WO2024138112A1 - Indazole compounds - Google Patents
Indazole compoundsDownload PDFInfo
- Publication number
- WO2024138112A1 WO2024138112A1PCT/US2023/085649US2023085649WWO2024138112A1WO 2024138112 A1WO2024138112 A1WO 2024138112A1US 2023085649 WUS2023085649 WUS 2023085649WWO 2024138112 A1WO2024138112 A1WO 2024138112A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- compound
- alkyl
- cancer
- alk
- compounds
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Classifications
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
Definitions
- the disclosurepertains to indazole compounds that are useful in treating cancer, pharmaceutical compositions that include one or more such indazole compounds, and methods of using such indazole compounds in treating cancer.
- kinase inhibitorshave been used to block the activity of kinases and thereby treat cancer (e.g., by inhibiting mitotic processes). These kinase inhibitors are often small molecules that target kinases to block the development, growth or spread of cancer.
- the compounds disclosed hereinprovide small molecule kinase inhibitors that are both efficacious and selective.
- XO, S, or NR
- R 1is H, optionally substituted C 1 -C 6 alkyl, or NH2;
- R 5ais H, halogen, -CN, -S(O) 2 C 1 -C 6 alkyl, OCF 3 , OC 1 -C 3 alkyl, or C 1 -C 3 alkyl;
- Q 5 , Q 6 , Q 7 , Q 8 , and Q 9are each independently N or CR 5 , wherein one or two of the Q 5 , Q 6 , Q 7 , Q 8 , and Q 9 is N and the remainder are CR 5 ;
- R 5is H, halogen, C 1 -Csalkyl, C 1 -Csalkoxyl, or cycloalkyl;
- R 6is C 1 -Cealkyl
- R 7is H, halogen, -C 1 -Cealkyl, -C 1 -Ce alkoxyl, or -cycloalkyl;
- R 8is H, halogen, -C 1 -Cealkyl, -C 1 -Ce alkoxyl, or -cycloalkyl.
- haloi.e., -F, -Cl, -Br, -I
- cyanoi.e., -OH, -C 1 -Cealkyl, C3- Cecycloalkyl, 3-7 membered heterocycloalkyl, -Cs-Cespirocycloalkyl, 3-7 membered spiroheterocycloalkyl, bridged cycloalkyl, bridged heterocycloalkyl, C2-Cealkenyl, C2-C6 alkynyl, C 1 -Cehaloalkyl (e.g., -CF3; -CHF2, -CH2CF3, and the like), -C 1 -Cealkoxy, -C 1 -Ce hal
- “optionally substituted,” or “substituted”means that the substituent may, but is not required to be, substituted with one or more of -C(O)(C 1 -C 6 haloalkyl), -NHSO 2 (C 1 -C 6 alkyl), -N(C 1 -C 6 alkyl)SO 2 (C 1 -C 6 alkyl), or - P(O)(C 1 -Cealkyl)2 (e.g., -P(O)(CH3)2).
- each of the above optional substituentsare themselves optionally substituted by one or two of these groups.
- C 1 -CeWhen a range of carbon atoms is used herein, for example, C 1 -Ce, all ranges, as well as individual numbers of carbon atoms are encompassed.
- C1-C3includes C1-C3, C1-C2, C2-C3, C 1 , C2, and C3.
- a “C 1 to C4 alkyl” grouprefers to all alkyl groups having from 1 to 4 carbons (e.g., 1, 2, 3, or 4), that is, CH3-, CH3CH2-, CH3CH2CH2-, (CH 3 ) 2 CH-, CH3CH2CH2CH2-, CH 3 CH2CH(CH 3 )- and (CH 3 ) 3 C-.
- a “C 1 to Ce alkyl” grouprefers to all alkyl groups having from 1 to 6 carbons (e.g., 1, 2, 3, 4, 5, or 6).
- alkylrefers to a fully saturated aliphatic hydrocarbon group.
- the alkyl moietymay be branched or straight chain. Examples of branched alkyl groups include, but are not limited to, iso-propyl, sec-butyl, t-butyl and the like. Examples of straight chain alkyl groups include, but are not limited to, methyl, ethyl, n- propyl, n-butyl, n-pentyl, n-hexyl, n-heptyl and the like.
- the alkyl groupmay have 1 to 30 carbon atoms (whenever it appears herein, a numerical range such as “1 to 30” refers to each integer in the given range; e.g., “1 to 30 carbon atoms” means that the alkyl group may consist of 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, or 30 carbon atoms, although the present definition also covers the occurrence of the term “alkyl” where no numerical range is designated).
- the “alkyl” groupmay also be a medium size alkyl having 1 to 12 carbon atoms.
- the “alkyl” groupcould also be a lower alkyl having 1 to 6 carbon atoms.
- alkyl groupmay be substituted or unsubstituted.
- C1-C5 alkylindicates that there are one to five carbon atoms in the alkyl chain, i.e., the alkyl chain is selected from methyl, ethyl, n-propyl, isopropyl, n-butyl, isobutyl, tert-butyl, pentyl (branched and straight-chained), etc.
- Typical alkyl groupsinclude, but are in no way limited to, methyl, ethyl, propyl, isopropyl, butyl, isobutyl, tertiary butyl, pentyl and hexyl.
- “Me”is methyl (e.g., CH3).
- Alkylene groupsi.e., fully saturated aliphatic hydrocarbon diradicals
- alkalkylene groups
- C 1 -Cealkrefers to an aliphatic hydrocarbon diradical that has between 1 and 6 carbon atoms.
- Co-Cealkrefers to an aliphatic hydrocarbon diradical that is either not present (i.e., Co), or has between 1 and 6 carbon atoms.
- alkenylrefers to an alkyl group that contains in the straight or branched hydrocarbon chain one or more double bonds.
- An alkenyl groupmay be unsubstituted or substituted.
- alkynylrefers to an alkyl group that contains in the straight or branched hydrocarbon chain one or more triple bonds.
- An alkynyl groupmay be unsubstituted or substituted.
- cycloalkylrefers to a completely saturated (no double or triple bonds) mono- or multi- cyclic hydrocarbon ring system. When composed of two or more rings, the rings may be joined together in a fused fashion. Cycloalkyl groups may contain between 3 and 12 carbon atoms. For example, a Cs-Cecycloalkyl group indicates that there three to six carbon atoms in the ring, that is, the ring is a cyclopropyl, cyclobutyl, cyclopentyl, or cyclohexyl group. A cycloalkyl group may be unsubstituted or substituted.
- spirocycloalkyl ringrefers to a cycloalkyl ring that shares one carbon atom with another cyclic ring.
- a 3-7 membered spirocycloalkyl ringindicates that there are 3, 4, 5, 6, or 7 carbon atoms in the cycloalkyl ring that shares a single carbon atom in common with another cyclic ring.
- shown beloware exemplary 3-7 membered spirocycloalkyl groups attached to a piperidine ring:
- arylrefers to a carbocyclic (all carbon) monocyclic or multicyclic aromatic ring system (including fused ring systems where two carbocyclic rings share a chemical bond) that has a fully delocalized pi-electron system throughout all the rings.
- the number of carbon atoms in an aryl groupcan vary.
- the aryl groupcan be a Ce-C 1 4 aryl group, a Ce-C 1 o aryl group, or a Ce aryl group.
- aryl groupsinclude, but are not limited to, benzene, naphthalene and azulene.
- An aryl groupmay be substituted or unsubstituted.
- heteroarylrefers to a monocyclic or multicyclic aromatic ring system (a ring system with fully delocalized pi-electron system) that contain(s) one or more heteroatoms, that is, an element other than carbon, including but not limited to, nitrogen, oxygen and sulfur.
- the number of atoms in the ring(s) of a heteroaryl groupcan vary.
- the heteroaryl groupcan contain 4 to 14 atoms in the ring(s), 5 to 10 atoms in the ring(s) or 5 to 6 atoms in the ring(s).
- heteroarylincludes fused ring systems where two rings, such as at least one aryl ring and at least one heteroaryl ring, or at least two heteroaryl rings, share at least one chemical bond.
- heteroaryl ringsinclude, but are not limited to, furan, furazan, thiophene, benzothiophene, phthalazine, pyrrole, oxazole, benzoxazole, 1,2,3-oxadiazole, 1,2,4-oxadiazole, thiazole, 1,2,3-thiadiazole, 1,2,4-thiadiazole, benzothiazole, imidazole, benzimidazole, indole, indazole, pyrazole, benzopyrazole, isoxazole, benzoisoxazole, isothiazole, triazole, benzotriazole, thiadiazole, tetrazole, pyridine, pyridazine, pyrimidine
- Heteroaryl ringsmay also include bridge head nitrogen atoms.
- bridge head nitrogen atomsFor example but not limited to: pyrazolo[l,5-a]pyridine, imidazo[l,2-a]pyridine, and pyrazolo[l,5-a]pyrimidine.
- a heteroaryl groupmay be substituted or unsubstituted.
- heterocycloalkylrefers to three-, four-, five-, six-, seven-, eight-, nine-, ten-, up to 18-membered monocyclic, bicyclic, and tricyclic ring system wherein carbon atoms together with from 1 to 5 heteroatoms constitute said ring system.
- a heterocycloalkylmay optionally contain one or more unsaturated bonds situated in such a way, however, that a fully delocalized pi-electron system does not occur throughout all the rings.
- the heteroatom(s)is an element other than carbon including, but not limited to, oxygen, sulfur, and nitrogen.
- heterocycloalkyl groupsinclude but are not limited to, 1,3-dioxin, 1,3-dioxane, 1,4-dioxane, 1,2-di oxolane, 1,3- dioxolane, 1,4-di oxolane, 1,3-oxathiane, 1,4-oxathiin, 1,3 -oxathiolane, 1 ,3-dithiole, 1,3- dithiolane, 1,4-oxathiane, tetrahydro- 1,4-thiazine, 2H-l,2-oxazine, maleimide, succinimide, barbituric acid, thiobarbituric acid, dioxopiperazine, hydantoin, dihydrouracil, trioxane, hexahydro-1, 3, 5-triazine, imidazoline, imidazolidine, isoxazoline, isoxazolidine, ox
- spiroheterocycloalkyl ringrefers to a heterocycloalkyl ring that shares one carbon atom with another cyclic ring.
- a 3- 7 membered spiroheterocycloalkyl ringindicates that there are 3, 4, 5, 6, or 7 atoms in the heterocycloalkyl ring, and only one of the carbon atoms in that heterocycloalkyl ring is also a member of another cyclic ring.
- shown beloware exemplary 3-7 membered spiroheterocycloalkyl groups attached to a piperidine ring:
- bridged bicyclic ringrefers to a ring system comprising two joined cycloalkyl or heterocycloalkyl rings that share at least three at least three atoms
- a 6-9 membered bridged bicyclic ringindicates that there are 6, 7, 8, or 9 atoms in the bridged bicyclic ring.
- shown beloware exemplary 6-9 membered bridged bicyclic rings:
- aminorefers to a -NH2 group.
- halogen atomrefers to fluorine, chlorine, bromine and iodine.
- saltrefers to a salt of a compound that does not cause significant irritation to an organism to which it is administered and does not abrogate the biological activity and properties of the compound.
- the saltis an acid addition salt of the compound.
- Pharmaceutical saltscan be obtained by reacting a compound with inorganic acids such as hydrohalic acid (e.g., hydrochloric acid or hydrobromic acid), sulfuric acid, nitric acid and phosphoric acid.
- Pharmaceutical saltscan also be obtained by reacting a compound with an organic acid such as aliphatic or aromatic carboxylic or sulfonic acids, for example formic, acetic, succinic, lactic, malic, tartaric, citric, ascorbic, nicotinic, methanesulfonic, ethanesulfonic, p-toluensulfonic, salicylic, trifluoroacetic acid, or naphthalenesulfonic acid.
- an organic acidsuch as aliphatic or aromatic carboxylic or sulfonic acids, for example formic, acetic, succinic, lactic, malic, tartaric, citric, ascorbic, nicotinic, methanesulfonic, ethanesulfonic, p-toluensulfonic, salicylic, trifluoroacetic acid, or naphthalenesulfonic acid.
- Pharmaceutical saltscan also be obtained by reacting a compound with a base to form a salt such as an ammonium salt, an alkali metal salt, such as a sodium or a potassium salt, an alkaline earth metal salt, such as a calcium or a magnesium salt, a salt of organic bases such as dicyclohexylamine, N-methyl-D-glucamine, tris(hydroxymethyl)methylamine, C1-C7 alkylamine, cyclohexylamine, triethanolamine, ethylenediamine, and salts with amino acids such as arginine and lysine.
- a saltsuch as an ammonium salt, an alkali metal salt, such as a sodium or a potassium salt, an alkaline earth metal salt, such as a calcium or a magnesium salt, a salt of organic bases such as dicyclohexylamine, N-methyl-D-glucamine, tris(hydroxymethyl)methylamine, C1-C7 alkylamine, cyclohexylamine
- each centermay independently be of R-configuration or S-configuration or a mixture thereof.
- the compounds provided hereinmay be enantiomerically pure, enantiomerically enriched, racemic mixture, diastereomerically pure, diastereomerically enriched, or a stereoisomeric mixture.
- each double bondmay independently be E or Z a mixture thereof.
- any instance of hydrogenmay include hydrogen-1 (protium), hydrogen-2 (deuterium), hydrogen-3 (tritium) or other isotopes;
- any instance of carbonmay include carbon-12, carbon-13, carbon-14, or other isotopes;
- any instance of oxygenmay include oxygen-16, oxygen-17, oxygen-18, or other isotopes;
- any instance of fluorinemay include one or more of fluorine- 18, fluorine- 19, or other isotopes;
- any instance of sulfurmay include one or more of sulfur-32, sulfur-34, sulfur-35, sulfur-36, or other isotopes.
- kinase inhibitormeans any compound, molecule or composition that inhibits or reduces the activity of a kinase.
- the inhibitioncan be achieved by, for example, blocking phosphorylation of the kinase (e.g., competing with adenosine triphosphate (ATP), a phosphorylating entity), by binding to a site outside the active site, affecting its activity by a conformational change, or by depriving kinases of access to the molecular chaperoning systems on which they depend for their cellular stability, leading to their ubiquitylation and degradation.
- ATPadenosine triphosphate
- subjectAs used herein, “subject,” “host,” “patient,” and “individual” are used interchangeably and shall be given its ordinary meaning and shall also refer to an organism that has FGFR proteins. This includes mammals, e.g., a human, a non-human primate, ungulates, canines, felines, equines, mice, rats, and the like. The term “mammal” includes both human and non-human mammals.
- sampleor “biological sample” shall be given its ordinary meaning and also encompasses a variety of sample types obtained from an organism and can be used in an imaging, a diagnostic, a prognostic, or a monitoring assay.
- the termencompasses blood and other liquid samples of biological origin, solid tissue samples, such as a biopsy specimen or tissue cultures or cells derived therefrom and the progeny thereof.
- the termencompasses samples that have been manipulated in any way after their procurement, such as by treatment with reagents, solubilization, or enrichment for certain components.
- the termencompasses a clinical sample, and also includes cells in cell culture, cell supernatants, cell lysates, serum, plasma, biological fluids, and tissue samples.
- treatmentshall be given its ordinary meaning and shall also include herein to generally refer to obtaining a desired pharmacologic and/or physiologic effect.
- the effectmay be prophylactic in terms of completely or partially preventing a disease or symptom thereof and/or may be therapeutic in terms of a partial or complete stabilization or cure for a disease and/or adverse effect attributable to the disease.
- Treatmentshall be given its ordinary meaning and shall also cover any treatment of a disease in a mammal, particularly a human, and includes: (a) preventing the disease or symptom from occurring in a subject which may be predisposed to the disease or symptom but has not yet been diagnosed as having it; (b) inhibiting the disease symptom, e.g., arresting its development; and/or (c) relieving the disease symptom, e.g., causing regression of the disease or symptom.
- cancerneoplasm
- tumorneoplasm
- tumorcells which exhibit relatively autonomous growth, so that they exhibit an aberrant growth phenotype characterized by a significant loss of control of cell proliferation.
- cells of interest for detection or treatment in the present applicationinclude precursors, precancerous e.g., benign), malignant, pre-metastatic, metastatic, and non-metastatic cells.
- FGFR related cancerdenotes those cancers that involve an increased activity in a mutant FGFR kinase, for example, the continued activation of FGFR.
- controlrefers shall be given its ordinary meaning and shall also include a sample or standard used for comparison with a sample which is being examined, processed, characterized, analyzed, etc.
- the controlis a sample obtained from a healthy patient or a non-tumor tissue sample obtained from a patient diagnosed with a tumor.
- the controlis a historical control or standard reference value or range of values.
- the controlis a comparison to a wild-type FGFR arrangement or scenario.
- the disclosureis directed to compounds of formula (I) or a pharmaceutically acceptable salt thereof.
- X in the compounds of formula (I)is O, S, or NR wherein
- Ris H or C 1 -Csalkyl.
- Xis O.
- Xis S. [0038] In some embodiments, X is NR.
- Ris H.
- Ris C 1 -C 3 alkyl, such as, for example, C 3 alkyl, C2alkyl, C 1 alkyl, methyl, ethyl, propyl, and the like.
- n in the compounds of formula (I)is 1 or 2.
- n1
- nis 2.
- m in the compounds of formula (I)is 1 or 2.
- mis 1.
- mis 2.
- nis 1 and m is 1.
- nis i and m is 2.
- nis 2 and m is 1.
- nis 2 and m is 2.
- R 1 in the compounds of formula (I)H, optionally substituted C 1 -C6alkyl, or NH2.
- R 1 in the compounds of formula (I)is H or C 1 -Cealkyl.
- R 1 in the compounds of formula (I)is H.
- R 1 in the compounds of formula (I)is NH2.
- R 1is C 1 -Cealkyl, such as, for example, C 1 -Cealkyl, C 1 -C 5 alkyl, C 1 -C 4 alkyl, C 1 -C 3 alkyl, C 1 -C 2 alkyl, C 1 alkyl, C 2 alkyl, C 3 alkyl, C 4 alkyl, C 5 alkyl, C6alkyl, methyl, ethyl, n-propyl, isopropyl, n-butyl, isosbutyl, sec-butyl, pentanyl, hexanyl, and the like.
- C 1 -Cealkylsuch as, for example, C 1 -Cealkyl, C 1 -C 5 alkyl, C 1 -C 4 alkyl, C 1 -C 3 alkyl, C 1 -C 2 alkyl, C 1 alkyl, C 2 alkyl, C 3 alkyl, C 4 alkyl, C
- R 1is -CH 3 .
- R 1 in the compounds of formula (I)is optionally substituted C 1 -Cealkyl.
- R 1is -CH2NH2.
- R 1is -CH2NHCH3.
- R 1is -CH2N(CH 3 )2.
- (I)are N and the others are each independently CR 5a .
- Q 1is N and Q 2 , Q 3 , and Q 4 are each independently
- Q 2is N and Q 1 , Q 3 , and Q 4 are each independently
- Q 3is N and Q 1 , Q 2 , and Q 4 are each independently CR 5a .
- Q 4is N and Q 1 , Q 2 , and Q 3 are each independently CR 5a .
- two of Q 1 , Q 2 , Q 3 , Q 4is N and the others are each independently CR 5a .
- Q 1 and Q 2are each N, and Q 3 , and Q 4 are each independently CR 5a .
- Q 1 and Q 3are each N, and Q 2 and Q 4 are each independently CR 5a .
- Q 1 and Q 4are each N, and Q 2 and Q 3 are each independently CR 5a .
- Q 2 and Q 3are each N, and Q 1 and Q 4 are each independently CR 5a .
- Q 2 and Q 4are each N, and Q 1 and Q 3 are each independently CR 5a .
- Q 3 and Q 4are each N, and Q 1 and Q 2 are each independently CR 5a .
- each R 5ais independently H, halogen, -CN, -S(O) 2 C 1 -C 3 alkyl, OCF 3 , OC 1 -C 3 alkyl, or C 1 -C 3 alkyl.
- At least one R 5ais H.
- At least one R 5ais halogen, z.e., -F, -Cl, -Br, or -I.
- At least one R 5ais -F.
- At least one R 5ais -CN.
- At least one R 5ais -SO 2 C 1 -Qalkyl, such as, for example, -SO 2 C 1 alkyl, -SO2C2alkyl, -SO 2 C 3 alkyl, -SO 2 CFBCH 3 , -SO2CH5, and the like. In some embodiments, at least one R 5a is -SO 2 CH 3 .
- R 5ais OCF 3 .
- At least one R 5ais OC 1 -C 3 alkyl, such as, for example, OC 1 -C 3 alkyl, OC 1 -C 2 alkyl, OC 1 alkyl, OC 2 alkyl, OC 3 alkyl, -OCH 3 , -OCH 2 CH 3 , -Opropyl, and the like. In some embodiments at least one R 5a is -OCH 3 .
- At least one R 5ais C 1 -C 3 alkyl, such as, for example, C 1 -C 3 alkyl, C 1 -C2alkyl, C 1 alkyl, C2alkyl, C 3 alkyl, -CH 3 , -CH 2 CH 3 , -propyl, and the like. In some embodiments at least one R 5a is -CH 3 .
- Q 5 , Q 6 , Q 7 , Q 8 , and Q 9 in the compounds of formula (I)are each independently N or CR 5 , wherein one or two of Q 5 , Q 6 , Q 7 , Q 8 , and Q 9 is N and the remainder are each independently CR 5 .
- one of Q 5 , Q 6 , Q 7 , Q 8 , or Q 9is N, and the remainder are each independently CR 5 .
- Q 5is N and Q 6 , Q 7 , Q 8 , and Q 9 are each independently CR 5 .
- Q 6is N and Q 5 , Q 7 , Q 8 , and Q 9 are each independently CR 5 .
- Q 7is N and Q 5 , Q 6 , Q 8 , and Q 9 are each independently CR 5 .
- Q 8is N and Q 5 , Q 6 , Q 7 , and Q 9 are each independently CR 5 .
- Q 9is N and Q 5 , Q 6 , Q 7 , and Q 8 are each independently CR 5 .
- two of Q 5 , Q 6 , Q 7 , Q 8 , or Q 9is each N, and the remainder are each independently CR 5 .
- Q 5 and Q 6are each N, and Q 7 , Q 8 , and Q 9 are each independently CR 5 .
- Q 5 and Q 7are each N, and Q 6 , Q 8 , and Q 9 are each independently CR 5 .
- Q 5 and Q 8are each N, and Q 6 , Q 7 , and Q 9 are each independently CR 5 .
- Q 5 and Q 9are each N, and Q 6 , Q 7 , and Q 8 are each independently CR 5 .
- Q 6 and Q 7are each N, and Q 5 , Q 8 , and Q 9 are each independently CR 5 .
- Q 6 and Q 8are each N, and Q 5 , Q 7 , and Q 9 are each independently CR 5 .
- Q 6 and Q 9are each N, and Q 5 , Q 7 , and Q 8 are each independently CR 5 .
- Q 7 and Q 8are each N, and Q 5 , Q 6 , and Q 9 are each independently CR 5 .
- Q 7 and Q 9are each N, and Q 5 , Q 6 , and Q 8 are each independently CR 5 .
- Q 8 and Q 9are each N, and Q 5 , Q 6 , and Q 7 are each independently CR 5 .
- each R 5 in the compounds of formula (I),is independently H, halogen, C 1 -Csalkyl, C 1 -Csalkoxyl, or cycloalkyl.
- At least one R 5is H.
- At least one R 5is halogen, such as, -F, -Cl, -Br, or -I.
- At least one R 5is -Cl.
- at least one R 5is C 1 -C 3 alkyl, such as, for example, C 1 -C 3 alkyl, C 1 -C2alkyl, C 1 alkyl, C2alkyl, Csalkyl, -CH3, - CH2CH3, -propyl, and the like.
- At least one R 5is -CH3.
- At least one R 5is C 1 -C 3 alkoxyl, such as, for example, C 1 -C 3 alkoxyl, C 1 -C2alkoxyl, C 1 alkoxyl, C2alkoxyl, Csalkoxyl, -OCH3, -OCH2CH3, -propoxyl, and the like.
- At least one R 5is -OCH3.
- At least one R 5is cycloalkyl, such as, for example, cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, or cycloheptyl, and the like.
- two R 5are halogen, and the remaining R 5 are H.
- Q 5 and Q 9are each independently CR 5 wherein each R 5 is independently a halogen; Q 6 and Q 8 are each independently CR 5 wherein each R 5 is H; and Q 7 is N.
- Q 5 , Q 8 , and Q 9are each independently CR 5 wherein each R 5 is independently a halogen; Q 6 is CR 5 wherein R 5 is H; and Q 7 is N.
- Q 5 and Q 9are each independently CR 5 wherein each R 5 is -Cl; Q 6 and Q 8 are each independently CR 5 wherein R 5 is H; and Q 7 is N.
- Q 5 and Q 9are each independently CR 5 wherein each R 5 is -Cl; Q 6 is CR 5 wherein R 5 is H; Q 8 is CR 5 wherein R 5 is -F; and Q 7 is N.
- Q 5 and Q 9are each independently CR 5 wherein each R 5 is independently halogen; Q 6 is CR 5 wherein R 5 is H and Q 8 is CR 5 wherein R 5 is C 1 -Csalkyl; and Q 7 is N.
- Q 5 and Q 9are each independently CR 5 wherein each R 5 is -Cl; Q 6 is CR 5 wherein R 5 is H and Q 8 is CR 5 wherein R 5 is -CH3; and Q 7 is N.
- Q 5 and Q 9are each independently CR 5 wherein each R 5 is halogen; Q 6 is CR 5 wherein R 5 is H and Q 8 is N; and Q 7 is CR 5 wherein R 5 is H.
- Q 5 and Q 9are each independently CR 5 wherein each R 5 is -Cl; Q 6 is CR 5 wherein R 5 is H and Q 8 is N; and Q 7 is CR 5 wherein R 5 is H.
- Q 5 and Q 9are each independently CR 5 wherein each R 5 is independently a halogen; Q 6 is CR 5 wherein R 5 is H and Q 8 is N; and Q 7 is N.
- Q 5 and Q 9are each independently CR 5 wherein each R 5 is -Cl; Q 6 is CR 5 wherein R 5 is H and Q 8 is N; and Q 7 is N.
- Q 5 and Q 9are each independently CR 5 wherein each R 5 is independently a C 1 -Csalkyl; Q 6 is CR 5 wherein R 5 is H and Q 8 is N; and Q 7 is N.
- Q 5 and Q 9are each independently CR 5 wherein each R 5 is -CH3; Q 6 is CR 5 wherein R 5 is H and Q 8 is N; and Q 7 is N.
- R 6 in the compounds of formula (I)is C 1 - Cealkyl, such as, for example, C 1 -Cealkyl, C 1 -Csalkyl, C 1 -C4alkyl, C 1 -Csalkyl, C 1 -C2alkyl, C 1 alkyl, C2alkyl, Csalkyl, C4alkyl, Csalkyl, Cealkyl, methyl, ethyl, n-propyl, isopropyl, n- butyl, isosbutyl, sec-butyl, pentanyl, hexanyl, and the like.
- R 6is -CH3.
- R 7 in the compounds of formula (I)is H, halogen, -C 1 - Cealkyl, -C 1 -Cealkoxyl, or -cycloalkyl.
- R 7 in the compounds of formula (I)is H.
- R 7 in the compounds of formula (I)is halogen, such as, for example, -F, -Cl, -Br, or -I.
- R 7 in the compounds of formula (I)is -F.
- R 7 in the compounds of formula (I)is -Cl.
- R 7 in the compounds of formula (I)is -C 1 -Cealkyl, such as, for example, substituted or unsubstituted: C 1 -Cealkyl, C 1 -Csalkyl, C 1 -C4alkyl, C 1 - C 3 alkyl, C 1 -C2alkyl, C 1 alkyl, C2alkyl, C2alkyl, C4alkyl, Csalkyl, Cealkyl, methyl, ethyl, n- propyl, isopropyl, n-butyl, isosbutyl, sec-butyl, pentanyl, hexanyl, and the like.
- R 7is -CH3.
- R 7 in the compounds of formula (I)is -C 1 -Ce alkoxyl, such as, for example, -C 1 -Cealkoxyl, -C 1 -C5alkoxyl, -C 1 -C4alkoxyl, -C 1 -Csalkoxyl, - C 1 -C2alkoxyl, -C 1 alkoxyl, -C2alkoxyl, -C3alkoxyl, -C4alkoxyl, -C 5 alkoxyl, -Cealkoxyl, - OCH3, -OCH2CH3, -propoxyl, and the like.
- R 7is -OCH3.
- R 7 in the compounds of formula (I)is -cycloalkyl, such as, for example, cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, or cycloheptyl, and the like.
- R 8is H, halogen, -C 1 -Cealkyl, -C 1 -Cealkoxyl, or - cycloalkyl.
- R 8 in the compounds of formula (I)is H.
- R 8 in the compounds of formula (I)is halogen, such as, for example, -F, -Cl, -Br, or -I.
- R 8 in the compounds of formula (I)is -F.
- R 8 in the compounds of formula (I)is -C 1 -Cealkyl, such as, for example, substituted or unsubstituted: C 1 -Cealkyl, C 1 -Csalkyl, C 1 -C4alkyl, C 1 - Csalkyl, C 1 -C2alkyl, C 1 alkyl, C2alkyl, Csalkyl, C4alkyl, Csalkyl, Cealkyl, methyl, ethyl, n- propyl, isopropyl, n-butyl, isosbutyl, sec-butyl, pentanyl, hexanyl, and the like.
- R 8is -CH3.
- R 8 in the compounds of formula (I)is -C 1 -Ce alkoxyl, such as, for example, -C 1 -Cealkoxyl, -C 1 -Csalkoxyl, -C 1 -C4alkoxyl, -C 1 -Csalkoxyl, - C 1 -C2alkoxyl, -C 1 alkoxyl, -C2alkoxyl, -Csalkoxyl, -C4alkoxyl, -Csalkoxyl, -Cealkoxyl, - OCH3, -OCH2CH3, -propoxyl, and the like.
- R 8is -OCH3.
- R 8 in the compounds of formula (I)is -cycloalkyl, such as, for example, cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, or cycloheptyl, and the like.
- R 7is H and R 8 is H.
- R 7is -F and R 8 is H.
- R 7is -Cl and R 8 is H.
- R 7is -CH3 and R 8 is H.
- R 7is -OCH3 and R 8 is H.
- R 7is -H and R 8 is - F.
- R 7is -H and R 8 is - Cl.
- R 7is -H and R 8 is - CH 3 .
- R 7is -H and R 8 is OCH3.
- the disclosureis directed to the compounds of formula (I) that are compounds of formula (IA): or a pharmaceutically acceptable salt thereof, wherein
- Q 2 and Q 4are each N, or one of Q 2 or Q 4 is N and the other is CR 5a ;
- R 5ais H, F, -SO2CH3, or -CN;
- R 5is H or CH3
- R 7is H, F, or OCH3, and R 1 and R 2 are as described above for formula (I).
- R 5is H.
- R 5is CH3.
- R 7is H.
- R 7is F.
- R 7is OCH3.
- Q 2 and Q 4are each N.
- one of Q 2 or Q 4is N and the other is CR 5a .
- R 5ais H or F.
- one of Q 2 or Q 4is N and the other is CR 5a , R 5a is H.
- one of Q 2 or Q 4is N and the other is CR 5a , R 5a is F.
- one of Q 2 or Q 4is N and the other is CR 5a , R 5a is CN or SO2CH3.
- one of Q 2 or Q 4is N and the other is CR 5a , R 5a is CN.
- one of Q 2 or Q 4is N and the other is CR 5a , R 5a is SO2CH3.
- the disclosureis directed to the compounds of formula (I) that are compounds of formula (IB):
- R 7is H or -OC 1 -ealkyl
- R 5ais halo, or -CN.
- the disclosureis directed to the compounds of formula (I) that are compounds of formula (IC): or a pharmaceutically acceptable salt thereof, wherein
- R 1is optionally substituted C 1 -Cealkyl, or NH2;
- R 7is H or -OC 1 -C6alkyl
- R 5ais halo, or -CN.
- R 7 in the compounds of formula (IB) or formula (IC)is H or -OC 1 -6alkyl.
- R 7 in the compounds of formula (IB) or formula (IC)is H.
- R 7 in the compounds of formula (IB) or formula (IC)is -OC 1 -Cealkyl, such as, for example, -OC 1 -Cealkyl, -OC 1 -Csalkyl, -OC 1 -C4alkyl, -OC 1 - C 3 alkyl, -OC 1 -C 2 alkyl, -OC 1 alkyl, -OC 2 alkyl, -OC 3 alkyl, -OC 4 alkyl, -OC 5 alkyl, -OC 6 alkyl, - OCH 3 , -OCH 2 CH 3 , -propoxyl, and the like.
- R 7 in the compounds of formula (IB) or formula (IC)is -OCH 3 .
- R 5a in the compounds of formula (IB) or formula (IC)is halo or -CN.
- R 5a in the compounds of formula (IB) or formula (IC)is -CN.
- R 5a in the compounds of formula (IB) or formula (IC)is halogen, such as, for example, -F, -Cl, -Br, or -I.
- R 5a in the compounds of formula (IB) or formula (IC)is F.
- R 5a in the compounds of formula (IB) or formula (IC)is Cl.
- R 5a in the compounds of formula (IB) or formula (IC)is Br.
- R 1 in the compounds of formula (I) or formula (IC)is optionally substituted C 1 -Cealkyl, orNH 2 .
- R 1is -CH 3 .
- R 1is -CH2NH2.
- R 1is -CH2NHCH3.
- R 1is -CH2N(CH3)2.
- R 1is -NH2.
- the compound of formula (I)is:
- the compound of formula (IB)is a compound of formula (IB1), or a pharmaceutically acceptable salt thereof.
- the compound of formula (IB)is a compound of formula
- the compound of formula (IB)is a compound of formula (IB3), or a pharmaceutically acceptable salt thereof.
- the compound of formula (IB)is a compound of formula
- the compound of formula (IB)is a compound of formula
- the compound of formula (IB)is a compound of formula (IB6), or a pharmaceutically acceptable salt thereof.
- the compound of formula (I)is: pharmaceutically acceptable salt thereof.
- the compound of formula (I)is: or a pharmaceutically acceptable salt thereof.
- the compound of formula (I)is: pharmaceutically acceptable salt thereof.
- the compound of formula (I)is:
- the compound of formula (I)is: or a pharmaceutically acceptable salt thereof.
- the compound of formula (I)is: or a pharmaceutically acceptable salt thereof.
- the compound of formula (I)is: pharmaceutically acceptable salt thereof.
- the disclosureis directed to the compounds shown in the
- references herein to formula (I) or subgenera thereofare meant to encompass the identified formula and any subgenera of those formula disclosed herein.
- references to formula (I)also encompass subgenera formula IA, IB, IB1, IB2, IB3, IB4, IB5, IB6 and IC.
- Stereoisomers of compounds of formula (I)are also contemplated by the present disclosure.
- the disclosureencompasses all stereoisomers and constitutional isomers of any compound disclosed or claimed herein, including all enantiomers and diastereomers, or mixtures thereof.
- compositions and methods of administrationare provided.
- the subject pharmaceutical compositionsare typically formulated to provide a therapeutically effective amount of a compound of the present disclosure as the active ingredient, or a pharmaceutically acceptable salt, ester, prodrug, solvate, hydrate or derivative thereof.
- the pharmaceutical compositionscontain a compound of the present disclosure or a pharmaceutically acceptable salt thereof, and one or more pharmaceutically acceptable excipients, carriers, including inert solid diluents and fillers, diluents, including sterile aqueous solution and various organic solvents, permeation enhancers, solubilizers and adjuvants.
- compositionscan be administered alone or in combination with one or more other agents, which are also typically administered in the form of pharmaceutical compositions.
- the one or more compounds of the invention and other agent(s)may be mixed into a preparation or both components may be formulated into separate preparations to use them in combination separately or at the same time.
- the concentration of one or more compounds provided in the pharmaceutical compositions of the present inventionis less than 100%, 90%, 80%, 70%, 60%, 50%, 40%, 30%, 20%, 19%, 18%, 17%, 16%, 15%, 14%, 13%, 12%, 11%, 10%, 9%, 8%, 7%, 6%, 5%, 4%, 3%, 2%, 1%, 0.9%, 0.8%, 0.7%, 0.6%, 0.5%, 0.4%, 0.3%, 0.2%, 0.1%, 0.09%, 0.08%, 0.07%, 0.06%, 0.05%, 0.04%, 0.03%, 0.02%, 0.01%, 0.009%, 0.008%, 0.007%, 0.006%, 0.005%, 0.004%, 0.003%, 0.002%, 0.001%, 0.0009%, 0.0008%, 0.0007%, 0.0006%, 0.0005%, 0.0004%, 0.0003%, 0.0002%, or 0.0001% (or a number in the range defined by and including any two numbers above)
- the concentration of one or more compounds of the inventionis greater than 90%, 80%, 70%, 60%, 50%, 40%, 30%, 20%, 19.75%, 19.50%, 19.25%, 19%, 18.75%, 18.50%, 18.25% 18%, 17.75%, 17.50%, 17.25% 17%, 16.75%, 16.50%, 16.25%, 16%, 15.75%, 15.50%, 15.25% 15%, 14.75%, 14.50%, 14.25% 14%, 13.75%, 13.50%, 13.25%, 13%, 12.75%, 12.50%, 12.25%, 12%, 11.75%, 11.50%, 11.25% 11%, 10.75%, 10.50%, 10.25% 10%, 9.75%, 9.50%, 9.25%, 9%, 8.75%, 8.50%, 8.25% 8%, 7.75%, 7.50%, 7.25%, 7%, 6.75%, 6.50%, 6.25%, 6%, 5.75%, 5.50%, 5.25%, 5%
- the concentration of one or more compounds of the inventionis in the range from approximately 0.0001% to approximately 50%, approximately 0.001% to approximately 40%, approximately 0.01% to approximately 30%, approximately 0.02% to approximately 29%, approximately 0.03% to approximately 28%, approximately 0.04% to approximately 27%, approximately 0.05% to approximately 26%, approximately 0.06% to approximately 25%, approximately 0.07% to approximately 24%, approximately 0.08% to approximately 23%, approximately 0.09% to approximately 22%, approximately 0.1% to approximately 21%, approximately 0.2% to approximately 20%, approximately 0.3% to approximately 19%, approximately 0.4% to approximately 18%, approximately 0.5% to approximately 17%, approximately 0.6% to approximately 16%, approximately 0.7% to approximately 15%, approximately 0.8% to approximately 14%, approximately 0.9% to approximately 12%, approximately 1% to approximately 10% w/w, w/v or v/v.
- the concentration of one or more compounds of the inventionis in the range from approximately 0.001% to approximately 10%, approximately 0.01% to approximately 5%, approximately 0.02% to approximately 4.5%, approximately 0.03% to approximately 4%, approximately 0.04% to approximately 3.5%, approximately 0.05% to approximately 3%, approximately 0.06% to approximately 2.5%, approximately 0.07% to approximately 2%, approximately 0.08% to approximately 1.5%, approximately 0.09% to approximately 1%, approximately 0.1% to approximately 0.9% w/w, w/v or v/v.
- the amount of one or more compounds of the inventionis equal to or less than 10 g, 9.5 g, 9.0 g, 8.5 g, 8.0 g, 7.5 g, 7.0 g, 6.5 g, 6.0 g, 5.5 g, 5.0 g, 4.5 g, 4.0 g, 3.5 g, 3.0 g, 2.5 g, 2.0 g, 1.5 g, 1.0 g, 0.95 g, 0.9 g, 0.85 g, 0.8 g, 0.75 g, 0.7 g, 0.65 g, 0.6 g, 0.55 g, 0.5 g, 0.45 g, 0.4 g, 0.35 g, 0.3 g, 0.25 g, 0.2 g, 0.15 g, 0.1 g, 0.09 g, 0.08 g, 0.07 g, 0.06 g, 0.05 g, 0.04 g, 0.03 g, 0.02 g, 0.01 g, 0.009
- the amount of one or more compounds of the inventionis more than 0.0001 g, 0.0002 g, 0.0003 g, 0.0004 g, 0.0005 g, 0.0006 g, 0.0007 g, 0.0008 g, 0.0009 g, 0.001 g, 0.0015 g, 0.002 g, 0.0025 g, 0.003 g, 0.0035 g, 0.004 g, 0.0045 g, 0.005 g, 0.0055 g, 0.006 g, 0.0065 g, 0.007 g, 0.0075 g, 0.008 g, 0.0085 g, 0.009 g, 0.0095 g, 0.01 g, 0.015 g, 0.02 g, 0.025 g, 0.03 g, 0.035 g, 0.04 g, 0.045 g, 0.05 g, 0.055 g, 0.06 g, 0.065 g, 0.07 g,
- the amount of one or more compounds of the inventionis in the range of 0.0001-10 g, 0.0005-9 g, 0.001-8 g, 0.005-7 g, 0.01-6 g, 0.05-5 g, 0.1-4 g, 0.5-4 g, or 1-3 g.
- the compounds according to the inventionare effective over a wide dosage range.
- dosagesfrom 0.01 to 1000 mg, from 0.5 to 100 mg, from 1 to 50 mg per day, and from 5 to 40 mg per day are examples of dosages that may be used.
- An exemplary dosageis 10 to 30 mg per day. The exact dosage will depend upon the route of administration, the form in which the compound is administered, the subject to be treated, the body weight of the subject to be treated, and the preference and experience of the attending physician.
- the amounts of the compounds described hereinare set forth on a free base basis. That is, the amounts indicate that amount of the compound administered, exclusive of, for example, solvent (such as in solvates) or counterions (such as in pharmaceutically acceptable salts).
- compositions for oral administrationare provided.
- the inventionprovides a pharmaceutical composition for oral administration containing a compound of the invention, and a pharmaceutical excipient suitable for oral administration.
- the inventionprovides a solid pharmaceutical composition for oral administration containing: (i) an effective amount of a compound of the invention; optionally (ii) an effective amount of a second agent; and (iii) a pharmaceutical excipient suitable for oral administration.
- the compositionfurther contains: (iv) an effective amount of a third agent.
- the pharmaceutical compositionmay be a liquid pharmaceutical composition suitable for oral consumption.
- Pharmaceutical compositions of the invention suitable for oral administrationcan be presented as discrete dosage forms, such as capsules, cachets, or tablets, or liquids or aerosol sprays each containing a predetermined amount of an active ingredient as a powder or in granules, a solution, or a suspension in an aqueous or non-aqueous liquid, an oil-in- water emulsion, or a water-in-oil liquid emulsion.
- Such dosage formscan be prepared by any of the methods of pharmacy, but all methods include the step of bringing the active ingredient into association with the carrier, which constitutes one or more necessary ingredients.
- compositionsare prepared by uniformly and intimately admixing the active ingredient with liquid carriers or finely divided solid carriers or both, and then, if necessary, shaping the product into the desired presentation.
- a tabletcan be prepared by compression or molding, optionally with one or more accessory ingredients.
- Compressed tabletscan be prepared by compressing in a suitable machine the active ingredient in a free- flowing form such as powder or granules, optionally mixed with an excipient such as, but not limited to, a binder, a lubricant, an inert diluent, and/or a surface active or dispersing agent.
- Molded tabletscan be made by molding in a suitable machine a mixture of the powdered compound moistened with an inert liquid diluent.
- This inventionfurther encompasses anhydrous pharmaceutical compositions and dosage forms comprising an active ingredient, since water can facilitate the degradation of some compounds.
- watermay be added (e.g., 5%) in the pharmaceutical arts as a means of simulating long-term storage in order to determine characteristics such as shelf- life or the stability of formulations over time.
- Anhydrous pharmaceutical compositions and dosage forms of the inventioncan be prepared using anhydrous or low moisture containing ingredients and low moisture or low humidity conditions.
- Pharmaceutical compositions and dosage forms of the invention which contain lactosecan be made anhydrous if substantial contact with moisture and/or humidity during manufacturing, packaging, and/or storage is expected.
- An anhydrous pharmaceutical compositionmay be prepared and stored such that its anhydrous nature is maintained.
- anhydrous compositionsmay be packaged using materials known to prevent exposure to water such that they can be included in suitable formulary kits.
- suitable packaginginclude, but are not limited to, hermetically sealed foils, plastic or the like, unit dose containers, blister packs, and strip packs.
- An active ingredientcan be combined in an intimate admixture with a pharmaceutical carrier according to conventional pharmaceutical compounding techniques.
- the carriercan take a wide variety of forms depending on the form of preparation desired for administration.
- any of the usual pharmaceutical mediacan be employed as carriers, such as, for example, water, glycols, oils, alcohols, flavoring agents, preservatives, coloring agents, and the like in the case of oral liquid preparations (such as suspensions, solutions, and elixirs) or aerosols; or carriers such as starches, sugars, micro-crystalline cellulose, diluents, granulating agents, lubricants, binders, and disintegrating agents can be used in the case of oral solid preparations, in some embodiments without employing the use of lactose.
- suitable carriersinclude powders, capsules, and tablets, with the solid oral preparations. If desired, tablets can be coated by standard aqueous or nonaqueous techniques.
- Binders suitable for use in pharmaceutical compositions and dosage formsinclude, but are not limited to, com starch, potato starch, or other starches, gelatin, natural and synthetic gums such as acacia, sodium alginate, alginic acid, other alginates, powdered tragacanth, guar gum, cellulose and its derivatives (e.g., ethyl cellulose, cellulose acetate, carboxymethyl cellulose calcium, sodium carboxymethyl cellulose), polyvinyl pyrrolidone, methyl cellulose, pre-gelatinized starch, hydroxypropyl methyl cellulose, microcrystalline cellulose, and mixtures thereof.
- natural and synthetic gumssuch as acacia, sodium alginate, alginic acid, other alginates, powdered tragacanth, guar gum, cellulose and its derivatives (e.g., ethyl cellulose, cellulose acetate, carboxymethyl cellulose calcium, sodium carboxymethyl cellulose), polyvinyl pyr
- suitable fillers for use in the pharmaceutical compositions and dosage forms disclosed hereininclude, but are not limited to, talc, calcium carbonate (e.g., granules or powder), microcrystalline cellulose, powdered cellulose, dextrates, kaolin, mannitol, silicic acid, sorbitol, starch, pre-gelatinized starch, and mixtures thereof.
- Disintegrantsmay be used in the compositions of the invention to provide tablets that disintegrate when exposed to an aqueous environment. Too much of a disintegrant may produce tablets which may disintegrate in the bottle. Too little may be insufficient for disintegration to occur and may thus alter the rate and extent of release of the active ingredient(s) from the dosage form. Thus, a sufficient amount of disintegrant that is neither too little nor too much to detrimentally alter the release of the active ingredient(s) may be used to form the dosage forms of the compounds disclosed herein. The amount of disintegrant used may vary based upon the type of formulation and mode of administration, and may be readily discernible to those of ordinary skill in the art.
- Disintegrantsthat can be used to form pharmaceutical compositions and dosage forms of the invention include, but are not limited to, agar-agar, alginic acid, calcium carbonate, microcrystalline cellulose, croscarmellose sodium, crospovidone, polacrilin potassium, sodium starch glycolate, potato or tapioca starch, other starches, pre-gelatinized starch, other starches, clays, other algins, other celluloses, gums or mixtures thereof.
- Lubricantswhich can be used to form pharmaceutical compositions and dosage forms of the invention include, but are not limited to, calcium stearate, magnesium stearate, mineral oil, light mineral oil, glycerin, sorbitol, mannitol, polyethylene glycol, other glycols, stearic acid, sodium lauryl sulfate, talc, hydrogenated vegetable oil (e.g., peanut oil, cottonseed oil, sunflower oil, sesame oil, olive oil, com oil, and soybean oil), zinc stearate, ethyl oleate, ethyl laureate, agar, or mixtures thereof.
- Additional lubricantsinclude, for example, a syloid silica gel, a coagulated aerosol of synthetic silica, or mixtures thereof.
- a lubricantcan optionally be added, in an amount of less than about 1 weight percent of the pharmaceutical composition.
- the active ingredient thereinmay be combined with various sweetening or flavoring agents, coloring matter or dyes and, if so desired, emulsifying and/or suspending agents, together with such diluents as water, ethanol, propylene glycol, glycerin and various combinations thereof.
- the tabletscan be uncoated or coated by known techniques to delay disintegration and absorption in the gastrointestinal tract and thereby provide a sustained action over a longer period.
- a time delay materialsuch as glyceryl monostearate or glyceryl distearate can be employed.
- Formulations for oral usecan also be presented as hard gelatin capsules wherein the active ingredient is mixed with an inert solid diluent, for example, calcium carbonate, calcium phosphate or kaolin, or as soft gelatin capsules wherein the active ingredient is mixed with water or an oil medium, for example, peanut oil, liquid paraffin or olive oil.
- Surfactantwhich can be used to form pharmaceutical compositions and dosage forms of the invention include, but are not limited to, hydrophilic surfactants, lipophilic surfactants, and mixtures thereof. That is, a mixture of hydrophilic surfactants may be employed, a mixture of lipophilic surfactants may be employed, or a mixture of at least one hydrophilic surfactant and at least one lipophilic surfactant may be employed.
- a suitable hydrophilic surfactantmay generally have an HLB value of at least 10, while suitable lipophilic surfactants may generally have an HLB value of or less than about 10.
- An empirical parameter used to characterize the relative hydrophilicity and hydrophobicity of non-ionic amphiphilic compoundsis the hydrophilic-lipophilic balance ("HLB" value).
- HLBhydrophilic-lipophilic balance
- Surfactants with lower HLB valuesare more lipophilic or hydrophobic, and have greater solubility in oils, while surfactants with higher HLB values are more hydrophilic, and have greater solubility in aqueous solutions.
- Hydrophilic surfactantsare generally considered to be those compounds having an HLB value greater than about 10, as well as anionic, cationic, or zwitterionic compounds for which the HLB scale is not generally applicable.
- lipophilic (i.e., hydrophobic) surfactantsare compounds having an HLB value equal to or less than about 10.
- HLB value of a surfactantis merely a rough guide generally used to enable formulation of industrial, pharmaceutical and cosmetic emulsions.
- Hydrophilic surfactantsmay be either ionic or non-ionic. Suitable ionic surfactants include, but are not limited to, alkylammonium salts; fusidic acid salts; fatty acid derivatives of amino acids, oligopeptides, and polypeptides; glyceride derivatives of amino acids, oligopeptides, and polypeptides; lecithins and hydrogenated lecithins; lysolecithins and hydrogenated lysolecithins; phospholipids and derivatives thereof; lysophospholipids and derivatives thereof; carnitine fatty acid ester salts; salts of alkylsulfates; fatty acid salts; sodium docusate; acyl lactylates; mono- and di-acetylated tartaric acid esters of mono- and di-glycerides; succinylated mono- and di-glycerides; citric acid esters of mono- and diglycerides;
- ionic surfactantsinclude, by way of example: lecithins, lysolecithin, phospholipids, lysophospholipids and derivatives thereof; carnitine fatty acid ester salts; salts of alkyl sulfates; fatty acid salts; sodium docusate; acylactylates; mono- and di-acetylated tartaric acid esters of mono- and di-glycerides; succinylated mono- and di-glycerides; citric acid esters of mono- and di-glycerides; and mixtures thereof.
- Ionic surfactantsmay be the ionized forms of lecithin, lysolecithin, phosphatidylcholine, phosphatidylethanolamine, phosphatidylglycerol, phosphatidic acid, phosphatidylserine, lysophosphatidylcholine, lysophosphatidylethanolamine, lysophosphatidylglycerol, lysophosphatidic acid, lysophosphatidylserine, PEG- phosphatidylethanolamine, PVP -phosphatidylethanolamine, lactylic esters of fatty acids, stearoyl-2-lactylate, stearoyl lactylate, succinylated monoglycerides, mono/diacetylated tartaric acid esters of mono/diglycerides, citric acid esters of mono/diglycerides, cholyl sarcosine, caproate,
- Hydrophilic non-ionic surfactantsmay include, but are not limited to, alkylglucosides; alkylmaltosides; alkylthioglucosides; lauryl macrogolglycerides; polyoxyalkylene alkyl ethers such as polyethylene glycol alkyl ethers; polyoxyalkylene alkylphenols such as polyethylene glycol alkyl phenols; polyoxyalkylene alkyl phenol fatty acid esters such as polyethylene glycol fatty acids monoesters and polyethylene glycol fatty acids diesters; polyethylene glycol glycerol fatty acid esters; polyglycerol fatty acid esters; polyoxyalkylene sorbitan fatty acid esters such as polyethylene glycol sorbitan fatty acid esters; hydrophilic transesterification products of a polyol with at least one member of the group consisting of glycerides, vegetable oils, hydrogenated vegetable oils, fatty acids, and sterols; polyoxyethylene glycol sorbit
- hydrophilic-non-ionic surfactantsinclude, without limitation, PEG- 10 laurate, PEG- 12 laurate, PEG-20 laurate, PEG-32 laurate, PEG-32 dilaurate, PEG- 12 oleate, PEG- 15 oleate, PEG-20 oleate, PEG-20 di oleate, PEG-32 oleate, PEG-200 oleate, PEG-400 oleate, PEG- 15 stearate, PEG-32 distearate, PEG-40 stearate, PEG- 100 stearate, PEG-20 dilaurate, PEG-25 glyceryl trioleate, PEG-32 dioleate, PEG-20 glyceryl laurate, PEG-30 glyceryl laurate, PEG-20 glyceryl stearate, PEG-20 glyceryl oleate, PEG-30 glyceryl oleate, PEG-30 gly
- Suitable lipophilic surfactantsinclude, by way of example only: fatty alcohols; glycerol fatty acid esters; acetylated glycerol fatty acid esters; lower alcohol fatty acids esters; propylene glycol fatty acid esters; sorbitan fatty acid esters; polyethylene glycol sorbitan fatty acid esters; sterols and sterol derivatives; polyoxyethylated sterols and sterol derivatives; polyethylene glycol alkyl ethers; sugar esters; sugar ethers; lactic acid derivatives of mono- and di-glycerides; hydrophobic transesterification products of a polyol with at least one member of the group consisting of glycerides, vegetable oils, hydrogenated vegetable oils, fatty acids and sterols; oil-soluble vitamins/vitamin derivatives; and mixtures thereof.
- preferred lipophilic surfactantsinclude glycerol fatty acid esters, propylene glycol fatty acid esters, and mixtures thereof, or are hydrophobic transesterification products of a polyol with at least one member of the group consisting of vegetable oils, hydrogenated vegetable oils, and triglycerides.
- the compositionmay include a solubilizer to ensure good solubilization and/or dissolution of the compound of the present invention and to minimize precipitation of the compound of the present invention. This can be especially important for compositions for non-oral use, e.g., compositions for injection.
- a solubilizermay also be added to increase the solubility of the hydrophilic drug and/or other components, such as surfactants, or to maintain the composition as a stable or homogeneous solution or dispersion.
- solubilizersinclude, but are not limited to, the following: alcohols and polyols, such as ethanol, isopropanol, butanol, benzyl alcohol, ethylene glycol, propylene glycol, butanediols and isomers thereof, glycerol, pentaerythritol, sorbitol, mannitol, transcutol, dimethyl isosorbide, polyethylene glycol, polypropylene glycol, polyvinylalcohol, hydroxypropyl methylcellulose and other cellulose derivatives, cyclodextrins and cyclodextrin derivatives; ethers of polyethylene glycols having an average molecular weight of about 200 to about 6000, such as tetrahydrofurfuryl alcohol PEG ether (glycofurol) or methoxy PEG ; amides and other nitrogen-containing compounds such as 2- pyrrolidone, 2-piperidone,
- solubilizersmay also be used. Examples include, but not limited to, triacetin, tri ethyl citrate, ethyl oleate, ethyl caprylate, dimethylacetamide, N- methylpyrrolidone, N-hydroxyethylpyrrolidone, polyvinylpyrrolidone, hydroxypropyl methylcellulose, hydroxypropyl cyclodextrins, ethanol, polyethylene glycol 200-100, glycofurol, transcutol, propylene glycol, and dimethyl isosorbide. Particularly preferred solubilizers include sorbitol, glycerol, triacetin, ethyl alcohol, PEG-400, glycofurol and propylene glycol.
- the amount of solubilizer that can be includedis not particularly limited.
- the amount of a given solubilizermay be limited to a bioacceptable amount, which may be readily determined by one of skill in the art.
- the solubilizercan be in a weight ratio of 10%, 25%o, 50%), 100%o, or up to about 200%> by weight, based on the combined weight of the drug, and other excipients.
- solubilizermay also be used, such as 5%>, 2%>, 1%) or even less.
- the solubilizermay be present in an amount of about 1%> to about 100%, more typically about 5%> to about 25%> by weight.
- the compositioncan further include one or more pharmaceutically acceptable additives and excipients.
- additives and excipientsinclude, without limitation, detackifiers, anti-foaming agents, buffering agents, polymers, antioxidants, preservatives, chelating agents, viscomodulators, tonicifiers, flavorants, colorants, odorants, opacifiers, suspending agents, binders, fillers, plasticizers, lubricants, and mixtures thereof.
- an acid or a basemay be incorporated into the composition to facilitate processing, to enhance stability, or for other reasons.
- pharmaceutically acceptable basesinclude amino acids, amino acid esters, ammonium hydroxide, potassium hydroxide, sodium hydroxide, sodium hydrogen carbonate, aluminum hydroxide, calcium carbonate, magnesium hydroxide, magnesium aluminum silicate, synthetic aluminum silicate, synthetic hydrocalcite, magnesium aluminum hydroxide, diisopropylethylamine, ethanolamine, ethylenediamine, triethanolamine, triethylamine, triisopropanolamine, trimethylamine, tri s(hydroxymethyl)aminom ethane (TRIS) and the like.
- basesthat are salts of a pharmaceutically acceptable acid, such as acetic acid, acrylic acid, adipic acid, alginic acid, alkanesulfonic acid, amino acids, ascorbic acid, benzoic acid, boric acid, butyric acid, carbonic acid, citric acid, fatty acids, formic acid, fumaric acid, gluconic acid, hydroquinosulfonic acid, isoascorbic acid, lactic acid, maleic acid, oxalic acid, parabromophenylsulfonic acid, propionic acid, p-toluenesulfonic acid, salicylic acid, stearic acid, succinic acid, tannic acid, tartaric acid, thioglycolic acid, toluenesulfonic acid, uric acid, and the like.
- a pharmaceutically acceptable acidsuch as acetic acid, acrylic acid, adipic acid, alginic acid, alkanesulfonic acid, amino acids,
- Salts of polyprotic acidssuch as sodium phosphate, disodium hydrogen phosphate, and sodium dihydrogen phosphate can also be used.
- the cationcan be any convenient and pharmaceutically acceptable cation, such as ammonium, alkali metals, alkaline earth metals, and the like.
- Examplemay include, but not limited to, sodium, potassium, lithium, magnesium, calcium and ammonium.
- Suitable acidsare pharmaceutically acceptable organic or inorganic acids.
- suitable inorganic acidsinclude hydrochloric acid, hydrobromic acid, hydriodic acid, sulfuric acid, nitric acid, boric acid, phosphoric acid, and the like.
- suitable organic acidsinclude acetic acid, acrylic acid, adipic acid, alginic acid, alkanesulfonic acids, amino acids, ascorbic acid, benzoic acid, boric acid, butyric acid, carbonic acid, citric acid, fatty acids, formic acid, fumaric acid, gluconic acid, hydroquinosulfonic acid, isoascorbic acid, lactic acid, maleic acid, methanesulfonic acid, oxalic acid, para-bromophenylsulfonic acid, propionic acid, p-toluenesulfonic acid, salicylic acid, stearic acid, succinic acid, tannic acid, tartaric acid, thioglycolic acid, toluenesulfonic acid, uric acid and the like.
- compositions for injectionare provided.
- the inventionprovides a pharmaceutical composition for injection containing a compound of the present invention and a pharmaceutical excipient suitable for injection.
- a pharmaceutical composition for injectioncontaining a compound of the present invention and a pharmaceutical excipient suitable for injection.
- Components and amounts of agents in the compositionsare as described herein.
- Aqueous solutions in salineare also conventionally used for injection. Ethanol, glycerol, propylene glycol, liquid polyethylene glycol, and the like (and suitable mixtures thereof), cyclodextrin derivatives, and vegetable oils may also be employed.
- the proper fluiditycan be maintained, for example, by the use of a coating, such as lecithin, for the maintenance of the required particle size in the case of dispersion and by the use of surfactants.
- a coatingsuch as lecithin
- surfactantsfor the maintenance of the required particle size in the case of dispersion and by the use of surfactants.
- the prevention of the action of microorganismscan be brought about by various antibacterial and antifungal agents, for example, parabens, chlorobutanol, phenol, sorbic acid, thimerosal, and the like.
- Sterile injectable solutionsare prepared by incorporating the compound of the present invention in the required amount in the appropriate solvent with various other ingredients as enumerated above, as required, followed by filtered sterilization.
- dispersionsare prepared by incorporating the various sterilized active ingredients into a sterile vehicle which contains the basic dispersion medium and the required other ingredients from those enumerated above.
- certain desirable methods of preparationare vacuum-drying and freeze- drying techniques which yield a powder of the active ingredient plus any additional desired ingredient from a previously sterile-filtered solution thereof.
- compositions for topicale.g. transdermal delivery.
- the inventionprovides a pharmaceutical composition for transdermal delivery containing a compound of the present invention and a pharmaceutical excipient suitable for transdermal delivery.
- compositions of the present inventioncan be formulated into preparations in solid, semisolid, or liquid forms suitable for local or topical administration, such as gels, water soluble jellies, creams, lotions, suspensions, foams, powders, slurries, ointments, solutions, oils, pastes, suppositories, sprays, emulsions, saline solutions, dimethylsulfoxide (DMSO)-based solutions.
- DMSOdimethylsulfoxide
- carriers with higher densitiesare capable of providing an area with a prolonged exposure to the active ingredients.
- a solution formulationmay provide more immediate exposure of the active ingredient to the chosen area.
- compositionsalso may comprise suitable solid or gel phase carriers or excipients, which are compounds that allow increased penetration of, or assist in the delivery of, therapeutic molecules across the stratum corneum permeability barrier of the skin.
- suitable solid or gel phase carriers or excipientswhich are compounds that allow increased penetration of, or assist in the delivery of, therapeutic molecules across the stratum corneum permeability barrier of the skin.
- penetration- enhancing moleculesknown to those trained in the art of topical formulation.
- humectantse.g., urea
- glycolse.g., propylene glycol
- alcoholse.g., ethanol
- fatty acidse.g., oleic acid
- surfactantse.g., isopropyl myristate and sodium lauryl sulfate
- pyrrolidonese.g., isopropyl myristate and sodium lauryl sulfate
- pyrrolidonese.glycerol monolaurate, sulfoxides, terpenes (e.g., menthol)
- aminesamides, alkanes, alkanols, water, calcium carbonate, calcium phosphate, various sugars, starches, cellulose derivatives, gelatin, and polymers such as polyethylene glycols.
- transdermal delivery devicespatches
- Such transdermal patchesmay be used to provide continuous or discontinuous infusion of a compound of the present invention in controlled amounts, either with or without another agent.
- transdermal patchesfor the delivery of pharmaceutical agents is well known in the art. See, e.g., U.S. Pat. Nos. 5,023,252, 4,992,445 and 5,001,139. Such patches may be constructed for continuous, pulsatile, or on demand delivery of pharmaceutical agents.
- compositions for inhalationare provided.
- compositions for inhalation or insufflationinclude solutions and suspensions in pharmaceutically acceptable, aqueous or organic solvents, or mixtures thereof, and powders.
- the liquid or solid compositionsmay contain suitable pharmaceutically acceptable excipients as described supra.
- the compositionsare administered by the oral or nasal respiratory route for local or systemic effect.
- Compositions in preferably pharmaceutically acceptable solventsmay be nebulized by use of inert gases. Nebulized solutions may be inhaled directly from the nebulizing device or the nebulizing device may be attached to a face mask tent, or intermittent positive pressure breathing machine. Solution, suspension, or powder compositions may be administered, preferably orally or nasally, from devices that deliver the formulation in an appropriate manner.
- compositionsmay also be prepared from compositions described herein and one or more pharmaceutically acceptable excipients suitable for sublingual, buccal, rectal, intraosseous, intraocular, intranasal, epidural, or intraspinal administration. Preparations for such pharmaceutical compositions are well-known in the art.
- Administration of the compounds or pharmaceutical composition of the present inventioncan be effected by any method that enables delivery of the compounds to the site of action. These methods include oral routes, intraduodenal routes, parenteral injection (including intravenous, intraarterial, subcutaneous, intramuscular, intravascular, intraperitoneal or infusion), topical (e.g. transdermal application), rectal administration, via local delivery by catheter or stent or through inhalation. Compounds can also be administered intraadiposally or intrathecally.
- an effective dosageis in the range of about 0.001 to about 100 mg per kg body weight per day, preferably about 1 to about 35 mg/kg/day, in single or divided doses. For a 70 kg human, this would amount to about 0.05 to 7 g/day, preferably about 0.05 to about 2.5 g/day. In some instances, dosage levels below the lower limit of the aforesaid range may be more than adequate, while in other cases still larger doses may be employed without causing any harmful side effect, e.g. by dividing such larger doses into several small doses for administration throughout the day.
- a compound of the inventionis administered in a single dose.
- a compound of the inventionis administered in multiple doses. Dosing may be about once, twice, three times, four times, five times, six times, or more than six times per day. Dosing may be about once a month, once every two weeks, once a week, or once every other day. In another embodiment a compound of the invention and another agent are administered together about once per day to about 6 times per day.
- the administration of a compound of the invention and an agentcontinues for less than about 7 days. In yet another embodiment the administration continues for more than about 6, 10, 14, 28 days, two months, six months, or one year. In some cases, continuous dosing is achieved and maintained as long as necessary.
- Administration of the compounds of the inventionmay continue as long as necessary.
- a compound of the inventionis administered for more than 1, 2, 3, 4, 5, 6, 7, 14, or 28 days.
- a compound of the inventionis administered for less than 28, 14, 7, 6, 5, 4, 3, 2, or 1 day.
- a compound of the inventionis administered chronically on an ongoing basis, e.g., for the treatment of chronic effects.
- An effective amount of a compound of the inventionmay be administered in either single or multiple doses by any of the accepted modes of administration of agents having similar utilities, including rectal, buccal, intranasal and transdermal routes, by intraarterial injection, intravenously, intraperitoneally, parenterally, intramuscularly, subcutaneously, orally, topically, or as an inhalant.
- compositions of the inventionmay also be delivered via an impregnated or coated device such as a stent, for example, or an artery -inserted cylindrical polymer.
- a method of administrationmay, for example, aid in the prevention or amelioration of restenosis following procedures such as balloon angioplasty.
- compounds of the inventionmay slow or inhibit the migration and proliferation of smooth muscle cells in the arterial wall which contribute to restenosis.
- a compound of the inventionmay be administered, for example, by local delivery from the struts of a stent, from a stent graft, from grafts, or from the cover or sheath of a stent.
- a compound of the inventionis admixed with a matrix.
- Such a matrixmay be a polymeric matrix, and may serve to bond the compound to the stent.
- Polymeric matrices suitable for such useinclude, for example, lactone-based polyesters or copolyesters such as polylactide, polycaprolactonglycolide, polyorthoesters, polyanhydrides, polyaminoacids, polysaccharides, polyphosphazenes, poly (ether-ester) copolymers (e.g. PEO-PLLA); polydimethylsiloxane, poly(ethylene-vinylacetate), acrylate-based polymers or copolymers (e.g.
- Compounds of the inventionmay be applied to the surface of the stent by various methods such as dip/spin coating, spray coating, dip-coating, and/or brush-coating.
- the compoundsmay be applied in a solvent and the solvent may be allowed to evaporate, thus forming a layer of compound onto the stent.
- the compoundmay be located in the body of the stent or graft, for example in microchannels or micropores.
- stentsWhen implanted, the compound diffuses out of the body of the stent to contact the arterial wall.
- stentsmay be prepared by dipping a stent manufactured to contain such micropores or microchannels into a solution of the compound of the invention in a suitable solvent, followed by evaporation of the solvent. Excess drug on the surface of the stent may be removed via an additional brief solvent wash.
- compounds of the inventionmay be covalently linked to a stent or graft.
- a covalent linkermay be used which degrades in vivo, leading to the release of the compound of the invention. Any bio-labile linkage may be used for such a purpose, such as ester, amide or anhydride linkages.
- Compounds of the inventionmay additionally be administered intravascularly from a balloon used during angioplasty. Extravascular administration of the compounds via the pericard or via advential application of formulations of the invention may also be performed to decrease restenosis.
- the compounds of the inventionmay be administered in dosages. It is known in the art that due to intersubject variability in compound pharmacokinetics, individualization of dosing regimen is necessary for optimal therapy. Dosing for a compound of the invention may be found by routine experimentation in light of the instant disclosure. [00294] When a compound of the invention is administered in a composition that comprises one or more agents, and the agent has a shorter half- life than the compound of the invention unit dose forms of the agent and the compound of the invention may be adjusted accordingly.
- the subject pharmaceutical compositionmay, for example, be in a form suitable for oral administration as a tablet, capsule, pill, powder, sustained release formulations, solution, suspension, for parenteral injection as a sterile solution, suspension or emulsion, for topical administration as an ointment or cream or for rectal administration as a suppository.
- the pharmaceutical compositionmay be in unit dosage forms suitable for single administration of precise dosages.
- the pharmaceutical compositionwill include a conventional pharmaceutical carrier or excipient and a compound according to the invention as an active ingredient. In addition, it may include other medicinal or pharmaceutical agents, carriers, adjuvants, etc.
- Exemplary parenteral administration formsinclude solutions or suspensions of active compound in sterile aqueous solutions, for example, aqueous propylene glycol or dextrose solutions. Such dosage forms can be suitably buffered, if desired.
- FGFR receptors(FGFR1, FGFR2, FGFR3, and FGFR4) share several structural features in common, including three extracellular immunoglobulin-like (Ig) domains, a hydrophobic transmembrane domain, and an intracellular tyrosine kinase domain split by a kinase insert domain, followed by a cytoplasmic c-terminal tail (Johnson et al., Adv. Cancer Res. 60: 1-40, 1993; and Wilkie et al., Curr. Biol. 5:500-507, 1995).
- Igimmunoglobulin-like domains
- a hydrophobic transmembrane domainan intracellular tyrosine kinase domain split by a kinase insert domain, followed by a cytoplasmic c-terminal tail
- a kinase insert domainspans positions 582 to 595 of the alpha Al isoform of FGFR1.
- the kinase insert domainspans positions 585 to 598 of the FGFR2 Ille isoform.
- the kinase insert domainspans positions 576 to 589 of the FGFR3 Ille isoform.
- the kinase insert domainspans positions 571 to 584 of FGFR4 isoform 1.
- the c- terminal tail of FGFRsbegins following the end of the tyrosine kinase domain and extends to the c-terminus of the protein.
- FGFR proteins and nucleic acids encoding FGFR proteinsare known in the art. Signaling by FGFRs regulates key biological processes including cell proliferation, survival, migration, and differentiation. Dysregulation of a FGFR gene, a FGFR protein, or expression or activity, or level of the same, has been associated with many types of cancer. For example, dysregulation of FGFRs can occur by multiple mechanisms, such as FGFR gene overexpression, FGFR gene amplification, activating mutations (e.g., point mutations or truncations), and chromosomal rearrangements that lead to FGFR fusion proteins. Dysregulation of a FGFR gene, a FGFR protein, or expression or activity, or level of the same, can result in (or cause in part) the development of a variety of different FGFR-associated cancers.
- FGFR fusion proteinsare known in the art. See, e.g., Baroy et al., PloS One; 1 l(9):e0163859. doi: 10.1371/journal. pone.0163859, 2016; Ren et al., Int. J. Cancer, 139(4):836-40, 2016; Marchwicka et al., Cell Biosci., 6:7. doi: 10.1186/sl3578-016-0075-9, 2016; PCT Patent Application Publication No. WO 2014/071419A2; U.S. Patent Application Publication No. 2015/0366866A1; PCT Patent Application Publication No. WO 2016/084883 Al; PCT Patent Application Publication No.
- FGFR point mutationsare known in the art. See, e.g., UniParc entry UPI00000534B8; UniParc entry UPI0000001COF; UniParc entry UPI000002A99A; UniParc entry UPI000012A72A; UniParc entry UPI000059D1C2; UniParc entry UPI000002A9AC; Uniparc entry UPI000012A72C; Uniparc entry UPI000012A72D; Uniparc entry UPI000013EOB8; Uniparc entry UPI0001CE06A3; Gen bank entry BAD92868.1; Ang et al., Diagn. Mo/. Patho/. Feb 24, 2014; U.S. Patent Application Publication No.
- EP2203449B1Yoza et al., Genes Cells., (10): 1049-1058, 2016; Bunney et al., EbioMedicine, 2(3): 194-204, 2015; Byron et al., Neop/asia, 15(8):975-88, 2013;
- VQRSPDWCCSTEGPLFWGDPVQNVSGPTRWDPVGQGAGPDMARPLPLHHGTSQGALG PSHTQSGe, et al, Am J Cancer Res. 7(7): 1540-1553, 2017. PMID: 28744403; Jiao et al, Nat Genet, 45(12): 1470-1473, 2013. doi: 10.1038/ng.2813; Jusakul et al, Cancer Discov. 7(10): 1116-1135, 2017. doi: 10.1158/2159-8290.CD-17-0368; Guyard et al, Respir Res., 18(1): 120, 2018.
- Compounds of the disclosurehave been found to inhibit FGFR1, FGFR2, FGFR3, and/or FGFR4 and are therefore believed to be useful for treating diseases and disorders which can be treated with an inhibitor of FGFR1, FGFR2, FGFR3 and/or FGFR4.
- compounds of the disclosurecan be useful in treating FGFR-associated diseases and disorders, e.g., proliferative disorders such as cancers, including hematological cancers and solid tumor, and angiogenesis-related disorders.
- Compounds of the disclosuremay also be useful in treating disorders arising from autosomal dominant mutations in FGFR, e.g., FGFR3, including, for example, developmental disorders.
- AchondroplasiaAchondroplasia
- HchHypochondroplasia
- SADDANSevere Achondroplasia with Developmental Delay and Acanthosis Nigricans
- TDThanatophoric dysplasia
- Non-limiting examples of FGFR-associated diseases and disordersinclude Acanthosis nigricans, Achondroplasia, Apert syndrome, Beare-Stevenson syndrome (BSS), Camptodactyly, tall stature, and hearing loss syndrome (CATSHL) syndrome, cleft lip and palate, congenital heart disease (e.g., associated with ambiguous genitalia), craniosynostosis, Crouzon syndrome, ectrodactyly, encephalocraniocutaneous lipomatosis, Hartsfield syndrome, hypochondroplasia, hypogonadoropic hypogonadism (e.g., hypogonadotropic hypogonadism 2 with or without anosmia, Kailman syndrome), ichthyosis vulgaris and/or atopic dermatitis, Jackson-Weiss syndrome, lethal pulmonary acinar dysplasia, microphthalmia, Muenke coronal craniosynostosis, osteoglophonic
- Non-limiting examples of FGFR1 associated diseases and disordersinclude congenital heart disease (e.g., associated with ambiguous genitalia), craniosynostosis, encephalocraniocutaneous lipomatosis, Hartsfield syndrome, hypogonadoropic hypogonadism (e.g., hypogonadotropic hypogonadism 2 with or without anosmia, Kailman syndrome), ichthyosis vulgaris and/or atopic dermatitis, Jackson-Weiss syndrome, osteoglophonic dysplasia, Pfeiffer syndrome, trigonocephaly 1 (also called metopic craniosynostosis), and tumor-induced osteomalacia.
- congenital heart diseasee.g., associated with ambiguous genitalia
- craniosynostosise.g., associated with ambiguous genitalia
- encephalocraniocutaneous lipomatosise.g., ambiguous genit
- Non-limiting examples of FGFR2 -associated diseases and disordersinclude Apert syndrome, Beare-Stevenson syndrome (BSS), Crouzon syndrome, ectrodactyly, Jackson-Weiss syndrome, lethal pulmonary acinar dysplasia, Pfeiffer syndrome, and syndactyly.
- Non-limiting examples of FGFR3 -associated diseases and disordersinclude acanthosis nigricans, achondroplasia, Camptodactyly, tall stature, and hearing loss syndrome (CATSHL) syndrome, cleft lip and palate, craniosynostosis, hypochondroplasia, microphthalmia, Muenke coronal craniosynostosis, seborrheic keratosis, and thanatophoric dysplasia (e.g., type I or type II). See also, See UniParc entry UPI00000534B8; UniParc entry UPI0000001COF;Uni Pare entry UP 1000002 A99A;UniParc entry
- angiogenesis-related disordermeans a disease characterized in part by an increased number or size of blood vessels in a tissue in a subject or patient, as compared to a similar tissue from a subject not having the disease.
- angiogenesis-related disordersinclude: cancer (e.g., any of the exemplary cancers described herein, such as prostate cancer, lung cancer, breast cancer, bladder cancer, renal cancer, colon cancer, gastric cancer, pancreatic cancer, ovarian cancer, melanoma, hepatoma, sarcoma, and lymphoma), exudative macular degeneration, proliferative diabetic retinopathy, ischemic retinopathy, retinopathy of prematurity, neovascular glaucoma, crizis rubeosis, corneal neovascularization, cyclitis, sickle cell retinopathy, and pterygium.
- cancere.g., any of the exemplary cancers described herein, such as prostate cancer, lung cancer, breast cancer
- Compounds of the disclosureinhibit wild-type FGFR1, FGFR2, FGFR3, and/or FGFR4. In other aspects, compounds of the disclosure inhibit a mutated FGFR1, FGFR2, FGFR3, and/or FGFR4. In other aspects, compounds of the disclosure inhibit FGFR1, FGFR2, FGFR3, and/or FGFR4 that includes an FGFR kinase inhibitor mutation.
- the cancere.g., FGFR-associated cancer
- the canceris a hematological cancer.
- the canceris a solid tumor.
- the cancere.g., FGFR-associated cancer
- a lung cancere.g., small cell lung carcinoma, nonsmall cell lung carcinoma, squamous cell carcinoma, lung adenocarcinoma, large cell carcinoma, mesothelioma, lung neuroendocrine carcinoma, smoking-associated lung cancer
- prostate cancercolorectal cancer (e.g., rectal adenocarcinoma)
- endometrial cancere.g., endometrioid endometrial cancer, endometrial adenocarcinoma
- breast cancere.g., hormone-receptor-positive breast cancer, triple-negative breast cancer, neuroendodrine carcinoma of the breast
- skin cancere.g., melanoma, cutaneous squamous cell carcinoma, basal cell carcinoma, large squamous cell carcinoma
- gallbladder cancere.g., dedifferenti
- the cancere.g., FGFR-associated cancer
- ALLacute lymphoblastic leukemia
- AMLacute myeloid leukemia
- cancer in adolescentsadrenocortical carcinoma
- anal cancerappendix cancer
- astrocytomaatypical teratoid/rhabdoid tumor
- basal cell carcinomabile duct cancer
- bladder cancerbone cancer
- brain stem gliomabrain tumor
- breast cancerbronchial tumor
- Burkitt lymphomacarcinoid tumor
- unknown primary carcinomacardiac tumors, cervical cancer, childhood cancers, chordoma
- CLLchronic lymphocytic leukemia
- CMLchronic myelogenous leukemia
- chronic myeloproliferative neoplasmsneoplasms by site, neoplasms, colon cancer, colorectal cancer, craniopharyngiom
- a hematological canceris selected from the group consisting of leukemias, lymphomas (non-Hodgkin's lymphoma), Hodgkin's disease (also called Hodgkin's lymphoma), and myeloma, for instance, acute lymphocytic leukemia (ALL), acute myeloid leukemia (AML), acute promyelocytic leukemia (APL), chronic lymphocytic leukemia (CLL), chronic myeloid leukemia (CML), chronic myelomonocytic leukemia (CMML), chronic neutrophilic leukemia (CNL), acute undifferentiated leukemia (AUL), anaplastic large-cell lymphoma (ALCL), prolymphocytic leukemia (PML), juvenile myelomonocyctic leukemia (JMML), adult Tcell ALL, AML
- ALLacute lymphocytic leukemia
- AMLacute myeloid leukemia
- APLacute promy
- hematological cancersinclude myeloproliferative disorders (MPD) such as polycythemia vera (PV), essential thrombocytopenia (ET) and idiopathic primary myelofibrosis (IMF/IPF/PMF).
- MPDmyeloproliferative disorders
- the hematological cancere.g., the hematological cancer that is a FGFR-associated cancer
- AMLor CMML.
- the cancere.g., the FGFR-associated cancer
- the canceris a solid tumor.
- solid tumorsexamples include, for example, lung cancer (e.g., lung adenocarcinoma, non-small-cell lung carcinoma, squamous cell lung cancer), bladder cancer, colorectal cancer, brain cancer, testicular cancer, bile duct cancer cervical cancer, prostate cancer, and sparmatocytic seminomas. See, for example, Turner and Grose, Nat. Rev. Cancer, 10(2): 116-129, 2010.
- the canceris selected from the group consisting of bladder cancer, brain cancer, breast cancer, cholangiocarcinoma, head and neck cancer, lung cancer, multiple myeloma, rhabdomyosarcoma, urethral cancer, and uterine cancer. In some embodiments, the cancer is selected from the group consisting of lung cancer, breast cancer, and brain cancer.
- a FGFRl-associated canceris selected from the group consisting of lung cancer, breast cancer, and brain cancer.
- the canceris selected from the group consisting of breast cancer, uterine cancer, cholangiocarcinoma, and lung cancer.
- a FGFR2-associated canceris selected from the group consisting of breast cancer, uterine cancer, cholangiocarcinoma, and lung cancer.
- the canceris selected from the group consisting of lung cancer, bladder cancer, urethral cancer, multiple myeloma, and head and neck cancer.
- a FGFR3 -associated canceris selected from the group consisting of lung cancer, bladder cancer, urethral cancer, multiple myeloma, and head and neck cancer.
- the canceris selected from lung cancer, rhabdomyosarcoma, and breast cancer.
- a FGFR4-associated canceris selected from hepatocellular carcinoma, lung cancer, rhabdomyosarcoma, and breast cancer.
- the compounds of the disclosureare useful in treating cancers associated with amplification or overexpression of FGFR1, for example, Breast cancer or carcinoma (e.g., hormone receptor-positive breast cancer, ductal carcinoma in situ (breast)), pancreatic ductal adenocarcinoma, pancreatic exocrine carcinoma, smoking- associated lung cancer, small cell lung cancer, lung adenocarcinoma, non-small cell lung cancer, squamous cell lung cancer or carcinoma, prostate cancer or carcinoma, ovarian cancer, fallopian tube carcinoma, bladder cancer, rhabdomyosarcoma, head and neck carcinoma (e.g., head and neck squamous cell carcinoma), esophageal cancer (e.g., esophageal squamous cell carcinoma), sarcoma (e.g., osteosarcoma), hepatocellular carcinoma, renal cell carcinoma, colorectal cancer (e.g., colorectal adenocar), e.g., color
- the compounds of the disclosureare useful in treating cancers associated with amplification of FGFR2, for example, Gastric cancer, gastroesophageal junction adenocarcinoma, breast cancer (e.g., triple negative breast cancer), colon cancer, colorectal cancer (e.g., colorectal adenocarcinoma), urothelial cancer, bladder adenocarcinoma, carcinoma of unknown primary, cholangiocarcinoma, endometrial adenocarcinoma, esophageal adenocarcinoma, gallbladder carcinoma, ovarian cancer, fallopian tube carcinoma, pancreatic exocrine carcinoma, sarcoma, squamous cell carcinoma.
- Gastric cancergastroesophageal junction adenocarcinoma
- breast cancere.g., triple negative breast cancer
- colon cancercolorectal cancer
- urothelial cancere.g., colorectal adenocarcinoma
- the compounds of the disclosureare useful in treating cancers associated with overexpression of FGFR2, for example, Myxoid lipocarcinoma, rectal cancer, renal cell carcinoma, breast cancer.
- the compounds of the disclosureare useful in treating cancers associated with upregulation of activity of FGFR3, for example, Colorectal cancer, hepatocellular carcinoma, pancreatic exocrine carcinoma. In some aspects, the compounds of the disclosure are useful in treating cancers associated with overexpression of activity of FGFR3, for example, Multiple myeloma, thyroid carcinoma.
- the compounds of the disclosureare useful in treating cancers associated with amplification of activity of FGFR3, for example, Bladder cancer and salivary adenoid cystic cancer, urothelial cancer, breast cancer, carcinoid, carcinoma of unknown primary, colorectal cancer (e.g., colorectal adenocarcinoma), gallbladder carcinoma, gastric cancer, gastroesophageal junction adenocarcinoma, glioma, mesothelioma, non-small cell lung carcinoma, small cell lung cancer, ovarian cancer, fallopian tube carcinoma, pancreatic exocrine carcinoma.
- colorectal cancere.g., colorectal adenocarcinoma
- gallbladder carcinomagastric cancer
- gastroesophageal junction adenocarcinomaglioma
- mesotheliomanon-small cell lung carcinoma
- small cell lung cancersmall cell lung cancer
- ovarian cancerfallopian tube carcinoma
- the compounds of the disclosureare useful in treating cancers associated with amplification of FGFR4, for example, Rhabdomyosarcoma, prostate cancer or carcinoma, breast cancer, urothelial cancer, carcinoid, carcinoma of unknown primary, esophageal adenocarcinoma, head and neck carcinoma, hepatocellular carcinoma, non-small cell lung carcinoma, ovarian cancer, fallopian tube carcinoma, peritoneal carcinoma, renal cell carcinoma.
- the compounds of the disclosureare useful in treating cancers associated with upregulation of activity of FGFR4, for example, Colorectal cancer, hepatocellular carcinoma, adrenal carcinoma, breast cancer.
- the compounds of the disclosureare useful in treating cancers associated with overexpression of activity of FGFR4, for example, Pancreatic intraepithelial neoplasia, and pancreatic ductal adenocarcinoma.
- the compounds of the disclosureare more selective for one FGFR than for another.
- the "selectivity" of a compound for a first target over a second targetmeans that the compound has more potent activity at the first target than the second target.
- a fold selectivitycan be calculated by any method known in the art. For example, a fold selectivity can be calculated by dividing the IC50 value (or Kd value) of a compound for the second target (e.g., FGFR1) by the IC50 value (or Kd value) of the same compound for the first target (e.g., FGFR2 or FGFR3).
- An IC50 value (or Kd value)can be determined by any method known in the art.
- a compoundis first determined to have an activity of less than 500 nM for the first target. In some embodiments, a compound is first determined to have an activity of less than 500 nM for the second target.
- the compounds of the disclosureare more selective for FGFR3 than for FGFR1.
- the compoundsare at least 3-fold more selective for FGFR3 than for FGFR1.
- the compoundsare 4, 5, 6, 7, 8, 9, 10, 15, 20, 30, 40, 50, 75, 100, 200, 500, or 1000 fold more selective for FGFR3 than for FGFR1.
- the compounds of the disclosureare more selective for FGFR4 than for FGFR1.
- the compoundsare at least 3 -fold more selective for FGFR4 than for FGFR1.
- the compoundsare 4, 5, 6, 7, 8, 9, 10, 15, 20, 30, 40, 50, 75, 100, 200, 500, or 1000 fold more selective for FGFR4 than for FGFR1.
- the compounds of the disclosureare more selective for FGFR2 than for FGFR1. In some aspects, the compounds are at least 3-fold more selective for FGFR2 than for FGFR1. In some aspects, the compounds are 4, 5, 6, 7, 8, 9, 10, 15, 20, 30, 40, 50, 75, 100, 200, 500, or 1000 fold more selective for FGFR2 than for FGFR1.
- the compounds of the disclosureare more selective for a first FGFR family member (e.g., FGFR2 or FGFR3) over a second FGFR family member (e.g., FGFR1 or FGFR4).
- the compounds of the disclosureare at least 3- fold more selective for a first FGFR family member over a second FGFR family member.
- the compoundsare at least 10, 20, 30, 40, 50, 60, 70, 80, 90, 200, 300, 400, 500, 600, 700, 800, 900, or at least 1000 fold more selective for a first FGFR family member over a second FGFR family member.
- the compounds of the disclosureare more selective for a first FGFR family member (e.g., FGFR4 or FGFR3) over a second FGFR family member (e.g., FGFR1 or FGFR2).
- the compounds of the disclosureare at least 3- fold more selective for a first FGFR family member over a second FGFR family member.
- the compoundsare at least 10, 20, 30, 40, 50, 60, 70, 80, 90, 200, 300, 400, 500, 600, 700, 800, 900, or at least 1000 fold more selective for a first FGFR family member over a second FGFR family member.
- the compounds of the disclosureare more selective for an FGFR kinase over another kinase that is not an FGFR kinase.
- the compounds of the disclosureare at least 3-fold more selective for an FGFR kinase over another kinase that is not an FGFR kinase.
- the compounds of the disclosureare at least 10, 20, 30, 40, 50, 60, 70, 80, 90, 200, 300, 400, 500, 600, 700, 800, 900, or at least 1000 fold more selective for an FGFR kinase over another kinase that is not an FGFR kinase.
- Kinases that are not FGFR kinasesinclude, for example, KDR kinase and Aurora B kinase.
- the compounds of the disclosureexhibit brain and/or central nervous system (CNS) penetrance. Such compounds are capable of crossing the blood brain barrier and inhibiting a FGFR kinase in the brain and/or other CNS structures.
- the compounds provided hereinare capable of crossing the blood brain barrier in a therapeutically effective amount.
- treatment of a subject with cancere.g., a FGFR-associated cancer such as a FGFR-associated brain or CNS cancer
- administratione.g., oral administration
- the compounds provided hereinare useful for treating a primary brain tumor or metastatic brain tumor.
- a FGFR-associated primary brain tumor or metastatic brain tumore.g., a FGFR-associated primary brain tumor or metastatic brain tumor.
- the compounds of the disclosureexhibit one or more of high GI absorption, low clearance, and low potential for drug-drug interactions.
- compounds of the disclosurecan be used for treating a subject diagnosed with (or identified as having) a FGFR-associated disease or disorder (e.g., a FGFR-associated cancer) that include administering to the subject a therapeutically effective amount of a compound of the disclosure. Also provided herein are methods for treating a subject identified or diagnosed as having a FGFR-associated disease or disorder (e.g., a FGFR-associated cancer) that include administering to the subject a therapeutically effective amount of a compound of the disclosure.
- a FGFR-associated disease or disordere.g., a FGFR-associated cancer
- the subject that has been identified or diagnosed as having a FGFR-associated disease or disordere.g., a FGFR- associated cancer
- a regulatory agency-approvede.g., FDA-approved test or assay for identifying dysregulation of a FGFR gene, a FGFR kinase, or expression or activity or level of any of the same, in a subject or a biopsy sample from the subject or by performing any of the non-limiting examples of assays described herein.
- the test or assayis provided as a kit.
- the FGFR- associated disease or disorderis a FGFR-associated cancer.
- the FGFR- associated cancercan be a cancer that includes one or more FGFR inhibitor resistance mutations.
- Some embodiments of these methodsfurther include administering to the subject an additional therapy or therapeutic agent (e.g., a second FGFR inhibitor, a second compound of the disclosure, or an immunotherapy.
- the subjectwas previously treated with a first FGFR inhibitor or previously treated with another treatment.
- the subjectis determined to have a FGFR- associated disease or disorder through the use of a regulatory agency-approved, e.g., FDA approved test or assay for identifying dysregulation of a FGFR gene, a FGFR kinase, or expression or activity or level of any of the same, in a subject or a biopsy sample from the subject or by performing any of the non-limiting examples of assays described herein.
- a regulatory agency-approvede.g., FDA approved test or assay for identifying dysregulation of a FGFR gene, a FGFR kinase, or expression or activity or level of any of the same, in a subject or a biopsy sample from the subject or by performing any of the non-limiting examples of assays described herein.
- the test or assayis provided as a kit.
- an additional therapy or therapeutic agente.g., a second FGFR inhibitor, a second compound of the disclosure, or an immunotherapy.
- the subjectwas previously treated with a first FGFR inhibitor or previously treated with another anticancer treatment, e.g., at least partial resection of the tumor or radiation therapy.
- the subjectis determined to have a FGFR-associated cancer through the use of a regulatory agency-approved, e.g., FDA-approved test or assay for identifying dysregulation of a FGFR gene, a FGFR kinase, or expression or activity or level of any of the same, in a subject or a biopsy sample from the subject or by performing any of the non -limiting examples of assays described herein.
- the test or assayis provided as a kit.
- the canceris a FGFR associated cancer.
- the FGFR-associated cancercan be a cancer that includes one or more FGFR inhibitor resistance mutations.
- the canceris a FGFR associated cancer.
- the FGFR- associated cancercan be a cancer that includes one or more FGFR activating mutations.
- Also provided are methods of treating a subjectthat include performing an assay on a sample obtained from the subject to determine whether the subject has a dysregulation of a FGFR gene, a FGFR kinase, or expression or activity or level of any of the same, and administering (e.g., specifically or selectively administering) a therapeutically effective amount of a compound of the disclosure or pharmaceutically acceptable salt or solvate thereof to the subject determined to have a dysregulation of a FGFR gene, a FGFR kinase, or expression or activity or level of any of the same.
- Some embodiments of these methodsfurther include administering to the subject an additional therapy or therapeutic agent (e.g., a second FGFR inhibitor, a second compound of the disclosure, or immunotherapy).
- an additional therapy or therapeutic agente.g., a second FGFR inhibitor, a second compound of the disclosure, or immunotherapy.
- the subjectwas previously treated with a first FGFR inhibitor or previously treated with another anticancer treatment, e.g., at least partial resection of a tumor or radiation therapy.
- the subjectis a subject suspected of having a FGFR-associated disease or disorder (e.g., a FGFR-associated cancer), a subject presenting with one or more symptoms of a FGFR-associated disease or disorder (e.g., a FGFR-associated cancer), or a subject having an elevated risk of developing a FGFR-associated disease or disorder (e.g., a FGFR-associated cancer).
- the assayutilizes next generation sequencing, pyrosequencing, immunohistochemistry, or break apart FISH analysis.
- the assayis a regulatory agency-approved assay, e.g., FDA-approved kit.
- the assayis a liquid biopsy.
- the dysregulation of a FGFR gene, a FGFR kinase, or expression or activity or level of any of the sameincludes one or more FGFR inhibitor resistance mutations.
- Also provided herein are methods of selecting a treatment for a subjectwherein the methods include a step of performing an assay on a sample obtained from the subject to determine whether the subject has a dysregulation of a FGFR gene, a FGFR kinase, or expression or activity or level of any of the same (e.g., one or more FGFR inhibitor resistance mutations), and identifying or diagnosing a subject determined to have a dysregulation of a FGFR gene, a FGFR kinase, or expression or activity or level of any of the same, as having a FGFR-associated cancer. Some embodiments further include administering the selected treatment to the subject identified or diagnosed as having a FGFR-associated cancer.
- the selected treatmentcan include administration of a therapeutically effective amount of a compound of the disclosure to the subject identified or diagnosed as having a FGFR-associated cancer.
- the assayis an in vitro assay.
- an assay that utilizes the next generation sequencing, immunohistochemistry, or break apart FISH analysisis included in the assay.
- the assayis a regulatory agency-approved, e.g., FDA-approved, kit.
- the assayis a liquid biopsy.
- Also provided herein are methods of treating a FGFR-associated cancer in a subjectthat include (a) administering one or more (e.g., two or more, three or more, four or more, five or more, or ten or more) doses of a first FGFR kinase inhibitor to a subject identified or diagnosed as having a FGFR associated cancer (e.g., any of the types of FGFR- associated cancers described herein) (e.g., identified or diagnosed as having a FGFR- associated cancer using any of the exemplary methods described herein or known in the art); (b) after step (a), determining a level of circulating tumor DNA in a biological sample (e.g., a biological sample comprising blood, serum, or plasma) obtained from the subject; (c) administering a therapeutically effective amount of a second FGFR inhibitor or a compound of the disclosure as a monotherapy or in conjunction with an additional therapy or therapeutic agent to a subject identified as having about the same or an elevated level of a biological sample (
- the reference level of circulating tumor DNAis a level of circulating tumor DNA in a biological sample obtained from the subject prior to step (a). Some embodiments of these methods further include determining the level of circulating tumor DNA in the biological sample obtained from the subject prior to step (a).
- the reference level of circulating tumor DNAis a threshold level of circulating tumor DNA (e.g., an average level of circulating tumor DNA in a population of subjects having a similar FGFR-associated cancer and having a similar stage of the FGFR-associated cancer, but receiving a non-effective treatment or a placebo, or not yet receiving therapeutic treatment, or a level of circulating tumor DNA in a subject having a similar FGFR-associated cancer and having a similar stage of the FGFR-associated cancer, but receiving a non-effective treatment or a placebo, or not yet receiving therapeutic treatment).
- a threshold level of circulating tumor DNAe.g., an average level of circulating tumor DNA in a population of subjects having a similar FGFR-associated cancer and having a similar stage of the FGFR-associated cancer, but receiving a non-effective treatment or a placebo, or not yet receiving therapeutic treatment.
- the first FGFR inhibitoris: ARQ-087, ASP5878, AZD4547, B-701, BAY1179470, BAY1 187982, BGJ398, brivanib, Debio 1347, dovitinib, E7090, erdafitinib, FPA144, HMPL-453, INCB054828, lenvatinib, lucitanib, LY3076226, MAX-40279, nintedanib, orantinib, pemigatinib, ponatinib, PRN1371, rogaratinib, sulfatinib, roblitinib, ICP-105, BIO- 1262, futibatinib, fisogatinib, LOXO-435, or RLY-4008.
- the additional therapy or therapeutic agentincludes one or more of radiation therapy, a chemotherapeutic agent (e.g., any of the exemplary chemotherapeutic agents described herein or known in the art), a checkpoint inhibitor (e.g., any of the exemplary checkpoint inhibitors described herein or known in the art), surgery (e.g., at least partial resection of the tumor), and one or more other kinase inhibitors (e.g., any of the kinase inhibitors described herein or known in the art).
- a chemotherapeutic agente.g., any of the exemplary chemotherapeutic agents described herein or known in the art
- a checkpoint inhibitore.g., any of the exemplary checkpoint inhibitors described herein or known in the art
- surgerye.g., at least partial resection of the tumor
- one or more other kinase inhibitorse.g., any of the kinase inhibitors described herein or known in the art.
- Compounds of the disclosuremay also be useful as adjuvants to cancer treatment, that is, they can be used in combination with one or more additional therapies or therapeutic agents, for example a chemotherapeutic agent that works by the same or by a different mechanism of action.
- a compound of the disclosurecan be used prior to administration of an additional therapeutic agent or additional therapy.
- a subject in need thereofcan be administered one or more doses of a compound of the disclosure for a period of time and then under go at least partial resection of the tumor.
- the treatment with one or more doses of a compound of the disclosurereduces the size of the tumor (e.g., the tumor burden) prior to the at least partial resection of the tumor.
- a subjecthas a cancer (e.g., a locally advanced or metastatic tumor) that is refractory or intolerant to standard therapy (e.g., administration of a chemotherapeutic agent, such as a first FGFR inhibitor or a multikinase inhibitor, immunotherapy, radiation, or a platinum-based agent (e.g., cisplatin)).
- a chemotherapeutic agentsuch as a first FGFR inhibitor or a multikinase inhibitor
- immunotherapye.g., radiation
- platinum-based agente.g., cisplatin
- a subjecthas a cancer (e.g., a locally advanced or metastatic tumor) that is refractory or intolerant to prior therapy (e.g., administration of a chemotherapeutic agent, such as a first FGFR inhibitor or a multikinase inhibitor, immunotherapy, radiation, or a platinum -based agent (e.g., cisplatin)).
- a chemotherapeutic agentsuch as a first FGFR inhibitor or a multikinase inhibitor, immunotherapy, radiation, or a platinum -based agent (e.g., cisplatin)
- the compound of the disclosureis administered in combination with a therapeutically effective amount of at least one additional therapeutic agent selected from one or more additional therapies or therapeutic (e.g., chemotherapeutic) agents.
- additional therapeutic agentsinclude: other FGFR-targeted therapeutic agents (i.e.
- a first or second FGFR kinase inhibitore.g., receptor tyrosine kinase targeted therapeutic agents (e.g., Trk inhibitors or EGFR inhibitors)), signal transduction pathway inhibitors, checkpoint inhibitors, modulators of the apoptosis pathway (e.g. obataclax); cytotoxic chemotherapeutics, angiogenesis-targeted therapies, immune-targeted agents, including immunotherapy, and radiotherapy.
- kinase inhibitorse.g., receptor tyrosine kinase targeted therapeutic agents (e.g., Trk inhibitors or EGFR inhibitors)
- signal transduction pathway inhibitorse.g., checkpoint inhibitors, modulators of the apoptosis pathway (e.g. obataclax); cytotoxic chemotherapeutics, angiogenesis-targeted therapies, immune-targeted agents, including immunotherapy, and radiotherapy.
- the compound of the disclosure, and the additional therapeutic agentare administered simultaneously as separate dosages.
- the compound of the disclosure, and the additional therapeutic agentare administered as separate dosages sequentially in any order, in jointly therapeutically effective amounts, e.g. in daily or intermittently dosages.
- the compound of the disclosure, and the additional therapeutic agentare administered simultaneously as a combined dosage.
- the disease or disorderis a FGFR-associated disease or disorder.
- the subjecthas been administered one or more doses of a compound of of the disclosure, prior to administration of the pharmaceutical composition.
- the treatment periodis at least 7 days (e.g., at least or about 8 days, at least or about 9 days, at least or about 10 days, at least or about 11 days, at least or about 12 days, at least or about 13 days, at least or about 14 days, at least or about 15 days, at least or about 16 days, at least or about 17 days, at least or about 18 days, at least or about 19 days, at least or about 20 days, at least or about 21 days, at least or about 22 days, at least or about 23 days, at least or about 24 days, at least or about 25 days, at least or about 26 days, at least or about 27 days, at least or about 28 days, at least or about 29 days, or at least or about 30 days).
- at least 7 dayse.g., at least or about 8 days, at least or about 9 days, at least or about 10 days, at least or about 11 days, at least or about 12 days, at least or about 13 days, at least or about 14 days, at least or about 15 days, at least or about 16 days, at least or
- the treatment periodis at least 21 days (e.g., at least or about 22 days, at least or about 23 days, at least or about 24 days, at least or about 25 days, at least or about 26 days, at least or about 27 days, at least or about 28 days, at least or about 29 days, at least or about 30 days, at least or about 31 days, at least or about 32 days, at least or about 33 days, at least or about 34 days, at least or about 35 days, at least or about 36 days, at least or about 37 days, at least or about 38 days, at least or about 39 days, or at least or about 40 days).
- at least 21 dayse.g., at least or about 22 days, at least or about 23 days, at least or about 24 days, at least or about 25 days, at least or about 26 days, at least or about 27 days, at least or about 28 days, at least or about 29 days, at least or about 30 days, at least or about 31 days, at least or about 32 days, at least or about 33 days, at least or about 34 days, at least or
- compositionsthat contain, as the active ingredient, a compound of the disclosure, in combination with one or more pharmaceutically acceptable carriers (excipients).
- the compositionis suitable for topical administration.
- the active ingredientis typically mixed with an excipient, diluted by an excipient or enclosed within such a carrier in the form of, for example, a capsule, sachet, paper, or other container.
- the excipientserves as a diluent, it can be a solid, semi-solid, or liquid material, which acts as a vehicle, carrier or medium for the active ingredient.
- compositionscan be in the form of tablets, pills, powders, lozenges, sachets, cachets, elixirs, suspensions, emulsions, solutions, syrups, aerosols (as a solid or in a liquid medium), ointments containing, for example, up to 10% by weight of the active compound, soft and hard gelatin capsules, suppositories, sterile injectable solutions, and sterile packaged powders.
- the compositionis formulated for oral administration.
- the compositionis formulated as a tablet or capsule.
- compositions comprising a compound of the disclosurecan be formulated in a unit dosage form, each dosage containing from about 5 to about 1,000 mg (1 g), more usually about 100 mg to about 500 mg, of the active ingredient.
- unit dosage formrefers to physically discrete units for human subjects and other subjects, each unit containing a predetermined quantity of active material (i.e., a compound of the disclosure) to produce the desired therapeutic effect, with a suitable pharmaceutical excipient.
- the compositions provided hereincontain from about 5 mg to about 50 mg of the active ingredient, i.e., the compound of the disclosure.
- the active ingredienti.e., the compound of the disclosure.
- the compositions provided hereincontain from about 50 mg to about 500 mg of the active ingredient.
- compositions provided hereincontain from about 500 mg to about 1,000 mg of the active ingredient.
- thisembodies compounds or compositions containing about 500 mg to about 550 mg, about 550 mg to about 600 mg, about 600 mg to about 650 mg, about 650 mg to about 700 mg, about 700 mg to about 750 mg, about 750 mg to about 800 mg, about 800 mg to about 850 mg, about 850 mg to about 900 mg, about 900 mg to about 950 mg, or about 950 mg to about 1,000 mg of the active ingredient.
- the active compoundmay be effective over a wide dosage range and is generally administered in a pharmaceutically effective amount. It will be understood, however, that the amount of the compound actually administered will usually be determined by a physician, according to the relevant circumstances, including the condition to be treated, the chosen route of administration, the actual compound administered, the age, weight, and response of the individual subject, the severity of the subject's symptoms, and the like.
- the compounds provided hereincan be administered in an amount ranging from about 1 mg/kg to about 100 mg/kg. In some embodiments, the compound provided herein can be administered in an amount of about 1 mg/kg to about 20 mg/kg, about 5 mg/kg to about 50 mg/kg, about 10 mg/kg to about 40 mg/kg, about 15 mg/kg to about 45 mg/kg, about 20 mg/kg to about 60 mg/kg, or about 40 mg/kg to about 70 mg/kg.
- such administrationcan be once-daily or twice-daily (BID) administration.
- Aspect 1A compound of formula (I): or a pharmaceutically acceptable salt thereof, wherein
- XO, S, or NR
- R 1is H or optionally substituted C 1 -Cealkyl
- R 5ais H, halogen, -CN, -S(O) 2 C 1 -C 6 alkyl, OCF 3 , OC 1 -C 3 alkyl, or C 1 -C 3 alkyl;
- Q 5 , Q 6 , Q 7 , Q 8 , and Q 9are each independently N or CR 5 , wherein one or two of the Q 5 , Q 6 , Q 7 , Q 8 , and Q 9 is N and the remainder are CR 5 ;
- R 5is H, halogen, C 1 -C 3 alkyl, C 1 -C 3 alkoxyl, or cycloalkyl;
- R 6is C 1 -Cealkyl
- R 7is H, halogen, -C 1 -Cealkyl, -C 1 -Ce alkoxyl, or -cycloalkyl;
- R 8is H, halogen, -C 1 -Cealkyl, -C 1 -Ce alkoxyl, or -cycloalkyl.
- Aspect 2The compound of aspect 1, wherein X is O.
- Aspect 3The compound of aspect 1 or aspect 2, wherein R 6 is CH3.
- Aspect 4The compound of any one of the preceding aspects, wherein R 8 is H.
- Aspect 6The compound of any one of the preceding aspects, wherein Q 1 and Q 3 are each CR 5a wherein R 5a is H.
- Aspect 7The compound of any one of the preceding aspects, wherein Q 2 and Q 4 are each N.
- Aspect 8The compound of any one of aspects 1-6, wherein Q 2 is N and Q 4 is CR 5a wherein R 5a is H, -CN, or F.
- Aspect 9The compound of any one of aspects 1-6, wherein Q 2 is N and Q 4 is CR 5a wherein R 5a is S(O)2C 1 -C6alkyl, OC 1 -C3alkyl, or C 1 -C3alkyl.
- Aspect 10The compound of any one of the preceding aspects, wherein Q 5 and Q 9 are each CR 5 wherein each R 5 is Cl; Q 7 is N; Q 6 is CR 5 wherein R 5 is H; and Q 8 is CR 5 wherein R 5 is H or CH3.
- Aspect 11The compound of any one of the preceding aspects, wherein n is i and m is 1.
- Q 2 and Q 4are each N, or one of Q 2 or Q 4 is N and the other is CR 5a ;
- R 5ais H, F, -SO2CH3, or -CN;
- R 5is H or CH3
- R 7is H, F, or OCH3.
- Aspect 13The compound of aspect 12, wherein R 5 is H.
- Aspect 14The compound of aspect 12, wherein R 5 is CH3.
- Aspect 15The compound of any one of aspects 12-14, wherein R 7 is H.
- Aspect 16The compound of any one of aspects 12-14, wherein R 7 is F.
- Aspect 17The compound of any one of aspects 12-14, wherein R 7 is OCH3.
- Aspect 18The compound of any one of aspects 12-17, wherein Q 2 and Q 4 are each N.
- Aspect 19The compound of any one of aspects 12-17, wherein one of Q 2 or Q 4 is N and the other is CR 5a .
- Aspect 20The compound of aspect 19, wherein R 5a is H or F.
- Aspect 21The compound of aspect 20, wherein R 5a is H.
- Aspect 22The compound of aspect 20, wherein R 5a is F.
- Aspect 23The compound of aspect 19, wherein R 5a is CN or SO2CH3.
- Aspect 24The compound of aspect 23, wherein R 5a is CN.
- Aspect 25The compound of aspect 23, wherein R 5a is SO2CH3.
- Aspect 26The compound of aspect 1, wherein R 5a is OCF3.
- Aspect 27The compound of any one of aspects 1-26, wherein R 1 is H.
- Aspect 28The compound of any one of aspects 1-26, wherein R 1 is CH3.
- NS(O)(C 1 -C 6 alkyl)(C 1 -C 6 alkyl);
- R 7is H or -OC 1 -Cealkyl
- R 5ais halo, or -CN.
- Aspect 30The compound of aspect 29, wherein R 7 is H.
- Aspect 31The compound of aspect 29, wherein R 7 is -OC 1 -Cealkyl.
- Aspect 32The compound of aspect 31, wherein R 7 is -OCH3.
- Aspect 33The compound of any one of aspects 29-32, R 5a is -CN.
- Aspect 34The compound of any one of aspects 29-32, R 5a is halo.
- Aspect 44The compound of aspect 29, or a pharmaceutically acceptable salt thereof, wherein the compound is:
- Aspect 45The compound of aspect 29, or a pharmaceutically acceptable salt thereof, wherein the compound is:
- Aspect 46The compound of aspect 29, or a pharmaceutically acceptable salt thereof, wherein the compound is:
- Aspect 48The compound of aspect 29, or a pharmaceutically acceptable salt thereof, wherein the compound is:
- Aspect 49The compound of aspect 29, or a pharmaceutically acceptable salt thereof, wherein the compound is:
- a pharmaceutical compositioncomprising a compound of any one of aspects 1-49, or a pharmaceutically acceptable salt thereof, and a pharmaceutically acceptable excipient.
- a method of treating a disease or disorder in a subject in need thereofcomprising administering to the subject a compound of any one of aspects 1 to 49, or a pharmaceutically acceptable salt thereof.
- Aspect 52The method of aspect 51, wherein the disease or disorder is cancer.
- Aspect 53The method of aspect 52, wherein the cancer is urothelial carcinoma, hepatocellular carcinoma, breast carcinoma, endometrial adenocarcinoma, ovarian carcinoma, primary glioma, cholangiocarcinoma, gastric adenocarcinoma, non-small cell lung carcinoma, pancreatic exocrine carcinoma, oral cancer, prostate cancer, bladder cancer, colorectal carcinoma, renal cell carcinoma, neuroendocrine carcinoma, myeloproliferative neoplasms, head and neck (squamous), melanoma, leiomyosarcoma, and/or sarcomas.
- the canceris urothelial carcinoma, hepatocellular carcinoma, breast carcinoma, endometrial adenocarcinoma, ovarian carcinoma, primary glioma, cholangiocarcinoma, gastric adenocarcinoma, non-small cell lung carcinoma, pancreatic exocrine carcinoma, oral cancer, prostate cancer,
- Aspect 54The method of aspect 53, wherein the cancer is bladder cancer.
- Aspect 55The method of aspect 53, wherein the cancer is urothelial carcinoma.
- Aspect 56The method of aspect 53, wherein the cancer is hepatocellular carcinoma.
- Aspect 57The method of any one of aspects 52 to 56, wherein the cancer is an FGFR- mutant cancer.
- Aspect 58The method of aspect 51, wherein the disease or disorder is a developmental disorder.
- Aspect 59The method of aspect 58, wherein the developmental disorder is achondroplasia.
- Aspect 60A method of inhibiting FGFR in a cell comprising contacting the cell with a compound of any one of aspects 1 to 49.
- the enantiomersmay be separated by conventional means (chiral chromatography, preparing diastereomeric salts, chiral derivatization, crystallization, enzymatic reactions, etc.).
- a chiral intermediate compoundis purified to prepare an enantiomerically pure (or substantially enantiomerically pure, enantiomerically enriched, etc.) intermediate.
- Step 1-Benzhydryl-3 -methylazeti din-3 -yl methanesulfonate.
- Methanesulfonyl chloride(6.2 mL, 80 mmol, 2 equiv) was added dropwise to a solution of 1- benzhydryl-3 -methyl-azeti din-3 -ol (10.13 g, 40 mmol, 1 equiv) and triethylamine (14.0 mL, 100 mmol, 2.5 equiv) in anhydrous dichloromethane (150 mL) at 0 °C.
- water50 mL was added, and the layers were separated.
- Step 2((l-Benzhydryl-3-methylazetidin-3-yl)imino)dimethyl-X6- sulfanone.
- Anhydrous sodium bicarbonate(1.9 g, 22.6 mmol, 1.5 equiv) was added to a mixture of compound 1 -benzhydryl-3 -m ethylazeti din-3 -yl methanesulfonate (5.0 g, 15.08 mmol, 1 equiv) and (dimethanesulfinylidene)amine (1.545 g, 16.6 mmol, 1.1 equiv) in acetonitrile (100 mL) at room temperature.
- Step 3ADimethyl((3-methylazeti din-3 -yl)imino)-X6-sulfanone hydrochloride.
- Step 3BA mixture of the product from step 3 A (4.8 g, 11.264 mmol, 1 equiv) and 20% palladium hydroxide on carbon (50% wet, 0.96 g, 20 w/w%) in methanol (250 mL) was hydrogenated (50 psi at room temperature) for 19 hours. LCMS analysis showed partial conversion. Additional 20% palladium hydroxide on carbon (50% wet, 0.96 g, 20 w/w%) was added and the hydrogenation was continued over a weekend. LCMS analysis showed that the reaction was complete. The mixture was filtered through celite and washed with methanol (3 x 50 mL).
- Step 45-[(lR)-l-(3,5-Dichloro-4-pyridyl)ethoxy]-3-(5,6-difluoro-3- pyridyl)-l -tetrahydropyran -2 -yl-indazole.
- Step 5((l-(5-(5-((R)-l-(3,5-Dichloropyridin-4-yl)ethoxy)-l-(tetrahydro- 2H-pyran-2-yl)-lH-indazol-3-yl)-3-fluoropyridin-2-yl)-3-methylazetidin-3- yl)imino)dimethyl-Z6-sulfanone.
- Step 6(R)-((l-(5-(5-(l-(3,5-Dichloropyridin-4-yl)ethoxy)-lH-indazol-3- yl)-3-fluoropyridin-2-yl)-3-methylazeti din-3 -yl)imino)dimethyl-X6-sulfanone.
- Trifluoroacetic acid(3.3 mL) was added to a solution of ((l-(5-(5-((R)-l-(3,5-dichloropyridin-4-yl)ethoxy)- l-(tetrahydro-2H-pyran-2-yl)-lH-indazol-3-yl)-3-fluoropyridin-2-yl)-3-methylazetidin-3- yl)imino)dimethyl-X6-sulfanone (330 mg, 0.50 mmol, 1.0 equiv) in dichloromethane (3.3 mL) at room temperature. After stirring at room temperature for 4 hours, the volatile material was removed under reduced pressure.
- the residuewas diluted with dichloromethane (20 mL) and water (10 mL) and adjusted to pH 9 with saturated sodium carbonate. The layers were separated, and the aqueous layer was extracted with di chloromethane (3 x 10 mL). The combined organic layers were washed with saturated brine (20 mL), dried over sodium sulfate, filtered, and concentrated under reduced pressure. The residue was purified on an Interchim automated chromatography system (Sorbtech 25 g silica gel column), eluting with a gradient of 0 to 10% methanol in di chloromethane to give a tan solid (178 mg, 63% yield).
- N,N-Diisopropylethylamine(0.87 mL, 5 mmol, 5 equiv) was added to a mixture of dimethyl((3-methylazeti din-3 -yl)imino)-X6-sulfanone hydrochloride (257 mg, 1 mmol, 1 equiv) in acetonitrile (10 mL). After stirring at room temperature for 10 minutes, 5-bromo-2-chloro-pyridine-3-carbonitrile (217 mg, 1 mmol, 1 equiv) was added and the mixture was stirred at room temperature for 2 days.
- Step 22-(3-((Dimethyl(oxo)-X6-sulfaneylidene)amino)-3-methylazetidin- l-yl)-5-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)nicotinonitrile.
- Step 35-(5-((R)-l-(3,5-Dichloropyridin-4-yl)ethoxy)-l-(tetrahydro-2H- pyran-2-yl)-lH-indazol-3-yl)-2-(3-((dimethyl(oxo)-X6-sulfaneylidene)amino)-3- methylazetidin-l-yl)nicotinonitrile.
- Step 4.(R)-5-(5-(l-(3,5-Dichloropyridin-4-yl)ethoxy)-lH-indazol-3-yl)-2- (3-((dimethyl(oxo)-X6-sulfaneylidene)amino)-3-methylazetidin-l-yl)nicotinonitrile.
- Trifluoroacetic acid(1.6 mL) was added to a solution of 5-(5-((R)-l-(3,5-dichloropyridin-4- yl)ethoxy)-l-(tetrahydro-2H-pyran-2-yl)-lH-indazol-3-yl)-2-(3-((dimethyl(oxo)-X6- sulfaneylidene)amino)-3-methylazetidin-l-yl)nicotinonitrile (163 mg, 0.249 mmol, 1.0 equiv) in dichloromethane (1.6 mL) at room temperature. After stirring at room temperature for 5 hours, the volatiles were removed under reduced pressure.
- the residuewas diluted with di chloromethane (20 mL) and water (10 mL) and adjusted to pH 9 with saturated sodium carbonate. The layers were separated, and the aqueous layer was extracted with di chloromethane (2 x 20 mL). The combined organic layers were washed with saturated brine (20 mL), dried over sodium sulfate, filtered, and concentrated under reduced pressure.
- the residuewas purified on an Interchim automated chromatography system (Sorbtech 25 g silica gel column), eluting with a gradient of 0 to 10% methanol in di chloromethane to give a pale-yellow solid (71 mg, 50% yield).
- Step 15-((R)-l-(3,5-Dichloropyridin-4-yl)ethoxy)-3-iodo-l-(tetrahydro- 2H-pyran-2-yl)-lH-indazole.
- Step 25-[5-[(lR)-l-(3,5-Dichloro-4-pyridyl)ethoxy]-l-tetrahydropyran-2- yl-indazol-3-yl]-2-fluoro-pyridine-3-carbonitrile.
- Step 35-[5-[(lR)-l-(3,5-Dichloro-4-pyridyl)ethoxy]-l-tetrahydropyran-2- yl-indazol-3-yl]-2-(3-hydroxy-3-methyl-azetidin-l-yl)pyridine-3-carbonitrile.
- Step 4[l-[3-Cyano-5-[5-[(lR)-l-(3,5-dichloro-4-pyridyl)ethoxy]-l- tetrahydropyran-2-yl-indazol-3-yl]-2-pyridyl]-3-methyl-azetidin-3-yl] methanesulfonate.
- TEAa solution of product step 3 (3.1 g, 5.35 mmol, 1.0 eq) in DCM (50 mL) was added TEA (1.08 g, 10.7 mmol, 2.0 eq).
- Step 52-((Dimethyl(oxo)-X6-sulfaneylidene)amino)acetonitrile.
- Bromoacetonitrile (7.09 g, 59.076 mmol, 1.1 equiv) and sodium bicarbonate (6.77 g, 80.558 mmol, 1.5 equiv)were added to a solution of (dimethanesulfinylidene) amine (5.0 g, 53.706 mmol, 1 equiv) in acetonitrile (200 mL) at room temperature. After refluxing for 15 hours, the mixture was cooled to room temperature and filtered. The filtrate was concentrated under reduced pressure.
- Step 6((2-Aminoethyl)imino)dimethyl-X6-sulfanone.
- LCMS analysisindicated that starting material remained. Additional Raney nickel (slurry in water, 2.1 g) was added, and the hydrogenation was continued over a weekend.
- Step 75-(5-((R)-l-(3,5-Dichloropyridin-4-yl)ethoxy)-l-(tetrahydro-2H- pyran-2-yl)-lH-indazol-3-yl)-2-(3-((2-((dimethyl(oxo)-X6- sulfaneylidene)amino)ethyl)amino)-3-methylazetidin-l-yl)nicotinonitrile.
- Step 85-[5-[(lR)-l-(3,5-dichloro-4-pyridyl)ethoxy]-lH-indazol-3-yl]-2- [3-[2-[[dimethyl(oxo)-k 6 -sulfanylidene]amino]ethylamino]-3-methyl-azetidin-l-yl]pyridine- 3-carbonitrile.
- Trifluoroacetic acid2.0 mL
- the residuewas diluted with di chloromethane (20 mL) and water (10 mL) then adjusted to pH 9 with saturated sodium carbonate. The layers were separated, and the aqueous layer was extracted with di chloromethane (3 x 20 mL). The combined organic layers were washed with saturated brine (20 mL), dried over sodium sulfate, filtered and concentrated under reduced pressure. The residue was dry loaded onto Celite (8 g) and purified on an InterChim automated chromatography system (RediSep Rf GOLD 50 g HP C18 column), eluting with a gradient of 0 to 100% acetonitrile in water to give a white solid after lyophilization (84 mg, 49% yield).
- Step 1(E)-N'-(2-Bromo-5-hydroxy-4-methoxybenzylidene)-4- methylbenzenesulfonohydrazide.
- p-Toluenesulfonyl hydrazide(0.56 g, 3.0 mmol, 1.0 equiv) was added to a solution of 2-bromo-5-hydroxy-4-methoxy -benzaldehyde (0.7 g, 3.0 mmol, 1.0 equiv) in methanol (7.0 mL) at rt.
- the resulting mixturewas heated at 60 °C for 2 h.
- the reactionwas cooled to rt and the solvent was removed under reduced pressure.
- Step 26-Methoxy-l-tosyl-lH-indazol-5-ol. Copper(I) oxide (0.22 g, 1.5 mmol, 0.5 equiv) was added to a solution of (E)-N'-(2-Bromo-5-hydroxy-4- methoxybenzylidene)-4-methylbenzenesulfonohydrazide (1.2 g, 3.0 mmol, 1.0 equiv) in isoamyl alcohol (30 mL) at room temperature. After heating at 132 °C for 2 hours, the mixture was cooled to room temperature and diluted with water (80 mL). The mixture was extract with ethyl acetate (4 x 50 mL).
- Step 3(R)-5-(l-(3,5-Dichloropyridin-4-yl)ethoxy)-6-methoxy-l-tosyl-lH- indazole.
- (lS)-l-(3,5-dichloro-4-pyridyl)ethyl] methanesulfonate (0.57 g, 2.1 mmol, 1.0 equiv) and cesium carbonate (1.03 g, 3.2 mmol, 1.5 equiv)were added to a solution of 6- methoxy-l-tosyl-lH-indazol-5-ol (0.67 g, 2.1 mmol, 1.0 equiv) in acetonitrile (21 mL) at room temperature.
- Step 4(R)-5-(l-(3,5-Dichloropyridin-4-yl)ethoxy)-6-methoxy-lH- indazole.
- IM Tetrabutylammonium fluoride in THF(7.2 mL, 7.2 mmol, 18.0 equiv) was added to a solution of (R)-5-(l-(3,5-dichloropyridin-4-yl)ethoxy)-6-methoxy-l-tosyl-lH- indazole (0.20 g, 0.4 mmol, 1.0 equiv) in tetrahydrofuran (4 mL) at room temperature.
- Step 5(R)-5-(l-(3,5-Dichloropyridin-4-yl)ethoxy)-3-iodo-6-methoxy-lH- indazole.
- Potassium hydroxide27.9 mg, 0.50 mmol, 2.25 equiv
- iodine84.1 mg, 0.33 mmol, 1.5 equiv
- Step 75-(5-((R)-l-(3,5-Dichloropyridin-4-yl)ethoxy)-6-methoxy-l- (tetrahy dro-2H-pyran-2-yl)- 1 H-indazol -3 -yl)-2-(3 -((dimethyl(oxo)-X6- sulfaneylidene)amino)-3-methylazetidin-l-yl)nicotinonitrile.
- Step 8(R)-5-(5-(l-(3,5-Dichloropyridin-4-yl)ethoxy)-6-methoxy-lH- indazol-3-yl)-2-(3-((dimethyl(oxo)-X6-sulfaneylidene)amino)-3-methylazetidin-l- yl)nicotinonitrile.
- Trifluoroacetic acid(2 mL) was added to a solution of product step 7 (92 mg, 0.134 mmol, 1.0 equiv) in di chloromethane (2 mL) at room temperature. After stirring at room temperature for 4 hours, the volatiles were removed under reduced pressure.
- Step 12-((l-Benzhydryl-3-methylazetidin-3-yl)(benzyl)amino)ethan-l-ol.
- (l-Benzhydryl-3-methyl-azetidin-3-yl) methanesulfonate (3.09 g, 9.3 mmol, 1 equiv)was treated with 2-benzylaminoethanol (2.82 g, 18.7 mmol, 2.0 equiv) and N,N- diisopropylethylamine (8.1 mL, 46.7 mmol, 5.0 equiv) in 1,4-dioxane (20 mL) at 100 °C for 20 hours.
- Step 3((2-((l-Benzhydryl-3-methylazeti din-3 - yl)(benzyl)amino)ethyl)imino)-dimethyl-X6-sulfanone.
- Step 4Dimethyl((2-((3-methylazeti din-3 -yl)amino)ethyl)imino)-X6- sulfanone dihydrochloride salt.
- 2M HC1 in diethyl ether(2.4 mL, 4.8 mmol, 5 equiv) was added to a solution of ((2-((l-benzhydryl-3-methylazetidin-3-yl)(benzyl)amino)ethyl)imino)- dimethyl-X6-sulfanone (0.44 g, 0.953 mmol, 1 equiv) in a mixture of acetonitrile (20 mL) and diethyl ether (10 mL) at room temperature.
- dihydrochloride salt(0.6 g) as a light-yellow solid.
- Additional 20% palladium hydroxide on carbon(50% wet, 0.12 g) was added, and the hydrogenation continued for 3 days.
- the mixturewas filtered through Celite, which was washed with methanol (3 x 50 mL).
- Step 5((2-((l-(5-(5-((R)-l-(3,5-Dichloropyridin-4-yl)ethoxy)-l- (tetrahydro-2H-pyran-2-yl)-lH-indazol-3-yl)-3-fluoropyridin-2-yl)-3-methylazetidin-3- yl)amino)ethyl)imino)dimethyl-X6-sulfanone.
- Step 6(R)-((2-((l-(5-(5-(l-(3,5-Dichloropyridin-4-yl)ethoxy)-lH-indazol- 3-yl)-3-fluoropyridin-2-yl)-3-methylazetidin-3-yl)amino)ethyl)imino)dimethyl-X6-sulfanone.
- Trifluoroacetic acid0.5 mL
- the residuewas diluted with dichloromethane (20 mL) and water (10 mL) then adjusted to pH 9 with saturated sodium carbonate.
- the aqueous layerwas extracted with dichloromethane (3 x 20 mL).
- the combined organic layerswere washed with saturated brine (20 mL), dried over sodium sulfate, filtered, and concentrated under reduced pressure.
- the residuewas dry loaded onto Celite (8 g) and purified on an InterChim automated chromatography system (RediSep Rf GOLD 50 g HP Cl 8 column), eluting with a gradient of 0 to 100% acetonitrile in water to give a white solid after lyophilization (7.0 mg, 44% yield).
- Stepl(l-(5-(5-((R)-l-(3,5-Dichloropyridin-4-yl)ethoxy)-l-(tetrahydro-2H- pyran-2-yl)-lH-indazol-3-yl)-3-fluoropyridin-2-yl)-3-methylazetidin-3-yl)methanol.
- Step 2(l-(5-(5-((R)-l-(3,5-Dichloropyridin-4-yl)ethoxy)-l-(tetrahydro- 2H-pyran-2-yl)-lH-indazol-3-yl)-3-fluoropyridin-2-yl)-3-methylazetidin-3-yl)methyl methanesulfonate.
- Methanesulfonic anhydride(240 mg, 1.38 mmol, 3 equiv) was added to a solution of product step 1 (270 mg, 0.46 mmol, 1 equiv) and N,N-diisopropylethylamine (178 mg, 1.38 mmol, 3 equiv) in dichloromethane (20 mL) at 0 °C. After stirring at room temperature for 16 hours, the reaction was quenched with water (20 mL) and extracted with di chloromethane (2 x 40 mL). The combined organic layers were dried over sodium sulfate, filtered, and concentrated under reduced pressure.
- Step 3((l-(5-(5-((R)-l-(3,5-Dichloropyridin-4-yl)ethoxy)-l-(tetrahydro- 2H-pyran-2-yl)-lH-indazol-3-yl)-3-fluoropyridin-2-yl)-3-methylazetidin-3- yl)methyl)imino)dimethyl-X6-sulfanone.
- N-Methyl-2- pyrrolidone(1 mL) was added and the mixture was heated at 80 °C for 3 days. After cooling to room temperature, the mixture was diluted with ethyl acetate (20 mL) and water (8 mL). The layers were separated, and the organic layer was washed with saturated brine (2 x 8 mL), dried over sodium sulfate, filtered and concentrated under reduced pressure.
- Step 4(R)-(((l-(5-(5-(l-(3,5-Dichloropyridin-4-yl)ethoxy)-lH-indazol-3- yl)-3-fluoropyridin-2-yl)-3-methylazeti din-3 -yl)methyl)imino)dimethyl-X6-sulfanone.
- Trifluoroacetic acid(2 mL) was added to a solution of compound 691-4 (73 mg, 0.11 mmol, 1.0 equiv) in di chloromethane (2 mL) at room temperature. After stirring at room temperature for 4 hours, the volatiles were removed under reduced pressure.
- Step 1Methyl 3-((tert-butoxycarbonyl)amino)-l-(5-(5-((R)-l-(3,5- dichloropyridin-4-yl)ethoxy)-l-(tetrahydro-2H-pyran-2-yl)-lH-indazol-3-yl)-3-fluoropyridin-
- Step 23-((tert-Butoxycarbonyl)amino)-l-(5-(5-((R)-l-(3,5- dichloropyridin-4-yl)ethoxy)-l-(tetrahydro-2H-pyran-2-yl)-lH-indazol-3-yl)-3-fluoropyridin- 2 -yl)azetidine-3 -carboxylic acid.
- Step 3tert-Butyl (l-(5-(5-((R)-l-(3,5-dichloropyridin-4-yl)ethoxy)-l- (tetrahydro-2H-pyran-2-yl)-lH-indazol-3-yl)-3-fluoropyridin-2-yl)-3-((dimethyl(oxo)-X6- sulfaneylidene)carbamoyl)azetidin-3-yl)carbamate.
- Step 4.(R)-3-Amino-l-(5-(5-(l-(3,5-dichloropyridin-4-yl)ethoxy)-lH- indazol-3-yl)-3-fluoropyridin-2-yl)-N-(dimethyl(oxo)-X6-sulfaneylidene)azetidine-3- carboxamide.
- the reaction mixturewas concentrated under reduced pressure and diluted with saturated sodium carbonate (50 mL) and ethyl acetate (20 mL). The layers were separated, and the aqueous layer was extracted with ethyl acetate (2 xlO mL). The combined organic layers were washed with saturated brine (25 mL), dried over magnesium sulfate, filtered, and concentrated under reduced pressure.
- the crude residuewas purified on a Biotage automated chromatography system (100 g RediSep Gold, C18 column), eluting with a gradient of 0 to 100% acetonitrile in water to give a white solid after lyophilization (83 mg, 25%).
- Example 8(R)-(((3-Amino-l-(5-(5-(l-(3,5-dichloropyridin-4- yl)ethoxy)-lH-indazol-3-yl)-3-fluoropyridin-2-yl)azetidin-3-yl)methyl)imino)dimethyl- k 6 -sulfanone.
- the reaction mixturewas quenched with a mixture of water (6 mL), methanol (3 mL) and trifluoroacetic acid, adjusting the pH to 2 at 5 °C.
- the mixturewas stirred at room temperature for 5 hours then neutralized with saturated sodium carbonate solution adjusting to pH 10.
- the mixturewas extracted with ethyl acetate (3 x 10 mL).
- the combined organic layerswere washed with saturated brine (10 mL), dried over magnesium sulfate, filtered, and concentrated under reduced pressure.
- the crude residuewas purified on a Biotage automated chromatography system (100 g RediSep Gold, Cl 8 column), eluting with a gradient of 0 to 100% acetonitrile in water to give a white solid after lyophilization (17 mg, 46%).
- Step 1((l-Benzhydryl-3-(pyrrolidin-l-ylmethyl)azetidin-3- yl)imino)dimethyl-X6-sulfanone (919-1): Pyrrolidine (421 mg, 5.9 mmol, 5 equiv) and N,N- diisopropylethylamine (1.53 g, 2.1 mL, 11.8 mmol, 10 equiv) were added to a solution of [1- benzhydryl-3-[[dimethyl(oxo)-X 6 -sulfanylidene]amino]azetidin-3-yl]methyl methanesulfonate (500 mg, 1.2 mmol, 1.0 equiv) in acetonitrile (30 mL) at room temperature.
- Step 2Dimethyl((3-(pyrrolidin-l-ylmethyl)azetidin-3-yl)imino)-X6- sulfanone trihydrochloride.
- Step 3((l-(5-(5-((R)-l-(3,5-Dichloropyridin-4-yl)ethoxy)-l-(tetrahydro- 2H-pyran-2-yl)-lH-indazol-3-yl)-3-fluoropyridin-2-yl)-3-(pyrrolidin-l-ylmethyl)azeti din-3- yl)imino)dimethyl-X6-sulfanone.
- Step 4(R)-((l-(5-(5-(l-(3,5-Dichloropyridin-4-yl)ethoxy)-lH-indazol-3- yl)-3-fluoropyridin-2-yl)-3-(pyrrolidin-l-ylmethyl)azeti din-3 -yl)imino)dimethyl-X6- sulfanone dihydrochloride.
- Example 10Example 10. 5-[5-[(lR)-l-(3,5-dichloro-4-pyridyl)ethoxy]-6-methoxy-lH-indazol-3-yl]-2- [3-[[dimethyl(oxo)-k 6 -sulfanylidene]amino]-3-(pyrrolidin-l-ylmethyl)azetidin-l- yl] pyridine-3-carbonitrile
- Step 15-(5-((R)-l-(3,5-Dichloropyridin-4-yl)ethoxy)-6-methoxy-l- (tetrahy dro-2H-pyran-2-yl)- 1 H-indazol -3 -yl)-2-(3 -((dimethyl(oxo)-X6- sulfaneylidene)amino)-3-(pyrrolidin-l-ylmethyl)azetidin-l-yl)nicotinonitrile.
- Step 25-[5-[(lR)-l-(3,5-dichloro-4-pyridyl)ethoxy]-6-methoxy-lH- indazol-3-yl]-2-[3-[[dimethyl(oxo)-X 6 -sulfanylidene]amino]-3-(pyrrolidin-l- ylmethyl)azetidin-l-yl]pyridine-3 -carbonitrile.
- Step 1l-Benzhydryl-3-(benzylamino)azetidine-3 -carbonitrile.
- 1- Benzhydrylazetidin-3-one (5.0 g, 21 mmol, 1 equiv) in methanol (55 mL)was sequentially treated with benzylamine (2.5 mL, 23.2 mmol, 1.1 equiv) and acetic acid (1.35 mL, 23.2 mmol, 1.1 equiv). After stirring for 1 hour, sodium cyanide (1.14 g, 23.2 mmol, 1.1 equiv) was added to the reaction.
- Step 2l-Benzhydryl-3-(benzylamino)azetidine-3 -carboxylic acid.
- 3M Sodium hydroxide(15 mL, 45 mmol, 2.9 equiv) was added to a suspension of 1-benzhydryl- 3-(benzylamino)azetidine-3-carbonitrile (5.40 g, 15.3 mmol, 1 equiv) in a mixture of ethanol (60 mL) and THF (30 mL). The mixture was heated at 60 °C for 3 days, cooled to room temperature then added to a stirred solution of acetic acid (10 mL, 175 mmol, 11 equiv) in water (500 mL). The resulting solids were filtered, washed with water (100 mL) and dried under vacuum at 40 °C overnight to give a white solid (4.85 g, 85%).
- LCMS m/z373 (M+H).
- Step 3l-Benzhydryl-3-(benzylamino)-N-(dimethyl(oxo) -X6- sulfaneylidene)-azetidine-3 -carboxamide.
- Step 4(((l-Benzhydryl-3-(benzylamino)azetidin-3- yl)methyl)imino)dimethyl-X6-sulfanone.
- IM Lithium aluminum hydride in THF(4 mL, 4 mmol, 1.1 equiv) was added to a suspension of l-Benzhydryl-3-(benzylamino)-N- (dimethyl(oxo) -X6-sulfaneylidene)-azetidine-3-carboxamide (1.62 g, 3.6 mmol, 1 equiv) in THF (20 mL) at 0 °C.
- Step 5(((3-Aminoazeti din-3 -yl (methyl )i mi no)di methyl -+6-sulfanone bis(2,2,2-trifluoroacetate).
- a solution of product mixture from step 4(0.300 g, 0.69 mmol, 1 equiv), in a mixture of methanol (20 mL) and trifluoroacetic acid (ImL, 13 mmol, 19 equiv) was added to 20% palladium hydroxide on carbon (50% wet) (0.150 g, 0.11 mmol, 0.15 equiv) in a 500 mL Parr flask.
- Step 7(R)-2-(3-Amino-3-(((dimethyl(oxo)-X6- sulfaneylidene)amino)methyl)azetidin-l-yl)-5-(5-(l-(3,5-dichloropyridin-4-yl)ethoxy)-6- methoxy-lH-indazol-3-yl)nicotinonitrile.
- Step 15-(5-((R)-l-(3,5-Dichloropyridin-4-yl)ethoxy)-6-methoxy-l- (tetrahy dro-2H-pyran-2-yl)- 1 H-indazol -3 -yl)-2-(3 -((dimethyl(oxo)-X6- sulfaneylidene)amino)-3-((dimethylamino)methyl)azetidin-l-yl)nicotinonitrile.
- Step 2(R)-5-(5-(l-(3,5-Dichloropyridin-4-yl)ethoxy)-6-methoxy-lH- indazol-3-yl)-2-(3-((dimethyl(oxo)-X6-sulfaneylidene)amino)-3-((dimethylamino)methyl) azetidin-l-yl)nicotinonitrile.
- Step 1Methyl l-benzhydryl-3-((methylsulfonyl)oxy)azetidine-3- carboxylate. N,N-Diisopropylethylamine (4.9 mL, 33.6 mmol, 2 equiv) was added to a solution of methyl l-benzhydryl-3-hydroxy-azetidine-3 -carboxylate (5.0 g, 16.8 mmol, 1.0 equiv) in dichloromethane (100 mL) at 0 °C. Methanesulfonic anhydride (4.1 g, 23.5 mmol, 1.4 equiv) was then added over 2 minutes.
- Step 3((l-Benzhydryl-3-(hydroxymethyl)azetidin-3-yl)imino)dimethyl- X6-sulfanone.
- Sodium borohydride(8.23 g, 217.5 mmol, 15 equiv) was added to a solution of methyl l-benzhydryl-3-((dimethyl(oxo)-X6-sulfaneylidene)amino)azetidine-3 -carboxylate (5.4 g, 14.5 mmol, 1.0 equiv) in methanol (100 mL) at 0 °C.
- the reactionwas stirred at 0 °C for 1 hour and at room temperature for 1 hour.
- Step 4(l-Benzhydryl-3-((dimethyl(oxo)-X6- sulfaneylidene)amino)azetidin-3-yl)methyl methanesulfonate.
- N,N-Diisopropylethylamine(0.51 mL, 2.9 mmol, 2 equiv) was added to a solution of ((l-benzhydryl-3- (hydroxymethyl)azetidin-3-yl)imino)dimethyl-X6-sulfanone (500 mg, 1.45 mmol, 1.0 equiv) in dichloromethane (30 mL) at 0 °C.
- Step 6((3-((Dimethylamino)methyl)azetidin-3-yl)imino)dimethyl-Z6- sulfanone trihydrochloride.
- 20% palladium hydroxide on activated carbon(50% wetted powder) (284 mg, 0.20 mmol, 0.15 equiv)
- concentrated HC10.5 mL, 6.0 mmol, 4.5 equiv
- methanol40 mL
- Step 7.((l-(5-(5-((R)-l-(3,5-Dichloropyridin-4-yl)ethoxy)-l-(tetrahydro- 2H-pyran-2-yl)-lH-indazol-3-yl)-3-fluoropyridin-2-yl)-3-((dimethylamino)methyl)azetidin- 3-yl)imino)dimethyl-Z6-sulfanone.
- Step 8(R)-((l-(5-(5-(l-(3,5-Dichloropyridin-4-yl)ethoxy)-lH-indazol-3- yl)-3-fluoropyridin-2-yl)-3-((dimethylamino)methyl)azeti din-3 -yl)imino)dimethyl-Z6- sulfanone.
- Kinase-tagged T7 phage strainswere prepared in an E. coli host derived from the BL21 strain. E. coli were grown to log -phase and infected with T7 phage and incubated with shaking at 32°C until lysis. The lysates were centrifuged and filtered to remove cell debris. Streptavidin-coated magnetic beads were treated with biotinylated small molecule ligands for 30 minutes at room temperature to generate affinity resins for kinase assays.
- Binding reactionswere assembled by combining kinases, liganded affinity beads, and test compounds in lx binding buffer (20% SeaBlock, 0.17x PBS, 0.05% Tween 20, 6 mM DTT).
- Test compoundswere prepared as 11 IX stocks in 100% DMSO. Kds were determined using an 11 -point 3-fold compound dilution series with three DMSO control points. All compounds for Kd measurements are distributed by acoustic transfer (non-contact dispensing) in 100% DMSO. The compounds were then diluted directly into the assays such that the final concentration of DMSO was 0.9%. All reactions performed in polypropylene 384-well plate. Each was a final volume of 0.02 ml. The assay plates were incubated at room temperature with shaking for 1 hour and the affinity beads were washed with wash buffer (lx PBS, 0.05% Tween 20).
- the beadswere then re-suspended in elution buffer (lx PBS, 0.05% Tween 20, 0.5 pM non-biotinylated affinity ligand) and incubated at room temperature with shaking for 30 minutes.
- elution bufferlx PBS, 0.05% Tween 20, 0.5 pM non-biotinylated affinity ligand
- Binding constantswere calculated with a standard dose-response curve using the Hill equation:
- Cell Viability Assay ProcedureCell Titer-Gio® 2.0 Luminescent cell viability assay reagent was purchased from Promega (Madison, WI). Ba/F3 cell lines were cultured in RPMI1640 media supplemented with 10% fetal bovine serum. Cultures were maintained at 37°C in a humidified atmosphere of 5% CO2 and 95% air.
- the cells in cell culture plateswere incubated with the compounds at 37 °C and 5% CO2 for 48 hours. Then 50 pl of Cell Titer Gio 2.0 reagent was added to each well of the cell culture plates. The contents were covered from light and mixed on an orbital shaker at room temperature for 10 min. Luminescence was recorded by a Synergy Hl Microplate Reader (Biotek, Winooski, VT ). Cells were assessed as a percentage of DMSO only treated control cells. Curves were plotted and IC50 values were calculated using the GraphPad Prism 8 program based on a sigmoidal dose-response equation (4 parameter).
- Luminescent cell viability assay reagentwas purchased from Promega (Madison, WI). KG-1, KATO-III, and MDA-MB-453 cell lines were purchased from American Type Culture Collection (Manassas, VA). RT112/84 cell line was purchased from Millipore-Sigma (St. Louis, MO). HuH7 cells were purchased from Seikisui Xenotech (Kansas City, KS). RT112/84 and MDA-MB-453 cells were cultured in RPMI1640 media supplemented with 10% fetal bovine serum. KG-1 and KATO-III cell lines were cultured in IMDM media supplemented with 20% FBS. HuH7 cells were cultured in IMDM media supplemented with 10% FBS. Cultures were maintained at 37°C in a humidified atmosphere of 5% CO2 and 95% air.
- Cell Viability Assay ProcedureCells were plated in 96-well clear bottom/white plates (Corning #3903) at a range of densities depending on the optimal assay window (5,000-20,000 cells/well in 1 OOJJ.1 of media), incubated overnight. The next day, test compound DMSO stock solutions were made at 10 mM and 2 pM final concentration. Compounds were then added to cells in a 9-dose, 4-fold dilution series starting at 3 pM with an HP 300e Digital Dispenser (each dose was applied in triplicate).
- DMSOwas backfilled to each well up to 301 nL total volume of test compound + DMSO, and a total of 301 nL DMSO was added to a control/no test compound well in triplicate.
- the cells in cell culture plateswere incubated with the compounds at 37 °C and 5% CO2 for 72 hours- 120 hours depending on the cell line. Then 50 pl of Cell Titer Gio 2.0 reagent was added to each well of the cell culture plates. The contents were covered from light and mixed on an orbital shaker at room temperature for minimum of 10 min. Luminescence was recorded by a Clariostar Plus Microplate Reader (BMG Labtech, Cary, NC ). Cells were assessed as a percentage of DMSO only treated control cells. Curves were plotted and IC50 values were calculated using the GraphPad Prism 9 program based on a sigmoidal dose-response equation (log (inhibitor) vs. response - Variable slope, 4-parameter).
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
Description
INDAZOLE COMPOUNDS
CROSS-REFERENCE TO RELATED APPLICATION
[0001] This application claims the benefit of United State Provisional Application No. 63/476,791, filed December 22, 2022, the entirety of which is incorporated by reference herein.
TECHNICAL FIELD
[0002] The disclosure pertains to indazole compounds that are useful in treating cancer, pharmaceutical compositions that include one or more such indazole compounds, and methods of using such indazole compounds in treating cancer.
BACKGROUND
[0003] Kinase inhibitors have been used to block the activity of kinases and thereby treat cancer (e.g., by inhibiting mitotic processes). These kinase inhibitors are often small molecules that target kinases to block the development, growth or spread of cancer.
[0004] However, although various inhibitors of kinases are known, there remains a need for selective inhibitors to be used for the treatment of diseases such as hyper- proliferative diseases, which offer one or more advantages over current compounds. Those advantages include: improved activity and/or efficacy; beneficial kinase selectivity profile according to the respective therapeutic need; improved side effect profile, such as fewer undesired side effects, lower intensity of side effects, or reduced (cyto)toxicity; improved targeting of mutant receptors in diseased cells; improved physicochemical properties, such as solubility/stability in water, body fluids, and/or pharmaceutical formulations; improved pharmacokinetic properties, allowing e.g. for dose reduction or an easier dosing scheme; easier drug substance manufacturing, e.g. by shorter synthetic routes or easier purification.
SUMMARY
[0005] The compounds disclosed herein provide small molecule kinase inhibitors that are both efficacious and selective.
[0006] In some aspects, the disclosure is directed to compounds of formula (I):

