WO2024227886A1 - Bispecific binding molecule - Google Patents
Bispecific binding moleculeDownload PDFInfo
- Publication number
- WO2024227886A1 WO2024227886A1PCT/EP2024/062141EP2024062141WWO2024227886A1WO 2024227886 A1WO2024227886 A1WO 2024227886A1EP 2024062141 WEP2024062141 WEP 2024062141WWO 2024227886 A1WO2024227886 A1WO 2024227886A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- seq
- binding molecule
- ifna
- antibody
- bispecific binding
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Classifications
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/24—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against cytokines, lymphokines or interferons
- C07K16/249—Interferons
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/20—Immunoglobulins specific features characterized by taxonomic origin
- C07K2317/24—Immunoglobulins specific features characterized by taxonomic origin containing regions, domains or residues from different species, e.g. chimeric, humanized or veneered
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/30—Immunoglobulins specific features characterized by aspects of specificity or valency
- C07K2317/31—Immunoglobulins specific features characterized by aspects of specificity or valency multispecific
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/70—Immunoglobulins specific features characterized by effect upon binding to a cell or to an antigen
- C07K2317/76—Antagonist effect on antigen, e.g. neutralization or inhibition of binding
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/90—Immunoglobulins specific features characterized by (pharmaco)kinetic aspects or by stability of the immunoglobulin
- C07K2317/92—Affinity (KD), association rate (Ka), dissociation rate (Kd) or EC50 value
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2318/00—Antibody mimetics or scaffolds
- C07K2318/20—Antigen-binding scaffold molecules wherein the scaffold is not an immunoglobulin variable region or antibody mimetics
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2319/00—Fusion polypeptide
Definitions
- the present disclosurerelates to a bispecific binding molecule having binding sites targeting interferon alpha (IFNa) and human complement component 5 (C5).
- the disclosurefurther relates to the use of such a bispecific binding molecule for the preparation of a pharmaceutical composition.
- the disclosurefurther relates to a method for the treatment or amelioration of a disease comprising administration of an effective amount of such a bispecific binding molecule.
- SLESystemic lupus erythematosus
- SLESystemic lupus erythematosus
- the immunopathogenesis of the diseasehas challenged clinicians and researchers alike for decades. Extensive studies have been able to establish several key role players involved in the complex interplay between the innate and adaptive immune system and complement system driving disease progression (Kaul et al, Nat. Rev. Dis. Prim. 2:16039, 2016).
- the disease pathologyis characterized by significant levels of autoantibodies towards nuclear self-antigens, produced by hyperactive autoreactive plasma cells and impaired clearance of apoptotic material as a result of a dysregulated immune system.
- ICsautoantibody-antigen immune complexes
- Type 1 interferonsin particular interferon alpha (IFNa) have been shown to host a fundamental role in the pathogenesis of SLE, overexpressed by plasmacytoid dendric cells (pDCs) upon stimuli of toll-like receptor 7 and 9 (TLR7 and TLR9) by ICs (Oon, Wilson and Wicks, Clin. Transl. Immunol. 5:29, 2016) and (Niewold et al, J. Biomed. Biotechnol. 2010:948364, 2010). Consequently, when expressed abundantly by pDCs, IFNa exerts an array of immune-modular effects promoting autoimmune disease progression via its interaction with the IFNaR.
- IFNainterferon alpha
- IFNainduced upregulation of costimulatory receptors CD80 and CD86 providing a platform for autoreactive CD4 + helper T cells to activate cytotoxic autoreactive CD8 + T cells with additional heightened perforin and granzymes expression due to direct stimuli by IFNa (Obermoser and Pascual, Lupus. 19:1012-1019, 2010).
- IFNaupregulates TLR7 and interferon regulating factor 7 (IRF7) in pDCs, mDCs and monocytes which subsequently upregulates the expression of IFNa itself.
- IRF7interferon regulating factor 7
- IFNainduces the expression of B cell activating factor (BAFF) which contributes to the survival of mature B cells and together with IL6 promotes differentiation of plasma blasts into mature autoantibody producing plasma cells (Jego et al, Immunity. 19:225-234, 2003, and Rdnnblom and Leonard, Lupus Sci. Med. 6:e000270, 2019).
- BAFFB cell activating factor
- Complement component Cq1 deficiencyis strongly related to SLE pathogenesis with detectable levels of C1q autoantibodies in specific patient cohorts (Leffler et al, Ann. Rheum. Dis., 73:1601-1606, 2014).
- Previous workhas shown the downregulation of IFNa expression upon stimuli of pDCs by C1q-autoantibody ICs.
- C1qis involved in clearing apoptotic material by opsonization, where autoantibodies contributing to lower levels of C1q directly affect the degradation and clearance of apoptotic material, in particular released neutrophil extracellular traps (NETs) from dying neutrophils. Consequently, the risk of hyperactive plasma cells to secret autoantibodies towards autoantigens from released cell material forming new ICs increases substantially when the levels of C1q are significantly low enough, tipping the balance in favor of progressive autoimmune reaction.
- the complement systemacts as a driving force in disease progression, contributing to hyper-inflammation by activation of complement factors triggered by ICs through mainly the classical pathway.
- Rituximabfailed to reach primary endpoints in two phase II trials, although post hoc analysis showed a reduced risk for a first severe flare compared to placebo (Tunnicliffe et al, Arthritis Care Res (Hoboken) 67(10): 1440-1452, 2015).
- the FDA-approved monoclonal antibody belimumabhas been of particular interest as it targets the aforementioned B cell activating factor (BAFF) and has been investigated in patients with continuous active SLE despite standard therapy regiments and show high levels of autoantibodies in tested plasma samples.
- BAFFB cell activating factor
- the drughas shown safety and efficacy in adult patients with active SLE during phase II and III trials, it does not address the hyperactive inflammatory severe cases of SLE involving lupus nephritis.
- Type 1 interferons and their related downstream pathwayshave been shown to correlate with SLE disease activity as previously described, promoting differentiation of cells involved in the adaptive immune system, therapies targeting the cytokine group and the IFNa receptor (IFNaR) have emerged as potential avenues for treating the disease.
- IFNaRIFNa receptor
- the monoclonal antibody rontalizumabwas developed toward the IFNa ligand in patients with moderate-severe SLE, where it did not meet primary or secondary endpoint in a phase II study evaluating the antibody efficacy (Kalunian et al, Ann Rheum Dis 75(1 ): 196-202, 2016).
- a recent phase III trialthe monoclonal antibody anifrolumab met its primary end point for the treatment of SLE, significantly reducing disease activity.
- this antibodytargets the IFNaR receptor with a blocking mechanism of action, in contrast to rontalizumab which binds the IFNa ligand itself and blocks it from receptor-ligand interaction (Morand et al, Arthritis Rheumatol 71 , Abstract L17 to ACR/ARP Annual Meeting 2019). Anifrolumab was approved by FDA for the treatment of moderate to severe SLE in 2021 (Deeks, Drugs 81 (15): 1795- 1802, 2021 ).
- the complement inhibiting antibody eculizumabhas shown to potentially be effective in a subgroup of patients with complement mediated thrombotic microangiopathy in lupus nephritis and poor response to GSCs (Coppo et al, Pediatr Nephrol 30(1 ): 167-172, 2015).
- RARheumatoid arthritis
- the complement systemhas been indicated as a main player, by initiation of the RA pathogenic mechanisms through interactions with citrullinated proteins and by mediating direct tissue damage and an association between complement system activation and disease activity is well established (Triggianese P, 2023).
- IFN-I and in particular IFNahave been implicated to be upregulated in RA and there is emerging data that IFNs may contribute to the transition from preclinical to sustained clinical disease.
- rheumatoid factordemonstrated a positive association with circulating IFNa levels in both established and early RA (Lin CHM, 2024).
- Atherosclerosisplaque formation in the arterial walls, can cause serious complications such as myocardial infarction (Ml) and stroke, which are the primary cause of death and morbidity worldwide.
- Mlmyocardial infarction
- strokestroke
- risk factorssuch as pro-inflammatory stimuli, cytokines and dysregulated lipids.
- the contribution of inflammation in atherosclerosisis well established and there is growing evidence of a central role of IFNa and the complement system in the progression (Kiss MG, 2022; Chen HJ, 2020).
- CNScentral nervous system
- Traumatic brain injury (TBI)is a leading cause of mortality in the modem world.
- TBITraumatic brain injury
- BBBblood brain barrier
- the complement systemhas a central role in secondary inflammation and elevated levels of complement related proteins such as C 1 q, C3, sC5b-9 and MBL have been demonstrated in human CSF models for TBI. Animal models of TBI have demonstrated that a reduction of complement system activity leads to reduced lesion sizes and improved neurological outcomes (Hammad, 2018).
- the interferon systemhas also been linked to the outcomes of TBI through knock-out mouse models, and neutralization by monoclonal antibodies suggests that IFNa, which peaks at 2 hours after TBI, may play a detrimental role in brain trauma (Roselli F, 2018).
- SCISpinal cord injury
- SAHsubarachnoid hemorrhage
- CSFcerebrospinal fluid
- NMONeuromyelitis Optica
- BBBblood brain barrier
- IFN-a levelswith clinical disease activity and seventy, suggesting a role for IFN-a in disease progression (Asgari N, 2013).
- NMOis also known to be associated with upregulation of the complement system and in a recent small trial, the treatment with the anti-C5 monoclonal antibody eculizumab decreased the number of neurological episodes (www.clinicaltrials.gov NCT00904826, Pittock SJ, 2013).
- autoimmune diseaseAs is understood by the background provided in the introduction above, a number of additional disease states, disorders, and injury such as autoimmune disease, cardiovascular disease, and central nervous system (CNS) disorders, disease and injury involve players of the adaptive, innate and complement systems.
- CNScentral nervous system
- SLEis characterized by the influence of a multitude of factors in the innate, adaptive and complement system governing the disease pathogenesis, driving disease progression. Many endeavors aiming at developing novel SLE therapies with targeted biologies have shown limited success in the clinic, and efficacious therapeutics have yet to be realized. Previous attempts to find SLE therapies have revealed the complexity of the disease, which is not governed by a single pathogenic mechanism. This reasoning also stands true for the other disease states, disorders and injury mentioned above.
- CNScentral nervous system
- compositionswhich comprise such bispecific binding molecules and are designed to be suited for treatment of various inflammatory, autoimmune diseases, cardiovascular disease, and/or central nervous system (CNS) diseases, disorders and injury; and treatment methods in which such molecules or pharmaceutical compositions are administered to a subject in need of such treatment.
- CNScentral nervous system
- a bispecific binding moleculecomprising at least one first moiety with at least one first binding site for human complement component 5 (C5), operably linked to at least one second moiety with at least one first binding site for interferon alpha (IFNa).
- C5human complement component 5
- IFNainterferon alpha
- bispecific binding moleculeis intended to refer to a binding molecule which has affinity for two separate targets.
- the bispecific binding moleculecomprises at least two moieties, at least one first moiety having affinity for C5 by virtue of its binding site for C5, and at least one second moiety having affinity for IFNa by virtue of its binding site for IFNa.
- first moietyis intended to refer a moiety having affinity for C5.
- second moietyis intended to refer to a moiety having affinity for IFNa.
- at least oneis intended to refer to the fact that the bispecific binding molecule may comprise one, or may comprise more than one “first moiety”.
- the term “at least one”is also intended to refer to the fact that the bispecific molecule may comprise one, or may comprise more than one “second moiety”. Therefore, the bispecific binding molecule, which has affinity for the above-mentioned targets simultaneously, may be able to bind one C5 while also simultaneously be able to bind one IFNa. By this same definition, the bispecific binding molecule may also bind at least one C5 while also simultaneously bind to at least one IFNa.
- first moiety and second moietymay for example be connected by covalent coupling using known organic chemistry methods, or, if one or both moieties are polypeptides, be expressed as one or more fusion polypeptides in a system for recombinant expression of polypeptides, or joined in any other fashion, directly or mediated by a linker comprising a number of amino acids.
- a bispecific binding moleculeas defined herein, wherein said at least one first moiety with at least one first binding site for human complement component 5 (C5) is a C5 binding polypeptide.
- the part of the bispecific binding molecule designated “first moiety”is a C5 binding polypeptide.
- the at least one first moietycomprises a binding polypeptide capable of selective interaction with at least one C5.
- a bispecific moleculeas defined herein, wherein said C5 binding polypeptide is selected from the group consisting of full-length antibodies, Fab fragments, Fab’ fragments, F(ab')2 fragments, single chain Fab (scFab) fragments, Fc fragments, Fv fragments, single chain Fv (scFv) fragments, (scFv)2, scFv-Fc constructs, domain antibodies, a chimeric antibody, a CDR-grafted antibody, a fully human antibody, a bivalent antibody construct, a synthetic antibody, a bivalent antibody, a trivalent antibody, a tetravalent antibody, bivalent single chain antibody, a trivalent single chain antibody, a multivalent single chain antibody, nanobodies, AlbudAbs, DARTs (dual affinity re-targeting), BiTEs (bispecific T-cell engager), TandAbs (tandem diabodies), DAFs (dual acting Fab), two
- the binding polypeptidecomprises a variant of protein Z, in turn derived from domain B of staphylococcal protein A and described in Nilsson B et al, Protein Engineering 1 : 107-133, 1987.
- variantshaving affinity for a number of different targets, have been selected from libraries and engineered further as described in numerous prior publications, for example but not limited to WO1 995/19374; Nord et al, Nat Biotech (1997) 15:772-777; and W02009/080811 , all incorporated herein by reference.
- C5 binding polypeptides useful as first moiety in the bispecific binding molecule of the disclosure and comprising a C5 binding motifare disclosed in WO201 3/126006 and WO2015/028558.
- WO2013/126006 and WO201 5/028558disclose a C5 binding motif, BM.
- the C5 binding polypeptidecomprises a C5 binding motif, BM, which motif consists of the amino acids sequence selected from: i) EX2X3X4A X6X7EIX10X11 LPNL X16X17X18QW X21AFIX25 X 2 6LX 28 D (SEQ ID NO:249), wherein, independently of each other, X2 is selected from H, Q, S, T and V; X3 is selected from I, L, M and V;
- X4is selected from A, D, E, H, K, L, N, Q, R, S, T and Y;
- Xeis selected from N and W;
- X7is selected from A, D, E, H, I, L, N, Q, R, S and T;
- X10is selected from D and E;
- X11is selected from A, E, G, H, K, L, Q, R, S, T and Y;
- Xis selected from N and T;
- X17is selected from I, L and V;
- X18is selected from A, D, E, H, K, N, Q, R, S and T;
- X21is selected from I, L and V;
- X25is selected from A, D, E, G, H, N, S and T;
- X26is selected from K and S;
- X28is selected from A, D, E, H, N, Q, S, T and Y; and ii) an amino acid sequence which has at least 86 % identity to the sequence defined in i), wherein the polypeptide binds to C5.
- X n ” and “X m ”are used to indicate amino acids in positions n and m in the sequence i) as defined above, wherein n and m are integers which indicate the position of an amino acid within said sequence as counted from the N-terminal end of said sequence.
- X4 and X7indicate the amino acid in position four and seven, respectively, from the N- terminal end of sequence i).
- the above defined class of sequence related polypeptides having a binding affinity for C5is derived from a common parent polypeptide sequence. More specifically, the definition of the class is based on an analysis of a large number of random polypeptide variants of the parent polypeptide that were selected for their interaction with C5 in selection experiments.
- the identified C5 binding motif, or “BM”corresponds to the target binding region of the parent scaffold, which region constitutes two alpha helices within a three-helical bundle protein domain.
- the varied amino acid residues of the two BM helicesconstitute a binding surface for interaction with the constant Fc part of antibodies. By random variation of binding surface residues and subsequent selection of variants, the Fc interaction capacity of the binding surface was replaced with a capacity for interaction with C5.
- the function of any polypeptideis dependent on the tertiary structure of the polypeptide. It is therefore possible to make minor changes to the amino acid sequence of a polypeptide without largely affecting the tertiary structure and the function thereof.
- the disclosureencompasses first moieties that are modified variants of a C5 binding polypeptide with retained C5 binding characteristics.
- the polypeptidecomprises modified variants of the BM of i), which are such that the resulting sequence is at least 86% identical to a sequence belonging to the class defined by i), such as at least 89% identical, such as at least 93 % identical, such as at least 96 % identical to a sequence belonging to the class defined by i).
- an amino acid residue belonging to a certain functional grouping of amino acid residuese.g. hydrophobic, hydrophilic, polar etc.
- such changesmay be made in any position of the sequence of the C5 binding polypeptide as defined herein. In other embodiments, such changes may be made only in the non-variable positions, also denoted scaffold amino acid residues. In such cases, changes are not allowed in the variable positions. In other embodiments, such changes may be only in the variable positions.
- selection of C5 binding polypeptide variantsmay for example be achieved by phage display for selection of naive variants of a protein scaffold optionally followed by affinity maturation and cell display for selection of affinity maturated C5 binding variants. It is however understood that any selection system, whether phage-based, bacterial-based, cell-based or other, may be used for selection of C5 binding polypeptides.
- C5 binding and ’’binding affinity for C5refers to a property of a polypeptide which may be tested for example by the use of surface plasmon resonance technology, such as in a Biacore instrument (Cytiva).
- C5 binding affinitymay e.g. be tested in an experiment in which C5 is immobilized on a sensor chip of a Biacore instrument, and the sample containing the polypeptide to be tested is passed over the chip.
- the polypeptide to be testedis immobilized on a sensor chip of the instrument, and a sample containing C5, or fragment thereof, is passed over the chip.
- Binding valuesmay for example be defined in a Biacore 2000 instrument (Cytiva). C5 is immobilized on a sensor chip of the measurement, and samples of the polypeptide whose affinity is to be determined are prepared by serial dilution and injected over the chip. KD values may then be calculated from the results using for example the 1 :1 Langmuir binding model of the BIAevaluation software provided by the instrument manufacturer.
- a bispecific binding moleculecomprising a C5 binding polypeptide wherein X n in sequence i) is independently selected from a group of possible residues.
- X nmay be selected from any one of the listed groups of possible residues and that this selection is independent from the selection of amino acids in X m , wherein n ⁇ m.
- any of the listed possible residues in position X nmay be independently combined with any of the listed possible residues in any other variable position.
- variable positionsare positions denoted with an “X” in sequence i) as defined above.
- “variable positions”are those positions that are randomized in a selection library of Z variants prior to selection, and may thus for example be positions 2, 3, 4, 6, 7, 10, 11 , 17, 18, 20, 21 , 25 and 28 in sequence i).
- This definition of “variable positions”does not include positions 16 and 26, which are scaffold positions in this context, albeit allowed to be either one of two alternatives in each position.
- the polypeptides disclosed in Nord et al. and Ldfblom et al.are also based on a scaffold from the Z derivative of domain B of protein A from Staphylococcus aureus, although directed to other targets.
- the amino acids in positions 23 (corresponding to position 16 in the instant C5 binding motif) and 33 (corresponding to position 26 in the instant C5 binding motif)are N and S, respectively.
- polypeptides with amino acid residues N and S in positions 23 and 33, respectively(corresponding to positions 16 and 26 in the instant C5 binding motif; see Figure 2 of Ldfblom et al.), and polypeptides with amino acid residues T and K in positions 23 and 33, respectively, all have a maintained basic structure and function.
- amino acid residues at positions 16 and 26form part of the common scaffold, and it is contemplated to have either N or T in scaffold position 16 and either S or K in scaffold position 26.
- X2is selected from H, T and V. In another embodiment, X2 is selected from T and V. In yet another embodiment, X2 is V.
- X3is selected from I, L and V. In another embodiment, X3 is selected from I and L. In yet another embodiment, X3 is I. In an alternative embodiment, X3 is L.
- X4is selected from A, D, E, K, L, Q and R. In another embodiment, X4 is selected from A, D, E, K and R. In yet another related embodiment, X4 is selected from D and E.
- Xeis W.
- X7is selected from A, D, N and T. In another embodiment, X7 is selected from D and N. In yet another related embodiment, X7 is D. In an alternative embodiment, X7 is N.
- Xuis selected from A, H, K, Q, R and S. In another embodiment, Xu is selected from A, H, K and R. In yet another related embodiment, Xu is selected from A, K and R. In yet another related embodiment, Xu is selected from K and R.
- Xis T.
- X17is selected from I and L. In another embodiment, X17 is I. In an alternative embodiment, X17 is L.
- X18is selected from A, D, E, N, Q, S and T. In another embodiment, X18 is selected from A, D, E, Q and S. In yet another related embodiment, X18 is selected from D, E and Q. In yet another related embodiment, X18 is selected from D and E. In yet another related embodiment, X18 is D. In an alternative embodiment, X18 is E.
- X21is selected from I and L. In another embodiment, X21 is I. In an alternative embodiment, X21 is L. In one embodiment, X2s is selected from E, H, N and T. In another embodiment, X25 is selected from E and N. In yet another related embodiment, X25 is N.
- X26is K.
- X28is selected from A, D, E, H, N, Q and S. In another embodiment, X28 is selected from A, D, E and S. In yet another related embodiment, X28 is selected from A, D and E. In yet another related embodiment, X28 is selected from D and E. In yet another related embodiment, X28 is D.
- X3X4is selected from LE and LD.
- X17X18is selected from IE and LD.
- X3is selected from I and L;
- X7is selected from D and N;
- V. X17is selected from I and L;
- the amino acid sequence i)fulfils at least five of the eight conditions l-VIII. More specifically, the amino acid sequence i) may fulfill at least six of the eight conditions l-VIII, such at least seven of the eight conditions l-VIII, such as all of the eight conditions l-VIII. As described in the examples of WO2013/126006 and
- the BM sequence i)is selected from any one of SEQ ID NO:1- 12, SEQ ID NQ:20, SEQ ID NO:23-24, SEQ ID NO:26-28, SEQ ID NO:32-35, SEQ ID NO:38-39, SEQ ID NO:41 , SEQ ID NO:46, SEQ ID NO:49, SEQ ID NO:56-57, SEQ ID NO:59, SEQ ID NO:66, SEQ ID NO:78-79, SEQ ID NO:87, SEQ ID NO:92, SEQ ID NQ:106, SEQ ID NQ:110, SEQ ID NO:119, SEQ ID NO:125, SEQ ID NO:141 , SEQ ID NO:151 , SEQ ID NO:161 , SEQ ID NO:166, SEQ ID NO:187, SEQ ID NO:197, SEQ ID NQ:203, SEQ ID NQ:205, SEQ ID NO:215 and SEQ ID NO:243.
- the BM sequence i)may be selected from any one of SEQ ID NO: 1-12, such as from SEQ ID NO:1 , SEQ ID NO:2, SEQ ID NO:3, SEQ ID NO:4 and SEQ ID NO:5.
- the BM sequence i)may be selected from SEQ ID NO:1 and SEQ ID NO:4.
- the BM sequence i)is SEQ ID NO:1.
- the BM sequence i)is SEQ ID NO:4.
- the C5 binding motif (BM) as defined hereinis flanked by additional amino acids N-terminally to the C5 binding motif. In other embodiments, the C5 binding motif is flanked by additional amino acids C-terminally to the C5 binding motif. In further embodiments, the C5 binding motif is flanked by additional amino acids both N-terminally and C-terminally to the C5 binding motif. Therefore, in some embodiments there is provided a bispecific binding molecule as defined herein, wherein the C5 binding polypeptide in the first moiety comprises additional amino acids at the C- terminal and/or N-terminal end.
- BMC5 binding motif
- the BMis thought to constitute two of the three helices of a three-helix bundle and can therefore replace such a two-helix motif within any three-helix bundle.
- the replacement of two helices of the three-helix bundle domain by the two BM heliceshas to be performed so as not to affect the basic structure of the polypeptide. That is, the overall folding of the Ca backbone of the polypeptide is substantially the same as that of the three- helix bundle protein domain of which it forms a part, e.g. having the same elements of secondary structure in the same order etc.
- a BM according to the present disclosure“forms part” of a three-helix bundle domain if the polypeptide according to this embodiment has the same fold as the original domain, implying that the basic structural properties are shared, those properties e.g. resulting in similar CD spectra.
- the skilled personis aware of other parameters that are relevant.
- the C5 binding motifthus forms part of a three-helix bundle protein domain.
- the BMmay essentially constitute two alpha helices with an interconnecting loop, within said three- helix bundle protein domain.
- said three-helix bundle protein domainis selected from domains of bacterial receptor proteins. Non-limiting examples of such domains are the five different three-helical domains of Protein A from Staphylococcus aureus, such as domain B, and derivatives thereof.
- the three-helical bundle protein domainis a variant of protein Z, which is derived from domain B of staphylococcal Protein A (Wahlberg E et al. 2003, PNAS 100(6):3185-3190).
- the bispecific binding molecule as defined hereincomprises an amino acid sequence selected from:
- AEAKFAK-[BM](SEQ ID NO:251 ); ADNNFNK-[BM] (SEQ ID NO:252);
- ADNKFNK-[BM](SEQ ID NO:253);
- VDNKFNK-[BM](SEQ ID NO:254)
- VDAKYAK-[BM](SEQ ID NO:255); and sequences having at least 86% identity thereto, wherein [BM] is as defined herein.
- the bispecific binding molecule as defined hereincomprises an amino acid sequence selected from:
- [BM]-DPSQSSELLAEAKKLNDAQAPK(SEQ ID NO:261 );
- [BM]-DPSQSSELLAEAKKLNKAQAPK(SEQ ID NO:266); and sequences having at least 86% identity thereto, wherein [BM] is as defined herein.
- the bispecific binding molecule as defined hereincomprises an amino acid sequence selected from:
- ADNKFNK-[BM]-DPSQSANLLAEAKKLNDAQAPK(SEQ ID NO:268); ADNKFNK-[BM]-DPSVSKEILAEAKKLNDAQAPK (SEQ ID NO:269); VDNKFNK-[BM]-DPSQSANLLAEAKKLNDAQAPK (SEQ ID NQ:270); AEAKYAK-[BM]-DPSESSELLSEAKKLNKSQAPK (SEQ ID NO:271 );
- VDAKYAK-[BM]-DPSQSSELLAEAKKLNDAQAPKSEQ ID NO:272
- VDAKYAK-[BM]-DPSQSSELLAEAKKLNDSQAPKSEQ ID NO:273
- AEAKYAK-[BM]-DPSQSSELLSEAKKLNDSQAPKSEQ ID NO:274; AEAKYAK-[BM]-DPSQSSELLSEAKKLSESQAPK (SEQ ID NO:275); AEAKFAK-[BM]-DPSQSSELLSEAKKLSESQAPK (SEQ ID NO:276); AEAKYAK-[BM]-DPSQSSELLSEAKKLESSQAPK (SEQ ID NO:277); VDAKYAK-[BM]-DPSQSSELLSEAKKLNDSQAPK (SEQ ID NO:278); VDAKYAK-[BM]-DPSQSSELLSEAKKLSESQAPK (SEQ ID NO:279); VDAKYAK-[BM]-DPSQSSELLSEAKKLESSQAPK (SEQ ID NQ:280); VDAKYAK-[BM]-DPSQSSELLAEAKKLNKAQAPK (SEQ ID NO:281 ); AEAKYAK-[BM]-DP
- the C5 binding polypeptidemay for example have a sequence which is at least 86%, such as at least 88 %, such as at least 90 %, such as at least 92 %, such as at least 94 %, such as at least 96 %, such as at least 98 % identical to a sequence as defined above.
- % identicalor “% identity”, as used in the specification and claims, is calculated as follows.
- the query sequenceis aligned to the target sequence using the CLUSTAL W algorithm (Thompson, J.D., Higgins, D.G. and Gibson, T.J., Nucleic Acids Research, 22: 4673-4680 (1994)).
- a comparisonis made over the window corresponding to the shortest of the aligned sequences.
- the shortest of the aligned sequencesmay in some instances be the target sequence, such as the albumin binding domain defined herein. In other instances, the query sequence may constitute the shortest of the aligned sequences.
- the query sequencemay for example consist of at least 10 amino acid residues, such as at least 20 amino acid residues, such as at least 30 amino acid residues, such as at least 40 amino acid residues, such as at least 50 amino acids, such as at least 60 amino acids, for example 45 amino acid residues.
- the amino acid residues at each positionare compared, and the percentage of positions in the query sequence that have identical correspondences in the target sequence is reported as % identity.
- the bispecific binding molecule of this aspect of the disclosureit comprises a C5 binding antibody or an antigen binding fragment thereof as first moiety.
- antibodiesare immunoglobulin molecules capable of specific binding to a target (an antigen), such as a carbohydrate, polynucleotide, lipid, polypeptide or other, through at least one antigen recognition site located in the variable region of the immunoglobulin molecule.
- a targetsuch as a carbohydrate, polynucleotide, lipid, polypeptide or other
- antibody or an antigen binding fragment thereofencompasses not only full-length or intact polyclonal or monoclonal antibodies, but also antigen binding fragments thereof, such as Fab fragments, Fab’ fragments, F(ab')2 fragments, single chain Fab (scFab) fragments, Fc fragments, Fv fragments, single chain Fv (scFv) fragments, (scFv)2, scFv-Fc constructs, domain antibodies, a chimeric antibody, a CDR-grafted antibody, a fully human antibody, a bivalent antibody construct, a synthetic antibody, a bivalent antibody, a trivalent antibody, a tetravalent antibody, bivalent single chain antibody, a trivalent single chain antibody, a multivalent single chain antibody; a microbody, a maxybody, an avimer, a small disulfide-bonded protein; and a binding protein derived from a scaffold selected from the group consisting of staphylococcal protein
- modified antibodies and antigen binding fragments thereofinclude nanobodies, AlbudAbs, DARTs (dual affinity re-targeting), BiTEs (bispecific T-cell engager), TandAbs (tandem diabodies), DAFs (dual acting Fab), two-in-one antibodies, SMIPs (small modular immunopharmaceuticals), FynomAbs (fynomers fused to antibodies), DVD-lgs (dual variable domain immunoglobulin), CovX-bodies (peptide modified antibodies), duobodies and triomAbs.
- This listing of variants of antibodies and antigen binding fragments thereofis not to be seen as limiting, and the skilled person is aware of other suitable variants.
- a full-length antibodycomprises two heavy chains and two light chains.
- Each heavy chaincontains a heavy chain variable region (VH) and first, second and third constant regions (CH1 , CH2 and CH3).
- Each light chaincontains a light chain variable region (VL) and a light chain constant region (CL).
- VHheavy chain variable region
- VLlight chain variable region
- CLlight chain constant region
- full-length antibodyrefers to an antibody of any class, such as IgD, IgE, IgG, IgA, IgM or IgY (or any sub-class thereof).
- the subunit structures and three-dimensional configurations of different classes of antibodiesare well known.
- An “antigen binding fragment”is a portion or region of an antibody molecule, or a derivative thereof, that retains all or a significant part of the antigen binding of the corresponding full-length antibody.
- An antigen binding fragmentmay comprise the heavy chain variable region (VH), the light chain variable region (VL), or both.
- VHheavy chain variable region
- VLlight chain variable region
- Each of the VH and VLtypically contains three complementarity determining regions CDR1 , CDR2 and CDR3.
- the three CDRs in VH or VLare flanked by framework regions (FR1 , FR2, FR3 and FR4).
