 Methyl 4-(1-methyl-4-(trifluoromethyl)-1H-imidazol-2-yl)-2-vinylbenzoate [1558] Methyl 2-bromo-4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]benzoate (2.00 g, 5.51 mmol), potassium trifluoro(vinyl)boron (1.10 g, 8.26 mmol), K
2CO
3 (1.52 g, 11.0 mmol, 665 μL) and bis(diphenylphosphino)ferrocene]dichloropalladium(II)-DCM (202 mg, 275 μmol) were mixed in degassed DMSO (20 mL). The reaction mixture was stirred at 100°C for 3 hr. The reaction mixture was cooled to room temperature, diluted with water (50 mL) and extracted with EtOAc (2×30 mL). The combined organic layers were washed with water (20 mL) and brine (20 mL), dried over anhydrous Na
2SO
4 and concentrated under reduced pressure to afford methyl 4-(1-methyl-4-(trifluoromethyl)-1H-imidazol-2-yl)-2-vinylbenzoate (1.50 g, crude) as a brown solid which was used in the next step without further purification. MS (ESI): [M+H]
+ m/z: calcd 311.1; found 311.0 Methyl 2-ethyl-4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]benzoate [1559] 10% Palladium on carbon (300 mg) was added to a solution of methyl 4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]-2-vinyl-benzoate (1.50 g, crude) in MeOH (60 mL). The reaction mixture was evacuated and backfilled with hydrogen. The reaction mixture was stirred at room temperature for 12 hr under hydrogen atmosphere. The reaction mixture was filtered. The filtrate was concentrated under reduced pressure to afford 2-ethyl-4-[1-methyl- 4-(trifluoromethyl)imidazol-2-yl]benzoate (1.50 g, crude) as a brown gum which was used in the next step without further purification. MS (ESI): [M+H]
+ m/z: calcd 313.12; found 313.0 [2-ethyl-4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methanol [1560] DIBAL (2.05 g, 14.4 mmol, 2.9 mL) was added dropwise to a stirred solution of methyl 2-ethyl-4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]benzoate (1.50 g, crude) in THF (30 mL) at -78 °C under argon atmosphere. The reaction mixture was stirred at ambient temperature for 12 hr. The reaction mixture was cooled to -20 °C and quenched by dropwise addition of an aqueous solution of NaOH (3 mL, 10%). To the obtained mixture NaOH (5 g, solid) was added. The resulting mixture was stirred at room temperature for 20 min. The organic solvent was decanted and concentrated under reduced pressure. The residue was subjected to flash column chromatography (silica, gradient Hexane - EtOAc) to afford [2- ethyl-4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methanol (450 mg, 1.58 mmol, 28.7% yield from methyl 2-bromo-4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]benzoate) as a beige solid.
1H NMR (400 MHz, CDCl
3) δ 1.23 (t, 3H), 2.70 (q, 2H), 3.74 (s, 3H), 4.64 – 4.78 (m, 3H), 7.29 (s, 1H), 7.38 (d, 1H), 7.44 – 7.48 (m, 2H). MS (ESI): [M+H]
+ m/z: calcd 285.12; found 285.2 4-cyclopropyl-5-[4-[[2-ethyl-4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidin-2-yl]-6-methoxy-pyrimidine [1561] NaH (13.5 mg, 338 μmol, 60% dispersion in mineral oil) was added to a solution of [2-ethyl-4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methanol (80.0 mg, 281 μmol) in THF (4.0 mL) at room temperature. The reaction mixture was stirred at room temperature for 15 min.4-cyclopropyl-6-methoxy-5-(4-methylsulfonylpyrimidin-2-yl)pyrimidine I-8 (86.2 mg, 281 μmol) was added to the reaction mixture. The resulting mixture was stirred at room temperature for 12 hr. The reaction mixture was diluted with water (5 mL) and extracted with EtOAc (2×5 mL). The combined organic layers were washed with brine (5 mL) and concentrated under reduced pressure. The residue was subjected to HPLC (1.5-9 min., 47-55% water – ACN, flow: 30 mL/min, column: SunFire C18 (R) 100×19 mm, 5 µm) to afford 4-cyclopropyl-5-[4-[[2-ethyl-4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidin-2-yl]-6-methoxy-pyrimidine (19.0 mg, 37.2 μmol, 13.2% yield) as a colorless gum.
1H NMR (600 MHz, DMSO-d
6) δ 0.86 – 0.91 (m, 2H), 1.03 – 1.07 (m, 2H), 1.19 (t, 3H), 1.69 – 1.75 (m, 1H), 2.75 (q, 2H), 3.79 (s, 3H), 3.86 (s, 3H), 5.53 (s, 2H), 7.05 (d, 1H), 7.52 – 7.57 (m, 2H), 7.60 (s, 1H), 7.94 (s, 1H), 8.68 (s, 1H), 8.70 (d, 1H). MS (ESI): [M+H]
+ m/z: calcd 511.24; found 511.2. Example 192 (Compound 123)
2,5-dichloro-4-methylsulfanyl-pyrimidine [1562] An aqueous solution of sodium thiomethoxide (3.64 g of 21% aqueous solution, 10.9 mmol) was added dropwise to a solution of 2,4,5-trichloropyrimidine (2.00 g, 10.9 mmol) in THF (30 mL) at -10 °C. The reaction mixture was stirred at room temperature for 16 hr. The obtained mixture was concentrated in vacuo. The residue was diluted with water (25 mL) and extracted with MTBE (4 × 30 mL). The combined organic layers were dried over anhydrous Na
2SO
4 and concentrated in vacuo to afford 2,5-dichloro-4-methylsulfanyl-pyrimidine (1.60 g, 8.20 mmol, 75.2% yield) as a light-yellow solid which was used in the next steps without further purification. MS (ESI): [M+H]
+ m/z: calcd 194.96; found 195.0. 5-chloro-2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-methylsulfanyl-pyrimidine [1563] A mixture of water (1.00 mL) and dioxane (10.0 mL) was evacuated and then backfilled with argon.2,5-dichloro-4-methylsulfanyl-pyrimidine (450 mg, 2.31 mmol), (4- cyclopropyl-6-methoxy-pyrimidin-5-yl)boronic acid I-5 (537 mg, 2.77 mmol), RuPhos Pd G4 (98.1 mg, 115 μmol) and potassium phosphate tribasic (1.47 g, 6.92 mmol) were added in an inert atmosphere at room temperature. The reaction mixture was stirred at 35 °C for 16 hr. The obtained mixture was cooled to room temperature and concentrated in vacuo. The residue was diluted with H
2O (10.0 mL) and extracted with EtOAc (2×15.0 mL). The combined organic layers were dried over anhydrous Na
2SO
4 and concentrated in vacuo. The residue was subjected to HPLC (2-10 min, 20-45% water – ACN, flow: 30 mL/min, column: SunFire C18100 ×19 mm, 5 µm) to afford 5-chloro-2-(4-cyclopropyl-6-methoxy-pyrimidin- 5-yl)-4-methylsulfanyl-pyrimidine (244 mg, 790 μmol, 34.3% yield) as a white solid. MS (ESI): [M+H]
+ m/z: calcd 309.06; found 309.0. 5-chloro-2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-methylsulfonyl-pyrimidine [1564] mCPBA (246 mg, 1.42 mmol, 75% purity) was added to a stirred solution of 5- chloro-2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-methylsulfanyl-pyrimidine (200 mg, 648 μmol) in DCM (25 mL). The resulting mixture was stirred at room temperature for 24 hr. The obtained mixture was washed with saturated aqueous NaHCO
3, dried over anhydrous Na
2SO
4 and concentrated in vacuo to afford 5-chloro-2-(4-cyclopropyl-6-methoxy-pyrimidin- 5-yl)-4-methylsulfonyl-pyrimidine (186 mg, 546 μmol, 84.3% yield) as a light-yellow solid which was used for the next step without further purification. MS (ESI): [M+H]
+ m/z: calcd 341.05; found 341.0. 5-chloro-2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[4-[1-isopropyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidine Synthesis of the starting [4-[1-isopropyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methanol is described for Compound 37. [1565] To a solution of [4-[1-isopropyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methanol (155 mg, 546 μmol) in DMF (5 mL) NaH (41.8 mg, 1.09 mmol, 60% dispersion in mineral oil) was added. The reaction mixture was stirred at room temperature for 30 min. To the obtained mixture 5-chloro-2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-methylsulfonyl- pyrimidine (186 mg, 546 μmol) was added and the mixture was stirred room temperature for 15 hr. The reaction mixture was poured into H
2O (10 mL) and extracted with EtOAc (20 mL). The organic layer was dried over anhydrous Na
2SO
4 and concentrated in vacuo. The residue was subjected to HPLC (2-10 min, 30% water+FA (0.1% vol.) - ACN+FA (0.1% vol.); flow: 30 mL/min, column: Waters SunFire C18, 100 x 19 mm, 5 µm) to afford 5- chloro-2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[4-[1-isopropyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidine (4.1 mg, 7.5 μmol, 1.4% yield) as yellow oil.
1H NMR (600 MHz, DMSO-d
6) δ 0.86 – 0.91 (m, 2H), 1.03 – 1.07 (m, 2H), 1.41 (d, 2H), 1.76 – 1.81 (m, 1H), 3.86 (s, 3H), 4.45 – 4.51 (m, 1H), 5.61 (s, 2H), 7.59 – 7.64 (m, 4H), 8.19 (s, 1H), 8.69 (s, 1H), 8.90 (s, 1H). MS (ESI): [M+H]
+ m/z: calcd 545.17; found 545.0. Example 193 (Compound 120)
2-chloro-4-methyl-6-methylsulfanyl-pyrimidine Synthesis of the starting [5-fluoro-6-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]-3- pyridyl]methanol is described for compound 48. [1566] Sodium methanethiolate (32.2 mmol, 10.8 mL, 21% wt. in water) was added to a stirred solution of 2,4-dichloro-6-methyl-pyrimidine (5.00 g, 30.7 mmol) in THF (50 mL) at 5 °C. The resulting mixture was stirred at room temperature for 16 hr. The reaction mixture was concentrated under reduced pressure. The residue was dissolved in MTBE (50 mL) and washed with water (3×10 mL). The combined organic layers were washed with brine, dried over anhydrous Na
2SO
4 and filtered. The filtrate was concentrated under reduced pressure. The residue was subjected to flash column chromatography (silica, gradient Hexane - MTBE) to afford 2-chloro-4-methyl-6-methylsulfanyl-pyrimidine (2.60 g, 14.9 mmol, 48.5% yield) as a white solid.
1H NMR (500 MHz, CDCl
3) δ 2.40 (s, 3H), 2.55 (s, 3H), 6.93 (s, 1H). MS (ESI): [M+H]
+ m/z: calcd 175.01; found 175.0 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-methyl-6-methylsulfanyl-pyrimidine [1567] 2-Chloro-4-methyl-6-methylsulfanyl-pyrimidine (2.00 g, 11.5 mmol), (4-cyclopropyl- 6-methoxy-pyrimidin-5-yl)boronic acid I-5 (3.33 g, 17.2 mmol), K
2CO
3 (4.75 g, 34.4 mmol,) and bis(diphenylphosphino)ferrocene]dichloropalladium(II)-DCM (935 mg, 1.15 mmol) were mixed in a degassed mixture of dioxane (35 mL) and water (2 mL) under argon atmosphere. The reaction mixture was stirred at 90 °C for 12 hr. The reaction mixture was cooled to room temperature and filtered. The filtrate was concentrated under reduced pressure to afford 2-(4- cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-methyl-6-methylsulfanyl-pyrimidine (3.20 g, 11.1 mmol, 97% yield) as a solid which was used in the next step without further purification. MS (ESI): [M+H]
+ m/z: calcd 289.11; found 289.0 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-methyl-6-methylsulfonyl-pyrimidine [1568] mCPBA (4.86 g, 23.9 mmol, 85% purity) was added to a stirred solution of 2-(4- cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-methyl-6-methylsulfanyl-pyrimidine (2.30 g, 7.98 mmol) in DCM (30 mL). The reaction mixture was stirred at room temperature for 12 hr. The reaction mixture was washed with saturated aqueous solution of NaHCO
3 (20 mL). The organic layer was separated, washed with water, brine, dried over anhydrous Na
2SO
4 and filtered. The filtrate was concentrated under reduced pressure. The residue was subjected to flash column chromatography (silica, gradient chloroform - acetonitrile) to afford 2-(4- cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-methyl-6-methylsulfonyl-pyrimidine (1.30 g, 4.06 mmol, 50.9% yield) as a light-yellow solid.
1H NMR (500 MHz, CDCl
3) δ 0.85 – 0.96 (m, 2H), 1.17 – 1.27 (m, 2H), 1.57 – 1.70 (m, 1H), 2.75 (s, 3H), 3.26 (s, 3H), 3.89 (s, 3H), 7.83 (s, 1H), 8.63 (s, 1H). MS (ESI): [M+H]
+ m/z: calcd 321.10; found 321.2 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[5-fluoro-6-[1-ethyl-4- (trifluoromethyl)imidazol-2-yl]-3-pyridyl]methoxy]-6-methyl-pyrimidine [1569] Sodium tert-butoxide (74.8 mg, 778 μmol) was added to a solution of [5-fluoro-6-[1- ethyl-4-(trifluoromethyl)imidazol-2-yl]-3-pyridyl]methanol (149 mg, 519 μmol) and 2-(4- cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-methyl-6-methylsulfonyl-pyrimidine (216 mg, 674 μmol) in THF (2 mL). The resulting mixture was stirred at room temperature for 13 hr. The reaction mixture was diluted with water (500 µL) and extracted with EtOAc (2×500 µL). The combined organic layers were washed with brine (700 µl), dried over anhydrous Na
2SO
4 and filtered. The filtrate was concentered under reduced pressure. The residue was subjected to HPLC (0-1.3-6.3 min, 40-40-90% water – MeOH, +0.1% vol. of 25% aq. NH
3; flow: 30 mL/min, column: XBridge BEH C18, 5 µm; then 0-1.3-6.3 min, 45-45-65% water+FA (0.1% vol.) - ACN+FA (0.1% vol.); flow: 30 mL/min, column: Chromatorex 18 SMB100-5T, 100×19 mm, 5 µm) to afford 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[5-fluoro-6-[1- ethyl-4-(trifluoromethyl)imidazol-2-yl]-3-pyridyl]methoxy]-6-methyl-pyrimidine (36.2 mg, 68.3 μmol, 13.6% yield) as a yellow gum.
1H NMR (600 MHz, DMSO-d
6) δ 0.87 – 0.92 (m, 2H), 1.02 – 1.06 (m, 2H), 1.33 (t, 3H), 1.63 – 1.68 (m, 1H), 2.47 (s, 3H), 3.85 (s, 3H), 4.25 (q, 2H), 5.55 (s, 2H), 6.97 (s, 1H), 8.03 (d, 1H), 8.15 (s, 1H), 8.67 (s, 2H). MS (ESI): [M+H]
+ m/z: calcd 530.22; found 530.2.
Isopropyl 5-fluoro-6-[1-isopropyl-4-(trifluoromethyl)imidazol-2-yl]pyridine-3-carboxylate Synthesis of the starting 5-fluoro-6-[4-(trifluoromethyl)-1H-imidazol-2-yl]pyridine-3- carboxylic acid was described for Compound 48. [1570] Methyl 5-fluoro-6-[4-(trifluoromethyl)-1H-imidazol-2-yl]pyridine-3-carboxylate (180 mg, 622 μmol), 2-bromopropane (230 mg, 1.87 mmol, 175 μL) and Cs
2CO
3 (608 mg, 1.87 mmol) were mixed in DMF (0.5 mL). The mixture was stirred at 80 °C for 12 hr. The mixture was cooled to room temperature and diluted with water (5 mL). The mixture was extracted with MTBE (10 mL). The organic layer was separated, washed with brine (5 mL) and concentrated under reduced pressure to afford isopropyl 5-fluoro-6-[1-isopropyl-4- (trifluoromethyl)imidazol-2-yl]pyridine-3-carboxylate (150 mg, 417 μmol, 67.1% yield) as a yellow oil which was used in the next steps without further purification. MS (ESI): [M+H]
+ m/z: calcd 360.13; found 360.1 [5-fluoro-6-[1-isopropyl-4-(trifluoromethyl)imidazol-2-yl]-3-pyridyl]methanol [1571] A solution of isopropyl 5-fluoro-6-[1-isopropyl-4-(trifluoromethyl)imidazol-2- yl]pyridine-3-carboxylate (150 mg, 417 μmol) in diethyl ether (5 mL) was added dropwise to a stirred suspension of lithium alumohydride (32 mg, 830 μmol) in diethyl ether (20 mL) at 0 °C. The reaction mixture was stirred at 0 °C for 20 min. The mixture was quenched with aqueous potassium hydroxide (150 μL, 30% wt.) and filtered. The filtrate was concentrated under reduced pressure to afford [5-fluoro-6-[1-isopropyl-4-(trifluoromethyl)imidazol-2-yl]- 3-pyridyl]methanol (121 mg, 396 μmol, 94.8% yield) as a yellow oil which was used in the next steps without further purification. MS (ESI): [M+H]
+ m/z: calcd 304.11; found 304.0 5-chloro-2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[5-fluoro-6-[1-isopropyl-4- (trifluoromethyl)imidazol-2-yl]-3-pyridyl]methoxy]pyrimidine Synthesis of the starting 5-chloro-2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4- methylsulfonyl-pyrimidine is described for Compound 123. [1572] Potassium tert-butoxide (36.1 mg, 322 μmol) was added to a stirred solution of [5- fluoro-6-[1-isopropyl-4-(trifluoromethyl)imidazol-2-yl]-3-pyridyl]methanol (65.0 mg, 214 μmol) and 5-chloro-2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-methylsulfonyl- pyrimidine (73.0 mg, 214 μmol) in THF (1 mL) at 0 °C. The reaction mixture was stirred at 0 °C for 1 hr. The reaction mixture was quenched by addition of water (0.1 mL). The resulting mixture was directly subjected to HPLC (0-2-9 min., 48-55-70% water – ACN, flow: 30 mL/min, column: Chromatorex C18 SMB100-5T 100×19 mm, 5 µm) to afford 5-chloro-2-(4- cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[5-fluoro-6-[1-isopropyl-4- (trifluoromethyl)imidazol-2-yl]-3-pyridyl]methoxy]pyrimidine (9.00 mg, 16.0 μmol, 7.5% yield) as a yellow solid.
1H NMR (600 MHz, DMSO-d
6) δ 0.89 – 0.93 (m, 2H), 1.04 – 1.08 (m, 2H), 1.42 (d, 6H), 1.77 – 1.83 (m, 1H), 3.86 (s, 3H), 4.77 – 4.84 (m, 1H), 5.67 (s, 2H), 8.06 (d, 1H), 8.31 (s, 1H), 8.70 (s, 1H), 8.71 (s, 1H), 8.92 (s, 1H). MS (ESI): [M+H]
+ m/z: calcd 564.18; found 564.2.
Methyl 6-[1-ethyl-4-(trifluoromethyl)imidazol-2-yl]-5-fluoro-pyridine-3-carboxylate Synthesis of the starting 5-chloro-2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4- methylsulfonyl-pyrimidine is described for Compound 123. Synthesis of the starting methyl 5-fluoro-6-[4-(trifluoromethyl)-1H-imidazol-2- yl]pyridine-3-carboxylate is described for Compound 43. [1573] A mixture of methyl 5-fluoro-6-[4-(trifluoromethyl)-1H-imidazol-2-yl]pyridine-3- carboxylate (0.500 g, 1.73 mmol), iodoethane (539 mg, 3.46 mmol, 278 μL) and K
2CO
3 (478 mg, 3.46 mmol) in DMF (7 mL) was stirred at room temperature for 12 hr. The mixture was poured into cold water (50 mL) and extracted with EA (2×30 mL). Combined organic layers were washed with brine (30 mL), dried over anhydrous Na
2SO
4 and concentrated under reduced pressure to afford methyl 6-[1-ethyl-4-(trifluoromethyl)imidazol-2-yl]-5-fluoro- pyridine-3-carboxylate (480 mg, 1.51 mmol, 87.5% yield) as a yellow gummy solid which was used in the next step without further purification.
1H NMR (400 MHz, CDCl
3) δ 1.45 (t, 3H), 3.98 (s, 3H), 4.37 (q, 2H), 7.44 (s, 1H), 8.12 (d, 1H), 9.06 (s, 1H). MS (ESI): [M+H]
+ m/z: calcd 318.09; found 318.0 [6-[1-ethyl-4-(trifluoromethyl)imidazol-2-yl]-5-fluoro-3-pyridyl]methanol [1574] DIBAL (2.98 mL, 3.19 mmol, 1.07 M) was added dropwise to a solution of methyl 6- [1-ethyl-4-(trifluoromethyl)imidazol-2-yl]-5-fluoro-pyridine-3-carboxylate (460 mg, 1.45 mmol) in THF (10 mL) at -50 °C. The mixture was allowed to warm to room temperature and stirred for 12 hr. The reaction mixture was cooled to 0 °C and quenched by dropwise addition of aqueous sodium hydroxide solution (0.45 mL, 28.5% wt.). Solid precipitate was filtered out. The filtrate was concentrated under reduced pressure. The residue was subjected to HPLC (0.5-6.5 min, 15-40% water – ACN, flow: 30 mL/min, column: CROMATOREX C18 100 ×19 mm, 5 µm) to afford [6-[1-ethyl-4-(trifluoromethyl)imidazol-2-yl]-5-fluoro-3- pyridyl]methanol (143 mg, 494 μmol, 34.1% yield) as an off-white solid. MS (ESI): [M+H]
+ m/z: calcd 290.09; found 290.0 5-chloro-2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[6-[1-ethyl-4- (trifluoromethyl)imidazol-2-yl]-5-fluoro-3-pyridyl]methoxy]pyrimidine Synthesis of the starting 5-chloro-2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4- methylsulfonyl-pyrimidine is described for Compound 123. [1575] Potassium tert-butoxide (58.2 mg, 519 μmol) was added to a solution of [6-[1-ethyl-4- (trifluoromethyl)imidazol-2-yl]-5-fluoro-3-pyridyl]methanol (100 mg, crude) and 5-chloro-2- (4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-methylsulfonyl-pyrimidine (118 mg, 346 μmol) in THF (1 mL) at 0 °C. The reaction mixture was stirred at 0 °C for 1 hr. The reaction mixture was quenched by addition of water (0.1 mL). The resulting mixture was directly subjected to HPLC (0-2-9 min., 43-50-65% water – ACN, flow: 30 mL/min, column: Chromatorex C18 SMB100-5T 100×19 mm, 5 µm) to afford 5-chloro-2-(4-cyclopropyl-6- methoxy-pyrimidin-5-yl)-4-[[6-[1-ethyl-4-(trifluoromethyl)imidazol-2-yl]-5-fluoro-3- pyridyl]methoxy]pyrimidine (15.0 mg, 27.3 μmol, 7.9% yield) as a yellow solid.
1H NMR (500 MHz, DMSO-d
6) δ 0.89 – 0.94 (m, 2H), 1.04 – 1.09 (m, 2H), 1.34 (t, 3H), 1.77 – 1.84 (m, 1H), 3.86 (s, 3H), 4.26 (q, 2H), 5.67 (s, 2H), 8.06 (d, 1H), 8.16 (s, 1H), 8.68 – 8.72 (m, 2H), 8.92 (s, 1H). MS (ESI): [M+H]
+ m/z: calcd 550.16; found 550.2.
Methyl 3-fluoro-4-[4-(trifluoromethyl)-1H-imidazol-2-yl]benzoate [1576] A mixture of 3,3-dibromo-1,1,1-trifluoro-propan-2-one (12.7 g, 47.1 mmol), sodium acetate (7.03 g, 85.6 mmol) and water (10 mL) was stirred at 100 °C for 45 min. The mixture was cooled to ambient temperature. Methyl 3-fluoro-4-formyl-benzoate (7.80 g, 42.8 mmol) and a solution of ammonia (35.0 g, 25% wt. in water) in MeOH (100 mL) were added to the mixture. The resulting mixture was stirred at room temperature for 12 hr. The reaction mixture was concentrated under reduced pressure to a half volume. The residue was extracted with DCM (50 mL). The organic layer was separated, washed with water (20 mL), dried over anhydrous Na
2SO
4 and filtered. The filtrate was concentrated under reduced pressure. The residue was triturated with hexane. The solid formed was collected by filtration and air-dried to afford methyl 3-fluoro-4-[4-(trifluoromethyl)-1H-imidazol-2-yl]benzoate (7.0 g, 24.3 mmol, 56.7% yield) as a yellow solid which was used in the next steps without further purification.
1H NMR (400 MHz, CDCl
3) δ 3.94 (s, 3H), 7.52 (s, 1H), 7.83 (d, 1H), 7.93 (d, 1H), 8.37 (t, 1H), 10.20 (br. s., 1H). MS (ESI): [M+H]
+ m/z: calcd 289.06; found 289.1 Methyl 4-[1-cyclopropyl-4-(trifluoromethyl)imidazol-2-yl]-3-fluoro-benzoate [1577] Methyl 3-fluoro-4-[4-(trifluoromethyl)-1H-imidazol-2-yl]benzoate (2.00 g, 6.94 mmol) was suspended in dioxane (27.7 mL) and stirred at room temperature for 30 min. Cyclopropylboronic acid (8.94 g, 104 mmol), copper bromide (697 mg, 4.86 mmol) and pyridine (7.68 g, 97.2 mmol, 7.9 mL) were added to the reaction mixture. The reaction mixture was vigorously stirred at room temperature for 144 hr with air access. The reaction mixture was concentrated under reduced pressure. The residue was subjected to flash column chromatography (silica, gradient DCM - EtOAc) to afford methyl 4-[1-cyclopropyl-4- (trifluoromethyl)imidazol-2-yl]-3-fluoro-benzoate (750 mg, 2.28 mmol, 32.9% yield) as a colorless oil.
1H NMR (400 MHz, CDCl
3) δ 0.67 – 0.73 (m, 2H), 0.86 – 0.94 (m, 2H), 3.46 – 3.52 (m, 1H), 3.95 (s, 3H), 7.41 (s, 1H), 7.69 (t, 1H), 7.83 (d, 1H), 7.92 (d, 1H). MS (ESI): [M+H]
+ m/z: calcd 329.09; found 329.2 [4-[1-cyclopropyl-4-(trifluoromethyl)imidazol-2-yl]-3-fluoro-phenyl]methanol [1578] A solution of methyl 4-[1-cyclopropyl-4-(trifluoromethyl)imidazol-2-yl]-3-fluoro- benzoate (0.75 g, 2.28 mmol) in Et
2O (5 mL) was added dropwise to a stirred suspension of LAH (261 mg, 6.85 mmol) in Et
2O (30 mL) at 0 °C. The reaction mixture was stirred at 0 °C for 20 min. The reaction mixture was quenched by dropwise addition of an aqueous KOH (1.2 mL, 30% wt.). The solids were filtered and the filtrate was concentrated under reduced pressure to afford [4-[1-cyclopropyl-4-(trifluoromethyl)imidazol-2-yl]-3-fluoro- phenyl]methanol (650 mg, 2.16 mmol, 94.8% yield) as a yellow oil which was used in the next step without further purification. MS (ESI): [M+H]
+ m/z: calcd 301.10; found 301.1 2-chloro-4-[[4-[1-cyclopropyl-4-(trifluoromethyl)imidazol-2-yl]-3-fluoro-phenyl]methoxy]- 5-methoxy-pyrimidine [1579] A mixture of [4-[1-cyclopropyl-4-(trifluoromethyl)imidazol-2-yl]-3-fluoro- phenyl]methanol (100 mg, 333 μmol) and potassium tert-butoxide (56.1 mg, 500 μmol) in THF (2 mL) was stirred at room temperature for 1 hr under argon atmosphere.2,4-dichloro- 5-methoxy-pyrimidine (119 mg, 666 μmol) was added to the mixture at -20 °C. The reaction mixture was stirred at -20 °C for 1 hr. The reaction mixture was allowed to warm to room temperature and concentrated under reduced pressure. The residue was diluted with MTBE (30 mL) and washed with water (15 mL). The organic layer was separated, washed with brine (20 mL), dried over anhydrous Na
2SO
4 and concentrated under reduced pressure. The residue was subjected to flash column chromatography (silica, gradient hexane - EtOAc) to afford 2- chloro-4-[[4-[1-cyclopropyl-4-(trifluoromethyl)imidazol-2-yl]-3-fluoro-phenyl]methoxy]-5- methoxy-pyrimidine (100 mg, 226 μmol, 67.8% yield) as a white solid. MS (ESI): [M+H]
+ m/z: calcd 443.09; found 443.2 2-[4-cyclopropyl-6-(difluoromethoxy)pyrimidin-5-yl]-4-[[4-[1-cyclopropyl-4- (trifluoromethyl)imidazol-2-yl]-3-fluoro-phenyl]methoxy]-5-methoxy-pyrimidine [1580] 2-Chloro-4-[[4-[1-cyclopropyl-4-(trifluoromethyl)imidazol-2-yl]-3-fluoro- phenyl]methoxy]-5-methoxy-pyrimidine (60.0 mg, 136 μmol), 4-cyclopropyl-6- (difluoromethoxy)-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyrimidine I-10 (63.4 mg, 203 μmol), potassium phosphate tribasic (57.5 mg, 271 μmol) and XPhos Pd G3 (5.7 mg, 6.78 μmol) were mixed in a degassed mixture of dioxane (2 mL) and water (200 µL). The reaction mixture was stirred at 90 °C for 5 hr. The reaction mixture was cooled to room temperature, filtered and subjected to HPLC (0-2-9 min., 43-50-60% water – ACN, +0.1% vol. of 25% aq. NH
3, flow: 30 mL/min, column: XBridge BEH 100×19 mm, 5 µm) to afford 2-[4-cyclopropyl-6-(difluoromethoxy)pyrimidin-5-yl]-4-[[4-[1-cyclopropyl-4- (trifluoromethyl)imidazol-2-yl]-3-fluoro-phenyl]methoxy]-5-methoxy-pyrimidine (23.0 mg, 38.8 μmol, 28.7% yield) as a white solid.