X = O, S, or NR;
R is H or C1-C3alkyl; n = 1 or 2; m = 1 or 2;
R1 is H, optionally substituted C1-C6alkyl, or NH2;
R2 is -Co-C6alkN=S(0)(C1-C6alkyl)(C1-C6alkyl), -NH-C1-C6alk-N=S(O)(C1- C6alkyl)(C1-C6alkyl), or -Co-C3alk-C(=0)-Co-C3alk-N=S(0)(C1-C6alkyl)(C1-C6alkyl); one or two of Q1, Q2, Q3, Q4 is N and the others are each independently CR5a;
R5a is H, halogen, -CN, -S(O)2C1-C6alkyl, OCF3, OC1-C3alkyl, or C1-C3alkyl;
Q5, Q6, Q7, Q8, and Q9 are each independently N or CR5, wherein one or two of the Q5, Q6, Q7, Q8, and Q9 is N and the remainder are CR5;
R5 is H, halogen, C1-Csalkyl, C1-Csalkoxyl, or cycloalkyl;
R6 is C1-Cealkyl;
R7 is H, halogen, -C1-Cealkyl, -C1-Ce alkoxyl, or -cycloalkyl; and
R8 is H, halogen, -C1-Cealkyl, -C1-Ce alkoxyl, or -cycloalkyl.
[0007] Stereoisomers of the compounds of formula (I), and the pharmaceutical salts and solvates thereof, are also described. Methods of using compounds of formula (I) are described, as well as pharmaceutical compositions including the compounds of formula (I).
DETAILED DESCRIPTION OF ILLUSTRATIVE EMBODIMENTS [0008] The disclosure may be more fully appreciated by reference to the following description, including the following definitions and examples. Certain features of the disclosed compositions and methods which are described herein in the context of separate aspects, may also be provided in combination in a single aspect. Alternatively, various features of the disclosed compositions and methods that are, for brevity, described in the context of a single aspect, may also be provided separately or in any subcombination.
[0009] The term “optionally substituted,” or “substituted” as used herein to describe a substituent defined herein, means that the substituent may, but is not required to be, substituted with one or more of: halo (i.e., -F, -Cl, -Br, -I), cyano, -OH, -C1-Cealkyl, C3- Cecycloalkyl, 3-7 membered heterocycloalkyl, -Cs-Cespirocycloalkyl, 3-7 membered spiroheterocycloalkyl, bridged cycloalkyl, bridged heterocycloalkyl, C2-Cealkenyl, C2-C6 alkynyl, C1-Cehaloalkyl (e.g., -CF3; -CHF2, -CH2CF3, and the like), -C1-Cealkoxy, -C1-Ce haloalkoxy (e.g., -OCF3; -OCHF2, -OCH2CF3, and the like), C1-Cealkylthio (e.g., -SCH3; - SCH2CH3, and the like), C1-Ce alkylamino (e.g., -CH2NH2; -CH2CH2NH2, and the like), - NH2, -NH(CI-C6 alkyl), -N(CI-C-6 alkyl)2, -NH(CI-C6 alkoxy), -C(O)NHC1-C6alkyl, - C(O)N(CI-C6 alkyl)2, -COOH, -C1-C6alkylCOOH .C3-C6cycloalkylCOOH, -C(O)NH2, -C1- C6alkylCONH2, -C3-C6cycloalkylCONH2, -C1-C6alkylCONHC1-C6alkyl, -C1-C6alkylCON(C1- C6alkyl)2, -C(O)C1-C6 alkyl, -C(O)OC1-C6 alkyl, -NHCO(CI-C6 alkyl), -N(CI-C6 alkyl)C(O)(C1-Ce alkyl), -S(O)C1-Ce alkyl, -S(O)2C1-Ce alkyl, oxo (i.e., =0), 6-12 membered aryl, or 5 to 12 membered heteroaryl groups. In other embodiments, “optionally substituted,” or “substituted” means that the substituent may, but is not required to be, substituted with one or more of -C(O)(C1-C6haloalkyl), -NHSO2(C1-C6alkyl), -N(C1-C6alkyl)SO2(C1-C6alkyl), or - P(O)(C1-Cealkyl)2 (e.g., -P(O)(CH3)2). In some embodiments, each of the above optional substituents are themselves optionally substituted by one or two of these groups.
[0010] When a range of carbon atoms is used herein, for example, C1-Ce, all ranges, as well as individual numbers of carbon atoms are encompassed. For example, “C1-C3” includes C1-C3, C1-C2, C2-C3, C1, C2, and C3. Thus, for example, a “C1 to C4 alkyl” group refers to all alkyl groups having from 1 to 4 carbons (e.g., 1, 2, 3, or 4), that is, CH3-, CH3CH2-, CH3CH2CH2-, (CH3)2CH-, CH3CH2CH2CH2-, CH3CH2CH(CH3)- and (CH3)3C-. A “C1 to Ce alkyl” group refers to all alkyl groups having from 1 to 6 carbons (e.g., 1, 2, 3, 4, 5, or 6). [0011] As used herein, the term “alkyl” refers to a fully saturated aliphatic hydrocarbon group. The alkyl moiety may be branched or straight chain. Examples of branched alkyl groups include, but are not limited to, iso-propyl, sec-butyl, t-butyl and the like. Examples of straight chain alkyl groups include, but are not limited to, methyl, ethyl, n- propyl, n-butyl, n-pentyl, n-hexyl, n-heptyl and the like. The alkyl group may have 1 to 30 carbon atoms (whenever it appears herein, a numerical range such as “1 to 30” refers to each integer in the given range; e.g., “1 to 30 carbon atoms” means that the alkyl group may consist of 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, or 30 carbon atoms, although the present definition also covers the occurrence of the term “alkyl” where no numerical range is designated). The “alkyl” group may also be a medium size alkyl having 1 to 12 carbon atoms. The “alkyl” group could also be a lower alkyl having 1 to 6 carbon atoms. An alkyl group may be substituted or unsubstituted. By way of example only, “C1-C5 alkyl” indicates that there are one to five carbon atoms in the alkyl chain, i.e., the alkyl chain is selected from methyl, ethyl, n-propyl, isopropyl, n-butyl, isobutyl, tert-butyl, pentyl (branched and straight-chained), etc. Typical alkyl groups include, but are in no way limited to, methyl, ethyl, propyl, isopropyl, butyl, isobutyl, tertiary butyl, pentyl and hexyl. In several embodiments, “Me” is methyl (e.g., CH3).
[0012] Alkylene groups, i.e., fully saturated aliphatic hydrocarbon diradicals, may be identified herein using the term “alk.” For example, the term “C1-Cealk” refers to an aliphatic hydrocarbon diradical that has between 1 and 6 carbon atoms. The term “Co-Cealk” refers to an aliphatic hydrocarbon diradical that is either not present (i.e., Co), or has between 1 and 6 carbon atoms.
[0013] As used herein, “alkenyl” refers to an alkyl group that contains in the straight or branched hydrocarbon chain one or more double bonds. An alkenyl group may be unsubstituted or substituted.
[0014] As used herein, “alkynyl” refers to an alkyl group that contains in the straight or branched hydrocarbon chain one or more triple bonds. An alkynyl group may be unsubstituted or substituted.
[0015] As used herein, “cycloalkyl” refers to a completely saturated (no double or triple bonds) mono- or multi- cyclic hydrocarbon ring system. When composed of two or more rings, the rings may be joined together in a fused fashion. Cycloalkyl groups may contain between 3 and 12 carbon atoms. For example, a Cs-Cecycloalkyl group indicates that there three to six carbon atoms in the ring, that is, the ring is a cyclopropyl, cyclobutyl, cyclopentyl, or cyclohexyl group. A cycloalkyl group may be unsubstituted or substituted.
[0016] As used herein, the term “spirocycloalkyl ring” refers to a cycloalkyl ring that shares one carbon atom with another cyclic ring. For example, a 3-7 membered spirocycloalkyl ring indicates that there are 3, 4, 5, 6, or 7 carbon atoms in the cycloalkyl ring that shares a single carbon atom in common with another cyclic ring. By way of example, shown below are exemplary 3-7 membered spirocycloalkyl groups attached to a piperidine ring:

[0017] As used herein, “aryl” refers to a carbocyclic (all carbon) monocyclic or multicyclic aromatic ring system (including fused ring systems where two carbocyclic rings share a chemical bond) that has a fully delocalized pi-electron system throughout all the rings. The number of carbon atoms in an aryl group can vary. For example, the aryl group can be a Ce-C14 aryl group, a Ce-C1o aryl group, or a Ce aryl group. Examples of aryl groups include, but are not limited to, benzene, naphthalene and azulene. An aryl group may be substituted or unsubstituted.
[0018] As used herein, “heteroaryl” refers to a monocyclic or multicyclic aromatic ring system (a ring system with fully delocalized pi-electron system) that contain(s) one or more heteroatoms, that is, an element other than carbon, including but not limited to, nitrogen, oxygen and sulfur. The number of atoms in the ring(s) of a heteroaryl group can vary. For example, the heteroaryl group can contain 4 to 14 atoms in the ring(s), 5 to 10 atoms in the ring(s) or 5 to 6 atoms in the ring(s). Furthermore, the term “heteroaryl” includes fused ring systems where two rings, such as at least one aryl ring and at least one heteroaryl ring, or at least two heteroaryl rings, share at least one chemical bond. Examples of heteroaryl rings include, but are not limited to, furan, furazan, thiophene, benzothiophene, phthalazine, pyrrole, oxazole, benzoxazole, 1,2,3-oxadiazole, 1,2,4-oxadiazole, thiazole, 1,2,3-thiadiazole, 1,2,4-thiadiazole, benzothiazole, imidazole, benzimidazole, indole, indazole, pyrazole, benzopyrazole, isoxazole, benzoisoxazole, isothiazole, triazole, benzotriazole, thiadiazole, tetrazole, pyridine, pyridazine, pyrimidine, pyrazine, purine, pteridine, quinoline, isoquinoline, quinazoline, quinoxaline, cinnoline, and triazine. Heteroaryl rings may also include bridge head nitrogen atoms. For example but not limited to: pyrazolo[l,5-a]pyridine, imidazo[l,2-a]pyridine, and pyrazolo[l,5-a]pyrimidine. A heteroaryl group may be substituted or unsubstituted.
[0019] As used herein, “heterocycloalkyl” refers to three-, four-, five-, six-, seven-, eight-, nine-, ten-, up to 18-membered monocyclic, bicyclic, and tricyclic ring system wherein carbon atoms together with from 1 to 5 heteroatoms constitute said ring system. A heterocycloalkyl may optionally contain one or more unsaturated bonds situated in such a way, however, that a fully delocalized pi-electron system does not occur throughout all the rings. The heteroatom(s) is an element other than carbon including, but not limited to, oxygen, sulfur, and nitrogen. A heterocycloalkyl may further contain one or more carbonyl or thiocarbonyl functionalities, so as to make the definition include oxo-systems and thiosystems such as lactams, lactones, cyclic imides, cyclic thioimides and cyclic carbamates. When composed of two or more rings, the rings may be joined together in a fused fashion. Additionally, any nitrogens in a heterocycloalkyl may be quatemized. Heterocycloalkyl groups may be unsubstituted or substituted. Examples of such “heterocycloalkyl” groups include but are not limited to, 1,3-dioxin, 1,3-dioxane, 1,4-dioxane, 1,2-di oxolane, 1,3- dioxolane, 1,4-di oxolane, 1,3-oxathiane, 1,4-oxathiin, 1,3 -oxathiolane, 1 ,3-dithiole, 1,3- dithiolane, 1,4-oxathiane, tetrahydro- 1,4-thiazine, 2H-l,2-oxazine, maleimide, succinimide, barbituric acid, thiobarbituric acid, dioxopiperazine, hydantoin, dihydrouracil, trioxane, hexahydro-1, 3, 5-triazine, imidazoline, imidazolidine, isoxazoline, isoxazolidine, oxazoline, oxazolidine, oxazolidinone, thiazoline, thiazolidine, morpholine, oxirane, piperidine A-Oxide, piperidine, piperazine, pyrrolidine, pyrrolidone, pyrrolidione, 4-piperidone, pyrazoline, pyrazolidine, 2-oxopyrrolidine, tetrahydropyran, 4H-pyran, tetrahydrothiopyran, thiamorpholine, thiamorpholine sulfoxide, thiamorpholine sulfone, and their benzo-fused analogs (e.g., benzimidazolidinone, tetrahydroquinoline, 3, 4-m ethylenedi oxyphenyl).
[0020] As used herein, the term “spiroheterocycloalkyl ring” refers to a heterocycloalkyl ring that shares one carbon atom with another cyclic ring. For example, a 3- 7 membered spiroheterocycloalkyl ring indicates that there are 3, 4, 5, 6, or 7 atoms in the heterocycloalkyl ring, and only one of the carbon atoms in that heterocycloalkyl ring is also a member of another cyclic ring. By way of example, shown below are exemplary 3-7 membered spiroheterocycloalkyl groups attached to a piperidine ring:

[0021] As used herein, the term “bridged bicyclic ring”, refers to a ring system comprising two joined cycloalkyl or heterocycloalkyl rings that share at least three at least three atoms For example, a 6-9 membered bridged bicyclic ring indicates that there are 6, 7, 8, or 9 atoms in the bridged bicyclic ring. By way of example, shown below are exemplary 6-9 membered bridged bicyclic rings:

6-membered 7-membered 8-membered 9-membered
[0022] As used herein, the term “amino” refers to a -NH2 group.
[0023] As used herein, the term “hydroxy” refers to a -OH group.
[0024] As used herein, the term “halogen atom” or “halogen” refers to fluorine, chlorine, bromine and iodine.
[0025] The term “pharmaceutically acceptable salt” refers to a salt of a compound that does not cause significant irritation to an organism to which it is administered and does not abrogate the biological activity and properties of the compound. In several embodiments, the salt is an acid addition salt of the compound. Pharmaceutical salts can be obtained by reacting a compound with inorganic acids such as hydrohalic acid (e.g., hydrochloric acid or hydrobromic acid), sulfuric acid, nitric acid and phosphoric acid. Pharmaceutical salts can also be obtained by reacting a compound with an organic acid such as aliphatic or aromatic carboxylic or sulfonic acids, for example formic, acetic, succinic, lactic, malic, tartaric, citric, ascorbic, nicotinic, methanesulfonic, ethanesulfonic, p-toluensulfonic, salicylic, trifluoroacetic acid, or naphthalenesulfonic acid. Pharmaceutical salts can also be obtained by reacting a compound with a base to form a salt such as an ammonium salt, an alkali metal salt, such as a sodium or a potassium salt, an alkaline earth metal salt, such as a calcium or a magnesium salt, a salt of organic bases such as dicyclohexylamine, N-methyl-D-glucamine, tris(hydroxymethyl)methylamine, C1-C7 alkylamine, cyclohexylamine, triethanolamine, ethylenediamine, and salts with amino acids such as arginine and lysine.
[0026] It is understood that, in any compound described herein having one or more chiral centers, if an absolute stereochemistry is not expressly indicated, then each center may independently be of R-configuration or S-configuration or a mixture thereof. Thus, the compounds provided herein may be enantiomerically pure, enantiomerically enriched, racemic mixture, diastereomerically pure, diastereomerically enriched, or a stereoisomeric mixture. In addition, it is understood that, in any compound described herein having one or more double bond(s) generating geometrical isomers that can be defined as E or Z, each double bond may independently be E or Z a mixture thereof. It is understood that, in any compound described herein having one or more chiral centers, all possible diastereomers are also envisioned. It is understood that, in any compound described herein all tautomers are envisioned. It is also understood that, in any compound described herein, all isotopes of the included atoms are envisioned. For example, any instance of hydrogen, may include hydrogen-1 (protium), hydrogen-2 (deuterium), hydrogen-3 (tritium) or other isotopes; any instance of carbon may include carbon-12, carbon-13, carbon-14, or other isotopes; any instance of oxygen may include oxygen-16, oxygen-17, oxygen-18, or other isotopes; any instance of fluorine may include one or more of fluorine- 18, fluorine- 19, or other isotopes; any instance of sulfur may include one or more of sulfur-32, sulfur-34, sulfur-35, sulfur-36, or other isotopes.
[0027] As used herein, the term “kinase inhibitor” means any compound, molecule or composition that inhibits or reduces the activity of a kinase. The inhibition can be achieved by, for example, blocking phosphorylation of the kinase (e.g., competing with adenosine triphosphate (ATP), a phosphorylating entity), by binding to a site outside the active site, affecting its activity by a conformational change, or by depriving kinases of access to the molecular chaperoning systems on which they depend for their cellular stability, leading to their ubiquitylation and degradation. [0028] As used herein, “subject,” “host,” “patient,” and “individual” are used interchangeably and shall be given its ordinary meaning and shall also refer to an organism that has FGFR proteins. This includes mammals, e.g., a human, a non-human primate, ungulates, canines, felines, equines, mice, rats, and the like. The term “mammal” includes both human and non-human mammals.
[0029] The term “sample” or “biological sample” shall be given its ordinary meaning and also encompasses a variety of sample types obtained from an organism and can be used in an imaging, a diagnostic, a prognostic, or a monitoring assay. The term encompasses blood and other liquid samples of biological origin, solid tissue samples, such as a biopsy specimen or tissue cultures or cells derived therefrom and the progeny thereof. The term encompasses samples that have been manipulated in any way after their procurement, such as by treatment with reagents, solubilization, or enrichment for certain components. The term encompasses a clinical sample, and also includes cells in cell culture, cell supernatants, cell lysates, serum, plasma, biological fluids, and tissue samples.
[0030] The terms “treatment,” “treating,” “treat” and the like shall be given its ordinary meaning and shall also include herein to generally refer to obtaining a desired pharmacologic and/or physiologic effect. The effect may be prophylactic in terms of completely or partially preventing a disease or symptom thereof and/or may be therapeutic in terms of a partial or complete stabilization or cure for a disease and/or adverse effect attributable to the disease. “Treatment” as used herein shall be given its ordinary meaning and shall also cover any treatment of a disease in a mammal, particularly a human, and includes: (a) preventing the disease or symptom from occurring in a subject which may be predisposed to the disease or symptom but has not yet been diagnosed as having it; (b) inhibiting the disease symptom, e.g., arresting its development; and/or (c) relieving the disease symptom, e.g., causing regression of the disease or symptom.
[0031] The terms “cancer,” “neoplasm,” and “tumor” are used interchangeably herein, shall be given its ordinary meaning and shall also refer to cells which exhibit relatively autonomous growth, so that they exhibit an aberrant growth phenotype characterized by a significant loss of control of cell proliferation. In general, cells of interest for detection or treatment in the present application include precursors, precancerous e.g., benign), malignant, pre-metastatic, metastatic, and non-metastatic cells. As used herein, “FGFR related cancer” denotes those cancers that involve an increased activity in a mutant FGFR kinase, for example, the continued activation of FGFR.
[0032] The term “control” refers shall be given its ordinary meaning and shall also include a sample or standard used for comparison with a sample which is being examined, processed, characterized, analyzed, etc. In several embodiments, the control is a sample obtained from a healthy patient or a non-tumor tissue sample obtained from a patient diagnosed with a tumor. In several embodiments, the control is a historical control or standard reference value or range of values. In several embodiments, the control is a comparison to a wild-type FGFR arrangement or scenario.
[0033] With respect to the use of substantially any plural and/or singular terms herein, those having skill in the art can translate from the plural to the singular and/or from the singular to the plural as is appropriate to the context and/or application. The various singular/plural permutations may be expressly set forth herein for sake of clarity. The indefinite article “a” or “an” does not exclude a plurality. The mere fact that certain measures are recited in mutually different dependent claims does not indicate that a combination of these measures cannot be used to advantage. Any reference signs in the claims should not be construed as limiting the scope.
[0034] In some aspects, the disclosure is directed to compounds of formula (I) or a pharmaceutically acceptable salt thereof.
or a pharmaceutically acceptable salt thereof.

[0035] In some aspects, X in the compounds of formula (I) is O, S, or NR wherein
R is H or C1-Csalkyl.
[0036] In some embodiments, X is O.
[0037] In other embodiments, X is S. [0038] In some embodiments, X is NR.
[0039] In some embodiments, R is H.
[0040] In other embodiments, R is C1-C3alkyl, such as, for example, C3alkyl, C2alkyl, C1alkyl, methyl, ethyl, propyl, and the like.
[0041] In some aspects, n in the compounds of formula (I) is 1 or 2.
[0042] In some embodiments, n is 1.
[0043] In some embodiments, n is 2.
[0044] In some aspects, m in the compounds of formula (I) is 1 or 2.
[0045] In some embodiments, m is 1.
[0046] In some embodiments, m is 2.
[0047] In some embodiments, n is 1 and m is 1.
[0048] In some embodiments, n is i and m is 2.
[0049] In some embodiments, n is 2 and m is 1.
[0050] In some embodiments, n is 2 and m is 2.
[0051] In some aspects, R1 in the compounds of formula (I) H, optionally substituted C1-C6alkyl, or NH2.
[0052] In some aspects, R1 in the compounds of formula (I) is H or C1-Cealkyl.
[0053] In some aspects, R1 in the compounds of formula (I) is H.
[0054] In some aspects, R1 in the compounds of formula (I) is NH2.
[0055] In some embodiments, R1 is C1-Cealkyl, such as, for example, C1-Cealkyl, C1-C5alkyl, C1-C4alkyl, C1-C3alkyl, C1-C2alkyl, C1alkyl, C2alkyl, C3alkyl, C4alkyl, C5alkyl, C6alkyl, methyl, ethyl, n-propyl, isopropyl, n-butyl, isosbutyl, sec-butyl, pentanyl, hexanyl, and the like.
[0056] In some embodiments, R1 is -CH3.
[0057] In some aspects, R1 in the compounds of formula (I) is optionally substituted C1-Cealkyl.
[0058] In some embodiments, R1 is -CH2NH2.
[0059] In some embodiments, R1 is -CH2NHCH3.
[0060] In some embodiments, R1 is -CH2N(CH3)2.
[0061] In some embodiments, [0062] In some aspects, R2 in the compounds of formula (I) is -Co-C6alkN=S(0)(C1- C6alkyl)(C1-C6alkyl), -NH-C1-C6alk-N=S(O)(C1-C6alkyl)(C1-C6alkyl), or -Co-C3alk-C(=0)- Co-C3alk-N=S(0)(C1-C6alkyl)(C1-C6alkyl).
[0062] In some aspects, R2 in the compounds of formula (I) is -Co-C6alkN=S(0)(C1- C6alkyl)(C1-C6alkyl), -NH-C1-C6alk-N=S(O)(C1-C6alkyl)(C1-C6alkyl), or -Co-C3alk-C(=0)- Co-C3alk-N=S(0)(C1-C6alkyl)(C1-C6alkyl).

