WO2024168022A2 - Degarelix polymeric formulations - Google Patents
Degarelix polymeric formulationsDownload PDFInfo
- Publication number
- WO2024168022A2 WO2024168022A2PCT/US2024/014780US2024014780WWO2024168022A2WO 2024168022 A2WO2024168022 A2WO 2024168022A2US 2024014780 WUS2024014780 WUS 2024014780WWO 2024168022 A2WO2024168022 A2WO 2024168022A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- composition
- degarelix
- less
- nmp
- kda
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Classifications
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/0012—Galenical forms characterised by the site of application
- A61K9/0019—Injectable compositions; Intramuscular, intravenous, arterial, subcutaneous administration; Compositions to be administered through the skin in an invasive manner
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
- A61K38/04—Peptides having up to 20 amino acids in a fully defined sequence; Derivatives thereof
- A61K38/08—Peptides having 5 to 11 amino acids
- A61K38/09—Luteinising hormone-releasing hormone [LHRH], i.e. Gonadotropin-releasing hormone [GnRH]; Related peptides
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/02—Inorganic compounds
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/06—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite
- A61K47/22—Heterocyclic compounds, e.g. ascorbic acid, tocopherol or pyrrolidones
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/30—Macromolecular organic or inorganic compounds, e.g. inorganic polyphosphates
- A61K47/34—Macromolecular compounds obtained otherwise than by reactions only involving carbon-to-carbon unsaturated bonds, e.g. polyesters, polyamino acids, polysiloxanes, polyphosphazines, copolymers of polyalkylene glycol or poloxamers
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/0012—Galenical forms characterised by the site of application
- A61K9/0019—Injectable compositions; Intramuscular, intravenous, arterial, subcutaneous administration; Compositions to be administered through the skin in an invasive manner
- A61K9/0024—Solid, semi-solid or solidifying implants, which are implanted or injected in body tissue
Definitions
- Degarelixacts by directly blocking GnRH action on the pituitary by competitively and reversibly binding to GnRH receptors and produces a fast suppression of testosterone with no initial surge or minimal surge.
- the currently marketed drug-delivery technology for degarelix in the United States and Europee.g., FIRMAGON® (degarelix for injection), Ferring Pharmaceuticals Inc. or Ferring GmbH
- FIRMAGON®degarelix for injection
- Ferring Pharmaceuticals Inc. or Ferring GmbHhas significant limitations, including administration of a starting dose given as two high volume injections (3 mL each), followed by maintenance doses administered as a single high volume injection (1 x 4 mL) every 28 days.
- degarelixis supplied as a powder (lyophilized, contained mannitol as a bulking agent), and once it is reconstituted with water the degarelix molecules aggregate and cross-link in a gel-forming network, resulting in a hydrogel. Because of its self-aggregation property and chemical instability in aqueous media, it has to be separated out from the aqueous vehicle and needs to be kept in a dried form by lyophilization. Therefore, it needs to be reconstituted at the time of administration. Since the rate and extent of self-aggregation/gel formation of peptide in aqueous media is concentration dependent, it is challenging to prepare a ready-to-inject formulation of degarelix for injection.
- degarelixis manufactured by aseptic processing and involves multiple steps for reconstitution.
- an injectable drug formulation of degarelix in a single syringe systemthat is ready-to-inject, and provides in a single injection, extended-release of a therapeutically effective amount of degarelix for more than a month within a low injection volume ( ⁇ 2.5 mL or 2 mL).
- a compositioncomprising a therapeutically effective amount of degarelix or a pharmaceutically acceptable salt thereof.
- the compositioncomprises a biodegradable polymer; and a biocompatible solvent, wherein the composition is formulated for subcutaneous injection into a subject; wherein a single dose of the composition is about 2.5 mL or less, or about 2.0 mL or less; and upon injection into the subject, the composition forms an in situ depot that releases the degarelix or pharmaceutically acceptable salt thereof over a time period of from about 1 month to about 6 months.
- the in situ depotreleases the degarelix or pharmaceutically acceptable salt thereof over a time period selected from the group consisting of: at least about 1 month, at least about 1.5 months, at least about 2 months, at least about 2.5 months, at least about 3 months, at least about 3.5 months, at least about 4 months, at least about 4.5 months, and at least about 5 months.
- the pharmaceutically acceptable salt of the degarelixis selected from degarelix acetate, degarelix citrate, degarelix pamoate, degarelix palmitate, and degarelix mesylate.
- the biodegradable polymercomprises monomer residues selected from the group consisting of lactide, glycolide, caprolactone, p- dioxanone, trimethylene carbonate, thylene oxide, propylene oxide, sebacic anhydride, diketene acetals/diols, and combinations thereof.
- the biodegradable polymercomprises lactide and glycolide monomer residues. In one aspect, a molar ratio of the lactide to glycolide monomer residues is from about 45:55 to about 99:1.
- a molar ratio of the lactide to glycolide monomer residuesis from about 50:50 to about 90:10.
- the biodegradable polymercomprises: lactide monomer residues and monomer residues selected from the group consisting of ⁇ - caprolactone, trimethylene carbonate, and combinations thereof.
- a molar ratio of the lactide monomer residues to the ⁇ -caprolactone and/or trimethylene carbonate monomer residuesis from about 10:90 to about 90:10.
- a molar ratio of the lactide monomer residues to the ⁇ -caprolactone and/or trimethylene carbonate monomer residuesis from about 25:75 to about 75:25. In yet another aspect, a molar ratio of the lactide monomer residues to the ⁇ -caprolactone and/or trimethylene carbonate monomer residues is about 75:25.
- the biodegradable polymercomprises at least one carboxylic acid end group, at least one hydroxyl end group, or at least one hydroxy end group and is substantially free of terminal carboxy end groups.
- the biodegradable polymeris poly(D,L-lactide- co-glycolide) (PLG), poly(D,L-lactide) (PLA), or poly(D,L-lactide-co- ⁇ -caprolactone) (PLC).
- the biodegradable polymeris poly(D,L-lactide-co- ⁇ -caprolactone) (PLC).
- the PLCcomprises at least one carboxylic acid end group.
- the PLCis a carboxylic acid-initiated PLC.
- the carboxylic acidis glycolic acid or lactic acid.
- the biodegradable polymeris acid-initiated 75:25 PLC.
- the biodegradable polymeris PLG.
- the PLGcomprises at least one hydroxyl end group.
- the PLGis a core- diol-initiated PLG.
- the core diolis 1,6-hexane-diol.
- the PLGis a mono functional alcohol-initiated PLG.
- the alcoholis dodecanol.
- the biodegradable polymeris acid-initiated 50:50 PLG.
- the biodegradable polymeris 1,6-hexane-diol-initiated 75:25 PLG. In another aspect, the biodegradable polymer is 1,6-hexane-diol-initiated 85:15 PLG. [0016] In one aspect of the composition, the biodegradable polymer is dodecanol- initiated PLA. [0017] In one aspect of the composition, the biocompatible polymer has a weight average molecular weight of from about 5 kDa to about 60 kDa. [0018] In one aspect of the composition, the biocompatible polymer has a weight average molecular weight of from about 5 kDa to about 35 kDa.
- the biocompatible polymercomprises less than about 0.5 wt%, or less than about 0.4 wt%, or less than about 0.3 wt%, or less than about 0.2 wt% or less than about 0.1 wt% unreacted lactide monomer.
- the amount of biodegradable polymer in the compositionis from about 10% to about 60% by weight of the composition.
- the amount of biocompatible solvent in the compositionis from about 10% to about 85% by weight of the composition.
- the therapeutically effective amount of degarelix or pharmaceutically acceptable salt in the compositionis from about 10% to about 40% by weight of the composition.
- the therapeutically effective amount of degarelix or pharmaceutically acceptable salt thereofis from about 40 mg – 500 mg.
- the therapeutically effective amount of degarelix or pharmaceutically acceptable salt thereofis from about 80 mg – 500 mg.
- the therapeutically effective amount of degarelix or pharmaceutically acceptable salt thereofis from about 120 mg – 500 mg.
- the biocompatible organic solventis selected from the group consisting of N-methyl-2-pyrrolidone (NMP), dimethyl sulfoxide (DMSO), dimethylacetamide (DMA), benzyl benzoate (BnBzO), polyethylene glycol 15 hydroxystearate, methyl ethyl ketone, methyl lactate, benzyl alcohol, propylene carbonate (PC), triacetin, tributyl citrate, acetyl tributyl citrate, acetyl triethyl citrate, triethyl citrate, diethylene glycol monomethyl ether, ethyl acetate, N-ethyl-2-pyrrolidone, glycofurol, and combinations thereof.
- NMPN-methyl-2-pyrrolidone
- DMSOdimethyl sulfoxide
- DMAdimethylacetamide
- BnBzObenzyl benzoate
- PCpropylene carbonate
- triacetin
- the biocompatible organic solventis NMP or DMSO.
- a single dose of the compositionis about 2.5 mL or less, about 2.4 mL or less, about 2.3 mL or less, about 2.2 mL or less, about 2.1 mL or less, about 2.0 mL or less, about 1.9 mL or less, about 1.8 mL or less, about 1.7 mL or less, about 1.6 mL or less, about 1.5 mL or less, about 1.4 mL or less, about 1.3 mL or less, about 1.2 mL or less, about 1.1 mL or less, about 1 mL or less, about 0.75 mL or less, about 0.5 mL or less, or about 0.375 mL or less.
- the compositionhas been terminally sterilized or sterile filtered. In one aspect, wherein the composition has been terminally sterilized by e-beam. [0029] In one aspect of the composition, the composition further comprises one or more additives.
- the additiveis selected from the group consisting of polysorbate 20, polysorbate 80, poloxamer 188, sorbitan trioleate, lecithin (e.g., soya or egg), polyethylene glycol (PEG), PEG 300, 2-pyrrolidone, alpha-tocopherol, Vitamin E TPGS, sucrose cocoate, sucrose stearate, sucrose laurate, proline, arginine, sodium metabisulfite butylated hydroxyanisole, butylated hydroxyquinone, butylhydroxyanisol, hydroxycoumarin, butylated hydroxytoluene, cephalm, ethyl gallate, propyl gallate, octyl gallate, lauryl gallate, propyl-hydroxybenzoate, trihydroxybutylrophenone, vitamin E, lecithin, ethanolamine, ZnCl 2 , MgCl 2 , CaCl 2 , DL-methioninecitric
- the degarelixis lyophilized, and the biodegradable polymer is dissolved in the biocompatible solvent.
- the degarelixis dissolved in a first biocompatible solvent, and the biodegradable polymer is dissolved in a second biocompatible solvent.
- the first and second biocompatible solventare the same.
- a pharmaceutical compositioncomprising about 10-30 wt% of degarelix acetate; about 20-80 wt% of N-methyl-2-pyrrolidone (NMP); and about 10-50 wt% of 75:25 poly(lactide-co- ⁇ -caprolactone) (PLC) copolymer having at least one carboxylic acid end group and having a weight average molecular weight of about 5-30 kDa is disclosed.
- NMPN-methyl-2-pyrrolidone
- PLCpoly(lactide-co- ⁇ -caprolactone) copolymer having at least one carboxylic acid end group and having a weight average molecular weight of about 5-30 kDa
- the compositioncomprises about 16 wt% of degarelix acetate; about 57 wt% of N-methyl-2-pyrrolidone (NMP); and about 27 wt% of 75:25 poly(lactide-co- ⁇ -caprolactone) (PLC) copolymer having at least one carboxylic acid end group and having a weight average molecular weight of about 17 kDa.
- the compositioncomprises about 16 wt% of degarelix acetate; about 57 wt% of NMP; and about 27 wt% of 75:25 PLC copolymer having at least one carboxylic acid end group and having a weight average molecular weight of about 8 kDa.
- unreacted lactide and caprolactone monomers in the PLC copolymerare less than about 1.0 wt%. In yet another aspect, unreacted lactide and caprolactone monomers in the PLC copolymer are less than about 0.5 wt%, than about 0.4 wt %, less than about 0.3 wt %, less than about 0.2 wt% and less than about 0.1 wt%.
- a pharmaceutical compositioncomprising a first container comprising 30-70 wt% of 75:25 PLC copolymer having at least one carboxylic acid end group and having a weight average molecular weight of 5-30 kDa, and 30-70 wt% of N- methyl-2-pyrrolidone (NMP); and a second container comprising 20-40 wt% of degarelix acetate and 60-80 wt% NMP is disclosed.
- NMPN- methyl-2-pyrrolidone
- the first containercomprises 50 wt% of 75:25 PLC copolymer having at least one carboxylic acid end group and having a weight average molecular weight of 17 kDa, and 50 wt% of N- methyl-2-pyrrolidone (NMP); and the second container comprises 35 wt% of degarelix acetate and 65 wt% NMP.
- the first containercomprises 70 wt% of 75:25 PLC copolymer having at least one carboxylic acid end group and having a weight average molecular weight of 8 kDa, and 30 wt% of NMP; and the second container comprising 35 wt% of degarelix acetate and 65 wt% NMP.
- unreacted lactide and caprolactone monomers in the PLC copolymerare less than about 1.0 wt%.
- unreacted lactide and caprolactone monomers in the PLC copolymerare less than about 0.5 wt%, than about 0.4 wt %, less than about 0.3 wt %, less than about 0.2 wt% and less than about 0.1 wt%.
- a pharmaceutical compositioncomprising about 10-30 wt% of degarelix acetate; about 20-80 wt% of NMP; and about 10-50 wt% of poly(D,L-lactide) (PLA) polymer having at least one carboxylic acid end group and having a weight average molecular weight of about 5-30 kDa is disclosed.
- the compositioncomprises about 16 wt% of degarelix acetate; about 57 wt% of NMP; and about 27 wt% of poly(D,L-lactide) (PLA) polymer having at least one carboxylic acid end group and having a weight average molecular weight of about 23 kDa.
- unreacted lactide monomers in the PLA polymerare less than about 1.0 wt%.
- unreacted lactide monomers in the PLA polymerare less than about 0.5 wt%, than about 0.4 wt %, less than about 0.3 wt %, less than about 0.2 wt% and less than about 0.1 wt%.
- a pharmaceutical compositioncomprising a first container comprising 30-70 wt% of PLA polymer having at least one carboxylic acid end group and having a weight average molecular weight of 5-30 kDa, and 30-70 wt% of NMP; and a second container comprising 20-40 wt% of degarelix acetate and 60-80 wt% NMP is disclosed.
- the first containercomprising 50 wt% of PLA polymer having at least one carboxylic acid end group and having a weight average molecular weight of 23 kDa, and 50 wt% of NMP; and the second container comprising 35 wt% of degarelix acetate and 65 wt% NMP.
- unreacted lactide monomers in the PLA polymerare less than about 1.0 wt%. In still another aspect, unreacted lactide monomers in the PLA polymer are less than about 0.5 wt%, than about 0.4 wt %, less than about 0.3 wt %, less than about 0.2 wt% and less than about 0.1 wt%.
- a pharmaceutical compositioncomprising about 10-30 wt% of degarelix acetate; about 20-80 wt% of NMP; and about 10-50 wt% of 75:25 poly(D,L- lactide-co-glycolide) (PLG) copolymer having at least one carboxylic acid end group and having a weight average molecular weight of about 5-30 kDa is disclosed.
- PLGpoly(D,L- lactide-co-glycolide)
- the compositioncomprises about 16 wt% of degarelix acetate; about 57 wt% of NMP; and about 27 wt% of 75:25 poly(D,L-lactide-co-glycolide) (PLG) copolymer having at least one carboxylic acid end group and having a weight average molecular weight of about 17 kDa.
- PLGpoly(D,L-lactide-co-glycolide) copolymer having at least one carboxylic acid end group and having a weight average molecular weight of about 17 kDa.
- unreacted lactide and glycolide monomers in the PLG copolymerare less than about 1.0 wt%.
- unreacted lactide and glycolide monomers in the PLG copolymerare less than about 0.5 wt%, than about 0.4 wt %, less than about 0.3 wt %, less than about 0.2 wt% and less than about 0.1 wt%.
- a pharmaceutical compositioncomprising a first container comprising 30-70 wt% of 75:25 PLG copolymer having at least one carboxylic acid end group and having a weight average molecular weight of 5-30 kDa, and 30-70 wt% of NMP; and a second container comprising 20-40 wt% of degarelix acetate and 60-80 wt% NMP is disclosed.
- the first containercomprises 50 wt% of 75:25 PLG copolymer having at least one carboxylic acid end group and having a weight average molecular weight of 17 kDa, and 50 wt% of NMP; and the second container comprises 35 wt% of degarelix acetate and 65 wt% NMP.
- unreacted lactide and glycolide monomers in the PLG copolymerare less than about 1.0 wt%.
- unreacted lactide and glycolide monomers in the PLG copolymerare less than about 0.5 wt%, than about 0.4 wt %, less than about 0.3 wt %, less than about 0.2 wt% and less than about 0.1 wt%.
- a pharmaceutical compositioncomprising about 10-30 wt% of degarelix acetate; about 20-80 wt% of NMP; and about 10-50 wt% of 75:25 PLG copolymer having at least one hydroxyl end group and having a weight average molecular weight of about 5-30 kDa is disclosed.
- the compositioncomprises about 16 wt% of degarelix acetate; about 57 wt% of NMP; and about 27 wt% of 75:25 PLG copolymer having at least one hydroxyl end group and having a weight average molecular weight of about 19 kDa.
- a pharmaceutical compositioncomprising a first container comprising 30-70 wt% of 75:25 PLG copolymer having at least one hydroxyl end group and having a weight average molecular weight of 5-30 kDa, and 30-70 wt% of NMP; and a second container comprising 20-40 wt% of degarelix acetate and 60-80 wt% NMP.
- the first containercomprises 50 wt% of 75:25 PLG copolymer having at least one hydroxyl end group and having a weight average molecular weight of 19 kDa, and 50 wt% of NMP; and the second container comprises 35 wt% of degarelix acetate and 65 wt% NMP.
- a pharmaceutical compositioncomprising about 10-30 wt% of degarelix acetate; about 10-40 wt% of NMP; about 10-40 wt% of DMSO; and (d) about 10-50 wt% of 75:25 PLG copolymer having at least one hydroxyl end group and having a weight average molecular weight of about 5-30 kDa is disclosed.
- the compositioncomprises about 16 wt% of degarelix acetate; about 30 wt% of NMP; about 27 wt% of DMSO; and (d) about 27 wt% of 75:25 PLG copolymer having at least one hydroxyl end group and having a weight average molecular weight of about 19 kDa.
- a pharmaceutical compositioncomprising a first container comprising 30-70 wt% of 75:25 PLG copolymer having at least one hydroxyl end group and having a weight average molecular weight of 5-30 kDa, and 30-70 wt% of NMP; and a second container comprising 20-40 wt% of degarelix acetate and 60-80 wt% DMSO is disclosed.
- the first containercomprises 50 wt% of 75:25 PLG copolymer having at least one hydroxyl end group and having a weight average molecular weight of 19 kDa, and 50 wt% of NMP; and the second container comprises 35 wt% of degarelix acetate and 65 wt% DMSO.
- a pharmaceutical compositioncomprising about 10-30wt% of degarelix citrate; about 20-80 wt% of NMP; and about 10-50 wt% of 75:25 PLG copolymer having at least one hydroxyl end group and having a weight average molecular weight of about 5-30 kDa is disclosed.
- the compositioncomprises about 16 wt% of degarelix citrate; about 57 wt% of NMP; and about 27 wt% of 75:25 PLG copolymer having at least one hydroxyl end group and having a weight average molecular weight of about 19 kDa.
- a pharmaceutical compositioncomprising a first container comprising 30-70 wt% of 75:25 PLG copolymer having at least one hydroxyl end group and having a weight average molecular weight of 19 kDa, and 50 wt% of NMP; and a second container comprising 20-40 wt% of degarelix citrate and 60-80 wt% NMP is disclosed.
- the first containercomprises 50 wt% of 75:25 PLG copolymer having at least one hydroxyl end group and having a weight average molecular weight of 19 kDa, and 50 wt% of NMP; and the second container comprises 35 wt% of degarelix citrate and 65 wt% NMP.
- the amount of degarelix or the pharmaceutically acceptable salt thereof in the pharmaceutical compositionis from about 8 wt% to about 39 wt% degarelix free base equivalent. In one aspect, the amount of degarelix or the pharmaceutically acceptable salt thereof in the pharmaceutical composition is from about 17 wt% to about 39 wt% degarelix free base equivalent.
- the amount of degarelix or the pharmaceutically acceptable salt thereof in the pharmaceutical compositionis from about 8 wt% to about 29 wt% degarelix free base equivalent. In one aspect, the amount of degarelix or the pharmaceutically acceptable salt thereof in the pharmaceutical composition is from about 8 wt% to about 20 wt% degarelix free base equivalent. In one aspect, the amount of degarelix or the pharmaceutically acceptable salt thereof in the pharmaceutical composition is from about 10 wt% to about 16 wt% degarelix free base equivalent. In one aspect, the amount of degarelix or the pharmaceutically acceptable salt thereof in the pharmaceutical composition is from about 14 wt% to about 15.5 wt% degarelix free base equivalent.
- the amount of degarelix or a pharmaceutically acceptable salt thereof (degarelix API) in a composition of the inventionis from about 30 wt% to about 35 wt% degarelix free base equivalent. In one aspect, the amount of degarelix API in the pharmaceutical composition is from about 31 wt% to about 34 wt% degarelix free base equivalent. In one aspect, the amount of degarelix API in the pharmaceutical composition is from about 31 wt% to about 33 wt% degarelix free base equivalent. In one aspect, the amount of degarelix API in the pharmaceutical composition is from about 31 wt% to about 32 wt% degarelix free base equivalent.
- a target dose to be administered to a subjectwhen a target dose to be administered to a subject is calculated based on degarelix free base equivalent in the composition, the percent by weight of solvent, and the percent by weight of additive if included, may be modified accordingly.
- methods of treating prostate cancer and central precocious puberty (CPP) as well as methods of reducing serum testosterone levels and suppressing ovarian function by subcutaneously administering the compositions disclosed herein to subjects in need thereofis also contemplated.
- a method of treating prostate cancer in a subjectcomprising subcutaneously administering the compositions disclosed herein to the subject is disclosed.
- the prostate canceris advanced prostate cancer.
- the dose amount of degarelix or pharmaceutically acceptable salt in the compositionis administered in a dose amount of about 40 mg to about 500 mg.
- a method of reducing serum testosterone levels in a subject to 50 ng/dL or less, comprising subcutaneously administering the compositions disclosed herein to the subjectis disclosed.
- the serum testosterone levelis below 20 ng/dL.
- the serum testosterone levelis below 10 ng/dL.
- a method of suppressing ovarian function in a subject with hormone-receptor positive breast cancercomprising subcutaneously administering the compositions disclosed herein to the subject is disclosed.
- the hormone receptor positive breast canceris estrogen receptor (ER) positive breast cancer.
- estradiol (E2) production levelis suppressed to a level less than about 20 pg/mL to about less than about 2 pg/mL.
- the subject’s follicle stimulating hormone (FSH) levelis suppressed to a level less than about 40 IU/L.
- the subject’s luteinizing hormone (LH) levelis suppressed to a level less than about 4 IU/L.
- CPPcentral precocious puberty
- the subject’s CPP blood serum LH concentration levelis reduced to a pre-pubertal concentration level of less than about 4 IU/L.
- the compositionis administered about once every 1 month, about once every 2 months, about once every 3 months, about once every 4 months, about once every 5 months, or about once every six months.
- the compositionis administered to the subject about once every three months.
- the compositionis administered as a loading dose followed by a maintenance dose of the composition about 1 month, 2 months or 3 months after the loading dose has been administered.
- a loading dosewhen a loading dose is administered, the maintenance dose is administered every 1 month, every 2 months or every 3 months thereafter. In one embodiment, no loading dose is administered, and the composition is administered about once every 1 month, about once every 2 months, about once every 3 months, about once every 4 months, about once every 5 months, or about once every six months.
- a delivery systemcomprising a syringe or syringes prefilled with a composition disclosed herein, in some embodiments together with instructions suitable for using the delivery system to administer the composition to a subject.
- a prefilled syringe system for administration of the compositions disclosed herein, comprising a single syringe containing the composition disclosed herein,is described.
- the syringeis a mixing syringe. In one aspect, the syringe is an autoinjector syringe. In still another aspect, the syringe is a dual-chambered syringe, and the composition disclosed herein is contained within one chamber of the syringe. In yet another aspect, the syringe is a dual-chambered syringe, and the biodegradable polymer and biocompatible solvent are contained in one chamber of the syringe and the degarelix or pharmaceutically acceptable salt thereofis contained in the second chamber of the syringe. In one aspect, the degarelix is lyophilized.
- the degarelix or or pharmaceutically acceptable salt thereofis dissolved or suspended in the biocompatible solvent.
- a prefilled syringe system for administration of the compositions disclosed herein, comprising a first syringe containing the biodegradable polymer and the biocompatible solvent, and a second syringe containing the degarelixis described.
- the first syringe and the second syringeare coupled together.
- the degarelix or pharmaceutically acceptable salt thereofis lyophilized.
- the degarelix or pharmaceutically acceptable salt thereofis dissolved or suspended in the biocompatible solvent.
- kitscomprising the prefilled syringe system disclosed herein and instructions for use of the prefilled syringe system to administer a composition contained therein are disclosed.
- a productcomprising the compositions disclosed herein for use in a method of treating prostate cancer or CPP is disclosed.
- a productcomprising the compositions disclosed herein for use in a method of reducing serum testosterone levels below to 50 ng/dL is disclosed.
- a productcomprising the compositions disclosed herein for use in a method of suppressing ovarian function in a subject with hormone-receptor positive breast cancer is disclosed.
- Fig. 1shows the percentage recovery of degarelix from the degarelix/organic solvent formulation samples (formulas 1, 2 and 5 (F1, F2 and F5) )(prefilled syringes) stored at 25°C for 0 months, 1 month, 3 months and 6 months.
- F1Degarelix acetate drug substance (DgA-DS)/N-methyl-2-pyrrolidone (NMP) (65%/35%);
- F2DgA-DS/dimethyl sulfoxide (DMSO) (65%/35%);
- F5DgA-DS/DMSO (60%/35%).
- FIG. 2shows the in vivo degarelix levels in plasma over the first 3 days post- injection using a rat model as described in section 19 of the Example section.
- Fig.3shows the in vivo degarelix levels in plasma over 28 days post-injection using a rat model as described in section 19 of the Example section.
- Fig.4shows the in vivo degarelix levels in plasma over 63 days post-injection using a rat model as described in section 19 of the Example section.
- Fig.5shows a magnified view of the in vivo degarelix levels in plasma over 63 days post-injection using a rat model as described in section 19 of the Example section.
- Fig.6shows the in vivo degarelix levels in plasma over 119 days post-injection using a rat model as described in section 19 of the Example section.
- Fig.7shows a magnified view of the in vivo degarelix levels in plasma over 119 days post-injection using a rat model as described in section 19 of the Example section.
- Fig. 8shows a semi-log view of the in vivo testosterone levels in plasma over 105 days post-injection using a rat model as described in section 19 of the Example section.
- active pharmaceutical ingredientAs used herein, the terms “active pharmaceutical ingredient”, abbreviated as “API”, and “drug” can be used interchangeably and generally refer to a biologically active compound that has therapeutic effects on the body.
- the “active pharmaceutical ingredient”may refer to an active drug, or a pharmaceutically acceptable salt of an active drug. As used herein, these terms may be used to refer to degarelix or a pharmaceutically acceptable salt thereof.
- Decorated drugAs used herein, these terms may be used to refer to degarelix or a pharmaceutically acceptable salt thereof.
- “Degarelix API”(whether capitalized or not) as used herein refers to degarelix or any pharmaceutically acceptable salt thereof.
- esteerrefers to a chemical functional group, C(O)OR’ where R’ represents an alkyl as defined herein.
- antioxidantrefers to compounds which are used to extend the shelf-life of products by preventing or inhibiting the oxidation of active substances and excipients.
- An antioxidantmay react with free radicals thereby blocking or inhibiting free radical chain reactions, or the antioxidant may have a lower redox potential than the active substances and excipients in the formulation. Additionally, or alternatively, a synergist antioxidant may enhance the effects of other antioxidants.
- biocompatiblemeans “not harmful to living tissue” or “safe for injection within a human body”.
- biodegradablerefers to any material that is converted, breaks down, or degrades, under physiological conditions into innocuous or natural byproducts, such as (but not limited to) water, gas, biomass, and/or organic salts, without regard to any specific degradation mechanism or process.
- GPCgel permeation chromatography
- the terms “patient” and “subject”are interchangeable and generally refer to an animal or a human to which a composition disclosed herein is administered or is to be administered.
- the term “pharmaceutically acceptable salt”refers to a salt of a compound, which possesses the desired pharmacological activity of the parent compound.
- Such saltsinclude, but are not limited to: (1) acid addition salts, formed with inorganic acids such as hydrochloric acid, hydrobromic acid, sulfuric acid, nitric acid, phosphoric acid, and the like; or formed with organic acids such as acetic acid, propionic acid, hexanoic acid, cyclopentanepropionic acid, glycolic acid, pyruvic acid, lactic acid, lauric acid, malonic acid, succinic acid, malic acid, maleic acid, fumaric acid, tartaric acid, citric acid, benzoic acid, 3-(4-hydroxybenzoyl)benzoic acid, cinnamic acid, mandelic acid, methanesulfonic acid, ethanesulfonic acid, 1,2-ethane-disulfonic acid, 2-hydroxyethan
- solventrefers to a liquid that dissolves a solid or liquid solute, or to a liquid external phase of a suspension throughout which solid or liquid substances may be suspended or dispersed.
- biocompatible solventmay be used interchangeably with the term “solvent”.
- co-solventrefers to a substance added to a solvent to increase or modify the solubility of a solute in the solvent.
- a co-solventmay increase or decrease solubility of a solute in a primary solvent and/or may impart other desirable characteristics onto a formulation (e.g., the extent of water insolubility of the solvents and co-solvents in a solvent system can impact the desired rate of diffusion into bodily fluids for controlling the rate and scope of degarelix API gelation; or solvent/co- solvents can control the viscosity of the compositions of the invention, which aids in preparing and administering the extended release composition to a subject).
- solvent/co- solventscan control the viscosity of the compositions of the invention, which aids in preparing and administering the extended release composition to a subject.
- the term “solubilizer”refers to a compound that increases the solubility of another substance.
- the term “surfactant”refers to a compound that lowers the surface tension between two liquids, between a gas and a liquid, or between a liquid and a solid.
- a surfactantcan act as a wetting agent, which aids in dispersing an active pharmaceutical ingredient in a liquid vehicle, or as a solubilizer under some circumstances.
- the term “therapeutically effective amount”means the amount of a compound or drug product that, when administered to a patient for treating a disease and/or treating or preventing one or more symptoms of a disease, is sufficient to affect such treatment or prevention for the disease.
- the “therapeutically effective amount”can vary depending on, for example, the compound or drug product, disease progression, the disease or condition to be treated, whether the therapy is an adjuvant therapy or a primary or curative therapy, and/or the age, weight, etc., of the patient to be treated.
- a therapeutically effective amount of degarelixcan include an amount effective to: (1) treat a disease including, but not limited to, advanced prostate cancer; (2) decrease plasma levels of luteinizing hormone (LH), reduce plasma level of follicle stimulating hormone (FSH), and/or reduce and maintain serum testosterone levels below castration level of 50 ng/dL; and/or (3) provide a plasma level of degarelix in a subject that is sufficient to achieve any of the results in (2).
- Therapeutically effective amounts of degarelix or a pharmaceutically acceptable salt thereof in a variety of diseases and conditionsare described in more detail below.
- area under the curveis the area under a plot of the concentration of a drug in the blood plasma as a function of time, and reflects the actual body exposure to a drug after administration of a dose of the drug. AUC is dependent on the rate of elimination of the drug from the body and the dose administered.
- the terms "at least one”, “one or more”, and “and/or”are open- ended expressions that are both conjunctive and disjunctive in operation.
- each of the expressions "at least one of A, B and C", “at least one of A, B, or C", “one or more of A, B, and C", “one or more of A, B, or C", “A, B, and/or C", and "A, B, or C”means A alone, B alone, C alone, A and B together, A and C together, B and C together, or A, B and C together.
- each one of A, B, and C in the above expressionsrefers to an element, such as X, Y, and Z, or class of elements, such as X1-Xn, Y1-Ym, and Z1-Zo
- the phraseis intended to refer to a single element selected from X, Y, and Z, a combination of elements selected from the same class (e.g., X1 and X2) as well as a combination of elements selected from two or more classes (e.g., Y1 and Zo).
- the term “depot”refers to the degarelix and/or biodegradable polymer in a composition/formulation disclosed herein once exposed to an aqueous environment (e.g., including a physiological environment in a body of an animal) in which, upon dissipation of the solvent in situ, a solid, semi-solid, or liquid depot is formed comprising the biodegradable polymer and the degarelix API which self-aggregates (e.g., as fibrils or a gel). Together, the biodegradable polymer and the degarelix API create a drug reservoir from which the degarelix is released over an extended period of time.
- an aqueous environmente.g., including a physiological environment in a body of an animal
- Every numerical range given throughout this disclosureis deemed to include both terminal values as well as each and every narrower numerical range that falls within such broader numerical range, as if such narrower numerical ranges were all expressly written herein.
- the phrase from about 2 to about 4includes the whole number and/or integer ranges from about 2 to about 3, from about 3 to about 4 and each possible range based on real (e.g., irrational and/or rational) numbers, such as from about 2.1 to about 4.9, from about 2.1 to about 3.4, and so on.
- Degarelixis a decapeptide, selective gonadotropin-releasing hormone (GnRH) receptor antagonist (blocker) that competitively and reversibly binds to GnRH receptors in the pituitary. This results in a significant reduction of follicle-stimulating hormone (FSH) and luteinizing hormone (LH), thus reducing testosterone levels in males, or estrogen levels in females.
- GnRHselective gonadotropin-releasing hormone
- FSHfollicle-stimulating hormone
- LHluteinizing hormone
- degarelix compositions of the inventionare indicated for LH suppression or testosterone suppression in adult male patients with advanced hormone-dependent prostate cancer in whom androgen deprivation is warranted.
- Degarelix compositions of the inventionare also useful for LH suppression or estrogen (e.g., estradiol) suppression in adult female patients with pre-menopausal or perimenopausal breast cancer, and are also useful for conditions including, but not limited to, endometriosis or uterine fibroids.
- Degarelix compositionsare also useful for LH suppression or sex hormone suppression (testosterone or estrogen, depending on whether the patient is male or female) in children with central precocious puberty (CPP).
- CPPcentral precocious puberty
- a long-acting injectable formulation of degarelixthat (1) is easy to prepare and administer (e.g., by a health care provider); (2) provides sustained or extended release of degarelix for more than a month after a single dose, and in one embodiment, for at least three months or longer; and (3) is administered in a total injection volume of about 2.5 mL or less, or about 2 mL or less.
