Unser Zeichen/Our Ref. 11.05.2023 CU01P377W01 CureVac SE Therapeutic nucleic acid for the treatment of ophthalmic diseases Introduction The invention relates inter alia to a composition comprising therapeutic nucleic acids, preferably RNA comprising at least one coding sequence encoding at least one peptide or protein for use in treatment or prevention of an ophthalmic disease, disorder or condition, wherein said composition is administered into the anterior segment of the eye of a subject in need thereof, preferably wherein the composition is administered to the cornea, the ciliary body, or the conjunctiva of the eye of a subject. Suitably, the therapeutic nucleic acid, preferably RNA of the composition is formulated in lipid-based carriers. Further aspects inter alia relate to eye drop formulations, contact lenses, and medical devices suitable for administering a therapeutic nucleic acid/ RNA composition into the anterior segment of the eye. Ophthalmic diseases are a major health problem worldwide. An ophthalmic disease can occur in many tissues of the eye (e.g., cornea, sclera, iris and pupil, lens, ciliary muscle, ciliary body, choroid, vitreous, retina and optic nerve, macula, etc.). Ocular diseases can be caused by infections (e.g., viruses, parasites, bacteria, and fungi), cancer, genetic causes, injuries, inflammations, post-surgical complications etc. Many diseases are associated with cells and/or tissues of the anterior segment or anterior cavity of the eye. The anterior segment or anterior cavity of the eye is constantly exposed to minor trauma. It consists of the front third of the eye that includes the structures in front of the vitreous humour: Cornea, sclera and conjunctiva, iris, ciliary body, trabecular meshwork and lens. The precorneal tear film, corneal epithelium, and conjunctival epithelium provide protection against this continued mild trauma; however, significant trauma can occur which requires ophthalmic examination and treatment. Most common conditions appearing in the anterior segment include Injected Conjunctiva, Conjunctivitis, Conjunctival Lymphoma, Conjunctival Haemangioma, Conjunctivochalasis, Chemosis, Conjunctival Haemorrhage, Corneal neovascularization, Keratitis, Keratoconus, Corneal ulcer, Ciliary body melanoma, Hyphema, Rubeosis iridis, Synechia, Uveitis-glaucoma-hyphema syndrome and Glaucoma. RNA-based therapeutics can be used in e.g. passive and active immunotherapy, protein replacement therapy, or genetic engineering. Accordingly, RNA has the potential to provide highly specific and individual treatment options for the therapy of a large variety of ophthalmic diseases, disorders, or conditions. However, the effectiveness of RNA therapeutics is limited by the delivery of the RNA to the site of disease in a spatially and temporally controlled manner. There are various pathological conditions which would highly profit if the RNA drug could be delivered to the anterior segment of the eye. Many inflammatory and proliferative diseases are associated with cells and tissues in the anterior region of the eye. Several hurdles of ophthalmic drug administration exist in implementing an effective treatment strategy for ocular diseases and disorders, mainly due to the unique anatomy and physiology of the eye. The combination of static barriers such as different layers and regions of the eye, and dynamic barriers such as blood flow, lymphatic clearance, tear dilution, or ribonucleases, pose a significant challenge for ocular RNA drug delivery. Traditional ophthalmic delivery routes of RNA therapeutics such as intravitreal administration as described in W02020097511, administration into the suprachoroidal space (W02020161342) orsubretinal injection (W02009105690) often limit the bioavailability of the expressed protein/peptide in the anterior segment of the eye. Especially, for intravitreal injections and subretinal injection, the injected RNA has to diffuse through the vitreous to reach the therapeutically relevant cells. Via these administration routes it is particularly challenging to reach cells and/or tissues in the anterior segment of the eye to e.g. treat diseases of the cornea. Accordingly, an object of the invention is to provide delivery and application methods for therapeutic nucleic acids, preferably RNA or compositions comprising therapeutic nucleic acids, preferably RNA to the anterior segment of the eye of a subject. A further object of the invention is to provide therapeutic nucleic acid, preferably RNA and compositions comprising said therapeutic nucleic acid, preferably RNA suitable for ocular delivery, e.g. as eye drop formulations, gel or oinment or coated on contact lenses. The objects mentioned above are solved by the underlying description and the accompanying claims. Definitions For the sake of clarity and readability the following definitions are provided. Any technical feature mentioned for these definitions may be read on each and every embodiment of the invention. Additional definitions and explanations may be specifically provided in the context of these embodiments. Percentages in the context of numbers should be understood as relative to the total number of the respective items. In other cases, and unless the context dictates otherwise, percentages should be understood as percentages by weight (wt.-%). About: The term "about" is used when determinants do not need to be identical, i.e.100% the same. Accordingly, "about" means, that a determinants may diverge by 0.1% to 20%, preferably by 0.1% to 10%; in particular, by 0.5%, 1%, 2%, 3%, 4%, 5%, 6%, 7%, 8%, 9%, 10%, 11%, 12%, 13%, 14%, 15%, 16%, 17%, 18%, 19%, 20%. The skilled person will know that e.g. certain parameters or determinants may slightly vary based on the method how the parameter was determined. Anterior Segment Developmental Anomalies (ASDA) and Anterior Segment Dysaenesis (ASP): as used herein, refer to a spectrum of disorders that affect the development of the front of the eye (the anterior segment), which includes the cornea, iris, ciliary body, and lens. The specific eye abnormalities (alone or in combination) vary depending on the subtype of ASD and genetic cause, and some types may also be associated with neurological abnormalities. Glaucoma develops in approximately 60% of people with ASD, during infancy or much later. Specific eye signs and symptoms of ASD may include: Underdevelopment of the iris (iris hypoplasia). An enlarged or reduced cornea diameter. Growth of new blood vessels (vascularization) and opacity in the cornea. Posterior embryotoxon (a thickened and displaced Schwalbe's line). Corectopia (displacement of the pupil). Polycoria (more than one pupillary opening). An abnormal iridocorneal angle (the angle formed by the iris and cornea). Ectopia lentis (displacement of the lens). Aphakia (absent lens). Cataracts. Anterior synechiae (when the iris adheres to the cornea). Posterior keratoconus (thinning of the cornea). Individual disorders within the ASD spectrum include Axenfeld-Rieger syndrome (which includes disorders formerly known as Axenfeld anomaly, Axenfeld syndrome, Rieger anomaly, Rieger syndrome, iridogoniodysgenesis and Peters anomaly. ASD may be caused by genetic changes in any of several genes and inheritance can be autosomal dominant or autosomal recessive, depending on the responsible gene. Antibody, antibody fragment: As used herein, the term "antibody" includes both an intact antibody and an antibody fragment. Typically, an intact "antibody" is an immunoglobulin that specifically binds to a particular antigen. An antibody may be a member of any immunoglobulin class, including any of the human classes: IgG, IgM, IgE, IgA and IgD. Typically, an intact antibody is a tetramer. Each tetramer consists of two identical pairs of polypeptide chains, each pair having a "light" chain and a "heavy" chain. An "antibody fragment" includes a portion of an intact antibody, such as the antigen-binding or variable region of an antibody. Examples of antibody fragments include Fab, Fab', F (ab') 2 and Fv fragments; the tribes; Tetra; linear antibodies; single-chain antibody molecules; and multi specific antibodies formed from antibody fragments. E.g., the antibody fragments comprise isolated fragments, "Fv" fragments consisting of heavy and light chain variable regions, recombinant single chain polypeptide molecules in which the light and heavy chain variable regions are linked together by a peptide linker ("ScFv Proteins") and minimal recognition units consisting of amino acid residues that mimic the hypen/ariable region. Examples of antigen-binding fragments of an antibody include, but are not limited to, Fab fragment, Fab 'fragment, F (ab') 2 fragment, scFv fragment, Fv fragment, dsFv diabody, dAb fragment, fragment Fd ', Fd fragment and an isolated complementarity determining region (CDR). Suitable antibodies that may be encoded by the therapeutic RNA of the invention include monoclonal antibodies, polyclonal antibodies, antibody mixtures or cocktails, human or humanized antibodies, chimeric antibodies, Fab fragments, or bispecific antibodies. Cationic, cationisable: Unless a different meaning is clear from the specific context, the term "cationic" means that the respective structure bears a positive charge, either permanently or not permanently but in response to certain conditions such as e.g. pH. Thus, the term "cationic" covers both "permanently cationic" and "cationisable". The term "cationisable" as used herein means that a compound, or group or atom, is positively charged at a lower pH and uncharged at a higher pH of its environment. Also in non-aqueous environments where no pH value can be determined, a cationisable compound, group or atom is positively charged at a high hydrogen ion concentration and uncharged at a low concentration or activity of hydrogen ions. It depends on the individual properties of the cationisable or polycationisable compound, in particular the pKa of the respective cationisable group or atom, at which pH or hydrogen ion concentration it is charged or uncharged. In diluted aqueous environments, the fraction of cationisable compounds, groups or atoms bearing a positive charge may be estimated using the so-called Henderson-Hasselbalch equation which is well-known to a person skilled in the art. E.g., if a compound or moiety is cationisable, it is preferred that it is positively charged at a phi value of about 1 to 9, preferably 4to 9, 5to 8 or even 6 to 8, more preferably of a pH value of or below 9, of or below 8, of or below 7, most preferably at physiological pH values, e.g. about 7.3 to 7.4, i.e. under physiological conditions, particularly under physiological salt conditions of the cell in vivo. In embodiments, it is preferred that the cationisable compound or moiety is predominantly neutral at physiological phi values, e.g. about 7.0-7.4, but becomes positively charged at lower pH values. In some embodiments, the preferred range of pKa for the cationisable compound or moiety is about 5 to about 7. CRISPR-associated protein: The term "CRISPR-associated protein" will be recognized and understood by the person of ordinary skill in the art. The term "CRISPR-associated protein" refers to RNA-guided endonucleases that are part of a CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats) system (and their homologs, variants, fragments or derivatives), which is used by prokaryotes to confer adaptive immunity against foreign DNA elements. CRISPR- associated proteins include, without limitation, Cas9, Cpf1 (Cas12), C2c1, C2c3, C2c2, Cas13, CasX and CasY. As used herein, the term "CRISPR-associated protein" includes wild-type proteins as well as homologs, variants, fragments and derivatives thereof. Therefore, when referring to artificial nucleic acid molecules encoding Cas9, Cpf1 (Cas12), C2c1, C2c3, and C2c2, Cas13, CasX and CasY, said artificial nucleic acid molecules may encode the respective wild- type proteins, or homologs, variants, fragments and derivatives thereof. Besides Cas9 and Cas12 (Cpf1), several other CRISPR-associated protein exist that are suitable for genetic engineering in the context of the invention, including Cas13, CasX and CasY; e.g. Cas13 i.e. WP15770004, WP18451595, WP21744063, WP21746774, ERK53440, WP31473346, CVRQ01000008, CRZ35554, WP22785443, WP36091002, WP12985477, WP13443710, ETD76934, WP38617242, WP2664492, WP4343973, WP44065294, ADAR2DD, WP47447901, ERI81700, WP34542281, WP13997271, WP41989581, WP47431796, WP14084666, WP60381855, WP14165541, WP63744070, WP65213424, WP45968377, EH006562, WP6261414, EKB06014, WP58700060, WP13446107, WP44218239, WP12458151, ERJ81987, ERJ65637, WP21665475, WP61156637, WP23846767, ERJ87335, WP5873511, WP39445055, WP52912312, WP53444417, WP12458414, WP39417390, EOA10535, WP61156470, WP13816155, WP5874195, WP39437199, WP39419792, WP39431778, WP46201018, WP39442171, WP39426176, WP39418912, WP39434803, WP39428968, WP25000926, EFU31981, WP4343581, WP36884929, BAU18623, AFJ07523, WP14708441, WP36860899, WP61868553, KJJ86756, EGQ18444, EKY00089, WP36929175, WP7412163, WP44072147, WP42518169, WP44074780, WP15024765, WP49354263, WP4919755, WP64970887, WP61710138); CasX (i.e. OGP07438, OHB99618); CasY( i.e. OJI08769, OGY82221, OJI06454, APG80656, OJI07455, OJI09436, PIP58309). Derived from: The term "derived from" as used throughout the present specification in the context of a nucleic acid, i.e. for a nucleic acid "derived from" (another) nucleic acid, means that the nucleic acid, which is derived from (another) nucleic acid, shares e.g. at least about 70%, 80%, 85%, 86%, 87%, 88%, 89%, 90%, 91 %, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or about 99% sequence identity with the nucleic acid from which it is derived. The skilled person is aware that sequence identity is typically calculated for the same types of nucleic acids, i.e. for DNA sequences or for RNA sequences. Thus, it is understood, if a DNA is "derived from" an RNA or if an RNA is "derived from" a DNA, in a first step the RNA sequence is converted into the corresponding DNA sequence (in particular by replacing the uracils (U) by thymidines (T) throughout the sequence) or, vice versa, the DNA sequence is converted into the corresponding RNA sequence (in particular by replacing the T by U throughout the sequence). Thereafter, the sequence identity of the DNA sequences or the sequence identity of the RNA sequences is determined. Preferably, a nucleic acid "derived from" a nucleic acid also refers to nucleic acid, which is modified in comparison to the nucleic acid from which it is derived, e.g. in order to increase RNA stability even further and/orto prolong and/or increase protein production. In the context of amino acid sequences, the term "derived from" means that the amino acid sequence, which is derived from (another) amino acid sequence, shares e.g. at least about 70%, 80%, 85%, 86%, 87%, 88%, 89%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or about 99% sequence identity with the amino acid sequence from which it is derived. Thus, it is understood, if e.g. a protein is "derived from" a certain protein, the protein that is "derived from" may represent a variant or fragment of said respective protein, sharing a certain percentage of sequence identity. Glaucoma: As used herein is a group of eye diseases that can cause vision loss and blindness by damaging a nerve in the back of your eye called the optic nerve. The main cause of damage to the optic nerve is intraocular pressure (IOP), excessive fluid pressure within the eye, which can be caused by factors such as blockage of drainage ducts and narrowing or closure of the angle between the iris and cornea. From here, the trabecular meshwork drains aqueous humor via the scleral venous sinus (Schlemm's canal) into scleral plexuses and general blood circulation. Glaucoma is primarily categorized as either open-angle or closed-angle (or angle-closure). In open-angle glaucoma, the iris meets the cornea normally, allowing the fluid from inside the eye to drain, thus relieving the internal pressure. When this angle is narrowed or closed, pressure increases over time, causing damage to the optic nerve and leading to blindness. Glaucoma filtration surgery (Trabeculectomv): As used herein is a procedure to treat glaucoma. The goal of a trabeculectomy is to lower eye pressure. By lowering eye pressure, it is hoped that the operated eye will be spared further glaucoma damage and can maintain its vision. During this operation, a tiny piece of the wall of the eye, which may include the trabecular meshwork (the natural drain), is removed by the surgeon. This opens a new drain which creates a bypass for the trabecular meshwork to reduce eye pressure. The eye pressure is reduced because fluid can now drain with relative ease through the new opening into a reservoir (bleb) underneath the conjunctiva (which comprises the surface of the eye). The fluid is then absorbed by the body. Guide RNA: As used herein, the term "guide RNA" (gRNA) thus relates to any RNA molecule capable of targeting a CRISPR-associated protein as defined above to a target DNA sequence of interest. In the context of the invention, the term guide RNA has to be understood in its broadest sense and may comprise two-molecule gRNAs ("tracrRNA/crRNA") comprising crRNA ("CRISPR RNA" or "targeter-RNA" or "crRNA" or "crRNA repeat") and a corresponding tracrRNA ("trans-acting CRISPR RNA" or "activator-RNA" or "tracrRNA") molecule, or single-molecule gRNAs. A "sgRNA" typically comprises a crRNA connected at its 3' end to the 5' end of a tracrRNA through a "loop" sequence. Fibrosis: The term "fibrosis" will be recognized and understood by the person of ordinary skill in the art, and inter alia relates to pathological wound healing in which e.g. connective tissue replaces normal parenchymal tissue to the extent that it goes unchecked, leading to considerable tissue remodelling and the formation of permanent scar tissue. Repeated injuries, chronic inflammation and repair are typically susceptible to fibrosis where an accidental excessive accumulation of extracellular matrix components, such as the collagen is produced by fibroblasts, leading to the formation of a permanent fibrotic scar. In response to injury, this is called scarring, and if fibrosis arises from a single cell line, this is called a fibroma. Physiologically, fibrosis acts to deposit connective tissue, which can interfere with or totally inhibit the normal architecture and function of the underlying organ or tissue. Fibrosis can be used to describe the pathological state of excess deposition of fibrous tissue, as well as the process of connective tissue deposition in healing. Defined by the pathological accumulation of extracellular matrix (ECM) proteins, fibrosis results in scarring and thickening of the affected tissue. It is in essence an exaggerated wound healing response which interferes with normal organ function. From the physiological perspective, fibrosis is similar to the process of scarring, in that both involve stimulated fibroblasts laying down connective tissue, including collagen and glycosaminoglycans. The process is initiated when immune cells such as macrophages release soluble factors that stimulate fibroblasts. The most well characterized pro-fibrotic mediator is TGFbeta, which is released by macrophages as well as any damaged tissue between surfaces called interstitium. Other soluble mediators offibrosis include CTGF, platelet-derived growth factor (PDGF), and interleukin 10 (IL-10). These initiate signal transduction pathways such as the AKT/mTOR and SMAD pathways that ultimately lead to the proliferation and activation offibroblasts, which deposit extracellular matrix into the surrounding connective tissue. This process of tissue repair is a complex one, with tight regulation of extracellular matrix (ECM) synthesis and degradation ensuring maintenance of normal tissue architecture. hlowever, the entire process, although necessary, can lead to a progressive irreversible fibrotic response if tissue injury is severe or repetitive, or if the wound healing response itself becomes deregulated. Fibrosis can occur in many tissues within the body, typically as a result of inflammation or damage, and examples include pathologies in the lung (e.g. cystic fibrosis, idiopathic pulmonary fibrosis), pathologies in the liver (e.g. cirrhosis), or pathologies in the heart (e.g. myocardial fibrosis). Fragment: The term "fragment" as used throughout the present specification in the context of a nucleic acid sequence or an amino acid (aa) sequence may typically be a shorter portion of a full-length sequence of e.g. a nucleic acid sequence or an amino acid sequence. A fragment typically consists of a sequence that is identical to the corresponding stretch within the full-length sequence. The term "fragment" as used throughout the present specification in the context of proteins or peptides may, typically, comprise a sequence of a protein or peptide as defined herein, which is, with regard to its amino acid sequence (or its encoded nucleic acid molecule), N-terminally and/or C-terminally truncated compared to the amino acid sequence of the original (native) protein (or its encoded nucleic acid molecule). Such truncation may thus occur either on the aa level or correspondingly on the nucleic acid level. A sequence identity with respect to such a fragment as defined herein may therefore preferably refer to the entire protein or peptide as defined herein or to the entire (coding) nucleic acid molecule of such a protein or peptide. Fragments of antigenic proteins or peptides may comprise at least one epitope of those proteins or peptides. Furthermore also domains of a protein, like the extracellular domain, the intracellular domain or the transmembrane domain and shortened or truncated versions of a protein may be understood to comprise a fragment of a protein. Heterologous: The terms "heterologous" or "heterologous sequence" as used throughout the present specification in the context of a nucleic acid sequence or an amino acid sequence refers to a sequence (e.g. DNA, RNA, amino acid) will be recognized and understood by the person of ordinary skill in the art, and is intended to refer to a sequence that is derived from another gene, from another allele, from another species. Two sequences are typically understood to be "heterologous" if they are not derivable from the same gene or in the same allele. I.e., although heterologous sequences may be derivable from the same organism, they naturally (in nature) do not occur in the same nucleic acid molecule, such as e.g. in the same RNA or protein. Identity (of a sequence): The term "identity" as used throughout the present specification in the context of a nucleic acid sequence or an amino acid sequence will be recognized and understood by the person of ordinary skill in the art, and is e.g. intended to refer to the percentage to which two sequences are identical. To determine the percentage to which two sequences are identical, e.g. nucleic acid sequences or aa sequences as defined herein, preferably the aa sequences encoded by the nucleic acid sequence as defined herein or the aa sequences themselves, the sequences can be aligned in order to be subsequently compared to one another. Therefore, e.g. a position of a first sequence may be compared with the corresponding position of the second sequence. If a position in the first sequence is occupied by the same residue as is the case at a position in the second sequence, the two sequences are identical at this position. If this is not the case, the sequences differ at this position. If insertions occur in the second sequence in comparison to the first sequence, gaps can be inserted into the first sequence to allow a further alignment. If deletions occur in the second sequence in comparison to the first sequence, gaps can be inserted into the second sequence to allow a further alignment. The percentage to which two sequences are identical is then a function of the number of identical positions divided by the total number of positions including those positions which are only occupied in one sequence.The percentage to which two sequences are identical can be determined using an algorithm, e.g. an algorithm integrated in the BLAST program. Nucleic acid: The terms "nucleic acid" or "nucleic acid molecule" will be recognized and understood by the person of ordinary skill in the art, and are e.g. intended to refer to a molecule comprising, preferably consisting of nucleic acid components. The term nucleic acid molecule preferably refers to DNA or RNA. It is preferably used synonymous with the term polynucleotide. Preferably, a nucleic acid or a nucleic acid molecule is a polymer comprising or consisting of nudeotide monomers (natural and/or modified), which are covalently linked to each other by phosphodiester-bonds of a sugar/phosphate-backbone. The term "nucleic acid molecule" also encompasses modified nucleic acid molecules, such as base-modified, sugar-modified or backbone-modified DNA or RNA (e.g. therapeutic RNA) molecules as defined herein. Nucleic acid sequence/ RNA sequence/ amino acid sequence: The terms "nucleic acid sequence", "RNA sequence" or "amino acid sequence" will be recognized and understood by the person of ordinary skill in the art, and are e.g. intended to refer to particular and individual order of the succession of its nucleotides or amino acids respectively. "Ophthalmic disease disorder or condition" or "ocular disease, disorder or condition": The terms "Ophthalmic disease disorder or condition" or "ocular disease, disorder or condition" will be recognized and understood by the person of ordinary skill in the art, and are e.g. intended to refer to a disease, disorder or condition affecting the eye and/or vision. Ocular/ophthalmic diseases, disorders or conditions can affect one or more of the following parts of the eye: eyelid, lacrimal system and orbit; conjunctiva; sclera, cornea, iris and ciliary body; lens; choroid and retina; vitreous body and globe; optic nerve and visual pathways; and ocular muscles. For example, an ocular or ophthalmic disease, disorder or condition may be caused by a protein deficiency or dysfunctions in the eye or parts of the anatomy associated with vision. An ocular/ophthalmic disease, disorder or condition may be caused by a protein surplus, over expression, and/or over activation in the eye or parts of the anatomy associated with vision. RNA in vitro transcription: The terms "RNA in vitro transcription" or "in vitro transcription" relate to a process wherein RNA is synthesized in a cell-free system in vitro. RNA may be obtained by DNA-dependent in vitro transcription of an appropriate DNA template, which is typically a linear DNA template (e.g. linearized plasmid DNA or PCR product). The promoter for controlling RNA in vitro transcription can be any promoter for any DNA-dependent RNA polymerase. Particular examples of DNA-dependent RNA polymerases are the T7, T3, SP6, or Syn5 RNA polymerases. In the context of the invention, the DNA template is typically linearized with a suitable restriction enzyme before it is subjected 5 to RNA in vitro transcription. Reagents typically used in RNA in vitro transcription include: a DNA template (linearized plasmid DNA or PCR product) with a promoter sequence that has a high binding affinity for its respective RNA polymerase such as bacteriophage-encoded RNA polymerases fT7, T3, SP6, or Syn5); ribonucleotide triphosphates (NTPs) for the four bases (adenine, cytosine, guanine and uracil); optionally, a cap analogue as defined herein; optionally, modified nucleotides as defined herein; a DNA-dependent RNA polymerase capable of binding to the 10 promoter sequence within the DNA template (e.g. T7, T3, SP6, or Syn5 RNA polymerase); optionally, a ribonuclease (RNase) inhibitor to inactivate any potentially contaminating RNase; optionally, pyrophosphatase; MgCl2; a buffer CTRIS or hlEPES) to maintain a suitable pH value, which can also contain antioxidants (e.g. DTT), and/or polyamines such as spermidine. 15 Variant (of a sequence): The term "variant" as used throughout the present specification in the context of a nucleic acid sequence will be recognized and understood by the person of ordinary skill in the art, and is e.g. intended to refer to a variant of a nucleic acid sequence derived from another nucleic acid sequence. E.g., a variant of a nucleic acid sequence may exhibit one or more nucleotide deletions, insertions, additions and/or substitutions compared to the nucleic acid sequence from which the variant is derived. A variant of a nucleic acid sequence may at least 50%, 60%, 2070%, 80%, 90%, or 95% identical to the nucleic add sequence the variant is derived from. The variant is a functional variant in the sense that the variant has retained at least 50%, 60%, 70%, 80%, 90%, or 95% or more of the function of the sequence where it is derived from. A "variant" of a nucleic acid sequence may have at least 70%, 75%, 80%, 85%, 90%, 95%, 98% or 99% nucleotide identity over a stretch of at least 10, 20,30,50, 75 or 100 nucleotide of such nucleic acid sequence. 25 The term "variant" as used throughout the present specification in the context of proteins or peptides will be recognized and understood by the person of ordinary skill in the art, and is e.g. intended to refer to a proteins or peptide variant having an amino acid sequence which differs from the original sequence in one or more mutation(s), such as one or more substituted, inserted and/or deleted amino acid(s). Preferably, these fragments and/or variants have the same biological function or specific activity compared to the full-length native protein, e.g. its specific antigenic property. 30 "Variants" of proteins or peptides as defined herein may comprise conservative amino acid substitution(s) compared to their native, i.e. non-mutated physiological, sequence. Those amino acid sequences as well as their encoding nucleotide sequences in particular fall under the term variants as defined herein. Substitutions in which amino acids, which originate from the same class, are exchanged for one another are called conservative substitutions. In particular, these are amino acids having aliphatic side chains, positively or negatively charged side chains, aromatic groups in the 35 side chains or amino acids, the side chains of which can enter into hydrogen bridges, e.g. side chains which have a hydroxyl function. This means that e.g. an amino acid having a polar side chain is replaced by another amino acid having a likewise polar side chain, or, e.g., an amino acid characterized by a hydrophobia side chain is substituted by another amino acid having a likewise hydrophobic side chain (e.g. serine (threonine) by threonine (serine) or leucine (isoleudne) by isoleucine (leucine)). Insertions and substitutions are possible, in particular, at those sequence positions 0 which cause no modification to the three-dimensional structure or do not affect the binding region. Modifications to a three-dimensional structure by insertion(s) or deletion(s) can easily be determined e.g. using CD spectra (circular dichroism spectra). A "variant" of a protein or peptide may have at least 70%, 75%, 80%, 85%, 90%, 95%, 98% or 99% amino acid identity over a stretch of at least 10,20,30,50,75 or 100 amino acids of such protein or peptide. Preferably, a variant of a protein comprises a functional variant of the protein, which means that the variant exerts the same effect 5 or functionality or at least 40%, 50%, 60%, 70%, 80%, 90%, or 95% of the effect or functionality as the protein it is derived from. Short description of the invention The present invention provides delivery and application methods for therapeutic nucleic acid, preferably RNA, or compositions comprising therapeutic nucleic acid, preferably RNA to the anterior segment of the eye of a subject. The present invention is based on the surprising finding that a protein, e.g. a therapeutic protein, encoded by a therapeutic nucleic acid, preferably RNA of the invention can efficiently be expressed in a mammalian eye upon administration of said nucleic acid/RNA into the anterior segment of the eye of a subject. Exemplary application routes for the administration were selected from cornea, the conjunctiva or the ciliary body administration. We were able to show that an administration of a composition comprising (therapeutic) nucleic acid, preferably RNA encoding a protein resulted in expression and activity of an encoded protein in the cells of the eye after administration to the cornea, conjunctiva or ciliary body (see Examples 3 to 6). As described herein including in the examples, the inventors have successfully delivered nucleic acid/RNA to the anterior segment of the eye (cornea, conjunctiva and ciliary body), resulting in robust protein expression in the eye. The inventors have demonstrated, for the first time, that delivery into the anterior segment of the eye may be used to effectively deliver nucleic acid/RNA encoding therapeutic proteins or antigenic proteins into the eye, providing an effective yet unexpected solution for this difficult problem of site- specific (local) ocular nucleic add/RNA delivery. Moreover, the finding that therapeutically relevant cell types of the anterior segment of the eye can be transfected with therapeutic nucleic acid/RNA opens yet unexpected treatment options for various diseases like Glaucoma or Conjunctivitis associated with defects or deficiencies of surrounding cells. For example, corneal administration of nucleic acid/RNA also display an advantage to known medication which, when topically administered, get quickly diluted by tears and removed from the ocular surface through blinking and turnover of the tears. However, in case of nucleic acid/RNA administration to e.g. the cornea, cornea! cells get transfected with the RNA and produce the encoded protein over an extended period of time. In a first aspect, the present invention relates to a composition comprising a therapeutic nucleic acid, preferably RNA comprising at least one coding sequence encoding at least one peptide or protein for use in treatment or prevention of an ophthalmic disease, disorder or condition, wherein the therapeutic nucleic acid, preferably RNA is administered to the anterior segment of the eye/ocular tissue of a subject. Preferably, the composition is administered to the cornea, the ciliary body, or the conjunctiva of the eye of a subject. In a second aspect, the present invention relates to a kit or kit of parts, preferably for use in treatment or prevention of an ophthalmic disease, disorder or condition, wherein said kit or kit of parts is administered to the anterior segment of the eye of a subject in need of treatment. Said kit or kit of parts comprises at least one composition as specified in the context of the first aspect, and optionally, a liquid vehicle for solubilising, and/or technical instructions providing information on administration and dosage of the components. In a third aspect, the invention relates to a method of treating or preventing of an ophthalmic disease, disorder or condition, wherein the method comprises applying or administering to a subject in need thereof the composition of the first aspect, or the kit or kit of parts of the second aspect via administration to the anterior segment of the eye of a subject. In a fourth aspect, the present invention relates to a method of delivering a therapeutic nucleic acid, preferably RNA molecule to cells and/or tissues of the anterior segment of the eye of a subject, wherein delivering is performed via administration to the cornea, the ciliary body, or the conjunctiva of the eye of a subject. Further aspects inter alia relate to eye drop formulation, contact lenses, medical devices or gels or ointments for use in treatment or prevention of an ophthalmic disease, disorder or condition, wherein the eye drop formulation, contact lenses, medical devices, and gels or ointments are administered to the anterior segment of the eye of a subject. Detailed description of the invention Where reference is made to "SEQ ID NOs" of other patent applications or patents, said sequences, e.g. amino acid sequences or nucleic acid sequences, are explicitly incorporated herein by reference. For "SEQ ID NOs" provided herein, information provided under "feature key", i.e. "source" (for nucleic acids or proteins) or "misc_feature" (for nucleic acids) or "REGION" (for proteins). (in the sequence listing according to WIPO ST.26 Standard) is also explicitly included herein in its entirety. Where reference is made to "SEQ ID NOs" in the context of RNA sequences, the skilled person will be able to derive RNA sequences from the referenced SEQ ID NOs also in cases where DNA sequences are provided. Where reference is made to "SEQ ID NOs" in the context of DNA sequences, the skilled person will be able to derive respective DNA sequences from the referenced SEQ ID NOs also in cases where RNA sequences are provided. Composition in treatment or prevention of an ophthalmic disease, disorder, or condition The anterior segment of the eye encompasses ofsclera, conjunctiva, cornea, ciliary body, iris, pupil, aqueous humor, trabecular meshwork and lens. Next to infection, injuries or inflammation, developmental disorders of the eye related to this anatomical section of the eye are grouped under the term Anterior Segment Developmental Anomalies (ASDA) and referred to as Anterior Segment Dysgenesis (ASD). Some symptoms overlap, such as the tendency to develop increased intraocular pressure (IOP). The ciliary body of the iris produces the aqueous humor leading to IOP. This extra fluid must be drained via the trabecular meshwork into Schlemm's canal and by the uveoscleral outflow pathway. Disruptions in this process are frequent in ASD, and thus secondary glaucoma is a common complication. Additionally, conjunctival and subconjunctival fibrogenesis and inflammation or corneal scarring are typical side effects which often occur subsequent to glaucoma filtration surgery, which is performed to reduce IOP. In a first aspect, the present invention relates to a composition comprising at least one therapeutic nucleic acid, comprising at least one coding sequence encoding at least one peptide or protein. Preferably, said composition is for use in treatment or prevention of an ophthalmic disease, disorder or condition, wherein the therapeutic nucleic acid is administered to the anterior segment of the eye/ocular tissue of a subject. It has to be noted that specific features and embodiments that are described in the context of the first aspect of the invention, that is in particular structural and functional features that describe the composition comprising a therapeutic nucleic acid of the invention, are likewise applicable to the second aspect (kit or kit of parts of the invention), the third aspect (method of treating or preventing of an ophthalmic disease, disorder or condition), or further aspects including medical uses and method of delivery. In preferred embodiments, the therapeutic nucleic acid of the invention is an isolated nucleic acid. The term "isolated nucleic acid" does not comprise a cell or a subject that comprises said nucleic acid but relates to the artificial nucleic acid as an isolated molecule or ensemble of isolated molecules. For example, the "isolated nucleic acid" can be an artificial nucleic acid isolated or purified from a cell (e.g. cell culture, bacterial culture), or can be an artificial nucleic acid (e.g. RNA) isolated from an RNA in vitro transcription. The term "artificial nucleic acid" as used herein is intended to refer to a nucleic acid that does not occur naturally. In other words, an artificial nucleic acid may be understood as a non-natural RNA molecule. Such molecules may be non- natural due to their individual sequence (e.g. G/C content modified coding sequence, UTRs) and/or due to other modifications, e.g. structural modifications of modified nucleotides. Artificial nucleic acids may be designed and/or generated by genetic engineering to correspond to a desired artificial sequence of nucleotides. In this context an artificial RNA is a sequence that may not occur naturally, i.e. it differs from the wild type sequence by at least one nucleotide/modifi cation. 