- examples of antigen binding fragmentsinclude, but are not limited to: (1) a Fab fragment, which is a monovalent fragment having a VL-CL chain and a VH-CH1 chain; (2) a Fab’ fragment, which is a Fab fragment with the heavy chain hinge region, (3) a F(ab')2 fragment, which is a dimer of Fab’ fragments joined by the heavy chain hinge region, for example linked by a disulfide bridge at the hinge region; (4) an Fc fragment; (5) an Fv fragment, which is the minimum antibody fragment having the VLand VH domains of a single arm of an antibody; (6) a single chain Fv (scFv) fragment, which is a single polypeptide chain in which the VH and VL domains of an scFv are linked by a peptide linker; (7) an (scFv)2, which comprises two VH domains and two VL domains, which are associated through the two VH domains via disulfide bridges and
- Antigen binding fragmentscan be prepared via routine methods.
- F(ab')2 fragmentscan be produced by pepsin digestion of a full- length antibody molecule, and Fab fragments can be generated by reducing the disulfide bridges of F(ab')2 fragments.
- fragmentscan be prepared via recombinant technology by expressing the heavy and light chain fragments in suitable host cells (e.g., E. coli, yeast, mammalian, plant or insect cells) and having them assembled to form the desired antigen-binding fragments either in vivo or in vitro.
- a single-chain antibodycan be prepared via recombinant technology by linking a nucleotide sequence coding for a heavy chain variable region and a nucleotide sequence coding for a light chain variable region.
- a flexible linkermay be incorporated between the two variable regions. The skilled person is aware of methods for the preparation of both full-length antibodies and antigen binding fragments thereof.
- the present aspect of the disclosureprovides a bispecific binding molecule, wherein said C5 binding polypeptide is selected from the group consisting of full-length antibodies, Fab fragments, Fab’ fragments, F(ab')2 fragments, single chain Fab (scFab) fragments, Fc fragments, Fv fragments, single chain Fv (scFv) fragments, (scFv)2, scFv-Fc constructs and domain antibodies.
- said C5 binding polypeptideis selected from the group consisting of full-length antibodies, Fab fragments, Fab’ fragments, F(ab')2 fragments, single chain Fab (scFab) fragments, Fc fragments, Fv fragments, single chain Fv (scFv) fragments, (scFv)2, scFv-Fc constructs and domain antibodies.
- the C5 binding polypeptideis an antibody, in particular a monoclonal antibody.
- monoclonal antibodyis meant an antibody preparation consisting of a single antibody species, i.e. all antibodies in the preparation have the same amino acid sequences, including the same CDRs, and thus bind the same epitope on their target antigen (by “target antigen” is meant the antigen containing the epitope bound by a particular antibody) with the same effect.
- target antigenis meant the antigen containing the epitope bound by a particular antibody
- the antibody of the disclosureis preferably not part of a polyclonal mix of antibodies.
- the antibodymay be of any isotype and subtype. Thus it may be an IgA, IgD, IgE, IgG, or IgM antibody.
- the heavy-chain constant domains that correspond to the different isotypes of immunoglobulinsare termed a, 5, E, y and p, respectively.
- the subunit structures and three-dimensional configurations of different isotypes of immunoglobulinsare well known.
- the antibodyis an IgG antibody.
- the IgG anti-C5 antibody of the disclosuremay be of any IgG subtype, i.e. it may be an lgG1 , lgG2, lgG3 or lgG4 antibody.
- the C5 binding polypeptideis a binding fragment of an antibody (i.e. an antibody fragment), that is a fragment which retains the ability of the antibody to bind specifically to its target antigen.
- an antibody fragmenti.e. an antibody fragment
- Such fragmentsare well known and examples include Fab’, Fab, F(ab’)2, Fv, Fd, or dAb fragments, which may be prepared according to techniques well known in the art.
- a Fab fragmentconsists of the antigen binding domain of an antibody, i.e. an individual antibody may be seen to contain two Fab fragments, each consisting of a light chain and its conjoined N-terminal section of a heavy chain. Thus a Fab fragment contains an entire light chain and the VH and CH1 domains of the heavy chain to which it is bound. Fab fragments may be obtained by digesting an antibody with papain.
- a F(ab’)2 fragmentconsists of the two Fab fragments of an antibody, plus the hinge regions of the heavy domains, including the disulphide bonds linking the two heavy chains together.
- a F(ab’)2 fragmentcan be seen as two covalently joined Fab fragments.
- F(ab’)2 fragmentsmay be obtained by digesting an antibody with pepsin. Reduction of F(ab’)2 fragments yields two Fab’ fragments, which can be seen as Fab fragments containing an additional sulfhydryl group which can be useful for conjugation of the fragment to other molecules.
- the C5 binding polypeptideis a synthetic or artificial construct, i.e. an antibody-like molecule which comprises a binding domain, but which is genetically engineered or artificially constructed.
- constructsinclude chimeric or CDR-grafted antibodies, as well as single chain antibodies and other constructs, e.g. scFv, dsFv, ds-scFv, dimers, minibodies, diabodies, single domain antibodies (DABs), TandAbs dimers and heavy chain antibodies such as VHH, etc.
- the C5 binding moleculeis a single chain variable fragment (scFv).
- scFvis a fusion protein in which a single polypeptide comprises both the VH and VL domains of an antibody.
- scFv fragmentsgenerally include a peptide linker covalently joining the VH and VL regions, which contributes to the stability of the molecule.
- the linkermay comprise from 1 to 20 amino acids, such as for example 1 , 2, 3 or 4 amino acids, 5, 10 or 15 amino acids, or other intermediate numbers in the range 1 to 20 as convenient.
- the peptide linkermay be formed from any generally convenient amino acid residues, such as glycine and/or serine.
- the linkeris a (G4S)4 linker (SEQ ID NO:283).
- G4Sa linker
- the VL domainmay instead be linked directly to the VH domain by a peptide bond.
- An scFvtypically comprises, N-terminal to C-terminal, a VH region linked to a VL region by a linker sequence.
- the scFvcomprises a peptide linker joining the C terminus of the heavy chain variable region (VH) to the N terminus of the light chain variable region (VL).
- a bispecific binding moleculeas described herein, wherein said C5 binding polypeptide is an antibody fragment which is an scFv.
- a complex as defined hereinwherein said C5 binding polypeptide and/or said C5 binding antibody or antigen binding fragment thereof or said bispecific binding molecule as a whole comprises additional amino acids at least one C-terminal and/or N- terminal end.
- Such a complexshould be understood as a complex having one or more additional amino acid residues at an N-terminal and/or C-terminal position in the polypeptide chain of the C5 binding polypeptide and/or of the C5 binding antibody or antigen binding fragment thereof.
- said bispecific binding moleculeshould be understood as having one or more additional amino acid residues at the N-terminal and/or C-terminal position of the molecule as a whole.
- said bispecific binding moleculemay comprise any suitable number of additional amino acid residues, for example at least one additional amino acid residue.
- Each additional amino acid residuemay individually or collectively be added in order to, for example, improve production, purification, stabilization in vivo or in vitro, coupling or detection of the complex.
- Such additional amino acid residuesmay comprise one or more amino acid residues added for the purpose of chemical coupling.
- Thisis the addition of a cysteine residue.
- Additional amino acid residuesmay also provide a ’’tag” for purification or detection of the polypeptide, such as a His6 tag, a (HisGlu)3 tag (“HEHEHE” tag) or a ”myc” (c-myc) tag or a ’’FLAG” tag for interaction with antibodies specific to the tag or immobilized metal affinity chromatography (IMAC) in the case of a His6- tag.
- a His6 tagsuch as a His6 tag, a (HisGlu)3 tag (“HEHEHE” tag) or a ”myc” (c-myc) tag or a ’’FLAG” tag for interaction with antibodies specific to the tag or immobilized metal affinity chromatography (IMAC) in the case of a His6- tag.
- IMACimmobilized metal affinity chromatography
- the first moiety of the bispecific binding molecule of the first aspect of the disclosureis a C5 binding polypeptide in multimeric form.
- Said multimeris understood to comprise at least two C5 binding polypeptides as defined herein as monomer units, the amino acid sequences of which may be the same or different.
- Multimeric forms of the C5 binding polypeptidesmay comprise a suitable number of domains, each having a C5 binding motif, and each forming a monomer within the multimer. These domains may have the same amino acid sequence, but alternatively, they may have different amino acid sequences.
- the C5 binding polypeptidemay form homo- or heteromultimers, for example homo- or heterodimers.
- a bispecific binding moleculeas described herein, comprising at least two first moieties, wherein each first moiety each individually comprises at least one binding site for human complement component 5 (C5), wherein the amino acid sequences for the said binding sites may be the same or different.
- said at least two first moietiesare not coupled together.
- C5 binding polypeptideis used to encompass C5 binding polypeptides in all forms, i.e. monomeric and multimeric forms.
- a bispecific binding moleculeas described herein and comprising a C5 binding polypeptide as first moiety, wherein said C5 binding polypeptide binds to C5 such that the KD value of the interaction is at most 1 x 10’ 6 M, such as at most 1 x 10’ 7 M, at most 1 x 10’ 8 M, or at most 1 x 10’ 9 M.
- Second moietyproviding a binding site for IFNo
- the bispecific binding molecule as defined hereinseparately targets IFNa in addition to being capable of binding C5.
- a bispecific binding molecule as defined hereinwherein said at least one second moiety with at least one first binding site for interferon alpha (IFNa) is an IFNa binding polypeptide.
- This second moietymay exhibit any one or more of the general properties, features, characteristics and/or embodiments described above in connection with the first moiety, in any combination.
- the second moietymay exhibit any one or more of the general properties, features, characteristics and/or embodiments described above but in relation to the target of the second moiety, being IFNa, and not in relation to the target of the first moiety, being C5.
- this informationwill not be repeated verbatim in connection with the second moiety, but is incorporated through this reference to the above disclosure.
- a bispecific binding moleculeas defined herein comprising an IFNa binding polypeptide, wherein said IFNa binding polypeptide is selected from the group consisting of full-length antibodies, Fab fragments, Fab’ fragments, F(ab')2 fragments, single chain Fab (scFab) fragments, Fc fragments, Fv fragments, single chain Fv (scFv) fragments, (scFv)2, scFv-Fc constructs, domain antibodies, a chimeric antibody, a CDR-grafted antibody, a fully human antibody, a bivalent antibody construct, a synthetic antibody, a bivalent antibody, a trivalent antibody, a tetravalent antibody, bivalent single chain antibody, a trivalent single chain antibody, a multivalent single chain antibody, nanobodies, AlbudAbs, DARTs (dual affinity re-targeting), BiTEs (bispecific T-cell engager), TandAbs (tandem diabodies), DAF
- a bispecific binding moleculeas defined herein, wherein said IFNa binding polypeptide is selected from the group consisting of full-length antibodies, Fab fragments, Fab’ fragments, F(ab')2 fragments, single chain Fab (scFab) fragments, Fc fragments, Fv fragments, single chain Fv (scFv) fragments, (scFv)2, scFv-Fc constructs and domain antibodies.
- said IFNa binding polypeptideis an IFNa binding antibody or antigen binding fragment thereof.
- the binding domaincan be defined according the amino acid sequence comprising the complementarity determining domains (CDRs). Therefore, in an embodiment of the present aspect there is provided a bispecific binding molecule as defined herein comprising an IFNa binding polypeptide, wherein said IFNa binding polypeptide comprises a binding domain of an antibody, the binding domain comprising a heavy chain variable region and a light chain variable region, each comprising six complementarity determining domains (CDRs), wherein:
- VLCDR1has the sequence set forth in SEQ ID NO:286;
- VLCDR2has the sequence set forth in SEQ ID NO:287;
- VLCDR3has the sequence set forth in SEQ ID NO:288;
- VHCDR1has the sequence set forth in SEQ ID NO:289;
- VHCDR2has the sequence set forth in SEQ ID NQ:290;
- VHCRD3has the sequence set forth in SEQ ID NO:291 .
- said antibody or antigen binding fragment thereofcomprises a light chain variable region comprising the amino acid sequence SEQ ID NO:284, or an amino acid sequence having at least 80% sequence identity thereto.
- a bispecific binding molecule as defined hereinwherein said antibody or antigen binding fragment thereof comprises a heavy chain variable region comprising the amino acid sequence SEQ ID NO:285, or an amino acid sequence having at least 80% sequence identity thereto.
- said antibody or antigen binding fragment thereofcomprises a light chain variable region comprising the amino acid sequence SEQ ID NO:284, or an amino acid sequence having at least 80% sequence identity thereto, and a heavy chain variable region comprising the amino acid sequence SEQ ID NO:285, or an amino acid sequence having at least 80% sequence identity thereto.
- said IFNa binding antibody or antigen binding fragment thereofis rontalizumab or a variant thereof.
- said IFNa binding antibody or antigen binding fragment thereofmay comprise a light chain comprising the amino acid sequence SEQ ID NO:293 or a sequence having at least 80% identity thereto, and/or a heavy chain comprising the amino acid sequence SEQ ID NO:294 or a sequence having at least 80% identity thereto.
- said IFNa binding antibody or antigen binding fragment thereofis rontalizumab, i.e. an antibody comprising a light chain comprising the amino acid sequence SEQ ID NO:293 and a heavy chain comprising the amino acid sequence SEQ ID NO:294.
- the CH2 regionmay comprise the amino acid substitution N297A.
- the N297A amino acid substitutionis introduced into the design to abrogate FcyR interactions, mitigating unwanted effector functions. Therefore, in one embodiment, there is provided a bispecific binding molecule as defined herein, wherein said IFNa binding polypeptide is an IFNa binding antibody or antigen binding fragment thereof, comprising an amino acid substitution N297A in the CH2 region of the antibody or antibody fragment thereof.
- the bispecific molecule as described hereinmay for example be present in the form of a fusion protein or a protein conjugate, thus, said at least one first moiety with at least one binding site for human complement component 5 (C5) as defined herein, is operably linked to at least one second moiety with at least one first binding site for interferon alpha (IFNa) as defined herein. Therefore, in an embodiment there is provided a bispecific binding molecule, comprising at least one first moiety with at least one first binding site for human complement component 5 (C5) as defined herein, operably linked to at least one second moiety with at least one first binding site for interferon alpha (IFNa) as defined herein.
- said at least one first moiety and said at least one second moietymay be coupled by means of chemical conjugation (using known organic chemistry methods) or by any other means, such as expression of the complex as a fusion protein or joined in any other fashion, either directly or via a linker, for example an amino acid linker.
- a bispecific binding moleculeas defined herein, wherein said bispecific binding molecule is a fusion protein or a protein conjugate.
- said bispecific binding moleculeis a fusion protein.
- said bispecific binding moleculeis a protein conjugate.
- said at least one first moiety as defined hereinis attached to the N-terminus or C-terminus of the heavy chain of said at least one second moiety which is an antibody or antigen binding fragment thereof.
- said at least one first moiety as defined hereinis attached to the N-terminus or C-terminus of the light chain of said at least one second moiety, which is an antibody or antigen binding fragment thereof.
- said at least one first moiety as defined hereinis attached to the N-terminus and/or C-terminus of the light chain and heavy chain of said at least one second moiety, which is an antibody or antigen binding fragment thereof.
- the at least one first moietymay be attached to only the N-terminus of the heavy chain(s), only the N-terminus of the light chain(s), only the C-terminus of the heavy chain(s), only the C-terminus of the light chain(s), both the N-terminus and the C-terminus of the heavy chain(s), both the N-terminus and the C-terminus of the light chain(s), only the C-terminus of the light chain(s) and the N-terminus of the heavy chain(s), only the C- terminus of the heavy chain(s) and the N-terminus of the light chain(s), of said at least one second moiety, which is an antibody or antigen binding fragment thereof.
- a bispecific binding molecule as defined hereinwherein said at least one first moiety and said at least one second moiety are covalently linked.
- a bispecific binding molecule as defined hereinwherein said at least one first moiety is covalently linked to: i) a CH3 domain of the heavy chain of an antibody or antibody fragment thereof as defined herein; ii) a CL domain of the light chain of an antibody or antibody fragment thereof as defined herein; iii) a VL domain of the light chain of an antibody or antibody fragment thereof as defined herein; or iv) a VH domain of the heavy chain of an antibody or antibody fragment thereof as defined herein.
- bispecific binding moleculeas defined herein, wherein the bispecific molecule comprises
- first moietyis covalently linked to a CH3 domain of each heavy chain of said antibody or antibody fragment thereof; ii) one first moiety is covalently linked to a CL domain of each light chain of said antibody or antibody fragment thereof; iii) one first moiety is covalently linked to a VH domain of each heavy chain of said antibody or antibody fragment thereof; or iv) one first moiety is covalently linked to a VL domain of each light chain of said antibody or antibody fragment thereof.
- said bispecific binding moleculefurther comprises at least one linker, such as at least one linker selected from flexible amino acid linkers, rigid amino acid linkers and cleavable amino acid linkers.
- said linkeris arranged between said first moiety as defined herein and said second moiety as defined herein.
- a bispecific binding moleculeas defined herein, wherein said at least one first moiety is operably bound to said at least one second moiety via at least one linker, such as at least one linker selected from flexible amino acid linkers, rigid amino acid linkers and cleavable amino acid linkers.
- Flexible linkersare often used in the art when the joined domains require a certain degree of movement or interaction, and may be particularly useful in some embodiments of the complex.
- Such linkersare generally composed of small, non-polar (for example G) or polar (for example S or T) amino acids.
- Some flexible linkersprimarily consist of stretches of G and S residues, for example (GGGGS)p. Adjusting the copy number “p” allows for optimization of linker in order to achieve appropriate separation between the functional moieties or to maintain necessary inter-moiety interaction.
- G and S linkersother flexible linkers are known in the art, such as G and S linkers containing additional amino acid residues, such as T and A, to maintain flexibility, as well as polar amino acid residues to improve solubility.
- said linkeris a flexible linker comprising glycine (G), serine (S) and/or threonine (T) residues.
- n1-5.
- m0-5.
- p1-5.
- said linkeris (GGGGS)3 (SEQ ID NO:303).
- said linkeris GGGGS (SEQ ID NO:304). In another specific embodiment, said linker is VDGS (SEQ ID NQ:305). In another specific embodiment, said linker is ASGS (SEQ ID NQ:306). In a preferred embodiment, said flexible amino acid linker comprises the amino acid sequence SEQ ID NO:292.
- a bispecific binding molecule of the first aspectcomprises a pair of polypeptide chains, said pair being selected from the group consisting of the pairs
- a bispecific binding moleculeas defined herein, comprising the pair of polypeptide chains represented by SEQ ID NO:295 and SEQ ID NO:296.
- a polynucleotideencoding at least one polypeptide forming part of a bispecific binding molecule according to the first aspect.
- at least one polynucleotidesuch as at least two polynucleotides, each polynucleotide independently encoding any one or more of the sub-components or polypeptide parts of said bispecific binding molecule, e.g.
- an expression vectorcomprising said polynucleotide.
- at least one vectorsuch as at least two vectors, comprising said at least one polypeptide(s).
- a host cellcomprising said expression vector.
- a host cellcomprising said at least one vector, such as comprising said at least two vectors.
- a method of producing a bispecific binding molecule as described abovecomprising culturing said host cell under conditions permissive of expression of said polypeptide from its expression vector(s), and isolating the bispecific binding molecule.
- a method of producing a bispecific binding molecule as described above or any one or more of the sub-components or polypeptide parts of said bispecific binding moleculecomprising culturing said host cell under conditions permissive of expression of said polypeptides from the at least one expression vector(s), and isolating the said sub-components or polypeptide parts of said bispecific binding molecule.
- any one or more of the sub-components or polypeptide parts of said bispecific binding moleculeit is meant any one or more of the sub-components or polypeptide parts which comprise the bispecific binding molecule, such as at least one heavy chain component of a first moiety, such as at least one light chain component of a first moiety, such as at least one heavy chain component of a second moiety, such as at least one light chain component of a second moiety.
- the bispecific binding molecule of the present disclosuremay alternatively be produced by non-biological peptide synthesis. Therefore, in another embodiment there is provided a method of producing a bispecific binding molecule as defined herein by non-biological peptide synthesis using amino acids and/or amino acid derivatives having protected reactive side-chains, the non-biological peptide synthesis comprising
- Said bispecific binding moleculemay also be produced by the conjugation of at least one first moiety to at least one second moiety thereof as described herein.
- conjugation methodsknown in the art, such as conventional chemical conjugation methods for example using charged succinimidyl esters or carbodiimides.
- compositioncomprising a bispecific binding molecule as described herein and at least one pharmaceutically acceptable excipient or carrier.
- said compositionfurther comprises at least one additional active agent, such as at least two additional active agents, such as at least three additional active agents.
- additional active agentsthat may prove useful in such a composition are immune response modifying agents.
- Non limiting examples of immune response modifying agentsthat can be used as additional active agent in embodiments of the composition according to this aspect include immunosuppressive and immunomodulating agents, and other anti-inflammatory agents.
- the bispecific binding molecule as described hereinmay be used in combination with an agent selected from the group consisting of disease-modifying antirheumatic drugs (DMARDs), such as gold salts, azathioprine, methotrexate and leflunomide; calcineurin inhibitors, such as cyclosporin A or FK 506; modulators of lymphocyte recirculation; mTOR inhibitors, such as rapamycin; an ascomycin having immuno-suppressive properties; glucocorticoids; corticosteroids; cyclophosphamide; immunosuppressive monoclonal antibodies; adhesion molecule inhibitors, such as LFA-1 antagonists, ICAM-1 or -3 antagonists, VCAM-4 antagonists or VLA-4 antagonists; blockers of proinflam matory cytokines; IL-1
- bispecific binding molecule as defined herein or a composition comprising said bispecific binding moleculemay be administered to a subject using standard administration techniques, such as including oral, respiratory, topical, intravenous, intraperitoneal, subcutaneous, pulmonary, transdermal, intramuscular, intranasal, buccal, sublingual or suppository administration.
- a bispecific binding molecule as defined herein or a composition as defined herein for oral, respiratory, topical, intravenous, intraperitoneal, subcutaneous, pulmonary, transdermal, intramuscular, intranasal, buccal, sublingual or suppository administrationsuch as for subcutaneous administration, such as for intravenous administration.
- a bispecific binding molecule or composition as described hereinfor use as a medicament.
- said bispecific binding molecule or compositionmodulates C5 function and IFNa function.
- a bispecific binding molecule or a composition as defined hereinfor use as a medicament to modulate C5 and IFNa function in vivo.
- the term “modulate”refers to changing the activity, such as rendering C5 function and the function of IFNa, partially inhibiting or fully inhibiting C5 function and the function of IFNa.
- partially or fully inhibiting C5 functionit is meant that cleavage of C5 into C5a and C5b is inhibited.
- IFNa functionIFNa interaction with interferon alpha receptor (IFNaR) is partially or fully inhibited.
- IFNaRinterferon alpha receptor
- a bispecific binding molecule or a composition as defined hereinfor use as a medicament to modulate C5 and IFNa function in vivo wherein said at least one first moiety inhibits the cleavage of C5 into C5a and C5b and said at least one second moiety inhibits IFNa interaction with INFaR.
- a bispecific binding molecule or composition as described herein for use in the treatment of a disease or disorder related to C5 and IFNaIn another embodiment, there is provided a bispecific binding molecule or composition as described herein for use as described herein, wherein said disease or disorder related to C5 and IFNa is selected from inflammatory and autoimmune diseases. In another embodiment, there is provided a bispecific binding molecule or composition as described herein for use as described herein, wherein said disease or disorder related to C5 and IFNa is selected from cardiovascular diseases. In another embodiment, there is provided a bispecific binding molecule or composition as described herein for use as described herein, wherein said disease or disorder related to C5 and IFNa is selected from a CNS disorder, disease and injury.
- C5 and IFNa related disorderrefers to any disorder, disease or condition in which C5 and IFNa play a regulatory role in the signaling pathway.
- C5 and IFNa related diseases or disordersfor the treatment of which the bispecific binding molecule or composition as described herein may be useful, include inflammatory diseases and autoimmune diseases, such as rheumatoid arthritis (RA), dermatomyositis, primary Sjogren’s syndrome, systemic sclerosis, psoriasis and type I interferonopathies, such as Aicardi-Goutieres syndrome, familial Chilblain lupus, systemic lupus erythematosus (SLE), lupus nephritis, in particular cohorts with patients displaying signs of activated complement system, cardiovascular disease, such as atherosclerosis, and central nervous system (CNS) disorders, disease and injury such as traumatic brain injury (TBI), spinal cord injury (SCI), stroke, such
- RArheumatoid arthritis
- said disorder or disease related to C5 and IFNais selected from the group consisting of rheumatoid arthritis (RA), dermatomyositis, primary Sjogren’s syndrome, systemic sclerosis, psoriasis and type I interferonopathies, such as Aicardi-Goutieres syndrome, familial Chilblain lupus, systemic lupus erythematosus (SLE), lupus nephritis, atherosclerosis, traumatic brain injury (TBI), spinal cord injury (SCI), stroke, such as subarachnoid hemorrhage (SAH), amyotrophic lateral sclerosis (ALS), neuromyelitis optica, and Alzheimer’s disease.
- RArheumatoid arthritis
- dermatomyositisprimary Sjogren’s syndrome
- systemic sclerosispsoriasis and type I interferonopathies
- type I interferonopathiessuch as Aicardi-
- the disorder or diseaseis selected from the group consisting of systemic lupus erythematosus (SLE), lupus nephritis and rheumatoid arthritis (RA).
- the disorder or diseaseis selected from the group consisting of traumatic brain injury (TBI), spinal cord injury (SCI), stroke, such as subarachnoid hemorrhage (SAH), amyotrophic lateral sclerosis (ALS), neuromyelitis optica, and Alzheimer’s disease.
- said disorder or disease related to C5 and IFNais systemic lupus erythematosus (SLE).
- said disorder or disease related to C5 and IFNais lupus nephritis. In another specific embodiment, said disorder or disease related to C5 and IFNa is rheumatoid arthritis (RA). In another specific embodiment, said disorder or disease related to C5 and IFNa is atherosclerosis. In another specific embodiment, said disorder or disease related to C5 and IFNa is traumatic brain injury (TBI). In another specific embodiment, said disorder or disease related to C5 and IFNa is spinal cord injury (SCI). In another specific embodiment, said disorder or disease related to C5 and IFNa is subarachnoid hemorrhage (SAH).
- RArheumatoid arthritis
- TBItraumatic brain injury
- SCIspinal cord injury
- SAHsubarachnoid hemorrhage
- said disorder or disease related to C5 and IFNais amyotrophic lateral sclerosis (ALS). In another specific embodiment, said disorder or disease related to C5 and IFNa is neuromyelitis optica. In another specific embodiment, said disorder or disease related to C5 and IFNa is Alzheimer’s disease. In a further embodiment, said bispecific binding molecule or composition alleviates the inflammatory profile in SLE, lupus nephritis and/or rheumatoid arthritis (RA).
- RArheumatoid arthritis
- said bispecific binding molecule or compositionalleviates the inflammatory profile in traumatic brain injury (TBI), spinal cord injury (SCI), stroke, such as subarachnoid hemorrhage (SAH), amyotrophic lateral sclerosis (ALS), neuromyelitis optica, and/or Alzheimer’s disease.
- TBItraumatic brain injury
- SCIspinal cord injury
- strokesuch as subarachnoid hemorrhage (SAH), amyotrophic lateral sclerosis (ALS), neuromyelitis optica, and/or Alzheimer’s disease.
- said bispecific binding molecule or compositionalleviates the inflammatory profile in atherosclerosis.
- administrationis selected from the group consisting of: oral, topical, intravenous, intraperitoneal, subcutaneous, pulmonary, transdermal, intramuscular, intranasal, buccal, sublingual or suppository administration, such as subcutaneous administration, such as intravenous administration.
- a method of treatment of disorder or disease related to C5 and IFNacomprising administering to a subject in need thereof an effective amount of a bispecific binding molecule or composition as described herein.
- the complex or composition as described hereinmodulates C5 and IFNa function in vivo. It may be beneficial to administer a therapeutically effective amount of a bispecific binding molecule as defined herein or composition as defined herein and at least one second drug substance, such as an immune response modulating agent as described above.
- co-administrationencompasses both concomitant and sequential administration.
- a method as defined abovefurther comprising co-administration of an immune response modulating agent as described above.
- said disorder or disease related to C5 and IFNais selected from inflammatory and autoimmune diseases.
- said disorder or disease related to C5 and IFNais selected from the group consisting of rheumatoid arthritis (RA), dermatomyositis, primary Sjogren’s syndrome, systemic sclerosis, psoriasis and type I interferonopathies, such as Aicardi-Goutieres syndrome, familial Chilblain lupus, systemic lupus erythematosus (SLE) and lupus nephritis.
- RArheumatoid arthritis
- dermatomyositisprimary Sjogren’s syndrome
- systemic sclerosispsoriasis
- type I interferonopathiessuch as Aicardi-Goutieres syndrome, familial Chilblain lupus, systemic lupus erythematosus (SLE) and lupus nephritis.
- the disorder or diseaseis selected from the group consisting of systemic lupus erythematosus (SLE) and lupus nephritis.
- said disorder or disease related to C5 and IFNais systemic lupus erythematosus (SLE).
- said disorder or disease related to C5 and IFNais lupus nephritis.
- said subjectis a mammalian subject, such as a human subject.
- said subjectis a human subject.
- said disorder or disease related to C5 and IFNais selected from central nervous system (CNS) diseases, disorders and injury.
- said disorder or disease related to C5 and IFNais selected from the group consisting of traumatic brain injury (TBI), spinal cord injury (SCI), stroke, such as subarachnoid hemorrhage (SAH), amyotrophic lateral sclerosis (ALS), neuromyelitis optica, and/or Alzheimer’s disease.
- TBItraumatic brain injury
- SCIspinal cord injury
- SCIsubarachnoid hemorrhage
- ALSamyotrophic lateral sclerosis
- neuromyelitis opticaand/or Alzheimer’s disease.
- said disorder or disease related to C5 and IFNais traumatic brain injury (TBI).
- said disorder or disease related to C5 and IFNais spinal cord injury (SCI).
- said disorder or disease related to C5 and IFNais stroke, such as subarachnoid hemorrhage (SAH).
- said disorder or disease related to C5 and IFNais amyotrophic lateral sclerosis (ALS).
- said disorder or disease related to C5 and IFNais neuromyelitis optica.
- said disorder or disease related to C5 and IFNais Alzheimer’s disease.
- said subjectis a mammalian subject, such as a human subject.
- said subjectis a human subject.
- Luo CClinical Value of Inflammatory Cytokines in Patients with Aneurysmal Subarachnoid Hemorrhage, Clinical Interventions in Aging 2022:17 615-626.
- Figure 1shows AffiMab primary developability screening: A) schematic illustration of AffiMab constructs; B) developability data of AffiMabs based on titers in small scale production cultures of 2.5 mL vs purified and SEC analyzed proteins for monomeric content shown in percentage of total detected peaks in apprehended chromatograms; C) simultaneous binding to IFNa and C5 by the AffiMabs, as evaluated with BLI. Data in (B) is presented as mean ⁇ SD of three replicates.
- Figure 2illustrates SDS-PAGE post purification of purified AffiMab constructs and the naked antibody rontalizumab serving as a benchmark.
- Figure 3shows the results of evaluation of C5 cleavage inhibition by AffiMabs in a whole blood hemolysis assay. Data is presented as mean ⁇ SD of two replicates.
- Figure 4shows the results of evaluation of inhibition of IFNa by AffiMab constructs in an in vitro reporter assay, showing A) neutralization of recombinant IFNa2b signaling by AffiMabs; B) neutralization of IFNa2b signaling in presence of excess C5 (100 nM). Data in (B-C) are presented as mean ⁇ SD of two replicates.