- a long acting, injectable composition or formulation(used interchangeably and which may also be referred to as a “drug product” “pharmaceutical product” “product” or “finished drug product”) that comprises degarelix or a pharmaceutically acceptable salt thereof, in polymer-based drug delivery system.
- the formulationcomprises a biodegradable polymer and a biocompatible, water-miscible solvent, and can optionally further include an additive as disclosed herein.
- the degarelix polymeric composition/formulationis a flowable solution/suspension where, upon delivery (i.e., injection) into an aqueous environment (e.g., the body of a human), the water-miscible solvent exchanges with surrounding aqueous body fluids, which results in the formation of a polymer-drug depot, that acts as a drug reservoir for extended release of the drug (i.e. release of the drug over an extended period of time).
- composition/formulations disclosed hereinprovide sustained or extended release of degarelix or a pharmaceutically acceptable salt thereof for more than a month when injected at low volumes (e.g., ⁇ 2.5 mL or ⁇ 2 mL) via subcutaneous injection.
- the long-acting injectable compositions suitable for use in the methods of this disclosurewhich may also be referred to as pharmaceutical compositions or formulations, extended-release compositions or formulations, or controlled release compositions or formulations, provide a biodegradable or bioerodible microporous in situ formed depot of biodegradable polymer and self-aggregated degarelix in a subject, from which the degarelix or a pharmaceutically acceptable salt thereof, is released over a period of one month or longer, and preferably at least three months or longer.
- the composition/formulationcan be supplied in a prefilled single or dual chamber syringe, or in a two-syringe mixing system.
- the compositionis administered from the syringe system in injection volumes of less than or equal to 2.5 mL, less than or equal to 2.4 mL, less than or equal to 2.3 mL, less than or equal to 2.2 mL, less than or equal to 2.1 mL, less than or equal to 2.0 mL, less than or equal to 1.9 mL, less than or equal to 1.8 mL, less than or equal to 1.7 mL, less than or equal to 1.6 mL, or less than equal to 1.5 mL and is suitable for subcutaneous injection.
- This compositionsolves the problem of the multiple, high-volume injections provided by the current degarelix product on the market.
- degarelix or a pharmaceutically acceptable salt thereofis released from the polymer depot over a period of at least about 1 month or longer, at least about 1.5 months or longer, at least about 2 months or longer, at least about 2.5 months or longer, at least about 3 months or longer, at least about 3.5 months or longer, at least about 4 months or longer, at least about 4.5 months or longer, at least about 5 months or longer, or at least about 6 months or longer.
- compositions/formulations disclosed hereincomprise degarelix or a pharmaceutically acceptable salt thereof in polymer-based drug delivery system.
- the pharmaceutically acceptable salts of the degarelixinclude, but are not limited to, degarelix acetate, degarelix citrate, degarelix pamoate, and degarelix mesylate.
- “Degarelix API”(whether capitalized or not) as used herein refers to degarelix or any pharmaceutically acceptable salt thereof.
- the disclosed compositionscomprise a biodegradable polymer, a biocompatible solvent, and degarelix or a pharmaceutically acceptable salt thereof.
- the biodegradable polymer or copolymersare: substantially insoluble in water and body fluid, biocompatible, and biodegradable and/or bioerodible within the body of a subject.
- the pharmaceutical compositionsare administered to a patient by subcutaneous injection as a liquid or gel wherein a solid, semi-solid, or liquid depot is formed in situ upon dissipation of the solvent.
- the depot so formedreleases degarelix or a pharmaceutically acceptable salt thereof, in a controlled, or extended, release manner.
- concentration of the degarelix or a pharmaceutically acceptable salt thereof, in the disclosed compositionscan vary and may range from about 1% to about 40% by weight of the final formulation or composition (i.e., the formulation or composition in its ready-to-inject state), including any whole number percent to any other whole number percent within the range of from about 1 percent to about 40 percent by weight.
- the total weight percent of each of the components in the final formulation/compositionequals 100% (e.g., if the amount of degarelix salt in the formulation is 20 wt%, then the total amount of polymer and solvent in the formulation, absent any additives, is 80 wt%).
- the concentration of the degarelix or pharmaceutically acceptable salt thereof in the compositionmay be about 5% by weight of the composition, or about 10% by weight of the composition, or about 15% by weight of the composition, or about 20% by weight of the composition, or about 25% by weight of the composition, or about 30% by weight of the composition, or about 35% by weight of the composition, or about 40% by weight of the composition.
- the concentration of degarelix or pharmaceutically acceptable salt thereofis no more than about 25% by weight of the composition.
- the amount of degarelix API in compositions of the inventionmay range from any tenth of a percent to any other tenth of a percent within the range of from about 1% to about 40% by weight.
- the amount of degarelix API in a composition of the inventionis referenced with respect to the amount of a pharmaceutically acceptable salt of degarelix, such as degarelix acetate or other salt, in the composition.
- the amount of degarelix API in a composition of the inventionis referenced with respect to the amount of degarelix free base equivalent in the composition (which may also simply be referred to as the amount of degarelix in the composition).
- determination of the dose amount of degarelix API to be administered to a subjectmay be calculated based on the amount of degarelix free base equivalent in the composition rather than the amount of degarelix salt in order to account for differences in purity, water, or acetic acid content (e.g., in the case of degarelix acetate), among various API lots. Calculation of the free base equivalent of degarelix in a composition is well understood within the art.
- the percentage (by weight) of degarelix as free base in a composition where the degarelix API is degarelix acetatecan be calculated in a simple formula such as: 100 – (wt% drug substance water content – wt% drug substance acetic acid content) * degarelix purity by assay (anhydrous/acid free base) * wt% degarelix acetate in the composition.
- the free base equivalentmay also be provided from the USP Monograph for a drug substance.
- the amount of degarelix or the pharmaceutically acceptable salt thereof in the pharmaceutical compositionis from about 8 wt% to about 39 wt% degarelix free base equivalent.
- the amount of degarelix or the pharmaceutically acceptable salt thereof in the pharmaceutical compositionis from about 17 wt% to about 39 wt% degarelix free base equivalent. In one aspect, the amount of degarelix or the pharmaceutically acceptable salt thereof in the pharmaceutical composition is from about 8 wt% to about 29 wt% degarelix free base equivalent. In one aspect, the amount of degarelix or the pharmaceutically acceptable salt thereof in the pharmaceutical composition is from about 8 wt% to about 20 wt% degarelix free base equivalent. In one aspect, the amount of degarelix or the pharmaceutically acceptable salt thereof in the pharmaceutical composition is from about 10 wt% to about 16 wt% degarelix free base equivalent.
- the amount of degarelix or the pharmaceutically acceptable salt thereof in the pharmaceutical compositionis from about 14 wt% to about 15.5 wt% degarelix free base equivalent. In one aspect, the amount of degarelix or a pharmaceutically acceptable salt thereof (degarelix API) in a composition of the invention is from about 30 wt% to about 35 wt% degarelix free base equivalent. In one aspect, the amount of degarelix API in the pharmaceutical composition is from about 31 wt% to about 34 wt% degarelix free base equivalent. In one aspect, the amount of degarelix API in the pharmaceutical composition is from about 31 wt% to about 33 wt% degarelix free base equivalent.
- the amount of degarelix API in the pharmaceutical compositionis from about 31 wt% to about 32 wt% degarelix free base equivalent.
- the percent by weight of solvent, and the percent by weight of additive if includedmay be modified accordingly.
- Biocompatible/biodegradable Polymersinclude polymers, copolymers and/or terpolymers formed of repeating units, which can be linear, branched, grafted and/or star-shaped.
- the polymerscan comprise monomer residues, such as lactide, glycolide, caprolactone, p-dioxanone, trimethylene carbonate, 1,5-dioxepan-2-one, 1,4-dioxepan-2-one, ethylene oxide, propylene oxide, sebacic anhydride, diketene acetals/diols, lactic acid, and combinations thereof.
- monomer residuessuch as lactide, glycolide, caprolactone, p-dioxanone, trimethylene carbonate, 1,5-dioxepan-2-one, 1,4-dioxepan-2-one, ethylene oxide, propylene oxide, sebacic anhydride, diketene acetals/diols, lactic acid, and combinations thereof.
- Biodegradable polymers suitable for use with the disclosed compositionsinclude, but are not limited to, polylactic acid, polyglycolic acid, polylactide (D-lactide, L- lactide), poly(D,L-lactide) (PLA), polyglycolide, polycaprolactones, poly(D,L-lactide-co- glycolide) (PLG), poly(D,L-lactide-co- ⁇ -caprolactone) (PLC), polyanhydrides, polyamides, polyurethanes, polyesteramides, polyorthoesters, polydioxanones, polyacetals, polyketals, polycarbonates, polyphosphazenes, polyhydroxybutyrates, polyhydroxyvalerates, polyalkylene oxalates, polyalkylene succinates, poly(malic acid), polyglutamic acids, poly(alkyl cyanoacrylates) polyethylene glycol, hyaluronic acid, alginate, collagen, chi
- the biodegradable polymermay be a copolymer of two monomers having a molar ratio of any two whole numbers X to Y, such that the sum of X and Y is 100.
- the biodegradable polymermay be a copolymer of two monomers having a molar ratio of any two whole numbers X to Y, where X and Y are each at least about 10 and no more than about 90, such that the sum of X and Y is 100, e.g., a copolymer comprising a 10:90 to 90:10 molar ratio of X:Y.
- X and Ymay be at least about 15 to no more than about 85 such that the sum of X and Y is 100, e.g., a copolymer comprising a 15:85 to 85:15 molar ratio of X:Y.
- X and Ymay be at least about 25 to no more than about 75 such that the sum of X and Y is 100, e.g., a copolymer comprising a 25:75 to 75:25 molar ratio of X:Y.
- both X and Ymay be about 50, e.g., a copolymer comprising a 50:50 molar ratio of X:Y.
- the biodegradable polymeris a copolymer comprising (a) D,L-lactide, D-lactide, L-lactide or glycolide monomer residues and (b) caprolactone monomer residues.
- the biodegradable polymeris a copolymer comprising lactide, preferably D,L-lactide, and caprolactone monomer residues.
- the biodegradable polymeris poly(D,L-lactide-co- ⁇ -caprolactone) (PLC).
- PLCcomprises at least one carboxylic acid end group.
- the PLCis a carboxylic acid-initiated PLC.
- the carboxylic acidis glycolic acid or lactic acid.
- the polymeris a copolymer comprising a molar ratio of lactide (or glycolide) to ⁇ -caprolactone ranging from about 10:90 to about 90:10, from about 20:80 to about 80:20, from about 25:75 to about 75:25, or from about 30:70 to about 70:30.
- the ratio of lactide (or glycolide) to ⁇ -caprolactoneis from about 25:75 to about 75:25.
- the ratio of lactide (or glycolide) to ⁇ -caprolactoneis from about 50:50 to about 75:25.
- the ratio of lactide (or glycolide) to ⁇ - caprolactoneis about 50:50. In still another aspect, the ratio of lactide (or glycolide) to ⁇ - caprolactone is about 75:25. In one aspect, the biodegradable polymer is acid-initiated 75:25 poly(D,L-lactide-co- ⁇ -caprolactone). [00100] In some embodiments, the biodegradable polymer is a copolymer comprising (a) D,L-lactide, D-lactide, L-lactide or glycolide monomer residues and (b) trimethylene carbonate (TMC) monomer residues.
- TMCtrimethylene carbonate
- the biodegradable polymeris a copolymer comprising lactide, preferably D,L-lactide, and TMC monomer residues.
- the biodegradable polymeris a copolymer comprising a molar ratio of lactide (or glycolide) to TMC ranging from about 10:90 to about 90:10, from about 20:80 to about 80:20, from about 25:75 to about 75:25, or from about 30:70 to about 70:30.
- the ratio of lactide (or glycolide) to TMCis from about 25:75 to about 75:25.
- the ratio of lactide (or glycolide) to TMCis about 50:50.
- the ratio of lactide (or glycolide) to TMCis about 75:25.
- the biodegradable polymeris a copolymer comprising (a) D,L-lactide, D-lactide, or L-lactide monomer residues and (b) glycolide monomer residues.
- the biodegradable polymeris poly(DL-lactide-co-glycolide) (PLG).
- PLGcomprises at least one carboxylic acid end group.
- the PLGis a core-diol-initiated PLG.
- the core diolis 1,6-hexane diol.
- the PLGwas initiated with an uncapped polyethylene glycol (PEG).
- the PLGis a mono functional alcohol-initiated PLG.
- the mono functional alcoholis dodecanol.
- the PLGwas initiated with a mono- capped PEG (mPEG).
- the PLGhas a molar ratio of lactide monomers to glycolide monomers from about 50:50 to about 90:10.
- the PLGhas a molar ratio of lactide to glycolide monomers from about 45:55 to about 99:1.
- the PLG copolymermay comprise a lactide to glycolide monomer molar ratio from about 50:50 to about 90:10, from about 50:50 to about 80:20, from about 50:50 to about 70:30, or from about 50:50 to about 75:25. In one aspect the molar ratio of lactide monomers to glycolide monomers is 50:50, 75:25 or 85:15. In one aspect, the biodegradable polymer is acid-initiated 50:50 poly(D,L-lactide-co-glycolide). In another aspect, the biodegradable polymer is 1,6-hexane diol initiated 75:25 poly(D,L-lactide-co-glycolide).
- the biodegradable polymeris 1,6-hexane diol initiated 85:15 poly(D,L- lactide-co-glycolide). In yet a further aspect, the biodegradable polymer is dodecanol- initiated poly(D,L-lactide). [00102] In some embodiments, the biodegradable polymer is poly(D,L-lactide) (PLA). In one aspect, the (PLA) comprises at least one carboxylic acid end group.
- suitable polymersinclude biodegradable polymers comprising a copolymer with lactide (including D,L-lactide, D-lactide, and/or L- lactide) and/or glycolide residues, wherein the molar percentage of the lactide and/or glycolide residues make up greater than about 5% and less than about 95% of the polymer.
- lactideincluding D,L-lactide, D-lactide, and/or L- lactide
- glycolide residueswherein the molar percentage of the lactide and/or glycolide residues make up greater than about 5% and less than about 95% of the polymer.
- the lactide and/or glycolide monomer residuesmake up at least about 5%, at least about 10%, at least about 15%, at least about 20%, at least about 25%, at least about 30%, at least about 35%, at least about 40%, at least about 45%, at least about 50%, at least about 55%, at least about 60%, at least about 65%, at least about 70%, at least about 75%, at least about 80%, at least about 85%, at least about 90%, or at least about 95% of total monomer residues of the copolymer.
- suitable polymers of the inventioninclude biodegradable polymers comprising a copolymer with caprolactone and/or TMC residues, wherein the caprolactone and/or trimethylene carbonate residues make up in an amount greater than about 5% and less than about 95% of the polymer.
- the caprolactone and/or TMC monomer residuesmake up at least about 5%, at least about 10%, at least about 15%, at least about 20%, at least about 25%, at least about 30%, at least about 35%, at least about 40%, at least about 45%, at least about 50%, at least about 55%, at least about 60%, at least about 65%, at least about 70%, at least about 75%, at least about 80%, at least about 85%, at least about 90%, or at least about 95% of total monomers of the copolymer.
- the biodegradable polymermay be formed using an initiator selected to provide a desired structure or functionality to the polymer in the form of a particular polymer block structure or end group structure which is introduced and/or incorporated into/onto the biodegradable polymer.
- the polymermay be formed using an organic acid (e.g., a hydroxy acid such as, for instance, glycolic acid) as the initiator which may result in the formation of a polymer comprising at least one carboxylic acid end group.
- the polymermay be formed using a monofunctional alcohol (e.g., dodecanol, or monomethoxy endcapped monofunctional polyethylene glycols (PEGs)) as the initiator which may result in the formation of a polymer comprising at least one hydroxy end group.
- a monofunctional alcohole.g., dodecanol, or monomethoxy endcapped monofunctional polyethylene glycols (PEGs)
- PEGsmonomethoxy endcapped monofunctional polyethylene glycols
- the polymermay be formed using a diol (e.g., hexanediol, PEG300, PEG400, PEG600) as the initiator which may result in the formation of a polymer comprising at least one hydroxy end group and that is also substantially free of terminal carboxy end groups.
- the biodegradable polymermay be formed using an initiator selected to provide a desired structure, and in particular a desired polymer block structure or end group structure, to the biodegradable polymer.
- the polymermay be formed using a low-molecular weight PEG (e.g., the above-mentioned PEG300, PEG400 or PEG600) as an initiator, which may result in the formation of a block copolymer comprising a low-molecular weight PEG block.
- block Amay comprise first monomer residues selected from D,L- lactide, D-lactide, or L-lactide, and second monomer residues selected from glycolide, ⁇ - caprolactone, or trimethylene carbonate.
- Block Bcomprises a low-molecular weight polyethylene glycol (PEG).
- the blocksmay be arranged in any number or order (e.g. as a di-block copolymer A-B, or a tri-block copolymer A-B-A or B-A-B). Such polymers are formed by initiation of the first and second monomer residues with a low-molecular weight PEG initiator.
- biodegradable polymers suitable for use in formulations according to the present inventionmay, generally, comprise a weight average molecular weight ranging from about 1 kDa and about 100 kDa.
- the biodegradable polymermay comprise a weight average molecular weight ranging from about 1 kDa to about 5 kDa, from about 1 kDa to about 10 kDa, from about 1 kDa to about 15 kDa, from about 1 kDa to about 20 kDa, from about 1 kDa to about 25 kDa, from about 1 kDa to about 30 kDa, from about 1 kDa to about 40 kDa, from about 1 kDa to about 50 kDa, from about 1 kDa to about 60 kDa, from about 1 kDa to about 70 kDa, from about 1 kDa to about 80 kDa, from about 1 kDa to about 90
- the weight average molecular weightis from about 5 kDa to about 60 kDa. In one aspect, the weight average molecular weight is from about 5 kDa to about 55 kDa. In one aspect, the weight average molecular weight is from about 5 kDa to about 50 kDa. In one aspect, the weight average molecular weight is from about 5 kDa to about 45 kDa. In one aspect, the weight average molecular weight is from about 5 kDa to about 40 kDa. In still another aspect, the weight average molecular weight is from about 5 kDa to 35 kDa.
- the biodegradable polymermay make up about 10 wt%, about 15 wt%, about 20 wt%, about 25 wt%, about 30 wt%, about 35 wt%, about 40 wt%, about 45 wt%, about 50 wt%, about 55 wt%, or about 60 wt% of the composition.
- the biodegradable polymermay make up about 20 wt%, about 25 wt%, or about 30 wt% of the composition.
- the biodegradable polymermay make up any whole-number weight percentage of the composition from about 10 wt% to about 60 wt%.
- the biodegradable polymermay make up any tenth of a whole number percent of the composition from about 10 wt% to about 60 wt%.
- the biodegradable polymersmay optionally be purified prior to use in the long-acting formulation to remove low-molecular weight oligomers and/or unreacted monomers, and/or catalyst.
- purifying polymersinclude the methods described in U.S. Pat. No. 4,810,775, U.S. Patent No. 7,019,106, and U.S. Patent No. 9,187,593, among others.
- the biocompatible polymercomprises less than about 0.5%, or less than about 0.4%, or less than about 0.3%, or less than about 0.2% or less than about 0.1% unreacted monomers. In some embodiments of the composition, the biocompatible polymer comprises less than about 0.5%, or less than about 0.4%, or less than about 0.3%, or less than about 0.2% or less than about 0.1% unreacted lactide monomer.
- Biocompatible SolventsAny suitable water-miscible solvent can be employed, provided the solvent is biocompatible, and miscible to dispersible in aqueous medium or body fluid. Examples of suitable solvents are disclosed, e.g., in Aldrich Handbook of Fine Chemicals and Laboratory Equipment, Milwaukee, Wis.
- the solventis able to diffuse into body fluid so that the flowable polymer-based composition coagulates, gels or solidifies.
- the solventmay or may not dissolve the polymer, although it is preferred that the solvent dissolves the polymer.
- Solvents and co-solvents that may be used in the disclosed compositionsare preferably biocompatible, non-toxic, solvents, which may be either hydrophilic or hydrophobic solvents, or may be a combination of hydrophilic solvents, hydrophobic solvents or hydrophilic and hydrophobic solvents, depending upon the desired release profile and the solubility of the polymer and/or the degarelix API in the polymer/solvent composition.
- the solvent and/or co-solventis an organic solvent.
- the solvent and/or co-solventsis a polar aprotic solvent.
- Suitable solvents and co-solventsmay comprise one or more solvents selected from the group consisting of amides, acids, alcohols, esters of monobasic acids, ether alcohols, sulfoxides, lactones, polyhydroxy alcohols, esters of polyhydroxy alcohols, ketones, and ethers.
- the solvents or cosolventsmay comprise at least one of N-methyl-2-pyrrolidone (NMP), dimethyl sulfoxide (DMSO), dimethylacetamide (DMA), acetone, benzyl benzoate (BnBzO), polyethylene glycol 15 hydroxystearate, ethyl acetate, glycofurol, N-hydroxyethyl-2-pyrrolidone, polyethylene glycol (PEG), benzyl alcohol, propylene carbonate (PC), propylene glycol, 2-pyrrolidone, ⁇ -tocopherol, triacetin, tributyl citrate, acetyl tributyl citrate, acetyl triethyl citrate, triethyl citrate, and combinations thereof.
- NMPN-methyl-2-pyrrolidone
- DMSOdimethyl sulfoxide
- DMAdimethylacetamide
- acetonebenzyl benzoate
- a suitable solventis selected from N-methyl-2- pyrrolidone (NMP) and/or dimethyl sulfoxide (DMSO).
- NMPN-methyl-2- pyrrolidone
- DMSOdimethyl sulfoxide
- the biodegradable polymeris dissolved in the biocompatible solvent or combination or mixture of solvents and/or co-solvents.
- the degarelix or pharmaceutically acceptable salt thereofmay be dissolved in a first biocompatible solvent and the biodegradable polymer may be dissolved in a second biocompatible solvent.
- the first and second biocompatible solventsare the same.
- the biodegradable polymeris dissolved in the biocompatible solvent, and the degarelix or pharmaceutically acceptable salt thereof is not dissolved in the solvent, but is provided in lyophilized or other dry form.
- the biocompatible solvent, or combination or mixture of solvents and/or co- solvents used in a composition of the inventionwill generally comprise between about 20 wt% and about 80 wt% of the final formulation, or between about 30 wt% and about 70 wt% of the final formulation, or between about 40 wt% and about 60 wt% of the final formulation, or alternatively the solvent or combination or mixture of solvents and/or co- solvents can range from any whole number percentage by weight of the formulation to any other whole number percentage by weight of the final formulation between about 20 wt% and about 80 wt%.
- the pharmaceutical compositions disclosed hereinmay comprise various additives (which may also be referred to as “excipients”) to improve the stability, the injectability, or/and other properties of the composition.
- the pharmaceutical compositionsmay comprise one or more of antioxidants, chelating agents, surfactants, co-solvents (also discussed above), stabilizers, complexing agents, antioxidants, and solubilizers.
- the pharmaceutical compositionsmay comprise one or more solubilizers to increases the solubility of one or more other component of the composition.
- Solubilizers useful in the disclosed compositionsinclude any solubilizer useful for parenteral injection, and include, but are not limited to, surfactants which lower the surface tension between two liquids, between a gas and a liquid, or between a liquid and a solid and other solubilizers.
- solubilizers and/or surfactantsfor use in the invention include, but are not limited to, polysorbate 20, polysorbate 80, poloxamer 188, sorbitan trioleate, lecithin (e.g., soya or egg), Vitamin E TPGS, sugar based esters or ethers (e.g., sugar acid esters of fatty alcohols or sugar alcohol esters of fatty acids, including, but not limited to, sucrose cocoate, sucrose stearate, sucrose laurate, etc.), amino acid-based solubility enhancers (e.g., proline, arginine, DL-methionine), protein-based solubility enhancers (e.g., hydrophobins), and others.
- sugar based esters or etherse.g., sugar acid esters of fatty alcohols or sugar alcohol esters of fatty acids, including, but not limited to, sucrose cocoate, sucrose stearate, sucrose laurate, etc.
- the pharmaceutical compositionsmay comprise one or more antioxidants to inhibit oxidation of the API and improve the stability of the formulation.
- suitable antioxidants for use in the inventioninclude, but are not limited to, citric acid, methanesulfonic acid, ascorbic acid, ethylenediaminotetraacetic acid (EDTA), mercaptoethanol, sodium metabisulfite butylated hydroxyanisole, butylated hydroxyquinone, butylhydroxyanisol, hydroxycoumarin, butylated hydroxytoluene, cephalm, ethyl gallate, propyl gallate, octyl gallate, lauryl gallate, propyl-hydroxybenzoate, trihydroxybutylrophenone, dimethylphenol, dibutylphenol, vitamin E, lecithin and ethanolamine.
- the pharmaceutical compositionsmay comprise one or more complexing agents to inhibit oxidation and/or degradation of the API and improve the stability of the formulation.
- suitable complexing agents for use in the inventioninclude, but are not limited to, ethylenediaminotetraacetic acid (EDTA), divalent metal salts (ZnCl 2 , MgCl 2 , CaCl 2 ), nitrilotriacetic acid, n-hydroxyethylethylenediaminetriacetic acid (HEDTA), ethylene glycol-bis( ⁇ -aminoethyl ether)-N,N,N′,N′-tetraacetic acid (EGTA) as well as several simple organic acids, such as, polycarboxylic acid (citric acid), hydrochloric acid, sulfuric acid, pamoic acid, and palmitic acid.
- EDTAethylenediaminotetraacetic acid
- ZnCl 2 , MgCl 2 , CaCl 2divalent metal salts
- HEDTAn
- the pharmaceutical compositionsmay comprise one or more stabilizing agents to prevent drug and/or polymer degradation (physical or chemical) and improve the stability of the formulation and increase shelf life.
- stabilizing agentsinclude, but are not limited to: surfactants (e.g., polysorbate 20, polysorbate 80, poloxamer 188), complexing agents (e.g., divalent metal salts), acid additives (e.g., acetic acid, citric acid, succinic acid, methane sulfonic acid, sulfuric acid, hydrochloric acid, pamoic acid, palmitic acid, hydrobromic acid, nitric acid, chromic acid, trifluoroethanes sulfonic acid, trichloroacetic acid, dichloroacetic acid, bromoacetic acid, chloroacetic acid, cyanoacetic acid, 2-chloropropanoic acid, 4-cyanobutanoic acid, perchloric acid, phosphoric acid, hydrogen iodide, and combinations
- compositionscomprising degarelix or a pharmaceutically acceptable salt thereof, a biocompatible and biodegradable polymer, a biocompatible solvent, and optionally one or more additives. All such compositions are contemplated for administration to a subject to treat a disease or condition for which administration of a GnRH agonist or GnRH antagonist may be useful.
- compositions of the inventionare contemplated for use to treat prostate cancer, including advanced prostate cancer; to treat central precocious puberty (CPP); to treat pre-menopausal or peri-menopausal breast cancer; or to treat other conditions, including, but not limited to endometriosis or uterine fibroids.
- compositionsare contemplated for administration to a subject to reduce luteinizing hormone (LH) levels in a subject, to reduce follicle stimulating hormone (FSH) levels in a subject, and in males, to reduce serum testosterone levels, and in females, to reduce serum estrogen levels.
- LHluteinizing hormone
- FSHfollicle stimulating hormone
- the degarelix or a pharmaceutically acceptable salt thereofin the disclosed compositions may form a monophasic mixture (e.g., a solution) or a biphasic mixture (e.g., a suspension or a dispersion) within the composition.
- the extended release compositions comprising the degarelix or pharmaceutically acceptable salt thereof, according to the present inventionmay appropriately be either a “solution”, or a “dispersion”, or a “suspension” of the degarelix or pharmaceutically acceptable salt thereof, in the solvent or in the combination of the biodegradable polymer and the solvent.
- the degarelix or pharmaceutically acceptable salt thereofmay in some embodiments be dissolved in solvent (as in a solution), or in the combination of polymer and solvent (as in a solution), or form solid particles sufficiently large for suspension and sedimentation as part of a heterogeneous mixture (as in a suspension or dispersion).
- the degarelix or pharmaceutically acceptable salt thereofis dissolved in the solvent when in the absence of the biocompatible polymer, and, when mixed in combination with the solvent(s) and the biocompatible polymer, may be in suspension or dispersion, or partial suspension, where some of the degarelix or pharmaceutically acceptable salt remains in solution and some is in suspension or dispersion.
- the polymer- solvent system compositionmay be a liquid-liquid dispersion.
- types of dispersionsmay include liquid-in oil, oil-in liquid, or oil-in-oil dispersions.
- the polymer-solvent system compositionmay be an emulsion.
- the degarelix APIis a stable solution and stays in solution in the biocompatible solvent and thus little or no reconstitution or mixing is needed to maintain the stable solution.
- the present inventionprovides a pharmaceutical composition
- a pharmaceutical compositioncomprising in the final formulation: (a) between about 10 wt% and about 30 wt% of a pharmaceutically acceptable salt of degarelix; (b) between about 20 wt% and about 80 wt% of N-methyl-2-pyrrolidone (NMP); and (c) between about 10 wt% and about 50 wt% of 75:25 poly(lactide-co- ⁇ -caprolactone) (PLC) copolymer, where the total weight percent of each of the components in the formulation equals 100% (e.g., if the amount of degarelix salt in the formulation is 20 wt%, then the total amount of polymer and solvent in the formulation, absent any additives, is 80 wt%).
- PLCpoly(lactide-co- ⁇ -caprolactone)
- the PLC copolymerhas at least one carboxylic acid end group. In one aspect, the PLC copolymer has a weight average molecular weight of between about 5 kDa and about 30 kDa. In one aspect, the pharmaceutically acceptable salt of degarelix is selected from degarelix acetate and degarelix citrate.
- the present disclosureprovides a pharmaceutical composition
- a pharmaceutical compositioncomprising about 16 wt% of degarelix acetate; about 57 wt% of N-methyl-2- pyrrolidone (NMP); and about 27 wt% of 75:25 poly(lactide-co- ⁇ -caprolactone) (PLC) copolymer having at least one carboxylic acid end group and having a weight average molecular weight of about 17 kDa.
- the present disclosureprovides a pharmaceutical composition
- a pharmaceutical compositioncomprising about 16 wt% of degarelix acetate; about 57 wt% of NMP; and about 27 wt% of 75:25 PLC copolymer having at least one carboxylic acid end group and having a weight average molecular weight of about 8 kDa.
- the present inventionprovides a pharmaceutical composition
- a pharmaceutical compositioncomprising a first container comprising 30-70 wt% of 75:25 PLC copolymer having at least one carboxylic acid end group and having a weight average molecular weight of 5-30 kDa, and 30-70 wt% of N-methyl-2-pyrrolidone (NMP); and a second container comprising 20-40 wt% of a pharmaceutically acceptable salt of degarelix and 60-80 wt% NMP.
- the pharmaceutically acceptable salt of degarelixis selected from degarelix acetate and degarelix citrate.
- the first and second containersare a first and second syringe, respectively.
- the first and second containersare a first and second chamber of a dual-chambered syringe, respectively.
- the present disclosureprovides a pharmaceutical composition comprising a first container comprising 50 wt% of 75:25 PLC copolymer having at least one carboxylic acid end group and having a weight average molecular weight of 17 kDa, and 50 wt% of N-methyl-2-pyrrolidone (NMP); and a second container comprising 35 wt% of degarelix acetate and 65 wt% NMP.
- the first and second containersare a first and second syringe, respectively.
- the first and second containersare a first and second chamber of a dual-chambered syringe, respectively.
- the present disclosureprovides a pharmaceutical composition comprising: a first container comprising 70 wt% of 75:25 PLC copolymer having at least one carboxylic acid end group and having a weight average molecular weight of 8 kDa, and 30 wt% of NMP; and a second container comprising 35 wt% of degarelix acetate and 65 wt% NMP.
- the first and second containersare a first and second syringe, respectively.
- the first and second containersare a first and second chamber of a dual-chambered syringe, respectively.
- the present inventionprovides a pharmaceutical composition comprising in the final formulation between about 10 wt% and about 30 wt% of a pharmaceutically acceptable salt of degarelix; between about 20 wt% and about 80 wt% of N-methyl-2-pyrrolidone (NMP); and between about 10 wt% and about 50 wt% of 75:25 poly(D,L-lactide) (PLA) polymer, where the total weight percent of each of the components in the formulation equals 100% (e.g., if the amount of degarelix salt in the formulation is 20 wt%, then the total amount of polymer and solvent in the formulation, absent any additives, is 80 wt%).
- NMPN-methyl-2-pyrrolidone
- PLApoly(D,L-lactide)
- the PLA polymerhas at least one carboxylic acid end group. In one aspect, the PLA polymer has a weight average molecular weight of between about 5 kDa and about 30 kDa. In one aspect, the pharmaceutically acceptable salt of degarelix is selected from degarelix acetate and degarelix citrate. [00127] In some embodiments, the present disclosure provides a pharmaceutical composition comprising about 16 wt% of degarelix acetate; about 57 wt% of NMP; and about 27 wt% of poly(D,L-lactide) (PLA) polymer having at least one carboxylic acid end group and having a weight average molecular weight of about 23 kDa.
- PLApoly(D,L-lactide)
- the present inventionprovides a pharmaceutical composition
- a pharmaceutical compositioncomprising a first container comprising 30-70 wt% of 75:25 PLA polymer having at least one carboxylic acid end group and having a weight average molecular weight of 5-30 kDa, and 30-70 wt% of N-methyl-2-pyrrolidone (NMP); and a second container comprising 20-40 wt% of a pharmaceutically acceptable salt of degarelix and 60-80 wt% NMP.
- the pharmaceutically acceptable salt of degarelixis selected from degarelix acetate and degarelix citrate.
- the first and second containersare a first and second syringe, respectively.
- the first and second containersare a first and second chamber of a dual-chambered syringe, respectively.
- the present disclosureprovides a pharmaceutical composition comprising a first container comprising 50 wt% of PLA polymer having at least one carboxylic acid end group and having a weight average molecular weight of 23 kDa, and 50 wt% of NMP; and a second container comprising 35 wt% of degarelix acetate and 65 wt% NMP.
- the first and second containersare a first and second syringe, respectively.
- the first and second containersare a first and second chamber of a dual-chambered syringe, respectively.
- the present inventionprovides a pharmaceutical composition
- a pharmaceutical compositioncomprising in the final formulation between about 10 wt% and about 30 wt% of a pharmaceutically acceptable salt of degarelix; between about 20 wt% and about 80 wt% of N-methyl-2-pyrrolidone (NMP); and between about 10 wt% and about 50 wt% of 75:25 poly(D,L-lactide-co-glycolide) (PLG) copolymer, where the total weight percent of each of the components in the formulation equals 100% (e.g., if the amount of degarelix salt in the formulation is 20 wt%, then the total amount of polymer and solvent in the formulation, absent any additives, is 80 wt%).