5 In an embodiment, the therapeutic nucleic acid of this invention is selected from DNA. The DNA may be any type of DMA that comprises a coding sequence as defined herein including any type of single stranded DNA, any type of double stranded DNA, any type of linear DNA, and any type of circular DNA. A suitable DNA in the context of the invention may be selected from bacterial plasmid, an adenovirus, a poxvirus, a 10 parapoxivirus (orf virus), a vaccinia virus, a fowlpox vims, a herpes virus, an adeno-associated virus (AAV), an alphavirus, a lentivirus, a lambda phage, a lymphocytic choriomeningitis virus and a Listeria sp, Salmonella sp. In preferred embodiments, the DNA a viral DNA, preferably an adeno-associated virus DNA. 15 In particularly preferred embodiments, the therapeutic nucleic acid of the invention is an RNA. In particularly preferred embodiments, the RNA is selected from an mRNA. Accordingly, the therapeutic nucleic acid of the invention is an mRNA, suitably an isolated mRNA. Preferably, the therapeutic nucleic acid, preferably the RNA, comprises about 50 to about 20000 nucleotides, or about 20500 to about 10000 nucleotides, or about 1000 to about 10000 nucleotides, or preferably about 1000 to about 5000 nucleotides, or even more preferably about 2000 to about 5000 nucleotides. The term "therapeutic RNA" or "therapeutic nucleic acid" relates to any nucleic acid, in particular any RNA as defined above, providing a therapeutic modality. The term "therapeutic" in that context has to be understood as "providing a 25 therapeutic function" or as "being suitable for therapy or administration". However, "therapeutic" in that context should not at all to be understood as being limited to a certain therapeutic modality. Examples for therapeutic modalities may be the provision of a coding sequence (via said therapeutic RNA) that encodes for a peptide or protein (wherein said peptide or protein has a certain therapeutic function, e.g. an antigen for a vaccine, or an enzyme for protein replacement therapies). A further therapeutic modality may be genetic engineering, wherein the RNA provides or orchestrates factors 30 to e.g. manipulate DNA and or RNA. Typically, the term "therapeutic RNA" does not include natural RNA extracts or RNA preparations (e.g. obtained from bacteria, or obtained from plants) that are not suitable for administration to a subject (e.g. animal, human). For being suitable for a therapeutic purpose, the RNA of the invention may be an artificial, non-natural RNA. 35 The term "treatment" or "treating" of a disease, disorder or condition includes preventing or protecting against the disease, disorder or condition (that is, causing the clinical symptoms not to develop); inhibiting the disease, disorder or condition (i.e., arresting/suppressing the development of clinical symptoms); and/or relieving the disease, disorder or condition (i.e., causing the regression of clinical symptoms). As will be appreciated, it is not always possible to distinguish between "preventing" and "suppressing" a disease, disorder or condition since the ultimate inductive event or 40 events may be unknown or latent. Accordingly, the term "prophylaxis" will be understood to constitute a type of "treatment" that encompasses "preventing" and "suppressing." The term "treatment" thus also includes "prophylaxis". The term "treatment" also includes prevention". As used herein, "ocular tissue" and "eye" include both the anterior segment of the eye (i.e., the portion of the eye in front 45 of the lens) and the posterior segment of the eye (i.e., the portion of the eye behind the lens) as shown in Figure 1. The eye includes both, an anterior region or segment 12 (the portion of the eye in front of and including the lens) and a posterior region or segment 14 (the portion of the eye behind the lens). The anterior segment 12 is bounded by the cornea 16 and the lens 18, while the posterior segment 14 is bounded by the sclera 20 and the lens 18. The anterior segment 12 is further subdivided into the anterior chamber 22, between the iris 24 and the cornea 16, and the posterior chamber 26, between the lens 18 and the iris 24. The cornea 16 and the sclera 20 collectively form a limbus 38 at the point at which they meet. The exposed portion of the sclera 20 on the anterior segment 12 of the eye is protected by a clear membrane referred to as the conjunctiva 45. Underlying the sclera 20 is the choroid 28 and the retina 27, collectively referred to as retina choroidal tissue. A vitreous humour 30 (also referred to as the "vitreous") is disposed between a ciliary body 32 (including a ciliary muscle and a ciliary process) and the retina 27. The anterior portion of the retina 27 forms an ora serrata 34. The loose connective tissue, or potential space, between the choroid 28 and the sclera 20 is referred to as the suprachoroid, comprising the suprachoroidal space (SCS) 36. Preferably, the composition for use in treatment comprising a therapeutic nucleic acid, preferably RNA, is administered to the anterior segment of the eye/ocular tissue of a subject. The term anterior segment or anterior cavity of the eye/ocular tissue of a subject refers to the front-most region of the eye and encompasses, without limitation sclera, conjunctiva, cornea, ciliary body, iris, pupil, aqueous humor, trabecular meshwork and lens. Accordingly, cells and tissues located in the anterior segment of the eye that may be addressed via delivery to the cornea, conjunctiva or ciliary body of the composition and may comprise Bowman's Layer, Descemet's membrane, Corneal stroma, Corneal epithelial cells, keratocytes, connective tissue, vessels, epithelial cells, columnar cells or conjunctival goblet cells and (smooth) muscle cells (ciliary muscle cells), ciliary vessels, ciliary nerve branches, fibroblasts, columnar cells or endothelial cells. Within the anterior segment are two fluid-filled spaces: the anterior chamber between the posterior surface of the cornea (i.e. the corneal endothelium) and the iris and the posterior chamber between the iris and the front face of the vitreous. Aqueous humour fills these spaces within the anterior segment and provides nutrients to the surrounding structures. Ocular drug delivery has been a major challenge due to the unique anatomy and physiology of the eye. Structural variation of each layer of ocular tissue can present a significant barrier following drug administration by any route. In theory, topical application of conventional medications is suitable for management of the anterior segment, and intravitreal injections or systemic medications are generally used for posterior infections (Chen and Penm Encyclopedia of Pharmacy Practice and Clinical Pharmacy, 2019). However, topical administration of conventional medications (e.g. peptides, proteins, small molecules) often show higher tear dilution of drug and turnover rate as well as facing the corneal barrier. In most applications that require a local administration of nucleic acid, e.g. RNA, tissue distribution may be a limiting factor. Improved tissue distribution may enable the uptake of the RNA drug by higher number of therapeutically relevant cells, which will then produce the protein encoded by the RNA (e.g. therapeutic protein). Usually, the levels of protein production relate closely with the numbers oftransfected cells and, as a result, with the observed activity or therapeutic effect. Improved tissue distribution is particularly relevant for medical treatments of the eye, where established methods of administration bring along some drawbacks e.g. in terms of distribution of the injected RNA within the eye. Therapeutic of nucleic acid, e.g. RNA administered to the anterior segment of the eye allows for a transient and localized production of therapeutic proteins, which reduces the danger of unwanted side effects. In preferred embodiments, the composition for use in treatment comprising the therapeutic nucleic acid, preferably the RNA, is administered to the cornea, the ciliary body, or the conjunctiva of the eye of a subject. Corneal administration In most preferred embodiments, the composition for use in treatment comprising the therapeutic nucleic acid, preferably the RNA, is administered to the cornea of the eye of a subject. The cornea of the eye is described as the transparent part of the eye that covers the iris and the pupil and allows light to enter the inside. The human cornea borders with the sclera at the corneal limbus and has unmyelinated nerve endings sensitive to touch, temperature and chemicals. Because transparency is of prime importance, the healthy cornea does not have or need blood vessels within it. Instead, oxygen dissolves in tears and then diffuses throughout the cornea to keep it healthy. Nutrients are transported via diffusion from the tear fluid through the outside surface and the aqueous humour through the inside surface. The cornea is a mechanical barrier and thus restricts the entry of any exogenous molecules into the eye and protects the ocular tissues. The three main parts present in the cornea are epithelium, stroma, and endothelium. The epithelium layer of the cornea is made up of lipid, and restricts the entry of topically administered hydrophilic drugs, acting as a rate limiting factor for the drug administration to the eye. Each layer offers a different polarity. The two layers of cornea, that is, epithelium and stroma, are considered as chief barriers for ophthalmic drug delivery. In embodiments, the composition for use in treatment comprising the therapeutic nucleic acid, preferably the RNA is administered to the cornea via topical administration or injection. The term "topical administration" has to be understood as delivery of the composition comprising at least one therapeutic nucleic acid, preferably RNA according to this invention, wherein the composition is applied on the cornea. In general, topical administration of medical drugs are applied to a particular place on or in the body surfaces such as the skin or mucous membranes. There are numerous dosage forms that can be used topically, including cream, drops, ointment, lotion, patches, dusting powder etc. Topical drug absorption depends on two major factors - biological and physicochemical properties. Low-cost, patient compliance, and ease of administration are reasons for choosing the topical route. Delivery systems such as eye drops, suspensions, and ointments may be used for the treatment of ocular diseases affecting anterior seg ment of the eye. Preferably, topical administration of the composition comprising the therapeutic nucleic acid, preferably the RNA to the cornea leads to a local effect of the therapeutic RNA (or its encoded peptide or protein) on the corneal surface or in cells of the cornea. Anatomical and physiological barriers of the eye prevent the efficiency of drugs to entry into the posterior segment, which in this case is advantageous. Thus, topical ocular delivery mostly results in high ocular bioavailability in the anterior segment of the eye and low ocular bioavailability in the posterior segment of the eye. In general, most of the applied dose of ocular medication which is administered to the cornea is lost due to tear turnover, tear dilution, and nasolacrimal drainage which is preferred if local treatment of the anterior segment is necessary. However, the therapeutic nucleic acid, preferably the RNA directly transfects surrounding cornea cells/tissue which leads to local expression of therapeutic proteins or peptides and thus improves the efficiency of the desired therapeutic effect. Ointment, eye drops, and gel are pharmaceutical dosage forms administrated by the topical route. The most preferred dosage form is eye drops which are made up of solution, emulsion, and suspension. Basically, the short-contact time of drugs with the eye surface following application of conventional topical dosage forms, the permeability problems of drugs across anatomical barriers, and the elimination of drugs by the physiological barriers are general limitations of the topical route which might be overcome by using RNA therapeutics. In other embodiments, the composition for use in treatment comprising the therapeutic nucleic acid, preferably the RNA, is administered to the cornea via injection. The cornea is one of the main barriers for drug diffusion because of its highly impermeable nature. Its continuous irrigation with a tear fluid may result in poor retention of the drugs on the ocular surface. Injection of the composition comprising at least one therapeutic nucleic acid, preferably RNA into the cornea may increase and/or prolong the translation efficiency of therapeutic nucleic acid, preferably RNA and thus also in addition of topical administration enhance the effect of the therapeutic nucleic acid, preferably RNA. In detail, a portion of the administered composition comprising the therapeutic nucleic acid, preferably RNA, administered to the cornea leads to a depot release of the encoded peptide or protein, optionally wherein the encoded peptide or protein is selected from a secreted protein. A depot-release of a medication (e.g the composition for use in treatment comprising the therapeutic nucleic acid) which is described herein releases slowly over time to permit less frequent administration of a medication. In embodiments, the composition comprising the therapeutic nucleic acid, preferably RNA, remains in the cornea or in tissue underlaying the cornea, near the microneedle insertion site, which allows for a slow release and a gradually transfection of surrounding cells. Thus, a slow release of the composition comprising the therapeutic nucleic acid, preferably RNA leads to a slow expression of the therapeutic protein or peptide. In preferred embodiments, the composition comprising the therapeutic nucleic acid, preferably the RNA, is administered to the cornea as part of an eye drop formulation, gels, cream, foam, lotion, paste, powder, sponge, solutions, tincture, spray, suspensions, oinment or coated on an applicator. The term "applicator" as used herein describes a (medical) device used for inserting and applying a substance to a surface. In this invention the composition comprising the therapeutic nucleic acid, preferably the RNA may be coated on an applicator before administration to/on the cornea. An applicator coated with a sustained-release medication/drugs would obviate the need for daily patient administration in favor of device application every few months or years. In embodiments, the composition comprising at least one therapeutic nucleic acid, preferably RNA according to this invention may also be co-extruded within polymer matrices on an applicator to achieve long-term release with desired release kinetics. Hereby, PLGA (Poly(lactic-co-glycolic) Acid) as a Slow-Release Drug-Carrying Matrix has been shown to be suitable sprayed on intraocular lenses (lOLs) as a drug-delivery implant. The material selection for long-acting, sustained-release applicators is crucial fortuning the release kinetics, degradation rate, and biocompatibility of the applicator. In some embodiments, the composition comprising at least one therapeutic nucleic acid, preferably RNA is coated as lipid-based formulation (e.g. liposomes, LNPs) on an applicator. In some embodiments, the composition comprising the therapeutic nucleic acid, preferably RNA is administered to the cornea as part of an eye gel. Gels are semisolid materials and display a nonfluid colloidal network or polymer network that is expanded throughout its whole volume by a fluid. Virtually any fluid can be used as an extender including water (hydrogels), oil, and air (aerogel). Both by weight and volume, gels are mostly fluid in composition and thus exhibit densities similar to those of their constituent liquids. Gels for the treatment of eye diseases are further described in CN 103040986, EP0562445, EP 1275376 or US2020390724. Eye gels comprising the composition comprising the therapeutic nucleic acid, preferably RNA may additionally comprise further active agents (e.g. an immune suppressant, an immune mediator, an antimuscarinic agent, a VGF inhibitor, an antibiotic and combinations thereof). In some embodiments the composition comprising the therapeutic nucleic acid, preferably RNA is administered to the cornea as part of an eye oinment. Oinments are semi-solid preparations of hydrocarbonsh omogeneous, viscous, most commonly a greasy, thick oil (oil 80% - water 20%) with a high viscosity, that is intended for external application to the skin or mucous membranes. Eye ointments are used to treat conditions such as dry eyes or eye infections (such as conjunctivitis). Ointments are thicker than drops which means they can stay on the eye cornea longer. In some preferred embodiments the applicator is a coated contact lens. A contact lens, or contact, is a small, thin plastic or silicone disc shaped medical device worn directly on the cornea of the eye, where they float on a film of tears in front of the cornea. In embodiments of this invention, the composition comprising at least one therapeutic nucleic acid, preferably RNA is be coated or part of a coated contact lens. Contact lenses coated with active substances, like the composition comprising at least one therapeutic nucleic acid, preferably RNA according to this invention, gradually release the active substance (e.g. the therapeutic RNA) into the eye, offer a promising alternative to eye drops, which can be unpleasant and hard for patients to properly administer. Additionally, using contact lenses for drug delivery (e.g delivery of therapeutic mRNA) can provide sustained high concentration in the anterior chamber postoperatively. Coating strategies of contact lenses are further described by US3389012 or W02008079495. The therapeutic nucleic acid, preferably RNA coated on the contact lens would directly transfect surrounding cells and/or tissue of the cornea which leads to translation of the encoded peptide or protein in desired cells and/or tissue. In some embodiments, the contact lens coated with the composition comprising the therapeutic nucleic acid, preferably RNA is a dissolving/biodegradable contact lens. In other embodiments, the applicator is a natural corneal transplant or an artificial cornea. A natural cornea transplant is often referred to as keratoplasty or a corneal graft of a healthy (human) donor tissue. Hereby, in embodiments, the composition comprising the therapeutic nucleic acid, preferably RNA according to this invention is coated on a natural corneal transplant. This may be advantageous in reducing graft failure due to infection. The application of therapeutic nucleic acid, preferably RNA may also reduce post-surgery medication and medication- related or off-target side effects. In other embodiments, the composition comprising the therapeutic nucleic acid, preferably RNA according to this invention is coated on an artificial corneal transplant. Hereby, an artificial or prosthetic cornea is known as a keratoprosthesis and is often made from polymers. In most preferred embodiments, the administration of the composition comprising the therapeutic nucleic acid, preferably RNA to the cornea leads to translation of the encoded peptide or protein in cells and/or tissue of the eye. The composition comprising the therapeutic nucleic acid, preferably RNA that is to be administered to the cornea of the subject may be most effective as the therapeutic nucleic acid, preferably RNA can be delivered directly, close to, or adjacent to the part of the eye that requires a therapeutic treatment, such as the cornea itself or the surrounding tissue of the anterior segment of the eye (e.g., for targeting diseases of the anterior segment or region, see for reference Figure 1). In preferred embodiments, the administration of the therapeutic nucleic acid, preferably RNA to the cornea results in (improved) expression and/or (improved) activity of the encoded peptide or protein in cells and/or tissues of the eye. In preferred embodiments, the administration of the composition comprising the therapeutic nucleic acid, preferably RNA to the cornea results in expression of the therapeutic nucleic acid, preferably RNA and hence to a measurable amount of peptide or protein in cells and/or tissues of the eye. The term "expression" has to be understood as translation of the administered therapeutic nucleic acid, preferably RNA of the composition into measurable amounts of protein. Protein levels may be detected by various methods in the art, including antibody-based detection methods or mass spectrometry. In embodiments, expression of the encoded peptide or protein is essentially restricted to the eye. Accordingly, it is preferred that administration of the composition to the cornea results in expression and hence to a measurable amount of peptide or protein in the eye. In preferred embodiments, the expression of the encoded peptide or protein is essentially restricted to the anterior segment eye. The term "activity" has to be understood as a result of the expressed therapeutic nucleic acid, preferably RNA (that is provided by the composition of the first aspect). In the context of the invention, activity may be determined by biological assays or animal studies. E.g., the activity of a Cas9 protein may be determined by analyzing the occurrence of genetic repair in the treated eye. In embodiments, activity of the encoded peptide or protein is essentially restricted to the eye. Accordingly, it is preferred that administration of the composition to the cornea results in activity of the encoded peptide or protein in the eye. Preferably, expression and/or activity may be in cells or tissues in the front of the eye or the anterior segment. In embodiments, the cells and/or tissue of the cornea encompasses Bowman's Layer, Descemet's membrane, Corneal stroma, Corneal epithelial cells, keratocytes, and endothelial cells. In preferred embodiments, administration of the composition comprising the therapeutic nucleic acid, preferably RNA to the cornea results in expression of the peptide or protein (provided by the therapeutic nucleic acid, preferably RNA of the composition) mainly located or essentially restricted to therapeutically relevant cells or tissues, including e.g. Bowman's Layer, Descemet's membrane, Corneal stroma, Corneal epithelial cells, keratocytes, and endothelial cells, while maintaining low levels of expression in other tissues or cells in the eye. In embodiments, administration of the composition comprising the therapeutic nucleic acid, preferably RNA to the cornea results in expression of the peptide or protein that is substantially increased compared to intravitreal and/or sub- retinal administration. In particular, the peptide or protein expression is substantially increased in the following cell types: Bowman's Layer, Descemet's membrane, Corneal stroma, Corneal epithelial cells, keratocytes, and endothelial cells In embodiments, administration of the composition comprising the therapeutic nucleic acid, preferably RNA to the cornea results in expression of the encoded peptide or protein that is more restricted to the therapeutically relevant cells (e.g. cells and/or tissue of the cornea) compared to intravitreal administration and/or sub-retinal administration. In preferred embodiments, the composition the therapeutic nucleic acid, preferably RNA is administered to or into the cornea, wherein the administration of the composition leads to translation of the encoded peptide or protein, thereby exerting a therapeutic effect. Therapeutic effects in that context may be reduced fibrosis, reduced neovascularization, reduced inflammation, neuroprotection, complement inhibition, reduced drusen formation and/or reduced scar formation. In preferred embodiments, the invention relates to a composition comprising at least one therapeutic nucleic acid, preferably RNA comprising at least one coding sequence encoding at least one peptide or protein for use in treatment or prevention of an ophthalmic disease, disorder or condition, wherein said composition is administered to the cornea of a subject in need thereof, wherein administration of said composition leads to reduced neovascularization. In some embodiments, the invention relates to a composition comprising the therapeutic nucleic acid, preferably RNA comprising at least one coding sequence encoding at least one peptide or protein for use in treatment or prevention of an ophthalmic disease, disorder or condition, wherein said composition is administered to the cornea of a subject in need thereof, wherein administration of said composition leads to reduced choroidal neovascularization (CNV) is the creation of new blood vessels in the choroid layer of the eye. Choroidal neovascularization is a common cause of neovascular degenerative maculopathy (i.e. 'wet' macular degeneration) commonly exacerbated by extreme myopia, malignant myopic degeneration, or age-related developments. In preferred embodiments, the invention relates to a composition comprising at least one therapeutic nucleic acid, preferably RNA comprising at least one coding sequence encoding at least one therapeutic peptide or protein for use in treatment or prevention of an ophthalmic disease, disorder or condition, wherein said composition is administered to the cornea of a subject in need thereof, wherein administration of said composition leads to reduced inflammation. In preferred embodiments, the invention relates to a composition comprising at least one therapeutic nucleic acid, preferably RNA comprising at least one coding sequence encoding at least one therapeutic peptide or protein for use in treatment or prevention of an ophthalmic disease, disorder or condition, wherein said composition is administered to the cornea a subject in need thereof, wherein administration of said composition leads to neuroprotection. In preferred embodiments, the invention relates to a composition comprising at least one therapeutic nucleic acid, preferably RNA comprising at least one coding sequence encoding at least one therapeutic peptide or protein for use in treatment or prevention of an ophthalmic disease, disorder or condition, wherein said composition is administered to the cornea of a subject in need thereof, wherein administration of said composition leads to complement inhibition. In preferred embodiments, the invention relates to a composition comprising at least one therapeutic nucleic acid, preferably RNA comprising at least one coding sequence encoding at least one therapeutic peptide or protein for use in treatment or prevention of an ophthalmic disease, disorder or condition, wherein said composition is administered to the cornea of a subject in need thereof, wherein administration of said composition leads to reduced drusen formation. In preferred embodiments, the invention relates to a composition comprising at least one therapeutic nucleic acid, preferably RNA comprising at least one coding sequence encoding at least one therapeutic peptide or protein for use in treatment or prevention of an ophthalmic disease, disorder or condition, wherein said composition is administered to the cornea of subject in need thereof, wherein administration of said composition leads to reduced scar formation. In preferred embodiments, the invention relates to a composition comprising at least one therapeutic nucleic acid, preferably RNA comprising at least one coding sequence encoding at least one therapeutic peptide or protein for use in treatment or prevention of an ophthalmic disease, disorder or condition, wherein said composition is administered to the cornea of subject in need thereof, wherein administration of said composition leads to reduced fibrosis. In some embodiments, the composition comprising the therapeutic nucleic acid, preferably RNA which is administered to the cornea comprises additional therapeutic proteins and/or peptides. This co-administration of therapeutic proteins and/or peptides may increase and/or prolong the therapeutic effect of the composition of this invention. Suitably, the co-administered therapeutic protein and/or peptide is selected or derived from the same therapeutic protein and/or peptide that is encoded on the therapeutic nucleic acid/RNA. Administration to the conjunctiva In preferred embodiments, the composition comprising the therapeutic nucleic acid, preferably RNA is administered to the conjunctiva of the eye of a subject via sub-conjunctival administration. The conjunctiva is a mucosal tissue that extends from the mucocutaneous junction at the lid margin to the limbal region next to the peripheral cornea and rests on the sclera. In essence, the role of the conjunctiva is to protect the transparency of the cornea, a much more vulnerable tissue that lacks blood and lymphatic vessels, as well as a sufficiently strong in situ immune response for full protection from foreign invaders. The primary function of the conjunctiva is to lubricate the eye by producing mucus and tears. Together, these fluids form a layer, called the tear film, which consists of the innermost mucus layer, the middle watery layer, and the outer oily layer. The conjunctiva is typically divided into three parts: (1) the tarsal, or palpebral, which lines the inner surface of the eyelids; (2) the forniceal, which lines the upper and lower fornices; and (3) the bulbar, which overlays the sclera on the anterior portion of the globe. These three regions are specialized in different functions, ranging from trapping small foreign objects in a net of secreted mucins and facilitating their removal by blinking to providing immune protection to the cornea by the local presence of lymphoid tissue. In preferred embodiments, the composition comprising the therapeutic nucleic acid, preferably RNA is administered sub-conjunctival. The term "sub-conjunctival injection" herein is a type of periocular route of injection for ocular drug administration by administration of a medication either under the conjunctiva (epibulbar) or underneath the conjunctiva lining the eyelid (subpalpebral). Within this invention the administration the composition comprising the therapeutic nucleic acid, preferably RNA is performed sub-conjunctival. Subconjunctival injections are indicated for treatment of lesions in the cornea, sclera, anterior uvea (vascular middle layer of the eye) and vitreous. The periocular region is the feature rich region around the eye which includes features like eyelids, eyelashes, eyebrows, tear duct, eye shape, skin texture etc. Periocular injections can be administered via the posterior subtenon (PST), retrobulbar (RB), subconjunctival (SC), and peribulbar routes. A depot of drug injected periocularly can reach the posterior segment in three ways: transsclerally, hematogenously, or via the anterior segment. In preferred embodiments, the sub-conjunctival administration of the composition comprising the therapeutic nucleic acid, preferably RNA is performed under the eyeball of the conjunctiva (epibulbar) or underneath the conjunctiva lining the eyelid (subpalpebral) via injection or insertion of an applicator. Injection underneath the conjunctiva allows drugs to bypass the epithelium, one of the main barriers that limit drug entry. Subconjunctival injection can be used in severe ophthalmic disease, disorder or condition that require high drug concentrations and also in animals that are difficult to treat. Significant systemic drug exposure occurs due to rapid absorption of sub-conjunctively injected drugs into the ocular venous circulation. Additionally, the subconjunctival injection bypasses the fatty layers of the bulbous conjunctiva and putting medications adjacent to sclera that is permeable to water, this will increase the penetration of water-soluble drugs into the eye. In preferred embodiments, a portion of the administered composition comprising the therapeutic nucleic acid, preferably RNA remains in the conjunctiva as a depot, or remain in tissue underlaying the conjunctiva, near the microneedle insertion site, serving as additional depot of the composition according to this invention. In detail, a portion of the administered composition comprising the therapeutic nucleic acid, preferably RNA, administered to the conjunctiva leads to a depot release of the encoded peptide or protein, optionally wherein the encoded peptide or protein is selected from a secreted protein. A depot-release of a medication (e.g the composition for use in treatment comprising the therapeutic nucleic acid) which is described herein releases slowly over time to permit less frequent administration of a medication. Thus, a slow release of the composition comprising the therapeutic nucleic acid, preferably RNA leads to a slow expression of the therapeutic protein or peptide. The term "applicator" as used herein describes a device used for inserting or for applying a substance to the conjunctiva. In this invention the composition comprising the therapeutic nucleic acid, preferably RNA is coated on an applicator before administration to the conjunctiva or sub-conjunctively injection. A sustained release of the therapeutic nucleic acid/RNA from the applicator obviates the need for daily patient administration in favor of device application every few months or years. In some embodiments, the composition comprising the therapeutic nucleic acid/RNA is coated on punctal plug. Punctal plugs are a type of occlusion device inserted into the tear duct (puncta) of an eye to prevent drainage of tears. Tears consist of aqueous, lipid and mucin layers, containing proteins, glycoproteins and lipids, and are involved in lubrication and protection of the ocular surface. Punctal plugs come in temporary or permanent options. Temporary plugs are made of a collagen material that can dissolve in a matter of weeks or months, while more permanent plugs are made of a more durable material that can last for years at a time. In preferred embodiments, the sub-conjunctival administration of the composition comprising the therapeutic nucleic acid, preferably RNA is performed using an injection needle, a microneedle, an injection device, a catheter, an implant delivery device, or a microcannula. In preferred embodiments, the composition comprising the therapeutic nucleic acid, preferably RNA is administered sub-conjunctival using a microinjection device. A suitable injection device for sub-conjunctival administration is a microinjection device. Microinjection is a direct method to introduce DNA or RNA into either cytoplasm or nucleus. It is a microsurgical procedure conducted on a single cell, using a glass needle (i.e., a fine, glass microcapillary pipette), a precision positioning device (a micromanipulator) to control the movement of the micropipette, and a microinjector. Extrusion of fluid containing the genetic material through the micropipette uses hydrostatic pressure. Injections are typically carried out under direct visual control, using a microscope. The small tip diameters of these micropipettes, combined with the high precision of the micromanipulator, allow accurate and precise DNA/RNA delivery. In preferred embodiments, the sub-conjunctival administration of the composition of the therapeutic nucleic acid, preferably RNA leads to translation of the encoded peptide or protein in cells and/or tissue of the eye. The composition comprising the therapeutic nucleic acid, preferably RNA that is to be administered sub-conjunctival to the eye of a subject may be most effective as the therapeutic nucleic acid, preferably RNA can be delivered directly, close to, or adjacent to the part of the eye that requires a therapeutic treatment, such as the conjunctiva itself or the surrounding tissue of the anterior segment of the eye (e.g., for targeting diseases of the anterior segment or region, see for reference Figure 1). In preferred embodiments, the administration of the therapeutic nucleic acid, preferably RNA to the conjunctiva and/or sub-conjunctival results in (improved) expression and/or (improved) activity of the encoded peptide or protein in cells and/or tissues of the eye. In preferred embodiments, the administration of the therapeutic nucleic acid, preferably RNA to the conjunctiva and/or sub-conjunctival results in expression of the therapeutic nucleic acid, preferably RNA and hence to a measurable amount ofpeptide or protein in cells and/or tissues of the eye. In embodiments, expression of the encoded peptide or protein is essentially restricted to the eye. Accordingly, it is preferred that administration of the composition to the conjunctiva and/or sub-conjunctival results in expression and hence to a measurable amount of peptide or protein in the eye. Accordingly, it is preferred that administration of the composition to the conjunctiva and/or sub-conjunctival results in activity of the encoded peptide or protein in the eye. In embodiments, the cells and/or tissue of the eye are selected from (mucin-secreting) epithelial cells, fibroblasts, melanocytes, dendritic cells, lymphocytes, eosinophils, neutrophils, and mast cells, mesenchymal stem cells (MSCs), connective tissue, vessels, columnar cells or conjunctival goblet cells. In preferred embodiments, administration of the composition comprising the therapeutic nucleic acid, preferably RNA to the conjunctiva and/or sub-conjunctival results in expression of the peptide or protein (provided by the therapeutic nucleic acid/RNA of the composition) mainly located or essentially restricted to therapeutically relevant cells or tissues, including e.g. (mudn-secreting) epithelial cells, fibroblasts, melanocytes, dendritic cells, lymphocytes, eosinophils, neutrophils, and mast cells, mesenchymal stem cells (MSCs), connective tissue, vessels, columnar cells or conjunctival goblet cells, while maintaining low levels of expression in other tissues or cells in the eye. In embodiments, administration of the composition comprising the therapeutic nucleic acid, preferably RNA to the conjunctiva and/or sub-conjunctival results in expression of the peptide or protein that is substantially increased compared to intravitreal and/or sub-retinal administration. In embodiments, administration of the composition comprising the therapeutic nucleic acid, preferably RNA to the conjunctiva and/or sub-conjunctival results in expression of the encoded peptide or protein that is more restricted to the therapeutically relevant cells (e.g. (mucin-secreting) epithelial cells, fibroblasts, melanocytes, dendritic cells, lymphocytes, eosinophils, neutrophils, and mast cells, mesenchymal stem cells (MSCs), connective tissue, vessels, columnar cells or conjunctival goblet cells) compared to intravitreal administration and/or sub-retinal administration. In preferred embodiments, the composition comprising the therapeutic nucleic acid, preferably RNA is administered to the conjunctiva and/or sub-conjunctival, wherein administration of the composition leads to translation of the encoded peptide or protein, thereby exerting a therapeutic effect. Therapeutic effects in that context may be reduced fibrosis, reduced neovascularization, reduced inflammation, neuroprotection, complement inhibition, reduced drusen formation and/or reduced scar formation. In preferred embodiments, the invention relates to a composition comprising the therapeutic nucleic acid, preferably RNA comprising at least one coding sequence encoding at least one peptide or protein for use in treatment or prevention of an ophthalmic disease, disorder or condition, wherein said composition is administered to the conjunctiva and/or sub-conjunctival of a subject in need thereof, wherein administration of said composition leads to reduced neovascularization (e.g. choroidal neovascularization). In preferred embodiments, the invention relates to a composition comprising the therapeutic nucleic acid, preferably RNA comprising at least one coding sequence encoding at least one therapeutic peptide or protein for use in treatment or prevention of an ophthalmic disease, disorder or condition, wherein said composition is administered to the conjunctiva and/or sub-conjunctival of a subject in need thereof, wherein administration of said composition leads to reduced inflammation. In preferred embodiments, the invention relates to a composition comprising the therapeutic nucleic acid, preferably RNA comprising at least one coding sequence encoding at least one therapeutic peptide or protein for use in treatment or prevention of an ophthalmic disease, disorder or condition, wherein said composition is administered to the conjunctiva and/or sub- conjunctiva! a subject in need thereof, wherein administration of said composition leads to neuroprotection. In preferred embodiments, the invention relates to a composition comprising the therapeutic nucleic acid, preferably RNA comprising at least one coding sequence encoding at least one therapeutic peptide or protein for use in treatment or prevention of an ophthalmic disease, disorder or condition, wherein said composition is administered to to the conjunctiva and/or sub-conjunctival of a subject in need thereof, wherein administration of said composition leads to complement inhibition. In preferred embodiments, the invention relates to a composition comprising the therapeutic nucleic acid, preferably RNA comprising at least one coding sequence encoding at least one therapeutic peptide or protein for use in treatment or prevention of an ophthalmic disease, disorder or condition, wherein said composition is administered to the conjunctiva and/or sub-conjunctival of a subject in need thereof, wherein administration of said composition leads to reduced drusen formation. In preferred embodiments, the invention relates to a composition comprising the therapeutic nucleic acid, preferably RNA comprising at least one coding sequence encoding at least one therapeutic peptide or protein for use in treatment or prevention of an ophthalmic disease, disorder or condition, wherein said composition is administered to the to the conjunctiva and/or sub-conjunctival of subject in need thereof, wherein administration of said composition leads to reduced scar formation. In preferred embodiments, the invention relates to a composition comprising the therapeutic nucleic acid, preferably RNA comprising at least one coding sequence encoding at least one therapeutic peptide or protein for use in treatment or prevention of an ophthalmic disease, disorder or condition, wherein said composition is administered to the to the conjunctiva and/or sub-conjunctival of subject in need thereof, wherein administration of said composition leads to reduced fibrosis. In some embodiments, the composition comprising the therapeutic nucleic acid, preferably RNA which is administered to the conjunctiva and/or sub-conjunctival additionally comprises therapeutic proteins and/or peptides. This co-administration of therapeutic proteins and/or peptides may increase and/or prolong the therapeutic effect of the composition of this invention. Suitably, the co-administered therapeutic protein and/or peptide is selected or derived from the same therapeutic protein and/or peptide that is encoded on the therapeutic nucleic acid/RNA. Administration to the ciliary body In preferred embodiments, the composition for use in treatment comprising the therapeutic nucleic acid, preferably RNA is administered to the ciliary body of the eye of a subject via injection or electrotransfection. The ciliary body is a ring-like thickening located between the anterior border of the choroid and the posterior aspect of the iris. On the cross-section, the ciliary body is triangular with its base near the iris and the apex near the choroid. Together with the iris and choroid, the ciliary body comprises the uveal tract. This tract is sandwiched between the sclera and the retina. There are three specialized structures that make up the largest portion ciliary body. These structures include the (1) Ciliary muscle, (2) Ciliary processes and (3) Ciliary epithelium. The