1H NMR (500 MHz, DMSO-d
6) δ 0.81 – 0.86 (m, 4H), 0.96 – 1.02 (m, 2H), 1.09 – 1.13 (m, 2H), 1.87 – 1.94 (m, 1H), 3.44 – 3.50 (m, 1H), 3.99 (s, 3H), 5.55 (s, 2H), 7.45 (d, 1H), 7.49 (d, 1H), 7.65 (t, 1H), 7.81 (t, 1H, CHF
2), 8.01 (s, 1H), 8.52 (s, 1H), 8.79 (s, 1H). MS (ESI): [M+H]
+ m/z: calcd 593.20; found 593.2. Example 197 (Compound 116)
2-chloro-5-ethoxy-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidine Synthesis of the starting 2-chloro-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidin-5-ol is described for Compound 122. [1581] Iodoethane (243 mg, 1.56 mmol, 125 μL) was added to a stirred mixture of 2-chloro- 4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidin-5-ol (400 mg, 1.04 mmol) and K
2CO
3 (287 mg, 2.08 mmol) in DMF (15 mL). The resulting mixture was stirred at 40 °C for 13 hr. The reaction mixture was cooled to room temperature, poured into water (50 mL) and extracted with EtOAc (4×25 mL). The combined organic layers were separated, washed with water (2×5 mL) and brine (2×5 mL), dried over anhydrous Na
2SO
4 and concentrated under reduced pressure. The residue was triturated with hexane and filtered to afford 2-chloro-5-ethoxy-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidine (350 mg, 848 μmol, 81.6% yield) as a white solid.
1H NMR (500 MHz, DMSO-d
6) δ 1.33 (t, 1H), 3.80 (s, 3H), 4.25 (q, 2H), 5.51 (s, 2H), 7.61 (d, 2H), 7.77 (d, 2H), 7.94 (s, 1H), 8.22 (s, 1H). 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-5-ethoxy-4-[[4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidine [1582] 2-Chloro-5-ethoxy-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidine (200 mg, 485 μmol), (4-cyclopropyl-6-methoxy-pyrimidin-5- yl)boronic acid I-5 (141 mg, 727 μmol), potassium phosphate tribasic (309 mg, 1.45 mmol) and XPhos Pd G3 (20.5 mg, 24.2 μmol) were mixed in degassed mixture of dioxane (12 mL) and water (2 mL) under argon atmosphere. The resulting mixture was evacuated and backfilled with argon. The reaction mixture was stirred at 60 °C for 6 hr. The reaction mixture was cooled to room temperature and concentrated under reduced pressure. The residue was diluted with EtOAc (25 mL) and washed with water (10 mL). The organic layer was separated, washed with brine (2×20 mL), dried over anhydrous Na
2SO
4 and concentrated under reduced pressure. The residue was subjected to HPLC (2-10 min, 40-65% water - ACN; flow: 30 mL/min, column: Chromatorex, 100×19 mm, 5 µm), then repurified by HPLC (2-10 min, 30-50% water+FA (0.1% vol.) - ACN+FA (0.1% vol.); flow: 30 mL/min, column: Chromatorex, 100×19 mm, 5 µm) to afford 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-5- ethoxy-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidine (61.0 mg, 116 μmol, 23.9% yield) as a light-yellow solid.
1H NMR (600 MHz, DMSO-d
6) δ 0.84 – 0.88 (m, 2H), 1.00 – 1.04 (m, 2H), 1.38 (t, 3H), 1.68 – 1.74 (m, 1H), 3.79 (s, 3H), 3.84 (s, 3H), 4.23 (q, 2H), 5.52 (s, 2H), 7.57 (d, 2H), 7.74 (d, 2H), 7.94 (s, 1H), 8.42 (s, 1H), 8.65 (s, 1H). MS (ESI): [M+H]
+ m/z: calcd 527.23; found 527.2. Example 198 (Compound 115)
The synthesis of starting 2-chloro-5-methoxy-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidine is described for Compound 12 and compound 132. 4,6-dicyclopropylpyrimidine [1583] Tris(acetylacetonato)iron(III) (4.27 g, 12.1 mmol) was added to a solution of 4,6- dichloropyrimidine (9.00 g, 60.4 mmol) in NMP (25 mL) and THF (180 mL). The resulting mixture was cooled to -20 °C. Bromo(cyclopropyl)magnesium (21.9 g, 151 mmol) was added to the mixture. The reaction mixture was stirred at room temperature for 3 hr. The reaction mixture was poured into saturated aqueous NH
4Cl (75 mL) and extracted with MTBE (100 mL). The organic layer was separated, washed with brine (50 mL) and concentrated under reduced pressure. The residue was subjected to flash column chromatography (SiO
2, gradient hexane - MTBE) to afford 4,6-dicyclopropylpyrimidine (7.50 g, 46.8 mmol, 77.5% yield) as a brown solid.
1H NMR (400 MHz, CDCl
3) δ 1.00 – 1.14 (m, 8H), 1.85 – 1.93 (m, 2H), 6.98 (s, 1H), 8.76 (s, 1H). MS (ESI): [M+H]
+ m/z: calcd 161.11; found 161.2. 5-bromo-4,6-dicyclopropyl-pyrimidine [1584] Bromine (8.98 g, 56.2 mmol, 2.90 mL) was added to a solution of 4,6- dicyclopropylpyrimidine (7.50 g, 46.8 mmol) in AcOH (100 mL). The reaction mixture was stirred at room temperature for 15 hr. An additional portion of Br
2 (7.48 g, 46.8 mmol, 2.42 mL, 1 eq.) was added to the mixture and the resulting mixture was stirred at room temperature for 15 hr. The reaction mixture was concentrated under reduced pressure. The residue was diluted with saturated aqueous solution of NaHCO
3 (50 mL) to pH≈8 and extracted with DCM (150 mL). The organic layer was separated, dried over anhydrous Na
2SO
4 and concentrated under reduced pressure. The residue was subjected to flash column chromatography (SiO
2, n-hexane - MTBE, gradient 9:1→8:2) to afford 5-bromo-4,6- dicyclopropyl-pyrimidine (7.00 g, 29.3 mmol, 62.5% yield) as a brown solid.
1H NMR (500 MHz, CDCl
3) δ 1.05 – 1.21 (m, 8H), 2.51 – 2.60 (m, 2H), 8.59 (s, 1H). MS (ESI): [M+H]
+ m/z: calcd 239.02; found 239.0 4,6-dicyclopropyl-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyrimidine [1585] 5-Bromo-4,6-dicyclopropyl-pyrimidine (500 mg, 2.09 mmol), 4,4,5,5-tetramethyl-2- (4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1,3,2-dioxaborolane (797 mg, 3.14 mmol), Cesium pivalate (1.22 g, 5.23 mmol) and bis(diphenylphosphino)ferrocene]dichloropalladium(II)-DCM (128 mg, 157 μmol) were mixed in degassed dioxane (10 mL) under argon atmosphere at room temperature. The reaction mixture was stirred at 80 °C for 15 hr. The reaction mixture was cooled to room temperature and concentrated under reduced pressure. The residue was diluted with water (10 mL) and extracted with EtOAc (3×20 mL). The combined organic layers were separated, dried over anhydrous Na
2SO
4 and concentrated under reduced pressure to afford 4,6- dicyclopropyl-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyrimidine (600 mg, crude) as a brown solid which was used in the next step without further purification. 4,6-dicyclopropyl-5-[5-methoxy-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidin-2-yl]pyrimidine [1586] 2-Chloro-5-methoxy-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidine (100 mg, 251 μmol), 4,6-dicyclopropyl-5-(4,4,5,5- tetramethyl-1,3,2-dioxaborolan-2-yl)pyrimidine (205 mg, crude from previous step), potassium phosphate tribasic anhydrous (213 mg, 1.00 mmol) and XPhos Pd G3 (15.9 mg, 18.8 μmol) were mixed in degassed mixture of water (1 mL) and dioxane (5 mL) under argon atmosphere at room temperature. The reaction mixture was stirred at 75 °C for 15 hr. The reaction mixture was cooled to room temperature, diluted with water (10 mL) and extracted with EtOAc (3×50 mL). The combined organic layers were separated, dried over anhydrous Na
2SO
4 and concentrated under reduced pressure. The residue was subjected to HPLC (0.5- 6.5 min, 20-35% water+FA (0.1% vol.) - ACN+FA (0.1% vol.); flow: 30 mL/min, column: SunFire C18, 100×19 mm, 5 µm) to afford 4,6-dicyclopropyl-5-[5-methoxy-4-[[4-[1-methyl- 4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidin-2-yl]pyrimidine (20.0 mg, 38.3 μmol, 15.3% yield) as an off-white solid.
1H NMR (500 MHz, DMSO-d
6) δ 0.83 – 0.89 (m, 4H), 0.98 - 1.04 (m, 4H), 1.64 – 1.73 (m, 2H), 3.77 (s, 3H), 3.95 (s, 3H), 5.52 (s, 2H), 7.56 (d, 2H), 7.72 (d, 2H), 7.92 (s, 1H), 8.48 (s, 1H), 8.77 (s, 1H). MS (ESI): [M+H]
+ m/z: calcd 523.24; found 523.2. Example 199 (Compound 112)
Compound 112 2-chloro-4,5-dimethyl-6-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidine [1587] 4-[1-Methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methanol (750 mg, 2.93 mmol) and 2,4-dichloro-5,6-dimethyl-pyrimidine (621.8 mg, 3.51 mmol) were mixed in toluene (30 mL) at 0 °C. To the obtained mixture potassium tert-butoxide (411 mg, 3.66 mmol) was added portion wise and the reaction was stirred at ambient temperature for 12 hrs. The mixture was poured into saturated aqueous NH
4Cl solution (100 mL). The resulting mixture was extracted with MTBE (4 × 20 mL). The combined organic layers were dried over anhydrous Na
2SO
4 and concentrated under reduced pressure. The residue was subjected to flash-column chromatography (SiO
2, gradient hexane - EtOAc) to afford desired 2-chloro- 4,5-dimethyl-6-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidine (375 mg, 945 μmol, 32.3% yield) and 4-chloro-5,6-dimethyl-2-[[4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidine (80.0 mg, 202 μmol, 6.9% yield) as a light-yellow solids. MS (ESI): [M+H]
+ m/z: calcd 396.10; found 397.2.
1H NMR (500 MHz, CDCl
3) δ 2.14 (s, 3H), 2.43 (s, 3H), 3.79 (s, 3H), 5.48 (s, 2H), 7.32 (s, 1H), 7.55 (d, 2H), 7.66 (d, 2H). 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4,5-dimethyl-6-[[4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidine [1588] 4-Cyclopropyl-6-methoxy-pyrimidin-5-yl)boronic acid (528 mg, 2.72 mmol), 2- chloro-4,5-dimethyl-6-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidine (360 mg, 907.27 μmol) and Potassium phosphate tribasic (481 mg, 2.27 mmol) were mixed in a degassed mixture of water (3 mL) and dioxane (20 mL). The reaction vessel was evacuated and backfilled with argon. Then RuPhos Pd G4 (77.2 mg, 90.7 μmol) was added, and the reaction mixture was stirred at 90 °C for 15 hrs. The mixture was cooled to room temperature and concentrated under reduced pressure. The residue was diluted with water (10 mL) and extracted with EtOAc (4 × 20 mL). The combined organic layers were filtered through a thin pad of silica, dried over anhydrous Na
2SO
4 and concentrated under reduced pressure. The residue was dissolved in MeOH (20 mL) then SiliaMetS® Dimercaptotriazine (100 mg) was added to the resulting solution. The resulting mixture was stirred for 6 hrs. and filtered. The light-yellow filtrate was subjected to HPLC (2-10 min, 27-50% water – ACN, flow: 30 mL/min, column: SunFire C18100 ×19 mm, 5 µm) to afford 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4,5-dimethyl-6-[[4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidine (102 mg, 200 μmol, 22% yield) as a light-yellow solid. MS (ESI): [M+H]
+ m/z: calcd 511.21; found 511.2.
1H NMR (600 MHz, DMSO-d
6) δ 0.83 – 0.88 (m, 2H), 1.00 – 1.03 (m, 2H), 1.60 – 1.67 (m, 1H), 2.21 (s, 3H), 2.46 (s, 3H), 3.78 (s, 3H), 3.84 (s, 3H), 5.49 (s, 2H), 7.58 (d, 2H), 7.73 (d, 2H), 7.94 (s, 1H), 8.65 (s, 1H). Example 200 (Compound 106)
[1589] To a solution of (4-methoxyphenyl)methanol (1 g, 7.24 mmol, 898.47 μL) in THF (20 mL) was added NaH (347.41 mg, 8.69 mmol, 60% purity) at 0 °C, the resulting solution was stirred for 0.5 hour at 0 °C. Then to the mixture was added 2,4-dichloro-6-methyl-pyrimidine (1.42 g, 8.69 mmol) at 0 °C. Then the mixture was stirred at room temperature for 2 hrs. The reaction mixture was diluted with EtOAc (100 mL), washed with saturated NH
4Cl aqueous solution (50 mL x 3), dried over anhydrous Na
2SO
4. After filtration, the filtrate was concentrated under reduced pressure. The residue was subject to silica gel column chromatography, eluted with 21% EtOAc in PE to afford the mixture of regioisomers. The regioisomers was separated by Prep-Achiral-SFC with the following conditions: Column: DAICEL DCpak P4VP, 3x25 cm, 5 μm, Mobile Phase A: CO
2, Mobile Phase B: IPA (0.1% 2 M NH
3-MeOH); Flow rate: 60 mL/min; Gradient: isocratic 16 % B; Column Temperature (℃): 35; Wavelength: 254 nm. The collected fractions from the first eluted product were combined and concentrated to give 2-chloro-4-((4-methoxybenzyl)oxy)-6-methylpyrimidine (0.42 g, 1.59 mmol, 22% yield) as an colorless oil. MS: m/z = 265.05 [M + H]
+.
1H NMR (400 MHz, Chloroform-d) δ 7.41 - 7.34 (m, 2H), 6.98 - 6.87 (m, 2H), 6.50 (s, 1H), 5.34 (s, 2H), 3.81 (d, J = 5.2 Hz, 3H), 2.41 (s, 3H). The collected fractions from the second eluted product were combined and concentrated to give 4-chloro-2-((4-methoxybenzyl)oxy)-6-methylpyrimidine (0.36 g, 1.36 mmol, 19% yield) as an colorless oil.
1H NMR (400 MHz, Chloroform-d) δ 7.45 - 7.38 (m, 2H), 6.92 - 6.85 (m, 2H), 6.84 (s, 1H), 5.35 (s, 2H), 3.80 (d, J = 4.8 Hz, 3H), 2.43 (s, 3H). 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[(4-methoxyphenyl)methoxy]-6-methyl- pyrimidine To a solution of 2-chloro-4-[(4-methoxyphenyl)methoxy]-6-methyl-pyrimidine (0.4 g, 1.51 mmol) in 1,4-dioxane (12 mL) and water (2.4 mL) were added 1,1'- Bis(diphenylphosphino)ferrocene-palladium(II)dichloride (110.57 mg, 151.11 μmol) and potassium phosphate (641.5 mg, 3.02 mmol), then the mixture solution was added (4- cyclopropyl-6-methoxypyrimidin-5-yl)boronic acid I-5 (351.8 mg, 1.81 mmol). The mixture was stirred at 40 ℃ for 16 hrs. The reaction mixture was diluted with EtOAc (50 mL), washed with saturated NH
4Cl aqueous solution (30 mL x 3), dried over anhydrous Na
2SO
4. After filtration, the filtrate was concentrated under reduced pressure. The residue was purified by silica gel column chromatography, eluted with 48% EtOAc in PE to afford 2-(4-cyclopropyl- 6-methoxy-pyrimidin-5-yl)-4-[(4-methoxyphenyl)methoxy]-6-methyl-pyrimidine (89 mg, 235.19 μmol, 16% yield) as a light-yellow solid. MS: m/z = 379.25 [M + H]
+. 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-6-methyl-pyrimidin-4-ol To a solution of 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[(4-methoxyphenyl)methoxy]- 6-methyl-pyrimidine (79 mg, 208.76 μmol) in 1,4-dioxane (2.4 mL) was added HCl (2.4 mL, 4.0 M in 1,4-dioxane), then the solution was stirred at room temperature for 1 hr. The reaction was concentrated under reduced pressure. The residue was purified by reverse phase chromatography with the following conditions: Column: C18 spherical Column, 20-35 um, 100A, 40 g; Mobile Phase A: Water (5 mM aq. NH
4HCO
3), Mobile Phase B: MeCN; Flow rate: 40 mL/min; Gradient: 5% B hold 3 min, 5% B to 18% B in 3.6 min, 18% B hold 3 min, 18% B to 95% B in 21.4 min, 95% B hold 3 min; Flow rate: 60 mL/min;; Detector: UV 254& 210 nm. The collected fractions were combined, concentrated and then lyophilized to give 2-(4- cyclopropyl-6-methoxy-pyrimidin-5-yl)-6-methyl-pyrimidin-4-ol (30 mg, 116.16 μmol, 56% yield). MS: m/z = 259.10 [M + H]
+. 4-((6-(5-chloro-1-methyl-4-(trifluoromethyl)-1H-imidazol-2-yl)-5-fluoropyridin-3- yl)methoxy)-4'-cyclopropyl-6'-methoxy-6-methyl-2,5'-bipyrimidine The starting material [6-[5-chloro-1-methyl-4-(trifluoromethyl)imidazol-2-yl]-5-fluoro-3- pyridyl]methanol was prepared as described for Compound 223. [1590] To a solution of 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-6-methyl-pyrimidin-4-ol (30 mg, 116.16 μmol) in THF (3 mL) was added triphenylphosphine (91.40 mg, 348.47 μmol) and diisopropyl azodicarboxylate (70.5 mg, 348.47 μmol, 68.6 μL). Then the mixture was added [6-[5-chloro-1-methyl-4-(trifluoromethyl)imidazol-2-yl]-5-fluoro-3-pyridyl]methanol (36 mg, 116.2 μmol). The mixture was stirred at room temperature for 2 hrs. The mixture was concentrated under reduced pressure. The resulted residue was purified by Prep-HPLC with the following conditions: Column: XBridge Shield RP18 OBD Column, 19 x 250 mm, 10 μm; Mobile Phase A: Water (10 mM aq. NH
4HCO
3), Mobile Phase B: ACN; Flow rate: 25 mL/min; Gradient: 70% B to 85% B in 15 min; Wavelength: 254 nm / 220 nm The collected fractions of the desired compound were combined, concentrated and then lyophilized to give 4-((6-(5- chloro-1-methyl-4-(trifluoromethyl)-1H-imidazol-2-yl)-5-fluoropyridin-3-yl)methoxy)-4'- cyclopropyl-6'-methoxy-6-methyl-2,5'-bipyrimidine (6.4 mg, 11.64 μmol, 10% yield) as an off-white solid. MS: m/z = 550.05 [M + H]
+.
1H NMR (400 MHz, Chloroform-d) δ 8.64 (s, 1H), 8.60 (s, 1H), 7.71 (dd, J = 10.0, 2.0 Hz, 1H), 6.68 (s, 1H), 5.56 (s, 2H), 3.90 (s, 3H), 3.85 (s, 3H), 2.56 (s, 3H), 1.68 - 1.60 (m, 1H), 1.28 - 1.19 (m, 2H), 0.95 - 0.85 (m, 2H).
19F NMR (376 MHz, Chloroform-d) δ -61.90 (s, 3F), -118.78 (s, 1F). Example 201 (Compound 105)
Compound 105 2-chloro-4-methyl-6-methylsulfanyl-pyrimidine [1591] Sodium methanethiolate (64.4 mmol, 22 mL, 21% wt. in water) was added to a stirred solution of 2,4-dichloro-6-methyl-pyrimidine (10.0 g, 61.4 mmol) in THF (15 mL) at 5 °C. The resulting mixture was stirred at room temperature for 16 hr. The reaction mixture was concentrated under reduced pressure. The residue was diluted with water (20 mL) and extracted with MTBE (3×15 mL). The combined organic layers were washed with brine (20 mL), dried over anhydrous Na
2SO
4 and filtered. The filtrate was concentrated under reduced pressure. The residue was subjected to flash column chromatography (silica, gradient hexane – MTBE) to afford 2-chloro-4-methyl-6-methylsulfanyl-pyrimidine (6.0 g, 34.4 mmol, 56% yield) as a white solid.
1H NMR (500 MHz, CDCl
3) δ 2.40 (s, 3H), 2.55 (s, 3H), 6.93 (s, 1H). MS (ESI): [M+H]
+ m/z: calcd 175.01; found 175.0 2-[4-cyclopropyl-6-(difluoromethoxy)pyrimidin-5-yl]-4-methyl-6-methylsulfanyl-pyrimidine [1592] 2-Chloro-4-methyl-6-methylsulfanyl-pyrimidine (138 mg, 788 μmol), 4-cyclopropyl- 6-(difluoromethoxy)-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyrimidine I-10 (246 mg, 788 μmol), potassium phosphate tribasic (502 mg, 2.36 mmol) and bis(diphenylphosphino)ferrocene]dichloropalladium(II)-DCM (64.4 mg, 78.8 μmol) were mixed in a degassed mixture of dioxane (5 mL) and water (500 µL). The mixture was stirred at 80 °C for 12 hr under argon atmosphere. The reaction mixture was cooled to room temperature and diluted with EtOAc (20 mL). Anhydrous Na
2SO
4 (1 g) was added to the mixture and the resulting mixture was stirred for 5 min. The mixture was filtered. The filtrate was concentrated under reduced pressure. The residue was triturated with MTBE (20 mL). The solid precipitate formed was filtered out. The filtrate was concentrated under reduced pressure to afford 2-[4-cyclopropyl-6-(difluoromethoxy)pyrimidin-5-yl]-4-methyl-6- methylsulfanyl-pyrimidine (300 mg, crude) as a yellow gum which was used in next step without purification. MS (ESI): [M+H]
+ m/z: calcd 325.10; found 325.2 2-[4-cyclopropyl-6-(difluoromethoxy)pyrimidin-5-yl]-4-methyl-6-methylsulfonyl-pyrimidine [1593] A mixture of 2-[4-cyclopropyl-6-(difluoromethoxy)pyrimidin-5-yl]-4-methyl-6- methylsulfanyl-pyrimidine (300 mg, crude) and mCPBA (413 mg, 2.03 mmol, 85% purity) in DCM (15 mL) was stirred at room temperature for 12 hr. The reaction mixture was washed with a saturated aqueous solution of NaHCO
3 (30 mL), dried over anhydrous Na
2SO
4 and concentrated under reduced pressure to afford 2-[4-cyclopropyl-6- (difluoromethoxy)pyrimidin-5-yl]-4-methyl-6-methylsulfonyl-pyrimidine (300 mg, crude) as a yellow gum which was used in the next step without further purification. MS (ESI): [M+H]
+ m/z: calcd 357.09; found 357.2 2-[4-cyclopropyl-6-(difluoromethoxy)pyrimidin-5-yl]-4-[[4-[1-cyclopropyl-4- (trifluoromethyl)imidazol-2-yl]-3-fluoro-phenyl]methoxy]-6-methyl-pyrimidine Synthesis of the starting [4-[1-cyclopropyl-4-(trifluoromethyl)imidazol-2-yl]-3-fluoro- phenyl]methanol is described for Compound 154. [1594] A mixture of 2-[4-cyclopropyl-6-(difluoromethoxy)pyrimidin-5-yl]-4-methyl-6- methylsulfonyl-pyrimidine (100 mg, crude), [4-[1-cyclopropyl-4-(trifluoromethyl)imidazol- 2-yl]-3-fluoro-phenyl]methanol (84.3 mg, 281 μmol) and NaH (13.5 mg, 337 μmol, 60% dispersion in mineral oil) in THF (2 mL) was stirred at room temperature for 12 hr. The reaction mixture was directly subjected to HPLC (0.6-8.6 min., 42-50-80% water – ACN, flow: 30 mL/min, column: Chromatorex C18 SMB100-5T 100×19 mm, 5 µm) to afford 2-[4- cyclopropyl-6-(difluoromethoxy)pyrimidin-5-yl]-4-[[4-[1-cyclopropyl-4- (trifluoromethyl)imidazol-2-yl]-3-fluoro-phenyl]methoxy]-6-methyl-pyrimidine (9.7 mg, 16.8 μmol, 6% yield) as a yellow gum.
1H NMR (600 MHz, DMSO-d
6) δ 0.79 – 0.85 (m, 4H), 0.97 – 1.02 (m, 2H), 1.09 – 1.13 (m, 2H), 1.82 – 1.88 (m, 1H), 2.52 (s, 3H), 3.43 – 3.49 (m, 1H), 5.53 (s, 2H), 7.04 (s, 1H), 7.42 (d, 1H), 7.48 (d, 1H), 7.63 (t, 1 H), 7.80 (t, 1H, CHF
2), 8.00 (s, 1H), 8.80 (s, 1H). MS (ESI): [M+H]
+ m/z: calcd 577.21; found 577.2.

Methyl 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-6-[[4-[1-cyclopropyl-4- (trifluoromethyl)imidazol-2-yl]-3-fluoro-phenyl]methoxy]pyrimidine-4-carboxylate Synthesis of the starting methyl 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-6- methylsulfonyl-pyrimidine-4-carboxylate is described for Compound 58. Synthesis of the starting [4-[1-cyclopropyl-4-(trifluoromethyl)imidazol-2-yl]-3-fluoro- phenyl]methanol is described for Compound 154. [1595] DBU (49.5 mg, 325 μmol, 48.7 μL) was added to a stirred solution of [4-[1- cyclopropyl-4-(trifluoromethyl)imidazol-2-yl]-3-fluoro-phenyl]methanol (69.8 mg, 232 μmol) and methyl 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-6-methylsulfonyl- pyrimidine-4-carboxylate (84.7 mg, 232 μmol) in THF (2 mL). The reaction mixture was stirred at room temperature for 10 hr. The reaction mixture was quenched by addition of water (5 mL) and extracted with EtOAc (2×5 mL). The combined organic layers were washed with brine (5 mL) and concentrated under reduced pressure to afford methyl 2-(4- cyclopropyl-6-methoxy-pyrimidin-5-yl)-6-[[4-[1-cyclopropyl-4-(trifluoromethyl)imidazol-2- yl]-3-fluoro-phenyl]methoxy]pyrimidine-4-carboxylate (130 mg, crude) as a yellow oil which was used in the next step without further purification. MS (ESI): [M+H]
+ m/z: calcd 585.19; found 585.2 [2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-6-[[4-[1-cyclopropyl-4- (trifluoromethyl)imidazol-2-yl]-3-fluoro-phenyl]methoxy]pyrimidin-4-yl]methanol [1596] Lithium borohydride (9.69 mg, 445 μmol) was added to a stirred solution of methyl 2- (4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-6-[[4-[1-cyclopropyl-4-(trifluoromethyl)imidazol- 2-yl]-3-fluoro-phenyl]methoxy]pyrimidine-4-carboxylate (130 mg, 222 μmol) in THF (1 mL). The reaction mixture was stirred at room temperature for 12 hr. The reaction mixture was quenched with MeOH (0.2 mL) and filtered. The filtrate was subjected to HPLC (0-2-9 min., 33-40-55% water – ACN, flow: 30 mL/min, column: Chromatorex C18 SMB100-5T, 100×19 mm, 5 µm) to afford [2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-6-[[4-[1- cyclopropyl-4-(trifluoromethyl)imidazol-2-yl]-3-fluoro-phenyl]methoxy]pyrimidin-4- yl]methanol (10.0 mg, 18.0 μmol, 7.8% yield from [4-[1-cyclopropyl-4- (trifluoromethyl)imidazol-2-yl]-3-fluoro-phenyl]methanol) as a yellow solid.