- NMPN-methyl-2-pyrrolidone
- PLGpoly(D,L-lactide-co-glycolide) copolymer
- the PLG copolymerhas at least one carboxylic acid end group. In one aspect, the PLG copolymer has a weight average molecular weight of between about 5 kDa and about 30 kDa. In one aspect, the pharmaceutically acceptable salt of degarelix is selected from degarelix acetate and degarelix citrate.
- the present disclosureprovides a pharmaceutical composition
- a pharmaceutical compositioncomprising about 16 wt% of degarelix acetate; about 57 wt% of NMP; and about 27 wt% of 75:25 poly(D,L-lactide-co-glycolide) (PLG) copolymer having at least one carboxylic acid end group and having a weight average molecular weight of about 17 kDa.
- PLGpoly(D,L-lactide-co-glycolide)
- the present inventionprovides a pharmaceutical composition
- a pharmaceutical compositioncomprising a first container comprising 30-70 wt% of 75:25 PLG polymer having at least one carboxylic acid end group and having a weight average molecular weight of 5-30 kDa, and 30-70 wt% of N-methyl-2-pyrrolidone (NMP); and a second container comprising 20-40 wt% of a pharmaceutically acceptable salt of degarelix and 60-80 wt% NMP.
- the pharmaceutically acceptable salt of degarelixis selected from degarelix acetate and degarelix citrate.
- the first and second containersare a first and second syringe, respectively.
- the first and second containersare a first and second chamber of a dual-chambered syringe, respectively.
- the present disclosureprovides a pharmaceutical composition comprising: a first container comprising 50 wt% of 75:25 PLG copolymer having at least one carboxylic acid end group and having a weight average molecular weight of 17 kDa, and 50 wt% of NMP; and a second container comprising 35 wt% of degarelix acetate and 65 wt% NMP.
- the first and second containersare a first and second syringe, respectively.
- the first and second containersare a first and second chamber of a dual-chambered syringe, respectively.
- the present inventionprovides a pharmaceutical composition comprising in the final formulation between about 10 wt% and about 30 wt% of a pharmaceutically acceptable salt of degarelix; between about 20 wt% and about 80 wt% of N-methyl-2-pyrrolidone (NMP); and between about 10 wt% and about 50 wt% of 75:25 poly(D,L-lactide-co-glycolide) (PLG) copolymer, where the total weight percent of each of the components in the formulation equals 100% (e.g., if the amount of degarelix salt in the formulation is 20 wt%, then the total amount of polymer and solvent in the formulation, absent any additives, is 80 wt%).
- NMPN-methyl-2-pyrrolidone
- PLGpoly(D,L-lactide-co-glycolide) copolymer
- the PLG copolymerhas at least one hydroxyl end group. In one aspect, the PLG copolymer has a weight average molecular weight of between about 5 kDa and about 30 kDa. In one aspect, the pharmaceutically acceptable salt of degarelix is selected from degarelix acetate and degarelix citrate. [00135] In some embodiments, the present disclosure provides a pharmaceutical composition comprising about 16 wt% of degarelix acetate; about 57 wt% of NMP; and about 27 wt% of 75:25 PLG copolymer having at least one hydroxyl end group and having a weight average molecular weight of about 19 kDa.
- the present disclosureprovides a pharmaceutical composition
- a pharmaceutical compositioncomprising about 16 wt% of degarelix citrate; about 57 wt% of NMP; and about 27 wt% of 75:25 PLG copolymer having at least one hydroxyl end group and having a weight average molecular weight of about 19 kDa.
- the present inventionprovides a pharmaceutical composition
- a pharmaceutical compositioncomprising a first container comprising 30-70 wt% of 75:25 PLG polymer having at least one hydroxyl end group and having a weight average molecular weight of 5- 30 kDa, and 30-70 wt% of N-methyl-2-pyrrolidone (NMP); and a second container comprising 20-40 wt% of a pharmaceutically acceptable salt of degarelix and 60-80 wt% NMP.
- the pharmaceutically acceptable salt of degarelixis selected from degarelix acetate and degarelix citrate.
- the first and second containersare a first and second syringe, respectively.
- the first and second containersare a first and second chamber of a dual-chambered syringe, respectively.
- the present disclosureprovides a pharmaceutical composition comprising a first container comprising 50 wt% of 75:25 PLG copolymer having at least one hydroxyl end group and having a weight average molecular weight of 19 kDa, and 50 wt% of NMP; and a second container comprising 35 wt% of degarelix acetate and 65 wt% NMP.
- the present disclosureprovides a pharmaceutical composition
- a pharmaceutical compositioncomprising a first container comprising 50 wt% of 75:25 PLG copolymer having at least one hydroxyl end group and having a weight average molecular weight of 19 kDa, and 50 wt% of NMP; and a second container comprising 35 wt% of degarelix citrate and 65 wt% NMP.
- the first and second containersare a first and second syringe, respectively.
- the first and second containersare a first and second chamber of a dual-chambered syringe, respectively.
- the present inventionprovides a pharmaceutical composition
- a pharmaceutical compositioncomprising in the final formulation between about 10 wt% and about 30 wt% of a pharmaceutically acceptable salt of degarelix; between about 20 wt% and about 80 wt% of N-methyl-2-pyrrolidone (NMP); between about 10 wt% and 40 wt% of DMSO, and between about 10 wt% and about 50 wt% of 75:25 poly(D,L-lactide-co-glycolide) (PLG) copolymer, where the total weight percent of each of the components in the formulation equals 100% (e.g., if the amount of degarelix salt in the formulation is 20 wt%, then the total amount of polymer and solvents in the formulation, absent any additives, is 80 wt%).
- NMPN-methyl-2-pyrrolidone
- PLGpoly(D,L-lactide-co-glycolide) copolymer
- the PLG copolymerhas at least one hydroxyl end group. In one aspect, the PLG copolymer has a weight average molecular weight of between about 5 kDa and about 30 kDa. In one aspect, the pharmaceutically acceptable salt of degarelix is selected from degarelix acetate and degarelix citrate. [00141] In some embodiments, the present disclosure provides a pharmaceutical composition comprising about 16 wt% of degarelix acetate; about 30 wt% of NMP; about 27 wt% of DMSO; and about 27 wt% of 75:25 PLG copolymer having at least one hydroxyl end group and having a weight average molecular weight of about 19 kDa.
- the present inventionprovides a pharmaceutical composition
- a pharmaceutical compositioncomprising a first container comprising 30-70 wt% of 75:25 PLG polymer having at least one hydroxyl end group and having a weight average molecular weight of 5- 30 kDa, and 30-70 wt% of N-methyl-2-pyrrolidone (NMP); and a second container comprising 20-40 wt% of a pharmaceutically acceptable salt of degarelix and 60-80 wt% DMSO.
- the pharmaceutically acceptable salt of degarelixis selected from degarelix acetate and degarelix citrate.
- the first and second containersare a first and second syringe, respectively.
- the first and second containersare a first and second chamber of a dual-chambered syringe, respectively.
- the present disclosureprovides a pharmaceutical composition comprising a first container comprising 50 wt% of 75:25 PLG copolymer having at least one hydroxyl end group and having a weight average molecular weight of 19 kDa, and 50 wt% of NMP; and a second container comprising 35 wt% of degarelix acetate and 65 wt% DMSO.
- the first and second containersare a first and second syringe, respectively.
- the first and second containersare a first and second chamber of a dual-chambered syringe, respectively.
- the amount of degarelix or the pharmaceutically acceptable salt thereof in the pharmaceutical compositionis from about 8 wt% to about 39 wt% degarelix free base equivalent. In one aspect, the amount of degarelix or the pharmaceutically acceptable salt thereof in the pharmaceutical composition is from about 17 wt% to about 39 wt% degarelix free base equivalent. In one aspect, the amount of degarelix or the pharmaceutically acceptable salt thereof in the pharmaceutical composition is from about 8 wt% to about 29 wt% degarelix free base equivalent.
- the amount of degarelix or the pharmaceutically acceptable salt thereof in the pharmaceutical compositionis from about 8 wt% to about 20 wt% degarelix free base equivalent. In one aspect, the amount of degarelix or the pharmaceutically acceptable salt thereof in the pharmaceutical composition is from about 10 wt% to about 16 wt% degarelix free base equivalent. In one aspect, the amount of degarelix or the pharmaceutically acceptable salt thereof in the pharmaceutical composition is from about 14 wt% to about 15.5 wt% degarelix free base equivalent. In one aspect, the amount of degarelix or a pharmaceutically acceptable salt thereof (degarelix API) in a composition of the invention is from about 30 wt% to about 35 wt% degarelix free base equivalent.
- the amount of degarelix API in the pharmaceutical compositionis from about 31 wt% to about 34 wt% degarelix free base equivalent. In one aspect, the amount of degarelix API in the pharmaceutical composition is from about 31 wt% to about 33 wt% degarelix free base equivalent. In one aspect, the amount of degarelix API in the pharmaceutical composition is from about 31 wt% to about 32 wt% degarelix free base equivalent.
- unreacted lactide, caprolactone, and/or glycolide monomers in the polymers or copolymersare less than about 1.0 wt%, less than about 0.5 wt%, than about 0.4 wt %, less than about 0.3 wt %, less than about 0.2 wt% and less than about 0.1 wt%.
- the first and/or second containermay be a syringe as disclosed herein.
- the first and second containersare a first and second syringe, respectively.
- the first and second containersare a first and second chamber of a dual-chambered syringe, respectively.
- the methods of this disclosureare used in the treatment of diseases or conditions including prostate cancer, including advanced prostate cancer.
- the methods of this disclosureare used in reducing serum testosterone levels in a subject to a level below a castrate level of at least 50 ng/dL, or below 20 ng/dL or below 10 ng/dL.
- the compositions disclosed hereinsuppresses the subject’s luteinizing hormone (LH), which in one aspect, is reduced to a level less than about 4 IU/L.
- the methods of this disclosureare used in suppressing ovarian function in a subject with hormone-receptor positive breast cancer.
- the hormone receptor positive breast canceris pre-menopausal breast cancer. In one aspect, the hormone receptor positive breast cancer is peri-menopausal breast cancer. In one aspect, the hormone receptor positive breast cancer is estrogen receptor (ER) positive breast cancer. In one aspect, the compositions disclosed herein suppress a subject’s estradiol (E2) production to a level less than about 20 pg/mL, less than about 15 pg/mL, less than about 10 pg/mL, less than about 5 pg/mL, less than about 4 pg/ml, less than about 3 pg/mL, or less than about 2 pg/mL.
- E2estradiol
- the E2 production levelis reduced to about 2.7 pg/mL.
- the compositions disclosed hereinsuppresses the breast cancer subject’s follicle stimulating hormone (FSH) to a level less than about 40 IU/L.
- the compositions disclosed hereinsuppresses the breast cancer subject’s luteinizing hormone (LH) to a level less than about 4 IU/L.
- the methods of this disclosureare used in treating endometriosis or uterine fibroids.
- the methods of this disclosureare used in the treatment of central precocious puberty (CPP).
- CPPis defined by early sexual development prompted by production and release of gonadotropins and/or sex steroids from normal endogenous sources including the hypothalamus or pituitary. Aberrations in gonadotropin and/or sex hormone concentration levels in children with CPP can result from various sources, including, but not limited to, physical injury, infection, genetic disease, or associated tumors. CPP caused by a genetic or undetermined pathology is classified to be idiopathic in nature, while CPP caused by a central nervous system (CNS) tumor and/or lesion is classified as organic in nature. CPP is accompanied by advanced bone age, accelerated growth velocity, and Hypothalamic- Pituitary-Gonadal-axis activation.
- CNScentral nervous system
- the compositions disclosed hereinreduce a subject having CPP blood serum LH concentration to a pre-pubertal concentration levels of ⁇ 4 IU/L.
- the methodscomprise subcutaneously or intramuscularly administering a disclosed long-acting injectable composition to a subject/patient suffering from prostate cancer, advanced prostate cancer, CPP, pre-menopausal breast cancer, post-menopausal breast cancer, as well as those subjects in need of reducing serum testosterone levels, estradiol levels, or LH levels.
- an aqueous environmente.g.
- the solventdissipates in situ, and a solid, semi-solid, or liquid depot is formed comprising the biodegradable polymer and the degarelix API (self-aggregation or gelation of degarelix or pharmaceutically acceptable salt thereof occurs), together forming a drug reservoir or depot.
- the resulting depotwill release the degarelix or a pharmaceutically acceptable salt thereof, over a desired extended time period.
- the degarelix or a pharmaceutically acceptable salt thereofis released into a subject/patient, for example, for a period of at least about 30 days or longer, at least about 60 days or longer, at least about 90 days or longer, at least about 120 days or longer, at least about 150 days or longer, or 180 days or longer.
- the degarelix or a pharmaceutically acceptable salt thereofis released into a subject/patient, for example, for a period of at least about one month or longer, at least about two months or longer, at least about three months or longer, at least about four months or longer, at least about five months or longer, or six months or longer.
- the degarelix or a pharmaceutically acceptable salt thereofis released into a subject/patient for a period of at least about four weeks or longer, at least about eight weeks or longer, at least about twelve weeks or longer, at least about sixteen weeks or longer, at least about 20 weeks or longer, or at least about 24 weeks or longer.
- the long-acting compositionmay be administered to the patient/subject once about every 30 days, once about every 60 days, once about every 90 days, once about every 120 days, once about every 150 days, or once about every 180 days.
- the long-acting compositionmay be administered to the patient/subject once about every 1 month, once about every 2 months, once about every 3 months, once about every 4 months, once about every 5 months, or once about every 6 months.
- the compositionis administered once about every 3 months.
- the long-acting compositionmay be administered to the patient/subject once about every four weeks, once about every eight weeks, once about every twelve weeks, once about every sixteen weeks, once about every 20 weeks, or once about every 24 weeks.
- a “month”can have between 28 and 31 days.
- the amount of degarelix or a pharmaceutically acceptable salt thereof, that will be effective in the treatment or reduction of the above diseases or conditions,will depend on the nature/severity of the condition or symptom. In vitro or in vivo assays may optionally be employed to help identify optimal dosage ranges.
- the amount of degarelix or a pharmaceutically acceptable salt thereof, administeredwill, of course, be dependent on, among other factors, the subject being treated, the age/weight of the subject, the severity of the affliction, the manner of administration and the judgment of the prescribing physician.
- the dose amount of degarelix or a pharmaceutically acceptable salt thereof for treating prostate cancer and/or advanced prostate cancer, in the compositionis administered in a dose amount of about 20 mg, about 25 mg, about 30 mg, about 35 mg, 40 mg, about 45 mg, about 50 mg, about 55 mg, about 60 mg, about 65 mg, about 70 mg, about 75 mg, about 80 mg, about 85 mg, about 90 mg, about 95 mg, about 100 mg, about 110 mg, about 120 mg, 125 mg, about 130 mg, about 135 mg, about 140 mg, about 145 mg, about 150 mg, about 155 mg, about 160 mg, about 165 mg, about 170 mg, about 175 mg, about 180 mg, about 185 mg, about 190 mg, about 195 mg, about 200 mg, about 210 mg, about 220 mg, about 225, about 230 mg, about 235 mg, about 240 mg, about 245 mg, about 250 mg, about 255 mg, about 260 mg, about 265 mg, about 270 mg, about 275 mg, about
- a single dosemay be administered in one injection. In still another aspect, a single dose may be administered in more than one injection.
- the disclosed compositionsare to be administered to a subject/patient once in a dosing period with varying durations between dosing (e.g., one month, 2 months, 3 months, 4 months, 5 months, or 6 months). In one aspect, the disclosed compositions are to be administered to a subject/patient in an initial loading dose followed by one or more maintenance doses of the disclosed compositions. Dosing may be provided alone or in combination with other drugs and may continue as long as required for effective treatment of the disease state or disorder. In some embodiments, the disclosed compositions are formulated to provide doses between about 20 mg – 500 mg.
- the disclosed compositionsare formulated to provide single doses of between about 40 mg – 500 mg. In some embodiments, the disclosed compositions are formulated to provide single doses of between about 80 mg – 500 mg. In some embodiments, the disclosed compositions are formulated to provide single doses of between about 120 mg – 500 mg. In some embodiments, the disclosed compositions are formulated to provide single doses between about 240 mg – 500 mg. In each of these embodiments, the composition is delivered in a total volume or per injection volume not to exceed 2.5 mL or not to exceed 2 mL.
- the injection volumeis about 2.5 mL or less, about 2.4 mL or less, about 2.3 mL or less, about 2.2 mL or less, about 2.1 mL or less, about 2.0 mL or less, about 1.9 mL or less, about 1.8 mL or less, about 1.75 mL or less, about 1.7 mL or less, about 1.6 mL or less, 1.5 mL or less, about 1.4 mL or less, about 1.3 mL or less, about 1.2 mL or less, about 1.1 mL or less, about 1 mL or less, about 0.75 mL or less, about 0.5 mL or less, or about 0.375 mL.
- the injection volumeis about 0.375 ml, about 0.5 ml, about 0.75 ml, about 1 ml, about 1.5 ml, or about 1.75 mL, or about 2 mL, or about 2.2 mL, or about 2.5 mL .
- a single dose of the compositionis administered to the patient in a single injection (i.e., multiple injections are not required in a single dosing period), where the injection volume is 2.5 mL or less, 2 mL or less, about 1.75 mL or less, 1.5 mL or less, about 1 mL or less, about 0.5 mL or less, or about 0.375 mL.
- the compositionis terminally sterilized such as by e-beam or Gamma irradiation or X-ray. In yet another aspect, the composition is sterile filtered. [00158] In some embodiments, the long-acting composition is administered as a monotherapy to patients. The therapeutic methods of this embodiment may reduce or eliminate one or more symptoms of the disease and/or condition disclosed herein.
- the long-acting compositionmay be administered as a combination therapy, such as with chemotherapeutics, radiation therapy, surgery, endocrine therapies such as selective estrogen receptor modulators (SERMs; such as tamoxifen, toremifene, raloxifene, ospemifene, and apeledoxifene), selective estrogen receptor degraders (SERDs; such as fulvestrant), aromatase inhibitors (AIs; such as anastrozole, letrozole, exemestane, vorozole, formestane, and fadrozole); mammalian target of rapamycin (mTOR) inhibitors; such as temsirolimus, sirolimus, everolimus, and ridaforolimus); Phosphatidylinositol 3-kinases inhibitors (PI-3 kinase or PI3K; such as alpelisib, idelalisib, and buparlisib
- the disclosed long-acting compositionmay be provided as a part of a delivery system comprising a syringe system, wherein the composition or formulation is contained within a syringe.
- the syringeis a pre-filled syringe system containing the disclosed composition as a single dose or as multiple doses.
- the syringeis a mixing syringe (e.g., a syringe that provides a mechanism for mixing the formulation contained therein, as needed).
- the syringeis a dual-chambered syringe, and the disclosed composition is contained within one chamber of the syringe.
- the syringeis a dual-chambered syringe, and the biodegradable polymer and biocompatible solvent are contained in one chamber of the syringe and the degarelix or pharmaceutically acceptable salt thereof is contained in the second chamber of the syringe.
- the degarelix or a pharmaceutically acceptable salt thereofmay be in a lyophilized form or may be dissolved or suspended in a biocompatible solvent, which may be the same as the biocompatible solvent that is used to dissolve the biodegradable polymer, or which may be different than the biocompatible solvent that is used to dissolve the biodegradable polymer.
- a biocompatible solventwhich may be the same as the biocompatible solvent that is used to dissolve the biodegradable polymer, or which may be different than the biocompatible solvent that is used to dissolve the biodegradable polymer.
- Another pre-filled syringe delivery system, disclosed hereincomprises a first syringe containing the biodegradable polymer and the biocompatible solvent, and a second syringe containing the degarelix or a pharmaceutically acceptable salt thereof.
- the degarelix or a pharmaceutically acceptable salt thereofmay be in a lyophilized form or may be dissolved or suspended in the biocompatible solvent, which may be the same as the biocompatible solvent that is used to dissolve the biodegradable polymer, or which may be different than the biocompatible solvent that is used to dissolve the biodegradable polymer.
- the first syringe and the second syringeare coupled together to mix the contents of the first and second syringe.
- the contents of the two syringesare mixed together to form a pharmaceutical composition for injection prior to administering the pharmaceutical composition to the subject.
- the pharmaceutical compositionmay be administered to a patient by injection using a syringe system, including a syringe system as described herein.
- the compositionis injected by subcutaneous injection, although other parenteral routes, including intramuscular injection, are contemplated herein.
- the compositionmay be administered by manual injection though a syringe with, for example, a 18 to 32 gauge needle, a 22 to 25 gauge needle, a 18 to 24 gauge needle, or an 18 to 22 gauge needle, or an 18 to 20 gauge needle, or may be administered by injection using an autoinjector.
- a kitcomprising a prefilled syringe system disclosed herein.
- formulationsmay include solvent blends, other additives such as: co- solvents or stabilizer excipients such as poly-carboxylic acids or salts (e.g., citric acid), strong acid (methane sulfonic acid), hydrophobic acid or their salts (e.g., pamoic acid, palmitic acid), surfactants such as polysorbate 20, polysorbate 80, poloxamer 188, or any of these as release modifiers.
- co- solvents or stabilizer excipientssuch as poly-carboxylic acids or salts (e.g., citric acid), strong acid (methane sulfonic acid), hydrophobic acid or their salts (e.g., pamoic acid, palmitic acid), surfactants such as polysorbate 20, polysorbate 80, poloxamer 188, or any of these as release modifiers.
- co- solvents or stabilizer excipientssuch as poly-carboxylic acids or salts (e.g., citric acid), strong acid (
- Non-aqueous water-miscible pharmaceutically acceptable organic solventslike N-methyl- 2-pyrrolidone (NMP), dimethyl sulfoxide (DMSO), and N, N-dimethylacetamide (DMA) have shown superior solubility capacity (at least 35-40% w/w) of degarelix than aqueous- based solvents.
- NMP and DMSOcan dissolve higher amounts of DgA without inducing self-aggregation, as seen in aqueous solvents.
- Other solvents testedinclude— benzyl alcohol, PEG 300, propylene carbonate, triacetin, triethyl citrate, ethyl acetate, and benzyl benzoate.
- Degarelix citrate (DgC)-DS preparation[00168] Degarelix citrate was formed by dissolving the degarelix as degarelix acetate (purchased from the vendor) in water ( ⁇ 40 mg/mL concentration) and then adding the aqueous solution of anhydrous citric acid (1M) drop by drop. The molar ratio of degarelix free-base to citric acid in the solution was varied as desired. The resulting solution was mixed for 10-15 mins and placed in the -80 °C freezer.
- the contentswere lyophilized to remove the water (and freeze-drying may remove some amount of acetic acid formed as a result of ion exchange).
- the dried powderwas re-dissolved in water and freeze-dried again.
- the purity of the lyophilized degarelix citrate powderwas tested using HPLC analysis. See descriptions of different degarelix citrate drug substance powders in Table 1.
- Degarelix mesylate preparation[00169] Degarelix mesylate was formed by dissolving the degarelix as degarelix acetate (purchased from the vendor) in water ( ⁇ 40 mg/mL concentration) and then adding the aqueous solution of strong acid methane sulfonic acid (1M) drop by drop. The molar ratio of degarelix free-base to methane sulfonic acid in the solution was varied as desired. The resulting solution was mixed for 10-15 mins and placed in the -80 °C freezer. The contents were lyophilized to remove the water (and freeze-drying may remove some amount of acetic acid).
- Polymerswere either purchased from Sigma Aldrich (RESOMER® polymers) or synthesized in-house.
- Poly(D.L-lactide-co-glycolide) (PLG) copolymers, poly-DL- lactide (PLA) polymers, or poly(D.L-lactide-co-caprolactone) (PDLCL or PLC) copolymerswere produced using the following methods.
- the amounts of monomers(DL- lactide or glycolide or caprolactone) and initiator (e.g., glycolic acid, 1,6-Hexanediol, polyethylene glycol (PEG)) were selected to obtain a targeted initiator, monomer molar ratio, and weight average molecular weight for each investigated polymer.
- the monomer molar ratios and weight average molecular weights reported in individual examplesare targeted values unless specified as actual or experimental.
- the polymerization vesselwas lowered into a temperature-controlled oil bath.
- the vesselIn the vessel, appropriate amounts of monomers (DL-lactide and/or glycolide and/or caprolactone, and initiator (glycolic acid or 1,6-Hexanediol) were added, the vessel contents were placed under a nitrogen atmosphere. The temperature of the vessel was increased until the reagents melted. A catalyst solution was made with appropriate amounts of stannous octoate and toluene and added to the vessel. The vessel was then heated to about 140-170°C under a nitrogen atmosphere for about 4-18 hours (depending on the polymer of interest) with constant stirring. Then, the vessel was evacuated to remove unreacted monomers, and the monomers were vacuum- distilled out of the polymerization mixture.
- monomersDL-lactide and/or glycolide and/or caprolactone, and initiator (glycolic acid or 1,6-Hexanediol) were added, the vessel contents were placed under a nitrogen atmosphere. The temperature of the vessel was increased until the reagents
- syringe APrefilled syringes (Syringe A) containing polymer delivery system were prepared by weighing the required amounts of polymer solution into 1.2 mL female polypropylene syringe barrel with Luer lock and plunger tip and capped with male polypropylene syringe cap. Filled syringes were then packaged in labeled foil pouches with a desiccant pack and the pouches were sealed.
- the filled syringeswere stored under refrigerated conditions (e.g., 2-8 oC) or accelerated conditions ( ⁇ 25°C see individual experiments).
- the syringesmay be irradiated via e-beam irradiation (see individual experiment). 6.
- DgA-DS or DgC-DS/organic-solvent bulk solution and prefilled syringes[00172]
- the desired amount of the DgA-DS or DgC-DSwas combined with the solvent NMP or DMSO or combination of solvents with or without a co-solvent or other additives, in the indicated amounts (see individual experiments below).
- the API and solventwere combined in a glass vial or jar and blanketed with nitrogen. Jars were mixed using the jar mill, Turbula, or shaker at room temperature until homogeneous.
- DgA or DgCare very soluble in NMP or DMSO and can load higher concentrations (at least 40% w/w).
- the bulk solutionwas manually filled into syringes and capped with a tip cap.
- the syringes used for fillingwere either 1.2 mL male polypropylene syringes or 1 mL male cyclic olefin copolymer syringes. Filled syringes were then packaged in labeled foil pouches with a desiccant pack and the pouches were sealed.
- the filled syringeswere stored under refrigerated conditions (e.g., 2- 8 oC) or accelerated conditions ( ⁇ 25°C see individual experiments).
- the syringesmay be irradiated via e-beam or gamma irradiation (see individual experiment) for terminal sterilization.
- the stabilizer excipientcan be selected from a list of acids which include polycarboxylic acids or their salts (Ex. citric acid), strong acids (Ex. methanesulfonic), hydrophobic acids or their salts (Ex.
- DgA-DSdesired amounts of DgA-DS, a stabilizer excipient(s) (e.g., anhydrous citric acid), and liquid polymer solution (see section 5 for bulk polymer solution preparation) were weighed into a scintillation vial, and the vial was put on a horizontal roller mixer to roll for 24-48 h for mixing. After mixing, the single- syringe formulations (solution/suspension) were considered ready for testing. 10.
- XRD Characterization of the DgA-DS solutionsSamples were prepared and analysis was performed at ambient temperature using the Rigaku MiniFlex XRD instrument according to standard protocols. After a sequence of samples was run, the data was processed using PDXL2 software.
- Degarelix acetatedid not show any crystallinity as an API powder, or at a concentration of 66 mg/mL in water. At higher concentrations, the peptide forms a gel. The gel was not crystalline. In theory, the gel exhibits peaks indicating a higher order structure attributed to the stacking of beta-sheets forming aggregates resulting in the gel formation. The addition of salts can aid the gelation to form at a lower concentration than without the salts. No gelation occurred in the presence of NMP solvent even when water was present. 11.
- Degarelix/organic solvent solutionswere prepared for HPLC analysis by dilution to volume with mobile phase A (0.1% trifluoroacetic acid water) and mixing thoroughly by vortex. Dilutions were performed as needed using volumetric glassware and mixed thoroughly by vortex mixing. A second dilution of 2 mL to 20 mL was performed with mobile phase A to obtain a working sample. [00180] For the HPLC test sample preparation of reconstituted/single-syringe formulations that contain polymer, syringe A and syringe B were coupled together and mixed for 45 cycles.
- the reconstituted productwas dispensed from the final mixed single- syringe formulation content into a 50 mL volumetric flask and the weight was recorded.
- 2.0 mL of mobile phase B(0.1% trifluoroacetic acid acetonitrile) was added and mixed by swirling.
- 3.0 mL of mobile phase Awas added and mixed by swirling.
- Sampleswere placed in a water bath at 55 oC ⁇ 2 oC for 30 minutes and then cooled to room temperature. Samples were diluted to volume with mobile phase A and mixed thoroughly by vortex, followed by dilution to 20 mL with mobile phase A with thorough mixing. A second dilution of 2 mL to 20 mL was performed with mobile phase A to obtain a working sample.
- HPLC analysis for assay and related compoundswas performed using an Agilent AdvanceBio Peptide 3.0 x 100 mm, 2.7 um column and an Agilent AdvanceBio Peptide Map Guard 3.0 x 5mm 2.7 um guard column at 30°C with a flow rate of 0.75 mL/min.
- the runtimewas 15 minutes with a 10- ⁇ L injection for assay and related compounds for the solution gel depot formulation, and a 20- ⁇ L injection for assay and related compounds for the polymer depot formulation.
- the mobile phaseswere 0.1% trifluoroacetic acid water for mobile phase A and 0.1% trifluoroacetic acid acetonitrile for mobile phase B. Detection was performed with a diode array detector set at 220 nm. The method used a gradient. See Table 2 below for the gradient method for an example chromatogram. 12. Recovery of drug from different Degarelix/Organic solvent formulations [00182] Prefilled syringes of degarelix/organic solvent formulations were prepared as described in section 6. Recovery of drugs from different formulations was tested using the HPLC method (see section 11) and is listed in Table 3.
- F6 samplewas a control sample that does not contain any polymer or monomers and was included in the study as a control to understand if the solvent causes any degradation.
- the F6 sampleshowed minimal signs of degradation (1% drop in assay after 24 h at 50°C), indicating that the drug is stable in NMP, and any degradation observed in other samples could be attributed to the presence of polymers/monomers.
- Bold text in Table 7 for Samples F8, F10 and F15indicates that these samples exhibited the most promising drug stability (e.g., less degradation of drug over the tested time period).
- F16 and F17are samples where DgA-DS was mixed with monomer solutions (Lactide/NMP or Glycolide/NMP), and it was observed that almost 90-100% of the drug was degraded by 7 days at room temperature, and 86-99% in 24 h at 50°C. These results indicated that the presence of monomers could induce significant degradation of the degarelix. These results indicate that purification of polymers to remove the residual or unreacted monomers will improve the stability of the API. [00196] Among F8-F14 samples that contain unpurified polymers, glycolic acid (GA) terminated PLC containing samples F8 and F10 have shown better compatibility with the drug compared to the PLG or PLC-PEG polymers.
- monomer solutionsLactide/NMP or Glycolide/NMP
- F8 and F10 samplesshowed ⁇ 5% (after 24 h at 50°C) and ⁇ 1.2% (after 7 days at room temperature) degradation, whereas other samples showed significantly higher degradation of the degarelix.
- F15 sample that contains purified acid-terminated PLG polymeralso showed promise, 7.2% (after 24 h at 50°C) and 4.5% (after 7 days at room temperature) degradation.
- F9, F11, and F14that contain similar polymers (acid-terminated PLGs) with a different monomer composition, the higher stability of degarelix in F15 could be related to the use of purified polymer in F15 compared to the unpurified versions in F9, F11, and F14.
- Acid-terminated PLC polymershowed improved stability of degarelix compared to other polymers, even without purification. Based on these results, purified acid- terminated PLC polymer showed promising potential for use in formulating a single syringe formulation.
- Table 8Observations: Bold text in Table 8 for Samples F20, F24, F28 and F32 indicates that these samples exhibited the most promising drug stability (e.g., less degradation of drug over the tested time period).
- LC-MS analysis of the single syringe formulations[00201] The working samples in the volumetric flask were obtained from HPLC sample preparation followed by filtration through a 0.45 ⁇ m syringe filter into an amber HPLC vial. The liquid chromatography analysis was performed using Agilent AdvanceBio Peptide 3.0 x 100 mm, 2.7 um column and an Agilent AdvanceBio Peptide Map Guard 3.0 x 5mm 2.7 um guard column at 30 °C.
- DgA-DS containing formulations without citric acid F37(15.0% impurities) and F41(15.2% impurities) generated significantly higher amount of impurities after only 14 days compared to the formulations that contained DgC- DS ( ⁇ 5% impurities after 21 days) or citric acid as an excipient ( ⁇ 8% after 18 days).
- Lower levels of acylated impuritieswere observed for citric acid-based formulations than formulations that do not contain any citric acid.
- citric acidas a stabilizer excipient to the drug-polymer formulation or using it as part of the alternate salt form that is prepared by co-lyophilization of DgA-DS with citric acid, both resulted in a similar improvement of drug stability on accelerated storage, and the lower impurity levels were observed. Further, this preliminary data suggested that stabilizers such as citric acid can be just added as an excipient to the formulation to improve the drug stability in presence of polymer, and the additional process of lyophilization to make DgC-DS form may be eliminated to simplify the manufacturing process.
- Citric acidwas added as an excipient to the formulation at four different molar levels [0.5 (F48), 1 (F45), 2 (F46), and 3 (F47) moles to 1 mole of degarelix).
- F48 with 0.5 mol citric acidshowed 7.8 % total impurities at day 18 vs.
- F37 with no citric acidshowed 15.0% total impurities at day 14.
- F45 with 1 mol of citric acidshowed 3.6% total impurities at day 18.
- 0.5 moles of citric acidhelped in improving the stability of DgA-DS compared to no additive version, higher levels of citric acid showed much better improvement in drug stability.
- FIRMAGON ®120 mg vial of FIRMAGON ® was reconstituted by adding 3 mL of sterile water for injection provided with the FIRMAGON ® kit, and the resulting reconstituted solution was at 40 mg/mL concentration of degarelix.
- Table 12Non-Clinical Study Dosing Details (“F#” denotes “Formulation Number”) [00211] In vivo degarelix release in rats for polymer-based formulations of degarelix at 28-days post injection is presented in Figure 2.
- Figure 3shows a magnified version of day- 0 to day-3 levels to show the Cmax levels.
- FIG. 4In vivo degarelix release in rats for polymer-based formulations of degarelix at 63-days post injection is presented in Figure 4.
- Figure 5shows a magnified version of the same data in Figure 4 to better illustrate the release of the different formulations.