ciliary body is responsible for aqueous humour production and outflow, secretion of hyaluronic acid into the vitreous and lens accommodation. It is also a critical component of the blood aqueous barrier. The ciliary body is composed of muscle, vessels and epithelium, wherein the main mass of the is smooth muscle in mammals. The ciliary body is attached to the lens by connective tissue called the zonular fibers (fibers ofZinn). Relaxation of the ciliary muscle puts tension on these fibers and changes the shape of the lens in order to focus light on the retina. Preferably, injection of the composition comprising the therapeutic nucleic acid, preferably RNA is performed into the ciliary muscle. In other embodiments, the injection of the composition comprising the therapeutic nucleic acid, preferably RNA is performed into ciliary processes. In another embodiment, the composition comprising the therapeutic nucleic acid, preferably RNA is performed into the ciliary epithelium. Pigment granules in the ciliary body can behave as considerable reservoirs for lipophilic drugs. Therefore, injection of the composition comprising the therapeutic nucleic acid, preferably RNA into the ciliary muscle may increase and/or prolong the therapeutic effect of the composition of this invention. In some embodiments, the composition comprising the therapeutic nucleic acid, preferably RNA is performed into the ciliary muscle via electrotransfection /electroporation/ electropermeabilization. Plasmid electrotransfection has been shown to be superior to recombinant protein ocular delivery which often requires invasive and painful repeated injections. Electroporation, also called electropermeabilization, is defined as the application of voltage pulses that generate an electric field between two electrodes that disrupts the integrity of a cell membrane, allowing the formation of pores. Electrical disruption of a cell membrane causes the formation of pores through which nucleic acids, proteins, and other small molecules present in the environment surrounding the cells can permeate, gaining access to the intracellular space. In preferred embodiments, the composition comprising the therapeutic nucleic acid, preferably RNA is administered to the ciliary body, preferably into the ciliary muscle, using a microinjection device. In other preferred embodiments the composition comprising the therapeutic nucleic acid, preferably RNA is administered to the ciliary body is performed using an injection needle, a microneedle, an injection device, a catheter, an implant delivery device, or a microcannula. As described herein, a microcannula, simply referred to as a cannula, is a thin, flexible hollow tube with a smooth round tip on one end. The opposite end has a plastic hub, which is attached to a syringe. Unlike a needle that easily pierces the skin, a blunt-tip microcannula opens up a path between structures in tissue. A microcannula is similar in size to a needle, but instead of a sharp point at the end, there is a rounded end with an opening on the side. In preferred embodiments, the administration to the ciliary body of the composition of the therapeutic nucleic acid, preferably RNA leads to translation of the encoded peptide or protein in cells and/or tissue of the eye. The composition comprising the therapeutic nucleic acid, preferably RNA that is to be administered to the ciliary body, preferably into the ciliary muscle of the eye of a subject may be most effective as the therapeutic nucleic acid, preferably RNA can be delivered directly, close to, or adjacent to the part of the eye that requires a therapeutic treatment, such as the ciliary body itself or the surrounding tissue of the anterior segment of the eye (e.g., for targeting diseases of the anterior segment or region, see for reference Figure 1). In preferred embodiments, the administration of the therapeutic nucleic acid, preferably RNA to the ciliary body, preferably into the ciliary muscle, results in (improved) expression and/or (improved) activity of the encoded peptide or protein in cells and/or tissues of the eye. In preferred embodiments, the administration of the therapeutic nucleic acid, preferably RNA to the ciliary body, preferably into the ciliary muscle, results in expression of the therapeutic nucleic acid, preferably RNA and hence to a measurable amount of peptide or protein in cells and/or tissues of the eye. In embodiments, expression of the encoded peptide or protein is essentially restricted to the eye. Accordingly, it is preferred that administration of the composition to the ciliary body, preferably into the ciliary muscle, results in expression and hence to a measurable amount of peptide or protein in the eye. Accordingly, it is preferred that administration of the composition to the ciliary body, preferably into the ciliary muscle, results in activity of the encoded peptide or protein in the eye. In embodiments, the cells and/or tissue of the eye are selected from (smooth) muscle cells (ciliary muscle cells), ciliary vessels, ciliary nerve branches, melanocytes, epithelial cells, fibroblasts, columnar cells. In preferred embodiments, administration of the composition comprising the therapeutic nucleic acid, preferably RNA to the ciliary body, preferably into the ciliary muscle, results in expression of the peptide or protein (provided by the therapeutic RNA of the composition) mainly located or essentially restricted to therapeutically relevant cells or tissues, including e.g. (smooth) muscle cells (ciliary muscle cells), ciliary vessels, ciliary nerve branches, melanocytes, epithelial cells, fibroblasts, columnar cells, while maintaining low levels of expression in other tissues or cells in the eye. In embodiments, administration of the composition comprising the therapeutic nucleic acid, preferably RNA to the ciliary body, preferably into the ciliary muscle, results in expression of the peptide or protein that is substantially increased compared to intravitreal and/or sub-retinal administration. In embodiments, administration of the composition comprising the therapeutic nucleic acid, preferably RNA to the ciliary body, preferably into the ciliary muscle, results in expression of the encoded peptide or protein that is more restricted to the therapeutically relevant cells compared to intravitreal administration and/or sub-retinal administration. In preferred embodiments, the composition comprising the therapeutic nucleic acid, preferably RNA is administered to the ciliary body, preferably into the ciliary muscle, wherein administration of the composition leads to translation of the encoded peptide or protein, thereby exerting a therapeutic effect. Therapeutic effects in that context may be reduced fibrosis, neovascularization, reduced inflammation, neuroprotection, complement inhibition, reduced drusen formation and/or reduced scar formation. In preferred embodiments, the invention relates to a composition comprising the therapeutic nucleic acid, preferably RNA comprising at least one coding sequence encoding at least one peptide or protein for use in treatment or prevention of an ophthalmic disease, disorder or condition, wherein said composition is administered to the ciliary body of a subject in need thereof, wherein administration of said composition leads to reduced neovascularization ((e.g. choroidal neovascularization). In preferred embodiments, the invention relates to a composition comprising the therapeutic nucleic acid, preferably RNA comprising at least one coding sequence encoding at least one therapeutic peptide or protein for use in treatment or prevention of an ophthalmic disease, disorder or condition, wherein said composition is administered to the ciliary body of a subject in need thereof, wherein administration of said composition leads to reduced inflammation. In preferred embodiments, the invention relates to a composition comprising the therapeutic nucleic acid, preferably RNA comprising at least one coding sequence encoding at least one therapeutic peptide or protein for use in treatment or prevention of an ophthalmic disease, disorder or condition, wherein said composition is administered to the ciliary body of a subject in need thereof, wherein administration of said composition leads to neuroprotection. In preferred embodiments, the invention relates to a composition comprising the therapeutic nucleic acid, preferably RNA comprising at least one coding sequence encoding at least one therapeutic peptide or protein for use in treatment or prevention of an ophthalmic disease, disorder or condition, wherein said composition is administered to to the ciliary body of a subject in need thereof, wherein administration of said composition leads to complement inhibition. In preferred embodiments, the invention relates to a composition comprising the therapeutic nucleic acid, preferably RNA comprising at least one coding sequence encoding at least one therapeutic peptide or protein for use in treatment or prevention of an ophthalmic disease, disorder or condition, wherein said composition is administered to the ciliary body of a subject in need thereof, wherein administration of said composition leads to reduced drusen formation. In preferred embodiments, the invention relates to a composition comprising the therapeutic nucleic acid, preferably RNA comprising at least one coding sequence encoding at least one therapeutic peptide or protein for use in treatment or prevention of an ophthalmic disease, disorder or condition, wherein said composition is administered to the to the ciliary body of subject in need thereof, wherein administration of said composition leads to reduced scar formation. In preferred embodiments, the invention relates to a composition comprising the therapeutic nucleic acid, preferably RNA comprising at least one coding sequence encoding at least one therapeutic peptide or protein for use in treatment or prevention of an ophthalmic disease, disorder or condition, wherein said composition is administered to the to the ciliary body of subject in need thereof, wherein administration of said composition leads to reduced fibrosis. In some embodiments, the composition comprising the therapeutic nucleic acid, preferably RNA which is administered to the to the ciliary body may additionally comprise therapeutic proteins and/or peptides. This co-administration of therapeutic proteins and/or peptides may increase and/or prolong the therapeutic effect of the composition of this invention. In preferred embodiments, the administration of the therapeutic nucleic acid, preferably RNA to or into the ciliary body leads to a slow release of constant amounts of the encoded peptide or protein. Therapeutic nucleic acid In embodiments, the therapeutic nucleic acid of this invention can be selected from DNA or RNA. The DNA may be any type of DNA that comprises a coding sequence as defined herein including any type of single stranded DNA, any type of double stranded DNA, any type of linear DMA, and any type of circular DNA. A suitable DNA in the context of the invention may be selected from bacterial plasmid, an adenovirus, a poxvirus, a parapoxivirus (orf virus), a vaccinia virus, a fowlpox virus, a herpes virus, an adeno-assodated virus (AAV), an alphavirus, a lentivirus, a lambda phage, a lymphocytic choriomeningitis virus and a Listeria sp, Salmonella sp. In preferred embodiments, the DNA a viral DNA, preferably an adeno-associated virus DNA. In particularly preferred embodiments, the therapeutic nucleic acid of the invention is an RNA. Encoded peptide or protein In preferred embodiments, the invention relates to a composition comprising at least one therapeutic nucleic acid, preferably RNA comprising at least one coding sequence encoding at least one therapeutic peptide or protein for use in treatment or prevention of an ophthalmic disease, disorder or condition, wherein said composition is administered to the anterior segment of the eye of a subject in need thereof, wherein administration of said composition leads to reduced neovascularization. In preferred embodiments, the invention relates to a composition comprising at least one therapeutic nucleic acid, preferably RNA comprising at least one coding sequence encoding at least one therapeutic peptide or protein for use in treatment or prevention of an ophthalmic disease, disorder or condition, wherein said composition is administered to the anterior segment of the eye of a subject in need thereof, wherein administration of said composition leads to reduced inflammation. In preferred embodiments, the invention relates to a composition comprising at least one therapeutic nucleic acid, preferably RNA comprising at least one coding sequence encoding at least one therapeutic peptide or protein for use in treatment or prevention of an ophthalmic disease, disorder or condition, wherein said composition is administered to the anterior segment of the eye of a subject in need thereof, wherein administration of said composition leads to neuroprotection. In preferred embodiments, the invention relates to a composition comprising at least one therapeutic nucleic acid, preferably RNA comprising at least one coding sequence encoding at least one therapeutic peptide or protein for use in treatment or prevention of an ophthalmic disease, disorder or condition, wherein said composition is administered to the anterior segment of the eye of a subject in need thereof, wherein administration of said composition leads to complement inhibition. In preferred embodiments, the invention relates to a composition comprising at least one therapeutic nucleic acid, preferably RNA comprising at least one coding sequence encoding at least one therapeutic peptide or protein for use in treatment or prevention of an ophthalmic disease, disorder or condition, wherein said composition is administered to the anterior segment of the eye of a subject in need thereof, wherein administration of said composition leads to reduced drusen formation. In preferred embodiments, the invention relates to a composition comprising at least one therapeutic nucleic acid, preferably RNA comprising at least one coding sequence encoding at least one therapeutic peptide or protein for use in treatment or prevention of an ophthalmic disease, disorder or condition, wherein said composition is administered to the anterior segment of the eye of a subject in need thereof, wherein administration of said composition leads to reduced scar formation. In preferred embodiments, the invention relates to a composition comprising at least one therapeutic nucleic acid, preferably RNA comprising at least one coding sequence encoding at least one therapeutic peptide or protein for use in treatment or prevention of an ophthalmic disease, disorder or condition, wherein said composition is administered to the anterior segment of the eye of a subject in need thereof, wherein administration of said composition leads to reduced fibrosis. In embodiments, the composition comprising the therapeutic nucleic acid, preferably RNA comprises at least one coding sequence (cds) which encodes at least one peptide or protein peptide or protein which is or is derived from a therapeutic peptide or protein. In various embodiments, the length of the encoded peptide or protein may be at least or greater than about 20, 30,40, 50,60,70,80, 90, 100,150,200,250, 300, 400,500,600,700, 800, 900,1000, or 1500 amino acids. In embodiments, the at least one cds encodes at least one therapeutic peptide or protein, wherein said therapeutic peptide or protein may be derived from an antibody, an antibody fragment, an intrabody, a receptor, a binding protein, a CRISPR-associated endonuclease, a transcription factor, a transcription factor inhibitor, an enzyme, a growth factor, a structural protein, a cytoplasmic or cytoskeletal protein, or fragments, variants, or combinations of any of these. In embodiments, the at least one cds encodes at least one therapeutic peptide or protein, wherein said therapeutic peptide or protein is derived from a CRISPR-associated endonuclease, wherein said CRISPR-associated endonudease is selected from Cas9, Cpf1, C2c1, C2c3, and C2c2, Cas13, CasX and CasY. In preferred embodiments, the at least one cds encodes at least one therapeutic peptide or protein, wherein said therapeutic peptide or protein is derived from an antibody, an antibody fragment, wherein said antibody, or antibody fragment is against a platelet derived growth factor (PEDF) or a vascular endothelial growth factor (VEGF). In some embodiments, the cds encodes at least one therapeutic peptide or protein, wherein said therapeutic peptide or protein is derived from pigment epithelium-derived factor (PEDF). PEDF is a member of the serpin (serine protease inhibitor) family. It is a multifunctional secreted protein that has anti-angiogenic and anti-tumorigenic functions. It is being investigated as a therapeutic candidate for treatment of such conditions as choroidal neovascularization, heart disease, and cancer. PEDF may help suppress unwanted neovascularization of the eye. Therefore, may be a useful tool to prevent choroidal neovascularization. Hereby, Choroidal neovascularization (CNV) is the creation of new blood vessels in the choroid layer of the eye. Choroidal neovascularization is a common cause of neovascular degenerative maculopathy (i.e. 'wet' macular degeneration) commonly exacerbated by extreme myopia, malignant myopic degeneration, or age-related developments. Suitably, the therapeutic RNA comprised in the composition encodes at least one therapeutic protein as provided in List 1 below. Therein, Protein/Gene Symbol and the corresponding Name (Symbol: Name) are provided List 1: Suitable therapeutic proteins provided by the therapeutic RNA of the invention ABCA4: ATP binding cassette subfamily A member 4; ADRB1: adrenoceptor beta 1; ANGPT1 : angiopoietin 1; ANGPT2: angiopoietin 2; BEST1 : bestrophin 1; CCR3: C-C motif chemokine receptor 3; CD276: CD276 molecule; CD59: CD59 molecule (CD59 blood group); CFD: complement factor D; CHM: CHM, Rab escort protein 1;CHST4: carbohydrate sulfotransferase 4; CNR1: cannabinoid receptor 1; CNTF: ciliary neurotrophic factor; CRYAA: crystallin alpha A; CRYAB: crystallin alpha B; CSF3R: colony stimulating factor 3 receptor; DCN: decorin; DICER1 : dicer 1, ribonuclease III; DRD2: dopamine receptor D2; EGFR: epidermal growth factor receptor; EGLN1: egl-9 family hypoxia inducible factor 1; ENG: endoglin; FLT1: fms related tyrosine kinase 1; FLT1 (1 -758): fms related tyrosine kinase 1 (secreted); FLT1-iso3(sFlt1-14): fms related tyrosine kinase 1 isoform 3 (secreted; P17948-3); FLT1-iso2(sFlt1): fms related tyrosine kinase 1 isoform 2 (secreted; P17948-2); FLT1-iso4: fms related tyrosine kinase 1 isoform 4 (secreted; P17948-4); GUCY1A1: guanylate cyclase 1 soluble subunit alpha 1; GUCY1A2: guanylate cyclase 1 soluble subunit alpha 2; GUCY1B1: guanylate cyclase 1 soluble subunit beta 1; GUCY1B2: guanylate cyclase 1 soluble subunit beta 2 (pseudogene); GUCY2D: guanylate cyclase 2D, retinal; GUCY2F: guanylate cyclase 2F, retinal; HEY1: hes related family bHLH transcription factor with YRPW motif 1; HEPH: hephaestin; HEPHL1: hephaestin like 1; IL1 RN: interleukin 1 receptor antagonist; KDR: kinase insert domain receptor; MAG: myelin associated glycoprotein; MERTK: MER proto- oncogene, tyrosine kinase; MY07A: myosin VIIA; MYOC: myocilin; NOTCH4: notch 4; NR3C1: nuclear receptor subfamily 3 group C member 1; NXNL1: nudeoredoxin like 1; OPA1: OPA1, mitochondrial dynamin like GTPase; OPA3: OPA3, outer mitochondrial membrane lipid metabolism regulator; OPTN: optineurin; PDGFA: platelet derived growth factor subunit A; PDGFB: platelet derived growth factor subunit B; PDGFC: platelet derived growth factor C; PDGFD: platelet derived growth factor D; PDGFRA: platelet derived growth factor receptor alpha; PDGFRB: platelet derived growth factor receptor beta; PGF: placental growth factor; PDCND1 : plexin D1; PPP3CA: protein phosphatase 3 catalytic subunit alpha; PPP3CB: protein phosphatase 3 catalytic subunit beta; PPP3CC: protein phosphatase 3 catalytic subunit gamma; PPP3R1 : protein phosphatase 3 regulatory subunit B, alpha; PPP3R2: protein phosphatase 3 regulatory subunit B, beta; PRPH2: peripherin 2; PTGDR: prostaglandin D2 receptor; PTGDR2: prostaglandin D2 receptor 2; PTGER1: prostaglandin E receptor 1; PTGER2: prostaglandin E receptor 2; PTGER3: prostaglandin E receptors; PTGER4: prostaglandin E receptor 4; PTGFR: prostaglandin F receptor; PTGIR: prostaglandin 12 receptor; RHO: rhodopsin; RLBP1: retinaldehyde binding protein 1; ROCK1: Rho associated coiled-coil containing protein kinase 1; ROCK2: Rho associated coiled-coil containing protein kinase 2; RPE65: RPE65, retinoid isomerohydrolase; RPGR: retinitis pigmentosa GTPase regulator; RS1: retinoschisin 1; SEMA3E: semaphorin 3E; SEMA3: semaphorin 3; SERPINF1: serpin family F member 1; SIRT1: sirtuin 1; SIRT2: sirtuin 2; SIRT3: sirtuin 3; SIRT4: sirtuin 4; SIRT5: sirtuin 5; SIRT6: sirtuin 6; SIRT7: sirtuin 7; SOD1: superoxide dismutase 1; TBXA2R: thromboxane A2 receptor; TEK: TEK receptor tyrosine kinase; Tl E1:tyrosinekinase with immunoglobulin like and EOF like domains 1;TNF: tumor necrosis factor; TNFRSF10C: TNF receptor superfamily member 10c; UNC5B: unc-5 netrin receptor B; USH2A: usherin; VEGFA: vascular endothelial growth factor A; VEGFA-isoVEGF165: vascular endothelial growth factor A; VEGFA-isoVEGF165B: vascular endothelial growth factor A; VEGFB: vascular endothelial growth factor B; VEGFC: vascular endothelial growth factor C; VEGFD: vascular endothelial growth factor D; WFS1: wolframin ER transmembrane glycoprotein; aflibercept: aflibercept; etanercept: etanercept; bevacizumab-LC: bevacizumab; bevacizumab-HC: bevacizumab; bevacizumab-RKR: bevacizumab; ranibizumab-LC: ranibizumab; ranibizumab-HC: ranibizumab; ranibizumab-RKR: ranibizumab; anti-CCL11-LC: anti-CCL11; anti-CCL11-HC: anti-CCL11; anti-PDGFB- LC: anti-PDGFB; anti-PDGFB-HC: anti-PDGFB; anti-PDGFA-LC: anti-PDGFB1/anti-PDGFE1; anti-PDGFA-HC: anti- PDGFB1/anti-PDGFE1; bersanlimab-LC: bersanlimab; bersanlimab-HC: bersanlimab; clazakizumab-LC: clazakizumab; clazakizumab-HC: clazakizumab; olokizumab-LC: olokizumab; Preferred therapeutic peptide or proteins that may be encoded by the cds of the at least one therapeutic RNA are provided in Table 1. Additional information regarding each of these suitable amino acid sequences may also be derived from the sequence listing, e.g. the corresponding nucleic acid transcript IDs ("Ensembl Transcript ID") is provided therein under identifier <223> ofW02020161342 for each sequence. In preferred embodiments, the ophthalmic disease, disorder or condition may be any deficiency associated with any one of the proteins provided in List 1. Accordingly, in embodiments, the ophthalmic disease, disorder or condition may be an ABCA4 deficiency, an ADRB1 deficiency, an ANGPT1 deficiency, an ANGPT2 deficiency, a BEST1 deficiency, a CCR3 deficiency, a CD276 deficiency, a CD59 deficiency, a CFD deficiency, a ChlM deficiency, a CHST4 deficiency, a CNR1 deficiency, a CNTF deficiency, a CRYAA deficiency, a CRYAB deficiency, a CSF3R deficiency, a DCN deficiency, a DICER1 deficiency, a DRD2 deficiency, an EGFR deficiency, an EGLN1 deficiency, an ENG deficiency, a FLT1 deficiency, a GUCY1A1 deficiency, a GUCY1A2 deficiency, a GUCY1B1 deficiency, a GUCY1B2 deficiency, a GUCY2D deficiency, a GUCY2F deficiency, a HEY1 deficiency, a HEPH deficiency, a HEPHL1 deficiency, an IL1RN deficiency, a KDR deficiency, a MAG deficiency, a MERTK deficiency, a MY07A deficiency, a MYOC deficiency, a NOTCH4 deficiency, a NR3C1 deficiency, a NXNL1 deficiency, an OPA1 deficiency, an OPA3 deficiency, an OPTN deficiency, a PDGFA deficiency, a PDGFB deficiency, a PDGFC deficiency, a PDGFD deficiency, a PDGFRA deficiency, a PDGFRB deficiency, a PGF deficiency, a PD(ND1 deficiency, a PPP3CA deficiency, a PPP3CB deficiency, a PPP3CC deficiency, a PPP3R1 deficiency, a PPP3R2 deficiency, a PRPH2 deficiency, a PTGDR deficiency, a PTGDR2 deficiency, a PTGER1 deficiency, a PTGER2 deficiency, a PTGER3 deficiency, a PTGER4 deficiency, a PTGFR deficiency, a PTGIR deficiency, a RHO deficiency, a RLBP1 deficiency, a ROCK1 deficiency, a ROCK2 deficiency, a RPE65 deficiency, a RPGR deficiency, a RS1 deficiency, a SEMA3E deficiency, a SEMA3A deficiency, SERPINF1 deficiency, a SIRT1 deficiency, a SIRT2 deficiency, a SIRT3 deficiency, a SIRT4 deficiency, a SIRT5 deficiency, a SIRT6 deficiency, a SIRT7 deficiency, a SOD1 deficiency, a TBXA2R deficiency, a TEK deficiency, a TIE1 deficiency, a TNF deficiency, a TNFRSF10C deficiency, an UNC5B deficiency, an USH2A deficiency, a VEGFA deficiency, a VEGFA-isoVEGF165 deficiency, a VEGFA-isoVEGF165B deficiency, a VEGFB deficiency, a VEGFC deficiency, a VEGFD deficiency, a WFS1 deficiency, a CCL11 deficiency, a DPP4 deficiency, an IL6R deficiency, a TNFRSF10A deficiency, a TNFRSF10B deficiency, a TNFRSF1A deficiency, or a TNFRSF1 B deficiency. In Table 1 below, each row corresponds to suitable therapeutic proteins that may be encoded by the therapeutic RNA of the composition (row 1 to 125). Column A provides Gene/Protein Symbols (derived from Ensembl Gene ID) of the suitable therapeutic proteins. The "Gene/Protein Symbol" used to describe suitable therapeutic proteins of the invention are also used throughout the description of the invention (as described above) as well as in the ST25 sequence listing under identifier <223> ofW02020161342 or as in the ST26 sequence listing of this invention (SEQ ID NO: 127-490). Column B provides a further description of the respective therapeutic protein. Column C indicates the corresponding Ensembl Gene ID [ENSGOOOOO...]. Column D provides protein SEQ ID NOs ofW02020161342 (SEQ ID No 2-365 and 436) of the respective amino acid sequences of the indicated therapeutic proteins. These sequences correspond to sequences SEQ ID NO: 127-490 of the ST26 sequence listing of this invention. As an example, row 1 of Table 1 provides "ABCA4" (row 1, Column A), an "ATP binding cassette subfamily A member 4" (row 1, Column B), that can be identified by the corresponding Ensembl Gene ID "ENSGOOOOO"198691.1 (row 1, Column C), having the amino acid sequences according to SEQ ID NOs: 2 or 3 ofW02020161342 (row 1, Column D). Table 1: Suitable therapeutic proteins and sequences provided by the therapeutic RNA of the composition




Accordingly, in preferred embodiments, the peptide or protein is selected from ABCA4, ADRB1, ANGPT1 , ANGPT2, BEST1, CCR3, CD276, CD59, CFD, CHM, CHST4, CNR1, CNTF, CRYAA, CRYAB, CSF3R, DCN, DICER1, DRD2, EGFR, EGLN1, ENG, FLT1, FLT1 (1-758), FLT1-iso3(sFlt1-14), FLT1-iso2(sFlt1), FLT1-iso4, GUCY1A1, GUCY1A2, GUCY1B1, GUCY1B2, GUCY2D, GUCY2F, HEY1, HEPH, HEPHL1, IL1RN, KDR, MAG, MERTK, MY07A, MYOC, NOTCH4, NR3C1, NXNL1, OPA1, OPA3, OPTN, PDGFA, PDGFB, PDGFC, PDGFD, PDGFRA, PDGFRB, PGF, PD(ND1,PPP3CA, PPP3CB,PPP3CC,PPP3R1, PPP3R2, PRPH2, PTGDR, PTGDR2, PTGER1, PTGER2, PTGER3, PTGER4, PTGFR, PTGIR, RHO, RLBP1, ROCK1, ROCK2, RPE65, RPGR, RS1, SEMA3E, SEMA3A, SERPINF1, SIRT1, SIRT2, SIRT3, SIRT4, SIRT5, SIRT6, SIRT7, SOD1, TBXA2R, TEK, TIE1, TNF, TNFRSF10C, UNC5B, USH2A, VEGFA, VEGFA-isoVEGF165, VEGFA-isoVEGF165B, VEGFB, VEGFC, VEGFD, WFS1 , aflibercept, etanercept, bevacizumab, ranibizumab, anti-CCL11, anti-PDGFB, anti-PDGFA, bersanlimab, clazakizumab, olokizumab, sarilumab, siltuximab, sirukumab, DPP4-Fc, IL6R-Fc, PDGFRB-Fc, TNFRSFIOA-Fc, TNFRSFIOB-Fc, TNFRSF1A-FC, TNFRSFIB-Fc, UNC5B-FC, PDGFRB-trap(lgG4), PDGFRB-trap(lgGI), Cas9, Cpf1, C2c1, C2c3, and C2c2, Cas13, CasX and CasY, or a fragment or variant of any of these. Accordingly, in preferred embodiments, the at least one therapeutic nucleic acid, preferably RNA of the composition comprises at least one cds, wherein the at least one cds encodes at least one therapeutic peptide or protein comprising or consisting of an amino acid sequences being identical or at least 70%, 80%, 85%, 86%, 87%, 88%, 89%, 90%, 91 %, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identical to any one of SEQ ID NOs: 127^90 or a fragment or variant thereof of any of these sequences. In other preferred embodiments, the at least one therapeutic nucleic acid, preferably RNA of the composition comprises at least one cds, wherein the at least one cds encodes at least one CRISPR-assodated endonuclease comprising or consisting of an amino acid sequences being identical or at least 70%, 80%, 85%, 86%, 87%, 88%, 89%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identical to any one of SEQ ID NOs: 428-1345,10999-11001, of published PCT patent application W02018/172556. Accordingly, SEQ ID NOs: 428-1345, 10999-11001 of W02018/172556 are herewith incorporated by reference. In preferred embodiments, the therapeutic nucleic acid, preferably RNA of the composition which is administered to the cornea leads to the expression of secreted proteins or peptides, preferably therapeutic proteins or peptides. In embodiments, the at least one cds encodes at least one peptide or protein, wherein said at least one peptide or protein is a therapeutic peptide or protein. In other embodiments, the at least one cds encodes at least one antigenic peptide or protein, wherein said antigenic peptide or protein may be derived from tumor antigen, a viral antigen, a bacterial antigen, a fungal antigen, or a parasite antigen. The term "antigen" as used herein will be recognized and understood by the person of ordinary skill in the art,and is e.g. intended to refer to a substance which may be recognized by the immune system, preferably by the adaptive immune system, and is capable of triggering an antigen-specific immune response, e.g. by formation of antibodies and/or antigen-specific T cells as part of an adaptive immune response. Typically, an antigen may be or may comprise a peptide or protein which may be presented by the MHC to T-cells. Also fragments, variants and derivatives of peptides or proteins derived from e.g. cancer antigens comprising at least one epitope may be understood as antigens. In the context of the present invention, an antigen may be the product of translation of a provided RNA of the first aspect. The term "antigenic peptide or protein" will be recognized and understood by the person of ordinary skill in the art, and is e.g. intended to refer to a peptide or protein derived from a (antigenic) protein which may stimulate the body's adaptive immune system to provide an adaptive immune response. Therefore an "antigenic peptide or protein" comprises at least one epitope or antigen of the protein it is derived from (e.g. tumor specific antigen). Suitably, tumor antigens may be derived from antigens causing eyelid tumors, conjunctival/corneal cancers, uveal cancers, retinal cancers, and orbital/adnexal cancers, in particular Squamous cell carcinoma, squamous neoplasia, Intraocular Melanoma or Retinoblastoma, Sebaceous Carcinoma of the eyelid, Uveal melanoma, iris melanoma, ciliary body melanoma, and choroidal melanoma, or tumor antigens causing a cancer with metastases in the eye (e.g. choroidal metastases, ocular metastases, orbital metastases). According to other embodiments, the therapeutic nucleic acid, preferably RNA of the composition encodes at least one peptide or protein as defined above and additionally at least one further heterologous peptide or protein element. Suitably, the at least one further heterologous peptide or protein element may promote/improve secretion of the peptide or protein (e.g. via secretory signal peptides), promote/improve anchoring of the peptide or protein in the plasma membrane (e.g. via transmembrane elements), promote/improve formation of complexes (e.g. via multimerization domains), promote/improve virus-like particle formation (VLP forming sequence), a nuclear localization signal (NLS). In addition, the RNA according may encode peptide linker elements, self-cleaving peptides, immunologic adjuvant sequences or dendritic cell targeting sequences. Suitable multimerization domains may be selected from the list of amino acid sequences according to SEQ ID NOs: 1116-1167 of W02017/081082, or fragments or variants of these sequences. Suitable transmembrane elements may be selected from the list of amino acid sequences according to SEQ ID NOs: 1228-1343 of W02017/081082, or fragments or variants of these sequences. Suitable VLP forming sequences may be selected from the list of amino acid sequences according to SEQ ID NOs: 1168-1227 of W02017/081082, or fragments or variants of these sequences. Suitable peptide linkers may be selected from the list of amino acid sequences according to SEQ ID NOs: 1509-1565 of W02017/081082, or fragments or variants of these sequences. Suitable self-cleaving peptides may be selected from the list of amino acid sequences according to SEQ ID NOs: 1434-1508 of W02017/081082, or fragments or variants of these sequences. Suitable immunologic adjuvant sequences may be selected from the list ofamino acid sequences according to SEQ ID NOs: 1360-1421 of W02017/081082, or fragments or variants of these sequences. Suitable dendritic cell targeting sequences may be selected from the list ofamino acid sequences according to SEQ ID NOs: 1344-1359 of W02017/081082, or fragments or variants of these sequences. Suitable secretory signal peptides may be selected from the list of amino acid sequences according to SEQ ID NOs: 1-1115 and SEQ ID NO: 1728 of W02017/081082, or fragments or variants of these sequences. Suitable nuclear localization signal (NLS) sequences may be selected from the list ofamino acid sequences according to SEQ ID NOs:426,427,10575,381 , 382, 384,11957,11958-11964 of published PCT patent application W02018/172556 or fragments or variants of these sequences. According to preferred embodiments, the therapeutic nucleic acid, preferably RNA of the composition comprises at least 5 one cds encoding at least one peptide or protein as specified herein, or fragments and variants thereof. In that context, any cds encoding at least one peptide or protein as defined herein or fragments and variants thereof may be understood as suitable cds and may therefore be comprised in the therapeutic nucleic acid, preferably RNA. In embodiments, the length the cds may be at least or greater than about 30, 40,50,60,70, 80, 90,100,150,200, 250, 300,350,400,450,500, 600,700,800,900, 1000, 1100,1200,1300,1400,1500, 1600,1700,1800,1900,2000, 2500,3000,3500, 4000, 5000, or 6000 nucleotides. In embodiments, the length of the cds may be in a range of from 10 about 300 to about 2000 nudeotides. The terms "coding sequence" or "coding region" and "cds" as used herein will be recognized and understood by the person of ordinary skill in the art, and are e.g. intended to refer to a sequence of several nucleotides which may be translated into a peptide or protein. In the context of the present invention a cds is preferably an RNA sequence, consisting of a number of nucleotide triplets, starting with a start codon and preferably terminating with one stop codon. 15 In embodiments, the cds of the RNA may terminate with or two or more stop codons. The first stop codon of the two or more stop codons may be TGA or UGA and the second stop codon of the two or more stop codons may be selected from TAA, TGA, TAG, UAA, UGA or UAG. In preferred embodiments, the therapeutic nucleic acid, preferably RNA of the composition may comprise or consist of at least one cds encoding any one ofSEQ ID NOs: 127^90 or SEQ ID NOs: 428-441,10999-11001,442-1345 of 20 W02018/172556 or fragments of variants thereof. It has to be understood that, on nucleic acid level, any RNA sequence which encodes an amino acid sequences being identical to SEQ ID NOs: 127-490 orSEQ ID NOs: 428-441, 10999-11001, 442-1345 of W02018/172556 or fragments or variants thereof, or any RNA sequence which encodes amino acid sequences being at least 70%, 80%, 85%, 86%, 87%, 88%, 89%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identical to any one ofSEQ ID NOs:127- 25490 or SEQ ID NOs: 428^41, 10999-11001, 442-1345 of W02018/172556 or fragments or variants thereof, may be selected and may accordingly be understood as suitable cds, and may therefore be comprised in the therapeutic RNA of the composition. RNA In embodiments, the therapeutic RNA of the composition of this invention is an isolated RNA or an artificial RNA. 30 Inn preferred embodiments, the therapeutic RNA of the composition is an artificial RNA. The term "artificial RNA" as used herein is intended to refer to an RNA that does not occur naturally. In other words, an artificial RNA may be understood as a non-natural RNA molecule. Such RNA molecules may be non-natural due to its individual sequence (e.g. G/C content modified coding sequence, UTRs) and/or due to other modifications, e.g. structural modifications of modified nucleotides. Artificial RNA may be designed and/or generated by genetic 35 engineering to correspond to a desired artificial sequence of nucleotides. In this context an artificial RNA is a sequence that may not occur naturally, i.e. it differs from the wild type sequence by at least one nucleotide. The term "artificial RNA" is not restricted to mean "one single molecule" but is understood to comprise an ensemble of essentially identical molecules. Accordingly, it may relate to a plurality of essentially identical RNA molecules. In preferred embodiments, the therapeutic RNA of the composition is selected from mRNA, circular RNA, replicon RNA, 40 or viral RNA. In embodiments, the therapeutic RNA of the composition is a circular RNA. As used herein, "circular RNA" or "circRNAs" have to be understood as a circular polynucleotide construct that encode at least one antigenic peptide or protein as defined herein. Accordingly, in preferred embodiments, said circRNA comprises at least one cds encoding at least one peptide or protein as defined herein. The production of circRNA can be performed using various methods provided in the art. Accordingly, methods for producing circular RNA as provided in US6210931 , US5773244, W01992/001813, W02015/034925 and W02016/011222 are incorporated herewith by reference. In embodiments, the therapeutic RNA of the composition is a replicon RNA. The term "replicon RNA" will be recognized and understood by the person of ordinary skill in the art, and is e.g. intended to be an optimized self-replicating RNA. Such constructs may include replicase elements derived from e.g. alphaviruses (e.g. SFV, SIN, VEE, or RRV) and the substitution of the structural virus proteins with the nucleic acid of interest. Alternatively, the replicase may be provided on an independent therapeutic RNA construct. Downstream of the replicase may be a sub-genomic promoter that controls replication of the replicon RNA. In preferred embodiments, the therapeutic RNA of the composition is an mRNA. The terms "RNA" and "mRNA" will be recognized and understood by the person of ordinary skill in the art, and are e.g. intended to be a ribonucleic acid molecule, i.e. a polymer consisting of nucleotides. These nudeotides are usually adenosine-monophosphate, uridine-monophosphate, guanosine-monophosphate and cytidine-monophosphate monomers which are connected to each other along a so-called backbone. The backbone is formed by phosphodiester bonds between the sugar, i.e. ribose, of a first and a phosphate moiety of a second, adjacent monomer. The specific succession of the monomers is called the RNA-sequence. The mRNA (messenger RNA) usually provides the nucleotide sequence that may be translated into an amino-add sequence of a particular peptide or protein after administration to the anterior segment of the eye. In preferred embodiments, the therapeutic RNA of the composition, preferably the mRNA has a length of 10000 and 50000 nucleotides Preferably, the therapeutic RNA of the composition for use in treatment may comprise about 50 to about 20000 nucleotides, or about 500 to about 10000 nucleotides, or about 1000 to about 10000 nucleotides, or preferably about 1000 to about 5000 nucleotides, or even more preferably about 1000 to about 3000 nucleotides. In embodiments, the therapeutic RNA of the composition is a modified and/or stabilized RNA. According to preferred embodiments, the therapeutic RNA of the composition may thus be provided as a "stabilized artificial RNA" or "stabilized therapeutic RNA" that is to say an RNA showing improved resistance to in vivo degradation and/or an RNA showing improved stability in vivo (suitably after administration to the anterior segment of the eye), and/or an RNA showing improved translatability in vivo (suitably after administration to the anterior segment of the eye). In the following, modifications are described which are suitably to "stabilize" the therapeutic RNA of the composition. In embodiments, stabilization may be affected by providing a "dried RNA" and/or a "purified RNA" as specified herein A purified therapeutic RNA may be a HPLC purified therapeutic RNA, and/or a pharmaceutical grade therapeutic RNA. Alternatively or in addition to that, stabilization can be effected, e.g., by a modified phosphate backbone of the therapeutic RNA of the composition. A backbone modification may be a modification in which phosphates of the backbone of the nucleotides contained in RNA are chemically modified. Nucleotides that may be preferably used comprise e.g. a phosphorothioate-modified phosphate backbone, preferably at least one of the phosphate oxygens contained in the phosphate backbone being replaced by a sulfur atom. Stabilized RNAs may further include, e.g.: non- ionic phosphate analogues, such as, e.g., alkyl and aryl phosphonates, in which the charged phosphonate oxygen is replaced by an alkyl or aryl group, or phosphodiesters and alkylphosphotriesters, in which the charged oxygen residue is present in alkylated form. Such backbone modifications typically include modifications from the group consisting of methylphosphonates, phosphoramidates and phosphorothioates (e.g. cytidine-5'-0-(1-thiophosphate)). In the following, suitable modifications are described that are capable of "stabilizing" the therapeutic RNA of the composition. According to embodiments, the