- Figure 5shows the result of C5a ELISA on plasma samples post AffiMab treatment in whole blood ex vivo assay, with untreated (UT), Healthy control untreated blood (UT HC), AffiMab and control constructs treated blood as indicated. Data are presented as mean ⁇ SD of two technical replicates Comparisons between treatments analyzed by two-tailed paired t-test, statistical significance *p ⁇ 0.05.
- Figure 6shows the results of evaluation of cytokine and chemokine profile in plasma after AffiMab or mAb (rontalizumab) treatment in whole blood ex vivo assay with donor blood by OLINK PLA assay. Data are presented as mean ⁇ SD of two technical replicates. Comparisons between treatments for each protein analyzed by two-tailed paired t-test, statistical significance P ⁇ 0.05.
- bispecific binding moleculescomprising at least one first moiety with at least one binding site for human complement component 5 (C5) operably linked to at least one second moiety with at least one binding site for interferon alpha (IFNa).
- C5human complement component 5
- IFNainterferon alpha
- These novel bispecific binding moleculesare also referred to as IFNa-C5 targeting “AffiMab” constructs.
- the Examplesfurther describe the characterization of these selected bispecific binding molecules, i.e. “AffiMabs”, and demonstrate their in vitro and ex vivo functionality.
- AffiMabswere designed and constructed by recombinant fusion of a gene encoding a C5 binding Z variant to the N- or C-terminus of the heavy chain or light chain of the IFNo binding mAb rontalizumab via a 15-amino-acid glycine-serine linker (SS(G)4S(G)eSS).
- SS(G)4S(G)eSS15-amino-acid glycine-serine linker
- the heavy chain and light chain of the AffiMabswere cloned by conventional restriction cloning into a dual-expression cassette vector pKTH17 (Volk et al, 2021 , supra), for encoding both the heavy and light chain in one vector.
- ExpiCHOTM cells(ThermoFisher Scientifics Inc. Waltham, MA, USA) were transfected as suggested by the manufacturer (ExpiCHO Expression System User Guide, Pub. No. MAN0014337 Rev. C.0) with a total of 20 pg plasmid DNA per transfection of respective construct in 25 mL culture volumes. The cells were cultured for 8 days prior to harvest.
- IgG quantificationIgG concentrations in the supernatant were determined by biolayer interferometry measurements in an Octet® RED96e system (Fortebio Biologies by Molecular Devices, USA) with Dip and ReadTM Protein A biosensors (Fortebio Biologies by Molecular Devices) according to the manufacturer's instructions.
- the supernatant samples from day 5were diluted 1 :1 in 20 mM citric acid pH 4.0, 0.1 % BSA (w/v), 0.1 % Tween-20, 0.5 M NaCI.
- a standard curvewas prepared from an IgG with the respective concentrations from 700 to 1 pg/mL.
- IgG purificationFor size exclusion chromatography analysis, the expressed AffiMabs were purified by Protein A facilitated purification on an AktaSTART system (GE Healthcare, USA) using mAbSelect SuRe columns (GE Healthcare). A 20 mM sodium phosphate, 0.15 M sodium chloride (pH 7.3) buffer was used as binding and wash buffer, 0.1 M glycine (pH 2.5) as elution buffer and 1 M Tris-HCI (pH 8.5) as neutralization buffer. Endotoxin levels were measured with LAL Cartridges and Endosafe Nextgen-PTS system (Charles River, MA, USA) according to manufacturer’s instructions.
- Size exclusion chromatographyIn total, 25 pg of each AffiMab in 100 pl were injected onto a Superdex Increase 200 10/30 GL gel filtration column (GE Healthcare) coupled to an Agilent 1200 series HPLC system (Agilent Technologies, USA). SEC runs were performed at a 0.5 mL/min flow rate with PBS as a running buffer. Eluted protein fragments were detected by an online 280 nm absorption measurement. Data analysis and peak integrations were performed using GraphPad prism 8.0 (GraphPad Software, USA).
- Biolayer interferometryTo screen and validate AffiMab constructs’ functional simultaneous binding to both targets, constructs were screened using the Octet RED96 system (Sartorious, Gottingen, Germany). Biotinylated IFNa2b was immobilized to streptavidin coated octet probes (Sartorius, Gottingen, Germany).
- SPRSurface plasmon resonance
- AffiMab lead candidate(RONT_HC15_zC5; comprising SEQ ID NO:295 and SEQ ID NO:296) or rontalizumab (R0NT_WT; comprising SEQ ID NO:293 and SEQ ID NO:294) was immobilized on CM5 chips (Cytiva, Sweden) by amine coupling. Following immobilization, each analyte (C5 or IFNa) was injected in a serial dilution (diluted in PBSB 1 %) ranging from 50 nM to 3.125 nM in a dilution series of 5 concentration and dilution factor of 2. Sensorgrams were analyzed using GraphPad prism 8.0 (GraphPad Software, USA) for analysis of IFNa binding and B devaluation software (Cytiva, Sweden) for analysis of C5 binding. Results are shown in Table 3.
- the mode-of-actionwas investigated for each respective entity of the bispecific binding molecule. Therefore, inhibition of C5 cleavage, formation of C5b-9 MAC complex and subsequent hemolytic activity by the respective AffiMab construct were studied in a whole blood assay with spiked human C5.
- control Z varianteither fused to an albumin binding domain (monovalent; “zC5-ABD”) or to the Fc domain (bivalent; “zC5-Fc”).
- Ront_15HC_zC5showed the highest inhibitory effect amongst the AffiMab candidates with an IC50 of 1 .7 nM compared to the bivalent zC5-Fc fusion control with an IC50 of 1.1 nM.
- zC5-ABDshowed a 6.6- and 4.2-fold higher IC50 than the two aforementioned constructs respectively, indicating that the natural symmetry of the AffiMab constructs contributed to the bivalency and had an effect on overall potency.
- Ront_LC15_zC5exhibited the highest IC50 value amongst the AffiMab constructs of 244 nm, a 144-fold increase compared to the top construct.
- the naked mAb, Ront_WTdoes not exhibit any significant effect on inhibition of hemolytic activity.
- Detailed IC50 valuesare listed in Table 3.
- IFNa reporter assaywas performed to study if the generated AffiMabs had retained the parental mAb’s potency to neutralize IFNa stimuli via IFNaR.
- IFNa2b2 nM recombinant IFNa2b (Invivogen, California, US) was incubated with concentration series ranging from 100 nM to 3.25 nM of AffiMab constructs (IgG 1 ) or control treatment for 45 min at room temperature in a 96 well format.
- HEK-BLUE IFNa responsive cell line(Invivogen) was then subseguently seeded to the wells at a density of 280 000 cells/mL in 180 pL test medium and plates were incubated for 24h at 37°C in a humidified atmosphere with 5% CO2.
- IFNa reporter assaywas performed to study if the generated AffiMabs have retained the parental mAb’s (Ront_WT) potency to neutralize IFNa stimuli via IFNaR.
- the reporter cell linehad the JAK/STAT1 ,2/ISGF3 pathway stably integrated and allowed monitoring of bioactive IFNa by an inducible secreted alkaline phosphatase (SEAP) reporter gene under the control of the IFNa inducible ISG54 promoter.
- SEAPinducible secreted alkaline phosphatase
- Ront_15HC_zC5inhibits recombinant IFNa2b signaling in parity with the naked antibody rontalizumab (Ront_WT) exhibiting an IC50 value of 14.4 ng/mL and 15.8 ng/mL respectively (Figure 4A).
- zC5_LC15_Rontexhibited a 17-fold increase compared to rontalizumab, higher than any of the other constructs (Table 4).
- Complement component 5exhibits a molecular weight of 188 kDa, compared to the average total molecular weight of the AffiMab constructs ( ⁇ 166 kDa) and the molecular weight of IFNa (19.9 kDa).
- zC5_LC15_Rontcontinued to show poor neutralization potency in comparison to the other AffiMab constructs, with an 8-fold higher IC50 than Rontalizumab.
- Table 5IC50 values derived from inhibition of IFNa in the in vitro interferon reporter assay
- the AffiMab candidate Ront_15HC_zC5was chosen as the leading candidate to continue with for further evaluation in ex vivo whole blood assays.
- the simultaneous effects of blocking the two ligands (C5 and IFNa) by the AffiMab construct Ront_HC15_zC5was evaluated in an ex vivo whole blood model utilizing blood derived from SLE patients with ongoing inflammation. Blood was treated with AffiMab construct Ront_HC15_zC5 or control treatments for 24h post plasma collection and evaluation of cytokine or chemokine levels of interest was performed by ELISA and OLINK proximity extension assay.
- Ront_HC15_zC5The effect of the AffiMab construct Ront_HC15_zC5 was studied in whole blood donated from SLE patients with active inflammation. Blood from SLE patients or healthy controls was collected in heparin coated vacutainer tubes. Blood was subsequently transferred to 2 mL endotoxin free tube at a volume of 0.5 mL. The tubes were spun down at 1200 g for 10 min and plasma was collected for cytokine quantification. AffiMab (Ront_HC15_zC5) or control treatment with the native antibody rontalizumab (R0NT_WT) was added at 30 mg/mL incubated overnight prior to plasma collection.
- Plasma sampleswere analyzed by the proximity ligation assay utilizing commercially available 96 Inflammation OLINK Panel (Olink, Sweden) for cytokine and chemokine profiling, and ELISA for C5a quantification according to manufacturer’s instructions (Human C5a ELISA kit, ThermoFisher).
- cytokine and chemokine profile in patient derived whole bloodwas analyzed by the OLINK 96 inflammation panel (Figure 6). Focused evaluation of diseases related cytokines demonstrated a significant upregulation of pro- inflammatory cytokines (IFNg, CXCL10) in untreated SLE patient derived blood (UT) compared to healthy control (UT HC) and lower levels of chemokine CXCL5 (a known neutrophile activator observed to be downregulated in SLE patient blood whilst upregulated in healthy tissue contributing to local immune activation and inflammatory damage of healthy tissue).
- IL-10functions as an anti-inflammatory cytokine and was shown to be upregulated in patient derived blood, a consequence believed to be due to the anti-inflammatory treatments patients are undergoing at the time of sampling. Hence an increase in the levels of these proteins in plasma is postulated to contribute to a more beneficial effect to alleviate the inflammatory profile.
- cytokine and chemokine profile post AffiMab treatmentdemonstrated a significant effect in decreasing pro-inflammatory cytokines and chemokines closely related to the IFNa signature in SLE (IFNg, CXCL10) compared to blood treated with the native antibody (R0NT_WT) or untreated blood (UT).
- CXCL5increased after treatment with Ront_HC15_zC5 in parity with the R0NT_WT treatment, an effect in-line with the sought outcome in relation to the chemokines’ aforementioned role when shown to be present at higher levels in SLE patients and correlation with decreased disease activity.
- AffiMabdemonstrated a capability to beneficially decrease plasma levels of the two key targeted ligands IFNa and C5, whilst also affecting downstream related cytokines and chemokines in a favorable manner by either increasing or decreasing the levels of these proteins depending on their biological function, collectively resulting in an alleviated SLE inflammatory profile.
- Treatment with AffiMab lead constructis able to affect levels of IFNa (Figure 4), C5 ( Figures 3 and 5) and downstream related cytokines or chemokines of interest ( Figure 6) in a favorable manner, demonstrating signs of affecting key cytokines and chemokines in the inflammatory SLE profile ex vivo.
- CSFcerebrospinal fluid
- CSFcerebrospinal fluid
- CSFis treated with AffiMab construct Ront_HC15_zC5 or control treatments post CSF collection and evaluation of selected cytokines of relevance is performed by ELISA-based methods.
- Ront_HC15_zC5The effect of the AffiMab construct Ront_HC15_zC5 is studied in CSF donated from patients with CNS disorder, disease or injury.
- CSF from patients or healthy controlsis collected in polystyrene collection tubes.
- CSF samplesare aliquoted and exposed to AffiMab (Ront_HC15_zC5) or control treatment with the native antibody rontalizumab (R0NT_WT), monospecific bivalent C5 inhibitor counterpart (Fc-zC5), or other relevant comparators such as the C5 blocking antibody eculizimab and/or the IFNaR blocking antibody anifrolumab, added at relevant concentrations and incubated under controlled conditions.
- Tubesare then spun at 4 °C 2000g for 10 minutes. The supernatant is collected, aliquoted, and frozen in a -80°C freezer for subsequent analysis.
- Cytokine quantification of CSF samplesis then analyzed using a commercially available ELISA-based method, for example but not limited to kits marketed by CLINK, Mabtech and ThermoFisher/lnvitrogen.
- the cytokine profile in patient derived CSFis expected to show an upregulation of relevant pro-inflammatory cytokines and a down-regulation of relevant anti-inflammatory cytokines in untreated patient derived CSF compared to healthy control.
- Evaluation of the cytokine profile post AffiMab treatmentis expected to demonstrate decreased levels in pro-inflammatory cytokines and increased levels in anti-inflammatory cytokines closely related to the IFNa signature of CNS disorder patients compared to CSF treated with the native antibody (R0NT_WT) or untreated CSF.
- AffiMab lead construct(RONT_HC15_zC5) is able to affect levels of IFNa, C5 and downstream related cytokines of interest in a favorable manner, demonstrating signs of affecting key cytokines in the CNS disorder-induced CSF profile ex vivo to regain immune homeostasis.
- Ront_HC15_zC5The simultaneous effects of blocking the two ligands (C5 and IFNa) by the AffiMab construct Ront_HC15_zC5 is evaluated in an ex vivo whole blood model utilizing blood derived from patients with CNS disorders, disease or injury (such as traumatic brain injury, spinal cord injury, stroke, amyotrophic lateral sclerosis (ALS), neuromyelitis optica, and Alzheimer’s disease).
- CNS disorderssuch as traumatic brain injury, spinal cord injury, stroke, amyotrophic lateral sclerosis (ALS), neuromyelitis optica, and Alzheimer’s disease.
- Bloodis treated with AffiMab construct Ront_HC15_zC5 or control treatments post plasma collection and evaluation of selected cytokines of relevance is performed by an ELISA-based method.
- Ront_HC15_zC5The effect of the AffiMab construct Ront_HC15_zC5 is studied in whole blood donated from patients with CNS disorder, disease or injury. Blood from patients or healthy controls is collected in heparin coated vacutainer tubes. Blood is subsequently transferred to 2 mL endotoxin free tube at a volume of 0.5 mL. The tubes are spun down at 1200 g for 10 min and plasma is collected for cytokine quantification. Cells are stored in stabilizer solution (Cytodelics AB) for downstream mRNA cell isolation utilizing paxgene blood RNA isolation kit. Extracted mRNA is subsequently analyzed for ISG gene transcript levels using an in-house designed gene panel with Nanostring technologies cartridges and nCounter SPRINT instrument.
- AffiMab(Ront_HC15_zC5) or control treatment of blood samples is carried out by incubation with AffiMab or the native antibody rontalizumab (R0NT_WT), or monospecific bivalent counterpart (Fc-zC5), or other relevant comparators at relevant concentrations and incubated under controlled conditions prior to plasma collection. Cytokine quantification of CSF samples is then analyzed using a commercially available ELISA-based method, for example but not limited to kits marketed by OLINK, Mabtech and ThermoFisher/lnvitrogen.
- the cytokine profile in patient derived bloodis expected to show upregulation of relevant pro-inflammatory cytokines and down-regulation of relevant anti-inflammatory cytokines in untreated patient derived blood compared to healthy control.
- Evaluation of the cytokine profile post AffiMab treatmentis expected to demonstrate decreased levels in pro-inflammatory cytokines and increased levels in anti-inflammatory cytokines closely related to the IFNa signature of CNS disorder, disease or injury patients compared to controls.
- this exampleis expected to demonstrate that treatment with AffiMab lead construct (RONT_HC15_zC5) is able to affect levels of IFNa, C5 and downstream related cytokines of interest in a favorable manner, demonstrating signs of affecting key cytokines in the CNS disorder induced whole blood profile ex vivo.
- the simultaneous effects of blocking the two ligands (C5 and IFNa) by the AffiMab construct Ront_HC15_zC5is evaluated in an ex vivo whole blood model utilizing blood derived from patients with rheumatoid arthritis (RA). Blood is treated with AffiMab construct Ront_HC15_zC5 or control treatments post plasma collection and evaluation of selected cytokines of relevance is performed by an ELISA-based method.
- Ront_HC15_zC5The effect of the AffiMab construct Ront_HC15_zC5 is studied in whole blood donated from patients with RA. Blood from patients or healthy controls is collected in heparin coated vacutainer tubes. Blood is subsequently transferred to 2 mL endotoxin free tube at a volume of 0.5 mL. The tubes are spun down at 1200 g for 10 min and plasma is collected for cytokine quantification.
- AffiMab(Ront_HC15_zC5) or control treatment of blood samples is carried out by incubation with AffiMab or the native antibody rontalizumab (R0NT_WT), or monospecific bivalent counterpart (Fc-zC5), or other relevant comparators at relevant concentrations and incubated under controlled conditions prior to plasma collection.
- Cytokine quantification of CSF samplesis then analyzed using a commercially available ELISA-based method, for example but not limited to kits marketed by OLINK, Mabtech and ThermoFisher/lnvitrogen.
- the cytokine profile in patient derived bloodis expected to show an upregulation of relevant pro-inflammatory cytokines and a down-regulation of relevant anti-inflammatory cytokines in untreated patient derived blood compared to healthy control.
- this exampleis expected to demonstrate that treatment with AffiMab lead construct (RONT_HC15_zC5) is able to affect levels of IFNa, C5 and downstream related cytokines of interest in a favorable manner, demonstrating signs of affecting key cytokines in the RA-induced whole blood profile ex vivo.
- Ront_HC15_zC5The simultaneous effects of blocking the two ligands (C5 and IFNa) by the AffiMab construct Ront_HC15_zC5 is evaluated in an ex vivo whole blood model utilizing blood derived from patients with atherosclerosis.
- Bloodis treated with AffiMab construct Ront_HC15_zC5 or control treatments post plasma collection and evaluation of selected cytokines of relevance is performed by an ELISA-based method.
- Ront_HC15_zC5The effect of the AffiMab construct Ront_HC15_zC5 is studied in whole blood donated from patients with atherosclerosis. Blood from patients or healthy controls is collected in heparin coated vacutainer tubes. Blood is subsequently transferred to 2 mL endotoxin free tube at a volume of 0.5 mL. The tubes are spun down at 1200 g for 10 min and plasma is collected for cytokine quantification.
- AffiMab(Ront_HC15_zC5) or control treatment of blood samples is carried out by incubation with AffiMab or the native antibody rontalizumab (R0NT_WT), or monospecific bivalent counterpart (Fc-zC5), or other relevant comparators at relevant concentrations and incubated under controlled conditions prior to plasma collection.
- Cytokine quantification of CSF samplesis then analyzed using a commercially available ELISA-based method, for example but not limited to kits marketed by OLINK, Mabtech and ThermoFisher/lnvitrogen. Results
- the cytokine profile in patient derived bloodis expected to show an upregulation of relevant pro-inflammatory cytokines and a down-regulation of relevant anti-inflammatory cytokines in untreated patient derived blood compared to healthy control.
- Evaluation of the cytokine profile post AffiMab treatmentis expected to demonstrate decreased levels in pro-inflammatory cytokines and increased levels in anti-inflammatory cytokines closely related to the IFNa signature of atherosclerosis patients compared to controls.
- this exampleis expected to demonstrate that treatment with AffiMab lead construct (RONT_HC15_zC5) is able to affect levels of IFNa, C5 and downstream related cytokines of interest in a favorable manner, demonstrating signs of affecting key cytokines in the atherosclerosisjnduced whole blood profile ex vivo.
- a bispecific binding moleculecomprising at least one first moiety with at least one first binding site for human complement component 5 (C5), operably linked to at least one second moiety with at least one first binding site for interferon alpha (IFNa).
- C5human complement component 5
- IFNainterferon alpha
- a bispecific binding molecule according to item 1wherein said at least one first moiety with at least one first binding site for human complement component 5 (C5) is a C5 binding polypeptide.
- X2is selected from H, Q, S, T and V;
- X3is selected from I, L, M and V;
- X4is selected from A, D, E, H, K, L, N, Q, R, S, T and Y;
- Xeis selected from N and W;
- X7is selected from A, D, E, H, I, L, N, Q, R, S and T;
- X10is selected from D and E;
- X11is selected from A, E, G, H, K, L, Q, R, S, T and Y;
- Xis selected from N and T;
- X17is selected from I, L and V;
- X18is selected from A, D, E, H, K, N, Q, R, S and T;
- X21is selected from I, L and V;
- X25is selected from A, D, E, G, H, N, S and T;
- X26is selected from K and S;
- X28is selected from A, D, E, H, N, Q, S, T and Y; and ii) an amino acid sequence which has at least 86 % identity to the sequence defined in i), wherein the polypeptide binds to C5.
- a bispecific binding moleculeaccording to any one of items 4-7, wherein X3 is selected from I, L and V.
- a bispecific binding molecule according to item 8wherein X3 is selected from I and L.
- a bispecific binding moleculeaccording to any one of items 4-14, wherein X 6 is W.
- a bispecific binding moleculeaccording to any one of items 4-15, wherein X7 is selected from A, D, N and T.
- X11is selected from A, H, K, Q, R and S.
- a bispecific binding molecule according to item 21wherein Xu is selected from A, K and R.
- a bispecific binding moleculeaccording to any one of items 4-23, wherein X is T.
- a bispecific binding moleculeaccording to any one of items 4-24 wherein X17 is selected from I and L.
- a bispecific binding moleculeaccording to any one of items 4-27, wherein X18 is selected from A, D, E, N, Q, S and T.
- a bispecific binding molecule according to item 31wherein Xis is D.
- a bispecific binding molecule according to item 31wherein Xis is E.
- X21is selected from I and L.
- X25is selected from A, E, H, N and T.
- a bispecific binding moleculeaccording to any one of items 4-39, wherein X 26 is K.
- a bispecific binding moleculeaccording to any one of items 4-40, wherein X28 is selected from A, D, E, H, N, Q and S.
- a bispecific binding molecule according to item 41wherein X28 is selected from A, D, E and S.
- a bispecific binding moleculeaccording to any one of items 11 -14, wherein X3X4 is selected from LE and LD.
- a bispecific binding moleculeaccording to any one of items 25-33, wherein X17X18 is selected from IE and LD.
- a bispecific binding moleculeaccording to any one of items 4-47, wherein the amino acid sequence i) fulfills at least four of the following eight conditions l-VIII: X 2 is V;
- X3is selected from I and L; III. X 6 is W;
- IV. X?is selected from D and N;
- V. X17is selected from I and L;
- a bispecific binding molecule according to item 51wherein the amino acid sequence i) fulfills all of the eight conditions l-VIII.
- a bispecific binding moleculeaccording to any one of items 4-52, wherein the amino acid sequence is selected from any one of SEQ ID NO: 1-248.
- a bispecific binding molecule according to item 53wherein the amino acid sequence is selected from any one of SEQ ID NO: 1-12, SEQ ID NQ:20, SEQ ID NO:23-24, SEQ ID NO:26-28, SEQ ID NO:32-35, SEQ ID NO:38-39, SEQ ID NO:41 , SEQ ID NO:46, SEQ ID NO:49, SEQ ID NO:56-57, SEQ ID NO:59, SEQ ID NO:66, SEQ ID NO:78-79, SEQ ID NO:87, SEQ ID NO:92, SEQ ID NQ:106, SEQ ID NQ:110, SEQ ID NO:119, SEQ ID NO:125, SEQ ID NO:141 , SEQ ID NO:151 , SEQ ID NO:161 , SEQ ID NO:166, SEQ ID NO:187, SEQ ID NO:197, SEQ ID NQ:203, SEQ ID NQ:205, SEQ ID NO:215 and SEQ ID NO:243.
- a bispecific binding molecule according to item 54the amino acid sequence is selected from any one of SEQ ID NO: 1-12.
- a bispecific binding molecule according to item 55wherein the amino acid sequence is selected from any one of SEQ ID NO: 1 , SEQ ID NO:2, SEQ ID NO:3, SEQ ID NO:4 and SEQ ID NO:5.
- 58.A bispecific binding molecule according to item 57, wherein the amino acid sequence is SEQ ID NO:1.
- a bispecific binding moleculeaccording to any one of items 2-59, wherein the C5 binding polypeptide comprises additional amino acids at the C- terminal and/or N-terminal end.
- a bispecific binding moleculeaccording to any one of items 4-60, wherein said C5 binding motif (BM) forms part of a three-helix bundle protein domain.
- a bispecific binding moleculeaccording to any one of items 4-61 , comprising an amino acid sequence selected from:
- ADNNFNK-[BM](SEQ ID NO:252);
- ADNKFNK-[BM](SEQ ID NO:253);
- VDNKFNK-[BM](SEQ ID NO:254)
- VDAKYAK-[BM](SEQ ID NO:255); and sequences having at least 86% identity thereto, wherein [BM] is as defined in any one of items 4-59.
- a bispecific binding moleculeaccording to any one of items 4-61 , comprising an amino acid sequence selected from:
- [BM]-DPSQSSELLAEAKKLNDAQAPK(SEQ ID NO:261 );
- [BM]-DPSQSSELLAEAKKLNKAQAPK(SEQ ID NO:266); and sequences having at least 86% identity thereto, wherein [BM] is as defined in any one of items 4-59.
- a bispecific binding moleculeaccording to any one of items 4-61 , comprising an amino acid sequence selected from:
- ADNNFNK-[BM]-DPSQSANLLSEAKKLNESQAPK(SEQ ID NO:267); ADNKFNK-[BM]-DPSQSANLLAEAKKLNDAQAPK (SEQ ID NO:268); ADNKFNK-[BM]-DPSVSKEILAEAKKLNDAQAPK (SEQ ID NO:269); VDNKFNK-[BM]-DPSQSANLLAEAKKLNDAQAPK (SEQ ID NQ:270); AEAKYAK-[BM]-DPSESSELLSEAKKLNKSQAPK (SEQ ID NO:271 ); VDAKYAK-[BM]-DPSQSSELLAEAKKLNDAQAPK (SEQ ID NO:272); VDAKYAK-[BM]-DPSQSSELLAEAKKLNDSQAPK (SEQ ID NO:273); AEAKYAK-[BM]-DPSQSSELLSEAKKLNDSQAPK (SEQ ID NO:274); AEAKYAK
- said C5 binding polypeptideis selected from the group consisting of full-length antibodies, Fab fragments, Fab’ fragments, F(ab')2 fragments, single chain Fab (scFab) fragments, Fc fragments, Fv fragments, single chain Fv (scFv) fragments, (SCFV)2, SCFV-FC constructs and domain antibodies.
- a bispecific binding moleculecomprising at least two first moieties, wherein each at least two first moieties each individually comprise at least one binding site for human complement component 5 (C5), wherein the amino acid sequences for the said binding sites may be the same or different.
- a bispecific binding moleculeaccording to any one of items 1-67, comprising at least two first moieties according to item 67, wherein said at least two first moieties are not coupled together.
- a bispecific binding moleculeaccording to any one of items 2-68, wherein said C5 binding polypeptide binds to C5 such that the KD value of the interaction is at most 1 x 10’ 6 M, such as at most 1 x 10’ 7 M, at most 1 x 10’ 8 M, or at most 1 x 10’ 9 M.
- a bispecific binding molecule according to item 1wherein said at least one second moiety with at least one first binding site for interferon alpha (IFNa) is an IFNa binding polypeptide.
- a bispecific binding molecule according to item 71wherein said IFNa binding polypeptide is selected from the group consisting of full-length antibodies, Fab fragments, Fab’ fragments, F(ab')2 fragments, single chain Fab (scFab) fragments, Fc fragments, Fv fragments, single chain Fv (scFv) fragments, (scFv)2, scFv-Fc constructs and domain antibodies.
- said IFNa binding polypeptideis selected from the group consisting of full-length antibodies, Fab fragments, Fab’ fragments, F(ab')2 fragments, single chain Fab (scFab) fragments, Fc fragments, Fv fragments, single chain Fv (scFv) fragments, (scFv)2, scFv-Fc constructs and domain antibodies.
- a bispecific binding moleculeaccording to any one of items 70-73, wherein said IFNa binding polypeptide comprises a binding domain of an antibody, the binding domain comprising a heavy chain variable region and a light chain variable region, each comprising 6 complementarity determining domains (CDRs), wherein:
- VLCDR1has the sequence set forth in SEQ ID NO:286;
- VLCDR2has the sequence set forth in SEQ ID NO:287;
- VLCDR3has the sequence set forth in SEQ ID NO:288;
- VHCDR1has the sequence set forth in SEQ ID NO:289;
- VHCDR2has the sequence set forth in SEQ ID NQ:290; and VHCDR3 has the sequence set forth in SEQ ID NO:291 .
- a bispecific binding moleculeaccording to any one of items 70-74, wherein said antibody or antigen binding fragment thereof comprises a light chain variable region comprising the amino acid sequence SEQ ID NO:284, or an amino acid sequence having at least 80% sequence identity thereto.
- a bispecific binding moleculeaccording to any one of items 70-75, wherein said antibody or antigen binding fragment thereof comprises a heavy chain variable region comprising the amino acid sequence SEQ ID NO:285, or an amino acid sequence having at least 80% sequence identity thereto.
- a bispecific binding moleculeaccording to any one of items 75-76, wherein said antibody or antigen binding fragment thereof comprises a light chain variable region comprising the amino acid sequence SEQ ID NO:284, or an amino acid sequence having at least 80% sequence identity thereto, and a heavy chain variable region comprising the amino acid sequence SEQ ID NO:285, or an amino acid sequence having at least 80% sequence identity thereto.
- a bispecific binding moleculeaccording to any one of items 75-77, wherein said antibody or antigen binding fragment thereof is rontalizumab or a variant thereof or antigen binding fragment thereof.
- a bispecific binding molecule according to item 78wherein said IFNa binding antibody or antigen binding fragment thereof comprises a light chain comprising the amino acid sequence SEQ ID NO:293 or a sequence having at least 80% identity thereto, and/or a heavy chain comprising the amino acid sequence SEQ ID NO:294 or a sequence having at least 80% identity thereto, such as being an antibody comprising a light chain comprising the amino acid sequence SEQ ID NO:293 and a heavy chain comprising the amino acid sequence SEQ ID NO:294.
- a bispecific binding moleculeaccording to any one of items 73-79, comprising an amino acid substitution N297A in the CH2 region of the antibody or antibody fragment thereof.
- a bispecific binding moleculecomprising at least one first moiety with at least one first binding site for human complement component 5 (C5) according any one of items 2-69, operably linked to at least one second moiety with at least one first binding site for interferon alpha (IFNa) according to any one of items 70-80.
- a bispecific binding molecule according to item 81wherein the at least one first moiety and the at least one second moiety are covalently linked.
- a bispecific binding molecule according to item 82wherein said at least one first moiety is covalently linked to: i) a CH3 domain of the heavy chain of an antibody or antibody fragment thereof according to any one of items 73-80; ii) a CL domain of the light chain of an antibody or antibody fragment thereof according to any one of items 73-80; iii) a VL domain of the light chain of an antibody or antibody fragment thereof according to any one of items 73-80; or iv) a VH domain of the heavy chain of an antibody or antibody fragment thereof according to any one of items 73-80.