- Figures 6 and 7show that all of the formulations released drug that resulted in measurable levels of degarelix in vivo for greater than 60 days.
- Figure 6In vivo degarelix release in rats for polymer-based formulations of degarelix at 119-days post injection is presented in Figure 6.
- Figure 7shows a magnified version of the same data in Figure 6 to better illustrate the release of the different formulations.
- Figures 6 and 7show that all of the formulations released drug that resulted in measurable levels of degarelix in vivo for greater than 90 days (at least 119 days). Also, the formulation L-TA11 (delarelix citrate) showed notably higher levels of drug in vivo compared to other formulations throughout the testing period.
- In vivo testosterone concentration levels (ng/mL) in rats for the polymer-based formulations of degarelix API in this example at 105-days post injectionis presented in Figure 8. Together, the results show that all of the formulations had plasma concentration of greater than 1 ng/mL which is the minimum drug concentration to induce chemical castration. In addition, the testosterone level remained below the “historical testosterone baseline” level and testosterone suppression occurred by day 4.
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- General Health & Medical Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Epidemiology (AREA)
- Medicinal Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Dermatology (AREA)
- Inorganic Chemistry (AREA)
- Gastroenterology & Hepatology (AREA)
- Reproductive Health (AREA)
- Endocrinology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Oil, Petroleum & Natural Gas (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Biomedical Technology (AREA)
- Neurosurgery (AREA)
- Immunology (AREA)
- Medicinal Preparation (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
Abstract
Description
DEGARELIX POLYMERIC FORMULATIONS CROSS-REFERENCE TO RELATED APPLICATION [001] This application claims the benefit of priority of U.S. Provisional Patent Application 63/484,432, filed 10 February 2023 the entirety of which is incorporated herein by reference. FIELD [002] This invention relates generally to long-acting injectable degarelix polymeric formulations. BACKGROUND [003] Degarelix is a decapeptide gonadotropin-releasing hormone (GnRH) receptor antagonist indicated for the treatment of patients with advanced prostate cancer. Degarelix acts by directly blocking GnRH action on the pituitary by competitively and reversibly binding to GnRH receptors and produces a fast suppression of testosterone with no initial surge or minimal surge. [004] The currently marketed drug-delivery technology for degarelix in the United States and Europe (e.g., FIRMAGON® (degarelix for injection), Ferring Pharmaceuticals Inc. or Ferring GmbH) has significant limitations, including administration of a starting dose given as two high volume injections (3 mL each), followed by maintenance doses administered as a single high volume injection (1 x 4 mL) every 28 days. The only commercially available formulation with a dosing interval greater than once monthly is marketed only in Japan (GONAX® (degarelix for injection), Astellas Pharma Inc.), and has similar significant limitations, including administration of a starting dose given as two injections of 3 mL each, followed by a maintenance dose given as two injections of 4 mL each every 12-weeks. Thus, for all currently available degarelix products, multiple, high- volume injections (a total of 4-8 mLs per dosing period) are needed for extended therapy, which are associated with injection-related pain. Furthermore, degarelix has a tendency to self-aggregate in aqueous media. In the case of FIRMAGON®, degarelix is supplied as a powder (lyophilized, contained mannitol as a bulking agent), and once it is reconstituted with water the degarelix molecules aggregate and cross-link in a gel-forming network, resulting in a hydrogel. Because of its self-aggregation property and chemical instability in aqueous media, it has to be separated out from the aqueous vehicle and needs to be kept in a dried form by lyophilization. Therefore, it needs to be reconstituted at the time of administration. Since the rate and extent of self-aggregation/gel formation of peptide in aqueous media is concentration dependent, it is challenging to prepare a ready-to-inject formulation of degarelix for injection. In addition, the commercially available form of degarelix is manufactured by aseptic processing and involves multiple steps for reconstitution. [005] There is an unmet need for a long-acting GnRH antagonist formulation that is easy to administer, can be terminally sterilized during manufacturing, is administered less frequently than once per month, reduces the overall number of injections in a single dosing period, significantly reduces the injection volume, and reduces pain upon injection. Thus, it would be highly beneficial to have an injectable drug formulation of degarelix in a single syringe system that is ready-to-inject, and provides in a single injection, extended-release of a therapeutically effective amount of degarelix for more than a month within a low injection volume (<2.5 mL or 2 mL). SUMMARY [006] In various embodiments, a composition comprising a therapeutically effective amount of degarelix or a pharmaceutically acceptable salt thereof is disclosed. The composition comprises a biodegradable polymer; and a biocompatible solvent, wherein the composition is formulated for subcutaneous injection into a subject; wherein a single dose of the composition is about 2.5 mL or less, or about 2.0 mL or less; and upon injection into the subject, the composition forms an in situ depot that releases the degarelix or pharmaceutically acceptable salt thereof over a time period of from about 1 month to about 6 months. [007] In one aspect of the composition, the in situ depot releases the degarelix or pharmaceutically acceptable salt thereof over a time period selected from the group consisting of: at least about 1 month, at least about 1.5 months, at least about 2 months, at least about 2.5 months, at least about 3 months, at least about 3.5 months, at least about 4 months, at least about 4.5 months, and at least about 5 months. [008] In one aspect of the composition, the pharmaceutically acceptable salt of the degarelix is selected from degarelix acetate, degarelix citrate, degarelix pamoate, degarelix palmitate, and degarelix mesylate. [009] In one aspect of the composition, the biodegradable polymer comprises monomer residues selected from the group consisting of lactide, glycolide, caprolactone, p- dioxanone, trimethylene carbonate, thylene oxide, propylene oxide, sebacic anhydride, diketene acetals/diols, and combinations thereof. [0010] In one aspect of the composition, the biodegradable polymer comprises lactide and glycolide monomer residues. In one aspect, a molar ratio of the lactide to glycolide monomer residues is from about 45:55 to about 99:1. In yet another aspect, a molar ratio of the lactide to glycolide monomer residues is from about 50:50 to about 90:10. [0011] In still another aspect of the composition, the biodegradable polymer comprises: lactide monomer residues and monomer residues selected from the group consisting of ε- caprolactone, trimethylene carbonate, and combinations thereof. In one aspect, a molar ratio of the lactide monomer residues to the ε-caprolactone and/or trimethylene carbonate monomer residues is from about 10:90 to about 90:10. In still another aspect, a molar ratio of the lactide monomer residues to the ε-caprolactone and/or trimethylene carbonate monomer residues is from about 25:75 to about 75:25. In yet another aspect, a molar ratio of the lactide monomer residues to the ε-caprolactone and/or trimethylene carbonate monomer residues is about 75:25. [0012] In one aspect of the composition, the biodegradable polymer comprises at least one carboxylic acid end group, at least one hydroxyl end group, or at least one hydroxy end group and is substantially free of terminal carboxy end groups. [0013] In one aspect of the composition, the biodegradable polymer is poly(D,L-lactide- co-glycolide) (PLG), poly(D,L-lactide) (PLA), or poly(D,L-lactide-co-ε-caprolactone) (PLC). [0014] In still another aspect of the composition, the biodegradable polymer is poly(D,L-lactide-co-ε-caprolactone) (PLC). In one aspect, the PLC comprises at least one carboxylic acid end group. In one aspect, the PLC is a carboxylic acid-initiated PLC. In one aspect, the carboxylic acid is glycolic acid or lactic acid. In yet another aspect, the biodegradable polymer is acid-initiated 75:25 PLC. [0015] In one aspect of the composition, the biodegradable polymer is PLG. In one aspect, the PLG comprises at least one hydroxyl end group. In one aspect, the PLG is a core- diol-initiated PLG. In still another aspect, the core diol is 1,6-hexane-diol. In still another aspect, the PLG is a mono functional alcohol-initiated PLG. In one aspect, the alcohol is dodecanol. In yet another aspect, the biodegradable polymer is acid-initiated 50:50 PLG. Still yet in another aspect, the biodegradable polymer is 1,6-hexane-diol-initiated 75:25 PLG. In another aspect, the biodegradable polymer is 1,6-hexane-diol-initiated 85:15 PLG. [0016] In one aspect of the composition, the biodegradable polymer is dodecanol- initiated PLA. [0017] In one aspect of the composition, the biocompatible polymer has a weight average molecular weight of from about 5 kDa to about 60 kDa. [0018] In one aspect of the composition, the biocompatible polymer has a weight average molecular weight of from about 5 kDa to about 35 kDa. [0019] In still another aspect of the composition, the biocompatible polymer comprises less than about 0.5 wt%, or less than about 0.4 wt%, or less than about 0.3 wt%, or less than about 0.2 wt% or less than about 0.1 wt% unreacted lactide monomer. [0020] In one aspect of the composition, the amount of biodegradable polymer in the composition is from about 10% to about 60% by weight of the composition. [0021] In yet another aspect of the composition, the amount of biocompatible solvent in the composition is from about 10% to about 85% by weight of the composition. [0022] In one aspect of the composition, the therapeutically effective amount of degarelix or pharmaceutically acceptable salt in the composition is from about 10% to about 40% by weight of the composition. [0023] In one aspect of the composition, the therapeutically effective amount of degarelix or pharmaceutically acceptable salt thereof is from about 40 mg – 500 mg. [0024] In yet another aspect of the composition, the therapeutically effective amount of degarelix or pharmaceutically acceptable salt thereof is from about 80 mg – 500 mg. [0025] In still another aspect of the composition, the therapeutically effective amount of degarelix or pharmaceutically acceptable salt thereof is from about 120 mg – 500 mg. [0026] In one aspect of the composition, the biocompatible organic solvent is selected from the group consisting of N-methyl-2-pyrrolidone (NMP), dimethyl sulfoxide (DMSO), dimethylacetamide (DMA), benzyl benzoate (BnBzO), polyethylene glycol 15 hydroxystearate, methyl ethyl ketone, methyl lactate, benzyl alcohol, propylene carbonate (PC), triacetin, tributyl citrate, acetyl tributyl citrate, acetyl triethyl citrate, triethyl citrate, diethylene glycol monomethyl ether, ethyl acetate, N-ethyl-2-pyrrolidone, glycofurol, and combinations thereof. In one preferred aspect, the biocompatible organic solvent is NMP or DMSO. [0027] In one aspect of the composition, a single dose of the composition is about 2.5 mL or less, about 2.4 mL or less, about 2.3 mL or less, about 2.2 mL or less, about 2.1 mL or less, about 2.0 mL or less, about 1.9 mL or less, about 1.8 mL or less, about 1.7 mL or less, about 1.6 mL or less, about 1.5 mL or less, about 1.4 mL or less, about 1.3 mL or less, about 1.2 mL or less, about 1.1 mL or less, about 1 mL or less, about 0.75 mL or less, about 0.5 mL or less, or about 0.375 mL or less. [0028] In one aspect of the composition, the composition has been terminally sterilized or sterile filtered. In one aspect, wherein the composition has been terminally sterilized by e-beam. [0029] In one aspect of the composition, the composition further comprises one or more additives. In one aspect, the additive is selected from the group consisting of polysorbate 20, polysorbate 80, poloxamer 188, sorbitan trioleate, lecithin (e.g., soya or egg), polyethylene glycol (PEG), PEG 300, 2-pyrrolidone, alpha-tocopherol, Vitamin E TPGS, sucrose cocoate, sucrose stearate, sucrose laurate, proline, arginine, sodium metabisulfite butylated hydroxyanisole, butylated hydroxyquinone, butylhydroxyanisol, hydroxycoumarin, butylated hydroxytoluene, cephalm, ethyl gallate, propyl gallate, octyl gallate, lauryl gallate, propyl-hydroxybenzoate, trihydroxybutylrophenone, vitamin E, lecithin, ethanolamine, ZnCl2, MgCl2, CaCl2, DL-methioninecitric acid, dimethylphenol, dibutylphenol, ethylenediaminotetraacetic acid (EDTA), ethylene glycol-bis(β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid (EGTA), ascorbic acid, nitrilotriacetic acid, n- hydroxyethylethylenediaminetriacetic acid (HEDTA), mercaptoethanol, and combinations thereof. [0030] In another aspect of the composition, the degarelix is lyophilized, and the biodegradable polymer is dissolved in the biocompatible solvent. [0031] In yet another aspect of the composition, the degarelix is dissolved in a first biocompatible solvent, and the biodegradable polymer is dissolved in a second biocompatible solvent. In one aspect, the first and second biocompatible solvent are the same. [0032] In one embodiment, a pharmaceutical composition, comprising about 10-30 wt% of degarelix acetate; about 20-80 wt% of N-methyl-2-pyrrolidone (NMP); and about 10-50 wt% of 75:25 poly(lactide-co-ε-caprolactone) (PLC) copolymer having at least one carboxylic acid end group and having a weight average molecular weight of about 5-30 kDa is disclosed. In one aspect of this pharmaceutical composition, the composition comprises about 16 wt% of degarelix acetate; about 57 wt% of N-methyl-2-pyrrolidone (NMP); and about 27 wt% of 75:25 poly(lactide-co-ε-caprolactone) (PLC) copolymer having at least one carboxylic acid end group and having a weight average molecular weight of about 17 kDa. In still another aspect of this composition, the composition comprises about 16 wt% of degarelix acetate; about 57 wt% of NMP; and about 27 wt% of 75:25 PLC copolymer having at least one carboxylic acid end group and having a weight average molecular weight of about 8 kDa. In still another aspect, unreacted lactide and caprolactone monomers in the PLC copolymer are less than about 1.0 wt%. In yet another aspect, unreacted lactide and caprolactone monomers in the PLC copolymer are less than about 0.5 wt%, than about 0.4 wt %, less than about 0.3 wt %, less than about 0.2 wt% and less than about 0.1 wt%. [0033] In one embodiment, a pharmaceutical composition, comprising a first container comprising 30-70 wt% of 75:25 PLC copolymer having at least one carboxylic acid end group and having a weight average molecular weight of 5-30 kDa, and 30-70 wt% of N- methyl-2-pyrrolidone (NMP); and a second container comprising 20-40 wt% of degarelix acetate and 60-80 wt% NMP is disclosed. In one aspect of this composition the first container comprises 50 wt% of 75:25 PLC copolymer having at least one carboxylic acid end group and having a weight average molecular weight of 17 kDa, and 50 wt% of N- methyl-2-pyrrolidone (NMP); and the second container comprises 35 wt% of degarelix acetate and 65 wt% NMP. In still another aspect of this pharmaceutical composition, the first container comprises 70 wt% of 75:25 PLC copolymer having at least one carboxylic acid end group and having a weight average molecular weight of 8 kDa, and 30 wt% of NMP; and the second container comprising 35 wt% of degarelix acetate and 65 wt% NMP. In still another aspect, unreacted lactide and caprolactone monomers in the PLC copolymer are less than about 1.0 wt%. In yet another aspect, unreacted lactide and caprolactone monomers in the PLC copolymer are less than about 0.5 wt%, than about 0.4 wt %, less than about 0.3 wt %, less than about 0.2 wt% and less than about 0.1 wt%. [0034] In one embodiment, a pharmaceutical composition comprising about 10-30 wt% of degarelix acetate; about 20-80 wt% of NMP; and about 10-50 wt% of poly(D,L-lactide) (PLA) polymer having at least one carboxylic acid end group and having a weight average molecular weight of about 5-30 kDa is disclosed. In one aspect of this pharmaceutical composition, the composition comprises about 16 wt% of degarelix acetate; about 57 wt% of NMP; and about 27 wt% of poly(D,L-lactide) (PLA) polymer having at least one carboxylic acid end group and having a weight average molecular weight of about 23 kDa. In one aspect, unreacted lactide monomers in the PLA polymer are less than about 1.0 wt%. In still another aspect, unreacted lactide monomers in the PLA polymer are less than about 0.5 wt%, than about 0.4 wt %, less than about 0.3 wt %, less than about 0.2 wt% and less than about 0.1 wt%. [0035] In one embodiment, a pharmaceutical composition, comprising a first container comprising 30-70 wt% of PLA polymer having at least one carboxylic acid end group and having a weight average molecular weight of 5-30 kDa, and 30-70 wt% of NMP; and a second container comprising 20-40 wt% of degarelix acetate and 60-80 wt% NMP is disclosed. In one aspect, the first container comprising 50 wt% of PLA polymer having at least one carboxylic acid end group and having a weight average molecular weight of 23 kDa, and 50 wt% of NMP; and the second container comprising 35 wt% of degarelix acetate and 65 wt% NMP. In one aspect, unreacted lactide monomers in the PLA polymer are less than about 1.0 wt%. In still another aspect, unreacted lactide monomers in the PLA polymer are less than about 0.5 wt%, than about 0.4 wt %, less than about 0.3 wt %, less than about 0.2 wt% and less than about 0.1 wt%. [0036] In one embodiment, a pharmaceutical composition, comprising about 10-30 wt% of degarelix acetate; about 20-80 wt% of NMP; and about 10-50 wt% of 75:25 poly(D,L- lactide-co-glycolide) (PLG) copolymer having at least one carboxylic acid end group and having a weight average molecular weight of about 5-30 kDa is disclosed. In one aspect of this composition, the composition comprises about 16 wt% of degarelix acetate; about 57 wt% of NMP; and about 27 wt% of 75:25 poly(D,L-lactide-co-glycolide) (PLG) copolymer having at least one carboxylic acid end group and having a weight average molecular weight of about 17 kDa. In one aspect, unreacted lactide and glycolide monomers in the PLG copolymer are less than about 1.0 wt%. In still another aspect, unreacted lactide and glycolide monomers in the PLG copolymer are less than about 0.5 wt%, than about 0.4 wt %, less than about 0.3 wt %, less than about 0.2 wt% and less than about 0.1 wt%. [0037] In one embodiment, a pharmaceutical composition, comprising a first container comprising 30-70 wt% of 75:25 PLG copolymer having at least one carboxylic acid end group and having a weight average molecular weight of 5-30 kDa, and 30-70 wt% of NMP; and a second container comprising 20-40 wt% of degarelix acetate and 60-80 wt% NMP is disclosed. In one aspect of this composition, the first container comprises 50 wt% of 75:25 PLG copolymer having at least one carboxylic acid end group and having a weight average molecular weight of 17 kDa, and 50 wt% of NMP; and the second container comprises 35 wt% of degarelix acetate and 65 wt% NMP. In one aspect, unreacted lactide and glycolide monomers in the PLG copolymer are less than about 1.0 wt%. In still another aspect, unreacted lactide and glycolide monomers in the PLG copolymer are less than about 0.5 wt%, than about 0.4 wt %, less than about 0.3 wt %, less than about 0.2 wt% and less than about 0.1 wt%. [0038] In one embodiment, a pharmaceutical composition, comprising about 10-30 wt% of degarelix acetate; about 20-80 wt% of NMP; and about 10-50 wt% of 75:25 PLG copolymer having at least one hydroxyl end group and having a weight average molecular weight of about 5-30 kDa is disclosed. In one aspect of this composition, the composition comprises about 16 wt% of degarelix acetate; about 57 wt% of NMP; and about 27 wt% of 75:25 PLG copolymer having at least one hydroxyl end group and having a weight average molecular weight of about 19 kDa. [0039] In one embodiment, a pharmaceutical composition, comprising a first container comprising 30-70 wt% of 75:25 PLG copolymer having at least one hydroxyl end group and having a weight average molecular weight of 5-30 kDa, and 30-70 wt% of NMP; and a second container comprising 20-40 wt% of degarelix acetate and 60-80 wt% NMP. In one aspect of this composition, the first container comprises 50 wt% of 75:25 PLG copolymer having at least one hydroxyl end group and having a weight average molecular weight of 19 kDa, and 50 wt% of NMP; and the second container comprises 35 wt% of degarelix acetate and 65 wt% NMP. [0040] In one embodiment, a pharmaceutical composition, comprising about 10-30 wt% of degarelix acetate; about 10-40 wt% of NMP; about 10-40 wt% of DMSO; and (d) about 10-50 wt% of 75:25 PLG copolymer having at least one hydroxyl end group and having a weight average molecular weight of about 5-30 kDa is disclosed. In one aspect of this composition, the composition comprises about 16 wt% of degarelix acetate; about 30 wt% of NMP; about 27 wt% of DMSO; and (d) about 27 wt% of 75:25 PLG copolymer having at least one hydroxyl end group and having a weight average molecular weight of about 19 kDa. [0041] In one embodiment, a pharmaceutical composition, comprising a first container comprising 30-70 wt% of 75:25 PLG copolymer having at least one hydroxyl end group and having a weight average molecular weight of 5-30 kDa, and 30-70 wt% of NMP; and a second container comprising 20-40 wt% of degarelix acetate and 60-80 wt% DMSO is disclosed. In one aspect of this composition, the first container comprises 50 wt% of 75:25 PLG copolymer having at least one hydroxyl end group and having a weight average molecular weight of 19 kDa, and 50 wt% of NMP; and the second container comprises 35 wt% of degarelix acetate and 65 wt% DMSO. [0042] In one embodiment, a pharmaceutical composition, comprising about 10-30wt% of degarelix citrate; about 20-80 wt% of NMP; and about 10-50 wt% of 75:25 PLG copolymer having at least one hydroxyl end group and having a weight average molecular weight of about 5-30 kDa is disclosed. In one aspect of this composition, the composition comprises about 16 wt% of degarelix citrate; about 57 wt% of NMP; and about 27 wt% of 75:25 PLG copolymer having at least one hydroxyl end group and having a weight average molecular weight of about 19 kDa. [0043] In one embodiment, a pharmaceutical composition, comprising a first container comprising 30-70 wt% of 75:25 PLG copolymer having at least one hydroxyl end group and having a weight average molecular weight of 19 kDa, and 50 wt% of NMP; and a second container comprising 20-40 wt% of degarelix citrate and 60-80 wt% NMP is disclosed. In one aspect of this composition, the first container comprises 50 wt% of 75:25 PLG copolymer having at least one hydroxyl end group and having a weight average molecular weight of 19 kDa, and 50 wt% of NMP; and the second container comprises 35 wt% of degarelix citrate and 65 wt% NMP. [0044] In any of the above embodiments, in one aspect, the amount of degarelix or the pharmaceutically acceptable salt thereof in the pharmaceutical composition is from about 8 wt% to about 39 wt% degarelix free base equivalent. In one aspect, the amount of degarelix or the pharmaceutically acceptable salt thereof in the pharmaceutical composition is from about 17 wt% to about 39 wt% degarelix free base equivalent. In one aspect, the amount of degarelix or the pharmaceutically acceptable salt thereof in the pharmaceutical composition is from about 8 wt% to about 29 wt% degarelix free base equivalent. In one aspect, the amount of degarelix or the pharmaceutically acceptable salt thereof in the pharmaceutical composition is from about 8 wt% to about 20 wt% degarelix free base equivalent. In one aspect, the amount of degarelix or the pharmaceutically acceptable salt thereof in the pharmaceutical composition is from about 10 wt% to about 16 wt% degarelix free base equivalent. In one aspect, the amount of degarelix or the pharmaceutically acceptable salt thereof in the pharmaceutical composition is from about 14 wt% to about 15.5 wt% degarelix free base equivalent. In one aspect, the amount of degarelix or a pharmaceutically acceptable salt thereof (degarelix API) in a composition of the invention is from about 30 wt% to about 35 wt% degarelix free base equivalent. In one aspect, the amount of degarelix API in the pharmaceutical composition is from about 31 wt% to about 34 wt% degarelix free base equivalent. In one aspect, the amount of degarelix API in the pharmaceutical composition is from about 31 wt% to about 33 wt% degarelix free base equivalent. In one aspect, the amount of degarelix API in the pharmaceutical composition is from about 31 wt% to about 32 wt% degarelix free base equivalent. In some aspects, when a target dose to be administered to a subject is calculated based on degarelix free base equivalent in the composition, the percent by weight of solvent, and the percent by weight of additive if included, may be modified accordingly. [0045] In various embodiments, methods of treating prostate cancer and central precocious puberty (CPP) as well as methods of reducing serum testosterone levels and suppressing ovarian function by subcutaneously administering the compositions disclosed herein to subjects in need thereof is also contemplated. [0046] In one embodiment, a method of treating prostate cancer in a subject, comprising subcutaneously administering the compositions disclosed herein to the subject is disclosed. In one aspect, the prostate cancer is advanced prostate cancer. In one aspect, the dose amount of degarelix or pharmaceutically acceptable salt in the composition is administered in a dose amount of about 40 mg to about 500 mg. [0047] In one embodiment, a method of reducing serum testosterone levels in a subject to 50 ng/dL or less, comprising subcutaneously administering the compositions disclosed herein to the subject is disclosed. In one aspect, the serum testosterone level is below 20 ng/dL. In still another aspect, the serum testosterone level is below 10 ng/dL. [0048] In one embodiment, a method of suppressing ovarian function in a subject with hormone-receptor positive breast cancer, comprising subcutaneously administering the compositions disclosed herein to the subject is disclosed. In one aspect, the hormone receptor positive breast cancer is estrogen receptor (ER) positive breast cancer. In one aspect, the subject’s estradiol (E2) production level is suppressed to a level less than about 20 pg/mL to about less than about 2 pg/mL. In one aspect, the subject’s follicle stimulating hormone (FSH) level is suppressed to a level less than about 40 IU/L. In one aspect, the subject’s luteinizing hormone (LH) level is suppressed to a level less than about 4 IU/L. [0049] In one embodiment, a method of treating central precocious puberty (CPP) in a subject comprising subcutaneously administering the compositions disclosed herein to the subject is disclosed. In one aspect, the subject’s CPP blood serum LH concentration level is reduced to a pre-pubertal concentration level of less than about 4 IU/L. [0050] In any aspects of the methods disclosed herein, the composition is administered about once every 1 month, about once every 2 months, about once every 3 months, about once every 4 months, about once every 5 months, or about once every six months. [0051] In any aspects of the methods disclosed herein, the composition is administered to the subject about once every three months. [0052] In any aspects of the methods disclosed herein, the composition is administered as a loading dose followed by a maintenance dose of the composition about 1 month, 2 months or 3 months after the loading dose has been administered. In one aspect, when a loading dose is administered, the maintenance dose is administered every 1 month, every 2 months or every 3 months thereafter. In one embodiment, no loading dose is administered, and the composition is administered about once every 1 month, about once every 2 months, about once every 3 months, about once every 4 months, about once every 5 months, or about once every six months. [0053] Another embodiment is a delivery system, comprising a syringe or syringes prefilled with a composition disclosed herein, in some embodiments together with instructions suitable for using the delivery system to administer the composition to a subject. In one embodiment, a prefilled syringe system for administration of the compositions disclosed herein, comprising a single syringe containing the composition disclosed herein, is described. In one aspect, the syringe is a mixing syringe. In one aspect, the syringe is an autoinjector syringe. In still another aspect, the syringe is a dual-chambered syringe, and the composition disclosed herein is contained within one chamber of the syringe. In yet another aspect, the syringe is a dual-chambered syringe, and the biodegradable polymer and biocompatible solvent are contained in one chamber of the syringe and the degarelix or pharmaceutically acceptable salt thereofis contained in the second chamber of the syringe. In one aspect, the degarelix is lyophilized. In still another aspect, the degarelix or or pharmaceutically acceptable salt thereof is dissolved or suspended in the biocompatible solvent. [0054] In one embodiment, a prefilled syringe system for administration of the compositions disclosed herein, comprising a first syringe containing the biodegradable polymer and the biocompatible solvent, and a second syringe containing the degarelix is described. In one aspect, the first syringe and the second syringe are coupled together. In yet another aspect, the degarelix or pharmaceutically acceptable salt thereof is lyophilized. In still another aspect, the degarelix or pharmaceutically acceptable salt thereof is dissolved or suspended in the biocompatible solvent. [0055] In one embodiment, a kit comprising the prefilled syringe system disclosed herein and instructions for use of the prefilled syringe system to administer a composition contained therein are disclosed. [0056] In one embodiment, a product comprising the compositions disclosed herein for use in a method of treating prostate cancer or CPP is disclosed. [0057] In one embodiment, a product comprising the compositions disclosed herein for use in a method of reducing serum testosterone levels below to 50 ng/dL is disclosed. [0058] In one embodiment, a product comprising the compositions disclosed herein for use in a method of suppressing ovarian function in a subject with hormone-receptor positive breast cancer is disclosed. BRIEF DESCRIPTION OF THE DRAWINGS [0059] Fig. 1 shows the percentage recovery of degarelix from the degarelix/organic solvent formulation samples (formulas 1, 2 and 5 (F1, F2 and F5) )(prefilled syringes) stored at 25°C for 0 months, 1 month, 3 months and 6 months. F1: Degarelix acetate drug substance (DgA-DS)/N-methyl-2-pyrrolidone (NMP) (65%/35%); F2: DgA-DS/dimethyl sulfoxide (DMSO) (65%/35%); F5: DgA-DS/DMSO (60%/35%). [0060] Fig. 2 shows the in vivo degarelix levels in plasma over the first 3 days post- injection using a rat model as described in section 19 of the Example section. [0061] Fig.3 shows the in vivo degarelix levels in plasma over 28 days post-injection using a rat model as described in section 19 of the Example section. [0062] Fig.4 shows the in vivo degarelix levels in plasma over 63 days post-injection using a rat model as described in section 19 of the Example section. [0063] Fig.5 shows a magnified view of the in vivo degarelix levels in plasma over 63 days post-injection using a rat model as described in section 19 of the Example section. [0064] Fig.6 shows the in vivo degarelix levels in plasma over 119 days post-injection using a rat model as described in section 19 of the Example section. [0065] Fig.7 shows a magnified view of the in vivo degarelix levels in plasma over 119 days post-injection using a rat model as described in section 19 of the Example section. [0066] Fig. 8 shows a semi-log view of the in vivo testosterone levels in plasma over 105 days post-injection using a rat model as described in section 19 of the Example section. DETAILED DESCRIPTION Definitions [0067] As used herein, the terms “active pharmaceutical ingredient”, abbreviated as “API”, and “drug” can be used interchangeably and generally refer to a biologically active compound that has therapeutic effects on the body. The “active pharmaceutical ingredient” may refer to an active drug, or a pharmaceutically acceptable salt of an active drug. As used herein, these terms may be used to refer to degarelix or a pharmaceutically acceptable salt thereof. “Degarelix API” (whether capitalized or not) as used herein refers to degarelix or any pharmaceutically acceptable salt thereof. [0068] As used herein, the term “ester” refers to a chemical functional group, C(O)OR’ where R’ represents an alkyl as defined herein. Representative examples include, but are not limited to, methoxycarbonyl, ethoxycarbonyl, propoxycarbonyl, butoxycarbonyl, and the like. [0069] As used herein, the term “antioxidant” refers to compounds which are used to extend the shelf-life of products by preventing or inhibiting the oxidation of active substances and excipients. An antioxidant may react with free radicals thereby blocking or inhibiting free radical chain reactions, or the antioxidant may have a lower redox potential than the active substances and excipients in the formulation. Additionally, or alternatively, a synergist antioxidant may enhance the effects of other antioxidants. [0070] As used herein, the term “biocompatible” means “not harmful to living tissue” or “safe for injection within a human body”. [0071] As used herein, the term “biodegradable” refers to any material that is converted, breaks down, or degrades, under physiological conditions into innocuous or natural byproducts, such as (but not limited to) water, gas, biomass, and/or organic salts, without regard to any specific degradation mechanism or process. [0072] As used herein, the terms “molecular weight” and “average molecular weight,” unless otherwise specified, refer to a weight-average molecular weight as measured by a conventional gel permeation chromatography (GPC) instrument (such as an Agilent 1260 Infinity Quaternary LC with Agilent G1362A Refractive Index Detector) utilizing polystyrene standards and tetrahydrofuran (THF) as the solvent. [0073] As used herein, the terms “patient” and “subject” are interchangeable and generally refer to an animal or a human to which a composition disclosed herein is administered or is to be administered. [0074] As used herein, the term “pharmaceutically acceptable salt” refers to a salt of a compound, which possesses the desired pharmacological activity of the parent compound. Such salts include, but are not limited to: (1) acid addition salts, formed with inorganic acids such as hydrochloric acid, hydrobromic acid, sulfuric acid, nitric acid, phosphoric acid, and the like; or formed with organic acids such as acetic acid, propionic acid, hexanoic acid, cyclopentanepropionic acid, glycolic acid, pyruvic acid, lactic acid, lauric acid, malonic acid, succinic acid, malic acid, maleic acid, fumaric acid, tartaric acid, citric acid, benzoic acid, 3-(4-hydroxybenzoyl)benzoic acid, cinnamic acid, mandelic acid, methanesulfonic acid, ethanesulfonic acid, 1,2-ethane-disulfonic acid, 2-hydroxyethanesulfonic acid, benzenesulfonic acid, 4-chlorobenzenesulfonic acid, 2-naphthalenesulfonic acid, 4- toluenesulfonic acid, camphorsulfonic acid, 4-methylbicyclo[2.2.2]-oct-2-ene-1-carboxylic acid, glucoheptonic acid, 3-phenylpropionic acid, trimethylacetic acid, tertiary butylacetic acid, lauryl sulfuric acid, gluconic acid, glutamic acid, hydroxynaphthoic acid, salicylic acid, stearic acid, muconic acid, pamoic acid, palmitic acid, and the like; or (2) salts formed when an acidic proton present in the parent compound is replaced by a metal ion, e.g., an alkali metal ion, an alkaline earth ion, or an aluminum ion; or coordinates with an organic base such as ethanolamine, diethanolamine, triethanolamine, N-methylglucamine and the like. [0075] As used herein, the term “solvent” refers to a liquid that dissolves a solid or liquid solute, or to a liquid external phase of a suspension throughout which solid or liquid substances may be suspended or dispersed. The term “biocompatible solvent” may be used interchangeably with the term “solvent”. [0076] As used herein, the term “co-solvent” refers to a substance added to a solvent to increase or modify the solubility of a solute in the solvent. Accordingly, a co-solvent may increase or decrease solubility of a solute in a primary solvent and/or may impart other desirable characteristics onto a formulation (e.g., the extent of water insolubility of the solvents and co-solvents in a solvent system can impact the desired rate of diffusion into bodily fluids for controlling the rate and scope of degarelix API gelation; or solvent/co- solvents can control the viscosity of the compositions of the invention, which aids in preparing and administering the extended release composition to a subject). [0077] As used herein, the term “solubilizer” refers to a compound that increases the solubility of another substance. [0078] As used herein, the term “surfactant” refers to a compound that lowers the surface tension between two liquids, between a gas and a liquid, or between a liquid and a solid. For example, a surfactant can act as a wetting agent, which aids in dispersing an active pharmaceutical ingredient in a liquid vehicle, or as a solubilizer under some circumstances. [0079] As used herein, the term “therapeutically effective amount” means the amount of a compound or drug product that, when administered to a patient for treating a disease and/or treating or preventing one or more symptoms of a disease, is sufficient to affect such treatment or prevention for the disease. The “therapeutically effective amount” can vary depending on, for example, the compound or drug product, disease progression, the disease or condition to be treated, whether the therapy is an adjuvant therapy or a primary or curative therapy, and/or the age, weight, etc., of the patient to be treated. By way of a non-limiting example, a therapeutically effective amount of degarelix, including a pharmaceutically acceptable salt thereof, can include an amount effective to: (1) treat a disease including, but not limited to, advanced prostate cancer; (2) decrease plasma levels of luteinizing hormone (LH), reduce plasma level of follicle stimulating hormone (FSH), and/or reduce and maintain serum testosterone levels below castration level of 50 ng/dL; and/or (3) provide a plasma level of degarelix in a subject that is sufficient to achieve any of the results in (2). Therapeutically effective amounts of degarelix or a pharmaceutically acceptable salt thereof in a variety of diseases and conditions are described in more detail below. [0080] As used herein, “area under the curve” or “AUC” in pharmacokinetics, is the area under a plot of the concentration of a drug in the blood plasma as a function of time, and reflects the actual body exposure to a drug after administration of a dose of the drug. AUC is dependent on the rate of elimination of the drug from the body and the dose administered. [0081] As used herein, the terms "at least one", "one or more", and "and/or" are open- ended expressions that are both conjunctive and disjunctive in operation. For example, each of the expressions "at least one of A, B and C", "at least one of A, B, or C", "one or more of A, B, and C", "one or more of A, B, or C", "A, B, and/or C", and "A, B, or C" means A alone, B alone, C alone, A and B together, A and C together, B and C together, or A, B and C together. When each one of A, B, and C in the above expressions refers to an element, such as X, Y, and Z, or class of elements, such as X1-Xn, Y1-Ym, and Z1-Zo, the phrase is intended to refer to a single element selected from X, Y, and Z, a combination of elements selected from the same class (e.g., X1 and X2) as well as a