1H NMR (500 MHz, DMSO-d
6) δ 0.79 – 0.85 (m, 4H), 0.86 – 0.92 (m, 2H), 1.01 – 1.06 (m, 2H), 1.63 – 1.69 (m, 1H), 3.44 – 3.50 (m, 1H), 3.84 (s, 3H), 4.56 (d, 2H), 5.54 (s, 2H), 5.68 (t, 1H), 7.05 (s, 1H), 7.44 (d, 1H), 7.50 (d, 1H), 7.65 (t, 1H), 8.01 (s, 1H), 8.67 (s, 1H). MS (ESI): [M+H]
+ m/z: calcd 557.22; found 557.2. Example 203 (Compound 103)
Synthesis of 3-fluoro-4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]benzonitrile Starting material 3-fluoro-4-[4-(trifluoromethyl)-1H-imidazol-2-yl]benzonitrile was prepared as described for Compound 190. [1597] A mixture of 3-fluoro-4-[4-(trifluoromethyl)-1H-imidazol-2-yl]benzonitrile (5 g, 19.6 mmol) in THF (50 mL) was stirred at -10 °C for 30 min then KO
tBu (1 M in THF, 30 mL, 30.0 mmol) was added dropwise followed by the addition of MeI (2 mL, 32.1 mmol) to the reaction mixture. The resulting mixture was stirred for 12 hrs at 20 °C. The reaction was quenched by addition of water (100 mL) and extracted with EtOAc (100 mL x 3). The combined organic layer was washed with brine (100 mL), dried over anhydrous Na
2SO
4, filtered and concentrated under reduced pressure to afford 3-fluoro-4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]benzonitrile (5 g, 94.8% yield) as yellow solid.
1H NMR (400 MHz, DMSO-d
6) δ ppm 8.03 - 8.16 (m, 2H), 7.80 - 7.89 (m, 2H), 3.65 (d, J = 1.43 Hz, 3 H); MS (ESI) [M+H]
+ m/z: calcd 270.1, found 270.0. 3-fluoro-4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]benzoic acid [1598] To a solution of 3-fluoro-4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]benzonitrile (5 g, 18.6 mmol) in EtOH (30 mL) was added a solution of KOH (2.19 g, 39.0 mmol) in H
2O (5 mL). The mixture was stirred at 80°C for 1 hour. The resulting mixture was concentrated under reduced pressure. The residue was diluted with H
2O (30 mL). The mixture was adjusted pH = 8 with saturated NaHCO
3 aqueous solution. The mixture was filtered. The filter cake was dried under reduced pressure to give 3-fluoro-4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]benzoic acid (5 g, 93.4% yield) as yellow solid, which was directly used without further purification. MS (ESI) [M+H]
+ m/z: calcd 289.1; found 288.9. [3-fluoro-4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methanol [1599] To a solution of LiAlH4 (988 mg, 26.0 mmol) in THF (30 mL) was added a solution of 3-fluoro-4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]benzoic acid (5 g, 17.4 mmol) in THF (30 mL) dropwise at 0 °C. The mixture was stirred at 20 °C for 3 hrs. After the reaction mixture was cooled to 0 °C, the reaction mixture was quenched by addition dropwise of H
2O (1 mL), 15% NaOH aqueous solution (1 mL), and then H
2O (3 mL). The mixture was dried over anhydrous MgSO
4, filtered and concentrated under reduced pressure. The residue was triturated with MTBE (10 mL). The mixture was filtered. The filter cake was dried under reduced pressure to give [3-fluoro-4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methanol (3 g, 63.1% yield) as white solid, which was directly used without further purification.
1H NMR (400 MHz, chloroform-d) δ ppm 7.51 (t, J = 7.7 Hz, 1H), 7.37 (s, 1H), 7.14 - 7.22 (m, 2H), 4.75 (s, 2H), 3.64 (d, J = 2.3 Hz, 3H);
19F NMR (376 MHz, chloroform- d) δ ppm -113.53, -62.66; MS (ESI) [M+H]
+ m/z: calcd 275.1; found 275.0. Methyl 2-chloro-6-methylsulfanyl-pyrimidine-4-carboxylate [1600] To a solution of methyl 2,6-dichloropyrimidine-4-carboxylate (10 g, 48.3 mmol) in THF (100 mL) was added NaSMe (20.3 g, 57.9 mmol, 20 wt% in H
2O) at 0 °C. The mixture was stirred at 20 °C for 12 hrs. The resulting mixture poured into 2N HCl aqueous solution (100 mL), and then adjusted pH = 8 with saturated NaHCO
3 aqueous solution. The mixture was extracted with EtOAc (100 mL x 3). The combined organic layer was dried over anhydrous Na
2SO
4, filtered and concentrated under reduced pressure to give methyl 2-chloro-6-methylsulfanyl-pyrimidine-4-carboxylate (8.3 g, 78.6% yield) as white solid, which was directly used without further purification.
1H NMR (400 MHz, DMSO-d
6) δ ppm 7.94 (s, 1H), 3.90 (s, 3H), 2.61 (s, 3H); MS (ESI) [M+H]
+ m/z: calcd 219.0; found 219.0. 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-6-methylsulfanyl-pyrimidine-4-carboxylic acid [1601] A mixture of methyl 2-chloro-6-methylsulfanyl-pyrimidine-4-carboxylate (4 g, 18.3 mmol), (4-cyclopropyl-6-methoxy-pyrimidin-5-yl)boronic acid I-5 (3.73 g, 19.2 mmol), XPhos Pd G
3 (776 mg, 0.917 mmol), XPhos (436 mg, 0.915 mmol), K
3PO
4 (9.71 g, 45.7 mmol) in dioxane (100 mL) and H
2O (20 mL) was stirred at 90 °C for 12 hrs. The resulting mixture was quenched by addition of water (30 mL) and extracted with EtOAc (30 mL x 3). The aqueous phase was adjusted pH = 4 with 2N HCl aqueous solution. The mixture was filtered and the filter cake was dried under reduced pressure to afford 2-(4-cyclopropyl- 6-methoxy-pyrimidin-5-yl)-6-methylsulfanyl-pyrimidine-4-carboxylic acid (4.9 g, crude) as off white solid, which was directly used without further purification. MS (ESI) [M+H]
+ m/z: calcd 319.1; found 319.0. Methyl 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-6-methylsulfanyl-pyrimidine-4- carboxylate [1602] To a mixture of 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-6-methylsulfanyl- pyrimidine-4-carboxylic acid (2.9 g, 9.11 mmol) in MeCN (100 mL) was added MeI (0.85 mL, 13.7 mmol). The mixture was stirred at 65 °C for 12 hrs. The resulting mixture was quenched by addition of water (50 mL) and extracted with EtOAc (50 mL x 3). The combined organic layer was washed with brine (100 mL), dried over anhydrous Na
2SO
4, filtered and concentrated under reduced pressure. The residue was purified by flash chromatography (Biotage®; 20 g AgelaFlash® Silica Flash Column, PE/EtOAc with EtOAc from 0-30%, 40 mL/min, 254nm) to afford methyl 2-(4-cyclopropyl-6-methoxy-pyrimidin-5- yl)-6-methylsulfanyl-pyrimidine-4-carboxylate (1.9 g, 62.8% yield) as brown solid. MS (ESI) [M+H]
+ m/z: calcd 333.1; found 332.9. Methyl 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-6-methylsulfonyl-pyrimidine-4- carboxylate [1603] To a solution of methyl 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-6- methylsulfanyl-pyrimidine-4-carboxylate (1.9 g, 5.72 mmol) in DCM (100 mL) was added mCPBA (2.38 g, 11.7 mmol, 85 wt%). The mixture was stirred at 20 °C for 12 hrs. The resulting mixture was filtered. The filtrate was washed with saturated Na
2SO
3 aqueous solution (100 mL), saturated NaHCO
3 aqueous solution (100 mL), dried over anhydrous Na
2SO
4, filtered and concentrated under reduced pressure to give methyl 2-(4-cyclopropyl-6- methoxy-pyrimidin-5-yl)-6-methylsulfonyl-pyrimidine-4-carboxylate (2.1 g, crude) as yellow solid, which was directly used without further purification. MS (ESI) [M+H]
+ m/z: calcd 365.1; found 365.0. Methyl 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-6-[[3-fluoro-4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidine-4-carboxylate and 2-(4- cyclopropyl-6-methoxy-pyrimidin-5-yl)-6-[[3-fluoro-4-[1-methyl-4-(trifluoromethyl)imidazol- 2-yl]phenyl]methoxy]pyrimidine-4-carboxylic acid [1604] To a solution of [3-fluoro-4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methanol (1.5 g, 5.47 mmol) in THF (20 mL) was added NaH (328 mg, 8.20 mmol, 60 wt% in mineral oil). The mixture was stirred at 20 °C for 30 min then methyl 2-(4- cyclopropyl-6-methoxy-pyrimidin-5-yl)-6-methylsulfonyl-pyrimidine-4-carboxylate (1.99 g, 5.47 mmol) was added, and the mixture was stirred at 45 °C for 12 hrs. The resulting mixture was poured into saturated NH
4Cl aqueous solution (100 mL) and extracted with EtOAc (100 mL x 3). The combined organic layer was dried over anhydrous Na
2SO
4, filtered and concentrated under reduced pressure. The residue was purified by flash chromatography (ISCO®; 20 g AgelaFlash® Silica Flash Column, PE/EtOAc with EtOAc from 0-40%, 50 mL/min, 254nm) to afford methyl 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-6-[[3-fluoro- 4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidine-4-carboxylate (1.1 g, 36% yield) as colorless oil. MS (ESI) [M+H]
+ m/z: calcd 559.2; found 559.1. The column was then eluted with 10:1 EtOAc/MeOH (150 mL) to give 2-(4-cyclopropyl-6-methoxy- pyrimidin-5-yl)-6-[[3-fluoro-4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidine-4-carboxylic acid (400 mg, 13.4% yield) as yellow solid, which was directly used without further purification. MS (ESI) [M+H]
+ m/z: calcd 545.1; found 545.1. 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-6-[[3-fluoro-4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidine-4-carboxylic acid [1605] 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-6-[[3-fluoro-4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidine-4-carboxylic acid (400 mg, 0.735 mmol) was purified by preparative HPLC (Instrument: Gilson GX-281 Liquid Handler, Gilson 322 Pump, Gilson 156 UV Detector; Column: Welch Xtimate C18100 × 25 mm × 3 μm; Mobile phase A: H
2O with NH
3-H
2O + NH
4HCO
3 (v%); Mobile phase B: MeCN; Gradient: B from 20% to 50% in 7.8 min, hold 100% B for 2 min; Flow Rate: 30 mL/min; Column Temperature: 30 °C; Wavelength: 220 nm, 254 nm) to give 2-(4-cyclopropyl-6- methoxy-pyrimidin-5-yl)-6-[[3-fluoro-4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidine-4-carboxylic acid (130 mg, 32.5% yield) as white dry powder.
1H NMR (400 MHz, methanol-d
4) δ ppm 8.58 (s, 1H), 7.76 (d, J = 1.0 Hz, 1H), 7.54 - 7.61 (m, 1H), 7.42 - 7.50 (m, 3H), 5.61 (s, 2H), 3.90 (s, 3H), 3.66 (d, J = 1.5 Hz, 3H), 1.68 (s, 1H), 1.12 (s, 2H), 0.90 (s, 2H);
19F NMR (376 MHz, methanol-d
4) δ ppm -115.16, -63.97; MS (ESI) [M+H]
+ m/z: calcd 545.1; found 545.1. Synthesis of [2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-6-[[3-fluoro-4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidin-4-yl]methanol (103) [1606] To a solution of methyl 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-6-[[3-fluoro-4- [1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidine-4-carboxylate (100 mg, 0.179 mmol) in THF (5 mL) was added LiAlH
4 (8 mg, 0.211 mmol) at 0 °C. The mixture was stirred at 20 °C for 1 hour. The mixture was quenched by addition of H
2O (0.08 mL), 15% NaOH aqueous solution (0.08 mL), and H
2O (0.24 mL). Then the mixture was dried over anhydrous MgSO
4, filtered and concentrated under reduced pressure. The residue was purified by preparative HPLC (Instrument: Gilson GX-281 Liquid Handler, Gilson 322 Pump, Gilson 156 UV Detector; Column: 2_Phenomenex Gemini C1875 × 40 mm × 3 μm; Mobile phase A: H
2O with NH
4HCO
3 (v%); Mobile phase B: MeCN; Gradient: B from 38% to 68% in 9.5 min, hold 100% B for 3 min; Flow Rate: 30 mL/min; Column Temperature: 30 °C; Wavelength: 220 nm, 254 nm) to give [2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-6- [[3-fluoro-4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidin-4- yl]methanol (6.9 mg, 7.3% yield) as white dry powder.
1H NMR (400 MHz, methanol- d
4) δ ppm 8.59 (s, 1H), 7.76 (d, J = 1.0 Hz, 1H), 7.54 - 7.60 (m, 1H), 7.41 - 7.48 (m, 2H), 7.11 (s, 1H), 5.59 (s, 2H), 4.67 (s, 2H), 3.91 (s, 3H), 3.66 (d, J = 1.3 Hz, 3H), 1.61 - 1.71 (m, 1H), 1.09 - 1.17 (m, 2H), 0.85 - 0.95 (m, 2H);
19F NMR (376 MHz, methanol-d
4) δ ppm - 115.20, -63.98; MS (ESI) [M+H]
+ m/z: calcd 531.2; found 531.1.
2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[3-fluoro-4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]-6-methyl-pyrimidine (102) Synthesis of the starting [3-fluoro-4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methanol is described for compound 97. [1607] To a solution of [3-fluoro-4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methanol (100 mg, 0.367 mmol) in THF (3 mL) was added NaH (18 mg, 0.450 mmol, 60 wt% in mineral oil). The mixture was stirred at 20 °C for 30 minutes.2-(4- cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-methyl-6-methylsulfonyl-pyrimidine (120 mg, 0.375 mmol) was added and stirred at 45 °C for 2 hrs. The resulting mixture was quenched by addition of water (20 mL) and extracted with EtOAc (30 mL x 3). The combined organic layer was dried over anhydrous Na
2SO
4, filtered and concentrated under reduced pressure. The residue was purified by preparative HPLC (Instrument: Gilson GX-281 Liquid Handler, Gilson 322 Pump, Gilson 156 UV Detector; Column: 2_Phenomenex Gemini C18, 75 × 40 mm, 3 μm; Mobile phase A: H
2O with 0.05% NH
3-H
2O + NH
4HCO
3 (v%); Mobile phase B: MeCN; Gradient: B from 47% to 77% in 7.8 min, hold 100% B for 1 min; Flow Rate: 30 mL/min; Column Temperature: 30 °C; Wavelength: 220 nm, 254 nm) to afford 2-(4- cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[3-fluoro-4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]-6-methyl-pyrimidine (50.6 mg, 27% yield) as white solid.
1H NMR (400 MHz, DMSO-d
6) δ ppm 8.66 (s, 1H), 8.01 (d, J = 1.2 Hz, 1H), 7.61 (t, J = 7.6 Hz, 1H), 7.49 (d, J = 11.2 Hz, 1H), 7.41 - 7.44 (m, 1H), 6.97 (s, 1H), 5.50 (s, 2H), 3.84 (s, 3H), 3.60 (d, J = 1.2 Hz, 3H), 2.46 (s, 3H), 1.60 - 1.68 (m, 1H), 1.03 (quin, J = 3.6 Hz, 2H), 0.85 - 0.90 (m, 2H);
19F NMR (377 MHz, DMSO-d
6) δ ppm -60.83, - 113.90; MS (ESI) [M+H]
+ m/z: calcd 515.2, found 515.1.
1-(2,6-dichloropyrimidin-4-yl)ethanone [1608] The mixture of methyl 2,6-dichloropyrimidine-4-carboxylate (8 g, 38.6 mmol) in THF (30 mL) was added MeMgBr (3 M in THF, 19 mL, 57.0 mmol) at -78 °C slowly. Then the mixture was stirred at -78 °C for 2 hrs under N
2. The mixture was quenched by addition of water (10 mL). The mixture was extracted with EtOAc (10 mL x 3). The combined organic layer was washed brine (10 mL), dried over anhydrous Na
2SO
4, filtered and concentrated under reduced pressure. The residue was subject to flash chromatography (ISCO
®; 40 g AgelaFlash
® Silica Flash Column, PE/EtOAc with EtOAc from 0-6%, flow rate = 60 mL/min, 254 nm) to afford 1-(2,6-dichloropyrimidin-4-yl)ethanone (1.3 g, 17.6% yield) as light-yellow oil.
1H NMR (400 MHz, chloroform-d) δ ppm 7.78 (s, 1H), 2.64 (s, 3H); MS (ESI) [M+H]
+ m/z: calcd 193.0, found 193.0. 1-(2-chloro-6-methylsulfanyl-pyrimidin-4-yl)ethanone [1609] A mixture of 1-(2,6-dichloropyrimidin-4-yl)ethanone (1.3 g, 6.81 mmol) and MeSNa (2.2 g, 6.31 mmol, 20 wt% in H
2O) in THF (15 mL) was stirred at -10 °C for 2 hrs. The resulting mixture was quenched by addition of water (10 mL) and extracted with EtOAc (10 mL x 3). The combined organic layer was dried over anhydrous Na
2SO
4, filtered and concentrated under reduced pressure. The residue was purified by flash chromatography (ISCO
®; 20 g AgelaFlash
® Silica Flash Column, PE/EtOAc with EtOAc from 0-60%, flow rate = 40 mL/min, 254 nm) to afford 1-(2-chloro-6-methylsulfanyl-pyrimidin-4-yl)ethanone (520 mg, 37.7% yield) as white solid.
1H NMR (400 MHz, chloroform-d) δ ppm 7.63 (d, J = 3.5 Hz, 1H), 2.63 - 2.69 (m, 3H), 2.61 (d, J = 3.3 Hz, 3H) MS (ESI) [M+H]
+ m/z calcd 203.0, found 202.9. 1-[2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-6-methylsulfanyl-pyrimidin-4-yl]ethanone [1610] A mixture of 1-(2-chloro-6-methylsulfanyl-pyrimidin-4-yl)ethanone (520 mg, 2.57 mmol), (4-cyclopropyl-6-methoxy-pyrimidin-5-yl)boronic acid I-5 (498 mg, 2.57 mmol), XPhos Pd G
3 (217 mg, 0.256 mmol), XPhos (122 mg, 0.256 mmol) and K
3PO
4 (670 mg, 7.69 mmol) in H
2O (1 mL) and dioxane (3 mL) was stirred at 95 °C for 12 hrs. The resulting mixture was quenched by addition of water (10 mL) and extracted with EtOAc (10 mL x 3). The combined organic layer was brine (10 mL), dried over anhydrous Na
2SO
4, filtered and concentrated under reduced pressure. The residue was purified by flash chromatography (ISCO
®; 20 g AgelaFlash
® Silica Flash Column, PE/EtOAc with EtOAc from 0-10%, flow rate = 30 mL/min,254 nm) to afford 1-[2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-6- methylsulfanyl-pyrimidin-4-yl]ethanone (410 mg, 50.5% yield) as yellow solid.
1H NMR (400 MHz, DMSO-d
6) δ ppm 8.71 (s, 1H), 7.75 (s, 1H), 3.88 (s, 3H), 2.60 (s, 3H), 2.59 (s, 3H), 1.81 (s, 1H), 1.04 - 1.11 (m, 2H), 0.89 - 1.00 (m, 2H); MS (ESI) [M+H]
+ m/z: calcd 317.1, found 317.0. 1-[2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-6-methylsulfonyl-pyrimidin-4-yl]ethanone [1611] A mixture of 1-[2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-6-methylsulfanyl- pyrimidin-4-yl]ethanone (100 mg, 0.316 mmol) and mCPBA (160 mg, 0.788 mmol, 85 wt%) in DCM (1 mL) was stirred at 25 °C for 12 hrs. The resulting mixture was quenched by addition of Na
2SO
3 (2 mL) and NaHCO
3 (2 mL) and extracted with DCM (5mL x 3). The combined organic layer was dried over anhydrous Na
2SO
4, filtered and concentrated under reduced pressure to afford 1-[2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-6-methylsulfonyl- pyrimidin-4-yl]ethanone (110 mg, crude) as white solid.
1H NMR (400 MHz, DMSO-d
6) δ ppm 8.78 (s, 1H), 8.26 (s, 1H), 3.91 (s, 3H), 3.49 (s, 3H), 2.69 (s, 3H), 1.91 - 1.96 (m, 1H), 1.13 (s, 2H), 0.95 (dd, J = 7.6, 3.2 Hz, 2H); MS (ESI) [M+H]
+ m/z: calcd 349.1, found 349.1. 1-[2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-6-[[3-fluoro-4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidin-4-yl]ethanone Synthesis of the starting [3-fluoro-4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methanol is described for compound 97. [1612] To a mixture of [3-fluoro-4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methanol (87 mg, 0.317 mmol) in THF (2 mL) was added NaH (15 mg, 0.375 mmol, 60 wt% in mineral oil) at 20 °C .The mixture was stirred at 20 °C for 30 min.1-[2-(4- cyclopropyl-6-methoxy-pyrimidin-5-yl)-6-methylsulfonyl-pyrimidin-4-yl]ethanone (110 mg, 0.316 mmol) was added to the mixture. The mixture was stirred at 45 °C for 2.5 hrs. The resulting mixture was quenched by addition of water (5 mL) and extracted with EtOAc (5 mL x 3). The combined organic layer was dried over anhydrous Na
2SO
4, filtered and concentrated under reduced pressure. The residue was purified by flash chromatography (ISCO
®; 20 g AgelaFlash
® Silica Flash Column, PE/EtOAc with EtOAc from 0-35%, flow rate = 40 mL/min, 254 nm) to afford 1-[2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-6-[[3- fluoro-4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidin-4- yl]ethanone (80 mg, 46.7% yield) as white oil. MS (ESI) [M+H]
+ m/z: calcd 543.2, found 543.1. 1-[2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-6-[[3-fluoro-4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidin-4-yl]ethanol (101) [1613] To a mixture of 1-[2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-6-[[3-fluoro-4-[1- methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidin-4-yl]ethanone (80 mg, 0.147 mmol) in MeOH (2 mL) was added NaBH
4 (20 mg, 0.528 mmol) at 0 ℃ slowly. The mixture was stirred at 25 °C for 1 hour
. The resulting mixture was quenched by addition of water (2 mL) and concentrated. The residue was purified by preparative HPLC (Instrument: Gilson GX-281 Liquid Handler, Gilson 322 Pump, Gilson 156 UV Detector; Column: 45% to 75% in 7.8 min, hold 100% B for 1 min; Flow Rate: 30 mL/min; Column Temperature: 30 °C; Wavelength: 220 nm, 254 nm) to afford 1-[2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)- 6-[[3-fluoro-4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidin-4- yl]ethanol (17 mg, 21.2% yield) as white solid.
1H NMR (400 MHz, DMSO-d
6) δ ppm 8.67 (s, 1H), 8.02 (s, 1H), 7.62 (t, J = 7.8 Hz, 1H), 7.52 (d, J = 11.0 Hz, 1H), 7.45 (d, J = 8.0 Hz, 1H), 7.06 (s, 1H), 5.66 (d, J = 5.0 Hz, 1H), 5.46 - 5.59 (m, 2H), 4.64 - 4.73 (m, 1H), 3.85 (s, 3H), 3.61 (s, 3H), 1.63 - 1.73 (m, 1H), 1.38 (d, J = 6.8 Hz, 3H), 1.01 - 1.07 (m, 2H), 0.88 (dd, J = 7.8, 3.3 Hz, 2H);
19F NMR (376 MHz, DMSO-d
6) δ ppm -60.83, -113.88; MS (ESI) [M+H]
+ m/z: calcd 545.2, found 545.2.
5-bromo-2-chloro-4-[[3-fluoro-4-[1-isopropyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidine The synthesis of the starting [3-fluoro-4-[1-isopropyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methanol was described in Example 76 (Compound 149). [1614] 5-Bromo-2,4-dichloro-pyrimidine (628 mg, 2.76 mmol) was added to a stirred mixture of [3-fluoro-4-[1-isopropyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methanol (500 mg, 1.65 mmol) and Cs
2CO
3 (1.08 g, 3.31 mmol) in ACN (10 mL). The reaction mixture was stirred at 65 °C for 12 hr. The reaction mixture was cooled to room temperature and concentrated under reduced pressure. The residue was diluted with water (30 mL) and extracted with EtOAc (40 mL). The organic layer was separated, washed with brine (30 mL), dried over anhydrous Na
2SO
4 and concentrated under reduced pressure to afford 5-bromo-2- chloro-4-[[3-fluoro-4-[1-isopropyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidine (850 mg, 1.72 mmol, 62.5% yield) as a red solid which was used in the next step without further purification. MS (ESI): [M+H]
+ m/z: calcd 495.00, 493.01; found 495.0, 493.0 1-[2-chloro-4-[[3-fluoro-4-[1-isopropyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidin-5-yl]ethanol [1615] Butyl lithium (0.460 mL, 1.15 mmol, 2.5M in hexane) was added dropwise to a solution of 5-bromo-2-chloro-4-[[3-fluoro-4-[1-isopropyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidine (380 mg, 770 μmol) in THF (5 mL) at -100 °C. Acetaldehyde (136 mg, 3.08 mmol, 173 μL) was added to the mixture at -90 °C. The mixture was stirred for 5 min then water (500 µL) was added to the mixture. The reaction mixture was allowed to warm to room temperature, diluted with MTBE (10 mL) and washed with brine (5 mL). The organic layer was separated and concentrated under reduced pressure to afford 1-[2-chloro-4- [[3-fluoro-4-[1-isopropyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidin-5- yl]ethanol (160 mg, 349 μmol, 45.3% yield) as a yellow oil which was used in the next step without further purification. MS (ESI): [M+H]
+ m/z: calcd 459.12; found 459.2 1-[2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[3-fluoro-4-[1-isopropyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidin-5-yl]ethanol [1616] 1-[2-chloro-4-[[3-fluoro-4-[1-isopropyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidin-5-yl]ethanol (160 mg, 349 μmol), (4-cyclopropyl-6-methoxy- pyrimidin-5-yl)boronic acid I-5 (203 mg, 1.05 mmol), potassium phosphate tribasic (222 mg, 1.05 mmol), RuPhos Pd G3 (14.8 mg, 17.4 μmol) were mixed in a degassed mixture of dioxane (4 mL) and water (400 µL) under argon atmosphere. The reaction mixture was stirred at 70 °C for 20 hr. Additional portions of (4-cyclopropyl-6-methoxy-pyrimidin-5- yl)boronic acid I-5 (203 mg, 1.05 mmol) and RuPhos Pd G3 (14.8 mg, 17.4 μmol) were added to the reaction mixture. The resulting mixture was stirred at 70 °C for 5 hr. The reaction mixture was cooled to room temperature, dried over anhydrous Na
2SO
4 and filtered. The filtrate was subjected to HPLC (0-2-10 min, 33-40-50% water - ACN; flow: 30 mL/min, column: Chromatorex C18 SMB100-5T, 100×19 mm, 5 µm) to afford 1-[2-(4-cyclopropyl-6- methoxy-pyrimidin-5-yl)-4-[[3-fluoro-4-[1-isopropyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidin-5-yl]ethanol (20.0 mg, 34.9 μmol, 10% yield) as a yellow solid. MS (ESI): [M+H]
+ m/z: calcd 573.26; found 573.2 rel-(S)-1-(4'-cyclopropyl-4-((3-fluoro-4-(1-isopropyl-4-(trifluoromethyl)-1H-imidazol-2- yl)benzyl)oxy)-6'-methoxy-[2,5'-bipyrimidin]-5-yl)ethanol (94) and rel-(R)-1-(4'-cyclopropyl-4-((3-fluoro-4-(1-isopropyl-4-(trifluoromethyl)-1H-imidazol-2- yl)benzyl)oxy)-6'-methoxy-[2,5'-bipyrimidin]-5-yl)ethanol (93) [1617] 1-[2-(4-Cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[3-fluoro-4-[1-isopropyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidin-5-yl]ethanol (20.0 mg, 34.9 μmol) was subjected to chiral HPLC (Column: CHIRALCEL OD-H (250×20 mm, 5 mkm)-I, mobile Phase: Hexane:IPA:MeOH, 90:5:5, flow rate: 12 mL/min) to afford rel-(S)-1-(4'- cyclopropyl-4-((3-fluoro-4-(1-isopropyl-4-(trifluoromethyl)-1H-imidazol-2-yl)benzyl)oxy)- 6'-methoxy-[2,5'-bipyrimidin]-5-yl)ethanol (7.00 mg, 12.2 μmol, 35.0% yield) and rel-(R)-1- (4'-cyclopropyl-4-((3-fluoro-4-(1-isopropyl-4-(trifluoromethyl)-1H-imidazol-2- yl)benzyl)oxy)-6'-methoxy-[2,5'-bipyrimidin]-5-yl)ethanol (7.00 mg, 12.2 μmol, 35.0% yield) as yellow oils. rel-(S)-1-(4'-cyclopropyl-4-((3-fluoro-4-(1-isopropyl-4-(trifluoromethyl)-1H- imidazol-2-yl)benzyl)oxy)-6'-methoxy-[2,5'-bipyrimidin]-5-yl)ethanol (94): 1H NMR (600 MHz, DMSO-d
6) δ 0.85 – 0.89 (m, 2H), 1.01 – 1.05 (m, 2H), 1.37 (d, 6H), 1.45 (d, 3H), 1.63 – 1.69 (m, 1H), 3.84 (s, 3H), 4.12 – 4.18 (m, 1H), 5.00 – 5.05 (m, 1H), 5.46 (br. s., 1H), 5.56 (s, 2H), 7.44 (d, 1H), 7.50 (d, 1H), 7.58 (t, 1H), 8.25 (s, 1H), 8.67 (s, 1H), 8.74 (s, 1H). MS (ESI): [M+H]
+ m/z: calcd 573.26; found 573.2 Enantiopurity: >99% (column: Chiralcel OD-H, 250 × 4.6 mm, 5 μm; mobile phase: Hexane-IPA-MeOH, 90-5-5; flow: 0.6 mL/min; RT=18.8 min). rel-(R)-1-(4'-cyclopropyl-4-((3-fluoro-4-(1-isopropyl-4-(trifluoromethyl)-1H- imidazol-2-yl)benzyl)oxy)-6'-methoxy-[2,5'-bipyrimidin]-5-yl)ethanol (93): 1H NMR (600 MHz, DMSO-d
6) δ 0.85 – 0.89 (m, 2H), 1.01 – 1.05 (m, 2H), 1.37 (d, 6H), 1.45 (d, 3H), 1.63 – 1.69 (m, 1H), 3.84 (s, 3H), 4.12 – 4.18 (m, 1H), 5.00 – 5.05 (m, 1H), 5.46 (br. s., 1H), 5.56 (s, 2H), 7.44 (d, 1H), 7.50 (d, 1H), 7.58 (t, 1H), 8.25 (s, 1H), 8.67 (s, 1H), 8.74 (s, 1H). MS (ESI): [M+H]
+ m/z: calcd 573.26; found 573.2 Enantiopurity: >99% (column: Chiralcel OD-H, 250 × 4.6 mm, 5 μm; mobile phase: Hexane-IPA-MeOH, 90-5-5; flow: 0.6 mL/min; RT=26.6 min).