[0063] In some aspects, R2 in the compounds of formula (I) is -Co-Cealk- N=S(O)(C1-C6alkyl)(C1-C6alkyl), or -NH-C1-C6alk-N=S(O)(C1-C6alkyl)(C1-C6alkyl).
[0064] In some embodiments, R2 is -Co-C6alk-N=S(0)(C1-C6alkyl)(C1-C6alkyl), such as, for example, -N=S(O)(C1-C6alkyl)(C1-C6alkyl), -C1-C6alk-N=S(O)(C1-C6alkyl)(C1- C6alkyl), -C1-C5alk-N=S(O)(C1-C6alkyl)(C1-C6alkyl), -C1-C4alk-N=S(O)(C1-C6alkyl)(C1- C6alkyl), -C1-C3alk-N=S(O)(C1-C6alkyl)(C1-C6alkyl), -C1-C2alk-N=S(O)(C1-C6alkyl)(C1- C6alkyl), -C1alk-N=S(O)(C1-C6alkyl)(C1-C6alkyl), -C2alk-N=S(O)(C1-C6alkyl)(C1-C6alkyl), - C3alk-N=S(O)(C1-C6alkyl)(C1-C6alkyl), -C4alk-N=S(O)(C1-C6alkyl)(C1-C6alkyl), -C5alk- N=S(O)(C1-C6alkyl)(C1-C6alkyl), -C6alk-N=S(O)(C1-C6alkyl)(C1-C6alkyl), -CH2- N=S(O)(C1-C6alkyl)(C1-C6alkyl), -CH2CH2-N=S(O)(C1-C6alkyl)(C1-C6alkyl), -CH2CH2CH2- N=S(O)(C1-C6alkyl)(C1-C6alkyl), -CH2(CH3)CH2-N=S(O)(C1-C6alkyl)(C1-C6alkyl), - CH2CH2CH2CH2-N=S(O)(C1-C6alkyl)(C1-C6alkyl), -CH2(CH3)CHCH2-N=S(O)(CI- C6alkyl)(C1-C6alkyl), -CH2CH2(CH3)CH-N=S(O)(C1-C6alkyl)(C1-C6alkyl), -CH2(CH3)2C- N=S(O)(C1-C6alkyl)(C1-C6alkyl), and the like.
[0065] In some embodiments, R2 is -Co-C6alk-N=S(0)(C1-C6alkyl)(C1-C6alkyl), such as, for example, -Co-C6alk-N=S(0)(C1-C5alkyl)(C1-C6alkyl), -Co-C6alk-N=S(0)(C1- C4alkyl)(C1-C6alkyl), -Co-C6alk-N=S(0)(C1-C3alkyl)(C1-C6alkyl), -Co-C6alk-N=S(0)(C1- C2alkyl)(C1-C6alkyl), -Co-C6alk-N=S(0)(C1alkyl)(C1-C6alkyl), -Co-C6alk- N=S(O)(C2alkyl)(C1-C6alkyl), -Co-C6alk-N=S(0)(C3alkyl)(C1-C6alkyl), -Co-C6alk- N=S(O)(C4alkyl)(C1-C6alkyl), -Co-C6alk-N=S(0)(C5alkyl)(C1-C6alkyl), -Co-C6alk- N=S(O)(C4alkyl)(C1-C6alkyl), -Co-C6alk-N=S(0)(CH3)(C1-C6alkyl), -Co-C6alk- N=S(O)(C2H5)(C1-C6alkyl), -C0-C6alk-N=S(O)(C3H7)(C1-C6alkyl), -Co-C6alk- N=S(O)(isopropyl)(C1-C6alkyl), -Co-C6alk-N=S(0)(C4H9)(C1-C6alkyl), -Co-C6alk- N=S(O)(isobutyl)(C1-C6alkyl), -C0-C6alk-N=S(O)(sec-butyl)(C1-C6alkyl), -Co-C6alk- N=S(O)(tert-butyl)(C1-C6alkyl), -Co-C6alk-N=S(0)(C5Hii)(C1-C6alkyl), -Co-C6alk- N=S(O)(C6Hi3)(C1-C6alkyl) and the like.
[0066] In some embodiments, R2 is -N=S(O)(CH3)2.
[0067] In some embodiments, R2 is -CH2-N=S(O)(CH3)2. [0068] In some embodiments, R2 is -NH-C1-C6alk-N=S(O)(C1-C6alkyl)(C1- Cealkyl), such as, for example, -NH-C1-C6alk-N=S(O)(C1-C6alkyl)(C1-C6alkyl), -NH-C1- C5alk-N=S(O)(C1-C6alkyl)(C1-C6alkyl), -NH-C1-C4alk-N=S(O)(C1-C6alkyl)(C1-C6alkyl), - NH-C1-C3alk-N=S(O)(C1-C6alkyl)(C1-C6alkyl), -NH-C1-C2alk-N=S(O)(C1-C6alkyl)(C1- C6alkyl), -NH-C1alk-N=S(O)(C1-C6alkyl)(C1-C6alkyl), -NH-C2alk-N=S(O)(C1-C6alkyl)(C1- C6alkyl), -NH-C3alk-N=S(O)(C1-C6alkyl)(C1-C6alkyl), -NH-C4alk-N=S(O)(C1-C6alkyl)(C1- C6alkyl), -NH-C5alk-N=S(O)(C1-C6alkyl)(C1-C6alkyl), -NH-C6alk-N=S(O)(C1-C6alkyl)(C1- C6alkyl), -NH-CH2-N=S(O)(C1-C6alkyl)(C1-C6alkyl), -NH-CH2CH2-N=S(O)(C1-C6alkyl)(C1- C6alkyl), -NH-CH2CH2CH2-N=S(O)(C1-C6alkyl)(C1-C6alkyl), -NH-CH2(CH3)CH2- N=S(O)(C1-C6alkyl)(C1-C6alkyl), -NH-CH2CH2CH2CH2-N=S(O)(C1-C6alkyl)(C1-C6alkyl), - NH-CH2(CH3)CHCH2-N=S(O)(C1-C6alkyl)(C1-C6alkyl), -NH-CH2CH2(CH3)CH- N=S(O)(C1-C6alkyl)(C1-C6alkyl), -NH-CH2(CH3)2C-N=S(O)(C1-C6alkyl)(C1-C6alkyl), and the like.
[0069] In some embodiments, R2 is -NH-C1-C6alk-N=S(O)(C1-C6alkyl)(C1- Cealkyl), such as, for example, -NH-C1-C6alk-N=S(O)(C1-C5alkyl)(C1-C6alkyl), -NH-C1- C6alk-N=S(O)(C1-C4alkyl)(C1-C6alkyl), -NH-C1-C6alk-N=S(O)(C1-C3alkyl)(C1-C6alkyl), - NH-C1-C6alk-N=S(O)(C1-C2alkyl)(C1-C6alkyl), -NH-C1-C6alk-N=S(O)(C1alkyl)(C1- C6alkyl), -NH-C1-C6alk-N=S(O)(C2alkyl)(C1-C6alkyl), -NH-C1-C6alk-N=S(O)(C3alkyl)(C1- C6alkyl), -NH-C1-C6alk-N=S(O)(C4alkyl)(C1-C6alkyl), -NH-C1-C6alk-N=S(O)(C5alkyl)(C1- C6alkyl), -NH-C1-C6alk-N=S(O)(C4alkyl)(C1-C6alkyl), -NH-C1-C6alk-N=S(O)(CH3)(C1- C6alkyl), -NH-C1-C6alk-N=S(O)(C2H5)(C1-C6alkyl), -NH-C1-C6alk-N=S(O)(C3H7)(C1- C6alkyl), -NH-C1-C6alk-N=S(O)(isopropyl)(C1-C6alkyl), -NH-C1-C6alk-N=S(O)(C4H9)(C1- C6alkyl), -NH-C1-C6alk-N=S(O)(isobutyl)(C1-C6alkyl), -NH-C1-C6alk-N=S(O)(sec- butyl)(C1-C6alkyl), -NH-C1-C6alk-N=S(O)(tert-butyl)(C1-C6alkyl), -NH-C1-C6alk- N=S(O)(C5Hii)(C1-C6alkyl), -NH-C1-C6alk-N=S(O)(C6Hi3)(C1-C6alkyl) and the like.
[0070] In some embodiments, R2 is -NH-CH2CH2-N=S(O)(CH3)2.
[0071] In some embodiments, R2 is -Co-C3alk-C(=0)-Co-C3alk-N=S(0)(C1- CealkylXC1-Cealkyl), such as, for example, -C(=0)-Co-C3alk-N=S(0)(C1-C6alkyl)(C1- C6alkyl), -C1alk-C(=O)-C0-C3alk-N=S(O)(C1-C6alkyl)(C1-C6alkyl), -C2alk-C(=O)-C0-C3alk- N=S(O)(C1-C6alkyl)(C1-C6alkyl), -C3alk-C(=0)-Co-C3alk-N=S(0)(C1-C6alkyl)(C1-C6alkyl), - Co-C3alk-C(=0)-N=S(0)(C1-C6alkyl)(C1-C6alkyl), -Co-C3alk-C(=0)-C1alk-N=S(0)(C1- C6alkyl)(C1-C6alkyl) -C0-C3alk-C(=O)-C2alk-N=S(O)(C1-C6alkyl)(C1-C6alkyl), -Co-C3alk- C(=O)-C3alk-N=S(O)(C1-C6alkyl)(C1-C6alkyl), -Co-C3alk-C(=0)-Co-C3alk-
N=S(O)(C1alkyl)(C1-C6alkyl), -Co-C3alk-C(=0)-Co-C3alk-N=S(0)(C2alkyl)(C1-C6alkyl), -Co-
C3alk-C(=0)-Co-C3alk-N=S(0)(C3alkyl)(C1-C6alkyl), -C0-C3alk-C(=O)-C0-C3alk-
N=S(O)(C4alkyl)(C1-C6alkyl), -Co-C3alk-C(=0)-Co-C3alk-N=S(0)(C5alkyl)(C1-C6alkyl), -Co-
C3alk-C(=0)-Co-C3alk-N=S(0)(C6alkyl)(C1-C6alkyl), -Co-C3alk-C(=0)-Co-C3alk-
N=S(O)(C1-C6alkyl)(C1alkyl), -Co-C3alk-C(=0)-Co-C3alk-N=S(0)(C1-C6alkyl)(C2alkyl), -Co-
C3alk-C(=0)-Co-C3alk-N=S(0)(C1-C6alkyl)(C3alkyl), -C0-C3alk-C(=O)-C0-C3alk-
N=S(O)(C1-C6alkyl)(C4alkyl), -Co-C3alk-C(=0)-Co-C3alk-N=S(0)(C1-C6alkyl)(C5alkyl), or -
Co-C3alk-C(=0)-Co-C3alk-N=S(0)(C1-C6alkyl)(C6alkyl).
[0072] In some embodiments, R2 is -C(=O)-N=S(O)(CH3)2.
[0073] In some aspects, one or two of Q1, Q2, Q3, Q4 in the compounds of formula
(I) are N and the others are each independently CR5a.
[0074] In some embodiments, Q1 is N and Q2, Q3, and Q4 are each independently
CR5a.
[0075] In some embodiments, Q2 is N and Q1, Q3, and Q4 are each independently
CR5a.
[0076] In some embodiments, Q3 is N and Q1, Q2, and Q4 are each independently CR5a.
[0077] In some embodiments, Q4 is N and Q1, Q2, and Q3 are each independently CR5a.
[0078] In other embodiments, two of Q1, Q2, Q3, Q4 is N and the others are each independently CR5a.
[0079] In some embodiments, Q1 and Q2 are each N, and Q3, and Q4 are each independently CR5a.
[0080] In some embodiments, Q1 and Q3 are each N, and Q2 and Q4 are each independently CR5a.
[0081] In some embodiments, Q1 and Q4 are each N, and Q2 and Q3 are each independently CR5a.
[0082] In some embodiments, Q2 and Q3 are each N, and Q1 and Q4 are each independently CR5a.
[0083] In some embodiments, Q2 and Q4 are each N, and Q1 and Q3 are each independently CR5a. [0084] In some embodiments, Q3 and Q4 are each N, and Q1 and Q2 are each independently CR5a.
[0085] In some aspects of the compounds of formula (I), each R5a is independently H, halogen, -CN, -S(O)2C1-C3alkyl, OCF3, OC1-C3alkyl, or C1-C3alkyl.
[0086] In some embodiments, at least one R5a is H.
[0087] In some embodiments, at least one R5a is halogen, z.e., -F, -Cl, -Br, or -I.
[0088] In some embodiments, at least one R5a is -F.
[0089] In some embodiments, at least one R5a is -CN.
[0090] In some embodiments at least one R5a is -SO2C1-Qalkyl, such as, for example, -SO2C1alkyl, -SO2C2alkyl, -SO2C3alkyl, -SO2CFBCH3, -SO2CH5, and the like. In some embodiments, at least one R5a is -SO2CH3.
[0091] In some embodiments, R5a is OCF3.
[0092] In some embodiments, at least one R5a is OC1-C3alkyl, such as, for example, OC1-C3alkyl, OC1-C2alkyl, OC1alkyl, OC2alkyl, OC3alkyl, -OCH3, -OCH2CH3, -Opropyl, and the like. In some embodiments at least one R5a is -OCH3.
[0093] In some embodiments, at least one R5a is C1-C3alkyl, such as, for example, C1-C3alkyl, C1-C2alkyl, C1alkyl, C2alkyl, C3alkyl, -CH3, -CH2CH3, -propyl, and the like. In some embodiments at least one R5a is -CH3.
[0094] In some aspects, Q5, Q6, Q7, Q8, and Q9 in the compounds of formula (I) are each independently N or CR5, wherein one or two of Q5, Q6, Q7, Q8, and Q9is N and the remainder are each independently CR5.
[0095] In some embodiments, one of Q5, Q6, Q7, Q8, or Q9 is N, and the remainder are each independently CR5.
[0096] In some embodiments, Q5 is N and Q6, Q7, Q8, and Q9 are each independently CR5.
[0097] In some embodiments, Q6 is N and Q5, Q7, Q8, and Q9 are each independently CR5.
[0098] In some embodiments, Q7 is N and Q5, Q6, Q8, and Q9 are each independently CR5.
[0099] In some embodiments, Q8 is N and Q5, Q6, Q7, and Q9 are each independently CR5. [00100] In some embodiments, Q9 is N and Q5, Q6, Q7, and Q8 are each independently CR5.
[00101] In other embodiments, two of Q5, Q6, Q7, Q8, or Q9 is each N, and the remainder are each independently CR5.
[00102] In some embodiments, Q5 and Q6 are each N, and Q7, Q8, and Q9 are each independently CR5.
[00103] In some embodiments, Q5 and Q7 are each N, and Q6, Q8, and Q9 are each independently CR5.
[00104] In some embodiments, Q5 and Q8 are each N, and Q6, Q7, and Q9 are each independently CR5.
[00105] In some embodiments, Q5 and Q9 are each N, and Q6, Q7, and Q8 are each independently CR5.
[00106] In some embodiments, Q6 and Q7 are each N, and Q5, Q8, and Q9 are each independently CR5.
[00107] In some embodiments, Q6 and Q8 are each N, and Q5, Q7, and Q9 are each independently CR5.
[00108] In some embodiments, Q6 and Q9 are each N, and Q5, Q7, and Q8 are each independently CR5.
[00109] In some embodiments, Q7 and Q8 are each N, and Q5, Q6, and Q9 are each independently CR5.
[00110] In some embodiments, Q7 and Q9 are each N, and Q5, Q6, and Q8 are each independently CR5.
[00111] In some embodiments, Q8 and Q9 are each N, and Q5, Q6, and Q7 are each independently CR5.
[00112] In some aspects of the disclosure, each R5 in the compounds of formula (I), is independently H, halogen, C1-Csalkyl, C1-Csalkoxyl, or cycloalkyl.
[00113] In some embodiments of the compounds of formula (I), at least one R5 is H.
[00114] In some embodiments of the compounds of formula (I), at least one R5 is halogen, such as, -F, -Cl, -Br, or -I.
[00115] In some embodiments, at least one R5 is -Cl. [00116] In some embodiments of the compounds of formula (I), at least one R5 is C1-C3alkyl, such as, for example, C1-C3alkyl, C1-C2alkyl, C1alkyl, C2alkyl, Csalkyl, -CH3, - CH2CH3, -propyl, and the like.
[00117] In some embodiments, at least one R5 is -CH3.
[00118] In some embodiments of the compounds of formula (I), at least one R5 is C1-C3alkoxyl, such as, for example, C1-C3alkoxyl, C1-C2alkoxyl, C1alkoxyl, C2alkoxyl, Csalkoxyl, -OCH3, -OCH2CH3, -propoxyl, and the like.
[00119] In some embodiments, at least one R5 is -OCH3.
[00120] In some embodiments of the compounds of formula (I), at least one R5 is cycloalkyl, such as, for example, cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, or cycloheptyl, and the like.
[00121] In some embodiments of the compounds of formula (I), two R5 are halogen, and the remaining R5 are H.
[00122] In other embodiments of the compounds of formula (I), two R5 are -Cl, and the remaining R5 are H.
[00123] In some embodiments of the compounds of formula (I), Q5 and Q9 are each independently CR5 wherein each R5 is independently a halogen; Q6 and Q8 are each independently CR5 wherein each R5 is H; and Q7 is N.
[00124] In some embodiments of the compounds of formula (I), Q5, Q8, and Q9 are each independently CR5 wherein each R5 is independently a halogen; Q6 is CR5 wherein R5 is H; and Q7 is N.
[00125] In some embodiments of the compounds of formula (I), Q5 and Q9 are each independently CR5 wherein each R5 is -Cl; Q6 and Q8 are each independently CR5 wherein R5 is H; and Q7 is N.
[00126] In some embodiments of the compounds of formula (I), Q5 and Q9 are each independently CR5 wherein each R5 is -Cl; Q6 is CR5 wherein R5 is H; Q8 is CR5 wherein R5 is -F; and Q7 is N.
[00127] In some embodiments of the compounds of formula (I), Q5 and Q9 are each independently CR5 wherein each R5 is independently halogen; Q6 is CR5 wherein R5 is H and Q8 is CR5 wherein R5 is C1-Csalkyl; and Q7 is N. [00128] In some embodiments of the compounds of formula (I), Q5 and Q9 are each independently CR5 wherein each R5 is -Cl; Q6 is CR5 wherein R5 is H and Q8 is CR5 wherein R5 is -CH3; and Q7 is N.
[00129] In some embodiments of the compounds of formula (I), Q5 and Q9 are each independently CR5 wherein each R5 is halogen; Q6 is CR5 wherein R5 is H and Q8 is N; and Q7 is CR5 wherein R5 is H.
[00130] In some embodiments of the compounds of formula (I), Q5 and Q9 are each independently CR5 wherein each R5 is -Cl; Q6 is CR5 wherein R5 is H and Q8 is N; and Q7 is CR5 wherein R5 is H.
[00131] In some embodiments of the compounds of formula (I), Q5 and Q9 are each independently CR5 wherein each R5 is independently a halogen; Q6 is CR5 wherein R5 is H and Q8 is N; and Q7 is N.
[00132] In some embodiments of the compounds of formula (I), Q5 and Q9 are each independently CR5 wherein each R5 is -Cl; Q6 is CR5 wherein R5 is H and Q8 is N; and Q7 is N.
[00133] In some embodiments of the compounds of formula (I), Q5 and Q9 are each independently CR5 wherein each R5 is independently a C1-Csalkyl; Q6 is CR5 wherein R5 is H and Q8 is N; and Q7 is N.
[00134] In some embodiments of the compounds of formula (I), Q5 and Q9 are each independently CR5 wherein each R5 is -CH3; Q6 is CR5 wherein R5 is H and Q8 is N; and Q7 is N.
[00135] In some aspects of the disclosure, R6 in the compounds of formula (I) is C1- Cealkyl, such as, for example, C1-Cealkyl, C1-Csalkyl, C1-C4alkyl, C1-Csalkyl, C1-C2alkyl, C1alkyl, C2alkyl, Csalkyl, C4alkyl, Csalkyl, Cealkyl, methyl, ethyl, n-propyl, isopropyl, n- butyl, isosbutyl, sec-butyl, pentanyl, hexanyl, and the like.
[00136] In some embodiments of the compounds of the disclosure, R6 is -CH3.
[00137] In some aspects, R7 in the compounds of formula (I) is H, halogen, -C1- Cealkyl, -C1-Cealkoxyl, or -cycloalkyl.
[00138] In some embodiments, R7 in the compounds of formula (I) is H.
[00139] In some embodiments, R7 in the compounds of formula (I) is halogen, such as, for example, -F, -Cl, -Br, or -I.
[00140] In some embodiments, R7 in the compounds of formula (I) is -F. [00141] In other embodiments, R7 in the compounds of formula (I) is -Cl.
[00142] In some embodiments, R7 in the compounds of formula (I) is -C1-Cealkyl, such as, for example, substituted or unsubstituted: C1-Cealkyl, C1-Csalkyl, C1-C4alkyl, C1- C3alkyl, C1-C2alkyl, C1alkyl, C2alkyl, C2alkyl, C4alkyl, Csalkyl, Cealkyl, methyl, ethyl, n- propyl, isopropyl, n-butyl, isosbutyl, sec-butyl, pentanyl, hexanyl, and the like.
[00143] In some embodiments, R7 is -CH3.
[00144] In some embodiments, R7 in the compounds of formula (I) is -C1-Ce alkoxyl, such as, for example, -C1-Cealkoxyl, -C1-C5alkoxyl, -C1-C4alkoxyl, -C1-Csalkoxyl, - C1-C2alkoxyl, -C1alkoxyl, -C2alkoxyl, -C3alkoxyl, -C4alkoxyl, -C5alkoxyl, -Cealkoxyl, - OCH3, -OCH2CH3, -propoxyl, and the like.
[00145] In some embodiments, R7is -OCH3.
[00146] In some embodiments, R7 in the compounds of formula (I) is -cycloalkyl, such as, for example, cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, or cycloheptyl, and the like.
[00147] In some aspects, R8 is H, halogen, -C1-Cealkyl, -C1-Cealkoxyl, or - cycloalkyl.
[00148] In some embodiments, R8 in the compounds of formula (I) is H.
[00149] In some embodiments, R8 in the compounds of formula (I) is halogen, such as, for example, -F, -Cl, -Br, or -I.
[00150] In some embodiments, R8 in the compounds of formula (I) is -F.
[00151] In some embodiments, R8 in the compounds of formula (I) is -C1-Cealkyl, such as, for example, substituted or unsubstituted: C1-Cealkyl, C1-Csalkyl, C1-C4alkyl, C1- Csalkyl, C1-C2alkyl, C1alkyl, C2alkyl, Csalkyl, C4alkyl, Csalkyl, Cealkyl, methyl, ethyl, n- propyl, isopropyl, n-butyl, isosbutyl, sec-butyl, pentanyl, hexanyl, and the like.
[00152] In some embodiments, R8 is -CH3.
[00153] In some embodiments, R8 in the compounds of formula (I) is -C1-Ce alkoxyl, such as, for example, -C1-Cealkoxyl, -C1-Csalkoxyl, -C1-C4alkoxyl, -C1-Csalkoxyl, - C1-C2alkoxyl, -C1alkoxyl, -C2alkoxyl, -Csalkoxyl, -C4alkoxyl, -Csalkoxyl, -Cealkoxyl, - OCH3, -OCH2CH3, -propoxyl, and the like.
[00154] In some embodiments, R8is -OCH3. [00155] In some embodiments, R8 in the compounds of formula (I) is -cycloalkyl, such as, for example, cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, or cycloheptyl, and the like.
[00156] In some embodiments of the compounds of formula (I), R7 is H and R8 is H.
[00157] In some embodiments of the compounds of formula (I), R7 is -F and R8 is H.
[00158] In some embodiments of the compounds of formula (I), R7 is -Cl and R8 is H.
[00159] In some embodiments of the compounds of formula (I), R7 is -CH3 and R8 is H.
[00160] In some embodiments of the compounds of formula (I), R7 is -OCH3 and R8 is H.
[00161] In some embodiments of the compounds of formula (I), R7 is -H and R8 is - F.
[00162] In some embodiments of the compounds of formula (I), R7 is -H and R8 is - Cl.
[00163] In some embodiments of the compounds of formula (I), R7 is -H and R8 is - CH3.
[00164] In some embodiments of the compounds of formula (I), R7 is -H and R8 is OCH3.
[00165] In some aspects, the disclosure is directed to the compounds of formula (I) that are compounds of formula (IA): or a pharmaceutically acceptable salt thereof, wherein
or a pharmaceutically acceptable salt thereof, wherein

Q2 and Q4 are each N, or one of Q2 or Q4 is N and the other is CR5a;
R5a is H, F, -SO2CH3, or -CN;
R5 is H or CH3; and
R7 is H, F, or OCH3, and R1 and R2 are as described above for formula (I).
[00166] In some embodiments of the compounds of formula (IA), R5 is H.
[00167] In some embodiments of the compounds of formula (IA), R5 is CH3.
[00168] In some embodiments of the compounds of formula (IA), R7 is H.
[00169] In some embodiments of the compounds of formula (IA), R7 is F.
[00170] In some embodiments of the compounds of formula (IA), R7 is OCH3.
[00171] In some embodiments of the compounds of formula (IA), Q2 and Q4 are each N.
[00172] In some embodiments of the compounds of formula (IA), one of Q2 or Q4 is N and the other is CR5a.
[00173] In some embodiments of the compounds of formula (IA) wherein one of Q2 or Q4 is N and the other is CR5a, R5a is H or F.
[00174] In some embodiments of the compounds of formula (IA) one of Q2 or Q4 is N and the other is CR5a, R5a is H.
[00175] In some embodiments of the compounds of formula (IA) one of Q2 or Q4 is N and the other is CR5a, R5a is F.
[00176] In some embodiments of the compounds of formula (IA) one of Q2 or Q4 is N and the other is CR5a, R5a is CN or SO2CH3.
[00177] In some embodiments of the compounds of formula (IA) one of Q2 or Q4 is N and the other is CR5a, R5a is CN.
[00178] In some embodiments of the compounds of formula (IA) one of Q2 or Q4 is N and the other is CR5a, R5a is SO2CH3.
[00179] In some aspects, the disclosure is directed to the compounds of formula (I) that are compounds of formula (IB):

R2 is -Co-C6alk-N=S(0)(C1-C6alkyl)(C1-C6alkyl), or -NHC1-C6alk-N=S(O)(C1- C6alkyl)C1-C6alkyl;
R7 is H or -OC1-ealkyl; and
R5a is halo, or -CN.
[00180] In other aspects, the disclosure is directed to the compounds of formula (I) that are compounds of formula (IC): or a pharmaceutically acceptable salt thereof, wherein
or a pharmaceutically acceptable salt thereof, wherein

R1 is optionally substituted C1-Cealkyl, or NH2;
R2 is -Co-C6alkN=S(0)(C1-C6alkyl)(C1-C6alkyl), -NH-C1-C6alk-N=S(O)(C1-
C6alkyl)(C1-C6alkyl), or -Co-C3alk-C(=0)-Co-C3alk-N=S(0)(C1-C6alkyl)(C1-C6alkyl);
R7is H or -OC1-C6alkyl; and
R5a is halo, or -CN.
[00181] In some aspects, R7 in the compounds of formula (IB) or formula (IC) is H or -OC1-6alkyl.
[00182] In some embodiments, R7 in the compounds of formula (IB) or formula (IC) is H. [00183] In some embodiments, R7 in the compounds of formula (IB) or formula (IC) is -OC1-Cealkyl, such as, for example, -OC1-Cealkyl, -OC1-Csalkyl, -OC1-C4alkyl, -OC1- C3alkyl, -OC1-C2alkyl, -OC1alkyl, -OC2alkyl, -OC3alkyl, -OC4alkyl, -OC5alkyl, -OC6alkyl, - OCH3, -OCH2CH3, -propoxyl, and the like.
[00184] In some embodiments, R7 in the compounds of formula (IB) or formula (IC) is -OCH3.
[00185] In some aspects, R5a in the compounds of formula (IB) or formula (IC) is halo or -CN.
[00186] In some embodiments, R5a in the compounds of formula (IB) or formula (IC) is -CN.
[00187] In some embodiments, R5a in the compounds of formula (IB) or formula (IC) is halogen, such as, for example, -F, -Cl, -Br, or -I.
[00188] In some embodiments, R5a in the compounds of formula (IB) or formula (IC) is F.
[00189] In some embodiments, R5a in the compounds of formula (IB) or formula (IC) is Cl.
[00190] In some embodiments, R5a in the compounds of formula (IB) or formula (IC) is Br.
[00191] In some aspects, R2 in the compounds of formula (IB) is -Co-Cealk- N=S(O)(C1-C6alkyl)(C1-C6alkyl), or -NHC1-C6alk-N=S(O)(C1-C6alkyl)(C1-C6alkyl).
[00192] In some aspects, R2 in the compounds of formula (I) is -Co-Cealk- N=S(O)(C1-C6alkyl)(C1-C6alkyl), or -NH-C1-C6alk-N=S(O)(C1-C6alkyl)(C1-C6alkyl).
[00193] In some aspects, R2 in the compounds of formula (I) or formula (IC) is -Co- C6alkN=S(O)(C1-C6alkyl)(C1-C6alkyl), -NH-C1-C6alk-N=S(O)(C1-C6alkyl)(C1-C6alkyl), or - Co-C3alk-C(=0)-Co-C3alk-N=S(0)(C1-C6alkyl)(C1-C6alkyl).
[00194] In some embodiments, R2 is -Co-C6alk-N=S(0)(C1-C6alkyl)(C1-C6alkyl), such as, for example, -N=S(O)(C1-C6alkyl)(C1-C6alkyl), -C1-C6alk-N=S(O)(C1-C6alkyl)(C1- C6alkyl), -C1-C5alk-N=S(O)(C1-C6alkyl)(C1-C6alkyl), -C1-C4alk-N=S(O)(C1-C6alkyl)(C1- C6alkyl), -C1-C3alk-N=S(O)(C1-C6alkyl)(C1-C6alkyl), -C1-C2alk-N=S(O)(C1-C6alkyl)(C1- C6alkyl), -C1alk-N=S(O)(C1-C6alkyl)(C1-C6alkyl), -C2alk-N=S(O)(C1-C6alkyl)(C1-C6alkyl), - C3alk-N=S(O)(C1-C6alkyl)(C1-C6alkyl), -C4alk-N=S(O)(C1-C6alkyl)(C1-C6alkyl), -C5alk- N=S(O)(C1-C6alkyl)(C1-C6alkyl), -C6alk-N=S(O)(C1-C6alkyl)(C1-C6alkyl), -CH2- N=S(O)(C1-C6alkyl)(C1-C6alkyl), -CH2CH2-N=S(O)(C1-C6alkyl)(C1-C6alkyl), -CH2CH2CH2- N=S(O)(C1-C6alkyl)(C1-C6alkyl), -CH2(CH3)CH2-N=S(O)(C1-C6alkyl)(C1-C6alkyl), - CH2CH2CH2CH2-N=S(O)(C1-C6alkyl)(C1-C6alkyl), -CH2(CH3)CHCH2-N=S(O)(CI- C6alkyl)(C1-C6alkyl), -CH2CH2(CH3)CH-N=S(O)(C1-C6alkyl)(C1-C6alkyl), -CH2(CH3)2C- N=S(O)(C1-C6alkyl)(C1-C6alkyl), and the like.
[00195] In some embodiments, R2 is -Co-C6alk-N=S(0)(C1-C6alkyl)(C1-C6alkyl), such as, for example, -Co-C6alk-N=S(0)(C1-C5alkyl)(C1-C6alkyl), -Co-C6alk-N=S(0)(C1- C4alkyl)(C1-C6alkyl), -Co-C6alk-N=S(0)(C1-C3alkyl)(C1-C6alkyl), -Co-C6alk-N=S(0)(C1- C2alkyl)(C1-C6alkyl), -Co-C6alk-N=S(0)(C1alkyl)(C1-C6alkyl), -Co-C6alk- N=S(O)(C2alkyl)(C1-C6alkyl), -Co-C6alk-N=S(0)(C3alkyl)(C1-C6alkyl), -Co-C6alk- N=S(O)(C4alkyl)(C1-C6alkyl), -Co-C6alk-N=S(0)(C5alkyl)(C1-C6alkyl), -Co-C6alk- N=S(O)(C4alkyl)(C1-C6alkyl), -Co-C6alk-N=S(0)(CH3)(C1-C6alkyl), -Co-C6alk- N=S(O)(C2H5)(C1-C6alkyl), -Co-C6alk-N=S(0)(C3H7)(C1-C6alkyl), -Co-C6alk- N=S(O)(isopropyl)(C1-C6alkyl), -Co-C6alk-N=S(0)(C4H9)(C1-C6alkyl), -Co-C6alk- N=S(O)(isobutyl)(C1-C6alkyl), -Co-C6alk-N=S(0)(sec-butyl)(C1-C6alkyl), -Co-C6alk- N=S(O)(tert-butyl)(C1-C6alkyl), -Co-C6alk-N=S(0)(C5Hii)(C1-C6alkyl), -Co-C6alk- N=S(O)(C6Hi3)(C1-C6alkyl) and the like.
[00196] In some embodiments, R2 is -N=S(O)(CH3)2.
[00197] In some embodiments, R2 is -CH2-N=S(O)(CH3)2.
[00198] In some embodiments, R2 is -NH-C1-C6alk-N=S(O)(C1-C6alkyl)(C1- Cealkyl), such as, for example, -NH-C1-C6alk-N=S(O)(C1-C6alkyl)(C1-C6alkyl), -NH-C1- C5alk-N=S(O)(C1-C6alkyl)(C1-C6alkyl), -NH-C1-C4alk-N=S(O)(C1-C6alkyl)(C1-C6alkyl), - NH-C1-C3alk-N=S(O)(C1-C6alkyl)(C1-C6alkyl), -NH-C1-C2alk-N=S(O)(C1-C6alkyl)(C1- C6alkyl), -NH-C1alk-N=S(O)(C1-C6alkyl)(C1-C6alkyl), -NH-C2alk-N=S(O)(C1-C6alkyl)(C1- C6alkyl), -NH-C3alk-N=S(O)(C1-C6alkyl)(C1-C6alkyl), -NH-C4alk-N=S(O)(C1-C6alkyl)(C1- C6alkyl), -NH-C5alk-N=S(O)(C1-C6alkyl)(C1-C6alkyl), -NH-C6alk-N=S(O)(C1-C6alkyl)(C1- C6alkyl), -NH-CH2-N=S(O)(C1-C6alkyl)(C1-C6alkyl), -NH-CH2CH2-N=S(O)(C1-C6alkyl)(C1- C6alkyl), -NH-CH2CH2CH2-N=S(O)(C1-C6alkyl)(C1-C6alkyl), -NH-CH2(CH3)CH2- N=S(O)(C1-C6alkyl)(C1-C6alkyl), -NH-CH2CH2CH2CH2-N=S(O)(C1-C6alkyl)(C1-C6alkyl), - NH-CH2(CH3)CHCH2-N=S(O)(C1-C6alkyl)(C1-C6alkyl), -NH-CH2CH2(CH3)CH- N=S(O)(C1-C6alkyl)(C1-C6alkyl), -NH-CH2(CH3)2C-N=S(O)(C1-C6alkyl)(C1-C6alkyl), and the like. [00199] In some embodiments, R2 is -NH-C1-C6alk-N=S(O)(C1-C6alkyl)(C1- Cealkyl), such as, for example, -NH-C1-C6alk-N=S(O)(C1-C5alkyl)(C1-C6alkyl), -NH-C1- C6alk-N=S(O)(C1-C4alkyl)(C1-C6alkyl), -NH-C1-C6alk-N=S(O)(C1-C3alkyl)(C1-C6alkyl), - NH-C1-C6alk-N=S(O)(C1-C2alkyl)(C1-C6alkyl), -NH-C1-C6alk-N=S(O)(C1alkyl)(C1- C6alkyl), -NH-C1-C6alk-N=S(O)(C2alkyl)(C1-C6alkyl), -NH-C1-C6alk-N=S(O)(C3alkyl)(C1- C6alkyl), -NH-C1-C6alk-N=S(O)(C4alkyl)(C1-C6alkyl), -NH-C1-C6alk-N=S(O)(C5alkyl)(C1- C6alkyl), -NH-C1-C6alk-N=S(O)(C4alkyl)(C1-C6alkyl), -NH-C1-C6alk-N=S(O)(CH3)(C1- C6alkyl), -NH-C1-C6alk-N=S(O)(C2H5)(C1-C6alkyl), -NH-C1-C6alk-N=S(O)(C3H7)(C1- C6alkyl), -NH-C1-C6alk-N=S(O)(isopropyl)(C1-C6alkyl), -NH-C1-C6alk-N=S(O)(C4H9)(C1- C6alkyl), -NH-C1-C6alk-N=S(O)(isobutyl)(C1-C6alkyl), -NH-C1-C6alk-N=S(O)(sec- butyl)(C1-C6alkyl), -NH-C1-C6alk-N=S(O)(tert-butyl)(C1-C6alkyl), -NH-C1-C6alk- N=S(O)(C5Hii)(C1-C6alkyl), -NH-C1-C6alk-N=S(OXC6Hi3)(C1-C6alkyl) and the like.
[00200] In some embodiments, R2 is -NH-CH2CH2-N=S(O)(CH3)2.
[00201] In some embodiments, R2 is -Co-C3alk-C(=0)-Co-C3alk-N=S(0)(C1- CealkylXC1-Cealkyl), such as, for example, -C(=0)-Co-C3alk-N=S(0)(C1-C6alkyl)(C1- C6alkyl), -C1alk-C(=O)-C0-C3alk-N=S(O)(C1-C6alkyl)(C1-C6alkyl), -C2alk-C(=O)-C0-C3alk- N=S(O)(C1-C6alkyl)(C1-C6alkyl), -C3alk-C(=O)-C0-C3alk-N=S(O)(C1-C6alkyl)(C1-C6alkyl), - Co-C3alk-C(=0)-N=S(0)(C1-C6alkyl)(C1-C6alkyl), -Co-C3alk-C(=0)-C1alk-N=S(0)(C1- C6alkyl)(C1-C6alkyl) -C0-C3alk-C(=O)-C2alk-N=S(O)(C1-C6alkyl)(C1-C6alkyl), -Co-C3alk- C(=O)-C3alk-N=S(O)(C1-C6alkyl)(C1-C6alkyl), -C0-C3alk-C(=O)-C0-C3alk- N=S(O)(C1alkyl)(C1-C6alkyl), -Co-C3alk-C(=0)-Co-C3alk-N=S(0)(C2alkyl)(C1-C6alkyl), -Co- C3alk-C(=O)-C0-C3alk-N=S(O)(C3alkyl)(C1-C6alkyl), -C0-C3alk-C(=O)-C0-C3alk- N=S(O)(C4alkyl)(C1-C6alkyl), -Co-C3alk-C(=0)-Co-C3alk-N=S(0)(C5alkyl)(C1-C6alkyl), -Co- C3alk-C(=O)-C0-C3alk-N=S(O)(C6alkyl)(C1-C6alkyl), -Co-C3alk-C(=0)-Co-C3alk- N=S(O)(C1-C6alkyl)(C1alkyl), -C0-C3alk-C(=O)-C0-C3alk-N=S(O)(C1-C6alkyl)(C2alkyl), -Co- C3alk-C(=O)-C0-C3alk-N=S(O)(C1-C6alkyl)(C3alkyl), -C0-C3alk-C(=O)-C0-C3alk- N=S(O)(C1-C6alkyl)(C4alkyl), -Co-C3alk-C(=0)-Co-C3alk-N=S(0)(C1-C6alkyl)(C5alkyl), or - Co-C3alk-C(=0)-Co-C3alk-N=S(0)(C1-C6alkyl)(C6alkyl).
[00202] In some embodiments, R2 is -C(=O)-N=S(O)(CH3)2.
[00203] In some aspects, R1 in the compounds of formula (I) or formula (IC) is optionally substituted C1-Cealkyl, orNH2.
[00204] In some embodiments, R1 is -CH3. [00205] In some embodiments, R1 is -CH2NH2.
[00206] In some embodiments, R1 is -CH2NHCH3.
[00207] In some embodiments, R1 is -CH2N(CH3)2.
[00208] In some embodiments,

[00209] In some embodiments, R1 is -NH2.
[00210] In some aspects, the compound of formula (I) is:


[00211] In some aspects, the compound of formula (IB) is a compound of formula (IB1), or a pharmaceutically acceptable salt thereof.

[00212] In some aspects, the compound of formula (IB) is a compound of formula
(IB2), or a pharmaceutically acceptable salt thereof. [00213] In some aspects, the compound of formula (IB) is a compound of formula (IB3), or a pharmaceutically acceptable salt thereof.
[00213] In some aspects, the compound of formula (IB) is a compound of formula (IB3), or a pharmaceutically acceptable salt thereof.


[00214] In some aspects, the compound of formula (IB) is a compound of formula
(IB4), or a pharmaceutically acceptable salt thereof.

[00215] In some aspects, the compound of formula (IB) is a compound of formula
(IB5), or a pharmaceutically acceptable salt thereof.

[00216] In some aspects, the compound of formula (IB) is a compound of formula (IB6), or a pharmaceutically acceptable salt thereof.

[00217] In some aspects, the compound of formula (I) is: pharmaceutically acceptable salt thereof.
pharmaceutically acceptable salt thereof.

[00218] In some aspects, the compound of formula (I) is: or a pharmaceutically acceptable salt thereof.
or a pharmaceutically acceptable salt thereof.

[00219] In some aspects, the compound of formula (I) is: pharmaceutically acceptable salt thereof.
pharmaceutically acceptable salt thereof.

[00220] In some aspects, the compound of formula (I) is:

[00224] In some aspects, the compound of formula (I) is: or a pharmaceutically acceptable salt thereof.
or a pharmaceutically acceptable salt thereof.

[00226] In some aspects, the compound of formula (I) is: or a pharmaceutically acceptable salt thereof.
or a pharmaceutically acceptable salt thereof.

[00227] In some aspects, the compound of formula (I) is: pharmaceutically acceptable salt thereof.
pharmaceutically acceptable salt thereof.