combination of elements selected from two or more classes (e.g., Y1 and Zo). [0082] As used herein, the term “depot” refers to the degarelix and/or biodegradable polymer in a composition/formulation disclosed herein once exposed to an aqueous environment (e.g., including a physiological environment in a body of an animal) in which, upon dissipation of the solvent in situ, a solid, semi-solid, or liquid depot is formed comprising the biodegradable polymer and the degarelix API which self-aggregates (e.g., as fibrils or a gel). Together, the biodegradable polymer and the degarelix API create a drug reservoir from which the degarelix is released over an extended period of time. [0083] Unless otherwise specified, all ratios between monomers in a copolymer disclosed herein are molar ratios. [0084] Unless otherwise specified, various amounts of API, biodegradable polymer, and solvents and co-solvents are reported in weight percentages of the solvent system, solvent system and biodegradable polymer, or pharmaceutical composition. [0085] Every maximum numerical limitation given throughout this disclosure is deemed to include each and every lower numerical limitation as an alternative, as if such lower numerical limitations were expressly written herein. Every minimum numerical limitation given throughout this disclosure is deemed to include each and every higher numerical limitation as an alternative, as if such higher numerical limitations were expressly written herein. Every numerical range given throughout this disclosure is deemed to include both terminal values as well as each and every narrower numerical range that falls within such broader numerical range, as if such narrower numerical ranges were all expressly written herein. By way of example, the phrase from about 2 to about 4 includes the whole number and/or integer ranges from about 2 to about 3, from about 3 to about 4 and each possible range based on real (e.g., irrational and/or rational) numbers, such as from about 2.1 to about 4.9, from about 2.1 to about 3.4, and so on. [0086] Reference will now be made in detail to particular embodiments of compounds, formulations, and methods. The disclosed embodiments are not intended to be limiting of the claims. [0087] Degarelix is a decapeptide, selective gonadotropin-releasing hormone (GnRH) receptor antagonist (blocker) that competitively and reversibly binds to GnRH receptors in the pituitary. This results in a significant reduction of follicle-stimulating hormone (FSH) and luteinizing hormone (LH), thus reducing testosterone levels in males, or estrogen levels in females. In one embodiment of the invention, degarelix compositions of the invention are indicated for LH suppression or testosterone suppression in adult male patients with advanced hormone-dependent prostate cancer in whom androgen deprivation is warranted. Degarelix compositions of the invention are also useful for LH suppression or estrogen (e.g., estradiol) suppression in adult female patients with pre-menopausal or perimenopausal breast cancer, and are also useful for conditions including, but not limited to, endometriosis or uterine fibroids. Degarelix compositions are also useful for LH suppression or sex hormone suppression (testosterone or estrogen, depending on whether the patient is male or female) in children with central precocious puberty (CPP). There is an unmet need for a long-acting injectable formulation of degarelix that (1) is easy to prepare and administer (e.g., by a health care provider); (2) provides sustained or extended release of degarelix for more than a month after a single dose, and in one embodiment, for at least three months or longer; and (3) is administered in a total injection volume of about 2.5 mL or less, or about 2 mL or less. [0088] Disclosed herein is a long acting, injectable composition or formulation (used interchangeably and which may also be referred to as a “drug product” “pharmaceutical product” “product” or “finished drug product”) that comprises degarelix or a pharmaceutically acceptable salt thereof, in polymer-based drug delivery system. The formulation comprises a biodegradable polymer and a biocompatible, water-miscible solvent, and can optionally further include an additive as disclosed herein. The degarelix polymeric composition/formulation is a flowable solution/suspension where, upon delivery (i.e., injection) into an aqueous environment (e.g., the body of a human), the water-miscible solvent exchanges with surrounding aqueous body fluids, which results in the formation of a polymer-drug depot, that acts as a drug reservoir for extended release of the drug (i.e. release of the drug over an extended period of time). [0089] The composition/formulations disclosed herein provide sustained or extended release of degarelix or a pharmaceutically acceptable salt thereof for more than a month when injected at low volumes (e.g., <2.5 mL or <2 mL) via subcutaneous injection. The long-acting injectable compositions suitable for use in the methods of this disclosure, which may also be referred to as pharmaceutical compositions or formulations, extended-release compositions or formulations, or controlled release compositions or formulations, provide a biodegradable or bioerodible microporous in situ formed depot of biodegradable polymer and self-aggregated degarelix in a subject, from which the degarelix or a pharmaceutically acceptable salt thereof, is released over a period of one month or longer, and preferably at least three months or longer. The composition/formulation can be supplied in a prefilled single or dual chamber syringe, or in a two-syringe mixing system. The composition is administered from the syringe system in injection volumes of less than or equal to 2.5 mL, less than or equal to 2.4 mL, less than or equal to 2.3 mL, less than or equal to 2.2 mL, less than or equal to 2.1 mL, less than or equal to 2.0 mL, less than or equal to 1.9 mL, less than or equal to 1.8 mL, less than or equal to 1.7 mL, less than or equal to 1.6 mL, or less than equal to 1.5 mL and is suitable for subcutaneous injection. This composition solves the problem of the multiple, high-volume injections provided by the current degarelix product on the market. [0090] After administration of the compositions disclosed herein, degarelix or a pharmaceutically acceptable salt thereof is released from the polymer depot over a period of at least about 1 month or longer, at least about 1.5 months or longer, at least about 2 months or longer, at least about 2.5 months or longer, at least about 3 months or longer, at least about 3.5 months or longer, at least about 4 months or longer, at least about 4.5 months or longer, at least about 5 months or longer, or at least about 6 months or longer. The duration of release from the depot can depend on one or a number of factors including, for example, the nature of the biodegradable polymer, the composition and molecular weight of the biodegradable polymer, the solvent, the amounts of the components in the composition, optional additives in the composition, and combinations of any of the foregoing, or other factors. Active Pharmaceutical Ingredient (API) [0091] The compositions/formulations disclosed herein comprise degarelix or a pharmaceutically acceptable salt thereof in polymer-based drug delivery system. The pharmaceutically acceptable salts of the degarelix include, but are not limited to, degarelix acetate, degarelix citrate, degarelix pamoate, and degarelix mesylate. “Degarelix API” (whether capitalized or not) as used herein refers to degarelix or any pharmaceutically acceptable salt thereof. [0092] Generally, the disclosed compositions comprise a biodegradable polymer, a biocompatible solvent, and degarelix or a pharmaceutically acceptable salt thereof. The biodegradable polymer or copolymers are: substantially insoluble in water and body fluid, biocompatible, and biodegradable and/or bioerodible within the body of a subject. The pharmaceutical compositions are administered to a patient by subcutaneous injection as a liquid or gel wherein a solid, semi-solid, or liquid depot is formed in situ upon dissipation of the solvent. The depot so formed releases degarelix or a pharmaceutically acceptable salt thereof, in a controlled, or extended, release manner. [0093] The concentration of the degarelix or a pharmaceutically acceptable salt thereof, in the disclosed compositions can vary and may range from about 1% to about 40% by weight of the final formulation or composition (i.e., the formulation or composition in its ready-to-inject state), including any whole number percent to any other whole number percent within the range of from about 1 percent to about 40 percent by weight. As disclosed herein the total weight percent of each of the components in the final formulation/composition equals 100% (e.g., if the amount of degarelix salt in the formulation is 20 wt%, then the total amount of polymer and solvent in the formulation, absent any additives, is 80 wt%). The concentration of the degarelix or pharmaceutically acceptable salt thereof in the composition may be about 5% by weight of the composition, or about 10% by weight of the composition, or about 15% by weight of the composition, or about 20% by weight of the composition, or about 25% by weight of the composition, or about 30% by weight of the composition, or about 35% by weight of the composition, or about 40% by weight of the composition. In some embodiments, the concentration of degarelix or pharmaceutically acceptable salt thereof is no more than about 25% by weight of the composition. In other embodiments, the amount of degarelix API in compositions of the invention may range from any tenth of a percent to any other tenth of a percent within the range of from about 1% to about 40% by weight. [0094] In some aspects, the amount of degarelix API in a composition of the invention is referenced with respect to the amount of a pharmaceutically acceptable salt of degarelix, such as degarelix acetate or other salt, in the composition. In some aspects, the amount of degarelix API in a composition of the invention is referenced with respect to the amount of degarelix free base equivalent in the composition (which may also simply be referred to as the amount of degarelix in the composition). By way of example, determination of the dose amount of degarelix API to be administered to a subject may be calculated based on the amount of degarelix free base equivalent in the composition rather than the amount of degarelix salt in order to account for differences in purity, water, or acetic acid content (e.g., in the case of degarelix acetate), among various API lots. Calculation of the free base equivalent of degarelix in a composition is well understood within the art. For example, the percentage (by weight) of degarelix as free base in a composition where the degarelix API is degarelix acetate can be calculated in a simple formula such as: 100 – (wt% drug substance water content – wt% drug substance acetic acid content) * degarelix purity by assay (anhydrous/acid free base) * wt% degarelix acetate in the composition. The free base equivalent may also be provided from the USP Monograph for a drug substance. [0095] In one aspect, the amount of degarelix or the pharmaceutically acceptable salt thereof in the pharmaceutical composition is from about 8 wt% to about 39 wt% degarelix free base equivalent. In one aspect, the amount of degarelix or the pharmaceutically acceptable salt thereof in the pharmaceutical composition is from about 17 wt% to about 39 wt% degarelix free base equivalent. In one aspect, the amount of degarelix or the pharmaceutically acceptable salt thereof in the pharmaceutical composition is from about 8 wt% to about 29 wt% degarelix free base equivalent. In one aspect, the amount of degarelix or the pharmaceutically acceptable salt thereof in the pharmaceutical composition is from about 8 wt% to about 20 wt% degarelix free base equivalent. In one aspect, the amount of degarelix or the pharmaceutically acceptable salt thereof in the pharmaceutical composition is from about 10 wt% to about 16 wt% degarelix free base equivalent. In one aspect, the amount of degarelix or the pharmaceutically acceptable salt thereof in the pharmaceutical composition is from about 14 wt% to about 15.5 wt% degarelix free base equivalent. In one aspect, the amount of degarelix or a pharmaceutically acceptable salt thereof (degarelix API) in a composition of the invention is from about 30 wt% to about 35 wt% degarelix free base equivalent. In one aspect, the amount of degarelix API in the pharmaceutical composition is from about 31 wt% to about 34 wt% degarelix free base equivalent. In one aspect, the amount of degarelix API in the pharmaceutical composition is from about 31 wt% to about 33 wt% degarelix free base equivalent. In one aspect, the amount of degarelix API in the pharmaceutical composition is from about 31 wt% to about 32 wt% degarelix free base equivalent. In some aspects, when a target dose to be administered to a subject is calculated based on degarelix free base equivalent in the composition, the percent by weight of solvent, and the percent by weight of additive if included, may be modified accordingly. Biocompatible/biodegradable Polymers [0096] Polymers suitable for use in the disclosed compositions include polymers, copolymers and/or terpolymers formed of repeating units, which can be linear, branched, grafted and/or star-shaped. Further, the polymers can comprise monomer residues, such as lactide, glycolide, caprolactone, p-dioxanone, trimethylene carbonate, 1,5-dioxepan-2-one, 1,4-dioxepan-2-one, ethylene oxide, propylene oxide, sebacic anhydride, diketene acetals/diols, lactic acid, and combinations thereof. [0097] Biodegradable polymers suitable for use with the disclosed compositions include, but are not limited to, polylactic acid, polyglycolic acid, polylactide (D-lactide, L- lactide), poly(D,L-lactide) (PLA), polyglycolide, polycaprolactones, poly(D,L-lactide-co- glycolide) (PLG), poly(D,L-lactide-co-ε-caprolactone) (PLC), polyanhydrides, polyamides, polyurethanes, polyesteramides, polyorthoesters, polydioxanones, polyacetals, polyketals, polycarbonates, polyphosphazenes, polyhydroxybutyrates, polyhydroxyvalerates, polyalkylene oxalates, polyalkylene succinates, poly(malic acid), polyglutamic acids, poly(alkyl cyanoacrylates) polyethylene glycol, hyaluronic acid, alginate, collagen, chitin and chitosan, and combinations or mixtures of the above materials. [0098] In some embodiments, the biodegradable polymer may be a copolymer of two monomers having a molar ratio of any two whole numbers X to Y, such that the sum of X and Y is 100. In some embodiments, the biodegradable polymer may be a copolymer of two monomers having a molar ratio of any two whole numbers X to Y, where X and Y are each at least about 10 and no more than about 90, such that the sum of X and Y is 100, e.g., a copolymer comprising a 10:90 to 90:10 molar ratio of X:Y. In some embodiments, X and Y may be at least about 15 to no more than about 85 such that the sum of X and Y is 100, e.g., a copolymer comprising a 15:85 to 85:15 molar ratio of X:Y. In some embodiments, X and Y may be at least about 25 to no more than about 75 such that the sum of X and Y is 100, e.g., a copolymer comprising a 25:75 to 75:25 molar ratio of X:Y. In some embodiments, both X and Y may be about 50, e.g., a copolymer comprising a 50:50 molar ratio of X:Y. [0099] In some embodiments, the biodegradable polymer is a copolymer comprising (a) D,L-lactide, D-lactide, L-lactide or glycolide monomer residues and (b) caprolactone monomer residues. In some embodiments, the biodegradable polymer is a copolymer comprising lactide, preferably D,L-lactide, and caprolactone monomer residues. In some embodiments, the biodegradable polymer is poly(D,L-lactide-co-ε-caprolactone) (PLC). In one aspect the PLC comprises at least one carboxylic acid end group. In one aspect, the PLC is a carboxylic acid-initiated PLC. In still another aspect, the carboxylic acid is glycolic acid or lactic acid. In some embodiments, the polymer is a copolymer comprising a molar ratio of lactide (or glycolide) to ε-caprolactone ranging from about 10:90 to about 90:10, from about 20:80 to about 80:20, from about 25:75 to about 75:25, or from about 30:70 to about 70:30. In one aspect, the ratio of lactide (or glycolide) to ε-caprolactone is from about 25:75 to about 75:25. In one aspect, the ratio of lactide (or glycolide) to ε-caprolactone is from about 50:50 to about 75:25. In one aspect, the ratio of lactide (or glycolide) to ε- caprolactone is about 50:50. In still another aspect, the ratio of lactide (or glycolide) to ε- caprolactone is about 75:25. In one aspect, the biodegradable polymer is acid-initiated 75:25 poly(D,L-lactide-co-ε-caprolactone). [00100] In some embodiments, the biodegradable polymer is a copolymer comprising (a) D,L-lactide, D-lactide, L-lactide or glycolide monomer residues and (b) trimethylene carbonate (TMC) monomer residues. In some embodiments, the biodegradable polymer is a copolymer comprising lactide, preferably D,L-lactide, and TMC monomer residues. In some embodiments, the biodegradable polymer is a copolymer comprising a molar ratio of lactide (or glycolide) to TMC ranging from about 10:90 to about 90:10, from about 20:80 to about 80:20, from about 25:75 to about 75:25, or from about 30:70 to about 70:30. In one aspect, the ratio of lactide (or glycolide) to TMC is from about 25:75 to about 75:25. In one aspect, the ratio of lactide (or glycolide) to TMC is about 50:50. In still another aspect, the ratio of lactide (or glycolide) to TMC is about 75:25. [00101] In some embodiments, the biodegradable polymer is a copolymer comprising (a) D,L-lactide, D-lactide, or L-lactide monomer residues and (b) glycolide monomer residues. In some embodiments, the biodegradable polymer is poly(DL-lactide-co-glycolide) (PLG). In one aspect the PLG comprises at least one carboxylic acid end group. In one aspect, the PLG is a core-diol-initiated PLG. In one aspect, the core diol is 1,6-hexane diol. In one aspect, the PLG was initiated with an uncapped polyethylene glycol (PEG). In still another aspect, the PLG is a mono functional alcohol-initiated PLG. In one aspect, the mono functional alcohol is dodecanol. In another aspect, the PLG was initiated with a mono- capped PEG (mPEG). In a non-limiting example, the PLG has a molar ratio of lactide monomers to glycolide monomers from about 50:50 to about 90:10. In one aspect, the PLG has a molar ratio of lactide to glycolide monomers from about 45:55 to about 99:1. In some instances, the PLG copolymer may comprise a lactide to glycolide monomer molar ratio from about 50:50 to about 90:10, from about 50:50 to about 80:20, from about 50:50 to about 70:30, or from about 50:50 to about 75:25. In one aspect the molar ratio of lactide monomers to glycolide monomers is 50:50, 75:25 or 85:15. In one aspect, the biodegradable polymer is acid-initiated 50:50 poly(D,L-lactide-co-glycolide). In another aspect, the biodegradable polymer is 1,6-hexane diol initiated 75:25 poly(D,L-lactide-co-glycolide). In yet another aspect, the biodegradable polymer is 1,6-hexane diol initiated 85:15 poly(D,L- lactide-co-glycolide). In yet a further aspect, the biodegradable polymer is dodecanol- initiated poly(D,L-lactide). [00102] In some embodiments, the biodegradable polymer is poly(D,L-lactide) (PLA). In one aspect, the (PLA) comprises at least one carboxylic acid end group. [00103] Further non-limiting examples of suitable polymers include biodegradable polymers comprising a copolymer with lactide (including D,L-lactide, D-lactide, and/or L- lactide) and/or glycolide residues, wherein the molar percentage of the lactide and/or glycolide residues make up greater than about 5% and less than about 95% of the polymer. In some embodiments, the lactide and/or glycolide monomer residues make up at least about 5%, at least about 10%, at least about 15%, at least about 20%, at least about 25%, at least about 30%, at least about 35%, at least about 40%, at least about 45%, at least about 50%, at least about 55%, at least about 60%, at least about 65%, at least about 70%, at least about 75%, at least about 80%, at least about 85%, at least about 90%, or at least about 95% of total monomer residues of the copolymer. Other non-limiting examples of suitable polymers of the invention include biodegradable polymers comprising a copolymer with caprolactone and/or TMC residues, wherein the caprolactone and/or trimethylene carbonate residues make up in an amount greater than about 5% and less than about 95% of the polymer. In some embodiments, the caprolactone and/or TMC monomer residues make up at least about 5%, at least about 10%, at least about 15%, at least about 20%, at least about 25%, at least about 30%, at least about 35%, at least about 40%, at least about 45%, at least about 50%, at least about 55%, at least about 60%, at least about 65%, at least about 70%, at least about 75%, at least about 80%, at least about 85%, at least about 90%, or at least about 95% of total monomers of the copolymer. [00104] In some embodiments, the biodegradable polymer may be formed using an initiator selected to provide a desired structure or functionality to the polymer in the form of a particular polymer block structure or end group structure which is introduced and/or incorporated into/onto the biodegradable polymer. By way of another non-limiting example, the polymer may be formed using an organic acid (e.g., a hydroxy acid such as, for instance, glycolic acid) as the initiator which may result in the formation of a polymer comprising at least one carboxylic acid end group. By way of another non-limiting example, the polymer may be formed using a monofunctional alcohol (e.g., dodecanol, or monomethoxy endcapped monofunctional polyethylene glycols (PEGs)) as the initiator which may result in the formation of a polymer comprising at least one hydroxy end group. By way of a further non-limiting example, the polymer may be formed using a diol (e.g., hexanediol, PEG300, PEG400, PEG600) as the initiator which may result in the formation of a polymer comprising at least one hydroxy end group and that is also substantially free of terminal carboxy end groups. In some embodiments, the biodegradable polymer may be formed using an initiator selected to provide a desired structure, and in particular a desired polymer block structure or end group structure, to the biodegradable polymer. By way of non- limiting example, the polymer may be formed using a low-molecular weight PEG (e.g., the above-mentioned PEG300, PEG400 or PEG600) as an initiator, which may result in the formation of a block copolymer comprising a low-molecular weight PEG block. By way of non-limiting example, block A may comprise first monomer residues selected from D,L- lactide, D-lactide, or L-lactide, and second monomer residues selected from glycolide, ε- caprolactone, or trimethylene carbonate. Block B comprises a low-molecular weight polyethylene glycol (PEG). The blocks may be arranged in any number or order (e.g. as a di-block copolymer A-B, or a tri-block copolymer A-B-A or B-A-B). Such polymers are formed by initiation of the first and second monomer residues with a low-molecular weight PEG initiator. [00105] In some embodiments, biodegradable polymers suitable for use in formulations according to the present invention may, generally, comprise a weight average molecular weight ranging from about 1 kDa and about 100 kDa. In some embodiments, the biodegradable polymer may comprise a weight average molecular weight ranging from about 1 kDa to about 5 kDa, from about 1 kDa to about 10 kDa, from about 1 kDa to about 15 kDa, from about 1 kDa to about 20 kDa, from about 1 kDa to about 25 kDa, from about 1 kDa to about 30 kDa, from about 1 kDa to about 40 kDa, from about 1 kDa to about 50 kDa, from about 1 kDa to about 60 kDa, from about 1 kDa to about 70 kDa, from about 1 kDa to about 80 kDa, from about 1 kDa to about 90 kDa, from about 1 kDa to about 100 kDa, or any value to any other value, in whole number increments, from about 1 kDa to about 100 kDa. In one aspect, the weight average molecular weight is from about 5 kDa to about 60 kDa. In one aspect, the weight average molecular weight is from about 5 kDa to about 55 kDa. In one aspect, the weight average molecular weight is from about 5 kDa to about 50 kDa. In one aspect, the weight average molecular weight is from about 5 kDa to about 45 kDa. In one aspect, the weight average molecular weight is from about 5 kDa to about 40 kDa. In still another aspect, the weight average molecular weight is from about 5 kDa to 35 kDa. [00106] In some embodiments of the composition, the biodegradable polymer may make up about 10 wt%, about 15 wt%, about 20 wt%, about 25 wt%, about 30 wt%, about 35 wt%, about 40 wt%, about 45 wt%, about 50 wt%, about 55 wt%, or about 60 wt% of the composition. In some embodiments, the biodegradable polymer may make up about 20 wt%, about 25 wt%, or about 30 wt% of the composition. Alternatively, the biodegradable polymer may make up any whole-number weight percentage of the composition from about 10 wt% to about 60 wt%. In other embodiments, the biodegradable polymer may make up any tenth of a whole number percent of the composition from about 10 wt% to about 60 wt%. [00107] In embodiments, the biodegradable polymers may optionally be purified prior to use in the long-acting formulation to remove low-molecular weight oligomers and/or unreacted monomers, and/or catalyst. Several methods of purifying polymers are known in the art, including the methods described in U.S. Pat. No. 4,810,775, U.S. Patent No. 7,019,106, and U.S. Patent No. 9,187,593, among others. In some embodiments of the composition, the biocompatible polymer comprises less than about 0.5%, or less than about 0.4%, or less than about 0.3%, or less than about 0.2% or less than about 0.1% unreacted monomers. In some embodiments of the composition, the biocompatible polymer comprises less than about 0.5%, or less than about 0.4%, or less than about 0.3%, or less than about 0.2% or less than about 0.1% unreacted lactide monomer. Biocompatible Solvents [00108] Any suitable water-miscible solvent can be employed, provided the solvent is biocompatible, and miscible to dispersible in aqueous medium or body fluid. Examples of suitable solvents are disclosed, e.g., in Aldrich Handbook of Fine Chemicals and Laboratory Equipment, Milwaukee, Wis. (2000); and in U.S. Pat. Nos. 5,324,519; 4,938,763; 5,702,716; 5,744,153; and 5,990,194. In various aspects, the solvent is able to diffuse into body fluid so that the flowable polymer-based composition coagulates, gels or solidifies. The solvent may or may not dissolve the polymer, although it is preferred that the solvent dissolves the polymer. Solvents and co-solvents that may be used in the disclosed compositions are preferably biocompatible, non-toxic, solvents, which may be either hydrophilic or hydrophobic solvents, or may be a combination of hydrophilic solvents, hydrophobic solvents or hydrophilic and hydrophobic solvents, depending upon the desired release profile and the solubility of the polymer and/or the degarelix API in the polymer/solvent composition. In one aspect, the solvent and/or co-solvent is an organic solvent. In still another aspect, the solvent and/or co-solvents is a polar aprotic solvent. [00109] Suitable solvents and co-solvents may comprise one or more solvents selected from the group consisting of amides, acids, alcohols, esters of monobasic acids, ether alcohols, sulfoxides, lactones, polyhydroxy alcohols, esters of polyhydroxy alcohols, ketones, and ethers. By way of non-limiting example, the solvents or cosolvents may comprise at least one of N-methyl-2-pyrrolidone (NMP), dimethyl sulfoxide (DMSO), dimethylacetamide (DMA), acetone, benzyl benzoate (BnBzO), polyethylene glycol 15 hydroxystearate, ethyl acetate, glycofurol, N-hydroxyethyl-2-pyrrolidone, polyethylene glycol (PEG), benzyl alcohol, propylene carbonate (PC), propylene glycol, 2-pyrrolidone, α-tocopherol, triacetin, tributyl citrate, acetyl tributyl citrate, acetyl triethyl citrate, triethyl citrate, and combinations thereof. [00110] In aspects of the invention, a suitable solvent is selected from N-methyl-2- pyrrolidone (NMP) and/or dimethyl sulfoxide (DMSO). [00111] In one aspect of the invention, the biodegradable polymer is dissolved in the biocompatible solvent or combination or mixture of solvents and/or co-solvents. In one aspect, the degarelix or pharmaceutically acceptable salt thereof may be dissolved in a first biocompatible solvent and the biodegradable polymer may be dissolved in a second biocompatible solvent. In one aspect, the first and second biocompatible solvents are the same. In one aspect, the biodegradable polymer is dissolved in the biocompatible solvent, and the degarelix or pharmaceutically acceptable salt thereof is not dissolved in the solvent, but is provided in lyophilized or other dry form. [00112] The biocompatible solvent, or combination or mixture of solvents and/or co- solvents used in a composition of the invention, will generally comprise between about 20 wt% and about 80 wt% of the final formulation, or between about 30 wt% and about 70 wt% of the final formulation, or between about 40 wt% and about 60 wt% of the final formulation, or alternatively the solvent or combination or mixture of solvents and/or co- solvents can range from any whole number percentage by weight of the formulation to any other whole number percentage by weight of the final formulation between about 20 wt% and about 80 wt%. Additives [00113] Optionally, the pharmaceutical compositions disclosed herein may comprise various additives (which may also be referred to as “excipients”) to improve the stability, the injectability, or/and other properties of the composition. For example, the pharmaceutical compositions may comprise one or more of antioxidants, chelating agents, surfactants, co-solvents (also discussed above), stabilizers, complexing agents, antioxidants, and solubilizers. [00114] In some embodiments, the pharmaceutical compositions may comprise one or more solubilizers to increases the solubility of one or more other component of the composition. Solubilizers useful in the disclosed compositions include any solubilizer useful for parenteral injection, and include, but are not limited to, surfactants which lower the surface tension between two liquids, between a gas and a liquid, or between a liquid and a solid and other solubilizers. Examples of suitable solubilizers and/or surfactants for use in the invention include, but are not limited to, polysorbate 20, polysorbate 80, poloxamer 188, sorbitan trioleate, lecithin (e.g., soya or egg), Vitamin E TPGS, sugar based esters or ethers (e.g., sugar acid esters of fatty alcohols or sugar alcohol esters of fatty acids, including, but not limited to, sucrose cocoate, sucrose stearate, sucrose laurate, etc.), amino acid-based solubility enhancers (e.g., proline, arginine, DL-methionine), protein-based solubility enhancers (e.g., hydrophobins), and others. [00115] In some embodiments, the pharmaceutical compositions may comprise one or more antioxidants to inhibit oxidation of the API and improve the stability of the formulation. Examples of suitable antioxidants for use in the invention include, but are not limited to, citric acid, methanesulfonic acid, ascorbic acid, ethylenediaminotetraacetic acid (EDTA), mercaptoethanol, sodium metabisulfite butylated hydroxyanisole, butylated hydroxyquinone, butylhydroxyanisol, hydroxycoumarin, butylated hydroxytoluene, cephalm, ethyl gallate, propyl gallate, octyl gallate, lauryl gallate, propyl-hydroxybenzoate, trihydroxybutylrophenone, dimethylphenol, dibutylphenol, vitamin E, lecithin and ethanolamine. [00116] In some embodiments, the pharmaceutical compositions may comprise one or more complexing agents to inhibit oxidation and/or degradation of the API and improve the stability of the formulation. Examples of suitable complexing agents for use in the invention include, but are not limited to, ethylenediaminotetraacetic acid (EDTA), divalent metal salts (ZnCl2, MgCl2, CaCl2), nitrilotriacetic acid, n-hydroxyethylethylenediaminetriacetic acid (HEDTA), ethylene glycol-bis(β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid (EGTA) as well as several simple organic acids, such as, polycarboxylic acid (citric acid), hydrochloric acid, sulfuric acid, pamoic acid, and palmitic acid. [00117] In some embodiments, the pharmaceutical compositions may comprise one or more stabilizing agents to prevent drug and/or polymer degradation (physical or chemical) and improve the stability of the formulation and increase shelf life. Examples include, but are not limited to: surfactants (e.g., polysorbate 20, polysorbate 80, poloxamer 188), complexing agents (e.g., divalent metal salts), acid additives (e.g., acetic acid, citric acid, succinic acid, methane sulfonic acid, sulfuric acid, hydrochloric acid, pamoic acid, palmitic acid, hydrobromic acid, nitric acid, chromic acid, trifluoroethanes sulfonic acid, trichloroacetic acid, dichloroacetic acid, bromoacetic acid, chloroacetic acid, cyanoacetic acid, 2-chloropropanoic acid, 4-cyanobutanoic acid, perchloric acid, phosphoric acid, hydrogen iodide, and combinations thereof), and alcohols (e.g., benzyl alcohol). Compositions [00118] In various aspects, the present disclosure provides long acting, injectable pharmaceutical compositions comprising degarelix or a pharmaceutically acceptable salt thereof, a biocompatible and biodegradable polymer, a biocompatible solvent, and optionally one or more additives. All such compositions are contemplated for administration to a subject to treat a disease or condition for which administration of a GnRH agonist or GnRH antagonist may be useful. In various aspects, the compositions of the invention are contemplated for use to treat prostate cancer, including advanced prostate cancer; to treat central precocious puberty (CPP); to treat pre-menopausal or peri-menopausal breast cancer; or to treat other conditions, including, but not limited to endometriosis or uterine fibroids. Further, such compositions are contemplated for administration to a subject to reduce luteinizing hormone (LH) levels in a subject, to reduce follicle stimulating hormone (FSH) levels in a subject, and in males, to reduce serum testosterone levels, and in females, to reduce serum estrogen levels. [00119] Depending upon the other components in the composition (i.e, the biodegradable polymer and/or solvent(s)), the degarelix or a pharmaceutically acceptable salt thereof, in the disclosed compositions may form a monophasic mixture (e.g., a solution) or a biphasic mixture (e.g., a suspension or a dispersion) within the composition. Accordingly, the extended release compositions comprising the degarelix or pharmaceutically acceptable salt thereof, according to the present invention may appropriately be either a “solution”, or a “dispersion”, or a “suspension” of the degarelix or pharmaceutically acceptable salt thereof, in the solvent or in the combination of the biodegradable polymer and the solvent. In other words, the degarelix or pharmaceutically acceptable salt thereof, may in some embodiments be dissolved in solvent (as in a solution), or in the combination of polymer and solvent (as in a solution), or form solid particles sufficiently large for suspension and sedimentation as part of a heterogeneous mixture (as in a suspension or dispersion). In some embodiments, the degarelix or pharmaceutically acceptable salt thereof is dissolved in the solvent when in the absence of the biocompatible polymer, and, when mixed in combination with the solvent(s) and the biocompatible polymer, may be in suspension or dispersion, or partial suspension, where some of the degarelix or pharmaceutically acceptable salt remains in solution and some is in suspension or dispersion. In some embodiments, the polymer- solvent system composition may be a liquid-liquid dispersion. As non-limiting examples, types of dispersions may include liquid-in oil, oil-in liquid, or oil-in-oil dispersions. In some embodiments, the polymer-solvent system composition may be an emulsion. In one aspect, the degarelix API is a stable solution and stays in solution in the biocompatible solvent and thus little or no reconstitution or mixing is needed to maintain the stable solution. [00120] In some embodiments, the present invention provides a pharmaceutical composition comprising in the final formulation: (a) between about 10 wt% and about 30 wt% of a pharmaceutically acceptable salt of degarelix; (b) between about 20 wt% and about 80 wt% of N-methyl-2-pyrrolidone (NMP); and (c) between about 10 wt% and about 50 wt% of 75:25 poly(lactide-co-ε-caprolactone) (PLC) copolymer, where the total weight percent of each of the components in the formulation equals 100% (e.g., if the amount of degarelix salt in the formulation is 20 wt%, then the total amount of polymer and solvent in the formulation, absent any additives, is 80 wt%). In one aspect, the PLC copolymer has at least one carboxylic acid end group. In one aspect, the PLC copolymer has a weight average molecular weight of between about 5 kDa and about 30 kDa. In one aspect, the pharmaceutically acceptable salt of degarelix is selected from degarelix acetate and degarelix citrate. [00121] In some embodiments, the present disclosure provides a pharmaceutical composition comprising about 16 wt% of degarelix acetate; about 57 wt% of N-methyl-2- pyrrolidone (NMP); and about 27 wt% of 75:25 poly(lactide-co-ε-caprolactone) (PLC) copolymer having at least one carboxylic acid end group and having a weight average molecular weight of about 17 kDa. [00122] In some embodiments, the present disclosure provides a pharmaceutical composition comprising about 16 wt% of degarelix acetate; about 57 wt% of NMP; and about 27 wt% of 75:25 PLC copolymer having at least one carboxylic acid end group and having a weight average molecular weight of about 8 kDa. [00123] In some embodiments, the present invention provides a pharmaceutical composition comprising a first container comprising 30-70 wt% of 75:25 PLC copolymer having at least one carboxylic acid end group and having a weight average molecular weight of 5-30 kDa, and 30-70 wt% of N-methyl-2-pyrrolidone (NMP); and a second container comprising 20-40 wt% of a pharmaceutically acceptable salt of degarelix and 60-80 wt% NMP. In one aspect, the pharmaceutically acceptable salt of degarelix is selected from degarelix acetate and degarelix citrate. In one embodiment, the first and second containers are a first and second syringe, respectively. In one embodiment, the first and second containers are a first and second chamber of a dual-chambered syringe, respectively. [00124] In some embodiments, the present disclosure provides a pharmaceutical composition comprising a first container comprising 50 wt% of 75:25 PLC copolymer having at least one carboxylic acid end group and having a weight average molecular weight of 17 kDa, and 50 wt% of N-methyl-2-pyrrolidone (NMP); and a second container comprising 35 wt% of degarelix acetate and 65 wt% NMP. In one embodiment, the first and second containers are a first and second syringe, respectively. In one embodiment, the first and second containers are a first and second chamber of a dual-chambered syringe, respectively. [00125] In some embodiments, the present disclosure provides a pharmaceutical composition comprising: a first container comprising 70 wt% of 75:25 PLC copolymer having at least one carboxylic acid end group and having a weight average molecular weight of 8 kDa, and 30 wt% of NMP; and a second container comprising 35 wt% of degarelix acetate and 65 wt% NMP. In one embodiment, the first and second containers are a first and second syringe, respectively. In one embodiment, the first and second containers are a first and second chamber of a dual-chambered syringe, respectively. [00126] In some embodiments, the present invention provides a pharmaceutical composition comprising in the final formulation between about 10 wt% and about 30 wt% of a pharmaceutically acceptable salt of degarelix; between about 20 wt% and about 80 wt% of N-methyl-2-pyrrolidone (NMP); and between about 10 wt% and about 50 wt% of 75:25 poly(D,L-lactide) (PLA) polymer, where the total weight percent of each of the components in the formulation equals 100% (e.g., if the amount of degarelix salt in the formulation is 20 wt%, then the total amount of polymer and solvent in the formulation, absent any additives, is 80 wt%). In