4-cyclopropyl-5-[4-[[3,5-difluoro-4-[1-isopropyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidin-2-yl]-6-methoxy-pyrimidine Starting material [3,5-difluoro-4-[1-isopropyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methanol was prepared as described for Compound 188. [1618] Sodium hydride (11.3 mg, 282 μmol, 60% dispersion in mineral oil) was added to a solution of [3,5-difluoro-4-[1-isopropyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methanol (129 mg, 282 μmol) in DMF (2.0 mL). The reaction mixture was stirred at room temperature for 15 min.4-cyclopropyl-6-methoxy-5-(4-methylsulfonylpyrimidin-2-yl)pyrimidine I-8 (86.4 mg, 282 μmol) was added to the reaction mixture. The reaction mixture was stirred at room temperature for 12 hr. The reaction mixture was diluted with water (2.0 mL), extracted with EtOAc (2×2.0 mL). The combined organic layers were washed with brine (3.0 mL) and concentrated under reduced pressure. The residue was subjected to HPLC (0.5-6.5 min., 50- 75% water – MeOH, +0.1% vol. of 25% aq. NH
3, flow: 30 mL/min, column: Chromatorex Phenyl SMB 100-5100 ×19 mm, 5 µm) to afford 4-cyclopropyl-5-[4-[[3,5-difluoro-4-[1- isopropyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidin-2-yl]-6-methoxy- pyrimidine (40.0 mg, 73.2 μmol, 26.0% yield) as a light-yellow solid. 1H NMR (600 MHz, DMSO-d
6) δ 0.85 – 0.90 (m, 2H), 1.00 – 1.05 (m, 2H), 1.34 (d, 6H), 1.64 – 1.71 (m, 1H), 3.83 (s, 3H), 4.05 – 4.11 (m, 1H), 5.51 (s, 2H), 7.11 (d, 1H), 7.44 (d, 2H), 8.32 (s, 1H), 8.66 (s, 1H), 8.72 (d, 1H). LCMS(ESI): [M+H]
+ m/z: calcd 547.22; found 547.4
4-cyclopropyl-5-[4-[[3,5-difluoro-4-[5-methyl-3-(trifluoromethyl)pyrazol-1- yl]phenyl]methoxy]pyrimidin-2-yl]-6-methoxy-pyrimidine Starting [3,5-difluoro-4-[5-methyl-3-(trifluoromethyl)pyrazol-1-yl]phenyl]methanol was prepared as described for Compound 108. [1619] Sodium hydride (24.1 mg, 630 μmol, 60% dispersion in mineral oil) was added to a solution of [3,5-difluoro-4-[5-methyl-3-(trifluoromethyl)pyrazol-1-yl]phenyl]methanol (160 mg, 548 μmol) and 4-cyclopropyl-6-methoxy-5-(4-methylsulfonylpyrimidin-2-yl)pyrimidine I-8 (176 mg, 575 μmol) in DMF (10 mL). The reaction mixture was stirred at room temperature for 15 hr. The reaction mixture was diluted with water (25 mL) and extracted with EtOAc (3×20 mL). The combined organic layers were concentrated under reduced pressure. The residue was dissolved in MeCN (8 mL). Metal scavenger SiliaMetS
® Dimercaptotriazine (50 mg) was added to this solution. The resulting mixture was stirred at room temperature for 10 hr. The mixture was filtered. The filtrate was subjected to HPLC (2- 10 min, 10-50% water+FA (0.1% vol.) - ACN+FA (0.1% vol.), flow: 30 mL/min, column: Waters SunFire C18, 100 x 19 mm, 5 µm ) to afford 4-cyclopropyl-5-[4-[[3,5-difluoro-4-[5- methyl-3-(trifluoromethyl)pyrazol-1-yl]phenyl]methoxy]pyrimidin-2-yl]-6-methoxy- pyrimidine (27.0 mg, 52.1 μmol, 9.51% yield) as a yellow solid. 1H NMR (600 MHz, DMSO-d
6) δ 0.86 – 0.90 (m, 2H), 1.01 – 1.05 (m, 2H), 1.64 – 1.69 (m, 1H), 2.18 (s, 3H), 3.83 (s, 3H), 5.52 (s, 2H), 6.84 (s, 1H), 7.13 (d, 1H), 7.57 (d, 2H), 8.66 (s, 1H), 8.74 (d, 1H). LCMS(ESI): [M+H]
+ m/z: calcd 519.18; found 519.2 Example 209 (Compound 53)

2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-5-methyl-4-[[3-methyl-4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidine Starting [3-methyl-4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methanol was prepared as described for Compound 144. [1620] Sodium hydride (8.27 mg, 207 μmol, 60% dispersion in mineral oil) was added to a vigorously stirred suspension of [3-methyl-4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methanol (106 mg, 392 μmol) in DMF (2.0 mL). The reaction mixture was stirred at room temperature for 1 hr.2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-5-methyl-4- methylsulfonyl-pyrimidine I-7 (62.8 mg, 196 μmol) was added to the reaction mixture. The reaction mixture was stirred at room temperature for 16 hr. The reaction mixture was diluted with water (5.0 mL) and extracted with EtOAc (20 mL). The organic layer was dried over anhydrous sodium sulfate and concentrated under reduced pressure. The residue was subjected to HPLC (0.5-6.5 min., 53-68% water – MeOH, +0.1% vol. of 25% aq. NH
3, flow: 30 mL/min, column: XBridge 100 ×19 mm, 5 µm) to afford 2-(4-cyclopropyl-6-methoxy- pyrimidin-5-yl)-5-methyl-4-[[3-methyl-4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidine (62.0 mg, 122 μmol, 31.1% yield) as a light-yellow solid. 1H NMR (600 MHz, DMSO-d
6) δ 0.83 – 0.87 (m, 2H), 0.99 – 1.03 (m, 2H), 1.65 – 1.69 (m, 1H), 2.15 (s, 3H), 2.22 (s, 3H), 3.49 (s, 3H), 3.83 (s, 3H), 5.47 (s, 2H), 7.38 (s, 2H), 7.44 (s, 1H), 7.92 (s, 1H), 8.54 (s, 1H), 8.65 (s, 1H). LCMS(ESI): [M+H]
+ m/z: calcd 511.24; found 511.0 Example 210 (Compound 158)
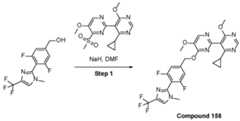
2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[3,5-difluoro-4-[1-methyl-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]-5-methoxy-pyrimidine Starting [3,5-difluoro-4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methanol was prepared as described for Compound 186. [1621] Sodium hydride (15.0 mg, 376 μmol, 60% dispersion in mineral oil) was added to a solution of [3,5-difluoro-4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methanol (100 mg, 342 μmol) in DMF (3.0 mL) under argon atmosphere. The reaction mixture was stirred at room temperature for 1 hr.2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-5-methoxy-4- methylsulfonyl-pyrimidine I-6(115 mg, 342 μmol) was added to the reaction mixture. The resulting mixture was stirred at room temperature for 48 hr. The reaction mixture was diluted with EtOAc (20 mL) and brine (20 mL). The organic layer was separated, washed with brine (2×15 mL) and dried over anhydrous sodium sulfate. To the resulting solution SiliaMetS
® Dimercaptotriazine (100 mg) was added, and the mixture was stirred for 30 min. The mixture was filtered. The filtrate was concentrated under reduced pressure. The residue was subjected to HPLC (2-10 min., 35-50% water – ACN, +0.1% vol. of 25% aq. NH
3, flow: 30 mL/min, column: SunFire 100 ×19 mm, 5 µm) to afford 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)- 4-[[3,5-difluoro-4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]-5-methoxy- pyrimidine (39.0 mg, 71.1 μmol, 20.8% yield) as a white solid. 1H NMR (600 MHz, DMSO-d
6) δ 0.84 – 0.88 (m, 2H), 0.99 – 1.03 (m, 2H), 1.67 – 1.72 (m, 1H), 3.57 (s, 3H), 3.82 (s, 3H), 3.96 (s, 3H), 5.51 (s, 2H), 7.40 (d, 2H), 8.06 (s, 1H), 8.45 (s, 1H), 8.64 (s, 1H). Example 211(Compound 184)
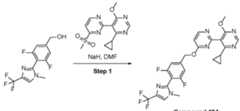
Compound 184 4-cyclopropyl-5-[4-[[3,5-difluoro-4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidin-2-yl]-6-methoxy-pyrimidine Starting [3,5-difluoro-4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methanol was prepared as described for Compound 186. [1622] Sodium hydride (45.2 mg, 1.13 mmol, 60% dispersion in mineral oil) was added to a solution of [3,5-difluoro-4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methanol (300 mg, 1.03 mmol) in DMF (10 mL) under argon atmosphere. The reaction mixture was stirred at room temperature for 1 hr.4-cyclopropyl-6-methoxy-5-(4-methylsulfonylpyrimidin-2- yl)pyrimidine I-8 (315 mg, 1.03 mmol) was added to the reaction mixture. The resulting mixture was stirred at room temperature for 24 hr. The reaction mixture was diluted with EtOAc (35 mL) and brine (20 mL). The organic layer was separated, washed with brine (2×15 mL) and dried over anhydrous sodium sulfate. To the resulting solution SiliaMetS
® Dimercaptotriazine (200 mg) was added, and the mixture was stirred for 30 min. The mixture was filtered. The filtrate was concentrated under reduced pressure. The residue was subjected to HPLC (2-10 min., 10-50% water – ACN, +0.1% vol. of 25% aq. NH
3, flow: 30 mL/min, column: SunFire 100 ×19 mm, 5 µm) to afford 4-cyclopropyl-5-[4-[[3,5-difluoro-4-[1- methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidin-2-yl]-6-methoxy- pyrimidine (155 mg, 299 μmol, 29.1% yield) as a light-yellow solid.
1H NMR (600 MHz, DMSO-d
6) δ 0.86 – 0.90 (m, 2H), 1.01 – 1.05 (m, 2H), 1.64 – 1.70 (m, 1H), 3.56 (s, 3H), 3.83 (s, 3H), 5.51 (s, 2H), 7.12 (d, 1H), 7.44 (d, 2H), 8.06 (s, 1H), 8.66 (s, 1H), 8.73 (d, 1H). LCMS(ESI): [M+H]
+ m/z: calcd 519.18; found 519.2 Example 212 (Compound 63)

methyl 2-[2-[4-[[tert-butyl(dimethyl)silyl]oxymethyl]phenyl]-4-(trifluoromethyl)imidazol-1- yl]propanoate [1623] Sodium hydride (185 mg, 4.63 mmol, 60% dispersion in mineral oil) was added to a solution of tert-butyl-dimethyl-[[4-[4-(trifluoromethyl)-1H-imidazol-2- yl]phenyl]methoxy]silane (1.50 g, 4.21 mmol) in DMF (5.0 mL). The reaction mixture was stirred at room temperature for 15 min. Methyl 2-chloropropanoate (670 mg, 5.47 mmol) was added to the reaction mixture. The resulting mixture was stirred at room temperature overnight. The reaction mixture was diluted with water (10 mL) and extracted with EtOAc (10 mL). The organic layer was washed with water (5.0 mL) and brine (5 mL) and concentrated under reduced pressure to afford methyl 2-[2-[4-[[tert- butyl(dimethyl)silyl]oxymethyl]phenyl]-4-(trifluoromethyl)imidazol-1-yl]propanoate (1.80 g, 4.07 mmol, 96.7% yield) as a light-yellow oil which was used in the next steps without further purification. LCMS(ESI): [M+H]
+ m/z: calcd 443.25; found 443.0 2-[2-[4-[[tert-butyl(dimethyl)silyl]oxymethyl]phenyl]-4-(trifluoromethyl)imidazol-1- yl]propan-1-ol [1624] A solution of methyl 2-[2-[4-[[tert-butyl(dimethyl)silyl]oxymethyl]phenyl]-4- (trifluoromethyl)imidazol-1-yl]propanoate (1.80 g, 4.07 mmol) in THF (20 mL) was added dropwise to a vigorously stirred suspension of LAH (168 mg, 4.94 mmol) in THF (50 mL) at 0°C. The reaction mixture was stirred at 0°C for 2 hr. An aqueous NaOH (1 mL, 10% wt.) was added dropwise to the reaction mixture. The resulting mixture was stirred for 30 min then solids were filtered out. The filtrate was concentrated under reduced pressure to afford 2-[2-[4-[[tert-butyl(dimethyl)silyl]oxymethyl]phenyl]-4-(trifluoromethyl)imidazol-1- yl]propan-1-ol (1.00 g, 2.41 mmol, 73.2% yield) as a light-yellow oil which was used in the next steps without further purification. 1H NMR (500 MHz, CDCl
3) δ 0.10 (s, 6H), 0.94 (s, 9H), 1.40 (d, 3H), 3.77 (d, 2H), 4.50 – 4.58 (m, 1H), 4.77 (s, 2H), 7.40 (d, 2H), 7.45 (s, 1H) 7.54 (d, 2H). [4-[1-(2-methoxy-1-methyl-ethyl)-4-(trifluoromethyl)imidazol-2-yl]phenyl]methanol [1625] Sodium hydride (191 mg, 4.78 mmol, 60% dispersion in mineral oil) was added to a solution of 2-[2-[4-[[tert-butyl(dimethyl)silyl]oxymethyl]phenyl]-4- (trifluoromethyl)imidazol-1-yl]propan-1-ol (1.80 g, 4.34 mmol) in DMF (10 mL). The reaction mixture was stirred at room temperature for 30 min. Iodomethane (801 mg, 5.64 mmol, 351 μL) was added to the reaction mixture. The resulting mixture was stirred at room temperature overnight. The reaction mixture was diluted with water (20 mL) and extracted with EtOAc (2×10 mL). Combined organic layers were washed with water (10 mL) and brine (10 mL) and concentrated under reduced pressure. The residue was subjected to flash column chromatography (SiO
2, gradient DCM - EtOAc) to afford [4-[1-(2-methoxy-1-methyl-ethyl)- 4-(trifluoromethyl)imidazol-2-yl]phenyl]methanol (240 mg, 764 μmol, 17.7% yield) as an yellowish oil. LCMS(ESI): [M+H]
+ m/z: calcd 315.16; found 315.0 2-chloro-4-[[4-[1-(2-methoxy-1-methyl-ethyl)-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]-5-methyl-pyrimidine [1626] Potassium tert-butoxide (66.0 mg, 588 μmol) was added to a solution of [4-[1-(2- methoxy-1-methyl-ethyl)-4-(trifluoromethyl)imidazol-2-yl]phenyl]methanol (240 mg, 764 μmol) in dioxane (10 mL). The reaction mixture was stirred at room temperature for 15 min. 2,4-dichloro-5-methyl-pyrimidine (87.1 mg, 535 μmol) was added to the reaction mixture. The resulting mixture was stirred at room temperature for 12 hr. The resulting mixture was diluted with water (15 mL) and extracted with EtOAc (20 mL). The resulting organic layer was washed with brine (5.0 mL), dried over anhydrous sodium sulfate and filtered. The filtrate was concentrated under reduced pressure. The residue was subjected to HPLC (0.5- 5.5-9.0 min, 45-45-48% water - ACN; flow: 30 mL/min, column: Waters SunFire C18, 100 x 19 mm, 5 µm) to afford 2-chloro-4-[[4-[1-(2-methoxy-1-methyl-ethyl)-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]-5-methyl-pyrimidine (94.0 mg, 213 μmol, 39.9% yield) as a light-yellow solid. LCMS(ESI): [M+H]
+ m/z: calcd 441.16; found 441.2 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[4-[1-(2-methoxy-1-methyl-ethyl)-4- (trifluoromethyl)imidazol-2-yl]phenyl]methoxy]-5-methyl-pyrimidine [1627] (4-Cyclopropyl-6-methoxy-pyrimidin-5-yl)boronic acid (33.0 mg, 170 μmol), 2- chloro-4-[[4-[1-(2-methoxy-1-methyl-ethyl)-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]-5-methyl-pyrimidine (50.0 mg, 113 μmol), potassium phosphate tribasic (72.2 mg, 340 μmol) and XPhos Pd G3 (1.13 μmol) were mixed in degassed dioxane (2.0 mL) and water (300 μL) under argon atmosphere. The reaction mixture was stirred at 95°C overnight. The reaction mixture was cooled to room temperature and concentrated under reduced pressure. The residue was diluted with EtOAc (20 mL) and washed with brine (10 mL). The organic layer was dried over anhydrous sodium sulfate and filtered. SiliaMetS
® Dimercaptotriazine (30.0 mg) was added to the filtrate. The resulting mixture was stirred at room temperature for 15 min, then solids were filtered out. The filtrate was concentrated under reduced pressure. The residue was subjected to HPLC (0.5-6.5min, 53%, water - ACN; flow 30ml/min; column SunFireC18100x19 mm 5 μm) to afford 2-(4-cyclopropyl-6- methoxy-pyrimidin-5-yl)-4-[[4-[1-(2-methoxy-1-methyl-ethyl)-4-(trifluoromethyl)imidazol- 2-yl]phenyl]methoxy]-5-methyl-pyrimidine (20.0 mg, 36.1 μmol, 31.8% yield) as a brown solid. LCMS(ESI): [M+H]
+ m/z: calcd 555.27; found 555.2 rel-(R)-4'-cyclopropyl-6'-methoxy-4-((4-(1-(1-methoxypropan-2-yl)-4-(trifluoromethyl)-1H- imidazol-2-yl)benzyl)oxy)-5-methyl-2,5'-bipyrimidine (Compound ent-63) and rel-(S)-4'- cyclopropyl-6'-methoxy-4-((4-(1-(1-methoxypropan-2-yl)-4-(trifluoromethyl)-1H-imidazol-2- yl)benzyl)oxy)-5-methyl-2,5'-bipyrimidine (Compound 63) [1628] Racemic 2-(4-cyclopropyl-6-methoxy-pyrimidin-5-yl)-4-[[4-[1-(2-methoxy-1-methyl- ethyl)-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]-5-methyl-pyrimidine (20.0 mg, 36.1 μmol) was subjected to chiral HPLC (column: Chiralpak AD-H V, 250×20 mm, 5 μm; mobile phase: Hexane-IPA-MeOH, 90-5-5; flow: 13 mL/min) to afford rel-(R)-4'- cyclopropyl-6'-methoxy-4-((4-(1-(1-methoxypropan-2-yl)-4-(trifluoromethyl)-1H-imidazol- 2-yl)benzyl)oxy)-5-methyl-2,5'-bipyrimidine (5.0 mg, 9.02 μmol, 25.0% yield) and rel-(S)-4'- cyclopropyl-6'-methoxy-4-((4-(1-(1-methoxypropan-2-yl)-4-(trifluoromethyl)-1H-imidazol- 2-yl)benzyl)oxy)-5-methyl-2,5'-bipyrimidine (5.0 mg, 9.02 μmol, 25.0% yield) as a white solids. rel-(R)-4'-cyclopropyl-6'-methoxy-4-((4-(1-(1-methoxypropan-2-yl)-4- (trifluoromethyl)-1H-imidazol-2-yl)benzyl)oxy)-5-methyl-2,5'-bipyrimidine (Compound ent- 63): 1H NMR (600 MHz, DMSO-d
6) δ 0.82 – 0.86 (m, 2H), 0.99 – 1.03 (m, 2H), 1.35 (d, 3H), 1.63 – 1.69 (m, 1H), 2.22 (s, 3H), 3.12 (s, 3H), 3.50 – 3.55 (m, 1H), 3.59 – 3.65 (m, 1H), 3.82 (s, 3H), 4.47 – 4.53 (m, 1H), 5.51 (s, 2H), 7.57 – 7.61 (m, 4H), 8.15 (s, 1H), 8.54 (s, 1H), 8.64 (s, 1H). LCMS(ESI): [M+H]+ m/z: calcd 555.27; found 555.2 Optical purity: 99% (column: Chiralpak AD-H, 250 × 4.6 mm, 5 μm; mobile phase: Hexane-IPA-MeOH, 90-5-5; flow: 0.6 mL/min; RT=47.32 min) rel-(S)-4'-cyclopropyl-6'-methoxy-4-((4-(1-(1-methoxypropan-2-yl)-4- (trifluoromethyl)-1H-imidazol-2-yl)benzyl)oxy)-5-methyl-2,5'-bipyrimidine (Compound 63): 1H NMR (600 MHz, DMSO-d
6) δ 0.82 – 0.86 (m, 2H), 0.99 – 1.03 (m, 2H), 1.35 (d, 3H), 1.63 – 1.69 (m, 1H), 2.22 (s, 3H), 3.12 (s, 3H), 3.50 – 3.55 (m, 1H), 3.59 – 3.65 (m, 1H), 3.82 (s, 3H), 4.47 – 4.53 (m, 1H), 5.51 (s, 2H), 7.57 – 7.61 (m, 4H), 8.15 (s, 1H), 8.54 (s, 1H), 8.64 (s, 1H). LCMS(ESI): [M+H]+ m/z: calcd 555.27; found 555.2 Optical purity: 100% (column: Chiralpak AD-H, 250 × 4.6 mm, 5 μm; mobile phase: Hexane-IPA-MeOH, 90-5-5; flow: 0.6 mL/min; RT=39.09 min) Example 213 (Compound 201) and (Compound 202)
Compound 202 4-chloro-1-isopropyl-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyrazole [1629] N-chlorosuccinimide (622 mg, 4.66 mmol) was added to 1-isopropyl-5-(4,4,5,5- tetramethyl-1,3,2-dioxaborolan-2-yl)pyrazole (1 g, 4.24 mmol) in THF (6 mL) and the mixture was stirred 3 hr at 60 °C. The solvent was evaporated, and the residue was treated with water (10 mL), then extracted with MTBE (20 mL). The organic phase was washed with water, filtered through silica pad and evaporated to give the title compound (0.5 g, 44% yield) which was used without further purification.
1H NMR (500 MHz, CDCl
3) δ 1.38 (s, 12H), 1.44 (d, 6H), 5.00 (sept, 1H), 7.42 (s, 1H). 2-(4-chloro-1-isopropyl-1H-pyrazol-5-yl)-4-((4-(1-methyl-4-(trifluoromethyl)-1H-imidazol-2- yl)benzyl)oxy)pyrimidine [1630] 2-chloro-4-[[4-[1-methyl-4-(trifluoromethyl)imidazol-2- yl]phenyl]methoxy]pyrimidine (60 mg, 163 μmol), 4-chloro-1-isopropyl-5-(4,4,5,5- tetramethyl-1,3,2-dioxaborolan-2-yl)pyrazole (88 mg, 325 μmol), potassium phosphate (tribasic anhydrous, 69 mg, 325 μmol), RuPhos Pd G3 (7 mg, 8.1 μmol) and dioxane-water (2 mL) were mixed under an argon atmosphere. The reaction mixture was stirred for 32 hr at ambient temperature. SiliaMetS® Dimercaptotriazine (100 mg) was added and the mixture was stirred for 3h. The mixture was diluted with MTBE (5 mL), filtered through a silica pad and evaporated to dryness. The resultant residue was subject to RP-HPLC purification (gradient elution: 50-55% ACN in water, 30 mL/min, Chromatorex column) to give rise to the title compound (49 mg, 63% yield).