[00228] In some aspects, the disclosure is directed to the compounds shown in the
Examples below, or pharmaceutically acceptable salts thereof.
[00229] References herein to formula (I) or subgenera thereof are meant to encompass the identified formula and any subgenera of those formula disclosed herein. For example, references to formula (I) also encompass subgenera formula IA, IB, IB1, IB2, IB3, IB4, IB5, IB6 and IC. [00230] Stereoisomers of compounds of formula (I) are also contemplated by the present disclosure. Thus, the disclosure encompasses all stereoisomers and constitutional isomers of any compound disclosed or claimed herein, including all enantiomers and diastereomers, or mixtures thereof.
[00231] Pharmaceutically acceptable salts and solvates of the compounds of formula (I) are also within the scope of the disclosure.
[00232] It is to be appreciated that certain features of the invention which are, for clarity, described herein in the context of separate embodiments, may also be provided in combination in a single embodiment. That is, unless obviously incompatible or specifically excluded, each individual embodiment is deemed to be combinable with any other embodiment s) and such a combination is considered to be another embodiment. Conversely, various features of the invention that are, for brevity, described in the context of a single embodiment, may also be provided separately or in any sub-combination. While an embodiment may be described as part of a series of steps or part of a more general structure, each said step may also be considered an independent embodiment in itself, combinable with others.
Pharmaceutical compositions and methods of administration
[00233] The subject pharmaceutical compositions are typically formulated to provide a therapeutically effective amount of a compound of the present disclosure as the active ingredient, or a pharmaceutically acceptable salt, ester, prodrug, solvate, hydrate or derivative thereof. In some embodiments, the pharmaceutical compositions contain a compound of the present disclosure or a pharmaceutically acceptable salt thereof, and one or more pharmaceutically acceptable excipients, carriers, including inert solid diluents and fillers, diluents, including sterile aqueous solution and various organic solvents, permeation enhancers, solubilizers and adjuvants.
[00234] The subject pharmaceutical compositions can be administered alone or in combination with one or more other agents, which are also typically administered in the form of pharmaceutical compositions. Where desired, the one or more compounds of the invention and other agent(s) may be mixed into a preparation or both components may be formulated into separate preparations to use them in combination separately or at the same time. [00235] In some embodiments, the concentration of one or more compounds provided in the pharmaceutical compositions of the present invention is less than 100%, 90%, 80%, 70%, 60%, 50%, 40%, 30%, 20%, 19%, 18%, 17%, 16%, 15%, 14%, 13%, 12%, 11%, 10%, 9%, 8%, 7%, 6%, 5%, 4%, 3%, 2%, 1%, 0.9%, 0.8%, 0.7%, 0.6%, 0.5%, 0.4%, 0.3%, 0.2%, 0.1%, 0.09%, 0.08%, 0.07%, 0.06%, 0.05%, 0.04%, 0.03%, 0.02%, 0.01%, 0.009%, 0.008%, 0.007%, 0.006%, 0.005%, 0.004%, 0.003%, 0.002%, 0.001%, 0.0009%, 0.0008%, 0.0007%, 0.0006%, 0.0005%, 0.0004%, 0.0003%, 0.0002%, or 0.0001% (or a number in the range defined by and including any two numbers above) w/w, w/v or v/v.
[00236] In some embodiments, the concentration of one or more compounds of the invention is greater than 90%, 80%, 70%, 60%, 50%, 40%, 30%, 20%, 19.75%, 19.50%, 19.25%, 19%, 18.75%, 18.50%, 18.25% 18%, 17.75%, 17.50%, 17.25% 17%, 16.75%, 16.50%, 16.25%, 16%, 15.75%, 15.50%, 15.25% 15%, 14.75%, 14.50%, 14.25% 14%, 13.75%, 13.50%, 13.25%, 13%, 12.75%, 12.50%, 12.25%, 12%, 11.75%, 11.50%, 11.25% 11%, 10.75%, 10.50%, 10.25% 10%, 9.75%, 9.50%, 9.25%, 9%, 8.75%, 8.50%, 8.25% 8%, 7.75%, 7.50%, 7.25%, 7%, 6.75%, 6.50%, 6.25%, 6%, 5.75%, 5.50%, 5.25%, 5%, 4.75%, 4.50%, 4.25%, 4%, 3.75%, 3.50%, 3.25%, 3%, 2.75%, 2.50%, 2.25%, 2%, 1.75%, 1.50%, 1.25% , 1%, 0.9%, 0.8%, 0.7%, 0.6%, 0.5%, 0.4%, 0.3%, 0.2%, 0.1%, 0.09%, 0.08%, 0.07%, 0.06%, 0.05%, 0.04%, 0.03%, 0.02%, 0.01%, 0.009%, 0.008%, 0.007%, 0.006%, 0.005%, 0.004%, 0.003%, 0.002%, 0.001%, 0.0009%, 0.0008%, 0.0007%, 0.0006%, 0.0005%, 0.0004%, 0.0003%, 0.0002%, or 0.0001% (or a number in the range defined by and including any two numbers above) w/w, w/v, or v/v.
[00237] In some embodiments, the concentration of one or more compounds of the invention is in the range from approximately 0.0001% to approximately 50%, approximately 0.001% to approximately 40%, approximately 0.01% to approximately 30%, approximately 0.02% to approximately 29%, approximately 0.03% to approximately 28%, approximately 0.04% to approximately 27%, approximately 0.05% to approximately 26%, approximately 0.06% to approximately 25%, approximately 0.07% to approximately 24%, approximately 0.08% to approximately 23%, approximately 0.09% to approximately 22%, approximately 0.1% to approximately 21%, approximately 0.2% to approximately 20%, approximately 0.3% to approximately 19%, approximately 0.4% to approximately 18%, approximately 0.5% to approximately 17%, approximately 0.6% to approximately 16%, approximately 0.7% to approximately 15%, approximately 0.8% to approximately 14%, approximately 0.9% to approximately 12%, approximately 1% to approximately 10% w/w, w/v or v/v.
[00238] In some embodiments, the concentration of one or more compounds of the invention is in the range from approximately 0.001% to approximately 10%, approximately 0.01% to approximately 5%, approximately 0.02% to approximately 4.5%, approximately 0.03% to approximately 4%, approximately 0.04% to approximately 3.5%, approximately 0.05% to approximately 3%, approximately 0.06% to approximately 2.5%, approximately 0.07% to approximately 2%, approximately 0.08% to approximately 1.5%, approximately 0.09% to approximately 1%, approximately 0.1% to approximately 0.9% w/w, w/v or v/v.
[00239] In some embodiments, the amount of one or more compounds of the invention is equal to or less than 10 g, 9.5 g, 9.0 g, 8.5 g, 8.0 g, 7.5 g, 7.0 g, 6.5 g, 6.0 g, 5.5 g, 5.0 g, 4.5 g, 4.0 g, 3.5 g, 3.0 g, 2.5 g, 2.0 g, 1.5 g, 1.0 g, 0.95 g, 0.9 g, 0.85 g, 0.8 g, 0.75 g, 0.7 g, 0.65 g, 0.6 g, 0.55 g, 0.5 g, 0.45 g, 0.4 g, 0.35 g, 0.3 g, 0.25 g, 0.2 g, 0.15 g, 0.1 g, 0.09 g, 0.08 g, 0.07 g, 0.06 g, 0.05 g, 0.04 g, 0.03 g, 0.02 g, 0.01 g, 0.009 g, 0.008 g, 0.007 g, 0.006 g, 0.005 g, 0.004 g, 0.003 g, 0.002 g, 0.001 g, 0.0009 g, 0.0008 g, 0.0007 g, 0.0006 g, 0.0005 g, 0.0004 g, 0.0003 g, 0.0002 g, or 0.0001 g (or a number in the range defined by and including any two numbers above).
[00240] In some embodiments, the amount of one or more compounds of the invention is more than 0.0001 g, 0.0002 g, 0.0003 g, 0.0004 g, 0.0005 g, 0.0006 g, 0.0007 g, 0.0008 g, 0.0009 g, 0.001 g, 0.0015 g, 0.002 g, 0.0025 g, 0.003 g, 0.0035 g, 0.004 g, 0.0045 g, 0.005 g, 0.0055 g, 0.006 g, 0.0065 g, 0.007 g, 0.0075 g, 0.008 g, 0.0085 g, 0.009 g, 0.0095 g, 0.01 g, 0.015 g, 0.02 g, 0.025 g, 0.03 g, 0.035 g, 0.04 g, 0.045 g, 0.05 g, 0.055 g, 0.06 g, 0.065 g, 0.07 g, 0.075 g, 0.08 g, 0.085 g, 0.09 g, 0.095 g, 0.1 g, , 0.15 g, 0.2 g, , 0.25 g, 0.3 g, , 0.35 g, 0.4 g, , 0.45 g, 0.5 g, 0.55 g, 0.6 g, , 0.65 g, 0.7 g, 0.75 g, 0.8 g, 0.85 g, 0.9 g, 0.95 g, 1 g, 1.5 g, 2 g, 2.5, 3 g, 3.5, 4 g, 4.5 g, 5 g, 5.5 g, 6 g, 6.5g, 7 g, 7.5g, 8 g, 8.5 g, 9 g, 9.5 g, or 10 g (or a number in the range defined by and including any two numbers above).
[00241] In some embodiments, the amount of one or more compounds of the invention is in the range of 0.0001-10 g, 0.0005-9 g, 0.001-8 g, 0.005-7 g, 0.01-6 g, 0.05-5 g, 0.1-4 g, 0.5-4 g, or 1-3 g.
[00242] In some embodiments, the compounds according to the invention are effective over a wide dosage range. For example, in the treatment of adult humans, dosages from 0.01 to 1000 mg, from 0.5 to 100 mg, from 1 to 50 mg per day, and from 5 to 40 mg per day are examples of dosages that may be used. An exemplary dosage is 10 to 30 mg per day. The exact dosage will depend upon the route of administration, the form in which the compound is administered, the subject to be treated, the body weight of the subject to be treated, and the preference and experience of the attending physician.
[00243] Unless otherwise noted, the amounts of the compounds described herein are set forth on a free base basis. That is, the amounts indicate that amount of the compound administered, exclusive of, for example, solvent (such as in solvates) or counterions (such as in pharmaceutically acceptable salts).
[00244] Described below are non- limiting exemplary pharmaceutical compositions and methods for preparing the same.
Pharmaceutical compositions for oral administration.
[00245] In some embodiments, the invention provides a pharmaceutical composition for oral administration containing a compound of the invention, and a pharmaceutical excipient suitable for oral administration.
[00246] In some embodiments, the invention provides a solid pharmaceutical composition for oral administration containing: (i) an effective amount of a compound of the invention; optionally (ii) an effective amount of a second agent; and (iii) a pharmaceutical excipient suitable for oral administration. In some embodiments, the composition further contains: (iv) an effective amount of a third agent.
[00247] In some embodiments, the pharmaceutical composition may be a liquid pharmaceutical composition suitable for oral consumption. Pharmaceutical compositions of the invention suitable for oral administration can be presented as discrete dosage forms, such as capsules, cachets, or tablets, or liquids or aerosol sprays each containing a predetermined amount of an active ingredient as a powder or in granules, a solution, or a suspension in an aqueous or non-aqueous liquid, an oil-in- water emulsion, or a water-in-oil liquid emulsion. Such dosage forms can be prepared by any of the methods of pharmacy, but all methods include the step of bringing the active ingredient into association with the carrier, which constitutes one or more necessary ingredients. In general, the compositions are prepared by uniformly and intimately admixing the active ingredient with liquid carriers or finely divided solid carriers or both, and then, if necessary, shaping the product into the desired presentation. For example, a tablet can be prepared by compression or molding, optionally with one or more accessory ingredients. Compressed tablets can be prepared by compressing in a suitable machine the active ingredient in a free- flowing form such as powder or granules, optionally mixed with an excipient such as, but not limited to, a binder, a lubricant, an inert diluent, and/or a surface active or dispersing agent. Molded tablets can be made by molding in a suitable machine a mixture of the powdered compound moistened with an inert liquid diluent.
[00248] This invention further encompasses anhydrous pharmaceutical compositions and dosage forms comprising an active ingredient, since water can facilitate the degradation of some compounds. For example, water may be added (e.g., 5%) in the pharmaceutical arts as a means of simulating long-term storage in order to determine characteristics such as shelf- life or the stability of formulations over time. Anhydrous pharmaceutical compositions and dosage forms of the invention can be prepared using anhydrous or low moisture containing ingredients and low moisture or low humidity conditions. Pharmaceutical compositions and dosage forms of the invention which contain lactose can be made anhydrous if substantial contact with moisture and/or humidity during manufacturing, packaging, and/or storage is expected. An anhydrous pharmaceutical composition may be prepared and stored such that its anhydrous nature is maintained. Accordingly, anhydrous compositions may be packaged using materials known to prevent exposure to water such that they can be included in suitable formulary kits. Examples of suitable packaging include, but are not limited to, hermetically sealed foils, plastic or the like, unit dose containers, blister packs, and strip packs.
[00249] An active ingredient can be combined in an intimate admixture with a pharmaceutical carrier according to conventional pharmaceutical compounding techniques. The carrier can take a wide variety of forms depending on the form of preparation desired for administration. In preparing the compositions for an oral dosage form, any of the usual pharmaceutical media can be employed as carriers, such as, for example, water, glycols, oils, alcohols, flavoring agents, preservatives, coloring agents, and the like in the case of oral liquid preparations (such as suspensions, solutions, and elixirs) or aerosols; or carriers such as starches, sugars, micro-crystalline cellulose, diluents, granulating agents, lubricants, binders, and disintegrating agents can be used in the case of oral solid preparations, in some embodiments without employing the use of lactose. For example, suitable carriers include powders, capsules, and tablets, with the solid oral preparations. If desired, tablets can be coated by standard aqueous or nonaqueous techniques.
[00250] Binders suitable for use in pharmaceutical compositions and dosage forms include, but are not limited to, com starch, potato starch, or other starches, gelatin, natural and synthetic gums such as acacia, sodium alginate, alginic acid, other alginates, powdered tragacanth, guar gum, cellulose and its derivatives (e.g., ethyl cellulose, cellulose acetate, carboxymethyl cellulose calcium, sodium carboxymethyl cellulose), polyvinyl pyrrolidone, methyl cellulose, pre-gelatinized starch, hydroxypropyl methyl cellulose, microcrystalline cellulose, and mixtures thereof.
[00251] Examples of suitable fillers for use in the pharmaceutical compositions and dosage forms disclosed herein include, but are not limited to, talc, calcium carbonate (e.g., granules or powder), microcrystalline cellulose, powdered cellulose, dextrates, kaolin, mannitol, silicic acid, sorbitol, starch, pre-gelatinized starch, and mixtures thereof.
[00252] Disintegrants may be used in the compositions of the invention to provide tablets that disintegrate when exposed to an aqueous environment. Too much of a disintegrant may produce tablets which may disintegrate in the bottle. Too little may be insufficient for disintegration to occur and may thus alter the rate and extent of release of the active ingredient(s) from the dosage form. Thus, a sufficient amount of disintegrant that is neither too little nor too much to detrimentally alter the release of the active ingredient(s) may be used to form the dosage forms of the compounds disclosed herein. The amount of disintegrant used may vary based upon the type of formulation and mode of administration, and may be readily discernible to those of ordinary skill in the art. About 0.5 to about 15 weight percent of disintegrant, or about 1 to about 5 weight percent of disintegrant, may be used in the pharmaceutical composition. Disintegrants that can be used to form pharmaceutical compositions and dosage forms of the invention include, but are not limited to, agar-agar, alginic acid, calcium carbonate, microcrystalline cellulose, croscarmellose sodium, crospovidone, polacrilin potassium, sodium starch glycolate, potato or tapioca starch, other starches, pre-gelatinized starch, other starches, clays, other algins, other celluloses, gums or mixtures thereof.
[00253] Lubricants which can be used to form pharmaceutical compositions and dosage forms of the invention include, but are not limited to, calcium stearate, magnesium stearate, mineral oil, light mineral oil, glycerin, sorbitol, mannitol, polyethylene glycol, other glycols, stearic acid, sodium lauryl sulfate, talc, hydrogenated vegetable oil (e.g., peanut oil, cottonseed oil, sunflower oil, sesame oil, olive oil, com oil, and soybean oil), zinc stearate, ethyl oleate, ethyl laureate, agar, or mixtures thereof. Additional lubricants include, for example, a syloid silica gel, a coagulated aerosol of synthetic silica, or mixtures thereof. A lubricant can optionally be added, in an amount of less than about 1 weight percent of the pharmaceutical composition.
[00254] When aqueous suspensions and/or elixirs are desired for oral administration, the active ingredient therein may be combined with various sweetening or flavoring agents, coloring matter or dyes and, if so desired, emulsifying and/or suspending agents, together with such diluents as water, ethanol, propylene glycol, glycerin and various combinations thereof.
[00255] The tablets can be uncoated or coated by known techniques to delay disintegration and absorption in the gastrointestinal tract and thereby provide a sustained action over a longer period. For example, a time delay material such as glyceryl monostearate or glyceryl distearate can be employed. Formulations for oral use can also be presented as hard gelatin capsules wherein the active ingredient is mixed with an inert solid diluent, for example, calcium carbonate, calcium phosphate or kaolin, or as soft gelatin capsules wherein the active ingredient is mixed with water or an oil medium, for example, peanut oil, liquid paraffin or olive oil.
[00256] Surfactant which can be used to form pharmaceutical compositions and dosage forms of the invention include, but are not limited to, hydrophilic surfactants, lipophilic surfactants, and mixtures thereof. That is, a mixture of hydrophilic surfactants may be employed, a mixture of lipophilic surfactants may be employed, or a mixture of at least one hydrophilic surfactant and at least one lipophilic surfactant may be employed.
[00257] A suitable hydrophilic surfactant may generally have an HLB value of at least 10, while suitable lipophilic surfactants may generally have an HLB value of or less than about 10. An empirical parameter used to characterize the relative hydrophilicity and hydrophobicity of non-ionic amphiphilic compounds is the hydrophilic-lipophilic balance (" HLB" value). Surfactants with lower HLB values are more lipophilic or hydrophobic, and have greater solubility in oils, while surfactants with higher HLB values are more hydrophilic, and have greater solubility in aqueous solutions. [00258] Hydrophilic surfactants are generally considered to be those compounds having an HLB value greater than about 10, as well as anionic, cationic, or zwitterionic compounds for which the HLB scale is not generally applicable. Similarly, lipophilic (i.e., hydrophobic) surfactants are compounds having an HLB value equal to or less than about 10. However, HLB value of a surfactant is merely a rough guide generally used to enable formulation of industrial, pharmaceutical and cosmetic emulsions.
[00259] Hydrophilic surfactants may be either ionic or non-ionic. Suitable ionic surfactants include, but are not limited to, alkylammonium salts; fusidic acid salts; fatty acid derivatives of amino acids, oligopeptides, and polypeptides; glyceride derivatives of amino acids, oligopeptides, and polypeptides; lecithins and hydrogenated lecithins; lysolecithins and hydrogenated lysolecithins; phospholipids and derivatives thereof; lysophospholipids and derivatives thereof; carnitine fatty acid ester salts; salts of alkylsulfates; fatty acid salts; sodium docusate; acyl lactylates; mono- and di-acetylated tartaric acid esters of mono- and di-glycerides; succinylated mono- and di-glycerides; citric acid esters of mono- and diglycerides; and mixtures thereof.
[00260] Within the aforementioned group, ionic surfactants include, by way of example: lecithins, lysolecithin, phospholipids, lysophospholipids and derivatives thereof; carnitine fatty acid ester salts; salts of alkyl sulfates; fatty acid salts; sodium docusate; acylactylates; mono- and di-acetylated tartaric acid esters of mono- and di-glycerides; succinylated mono- and di-glycerides; citric acid esters of mono- and di-glycerides; and mixtures thereof.
[00261] Ionic surfactants may be the ionized forms of lecithin, lysolecithin, phosphatidylcholine, phosphatidylethanolamine, phosphatidylglycerol, phosphatidic acid, phosphatidylserine, lysophosphatidylcholine, lysophosphatidylethanolamine, lysophosphatidylglycerol, lysophosphatidic acid, lysophosphatidylserine, PEG- phosphatidylethanolamine, PVP -phosphatidylethanolamine, lactylic esters of fatty acids, stearoyl-2-lactylate, stearoyl lactylate, succinylated monoglycerides, mono/diacetylated tartaric acid esters of mono/diglycerides, citric acid esters of mono/diglycerides, cholyl sarcosine, caproate, caprylate, caprate, laurate, myristate, palmitate, oleate, ricinoleate, linoleate, linolenate, stearate, lauryl sulfate, teracecyl sulfate, docusate, lauroyl carnitines, palmitoyl carnitines, myristoyl carnitines, and salts and mixtures thereof. [00262] Hydrophilic non-ionic surfactants may include, but are not limited to, alkylglucosides; alkylmaltosides; alkylthioglucosides; lauryl macrogolglycerides; polyoxyalkylene alkyl ethers such as polyethylene glycol alkyl ethers; polyoxyalkylene alkylphenols such as polyethylene glycol alkyl phenols; polyoxyalkylene alkyl phenol fatty acid esters such as polyethylene glycol fatty acids monoesters and polyethylene glycol fatty acids diesters; polyethylene glycol glycerol fatty acid esters; polyglycerol fatty acid esters; polyoxyalkylene sorbitan fatty acid esters such as polyethylene glycol sorbitan fatty acid esters; hydrophilic transesterification products of a polyol with at least one member of the group consisting of glycerides, vegetable oils, hydrogenated vegetable oils, fatty acids, and sterols; polyoxyethylene sterols, derivatives, and analogues thereof; polyoxyethylated vitamins and derivatives thereof; polyoxyethylene-polyoxypropylene block copolymers; and mixtures thereof; polyethylene glycol sorbitan fatty acid esters and hydrophilic transesterification products of a polyol with at least one member of the group consisting of triglycerides, vegetable oils, and hydrogenated vegetable oils. The polyol may be glycerol, ethylene glycol, polyethylene glycol, sorbitol, propylene glycol, pentaerythritol, or a saccharide.
[00263] Other hydrophilic-non-ionic surfactants include, without limitation, PEG- 10 laurate, PEG- 12 laurate, PEG-20 laurate, PEG-32 laurate, PEG-32 dilaurate, PEG- 12 oleate, PEG- 15 oleate, PEG-20 oleate, PEG-20 di oleate, PEG-32 oleate, PEG-200 oleate, PEG-400 oleate, PEG- 15 stearate, PEG-32 distearate, PEG-40 stearate, PEG- 100 stearate, PEG-20 dilaurate, PEG-25 glyceryl trioleate, PEG-32 dioleate, PEG-20 glyceryl laurate, PEG-30 glyceryl laurate, PEG-20 glyceryl stearate, PEG-20 glyceryl oleate, PEG-30 glyceryl oleate, PEG-30 glyceryl laurate, PEG-40 glyceryl laurate, PEG-40 palm kernel oil, PEG-50 hydrogenated castor oil, PEG-40 castor oil, PEG-35 castor oil, PEG-60 castor oil, PEG-40 hydrogenated castor oil, PEG-60 hydrogenated castor oil, PEG-60 corn oil, PEG-6 caprate/ capryl ate glycerides, PEG-8 caprate/caprylate glycerides, polyglyceryl- 10 laurate, PEG-30 cholesterol, PEG-25 phyto sterol, PEG-30 soya sterol, PEG-20 trioleate, PEG-40 sorbitan oleate, PEG-80 sorbitan laurate, polysorbate 20, polysorbate 80, POE-9 lauryl ether, POE-23 lauryl ether, POE- 10 oleyl ether, POE-20 oleyl ether, POE-20 stearyl ether, tocopheryl PEG- 100 succinate, PEG-24 cholesterol, polyglyceryl-lOoleate, Tween 40, Tween 60, sucrose monostearate, sucrose mono laurate, sucrose monopalmitate, PEG 10-100 nonyl phenol series, PEG 15-100 octyl phenol series, and poloxamers. [00264] Suitable lipophilic surfactants include, by way of example only: fatty alcohols; glycerol fatty acid esters; acetylated glycerol fatty acid esters; lower alcohol fatty acids esters; propylene glycol fatty acid esters; sorbitan fatty acid esters; polyethylene glycol sorbitan fatty acid esters; sterols and sterol derivatives; polyoxyethylated sterols and sterol derivatives; polyethylene glycol alkyl ethers; sugar esters; sugar ethers; lactic acid derivatives of mono- and di-glycerides; hydrophobic transesterification products of a polyol with at least one member of the group consisting of glycerides, vegetable oils, hydrogenated vegetable oils, fatty acids and sterols; oil-soluble vitamins/vitamin derivatives; and mixtures thereof. Within this group, preferred lipophilic surfactants include glycerol fatty acid esters, propylene glycol fatty acid esters, and mixtures thereof, or are hydrophobic transesterification products of a polyol with at least one member of the group consisting of vegetable oils, hydrogenated vegetable oils, and triglycerides.
[00265] In one embodiment, the composition may include a solubilizer to ensure good solubilization and/or dissolution of the compound of the present invention and to minimize precipitation of the compound of the present invention. This can be especially important for compositions for non-oral use, e.g., compositions for injection. A solubilizer may also be added to increase the solubility of the hydrophilic drug and/or other components, such as surfactants, or to maintain the composition as a stable or homogeneous solution or dispersion.
[00266] Examples of suitable solubilizers include, but are not limited to, the following: alcohols and polyols, such as ethanol, isopropanol, butanol, benzyl alcohol, ethylene glycol, propylene glycol, butanediols and isomers thereof, glycerol, pentaerythritol, sorbitol, mannitol, transcutol, dimethyl isosorbide, polyethylene glycol, polypropylene glycol, polyvinylalcohol, hydroxypropyl methylcellulose and other cellulose derivatives, cyclodextrins and cyclodextrin derivatives; ethers of polyethylene glycols having an average molecular weight of about 200 to about 6000, such as tetrahydrofurfuryl alcohol PEG ether (glycofurol) or methoxy PEG ; amides and other nitrogen-containing compounds such as 2- pyrrolidone, 2-piperidone, s-caprolactam, N-alkylpyrrolidone, N-hydroxyalkylpyrrolidone, N-alkylpiperidone, N-alkylcaprolactam, dimethylacetamide and polyvinylpyrrolidone; esters such as ethyl propionate, tributylcitrate, acetyl tri ethyl citrate, acetyl tributyl citrate, tri ethyl citrate, ethyl oleate, ethyl caprylate, ethyl butyrate, triacetin, propylene glycol monoacetate, propylene glycol diacetate, s-caprolactone and isomers thereof, 5- valerolactone and isomers thereof, P -butyrolactone and isomers thereof; and other solubilizers known in the art, such as dimethyl acetamide, dimethyl isosorbide, N-methyl pyrrolidones, monooctanoin, diethylene glycol monoethyl ether, and water.
[00267] Mixtures of solubilizers may also be used. Examples include, but not limited to, triacetin, tri ethyl citrate, ethyl oleate, ethyl caprylate, dimethylacetamide, N- methylpyrrolidone, N-hydroxyethylpyrrolidone, polyvinylpyrrolidone, hydroxypropyl methylcellulose, hydroxypropyl cyclodextrins, ethanol, polyethylene glycol 200-100, glycofurol, transcutol, propylene glycol, and dimethyl isosorbide. Particularly preferred solubilizers include sorbitol, glycerol, triacetin, ethyl alcohol, PEG-400, glycofurol and propylene glycol.
[00268] The amount of solubilizer that can be included is not particularly limited. The amount of a given solubilizer may be limited to a bioacceptable amount, which may be readily determined by one of skill in the art. In some circumstances, it may be advantageous to include amounts of solubilizers far in excess of bioacceptable amounts, for example to maximize the concentration of the drug, with excess solubilizer removed prior to providing the composition to a subject using conventional techniques, such as distillation or evaporation. Thus, if present, the solubilizer can be in a weight ratio of 10%, 25%o, 50%), 100%o, or up to about 200%> by weight, based on the combined weight of the drug, and other excipients. If desired, very small amounts of solubilizer may also be used, such as 5%>, 2%>, 1%) or even less. Typically, the solubilizer may be present in an amount of about 1%> to about 100%, more typically about 5%> to about 25%> by weight.
[00269] The composition can further include one or more pharmaceutically acceptable additives and excipients. Such additives and excipients include, without limitation, detackifiers, anti-foaming agents, buffering agents, polymers, antioxidants, preservatives, chelating agents, viscomodulators, tonicifiers, flavorants, colorants, odorants, opacifiers, suspending agents, binders, fillers, plasticizers, lubricants, and mixtures thereof.
[00270] In addition, an acid or a base may be incorporated into the composition to facilitate processing, to enhance stability, or for other reasons. Examples of pharmaceutically acceptable bases include amino acids, amino acid esters, ammonium hydroxide, potassium hydroxide, sodium hydroxide, sodium hydrogen carbonate, aluminum hydroxide, calcium carbonate, magnesium hydroxide, magnesium aluminum silicate, synthetic aluminum silicate, synthetic hydrocalcite, magnesium aluminum hydroxide, diisopropylethylamine, ethanolamine, ethylenediamine, triethanolamine, triethylamine, triisopropanolamine, trimethylamine, tri s(hydroxymethyl)aminom ethane (TRIS) and the like. Also suitable are bases that are salts of a pharmaceutically acceptable acid, such as acetic acid, acrylic acid, adipic acid, alginic acid, alkanesulfonic acid, amino acids, ascorbic acid, benzoic acid, boric acid, butyric acid, carbonic acid, citric acid, fatty acids, formic acid, fumaric acid, gluconic acid, hydroquinosulfonic acid, isoascorbic acid, lactic acid, maleic acid, oxalic acid, parabromophenylsulfonic acid, propionic acid, p-toluenesulfonic acid, salicylic acid, stearic acid, succinic acid, tannic acid, tartaric acid, thioglycolic acid, toluenesulfonic acid, uric acid, and the like. Salts of polyprotic acids, such as sodium phosphate, disodium hydrogen phosphate, and sodium dihydrogen phosphate can also be used. When the base is a salt, the cation can be any convenient and pharmaceutically acceptable cation, such as ammonium, alkali metals, alkaline earth metals, and the like. Example may include, but not limited to, sodium, potassium, lithium, magnesium, calcium and ammonium.
[00271] Suitable acids are pharmaceutically acceptable organic or inorganic acids. Examples of suitable inorganic acids include hydrochloric acid, hydrobromic acid, hydriodic acid, sulfuric acid, nitric acid, boric acid, phosphoric acid, and the like. Examples of suitable organic acids include acetic acid, acrylic acid, adipic acid, alginic acid, alkanesulfonic acids, amino acids, ascorbic acid, benzoic acid, boric acid, butyric acid, carbonic acid, citric acid, fatty acids, formic acid, fumaric acid, gluconic acid, hydroquinosulfonic acid, isoascorbic acid, lactic acid, maleic acid, methanesulfonic acid, oxalic acid, para-bromophenylsulfonic acid, propionic acid, p-toluenesulfonic acid, salicylic acid, stearic acid, succinic acid, tannic acid, tartaric acid, thioglycolic acid, toluenesulfonic acid, uric acid and the like.
Pharmaceutical compositions for injection.
[00272] In some embodiments, the invention provides a pharmaceutical composition for injection containing a compound of the present invention and a pharmaceutical excipient suitable for injection. Components and amounts of agents in the compositions are as described herein.
[00273] The forms in which the novel compositions of the present invention may be incorporated for administration by injection include aqueous or oil suspensions, or emulsions, with sesame oil, corn oil, cottonseed oil, or peanut oil, as well as elixirs, mannitol, dextrose, or a sterile aqueous solution, and similar pharmaceutical vehicles. [00274] Aqueous solutions in saline are also conventionally used for injection. Ethanol, glycerol, propylene glycol, liquid polyethylene glycol, and the like (and suitable mixtures thereof), cyclodextrin derivatives, and vegetable oils may also be employed. The proper fluidity can be maintained, for example, by the use of a coating, such as lecithin, for the maintenance of the required particle size in the case of dispersion and by the use of surfactants. The prevention of the action of microorganisms can be brought about by various antibacterial and antifungal agents, for example, parabens, chlorobutanol, phenol, sorbic acid, thimerosal, and the like.
[00275] Sterile injectable solutions are prepared by incorporating the compound of the present invention in the required amount in the appropriate solvent with various other ingredients as enumerated above, as required, followed by filtered sterilization. Generally, dispersions are prepared by incorporating the various sterilized active ingredients into a sterile vehicle which contains the basic dispersion medium and the required other ingredients from those enumerated above. In the case of sterile powders for the preparation of sterile injectable solutions, certain desirable methods of preparation are vacuum-drying and freeze- drying techniques which yield a powder of the active ingredient plus any additional desired ingredient from a previously sterile-filtered solution thereof.
[00276] Pharmaceutical compositions for topical (e.g. transdermal) delivery.
[00277] In some embodiments, the invention provides a pharmaceutical composition for transdermal delivery containing a compound of the present invention and a pharmaceutical excipient suitable for transdermal delivery.
[00278] Compositions of the present invention can be formulated into preparations in solid, semisolid, or liquid forms suitable for local or topical administration, such as gels, water soluble jellies, creams, lotions, suspensions, foams, powders, slurries, ointments, solutions, oils, pastes, suppositories, sprays, emulsions, saline solutions, dimethylsulfoxide (DMSO)-based solutions. In general, carriers with higher densities are capable of providing an area with a prolonged exposure to the active ingredients. In contrast, a solution formulation may provide more immediate exposure of the active ingredient to the chosen area.
[00279] The pharmaceutical compositions also may comprise suitable solid or gel phase carriers or excipients, which are compounds that allow increased penetration of, or assist in the delivery of, therapeutic molecules across the stratum corneum permeability barrier of the skin. There are many of these penetration- enhancing molecules known to those trained in the art of topical formulation.
[00280] Examples of such carriers and excipients include, but are not limited to, humectants (e.g., urea), glycols (e.g., propylene glycol), alcohols (e.g., ethanol), fatty acids (e.g., oleic acid), surfactants (e.g., isopropyl myristate and sodium lauryl sulfate), pyrrolidones, glycerol monolaurate, sulfoxides, terpenes (e.g., menthol), amines, amides, alkanes, alkanols, water, calcium carbonate, calcium phosphate, various sugars, starches, cellulose derivatives, gelatin, and polymers such as polyethylene glycols.
[00281] Another exemplary formulation for use in the methods of the present invention employs transdermal delivery devices ("patches"). Such transdermal patches may be used to provide continuous or discontinuous infusion of a compound of the present invention in controlled amounts, either with or without another agent.
The construction and use of transdermal patches for the delivery of pharmaceutical agents is well known in the art. See, e.g., U.S. Pat. Nos. 5,023,252, 4,992,445 and 5,001,139. Such patches may be constructed for continuous, pulsatile, or on demand delivery of pharmaceutical agents.
Pharmaceutical compositions for inhalation.
[00282] Compositions for inhalation or insufflation include solutions and suspensions in pharmaceutically acceptable, aqueous or organic solvents, or mixtures thereof, and powders. The liquid or solid compositions may contain suitable pharmaceutically acceptable excipients as described supra. Preferably the compositions are administered by the oral or nasal respiratory route for local or systemic effect. Compositions in preferably pharmaceutically acceptable solvents may be nebulized by use of inert gases. Nebulized solutions may be inhaled directly from the nebulizing device or the nebulizing device may be attached to a face mask tent, or intermittent positive pressure breathing machine. Solution, suspension, or powder compositions may be administered, preferably orally or nasally, from devices that deliver the formulation in an appropriate manner.
Other pharmaceutical compositions.
[00283] Pharmaceutical compositions may also be prepared from compositions described herein and one or more pharmaceutically acceptable excipients suitable for sublingual, buccal, rectal, intraosseous, intraocular, intranasal, epidural, or intraspinal administration. Preparations for such pharmaceutical compositions are well-known in the art. See, e.g., Anderson, Philip O.; Knoben, James E.; Troutman, William G, eds., Handbook of Clinical Drug Data, Tenth Edition, McGraw-Hill, 2002; Pratt and Taylor, eds., Principles of Drug Action, Third Edition, Churchill Livingston, New York, 1990; Katzung, ed., Basic and Clinical Pharmacology, Ninth Edition, McGraw Hill, 20037ybg; Goodman and Gilman, eds., The Pharmacological Basis of Therapeutics, Tenth Edition, McGraw Hill, 2001 ; Remingtons Pharmaceutical Sciences, 20th Ed., Lippincott Williams & Wilkins., 2000; Martindale, The Extra Pharmacopoeia, Thirty-Second Edition (The Pharmaceutical Press, London, 1999); all of which are incorporated by reference herein in their entirety.
[00284] Administration of the compounds or pharmaceutical composition of the present invention can be effected by any method that enables delivery of the compounds to the site of action. These methods include oral routes, intraduodenal routes, parenteral injection (including intravenous, intraarterial, subcutaneous, intramuscular, intravascular, intraperitoneal or infusion), topical (e.g. transdermal application), rectal administration, via local delivery by catheter or stent or through inhalation. Compounds can also be administered intraadiposally or intrathecally.
[00285] The amount of the compound administered will be dependent on the subject being treated, the severity of the disorder or condition, the rate of administration, the disposition of the compound and the discretion of the prescribing physician. However, an effective dosage is in the range of about 0.001 to about 100 mg per kg body weight per day, preferably about 1 to about 35 mg/kg/day, in single or divided doses. For a 70 kg human, this would amount to about 0.05 to 7 g/day, preferably about 0.05 to about 2.5 g/day. In some instances, dosage levels below the lower limit of the aforesaid range may be more than adequate, while in other cases still larger doses may be employed without causing any harmful side effect, e.g. by dividing such larger doses into several small doses for administration throughout the day.
[00286] In some embodiments, a compound of the invention is administered in a single dose.
[00287] Typically, such administration will be by injection, e.g., intravenous injection, in order to introduce the agent quickly. However, other routes may be used as appropriate. A single dose of a compound of the invention may also be used for treatment of an acute condition. [00288] In some embodiments, a compound of the invention is administered in multiple doses. Dosing may be about once, twice, three times, four times, five times, six times, or more than six times per day. Dosing may be about once a month, once every two weeks, once a week, or once every other day. In another embodiment a compound of the invention and another agent are administered together about once per day to about 6 times per day. In another embodiment the administration of a compound of the invention and an agent continues for less than about 7 days. In yet another embodiment the administration continues for more than about 6, 10, 14, 28 days, two months, six months, or one year. In some cases, continuous dosing is achieved and maintained as long as necessary.
[00289] Administration of the compounds of the invention may continue as long as necessary. In some embodiments, a compound of the invention is administered for more than 1, 2, 3, 4, 5, 6, 7, 14, or 28 days. In some embodiments, a compound of the invention is administered for less than 28, 14, 7, 6, 5, 4, 3, 2, or 1 day. In some embodiments, a compound of the invention is administered chronically on an ongoing basis, e.g., for the treatment of chronic effects.
[00290] An effective amount of a compound of the invention may be administered in either single or multiple doses by any of the accepted modes of administration of agents having similar utilities, including rectal, buccal, intranasal and transdermal routes, by intraarterial injection, intravenously, intraperitoneally, parenterally, intramuscularly, subcutaneously, orally, topically, or as an inhalant.
[00291] The compositions of the invention may also be delivered via an impregnated or coated device such as a stent, for example, or an artery -inserted cylindrical polymer. Such a method of administration may, for example, aid in the prevention or amelioration of restenosis following procedures such as balloon angioplasty. Without being bound by theory, compounds of the invention may slow or inhibit the migration and proliferation of smooth muscle cells in the arterial wall which contribute to restenosis. A compound of the invention may be administered, for example, by local delivery from the struts of a stent, from a stent graft, from grafts, or from the cover or sheath of a stent. In some embodiments, a compound of the invention is admixed with a matrix. Such a matrix may be a polymeric matrix, and may serve to bond the compound to the stent. Polymeric matrices suitable for such use, include, for example, lactone-based polyesters or copolyesters such as polylactide, polycaprolactonglycolide, polyorthoesters, polyanhydrides, polyaminoacids, polysaccharides, polyphosphazenes, poly (ether-ester) copolymers (e.g. PEO-PLLA); polydimethylsiloxane, poly(ethylene-vinylacetate), acrylate-based polymers or copolymers (e.g. polyhydroxyethyl methylmethacrylate, polyvinyl pyrrolidinone), fluorinated polymers such as polytetrafluoroethylene and cellulose esters. Suitable matrices may be nondegrading or may degrade with time, releasing the compound or compounds. Compounds of the invention may be applied to the surface of the stent by various methods such as dip/spin coating, spray coating, dip-coating, and/or brush-coating. The compounds may be applied in a solvent and the solvent may be allowed to evaporate, thus forming a layer of compound onto the stent. Alternatively, the compound may be located in the body of the stent or graft, for example in microchannels or micropores. When implanted, the compound diffuses out of the body of the stent to contact the arterial wall. Such stents may be prepared by dipping a stent manufactured to contain such micropores or microchannels into a solution of the compound of the invention in a suitable solvent, followed by evaporation of the solvent. Excess drug on the surface of the stent may be removed via an additional brief solvent wash. In yet other embodiments, compounds of the invention may be covalently linked to a stent or graft. A covalent linker may be used which degrades in vivo, leading to the release of the compound of the invention. Any bio-labile linkage may be used for such a purpose, such as ester, amide or anhydride linkages. Compounds of the invention may additionally be administered intravascularly from a balloon used during angioplasty. Extravascular administration of the compounds via the pericard or via advential application of formulations of the invention may also be performed to decrease restenosis.
[00292] A variety of stent devices which may be used as described are disclosed, for example, in the following references, all of which are hereby incorporated by reference: U.S. Pat. No. 5451233; U.S. Pat. No. 5040548; U.S. Pat. No. 5061273; U.S. Pat. No. 5496346; U.S. Pat. No. 5292331; U.S. Pat. No. 5674278; U.S. Pat. No. 3657744; U.S. Pat. No. 4739762; U.S. Pat. No. 5195984; U.S. Pat. No. 5292331; U.S. Pat. No. 5674278; U.S. Pat. No. 5879382; U.S. Pat. No. 6344053.
[00293] The compounds of the invention may be administered in dosages. It is known in the art that due to intersubject variability in compound pharmacokinetics, individualization of dosing regimen is necessary for optimal therapy. Dosing for a compound of the invention may be found by routine experimentation in light of the instant disclosure. [00294] When a compound of the invention is administered in a composition that comprises one or more agents, and the agent has a shorter half- life than the compound of the invention unit dose forms of the agent and the compound of the invention may be adjusted accordingly.
[00295] The subject pharmaceutical composition may, for example, be in a form suitable for oral administration as a tablet, capsule, pill, powder, sustained release formulations, solution, suspension, for parenteral injection as a sterile solution, suspension or emulsion, for topical administration as an ointment or cream or for rectal administration as a suppository. The pharmaceutical composition may be in unit dosage forms suitable for single administration of precise dosages. The pharmaceutical composition will include a conventional pharmaceutical carrier or excipient and a compound according to the invention as an active ingredient. In addition, it may include other medicinal or pharmaceutical agents, carriers, adjuvants, etc.
[00296] Exemplary parenteral administration forms include solutions or suspensions of active compound in sterile aqueous solutions, for example, aqueous propylene glycol or dextrose solutions. Such dosage forms can be suitably buffered, if desired.
Methods of use
[00297] The FGFR receptors (FGFR1, FGFR2, FGFR3, and FGFR4) share several structural features in common, including three extracellular immunoglobulin-like (Ig) domains, a hydrophobic transmembrane domain, and an intracellular tyrosine kinase domain split by a kinase insert domain, followed by a cytoplasmic c-terminal tail (Johnson et al., Adv. Cancer Res. 60: 1-40, 1993; and Wilkie et al., Curr. Biol. 5:500-507, 1995). In FGFRl, the kinase insert domain spans positions 582 to 595 of the alpha Al isoform of FGFR1. In FGFR2, the kinase insert domain spans positions 585 to 598 of the FGFR2 Ille isoform. In FGFR3, the kinase insert domain spans positions 576 to 589 of the FGFR3 Ille isoform. In FGFR4, the kinase insert domain spans positions 571 to 584 of FGFR4 isoform 1. The c- terminal tail of FGFRs begins following the end of the tyrosine kinase domain and extends to the c-terminus of the protein. Several isoforms of each FGFR have been identified and are the result of alternative splicing of their mRNAs (Johnson et al., Mol. Cell. Biol. 11 :4627-4634, 1995; and Chellaiah et al., J. Biol. Chem. 269: 11620-11627, 1994). [00298] A few of the receptor variants that result from this alternative splicing have different ligand binding specificities and affinities (Zimmer et al., J. Biol. Chem. 268:7899- 7903, 1993; Cheon et al., Proc. Natl. Acad. Sci. U.S.A. 91 :989-993, 1994; and Miki et al., Proc. Natl. Acad. Sci. U.S.A. 89:246-250, 1992). Protein sequences for FGFR proteins and nucleic acids encoding FGFR proteins are known in the art. Signaling by FGFRs regulates key biological processes including cell proliferation, survival, migration, and differentiation. Dysregulation of a FGFR gene, a FGFR protein, or expression or activity, or level of the same, has been associated with many types of cancer. For example, dysregulation of FGFRs can occur by multiple mechanisms, such as FGFR gene overexpression, FGFR gene amplification, activating mutations (e.g., point mutations or truncations), and chromosomal rearrangements that lead to FGFR fusion proteins. Dysregulation of a FGFR gene, a FGFR protein, or expression or activity, or level of the same, can result in (or cause in part) the development of a variety of different FGFR-associated cancers.
[00299] FGFR fusion proteins are known in the art. See, e.g., Baroy et al., PloS One; 1 l(9):e0163859. doi: 10.1371/journal. pone.0163859, 2016; Ren et al., Int. J. Cancer, 139(4):836-40, 2016; Marchwicka et al., Cell Biosci., 6:7. doi: 10.1186/sl3578-016-0075-9, 2016; PCT Patent Application Publication No. WO 2014/071419A2; U.S. Patent Application Publication No. 2015/0366866A1; PCT Patent Application Publication No. WO 2016/084883 Al; PCT Patent Application Publication No. WO 2016/030509 Al; PCT Patent Application Publication No. WO 2015/150900A2; PCT Patent Application Publication No. WO 2015/120094A2; Kasaian et al., BMC Cancer., 15:984, 2015; Vakil et al., NeuroOncology, 18:Supp. Supplement 3, pp. iii93. Abstract Number: LG-64, 17th International Symposium on Pediatric Neuro-Oncology, Liverpool, United Kingdom, 2016; Astsaturov et al., Journal of Clinical Oncology, 34:Supp. Supplement 15, Abstract Number: 11504, 2016 Annual Meeting of the American Society of Clinical Oncology, Chicago, IL; Heinrich et al., Journal of Clinical Oncology, 34:Supp. Supplement 15, Abstract Number: 11012, 2016 Annual Meeting of the American Society of Clinical Oncology, Chicago, IL; Hall et al., Molecular Cancer Therapeutics, Vol. 14, No. 12, Supp.2, Abstract Number: B151, AACR- NCI-EORTC International Conference: Molecular Targets and Cancer Therapeutics, 2015; Reuther et al., Journal of Molecular Diagnostics, Vol. 17, No. 6, pp. 813, Abstract Number: ST02, 2015 Annual Meeting of the Association for Molecular Pathology, Austin, TX; Moeini et al., Clin. Cancer. Res., 22(2):291-300, 2016; Schrock et al, J Thorac. Oneal. pii S1556- 0864(18)30674-9, 2018. doi: 10.1016/j.jtho.2018.05.027; Pekmezci et al, Acta Nur otapho/ Commun. 6(1):47. doi: 10.1186/s40478-018-0551-z; Lowery et al. Clin Cancer Res. pii: clincanres.0078.2018. doi: 10.1158/1078-0432.CCR-18-0078; Ryland et al. J Clin Patho/ pii: jclinpath-2018-205195, 2018. doi: 10.1136/jclinpath-2018-205195; Ferguson et al. J Neuropatho/ Exp Neural 77(6): 437-442, 2018. doi: 10.1093/jnen/nly022; Wu et al, BMC Cancer 18(1):343, 2018. doi: 10.1186/sl2885-018-4236-6; Shibata et al, Cancer Sci 109(5): 1282-1291, 2018. doi: 10.1111/cas.13582; Papdopoulos et al, Br J Cancer, 1117(11): 1592-1599, 2017. doi: 10.1038/bjc.2017.330; Hall et al, PLoS One,
1 l(9):el062594, 2016. doi: 10.1371/journal. pone.0162594; Johnson et al, Oncologist, 22(12): 1478-1490, 2017. doi: 10.1634/theoncologist.2017-0242; Yang et al, Am J Hum Genet, 98(5):843-856, 2016. doi: 10.1016/j.ajhg.2016.03.017; U.S. Patent Application Publication No. 2013/009621; Babina and Turner, Nat Rev Cancer 17(5):318-332, 2017. doi: 10.1038/nrc.2017.8; Ryland et al, J Clin Patho/., 2018 May 14. pii: jclinpath-2018-205195. doi: 10.1136/jclinpath-2018-205195; Kumar et al, Am JClinPatho/ 143(5):738-748, 2015. doi: 10.1309/AJCPUD6W1JLQQMNA; Grand et al, Genes Chromosomes Cancer40(l):78- 83, 2004. doi: 10.1002/gcc.20023; Reeser, et al, JMo/Diagn, 19(5):682-696, 2017. doi: 10.1016/j.jmoldx.2017.05.006; Basturk, et al, Mod Patho/ 30(12): 1760-1772, 2017. doi: 10.1038/modpathol.2017.60; Wang, et al, Cancer 123(20):3916-3924, 2017. doi: 10.1002/cncr.30837; Kim, et al, Oncotarget, 8(9): 15014-15022, 2017. doi: 10.18632/oncotarget.l4788; Busse, et al, Genes Chromosomes Cancer, 56(10):730-749, 2017. doi: 10.1002/gcc.22477; Shi, et al, J TranslMed., 14(1):339, 2016. doi: 10.1186/sl2967-016-1075-6, each of which is incorporated by reference herein.
FGFR point mutations are known in the art. See, e.g., UniParc entry UPI00000534B8; UniParc entry UPI0000001COF; UniParc entry UPI000002A99A; UniParc entry UPI000012A72A; UniParc entry UPI000059D1C2; UniParc entry UPI000002A9AC; Uniparc entry UPI000012A72C; Uniparc entry UPI000012A72D; Uniparc entry UPI000013EOB8; Uniparc entry UPI0001CE06A3; Gen bank entry BAD92868.1; Ang et al., Diagn. Mo/. Patho/. Feb 24, 2014; U.S. Patent Application Publication No. 2011/0008347; Gallo et al., Cytokine Growth Factor Rev. 26:425-449, 2015; Davies et al., J. Cancer Res. 65:7591, 2005; Kelleher et al., Carcinogenesis 34:2198, 2013; Cazier et al., Nat. Commun. 5:3756, 2014; Liu et al., Genet. Mo/. Res. 13: 1109, 2014; Trudel et al., Blood 107:4039, 2006; Gallo et al., Cytokine Growth Factor Rev. 26:425, 2015; Liao et al., Cancer Res. 73:5195-5205, 2013; Martincorena et al., Science 348:880 (2015); U.S. Patent Application Publication No. US2016/0235744 Al; U.S. Patent No. 9254288B2; U.S. Patent No. 9267176B2; U.S. Patent Application Publication No. S2016/0215350A1; European Patent Application Publication No. EP3023101 Al; PCT Patent Application Publication No. W02016105503A1; Rivera et al., Acta. Neuropatho/.,131(6):847-63, 2016; Lo lacono et al., Oncotarget., 7(12): 14394-404, 2016; Deeken et al., Journal of Clinical Oncology, 34:Supp. Supplement 15, pp. iii93. Abstract Number: el 7520, 2016 Annual Meeting of the American Society of Clinical Oncology, Chicago, IL; Sullivan et al., Journal of Clinical Oncology, 34:Supp. Supplement 15, pp. iii93. Abstract Number: 11596, 2016 Annual Meeting of the American Society of Clinical Oncology, Chicago, IL; Nguyen et al., Molecular Cancer Therapeutics, Vol. 14, No. 12, Supp.2, Abstract Number: C199, AACR-NCI-EORTC International Conference: Molecular Targets and Cancer Therapeutics, 2015; Li et al., Hum. Patho/., 55: 143-50, 2016; European Patent No. EP2203449B1; Yoza et al., Genes Cells., (10): 1049-1058, 2016; U.S. Patent No. 9,254,288B2; European Patent Application Publication No. 3023101A1; PCT Application Publication No. WO 2015/099127 Al;
European Patent No. EP2203449B1; Yoza et al., Genes Cells., (10): 1049-1058, 2016; Bunney et al., EbioMedicine, 2(3): 194-204, 2015; Byron et al., Neop/asia, 15(8):975-88, 2013;
European Patent Application Publication No. EP3023101A1; PCT Application Publication No. WO 2015/099127 Al; Thussbas et al., J. Clin. Oneal., 24(23): 3747-55, 2006; Chell et al., Oncogene, 32(25):3059-70, 2013; Tanizaki et al, Cancer Res. 75(15):3149-3146 doi: 10.1158/0008-5472. CAN-14-3771; Yang et al, EBioMedicine pii S2352-3964(18)30218-4. doi: 10.1016/j.ebiom.2018.06.011; Jakobsen, et al Oncotarget 9(40):26195-26208, 2018. doi: 10.18632/oncotarget.25490; Stone, et al Acta Neuropatho/ 135(1): 115-129, 2017. doi: 10.1007/s00401-017-1773-z; Pekmezci et al, Acta Nurotaphol. Commun. 6(1):47. doi: 10.1186/s40478-018-0551-z; De Mattos-Arruda et al, Oncotarget 9(29):20617-20630, 2018. doi: 10.18632/oncotarget.25041; Oliveira et al, J Exp Clin Cancer Res 37(1):84, 2018. doi: 10.1186/sl3046-018-0746-y; Cha et al, Mo/ Oneal 12(7): 993 -1003, 2018. doi: 10.1002/1878- 0261.12194; Ikeda et al, Oncologist, 23(5):586-593, 2018. doi: 10.1634/theoncologist.2017- 0479; Pelaez-Garda et al, PLoS One, 8(5):e63695, 2013. doi: 10.1371/joumal. pone.0063695; Shimada et al, Oncotarget, 8(55):93567-93579, 2017. doi: 10.18632/oncotarget.20510;
Welander et al, WorldJSurg, 42(2):482-489, 2018. doi: 10.1007 /s00268-01 7-4320-0;
Chandrani et al, Ann Oneal, 28(3):597-603, 2017. doi: 10.1093/annonc/mdw636; Dalin et al, Nat Commun, 8(1): 1197, 2017. doi: 10.1038/s41467-017-01178-z; Taurin et al, Inti Gyneco/ Cancer, 28(1): 152-160, 2018. doi: 10.1097/IGC.0000000000001129; Haugh et al, Jinvest Dermatol 138(2):384-393, 2018. doi: 10.1016/j .jid.2017.08.022; Babina and Turner, Nat Rev Cancer 17(5):318-332, 2017. doi: 10.1038/nrc.2017.8; Greenman et al, Nature 446(7132): 153-158, 2007. doi: 10.1038/nature05610; Helsten et al, Clin Cancer Res, 22(l):259-267, 2016. doi: 10.1158/1078-0432.CCR-14-3212; Kim et al, BMC Urol, 18:68, 2018. doi: 10.1186/s 12894-018-0380-1; Goyal et al, Cancer Discov, 7(3):252-263, 2017. doi: 10.1158/2159-8290. CD-16-1000; Premov et al, Oncogene, 36(22):3168-3177, 2017. doi: 10.1038/onc.2016.464; Geelvink et al, IntJMo/ Sci. 19(9): pii:E2548, 2018. doi: 10.3390/ijmsl9092548; Lee et al, Exp Ther Med. 16(2): 1343-1349, 2018. doi:
10.3892/etm.2018.6323; Kas et al, Cancer Res, 78(19):5668-5679, 2018. doi: 10.1158/0008- 5472.CAN-18-0757; Chesi et al, Blood, 97(3):729-736, 2001. PMID: 11157491. Note that the deletion ofFGFR3 isoform Hie residues 795-808 also deletes the stop codon, elongating the protein by 99 amino acids
(ATGPQQCEGSLAAHPAAGAQPLPGMRLSADGETATQSFGLCVCVCVCVCVCTSACACVR AHLASRCRGTLGVPAA
VQRSPDWCCSTEGPLFWGDPVQNVSGPTRWDPVGQGAGPDMARPLPLHHGTSQGALG PSHTQS); Ge, et al, Am J Cancer Res. 7(7): 1540-1553, 2017. PMID: 28744403; Jiao et al, Nat Genet, 45(12): 1470-1473, 2013. doi: 10.1038/ng.2813; Jusakul et al, Cancer Discov. 7(10): 1116-1135, 2017. doi: 10.1158/2159-8290.CD-17-0368; Guyard et al, Respir Res., 18(1): 120, 2018. doi: 10.1186/sl2931-017-0605-y; Paik et al, Clin Cancer Res., 23(18):5366- 5373, 2017. doi: 10.1158/1078-0432.CCR-17-0645; Roy et al, ModPatho/., 30(8): 1133- 1143, 2017. doi: 10.1038/modpathol.2017.33; Chakrabarty et al, Br J Cancer, 117(1): 136- 143, 2017. doi: 10.1038/bjc.2017.148; Hoang et al, Sci TranslMed, 5(197): 197ral02. doi: 10.1126/scitranslmed.3006200; Kim et al, Ann OneaL, 28(6): 1250-1259. doi: 10.1093/annonc/mdx098, each of which is incorporated by reference herein.
[00300] Compounds of the disclosure have been found to inhibit FGFR1, FGFR2, FGFR3, and/or FGFR4 and are therefore believed to be useful for treating diseases and disorders which can be treated with an inhibitor of FGFR1, FGFR2, FGFR3 and/or FGFR4. For example, compounds of the disclosure can be useful in treating FGFR-associated diseases and disorders, e.g., proliferative disorders such as cancers, including hematological cancers and solid tumor, and angiogenesis-related disorders. Compounds of the disclosure may also be useful in treating disorders arising from autosomal dominant mutations in FGFR, e.g., FGFR3, including, for example, developmental disorders. Developmental disorders to be treated with compounds of the disclosure include Achondroplasia (Ach) and related chondrodysplasia syndromes, including Hypochondroplasia (Hch), Severe Achondroplasia with Developmental Delay and Acanthosis Nigricans (SADDAN), and Thanatophoric dysplasia (TD).
[00301] Non-limiting examples of FGFR-associated diseases and disorders include Acanthosis nigricans, Achondroplasia, Apert syndrome, Beare-Stevenson syndrome (BSS), Camptodactyly, tall stature, and hearing loss syndrome (CATSHL) syndrome, cleft lip and palate, congenital heart disease (e.g., associated with ambiguous genitalia), craniosynostosis, Crouzon syndrome, ectrodactyly, encephalocraniocutaneous lipomatosis, Hartsfield syndrome, hypochondroplasia, hypogonadoropic hypogonadism (e.g., hypogonadotropic hypogonadism 2 with or without anosmia, Kailman syndrome), ichthyosis vulgaris and/or atopic dermatitis, Jackson-Weiss syndrome, lethal pulmonary acinar dysplasia, microphthalmia, Muenke coronal craniosynostosis, osteoglophonic dysplasia, Pfeiffer syndrome, seborrheic keratosis, syndactyly, thanatophoric dysplasia (e.g., type I or type II), trigonocephaly 1 (also called metopic craniosynostosis), and tumor-induced osteomalacia.
[00302] Non-limiting examples of FGFR1 associated diseases and disorders include congenital heart disease (e.g., associated with ambiguous genitalia), craniosynostosis, encephalocraniocutaneous lipomatosis, Hartsfield syndrome, hypogonadoropic hypogonadism (e.g., hypogonadotropic hypogonadism 2 with or without anosmia, Kailman syndrome), ichthyosis vulgaris and/or atopic dermatitis, Jackson-Weiss syndrome, osteoglophonic dysplasia, Pfeiffer syndrome, trigonocephaly 1 (also called metopic craniosynostosis), and tumor-induced osteomalacia.
[00303] Non-limiting examples of FGFR2 -associated diseases and disorders include Apert syndrome, Beare-Stevenson syndrome (BSS), Crouzon syndrome, ectrodactyly, Jackson-Weiss syndrome, lethal pulmonary acinar dysplasia, Pfeiffer syndrome, and syndactyly. Non-limiting examples of FGFR3 -associated diseases and disorders include acanthosis nigricans, achondroplasia, Camptodactyly, tall stature, and hearing loss syndrome (CATSHL) syndrome, cleft lip and palate, craniosynostosis, hypochondroplasia, microphthalmia, Muenke coronal craniosynostosis, seborrheic keratosis, and thanatophoric dysplasia (e.g., type I or type II). See also, See UniParc entry UPI00000534B8; UniParc entry UPI0000001COF;Uni Pare entry UP 1000002 A99A;UniParc entry
UPI000012 A72 A; Yong-Xing et al., Hum. Mol. Genet. 9(13):2001-2008, 2000; Eeva-Maria Laitinen et al., PLoS One 7(6):e39450, 2012; Hart et al., Oncogene 19(29):3309-3320, 2000; Shiang et al., Cell 76:335-342, 1994; Rosseau et al., Nature 371 :252-254, 1994; Tavormina et al., Nature Genet. 9:321-328, 1995; Bellus et al., Nature Genet. 10:357-359, 1995; Muenke et al., Nature Genet. 8:269-274, 1994; Rutland et al., Nature Genet. 9: 173-176, 1995; Reardon et al., Nature Genet. 8:98-103, 1994; Wilkie et al., Nature Genet. 9: 165-172, 1995; Jabs et al., Nature Genet. 8:275-279, 1994; Japanese Patent No. JP05868992B2; Ye et al., Plast.
Reconstr. Surg., 137(3):952-61, 2016; U.S. Patent No. 9447098B2; Bellus et al., Am. J. Med. Genet. 85(l):53-65, 1999; PCT Patent Application Publication No. WO2016139227A1;
Australian Patent Application Publication No. AU2014362227A1; Chinese Patent No. CN102741256B; Ohishi et al., Am. J. Med. Genet. A., doi: 10.1002/ajmg.a.37992, 2016; Nagahara et al., Clin. Pediatr. Endocrinol., 25(3): 103-106, 2016; Hibberd et al., Am. J. Med. Genet. A., doi: 10.1002/ajmg.a.37862, 2016; Dias et al., Exp. Mol. Pathol., 101(1): 116-23, 2016; Lin et al., Mol. Med. Rep., 14(3): 1941 -6, 2016; Barnett et al., Hum. Mutat., 37(9):955- 63, 2016; Krstevska-Konstantinova et al., Med. Arch., 70(2): 148-50, 2016; Kuentz et al., Br. J. Dermatol., doi: 10.1111/bjd.14681, 2016; Ron et al., Am. J. Case Rep., 15;17:254-8, 2016;
Fernandes et al., Am. J. Med. Genet. A., 170(6): 1532-7, 2016; Lindy et al., Am. J. Med. Genet. A., 170(6): 1573-9, 2016; Bennett et al., Am. J. Hum. Genet., 98(3):579-87, 2016; Ichiyama et al., J. Eur. Acad. Dermatol. Venereal., 30(3):442-5, 2016; Zhao et al., Int. J. Clin. Exp. Med., 8(10): 19241-9, 2015; Hasegawa et al., Am. J. Med. Genet. A., 170A(5): 1370-2, 2016; Legeai-Mallet, Endocr. Dev., 30:98-105, 2016; Takagi, Am. J. Med. Genet. A., 167A(ll):2851-4, 2015; Goncalves, Fertil. Steril., 104(5): 1261-7.el, 2015; Miller et al., Journal of Clinical Oncology, 34:Supp. Supplement 15, pp. iii93. AbstractNumber: e22500, 2016 Annual Meeting of the American Society of Clinical Oncology, Chicago, IL;
Sarabipour et al., J. Mol. Biol., 428(20):3903-3910, 2016; Escobar et al., Am. J. Med. Genet. A., 170(7): 1908-11, 2016; Mazen et al., Sex Dev., 10(1): 16-22, 2016; Taylan et al., J Allergy Clin Immunol, 136(2):507-9, 2015. doi: 10.1016/j .jaci.2015.02.010; Kant et al, EuroJourn Endocrinol, 172(6):763-770, 2015. doi: 10.1530ZEJE-14-0945; Gonzalez-Del Angel et al, Am JmedGenetA, 176(1 ): 161 - 166, 2018. doi: 10.1002/ajmg.a.38526; Lei and Deng, Int J Biol Sci 13(9):1163: 1171, 2017. doi: 10.7150/ijbs.20792; Lajeunie et al, Eur J Hum Genet, 14(3):289-298, 2006. doi: 10.1038/sj.ejhg.5201558; Karadimas et al, Prenat Diagn, 26(3):258-261, 2006. doi: 10.1002/pd.l392; Ibrahimi et al, Hum Mo/ Genet 13(19):2313- 2324, 2004. doi: 10.1093/hmg/ddh235; Trarbach et al, J Clin Endocrinol Metab., 91(10):4006-4012, 2006. doi: 10.1210/jc.2005-2793; Dode et al, Nat Genet, 33(4):463-465, 2003. doi: 10.1038/ngl 122, each of which is incorporated by reference herein.
[00304] The term "angiogenesis-related disorder" means a disease characterized in part by an increased number or size of blood vessels in a tissue in a subject or patient, as compared to a similar tissue from a subject not having the disease. Non-limiting examples of angiogenesis-related disorders include: cancer ( e.g., any of the exemplary cancers described herein, such as prostate cancer, lung cancer, breast cancer, bladder cancer, renal cancer, colon cancer, gastric cancer, pancreatic cancer, ovarian cancer, melanoma, hepatoma, sarcoma, and lymphoma), exudative macular degeneration, proliferative diabetic retinopathy, ischemic retinopathy, retinopathy of prematurity, neovascular glaucoma, iritis rubeosis, corneal neovascularization, cyclitis, sickle cell retinopathy, and pterygium.
[00305] Compounds of the disclosure inhibit wild-type FGFR1, FGFR2, FGFR3, and/or FGFR4. In other aspects, compounds of the disclosure inhibit a mutated FGFR1, FGFR2, FGFR3, and/or FGFR4. In other aspects, compounds of the disclosure inhibit FGFR1, FGFR2, FGFR3, and/or FGFR4 that includes an FGFR kinase inhibitor mutation.
[00306] In some embodiments of any of the methods or uses described herein, the cancer (e.g., FGFR-associated cancer) is a hematological cancer. In some embodiments of any of the methods or uses described herein, the cancer (e.g., FGFR-associated cancer) is a solid tumor.
[00307] In some embodiments of any of the methods or uses described herein, the cancer (e.g., FGFR-associated cancer) is a lung cancer (e.g., small cell lung carcinoma, nonsmall cell lung carcinoma, squamous cell carcinoma, lung adenocarcinoma, large cell carcinoma, mesothelioma, lung neuroendocrine carcinoma, smoking-associated lung cancer), prostate cancer, colorectal cancer (e.g., rectal adenocarcinoma), endometrial cancer (e.g., endometrioid endometrial cancer, endometrial adenocarcinoma), breast cancer (e.g., hormone-receptor-positive breast cancer, triple-negative breast cancer, neuroendodrine carcinoma of the breast), skin cancer (e.g., melanoma, cutaneous squamous cell carcinoma, basal cell carcinoma, large squamous cell carcinoma), gallbladder cancer, liposarcoma (e.g., dedifferentiated liposarcoma, myxoid liposarcoma), pheochromocytoma, myoepithelial carcinoma, urothelial carcinoma, spermatocytic seminoma, stomach cancer, head and neck cancer (e.g., head and neck (squamous) carcinoma, head and neck adenoid cystic adenocarcinoma), brain cancer (e.g., glialneural tumors, glioma, neuroblastoma, glioblastoma, pilocytic astrocytoma, Rosette forming glioneural tumor, dysembryoplastic neuroepithelial tumor, anaplastic astrocytoma, medulloblastoma, ganglioglioma, oligodendroglioma), malignant peripheral nerve sheath tumor, sarcoma (e.g., soft tissue sarcoma (e.g., leiomyosarcoma), osteosarcoma), esophageal cancer (e.g., esophageal adenocarcinoma), lymphoma, bladder cancer (e.g., bladder urothelial (transition cell) carcinoma), cervical cancer (e.g., cervical squamous cell carcinoma, cervical adenocarcinoma), fallopian tube cancer (e.g., fallopian tube carcinoma), ovarian cancer (e.g., ovarian serous cancer, ovarian mucinous carcinoma), cholangiocarcinoma, adenoid cystic carcinoma, pancreatic cancer (e.g., pancreatic exocrine carcinoma, pancreatic ductal adenocarcinoma, pancreatic cancer intraepithelial neoplasia), salivary gland cancer (e.g., pleomorphic salivary gland adenocarcinoma, salivary adenoid cystic cancer), oral cancer (e.g., oral squamous cell carcinoma), uterine cancer, gastric or stomach cancer (e.g., gastric adenocarcinoma), gastrointestinal stromal tumors, myeloma (e.g., multiple myeloma), lymphoepithelioma, anal cancer (e.g., anal squamous cell carcinoma), prostate cancer (e.g., prostate adenocarcinoma), renal cell carcinoma, thymic cancer, gastroesophogeal junction adenocarcinoma, testicular cancer, rhabdomyosarcoma (e.g., alveolar rhabdomyosarcoma, embryonic rhabomyosarcoma), renal papillary carcinoma, liver cancer (e.g., hepatocellular carcinoma, intrahepatic cholangiocarcinoma), carcinoid, myeloid proliferative disorders (also called myeloid proliferative neoplasms (MPN); e.g., 8pll myeloproliferative syndrome (EMS, also called stem cell leukemia/lymphoma), acute myeloid leukemia (AML), chronic myeloid leukemia (CML)), lymphoma (e.g., T-cell lymphoma, T-lymphoblastic lymphoma, acute lymphoblastic leukemia (ALL), B-cell lymphoma), myeloid and lymphoid neoplasms, chronic neutrophilic leukemia, phosphaturic mesenchymal tumor, thyroid cancer (e.g. anaplastic thyroid carcinoma), or biliary duct cancer.
[00308] In some embodiments of any of the methods or uses described herein, the cancer (e.g., FGFR-associated cancer) is selected from the group of: acute lymphoblastic leukemia (ALL), acute myeloid leukemia (AML), cancer in adolescents, adrenocortical carcinoma, anal cancer, appendix cancer, astrocytoma, atypical teratoid/rhabdoid tumor, basal cell carcinoma, bile duct cancer, bladder cancer, bone cancer, brain stem glioma, brain tumor, breast cancer, bronchial tumor, Burkitt lymphoma, carcinoid tumor, unknown primary carcinoma, cardiac tumors, cervical cancer, childhood cancers, chordoma, chronic lymphocytic leukemia (CLL), chronic myelogenous leukemia (CML), chronic myeloproliferative neoplasms, neoplasms by site, neoplasms, colon cancer, colorectal cancer, craniopharyngioma, cutaneous T-cell lymphoma, cutaneous angiosarcoma, bile duct cancer, ductal carcinoma in situ, embryonal tumors, endometrial cancer, ependymoma, esophageal cancer, esthesioneuroblastoma, Ewing sarcoma, extracranial germ cell tumor, extragonadal germ cell tumor, extrahepatic bile duct cancer, eye cancer, fallopian tube cancer, fibrous histiocytoma of bone, gallbladder cancer, gastric cancer, gastrointestinal carcinoid tumor, gastrointestinal stromal tumors (GIST), germ cell tumor, gestational trophoblastic disease, glioma, hairy cell tumor, hairy cell leukemia, head and neck cancer, thoracic neoplasms, head and neck neoplasms, CNS tumor, primary CNS tumor, heart cancer, hepatocellular cancer, histiocytosis, Hodgkin's lymphoma, hypopharyngeal cancer, intraocular melanoma, islet cell tumors, pancreatic neuroendocrine tumors, Kaposi sarcoma, kidney cancer, Langerhans cell histiocytosis, laryngeal cancer, leukemia, lip and oral cavity cancer, liver cancer, lung cancer, lymphoma, macroglobulinemia, malignant fibrous histiocytoma of bone, osteocarcinoma, melanoma, Merkel cell carcinoma, mesothelioma, metastatic squamous neck cancer, midline tract carcinoma, mouth cancer, multiple endocrine neoplasia syndromes, multiple myeloma, mycosis fungoides, myelodysplastic syndromes, myelodysplastic/myeloproliferative neoplasms, neoplasms by site, neoplasms, myelogenous leukemia, myeloid leukemia, multiple myeloma, myeloproliferative neoplasms, nasal cavity and para nasal sinus cancer, nasopharyngeal cancer, neuroblastoma, non-Hodgkin's lymphoma, non-small cell lung cancer, lung neoplasm, pulmonary cancer, pulmonary neoplasms, respiratory tract neoplasms, bronchogenic carcinoma, bronchial neoplasms, oral cancer, oral cavity cancer, lip cancer, oropharyngeal cancer, osteosarcoma, ovarian cancer, pancreatic cancer, papillomatosis, paraganglioma, para nasal sinus and nasal cavity cancer, parathyroid cancer, penile cancer, pharyngeal cancer, pheochromosytoma, pituitary cancer, plasma cell neoplasm, pleuropulmonary blastoma, pregnancy-associated breast cancer, primary central nervous system lymphoma, primary peritoneal cancer, prostate cancer, rectal cancer, colon cancer, colonic neoplasms, renal cell cancer, retinoblastoma, rhabdomyosarcoma, salivary gland cancer, sarcoma, Sezary syndrome, skin cancer, Spitz tumors, small cell lung cancer, small intestine cancer, soft tissue sarcoma, squamous cell carcinoma, squamous neck cancer, stomach cancer, T-cell lymphoma, testicular cancer, throat cancer, thymoma and thymic carcinoma, thyroid cancer, transitional cell cancer of the renal pelvis and ureter, unknown primary carcinoma, urethral cancer, uterine cancer, uterine sarcoma, vaginal cancer, vulvar cancer, and Wilms' tumor.
[00309] In some embodiments, a hematological cancer (e.g., hematological cancers that are FGFR associated cancers) is selected from the group consisting of leukemias, lymphomas (non-Hodgkin's lymphoma), Hodgkin's disease (also called Hodgkin's lymphoma), and myeloma, for instance, acute lymphocytic leukemia (ALL), acute myeloid leukemia (AML), acute promyelocytic leukemia (APL), chronic lymphocytic leukemia (CLL), chronic myeloid leukemia (CML), chronic myelomonocytic leukemia (CMML), chronic neutrophilic leukemia (CNL), acute undifferentiated leukemia (AUL), anaplastic large-cell lymphoma (ALCL), prolymphocytic leukemia (PML), juvenile myelomonocyctic leukemia (JMML), adult Tcell ALL, AML with trilineage myelodysplasia (AML/TMDS), mixed lineage leukemia (MLL), myelodysplastic syndromes (MDSs), myeloproliferative disorders (MPD), and multiple myeloma (MM).
[00310] Additional examples of hematological cancers include myeloproliferative disorders (MPD) such as polycythemia vera (PV), essential thrombocytopenia (ET) and idiopathic primary myelofibrosis (IMF/IPF/PMF). In some embodiments, the hematological cancer (e.g., the hematological cancer that is a FGFR-associated cancer) is AML or CMML. In some embodiments, the cancer (e.g., the FGFR-associated cancer) is a solid tumor. Examples of solid tumors (e.g., solid tumors that are FGFR-associated cancers) include, for example, lung cancer (e.g., lung adenocarcinoma, non-small-cell lung carcinoma, squamous cell lung cancer), bladder cancer, colorectal cancer, brain cancer, testicular cancer, bile duct cancer cervical cancer, prostate cancer, and sparmatocytic seminomas. See, for example, Turner and Grose, Nat. Rev. Cancer, 10(2): 116-129, 2010.
[00311] In some embodiments, the cancer is selected from the group consisting of bladder cancer, brain cancer, breast cancer, cholangiocarcinoma, head and neck cancer, lung cancer, multiple myeloma, rhabdomyosarcoma, urethral cancer, and uterine cancer. In some embodiments, the cancer is selected from the group consisting of lung cancer, breast cancer, and brain cancer.
[00312] In some embodiments, a FGFRl-associated cancer is selected from the group consisting of lung cancer, breast cancer, and brain cancer. [00313] In some embodiments, the cancer is selected from the group consisting of breast cancer, uterine cancer, cholangiocarcinoma, and lung cancer.
[00314] In some embodiments, a FGFR2-associated cancer is selected from the group consisting of breast cancer, uterine cancer, cholangiocarcinoma, and lung cancer. In some embodiments, the cancer is selected from the group consisting of lung cancer, bladder cancer, urethral cancer, multiple myeloma, and head and neck cancer.
[00315] In some embodiments, a FGFR3 -associated cancer is selected from the group consisting of lung cancer, bladder cancer, urethral cancer, multiple myeloma, and head and neck cancer.
[00316] In some embodiments, the cancer is selected from lung cancer, rhabdomyosarcoma, and breast cancer.
[00317] In some embodiments, a FGFR4-associated cancer is selected from hepatocellular carcinoma, lung cancer, rhabdomyosarcoma, and breast cancer.
[00318] In some aspects, the compounds of the disclosure are useful in treating cancers associated with amplification or overexpression of FGFR1, for example, Breast cancer or carcinoma (e.g., hormone receptor-positive breast cancer, ductal carcinoma in situ (breast)), pancreatic ductal adenocarcinoma, pancreatic exocrine carcinoma, smoking- associated lung cancer, small cell lung cancer, lung adenocarcinoma, non-small cell lung cancer, squamous cell lung cancer or carcinoma, prostate cancer or carcinoma, ovarian cancer, fallopian tube carcinoma, bladder cancer, rhabdomyosarcoma, head and neck carcinoma (e.g., head and neck squamous cell carcinoma), esophageal cancer (e.g., esophageal squamous cell carcinoma), sarcoma (e.g., osteosarcoma), hepatocellular carcinoma, renal cell carcinoma, colorectal cancer (e.g., colorectal adenocarcinoma), prostate cancer, salivary gland tumors, glioblastoma multiforme, urinary bladder cancer, urothelial carcinoma, carcinoma of unknown primary, squamous non-lung tumors, gastric cancer, gastroesophageal junction carcinoma, adenoid cystic carcinoma, anal squamous cell carcinoma, oral squamous cell carcinoma, cholangiocarcinoma, hemangioendothelioma, leiomyosarcoma, melanoma, neuroendocrine carcinoma, squamous cell carcinoma, uterine carcinosarcoma.
[00319] In some aspects, the compounds of the disclosure are useful in treating cancers associated with amplification of FGFR2, for example, Gastric cancer, gastroesophageal junction adenocarcinoma, breast cancer (e.g., triple negative breast cancer), colon cancer, colorectal cancer (e.g., colorectal adenocarcinoma), urothelial cancer, bladder adenocarcinoma, carcinoma of unknown primary, cholangiocarcinoma, endometrial adenocarcinoma, esophageal adenocarcinoma, gallbladder carcinoma, ovarian cancer, fallopian tube carcinoma, pancreatic exocrine carcinoma, sarcoma, squamous cell carcinoma.
[00320] In some aspects, the compounds of the disclosure are useful in treating cancers associated with overexpression of FGFR2, for example, Myxoid lipocarcinoma, rectal cancer, renal cell carcinoma, breast cancer.
[00321] In some aspects, the compounds of the disclosure are useful in treating cancers associated with upregulation of activity of FGFR3, for example, Colorectal cancer, hepatocellular carcinoma, pancreatic exocrine carcinoma. In some aspects, the compounds of the disclosure are useful in treating cancers associated with overexpression of activity of FGFR3, for example, Multiple myeloma, thyroid carcinoma. In some aspects, the compounds of the disclosure are useful in treating cancers associated with amplification of activity of FGFR3, for example, Bladder cancer and salivary adenoid cystic cancer, urothelial cancer, breast cancer, carcinoid, carcinoma of unknown primary, colorectal cancer (e.g., colorectal adenocarcinoma), gallbladder carcinoma, gastric cancer, gastroesophageal junction adenocarcinoma, glioma, mesothelioma, non-small cell lung carcinoma, small cell lung cancer, ovarian cancer, fallopian tube carcinoma, pancreatic exocrine carcinoma.
[00322] In some aspects, the compounds of the disclosure are useful in treating cancers associated with amplification of FGFR4, for example, Rhabdomyosarcoma, prostate cancer or carcinoma, breast cancer, urothelial cancer, carcinoid, carcinoma of unknown primary, esophageal adenocarcinoma, head and neck carcinoma, hepatocellular carcinoma, non-small cell lung carcinoma, ovarian cancer, fallopian tube carcinoma, peritoneal carcinoma, renal cell carcinoma.
[00323] In some aspects, the compounds of the disclosure are useful in treating cancers associated with upregulation of activity of FGFR4, for example, Colorectal cancer, hepatocellular carcinoma, adrenal carcinoma, breast cancer.
[00324] In some aspects, the compounds of the disclosure are useful in treating cancers associated with overexpression of activity of FGFR4, for example, Pancreatic intraepithelial neoplasia, and pancreatic ductal adenocarcinoma.
[00325] In some aspects, the compounds of the disclosure are more selective for one FGFR than for another. As used herein, the "selectivity" of a compound for a first target over a second target means that the compound has more potent activity at the first target than the second target. A fold selectivity can be calculated by any method known in the art. For example, a fold selectivity can be calculated by dividing the IC50 value (or Kd value) of a compound for the second target (e.g., FGFR1) by the IC50 value (or Kd value) of the same compound for the first target (e.g., FGFR2 or FGFR3). An IC50 value (or Kd value) can be determined by any method known in the art. In some embodiments, a compound is first determined to have an activity of less than 500 nM for the first target. In some embodiments, a compound is first determined to have an activity of less than 500 nM for the second target.
[00326] For example, in some aspects, the compounds of the disclosure are more selective for FGFR3 than for FGFR1. In some aspects, the compounds are at least 3-fold more selective for FGFR3 than for FGFR1. In some aspects, the compounds are 4, 5, 6, 7, 8, 9, 10, 15, 20, 30, 40, 50, 75, 100, 200, 500, or 1000 fold more selective for FGFR3 than for FGFR1.
[00327] For example, in some aspects, the compounds of the disclosure are more selective for FGFR4 than for FGFR1. In some aspects, the compounds are at least 3 -fold more selective for FGFR4 than for FGFR1. In some aspects, the compounds are 4, 5, 6, 7, 8, 9, 10, 15, 20, 30, 40, 50, 75, 100, 200, 500, or 1000 fold more selective for FGFR4 than for FGFR1.
[00328] In some aspects, the compounds of the disclosure are more selective for FGFR2 than for FGFR1. In some aspects, the compounds are at least 3-fold more selective for FGFR2 than for FGFR1. In some aspects, the compounds are 4, 5, 6, 7, 8, 9, 10, 15, 20, 30, 40, 50, 75, 100, 200, 500, or 1000 fold more selective for FGFR2 than for FGFR1.
[00329] In some aspects, the compounds of the disclosure are more selective for a first FGFR family member (e.g., FGFR2 or FGFR3) over a second FGFR family member (e.g., FGFR1 or FGFR4). In some aspects, the compounds of the disclosure are at least 3- fold more selective for a first FGFR family member over a second FGFR family member. In some aspects, the compounds are at least 10, 20, 30, 40, 50, 60, 70, 80, 90, 200, 300, 400, 500, 600, 700, 800, 900, or at least 1000 fold more selective for a first FGFR family member over a second FGFR family member.
[00330] In some aspects, the compounds of the disclosure are more selective for a first FGFR family member (e.g., FGFR4 or FGFR3) over a second FGFR family member (e.g., FGFR1 or FGFR2). In some aspects, the compounds of the disclosure are at least 3- fold more selective for a first FGFR family member over a second FGFR family member. In some aspects, the compounds are at least 10, 20, 30, 40, 50, 60, 70, 80, 90, 200, 300, 400, 500, 600, 700, 800, 900, or at least 1000 fold more selective for a first FGFR family member over a second FGFR family member.
[00331] In some aspects, the compounds of the disclosure are more selective for an FGFR kinase over another kinase that is not an FGFR kinase. For example, the compounds of the disclosure are at least 3-fold more selective for an FGFR kinase over another kinase that is not an FGFR kinase. In some aspects, the compounds of the disclosure are at least 10, 20, 30, 40, 50, 60, 70, 80, 90, 200, 300, 400, 500, 600, 700, 800, 900, or at least 1000 fold more selective for an FGFR kinase over another kinase that is not an FGFR kinase. Kinases that are not FGFR kinases include, for example, KDR kinase and Aurora B kinase.
[00332] In some embodiments, the compounds of the disclosure exhibit brain and/or central nervous system (CNS) penetrance. Such compounds are capable of crossing the blood brain barrier and inhibiting a FGFR kinase in the brain and/or other CNS structures. In some embodiments, the compounds provided herein are capable of crossing the blood brain barrier in a therapeutically effective amount. For example, treatment of a subject with cancer (e.g., a FGFR-associated cancer such as a FGFR-associated brain or CNS cancer) can include administration (e.g., oral administration) of the compound to the subject. In some such embodiments, the compounds provided herein are useful for treating a primary brain tumor or metastatic brain tumor. For example, a FGFR-associated primary brain tumor or metastatic brain tumor.
[00333] In some embodiments, the compounds of the disclosure, exhibit one or more of high GI absorption, low clearance, and low potential for drug-drug interactions.
[00334] In some aspects, compounds of the disclosure can be used for treating a subject diagnosed with (or identified as having) a FGFR-associated disease or disorder (e.g., a FGFR-associated cancer) that include administering to the subject a therapeutically effective amount of a compound of the disclosure. Also provided herein are methods for treating a subject identified or diagnosed as having a FGFR-associated disease or disorder (e.g., a FGFR-associated cancer) that include administering to the subject a therapeutically effective amount of a compound of the disclosure. In some embodiments, the subject that has been identified or diagnosed as having a FGFR-associated disease or disorder (e.g., a FGFR- associated cancer) through the use of a regulatory agency-approved, e.g., FDA-approved test or assay for identifying dysregulation of a FGFR gene, a FGFR kinase, or expression or activity or level of any of the same, in a subject or a biopsy sample from the subject or by performing any of the non-limiting examples of assays described herein. In some embodiments, the test or assay is provided as a kit. In some embodiments, the FGFR- associated disease or disorder is a FGFR-associated cancer. For example, the FGFR- associated cancer can be a cancer that includes one or more FGFR inhibitor resistance mutations.
[00335] Also provided are methods for treating a disease or disorder in a subject in need thereof, the method comprising: (a) detecting a FGFR-associated disease or disorder in the subject; and (b) administering to the subject a therapeutically effective amount of a compound of the disclosure. Some embodiments of these methods further include administering to the subject an additional therapy or therapeutic agent (e.g., a second FGFR inhibitor, a second compound of the disclosure, or an immunotherapy. In some embodiments, the subject was previously treated with a first FGFR inhibitor or previously treated with another treatment. In some embodiments, the subject is determined to have a FGFR- associated disease or disorder through the use of a regulatory agency-approved, e.g., FDA approved test or assay for identifying dysregulation of a FGFR gene, a FGFR kinase, or expression or activity or level of any of the same, in a subject or a biopsy sample from the subject or by performing any of the non-limiting examples of assays described herein. In some embodiments, the test or assay is provided as a kit.
[00336] Also provided are methods for treating cancer in a subject in need thereof, the method comprising: (a) detecting a FGFR-associated cancer in the subject ; and (b) administering to the subject a therapeutically effective amount of a compound of the disclosure. Some embodiments of these methods further include administering to the subject an additional therapy or therapeutic agent (e.g., a second FGFR inhibitor, a second compound of the disclosure, or an immunotherapy). In some embodiments, the subject was previously treated with a first FGFR inhibitor or previously treated with another anticancer treatment, e.g., at least partial resection of the tumor or radiation therapy. In some embodiments, the subject is determined to have a FGFR-associated cancer through the use of a regulatory agency-approved, e.g., FDA-approved test or assay for identifying dysregulation of a FGFR gene, a FGFR kinase, or expression or activity or level of any of the same, in a subject or a biopsy sample from the subject or by performing any of the non -limiting examples of assays described herein. In some embodiments, the test or assay is provided as a kit. In some embodiments, the cancer is a FGFR associated cancer. For example, the FGFR-associated cancer can be a cancer that includes one or more FGFR inhibitor resistance mutations. In some embodiments, the cancer is a FGFR associated cancer. For example, the FGFR- associated cancer can be a cancer that includes one or more FGFR activating mutations.
[00337] Also provided are methods of treating a subject that include performing an assay on a sample obtained from the subject to determine whether the subject has a dysregulation of a FGFR gene, a FGFR kinase, or expression or activity or level of any of the same, and administering (e.g., specifically or selectively administering) a therapeutically effective amount of a compound of the disclosure or pharmaceutically acceptable salt or solvate thereof to the subject determined to have a dysregulation of a FGFR gene, a FGFR kinase, or expression or activity or level of any of the same. Some embodiments of these methods further include administering to the subject an additional therapy or therapeutic agent (e.g., a second FGFR inhibitor, a second compound of the disclosure, or immunotherapy). In some embodiments of these methods, the subject was previously treated with a first FGFR inhibitor or previously treated with another anticancer treatment, e.g., at least partial resection of a tumor or radiation therapy. In some embodiments, the subject is a subject suspected of having a FGFR-associated disease or disorder (e.g., a FGFR-associated cancer), a subject presenting with one or more symptoms of a FGFR-associated disease or disorder (e.g., a FGFR-associated cancer), or a subject having an elevated risk of developing a FGFR-associated disease or disorder (e.g., a FGFR-associated cancer). In some embodiments, the assay utilizes next generation sequencing, pyrosequencing, immunohistochemistry, or break apart FISH analysis. In some embodiments, the assay is a regulatory agency-approved assay, e.g., FDA-approved kit. In some embodiments, the assay is a liquid biopsy. Additional, non-limiting assays that may be used in these methods are described herein. Additional assays are also known in the art. In some embodiments, the dysregulation of a FGFR gene, a FGFR kinase, or expression or activity or level of any of the same includes one or more FGFR inhibitor resistance mutations.
[00338] Also provided herein are methods of selecting a treatment for a subject, wherein the methods include a step of performing an assay on a sample obtained from the subject to determine whether the subject has a dysregulation of a FGFR gene, a FGFR kinase, or expression or activity or level of any of the same (e.g., one or more FGFR inhibitor resistance mutations), and identifying or diagnosing a subject determined to have a dysregulation of a FGFR gene, a FGFR kinase, or expression or activity or level of any of the same, as having a FGFR-associated cancer. Some embodiments further include administering the selected treatment to the subject identified or diagnosed as having a FGFR-associated cancer. For example, in some embodiments, the selected treatment can include administration of a therapeutically effective amount of a compound of the disclosure to the subject identified or diagnosed as having a FGFR-associated cancer. In some embodiments, the assay is an in vitro assay. For example, an assay that utilizes the next generation sequencing, immunohistochemistry, or break apart FISH analysis. In some embodiments, the assay is a regulatory agency-approved, e.g., FDA-approved, kit. In some embodiments, the assay is a liquid biopsy.
[00339] Also provided herein are methods of treating a FGFR-associated cancer in a subject that include (a) administering one or more (e.g., two or more, three or more, four or more, five or more, or ten or more) doses of a first FGFR kinase inhibitor to a subject identified or diagnosed as having a FGFR associated cancer ( e.g., any of the types of FGFR- associated cancers described herein) (e.g., identified or diagnosed as having a FGFR- associated cancer using any of the exemplary methods described herein or known in the art); (b) after step (a), determining a level of circulating tumor DNA in a biological sample (e.g., a biological sample comprising blood, serum, or plasma) obtained from the subject; (c) administering a therapeutically effective amount of a second FGFR inhibitor or a compound of the disclosure as a monotherapy or in conjunction with an additional therapy or therapeutic agent to a subject identified as having about the same or an elevated level of circulating tumor DNA as compared to a reference level of circulating tumor DNA (e.g., any of the reference levels of circulating tumor DNA described herein). In some examples of these methods, the reference level of circulating tumor DNA is a level of circulating tumor DNA in a biological sample obtained from the subject prior to step (a). Some embodiments of these methods further include determining the level of circulating tumor DNA in the biological sample obtained from the subject prior to step (a). In some examples of these methods, the reference level of circulating tumor DNA is a threshold level of circulating tumor DNA (e.g., an average level of circulating tumor DNA in a population of subjects having a similar FGFR-associated cancer and having a similar stage of the FGFR-associated cancer, but receiving a non-effective treatment or a placebo, or not yet receiving therapeutic treatment, or a level of circulating tumor DNA in a subject having a similar FGFR-associated cancer and having a similar stage of the FGFR-associated cancer, but receiving a non-effective treatment or a placebo, or not yet receiving therapeutic treatment). In some examples of these methods, the first FGFR inhibitor is: ARQ-087, ASP5878, AZD4547, B-701, BAY1179470, BAY1 187982, BGJ398, brivanib, Debio 1347, dovitinib, E7090, erdafitinib, FPA144, HMPL-453, INCB054828, lenvatinib, lucitanib, LY3076226, MAX-40279, nintedanib, orantinib, pemigatinib, ponatinib, PRN1371, rogaratinib, sulfatinib, roblitinib, ICP-105, BIO- 1262, futibatinib, fisogatinib, LOXO-435, or RLY-4008.
[00340] Compounds of the disclosure can also be administered with additional therapy or therapeutic agents. In some aspects, the additional therapy or therapeutic agent includes one or more of radiation therapy, a chemotherapeutic agent ( e.g., any of the exemplary chemotherapeutic agents described herein or known in the art), a checkpoint inhibitor (e.g., any of the exemplary checkpoint inhibitors described herein or known in the art), surgery (e.g., at least partial resection of the tumor), and one or more other kinase inhibitors (e.g., any of the kinase inhibitors described herein or known in the art).
[00341] Compounds of the disclosure may also be useful as adjuvants to cancer treatment, that is, they can be used in combination with one or more additional therapies or therapeutic agents, for example a chemotherapeutic agent that works by the same or by a different mechanism of action. In some embodiments, a compound of the disclosure can be used prior to administration of an additional therapeutic agent or additional therapy. For example, a subject in need thereof can be administered one or more doses of a compound of the disclosure for a period of time and then under go at least partial resection of the tumor. In some embodiments, the treatment with one or more doses of a compound of the disclosure reduces the size of the tumor (e.g., the tumor burden) prior to the at least partial resection of the tumor. In some embodiments, a subject has a cancer (e.g., a locally advanced or metastatic tumor) that is refractory or intolerant to standard therapy (e.g., administration of a chemotherapeutic agent, such as a first FGFR inhibitor or a multikinase inhibitor, immunotherapy, radiation, or a platinum-based agent (e.g., cisplatin)). In some embodiments, a subject has a cancer (e.g., a locally advanced or metastatic tumor) that is refractory or intolerant to prior therapy (e.g., administration of a chemotherapeutic agent, such as a first FGFR inhibitor or a multikinase inhibitor, immunotherapy, radiation, or a platinum -based agent (e.g., cisplatin)). [00342] In some embodiments of any the methods described herein, the compound of the disclosure is administered in combination with a therapeutically effective amount of at least one additional therapeutic agent selected from one or more additional therapies or therapeutic (e.g., chemotherapeutic) agents. Non-limiting examples of additional therapeutic agents include: other FGFR-targeted therapeutic agents (i.e. a first or second FGFR kinase inhibitor), other kinase inhibitors (e.g., receptor tyrosine kinase targeted therapeutic agents (e.g., Trk inhibitors or EGFR inhibitors)), signal transduction pathway inhibitors, checkpoint inhibitors, modulators of the apoptosis pathway (e.g. obataclax); cytotoxic chemotherapeutics, angiogenesis-targeted therapies, immune-targeted agents, including immunotherapy, and radiotherapy.
[00343] Also provided herein are methods of treating a disease or disorder, comprising administering to a subject in need thereof a pharmaceutical combination for treating the disease or disorder which comprises (a) a compound of the disclosure, (b) an additional therapeutic agent, and (c) optionally at least one pharmaceutically acceptable carrier for simultaneous, separate or sequential use for the treatment of the disease or disorder, wherein the amounts of the compound of the disclosure and the additional therapeutic agent are together effective in treating the disease or disorder. In some embodiments, the compound of the disclosure, and the additional therapeutic agent are administered simultaneously as separate dosages. In some embodiments, the compound of the disclosure, and the additional therapeutic agent are administered as separate dosages sequentially in any order, in jointly therapeutically effective amounts, e.g. in daily or intermittently dosages. In some embodiments, the compound of the disclosure, and the additional therapeutic agent are administered simultaneously as a combined dosage. In some embodiments, the disease or disorder is a FGFR-associated disease or disorder. In some embodiments, the subject has been administered one or more doses of a compound of of the disclosure, prior to administration of the pharmaceutical composition.
[00344] In some embodiments, the treatment period is at least 7 days (e.g., at least or about 8 days, at least or about 9 days, at least or about 10 days, at least or about 11 days, at least or about 12 days, at least or about 13 days, at least or about 14 days, at least or about 15 days, at least or about 16 days, at least or about 17 days, at least or about 18 days, at least or about 19 days, at least or about 20 days, at least or about 21 days, at least or about 22 days, at least or about 23 days, at least or about 24 days, at least or about 25 days, at least or about 26 days, at least or about 27 days, at least or about 28 days, at least or about 29 days, or at least or about 30 days).
[00345] In some embodiments, the treatment period is at least 21 days (e.g., at least or about 22 days, at least or about 23 days, at least or about 24 days, at least or about 25 days, at least or about 26 days, at least or about 27 days, at least or about 28 days, at least or about 29 days, at least or about 30 days, at least or about 31 days, at least or about 32 days, at least or about 33 days, at least or about 34 days, at least or about 35 days, at least or about 36 days, at least or about 37 days, at least or about 38 days, at least or about 39 days, or at least or about 40 days).
[00346] Also provided herein are pharmaceutical compositions that contain, as the active ingredient, a compound of the disclosure, in combination with one or more pharmaceutically acceptable carriers (excipients). In some embodiments, the composition is suitable for topical administration. In making the compositions provided herein, the active ingredient is typically mixed with an excipient, diluted by an excipient or enclosed within such a carrier in the form of, for example, a capsule, sachet, paper, or other container. When the excipient serves as a diluent, it can be a solid, semi-solid, or liquid material, which acts as a vehicle, carrier or medium for the active ingredient. Thus, the compositions can be in the form of tablets, pills, powders, lozenges, sachets, cachets, elixirs, suspensions, emulsions, solutions, syrups, aerosols (as a solid or in a liquid medium), ointments containing, for example, up to 10% by weight of the active compound, soft and hard gelatin capsules, suppositories, sterile injectable solutions, and sterile packaged powders. In some embodiments, the composition is formulated for oral administration. In some embodiments, the composition is formulated as a tablet or capsule.
[00347] The compositions comprising a compound of the disclosure can be formulated in a unit dosage form, each dosage containing from about 5 to about 1,000 mg (1 g), more usually about 100 mg to about 500 mg, of the active ingredient. The term "unit dosage form" refers to physically discrete units for human subjects and other subjects, each unit containing a predetermined quantity of active material (i.e., a compound of the disclosure) to produce the desired therapeutic effect, with a suitable pharmaceutical excipient.
[00348] In some embodiments, the compositions provided herein contain from about 5 mg to about 50 mg of the active ingredient, i.e., the compound of the disclosure. One having ordinary skill in the art will appreciate that this embodies compounds or compositions containing about 5 mg to about 10 mg, about 10 mg to about 15 mg, about 15 mg to about 20 mg, about 20 mg to about 25 mg, about 25 mg to about 30 mg, about 30 mg to about 35 mg, about 35 mg to about 40 mg, about 40 mg to about 45 mg, or about 45 mg to about 50 mg of the active ingredient. In some embodiments, the compositions provided herein contain from about 50 mg to about 500 mg of the active ingredient. One having ordinary skill in the art will appreciate that this embodies compounds or compositions containing about 50 mg to about 100 mg, about 100 mg to about 150 mg, about 150 mg to about 200 mg, about 200 mg to about 250 mg, about 250 mg to about 300 mg, about 350 mg to about 400 mg, or about 450 mg to about 500 mg of the active ingredient. In some embodiments, the compositions provided herein contain from about 500 mg to about 1,000 mg of the active ingredient. One having ordinary skill in the art will appreciate that this embodies compounds or compositions containing about 500 mg to about 550 mg, about 550 mg to about 600 mg, about 600 mg to about 650 mg, about 650 mg to about 700 mg, about 700 mg to about 750 mg, about 750 mg to about 800 mg, about 800 mg to about 850 mg, about 850 mg to about 900 mg, about 900 mg to about 950 mg, or about 950 mg to about 1,000 mg of the active ingredient.
[00349] The active compound may be effective over a wide dosage range and is generally administered in a pharmaceutically effective amount. It will be understood, however, that the amount of the compound actually administered will usually be determined by a physician, according to the relevant circumstances, including the condition to be treated, the chosen route of administration, the actual compound administered, the age, weight, and response of the individual subject, the severity of the subject's symptoms, and the like.
[00350] In some embodiments, the compounds provided herein can be administered in an amount ranging from about 1 mg/kg to about 100 mg/kg. In some embodiments, the compound provided herein can be administered in an amount of about 1 mg/kg to about 20 mg/kg, about 5 mg/kg to about 50 mg/kg, about 10 mg/kg to about 40 mg/kg, about 15 mg/kg to about 45 mg/kg, about 20 mg/kg to about 60 mg/kg, or about 40 mg/kg to about 70 mg/kg. For example, about 5 mg/kg, about 10 mg/kg, about 15 mg/kg, about 20 mg/kg, about 25 mg/kg, about 30 mg/kg, about 35 mg/kg, about 40 mg/kg, about 45 mg/kg, about 50 mg/kg, about 55 mg/kg, about 60 mg/kg, about 65 mg/kg, about 70 mg/kg, about 75 mg/kg, about 80 mg/kg, about 85 mg/kg, about 90 mg/kg, about 95 mg/kg, or about 100 mg/kg. In some embodiments, such administration can be once-daily or twice-daily (BID) administration.
[00351] The disclosure is also directed to the following aspects: Aspect 1. A compound of formula (I): or a pharmaceutically acceptable salt thereof, wherein
or a pharmaceutically acceptable salt thereof, wherein