therapeutic RNA of the composition is a modified therapeutic RNA, wherein the modification refers to chemical modifications comprising backbone modifications as well as sugar modifications or base modifications. A modified therapeutic RNA may comprise nucleotide analogues/modifications, e.g. backbone modifications, sugar modifications or base modifications. A backbone modification in the context of the invention is a modification, in which phosphates of the backbone of the nucleotides are chemically modified. A sugar modification in the context of the invention is a chemical modification of the sugar of the nucleotides of the RNA. Furthermore, a base modification in the context of the invention is a chemical modification of the base moiety of the nucleotides of the RNA. In this context, nucleotide analogues or modifications are preferably selected from nucleotide analogues which are applicable for transcription and/or translation. In some embodiments, at least one chemical modification is selected from pseudouridine, Nl-methylpseudouridine, N1- ethylpseudouridine, 2-thiouridine, 4'-thiouridine, 5-methylcytosine, 5-methyluridine, 2-thio-1-methyl-1-deaza- pseudouridine, 2-thio-l-methyl-pseudouridine, 2-thio-5-aza-uridine, 2-thio-dihydropseudouridine, 2-thio- dihydrouridine, 2-thio-pseudouridine, 4-methoxy-2-thio-pseudouridine, 4-methoxy- pseudouridine, 4-thio-l-methyl-pseudouridine, 4- thio-pseudouridine, 5-aza-uridine, dihydropseudouridine, 5-methoxyuridine and 2'-0-methyl uridine. In some embodiments, 100% of the uracil in the cds have a chemical modification, preferably a chemical modification is in the 5-position of the uracil. Particularly preferred in the context of the invention are pseudouridine (4;), Nl-methylpseudouridine (mlip), 5- methylcytosine, and 5-methoxyuridine. Accordingly, the therapeutic RNA or the coding sequence may comprise at least one modified nucleotide selected from pseudouridine (ip), N1- methylpseudouridine (m1qj), 5-methylcytosine, and 5- methoxyuridine. In preferred embodiments, the therapeutic RNA of the composition, preferably the mRNA, comprises at least one modified nucleotide, preferably selected from pseudouridine (ip) or Nl-methylpseudouridine (m1t|;). In preferred embodiments, the therapeutic RNA of the composition, preferably the mRNA comprises N1- methylpseudouridine (m1i(J). In some embodiments, the therapeutic RNA of the composition comprises at least one coding sequence. In preferred embodiments, at least one coding sequence of the therapeutic RNA is a codon modified coding sequence, wherein the amino acid sequence encoded by the at least one codon modified coding sequence is preferably not being modified compared to the amino acid sequence encoded by the corresponding wild type coding sequence. The term "codon modified coding sequence" relates to coding sequences that differ in at least one codon (triplets of nucleotides coding for one amino acid) compared to the corresponding wild type cds. Suitably, a codon modified cds in the context of the invention may show improved resistance to in vivo degradation and/or improved stability in vivo, and/or improved translatability in vivo. Codon modifications in the broadest sense make use of the degeneracy of the genetic code wherein multiple codons encoding the same amino acid may be used interchangeably (cf. Table 2 of W02020161342) to optimize/modify the cds for in vivo applications as outlined above. In preferred embodiments, the at least one codon modified coding sequence of the therapeutic RNA is selected from C maximized coding sequence, CAI maximized coding sequence, human codon usage adapted coding sequence, G/C content modified coding sequence, and G/C optimized coding sequence, or any combination thereof. In particularly preferred embodiments, the at least one cds is a codon modified cds, wherein the codon modified cds is selected from C maximized cds, CAI maximized cds, human codon usage adapted cds, G/C content modified cds, and G/C optimized cds, or any combination thereof. Accordingly, the cds of the therapeutic RNA encoding e.g. amino acid sequences according to SEQ ID NOs: 127-490 may be a C maximized cds, a CAI maximized cds, a human codon usage adapted cds, a G/C content modified cds, and a G/C optimized cds, or any combination thereof. In preferred embodiments, the therapeutic RNA of the composition may be modified, wherein the C content of the at least one cds may be increased, preferably maximized, compared to the C content of the corresponding wild type cds (herein referred to as "C maximized coding sequence"). The amino acid sequence encoded by the C maximized cds of the RNA is preferably not modified as compared to the amino acid sequence encoded by the respective wild type nucleic acid cds. The generation of a C maximized nucleic acid sequences may be carried out using a method according to W02015/062738, the disclosure of W02015/062738 included herewith by reference. Accordingly, the therapeutic RNA of the composition may comprise or consist of at least one cds encoding at least one peptide or protein having an amino acid sequence being at least 70%, 80%, 85%, 86%, 87%, 88%, 89%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identical to any one ofSEQ ID NOs: 127-490 wherein the cds is a C maximized coding sequence as explained above. In preferred embodiments, the therapeutic RNA of the composition may be modified, wherein the G/C content of the at least one cds may be optimized compared to the G/C content of the corresponding wild type cds (herein referred to as "G/C content optimized coding sequence"). "Optimized" in that context refers to a cds wherein the G/C content is preferably increased to the essentially highest possible G/C content. The amino acid sequence encoded by the G/C content optimized cds of the RNA is preferably not modified as compared to the amino acid sequence encoded by the respective wild type cds. The generation of a G/C content optimized RNA sequence may be carried out using a G/C content optimization method according to W02002/098443. In this context, the disclosure of W02002/098443 is included in its full scope in the present invention. Accordingly, the therapeutic RNA of the composition may comprise or consist of at least one cds encoding at least one peptide or protein having an amino acid sequences being at least 70%, 80%, 85%, 86%, 87%, 88%, 89%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identical to any one of SEQ ID NOs: 127-490 wherein the cds is a "G/C content optimized coding sequence" as explained above. In embodiments, the therapeutic RNA of the composition may be modified, wherein the codons in the at least one cds may be adapted to human codon usage (herein referred to as "human codon usage adapted coding sequence"). Codons encoding the same amino acid occur at different frequencies in a subject, e.g. a human. Accordingly, the cds of the RNA is preferably modified such that the frequency of the codons encoding the same amino acid corresponds to the naturally occurring frequency of that codon according to the human codon usage e.g. as shown in Table 2 of W02020161342. E.g., in the case of the amino acid Ala, the wild type cds is preferably adapted in a way that the codon "GCC" is used with a frequency of 0.40, the codon "GCT" is used with a frequency of 0.28, the codon "GCA" is used with a frequency of 0.22 and the codon "GCG" is used with a frequency of 0.10 etc. (see Table 2 ofW02020161342). Accordingly, such a procedure (as exemplified for Ala) is applied for each amino acid encoded by the cds of the RNA to obtain sequences adapted to human codon usage. Accordingly, the therapeutic RNA of the composition may comprise or consist of at least one cds encoding at least one peptide or protein having an amino acid sequences being at least 70%, 80%, 85%, 86%, 87%, 88%, 89%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identical to any one ofSEQ ID NOs: 127-490 wherein the cds is a "human codon usage adapted coding sequence" as explained above. In embodiments, the therapeutic RNA of the composition may be modified, wherein the codon adaptation index (CAI) may be increased or preferably maximised in the at least one cds (herein referred to as "CAI maximized coding sequence"). Accordingly, it is preferred that all codons of the wild type nucleic acid sequence that are relatively rare in e.g. a human cell are exchanged for a respective codon that is frequent in the e.g. a human cell, wherein the frequent 5 codon encodes the same amino acid as the relatively rare codon. Suitably, the most frequent codons are used for each encoded amino acid (see Table 2 ofW02020161342, most frequent human codons are marked with asterisks). Suitably, the RNA comprises at least one cds, wherein the codon adaptation index (CAI) of the at least one cds is at least 0.5, at least 0.8, at least 0.9 or at least 0.95. Most preferably, the codon adaptation index (CAI) of the at least one cds is 1. E.g., in the case of the amino acid Ala, the wild type cds is adapted in a way that the most frequent human 10 codon "GCC" is always used for said amino acid. Accordingly, such a procedure (as exemplified for Ala) is applied for each amino acid encoded by the cds of the RNA to obtain a CAI maximized cds. Accordingly, the therapeutic RNA of the composition may comprise or consist of at least one cds encoding at least one peptide or protein having an amino acid sequences being at least 70%, 80%, 85%, 86%, 87%, 88%, 89%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identical to any one ofSEQ ID NOs: 127-490 wherein the cds is a "CAI maximized coding sequence" as 15 explained above. In certain embodiments, the therapeutic RNA of the composition comprises a cds encoding a CRISPR-assodated endonuclease as defined herein. In embodiments, the therapeutic RNA of the composition comprises at least one cds comprising or consisting of a nucleic acid sequence being at least 70%, 80%, 85%, 86%, 87%, 88%, 89%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identical to any one of SEQ ID NO: 411; 2540-2553; 11117-11119; 2011355-11357; 2554-3457; 1380-1393; 3700-3713; 4860-4873; 6020-6033; 7180-7193; 8340-8353; 11237-11239; 11473-11475; 11591-11593; 11709-11711; 11827-11829; 11945-11947; 1394-2297; 3714-4617; 4874-5777:6034- 6937; 7194-8097; 8354-9257; 412; 3474-3887; 2314-2327; 4634-4647; 5794-5807; 6954-6967; 8114-8127; 413-425; 3490-3503; 3506-3519; 3522-3535; 3538-3551; 3554-3567; 3570-3583; 3586-3599; 3602-3615; 3618-3631; 3634- 3647; 3650-3663; 3666-3679; 3682-3695; 9514-9527; 9626-9639; 9738-9751; 9850-9863; 9962-9975,10074-10087; 2510186-10199; 10298-10311 ; 2330-2343; 2346-2359; 2362-2375; 2378-2391 ; 2394-2407; 2410-2423; 2426-2439; 2442-2455; 2458-2471; 2474-2487; 2490-2503; 2506-2519; 2522-2535; 9498-9511; 9610-9623; 9722-9735; 9834- 9847; 9946-9959; 10058-10071; 10170-10183-10282-10295; 4650-4663; 4666-4679; 4682-4695; 4698-4711; 4714- 4727; 4730-4743; 4746-4759; 4762-4775; 4778-4791; 4794-4807; 4810-4823; 4826-4839; 4842-4855; 9530-9543; 9642-9655;9754-9767;9866-9879; 9978-9991; 10090-10103; 10202-10215; 10314-10327; 5810-5823; 5826-5839; 305842-5855; 5858-5871; 5874-5887; 5890-5903; 5906-5919; 5922-5935; 5938-5951 ; 5954-5967; 5970-5983, 5986- 5999; 6002-6015; 9546-9559; 9658-9671; 9770-9783; 9882-9895; 9994-10007; 10106-10119; 10218-10231; 10330- 10343; 6970-6983; 6986-6999; 7002-7015; 7018-7031; 7034-7047; 7050-7063; 7066-7079; 7082-7095; 7098-7111; 7114-7127; 7130-7143; 7146-7159; 7162-7175; 9562-9575; 9674-9687; 9786-9799; 9898-9911; 10010-10023; 10122- 10135; 10234-10247; 10346-10359; 8130-8143; 8146-8159; 8162-8175; 8178-8191; 8194-8207; 8210-8223; 8226- 358239; 8242-8255; 8258-8271; 8274-8287; 8290-8302; 8306-8319; 8322-8335; 9578-9591 ; 9690-9703; 9802-9815; 9914-9927; 10026-10039; 10138-10151; 10250-10263; 10362-10375; 9290-9303; 9306-9319; 9322-9335; 9338-9351; 9354-9367; 9370-9383; 9386-9399; 9402-9415; 9418-9431; 9434-9447; 9450-9463; 9466-9479; 9482-9495; 9594 - 9607; 9706-9719; 9818-9831; 9930-9943; 10042-10055; 10154-10167; 10266-10279; 10378-10391 of published PCT patent application WO 2018/172556, said sequences herewith incorporated by reference. 40 In other embodiments, the therapeutic RNA of the composition comprises at least one cds comprising or consisting of a nucleic acid sequence being at least 70%, 80%, 85%, 86%, 87%, 88%, 89%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identical to any one of SEQ ID NOs: 10552; 3458-3459; 3460-3473; 2298-2299; 4618-4619; 5778- 5779; 6938-6939; 8098-8099; 9258-9259 ; 2300-2313; 4620-4633; 5780-5793; 6940-6953; 8100-8113; 9260-9273; 3488-3489; 10396; 2328-2329; 10395; 4648-4649; 10397; 5808-5809; 10398; 6968-6969; 10399; 8128-8129; 10400; 459274-9287; 3504-3505; 3520-3521; 3536-3537; 3552-3553; 3568-3669; 3584-3585; 3600-3601 ; 3616-3617; 3632- 3633; 3648-3649; 3664-3665; 3680-3681; 3696-3697; 9528-9529; 9640-9641; 9752-9753; 9864-9865; 9976-9977; 10088-10089; 10200-10201; 10312-10313; 10403; 10410; 10417;10424;10431;10438;10445; 10452;10459; 10466; 10473;10480;10487;10494; 10501; 10508;10515;10522;10529;10536; 10543; 2344-2345; 2360-2361; 2376-2377; 2392-2393; 2408-2409; 2424-2425; 2440-2441; 2456-2457; 2472-2473; 2489-2490; 2504-2505; 2520-2521; 2536- 52537; 9512-9513; 9624-9625; 9736-9737; 9848-9849; 9960-9961; 10072-10073; 10184-10185; 10296-10297; 10402; 10409;10416;10423;10430;10437; 10444;10451;10458;10465;10472; 10479;10486;10493;10500;10507; 10514;10521;10528;10535; 10542; 4664^665; 4680-4681; 4696-4697; 4712-4713; 4728-4729; 4744-4745; 4760- 4761; 4776-4777; 4792-4793; 4808-4809; 4824-4825; 4840-4841; 4856-4857; 9544-9545; 9656-9657; 9768-9769; 9880-9881; 9992-9993; 10104-10105; 10216-10217; 10328-10329; 10404; 10411; 10418;10425;10432;10439; 1010446;10453;10460; 10467; 10474;10481;10488;10495;10502; 10509;10516;10523;10530;10537; 10544;5824- 5825; 5840-5841; 5856-5857; 5872-5873; 5888-5889; 5904-5905; 5920-5921; 5936-5937; 5952-5953; 5968-5969; 5984-5985; 6000-6001; 6016-6017; 9560-9561; 9672-9673; 9784-9785; 9896-9897; 10008-10009; 10120-10121; 10232-10233; 10344-10345; 10405; 10412; 10419; 10426;10433;10440;10447;10454; 10461;10468;10475; 10482; 10489;10496;10503;10510; 10517; 10524;10531;10538;10545;7033;7048-7049; 7064-7065; 7080-7081; 7096- 157097; 7112-7113; 7128-7129; 7144-7145; 7160-7161; 7176-7177; 9576-9577; 9688-9689; 9800-9801; 9912-9913; 10024-10025; 10136-10137; 10248-10249; 10360-10361; 10406; 10413;10420;10427;10434;10441; 10448;10455; 10462;10469;10476;10483; 10490; 10497;10504;10511;10518;10525; 10532; 10539; 10546; 8144-8145; 8160- 8160; 8176-8177; 8192-8193; 8208-8209; 8224-8225; 8240-8241; 8256-8257; 8272-8273; 8288 -8289; 8304-8305; 8320-8321; 8336-8337; 9592-9593; 9704-9705; 9816-9817; 9928-9929; 10040-10041; 10152-10153; 10264-10265; 2010376-10377; 10407; 10414; 10421; 10428;10435;10442;10449;10456; 10463;10470;10477;10484;10491; 10498;10505;10512; 10519; 10526; 10533; 10540; 10547; 9288-9289; 10401; 10553; 10582-10583;10579-10580; 10585-10586; 10588-10589; 10591-10592; 10594-10595; 10597-10598; 10554-10574;10601;10602;10615; 10616; 10629;10630;10643;10644; 10657; 10658;10671;10672;10685;10686; 10699;10700;10713;10714;10727; 10728;10741;10742;10755; 10756; 10769;10770;10783;10784;10797; 10798;10811;10812;10825;10826; 2510839;10840;10853;10854; 10867;10868;10881;10882;10603;10604; 10617;10618;10631;10632;10645; 10646;10659;10660;10673;10674; 10687;10688;10701;10702; 10715; 10716;10729;10730;10743;10744; 10757;10758;10771;10772;10785;10786; 10799;10800;10813;10814; 10827; 10828;10841;10842;10855; 10856;10869;10870;10883; 10884; 10605;10606;10619;10620;10633; 10634;10647;10648;10661;10662; 10675;10676; 10689; 10690; 10703; 10704;10717;10718;10731;10732; 10745;10746;10759;10760;10773; 3010774;10787;10788;10801; 10802;10815;10816;10829; 10830; 10843;10844;10857;10858; 10871; 10872; 10885;10886;10607;10608;10621; 10622;10635;10636;10649;10650; 10663;10664;10677;10678;10691; 10692;10705;10706;10719;10720; 10733;10734;10747;10748;10761; 10762;10775;10776;10789;10790; 10803;10804;10817;10818; 10831; 10832;10845;10846;10859; 10860; 10873;10874;10887;10888;10609; 10610;10623;10624;10637; 10638; 10651;10652;10665;10666;10679; 10680;10693;10694;10707;10708; 3510721;10722;10735;10736; 10749;10750;10763;10764; 10777; 10778;10791;10792;10805;10806; 10819; 10820;10833;10834;10847; 10848; 10861;10862;10875;10876;10889; 10890;10611;10612;10625;10626; 10639;10640;10653;10654;10667; 10668;10681;10682;10695;10696; 10709; 10710;10723;10724;10737; 10738;10751;10752;10765; 10766; 10779;10780;10793;10794; 10807; 10808;10821;10822;10835;10836; 10849;10850;10863;10864; 10877; 10878; 10891; 10892; 9304-9305; 9320-9321; 9336-9337; 9352-9353; 9368- 409369; 9384-9385; 9400-9401; 9416-9417; 9432-9433; 9448-9449; 9464-9465; 9480-9481; 9496-9497; 9608-9609; 9720-9721; 9832-9833; 9944-9945; 10056-10057; 10168-10169; 10280-10281; 10392-10393;10408;10415;10422; 10429;10436;10443;10450;10457; 10464;10471;10478;10485;10492;10499; 10506;10513;10520;10527; 10534; 10541; 10548 of published PCT patent application WO 2018/172556, said sequences herewith incorporated by reference. 45
In embodiments, the therapeutic RNA of the composition may be monocistronic, bicistronic, or multicistronic. The term "monocistronic" will be recognized and understood by the person of ordinary skill in the art, and is e.g. intended to refer to an RNA that comprises only one coding sequences. The terms "bicistronic", or "multicistronic" as used herein will be recognized and understood by the person of ordinary skill in the art, and are e.g. intended to refer to an RNA that may have two (bicistronic) or more (multicistronic) coding sequences. In preferred embodiments, the therapeutic RNA of the composition is monocistronic. In embodiments, the therapeutic RNA of the composition is monocistronic and the cds of said RNA encodes at least two different proteins as defined above (e.g. derived from List 1 or Table 1). Accordingly, said cds may e.g. encode at least two, three, four, five, six, seven, eight and more therapeutic proteins, linked with or without an amino acid linker sequence, wherein said linker sequence can comprise rigid linkers, flexible linkers, cleavable linkers, or a combination thereof. Such constructs are herein referred to as "multi-protein-constructs". In embodiments, the therapeutic RNA of the composition may be bicistronic or multicistronic and comprises at least two coding sequences, wherein the at least two coding sequences encode two or more proteins as defined above (e.g. derived from List 1 or Table 1). Accordingly, the coding sequences in a bicistronic or multicistronic RNA suitably encode distinct proteins or peptides as defined herein. Preferably, the coding sequences in said bicistronic or multicistronic constructs may be separated by at least one IRES (internal ribosomal entry site) sequence. In that context, suitable IRES sequences may be selected from the list of nucleic acid sequences according to SEQ ID NOs: 1566-1662 of the patent application W02017/081082, or fragments or variants of these sequences. In this context, the disclosure of W02017/081082 relating to IRES sequences is herewith incorporated by reference. UTR In embodiments, the composition comprising the therapeutic nucleic acid, preferably RNA comprises at least one heterologous untranslated region (UTR) which is selected from at least one heterologous 5'-UTR and/or at least one heterologous 3'-UTR. The therapeutic nucleic acid, preferably RNA of the composition may be composed of a protein-coding region ("coding sequence" or "cds"), and 5'- UTR and/or 3'- UTR. UTRs may harbor regulatory sequence elements or motifs that determine RNA turnover, stability, and/or localization. Moreover, UTRs may harbor sequence elements or motifs that enhance translation. In medical application of RNA, translation of the RNA cds into at least one peptide or protein is of paramount importance to therapeutic efficacy. Certain combinations of 3'-UTRs and/or 5'-UTRs may enhance the expression of operably linked coding sequences encoding peptides or proteins as defined above. Therapeutic RNA harboring said UTR combinations advantageously enable rapid and transient expression of encoded peptides or proteins after administration to a subject, preferably after administration to the anterior segment of the eye of a subject. Accordingly, the therapeutic RNA of the composition may comprise certain combinations of3'-UTRs and/or 5'-UTRs, resulting in translation of the protein (e.g. therapeutic protein, CRISPR-associated endonuclease, as defined above), and hence, in expression of the protein in therapeutically relevant cells or tissues in the eye (e.g., cells of the anterior segment of the eye as defined above). Suitably, the therapeutic nucleic acid, preferably RNA of the composition comprises at least one heterologous 5'-UTR and/or at least one heterologous 3'-UTR. Said heterologous 5'-UTRs or 3'-UTRs may be derived from naturally occurring genes or may be synthetically engineered. In preferred embodiments, the therapeutic RNA comprises at least one cds operably linked to at least one (heterologous) 3'-UTR and/or at least one (heterologous) 5'-UTR. In preferred embodiments, the therapeutic nucleic acid, preferably RNA of the composition comprises at least one heterologous 3'-UTR. Preferably, at least one heterologous 3'-UTR comprises or consists of a nucleic acid sequence derived from a 3'-UTR of a gene selected from PSMB3, ALB7, alpha-globin, CASP1, COX6B1, GNAS, NDUFA1 and RPS9, or from a homolog, a fragment or a variant of any one of these genes, preferably wherein the at least one heterologous 3'-UTR comprises 5 or consists of a nucleic acid sequence being identical or at least 70%, 80%, 85%, 86%, 87%, 88%, 89%, 90%, 91 %, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identical to SEQ ID NOs: 64-121 , or a fragment or a variant of any of these. In preferred embodiments, at least one heterologous 3'-UTR of the therapeutic RNA comprises or consists of a nucleic acid sequence being identical or at least 70%, 80%, 85%, 86%, 87%, 88%, 89%, 90%, 91%, 92%, 93%, 94%, 95%, 1096%, 97%, 98%, or 99% identical to SEQ ID NO: 64 or 65, or a fragment or a variant thereof. The term "S'-untranslated region" or "3'-UTR" or "3'-UTR element" will be recognized and understood by the person of ordinary skill in the art, and are e.g. intended to refer to a part of the therapeutic RNA, located 3' (i.e. downstream) of a cds, which is not translated into protein. A 3'-UTR may be part of an RNA, e.g. an mRNA, located between a cds and a 15 terminal poly(A) sequence. A 3'-UTR may comprise elements for controlling gene expression, also called regulatory elements. Such regulatory elements may be, e.g., ribosomal binding sites, miRNA binding sites etc. Preferably, the therapeutic nucleic acid, preferably RNA of the composition comprises a 3'-UTR, which may be derivable from a gene that relates to an RNA with enhanced half-life (i.e. that provides a stable RNA). In some embodiments, a 3'-UTR comprises one or more of a polyadenylation signal, a binding site for proteins that 20 affect an RNA stability of location in a cell, or one or more miRNA or binding sites for miRNAs. MicroRNAs (or miRNA) are 19-25 nucleotide long noncoding RNAs that bind to the 3'-UTR of nucleic acid molecules and down-regulate gene expression either by reducing nucleic acid molecule stability or by inhibiting translation. E.g., microRNAs are known to regulate RNA, and thereby protein expression, e.g. in liver (miR-122), heart (miR-ld, miR- 149), endothelial cells (miR-17-92, miR-126), adipose tissue (let-7, miR-30c), kidney (miR-192, miR-194, miR-204), 5 myeloid cells (miR-142-3p, miR-142-5p, miR-16, miR-21, miR-223, miR-24, miR-27), muscle (miR-133, miR-206, miR- 208), and lung epithelial cells (let-7, miR-133, miR-126). The RNA of the invention may comprise one or more microRNA target sequences, microRNA sequences, or microRNA seeds. Such sequences may e.g. correspond to any known microRNA such as those taught in US Publications US2005/0261218 and US2005/0059005, the contents of which are incorporated herein by reference. 0 Accordingly, miRNA, or binding sites for miRNAs as defined above may be removed from the 3'-UTR or introduced into the 3'-UTR in order to tailor the expression of the RNA expression to desired cell types or tissues, preferably to desired cell types or tissues in the eye. In preferred embodiments, the therapeutic nucleic acid, preferably RNA of the composition comprises at least one heterologous 3'-UTR, wherein the at least one heterologous 3'-UTR comprises a nucleic acid sequence derived from a53'-UTR of a gene selected from PSMB3, ALB7, alpha-globin (referred to as "muag"), CASP1 , COX6B1, GNAS, NDUFA1 and RPS9, or from a homolog, a fragment or variant of any one of these genes. In embodiments, the therapeutic nucleic acid, preferably RNA of the composition may comprise a 3'-UTR derived from a ALB7 gene, wherein said 3'-UTR derived from an ALB7 gene comprises or consists of a nucleic acid sequence being identical or at least 70%, 80%, 85%, 86%, 87%, 88%, 89%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% 0 identical to SEQ ID NOs:68 or 69 or a fragment or a variant thereof. In embodiments, the therapeutic RNA of the composition may comprise a 3'-UTR derived from a alpha-globin gene, wherein said 3'-UTR derived from a alpha- globin gene ("muag") comprises or consists of a nucleic acid sequence being identical or at least 70%, 80%, 85%, 86%, 87%, 88%, 89%, 90%, 91 %, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identical to SEQ ID NOs: 66 or 67 or a fragment or a variant thereof. In embodiments, the therapeutic RNA of the composition may comprise a 3'-UTR derived from a PSMB3 gene, wherein said 3'-UTR derived from a PSMB3 gene comprises or consists of a nucleic acid sequence being identical or at least 70%, 80%, 85%, 86%, 87%, 88%, 89%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identical to SEQ ID NOs: 64 or 65 or a fragment or a variant thereof. In embodiments, the therapeutic RNA of the composition may comprise a 3'-UTR derived from a CASP1 gene, wherein said 3'-UTR derived from a CASP1 gene comprises or consists of a nucleic acid sequence being identical or at least 70%, 80%, 85%, 86%, 87%, 88%, 89%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identical to SEQ ID N0s:72 or 73 or a fragment or a variant thereof. In embodiments, the therapeutic RNA of the composition may comprise a 3'-UTR derived from a COX6B1 gene, wherein said 3'-UTR derived from a COX6B1 gene comprises or consists of a nucleic acid sequence being identical or at least 70%, 80%, 85%, 86%, 87%, 88%, 89%, 90%, 91 %, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identical to SEQ ID NOs: 74 or 75 or a fragment or a variant thereof. In embodiments, the therapeutic RNA of the composition may comprise a 3'-UTR derived from a GNAS gene, wherein said 3'-UTR derived from a GNAS gene comprises or consists of a nucleic acid sequence being identical or at least 70%, 80%, 85%, 86%, 87%, 88%, 89%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identical to SEQ ID NOs: 78 or 79 or a fragment or a variant thereof. In embodiments, the therapeutic RNA of the composition may comprise a 3'-UTR derived from a NDUFA1 gene, wherein said 3'-UTR derived from a NDUFA1 gene comprises or consists of a nucleic acid sequence being identical or at least 70%, 80%, 85%, 86%, 87%, 88%, 89%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identical to SEQ ID NOs: 80 or 81 or a fragment or a variant thereof. In embodiments, the therapeutic RNA of the composition may comprise a 3'-UTR derived from a RPS9 gene, wherein said 3'-UTR derived from a RPS9 gene comprises or consists of a nucleic acid sequence being identical or at least 70%, 80%, 85%, 86%, 87%, 88%, 89%, 90%, 91 %, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identical to SEQ ID NOs: 82 or 83 or a fragment or a variant thereof. Accordingly, the therapeutic nucleic acid, preferably RNA of the composition may suitably comprise at least one 3'-UTR comprising or consisting of a nucleic acid sequence being identical or at least 70%, 80%, 85%, 86%, 87%, 88%, 89%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identical to SEQ ID NOs: 110-121 or a fragment or a variant thereof. In other embodiments, the therapeutic RNA of the composition comprises a 3'-UTR as described in W02016/107877, the disclosure of W02016/107877 relating to 3'-UTR sequences herewith incorporated by reference. Suitable 3'-UTRs are SEQ ID NOs: 1 to 24 and SEQ ID NOs: 49 to 318 of W02016/107877, or fragments or variants of these sequences. Accordingly, the 3'-UTRs of the RNA may comprise or consist of a corresponding RNA sequence of the nucleic acid sequence according SEQ ID NOs: 1 to 24 and SEQ ID NOs: 49 to 318ofW02016/107877. In other embodiments, the therapeutic RNA comprises a 3'-UTR as described in W02017/036580, the disclosure of W02017/036580 relating to 3'-UTR sequences herewith incorporated by reference. Suitable 3'-UTRs are SEQ ID NOs: 152 to 204 of W02017/036580, or fragments or variants of these sequences. Accordingly, the 3'-UTR of the RNA may comprise or consist of a corresponding RNA sequence of the nucleic acid sequence according SEQ ID NOs: 152 to 204 of W02017/036580. In other embodiments, the therapeutic RNA of the composition comprises a 3'-UTR as described in W02016022914, the disclosure ofW02016022914 relating to 3'-UTR sequences herewith incorporated by reference. Particularly preferred 3'-UTRs are nucleic acid sequences according to SEQ ID NOs: 20 to 36 of W02016022914, or fragments or variants of these sequences. In this context, it is particularly preferred that the 3'-UTR of the RNA comprises or consists of a corresponding RNA sequence of the nucleic acid sequence according to SEQ ID NOs: 20 to 36 of W02016022914. In preferred embodiments, the therapeutic nucleic acid, preferably RNA of the composition comprises at least one heterologous 5'-UTR. Preferably, at least one heterologous 5'-UTR comprises or consists of a nucleic acid sequence derived from a 5'-UTR of a gene selected from HSD17B4, RPL32, ASAH1, ATP5A1 , MP68, NDUFA4, NOSIP, RPL31 , SLC7A3, TUBB4B and UBQLN2, or from a homolog, a fragment or variant of any one of these genes, preferably selected from HSD17B4, preferably wherein the at least one heterologous 3'-UTR comprises or consists of a nucleic acid sequence being identical or at least 70%, 80%, 85%, 86%, 87%, 88%, 89%, 90%, 91 %, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identical to SEQ ID NOs: 10-63, or a fragment or a variant of any of these. In preferred embodiments, at least one heterologous 5'-UTR of the therapeutic nucleic acid, preferably RNA comprises or consists of a nucleic acid sequence being identical or at least 70%, 80%, 85%, 86%, 87%, 88%, 89%, 90%, 91 %, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identical to SEQ ID NO: 10 or 11, ora fragment or a variant thereof. The terms "S'-untranslated region" or "5'-UTR" or "5'-UTR element" will be recognized and understood by the person of ordinary skill in the art, and are e.g. intended to refer to a part the RNA, located 5' (i.e. "upstream") of a cds, which is not translated into protein. A 5'-UTR may be part of an RNA located 5' of the cds. Typically, a 5'-UTR starts with the transcriptional start site and ends before the start codon of the cds. A 5'-UTR may comprise elements for controlling gene expression, called regulatory elements. Such regulatory elements may be, e.g., ribosomal binding sites, miRNA binding sites etc. The 5'-UTR may be post-transcriptionally modified, e.g. by enzymatic or post-transcriptional addition of a S'-cap structure (see above). Preferably, the therapeutic nucleic acid, preferably RNA of the composition comprises a 5'-UTR, which may be derivable from a gene that relates to an RNA with enhanced half-life (i.e. that provides a stable RNA). In some embodiments, a 5'-UTR comprises one or more of a binding site for proteins that affect an RNA stability of location in a cell, or one or more miRNA or binding sites for miRNAs (as defined above). Accordingly, miRNA or binding sites for miRNAs as defined above may be removed from the 5'-UTR or introduced into the 5'-UTR in order to tailor the expression of the RNA expression to desired cell types or tissues, preferably in desired cell types or tissues in the eye. In preferred embodiments, the therapeutic nucleic acid, preferably RNA of the composition comprises at least one heterologous 5'-UTR, wherein the at least one heterologous 5'-UTR comprises a nucleic acid sequence derived from a 5'-UTR of gene selected from HSD17B4, RPL32, ASAH1, ATP5A1, MP68, NDUFA4, NOSIP, RPL31, SLC7A3, TUBB4B,and UBQLN2, orfrom ahomolog, a fragment or variant of any one of these genes. In embodiments, the therapeutic nucleic acid, preferably RNA of the composition may comprise a 5'-UTR derived from a RPL32 gene, wherein said 5'-UTR derived from a RPL32 gene comprises or consists of a nucleic acid sequence being identical or at least 70%, 80%, 85%, 86%, 87%, 88%, 89%, 90%, 91 %, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identical to SEQ ID NOs: 12 or 13 ora fragment or a variant thereof. In embodiments, the therapeutic nucleic acid, preferably RNA of the composition may comprise a 5'-UTR derived from a HSD17B4 gene, wherein said 5'-UTR derived from a HSD17B4 gene comprises or consists of a nucleic acid sequence being identical or at least 70%, 80%, 85%, 86%, 87%, 88%, 89%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identical to SEQ ID NOs: 10 or 11 or a fragment or a variant thereof. In embodiments, the therapeutic nucleic acid, preferably RNA of the composition may comprise a 5'-UTR derived from a ASAH1 gene, wherein said 5'-UTR derived from a ASAH1 gene comprises or consists of a nucleic acid sequence being identical or at least 70%, 80%, 85%, 86%, 87%, 88%, 89%, 90%, 91 %, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identical to SEQ ID NOs: 16 or 17 ora fragment or a variant thereof. In embodiments, the therapeutic nucleic acid, preferably RNA of the composition may comprise a 5'- UTR derived from a ATP5A1 gene, wherein said 5'-UTR derived from a ATP5A1 gene comprises or consists of a nucleic acid sequence being identical or at least 70%, 80%, 85%, 86%, 87%, 88%, 89%, 90%, 91 %, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identical to SEQ ID NOs: 18 or 19 ora fragment or a variant thereof. In embodiments, the therapeutic nucleic acid, preferably RNA of the composition may comprise a 5'-UTR derived from a MP68 gene, wherein said 5'-UTR derived from a MP68 gene comprises or consists of a nucleic acid sequence being identical or at least 70%, 80%, 85%, 86%, 87%, 88%, 89%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identical to SEQ ID NOs: 26 or 27 or a fragment or a variant thereof. In embodiments, the therapeutic nucleic acid, preferably RNA of the composition may comprise a 5'-UTR derived from a NDUFA4 gene, wherein said 5'-UTR derived from a NDUFA4 gene comprises or consists of a nucleic acid sequence being identical or at least 70%, 80%, 85%, 86%, 87%, 88%, 89%, 90%, 91 %, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identical to SEQ ID NOs: 28 or 29 or a fragment or a variant thereof. In embodiments, the therapeutic RNA of the composition may comprise a 5'-UTR derived from a NOSIP gene, wherein said 5'-UTR derived from a NOSIP gene comprises or consists of a nucleic acid sequence being identical or at least 70%, 80%, 85%, 86%, 87%, 88%, 89%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identical to SEQ ID NOs: 30 or 31 or a fragment or a variant thereof. In embodiments, the therapeutic nucleic acid, preferably RNA may comprise a 5'-UTR derived from a RPL31 gene, wherein said 5'-UTR derived from a RPL31 gene comprises or consists of a nucleic acid sequence being identical or at least 70%, 80%, 85%, 86%, 87%, 88%, 89%, 90%, 91 %, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identical to SEQ ID NOs: 32 or 33 or a fragment or a variant thereof. In embodiments, the therapeutic RNA of the composition may comprise a 5'-UTR derived from a SLC7A3 gene, wherein said 5'-UTR derived from a SLC7A3 gene comprises or consists of a nucleic acid sequence being identical or at least 70%, 80%, 85%, 86%, 87%, 88%, 89%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identical to SEQ ID NOs: 36 or 37 or a fragment or a variant thereof. In embodiments, the therapeutic nucleic acid, preferably RNA of the composition may comprise a 5'-UTR derived from a TUBB4B gene, wherein said 5'-UTR derived from a TUBB4B gene comprises or consists of a nucleic acid sequence being identical or at least 70%, 80%, 85%, 86%, 87%, 88%, 89%, 90%, 91 %, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identical to SEQ ID NOs: 38 or 39 or a fragment or a variant thereof. In embodiments, the therapeutic nucleic acid, preferably RNA of the composition may comprise a 5'-UTR derived from a UBQLN2 gene, wherein said 5'-UTR derived from a UBQLN2 gene comprises or consists of a nucleic acid sequence being identical or at least 70%, 80%, 85%, 86%, 87%, 88%, 89%, 90%, 91 %, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identical to SEQ ID NO: 40 or 41 or a fragment or a variant thereof. Accordingly, the therapeutic nucleic acid, preferably RNA of the composition may suitably comprise at least one 5'-UTR comprising or consisting of a nucleic acid sequence being identical or at least 70%, 80%, 85%, 86%, 87%, 88%, 89%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identical to SEQ ID NOs: 10-63 or a fragment or a variant thereof. In other embodiments, the therapeutic nucleic acid, preferably RNA of the composition comprises a 5'-UTR as described in W02013/143700, the disclosure of W02013/143700 relating to 5'-UTR sequences herewith incorporated by reference. Particularly preferred 5'-UTRs are nucleic acid sequences derived from SEQ ID NOs: 1-1363, SEQ ID NO: 1395, SEQ ID NO: 1421 and SEQ ID NO: 1422 of W02013/143700, or fragments or variants of these sequences. In this context, it is preferred that the 5'-UTR of the RNA comprises or consists of a corresponding RNA sequence of the nucleic acid sequence according SEQ ID NOs: 1-1363, SEQ ID NO: 1395, SEQ ID NO: 1421 and SEQ ID NO: 1422 of W02013/143700. In other embodiments, the therapeutic RNA of the composition comprises a 5'-UTR as described in W02016/107877, the disclosure of W02016/107877 relating to 5'-UTR sequences herewith incorporated by reference. Particularly preferred 5'-UTRs are nucleic acid sequences according to SEQ ID NOs: 25 to 30 and SEQ ID NOs: 319 to 382 of W02016/107877, or fragments or variants of these sequences. In this context, it is particularly preferred that the 5'-UTR of the RNA comprises or consists of a corresponding RNA sequence of the nucleic acid sequence according SEQ ID NOs: 25 to 30 and SEQ ID NOs: 319 to 382 of W02016/107877. In other embodiments, the therapeutic RNA of the composition comprises a 5'-UTR as described in W02017/036580, the disclosure of W02017/036580 relating to 5'-UTR sequences herewith incorporated by reference. Particularly preferred 5'-UTRs are nucleic acid sequences according to SEQ ID NOs: 1 to 151 of W02017/036580, or fragments or variants of these sequences. In this context, it is particularly preferred that the 5'-UTR of the RNA comprises or consists of a corresponding RNA sequence of the nucleic acid sequence according to SEQ ID NOs: 1 to 151 of W02017/036580. In other embodiments, the therapeutic RNA of the composition comprises a 5'-UTR as described in W02016022914, the disclosure of W02016022914 relating to 5'-UTR sequences herewith incorporated by reference. Particularly preferred 5'-UTRs are nucleic acid sequences according to SEQ ID NOs: 3 to 19 of W02016022914, or fragments or variants of these sequences. In this context, it is particularly preferred that the 5'-UTR of the RNA comprises or consists of a corresponding RNA sequence of the nucleic acid sequence according to SEQ ID NOs: 3 to 19 of W02016022914. Suitably, in preferred embodiments, the therapeutic nucleic acid, preferably RNA of the composition comprises at least one cds encoding at least one peptide or protein as specified herein (e.g. therapeutic protein, CRISPR-associated endonuclease), operably linked to a 3'-UTR and/or a 5'-UTR selected from the following 5'-UTR/3'-UTR combinations: a-1 (HSD17B4/PSMB3), a-2 (NDUFA4/PSMB3), a-3 (SLC7A3/PSMB3), a-4 (NOSIP/PSMB3), a-5 (MP68/PSMB3), b-1 (UBQLN2/RPS9), b-2 (ASAH1/RPS9), b-3 (HSD17B4/RPS9), b-4 (HSD17B4/CASP1), b-5 (NOSIP/COX6B1), c-1 (NDUFA4/RPS9), c-2 (NOSIP/NDUFA1), c-3 (NDUFA4/COX6B1), c-4 (NDUFA4 /NDUFA1), c-5 (ATP5A1/PSMB3), d- 1 (Rpl31/PSMB3), d-2 (ATP5A1/CASP1), d-3 (SLC7A3/GNAS), d-4 (HSD17B4/NDUFA1), d-5 (Slc7a3/Ndufa1), e-1 CTUBB4B/RPS9), e-2 (RPL31/RPS9), e-3 (MP68/RPS9), e-4 (NOSIP/RPS9), e-5 (ATP5A1/RPS9), e-6 (ATP5A1/COX6B1), f-1 (ATP5A1/GNAS), f-2 (ATP5A1/NDUFA1), f-3 (HSD17B4/COX6B1), f-4 (HSD17B4/GNAS), f-5 (MP68/COX6B1), g-1 (MP68/NDUFA1), g-2 (NDUFA4/CASP1), g-3 (NDUFA4/GNAS), g-4 (NOSIP/CASP1), g-5 (RPL31/CASP1), h-1 (RPL31/COX6B1), h-2 (RPL31/GNAS), h-3 (RPL31/NDUFA1), h-4 (Slc7a3/CASP1), h-5 (SLC7A3/COX6B1), i-1 (SLC7A3/RPS9), i-2 (RPL32/ALB7), i-2 (RPL32/ALB7), or i-3 (a-globin gene/-). In preferred embodiments, the therapeutic nucleic acid, preferably RNA of the composition comprises at least one heterologous 5'-UTR is selected from HSD17B4 and the at least one heterologous 3' UTR is selected from PSMB3. CAP^A/Ucontentand Kozak In some embodiments, the therapeutic nucleic acid, preferably RNA of the composition comprises a 5'-cap structure, which preferably stabilizes the RNA and/or enhances expression of the encoded peptide or protein. A 5'-cap structure is of particular importance in embodiments where the therapeutic RNA is linear, e.g. a linear mRNA or a linear replicon RNA. In preferred embodiments, the therapeutic nucleic acid, in particular the mRNA of the composition comprises a 5'-cap structure, preferably m7G (m7G(5')ppp(5')G), capO, cap1, cap2, a modified capO or a modified cap1 structure. The term "5'-cap structure" as used herein will be recognized and understood by the person