1H NMR (600 MHz, DMSO-d
6) δ 1.38 (d, 6H), 3.77 (s, 3H), 5.27 (sept, 1H), 5.59 (d, 2H), 7.09 (d, 1H), 7.61 (d, 2H), 7.70 – 7.78 (m, 3H), 7.93 (d, 1H), 8.75 (d, 1H). LCMS(ESI): [M+H]
+ m/z: calcd 477.2; found 477.2 2-(4-chloro-1-isopropyl-1H-pyrazol-5-yl)-5-methoxy-4-((4-(1-methyl-4-(trifluoromethyl)-1H- imidazol-2-yl)benzyl)oxy)pyrimidine [1631] Prepared similarly to Compound 201, starting from 2-chloro-5-methoxy-4-[[4-[1- methyl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methoxy]pyrimidine, prepared as described for Compound 12 and compound 132. 1H NMR (600 MHz, DMSO-d6) δ 1.34 (d, 6H), 3.77 (s, 3H), 3.96 (s, 3H), 5.19 (sept, 1H), 5.59 (d, 2H), 7.57 (d, 2H), 7.65 (s, 1H), 7.75 (d, 2H), 7.96 (s, 1H), 8.51 (s, 1H). LCMS(ESI): [M+H]+ m/z: calcd 507.2; found 507.2 Example 214 The compounds in Table 2 were prepared in a manner similar to Examples 1-214, using methods described in Schemes 1-5 or variations thereof. Table 2

Biology Example 1. Measurement of inhibition of deubiquitinase activity by exemplary compounds Deubiquitinase activity of USP1-UAF1 was measured using Ubiquitin-rhodamine 110 as a substrate. Cleavage of amide bond between rhodamine and C-terminal Glycine of Ubiquitin peptide yields Rhodamine 110-Gly, leading to an increase of fluorescence signal. The assay buffer consisted of 50 mM HEPES (pH 7.0), 1% DMSO, 0.01% Bovine Serum Albumin, 1 mM TCEP, 0.005% Tween-20. Total assay volume was 20 µL. Compounds depicted below were dissolved in 10 mM DMSO stock and enzyme inhibition was measured in dose response format with top concentration of 10 µM in final assay well. 10 µL of enzyme buffer mix consisting of 1 nM USP1-UAF1 in the assay buffer describe above was added to compounds and incubated at ambient temperature for 30 min. 10 µL of substrate mix consisting of 200 nM Ubiquitin-Rho110 was added to initiate the deubiquitinate reaction catalyzed by USP1/UAF1. End point fluorescence intensity of USP1/UAF1 deubiquitinase product, Rhodamine 110-Gly, was measured at Excitation of 480 nm/Emission at 540 nm. [1632] Percentage of activity was calculated by normalization of fluorescence intensity to control wells using the following equations: % Activity = 100*((FI
observed – Min)/(Max-Min)- 1) where FI
observed is the fluorescence intensity read out of the compound of interest samples, Min and Max is the fluorescence intensity of control well samples consisting of 1 mM of known USP1-UAF1 inhibitor probe ML-323 and DMSO controls respectively. IC
50 values were calculated using the standard dose response fit in Genedata Screener® where the top and bottom were fixed to 0 and -100 respectively. Biology Example 2. Cellular viability assay [1633] For short-term viability assays, cells were seeded in triplicates in 384-well plates one day prior to compound addition. Cells were incubated for 10 days with DMSO and increasing concentration of compounds. Cell viability was determined at end of the assay using Cell Titer-Glo Luminescence Assay (Promega) following manufacturer’s instructions using an EnVision plate reader (Perkin Elmer). Measured values were normalized using DMSO control wells (100 %) and complete cell kill control (0%; 10µM MG132). [1634] For long-term colony formation assays, cells were seeded in 12-well or 6-well plates at a very low density one day prior to compound addition. Cells were incubated for 7-21 days, depending on the cell line doubling time, with DMSO and increasing concentration of compounds. Media containing fresh compound was replenished every 3-4 days. At end of the incubation period, cells were stained with 0.1% crystal violet in 10% methanol for 10 minutes at room temperature. Stained plates were scanned and quantified using the Li-Cor Odyssey imaging system. Biology Example 3. Cellular PD AlphaLISA assay [1635] Deubiquitinase activity of USP1-UAF1 in MDA-MB-436 cells was measured using an AlphaLISA assay (Perkin Elmer) developed for ubiquitinated-PCNA (ub-PCNA) as the analyte. Inhibition of USP1-UAF1 function leads to accumulation of ub-PCNA and the resulting fluorescence intensity generated in the assay is proportional to the amount of ub- PCNA. The assay employs an anti-Ub-PCNA antibody linked to acceptor bead using the CaptSure technology, streptavidin-coated donor beads, a biotinylated anti-PCNA (total PCNA) antibody, and AlphaLISA Lysis buffer and Surefire Ultra (SFU) dilution buffer (obtained from the manufacturer). Test compounds were dissolved in 10 mM DMSO stock solution and USP1 inhibition was measured in a dose-response format with top concentration of 10 µM in final assay well. The AlphaLISA signal is detected with an EnVision plate reader (Perkin Elmer) with measured values being normalized to DMSO control wells (0 %) and USP1 inhibitor (100%; I-138, 1 uM, see Simoneau, A. et al., Mol Cancer Ther, 22(2): 215- 226 (2023)). The dose-response data were fitted using the Smart Fit algorithm with variable Hill slope (Genedata). [1636] In brief, for the ub-PCNA AlphaLISA assay, cells were seeded onto 384-well plates (Corning) and allowed to adhere overnight. Test compounds were added using a Tecan Digital Dispenser (Hewlett-Packard). After 24 hours (5% CO
2 atmosphere at 37 °C), the media was aspirated, cells were washed with HBSS buffer. HBSS was then aspirated and 10 µL of AlphaLISA lysis buffer was added to each well. Plates were sealed with TOPSeal A- Plus and incubated for 30 min with agitation (plate shaker at 350 rpm). Antibodies were diluted to optimized concentrations in SFU dilution buffer and 5 µL SFU containing both CaptSure-conjugated ub-PCNA and biotin-conjugated PCNA antibodies was added to each well. Plates were then incubated for 1 hour at room temperature. AlphaLISA CaptSure acceptor beads were then diluted to optimized concentration in SFU buffer and 5 µL was added to each well and plates were incubated for 1 hour at room temperature in the dark. Streptavidin donor beads were then diluted to optimized concentration in SFU buffer and 5 µL was added to each well and plates were incubated for 1 hour at room temperature in the dark. AlphaLISA signals were read on an EnVision plate reader (Perkin Elmer) with Mirror 444 (D640), filter 203 (615 nm) with an excitation time of 180 ms and total measurement time of 550 ms. Other Embodiments [1637] In the claims articles such as “a,” “an,” and “the” may mean one or more than one unless indicated to the contrary or otherwise evident from the context. Claims or descriptions that include “or” between one or more members of a group are considered satisfied if one, more than one, or all of the group members are present in, employed in, or otherwise relevant to a given product or process unless indicated to the contrary or otherwise evident from the context. The invention includes embodiments in which exactly one member of the group is present in, employed in, or otherwise relevant to a given product or process. The invention includes embodiments in which more than one, or all of the group members are present in, employed in, or otherwise relevant to a given product or process. [1638] Furthermore, the invention encompasses all variations, combinations, and permutations in which one or more limitations, elements, clauses, and descriptive terms from one or more of the listed claims is introduced into another claim. For example, any claim that is dependent on another claim can be modified to include one or more limitations found in any other claim that is dependent on the same base claim. Where elements are presented as lists, e.g., in Markush group format, each subgroup of the elements is also disclosed, and any element(s) can be removed from the group. It should it be understood that, in general, where the invention, or aspects of the invention, is/are referred to as comprising particular elements and/or features, an embodiment of the invention or aspects of the invention consist, or consist essentially of, such elements and/or features. For purposes of simplicity, those embodiments have not been specifically set forth in haec verba herein. It is also noted that the terms “comprising” and “containing” are intended to be open and permits the inclusion of additional elements or steps. Where ranges are given, endpoints are included. Furthermore, unless otherwise indicated or otherwise evident from the context and understanding of one of ordinary skill in the art, values that are expressed as ranges can assume any specific value or sub–range within the stated ranges in different embodiments of the invention, to the tenth of the unit of the lower limit of the range, unless the context clearly dictates otherwise. [1639] This application refers to various issued patents, published patent applications, journal articles, and other publications, all of which are incorporated herein by reference. If there is a conflict between any of the incorporated references and the instant specification, the specification shall control. In addition, any particular embodiment disclosed herein that falls within the prior art may be explicitly excluded from any one or more of the claims. Because such embodiments are deemed to be known to one of ordinary skill in the art, they may be excluded even if the exclusion is not set forth explicitly herein. Any particular embodiment of the invention can be excluded from any claim, for any reason, whether or not related to the existence of prior art. [1640] Those skilled in the art will recognize or be able to ascertain using no more than routine experimentation many equivalents to the specific embodiments described herein. The scope of the present embodiments described herein is not intended to be limited to the above Description, but rather is as set forth in the appended claims. Those of ordinary skill in the art will appreciate that various changes and modifications to this description may be made without departing from the spirit or scope disclosed herein, as defined in the following claims.
 andX3 wherein X3 is CH or N and wherein the left-side attachment point connects to CRcRc’ and the right attachment point connects to Ring B; L is selected from the group consisting of –O– and –S–; Ring B is a 5-10 membered heteroaryl or a 3-10 membered heterocyclyl; each RA is independently selected from the group consisting of –D, oxo, halo, – CN, –C1–C6 alkyl, –C1–C6 haloalkyl, –C1–C6 hydroxyalkyl, –C3–C10 cycloalkyl, –ORA1 and – N(RA1)2; each RA1 is independently selected from the group consisting of H, –C1–C6 alkyl, –C1–C6 haloalkyl and C3–C9 cycloalkyl wherein each hydrogen atom of each alkyl and cycloalkyl can be replaced by deuterium; or two instances of RA1 on a nitrogen atom can be taken together with the nitrogen to which they are attached to form a 4-6 member monocyclic heterocycle containing one nitrogen atom and 0-1 additional heteroatoms selected from N and O; each RB is independently selected from the group consisting of halo, –CN, –C1–C6 alkyl, –C1–C6 alkenyl, –heteroC1-C4 alkyl, –C1–C6 haloalkyl, –C1–C6 hydroxyalkyl, –C3–C10 cycloalkyl, 3-10 membered heterocyclyl, C3-C9 cycloalkylalkyl, –ORB1, –N(RB1)2, – C(=O)RB1, –C(=O)ORB1, –NRB1C(=O)RB1, –NRB1C(=O)ORB1, –C(=O)N(RB1)2, – OC(=O)N(RB1)2, –S(=O)RB1, –S(=O)2RB1, –SRB1, –S(=O)(=NRB1)RB1, –NRB1S(=O)2RB1 and –S(=O)2N(RB1)2, wherein each alkyl, alkenyl, cycloalkyl, heterocyclyl and cycloalkylalkyl of RB can be partially or fully deuterated and can be substituted at any available position with 0, 1 or 2 substituents selected from –F, –Cl, –OH– and –CH3; each RB1 is independently selected from the group consisting of H, –C1–C6 alkyl, –heteroC1-C4 alkyl, –C1–C6 haloalkyl, –C3–C9 cycloalkyl and 3-7 membered heterocyclyl wherein each hydrogen atom of each alkyl, cycloalkyl and heterocyclyl can be replaced by deuterium; or two instances of RB1 on a nitrogen atom can be taken together with the nitrogen to which they are attached to form a 4-6 member monocyclic heterocycle containing one nitrogen atom and 0-1 additional heteroatoms selected from N and O; each RC is independently selected from the group consisting of H, –D, oxo, halo, – CN, –ORC1, –SRC1 –NRC12, –C1–C6 alkyl, –C1–C6 hydroxyalkyl, –C1–C6 haloalkyl, –C3–C9 cycloalkyl, 3-10 member heterocyclyl and –heteroC1-C4 alkyl wherein each alkyl, cycloalkyl and heterocyclyl of RC can partially or fully deuterated and can be substituted at any available position with 0, 1 or 2 substituents selected from –F, –Cl, –OH– and –CH3; each RC1 is independently selected from the group consisting of H and –C1–C6 alkyl, wherein each hydrogen atom of the alkyl can be independently replaced by deuterium; or two instances of RC1 on a nitrogen atom can be taken together with the nitrogen to which they are attached to form a 4-6 member monocyclic heterocycle containing one nitrogen atom and 0-1 additional heteroatoms selected from N and O; each RD is independently selected from the group consisting of –D, halo, –CN, – C1–C6 alkyl, –heteroC1-C4 alkyl, –C1–C6 haloalkyl, –C1–C6 hydroxyalkyl, –C3–C10 cycloalkyl, 3-10 membered heterocyclyl, –C6-C10 aryl, –ORD1, –N(RD1)2, –C(=O)RD1, – C(=O)ORD1, –NRD1C(=O)RD1, –NRD1C(=O)ORD1, –C(=O)N(RD1)2, –OC(=O)N(RD1)2, – S(=O)RD1, –S(=O)2RD1, –SRD1, –S(=O)(=NRD1)RD1, –NRD1S(=O)2RD1 and –S(=O)2N(RD1)2 , wherein each alkyl, cycloalkyl, heterocyclyl and aryl of RD can be substituted at any available position with 0, 1 or 2 substituents selected from –F, –Cl, –OH– and –CH3; each RD1 is independently selected from the group consisting of H, –C1–C6 alkyl, –heteroC1-C4 alkyl, –C1–C6 haloalkyl, –C3–C9 cycloalkyl and 3-7 membered heterocyclyl, wherein each hydrogen atom of each alkyl, cycloalkyl and heterocyclyl can be replaced by deuterium; or two instances of RD1 on a nitrogen atom can be taken together with the nitrogen to which they are attached to form a 4-6 member monocyclic heterocycle containing one nitrogen atom and 0-1 additional heteroatoms selected from N and O; RE is selected from H, –D, halo, –CN, –C1–C6 alkyl, –C1-C6 alkynyl, –heteroC1– C6 alkyl, –C1–C6 haloalkyl, –C1–C6 hydroxyalkyl, –C3–C10 cycloalkyl, 3-10 membered heterocyclyl, –C6-C10 aryl, 5-10 member heteroaryl, heterocyclylalkyl, heteroarylalkyl, arylalkyl, cycloalkylalkyl, –ORE1, –SRE1, –N(RE1)2, –C1–C6 alkylene–NRE1C(=O)ORE1, – C(=O)RE1, –C(=O)ORE1, –NRE1C(=O)RE1, –NRE1C(=O)ORE1, –C(=O)N(RE1)2, and – OC(=O)N(RE1)2, wherein each alkyl, alkynyl, heteroalkyl, haloalkyl, cycloalkyl, heterocyclyl, aryl, heteroaryl, cycloalkylalkyl, heterocyclylalkyl, arylalkyl and heteroarylalkyl is optionally substituted at any available position; each RE1 is independently selected from H, –C1–C6 alkyl (wherein each hydrogen can be independently replaced by deuterium), –heteroC1–C6 alkyl, –C1–C6 haloalkyl, –C3–C9 cycloalkyl, 3-7 membered heterocyclyl, cycloalkylalkyl, heterocyclylalkyl, aryl, 5-6 membered heteroaryl, arylalkyl and heteroarylalkyl wherein each alkyl, haloalkyl, cycloalkyl, heterocyclyl, aryl, heteroaryl, cycloalkylalkyl, heterocyclylalkyl, arylalkyl and heteroarylalkyl is optionally substituted at any available position; each Rc and Rc’ is independently selected from the group consisting of H, –D, – C1–C6 alkyl, –heteroC1-C4 alkyl and –C1–C6 haloalkyl or Rc and Rc’ can be taken together with the atom to which they are attached to form a –C3–C9 cycloalkyl; n is 0, 1, 2,3 or 4; m is 0, 1, 2 or 3; and p is 0, 1, 2 or 3. [0009] In an embodiment, provided are compounds selected from the compounds of Table 1, or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof. [0010] In an embodiment, provided is a pharmaceutical composition comprising a compound as described herein or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof and a pharmaceutically acceptable carrier. [0011] In an embodiment, the pharmaceutical composition comprises a second therapeutic agent. [0012] In one aspect provided is a method for treating or preventing a disease or disorder sensitive to inhibition of USP1 comprising administering to a patient in need thereof a therapeutically effective amount of a compound of Formula (I) or Formula (II) or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof. [0013] In one aspect provided is a method of treating a disease or disorder sensitive to the inhibition of USP1 comprising administering to a patient in need thereof a therapeutically effective amount of a compound of Formula (I) or Formula (II) or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof. [0014] In one aspect provided is a method for inhibiting USP1 comprising administering to a patient in need thereof an effective amount of a compound of Formula (I) or Formula (II) or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof. [0015] In one aspect provided is a method for treating or preventing cancer in a patient in need thereof comprising administering to the patient in need thereof a therapeutically effective amount of a compound of Formula (I) or Formula (II) or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof. [0016] In one aspect provided is a method for treating cancer in a patient in need thereof comprising administering to the patient in need thereof a therapeutically effective amount of a compound of Formula (I) or Formula (II) or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof. [0017] In one aspect provided is a method for treating or preventing a disease or disorder associated with DNA damage comprising administering to a patient in need of a treatment for diseases or disorders associated with DNA damage an effective amount of a compound of Formula (I) or Formula (II) or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof. [0018] In one aspect provided is a method for treating a disease or disorder associated with DNA damage comprising administering to a patient in need of a treatment for diseases or disorders associated with DNA damage an effective amount of a compound of Formula (I) or Formula (II) or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof. [0019] In one aspect provided is a method of inhibiting, modulating or reducing DNA repair activity exercised by USP1 comprising administering to a patient in need thereof an effective amount of a compound of Formula (I) or Formula (II) or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof. [0020] In one aspect provided is a compound of Formula (I) or Formula (II) or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof for use in a method for treating or preventing a disease or disorder sensitive to the inhibition of USP1 comprising administering to a patient in need thereof a therapeutically effective amount of the compound or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof. [0021] In one aspect provided is a compound of Formula (I) or Formula (II) or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof for use in a method of treating a disease or disorder sensitive to the inhibition of USP1 comprising administering to a patient in need thereof a therapeutically effective amount of the compound or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof. [0022] In one aspect provided is a compound of Formula (I) or Formula (II) or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof for use in a method for inhibiting USP1 comprising administering to a patient in need thereof an effective amount of the compound or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof. [0023] In one aspect provided is a compound of Formula (I) or Formula (II) or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof for use in a method for treating or preventing cancer in a patient in need thereof comprising administering to the patient in need thereof a therapeutically effective amount of the compound or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof. [0024] In one aspect provided is a compound of Formula (I) or Formula (II) or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof for use in a method for treating cancer in a patient in need thereof comprising administering to the patient in need thereof a therapeutically effective amount of the compound or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof. [0025] In one aspect provided is a compound of Formula (I) or Formula (II) or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof for use in a method for treating or preventing a disease or disorder associated with DNA damage. [0026] In one aspect provided is a compound of Formula (I) or Formula (II) or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof for use in a method for treating a disease or disorder associated with DNA damage. [0027] In one aspect provided is a compound of Formula (I) or Formula (II) or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof for use in a method of inhibiting, modulating or reducing DNA repair activity exercised by USP1 comprising administering to a patient in need thereof an effective amount of the compound or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof. [0028] In one aspect, provided is a use of an effective amount of a compound of Formula (I) or Formula (II) or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof in the manufacturing of a medicament for treating or preventing a disease or disorder sensitive to the inhibition of USP1. [0029] In one aspect, provided is a use of an effective amount of a compound of Formula (I) or Formula (II) or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof in the manufacturing of a medicament for treating a disease or disorder sensitive to the inhibition of USP1. [0030] In one aspect, provided is a use of an effective amount of a compound of Formula (I) or Formula (II) or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof in the manufacturing of a medicament for inhibiting USP1. [0031] In one aspect, provided is a use of an effective amount of a compound of Formula (I) or Formula (II) or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof in the manufacturing of a medicament for treating or preventing cancer in a patient in need thereof. [0032] In one aspect, provided is a use of an effective amount of a compound of Formula (I) or Formula (II) or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof in the manufacturing of a medicament for treating cancer in a patient in need thereof. [0033] In one aspect, provided is a use of an effective amount of a compound of Formula (I) or Formula (II) or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof in the manufacturing of a medicament for treating or preventing a disease or disorder associated with DNA damage. [0034] In one aspect, provided is a use of an effective amount of a compound of Formula (I) or Formula (II) or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof in the manufacturing of a medicament for treating a disease or disorder associated with DNA damage. [0035] In one aspect, provided is a use of an effective amount of a compound of Formula (I) or Formula (II) or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof for inhibiting, modulating or reducing DNA repair activity exercised by USP1. [0036] In one aspect, provided is a use of an effective amount of a compound of Formula (I) or Formula (II) or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof for treating or preventing a disease or disorder sensitive to the inhibition of USP1. [0037] In one aspect, provided is a use of an effective amount of a compound of Formula (I) or Formula (II) or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof for treating a disease or disorder sensitive to the inhibition of USP1. [0038] In one aspect, provided is a use of an effective amount of a compound of Formula (I) or Formula (II) or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof for inhibiting USP1. [0039] In one aspect, provided is a use of an effective amount of a compound of Formula (I) or Formula (II) or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof for treating or preventing cancer in a patient in need thereof. [0040] In one aspect, provided is a use of an effective amount of a compound of Formula (I) or Formula (II) or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof for treating cancer in a patient in need thereof. [0041] In one aspect, provided is a use of an effective amount of a compound of Formula (I) or Formula (II) or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof for treating or preventing a disease or disorder associated with DNA damage. [0042] In one aspect, provided is a use of an effective amount of a compound of Formula (I) or Formula (II) or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof for treating a disease or disorder associated with DNA damage. [0043] In one aspect, provided is a use of an effective amount of a compound of Formula (I) or Formula (II) or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof for inhibiting, modulating or reducing DNA repair activity exercised by USP1. Detailed Description [0044] The disclosure herein sets forth exemplary methods, parameters and the like. It should be recognized, however, that such description is not intended as a limitation on the scope of the present disclosure but is instead provided as a description of exemplary embodiments. Definitions [0045] As used in the present disclosure, the following words and phrases are generally intended to have the meanings as set forth below unless expressly indicated otherwise or the context in which they are used indicates otherwise. Chemical Definitions [0046] Definitions of specific functional groups and chemical terms are described in more detail below. The chemical elements are identified in accordance with the Periodic Table of the Elements, CAS version, Handbook of Chemistry and Physics, 75th Ed., inside cover, and specific functional groups are generally defined as described therein. Additionally, general principles of organic chemistry, as well as specific functional moieties and reactivity, are described in Thomas Sorrell, Organic Chemistry, University Science Books, Sausalito, 1999; Smith and March, March’s Advanced Organic Chemistry, 5th Edition, John Wiley & Sons, Inc., New York, 2001; Larock, Comprehensive Organic Transformations, VCH Publishers, Inc., New York, 1989; and Carruthers, Some Modern Methods of Organic Synthesis, 3rd Edition, Cambridge University Press, Cambridge, 1987. [0047] Compounds described herein can comprise one or more asymmetric centers, and thus can exist in various isomeric forms, e.g., enantiomers and/or diastereomers. For example, the compounds described herein can be in the form of an individual enantiomer, diastereomer or geometric isomer, or can be in the form of a mixture of stereoisomers, including racemic mixtures and mixtures enriched in one or more stereoisomer. Unless stereochemistry is explicitly indicated in a structure, the structure is intended to embrace all possible stereoisomers of the compound depicted. If stereochemistry is explicitly indicated for one portion or portions of a molecule, but not for another portion or portions of a molecule, the structure is intended to embrace all possible stereoisomers for the portion or portions where stereochemistry is not explicitly indicated. Isomers can be isolated from mixtures by methods known to those skilled in the art, including chiral high-pressure liquid chromatography (HPLC) and the formation and crystallization of chiral salts; or preferred isomers can be prepared by asymmetric syntheses. See, for example, Jacques et al., Enantiomers, Racemates and Resolutions (Wiley Interscience, New York, 1981); Wilen et al., Tetrahedron 33:2725 (1977); Eliel, Stereochemistry of Carbon Compounds (McGraw–Hill, NY, 1962); and Wilen, Tables of Resolving Agents and Optical Resolutions p.268 (E.L. Eliel, Ed., Univ. of Notre Dame Press, Notre Dame, IN 1972). The invention additionally encompasses compounds described herein as individual isomers substantially free of other isomers, and alternatively, as mixtures of various isomers. [0048] The “enantiomeric excess” (“e.e.”) or “% enantiomeric excess” (“%e.e.”) of a composition as used herein refers to an excess of one enantiomer relative to the other enantiomer present in the composition. For example, a composition can contain 90% of one enantiomer, e.g., the S enantiomer, and 10% of the other enantiomer, i.e., the R enantiomer. e.e. = (90-10)/100 = 80%. [0049] Thus, a composition containing 90% of one enantiomer and 10% of the other enantiomer is said to have an enantiomeric excess