one aspect, the PLA polymer has at least one carboxylic acid end group. In one aspect, the PLA polymer has a weight average molecular weight of between about 5 kDa and about 30 kDa. In one aspect, the pharmaceutically acceptable salt of degarelix is selected from degarelix acetate and degarelix citrate. [00127] In some embodiments, the present disclosure provides a pharmaceutical composition comprising about 16 wt% of degarelix acetate; about 57 wt% of NMP; and about 27 wt% of poly(D,L-lactide) (PLA) polymer having at least one carboxylic acid end group and having a weight average molecular weight of about 23 kDa. [00128] In some embodiments, the present invention provides a pharmaceutical composition comprising a first container comprising 30-70 wt% of 75:25 PLA polymer having at least one carboxylic acid end group and having a weight average molecular weight of 5-30 kDa, and 30-70 wt% of N-methyl-2-pyrrolidone (NMP); and a second container comprising 20-40 wt% of a pharmaceutically acceptable salt of degarelix and 60-80 wt% NMP. In one aspect, the pharmaceutically acceptable salt of degarelix is selected from degarelix acetate and degarelix citrate. In one embodiment, the first and second containers are a first and second syringe, respectively. In one embodiment, the first and second containers are a first and second chamber of a dual-chambered syringe, respectively. [00129] In some embodiments, the present disclosure provides a pharmaceutical composition comprising a first container comprising 50 wt% of PLA polymer having at least one carboxylic acid end group and having a weight average molecular weight of 23 kDa, and 50 wt% of NMP; and a second container comprising 35 wt% of degarelix acetate and 65 wt% NMP. In one embodiment, the first and second containers are a first and second syringe, respectively. In one embodiment, the first and second containers are a first and second chamber of a dual-chambered syringe, respectively. [00130] In some embodiments, the present invention provides a pharmaceutical composition comprising in the final formulation between about 10 wt% and about 30 wt% of a pharmaceutically acceptable salt of degarelix; between about 20 wt% and about 80 wt% of N-methyl-2-pyrrolidone (NMP); and between about 10 wt% and about 50 wt% of 75:25 poly(D,L-lactide-co-glycolide) (PLG) copolymer, where the total weight percent of each of the components in the formulation equals 100% (e.g., if the amount of degarelix salt in the formulation is 20 wt%, then the total amount of polymer and solvent in the formulation, absent any additives, is 80 wt%). In one aspect, the PLG copolymer has at least one carboxylic acid end group. In one aspect, the PLG copolymer has a weight average molecular weight of between about 5 kDa and about 30 kDa. In one aspect, the pharmaceutically acceptable salt of degarelix is selected from degarelix acetate and degarelix citrate. [00131] In some embodiments, the present disclosure provides a pharmaceutical composition comprising about 16 wt% of degarelix acetate; about 57 wt% of NMP; and about 27 wt% of 75:25 poly(D,L-lactide-co-glycolide) (PLG) copolymer having at least one carboxylic acid end group and having a weight average molecular weight of about 17 kDa. [00132] In some embodiments, the present invention provides a pharmaceutical composition comprising a first container comprising 30-70 wt% of 75:25 PLG polymer having at least one carboxylic acid end group and having a weight average molecular weight of 5-30 kDa, and 30-70 wt% of N-methyl-2-pyrrolidone (NMP); and a second container comprising 20-40 wt% of a pharmaceutically acceptable salt of degarelix and 60-80 wt% NMP. In one aspect, the pharmaceutically acceptable salt of degarelix is selected from degarelix acetate and degarelix citrate. In one embodiment, the first and second containers are a first and second syringe, respectively. In one embodiment, the first and second containers are a first and second chamber of a dual-chambered syringe, respectively. [00133] In some embodiments, the present disclosure provides a pharmaceutical composition comprising: a first container comprising 50 wt% of 75:25 PLG copolymer having at least one carboxylic acid end group and having a weight average molecular weight of 17 kDa, and 50 wt% of NMP; and a second container comprising 35 wt% of degarelix acetate and 65 wt% NMP. In one embodiment, the first and second containers are a first and second syringe, respectively. In one embodiment, the first and second containers are a first and second chamber of a dual-chambered syringe, respectively. [00134] In some embodiments, the present invention provides a pharmaceutical composition comprising in the final formulation between about 10 wt% and about 30 wt% of a pharmaceutically acceptable salt of degarelix; between about 20 wt% and about 80 wt% of N-methyl-2-pyrrolidone (NMP); and between about 10 wt% and about 50 wt% of 75:25 poly(D,L-lactide-co-glycolide) (PLG) copolymer, where the total weight percent of each of the components in the formulation equals 100% (e.g., if the amount of degarelix salt in the formulation is 20 wt%, then the total amount of polymer and solvent in the formulation, absent any additives, is 80 wt%). In one aspect, the PLG copolymer has at least one hydroxyl end group. In one aspect, the PLG copolymer has a weight average molecular weight of between about 5 kDa and about 30 kDa. In one aspect, the pharmaceutically acceptable salt of degarelix is selected from degarelix acetate and degarelix citrate. [00135] In some embodiments, the present disclosure provides a pharmaceutical composition comprising about 16 wt% of degarelix acetate; about 57 wt% of NMP; and about 27 wt% of 75:25 PLG copolymer having at least one hydroxyl end group and having a weight average molecular weight of about 19 kDa. [00136] In some embodiments, the present disclosure provides a pharmaceutical composition comprising about 16 wt% of degarelix citrate; about 57 wt% of NMP; and about 27 wt% of 75:25 PLG copolymer having at least one hydroxyl end group and having a weight average molecular weight of about 19 kDa. [00137] In some embodiments, the present invention provides a pharmaceutical composition comprising a first container comprising 30-70 wt% of 75:25 PLG polymer having at least one hydroxyl end group and having a weight average molecular weight of 5- 30 kDa, and 30-70 wt% of N-methyl-2-pyrrolidone (NMP); and a second container comprising 20-40 wt% of a pharmaceutically acceptable salt of degarelix and 60-80 wt% NMP. In one aspect, the pharmaceutically acceptable salt of degarelix is selected from degarelix acetate and degarelix citrate. In one embodiment, the first and second containers are a first and second syringe, respectively. In one embodiment, the first and second containers are a first and second chamber of a dual-chambered syringe, respectively. [00138] In some embodiments, the present disclosure provides a pharmaceutical composition comprising a first container comprising 50 wt% of 75:25 PLG copolymer having at least one hydroxyl end group and having a weight average molecular weight of 19 kDa, and 50 wt% of NMP; and a second container comprising 35 wt% of degarelix acetate and 65 wt% NMP. [00139] In some embodiments, the present disclosure provides a pharmaceutical composition comprising a first container comprising 50 wt% of 75:25 PLG copolymer having at least one hydroxyl end group and having a weight average molecular weight of 19 kDa, and 50 wt% of NMP; and a second container comprising 35 wt% of degarelix citrate and 65 wt% NMP. In one embodiment, the first and second containers are a first and second syringe, respectively. In one embodiment, the first and second containers are a first and second chamber of a dual-chambered syringe, respectively. [00140] In some embodiments, the present invention provides a pharmaceutical composition comprising in the final formulation between about 10 wt% and about 30 wt% of a pharmaceutically acceptable salt of degarelix; between about 20 wt% and about 80 wt% of N-methyl-2-pyrrolidone (NMP); between about 10 wt% and 40 wt% of DMSO, and between about 10 wt% and about 50 wt% of 75:25 poly(D,L-lactide-co-glycolide) (PLG) copolymer, where the total weight percent of each of the components in the formulation equals 100% (e.g., if the amount of degarelix salt in the formulation is 20 wt%, then the total amount of polymer and solvents in the formulation, absent any additives, is 80 wt%). In one aspect, the PLG copolymer has at least one hydroxyl end group. In one aspect, the PLG copolymer has a weight average molecular weight of between about 5 kDa and about 30 kDa. In one aspect, the pharmaceutically acceptable salt of degarelix is selected from degarelix acetate and degarelix citrate. [00141] In some embodiments, the present disclosure provides a pharmaceutical composition comprising about 16 wt% of degarelix acetate; about 30 wt% of NMP; about 27 wt% of DMSO; and about 27 wt% of 75:25 PLG copolymer having at least one hydroxyl end group and having a weight average molecular weight of about 19 kDa. [00142] In some embodiments, the present invention provides a pharmaceutical composition comprising a first container comprising 30-70 wt% of 75:25 PLG polymer having at least one hydroxyl end group and having a weight average molecular weight of 5- 30 kDa, and 30-70 wt% of N-methyl-2-pyrrolidone (NMP); and a second container comprising 20-40 wt% of a pharmaceutically acceptable salt of degarelix and 60-80 wt% DMSO. In one aspect, the pharmaceutically acceptable salt of degarelix is selected from degarelix acetate and degarelix citrate. In one embodiment, the first and second containers are a first and second syringe, respectively. In one embodiment, the first and second containers are a first and second chamber of a dual-chambered syringe, respectively. [00143] In some embodiments, the present disclosure provides a pharmaceutical composition comprising a first container comprising 50 wt% of 75:25 PLG copolymer having at least one hydroxyl end group and having a weight average molecular weight of 19 kDa, and 50 wt% of NMP; and a second container comprising 35 wt% of degarelix acetate and 65 wt% DMSO. In one embodiment, the first and second containers are a first and second syringe, respectively. In one embodiment, the first and second containers are a first and second chamber of a dual-chambered syringe, respectively. [00144] In any of the above embodiments, in one aspect, the amount of degarelix or the pharmaceutically acceptable salt thereof in the pharmaceutical composition is from about 8 wt% to about 39 wt% degarelix free base equivalent. In one aspect, the amount of degarelix or the pharmaceutically acceptable salt thereof in the pharmaceutical composition is from about 17 wt% to about 39 wt% degarelix free base equivalent. In one aspect, the amount of degarelix or the pharmaceutically acceptable salt thereof in the pharmaceutical composition is from about 8 wt% to about 29 wt% degarelix free base equivalent. In one aspect, the amount of degarelix or the pharmaceutically acceptable salt thereof in the pharmaceutical composition is from about 8 wt% to about 20 wt% degarelix free base equivalent. In one aspect, the amount of degarelix or the pharmaceutically acceptable salt thereof in the pharmaceutical composition is from about 10 wt% to about 16 wt% degarelix free base equivalent. In one aspect, the amount of degarelix or the pharmaceutically acceptable salt thereof in the pharmaceutical composition is from about 14 wt% to about 15.5 wt% degarelix free base equivalent. In one aspect, the amount of degarelix or a pharmaceutically acceptable salt thereof (degarelix API) in a composition of the invention is from about 30 wt% to about 35 wt% degarelix free base equivalent. In one aspect, the amount of degarelix API in the pharmaceutical composition is from about 31 wt% to about 34 wt% degarelix free base equivalent. In one aspect, the amount of degarelix API in the pharmaceutical composition is from about 31 wt% to about 33 wt% degarelix free base equivalent. In one aspect, the amount of degarelix API in the pharmaceutical composition is from about 31 wt% to about 32 wt% degarelix free base equivalent. [00145] In some embodiments, unreacted lactide, caprolactone, and/or glycolide monomers in the polymers or copolymers are less than about 1.0 wt%, less than about 0.5 wt%, than about 0.4 wt %, less than about 0.3 wt %, less than about 0.2 wt% and less than about 0.1 wt%. [00146] In various aspects the first and/or second container may be a syringe as disclosed herein. In one embodiment, the first and second containers are a first and second syringe, respectively. In one embodiment, the first and second containers are a first and second chamber of a dual-chambered syringe, respectively. Treatment Methods, Uses and Administration [00147] The methods of this disclosure are used in the treatment of diseases or conditions including prostate cancer, including advanced prostate cancer. [00148] Still further, the methods of this disclosure are used in reducing serum testosterone levels in a subject to a level below a castrate level of at least 50 ng/dL, or below 20 ng/dL or below 10 ng/dL. In one aspect, the compositions disclosed herein suppresses the subject’s luteinizing hormone (LH), which in one aspect, is reduced to a level less than about 4 IU/L. [00149] Further, the methods of this disclosure are used in suppressing ovarian function in a subject with hormone-receptor positive breast cancer. In one aspect, the hormone receptor positive breast cancer is pre-menopausal breast cancer. In one aspect, the hormone receptor positive breast cancer is peri-menopausal breast cancer. In one aspect, the hormone receptor positive breast cancer is estrogen receptor (ER) positive breast cancer. In one aspect, the compositions disclosed herein suppress a subject’s estradiol (E2) production to a level less than about 20 pg/mL, less than about 15 pg/mL, less than about 10 pg/mL, less than about 5 pg/mL, less than about 4 pg/ml, less than about 3 pg/mL, or less than about 2 pg/mL. In a preferred aspect, the E2 production level is reduced to about 2.7 pg/mL. In still another aspect, the compositions disclosed herein suppresses the breast cancer subject’s follicle stimulating hormone (FSH) to a level less than about 40 IU/L. In yet another aspect, the compositions disclosed herein suppresses the breast cancer subject’s luteinizing hormone (LH) to a level less than about 4 IU/L. [00150] Still further, the methods of this disclosure are used in treating endometriosis or uterine fibroids. [00151] The methods of this disclosure are used in the treatment of central precocious puberty (CPP). CPP is defined by early sexual development prompted by production and release of gonadotropins and/or sex steroids from normal endogenous sources including the hypothalamus or pituitary. Aberrations in gonadotropin and/or sex hormone concentration levels in children with CPP can result from various sources, including, but not limited to, physical injury, infection, genetic disease, or associated tumors. CPP caused by a genetic or undetermined pathology is classified to be idiopathic in nature, while CPP caused by a central nervous system (CNS) tumor and/or lesion is classified as organic in nature. CPP is accompanied by advanced bone age, accelerated growth velocity, and Hypothalamic- Pituitary-Gonadal-axis activation. In one aspect, the compositions disclosed herein reduce a subject having CPP blood serum LH concentration to a pre-pubertal concentration levels of <4 IU/L. [00152] The methods comprise subcutaneously or intramuscularly administering a disclosed long-acting injectable composition to a subject/patient suffering from prostate cancer, advanced prostate cancer, CPP, pre-menopausal breast cancer, post-menopausal breast cancer, as well as those subjects in need of reducing serum testosterone levels, estradiol levels, or LH levels. Upon injection of the pharmaceutical composition into the body and contact of the composition with an aqueous environment (e.g. a bodily fluid), the solvent dissipates in situ, and a solid, semi-solid, or liquid depot is formed comprising the biodegradable polymer and the degarelix API (self-aggregation or gelation of degarelix or pharmaceutically acceptable salt thereof occurs), together forming a drug reservoir or depot. The resulting depot will release the degarelix or a pharmaceutically acceptable salt thereof, over a desired extended time period. In various embodiments, the degarelix or a pharmaceutically acceptable salt thereof is released into a subject/patient, for example, for a period of at least about 30 days or longer, at least about 60 days or longer, at least about 90 days or longer, at least about 120 days or longer, at least about 150 days or longer, or 180 days or longer. In still other embodiments, the degarelix or a pharmaceutically acceptable salt thereof is released into a subject/patient, for example, for a period of at least about one month or longer, at least about two months or longer, at least about three months or longer, at least about four months or longer, at least about five months or longer, or six months or longer. In still other embodiments, the degarelix or a pharmaceutically acceptable salt thereof is released into a subject/patient for a period of at least about four weeks or longer, at least about eight weeks or longer, at least about twelve weeks or longer, at least about sixteen weeks or longer, at least about 20 weeks or longer, or at least about 24 weeks or longer. [00153] The long-acting composition may be administered to the patient/subject once about every 30 days, once about every 60 days, once about every 90 days, once about every 120 days, once about every 150 days, or once about every 180 days. In another aspect, the long-acting composition may be administered to the patient/subject once about every 1 month, once about every 2 months, once about every 3 months, once about every 4 months, once about every 5 months, or once about every 6 months. In a preferred aspect, the composition is administered once about every 3 months. In another aspect, the long-acting composition may be administered to the patient/subject once about every four weeks, once about every eight weeks, once about every twelve weeks, once about every sixteen weeks, once about every 20 weeks, or once about every 24 weeks. According to the present invention, for clarity, a “month” can have between 28 and 31 days. [00154] The amount of degarelix or a pharmaceutically acceptable salt thereof, that will be effective in the treatment or reduction of the above diseases or conditions, will depend on the nature/severity of the condition or symptom. In vitro or in vivo assays may optionally be employed to help identify optimal dosage ranges. The amount of degarelix or a pharmaceutically acceptable salt thereof, administered will, of course, be dependent on, among other factors, the subject being treated, the age/weight of the subject, the severity of the affliction, the manner of administration and the judgment of the prescribing physician. [00155] The dose amount of degarelix or a pharmaceutically acceptable salt thereof for treating prostate cancer and/or advanced prostate cancer, in the composition is administered in a dose amount of about 20 mg, about 25 mg, about 30 mg, about 35 mg, 40 mg, about 45 mg, about 50 mg, about 55 mg, about 60 mg, about 65 mg, about 70 mg, about 75 mg, about 80 mg, about 85 mg, about 90 mg, about 95 mg, about 100 mg, about 110 mg, about 120 mg, 125 mg, about 130 mg, about 135 mg, about 140 mg, about 145 mg, about 150 mg, about 155 mg, about 160 mg, about 165 mg, about 170 mg, about 175 mg, about 180 mg, about 185 mg, about 190 mg, about 195 mg, about 200 mg, about 210 mg, about 220 mg, about 225, about 230 mg, about 235 mg, about 240 mg, about 245 mg, about 250 mg, about 255 mg, about 260 mg, about 265 mg, about 270 mg, about 275 mg, about 280 mg, about 285 mg, about 290 mg, about 295 mg, about 300 mg, about 310 mg, about 320 mg, about 330 mg, about 340 mg, about 350 mg, about 360 mg, about 370 mg, about 380 mg, about 390 mg, about 400, about 410 mg, about 420 mg, about 430 mg, about 440 mg, about 450 mg, about 460 mg, about 470 mg, about 480 mg, about 490 mg, or about 500 mg. In one aspect, a single dose may be administered in one injection. In still another aspect, a single dose may be administered in more than one injection. [00156] In one aspect, the disclosed compositions are to be administered to a subject/patient once in a dosing period with varying durations between dosing (e.g., one month, 2 months, 3 months, 4 months, 5 months, or 6 months). In one aspect, the disclosed compositions are to be administered to a subject/patient in an initial loading dose followed by one or more maintenance doses of the disclosed compositions. Dosing may be provided alone or in combination with other drugs and may continue as long as required for effective treatment of the disease state or disorder. In some embodiments, the disclosed compositions are formulated to provide doses between about 20 mg – 500 mg. In some embodiments, the disclosed compositions are formulated to provide single doses of between about 40 mg – 500 mg. In some embodiments, the disclosed compositions are formulated to provide single doses of between about 80 mg – 500 mg. In some embodiments, the disclosed compositions are formulated to provide single doses of between about 120 mg – 500 mg. In some embodiments, the disclosed compositions are formulated to provide single doses between about 240 mg – 500 mg. In each of these embodiments, the composition is delivered in a total volume or per injection volume not to exceed 2.5 mL or not to exceed 2 mL. In some embodiments, the injection volume is about 2.5 mL or less, about 2.4 mL or less, about 2.3 mL or less, about 2.2 mL or less, about 2.1 mL or less, about 2.0 mL or less, about 1.9 mL or less, about 1.8 mL or less, about 1.75 mL or less, about 1.7 mL or less, about 1.6 mL or less, 1.5 mL or less, about 1.4 mL or less, about 1.3 mL or less, about 1.2 mL or less, about 1.1 mL or less, about 1 mL or less, about 0.75 mL or less, about 0.5 mL or less, or about 0.375 mL. In some embodiments, the injection volume is about 0.375 ml, about 0.5 ml, about 0.75 ml, about 1 ml, about 1.5 ml, or about 1.75 mL, or about 2 mL, or about 2.2 mL, or about 2.5 mL . In some embodiments, a single dose of the composition is administered to the patient in a single injection (i.e., multiple injections are not required in a single dosing period), where the injection volume is 2.5 mL or less, 2 mL or less, about 1.75 mL or less, 1.5 mL or less, about 1 mL or less, about 0.5 mL or less, or about 0.375 mL. [00157] In some embodiments, the composition is terminally sterilized such as by e-beam or Gamma irradiation or X-ray. In yet another aspect, the composition is sterile filtered. [00158] In some embodiments, the long-acting composition is administered as a monotherapy to patients. The therapeutic methods of this embodiment may reduce or eliminate one or more symptoms of the disease and/or condition disclosed herein. In other embodiments, the long-acting composition may be administered as a combination therapy, such as with chemotherapeutics, radiation therapy, surgery, endocrine therapies such as selective estrogen receptor modulators (SERMs; such as tamoxifen, toremifene, raloxifene, ospemifene, and bazedoxifene), selective estrogen receptor degraders (SERDs; such as fulvestrant), aromatase inhibitors (AIs; such as anastrozole, letrozole, exemestane, vorozole, formestane, and fadrozole); mammalian target of rapamycin (mTOR) inhibitors; such as temsirolimus, sirolimus, everolimus, and ridaforolimus); Phosphatidylinositol 3-kinases inhibitors (PI-3 kinase or PI3K; such as alpelisib, idelalisib, and buparlisib); cyclin- dependent kinases 4 and 6 inhibitors (CDK4/6 inhibitors; such as abemaciclib, palbociclib, and ribociclib); LHRH agonists (such as leuprolide, gonadorelin, goserelin, histrelin, nafarelin, buserelin, and triptorelin and their pharmaceutically acceptable salts thereof), immuno-therapy, and gene therapy. [00159] The disclosed long-acting composition may be provided as a part of a delivery system comprising a syringe system, wherein the composition or formulation is contained within a syringe. In some embodiments, the syringe is a pre-filled syringe system containing the disclosed composition as a single dose or as multiple doses. In one aspect of the pre- filled syringe system, the syringe is a mixing syringe (e.g., a syringe that provides a mechanism for mixing the formulation contained therein, as needed). In one aspect of the pre-filled syringe system, the syringe is a dual-chambered syringe, and the disclosed composition is contained within one chamber of the syringe. In yet another aspect of the pre-filled syringe system, the syringe is a dual-chambered syringe, and the biodegradable polymer and biocompatible solvent are contained in one chamber of the syringe and the degarelix or pharmaceutically acceptable salt thereof is contained in the second chamber of the syringe. The degarelix or a pharmaceutically acceptable salt thereof may be in a lyophilized form or may be dissolved or suspended in a biocompatible solvent, which may be the same as the biocompatible solvent that is used to dissolve the biodegradable polymer, or which may be different than the biocompatible solvent that is used to dissolve the biodegradable polymer. [00160] Another pre-filled syringe delivery system, disclosed herein comprises a first syringe containing the biodegradable polymer and the biocompatible solvent, and a second syringe containing the degarelix or a pharmaceutically acceptable salt thereof. The degarelix or a pharmaceutically acceptable salt thereof may be in a lyophilized form or may be dissolved or suspended in the biocompatible solvent, which may be the same as the biocompatible solvent that is used to dissolve the biodegradable polymer, or which may be different than the biocompatible solvent that is used to dissolve the biodegradable polymer. In one aspect of this system, the first syringe and the second syringe are coupled together to mix the contents of the first and second syringe. The contents of the two syringes are mixed together to form a pharmaceutical composition for injection prior to administering the pharmaceutical composition to the subject. [00161] In some embodiments, the pharmaceutical composition may be administered to a patient by injection using a syringe system, including a syringe system as described herein. In aspects, the composition is injected by subcutaneous injection, although other parenteral routes, including intramuscular injection, are contemplated herein. [00162] The composition may be administered by manual injection though a syringe with, for example, a 18 to 32 gauge needle, a 22 to 25 gauge needle, a 18 to 24 gauge needle, or an 18 to 22 gauge needle, or an 18 to 20 gauge needle, or may be administered by injection using an autoinjector. [00163] Also contemplated herein is a kit comprising a prefilled syringe system disclosed herein. [00164] The following experimental results are provided for purposes of illustration and are not intended to limit the scope of the invention. EXAMPLES [00165] This example describes methods used to prepare and test the extended-release compositions comprising degarelix acetate (DgA) API, or other forms of degarelix where the counter ions are formed with other acids such as poly-carboxylic acids (citric acid), strong acid (methane sulfonic acid), or hydrophobic acid (pamoic acid, palmitic acid). The following examples specifically discuss the preparation and use of the degarelix citrate form of counter ion. [00166] These formulations may include solvent blends, other additives such as: co- solvents or stabilizer excipients such as poly-carboxylic acids or salts (e.g., citric acid), strong acid (methane sulfonic acid), hydrophobic acid or their salts (e.g., pamoic acid, palmitic acid), surfactants such as polysorbate 20, polysorbate 80, poloxamer 188, or any of these as release modifiers. 1. Preliminary screening DgA-Drug Substance (DS) solubility in various water miscible pharmaceutically acceptable organic solvents: [00167] Various organic solvents were screened for the maximal solubility of DgA-DS. Non-aqueous water-miscible pharmaceutically acceptable organic solvents like N-methyl- 2-pyrrolidone (NMP), dimethyl sulfoxide (DMSO), and N, N-dimethylacetamide (DMA) have shown superior solubility capacity (at least 35-40% w/w) of degarelix than aqueous- based solvents. NMP and DMSO can dissolve higher amounts of DgA without inducing self-aggregation, as seen in aqueous solvents. Other solvents tested include– benzyl alcohol, PEG 300, propylene carbonate, triacetin, triethyl citrate, ethyl acetate, and benzyl benzoate. In the majority of these solvents, the drug is either insoluble or has solubility limits much lower than NMP, DMSO or DMA. 2. Degarelix citrate (DgC)-DS preparation [00168] Degarelix citrate was formed by dissolving the degarelix as degarelix acetate (purchased from the vendor) in water (~40 mg/mL concentration) and then adding the aqueous solution of anhydrous citric acid (1M) drop by drop. The molar ratio of degarelix free-base to citric acid in the solution was varied as desired. The resulting solution was mixed for 10-15 mins and placed in the -80 °C freezer. The contents were lyophilized to remove the water (and freeze-drying may remove some amount of acetic acid formed as a result of ion exchange). The dried powder was re-dissolved in water and freeze-dried again. The purity of the lyophilized degarelix citrate powder was tested using HPLC analysis. See descriptions of different degarelix citrate drug substance powders in Table 1.
Table 1 Description of Degarelix Citrate (DgC)-Drug Substance Powders 3. Degarelix mesylate preparation [00169] Degarelix mesylate was formed by dissolving the degarelix as degarelix acetate (purchased from the vendor) in water (~40 mg/mL concentration) and then adding the aqueous solution of strong acid methane sulfonic acid (1M) drop by drop. The molar ratio of degarelix free-base to methane sulfonic acid in the solution was varied as desired. The resulting solution was mixed for 10-15 mins and placed in the -80 °C freezer. The contents were lyophilized to remove the water (and freeze-drying may remove some amount of acetic acid). The dried powder was re-dissolved in water and freeze-dried again. The purity of the lyophilized degarelix mesylate powder was tested using HPLC analysis. 4. Preparation of Polymers [00170] Polymers were either purchased from Sigma Aldrich (RESOMER® polymers) or synthesized in-house. Poly(D.L-lactide-co-glycolide) (PLG) copolymers, poly-DL- lactide (PLA) polymers, or poly(D.L-lactide-co-caprolactone) (PDLCL or PLC) copolymers were produced using the following methods. The amounts of monomers (DL- lactide or glycolide or caprolactone) and initiator (e.g., glycolic acid, 1,6-Hexanediol, polyethylene glycol (PEG)) were selected to obtain a targeted initiator, monomer molar ratio, and weight average molecular weight for each investigated polymer. The monomer molar ratios and weight average molecular weights reported in individual examples are targeted values unless specified as actual or experimental. The polymerization vessel was lowered into a temperature-controlled oil bath. In the vessel, appropriate amounts of monomers (DL-lactide and/or glycolide and/or caprolactone, and initiator (glycolic acid or 1,6-Hexanediol) were added, the vessel contents were placed under a nitrogen atmosphere. The temperature of the vessel was increased until the reagents melted. A catalyst solution was made with appropriate amounts of stannous octoate and toluene and added to the vessel. The vessel was then heated to about 140-170°C under a nitrogen atmosphere for about 4-18 hours (depending on the polymer of interest) with constant stirring. Then, the vessel was evacuated to remove unreacted monomers, and the monomers were vacuum- distilled out of the polymerization mixture. The hot melt was then extruded into cooling pans. After cooling, the solid mass was broken up into smaller pieces. The polymer was purified as needed using the solvent/non-solvent-induced phase separation method. In the notation, PLC-H-P or PLA-H-P or PLG-H-P used here ‘H’ denotes if the polymer is acid terminated and ‘P’ denotes additional polymer purification step by solvent/non-solvent induced phase separation method. 5. Preparation of bulk polymer solutions and prefilled syringes (Syringe A) of polymer solution [00171] Polymer bulk solutions were prepared by weighing a known amount of each polymer material into individual Flack-Tek jars of the desired size. A known amount of NMP was added to each polymer, blanketed with nitrogen and the containers were placed on a horizontal jar mill or Turbula for mixing at room temperature. Jars were mixed until a visually clear homogenous polymer solution is obtained indicating the complete dissolution of the polymer in the solvent. Prefilled syringes (Syringe A) containing polymer delivery system were prepared by weighing the required amounts of polymer solution into 1.2 mL female polypropylene syringe barrel with Luer lock and plunger tip and capped with male polypropylene syringe cap. Filled syringes were then packaged in labeled foil pouches with a desiccant pack and the pouches were sealed. After filling the syringes with the formulations, the filled syringes were stored under refrigerated conditions (e.g., 2-8 ºC) or accelerated conditions (≥25°C see individual experiments). The syringes may be irradiated via e-beam irradiation (see individual experiment). 6. Preparation of DgA-DS or DgC-DS/organic-solvent bulk solution and prefilled syringes [00172] To produce the drug/solvent bulk solution comprising the API (DgA or DgC), the desired amount of the DgA-DS or DgC-DS was combined with the solvent NMP or DMSO or combination of solvents with or without a co-solvent or other additives, in the indicated amounts (see individual experiments below). The API and solvent were combined in a glass vial or jar and blanketed with nitrogen. Jars were mixed using the jar mill, Turbula, or shaker at room temperature until homogeneous. DgA or DgC are very soluble in NMP or DMSO and can load higher concentrations (at least 40% w/w). After dissolving the API in the organic solvent, the bulk solution was manually filled into syringes and capped with a tip cap. The syringes used for filling were either 1.2 mL male polypropylene syringes or 1 mL male cyclic olefin copolymer syringes. Filled syringes were then packaged in labeled foil pouches with a desiccant pack and the pouches were sealed. After filling the syringes with the formulations, the filled syringes were stored under refrigerated conditions (e.g., 2- 8 ºC) or accelerated conditions (≥25°C see individual experiments). The syringes may be irradiated via e-beam or gamma irradiation (see individual experiment) for terminal sterilization. 7. Preparation of Reconstituted Formulation for testing: [00173] Immediately before testing, “A” and “B” syringes were coupled and mixed by cycling the contents from one syringe to the other for 30-45 cycles. The mixed formulation was finally transferred to the male dosing syringe for delivery and testing. A safety needle of 20 g was used for delivering the formulation when needed. 8. Preparation methods for Single-Syringe Formulation [00174] Selected degarelix (acetate or citrate form) was dissolved in organic solvent (Ex: NMP or DMSO) at desired amounts and then mixed with the desired amount of liquid polymer solution (see section 5 for bulk polymer solution preparation) in a scintillation vial, and the vial was put on a horizontal roller mixer to roll for 24-48 h for mixing. After mixing, the single-syringe formulations (solution/suspension) were considered ready for testing. [00175] As an alternative to the above method of preparation, selected degarelix (acetate or citrate form) and liquid polymer solution (see section 7 for bulk polymer solution preparation) were weighed into a scintillation vial, and the vial was put on a horizontal roller mixer to roll for 24-48 h for mixing. After mixing, the single-syringe formulations (solution/suspension) were considered ready for testing. 9. Preparation of Single-Syringe Formulation with added stabilizers [00176] The stabilizer excipient can be selected from a list of acids which include polycarboxylic acids or their salts (Ex. citric acid), strong acids (Ex. methanesulfonic), hydrophobic acids or their salts (Ex. pamoic acid, palmitic acid, lauric acid, sodium dodecyl sulfate). Degarelix acetate powder, a stabilizer excipient(s) (anhydrous citric acid), and organic solvent (e.g., NMP or DMSO) were weighed into a scintillation vial, and the vial was put on a horizontal roller mixer to roll for 24-48 h for mixing. After mixing, desired amounts of polymer bulk solution (see section 5) were added to the drug solution vial, and the vial was put on a horizontal roller mixer and continued to mix for 24-48 h. After mixing, the single-syringe formulations were considered ready for testing. [00177] As an alternative preparation method to the above, desired amounts of DgA-DS, a stabilizer excipient(s) (e.g., anhydrous citric acid), and liquid polymer solution (see section 5 for bulk polymer solution preparation) were weighed into a scintillation vial, and the vial was put on a horizontal roller mixer to roll for 24-48 h for mixing. After mixing, the single- syringe formulations (solution/suspension) were considered ready for testing. 10. XRD Characterization of the DgA-DS solutions [00178] Samples were prepared and analysis was performed at ambient temperature using the Rigaku MiniFlex XRD instrument according to standard protocols. After a sequence of samples was run, the data was processed using PDXL2 software. Degarelix acetate did not show any crystallinity as an API powder, or at a concentration of 66 mg/mL in water. At higher concentrations, the peptide forms a gel. The gel was not crystalline. In theory, the gel exhibits peaks indicating a higher order structure attributed to the stacking of beta-sheets forming aggregates resulting in the gel formation. The addition of salts can aid the gelation to form at a lower concentration than without the salts. No gelation occurred in the presence of NMP solvent even when water was present. 11. Analysis of degarelix (assay) and Total related compounds in the formulations [00179] Degarelix/organic solvent solutions were prepared for HPLC analysis by dilution to volume with mobile phase A (0.1% trifluoroacetic acid water) and mixing thoroughly by vortex. Dilutions were performed as needed using volumetric glassware and mixed thoroughly by vortex mixing. A second dilution of 2 mL to 20 mL was performed with mobile phase A to obtain a working sample. [00180] For the HPLC test sample preparation of reconstituted/single-syringe formulations that contain polymer, syringe A and syringe B were coupled together and mixed for 45 cycles. The reconstituted product was dispensed from the final mixed single- syringe formulation content into a 50 mL volumetric flask and the weight was recorded. 2.0 mL of mobile phase B (0.1% trifluoroacetic acid acetonitrile) was added and mixed by swirling. 3.0 mL of mobile phase A was added and mixed by swirling. Samples were placed in a water bath at 55 ºC ± 2 ºC for 30 minutes and then cooled to room temperature. Samples were diluted to volume with mobile phase A and mixed thoroughly by vortex, followed by dilution to 20 mL with mobile phase A with thorough mixing. A second dilution of 2 mL to 20 mL was performed with mobile phase A to obtain a working sample. The sample solutions were filtered through a 0.45 um PTFE syringe filter into amber HPLC vials before collection into the HPLC vial. [00181] HPLC analysis for assay and related compounds was performed using an Agilent AdvanceBio Peptide 3.0 x 100 mm, 2.7 um column and an Agilent AdvanceBio Peptide Map Guard 3.0 x 5mm 2.7 um guard column at 30°C with a flow rate of 0.75 mL/min. The runtime was 15 minutes with a 10-µL injection for assay and related compounds for the solution gel depot formulation, and a 20-µL injection for assay and related compounds for the polymer depot formulation. The mobile phases were 0.1% trifluoroacetic acid water for mobile phase A and 0.1% trifluoroacetic acid acetonitrile for mobile phase B. Detection was performed with a diode array detector set at 220 nm. The method used a gradient. See Table 2 below for the gradient method for an example chromatogram.