X = O, S, or NR;
R is H or C1-C3alkyl; n = 1 or 2; m = 1 or 2;
R1 is H or optionally substituted C1-Cealkyl;
R2 is -Co-C6alkN=S(0)(C1-C6alkyl)(C1-C6alkyl), or -NH-C1-C6alk- N=S(O)(C1-C6alkyl)(C1-C6alkyl); one or two of Q1, Q2, Q3, Q4 is N and the others are each independently CR5a;
R5a is H, halogen, -CN, -S(O)2C1-C6alkyl, OCF3, OC1-C3alkyl, or C1-C3alkyl;
Q5, Q6, Q7, Q8, and Q9 are each independently N or CR5, wherein one or two of the Q5, Q6, Q7, Q8, and Q9 is N and the remainder are CR5;
R5 is H, halogen, C1-C3alkyl, C1-C3alkoxyl, or cycloalkyl;
R6 is C1-Cealkyl;
R7 is H, halogen, -C1-Cealkyl, -C1-Ce alkoxyl, or -cycloalkyl; and
R8 is H, halogen, -C1-Cealkyl, -C1-Ce alkoxyl, or -cycloalkyl.
Aspect 2. The compound of aspect 1, wherein X is O. Aspect 3. The compound of aspect 1 or aspect 2, wherein R6 is CH3.
Aspect 4. The compound of any one of the preceding aspects, wherein R8 is H.
Aspect 5. The compound of any one of the preceding aspects, wherein R7 is H, F, or
OCH3.
Aspect 6. The compound of any one of the preceding aspects, wherein Q1 and Q3 are each CR5a wherein R5a is H.
Aspect 7. The compound of any one of the preceding aspects, wherein Q2 and Q4 are each N.
Aspect 8. The compound of any one of aspects 1-6, wherein Q2 is N and Q4 is CR5a wherein R5a is H, -CN, or F.
Aspect 9. The compound of any one of aspects 1-6, wherein Q2 is N and Q4 is CR5a wherein R5a is S(O)2C1-C6alkyl, OC1-C3alkyl, or C1-C3alkyl.
Aspect 10. The compound of any one of the preceding aspects, wherein Q5 and Q9 are each CR5 wherein each R5 is Cl; Q7 is N; Q6 is CR5 wherein R5 is H; and Q8 is CR5 wherein R5 is H or CH3.
Aspect 11. The compound of any one of the preceding aspects, wherein n is i and m is 1.
Aspect 12. The compound of any one of the preceding aspects, wherein the compound of formula (I) is a compound of formula (IA):

Q2 and Q4 are each N, or one of Q2 or Q4 is N and the other is CR5a;
R5a is H, F, -SO2CH3, or -CN;
R5 is H or CH3; and
R7 is H, F, or OCH3.
Aspect 13. The compound of aspect 12, wherein R5 is H.
Aspect 14. The compound of aspect 12, wherein R5 is CH3.
Aspect 15. The compound of any one of aspects 12-14, wherein R7 is H.
Aspect 16. The compound of any one of aspects 12-14, wherein R7 is F.
Aspect 17. The compound of any one of aspects 12-14, wherein R7 is OCH3.
Aspect 18. The compound of any one of aspects 12-17, wherein Q2 and Q4 are each N.
Aspect 19. The compound of any one of aspects 12-17, wherein one of Q2 or Q4 is N and the other is CR5a.
Aspect 20. The compound of aspect 19, wherein R5a is H or F.
Aspect 21. The compound of aspect 20, wherein R5a is H.
Aspect 22. The compound of aspect 20, wherein R5a is F. Aspect 23. The compound of aspect 19, wherein R5a is CN or SO2CH3.
Aspect 24. The compound of aspect 23, wherein R5a is CN.
Aspect 25. The compound of aspect 23, wherein R5a is SO2CH3.
Aspect 26. The compound of aspect 1, wherein R5a is OCF3.
Aspect 27. The compound of any one of aspects 1-26, wherein R1 is H.
Aspect 28. The compound of any one of aspects 1-26, wherein R1 is CH3.
Aspect 29. The compound of aspect 1, wherein the compound of formula (I) is a compound of formula (IB): or a pharmaceutically acceptable salt thereof, wherein R2 is -Co-C6alk-N=S(0)(C1-C6alkyl)(C1-C6alkyl), or -NH-C1-C6alk-
or a pharmaceutically acceptable salt thereof, wherein R2 is -Co-C6alk-N=S(0)(C1-C6alkyl)(C1-C6alkyl), or -NH-C1-C6alk-

N=S(O)(C1-C6alkyl)(C1-C6alkyl);
R7is H or -OC1-Cealkyl; and
R5a is halo, or -CN.
Aspect 30. The compound of aspect 29, wherein R7 is H.
Aspect 31. The compound of aspect 29, wherein R7 is -OC1-Cealkyl.
Aspect 32. The compound of aspect 31, wherein R7 is -OCH3.
Aspect 33. The compound of any one of aspects 29-32, R5a is -CN.
Aspect 34. The compound of any one of aspects 29-32, R5a is halo. Aspect 35. The compound of aspect 34, R5a is F, Cl, or Br.
Aspect 36. The compound of aspect 35, R5a is F.
Aspect 37. The compound of aspect 35, R5a is Cl.
Aspect 38. The compound of aspect 35, R5a is Br.
Aspect 39. The compound of any one of aspects 29-38, wherein R2 is -Co-Cealk- N=S(O)(C1-C6alkyl)(C1-C6alkyl).
Aspect 40. The compound of aspect 39, wherein R2 is -N=S(O)(CH3)2.
Aspect 41. The compound of aspect 39, wherein R2 is -CH2-N=S(O)(CH3)2.
Aspect 42. The compound of any one of aspects 29-38, wherein R2 is -NH-C1-Cealk- N=S(O)(C1-C6alkyl)(C1-C6alkyl).
Aspect 43. The compound of aspect 42, wherein R2 is -NH-CH2CH2-N=S(O)(CH3)2.
Aspect 44. The compound of aspect 29, or a pharmaceutically acceptable salt thereof, wherein the compound is:

Aspect 45. The compound of aspect 29, or a pharmaceutically acceptable salt thereof, wherein the compound is:

Aspect 46. The compound of aspect 29, or a pharmaceutically acceptable salt thereof, wherein the compound is:

Aspect 47. The compound of aspect 29, or a pharmaceutically acceptable salt thereof, wherein the compound is:

Aspect 48. The compound of aspect 29, or a pharmaceutically acceptable salt thereof, wherein the compound is:

Aspect 49. The compound of aspect 29, or a pharmaceutically acceptable salt thereof, wherein the compound is:

Aspect 50. A pharmaceutical composition comprising a compound of any one of aspects 1-49, or a pharmaceutically acceptable salt thereof, and a pharmaceutically acceptable excipient.
Aspect 51. A method of treating a disease or disorder in a subject in need thereof comprising administering to the subject a compound of any one of aspects 1 to 49, or a pharmaceutically acceptable salt thereof.
Aspect 52. The method of aspect 51, wherein the disease or disorder is cancer.
Aspect 53. The method of aspect 52, wherein the cancer is urothelial carcinoma, hepatocellular carcinoma, breast carcinoma, endometrial adenocarcinoma, ovarian carcinoma, primary glioma, cholangiocarcinoma, gastric adenocarcinoma, non-small cell lung carcinoma, pancreatic exocrine carcinoma, oral cancer, prostate cancer, bladder cancer, colorectal carcinoma, renal cell carcinoma, neuroendocrine carcinoma, myeloproliferative neoplasms, head and neck (squamous), melanoma, leiomyosarcoma, and/or sarcomas.
Aspect 54. The method of aspect 53, wherein the cancer is bladder cancer.
Aspect 55. The method of aspect 53, wherein the cancer is urothelial carcinoma.
Aspect 56. The method of aspect 53, wherein the cancer is hepatocellular carcinoma.
Aspect 57. The method of any one of aspects 52 to 56, wherein the cancer is an FGFR- mutant cancer.
Aspect 58. The method of aspect 51, wherein the disease or disorder is a developmental disorder.
Aspect 59. The method of aspect 58, wherein the developmental disorder is achondroplasia.
Aspect 60. A method of inhibiting FGFR in a cell comprising contacting the cell with a compound of any one of aspects 1 to 49.
Examples
[00352] The examples and preparations provided below further illustrate and exemplify the compounds of the present invention and methods of preparing such compounds. It is to be understood that the scope of the present invention is not limited in any way by the scope of the following examples and preparations.
[00353] In several embodiments, where single enantiomers are provided, the enantiomers may be separated by conventional means (chiral chromatography, preparing diastereomeric salts, chiral derivatization, crystallization, enzymatic reactions, etc.). In several embodiments, a chiral intermediate compound is purified to prepare an enantiomerically pure (or substantially enantiomerically pure, enantiomerically enriched, etc.) intermediate.

Example 1. (R)-((l-(5-(5-(l-(3,5-Dichloropyridin-4-yl)ethoxy)-lH-indazol-3-yl)-3- fluoropyridin-2-yl)-3-methylazetidin-3-yl)imino)dimethyl-L6-sulfanone
[00354] Step 1. 1 -Benzhydryl-3 -methylazeti din-3 -yl methanesulfonate. Methanesulfonyl chloride (6.2 mL, 80 mmol, 2 equiv) was added dropwise to a solution of 1- benzhydryl-3 -methyl-azeti din-3 -ol (10.13 g, 40 mmol, 1 equiv) and triethylamine (14.0 mL, 100 mmol, 2.5 equiv) in anhydrous dichloromethane (150 mL) at 0 °C. After stirring at 0 to 5 °C for 6 hours, water (50 mL) was added, and the layers were separated. The aqueous layer was extracted with dichloromethane (3 x 50 mL). The combined organic layers were washed with saturated brine (80 mL), dried over sodium sulfate, filtered, and concentrated under reduced pressure. The residue was purified on an Interchim automated chromatography system (220 g Sorbtech silica gel column), eluting with a gradient of 0 to 40% ethyl acetate in heptanes to give a white solid (4.63 g, 35% yield). LCMS m/z = 332 (M+H).
[00355] Step 2. ((l-Benzhydryl-3-methylazetidin-3-yl)imino)dimethyl-X6- sulfanone. Anhydrous sodium bicarbonate (1.9 g, 22.6 mmol, 1.5 equiv) was added to a mixture of compound 1 -benzhydryl-3 -m ethylazeti din-3 -yl methanesulfonate (5.0 g, 15.08 mmol, 1 equiv) and (dimethanesulfinylidene)amine (1.545 g, 16.6 mmol, 1.1 equiv) in acetonitrile (100 mL) at room temperature. After heating at 80 °C for 16 hours, the reaction was cooled to room temperature. The solid was filtered off and the filtrate was concentrated under reduced pressure to give a brown oil. This crude material was purified on a Biotage automated chromatography system (Sorbtech 120 g silica gel column), eluting with a gradient of 0 to 10% methanol in di chloromethane to give a yellow solid (3.7 g, 75% yield). LCMS m/z = 329 (M+H). [00356] Step 3A. Dimethyl((3-methylazeti din-3 -yl)imino)-X6-sulfanone hydrochloride. A 2M HC1 in diethyl ether solution (16.9 mL, 3 equiv) was added to a solution of ((l-benzhydryl-3-methylazetidin-3-yl)imino)dimethyl-X6-sulfanone (3.7 g, 11.26 mmol, 1 equiv) in acetonitrile (75 mL) at room temperature. After stirring for 1 hour, the mixture was concentrated under reduced pressure to give the hydrochloride salt (4.8 g) as an off-white solid.
[00357] Step 3B. A mixture of the product from step 3 A (4.8 g, 11.264 mmol, 1 equiv) and 20% palladium hydroxide on carbon (50% wet, 0.96 g, 20 w/w%) in methanol (250 mL) was hydrogenated (50 psi at room temperature) for 19 hours. LCMS analysis showed partial conversion. Additional 20% palladium hydroxide on carbon (50% wet, 0.96 g, 20 w/w%) was added and the hydrogenation was continued over a weekend. LCMS analysis showed that the reaction was complete. The mixture was filtered through celite and washed with methanol (3 x 50 mL). The filtrate was concentrated under reduced pressure and the residue was triturated with heptanes (3 x 50 mL) at 35 °C to give a yellow solid (2.83 g, >100% yield) containing diphenylmethane. LCMS m/z = 163 (free base) (M+H).
[00358] Step 4. 5-[(lR)-l-(3,5-Dichloro-4-pyridyl)ethoxy]-3-(5,6-difluoro-3- pyridyl)-l -tetrahydropyran -2 -yl-indazole. To a solution of 5-[(lR)-l-(3,5-dichloro-4- pyridyl)ethoxy]-3-iodo-l-tetrahydropyran-2-yl-indazole (4.50 g, 8.68 mmol, 1.0 eq) and 2,3- difhioro-5-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)pyridine (3.14 g, 13.03 mmol, 1.5 eq) in dioxane (50 mL) and water (5 mL) was added K2CO3 (3.60 g, 26.05 mmol, 3.0 eq) and Pd(dppf)C12 (708 mg, 0.87 mmol, 0.1 eq). The reaction mixture was stirred for 3 h at 90 °C under N2 protection. After the reaction was completed, the solid was filtered out and the filtrate was concentrated in vacuum. The crude product was purified by silica gel column (Petroleum Ether/EtOAc = 10/1) to give a white solid (2.3 g, 52% yield). LCMS m/z = 505.2 (M+H).
[00359] Step 5. ((l-(5-(5-((R)-l-(3,5-Dichloropyridin-4-yl)ethoxy)-l-(tetrahydro- 2H-pyran-2-yl)-lH-indazol-3-yl)-3-fluoropyridin-2-yl)-3-methylazetidin-3- yl)imino)dimethyl-Z6-sulfanone. Dimethyl((3-methylazeti din-3 -yl)imino)-+6-sulfanone hydrochloride (255 mg, 1.28 mmol, 2.5 equiv) and potassium carbonate (276 mg, 2.0 mmol, 4 equiv) were added to a solution of 5-[(lR)-l-(3,5-dichloro-4-pyridyl)ethoxy]-3-(5,6- difluoro-3 -pyridyl)- l-tetrahydropyran-2-yl-indazole (252 mg, 0.50 mmol, 1 equiv) in acetonitrile (10 mL). After stirring at 70 °C for 18 hours, the reaction was cooled to room temperature and diluted with ethyl acetate (40 mL) and water (20 mL). The layers were separated, and the aqueous layer was extracted with ethyl acetate (2 x 20 mL). The combined organic layers were washed with saturated brine (40 mL), dried over sodium sulfate, filtered, and concentrated under reduced pressure to give a white solid (330 mg). LCMS m/z = 647 (M+H).
[00360] Step 6. (R)-((l-(5-(5-(l-(3,5-Dichloropyridin-4-yl)ethoxy)-lH-indazol-3- yl)-3-fluoropyridin-2-yl)-3-methylazeti din-3 -yl)imino)dimethyl-X6-sulfanone. Trifluoroacetic acid (3.3 mL) was added to a solution of ((l-(5-(5-((R)-l-(3,5-dichloropyridin-4-yl)ethoxy)- l-(tetrahydro-2H-pyran-2-yl)-lH-indazol-3-yl)-3-fluoropyridin-2-yl)-3-methylazetidin-3- yl)imino)dimethyl-X6-sulfanone (330 mg, 0.50 mmol, 1.0 equiv) in dichloromethane (3.3 mL) at room temperature. After stirring at room temperature for 4 hours, the volatile material was removed under reduced pressure. The residue was diluted with dichloromethane (20 mL) and water (10 mL) and adjusted to pH 9 with saturated sodium carbonate. The layers were separated, and the aqueous layer was extracted with di chloromethane (3 x 10 mL). The combined organic layers were washed with saturated brine (20 mL), dried over sodium sulfate, filtered, and concentrated under reduced pressure. The residue was purified on an Interchim automated chromatography system (Sorbtech 25 g silica gel column), eluting with a gradient of 0 to 10% methanol in di chloromethane to give a tan solid (178 mg, 63% yield). LCMS m/z = 563.1 (M+H); 1H NMR (400 MHz, DMSO-d6) 5 = 13.11 (s, 1H), 8.57 (s, 2H), 8.37 (t, J = 1.7 Hz, 1H), 7.70 (dd, J = 1.8, 13.1 Hz, 1H), 7.47 (d, J = 9.0 Hz, 1H), 7.21 (d, J = 2.1 Hz, 1H), 7.10 (dd, J = 2.3, 9.0 Hz, 1H), 6.14 (q, J = 6.7 Hz, 1H), 4.13 (d, J = 7.3 Hz, 2H), 4.02 (dd, J = 2.1, 8.3 Hz, 2H), 3.07 (s, 6H), 1.76 (d, J = 6.6 Hz, 3H), 1.66 (s, 3H).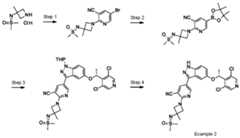

Example 2. 5-[5-[(lR)-l-(3,5-dichloro-4-pyridyl)ethoxy]-lH-indazol-3-yl]-2-[3-
[[dimethyl(oxo)-k6-sulfanylidene]amino]-3-methyl-azetidin-l-yl]pyridine-3-carbonitrile [00361] Step 1. 5-Bromo-2-(3-((Dimethyl(oxo)-X6-sulfaneylidene)amino)-3- methylazetidin-l-yl)nicotinonitrile. N,N-Diisopropylethylamine (0.87 mL, 5 mmol, 5 equiv) was added to a mixture of dimethyl((3-methylazeti din-3 -yl)imino)-X6-sulfanone hydrochloride (257 mg, 1 mmol, 1 equiv) in acetonitrile (10 mL). After stirring at room temperature for 10 minutes, 5-bromo-2-chloro-pyridine-3-carbonitrile (217 mg, 1 mmol, 1 equiv) was added and the mixture was stirred at room temperature for 2 days. The mixture was absorbed onto Celite (10 g) under reduced pressure and purified on an Interchim automated chromatography system (RediSep Rf GOLD 100 g HP Cl 8 column), eluting with a gradient of 0 to 100% acetonitrile in water to give a pale-yellow solid (218 mg, 64% yield). LCMS m/z = 344 (M+H).
[00362] Step 2. 2-(3-((Dimethyl(oxo)-X6-sulfaneylidene)amino)-3-methylazetidin- l-yl)-5-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)nicotinonitrile. A mixture of 5-bromo-2- (3-((dimethyl(oxo)-X6-sulfaneylidene)amino)-3-methylazetidin-l-yl)nicotinonitrile (218 mg, 0.635 mmol, 1 equiv), bis(pinacolato)diboron (242 mg, 0.953 mmol, 1.5 equiv), potassium acetate (125 mg, 1.270 mmol, 2 equiv) and [l,l'-bis(diphenylphosphino)ferrocene]- dichloropalladium(II) (47 mg, 0.0635 mmol, 0.1 equiv) in 1,4-dioxane (9 mL) was sparged with nitrogen for 10 minutes, and then heated at 90 °C for 18 hours. LCMS analysis indicated that all starting material was consumed, and the product was formed. The crude reaction mixture was used subsequently in next step. LCMS m/z = 309 (boronic acid) and 391 (M+H).
[00363] Step 3. 5-(5-((R)-l-(3,5-Dichloropyridin-4-yl)ethoxy)-l-(tetrahydro-2H- pyran-2-yl)-lH-indazol-3-yl)-2-(3-((dimethyl(oxo)-X6-sulfaneylidene)amino)-3- methylazetidin-l-yl)nicotinonitrile. The mixture from step 2 (0.635 mmol, 1.5 equiv) in 1,4- dioxane was treated with 5-[(lR)-l-(3,5-dichloro-4-pyridyl)ethoxy]-3-iodo-l- tetrahydropyran-2-yl-indazole (219 mg, 0.423 mmol, 1 equiv), [l,l'-bis(diphenylphosphino) ferrocene]dichloropalladium(II) (31 mg, 0.0423 mmol, 0.1 equiv) and potassium carbonate (117 mg, 0.846 mmol, 2 equiv) and water (1.0 mL). After sparging with nitrogen for 10 minutes, the reaction was heated at 90 °C for 18 hours. The mixture was cooled to room temperature and diluted with ethyl acetate (50 mL) and water (20 mL). The layers were separated, and the aqueous layer was extracted with ethyl acetate (3 x 20 mL). The combined organic layers were washed with saturated brine (20 mL), dried over sodium sulfate, filtered, and concentrated under reduced pressure. The crude product was purified on an Interchim automated chromatography system (Sorbtech 25 g silica gel column), eluting with a gradient of 0 to 10% methanol in ethyl acetate to give a brown solid (163 mg, 38% yield, 2 steps). LCMS m/z = 654 (M+H).
[00364] Step 4. (R)-5-(5-(l-(3,5-Dichloropyridin-4-yl)ethoxy)-lH-indazol-3-yl)-2- (3-((dimethyl(oxo)-X6-sulfaneylidene)amino)-3-methylazetidin-l-yl)nicotinonitrile. Trifluoroacetic acid (1.6 mL) was added to a solution of 5-(5-((R)-l-(3,5-dichloropyridin-4- yl)ethoxy)-l-(tetrahydro-2H-pyran-2-yl)-lH-indazol-3-yl)-2-(3-((dimethyl(oxo)-X6- sulfaneylidene)amino)-3-methylazetidin-l-yl)nicotinonitrile (163 mg, 0.249 mmol, 1.0 equiv) in dichloromethane (1.6 mL) at room temperature. After stirring at room temperature for 5 hours, the volatiles were removed under reduced pressure. The residue was diluted with di chloromethane (20 mL) and water (10 mL) and adjusted to pH 9 with saturated sodium carbonate. The layers were separated, and the aqueous layer was extracted with di chloromethane (2 x 20 mL). The combined organic layers were washed with saturated brine (20 mL), dried over sodium sulfate, filtered, and concentrated under reduced pressure. The residue was purified on an Interchim automated chromatography system (Sorbtech 25 g silica gel column), eluting with a gradient of 0 to 10% methanol in di chloromethane to give a pale-yellow solid (71 mg, 50% yield). LCMS m/z = 570.1 (M+H); 1H NMR (400 MHz, DMSO-d6) 5 = 13.16 (s, 1H), 8.76 (d, J = 2.3 Hz, 1H), 8.57 (s, 2H), 8.14 (d, J = 2.3 Hz, 1H), 7.49 (d, J = 8.9 Hz, 1H), 7.19 - 7.09 (m, 2H), 6.14 (q, J = 6.7 Hz, 1H), 4.31 - 4.17 (m, 4H), 3.09 (s, 6H), 1.76 (d, J = 6.6 Hz, 3H), 1.66 (s, 3H). Example 3. 5-[5-[(lR)-l-(3,5-dichloro-4-pyridyl)ethoxy]-lH-indazol-3-yl]-2-[3-[2- [[dimethyl(oxo)-k6-sulfanylidene]amino]ethylamino]-3-methyl-azetidin-l-yl]pyridine-3- carbonitrile
Example 3. 5-[5-[(lR)-l-(3,5-dichloro-4-pyridyl)ethoxy]-lH-indazol-3-yl]-2-[3-[2- [[dimethyl(oxo)-k6-sulfanylidene]amino]ethylamino]-3-methyl-azetidin-l-yl]pyridine-3- carbonitrile

[00365] Step 1. 5-((R)-l-(3,5-Dichloropyridin-4-yl)ethoxy)-3-iodo-l-(tetrahydro- 2H-pyran-2-yl)-lH-indazole. A mixture of 3-iodo-l-tetrahydropyran-2-yl-indazol-5-ol (1.0 g, 2.90 mmol, 1.0 equiv), [(lS)-l-(3,5-dichloro-4-pyridyl)ethyl] methanesulfonate (780 mg, 2.90 mmol, 1.0 equiv) and cesium carbonate (1.41 g, 14.45 mmol, 1.5 equiv) in N,N- dimethylformamide (20 mL) was heated at 130 °C for 16 h. The volatiles were removed under reduced pressure and the residue was suspended in saturated ammonium chloride (50 mL). The solution was extracted with ethyl acetate (3 x 50 mL). The combined organic layers were dried over sodium sulfate, filtered, and concentrated under reduced pressure. The crude product was purified on a Biichi automated chromatography system (Sorbtech 40 g silica gel column), eluting with a gradient of 0 to 30% ethyl acetate in heptanes to give a white solid (1.01 g, 88% yield). LCMS m/z = 517.2 (M+H).
[00366] Step 2. 5-[5-[(lR)-l-(3,5-Dichloro-4-pyridyl)ethoxy]-l-tetrahydropyran-2- yl-indazol-3-yl]-2-fluoro-pyridine-3-carbonitrile. To a solution of 5-((R)-l-(3,5- dichloropyridin-4-yl)ethoxy)-3-iodo-l-(tetrahydro-2H-pyran-2-yl)-lH-indazole (5.0 g, 9.65 mmol, 1.0 eq) and 2-fluoro-5-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)pyridine-3- carbonitrile (3.11 g, 12.54 mmol, 1.3 eq) in dioxane (100 mL) and water (10 mL) was added K2CO3 (2.66 g, 19.30 mmol, 1.0 eq) and Pd(dppf)C12 (705 mg, 0.965 mmol, 0.1 eq) at 25 °C. The resulting mixture was stirred at 90 °C for 2 h under N2. After cooling to rt, the mixture was quenched with water (50 mL) and extracted with ethyl acetate (50 mL x 3). The combined organic phases were washed with brine (100 mL), dried over anhydrous Na2SO4, filtered and concentrated. The crude product was purified by silica gel chromatography (Petroleum Ether/EtOAc = 3/1) to give a white solid (4.8 g, 97% yield). LCMS m/z = 512.2 (M+H).
[00367] Step 3. 5-[5-[(lR)-l-(3,5-Dichloro-4-pyridyl)ethoxy]-l-tetrahydropyran-2- yl-indazol-3-yl]-2-(3-hydroxy-3-methyl-azetidin-l-yl)pyridine-3-carbonitrile. To a solution of 5-[5-[(lR)-l-(3,5-dichloro-4-pyridyl)ethoxy]-l-tetrahydropyran-2-yl-indazol-3-yl]-2- fluoro-pyridine-3 -carbonitrile (3.2 g, 6.25 mmol, 1.0 eq) and 3-methylazetidin-3-ol (1.54 g, 12.49 mmol, 2.0 eq) in DMSO (10 mL) was added DIEA (2.42 g, 18.74 mmol, 3.0 eq) at 25 °C. The resulting solution was stirred at 90 °C for 3 h, cooled to room temperature, quenched with water (20 mL), and extracted with ethyl acetate (20 mL x 3). The combined organic phases were washed with brine (30 mL), dried with anhydrous Na2SO4, filtered and concentrated. The crude product was purified by silica gel chromatography (Petroleum Ether/EtOAc = 2/1) to give a white solid (3.1 g, 86% yield). LCMS m/z = 579.3 (M+H).
[00368] Step 4. [l-[3-Cyano-5-[5-[(lR)-l-(3,5-dichloro-4-pyridyl)ethoxy]-l- tetrahydropyran-2-yl-indazol-3-yl]-2-pyridyl]-3-methyl-azetidin-3-yl] methanesulfonate. To a solution of product step 3 (3.1 g, 5.35 mmol, 1.0 eq) in DCM (50 mL) was added TEA (1.08 g, 10.7 mmol, 2.0 eq). Methanesulfonyl chloride (738 mg, 6.42 mmol, 1.2 eq) was added dropwise at 0 °C. The resulting solution was stirred at rt for 2 h and was concentrated. The crude product was purified by silica gel chromatography (Petroleum Ether/EtOAc = 3/1) to give a white solid (3.3 g, 94% yield). LCMS m/z = 657.2 (M + 1).
[00369] Step 5. 2-((Dimethyl(oxo)-X6-sulfaneylidene)amino)acetonitrile. Bromoacetonitrile (7.09 g, 59.076 mmol, 1.1 equiv) and sodium bicarbonate (6.77 g, 80.558 mmol, 1.5 equiv) were added to a solution of (dimethanesulfinylidene) amine (5.0 g, 53.706 mmol, 1 equiv) in acetonitrile (200 mL) at room temperature. After refluxing for 15 hours, the mixture was cooled to room temperature and filtered. The filtrate was concentrated under reduced pressure. The residue was purified on an Interchim automated chromatography system (Sorbtech 120 g silica gel column), eluting with a gradient of 0 to 10% methanol in di chloromethane, to give a pale-yellow oil (3.14 g, 44%). LCMS m/z = 133.1 (M+H).
[00370] Step 6. ((2-Aminoethyl)imino)dimethyl-X6-sulfanone. A mixture of 2- ((dimethyl(oxo)-X6-sulfaneylidene)amino)acetonitrile (3.14 g, 23.755 mmol, 1 equiv) and Raney nickel (slurry in water, 1.68 g) in 2M ammonia in methanol (140 mL) was hydrogenated (20 psi at room temperature) for 27 hours. LCMS analysis indicated that starting material remained. Additional Raney nickel (slurry in water, 2.1 g) was added, and the hydrogenation was continued over a weekend. The mixture was filtered through Celite and was washed with methanol (3 x 100 mL). The filtrate was concentrated under reduced pressure, dried under vacuum at 40 °C to give a dark brown oil (3.0 g, 93%), that was used directly in the next step. LCMS m/z = 137.1 (M+H).
[00371] Step 7. 5-(5-((R)-l-(3,5-Dichloropyridin-4-yl)ethoxy)-l-(tetrahydro-2H- pyran-2-yl)-lH-indazol-3-yl)-2-(3-((2-((dimethyl(oxo)-X6- sulfaneylidene)amino)ethyl)amino)-3-methylazetidin-l-yl)nicotinonitrile. [l-[3-Cyano-5-[5- [(lR)-l-(3,5-dichloro-4-pyridyl)ethoxy]-l-tetrahydropyran-2-yl-indazol-3-yl]-2-pyridyl]-3- methyl-azetidin-3-yl] methanesulfonate (329 mg, 0.5 mmol, 1 equiv) was added to a solution of ((2-aminoethyl)imino)dimethyl-X6-sulfanone (170 mg, 1.25 mmol, 2.5 equiv) in acetonitrile (5 mL) in a pressure tube, followed by potassium tert-butoxide (144 mg, 1.5 mmol, 3 equiv). After stirring at 70 °C for 3 days, the reaction was cooled to room temperature and quenched with water (30 mL). The mixture was extracted with di chloromethane (4 x 30 mL). The combined organic layers were dried over sodium sulfate, filtered, and concentrated under reduced pressure. The residue was purified on an Biotage automated chromatography system (Biotage 25 g silica gel column), eluting with a gradient of 0 to 10% methanol in di chloromethane to give a dark oil (197 mg, 43% yield). LCMS m/z = 697.2 (M+H).
[00372] Step 8. 5-[5-[(lR)-l-(3,5-dichloro-4-pyridyl)ethoxy]-lH-indazol-3-yl]-2- [3-[2-[[dimethyl(oxo)-k6-sulfanylidene]amino]ethylamino]-3-methyl-azetidin-l-yl]pyridine- 3-carbonitrile. Trifluoroacetic acid (2.0 mL) was added to a solution of product step 7 (197 mg, 0.282 mmol, 1.0 equiv) in dichloromethane (2.0 mL) at room temperature. After stirring at room temperature for 4 hours, the volatiles were removed under reduced pressure. The residue was diluted with di chloromethane (20 mL) and water (10 mL) then adjusted to pH 9 with saturated sodium carbonate. The layers were separated, and the aqueous layer was extracted with di chloromethane (3 x 20 mL). The combined organic layers were washed with saturated brine (20 mL), dried over sodium sulfate, filtered and concentrated under reduced pressure. The residue was dry loaded onto Celite (8 g) and purified on an InterChim automated chromatography system (RediSep Rf GOLD 50 g HP C18 column), eluting with a gradient of 0 to 100% acetonitrile in water to give a white solid after lyophilization (84 mg, 49% yield). LCMS m/z = 613.2 (M+H); 1H NMR (400 MHz, DMSO-d6) 5 = 13.16 (s, 1H), 8.76 (d, J = 2.3 Hz, 1H), 8.58 (s, 2H), 8.13 (d, J = 2.3 Hz, 1H), 7.49 (d, J = 8.9 Hz, 1H), 7.16 (d, J = 2.1 Hz, 1H), 7.10 (dd, J = 2.3, 9.0 Hz, 1H), 6.17 - 6.11 (m, 1H), 4.16 - 4.09 (m, 2H), 4.08 - 4.01 (m, 2H), 3.05 - 2.98 (m, 8H), 2.62 (t, J = 6.4 Hz, 2H), 2.48 - 2.21 (m, 1H), 1.76 (d, J = 6.6 Hz, 3H), 1.43 (s, 3H).