of 80%. [0050] The “diastereomeric excess” (“d.e.”) or “% diastereomeric excess” (“%d.e.”) of a composition as used herein refers to an excess of one diastereomer relative to one or more different diastereomers present in the composition. For example, a composition can contain 90% of one diastereomer, and 10% of one or more different diastereomers. d.e. = (90-10)/100 = 80%. [0051] Thus, a composition containing 90% of one diastereomers and 10% of one or more different diastereomers is said to have a diastereomeric excess of 80%. [0052] In an alternative embodiment, compounds described herein may also comprise one or more isotopic substitutions. For example, hydrogen may be2H (D or deuterium) or3H (T or tritium); carbon may be, for example,13C or14C; oxygen may be, for example,18O; nitrogen may be, for example,15N, and the like. In other embodiments, a particular isotope (e.g.,3H,13C,14C,18O, or15N) can represent at least 1%, at least 5%, at least 10%, at least 15%, at least 20%, at least 25%, at least 30%, at least 35%, at least 40%, at least 45%, at least 50%, at least 60%, at least 65%, at least 70%, at least 75%, at least 80%, at least 85%, at least 90%, at least 95%, at least 99%, or at least 99.9% of the total isotopic abundance of an element that occupies a specific site of the compound. [0053] In a formula, is a single bond where the stereochemistry of the moieties immediately attached thereto is not specified. [0054] When a range of values is listed, it is intended to encompass each value and sub– range within the range. For example, “C1–6 alkyl” is intended to encompass, C1, C2, C3, C4, C5, C6, C1–6, C1–5, C1–4, C1–3, C1–2, C2–6, C2–5, C2–4, C2–3, C3–6, C3–5, C3–4, C4–6, C4–5, and C5–6 alkyl. [0055] The following terms are intended to have the meanings presented therewith below and are useful in understanding the description and intended scope of the present invention. When describing the invention, which may include compounds, pharmaceutical compositions containing such compounds and methods of using such compounds and compositions, the following terms, if present, have the following meanings unless otherwise indicated. It should also be understood that when described herein any of the moieties defined forth below may be substituted with a variety of substituents, and that the respective definitions are intended to include such substituted moieties within their scope as set out below. Unless otherwise stated, the term “substituted” is to be defined as set out below. It should be further understood that the terms “groups” and “radicals” can be considered interchangeable when used herein. The articles “a” and “an” may be used herein to refer to one or to more than one (i.e. at least one) of the grammatical objects of the article. By way of example “an analogue” means one analogue or more than one analogue. [0056] The term “unsaturated bond” refers to a double or triple bond. [0057] The term “unsaturated” or “partially unsaturated” refers to a moiety that includes at least one double or triple bond. [0058] The term “saturated” refers to a moiety that does not contain a double or triple bond, i.e., the moiety only contains single bonds. [0059] Affixing the suffix “-ene” to a group indicates the group is a divalent moiety, e.g., alkylene is the divalent moiety of alkyl, alkenylene is the divalent moiety of alkenyl, alkynylene is the divalent moiety of alkynyl, heteroalkylene is the divalent moiety of heteroalkyl, heteroalkenylene is the divalent moiety of heteroalkenyl, heteroalkynylene is the divalent moiety of heteroalkynyl, carbocyclylene is the divalent moiety of carbocyclyl, heterocyclylene is the divalent moiety of heterocyclyl, arylene is the divalent moiety of aryl, and heteroarylene is the divalent moiety of heteroaryl. [0060] The term “azido” refers to the radical –N3. [0061] “Aliphatic” refers to an alkyl, alkenyl, alkynyl, or carbocyclyl group, as defined herein. [0062] “Cycloalkylalkyl” refers to an alkyl radical in which the alkyl group is substituted with a cycloalkyl group. Typical cycloalkylalkyl groups include, but are not limited to, cyclopropylmethyl, cyclobutylmethyl, cyclopentylmethyl, cyclohexylmethyl, cycloheptylmethyl, cyclooctylmethyl, cyclopropylethyl, cyclobutylethyl, cyclopentylethyl, cyclohexylethyl, cycloheptylethyl, and cyclooctylethyl, and the like. [0063] “Heterocyclylalkyl” refers to an alkyl radical in which the alkyl group is substituted with a heterocyclyl group. Typical heterocyclylalkyl groups include, but are not limited to, pyrrolidinylmethyl, piperidinylmethyl, piperazinylmethyl, morpholinylmethyl, pyrrolidinylethyl, piperidinylethyl, piperazinylethyl, morpholinylethyl, and the like. [0064] “Aralkyl” or “arylalkyl” is a subset of alkyl and aryl, as defined herein, and refers to an optionally substituted alkyl group substituted by an optionally substituted aryl group. [0065] “Alkyl” refers to a radical of a straight–chain or branched saturated hydrocarbon group having from 1 to 20 carbon atoms (“C1–20 alkyl”). In an embodiment, an alkyl group has 1 to 12 carbon atoms (“C1–12 alkyl”). In an embodiment, an alkyl group has 1 to 10 carbon atoms (“C1–10 alkyl”). In an embodiment, an alkyl group has 1 to 9 carbon atoms (“C1–9 alkyl”). In an embodiment, an alkyl group has 1 to 8 carbon atoms (“C1–8 alkyl”). In an embodiment, an alkyl group has 1 to 7 carbon atoms (“C1–7 alkyl”). In an embodiment, an alkyl group has 1 to 6 carbon atoms (“C1–6 alkyl”, also referred to herein as “lower alkyl”). In an embodiment, an alkyl group has 1 to 5 carbon atoms (“C1–5 alkyl”). In an embodiment, an alkyl group has 1 to 4 carbon atoms (“C1–4 alkyl”). In an embodiment, an alkyl group has 1 to 3 carbon atoms (“C1–3 alkyl”). In an embodiment, an alkyl group has 1 to 2 carbon atoms (“C1–2 alkyl”). In an embodiment, an alkyl group has 1 carbon atom (“C1 alkyl”). In an embodiment, an alkyl group has 2 to 6 carbon atoms (“C2–6 alkyl”). Examples of C1–6 alkyl groups include methyl (C1), ethyl (C2), n–propyl (C3), isopropyl (C3), n–butyl (C4), tert–butyl (C4), sec–butyl (C4), iso–butyl (C4), n–pentyl (C5), 3–pentanyl (C5), amyl (C5), neopentyl (C5), 3–methyl–2–butanyl (C5), tertiary amyl (C5), and n–hexyl (C6). Additional examples of alkyl groups include n–heptyl (C7), n–octyl (C8) and the like. Unless otherwise specified, each instance of an alkyl group is independently optionally substituted, i.e., unsubstituted (an “unsubstituted alkyl”) or substituted (a “substituted alkyl”) with one or more substituents; e.g., for instance from 1 to 5 substituents, 1 to 3 substituents, or 1 substituent. In an embodiment, the alkyl group is unsubstituted C1–10 alkyl (e.g., –CH3). In an embodiment, the alkyl group is substituted C1–10 alkyl. Common alkyl abbreviations include Me (–CH3), Et (– CH2CH3),iPr (–CH(CH3)2),nPr (–CH2CH2CH3),nBu (–CH2CH2CH2CH3), oriBu (– CH2CH(CH3)2). [0066] “Alkylene” refers to an alkyl group wherein two hydrogens are removed to provide a divalent radical, and which may be substituted or unsubstituted. Unsubstituted alkylene groups include, but are not limited to, methylene (–CH2-), ethylene (–CH2CH2-), propylene (– CH2CH2CH2-), butylene (–CH2CH2CH2CH2-), pentylene (–CH2CH2CH2CH2CH2-), hexylene (–CH2CH2CH2CH2CH2CH2-), and the like. Exemplary substituted alkylene groups, e.g., substituted with one or more alkyl (methyl) groups, include but are not limited to, substituted methylene (–CH(CH3)-, (–C(CH3)2-), substituted ethylene (–CH(CH3)CH2-,–CH2CH(CH3)-, –C(CH3)2CH2-,–CH2C(CH3)2-), substituted propylene (–CH(CH3)CH2CH2-, – CH2CH(CH3)CH2-, –CH2CH2CH(CH3)-, –C(CH3)2CH2CH2-, –CH2C(CH3)2CH2-, – CH2CH2C(CH3)2-), and the like. When a range or number of carbons is provided for a particular alkylene group, it is understood that the range or number refers to the range or number of carbons in the linear carbon divalent chain. Alkylene groups may be substituted or unsubstituted with one or more substituents as described herein. [0067] “Alkenyl” refers to a radical of a straight–chain or branched hydrocarbon group having from 2 to 20 carbon atoms, one or more carbon–carbon double bonds (e.g., 1, 2, 3, or 4 carbon–carbon double bonds), and optionally one or more carbon–carbon triple bonds (e.g., 1, 2, 3, or 4 carbon–carbon triple bonds) (“C2–20 alkenyl”). In an embodiment, alkenyl does not contain any triple bonds. In an embodiment, an alkenyl group has 2 to 10 carbon atoms (“C2–10 alkenyl”). In an embodiment, an alkenyl group has 2 to 9 carbon atoms (“C2–9 alkenyl”). In an embodiment, an alkenyl group has 2 to 8 carbon atoms (“C2–8 alkenyl”). In an embodiment, an alkenyl group has 2 to 7 carbon atoms (“C2–7 alkenyl”). In an embodiment, an alkenyl group has 2 to 6 carbon atoms (“C2–6 alkenyl”). In an embodiment, an alkenyl group has 2 to 5 carbon atoms (“C2–5 alkenyl”). In an embodiment, an alkenyl group has 2 to 4 carbon atoms (“C2–4 alkenyl”). In an embodiment, an alkenyl group has 2 to 3 carbon atoms (“C2–3 alkenyl”). In an embodiment, an alkenyl group has 2 carbon atoms (“C2 alkenyl”). The one or more carbon–carbon double bonds can be internal (such as in 2– butenyl) or terminal (such as in 1–butenyl). Examples of C2–4 alkenyl groups include ethenyl (C2), 1–propenyl (C3), 2–propenyl (C3), 1–butenyl (C4), 2–butenyl (C4), butadienyl (C4), and the like. Examples of C2–6 alkenyl groups include the aforementioned C2–4 alkenyl groups as well as pentenyl (C5), pentadienyl (C5), hexenyl (C6), and the like. Additional examples of alkenyl include heptenyl (C7), octenyl (C8), octatrienyl (C8), and the like. Unless otherwise specified, each instance of an alkenyl group is independently optionally substituted, i.e., unsubstituted (an “unsubstituted alkenyl”) or substituted (a “substituted alkenyl”) with one or more substituents e.g., for instance from 1 to 5 substituents, 1 to 3 substituents, or 1 substituent. In an embodiment, the alkenyl group is unsubstituted C2–10 alkenyl. In an embodiment, the alkenyl group is substituted C2–10 alkenyl. [0068] “Alkynyl” refers to a radical of a straight–chain or branched hydrocarbon group having from 2 to 20 carbon atoms, one or more carbon–carbon triple bonds (e.g., 1, 2, 3, or 4 carbon–carbon triple bonds), and optionally one or more carbon–carbon double bonds (e.g., 1, 2, 3, or 4 carbon–carbon double bonds) (“C2–20 alkynyl”). In an embodiment, alkynyl does not contain any double bonds. In an embodiment, an alkynyl group has 2 to 10 carbon atoms (“C2–10 alkynyl”). In an embodiment, an alkynyl group has 2 to 9 carbon atoms (“C2–9 alkynyl”). In an embodiment, an alkynyl group has 2 to 8 carbon atoms (“C2–8 alkynyl”). In an embodiment, an alkynyl group has 2 to 7 carbon atoms (“C2–7 alkynyl”). In an embodiment, an alkynyl group has 2 to 6 carbon atoms (“C2–6 alkynyl”). In an embodiment, an alkynyl group has 2 to 5 carbon atoms (“C2–5 alkynyl”). In an embodiment, an alkynyl group has 2 to 4 carbon atoms (“C2–4 alkynyl”). In an embodiment, an alkynyl group has 2 to 3 carbon atoms (“C2–3 alkynyl”). In an embodiment, an alkynyl group has 2 carbon atoms (“C2 alkynyl”). The one or more carbon–carbon triple bonds can be internal (such as in 2– butynyl) or terminal (such as in 1–butynyl). Examples of C2–4 alkynyl groups include, without limitation, ethynyl (C2), 1–propynyl (C3), 2–propynyl (C3), 1–butynyl (C4), 2– butynyl (C4), and the like. Examples of C2–6 alkenyl groups include the aforementioned C2–4 alkynyl groups as well as pentynyl (C5), hexynyl (C6), and the like. Additional examples of alkynyl include heptynyl (C7), octynyl (C8), and the like. Unless otherwise specified, each instance of an alkynyl group is independently optionally substituted, i.e., unsubstituted (an “unsubstituted alkynyl”) or substituted (a “substituted alkynyl”) with one or more substituents; e.g., for instance from 1 to 5 substituents, 1 to 3 substituents, or 1 substituent. In an embodiment, the alkynyl group is unsubstituted C2–10 alkynyl. In an embodiment, the alkynyl group is substituted C2–10 alkynyl. [0069] The term “heteroalkyl,” as used herein, refers to an alkyl group, as defined herein, which further comprises 1 or more (e.g., 1, 2, 3, or 4) heteroatoms (e.g., oxygen, sulfur, nitrogen, boron, silicon, phosphorus and oxidized forms thereof, e.g., SO2) within the parent chain, wherein the one or more heteroatoms is inserted between adjacent carbon atoms within the parent carbon chain and/or one or more heteroatoms is inserted between a carbon atom and the parent molecule, i.e., between the point of attachment. In an embodiment, the heteroalkyl contains 1-2 heteroatoms independently selected from N, O and S and oxidized forms thereof (e.g., SO2). In an embodiment, the heteroalkyl contains 1 heteroatom independently selected from N, O and S and oxidized forms thereof (e.g., SO2). In an embodiment, the heteroalkyl contains 2 heteroatoms independently selected from N, O and S and oxidized forms thereof (e.g., SO2). In an embodiment, a heteroalkyl group refers to a saturated group having from 1 to 10 carbon atoms and 1, 2, 3, or 4 heteroatoms (“heteroC1–10 alkyl”). In an embodiment, a heteroalkyl group is a saturated group having 1 to 9 carbon atoms and 1, 2, 3, or 4 heteroatoms (“heteroC1–9 alkyl”). In an embodiment, a heteroalkyl group is a saturated group having 1 to 8 carbon atoms and 1, 2, 3, or 4 heteroatoms (“heteroC1–8 alkyl”). In an embodiment, a heteroalkyl group is a saturated group having 1 to 7 carbon atoms and 1, 2, 3, or 4 heteroatoms (“heteroC1–7 alkyl”). In an embodiment, a heteroalkyl group is a group having 1 to 6 carbon atoms and 1, 2, or 3 heteroatoms (“heteroC1–6 alkyl”). In an embodiment, a heteroalkyl group is a saturated group having 1 to 5 carbon atoms and 1 or 2 heteroatoms (“heteroC1–5 alkyl”). In an embodiment, a heteroalkyl group is a saturated group having 1 to 4 carbon atoms and 1 or 2 heteroatoms (“heteroC1–4 alkyl”). In an embodiment, a heteroalkyl group is a saturated group having 1 to 3 carbon atoms and 1 heteroatom (“heteroC1–3 alkyl”). In an embodiment, a heteroalkyl group is a saturated group having 1 to 2 carbon atoms and 1 heteroatom (“heteroC1–2 alkyl”). In an embodiment, a heteroalkyl group is a saturated group having 1 carbon atom and 1 heteroatom (“heteroC1 alkyl”). In an embodiment, a heteroalkyl group is a saturated group having 2 to 6 carbon atoms and 1 or 2 heteroatoms (“heteroC2–6 alkyl”). Unless otherwise specified, each instance of a heteroalkyl group is independently unsubstituted (an “unsubstituted heteroalkyl”) or substituted (a “substituted heteroalkyl”) with one or more substituents. In an embodiment, the heteroalkyl group is an unsubstituted heteroC1–10 alkyl. In an embodiment, the heteroalkyl group is a substituted heteroC1–10 alkyl. Exemplary heteroalkyl groups include: –CH2OH, –CH2OCH3, –CH2NH2, –CH2NH(CH3), –CH2N(CH3)2, –CH2CH2OH, – CH2CH2OCH3, –CH2CH2NH2, –CH2CH2NH(CH3), –CH2CH2N(CH3)2, –CH2CH2S(O)2CH3. [0070] “Aryl” refers to a radical of a monocyclic or polycyclic (e.g., bicyclic or tricyclic) 4n+2 aromatic ring system (e.g., having 6, 10, or 14 π electrons shared in a cyclic array) having 6–14 ring carbon atoms and zero heteroatoms provided in the aromatic ring system (“C6–14 aryl”). In an embodiment, an aryl group has six ring carbon atoms (“C6 aryl”; e.g., phenyl). In an embodiment, an aryl group has ten ring carbon atoms (“C10 aryl”; e.g., naphthyl such as 1–naphthyl and 2–naphthyl). In an embodiment, an aryl group has fourteen ring carbon atoms (“C14 aryl”; e.g., anthracyl). “Aryl” also includes ring systems wherein the aryl ring, as defined above, is fused with one or more carbocyclyl or heterocyclyl groups wherein the radical or point of attachment is on the aryl ring, and in such instances, the number of carbon atoms continue to designate the number of carbon atoms in the aryl ring system. Particularly aryl groups include phenyl, naphthyl, indenyl, and tetrahydronaphthyl. Unless otherwise specified, each instance of an aryl group is independently optionally substituted, i.e., unsubstituted (an “unsubstituted aryl”) or substituted (a “substituted aryl”) with one or more substituents. In an embodiment, the aryl group is unsubstituted C6–14 aryl. In an embodiment, the aryl group is substituted C6–14 aryl. [0071] In an embodiment, an aryl group is substituted with one or more of groups selected from halo, C1–C8 alkyl, C1–C8 haloalkyl, cyano, hydroxy, C1–C8 alkoxy, and amino. [0072] Examples of representative substituted aryls include the following:
andX3 wherein X3 is CH or N and wherein the left-side attachment point connects to CRcRc’ and the right attachment point connects to Ring B; L is selected from the group consisting of –O– and –S–; Ring B is a 5-10 membered heteroaryl or a 3-10 membered heterocyclyl; each RA is independently selected from the group consisting of –D, oxo, halo, – CN, –C1–C6 alkyl, –C1–C6 haloalkyl, –C1–C6 hydroxyalkyl, –C3–C10 cycloalkyl, –ORA1 and – N(RA1)2; each RA1 is independently selected from the group consisting of H, –C1–C6 alkyl, –C1–C6 haloalkyl and C3–C9 cycloalkyl wherein each hydrogen atom of each alkyl and cycloalkyl can be replaced by deuterium; or two instances of RA1 on a nitrogen atom can be taken together with the nitrogen to which they are attached to form a 4-6 member monocyclic heterocycle containing one nitrogen atom and 0-1 additional heteroatoms selected from N and O; each RB is independently selected from the group consisting of halo, –CN, –C1–C6 alkyl, –C1–C6 alkenyl, –heteroC1-C4 alkyl, –C1–C6 haloalkyl, –C1–C6 hydroxyalkyl, –C3–C10 cycloalkyl, 3-10 membered heterocyclyl, C3-C9 cycloalkylalkyl, –ORB1, –N(RB1)2, – C(=O)RB1, –C(=O)ORB1, –NRB1C(=O)RB1, –NRB1C(=O)ORB1, –C(=O)N(RB1)2, – OC(=O)N(RB1)2, –S(=O)RB1, –S(=O)2RB1, –SRB1, –S(=O)(=NRB1)RB1, –NRB1S(=O)2RB1 and –S(=O)2N(RB1)2, wherein each alkyl, alkenyl, cycloalkyl, heterocyclyl and cycloalkylalkyl of RB can be partially or fully deuterated and can be substituted at any available position with 0, 1 or 2 substituents selected from –F, –Cl, –OH– and –CH3; each RB1 is independently selected from the group consisting of H, –C1–C6 alkyl, –heteroC1-C4 alkyl, –C1–C6 haloalkyl, –C3–C9 cycloalkyl and 3-7 membered heterocyclyl wherein each hydrogen atom of each alkyl, cycloalkyl and heterocyclyl can be replaced by deuterium; or two instances of RB1 on a nitrogen atom can be taken together with the nitrogen to which they are attached to form a 4-6 member monocyclic heterocycle containing one nitrogen atom and 0-1 additional heteroatoms selected from N and O; each RC is independently selected from the group consisting of H, –D, oxo, halo, – CN, –ORC1, –SRC1 –NRC12, –C1–C6 alkyl, –C1–C6 hydroxyalkyl, –C1–C6 haloalkyl, –C3–C9 cycloalkyl, 3-10 member heterocyclyl and –heteroC1-C4 alkyl wherein each alkyl, cycloalkyl and heterocyclyl of RC can partially or fully deuterated and can be substituted at any available position with 0, 1 or 2 substituents selected from –F, –Cl, –OH– and –CH3; each RC1 is independently selected from the group consisting of H and –C1–C6 alkyl, wherein each hydrogen atom of the alkyl can be independently replaced by deuterium; or two instances of RC1 on a nitrogen atom can be taken together with the nitrogen to which they are attached to form a 4-6 member monocyclic heterocycle containing one nitrogen atom and 0-1 additional heteroatoms selected from N and O; each RD is independently selected from the group consisting of –D, halo, –CN, – C1–C6 alkyl, –heteroC1-C4 alkyl, –C1–C6 haloalkyl, –C1–C6 hydroxyalkyl, –C3–C10 cycloalkyl, 3-10 membered heterocyclyl, –C6-C10 aryl, –ORD1, –N(RD1)2, –C(=O)RD1, – C(=O)ORD1, –NRD1C(=O)RD1, –NRD1C(=O)ORD1, –C(=O)N(RD1)2, –OC(=O)N(RD1)2, – S(=O)RD1, –S(=O)2RD1, –SRD1, –S(=O)(=NRD1)RD1, –NRD1S(=O)2RD1 and –S(=O)2N(RD1)2 , wherein each alkyl, cycloalkyl, heterocyclyl and aryl of RD can be substituted at any available position with 0, 1 or 2 substituents selected from –F, –Cl, –OH– and –CH3; each RD1 is independently selected from the group consisting of H, –C1–C6 alkyl, –heteroC1-C4 alkyl, –C1–C6 haloalkyl, –C3–C9 cycloalkyl and 3-7 membered heterocyclyl, wherein each hydrogen atom of each alkyl, cycloalkyl and heterocyclyl can be replaced by deuterium; or two instances of RD1 on a nitrogen atom can be taken together with the nitrogen to which they are attached to form a 4-6 member monocyclic heterocycle containing one nitrogen atom and 0-1 additional heteroatoms selected from N and O; RE is selected from H, –D, halo, –CN, –C1–C6 alkyl, –C1-C6 alkynyl, –heteroC1– C6 alkyl, –C1–C6 haloalkyl, –C1–C6 hydroxyalkyl, –C3–C10 cycloalkyl, 3-10 membered heterocyclyl, –C6-C10 aryl, 5-10 member heteroaryl, heterocyclylalkyl, heteroarylalkyl, arylalkyl, cycloalkylalkyl, –ORE1, –SRE1, –N(RE1)2, –C1–C6 alkylene–NRE1C(=O)ORE1, – C(=O)RE1, –C(=O)ORE1, –NRE1C(=O)RE1, –NRE1C(=O)ORE1, –C(=O)N(RE1)2, and – OC(=O)N(RE1)2, wherein each alkyl, alkynyl, heteroalkyl, haloalkyl, cycloalkyl, heterocyclyl, aryl, heteroaryl, cycloalkylalkyl, heterocyclylalkyl, arylalkyl and heteroarylalkyl is optionally substituted at any available position; each RE1 is independently selected from H, –C1–C6 alkyl (wherein each hydrogen can be independently replaced by deuterium), –heteroC1–C6 alkyl, –C1–C6 haloalkyl, –C3–C9 cycloalkyl, 3-7 membered heterocyclyl, cycloalkylalkyl, heterocyclylalkyl, aryl, 5-6 membered heteroaryl, arylalkyl and heteroarylalkyl wherein each alkyl, haloalkyl, cycloalkyl, heterocyclyl, aryl, heteroaryl, cycloalkylalkyl, heterocyclylalkyl, arylalkyl and heteroarylalkyl is optionally substituted at any available position; each Rc and Rc’ is independently selected from the group consisting of H, –D, – C1–C6 alkyl, –heteroC1-C4 alkyl and –C1–C6 haloalkyl or Rc and Rc’ can be taken together with the atom to which they are attached to form a –C3–C9 cycloalkyl; n is 0, 1, 2,3 or 4; m is 0, 1, 2 or 3; and p is 0, 1, 2 or 3. [0009] In an embodiment, provided are compounds selected from the compounds of Table 1, or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof. [0010] In an embodiment, provided is a pharmaceutical composition comprising a compound as described herein or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof and a pharmaceutically acceptable carrier. [0011] In an embodiment, the pharmaceutical composition comprises a second therapeutic agent. [0012] In one aspect provided is a method for treating or preventing a disease or disorder sensitive to inhibition of USP1 comprising administering to a patient in need thereof a therapeutically effective amount of a compound of Formula (I) or Formula (II) or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof. [0013] In one aspect provided is a method of treating a disease or disorder sensitive to the inhibition of USP1 comprising administering to a patient in need thereof a therapeutically effective amount of a compound of Formula (I) or Formula (II) or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof. [0014] In one aspect provided is a method for inhibiting USP1 comprising administering to a patient in need thereof an effective amount of a compound of Formula (I) or Formula (II) or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof. [0015] In one aspect provided is a method for treating or preventing cancer in a patient in need thereof comprising administering to the patient in need thereof a therapeutically effective amount of a compound of Formula (I) or Formula (II) or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof. [0016] In one aspect provided is a method for treating cancer in a patient in need thereof comprising administering to the patient in need thereof a therapeutically effective amount of a compound of Formula (I) or Formula (II) or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof. [0017] In one aspect provided is a method for treating or preventing a disease or disorder associated with DNA damage comprising administering to a patient in need of a treatment for diseases or disorders associated with DNA damage an effective amount of a compound of Formula (I) or Formula (II) or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof. [0018] In one aspect provided is a method for treating a disease or disorder associated with DNA damage comprising administering to a patient in need of a treatment for diseases or disorders associated with DNA damage an effective amount of a compound of Formula (I) or Formula (II) or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof. [0019] In one aspect provided is a method of inhibiting, modulating or reducing DNA repair activity exercised by USP1 comprising administering to a patient in need thereof an effective amount of a compound of Formula (I) or Formula (II) or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof. [0020] In one aspect provided is a compound of Formula (I) or Formula (II) or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof for use in a method for treating or preventing a disease or disorder sensitive to the inhibition of USP1 comprising administering to a patient in need thereof a therapeutically effective amount of the compound or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof. [0021] In one aspect provided is a compound of Formula (I) or Formula (II) or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof for use in a method of treating a disease or disorder sensitive to the inhibition of USP1 comprising administering to a patient in need thereof a therapeutically effective amount of the compound or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof. [0022] In one aspect provided is a compound of Formula (I) or Formula (II) or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof for use in a method for inhibiting USP1 comprising administering to a patient in need thereof an effective amount of the compound or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof. [0023] In one aspect provided is a compound of Formula (I) or Formula (II) or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof for use in a method for treating or preventing cancer in a patient in need thereof comprising administering to the patient in need thereof a therapeutically effective amount of the compound or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof. [0024] In one aspect provided is a compound of Formula (I) or Formula (II) or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof for use in a method for treating cancer in a patient in need thereof comprising administering to the patient in need thereof a therapeutically effective amount of the compound or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof. [0025] In one aspect provided is a compound of Formula (I) or Formula (II) or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof for use in a method for treating or preventing a disease or disorder associated with DNA damage. [0026] In one aspect provided is a compound of Formula (I) or Formula (II) or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof for use in a method for treating a disease or disorder associated with DNA damage. [0027] In one aspect provided is a compound of Formula (I) or Formula (II) or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof for use in a method of inhibiting, modulating or reducing DNA repair activity exercised by USP1 comprising administering to a patient in need thereof an effective amount of the compound or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof. [0028] In one aspect, provided is a use of an effective amount of a compound of Formula (I) or Formula (II) or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof in the manufacturing of a medicament for treating or preventing a disease or disorder sensitive to the inhibition of USP1. [0029] In one aspect, provided is a use of an effective amount of a compound of Formula (I) or Formula (II) or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof in the manufacturing of a medicament for treating a disease or disorder sensitive to the inhibition of USP1. [0030] In one aspect, provided is a use of an effective amount of a compound of Formula (I) or Formula (II) or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof in the manufacturing of a medicament for inhibiting USP1. [0031] In one aspect, provided is a use of an effective amount of a compound of Formula (I) or Formula (II) or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof in the