- a bispecific binding molecule according to item 83wherein the bispecific molecule comprises one antibody or antibody fragment thereof and two first moieties, and: i) one first moiety is covalently linked to a CH3 domain of each heavy chain of said antibody or antibody fragment thereof; ii) one first moiety is covalently linked to a CL domain of each light chain of said antibody or antibody fragment thereof; iii) one first moiety is covalently linked to a VH domain of each heavy chain of said antibody or antibody fragment thereof; or iv) one first moiety is covalently linked to a VL domain of each light chain of said antibody or antibody fragment thereof.
- a bispecific binding moleculeaccording to any one of items 81-84, wherein said at least one first moiety is operably bound to said at least one second moiety via at least one linker, such as at least one linker selected from flexible amino acid linkers, rigid amino acid linkers and cleavable amino acid linkers.
- a bispecific binding moleculecomprising a pair of polypeptide chains, said pair being selected from the group consisting of the pairs
- a bispecific binding molecule according to item 87comprising the pair of polypeptide chains represented by SEQ ID NO:295 and SEQ ID NO:296.
- Host cellcomprising an expression vector according to item 90.
- Method of producing a bispecific binding molecule according to any one of items 1-88comprising
- compositioncomprising a bispecific binding molecule according to any one of items 1-88 and at least one pharmaceutically acceptable excipient or carrier.
- Bispecific binding moleculeaccording to any one of items 1-88 or a composition according item 94 for oral, respiratory, topical, intravenous, intraperitoneal, subcutaneous, pulmonary, transdermal, intramuscular, intranasal, buccal, sublingual or suppository administration, such as for subcutaneous administration, such as intravenous administration.
- Bispecific binding moleculeaccording to any one of items 1-88 or a composition according to item 94 for use as a medicament.
- Bispecific binding molecule or composition for use as a medicament according to item 96wherein said bispecific binding molecule or composition modulates C5 and IFNa function in vivo.
- Bispecific binding molecule or composition for use according to item 98wherein said disorder or disease related to C5 and IFNa is selected from inflammatory and autoimmune diseases.
- Bispecific binding molecule or composition for use according to any one of items 98-99wherein said disorder or disease related to C5 and IFNa is selected from the group consisting of rheumatoid arthritis (RA), dermatomyositis, primary Sjogren’s syndrome, systemic sclerosis, psoriasis and type I interferonopathies, such as Aicardi-Goutieres syndrome, familial Chilblain lupus, systemic lupus erythematosus (SLE) and lupus nephritis.
- RArheumatoid arthritis
- SLEsystemic lupus erythematosus
- SLEsystemic lupus erythematosus
- lupus nephritisfor example systemic lupus erythematosus, for example lupus nephritis.
- Bispecific binding molecule or composition for use according to item 101wherein said bispecific binding molecule or composition alleviates the inflammatory profile in SLE.
- Method of treatment of a disorder or disease related to C5 and IFNacomprising administering to a subject in need thereof an effective amount of a bispecific binding molecule according to any one of items 1-88 or a composition according to item 94.
- Method according to item 106wherein said disorder or disease related to C5 and IFNa is selected from the group consisting of rheumatoid arthritis (RA), dermatomyositis, primary Sjogren’s syndrome, systemic sclerosis, psoriasis and type I interferonopathies, such as Aicardi-Goutieres syndrome, familial Chilblain lupus, systemic lupus erythematosus (SLE) and lupus nephritis.
- RArheumatoid arthritis
- dermatomyositisprimary Sjogren’s syndrome
- systemic sclerosispsoriasis
- type I interferonopathiessuch as Aicardi-Goutieres syndrome, familial Chilblain lupus, systemic lupus erythematosus (SLE) and lupus nephritis.
- Method according to item 107wherein said disorder or disease related to C5 and IFNa is systemic lupus erythematosus (SLE) or lupus nephritis, for example systemic lupus erythematosus, for example lupus nephritis.
- SLEsystemic lupus erythematosus
- lupus nephritisfor example systemic lupus erythematosus, for example lupus nephritis.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Biochemistry (AREA)
- Biophysics (AREA)
- Life Sciences & Earth Sciences (AREA)
- Genetics & Genomics (AREA)
- Medicinal Chemistry (AREA)
- Molecular Biology (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Immunology (AREA)
- Peptides Or Proteins (AREA)
Abstract
Description
BISPECIFIC BINDING MOLECULE
Field
The present disclosure relates to a bispecific binding molecule having binding sites targeting interferon alpha (IFNa) and human complement component 5 (C5). The disclosure further relates to the use of such a bispecific binding molecule for the preparation of a pharmaceutical composition. The disclosure further relates to a method for the treatment or amelioration of a disease comprising administration of an effective amount of such a bispecific binding molecule.
Background
Systemic lupus erythematosus (SLE) is a complex multisystemic chronic autoimmune disease characterized by a cyclic disease progression between remission and active state involving a self-damaging hyperactive immune system. Consequently, the disease presents itself in a multitude of ways, though in the most severe cases chronic inflammatory organ damage and mortality (Obermoser and Pascual, Lupus. 19:1012-1019, 2010). A majority of patients suffer from impaired kidney functions and in more aggressive disease states develop severe lupus nephritis, i.e. hyperinflammation of the kidneys (Almaani, Meara and Rovin, Clin. J. Am. Soc. Nephrol. 12:825-835, 2017). The immunopathogenesis of the disease has challenged clinicians and researchers alike for decades. Extensive studies have been able to establish several key role players involved in the complex interplay between the innate and adaptive immune system and complement system driving disease progression (Kaul et al, Nat. Rev. Dis. Prim. 2:16039, 2016). The disease pathology is characterized by significant levels of autoantibodies towards nuclear self-antigens, produced by hyperactive autoreactive plasma cells and impaired clearance of apoptotic material as a result of a dysregulated immune system. These autoantibody-antigen immune complexes (ICs) can subsequently push the autoimmunity cycle further in a multitude of ways affecting the adaptive, innate and complement system (Oon, Wilson and Wicks, Clin. Transl. Immunol. 5:29, 2016). Type 1 interferons, in particular interferon alpha (IFNa) have been shown to host a fundamental role in the pathogenesis of SLE, overexpressed by plasmacytoid dendric cells (pDCs) upon stimuli of toll-like receptor 7 and 9 (TLR7 and TLR9) by ICs (Oon, Wilson and Wicks, Clin. Transl. Immunol. 5:29, 2016) and (Niewold et al, J. Biomed. Biotechnol. 2010:948364, 2010). Consequently, when expressed abundantly by pDCs, IFNa exerts an array of immune-modular effects promoting autoimmune disease progression via its interaction with the IFNaR. In the adaptive immune system, IFNa induced upregulation of costimulatory receptors CD80 and CD86 providing a platform for autoreactive CD4+ helper T cells to activate cytotoxic autoreactive CD8+ T cells with additional heightened perforin and granzymes expression due to direct stimuli by IFNa (Obermoser and Pascual, Lupus. 19:1012-1019, 2010). In its arguably most pivotal role as one of the main villains of SLE pathogenesis, IFNa upregulates TLR7 and interferon regulating factor 7 (IRF7) in pDCs, mDCs and monocytes which subsequently upregulates the expression of IFNa itself. Thus, contributing to a feed-back loop by increasing pDCs sensitivity towards stimuli by ICs, resulting in further augmentation of IFNa expression and progression of disease activity (Obermoser and Pascual, Lupus. 19: 1012-1019, 2010) and (Ganguly et al, J. Exp. Med. 206:1983-1994, 2009). Further, IFNa induces the expression of B cell activating factor (BAFF) which contributes to the survival of mature B cells and together with IL6 promotes differentiation of plasma blasts into mature autoantibody producing plasma cells (Jego et al, Immunity. 19:225-234, 2003, and Rdnnblom and Leonard, Lupus Sci. Med. 6:e000270, 2019). This in turn plays back to the vicious IFNa positive feed-back loop through the formation of new ICs with secreted autoantibodies engaging TLR7/9 further promoting IFNa production.
The complement system has been identified as having paradoxical roles in the pathogenesis of SLE, as it both suppresses and drives disease progression. Complement component Cq1 deficiency is strongly related to SLE pathogenesis with detectable levels of C1q autoantibodies in specific patient cohorts (Leffler et al, Ann. Rheum. Dis., 73:1601-1606, 2014). Previous work has shown the downregulation of IFNa expression upon stimuli of pDCs by C1q-autoantibody ICs. Further, C1q is involved in clearing apoptotic material by opsonization, where autoantibodies contributing to lower levels of C1q directly affect the degradation and clearance of apoptotic material, in particular released neutrophil extracellular traps (NETs) from dying neutrophils. Consequently, the risk of hyperactive plasma cells to secret autoantibodies towards autoantigens from released cell material forming new ICs increases substantially when the levels of C1q are significantly low enough, tipping the balance in favor of progressive autoimmune reaction. Hereafter the complement system acts as a driving force in disease progression, contributing to hyper-inflammation by activation of complement factors triggered by ICs through mainly the classical pathway. The clinical manifestation of these effects is shown in the form of IC depositions in kidneys that activate the complement and cause severe inflammation in patients with lupus nephritis (Park et al, Blood Adv 2(16):2090-2094, 2018; Pang et al, Immunobiology 219(12):980-989, 2014).
Current standard therapies for SLE are dictated by the disease activity state (remission vs active state) and multisystemic severity. The rule of practice is to start treatment with glucocorticosteroids (GSCs) and introduction of immunosuppressive drug treatment in more active and persistent disease states. Several strategies to target constituents of the complex immunopathogenesis of SLE involving biological drugs have been or are in clinical trials. In regard to antibody therapy, the anti-CD20 B cell depleting antibody rituximab was investigated for treatment of SLE patients with lupus nephritis that did not answer to first line therapy with GSCs. The rationale for anti-CD20 treatment stems from the ample evidence that hyperactive B cell lymphocytes are strongly related to the disease progression of SLE. Rituximab failed to reach primary endpoints in two phase II trials, although post hoc analysis showed a reduced risk for a first severe flare compared to placebo (Tunnicliffe et al, Arthritis Care Res (Hoboken) 67(10): 1440-1452, 2015).
The FDA-approved monoclonal antibody belimumab has been of particular interest as it targets the aforementioned B cell activating factor (BAFF) and has been investigated in patients with continuous active SLE despite standard therapy regiments and show high levels of autoantibodies in tested plasma samples. Though the drug has shown safety and efficacy in adult patients with active SLE during phase II and III trials, it does not address the hyperactive inflammatory severe cases of SLE involving lupus nephritis. As increased levels of Type 1 interferons and their related downstream pathways have been shown to correlate with SLE disease activity as previously described, promoting differentiation of cells involved in the adaptive immune system, therapies targeting the cytokine group and the IFNa receptor (IFNaR) have emerged as potential avenues for treating the disease. The monoclonal antibody rontalizumab was developed toward the IFNa ligand in patients with moderate-severe SLE, where it did not meet primary or secondary endpoint in a phase II study evaluating the antibody efficacy (Kalunian et al, Ann Rheum Dis 75(1 ): 196-202, 2016). A recent phase III trial, the monoclonal antibody anifrolumab met its primary end point for the treatment of SLE, significantly reducing disease activity. Interestingly, this antibody targets the IFNaR receptor with a blocking mechanism of action, in contrast to rontalizumab which binds the IFNa ligand itself and blocks it from receptor-ligand interaction (Morand et al, Arthritis Rheumatol 71 , Abstract L17 to ACR/ARP Annual Meeting 2019). Anifrolumab was approved by FDA for the treatment of moderate to severe SLE in 2021 (Deeks, Drugs 81 (15): 1795- 1802, 2021 ).
With regard to the complement system and SLE, the complement inhibiting antibody eculizumab has shown to potentially be effective in a subgroup of patients with complement mediated thrombotic microangiopathy in lupus nephritis and poor response to GSCs (Coppo et al, Pediatr Nephrol 30(1 ): 167-172, 2015).
Rheumatoid arthritis (RA), another chronic autoimmune disease, affects the lining of joints, and causes painful swelling that can ultimately result in bone erosion and deformed joints. In RA, the complement system has been indicated as a main player, by initiation of the RA pathogenic mechanisms through interactions with citrullinated proteins and by mediating direct tissue damage and an association between complement system activation and disease activity is well established (Triggianese P, 2023). Also, IFN-I and in particular IFNa have been implicated to be upregulated in RA and there is emerging data that IFNs may contribute to the transition from preclinical to sustained clinical disease. In early RA cohorts, evidence of increased IFNa signaling has been observed and this, together with other parameters, was able to differentiate progressors with a median of 4.1 years before symptom onset. Conversely, other studies have found rheumatoid factor (RF) demonstrated a positive association with circulating IFNa levels in both established and early RA (Lin CHM, 2024).
There is also evidence to suggest that a simultaneous up-regulation of the interferon and complement systems may play part also in cardiovascular disease. Atherosclerosis, plaque formation in the arterial walls, can cause serious complications such as myocardial infarction (Ml) and stroke, which are the primary cause of death and morbidity worldwide. The disease is driven by risk factors such as pro-inflammatory stimuli, cytokines and dysregulated lipids. The contribution of inflammation in atherosclerosis is well established and there is growing evidence of a central role of IFNa and the complement system in the progression (Kiss MG, 2022; Chen HJ, 2020).
There is also evidence to suggest the involvement of the interferon and complement systems in central nervous system (CNS) disease, disorders and injury. Traumatic brain injury (TBI) is a leading cause of mortality in the modem world. Subsequent to this primary injury, the blood brain barrier (BBB) becomes compromised resulting in a significant influx of cells, inflammatory mediators and plasma proteins, including complement proteins, that drive the delayed secondary inflammation cascade, which is the major determinant of clinical outcome and thus recovery and survival (Carpanini, 2019).
The complement system has a central role in secondary inflammation and elevated levels of complement related proteins such as C 1 q, C3, sC5b-9 and MBL have been demonstrated in human CSF models for TBI. Animal models of TBI have demonstrated that a reduction of complement system activity leads to reduced lesion sizes and improved neurological outcomes (Hammad, 2018).
The interferon system has also been linked to the outcomes of TBI through knock-out mouse models, and neutralization by monoclonal antibodies suggests that IFNa, which peaks at 2 hours after TBI, may play a detrimental role in brain trauma (Roselli F, 2018).
Spinal cord injury (SCI) results in dysfunction/complete loss of function below the lesion site and symptoms are often life-long. Like in TBI, the primary pathology in SCI is caused by a mechanical force that damages the neural tissue, followed by post-injury inflammation where the production of pro-inflammatory mediators and infiltration of immune cells result in secondary pathology. Studies have demonstrated elevated C3, C4, and C5 levels in plasma of patients post-SCI, and a number of animal model experiments have demonstrated that pharmacological complement inhibition resulted in improved outcomes (Carpanini, 2019; Li L, Neurobiol Dis, 2014). Moreover, an IFNa overexpression in the secondary inflammation has been suggested, and a study using a rat SCI model that evaluated the effect of bone marrow derived mesenchymal stem cells (BMSCs) transplantation plus hyperbaric oxygen reported a larger reduction of IFNa levels along with behavioral improvement and axon recovery in rats having revived both treatments compared to controls (Geng CK et al, 2015; Zindler E et al, 2010).
Another CNS injury, subarachnoid hemorrhage (SAH), a subtype of stroke caused by rupture of an aneurysm of an intracranial artery in the subarachnoid space, also has an upregulation of IFN-a signaling. A retrospective study on serum from SAH patients demonstrated higher IL-1 (3, IL-5, IL-2, IL-6, IL-8, IL-10, IFN-a, IFN-y, and TNF levels in the poor prognosis group than in the good prognosis group suggesting a prognostic value (P < 0.05) (Luo C, 2022).
Experimental model experiments have shown that systemic treatment through intraperitoneal injection with C5-specific antibodies, which prevent the formation of the pro-inflammatory peptide C5a and the membrane attack complex, reduced microglial activation and cell death by 40%. An analysis of patients’ CSF showed that C5a was >1400-fold increased 1 day after aneurysmal SAH and then gradually decreased (van Dijk, 2020). A recent open-label Ph 2a study investigated the effect of high-dose intravenous infusion of C5-inhibiting antibody eculizumab into aSAH patients. A large reduction in complement activity in serum was observed, however the primary outcome of >55% reduction of C5a in the cerebrospinal fluid (CSF) was not met, potentially due to study design flaws and large patient variability (Koopman I. et al, European Stroke Journal 2023).
Neuromyelitis Optica (NMO) is a rare autoimmune condition characterized by blood brain barrier (BBB) disruption and inflammation/degeneration of the optic nerve and spinal cord. Studies have demonstrated association of IFN-a levels with clinical disease activity and seventy, suggesting a role for IFN-a in disease progression (Asgari N, 2013). NMO is also known to be associated with upregulation of the complement system and in a recent small trial, the treatment with the anti-C5 monoclonal antibody eculizumab decreased the number of neurological episodes (www.clinicaltrials.gov NCT00904826, Pittock SJ, 2013).
As is understood by the background provided in the introduction above, a number of additional disease states, disorders, and injury such as autoimmune disease, cardiovascular disease, and central nervous system (CNS) disorders, disease and injury involve players of the adaptive, innate and complement systems.
To summarize, SLE is characterized by the influence of a multitude of factors in the innate, adaptive and complement system governing the disease pathogenesis, driving disease progression. Many endeavors aiming at developing novel SLE therapies with targeted biologies have shown limited success in the clinic, and efficacious therapeutics have yet to be realized. Previous attempts to find SLE therapies have revealed the complexity of the disease, which is not governed by a single pathogenic mechanism. This reasoning also stands true for the other disease states, disorders and injury mentioned above.
As such, there is a need for a therapeutic approach which is able to achieve significant efficacy and thereby meet the unmet need of affected individuals. Disclosure of the invention
The different aspects of the present disclosure address this need by providing an innovative and broadened therapeutic approach targeting both the complement and interferon pathways.
It is an object of the present disclosure to provide a new multispecific agent, such as a bispecific binding molecule, which could for example be used for therapeutic applications.
It is an object of the present disclosure to provide a bispecific binding molecule allowing for efficient therapy targeting various forms of inflammatory, autoimmune diseases, cardiovascular disease, and/or central nervous system (CNS) diseases, disorders and injury while alleviating the abovementioned and other drawbacks of current therapeutic approaches.
These disease states, disorders, and injury such as autoimmune disease, cardiovascular disease, and central nervous system (CNS) disorders, disease and injury involve players of the adaptive, innate and complement systems. Therefore, they could benefit from therapies which address the involvement of and the complicated relationship between the adaptive, innate and complement systems.
It is an object of the present disclosure to provide pharmaceutical compositions which comprise such bispecific binding molecules and are designed to be suited for treatment of various inflammatory, autoimmune diseases, cardiovascular disease, and/or central nervous system (CNS) diseases, disorders and injury; and treatment methods in which such molecules or pharmaceutical compositions are administered to a subject in need of such treatment.
These objects, and other objects which are evident to the skilled person from the disclosure, are met by the different aspects of the invention as claimed in the appended claims and as generally disclosed herein.
Bispecific binding molecule
Thus, in a first aspect of the disclosure, there is provided a bispecific binding molecule, comprising at least one first moiety with at least one first binding site for human complement component 5 (C5), operably linked to at least one second moiety with at least one first binding site for interferon alpha (IFNa).
When used herein to denote the first aspect of the disclosure, the term “bispecific binding molecule” is intended to refer to a binding molecule which has affinity for two separate targets. The bispecific binding molecule comprises at least two moieties, at least one first moiety having affinity for C5 by virtue of its binding site for C5, and at least one second moiety having affinity for IFNa by virtue of its binding site for IFNa. The term “first moiety” is intended to refer a moiety having affinity for C5. The term “second moiety” is intended to refer to a moiety having affinity for IFNa. The term “at least one” is intended to refer to the fact that the bispecific binding molecule may comprise one, or may comprise more than one “first moiety”. Furthermore, the term “at least one” is also intended to refer to the fact that the bispecific molecule may comprise one, or may comprise more than one “second moiety”. Therefore, the bispecific binding molecule, which has affinity for the above-mentioned targets simultaneously, may be able to bind one C5 while also simultaneously be able to bind one IFNa. By this same definition, the bispecific binding molecule may also bind at least one C5 while also simultaneously bind to at least one IFNa. The two moieties, referred to as “first moiety” and “second moiety”, may for example be connected by covalent coupling using known organic chemistry methods, or, if one or both moieties are polypeptides, be expressed as one or more fusion polypeptides in a system for recombinant expression of polypeptides, or joined in any other fashion, directly or mediated by a linker comprising a number of amino acids.
First moiety, providing a binding site for C5
In one embodiment of the disclosure, there is provided a bispecific binding molecule as defined herein, wherein said at least one first moiety with at least one first binding site for human complement component 5 (C5) is a C5 binding polypeptide. In this embodiment, the part of the bispecific binding molecule designated “first moiety” is a C5 binding polypeptide. In one embodiment, the at least one first moiety comprises a binding polypeptide capable of selective interaction with at least one C5. In one embodiment, there is also provided a bispecific molecule as defined herein, wherein said at least one first moiety is two, three, or four first moieties, wherein each first moiety is a C5 binding polypeptide. It follows that in this particular embodiment that each C5 binding polypeptide is capable of binding to an individual C5, which means that in this particular embodiment the bispecific molecule targets two individual C5 simultaneously, or three individual C5 simultaneously, or four individual C5 simultaneously.
In another embodiment of the disclosure, there is provided a bispecific molecule as defined herein, wherein said C5 binding polypeptide is selected from the group consisting of full-length antibodies, Fab fragments, Fab’ fragments, F(ab')2 fragments, single chain Fab (scFab) fragments, Fc fragments, Fv fragments, single chain Fv (scFv) fragments, (scFv)2, scFv-Fc constructs, domain antibodies, a chimeric antibody, a CDR-grafted antibody, a fully human antibody, a bivalent antibody construct, a synthetic antibody, a bivalent antibody, a trivalent antibody, a tetravalent antibody, bivalent single chain antibody, a trivalent single chain antibody, a multivalent single chain antibody, nanobodies, AlbudAbs, DARTs (dual affinity re-targeting), BiTEs (bispecific T-cell engager), TandAbs (tandem diabodies), DAFs (dual acting Fab), two-in-one antibodies, SMIPs (small modular immunopharmaceuticals), FynomAbs (fynomers fused to antibodies), DVD-lgs (dual variable domain immunoglobulin), CovX-bodies (peptide modified antibodies), duobodies and triomAbs; a microbody, a maxybody, an avimer, a small disulfide-bonded protein; and a binding protein derived from a scaffold selected from the group consisting of staphylococcal protein A and domains thereof, other three helix domains, lipocalins, ankyrin repeat domains, cellulose binding domains, y crystallines, green fluorescent protein, human cytotoxic T lymphocyte- associated antigen 4, protease inhibitors such as Kunitz domains, PDZ domains, SH3 domains, peptide aptamers, staphylococcal nuclease, tendamistats, fibronectin type III domain, transferrin, zinc fingers and conotoxins.
In some examples of such an embodiment, the binding polypeptide comprises a variant of protein Z, in turn derived from domain B of staphylococcal protein A and described in Nilsson B et al, Protein Engineering 1 : 107-133, 1987. Such variants, having affinity for a number of different targets, have been selected from libraries and engineered further as described in numerous prior publications, for example but not limited to WO1 995/19374; Nord et al, Nat Biotech (1997) 15:772-777; and W02009/080811 , all incorporated herein by reference.
C5 binding polypeptides useful as first moiety in the bispecific binding molecule of the disclosure and comprising a C5 binding motif are disclosed in WO201 3/126006 and WO2015/028558. In particular, WO2013/126006 and WO201 5/028558 disclose a C5 binding motif, BM. In one embodiment, the C5 binding polypeptide comprises a C5 binding motif, BM, which motif consists of the amino acids sequence selected from: i) EX2X3X4A X6X7EIX10X11 LPNL X16X17X18QW X21AFIX25 X26LX28D (SEQ ID NO:249), wherein, independently of each other, X2 is selected from H, Q, S, T and V; X3 is selected from I, L, M and V;
X4 is selected from A, D, E, H, K, L, N, Q, R, S, T and Y;
Xe is selected from N and W;
X7 is selected from A, D, E, H, I, L, N, Q, R, S and T;
X10 is selected from D and E;
X11 is selected from A, E, G, H, K, L, Q, R, S, T and Y;
X is selected from N and T;
X17 is selected from I, L and V;
X18 is selected from A, D, E, H, K, N, Q, R, S and T;
X21 is selected from I, L and V;
X25 is selected from A, D, E, G, H, N, S and T;
X26 is selected from K and S;
X28 is selected from A, D, E, H, N, Q, S, T and Y; and ii) an amino acid sequence which has at least 86 % identity to the sequence defined in i), wherein the polypeptide binds to C5. As used herein, “Xn” and “Xm” are used to indicate amino acids in positions n and m in the sequence i) as defined above, wherein n and m are integers which indicate the position of an amino acid within said sequence as counted from the N-terminal end of said sequence. For example, X4 and X7 indicate the amino acid in position four and seven, respectively, from the N- terminal end of sequence i).
Examples of specific C5 binding motifs as previously disclosed in WO201 3/126006 and WO2015/028558 are disclosed as SEQ ID NO: 1-248 herein.
The above defined class of sequence related polypeptides having a binding affinity for C5 is derived from a common parent polypeptide sequence. More specifically, the definition of the class is based on an analysis of a large number of random polypeptide variants of the parent polypeptide that were selected for their interaction with C5 in selection experiments. The identified C5 binding motif, or “BM”, corresponds to the target binding region of the parent scaffold, which region constitutes two alpha helices within a three-helical bundle protein domain. In the parent scaffold, the varied amino acid residues of the two BM helices constitute a binding surface for interaction with the constant Fc part of antibodies. By random variation of binding surface residues and subsequent selection of variants, the Fc interaction capacity of the binding surface was replaced with a capacity for interaction with C5.
As the skilled person will realize, the function of any polypeptide, such as the C5 binding capacity of the polypeptides of this embodiment, is dependent on the tertiary structure of the polypeptide. It is therefore possible to make minor changes to the amino acid sequence of a polypeptide without largely affecting the tertiary structure and the function thereof. Thus, the disclosure encompasses first moieties that are modified variants of a C5 binding polypeptide with retained C5 binding characteristics. In one embodiment, the polypeptide comprises modified variants of the BM of i), which are such that the resulting sequence is at least 86% identical to a sequence belonging to the class defined by i), such as at least 89% identical, such as at least 93 % identical, such as at least 96 % identical to a sequence belonging to the class defined by i). For example, it is possible that an amino acid residue belonging to a certain functional grouping of amino acid residues (e.g. hydrophobic, hydrophilic, polar etc.) could be exchanged for another amino acid residue from the same functional group.
In some embodiments, such changes may be made in any position of the sequence of the C5 binding polypeptide as defined herein. In other embodiments, such changes may be made only in the non-variable positions, also denoted scaffold amino acid residues. In such cases, changes are not allowed in the variable positions. In other embodiments, such changes may be only in the variable positions.
As described in WO2013/126006 and WO2015/028558, selection of C5 binding polypeptide variants may for example be achieved by phage display for selection of naive variants of a protein scaffold optionally followed by affinity maturation and cell display for selection of affinity maturated C5 binding variants. It is however understood that any selection system, whether phage-based, bacterial-based, cell-based or other, may be used for selection of C5 binding polypeptides.
The terms “C5 binding” and ’’binding affinity for C5” as used in this specification refers to a property of a polypeptide which may be tested for example by the use of surface plasmon resonance technology, such as in a Biacore instrument (Cytiva). C5 binding affinity may e.g. be tested in an experiment in which C5 is immobilized on a sensor chip of a Biacore instrument, and the sample containing the polypeptide to be tested is passed over the chip. Alternatively, the polypeptide to be tested is immobilized on a sensor chip of the instrument, and a sample containing C5, or fragment thereof, is passed over the chip. The skilled person may then interpret the results obtained by such experiments to establish at least a qualitative measure of the binding of the polypeptide to C5. If a quantitative measure is desired, for example to determine the apparent equilibrium dissociation constant KD for the interaction, surface plasmon resonance methods may also be used. Binding values may for example be defined in a Biacore 2000 instrument (Cytiva). C5 is immobilized on a sensor chip of the measurement, and samples of the polypeptide whose affinity is to be determined are prepared by serial dilution and injected over the chip. KD values may then be calculated from the results using for example the 1 :1 Langmuir binding model of the BIAevaluation software provided by the instrument manufacturer.
In embodiments according to the first aspect, there is provided a bispecific binding molecule comprising a C5 binding polypeptide wherein Xn in sequence i) is independently selected from a group of possible residues. The skilled person will appreciate that Xn may be selected from any one of the listed groups of possible residues and that this selection is independent from the selection of amino acids in Xm, wherein n^m. Thus, any of the listed possible residues in position Xn may be independently combined with any of the listed possible residues in any other variable position.
According to one definition of such “variable positions”, these are positions denoted with an “X” in sequence i) as defined above. According to another definition, “variable positions” are those positions that are randomized in a selection library of Z variants prior to selection, and may thus for example be positions 2, 3, 4, 6, 7, 10, 11 , 17, 18, 20, 21 , 25 and 28 in sequence i). This definition of “variable positions” does not include positions 16 and 26, which are scaffold positions in this context, albeit allowed to be either one of two alternatives in each position. Reference is made to Nord et al. (1995), Prot Eng 8:601-608, and Ldfblom et al. (2010), FEBS Letters, 584:2670-2680. Like the C5 binding polypeptides, or Z variants, of the present disclosure, the polypeptides disclosed in Nord et al. and Ldfblom et al. are also based on a scaffold from the Z derivative of domain B of protein A from Staphylococcus aureus, although directed to other targets. As shown in Nord et al. (see for example Figure 4), the amino acids in positions 23 (corresponding to position 16 in the instant C5 binding motif) and 33 (corresponding to position 26 in the instant C5 binding motif) are N and S, respectively. As also shown in Ldfblom et al., polypeptides with amino acid residues N and S in positions 23 and 33, respectively (corresponding to positions 16 and 26 in the instant C5 binding motif; see Figure 2 of Ldfblom et al.), and polypeptides with amino acid residues T and K in positions 23 and 33, respectively, all have a maintained basic structure and function. Thus, in the context of this definition of “variable positions”, the amino acid residues at positions 16 and 26 form part of the common scaffold, and it is contemplated to have either N or T in scaffold position 16 and either S or K in scaffold position 26.
In one embodiment of a bispecific binding molecule according to the present disclosure comprising a C5 binding polypeptide, X2 is selected from H, T and V. In another embodiment, X2 is selected from T and V. In yet another embodiment, X2 is V.
In one embodiment, X3 is selected from I, L and V. In another embodiment, X3 is selected from I and L. In yet another embodiment, X3 is I. In an alternative embodiment, X3 is L.
In one embodiment, X4 is selected from A, D, E, K, L, Q and R. In another embodiment, X4 is selected from A, D, E, K and R. In yet another related embodiment, X4 is selected from D and E.
In one embodiment of the C5 binding polypeptide according to the present invention, Xe is W.
In one embodiment, X7 is selected from A, D, N and T. In another embodiment, X7 is selected from D and N. In yet another related embodiment, X7 is D. In an alternative embodiment, X7 is N.
In one embodiment, Xu is selected from A, H, K, Q, R and S. In another embodiment, Xu is selected from A, H, K and R. In yet another related embodiment, Xu is selected from A, K and R. In yet another related embodiment, Xu is selected from K and R.