of ordinary skill in the art, and is e.g. intended to refer to a 5' modified nucleotide, particularly a guanine nucleotide, positioned at the 5'-end of an RNA molecule, e.g. an mRNA molecule. Preferably, the 5'-cap structure is connected via a 5'-5'-triphosphate linkage to the RNA. 5'-cap structures which may be suitable in the context of the present invention are capO (methylation of the first nucleobase, e.g. m7GpppN), cap1 (additional methylation of the ribose of the adjacent nudeotide ofm7GpppN), cap2 (additional methylation of the ribose of the 2nd nucleotide downstream of the m7GpppN), cap3 (additional methylation of the ribose of the 3rd nucleotide downstream of the m7GpppN), cap4 (additional methylation of the ribose of the 4th nucleotide downstream of the m7GpppN), ARCA (anti-reverse cap analogue), modified ARCA (e.g. phosphothioate modified ARCA), inosine, Nl-methyl-guanosine, 2'-fluoro-guanosine, 7-deaza-guanosine, 8-oxo-guanosine, 2-amino- guanosine, LNA-guanosine, and 2-azido-guanosine. A 5'-cap (capO or cap1) structure may be formed in chemical RNA synthesis or RNA in vitro transcription (co- transcriptional capping) using cap analogues. The term "cap analogue" as used herein will be recognized and understood by the person of ordinary skill in the art,and is e.g. intended to refer to a non-polymerizable di-nucleotide or tri-nucleotide that has cap functionality in that it facilitates translation or localization, and/or prevents degradation of a nucleic acid molecule, particularly of an RNA molecule, when incorporated at the 5'-end of the nucleic acid molecule. Non-polymerizable means that the cap analogue will be incorporated only at the S'-terminus because it does not have a 5' triphosphate and therefore cannot be extended in the S'-direction by a template-dependent polymerase, particularly, by template-dependent RNA polymerase. Examples of cap analogues include, but are not limited to, a chemical structure selected from the group consisting ofm7GpppG, m7GpppA, m7GpppC; unmethylated cap analogues (e.g. GpppG); dimethylated cap analogue (e.g. m2,7GpppG), trimethylated cap analogue (e.g. m2,2,7GpppG), dimethylated symmetrical cap analogues (e.g. m7Gpppm7G), or anti reverse cap analogues (e.g. ARCA; m7,2'OmeGpppG, m7,2'dGpppG, m7,3'OmeGpppG, m7,3'dGpppG and their tetraphosphate derivatives). Further cap analogues have been described previously (W02008/016473, W02008/157688, W02009/149253, W02011/015347, and W02013/059475). Further suitable cap analogues in that context are described in W02017/066793, W02017/066781, W02017/066791, W02017/066789, W02017/053297, W02017/066782, W02018075827 and W02017/066797 wherein the disclosures referring to cap analogues are incorporated herewith by reference. In embodiments, a modified cap1 structure is generated using tri-nucleotide cap analogue as disclosed in W02017/053297, W02017/066793, W02017/066781, W02017/066791, W02017/066789, W02017/066782, W02018075827, W02017/066797, and W02023007019. In particular, any cap structures derivable from the structure disclosed in claim 1-5 of W02017/053297 may be suitably used to co-transcriptionally generate a modified cap1 structure. Further, any cap structures derivable from the structure defined in claim 1 or claim 21 of W02018075827 may be suitably used to co-transcriptionally generate a modified cap1 structure. In preferred embodiments, the 5'-cap structure may suitably be added co-transcriptionally using tri-nudeotide cap analogue as defined herein in an RNA in vitro transcription reaction as defined herein. Preferred cap-analogues are the di-nucleotide cap analogues m7G(5')ppp(5')G (m7G) or 3'-0-Me-m7G(5')ppp(5')G to co-transcriptionally generate capO structures. Further preferred cap-analogues are the tri-nucleotide cap analogues m7G(5')ppp(5')(2'OMeA)pG or m7G(5')ppp(5')(2'OMeG)pG to co-transcriptionally generate cap1 structures. In other embodiments, the 5'-cap structure is formed via enzymatic capping using capping enzymes (e.g. vaccinia virus capping enzymes and/or cap-dependent 2'-0 methyltransferases) to generate capO or cap1 or cap2 structures. In embodiments, the 5'-cap structure (capO or cap1) may be added using immobilized capping enzymes and/or cap- dependent 2'-0 methyltransferases using methods and means disclosed in W02016/193226. In a particularly preferred embodiment, the therapeutic nucleic acid, preferably RNA of the composition comprises a cap1 structure, wherein said cap1 structure may be formed enzymatically or co-transcriptionally (e.g. using m7G(5')ppp(5')(2'OMeA), or m7G(5')ppp(5')(2'OMeG) analogues). In preferred embodiments, the therapeutic nucleic acid, preferably RNA of the composition comprises an m7G(5')ppp(5')(2'OMeA) cap structure. In such embodiments, the therapeutic RNA comprises a 5' terminal m7G cap, and an additional methylation of the ribose of the adjacent nucleotide of m7GpppN, in that case, a 2'0 methylated adenosine. In other preferred embodiments, the therapeutic nucleic acid, preferably RNA of the composition comprises an m7G(5')ppp(5')(2'OMeG) cap structure. In such embodiments, the therapeutic RNA comprises a 5' terminal m7G cap, and an additional methylation of the ribose of the adjacent nucleotide, in that case, a 2'0 methylated guanosine. Accordingly, whenever reference is made to suitable RNA or mRNA sequences in the context of the invention, the first nucleotide of said RNA or mRNA sequence, that is, the nucleotide downstream of the m7G(5')ppp structure, may be a 2'0 methylated guanosine or a 2'0 methylated adenosine. In embodiments, the A/U content in the environment of the ribosome binding site of the therapeutic RNA of the composition may be increased compared to the A/U content in the environment of the ribosome binding site of its respective wild type nucleic acid. This modification (an increased A/U content around the ribosome binding site) increases the efficiency of ribosome binding to the nucleic acid, preferably the RNA. An effective binding of the ribosomes to the ribosome binding site in turn has the effect of an efficient translation of the RNA. Accordingly, in a particularly preferred embodiment, the therapeutic nucleic acid, preferably RNA of the composition comprises a ribosome binding site, also referred to as "Kozak sequence" identical to or at least 80%, 85%, 90%, 95% identical to any one of the sequences GCCGCCACCATGG, GCCGCCACCAUGG, GCCGCCACC, GCCGCCACC, ACC or fragments or variants thereof. PolvA, PolvC and Histon-Stem-Loop In preferred embodiments, the therapeutic nucleic acid, preferably RNA of the composition comprises at least one poly(N) sequence, e.g. at least one poly(A) sequence, at least one poly(U) sequence, at least one poly(C) sequence, or combinations thereof. In preferred embodiments, the therapeutic nucleic acid, preferably RNA of the composition comprises at least one poly(A) sequence. The terms "poly(A) sequence", "poly(A) tail" or "3'-poly(A) tail" as used herein will be recognized and understood by the person of ordinary skill in the art, and are e.g. intended to be a sequence of adenosine nucleotides, typically located at the S'-end of a therapeutic RNA, of up to about 1000 adenosine nucleotides. Said poly(A) sequence is essentially homopolymeric, e.g. a poly(A) sequence of e.g.100 adenosine nucleotides has essentially the length of 100 nucleotides. In other embodiments, the poly(A) sequence may be interrupted by at least one nucleotide different from an adenosine nudeotide. Preferably, at least one poly(A) sequence comprises about 100 adenosine nucleotides. The poly(A) sequence, suitable located downstream of a 3' UTR as defined herein, may comprise about 10 to about 500 adenosine nucleotides, about 10 to about 200 adenosine nucleotides, about 40 to about 200 adenosine nudeotides, or about 40 to about 150 adenosine nucleotides. Suitably, the length of the poly(A) sequence may be at least about or even more than about 10, 50, 64, 75,100,200,300,400, or 500 adenosine nucleotides. Suitably, the poly(A) sequence of the therapeutic RNA may be long enough to bind at least 2, 3,4, 5 or more monomers of PolyA Binding Proteins. In preferred embodiments, the poly(A) sequence comprises about 50 to about 250 adenosines. In a particularly preferred embodiment, the poly(A) sequence comprises about 64 adenosine nucleotides. Preferably, at least one poly(A) sequence is located at the 3' terminus, optionally, wherein the 3' terminal nucleotide is an adenosine. Preferably, the poly(A) sequence of the therapeutic RNA is obtained from a DNA template during RNA in vitro transcription. In other embodiments, the poly(A) sequence is obtained in vitro by common methods of chemical synthesis without being necessarily transcribed from a DNA template. In other embodiments, poly(A) sequences are generated by enzymatic polyadenylation of the RNA (after RNA in vitro transcription) using commercially available polyadenylation kits and corresponding protocols known in the art, or alternatively, by using immobilized poly(A)polymerases e.g. using a methods and means as described in W02016/174271. In embodiments, the therapeutic nucleic acid, preferably RNA of the composition may comprise a poly(A) sequence derived from a template DNA and may comprise at least one additional poly(A) sequence generated by enzymatic polyadenylation, e.g. as described in W02016/091391. In embodiments where enzymatic polyadenylation of RNA is used, it has to be understood that RNA or mRNA sequences as described herein, may additionally comprise about 30 to about 500 adenosine nucleotides located at the 3' terminus / the 3'-end. In preferred embodiments, the therapeutic nucleic acid, preferably RNA of the composition may comprise at least one poly(C) sequence. The term "poly(C) sequence" as used herein will be recognized and understood by the person of ordinary skill in the art, and are e.g. intended to be a sequence of cytosine nucleotides, typically located at the 3'-end of an RNA, of up to about 200 cytosine nucleotides. In preferred embodiments, the poly(C) sequence, suitable located at the 3' terminus downstream of the 3' UTR as defined herein, comprises about 10 to about 200 cytosine nucleotides, about 10 to about 100 cytosine nucleotides, about 20 to about 70 cytosine nucleotides, about 20 to about 60 cytosine nucleotides, or about 10 to about 40 cytosine nudeotides. In a particularly preferred embodiment, the poly(C) sequence comprises about 30 cytosine nucleotides. Preferably, the poly(C) sequence in the RNA sequence of the present invention is derived from a DNA template by RNA in vitro transcription. In other embodiments, the poly(C) sequence is obtained in vitro by common methods of chemical synthesis, or enzymatically, without being necessarily transcribed from a DNA template. In preferred embodiments, the therapeutic nucleic add, preferably RNA of the composition comprises at least one histone stem-loop. The term "histone stem-loop" (abbreviated as "hsl") as used herein will be recognized and understood by the person of ordinary skill in the art, and is e.g. intended to refer to nucleic acid sequences predominantly found in histone mRNAs. Histone stem-loop sequences/structures may suitably be selected from histone stem-loop sequences as disclosed in W02012/019780, the disclosure relating to histone stem-loop sequences/histone stem-loop structures incorporated herewith by reference. A histone stem-loop sequence that may be used within the present invention may preferably be derived from formulae (I) or (II) of W02012/019780. According to a further preferred embodiment the therapeutic nucleic acid, preferably RNA may comprise at least one histone stem-loop sequence derived from at least one of the specific formulae (la) or (lla) of the patent application W02012/019780. In particularly preferred embodiment, the therapeutic nucleic acid, preferably RNA of the composition comprises at least one histone stem-loop sequence, wherein said histone stem-loop sequence comprises a nucleic acid sequence identical or at least 70%, 80%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identical to SEQ ID NOs: 1 or 2, or fragments or variants thereof. In embodiments, the therapeutic nucleic acid, preferably RNA of the composition comprises a S'-terminal sequence element. Said 3'-terminal sequence element comprises a poly(A)sequence and a histone-stem-loop sequence, wherein said sequence element is located at the 3' terminus of the RNA of the invention. Accordingly, the therapeutic nucleic acid, preferably RNA of the composition may comprise a S'-terminal sequence element comprising or consisting of a nucleic acid sequence being identical or at least 70%, 80%, 90%, 91 %, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identical to SEQ ID NOs: 3-9, or a fragment or variant thereof. In various embodiments, the therapeutic nucleic acid, preferably RNA of the composition may comprise a 5'-terminal sequence element according to GGGAG, AGGAGA, GGGAAA, AGAATA, AGATTA, GATGGG or GGGCG or a fragment or variant thereof. Such a 5'-terminal sequence element comprises e.g. a binding site for T7 RNA polymerase. Further, the first nucleotide of said S'-terminal start sequence may preferably comprise a 2'0 methylation, e.g.2'0 methylated guanosine or a 2'0 methylated adenosine. Elements of therapeutic RNA In various embodiments, the therapeutic nucleic acid, preferably RNA of the composition comprises the following elements, preferably in 5'- to S'-direction: A) 5'-cap structure, preferably as specified herein; B) 5'-terminal start element, preferably as specified herein; C) optionally, a cleavage site for a catalytic nucleic acid molecule, preferably as specified herein; D) optionally, S'-UTR, preferably as specified herein; E) a ribosome binding site, preferably as specified herein; F) at least one coding sequence, preferably as specified herein; G) 3'-UTR, preferably as specified herein; H) optionally, poly(A) sequence, preferably as specified herein; I) optionally, poly(C) sequence, preferably as specified herein; J) optionally, histone stem-loop preferably as specified herein; K) optionally, S'-terminal sequence element, preferably as specified herein. In particularly preferred embodiments the therapeutic nucleic acid, preferably RNA of the composition comprises the following elements A to G in 5' to 3' direction: A) 5'-cap structure selected from m7G(5'), m7G(5')ppp(5')(2'OMeA), or m7G(5')ppp(5')(2'OMeG); B) S'-terminal start element or fragments or variants thereof; C) 3'-UTR as specified herein; D) a ribosome binding site or fragments or variants thereof; E) at least one coding sequence encoding at least one protein selected from SEQ ID NOs: 127-490 or fragments or variants thereof; F) 5'-UTR as specified herein; G) poly(A) sequence comprising about 50 to about 500 adenosines, preferably about 64 adenosines; and wherein the 3'-UTR and/or 5'-UTR element is selected from UTR combinations according to a-1, a-2, a-3, a-4, a-5, b-1 , b-2, b-3, b-4, b-5, c-1 , c-2, c-3, c-4, c-5, d-1, d-2, d-3, d-4, d-5, e-1, e-2, e-3, e-4, e-5, e-6, f-1, f-2, f-3, f-4, f-5, g-1 , g-2, g- 3, g-4, g-5, h-1, h-2, h-3, h-4, h-5, i-1 , i-2, or i-3, as specified herein, wherein said RNA optionally comprises a poly(C) sequence comprising about 10 to about 100 cytosines, wherein said RNA optionally comprises a histone stem-loop selected from SEQ ID NOs: 1 or 2. In preferred embodiments, the therapeutic nucleic acid, preferably RNA of the composition comprises or consists of an RNA sequence which is identical or at least 70%, 80%, 85%, 86%, 87%, 88%, 89%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identical to a nucleic acid sequence selected from the group consisting of SEQ ID NOs: 11011-11042; 11249-11280; 11131-11162; 11367-11398; 11485-11516; 11603-11634; 11721-11752; 11839-11870; 11044-11116; 11282-11354; 11164-11236; 11400-11472; 11518-11590; 11636-11708; 11754-11826; 11872-11944; 11011-11042; 11249-11280; 11044-11116; 11282-11354; 11131-11162; 11367-11398; 11485-11516; 11603-11634; 11721-11752; 11839-11870; 11164-11236; 11400-11472; 11518-11590; 11636-11708; 11754-11826; 11872-11944; 11120-11122;11240;11241;11358;11359;11476; 11477;11594;11595;11712;11713;11830;11831;11948; 11949; 11123-11130; 11360-11366; 11242-11248; 11478-11484; 11596-11602; 11714-11720; 11832-11838:11950- 11956 of published PCT patent application W02018/172556, said RNA sequences from W02018/172556 herewith incorporated by reference. In embodiments, the composition may comprise a plurality or at least more than one of the therapeutic nucleic acid, preferably RNA species as defined above, wherein each therapeutic nucleic acid, preferably RNA species may encode a different peptide or protein. In embodiments, the composition comprises more than one or a plurality, e.g.2, 3,4, 5, 6, 7, 8, 9,10,11,12,13, 14,15 of different therapeutic nucleic acid preferably RNAs as defined above. In a further embodiment, the composition comprises at least one therapeutic nucleic acid, preferably RNA encoding a CRISPR-assodated protein, preferably at least one RNA which is identical or at least 70%, 80%, 85%, 86%, 87%, 88%, 589%, 90%, 91 %, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identical to a nucleic acid sequence selected from the group consisting of SEQ ID NOs: 11011-11042; 11249-11280; 11131-11162; 11367-11398; 11485-11516;11603- 11634; 11721-11752; 11839-11870; 11044-11116; 11282-11354; 11164-11236; 11400-11472; 11518-11590; 11636- 11708; 11754-11826; 11872-11944; 11011-11042; 11249-11280; 11044-11116; 11282-11354; 11131-11162; 11367- 11398; 11485-11516; 11603-11634; 11721-11752; 11839-11870; 11164-11236; 11400-11472; 11518-11590; 11636- 1011708; 11754-11826; 11872-11944; 11120-11122; 11240; 11241;11358;11359;11476;11477;11594;11595;11712; 11713; 11830; 11831; 11948; 11949; 11123-11130; 11360-11366; 11242-11248; 11478-11484; 11596-11602; 11714- 11720;11832-11838; 11950-11956, of published PCT patent application W02018/172556, and additionally, at least one guide RNA. Suitably, administration of the composition leads to expression of the encoded CRISPR-associated protein (provided by the therapeutic RNA) in the eye, preferably in therapeutically relevant target cells of the eye (as 15 defined above), preferably amenable to treatment by knocking in, knocking out, manipulating or modulating the expression of a gene of interest, well-orchestrated by the guide RNA of the composition. The respective guide RNA of the composition may be chosen based on the gene of interest associated with a deficiency, disease or disorder as defined above. For the production of a composition comprising at least 2,3,4,5, 6, 7, 8, 9,10 RNA constructs, methods as disclosed in 20 published patent application W02017/1090134 are preferably used and adapted accordingly. In embodiments where guide RNA is comprised in the composition, said guide RNA may alternatively be produced by common chemical RNA synthesis methods. Compositions of the present invention are suitably sterile and/or pyrogen-free. In embodiments, the composition may further comprise an anti-inflammatory drug, a vascular endothelial growth factor (VEGF) modulator (e.g., anti-VEGF antibody), a platelet derived growth factor (PDGF) modulator, an angiogenesis 25 inhibitor, an immunosuppressive agent, a vascular permeability inhibitor, or a combination thereof. In embodiments, the composition may comprise modulators and/or activators of any of the provided protein targets of List 1. In other embodiments, the composition may comprise at least one of the agents provided in paragraphs [1263] to [1276]of published PCT application W02017/192565, the content herewith incorporated by reference. In embodiments, the therapeutic nucleic acid, preferably the RNA comprised in a composition is provided in an amount 30 of about100ng to about 500pg,about 1pg to about 500|jg, about 1 pg to about 400|jg, about 1 pg to about SOOpg, about 1 pg to about 200|jg, about 1 pg to about 10Opg,about 1pg to about 50|jg, about 1 ^ig to about 25pg,about 1 pg to about 10pg, specifically, in an amount of about 1pg, about 2pg,about 3pg,about 4|jg, about 5|jg, about 6|jg, about 7|jg, about 8|jg, about 9|jg, about 10|jg, about 15pg, about 20pg, about 25|jg, about 30|jg, about 35|jg, about 40|jg, about 45pg,about 50^9,about SOpg, about 70|jg, about 80pg, about 90pg, about lOOpg, about 200|jg, about 300pg,about 5400|jg, about SOOpg. The composition may comprise a safe and effective amount of the therapeutic nucleic acid, preferably the RNA as specified herein. As used herein, "safe and effective amount" means an amount of the therapeutic RNA that is sufficient to results in expression and/or activity of the encoded protein in the eye after administration to the anterior part of the eye of a subject. At the same time, a "safe and effective amount" is small enough to avoid serious side-effects of the 0 therapeutic RNA/of the composition via administration to the anterior part of the eye of a subject. A "safe and effective amount" of the therapeutic nucleic acid, preferably the RNA of the composition will furthermore vary in connection with the particular condition to be treated and also with the age and physical condition of the patient to be treated, the severity of the condition, the duration of the treatment, the nature of the accompanying therapy, of the particular pharmaceutically acceptable carrier used etc. Moreover, the "safe and effective amount" of the RNA or the composition as described herein may depend from application route (e.g. to the ciliary body, to the cornea, sub- conjunctival), application device (needle injection, injection device), and/or complexation/formulation (e.g. RNA in association with a polymeric carrier or LNP). Moreover, the "safe and effective amount" of the RNA or the composition may depend from the condition of the treated subject (infant, immunocompromised human subject etc.). In the context of the invention, a "composition" refers to any type of composition in which the specified ingredients (e.g. RNA encoding peptide or protein as specified above e.g. in association with a polymeric carrier or LNP), may be incorporated, optionally along with any further constituents, usually with at least one pharmaceutically acceptable carrier or exdpient. The composition may be a dry composition such as a powder or granules, or a solid unit such as a lyophilized form. Alternatively, the composition may be in liquid form, and each constituent may be independently incorporated in dissolved or dispersed (e.g. suspended or emulsified) form. Carrier/LNP In preferred embodiments, the composition comprises at least one therapeutic nucleic acid, preferably RNA and at least one pharmaceutically acceptable carrier or excipient. Accordingly, the composition is preferably pharmaceutical composition. Preferably, the carrier or excipient is suitable for ocular administration, preferably for administration to the anterior segment of the eye of a subject in need of treatment. The term "pharmaceutically acceptable carrier" or "pharmaceutically acceptable excipient" as used herein preferably includes the liquid or non-liquid basis of the composition for administration. If the composition is provided in liquid form, the carrier may be water, e.g. pyrogen-free water; isotonic saline or buffered (aqueous) solutions, e.g. phosphate, citrate etc. buffered solutions. Water or preferably a buffer, more preferably an aqueous buffer, may be used, containing a sodium salt, preferably at least 50mM of a sodium salt, a calcium salt, preferably at least 0.01 mM of a calcium salt, and optionally a potassium salt, preferably at least 3mM of a potassium salt. Accordingly, in embodiments, the composition may comprise pharmaceutically acceptable carriers or excipients using one or more pharmaceutically acceptable carriers or excipients to e.g. increase stability, increase cell transfection, permit the sustained or delayed, increase the translation of encoded protein in vivo, and/or alter the release profile of encoded protein in vivo. In addition to traditional excipients such as any and all solvents, dispersion media, diluents, or other liquid vehicles, dispersion or suspension aids, surface active agents, isotonic agents, thickening or emulsifying agents, preservatives, exdpients of the present invention can include, without limitation, lipidoids, liposomes, lipid nanoparticles, polymers, lipoplexes, core-shell nanoparticles, peptides, proteins, cells transfected with polynudeotides, hyaluronidase, nanopartide mimics and combinations thereof. In embodiments, one or more compatible solid or liquid fillers or diluents or encapsulating compounds may be used as well, which are suitable/compatible for administration to a subject. The term "compatible" as used herein means that the constituents of the composition are capable of being mixed with the at least one therapeutic nucleic acid and, optionally, a plurality of therapeutic nucleic acids of the composition, in such a manner that no interaction occurs, which would substantially reduce the biological activity or the pharmaceutical effectiveness of the composition under typical use conditions (e.g., administration to the anterior segment of the eye). Pharmaceutically acceptable carriers or exdpients must have sufficiently high purity and sufficiently low toxicity to make them suitable for administration to a subject to be treated. Compounds which may be used as pharmaceutically acceptable carriers or excipients may be sugars, such as, for example, lactose, glucose, trehalose, mannose, and sucrose; starches, such as, for example, corn starch or potato starch; dextrose; cellulose and its derivatives, such as, for example, sodium carboxymethylcellulose, ethylcellulose, cellulose acetate; powdered tragacanth; malt; gelatin; tallow; solid glidants, such as, for example, stearic acid, magnesium stearate; calcium sulfate; vegetable oils, such as, for example, groundnut oil, cottonseed oil, sesame oil, olive oil, corn oil and oil from theobroma; polyols, such as, for example, polypropylene glycol, glycerol, sorbitol, mannitol and polyethylene glycol; alginicacid. The term "subject", "patient" or "individual" as used herein generally includes humans and non-human animals and preferably mammals, including chimeric and transgenic animals and disease models. Subjects to which administration of the compositions, preferably the composition comprising the therapeutic RNA, is contemplated include, but are not limited to, humans and/or other primates; mammals, including commercially relevant mammals such as cattle, pigs, 5 horses, sheep, cats, dogs; and/or birds, including commercially relevant birds such as poultry, chickens, ducks, geese, and/or turkeys. Preferably, the term "subject" refers a non-human primate or a human, most preferably a human. In preferred embodiments, a "subject in need of treatment", or a "subject in need thereof in the context of the invention is a human subject. Cationic or polycationic compound 10 In a preferred embodiment, the at least one therapeutic nucleic acid, preferably the RNA of the composition is complexed or associated with, or at least partially complexed or partially associated with one or more cationic or polycationic compound, preferably cationic or polycationic polymer, cationic or polycationic polysaccharide, cationic or polycationic lipid, cationic or polycationic protein, or cationic or polycationic peptide, or any combinations thereof. The term "cationic or polycationic compound" as used herein will be recognized and understood by the person of 15 ordinary skill in the art, and are e.g. intended to refer to a charged molecule, which is positively charged at a pH value ranging from about 1 to 9, at a pH value ranging from about 3 to 8, at a pH value ranging from about 4 to 8, at a pH value ranging from about 5 to 8, more preferably at a pH value ranging from about 6 to 8, even more preferably at a pH value ranging from about 7 to 8, most preferably at a physiological pH, e.g. ranging from about 7.2 to about 7.5. Accordingly, a cationic component, e.g. a cationic peptide, cationic protein, cationic polymer, cationic polysaccharide, 20 cationic lipid may be any positively charged compound or polymer which is positively charged under physiological conditions. A "cationic or polycationic peptide or protein" may contain at least one positively charged amino acid, or more than one positively charged amino acid, e.g. selected from Arg, His, Lys or Orn. Accordingly, "polycationic" components are also within the scope exhibiting more than one positive charge under the given conditions. In this context it is particularly preferred that the therapeutic nucleic acid, preferably the RNA of the composition is 25 complexed or at least partially complexed with a cationic or polycationic compound and/or a polymeric carrier, preferably cationic proteins or peptides. In this context, the disclosure of W02010/037539 and W02012/113513 is incorporated herewith by reference. Partially means that only a part of the RNA of the composition is complexed with a cationic compound and that the rest of the RNA is present in uncomplexed form ("free"). Further preferred cationic or polycationic proteins or peptides that may be used for complexation can be derived from 30 formula (Arg)l;(Lys)m;(His)n;(Orn)o;(Xaa)x of the patent application W02009/030481 or W02011/026641, the disclosure ofW02009/030481 orW02011/026641 relating thereto incorporated herewith by reference. In preferred embodiments, the at least one therapeutic nucleic acid, preferably the RNA is complexed or at least partially complexed with at least one cationic or polycationic proteins or peptides preferably selected from SEQ ID NOs: 35127-490 or any combinations thereof. Polymeric carrier According to embodiments, the composition comprises at least one therapeutic RNA as defined herein, and a polymeric carrier. 0 The term "polymeric carrier" as used herein will be recognized and understood by the person of ordinary skill in the art, and is e.g. intended to refer to a compound that facilitates transport and/or complexation of another compound (e.g. RNA cargo). A polymeric carrier is typically a carrier that is formed of a polymer. A polymeric carrier may be associated to its cargo (e.g. RNA) by covalent or non-covalent interaction. A polymer may be based on different subunits, such as a copolymer. In some embodiments, the polymeric carrier comprises PEI. In some embodiments, PEI is branched PEI. PEI may be a branched PEI of a molecular weight ranging from 10 to 40 kDA, e.g., 25 kDa. In some embodiments, PEI is linear PEI. In some embodiments, the PEI nanoparticle has a mean diameter of or less than about 60nm (e.g., of or less than about 55nm, of or less than about 50nm, of or less than about 45nm, of or less than about 40nm, of or less than about 35nm, of or less than about 30nm, or of or less than about 25nm). Suitable nanoparticles may be in the range of25nm to 60nm, e.g.30nm to 50nm. As used herein, the mean diameter may be represented by the z-average as determined by dynamic light scattering as commonly known in the art. When PEI is present, it may be branched PEI of a molecular weight ranging from 10 to 40 kDA, e.g., 25 kDa branched PEI (Sigma #408727 In a preferred embodiment, the at least one therapeutic nucleic acid, preferably RNA of the composition is complexed or associated with a polyethylene glycol/peptide polymer comprising HO-PEG5000-S-(S-CHHHHHHRRRRHHHHHHC-S- )7-S-PEG5000-OH (SEQ ID NO: 125 as peptide monomer). In particulariy preferred embodiments, the at least one therapeutic nucleic acid, preferably RNA of the composition is complexed or associated with a polyethylene glycol/peptide polymer comprising HO-PEG5000-S-(S- CGHHHHHRRRRHHHHHGC-S-)4-S-PEG5000-OH (SEQ ID N0:126 as peptide monomer). Lipidoid In other embodiments, the composition comprises at least one therapeutic nucleic acid, preferably RNA complexed or associated with polymeric carriers and, optionally, with at least one lipid or lipidoid component as described in published PCT applications W02017/212008A1 , W02017/212006A1, W02017/212007A1, and W02017/212009A1. In this context, the disclosures of W02017/212008A1, W02017/212006A1, W02017/212007A1, and W02017/212009A1 are herewith incorporated by reference. In a preferred embodiment, the polymeric carrier is a peptide polymer, preferably a polyethylene glycol/peptide polymer as defined above, and a lipid component, preferably a lipidoid component, more preferably lipidoid component. A lipidoid compound, also simply referred to as lipidoid (or lipidoit), is a lipid-like compound, i.e. an amphiphilic compound with lipid-like physical properties. The lipidoid compound is preferably a compound which comprises two or more cationic nitrogen atoms and at least two lipophilic tails. In contrast to many conventional cationic lipids, the lipidoid compound may be free of a hydrolysable linking group, in particular linking groups comprising hydrolysable ester, amide or carbamate groups. The cationic nitrogen atoms of the lipidoid may be cationisable or permanently cationic, or both types of cationic nitrogens may be present in the compound. In the context of the present invention the term lipid is considered to also encompass lipidoid compounds. In some embodiments of the inventions, the lipidoid compound comprises a PEG moiety. In a embodiment, at least one lipidoid component is selected from 3-C12-OH, 3-C12-OH-cat, or3-C12-C3-OH. Further suitable lipidoid compounds may be derived from published PCT patent application W02010/053572. In particular, lipidoid compounds derivable from claims 1 to 297 of published PCT patent application W02010/053572 may be used in the context of the invention, e.g. incorporated into the peptide polymer as described herein, or e.g. incorporated into the lipid nanoparticle (as described below). Accordingly, claims 1 to 297 of published PCT patent application W02010053572, and the specific disclosure relating thereto, is herewith incorporated by reference. Lipid-based carriers In preferred embodiments, the at least one therapeutic nucleic acid, preferably RNA of the composition is complexed, partially complexed, encapsulated, partially encapsulated, or associated with one or more lipids (e.g. cationic lipids and/or neutral lipids), thereby forming liposomes, lipid nanoparticles (LNPs), lipoplexes, and/or nanoliposomes. In preferred embodiments, at least one therapeutic nucleic acid, preferably RNA is formulated in lipid-based carriers. In the context of the invention, the term "lipid-based carriers" encompass lipid-based delivery systems for therapeutic RNA (e.g. mRNA) that comprise a lipid component. A lipid-based carrier may additionally comprise other components suitable for encapsulating/incorporating/complexing a therapeutic RNA (e.g. mRNA) including a cationic or polycationic polymer, a cationic or polycationic polysaccharide, a cationic or polycationic protein, a cationic or polycationic peptide, or any combinations thereof. In the context of the invention, a typical "lipid-based carrier" is selected from liposomes, lipid nanoparticles (LNPs), lipoplexes, solid lipid nanoparticles, and/or nanoliposomes. The therapeutic nucleic add/RNA, preferably the mRNA of the composition may completely or partially incorporated or encapsulated in a lipid-based carrier, wherein the therapeutic RNA (e.g. mRNA) may be located in the interior space of the lipid-based carrier, within the lipid layer/membrane of the lipid-based carrier, or associated with the exterior surface of the lipid-based carrier. The incorporation of therapeutic RNA, preferably the mRNA into lipid-based carriers may be referred to as "encapsulation". A "lipid-based carrier" is not restricted to any particular morphology, and include any morphology generated when e.g. an aggregation reducing lipid and at least one further lipid are combined, e.g. in an aqueous environment in the presence of therapeutic RNA (e.g. mRNA). For example, an LNP, a liposome, a lipid complex, a lipoplex and the like are within the scope of the term "lipid-based carrier". Lipid-based carriers can be of different sizes such as, but not limited to, a multilamellar vesicle (MLV) which may be hundreds of nanometers in diameter and may contain a series of concentric bilayers separated by narrow aqueous compartments, a small unicellular vesicle (SUV) which may be smaller than 50nm in diameter, and a large unilamellar vesicle (LUV) which may be between 50nm and 500nm in diameter. Liposomes, a specific type of lipid-based carrier, are characterized as microscopic vesicles having an interior aqua space sequestered from an outer medium by a membrane of one or more bilayers. In a liposome, the at least one therapeutic RNA (e.g. mRNA) is typically located in the interior aqueous space enveloped by some or the entire lipid portion of the liposome. Bilayer membranes of liposomes are typically formed by amphiphilic molecules, such as lipids of synthetic or natural origin that comprise spatially separated hydrophilic and hydrophobic domains. Lipid nanoparticles (LNPs), a specific type of lipid-based carrier, are characterized as microscopic lipid particles having a solid core or partially solid core. Typically, an LNP does not comprise an interior aqua space sequestered from an outer medium by a bilayer. In an LNP, the at least one therapeutic RNA (e.g.mRNA) may be encapsulated or incorporated in the lipid portion of the LNP enveloped by some or the entire lipid portion of the LNP. An LNP may comprise any lipid capable of forming a particle to which the therapeutic RNA (e.g. mRNA) may be attached, or in which the therapeutic RNA may be encapsulated. Preferably, said lipid-based carriers are particularly suitable for ocular administration. In preferred embodiments, the lipid-based carriers of the composition are selected from liposomes, lipid nanoparticles, lipoplexes, solid lipid nanoparticles, lipo-polylexes, and/or nanoliposomes. In preferred embodiments, the therapeutic nucleic acid/RNA of the composition is formulated or encapsulated or complexed or associated with at least one lipid- based carrier. In most preferred embodiments, the lipid-based carriers are lipid nanoparticles, preferably wherein the lipid nanoparticles encapsulate the therapeutic nucleic acid/RNA. The term "encapsulated", e.g. incorporated, complexed, encapsulated, partially encapsulated, associated, partially associated, refers to the essentially stable combination of therapeutic nucleic acid/RNA, preferably mRNA with one or more lipids into lipid-based carriers (e.g. larger complexes or assemblies) preferably without covalent binding of the therapeutic RNA. The lipid-based carriers - encapsulated therapeutic nucleic acid/RNA (e.g. mRNA) may be completely or partially located in the interior of the lipid-based carrier (e.g. the lipid portion and/or an interior space) and/or within the lipid layer/membrane of the lipid-based carriers. The encapsulation of a therapeutic RNA (e.g. mRNA) into lipid-based carriers is also referred to herein as "incorporation" as the therapeutic nucleic acid/RNA (e.g.m RNA) is preferably contained within the interior of the lipid-based carriers. Without wishing to be bound to theory, the purpose of incorporating or encapsulating the therapeutic nucleic acid/RNA into lipid-based carriers may be to protect the therapeutic nucleic acid/RNA from an environment which may contain enzymes, chemicals, or conditions that degrade the therapeutic nucleic acid/RNA (e.g. mRNA). Moreover, incorporating the therapeutic RNA into lipid-based carriers may promote the uptake of the therapeutic nucleic acid/RNA and their release from the endosomal compartment, and hence, may enhance the therapeutic effect of the therapeutic nucleic acid/RNA (e.g. mRNA) when administered to a cell or a subject. In this context, the terms "complexed" or "associated" refer to the essentially stable combination of therapeutic nucleic acid/RNA as defined herein with one or more lipids into larger complexes or assemblies without covalent binding. The term "lipid nanoparticle", also referred to as "LNP", is not restricted to any particular morphology, and include any morphology generated when a cationic lipid and optionally one or more further lipids are combined, e.g. in an aqueous environment and/or in the presence of RNA. E.g., a liposome, a lipid complex, a lipoplex and the like are within the scope of an LNP. Liposomes, lipid nanoparticles (LNPs), lipoplexes, and/or nanoliposomes can be of different sizes such as, but not limited to, a multilamellar vesicle (MLV) which may be hundreds of nanometers in diameter and may contain a series of concentric bilayers separated by narrow aqueous compartments, a small unicellular vesicle (SUV) which may be smaller than 50nm in diameter, and a large unilamellar vesicle (LUV) which may be between 50nm and 500nm in diameter. LNPs of the invention are suitably characterized as microscopic vesicles having an interior aqua space sequestered from an outer medium by a membrane of one or more bilayers. Bilayer membranes of LNPs are typically formed by amphiphilic molecules, such as lipids of synthetic or natural origin that comprise spatially separated hydrophilic and hydrophobic domains. Bilayer membranes of the liposomes can also be formed by amphophilic polymers and surfactants (e.g., polymerosomes, niosomes, etc.). In the context of the present invention, an LNP typically serves to transport the therapeutic RNA of the composition to a target cell or tissue in the eye. Accordingly, in preferred embodiments, the at least one therapeutic RNA of the composition is complexed with one or more lipids thereby forming lipid nanoparticles (LNP). In preferred embodiments, the lipid-based carriers of the composition comprise at least one or more lipids selected from at least one aggregation-reducing lipid, at least one cationic lipid, at least one neutral lipid or phospholipid, or at least one steroid or steroid analog, or any combinations thereof. In preferred embodiments, the lipid-based carriers of the composition comprise (i) an aggregation-reducing lipid, (ii) a cationic lipid or ionizable lipid, and (iii) a neutral lipid/phospholipid