Example 4. 5-[5-[(lR)-l-(3,5-dichloro-4-pyridyl)ethoxy]-6-methoxy-lH-indazol-3-yl]-2-
[3-[[dimethyl(oxo)-k6-sulfanylidene]amino]-3-methyl-azetidin-l-yl]pyridine-3- carbonitrile
[00373] Step 1. (E)-N'-(2-Bromo-5-hydroxy-4-methoxybenzylidene)-4- methylbenzenesulfonohydrazide. p-Toluenesulfonyl hydrazide (0.56 g, 3.0 mmol, 1.0 equiv) was added to a solution of 2-bromo-5-hydroxy-4-methoxy -benzaldehyde (0.7 g, 3.0 mmol, 1.0 equiv) in methanol (7.0 mL) at rt. The resulting mixture was heated at 60 °C for 2 h. The reaction was cooled to rt and the solvent was removed under reduced pressure. The residue was dissolved in ethyl acetate (20 mL), then heptanes (80 mL) was added to give a lightyellow solid (1.21 g, 100%). LCMS m/z = 399.0 (M+H).
[00374] Step 2. 6-Methoxy-l-tosyl-lH-indazol-5-ol. Copper(I) oxide (0.22 g, 1.5 mmol, 0.5 equiv) was added to a solution of (E)-N'-(2-Bromo-5-hydroxy-4- methoxybenzylidene)-4-methylbenzenesulfonohydrazide (1.2 g, 3.0 mmol, 1.0 equiv) in isoamyl alcohol (30 mL) at room temperature. After heating at 132 °C for 2 hours, the mixture was cooled to room temperature and diluted with water (80 mL). The mixture was extract with ethyl acetate (4 x 50 mL). The combined organic layers were dried over sodium sulfate and filtered. The filtrate was concentrated onto silica gel (8.0 g) and purified on a Biotage automated purification system (Biotage Sfar Silica, 50 g; 0% to 100% ethyl acetate in heptanes) to give a light-yellow solid (0.66 g, 70% yield). LCMS m/z = 319.1 (M+H).
[00375] Step 3. (R)-5-(l-(3,5-Dichloropyridin-4-yl)ethoxy)-6-methoxy-l-tosyl-lH- indazole. (lS)-l-(3,5-dichloro-4-pyridyl)ethyl] methanesulfonate (0.57 g, 2.1 mmol, 1.0 equiv) and cesium carbonate (1.03 g, 3.2 mmol, 1.5 equiv) were added to a solution of 6- methoxy-l-tosyl-lH-indazol-5-ol (0.67 g, 2.1 mmol, 1.0 equiv) in acetonitrile (21 mL) at room temperature. After heating at 90 °C overnight, the mixture was cooled to room temperature and concentrated onto silica gel (6.0 g) under reduced pressure. The product was purified on a Biotage automated purification system (Sorbtech silica, 40 g), eluting with a gradient of 0% to 60% ethyl acetate in heptanes to give a white solid (0.71 g, 70% yield). LCMS m/z = 492.1 (M+H).
[00376] Step 4. (R)-5-(l-(3,5-Dichloropyridin-4-yl)ethoxy)-6-methoxy-lH- indazole. IM Tetrabutylammonium fluoride in THF (7.2 mL, 7.2 mmol, 18.0 equiv) was added to a solution of (R)-5-(l-(3,5-dichloropyridin-4-yl)ethoxy)-6-methoxy-l-tosyl-lH- indazole (0.20 g, 0.4 mmol, 1.0 equiv) in tetrahydrofuran (4 mL) at room temperature. After heating at 50 °C for 4 days, the solvent was removed under reduced pressure. The residue was concentrated onto silica gel (2.0 g) and purified on a Biotage automated purification system (Sorbtech silica, 12 g), eluting with a gradient of 0% to 100% ethyl acetate in heptanes to give a white solid (74.7 mg, 54%). LCMS m/z = 338.0 (M+H).
[00377] Step 5. (R)-5-(l-(3,5-Dichloropyridin-4-yl)ethoxy)-3-iodo-6-methoxy-lH- indazole. Potassium hydroxide (27.9 mg, 0.50 mmol, 2.25 equiv) and iodine (84.1 mg, 0.33 mmol, 1.5 equiv) were added to a solution of (R)-5-(l-(3,5-dichloropyridin-4-yl)ethoxy)-6- methoxy-lH-indazole (74.7 mg, 0.22 mmol, 1.0 equiv) in N,N-dimethylformamide (2.2 mL) at 0 °C. The resulting mixture was allowed to warm up to room temperature and stirred overnight. The reaction was diluted with ethyl acetate (10 mL) and washed with water (4 x 5 mL). The organic layer was dried over sodium sulfate and concentrated onto silica gel (1.5 g) under reduced pressure. The product was purified on a Biotage automated purification system (Sorbtech silica, 12 g), eluting with a gradient of 0% to 100% ethyl acetate in heptanes to give an off-white solid (80 mg, 77%). LCMS m/z = 463.9 (M+H). [00378] Step 6. 5-((R)-l-(3,5-Dichloropyridin-4-yl)ethoxy)-3-iodo-6-methoxy-l- (tetrahydro-2H-pyran-2-yl)-lH-indazole. 5-[(lR)-l-(3,5-Dichloro-4-pyridyl)ethoxy]-3-iodo- 6-methoxy-lH-indazole (0.5 g, 1.1 mmol, 1 equiv) was treated with 3,4-dihydro-2H-pyran (0.2 mL, 2.2 mmol, 2 equiv) and p-toluenesulfonic acid monohydrate (10 mg, 0.05 mmol, 0.05 equiv) in anhydrous dichloromethane (6 mL) at room temperature overnight. The mixture was diluted with dichloromethane (8 mL) and washed with water (8 mL). The organic layer was dried over sodium sulfate, filtered and concentrated under reduced pressure onto silica gel (6 g). The residue was purified on a Biotage automated chromatography system (Sorbtech, 12 g silica gel column), eluting with a gradient of 0 to 20% ethyl acetate in heptanes to give a white solid (510 mg, 86% yield), m/z = 548 (M+H).
[00379] Step 7. 5-(5-((R)-l-(3,5-Dichloropyridin-4-yl)ethoxy)-6-methoxy-l- (tetrahy dro-2H-pyran-2-yl)- 1 H-indazol -3 -yl)-2-(3 -((dimethyl(oxo)-X6- sulfaneylidene)amino)-3-methylazetidin-l-yl)nicotinonitrile. The above crude mixture of 2- [3-[[dimethyl(oxo)-X6-sulfanylidene]amino]-3-methyl-azetidin-l-yl]-5-(4,4,5,5-tetramethyl- l,3,2-dioxaborolan-2-yl)pyridine-3-carbonitrile (0.35 mmol, 1.3 equiv) in 1,4-di oxane was treated with 5-((R)-l-(3,5-dichloropyridin-4-yl)ethoxy)-3-iodo-6-methoxy-l-(tetrahydro-2H- pyran-2-yl)-lH-indazole (148 mg, 0.27 mmol, 1 equiv), [l,l'-bis(diphenylphosphino) ferrocene]dichloropalladium(II) (20 mg, 0.027 mmol, 0.1 equiv) and potassium carbonate (75 mg, 0.54 mmol, 2 equiv) and water (1.0 mL). After sparging with nitrogen for 10 minutes, the reaction was heated at 90 °C for 22 hours. The mixture was cooled to room temperature and diluted with ethyl acetate (50 mL) and water (20 mL). The layers were separated, and the aqueous layer was extracted with ethyl acetate (3 x 20 mL). The combined organic layers were washed with saturated brine (20 mL), dried over sodium sulfate, filtered, and concentrated under reduced pressure. The crude product was purified on an Interchim automated chromatography system (Sorbtech 25 g silica gel column), eluting with a gradient of 0 to 10% methanol in ethyl acetate to give a brown solid (92 mg, 50% yield for 2 steps). LCMS m/z = 684.2 (M+H).
[00380] Step 8. (R)-5-(5-(l-(3,5-Dichloropyridin-4-yl)ethoxy)-6-methoxy-lH- indazol-3-yl)-2-(3-((dimethyl(oxo)-X6-sulfaneylidene)amino)-3-methylazetidin-l- yl)nicotinonitrile. Trifluoroacetic acid (2 mL) was added to a solution of product step 7 (92 mg, 0.134 mmol, 1.0 equiv) in di chloromethane (2 mL) at room temperature. After stirring at room temperature for 4 hours, the volatiles were removed under reduced pressure. The residue was diluted with dichloromethane (20 mL) and water (10 mL), then adjusted to pH 9 saturated sodium carbonate. The layers were separated, and the aqueous layer was extracted with di chloromethane (2 x 20 mL). The combined organic layers were washed with saturated brine (20 mL), dried over sodium sulfate, filtered, and concentrated under reduced pressure. The residue was purified on a Biotage automated chromatography system (Sorbtech 25 g silica gel column), eluting with a gradient of 0 to 10% methanol in di chloromethane to give a white solid (42 mg, 52% yield). LCMS m/z = 600.1 (M+H); 1H NMR (400 MHz, DMSO-d6) 5 = 12.96 (s, 1H), 8.72 (d, J = 2.3 Hz, 1H), 8.59 (s, 2H), 8.07 (d, J = 2.2 Hz, 1H), 7.05 (s, 1H), 7.01 (s, 1H), 6.00 (q, J = 6.7 Hz, 1H), 4.30 - 4.17 (m, 4H), 3.86 (s, 3H), 3.08 (s, 6H), 1.76 (d, J = 6.7 Hz, 3H), 1.65 (s, 3H).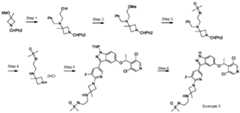

Example 5. l-[5-[5-[(lR)-l-(3,5-Dichloro-4-pyridyl)ethoxy]-lH-indazol-3-yl]-3-fluoro-2- pyridyl]-N-[2-[[dimethyl(oxo)-k6-sulfanylidene]amino]ethyl]-3-methyl-azetidin-3-amine
[00381] Step 1. 2-((l-Benzhydryl-3-methylazetidin-3-yl)(benzyl)amino)ethan-l-ol. (l-Benzhydryl-3-methyl-azetidin-3-yl) methanesulfonate (3.09 g, 9.3 mmol, 1 equiv) was treated with 2-benzylaminoethanol (2.82 g, 18.7 mmol, 2.0 equiv) and N,N- diisopropylethylamine (8.1 mL, 46.7 mmol, 5.0 equiv) in 1,4-dioxane (20 mL) at 100 °C for 20 hours. After cooling to room temperature, the reaction was diluted with water (20 mL) and extracted with ethyl acetate (2 x 20 mL). The combined organic layers were dried over sodium sulfate, filtered, and concentrated under reduced pressure onto Celite (10 g). The residue was purified on a Biotage automated chromatography system (Biotage 50 g silica gel column), eluting with a gradient of 5 to 25% ethyl acetate in hexanes to give a white solid (2.56 g, 71% yield). LCMS m/z = 387 (M+H). [00382] Step 2. 2-((l-Benzhydryl-3-methylazetidin-3-yl)(benzyl)amino)ethyl methane-sulfonate. Methanesulfonic anhydride (0.57 g, 3.26 mmol, 1.5 equiv) was added to a solution of 2-((l-Benzhydryl-3-methylazetidin-3-yl)(benzyl)amino)ethan-l-ol (0.84 g, 2.17 mmol, 1 equiv) in di chloromethane (40 mL) at 0 °C. After stirring at 0 °C for 2 hours, water (15 mL) was added to quench the reaction. The layers were separated, and the aqueous layer was extracted with dichloromethane (3 x 20 mL). The combined organic layers were washed with saturated brine (50 mL), dried over sodium sulfate, filtered, and concentrated under reduced pressure to give a crude colorless oil (1.09 g, >100% yield), which was used subsequently in the next step. LCMS m/z = 465.2 (M+H).
[00383] Step 3. ((2-((l-Benzhydryl-3-methylazeti din-3 - yl)(benzyl)amino)ethyl)imino)-dimethyl-X6-sulfanone. 2-((l-Benzhydryl-3-methylazeti din-3 - yl)(benzyl)amino)ethyl methane-sulfonate (1.09 g, 2.17 mmol, 1.0 equiv) and sodium bicarbonate (0.365 g, 4.35 mmol, 2.0 equiv) were added to a solution of (dimethanesulfmylidene)amine (0.20 g, 2.17 mmol, 1.0 equiv) in acetonitrile (50 mL) at room temperature. After refluxing 16 hours, the reaction was cooled to room temperature and filtered. The filtrate was concentrated under reduced pressure. The residue was purified on a Biotage automated chromatography system (Sorbtech 40 g silica gel column), eluting with a gradient of 0 to 10% methanol in dichloromethane to give a yellow oil (0.24 g, 24% yield). LCMS m/z = 462.2 (M+H).
[00384] Step 4. Dimethyl((2-((3-methylazeti din-3 -yl)amino)ethyl)imino)-X6- sulfanone dihydrochloride salt. 2M HC1 in diethyl ether (2.4 mL, 4.8 mmol, 5 equiv) was added to a solution of ((2-((l-benzhydryl-3-methylazetidin-3-yl)(benzyl)amino)ethyl)imino)- dimethyl-X6-sulfanone (0.44 g, 0.953 mmol, 1 equiv) in a mixture of acetonitrile (20 mL) and diethyl ether (10 mL) at room temperature. After stirring for 1 hour, the mixture was concentrated under reduced pressure to give dihydrochloride salt (0.6 g) as a light-yellow solid. A mixture of dihydrochloride salt (0.6 g, 0.953 mmol) and 20% palladium hydroxide on carbon (50% wet, 0.12 g) in methanol was hydrogenated (50 psi at room temperature) for 22 hours. LCMS analysis indicated a mixture of desired product (m/z = 205) and partially debenzylated product (m/z = 371) were formed. Additional 20% palladium hydroxide on carbon (50% wet, 0.12 g) was added, and the hydrogenation continued for 3 days. The mixture was filtered through Celite, which was washed with methanol (3 x 50 mL). The filtrate was concentrated under reduced pressure and the residue was triturated with heptanes (3 x 10 mL) to give a yellow solid (0.33 g, >100% yield) that contained diphenylmethane, and was used in the next step without further purification. LCMS m/z = 206.1 (M+H).
[00385] Step 5. ((2-((l-(5-(5-((R)-l-(3,5-Dichloropyridin-4-yl)ethoxy)-l- (tetrahydro-2H-pyran-2-yl)-lH-indazol-3-yl)-3-fluoropyridin-2-yl)-3-methylazetidin-3- yl)amino)ethyl)imino)dimethyl-X6-sulfanone. Dimethyl((2-((3-methylazeti din-3 - yl)amino)ethyl)imino)-X6-sulfanone dihydrochloride salt (0.33 g, -0.953 mmol, 2 equiv) and potassium carbonate (0.329 g, 2.383 mmol, 5 equiv) were added to a solution of 5-[(lR)-l- (3,5-dichloro-4-pyridyl)ethoxy]-3-(5,6-difluoro-3-pyridyl)-l-tetrahydropyran-2-yl-indazole (0.241 g, 0.477 mmol, 1 equiv) in acetonitrile (20 mL). After stirring at 70 °C for 48 hours, the reaction was cooled to room temperature and diluted with ethyl acetate (40 mL) and water (20 mL). The layers were separated, and the aqueous layer was extracted with ethyl acetate (2 x 20 mL). The combined organic layers were washed with saturated brine (40 mL), dried over sodium sulfate, filtered, and concentrated under reduced pressure. The residue was purified on an Interchim automated chromatography system (Sorbtech 25 g silica gel column), eluting with a gradient of 0 to 10% methanol in di chloromethane, to give a paleyellow oil (18 mg, 5.5% yield). LCMS m/z = 690.2 (M+H).
[00386] Step 6. (R)-((2-((l-(5-(5-(l-(3,5-Dichloropyridin-4-yl)ethoxy)-lH-indazol- 3-yl)-3-fluoropyridin-2-yl)-3-methylazetidin-3-yl)amino)ethyl)imino)dimethyl-X6-sulfanone. Trifluoroacetic acid (0.5 mL) was added to a solution of product step 5 (18 mg, 0.0261 mmol, 1.0 equiv) in dichloromethane (2.0 mL) at room temperature. After stirring at room temperature for 4 hours, the volatiles were removed under reduced pressure. The residue was diluted with dichloromethane (20 mL) and water (10 mL) then adjusted to pH 9 with saturated sodium carbonate. The aqueous layer was extracted with dichloromethane (3 x 20 mL). The combined organic layers were washed with saturated brine (20 mL), dried over sodium sulfate, filtered, and concentrated under reduced pressure. The residue was dry loaded onto Celite (8 g) and purified on an InterChim automated chromatography system (RediSep Rf GOLD 50 g HP Cl 8 column), eluting with a gradient of 0 to 100% acetonitrile in water to give a white solid after lyophilization (7.0 mg, 44% yield). LCMS m/z = 606.2 (M+H); 1H NMR (400 MHz, DMSO-d6) 5 = 13.11 (br s, 1H), 8.57 (s, 2H), 8.36 (t, J = 1.5 Hz, 1H), 7.69 (dd, J = 1.8, 13.2 Hz, 1H), 7.47 (d, J = 9.0 Hz, 1H), 7.43 - 7.23 (m, 1H), 7.19 (d, J = 2.1 Hz, 1H), 7.10 (dd, J = 2.3, 9.0 Hz, 1H), 6.14 (d, J = 6.7 Hz, 1H), 3.98 (br d, J = 8.2 Hz, 2H), 3.88 (br d, J = 8.1 Hz, 2H), 3.53 - 3.41 (m, 3H), 3.11 - 2.81 (m, 5H), 2.70 - 2.56 (m, 2H), 1.76 (d, J = 6.7 Hz, 3H), 1.43 (s, 3H).

Example 6. [!-[5-[5-[(lR)-l-(3,5-dichloro-4-pyridyl)ethoxy]-lH-indazol-3-yl]-3-fluoro-2- pyridyl]-3-methyl-azetidin-3-yl]methylimino-dimethyl-oxo-k6-sulfane
[00387] Stepl . (l-(5-(5-((R)-l-(3,5-Dichloropyridin-4-yl)ethoxy)-l-(tetrahydro-2H- pyran-2-yl)-lH-indazol-3-yl)-3-fluoropyridin-2-yl)-3-methylazetidin-3-yl)methanol. 5-[(lR)- l-(3,5-Dichloro-4-pyridyl)ethoxy]-3-(5,6-difluoro-3-pyridyl)-l-tetrahydropyran-2-yl- indazole (206 mg, 1.5 mmol, 3 equiv) and potassium carbonate (622 mg, 4.5 mmol, 9 equiv) were added to a solution of (3 -methylazeti din-3 -yl)m ethanol hydrochloride (253 mg, 0.5 mmol, 1 equiv) in acetonitrile (10 mL). After stirring at 80 °C for 16 hours, the reaction was cooled to room temperature and diluted with ethyl acetate (20 mL) and water (10 mL). The layers were separated, and the aqueous layer was extracted with ethyl acetate (2 x 20 mL). The combined organic layers were washed with saturated brine (20 mL), dried over sodium sulfate, filtered and concentrated under reduced pressure. The residue was purified on a Biotage automated chromatography system (Sorbtech 25 g silica gel column), eluting with a gradient of 0 to 100% ethyl acetate in hexanes to give a white solid (270 mg, 92% yield). LCMS m/z = 586.2 (M+H).
[00388] Step 2. (l-(5-(5-((R)-l-(3,5-Dichloropyridin-4-yl)ethoxy)-l-(tetrahydro- 2H-pyran-2-yl)-lH-indazol-3-yl)-3-fluoropyridin-2-yl)-3-methylazetidin-3-yl)methyl methanesulfonate. Methanesulfonic anhydride (240 mg, 1.38 mmol, 3 equiv) was added to a solution of product step 1 (270 mg, 0.46 mmol, 1 equiv) and N,N-diisopropylethylamine (178 mg, 1.38 mmol, 3 equiv) in dichloromethane (20 mL) at 0 °C. After stirring at room temperature for 16 hours, the reaction was quenched with water (20 mL) and extracted with di chloromethane (2 x 40 mL). The combined organic layers were dried over sodium sulfate, filtered, and concentrated under reduced pressure. The residue was purified on an Biotage automated chromatography system (Sorbtech 12 g silica gel column), eluting with a gradient of 0 to 100% ethyl acetate in hexanes to give an off-white solid (218 mg, 71% yield). LCMS m/z = 664.1 (M+H).
[00389] Step 3. ((l-(5-(5-((R)-l-(3,5-Dichloropyridin-4-yl)ethoxy)-l-(tetrahydro- 2H-pyran-2-yl)-lH-indazol-3-yl)-3-fluoropyridin-2-yl)-3-methylazetidin-3- yl)methyl)imino)dimethyl-X6-sulfanone. Dimethyl sulfoximine (153 mg, 1.64 mmol, 5 equiv) was added to a solution of (l-(5-(5-((R)-l-(3,5-dichloropyridin-4-yl)ethoxy)-l-(tetrahydro- 2H-pyran-2-yl)-lH-indazol-3-yl)-3-fluoropyridin-2-yl)-3-methylazetidin-3-yl)methyl methanesulfonate (218 mg, 0.33 mmol, 1 equiv) in acetonitrile (5 mL) in a pressure tube, followed by addition of sodium tert-butoxide (95 mg, 0.98 mmol, 3 equiv). N-Methyl-2- pyrrolidone (1 mL) was added and the mixture was heated at 80 °C for 3 days. After cooling to room temperature, the mixture was diluted with ethyl acetate (20 mL) and water (8 mL). The layers were separated, and the organic layer was washed with saturated brine (2 x 8 mL), dried over sodium sulfate, filtered and concentrated under reduced pressure. The residue was purified on an Biotage automated chromatography system (Sorbtech 12 g silica gel column), eluting with a gradient of 0 to 100% ethyl acetate in hexanes followed by 10% methanol in ethyl acetate to give an off-white solid (73 mg, 34% yield). LCMS m/z = 661.1 (M+H).
[00390] Step 4. (R)-(((l-(5-(5-(l-(3,5-Dichloropyridin-4-yl)ethoxy)-lH-indazol-3- yl)-3-fluoropyridin-2-yl)-3-methylazeti din-3 -yl)methyl)imino)dimethyl-X6-sulfanone. Trifluoroacetic acid (2 mL) was added to a solution of compound 691-4 (73 mg, 0.11 mmol, 1.0 equiv) in di chloromethane (2 mL) at room temperature. After stirring at room temperature for 4 hours, the volatiles were removed under reduced pressure. The residue was diluted with ethyl acetate (20 mL) and water 10 mL), then adjusted to pH 9 with saturated sodium carbonate. The layers were separated and the aqueous layer was extracted with ethyl acetate (3 x 10 mL). The combined organic layers were washed with saturated brine (20 mL), dried over sodium sulfate, filtered, and concentrated under reduced pressure. The residue was dissolved in dimethyl sulfoxide (10 mL) and purified on a Biotage automated chromatography system (RediSep Rf GOLD 100 g HP C18 column), eluting with a gradient of 0 to 100% acetonitrile in water to give an off-white solid after lyophilization (36 mg, 56% yield). LCMS m/z = 577.1 (M+H); 1H NMR (400 MHz, DMSO-d6) 5 = 13.10 (br s, 1H), 8.57 (s, 2H), 8.34 (t, J = 1.5 Hz, 1H), 7.66 (dd, J = 1.7, 13.3 Hz, 1H), 7.47 (d, J = 9.0 Hz, 1H), 7.20 (d, J = 2.0 Hz, 1H), 7.09 (dd, J = 2.3, 9.0 Hz, 1H), 6.13 (q, J = 6.6 Hz, 1H), 3.98 (dd, J = 1.6, 7.9 Hz, 2H), 3.74 (dd, J = 1.6, 7.9 Hz, 2H), 3.09 (s, 2H), 3.01 (s, 6H), 1.76 (d, J = 6.6 Hz, 3H), 1.31 (s, 3H).

Example 7. (R)-3-Amino-l-(5-(5-(l-(3,5-dichloropyridin-4-yl)ethoxy)-lH-indazol-3-yl)- 3-fluoropyridin-2-yl)-N-(dimethyl(oxo)-k6-sulfaneylidene)azetidine-3-carboxamide.
[00391] Step 1. Methyl 3-((tert-butoxycarbonyl)amino)-l-(5-(5-((R)-l-(3,5- dichloropyridin-4-yl)ethoxy)-l-(tetrahydro-2H-pyran-2-yl)-lH-indazol-3-yl)-3-fluoropyridin-
2 -yl)azetidine-3 -carboxylate. A mixture of 5-[(lR)-l-(3,5-dichloro-4-pyridyl)ethoxy]-3-(5,6- difluoro-3 -pyridyl)- l-tetrahydropyran-2-yl-indazole (3.16 g, 6.25 mmol, 1.0 equiv), methyl
3-(tert-butoxycarbonylamino)azetidine-3-carboxylate (2.0 g, 7.5 mmol, 1.2 equiv), and N,N- diisopropylethylamine (3.27 mL, 18.75 mmol, 3.0 equiv) in acetonitrile (60 mL) was heated at 80 °C for 30 hours. The reaction mixture was cooled to room temperature, diluted with water (250 mL) and extracted with ethyl acetate (3 x 250 mL). The combined organic layers were dried over sodium sulfate, filtered, and concentrated under reduced pressure. The residue was purified on a Biotage automated chromatography system (Biotage Sfar 50 g silica-gel column) eluting with a gradient of 0 to 55% ethyl acetate in hexanes to give an off- white foam (3.7 g, 82%).LCMS m/z = 715.1 (M+H).
[00392] Step 2. 3-((tert-Butoxycarbonyl)amino)-l-(5-(5-((R)-l-(3,5- dichloropyridin-4-yl)ethoxy)-l-(tetrahydro-2H-pyran-2-yl)-lH-indazol-3-yl)-3-fluoropyridin- 2 -yl)azetidine-3 -carboxylic acid. A solution of lithium hydroxide monohydrate (650 mg, 15.5 mmol, 3.0 equiv) in water (11 mL) was added dropwise over 5 minutes to a solution of methyl 3-((tert-butoxycarbonyl)amino)-l-(5-(5-((R)-l-(3,5-dichloropyridin-4-yl)ethoxy)-l- (tetrahydro-2H-pyran-2-yl)-lH-indazol-3-yl)-3-fluoropyridin-2-yl)azetidine-3-carboxylate (3.7 g) in methanol (33 mL) and THF (22 mL) at room temperature. The resulting mixture was stirred at room temperature for 16 hours. The reaction mixture was adjusted to pH 2 with 2M HC1 (~13 mL), and then was extracted with ethyl acetate (500 mL). The organic extract was washed with water (200 mL) and saturated brine (200 mL), dried over sodium sulfate, filtered, and concentrated under reduced pressure to give a white solid (1.58 g, 71%), which was used directly in the next step. LCMS m/z = 701.2 (M+H).
[00393] Step 3. tert-Butyl (l-(5-(5-((R)-l-(3,5-dichloropyridin-4-yl)ethoxy)-l- (tetrahydro-2H-pyran-2-yl)-lH-indazol-3-yl)-3-fluoropyridin-2-yl)-3-((dimethyl(oxo)-X6- sulfaneylidene)carbamoyl)azetidin-3-yl)carbamate. N,N-diisopropylethylamine (0.65 mL, 3.71 mmol, 13 equiv) and l-[bis(dimethylamino)methylene]-lH-l,2,3-triazolo[4,5- b]pyridinium 3-oxid hexafluorophosphate (163 mg, 0.43 mmol, 1.5 equiv) were sequentially added to a solution of 3-((tert-butoxycarbonyl)amino)-l-(5-(5-((R)-l-(3,5-dichloropyridin-4- yl)ethoxy)-l-(tetrahydro-2H-pyran-2-yl)-lH-indazol-3-yl)-3-fluoropyridin-2-yl)azetidine-3- carboxylic acid (200 mg, 0.29 mmol, 1.0 equiv) at 5 °C in N,N-dimethylacetamide (3 mL). After stirring for 20 minutes, iminodimethyl -X6-sulfanone (53 mg, 0.57 mmol, 2.0 equiv) was added. The mixture was stirred at room temperature for 16 hours, then quenched with water (120 mL). The resulting precipitate was stirred at room temperature for 1 hour, filtered, washed with water (2 x 10 mL) and dried at 40 to 50 °C under vacuum for 20 hours. The crude residue was purified on a Biotage automated chromatography system (12 g Sorbtech, 60 pm silica gel column), eluting with a gradient of 0 to 100% ethyl acetate in hexanes to give an off-white solid (176 mg, 79%). LCMS m/z = 776 (M+H).
[00394] Step 4. (R)-3-Amino-l-(5-(5-(l-(3,5-dichloropyridin-4-yl)ethoxy)-lH- indazol-3-yl)-3-fluoropyridin-2-yl)-N-(dimethyl(oxo)-X6-sulfaneylidene)azetidine-3- carboxamide. A mixture of tert-butyl (l-(5-(5-((R)-l-(3,5-dichloropyridin-4-yl)ethoxy)-l- (tetrahydro-2H-pyran-2-yl)-lH-indazol-3-yl)-3-fluoropyridin-2-yl)-3-((dimethyl(oxo)-X6- sulfaneylidene)carbamoyl)azetidin-3-yl)carbamate (160 mg, 0.21 mmol, 1 equiv) and trifluoroacetic acid (3 mL, 38.94 mmol, 189 equiv) in dichloromethane (3 mL) was stirred at room temperature for 4 hours. The reaction mixture was concentrated under reduced pressure and diluted with saturated sodium carbonate (50 mL) and ethyl acetate (20 mL). The layers were separated, and the aqueous layer was extracted with ethyl acetate (2 xlO mL). The combined organic layers were washed with saturated brine (25 mL), dried over magnesium sulfate, filtered, and concentrated under reduced pressure. The crude residue was purified on a Biotage automated chromatography system (100 g RediSep Gold, C18 column), eluting with a gradient of 0 to 100% acetonitrile in water to give a white solid after lyophilization (83 mg, 25%). LCMS m/z = 592.1 (M+H); 1H NMR (400 MHz, DMSO-d6) 5 = 13.10 (br s, 1H), 8.57 (s, 2H), 8.37 (s, 1H), 7.70 (dd, J = 1.7, 13.3 Hz, 1H), 7.47 (d, J = 8.9 Hz, 1H), 7.20 (s, 1H), 7.10 (dd, J = 2.2, 9.0 Hz, 1H), 6.14 (q, J = 6.7 Hz, 1H), 4.45 (br d, J = 7.1 Hz, 2H), 3.94 (d, J = 7.6 Hz, 2H), 3.39 (s, 6H), 1.76 (d, J = 6.6 Hz, 3H); 19F NMR (376 MHz, DMSO-d6) 5 = -139.77 (br d, J = 13.6 Hz, IF).
[00395] Example 8. (R)-(((3-Amino-l-(5-(5-(l-(3,5-dichloropyridin-4- yl)ethoxy)-lH-indazol-3-yl)-3-fluoropyridin-2-yl)azetidin-3-yl)methyl)imino)dimethyl- k6-sulfanone. To a solution of (R)-3-amino-l-(5-(5-(l-(3,5-dichloropyridin-4-yl)ethoxy)-lH- indazol-3-yl)-3-fluoropyridin-2-yl)-N-(dimethyl(oxo)-X6-sulfaneylidene)azetidine-3- carboxamide (33 mg, 0.05 mmol, 1.0 equiv) in THF (3 mL) at 5 °C was added IM borane THF solution in THF (0.67 mL, 0.67 mmol, 12 equiv). After stirring for 10 minutes, the reaction mixture was warmed to room temperature and stirred for 4 hours. The reaction mixture was quenched with a mixture of water (6 mL), methanol (3 mL) and trifluoroacetic acid, adjusting the pH to 2 at 5 °C. The mixture was stirred at room temperature for 5 hours then neutralized with saturated sodium carbonate solution adjusting to pH 10. The mixture was extracted with ethyl acetate (3 x 10 mL). The combined organic layers were washed with saturated brine (10 mL), dried over magnesium sulfate, filtered, and concentrated under reduced pressure. The crude residue was purified on a Biotage automated chromatography system (100 g RediSep Gold, Cl 8 column), eluting with a gradient of 0 to 100% acetonitrile in water to give a white solid after lyophilization (17 mg, 46%). LCMS m/z = 578.2 (M+H); 1H NMR (400 MHz, DMSO-d6) 5 = 13.09 (br s, 1H), 8.57 (s, 2H), 8.36 (t, J = 1.6 Hz, 1H), 7.67 (dd, J = 1.8, 13.3 Hz, 1H), 7.47 (d, J = 9.0 Hz, 1H), 7.20 (d, J = 2.2 Hz, 1H), 7.10 (dd, J = 2.3, 9.0 Hz, 1H), 6.14 (q, J = 6.7 Hz, 1H), 4.25 - 3.98 (m, 2H), 3.81 (d, J = 7.5 Hz, 2H), 3.85 - 3.62 (m, 1H), 3.17 (s, 2H), 3.04 (s, 6H), 1.76 (d, J = 6.6 Hz, 3H); 19F NMR (376 MHz, DMSO-d6) 5 = -139.22 (br d, J = 13.6 Hz, IF).

Example 9
Example 9. [l-[5-[5-[(lR)-l-(3,5-dichloro-4-pyridyl)ethoxy]-lH-indazol-3-yl]-3-fluoro-2- pyridyl]-3-(pyrrolidin-l-ylmethyl)azetidin-3-yl]imino-dimethyl-oxo-k6-sulfane
[00396] Step 1. ((l-Benzhydryl-3-(pyrrolidin-l-ylmethyl)azetidin-3- yl)imino)dimethyl-X6-sulfanone (919-1): Pyrrolidine (421 mg, 5.9 mmol, 5 equiv) and N,N- diisopropylethylamine (1.53 g, 2.1 mL, 11.8 mmol, 10 equiv) were added to a solution of [1- benzhydryl-3-[[dimethyl(oxo)-X6-sulfanylidene]amino]azetidin-3-yl]methyl methanesulfonate (500 mg, 1.2 mmol, 1.0 equiv) in acetonitrile (30 mL) at room temperature. The mixture was heated at 80 °C for 3.5 hours then cooled to room temperature and concentrated under reduced pressure. The residue was purified on a Biotage automated chromatography system (Biotage Sfar KP-Amino D Duo 50 pm 28 g column x 2, stacked), eluting with a gradient of 0 to 100% ethyl acetate in hexanes to give an off-white solid (400 mg, 85%). LCMS m/z = 423.1 (M+H).
[00397] Step 2. Dimethyl((3-(pyrrolidin-l-ylmethyl)azetidin-3-yl)imino)-X6- sulfanone trihydrochloride. A mixture of ((l-benzhydryl-3-(pyrrolidin-l-ylmethyl)azeti din-3 - yl)imino)dimethyl-Z6-sulfanone (400 mg, 1.01 mmol), 20% palladium hydroxide on activated carbon (50% wetted powder) (212 mg, 0.15 mmol, 0.15 equiv) and concentrated HC1 (0.38 mL, 4.53 mmol, 4.5 equiv) in methanol (40 mL) was hydrogenated at 50 psi at room temperature for 70 hours in a Parr shaker. The reaction mixture was filtered through a pad of Celite, which was washed with 10% water in methanol (40 mL). The filtrate was concentrated under reduced pressure. The residue was azeotroped with toluene (2 x 50 mL) and dried under vacuum at 40 °C for 3 hours to give a brown oil (342 mg, 99%). LCMS m/z = 232.2 (M+H) free base.
[00398] Step 3. ((l-(5-(5-((R)-l-(3,5-Dichloropyridin-4-yl)ethoxy)-l-(tetrahydro- 2H-pyran-2-yl)-lH-indazol-3-yl)-3-fluoropyridin-2-yl)-3-(pyrrolidin-l-ylmethyl)azeti din-3- yl)imino)dimethyl-X6-sulfanone. A mixture of 5-[(lR)-l-(3,5-dichloro-4-pyridyl)ethoxy]-3- (5,6-difluoro-3-pyridyl)-l-tetrahydropyran-2-yl-indazole (230 mg, 0.46 mmol, 1.0 equiv), dimethyl((3-(pyrrolidin-l-ylmethyl)azeti din-3 -yl)imino)-X6-sulfanone trihydrochloride (171 mg, 0.50 mmol, 1.1 equiv) and N,N-diisopropylethylamine (1.77 g, 2.4 mL, 13.7 mmol, 30 equiv) in acetonitrile (20 mL) was heated at 80 °C for 16 hours. The reaction was cooled to room temperature and concentrated under reduced pressure. The residue was purified on a Biotage automated chromatography system (Biotage® Sfar KP -Amino D Duo 50 pm 28 g column), eluting with a gradient of 0 to 10% methanol in ethyl acetate to give an off-white solid (290 mg, 89%). LCMS m/z = 716.2 (M+H).
[00399] Step 4. (R)-((l-(5-(5-(l-(3,5-Dichloropyridin-4-yl)ethoxy)-lH-indazol-3- yl)-3-fluoropyridin-2-yl)-3-(pyrrolidin-l-ylmethyl)azeti din-3 -yl)imino)dimethyl-X6- sulfanone dihydrochloride. ((l-(5-(5-((R)-l-(3,5-Dichloropyridin-4-yl)ethoxy)-l-(tetrahydro- 2H-pyran-2-yl)-lH-indazol-3-yl)-3-fluoropyridin-2-yl)-3-(pyrrolidin-l-ylmethyl)azeti din-3- yl)imino)dimethyl-X6-sulfanone (145 mg, 0.20 mmol, 1.0 equiv) in ethanol (3 mL) was treated with 6M HC1 (0.27 mL, 1.62 mmol, 8 equiv) at 65 °C. After 4 hours, the mixture was cooled to room temperature. The solids were filtered, washed with methyl tert-butyl ether (70 mL) and dried at 40 °C under vacuum for 16 hours to give a yellow solid as the dihydrochloride(120 mg, 84%). LCMS m/z= 632.1 (M+H) free base; 1H NMR (400 MHz, DMSO-d6) 5 = 9.71 (br s, 1H), 8.57 (s, 2H), 8.39 (s, 1H), 7.78 (dd, J = 1.7, 13.0 Hz, 1H), 7.49 (d, J = 8.9 Hz, 1H), 7.20 (d, J = 2.1 Hz, 1H), 7.11 (dd, J = 2.2, 9.0 Hz, 1H), 6.14 (q, J = 6.6 Hz, 1H), 4.40 - 4.29 (m, 4H), 3.62 (br d, J = 5.4 Hz, 4H), 3.30 (s, 6H), 3.13 (br s, 2H), 1.99 (br s, 4H), 1.76 (d, J = 6.6 Hz, 3H).

Example 10 Example 10. 5-[5-[(lR)-l-(3,5-dichloro-4-pyridyl)ethoxy]-6-methoxy-lH-indazol-3-yl]-2- [3-[[dimethyl(oxo)-k6-sulfanylidene]amino]-3-(pyrrolidin-l-ylmethyl)azetidin-l- yl] pyridine-3-carbonitrile
[00400] Step 1. 5-(5-((R)-l-(3,5-Dichloropyridin-4-yl)ethoxy)-6-methoxy-l- (tetrahy dro-2H-pyran-2-yl)- 1 H-indazol -3 -yl)-2-(3 -((dimethyl(oxo)-X6- sulfaneylidene)amino)-3-(pyrrolidin-l-ylmethyl)azetidin-l-yl)nicotinonitrile. A mixture of 5- [5-[(lR)-l-(3,5-dichloro-4-pyridyl)ethoxy]-6-methoxy-l-tetrahydropyran-2-yl-indazol-3-yl]- 2-fluoro-pyridine-3-carbonitrile (248 mg, 0.46 mmol, 1.0 equiv), dimethyl-oxo-[3- (pyrrolidin-l-ylmethyl)azeti din-3 -yl]imino-X6-sulfane (171 mg, 0.50 mmol, 1.1 equiv) and
N,N-diisopropylethylamine (2.4 mL, 13.7 mmol, 30 equiv) in acetonitrile (20 mL) was stirred at room temperature for 16 hours. The reaction was concentrated under reduced pressure. The residue was purified on a Biotage automated chromatography system (Biotage® Sfar KP- Amino D Duo 50 pm 28 g column), eluting with a gradient of 0 to 10% methanol in ethyl acetate to give an off-white solid (346 mg, 99%). LCMS m/z = 753.2 (M+H).
[00401] Step 2. 5-[5-[(lR)-l-(3,5-dichloro-4-pyridyl)ethoxy]-6-methoxy-lH- indazol-3-yl]-2-[3-[[dimethyl(oxo)-X6-sulfanylidene]amino]-3-(pyrrolidin-l- ylmethyl)azetidin-l-yl]pyridine-3 -carbonitrile. 5-(5-((R)-l-(3,5-Dichloropyridin-4- yl)ethoxy)-6-m ethoxy- 1 -(tetrahydro-2H-pyran-2-yl)- 1 H-indazol -3 -yl)-2-(3 -((dimethyl(oxo)- X6-sulfaneylidene)amino)-3-(pyrrolidin-l-ylmethyl)azetidin-l-yl)nicotinonitrile (173 mg,
O.23 mmol, 1.0 equiv) in ethanol (3.5 mL) was treated with 6M HC1 (0.31 mL, 1.8 mmol, 8 equiv) at 40 °C. After 16 hours, the mixture was cooled to room temperature. The mixture was diluted with ethyl acetate (20 mL) and saturated sodium carbonate solution (8 mL). The aqueous phase was extracted with ethyl acetate (10 mL). The combined organic layers were dried over sodium sulfate, filtered, and concentrated under reduced pressure. The residue was dissolved in dimethyl sulfoxide (6 mL) and purified on a Biotage automated chromatography system (RediSep Rf GOLD 100 g HP Cl 8 column), eluting with a gradient of 0 to 100% acetonitrile in water. Product containing fractions were lyophilized to give an off-white solid (79 mg, 51%). LCMS: m/z = 669.2 (M+H); 1H NMR (400 MHz, DMSO-d6) 5 = 12.96 (br s, 1H), 8.70 (d, J = 2.2 Hz, 1H), 8.58 (s, 2H), 8.06 (d, J = 2.2 Hz, 1H), 7.04 (s, 1H), 7.00 (s, 1H), 5.99 (q, J = 6.7 Hz, 1H), 4.35 - 4.24 (m, 4H), 3.86 (s, 3H), 3.10 (s, 6H), 2.84 (s, 2H), 2.63 (br s, 4H), 1.76 (d, J = 6.6 Hz, 3H), 1.66 (br s, 4H).

Example 11. 2-[3-amino-3-[[[dimethyl(oxo)-k6-sulfanylidene]amino]-methyl]azetidin-l- yl]-5-[5-[(lR)-l-(3,5-dichloro-4-pyridyl)ethoxy]-6-methoxy-lH-indazol-3-yl]pyridine-3- carbonitrile
[00402] Step 1. l-Benzhydryl-3-(benzylamino)azetidine-3 -carbonitrile. 1- Benzhydrylazetidin-3-one (5.0 g, 21 mmol, 1 equiv) in methanol (55 mL) was sequentially treated with benzylamine (2.5 mL, 23.2 mmol, 1.1 equiv) and acetic acid (1.35 mL, 23.2 mmol, 1.1 equiv). After stirring for 1 hour, sodium cyanide (1.14 g, 23.2 mmol, 1.1 equiv) was added to the reaction. After heating at 60 °C for 24 hours, the reaction was cooled to room temperature and the solids were filtered, washed with cold methanol (15 mL) and dried under vacuum at 40 °C overnight to give an off-white solid (6.66 g, 89%). LCMS m/z = 354 (M+H).
[00403] Step 2. l-Benzhydryl-3-(benzylamino)azetidine-3 -carboxylic acid. 3M Sodium hydroxide (15 mL, 45 mmol, 2.9 equiv) was added to a suspension of 1-benzhydryl- 3-(benzylamino)azetidine-3-carbonitrile (5.40 g, 15.3 mmol, 1 equiv) in a mixture of ethanol (60 mL) and THF (30 mL). The mixture was heated at 60 °C for 3 days, cooled to room temperature then added to a stirred solution of acetic acid (10 mL, 175 mmol, 11 equiv) in water (500 mL). The resulting solids were filtered, washed with water (100 mL) and dried under vacuum at 40 °C overnight to give a white solid (4.85 g, 85%). LCMS m/z = 373 (M+H).
[00404] Step 3. l-Benzhydryl-3-(benzylamino)-N-(dimethyl(oxo) -X6- sulfaneylidene)-azetidine-3 -carboxamide. A suspension l-Benzhydryl-3- (benzylamino)azetidine-3-carboxylic acid (2.5 g, 6.7 mmol, 1 equiv), dimethyl sulfoximine (1.25 g, 13.4 mmol, 2 equiv) in a mixture of THF (36 mL) and N,N-diisopropylethylamine (5.0 mL, 28.7 mmol, 4.3 equiv) in N,N-dimethylacetamide (12 mL) was treated with HATU (3.83 g, 10.1 mmol, 1.5 equiv) at room temperature. The reaction slowly produced a yellow solution as the reaction progressed. After stirring overnight, the mixture was diluted with ethyl acetate (100 mL) and washed with water (2 x 50 mL). The organic layer was dried over sodium sulfate, filtered, and concentrated under reduced pressure onto Celite. The residue was purified on a Biotage automated chromatography system (Sorbtech 120 g silica gel column), eluting with a gradient of 0 to 100% ethyl acetate in hexanes. Fractions with product were concentrated under reduced pressure, combined with water (25 mL) and ethanol (15 mL) and heated at 50 °C for 4 hours. After cooling to room temperature, the resulting solid was filtered, washed with water (50 mL) and dried at 40 °C overnight under vacuum to give a beige solid (1.78 g, 59%). LCMS m/z = 448 (M+H).
[00405] Step 4. (((l-Benzhydryl-3-(benzylamino)azetidin-3- yl)methyl)imino)dimethyl-X6-sulfanone. IM Lithium aluminum hydride in THF (4 mL, 4 mmol, 1.1 equiv) was added to a suspension of l-Benzhydryl-3-(benzylamino)-N- (dimethyl(oxo) -X6-sulfaneylidene)-azetidine-3-carboxamide (1.62 g, 3.6 mmol, 1 equiv) in THF (20 mL) at 0 °C. After stirring for 2 hours at 0 °C, the reaction was quenched by the dropwise addition of saturated sodium potassium tartrate (10 mL). The mixture was stirred at room temperature for 1 hour, then extract with ethyl acetate (2 x 25 mL). The combined organic layers were washed with saturated sodium potassium tartrate (15 mL) then concentrated under reduced pressure onto Celite. The residue was purified on a Biotage automated chromatography system (Yamazen 55 g column), eluting with a gradient of 0 to 3% methanol in dichloromethane to give a crude yellow oil (0.30 g, 19%) that was used directly in the next step (LCMS m/z = 434 (M+H)). The material also contained amide reduction to alcohol product (0.85 g, 65%, m/z = 359).
[00406] Step 5. (((3-Aminoazeti din-3 -yl (methyl )i mi no)di methyl -+6-sulfanone bis(2,2,2-trifluoroacetate). A solution of product mixture from step 4 (0.300 g, 0.69 mmol, 1 equiv), in a mixture of methanol (20 mL) and trifluoroacetic acid (ImL, 13 mmol, 19 equiv) was added to 20% palladium hydroxide on carbon (50% wet) (0.150 g, 0.11 mmol, 0.15 equiv) in a 500 mL Parr flask. The mixture was hydrogenated at 50 psi for 20 hours, then filtered through Celite, and was rinsed with methanol. The filtrate was concentrated under reduced pressure to give a crude a yellow oil (0.40 g, 100%), used directly in the next step. LCMS m/z = 178 (M+H). [00407] Step 6. 2-(3-Amino-3-(((dimethyl(oxo)-X6- sulfaneylidene)amino)methyl)azetidin-l-yl)-5-(5-((R)-l-(3,5-dichloropyridin-4-yl)ethoxy)-6- methoxy- 1 -(tetrahydro-2H-pyran-2-yl)- lH-indazol-3 -yl)nicotinonitrile. Crude (((3 - aminoazetidin-3-yl)methyl)imino)dimethyl-X6-sulfanone bis(2,2,2-trifluoroacetate) from step 5 (400 mg, <0.69 mmol, 1.5 equiv) in acetonitrile (6 mL) was treated with N,N- diisopropylethylamine (2.0 mL, 11.5 mmol, 25 equiv) and 5-[5-[(lR)-l-(3,5-dichloro-4- pyridyl)ethoxy]-6-methoxy-l-tetrahydropyran-2-yl-indazol-3-yl]-2-fluoro-pyridine-3- carbonitrile (250 mg, 0.46 mmol, 1 equiv) at room temperature for 3 days. The reaction was diluted with water (25 mL) and extracted with dichloromethane (3 x 25 mL). The combined organic layers were washed with saturated brine (10 mL), dried over sodium sulfate, then concentrated under reduced pressure onto Celite (8 g). The residue was purified on a Biotage automated chromatography system (Biotage KP-amino 28 g column), eluting with a gradient of 0 to 80% acetone in hexanes to give an amber oil (78 mg, 24%). LCMS m/z = 699 (M+H).
[00408] Step 7. (R)-2-(3-Amino-3-(((dimethyl(oxo)-X6- sulfaneylidene)amino)methyl)azetidin-l-yl)-5-(5-(l-(3,5-dichloropyridin-4-yl)ethoxy)-6- methoxy-lH-indazol-3-yl)nicotinonitrile. A solution of 2-(3-amino-3-(((dimethyl(oxo)-X6- sulfaneylidene)amino)methyl)azetidin-l-yl)-5-(5-((R)-l-(3,5-dichloropyridin-4-yl)ethoxy)-6- methoxy-l-(tetrahydro-2H-pyran-2-yl)-lH-indazol-3-yl)nicotinonitrile 1 (75 mg, 0.11 mmol) in dichloromethane (2 mL) was treated with trifluoroacetic acid (1 mL) at room temperature. After 16 hours, the mixture was concentrated under reduced pressure. The residue was treated with saturated sodium carbonate (5 mL) and extracted with dichloromethane (3 x 5 mL). The combined organic layers were concentrated under reduced pressure. The residue was dissolved in dimethyl sulfoxide (4 mL) and purified on a Biotage automated chromatography system (RediSep Rf GOLD 100 g HP Cl 8 column) eluting with a gradient of 0 to 100% acetonitrile in water. The product containing fractions were lyophilized to give an off-white solid (31 mg, 47%). LCMS m/z = 615 (M+H); 1H NMR (400 MHz, DMSO-d6) 5 = 12.95 (br s, 1H), 8.70 (d, J = 2.0 Hz, 1H), 8.59 (s, 2H), 8.04 (d, J = 2.1 Hz, 1H), 7.04 (s, 1H), 7.00 (s, 1H), 5.99 (q, J = 6.6 Hz, 1H), 4.19 (d, J = 8.6 Hz, 2H), 3.94 (d, J = 8.6 Hz, 2H), 3.86 (s, 3H), 3.13 (s, 2H), 3.04 (s, 6H), 2.19 (br s, 2H), 1.76 (d, J = 6.7 Hz, 3H).