In one embodiment, X is T.
In one embodiment, X17 is selected from I and L. In another embodiment, X17 is I. In an alternative embodiment, X17 is L.
In one embodiment, X18 is selected from A, D, E, N, Q, S and T. In another embodiment, X18 is selected from A, D, E, Q and S. In yet another related embodiment, X18 is selected from D, E and Q. In yet another related embodiment, X18 is selected from D and E. In yet another related embodiment, X18 is D. In an alternative embodiment, X18 is E.
In one embodiment, X21 is selected from I and L. In another embodiment, X21 is I. In an alternative embodiment, X21 is L. In one embodiment, X2s is selected from E, H, N and T. In another embodiment, X25 is selected from E and N. In yet another related embodiment, X25 is N.
In one embodiment, X26 is K.
In one embodiment, X28 is selected from A, D, E, H, N, Q and S. In another embodiment, X28 is selected from A, D, E and S. In yet another related embodiment, X28 is selected from A, D and E. In yet another related embodiment, X28 is selected from D and E. In yet another related embodiment, X28 is D.
In one embodiment, X3X4 is selected from LE and LD.
In one embodiment, X17X18 is selected from IE and LD.
In the above embodiments of the first aspect, examples of C5 binding polypeptides falling within the defined class of polypeptides are identified. It is contemplated that the individual embodiments may be combined in all conceivable ways and still fall within the contemplated scope. Such combinations of individual embodiments define a restricted amino acid sequence, in one or more of the positions X2-X28, as compared to the amino acid definition in i). The above embodiments may for example be combined such that the amino acid sequence i) fulfils at least four of the following eight conditions l-VIII:
I. X2 is V;
II. X3 is selected from I and L;
III. X6 is W;
IV. X7 is selected from D and N;
V. X17 is selected from I and L;
VI. X21 is L;
VII. X25 is N;
VIII. X28 is D.
In some embodiments, the amino acid sequence i) fulfils at least five of the eight conditions l-VIII. More specifically, the amino acid sequence i) may fulfill at least six of the eight conditions l-VIII, such at least seven of the eight conditions l-VIII, such as all of the eight conditions l-VIII. As described in the examples of WO2013/126006 and
WO201 5/028558, the selection of C5 binding variants led to the identification of individual C5 binding motif (BM) sequences. These sequences constitute individual embodiments of C5 binding polypeptides useful as first moiety in the bispecific binding molecule according to this aspect. The sequences of individual C5 binding motifs are presented as SEQ ID NO: 1-248. In one embodiment, the BM sequence i) is selected from any one of SEQ ID NO:1- 12, SEQ ID NQ:20, SEQ ID NO:23-24, SEQ ID NO:26-28, SEQ ID NO:32-35, SEQ ID NO:38-39, SEQ ID NO:41 , SEQ ID NO:46, SEQ ID NO:49, SEQ ID NO:56-57, SEQ ID NO:59, SEQ ID NO:66, SEQ ID NO:78-79, SEQ ID NO:87, SEQ ID NO:92, SEQ ID NQ:106, SEQ ID NQ:110, SEQ ID NO:119, SEQ ID NO:125, SEQ ID NO:141 , SEQ ID NO:151 , SEQ ID NO:161 , SEQ ID NO:166, SEQ ID NO:187, SEQ ID NO:197, SEQ ID NQ:203, SEQ ID NQ:205, SEQ ID NO:215 and SEQ ID NO:243. More specifically, the BM sequence i) may be selected from any one of SEQ ID NO: 1-12, such as from SEQ ID NO:1 , SEQ ID NO:2, SEQ ID NO:3, SEQ ID NO:4 and SEQ ID NO:5. In particular, the BM sequence i) may be selected from SEQ ID NO:1 and SEQ ID NO:4. In a preferred embodiment, the BM sequence i) is SEQ ID NO:1. In a another preferred embodiment, the BM sequence i) is SEQ ID NO:4.
In some embodiments, the C5 binding motif (BM) as defined herein is flanked by additional amino acids N-terminally to the C5 binding motif. In other embodiments, the C5 binding motif is flanked by additional amino acids C-terminally to the C5 binding motif. In further embodiments, the C5 binding motif is flanked by additional amino acids both N-terminally and C-terminally to the C5 binding motif. Therefore, in some embodiments there is provided a bispecific binding molecule as defined herein, wherein the C5 binding polypeptide in the first moiety comprises additional amino acids at the C- terminal and/or N-terminal end.
These additional amino acid residues may play a role in enhancing the binding of C5 by the binding domain, improving the conformational stability of the folded C5 binding domain, or other purposes, related for example to one or more of production, purification, or stabilization in vivo or in vitro. In some embodiments, there is provided a bispecific binding molecule as defined herein, wherein the C5 binding motif (BM) “forms part of” a three- helix bundle protein domain. This is understood to mean that the sequence of the BM is “inserted” into or “grafted” onto the sequence of the original three- helix bundle domain, such that the BM replaces a similar structural motif in the original domain. For example, without wishing to be bound by theory, the BM is thought to constitute two of the three helices of a three-helix bundle and can therefore replace such a two-helix motif within any three-helix bundle. As the skilled person will realize, the replacement of two helices of the three-helix bundle domain by the two BM helices has to be performed so as not to affect the basic structure of the polypeptide. That is, the overall folding of the Ca backbone of the polypeptide is substantially the same as that of the three- helix bundle protein domain of which it forms a part, e.g. having the same elements of secondary structure in the same order etc. Thus, a BM according to the present disclosure “forms part” of a three-helix bundle domain if the polypeptide according to this embodiment has the same fold as the original domain, implying that the basic structural properties are shared, those properties e.g. resulting in similar CD spectra. The skilled person is aware of other parameters that are relevant.
In particular embodiments, the C5 binding motif (BM) thus forms part of a three-helix bundle protein domain. For example, the BM may essentially constitute two alpha helices with an interconnecting loop, within said three- helix bundle protein domain. In particular embodiments, said three-helix bundle protein domain is selected from domains of bacterial receptor proteins. Non-limiting examples of such domains are the five different three-helical domains of Protein A from Staphylococcus aureus, such as domain B, and derivatives thereof. In some embodiments, the three-helical bundle protein domain is a variant of protein Z, which is derived from domain B of staphylococcal Protein A (Wahlberg E et al. 2003, PNAS 100(6):3185-3190).
In some embodiments, the bispecific binding molecule as defined herein, comprises an amino acid sequence selected from:
AEAKYAK-[BM] (SEQ ID NO:250);
AEAKFAK-[BM] (SEQ ID NO:251 ); ADNNFNK-[BM] (SEQ ID NO:252);
ADNKFNK-[BM] (SEQ ID NO:253);
VDNKFNK-[BM] (SEQ ID NO:254);
VDAKYAK-[BM] (SEQ ID NO:255); and sequences having at least 86% identity thereto, wherein [BM] is as defined herein.
In other embodiments, the bispecific binding molecule as defined herein, comprises an amino acid sequence selected from:
[BM]-DPSQSANLLSEAKKLNESQAPK (SEQ ID NO:256);
[BM]-DPSQSANLLAEAKKLNDAQAPK (SEQ ID NO:257);
[BM]-DPSVSKEILAEAKKLNDAQAPK (SEQ ID NO:258);
[BM]-DPSQSANLLAEAKKLNDAQAPK (SEQ ID NO:259);
[BM]-DPSESSELLSEAKKLNKSQAPK (SEQ ID NQ:260);
[BM]-DPSQSSELLAEAKKLNDAQAPK (SEQ ID NO:261 );
[BM]-DPSQSSELLAEAKKLNDSQAPK (SEQ ID NO:262);
[BM]-DPSQSSELLSEAKKLNDSQAPK (SEQ ID NO:263);
[BM]-DPSQSSELLSEAKKLSESQAPK (SEQ ID NO:264);
[BM]-DPSQSSELLSEAKKLESSQAPK (SEQ ID NO:265);
[BM]-DPSQSSELLAEAKKLNKAQAPK (SEQ ID NO:266); and sequences having at least 86% identity thereto, wherein [BM] is as defined herein.
In particular embodiments, the bispecific binding molecule as defined herein, comprises an amino acid sequence selected from:
ADNNFNK-[BM]-DPSQSANLLSEAKKLNESQAPK (SEQ ID NO:267);
ADNKFNK-[BM]-DPSQSANLLAEAKKLNDAQAPK (SEQ ID NO:268); ADNKFNK-[BM]-DPSVSKEILAEAKKLNDAQAPK (SEQ ID NO:269); VDNKFNK-[BM]-DPSQSANLLAEAKKLNDAQAPK (SEQ ID NQ:270); AEAKYAK-[BM]-DPSESSELLSEAKKLNKSQAPK (SEQ ID NO:271 );
VDAKYAK-[BM]-DPSQSSELLAEAKKLNDAQAPK (SEQ ID NO:272);
VDAKYAK-[BM]-DPSQSSELLAEAKKLNDSQAPK (SEQ ID NO:273);
AEAKYAK-[BM]-DPSQSSELLSEAKKLNDSQAPK (SEQ ID NO:274); AEAKYAK-[BM]-DPSQSSELLSEAKKLSESQAPK (SEQ ID NO:275); AEAKFAK-[BM]-DPSQSSELLSEAKKLSESQAPK (SEQ ID NO:276); AEAKYAK-[BM]-DPSQSSELLSEAKKLESSQAPK (SEQ ID NO:277); VDAKYAK-[BM]-DPSQSSELLSEAKKLNDSQAPK (SEQ ID NO:278); VDAKYAK-[BM]-DPSQSSELLSEAKKLSESQAPK (SEQ ID NO:279); VDAKYAK-[BM]-DPSQSSELLSEAKKLESSQAPK (SEQ ID NQ:280); VDAKYAK-[BM]-DPSQSSELLAEAKKLNKAQAPK (SEQ ID NO:281 ); AEAKYAK-[BM]-DPSQSSELLAEAKKLNKAQAPK (SEQ ID NO:282); and sequences having at least 86% identity thereto, wherein [BM] is as defined herein.
As discussed above, polypeptides comprising minor changes as compared to the above amino acid sequences without largely affecting the tertiary structure and the function thereof also fall within the scope of the present disclosure. Thus, in some embodiments, the C5 binding polypeptide may for example have a sequence which is at least 86%, such as at least 88 %, such as at least 90 %, such as at least 92 %, such as at least 94 %, such as at least 96 %, such as at least 98 % identical to a sequence as defined above.
The term “% identical” or “% identity”, as used in the specification and claims, is calculated as follows. The query sequence is aligned to the target sequence using the CLUSTAL W algorithm (Thompson, J.D., Higgins, D.G. and Gibson, T.J., Nucleic Acids Research, 22: 4673-4680 (1994)). A comparison is made over the window corresponding to the shortest of the aligned sequences. The shortest of the aligned sequences may in some instances be the target sequence, such as the albumin binding domain defined herein. In other instances, the query sequence may constitute the shortest of the aligned sequences. The query sequence may for example consist of at least 10 amino acid residues, such as at least 20 amino acid residues, such as at least 30 amino acid residues, such as at least 40 amino acid residues, such as at least 50 amino acids, such as at least 60 amino acids, for example 45 amino acid residues. The amino acid residues at each position are compared, and the percentage of positions in the query sequence that have identical correspondences in the target sequence is reported as % identity. In one embodiment of the bispecific binding molecule of this aspect of the disclosure, it comprises a C5 binding antibody or an antigen binding fragment thereof as first moiety. As is well known, antibodies are immunoglobulin molecules capable of specific binding to a target (an antigen), such as a carbohydrate, polynucleotide, lipid, polypeptide or other, through at least one antigen recognition site located in the variable region of the immunoglobulin molecule. As used herein, the term “antibody or an antigen binding fragment thereof” encompasses not only full-length or intact polyclonal or monoclonal antibodies, but also antigen binding fragments thereof, such as Fab fragments, Fab’ fragments, F(ab')2 fragments, single chain Fab (scFab) fragments, Fc fragments, Fv fragments, single chain Fv (scFv) fragments, (scFv)2, scFv-Fc constructs, domain antibodies, a chimeric antibody, a CDR-grafted antibody, a fully human antibody, a bivalent antibody construct, a synthetic antibody, a bivalent antibody, a trivalent antibody, a tetravalent antibody, bivalent single chain antibody, a trivalent single chain antibody, a multivalent single chain antibody; a microbody, a maxybody, an avimer, a small disulfide-bonded protein; and a binding protein derived from a scaffold selected from the group consisting of staphylococcal protein A and domains thereof, other three helix domains, lipocalins, ankyrin repeat domains, cellulose binding domains, y crystallines, green fluorescent protein, human cytotoxic T lymphocyte-associated antigen 4, protease inhibitors such as Kunitz domains, PDZ domains, SH3 domains, peptide aptamers, staphylococcal nuclease, tendamistats, fibronectin type III domain, transferrin, zinc fingers and conotoxins. Further examples of modified antibodies and antigen binding fragments thereof include nanobodies, AlbudAbs, DARTs (dual affinity re-targeting), BiTEs (bispecific T-cell engager), TandAbs (tandem diabodies), DAFs (dual acting Fab), two-in-one antibodies, SMIPs (small modular immunopharmaceuticals), FynomAbs (fynomers fused to antibodies), DVD-lgs (dual variable domain immunoglobulin), CovX-bodies (peptide modified antibodies), duobodies and triomAbs. This listing of variants of antibodies and antigen binding fragments thereof is not to be seen as limiting, and the skilled person is aware of other suitable variants. A full-length antibody comprises two heavy chains and two light chains. Each heavy chain contains a heavy chain variable region (VH) and first, second and third constant regions (CH1 , CH2 and CH3). Each light chain contains a light chain variable region (VL) and a light chain constant region (CL). Depending on the amino acid sequence of the constant domain of its heavy chains, antibodies are assigned to different classes. There are six major classes of antibodies: IgA, IgD, IgE, IgG, IgM and IgY, and several of these may be further divided into subclasses (isotypes), e.g., lgG1 , lgG2, lgG3, lgG-4, lgA1 and lgA2. The term “full-length antibody” as used herein refers to an antibody of any class, such as IgD, IgE, IgG, IgA, IgM or IgY (or any sub-class thereof). The subunit structures and three-dimensional configurations of different classes of antibodies are well known.
An “antigen binding fragment” is a portion or region of an antibody molecule, or a derivative thereof, that retains all or a significant part of the antigen binding of the corresponding full-length antibody. An antigen binding fragment may comprise the heavy chain variable region (VH), the light chain variable region (VL), or both. Each of the VH and VL typically contains three complementarity determining regions CDR1 , CDR2 and CDR3. The three CDRs in VH or VL are flanked by framework regions (FR1 , FR2, FR3 and FR4). As briefly listed above, examples of antigen binding fragments include, but are not limited to: (1) a Fab fragment, which is a monovalent fragment having a VL-CL chain and a VH-CH1 chain; (2) a Fab’ fragment, which is a Fab fragment with the heavy chain hinge region, (3) a F(ab')2 fragment, which is a dimer of Fab’ fragments joined by the heavy chain hinge region, for example linked by a disulfide bridge at the hinge region; (4) an Fc fragment; (5) an Fv fragment, which is the minimum antibody fragment having the VLand VH domains of a single arm of an antibody; (6) a single chain Fv (scFv) fragment, which is a single polypeptide chain in which the VH and VL domains of an scFv are linked by a peptide linker; (7) an (scFv)2, which comprises two VH domains and two VL domains, which are associated through the two VH domains via disulfide bridges and (8) domain antibodies, which can be antibody single variable domain (VH or VL) polypeptides that specifically bind antigens. Antigen binding fragments can be prepared via routine methods. For example, F(ab')2 fragments can be produced by pepsin digestion of a full- length antibody molecule, and Fab fragments can be generated by reducing the disulfide bridges of F(ab')2 fragments. Alternatively, fragments can be prepared via recombinant technology by expressing the heavy and light chain fragments in suitable host cells (e.g., E. coli, yeast, mammalian, plant or insect cells) and having them assembled to form the desired antigen-binding fragments either in vivo or in vitro. A single-chain antibody can be prepared via recombinant technology by linking a nucleotide sequence coding for a heavy chain variable region and a nucleotide sequence coding for a light chain variable region. For example, a flexible linker may be incorporated between the two variable regions. The skilled person is aware of methods for the preparation of both full-length antibodies and antigen binding fragments thereof.
Thus, in one embodiment, the present aspect of the disclosure provides a bispecific binding molecule, wherein said C5 binding polypeptide is selected from the group consisting of full-length antibodies, Fab fragments, Fab’ fragments, F(ab')2 fragments, single chain Fab (scFab) fragments, Fc fragments, Fv fragments, single chain Fv (scFv) fragments, (scFv)2, scFv-Fc constructs and domain antibodies.
In one embodiment, the C5 binding polypeptide is an antibody, in particular a monoclonal antibody. By “monoclonal antibody” is meant an antibody preparation consisting of a single antibody species, i.e. all antibodies in the preparation have the same amino acid sequences, including the same CDRs, and thus bind the same epitope on their target antigen (by “target antigen” is meant the antigen containing the epitope bound by a particular antibody) with the same effect. In other words, the antibody of the disclosure is preferably not part of a polyclonal mix of antibodies.
If the C5 binding polypeptide is an antibody, the antibody may be of any isotype and subtype. Thus it may be an IgA, IgD, IgE, IgG, or IgM antibody. The heavy-chain constant domains that correspond to the different isotypes of immunoglobulins are termed a, 5, E, y and p, respectively. The subunit structures and three-dimensional configurations of different isotypes of immunoglobulins are well known. Preferably the antibody is an IgG antibody. As noted above, there are four subtypes of IgG antibody: IgG 1 , lgG2, lgG3 and lgG4. The IgG anti-C5 antibody of the disclosure may be of any IgG subtype, i.e. it may be an lgG1 , lgG2, lgG3 or lgG4 antibody.
In one embodiment, the C5 binding polypeptide is a binding fragment of an antibody (i.e. an antibody fragment), that is a fragment which retains the ability of the antibody to bind specifically to its target antigen. Such fragments are well known and examples include Fab’, Fab, F(ab’)2, Fv, Fd, or dAb fragments, which may be prepared according to techniques well known in the art.
A Fab fragment consists of the antigen binding domain of an antibody, i.e. an individual antibody may be seen to contain two Fab fragments, each consisting of a light chain and its conjoined N-terminal section of a heavy chain. Thus a Fab fragment contains an entire light chain and the VH and CH1 domains of the heavy chain to which it is bound. Fab fragments may be obtained by digesting an antibody with papain.
A F(ab’)2 fragment consists of the two Fab fragments of an antibody, plus the hinge regions of the heavy domains, including the disulphide bonds linking the two heavy chains together. In other words, a F(ab’)2 fragment can be seen as two covalently joined Fab fragments. F(ab’)2 fragments may be obtained by digesting an antibody with pepsin. Reduction of F(ab’)2 fragments yields two Fab’ fragments, which can be seen as Fab fragments containing an additional sulfhydryl group which can be useful for conjugation of the fragment to other molecules.
In an alternative embodiment, the C5 binding polypeptide is a synthetic or artificial construct, i.e. an antibody-like molecule which comprises a binding domain, but which is genetically engineered or artificially constructed. Such constructs include chimeric or CDR-grafted antibodies, as well as single chain antibodies and other constructs, e.g. scFv, dsFv, ds-scFv, dimers, minibodies, diabodies, single domain antibodies (DABs), TandAbs dimers and heavy chain antibodies such as VHH, etc. In a particular embodiment, the C5 binding molecule is a single chain variable fragment (scFv). An scFv is a fusion protein in which a single polypeptide comprises both the VH and VL domains of an antibody. scFv fragments generally include a peptide linker covalently joining the VH and VL regions, which contributes to the stability of the molecule. The linker may comprise from 1 to 20 amino acids, such as for example 1 , 2, 3 or 4 amino acids, 5, 10 or 15 amino acids, or other intermediate numbers in the range 1 to 20 as convenient. As is well known to the person of skill in the art, the peptide linker may be formed from any generally convenient amino acid residues, such as glycine and/or serine. In a particular embodiment in which the C5 binding polypeptide in the bispecific binding molecule of the disclosure is an scFv, the linker is a (G4S)4 linker (SEQ ID NO:283). However, it is not essential that a linker be present, and the VL domain may instead be linked directly to the VH domain by a peptide bond. An scFv typically comprises, N-terminal to C-terminal, a VH region linked to a VL region by a linker sequence. In other words, in such an embodiment, the scFv comprises a peptide linker joining the C terminus of the heavy chain variable region (VH) to the N terminus of the light chain variable region (VL).
In a particular embodiment, there is provided a bispecific binding molecule as described herein, wherein said C5 binding polypeptide is an antibody fragment which is an scFv.
The skilled person will understand that various modifications and/or additions can be made to an C5 binding polypeptide as defined herein, to an antibody or antigen binding fragment thereof, or to the bispecific binding molecule as a whole as defined herein in order to tailor the bispecific binding molecule to a specific application without departing from the scope of the present disclosure.
Thus, in one embodiment there is provided a complex as defined herein, wherein said C5 binding polypeptide and/or said C5 binding antibody or antigen binding fragment thereof or said bispecific binding molecule as a whole comprises additional amino acids at least one C-terminal and/or N- terminal end. Such a complex should be understood as a complex having one or more additional amino acid residues at an N-terminal and/or C-terminal position in the polypeptide chain of the C5 binding polypeptide and/or of the C5 binding antibody or antigen binding fragment thereof. Alternatively, if the bispecific binding molecule is expressed as a fusion protein, said bispecific binding molecule should be understood as having one or more additional amino acid residues at the N-terminal and/or C-terminal position of the molecule as a whole. Thus, said bispecific binding molecule may comprise any suitable number of additional amino acid residues, for example at least one additional amino acid residue. Each additional amino acid residue may individually or collectively be added in order to, for example, improve production, purification, stabilization in vivo or in vitro, coupling or detection of the complex. Such additional amino acid residues may comprise one or more amino acid residues added for the purpose of chemical coupling. One example of this is the addition of a cysteine residue. Additional amino acid residues may also provide a ’’tag” for purification or detection of the polypeptide, such as a His6 tag, a (HisGlu)3 tag (“HEHEHE” tag) or a ”myc” (c-myc) tag or a ’’FLAG” tag for interaction with antibodies specific to the tag or immobilized metal affinity chromatography (IMAC) in the case of a His6- tag.
In a further embodiment, the first moiety of the bispecific binding molecule of the first aspect of the disclosure is a C5 binding polypeptide in multimeric form. Said multimer is understood to comprise at least two C5 binding polypeptides as defined herein as monomer units, the amino acid sequences of which may be the same or different. Multimeric forms of the C5 binding polypeptides may comprise a suitable number of domains, each having a C5 binding motif, and each forming a monomer within the multimer. These domains may have the same amino acid sequence, but alternatively, they may have different amino acid sequences. In other words, the C5 binding polypeptide may form homo- or heteromultimers, for example homo- or heterodimers. Therefore, in one embodiment there is provided a bispecific binding molecule as described herein, comprising at least two first moieties, wherein each first moiety each individually comprises at least one binding site for human complement component 5 (C5), wherein the amino acid sequences for the said binding sites may be the same or different. In another embodiment, said at least two first moieties are not coupled together. For the sake of clarity, throughout this disclosure, the term “C5 binding polypeptide” is used to encompass C5 binding polypeptides in all forms, i.e. monomeric and multimeric forms.
In one embodiment, there is provided a bispecific binding molecule as described herein and comprising a C5 binding polypeptide as first moiety, wherein said C5 binding polypeptide binds to C5 such that the KD value of the interaction is at most 1 x 10’6 M, such as at most 1 x 10’7 M, at most 1 x 10’8 M, or at most 1 x 10’9 M.
Second moiety, providing a binding site for IFNo
The bispecific binding molecule as defined herein separately targets IFNa in addition to being capable of binding C5. In one embodiment, there is provided a bispecific binding molecule as defined herein, wherein said at least one second moiety with at least one first binding site for interferon alpha (IFNa) is an IFNa binding polypeptide. This second moiety may exhibit any one or more of the general properties, features, characteristics and/or embodiments described above in connection with the first moiety, in any combination. Naturally, what is intended by the previous sentence is to say that the second moiety may exhibit any one or more of the general properties, features, characteristics and/or embodiments described above but in relation to the target of the second moiety, being IFNa, and not in relation to the target of the first moiety, being C5. For the sake of brevity, this information will not be repeated verbatim in connection with the second moiety, but is incorporated through this reference to the above disclosure.
In one embodiment, there is provided a bispecific binding molecule as defined herein comprising an IFNa binding polypeptide, wherein said IFNa binding polypeptide is selected from the group consisting of full-length antibodies, Fab fragments, Fab’ fragments, F(ab')2 fragments, single chain Fab (scFab) fragments, Fc fragments, Fv fragments, single chain Fv (scFv) fragments, (scFv)2, scFv-Fc constructs, domain antibodies, a chimeric antibody, a CDR-grafted antibody, a fully human antibody, a bivalent antibody construct, a synthetic antibody, a bivalent antibody, a trivalent antibody, a tetravalent antibody, bivalent single chain antibody, a trivalent single chain antibody, a multivalent single chain antibody, nanobodies, AlbudAbs, DARTs (dual affinity re-targeting), BiTEs (bispecific T-cell engager), TandAbs (tandem diabodies), DAFs (dual acting Fab), two-in-one antibodies, SMIPs (small modular immunopharmaceuticals), FynomAbs (fynomers fused to antibodies), DVD-lgs (dual variable domain immunoglobulin), CovX-bodies (peptide modified antibodies), duobodies and triomAbs; a microbody, a maxybody, an avimer, a small disulfide-bonded protein; and a binding protein derived from a scaffold selected from the group consisting of staphylococcal protein A and domains thereof, other three helix domains, lipocalins, ankyrin repeat domains, cellulose binding domains, y crystallines, green fluorescent protein, human cytotoxic T lymphocyte-associated antigen 4, protease inhibitors such as Kunitz domains, PDZ domains, SH3 domains, peptide aptamers, staphylococcal nuclease, tendamistats, fibronectin type III domain, transferrin, zinc fingers and conotoxins.
In another embodiment there is provided a bispecific binding molecule as defined herein, wherein said IFNa binding polypeptide is selected from the group consisting of full-length antibodies, Fab fragments, Fab’ fragments, F(ab')2 fragments, single chain Fab (scFab) fragments, Fc fragments, Fv fragments, single chain Fv (scFv) fragments, (scFv)2, scFv-Fc constructs and domain antibodies. In another embodiment, there is provided a bispecific binding molecule as defined herein, wherein said IFNa binding polypeptide is an IFNa binding antibody or antigen binding fragment thereof.
In any of the contemplated embodiments wherein said at least one second moiety comprises the binding domain of an antibody, the binding domain can be defined according the amino acid sequence comprising the complementarity determining domains (CDRs). Therefore, in an embodiment of the present aspect there is provided a bispecific binding molecule as defined herein comprising an IFNa binding polypeptide, wherein said IFNa binding polypeptide comprises a binding domain of an antibody, the binding domain comprising a heavy chain variable region and a light chain variable region, each comprising six complementarity determining domains (CDRs), wherein:
VLCDR1 has the sequence set forth in SEQ ID NO:286; VLCDR2 has the sequence set forth in SEQ ID NO:287; VLCDR3 has the sequence set forth in SEQ ID NO:288;
VHCDR1 has the sequence set forth in SEQ ID NO:289;
VHCDR2 has the sequence set forth in SEQ ID NQ:290; and
VHCRD3 has the sequence set forth in SEQ ID NO:291 .
In one embodiment, said antibody or antigen binding fragment thereof comprises a light chain variable region comprising the amino acid sequence SEQ ID NO:284, or an amino acid sequence having at least 80% sequence identity thereto. An another embodiment, there is provided a bispecific binding molecule as defined herein, wherein said antibody or antigen binding fragment thereof comprises a heavy chain variable region comprising the amino acid sequence SEQ ID NO:285, or an amino acid sequence having at least 80% sequence identity thereto.
In another embodiment, said antibody or antigen binding fragment thereof comprises a light chain variable region comprising the amino acid sequence SEQ ID NO:284, or an amino acid sequence having at least 80% sequence identity thereto, and a heavy chain variable region comprising the amino acid sequence SEQ ID NO:285, or an amino acid sequence having at least 80% sequence identity thereto.
In one embodiment, said IFNa binding antibody or antigen binding fragment thereof is rontalizumab or a variant thereof. For example, said IFNa binding antibody or antigen binding fragment thereof may comprise a light chain comprising the amino acid sequence SEQ ID NO:293 or a sequence having at least 80% identity thereto, and/or a heavy chain comprising the amino acid sequence SEQ ID NO:294 or a sequence having at least 80% identity thereto. In a specific embodiment, said IFNa binding antibody or antigen binding fragment thereof is rontalizumab, i.e. an antibody comprising a light chain comprising the amino acid sequence SEQ ID NO:293 and a heavy chain comprising the amino acid sequence SEQ ID NO:294.
In any of the contemplated embodiments wherein said at least one second moiety comprises an IFNa binding antibody or antigen binding fragment thereof, the CH2 region may comprise the amino acid substitution N297A. The N297A amino acid substitution is introduced into the design to abrogate FcyR interactions, mitigating unwanted effector functions. Therefore, in one embodiment, there is provided a bispecific binding molecule as defined herein, wherein said IFNa binding polypeptide is an IFNa binding antibody or antigen binding fragment thereof, comprising an amino acid substitution N297A in the CH2 region of the antibody or antibody fragment thereof.
The bispecific molecule as described herein may for example be present in the form of a fusion protein or a protein conjugate, thus, said at least one first moiety with at least one binding site for human complement component 5 (C5) as defined herein, is operably linked to at least one second moiety with at least one first binding site for interferon alpha (IFNa) as defined herein. Therefore, in an embodiment there is provided a bispecific binding molecule, comprising at least one first moiety with at least one first binding site for human complement component 5 (C5) as defined herein, operably linked to at least one second moiety with at least one first binding site for interferon alpha (IFNa) as defined herein. Furthermore, said at least one first moiety and said at least one second moiety may be coupled by means of chemical conjugation (using known organic chemistry methods) or by any other means, such as expression of the complex as a fusion protein or joined in any other fashion, either directly or via a linker, for example an amino acid linker.
Thus in one embodiment, there is provided a bispecific binding molecule as defined herein, wherein said bispecific binding molecule is a fusion protein or a protein conjugate. In one embodiment, said bispecific binding molecule is a fusion protein. In another embodiment, said bispecific binding molecule is a protein conjugate. In one embodiment of said bispecific binding molecule, said at least one first moiety as defined herein is attached to the N-terminus or C-terminus of the heavy chain of said at least one second moiety which is an antibody or antigen binding fragment thereof. In another embodiment, said at least one first moiety as defined herein is attached to the N-terminus or C-terminus of the light chain of said at least one second moiety, which is an antibody or antigen binding fragment thereof. In one embodiment, said at least one first moiety as defined herein is attached to the N-terminus and/or C-terminus of the light chain and heavy chain of said at least one second moiety, which is an antibody or antigen binding fragment thereof. For example, the at least one first moiety may be attached to only the N-terminus of the heavy chain(s), only the N-terminus of the light chain(s), only the C-terminus of the heavy chain(s), only the C-terminus of the light chain(s), both the N-terminus and the C-terminus of the heavy chain(s), both the N-terminus and the C-terminus of the light chain(s), only the C-terminus of the light chain(s) and the N-terminus of the heavy chain(s), only the C- terminus of the heavy chain(s) and the N-terminus of the light chain(s), of said at least one second moiety, which is an antibody or antigen binding fragment thereof.