or a steroid/steroid analog. In particularly preferred embodiments, the lipid-based carriers of the composition comprise an (i) aggregation-reducing lipid, (ii) a cationic lipid or ionizable lipid, (iii) a neutral lipid or phospholipid, (iv) and a steroid or steroid analog. In some embodiments, the cationic lipid or ionizable lipid is selected from an amino lipid, preferably wherein the amino lipid comprises a tertiary amine group. Cationic lipid of LNP LNPs typically comprise a cationic lipid and one or more excipient selected from neutral lipids, charged lipids, steroids and polymer conjugated lipids (e.g. PEGylated lipid). The at least one therapeutic RNA may be encapsulated in the lipid portion of the LNP or an aqueous space enveloped by some or the entire lipid portion of the LNP. The at least one therapeutic RNA or a portion thereof may also be associated and complexed with the LNP. An LNP may comprise any lipid capable of forming a particle to which the therapeutic RNA are attached, or in which the one or therapeutic RNAs are encapsulated. Preferably, the LNP comprising therapeutic RNA comprises one or more cationic lipids, and one or more stabilizing lipids. Stabilizing lipids include neutral lipids and PEGylated lipids.. The cationic lipid of an LNP may be cationisable, i.e. it becomes protonated as the pH is lowered below the pK of the ionizable group of the lipid, but is progressively more neutral at higher pH values. At pH values below the pK, the lipid is then able to associate with negatively charged nucleic acids. In certain embodiments, the cationic lipid comprises a zwitterionic lipid that assumes a positive charge on pH decrease. The LNP may comprise any further cationic or cationisable lipid, i.e. any of a number of lipid species which carry a net positive charge at a selective pH, such as physiological pH. Such lipids include, but are not limited to, DSDMA, N,N-dioleyl-N,N-dimethylammonium chloride (DODAC), N,N- distearyl-N,N-dimethylammonium bromide (DDAB), 1,2-dioleoyltrimethyl ammonium propane chloride (DOTAP) (also known as N-(2,3-dioleoyloxy)propyl)-N,N,N-trimethylammonium chloride and 1,2-Dioleyloxy-3-trimethylaminopropane chloride salt), N-(1-(2,3-dioleyloxy)propyl)-N,N,N-trimethylammonium chloride (DOTMA), N,N-dimethyl-2,3- dioleyloxy)propylamine (DODMA), ckk-E12, ckk, 1,2-DiLinoleyloxy-N,N-dimethylaminopropane (DLinDMA), 1,2- Dilinolenyloxy-N,N-dimethylaminopropane (DLenDMA), 1,2-di-y-linolenyloxy-N,N-dimethylaminopropane (y-DLenDMA), 98N12-5,1,2-Dilinoleylcarbamoyloxy-3-dimethylaminopropane (DLin-C-DAP), 1,2-Dilinoleyoxy-3- (dimethylamino)acetoxypropane (DLin-DAC), 1,2-Dilinoleyoxy-3-morpholinopropane (DLin-MA), 1,2-Dilinoleoyl-3- dimethylaminopropane (DLinDAP), 1,2-Dilinoleylthio-3-dimethylaminopropane (DLin-S- DMA), l-Linoleoyl-2-linoleyloxy- 3-dimethylaminopropane (DLin-2-DMAP), 1,2-Dilinoleyloxy-3-trimethylaminopropane chloride salt (DLin-TMA.CI), ICE (Imidazol-based), HGT5000, HGT5001, DMDMA, CLinDMA, CpLinDMA, DMOBA, DOcarbDAP, DLincarbDAP, DLinCDAP, KLin-K-DMA, DLin-K-XTC2-DMA, XTC (2,2-Dilinoleyl-4-dimethylaminoethyl-[l,3]-dioxolane) HGT4003,1,2- Dilinoleoyl-3-trimethylaminopropane chloride salt (DLin-TAP.CI), 1,2-Dilinoleyloxy-3-(N- methylpiperazino)propane (DLin-MPZ), or3-(N,N-Dilinoleylamino)-1,2-propanediol (DLinAP), 3-(N,N-Dioleylamino)-1,2-propanedio (DOAP), 1,2- Dilinoleyloxo-3-(2-N,N- dimethylamino)ethoxypropane (DLin-EG-DM A), 2,2-Dilinoleyl-4-dimethylaminomethyl-[1,3]- dioxolane (DLin-K-DMA) or analogs thereof, (3aR,5s,6aS)-N,N-dimethyl-2,2-di((9Z,12Z)-octadeca-9,12- dienyl)tetrahydro-3aH-cyclopenta[d][1,3]dioxol-5-amine,(6Z,9Z,28Z,31Z)-heptatriaconta-6,9,28,31-tetraen-19-yl-4- (dimethylamino)butanoate (MC3), ALNY-100 ((3aR,5s,6aS)- N,N-dimethyl-2,2-di((9Z,12Z)-octadeca-9,12- dienyl)tetrahydro-3aH-cyclopenta[d] [1 ,3]dioxol- 5-amine)), 1,1 '-(2-(4-(2-((2-(bis(2-hydroxydodecyl)amino)ethyl)(2- hydroxydodecyl)amino)ethyl)piperazin-1-yl)ethylazanediyl)didodecan-2-ol(C12-200),2,2-dilinoleyl-4-(2- dimethylaminoethyl)-[1,3]-dioxolane (DLin-K-C2-DMA), 2,2-dilinoleyl-4-dimethylaminomethyl-[1 ,3]-dioxolane (DLin-K- DMA), N098-5(4,7,13-tris(3-oxo-3-(undecylamino)propyl)-NI ,N 16-diundecyl-4,7, 10,13-tetraazahexadecane-l,16- diamide), (6Z,9Z,28Z,31Z)-heptatriaconta-6,9,28,31-tetraen-19-yl4-(dimethylamino) butanoate (DLin-M-CS-DMA), 3- ((6Z,9Z,28Z,31Z)-heptatriaconta-6,9,28,31-tetraen-19-yloxy)-N,N-dimethylpropan-1-amine(MC3 Ether), 4- ((6Z,9Z,28Z,31Z)-heptatriaconta-6,9,28,31-tetraen-19-yloxy)-N,N-dimethylbutan-l-amine(MC4 Ether), LIPOFECTIN® (commercially available cationic liposomes comprising DOTMA and 1,2-dioleoyl-sn-3phosphoethanolamine (DOPE), from GIBCO/BRL, Grand Island, N.Y.); LIPOFECTAMINE® (commercially available cationic liposomes comprising N- (1-(2,3dioleyloxy)propyl)-N-(2-(sperminecarboxamido)ethyl)-N,N-dimethylammoniumtrifluoroacetate(DOSPA)and (DOPE), from GIBCO/BRL); and TRANSFECTAM® (commercially available cationic lipids comprising dioctadecylamidoglycyl carboxyspermine (DOGS) in ethanol from Promega Corp., Madison, Wis.) or any combination of any of the foregoing. Further suitable cationic lipids for use in the compositions and methods of the invention include those described in international patent publications W02010/053572 (and particularly, Cl 2-200 described at paragraph [00225]) and W02012/170930, both of which are incorporated herein by reference, HGT4003, HGT5000, HGTS001, HGT5001, HGT5002 (see US20150140070A1). In some embodiments, the lipid is selected from the group consisting of98N12-5, C12-200, and ckk-E12. In one embodiment, the further cationic lipid is an amino lipid. Representative amino lipids include, but are not limited to, 1 ,2-dilinoleyoxy-3-(dimethylamino)acetoxypropane (DLin- DAC), 1,2-dilinoleyoxy-3morpholinopropane (DLin-MA), 1,2-dilinoleoyl-3-dimethylaminopropane (DLinDAP), 1,2- dilinoleylthio-3-dimethylaminopropane (DLin-S-DMA), 1-linoleoyl-2-linoleyloxy-3dimethylaminopropane (DLin-2-DMAP), 1,2-dilinoleyloxy-3-trimethylaminopropane chloride salt (DLin-TMA.CI), 1,2-dilinoleoyl-3-trimethylaminopropane chloride salt (DLin-TAP.CI), 1,2-dilinoleyloxy-3-(N-methylpiperazino)propane (DLin-MPZ), 3-(N,Ndilinoleylamino)-1,2- propanediol (DLinAP), 3-(N,N-dioleylamino)-1,2-propanediol (DOAP), 1,2-dilinoleyloxo-3-(2-N,N- dimethylamino)ethoxypropane (DLin-EG-DMA), and 2,2-dilinoleyl-4-dimethylaminomethyl-[1,3]-dioxolane (DLin-K- DMA), 2,2-dilinoleyl-4-(2-dimethylaminoethyl)-[l,3]-dioxolane (DLin-KC2-DMA); dilinoleyl-methyl-4- dimethylaminobutyrate (DLin-MCS-DMA); MC3 (US20100324120). In one embodiment, the at least one therapeutic nucleic acid/RNA may be formulated in an aminoalcohol lipidoid. Aminoalcohol lipidoids which may be used in the present invention may be prepared by the methods described in U.S. Patent No.8,450,298, herein incorporated by reference in its entirety. Suitable (ionizable) lipids can also be the compounds as disclosed in Tables 1, 2 and 3 and as defined in claims 1-24 of published PCT patent application W02017/075531 A1, the specific disclosure hereby incorporated by reference. In another embodiment, ionizable lipids can also be the compounds as disclosed in published PCT patent application W02015/074085A1 (i.e. ATX-001 to ATX-032 or the compounds as specified in claims 1-26), U.S. Appl. Nos. 61/905,724 and 15/614,499 or U.S. Patent Nos.9,593,077 and 9,567,296, the specific disclosure hereby incorporated by reference. In that context, any lipid derived from generic formula (X1)
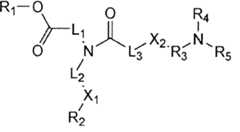
wherein, Ri and R2 are the same or different, each a linear or branched alkyl consisting of 1 to 9 carbons, an alkenyl or alkynyl consisting of2 to 11 carbons, Li and L2 are the same or different, each a linear alkylene or alkenylene consisting of 5 to 18 carbons, or forming a heterocycle with N, Xi is a bond, or is -CO-0- whereby -L2-CO-0-R2 is formed, X2 is S or 0, L3 is a bond or a linear or branched alkylene consisting of 1 to 6 carbons, or forming a heterocycle with N, R3 is a linear or branched alkylene consisting of 1 to 6 carbons, and R4 and R 5 are the same or different, each hydrogen or a linear or branched alkyl consisting of 1 to 6 carbons; or a pharmaceutically acceptable salt thereof may be suitably used. In other embodiments, suitable cationic lipids can also be the compounds as disclosed in published PCT patent application W02017/117530A1 (i.e. lipids 13,14,15,16,17,18,19,20, or the compounds as specified in the claims), the specific disclosure hereby incorporated by reference. In that context, any lipid derived from generic formula (X2)
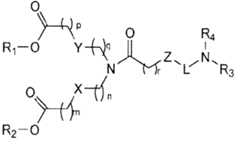
wherein X is a linear or branched alkylene or alkenylene, monocydic, bicyclic, or tricyclic arene or heteroarene; Y is a bond, an ethene, or an unsubstituted or substituted aromatic or heteroaromatic ring; Z is S or 0; 5 L is a linear or branched alkylene of 1 to 6 carbons; R-3 and R4 are independently a linear or branched alkyl of 1 to 6 carbons; Ri and R2 are independently a linear or branched alkyl or alkenyl of 1 to 20 carbons; r is 0 to 6; and m,n,p,and q are independently 1 to 18; wherein when n=q, m=p, and Ri=R2, then X and Y differ; 10 wherein when X=Y, n=q, m=p, then Ri and R2 differ; wherein when X=Y, n=q, and Ri=R2, then m and p differ; and wherein when X=Y, m=p, and Ri=R2, then n and q differ; or a pharmaceutically acceptable salt thereof. 15 In preferred embodiments, a lipid may be used derived from formula (X2), wherein, X is a bond, linear or branched alkylene, alkenylene, or monocyclic, bicyclic, or tricydic arene or heteroarene; Y is a monocyclic, bicyclic, or tricyclic arene or heteroarene; Z is S orO; Lisa linear or branched alkylene of 1 to 6 carbons; R3 and R4 are independently a linear or branched alkyl of 1 to 6 carbons; Ri and R2 are independently a linear or branched alkyl or alkenyl of 1 to 20 carbons; r is 0 to 6; and m, n,p,and q are independently 1 to 18; ora pharmaceutically acceptable salt thereof may be 20 suitably used. In further embodiments, the lipid formulation comprises cationic or ionizable lipids as defined in Formula I of paragraph [00251] ofW02021222801 or a lipid selected from the disclosure of paragraphs [00260] or [00261] of W02021222801. In other embodiments, the lipid formulation comprises cationic or ionizable lipids selected from the group consisting of ATX-001 toATX-132 as disclosed in claim 90 of W02021183563, preferably ATX-0126. The disclosure of 25 W02021222801 and W02021183563, especially aforementioned lipids, are incorporated herewith by reference. In embodiments, cationic or ionizable lipids may be selected from the lipids disclosed in W02018078053 (i.e. lipids derived from formula I, II, and III of W02018078053, or lipids as specified in claims 1 to 12 of W02018078053), the disclosure of W02018078053 hereby incorporated by reference in its entirety. In that context, lipids disclosed in Table 7 of W02018078053 (e.g. lipids derived from formula 1-1 to 1-41) and lipids disclosed in Table 8 of W02018078053 (e.g. 30 lipids derived from formula 11-1 to II-36) may be suitably used in the context of the invention. Accordingly, formula 1-1 to formula 1-41 and formula 11-1 to formula II-36 ofW02018078053A, and the specific disclosure relating thereto, are herewith incorporated by reference. In preferred embodiments, the lipid-based carriers of the composition comprise a cationic lipid selected or derived from structures 111-1 to III-36 of Table 9 of published PCT patent application W02018078053. Accordingly, formula 111-1 to III- 3536 of W02018078053, and the specific disclosure relating thereto, are herewith incorporated by reference. In preferred embodiments, the lipid-based carriers comprises a cationic lipid selected or derived from formula 111-3:
(111-3) The lipid of formula 111-3 as suitably used herein has the chemical term ((4-hydroxybutyl)azanediyl)bis(hexane-6,1- diyl)bis(2-hexyldecanoate), also referred to as ALC-0315 i.e. CAS Number 2036272-55-4. Further suitable cationic lipids may be selected or derived from cationic lipids according to PCT claims 1 to 14 of published patent application W02021123332, or table 1 ofW02021123332, the disclosure relating to claims 1 to 14 or table 1 of W02021123332 herewith incorporated by reference. Accordingly, suitable cationic lipids may be selected or derived from cationic lipids according to Compound 1 to Compound 27 (C1 - C27) of Table 1 ofW02021123332. In other preferred embodiments, the lipid-based carriers of the composition comprise a cationic lipid selected or derived from (COATSOME®SS-EC) SS-33/4PE-15 (see C23 in Table 1 of W02021123332). In other preferred embodiments, the lipid-based carriers of the composition comprise a cationic lipid selected or derived from HEXA-C5DE-PipSS (see C2 in Table 1 ofW02021123332). In most preferred embodiments, the lipid-based carriers of the composition comprise a cationic lipid selected or derived from compound C26 as disclosed in Table 1 of W02021123332:

In other embodiments, the lipid-based carriers of the I composition comprise a cationic lipid selected or derived from 9- Heptadecanyl 8-{(2-hydroxyethyl)[6-oxo-6-(undecyloxy)hexyl]amino}octanoate, also referred to as SM-102. Other preferred lipid-based carriers (e.g. LNPs) of the composition comprise a squaramide ionizable amino lipid, more preferably a cationic lipid selected from the group consisting of formulas (M1) and (M2):

wherein the substituents (e.g. Ri, R2, Rs, R5, Re, R?, Rio, M, Mi, m, n, o, I) are defined in claims 1 to 13 of US10392341 B2; US10392341 B2 being incorporated herein in its entirety. Accordingly, in embodiments, the lipid-based carriers of the composition comprise a cationic lipid selected or derived from above mentioned ALC-0315, SM-102, SS-33/4PE-15, HEXA-C5DE-PipSS, or compound C26 (see C26 in Table 1 of W02021123332). In certain embodiments, the cationic lipid is present in the LNP in an amount from about 30 to about 95 mole percent, relative to the total lipid content of the LNP. If more than one cationic lipid is incorporated within the LNP, such percentages apply to the combined cationic lipids. In one embodiment, the cationic lipid is present in the LNP in an amount from about 30 to about 70 mole percent. In one embodiment, the cationic lipid is present in the LNP in an amount from about 40 to about 60 mole percent, such as about 40,41,42, 43,44,45,46, 47, 48,49,50,51, 52,53,54,55,56, 57, 58, 59 or 60 mole percent, respectively. In embodiments, the cationic lipid is present in the LNP in an amount from about 47 to about 48 mole percent, such as about 47.0,47.1, 47.2, 47.3, 47.4, 47.5, 47.6, 47.7, 47.8, 47.9, 50.0 mole percent, respectively, wherein 47.7 mole percent are particularly preferred. In some embodiments, the cationic lipid is present in a ratio of from about 20mol% to about 70 or 75mol% or from about 45 to about 65mol% or about 20,25,30,35, 40,45,50,55, 60, 65, or about 70mol% of the total lipid present in the LNP. In further embodiments, the LNPs comprise from about 25% to about 75% on a molar basis ofcationic lipid, e.g., from about 20 to about 70%, from about 35 to about 65%, from about 45 to about 65%, about 60%, about 57.5%, about 57.1 %,about 50% or about 40% on a molar basis (based upon 100% total moles of lipid in the LNP). In some embodiments, the ratio of cationic lipid to RNA is from about 3 to about 15, such as from about 5 to about 13 or from about 7 to about 11. In some embodiments of the invention the LNP comprises a combination or mixture of any the lipids described above. Other suitable (cationic or ionizable) lipids are disclosed in published patent applications W02009/086558, W02009/127060, W02010/048536, W02010/054406, W02010/088537, W02010/129709, W02011/153493, WO 2013/063468, US2011/0256175, US2012/0128760, US2012/0027803, US8158601, W02016/118724, W02016/118725, W02017/070613, W02017/070620, W02017/099823, W02012/040184, W02011/153120, W02011/149733, W02011/090965, W02011/043913, W02011/022460, W02012/061259, W02012/054365, W02012/044638, W02010/080724, W02010/21865, W02008/103276, W02013/086373, W02013/086354, and US Patent Nos.7,893,302, 7,404,969, 8,283,333, 8,466,122 and 8,569,256 and US Patent Publication No. US2010/0036115, US2012/0202871, US2013/0064894, US2013/0129785, US2013/0150625, US20130178541, US2013/0225836, US2014/0039032 and W02017/112865. In that context, the disclosures ofW02009/086558, W02009/127060, W02010/048536, W02010/054406, W02010/088537, W02010/129709, W02011/153493, WO 2013/063468, US2011/0256175, US2012/0128760, US2012/0027803, US8158601,W02016/118724, W02016/118725, W02017/070613, W02017/070620, W02017/099823, W02012/040184, W02011/153120, W02011/149733, W02011/090965, W02011/043913, W02011/022460, W02012/061259, W02012/054365, W02012/044638, W02010/080724, W02010/21865, W02008/103276, W02013/086373, W02013/086354, US Patent Nos.7,893,302, 7,404,969, 8,283,333, 8,466,122 and 8,569,256 and US Patent Publication No. US2010/0036115, US2012/0202871, US2013/0064894, US2013/0129785, US2013/0150625, US20130178541, US2013/0225836 and US2014/0039032 and W02017/112865 specifically relating to (cationic) lipids suitable for LNPs are incorporated herewith by reference. In some embodiments, amino or cationic lipids as defined herein have at least one protonatable or deprotonatable group, such that the lipid is positively charged at a pH at or below physiological pH (e.g. pH 7.4), and neutral at a second pH, preferably at or above physiological pH. It will, of course, be understood that the addition or removal of protons as a function of pH is an equilibrium process, and that the reference to a charged or a neutral lipid refers to the nature of the predominant species and does not require that all of lipids have to be present in the charged or neutral form. Lipids having more than one protonatable or deprotonatable group, or which are zwitterionic, are not excluded and may likewise suitable in the context of the present invention. In some embodiments, the protonatable lipids have a pKa of the protonatable group in the range of about 4 to about 11, e.g., a pKa of about 5 to about 7. LNPs can comprise two or more (different) cationic lipids. The cationic lipids may be selected to contribute different advantageous properties. E.g., cationic lipids that differ in properties such as amine pKa, chemical stability, half-life in circulation, half-life in tissue, net accumulation in tissue, ortoxicity can be used in the LNP. In particular, the cationic lipids can be chosen so that the properties of the mixed-LNP are more desirable than the properties ofasingle-LNP of individual lipids. The amount of the permanently cationic lipid or lipidoid may be selected taking the amount of the nucleic acid cargo into account. In one embodiment, these amounts are selected such as to result in an N/P ratio of the nanoparticle(s) or of the composition in the range from about 0.1 to about 20. In this context, the N/P ratio is defined as the mole ratio of the nitrogen atoms ("N") of the basic nitrogen-containing groups of the lipid or lipidoid to the phosphate groups ("P") of the RNA which is used as cargo. The N/P ratio may be calculated on the basis that e.g.1 pg RNA typically contains about Snmol phosphate residues, provided that the RNA exhibits a statistical distribution of bases. The "N"-value of the lipid or lipidoid may be calculated based on its molecular weight and the relative content of permanently cationic and - if present - cationisable groups. In preferred embodiments, the lipid-based carriers comprise a cationic lipid selected or derived from SM-102, SS- 33/4PE-15, HEXA-C5DE-PipSS, or compound C26 Neutral Lipid In preferred embodiments, the therapeutic nucleic acid/RNA of the composition is complexed with one or more lipids thereby forming LNPs, wherein the LNP additionally comprises one or more neutral lipid and/or one or more steroid or steroid analogue. Suitable stabilizing lipids include neutral lipids and anionic lipids. The term "neutral lipid" refers to any one of a number of lipid species that exist in either an uncharged or neutral zwitterionic form at physiological pH. Suitable neutral lipids include diacylphosphatidylcholines, diacylphosphatidylethanolamines, ceramides, sphingomyelins, dihydrosphingomyelins, cephalins, and cerebrosides. The selection of neutral lipids for use in the particles described herein is generally guided by consideration of, e.g., lipid particle size and stability of the lipid particle in the bloodstream. Preferably, the neutral lipid is a lipid having two acyl groups (e.g., diacylphosphatidylcholine and diacylphosphatidylethanolamine). In one embodiment, the neutral lipids contain saturated fatty acids with carbon chain lengths in the range of C10 to C20. In another embodiment, neutral lipids with mono or diunsaturated fatty acids with carbon chain lengths in the range ofC10 to C20 are used. Additionally, neutral lipids having mixtures of saturated and unsaturated fatty acid chains can be used. In embodiments, the LNP additionally comprises one or more neutral lipids, wherein the neutral lipid is selected from the group comprising distearoylphosphatidylcholine (DSPC), dioleoylphosphatidylcholine (DOPC), dipalmitoylphosphatidylcholine (DPPC), dioleoylphosphatidylglycerol (DOPG), dipalmitoylphosphatidylglycerol (DPPG), dioleoyl-phosphatidylethanolamine (DOPE), palmitoyloleoylphosphatidylcholine (POPC), palmitoyloleoyl- phosphatidylethanolamine (POPE) and dioleoyl-phosphatidylethanolamine 4-(N-maleimidomethyl)-cydohexane- lcarboxylate (DOPE-mal), dipalmitoyl phosphatidyl ethanolamine (DPPE), dimyristoylphosphoethanolamine (DMPE), distearoyl-phosphatidylethanolamine (DSPE), 16-0-monomethyl PE, 16-0-dimethyl PE, 18-1-trans PE, l-stearioyl-2- oleoylphosphatidyethanol amine (SOPE), and 1,2-dielaidoyl-sn-glycero-3-phophoethanolamine (transDOPE), 1,2- diphytanoyl-sn-glycero-3-phosphoethanolamine (DPhyPE), or mixtures thereof. In preferred embodiments, the neutral lipid of the lipid-based carriers of the composition is selected or derived from distearoylphosphatidylcholine (DSPC) In preferred embodiments, the neutral lipid of the lipid-based carriers of the composition is selected or derived from 1,2- diheptanoyl-sn-glycero-3-phosphocholine(DHPC). In other preferred embodiments, the neutral lipid of the lipid-based carriers of the composition is selected or derived from 1,2-diphytanoyl-sn-glycero-3-phosphoethanolamine (DPhyPE). Accordingly, in preferred embodiments, the lipid-based carriers of the composition comprise a neutral lipid selected or derived from DSPC, DHPC, or DPhyPE. Therefore, in another embodiment, the invention is related to the use ofalipid with high fusogenicity in a lipid-based carrier or nucleic acid-lipid particle, preferably DPhyPE, as depicted here:

(
DPhyPE). In various embodiments, the molar ratio of the cationic lipid to the neutral lipid in the lipid-based carriers ranges from about 2:1 to about 8:1. The neutral lipid is preferably from about 5mol% to about 90mol%, about 5mol% to about 10mol%, about 5,10,15,20, 25,30,35,40,45, 50,55,60,65,70,75, 80, 85, or about 90mol% of the total lipid present in the lipid-based carrier. In 5 one embodiment, the lipid-based carrier includes from about 0% to about 15% or 45% on a molar basis of neutral lipid, e.g., from about 3% to about 12% or from about 5% to about 10%. For instance, the lipid-based carrier may include about 15%, about 10%, about 7.5%, or about 7.1% of neutral lipid on a molar basis (based upon 100% total moles of lipid in the lipid-based carrier). In a most preferred embodiment, the lipid-based carrier includes from about 10% on a molar basis of neutral lipid. 10 In some embodiments, the LNPs comprise a neutral lipid selected from DSPC, DPPC, DMPC, DOPC,POPC, DOPE and SM. In various embodiments, the molar ratio of the cationic lipid to the neutral lipid ranges from about 2:1 to about 8:1. In preferred embodiments, the neutral lipid is 1,2-distearoyl-sn-glycero-3-phosphocholine (DSPC). The molar ratio of the cationic lipid to DSPC may be in the range from about 2:1 to 8:1. In preferred embodiments, the steroid is cholesterol. The molar ratio of the cationic lipid to cholesterol may be in the range from about 2:1 to 1:1. In some 15 embodiments, the cholesterol may be PEGylated. Stemids, steroid_analogs orsterols: In some embodiments, the lipid-based carriers of the composition comprise a steroid, steroid analog orsterol. Suitably, the steroid, steroid analog or sterol may be derived or selected from cholesterol, cholesteryl hemisuccinate (CHEMS) and a derivate thereof. In other embodiments, the lipid-based carriers of the composition comprise a steroid, 20 steroid analog or sterol derived from a phytosterol (e.g., a sitosterol, such as beta-sitosterol), preferably from a compound having the structure of Formula I as disclosed in claim 1 ofW02020061332; the disclosure of W02020061332, especially the disclosure of Formula I and phytosterols being incorporated herewith by reference. In a further embodiment, the steroid is an imidazole cholesterol ester or "ICE" as disclosed in paragraphs [0320] and [0339]- [0340] ofW02019226925A1;W02019226925A1 being incorporated herein by reference in its entirety. 25 The sterol can be about 10 mol % to about 60 mol % or about 25 mol % to about 40 mol % of the lipid particle. In one embodiment, the sterol is about 10,15,20,25,30, 35, 40,45,50,55, or about 60 mol % of the total lipid present in the lipid particle. In another embodiment, the LNPs include from about 5% to about 50% on a molar basis of the sterol, e.g., about 15% to about 45%, about 20% to about 40%, about 48%, about 40%, about 38.5%, about 35%, about 34.4%, about 31.5% or about 31% on a molar basis (based upon 100% total moles oflipid in the LNP). 30 In preferred embodiments, the lipid-based carriers comprise a steroid or steroid analog selected or derived from cholesterol, cholesteryl hemisuccinate (ChlEMS), preferably cholesterol. In particularly preferred embodiments, the lipid-based carriers of the composition comprise cholesterol. The molar ratio of the cationic lipid to cholesterol in the lipid-based carriers may be in the range from about 2:1 to about 1:1. In some embodiments, the cholesterol may be PEGylated. 35 In some embodiments, the lipid-based carrier comprises about 10mol% to about 60mol% or about 25mol% to about 40mol% sterol (based on 100% total moles oflipids in the lipid-based carrier). In one embodiment, the sterol is about 10,15,20,25,30, 35, 40, 45, 50, 55, or about 60mol% of the total lipid present in the lipid-based carrier. In another embodiment, the lipid-based carriers include from about 5% to about 50% on a molar basis of the sterol, e.g., about 15% to about 45%, about 20% to about 40%, about 48%, about 40%, about 38.5%, about 35%, about 34.4%, about 4031.5% or about 30% on a molar basis (based upon 100% total moles of lipid in the lipid-based carrier). In preferred embodiments, the lipid-based carrier comprises about 28%, about 29% or about 30% sterol (based on 100% total moles oflipids in the lipid-based carrier). In most preferred embodiments, the lipid-based carrier comprises about 40.9% sterol (based on 100% total moles of lipids in the lipid-based carrier). In another most preferred embodiment, the lipid- based carrier includes from about 28.5% on a molar basis ofsterol, preferably cholesterol. Preferably, LNPs comprise: (a) at least one therapeutic RNA, (b) a cationic lipid, (c) an aggregation reducing agent or aggregation-reducing lipid (such as polyethylene glycol (PEG) lipid or PEG-modified lipid), (d) optionally a non-cationic lipid (such as a neutral lipid), and (e) optionally, a sterol. Aaareaation reducing lipids /polymer conjugated lipids (PEG and PMOZ): In preferred embodiments, the lipid-based carriers comprise at least one aggregation reducing lipid or moiety. The term "aggregation reducing moiety" refers to a molecule comprising a moiety suitable of reducing or preventing aggregation of the lipid-based carriers. The term "aggregation reducing lipid" refers to a molecule comprising both a lipid portion and a moiety suitable of reducing or preventing aggregation of the lipid-based carriers. Under storage conditions or during formulation, the lipid- based carriers may undergo charge-induced aggregation, a condition which can be undesirable for the stability of the lipid-based carriers. Therefore, it can be desirable to include a compound or moiety which can reduce aggregation, for example by sterically stabilizing the lipid-based carriers. Such a steric stabilization may occur when a compound having a sterically bulky but uncharged moiety that shields or screens the charged portions of a lipid-based carriers from close approach to other lipid-based carriers in the composition. In the context of the invention, stabilization of the lipid-based carriers is achieved by including lipids which may comprise a lipid bearing a sterically bulky group which, after formation of the lipid-based carrier, is preferably located on the exterior of the lipid-based carrier. Suitable aggregation reducing groups include hydrophilic groups, e.g. monosialoganglioside GM1, polyamide oligomers (PAO), or certain polymers, such as poly(oxyalkylenes), e.g., poly(ethylene glycol) or poly(propylene glycol). In preferred embodiments, the aggregation reducing lipid selected from a polymer conjugated lipid. Lipids comprising a polymer as aggregation reducing group are herein referred to as "polymer conjugated lipid". The term "polymer conjugated lipid" refers to a molecule comprising both a lipid portion and a polymer portion, wherein the polymer is suitable of reducing or preventing aggregation of lipid-based carriers comprising the therapeutic RNA. A polymer has to be understood as a substance or material consisting of very large molecules, or macromolecules, composed of many repeating subunits. A suitable polymer in the context of the invention may be a hydrophilic polymer. An example of a polymer conjugated lipid is a PEGylated or PEG-conjugated lipid. In preferred embodiments, the lipid-based carriers of the composition comprise an aggregation reducing lipid selected from a polymer conjugated lipid. LNP in viva characteristics and behavior can be modified by addition of a hydrophilic polymer coating, e.g. polyethylene glycol (PEG), to the LNP surface to confer steric stabilization. Furthermore, LNPs can be used for specific targeting by attaching ligands (e.g. antibodies, peptides, and carbohydrates) to its surface or to the terminal end of the attached PEG chains (e.g. via PEGylated lipids or PEGylated cholesterol). In some embodiments, the LNPs comprise a polymer conjugated lipid. The term "polymer conjugated lipid" refers to a molecule comprising both a lipid portion and a polymer portion. An example of a polymer conjugated lipid is a PEGylated lipid. The term "PEGylated lipid" refers to a molecule comprising both a lipid portion and a polyethylene glycol portion. PEGylated lipids are known in the art and include 1-(monomethoxy-polyethyleneglycol)-2,3- dimyristoylglycerol (PEG-s- DMG) and the like. In preferred embodiments, the polymer conjugated lipid is selected from a PEG-conjugated lipid or a PEG-free lipid. In certain embodiments, the LNP comprises an additional, stabilizing-lipid which is a polyethylene glycol-lipid (PEGylated lipid). Suitable polyethylene glycol-lipids include PEG-modified phosphatidylethanolamine, PEG-modified phosphatidic acid, PEG-modified ceramides (e.g. PEG-CerC14 or PEG-CerC20), PEG-modified dialkylamines, PEG- modified diacylglycerols, PEG-modified dialkylglycerols. Representative polyethylene glycol-lipids include PEG-c- DOMG, PEG-c-DMA, PEG-s-DMG, PEG-DMG, PEG-DSG, PEG-DSPE, PEG-DOMG. In one embodiment, the polyethylene glycol-lipid is N-[(methoxy poly(ethylene glycol)2000)carbamyl]-1,2-dimyristyloxlpropyl-3-amine (PEG-c- DMA). In a preferred embodiment, the polyethylene glycol-lipid is PEG-2000-DMG. In one embodiment, the polyethylene glycol-lipid is PEG-c-DOMG). In other embodiments, the LNPs comprise a PEGylated diacylglycerol (PEG-DAG) such as 1-(monomethoxy-polyethyleneglycol)-2,3-dimyristoylglycerol (PEG-DMG), a PEGylated phosphatidylethanoloamine (PEG-PE), a PEG succinate diacylglycerol (PEG-S-DAG) such as 4-0-(2',3'- di(tetradecanoyloxy)propyl-1-0-(u-methoxy(polyethoxy)ethyl)butanedioate (PEG-S-DMG), a PEGylated ceramide (PEG-cer), or a PEG dialkoxypropylcarbamate such as to-methoxy(polyethoxy)ethyl-N- (2,3di(tetradecanoxy)propyl)carbamateor2,3-di(tetradecanoxy)propyl-N-(u-methoxy(polyethoxy)ethyl)carbamate. In preferred embodiments the at least one therapeutic RNA of the composition is complexed with one or more lipids thereby forming LNPs, wherein the LNP additionally comprises a PEGylated lipid that is preferably derived from formula (IV) of published PCT patent application W02018/078053A1. Accordingly, PEGylated lipid derived from formula (IV) of published PCT patent application W02018/078053A1, and the respective disclosure relating thereto, is herewith incorporated by reference. In a particularly preferred embodiments, the at least one therapeutic RNA of the composition is complexed with one or more lipids thereby forming LNPs, wherein the LNP additionally comprises a PEGylated lipid, wherein the PEG lipid is preferably derived from formula (IVa) of published PCT patent application W02018/078053A1. Accordingly, PEGylated lipid derived from formula (IVa) of published PCT patent application W02018/078053A1, and the respective disclosure relating thereto, is herewith incorporated by reference. In a embodiment the PEG lipid is of formula (IVa)
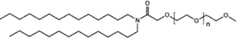
(IVa), preferably wherein n has a mean value ranging from 30 to 60, such as about 30±2,32±2,34±2, 36±2, 38±2, 40±2, 42±2,44±2,46±2,48±2, 50±2, 52±2,54±2, 56±2, 58±2, or60±2. In a most preferred embodiment n is about 49. In another very preferred embodiment n is 45. In further preferred embodiments said PEG lipid is of formula (IVa), wherein n is an integer selected such that the average molecular weight of the PEG lipid is about 2000g/mol to about SOOOg/mol or about 2300g/mol to about 2700g/mol. In another preferred embodiment said PEG lipid is of formula (IVa), wherein n is an integer selected such that the average molecular weight of the PEG lipid is about 2000g/mol. The PEG-conjugated lipid of formula IVa as suitably used herein has the chemical 2[(polyethylene glycol)-2000]-N,N- ditetradecylacetamide, also referred to as ALC-0159. Accordingly, in embodiments, the aggregation reducing lipid is a PEG-conjugated lipid preferably selected or derived from DMG-PEG 2000, C10-PEG2K, Cer8-PEG2K, orALC-0159. Further examples ofPEG-lipids suitable in that context are provided in US2015/0376115A1 and W02015/199952, each of which is incorporated by reference in its entirety. In other preferred embodiments, the lipid-based carriers of the composition comprise an aggregation reducing lipid, wherein the aggregation reducing lipid is not a PEG-conjugated lipid. Accordingly, the aggregation reducing lipid may suitably be selected from a PEG-less lipid, e.g. a PEG-less polymer conjugated lipid. In some embodiments, lipid-based carriers include less than about 3, 2, or 1 mole percent of aggregation reducing lipid, based on the total moles of lipid in the lipid-based carrier. In further embodiments, lipid-based carriers comprise from about 0.1% to about 10% of the aggregation reducing lipid on a molar basis, e.g., about 0.5 to about 10%, about 0.5 to about 5%, about 10%, about 5%, about 4%, about 3%, about 2%, about 1.5%, about 1 %, about 0.5%, or about 0.3% on a molar basis (based on 100% total moles of lipids in the lipid-based carrier). In other preferred embodiments, lipid- based carriers comprise from about 1.0% to about 2.0% of the aggregation reducing lipid on a molar basis, e.g., about 1.2 to about 1.9%, about 1.2 to about 1.8%, about 1.3 to about 1.8%, about 1.4 to about 1.8%, about 1.5 to about 1.8%, about 1.6 to about 1.8%, in particular about 1.4%, about 1.5%, about 1.6%, about 1.7%, about 1.8%, about 1.9%, most preferably 1.7% (based on 100% total moles of lipids in the lipid-based carrier). In other preferred embodiments, lipid- based carriers comprise about 2.5%, 3%, 3.5%, 4%, 4.5% or 5%, preferably 2.5% of the aggregation reducing lipid on a molar basis (based on 100% total moles of lipids in the lipid-based carrier). In various embodiments, the molar ratio of the cationic lipid to the aggregation reducing lipid ranges from about 100:1 to about 25:1. In preferred embodiments, the lipid-based carriers of the composition comprise an aggregation reducing lipid, wherein the aggregation reducing lipid is not a PEG-conjugated lipid. In preferred embodiments, the aggregation reducing lipid (or polymer conjugated lipid) is a PEG-free lipid that comprises a polymer different from PEG. A PEG-free lipid in the context of the invention may be selected or derived from a POZ-lipid. In preferred embodiments, the POZ lipids or respectively preferred polymer conjugated lipids are described in PCT/EP2022/074439, the full disclosure herewith incorporated by reference. In particular, the disclosure relating to polymer conjugated lipids as shown in any one of claims 1 to 8 the disclosure relating to polymer conjugated lipids as shown in any one of claims 9 to 46 of PCT/EP2022/074439 are incorporated by reference. In preferred embodiments, the lipid-based carriers of the composition do not comprise a PEG-conjugated lipid. Accordingly, in further preferred embodiments, the polymer conjugated lipid is a POZ-lipid, which is defined as a compound according to formula (POZ): [H] - [linker] - [M] wherein [H] is a homopolymer moiety comprising at least one polyoxazoline (POZ) monomer unit

wherein R is C1-9 alkyl or C2-9 alkenyl and n has a mean value ranging from 2 to 200, preferably from 20 to 100, more preferably from 24 to 26 or 45 to 50 [linker] is an optional linker group, and [M] is a lipid moiety. In an embodiment in that context, [H] is a heteropolymer moiety or homopolymer moiety comprising multiple monomer units selected from the group consisting of poly(2-methyl-2-oxazoline)(PMOZ)
poly(2-ethyl-2-oxazoline) (PEOZ) :
poly(2-propyl-2-oxazoline)(PPOZ)
poly(2-isopropy 2-oxazoline)(PIPOZ) poly(2-methoxymethyl-2-oxazoline) (PMeOMeOx), and poly(2-dimethylamino-2-oxazoline)(PDMAOx), preferably wherein [H] is a homopolymer moiety comprising multiple PMOZ or PEOZ monomer units, more preferably wherein [H] comprises or preferably consists of multiple PMOZ monomer units, wherein (i) n has a mean value ranging from 2 to 200, preferably from 20 to 100, more preferably from 24 to 26 or 45 to 50 or wherein (ii) n is selected such that the [hi] moiety has an average molecular weight of 1.5 to 22 kDa, more preferably of2 to 19 kDa, even more preferably of about 7.5 kDa or of about 15 kDa, preferably from 1 to 15 kDa, more preferably of 2 to 12.5 kDa, even more preferably of about 5 kDa or of about 10 kDa. In another embodiment in that context, [H] is a heteropolymer moiety or homopolymer moiety comprising multiple monomer units selected from the group consisting of

A A A In yet another embodiment, the [H] from the polymer conjugated lipid according to formula (POZ) is selected from the group consisting of poly(2-methoxymethyl-2-oxazoline) (PMeOMeOx) and poly(2-dimethylamino-2-oxazoline) (PDMAOx). In one embodiment, the lipid moiety [M] as shown in formula (POZ) comprises at least one straight or branched, saturated or unsaturated alkyl chain containing from 6 to 30 carbon atoms, preferably wherein the lipid moiety [M] comprises at least one straight or branched saturated alkyl chain, wherein the alkyl chain is optionally interrupted by one or more biodegradable group(s) and/or optionally comprises one terminal biodegradable group, wherein the biodegradable group is selected from the group consisting of but not limited to a pH-sensitive moiety, a zwitterionic linker, non-ester containing linker moieties and ester-containing linker moieties (-C(O)O— or —OC(0)—), amido (— C(O)NH-), disulfide (—S—S—), carbonyl (-C(O)—), ether (—0—), thioether (—S—), oxime (e.g., —C(H)=N—0— or—0— N=C(H)—), carbamate (-NHC(O)O-), urea (-NHC(O)NH-), succinyl (—(0)CCH2CH2C(0)—), sucdnamidyl (—NHC(0)CH2CH2C(0)NH—), —C(R5)=N—, —N=C(R5)—, —C(R5)=N—0— —0—N=C(R5)—, — 0—c(0)0-, —C(0)N(R5), —N(R5)C(0)—, —C(S)(NR5)—, (NR5)C(S)—, —N(R5)C(0)N(R5)—, —C(0)S—, — SC(0)—, —C(S)0—, —OC(S)—, —OSi(R5)20—, —C(0)(CR3R4)C(0)0—, or —OC(0)(CR3R4)C(0)—, carbonate (—0c(0)0—), succinoyl, phosphate esters (—0—(O)POH—0—), cyclic compound, heterocyclic compound, piperidine, pyrazine, pyridine, piperazine, and sulfonate esters, as well as combinations thereof, wherein R3, R4 and R5 are, independently H or alkyl (e.g. C1-C4 alkyl). In another embodiment, the lipid moiety [M] comprises at least one straight or branched, saturated or unsaturated alkyl chain comprising 6, 7,8, 9,10,11,12,13,14,15,16,17,18,19, 20, 21,22,23,24,25, 26,27,28, 29, or 30 carbon atoms, preferably in the range of 10 to 20 carbon atoms, more preferably in the range of 12 to 18 carbon atoms, even more preferably 14, 16 or 18 carbon atoms, even more preferably 16 or 18 carbon atoms, most preferably 14 carbon atoms, wherein all selections are independent of one another. In one embodiment, the linker group [linker] as shown in formula (POZ) is selected from the group consisting of but not limited to a pH-sensitive moiety, a zwitterionic linker, non-ester containing linker moieties and ester-containing linker moieties (-C(O)O— or-OC(O)—), amido (-C(O)NH-), disulfide (—S—S—), carbonyl (-C(O)—), ether (—0—), thioether (—S—), oxime (e.g., —C(H)=N—0—or—0— N=C(H)—), carbamate (—NHC(O)O-), urea (— NHC(O)NH-), succinyl (—(0)CCH2CH2C(0)—), succinamidyl (—NHC(0)CH2CH2C(0)NH—), —C(R5)=N— — N=C(R5)—, —C(R5)=N—0—, —0—N=C(R5)—, —0—C(0)0—, —C(0)N(R5), —N(R5)C(0)—, —C(S)(NR5)—, (NR5)C(S)—, —N(R5)C(0)N(R5)—, —C(0)S—, —SC(0)—, —C(S)0—, —OC(S)—, —OSi(R5)20—, — C(0)(CR3R4)C(0)0—, or—OC(0)(CR3R4)C(0)—, carbonate (-OC(O)O-), succinoyl, phosphate esters (—0— (O)POH-O—), and sulfonate esters, as well as combinations thereof, wherein R3, R4 and R5 are, independently H or alkyl(e.g.C1-C4alkyl).