3. Degarelix mesylate preparation [00169] Degarelix mesylate was formed by dissolving the degarelix as degarelix acetate (purchased from the vendor) in water (~40 mg/mL concentration) and then adding the aqueous solution of strong acid methane sulfonic acid (1M) drop by drop. The molar ratio of degarelix free-base to methane sulfonic acid in the solution was varied as desired. The resulting solution was mixed for 10-15 mins and placed in the -80 °C freezer. The contents were lyophilized to remove the water (and freeze-drying may remove some amount of acetic acid). The dried powder was re-dissolved in water and freeze-dried again. The purity of the lyophilized degarelix mesylate powder was tested using HPLC analysis. 4. Preparation of Polymers [00170] Polymers were either purchased from Sigma Aldrich (RESOMER® polymers) or synthesized in-house. Poly(D.L-lactide-co-glycolide) (PLG) copolymers, poly-DL- lactide (PLA) polymers, or poly(D.L-lactide-co-caprolactone) (PDLCL or PLC) copolymers were produced using the following methods. The amounts of monomers (DL- lactide or glycolide or caprolactone) and initiator (e.g., glycolic acid, 1,6-Hexanediol, polyethylene glycol (PEG)) were selected to obtain a targeted initiator, monomer molar ratio, and weight average molecular weight for each investigated polymer. The monomer molar ratios and weight average molecular weights reported in individual examples are targeted values unless specified as actual or experimental. The polymerization vessel was lowered into a temperature-controlled oil bath. In the vessel, appropriate amounts of monomers (DL-lactide and/or glycolide and/or caprolactone, and initiator (glycolic acid or 1,6-Hexanediol) were added, the vessel contents were placed under a nitrogen atmosphere. The temperature of the vessel was increased until the reagents melted. A catalyst solution was made with appropriate amounts of stannous octoate and toluene and added to the vessel. The vessel was then heated to about 140-170°C under a nitrogen atmosphere for about 4-18 hours (depending on the polymer of interest) with constant stirring. Then, the vessel was evacuated to remove unreacted monomers, and the monomers were vacuum- distilled out of the polymerization mixture. The hot melt was then extruded into cooling pans. After cooling, the solid mass was broken up into smaller pieces. The polymer was purified as needed using the solvent/non-solvent-induced phase separation method. In the notation, PLC-H-P or PLA-H-P or PLG-H-P used here ‘H’ denotes if the polymer is acid terminated and ‘P’ denotes additional polymer purification step by solvent/non-solvent induced phase separation method. 5. Preparation of bulk polymer solutions and prefilled syringes (Syringe A) of polymer solution [00171] Polymer bulk solutions were prepared by weighing a known amount of each polymer material into individual Flack-Tek jars of the desired size. A known amount of NMP was added to each polymer, blanketed with nitrogen and the containers were placed on a horizontal jar mill or Turbula for mixing at room temperature. Jars were mixed until a visually clear homogenous polymer solution is obtained indicating the complete dissolution of the polymer in the solvent. Prefilled syringes (Syringe A) containing polymer delivery system were prepared by weighing the required amounts of polymer solution into 1.2 mL female polypropylene syringe barrel with Luer lock and plunger tip and capped with male polypropylene syringe cap. Filled syringes were then packaged in labeled foil pouches with a desiccant pack and the pouches were sealed. After filling the syringes with the formulations, the filled syringes were stored under refrigerated conditions (e.g., 2-8 ºC) or accelerated conditions (≥25°C see individual experiments). The syringes may be irradiated via e-beam irradiation (see individual experiment). 6. Preparation of DgA-DS or DgC-DS/organic-solvent bulk solution and prefilled syringes [00172] To produce the drug/solvent bulk solution comprising the API (DgA or DgC), the desired amount of the DgA-DS or DgC-DS was combined with the solvent NMP or DMSO or combination of solvents with or without a co-solvent or other additives, in the indicated amounts (see individual experiments below). The API and solvent were combined in a glass vial or jar and blanketed with nitrogen. Jars were mixed using the jar mill, Turbula, or shaker at room temperature until homogeneous. DgA or DgC are very soluble in NMP or DMSO and can load higher concentrations (at least 40% w/w). After dissolving the API in the organic solvent, the bulk solution was manually filled into syringes and capped with a tip cap. The syringes used for filling were either 1.2 mL male polypropylene syringes or 1 mL male cyclic olefin copolymer syringes. Filled syringes were then packaged in labeled foil pouches with a desiccant pack and the pouches were sealed. After filling the syringes with the formulations, the filled syringes were stored under refrigerated conditions (e.g., 2- 8 ºC) or accelerated conditions (≥25°C see individual experiments). The syringes may be irradiated via e-beam or gamma irradiation (see individual experiment) for terminal sterilization. 7. Preparation of Reconstituted Formulation for testing: [00173] Immediately before testing, “A” and “B” syringes were coupled and mixed by cycling the contents from one syringe to the other for 30-45 cycles. The mixed formulation was finally transferred to the male dosing syringe for delivery and testing. A safety needle of 20 g was used for delivering the formulation when needed. 8. Preparation methods for Single-Syringe Formulation [00174] Selected degarelix (acetate or citrate form) was dissolved in organic solvent (Ex: NMP or DMSO) at desired amounts and then mixed with the desired amount of liquid polymer solution (see section 5 for bulk polymer solution preparation) in a scintillation vial, and the vial was put on a horizontal roller mixer to roll for 24-48 h for mixing. After mixing, the single-syringe formulations (solution/suspension) were considered ready for testing. [00175] As an alternative to the above method of preparation, selected degarelix (acetate or citrate form) and liquid polymer solution (see section 7 for bulk polymer solution preparation) were weighed into a scintillation vial, and the vial was put on a horizontal roller mixer to roll for 24-48 h for mixing. After mixing, the single-syringe formulations (solution/suspension) were considered ready for testing. 9. Preparation of Single-Syringe Formulation with added stabilizers [00176] The stabilizer excipient can be selected from a list of acids which include polycarboxylic acids or their salts (Ex. citric acid), strong acids (Ex. methanesulfonic), hydrophobic acids or their salts (Ex. pamoic acid, palmitic acid, lauric acid, sodium dodecyl sulfate). Degarelix acetate powder, a stabilizer excipient(s) (anhydrous citric acid), and organic solvent (e.g., NMP or DMSO) were weighed into a scintillation vial, and the vial was put on a horizontal roller mixer to roll for 24-48 h for mixing. After mixing, desired amounts of polymer bulk solution (see section 5) were added to the drug solution vial, and the vial was put on a horizontal roller mixer and continued to mix for 24-48 h. After mixing, the single-syringe formulations were considered ready for testing. [00177] As an alternative preparation method to the above, desired amounts of DgA-DS, a stabilizer excipient(s) (e.g., anhydrous citric acid), and liquid polymer solution (see section 5 for bulk polymer solution preparation) were weighed into a scintillation vial, and the vial was put on a horizontal roller mixer to roll for 24-48 h for mixing. After mixing, the single- syringe formulations (solution/suspension) were considered ready for testing. 10. XRD Characterization of the DgA-DS solutions [00178] Samples were prepared and analysis was performed at ambient temperature using the Rigaku MiniFlex XRD instrument according to standard protocols. After a sequence of samples was run, the data was processed using PDXL2 software. Degarelix acetate did not show any crystallinity as an API powder, or at a concentration of 66 mg/mL in water. At higher concentrations, the peptide forms a gel. The gel was not crystalline. In theory, the gel exhibits peaks indicating a higher order structure attributed to the stacking of beta-sheets forming aggregates resulting in the gel formation. The addition of salts can aid the gelation to form at a lower concentration than without the salts. No gelation occurred in the presence of NMP solvent even when water was present. 11. Analysis of degarelix (assay) and Total related compounds in the formulations [00179] Degarelix/organic solvent solutions were prepared for HPLC analysis by dilution to volume with mobile phase A (0.1% trifluoroacetic acid water) and mixing thoroughly by vortex. Dilutions were performed as needed using volumetric glassware and mixed thoroughly by vortex mixing. A second dilution of 2 mL to 20 mL was performed with mobile phase A to obtain a working sample. [00180] For the HPLC test sample preparation of reconstituted/single-syringe formulations that contain polymer, syringe A and syringe B were coupled together and mixed for 45 cycles. The reconstituted product was dispensed from the final mixed single- syringe formulation content into a 50 mL volumetric flask and the weight was recorded. 2.0 mL of mobile phase B (0.1% trifluoroacetic acid acetonitrile) was added and mixed by swirling. 3.0 mL of mobile phase A was added and mixed by swirling. Samples were placed in a water bath at 55 ºC ± 2 ºC for 30 minutes and then cooled to room temperature. Samples were diluted to volume with mobile phase A and mixed thoroughly by vortex, followed by dilution to 20 mL with mobile phase A with thorough mixing. A second dilution of 2 mL to 20 mL was performed with mobile phase A to obtain a working sample. The sample solutions were filtered through a 0.45 um PTFE syringe filter into amber HPLC vials before collection into the HPLC vial. [00181] HPLC analysis for assay and related compounds was performed using an Agilent AdvanceBio Peptide 3.0 x 100 mm, 2.7 um column and an Agilent AdvanceBio Peptide Map Guard 3.0 x 5mm 2.7 um guard column at 30°C with a flow rate of 0.75 mL/min. The runtime was 15 minutes with a 10-µL injection for assay and related compounds for the solution gel depot formulation, and a 20-µL injection for assay and related compounds for the polymer depot formulation. The mobile phases were 0.1% trifluoroacetic acid water for mobile phase A and 0.1% trifluoroacetic acid acetonitrile for mobile phase B. Detection was performed with a diode array detector set at 220 nm. The method used a gradient. See Table 2 below for the gradient method for an example chromatogram. 12. Recovery of drug from different Degarelix/Organic solvent formulations [00182] Prefilled syringes of degarelix/organic solvent formulations were prepared as described in section 6. Recovery of drugs from different formulations was tested using the HPLC method (see section 11) and is listed in Table 3.
12. Recovery of drug from different Degarelix/Organic solvent formulations [00182] Prefilled syringes of degarelix/organic solvent formulations were prepared as described in section 6. Recovery of drugs from different formulations was tested using the HPLC method (see section 11) and is listed in Table 3.


Table 3 Drug recovery testing from prefilled syringes of organic-solvent based formulations (“F#” denotes “Formulation Number”) [00183] Table 3 Observations: All the formulations showed good recoveries (90.0- 110.0%), indicating the applicability of the method and the initial stability of the API in different solvent systems. 13. Stability of degarelix in organic solvents (Syringe B) [00184] Prefilled syringes of DgA-DS/NMP or DgA-DS/DMSO solutions were prepared as described in section 6. The samples were not terminally sterilized. The composition of the stability samples is shown in Table 4. The sealed foil pouches containing prefilled syringes were paced at accelerated storage conditions (25±2°C) for 6 months and tested for drug assay or recovery and related compounds (see section 11 for procedure) at each time pull (0, 1, 3 and 6 months). The results of this study are presented in Figure 1. No degradation of the drug was observed at 6 months, and a slight increase in percentage recoveries over time could be attributed to the solvent loss/absorption in prefilled syringes at accelerated conditions. Table 4 Compositions Of Formulations
(“F#” denotes “Formulation Number”) [00183] Table 3 Observations: All the formulations showed good recoveries (90.0- 110.0%), indicating the applicability of the method and the initial stability of the API in different solvent systems. 13. Stability of degarelix in organic solvents (Syringe B) [00184] Prefilled syringes of DgA-DS/NMP or DgA-DS/DMSO solutions were prepared as described in section 6. The samples were not terminally sterilized. The composition of the stability samples is shown in Table 4. The sealed foil pouches containing prefilled syringes were paced at accelerated storage conditions (25±2°C) for 6 months and tested for drug assay or recovery and related compounds (see section 11 for procedure) at each time pull (0, 1, 3 and 6 months). The results of this study are presented in Figure 1. No degradation of the drug was observed at 6 months, and a slight increase in percentage recoveries over time could be attributed to the solvent loss/absorption in prefilled syringes at accelerated conditions. Table 4 Compositions Of Formulations [00185] As shown in Figure 1 degarelix acetate was found to be stable in NMP and DMSO (at studied concentrations) at 25°C for 6 months (accelerated conditions). There was no significant increase in the related compounds. At 6 months, the total related compounds were below 2%. The observed slight increase in percentage recoveries over time for the F1, F2, and F5 formulations on storage could be attributed to the solvent loss over time at accelerated storage conditions from the prefilled syringes or stopper absorption. No gelation was observed in the formulations F1, F2, and F5. F5 formulation was made at a higher concentration than F2, and both were found to be stable and did not show any gelation. This supports that higher concentration, such as 40% w/w, is also feasible. 14. Terminal sterilization feasibility of degarelix solutions via e-beam or Gamma irradiation. [00186] The prefilled syringes of degarelix/organic solvent were exposed to e-beam or gamma radiation at different doses and tested the drug concentrations or recoveries for pre and post-irradiation samples using HPLC (see section 11) and the results are presented in Table 5.
[00185] As shown in Figure 1 degarelix acetate was found to be stable in NMP and DMSO (at studied concentrations) at 25°C for 6 months (accelerated conditions). There was no significant increase in the related compounds. At 6 months, the total related compounds were below 2%. The observed slight increase in percentage recoveries over time for the F1, F2, and F5 formulations on storage could be attributed to the solvent loss over time at accelerated storage conditions from the prefilled syringes or stopper absorption. No gelation was observed in the formulations F1, F2, and F5. F5 formulation was made at a higher concentration than F2, and both were found to be stable and did not show any gelation. This supports that higher concentration, such as 40% w/w, is also feasible. 14. Terminal sterilization feasibility of degarelix solutions via e-beam or Gamma irradiation. [00186] The prefilled syringes of degarelix/organic solvent were exposed to e-beam or gamma radiation at different doses and tested the drug concentrations or recoveries for pre and post-irradiation samples using HPLC (see section 11) and the results are presented in Table 5.


Table 5 Results Of Irradiation Feasibility Studies (“F#” denotes “Formulation Number”) [00187] Table 5 Observations: These results showed that depending on the irradiation type, irradiation dose, concentration of the drug, salt form, and stabilizer, the degarelix solutions in an organic solvent are feasible for terminal sterilization by irradiation. [00188] F4-e25-2 formulation of degarelix citrate showed better stability upon e-beam irradiation compared to degarelix acetate formulation F1-e25-2. [00189] In general, 35% w/w drug solution samples (F1-e34, F2-e34, F1-g22) showed better stability upon irradiation compared to 40% w/w drug solution formulations (F6-e34, 5-e34, F6-g22). [00190] Formulations F1-e25, F2-e25, F3-e25, and F4-e25 were tested for total related compounds, no significant increase was observed post-irradiation of the samples via e- beam. 15. Stability of Irradiated samples in accelerated storage conditions [00191] Irradiated prefilled syringes of degarelix/organic solvent formulations were stored at different accelerated storage conditions (25°C) and tested for assay at 30 and 120 days using the HPLC method (see section 11). Results are presented in Table 6. Table 6 Accelerated Storage Stability Testing Of Irradiated Prefilled Syringes Of Degarelix Solution Formulations
(“F#” denotes “Formulation Number”) [00187] Table 5 Observations: These results showed that depending on the irradiation type, irradiation dose, concentration of the drug, salt form, and stabilizer, the degarelix solutions in an organic solvent are feasible for terminal sterilization by irradiation. [00188] F4-e25-2 formulation of degarelix citrate showed better stability upon e-beam irradiation compared to degarelix acetate formulation F1-e25-2. [00189] In general, 35% w/w drug solution samples (F1-e34, F2-e34, F1-g22) showed better stability upon irradiation compared to 40% w/w drug solution formulations (F6-e34, 5-e34, F6-g22). [00190] Formulations F1-e25, F2-e25, F3-e25, and F4-e25 were tested for total related compounds, no significant increase was observed post-irradiation of the samples via e- beam. 15. Stability of Irradiated samples in accelerated storage conditions [00191] Irradiated prefilled syringes of degarelix/organic solvent formulations were stored at different accelerated storage conditions (25°C) and tested for assay at 30 and 120 days using the HPLC method (see section 11). Results are presented in Table 6. Table 6 Accelerated Storage Stability Testing Of Irradiated Prefilled Syringes Of Degarelix Solution Formulations [00192] Table 6 Observations: No pronounced degradation of the degarelix was observed in the irradiated samples over the studied period and a slight increase in percentage recoveries over time could be attributed to the solvent loss or absorption in prefilled syringes at accelerated storage conditions. 16. Compatibility of degarelix acetate with a Polymer solution or monomer solution [00193] A known amount of DgA-DS was combined with known amounts of selected polymer solutions or monomer solution and incubated at 50 °C and/or at room temperature, and tested for drug recovery at selected time points. See Table 7 and Table 8 for details. Table 7 Degarelix Acetate Polymer Or Monomer Solution Compatibility Testing
[00192] Table 6 Observations: No pronounced degradation of the degarelix was observed in the irradiated samples over the studied period and a slight increase in percentage recoveries over time could be attributed to the solvent loss or absorption in prefilled syringes at accelerated storage conditions. 16. Compatibility of degarelix acetate with a Polymer solution or monomer solution [00193] A known amount of DgA-DS was combined with known amounts of selected polymer solutions or monomer solution and incubated at 50 °C and/or at room temperature, and tested for drug recovery at selected time points. See Table 7 and Table 8 for details. Table 7 Degarelix Acetate Polymer Or Monomer Solution Compatibility Testing (“F#” denotes “Formulation Number”). NA indicates: Not Available Table 8 Degarelix Acetate Polymer Or Monomer Solution Compatibility Testing
(“F#” denotes “Formulation Number”). NA indicates: Not Available Table 8 Degarelix Acetate Polymer Or Monomer Solution Compatibility Testing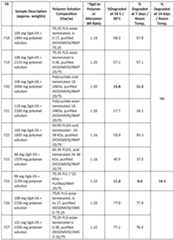
 (“F#” denotes “Formulation Number”) NA indicates: Not Available [00194] Table 7 Observations: F6 sample was a control sample that does not contain any polymer or monomers and was included in the study as a control to understand if the solvent causes any degradation. The F6 sample showed minimal signs of degradation (1% drop in assay after 24 h at 50°C), indicating that the drug is stable in NMP, and any degradation observed in other samples could be attributed to the presence of polymers/monomers. Bold text in Table 7 for Samples F8, F10 and F15 indicates that these samples exhibited the most promising drug stability (e.g., less degradation of drug over the tested time period). [00195] F16 and F17 are samples where DgA-DS was mixed with monomer solutions (Lactide/NMP or Glycolide/NMP), and it was observed that almost 90-100% of the drug was degraded by 7 days at room temperature, and 86-99% in 24 h at 50°C. These results indicated that the presence of monomers could induce significant degradation of the degarelix. These results indicate that purification of polymers to remove the residual or unreacted monomers will improve the stability of the API. [00196] Among F8-F14 samples that contain unpurified polymers, glycolic acid (GA) terminated PLC containing samples F8 and F10 have shown better compatibility with the drug compared to the PLG or PLC-PEG polymers. F8 and F10 samples showed ≤5% (after 24 h at 50°C) and ≤1.2% (after 7 days at room temperature) degradation, whereas other samples showed significantly higher degradation of the degarelix. [00197] F15 sample that contains purified acid-terminated PLG polymer also showed promise, 7.2% (after 24 h at 50°C) and 4.5% (after 7 days at room temperature) degradation. Although there are other samples such as F9, F11, and F14 that contain similar polymers (acid-terminated PLGs) with a different monomer composition, the higher stability of degarelix in F15 could be related to the use of purified polymer in F15 compared to the unpurified versions in F9, F11, and F14. As discussed earlier, the presence of residual monomers could be detrimental to the API stability, so purification of the polymer helped in improving the stability. [00198] Acid-terminated PLC polymer showed improved stability of degarelix compared to other polymers, even without purification. Based on these results, purified acid- terminated PLC polymer showed promising potential for use in formulating a single syringe formulation. [00199] Table 8 Observations: Bold text in Table 8 for Samples F20, F24, F28 and F32 indicates that these samples exhibited the most promising drug stability (e.g., less degradation of drug over the tested time period). Based on the results from Table 8, if the stability trends of degarelix in polymer solutions are to be ranked with respect to the polymer composition, the rank was PLC>PLA>PLG, and acid-terminated>ester-terminated polymer. These results confirmed earlier observations, that purified acid-terminated PLC polymers are more compatible with degarelix and showed promising potential as a polymer of choice for a single syringe formulation. In addition to purified acid-terminated PLC, purified acid- terminated PLA polymer also showed promising stability for a single syringe formulation. [00200] Degradation of DgA-DS was observed in presence of polymer solution. Therefore, additional studies were conducted where modified degarelix (DgC-DS instead of DgA-DS) or the use of stabilizer excipient were evaluated to further improve the stability of the degarelix with a goal of making a stable single syringe formulation. 17. LC-MS analysis of the single syringe formulations [00201] The working samples in the volumetric flask were obtained from HPLC sample preparation followed by filtration through a 0.45 µm syringe filter into an amber HPLC vial. The liquid chromatography analysis was performed using Agilent AdvanceBio Peptide 3.0 x 100 mm, 2.7 um column and an Agilent AdvanceBio Peptide Map Guard 3.0 x 5mm 2.7 um guard column at 30 °C. The flow rate was set at 0.35mL/min with 20µL injection and the gradient is listed below in Table 9. Mobile phase A was 0.1% trifluoroacetic acid water and mobile phase B was 0.1% trifluoroacetic acid acetonitrile. Both UV (at 220nm) and mass spec signals are collected. Table 9 Gradient Method For LC Run
(“F#” denotes “Formulation Number”) NA indicates: Not Available [00194] Table 7 Observations: F6 sample was a control sample that does not contain any polymer or monomers and was included in the study as a control to understand if the solvent causes any degradation. The F6 sample showed minimal signs of degradation (1% drop in assay after 24 h at 50°C), indicating that the drug is stable in NMP, and any degradation observed in other samples could be attributed to the presence of polymers/monomers. Bold text in Table 7 for Samples F8, F10 and F15 indicates that these samples exhibited the most promising drug stability (e.g., less degradation of drug over the tested time period). [00195] F16 and F17 are samples where DgA-DS was mixed with monomer solutions (Lactide/NMP or Glycolide/NMP), and it was observed that almost 90-100% of the drug was degraded by 7 days at room temperature, and 86-99% in 24 h at 50°C. These results indicated that the presence of monomers could induce significant degradation of the degarelix. These results indicate that purification of polymers to remove the residual or unreacted monomers will improve the stability of the API. [00196] Among F8-F14 samples that contain unpurified polymers, glycolic acid (GA) terminated PLC containing samples F8 and F10 have shown better compatibility with the drug compared to the PLG or PLC-PEG polymers. F8 and F10 samples showed ≤5% (after 24 h at 50°C) and ≤1.2% (after 7 days at room temperature) degradation, whereas other samples showed significantly higher degradation of the degarelix. [00197] F15 sample that contains purified acid-terminated PLG polymer also showed promise, 7.2% (after 24 h at 50°C) and 4.5% (after 7 days at room temperature) degradation. Although there are other samples such as F9, F11, and F14 that contain similar polymers (acid-terminated PLGs) with a different monomer composition, the higher stability of degarelix in F15 could be related to the use of purified polymer in F15 compared to the unpurified versions in F9, F11, and F14. As discussed earlier, the presence of residual monomers could be detrimental to the API stability, so purification of the polymer helped in improving the stability. [00198] Acid-terminated PLC polymer showed improved stability of degarelix compared to other polymers, even without purification. Based on these results, purified acid- terminated PLC polymer showed promising potential for use in formulating a single syringe formulation. [00199] Table 8 Observations: Bold text in Table 8 for Samples F20, F24, F28 and F32 indicates that these samples exhibited the most promising drug stability (e.g., less degradation of drug over the tested time period). Based on the results from Table 8, if the stability trends of degarelix in polymer solutions are to be ranked with respect to the polymer composition, the rank was PLC>PLA>PLG, and acid-terminated>ester-terminated polymer. These results confirmed earlier observations, that purified acid-terminated PLC polymers are more compatible with degarelix and showed promising potential as a polymer of choice for a single syringe formulation. In addition to purified acid-terminated PLC, purified acid- terminated PLA polymer also showed promising stability for a single syringe formulation. [00200] Degradation of DgA-DS was observed in presence of polymer solution. Therefore, additional studies were conducted where modified degarelix (DgC-DS instead of DgA-DS) or the use of stabilizer excipient were evaluated to further improve the stability of the degarelix with a goal of making a stable single syringe formulation. 17. LC-MS analysis of the single syringe formulations [00201] The working samples in the volumetric flask were obtained from HPLC sample preparation followed by filtration through a 0.45 µm syringe filter into an amber HPLC vial. The liquid chromatography analysis was performed using Agilent AdvanceBio Peptide 3.0 x 100 mm, 2.7 um column and an Agilent AdvanceBio Peptide Map Guard 3.0 x 5mm 2.7 um guard column at 30 °C. The flow rate was set at 0.35mL/min with 20µL injection and the gradient is listed below in Table 9. Mobile phase A was 0.1% trifluoroacetic acid water and mobile phase B was 0.1% trifluoroacetic acid acetonitrile. Both UV (at 220nm) and mass spec signals are collected. Table 9 Gradient Method For LC Run [00202] The mass spec signals were collected using ESI resource at positive mode. The parameter settings of the ESI source are listed below in Table 10. Table 10 Esi Source Setting Of Mass Spectrometer
[00202] The mass spec signals were collected using ESI resource at positive mode. The parameter settings of the ESI source are listed below in Table 10. Table 10 Esi Source Setting Of Mass Spectrometer 18. Improved stability of degarelix using DgC-DS or Citric acid stabilizer excipients [00203] Single-syringe formulations of DgA-DS and DgC-DS (different moles, see section 2 Table 1) were prepared as per the procedure in section 8. Single-syringe formulations of DgA-DS with added citric acid excipient were prepared as per the procedure in section 9. Single-syringe formulations of DgA-DS with and without stabilizer (different moles of anhydrous citric acid), and DgC-DS were tested for stability under accelerated storage conditions at 50°C using LC-MS. The m/z values of the molecular ion for the main and related compound peaks are summarized in Table 11. Table 11 Degradation And Impurities Of Single Syringe Formulations
18. Improved stability of degarelix using DgC-DS or Citric acid stabilizer excipients [00203] Single-syringe formulations of DgA-DS and DgC-DS (different moles, see section 2 Table 1) were prepared as per the procedure in section 8. Single-syringe formulations of DgA-DS with added citric acid excipient were prepared as per the procedure in section 9. Single-syringe formulations of DgA-DS with and without stabilizer (different moles of anhydrous citric acid), and DgC-DS were tested for stability under accelerated storage conditions at 50°C using LC-MS. The m/z values of the molecular ion for the main and related compound peaks are summarized in Table 11. Table 11 Degradation And Impurities Of Single Syringe Formulations (“F#” denotes “Formulation Number”) [00204] Table 11 Observations: This accelerated stability at 50°C evaluated the effect of i) citric acid excipient, and ii) degarelix citrate form over degarelix acetate form on the stability of degarelix in the polymer-based single-syringe formulations. [00205] If the total impurities (area sum%) of F37 vs. (F34, F35, F36, F45, F46, F47, F48), and F41 vs. (F38, F39, F40) is compared, DgA-DS containing formulations without citric acid F37 (15.0% impurities) and F41(15.2% impurities) generated significantly higher amount of impurities after only 14 days compared to the formulations that contained DgC- DS (<5% impurities after 21 days) or citric acid as an excipient (<8% after 18 days). Lower levels of acylated impurities were observed for citric acid-based formulations than formulations that do not contain any citric acid. [00206] Addition of citric acid as a stabilizer excipient to the drug-polymer formulation or using it as part of the alternate salt form that is prepared by co-lyophilization of DgA-DS with citric acid, both resulted in a similar improvement of drug stability on accelerated storage, and the lower impurity levels were observed. Further, this preliminary data suggested that stabilizers such as citric acid can be just added as an excipient to the formulation to improve the drug stability in presence of polymer, and the additional process of lyophilization to make DgC-DS form may be eliminated to simplify the manufacturing process. [00207] Citric acid was added as an excipient to the formulation at four different molar levels [0.5 (F48), 1 (F45), 2 (F46), and 3 (F47) moles to 1 mole of degarelix). [00208] F48 with 0.5 mol citric acid showed 7.8 % total impurities at day 18 vs. F37 with no citric acid showed 15.0% total impurities at day 14. F45 with 1 mol of citric acid showed 3.6% total impurities at day 18. Although 0.5 moles of citric acid helped in improving the stability of DgA-DS compared to no additive version, higher levels of citric acid showed much better improvement in drug stability. This set of results indicates that higher than 0.5 moles of citric acid to degarelix is needed to achieve better drug stability. The addition of 1:1 or molar excess of citric acid to the degarelix showed promising results. 19. Animal dosing [00209] Prefilled syringes of liquid polymer solutions (Syringe A) and degarelix/organic solvent solutions (Syringe B) were prepared as described in section 5 and section 6. The samples were not e-beamed. Degarelix release rates of these formulations were obtained using a rat model. Male rats were each injected with a single subcutaneous injection of the reconstituted formulations obtained by mixing Syringe A and Syringe B versions of different formulations. The composition of the formulations and the dosing details are listed in Table 12. [00210] At predetermined time points, rats were bled and plasma degarelix levels were determined using liquid chromatography with tandem mass spectrometry (LC-MS/MS). Each data point is based on an average plasma degarelix concentration. Six rats were dosed for each group, with sparse blood sampling performed for early time points. FIRMAGON® (degarelix for injection) was included in the study as a control. The control was injected on day 0 and Day 28 at 15 mg/kg to understand steady state and accumulation, whereas test groups received a single injection of 45 mg/kg. A 120 mg vial of FIRMAGON® was reconstituted by adding 3 mL of sterile water for injection provided with the FIRMAGON® kit, and the resulting reconstituted solution was at 40 mg/mL concentration of degarelix. Table 12 Non-Clinical Study Dosing Details
(“F#” denotes “Formulation Number”) [00204] Table 11 Observations: This accelerated stability at 50°C evaluated the effect of i) citric acid excipient, and ii) degarelix citrate form over degarelix acetate form on the stability of degarelix in the polymer-based single-syringe formulations. [00205] If the total impurities (area sum%) of F37 vs. (F34, F35, F36, F45, F46, F47, F48), and F41 vs. (F38, F39, F40) is compared, DgA-DS containing formulations without citric acid F37 (15.0% impurities) and F41(15.2% impurities) generated significantly higher amount of impurities after only 14 days compared to the formulations that contained DgC- DS (<5% impurities after 21 days) or citric acid as an excipient (<8% after 18 days). Lower levels of acylated impurities were observed for citric acid-based formulations than formulations that do not contain any citric acid. [00206] Addition of citric acid as a stabilizer excipient to the drug-polymer formulation or using it as part of the alternate salt form that is prepared by co-lyophilization of DgA-DS with citric acid, both resulted in a similar improvement of drug stability on accelerated storage, and the lower impurity levels were observed. Further, this preliminary data suggested that stabilizers such as citric acid can be just added as an excipient to the formulation to improve the drug stability in presence of polymer, and the additional process of lyophilization to make DgC-DS form may be eliminated to simplify the manufacturing process. [00207] Citric acid was added as an excipient to the formulation at four different molar levels [0.5 (F48), 1 (F45), 2 (F46), and 3 (F47) moles to 1 mole of degarelix). [00208] F48 with 0.5 mol citric acid showed 7.8 % total impurities at day 18 vs. F37 with no citric acid showed 15.0% total impurities at day 14. F45 with 1 mol of citric acid showed 3.6% total impurities at day 18. Although 0.5 moles of citric acid helped in improving the stability of DgA-DS compared to no additive version, higher levels of citric acid showed much better improvement in drug stability. This set of results indicates that higher than 0.5 moles of citric acid to degarelix is needed to achieve better drug stability. The addition of 1:1 or molar excess of citric acid to the degarelix showed promising results. 19. Animal dosing [00209] Prefilled syringes of liquid polymer solutions (Syringe A) and degarelix/organic solvent solutions (Syringe B) were prepared as described in section 5 and section 6. The samples were not e-beamed. Degarelix release rates of these formulations were obtained using a rat model. Male rats were each injected with a single subcutaneous injection of the reconstituted formulations obtained by mixing Syringe A and Syringe B versions of different formulations. The composition of the formulations and the dosing details are listed in Table 12. [00210] At predetermined time points, rats were bled and plasma degarelix levels were determined using liquid chromatography with tandem mass spectrometry (LC-MS/MS). Each data point is based on an average plasma degarelix concentration. Six rats were dosed for each group, with sparse blood sampling performed for early time points. FIRMAGON® (degarelix for injection) was included in the study as a control. The control was injected on day 0 and Day 28 at 15 mg/kg to understand steady state and accumulation, whereas test groups received a single injection of 45 mg/kg. A 120 mg vial of FIRMAGON® was reconstituted by adding 3 mL of sterile water for injection provided with the FIRMAGON® kit, and the resulting reconstituted solution was at 40 mg/mL concentration of degarelix. Table 12 Non-Clinical Study Dosing Details (“F#” denotes “Formulation Number”) [00211] In vivo degarelix release in rats for polymer-based formulations of degarelix at 28-days post injection is presented in Figure 2. Figure 3 shows a magnified version of day- 0 to day-3 levels to show the Cmax levels. As seen in Figures 2 and 3, all the polymer-based formulations (Groups F-TA5 to L-TA11) showed higher initial release (Cmax) than control (Group A-CA1) which could be attributed to the higher injection dose. Groups F-TA5 to L- TA11 were injected at 3 times higher doses (45 mg/kg) than control A-CA1 (15 mg/kg). [00212] Figures 2 and 3 show that, although polymer-based formulations were injected with a 3 times higher dose than the control, the initial Cmax numbers observed for polymer- based test articles were much less than the 3*Cmax of the control, which indicates that the polymer-based formulations may have resulted in a better encapsulation of the drug and lower release of the drug initially. [00213] In vivo degarelix release in rats for polymer-based formulations of degarelix at 63-days post injection is presented in Figure 4. Figure 5 shows a magnified version of the same data in Figure 4 to better illustrate the release of the different formulations. Figures 6 and 7 show that all of the formulations released drug that resulted in measurable levels of degarelix in vivo for greater than 60 days. [00214] In vivo degarelix release in rats for polymer-based formulations of degarelix at 119-days post injection is presented in Figure 6. Figure 7 shows a magnified version of the same data in Figure 6 to better illustrate the release of the different formulations. Figures 6 and 7 show that all of the formulations released drug that resulted in measurable levels of degarelix in vivo for greater than 90 days (at least 119 days). Also, the formulation L-TA11 (delarelix citrate) showed notably higher levels of drug in vivo compared to other formulations throughout the testing period. [00215] In vivo testosterone concentration levels (ng/mL) in rats for the polymer-based formulations of degarelix API in this example at 105-days post injection is presented in Figure 8. Together, the results show that all of the formulations had plasma concentration of greater than 1 ng/mL which is the minimum drug concentration to induce chemical castration. In addition, the testosterone level remained below the “historical testosterone baseline” level and testosterone suppression occurred by day 4. Lastly, a second dose of FIRMAGON® (A-CA1) at Day 28 did not suppress testosterone concentration levels any more than the other formulations dosed once on Day 0. [00216] Table 13 shows the PK data for the experiment through Day 119 post-injection described above. The Cmax for all polymer-based formulations occurred earlier based on Tmax compared to Firmagon; this could lead to a faster reduction in testosterone levels. Table 13 PK Data
(“F#” denotes “Formulation Number”) [00211] In vivo degarelix release in rats for polymer-based formulations of degarelix at 28-days post injection is presented in Figure 2. Figure 3 shows a magnified version of day- 0 to day-3 levels to show the Cmax levels. As seen in Figures 2 and 3, all the polymer-based formulations (Groups F-TA5 to L-TA11) showed higher initial release (Cmax) than control (Group A-CA1) which could be attributed to the higher injection dose. Groups F-TA5 to L- TA11 were injected at 3 times higher doses (45 mg/kg) than control A-CA1 (15 mg/kg). [00212] Figures 2 and 3 show that, although polymer-based formulations were injected with a 3 times higher dose than the control, the initial Cmax numbers observed for polymer- based test articles were much less than the 3*Cmax of the control, which indicates that the polymer-based formulations may have resulted in a better encapsulation of the drug and lower release of the drug initially. [00213] In vivo degarelix release in rats for polymer-based formulations of degarelix at 63-days post injection is presented in Figure 4. Figure 5 shows a magnified version of the same data in Figure 4 to better illustrate the release of the different formulations. Figures 6 and 7 show that all of the formulations released drug that resulted in measurable levels of degarelix in vivo for greater than 60 days. [00214] In vivo degarelix release in rats for polymer-based formulations of degarelix at 119-days post injection is presented in Figure 6. Figure 7 shows a magnified version of the same data in Figure 6 to better illustrate the release of the different formulations. Figures 6 and 7 show that all of the formulations released drug that resulted in measurable levels of degarelix in vivo for greater than 90 days (at least 119 days). Also, the formulation L-TA11 (delarelix citrate) showed notably higher levels of drug in vivo compared to other formulations throughout the testing period. [00215] In vivo testosterone concentration levels (ng/mL) in rats for the polymer-based formulations of degarelix API in this example at 105-days post injection is presented in Figure 8. Together, the results show that all of the formulations had plasma concentration of greater than 1 ng/mL which is the minimum drug concentration to induce chemical castration. In addition, the testosterone level remained below the “historical testosterone baseline” level and testosterone suppression occurred by day 4. Lastly, a second dose of FIRMAGON® (A-CA1) at Day 28 did not suppress testosterone concentration levels any more than the other formulations dosed once on Day 0. [00216] Table 13 shows the PK data for the experiment through Day 119 post-injection described above. The Cmax for all polymer-based formulations occurred earlier based on Tmax compared to Firmagon; this could lead to a faster reduction in testosterone levels. Table 13 PK Data *Dosed once on Day 0 and once on Day 28. Dose normalized parameters are calculated off total dose. [00217] Various modifications of the above-described invention will be evident to those skilled in the art. It is intended that such modifications are included within the scope of the following claims.