In a further embodiment, there is provided a bispecific binding molecule as defined herein, wherein said at least one first moiety and said at least one second moiety are covalently linked. In another embodiment, there is provided a bispecific binding molecule as defined herein, wherein said at least one first moiety is covalently linked to: i) a CH3 domain of the heavy chain of an antibody or antibody fragment thereof as defined herein; ii) a CL domain of the light chain of an antibody or antibody fragment thereof as defined herein; iii) a VL domain of the light chain of an antibody or antibody fragment thereof as defined herein; or iv) a VH domain of the heavy chain of an antibody or antibody fragment thereof as defined herein.
In another embodiment there is provided a bispecific binding molecule as defined herein, wherein the bispecific molecule comprises
- one antibody or antibody fragment thereof as second moiety, and
- two first moieties, and: i) one first moiety is covalently linked to a CH3 domain of each heavy chain of said antibody or antibody fragment thereof; ii) one first moiety is covalently linked to a CL domain of each light chain of said antibody or antibody fragment thereof; iii) one first moiety is covalently linked to a VH domain of each heavy chain of said antibody or antibody fragment thereof; or iv) one first moiety is covalently linked to a VL domain of each light chain of said antibody or antibody fragment thereof.
The skilled person is aware that the construction of a fusion protein often involves the use of linkers between the functional moieties to be fused, and there are different kinds of linkers with different properties, such as flexible amino acid linkers, rigid amino acid linkers and cleavable amino acid linkers. Linkers have been used to for example increase stability or improve folding of fusion proteins, to increase expression, improve biological activity, enable targeting and alter pharmacokinetics of fusion proteins. Thus, in one embodiment, said bispecific binding molecule further comprises at least one linker, such as at least one linker selected from flexible amino acid linkers, rigid amino acid linkers and cleavable amino acid linkers. In one embodiment, said linker is arranged between said first moiety as defined herein and said second moiety as defined herein. Therein, in another embodiment, there is provided a bispecific binding molecule as defined herein, wherein said at least one first moiety is operably bound to said at least one second moiety via at least one linker, such as at least one linker selected from flexible amino acid linkers, rigid amino acid linkers and cleavable amino acid linkers.
Flexible linkers are often used in the art when the joined domains require a certain degree of movement or interaction, and may be particularly useful in some embodiments of the complex. Such linkers are generally composed of small, non-polar (for example G) or polar (for example S or T) amino acids. Some flexible linkers primarily consist of stretches of G and S residues, for example (GGGGS)p. Adjusting the copy number “p” allows for optimization of linker in order to achieve appropriate separation between the functional moieties or to maintain necessary inter-moiety interaction. Apart from G and S linkers, other flexible linkers are known in the art, such as G and S linkers containing additional amino acid residues, such as T and A, to maintain flexibility, as well as polar amino acid residues to improve solubility.
Thus in one embodiment, said linker is a flexible linker comprising glycine (G), serine (S) and/or threonine (T) residues. In one embodiment, said linker has a general formula selected from (GnSm)p and (SmGn)p, wherein, independently, n = 1-7, m = 0-7, n + m < 8 and p = 1-7. In one embodiment, n = 1-5. In one embodiment, m = 0-5. In one embodiment, p = 1-5. In a more specific embodiment, n = 4, m = 1 and p = 1-4. In an even more specific embodiment, said linker is (GGGGS)3 (SEQ ID NO:303). In another specific embodiment, said linker is GGGGS (SEQ ID NO:304). In another specific embodiment, said linker is VDGS (SEQ ID NQ:305). In another specific embodiment, said linker is ASGS (SEQ ID NQ:306). In a preferred embodiment, said flexible amino acid linker comprises the amino acid sequence SEQ ID NO:292.
In specific embodiments of a bispecific binding molecule of the first aspect, it comprises a pair of polypeptide chains, said pair being selected from the group consisting of the pairs
SEQ ID NO:295 and SEQ ID NO:296, SEQ ID NO:297 and SEQ ID NO:298, SEQ ID NO:299 and SEQ ID NQ:300, and SEQ ID NQ:301 and SEQ ID NQ:302.
In a preferred embodiment, there is provided a bispecific binding molecule as defined herein, comprising the pair of polypeptide chains represented by SEQ ID NO:295 and SEQ ID NO:296.
In a further aspect of the present disclosure, there is provided a polynucleotide encoding at least one polypeptide forming part of a bispecific binding molecule according to the first aspect. In an embodiment of this aspect, there is provided at least one polynucleotide, such as at least two polynucleotides, each polynucleotide independently encoding any one or more of the sub-components or polypeptide parts of said bispecific binding molecule, e.g.
- one heavy chain component of a second moiety antibody fused to one first moiety and one light chain component of a second moiety antibody, or
- one light chain component of a second moiety antibody fused to one first moiety and one heavy chain component of a second moiety antibody.
In another aspect of the present disclosure, there is provided an expression vector comprising said polynucleotide. In an embodiment of this aspect, there is provided at least one vector, such as at least two vectors, comprising said at least one polypeptide(s).
In another aspect of the present disclosure, there is provided a host cell comprising said expression vector. In an embodiment of this aspect, there is provided a host cell comprising said at least one vector, such as comprising said at least two vectors.
Also encompassed by this disclosure, in another aspect, is a method of producing a bispecific binding molecule as described above, comprising culturing said host cell under conditions permissive of expression of said polypeptide from its expression vector(s), and isolating the bispecific binding molecule. In an embodiment of this aspect, there is provided a method of producing a bispecific binding molecule as described above or any one or more of the sub-components or polypeptide parts of said bispecific binding molecule, comprising culturing said host cell under conditions permissive of expression of said polypeptides from the at least one expression vector(s), and isolating the said sub-components or polypeptide parts of said bispecific binding molecule. By “any one or more of the sub-components or polypeptide parts of said bispecific binding molecule” it is meant any one or more of the sub-components or polypeptide parts which comprise the bispecific binding molecule, such as at least one heavy chain component of a first moiety, such as at least one light chain component of a first moiety, such as at least one heavy chain component of a second moiety, such as at least one light chain component of a second moiety.
The bispecific binding molecule of the present disclosure, or any one or more of its sub-component, polypeptide parts, may alternatively be produced by non-biological peptide synthesis. Therefore, in another embodiment there is provided a method of producing a bispecific binding molecule as defined herein by non-biological peptide synthesis using amino acids and/or amino acid derivatives having protected reactive side-chains, the non-biological peptide synthesis comprising
- step-wise coupling of the amino acids and/or the amino acid derivatives to form a derivatives to form a bispecific binding molecule as described herein in the form of a polypeptide or fusion protein having protected reactive sidechains,
- removal of the protecting groups from the reactive side-chains of the polypeptide or fusion protein, and
- folding of the polypeptide or fusion protein in aqueous solution.
Said bispecific binding molecule may also be produced by the conjugation of at least one first moiety to at least one second moiety thereof as described herein. The skilled person is aware of conjugation methods known in the art, such as conventional chemical conjugation methods for example using charged succinimidyl esters or carbodiimides.
In another aspect, there is provided a composition comprising a bispecific binding molecule as described herein and at least one pharmaceutically acceptable excipient or carrier. In one embodiment, said composition further comprises at least one additional active agent, such as at least two additional active agents, such as at least three additional active agents. Non-limiting examples of additional active agents that may prove useful in such a composition are immune response modifying agents.
Non limiting examples of immune response modifying agents that can be used as additional active agent in embodiments of the composition according to this aspect include immunosuppressive and immunomodulating agents, and other anti-inflammatory agents. For example, the bispecific binding molecule as described herein may be used in combination with an agent selected from the group consisting of disease-modifying antirheumatic drugs (DMARDs), such as gold salts, azathioprine, methotrexate and leflunomide; calcineurin inhibitors, such as cyclosporin A or FK 506; modulators of lymphocyte recirculation; mTOR inhibitors, such as rapamycin; an ascomycin having immuno-suppressive properties; glucocorticoids; corticosteroids; cyclophosphamide; immunosuppressive monoclonal antibodies; adhesion molecule inhibitors, such as LFA-1 antagonists, ICAM-1 or -3 antagonists, VCAM-4 antagonists or VLA-4 antagonists; blockers of proinflam matory cytokines; IL-1 blockers such as anakinra or IL-1 trap; IL-17 blockers; chemokine blockers; non-steroidal anti-inflammatory drugs (NSAIDs) such as aspirin; and anti-infectious agents and other immune response modulating agents, as well as combinations thereof.
The skilled person will appreciate that the bispecific binding molecule as defined herein or a composition comprising said bispecific binding molecule may be administered to a subject using standard administration techniques, such as including oral, respiratory, topical, intravenous, intraperitoneal, subcutaneous, pulmonary, transdermal, intramuscular, intranasal, buccal, sublingual or suppository administration. Thus, in one embodiment there is provided a bispecific binding molecule as defined herein or a composition as defined herein for oral, respiratory, topical, intravenous, intraperitoneal, subcutaneous, pulmonary, transdermal, intramuscular, intranasal, buccal, sublingual or suppository administration, such as for subcutaneous administration, such as for intravenous administration.
Hence, in another aspect of the present disclosure, there is provided a bispecific binding molecule or composition as described herein for use as a medicament. In one embodiment, said bispecific binding molecule or composition modulates C5 function and IFNa function. In one embodiment, there is provided a bispecific binding molecule or a composition as defined herein, for use as a medicament to modulate C5 and IFNa function in vivo. As used herein, the term “modulate” refers to changing the activity, such as rendering C5 function and the function of IFNa, partially inhibiting or fully inhibiting C5 function and the function of IFNa. By “partially or fully inhibiting C5 function” it is meant that cleavage of C5 into C5a and C5b is inhibited. By “partially or fully inhibiting IFNa function” it is meant that IFNa interaction with interferon alpha receptor (IFNaR) is partially or fully inhibited. Thus, in another embodiment there is provided a bispecific binding molecule or a composition as defined herein, for use as a medicament to modulate C5 and IFNa function in vivo wherein said at least one first moiety inhibits the cleavage of C5 into C5a and C5b and said at least one second moiety inhibits IFNa interaction with INFaR.
In one embodiment, there is provided a bispecific binding molecule or composition as described herein for use in the treatment of a disease or disorder related to C5 and IFNa. In another embodiment, there is provided a bispecific binding molecule or composition as described herein for use as described herein, wherein said disease or disorder related to C5 and IFNa is selected from inflammatory and autoimmune diseases. In another embodiment, there is provided a bispecific binding molecule or composition as described herein for use as described herein, wherein said disease or disorder related to C5 and IFNa is selected from cardiovascular diseases. In another embodiment, there is provided a bispecific binding molecule or composition as described herein for use as described herein, wherein said disease or disorder related to C5 and IFNa is selected from a CNS disorder, disease and injury. As used herein, the term “C5 and IFNa related disorder” refers to any disorder, disease or condition in which C5 and IFNa play a regulatory role in the signaling pathway. A non-limiting list of C5 and IFNa related diseases or disorders, for the treatment of which the bispecific binding molecule or composition as described herein may be useful, include inflammatory diseases and autoimmune diseases, such as rheumatoid arthritis (RA), dermatomyositis, primary Sjogren’s syndrome, systemic sclerosis, psoriasis and type I interferonopathies, such as Aicardi-Goutieres syndrome, familial Chilblain lupus, systemic lupus erythematosus (SLE), lupus nephritis, in particular cohorts with patients displaying signs of activated complement system, cardiovascular disease, such as atherosclerosis, and central nervous system (CNS) disorders, disease and injury such as traumatic brain injury (TBI), spinal cord injury (SCI), stroke, such as subarachnoid hemorrhage (SAH), amyotrophic lateral sclerosis (ALS), neuromyelitis optica, and Alzheimer’s disease. Thus, in one embodiment, said disorder or disease related to C5 and IFNa is selected from the group consisting of rheumatoid arthritis (RA), dermatomyositis, primary Sjogren’s syndrome, systemic sclerosis, psoriasis and type I interferonopathies, such as Aicardi-Goutieres syndrome, familial Chilblain lupus, systemic lupus erythematosus (SLE), lupus nephritis, atherosclerosis, traumatic brain injury (TBI), spinal cord injury (SCI), stroke, such as subarachnoid hemorrhage (SAH), amyotrophic lateral sclerosis (ALS), neuromyelitis optica, and Alzheimer’s disease. In one embodiment, the disorder or disease is selected from the group consisting of systemic lupus erythematosus (SLE), lupus nephritis and rheumatoid arthritis (RA). In one embodiment, the disorder or disease is selected from the group consisting of traumatic brain injury (TBI), spinal cord injury (SCI), stroke, such as subarachnoid hemorrhage (SAH), amyotrophic lateral sclerosis (ALS), neuromyelitis optica, and Alzheimer’s disease. In a specific embodiment, said disorder or disease related to C5 and IFNa is systemic lupus erythematosus (SLE). In another specific embodiment, said disorder or disease related to C5 and IFNa is lupus nephritis. In another specific embodiment, said disorder or disease related to C5 and IFNa is rheumatoid arthritis (RA). In another specific embodiment, said disorder or disease related to C5 and IFNa is atherosclerosis. In another specific embodiment, said disorder or disease related to C5 and IFNa is traumatic brain injury (TBI). In another specific embodiment, said disorder or disease related to C5 and IFNa is spinal cord injury (SCI). In another specific embodiment, said disorder or disease related to C5 and IFNa is subarachnoid hemorrhage (SAH). In another specific embodiment, said disorder or disease related to C5 and IFNa is amyotrophic lateral sclerosis (ALS). In another specific embodiment, said disorder or disease related to C5 and IFNa is neuromyelitis optica. In another specific embodiment, said disorder or disease related to C5 and IFNa is Alzheimer’s disease. In a further embodiment, said bispecific binding molecule or composition alleviates the inflammatory profile in SLE, lupus nephritis and/or rheumatoid arthritis (RA). In another further embodiment, said bispecific binding molecule or composition alleviates the inflammatory profile in traumatic brain injury (TBI), spinal cord injury (SCI), stroke, such as subarachnoid hemorrhage (SAH), amyotrophic lateral sclerosis (ALS), neuromyelitis optica, and/or Alzheimer’s disease. In another further embodiment, said bispecific binding molecule or composition alleviates the inflammatory profile in atherosclerosis. In a further embodiment, administration is selected from the group consisting of: oral, topical, intravenous, intraperitoneal, subcutaneous, pulmonary, transdermal, intramuscular, intranasal, buccal, sublingual or suppository administration, such as subcutaneous administration, such as intravenous administration.
In a related aspect, there is provided a method of treatment of disorder or disease related to C5 and IFNa, comprising administering to a subject in need thereof an effective amount of a bispecific binding molecule or composition as described herein. In a more specific embodiment of said method, the complex or composition as described herein modulates C5 and IFNa function in vivo. It may be beneficial to administer a therapeutically effective amount of a bispecific binding molecule as defined herein or composition as defined herein and at least one second drug substance, such as an immune response modulating agent as described above.
As used herein, the term “co-administration” encompasses both concomitant and sequential administration. Thus, in one embodiment there is provided a method as defined above further comprising co-administration of an immune response modulating agent as described above.
In one embodiment there is provided a method of treatment as defined herein, wherein said disorder or disease related to C5 and IFNa is selected from inflammatory and autoimmune diseases. In another embodiment, said disorder or disease related to C5 and IFNa is selected from the group consisting of rheumatoid arthritis (RA), dermatomyositis, primary Sjogren’s syndrome, systemic sclerosis, psoriasis and type I interferonopathies, such as Aicardi-Goutieres syndrome, familial Chilblain lupus, systemic lupus erythematosus (SLE) and lupus nephritis. In one embodiment, the disorder or disease is selected from the group consisting of systemic lupus erythematosus (SLE) and lupus nephritis. In a specific embodiment, said disorder or disease related to C5 and IFNa is systemic lupus erythematosus (SLE). In another specific embodiment, said disorder or disease related to C5 and IFNa is lupus nephritis. In one embodiment of such a method, said subject is a mammalian subject, such as a human subject. In another embodiment of such a method, said subject is a human subject.
In one embodiment there is provided a method of treatment as defined herein, wherein said disorder or disease related to C5 and IFNa is selected from central nervous system (CNS) diseases, disorders and injury. In another embodiment, said disorder or disease related to C5 and IFNa is selected from the group consisting of traumatic brain injury (TBI), spinal cord injury (SCI), stroke, such as subarachnoid hemorrhage (SAH), amyotrophic lateral sclerosis (ALS), neuromyelitis optica, and/or Alzheimer’s disease. In a specific embodiment, said disorder or disease related to C5 and IFNa is traumatic brain injury (TBI). In another specific embodiment, said disorder or disease related to C5 and IFNa is spinal cord injury (SCI). In another specific embodiment, said disorder or disease related to C5 and IFNa is stroke, such as subarachnoid hemorrhage (SAH). In another specific embodiment, said disorder or disease related to C5 and IFNa is amyotrophic lateral sclerosis (ALS). In another specific embodiment, said disorder or disease related to C5 and IFNa is neuromyelitis optica. In another specific embodiment, said disorder or disease related to C5 and IFNa is Alzheimer’s disease. In one embodiment of such a method, said subject is a mammalian subject, such as a human subject. In another embodiment of such a method, said subject is a human subject.
While the invention has been described with reference to various exemplary aspects and embodiments, it will be understood by those skilled in the art that various changes may be made and equivalents may be substituted for elements thereof without departing from the scope of the invention. Therefore, it is intended that the invention not be limited to any particular embodiment contemplated, but that the invention will include all embodiments falling within the scope of the appended claims. Other features and advantages of the instant invention will be apparent from the following detailed description and examples, which should not be construed as limiting. The contents of all references, patents and published patent applications cited throughout this application are expressly incorporated herein by reference.
References
Almaani, Meara and Rovin, Clin. J. Am. Soc. Nephrol. 12:825-835, 2017.
Asgari N, Interferon Alpha Association with Neuromyelitis Optica, Clin Dev Immunol. 2013; 2013: 713519.
Brendan SMH et al, The Role of Interferon- in Neurodegenerative Diseases: A Systematic Review, Journal of Alzheimer’s Disease 94 (2023) S45-S66. Chen HJ, Tas SW, de Winther MPJ. “Type-I interferons in atherosclerosis.", J Exp Med. 2020 Jan 6;217(1 ):e20190459.
Ganguly et al, J. Exp. Med. 206:1983-1994, 2009.
Geng CK et al, Effect of mesenchymal stem cells transplantation combining with hyperbaric oxygen therapy on rehabilitation of rat spinal cord injury, Asian Pacific Journal of Tropical Medicine 2015; 8(6): 468-473.
Hammad et al. The role of the complement system in traumatic brain injury: a review, Journal of Neuroinflammation (2018) 15:24.
Jego et al, Immunity. 19:225-234, 2003.
Kaul et al, Nat. Rev. Dis. Prim. 2:16039, 2016.
Kjeldgaard AL et al, Complement Profiles in Patients with Amyotrophic Lateral Sclerosis: A Prospective Observational Cohort Stud, Journal of Inflammation Research 2021 :14 1043-1053.
Koopman I. et al, Safety and pharmacodynamic efficacy of eculizumab in aneurysmal subarachnoid hemorrhage (CLASH): A phase 2a randomized clinical trial, European Stroke Journal 2023, Vol. 8(4) 1097-1106.
Koopman I. et al. Complement C5 Antibodies for decreasing brain injury after aneurysmal Subarachnoid Haemorrhage (CLASH): study protocol for a randomised controlled phase II clinical trial, Trials (2020) 21 :969.
Leffler et al, Ann. Rheum. Dis., 73:1601-1606, 2014.
Li L, Xiong Z-Y, Qian ZM, Zhao T-Z, Feng H, Hu S, et al. Complement C5a is detrimental to histological and functional locomotor recovery after spinal cord injury in mice. Neurobiol Dis. (2014) 66:74-82. doi:10.1016/j.nbd.2014.02.008
Lin CHM et al, Role of IFN-a in Rheumatoid Arthritis, Curr Rheumatol Rep (2024) 26:37-52.
Luo C, Clinical Value of Inflammatory Cytokines in Patients with Aneurysmal Subarachnoid Hemorrhage, Clinical Interventions in Aging 2022:17 615-626.
Ldfblom et al. (2010), FEBS Letters, 584:2670-2680.
MG Kiss, CJ Binder., “The multifaceted impact of complement on atherosclerosis.”, Atherosclerosis, 2022 Jun:351 :29-40.
Niewold et al, J. Biomed. Biotechnol. 2010:948364, 2010.
Nord et al. (1995), Prot Eng 8:601-608. Obermoser and Pascual, Lupus. 19:1012-1019, 2010.
Oon, Wilson and Wicks, Clin. Transl. Immunol. 5:29, 2016.
Pang et al, Immunobiology 219(12):980-989, 2014.
Park et al, Blood Adv 2(16):2090-2094, 2018.
Pittock SJ, Lennon VA, McKeon A, Mandrekar J, Weinshenker BG, Lucchinetti CF, et al. Eculizumab in AQP4-lgG-positive relapsing neuromyelitis optica spectrum disorders: an open-label pilot study. Lancet Neurol. (2013) 12:554-62. doi: 10.1016/S 1474-4422 (13)70076-0
Roselli F, Chandrasekar A and Morganti-Kossmann MC (2018) Interferons in Traumatic Brain and Spinal Cord Injury: Current Evidence for Translational Application. Front. Neurol. 9:458. doi: 10.3389/fneur.2018.00458
Rdnnblom and Leonard, Lupus Sci. Med. 6:e000270, 2019.
Sanford SAI and McEwan WA (2022) Type-I Interferons in Alzheimer’s Disease and Other Tauopathies. Front. Cell. Neurosci. 16:949340.
Triggianese P, Conigliaro P, De Martino E, Monosi B, Chimenti MS .
Overview on the Link Between the Complement System and Auto-Immune Articular and Pulmonary Disease. Rheumatology: Research and Reviews 2023:15 65-79. van Dijk BJ et al, Complement C5 contributes to brain injury after subarachnoid hemorrhage. Transl Stroke Res 2020; 11 : 678-688.
Zindler E et al, Neuronal injury in chronic CNS inflammation, Best Pract Res Clin Anaesthesiol, 2010 Dec;24(4):551-62.
Brief description of the figures
Figure 1 shows AffiMab primary developability screening: A) schematic illustration of AffiMab constructs; B) developability data of AffiMabs based on titers in small scale production cultures of 2.5 mL vs purified and SEC analyzed proteins for monomeric content shown in percentage of total detected peaks in apprehended chromatograms; C) simultaneous binding to IFNa and C5 by the AffiMabs, as evaluated with BLI. Data in (B) is presented as mean ± SD of three replicates. Figure 2 illustrates SDS-PAGE post purification of purified AffiMab constructs and the naked antibody rontalizumab serving as a benchmark.
Figure 3 shows the results of evaluation of C5 cleavage inhibition by AffiMabs in a whole blood hemolysis assay. Data is presented as mean ± SD of two replicates.
Figure 4 shows the results of evaluation of inhibition of IFNa by AffiMab constructs in an in vitro reporter assay, showing A) neutralization of recombinant IFNa2b signaling by AffiMabs; B) neutralization of IFNa2b signaling in presence of excess C5 (100 nM). Data in (B-C) are presented as mean ± SD of two replicates.
Figure 5 shows the result of C5a ELISA on plasma samples post AffiMab treatment in whole blood ex vivo assay, with untreated (UT), Healthy control untreated blood (UT HC), AffiMab and control constructs treated blood as indicated. Data are presented as mean ± SD of two technical replicates Comparisons between treatments analyzed by two-tailed paired t-test, statistical significance *p<0.05.
Figure 6 shows the results of evaluation of cytokine and chemokine profile in plasma after AffiMab or mAb (rontalizumab) treatment in whole blood ex vivo assay with donor blood by OLINK PLA assay. Data are presented as mean ± SD of two technical replicates. Comparisons between treatments for each protein analyzed by two-tailed paired t-test, statistical significance P<0.05.
Examples
Summary
The following Examples disclose the development of novel bispecific binding molecules, i.e. bispecific binding molecules comprising at least one first moiety with at least one binding site for human complement component 5 (C5) operably linked to at least one second moiety with at least one binding site for interferon alpha (IFNa). These novel bispecific binding molecules are also referred to as IFNa-C5 targeting “AffiMab” constructs. The Examples further describe the characterization of these selected bispecific binding molecules, i.e. “AffiMabs”, and demonstrate their in vitro and ex vivo functionality.
Example 1
Design, cloning, production and purification of IFNa-C5 targeting AffiMab constructs
AffiMabs were designed and constructed by recombinant fusion of a gene encoding a C5 binding Z variant to the N- or C-terminus of the heavy chain or light chain of the IFNo binding mAb rontalizumab via a 15-amino-acid glycine-serine linker (SS(G)4S(G)eSS). In total, eight AffiMab constructs were constructed divided in two IgG subclass scaffolds (lgG1 and lgG4) with analogous fusion points as described above. Designed and synthesized heavy and light chain for respective AffiMab construct were cloned into the inhouse bicistronic vector framework pKTH 17 (Volk et al, Drugs R D 21 : 157- 168, 2021 ) via restriction cloning. Quality of the constructs was assessed by SDS-PAGE and size exclusion chromatography (SEC), and target binding analysis was assessed by biolayer interferometry (BLI).
Materials and methods
Design: Constructs were designed by recombinantly fusing the C5 binding Z variant zC5 (SEQ ID NO:307) to either chain and termini end of the antibody architecture (Table 1 , Figure 1A). The S2G4SG6S2 flexible linker (SEQ ID NO:292) was utilized in all constructs for the recombinant production. Furthermore, a previously described mutation to the Fc region to abrogate Fc mediated effector functions of lgG1 s was introduced to the design (N297A, disruption of vital glycan in CH2 needed for FcgR engagement). The heavy chain and light chain of the AffiMabs were cloned by conventional restriction cloning into a dual-expression cassette vector pKTH17 (Volk et al, 2021 , supra), for encoding both the heavy and light chain in one vector.
Transfection: ExpiCHO™ cells (ThermoFisher Scientifics Inc. Waltham, MA, USA) were transfected as suggested by the manufacturer (ExpiCHO Expression System User Guide, Pub. No. MAN0014337 Rev. C.0) with a total of 20 pg plasmid DNA per transfection of respective construct in 25 mL culture volumes. The cells were cultured for 8 days prior to harvest.
IgG quantification: IgG concentrations in the supernatant were determined by biolayer interferometry measurements in an Octet® RED96e system (Fortebio Biologies by Molecular Devices, USA) with Dip and Read™ Protein A biosensors (Fortebio Biologies by Molecular Devices) according to the manufacturer's instructions. The supernatant samples from day 5 (culture continued for total of 8 days) were diluted 1 :1 in 20 mM citric acid pH 4.0, 0.1 % BSA (w/v), 0.1 % Tween-20, 0.5 M NaCI. A standard curve was prepared from an IgG with the respective concentrations from 700 to 1 pg/mL.
IgG purification: For size exclusion chromatography analysis, the expressed AffiMabs were purified by Protein A facilitated purification on an AktaSTART system (GE Healthcare, USA) using mAbSelect SuRe columns (GE Healthcare). A 20 mM sodium phosphate, 0.15 M sodium chloride (pH 7.3) buffer was used as binding and wash buffer, 0.1 M glycine (pH 2.5) as elution buffer and 1 M Tris-HCI (pH 8.5) as neutralization buffer. Endotoxin levels were measured with LAL Cartridges and Endosafe Nextgen-PTS system (Charles River, MA, USA) according to manufacturer’s instructions.
SDS-PAGE analysis: Total of 4 pg of the purified AffiMab samples were mixed with 3x loading buffer (0.1 M Tris-HCI, 45% glycerol, 0.03% bromophenol blue, 0.3% SDS) for non-reducing conditions and for the reducing analysis mixed with 3x loading buffer containing 0.15 M Tris 2- carboxyethyl-phosphine hydrochloride and incubation at 95°C for 7 min. The samples were run on a 4-20% Criterion™ TGX Stain-Free™ protein gel (BioRad Laboratories) according to the company's protocol. The bands were visualized by staining the gel in GelCode™ Blue Safe protein stain (Thermo Fisher Scientific) for 1 h at room temperature and gentle shaking.
Size exclusion chromatography: In total, 25 pg of each AffiMab in 100 pl were injected onto a Superdex Increase 200 10/30 GL gel filtration column (GE Healthcare) coupled to an Agilent 1200 series HPLC system (Agilent Technologies, USA). SEC runs were performed at a 0.5 mL/min flow rate with PBS as a running buffer. Eluted protein fragments were detected by an online 280 nm absorption measurement. Data analysis and peak integrations were performed using GraphPad prism 8.0 (GraphPad Software, USA).
Biolayer interferometry: To screen and validate AffiMab constructs’ functional simultaneous binding to both targets, constructs were screened using the Octet RED96 system (Sartorious, Gottingen, Germany). Biotinylated IFNa2b was immobilized to streptavidin coated octet probes (Sartorius, Gottingen, Germany). Different concentrations of each AffiMab construct (100 nM, 50 nM and 25 nM) were then subsequently allowed to 1) associate to the respective target for 300 s, 2) stabilize association in PBS for 30 s, 3) associate to the second unbiotinylated target (100 nM) for 300 s, and 4) stabilize and dissociate in PBS for 100 s. Sensorgrams were obtained for each concentration and analyzed using GraphPad prism 8.0 (GraphPad Software, USA).
Surface plasmon resonance (SPR): AffiMab lead candidate (RONT_HC15_zC5; comprising SEQ ID NO:295 and SEQ ID NO:296) or rontalizumab (R0NT_WT; comprising SEQ ID NO:293 and SEQ ID NO:294) was immobilized on CM5 chips (Cytiva, Sweden) by amine coupling. Following immobilization, each analyte (C5 or IFNa) was injected in a serial dilution (diluted in PBSB 1 %) ranging from 50 nM to 3.125 nM in a dilution series of 5 concentration and dilution factor of 2. Sensorgrams were analyzed using GraphPad prism 8.0 (GraphPad Software, USA) for analysis of IFNa binding and B devaluation software (Cytiva, Sweden) for analysis of C5 binding. Results are shown in Table 3.
Results
Developability comparisons of AffiMab variants revealed production variabilities and favorable candidates. Comparison of measured titers 5 days post transfection in ExpiCHO cells and the monomeric content post purification analyzed by SEC demonstrated a variable production output for the different candidates (Figure 1 B). The SEC results showed lower populations of heavier and lighter non-native species for Ront_HC15_zC5 and zC5_HC15_Ront than for any of the other constructs resulting in 98% full length AffiMabs compared to their respective light chain counterparts. Therefore, generally, heavy chain fusions (Ront_HC15_zC5 and zC5_HC15_Ront) demonstrated a higher level of monomeric content and titers than light chain fusions (Ront_LC15_zC5 and zC5_LC15_Ront) (Figure 1 B, Table 2). A reducing SDS-PAGE of the AffiMab candidates appeared to 5 indicate a chemical sensitivity to reducing conditions of the heavy chain N- terminally fused AffiMab, i.e. zC5_HC15_Ront, as shown by an additional unexpected band appearing at a size corresponding a heavy chain without zC5 fused to it (approx. 50-55 kDa) (Figure 2). All candidates were confirmed to bind both targets simultaneously by BLI, with the C-terminal fusion
10 candidates (Ront_HC15_zC5 and Ront_LC15_zC5) demonstrating faster on- rate than the N-terminal fusion candidates (zC5_HC15_Ront and zC5_LC15_Ront) (Figure 1 C).