In a very preferred embodiment, the polymer conjugated lipid has the structure of
In another very preferred embodiment, the polymer conjugated lipid has the structure of In another very preferred embodiment, the polymer conjugated lipid has the structure of "PMOZ 2" with n = 50 i.e. having 50 monomer repeats. In an even further preferred embodiment, the polymer conjugated lipid has the structure of In another preferred embodiment, the polymer conjugated lipid has the structure of ["PMOZ 5"]. In an even further very preferred embodiment, the polymer conjugated lipid has the structure of
[-PMOZ 4"], preferably with n = 50 i.e. having 50 monomer repeats, i.e.

["PMOZ 4" with n = 50 i.e. having 50 monomer repeats]. For "PMOZ 1" to "PMOZ 5", preferably n has a mean value ranging from 2 to 200, preferably from 20 to 100, more preferably from 24 to 26, even more preferably about 100, or further even more preferably from 45 to 50, most preferably 50 or wherein n is selected such that the [P] moiety has an average molecular weight of about 4.2 kDa to about 4.4 kDa, or most preferably about 4.3 kDa. In another very preferred embodiment, the linker group [linker] comprises preferably an amide linker moiety. In a further very preferred embodiment, the linker group [linker] comprises preferably an ester linker moiety. In a further very preferred embodiment, the linker group [linker] comprises preferably a succinate linker moiety. In another very preferred embodiment, the linker group [linker] comprises both an ester linker and an amid linker moiety. In another preferred embodiment, the linker group [linker] comprises both an ester linker, an amine linker and an amid linker moiety. It is noted herein, that all chemical compounds mentioned throughout the whole specification may be produced via processes known to a skilled worker; starting materials and/or reagents used in the processes are obtainable through routine knowledge of a skilled worker on the basis of common general knowledge (e.g. from text books or from e.g. patent applications W02022173667, W02009043027, W02013067199, W02010006282, W02009089542, W02016019340, W02008106186, W02020264505, and W02020023947, the complete disclosure of said patent applications is incorporated by reference herein). In yet a further embodiment, the lipid nanoparticle does not comprise a polyethylene glycol-(PEG)-lipid conjugate or a conjugate of PEG and a lipid-like material, and preferably do not comprise PEG and/or (ii) the polymer conjugated lipid of the invention does not comprise a sulphur group (-S-), a terminating nucleophile, and/or is covalently coupled to a biologically active ingredient is a therapeutic RNA compound selected from the group consisting of RNA, an artificial mRNA, chemically modified or unmodified messenger RNA (mRNA) comprising at least one coding sequence, self- replicating RNA, circular RNA, viral RNA, and replicon RNA; or any combination thereof, preferably wherein the biologically active ingredient is chemically modified mRNA or chemically unmodified mRNA, more preferably wherein the biologically active ingredient is chemically unmodified mRNA. In another very preferred embodiment, the polymer conjugated lipid of the invention does not comprise sulphur (S) or a sulphur group (-S-). In further preferred embodiments, lipid nanoparticles and/or polymer conjugated lipids may be selected from the lipid nanoparticles and/or lipids as disclosed in PCT/EP2022/074439 (i.e. lipids derived from formula I, II, and III of PCT/EP2022/074439, or lipid nanoparticles and/or lipids as specified in Claims 1 to 46 of PCT/EP2022/074439), the disclosure of PCT/EP2022/074439 hereby incorporated by reference in its entirety. In preferred embodiments, the at least one aggregation-reducing lipid, preferably the PEG-conjugated lipid, is selected or derived from ALC-0159, DMG-PEG 2000, C10-PEG2K, Cer8-PEG2K. In particularly preferred embodiments, the aggregation-reducing lipid isALC-0159. Preferably, the aggregation reducing lipid is selected or derived of a POZ-lipid, which is defined as a compound according to formula (POZ) as defined herein. In some embodiments, lipid-based carriers include less than about 3mol%, 2mol%, or 1 mol% of aggregation reducing lipid, based on the total moles of lipid in the lipid-based carrier. In further embodiments, lipid-based carriers comprise from about 0.1% to about 10% of the aggregation reducing lipid on a molar basis, e.g. about 0.5% to about 10%, about 0.5% to about 5%, about 10%, about 5%, about 4%, about 3%, about 2%, about 1.5%, about 1 %, about 0.5%, or about 0.3% on a molar basis (based on 100% total moles of lipids in the lipid-based carrier). In other preferred embodiments, lipid-based carriers comprise from about 1.0% to about 2.0% of the aggregation reducing lipid on a molar basis, e.g. about 1.2% to about 1.9%, about 1.2% to about 1.8%, about 1.3% to about 1.8%, about 1.4% to about 1.8%, about 1.5% to about 1.8%, about 1.6% to about 1.8%, in particular about 1.4%, about 1.5%, about 1.6%, about 1.7%, about 1.8%, about 1.9%, most preferably 1.7% (based on 100% total moles of lipids in the lipid-based carrier). In other preferred embodiments, lipid-based carriers comprise about 2.5%, 3%, 3.5%, 4%, 4.5% or 5%, preferably 2.5% of the aggregation reducing lipid on a molar basis (based on 100% total moles of lipids in the lipid-based carrier). In various embodiments, the molar ratio of the cationic lipid to the aggregation reducing lipid ranges from about 100:1 to about 25:1. Accordingly, in a very preferred embodiment, the lipid-based carriers of the composition preferably comprises 59 mol% ofcationic lipid "C26" (described herein above and/or below), 10 mol% DPhyPE, 28.5 mol% cholesterol and 2.5 mol% PMOZ 4 (described herein above and/or below). In embodiments, the liposomes, lipid nanoparticles, lipoplexes, and/or nanoliposomes preferably consist of (i) at least one cationic lipid; (ii) at least one neutral lipid; (iii) at least one steroid or steroid analogue; and (iv) at least one aggregation reducing-lipid, wherein, preferably, (i) to (iv) are in a molar ratio of about 20-60% cationic lipid, 5-25% neutral lipid, 25-55% sterol, and 0.5-15% PEG-lipid. In embodiments, the at least one therapeutic nucleic acid/RNA of the composition is complexed with one or more lipids thereby forming LNPs, wherein the LNP essentially consists of (i) at least one cationic lipid as defined herein, preferably lipid 111-3; (ii) a neutral lipid as defined herein, preferably 1,2-distearoyl-sn-glycero-3-phosphocholine (DSPC); (iii) a steroid or steroid analogue as defined herein, preferably cholesterol; and (iv) a PEG-lipid as defined herein, e.g. PEG-DMG or PEG-cDMA, preferably a PEGylated lipid of formula (IVa), wherein (i) to (iv) are in a molar ratio of about 20-60% cationic lipid: 5-25% neutral lipid: 25-55% sterol; 0.5-15% PEG- lipid. In embodiments, the at least one therapeutic nucleic acid/RNA of the composition is complexed with one or more lipids thereby forming LNPs, wherein the LNP comprises (i) at least one cationic lipid selected from SM-102; (ii) at least one neutral lipid selected from DSPC; (iii) at least one steroid or steroid analogue selected from cholesterol; and (iv) at least one aggregation reducing lipid selected from DMG-PEG 2000 or "PMOZ 4"; and wherein the lipid-based carriers encapsulate the therapeutic nucleic acid/RNA of the composition, preferably wherein i) to (iv) are n a weight ratio of about 50% cationic lipid, about 10% neutral lipid, about 38.5% steroid or steroid analogue, and about 1.5% aggregation reducing lipid, preferably wherein the lipid-based carriers encapsulate the therapeutic nucleic acid/RNA. Such LNPs are herein referred to as SM-102-LNPs. In preferred embodiments the at least one therapeutic nucleic acid/RNA of the composition is complexed with one or more lipids thereby forming LNPs, wherein the LNP comprises (i) at least one cationic lipid selected from SM-102; (ii) at least one neutral lipid selected from DSPC; (iii) at least one steroid or steroid analogue selected from cholesterol; and (iv) at least one aggregation reducing lipid selected from DMG-PEG 2000 or "PMOZ 4"; and wherein the lipid-based carriers encapsulate the therapeutic nucleic acid/RNA of the composition, preferably wherein i) to (iv) are n a weight ratio of about 48.5% cationic lipid, about 11.1% neutral lipid, about 38.9% steroid or steroid analogue, and about 1.5% aggregation reducing lipid, preferably wherein the lipid-based carriers encapsulate the nucleic acid. A preferred N/P ratio for this formulation is about 4.85 (lipid to RNA mol ratio). Such LNPs are herein referred to as SM-102-LNPs. In preferred embodiments, the at least one therapeutic nucleic acid/RNA of the composition is complexed with one or more lipids thereby forming LNPs, wherein the LNP comprises (i) at least one cationic lipid selected from SS-33/4PE-15, HEXA-C5DE-PipSS or compound C26 (see C26 in Table 1 of W02021123332); (ii) at least one neutral lipid selected from DPhyPE; (iii) at least one steroid or steroid analog selected from cholesterol; and (iv) at least one aggregation reducing lipid selected from DMG-PEG 2000 or "PMOZ 4" and wherein the lipid-based carriers encapsulate the therapeutic nucleic acid/RNA of the composition, preferably wherein the lipid-based carriers encapsulate the nucleic acid (e.g. the RNA). Such LNPs are herein referred to as GN- LNPs. In a preferred embodiment, the at least one therapeutic nucleic acid, preferably RNA of the composition is complexed with one or more lipids thereby forming LNPs, wherein LNPs have a molar ratio of approximately 50:10:38.5:1.5, preferably 47.5:10:40.8:1.7 or more preferably 47.4:10:40.9:1.7 (i.e. proportion (mol%) of cationic lipid (preferably lipid 111-3), DSPC, cholesterol and PEG-lipid ((preferably PEG-lipid of formula (IVa) with n = 49); solubilized in ethanol). In preferred embodiments, the lipid-based carriers comprise about 20-60% cationic lipid, about 5-25% neutral lipid, about 25-55% steroid or steroid analogue, and about 0.5-15% aggregation reducing lipid. In other preferred embodiments, the wt/wt ratio of lipid to nucleic acid in the lipid-based carrier is from about 10:1 to about 60:1, preferably from about 20:1 to about 30:1. The total amount of therapeutic nucleic acid, preferably RNA in the LNPs may vary and is defined depending on the e.g. RNA to total lipid w/w ratio. In one embodiment of the invention the RNA to total lipid ratio is less than 0.06 w/w, preferably between 0.03 w/w and 0.04 w/w. Preferably, the N/P ratio of the lipid-based carriers encapsulating the nucleic acid is in a range from about 1 to about 10, preferably in a range from about 5 to about 7. In various embodiments, the LNP as defined herein have a mean diameter of from about 50nm to about 200nm, from about 60nm to about 200nm, from about 70nm to about 200nm, from about 80nm to about 200nm, from about 90nm to about 200nm, from about 90nm to about 190nm, from about 90nm to about 180nm, from about 90nm to about 170nm, from about 90nm to about 160nm, from about 90nm to about 150nm, from about 90nm to about 140nm, from about 90nm to about 130nm, from about 90nm to about 120nm, from about 90nm to about 10Onm, from about 70nm to about 90nm, from about 80nm to about 90nm, from about 70nm to about 80nm, or about 30nm, 35nm, 40nm, 45nm, 50nm, 55nm, 60nm, 65nm, 70nm, 75nm, 80nm, 85nm, 90nm, 95nm, 100nm, 105nm, 110nm, 115nm, 120nm, 125nm, 130nm, 135nm, 140nm, 145nm, 150nm, 160nm, 170nm, 180nm, 190nm, or 200nm and are substantially non-toxic. As used herein, the mean diameter may be represented by the z- average as determined by dynamic light scattering as commonly known in the art. In embodiments, the lipid-based carriers have a Z-average size in a range of about 50nm to about 120nm. The polydispersity index (PDI) of the nanoparticles is typically in the range of0.1 to 0.5. In a particular embodiment, a PDI is below 0.2. Typically, the PDI is determined by dynamic light scattering as commonly known in the art. In another preferred embodiment of the invention the lipid nanoparticles have a hydrodynamic diameter in the range from about 50nm to about 300nm, or from about 60nm to about 250nm, from about 60nm to about 150nm, or from about 60nm to about 120nm, respectively. Other suitable cationic orionizable, neutral, steroid/sterol or aaareaation reducing lipids: Other suitable cationic or ionizable, neutral, steroid/sterol or aggregation reducing lipids are disclosed in W02010053572, W02011068810, W02012170889, W02012170930, W02013052523, W02013090648, W02013149140, W02013149141, W02013151663, W02013151664, W02013151665, W02013151666, W02013151667, W02013151668, W02013151669, W02013151670, W02013151671, W02013151672, W02013151736, W02013185069, W02014081507, W02014089486, W02014093924, W02014144196, W02014152211, W02014152774, W02014152940, W02014159813, W02014164253, W02015061461, W02015061467, W02015061500, W02015074085, W02015105926, W02015148247, W02015164674, W02015184256, W02015199952, W02015200465, W02016004318, W02016022914, W02016036902, W02016081029, W02016118724, W02016118725, W02016176330, W02017004143, W02017019935, W02017023817, W02017031232, W02017049074, W02017049245, W02017070601, W02017070613, W02017070616, W02017070618, W02017070620, W02017070622, W02017070623, W02017070624, W02017070626, W02017075038, W02017075531, W02017099823, W02017106799, W02017112865, W02017117528, W02017117530, W02017180917, W02017201325, W02017201340, W02017201350, W02017201352, W02017218704, W02017223135, W02018013525, W02018081480, W02018081638, W02018089540, W02018089790, W02018089801, W02018089851, W02018107026, W02018118102, W02018119163, W02018157009, W02018165257, W02018170245, W02018170306, W02018170322, W02018170336, W02018183901, W02018187590, W02018191657, W02018191719, W02018200943, W02018231709, W02018231990, W02018232120, W02018232357, W02019036000, W02019036008, W02019036028, W02019036030, W02019040590, W02019089818, W02019089828, W02019140102, W02019152557, W02019152802, W02019191780, W02019222277, W02019222424, W02019226650, W02019226925, W02019232095, W02019232097, W02019232103, W02019232208, W02020061284, W02020061295, W02020061332, W02020061367, W02020081938, W02020097376, W02020097379, W02020097384, W02020102172, W02020106903, W02020146805, W02020214946, W02020219427, W02020227085, W02020232276, W02020243540, W02020257611, W02020257716, W02021007278, W02021016430, W02021022173, W02021026358, W02021030701, W02021046260, W02021050986, W02021055833, W02021055835, W02021055849, W02021127394, W02021127641 , W02021202694, W02021231697, W02021231901, W02008103276, W02009086558, W02009127060, W02010048536, W02010054406, W02010080724, W02010088537, W02010129709, W0201021865, W02011022460, W02011043913, W02011090965, W02011149733, W02011153120, W02011153493, W02012040184, W02012044638, W02012054365, W02012061259, W02013063468, W02013086354, W02013086373, US7893302B2, US7404969B2, US8158601B2, US8283333B2, US8466122B2, US8569256B2, US20100036115, US20110256175, US20120202871, US20120027803, US20120128760, US20130064894, US20130129785, US20130150625, US20130178541, US20130225836, and US20140039032; the disclosures specifically relating to cationic or ionizable, neutral, sterol or aggregation reducing lipids suitable for lipid-based carriers of the foregoing publications are incorporated herewith by reference. For example, suitable cationic lipids or cationisable or ionizable lipids include, but are not limited to, DSDMA, N,N- dioleyl-N,N-dimethylammonium chloride (DODAC), N,N-distearyl-N,N-dimethylammonium bromide (DDAB), 1,2- dioleoyltrimethyl ammonium propane chloride (DOTAP) (also known as N-(2,3-dioleoyloxy)propyl)-N,N,N- trimethylammonium chloride and 1,2-Dioleyloxy-3-trimethylaminopropane chloride salt), N-(1-(2,3-dioleyloxy)propyl)- N,N,N-trimethylammonium chloride (DOTMA), N,N-dimethyl-2,3-dioleyloxy)propylamine (DODMA), ckk-E12 (W02015200465), 1,2-DiLinoleyloxy-N,N-dimethylaminopropane (DLinDMA), 1,2-Dilinolenyloxy-N,N- dimethylaminopropane (DLenDMA), 1,2-di-y-linolenyloxy-N,N-dimethylaminopropane (y-DLenDMA), 98N12-5, 1,2- Dilinoleylcarbamoyloxy-3-dimethylaminopropane (DLin-C-DAP), ICE (Imidazol-based), HGT5000, HGT5001, DMDMA, CLinDMA, CpLinDMA, DMOBA, DOcarbDAP, DLincarbDAP, DLinCDAP, KLin-K-DMA, DLin-K-XTC2-DMA, XTC (2,2- Dilinoleyl-4-dimethylaminoethyl-[1,3]-dioxolane) HGT4003,1,2-Dilinoleoyl-3-trimethylaminopropane chloride salt (DLin- TAP.CI), 1,2-Dilinoleyloxo-3-(2-N,N-dimethylamino)ethoxypropane (DLin-EG-DM A), 2,2-Dilinoleyl-4- dimethylaminomethyl-[1,3]-dioxolane(DLin-K-DMA),(6Z,9Z,28Z,31Z)-heptatriaconta-6,9,28,31-tetraen-19-yl-4- (dimethylamino)butanoate (MC3, US20100324120), ALNY-100 ((3aR,5s,6aS)-N,N-dimethyl-2,2-di((9Z,12Z)-octadeca- 9,12-dienyl)tetrahydro-3aH-cydopenta[d] [1,3]dioxol-5-amine)), NC98-5 (4,7,13-tris(3-oxo-3-(undecylamino)propyl)-N,N 16-diundecyl-4,7,10,13-tetraazahexadecane-l,16-diamide),(6Z,9Z,28Z,31Z)-heptatriaconta-6,9,28,31-tetraen-19-yl4- (dimethylamino)butanoate(DLin-M-C3-DMA),3-((6Z,9Z,28Z,31Z)-heptatriaconta-6,9,28,31-tetraen-19-yloxy)-N,N- dimethylpropan-1-amine(MC3 Ether), 4-((6Z,9Z,28Z,31Z)-heptatriaconta-6,9,28,31-tetraen-19-yloxy)-N,N- dimethylbutan-1-amine (MC4 Ether), LIPOFECTIN® (commercially available cationic liposomes comprising DOTMA and 1,2-dioleoyl-sn-3phosphoethanolamine (DOPE), from GIBCO/BRL, Grand Island, N.Y.); LIPOFECTAMINE® (commercially available cationic liposomes comprising N-(1-(2,3dioleyloxy)propyl)-N-(2-(sperminecarboxamido)ethyl)- N,N-dimethylammonium trifluoroacetate (DOSPA) and (DOPE), from GIBCO/BRL); and TRANSFECTAM® (commercially available cationic lipids comprising dioctadecylamidoglycyl carboxyspermine (DOGS) in ethanol from Promega Corp., Madison, Wis.) or any combination of any of the foregoing. Further suitable cationic or ionizable lipids include those described in international patent publications W02010/053572 (and particularly, 1,1'-(2-(4-(2-((2-(bis(2- hydroxydodecyl)amino)ethyl)(2-hydroxydodecyl)amino)ethyl)piperazin-1-yl)ethylazanediyl)didodecan-2-ol(C12-200) described at paragraph [00225] of W02010053572) and W02012170930, both of which are incorporated herein by reference, HGT4003, HGT5000, HGTS001, HGT5001, HGT5002 (see US2015140070), 1,2-dilinoleyoxy-3- (dimethylamino)acetoxypropane (DLin-DAC), 1,2-dilinoleyoxy-3morpholinopropane (DLin-MA), 1,2-dilinoleoyl-3- dimethylaminopropane (DLinDAP), 1,2-dilinoleylthio-3-dimethylaminopropane (DLin-S-DMA), l-linoleoyl-2-linoleyloxy- Sdimethylaminopropane (DLin-2-DMAP), 1,2-dilinoleyloxy-3-trimethylaminopropane chloride salt (DLin-TMA.CI), 1,2- dilinoleyloxy-3-(N-methylpiperazino)propane (DLin-MPZ), 3-(N,N-dilinoleylamino)-1,2-propanediol (DLinAP), 3-(N,N- dioleylamino)-1,2-propanediol (DOAP), 1,2-dilinoleyloxo-3-(2-N,N-dimethylamino)ethoxypropane (DLin-EG-DMA), 2,2- dilinoleyl-4-(2-dimethylaminoethyl)-[1,3]-dioxolane (DLin-KC2-DMA, W02010042877); dilinoleyl-methyl-4- dimethylaminobutyrate(DLin-MC3-DMA). According to various suitable embodiments, suitable carriers for delivery to the anterior segment of the eye may include polymer based carriers, such as polyethyleneimine (PEI), lipid nanoparticles and liposomes, nanoliposomes, ceramide- containing nanoliposomes, proteoliposomes, both natural and synthetically-derived exosomes, natural, synthetic and semisynthetic lamellar bodies, nanoparticulates, calcium phosphor-silicate nanoparticulates, calcium phosphate nanoparticulates, silicon dioxide nanoparticulates, nanocrystalline particulates, semiconductor nanoparticulates, poly(D- arginine), sol-gels, nanodendrimers, starch-based delivery systems, micelles, emulsions, niosomes, multi-domain-block polymers (vinyl polymers, polypropyl acrylic acid polymers, dynamic poly conjugates). In other embodiments, the at least one therapeutic nucleic acid/RNA of the composition may be formulated in amphiphilic macromolecules (AMs). AMs comprise biocompatible amphiphilic polymers which have an alkylated sugar backbone covalently linked to poly(ethylene glycol). In aqueous solution, the AMs self-assemble to form micelles. Non- limiting examples of methods of forming AMs and AMs are described in US Patent Publication No. US20130217753, the contents of which are herein incorporated by reference in its entirety. In other embodiments, the at least one therapeutic nucleic acid/RNA of the composition may be formulated in inorganic nanoparticles (U.S. Pat. No.8,257,745, herein incorporated by reference in its entirety). The inorganic nanoparticles may include, but are not limited to, clay substances that are water swellable. As a non-limiting example, the inorganic nanoparticle may include synthetic smectite clays which are made from simple silicates (See e.g., U.S. Pat. No. 5,585,108 and 8,257,745 each of which are herein incorporated by reference in their entirety). In other embodiments, the at least one therapeutic nucleic acid/RNA of the composition may be formulated in water- dispersible nanoparticle comprising a semiconductive or metallic material (U.S. Pub. No.20120228565; herein incorporated by reference in its entirety) or formed in a magnetic nanoparticle (U.S. Pub. No.20120265001 and 20120283503; each of which is herein incorporated by reference in its entirety). The water-dispersible nanoparticles may be hydrophobic nanoparticles or hydrophilic nanoparticles. In other embodiments, the at least one therapeutic nucleic acid/RNA of the composition may be formulated in high density lipoprotein-nucleic acid particles. As a non-limiting example, the particles may comprise a nucleic acid component and a polypeptide comprising a positively charged region which associates with the nucleic acid component as described in US Patent No.8,734,853, the contents of which is herein incorporated by reference in its entirety. In other embodiments, the at least one therapeutic nucleic acid/RNA of the composition may be formulated in a micelle or coated on a micelle for delivery or may be encapsulated into any hydrogel known in the art which may form a gel when injected into a subject or may be formulated in and/or delivered using a nanolipogel. In other embodiments, the at least one therapeutic nucleic acid/RNA of the composition may be formulated in exosomes. The exosomes may be loaded with at least one polynucleotide and delivered to cells, tissues and/or organisms. As a non-limiting example, the RNA may be loaded in the exosomes described in International Publication No. W02013084000, herein incorporated by reference in its entirety. In embodiments, the exosomes are obtained from cells that have been induced to undergo oxidative stress such as, but not limited to, the exosomes described in International Patent Publication No. W02014028763, the contents of which are herein incorporated by reference in its entirety. Accordingly, in preferred embodiments, the at least one therapeutic nucleic acid/RNA of the composition is complexed or associated with or at least partially complexed or partially associated with one or more exosomes. Accordingly, the pharmaceutically acceptable carrier as used herein preferably includes the liquid or non-liquid basis of the inventive composition. If the inventive composition is provided in liquid form, the carrier will be water, typically pyrogen-free water; isotonic saline or buffered (aqueous) solutions, e.g. phosphate, citrate etc. buffered solutions. Preferably, Ringer- or Ringer-Lactate solution as described in W02006/122828 is used as a liquid basis for the composition according to the invention. In embodiments, the composition comprising the therapeutic nucleic acid, preferably RNA is administered in Ringer- or Ringer-Lactate solution. 5 In embodiments, the composition may be provided in lyophilized or dried form (using e.g. lyophilisation or drying methods as described in W02016/165831 , W02011/069586, W02016/184575 or W02016/184576). Preferably, the lyophilized or dried composition is reconstituted in a suitable buffer, advantageously based on an aqueous carrier, prior to administration to the anterior segment of the eye, e.g. Ringer- or Ringer-Lactate solution or a phosphate buffer solution. 10 In embodiments the composition comprises additionally at least one antagonist of at least one RNA sensing pattern recognition receptor selected from a Toll-like receptor, preferably a TLR7 antagonist and/or a TLR8 antagonist. Suitable antagonist of at least one RNA sensing pattern recognition receptor are disclosed in published PCT patent application W02021028439, the full disclosure herewith incorporated by reference. In particular, the disclosure relating to suitable antagonist of at least one RNA sensing pattern recognition receptors as defined in any one of the claims 1 to 1594 of W02021028439 are incorporated by reference. In preferred embodiments, the at least one antagonist of at least one RNA sensing pattern recognition receptor is a single stranded oligonucleotide that comprises or consists of a therapeutic RNA sequence being identical or at least 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identical to a therapeutic RNA sequence selected from the group consisting ofSEQ ID NOs: 85-212 of W02021028439, or fragments of any of these sequences. A particularly 20 preferred antagonist in that context is 5'-GAG CGmG CCA-3' (SEQ ID NO: 85 of W02021028439), or a fragment or variant thereof. In embodiments, the at least one antagonist of at least one RNA sensing pattern recognition receptor and the at least one RNA encoding are separately formulated in the lipid-based carriers as defined herein or co-formulated in the lipid- based carriers as defined herein. 25 In other embodiments, the composition additionally comprises an anti-proliferative drug or an anti-fibrotic drug. In embodiments, the composition additionally comprises an anti-inflammatory agent, wherein the anti-inflammatory agent may comprise a steroid or a nonsteroidal anti-inflammatory drug (NSAID). Preferably, the administration of further substances (e.g. an anti-proliferative drug, an anti-fibrotic drug, an anti- 30 inflammatory drug, a vascular endothelial growth factor modulator, a platelet derived growth factor modulator, an angiogenesis inhibitor, an immunosuppressive agent, a vascular permeability inhibitor, or a combination thereof) may enhance and/or prolong the therapeutic effect of the composition comprising the therapeutic RNA. In some embodiments, the composition may additionally comprise the anti-proliferative drugs mitomycin C (MMC) or 5- fluorouracil (5-FU). 5 In embodiments, the ophthalmic disease, disorder or condition is caused or associated with (aberrant) fibrosis, toxic anterior segment syndrome, cataract, neovascularization, inflammation, neuronal damage, complement activation, drusen formation, scar formation, (chronic) intraocular pressure (IOP), Anterior Segment Dysgenesis (ASD) or Anterior Segment Developmental Anomalies (ASDA). 0 Developmental disorders of the eye related to the anterior segment of the eye are grouped under the term Anterior Segment Developmental Anomalies (ASDA) and referred to as Anterior Segment Dysgenesis (ASD). Anterior segment dysgenesis (ASD) is a failure of the normal development of the tissues of the anterior segment of the eye. It leads to anomalies in the structure of the mature anterior segment, associated with an increased risk of glaucoma and corneal opacity. These disorders show vast phenotypic and genetic heterogeneity a. Some symptoms overlap, such as the tendency to develop increased intraocular pressure (IOP). The ciliary body of the iris produces the aqueous humor which must be drained via the trabecular meshwork into Schlemm's canal and by the uveoscleral outflow pathway. Disruptions in this process are frequent in ASD, and thus secondary glaucoma is a common complication. ASD includes Posterior embryotoxon, Axenfeld-Rieger syndrome, Peters anomaly/Peters Plus syndrome, primary congenital glaucoma, Aniridia, Congenital hereditary endothelial dystrophy, Posterior Polymorphous Corneal Dystrophy, Sclerocornea, Megalocornea, Iridocorneal Endothelial Syndrome, Iridogoniodysgenesis syndrome, Congenital Iris Ectropion Syndrome, and Posterior Keratoconus. embodiments, the ophthalmic disease, disorder or condition is caused or associated with toxic anterior segment syndrome (TASS or sterile endophthalmitis or postoperative uveitis). TASS is an acute severe intraocular inflammation accompanied by diffuse corneal edema within 1-2 days of anterior segment surgery which is most commonly associated with cataract surgery. In other embodiments, the ophthalmic disease, disorder or condition is caused or associated with ocular fibrosis- associated proliferation, conjunctiva! fibrosis, ocular cicatricial pemphigoid, corneal scarring, corneal epithelial down growth, or aberrant post-surgical fibrosis. Preferably, wherein the ophthalmic disease, disorder or condition is selected from toxic anterior segment syndrome, Anterior blepharitis, keratitis, cataract, neovascularization, dry eye, diabetic eye disease, retinal detachment, optic nen/e disease, Keratoconus (KCN), Blepharitis, endocrine disorders, cancer disease, chalazion, infectious disease, parasitic disease, corneal scaring, pigmentary uveitis, branch retinal vein occlusion, central retinal vein occlusion, macular edema, cystoid macular edema, uveitic macular edema, cytomegalovirus retinitis, endophthalmitis, scleritis, choriotetinitis, dry eye syndrome, Norris disease, Coat's disease, choroidal neovascularization (CNV), persistent hyperplastic primary vitreous, familial exudative vitreoretinopathy, Leber congenital amaurosis, X-linked retinoschisis, Leber's hereditary optic neurophathy, uveitis, refraction and accommodation disorders, keratoconus, Meibomian Gland Dysfunction (MGD), amblyopia, conjunctivitis, pterygium, corneal ulcers, dacryocystitis, Duane retraction syndrome, optic neuritis, ocular inflammation, glaucoma, macular degeneration, Uveitis-glaucoma-hyphaema (UGH) syndrome, Corneal Edema, Keratoconus, Trachoma, Fuchs Dystrophy, Anterior Capsular Contraction Syndrome, Anterior Segment Dysgenesis (ASD) or Anterior Segment Developmental Anomalies (ASDA) and uveitis, or any disease, disorder or condition related or associated thereto. In some embodiments, the ophthalmic disease, disorder or condition is selected from macular degeneration (e.g. wet age-related macular degeneration or wet-AMD). In some embodiments, the ophthalmic disease, disorder or condition is selected from diabetic eye disease (e.g.diabetic retinopathy). In preferred embodiments, the ophthalmic disease, disorder or condition is selected from glaucoma and glaucoma-associated complications. Hereby, glaucoma-associated complications may occur during or after glaucoma filtration surgery (Trabeculectomy). Additionally, conjunctival and subconjunctival fibrogenesis (and inflammation) may occur as a side effect subsequent to glaucoma filtration surgery. Glaucoma is a group of ocular disorders characterized by high intraocular pressure-associated neuropathies and is one of the leading causes of blindness in the general population. Its most frequent cause is an obstruction of aqueous humor outflow through the trabecular meshwork. It can also be caused by the increased production of aqueous humor. The abnormal collection of aqueous humor increases the intraocular pressure which leads to the atrophy of the optic nerve and progressive loss of vision. Intraocular pressure depends on the levels of production and resorption of aqueous humour. Because the ciliary body produces aqueous humour, it is the main target of many medications against glaucoma. Its inhibition leads to the lowering of aqueous humour production and causes a subsequent drop in the intraocular pressure. Current treatment options like beta-blockers, Alpha-adrenergic agonists and Carbonic anhydrase inhibitors decrease fluid production but may also lead to unwanted side effects. A treatment option is a trabeculectomy (glaucoma filtration surgery), which is a filtering surgery where an ostium is created into the anterior chamber from underneath a partial thickness scleral flap to allow for aqueous flow out of the eye. The aqueous flows into the subconjunctival space, usually leading to an elevation of the conjunctiva, referred to as a filtering bleb. In embodiments, the composition comprising the therapeutic nucleic acid/RNA administered to the anterior segment of the eye of a subject lead to translation of an encoded peptide or protein, thereby exerting a therapeutic effect. Hereby, the local administration of the therapeutic nucleic acid/RNA may reduce fibrogenesis and/or corneal scaring followed glaucoma surgery or treatment. Hereby, the local administration of the therapeutic RNA may reduce inflammation followed glaucoma surgery or treatment. In preferred embodiments, the composition comprising the therapeutic nucleic acid/RNA, preferably the mRNA, administered to the cornea leads to reduced fibrosis, reduced inflammation, complement inhibition, reduced drusen formation, reduced scar formation, reduced (chronic) intraocular pressure (IOP) and/or reduction of Corneal Ulcer, Corneal Edema, Keratoconus, Corneal Stromal Dystrophies, Cornea Farinata, cornea plana, cornea transplantation- induced glaucoma, Cornea Verticillata, Corneal Allograft Rejection and Failure, Fuchs Dystrophy Corneal Dellen, Megalocornea, Corneal Epithelial Defect, iridocorneal endothelial (ICE) syndrome, Corneal hypoesthesia/hypesthesia, Corneal Keloids or Corneal Neovascularization. In some embodiments, the composition comprising the therapeutic RNA, preferably the mRNA, administered to the cornea leads to reduced allergies-associated symptoms or conditions. In most preferred embodiments, the composition comprising the therapeutic nucleic acid/RNA administered to the anterior segment of the eye of a subject lead to a reduction of the intraocular pressure (IOP). In other preferred embodiments, the composition comprising the therapeutic nucleic acid/RNA, preferably the mRNA, administered to the conjunctiva preferably via sub-conjunctival injection, leads to reduced ocular fibrosis-associated proliferation, conjunctival fibrosis, ocular dcatricial pemphigoid, comeal scarring, corneal epithelial down growth, or aberrant post-surgical fibrosis, reduced inflammation, complement inhibition, reduced drusen formation, reduced scar formation, reduced (chronic) intraocular pressure (IOP) and/or reduction of conjunctivitis, Injected Conjunctiva, Conjunctival Hemorrhage, Conjunctival Lymphoma, Conjunctiva] Hemangioma, Conjunctivochalasis, Pterygium, Pinguecula or Chemosis. In other embodiments, the composition comprising the therapeutic nucleic acid/RNA, preferably the mRNA, administered to the conjunctiva preferably via sub-conjunctival injection, leads to reduction of conjunctival diseases which are disclosed within W02014158863. In other preferred embodiments, the composition comprising the therapeutic nucleic acid/RNA, preferably the mRNA, administered to the ciliary body leads to reduced fibrosis, reduced inflammation, complement inhibition, reduced drusen