manufacturing of a medicament for treating or preventing cancer in a patient in need thereof. [0032] In one aspect, provided is a use of an effective amount of a compound of Formula (I) or Formula (II) or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof in the manufacturing of a medicament for treating cancer in a patient in need thereof. [0033] In one aspect, provided is a use of an effective amount of a compound of Formula (I) or Formula (II) or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof in the manufacturing of a medicament for treating or preventing a disease or disorder associated with DNA damage. [0034] In one aspect, provided is a use of an effective amount of a compound of Formula (I) or Formula (II) or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof in the manufacturing of a medicament for treating a disease or disorder associated with DNA damage. [0035] In one aspect, provided is a use of an effective amount of a compound of Formula (I) or Formula (II) or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof for inhibiting, modulating or reducing DNA repair activity exercised by USP1. [0036] In one aspect, provided is a use of an effective amount of a compound of Formula (I) or Formula (II) or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof for treating or preventing a disease or disorder sensitive to the inhibition of USP1. [0037] In one aspect, provided is a use of an effective amount of a compound of Formula (I) or Formula (II) or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof for treating a disease or disorder sensitive to the inhibition of USP1. [0038] In one aspect, provided is a use of an effective amount of a compound of Formula (I) or Formula (II) or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof for inhibiting USP1. [0039] In one aspect, provided is a use of an effective amount of a compound of Formula (I) or Formula (II) or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof for treating or preventing cancer in a patient in need thereof. [0040] In one aspect, provided is a use of an effective amount of a compound of Formula (I) or Formula (II) or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof for treating cancer in a patient in need thereof. [0041] In one aspect, provided is a use of an effective amount of a compound of Formula (I) or Formula (II) or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof for treating or preventing a disease or disorder associated with DNA damage. [0042] In one aspect, provided is a use of an effective amount of a compound of Formula (I) or Formula (II) or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof for treating a disease or disorder associated with DNA damage. [0043] In one aspect, provided is a use of an effective amount of a compound of Formula (I) or Formula (II) or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof for inhibiting, modulating or reducing DNA repair activity exercised by USP1. Detailed Description [0044] The disclosure herein sets forth exemplary methods, parameters and the like. It should be recognized, however, that such description is not intended as a limitation on the scope of the present disclosure but is instead provided as a description of exemplary embodiments. Definitions [0045] As used in the present disclosure, the following words and phrases are generally intended to have the meanings as set forth below unless expressly indicated otherwise or the context in which they are used indicates otherwise. Chemical Definitions [0046] Definitions of specific functional groups and chemical terms are described in more detail below. The chemical elements are identified in accordance with the Periodic Table of the Elements, CAS version, Handbook of Chemistry and Physics, 75th Ed., inside cover, and specific functional groups are generally defined as described therein. Additionally, general principles of organic chemistry, as well as specific functional moieties and reactivity, are described in Thomas Sorrell, Organic Chemistry, University Science Books, Sausalito, 1999; Smith and March, March’s Advanced Organic Chemistry, 5th Edition, John Wiley & Sons, Inc., New York, 2001; Larock, Comprehensive Organic Transformations, VCH Publishers, Inc., New York, 1989; and Carruthers, Some Modern Methods of Organic Synthesis, 3rd Edition, Cambridge University Press, Cambridge, 1987. [0047] Compounds described herein can comprise one or more asymmetric centers, and thus can exist in various isomeric forms, e.g., enantiomers and/or diastereomers. For example, the compounds described herein can be in the form of an individual enantiomer, diastereomer or geometric isomer, or can be in the form of a mixture of stereoisomers, including racemic mixtures and mixtures enriched in one or more stereoisomer. Unless stereochemistry is explicitly indicated in a structure, the structure is intended to embrace all possible stereoisomers of the compound depicted. If stereochemistry is explicitly indicated for one portion or portions of a molecule, but not for another portion or portions of a molecule, the structure is intended to embrace all possible stereoisomers for the portion or portions where stereochemistry is not explicitly indicated. Isomers can be isolated from mixtures by methods known to those skilled in the art, including chiral high-pressure liquid chromatography (HPLC) and the formation and crystallization of chiral salts; or preferred isomers can be prepared by asymmetric syntheses. See, for example, Jacques et al., Enantiomers, Racemates and Resolutions (Wiley Interscience, New York, 1981); Wilen et al., Tetrahedron 33:2725 (1977); Eliel, Stereochemistry of Carbon Compounds (McGraw–Hill, NY, 1962); and Wilen, Tables of Resolving Agents and Optical Resolutions p.268 (E.L. Eliel, Ed., Univ. of Notre Dame Press, Notre Dame, IN 1972). The invention additionally encompasses compounds described herein as individual isomers substantially free of other isomers, and alternatively, as mixtures of various isomers. [0048] The “enantiomeric excess” (“e.e.”) or “% enantiomeric excess” (“%e.e.”) of a composition as used herein refers to an excess of one enantiomer relative to the other enantiomer present in the composition. For example, a composition can contain 90% of one enantiomer, e.g., the S enantiomer, and 10% of the other enantiomer, i.e., the R enantiomer. e.e. = (90-10)/100 = 80%. [0049] Thus, a composition containing 90% of one enantiomer and 10% of the other enantiomer is said to have an enantiomeric excess of 80%. [0050] The “diastereomeric excess” (“d.e.”) or “% diastereomeric excess” (“%d.e.”) of a composition as used herein refers to an excess of one diastereomer relative to one or more different diastereomers present in the composition. For example, a composition can contain 90% of one diastereomer, and 10% of one or more different diastereomers. d.e. = (90-10)/100 = 80%. [0051] Thus, a composition containing 90% of one diastereomers and 10% of one or more different diastereomers is said to have a diastereomeric excess of 80%. [0052] In an alternative embodiment, compounds described herein may also comprise one or more isotopic substitutions. For example, hydrogen may be2H (D or deuterium) or3H (T or tritium); carbon may be, for example,13C or14C; oxygen may be, for example,18O; nitrogen may be, for example,15N, and the like. In other embodiments, a particular isotope (e.g.,3H,13C,14C,18O, or15N) can represent at least 1%, at least 5%, at least 10%, at least 15%, at least 20%, at least 25%, at least 30%, at least 35%, at least 40%, at least 45%, at least 50%, at least 60%, at least 65%, at least 70%, at least 75%, at least 80%, at least 85%, at least 90%, at least 95%, at least 99%, or at least 99.9% of the total isotopic abundance of an element that occupies a specific site of the compound. [0053] In a formula, is a single bond where the stereochemistry of the moieties immediately attached thereto is not specified. [0054] When a range of values is listed, it is intended to encompass each value and sub– range within the range. For example, “C1–6 alkyl” is intended to encompass, C1, C2, C3, C4, C5, C6, C1–6, C1–5, C1–4, C1–3, C1–2, C2–6, C2–5, C2–4, C2–3, C3–6, C3–5, C3–4, C4–6, C4–5, and C5–6 alkyl. [0055] The following terms are intended to have the meanings presented therewith below and are useful in understanding the description and intended scope of the present invention. When describing the invention, which may include compounds, pharmaceutical compositions containing such compounds and methods of using such compounds and compositions, the following terms, if present, have the following meanings unless otherwise indicated. It should also be understood that when described herein any of the moieties defined forth below may be substituted with a variety of substituents, and that the respective definitions are intended to include such substituted moieties within their scope as set out below. Unless otherwise stated, the term “substituted” is to be defined as set out below. It should be further understood that the terms “groups” and “radicals” can be considered interchangeable when used herein. The articles “a” and “an” may be used herein to refer to one or to more than one (i.e. at least one) of the grammatical objects of the article. By way of example “an analogue” means one analogue or more than one analogue. [0056] The term “unsaturated bond” refers to a double or triple bond. [0057] The term “unsaturated” or “partially unsaturated” refers to a moiety that includes at least one double or triple bond. [0058] The term “saturated” refers to a moiety that does not contain a double or triple bond, i.e., the moiety only contains single bonds. [0059] Affixing the suffix “-ene” to a group indicates the group is a divalent moiety, e.g., alkylene is the divalent moiety of alkyl, alkenylene is the divalent moiety of alkenyl, alkynylene is the divalent moiety of alkynyl, heteroalkylene is the divalent moiety of heteroalkyl, heteroalkenylene is the divalent moiety of heteroalkenyl, heteroalkynylene is the divalent moiety of heteroalkynyl, carbocyclylene is the divalent moiety of carbocyclyl, heterocyclylene is the divalent moiety of heterocyclyl, arylene is the divalent moiety of aryl, and heteroarylene is the divalent moiety of heteroaryl. [0060] The term “azido” refers to the radical –N3. [0061] “Aliphatic” refers to an alkyl, alkenyl, alkynyl, or carbocyclyl group, as defined herein. [0062] “Cycloalkylalkyl” refers to an alkyl radical in which the alkyl group is substituted with a cycloalkyl group. Typical cycloalkylalkyl groups include, but are not limited to, cyclopropylmethyl, cyclobutylmethyl, cyclopentylmethyl, cyclohexylmethyl, cycloheptylmethyl, cyclooctylmethyl, cyclopropylethyl, cyclobutylethyl, cyclopentylethyl, cyclohexylethyl, cycloheptylethyl, and cyclooctylethyl, and the like. [0063] “Heterocyclylalkyl” refers to an alkyl radical in which the alkyl group is substituted with a heterocyclyl group. Typical heterocyclylalkyl groups include, but are not limited to, pyrrolidinylmethyl, piperidinylmethyl, piperazinylmethyl, morpholinylmethyl, pyrrolidinylethyl, piperidinylethyl, piperazinylethyl, morpholinylethyl, and the like. [0064] “Aralkyl” or “arylalkyl” is a subset of alkyl and aryl, as defined herein, and refers to an optionally substituted alkyl group substituted by an optionally substituted aryl group. [0065] “Alkyl” refers to a radical of a straight–chain or branched saturated hydrocarbon group having from 1 to 20 carbon atoms (“C1–20 alkyl”). In an embodiment, an alkyl group has 1 to 12 carbon atoms (“C1–12 alkyl”). In an embodiment, an alkyl group has 1 to 10 carbon atoms (“C1–10 alkyl”). In an embodiment, an alkyl group has 1 to 9 carbon atoms (“C1–9 alkyl”). In an embodiment, an alkyl group has 1 to 8 carbon atoms (“C1–8 alkyl”). In an embodiment, an alkyl group has 1 to 7 carbon atoms (“C1–7 alkyl”). In an embodiment, an alkyl group has 1 to 6 carbon atoms (“C1–6 alkyl”, also referred to herein as “lower alkyl”). In an embodiment, an alkyl group has 1 to 5 carbon atoms (“C1–5 alkyl”). In an embodiment, an alkyl group has 1 to 4 carbon atoms (“C1–4 alkyl”). In an embodiment, an alkyl group has 1 to 3 carbon atoms (“C1–3 alkyl”). In an embodiment, an alkyl group has 1 to 2 carbon atoms (“C1–2 alkyl”). In an embodiment, an alkyl group has 1 carbon atom (“C1 alkyl”). In an embodiment, an alkyl group has 2 to 6 carbon atoms (“C2–6 alkyl”). Examples of C1–6 alkyl groups include methyl (C1), ethyl (C2), n–propyl (C3), isopropyl (C3), n–butyl (C4), tert–butyl (C4), sec–butyl (C4), iso–butyl (C4), n–pentyl (C5), 3–pentanyl (C5), amyl (C5), neopentyl (C5), 3–methyl–2–butanyl (C5), tertiary amyl (C5), and n–hexyl (C6). Additional examples of alkyl groups include n–heptyl (C7), n–octyl (C8) and the like. Unless otherwise specified, each instance of an alkyl group is independently optionally substituted, i.e., unsubstituted (an “unsubstituted alkyl”) or substituted (a “substituted alkyl”) with one or more substituents; e.g., for instance from 1 to 5 substituents, 1 to 3 substituents, or 1 substituent. In an embodiment, the alkyl group is unsubstituted C1–10 alkyl (e.g., –CH3). In an embodiment, the alkyl group is substituted C1–10 alkyl. Common alkyl abbreviations include Me (–CH3), Et (– CH2CH3),iPr (–CH(CH3)2),nPr (–CH2CH2CH3),nBu (–CH2CH2CH2CH3), oriBu (– CH2CH(CH3)2). [0066] “Alkylene” refers to an alkyl group wherein two hydrogens are removed to provide a divalent radical, and which may be substituted or unsubstituted. Unsubstituted alkylene groups include, but are not limited to, methylene (–CH2-), ethylene (–CH2CH2-), propylene (– CH2CH2CH2-), butylene (–CH2CH2CH2CH2-), pentylene (–CH2CH2CH2CH2CH2-), hexylene (–CH2CH2CH2CH2CH2CH2-), and the like. Exemplary substituted alkylene groups, e.g., substituted with one or more alkyl (methyl) groups, include but are not limited to, substituted methylene (–CH(CH3)-, (–C(CH3)2-), substituted ethylene (–CH(CH3)CH2-,–CH2CH(CH3)-, –C(CH3)2CH2-,–CH2C(CH3)2-), substituted propylene (–CH(CH3)CH2CH2-, – CH2CH(CH3)CH2-, –CH2CH2CH(CH3)-, –C(CH3)2CH2CH2-, –CH2C(CH3)2CH2-, – CH2CH2C(CH3)2-), and the like. When a range or number of carbons is provided for a particular alkylene group, it is understood that the range or number refers to the range or number of carbons in the linear carbon divalent chain. Alkylene groups may be substituted or unsubstituted with one or more substituents as described herein. [0067] “Alkenyl” refers to a radical of a straight–chain or branched hydrocarbon group having from 2 to 20 carbon atoms, one or more carbon–carbon double bonds (e.g., 1, 2, 3, or 4 carbon–carbon double bonds), and optionally one or more carbon–carbon triple bonds (e.g., 1, 2, 3, or 4 carbon–carbon triple bonds) (“C2–20 alkenyl”). In an embodiment, alkenyl does not contain any triple bonds. In an embodiment, an alkenyl group has 2 to 10 carbon atoms (“C2–10 alkenyl”). In an embodiment, an alkenyl group has 2 to 9 carbon atoms (“C2–9 alkenyl”). In an embodiment, an alkenyl group has 2 to 8 carbon atoms (“C2–8 alkenyl”). In an embodiment, an alkenyl group has 2 to 7 carbon atoms (“C2–7 alkenyl”). In an embodiment, an alkenyl group has 2 to 6 carbon atoms (“C2–6 alkenyl”). In an embodiment, an alkenyl group has 2 to 5 carbon atoms (“C2–5 alkenyl”). In an embodiment, an alkenyl group has 2 to 4 carbon atoms (“C2–4 alkenyl”). In an embodiment, an alkenyl group has 2 to 3 carbon atoms (“C2–3 alkenyl”). In an embodiment, an alkenyl group has 2 carbon atoms (“C2 alkenyl”). The one or more carbon–carbon double bonds can be internal (such as in 2– butenyl) or terminal (such as in 1–butenyl). Examples of C2–4 alkenyl groups include ethenyl (C2), 1–propenyl (C3), 2–propenyl (C3), 1–butenyl (C4), 2–butenyl (C4), butadienyl (C4), and the like. Examples of C2–6 alkenyl groups include the aforementioned C2–4 alkenyl groups as well as pentenyl (C5), pentadienyl (C5), hexenyl (C6), and the like. Additional examples of alkenyl include heptenyl (C7), octenyl (C8), octatrienyl (C8), and the like. Unless otherwise specified, each instance of an alkenyl group is independently optionally substituted, i.e., unsubstituted (an “unsubstituted alkenyl”) or substituted (a “substituted alkenyl”) with one or more substituents e.g., for instance from 1 to 5 substituents, 1 to 3 substituents, or 1 substituent. In an embodiment, the alkenyl group is unsubstituted C2–10 alkenyl. In an embodiment, the alkenyl group is substituted C2–10 alkenyl. [0068] “Alkynyl” refers to a radical of a straight–chain or branched hydrocarbon group having from 2 to 20 carbon atoms, one or more carbon–carbon triple bonds (e.g., 1, 2, 3, or 4 carbon–carbon triple bonds), and optionally one or more carbon–carbon double bonds (e.g., 1, 2, 3, or 4 carbon–carbon double bonds) (“C2–20 alkynyl”). In an embodiment, alkynyl does not contain any double bonds. In an embodiment, an alkynyl group has 2 to 10 carbon atoms (“C2–10 alkynyl”). In an embodiment, an alkynyl group has 2 to 9 carbon atoms (“C2–9 alkynyl”). In an embodiment, an alkynyl group has 2 to 8 carbon atoms (“C2–8 alkynyl”). In an embodiment, an alkynyl group has 2 to 7 carbon atoms (“C2–7 alkynyl”). In an embodiment, an alkynyl group has 2 to 6 carbon atoms (“C2–6 alkynyl”). In an embodiment, an alkynyl group has 2 to 5 carbon atoms (“C2–5 alkynyl”). In an embodiment, an alkynyl group has 2 to 4 carbon atoms (“C2–4 alkynyl”). In an embodiment, an alkynyl group has 2 to 3 carbon atoms (“C2–3 alkynyl”). In an embodiment, an alkynyl group has 2 carbon atoms (“C2 alkynyl”). The one or more carbon–carbon triple bonds can be internal (such as in 2– butynyl) or terminal (such as in 1–butynyl). Examples of C2–4 alkynyl groups include, without limitation, ethynyl (C2), 1–propynyl (C3), 2–propynyl (C3), 1–butynyl (C4), 2– butynyl (C4), and the like. Examples of C2–6 alkenyl groups include the aforementioned C2–4 alkynyl groups as well as pentynyl (C5), hexynyl (C6), and the like. Additional examples of alkynyl include heptynyl (C7), octynyl (C8), and the like. Unless otherwise specified, each instance of an alkynyl group is independently optionally substituted, i.e., unsubstituted (an “unsubstituted alkynyl”) or substituted (a “substituted alkynyl”) with one or more substituents; e.g., for instance from 1 to 5 substituents, 1 to 3 substituents, or 1 substituent. In an embodiment, the alkynyl group is unsubstituted C2–10 alkynyl. In an embodiment, the alkynyl group is substituted C2–10 alkynyl. [0069] The term “heteroalkyl,” as used herein, refers to an alkyl group, as defined herein, which further comprises 1 or more (e.g., 1, 2, 3, or 4) heteroatoms (e.g., oxygen, sulfur, nitrogen, boron, silicon, phosphorus and oxidized forms thereof, e.g., SO2) within the parent chain, wherein the one or more heteroatoms is inserted between adjacent carbon atoms within the parent carbon chain and/or one or more heteroatoms is inserted between a carbon atom and the parent molecule, i.e., between the point of attachment. In an embodiment, the heteroalkyl contains 1-2 heteroatoms independently selected from N, O and S and oxidized forms thereof (e.g., SO2). In an embodiment, the heteroalkyl contains 1 heteroatom independently selected from N, O and S and oxidized forms thereof (e.g., SO2). In an embodiment, the heteroalkyl contains 2 heteroatoms independently selected from N, O and S and oxidized forms thereof (e.g., SO2). In an embodiment, a heteroalkyl group refers to a saturated group having from 1 to 10 carbon atoms and 1, 2, 3, or 4 heteroatoms (“heteroC1–10 alkyl”). In an embodiment, a heteroalkyl group is a saturated group having 1 to 9 carbon atoms and 1, 2, 3, or 4 heteroatoms (“heteroC1–9 alkyl”). In an embodiment, a heteroalkyl group is a saturated group having 1 to 8 carbon atoms and 1, 2, 3, or 4 heteroatoms (“heteroC1–8 alkyl”). In an embodiment, a heteroalkyl group is a saturated group having 1 to 7 carbon atoms and 1, 2, 3, or 4 heteroatoms (“heteroC1–7 alkyl”). In an embodiment, a heteroalkyl group is a group having 1 to 6 carbon atoms and 1, 2, or 3 heteroatoms (“heteroC1–6 alkyl”). In an embodiment, a heteroalkyl group is a saturated group having 1 to 5 carbon atoms and 1 or 2 heteroatoms (“heteroC1–5 alkyl”). In an embodiment, a heteroalkyl group is a saturated group having 1 to 4 carbon atoms and 1 or 2 heteroatoms (“heteroC1–4 alkyl”). In an embodiment, a heteroalkyl group is a saturated group having 1 to 3 carbon atoms and 1 heteroatom (“heteroC1–3 alkyl”). In an embodiment, a heteroalkyl group is a saturated group having 1 to 2 carbon atoms and 1 heteroatom (“heteroC1–2 alkyl”). In an embodiment, a heteroalkyl group is a saturated group having 1 carbon atom and 1 heteroatom (“heteroC1 alkyl”). In an embodiment, a heteroalkyl group is a saturated group having 2 to 6 carbon atoms and 1 or 2 heteroatoms (“heteroC2–6 alkyl”). Unless otherwise specified, each instance of a heteroalkyl group is independently unsubstituted (an “unsubstituted heteroalkyl”) or substituted (a “substituted heteroalkyl”) with one or more substituents. In an embodiment, the heteroalkyl group is an unsubstituted heteroC1–10 alkyl. In an embodiment, the heteroalkyl group is a substituted heteroC1–10 alkyl. Exemplary heteroalkyl groups include: –CH2OH, –CH2OCH3, –CH2NH2, –CH2NH(CH3), –CH2N(CH3)2, –CH2CH2OH, – CH2CH2OCH3, –CH2CH2NH2, –CH2CH2NH(CH3), –CH2CH2N(CH3)2, –CH2CH2S(O)2CH3. [0070] “Aryl” refers to a radical of a monocyclic or polycyclic (e.g., bicyclic or tricyclic) 4n+2 aromatic ring system (e.g., having 6, 10, or 14 π electrons shared in a cyclic array) having 6–14 ring carbon atoms and zero heteroatoms provided in the aromatic ring system (“C6–14 aryl”). In an embodiment, an aryl group has six ring carbon atoms (“C6 aryl”; e.g., phenyl). In an embodiment, an aryl group has ten ring carbon atoms (“C10 aryl”; e.g., naphthyl such as 1–naphthyl and 2–naphthyl). In an embodiment, an aryl group has fourteen ring carbon atoms (“C14 aryl”; e.g., anthracyl). “Aryl” also includes ring systems wherein the aryl ring, as defined above, is fused with one or more carbocyclyl or heterocyclyl groups wherein the radical or point of attachment is on the aryl ring, and in such instances, the number of carbon atoms continue to designate the number of carbon atoms in the aryl ring system. Particularly aryl groups include phenyl, naphthyl, indenyl, and tetrahydronaphthyl. Unless otherwise specified, each instance of an aryl group is independently optionally substituted, i.e., unsubstituted (an “unsubstituted aryl”) or substituted (a “substituted aryl”) with one or more substituents. In an embodiment, the aryl group is unsubstituted C6–14 aryl. In an embodiment, the aryl group is substituted C6–14 aryl. [0071] In an embodiment, an aryl group is substituted with one or more of groups selected from halo, C1–C8 alkyl, C1–C8 haloalkyl, cyano, hydroxy, C1–C8 alkoxy, and amino. [0072] Examples of representative substituted aryls include the following: wherein one of R56 and R57 may be hydrogen and at least one of R56 and R57 is each independently selected from C1–C8 alkyl, C1–C8 haloalkyl, 4-10 membered heterocyclyl, alkanoyl, C1–C8 alkoxy, heteroaryloxy, alkylamino, arylamino, heteroarylamino, NR58COR59, NR58SOR59 NR58SO2R59, COOalkyl, COOaryl, CONR58R59, CONR58OR59, NR58R59, SO2NR58R59, S-alkyl, SOalkyl, SO2alkyl, Saryl, SOaryl, SO2aryl; or R56 and R57 may be joined to form a cyclic ring (saturated or unsaturated) from 5 to 8 atoms, optionally containing one or more heteroatoms selected from the group N, O, or S. R60 and R61 are independently hydrogen, C1–C8 alkyl, C1–C4 haloalkyl, C3–C10 cycloalkyl, 4-10 membered heterocyclyl, C6–C10 aryl, substituted C6–C10 aryl, 5-10 membered heteroaryl, or substituted 5-10 membered heteroaryl. [0073] “Fused aryl” refers to an aryl having two of its ring carbons in common with a second aryl or heteroaryl ring or with a carbocyclyl or heterocyclyl ring. [0074] “Heteroaryl” refers to a radical of a 5–10 membered monocyclic or bicyclic 4n+2 aromatic ring system (e.g., having 6 or 10 π electrons shared in a cyclic array) having ring carbon atoms and 1–4 ring heteroatoms provided in the aromatic ring system, wherein each heteroatom is independently selected from nitrogen, oxygen and sulfur (“5–10 membered heteroaryl”). In heteroaryl groups that contain one or more nitrogen atoms, the point of attachment can be a carbon or nitrogen atom, as valency permits. Heteroaryl bicyclic ring systems can include one or more heteroatoms in one or both rings. “Heteroaryl” includes ring systems wherein the heteroaryl ring, as defined above, is fused with one or more carbocyclyl or heterocyclyl groups wherein the point of attachment is on the heteroaryl ring, and in such instances, the number of ring members continue to designate the number of ring members in the heteroaryl ring system. “Heteroaryl” also includes ring systems wherein the heteroaryl ring, as defined above, is fused with one or more aryl groups wherein the point of attachment is either on the aryl or heteroaryl ring, and in such instances, the number of ring members designates the number of ring members in the fused (aryl/heteroaryl) ring system. Bicyclic heteroaryl groups wherein one ring does not contain a heteroatom (e.g., indolyl, quinolinyl, carbazolyl, and the like) the point of attachment can be on either ring, i.e., either the ring bearing a heteroatom (e.g., 2–indolyl) or the ring that does not contain a heteroatom (e.g., 5– indolyl). [0075] In an embodiment, a heteroaryl group is a 5–10 membered aromatic ring system having ring carbon atoms and 1–4 ring heteroatoms provided in the aromatic ring system, wherein each heteroatom is independently selected from nitrogen, oxygen, and sulfur (“5–10 membered heteroaryl”). In an embodiment, a heteroaryl group is a 5–8 membered aromatic ring system having ring carbon atoms and 1–4 ring heteroatoms provided in the aromatic ring system, wherein each heteroatom is independently selected from nitrogen, oxygen, and sulfur (“5–8 membered heteroaryl”). In an embodiment, a heteroaryl group is a 5–6 membered aromatic ring system having ring carbon atoms and 1–4 ring heteroatoms provided in the aromatic ring system, wherein each heteroatom is independently selected from nitrogen, oxygen, and sulfur (“5–6 membered heteroaryl”). In an embodiment, the 5–6 membered heteroaryl has 1–3 ring heteroatoms selected from nitrogen, oxygen, and sulfur. In an embodiment, the 5–6 membered heteroaryl has 1–2 ring heteroatoms selected from nitrogen, oxygen, and sulfur. In an embodiment, the 5–6 membered heteroaryl has 1 ring heteroatom selected from nitrogen, oxygen, and sulfur. Unless otherwise specified, each instance of a heteroaryl group is independently optionally substituted, i.e., unsubstituted (an “unsubstituted heteroaryl”) or substituted (a “substituted heteroaryl”) with one or more substituents. In an embodiment, the heteroaryl group is unsubstituted 5–14 membered heteroaryl. In an embodiment, the heteroaryl group is substituted 5–14 membered heteroaryl. In an embodiment, a heteroaryl group is a bicyclic 8-12 membered aromatic ring system having ring carbon atoms and 1-6 ring heteroatoms provided in the aromatic ring system, wherein each heteroatom is independently selected from nitrogen, oxygen, and sulfur (“8-12 membered bicyclic heteroaryl”). In an embodiment, a heteroaryl group is an 8-10 membered bicyclic aromatic ring system having ring carbon atoms and 1-6 ring heteroatoms provided in the aromatic ring system, wherein each heteroatom is independently selected from nitrogen, oxygen, and sulfur (“8-10 membered bicyclic heteroaryl”). In an embodiment, a heteroaryl group is a 9-10 membered bicyclic aromatic ring system having ring carbon atoms and 1-6 ring heteroatoms provided in the aromatic ring system, wherein each heteroatom is independently selected from nitrogen, oxygen, and sulfur (“9-10 membered bicyclic heteroaryl”). Unless otherwise specified, each instance of a heteroaryl group is independently unsubstituted (an “unsubstituted heteroaryl”) or substituted (a “substituted heteroaryl”) with one or more substituents. In an embodiment, the heteroaryl group is an unsubstituted 5-14 membered heteroaryl. In an embodiment, the heteroaryl group is a substituted 5-14 membered heteroaryl. [0076] Exemplary 5–membered heteroaryl groups containing one heteroatom include, without limitation, pyrrolyl, furanyl and thiophenyl. Exemplary 5–membered heteroaryl groups containing two heteroatoms include, without limitation, imidazolyl, pyrazolyl, oxazolyl, isoxazolyl, thiazolyl, and isothiazolyl. Exemplary 5–membered heteroaryl groups containing three heteroatoms include, without limitation, triazolyl, oxadiazolyl, and thiadiazolyl. Exemplary 5–membered heteroaryl groups containing four heteroatoms include, without limitation, tetrazolyl. Exemplary 6–membered heteroaryl groups containing one heteroatom include, without limitation, pyridinyl. Exemplary 6–membered heteroaryl groups containing two heteroatoms include, without limitation, pyridazinyl, pyrimidinyl, and pyrazinyl. Exemplary 6–membered heteroaryl groups containing three or four heteroatoms include, without limitation, triazinyl and tetrazinyl, respectively. Exemplary 7–membered heteroaryl groups containing one heteroatom include, without limitation, azepinyl, oxepinyl, and thiepinyl. Exemplary 5,6–bicyclic heteroaryl groups include, without limitation, indolyl, isoindolyl, indazolyl, benzotriazolyl, benzothiophenyl, isobenzothiophenyl, benzofuranyl, benzoisofuranyl, benzimidazolyl, benzoxazolyl, benzisoxazolyl, benzoxadiazolyl, benzthiazolyl, benzisothiazolyl, benzthiadiazolyl, indolizinyl, and purinyl. Exemplary 6,6– bicyclic heteroaryl groups include, without limitation, naphthyridinyl, pteridinyl, quinolinyl, isoquinolinyl, cinnolinyl, quinoxalinyl, phthalazinyl, and quinazolinyl. [0077] Examples of representative heteroaryls include the following:
wherein one of R56 and R57 may be hydrogen and at least one of R56 and R57 is each independently selected from C1–C8 alkyl, C1–C8 haloalkyl, 4-10 membered heterocyclyl, alkanoyl, C1–C8 alkoxy, heteroaryloxy, alkylamino, arylamino, heteroarylamino, NR58COR59, NR58SOR59 NR58SO2R59, COOalkyl, COOaryl, CONR58R59, CONR58OR59, NR58R59, SO2NR58R59, S-alkyl, SOalkyl, SO2alkyl, Saryl, SOaryl, SO2aryl; or R56 and R57 may be joined to form a cyclic ring (saturated or unsaturated) from 5 to 8 atoms, optionally containing one or more heteroatoms selected from the group N, O, or S. R60 and R61 are independently hydrogen, C1–C8 alkyl, C1–C4 haloalkyl, C3–C10 cycloalkyl, 4-10 membered heterocyclyl, C6–C10 aryl, substituted C6–C10 aryl, 5-10 membered heteroaryl, or substituted 5-10 membered heteroaryl. [0073] “Fused aryl” refers to an aryl having two of its ring carbons in common with a second aryl or heteroaryl ring or with a carbocyclyl or heterocyclyl ring. [0074] “Heteroaryl” refers to a radical of a 5–10 membered monocyclic or bicyclic 4n+2 aromatic ring system (e.g., having 6 or 10 π electrons shared in a cyclic array) having ring carbon atoms and 1–4 ring heteroatoms provided in the aromatic ring system, wherein each heteroatom is independently selected from nitrogen, oxygen and sulfur (“5–10 membered heteroaryl”). In heteroaryl groups that contain one or more nitrogen atoms, the point of attachment can be a carbon or nitrogen atom, as valency permits. Heteroaryl bicyclic ring systems can include one or more heteroatoms in one or both rings. “Heteroaryl” includes ring systems wherein the heteroaryl ring, as defined above, is fused with one or more carbocyclyl or heterocyclyl groups wherein the point of attachment is on the heteroaryl ring, and in such instances, the number of ring members continue to designate the number of ring members in the heteroaryl ring system. “Heteroaryl” also includes ring systems wherein the heteroaryl ring, as defined above, is fused with one or more aryl groups wherein the point of attachment is either on the aryl or heteroaryl ring, and in such instances, the number of ring members designates the number of ring members in the fused (aryl/heteroaryl) ring system. Bicyclic heteroaryl groups wherein one ring does not contain a heteroatom (e.g., indolyl, quinolinyl, carbazolyl, and the like) the point of attachment can be on either ring, i.e., either the ring bearing a heteroatom (e.g., 2–indolyl) or the ring that does not contain a heteroatom (e.g., 5– indolyl). [0075] In an embodiment, a heteroaryl group is a 5–10 membered aromatic ring system having ring carbon atoms and 1–4 ring heteroatoms provided in the aromatic ring system, wherein each heteroatom is independently selected from nitrogen, oxygen, and sulfur (“5–10 membered heteroaryl”). In an embodiment, a heteroaryl group is a 5–8 membered aromatic ring system having ring carbon atoms and 1–4 ring heteroatoms provided in the aromatic ring system, wherein each heteroatom is independently selected from nitrogen, oxygen, and sulfur (“5–8 membered heteroaryl”). In an embodiment, a heteroaryl group is a 5–6 membered aromatic ring system having ring carbon atoms and 1–4 ring heteroatoms provided in the aromatic ring system, wherein each heteroatom is independently selected from nitrogen, oxygen, and sulfur (“5–6 membered heteroaryl”). In an embodiment, the 5–6 membered heteroaryl has 1–3 ring heteroatoms selected from nitrogen, oxygen, and sulfur. In an embodiment, the 5–6 membered heteroaryl has 1–2 ring heteroatoms selected from nitrogen, oxygen, and sulfur. In an embodiment, the 5–6 membered heteroaryl has 1 ring heteroatom selected from nitrogen, oxygen, and sulfur. Unless otherwise specified, each instance of a heteroaryl group is independently optionally substituted, i.e., unsubstituted (an “unsubstituted heteroaryl”) or substituted (a “substituted heteroaryl”) with one or more substituents. In an embodiment, the heteroaryl group is unsubstituted 5–14 membered heteroaryl. In an embodiment, the heteroaryl group is substituted 5–14 membered heteroaryl. In an embodiment, a heteroaryl group is a bicyclic 8-12 membered aromatic ring system having ring carbon atoms and 1-6 ring heteroatoms provided in the aromatic ring system, wherein each heteroatom is independently selected from nitrogen, oxygen, and sulfur (“8-12 membered bicyclic heteroaryl”). In an embodiment, a heteroaryl group is an 8-10 membered bicyclic aromatic ring system having ring carbon atoms and 1-6 ring heteroatoms provided in the aromatic ring system, wherein each heteroatom is independently selected from nitrogen, oxygen, and sulfur (“8-10 membered bicyclic heteroaryl”). In an embodiment, a heteroaryl group is a 9-10 membered bicyclic aromatic ring system having ring carbon atoms and 1-6 ring heteroatoms provided in the aromatic ring system, wherein each heteroatom is independently selected from nitrogen, oxygen, and sulfur (“9-10 membered bicyclic heteroaryl”). Unless otherwise specified, each instance of a heteroaryl group is independently unsubstituted (an “unsubstituted heteroaryl”) or substituted (a “substituted heteroaryl”) with one or more substituents. In an embodiment, the heteroaryl group is an unsubstituted 5-14 membered heteroaryl. In an embodiment, the heteroaryl group is a substituted 5-14 membered heteroaryl. [0076] Exemplary 5–membered heteroaryl groups containing one heteroatom include, without limitation, pyrrolyl, furanyl and thiophenyl. Exemplary 5–membered heteroaryl groups containing two heteroatoms include, without limitation, imidazolyl, pyrazolyl, oxazolyl, isoxazolyl, thiazolyl, and isothiazolyl. Exemplary 5–membered heteroaryl groups containing three heteroatoms include, without limitation, triazolyl, oxadiazolyl, and thiadiazolyl. Exemplary 5–membered heteroaryl groups containing four heteroatoms include, without limitation, tetrazolyl. Exemplary 6–membered heteroaryl groups containing one heteroatom include, without limitation, pyridinyl. Exemplary 6–membered heteroaryl groups containing two heteroatoms include, without limitation, pyridazinyl, pyrimidinyl, and pyrazinyl. Exemplary 6–membered heteroaryl groups containing three or four heteroatoms include, without limitation, triazinyl and tetrazinyl, respectively. Exemplary 7–membered heteroaryl groups containing one heteroatom include, without limitation, azepinyl, oxepinyl, and thiepinyl. Exemplary 5,6–bicyclic heteroaryl groups include, without limitation, indolyl, isoindolyl, indazolyl, benzotriazolyl, benzothiophenyl, isobenzothiophenyl, benzofuranyl, benzoisofuranyl, benzimidazolyl, benzoxazolyl, benzisoxazolyl, benzoxadiazolyl, benzthiazolyl, benzisothiazolyl, benzthiadiazolyl, indolizinyl, and purinyl. Exemplary 6,6– bicyclic heteroaryl groups include, without limitation, naphthyridinyl, pteridinyl, quinolinyl, isoquinolinyl, cinnolinyl, quinoxalinyl, phthalazinyl, and quinazolinyl. [0077] Examples of representative heteroaryls include the following:












































































































































































































































































































































































































































































 ; X1 and X2 are each independently CH or N; Ring A is selected from the group consisting of:
; X1 and X2 are each independently CH or N; Ring A is selected from the group consisting of: wherein X3 is CH or N and wherein the left-side attachment point connects to CRcRc’ and the right attachment point connects to Ring B; L is selected from the group consisting of –O– and –S–; Ring B is a 5-10 membered heteroaryl or a 3-10 membered heterocyclyl; each RA is independently selected from the group consisting of –D, oxo, halo, – CN, –C1–C6 alkyl, –C1–C6 haloalkyl, –C1–C6 hydroxyalkyl, –C3–C10 cycloalkyl, –ORA1 and – N(RA1)2; each RA1 is independently selected from the group consisting of H, –C1–C6 alkyl, – C1–C6 haloalkyl and C3–C9 cycloalkyl wherein each hydrogen atom of each alkyl and cycloalkyl can be replaced by deuterium; or two instances of RA1 on a nitrogen atom can be taken together with the nitrogen to which they are attached to form a 4-6 member monocyclic heterocycle containing one nitrogen atom and 0-1 additional heteroatoms selected from N and O; each RB is independently selected from the group consisting of halo, –CN, –C1–C6 alkyl, –C1–C6 alkenyl, –heteroC1-C4 alkyl, –C1–C6 haloalkyl, –C1–C6 hydroxyalkyl, –C3–C10 cycloalkyl, 3-10 membered heterocyclyl, C3-C9 cycloalkylalkyl, –ORB1, –N(RB1)2, – C(=O)RB1, –C(=O)ORB1, –NRB1C(=O)RB1, –NRB1C(=O)ORB1, –C(=O)N(RB1)2, – OC(=O)N(RB1)2, –S(=O)RB1, –S(=O)2RB1, –SRB1, –S(=O)(=NRB1)RB1, –NRB1S(=O)2RB1 and –S(=O)2N(RB1)2, wherein each alkyl, alkenyl, cycloalkyl, heterocyclyl and cycloalkylalkyl of RB can be partially or fully deuterated and can be substituted at any available position with 0, 1 or 2 substituents selected from –F, –Cl, –OH– and –CH3; each RB1 is independently selected from the group consisting of H, –C1–C6 alkyl, – heteroC1-C4 alkyl, –C1–C6 haloalkyl, –C3–C9 cycloalkyl and 3-7 membered heterocyclyl wherein each hydrogen atom of each alkyl, cycloalkyl and heterocyclyl can be replaced by deuterium; or two instances of RB1 on a nitrogen atom can be taken together with the nitrogen to which they are attached to form a 4-6 member monocyclic heterocycle containing one nitrogen atom and 0-1 additional heteroatoms selected from N and O; each RC is independently selected from the group consisting of H, –D, oxo, halo, – CN, –ORC1, –SRC1 –NRC1 2, –C1–C6 alkyl, –C1–C6 hydroxyalkyl, –C1–C6 haloalkyl, –C3–C9 cycloalkyl, 3-10 member heterocyclyl and –heteroC1-C4 alkyl wherein each alkyl, cycloalkyl and heterocyclyl of RC can partially or fully deuterated and can be substituted at any available position with 0, 1 or 2 substituents selected from –F, –Cl, –OH– and –CH3; each RC1 is independently selected from the group consisting of H and –C1–C6 alkyl, wherein each hydrogen atom of the alkyl can be independently replaced by deuterium; or two instances of RC1 on a nitrogen atom can be taken together with the nitrogen to which they are attached to form a 4-6 member monocyclic heterocycle containing one nitrogen atom and 0-1 additional heteroatoms selected from N and O; each RD is independently selected from the group consisting of –D, halo, –CN, – C1–C6 alkyl, –heteroC1-C4 alkyl, –C1–C6 haloalkyl, –C1–C6 hydroxyalkyl, –C3–C10 cycloalkyl, 3-10 membered heterocyclyl, –C6-C10 aryl, –ORD1, –N(RD1)2, –C(=O)RD1, – C(=O)ORD1, –NRD1C(=O)RD1, –NRD1C(=O)ORD1, –C(=O)N(RD1)2, –OC(=O)N(RD1)2, – S(=O)RD1, –S(=O)2RD1, –SRD1, –S(=O)(=NRD1)RD1, –NRD1S(=O)2RD1 and –S(=O)2N(RD1)2 , wherein each alkyl, cycloalkyl, heterocyclyl and aryl of RD can be substituted at any available position with 0, 1 or 2 substituents selected from –F, –Cl, –OH– and –CH3; each RD1 is independently selected from the group consisting of H, –C1–C6 alkyl, – heteroC1-C4 alkyl, –C1–C6 haloalkyl, –C3–C9 cycloalkyl and 3-7 membered heterocyclyl, wherein each hydrogen atom of each alkyl, cycloalkyl and heterocyclyl can be replaced by deuterium; or two instances of RD1 on a nitrogen atom can be taken together with the nitrogen to which they are attached to form a 4-6 member monocyclic heterocycle containing one nitrogen atom and 0-1 additional heteroatoms selected from N and O; RE is selected from H, –D, halo, –CN, –C1–C6 alkyl, –C1-C6 alkynyl, –heteroC1–C6 alkyl, –C1–C6 haloalkyl, –C1–C6 hydroxyalkyl, –C3–C10 cycloalkyl, 3-10 membered heterocyclyl, –C6-C10 aryl, 5-10 member heteroaryl, heterocyclylalkyl, heteroarylalkyl, arylalkyl, cycloalkylalkyl, –ORE1, –SRE1, –N(RE1)2, –C1–C6 alkylene–NRE1C(=O)ORE1, – C(=O)RE1, –C(=O)ORE1, –NRE1C(=O)RE1, –NRE1C(=O)ORE1, –C(=O)N(RE1)2, and – OC(=O)N(RE1)2, wherein each alkyl, alkynyl, heteroalkyl, haloalkyl, cycloalkyl, heterocyclyl, aryl, heteroaryl, cycloalkylalkyl, heterocyclylalkyl, arylalkyl and heteroarylalkyl is optionally substituted at any available position; each RE1 is independently selected from H, –C1–C6 alkyl (wherein each hydrogen can be independently replaced by deuterium), –heteroC1–C6 alkyl, –C1–C6 haloalkyl, –C3–C9 cycloalkyl, 3-7 membered heterocyclyl, cycloalkylalkyl, heterocyclylalkyl, aryl, 5-6 membered heteroaryl, arylalkyl and heteroarylalkyl wherein each alkyl, haloalkyl, cycloalkyl, heterocyclyl, aryl, heteroaryl, cycloalkylalkyl, heterocyclylalkyl, arylalkyl and heteroarylalkyl is optionally substituted at any available position; each Rc and Rc’ is independently selected from the group consisting of H, –D, – C1–C6 alkyl, –heteroC1-C4 alkyl and –C1–C6 haloalkyl or Rc and Rc’ can be taken together with the atom to which they are attached to form a –C3–C9 cycloalkyl; n is 0, 1, 2, 3 or 4; m is 0, 1, 2 or 3; and p is 0, 1, 2 or 3.
wherein X3 is CH or N and wherein the left-side attachment point connects to CRcRc’ and the right attachment point connects to Ring B; L is selected from the group consisting of –O– and –S–; Ring B is a 5-10 membered heteroaryl or a 3-10 membered heterocyclyl; each RA is independently selected from the group consisting of –D, oxo, halo, – CN, –C1–C6 alkyl, –C1–C6 haloalkyl, –C1–C6 hydroxyalkyl, –C3–C10 cycloalkyl, –ORA1 and – N(RA1)2; each RA1 is independently selected from the group consisting of H, –C1–C6 alkyl, – C1–C6 haloalkyl and C3–C9 cycloalkyl wherein each hydrogen atom of each alkyl and cycloalkyl can be replaced by deuterium; or two instances of RA1 on a nitrogen atom can be taken together with the nitrogen to which they are attached to form a 4-6 member monocyclic heterocycle containing one nitrogen atom and 0-1 additional heteroatoms selected from N and O; each RB is independently selected from the group consisting of halo, –CN, –C1–C6 alkyl, –C1–C6 alkenyl, –heteroC1-C4 alkyl, –C1–C6 haloalkyl, –C1–C6 hydroxyalkyl, –C3–C10 cycloalkyl, 3-10 membered heterocyclyl, C3-C9 cycloalkylalkyl, –ORB1, –N(RB1)2, – C(=O)RB1, –C(=O)ORB1, –NRB1C(=O)RB1, –NRB1C(=O)ORB1, –C(=O)N(RB1)2, – OC(=O)N(RB1)2, –S(=O)RB1, –S(=O)2RB1, –SRB1, –S(=O)(=NRB1)RB1, –NRB1S(=O)2RB1 and –S(=O)2N(RB1)2, wherein each alkyl, alkenyl, cycloalkyl, heterocyclyl and cycloalkylalkyl of RB can be partially or fully deuterated and can be substituted at any available position with 0, 1 or 2 substituents selected from –F, –Cl, –OH– and –CH3; each RB1 is independently selected from the group consisting of H, –C1–C6 alkyl, – heteroC1-C4 alkyl, –C1–C6 haloalkyl, –C3–C9 cycloalkyl and 3-7 membered heterocyclyl wherein each hydrogen atom of each alkyl, cycloalkyl and heterocyclyl can be replaced by deuterium; or two instances of RB1 on a nitrogen atom can be taken together with the nitrogen to which they are attached to form a 4-6 member monocyclic heterocycle containing one nitrogen atom and 0-1 additional heteroatoms selected from N and O; each RC is independently selected from the group consisting of H, –D, oxo, halo, – CN, –ORC1, –SRC1 –NRC1 2, –C1–C6 alkyl, –C1–C6 hydroxyalkyl, –C1–C6 haloalkyl, –C3–C9 cycloalkyl, 3-10 member heterocyclyl and –heteroC1-C4 alkyl wherein each alkyl, cycloalkyl and heterocyclyl of RC can partially or fully deuterated and can be substituted at any available position with 0, 1 or 2 substituents selected from –F, –Cl, –OH– and –CH3; each RC1 is independently selected from the group consisting of H and –C1–C6 alkyl, wherein each hydrogen atom of the alkyl can be independently replaced by deuterium; or two instances of RC1 on a nitrogen atom can be taken together with the nitrogen to which they are attached to form a 4-6 member monocyclic heterocycle containing one nitrogen atom and 0-1 additional heteroatoms selected from N and O; each RD is independently selected from the group consisting of –D, halo, –CN, – C1–C6 alkyl, –heteroC1-C4 alkyl, –C1–C6 haloalkyl, –C1–C6 hydroxyalkyl, –C3–C10 cycloalkyl, 3-10 membered heterocyclyl, –C6-C10 aryl, –ORD1, –N(RD1)2, –C(=O)RD1, – C(=O)ORD1, –NRD1C(=O)RD1, –NRD1C(=O)ORD1, –C(=O)N(RD1)2, –OC(=O)N(RD1)2, – S(=O)RD1, –S(=O)2RD1, –SRD1, –S(=O)(=NRD1)RD1, –NRD1S(=O)2RD1 and –S(=O)2N(RD1)2 , wherein each alkyl, cycloalkyl, heterocyclyl and aryl of RD can be substituted at any available position with 0, 1 or 2 substituents selected from –F, –Cl, –OH– and –CH3; each RD1 is independently selected from the group consisting of H, –C1–C6 alkyl, – heteroC1-C4 alkyl, –C1–C6 haloalkyl, –C3–C9 cycloalkyl and 3-7 membered heterocyclyl, wherein each hydrogen atom of each alkyl, cycloalkyl and heterocyclyl can be replaced by deuterium; or two instances of RD1 on a nitrogen atom can be taken together with the nitrogen to which they are attached to form a 4-6 member monocyclic heterocycle containing one nitrogen atom and 0-1 additional heteroatoms selected from N and O; RE is selected from H, –D, halo, –CN, –C1–C6 alkyl, –C1-C6 alkynyl, –heteroC1–C6 alkyl, –C1–C6 haloalkyl, –C1–C6 hydroxyalkyl, –C3–C10 cycloalkyl, 3-10 membered heterocyclyl, –C6-C10 aryl, 5-10 member heteroaryl, heterocyclylalkyl, heteroarylalkyl, arylalkyl, cycloalkylalkyl, –ORE1, –SRE1, –N(RE1)2, –C1–C6 alkylene–NRE1C(=O)ORE1, – C(=O)RE1, –C(=O)ORE1, –NRE1C(=O)RE1, –NRE1C(=O)ORE1, –C(=O)N(RE1)2, and – OC(=O)N(RE1)2, wherein each alkyl, alkynyl, heteroalkyl, haloalkyl, cycloalkyl, heterocyclyl, aryl, heteroaryl, cycloalkylalkyl, heterocyclylalkyl, arylalkyl and heteroarylalkyl is optionally substituted at any available position; each RE1 is independently selected from H, –C1–C6 alkyl (wherein each hydrogen can be independently replaced by deuterium), –heteroC1–C6 alkyl, –C1–C6 haloalkyl, –C3–C9 cycloalkyl, 3-7 membered heterocyclyl, cycloalkylalkyl, heterocyclylalkyl, aryl, 5-6 membered heteroaryl, arylalkyl and heteroarylalkyl wherein each alkyl, haloalkyl, cycloalkyl, heterocyclyl, aryl, heteroaryl, cycloalkylalkyl, heterocyclylalkyl, arylalkyl and heteroarylalkyl is optionally substituted at any available position; each Rc and Rc’ is independently selected from the group consisting of H, –D, – C1–C6 alkyl, –heteroC1-C4 alkyl and –C1–C6 haloalkyl or Rc and Rc’ can be taken together with the atom to which they are attached to form a –C3–C9 cycloalkyl; n is 0, 1, 2, 3 or 4; m is 0, 1, 2 or 3; and p is 0, 1, 2 or 3.

 5. The compound of claim 1 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein RE is selected from H, –D, halo, –C1–C6 alkyl (substituted with 0 or 1 instances of –CN), –C1–C6 haloalkyl (substituted with 0 or 1 instances of –NH2 or –NHCH3), heteroC1–C4alkyl, –C1–C6 hydroxyalkyl (substituted with 0 or 1 instances of –CF3), –C1–C6 alkylene–NHC(=O)O(C1-C6alkyl), –C3–C10 cycloalkyl (substituted with 0 or 1 instances of –CF3), 3-10 member heterocyclyl (substituted with 0 or 1 instances of –F),
5. The compound of claim 1 or a pharmaceutically acceptable salt, hydrate, solvate, prodrug, stereoisomer, or tautomer thereof, wherein RE is selected from H, –D, halo, –C1–C6 alkyl (substituted with 0 or 1 instances of –CN), –C1–C6 haloalkyl (substituted with 0 or 1 instances of –NH2 or –NHCH3), heteroC1–C4alkyl, –C1–C6 hydroxyalkyl (substituted with 0 or 1 instances of –CF3), –C1–C6 alkylene–NHC(=O)O(C1-C6alkyl), –C3–C10 cycloalkyl (substituted with 0 or 1 instances of –CF3), 3-10 member heterocyclyl (substituted with 0 or 1 instances of –F),
 , , , ,
, , , , Ocyclopropyl, –O-oxetan-3-yl, –O-tetrahydrofuran-3-yl, –OCH2CH(NH2)CF3, –OCF3, – OCH2CHF2, –OCH2F, –OCHF2, –OCH2CF3, –OMe, –OCD3, –OEt and –OiPr.
Ocyclopropyl, –O-oxetan-3-yl, –O-tetrahydrofuran-3-yl, –OCH2CH(NH2)CF3, –OCF3, – OCH2CHF2, –OCH2F, –OCHF2, –OCH2CF3, –OMe, –OCD3, –OEt and –OiPr.

 is selected from the group consisting
is selected from the group consisting
 wherein: each R1 is independently selected from the group consisting of –D, halo, –CN, –C1–C6 alkyl, –heteroC1-C4 alkyl, –C1–C6 haloalkyl, –C1–C6 hydroxyalkyl, –C3–C10 cycloalkyl, 3-10 membered heterocyclyl, –C6-C10 aryl, –ORa1, –N(Ra1)2, –C(=O)Ra1, –C(=O)ORa1, – NRa1C(=O)Ra1, –NRa1C(=O)ORa1, –C(=O)N(Ra1)2, –OC(=O)N(Ra1)2, –S(=O)Ra1, –S(=O)2Ra1, –SRa1, –S(=O)(=NRa1)Ra1, –NRa1S(=O)2Ra1 and –S(=O)2N(Ra1)2 , wherein each alkyl, cycloalkyl, heterocyclyl and aryl of R1 can be substituted at any available position with 0, 1 or 2 substituents selected from –F, –Cl, –OH– and –CH3; each Ra1 is independently selected from the group consisting of H, –C1–C6 alkyl, – heteroC1-C4 alkyl, –C1–C6 haloalkyl, –C3–C9 cycloalkyl and 3-7 membered heterocyclyl, wherein each hydrogen atom of each alkyl, cycloalkyl heterocyclyl can be replaced by deuterium; or two instances of Ra1 on a nitrogen atom can be taken together with the nitrogen to which they are attached to form a 4-6 member monocyclic heterocycle containing one nitrogen atom and 0-1 additional heteroatoms selected from N and O; each R2 is independently selected from the group consisting of –D, halo, –CN, – C1–C6 alkyl, –heteroC1-C4 alkyl, –C1–C6 haloalkyl, –C1–C6 hydroxyalkyl, –C3–C10 cycloalkyl, 3-10 membered heterocyclyl, –C6-C10 aryl, –ORa2, –N(Ra2)2, –C(=O)Ra2, – C(=O)ORa2, –NRa2C(=O)Ra2, –NRa2C(=O)ORa2, –C(=O)N(Ra2)2, –OC(=O)N(Ra2)2, – S(=O)Ra2, –S(=O)2Ra2, –SRa2, –S(=O)(=NRa2)Ra2, –NRa2S(=O)2Ra2 and –S(=O)2N(Ra2)2 , wherein each alkyl, cycloalkyl, heterocyclyl and aryl of R2 can be substituted at any available position with 0, 1 or 2 substituents selected from –F, –Cl, –OH– and –CH3; each Ra2 is independently selected from the group consisting of H, –C1–C6 alkyl, – heteroC1-C4 alkyl, –C1–C6 haloalkyl, –C3–C9 cycloalkyl and 3-7 membered heterocyclyl, wherein each hydrogen atom of each alkyl, cycloalkyl heterocyclyl can be replaced by deuterium; or two instances of Ra2 on a nitrogen atom can be taken together with the nitrogen to which they are attached to form a 4-6 member monocyclic heterocycle containing one nitrogen atom and 0-1 additional heteroatoms selected from N and O.
wherein: each R1 is independently selected from the group consisting of –D, halo, –CN, –C1–C6 alkyl, –heteroC1-C4 alkyl, –C1–C6 haloalkyl, –C1–C6 hydroxyalkyl, –C3–C10 cycloalkyl, 3-10 membered heterocyclyl, –C6-C10 aryl, –ORa1, –N(Ra1)2, –C(=O)Ra1, –C(=O)ORa1, – NRa1C(=O)Ra1, –NRa1C(=O)ORa1, –C(=O)N(Ra1)2, –OC(=O)N(Ra1)2, –S(=O)Ra1, –S(=O)2Ra1, –SRa1, –S(=O)(=NRa1)Ra1, –NRa1S(=O)2Ra1 and –S(=O)2N(Ra1)2 , wherein each alkyl, cycloalkyl, heterocyclyl and aryl of R1 can be substituted at any available position with 0, 1 or 2 substituents selected from –F, –Cl, –OH– and –CH3; each Ra1 is independently selected from the group consisting of H, –C1–C6 alkyl, – heteroC1-C4 alkyl, –C1–C6 haloalkyl, –C3–C9 cycloalkyl and 3-7 membered heterocyclyl, wherein each hydrogen atom of each alkyl, cycloalkyl heterocyclyl can be replaced by deuterium; or two instances of Ra1 on a nitrogen atom can be taken together with the nitrogen to which they are attached to form a 4-6 member monocyclic heterocycle containing one nitrogen atom and 0-1 additional heteroatoms selected from N and O; each R2 is independently selected from the group consisting of –D, halo, –CN, – C1–C6 alkyl, –heteroC1-C4 alkyl, –C1–C6 haloalkyl, –C1–C6 hydroxyalkyl, –C3–C10 cycloalkyl, 3-10 membered heterocyclyl, –C6-C10 aryl, –ORa2, –N(Ra2)2, –C(=O)Ra2, – C(=O)ORa2, –NRa2C(=O)Ra2, –NRa2C(=O)ORa2, –C(=O)N(Ra2)2, –OC(=O)N(Ra2)2, – S(=O)Ra2, –S(=O)2Ra2, –SRa2, –S(=O)(=NRa2)Ra2, –NRa2S(=O)2Ra2 and –S(=O)2N(Ra2)2 , wherein each alkyl, cycloalkyl, heterocyclyl and aryl of R2 can be substituted at any available position with 0, 1 or 2 substituents selected from –F, –Cl, –OH– and –CH3; each Ra2 is independently selected from the group consisting of H, –C1–C6 alkyl, – heteroC1-C4 alkyl, –C1–C6 haloalkyl, –C3–C9 cycloalkyl and 3-7 membered heterocyclyl, wherein each hydrogen atom of each alkyl, cycloalkyl heterocyclyl can be replaced by deuterium; or two instances of Ra2 on a nitrogen atom can be taken together with the nitrogen to which they are attached to form a 4-6 member monocyclic heterocycle containing one nitrogen atom and 0-1 additional heteroatoms selected from N and O.







 Formula (I-b); wherein Ra2 is selected from the group consisting of CH3, –CHF2 and –CD3.
Formula (I-b); wherein Ra2 is selected from the group consisting of CH3, –CHF2 and –CD3.
 wherein the left-side attachment point connects to CRcRc’ and the right attachment point connects to Ring B.
wherein the left-side attachment point connects to CRcRc’ and the right attachment point connects to Ring B.
 wherein the left-side attachment point connects to CRcRc’ and the right attachment point connects to Ring B.
wherein the left-side attachment point connects to CRcRc’ and the right attachment point connects to Ring B.
 selected from the group consisting of
selected from the group consisting of , ,
, ,





 is selected from the group consisting of
is selected from the group consisting of , ,
, , wherein the left-side attachment point connects to CRcRc’ and the right attachment point connects to Ring B.
wherein the left-side attachment point connects to CRcRc’ and the right attachment point connects to Ring B.


 Formula (I-c); wherein Ra2 is selected from the group consisting of CH3, –CHF2 and –CD3.
Formula (I-c); wherein Ra2 is selected from the group consisting of CH3, –CHF2 and –CD3.
 Formula (I-d); wherein Ra2 is selected from the group consisting of CH3, –CHF2 and –CD3.
Formula (I-d); wherein Ra2 is selected from the group consisting of CH3, –CHF2 and –CD3.
 Formula (I-d-1); wherein Ra2 is selected from the group consisting of CH3, –CHF2 and –CD3.
Formula (I-d-1); wherein Ra2 is selected from the group consisting of CH3, –CHF2 and –CD3.


 selected from the group consisting
selected from the group consisting ,
,



 wherein RA is F; Ra2 is selected from the group consisting of CH3,iPr, –CHF2 and –CD3; and RB is selected from the group consisting of –Me, –Et and cyclopropyl.
wherein RA is F; Ra2 is selected from the group consisting of CH3,iPr, –CHF2 and –CD3; and RB is selected from the group consisting of –Me, –Et and cyclopropyl.
 Formula (I-f); wherein Ra2 is selected from the group consisting of CH3, –CHF2 and –CD3; and RB is selected from the group consisting of –Me, –Et and cyclopropyl.
Formula (I-f); wherein Ra2 is selected from the group consisting of CH3, –CHF2 and –CD3; and RB is selected from the group consisting of –Me, –Et and cyclopropyl.
 Formula (I-f-1); wherein Ra2 is selected from the group consisting of CH3, –CHF2 and –CD3; and RB is selected from the group consisting of –Me, –Et and cyclopropyl.
Formula (I-f-1); wherein Ra2 is selected from the group consisting of CH3, –CHF2 and –CD3; and RB is selected from the group consisting of –Me, –Et and cyclopropyl.








