Table 1 : Overview of AffiMab construct nomenclature

15
Table 2: Monomeric content of AffiMab constructs after purification by SEC

Table 3: SPR affinity measurements Example 2
Example 2

Blocking of C5 and complement driven hemolysis by AffiMabs in an in vitro whole blood assay
The mode-of-action was investigated for each respective entity of the bispecific binding molecule. Therefore, inhibition of C5 cleavage, formation of C5b-9 MAC complex and subsequent hemolytic activity by the respective AffiMab construct were studied in a whole blood assay with spiked human C5.
Materials and methods
In vitro whole blood assay: Blocking of C5 and complement driven hemolysis were studied using antibody (Rabbit anti-sheep RBC stroma, Sigma #S1389) coated sheep red blood cells. 5 x 106 cells in 50 pL GVBp (0.15 mM CaCI2, 0.5 mM MgCI2, 3 mM NaN3, 138 mM NaCI, 0.1 % gelatine, 1 .8 mM sodium barbital and 3.1 mM barbituric acid, pH 7.3-7.4) and the hemolytic activity quantified by measuring release of hemoglobin at absorbance 415 nm. C5 deficient human serum was spiked with human C5 at 0.023 nM. The ability of blocking hemolytic activity by the AffiMab constructs was studied at a concentration series of each and equimolar amounts of control Z variant either fused to an albumin binding domain (monovalent; “zC5-ABD”) or to the Fc domain (bivalent; “zC5-Fc”).
Results
Analysis utilizing in vitro whole blood assay demonstrated that all AffiMab constructs were able to inhibit hemolytic activity in a concentration dependent manner (Figure 3). Ront_15HC_zC5 showed the highest inhibitory effect amongst the AffiMab candidates with an IC50 of 1 .7 nM compared to the bivalent zC5-Fc fusion control with an IC50 of 1.1 nM. zC5-ABD showed a 6.6- and 4.2-fold higher IC50 than the two aforementioned constructs respectively, indicating that the natural symmetry of the AffiMab constructs contributed to the bivalency and had an effect on overall potency. Ront_LC15_zC5 exhibited the highest IC50 value amongst the AffiMab constructs of 244 nm, a 144-fold increase compared to the top construct. The naked mAb, Ront_WT, does not exhibit any significant effect on inhibition of hemolytic activity. Detailed IC50 values are listed in Table 3.
Table 4: IC50 values from C5 inhibition in in vitro whole blood hemolysis

Example 3 Neutralization of IFNa signaling by AffiMabs in an in vitro cell reporter assay
An IFNa reporter assay was performed to study if the generated AffiMabs had retained the parental mAb’s potency to neutralize IFNa stimuli via IFNaR.
Materials and methods
2 nM recombinant IFNa2b (Invivogen, California, US) was incubated with concentration series ranging from 100 nM to 3.25 nM of AffiMab constructs (IgG 1 ) or control treatment for 45 min at room temperature in a 96 well format. HEK-BLUE IFNa responsive cell line (Invivogen) was then subseguently seeded to the wells at a density of 280 000 cells/mL in 180 pL test medium and plates were incubated for 24h at 37°C in a humidified atmosphere with 5% CO2. Supernatant from the cell cultures were then incubated with pre-wared BLUE-substrate for 1 ,5h at 37°C in a humidified atmosphere with 5% CO2 according to manufacturer’s instruction for the quantification of secreted alkaline phosphatases in response to IFNa stimulation. Color metric read out was performed by absorbance measurements at OD 655 nm utilizing Clariostar system (BMG Labtech, Germany).
Results
An IFNa reporter assay was performed to study if the generated AffiMabs have retained the parental mAb’s (Ront_WT) potency to neutralize IFNa stimuli via IFNaR. The reporter cell line had the JAK/STAT1 ,2/ISGF3 pathway stably integrated and allowed monitoring of bioactive IFNa by an inducible secreted alkaline phosphatase (SEAP) reporter gene under the control of the IFNa inducible ISG54 promoter. The results showed that the Ront_15HC_zC5 inhibits recombinant IFNa2b signaling in parity with the naked antibody rontalizumab (Ront_WT) exhibiting an IC50 value of 14.4 ng/mL and 15.8 ng/mL respectively (Figure 4A). zC5_LC15_Ront exhibited a 17-fold increase compared to rontalizumab, higher than any of the other constructs (Table 4). Complement component 5 exhibits a molecular weight of 188 kDa, compared to the average total molecular weight of the AffiMab constructs (~166 kDa) and the molecular weight of IFNa (19.9 kDa). Consequently, the larger size of the C5 target protein is thought to have contributed to sterically hindering binding to IFNa by the AffiMab constructs and subsequently affected the potency to neutralize the target. Therefore, an equivalent IFNa neutralization assay was performed, though in presence of excess C5 protein (100 nM). Interestingly, a general increase in IC50 was observed for all AffiMab constructs (Table 4). The top performing AffiMab construct without the presence of C5, Ront_15HC_zC5, continues to be most potent construct at neutralizing IFNa in the presence of excess C5 in comparison to the naked antibody though not dramatically different from zC5_15HC_Ront (Figure 4B, Table 4). zC5_LC15_Ront continued to show poor neutralization potency in comparison to the other AffiMab constructs, with an 8-fold higher IC50 than Rontalizumab. Table 5: IC50 values derived from inhibition of IFNa in the in vitro interferon reporter assay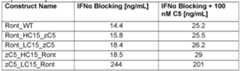

Conclusion
Based on the production screening and primary in vitro analysis of the constructs, the AffiMab candidate Ront_15HC_zC5 was chosen as the leading candidate to continue with for further evaluation in ex vivo whole blood assays.
Example 4
Ex vivo whole blood assay
The simultaneous effects of blocking the two ligands (C5 and IFNa) by the AffiMab construct Ront_HC15_zC5 was evaluated in an ex vivo whole blood model utilizing blood derived from SLE patients with ongoing inflammation. Blood was treated with AffiMab construct Ront_HC15_zC5 or control treatments for 24h post plasma collection and evaluation of cytokine or chemokine levels of interest was performed by ELISA and OLINK proximity extension assay.
Materials and methods
The effect of the AffiMab construct Ront_HC15_zC5 was studied in whole blood donated from SLE patients with active inflammation. Blood from SLE patients or healthy controls was collected in heparin coated vacutainer tubes. Blood was subsequently transferred to 2 mL endotoxin free tube at a volume of 0.5 mL. The tubes were spun down at 1200 g for 10 min and plasma was collected for cytokine quantification. AffiMab (Ront_HC15_zC5) or control treatment with the native antibody rontalizumab (R0NT_WT) was added at 30 mg/mL incubated overnight prior to plasma collection. Plasma samples were analyzed by the proximity ligation assay utilizing commercially available 96 Inflammation OLINK Panel (Olink, Sweden) for cytokine and chemokine profiling, and ELISA for C5a quantification according to manufacturer’s instructions (Human C5a ELISA kit, ThermoFisher).
Results
Evaluation of C5a levels post AffiMab (Ront_HC15_zC5) incubation demonstrated a significant decrease of the cleaved ligand in AffiMab treated blood (Figure 5). Moreover, the data indicated a stronger effect by Ront_HC15_zC5 than the control treatment with a monospecific bivalent counterpart (Fc-zC5).
The cytokine and chemokine profile in patient derived whole blood was analyzed by the OLINK 96 inflammation panel (Figure 6). Focused evaluation of diseases related cytokines demonstrated a significant upregulation of pro- inflammatory cytokines (IFNg, CXCL10) in untreated SLE patient derived blood (UT) compared to healthy control (UT HC) and lower levels of chemokine CXCL5 (a known neutrophile activator observed to be downregulated in SLE patient blood whilst upregulated in healthy tissue contributing to local immune activation and inflammatory damage of healthy tissue). IL-10 functions as an anti-inflammatory cytokine and was shown to be upregulated in patient derived blood, a consequence believed to be due to the anti-inflammatory treatments patients are undergoing at the time of sampling. Hence an increase in the levels of these proteins in plasma is postulated to contribute to a more beneficial effect to alleviate the inflammatory profile.
Evaluation of the cytokine and chemokine profile post AffiMab treatment demonstrated a significant effect in decreasing pro-inflammatory cytokines and chemokines closely related to the IFNa signature in SLE (IFNg, CXCL10) compared to blood treated with the native antibody (R0NT_WT) or untreated blood (UT). Conversely, CXCL5 increased after treatment with Ront_HC15_zC5 in parity with the R0NT_WT treatment, an effect in-line with the sought outcome in relation to the chemokines’ aforementioned role when shown to be present at higher levels in SLE patients and correlation with decreased disease activity. Furthermore, a significant increase in the antiinflammatory cytokine IL10 is observed for AffiMab treated blood, however not to the same extent as the naked antibody (R0NT_WT). In conclusion, the AffiMab demonstrated a capability to beneficially decrease plasma levels of the two key targeted ligands IFNa and C5, whilst also affecting downstream related cytokines and chemokines in a favorable manner by either increasing or decreasing the levels of these proteins depending on their biological function, collectively resulting in an alleviated SLE inflammatory profile.
Conclusion
Treatment with AffiMab lead construct (RONT_HC15_zC5) is able to affect levels of IFNa (Figure 4), C5 (Figures 3 and 5) and downstream related cytokines or chemokines of interest (Figure 6) in a favorable manner, demonstrating signs of affecting key cytokines and chemokines in the inflammatory SLE profile ex vivo.
Example 5
Ex vivo cerebrospinal fluid (CSF) assay
The simultaneous effects of blocking the two ligands (C5 and IFNa) by the AffiMab construct Ront_HC15_zC5 is evaluated in an ex vivo cerebrospinal fluid (CSF) model utilizing CSF derived from patients with central nervous system (CNS) disorders, disease or injury (such as traumatic brain injury, spinal cord injury, stroke, amyotrophic lateral sclerosis (ALS), neuromyelitis optica, and Alzheimer’s disease).
CSF is treated with AffiMab construct Ront_HC15_zC5 or control treatments post CSF collection and evaluation of selected cytokines of relevance is performed by ELISA-based methods.
Materials and methods The effect of the AffiMab construct Ront_HC15_zC5 is studied in CSF donated from patients with CNS disorder, disease or injury. CSF from patients or healthy controls is collected in polystyrene collection tubes. CSF samples are aliquoted and exposed to AffiMab (Ront_HC15_zC5) or control treatment with the native antibody rontalizumab (R0NT_WT), monospecific bivalent C5 inhibitor counterpart (Fc-zC5), or other relevant comparators such as the C5 blocking antibody eculizimab and/or the IFNaR blocking antibody anifrolumab, added at relevant concentrations and incubated under controlled conditions. Tubes are then spun at 4 °C 2000g for 10 minutes. The supernatant is collected, aliquoted, and frozen in a -80°C freezer for subsequent analysis.
Cytokine quantification of CSF samples is then analyzed using a commercially available ELISA-based method, for example but not limited to kits marketed by CLINK, Mabtech and ThermoFisher/lnvitrogen.
Results
Evaluation of C5a levels post AffiMab (Ront_HC15_zC5) incubation is expected to show a decrease of the cleaved ligand in AffiMab treated CSF. Moreover, the data is expected to indicate equal or stronger effect by Ront_HC15_zC5 than the control treatment with a monospecific bivalent counterpart (Fc-zC5).
The cytokine profile in patient derived CSF is expected to show an upregulation of relevant pro-inflammatory cytokines and a down-regulation of relevant anti-inflammatory cytokines in untreated patient derived CSF compared to healthy control.
Evaluation of the cytokine profile post AffiMab treatment is expected to demonstrate decreased levels in pro-inflammatory cytokines and increased levels in anti-inflammatory cytokines closely related to the IFNa signature of CNS disorder patients compared to CSF treated with the native antibody (R0NT_WT) or untreated CSF.
Conclusion
In conclusion, this example is expected to demonstrate that treatment with
AffiMab lead construct (RONT_HC15_zC5) is able to affect levels of IFNa, C5 and downstream related cytokines of interest in a favorable manner, demonstrating signs of affecting key cytokines in the CNS disorder-induced CSF profile ex vivo to regain immune homeostasis.
Example 6 Ex vivo whole blood assay
The simultaneous effects of blocking the two ligands (C5 and IFNa) by the AffiMab construct Ront_HC15_zC5 is evaluated in an ex vivo whole blood model utilizing blood derived from patients with CNS disorders, disease or injury (such as traumatic brain injury, spinal cord injury, stroke, amyotrophic lateral sclerosis (ALS), neuromyelitis optica, and Alzheimer’s disease).
Blood is treated with AffiMab construct Ront_HC15_zC5 or control treatments post plasma collection and evaluation of selected cytokines of relevance is performed by an ELISA-based method.
Materials and methods
The effect of the AffiMab construct Ront_HC15_zC5 is studied in whole blood donated from patients with CNS disorder, disease or injury. Blood from patients or healthy controls is collected in heparin coated vacutainer tubes. Blood is subsequently transferred to 2 mL endotoxin free tube at a volume of 0.5 mL. The tubes are spun down at 1200 g for 10 min and plasma is collected for cytokine quantification. Cells are stored in stabilizer solution (Cytodelics AB) for downstream mRNA cell isolation utilizing paxgene blood RNA isolation kit. Extracted mRNA is subsequently analyzed for ISG gene transcript levels using an in-house designed gene panel with Nanostring technologies cartridges and nCounter SPRINT instrument.
AffiMab (Ront_HC15_zC5) or control treatment of blood samples is carried out by incubation with AffiMab or the native antibody rontalizumab (R0NT_WT), or monospecific bivalent counterpart (Fc-zC5), or other relevant comparators at relevant concentrations and incubated under controlled conditions prior to plasma collection. Cytokine quantification of CSF samples is then analyzed using a commercially available ELISA-based method, for example but not limited to kits marketed by OLINK, Mabtech and ThermoFisher/lnvitrogen.
Results
Evaluation of C5a levels post AffiMab (Ront_HC15_zC5) incubation is expected to show a decrease of the cleaved ligand in AffiMab treated blood. Moreover, the data is expected to indicate equal or stronger effect by Ront_HC15_zC5 than the control treatment with a monospecific bivalent counterpart (Fc-zC5).
The cytokine profile in patient derived blood is expected to show upregulation of relevant pro-inflammatory cytokines and down-regulation of relevant anti-inflammatory cytokines in untreated patient derived blood compared to healthy control.
Evaluation of the cytokine profile post AffiMab treatment is expected to demonstrate decreased levels in pro-inflammatory cytokines and increased levels in anti-inflammatory cytokines closely related to the IFNa signature of CNS disorder, disease or injury patients compared to controls.
Conclusion
In conclusion, this example is expected to demonstrate that treatment with AffiMab lead construct (RONT_HC15_zC5) is able to affect levels of IFNa, C5 and downstream related cytokines of interest in a favorable manner, demonstrating signs of affecting key cytokines in the CNS disorder induced whole blood profile ex vivo.
Example 7 Ex vivo whole blood assay
The simultaneous effects of blocking the two ligands (C5 and IFNa) by the AffiMab construct Ront_HC15_zC5 is evaluated in an ex vivo whole blood model utilizing blood derived from patients with rheumatoid arthritis (RA). Blood is treated with AffiMab construct Ront_HC15_zC5 or control treatments post plasma collection and evaluation of selected cytokines of relevance is performed by an ELISA-based method.
Materials and methods
The effect of the AffiMab construct Ront_HC15_zC5 is studied in whole blood donated from patients with RA. Blood from patients or healthy controls is collected in heparin coated vacutainer tubes. Blood is subsequently transferred to 2 mL endotoxin free tube at a volume of 0.5 mL. The tubes are spun down at 1200 g for 10 min and plasma is collected for cytokine quantification.
AffiMab (Ront_HC15_zC5) or control treatment of blood samples is carried out by incubation with AffiMab or the native antibody rontalizumab (R0NT_WT), or monospecific bivalent counterpart (Fc-zC5), or other relevant comparators at relevant concentrations and incubated under controlled conditions prior to plasma collection.
Cytokine quantification of CSF samples is then analyzed using a commercially available ELISA-based method, for example but not limited to kits marketed by OLINK, Mabtech and ThermoFisher/lnvitrogen.
Results
Evaluation of C5a levels post AffiMab (Ront_HC15_zC5) incubation is expected to show a decrease of the cleaved ligand in AffiMab treated blood. Moreover, the data is expected to indicate equal or stronger effect by Ront_HC15_zC5 than the control treatment with a monospecific bivalent counterpart (Fc-zC5).
The cytokine profile in patient derived blood is expected to show an upregulation of relevant pro-inflammatory cytokines and a down-regulation of relevant anti-inflammatory cytokines in untreated patient derived blood compared to healthy control.
Evaluation of the cytokine profile post AffiMab treatment is expected to demonstrate decreased levels in pro-inflammatory cytokines and increased levels in anti-inflammatory cytokines closely related to the IFNa signature of RA patients compared to controls. Conclusion
In conclusion, this example is expected to demonstrate that treatment with AffiMab lead construct (RONT_HC15_zC5) is able to affect levels of IFNa, C5 and downstream related cytokines of interest in a favorable manner, demonstrating signs of affecting key cytokines in the RA-induced whole blood profile ex vivo.
Example 8 Ex vivo whole blood assay
The simultaneous effects of blocking the two ligands (C5 and IFNa) by the AffiMab construct Ront_HC15_zC5 is evaluated in an ex vivo whole blood model utilizing blood derived from patients with atherosclerosis.
Blood is treated with AffiMab construct Ront_HC15_zC5 or control treatments post plasma collection and evaluation of selected cytokines of relevance is performed by an ELISA-based method.
Materials and methods
The effect of the AffiMab construct Ront_HC15_zC5 is studied in whole blood donated from patients with atherosclerosis. Blood from patients or healthy controls is collected in heparin coated vacutainer tubes. Blood is subsequently transferred to 2 mL endotoxin free tube at a volume of 0.5 mL. The tubes are spun down at 1200 g for 10 min and plasma is collected for cytokine quantification.
AffiMab (Ront_HC15_zC5) or control treatment of blood samples is carried out by incubation with AffiMab or the native antibody rontalizumab (R0NT_WT), or monospecific bivalent counterpart (Fc-zC5), or other relevant comparators at relevant concentrations and incubated under controlled conditions prior to plasma collection.
Cytokine quantification of CSF samples is then analyzed using a commercially available ELISA-based method, for example but not limited to kits marketed by OLINK, Mabtech and ThermoFisher/lnvitrogen. Results
Evaluation of C5a levels post AffiMab (Ront_HC15_zC5) incubation is expected to show a decrease of the cleaved ligand in AffiMab treated blood. Moreover, the data is expected to indicate equal or stronger effect by Ront_HC15_zC5 than the control treatment with a monospecific bivalent counterpart (Fc-zC5).
The cytokine profile in patient derived blood is expected to show an upregulation of relevant pro-inflammatory cytokines and a down-regulation of relevant anti-inflammatory cytokines in untreated patient derived blood compared to healthy control.
Evaluation of the cytokine profile post AffiMab treatment is expected to demonstrate decreased levels in pro-inflammatory cytokines and increased levels in anti-inflammatory cytokines closely related to the IFNa signature of atherosclerosis patients compared to controls.
Conclusion
In conclusion, this example is expected to demonstrate that treatment with AffiMab lead construct (RONT_HC15_zC5) is able to affect levels of IFNa, C5 and downstream related cytokines of interest in a favorable manner, demonstrating signs of affecting key cytokines in the atherosclerosisjnduced whole blood profile ex vivo.
ITEMIZED LISTING OF EMBODIMENTS
1 . A bispecific binding molecule, comprising at least one first moiety with at least one first binding site for human complement component 5 (C5), operably linked to at least one second moiety with at least one first binding site for interferon alpha (IFNa).
2. A bispecific binding molecule according to item 1 , wherein said at least one first moiety with at least one first binding site for human complement component 5 (C5) is a C5 binding polypeptide.
3. A bispecific binding molecule according to item 2, wherein said C5 binding polypeptide is selected from the group consisting of full-length antibodies, Fab fragments, Fab’ fragments, F(ab')2 fragments, single chain Fab (scFab) fragments, Fc fragments, Fv fragments, single chain Fv (scFv) fragments, (scFv)2, scFv-Fc constructs, domain antibodies, a chimeric antibody, a CDR- grafted antibody, a fully human antibody, a bivalent antibody construct, a synthetic antibody, a bivalent antibody, a trivalent antibody, a tetravalent antibody, bivalent single chain antibody, a trivalent single chain antibody, a multivalent single chain antibody, nanobodies, AlbudAbs, DARTs (dual affinity re-targeting), BiTEs (bispecific T-cell engager), TandAbs (tandem diabodies), DAFs (dual acting Fab), two-in-one antibodies, SMIPs (small modular immunopharmaceuticals), FynomAbs (fynomers fused to antibodies), DVD- Igs (dual variable domain immunoglobulin), CovX-bodies (peptide modified antibodies), duobodies and triomAbs; a microbody, a maxybody, an avimer, a small disulfide-bonded protein; and a binding protein derived from a scaffold selected from the group consisting of staphylococcal protein A and domains thereof, other three helix domains, lipocalins, ankyrin repeat domains, cellulose binding domains, y crystallines, green fluorescent protein, human cytotoxic T lymphocyte-associated antigen 4, protease inhibitors such as Kunitz domains, PDZ domains, SH3 domains, peptide aptamers, staphylococcal nuclease, tendamistats, fibronectin type III domain, transferrin, zinc fingers and conotoxins.
4. A bispecific binding molecule according to item 3, wherein said C5 binding polypeptide comprises a C5 binding motif, BM, which motif consists of the amino acids sequence selected from: i) EX2X3X4A X6X7EIX10X11LPNL X16X17X18QW X21AFIX25 X26LX28D (SEQ ID NO:249); wherein, independently of each other,
X2 is selected from H, Q, S, T and V;
X3 is selected from I, L, M and V;
X4 is selected from A, D, E, H, K, L, N, Q, R, S, T and Y;
Xe is selected from N and W;
X7 is selected from A, D, E, H, I, L, N, Q, R, S and T;
X10 is selected from D and E;
X11 is selected from A, E, G, H, K, L, Q, R, S, T and Y;
X is selected from N and T;
X17 is selected from I, L and V;
X18 is selected from A, D, E, H, K, N, Q, R, S and T;
X21 is selected from I, L and V;
X25 is selected from A, D, E, G, H, N, S and T;
X26 is selected from K and S;
X28 is selected from A, D, E, H, N, Q, S, T and Y; and ii) an amino acid sequence which has at least 86 % identity to the sequence defined in i), wherein the polypeptide binds to C5.
5. A bispecific binding molecule according to item 3, wherein X2 is selected from H, T and V.
6. A bispecific binding molecule according to item 5, wherein X2 is selected from T and V.
7. A bispecific binding molecule according to item 6, wherein X2 is V.
8. A bispecific binding molecule according to any one of items 4-7, wherein X3 is selected from I, L and V.
9. A bispecific binding molecule according to item 8, wherein X3 is selected from I and L.
10. A bispecific binding molecule according to item 9, wherein X3 is I.
11 . A bispecific binding molecule according to item 9, wherein X3 is L.
12. A bispecific binding molecule according to any one of items 4-11 , wherein X4 is selected from A, D, E, K, L, Q and R. 13. A bispecific binding molecule according to item 12, wherein X4 is selected from A, D, E, K and R.
14. A bispecific binding molecule according to item 13, wherein X4 is selected from D and E.
15. A bispecific binding molecule according to any one of items 4-14, wherein X6 is W.
16. A bispecific binding molecule according to any one of items 4-15, wherein X7 is selected from A, D, N and T.
17. A bispecific binding molecule according to item 16, wherein X7 is selected from D and N.
18. A bispecific binding molecule according to item 17, wherein X7 is D.
19. A bispecific binding molecule according to item 17, wherein X7 is N.
20. A bispecific binding molecule according to any one of items 4-19, wherein
X11 is selected from A, H, K, Q, R and S.
21 . A bispecific binding molecule according to item 20, wherein Xu is selected from A, H, K and R.
22. A bispecific binding molecule according to item 21 , wherein Xu is selected from A, K and R.
23. A bispecific binding molecule according to item 22, wherein Xu is selected from K and R
24. A bispecific binding molecule according to any one of items 4-23, wherein X is T.
25. A bispecific binding molecule according to any one of items 4-24 wherein X17 is selected from I and L.
26. A bispecific binding molecule according to item 25, wherein X17 is I.
27. A bispecific binding molecule according to item 25, wherein X17 is L.
28. A bispecific binding molecule according to any one of items 4-27, wherein X18 is selected from A, D, E, N, Q, S and T.
29. A bispecific binding molecule according to item 28, wherein X18 is selected from A, D, E, Q and S.
30. A bispecific binding molecule according to item 29, wherein X18 is selected from D, E and Q. 31 . A bispecific binding molecule according to item 30, wherein Xis is selected from D and E.
32. A bispecific binding molecule according to item 31 , wherein Xis is D.
33. A bispecific binding molecule according to item 31 , wherein Xis is E.
34. A bispecific binding molecule according to any one of items 4-33, wherein
X21 is selected from I and L.
35. A bispecific binding molecule according to item 34, wherein X21 is I.
36. A bispecific binding molecule according to item 34, wherein X21 is L.
37. A bispecific binding molecule according to any one of items 4-36, wherein
X25 is selected from A, E, H, N and T.
38. A bispecific binding molecule according to item 37, wherein X25 is selected from E and N.
39. A bispecific binding molecule according to item 38, wherein X25 is N.
40. A bispecific binding molecule according to any one of items 4-39, wherein X26 is K.
41 . A bispecific binding molecule according to any one of items 4-40, wherein X28 is selected from A, D, E, H, N, Q and S.
42. A bispecific binding molecule according to item 41 , wherein X28 is selected from A, D, E and S.
43. A bispecific binding molecule according to item 42, wherein X28 is selected from A, D and E.
44. A bispecific binding molecule according to item 43, wherein X28 is selected from D and E.
45. A bispecific binding molecule according to item 44, wherein X28 is D.
46. A bispecific binding molecule according to any one of items 11 -14, wherein X3X4 is selected from LE and LD.
47. A bispecific binding molecule according to any one of items 25-33, wherein X17X18 is selected from IE and LD.
48. A bispecific binding molecule according to any one of items 4-47, wherein the amino acid sequence i) fulfills at least four of the following eight conditions l-VIII: X2 is V;
X2 is V;

II. X3 is selected from I and L; III. X6 is W;
IV. X? is selected from D and N;
V. X17 is selected from I and L;
VI. X21 is L;
VII. X25 is N;
VIII. X28 is D.
49. A bispecific binding molecule according to item 48, wherein the amino acid sequence i) fulfills at least five of the eight conditions l-VIII.
50. A bispecific binding molecule according to item 49, wherein the amino acid sequence i) fulfills at least six of the eight conditions l-VIII.
51 . A bispecific binding molecule according to item 50, wherein the amino acid sequence i) fulfills at least seven of the eight conditions l-VIII.
52. A bispecific binding molecule according to item 51 , wherein the amino acid sequence i) fulfills all of the eight conditions l-VIII.
53. A bispecific binding molecule according to any one of items 4-52, wherein the amino acid sequence is selected from any one of SEQ ID NO: 1-248.
54. A bispecific binding molecule according to item 53, wherein the amino acid sequence is selected from any one of SEQ ID NO: 1-12, SEQ ID NQ:20, SEQ ID NO:23-24, SEQ ID NO:26-28, SEQ ID NO:32-35, SEQ ID NO:38-39, SEQ ID NO:41 , SEQ ID NO:46, SEQ ID NO:49, SEQ ID NO:56-57, SEQ ID NO:59, SEQ ID NO:66, SEQ ID NO:78-79, SEQ ID NO:87, SEQ ID NO:92, SEQ ID NQ:106, SEQ ID NQ:110, SEQ ID NO:119, SEQ ID NO:125, SEQ ID NO:141 , SEQ ID NO:151 , SEQ ID NO:161 , SEQ ID NO:166, SEQ ID NO:187, SEQ ID NO:197, SEQ ID NQ:203, SEQ ID NQ:205, SEQ ID NO:215 and SEQ ID NO:243.
55. A bispecific binding molecule according to item 54, the amino acid sequence is selected from any one of SEQ ID NO: 1-12.
56. A bispecific binding molecule according to item 55, wherein the amino acid sequence is selected from any one of SEQ ID NO: 1 , SEQ ID NO:2, SEQ ID NO:3, SEQ ID NO:4 and SEQ ID NO:5.
57. A bispecific binding molecule according to item 56, wherein the amino acid sequence is selected from SEQ ID NO:1 and SEQ ID NO:4. 58. A bispecific binding molecule according to item 57, wherein the amino acid sequence is SEQ ID NO:1.
59. A bispecific binding molecule according to item 57, wherein the amino acid sequence is SEQ ID NO:4.
60. A bispecific binding molecule according to any one of items 2-59, wherein the C5 binding polypeptide comprises additional amino acids at the C- terminal and/or N-terminal end.
61 . A bispecific binding molecule according to any one of items 4-60, wherein said C5 binding motif (BM) forms part of a three-helix bundle protein domain.
62. A bispecific binding molecule according to any one of items 4-61 , comprising an amino acid sequence selected from:
AEAKYAK-[BM] (SEQ ID NQ:250);
AEAKFAK-[BM] (SEQ ID NO:251 );
ADNNFNK-[BM] (SEQ ID NO:252);
ADNKFNK-[BM] (SEQ ID NO:253);
VDNKFNK-[BM] (SEQ ID NO:254);
VDAKYAK-[BM] (SEQ ID NO:255); and sequences having at least 86% identity thereto, wherein [BM] is as defined in any one of items 4-59.
63. A bispecific binding molecule according to any one of items 4-61 , comprising an amino acid sequence selected from:
[BM]-DPSQSANLLSEAKKLNESQAPK (SEQ ID NO:256);
[BM]-DPSQSANLLAEAKKLNDAQAPK (SEQ ID NO:257);
[BM]-DPSVSKEILAEAKKLNDAQAPK (SEQ ID NO:258);
[BM]-DPSQSANLLAEAKKLNDAQAPK (SEQ ID NO:259);
[BM]-DPSESSELLSEAKKLNKSQAPK (SEQ ID NQ:260);
[BM]-DPSQSSELLAEAKKLNDAQAPK (SEQ ID NO:261 );
[BM]-DPSQSSELLAEAKKLNDSQAPK (SEQ ID NO:262);
[BM]-DPSQSSELLSEAKKLNDSQAPK (SEQ ID NO:263);
[BM]-DPSQSSELLSEAKKLSESQAPK (SEQ ID NO:264);
[BM]-DPSQSSELLSEAKKLESSQAPK (SEQ ID NO:265);
[BM]-DPSQSSELLAEAKKLNKAQAPK (SEQ ID NO:266); and sequences having at least 86% identity thereto, wherein [BM] is as defined in any one of items 4-59.