formation, reduced scar formation, reduced (chronic) intraocular pressure (IOP) and/or reduction of Ciliary body melanoma, Primary Ciliary Body Non-Hodgkin Lymphoma, Metastatic Malignant Neoplasm in the Ciliary Body, Primary Iridocyclitis or Hyphema. Kit of parts In a second aspect, the present invention provides a kit or kit of parts, preferably a kit or kit of parts for use in treatment or prevention of an ophthalmic disease, disorder or condition, wherein said kit or kit of parts is administered to the anterior segment of the eye of a subject in need of treatment. Notably, embodiments relating to the first aspect of the invention are likewise applicable to embodiments of the second aspect of the invention, and embodiments relating to the second aspect of the invention are likewise applicable to embodiments of the first aspect of the invention. The kit or kit of parts for use may comprise at least one therapeutic nucleic acid/RNA as defined in the context of the first aspect, and/orthe composition of the first aspect, and optionally, a liquid vehicle for solubilising, and optionally technical instructions providing information on administration and dosage of the components. Embodiments and features disclosed in the context of therapeutic nucleic acid or RNA suitable for ocular delivery or embodiments and features disclosed in the context of the composition (e.g., LNP formulation, delivery devices) are likewise applicable for the RNA and/or the composition of the kit or the kit of parts. The kit or kit of parts for use may further comprise additional components as described in the context of the composition, in particular, pharmaceutically acceptable carriers, excipients, buffers and the like. The kit or kit of parts for use may further comprise an anti-inflammatory drug, a vascular endothelial growth factor (VEGF) modulator, a platelet derived growth factor (PDGF) modulator, an angiogenesis inhibitor, an immunosuppressive agent, a vascular permeability inhibitor, or a combination thereof, to the anterior segment of the eye of the patient in need of treatment. Further, the kit or kit of parts for use may comprise modulators and/or activators of any of the provided protein targets of List 1. In other embodiments, the kit or kit of parts for use may comprise at least one of the agents provided in paragraph [1263] to [1276] of published PCT application W02017/192565. The technical instructions of said kit or kit of parts for use may comprise information about administration and dosage and patient groups. Such kits, preferably kits of parts, may be applied e.g. for any of the applications or medical uses mentioned herein. Preferably, the individual components of the kit or kit of parts for use may be provided in lyophilised form. The kit may further contain as a part a vehicle (e.g. pharmaceutically acceptable buffer solution) for solubilising the therapeutic nucleic acid or RNA or the composition of the first aspect. In preferred embodiments, the kit or kit of parts for use comprises Ringer- or Ringer lactate solution. In preferred embodiments, the kit or kit of parts for may comprise an applicator for delivery to the anterior segment of the eye, an injection needle suitable for delivery to the anterior segment of the eye, a microneedle suitable for delivery to the anterior segment of the eye, an injection device suitable for delivery to the anterior segment of the eye, a catheter suitable for delivery to the anterior segment of the eye, an implant delivery device suitable for delivery to the anterior segment of the eye, or a microcannula suitable for delivery to the anterior segment of the eye. Any of the above kits may be used in applications or medical uses as defined in the context of the invention. Methods of treatment and delivery In a third aspect, the invention relates to a method of treating or preventing of an ophthalmic disease, disorder or condition, wherein the method comprises applying or administering to a subject in need thereof the composition as defined in the context of the first aspect, or the kit or kit of parts of the second aspect via administration to the anterior segment of the eye of a subject. Notably, embodiments relating to the first or second aspect of the invention are likewise applicable to embodiments of the third aspect, and embodiments relating to the third aspect of the invention are likewise applicable to embodiments relating to the first or second aspect of the invention. In preferred embodiments of the third aspect, the method comprises a step of applying or administering to a subject the composition as defined in the context of the first aspect, or the kit or kit of parts of the second aspect, wherein said composition or kit or kit of parts is administered into the to the anterior segment of the eye of a subject. In preferred embodiments, the therapeutic nucleic acid/RNA of the composition and/or the encoded peptide (provided by the therapeutic nucleic acid/RNA) is substantially localized to (therapeutically relevant) cells and/or tissues, preferably to (therapeutically relevant) cells and/or tissues of the anterior segment of the eye upon administration to the cornea, conjunctiva and/or ciliary body. In particular, such method may comprise the steps of: a) providing the composition of the first aspect, or the kit or kit of parts of the second aspect; b) administering said composition or kit or kit of parts to the anterior segment of the eye; c) optionally, administering a further substance (e.g. an anti-proliferative drug, an anti-fibrotic drug, an anti- inflammatory drug, a vascular endothelial growth factor modulator, a platelet derived growth factor modulator, an angiogenesis inhibitor, an immunosuppressive agent, a vascular permeability inhibitor, or a combination thereof) preferably to the anterior segment of the eye; d) optionally, administering a further therapeutic nucleic acid/RNA and/or guide RNA, preferably to the anterior segment of the eye. In a preferred embodiment of the third aspect, the invention relates to a method for gene therapy of an ophthalmic disease, disorder, or condition. In embodiments, the method for gene therapy comprises a step of applying or administering to a subject a composition of the first aspect, or the kit or kit of parts of the second aspect, wherein said composition or kit or kit of parts is administered to the anterior segment of the eye, wherein said composition or kit or kit of parts comprises a therapeutic nucleic acid/RNA encoding at least one CRISPR-associated endonuclease and at least one guide RNA. In embodiments, the therapeutic nucleic add/RNA encoding at least one CRISP-associated endonuclease and/or the CRISP-associated endonuclease is substantially localized to (therapeutically relevant) cells and/or tissues, preferably to (therapeutically relevant) cells and/or tissues of the anterior segment of the eye upon administration to the cornea and/or the conjunctiva and/or to the ciliary body. In particular, such the method for gene therapy may comprise the steps of: a) providing a composition, or the kit or kit of parts, comprising at least one therapeutic RNA encoding at least one CRISP-associated endonuclease and, optionally, at least one guide RNA; b) applying or administering said composition, or kit or kit of parts to the eye of a subject in need of gene therapy, wherein applying or administering is performed by administering to the anterior segment of the eye; c) optionally, administering a further substance (e.g. an anti-inflammatory drug, a vascular endothelial growth factor modulator, a platelet derived growth factor modulator, an angiogenesis inhibitor, an immunosuppressive agent, a vascular permeability inhibitor, or a combination thereof), preferably to the anterior segment of the eye or intravitreally; d) optionally, administering a further therapeutic RNA and/or guideRNA, preferably to the anterior segment of the eye or intravitreally, "Gene therapy" preferably involves modulating (i.e. restoring, enhancing, decreasing or inhibiting) gene expression in a subject in order to achieve a therapeutic effect. To this end, gene therapy typically encompasses the introduction of nucleic acids into cells. The term generally refers to the manipulation of a genome for therapeutic purposes and includes the use of genome-editing technologies (e.g. CRISPR/Cas) for correction of mutations that cause disease, the addition of therapeutic genes to the genome, the removal of deleterious genes or genome sequences, and the modulation of gene expression. Gene therapy may involve in vivo or ex vivo transformation of the host cells. In a fourth aspect, the present invention relates to a method of delivering a therapeutic nucleic acid/RNA to cells and/or tissues of the anterior segment of the eye of a subject, wherein delivering is performed via administration to the cornea, the ciliary body, or the conjunctiva of the eye of a subject. Said method comprising the steps of providing a composition comprising said therapeutic nucleic acid/RNA preferably as defined herein, or a composition, preferably as defined herein, or kit or kit of parts preferably as defined herein, and delivering said therapeutic nucleic acid/RNA, composition, or kit or kit of parts to cells and/or tissues of the eye of the subject, wherein delivering is performed by administering said composition to the anterior segment of the eye of a subject. Notably, embodiments relating to the first, second, or third aspect of the invention are likewise applicable to embodiments of the fourth aspect, and embodiments relating to the fourth aspect of the invention are likewise applicable to embodiments relating to the first, second, or third aspect of the invention In some embodiments, the administration of the therapeutic nucleic acid/RNA may be performed prior, after and/or during eye surgery. Preferably, the composition comprising the therapeutic RNA is administered prior, during or after glaucoma surgery, cataract surgery, or Laser-Assisted In-Situ Keratomileusis (LASIK). In preferred embodiments of the fourth aspect, delivering to the anterior segment of the eye leads to expression, that is translation, of the encoded protein in cells and/or tissues of the eye, preferably in therapeutically relevant cells or tissues of the eye (as defined above). In preferred embodiments of the third and fourth aspect, administration to the anterior segment of the eye is preferably performed in a non-surgical manner, e.g. via an injection needle, a microneedle, an injection device, a microinjection device, a catheter, an implant delivery device, or a microcannula to a subject, preferably to a subject in need of treatment. In preferred embodiments of the third and fourth aspect, the administering step comprises inserting a hollow microneedle into the cornea at an insertion site, the microneedle having a tip end with an opening, and infusing the composition as defined in the context of the first aspect into the cornea through the inserted microneedle. In preferred embodiments of the third and fourth aspect, the administering step comprises inserting a hollow microneedle under the conjunctiva at an insertion site, the microneedle having a tip end with an opening, and infusing the composition as defined in the context of the first aspect under the conjunctiva (sub-conjunctival) through the inserted microneedle. In preferred embodiments of the third and fourth aspect, the administering step comprises inserting a hollow microneedle into the ciliary body at an insertion site, the microneedle having a tip end with an opening, and infusing the composition as defined in the context of the first aspect into the ciliary body through the inserted microneedle. Preferably, the composition of the invention is applied or administered more than once, for example two times, three times, or four times, for example periodically. The primary objectives of any ocular drug-delivery system are to maintain therapeutic drug concentrations at the target site, reduce dosage frequency, and overcome various dynamic and static ocular barriers. Most importantly, the drug-delivery system should cause no adverse ocular reactions and aim to achieve enhanced drug bioavailability. The methods outlined in the context of the third and fourth aspect may be applied for laboratory, for research, for diagnostic, for commercial purposes and/or for therapeutic purposes. Preferably, the methods may be carried out in the context of the treatment or prevention of ophthalmic diseases, disorders or conditions as defined herein. In embodiments of the third and fourth aspect, the composition comprising the therapeutic nucleic acid, preferably RNA is administered to a subject that has suffered a trauma to the eye, corneal diseases, conjunctival diseases, disorders of ciliary body or has undergone an ocular surgery. In embodiments, the composition comprising the therapeutic nucleic acid, preferably RNA is administered as an eye drop formulation, wherein the eye drop formulation is administered to the anterior segment of the eye of a subject. Common eye drops are accessible in the forms of water and oil solutions, emulsions, or suspensions of one or more active ingredients, which may contain preservatives if stored in multiuse packaging. These forms are sterile and isotonic. The optimum pH for eye drops equals that of tear fluid and is about 7.4. Depending on the condition being treated, they may contain steroids, antihistamines, sympathomimetics, beta receptor blockers, parasympathomimetics, parasympatholytics, prostaglandins, nonsteroidal anti-inflammatory drugs (NSAIDs), antibiotics, antifungals, or topical anesthetics. Preferably, the eye drop formulation of this invention is administered to the cornea of the eye of a subject via topical administration as described above. In other embodiments, the composition comprising the therapeutic nucleic acid, preferably RNA is administered via a coated contact lens wherein the contact lens is coated with /or comprising at least one therapeutic nucleic acid, preferably RNA comprising at least one coding sequence encoding at least one peptide or protein for use in treatment or prevention of an ophthalmic disease, disorder or condition, wherein the contact lens is administered to the anterior segment of the eye of a subject. In preferred embodiments, the coated contact lens is administered to the cornea of the eye of a subject. In other embodiments, the composition comprising the therapeutic nucleic acid, preferably RNA is administered via a medical device coated with at least one therapeutic nucleic acid, preferably RNA comprising at least one coding sequence encoding at least one peptide or protein for use in treatment or prevention of an ophthalmic disease, disorder or condition, wherein the medical device is administered to the anterior segment of the eye of a subject. In preferred embodiments, the medical device coated with or comprising the therapeutic nucleic acid/RNA is an ocular implant (e.g ocular stent), preferably an eye stent. Eye stents are used as a treatment for glaucoma and, in children, NLD (nasolacrimal duct) obstructions. Stents are very small tubes usually made of plastic, fabric, or metal, which are surgically inserted to relieve obstructions and keep a path open so blood or other fluids can pass. Stenting to help treat glaucoma is used in combination with cataract surgery to reduce pressure inside the eye in certain adult patients who have mild or moderate open-angle glaucoma. Clear fluid in a healthy eye flow freely through the front chamber of the eye and drains out through a mesh of tissue and exit via into a canal at the edge of iris and cornea. If the meshwork becomes blocked or drains slowly, pressure builds up inside the eye to a level that may cause vision loss. A stent creates an opening to allow better drainage. In a healthy child, the nasolacrimal duct allows tears to exit the eye by draining into the nasal passages. In NLD, that duct is blocked or did not open at birth. Current inventions for anterior ocular segment disorders such as punctum plugs, ocular implants, drug-eluting contact lenses, and ocular iontophoresis represent state-of-the-art inventions for sustained and controlled drug release. Ocular implants involve introducing a foreign body into the eye and can result in implant-related ocular side effects. The release of the composition comprising the therapeutic nucleic acid/RNA coated on a medical device according to this invention may reduce implant-associate side effects. In preferred embodiments, the composition comprising the therapeutic nucleic acid/RNA is controlled- release/sustain- release from the coating of the medical device. In embodiments, the composition comprising the therapeutic nucleic acid/RNA is coated on a medical device (e.g contact lens or ocular implant) using standard coating method including dip coating, electro-treated coating, spray coating or plasma-treated coating. In preferred embodiments, the applicator coated with or comprising the therapeutic nucleic acid/RNA is implanted in the conjunctiva or the ciliary muscle eye of a subject. In other embodiments, the composition comprising the therapeutic nucleic acid/RNA is administered as a gel or oinment for use in treatment or prevention of an ophthalmic disease, disorder or condition, wherein the gel or oinment is administered to the anterior segment of the eye of a subject. Gels are mostly used to treat dry eye. Eye gels can either be a more viscous type of eye drop or can come as an insert that will dissolve over time when placed in between your eyelid and eyeball. Gels will perform a similar role to eye drops when treating dry eye but will take more time to become effective in the process of lubricating the surface of your eye and can cause blurry vision. Overall, gels will be more effective than eye drops when used to treat more severe symptoms caused by dry eye. Ointments are typically more effective at treating symptoms than eye drops and can treat more symptoms when compared to gels. Similar to gels, ointments take more time to become effective and can cause blurry vision when they are applied. The gel or oinment comprising the composition comprising the therapeutic nucleic add/RNA of this invention is preferably administered topical to the cornea of the eye of a subject. Brief description of list and tables List 1: Suitable therapeutic proteins provided by the therapeutic RNA of the invention Table 1: Suitable therapeutic proteins and sequences provided by the therapeutic RNA of the composition Table 2: RNA constructs used in the Examples Table 3: SM-102-LNP variants for cornea application used in Example 3 Table 4: RNA constructs used in Example 5 Table 5: RNA constructs used in Example 6 Brief description of the drawings Figure 1: cross-sectional view of an illustration of the human eye, comprising anterior region or segment 12, posterior region or segment 14, cornea 16, lens 18, sclera 20, anterior chamber 22, iris 24, cornea 16, posterior chamber 26, limbus 38, conjunctiva 45, choroid 28, retina 27, retinachoroidal tissue (choroid 28, retina 27), vitreous 30, ciliary body 32, ora serrata 34, suprachoroidal space 36. Further explanations are provided in the description. Figure 2: shows that LNP-formulated luciferase mRNA can be delivered intravitreally (A) or via contact lenses ex vivo (B) measured by luminescence assay. Further information is provided in the Example section, Example 3. Figure 3: shows that the in vitro system EpiOcular™ which resembles the human cornea (A) can be successfully transfected with GFP mRNA formulated in SM-102 N/P 8 LNPs or (B) formulated with SM-102 N/P 6 LNPs (C). EpiOcular™ cells transfected with LNP formulated mRNA encoding pigment epithelium-derived factor (PEDF) or luciferase show no toxicities related to the transfections (D). E shows a reduction of cells transfected with PEDF mRNA. Further information is provided in the Example section, Example 4. Figure 4: shows that sub-conjunctival injection with is displayed in A (Koutsoviti et al Materials, 2021) of LNP formulated luciferase mRNA leads to a production of high levels of active protein in vivo (B). Further information is provided in the Example section, Example 5. Figure 5: shows that injection of CVCM®-formulated luciferase mRNA lead to a production of high levels of active protein in vivo. Further information is provided in the Example section, Example 6. Examples In the following, examples illustrating various embodiments and aspects of the invention are presented. However, the present invention shall not to be limited in scope by the specific embodiments presented herein and should rather be understood as being applicable to other compositions or uses as for example defined in the specification. Accordingly, the following preparations and examples are given to enable those skilled in the art to more clearly understand and to practice the present invention. Indeed, various modifications of the invention in addition to those described herein will become readily apparent to those skilled in the art from the foregoing description, accompanying figures and the examples below. Example 1: Preparation of compositions for in vivo and ex vivo experiments The present example provides methods of obtaining the RNA as well as methods of generating composition of the invention comprising the RNA formulated in lipid-based carriers. 1.1. Preparation of DNA templates for RNA in vitro transcription: DNA sequences encoding luciferase, GFP and PEDF (Pigment epithelium-derived factor) were prepared and used for subsequent RNA in vitro transcription reactions. Some DNA sequences were prepared by modifying the wild type or reference encoding DNA sequences by introducing a G/C optimized coding sequence for stabilization and expression optimization. Sequences were introduced into a pUC derived DNA vector to comprise a stabilizing heterologous UTR sequence and a stretch ofadenosines and optionally a histone stem-loop (hSL). The obtained plasmid DNA templates were transformed and propagated in bacteria using common protocols known in the art. Eventually, the plasmid DNA templates were extracted, purified, and linearized using a restriction enzyme. The herein used RNA constructs and coding sequences are provided in Table 2. Table 2: RNA constructs used in the Examples

1.2^RNA_/n v/fro transcription from plasmid DNA templates: Linearized DNA templates were used for DNA dependent RNA in vitro transcription (IVT) using T7 RNA polymerase in the presence of a sequence optimized nucleotide mixture (ATP/GTP/CTP/UTP) and cap analog (for Cap1: m7G(5')ppp(5')(2'OMeA)pG; TriLink) under suitable buffer conditions. After RNA in vitro transcription, the obtained RNA IVT reaction was subjected to purification steps comprising RP-HPLC. 1.3. Preparation of Dolvmer-lipidoid complexes carrying mRNA (CVCM®s): 20mg of peptide (CGH5R4H5GC-NH2) was dissolved in N-methylpyrrolidone. After addition of 2ml of borate buffer (pH 8.5), the solution was stirred at RT for >18h. Subsequently, 12.6mgofPEG-SH 5000, dissolved in N-methylpyrrolidone, was added to the peptide solution and the vial filled up to 3ml with borate buffer (pH 8.5). After 18h, the reaction mixture was purified and further concentrated using Centricon filter units (MWCO 10kDa) and subsequently lyophilized. The obtained polyethylene glycol/peptide polymers (HO-PEG 5000-S-(S-CGHsR4H5GC-S-)7-S-PEG 5000-OH), dissolved in water, were used to prepare RNA formulations. RNA was complexed with the polymer at the 1:2 ratio (W/W) and 3- C12-C3-OH lipidoid was used. Formulated RNA was lyophilized and stored at -80°C. 1,4. Preparation of lipid-based carriers encapsulating the mRNA: An ethanolic lipid solution is prepared by solubilizing a cationic lipid, a neutral lipid, cholesterol, and an aggregation reducing lipid in ethanol. An aqueous RNA solution is prepared by adjusting the purified RNA (obtained according to Example 1.2) to a target concentration in 50mM citrate buffer, pH 4.0. Lipid-based carriers are prepared essentially according to the procedures described in W02015199952, W02017004143 and W02017075531. In short, lipid-based carriers are prepared at a ratio of RNA to total Lipid of about 0.03-0.04 w/w. Pumps are used to combine the ethanolic lipid solution with a flow rate F1 and the RNA aqueous solution with a flow rate F2 at a ratio of about 1:5 to 1:3 (vol/vol) in a T-piece system. F1 and/or F2 are adjusted to flow rates above 15ml/min to allow the formation of LNPs encapsulating the RNA that have a Z-average size in a range from about 50nm to about 120nm. After formulation, the ethanol is removed by at least one TFF step and at least one clarifying filtration step. After clarifying filtration, the filtrate is adjusted to a desired concentration (typically 1 g/l RNA) using a buffer comprising sucrose, sodium chloride, and a buffering agent. Subsequently, the resulting formulation is filtered through sterilizing filters to reduce bioburden as used for in vitro and in vivo studies. Other LNPs used in the working examples were also prepared using the NanoAssemblrTM microfluidic system (Precision NanoSystems Inc., Vancouver, BC) according to standard protocols which enables controlled, bottom-up, molecular self-assembly of nanoparticles via custom-engineered microfluidic mixing chips that enable millisecond mixing of nanoparticle components at a nanolitre scale. The respective lipids were solubilized in alcoholic solution (ethanol) according to standard procedures. Subsequently, this ethanol mix of lipids was combined with an aqueous phase containing appropriate amounts of mRNA. Syringe pumps were installed into inlet parts of the NanoAssemblrTM and used to mix the ethanolic lipid solution with the mRNA aqueous solution at a ratio of about 1:5 to 1 :2 (vol/vol) with total flow rates from about 17 ml/min to about 23 ml/min. The ethanol was then removed and the external buffer replaced with PBS/sucrose buffer. Finally, the lipid nanoparticles were filtered through a 0.2 pm pore sterile filter The herein tested SM-102 LNPswere composed of38.5mol% or40.9mol% cholesterol, 10mol% DSPC, 50mol% SM- 102, and 1.5mol% DMG-PEG 2000. Example 2: Methods used in the experiments The present example provides methods used for testing the RNA formulations (obtained according to Example 1). 2.1. Luciferase Assay: Frozen rabbit eyes were dissected and homogenized in lysis buffer, subsequently 20 pl of lysate are added to each well of a 96 well plate and before the plate was read using the Luminometer, 100 pl of ludferase assay reagent was added to each well and the plate was read immediately. Statistical analysis was carried out using a one-way ANOVA with Tukey's multiple comparison correction. 2.2 Proliferation assay A proliferation assay, a lactate dehydrogenase (LDH) assay (Promega, G1780) was used to assess toxicity of the formulations according to the manufacturer's guidelines. 2.3 Cell viability assay MTT ((3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide)-assay (MatTek, Slovak Republic) was performed to measure cell viability and cell proliferation according to manufacturer's description. Example 3: Ex vivo application ofSM-102-LNP formulated luciferase mRNA in rabbit corneas The aim of the experiment was to test the effect of LNP formulated luciferase mRNA coated on contact lenses for topical transfection of the eye cornea. Additionally, as comparison, LNP formulated luciferase mRNA was injected (intravitreally) into the vitreous of the rabbit eye of New Zealand white rabbits. Rabbit corneas of New Zealand white rabbits were transfected ex vivo with SM-102- LNP formulated mRNA encoding luciferase (R8730, SEQ ID NO.493). RNA formulations used in the experiment were prepared according to Example 1. The formulations F1-5 were applied in acetate buffer intravitreally (Figure 2A) or were applied under contact lenses before being placed on the cornea (Figure 2B). Hereby, 10 pg of luciferase mRNA in 50 pl were injected or applied per eye. Acetate buffer was used as a control.24 h after administration, the rabbit eyes were homogenized and luminescence of the eye tissue was evaluated (according to 2.1). Table 3: SM-102-LNP variants for cornea application used in Example 3

Summary Surprisingly, the results show that corneal administration of RNA formulated in a lipid-based carrier (LNP) via coated contact lens successfully transfected cells in the eye (Figure 2B). A comparable luminescence level was detected for intravitreal administration of RNA formulated in SM-102-based LNPs (Figure 2A). Example 4: Transfection of Zs GFP and PEDF mRNA in in vitro human cornea system EpiOcular™ The aim of this experiment was the transfection efficiency of LNP formulated Zs GFP mRNA using the in vitro human cornea system EpiOcular™. Additionally, the anti-proliferative effect of Pigment epithelium-derived factor (PEDF) was shown using this cornea cell system. PEDF also known as serpin F1 (SERPINF1), is a multifunctional ocular protein secreted by the retinal pigment epithelium that has anti-angiogenic, anti-tumorigenic, and neurotrophic functions. PEDF is known to suppress retinal neovascularization and endothelial cell proliferation. It is being investigated as a therapeutic candidate for treatment of such conditions as choroidal neovascularization, heart disease, and cancer. PEDF may help suppress unwanted neovascularization of the eye. Therefore, may be a useful tool to prevent choroidal neovascularization. EpiOcular™ from MatTek cooperation is an in vitro system, which very closely resembles a human cornea (Figure 3A). This reconstructed ocular tissue model containing normal human keratinocytes is used to evaluate possible toxicities of different compounds to be tested in the clinic. It has the ability to differentiate materials that are ocular non-imtants from materials that are ocular irritants. The EpiOcular™ Tissue from MatTek cooperation was used according to manufacturer's description. To analyze transfection efficiency, EpiOcular™ keratinocytes were transfected with formulated mRNA encoding Zs GFP (R8980, SEQ ID NO: 497) (200ng/well). Because membranes have a very strong autofluorescence, the cells were detached and replated for imaging after 24 h, seeding in 8-well IbiTreat chambers and incubation overnight. LNPs used: SM-102 N/P 6 and SM-102 N/P 8 (SM-102: 50%, Cholesterol: 38.5%, DSPC: 10%, mPEG2000-DMG: 1.5%). For the toxicity assay EpiOcular cells were transfected with 200 ng SM-102-LNP-formulated mRNA encoding PEDF (R8719, SEQ ID NO: 499, Pigment epithelium-derived factor) or luciferase (R8730, SEQ ID NO: 493). Levels of LDH 24 h were evaluated after transfecting cells. RNA formulations used in the experiment were prepared according to Example 1, methods used herein are described in Example 2. Cell viability was measured using a lactate dehydrogenase (LDH) assay (Promega, G1780). Furthermore, the number of cells was determined via MTT Assay after stimulation of cells with TGFp (transforming growth factor-beta). Here, cells were stimulated with TGFp (50 ng/mL) 2 hours before transfection with 200ng of SM- 102-LNP-formulated mRNA encoding PEDF mRNA formulation (R8719, SEQ ID NO: 499) or mRNA encoding luciferase (R8730, SEQ ID N0:493). MTT ((3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide)-assay was performed 24 h after transfection. Summary Within this experiment, it could be shown that corneal administration of LNP formulated mRNA leads to protein expression and also exhibit an therapeutic effect: GFP-positive cells could be detected 24 h after transfection (FigureB and C). No influence on cell viability was demonstrated for expression of pigment epithelium-derived factor (PEDF) and luciferase (Figure 3D). A clear reduction in the number of cells transfected with PEDF was shown as determined by an MTT assay (Figure 3E). Since there are no toxicities related to transfections (LDH) it is likely that the cell proliferation was inhibited in the presence and absence ofTGFp. Examele 5: In vivo application of LNP formulated luciferase mRNA in injected sub-conjunctivelv into rabbit The aim of this experiment was to test the effect of different LNP formulated luciferase mRNA injected under the conjunctiva of the rabbit eye. Sub-conjunctival injection is a type of periocular route of injection suitable for ocular drug administration (Figure 4A). Medications are applied either under the conjunctiva or underneath the conjunctiva lining the eyelid. The sub-conjunctival injection bypasses the fatty layers of the bulbous conjunctiva, putting medications adjacent to sclera that is permeable to water. This increases the penetration of water-soluble drugs into the eye. Hereby, 9 weeks old female New Zealand white rabbits were injected sub-conjunctively with 5 pg in 25 pL of LNP- formulated mRNA encoding luciferase (R8730, SEQ ID N0:493, Table 4). The injections were done unilaterally in the right eye. Luminescence was evaluated after 24h after transfection. Table 4: RNA constructs used in Example 5

Summary As demonstrated in Figure 4B for the first time, sub-conjunctively injection of different LNP- formulated mRNA works effectively and leads to the production of high levels of active protein (luciferase). Example 6: In vivo application of polymer-lipidoid complexes (CVCM®) -formulated mRNA into the ciliary muscle The goal of this experiment was to test the effect of polymer-lipidoid complexes (CVCM®)- formulated luciferase mRNA injected into the ciliary muscle of the rabbit eye. Injection into the ciliary muscle is considered less invasive than sub- retinal injections. Hereby, the ciliary body of 9 weeks old female New Zealand white rabbits was injected with 2 (Jg/pl in 5 pl of CVCM® - formulated mRNA encoding luciferase (R6651, SEQ ID N0:495). Three right eyes of New Zealand white rabbits were injected per formulation. Luminescence was evaluated after 24h after transfection. Table 5: RNA constructs used in Example 6 '
3

Summary As demonstrated in Figure 5, injection of CVCM®- formulated luciferase mRNA into the ciliary body works effectively and leads to the production of high levels of active protein. Summary of the findings of examples 1 to 6: As demonstrated in the present examples, nucleic acid, in particular RNA was successfully and effectively delivered to the anterior segment of the eye. Corneal administration of luciferase mRNA coated on contact lenses or injected intravitreally in rabbit corneas showed high levels of protein expression (Figure 2). Transfection of mRNA into the in vitro cornea system EpiOcular™ showed high levels of GFP protein expression as well a therapeutic effect of pigment epithelium-derived factor (PEDF) (Figure 3). Additionally, LNP-formulated luciferase mRNA leads to high levels of protein expression injected sub-conjunctively into rabbit eyes (Figure 4). A high transfection efficiency and protein expression using polymer-lipidoid complexes (CVCM®)- formulated luciferase mRNA injected into the ciliary body of rabbit eyes could be shown as well (Figure 5).
