Example 12. 5-[5-[(lR)-l-(3,5-dichloro-4-pyridyl)ethoxy]-6-methoxy-lH-indazol-3-yl]-2- [3-[(dimethylamino)methyl]-3-[[dimethyl(oxo)-k6-sulfanylidene]amino]azetidin-l- yl] pyridine-3-carbonitrile.
[00409] Step 1. 5-(5-((R)-l-(3,5-Dichloropyridin-4-yl)ethoxy)-6-methoxy-l- (tetrahy dro-2H-pyran-2-yl)- 1 H-indazol -3 -yl)-2-(3 -((dimethyl(oxo)-X6- sulfaneylidene)amino)-3-((dimethylamino)methyl)azetidin-l-yl)nicotinonitrile. A mixture of 5-[5-[(lR)-l-(3,5-dichloro-4-pyridyl)ethoxy]-6-methoxy-l-tetrahydropyran-2-yl-indazol-3- yl]-2-fluoro-pyridine-3-carbonitrile (332 mg, 0.61 mmol, 1.0 equiv), l-[3-[[dimethyl(oxo)-X6- sulfanylidene]amino]azetidin-3-yl]-N,N-dimethyl-methanamine (212 mg, 0.67 mmol, 1.1 equiv) and N,N-diisopropylethylamine (3.2 mL, 18.4 mmol, 30 equiv) in acetonitrile (35 mL) was stirred at room temperature for 16 hours. The reaction was concentrated under reduced pressure. The residue was purified on a Biotage automated chromatography system (Biotage Sfar KP -Amino D Duo 50 pm 28 g column), eluting with a gradient of 0 to 10% methanol in ethyl acetate to give an off-white solid (450 mg, 99%). LCMS m/z = 727.2 (M+H).
[00410] Step 2. (R)-5-(5-(l-(3,5-Dichloropyridin-4-yl)ethoxy)-6-methoxy-lH- indazol-3-yl)-2-(3-((dimethyl(oxo)-X6-sulfaneylidene)amino)-3-((dimethylamino)methyl) azetidin-l-yl)nicotinonitrile. 5-(5-((R)-l-(3,5-Dichloropyridin-4-yl)ethoxy)-6-methoxy-l- (tetrahy dro-2H-pyran-2-yl)- 1 H-indazol -3 -yl)-2-(3 -((dimethyl(oxo)-X6- sulfaneylidene)amino)-3-((dimethylamino)methyl)azetidin-l-yl)nicotinonitrile (225 mg, 0.31 mmol, 1.0 equiv) in ethanol (4.6 mL) was treated with 6M HC1 (0.41 mL, 2.5 mmol, 8 equiv) at 40 °C. After 16 hours, the mixture was cooled to room temperature, diluted with ethyl acetate (20 mL) and saturated sodium carbonate solution (8 mL). The layers were separated, and the aqueous phase was extracted with ethyl acetate (10 mL). The combined organic layers were dried over sodium sulfate, filtered, and concentrated under reduced pressure. The residue was dissolved in dimethyl sulfoxide (6 mL) and purified on a Biotage automated chromatography system (RediSep Rf GOLD 100 g HP C18 column), eluting with a gradient of 0 to 100% acetonitrile in water. Fractions containing clean product were lyophilized to give an off-white solid (113 mg, 57%). LCMS: m/z = 643.2 (M+H)+; 1H NMR (400 MHz, DMSO-d6) 5 = 12.96 (br s, 1H), 8.71 (d, J = 2.3 Hz, 1H), 8.59 (s, 2H), 8.06 (d, J = 2.3 Hz, 1H), 7.05 (s, 1H), 7.00 (s, 1H), 6.03 - 5.96 (m, 1H), 4.37 - 4.22 (m, 4H), 3.86 (s, 3H), 3.11 (s, 6H), 2.66 (s, 2H), 2.29 (s, 6H), 1.76 (d, J = 6.7 Hz, 3H).

Example 13
Example 13. l-[l-[5-[5-[(lR)-l-(3,5-dichloro-4-pyridyl)ethoxy]-lH-indazol-3-yl]-3- fluoro-2-pyridyl]-3-[[dimethyl(oxo)-k6-sulfanylidene]amino]azetidin-3-yl]-N,N-dimethyl- methanamine
[00411] Step 1. Methyl l-benzhydryl-3-((methylsulfonyl)oxy)azetidine-3- carboxylate. N,N-Diisopropylethylamine (4.9 mL, 33.6 mmol, 2 equiv) was added to a solution of methyl l-benzhydryl-3-hydroxy-azetidine-3 -carboxylate (5.0 g, 16.8 mmol, 1.0 equiv) in dichloromethane (100 mL) at 0 °C. Methanesulfonic anhydride (4.1 g, 23.5 mmol, 1.4 equiv) was then added over 2 minutes. After 16 hours at room temperature, the mixture was washed with water (2 x 50 mL). The organic layer was dried over sodium sulfate, filtered, and concentrated under reduced pressure. The residue was dried at 35 °C under vacuum for 90 minutes to give an off-white solid (7.1 g, 100%). LCMS m/z = 376.1 (M+H). [00412] Step 2. Methyl l-benzhydryl-3-((dimethyl(oxo)-X6- sulfaneylidene)amino)azetidine-3-carboxylate. Dimethyl sulfoximine (2.44 g, 26.2 mmol, 1.4 equiv) and sodium bicarbonate (2.38 g, 28.4 mmol, 1.5 equiv) were added to a solution of methyl l-benzhydryl-3-((methylsulfonyl)oxy)azetidine-3-carboxylate (7.1 g, 16.8 mmol, 1.0 equiv) in acetonitrile (100 mL) at room temperature. The mixture was heated at 80 °C for 16 hours then cooled to room temperature, and concentrated under reduced pressure. The residue was diluted with ethyl acetate (200 mL) and water (50 mL). The layers were separated, and the aqueous layer was extracted with ethyl acetate (100 mL). The combined organic layers were dried over sodium sulfate, filtered, and concentrated under reduced pressure. The residue was purified on a Biotage automated chromatography system (Sorbtech 120 g column), eluting with a gradient of 0 to 100% ethyl acetate containing 2% methanol in hexanes to give a yellow oil (5.4 g, 86% yield over two steps). LCMS m/z = 373.2 (M+H).
[00413] Step 3. ((l-Benzhydryl-3-(hydroxymethyl)azetidin-3-yl)imino)dimethyl- X6-sulfanone. Sodium borohydride (8.23 g, 217.5 mmol, 15 equiv) was added to a solution of methyl l-benzhydryl-3-((dimethyl(oxo)-X6-sulfaneylidene)amino)azetidine-3 -carboxylate (5.4 g, 14.5 mmol, 1.0 equiv) in methanol (100 mL) at 0 °C. The reaction was stirred at 0 °C for 1 hour and at room temperature for 1 hour. The mixture was diluted with ethyl acetate (600 mL) and sequentially washed with saturated sodium bicarbonate (100 mL), water (100 mL) and saturated brine (2 x 100 mL). The organic layer was dried over sodium sulfate, filtered, and concentrated under reduced pressure. The residue was purified on a Biotage automated chromatography system (Biotage Sfar KP-Amino D Duo 50 pm 110 g column), eluting with a gradient of 0 to 100% ethyl acetate in hexanes to give a white solid (3.9 g, 78%). LCMS m/z = 345.2 (M+H).
[00414] Step 4. (l-Benzhydryl-3-((dimethyl(oxo)-X6- sulfaneylidene)amino)azetidin-3-yl)methyl methanesulfonate. N,N-Diisopropylethylamine (0.51 mL, 2.9 mmol, 2 equiv) was added to a solution of ((l-benzhydryl-3- (hydroxymethyl)azetidin-3-yl)imino)dimethyl-X6-sulfanone (500 mg, 1.45 mmol, 1.0 equiv) in dichloromethane (30 mL) at 0 °C. Methanesulfonic anhydride (380 mg, 2.18 mmol, 1.5 equiv) was added. The reaction was warmed to room temperature, stirred for 6 hours, then washed with water (2 x 15 mL). The organic layer was dried over sodium sulfate, filtered, and concentrated under reduced pressure. The residue was dried at 35 °C under vacuum for 90 minutes to give an off-white solid (700 mg, 100%). LCMS m/z = 345.1 (M+H), [00415] Step 5. ((l-Benzhydryl-3-((dimethylamino)methyl)azetidin-3- yl)imino)dimethyl-X6-sulfanone. 2M Dimethylamine in THF (8.3 mL, 16.6 mmol, 10 equiv) and N,N-diisopropylethylamine (2.14 g, 2.9 mL, 16.6 mmol, 10 equiv) were added to a solution of crude compound 910-5 (700 mg, <1.45 mmol, 1.0 equiv) in acetonitrile (40 mL) at room temperature. The mixture was heated at 66 °C for 3.5 hours then cooled to room temperature and concentrated under reduced pressure. The residue was purified on a Biotage automated chromatography system (Biotage Sfar KP-Amino D Duo 50 pm 28 g column x 2, stacked), eluting with a gradient of 0 to 10% methanol in ethyl acetate to give an off-white solid (500 mg, 92% yield over two steps). LCMS m/z = 372.2 (M+H).
[00416] Step 6. ((3-((Dimethylamino)methyl)azetidin-3-yl)imino)dimethyl-Z6- sulfanone trihydrochloride. A mixture of ((l-benzhydryl-3-((dimethylamino)methyl)azetidin- 3-yl)imino)dimethyl-Z6-sulfanone (500 mg, 1.35 mmol), 20% palladium hydroxide on activated carbon (50% wetted powder) (284 mg, 0.20 mmol, 0.15 equiv) and concentrated HC1 (0.5 mL, 6.0 mmol, 4.5 equiv) in methanol (40 mL) was hydrogenated at 50 psi at room temperature for 70 hours in a Parr shaker. The reaction mixture was filtered through a pad of Celite, washed with 10% water in methanol (40 mL), and then concentrated under reduced pressure. The residue was azeotroped with toluene (2 x 50 mL) and dried under vacuum at 40 °C for 3 hours to give the HC1 salt as a brown oil (424 mg, 99%) LCMS m/z = 206.1 (M+H) free base.
[00417] Step 7. ((l-(5-(5-((R)-l-(3,5-Dichloropyridin-4-yl)ethoxy)-l-(tetrahydro- 2H-pyran-2-yl)-lH-indazol-3-yl)-3-fluoropyridin-2-yl)-3-((dimethylamino)methyl)azetidin- 3-yl)imino)dimethyl-Z6-sulfanone. A mixture of 5-[(lR)-l-(3,5-dichloro-4-pyridyl)ethoxy]- 3-(5,6-difhioro-3-pyridyl)-l-tetrahydropyran-2-yl-indazole (310 mg, 0.61 mmol, 1.0 equiv), ((3-((Dimethylamino)methyl)azetidin-3-yl)imino)dimethyl-Z6-sulfanone trihydrochloride (212 mg, 0.67 mmol, 1.1 equiv) and N,N-diisopropylethylamine (2.38 g, 3.2 mL, 18.4 mmol, 30 equiv) in acetonitrile (35 mL) was heated at 80 °C for 16 hours. The reaction was cooled to room temperature and concentrated under reduced pressure. The residue was purified on a Biotage automated chromatography system (Biotage Sfar KP-Amino D Duo 50 pm 28 g column), eluting with a gradient of 0 to 10% methanol in ethyl acetate to give an off-white solid (386 mg, 91%). LCMS m/z = 690.2 (M+H).
[00418] Step 8. (R)-((l-(5-(5-(l-(3,5-Dichloropyridin-4-yl)ethoxy)-lH-indazol-3- yl)-3-fluoropyridin-2-yl)-3-((dimethylamino)methyl)azeti din-3 -yl)imino)dimethyl-Z6- sulfanone. ((l-(5-(5-((R)-l-(3,5-Dichloropyridin-4-yl)ethoxy)-l-(tetrahydro-2H-pyran-2-yl)- lH-indazol-3-yl)-3-fluoropyridin-2-yl)-3-((dimethylamino)methyl)azetidin-3- yl)imino)dimethyl-X6-sulfanone (234 mg, 0.34 mmol, 1.0 equiv) in ethanol (5 mL) was treated with 6M HC1 (0.45 mL, 2.7 mmol, 8 equiv) at 65 °C. After 4 hours, the mixture was cooled to room temperature. The mixture was diluted with ethyl acetate (20 mL) and saturated sodium carbonate solution (8 mL). The layers were separated, and the aqueous phase was extracted with ethyl acetate (10 mL). The combined organic layers were dried over sodium sulfate, filtered, and concentrated under reduced pressure. The residue was dissolved in dimethyl sulfoxide (6 mL) and purified on a Biotage automated chromatography system (RediSep Rf GOLD 100 g HP Cl 8 column), eluting with a gradient of 0 to 100% acetonitrile in water. Product containing fractions were lyophilized to give a white solid as the dihydrochloride (110 mg, 54%). LCMS: m/z = 606.1 (M+H); 1H NMR (400 MHz, DMSO- d6) 5 = 13.10 (br s, 1H), 8.57 (s, 2H), 8.36 (t, J = 1.6 Hz, 1H), 7.69 (dd, J = 1.8, 13.2 Hz, 1H), 7.47 (d, J = 8.9 Hz, 1H), 7.20 (d, J = 2.1 Hz, 1H), 7.10 (dd, J = 2.2, 9.0 Hz, 1H), 6.14 (q, J = 6.6 Hz, 1H), 4.19 - 4.10 (m, 4H), 3.09 (s, 6H), 2.66 (s, 2H), 2.29 (s, 6H), 1.76 (d, J = 6.6 Hz, 3H).
[00419] The Examples in the table below were synthesized as described above, or using analogous methods.







- Ill - Kinase Assays
[00420] Kinase-tagged T7 phage strains were prepared in an E. coli host derived from the BL21 strain. E. coli were grown to log -phase and infected with T7 phage and incubated with shaking at 32°C until lysis. The lysates were centrifuged and filtered to remove cell debris. Streptavidin-coated magnetic beads were treated with biotinylated small molecule ligands for 30 minutes at room temperature to generate affinity resins for kinase assays. The liganded beads were blocked with excess biotin and washed with blocking buffer SeaBlock (Pierce), 1% BSA, 0.05% Tween 20, 1 mM DTT to remove unbound ligand and to reduce non-specific binding. Binding reactions were assembled by combining kinases, liganded affinity beads, and test compounds in lx binding buffer (20% SeaBlock, 0.17x PBS, 0.05% Tween 20, 6 mM DTT).
[00421] Test compounds were prepared as 11 IX stocks in 100% DMSO. Kds were determined using an 11 -point 3-fold compound dilution series with three DMSO control points. All compounds for Kd measurements are distributed by acoustic transfer (non-contact dispensing) in 100% DMSO. The compounds were then diluted directly into the assays such that the final concentration of DMSO was 0.9%. All reactions performed in polypropylene 384-well plate. Each was a final volume of 0.02 ml. The assay plates were incubated at room temperature with shaking for 1 hour and the affinity beads were washed with wash buffer (lx PBS, 0.05% Tween 20). The beads were then re-suspended in elution buffer (lx PBS, 0.05% Tween 20, 0.5 pM non-biotinylated affinity ligand) and incubated at room temperature with shaking for 30 minutes. The kinase concentration in the eluates was measured by qPCR.
[00422] Binding constants (Kds)
[00423] Binding constants were calculated with a standard dose-response curve using the Hill equation:

[00424] The Hill Slope was set to -1. Curves were fitted using a non-linear least square fit with the Levenberg-Marquardt algorithm.
Table 1.
A= 1-10 nM
B= >10 to 20 nM
C= >20 to 30 nM D= >30 to 100 nM

Cell Viability Assays
1. Ba/F3 Cell Viability Assay

[00425] Experimental Purpose: Recombinant kinase fusions are transduced into parental Ba/F3, which becomes dependent upon this constitutive kinase activity for IL3- independent survival. Inhibition of kinase activity leads to cell death, which is monitored using CellTiter-Glo® 2.0 (Promega) which measures intracellular ATP concentration that in turn serves as a marker for viability. BCR-FGFR1 Ba/F3, BCR-FGFR2 and FGFR3- BAIAP2L1 Ba/F3 were obtained from Advanced Cellular Dynamics (Seattle, WA). ETV6- FGFR4 was obtained from Kyinno (Waltham, MA). [00426] Cell Viability Assay Procedure: Cell Titer-Gio® 2.0 Luminescent cell viability assay reagent was purchased from Promega (Madison, WI). Ba/F3 cell lines were cultured in RPMI1640 media supplemented with 10% fetal bovine serum. Cultures were maintained at 37°C in a humidified atmosphere of 5% CO2 and 95% air.
[00427] Cells were plated in 96-well clear bottom/white plates (Corning #3903) at 10,000 cells/well in lOOpl of media, incubated overnight. The next day, test compound DMSO stock solutions were made at 10 mM and 2 pM final concentration. Compounds were then added to cells in a 9-dose, 10-fold dilution series starting at 30 pM with an HP 300e Digital Dispenser (each dose was applied in triplicate). DMSO was backfilled to each well up to 301 nL total volume of test compound + DMSO, and a total of 301 nL DMSO was added to a control/no test compound well in triplicate. The cells in cell culture plates were incubated with the compounds at 37 °C and 5% CO2 for 48 hours. Then 50 pl of Cell Titer Gio 2.0 reagent was added to each well of the cell culture plates. The contents were covered from light and mixed on an orbital shaker at room temperature for 10 min. Luminescence was recorded by a Synergy Hl Microplate Reader (Biotek, Winooski, VT ). Cells were assessed as a percentage of DMSO only treated control cells. Curves were plotted and IC50 values were calculated using the GraphPad Prism 8 program based on a sigmoidal dose-response equation (4 parameter).
Table 2. Ba/F3 Cell data
A = 0.1 - 50 nM B = >50 - 200nM C = >200 - 1000 nM D = > 1000 nM



2. Cancer Cell Line Cell Viability Assays

[00428] Experimental Purpose: To detect the change of intracellular ATP by Cell Titer-Gio® and to evaluate the inhibitory effect of the compounds on cancer cell lines by determining the in vitro IC50 value of the compounds.
[00429] Cell Titer-Gio® 2.0 Luminescent cell viability assay reagent was purchased from Promega (Madison, WI). KG-1, KATO-III, and MDA-MB-453 cell lines were purchased from American Type Culture Collection (Manassas, VA). RT112/84 cell line was purchased from Millipore-Sigma (St. Louis, MO). HuH7 cells were purchased from Seikisui Xenotech (Kansas City, KS). RT112/84 and MDA-MB-453 cells were cultured in RPMI1640 media supplemented with 10% fetal bovine serum. KG-1 and KATO-III cell lines were cultured in IMDM media supplemented with 20% FBS. HuH7 cells were cultured in IMDM media supplemented with 10% FBS. Cultures were maintained at 37°C in a humidified atmosphere of 5% CO2 and 95% air.
[00430] Cell Viability Assay Procedure: Cells were plated in 96-well clear bottom/white plates (Corning #3903) at a range of densities depending on the optimal assay window (5,000-20,000 cells/well in 1 OOJJ.1 of media), incubated overnight. The next day, test compound DMSO stock solutions were made at 10 mM and 2 pM final concentration. Compounds were then added to cells in a 9-dose, 4-fold dilution series starting at 3 pM with an HP 300e Digital Dispenser (each dose was applied in triplicate). DMSO was backfilled to each well up to 301 nL total volume of test compound + DMSO, and a total of 301 nL DMSO was added to a control/no test compound well in triplicate. The cells in cell culture plates were incubated with the compounds at 37 °C and 5% CO2 for 72 hours- 120 hours depending on the cell line. Then 50 pl of Cell Titer Gio 2.0 reagent was added to each well of the cell culture plates. The contents were covered from light and mixed on an orbital shaker at room temperature for minimum of 10 min. Luminescence was recorded by a Clariostar Plus Microplate Reader (BMG Labtech, Cary, NC ). Cells were assessed as a percentage of DMSO only treated control cells. Curves were plotted and IC50 values were calculated using the GraphPad Prism 9 program based on a sigmoidal dose-response equation (log (inhibitor) vs. response - Variable slope, 4-parameter).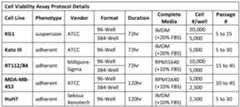

Table 3. Cancer Cell Data
A = 0.1 - 50 nM
B = >50 - 200nM C = >200 - 1000 nM
D = > 1000 nM



Claims
1. A compound of formula (I): or a pharmaceutically acceptable salt thereof, wherein
or a pharmaceutically acceptable salt thereof, wherein

X = O, S, or NR;
R is H or C1-C3alkyl; n = 1 or 2; m = 1 or 2;
R1 is H, optionally substituted C1-C6alkyl, or NH2;
R2 is -Co-C6alkN=S(0)(C1-C6alkyl)(C1-C6alkyl), -NH-C1-C6alk-N=S(O)(C1- C6alkyl)(C1-C6alkyl), or -Co-C3alk-C(=0)-Co-C3alk-N=S(0)(C1-C6alkyl)(C1-C6alkyl); one or two of Q1, Q2, Q3, Q4 is N and the others are each independently CR5a;
R5a is H, halogen, -CN, -S(O)2C1-C6alkyl, OCF3, OC1-C3alkyl, or C1-C3alkyl;
Q5, Q6, Q7, Q8, and Q9 are each independently N or CR5, wherein one or two of the Q5, Q6, Q7, Q8, and Q9 is N and the remainder are CR5;
R5 is H, halogen, C1-C3alkyl, C1-C3alkoxyl, or cycloalkyl;
R6 is C1-Cealkyl;
R7 is H, halogen, -C1-Cealkyl, -C1-C6 alkoxyl, or -cycloalkyl; and
R8 is H, halogen, -C1-Cealkyl, -C1-C6 alkoxyl, or -cycloalkyl.
2. The compound of claim 1, wherein X is O.
3. The compound of claim 1 or claim 2, wherein R6 is CH3.
4. The compound of any one of the preceding claims, wherein R8 is H.
5. The compound of any one of the preceding claims, wherein R7 is H, F, or OCH3.
6. The compound of any one of the preceding claims, wherein Q1 and Q3 are each CR5a wherein R5a is H.
7. The compound of any one of the preceding claims, wherein Q2 and Q4 are each N.
8. The compound of any one of claims 1-6, wherein Q2 is N and Q4 is CR5a wherein R5a is H, -CN, or F.
9. The compound of any one of claims 1-6, wherein Q2 is N and Q4 is CR5a wherein R5a is S(O)2C1-C6alkyl, OC1-C3alkyl, or C1-C3alkyl.
10. The compound of any one of the preceding claims, wherein Q5 and Q9 are each CR5 wherein each R5 is Cl; Q7 is N; Q6 is CR5 wherein R5 is H; and Q8 is CR5 wherein R5 is H or CH3.
11. The compound of any one of the preceding claims, wherein n is i and m is 1.
12. The compound of any one of the preceding claims, wherein the compound of formula (I) is a compound of formula (IA): or a pharmaceutically acceptable salt thereof, wherein
or a pharmaceutically acceptable salt thereof, wherein

Q2 and Q4 are each N, or one of Q2 or Q4 is N and the other is CR5a;
R5a is H, F, -SO2CH3, or -CN;
R5 is H or CH3; and
R7 is H, F, or OCH3.
13. The compound of claim 12, wherein R5 is H.
14. The compound of claim 12, wherein R5 is CH3.
15. The compound of any one of claims 12-14, wherein R7 is H.
16. The compound of any one of claims 12-14, wherein R7 is F.
17. The compound of any one of claims 12-14, wherein R7 is OCH3.
18. The compound of any one of claims 12-17, wherein Q2 and Q4 are each N.
19. The compound of any one of claims 12-17, wherein one of Q2 or Q4 is N and the other is CR5a.
20. The compound of claim 19, wherein R5a is H or F.
21. The compound of claim 20, wherein R5a is H.
22. The compound of claim 20, wherein R5a is F.
23. The compound of claim 19, wherein R5a is CN or SO2CH3.
24. The compound of claim 23, wherein R5a is CN.
25. The compound of claim 23, wherein R5a is SO2CH3.
26. The compound of claim 1, wherein R5a is OCF3.
27. The compound of any one of claims 1-26, wherein R1 is H.
28. The compound of any one of claims 1-26, wherein R1 is CH3.
29. The compound of claim 1, wherein the compound of formula (I) is a compound of formula (IC): or a pharmaceutically acceptable salt thereof, wherein
or a pharmaceutically acceptable salt thereof, wherein

R1 is optionally substituted C1-Cealkyl, or NFF;
R2 is -Co-C6alkN=S(0)(C1-C6alkyl)(C1-C6alkyl), -NH-C1-C6alk-N=S(O)(C1- C6alkyl)(C1-C6alkyl), or -Co-C3alk-C(=0)-Co-C3alk-N=S(0)(C1-C6alkyl)(C1-C6alkyl);
R7is H or -OC1-Cealkyl; and R5a is halo, or -CN.
30. The compound of claim 29, wherein R7 is H.
31. The compound of claim 29, wherein R7 is -OC1-Cealkyl.
32. The compound of claim 31, wherein R7 is -OCH3.
33. The compound of any one of claims 29-32, wherein R5a is -CN.
34. The compound of any one of claims 29-32, wherein R5a is halo.
35. The compound of claim 34, wherein R5a is F, Cl, or Br.
36. The compound of claim 35, wherein R5a is F.
37. The compound of claim 35, wherein R5a is Cl.
38. The compound of claim 35, wherein R5a is Br.
39. The compound of any one of claims 29-38, wherein R2 is -Co-C6alk-N=S(0)(C1- C6alkyl)(C1-C6alkyl).
40. The compound of claim 39, wherein R2 is -N=S(O)(CH3)2.
41. The compound of claim 39, wherein R2 is -CH2-N=S(O)(CH3)2.
42. The compound of any one of claims 29-38, wherein R2 is -NH-C1-C6alk-N=S(O)(C1- C6alkyl)(C1-C6alkyl).
43. The compound of claim 42, wherein R2 is -NH-CH2CH2-N=S(O)(CH3)2.
44. The compound of any one of claims 29-38, wherein R2 is -Co-C3alk-C(=0)-Co-C3alk- N=S(O)(C1-C6alkyl)(C1-C6alkyl).
45. The compound of claim 44, wherein R2 is -C(=O)-N=S(O)(CH3)2.
46. The compound of any one of claims 29-45, wherein R1 is optionally substituted C1- Cealkyl.
47. The compound of claim 46, wherein R1 is -CH3.
48. The compound of claim 46, wherein R1 is -CH2NH2.
49. The compound of claim 46, wherein R1 is -CH2NHCH3.
50. The compound of claim 46, wherein R1 is -CH2N(CH3)2.
51. The compound of claim 46, wherein R1 is

52. The compound of any one of claims 29-45, wherein R1 is -NH2.
53. The compound of claim 1, or a pharmaceutically acceptable salt thereof, wherein the compound is:


54. A pharmaceutical composition comprising a compound of any one of claims 1-53, or a pharmaceutically acceptable salt thereof, and a pharmaceutically acceptable excipient.
55. A method of treating a disease or disorder in a subject in need thereof comprising administering to the subject a compound of any one of claims 1 to 53, or a pharmaceutically acceptable salt thereof.
56. The method of claim 55, wherein the disease or disorder is cancer.
57. The method of claim 56, wherein the cancer is urothelial carcinoma, hepatocellular carcinoma, breast carcinoma, endometrial adenocarcinoma, ovarian carcinoma, primary glioma, cholangiocarcinoma, gastric adenocarcinoma, non-small cell lung carcinoma, pancreatic exocrine carcinoma, oral cancer, prostate cancer, bladder cancer, colorectal carcinoma, renal cell carcinoma, neuroendocrine carcinoma, myeloproliferative neoplasms, head and neck (squamous), melanoma, leiomyosarcoma, and/or sarcomas.
58. The method of claim 57, wherein the cancer is bladder cancer.
59. The method of claim 57, wherein the cancer is urothelial carcinoma.
60. The method of claim 57, wherein the cancer is hepatocellular carcinoma.
61. The method of any one of claims 56 to 60, wherein the cancer is an FGFR-mutant cancer.
62. The method of claim 55, wherein the disease or disorder is a developmental disorder.
63. The method of claim 62, wherein the developmental disorder is achondroplasia.
64. A method of inhibiting FGFR in a cell comprising contacting the cell with a compound of any one of claims 1 to 53.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US202263476791P | 2022-12-22 | 2022-12-22 | |
| US63/476,791 | 2022-12-22 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2024138112A1true WO2024138112A1 (en) | 2024-06-27 |
Family
ID=89900743
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2023/085649PendingWO2024138112A1 (en) | 2022-12-22 | 2023-12-22 | Indazole compounds |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2024138112A1 (en) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2025061029A1 (en)* | 2023-09-18 | 2025-03-27 | 3H Pharmaceuticals Co., Ltd. | Fgfr inhibitors and methods of use thereof |
Citations (35)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3657744A (en) | 1970-05-08 | 1972-04-25 | Univ Minnesota | Method for fixing prosthetic implants in a living body |
| US4739762A (en) | 1985-11-07 | 1988-04-26 | Expandable Grafts Partnership | Expandable intraluminal graft, and method and apparatus for implanting an expandable intraluminal graft |
| US4992445A (en) | 1987-06-12 | 1991-02-12 | American Cyanamid Co. | Transdermal delivery of pharmaceuticals |
| US5001139A (en) | 1987-06-12 | 1991-03-19 | American Cyanamid Company | Enchancers for the transdermal flux of nivadipine |
| US5023252A (en) | 1985-12-04 | 1991-06-11 | Conrex Pharmaceutical Corporation | Transdermal and trans-membrane delivery of drugs |
| US5040548A (en) | 1989-06-01 | 1991-08-20 | Yock Paul G | Angioplasty mehtod |
| US5061273A (en) | 1989-06-01 | 1991-10-29 | Yock Paul G | Angioplasty apparatus facilitating rapid exchanges |
| US5195984A (en) | 1988-10-04 | 1993-03-23 | Expandable Grafts Partnership | Expandable intraluminal graft |
| US5292331A (en) | 1989-08-24 | 1994-03-08 | Applied Vascular Engineering, Inc. | Endovascular support device |
| US5451233A (en) | 1986-04-15 | 1995-09-19 | Yock; Paul G. | Angioplasty apparatus facilitating rapid exchanges |
| US5496346A (en) | 1987-01-06 | 1996-03-05 | Advanced Cardiovascular Systems, Inc. | Reinforced balloon dilatation catheter with slitted exchange sleeve and method |
| US5674278A (en) | 1989-08-24 | 1997-10-07 | Arterial Vascular Engineering, Inc. | Endovascular support device |
| US6344053B1 (en) | 1993-12-22 | 2002-02-05 | Medtronic Ave, Inc. | Endovascular support device and method |
| US20110008347A1 (en) | 2006-12-01 | 2011-01-13 | Agency For Science ,Technology And Research | Cancer-related protein kinases |
| CN102741256A (en) | 2010-01-29 | 2012-10-17 | 韩美药品株式会社 | Bicyclic heteroaryl derivatives having inhibitory activity for protein kinase |
| US20130009621A1 (en) | 2011-07-07 | 2013-01-10 | Silergy Semiconductor Technology (Hangzhou) Ltd | Low offset, fast response voltage controlled current source and controlling method thereof |
| WO2014071419A2 (en) | 2012-11-05 | 2014-05-08 | Foundation Medicine, Inc. | Novel fusion molecules and uses thereof |
| WO2015099127A1 (en) | 2013-12-27 | 2015-07-02 | 中外製薬株式会社 | Fgfr gatekeeper mutant gene and drug targeting same |
| WO2015120094A2 (en) | 2014-02-04 | 2015-08-13 | Mayo Foundation For Medical Education And Research | Method of identifying tyrosine kinase receptor rearrangements in patients |
| WO2015150900A2 (en) | 2014-03-31 | 2015-10-08 | Debiopharm International Sa | Fgfr fusions |
| US20150366866A1 (en) | 2013-01-18 | 2015-12-24 | Foundation Medicine, Inc. | Methods of treating cholangiocarcinoma |
| US9254288B2 (en) | 2012-05-07 | 2016-02-09 | The Translational Genomics Research Institute | Susceptibility of tumors to tyrosine kinase inhibitors and treatment thereof |
| US9267176B2 (en) | 2011-08-05 | 2016-02-23 | Astellas Pharma Inc. | Method for detecting novel FGFR4 mutant |
| JP5868992B2 (en) | 2010-11-29 | 2016-02-24 | アステックス、セラピューティックス、リミテッドAstex Therapeutics Limited | Substituted benzopyrazine derivatives as FGFR kinase inhibitors for the treatment of cancer diseases |
| WO2016030509A1 (en) | 2014-08-28 | 2016-03-03 | Oncoethix Gmbh | Methods of treating acute myeloid leukemia or acute lymphoid leukemia using pharmaceutical compositions containing thienotriazolodiazepine compounds |
| EP2203449B1 (en) | 2007-10-12 | 2016-04-27 | Astex Therapeutics Limited | Bicyclic heterocyclic compounds as protein tyrosine kinase inhibitors |
| EP3023101A1 (en) | 2013-07-18 | 2016-05-25 | Taiho Pharmaceutical Co., Ltd. | Therapeutic agent for fgfr inhibitor-resistant cancer |
| WO2016084883A1 (en) | 2014-11-26 | 2016-06-02 | 国立研究開発法人国立がん研究センター | Novel therapeutic target fusion gene in biliary tract cancer |
| AU2014362227A1 (en) | 2013-12-11 | 2016-06-23 | Accuragen Holdings Limited | Compositions and methods for detecting rare sequence variants |
| WO2016105503A1 (en) | 2014-12-24 | 2016-06-30 | Genentech, Inc. | Therapeutic, diagnostic and prognostic methods for cancer of the bladder |
| US20160215350A1 (en) | 2013-09-09 | 2016-07-28 | Nantomics, Llc | Proteomics Analysis and Discovery through DNA and RNA Sequencing, Systems and Methods |
| US20160235744A1 (en) | 2011-10-28 | 2016-08-18 | Astex Therapeutics Limited | SUBSTITUTED PYRIDO[2,3-b]PYRAZINES AS FGFR KINASE INHIBITORS |
| WO2016139227A1 (en) | 2015-03-03 | 2016-09-09 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Fgfr3 antagonists |
| US9447098B2 (en) | 2012-05-30 | 2016-09-20 | Astex Therapeutics Ltd | Pteridines as FGFR inhibitors |
| WO2022147246A1 (en)* | 2020-12-30 | 2022-07-07 | Tyra Biosciences, Inc. | Indazole compounds as kinase inhibitors |
- 2023
- 2023-12-22WOPCT/US2023/085649patent/WO2024138112A1/enactivePending
Patent Citations (37)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3657744A (en) | 1970-05-08 | 1972-04-25 | Univ Minnesota | Method for fixing prosthetic implants in a living body |
| US4739762A (en) | 1985-11-07 | 1988-04-26 | Expandable Grafts Partnership | Expandable intraluminal graft, and method and apparatus for implanting an expandable intraluminal graft |
| US4739762B1 (en) | 1985-11-07 | 1998-10-27 | Expandable Grafts Partnership | Expandable intraluminal graft and method and apparatus for implanting an expandable intraluminal graft |
| US5023252A (en) | 1985-12-04 | 1991-06-11 | Conrex Pharmaceutical Corporation | Transdermal and trans-membrane delivery of drugs |
| US5451233A (en) | 1986-04-15 | 1995-09-19 | Yock; Paul G. | Angioplasty apparatus facilitating rapid exchanges |
| US5496346A (en) | 1987-01-06 | 1996-03-05 | Advanced Cardiovascular Systems, Inc. | Reinforced balloon dilatation catheter with slitted exchange sleeve and method |
| US5001139A (en) | 1987-06-12 | 1991-03-19 | American Cyanamid Company | Enchancers for the transdermal flux of nivadipine |
| US4992445A (en) | 1987-06-12 | 1991-02-12 | American Cyanamid Co. | Transdermal delivery of pharmaceuticals |
| US5195984A (en) | 1988-10-04 | 1993-03-23 | Expandable Grafts Partnership | Expandable intraluminal graft |
| US5040548A (en) | 1989-06-01 | 1991-08-20 | Yock Paul G | Angioplasty mehtod |
| US5061273A (en) | 1989-06-01 | 1991-10-29 | Yock Paul G | Angioplasty apparatus facilitating rapid exchanges |
| US5292331A (en) | 1989-08-24 | 1994-03-08 | Applied Vascular Engineering, Inc. | Endovascular support device |
| US5674278A (en) | 1989-08-24 | 1997-10-07 | Arterial Vascular Engineering, Inc. | Endovascular support device |
| US5879382A (en) | 1989-08-24 | 1999-03-09 | Boneau; Michael D. | Endovascular support device and method |
| US6344053B1 (en) | 1993-12-22 | 2002-02-05 | Medtronic Ave, Inc. | Endovascular support device and method |
| US20110008347A1 (en) | 2006-12-01 | 2011-01-13 | Agency For Science ,Technology And Research | Cancer-related protein kinases |
| EP2203449B1 (en) | 2007-10-12 | 2016-04-27 | Astex Therapeutics Limited | Bicyclic heterocyclic compounds as protein tyrosine kinase inhibitors |
| CN102741256A (en) | 2010-01-29 | 2012-10-17 | 韩美药品株式会社 | Bicyclic heteroaryl derivatives having inhibitory activity for protein kinase |
| JP5868992B2 (en) | 2010-11-29 | 2016-02-24 | アステックス、セラピューティックス、リミテッドAstex Therapeutics Limited | Substituted benzopyrazine derivatives as FGFR kinase inhibitors for the treatment of cancer diseases |
| US20130009621A1 (en) | 2011-07-07 | 2013-01-10 | Silergy Semiconductor Technology (Hangzhou) Ltd | Low offset, fast response voltage controlled current source and controlling method thereof |
| US9267176B2 (en) | 2011-08-05 | 2016-02-23 | Astellas Pharma Inc. | Method for detecting novel FGFR4 mutant |
| US20160235744A1 (en) | 2011-10-28 | 2016-08-18 | Astex Therapeutics Limited | SUBSTITUTED PYRIDO[2,3-b]PYRAZINES AS FGFR KINASE INHIBITORS |
| US9254288B2 (en) | 2012-05-07 | 2016-02-09 | The Translational Genomics Research Institute | Susceptibility of tumors to tyrosine kinase inhibitors and treatment thereof |
| US9447098B2 (en) | 2012-05-30 | 2016-09-20 | Astex Therapeutics Ltd | Pteridines as FGFR inhibitors |
| WO2014071419A2 (en) | 2012-11-05 | 2014-05-08 | Foundation Medicine, Inc. | Novel fusion molecules and uses thereof |
| US20150366866A1 (en) | 2013-01-18 | 2015-12-24 | Foundation Medicine, Inc. | Methods of treating cholangiocarcinoma |
| EP3023101A1 (en) | 2013-07-18 | 2016-05-25 | Taiho Pharmaceutical Co., Ltd. | Therapeutic agent for fgfr inhibitor-resistant cancer |
| US20160215350A1 (en) | 2013-09-09 | 2016-07-28 | Nantomics, Llc | Proteomics Analysis and Discovery through DNA and RNA Sequencing, Systems and Methods |
| AU2014362227A1 (en) | 2013-12-11 | 2016-06-23 | Accuragen Holdings Limited | Compositions and methods for detecting rare sequence variants |
| WO2015099127A1 (en) | 2013-12-27 | 2015-07-02 | 中外製薬株式会社 | Fgfr gatekeeper mutant gene and drug targeting same |
| WO2015120094A2 (en) | 2014-02-04 | 2015-08-13 | Mayo Foundation For Medical Education And Research | Method of identifying tyrosine kinase receptor rearrangements in patients |
| WO2015150900A2 (en) | 2014-03-31 | 2015-10-08 | Debiopharm International Sa | Fgfr fusions |
| WO2016030509A1 (en) | 2014-08-28 | 2016-03-03 | Oncoethix Gmbh | Methods of treating acute myeloid leukemia or acute lymphoid leukemia using pharmaceutical compositions containing thienotriazolodiazepine compounds |
| WO2016084883A1 (en) | 2014-11-26 | 2016-06-02 | 国立研究開発法人国立がん研究センター | Novel therapeutic target fusion gene in biliary tract cancer |
| WO2016105503A1 (en) | 2014-12-24 | 2016-06-30 | Genentech, Inc. | Therapeutic, diagnostic and prognostic methods for cancer of the bladder |
| WO2016139227A1 (en) | 2015-03-03 | 2016-09-09 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Fgfr3 antagonists |
| WO2022147246A1 (en)* | 2020-12-30 | 2022-07-07 | Tyra Biosciences, Inc. | Indazole compounds as kinase inhibitors |
Non-Patent Citations (122)
| Title |
|---|
| "Handbook of Clinical Drug Data", 2002, MCGRAW-HILL |
| "Principles of Drug Action", 1990, CHURCHILL LIVINGSTON |
| ANG ET AL., DIAGN. MOL. PATHOL., 24 February 2014 (2014-02-24) |
| ASTSATUROV ET AL., JOURNAL OF CLINICAL ONCOLOGY, vol. 34, no. 11012, 2016, pages iii93 |
| BABINATURNER, NAT REV CANCER, vol. 17, no. 5, 2017, pages 318 - 332 |
| BARNETT ET AL., HUM. MUTAT., vol. 37, no. 9, 2016, pages 955 - 63 |
| BAROY ET AL., PLOS ONE, vol. 11, no. 9, 2016, pages e0163859 |
| BASTURK ET AL., MOD PATHOL, vol. 30, no. 12, 2017, pages 1760 - 1772 |
| BELLUS ET AL., AM. J. MED. GENET., vol. 85, no. 1, 1999, pages 53 - 65 |
| BENNETT ET AL., AM. J. HUM. GENET., vol. 98, no. 3, 2016, pages 579 - 87 |
| BUNNEY ET AL., EBIOMEDICINE, vol. 2, no. 3, 2015, pages 194 - 204 |
| BUSSE ET AL., GENES CHROMOSOMES CANCER, vol. 56, no. 10, 2017, pages 730 - 749 |
| BYRON ET AL., NEOPLASIA, vol. 15, no. 8, 2013, pages 975 - 88 |
| CAZIER ET AL., NAT. COMMUN., vol. 5, 2014, pages 3756 |
| CHA ET AL., MO/ ONEAL, vol. 12, no. 7, 2018, pages 993 - 1003 |
| CHAKRABARTY ET AL., BRJ CANCER, vol. 1117, no. 11, 2017, pages 1592 - 1599 |
| CHANDRANI ET AL., ANN ONEAL, vol. 28, no. 3, 2017, pages 597 - 603 |
| CHELL ET AL., ONCOGENE, vol. 32, no. 25, 2013, pages 3059 - 70 |
| CHELLAIAH ET AL., J. BIOL. CHEM., vol. 269, 1994, pages 11620 - 11627 |
| CHEON ET AL., PROC. NATL. ACAD. SCI. U.S.A., vol. 91, 1994, pages 989 - 993 |
| CHESI ET AL., BLOOD, vol. 97, no. 3, 2001, pages 729 - 736 |
| DALIN ET AL., NAT COMMUN, vol. 8, no. 1, 2017, pages 1197 |
| DAVIES ET AL., J. CANCER RES., vol. 65, 2005, pages 7591 |
| DE MATTOS-ARRUDA ET AL., ONCOTARGET, vol. 9, no. 29, 2018, pages 20617 - 20630 |
| DIAS ET AL., EXP. MOL. PATHOL., vol. 101, no. 1, 2016, pages 116 - 23 |
| DODE ET AL., NAT GENET, vol. 33, no. 4, 2003, pages 463 - 465 |
| EEVA-MARIA LAITINEN ET AL., PLOS ONE, vol. 7, no. 6, 2012, pages e39450 |
| FERGUSON ET AL., J NEUROPATHOLEXP NEURAL, vol. 77, no. 6, 2018, pages 437 - 442 |
| FERNANDES ET AL., AM. J. MED. GENET. A., vol. 170A, no. 7, 2016, pages 1908 - 11 |
| GALLO ET AL., CYTOKINE GROWTH FACTOR REV, vol. 26, 2015, pages 425 - 449 |
| GALLO ET AL., CYTOKINE GROWTH FACTOR REV., vol. 26, 2015, pages 425 |
| GE ET AL., AM J CANCER RES, vol. 7, no. 7, 2017, pages 1540 - 1553 |
| GEELVINK ET AL., INT J MOL SCI, vol. 19, no. 9, 2018, pages E2548 |
| GONCALVES, FERTIL. STERIL., vol. 104, no. 5, 2015, pages 1261 - 7 |
| GONZALEZ-DEL ANGEL ET AL., AM J MED GENET A, vol. 176, no. 1, 2018, pages 161 - 166 |
| GRAND ET AL., GENES CHROMOSOMES CANCER, vol. 40, no. 1, 2004, pages 78 - 83 |
| GREENMAN ET AL., NATURE, vol. 446, no. 7132, 2007, pages 153 - 158 |
| GUYARD ET AL., RESPIR RES., vol. 18, no. 1, 2018, pages 120 |
| HALL ET AL., MOLECULAR CANCER THERAPEUTICS, vol. 14, no. 12, 2015 |
| HALL ET AL., PLOS ONE, vol. 11, no. 9, 2016, pages e1062594 |
| HART ET AL., ONCOGENE, vol. 19, no. 29, 2000, pages 3309 - 3320 |
| HAUGH ET AL., J INVEST DERMATOL, vol. 138, no. 2, 2018, pages 384 - 393 |
| HELSTEN ET AL., CLIN CANCER RES, vol. 22, no. 1, 2016, pages 259 - 267 |
| HOANG ET AL., SCI TRANSLMED., vol. 5, no. 197, pages 197 - 102 |
| IKEDA ET AL., ONCOLOGIST, vol. 23, no. 5, 2018, pages 586 - 593 |
| JIAO ET AL., NAT GENET, vol. 45, no. 12, 2013, pages 1470 - 1473 |
| JOHNSON ET AL., ADV. CANCER RES., vol. 60, 1993, pages 1 - 40 |
| JOHNSON ET AL., MOL. CELL. BIOL., vol. 11, 1995, pages 4627 - 4634 |
| JOHNSON ET AL., ONCOLOGIST, vol. 22, no. 12, 2017, pages 1478 - 1490 |
| JUSAKUL ET AL., CANCER DISCOV, vol. 7, no. 10, 2017, pages 1116 - 1135 |
| KANT ET AL., EUROJOURN ENDOCRINOL, vol. 172, no. 6, 2015, pages 763 - 770 |
| KARADIMAS ET AL., PRENAT DIAGN, vol. 26, no. 3, 2006, pages 258 - 261 |
| KAS ET AL., CANCER RES, vol. 78, no. 19, 2018, pages 5668 - 5679 |
| KASAIAN ET AL., BMC CANCER., vol. 15, 2015, pages 984 |
| KELLEHER ET AL., CARCINOGENESIS, vol. 34, 2013, pages 2198 |
| KIM ET AL., ANN ONEAL., vol. 28, no. 6, pages 1250 - 1259 |
| KIM ET AL., BMC UROL, vol. 18, 2018, pages 68 |
| KRSTEVSKA-KONSTANTINOVA ET AL., MED. ARCH., vol. 70, no. 2, 2016, pages 148 - 50 |
| KUENTZ ET AL., BR. J. DERMATOL., 2016 |
| KUMAR ET AL., AM JCLINPATHOL, vol. 143, no. 5, 2015, pages 738 - 748 |
| LAJEUNIE ET AL., EURJ HUM GENET, vol. 14, no. 3, 2006, pages 289 - 298 |
| LBRAHIMI ET AL., HUM MOL GENET, vol. 13, no. 19, 2004, pages 2313 - 2324 |
| LCHIYAMA ET AL., J. EUR. ACAD. DERMATOL. VENEREAL., vol. 30, no. 3, 2016, pages 442 - 5 |
| LEE ET AL., EXP THER MED, vol. 16, no. 2, 2018, pages 1343 - 1349 |
| LEGEAI-MALLET, ENDOCR. DEV., vol. 30, 2016, pages 98 - 105 |
| LEIDENG, INT J BIOL SCI, vol. 13, no. 9, 2017, pages 1163 - 1171 |
| LI ET AL., HUM. PATHOL., vol. 55, 2016, pages 143 - 50 |
| LIAO ET AL., CANCER RES, vol. 73, 2013, pages 5195 - 5205 |
| LIN ET AL., MOL. MED. REP., vol. 14, no. 3, 2016, pages 1941 - 6 |
| LIU ET AL., GENET. MOL. RES., vol. 13, 2014, pages 1109 |
| LO IACONO ET AL., ONCOTARGET., vol. 7, no. 12, 2016, pages 14394 - 404 |
| LOWERY ET AL., CLIN CANCER RES., vol. 23, no. 18, 2017, pages 5366 - 5373 |
| MARCHWICKA ET AL., CELL BIOSCI., vol. 6, no. 7, 2016 |
| MARTINCORENA ET AL., SCIENCE, vol. 348, 2015, pages 880 |
| MAZEN ET AL., SEX DEV., vol. 10, no. 1, 2016, pages 16 - 22 |
| MIKI ET AL., PROC. NATL. ACAD. SCI. U.S.A., vol. 89, 1992, pages 246 - 250 |
| MOEINI ET AL., CLIN. CANCER. RES., vol. 22, no. 2, 2016, pages 291 - 300 |
| NAGAHARA ET AL., CLIN. PEDIATR. ENDOCRINOL., vol. 25, no. 3, 2016, pages 103 - 106 |
| NGUYEN ET AL.: "Molecular Cancer Therapeutics", AACR-NCI-EORTC INTERNATIONAL CONFERENCE: MOLECULAR TARGETS AND CANCER THERAPEUTICS, vol. 14, no. 12, 2015 |
| OLIVEIRA ET AL., J EXP CLIN CANCER RES, vol. 37, no. 1, 2018, pages 84 |
| PEKMEZCI ET AL., ACTA NUROTAPHOL. COMMUN., vol. 6, no. 1, pages 47 |
| PELAEZ-GARDA ET AL., PLOS ONE, vol. 8, no. 5, 2013, pages e63695 |
| PREMOV ET AL., ONCOGENE, vol. 36, no. 22, 2017, pages 3168 - 3177 |
| REARDON ET AL., NATURE GENET, vol. 8, 1994, pages 275 - 279 |
| REESER ET AL., JMOLDIAGN, vol. 19, no. 5, 2017, pages 682 - 696 |
| REN ET AL., INT. J. CANCER, vol. 139, no. 4, 2016, pages 836 - 40 |
| REUTHER ET AL., JOURNAL OF MOLECULAR DIAGNOSTICS, vol. 17, no. 6, pages 813 |
| RIVERA ET AL., ACTA. NEUROPATHOL, vol. 131, no. 6, 2016, pages 847 - 63 |
| RON ET AL., AM. J. CASE REP., vol. 15, no. 17, 2016, pages 254 - 8 |
| ROSSEAU ET AL., NATURE, vol. 371, 1994, pages 252 - 254 |
| ROY ET AL., MODPATHOL., vol. 30, no. 8, 2017, pages 1133 - 1143 |
| RYLAND ET AL., J CLIN PATHOL., 14 May 2018 (2018-05-14) |
| RYLAND ET AL., J CLINPATHOL, 2018 |
| SARABIPOUR ET AL., J. MOL. BIOL., vol. 428, no. 20, 2016, pages 3903 - 3910 |
| SCHROCK ET AL., J THORAC. ONEAL., 2018 |
| SHI ET AL., J TRANSL MED., vol. 14, no. 1, 2016, pages 339 |
| SHIANG ET AL., CELL, vol. 76, 1994, pages 335 - 342 |
| SHIBATA ET AL., CANCER SCI, vol. 109, no. 5, 2018, pages 1282 - 1291 |
| SHIMADA ET AL., ONCOTARGET, vol. 8, no. 55, 2017, pages 93567 - 93579 |
| STONE ET AL., ACTA NEUROPATHOL, vol. 135, no. 1, 2017, pages 115 - 129 |
| SULLIVAN ET AL., JOURNAL OF CLINICAL ONCOLOGY, vol. 34, pages iii93 |
| TAKAGI, AM. J. MED. GENET. A., vol. 167A, no. 11, 2015, pages 2851 - 4 |
| TANIZAKI ET AL., CANCER RES., vol. 75, no. 15, pages 3149 - 3146 |
| TAURIN ET AL., INTL GYNECOL CANCER, vol. 28, no. 1, 2018, pages 152 - 160 |
| TAVORMINA ET AL., NATURE GENET, vol. 10, 1995, pages 165 - 172 |
| TAYLAN ET AL., J ALLERGY CLIN IMMUNOL, vol. 136, no. 2, 2015, pages 507 - 9 |
| THUSSBAS ET AL., J. CLIN. ONEAL., vol. 24, no. 23, 2006, pages 3747 - 55 |
| TRARBACH ET AL., J CLIN ENDOCRINOL METAB., vol. 91, no. 10, 2006, pages 4006 - 4012 |
| TRUDEL ET AL., BLOOD, vol. 107, 2006, pages 4039 |
| TURNERGROSE, NAT. REV. CANCER, vol. 10, no. 2, 2010, pages 116 - 129 |
| VAKIL ET AL.: "17th International Symposium on Pediatric Neuro-Oncology, Liverpool, United Kingdom", vol. 18, 2016, article "Neuro-Oncology", pages: iii93 |
| WANG ET AL., CANCER, vol. 123, no. 20, 2017, pages 3916 - 3924 |
| WELANDER ET AL., WORLD J SURG, vol. 42, no. 2, 2018, pages 482 - 489 |
| WILKIE ET AL., CURR. BIOL., vol. 5, 1995, pages 500 - 507 |
| WU ET AL., BMC CANCER, vol. 18, no. 1, 2018, pages 343 |
| YANG ET AL., AM J HUM GENET, vol. 98, no. 5, 2016, pages 843 - 856 |
| YANG ET AL., EBIOMEDICINE |
| YE ET AL., PLAST. RECONSTR. SURG., vol. 137, no. 3, 2016, pages 952 - 61 |
| YONG-XING ET AL., HUM. MOL. GENET., vol. 9, no. 13, 2000, pages 2001 - 2008 |
| YOZA ET AL., GENES CELLS., no. 10, 2016, pages 1049 - 1058 |
| ZHAO ET AL., INT. J. CLIN. EXP. MED., vol. 8, no. 10, 2015, pages 19241 - 9 |
| ZIMMER ET AL., J. BIOL. CHEM., vol. 268, 1993, pages 7899 - 7903 |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2025061029A1 (en)* | 2023-09-18 | 2025-03-27 | 3H Pharmaceuticals Co., Ltd. | Fgfr inhibitors and methods of use thereof |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US12071428B2 (en) | Indazole compounds as kinase inhibitors | |
| US11673893B2 (en) | CDK inhibitors and their use as pharmaceuticals | |
| US20240316022A1 (en) | Indazole compounds | |
| WO2021138391A1 (en) | Indazole compounds | |
| WO2024006897A1 (en) | Indazole compounds | |
| WO2024006883A1 (en) | Polymorphic compounds and uses thereof | |
| WO2024138112A1 (en) | Indazole compounds | |
| AU2022226671B2 (en) | Aminopyrimidine compounds and methods of their use | |
| US20230144528A1 (en) | CDK Inhibitors And Their Use As Pharmaceuticals | |
| CN116761798A (en) | Indazoles as kinase inhibitors | |
| WO2025129014A1 (en) | Indazole compounds for the treatment of cancer | |
| US12441707B2 (en) | Indazole compounds | |
| EA050408B1 (en) | INDAZOLE COMPOUND AS KINASE INHIBITORS | |
| US20250059175A1 (en) | Mutant Pi3k-Alpha Inhibitors And Their Use As Pharmaceuticals | |
| KR20240127909A (en) | Azolylpyridine pyridazinone amides as sos1 inhibitors |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | Ref document number:23853599 Country of ref document:EP Kind code of ref document:A1 | |
| NENP | Non-entry into the national phase | Ref country code:DE |