64. A bispecific binding molecule according to any one of items 4-61 , comprising an amino acid sequence selected from:
ADNNFNK-[BM]-DPSQSANLLSEAKKLNESQAPK (SEQ ID NO:267); ADNKFNK-[BM]-DPSQSANLLAEAKKLNDAQAPK (SEQ ID NO:268); ADNKFNK-[BM]-DPSVSKEILAEAKKLNDAQAPK (SEQ ID NO:269); VDNKFNK-[BM]-DPSQSANLLAEAKKLNDAQAPK (SEQ ID NQ:270); AEAKYAK-[BM]-DPSESSELLSEAKKLNKSQAPK (SEQ ID NO:271 ); VDAKYAK-[BM]-DPSQSSELLAEAKKLNDAQAPK (SEQ ID NO:272); VDAKYAK-[BM]-DPSQSSELLAEAKKLNDSQAPK (SEQ ID NO:273); AEAKYAK-[BM]-DPSQSSELLSEAKKLNDSQAPK (SEQ ID NO:274); AEAKYAK-[BM]-DPSQSSELLSEAKKLSESQAPK (SEQ ID NO:275); AEAKFAK-[BM]-DPSQSSELLSEAKKLSESQAPK (SEQ ID NO:276); AEAKYAK-[BM]-DPSQSSELLSEAKKLESSQAPK (SEQ ID NO:277); VDAKYAK-[BM]-DPSQSSELLSEAKKLNDSQAPK (SEQ ID NO:278); VDAKYAK-[BM]-DPSQSSELLSEAKKLSESQAPK (SEQ ID NO:279); VDAKYAK-[BM]-DPSQSSELLSEAKKLESSQAPK (SEQ ID NQ:280); VDAKYAK-[BM]-DPSQSSELLAEAKKLNKAQAPK (SEQ ID NO:281 ); AEAKYAK-[BM]-DPSQSSELLAEAKKLNKAQAPK (SEQ ID NO:282); and sequences having at least 86% identity thereto, wherein [BM] is as defined in any one of items 4-59.
65. A bispecific binding molecule according to item 3, wherein said C5 binding polypeptide is selected from the group consisting of full-length antibodies, Fab fragments, Fab’ fragments, F(ab')2 fragments, single chain Fab (scFab) fragments, Fc fragments, Fv fragments, single chain Fv (scFv) fragments, (SCFV)2, SCFV-FC constructs and domain antibodies.
66. A bispecific binding molecule according to item 65, wherein said C5 binding polypeptide is an antibody fragment which is an scFv.
67. A bispecific binding molecule according to any preceding item, comprising at least two first moieties, wherein each at least two first moieties each individually comprise at least one binding site for human complement component 5 (C5), wherein the amino acid sequences for the said binding sites may be the same or different.
68. A bispecific binding molecule according to any one of items 1-67, comprising at least two first moieties according to item 67, wherein said at least two first moieties are not coupled together.
69. A bispecific binding molecule according to any one of items 2-68, wherein said C5 binding polypeptide binds to C5 such that the KD value of the interaction is at most 1 x 10’6 M, such as at most 1 x 10’7 M, at most 1 x 10’8 M, or at most 1 x 10’9 M.
70. A bispecific binding molecule according to item 1 , wherein said at least one second moiety with at least one first binding site for interferon alpha (IFNa) is an IFNa binding polypeptide.
71. A bispecific binding molecule according to item 70, wherein said IFNa binding polypeptide is selected from the group consisting of full-length antibodies, Fab fragments, Fab’ fragments, F(ab')2 fragments, single chain Fab (scFab) fragments, Fc fragments, Fv fragments, single chain Fv (scFv) fragments, (scFv)2, scFv-Fc constructs, domain antibodies, a chimeric antibody, a CDR-grafted antibody, a fully human antibody, a bivalent antibody construct, a synthetic antibody, a bivalent antibody, a trivalent antibody, a tetravalent antibody, bivalent single chain antibody, a trivalent single chain antibody, a multivalent single chain antibody, nanobodies, AlbudAbs, DARTs (dual affinity re-targeting), BiTEs (bispecific T-cell engager), TandAbs (tandem diabodies), DAFs (dual acting Fab), two-in-one antibodies, SMIPs (small modular immunopharmaceuticals), FynomAbs (fynomers fused to antibodies), DVD-lgs (dual variable domain immunoglobulin), CovX-bodies (peptide modified antibodies), duobodies and triomAbs; a microbody, a maxybody, an avimer, a small disulfide-bonded protein; and a binding protein derived from a scaffold selected from the group consisting of staphylococcal protein A and domains thereof, other three helix domains, lipocalins, ankyrin repeat domains, cellulose binding domains, y crystallines, green fluorescent protein, human cytotoxic T lymphocyte-associated antigen 4, protease inhibitors such as Kunitz domains, PDZ domains, SH3 domains, peptide aptamers, staphylococcal nuclease, tendamistats, fibronectin type III domain, transferrin, zinc fingers and conotoxins.
72. A bispecific binding molecule according to item 71 , wherein said IFNa binding polypeptide is selected from the group consisting of full-length antibodies, Fab fragments, Fab’ fragments, F(ab')2 fragments, single chain Fab (scFab) fragments, Fc fragments, Fv fragments, single chain Fv (scFv) fragments, (scFv)2, scFv-Fc constructs and domain antibodies.
73. A bispecific binding molecule according to item 72, wherein said IFNa binding polypeptide is an IFNa binding antibody or antigen binding fragment thereof.
74. A bispecific binding molecule according to any one of items 70-73, wherein said IFNa binding polypeptide comprises a binding domain of an antibody, the binding domain comprising a heavy chain variable region and a light chain variable region, each comprising 6 complementarity determining domains (CDRs), wherein:
VLCDR1 has the sequence set forth in SEQ ID NO:286;
VLCDR2 has the sequence set forth in SEQ ID NO:287;
VLCDR3 has the sequence set forth in SEQ ID NO:288;
VHCDR1 has the sequence set forth in SEQ ID NO:289;
VHCDR2 has the sequence set forth in SEQ ID NQ:290; and VHCDR3 has the sequence set forth in SEQ ID NO:291 .
75. A bispecific binding molecule according to any one of items 70-74, wherein said antibody or antigen binding fragment thereof comprises a light chain variable region comprising the amino acid sequence SEQ ID NO:284, or an amino acid sequence having at least 80% sequence identity thereto.
76. A bispecific binding molecule according to any one of items 70-75, wherein said antibody or antigen binding fragment thereof comprises a heavy chain variable region comprising the amino acid sequence SEQ ID NO:285, or an amino acid sequence having at least 80% sequence identity thereto.
77. A bispecific binding molecule according to any one of items 75-76, wherein said antibody or antigen binding fragment thereof comprises a light chain variable region comprising the amino acid sequence SEQ ID NO:284, or an amino acid sequence having at least 80% sequence identity thereto, and a heavy chain variable region comprising the amino acid sequence SEQ ID NO:285, or an amino acid sequence having at least 80% sequence identity thereto.
78. A bispecific binding molecule according to any one of items 75-77, wherein said antibody or antigen binding fragment thereof is rontalizumab or a variant thereof or antigen binding fragment thereof.
79. A bispecific binding molecule according to item 78, wherein said IFNa binding antibody or antigen binding fragment thereof comprises a light chain comprising the amino acid sequence SEQ ID NO:293 or a sequence having at least 80% identity thereto, and/or a heavy chain comprising the amino acid sequence SEQ ID NO:294 or a sequence having at least 80% identity thereto, such as being an antibody comprising a light chain comprising the amino acid sequence SEQ ID NO:293 and a heavy chain comprising the amino acid sequence SEQ ID NO:294.
80. A bispecific binding molecule according to any one of items 73-79, comprising an amino acid substitution N297A in the CH2 region of the antibody or antibody fragment thereof.
81 . A bispecific binding molecule, comprising at least one first moiety with at least one first binding site for human complement component 5 (C5) according any one of items 2-69, operably linked to at least one second moiety with at least one first binding site for interferon alpha (IFNa) according to any one of items 70-80.
82. A bispecific binding molecule according to item 81 , wherein the at least one first moiety and the at least one second moiety are covalently linked.
83. A bispecific binding molecule according to item 82, wherein said at least one first moiety is covalently linked to: i) a CH3 domain of the heavy chain of an antibody or antibody fragment thereof according to any one of items 73-80; ii) a CL domain of the light chain of an antibody or antibody fragment thereof according to any one of items 73-80; iii) a VL domain of the light chain of an antibody or antibody fragment thereof according to any one of items 73-80; or iv) a VH domain of the heavy chain of an antibody or antibody fragment thereof according to any one of items 73-80.
84. A bispecific binding molecule according to item 83, wherein the bispecific molecule comprises one antibody or antibody fragment thereof and two first moieties, and: i) one first moiety is covalently linked to a CH3 domain of each heavy chain of said antibody or antibody fragment thereof; ii) one first moiety is covalently linked to a CL domain of each light chain of said antibody or antibody fragment thereof; iii) one first moiety is covalently linked to a VH domain of each heavy chain of said antibody or antibody fragment thereof; or iv) one first moiety is covalently linked to a VL domain of each light chain of said antibody or antibody fragment thereof.
85. A bispecific binding molecule according to any one of items 81-84, wherein said at least one first moiety is operably bound to said at least one second moiety via at least one linker, such as at least one linker selected from flexible amino acid linkers, rigid amino acid linkers and cleavable amino acid linkers.
86. A bispecific binding molecule according to item 85, wherein said flexible amino acid linker comprises the amino acid sequence SEQ ID NO: 292.
87. A bispecific binding molecule according to any preceding item, comprising a pair of polypeptide chains, said pair being selected from the group consisting of the pairs
SEQ ID NO:295 and SEQ ID NO:296,
SEQ ID NO:297 and SEQ ID NO:298,
SEQ ID NO:299 and SEQ ID NQ:300, and
SEQ ID NQ:301 and SEQ ID NQ:302.
88. A bispecific binding molecule according to item 87 comprising the pair of polypeptide chains represented by SEQ ID NO:295 and SEQ ID NO:296.
89. Polynucleotide encoding a bispecific binding molecule according to any preceding item.
90. Expression vector encoding a polynucleotide according to item 89.
91 . Host cell comprising an expression vector according to item 90. 92. Method of producing a bispecific binding molecule according to any one of items 1-88 comprising
- culturing a host cell according to item 90 under conditions permissive of expression of said polynucleotide from said expression vector; and
- isolating said bispecific binding molecule.
93. Method of producing a bispecific binding molecule according to any one of items 1-88 by non-biological peptide synthesis using amino acids and/or amino acid derivatives having protected reactive side-chains, the non- biological peptide synthesis comprising
- step-wise coupling of the amino acids and/or the amino acid derivatives to form a bispecific binding molecule according to any one of items 1-88 in the form of a polypeptide or fusion protein having protected reactive side-chains,
- removal of the protecting groups from the reactive side-chains of the polypeptide or fusion protein, and
- folding of the bispecific binding molecule in aqueous solution.
94. Composition comprising a bispecific binding molecule according to any one of items 1-88 and at least one pharmaceutically acceptable excipient or carrier.
95. Bispecific binding molecule according to any one of items 1-88 or a composition according item 94 for oral, respiratory, topical, intravenous, intraperitoneal, subcutaneous, pulmonary, transdermal, intramuscular, intranasal, buccal, sublingual or suppository administration, such as for subcutaneous administration, such as intravenous administration.
96. Bispecific binding molecule according to any one of items 1-88 or a composition according to item 94 for use as a medicament.
97. Bispecific binding molecule or composition for use as a medicament according to item 96, wherein said bispecific binding molecule or composition modulates C5 and IFNa function in vivo.
98. Bispecific binding molecule or composition for use according to any one of items 96-97 in the treatment of a disorder or disease related to C5 and IFNa.
99. Bispecific binding molecule or composition for use according to item 98, wherein said disorder or disease related to C5 and IFNa is selected from inflammatory and autoimmune diseases. 100. Bispecific binding molecule or composition for use according to any one of items 98-99, wherein said disorder or disease related to C5 and IFNa is selected from the group consisting of rheumatoid arthritis (RA), dermatomyositis, primary Sjogren’s syndrome, systemic sclerosis, psoriasis and type I interferonopathies, such as Aicardi-Goutieres syndrome, familial Chilblain lupus, systemic lupus erythematosus (SLE) and lupus nephritis.
101 . Bispecific binding molecule or composition for use according to any one of items 98-100, wherein said disorder or disease related to C5 and IFNa is systemic lupus erythematosus (SLE) or lupus nephritis, for example systemic lupus erythematosus, for example lupus nephritis.
102. Bispecific binding molecule or composition for use according to item 101 , wherein said bispecific binding molecule or composition alleviates the inflammatory profile in SLE.
103. Bispecific binding molecule or composition for use according to any one of items 96-102 wherein administration is selected from the group consisting of: oral, topical, intravenous, intraperitoneal, subcutaneous, pulmonary, transdermal, intramuscular, intranasal, buccal, sublingual or suppository administration, such as subcutaneous administration, such as intravenous administration.
104. Method of treatment of a disorder or disease related to C5 and IFNa, comprising administering to a subject in need thereof an effective amount of a bispecific binding molecule according to any one of items 1-88 or a composition according to item 94.
105. Method according to item 104, wherein said bispecific binding molecule modulates C5 and IFNa function in vivo.
106. Method according to any one of items 104-105, wherein said disorder or disease related to C5 and IFNa is selected from inflammatory and autoimmune diseases.
107. Method according to item 106, wherein said disorder or disease related to C5 and IFNa is selected from the group consisting of rheumatoid arthritis (RA), dermatomyositis, primary Sjogren’s syndrome, systemic sclerosis, psoriasis and type I interferonopathies, such as Aicardi-Goutieres syndrome, familial Chilblain lupus, systemic lupus erythematosus (SLE) and lupus nephritis.
108. Method according to item 107, wherein said disorder or disease related to C5 and IFNa is systemic lupus erythematosus (SLE) or lupus nephritis, for example systemic lupus erythematosus, for example lupus nephritis.
109. Method according to any one of items 104-108, wherein said subject is a mammalian subject, such as a human subject.
Claims
1 . A bispecific binding molecule, comprising at least one first moiety with at least one first binding site for human complement component 5 (C5), operably linked to at least one second moiety with at least one first binding site for interferon alpha (IFNa).
2. A bispecific binding molecule according to claim 1 , wherein said at least one first moiety with at least one first binding site for human complement component 5 (C5) is a C5 binding polypeptide.
3. A bispecific binding molecule according to claim 2, wherein said C5 binding polypeptide comprises a C5 binding motif, BM, which motif consists of the amino acids sequence selected from: i) EX2X3X4A X6X7EIX10X11LPNL X16X17X18QW X21AFIX25 X26LX28D (SEQ ID NO:249); wherein, independently of each other,
X2 is selected from H, Q, S, T and V;
X3 is selected from I, L, M and V;
X4 is selected from A, D, E, H, K, L, N, Q, R, S, T and Y;
Xe is selected from N and W;
X7 is selected from A, D, E, H, I, L, N, Q, R, S and T;
X10 is selected from D and E;
X11 is selected from A, E, G, H, K, L, Q, R, S, T and Y;
X is selected from N and T;
X17 is selected from I, L and V;
X18 is selected from A, D, E, H, K, N, Q, R, S and T;
X21 is selected from I, L and V;
X25 is selected from A, D, E, G, H, N, S and T;
X26 is selected from K and S;
X28 is selected from A, D, E, H, N, Q, S, T and Y; and ii) an amino acid sequence which has at least 86 % identity to the sequence defined in i), wherein the polypeptide binds to C5.
4. A bispecific binding molecule according to claim 3, wherein the amino acid sequence is selected from any one of SEQ ID NO: 1-248.
5. A bispecific binding molecule according to any preceding claim, wherein said at least one second moiety with at least one first binding site for interferon alpha (IFNa) is an IFNa binding polypeptide.
6. A bispecific binding molecule according to claim 5, wherein said IFNa binding polypeptide is an IFNa binding antibody or antigen binding fragment thereof.
7. A bispecific binding molecule according to claim 6, wherein said IFNa binding polypeptide comprises a binding domain of an antibody, the binding domain comprising a heavy chain variable region and a light chain variable region, each comprising 6 complementarity determining domains (CDRs), wherein:
VLCDR1 has the sequence set forth in SEQ ID NO:286;
VLCDR2 has the sequence set forth in SEQ ID NO:287;
VLCDR3 has the sequence set forth in SEQ ID NO:288;
VHCDR1 has the sequence set forth in SEQ ID NO:289;
VHCDR2 has the sequence set forth in SEQ ID NQ:290; and VHCDR3 has the sequence set forth in SEQ ID NO:291 .
8. A bispecific binding molecule according to any one of claims 6-7, wherein said antibody or antigen binding fragment thereof comprises a light chain variable region comprising the amino acid sequence SEQ ID NO:284, or an amino acid sequence having at least 80% sequence identity thereto, and a heavy chain variable region comprising the amino acid sequence SEQ ID NO:285, or an amino acid sequence having at least 80% sequence identity thereto.
9. A bispecific binding molecule according to any one of claims 6-8, wherein said antibody or antigen binding fragment thereof is rontalizumab or a variant thereof or antigen binding fragment thereof.
10. A bispecific binding molecule according to any one of claims 6-9, wherein said IFNa binding antibody or antigen binding fragment thereof comprises a light chain comprising the amino acid sequence SEQ ID NO:293 or a sequence having at least 80% identity thereto, and/or a heavy chain comprising the amino acid sequence SEQ ID NO:294 or a sequence having at least 80% identity thereto, such as being an antibody comprising a light chain comprising the amino acid sequence SEQ ID NO:293 and a heavy chain comprising the amino acid sequence SEQ ID NO:294.
11 . A bispecific binding molecule, comprising at least one first moiety with at least one first binding site for human complement component 5 (C5) according any one of claims 2-4, operably linked to at least one second moiety with at least one first binding site for interferon alpha (IFNa) according to any one of claims 5-10.
12. A bispecific binding molecule according to any preceding claim, comprising a pair of polypeptide chains, said pair being selected from the group consisting of the pairs
SEQ ID NO:295 and SEQ ID NO:296,
SEQ ID NO:297 and SEQ ID NO:298, SEQ ID NO:299 and SEQ ID NQ:300, and SEQ ID NQ:301 and SEQ ID NQ:302.
13. Composition comprising a bispecific binding molecule according to any one of claims 1-12 and at least one pharmaceutically acceptable excipient or carrier.
14. Bispecific binding molecule according to any one of claims 1 -12 or a composition according to claim 13 for use as a medicament, for example for use in the treatment of a disorder or disease related to C5 and IFNa, for example wherein said disorder or disease related to C5 and IFNa is systemic lupus erythematosus (SLE) or lupus nephritis, for example systemic lupus erythematosus, for example lupus nephritis.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP23171076 | 2023-05-02 | ||
| EP23171076.5 | 2023-05-02 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2024227886A1true WO2024227886A1 (en) | 2024-11-07 |
Family
ID=86328535
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2024/062141PendingWO2024227886A1 (en) | 2023-05-02 | 2024-05-02 | Bispecific binding molecule |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2024227886A1 (en) |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1995019374A1 (en) | 1994-01-14 | 1995-07-20 | Pharmacia Ab | Bacterial receptor structures |
| WO2009080811A1 (en) | 2007-12-21 | 2009-07-02 | Affibody Ab | Polypeptide libraries with a predetermined scaffold |
| WO2013101771A2 (en)* | 2011-12-30 | 2013-07-04 | Genentech, Inc. | Compositions and method for treating autoimmune diseases |
| WO2013126006A1 (en) | 2012-02-20 | 2013-08-29 | Swedish Orphan Biovitrum Ab (Publ) | Polypeptides binding to human complement c5 |
| WO2015028558A1 (en) | 2013-08-28 | 2015-03-05 | Swedish Orphan Biovitrum Ab (Publ) | Stable polypeptides binding to human complement c5 |
- 2024
- 2024-05-02WOPCT/EP2024/062141patent/WO2024227886A1/enactivePending
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1995019374A1 (en) | 1994-01-14 | 1995-07-20 | Pharmacia Ab | Bacterial receptor structures |
| WO2009080811A1 (en) | 2007-12-21 | 2009-07-02 | Affibody Ab | Polypeptide libraries with a predetermined scaffold |
| WO2013101771A2 (en)* | 2011-12-30 | 2013-07-04 | Genentech, Inc. | Compositions and method for treating autoimmune diseases |
| WO2013126006A1 (en) | 2012-02-20 | 2013-08-29 | Swedish Orphan Biovitrum Ab (Publ) | Polypeptides binding to human complement c5 |
| WO2015028558A1 (en) | 2013-08-28 | 2015-03-05 | Swedish Orphan Biovitrum Ab (Publ) | Stable polypeptides binding to human complement c5 |
Non-Patent Citations (45)
| Title |
|---|
| ALMAANIMEARAROVIN, CLIN. J. AM. SOC. NEPHROL., vol. 12, 2017, pages 825 - 835 |
| ASGARI N: "Interferon Alpha Association with Neuromyelitis Optica", CLIN DEV IMMUNOL., vol. 2013, 2013, pages 713519 |
| BERGLUND MAGNUS M. ET AL: "The clinical potential of Affibody-based inhibitors of C5 for therapeutic complement disruption", vol. 13, no. 3, 3 March 2016 (2016-03-03), GB, pages 241 - 243, XP055891543, ISSN: 1478-9450, Retrieved from the Internet <URL:http://dx.doi.org/10.1586/14789450.2016.1148604> DOI: 10.1586/14789450.2016.1148604* |
| BRENDAN SMH ET AL.: "The Role of Interferon- in Neurodegenerative Diseases: A Systematic Review", JOURNAL OF ALZHEIMER'S DISEASE, vol. 94, 2023, pages S45 - S66 |
| CHEN HJTAS SWDE WINTHER MPJ: "Type-I interferons in atherosclerosis", J EXP MED., vol. 217, no. 1, 6 January 2020 (2020-01-06), pages e20190459 |
| COPPO ET AL., PEDIATR NEPHROL, vol. 30, no. 1, 2015, pages 167 - 172 |
| DEEKS, DRUGS, vol. 81, no. 15, 2021, pages 1795 - 1802 |
| FEIFAN YU ET AL: "An affibody-adalimumab hybrid blocks combined IL-6 and TNF-triggered serum amyloid A secretion in vivo", MABS, vol. 6, no. 6, 1 November 2014 (2014-11-01), US, pages 1598 - 1607, XP055457021, ISSN: 1942-0862, DOI: 10.4161/mabs.36089* |
| GANGULY ET AL., J. EXP. MED., vol. 206, 2009, pages 1983 - 1994 |
| GENG CK ET AL.: "Effect of mesenchymal stem cells transplantation combining with hyperbaric oxygen therapy on rehabilitation of rat spinal cord injury", ASIAN PACIFIC JOURNAL OF TROPICAL MEDICINE, vol. 8, no. 6, 2015, pages 468 - 473 |
| GILKESON GARY S.: "Complement-Targeted Therapies in Lupus", vol. 1, no. 1, 22 January 2015 (2015-01-22), pages 10 - 18, XP093082662, Retrieved from the Internet <URL:https://link.springer.com/content/pdf/10.1007/s40674-014-0009-9.pdf?pdf=button> DOI: 10.1007/s40674-014-0009-9* |
| HAMMAD ET AL.: "The role of the complement system in traumatic brain injury: a review", JOURNAL OF NEUROINFLAMMATION, vol. 15, 2018, pages 24 |
| JEGO ET AL., IMMUNITY, vol. 19, 2003, pages 225 - 234 |
| KALUNIAN ET AL., ANN RHEUM DIS, vol. 75, no. 1, 2016, pages 196 - 202 |
| KALUNIAN KENNETH C ET AL: "A Phase II study of the efficacy and safety of rontalizumab (rhuMAb interferon-a) in patients with systemic lupus erythematosus (ROSE)", ANNALS OF THE RHEUMATIC DISEASES, B M J GROUP, GB, vol. 75, no. 1, 1 January 2016 (2016-01-01), pages 196 - 202, XP009191976, ISSN: 1468-2060* |
| KAUL ET AL., NAT. REV. DIS. PRIM., vol. 2, 2016, pages 16039 |
| KJELDGAARD AL ET AL.: "Complement Profiles in Patients with Amyotrophic Lateral Sclerosis: A Prospective Observational Cohort Stud", JOURNAL OF INFLAMMATION RESEARCH, vol. 14, 2021, pages 1043 - 1053 |
| KOOPMAN I ET AL.: "Complement C5 Antibodies for decreasing brain injury after aneurysmal Subarachnoid Haemorrhage (CLASH): study protocol for a randomised controlled phase II clinical trial", TRIALS, vol. 21, 2020, pages 969 |
| KOOPMAN I ET AL.: "Safety and pharmacodynamic efficacy of eculizumab in aneurysmal subarachnoid hemorrhage (CLASH): A phase 2a randomized clinical trial", EUROPEAN STROKE JOURNAL, vol. 8, no. 4, 2023, pages 1097 - 1106 |
| LEFFLER ET AL., ANN. RHEUM. DIS., vol. 73, 2014, pages 1601 - 1606 |
| LI LXIONG Z-YQIAN ZMZHAO T-ZFENG HHU S ET AL.: "Complement C5a is detrimental to histological and functional locomotor recovery after spinal cord injury in mice", NEUROBIOL DIS, vol. 66, 2014, pages 74 - 82 |
| LIN CHM ET AL.: "Role of IFN-a in Rheumatoid Arthritis", CURR RHEUMATOL REP, vol. 26, 2024, pages 37 - 52 |
| LOFBLOM ET AL., FEBS LETTERS, vol. 584, 2010, pages 2670 - 2680 |
| LUO C: "Clinical Value of Inflammatory Cytokines in Patients with Aneurysmal Subarachnoid Hemorrhage", CLINICAL INTERVENTIONS IN AGING, vol. 17, 2022, pages 615 - 626 |
| MG KISSCJ BINDER: "The multifaceted impact of complement on atherosclerosis", ATHEROSCLEROSIS, vol. 351, June 2022 (2022-06-01), pages 29 - 40, XP087085422, DOI: 10.1016/j.atherosclerosis.2022.03.014 |
| MORAND ET AL., ARTHRITIS RHEUMATOL |
| NIEWOLD ET AL., J. BIOMED. BIOTECHNOL., vol. 2010, 2010, pages 948364 |
| NILSSON B ET AL., PROTEIN ENGINEERING, vol. 1, 1987, pages 107 - 133 |
| NORD ET AL., NAT BIOTECH, vol. 15, 1997, pages 772 - 777 |
| NORD ET AL., PROT ENG, vol. 8, 1995, pages 601 - 608 |
| OBERMOSERPASCUAL, LUPUS, vol. 19, 2010, pages 1012 - 1019 |
| OONWILSONWICKS, CLIN. TRANSL. IMMUNOL., vol. 5, 2016, pages 29 |
| PANG ET AL., IMMUNOBIOLOGY, vol. 219, no. 12, 2014, pages 980 - 989 |
| PARK ET AL., BLOOD ADV, vol. 2, no. 16, 2018, pages 2090 - 2094 |
| PITTOCK SJLENNON VAMCKEON AMANDREKAR JWEINSHENKER BGLUCCHINETTI CF ET AL.: "Eculizumab in AQP4-IgG-positive relapsing neuromyelitis optica spectrum disorders: an open-label pilot study", LANCET NEUROL, vol. 12, 2013, pages 554 - 62, XP002734555, DOI: 10.1016/S1474-4422(13)70076-0 |
| RÖNNBLOMLEONARD, LUPUS SCI. MED., vol. 6, 2019, pages e000270 |
| ROSELLI FCHANDRASEKAR AMORGANTI-KOSSMANN MC: "Interferons in Traumatic Brain and Spinal Cord Injury: Current Evidence for Translational Application", FRONT. NEUROL., vol. 9, 2018, pages 458 |
| SANFORD SAIMCEWAN WA: "Type-I Interferons in Alzheimer's Disease and Other Tauopathies", FRONT. CELL. NEUROSCI., vol. 16, 2022, pages 949340 |
| THOMPSON, J.D.HIGGINS, D.G.GIBSON, T.J., NUCLEIC ACIDS RESEARCH, vol. 22, 1994, pages 4673 - 4680 |
| TRIGGIANESE PCONIGLIARO PDE MARTINO EMONOSI BCHIMENTI MS: "Overview on the Link Between the Complement System and Auto-Immune Articular and Pulmonary Disease", RHEUMATOLOGY: RESEARCH AND REVIEWS, vol. 15, 2023, pages 65 - 79 |
| TUNNICLIFFE ET AL., ARTHRITIS CARE RES (HOBOKEN, vol. 67, no. 10, 2015, pages 1440 - 1452 |
| VAN DIJK BJ ET AL.: "Complement C5 contributes to brain injury after subarachnoid hemorrhage", TRANSL STROKE RES, vol. 11, 2020, pages 678 - 688, XP037186169, DOI: 10.1007/s12975-019-00757-0 |
| VOLK ET AL., DRUGS R D, vol. 21, 2021, pages 157 - 168 |
| WAHLBERG E ET AL., PNAS, vol. 100, no. 6, 2003, pages 3185 - 3190 |
| ZINDLER E ET AL.: "Neuronal injury in chronic CNS inflammation", BEST PRACT RES CLIN ANAESTHESIOL, vol. 24, no. 4, December 2010 (2010-12-01), pages 551 - 62, XP027523154 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US12428497B2 (en) | Multispecific antibody constructs | |
| JP7227312B2 (en) | Anti-N3pGlu amyloid beta peptide antibody and uses thereof | |
| JP7019198B2 (en) | C5 Antibodies and Methods for Prevention and Treatment of Complement-Related Diseases | |
| JP2023093549A (en) | Pd-1 agonist antibodies and uses thereof | |
| US20170198062A1 (en) | Multi-specific antibody constructs | |
| JP6105146B2 (en) | Pan-ELR + CXC chemokine antibody | |
| US20230279129A1 (en) | Method of treating or preventing ischemia-reperfusion injury | |
| TW201444867A (en) | Anti-TNF-anti-IL-17 bispecific antibodies | |
| KR20250110365A (en) | Site directed mutagenesis of trem-1 antibodies for decreasing viscosity | |
| CN111763259B (en) | anti-CD 40 antibodies and uses thereof | |
| WO2023061390A1 (en) | Treatment of ige-mediated diseases | |
| CN106573972A (en) | Humanized anti-IgE antibodies that cross-link CD23 of B lymphocytes but do not sensitize mast cells | |
| KR20240031229A (en) | Antibodies for the treatment of alpha-synucleinopathy | |
| WO2024227886A1 (en) | Bispecific binding molecule | |
| WO2013103783A1 (en) | Murine il-13 antibodies | |
| JP2022169737A (en) | Anti-RAMP2 antibody | |
| WO2018207638A1 (en) | Anti-cldn-5 antibody, and drug containing said antibody | |
| JP7755895B2 (en) | Isolated antigen-binding proteins and uses thereof | |
| US20250154242A1 (en) | Anti-tnf-alpha antibodies and compositions | |
| JP2024074278A (en) | LAG-3 and PD-1/LAG-3 Antibodies | |
| TW202317616A (en) | Isolated antigen-binding protein and use thereof | |
| JP2025526279A (en) | Anti-TNFα antibodies and compositions | |
| WO2023023150A2 (en) | Methods and compositions related to neutralizing antibodies against human coronavirus | |
| TW202509059A (en) | Bispecific anti-pseudomonas antibodies with modified fc regions and methods of use thereof | |
| WO2024194298A1 (en) | Humanised antibody against amyloid beta 42 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | Ref document number:24723843 Country of ref document:EP Kind code of ref document:A1 |