*Dosed once on Day 0 and once on Day 28. Dose normalized parameters are calculated off total dose. [00217] Various modifications of the above-described invention will be evident to those skilled in the art. It is intended that such modifications are included within the scope of the following claims.










Claims
CLAIMS 1. A composition, comprising: a therapeutically effective amount of degarelix or a pharmaceutically acceptable salt thereof; a biodegradable polymer; and a biocompatible solvent, wherein: the composition is formulated for subcutaneous injection into a subject; a single dose of the composition is about 2.5 mL or less or about 2.0 mL or less; and upon injection into the subject, the composition forms an in situ depot that releases the degarelix or pharmaceutically acceptable salt thereof over a time period of from about 1 month to about 6 months.
2. The composition of claim 1, wherein the in situ depot releases the degarelix or pharmaceutically acceptable salt thereof over a time period selected from the group consisting of: at least about 1 month, at least about 1.5 months, at least about 2 months, at least about 2.5 months, at least about 3 months, at least about 3.5 months, at least about 4 months, at least about 4.5 months, and at least about 5 months.
3. The composition of claim 1, wherein the pharmaceutically acceptable salt of the degarelix is selected from degarelix acetate, degarelix citrate, degarelix pamoate, degarelix palmitate, and degarelix mesylate.
4. The composition of claim 1, wherein the biodegradable polymer comprises monomer residues selected from the group consisting of lactide, glycolide, caprolactone, p- dioxanone, trimethylene carbonate, thylene oxide, propylene oxide, sebacic anhydride, diketene acetals/diols, and combinations thereof.
5. The composition of claim 4, wherein the biodegradable polymer comprises lactide and glycolide monomer residues.
6. The composition of claim 5, wherein a molar ratio of the lactide to glycolide monomer residues is from about 45:55 to about 99:1.
7. The composition of claim 7, wherein a molar ratio of the lactide to glycolide monomer residues is from about 50:50 to about 90:10.
8. The composition of 4, wherein the biodegradable polymer comprises: lactide monomer residues and monomer residues selected from the group consisting of ε- caprolactone, trimethylene carbonate, and combinations thereof.
9. The composition of claim 8, wherein a molar ratio of the lactide monomer residues to the ε-caprolactone and/or trimethylene carbonate monomer residues is from about 10:90 to about 90:10.
10. The composition of claim 9, wherein a molar ratio of the lactide monomer residues to the ε-caprolactone and/or trimethylene carbonate monomer residues is from about 25:75 to about 75:25.
11. The composition of claim 10, wherein a molar ratio of the lactide monomer residues to the ε-caprolactone and/or trimethylene carbonate monomer residues is about 75:25.
12. The composition of any one of claims above, wherein the biodegradable polymer comprises at least one carboxylic acid end group, at least one hydroxyl end group, or at least one hydroxy end group and is substantially free of terminal carboxy end groups.
13. The composition of any one of claims 8-11, wherein the biodegradable polymer is poly(D,L-lactide-co-glycolide) (PLG), poly(D,L-lactide) (PLA), or poly(D,L-lactide-co-ε- caprolactone) (PLC).
14. The composition of any one of claims 8-11, wherein the biodegradable polymer is poly(D,L-lactide-co-ε-caprolactone) (PLC).
15. The composition of claim 14, wherein the PLC comprises at least one carboxylic acid end group.
16. The composition of claim 15, wherein the PLC is a carboxylic acid-initiated PLC.
17. The composition of claim 16, wherein the carboxylic acid is glycolic acid or lactic acid.
18. The composition of claim 14, wherein the biodegradable polymer is acid-initiated 75:25 PLC.
19. The composition of any one of claims 5-7, wherein the biodegradable polymer is PLG.
20. The composition of claim 19, wherein the PLG comprises at least one hydroxyl end group.
21. The composition of 20, wherein the PLG is a core-diol-initiated PLG.
22. The composition of claim 21, wherein the core diol is 1,6-hexane-diol.
23. The composition of claim 19, wherein the PLG is a mono functional alcohol-initiated PLG.
24. The composition of claim 23, wherein the alcohol is dodecanol.
25. The composition of claim 19, wherein the biodegradable polymer is acid-initiated 50:50 PLG.
26. The composition of claim 22, wherein the biodegradable polymer is 1,6-hexane-diol- initiated 75:25 PLG.
27. The composition of claim 22, wherein the biodegradable polymer is 1,6-hexane-diol- initiated 85:15 PLG.
28. The composition of claim 13 wherein the biodegradable polymer is dodecanol- initiated PLA.
29. The composition of claim 1, wherein the biocompatible polymer has a weight average molecular weight of from about 5 kDa to about 60 kDa.
30. The composition of claim 1, wherein the biocompatible polymer has a weight average molecular weight of from about 5 kDa to about 35 kDa.
31. The composition of claim 1, wherein the biocompatible polymer comprises less than about 0.5 wt%, or less than about 0.4 wt%, or less than about 0.3 wt%, or less than about 0.2 wt% or less than about 0.1 wt% unreacted lactide monomer.
32. The composition of claim 1, wherein the amount of biodegradable polymer in the composition is from about 10% to about 60% by weight of the composition.
33. The composition of claim 1, wherein the amount of biocompatible solvent in the composition is from about 10% to about 85% by weight of the composition.
34. The composition of claim 1, wherein the therapeutically effective amount of degarelix or pharmaceutically acceptable salt in the composition is from about 10% to about 40% by weight of the composition.
35. The composition of claim 1, wherein the therapeutically effective amount of degarelix or pharmaceutically acceptable salt thereof in the composition is from about 8 wt% to about 39 wt% degarelix free base equivalent.
36. The composition of claim 1, wherein the therapeutically effective amount of degarelix or pharmaceutically acceptable salt thereof is from about 40 mg – 500 mg.
37. The composition of claim 1, wherein the therapeutically effective amount of degarelix or pharmaceutically acceptable salt thereof is from about 80 mg – 500 mg.
38. The composition of claim 1, wherein the therapeutically effective amount of degarelix or pharmaceutically acceptable salt thereof is from about 120 mg – 500 mg.
39. The composition of claim 1, wherein the biocompatible solvent is selected from the group consisting of: N-methyl-2-pyrrolidone (NMP), dimethyl sulfoxide (DMSO), dimethylacetamide (DMA), benzyl benzoate (BnBzO), polyethylene glycol 15 hydroxystearate, methyl ethyl ketone, methyl lactate, benzyl alcohol, propylene carbonate (PC), triacetin, tributyl citrate, acetyl tributyl citrate, acetyl triethyl citrate, triethyl citrate, diethylene glycol monomethyl ether, ethyl acetate, N-ethyl-2-pyrrolidone, glycofurol and combinations thereof.
40. The composition of claim 39, wherein the biocompatible solvent is NMP or DMSO.
41. The composition of claim 1, further comprising one or more additives selected from the group consisting of polysorbate 20, polysorbate 80, poloxamer 188, sorbitan trioleate, lecithin (e.g., soya or egg), polyethylene glycol (PEG), PEG 300, 2-pyrrolidone, alpha- tocopherol, Vitamin E TPGS, sucrose cocoate, sucrose stearate, sucrose laurate, proline, arginine, sodium metabisulfite butylated hydroxyanisole, butylated hydroxyquinone, butylhydroxyanisol, hydroxycoumarin, butylated hydroxytoluene, cephalm, ethyl gallate, propyl gallate, octyl gallate, lauryl gallate, propyl-hydroxybenzoate, trihydroxybutylrophenone, vitamin E, lecithin, ethanolamine, ZnCl2, MgCl2, CaCl2, DL- methioninecitric acid, dimethylphenol, dibutylphenol, ethylenediaminotetraacetic acid (EDTA), ethylene glycol-bis(β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid (EGTA), ascorbic acid, , nitrilotriacetic acid, n-hydroxyethylethylenediaminetriacetic acid (HEDTA), mercaptoethanol, and combinations thereof.
42. The composition of claim 1, wherein a single dose of the composition is about 2.5 mL or less, about 2.4 mL or less, about 2.3 mL or less, about 2.2 mL or less, about 2.1 mL or less, about 2.0 mL or less, about 1.9 mL or less, about 1.8 mL or less, about 1.7 mL or less, about 1.6 mL or less, about 1.5 mL or less, about 1.4 mL or less, about 1.3 mL or less, about 1.2 mL or less, about 1.1 mL or less, about 1 mL or less, about 0.75 mL or less, about 0.5 mL or less, or about 0.375 mL or less.
43. The composition of claim 1, wherein the degarelix or pharmaceutically acceptable salt thereof is lyophilized, and wherein the biodegradable polymer is dissolved in the biocompatible solvent.
44. The composition of claim 1, wherein the degarelix or pharmaceutically acceptable salt thereof is dissolved in a first biocompatible solvent, and wherein the biodegradable polymer is dissolved in a second biocompatible solvent.
45. The composition of claim 44, wherein the first and second biocompatible solvent are the same.
46. The composition of any one of claims 1-45, wherein the composition has been terminally sterilized or sterile filtered.
47. The composition of claim 46, wherein the composition has been terminally sterilized by e-beam.
48. A pharmaceutical composition, comprising: (a) about 10-30 wt% of degarelix acetate; (b) about 20-80 wt% of N-methyl-2-pyrrolidone (NMP); and (c) about 10-50 wt% of 75:25 poly(lactide-co-ε-caprolactone) (PLC) copolymer having at least one carboxylic acid end group and having a weight average molecular weight of about 5-30 kDa.
49. The pharmaceutical composition of claim 48, wherein the composition comprises: (a) about 16 wt% of degarelix acetate; (b) about 57 wt% of N-methyl-2-pyrrolidone (NMP); and (c) about 27 wt% of 75:25 poly(lactide-co-ε-caprolactone) (PLC) copolymer having at least one carboxylic acid end group and having a weight average molecular weight of about 17 kDa.
50. The pharmaceutical composition of claim 48, wherein the composition comprises: (a) about 16 wt% of degarelix acetate; (b) about 57 wt% of NMP; and (c) about 27 wt% of 75:25 PLC copolymer having at least one carboxylic acid end group and having a weight average molecular weight of about 8 kDa.
51. A pharmaceutical composition, comprising: (a) a first container comprising 30-70 wt% of 75:25 PLC copolymer having at least one carboxylic acid end group and having a weight average molecular weight of 5-30 kDa, and 30-70 wt% of N-methyl-2- pyrrolidone (NMP); and (b) a second container comprising 20-40 wt% of degarelix acetate and 60-80 wt% NMP.
52. The pharmaceutical composition of claim 51, wherein: (a) the first container comprises 50 wt% of 75:25 PLC copolymer having at least one carboxylic acid end group and having a weight average molecular weight of 17 kDa, and 50 wt% of N-methyl-2-pyrrolidone (NMP); and (b) the second container comprises 35 wt% of degarelix acetate and 65 wt% NMP.
53. The pharmaceutical composition of claim 51, wherein: (a) the first container comprises 70 wt% of 75:25 PLC copolymer having at least one carboxylic acid end group and having a weight average molecular weight of 8 kDa, and 30 wt% of NMP; and (b) the second container comprising 35 wt% of degarelix acetate and 65 wt% NMP.
54. The pharmaceutical composition of any one of claims 48-53, wherein unreacted lactide and caprolactone monomers in the PLC copolymer are less than about 1.0 wt%.
55. The pharmaceutical composition of any one of claims 48-53, wherein unreacted lactide and caprolactone monomers in the PLC copolymer are less than about 0.5 wt%, than about 0.4 wt %, less than about 0.3 wt %, less than about 0.2 wt% and less than about 0.1 wt%.
56. A pharmaceutical composition, comprising: (a) about 10-30 wt% of degarelix acetate; (b) about 20-80 wt% of NMP; and (c) about 10-50 wt% of poly(D,L-lactide) (PLA) polymer having at least one carboxylic acid end group and having a weight average molecular weight of about 5-30 kDa.
57. The pharmaceutical composition of claim 56, wherein the composition comprises: (a) about 16 wt% of degarelix acetate; (b) about 57 wt% of NMP; and (c) about 27 wt% of poly(D,L-lactide) (PLA) polymer having at least one carboxylic acid end group and having a weight average molecular weight of about 23 kDa.
58. A pharmaceutical composition, comprising: (a) a first container comprising 30-70 wt% of PLA polymer having at least one carboxylic acid end group and having a weight average molecular weight of 5-30 kDa, and 30-70 wt% of NMP; and (b) a second container comprising 20-40 wt% of degarelix acetate and 60-80 wt% NMP.
59. The pharmaceutical composition of claim 58, wherein: (a) the first container comprising 50 wt% of PLA polymer having at least one carboxylic acid end group and having a weight average molecular weight of 23 kDa, and 50 wt% of NMP; and (b) the second container comprising 35 wt% of degarelix acetate and 65 wt% NMP.
60. The pharmaceutical composition of any one of claims 56-59, wherein unreacted lactide monomers in the PLA polymer are less than about 1.0 wt%.
61. The pharmaceutical composition of any one of claims 56-59, wherein unreacted lactide monomers in the PLA polymer are less than about 0.5 wt%, than about 0.4 wt %, less than about 0.3 wt %, less than about 0.2 wt% and less than about 0.1 wt%.
62. A pharmaceutical composition, comprising: (a) about 10-30 wt% of degarelix acetate; (b) about 20-80 wt% of NMP; and (c) about 10-50 wt% of 75:25 poly(D,L-lactide-co-glycolide) (PLG) copolymer having at least one carboxylic acid end group and having a weight average molecular weight of about 5-30 kDa.
63. The pharmaceutical composition of claim 62, wherein the composition comprises: (a) about 16 wt% of degarelix acetate; (b) about 57 wt% of NMP; and (c) about 27 wt% of 75:25 poly(D,L-lactide-co-glycolide) (PLG) copolymer having at least one carboxylic acid end group and having a weight average molecular weight of about 17 kDa.
64. A pharmaceutical composition, comprising: (a) a first container comprising 30-70 wt% of 75:25 PLG copolymer having at least one carboxylic acid end group and having a weight average molecular weight of 5-30 kDa, and 30-70 wt% of NMP; and (b) a second container comprising 20-40 wt% of degarelix acetate and 60-80 wt% NMP.
65. The pharmaceutical composition of claim 64 wherein: (a) the first container comprises 50 wt% of 75:25 PLG copolymer having at least one carboxylic acid end group and having a weight average molecular weight of 17 kDa, and 50 wt% of NMP; and (b) the second container comprises 35 wt% of degarelix acetate and 65 wt% NMP.
66. The pharmaceutical composition of any one of claims 62-65, wherein unreacted lactide and glycolide monomers in the PLG copolymer are less than about 1.0 wt%.
67. The pharmaceutical composition of any one of claims 62-65, wherein unreacted lactide and glycolide monomers in the PLG copolymer are less than about 0.5 wt%, than about 0.4 wt %, less than about 0.3 wt %, less than about 0.2 wt% and less than about 0.1 wt%.
68. A pharmaceutical composition, comprising: (a) about 10-30 wt% of degarelix acetate; (b) about 20-80 wt% of NMP; and (c) about 10-50 wt% of 75:25 PLG copolymer having at least one hydroxyl end group and having a weight average molecular weight of about 5-30 kDa.
69. The pharmaceutical composition of claim 68, wherein the composition comprises: (a) about 16 wt% of degarelix acetate; (b) about 57 wt% of NMP; and (c) about 27 wt% of 75:25 PLG copolymer having at least one hydroxyl end group and having a weight average molecular weight of about 19 kDa.
70. A pharmaceutical composition, comprising: (a) a first container comprising 30-70 wt% of 75:25 PLG copolymer having at least one hydroxyl end group and having a weight average molecular weight of 5-30 kDa, and 30-70 wt% of NMP; and (b) a second container comprising 20-40 wt% of degarelix acetate and 60-80 wt% NMP.
71. The pharmaceutical composition of claim 70, wherein: (a) the first container comprises 50 wt% of 75:25 PLG copolymer having at least one hydroxyl end group and having a weight average molecular weight of 19 kDa, and 50 wt% of NMP; and (b) the second container comprises 35 wt% of degarelix acetate and 65 wt% NMP.
72. A pharmaceutical composition, comprising: (a) about 10-30 wt% of degarelix acetate; (b) about 10-40 wt% of NMP; (c) about 10-40 wt% of DMSO; and (d) about 10-50 wt% of 75:25 PLG copolymer having at least one hydroxyl end group and having a weight average molecular weight of about 5-30 kDa.
73. The pharmaceutical composition of claim 72, wherein the composition comprises: (a) about 16 wt% of degarelix acetate; (b) about 30 wt% of NMP; (c) about 27 wt% of DMSO; and (d) about 27 wt% of 75:25 PLG copolymer having at least one hydroxyl end group and having a weight average molecular weight of about 19 kDa.
74. A pharmaceutical composition, comprising: (a) a first container comprising 30-70 wt% of 75:25 PLG copolymer having at least one hydroxyl end group and having a weight average molecular weight of 5-30 kDa, and 30-70 wt% of NMP; and (b) a second container comprising 20-40 wt% of degarelix acetate and 60-80 wt% DMSO.
75. The pharmaceutical composition of claim 74, wherein: (a) the first container comprises 50 wt% of 75:25 PLG copolymer having at least one hydroxyl end group and having a weight average molecular weight of 19 kDa, and 50 wt% of NMP; and (b) the second container comprises 35 wt% of degarelix acetate and 65 wt% DMSO.
76. A pharmaceutical composition, comprising: (a) about 10-30wt% of degarelix citrate; (b) about 20-80 wt% of NMP; and (c) about 10-50 wt% of 75:25 PLG copolymer having at least one hydroxyl end group and having a weight average molecular weight of about 5-30 kDa.
77. The pharmaceutical composition of claim 76, wherein the composition comprises: (a) about 16 wt% of degarelix citrate; (b) about 57 wt% of NMP; and (c) about 27 wt% of 75:25 PLG copolymer having at least one hydroxyl end group and having a weight average molecular weight of about 19 kDa.
78. A pharmaceutical composition, comprising: (a) a first container comprising 30-70 wt% of 75:25 PLG copolymer having at least one hydroxyl end group and having a weight average molecular weight of 19 kDa, and 50 wt% of NMP; and (b) a second container comprising 20-40 wt% of degarelix citrate and 60-80 wt% NMP.
79. The pharmaceutical composition of claim 78, wherein: (a) the first container comprises 50 wt% of 75:25 PLG copolymer having at least one hydroxyl end group and having a weight average molecular weight of 19 kDa, and 50 wt% of NMP; and (b) the second container comprises 35 wt% of degarelix citrate and 65 wt% NMP.
80. A method of treating prostate cancer in a subject, comprising subcutaneously administering the composition of any one of claims 1-79 to the subject.
81. The method of claim 80, wherein the prostate cancer is advanced prostate cancer.
82. The method of claim 80 or 81, wherein the dose amount of degarelix or pharmaceutically acceptable salt in the composition is administered in a dose amount of about 40 mg to about 500 mg.
83. A method of reducing serum testosterone levels in a subject to 50 ng/dL or less, comprising subcutaneously administering the composition of any one of claims 1-79 to the subject.
84. The method of claim 83, wherein serum testosterone levels are reduced to 20 ng/dL or less.
85. The method of claim 83, wherein serum testosterone levels are reduced to 10 ng/dL or less.
86. A method of suppressing ovarian function in a subject with hormone-receptor positive breast cancer, comprising subcutaneously administering the composition of any one of claims 1-79 to the subject.
87. The method of claim 86, wherein the hormone receptor positive breast cancer is estrogen receptor (ER) positive breast cancer.
88. The method of claim 87, wherein the subject’s estradiol (E2) production level is suppressed to a level less than about 20 pg/mL to about less than about 2 pg/mL.
89. The method of claim 86, wherein the subject’s follicle stimulating hormone (FSH) level is suppressed to a level less than about 40 IU/L.
90. The method of claim 86, wherein the subject’s luteinizing hormone (LH) level is suppressed to a level less than about 4 IU/L.
91. A method of treating central precocious puberty (CPP) in a subject comprising subcutaneously administering the composition of any one of claims 1-79 to the subject.
92. The method of claim 91, wherein the subject’s CPP blood serum LH concentration level is reduced to a pre-pubertal concentration level of less than about 4 IU/L.
93. The method of any one of claims 80-92, wherein the composition is administered about once every 1 month, about once every 2 months, about once every 3 months, about once every 4 months, about once every 5 months, or about once every six months.
94. The method of any one of claims 80-92, wherein the composition is administered to the subject about once every three months.
95. The method of any one of claims 80-92, wherein the composition is administered as a loading dose followed by a maintenance dose of the composition about 1 month, 2 months or 3 months after the loading dose has been administered.
96. The method of claim 95, when a loading dose is administered, the maintenance dose is administered every 1 month, every 2 months or every 3 months thereafter.
97. A prefilled syringe system for administration of the composition of claim 1, comprising a single syringe containing the composition of any one of claims 1-79.
98. The prefilled syringe system of claim 97, wherein the syringe is a mixing syringe.
99. The prefilled syringe system of claim 97, wherein the syringe is a dual-chambered syringe, and the composition of claim 1 is contained within one chamber of the syringe.
100. The prefilled syringe system of claim 97, wherein the syringe is a dual-chambered syringe, and wherein the biodegradable polymer and biocompatible solvent are contained in one chamber of the syringe and the degarelix or pharmaceutically acceptable salt is contained in the second chamber of the syringe.
101. The prefilled syringe system of claim 100, wherein the degarelix or pharmaceutically acceptable salt is lyophilized.
102. The prefilled syringe system of claim 100, wherein the degarelix or pharmaceutically acceptable salt is dissolved or suspended in the biocompatible solvent.
103. A prefilled syringe system for administration of the composition of any one of claims 1-79, comprising a first syringe containing the biodegradable polymer and the biocompatible solvent, and a second syringe containing the degarelix.
104. The prefilled syringe system of claim 103, wherein the first syringe and the second syringe are coupled together.
105. The prefilled syringe system of claim 103, wherein the degarelix or pharmaceutically acceptable salt is lyophilized.
106. The prefilled syringe system of claim 103, wherein the degarelix or pharmaceutically acceptable salt is dissolved or suspended in the biocompatible solvent.
107. A kit comprising the prefilled syringe system of any one of claims 97-106 and instructions.
108. A product comprising the composition of any one of claims 1-79 for use in a method of treating prostate cancer or CPP.
109. A product comprising the composition of any one of claims 1-79 for use in a method of reducing serum testosterone levels to 50 ng/dL or less.
110. A product comprising the composition of any one of claims 1-79 for use in a method of suppressing ovarian function in a subject with hormone-receptor positive breast cancer.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US202363484432P | 2023-02-10 | 2023-02-10 | |
| US63/484,432 | 2023-02-10 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2024168022A2true WO2024168022A2 (en) | 2024-08-15 |
| WO2024168022A3 WO2024168022A3 (en) | 2024-10-03 |
Family
ID=90362495
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2024/014780PendingWO2024168022A2 (en) | 2023-02-10 | 2024-02-07 | Degarelix polymeric formulations |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2024168022A2 (en) |
Citations (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4810775A (en) | 1987-03-19 | 1989-03-07 | Boehringer Ingelheim Kg | Process for purifying resorbable polyesters |
| US4938763A (en) | 1988-10-03 | 1990-07-03 | Dunn Richard L | Biodegradable in-situ forming implants and methods of producing the same |
| US5324519A (en) | 1989-07-24 | 1994-06-28 | Atrix Laboratories, Inc. | Biodegradable polymer composition |
| US5702716A (en) | 1988-10-03 | 1997-12-30 | Atrix Laboratories, Inc. | Polymeric compositions useful as controlled release implants |
| US5744153A (en) | 1994-04-08 | 1998-04-28 | Atrix Laboratories, Inc. | Liquid delivery compositions |
| US7019106B2 (en) | 2000-08-07 | 2006-03-28 | Wako Pure Chemical Industries, Ltd. | Lactic acid polymer and process for producing the same |
| US9187593B2 (en) | 2007-02-15 | 2015-11-17 | Tolmar Therapeutics, Inc. | Low burst polymers and methods to produce polymer |
Family Cites Families (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2009148583A2 (en)* | 2008-06-03 | 2009-12-10 | Qlt Usa, Inc. | Method for improvement of octreotide bioavailability |
| MX2016004444A (en)* | 2013-10-08 | 2016-11-10 | Ferring Bv | Microparticles comprising gnrh made by pgss. |
| PT3331495T (en)* | 2015-08-03 | 2020-12-07 | Tolmar International Ltd | Liquid polymer delivery system for extended administration of drugs |
| CN107773528A (en)* | 2016-08-24 | 2018-03-09 | 南京星银药业集团有限公司 | A kind of acetic acid Ac-D-2Nal-D-4Cpa-D-3Pal-Ser-4Aph(Hor)-D-4Aph(Cbm)-Leu-Lys(iPr)-Pro-D-Ala-NH2 injection-type sustained-release implant |
| ES2914314T3 (en)* | 2017-01-31 | 2022-06-09 | Veru Inc | Compositions and methods for the long-term release of gonadotropin-releasing hormone (GnRH) antagonists |
- 2024
- 2024-02-07WOPCT/US2024/014780patent/WO2024168022A2/enactivePending
Patent Citations (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4810775A (en) | 1987-03-19 | 1989-03-07 | Boehringer Ingelheim Kg | Process for purifying resorbable polyesters |
| US4938763A (en) | 1988-10-03 | 1990-07-03 | Dunn Richard L | Biodegradable in-situ forming implants and methods of producing the same |
| US4938763B1 (en) | 1988-10-03 | 1995-07-04 | Atrix Lab Inc | Biodegradable in-situ forming implants and method of producing the same |
| US5702716A (en) | 1988-10-03 | 1997-12-30 | Atrix Laboratories, Inc. | Polymeric compositions useful as controlled release implants |
| US5990194A (en) | 1988-10-03 | 1999-11-23 | Atrix Laboratories, Inc. | Biodegradable in-situ forming implants and methods of producing the same |
| US5324519A (en) | 1989-07-24 | 1994-06-28 | Atrix Laboratories, Inc. | Biodegradable polymer composition |
| US5744153A (en) | 1994-04-08 | 1998-04-28 | Atrix Laboratories, Inc. | Liquid delivery compositions |
| US7019106B2 (en) | 2000-08-07 | 2006-03-28 | Wako Pure Chemical Industries, Ltd. | Lactic acid polymer and process for producing the same |
| US9187593B2 (en) | 2007-02-15 | 2015-11-17 | Tolmar Therapeutics, Inc. | Low burst polymers and methods to produce polymer |
Also Published As
| Publication number | Publication date |
|---|---|
| WO2024168022A3 (en) | 2024-10-03 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US20220354956A1 (en) | Biodegradable drug delivery for hydrophobic compositions | |
| US11801217B2 (en) | Biodegradable block copolymer drug delivery composition | |
| CN103491946A (en) | Biodegradable drug delivery compositions | |
| CN101336890A (en) | Anticancer sustained-release gel injection | |
| EP3713986A1 (en) | Composition | |
| US20200323987A1 (en) | Temperature-responsive degradable hydrogels | |
| WO2024168022A2 (en) | Degarelix polymeric formulations | |
| WO2019084197A1 (en) | Temperature-responsive degradable hydrogels | |
| WO2025008759A1 (en) | Polymeric leuprolide acetate formulations | |
| WO2024168024A1 (en) | Degarelix organic solvent formulations | |
| US20240000797A1 (en) | Biodegradable polymer delivery system for extended delivery of testosterone | |
| US12214105B2 (en) | Long lasting biodegradable polymer based in-situ forming implant for treatment of bone injuries | |
| TW202440148A (en) | Injectable composition of gnrh antagonists and its preparation method and application |